User login
Prevention and treatment of influenza in the primary care office
Every year, 5% to 20% of US residents contract the flu, 200,000 are hospitalized for it, and 36,000 die of influenza-related complications. The economic impact, including direct medical costs and lost earnings, exceeds $87 billion.1 Despite this, less than half of eligible US residents were vaccinated in the 2012–2013 season, with uninsured people more than twice as likely to forgo vaccination.2,3
Several studies have shown that influenza vaccination reduces the need for outpatient encounters and hospitalizations and lowers the incidence of death from acute myocardial infarction, the rate of all-cause mortality, and even the incidence of therapies administered by implantable defibrillators.4–6 In the 2012–2013 influenza season, vaccination prevented an estimated 3.2 million medically attended illnesses and almost 80,000 hospitalizations; 70% of hospitalizations prevented were in children age 6 months to 4 years and in adults over age 65.7
After the 2009 H1N1 pandemic, which disproportionately killed previously healthy adults, the US Centers for Disease Control and Prevention (CDC) expanded its vaccination recommendations to include everyone above the age of 6 months, with few contraindications.8
In addition, recent years have seen a great expansion in vaccine options, changes in the at-risk demographics, and continued widespread resistance to certain antiviral agents, with implications for practice in primary care.
Here, we review the barriers and the new options for treatment and prevention of influenza.
HEMAGGLUTININ AND NEURAMINIDASE
Influenza infection is caused by one of the circulating strains of influenza virus A or B.
The major viral surface glycoproteins are hemagglutinin and neuraminidase. Hemagglutinin plays an important role in viral attachment to host cells and is the major immunogen in the influenza vaccine. Neuraminidase contains an active enzymatic site that cleaves the newly formed budding influenza viruses from host-cell sialic acid residues and allows them to be released from the cell membrane to infect other respiratory epithelial cells. It is the target of currently recommended antiviral drugs.
VACCINE PRODUCTION
Throughout the year, 130 influenza centers around the world sample circulating strains and share their data with five World Health Organization (WHO) Collaborating Centers for Reference and Research on Influenza. The WHO analyzes the circulation patterns, predicts the strains most likely to be circulating in the next influenza season, and shares these strains with manufacturers of the vaccine.
Pharmaceutical companies then begin an elaborate process of producing and distributing hundreds of millions of doses of vaccine worldwide. The production traditionally uses millions of fertilized chicken eggs to produce strain-specific influenza hemagglutinin. Individual vaccine strains are combined into the final product after being inactivated by chemical or physical splitting of the viral envelope with or without subsequent purification of the hemagglutinin particles.
Before 2013, the WHO’s yearly recommendations included two strains of influenza A and a single strain of influenza B. In 2013, new quadrivalent vaccines that include protection against a second strain of influenza B were approved.
The WHO strain-selection process allows manufacturers about 6 months to produce the vaccine. In a typical year, the worldwide demand is about 400 million doses. The theoretical maximal annual worldwide capacity, given current techniques, is fewer than 1 billion doses, which is well short of the 10 billion doses necessary to allow for the double vaccination needed in a pandemic.9 Newly approved recombinant manufacturing techniques offer greater production efficiency, while novel methods of intradermal administration increase vaccine immunogenicity, decreasing the amount of viral antigens used per dose.
INACTIVATED VS LIVE-ATTENUATED
In addition to intramuscular inactivated influenza vaccine, a live-attenuated vaccine in the form of an intranasal spray (FluMist) became available in 2003. This form is generally favored in children, as it avoids the discomfort of an injection. It contains live, weakened, cold-adapted influenza strains that reproduce in the relatively colder temperatures of the exterior nares but cannot survive in the warmer temperatures of the lung and proximal airways. It is approved for healthy people 2 to 49 years of age, and some evidence suggests that it may be more effective than inactivated influenza vaccine in children,10 although its utility is limited by multiple contraindications (see below).
INFLUENZA VACCINE INDICATIONS AND CONTRAINDICATIONS
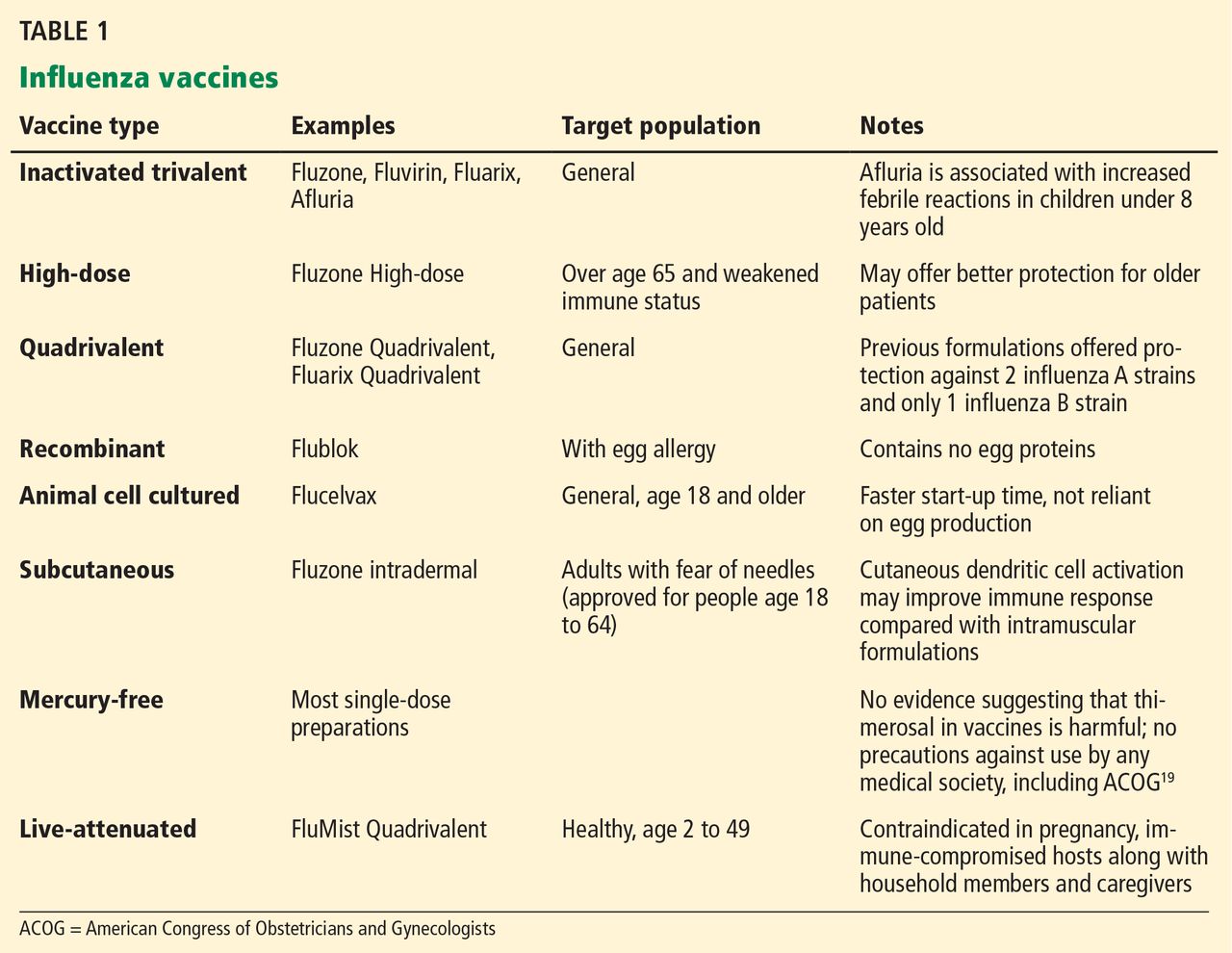
Vaccination for influenza is recommended for all persons 6 months of age and older, an expansion from pre-2009 guidelines that did not recommend vaccination for healthy adults age 19 to 49 who were not in contact with people at high risk of influenza-related complications.8 Many new vaccine formulations have become available in recent years, each with specific benefits, risks, and target populations (Table 1).
Contraindications to inactivated vaccine
The only firm contraindication to inactivated influenza vaccine is previous severe allergic reaction to influenza vaccine or any of its components. Those with moderate to severe acute illness are advised to wait until their condition improves before being vaccinated. People who have had Guillain-Barré syndrome and those with egg allergy are discussed in MISAPPREHENSIONS THAT POSE BARRIERS TO VACCINATION, below. There is no risk of influenza infection from inactivated influenza vaccine.
Contraindications to live-attenuated influenza vaccine
Unlike inactivated influenza vaccine, the live-attenuated vaccine does result in shedding of vaccine-strain virus from the vaccinated host, with the theoretical potential for transmission of the virus from the vaccine recipient to other people, as well as the potential for influenza-like illness in vaccine recipients.11,12 Based on reported events, the former is estimated to occur in 10 to 20 per 1 million vaccinations, although these cases have never been proven to be caused by a cold-adapted vaccine-strain rather than by coincidental transmission of circulating wild-type viral strains.13
Despite this exceedingly small risk of viral transmission, live-attenuated influenza vaccine has multiple contraindications, including age less than 2 years and more than 49 years, disease- or drug-related compromised immune status, pregnancy, egg allergy, and history of allergic reaction to the formulation. These limit its use and are important to review in detail before prescribing.14
Use of neuraminidase inhibitors within 2 days before or 2 weeks after receiving live-attenuated influenza vaccine may interfere with replication of the cold-adapted strain and decrease the vaccine’s effectiveness.14
EFFECTIVENESS OF INFLUENZA VACCINATION IN OLDER ADULTS
The effectiveness of influenza vaccination depends on the age and health status of the person being vaccinated, as well as on the quality of the match between the vaccine and the circulating influenza viruses.
In the 2012–2013 season, the adjusted vaccine effectiveness was 56% overall, 47% for influenza A H3N2, and 67% for influenza B. However, in people age 65 and older, the overall adjusted vaccine effectiveness was 27%, and only 9% for influenza A H3N2.15 Thus, even though the vaccine-virus match was considered good, the vaccine was suboptimally effective in the older group. This may be an argument for using the recently approved high-dose vaccine in that age group. Although the high-dose vaccine has been shown to be significantly more immunogenic in older adults, it is too early to know if it is clinically more effective in preventing influenza in this age group.
Despite the lower-than-expected effectiveness in preventing influenza in the 2012–2013 season in people age 65 and older, several well-designed studies found that influenza vaccination prevented severe disease, including one study that found vaccination to be 89% effective in reducing influenza-associated hospitalizations in the 2010–2011 flu season.4,16

The limited effectiveness of vaccination in the older age group reminds us of the importance of early recognition and treatment of patients at high risk of influenza-related complications (see Table 2). It is also a call for greater compliance with vaccination in younger people, with a goal of achieving the 80% vaccination rate that has been calculated as adequate to achieve herd immunity.17
MISAPPREHENSIONS THAT POSE BARRIERS TO VACCINATION
Concern about potential adverse effects is the most common reason for refusing influenza vaccination, even among health care workers.18 However, the only commonly encountered adverse effect of the intramuscular inactivated influenza vaccine is injection-site pain.
‘Catching the flu from a flu shot’
Many people think that they can “catch the flu from a flu shot” (or think that they actually did), but vaccine-acquired influenza is not possible with the inactivated influenza vaccine,19 and it is only a theoretical, undocumented consideration with the live-attenuated vaccine.
Various respiratory viruses other than influenza also cause viral upper-respiratory infections during the influenza season. These infections may coincide with influenza vaccination and are frequently misconstrued as a side effect of the influenza vaccine or as evidence of vaccine ineffectiveness.
Unnecessary concerns about simultaneous vaccinations
Patients and doctors are often concerned about simultaneous administration of multiple vaccines and choose to spread out indicated vaccinations over multiple visits. This practice increases patients’ risk of illness from vaccine-preventable diseases. Research shows that simultaneous administration does not alter the safety or effectiveness of vaccination.20–22 The CDC recommends simultaneous administration of all indicated live and inactivated vaccinations in order to reduce barriers to vaccination.20
Fear of Guillain-Barré syndrome
Guillain-Barré syndrome, an acute ascending polyneuropathy, has been blamed on influenza vaccination in cases that developed after the 1976 influenza A (H1N1) epidemic.
Most cases are self-limiting but require intensive treatment and supportive care. Full recovery occurs in 60% of cases, though some people experience persistent symptoms. The mortality rate is less than 5%.23
After the 1976 influenza pandemic, approximately 400 cases of Guillain-Barré syndrome arose in 45 million vaccine recipients, or about 1 case per 100,000 people.24 Multiple subsequent population analyses concluded that the actual incidence of Guillain-Barré syndrome attributable to influenza vaccination is negligible, at less than 1 case in 1 million vaccinations. Against this, we should compare the real risk of illness and death from influenza infection, which itself is a risk factor for Guillain-Barré syndrome.25
Should a person with a history of Guillain-Barré syndrome be revaccinated against influenza? The risk was evaluated in a large retrospective analysis of cases identified in the Kaiser Permanente Northern California Database from 1995 to 2006.26 Five hundred fifty cases of Guillain-Barré syndrome were identified, of which 18 had arisen within 6 weeks of the patient receiving a flu shot. Four hundred five doses of inactivated influenza vaccine were subsequently given to 105 patients who had a history of Guillain-Barré syndrome, two of whom had developed the syndrome within 6 weeks of receiving the shot. There were no documented episodes of recurrent Guillain-Barré syndrome in any of these patients. Only 6 of 550 patients with a history of the disease developed it again; none of these 6 had received the influenza vaccine in the preceding 2 months, and only 1 had been exposed to the measles-mumps-rubella vaccine in the 4 months before vaccination.
Nevertheless, expert opinion recommends lifelong avoidance of any immunization that had been given within 6 weeks before the onset of symptoms of Guillain-Barré syndrome.27
Overstated concern about egg allergy
Anaphylactic reactions can occur after influenza vaccination in people who have severe egg allergy, and concern about these reactions unfortunately prevents many otherwise eligible people with mild allergy from being vaccinated.
These reactions are much less common than feared. In a well-designed prospective cohort study of 367 patients with a history of egg allergy and positive skin-prick tests, including 132 with a history of severe allergy and 4 with a history of mild allergic symptoms arising in response to previous influenza vaccinations, none developed anaphylaxis.28
The same authors reviewed 26 studies in more than 4,000 egg-allergic patients, of whom more than 500 had a history of severe egg-associated reactions, and likewise found no cases of influenza vaccine-associated anaphylaxis. They concluded that the inactivated influenza vaccine is safer than the egg-derived mumps-measles-rubella vaccine, for which precautions for egg allergy no longer exist.28
People with a history of more serious reactions, ranging from stomach upset to anaphylaxis, can be safely vaccinated with a recombinant vaccine or referred to an allergist for further testing. People who experience hives as their only reaction to egg exposure should receive full-dose vaccination but then be observed for a half hour afterward.
The recombinant trivalent influenza vaccine Flublok was approved in 2013 for people age 18 to 49. It is the first commercially available influenza vaccine produced in a continuous insect cell line using a baculovirus vector. No eggs are used in its production, and it is approved for use in patients with egg allergy of any severity.
People who have a history of more serious reactions, including abdominal pain, nausea, vomiting, dizziness, or wheezing can be vaccinated with the recombinant vaccine or referred to an allergy specialist.
Despite this new option, understanding of alternative immunization guidelines for people with egg allergies, available on the CDC website29 remains important, as the availability of the recombinant trivalent influenza vaccine remains limited in the 2013–2014 influenza season.
Misconception about mercury toxicity
Thimerosal is an ethylmercury-containing preservative used in multidose antiviral vaccines, including some influenza vaccines.30 It is designed to prevent bacterial and fungal colonization of the vaccine vial while not reducing vaccine effectiveness or causing toxicity.
Contemporary understanding of mercury neurotoxicity is based largely on studies of methylmercury, including long-term, low-dose exposure in remote communities in the Faroe Islands and the Seychelles through regular consumption of fish and whale meat.31,32 These exposure studies had conflicting results: those in the Faroe Islands demonstrated toxicity, but the Seychelles studies actually showed better neurologic test scores at higher mercury levels, a trend the authors attributed to the beneficial effects of maternal fish consumption.
The results of the methylmercury studies have been extrapolated to ethylmercury (contained in thimerosal), although the two chemicals have vastly different pharmacologic properties. For example, methylmercury has a longer half-life and greater transport across the blood-brain barrier.33 A direct comparison found that ethylmercury is less toxic than methylmercury, although an increase in ethylmercury concentration of only 20% resulted in similar toxicity profiles.34 These studies were performed at concentrations of mercury thousands of times higher than those resulting from vaccination: nearly 150,000 times greater than those in an average adult or 15,000 times greater than those in a 1-year-old child from the typical 25-μg thimerosal dose allowed in contemporary influenza vaccines.
Despite much negative publicity, no link has been shown between thimerosal and autism.30 Multiple regulatory, scientific, and medical organizations including the US Food and Drug Administration (FDA), the WHO, the National Institutes of Health, the CDC, the American Academy of Pediatrics, and the American Congress of Obstetricians and Gynecologists (ACOG) have evaluated the data on the safety of thimerosal in vaccines and have agreed that it is safe. However, most of them urged vaccine manufacturers to eliminate mercury from vaccines as a precaution.30,35 Thimerosal has subsequently been eliminated from all childhood vaccines except for influenza vaccine, with no resulting decrease in childhood autism diagnoses.36
Considering that no harm from thimerosal at FDA-approved doses has been documented, and considering the real risk of influenza-related complications, particularly in young children and pregnant women, we recommend vaccination using whatever vaccine formulation is locally available for all patients, including children age 6 months and older and pregnant women. Nevertheless, given that mercury is being eliminated from childhood vaccines and that preservative-free single-dose vials are increasingly available in the United States, it seems reasonable to use thimerosal-free formulations for children, expectant mothers, and patients concerned about exposure if these formulations are readily available. Influenza vaccination should not be delayed if a thimerosal-free formulation is not readily available.
NEW VACCINE FORMULATIONS
Recent years have seen a dramatic expansion in influenza vaccine options (Table 1).
Quadrivalent vaccines
Quadrivalent vaccines protect against two strains of influenza A and two strains of influenza B, whereas earlier formulations included only one influenza B strain. Vaccination against either influenza B strain offers only limited cross-protection against the other B strain, and previous formulations involved assumptions about which strain would predominate in any given year. The CDC estimates that switching to quadrivalent vaccines will prevent up to 970,000 cases of influenza, 8,200 hospitalizations, and 485 deaths per year.37
Intradermal vaccine
The newly available Fluzone Intradermal vaccine contains smaller doses of hemagglutinin but is still effective because antigen-presenting dendritic cells in the skin reduce the required amount of vaccine antigen necessary for inducing protection.38 This may provide an advantage in the event of vaccine shortage. Also, since it is given in needles only 1.5 mm long, it may appeal to people who are afraid of needles.
The stronger immune reaction with intradermal administration causes more redness, induration, and tenderness at the injection site than with intramuscular administration.39 Patients should not be surprised by this reaction and can be advised to apply ice packs for symptomatic relief.
High-dose vaccine
A high-dose vaccine was approved in 2009 for use in adults age 65 and older. It contains 60 μg of hemagglutinin, compared with 15 μg in standard-dose vaccines, and has been shown to improve seroconversion rates. It remains to be seen if this translates into better clinical outcomes in older adults.40 Further studies will be necessary before we can recommend high-dose vaccines to other people with weakened immune response, such as those undergoing chemotherapy or those infected with human immunodeficiency virus (HIV).
Cell-based vaccines
Flucelvax was the first cell-based influenza vaccine. However, unlike the recombinant trivalent influenza vaccine, which uses no eggs in its manufacturing process, Flucelvax production starts with egg-derived influenza strains that are subsequently propagated in liquid culture of animal cells. It may therefore contain traces of egg protein, and it has not been studied in people with egg allergy.41
An advantage of the cell-based production technique is the use of fewer or no eggs at all, which may result in greater manufacturing efficiency. Also, it is a closed process that reduces the risk of bacterial contamination as well as reliance on antibiotics or preservatives, such as thimerosal, in the manufacturing process.42
CHEMOPROPHYLAXIS WITH NEURAMINIDASE INHIBITORS
The mainstays of influenza prevention are seasonal vaccination and appropriate infection-prevention practices. In addition, in patients at high risk of influenza-related complications (Table 2),43 postexposure chemoprophylaxis with a neuraminidase inhibitor, ie, oseltamivir (Tamiflu) or zanamivir (Relenza), is an effective preventive strategy, especially in years when the match between vaccine and circulating virus strains is suboptimal.44,45
Neuraminidase inhibitors are competitive inhibitors of the active site of the influenza glycoprotein neuraminidase, responsible for viral release from infected respiratory epithelial cells. Rates of resistance to neuraminidase inhibitors have been less than 1% in the United States in recent years, while resistance to the adamantanes amantadine (Symmetrel) and rimantadine (Flumadine) can be as high as 92%, depending on the virus isolate. Thus, their use for treatment or prophylaxis of influenza is not currently recommended by the CDC.46
Chemoprophylaxis with any agent may promote emergence of resistant strains, can cause adverse reactions, and should never be considered a substitute for vaccination.
ANTI-INFLUENZA AGENTS
Two neuraminidase inhibitors, oseltamivir and zanamivir, are approved by the FDA for preventing and treating uncomplicated influenza. Treatment must be instituted within 2 days of onset of symptoms to be effective.
Oseltamivir is available as an oral capsule or powder for liquid suspension. Its most common adverse effects are gastrointestinal upset including diarrhea, nausea, and vomiting.44
Zanamivir is only available in the form of a dry powder inhaler because of the drug’s poor oral bioavailability, and only 4% to 17% of the inhaled dose is systemically absorbed.45 There is a theoretical benefit in targeted delivery of zanamivir to the primary organ affected by influenza, and gastrointestinal side effects are less common with this drug.44,45 Unfortunately, the zanamivir inhaler requires complicated assembly and dexterity for administration (see the video on YouTube47), which may make it unreliable in certain patient groups, especially handicapped and elderly patients. Administration has been associated with bronchospasm, resulting in a more than 20% reduction in the forced expiratory volume in 1 second, and it is contraindicated in patients with underlying reactive airway disease such as chronic obstructive pulmonary disease or asthma.45

Table 3 lists the doses and duration of therapy for oseltamivir and zanamivir in adults with normal renal function, as well as approximate costs. No generic formulations of neuraminidase inhibitors are currently available, and outpatient use may not be covered by medical insurance. Several other neuraminidase inhibitors are either under development or at various stages in the FDA approval process.
EFFECTIVENESS OF ANTI-INFLUENZA DRUGS
Treatment with oseltamivir has been shown to reduce the duration of symptoms by approximately 1 day if initiated within 36 hours of onset of illness and 1.5 to 2 days if initiated within 24 hours.48,49 Trials and meta-analyses of zanamivir show similar effectiveness, though some suggest that symptoms were alleviated as much as 3 days sooner than in controls in a subgroup of patients who were febrile at presentation.50,51 Dual neuraminidase inhibitor therapy in an attempt to prevent emergence of resistance seems logical but was actually found to be less effective than monotherapy, according to a 2010 study.52
The effectiveness of neuraminidase inhibitors in reducing influenza-related complications and mortality rates has been controversial in recent years, as these outcomes were not addressed in initial studies that secured FDA approval. Several meta-analyses differ in their assessments of available data quality and conclusions. A 2009 Cochrane review questioned the completeness and the veracity of the data from manufacturer-funded trial data, much of which was unpublished and not made available to reviewers, and it concluded that a reduction of complications could not be supported by the available data.53 Hernán and Lipsitch,54 in a 2011 review, calculated that oseltamivir reduces the risk of lower respiratory tract complications by 28% in patients with influenza-like symptoms and by 37% in patients with confirmed influenza infection.
Additional trials and better access to available data are needed to settle the question of the effectiveness of neuraminidase inhibitors in reducing complications of influenza. Meanwhile, they remain strongly recommended by major health organizations, including the CDC and the WHO, which lists oseltamivir on its “model list of essential medicines.”
VIRAL RESISTANCE TO NEURAMINIDASE INHIBITORS
Viral resistance to neuraminidase inhibitors occurs through multiple mechanisms and may arise without selective pressure from exposure to these drugs.55
Oseltamivir possesses a hydrophobic moiety that requires viral neuraminidase to undergo a complex reconfiguration to expose the active site prior to binding. Any mutation affecting its ability to undergo this structural rearrangement can promote resistance by decreased oseltamivir access to the active site.
Zanamivir has a structural homology to the neuraminidase active site and requires no such reconfiguration. Additionally, mutations promoting resistance to zanamivir may actually decrease viral fitness; thus, resistance to zanamivir is significantly less common than to oseltamivir.55
About 2,000 influenza virus isolates currently circulating in the United States were tested for resistance; only 1% of the 2009 influenza A H1N1 isolates demonstrated resistance to oseltamivir, and none to zanamivir.56
The CDC regularly updates the resistance patterns of circulating influenza strains at www.cdc.gov/flu/weekly/index.htm.
SPECIAL CONSIDERATIONS
Pregnancy
Pregnant women may be at higher risk of severe influenza complications. This was especially true during the 2009 H1N1 pandemic, when pregnant women had a five times higher risk of death from influenza-related complications. Additionally, fever during pregnancy is an independent risk factor for adverse outcomes in the offspring.57 Maternal vaccination against influenza effectively protects the infant for the first 6 months of life, when vaccination is not recommended because of a poor immune response.58
Live-attenuated influenza vaccine is contraindicated during pregnancy. Given the documented risks to the mother from influenza and no documented harm from preservatives in multiuse vaccine vials, the Advisory Committee on Immunization Practices (ACIP) and ACOG do not state a preference for thimerosal-containing or thimerosal-free vaccine for any group, including pregnant women. Pregnant women should be vaccinated with whatever inactivated influenza vaccine formulation is available at the earliest opportunity in the beginning of the influenza season, regardless of the trimester of pregnancy.
Pregnant women are at high risk of influenza-related complications and should be considered for postexposure antiviral prophylaxis or early treatment with a neuraminidase inhibitor. However, both of the approved neuraminidase inhibitors are in pregnancy safety category C, indicating possible adverse effects in animal studies and a lack of safety data in pregnant humans. As with all category C medications, the risks and benefits must be considered, taking into account maternal comorbidities, vaccination status, effectiveness of the season’s influenza vaccine, and the virulence of circulating influenza strains.
As oseltamivir is associated with nausea and gastrointestinal side effects and as zanamivir has less systemic absorption, it may be reasonable to prescribe zanamivir for women already experiencing severe pregnancy-related nausea.
Immunocompromised people
Inactivated influenza vaccine is recommended and live-attenuated influenza vaccine is contraindicated for all immunocompromised people. Generally speaking, any form of immune compromise will decrease the immunogenicity of the vaccine. Additional considerations vary depending on the cause and severity of the immunocompromised status.
HIV-infected patients have higher seroconversion rates when vaccinated with the high-dose vaccine than with the standard-dose vaccine; however, as in adults over age 65, the clinical benefit has yet to be evaluated.59 The efficacy of vaccination is predictably related to the CD4 cell count, as T cells are necessary to mount a response.60 No documented benefit is gained from booster influenza vaccination in this group of patients.
Cancer patients should receive inactivated influenza vaccine every year. Postexposure chemoprophylaxis should be considered, and early treatment with a neuraminidase inhibitor is recommended in patients undergoing chemotherapy.
Solid-organ transplant recipients face a risk of organ rejection if they contract influenza infection, in addition to a higher risk of influenza-related complications.61 Transplant recipients should receive inactivated influenza vaccine as soon as it becomes available at the beginning of every influenza season. Additional research is necessary to evaluate the safety and effectiveness of the high-dose influenza vaccine in this patient group.
MORE OPTIONS, GREAT BENEFIT
Influenza remains a significant source of morbidity and mortality in the United States, and emerging pandemic strains as well as the aging population pose the risk of increased disease burden. New vaccine options offer hope of greater safety, improved efficacy, and higher vaccination rates though broader appeal to individuals. The actual differences in protection between various vaccine options are insignificant relative to the overall benefit of vaccination.
Health care providers should inquire about patients’ understanding and address their concerns about vaccination. Giving an available influenza vaccine within approved indications should not be delayed if alternative vaccine options are not readily available.
In addition to vaccination, patients at high risk of complications should be advised early in the influenza season to inform their doctors about potential exposure to influenza or the development of flu-like symptoms for consideration of early treatment or postexposure prophylaxis with a neuraminidase inhibitor.
- Molinari NA, Ortega-Sanchez IR, Messonnier ML, et al. The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine 2007; 25:5086–5096.
- Soni A. Influenza Immunization Rates for Selected at Risk Populations among the US Adult Civilian Noninstitutionalized Population, 2006. Statistical Brief #226. December 2008. Agency for Healthcare Research and Quality, Rockville, MD. http://meps.ahrq.gov/data_files/publications/st226/stat226.pdf. Accessed January 31, 2014.
- Centers for Disease Control and Prevention (CDC). Flu vaccination coverage, United States, 2012–13 Influenza Season. http://www.cdc.gov/flu/fluvaxview/coverage-1213estimates.htm - age-group-adults. Accessed January 31, 2014.
- Castilla J, Godoy P, Domínguez A, et al; CIBERESP Cases and Controls in Influenza Working Group Spain. Influenza vaccine effectiveness in preventing outpatient, inpatient, and severe cases of laboratory-confirmed influenza. Clin Infect Dis 2013; 57:167–175.
- Talbot HK, Zhu Y, Chen Q, Williams JV, Thompson MG, Griffin MR. Effectiveness of influenza vaccine for preventing laboratory-confirmed influenza hospitalizations in adults, 2011–2012 influenza season. Clin Infect Dis 2013; 56:1774–1777.
- Udell JA, Zawi R, Bhatt DL, et al. Association between influenza vaccination and cardiovascular outcomes in high-risk patients: a meta-analysis. JAMA 2013; 310:1711–1720.
- Centers for Disease Control and Prevention (CDC). Estimated influenza illnesses and hospitalizations averted by influenza vaccination—United States, 2012–13 influenza season. MMWR Morb Mortal Wkly Rep 2013; 62:997–1000.
- Centers for Disease Control and Prevention (CDC). Prevention and control of seasonal influenza with vaccines. Recommendations of the Advisory Committee on Immunization Practices—United States, 2013–2014. MMWR Recomm Rep 2013; 62:1–43.
- Friede M. Snapshot of influenza vaccine manufacturing capacity worldwide and summary of WHO-HHS activities to promote technology transfer. World Health Organization Global Action Plan for Influenza II Meeting 2011. www.who.int/phi/Session1B_Current_Manufacturing_Capacity_Worldwide_Friede.pdf. Accessed February 5, 2014.
- Ashkenazi S, Vertruyen A, Arístegui J, et al., CAIV-T Study Group. Superior relative efficacy of live attenuated influenza vaccine compared with inactivated influenza vaccine in young children with recurrent respiratory tract infections. Pediatr Infect Dis J 2006; 25:870–879.
- Izurieta HS, Haber P, Wise RP, et al. Adverse events reported following live, cold-adapted, intranasal influenza vaccine. JAMA 2005; 294:2720–2725.
- Vesikari T, Karvonen A, Korhonen T, et al; CAIV-T Transmission Study Group. A randomized, double-blind study of the safety, transmissibility and phenotypic and genotypic stability of cold-adapted influenza virus vaccine. Pediatr Infect Dis J 2006; 25:590–595.
- Kamboj M, Sepkowitz KA. Risk of transmission associated with live attenuated vaccines given to healthy persons caring for or residing with an immunocompromised patient. Infect Control Hosp Epidemiol 2007; 28:702–707.
- Centers for Disease Control and Prevention (CDC). Live Attenuated Influenza Vaccine [LAIV] (The Nasal Spray Flu Vaccine). http://www.cdc.gov/flu/about/qa/nasalspray.htm. Accessed February 3, 2014.
- Centers for Disease Control and Prevention (CDC). Interim adjusted estimates of seasonal influenza vaccine effectiveness—United States, February 2013. MMWR Morb Mortal Wkly Rep 2013; 62:119–123.
- Voordouw AC, Sturkenboom MC, Dieleman JP, et al. Annual revaccination against influenza and mortality risk in community-dwelling elderly persons. JAMA 2004; 292:2089–2095.
- Plans-Rubió P. The vaccination coverage required to establish herd immunity against influenza viruses. Prev Med 2012; 55:72–77.
- Aziz NA, Muhamad S, Manaf MR, Hamid MZ. Factors Influencing H1N1 vaccination among primary health care workers: a cross-sectional study. Int J Prev Med 2013; 4:664–670.
- Nichol KL, Margolis KL, Lind A, et al. Side effects associated with influenza vaccination in healthy working adults. A randomized, placebo-controlled trial. Arch Intern Med 1996; 156:1546–1550.
- National Center for Immunization and Respiratory Diseases. General recommendations on immunization—recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2011; 60( 2):1–64.
- Tseng HF, Smith N, Sy LS, Jacobsen LJ. Evaluation of the incidence of herpes zoster after concomitant administration of zoster vaccine and polysaccharide pneumococcal vaccine. Vaccine 2011; 29:3628–3632.
- Offit PA, Quarles J, Gerber MA, et al. Addressing parents’ concerns: do multiple vaccines overwhelm or weaken the infant’s immune system? Pediatrics 2002; 109:124–129.
- Rajabally YA, Uncini A. Outcome and its predictors in Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry 2012; 83:711–718.
- Schonberger LB, Bregman DJ, Sullivan-Bolyai JZ, et al. Guillain-Barré syndrome following vaccination in the National Influenza Immunization Program, United States, 1976—1977. Am J Epidemiol 1979; 110:105–123.
- Lehmann HC, Hartung HP, Kieseier BC, Hughes RA. Guillain-Barré syndrome after exposure to influenza virus. Lancet Infect Dis 2010; 10:643–651.
- Baxter R, Lewis N, Bakshi N, Vellozzi C, Klein NP, Network C. Recurrent Guillain-Barré syndrome following vaccination. Clin Infect Dis 2012; 54:800–804.
- Hughes RA, Wijdicks EF, Benson E, et al. Supportive care for patients with Guillain-Barré syndrome. Arch Neurol 2005; 62:1194–1198.
- Des Roches A, Paradis L, Gagnon R, et al. Egg-allergic patients can be safely vaccinated against influenza. J Allergy Clin Immunol 2012; 130:1213–1216.e1.
- US Centers for Disease Control and Prevention. Influenza vaccination of people with a history of egg allergy. www.immunize.org/catg.d/p3094.pdf. Accessed February 3, 2014.
- US Food Drug Administration. Thimerosal in vaccines. www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/VaccineSafety/UCM096228. Accessed February 3, 2014.
- Davidson PW, Kost J, Myers GJ, Cox C, Clarkson TW, Shamlaye CF. Methylmercury and neurodevelopment: reanalysis of the Seychelles Child Development Study outcomes at 66 months of age. JAMA 2001; 285:1291–1293.
- Grandjean P, Weihe P, White RF, et al. Cognitive deficit in 7-year-old children with prenatal exposure to methylmercury. Neurotoxicol Teratol 1997; 19:417–428.
- Nelson KB, Bauman ML. Thimerosal and autism? Pediatrics 2003; 111:674–679.
- Magos L, Brown AW, Sparrow S, Bailey E, Snowden RT, Skipp WR. The comparative toxicology of ethyl- and methylmercury. Arch Toxicol 1985; 57:260–267.
- American Congress of Obstetricians and Gynecologists. Influenza vaccination during pregnancy. www.acog.org/Resources_And_Publications/Committee_Opinions/Committee_on_Obstetric_Practice/Influenza_Vaccination_During_Pregnancy. Accessed February 3, 2014.
- US Centers for Disease Control and Prevention. Understanding thimerosal, mercury, and vaccine safety. www.cdc.gov/vaccines/hcp/patient-ed/conversations/downloads/vacsafe-thimerosal-color-office.pdf. Accessed February 3, 2014.
- Reed C, Meltzer MI, Finelli L, Fiore A. Public health impact of including two lineages of influenza B in a quadrivalent seasonal influenza vaccine. Vaccine 2012; 30:1993–1998.
- Tsang P, Gorse GJ, Strout CB, et al. Immunogenicity and safety of Fluzone intradermal and high-dose influenza vaccines in older adults ≥65 years of age: a randomized, controlled, phase II trial. Vaccine 2013. doi: 10.1016/j.vaccine.2013.09.074. [Epub ahead of print]
- Sanofi Pasteur. Fluzone package insert. www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM305080.pdf. Accessed February 3, 2014.
- Falsey AR, Treanor JJ, Tornieporth N, Capellan J, Gorse GJ. Randomized, double-blind controlled phase 3 trial comparing the immunogenicity of high-dose and standard-dose influenza vaccine in adults 65 years of age and older. J Infect Dis 2009; 200:172–180.
- US Food Drug Administration. Flucelvax FDA application. www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM332069.pdf. Accessed February 3, 2014.
- Novartis. Flucelvax (influenza virus vaccine) fact sheet. www.novartis-vaccines.com/downloads/flucelvax/Flucelvax_Fact_Sheet.pdf. Accessed February 3, 2014.
- US Centers for Disease Control and Prevention. People at high risk for developing flu-related complications. www.cdc.gov/flu/about/disease/high_risk.htm. Accessed February 3, 2014.
- Roche Pharmaceuticals. Tamiflu package insert. http://www.gene.com/download/pdf/tamiflu_prescribing.pdf. Accessed February 3, 2014.
- GlaxoSmithKline. Relenza package insert. http://us.gsk.com/products/assets/us_relenza.pdf. Accessed February 3, 2014.
- Fiore AE, Fry A, Shay D, et al. Antiviral agents for the treatment and chemoprophylaxis of influenza—recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2011; 60:1–24.
- Administration technique for zanamivir (Relenza) Diskhaler. YouTube. 2009. www.youtube.com/watch?v=sQI0a0ToSPo. Accessed February 6, 2014.
- Nicholson KG, Aoki FY, Osterhaus AD, et al. Efficacy and safety of oseltamivir in treatment of acute influenza: a randomised controlled trial. Neuraminidase Inhibitor Flu Treatment Investigator Group. Lancet 2000; 355:1845–1850.
- Treanor JJ, Hayden FG, Vrooman PS, et al. Efficacy and safety of the oral neuraminidase inhibitor oseltamivir in treating acute influenza: a randomized controlled trial. US Oral Neuraminidase Study Group. JAMA 2000; 283:1016–1624.
- Cooper NJ, Sutton AJ, Abrams KR, Wailoo A, Turner D, Nicholson KG. Effectiveness of neuraminidase inhibitors in treatment and prevention of influenza A and B: systematic review and meta-analyses of randomised controlled trials. BMJ 2003; 326:1235.
- Hayden FG, Osterhaus AD, Treanor JJ, et al. Efficacy and safety of the neuraminidase inhibitor zanamivir in the treatment of influenzavirus infections. GG167 Influenza Study Group. N Engl J Med 1997; 337:874–880.
- Duval X, van der Werf S, Blanchon T, et al. Efficacy of oseltamivir-zanamivir combination compared to each monotherapy for seasonal influenza: a randomized placebo-controlled trial. PLoS Med 2010; 7:e1000362.
- Jefferson T, Jones M, Doshi P, Del Mar C. Neuraminidase inhibitors for preventing and treating influenza in healthy adults: systematic review and meta-analysis. BMJ 2009; 339:b5106.
- Hernán MA, Lipsitch M. Oseltamivir and risk of lower respiratory tract complications in patients with flu symptoms: a meta-analysis of eleven randomized clinical trials. Clin Infect Dis 2011; 53:277–279.
- Samson M, Pizzorno A, Abed Y, Boivin G. Influenza virus resistance to neuraminidase inhibitors. Antiviral Res 2013; 98:174–185.
- US Centers for Disease Control and Prevention. FluView. www.cdc.gov/flu/weekly. Accessed February 3, 2014.
- Acs N, Bánhidy F, Puhó E, Czeizel AE. Maternal influenza during pregnancy and risk of congenital abnormalities in offspring. Birth Defects Res A Clin Mol Teratol 2005; 73:989–996.
- Zaman K, Roy E, Arifeen SE, et al. Effectiveness of maternal influenza immunization in mothers and infants. N Engl J Med 2008; 359:1555–1564.
- McKittrick N, Frank I, Jacobson JM, et al. Improved immunogenicity with high-dose seasonal influenza vaccine in HIV-infected persons: a single-center, parallel, randomized trial. Ann Intern Med 2013; 158:19–26.
- Kroon FP, van Dissel JT, de Jong JC, van Furth R. Antibody response to influenza, tetanus and pneumococcal vaccines in HIV-seropositive individuals in relation to the number of CD4+ lymphocytes. AIDS 1994; 8:469–476.
- Vilchez RA, McCurry K, Dauber J, et al. Influenza virus infection in adult solid organ transplant recipients. Am J Transplant 2002; 2:287–291.
Every year, 5% to 20% of US residents contract the flu, 200,000 are hospitalized for it, and 36,000 die of influenza-related complications. The economic impact, including direct medical costs and lost earnings, exceeds $87 billion.1 Despite this, less than half of eligible US residents were vaccinated in the 2012–2013 season, with uninsured people more than twice as likely to forgo vaccination.2,3
Several studies have shown that influenza vaccination reduces the need for outpatient encounters and hospitalizations and lowers the incidence of death from acute myocardial infarction, the rate of all-cause mortality, and even the incidence of therapies administered by implantable defibrillators.4–6 In the 2012–2013 influenza season, vaccination prevented an estimated 3.2 million medically attended illnesses and almost 80,000 hospitalizations; 70% of hospitalizations prevented were in children age 6 months to 4 years and in adults over age 65.7
After the 2009 H1N1 pandemic, which disproportionately killed previously healthy adults, the US Centers for Disease Control and Prevention (CDC) expanded its vaccination recommendations to include everyone above the age of 6 months, with few contraindications.8
In addition, recent years have seen a great expansion in vaccine options, changes in the at-risk demographics, and continued widespread resistance to certain antiviral agents, with implications for practice in primary care.
Here, we review the barriers and the new options for treatment and prevention of influenza.
HEMAGGLUTININ AND NEURAMINIDASE
Influenza infection is caused by one of the circulating strains of influenza virus A or B.
The major viral surface glycoproteins are hemagglutinin and neuraminidase. Hemagglutinin plays an important role in viral attachment to host cells and is the major immunogen in the influenza vaccine. Neuraminidase contains an active enzymatic site that cleaves the newly formed budding influenza viruses from host-cell sialic acid residues and allows them to be released from the cell membrane to infect other respiratory epithelial cells. It is the target of currently recommended antiviral drugs.
VACCINE PRODUCTION
Throughout the year, 130 influenza centers around the world sample circulating strains and share their data with five World Health Organization (WHO) Collaborating Centers for Reference and Research on Influenza. The WHO analyzes the circulation patterns, predicts the strains most likely to be circulating in the next influenza season, and shares these strains with manufacturers of the vaccine.
Pharmaceutical companies then begin an elaborate process of producing and distributing hundreds of millions of doses of vaccine worldwide. The production traditionally uses millions of fertilized chicken eggs to produce strain-specific influenza hemagglutinin. Individual vaccine strains are combined into the final product after being inactivated by chemical or physical splitting of the viral envelope with or without subsequent purification of the hemagglutinin particles.
Before 2013, the WHO’s yearly recommendations included two strains of influenza A and a single strain of influenza B. In 2013, new quadrivalent vaccines that include protection against a second strain of influenza B were approved.
The WHO strain-selection process allows manufacturers about 6 months to produce the vaccine. In a typical year, the worldwide demand is about 400 million doses. The theoretical maximal annual worldwide capacity, given current techniques, is fewer than 1 billion doses, which is well short of the 10 billion doses necessary to allow for the double vaccination needed in a pandemic.9 Newly approved recombinant manufacturing techniques offer greater production efficiency, while novel methods of intradermal administration increase vaccine immunogenicity, decreasing the amount of viral antigens used per dose.
INACTIVATED VS LIVE-ATTENUATED
In addition to intramuscular inactivated influenza vaccine, a live-attenuated vaccine in the form of an intranasal spray (FluMist) became available in 2003. This form is generally favored in children, as it avoids the discomfort of an injection. It contains live, weakened, cold-adapted influenza strains that reproduce in the relatively colder temperatures of the exterior nares but cannot survive in the warmer temperatures of the lung and proximal airways. It is approved for healthy people 2 to 49 years of age, and some evidence suggests that it may be more effective than inactivated influenza vaccine in children,10 although its utility is limited by multiple contraindications (see below).
INFLUENZA VACCINE INDICATIONS AND CONTRAINDICATIONS
Vaccination for influenza is recommended for all persons 6 months of age and older, an expansion from pre-2009 guidelines that did not recommend vaccination for healthy adults age 19 to 49 who were not in contact with people at high risk of influenza-related complications.8 Many new vaccine formulations have become available in recent years, each with specific benefits, risks, and target populations (Table 1).
Contraindications to inactivated vaccine
The only firm contraindication to inactivated influenza vaccine is previous severe allergic reaction to influenza vaccine or any of its components. Those with moderate to severe acute illness are advised to wait until their condition improves before being vaccinated. People who have had Guillain-Barré syndrome and those with egg allergy are discussed in MISAPPREHENSIONS THAT POSE BARRIERS TO VACCINATION, below. There is no risk of influenza infection from inactivated influenza vaccine.
Contraindications to live-attenuated influenza vaccine
Unlike inactivated influenza vaccine, the live-attenuated vaccine does result in shedding of vaccine-strain virus from the vaccinated host, with the theoretical potential for transmission of the virus from the vaccine recipient to other people, as well as the potential for influenza-like illness in vaccine recipients.11,12 Based on reported events, the former is estimated to occur in 10 to 20 per 1 million vaccinations, although these cases have never been proven to be caused by a cold-adapted vaccine-strain rather than by coincidental transmission of circulating wild-type viral strains.13
Despite this exceedingly small risk of viral transmission, live-attenuated influenza vaccine has multiple contraindications, including age less than 2 years and more than 49 years, disease- or drug-related compromised immune status, pregnancy, egg allergy, and history of allergic reaction to the formulation. These limit its use and are important to review in detail before prescribing.14
Use of neuraminidase inhibitors within 2 days before or 2 weeks after receiving live-attenuated influenza vaccine may interfere with replication of the cold-adapted strain and decrease the vaccine’s effectiveness.14
EFFECTIVENESS OF INFLUENZA VACCINATION IN OLDER ADULTS
The effectiveness of influenza vaccination depends on the age and health status of the person being vaccinated, as well as on the quality of the match between the vaccine and the circulating influenza viruses.
In the 2012–2013 season, the adjusted vaccine effectiveness was 56% overall, 47% for influenza A H3N2, and 67% for influenza B. However, in people age 65 and older, the overall adjusted vaccine effectiveness was 27%, and only 9% for influenza A H3N2.15 Thus, even though the vaccine-virus match was considered good, the vaccine was suboptimally effective in the older group. This may be an argument for using the recently approved high-dose vaccine in that age group. Although the high-dose vaccine has been shown to be significantly more immunogenic in older adults, it is too early to know if it is clinically more effective in preventing influenza in this age group.
Despite the lower-than-expected effectiveness in preventing influenza in the 2012–2013 season in people age 65 and older, several well-designed studies found that influenza vaccination prevented severe disease, including one study that found vaccination to be 89% effective in reducing influenza-associated hospitalizations in the 2010–2011 flu season.4,16
The limited effectiveness of vaccination in the older age group reminds us of the importance of early recognition and treatment of patients at high risk of influenza-related complications (see Table 2). It is also a call for greater compliance with vaccination in younger people, with a goal of achieving the 80% vaccination rate that has been calculated as adequate to achieve herd immunity.17
MISAPPREHENSIONS THAT POSE BARRIERS TO VACCINATION
Concern about potential adverse effects is the most common reason for refusing influenza vaccination, even among health care workers.18 However, the only commonly encountered adverse effect of the intramuscular inactivated influenza vaccine is injection-site pain.
‘Catching the flu from a flu shot’
Many people think that they can “catch the flu from a flu shot” (or think that they actually did), but vaccine-acquired influenza is not possible with the inactivated influenza vaccine,19 and it is only a theoretical, undocumented consideration with the live-attenuated vaccine.
Various respiratory viruses other than influenza also cause viral upper-respiratory infections during the influenza season. These infections may coincide with influenza vaccination and are frequently misconstrued as a side effect of the influenza vaccine or as evidence of vaccine ineffectiveness.
Unnecessary concerns about simultaneous vaccinations
Patients and doctors are often concerned about simultaneous administration of multiple vaccines and choose to spread out indicated vaccinations over multiple visits. This practice increases patients’ risk of illness from vaccine-preventable diseases. Research shows that simultaneous administration does not alter the safety or effectiveness of vaccination.20–22 The CDC recommends simultaneous administration of all indicated live and inactivated vaccinations in order to reduce barriers to vaccination.20
Fear of Guillain-Barré syndrome
Guillain-Barré syndrome, an acute ascending polyneuropathy, has been blamed on influenza vaccination in cases that developed after the 1976 influenza A (H1N1) epidemic.
Most cases are self-limiting but require intensive treatment and supportive care. Full recovery occurs in 60% of cases, though some people experience persistent symptoms. The mortality rate is less than 5%.23
After the 1976 influenza pandemic, approximately 400 cases of Guillain-Barré syndrome arose in 45 million vaccine recipients, or about 1 case per 100,000 people.24 Multiple subsequent population analyses concluded that the actual incidence of Guillain-Barré syndrome attributable to influenza vaccination is negligible, at less than 1 case in 1 million vaccinations. Against this, we should compare the real risk of illness and death from influenza infection, which itself is a risk factor for Guillain-Barré syndrome.25
Should a person with a history of Guillain-Barré syndrome be revaccinated against influenza? The risk was evaluated in a large retrospective analysis of cases identified in the Kaiser Permanente Northern California Database from 1995 to 2006.26 Five hundred fifty cases of Guillain-Barré syndrome were identified, of which 18 had arisen within 6 weeks of the patient receiving a flu shot. Four hundred five doses of inactivated influenza vaccine were subsequently given to 105 patients who had a history of Guillain-Barré syndrome, two of whom had developed the syndrome within 6 weeks of receiving the shot. There were no documented episodes of recurrent Guillain-Barré syndrome in any of these patients. Only 6 of 550 patients with a history of the disease developed it again; none of these 6 had received the influenza vaccine in the preceding 2 months, and only 1 had been exposed to the measles-mumps-rubella vaccine in the 4 months before vaccination.
Nevertheless, expert opinion recommends lifelong avoidance of any immunization that had been given within 6 weeks before the onset of symptoms of Guillain-Barré syndrome.27
Overstated concern about egg allergy
Anaphylactic reactions can occur after influenza vaccination in people who have severe egg allergy, and concern about these reactions unfortunately prevents many otherwise eligible people with mild allergy from being vaccinated.
These reactions are much less common than feared. In a well-designed prospective cohort study of 367 patients with a history of egg allergy and positive skin-prick tests, including 132 with a history of severe allergy and 4 with a history of mild allergic symptoms arising in response to previous influenza vaccinations, none developed anaphylaxis.28
The same authors reviewed 26 studies in more than 4,000 egg-allergic patients, of whom more than 500 had a history of severe egg-associated reactions, and likewise found no cases of influenza vaccine-associated anaphylaxis. They concluded that the inactivated influenza vaccine is safer than the egg-derived mumps-measles-rubella vaccine, for which precautions for egg allergy no longer exist.28
People with a history of more serious reactions, ranging from stomach upset to anaphylaxis, can be safely vaccinated with a recombinant vaccine or referred to an allergist for further testing. People who experience hives as their only reaction to egg exposure should receive full-dose vaccination but then be observed for a half hour afterward.
The recombinant trivalent influenza vaccine Flublok was approved in 2013 for people age 18 to 49. It is the first commercially available influenza vaccine produced in a continuous insect cell line using a baculovirus vector. No eggs are used in its production, and it is approved for use in patients with egg allergy of any severity.
People who have a history of more serious reactions, including abdominal pain, nausea, vomiting, dizziness, or wheezing can be vaccinated with the recombinant vaccine or referred to an allergy specialist.
Despite this new option, understanding of alternative immunization guidelines for people with egg allergies, available on the CDC website29 remains important, as the availability of the recombinant trivalent influenza vaccine remains limited in the 2013–2014 influenza season.
Misconception about mercury toxicity
Thimerosal is an ethylmercury-containing preservative used in multidose antiviral vaccines, including some influenza vaccines.30 It is designed to prevent bacterial and fungal colonization of the vaccine vial while not reducing vaccine effectiveness or causing toxicity.
Contemporary understanding of mercury neurotoxicity is based largely on studies of methylmercury, including long-term, low-dose exposure in remote communities in the Faroe Islands and the Seychelles through regular consumption of fish and whale meat.31,32 These exposure studies had conflicting results: those in the Faroe Islands demonstrated toxicity, but the Seychelles studies actually showed better neurologic test scores at higher mercury levels, a trend the authors attributed to the beneficial effects of maternal fish consumption.
The results of the methylmercury studies have been extrapolated to ethylmercury (contained in thimerosal), although the two chemicals have vastly different pharmacologic properties. For example, methylmercury has a longer half-life and greater transport across the blood-brain barrier.33 A direct comparison found that ethylmercury is less toxic than methylmercury, although an increase in ethylmercury concentration of only 20% resulted in similar toxicity profiles.34 These studies were performed at concentrations of mercury thousands of times higher than those resulting from vaccination: nearly 150,000 times greater than those in an average adult or 15,000 times greater than those in a 1-year-old child from the typical 25-μg thimerosal dose allowed in contemporary influenza vaccines.
Despite much negative publicity, no link has been shown between thimerosal and autism.30 Multiple regulatory, scientific, and medical organizations including the US Food and Drug Administration (FDA), the WHO, the National Institutes of Health, the CDC, the American Academy of Pediatrics, and the American Congress of Obstetricians and Gynecologists (ACOG) have evaluated the data on the safety of thimerosal in vaccines and have agreed that it is safe. However, most of them urged vaccine manufacturers to eliminate mercury from vaccines as a precaution.30,35 Thimerosal has subsequently been eliminated from all childhood vaccines except for influenza vaccine, with no resulting decrease in childhood autism diagnoses.36
Considering that no harm from thimerosal at FDA-approved doses has been documented, and considering the real risk of influenza-related complications, particularly in young children and pregnant women, we recommend vaccination using whatever vaccine formulation is locally available for all patients, including children age 6 months and older and pregnant women. Nevertheless, given that mercury is being eliminated from childhood vaccines and that preservative-free single-dose vials are increasingly available in the United States, it seems reasonable to use thimerosal-free formulations for children, expectant mothers, and patients concerned about exposure if these formulations are readily available. Influenza vaccination should not be delayed if a thimerosal-free formulation is not readily available.
NEW VACCINE FORMULATIONS
Recent years have seen a dramatic expansion in influenza vaccine options (Table 1).
Quadrivalent vaccines
Quadrivalent vaccines protect against two strains of influenza A and two strains of influenza B, whereas earlier formulations included only one influenza B strain. Vaccination against either influenza B strain offers only limited cross-protection against the other B strain, and previous formulations involved assumptions about which strain would predominate in any given year. The CDC estimates that switching to quadrivalent vaccines will prevent up to 970,000 cases of influenza, 8,200 hospitalizations, and 485 deaths per year.37
Intradermal vaccine
The newly available Fluzone Intradermal vaccine contains smaller doses of hemagglutinin but is still effective because antigen-presenting dendritic cells in the skin reduce the required amount of vaccine antigen necessary for inducing protection.38 This may provide an advantage in the event of vaccine shortage. Also, since it is given in needles only 1.5 mm long, it may appeal to people who are afraid of needles.
The stronger immune reaction with intradermal administration causes more redness, induration, and tenderness at the injection site than with intramuscular administration.39 Patients should not be surprised by this reaction and can be advised to apply ice packs for symptomatic relief.
High-dose vaccine
A high-dose vaccine was approved in 2009 for use in adults age 65 and older. It contains 60 μg of hemagglutinin, compared with 15 μg in standard-dose vaccines, and has been shown to improve seroconversion rates. It remains to be seen if this translates into better clinical outcomes in older adults.40 Further studies will be necessary before we can recommend high-dose vaccines to other people with weakened immune response, such as those undergoing chemotherapy or those infected with human immunodeficiency virus (HIV).
Cell-based vaccines
Flucelvax was the first cell-based influenza vaccine. However, unlike the recombinant trivalent influenza vaccine, which uses no eggs in its manufacturing process, Flucelvax production starts with egg-derived influenza strains that are subsequently propagated in liquid culture of animal cells. It may therefore contain traces of egg protein, and it has not been studied in people with egg allergy.41
An advantage of the cell-based production technique is the use of fewer or no eggs at all, which may result in greater manufacturing efficiency. Also, it is a closed process that reduces the risk of bacterial contamination as well as reliance on antibiotics or preservatives, such as thimerosal, in the manufacturing process.42
CHEMOPROPHYLAXIS WITH NEURAMINIDASE INHIBITORS
The mainstays of influenza prevention are seasonal vaccination and appropriate infection-prevention practices. In addition, in patients at high risk of influenza-related complications (Table 2),43 postexposure chemoprophylaxis with a neuraminidase inhibitor, ie, oseltamivir (Tamiflu) or zanamivir (Relenza), is an effective preventive strategy, especially in years when the match between vaccine and circulating virus strains is suboptimal.44,45
Neuraminidase inhibitors are competitive inhibitors of the active site of the influenza glycoprotein neuraminidase, responsible for viral release from infected respiratory epithelial cells. Rates of resistance to neuraminidase inhibitors have been less than 1% in the United States in recent years, while resistance to the adamantanes amantadine (Symmetrel) and rimantadine (Flumadine) can be as high as 92%, depending on the virus isolate. Thus, their use for treatment or prophylaxis of influenza is not currently recommended by the CDC.46
Chemoprophylaxis with any agent may promote emergence of resistant strains, can cause adverse reactions, and should never be considered a substitute for vaccination.
ANTI-INFLUENZA AGENTS
Two neuraminidase inhibitors, oseltamivir and zanamivir, are approved by the FDA for preventing and treating uncomplicated influenza. Treatment must be instituted within 2 days of onset of symptoms to be effective.
Oseltamivir is available as an oral capsule or powder for liquid suspension. Its most common adverse effects are gastrointestinal upset including diarrhea, nausea, and vomiting.44
Zanamivir is only available in the form of a dry powder inhaler because of the drug’s poor oral bioavailability, and only 4% to 17% of the inhaled dose is systemically absorbed.45 There is a theoretical benefit in targeted delivery of zanamivir to the primary organ affected by influenza, and gastrointestinal side effects are less common with this drug.44,45 Unfortunately, the zanamivir inhaler requires complicated assembly and dexterity for administration (see the video on YouTube47), which may make it unreliable in certain patient groups, especially handicapped and elderly patients. Administration has been associated with bronchospasm, resulting in a more than 20% reduction in the forced expiratory volume in 1 second, and it is contraindicated in patients with underlying reactive airway disease such as chronic obstructive pulmonary disease or asthma.45
Table 3 lists the doses and duration of therapy for oseltamivir and zanamivir in adults with normal renal function, as well as approximate costs. No generic formulations of neuraminidase inhibitors are currently available, and outpatient use may not be covered by medical insurance. Several other neuraminidase inhibitors are either under development or at various stages in the FDA approval process.
EFFECTIVENESS OF ANTI-INFLUENZA DRUGS
Treatment with oseltamivir has been shown to reduce the duration of symptoms by approximately 1 day if initiated within 36 hours of onset of illness and 1.5 to 2 days if initiated within 24 hours.48,49 Trials and meta-analyses of zanamivir show similar effectiveness, though some suggest that symptoms were alleviated as much as 3 days sooner than in controls in a subgroup of patients who were febrile at presentation.50,51 Dual neuraminidase inhibitor therapy in an attempt to prevent emergence of resistance seems logical but was actually found to be less effective than monotherapy, according to a 2010 study.52
The effectiveness of neuraminidase inhibitors in reducing influenza-related complications and mortality rates has been controversial in recent years, as these outcomes were not addressed in initial studies that secured FDA approval. Several meta-analyses differ in their assessments of available data quality and conclusions. A 2009 Cochrane review questioned the completeness and the veracity of the data from manufacturer-funded trial data, much of which was unpublished and not made available to reviewers, and it concluded that a reduction of complications could not be supported by the available data.53 Hernán and Lipsitch,54 in a 2011 review, calculated that oseltamivir reduces the risk of lower respiratory tract complications by 28% in patients with influenza-like symptoms and by 37% in patients with confirmed influenza infection.
Additional trials and better access to available data are needed to settle the question of the effectiveness of neuraminidase inhibitors in reducing complications of influenza. Meanwhile, they remain strongly recommended by major health organizations, including the CDC and the WHO, which lists oseltamivir on its “model list of essential medicines.”
VIRAL RESISTANCE TO NEURAMINIDASE INHIBITORS
Viral resistance to neuraminidase inhibitors occurs through multiple mechanisms and may arise without selective pressure from exposure to these drugs.55
Oseltamivir possesses a hydrophobic moiety that requires viral neuraminidase to undergo a complex reconfiguration to expose the active site prior to binding. Any mutation affecting its ability to undergo this structural rearrangement can promote resistance by decreased oseltamivir access to the active site.
Zanamivir has a structural homology to the neuraminidase active site and requires no such reconfiguration. Additionally, mutations promoting resistance to zanamivir may actually decrease viral fitness; thus, resistance to zanamivir is significantly less common than to oseltamivir.55
About 2,000 influenza virus isolates currently circulating in the United States were tested for resistance; only 1% of the 2009 influenza A H1N1 isolates demonstrated resistance to oseltamivir, and none to zanamivir.56
The CDC regularly updates the resistance patterns of circulating influenza strains at www.cdc.gov/flu/weekly/index.htm.
SPECIAL CONSIDERATIONS
Pregnancy
Pregnant women may be at higher risk of severe influenza complications. This was especially true during the 2009 H1N1 pandemic, when pregnant women had a five times higher risk of death from influenza-related complications. Additionally, fever during pregnancy is an independent risk factor for adverse outcomes in the offspring.57 Maternal vaccination against influenza effectively protects the infant for the first 6 months of life, when vaccination is not recommended because of a poor immune response.58
Live-attenuated influenza vaccine is contraindicated during pregnancy. Given the documented risks to the mother from influenza and no documented harm from preservatives in multiuse vaccine vials, the Advisory Committee on Immunization Practices (ACIP) and ACOG do not state a preference for thimerosal-containing or thimerosal-free vaccine for any group, including pregnant women. Pregnant women should be vaccinated with whatever inactivated influenza vaccine formulation is available at the earliest opportunity in the beginning of the influenza season, regardless of the trimester of pregnancy.
Pregnant women are at high risk of influenza-related complications and should be considered for postexposure antiviral prophylaxis or early treatment with a neuraminidase inhibitor. However, both of the approved neuraminidase inhibitors are in pregnancy safety category C, indicating possible adverse effects in animal studies and a lack of safety data in pregnant humans. As with all category C medications, the risks and benefits must be considered, taking into account maternal comorbidities, vaccination status, effectiveness of the season’s influenza vaccine, and the virulence of circulating influenza strains.
As oseltamivir is associated with nausea and gastrointestinal side effects and as zanamivir has less systemic absorption, it may be reasonable to prescribe zanamivir for women already experiencing severe pregnancy-related nausea.
Immunocompromised people
Inactivated influenza vaccine is recommended and live-attenuated influenza vaccine is contraindicated for all immunocompromised people. Generally speaking, any form of immune compromise will decrease the immunogenicity of the vaccine. Additional considerations vary depending on the cause and severity of the immunocompromised status.
HIV-infected patients have higher seroconversion rates when vaccinated with the high-dose vaccine than with the standard-dose vaccine; however, as in adults over age 65, the clinical benefit has yet to be evaluated.59 The efficacy of vaccination is predictably related to the CD4 cell count, as T cells are necessary to mount a response.60 No documented benefit is gained from booster influenza vaccination in this group of patients.
Cancer patients should receive inactivated influenza vaccine every year. Postexposure chemoprophylaxis should be considered, and early treatment with a neuraminidase inhibitor is recommended in patients undergoing chemotherapy.
Solid-organ transplant recipients face a risk of organ rejection if they contract influenza infection, in addition to a higher risk of influenza-related complications.61 Transplant recipients should receive inactivated influenza vaccine as soon as it becomes available at the beginning of every influenza season. Additional research is necessary to evaluate the safety and effectiveness of the high-dose influenza vaccine in this patient group.
MORE OPTIONS, GREAT BENEFIT
Influenza remains a significant source of morbidity and mortality in the United States, and emerging pandemic strains as well as the aging population pose the risk of increased disease burden. New vaccine options offer hope of greater safety, improved efficacy, and higher vaccination rates though broader appeal to individuals. The actual differences in protection between various vaccine options are insignificant relative to the overall benefit of vaccination.
Health care providers should inquire about patients’ understanding and address their concerns about vaccination. Giving an available influenza vaccine within approved indications should not be delayed if alternative vaccine options are not readily available.
In addition to vaccination, patients at high risk of complications should be advised early in the influenza season to inform their doctors about potential exposure to influenza or the development of flu-like symptoms for consideration of early treatment or postexposure prophylaxis with a neuraminidase inhibitor.
Every year, 5% to 20% of US residents contract the flu, 200,000 are hospitalized for it, and 36,000 die of influenza-related complications. The economic impact, including direct medical costs and lost earnings, exceeds $87 billion.1 Despite this, less than half of eligible US residents were vaccinated in the 2012–2013 season, with uninsured people more than twice as likely to forgo vaccination.2,3
Several studies have shown that influenza vaccination reduces the need for outpatient encounters and hospitalizations and lowers the incidence of death from acute myocardial infarction, the rate of all-cause mortality, and even the incidence of therapies administered by implantable defibrillators.4–6 In the 2012–2013 influenza season, vaccination prevented an estimated 3.2 million medically attended illnesses and almost 80,000 hospitalizations; 70% of hospitalizations prevented were in children age 6 months to 4 years and in adults over age 65.7
After the 2009 H1N1 pandemic, which disproportionately killed previously healthy adults, the US Centers for Disease Control and Prevention (CDC) expanded its vaccination recommendations to include everyone above the age of 6 months, with few contraindications.8
In addition, recent years have seen a great expansion in vaccine options, changes in the at-risk demographics, and continued widespread resistance to certain antiviral agents, with implications for practice in primary care.
Here, we review the barriers and the new options for treatment and prevention of influenza.
HEMAGGLUTININ AND NEURAMINIDASE
Influenza infection is caused by one of the circulating strains of influenza virus A or B.
The major viral surface glycoproteins are hemagglutinin and neuraminidase. Hemagglutinin plays an important role in viral attachment to host cells and is the major immunogen in the influenza vaccine. Neuraminidase contains an active enzymatic site that cleaves the newly formed budding influenza viruses from host-cell sialic acid residues and allows them to be released from the cell membrane to infect other respiratory epithelial cells. It is the target of currently recommended antiviral drugs.
VACCINE PRODUCTION
Throughout the year, 130 influenza centers around the world sample circulating strains and share their data with five World Health Organization (WHO) Collaborating Centers for Reference and Research on Influenza. The WHO analyzes the circulation patterns, predicts the strains most likely to be circulating in the next influenza season, and shares these strains with manufacturers of the vaccine.
Pharmaceutical companies then begin an elaborate process of producing and distributing hundreds of millions of doses of vaccine worldwide. The production traditionally uses millions of fertilized chicken eggs to produce strain-specific influenza hemagglutinin. Individual vaccine strains are combined into the final product after being inactivated by chemical or physical splitting of the viral envelope with or without subsequent purification of the hemagglutinin particles.
Before 2013, the WHO’s yearly recommendations included two strains of influenza A and a single strain of influenza B. In 2013, new quadrivalent vaccines that include protection against a second strain of influenza B were approved.
The WHO strain-selection process allows manufacturers about 6 months to produce the vaccine. In a typical year, the worldwide demand is about 400 million doses. The theoretical maximal annual worldwide capacity, given current techniques, is fewer than 1 billion doses, which is well short of the 10 billion doses necessary to allow for the double vaccination needed in a pandemic.9 Newly approved recombinant manufacturing techniques offer greater production efficiency, while novel methods of intradermal administration increase vaccine immunogenicity, decreasing the amount of viral antigens used per dose.
INACTIVATED VS LIVE-ATTENUATED
In addition to intramuscular inactivated influenza vaccine, a live-attenuated vaccine in the form of an intranasal spray (FluMist) became available in 2003. This form is generally favored in children, as it avoids the discomfort of an injection. It contains live, weakened, cold-adapted influenza strains that reproduce in the relatively colder temperatures of the exterior nares but cannot survive in the warmer temperatures of the lung and proximal airways. It is approved for healthy people 2 to 49 years of age, and some evidence suggests that it may be more effective than inactivated influenza vaccine in children,10 although its utility is limited by multiple contraindications (see below).
INFLUENZA VACCINE INDICATIONS AND CONTRAINDICATIONS
Vaccination for influenza is recommended for all persons 6 months of age and older, an expansion from pre-2009 guidelines that did not recommend vaccination for healthy adults age 19 to 49 who were not in contact with people at high risk of influenza-related complications.8 Many new vaccine formulations have become available in recent years, each with specific benefits, risks, and target populations (Table 1).
Contraindications to inactivated vaccine
The only firm contraindication to inactivated influenza vaccine is previous severe allergic reaction to influenza vaccine or any of its components. Those with moderate to severe acute illness are advised to wait until their condition improves before being vaccinated. People who have had Guillain-Barré syndrome and those with egg allergy are discussed in MISAPPREHENSIONS THAT POSE BARRIERS TO VACCINATION, below. There is no risk of influenza infection from inactivated influenza vaccine.
Contraindications to live-attenuated influenza vaccine
Unlike inactivated influenza vaccine, the live-attenuated vaccine does result in shedding of vaccine-strain virus from the vaccinated host, with the theoretical potential for transmission of the virus from the vaccine recipient to other people, as well as the potential for influenza-like illness in vaccine recipients.11,12 Based on reported events, the former is estimated to occur in 10 to 20 per 1 million vaccinations, although these cases have never been proven to be caused by a cold-adapted vaccine-strain rather than by coincidental transmission of circulating wild-type viral strains.13
Despite this exceedingly small risk of viral transmission, live-attenuated influenza vaccine has multiple contraindications, including age less than 2 years and more than 49 years, disease- or drug-related compromised immune status, pregnancy, egg allergy, and history of allergic reaction to the formulation. These limit its use and are important to review in detail before prescribing.14
Use of neuraminidase inhibitors within 2 days before or 2 weeks after receiving live-attenuated influenza vaccine may interfere with replication of the cold-adapted strain and decrease the vaccine’s effectiveness.14
EFFECTIVENESS OF INFLUENZA VACCINATION IN OLDER ADULTS
The effectiveness of influenza vaccination depends on the age and health status of the person being vaccinated, as well as on the quality of the match between the vaccine and the circulating influenza viruses.
In the 2012–2013 season, the adjusted vaccine effectiveness was 56% overall, 47% for influenza A H3N2, and 67% for influenza B. However, in people age 65 and older, the overall adjusted vaccine effectiveness was 27%, and only 9% for influenza A H3N2.15 Thus, even though the vaccine-virus match was considered good, the vaccine was suboptimally effective in the older group. This may be an argument for using the recently approved high-dose vaccine in that age group. Although the high-dose vaccine has been shown to be significantly more immunogenic in older adults, it is too early to know if it is clinically more effective in preventing influenza in this age group.
Despite the lower-than-expected effectiveness in preventing influenza in the 2012–2013 season in people age 65 and older, several well-designed studies found that influenza vaccination prevented severe disease, including one study that found vaccination to be 89% effective in reducing influenza-associated hospitalizations in the 2010–2011 flu season.4,16
The limited effectiveness of vaccination in the older age group reminds us of the importance of early recognition and treatment of patients at high risk of influenza-related complications (see Table 2). It is also a call for greater compliance with vaccination in younger people, with a goal of achieving the 80% vaccination rate that has been calculated as adequate to achieve herd immunity.17
MISAPPREHENSIONS THAT POSE BARRIERS TO VACCINATION
Concern about potential adverse effects is the most common reason for refusing influenza vaccination, even among health care workers.18 However, the only commonly encountered adverse effect of the intramuscular inactivated influenza vaccine is injection-site pain.
‘Catching the flu from a flu shot’
Many people think that they can “catch the flu from a flu shot” (or think that they actually did), but vaccine-acquired influenza is not possible with the inactivated influenza vaccine,19 and it is only a theoretical, undocumented consideration with the live-attenuated vaccine.
Various respiratory viruses other than influenza also cause viral upper-respiratory infections during the influenza season. These infections may coincide with influenza vaccination and are frequently misconstrued as a side effect of the influenza vaccine or as evidence of vaccine ineffectiveness.
Unnecessary concerns about simultaneous vaccinations
Patients and doctors are often concerned about simultaneous administration of multiple vaccines and choose to spread out indicated vaccinations over multiple visits. This practice increases patients’ risk of illness from vaccine-preventable diseases. Research shows that simultaneous administration does not alter the safety or effectiveness of vaccination.20–22 The CDC recommends simultaneous administration of all indicated live and inactivated vaccinations in order to reduce barriers to vaccination.20
Fear of Guillain-Barré syndrome
Guillain-Barré syndrome, an acute ascending polyneuropathy, has been blamed on influenza vaccination in cases that developed after the 1976 influenza A (H1N1) epidemic.
Most cases are self-limiting but require intensive treatment and supportive care. Full recovery occurs in 60% of cases, though some people experience persistent symptoms. The mortality rate is less than 5%.23
After the 1976 influenza pandemic, approximately 400 cases of Guillain-Barré syndrome arose in 45 million vaccine recipients, or about 1 case per 100,000 people.24 Multiple subsequent population analyses concluded that the actual incidence of Guillain-Barré syndrome attributable to influenza vaccination is negligible, at less than 1 case in 1 million vaccinations. Against this, we should compare the real risk of illness and death from influenza infection, which itself is a risk factor for Guillain-Barré syndrome.25
Should a person with a history of Guillain-Barré syndrome be revaccinated against influenza? The risk was evaluated in a large retrospective analysis of cases identified in the Kaiser Permanente Northern California Database from 1995 to 2006.26 Five hundred fifty cases of Guillain-Barré syndrome were identified, of which 18 had arisen within 6 weeks of the patient receiving a flu shot. Four hundred five doses of inactivated influenza vaccine were subsequently given to 105 patients who had a history of Guillain-Barré syndrome, two of whom had developed the syndrome within 6 weeks of receiving the shot. There were no documented episodes of recurrent Guillain-Barré syndrome in any of these patients. Only 6 of 550 patients with a history of the disease developed it again; none of these 6 had received the influenza vaccine in the preceding 2 months, and only 1 had been exposed to the measles-mumps-rubella vaccine in the 4 months before vaccination.
Nevertheless, expert opinion recommends lifelong avoidance of any immunization that had been given within 6 weeks before the onset of symptoms of Guillain-Barré syndrome.27
Overstated concern about egg allergy
Anaphylactic reactions can occur after influenza vaccination in people who have severe egg allergy, and concern about these reactions unfortunately prevents many otherwise eligible people with mild allergy from being vaccinated.
These reactions are much less common than feared. In a well-designed prospective cohort study of 367 patients with a history of egg allergy and positive skin-prick tests, including 132 with a history of severe allergy and 4 with a history of mild allergic symptoms arising in response to previous influenza vaccinations, none developed anaphylaxis.28
The same authors reviewed 26 studies in more than 4,000 egg-allergic patients, of whom more than 500 had a history of severe egg-associated reactions, and likewise found no cases of influenza vaccine-associated anaphylaxis. They concluded that the inactivated influenza vaccine is safer than the egg-derived mumps-measles-rubella vaccine, for which precautions for egg allergy no longer exist.28
People with a history of more serious reactions, ranging from stomach upset to anaphylaxis, can be safely vaccinated with a recombinant vaccine or referred to an allergist for further testing. People who experience hives as their only reaction to egg exposure should receive full-dose vaccination but then be observed for a half hour afterward.
The recombinant trivalent influenza vaccine Flublok was approved in 2013 for people age 18 to 49. It is the first commercially available influenza vaccine produced in a continuous insect cell line using a baculovirus vector. No eggs are used in its production, and it is approved for use in patients with egg allergy of any severity.
People who have a history of more serious reactions, including abdominal pain, nausea, vomiting, dizziness, or wheezing can be vaccinated with the recombinant vaccine or referred to an allergy specialist.
Despite this new option, understanding of alternative immunization guidelines for people with egg allergies, available on the CDC website29 remains important, as the availability of the recombinant trivalent influenza vaccine remains limited in the 2013–2014 influenza season.
Misconception about mercury toxicity
Thimerosal is an ethylmercury-containing preservative used in multidose antiviral vaccines, including some influenza vaccines.30 It is designed to prevent bacterial and fungal colonization of the vaccine vial while not reducing vaccine effectiveness or causing toxicity.
Contemporary understanding of mercury neurotoxicity is based largely on studies of methylmercury, including long-term, low-dose exposure in remote communities in the Faroe Islands and the Seychelles through regular consumption of fish and whale meat.31,32 These exposure studies had conflicting results: those in the Faroe Islands demonstrated toxicity, but the Seychelles studies actually showed better neurologic test scores at higher mercury levels, a trend the authors attributed to the beneficial effects of maternal fish consumption.
The results of the methylmercury studies have been extrapolated to ethylmercury (contained in thimerosal), although the two chemicals have vastly different pharmacologic properties. For example, methylmercury has a longer half-life and greater transport across the blood-brain barrier.33 A direct comparison found that ethylmercury is less toxic than methylmercury, although an increase in ethylmercury concentration of only 20% resulted in similar toxicity profiles.34 These studies were performed at concentrations of mercury thousands of times higher than those resulting from vaccination: nearly 150,000 times greater than those in an average adult or 15,000 times greater than those in a 1-year-old child from the typical 25-μg thimerosal dose allowed in contemporary influenza vaccines.
Despite much negative publicity, no link has been shown between thimerosal and autism.30 Multiple regulatory, scientific, and medical organizations including the US Food and Drug Administration (FDA), the WHO, the National Institutes of Health, the CDC, the American Academy of Pediatrics, and the American Congress of Obstetricians and Gynecologists (ACOG) have evaluated the data on the safety of thimerosal in vaccines and have agreed that it is safe. However, most of them urged vaccine manufacturers to eliminate mercury from vaccines as a precaution.30,35 Thimerosal has subsequently been eliminated from all childhood vaccines except for influenza vaccine, with no resulting decrease in childhood autism diagnoses.36
Considering that no harm from thimerosal at FDA-approved doses has been documented, and considering the real risk of influenza-related complications, particularly in young children and pregnant women, we recommend vaccination using whatever vaccine formulation is locally available for all patients, including children age 6 months and older and pregnant women. Nevertheless, given that mercury is being eliminated from childhood vaccines and that preservative-free single-dose vials are increasingly available in the United States, it seems reasonable to use thimerosal-free formulations for children, expectant mothers, and patients concerned about exposure if these formulations are readily available. Influenza vaccination should not be delayed if a thimerosal-free formulation is not readily available.
NEW VACCINE FORMULATIONS
Recent years have seen a dramatic expansion in influenza vaccine options (Table 1).
Quadrivalent vaccines
Quadrivalent vaccines protect against two strains of influenza A and two strains of influenza B, whereas earlier formulations included only one influenza B strain. Vaccination against either influenza B strain offers only limited cross-protection against the other B strain, and previous formulations involved assumptions about which strain would predominate in any given year. The CDC estimates that switching to quadrivalent vaccines will prevent up to 970,000 cases of influenza, 8,200 hospitalizations, and 485 deaths per year.37
Intradermal vaccine
The newly available Fluzone Intradermal vaccine contains smaller doses of hemagglutinin but is still effective because antigen-presenting dendritic cells in the skin reduce the required amount of vaccine antigen necessary for inducing protection.38 This may provide an advantage in the event of vaccine shortage. Also, since it is given in needles only 1.5 mm long, it may appeal to people who are afraid of needles.
The stronger immune reaction with intradermal administration causes more redness, induration, and tenderness at the injection site than with intramuscular administration.39 Patients should not be surprised by this reaction and can be advised to apply ice packs for symptomatic relief.
High-dose vaccine
A high-dose vaccine was approved in 2009 for use in adults age 65 and older. It contains 60 μg of hemagglutinin, compared with 15 μg in standard-dose vaccines, and has been shown to improve seroconversion rates. It remains to be seen if this translates into better clinical outcomes in older adults.40 Further studies will be necessary before we can recommend high-dose vaccines to other people with weakened immune response, such as those undergoing chemotherapy or those infected with human immunodeficiency virus (HIV).
Cell-based vaccines
Flucelvax was the first cell-based influenza vaccine. However, unlike the recombinant trivalent influenza vaccine, which uses no eggs in its manufacturing process, Flucelvax production starts with egg-derived influenza strains that are subsequently propagated in liquid culture of animal cells. It may therefore contain traces of egg protein, and it has not been studied in people with egg allergy.41
An advantage of the cell-based production technique is the use of fewer or no eggs at all, which may result in greater manufacturing efficiency. Also, it is a closed process that reduces the risk of bacterial contamination as well as reliance on antibiotics or preservatives, such as thimerosal, in the manufacturing process.42
CHEMOPROPHYLAXIS WITH NEURAMINIDASE INHIBITORS
The mainstays of influenza prevention are seasonal vaccination and appropriate infection-prevention practices. In addition, in patients at high risk of influenza-related complications (Table 2),43 postexposure chemoprophylaxis with a neuraminidase inhibitor, ie, oseltamivir (Tamiflu) or zanamivir (Relenza), is an effective preventive strategy, especially in years when the match between vaccine and circulating virus strains is suboptimal.44,45
Neuraminidase inhibitors are competitive inhibitors of the active site of the influenza glycoprotein neuraminidase, responsible for viral release from infected respiratory epithelial cells. Rates of resistance to neuraminidase inhibitors have been less than 1% in the United States in recent years, while resistance to the adamantanes amantadine (Symmetrel) and rimantadine (Flumadine) can be as high as 92%, depending on the virus isolate. Thus, their use for treatment or prophylaxis of influenza is not currently recommended by the CDC.46
Chemoprophylaxis with any agent may promote emergence of resistant strains, can cause adverse reactions, and should never be considered a substitute for vaccination.
ANTI-INFLUENZA AGENTS
Two neuraminidase inhibitors, oseltamivir and zanamivir, are approved by the FDA for preventing and treating uncomplicated influenza. Treatment must be instituted within 2 days of onset of symptoms to be effective.
Oseltamivir is available as an oral capsule or powder for liquid suspension. Its most common adverse effects are gastrointestinal upset including diarrhea, nausea, and vomiting.44
Zanamivir is only available in the form of a dry powder inhaler because of the drug’s poor oral bioavailability, and only 4% to 17% of the inhaled dose is systemically absorbed.45 There is a theoretical benefit in targeted delivery of zanamivir to the primary organ affected by influenza, and gastrointestinal side effects are less common with this drug.44,45 Unfortunately, the zanamivir inhaler requires complicated assembly and dexterity for administration (see the video on YouTube47), which may make it unreliable in certain patient groups, especially handicapped and elderly patients. Administration has been associated with bronchospasm, resulting in a more than 20% reduction in the forced expiratory volume in 1 second, and it is contraindicated in patients with underlying reactive airway disease such as chronic obstructive pulmonary disease or asthma.45
Table 3 lists the doses and duration of therapy for oseltamivir and zanamivir in adults with normal renal function, as well as approximate costs. No generic formulations of neuraminidase inhibitors are currently available, and outpatient use may not be covered by medical insurance. Several other neuraminidase inhibitors are either under development or at various stages in the FDA approval process.
EFFECTIVENESS OF ANTI-INFLUENZA DRUGS
Treatment with oseltamivir has been shown to reduce the duration of symptoms by approximately 1 day if initiated within 36 hours of onset of illness and 1.5 to 2 days if initiated within 24 hours.48,49 Trials and meta-analyses of zanamivir show similar effectiveness, though some suggest that symptoms were alleviated as much as 3 days sooner than in controls in a subgroup of patients who were febrile at presentation.50,51 Dual neuraminidase inhibitor therapy in an attempt to prevent emergence of resistance seems logical but was actually found to be less effective than monotherapy, according to a 2010 study.52
The effectiveness of neuraminidase inhibitors in reducing influenza-related complications and mortality rates has been controversial in recent years, as these outcomes were not addressed in initial studies that secured FDA approval. Several meta-analyses differ in their assessments of available data quality and conclusions. A 2009 Cochrane review questioned the completeness and the veracity of the data from manufacturer-funded trial data, much of which was unpublished and not made available to reviewers, and it concluded that a reduction of complications could not be supported by the available data.53 Hernán and Lipsitch,54 in a 2011 review, calculated that oseltamivir reduces the risk of lower respiratory tract complications by 28% in patients with influenza-like symptoms and by 37% in patients with confirmed influenza infection.
Additional trials and better access to available data are needed to settle the question of the effectiveness of neuraminidase inhibitors in reducing complications of influenza. Meanwhile, they remain strongly recommended by major health organizations, including the CDC and the WHO, which lists oseltamivir on its “model list of essential medicines.”
VIRAL RESISTANCE TO NEURAMINIDASE INHIBITORS
Viral resistance to neuraminidase inhibitors occurs through multiple mechanisms and may arise without selective pressure from exposure to these drugs.55
Oseltamivir possesses a hydrophobic moiety that requires viral neuraminidase to undergo a complex reconfiguration to expose the active site prior to binding. Any mutation affecting its ability to undergo this structural rearrangement can promote resistance by decreased oseltamivir access to the active site.
Zanamivir has a structural homology to the neuraminidase active site and requires no such reconfiguration. Additionally, mutations promoting resistance to zanamivir may actually decrease viral fitness; thus, resistance to zanamivir is significantly less common than to oseltamivir.55
About 2,000 influenza virus isolates currently circulating in the United States were tested for resistance; only 1% of the 2009 influenza A H1N1 isolates demonstrated resistance to oseltamivir, and none to zanamivir.56
The CDC regularly updates the resistance patterns of circulating influenza strains at www.cdc.gov/flu/weekly/index.htm.
SPECIAL CONSIDERATIONS
Pregnancy
Pregnant women may be at higher risk of severe influenza complications. This was especially true during the 2009 H1N1 pandemic, when pregnant women had a five times higher risk of death from influenza-related complications. Additionally, fever during pregnancy is an independent risk factor for adverse outcomes in the offspring.57 Maternal vaccination against influenza effectively protects the infant for the first 6 months of life, when vaccination is not recommended because of a poor immune response.58
Live-attenuated influenza vaccine is contraindicated during pregnancy. Given the documented risks to the mother from influenza and no documented harm from preservatives in multiuse vaccine vials, the Advisory Committee on Immunization Practices (ACIP) and ACOG do not state a preference for thimerosal-containing or thimerosal-free vaccine for any group, including pregnant women. Pregnant women should be vaccinated with whatever inactivated influenza vaccine formulation is available at the earliest opportunity in the beginning of the influenza season, regardless of the trimester of pregnancy.
Pregnant women are at high risk of influenza-related complications and should be considered for postexposure antiviral prophylaxis or early treatment with a neuraminidase inhibitor. However, both of the approved neuraminidase inhibitors are in pregnancy safety category C, indicating possible adverse effects in animal studies and a lack of safety data in pregnant humans. As with all category C medications, the risks and benefits must be considered, taking into account maternal comorbidities, vaccination status, effectiveness of the season’s influenza vaccine, and the virulence of circulating influenza strains.
As oseltamivir is associated with nausea and gastrointestinal side effects and as zanamivir has less systemic absorption, it may be reasonable to prescribe zanamivir for women already experiencing severe pregnancy-related nausea.
Immunocompromised people
Inactivated influenza vaccine is recommended and live-attenuated influenza vaccine is contraindicated for all immunocompromised people. Generally speaking, any form of immune compromise will decrease the immunogenicity of the vaccine. Additional considerations vary depending on the cause and severity of the immunocompromised status.
HIV-infected patients have higher seroconversion rates when vaccinated with the high-dose vaccine than with the standard-dose vaccine; however, as in adults over age 65, the clinical benefit has yet to be evaluated.59 The efficacy of vaccination is predictably related to the CD4 cell count, as T cells are necessary to mount a response.60 No documented benefit is gained from booster influenza vaccination in this group of patients.
Cancer patients should receive inactivated influenza vaccine every year. Postexposure chemoprophylaxis should be considered, and early treatment with a neuraminidase inhibitor is recommended in patients undergoing chemotherapy.
Solid-organ transplant recipients face a risk of organ rejection if they contract influenza infection, in addition to a higher risk of influenza-related complications.61 Transplant recipients should receive inactivated influenza vaccine as soon as it becomes available at the beginning of every influenza season. Additional research is necessary to evaluate the safety and effectiveness of the high-dose influenza vaccine in this patient group.
MORE OPTIONS, GREAT BENEFIT
Influenza remains a significant source of morbidity and mortality in the United States, and emerging pandemic strains as well as the aging population pose the risk of increased disease burden. New vaccine options offer hope of greater safety, improved efficacy, and higher vaccination rates though broader appeal to individuals. The actual differences in protection between various vaccine options are insignificant relative to the overall benefit of vaccination.
Health care providers should inquire about patients’ understanding and address their concerns about vaccination. Giving an available influenza vaccine within approved indications should not be delayed if alternative vaccine options are not readily available.
In addition to vaccination, patients at high risk of complications should be advised early in the influenza season to inform their doctors about potential exposure to influenza or the development of flu-like symptoms for consideration of early treatment or postexposure prophylaxis with a neuraminidase inhibitor.
- Molinari NA, Ortega-Sanchez IR, Messonnier ML, et al. The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine 2007; 25:5086–5096.
- Soni A. Influenza Immunization Rates for Selected at Risk Populations among the US Adult Civilian Noninstitutionalized Population, 2006. Statistical Brief #226. December 2008. Agency for Healthcare Research and Quality, Rockville, MD. http://meps.ahrq.gov/data_files/publications/st226/stat226.pdf. Accessed January 31, 2014.
- Centers for Disease Control and Prevention (CDC). Flu vaccination coverage, United States, 2012–13 Influenza Season. http://www.cdc.gov/flu/fluvaxview/coverage-1213estimates.htm - age-group-adults. Accessed January 31, 2014.
- Castilla J, Godoy P, Domínguez A, et al; CIBERESP Cases and Controls in Influenza Working Group Spain. Influenza vaccine effectiveness in preventing outpatient, inpatient, and severe cases of laboratory-confirmed influenza. Clin Infect Dis 2013; 57:167–175.
- Talbot HK, Zhu Y, Chen Q, Williams JV, Thompson MG, Griffin MR. Effectiveness of influenza vaccine for preventing laboratory-confirmed influenza hospitalizations in adults, 2011–2012 influenza season. Clin Infect Dis 2013; 56:1774–1777.
- Udell JA, Zawi R, Bhatt DL, et al. Association between influenza vaccination and cardiovascular outcomes in high-risk patients: a meta-analysis. JAMA 2013; 310:1711–1720.
- Centers for Disease Control and Prevention (CDC). Estimated influenza illnesses and hospitalizations averted by influenza vaccination—United States, 2012–13 influenza season. MMWR Morb Mortal Wkly Rep 2013; 62:997–1000.
- Centers for Disease Control and Prevention (CDC). Prevention and control of seasonal influenza with vaccines. Recommendations of the Advisory Committee on Immunization Practices—United States, 2013–2014. MMWR Recomm Rep 2013; 62:1–43.
- Friede M. Snapshot of influenza vaccine manufacturing capacity worldwide and summary of WHO-HHS activities to promote technology transfer. World Health Organization Global Action Plan for Influenza II Meeting 2011. www.who.int/phi/Session1B_Current_Manufacturing_Capacity_Worldwide_Friede.pdf. Accessed February 5, 2014.
- Ashkenazi S, Vertruyen A, Arístegui J, et al., CAIV-T Study Group. Superior relative efficacy of live attenuated influenza vaccine compared with inactivated influenza vaccine in young children with recurrent respiratory tract infections. Pediatr Infect Dis J 2006; 25:870–879.
- Izurieta HS, Haber P, Wise RP, et al. Adverse events reported following live, cold-adapted, intranasal influenza vaccine. JAMA 2005; 294:2720–2725.
- Vesikari T, Karvonen A, Korhonen T, et al; CAIV-T Transmission Study Group. A randomized, double-blind study of the safety, transmissibility and phenotypic and genotypic stability of cold-adapted influenza virus vaccine. Pediatr Infect Dis J 2006; 25:590–595.
- Kamboj M, Sepkowitz KA. Risk of transmission associated with live attenuated vaccines given to healthy persons caring for or residing with an immunocompromised patient. Infect Control Hosp Epidemiol 2007; 28:702–707.
- Centers for Disease Control and Prevention (CDC). Live Attenuated Influenza Vaccine [LAIV] (The Nasal Spray Flu Vaccine). http://www.cdc.gov/flu/about/qa/nasalspray.htm. Accessed February 3, 2014.
- Centers for Disease Control and Prevention (CDC). Interim adjusted estimates of seasonal influenza vaccine effectiveness—United States, February 2013. MMWR Morb Mortal Wkly Rep 2013; 62:119–123.
- Voordouw AC, Sturkenboom MC, Dieleman JP, et al. Annual revaccination against influenza and mortality risk in community-dwelling elderly persons. JAMA 2004; 292:2089–2095.
- Plans-Rubió P. The vaccination coverage required to establish herd immunity against influenza viruses. Prev Med 2012; 55:72–77.
- Aziz NA, Muhamad S, Manaf MR, Hamid MZ. Factors Influencing H1N1 vaccination among primary health care workers: a cross-sectional study. Int J Prev Med 2013; 4:664–670.
- Nichol KL, Margolis KL, Lind A, et al. Side effects associated with influenza vaccination in healthy working adults. A randomized, placebo-controlled trial. Arch Intern Med 1996; 156:1546–1550.
- National Center for Immunization and Respiratory Diseases. General recommendations on immunization—recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2011; 60( 2):1–64.
- Tseng HF, Smith N, Sy LS, Jacobsen LJ. Evaluation of the incidence of herpes zoster after concomitant administration of zoster vaccine and polysaccharide pneumococcal vaccine. Vaccine 2011; 29:3628–3632.
- Offit PA, Quarles J, Gerber MA, et al. Addressing parents’ concerns: do multiple vaccines overwhelm or weaken the infant’s immune system? Pediatrics 2002; 109:124–129.
- Rajabally YA, Uncini A. Outcome and its predictors in Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry 2012; 83:711–718.
- Schonberger LB, Bregman DJ, Sullivan-Bolyai JZ, et al. Guillain-Barré syndrome following vaccination in the National Influenza Immunization Program, United States, 1976—1977. Am J Epidemiol 1979; 110:105–123.
- Lehmann HC, Hartung HP, Kieseier BC, Hughes RA. Guillain-Barré syndrome after exposure to influenza virus. Lancet Infect Dis 2010; 10:643–651.
- Baxter R, Lewis N, Bakshi N, Vellozzi C, Klein NP, Network C. Recurrent Guillain-Barré syndrome following vaccination. Clin Infect Dis 2012; 54:800–804.
- Hughes RA, Wijdicks EF, Benson E, et al. Supportive care for patients with Guillain-Barré syndrome. Arch Neurol 2005; 62:1194–1198.
- Des Roches A, Paradis L, Gagnon R, et al. Egg-allergic patients can be safely vaccinated against influenza. J Allergy Clin Immunol 2012; 130:1213–1216.e1.
- US Centers for Disease Control and Prevention. Influenza vaccination of people with a history of egg allergy. www.immunize.org/catg.d/p3094.pdf. Accessed February 3, 2014.
- US Food Drug Administration. Thimerosal in vaccines. www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/VaccineSafety/UCM096228. Accessed February 3, 2014.
- Davidson PW, Kost J, Myers GJ, Cox C, Clarkson TW, Shamlaye CF. Methylmercury and neurodevelopment: reanalysis of the Seychelles Child Development Study outcomes at 66 months of age. JAMA 2001; 285:1291–1293.
- Grandjean P, Weihe P, White RF, et al. Cognitive deficit in 7-year-old children with prenatal exposure to methylmercury. Neurotoxicol Teratol 1997; 19:417–428.
- Nelson KB, Bauman ML. Thimerosal and autism? Pediatrics 2003; 111:674–679.
- Magos L, Brown AW, Sparrow S, Bailey E, Snowden RT, Skipp WR. The comparative toxicology of ethyl- and methylmercury. Arch Toxicol 1985; 57:260–267.
- American Congress of Obstetricians and Gynecologists. Influenza vaccination during pregnancy. www.acog.org/Resources_And_Publications/Committee_Opinions/Committee_on_Obstetric_Practice/Influenza_Vaccination_During_Pregnancy. Accessed February 3, 2014.
- US Centers for Disease Control and Prevention. Understanding thimerosal, mercury, and vaccine safety. www.cdc.gov/vaccines/hcp/patient-ed/conversations/downloads/vacsafe-thimerosal-color-office.pdf. Accessed February 3, 2014.
- Reed C, Meltzer MI, Finelli L, Fiore A. Public health impact of including two lineages of influenza B in a quadrivalent seasonal influenza vaccine. Vaccine 2012; 30:1993–1998.
- Tsang P, Gorse GJ, Strout CB, et al. Immunogenicity and safety of Fluzone intradermal and high-dose influenza vaccines in older adults ≥65 years of age: a randomized, controlled, phase II trial. Vaccine 2013. doi: 10.1016/j.vaccine.2013.09.074. [Epub ahead of print]
- Sanofi Pasteur. Fluzone package insert. www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM305080.pdf. Accessed February 3, 2014.
- Falsey AR, Treanor JJ, Tornieporth N, Capellan J, Gorse GJ. Randomized, double-blind controlled phase 3 trial comparing the immunogenicity of high-dose and standard-dose influenza vaccine in adults 65 years of age and older. J Infect Dis 2009; 200:172–180.
- US Food Drug Administration. Flucelvax FDA application. www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM332069.pdf. Accessed February 3, 2014.
- Novartis. Flucelvax (influenza virus vaccine) fact sheet. www.novartis-vaccines.com/downloads/flucelvax/Flucelvax_Fact_Sheet.pdf. Accessed February 3, 2014.
- US Centers for Disease Control and Prevention. People at high risk for developing flu-related complications. www.cdc.gov/flu/about/disease/high_risk.htm. Accessed February 3, 2014.
- Roche Pharmaceuticals. Tamiflu package insert. http://www.gene.com/download/pdf/tamiflu_prescribing.pdf. Accessed February 3, 2014.
- GlaxoSmithKline. Relenza package insert. http://us.gsk.com/products/assets/us_relenza.pdf. Accessed February 3, 2014.
- Fiore AE, Fry A, Shay D, et al. Antiviral agents for the treatment and chemoprophylaxis of influenza—recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2011; 60:1–24.
- Administration technique for zanamivir (Relenza) Diskhaler. YouTube. 2009. www.youtube.com/watch?v=sQI0a0ToSPo. Accessed February 6, 2014.
- Nicholson KG, Aoki FY, Osterhaus AD, et al. Efficacy and safety of oseltamivir in treatment of acute influenza: a randomised controlled trial. Neuraminidase Inhibitor Flu Treatment Investigator Group. Lancet 2000; 355:1845–1850.
- Treanor JJ, Hayden FG, Vrooman PS, et al. Efficacy and safety of the oral neuraminidase inhibitor oseltamivir in treating acute influenza: a randomized controlled trial. US Oral Neuraminidase Study Group. JAMA 2000; 283:1016–1624.
- Cooper NJ, Sutton AJ, Abrams KR, Wailoo A, Turner D, Nicholson KG. Effectiveness of neuraminidase inhibitors in treatment and prevention of influenza A and B: systematic review and meta-analyses of randomised controlled trials. BMJ 2003; 326:1235.
- Hayden FG, Osterhaus AD, Treanor JJ, et al. Efficacy and safety of the neuraminidase inhibitor zanamivir in the treatment of influenzavirus infections. GG167 Influenza Study Group. N Engl J Med 1997; 337:874–880.
- Duval X, van der Werf S, Blanchon T, et al. Efficacy of oseltamivir-zanamivir combination compared to each monotherapy for seasonal influenza: a randomized placebo-controlled trial. PLoS Med 2010; 7:e1000362.
- Jefferson T, Jones M, Doshi P, Del Mar C. Neuraminidase inhibitors for preventing and treating influenza in healthy adults: systematic review and meta-analysis. BMJ 2009; 339:b5106.
- Hernán MA, Lipsitch M. Oseltamivir and risk of lower respiratory tract complications in patients with flu symptoms: a meta-analysis of eleven randomized clinical trials. Clin Infect Dis 2011; 53:277–279.
- Samson M, Pizzorno A, Abed Y, Boivin G. Influenza virus resistance to neuraminidase inhibitors. Antiviral Res 2013; 98:174–185.
- US Centers for Disease Control and Prevention. FluView. www.cdc.gov/flu/weekly. Accessed February 3, 2014.
- Acs N, Bánhidy F, Puhó E, Czeizel AE. Maternal influenza during pregnancy and risk of congenital abnormalities in offspring. Birth Defects Res A Clin Mol Teratol 2005; 73:989–996.
- Zaman K, Roy E, Arifeen SE, et al. Effectiveness of maternal influenza immunization in mothers and infants. N Engl J Med 2008; 359:1555–1564.
- McKittrick N, Frank I, Jacobson JM, et al. Improved immunogenicity with high-dose seasonal influenza vaccine in HIV-infected persons: a single-center, parallel, randomized trial. Ann Intern Med 2013; 158:19–26.
- Kroon FP, van Dissel JT, de Jong JC, van Furth R. Antibody response to influenza, tetanus and pneumococcal vaccines in HIV-seropositive individuals in relation to the number of CD4+ lymphocytes. AIDS 1994; 8:469–476.
- Vilchez RA, McCurry K, Dauber J, et al. Influenza virus infection in adult solid organ transplant recipients. Am J Transplant 2002; 2:287–291.
- Molinari NA, Ortega-Sanchez IR, Messonnier ML, et al. The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine 2007; 25:5086–5096.
- Soni A. Influenza Immunization Rates for Selected at Risk Populations among the US Adult Civilian Noninstitutionalized Population, 2006. Statistical Brief #226. December 2008. Agency for Healthcare Research and Quality, Rockville, MD. http://meps.ahrq.gov/data_files/publications/st226/stat226.pdf. Accessed January 31, 2014.
- Centers for Disease Control and Prevention (CDC). Flu vaccination coverage, United States, 2012–13 Influenza Season. http://www.cdc.gov/flu/fluvaxview/coverage-1213estimates.htm - age-group-adults. Accessed January 31, 2014.
- Castilla J, Godoy P, Domínguez A, et al; CIBERESP Cases and Controls in Influenza Working Group Spain. Influenza vaccine effectiveness in preventing outpatient, inpatient, and severe cases of laboratory-confirmed influenza. Clin Infect Dis 2013; 57:167–175.
- Talbot HK, Zhu Y, Chen Q, Williams JV, Thompson MG, Griffin MR. Effectiveness of influenza vaccine for preventing laboratory-confirmed influenza hospitalizations in adults, 2011–2012 influenza season. Clin Infect Dis 2013; 56:1774–1777.
- Udell JA, Zawi R, Bhatt DL, et al. Association between influenza vaccination and cardiovascular outcomes in high-risk patients: a meta-analysis. JAMA 2013; 310:1711–1720.
- Centers for Disease Control and Prevention (CDC). Estimated influenza illnesses and hospitalizations averted by influenza vaccination—United States, 2012–13 influenza season. MMWR Morb Mortal Wkly Rep 2013; 62:997–1000.
- Centers for Disease Control and Prevention (CDC). Prevention and control of seasonal influenza with vaccines. Recommendations of the Advisory Committee on Immunization Practices—United States, 2013–2014. MMWR Recomm Rep 2013; 62:1–43.
- Friede M. Snapshot of influenza vaccine manufacturing capacity worldwide and summary of WHO-HHS activities to promote technology transfer. World Health Organization Global Action Plan for Influenza II Meeting 2011. www.who.int/phi/Session1B_Current_Manufacturing_Capacity_Worldwide_Friede.pdf. Accessed February 5, 2014.
- Ashkenazi S, Vertruyen A, Arístegui J, et al., CAIV-T Study Group. Superior relative efficacy of live attenuated influenza vaccine compared with inactivated influenza vaccine in young children with recurrent respiratory tract infections. Pediatr Infect Dis J 2006; 25:870–879.
- Izurieta HS, Haber P, Wise RP, et al. Adverse events reported following live, cold-adapted, intranasal influenza vaccine. JAMA 2005; 294:2720–2725.
- Vesikari T, Karvonen A, Korhonen T, et al; CAIV-T Transmission Study Group. A randomized, double-blind study of the safety, transmissibility and phenotypic and genotypic stability of cold-adapted influenza virus vaccine. Pediatr Infect Dis J 2006; 25:590–595.
- Kamboj M, Sepkowitz KA. Risk of transmission associated with live attenuated vaccines given to healthy persons caring for or residing with an immunocompromised patient. Infect Control Hosp Epidemiol 2007; 28:702–707.
- Centers for Disease Control and Prevention (CDC). Live Attenuated Influenza Vaccine [LAIV] (The Nasal Spray Flu Vaccine). http://www.cdc.gov/flu/about/qa/nasalspray.htm. Accessed February 3, 2014.
- Centers for Disease Control and Prevention (CDC). Interim adjusted estimates of seasonal influenza vaccine effectiveness—United States, February 2013. MMWR Morb Mortal Wkly Rep 2013; 62:119–123.
- Voordouw AC, Sturkenboom MC, Dieleman JP, et al. Annual revaccination against influenza and mortality risk in community-dwelling elderly persons. JAMA 2004; 292:2089–2095.
- Plans-Rubió P. The vaccination coverage required to establish herd immunity against influenza viruses. Prev Med 2012; 55:72–77.
- Aziz NA, Muhamad S, Manaf MR, Hamid MZ. Factors Influencing H1N1 vaccination among primary health care workers: a cross-sectional study. Int J Prev Med 2013; 4:664–670.
- Nichol KL, Margolis KL, Lind A, et al. Side effects associated with influenza vaccination in healthy working adults. A randomized, placebo-controlled trial. Arch Intern Med 1996; 156:1546–1550.
- National Center for Immunization and Respiratory Diseases. General recommendations on immunization—recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2011; 60( 2):1–64.
- Tseng HF, Smith N, Sy LS, Jacobsen LJ. Evaluation of the incidence of herpes zoster after concomitant administration of zoster vaccine and polysaccharide pneumococcal vaccine. Vaccine 2011; 29:3628–3632.
- Offit PA, Quarles J, Gerber MA, et al. Addressing parents’ concerns: do multiple vaccines overwhelm or weaken the infant’s immune system? Pediatrics 2002; 109:124–129.
- Rajabally YA, Uncini A. Outcome and its predictors in Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry 2012; 83:711–718.
- Schonberger LB, Bregman DJ, Sullivan-Bolyai JZ, et al. Guillain-Barré syndrome following vaccination in the National Influenza Immunization Program, United States, 1976—1977. Am J Epidemiol 1979; 110:105–123.
- Lehmann HC, Hartung HP, Kieseier BC, Hughes RA. Guillain-Barré syndrome after exposure to influenza virus. Lancet Infect Dis 2010; 10:643–651.
- Baxter R, Lewis N, Bakshi N, Vellozzi C, Klein NP, Network C. Recurrent Guillain-Barré syndrome following vaccination. Clin Infect Dis 2012; 54:800–804.
- Hughes RA, Wijdicks EF, Benson E, et al. Supportive care for patients with Guillain-Barré syndrome. Arch Neurol 2005; 62:1194–1198.
- Des Roches A, Paradis L, Gagnon R, et al. Egg-allergic patients can be safely vaccinated against influenza. J Allergy Clin Immunol 2012; 130:1213–1216.e1.
- US Centers for Disease Control and Prevention. Influenza vaccination of people with a history of egg allergy. www.immunize.org/catg.d/p3094.pdf. Accessed February 3, 2014.
- US Food Drug Administration. Thimerosal in vaccines. www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/VaccineSafety/UCM096228. Accessed February 3, 2014.
- Davidson PW, Kost J, Myers GJ, Cox C, Clarkson TW, Shamlaye CF. Methylmercury and neurodevelopment: reanalysis of the Seychelles Child Development Study outcomes at 66 months of age. JAMA 2001; 285:1291–1293.
- Grandjean P, Weihe P, White RF, et al. Cognitive deficit in 7-year-old children with prenatal exposure to methylmercury. Neurotoxicol Teratol 1997; 19:417–428.
- Nelson KB, Bauman ML. Thimerosal and autism? Pediatrics 2003; 111:674–679.
- Magos L, Brown AW, Sparrow S, Bailey E, Snowden RT, Skipp WR. The comparative toxicology of ethyl- and methylmercury. Arch Toxicol 1985; 57:260–267.
- American Congress of Obstetricians and Gynecologists. Influenza vaccination during pregnancy. www.acog.org/Resources_And_Publications/Committee_Opinions/Committee_on_Obstetric_Practice/Influenza_Vaccination_During_Pregnancy. Accessed February 3, 2014.
- US Centers for Disease Control and Prevention. Understanding thimerosal, mercury, and vaccine safety. www.cdc.gov/vaccines/hcp/patient-ed/conversations/downloads/vacsafe-thimerosal-color-office.pdf. Accessed February 3, 2014.
- Reed C, Meltzer MI, Finelli L, Fiore A. Public health impact of including two lineages of influenza B in a quadrivalent seasonal influenza vaccine. Vaccine 2012; 30:1993–1998.
- Tsang P, Gorse GJ, Strout CB, et al. Immunogenicity and safety of Fluzone intradermal and high-dose influenza vaccines in older adults ≥65 years of age: a randomized, controlled, phase II trial. Vaccine 2013. doi: 10.1016/j.vaccine.2013.09.074. [Epub ahead of print]
- Sanofi Pasteur. Fluzone package insert. www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM305080.pdf. Accessed February 3, 2014.
- Falsey AR, Treanor JJ, Tornieporth N, Capellan J, Gorse GJ. Randomized, double-blind controlled phase 3 trial comparing the immunogenicity of high-dose and standard-dose influenza vaccine in adults 65 years of age and older. J Infect Dis 2009; 200:172–180.
- US Food Drug Administration. Flucelvax FDA application. www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM332069.pdf. Accessed February 3, 2014.
- Novartis. Flucelvax (influenza virus vaccine) fact sheet. www.novartis-vaccines.com/downloads/flucelvax/Flucelvax_Fact_Sheet.pdf. Accessed February 3, 2014.
- US Centers for Disease Control and Prevention. People at high risk for developing flu-related complications. www.cdc.gov/flu/about/disease/high_risk.htm. Accessed February 3, 2014.
- Roche Pharmaceuticals. Tamiflu package insert. http://www.gene.com/download/pdf/tamiflu_prescribing.pdf. Accessed February 3, 2014.
- GlaxoSmithKline. Relenza package insert. http://us.gsk.com/products/assets/us_relenza.pdf. Accessed February 3, 2014.
- Fiore AE, Fry A, Shay D, et al. Antiviral agents for the treatment and chemoprophylaxis of influenza—recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2011; 60:1–24.
- Administration technique for zanamivir (Relenza) Diskhaler. YouTube. 2009. www.youtube.com/watch?v=sQI0a0ToSPo. Accessed February 6, 2014.
- Nicholson KG, Aoki FY, Osterhaus AD, et al. Efficacy and safety of oseltamivir in treatment of acute influenza: a randomised controlled trial. Neuraminidase Inhibitor Flu Treatment Investigator Group. Lancet 2000; 355:1845–1850.
- Treanor JJ, Hayden FG, Vrooman PS, et al. Efficacy and safety of the oral neuraminidase inhibitor oseltamivir in treating acute influenza: a randomized controlled trial. US Oral Neuraminidase Study Group. JAMA 2000; 283:1016–1624.
- Cooper NJ, Sutton AJ, Abrams KR, Wailoo A, Turner D, Nicholson KG. Effectiveness of neuraminidase inhibitors in treatment and prevention of influenza A and B: systematic review and meta-analyses of randomised controlled trials. BMJ 2003; 326:1235.
- Hayden FG, Osterhaus AD, Treanor JJ, et al. Efficacy and safety of the neuraminidase inhibitor zanamivir in the treatment of influenzavirus infections. GG167 Influenza Study Group. N Engl J Med 1997; 337:874–880.
- Duval X, van der Werf S, Blanchon T, et al. Efficacy of oseltamivir-zanamivir combination compared to each monotherapy for seasonal influenza: a randomized placebo-controlled trial. PLoS Med 2010; 7:e1000362.
- Jefferson T, Jones M, Doshi P, Del Mar C. Neuraminidase inhibitors for preventing and treating influenza in healthy adults: systematic review and meta-analysis. BMJ 2009; 339:b5106.
- Hernán MA, Lipsitch M. Oseltamivir and risk of lower respiratory tract complications in patients with flu symptoms: a meta-analysis of eleven randomized clinical trials. Clin Infect Dis 2011; 53:277–279.
- Samson M, Pizzorno A, Abed Y, Boivin G. Influenza virus resistance to neuraminidase inhibitors. Antiviral Res 2013; 98:174–185.
- US Centers for Disease Control and Prevention. FluView. www.cdc.gov/flu/weekly. Accessed February 3, 2014.
- Acs N, Bánhidy F, Puhó E, Czeizel AE. Maternal influenza during pregnancy and risk of congenital abnormalities in offspring. Birth Defects Res A Clin Mol Teratol 2005; 73:989–996.
- Zaman K, Roy E, Arifeen SE, et al. Effectiveness of maternal influenza immunization in mothers and infants. N Engl J Med 2008; 359:1555–1564.
- McKittrick N, Frank I, Jacobson JM, et al. Improved immunogenicity with high-dose seasonal influenza vaccine in HIV-infected persons: a single-center, parallel, randomized trial. Ann Intern Med 2013; 158:19–26.
- Kroon FP, van Dissel JT, de Jong JC, van Furth R. Antibody response to influenza, tetanus and pneumococcal vaccines in HIV-seropositive individuals in relation to the number of CD4+ lymphocytes. AIDS 1994; 8:469–476.
- Vilchez RA, McCurry K, Dauber J, et al. Influenza virus infection in adult solid organ transplant recipients. Am J Transplant 2002; 2:287–291.
KEY POINTS
- Influenza vaccination is effective at preventing influenza-associated disease.
- Influenza vaccine is safe in people with a history of mild egg allergy.
- Many new vaccine formulations exist and may offer benefits to different patient groups.
- Neuraminidase inhibitors are recommended for treatment and postexposure prophylaxis in patients at high risk of influenza-related complications; however, they are not a substitute for vaccination.
Placenta fails to deliver: Mother dies of hemorrhage
PLACENTA FAILS TO DELIVER: MOTHER DIES OF HEMORRHAGE
After a 38-year-old woman gave birth, the placenta did not deliver. The ObGyn was unable remove the entire placenta and the mother began to hemorrhage. After an hour, the patient was given a blood transfusion. She could not be stabilized and died.
ESTATE’S CLAIM The ObGyn was negligent. He failed to remove the entire placenta and did not treat the hemorrhage in a timely manner. The hospital staff was negligent in failing to properly address the massive hemorrhage. A prompt transfusion would have saved the woman’s life, but the anesthesiologist who had to approve the procedure could not be located. Other procedures, including a hysterectomy, could have saved the mother’s life.
DEFENDANTS’ DEFENSE The ObGyn claimed that incomplete delivery of the placenta and postpartum hemorrhage are known complications of a delivery. The hospital claimed that the staff had acted appropriately and that it was not responsible for the actions of the anesthesiologist, an independent contractor. The anesthesiologist denied negligence.
VERDICT A $2 million New York settlement was reached that included $200,000 from the hospital and $1.8 million from the physicians’ insurers.
Related Article: Postpartum hemorrhage: 11 critical questions, answered by an expert Haywood L. Brown, MD (January 2011)
DECREASED FETAL MOVEMENT OVERLOOKED; SEVERE INJURY TO BABY
At her 39th-week prenatal visit at a clinic, the mother reported decreased fetal movement. Acoustic stimulation of the fetus was attempted twice without response. The fetal heart-rate monitor identified a normal heart rate without variability or accelerations. The mother was taken by wheelchair to the hospital next door. A note explaining the nonreassuring findings allegedly accompanied her.
The mother waited to be admitted. When a fetal heart-rate monitor was connected 30 minutes after admission, results were still nonreassuring.
A resident examined the mother 45 minutes later. He called the attending ObGyn, and they decided to postpone cesarean delivery because the mother had eaten breakfast.
When the fetal heart rate crashed 4 hours later, a second-year resident began emergency cesarean delivery. The ObGyn, who had never examined the patient, observed some of the procedure in the OR.
The baby was born with catastrophic brain damage, and has spastic quadriplegia cerebral palsy, feeding problems, and significant cognitive and developmental delays.
PARENTS’ CLAIM A cesarean delivery should have been performed immediately after the mother’s admission. Even if the cesarean had been begun 15 to 20 minutes earlier, the injury could have been avoided. The ObGyn never examined the mother nor did he participate in the cesarean delivery.
DEFENDANTS’ DEFENSE The ObGyn and hospital denied negligence. The note was not attached to the patient’s chart. At trial, the ObGyn admitted that a delivery 15 to 20 minutes earlier might have avoided the injury.
VERDICT A $33,591,900 Tennessee verdict was returned.
WOMAN BECOMES PREGNANT AFTER TUBAL LIGATION
A 32-year-old woman requested sterilization after the birth of her third child. A Falope ring tubal ligation procedure was performed by a gynecologist in April 2006. During surgery, the device used by the gynecologist ejected 2 silastic bands on the right side instead of one.
The patient learned she was pregnant in March 2007. Her high-risk pregnancy ended with cesarean delivery in September 2007. The delivering ObGyn found the patient’s right fallopian tube in its natural, unscarred state. A silastic band was applied to the right ovarian ligament, not the right fallopian tube.
PATIENT’S CLAIM The gynecologist banded the ovarian ligament instead of the fallopian tube.
PHYSICIAN’S DEFENSE The procedure was properly performed. The rings initially enclosed the fallopian tube and ovarian ligament, but the top ring subsequently migrated off the structures, allowing the fallopian tube to slip out of the attachment. Failure to sterilize is a known risk of the procedure.
VERDICT An Illinois defense verdict was returned.
ABORTION ATTEMPTED BUT PREGNANCY IS ECTOPIC
A 14-year-old patient went to a clinic for elective abortion at 8 weeks’ gestation. Ultrasonography (US) prior to the procedure showed an intrauterine pregnancy. After dilating the cervix, the ObGyn inserted a semi-rigid vacuum aspiration curette to suction the uterine contents, but received nothing. A second US confirmed an intrauterine pregnancy. The ObGyn was able to locate the pregnancy and indent the gestational sac with 3 different dilators and the curette. The pregnancy decreased in size on US after the suction was applied. However, the patient’s vital signs dropped dramatically, and she was rushed to the hospital. During emergency surgery, severe pelvic adhesive disease complicated the ability to stop the hemorrhage. Four physicians concurred that supracervical hysterectomy was needed to save the patient’s life. Postoperative pathology identified a cornual or interstitial ectopic pregnancy.
PATIENT’S CLAIM The ObGyn failed to heed several warning signs of ectopic pregnancy. Further testing should have been done before the second round of vacuum. If ectopic pregnancy had been discovered earlier, the patient could have undergone surgery that would have preserved her uterus and allowed her to bear children. The ObGyn tore the uterus multiple times when he turned on the suction, causing massive hemorrhage.
PHYSICIAN’S DEFENSE Ultrasonography clearly showed an intrauterine pregnancy. There was nothing to cause suspicion that the pregnancy was ectopic. She might be able to have a child through surrogacy.
VERDICT A $950,000 Illinois verdict was returned.
Related Article: Is the hCG discriminatory zone a reliable indicator of intrauterine or ectopic pregnancy? Andrew M. Kaunitz, MD (Examining the Evidence, February 2012)
MACROSOMIC FETUS: MOTHER AND BABY BOTH INJURED
When prenatal ultrasonography indicated the fetal weight was 10 lbs, the patient and her mother expressed concern over delivery of such a large baby. The ObGyn reassured them that it would not be a problem.
Four days later, the mother went into labor. She was 9-cm dilated 4.5 hours later, but only progressed to 9.5 cm over the next 7 hours. She was told to begin to push, but, after 2 hours, birth had not occurred. The ObGyn used forceps to deliver the head 45 minutes later. Shoulder dystocia was encountered and there was a 3.5-minute delivery delay. The baby suffered oxygen deprivation and the mother experienced a 4th-degree perineal tear.
After the NICU team resuscitated the baby, she was transferred to another hospital, where she underwent “head cooling” in an attempt to mitigate her injuries. The child has mild cerebral palsy, with right hemiparesis, speech delay, and additional neurologic injuries.
PARENTS' CLAIM Cesarean delivery was unnecessarily delayed. The ObGyn was negligent in not performing an emergency cesarean delivery after 2 hours of pushing was not effective. The ObGyn never suggested a cesarean delivery, it was not noted in the chart, and no one else present at the time remembered the option being offered.
PHYSICIAN’S DEFENSE There was nothing during labor to contraindicate a vaginal birth. The ObGyn claimed that he offered a cesarean delivery after 2 hours of pushing. The baby’s blood gas reading at delivery was normal. Any brain injuries to the baby were from resuscitation.
VERDICT A $4,080,500 Pennsylvania verdict was returned.
Related Articles:
When macrosomia is suspected at term, does induction of labor lower the risk of cesarean delivery? Jennifer T. Ahn, MD (Examining the Evidence, May 2012)
Develop and use a checklist for 3rd- and 4th-degree perinatal lacerations Robert L. Barbieri, MD (Editorial, August 2013)
BOWEL INJURY DURING CESAREAN DELIVERY
During cesarean delivery, the mother suffered a bowel injury that led to infection and several abdominal abscesses. She required two procedures for drain placement plus two additional operations.
PATIENT’S CLAIM The ObGyn was negligent in how he performed the cesarean delivery and for not treating the injury and subsequent infection in a timely manner. The abscesses took 3 years to resolve; additional procedures left scarring and aggravated a spinal injury.
PHYSICIAN’S DEFENSE Bowel perforation is a known complication of cesarean delivery. It probably occurred during manipulation of the uterus in an area that was not visible.
VERDICT A $750,000 New Jersey verdict was returned.
Related Article: How to avoid intestinal and urinary tract injuries during gynecologic laparoscopy Michael Baggish, MD (Surgical Technique, October 2012)
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
TELL US WHAT YOU THINK!
Share your thoughts on this article or on any topic relevant to ObGyns and women’s health practitioners. Tell us which topics you’d like to see covered in future issues, and what challenges you face in daily practice. We will consider publishing your letter and in a future issue.
Send your letter to: [email protected] Please include the city and state in which you practice.
Stay in touch! Your feedback is important to us!
PLACENTA FAILS TO DELIVER: MOTHER DIES OF HEMORRHAGE
After a 38-year-old woman gave birth, the placenta did not deliver. The ObGyn was unable remove the entire placenta and the mother began to hemorrhage. After an hour, the patient was given a blood transfusion. She could not be stabilized and died.
ESTATE’S CLAIM The ObGyn was negligent. He failed to remove the entire placenta and did not treat the hemorrhage in a timely manner. The hospital staff was negligent in failing to properly address the massive hemorrhage. A prompt transfusion would have saved the woman’s life, but the anesthesiologist who had to approve the procedure could not be located. Other procedures, including a hysterectomy, could have saved the mother’s life.
DEFENDANTS’ DEFENSE The ObGyn claimed that incomplete delivery of the placenta and postpartum hemorrhage are known complications of a delivery. The hospital claimed that the staff had acted appropriately and that it was not responsible for the actions of the anesthesiologist, an independent contractor. The anesthesiologist denied negligence.
VERDICT A $2 million New York settlement was reached that included $200,000 from the hospital and $1.8 million from the physicians’ insurers.
Related Article: Postpartum hemorrhage: 11 critical questions, answered by an expert Haywood L. Brown, MD (January 2011)
DECREASED FETAL MOVEMENT OVERLOOKED; SEVERE INJURY TO BABY
At her 39th-week prenatal visit at a clinic, the mother reported decreased fetal movement. Acoustic stimulation of the fetus was attempted twice without response. The fetal heart-rate monitor identified a normal heart rate without variability or accelerations. The mother was taken by wheelchair to the hospital next door. A note explaining the nonreassuring findings allegedly accompanied her.
The mother waited to be admitted. When a fetal heart-rate monitor was connected 30 minutes after admission, results were still nonreassuring.
A resident examined the mother 45 minutes later. He called the attending ObGyn, and they decided to postpone cesarean delivery because the mother had eaten breakfast.
When the fetal heart rate crashed 4 hours later, a second-year resident began emergency cesarean delivery. The ObGyn, who had never examined the patient, observed some of the procedure in the OR.
The baby was born with catastrophic brain damage, and has spastic quadriplegia cerebral palsy, feeding problems, and significant cognitive and developmental delays.
PARENTS’ CLAIM A cesarean delivery should have been performed immediately after the mother’s admission. Even if the cesarean had been begun 15 to 20 minutes earlier, the injury could have been avoided. The ObGyn never examined the mother nor did he participate in the cesarean delivery.
DEFENDANTS’ DEFENSE The ObGyn and hospital denied negligence. The note was not attached to the patient’s chart. At trial, the ObGyn admitted that a delivery 15 to 20 minutes earlier might have avoided the injury.
VERDICT A $33,591,900 Tennessee verdict was returned.
WOMAN BECOMES PREGNANT AFTER TUBAL LIGATION
A 32-year-old woman requested sterilization after the birth of her third child. A Falope ring tubal ligation procedure was performed by a gynecologist in April 2006. During surgery, the device used by the gynecologist ejected 2 silastic bands on the right side instead of one.
The patient learned she was pregnant in March 2007. Her high-risk pregnancy ended with cesarean delivery in September 2007. The delivering ObGyn found the patient’s right fallopian tube in its natural, unscarred state. A silastic band was applied to the right ovarian ligament, not the right fallopian tube.
PATIENT’S CLAIM The gynecologist banded the ovarian ligament instead of the fallopian tube.
PHYSICIAN’S DEFENSE The procedure was properly performed. The rings initially enclosed the fallopian tube and ovarian ligament, but the top ring subsequently migrated off the structures, allowing the fallopian tube to slip out of the attachment. Failure to sterilize is a known risk of the procedure.
VERDICT An Illinois defense verdict was returned.
ABORTION ATTEMPTED BUT PREGNANCY IS ECTOPIC
A 14-year-old patient went to a clinic for elective abortion at 8 weeks’ gestation. Ultrasonography (US) prior to the procedure showed an intrauterine pregnancy. After dilating the cervix, the ObGyn inserted a semi-rigid vacuum aspiration curette to suction the uterine contents, but received nothing. A second US confirmed an intrauterine pregnancy. The ObGyn was able to locate the pregnancy and indent the gestational sac with 3 different dilators and the curette. The pregnancy decreased in size on US after the suction was applied. However, the patient’s vital signs dropped dramatically, and she was rushed to the hospital. During emergency surgery, severe pelvic adhesive disease complicated the ability to stop the hemorrhage. Four physicians concurred that supracervical hysterectomy was needed to save the patient’s life. Postoperative pathology identified a cornual or interstitial ectopic pregnancy.
PATIENT’S CLAIM The ObGyn failed to heed several warning signs of ectopic pregnancy. Further testing should have been done before the second round of vacuum. If ectopic pregnancy had been discovered earlier, the patient could have undergone surgery that would have preserved her uterus and allowed her to bear children. The ObGyn tore the uterus multiple times when he turned on the suction, causing massive hemorrhage.
PHYSICIAN’S DEFENSE Ultrasonography clearly showed an intrauterine pregnancy. There was nothing to cause suspicion that the pregnancy was ectopic. She might be able to have a child through surrogacy.
VERDICT A $950,000 Illinois verdict was returned.
Related Article: Is the hCG discriminatory zone a reliable indicator of intrauterine or ectopic pregnancy? Andrew M. Kaunitz, MD (Examining the Evidence, February 2012)
MACROSOMIC FETUS: MOTHER AND BABY BOTH INJURED
When prenatal ultrasonography indicated the fetal weight was 10 lbs, the patient and her mother expressed concern over delivery of such a large baby. The ObGyn reassured them that it would not be a problem.
Four days later, the mother went into labor. She was 9-cm dilated 4.5 hours later, but only progressed to 9.5 cm over the next 7 hours. She was told to begin to push, but, after 2 hours, birth had not occurred. The ObGyn used forceps to deliver the head 45 minutes later. Shoulder dystocia was encountered and there was a 3.5-minute delivery delay. The baby suffered oxygen deprivation and the mother experienced a 4th-degree perineal tear.
After the NICU team resuscitated the baby, she was transferred to another hospital, where she underwent “head cooling” in an attempt to mitigate her injuries. The child has mild cerebral palsy, with right hemiparesis, speech delay, and additional neurologic injuries.
PARENTS' CLAIM Cesarean delivery was unnecessarily delayed. The ObGyn was negligent in not performing an emergency cesarean delivery after 2 hours of pushing was not effective. The ObGyn never suggested a cesarean delivery, it was not noted in the chart, and no one else present at the time remembered the option being offered.
PHYSICIAN’S DEFENSE There was nothing during labor to contraindicate a vaginal birth. The ObGyn claimed that he offered a cesarean delivery after 2 hours of pushing. The baby’s blood gas reading at delivery was normal. Any brain injuries to the baby were from resuscitation.
VERDICT A $4,080,500 Pennsylvania verdict was returned.
Related Articles:
When macrosomia is suspected at term, does induction of labor lower the risk of cesarean delivery? Jennifer T. Ahn, MD (Examining the Evidence, May 2012)
Develop and use a checklist for 3rd- and 4th-degree perinatal lacerations Robert L. Barbieri, MD (Editorial, August 2013)
BOWEL INJURY DURING CESAREAN DELIVERY
During cesarean delivery, the mother suffered a bowel injury that led to infection and several abdominal abscesses. She required two procedures for drain placement plus two additional operations.
PATIENT’S CLAIM The ObGyn was negligent in how he performed the cesarean delivery and for not treating the injury and subsequent infection in a timely manner. The abscesses took 3 years to resolve; additional procedures left scarring and aggravated a spinal injury.
PHYSICIAN’S DEFENSE Bowel perforation is a known complication of cesarean delivery. It probably occurred during manipulation of the uterus in an area that was not visible.
VERDICT A $750,000 New Jersey verdict was returned.
Related Article: How to avoid intestinal and urinary tract injuries during gynecologic laparoscopy Michael Baggish, MD (Surgical Technique, October 2012)
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
TELL US WHAT YOU THINK!
Share your thoughts on this article or on any topic relevant to ObGyns and women’s health practitioners. Tell us which topics you’d like to see covered in future issues, and what challenges you face in daily practice. We will consider publishing your letter and in a future issue.
Send your letter to: [email protected] Please include the city and state in which you practice.
Stay in touch! Your feedback is important to us!
PLACENTA FAILS TO DELIVER: MOTHER DIES OF HEMORRHAGE
After a 38-year-old woman gave birth, the placenta did not deliver. The ObGyn was unable remove the entire placenta and the mother began to hemorrhage. After an hour, the patient was given a blood transfusion. She could not be stabilized and died.
ESTATE’S CLAIM The ObGyn was negligent. He failed to remove the entire placenta and did not treat the hemorrhage in a timely manner. The hospital staff was negligent in failing to properly address the massive hemorrhage. A prompt transfusion would have saved the woman’s life, but the anesthesiologist who had to approve the procedure could not be located. Other procedures, including a hysterectomy, could have saved the mother’s life.
DEFENDANTS’ DEFENSE The ObGyn claimed that incomplete delivery of the placenta and postpartum hemorrhage are known complications of a delivery. The hospital claimed that the staff had acted appropriately and that it was not responsible for the actions of the anesthesiologist, an independent contractor. The anesthesiologist denied negligence.
VERDICT A $2 million New York settlement was reached that included $200,000 from the hospital and $1.8 million from the physicians’ insurers.
Related Article: Postpartum hemorrhage: 11 critical questions, answered by an expert Haywood L. Brown, MD (January 2011)
DECREASED FETAL MOVEMENT OVERLOOKED; SEVERE INJURY TO BABY
At her 39th-week prenatal visit at a clinic, the mother reported decreased fetal movement. Acoustic stimulation of the fetus was attempted twice without response. The fetal heart-rate monitor identified a normal heart rate without variability or accelerations. The mother was taken by wheelchair to the hospital next door. A note explaining the nonreassuring findings allegedly accompanied her.
The mother waited to be admitted. When a fetal heart-rate monitor was connected 30 minutes after admission, results were still nonreassuring.
A resident examined the mother 45 minutes later. He called the attending ObGyn, and they decided to postpone cesarean delivery because the mother had eaten breakfast.
When the fetal heart rate crashed 4 hours later, a second-year resident began emergency cesarean delivery. The ObGyn, who had never examined the patient, observed some of the procedure in the OR.
The baby was born with catastrophic brain damage, and has spastic quadriplegia cerebral palsy, feeding problems, and significant cognitive and developmental delays.
PARENTS’ CLAIM A cesarean delivery should have been performed immediately after the mother’s admission. Even if the cesarean had been begun 15 to 20 minutes earlier, the injury could have been avoided. The ObGyn never examined the mother nor did he participate in the cesarean delivery.
DEFENDANTS’ DEFENSE The ObGyn and hospital denied negligence. The note was not attached to the patient’s chart. At trial, the ObGyn admitted that a delivery 15 to 20 minutes earlier might have avoided the injury.
VERDICT A $33,591,900 Tennessee verdict was returned.
WOMAN BECOMES PREGNANT AFTER TUBAL LIGATION
A 32-year-old woman requested sterilization after the birth of her third child. A Falope ring tubal ligation procedure was performed by a gynecologist in April 2006. During surgery, the device used by the gynecologist ejected 2 silastic bands on the right side instead of one.
The patient learned she was pregnant in March 2007. Her high-risk pregnancy ended with cesarean delivery in September 2007. The delivering ObGyn found the patient’s right fallopian tube in its natural, unscarred state. A silastic band was applied to the right ovarian ligament, not the right fallopian tube.
PATIENT’S CLAIM The gynecologist banded the ovarian ligament instead of the fallopian tube.
PHYSICIAN’S DEFENSE The procedure was properly performed. The rings initially enclosed the fallopian tube and ovarian ligament, but the top ring subsequently migrated off the structures, allowing the fallopian tube to slip out of the attachment. Failure to sterilize is a known risk of the procedure.
VERDICT An Illinois defense verdict was returned.
ABORTION ATTEMPTED BUT PREGNANCY IS ECTOPIC
A 14-year-old patient went to a clinic for elective abortion at 8 weeks’ gestation. Ultrasonography (US) prior to the procedure showed an intrauterine pregnancy. After dilating the cervix, the ObGyn inserted a semi-rigid vacuum aspiration curette to suction the uterine contents, but received nothing. A second US confirmed an intrauterine pregnancy. The ObGyn was able to locate the pregnancy and indent the gestational sac with 3 different dilators and the curette. The pregnancy decreased in size on US after the suction was applied. However, the patient’s vital signs dropped dramatically, and she was rushed to the hospital. During emergency surgery, severe pelvic adhesive disease complicated the ability to stop the hemorrhage. Four physicians concurred that supracervical hysterectomy was needed to save the patient’s life. Postoperative pathology identified a cornual or interstitial ectopic pregnancy.
PATIENT’S CLAIM The ObGyn failed to heed several warning signs of ectopic pregnancy. Further testing should have been done before the second round of vacuum. If ectopic pregnancy had been discovered earlier, the patient could have undergone surgery that would have preserved her uterus and allowed her to bear children. The ObGyn tore the uterus multiple times when he turned on the suction, causing massive hemorrhage.
PHYSICIAN’S DEFENSE Ultrasonography clearly showed an intrauterine pregnancy. There was nothing to cause suspicion that the pregnancy was ectopic. She might be able to have a child through surrogacy.
VERDICT A $950,000 Illinois verdict was returned.
Related Article: Is the hCG discriminatory zone a reliable indicator of intrauterine or ectopic pregnancy? Andrew M. Kaunitz, MD (Examining the Evidence, February 2012)
MACROSOMIC FETUS: MOTHER AND BABY BOTH INJURED
When prenatal ultrasonography indicated the fetal weight was 10 lbs, the patient and her mother expressed concern over delivery of such a large baby. The ObGyn reassured them that it would not be a problem.
Four days later, the mother went into labor. She was 9-cm dilated 4.5 hours later, but only progressed to 9.5 cm over the next 7 hours. She was told to begin to push, but, after 2 hours, birth had not occurred. The ObGyn used forceps to deliver the head 45 minutes later. Shoulder dystocia was encountered and there was a 3.5-minute delivery delay. The baby suffered oxygen deprivation and the mother experienced a 4th-degree perineal tear.
After the NICU team resuscitated the baby, she was transferred to another hospital, where she underwent “head cooling” in an attempt to mitigate her injuries. The child has mild cerebral palsy, with right hemiparesis, speech delay, and additional neurologic injuries.
PARENTS' CLAIM Cesarean delivery was unnecessarily delayed. The ObGyn was negligent in not performing an emergency cesarean delivery after 2 hours of pushing was not effective. The ObGyn never suggested a cesarean delivery, it was not noted in the chart, and no one else present at the time remembered the option being offered.
PHYSICIAN’S DEFENSE There was nothing during labor to contraindicate a vaginal birth. The ObGyn claimed that he offered a cesarean delivery after 2 hours of pushing. The baby’s blood gas reading at delivery was normal. Any brain injuries to the baby were from resuscitation.
VERDICT A $4,080,500 Pennsylvania verdict was returned.
Related Articles:
When macrosomia is suspected at term, does induction of labor lower the risk of cesarean delivery? Jennifer T. Ahn, MD (Examining the Evidence, May 2012)
Develop and use a checklist for 3rd- and 4th-degree perinatal lacerations Robert L. Barbieri, MD (Editorial, August 2013)
BOWEL INJURY DURING CESAREAN DELIVERY
During cesarean delivery, the mother suffered a bowel injury that led to infection and several abdominal abscesses. She required two procedures for drain placement plus two additional operations.
PATIENT’S CLAIM The ObGyn was negligent in how he performed the cesarean delivery and for not treating the injury and subsequent infection in a timely manner. The abscesses took 3 years to resolve; additional procedures left scarring and aggravated a spinal injury.
PHYSICIAN’S DEFENSE Bowel perforation is a known complication of cesarean delivery. It probably occurred during manipulation of the uterus in an area that was not visible.
VERDICT A $750,000 New Jersey verdict was returned.
Related Article: How to avoid intestinal and urinary tract injuries during gynecologic laparoscopy Michael Baggish, MD (Surgical Technique, October 2012)
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
TELL US WHAT YOU THINK!
Share your thoughts on this article or on any topic relevant to ObGyns and women’s health practitioners. Tell us which topics you’d like to see covered in future issues, and what challenges you face in daily practice. We will consider publishing your letter and in a future issue.
Send your letter to: [email protected] Please include the city and state in which you practice.
Stay in touch! Your feedback is important to us!
Antithrombotic Therapy Management
The periprocedural management of antithrombotic medications is a common challenge for hospitalists, for which there is limited high‐quality evidence to guide clinical decision making. The introduction of third‐generation antiplatelet agents (prasugrel and ticagrelor) and the new oral anticoagulants (rivaroxaban, apixaban, and dabigatran), has added an additional layer of complexity to clinical management.
This article will provide a conceptual framework for the periprocedural management of antithrombotic therapy, with a particular focus on procedures that are considered core competencies by the Society of Hospital Medicine; these include: arthrocentesis, lumbar puncture, paracentesis, thoracentesis, and central line placement (Table 1).[1, 2] The recommendations in this article are based on a review of published guidelines and consensus statements and their supporting literature.[3, 4, 5, 6, 7, 8] Additional articles were identified by performing a PubMed keyword search using the terms perioperative management or periprocedural management and anticoagulation or antithrombotic or antiplatelet in combination with keywords relevant to the content areas (eg, arthrocentesis, lumbar puncture). Articles for inclusion were chosen based on methodological quality and relevance to hospital medicine.
There are several questions that must be addressed when developing a periprocedural antithrombotic management strategy:
- What is the patient's risk of bleeding if antithrombotic therapy is continued?
- What is the patient's risk of thromboembolism if antithrombotic therapy is interrupted?
- Are there interventions that can decrease the risk of periprocedural bleeding and/or thromboembolism?
WHAT IS THE PATIENT'S RISK OF BLEEDING IF ANTITHROMBOTIC THERAPY IS CONTINUED?
Although the risk of bleeding is well described for many procedures, there are limited data on how that risk is affected by coagulopathy in general and antithrombotic medications in particular. When these data are available, they are largely derived from case series or bridging registries, which include heterogeneous patient populations and nonstandardized definitions of bleeding.[8, 9, 10] As such, few procedural or surgical professional societies have published guidelines on the periprocedural management of antithrombotic therapy,[3, 4, 5, 11]and guidelines from the American College of Chest Physicians (ACCP), the American College of Cardiology (ACC), and American Heart Association (AHA) only provide specific recommendations regarding minor ambulatory procedures.[6, 7, 8]
Procedures can be categorized as low or high risk for bleeding based on the following considerations: the extent of associated tissue injury, proximity to vital organs or vascular structures, the ability to readily detect and control bleeding, and the morbidity associated with a bleeding complication (eg, a small bleed into the epidural space is potentially catastrophic, whereas a large bleed from the colon often results in no permanent harm). For procedures with a high risk or consequence of bleeding, anticoagulants must be stopped, whereas in some cases antiplatelet agents can be safely continued. For procedures with a low risk or consequence of bleeding, it may be possible to continue both anticoagulant and antiplatelet agents.
| Procedure | Antithrombotic Therapy | |||||
|---|---|---|---|---|---|---|
| Aspirin | Thienopyridines | Prophylactic UFH or LWMH | Therapeutic UFH or LMWH | Warfarin | NOACs | |
| ||||||
| Arthrocentesis[12, 13, 14, 15] | + | + | + | + | + | + |
| Lumbar puncture[3] | + | 5000 units UFH BID | ||||
| Paracentesis[28, 29, 30] | + | + | + | |||
| Thoracentesis[37, 38, 39, 40, 41, 42] | + | + | + | |||
| Central venous catheter insertion[48, 49, 50, 51, 52, 53] | + | + | + | |||
Because procedures in hospitalized patients are most often performed for the purpose of diagnosing or treating an emergent condition, the risk of delaying the procedure while antithrombotic medications are held must be part of the overall risk‐benefit calculation.
Arthrocentesis
Bleeding complications from arthrocentesis are very rare, and there are few data on the additional risk associated with antithrombotic therapy.[12, 13, 14] In a retrospective cohort study, investigators determined the incidence of clinically significant bleeding (defined as bleeding requiring reversal of anticoagulation, prolonged manual pressure, surgical intervention, hospital admission, or delay in hospital discharge) and procedure‐related pain among 514 patients on antithrombotic therapy referred for arthrocentesis or injection of the hip, shoulder, or knee. Four hundred fifty‐six procedures were performed in patients without interrupting warfarin therapy, all of whom maintained an international normalized ratio (INR)2, and 184 procedures were performed in patients who had stopped their warfarin to achieve an INR <2. Antiplatelet therapy was routinely continued in both groups, with 48% of patients taking aspirin and 9% clopidogrel. There was 1 bleeding complication (0.2%) in a patient with an INR of 2.3 who was also taking aspirin, and 2 patients developed procedure‐related pain (INR 3.3 and 5.3, neither taking antiplatelet medications).[15]
Based on the available evidence, arthrocentesis appears to be safe in patients on therapeutic warfarin, with or without aspirin and/or clopidogrel. At present, there are no published studies that address the risk of arthrocentesis in patients taking other antiplatelet or anticoagulant medications, but given the low overall risk of this procedure, it is reasonable to infer that these medications can also be safely continued.
Lumbar Puncture
The incidence of bleeding complications from diagnostic lumbar puncture is unknown, but is likely similar to that seen with spinal anesthesia, where in a large retrospective observational study, spinal hematoma occurred in 1:165,000 spinal block procedures.[16] Factors associated with an increased risk of spinal hematoma include traumatic tap, advanced age, female gender, spinal cord or vertebral column abnormalities, coagulopathy, and not allowing sufficient time between stopping and restarting antithrombotic therapy.[3, 17, 18, 19, 20]
Therapeutic anticoagulation must be stopped and prophylactic anticoagulation delayed before performing a lumbar puncture. The 1 exception is low‐dose unfractionated heparin (UFH), which the American Society for Regional Anesthesia (ARSA) recommends continuing in patients undergoing neuraxial procedures, provided the total dose is 5000 U twice daily. This assessment is based on observational data, surveys of practice patterns, and decades of use without evidence of complications; in fact, there are only 5 case reports of spinal hematomas in this population.[3] However, because these data are from surgical populations, in which heparin thromboprophylaxis is typically dosed at 5000 units twice daily, there are limited data on the safety of higher or more frequent doses of heparin. In a retrospective cohort study of 928 patients who received thoracic epidural analgesia in conjunction with UFH dosed at 5000 U, 3 times daily, there were no cases of neuraxial bleeding, but given the rarity of neuraxial hematoma, it is not possible to draw any conclusions from this relatively small sample size.[21]
In November 2013, based on surveillance data showing increased risk for spinal or epidural hematoma associated with low‐molecular‐weight heparin (LMWH), the US Food and Drug Administration (FDA) issued a drug safety communication recommending that neuraxial procedures be delayed for 12 hours after prophylactic LMWH and 24 hours after therapeutic LMWH, and that LMWH not be restarted for at least 4 hours after catheter removal.[20] These recommendations are largely consistent with existing guidelines[3, 22] but are not explicitly stated in the package insert for any of the LMWHs available in the United States,[23, 24, 25] and the FDA is working with the manufacturers to add this information.
Nonsteroidal anti‐inflammatory drugs (NSAIDs), dipyridamole, and aspirin do not appear to increase the risk of spinal hematoma and are considered safe to continue.[11, 26] There are limited data on the safety of thienopyridine medications in neuraxial anesthesia, but based on case reports and increased bleeding rates seen in surgical populations, it is generally recommended that these medications be discontinued before performing a lumbar puncture.[3, 22, 27]
The optimal time to restart anticoagulation after a lumbar puncture is unknown. The ARSA recommends a minimum of 1 hour for UFH and 2 hours for LMWH after neuraxial catheter removal, and provides no specific guidance about other anticoagulants,[3] whereas the European Society of Anesthesiology recommends a minimum of 1 hour for UFH, 4 hours for LMWH, 4 to 6 hours for rivaroxaban and apixiban, and 6 hours for dabigatran and fondaparinux.[22] Longer time periods should be considered after a traumatic tap, and postprocedure monitoring of neurological function is recommended for all patients.
The available evidence suggests that lumbar puncture can be safely performed in patients being treated with aspirin, NSAIDs, and UFH dosed at 5000 U twice daily; the safety of higher or more frequent doses of UFH is not known. Lumbar puncture should be delayed 12 hours after prophylactic LMWH and 24 hours after therapeutic LMWH, and LMWH should not be restarted for at least 4 hours after the procedure.[20] There are limited data on the safety of thienopyridines, but they should generally be discontinued, and all other prophylactic or therapeutic anticoagulation must be stopped prior to the procedure.
Paracentesis
Bleeding complications from paracentesis are uncommon, with abdominal wall hematoma and hemoperitoneum complicating 1% and 0.01% of procedures, respectively.[28, 29, 30] Whether antithrombotic therapy increases the risk of bleeding during paracentesis is unknown, primarily because most patients for whom the procedure is indicated have coagulopathy and thrombocytopenia from liver disease, and are therefore rarely treated with these medications.
Although patients with liver disease often have an elevated INR due to impaired hepatic synthesis of clotting factors, it is incorrect to generalize the observed rate of bleeding in this population to patients with an elevated INR from warfarin therapy who may require paracentesis for reasons unrelated to liver disease (eg, malignancy or infection). The coagulopathy of liver disease reflects deficiencies in the hepatic production of both pro‐ and anticoagulant proteins, and these patients develop both thrombotic and hemorrhagic complications irrespective of their in vitro coagulation indices.[31]
Although the available evidence suggests that paracentesis can be safely performed in patients with coagulopathy from liver disease, regardless of the INR,[30] little is known about the bleeding risk in other patients, with or without antithrombotic therapy. Based on indirect evidence, it is reasonable to assume that prophylactic UFH or LWMH or antiplatelet therapy would confer minimal additional risk, whereas the safety of continuing therapeutic anticoagulation is unknown.
Thoracentesis
Bleeding complications from thoracentesis are uncommon, generally occurring in <1% of procedures.[32, 33, 34] Factors associated with increased risk of overall complications include operator inexperience, large volume drainage, and lack of ultrasound guidance.[34, 35, 36] There are no studies that specifically address the risk of bleeding in patients on anticoagulant therapy, but such patients are included in studies on the risk of bleeding with coagulopathy.[37, 38, 39, 40]
In a retrospective cohort study of 1076 ultrasound‐guided thoracenteses performed by radiologists on patients with coagulopathy (defined as thrombocytopenia or an elevated INR from any cause), there were no bleeding complications (defined as anything other than minimal symptoms not requiring intervention). Among the patients in this study, 497 (46%) patients had a preprocedure INR >1.5; 198 (24%) had an INR between 2 and 3, and 32 (4%) had an INR >3.[39]
A similar study, which compared outcomes in patients with corrected and uncorrected coagulopathy, included 744 patients with an INR >1.6 (from any cause), of which 167 received preprocedural fresh‐frozen plasma (FFP) and 577 did not. There was 1 (0.1%) bleeding complication in a patient who received prophylactic FFP and none in the group that was not transfused.[38]
In a prospective cohort of 312 patients at increased risk for bleeding (from coagulopathy or antithrombotic medications) who underwent ultrasound‐guided thoracentesis by a pulmonologist or physician's assistant, 44 (34%) had an INR >1.5 (secondary to liver disease or warfarin therapy), 15 (12%) were taking clopidogrel, and 14 (11%) were treated with therapeutic LMWH within 12 hours or therapeutic UFH within 4.5 hours of the procedure. There were no bleeding complications in any of the patients (defined as mean change in hematocrit, chest x‐ray abnormalities, hemothorax, or requirement for transfusion).[37]
Although there are no studies that specifically address the use of aspirin and bleeding complications in thoracentesis, it is generally considered safe to continue this medication,[5] and there are small studies that show that thoracentesis and small‐bore chest tubes can be safely placed in patients taking clopidogrel.[41, 42]
Thoracentesis is associated with a low rate of bleeding complications, and when performed by an experienced operator using ultrasound, warfarin does not appear to increase this risk. However, given the low overall complication rate, it is not known whether patients on warfarin would have worse outcomes in the event of more serious complications (eg, intercostal artery laceration). At present, there are no published studies that address the risk of thoracentesis in patients taking new oral anticoagulants (NOACs).
Central Venous Catheter Insertion
The incidence of bleeding complications from central venous catheter (CVC) placement varies depending on the site of insertion and definition of bleeding, with hematoma and hemothorax occurring in 0.1% to 6.9%, and 0.4% to 1.3% of procedures, respectively.[43, 44, 45] Factors that increase the likelihood of complications include operator inexperience, multiple needle passes, and lack of ultrasound guidance.[46, 47] There are no studies that specifically address the risk of bleeding from CVC placement in patients on anticoagulant therapy, but such patients are included in studies of CVC placement in patients with coagulopathy, which report similar complication rates as seen in patients with normal hemostasis.[48, 49, 50, 51, 52, 53]
In a retrospective cohort study, investigators collected information on CVC‐associated bleeding complications in 281 medical and surgical intensive care patients with coagulopathy (INR 1.5 from any cause) after they adopted a more conservative approach to plasma transfusion in their intensive care unit; specifically, the routine use of prophylactic FFP to correct coagulopathy was discouraged for patients with an INR <3 (vs usual practice using an INR cutoff of 1.5), but the final decision was left to the discretion of the attending performing or supervising the procedure. Bleeding was defined as insertion‐site hematoma, interventions other than local manual pressure, and the need for blood transfusion. One case of bleeding (hematoma) was observed in a patient with an INR of 3.9, who received FFP before the procedure. There were no complications among those with uncorrected coagulopathy, including 66 patients with an INR between 1.5 and 2.9, and 6 with an INR 3.0. Ultrasound guidance was used in 50% of CVCs placed in the internal jugular vein.[54]
Although there are no studies that specifically address the use of antiplatelet drugs and bleeding complications in CVC placement, aspirin is generally considered safe to continue,[5] and by inference, thienopyridines are expected to add minimal additional risk.
CVC placement is associated with a variable rate of bleeding complications, with hematoma being relatively common. Based on the available literature, warfarin does not appear to increase this risk, but there are limited data from which to draw firm conclusions. A femoral or jugular approach may be preferable because they allow for ultrasound visualization and are amenable to manual compression. There are no published studies that address the risk of CVC placement in patients taking NOACs, and although the risk of bleeding is probably similar to patients receiving warfarin, the lack of effective reversal agents for these medications should be part of any risk‐benefit calculation.[55]
WHAT IS THE PATIENT'S RISK OF THROMBOEMBOLISM IF ANTITHROMBOTIC THERAPY IS INTERRUPTED?
Anticoagulants
If it is determined that a procedure cannot safely be performed while continuing antithrombotic therapy, one must then consider the patient's risk of thromboembolism if these therapies are temporarily interrupted. Unfortunately, there are few robust clinical studies from which to make this assessment, and therefore most clinicians rely on the risk stratification model proposed by the ACCP, which divides patients into 3 tiers (low, moderate, high), based on their indication for anticoagulation and risk factors for thromboembolism (Table 2)[8]. The ACCP model is largely based on indirect evidence from antithrombotic therapy trials in nonoperative patients, and its application to perioperative patients necessitates several assumptions that may not hold true in practice.
| Indication for Anticoagulant Therapy | |||
|---|---|---|---|
| Risk Stratum | Mechanical Heart Valve | Atrial Fibrillation | VTE |
| |||
| High Thrombotic Risk |
|
|
|
| Moderate Thrombotic Risk |
|
|
|
| Low Thrombotic Risk |
|
|
|
First, it assumes that the annualized risk of a thrombotic event in nonoperative patients can be prorated to determine the short‐term risk of discontinuing antithrombotic therapy in the perioperative period. For example, it has been estimated that the risk for perioperative stroke in a patient with atrial fibrillation who temporarily interrupts anticoagulation for 1 week would be 0.1% (5% per year 52 weeks),[56, 57]and yet we know from observational data that the actual risk of perioperative stroke in similar patients is 5 to 7 times higher.[58, 59] Second, it assumes that bridging therapy will decrease the risk of thromboembolism in high‐risk patients when warfarin therapy is interrupted, a premise that is logical but has not been subject to randomized controlled trials.[60] Third, it does not take into account the surgery‐specific risk for thromboembolism, which varies significantly, with arterial thromboembolism being more common in cardiac valve, vascular, and neurologic procedures, and venous thromboembolism (VTE) being more likely in orthopedic, trauma, and cancer surgery.[61, 62] These limitations notwithstanding, the ACCP model still offers the best available framework for thrombotic risk assessment and a reasonable starting point for clinical decision making.
Antiplatelet Agents
Patients with coronary artery stents who undergo noncardiac surgery are at increased risk for adverse cardiovascular events, including acute stent thrombosis, which carries a risk of myocardial infarction and death of 70% and 30%, respectively.[63] This risk is highest during the period between stent implantation and endothelialization, a process that takes 4 to 6 weeks for bare‐metal stents (BMS) and 6 to 12 months for drug‐eluting stents (DES). Premature discontinuation of dual antiplatelet therapy is the most important risk factor for stent thrombosis during this time.[64] Although the optimal perioperative strategy for these patients is unknown, there is general agreement that elective surgery should be delayed for at least 4 weeks in patients with a BMS and 12 months for patients with a DES. If a procedure or surgery is required during this time period, every effort should be made to continue dual antiplatelet therapy; if this is not possible, aspirin should be continued, and thienopyridine therapy should be interrupted as briefly as possible (Table 3).
| Recommended Interval Between Last Dose of Medication and Procedure | Recommended Interval Between Procedure and First Dose of Medication, h | ||
|---|---|---|---|
| Low Risk or Consequence of Postprocedure Bleeding | High Risk or Consequence of Postprocedure Bleeding | ||
| |||
| Antiplatelet Medicationsa | |||
| Aspirin (81325 mg dailydipyridamole) | 710 days (skip 69 doses) | 24 | 48 |
| Ticlodipine (250 mg twice daily) | 1014 days (skip 1926 doses) | 24 | 48 |
| Clopidogrel (75 mg once daily) | 710 days (skip 69 doses)b | 24 | 48 |
| Prasugrel (10 mg once daily) | 710 days (skip 69 dose)c | 24 | 48 |
| Ticagrelor (90 mg twice daily; t =8 hours) | 5 days (skip 8 doses) | 24 | 48 |
| Cilostazol (100 mg twice daily; t =11 hours) | 3 days (skip 4 doses) | 24 | 48 |
| Anticoagulant Medicationse | |||
| Warfarin (t =3642 hours, but highly variable) | 6 days (skip 5 doses)f | 12 | 24 |
| Intravenous UFH (t 60 minutes) | 46 hours | 24 | 4872 |
| LMWH (t =37 hours) | |||
| Prophylactic dosing | 12 hours# | 12 | 2436 |
| Therapeutic dosing | |||
| Once daily | 24 hours (give 50% of last total dose)# | 24 | 4872 |
| Twice daily | 24 hours (skip 1 dose)# | 24 | 4872 |
| Fondaparinux (t =17 hours, any dose) | 34 days (skip 23 doses)h | 24 | 4872 |
| Dabigatran (150 mg twice daily) | |||
| CrCl>50 mL/min (t =1417 hours) | 3 days (skip 4 doses) | 24 | 4872 |
| CrCl 3050 mL/min (t =1618 hours) | 45 days (skip 68 doses) | 24 | 4872 |
| CrCl 1530 mL/min (t =1618 hours)i | 45 days (skip 68 doses) | 24 | 4872 |
| Rivaroxaban (20 mg once daily) | |||
| CrCl>50 mL/min (t =89 hours) | 3 days (skip 2 doses) | 24 | 4872 |
| CrCl 3050 mL/min (t =9 hours) | 3 days (skip 2 doses) | 24 | 4872 |
| CrCl 1529.9 mL/min (t =910 hours)j | 4 days (skip 3 doses) | 24 | 4872 |
| Apixiban (5 mg twice daily) | |||
| CrCl>50 mL/min (t =78 hours) | 3 days (skip 4 doses) | 24 | 4872 |
| CrCl 3050 mL/min (t =1718 hours) | 4 days (skip 6 doses) | 24 | 4872 |
ARE THERE INTERVENTIONS THAT CAN DECREASE THE RISK OF PERIPROCEDURAL BLEEDING AND/OR THROMBOEMBOLISM?
Mitigating the Risk of Bleeding
Bleeding complications can be reduced by allowing a sufficient time for the effects of antithrombotic medications to wear off before performing a procedure. This requires an understanding of the pharmacology of these medications, with particular attention to patients in whom these medications are less well studied, including the elderly, patients with renal insufficiency, and those with very high or low body mass index. Table 3 provides recommendations for when to stop antithrombotic therapy prior to an invasive procedure. The intervals are based on the time needed to achieve a minimal antithrombotic effect, which is generally 4 to 5 half‐lives for anticoagulants and 7 to 10 days for irreversible antiplatelet agents. Shorter intervals may be appropriate for procedures with low risk or consequence of bleeding, but there are insufficient data to make specific recommendations regarding this strategy.
It is equally important to ensure that there is adequate time for postoperative hemostasis prior to restarting antithrombotic therapy. Data from VTE prophylaxis trials and bridging studies consistently show that bleeding complications occur more frequently when anticoagulation is started too early, and antithrombotic therapy should generally be delayed 24 hours in patients at average risk and 48 to 72 hours in patients at high risk or consequence for postoperative bleeding.[8, 60, 65]
Aspirin increases the risk of surgical blood loss and transfusion by up to 20%, and by up to 50% when given in combination with clopidogrel, but with the exception of intracranial surgery, there does not appear to be an increase in perioperative morbidity or mortality with either of these agents.[66]
Mitigating the Risk of Thromboembolism
Once the decision has been made to temporarily discontinue warfarin, the next consideration is whether to bridge with a short acting anticoagulant (typically subcutaneous LMWH or intravenous UFH) during the period of time when the INR is subtherapeutic. Conceptually, one would expect this strategy would minimize the risk of thromboembolism, but its efficacy has never been clearly demonstrated. In fact, in a systematic review and meta‐analysis of 34 studies that compared the rates of thromboembolism among bridged and nonbridged patients, heparin therapy did not reduce the risk of thromboembolic events (odds ratio: 0.80; 95% confidence interval: 0.421.54), but did result in higher rates of periprocedural bleeding.[60]
The applicability of these results to clinical practice are limited by the heterogeneity of the data used in the analysis; specifically, bridging strategies varied (including therapeutic, intermediate, and prophylactic dose regimens), there was wide variation in the types of surgery (and therefore bleeding risk), and because the majority of studies were observational, there is a significant likelihood of confounding by indication (ie, patients at high risk for thromboembolism are more likely to receive bridging therapy), and thus the benefit of this strategy may be underestimated. It is also important to note that in the majority studies anticoagulation was restarted <24 hours after the procedure, which likely contributed to the increased rate of bleeding.
Therefore, although bridging therapy is not indicated for patients at low risk, it is premature to conclude that it should be avoided in patients at moderate or high risk for thromboembolism. The results of 2 ongoing, randomized, placebo‐controlled trials of bridging therapy in patients taking warfarin for atrial fibrillation (Effectiveness of Bridging Anticoagulation for Surgery [BRIDGE]) or mechanical heart values (A Double Blind Randomized Control Trial of Post‐Operative Low Molecular Weight Heparin Bridging Therapy Versus Placebo Bridging Therapy for Patients Who Are at High Risk for Arterial Thromboembolism [PERIOP‐2]) should help to answer this question.[67, 68]
The uncertainty regarding the benefits of bridging therapy is reflected in the changes to the most recent ACCP guidelines. In 2008, the ACCP recommended low‐dose LMWH or no bridging for patients at low risk (grade 2C), therapeutic‐dose bridging for patients at moderate risk (grade 2C), and therapeutic‐dose bridging for patients at high risk for thromboembolism (Grade 1C).[56] In 2012, the ACCP recommended against bridging for low‐risk patients (grade 2C), made no specific recommendation regarding moderate‐risk patients, and offered a less robust recommendation for bridging in high‐risk patients (grade 2C).[8]
Until the results of the BRIDGE and PERIOP‐2 trials are available, the author still favors therapeutic bridging for patients at high risk and selected patients at moderate risk for thromboembolism, provided sufficient time is allowed for postoperative hemostasis before anticoagulation is restarted. For procedures with a high risk or consequence of bleeding, intravenous UFH (without a bolus) is a reasonable initial postoperative strategy to insure that anticoagulation is tolerated before committing to LMWH. Indirect evidence supports the use of prophylactic or intermediate‐dose bridging regimens in patients for whom the primary consideration is the prevention of recurrent VTE, but data to show that this strategy is effective for the prevention of arterial thromboembolism are lacking.
Intravenous glycoprotein IIb/IIIa inhibitors are sometimes used to bridge high‐risk patients with coronary artery stents who must stop antiplatelet therapy prior to a procedure, but the data to support this practice are limited and observational in nature.[69, 70]
STARTING AND STOPPING ANTITHROMBOTIC THERAPY
Warfarin
For patients on warfarin, the INR at which it is safe to perform invasive procedures is unknown. Normal hemostasis requires clotting factor levels of approximately 20% to 40% of normal,[71] which generally corresponds to an INR of <1.5, whereas for most indications, therapeutic anticoagulation is achieved when the INR is between 2.0 and 3.5. However, because the relationship between the INR and the levels of clotting factors is nonlinear, for a given patient, the INR may be abnormal (ie, >1) despite levels of clotting factors that are sufficient for periprocedural hemostasis.[72, 73, 74, 75] Because of its relatively long half‐life (3642 hours), warfarin should be stopped 6 days (skip 5 doses) prior to a procedure to achieve an INR of <1.5, but can safely be restarted the same day in most patients.
Heparins
The half‐life of intravenous heparin is dose dependent, and at therapeutic levels is approximately 60 minutes; therefore, it should be discontinued 4 to 6 hours (5 half‐lives) before performing an invasive procedure.[76] The half‐life of subcutaneous LMWHs ranges from 3 to 7 hours in healthy volunteers,[23, 24, 25] and is often longer in patients for whom these medications are commonly prescribed.[77, 78] Therefore, when administered at therapeutic doses twice daily, the last dose should be given in the morning the day before the procedure, and for therapeutic once‐daily regimens, the last dose should be reduced by 50%.[8] The optimal time to discontinue prophylactic doses of LWMH prior to an invasive procedure is unclear, but a minimum of 12 hours is recommended.[22, 79] Because LWMHs are renally cleared, longer intervals are needed for patients with impaired renal function.[76, 80]
New Oral Anticoagulants
The manufacturer of rivaroxaban recommends that if anticoagulation must be discontinued, it be stopped at least 24 hours before the procedure.[81] Although this may be sufficient for procedures with a low risk or consequence of bleeding, the half‐life of rivaroxaban is between 8 and 10 hours, and therefore 48 hours (45 half‐lives) is required to ensure minimal residual anticoagulant effect.
Apixaban has a clearance half‐life of 6 hours, but displays prolonged absorption such that its effective half‐life is 12 hours after repeated dosing. The manufacturer recommends that it be stopped at least 24 hours prior to a procedure with a low risk or consequence of bleeding, and 48 hours prior to a procedure with a high risk or consequence of bleeding.[82]
The manufacturer of dabigatran recommends that the drug be discontinued 1 to 2 days (creatinine clearance (CrCl) 50 mL/min) or 3 to 5 days (CrCl <50 mL/min) before invasive or surgical procedures, and that longer times be considered when complete hemostasis is required.[83] Given that the half‐life of dabigatran is 14 to 17 hours, the author recommends that it be stopped at least 2 days (3 half‐lives) prior to a procedure with a low risk or consequence of bleeding, and 3 days (45 half‐lives) prior to a procedure with a high risk or consequence of bleeding.
The clearance of all the NOACs is significantly prolonged in patients with renal impairment, and a longer interval between the last dose and the procedure is necessary in patients with renal failure to ensure normal hemostasis (Table 3).
The effect of the NOACs on the standard clotting assays are complex and vary depending on drug dose, the type of reagents used, and the calibration of the equipment. For dabigatran, the activated partial thromboplastin time (aPTT) and the thrombin time (TT) are sufficiently sensitive to allow for a qualitative assessment of drug effect, such that a normal aPTT indicates the absence, or a very low level of an anticoagulant effect, and a normal TT essentially rules out an effect. Accurate quantitative testing of dabigatran requires an appropriately calibrated dilute thrombin test or ecarin clotting time assay.[84, 85]
Depending on the thromboplastin reagent used, the prothrombin time (PT) may be sufficiently sensitive to rivaroxaban that a normal level rules out a residual drug effect,[86] but this does not hold true for apixaban, which has minimal effect on the PT at therapeutic concentrations. The aPTT is insensitive to both rivaroxaban and apixaban and cannot be used for assessing residual drug effect. Accurate quantitative testing of rivaroxaban or apixaban requires an anti‐factor Xa assay calibrated for use with these agents.[84]
Antiplatelet Agents
Aspirin irreversibly inhibits platelet cyclooxygenase activity, and the thienopyridines clopidogrel and prasugrel, irreversibly inhibit the platelet P2Y12 receptor. As such, the biological effects of these medications persist until the platelet pool has turned over, a process that occurs at 10% to 12% per day and takes 7 to 10 days to complete.[87] The minimum number of functional platelets required to ensure adequate periprocedural hemostasis is unknown, but is likely between 50 and 100,000/L.[88] Therefore, assuming a platelet pool of 200,000/L, most patients will regenerate an adequate number of functional platelets by 5 days after discontinuing therapy, and nearly all will have normal platelet function by 10 days. Determining the risk of bleeding prior to complete turnover of the platelet pool is further complicated by genetic variability between patients in drug metabolism and the degree of platelet inhibition by these agents.[89]
Owing to this complexity, guidelines and prescribing recommendations are inconsistent. The ACCP recommends stopping antiplatelet agents 7 to 10 days prior to an invasive procedure, and the ACC/AHA makes no specific recommendations at all.[90] Based on data from patients undergoing cardiac bypass surgery, it is recommended that clopidogrel be stopped 5 days, and prasugrel 7 days, prior to an invasive procedure.[91, 92] The elimination half‐life of ticlodipine is sufficiently long (up to 96 hours after repeated dosing) that it should be stopped 10 to 14 days prior to an invasive procedure.[87] Ticagrelor is a reversible P2Y12 receptor inhibitor with a half‐life of approximately 8 hours and should therefore have minimal effect by 3 days after discontinuation; however, the manufacturer recommends that it be stopped 5 days prior to an invasive procedure.[93]
The optimal time to restart antiplatelet agents after an invasive procedure is also unknown. The 2008 ACCP guidelines recommended restarting aspirin and/or clopidogrel in 24 hours, or as hemostasis allows,[56] whereas neither the 2007 or 2009 ACC/AHA guidelines,[90] or the most recent 2012 ACCP guidelines,[8] offer specific recommendations. Aspirin, prasugrel, and ticagrelor have a rapid onset of action, whereas the full antiplatelet effect of clopidogrel does not occur for several days, and for patients in whom more rapid platelet inhibition is desired, a loading dose (300600 mg) may be appropriate.[87]
CONCLUSIONS
Deciding on an optimal periprocedural antithrombotic management strategy is a common challenge for hospitalists that requires careful consideration of both patient and procedure related‐risk factors for bleeding and thrombosis, as well as the consequences of delaying or forgoing the procedure altogether. For many procedures, there is evidence that antithrombotic therapy can be safely continued, thereby obviating the risk associated with interrupting therapy. When antithrombotic therapy must be stopped, it should be done in a manner that appropriately balances the risks and consequence of periprocedural bleeding and thromboembolism. Strategies to decrease the risk of perioperative bleeding include allowing sufficient time for the effects of antithrombotic therapy to subside before starting the procedure, and ensuring adequate time for hemostasis before restarting antithrombotic therapy. Bridging therapy may provide net clinical benefit for patients at moderate to high risk for thromboembolism, but this will not be clear until the results of several ongoing bridging trials are available. The periprocedural antithrombotic management strategy should be developed in collaboration with the relevant providers and with active participation by the patient in all decisions and treatment plans. Standardized protocols and documentation can help to minimize unintended variation in practice and improve information transfer during transitions of care.
Acknowledgements
The author would like to thank Shoshana and Lola Herzig for their support in the design and preparation of the manuscript.
Disclosure: Nothing to report.
- , , , , . Core competencies in hospital medicine: development and methodology. J Hosp Med. 2006;1(suppl 1):48–56.
- , , , , . Procedures performed by hospitalist and non‐hospitalist general internists. J Gen Int Med. 2010;25(5):448–452.
- , , , et al. Regional anesthesia in the patient receiving antithrombotic or thrombolytic therapy: American Society of Regional Anesthesia and Pain Medicine evidence‐based guidelines (Third Edition). Reg Anesth Pain Med. 2010;35(1):64–101.
- ASGE Standards of Practice Committee, , , , et al. Management of antithrombotic agents for endoscopic procedures. Gastrointest Endosc. 2009;70(6):1060–1070.
- , , , et al. Consensus guidelines for periprocedural management of coagulation status and hemostasis risk in percutaneous image‐guided interventions. J Vasc Interv Radiol. 2012;23(6):727–736.
- , , , et al. 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation. 2008;118(15):e523–e661.
- , , , et al. Prevention of premature discontinuation of dual antiplatelet therapy in patients with coronary artery stents: a science advisory from the American Heart Association, American College of Cardiology, Society for Cardiovascular Angiography and Interventions, American College of Surgeons, and American Dental Association, with representation from the American College of Physicians. J Am Coll Cardiol. 2007;49(6):734–739.
- , , , et al. Perioperative management of antithrombotic therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 Suppl):e326S–e350S.
- , , , et al. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in surgical patients. J Thromb Haemost. 2010;8(1):202–204.
- , , , et al. Clinical outcomes with unfractionated heparin or low‐molecular‐weight heparin as bridging therapy in patients on long‐term oral anticoagulants: the REGIMEN registry. J Thromb Haemost. 2006;4(6):1246–1252.
- , , , et al. Assessment of bleeding risk of interventional techniques: a best evidence synthesis of practice patterns and perioperative management of anticoagulant and antithrombotic therapy. Pain Physician. 2013;16(2 suppl):Se261–Se318.
- , . Perioperative management of patients receiving oral anticoagulants: a systematic review. Arch Intern Med. 2003;163(8):901–908.
- , , , et al. Frequency of the bleeding risk in patients receiving warfarin submitted to arthrocentesis of the knee [in Italian]. Reumatismo. 2003;55(3):159–163.
- , . A prospective study of the safety of joint and soft tissue aspirations and injections in patients taking warfarin sodium. Arthritis Rheum. 1998;41(4):736–739.
- , . Safety of arthrocentesis and joint injection in patients receiving anticoagulation at therapeutic levels. Am J Med. 2012;125(3):265–269.
- , , . Severe neurological complications after central neuraxial blockades in Sweden 1990–1999. Anesthesiology. 2004;101(4):950–959.
- , . Managing anticoagulated patients during neuraxial anaesthesia. Br J Haematol. 2010;149(2):195–208.
- . Impaired haemostasis and regional anaesthesia. Can J Anaesth. 1996;43(5 pt 2):R129–R141.
- , , . Cauda equina syndrome following a lumbar puncture. J Clin Neurosci. 2009;16(5):714–716.
- United States Food and Drug Safety Communication: updated recommendations to decrease risk of spinal column bleeding and paralysis in patients on low molecular weight heparins. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm373595.htm. Accessed January 6, 2014.
- , , , , . Three‐times‐daily subcutaneous unfractionated heparin and neuraxial anesthesia: a retrospective review of 928 cases. Reg Anesth Pain Med. 2012;37(6):623–626.
- , , , et al. Regional anaesthesia and antithrombotic agents: recommendations of the European Society of Anaesthesiology. Eur J Anaesthesiol. 2010;27(12):999–1015.
- Eisai Inc. Fragmin (dalteparin sodium injection) full prescribing information. 2009. Available at: http://us.eisai.com/wps/wcm/connect/Eisai/Home/Our+Products/FRAGMIN. Accessed January 6, 2014
- Sanofi‐Aventis. Lovenox (enoxaparin sodium injection) full prescribing information. 2013. Available at: http://products.sanofi.us/lovenox/lovenox.html. Accessed January 6, 2014.
- LEO Pharmaceutical Products. Innohep (tinzaparin sodium injection) full prescribing information. 2008. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020484s011lbl.pdf. Accessed January 6, 2014.
- , , , et al. Preoperative antiplatelet therapy does not increase the risk of spinal hematoma associated with regional anesthesia. Anesth Analg. 1995;80(2):303–309.
- , , , et al. Addendum of newer anticoagulants to the SIR consensus guideline. J Vasc Interv Radiol. 2013;24(5):641–645.
- , . Severe haemorrhage following abdominal paracentesis for ascites in patients with liver disease. Aliment Pharmacol Ther. 2005;21(5):525–529.
- , . Ultrasound guidance decreases complications and improves the cost of care among patients undergoing thoracentesis and paracentesis. Chest. 2013;143(2):532–538.
- . Management of adult patients with ascites due to cirrhosis. Hepatology. 2004;39(3):841–856.
- , . The coagulopathy of chronic liver disease. N Engl J Med. 2011;365(2):147–156.
- , , , , . Necessity of routine chest roentgenography after thoracentesis. Ann Intern Med. 1996;124(9):816–820.
- , , , , . Pleural procedures and pleuroscopy. Respirology. 2009;14(6):796–807.
- , , . Ultrasonography guidance reduces complications and costs associated with thoracentesis procedures. J Clin Ultrasound. 2012;40(3):135–141.
- , , , . Reducing iatrogenic risk in thoracentesis: establishing best practice via experiential training in a zero‐risk environment. Chest. 2009;135(5):1315–1320.
- , . Improving the safety of thoracentesis. Curr Opin Pulm Med. 2011;17(4):232–236.
- , , , , . The safety of thoracentesis in patients with uncorrected bleeding risk. Ann Am Thorac Soc. 2013;10(4):336–341.
- , , , et al. Safety of ultrasound‐guided thoracentesis in patients with abnormal preprocedural coagulation parameters. Chest. 2013;144(2):456–463.
- , . Abnormal preprocedural international normalized ratio and platelet counts are not associated with increased bleeding complications after ultrasound‐guided thoracentesis. AJR Am J Roentgenol. 2011;197(1):W164–W168.
- , . Lack of increased bleeding after paracentesis and thoracentesis in patients with mild coagulation abnormalities. Transfusion. 1991;31(2):164–171.
- , , . Safety of ultrasound‐guided small‐bore chest tube insertion in patients on clopidogrel. J Bronchology Interv Pulmonol. 2013;20(1):16–20.
- , , , . Effect of routine clopidogrel use on bleeding complications after ultrasound‐guided thoracentesis. J Bronchology Interv Pulmonol. 2012;19(4):284–287.
- , . Preventing complications of central venous catheterization. N Engl J Med. 2003;348(12):1123–1133.
- , , . Complications of central venous catheters: internal jugular versus subclavian access—a systematic review. Crit Care Med. 2002;30(2):454–460.
- , , , . Risk factors for acute adverse events during ultrasound‐guided central venous cannulation in the emergency department. Acad Emerg Med. 2010;17(10):1055–1061.
- . Complications of central venous catheterization. J Am Coll Surg. 2007;204(4):681–696.
- , , , , , . Real‐time two‐dimensional ultrasound guidance for central venous cannulation: a meta‐analysis. Anesthesiology. 2013;118(2):361–375.
- , , . Central venous catheter placement in patients with disorders of hemostasis. Chest. 1996;110(1):185–188.
- , , , . Invasive line placement in critically ill patients: do hemostatic defects matter? Transfusion. 1996;36(9):827–831.
- , , , , , . Bleeding complications after central line insertions: relevance of pre‐procedure coagulation tests and institutional transfusion policy. Acta Anaesthesiol Scand. 2013;57(5):573–579.
- , , , et al. Low levels of prothrombin time (INR) and platelets do not increase the risk of significant bleeding when placing central venous catheters. Med Klin (Munich). 2009;104(5):331–335.
- , , , et al. Coagulation disorders in patients with cancer: nontunneled central venous catheter placement with US guidance—a single‐institution retrospective analysis. Radiology. 2009;253(1):249–252.
- , , . US‐guided placement of central vein catheters in patients with disorders of hemostasis. Eur J Radiol. 2008;65(2):253–256.
- , , . Central line placement in patients with and without prophylactic plasma. J Crit Care. 2012;27(5):529.e529–e513.
- , , . How I treat: target specific oral anticoagulant associated bleeding [published online ahead of print January 2, 2014]. Blood. doi: 10.1182/blood‐2013‐09‐529784.
- , , , et al. The perioperative management of antithrombotic therapy: American College of Chest Physicians evidence‐based clinical practice guidelines (8th edition). Chest. 2008;133(6 suppl):299S–339S.
- , . Management of anticoagulation before and after elective surgery. N Engl J Med. 1997;336(21):1506–1511.
- , , , et al. RIsk of thromboembolism with short‐term interruption of warfarin therapy. Arch Intern Med. 2008;168(1):63–69.
- , , , et al. Periprocedural bleeding and thromboembolic events with dabigatran compared with warfarin: results from the Randomized Evaluation of Long‐Term Anticoagulation Therapy (RE‐LY) randomized trial. Circulation. 2012;126(3):343–348.
- , , , , , . Periprocedural heparin bridging in patients receiving vitamin K antagonists: systematic review and meta‐analysis of bleeding and thromboembolic rates. Circulation. 2012;126(13):1630–1639.
- , , , et al. Prevention of VTE in nonorthopedic surgical patients: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 Suppl):e227S–277S.
- , , , et al. Periprocedural anticoagulation management of patients with venous thromboembolism. Arterioscler Thromb Vasc Biol. 2010;30(3):442–448.
- , . Late coronary stent thrombosis. Circulation. 2007;116(17):1952–1965.
- , , , et al. Predictors of coronary stent thrombosis: the Dutch Stent Thrombosis Registry. J Am Coll Cardiol. 2009;53(16):1399–1409.
- , , , et al. Predictors of major bleeding in peri‐procedural anticoagulation management. J Thromb Haemost. 2012;10(2):261–267.
- , , . Perioperative antiplatelet therapy: the case for continuing therapy in patients at risk of myocardial infarction. Br J Anaesth. 2007;99(3):316–328.
- , . Effectiveness of bridging anticoagulation for surgery (the BRIDGE Study). Available at: www.ClinicalTrials.gov. Identifier: NCT00786474. Accessed October 22, 2013.
- . A safety and effectiveness study of LMWH bridging therapy versus placebo bridging therapy for patients on long term warfarin and require temporary interruption of their warfarin (PERIOP2). Available at: www.ClinicalTrials.gov. Identifier: NCT00432796. Accessed October 20, 2013.
- , , , et al. Outcomes of a preoperative “bridging” strategy with glycoprotein IIb/IIIa inhibitors to prevent perioperative stent thrombosis in patients with drug‐eluting stents who undergo surgery necessitating interruption of thienopyridine administration. EuroIntervention. 2013;9(2):204–211.
- , , , et al. Safety of “bridging” with eptifibatide for patients with coronary stents before cardiac and non‐cardiac surgery. Am J Cardiol. 2012;110(4):485–490.
- . Hemostatic problems in surgical patients. In: Colman RW HJ, Marder VJ, Clowes AW, George JN, ed. Hemostasis and Thrombosis. 4th ed. Philadelphia, PA: Lippincott Williams 2001:1033.
- . Reversal of drug‐induced anticoagulation: old solutions and new problems. Transfusion. 2012;52:45S–55S.
- , , . A feasibility study of continuing dose‐reduced warfarin for invasive procedures in patients with high thromboembolic risk. Chest. 2005;127(3):922–927.
- , , , et al. A simple and safe nomogram for the management of oral anticoagulation prior to minor surgery. Clin Lab Haematol. 2003;25(2):127–130.
- , , , , . International normalized ratio versus plasma levels of coagulation factors in patients on vitamin K antagonist therapy. Arch Pathol Lab Med. 2011;135(4):490–494.
- , , , . Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College Of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 suppl):e24S–e43S.
- , , , . Bridging anticoagulation with low‐molecular‐weight heparin after interruption of warfarin therapy is associated with a residual anticoagulant effect prior to surgery. Thromb Haemost. 2005;94(3):528–531.
- , , , et al. Brief communication: Preoperative anticoagulant activity after bridging low‐molecular‐weight heparin for temporary interruption of warfarin. Ann Intern Med. 2007;146(3):184–187.
- . Regional anaesthesia in the patient receiving antithrombotic and antiplatelet therapy. Br J Anaesth. 2011;107(suppl 1):i96–i106.
- , , , . Meta‐analysis: low‐molecular‐weight heparin and bleeding in patients with severe renal insufficiency. Ann Intern Med. 2006;144(9):673–684.
- Janssen Pharmaceuticals, Inc. Xarelto (rivaroxaban) full prescribing information. 2013. Available at: http://www.xareltohcp.com/sites/default/files/pdf/xarelto_0.pdf#zoom=100. Accessed October 1, 2013.
- Bristol Meyers Squibb, Inc. Eliquis (apixaban) full prescribing information. 2013. Available at: http://packageinserts.bms.com/pi/pi_eliquis.pdf. Accessed October 1, 2013.
- Boehringer Ingelheim Pharmaceuticals I. Pradaxa (dabigatran etexilate mesylate) full prescribing information. Available at: http://bidocs.boehringer‐ingelheim.com/BIWebAccess/ViewServlet.ser?docBase= renetnt11(2):245–252.
- . The laboratory and the direct oral anticoagulants. Blood. 2013;121(20):4032–4035.
- , , , , , . Assessment of the impact of rivaroxaban on coagulation assays: laboratory recommendations for the monitoring of rivaroxaban and review of the literature. Thromb Res. 2012;130(6):956–966.
- , , , , . Antiplatelet drugs: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 suppl):e89S–119S.
- . Evidence‐based platelet transfusion guidelines. Hematology Am Soc Hematol Educ Program. 2007:172–178.
- , , . Platelet function testing and prediction of procedural bleeding risk. Thromb Haemost. 2013;109(5):817–824.
- , , , . Perioperative management of antiplatelet therapy in patients with a coronary stent who need non‐cardiac surgery: a systematic review of clinical practice guidelines. Chest. 2013;144(6):1848–1856.
- Eli Lilly Pharmaceuticals, Inc. Effient (prasugrel) full prescribing information. 2012. Available at: http://pi.lilly.com/us/effient.pdf. Accessed October 1, 2013.
- , , , . Antiplatelet therapy and cardiac surgery: review of recent evidence and clinical implications. Can J Cardiol. 2013;29(9):1042–1047.
- AstraZeneca. Brilinta (ticagrelor) full prescribing information. 2013. Available at: http://www1.astrazeneca‐us.com/pi/brilinta.pdf. Accessed October 1, 2013.
- , , , et al. 2012 update to the Society of Thoracic Surgeons guideline on use of antiplatelet drugs in patients having cardiac and noncardiac operations. Ann Thorac Surg. 2012;94(5):1761–1781.
- , . How I treat anticoagulated patients undergoing an elective procedure or surgery. Blood. 2012;120(15):2954–2962.
- , , , , American College of Chest Physicians. Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 suppl):e24S–e43S.
The periprocedural management of antithrombotic medications is a common challenge for hospitalists, for which there is limited high‐quality evidence to guide clinical decision making. The introduction of third‐generation antiplatelet agents (prasugrel and ticagrelor) and the new oral anticoagulants (rivaroxaban, apixaban, and dabigatran), has added an additional layer of complexity to clinical management.
This article will provide a conceptual framework for the periprocedural management of antithrombotic therapy, with a particular focus on procedures that are considered core competencies by the Society of Hospital Medicine; these include: arthrocentesis, lumbar puncture, paracentesis, thoracentesis, and central line placement (Table 1).[1, 2] The recommendations in this article are based on a review of published guidelines and consensus statements and their supporting literature.[3, 4, 5, 6, 7, 8] Additional articles were identified by performing a PubMed keyword search using the terms perioperative management or periprocedural management and anticoagulation or antithrombotic or antiplatelet in combination with keywords relevant to the content areas (eg, arthrocentesis, lumbar puncture). Articles for inclusion were chosen based on methodological quality and relevance to hospital medicine.
There are several questions that must be addressed when developing a periprocedural antithrombotic management strategy:
- What is the patient's risk of bleeding if antithrombotic therapy is continued?
- What is the patient's risk of thromboembolism if antithrombotic therapy is interrupted?
- Are there interventions that can decrease the risk of periprocedural bleeding and/or thromboembolism?
WHAT IS THE PATIENT'S RISK OF BLEEDING IF ANTITHROMBOTIC THERAPY IS CONTINUED?
Although the risk of bleeding is well described for many procedures, there are limited data on how that risk is affected by coagulopathy in general and antithrombotic medications in particular. When these data are available, they are largely derived from case series or bridging registries, which include heterogeneous patient populations and nonstandardized definitions of bleeding.[8, 9, 10] As such, few procedural or surgical professional societies have published guidelines on the periprocedural management of antithrombotic therapy,[3, 4, 5, 11]and guidelines from the American College of Chest Physicians (ACCP), the American College of Cardiology (ACC), and American Heart Association (AHA) only provide specific recommendations regarding minor ambulatory procedures.[6, 7, 8]
Procedures can be categorized as low or high risk for bleeding based on the following considerations: the extent of associated tissue injury, proximity to vital organs or vascular structures, the ability to readily detect and control bleeding, and the morbidity associated with a bleeding complication (eg, a small bleed into the epidural space is potentially catastrophic, whereas a large bleed from the colon often results in no permanent harm). For procedures with a high risk or consequence of bleeding, anticoagulants must be stopped, whereas in some cases antiplatelet agents can be safely continued. For procedures with a low risk or consequence of bleeding, it may be possible to continue both anticoagulant and antiplatelet agents.
| Procedure | Antithrombotic Therapy | |||||
|---|---|---|---|---|---|---|
| Aspirin | Thienopyridines | Prophylactic UFH or LWMH | Therapeutic UFH or LMWH | Warfarin | NOACs | |
| ||||||
| Arthrocentesis[12, 13, 14, 15] | + | + | + | + | + | + |
| Lumbar puncture[3] | + | 5000 units UFH BID | ||||
| Paracentesis[28, 29, 30] | + | + | + | |||
| Thoracentesis[37, 38, 39, 40, 41, 42] | + | + | + | |||
| Central venous catheter insertion[48, 49, 50, 51, 52, 53] | + | + | + | |||
Because procedures in hospitalized patients are most often performed for the purpose of diagnosing or treating an emergent condition, the risk of delaying the procedure while antithrombotic medications are held must be part of the overall risk‐benefit calculation.
Arthrocentesis
Bleeding complications from arthrocentesis are very rare, and there are few data on the additional risk associated with antithrombotic therapy.[12, 13, 14] In a retrospective cohort study, investigators determined the incidence of clinically significant bleeding (defined as bleeding requiring reversal of anticoagulation, prolonged manual pressure, surgical intervention, hospital admission, or delay in hospital discharge) and procedure‐related pain among 514 patients on antithrombotic therapy referred for arthrocentesis or injection of the hip, shoulder, or knee. Four hundred fifty‐six procedures were performed in patients without interrupting warfarin therapy, all of whom maintained an international normalized ratio (INR)2, and 184 procedures were performed in patients who had stopped their warfarin to achieve an INR <2. Antiplatelet therapy was routinely continued in both groups, with 48% of patients taking aspirin and 9% clopidogrel. There was 1 bleeding complication (0.2%) in a patient with an INR of 2.3 who was also taking aspirin, and 2 patients developed procedure‐related pain (INR 3.3 and 5.3, neither taking antiplatelet medications).[15]
Based on the available evidence, arthrocentesis appears to be safe in patients on therapeutic warfarin, with or without aspirin and/or clopidogrel. At present, there are no published studies that address the risk of arthrocentesis in patients taking other antiplatelet or anticoagulant medications, but given the low overall risk of this procedure, it is reasonable to infer that these medications can also be safely continued.
Lumbar Puncture
The incidence of bleeding complications from diagnostic lumbar puncture is unknown, but is likely similar to that seen with spinal anesthesia, where in a large retrospective observational study, spinal hematoma occurred in 1:165,000 spinal block procedures.[16] Factors associated with an increased risk of spinal hematoma include traumatic tap, advanced age, female gender, spinal cord or vertebral column abnormalities, coagulopathy, and not allowing sufficient time between stopping and restarting antithrombotic therapy.[3, 17, 18, 19, 20]
Therapeutic anticoagulation must be stopped and prophylactic anticoagulation delayed before performing a lumbar puncture. The 1 exception is low‐dose unfractionated heparin (UFH), which the American Society for Regional Anesthesia (ARSA) recommends continuing in patients undergoing neuraxial procedures, provided the total dose is 5000 U twice daily. This assessment is based on observational data, surveys of practice patterns, and decades of use without evidence of complications; in fact, there are only 5 case reports of spinal hematomas in this population.[3] However, because these data are from surgical populations, in which heparin thromboprophylaxis is typically dosed at 5000 units twice daily, there are limited data on the safety of higher or more frequent doses of heparin. In a retrospective cohort study of 928 patients who received thoracic epidural analgesia in conjunction with UFH dosed at 5000 U, 3 times daily, there were no cases of neuraxial bleeding, but given the rarity of neuraxial hematoma, it is not possible to draw any conclusions from this relatively small sample size.[21]
In November 2013, based on surveillance data showing increased risk for spinal or epidural hematoma associated with low‐molecular‐weight heparin (LMWH), the US Food and Drug Administration (FDA) issued a drug safety communication recommending that neuraxial procedures be delayed for 12 hours after prophylactic LMWH and 24 hours after therapeutic LMWH, and that LMWH not be restarted for at least 4 hours after catheter removal.[20] These recommendations are largely consistent with existing guidelines[3, 22] but are not explicitly stated in the package insert for any of the LMWHs available in the United States,[23, 24, 25] and the FDA is working with the manufacturers to add this information.
Nonsteroidal anti‐inflammatory drugs (NSAIDs), dipyridamole, and aspirin do not appear to increase the risk of spinal hematoma and are considered safe to continue.[11, 26] There are limited data on the safety of thienopyridine medications in neuraxial anesthesia, but based on case reports and increased bleeding rates seen in surgical populations, it is generally recommended that these medications be discontinued before performing a lumbar puncture.[3, 22, 27]
The optimal time to restart anticoagulation after a lumbar puncture is unknown. The ARSA recommends a minimum of 1 hour for UFH and 2 hours for LMWH after neuraxial catheter removal, and provides no specific guidance about other anticoagulants,[3] whereas the European Society of Anesthesiology recommends a minimum of 1 hour for UFH, 4 hours for LMWH, 4 to 6 hours for rivaroxaban and apixiban, and 6 hours for dabigatran and fondaparinux.[22] Longer time periods should be considered after a traumatic tap, and postprocedure monitoring of neurological function is recommended for all patients.
The available evidence suggests that lumbar puncture can be safely performed in patients being treated with aspirin, NSAIDs, and UFH dosed at 5000 U twice daily; the safety of higher or more frequent doses of UFH is not known. Lumbar puncture should be delayed 12 hours after prophylactic LMWH and 24 hours after therapeutic LMWH, and LMWH should not be restarted for at least 4 hours after the procedure.[20] There are limited data on the safety of thienopyridines, but they should generally be discontinued, and all other prophylactic or therapeutic anticoagulation must be stopped prior to the procedure.
Paracentesis
Bleeding complications from paracentesis are uncommon, with abdominal wall hematoma and hemoperitoneum complicating 1% and 0.01% of procedures, respectively.[28, 29, 30] Whether antithrombotic therapy increases the risk of bleeding during paracentesis is unknown, primarily because most patients for whom the procedure is indicated have coagulopathy and thrombocytopenia from liver disease, and are therefore rarely treated with these medications.
Although patients with liver disease often have an elevated INR due to impaired hepatic synthesis of clotting factors, it is incorrect to generalize the observed rate of bleeding in this population to patients with an elevated INR from warfarin therapy who may require paracentesis for reasons unrelated to liver disease (eg, malignancy or infection). The coagulopathy of liver disease reflects deficiencies in the hepatic production of both pro‐ and anticoagulant proteins, and these patients develop both thrombotic and hemorrhagic complications irrespective of their in vitro coagulation indices.[31]
Although the available evidence suggests that paracentesis can be safely performed in patients with coagulopathy from liver disease, regardless of the INR,[30] little is known about the bleeding risk in other patients, with or without antithrombotic therapy. Based on indirect evidence, it is reasonable to assume that prophylactic UFH or LWMH or antiplatelet therapy would confer minimal additional risk, whereas the safety of continuing therapeutic anticoagulation is unknown.
Thoracentesis
Bleeding complications from thoracentesis are uncommon, generally occurring in <1% of procedures.[32, 33, 34] Factors associated with increased risk of overall complications include operator inexperience, large volume drainage, and lack of ultrasound guidance.[34, 35, 36] There are no studies that specifically address the risk of bleeding in patients on anticoagulant therapy, but such patients are included in studies on the risk of bleeding with coagulopathy.[37, 38, 39, 40]
In a retrospective cohort study of 1076 ultrasound‐guided thoracenteses performed by radiologists on patients with coagulopathy (defined as thrombocytopenia or an elevated INR from any cause), there were no bleeding complications (defined as anything other than minimal symptoms not requiring intervention). Among the patients in this study, 497 (46%) patients had a preprocedure INR >1.5; 198 (24%) had an INR between 2 and 3, and 32 (4%) had an INR >3.[39]
A similar study, which compared outcomes in patients with corrected and uncorrected coagulopathy, included 744 patients with an INR >1.6 (from any cause), of which 167 received preprocedural fresh‐frozen plasma (FFP) and 577 did not. There was 1 (0.1%) bleeding complication in a patient who received prophylactic FFP and none in the group that was not transfused.[38]
In a prospective cohort of 312 patients at increased risk for bleeding (from coagulopathy or antithrombotic medications) who underwent ultrasound‐guided thoracentesis by a pulmonologist or physician's assistant, 44 (34%) had an INR >1.5 (secondary to liver disease or warfarin therapy), 15 (12%) were taking clopidogrel, and 14 (11%) were treated with therapeutic LMWH within 12 hours or therapeutic UFH within 4.5 hours of the procedure. There were no bleeding complications in any of the patients (defined as mean change in hematocrit, chest x‐ray abnormalities, hemothorax, or requirement for transfusion).[37]
Although there are no studies that specifically address the use of aspirin and bleeding complications in thoracentesis, it is generally considered safe to continue this medication,[5] and there are small studies that show that thoracentesis and small‐bore chest tubes can be safely placed in patients taking clopidogrel.[41, 42]
Thoracentesis is associated with a low rate of bleeding complications, and when performed by an experienced operator using ultrasound, warfarin does not appear to increase this risk. However, given the low overall complication rate, it is not known whether patients on warfarin would have worse outcomes in the event of more serious complications (eg, intercostal artery laceration). At present, there are no published studies that address the risk of thoracentesis in patients taking new oral anticoagulants (NOACs).
Central Venous Catheter Insertion
The incidence of bleeding complications from central venous catheter (CVC) placement varies depending on the site of insertion and definition of bleeding, with hematoma and hemothorax occurring in 0.1% to 6.9%, and 0.4% to 1.3% of procedures, respectively.[43, 44, 45] Factors that increase the likelihood of complications include operator inexperience, multiple needle passes, and lack of ultrasound guidance.[46, 47] There are no studies that specifically address the risk of bleeding from CVC placement in patients on anticoagulant therapy, but such patients are included in studies of CVC placement in patients with coagulopathy, which report similar complication rates as seen in patients with normal hemostasis.[48, 49, 50, 51, 52, 53]
In a retrospective cohort study, investigators collected information on CVC‐associated bleeding complications in 281 medical and surgical intensive care patients with coagulopathy (INR 1.5 from any cause) after they adopted a more conservative approach to plasma transfusion in their intensive care unit; specifically, the routine use of prophylactic FFP to correct coagulopathy was discouraged for patients with an INR <3 (vs usual practice using an INR cutoff of 1.5), but the final decision was left to the discretion of the attending performing or supervising the procedure. Bleeding was defined as insertion‐site hematoma, interventions other than local manual pressure, and the need for blood transfusion. One case of bleeding (hematoma) was observed in a patient with an INR of 3.9, who received FFP before the procedure. There were no complications among those with uncorrected coagulopathy, including 66 patients with an INR between 1.5 and 2.9, and 6 with an INR 3.0. Ultrasound guidance was used in 50% of CVCs placed in the internal jugular vein.[54]
Although there are no studies that specifically address the use of antiplatelet drugs and bleeding complications in CVC placement, aspirin is generally considered safe to continue,[5] and by inference, thienopyridines are expected to add minimal additional risk.
CVC placement is associated with a variable rate of bleeding complications, with hematoma being relatively common. Based on the available literature, warfarin does not appear to increase this risk, but there are limited data from which to draw firm conclusions. A femoral or jugular approach may be preferable because they allow for ultrasound visualization and are amenable to manual compression. There are no published studies that address the risk of CVC placement in patients taking NOACs, and although the risk of bleeding is probably similar to patients receiving warfarin, the lack of effective reversal agents for these medications should be part of any risk‐benefit calculation.[55]
WHAT IS THE PATIENT'S RISK OF THROMBOEMBOLISM IF ANTITHROMBOTIC THERAPY IS INTERRUPTED?
Anticoagulants
If it is determined that a procedure cannot safely be performed while continuing antithrombotic therapy, one must then consider the patient's risk of thromboembolism if these therapies are temporarily interrupted. Unfortunately, there are few robust clinical studies from which to make this assessment, and therefore most clinicians rely on the risk stratification model proposed by the ACCP, which divides patients into 3 tiers (low, moderate, high), based on their indication for anticoagulation and risk factors for thromboembolism (Table 2)[8]. The ACCP model is largely based on indirect evidence from antithrombotic therapy trials in nonoperative patients, and its application to perioperative patients necessitates several assumptions that may not hold true in practice.
| Indication for Anticoagulant Therapy | |||
|---|---|---|---|
| Risk Stratum | Mechanical Heart Valve | Atrial Fibrillation | VTE |
| |||
| High Thrombotic Risk |
|
|
|
| Moderate Thrombotic Risk |
|
|
|
| Low Thrombotic Risk |
|
|
|
First, it assumes that the annualized risk of a thrombotic event in nonoperative patients can be prorated to determine the short‐term risk of discontinuing antithrombotic therapy in the perioperative period. For example, it has been estimated that the risk for perioperative stroke in a patient with atrial fibrillation who temporarily interrupts anticoagulation for 1 week would be 0.1% (5% per year 52 weeks),[56, 57]and yet we know from observational data that the actual risk of perioperative stroke in similar patients is 5 to 7 times higher.[58, 59] Second, it assumes that bridging therapy will decrease the risk of thromboembolism in high‐risk patients when warfarin therapy is interrupted, a premise that is logical but has not been subject to randomized controlled trials.[60] Third, it does not take into account the surgery‐specific risk for thromboembolism, which varies significantly, with arterial thromboembolism being more common in cardiac valve, vascular, and neurologic procedures, and venous thromboembolism (VTE) being more likely in orthopedic, trauma, and cancer surgery.[61, 62] These limitations notwithstanding, the ACCP model still offers the best available framework for thrombotic risk assessment and a reasonable starting point for clinical decision making.
Antiplatelet Agents
Patients with coronary artery stents who undergo noncardiac surgery are at increased risk for adverse cardiovascular events, including acute stent thrombosis, which carries a risk of myocardial infarction and death of 70% and 30%, respectively.[63] This risk is highest during the period between stent implantation and endothelialization, a process that takes 4 to 6 weeks for bare‐metal stents (BMS) and 6 to 12 months for drug‐eluting stents (DES). Premature discontinuation of dual antiplatelet therapy is the most important risk factor for stent thrombosis during this time.[64] Although the optimal perioperative strategy for these patients is unknown, there is general agreement that elective surgery should be delayed for at least 4 weeks in patients with a BMS and 12 months for patients with a DES. If a procedure or surgery is required during this time period, every effort should be made to continue dual antiplatelet therapy; if this is not possible, aspirin should be continued, and thienopyridine therapy should be interrupted as briefly as possible (Table 3).
| Recommended Interval Between Last Dose of Medication and Procedure | Recommended Interval Between Procedure and First Dose of Medication, h | ||
|---|---|---|---|
| Low Risk or Consequence of Postprocedure Bleeding | High Risk or Consequence of Postprocedure Bleeding | ||
| |||
| Antiplatelet Medicationsa | |||
| Aspirin (81325 mg dailydipyridamole) | 710 days (skip 69 doses) | 24 | 48 |
| Ticlodipine (250 mg twice daily) | 1014 days (skip 1926 doses) | 24 | 48 |
| Clopidogrel (75 mg once daily) | 710 days (skip 69 doses)b | 24 | 48 |
| Prasugrel (10 mg once daily) | 710 days (skip 69 dose)c | 24 | 48 |
| Ticagrelor (90 mg twice daily; t =8 hours) | 5 days (skip 8 doses) | 24 | 48 |
| Cilostazol (100 mg twice daily; t =11 hours) | 3 days (skip 4 doses) | 24 | 48 |
| Anticoagulant Medicationse | |||
| Warfarin (t =3642 hours, but highly variable) | 6 days (skip 5 doses)f | 12 | 24 |
| Intravenous UFH (t 60 minutes) | 46 hours | 24 | 4872 |
| LMWH (t =37 hours) | |||
| Prophylactic dosing | 12 hours# | 12 | 2436 |
| Therapeutic dosing | |||
| Once daily | 24 hours (give 50% of last total dose)# | 24 | 4872 |
| Twice daily | 24 hours (skip 1 dose)# | 24 | 4872 |
| Fondaparinux (t =17 hours, any dose) | 34 days (skip 23 doses)h | 24 | 4872 |
| Dabigatran (150 mg twice daily) | |||
| CrCl>50 mL/min (t =1417 hours) | 3 days (skip 4 doses) | 24 | 4872 |
| CrCl 3050 mL/min (t =1618 hours) | 45 days (skip 68 doses) | 24 | 4872 |
| CrCl 1530 mL/min (t =1618 hours)i | 45 days (skip 68 doses) | 24 | 4872 |
| Rivaroxaban (20 mg once daily) | |||
| CrCl>50 mL/min (t =89 hours) | 3 days (skip 2 doses) | 24 | 4872 |
| CrCl 3050 mL/min (t =9 hours) | 3 days (skip 2 doses) | 24 | 4872 |
| CrCl 1529.9 mL/min (t =910 hours)j | 4 days (skip 3 doses) | 24 | 4872 |
| Apixiban (5 mg twice daily) | |||
| CrCl>50 mL/min (t =78 hours) | 3 days (skip 4 doses) | 24 | 4872 |
| CrCl 3050 mL/min (t =1718 hours) | 4 days (skip 6 doses) | 24 | 4872 |
ARE THERE INTERVENTIONS THAT CAN DECREASE THE RISK OF PERIPROCEDURAL BLEEDING AND/OR THROMBOEMBOLISM?
Mitigating the Risk of Bleeding
Bleeding complications can be reduced by allowing a sufficient time for the effects of antithrombotic medications to wear off before performing a procedure. This requires an understanding of the pharmacology of these medications, with particular attention to patients in whom these medications are less well studied, including the elderly, patients with renal insufficiency, and those with very high or low body mass index. Table 3 provides recommendations for when to stop antithrombotic therapy prior to an invasive procedure. The intervals are based on the time needed to achieve a minimal antithrombotic effect, which is generally 4 to 5 half‐lives for anticoagulants and 7 to 10 days for irreversible antiplatelet agents. Shorter intervals may be appropriate for procedures with low risk or consequence of bleeding, but there are insufficient data to make specific recommendations regarding this strategy.
It is equally important to ensure that there is adequate time for postoperative hemostasis prior to restarting antithrombotic therapy. Data from VTE prophylaxis trials and bridging studies consistently show that bleeding complications occur more frequently when anticoagulation is started too early, and antithrombotic therapy should generally be delayed 24 hours in patients at average risk and 48 to 72 hours in patients at high risk or consequence for postoperative bleeding.[8, 60, 65]
Aspirin increases the risk of surgical blood loss and transfusion by up to 20%, and by up to 50% when given in combination with clopidogrel, but with the exception of intracranial surgery, there does not appear to be an increase in perioperative morbidity or mortality with either of these agents.[66]
Mitigating the Risk of Thromboembolism
Once the decision has been made to temporarily discontinue warfarin, the next consideration is whether to bridge with a short acting anticoagulant (typically subcutaneous LMWH or intravenous UFH) during the period of time when the INR is subtherapeutic. Conceptually, one would expect this strategy would minimize the risk of thromboembolism, but its efficacy has never been clearly demonstrated. In fact, in a systematic review and meta‐analysis of 34 studies that compared the rates of thromboembolism among bridged and nonbridged patients, heparin therapy did not reduce the risk of thromboembolic events (odds ratio: 0.80; 95% confidence interval: 0.421.54), but did result in higher rates of periprocedural bleeding.[60]
The applicability of these results to clinical practice are limited by the heterogeneity of the data used in the analysis; specifically, bridging strategies varied (including therapeutic, intermediate, and prophylactic dose regimens), there was wide variation in the types of surgery (and therefore bleeding risk), and because the majority of studies were observational, there is a significant likelihood of confounding by indication (ie, patients at high risk for thromboembolism are more likely to receive bridging therapy), and thus the benefit of this strategy may be underestimated. It is also important to note that in the majority studies anticoagulation was restarted <24 hours after the procedure, which likely contributed to the increased rate of bleeding.
Therefore, although bridging therapy is not indicated for patients at low risk, it is premature to conclude that it should be avoided in patients at moderate or high risk for thromboembolism. The results of 2 ongoing, randomized, placebo‐controlled trials of bridging therapy in patients taking warfarin for atrial fibrillation (Effectiveness of Bridging Anticoagulation for Surgery [BRIDGE]) or mechanical heart values (A Double Blind Randomized Control Trial of Post‐Operative Low Molecular Weight Heparin Bridging Therapy Versus Placebo Bridging Therapy for Patients Who Are at High Risk for Arterial Thromboembolism [PERIOP‐2]) should help to answer this question.[67, 68]
The uncertainty regarding the benefits of bridging therapy is reflected in the changes to the most recent ACCP guidelines. In 2008, the ACCP recommended low‐dose LMWH or no bridging for patients at low risk (grade 2C), therapeutic‐dose bridging for patients at moderate risk (grade 2C), and therapeutic‐dose bridging for patients at high risk for thromboembolism (Grade 1C).[56] In 2012, the ACCP recommended against bridging for low‐risk patients (grade 2C), made no specific recommendation regarding moderate‐risk patients, and offered a less robust recommendation for bridging in high‐risk patients (grade 2C).[8]
Until the results of the BRIDGE and PERIOP‐2 trials are available, the author still favors therapeutic bridging for patients at high risk and selected patients at moderate risk for thromboembolism, provided sufficient time is allowed for postoperative hemostasis before anticoagulation is restarted. For procedures with a high risk or consequence of bleeding, intravenous UFH (without a bolus) is a reasonable initial postoperative strategy to insure that anticoagulation is tolerated before committing to LMWH. Indirect evidence supports the use of prophylactic or intermediate‐dose bridging regimens in patients for whom the primary consideration is the prevention of recurrent VTE, but data to show that this strategy is effective for the prevention of arterial thromboembolism are lacking.
Intravenous glycoprotein IIb/IIIa inhibitors are sometimes used to bridge high‐risk patients with coronary artery stents who must stop antiplatelet therapy prior to a procedure, but the data to support this practice are limited and observational in nature.[69, 70]
STARTING AND STOPPING ANTITHROMBOTIC THERAPY
Warfarin
For patients on warfarin, the INR at which it is safe to perform invasive procedures is unknown. Normal hemostasis requires clotting factor levels of approximately 20% to 40% of normal,[71] which generally corresponds to an INR of <1.5, whereas for most indications, therapeutic anticoagulation is achieved when the INR is between 2.0 and 3.5. However, because the relationship between the INR and the levels of clotting factors is nonlinear, for a given patient, the INR may be abnormal (ie, >1) despite levels of clotting factors that are sufficient for periprocedural hemostasis.[72, 73, 74, 75] Because of its relatively long half‐life (3642 hours), warfarin should be stopped 6 days (skip 5 doses) prior to a procedure to achieve an INR of <1.5, but can safely be restarted the same day in most patients.
Heparins
The half‐life of intravenous heparin is dose dependent, and at therapeutic levels is approximately 60 minutes; therefore, it should be discontinued 4 to 6 hours (5 half‐lives) before performing an invasive procedure.[76] The half‐life of subcutaneous LMWHs ranges from 3 to 7 hours in healthy volunteers,[23, 24, 25] and is often longer in patients for whom these medications are commonly prescribed.[77, 78] Therefore, when administered at therapeutic doses twice daily, the last dose should be given in the morning the day before the procedure, and for therapeutic once‐daily regimens, the last dose should be reduced by 50%.[8] The optimal time to discontinue prophylactic doses of LWMH prior to an invasive procedure is unclear, but a minimum of 12 hours is recommended.[22, 79] Because LWMHs are renally cleared, longer intervals are needed for patients with impaired renal function.[76, 80]
New Oral Anticoagulants
The manufacturer of rivaroxaban recommends that if anticoagulation must be discontinued, it be stopped at least 24 hours before the procedure.[81] Although this may be sufficient for procedures with a low risk or consequence of bleeding, the half‐life of rivaroxaban is between 8 and 10 hours, and therefore 48 hours (45 half‐lives) is required to ensure minimal residual anticoagulant effect.
Apixaban has a clearance half‐life of 6 hours, but displays prolonged absorption such that its effective half‐life is 12 hours after repeated dosing. The manufacturer recommends that it be stopped at least 24 hours prior to a procedure with a low risk or consequence of bleeding, and 48 hours prior to a procedure with a high risk or consequence of bleeding.[82]
The manufacturer of dabigatran recommends that the drug be discontinued 1 to 2 days (creatinine clearance (CrCl) 50 mL/min) or 3 to 5 days (CrCl <50 mL/min) before invasive or surgical procedures, and that longer times be considered when complete hemostasis is required.[83] Given that the half‐life of dabigatran is 14 to 17 hours, the author recommends that it be stopped at least 2 days (3 half‐lives) prior to a procedure with a low risk or consequence of bleeding, and 3 days (45 half‐lives) prior to a procedure with a high risk or consequence of bleeding.
The clearance of all the NOACs is significantly prolonged in patients with renal impairment, and a longer interval between the last dose and the procedure is necessary in patients with renal failure to ensure normal hemostasis (Table 3).
The effect of the NOACs on the standard clotting assays are complex and vary depending on drug dose, the type of reagents used, and the calibration of the equipment. For dabigatran, the activated partial thromboplastin time (aPTT) and the thrombin time (TT) are sufficiently sensitive to allow for a qualitative assessment of drug effect, such that a normal aPTT indicates the absence, or a very low level of an anticoagulant effect, and a normal TT essentially rules out an effect. Accurate quantitative testing of dabigatran requires an appropriately calibrated dilute thrombin test or ecarin clotting time assay.[84, 85]
Depending on the thromboplastin reagent used, the prothrombin time (PT) may be sufficiently sensitive to rivaroxaban that a normal level rules out a residual drug effect,[86] but this does not hold true for apixaban, which has minimal effect on the PT at therapeutic concentrations. The aPTT is insensitive to both rivaroxaban and apixaban and cannot be used for assessing residual drug effect. Accurate quantitative testing of rivaroxaban or apixaban requires an anti‐factor Xa assay calibrated for use with these agents.[84]
Antiplatelet Agents
Aspirin irreversibly inhibits platelet cyclooxygenase activity, and the thienopyridines clopidogrel and prasugrel, irreversibly inhibit the platelet P2Y12 receptor. As such, the biological effects of these medications persist until the platelet pool has turned over, a process that occurs at 10% to 12% per day and takes 7 to 10 days to complete.[87] The minimum number of functional platelets required to ensure adequate periprocedural hemostasis is unknown, but is likely between 50 and 100,000/L.[88] Therefore, assuming a platelet pool of 200,000/L, most patients will regenerate an adequate number of functional platelets by 5 days after discontinuing therapy, and nearly all will have normal platelet function by 10 days. Determining the risk of bleeding prior to complete turnover of the platelet pool is further complicated by genetic variability between patients in drug metabolism and the degree of platelet inhibition by these agents.[89]
Owing to this complexity, guidelines and prescribing recommendations are inconsistent. The ACCP recommends stopping antiplatelet agents 7 to 10 days prior to an invasive procedure, and the ACC/AHA makes no specific recommendations at all.[90] Based on data from patients undergoing cardiac bypass surgery, it is recommended that clopidogrel be stopped 5 days, and prasugrel 7 days, prior to an invasive procedure.[91, 92] The elimination half‐life of ticlodipine is sufficiently long (up to 96 hours after repeated dosing) that it should be stopped 10 to 14 days prior to an invasive procedure.[87] Ticagrelor is a reversible P2Y12 receptor inhibitor with a half‐life of approximately 8 hours and should therefore have minimal effect by 3 days after discontinuation; however, the manufacturer recommends that it be stopped 5 days prior to an invasive procedure.[93]
The optimal time to restart antiplatelet agents after an invasive procedure is also unknown. The 2008 ACCP guidelines recommended restarting aspirin and/or clopidogrel in 24 hours, or as hemostasis allows,[56] whereas neither the 2007 or 2009 ACC/AHA guidelines,[90] or the most recent 2012 ACCP guidelines,[8] offer specific recommendations. Aspirin, prasugrel, and ticagrelor have a rapid onset of action, whereas the full antiplatelet effect of clopidogrel does not occur for several days, and for patients in whom more rapid platelet inhibition is desired, a loading dose (300600 mg) may be appropriate.[87]
CONCLUSIONS
Deciding on an optimal periprocedural antithrombotic management strategy is a common challenge for hospitalists that requires careful consideration of both patient and procedure related‐risk factors for bleeding and thrombosis, as well as the consequences of delaying or forgoing the procedure altogether. For many procedures, there is evidence that antithrombotic therapy can be safely continued, thereby obviating the risk associated with interrupting therapy. When antithrombotic therapy must be stopped, it should be done in a manner that appropriately balances the risks and consequence of periprocedural bleeding and thromboembolism. Strategies to decrease the risk of perioperative bleeding include allowing sufficient time for the effects of antithrombotic therapy to subside before starting the procedure, and ensuring adequate time for hemostasis before restarting antithrombotic therapy. Bridging therapy may provide net clinical benefit for patients at moderate to high risk for thromboembolism, but this will not be clear until the results of several ongoing bridging trials are available. The periprocedural antithrombotic management strategy should be developed in collaboration with the relevant providers and with active participation by the patient in all decisions and treatment plans. Standardized protocols and documentation can help to minimize unintended variation in practice and improve information transfer during transitions of care.
Acknowledgements
The author would like to thank Shoshana and Lola Herzig for their support in the design and preparation of the manuscript.
Disclosure: Nothing to report.
The periprocedural management of antithrombotic medications is a common challenge for hospitalists, for which there is limited high‐quality evidence to guide clinical decision making. The introduction of third‐generation antiplatelet agents (prasugrel and ticagrelor) and the new oral anticoagulants (rivaroxaban, apixaban, and dabigatran), has added an additional layer of complexity to clinical management.
This article will provide a conceptual framework for the periprocedural management of antithrombotic therapy, with a particular focus on procedures that are considered core competencies by the Society of Hospital Medicine; these include: arthrocentesis, lumbar puncture, paracentesis, thoracentesis, and central line placement (Table 1).[1, 2] The recommendations in this article are based on a review of published guidelines and consensus statements and their supporting literature.[3, 4, 5, 6, 7, 8] Additional articles were identified by performing a PubMed keyword search using the terms perioperative management or periprocedural management and anticoagulation or antithrombotic or antiplatelet in combination with keywords relevant to the content areas (eg, arthrocentesis, lumbar puncture). Articles for inclusion were chosen based on methodological quality and relevance to hospital medicine.
There are several questions that must be addressed when developing a periprocedural antithrombotic management strategy:
- What is the patient's risk of bleeding if antithrombotic therapy is continued?
- What is the patient's risk of thromboembolism if antithrombotic therapy is interrupted?
- Are there interventions that can decrease the risk of periprocedural bleeding and/or thromboembolism?
WHAT IS THE PATIENT'S RISK OF BLEEDING IF ANTITHROMBOTIC THERAPY IS CONTINUED?
Although the risk of bleeding is well described for many procedures, there are limited data on how that risk is affected by coagulopathy in general and antithrombotic medications in particular. When these data are available, they are largely derived from case series or bridging registries, which include heterogeneous patient populations and nonstandardized definitions of bleeding.[8, 9, 10] As such, few procedural or surgical professional societies have published guidelines on the periprocedural management of antithrombotic therapy,[3, 4, 5, 11]and guidelines from the American College of Chest Physicians (ACCP), the American College of Cardiology (ACC), and American Heart Association (AHA) only provide specific recommendations regarding minor ambulatory procedures.[6, 7, 8]
Procedures can be categorized as low or high risk for bleeding based on the following considerations: the extent of associated tissue injury, proximity to vital organs or vascular structures, the ability to readily detect and control bleeding, and the morbidity associated with a bleeding complication (eg, a small bleed into the epidural space is potentially catastrophic, whereas a large bleed from the colon often results in no permanent harm). For procedures with a high risk or consequence of bleeding, anticoagulants must be stopped, whereas in some cases antiplatelet agents can be safely continued. For procedures with a low risk or consequence of bleeding, it may be possible to continue both anticoagulant and antiplatelet agents.
| Procedure | Antithrombotic Therapy | |||||
|---|---|---|---|---|---|---|
| Aspirin | Thienopyridines | Prophylactic UFH or LWMH | Therapeutic UFH or LMWH | Warfarin | NOACs | |
| ||||||
| Arthrocentesis[12, 13, 14, 15] | + | + | + | + | + | + |
| Lumbar puncture[3] | + | 5000 units UFH BID | ||||
| Paracentesis[28, 29, 30] | + | + | + | |||
| Thoracentesis[37, 38, 39, 40, 41, 42] | + | + | + | |||
| Central venous catheter insertion[48, 49, 50, 51, 52, 53] | + | + | + | |||
Because procedures in hospitalized patients are most often performed for the purpose of diagnosing or treating an emergent condition, the risk of delaying the procedure while antithrombotic medications are held must be part of the overall risk‐benefit calculation.
Arthrocentesis
Bleeding complications from arthrocentesis are very rare, and there are few data on the additional risk associated with antithrombotic therapy.[12, 13, 14] In a retrospective cohort study, investigators determined the incidence of clinically significant bleeding (defined as bleeding requiring reversal of anticoagulation, prolonged manual pressure, surgical intervention, hospital admission, or delay in hospital discharge) and procedure‐related pain among 514 patients on antithrombotic therapy referred for arthrocentesis or injection of the hip, shoulder, or knee. Four hundred fifty‐six procedures were performed in patients without interrupting warfarin therapy, all of whom maintained an international normalized ratio (INR)2, and 184 procedures were performed in patients who had stopped their warfarin to achieve an INR <2. Antiplatelet therapy was routinely continued in both groups, with 48% of patients taking aspirin and 9% clopidogrel. There was 1 bleeding complication (0.2%) in a patient with an INR of 2.3 who was also taking aspirin, and 2 patients developed procedure‐related pain (INR 3.3 and 5.3, neither taking antiplatelet medications).[15]
Based on the available evidence, arthrocentesis appears to be safe in patients on therapeutic warfarin, with or without aspirin and/or clopidogrel. At present, there are no published studies that address the risk of arthrocentesis in patients taking other antiplatelet or anticoagulant medications, but given the low overall risk of this procedure, it is reasonable to infer that these medications can also be safely continued.
Lumbar Puncture
The incidence of bleeding complications from diagnostic lumbar puncture is unknown, but is likely similar to that seen with spinal anesthesia, where in a large retrospective observational study, spinal hematoma occurred in 1:165,000 spinal block procedures.[16] Factors associated with an increased risk of spinal hematoma include traumatic tap, advanced age, female gender, spinal cord or vertebral column abnormalities, coagulopathy, and not allowing sufficient time between stopping and restarting antithrombotic therapy.[3, 17, 18, 19, 20]
Therapeutic anticoagulation must be stopped and prophylactic anticoagulation delayed before performing a lumbar puncture. The 1 exception is low‐dose unfractionated heparin (UFH), which the American Society for Regional Anesthesia (ARSA) recommends continuing in patients undergoing neuraxial procedures, provided the total dose is 5000 U twice daily. This assessment is based on observational data, surveys of practice patterns, and decades of use without evidence of complications; in fact, there are only 5 case reports of spinal hematomas in this population.[3] However, because these data are from surgical populations, in which heparin thromboprophylaxis is typically dosed at 5000 units twice daily, there are limited data on the safety of higher or more frequent doses of heparin. In a retrospective cohort study of 928 patients who received thoracic epidural analgesia in conjunction with UFH dosed at 5000 U, 3 times daily, there were no cases of neuraxial bleeding, but given the rarity of neuraxial hematoma, it is not possible to draw any conclusions from this relatively small sample size.[21]
In November 2013, based on surveillance data showing increased risk for spinal or epidural hematoma associated with low‐molecular‐weight heparin (LMWH), the US Food and Drug Administration (FDA) issued a drug safety communication recommending that neuraxial procedures be delayed for 12 hours after prophylactic LMWH and 24 hours after therapeutic LMWH, and that LMWH not be restarted for at least 4 hours after catheter removal.[20] These recommendations are largely consistent with existing guidelines[3, 22] but are not explicitly stated in the package insert for any of the LMWHs available in the United States,[23, 24, 25] and the FDA is working with the manufacturers to add this information.
Nonsteroidal anti‐inflammatory drugs (NSAIDs), dipyridamole, and aspirin do not appear to increase the risk of spinal hematoma and are considered safe to continue.[11, 26] There are limited data on the safety of thienopyridine medications in neuraxial anesthesia, but based on case reports and increased bleeding rates seen in surgical populations, it is generally recommended that these medications be discontinued before performing a lumbar puncture.[3, 22, 27]
The optimal time to restart anticoagulation after a lumbar puncture is unknown. The ARSA recommends a minimum of 1 hour for UFH and 2 hours for LMWH after neuraxial catheter removal, and provides no specific guidance about other anticoagulants,[3] whereas the European Society of Anesthesiology recommends a minimum of 1 hour for UFH, 4 hours for LMWH, 4 to 6 hours for rivaroxaban and apixiban, and 6 hours for dabigatran and fondaparinux.[22] Longer time periods should be considered after a traumatic tap, and postprocedure monitoring of neurological function is recommended for all patients.
The available evidence suggests that lumbar puncture can be safely performed in patients being treated with aspirin, NSAIDs, and UFH dosed at 5000 U twice daily; the safety of higher or more frequent doses of UFH is not known. Lumbar puncture should be delayed 12 hours after prophylactic LMWH and 24 hours after therapeutic LMWH, and LMWH should not be restarted for at least 4 hours after the procedure.[20] There are limited data on the safety of thienopyridines, but they should generally be discontinued, and all other prophylactic or therapeutic anticoagulation must be stopped prior to the procedure.
Paracentesis
Bleeding complications from paracentesis are uncommon, with abdominal wall hematoma and hemoperitoneum complicating 1% and 0.01% of procedures, respectively.[28, 29, 30] Whether antithrombotic therapy increases the risk of bleeding during paracentesis is unknown, primarily because most patients for whom the procedure is indicated have coagulopathy and thrombocytopenia from liver disease, and are therefore rarely treated with these medications.
Although patients with liver disease often have an elevated INR due to impaired hepatic synthesis of clotting factors, it is incorrect to generalize the observed rate of bleeding in this population to patients with an elevated INR from warfarin therapy who may require paracentesis for reasons unrelated to liver disease (eg, malignancy or infection). The coagulopathy of liver disease reflects deficiencies in the hepatic production of both pro‐ and anticoagulant proteins, and these patients develop both thrombotic and hemorrhagic complications irrespective of their in vitro coagulation indices.[31]
Although the available evidence suggests that paracentesis can be safely performed in patients with coagulopathy from liver disease, regardless of the INR,[30] little is known about the bleeding risk in other patients, with or without antithrombotic therapy. Based on indirect evidence, it is reasonable to assume that prophylactic UFH or LWMH or antiplatelet therapy would confer minimal additional risk, whereas the safety of continuing therapeutic anticoagulation is unknown.
Thoracentesis
Bleeding complications from thoracentesis are uncommon, generally occurring in <1% of procedures.[32, 33, 34] Factors associated with increased risk of overall complications include operator inexperience, large volume drainage, and lack of ultrasound guidance.[34, 35, 36] There are no studies that specifically address the risk of bleeding in patients on anticoagulant therapy, but such patients are included in studies on the risk of bleeding with coagulopathy.[37, 38, 39, 40]
In a retrospective cohort study of 1076 ultrasound‐guided thoracenteses performed by radiologists on patients with coagulopathy (defined as thrombocytopenia or an elevated INR from any cause), there were no bleeding complications (defined as anything other than minimal symptoms not requiring intervention). Among the patients in this study, 497 (46%) patients had a preprocedure INR >1.5; 198 (24%) had an INR between 2 and 3, and 32 (4%) had an INR >3.[39]
A similar study, which compared outcomes in patients with corrected and uncorrected coagulopathy, included 744 patients with an INR >1.6 (from any cause), of which 167 received preprocedural fresh‐frozen plasma (FFP) and 577 did not. There was 1 (0.1%) bleeding complication in a patient who received prophylactic FFP and none in the group that was not transfused.[38]
In a prospective cohort of 312 patients at increased risk for bleeding (from coagulopathy or antithrombotic medications) who underwent ultrasound‐guided thoracentesis by a pulmonologist or physician's assistant, 44 (34%) had an INR >1.5 (secondary to liver disease or warfarin therapy), 15 (12%) were taking clopidogrel, and 14 (11%) were treated with therapeutic LMWH within 12 hours or therapeutic UFH within 4.5 hours of the procedure. There were no bleeding complications in any of the patients (defined as mean change in hematocrit, chest x‐ray abnormalities, hemothorax, or requirement for transfusion).[37]
Although there are no studies that specifically address the use of aspirin and bleeding complications in thoracentesis, it is generally considered safe to continue this medication,[5] and there are small studies that show that thoracentesis and small‐bore chest tubes can be safely placed in patients taking clopidogrel.[41, 42]
Thoracentesis is associated with a low rate of bleeding complications, and when performed by an experienced operator using ultrasound, warfarin does not appear to increase this risk. However, given the low overall complication rate, it is not known whether patients on warfarin would have worse outcomes in the event of more serious complications (eg, intercostal artery laceration). At present, there are no published studies that address the risk of thoracentesis in patients taking new oral anticoagulants (NOACs).
Central Venous Catheter Insertion
The incidence of bleeding complications from central venous catheter (CVC) placement varies depending on the site of insertion and definition of bleeding, with hematoma and hemothorax occurring in 0.1% to 6.9%, and 0.4% to 1.3% of procedures, respectively.[43, 44, 45] Factors that increase the likelihood of complications include operator inexperience, multiple needle passes, and lack of ultrasound guidance.[46, 47] There are no studies that specifically address the risk of bleeding from CVC placement in patients on anticoagulant therapy, but such patients are included in studies of CVC placement in patients with coagulopathy, which report similar complication rates as seen in patients with normal hemostasis.[48, 49, 50, 51, 52, 53]
In a retrospective cohort study, investigators collected information on CVC‐associated bleeding complications in 281 medical and surgical intensive care patients with coagulopathy (INR 1.5 from any cause) after they adopted a more conservative approach to plasma transfusion in their intensive care unit; specifically, the routine use of prophylactic FFP to correct coagulopathy was discouraged for patients with an INR <3 (vs usual practice using an INR cutoff of 1.5), but the final decision was left to the discretion of the attending performing or supervising the procedure. Bleeding was defined as insertion‐site hematoma, interventions other than local manual pressure, and the need for blood transfusion. One case of bleeding (hematoma) was observed in a patient with an INR of 3.9, who received FFP before the procedure. There were no complications among those with uncorrected coagulopathy, including 66 patients with an INR between 1.5 and 2.9, and 6 with an INR 3.0. Ultrasound guidance was used in 50% of CVCs placed in the internal jugular vein.[54]
Although there are no studies that specifically address the use of antiplatelet drugs and bleeding complications in CVC placement, aspirin is generally considered safe to continue,[5] and by inference, thienopyridines are expected to add minimal additional risk.
CVC placement is associated with a variable rate of bleeding complications, with hematoma being relatively common. Based on the available literature, warfarin does not appear to increase this risk, but there are limited data from which to draw firm conclusions. A femoral or jugular approach may be preferable because they allow for ultrasound visualization and are amenable to manual compression. There are no published studies that address the risk of CVC placement in patients taking NOACs, and although the risk of bleeding is probably similar to patients receiving warfarin, the lack of effective reversal agents for these medications should be part of any risk‐benefit calculation.[55]
WHAT IS THE PATIENT'S RISK OF THROMBOEMBOLISM IF ANTITHROMBOTIC THERAPY IS INTERRUPTED?
Anticoagulants
If it is determined that a procedure cannot safely be performed while continuing antithrombotic therapy, one must then consider the patient's risk of thromboembolism if these therapies are temporarily interrupted. Unfortunately, there are few robust clinical studies from which to make this assessment, and therefore most clinicians rely on the risk stratification model proposed by the ACCP, which divides patients into 3 tiers (low, moderate, high), based on their indication for anticoagulation and risk factors for thromboembolism (Table 2)[8]. The ACCP model is largely based on indirect evidence from antithrombotic therapy trials in nonoperative patients, and its application to perioperative patients necessitates several assumptions that may not hold true in practice.
| Indication for Anticoagulant Therapy | |||
|---|---|---|---|
| Risk Stratum | Mechanical Heart Valve | Atrial Fibrillation | VTE |
| |||
| High Thrombotic Risk |
|
|
|
| Moderate Thrombotic Risk |
|
|
|
| Low Thrombotic Risk |
|
|
|
First, it assumes that the annualized risk of a thrombotic event in nonoperative patients can be prorated to determine the short‐term risk of discontinuing antithrombotic therapy in the perioperative period. For example, it has been estimated that the risk for perioperative stroke in a patient with atrial fibrillation who temporarily interrupts anticoagulation for 1 week would be 0.1% (5% per year 52 weeks),[56, 57]and yet we know from observational data that the actual risk of perioperative stroke in similar patients is 5 to 7 times higher.[58, 59] Second, it assumes that bridging therapy will decrease the risk of thromboembolism in high‐risk patients when warfarin therapy is interrupted, a premise that is logical but has not been subject to randomized controlled trials.[60] Third, it does not take into account the surgery‐specific risk for thromboembolism, which varies significantly, with arterial thromboembolism being more common in cardiac valve, vascular, and neurologic procedures, and venous thromboembolism (VTE) being more likely in orthopedic, trauma, and cancer surgery.[61, 62] These limitations notwithstanding, the ACCP model still offers the best available framework for thrombotic risk assessment and a reasonable starting point for clinical decision making.
Antiplatelet Agents
Patients with coronary artery stents who undergo noncardiac surgery are at increased risk for adverse cardiovascular events, including acute stent thrombosis, which carries a risk of myocardial infarction and death of 70% and 30%, respectively.[63] This risk is highest during the period between stent implantation and endothelialization, a process that takes 4 to 6 weeks for bare‐metal stents (BMS) and 6 to 12 months for drug‐eluting stents (DES). Premature discontinuation of dual antiplatelet therapy is the most important risk factor for stent thrombosis during this time.[64] Although the optimal perioperative strategy for these patients is unknown, there is general agreement that elective surgery should be delayed for at least 4 weeks in patients with a BMS and 12 months for patients with a DES. If a procedure or surgery is required during this time period, every effort should be made to continue dual antiplatelet therapy; if this is not possible, aspirin should be continued, and thienopyridine therapy should be interrupted as briefly as possible (Table 3).
| Recommended Interval Between Last Dose of Medication and Procedure | Recommended Interval Between Procedure and First Dose of Medication, h | ||
|---|---|---|---|
| Low Risk or Consequence of Postprocedure Bleeding | High Risk or Consequence of Postprocedure Bleeding | ||
| |||
| Antiplatelet Medicationsa | |||
| Aspirin (81325 mg dailydipyridamole) | 710 days (skip 69 doses) | 24 | 48 |
| Ticlodipine (250 mg twice daily) | 1014 days (skip 1926 doses) | 24 | 48 |
| Clopidogrel (75 mg once daily) | 710 days (skip 69 doses)b | 24 | 48 |
| Prasugrel (10 mg once daily) | 710 days (skip 69 dose)c | 24 | 48 |
| Ticagrelor (90 mg twice daily; t =8 hours) | 5 days (skip 8 doses) | 24 | 48 |
| Cilostazol (100 mg twice daily; t =11 hours) | 3 days (skip 4 doses) | 24 | 48 |
| Anticoagulant Medicationse | |||
| Warfarin (t =3642 hours, but highly variable) | 6 days (skip 5 doses)f | 12 | 24 |
| Intravenous UFH (t 60 minutes) | 46 hours | 24 | 4872 |
| LMWH (t =37 hours) | |||
| Prophylactic dosing | 12 hours# | 12 | 2436 |
| Therapeutic dosing | |||
| Once daily | 24 hours (give 50% of last total dose)# | 24 | 4872 |
| Twice daily | 24 hours (skip 1 dose)# | 24 | 4872 |
| Fondaparinux (t =17 hours, any dose) | 34 days (skip 23 doses)h | 24 | 4872 |
| Dabigatran (150 mg twice daily) | |||
| CrCl>50 mL/min (t =1417 hours) | 3 days (skip 4 doses) | 24 | 4872 |
| CrCl 3050 mL/min (t =1618 hours) | 45 days (skip 68 doses) | 24 | 4872 |
| CrCl 1530 mL/min (t =1618 hours)i | 45 days (skip 68 doses) | 24 | 4872 |
| Rivaroxaban (20 mg once daily) | |||
| CrCl>50 mL/min (t =89 hours) | 3 days (skip 2 doses) | 24 | 4872 |
| CrCl 3050 mL/min (t =9 hours) | 3 days (skip 2 doses) | 24 | 4872 |
| CrCl 1529.9 mL/min (t =910 hours)j | 4 days (skip 3 doses) | 24 | 4872 |
| Apixiban (5 mg twice daily) | |||
| CrCl>50 mL/min (t =78 hours) | 3 days (skip 4 doses) | 24 | 4872 |
| CrCl 3050 mL/min (t =1718 hours) | 4 days (skip 6 doses) | 24 | 4872 |
ARE THERE INTERVENTIONS THAT CAN DECREASE THE RISK OF PERIPROCEDURAL BLEEDING AND/OR THROMBOEMBOLISM?
Mitigating the Risk of Bleeding
Bleeding complications can be reduced by allowing a sufficient time for the effects of antithrombotic medications to wear off before performing a procedure. This requires an understanding of the pharmacology of these medications, with particular attention to patients in whom these medications are less well studied, including the elderly, patients with renal insufficiency, and those with very high or low body mass index. Table 3 provides recommendations for when to stop antithrombotic therapy prior to an invasive procedure. The intervals are based on the time needed to achieve a minimal antithrombotic effect, which is generally 4 to 5 half‐lives for anticoagulants and 7 to 10 days for irreversible antiplatelet agents. Shorter intervals may be appropriate for procedures with low risk or consequence of bleeding, but there are insufficient data to make specific recommendations regarding this strategy.
It is equally important to ensure that there is adequate time for postoperative hemostasis prior to restarting antithrombotic therapy. Data from VTE prophylaxis trials and bridging studies consistently show that bleeding complications occur more frequently when anticoagulation is started too early, and antithrombotic therapy should generally be delayed 24 hours in patients at average risk and 48 to 72 hours in patients at high risk or consequence for postoperative bleeding.[8, 60, 65]
Aspirin increases the risk of surgical blood loss and transfusion by up to 20%, and by up to 50% when given in combination with clopidogrel, but with the exception of intracranial surgery, there does not appear to be an increase in perioperative morbidity or mortality with either of these agents.[66]
Mitigating the Risk of Thromboembolism
Once the decision has been made to temporarily discontinue warfarin, the next consideration is whether to bridge with a short acting anticoagulant (typically subcutaneous LMWH or intravenous UFH) during the period of time when the INR is subtherapeutic. Conceptually, one would expect this strategy would minimize the risk of thromboembolism, but its efficacy has never been clearly demonstrated. In fact, in a systematic review and meta‐analysis of 34 studies that compared the rates of thromboembolism among bridged and nonbridged patients, heparin therapy did not reduce the risk of thromboembolic events (odds ratio: 0.80; 95% confidence interval: 0.421.54), but did result in higher rates of periprocedural bleeding.[60]
The applicability of these results to clinical practice are limited by the heterogeneity of the data used in the analysis; specifically, bridging strategies varied (including therapeutic, intermediate, and prophylactic dose regimens), there was wide variation in the types of surgery (and therefore bleeding risk), and because the majority of studies were observational, there is a significant likelihood of confounding by indication (ie, patients at high risk for thromboembolism are more likely to receive bridging therapy), and thus the benefit of this strategy may be underestimated. It is also important to note that in the majority studies anticoagulation was restarted <24 hours after the procedure, which likely contributed to the increased rate of bleeding.
Therefore, although bridging therapy is not indicated for patients at low risk, it is premature to conclude that it should be avoided in patients at moderate or high risk for thromboembolism. The results of 2 ongoing, randomized, placebo‐controlled trials of bridging therapy in patients taking warfarin for atrial fibrillation (Effectiveness of Bridging Anticoagulation for Surgery [BRIDGE]) or mechanical heart values (A Double Blind Randomized Control Trial of Post‐Operative Low Molecular Weight Heparin Bridging Therapy Versus Placebo Bridging Therapy for Patients Who Are at High Risk for Arterial Thromboembolism [PERIOP‐2]) should help to answer this question.[67, 68]
The uncertainty regarding the benefits of bridging therapy is reflected in the changes to the most recent ACCP guidelines. In 2008, the ACCP recommended low‐dose LMWH or no bridging for patients at low risk (grade 2C), therapeutic‐dose bridging for patients at moderate risk (grade 2C), and therapeutic‐dose bridging for patients at high risk for thromboembolism (Grade 1C).[56] In 2012, the ACCP recommended against bridging for low‐risk patients (grade 2C), made no specific recommendation regarding moderate‐risk patients, and offered a less robust recommendation for bridging in high‐risk patients (grade 2C).[8]
Until the results of the BRIDGE and PERIOP‐2 trials are available, the author still favors therapeutic bridging for patients at high risk and selected patients at moderate risk for thromboembolism, provided sufficient time is allowed for postoperative hemostasis before anticoagulation is restarted. For procedures with a high risk or consequence of bleeding, intravenous UFH (without a bolus) is a reasonable initial postoperative strategy to insure that anticoagulation is tolerated before committing to LMWH. Indirect evidence supports the use of prophylactic or intermediate‐dose bridging regimens in patients for whom the primary consideration is the prevention of recurrent VTE, but data to show that this strategy is effective for the prevention of arterial thromboembolism are lacking.
Intravenous glycoprotein IIb/IIIa inhibitors are sometimes used to bridge high‐risk patients with coronary artery stents who must stop antiplatelet therapy prior to a procedure, but the data to support this practice are limited and observational in nature.[69, 70]
STARTING AND STOPPING ANTITHROMBOTIC THERAPY
Warfarin
For patients on warfarin, the INR at which it is safe to perform invasive procedures is unknown. Normal hemostasis requires clotting factor levels of approximately 20% to 40% of normal,[71] which generally corresponds to an INR of <1.5, whereas for most indications, therapeutic anticoagulation is achieved when the INR is between 2.0 and 3.5. However, because the relationship between the INR and the levels of clotting factors is nonlinear, for a given patient, the INR may be abnormal (ie, >1) despite levels of clotting factors that are sufficient for periprocedural hemostasis.[72, 73, 74, 75] Because of its relatively long half‐life (3642 hours), warfarin should be stopped 6 days (skip 5 doses) prior to a procedure to achieve an INR of <1.5, but can safely be restarted the same day in most patients.
Heparins
The half‐life of intravenous heparin is dose dependent, and at therapeutic levels is approximately 60 minutes; therefore, it should be discontinued 4 to 6 hours (5 half‐lives) before performing an invasive procedure.[76] The half‐life of subcutaneous LMWHs ranges from 3 to 7 hours in healthy volunteers,[23, 24, 25] and is often longer in patients for whom these medications are commonly prescribed.[77, 78] Therefore, when administered at therapeutic doses twice daily, the last dose should be given in the morning the day before the procedure, and for therapeutic once‐daily regimens, the last dose should be reduced by 50%.[8] The optimal time to discontinue prophylactic doses of LWMH prior to an invasive procedure is unclear, but a minimum of 12 hours is recommended.[22, 79] Because LWMHs are renally cleared, longer intervals are needed for patients with impaired renal function.[76, 80]
New Oral Anticoagulants
The manufacturer of rivaroxaban recommends that if anticoagulation must be discontinued, it be stopped at least 24 hours before the procedure.[81] Although this may be sufficient for procedures with a low risk or consequence of bleeding, the half‐life of rivaroxaban is between 8 and 10 hours, and therefore 48 hours (45 half‐lives) is required to ensure minimal residual anticoagulant effect.
Apixaban has a clearance half‐life of 6 hours, but displays prolonged absorption such that its effective half‐life is 12 hours after repeated dosing. The manufacturer recommends that it be stopped at least 24 hours prior to a procedure with a low risk or consequence of bleeding, and 48 hours prior to a procedure with a high risk or consequence of bleeding.[82]
The manufacturer of dabigatran recommends that the drug be discontinued 1 to 2 days (creatinine clearance (CrCl) 50 mL/min) or 3 to 5 days (CrCl <50 mL/min) before invasive or surgical procedures, and that longer times be considered when complete hemostasis is required.[83] Given that the half‐life of dabigatran is 14 to 17 hours, the author recommends that it be stopped at least 2 days (3 half‐lives) prior to a procedure with a low risk or consequence of bleeding, and 3 days (45 half‐lives) prior to a procedure with a high risk or consequence of bleeding.
The clearance of all the NOACs is significantly prolonged in patients with renal impairment, and a longer interval between the last dose and the procedure is necessary in patients with renal failure to ensure normal hemostasis (Table 3).
The effect of the NOACs on the standard clotting assays are complex and vary depending on drug dose, the type of reagents used, and the calibration of the equipment. For dabigatran, the activated partial thromboplastin time (aPTT) and the thrombin time (TT) are sufficiently sensitive to allow for a qualitative assessment of drug effect, such that a normal aPTT indicates the absence, or a very low level of an anticoagulant effect, and a normal TT essentially rules out an effect. Accurate quantitative testing of dabigatran requires an appropriately calibrated dilute thrombin test or ecarin clotting time assay.[84, 85]
Depending on the thromboplastin reagent used, the prothrombin time (PT) may be sufficiently sensitive to rivaroxaban that a normal level rules out a residual drug effect,[86] but this does not hold true for apixaban, which has minimal effect on the PT at therapeutic concentrations. The aPTT is insensitive to both rivaroxaban and apixaban and cannot be used for assessing residual drug effect. Accurate quantitative testing of rivaroxaban or apixaban requires an anti‐factor Xa assay calibrated for use with these agents.[84]
Antiplatelet Agents
Aspirin irreversibly inhibits platelet cyclooxygenase activity, and the thienopyridines clopidogrel and prasugrel, irreversibly inhibit the platelet P2Y12 receptor. As such, the biological effects of these medications persist until the platelet pool has turned over, a process that occurs at 10% to 12% per day and takes 7 to 10 days to complete.[87] The minimum number of functional platelets required to ensure adequate periprocedural hemostasis is unknown, but is likely between 50 and 100,000/L.[88] Therefore, assuming a platelet pool of 200,000/L, most patients will regenerate an adequate number of functional platelets by 5 days after discontinuing therapy, and nearly all will have normal platelet function by 10 days. Determining the risk of bleeding prior to complete turnover of the platelet pool is further complicated by genetic variability between patients in drug metabolism and the degree of platelet inhibition by these agents.[89]
Owing to this complexity, guidelines and prescribing recommendations are inconsistent. The ACCP recommends stopping antiplatelet agents 7 to 10 days prior to an invasive procedure, and the ACC/AHA makes no specific recommendations at all.[90] Based on data from patients undergoing cardiac bypass surgery, it is recommended that clopidogrel be stopped 5 days, and prasugrel 7 days, prior to an invasive procedure.[91, 92] The elimination half‐life of ticlodipine is sufficiently long (up to 96 hours after repeated dosing) that it should be stopped 10 to 14 days prior to an invasive procedure.[87] Ticagrelor is a reversible P2Y12 receptor inhibitor with a half‐life of approximately 8 hours and should therefore have minimal effect by 3 days after discontinuation; however, the manufacturer recommends that it be stopped 5 days prior to an invasive procedure.[93]
The optimal time to restart antiplatelet agents after an invasive procedure is also unknown. The 2008 ACCP guidelines recommended restarting aspirin and/or clopidogrel in 24 hours, or as hemostasis allows,[56] whereas neither the 2007 or 2009 ACC/AHA guidelines,[90] or the most recent 2012 ACCP guidelines,[8] offer specific recommendations. Aspirin, prasugrel, and ticagrelor have a rapid onset of action, whereas the full antiplatelet effect of clopidogrel does not occur for several days, and for patients in whom more rapid platelet inhibition is desired, a loading dose (300600 mg) may be appropriate.[87]
CONCLUSIONS
Deciding on an optimal periprocedural antithrombotic management strategy is a common challenge for hospitalists that requires careful consideration of both patient and procedure related‐risk factors for bleeding and thrombosis, as well as the consequences of delaying or forgoing the procedure altogether. For many procedures, there is evidence that antithrombotic therapy can be safely continued, thereby obviating the risk associated with interrupting therapy. When antithrombotic therapy must be stopped, it should be done in a manner that appropriately balances the risks and consequence of periprocedural bleeding and thromboembolism. Strategies to decrease the risk of perioperative bleeding include allowing sufficient time for the effects of antithrombotic therapy to subside before starting the procedure, and ensuring adequate time for hemostasis before restarting antithrombotic therapy. Bridging therapy may provide net clinical benefit for patients at moderate to high risk for thromboembolism, but this will not be clear until the results of several ongoing bridging trials are available. The periprocedural antithrombotic management strategy should be developed in collaboration with the relevant providers and with active participation by the patient in all decisions and treatment plans. Standardized protocols and documentation can help to minimize unintended variation in practice and improve information transfer during transitions of care.
Acknowledgements
The author would like to thank Shoshana and Lola Herzig for their support in the design and preparation of the manuscript.
Disclosure: Nothing to report.
- , , , , . Core competencies in hospital medicine: development and methodology. J Hosp Med. 2006;1(suppl 1):48–56.
- , , , , . Procedures performed by hospitalist and non‐hospitalist general internists. J Gen Int Med. 2010;25(5):448–452.
- , , , et al. Regional anesthesia in the patient receiving antithrombotic or thrombolytic therapy: American Society of Regional Anesthesia and Pain Medicine evidence‐based guidelines (Third Edition). Reg Anesth Pain Med. 2010;35(1):64–101.
- ASGE Standards of Practice Committee, , , , et al. Management of antithrombotic agents for endoscopic procedures. Gastrointest Endosc. 2009;70(6):1060–1070.
- , , , et al. Consensus guidelines for periprocedural management of coagulation status and hemostasis risk in percutaneous image‐guided interventions. J Vasc Interv Radiol. 2012;23(6):727–736.
- , , , et al. 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation. 2008;118(15):e523–e661.
- , , , et al. Prevention of premature discontinuation of dual antiplatelet therapy in patients with coronary artery stents: a science advisory from the American Heart Association, American College of Cardiology, Society for Cardiovascular Angiography and Interventions, American College of Surgeons, and American Dental Association, with representation from the American College of Physicians. J Am Coll Cardiol. 2007;49(6):734–739.
- , , , et al. Perioperative management of antithrombotic therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 Suppl):e326S–e350S.
- , , , et al. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in surgical patients. J Thromb Haemost. 2010;8(1):202–204.
- , , , et al. Clinical outcomes with unfractionated heparin or low‐molecular‐weight heparin as bridging therapy in patients on long‐term oral anticoagulants: the REGIMEN registry. J Thromb Haemost. 2006;4(6):1246–1252.
- , , , et al. Assessment of bleeding risk of interventional techniques: a best evidence synthesis of practice patterns and perioperative management of anticoagulant and antithrombotic therapy. Pain Physician. 2013;16(2 suppl):Se261–Se318.
- , . Perioperative management of patients receiving oral anticoagulants: a systematic review. Arch Intern Med. 2003;163(8):901–908.
- , , , et al. Frequency of the bleeding risk in patients receiving warfarin submitted to arthrocentesis of the knee [in Italian]. Reumatismo. 2003;55(3):159–163.
- , . A prospective study of the safety of joint and soft tissue aspirations and injections in patients taking warfarin sodium. Arthritis Rheum. 1998;41(4):736–739.
- , . Safety of arthrocentesis and joint injection in patients receiving anticoagulation at therapeutic levels. Am J Med. 2012;125(3):265–269.
- , , . Severe neurological complications after central neuraxial blockades in Sweden 1990–1999. Anesthesiology. 2004;101(4):950–959.
- , . Managing anticoagulated patients during neuraxial anaesthesia. Br J Haematol. 2010;149(2):195–208.
- . Impaired haemostasis and regional anaesthesia. Can J Anaesth. 1996;43(5 pt 2):R129–R141.
- , , . Cauda equina syndrome following a lumbar puncture. J Clin Neurosci. 2009;16(5):714–716.
- United States Food and Drug Safety Communication: updated recommendations to decrease risk of spinal column bleeding and paralysis in patients on low molecular weight heparins. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm373595.htm. Accessed January 6, 2014.
- , , , , . Three‐times‐daily subcutaneous unfractionated heparin and neuraxial anesthesia: a retrospective review of 928 cases. Reg Anesth Pain Med. 2012;37(6):623–626.
- , , , et al. Regional anaesthesia and antithrombotic agents: recommendations of the European Society of Anaesthesiology. Eur J Anaesthesiol. 2010;27(12):999–1015.
- Eisai Inc. Fragmin (dalteparin sodium injection) full prescribing information. 2009. Available at: http://us.eisai.com/wps/wcm/connect/Eisai/Home/Our+Products/FRAGMIN. Accessed January 6, 2014
- Sanofi‐Aventis. Lovenox (enoxaparin sodium injection) full prescribing information. 2013. Available at: http://products.sanofi.us/lovenox/lovenox.html. Accessed January 6, 2014.
- LEO Pharmaceutical Products. Innohep (tinzaparin sodium injection) full prescribing information. 2008. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020484s011lbl.pdf. Accessed January 6, 2014.
- , , , et al. Preoperative antiplatelet therapy does not increase the risk of spinal hematoma associated with regional anesthesia. Anesth Analg. 1995;80(2):303–309.
- , , , et al. Addendum of newer anticoagulants to the SIR consensus guideline. J Vasc Interv Radiol. 2013;24(5):641–645.
- , . Severe haemorrhage following abdominal paracentesis for ascites in patients with liver disease. Aliment Pharmacol Ther. 2005;21(5):525–529.
- , . Ultrasound guidance decreases complications and improves the cost of care among patients undergoing thoracentesis and paracentesis. Chest. 2013;143(2):532–538.
- . Management of adult patients with ascites due to cirrhosis. Hepatology. 2004;39(3):841–856.
- , . The coagulopathy of chronic liver disease. N Engl J Med. 2011;365(2):147–156.
- , , , , . Necessity of routine chest roentgenography after thoracentesis. Ann Intern Med. 1996;124(9):816–820.
- , , , , . Pleural procedures and pleuroscopy. Respirology. 2009;14(6):796–807.
- , , . Ultrasonography guidance reduces complications and costs associated with thoracentesis procedures. J Clin Ultrasound. 2012;40(3):135–141.
- , , , . Reducing iatrogenic risk in thoracentesis: establishing best practice via experiential training in a zero‐risk environment. Chest. 2009;135(5):1315–1320.
- , . Improving the safety of thoracentesis. Curr Opin Pulm Med. 2011;17(4):232–236.
- , , , , . The safety of thoracentesis in patients with uncorrected bleeding risk. Ann Am Thorac Soc. 2013;10(4):336–341.
- , , , et al. Safety of ultrasound‐guided thoracentesis in patients with abnormal preprocedural coagulation parameters. Chest. 2013;144(2):456–463.
- , . Abnormal preprocedural international normalized ratio and platelet counts are not associated with increased bleeding complications after ultrasound‐guided thoracentesis. AJR Am J Roentgenol. 2011;197(1):W164–W168.
- , . Lack of increased bleeding after paracentesis and thoracentesis in patients with mild coagulation abnormalities. Transfusion. 1991;31(2):164–171.
- , , . Safety of ultrasound‐guided small‐bore chest tube insertion in patients on clopidogrel. J Bronchology Interv Pulmonol. 2013;20(1):16–20.
- , , , . Effect of routine clopidogrel use on bleeding complications after ultrasound‐guided thoracentesis. J Bronchology Interv Pulmonol. 2012;19(4):284–287.
- , . Preventing complications of central venous catheterization. N Engl J Med. 2003;348(12):1123–1133.
- , , . Complications of central venous catheters: internal jugular versus subclavian access—a systematic review. Crit Care Med. 2002;30(2):454–460.
- , , , . Risk factors for acute adverse events during ultrasound‐guided central venous cannulation in the emergency department. Acad Emerg Med. 2010;17(10):1055–1061.
- . Complications of central venous catheterization. J Am Coll Surg. 2007;204(4):681–696.
- , , , , , . Real‐time two‐dimensional ultrasound guidance for central venous cannulation: a meta‐analysis. Anesthesiology. 2013;118(2):361–375.
- , , . Central venous catheter placement in patients with disorders of hemostasis. Chest. 1996;110(1):185–188.
- , , , . Invasive line placement in critically ill patients: do hemostatic defects matter? Transfusion. 1996;36(9):827–831.
- , , , , , . Bleeding complications after central line insertions: relevance of pre‐procedure coagulation tests and institutional transfusion policy. Acta Anaesthesiol Scand. 2013;57(5):573–579.
- , , , et al. Low levels of prothrombin time (INR) and platelets do not increase the risk of significant bleeding when placing central venous catheters. Med Klin (Munich). 2009;104(5):331–335.
- , , , et al. Coagulation disorders in patients with cancer: nontunneled central venous catheter placement with US guidance—a single‐institution retrospective analysis. Radiology. 2009;253(1):249–252.
- , , . US‐guided placement of central vein catheters in patients with disorders of hemostasis. Eur J Radiol. 2008;65(2):253–256.
- , , . Central line placement in patients with and without prophylactic plasma. J Crit Care. 2012;27(5):529.e529–e513.
- , , . How I treat: target specific oral anticoagulant associated bleeding [published online ahead of print January 2, 2014]. Blood. doi: 10.1182/blood‐2013‐09‐529784.
- , , , et al. The perioperative management of antithrombotic therapy: American College of Chest Physicians evidence‐based clinical practice guidelines (8th edition). Chest. 2008;133(6 suppl):299S–339S.
- , . Management of anticoagulation before and after elective surgery. N Engl J Med. 1997;336(21):1506–1511.
- , , , et al. RIsk of thromboembolism with short‐term interruption of warfarin therapy. Arch Intern Med. 2008;168(1):63–69.
- , , , et al. Periprocedural bleeding and thromboembolic events with dabigatran compared with warfarin: results from the Randomized Evaluation of Long‐Term Anticoagulation Therapy (RE‐LY) randomized trial. Circulation. 2012;126(3):343–348.
- , , , , , . Periprocedural heparin bridging in patients receiving vitamin K antagonists: systematic review and meta‐analysis of bleeding and thromboembolic rates. Circulation. 2012;126(13):1630–1639.
- , , , et al. Prevention of VTE in nonorthopedic surgical patients: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 Suppl):e227S–277S.
- , , , et al. Periprocedural anticoagulation management of patients with venous thromboembolism. Arterioscler Thromb Vasc Biol. 2010;30(3):442–448.
- , . Late coronary stent thrombosis. Circulation. 2007;116(17):1952–1965.
- , , , et al. Predictors of coronary stent thrombosis: the Dutch Stent Thrombosis Registry. J Am Coll Cardiol. 2009;53(16):1399–1409.
- , , , et al. Predictors of major bleeding in peri‐procedural anticoagulation management. J Thromb Haemost. 2012;10(2):261–267.
- , , . Perioperative antiplatelet therapy: the case for continuing therapy in patients at risk of myocardial infarction. Br J Anaesth. 2007;99(3):316–328.
- , . Effectiveness of bridging anticoagulation for surgery (the BRIDGE Study). Available at: www.ClinicalTrials.gov. Identifier: NCT00786474. Accessed October 22, 2013.
- . A safety and effectiveness study of LMWH bridging therapy versus placebo bridging therapy for patients on long term warfarin and require temporary interruption of their warfarin (PERIOP2). Available at: www.ClinicalTrials.gov. Identifier: NCT00432796. Accessed October 20, 2013.
- , , , et al. Outcomes of a preoperative “bridging” strategy with glycoprotein IIb/IIIa inhibitors to prevent perioperative stent thrombosis in patients with drug‐eluting stents who undergo surgery necessitating interruption of thienopyridine administration. EuroIntervention. 2013;9(2):204–211.
- , , , et al. Safety of “bridging” with eptifibatide for patients with coronary stents before cardiac and non‐cardiac surgery. Am J Cardiol. 2012;110(4):485–490.
- . Hemostatic problems in surgical patients. In: Colman RW HJ, Marder VJ, Clowes AW, George JN, ed. Hemostasis and Thrombosis. 4th ed. Philadelphia, PA: Lippincott Williams 2001:1033.
- . Reversal of drug‐induced anticoagulation: old solutions and new problems. Transfusion. 2012;52:45S–55S.
- , , . A feasibility study of continuing dose‐reduced warfarin for invasive procedures in patients with high thromboembolic risk. Chest. 2005;127(3):922–927.
- , , , et al. A simple and safe nomogram for the management of oral anticoagulation prior to minor surgery. Clin Lab Haematol. 2003;25(2):127–130.
- , , , , . International normalized ratio versus plasma levels of coagulation factors in patients on vitamin K antagonist therapy. Arch Pathol Lab Med. 2011;135(4):490–494.
- , , , . Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College Of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 suppl):e24S–e43S.
- , , , . Bridging anticoagulation with low‐molecular‐weight heparin after interruption of warfarin therapy is associated with a residual anticoagulant effect prior to surgery. Thromb Haemost. 2005;94(3):528–531.
- , , , et al. Brief communication: Preoperative anticoagulant activity after bridging low‐molecular‐weight heparin for temporary interruption of warfarin. Ann Intern Med. 2007;146(3):184–187.
- . Regional anaesthesia in the patient receiving antithrombotic and antiplatelet therapy. Br J Anaesth. 2011;107(suppl 1):i96–i106.
- , , , . Meta‐analysis: low‐molecular‐weight heparin and bleeding in patients with severe renal insufficiency. Ann Intern Med. 2006;144(9):673–684.
- Janssen Pharmaceuticals, Inc. Xarelto (rivaroxaban) full prescribing information. 2013. Available at: http://www.xareltohcp.com/sites/default/files/pdf/xarelto_0.pdf#zoom=100. Accessed October 1, 2013.
- Bristol Meyers Squibb, Inc. Eliquis (apixaban) full prescribing information. 2013. Available at: http://packageinserts.bms.com/pi/pi_eliquis.pdf. Accessed October 1, 2013.
- Boehringer Ingelheim Pharmaceuticals I. Pradaxa (dabigatran etexilate mesylate) full prescribing information. Available at: http://bidocs.boehringer‐ingelheim.com/BIWebAccess/ViewServlet.ser?docBase= renetnt11(2):245–252.
- . The laboratory and the direct oral anticoagulants. Blood. 2013;121(20):4032–4035.
- , , , , , . Assessment of the impact of rivaroxaban on coagulation assays: laboratory recommendations for the monitoring of rivaroxaban and review of the literature. Thromb Res. 2012;130(6):956–966.
- , , , , . Antiplatelet drugs: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 suppl):e89S–119S.
- . Evidence‐based platelet transfusion guidelines. Hematology Am Soc Hematol Educ Program. 2007:172–178.
- , , . Platelet function testing and prediction of procedural bleeding risk. Thromb Haemost. 2013;109(5):817–824.
- , , , . Perioperative management of antiplatelet therapy in patients with a coronary stent who need non‐cardiac surgery: a systematic review of clinical practice guidelines. Chest. 2013;144(6):1848–1856.
- Eli Lilly Pharmaceuticals, Inc. Effient (prasugrel) full prescribing information. 2012. Available at: http://pi.lilly.com/us/effient.pdf. Accessed October 1, 2013.
- , , , . Antiplatelet therapy and cardiac surgery: review of recent evidence and clinical implications. Can J Cardiol. 2013;29(9):1042–1047.
- AstraZeneca. Brilinta (ticagrelor) full prescribing information. 2013. Available at: http://www1.astrazeneca‐us.com/pi/brilinta.pdf. Accessed October 1, 2013.
- , , , et al. 2012 update to the Society of Thoracic Surgeons guideline on use of antiplatelet drugs in patients having cardiac and noncardiac operations. Ann Thorac Surg. 2012;94(5):1761–1781.
- , . How I treat anticoagulated patients undergoing an elective procedure or surgery. Blood. 2012;120(15):2954–2962.
- , , , , American College of Chest Physicians. Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 suppl):e24S–e43S.
- , , , , . Core competencies in hospital medicine: development and methodology. J Hosp Med. 2006;1(suppl 1):48–56.
- , , , , . Procedures performed by hospitalist and non‐hospitalist general internists. J Gen Int Med. 2010;25(5):448–452.
- , , , et al. Regional anesthesia in the patient receiving antithrombotic or thrombolytic therapy: American Society of Regional Anesthesia and Pain Medicine evidence‐based guidelines (Third Edition). Reg Anesth Pain Med. 2010;35(1):64–101.
- ASGE Standards of Practice Committee, , , , et al. Management of antithrombotic agents for endoscopic procedures. Gastrointest Endosc. 2009;70(6):1060–1070.
- , , , et al. Consensus guidelines for periprocedural management of coagulation status and hemostasis risk in percutaneous image‐guided interventions. J Vasc Interv Radiol. 2012;23(6):727–736.
- , , , et al. 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation. 2008;118(15):e523–e661.
- , , , et al. Prevention of premature discontinuation of dual antiplatelet therapy in patients with coronary artery stents: a science advisory from the American Heart Association, American College of Cardiology, Society for Cardiovascular Angiography and Interventions, American College of Surgeons, and American Dental Association, with representation from the American College of Physicians. J Am Coll Cardiol. 2007;49(6):734–739.
- , , , et al. Perioperative management of antithrombotic therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 Suppl):e326S–e350S.
- , , , et al. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in surgical patients. J Thromb Haemost. 2010;8(1):202–204.
- , , , et al. Clinical outcomes with unfractionated heparin or low‐molecular‐weight heparin as bridging therapy in patients on long‐term oral anticoagulants: the REGIMEN registry. J Thromb Haemost. 2006;4(6):1246–1252.
- , , , et al. Assessment of bleeding risk of interventional techniques: a best evidence synthesis of practice patterns and perioperative management of anticoagulant and antithrombotic therapy. Pain Physician. 2013;16(2 suppl):Se261–Se318.
- , . Perioperative management of patients receiving oral anticoagulants: a systematic review. Arch Intern Med. 2003;163(8):901–908.
- , , , et al. Frequency of the bleeding risk in patients receiving warfarin submitted to arthrocentesis of the knee [in Italian]. Reumatismo. 2003;55(3):159–163.
- , . A prospective study of the safety of joint and soft tissue aspirations and injections in patients taking warfarin sodium. Arthritis Rheum. 1998;41(4):736–739.
- , . Safety of arthrocentesis and joint injection in patients receiving anticoagulation at therapeutic levels. Am J Med. 2012;125(3):265–269.
- , , . Severe neurological complications after central neuraxial blockades in Sweden 1990–1999. Anesthesiology. 2004;101(4):950–959.
- , . Managing anticoagulated patients during neuraxial anaesthesia. Br J Haematol. 2010;149(2):195–208.
- . Impaired haemostasis and regional anaesthesia. Can J Anaesth. 1996;43(5 pt 2):R129–R141.
- , , . Cauda equina syndrome following a lumbar puncture. J Clin Neurosci. 2009;16(5):714–716.
- United States Food and Drug Safety Communication: updated recommendations to decrease risk of spinal column bleeding and paralysis in patients on low molecular weight heparins. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm373595.htm. Accessed January 6, 2014.
- , , , , . Three‐times‐daily subcutaneous unfractionated heparin and neuraxial anesthesia: a retrospective review of 928 cases. Reg Anesth Pain Med. 2012;37(6):623–626.
- , , , et al. Regional anaesthesia and antithrombotic agents: recommendations of the European Society of Anaesthesiology. Eur J Anaesthesiol. 2010;27(12):999–1015.
- Eisai Inc. Fragmin (dalteparin sodium injection) full prescribing information. 2009. Available at: http://us.eisai.com/wps/wcm/connect/Eisai/Home/Our+Products/FRAGMIN. Accessed January 6, 2014
- Sanofi‐Aventis. Lovenox (enoxaparin sodium injection) full prescribing information. 2013. Available at: http://products.sanofi.us/lovenox/lovenox.html. Accessed January 6, 2014.
- LEO Pharmaceutical Products. Innohep (tinzaparin sodium injection) full prescribing information. 2008. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020484s011lbl.pdf. Accessed January 6, 2014.
- , , , et al. Preoperative antiplatelet therapy does not increase the risk of spinal hematoma associated with regional anesthesia. Anesth Analg. 1995;80(2):303–309.
- , , , et al. Addendum of newer anticoagulants to the SIR consensus guideline. J Vasc Interv Radiol. 2013;24(5):641–645.
- , . Severe haemorrhage following abdominal paracentesis for ascites in patients with liver disease. Aliment Pharmacol Ther. 2005;21(5):525–529.
- , . Ultrasound guidance decreases complications and improves the cost of care among patients undergoing thoracentesis and paracentesis. Chest. 2013;143(2):532–538.
- . Management of adult patients with ascites due to cirrhosis. Hepatology. 2004;39(3):841–856.
- , . The coagulopathy of chronic liver disease. N Engl J Med. 2011;365(2):147–156.
- , , , , . Necessity of routine chest roentgenography after thoracentesis. Ann Intern Med. 1996;124(9):816–820.
- , , , , . Pleural procedures and pleuroscopy. Respirology. 2009;14(6):796–807.
- , , . Ultrasonography guidance reduces complications and costs associated with thoracentesis procedures. J Clin Ultrasound. 2012;40(3):135–141.
- , , , . Reducing iatrogenic risk in thoracentesis: establishing best practice via experiential training in a zero‐risk environment. Chest. 2009;135(5):1315–1320.
- , . Improving the safety of thoracentesis. Curr Opin Pulm Med. 2011;17(4):232–236.
- , , , , . The safety of thoracentesis in patients with uncorrected bleeding risk. Ann Am Thorac Soc. 2013;10(4):336–341.
- , , , et al. Safety of ultrasound‐guided thoracentesis in patients with abnormal preprocedural coagulation parameters. Chest. 2013;144(2):456–463.
- , . Abnormal preprocedural international normalized ratio and platelet counts are not associated with increased bleeding complications after ultrasound‐guided thoracentesis. AJR Am J Roentgenol. 2011;197(1):W164–W168.
- , . Lack of increased bleeding after paracentesis and thoracentesis in patients with mild coagulation abnormalities. Transfusion. 1991;31(2):164–171.
- , , . Safety of ultrasound‐guided small‐bore chest tube insertion in patients on clopidogrel. J Bronchology Interv Pulmonol. 2013;20(1):16–20.
- , , , . Effect of routine clopidogrel use on bleeding complications after ultrasound‐guided thoracentesis. J Bronchology Interv Pulmonol. 2012;19(4):284–287.
- , . Preventing complications of central venous catheterization. N Engl J Med. 2003;348(12):1123–1133.
- , , . Complications of central venous catheters: internal jugular versus subclavian access—a systematic review. Crit Care Med. 2002;30(2):454–460.
- , , , . Risk factors for acute adverse events during ultrasound‐guided central venous cannulation in the emergency department. Acad Emerg Med. 2010;17(10):1055–1061.
- . Complications of central venous catheterization. J Am Coll Surg. 2007;204(4):681–696.
- , , , , , . Real‐time two‐dimensional ultrasound guidance for central venous cannulation: a meta‐analysis. Anesthesiology. 2013;118(2):361–375.
- , , . Central venous catheter placement in patients with disorders of hemostasis. Chest. 1996;110(1):185–188.
- , , , . Invasive line placement in critically ill patients: do hemostatic defects matter? Transfusion. 1996;36(9):827–831.
- , , , , , . Bleeding complications after central line insertions: relevance of pre‐procedure coagulation tests and institutional transfusion policy. Acta Anaesthesiol Scand. 2013;57(5):573–579.
- , , , et al. Low levels of prothrombin time (INR) and platelets do not increase the risk of significant bleeding when placing central venous catheters. Med Klin (Munich). 2009;104(5):331–335.
- , , , et al. Coagulation disorders in patients with cancer: nontunneled central venous catheter placement with US guidance—a single‐institution retrospective analysis. Radiology. 2009;253(1):249–252.
- , , . US‐guided placement of central vein catheters in patients with disorders of hemostasis. Eur J Radiol. 2008;65(2):253–256.
- , , . Central line placement in patients with and without prophylactic plasma. J Crit Care. 2012;27(5):529.e529–e513.
- , , . How I treat: target specific oral anticoagulant associated bleeding [published online ahead of print January 2, 2014]. Blood. doi: 10.1182/blood‐2013‐09‐529784.
- , , , et al. The perioperative management of antithrombotic therapy: American College of Chest Physicians evidence‐based clinical practice guidelines (8th edition). Chest. 2008;133(6 suppl):299S–339S.
- , . Management of anticoagulation before and after elective surgery. N Engl J Med. 1997;336(21):1506–1511.
- , , , et al. RIsk of thromboembolism with short‐term interruption of warfarin therapy. Arch Intern Med. 2008;168(1):63–69.
- , , , et al. Periprocedural bleeding and thromboembolic events with dabigatran compared with warfarin: results from the Randomized Evaluation of Long‐Term Anticoagulation Therapy (RE‐LY) randomized trial. Circulation. 2012;126(3):343–348.
- , , , , , . Periprocedural heparin bridging in patients receiving vitamin K antagonists: systematic review and meta‐analysis of bleeding and thromboembolic rates. Circulation. 2012;126(13):1630–1639.
- , , , et al. Prevention of VTE in nonorthopedic surgical patients: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 Suppl):e227S–277S.
- , , , et al. Periprocedural anticoagulation management of patients with venous thromboembolism. Arterioscler Thromb Vasc Biol. 2010;30(3):442–448.
- , . Late coronary stent thrombosis. Circulation. 2007;116(17):1952–1965.
- , , , et al. Predictors of coronary stent thrombosis: the Dutch Stent Thrombosis Registry. J Am Coll Cardiol. 2009;53(16):1399–1409.
- , , , et al. Predictors of major bleeding in peri‐procedural anticoagulation management. J Thromb Haemost. 2012;10(2):261–267.
- , , . Perioperative antiplatelet therapy: the case for continuing therapy in patients at risk of myocardial infarction. Br J Anaesth. 2007;99(3):316–328.
- , . Effectiveness of bridging anticoagulation for surgery (the BRIDGE Study). Available at: www.ClinicalTrials.gov. Identifier: NCT00786474. Accessed October 22, 2013.
- . A safety and effectiveness study of LMWH bridging therapy versus placebo bridging therapy for patients on long term warfarin and require temporary interruption of their warfarin (PERIOP2). Available at: www.ClinicalTrials.gov. Identifier: NCT00432796. Accessed October 20, 2013.
- , , , et al. Outcomes of a preoperative “bridging” strategy with glycoprotein IIb/IIIa inhibitors to prevent perioperative stent thrombosis in patients with drug‐eluting stents who undergo surgery necessitating interruption of thienopyridine administration. EuroIntervention. 2013;9(2):204–211.
- , , , et al. Safety of “bridging” with eptifibatide for patients with coronary stents before cardiac and non‐cardiac surgery. Am J Cardiol. 2012;110(4):485–490.
- . Hemostatic problems in surgical patients. In: Colman RW HJ, Marder VJ, Clowes AW, George JN, ed. Hemostasis and Thrombosis. 4th ed. Philadelphia, PA: Lippincott Williams 2001:1033.
- . Reversal of drug‐induced anticoagulation: old solutions and new problems. Transfusion. 2012;52:45S–55S.
- , , . A feasibility study of continuing dose‐reduced warfarin for invasive procedures in patients with high thromboembolic risk. Chest. 2005;127(3):922–927.
- , , , et al. A simple and safe nomogram for the management of oral anticoagulation prior to minor surgery. Clin Lab Haematol. 2003;25(2):127–130.
- , , , , . International normalized ratio versus plasma levels of coagulation factors in patients on vitamin K antagonist therapy. Arch Pathol Lab Med. 2011;135(4):490–494.
- , , , . Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College Of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 suppl):e24S–e43S.
- , , , . Bridging anticoagulation with low‐molecular‐weight heparin after interruption of warfarin therapy is associated with a residual anticoagulant effect prior to surgery. Thromb Haemost. 2005;94(3):528–531.
- , , , et al. Brief communication: Preoperative anticoagulant activity after bridging low‐molecular‐weight heparin for temporary interruption of warfarin. Ann Intern Med. 2007;146(3):184–187.
- . Regional anaesthesia in the patient receiving antithrombotic and antiplatelet therapy. Br J Anaesth. 2011;107(suppl 1):i96–i106.
- , , , . Meta‐analysis: low‐molecular‐weight heparin and bleeding in patients with severe renal insufficiency. Ann Intern Med. 2006;144(9):673–684.
- Janssen Pharmaceuticals, Inc. Xarelto (rivaroxaban) full prescribing information. 2013. Available at: http://www.xareltohcp.com/sites/default/files/pdf/xarelto_0.pdf#zoom=100. Accessed October 1, 2013.
- Bristol Meyers Squibb, Inc. Eliquis (apixaban) full prescribing information. 2013. Available at: http://packageinserts.bms.com/pi/pi_eliquis.pdf. Accessed October 1, 2013.
- Boehringer Ingelheim Pharmaceuticals I. Pradaxa (dabigatran etexilate mesylate) full prescribing information. Available at: http://bidocs.boehringer‐ingelheim.com/BIWebAccess/ViewServlet.ser?docBase= renetnt11(2):245–252.
- . The laboratory and the direct oral anticoagulants. Blood. 2013;121(20):4032–4035.
- , , , , , . Assessment of the impact of rivaroxaban on coagulation assays: laboratory recommendations for the monitoring of rivaroxaban and review of the literature. Thromb Res. 2012;130(6):956–966.
- , , , , . Antiplatelet drugs: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 suppl):e89S–119S.
- . Evidence‐based platelet transfusion guidelines. Hematology Am Soc Hematol Educ Program. 2007:172–178.
- , , . Platelet function testing and prediction of procedural bleeding risk. Thromb Haemost. 2013;109(5):817–824.
- , , , . Perioperative management of antiplatelet therapy in patients with a coronary stent who need non‐cardiac surgery: a systematic review of clinical practice guidelines. Chest. 2013;144(6):1848–1856.
- Eli Lilly Pharmaceuticals, Inc. Effient (prasugrel) full prescribing information. 2012. Available at: http://pi.lilly.com/us/effient.pdf. Accessed October 1, 2013.
- , , , . Antiplatelet therapy and cardiac surgery: review of recent evidence and clinical implications. Can J Cardiol. 2013;29(9):1042–1047.
- AstraZeneca. Brilinta (ticagrelor) full prescribing information. 2013. Available at: http://www1.astrazeneca‐us.com/pi/brilinta.pdf. Accessed October 1, 2013.
- , , , et al. 2012 update to the Society of Thoracic Surgeons guideline on use of antiplatelet drugs in patients having cardiac and noncardiac operations. Ann Thorac Surg. 2012;94(5):1761–1781.
- , . How I treat anticoagulated patients undergoing an elective procedure or surgery. Blood. 2012;120(15):2954–2962.
- , , , , American College of Chest Physicians. Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 suppl):e24S–e43S.
Once-weekly exenatide can be especially useful to treat diabetes in schizophrenia
The incidence of diabetes in people with schizophrenia is 2- to 3-fold that of the general population—which has been attributed to several variables, including adverse effects of antipsychotic drugs, susceptibility related to mental illness, lifestyle, and social health determinants.1 Controlling diabetes is important because cardiovascular consequences of the disease contribute to the shortened life expectancy seen in patients with schizophrenia.2
The dosing frequency of a newer formulation of exenatide, a glucose-lowering drug that has been available for almost a decade, can help manage type 2 diabetes mellitus in your patient with schizophrenia.
What is the new formulation and why is it appealing?
Exenatide is a glucagon-like peptide-1 (GLP-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2
diabetes mellitus. GLP-1 agonists lower the blood glucose level by enhancing glucose-dependent insulin secretion, suppressing glucagon secretion, slowing gastric emptying, and enhancing satiety. Exenatide was approved by the FDA
in 2005 as a twice-daily subcutaneous injection (brand name, Byetta); the once-weekly formulation, also for subcutaneous injection (brand name, Bydureon), was approved in 2012.
Practical use for psychiatric patients
Because psychiatric patients face medication adherence challenges, the once-
weekly formulation of exenatide is appealing. The patient or a member of his (her) care team can administer the once-weekly injection.
Practitioners might be concerned that patients with comorbid psychiatric illness and diabetes will overreact to an elevated blood glucose reading by overusing medications such as oral hypoglycemics and insulin. The fixed-dosage of weekly exenatide minimizes the risk that a patient will react to a single elevated blood glucose reading.
Exenatide can produce weight loss, which might benefit patients who suffer from the metabolic adverse effects of an atypical antipsychotic, including an elevated blood glucose level and weight gain.
Real-world application
We have used once-weekly exenatide successfully in a female patient with schizophrenia who was taking quetiapine and haloperidol, and had uncontrolled diabetes resulting from medication nonadherence and lack of insight into diabetes.
The patient’s hemoglobin A1c level remained at 8.8% (target A1c, <7%, as set by the American Diabetes Association), despite taking 3 oral diabetes medications at maximum dosage.
The care team determined that daily insulin injections were too risky, given the patient’s compulsive behavior; she had a history of medication overuse in response to significantly elevated blood glucose.
Once-weekly exenatide, however, was a feasible alternative. Three months after she was started on once-weekly exenatide, and with additional lifestyle modifications, her hemoglobin A1c level had fallen to 6.4%, without any hypoglycemic episodes.
Select patients carefully
Exenatide is not a first-line therapy because of its potential side effects (Table), route of administration, and cost. Consider the once-weekly formulation of the drug on a patient-by-patient basis for patients with schizophrenia whose diabetes otherwise cannot be controlled.

Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. De Hert M, van Winkel R, Van Eyck D, et al. Prevalence of diabetes, metabolic syndrome and metabolic abnormalities in schizophrenia over the course of the illness: a cross-sectional study. Clin Pract Epidemiol Ment Health. 2006;2:14.
2. Laursen TM, Munk-Olsen T, Vestergaard M. Life expectancy and cardiovascular mortality in persons with schizophrenia. Curr Opin Psychiatry. 2012;25(2):83-88.
3. Bydureon [package insert]. San Diego, CA: Amylin Pharmaceuticals, Inc.; 2012.
The incidence of diabetes in people with schizophrenia is 2- to 3-fold that of the general population—which has been attributed to several variables, including adverse effects of antipsychotic drugs, susceptibility related to mental illness, lifestyle, and social health determinants.1 Controlling diabetes is important because cardiovascular consequences of the disease contribute to the shortened life expectancy seen in patients with schizophrenia.2
The dosing frequency of a newer formulation of exenatide, a glucose-lowering drug that has been available for almost a decade, can help manage type 2 diabetes mellitus in your patient with schizophrenia.
What is the new formulation and why is it appealing?
Exenatide is a glucagon-like peptide-1 (GLP-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2
diabetes mellitus. GLP-1 agonists lower the blood glucose level by enhancing glucose-dependent insulin secretion, suppressing glucagon secretion, slowing gastric emptying, and enhancing satiety. Exenatide was approved by the FDA
in 2005 as a twice-daily subcutaneous injection (brand name, Byetta); the once-weekly formulation, also for subcutaneous injection (brand name, Bydureon), was approved in 2012.
Practical use for psychiatric patients
Because psychiatric patients face medication adherence challenges, the once-
weekly formulation of exenatide is appealing. The patient or a member of his (her) care team can administer the once-weekly injection.
Practitioners might be concerned that patients with comorbid psychiatric illness and diabetes will overreact to an elevated blood glucose reading by overusing medications such as oral hypoglycemics and insulin. The fixed-dosage of weekly exenatide minimizes the risk that a patient will react to a single elevated blood glucose reading.
Exenatide can produce weight loss, which might benefit patients who suffer from the metabolic adverse effects of an atypical antipsychotic, including an elevated blood glucose level and weight gain.
Real-world application
We have used once-weekly exenatide successfully in a female patient with schizophrenia who was taking quetiapine and haloperidol, and had uncontrolled diabetes resulting from medication nonadherence and lack of insight into diabetes.
The patient’s hemoglobin A1c level remained at 8.8% (target A1c, <7%, as set by the American Diabetes Association), despite taking 3 oral diabetes medications at maximum dosage.
The care team determined that daily insulin injections were too risky, given the patient’s compulsive behavior; she had a history of medication overuse in response to significantly elevated blood glucose.
Once-weekly exenatide, however, was a feasible alternative. Three months after she was started on once-weekly exenatide, and with additional lifestyle modifications, her hemoglobin A1c level had fallen to 6.4%, without any hypoglycemic episodes.
Select patients carefully
Exenatide is not a first-line therapy because of its potential side effects (Table), route of administration, and cost. Consider the once-weekly formulation of the drug on a patient-by-patient basis for patients with schizophrenia whose diabetes otherwise cannot be controlled.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
The incidence of diabetes in people with schizophrenia is 2- to 3-fold that of the general population—which has been attributed to several variables, including adverse effects of antipsychotic drugs, susceptibility related to mental illness, lifestyle, and social health determinants.1 Controlling diabetes is important because cardiovascular consequences of the disease contribute to the shortened life expectancy seen in patients with schizophrenia.2
The dosing frequency of a newer formulation of exenatide, a glucose-lowering drug that has been available for almost a decade, can help manage type 2 diabetes mellitus in your patient with schizophrenia.
What is the new formulation and why is it appealing?
Exenatide is a glucagon-like peptide-1 (GLP-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2
diabetes mellitus. GLP-1 agonists lower the blood glucose level by enhancing glucose-dependent insulin secretion, suppressing glucagon secretion, slowing gastric emptying, and enhancing satiety. Exenatide was approved by the FDA
in 2005 as a twice-daily subcutaneous injection (brand name, Byetta); the once-weekly formulation, also for subcutaneous injection (brand name, Bydureon), was approved in 2012.
Practical use for psychiatric patients
Because psychiatric patients face medication adherence challenges, the once-
weekly formulation of exenatide is appealing. The patient or a member of his (her) care team can administer the once-weekly injection.
Practitioners might be concerned that patients with comorbid psychiatric illness and diabetes will overreact to an elevated blood glucose reading by overusing medications such as oral hypoglycemics and insulin. The fixed-dosage of weekly exenatide minimizes the risk that a patient will react to a single elevated blood glucose reading.
Exenatide can produce weight loss, which might benefit patients who suffer from the metabolic adverse effects of an atypical antipsychotic, including an elevated blood glucose level and weight gain.
Real-world application
We have used once-weekly exenatide successfully in a female patient with schizophrenia who was taking quetiapine and haloperidol, and had uncontrolled diabetes resulting from medication nonadherence and lack of insight into diabetes.
The patient’s hemoglobin A1c level remained at 8.8% (target A1c, <7%, as set by the American Diabetes Association), despite taking 3 oral diabetes medications at maximum dosage.
The care team determined that daily insulin injections were too risky, given the patient’s compulsive behavior; she had a history of medication overuse in response to significantly elevated blood glucose.
Once-weekly exenatide, however, was a feasible alternative. Three months after she was started on once-weekly exenatide, and with additional lifestyle modifications, her hemoglobin A1c level had fallen to 6.4%, without any hypoglycemic episodes.
Select patients carefully
Exenatide is not a first-line therapy because of its potential side effects (Table), route of administration, and cost. Consider the once-weekly formulation of the drug on a patient-by-patient basis for patients with schizophrenia whose diabetes otherwise cannot be controlled.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. De Hert M, van Winkel R, Van Eyck D, et al. Prevalence of diabetes, metabolic syndrome and metabolic abnormalities in schizophrenia over the course of the illness: a cross-sectional study. Clin Pract Epidemiol Ment Health. 2006;2:14.
2. Laursen TM, Munk-Olsen T, Vestergaard M. Life expectancy and cardiovascular mortality in persons with schizophrenia. Curr Opin Psychiatry. 2012;25(2):83-88.
3. Bydureon [package insert]. San Diego, CA: Amylin Pharmaceuticals, Inc.; 2012.
1. De Hert M, van Winkel R, Van Eyck D, et al. Prevalence of diabetes, metabolic syndrome and metabolic abnormalities in schizophrenia over the course of the illness: a cross-sectional study. Clin Pract Epidemiol Ment Health. 2006;2:14.
2. Laursen TM, Munk-Olsen T, Vestergaard M. Life expectancy and cardiovascular mortality in persons with schizophrenia. Curr Opin Psychiatry. 2012;25(2):83-88.
3. Bydureon [package insert]. San Diego, CA: Amylin Pharmaceuticals, Inc.; 2012.
Key steps to take when a patient commits suicide
The suicide of a patient is a relatively frequent occurrence in psychiatry. As many as 68% of consultant psychiatrists acknowledge the loss of a patient to suicide.1 Conservative estimates are that as many as 54% of psychiatry resident trainees experience patient suicide.2
Up to 57% of psychiatrists who have experienced a patient’s suicide have developed symptoms of posttraumatic stress disorder.3 There are steps you can take personally, with your staff, and with the patient’s family to mitigate social,
ethical, and legal consequences of a patient committing suicide, and to improve risk management.
Steps to take for yourself
1. In an inpatient psychiatric facility, be aware of standard operating procedures after a suicide; inform only an immediate supervisor if you learn of a suicide. In a group practice, inform the owner of the practice and receive advice on how to proceed. Do not contact the coroner’s office, the police, the deceased’s family, or legal counsel until advised to do so by a direct supervisor.
2. Be prepared to work with the coroner’s or medical examiner’s office. Write a detailed note summarizing the patient’s clinical history before the suicide; describe the clinical team’s work with the patient, the treatment plan, and an estimate of suicide risk.
3. Contact a trusted colleague or mentor; seeking formal and informal support from colleagues has shown to be helpful in coping with patient suicide.4 Group
support helps diminish feelings of pain and loneliness and helps one regain a sense of empowerment and willingness to treat other suicidal patients.
4. If possible, attend the patient’s funeral. This gesture often is welcomed by the family and facilitates the grieving process. Attending the funeral is not an admission of responsibility for the suicide.
5. Participate in the audit process (ie, what went wrong?, Could something have been done differently?).
Steps to take with the patient’s family
1. Once standard operating procedure allows, and, preferably within 24 hours of the suicide, contact the patient’s family to express your grief; give the family an opportunity to ask questions. Early communication and support reduces anger displaced on the psychiatrist. Initial contact can be used to provide support and as an opportunity to share and communicate.
2. When speaking with the family, discuss treatment efforts and emphasize that all realistic efforts were made to help the patient. Let family members vent their anger and hostility; the grieving process is hard, complex, and painful when a loved one has committed suicide.
3. Support the family’s decisions about mourning rituals specific to their culture and needs; involving the clergy early on can be helpful. Discussing the autopsy report with the family can be another way to show support.
4. Continue to offer support through stressful times, such as anniversaries and birthdays.
Steps to take with staff
1. Make staff aware of the death as a group; encourage them to attend funeral services.
2. Avoid placing blame; encourage group support and venting of emotions.
3. Be available to the staff so that they can share feelings of hurt and disappointment with you.
4. Maintain the schedule on unit, restoring a sense of stability and normalcy.
5. A so-called psychological autopsy exercise is recommended, in which you can emphasize the learning experience and focus on improvements4 that can help formulate policy reforms for providing better care.
Steps to improve risk management
1. If you work in a hospital, immediately contact the risk management team.
2. Seek legal counsel as soon as possible and involve counsel at all stages.
3. Notify your malpractice insurance carrier.
4. Complete the patient’s medical record and describe the facts as they occurred. Date the records accurately with clarification on notes entered after the suicide. Avoid drawing conclusions. Do not apologize for, or justify, your treatment decisions.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Alexander DA, Klein S, Gray NM, et al. Suicide by patients: questionnaire study of its effect on consultant psychiatrists. BMJ. 2000;320(7249):1571-1574.
2. Courtenay KP, Stephens JP. The experience of patient suicide among trainees in psychiatry. The Psychiatrist. 2001;25:51-52.
3. Chemtob CM, Hamada RS, Bauer G, et al. Patients’ suicides: frequency and impact on psychiatrists. Am J Psychiatry. 1988;145(2):224-228.
4. Kaye NS, Soreff SM. The psychiatrist’s role, responses, and responsibilities when a patient commits suicide. Am J Psychiatry. 1991;148(6):739-743.
The suicide of a patient is a relatively frequent occurrence in psychiatry. As many as 68% of consultant psychiatrists acknowledge the loss of a patient to suicide.1 Conservative estimates are that as many as 54% of psychiatry resident trainees experience patient suicide.2
Up to 57% of psychiatrists who have experienced a patient’s suicide have developed symptoms of posttraumatic stress disorder.3 There are steps you can take personally, with your staff, and with the patient’s family to mitigate social,
ethical, and legal consequences of a patient committing suicide, and to improve risk management.
Steps to take for yourself
1. In an inpatient psychiatric facility, be aware of standard operating procedures after a suicide; inform only an immediate supervisor if you learn of a suicide. In a group practice, inform the owner of the practice and receive advice on how to proceed. Do not contact the coroner’s office, the police, the deceased’s family, or legal counsel until advised to do so by a direct supervisor.
2. Be prepared to work with the coroner’s or medical examiner’s office. Write a detailed note summarizing the patient’s clinical history before the suicide; describe the clinical team’s work with the patient, the treatment plan, and an estimate of suicide risk.
3. Contact a trusted colleague or mentor; seeking formal and informal support from colleagues has shown to be helpful in coping with patient suicide.4 Group
support helps diminish feelings of pain and loneliness and helps one regain a sense of empowerment and willingness to treat other suicidal patients.
4. If possible, attend the patient’s funeral. This gesture often is welcomed by the family and facilitates the grieving process. Attending the funeral is not an admission of responsibility for the suicide.
5. Participate in the audit process (ie, what went wrong?, Could something have been done differently?).
Steps to take with the patient’s family
1. Once standard operating procedure allows, and, preferably within 24 hours of the suicide, contact the patient’s family to express your grief; give the family an opportunity to ask questions. Early communication and support reduces anger displaced on the psychiatrist. Initial contact can be used to provide support and as an opportunity to share and communicate.
2. When speaking with the family, discuss treatment efforts and emphasize that all realistic efforts were made to help the patient. Let family members vent their anger and hostility; the grieving process is hard, complex, and painful when a loved one has committed suicide.
3. Support the family’s decisions about mourning rituals specific to their culture and needs; involving the clergy early on can be helpful. Discussing the autopsy report with the family can be another way to show support.
4. Continue to offer support through stressful times, such as anniversaries and birthdays.
Steps to take with staff
1. Make staff aware of the death as a group; encourage them to attend funeral services.
2. Avoid placing blame; encourage group support and venting of emotions.
3. Be available to the staff so that they can share feelings of hurt and disappointment with you.
4. Maintain the schedule on unit, restoring a sense of stability and normalcy.
5. A so-called psychological autopsy exercise is recommended, in which you can emphasize the learning experience and focus on improvements4 that can help formulate policy reforms for providing better care.
Steps to improve risk management
1. If you work in a hospital, immediately contact the risk management team.
2. Seek legal counsel as soon as possible and involve counsel at all stages.
3. Notify your malpractice insurance carrier.
4. Complete the patient’s medical record and describe the facts as they occurred. Date the records accurately with clarification on notes entered after the suicide. Avoid drawing conclusions. Do not apologize for, or justify, your treatment decisions.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
The suicide of a patient is a relatively frequent occurrence in psychiatry. As many as 68% of consultant psychiatrists acknowledge the loss of a patient to suicide.1 Conservative estimates are that as many as 54% of psychiatry resident trainees experience patient suicide.2
Up to 57% of psychiatrists who have experienced a patient’s suicide have developed symptoms of posttraumatic stress disorder.3 There are steps you can take personally, with your staff, and with the patient’s family to mitigate social,
ethical, and legal consequences of a patient committing suicide, and to improve risk management.
Steps to take for yourself
1. In an inpatient psychiatric facility, be aware of standard operating procedures after a suicide; inform only an immediate supervisor if you learn of a suicide. In a group practice, inform the owner of the practice and receive advice on how to proceed. Do not contact the coroner’s office, the police, the deceased’s family, or legal counsel until advised to do so by a direct supervisor.
2. Be prepared to work with the coroner’s or medical examiner’s office. Write a detailed note summarizing the patient’s clinical history before the suicide; describe the clinical team’s work with the patient, the treatment plan, and an estimate of suicide risk.
3. Contact a trusted colleague or mentor; seeking formal and informal support from colleagues has shown to be helpful in coping with patient suicide.4 Group
support helps diminish feelings of pain and loneliness and helps one regain a sense of empowerment and willingness to treat other suicidal patients.
4. If possible, attend the patient’s funeral. This gesture often is welcomed by the family and facilitates the grieving process. Attending the funeral is not an admission of responsibility for the suicide.
5. Participate in the audit process (ie, what went wrong?, Could something have been done differently?).
Steps to take with the patient’s family
1. Once standard operating procedure allows, and, preferably within 24 hours of the suicide, contact the patient’s family to express your grief; give the family an opportunity to ask questions. Early communication and support reduces anger displaced on the psychiatrist. Initial contact can be used to provide support and as an opportunity to share and communicate.
2. When speaking with the family, discuss treatment efforts and emphasize that all realistic efforts were made to help the patient. Let family members vent their anger and hostility; the grieving process is hard, complex, and painful when a loved one has committed suicide.
3. Support the family’s decisions about mourning rituals specific to their culture and needs; involving the clergy early on can be helpful. Discussing the autopsy report with the family can be another way to show support.
4. Continue to offer support through stressful times, such as anniversaries and birthdays.
Steps to take with staff
1. Make staff aware of the death as a group; encourage them to attend funeral services.
2. Avoid placing blame; encourage group support and venting of emotions.
3. Be available to the staff so that they can share feelings of hurt and disappointment with you.
4. Maintain the schedule on unit, restoring a sense of stability and normalcy.
5. A so-called psychological autopsy exercise is recommended, in which you can emphasize the learning experience and focus on improvements4 that can help formulate policy reforms for providing better care.
Steps to improve risk management
1. If you work in a hospital, immediately contact the risk management team.
2. Seek legal counsel as soon as possible and involve counsel at all stages.
3. Notify your malpractice insurance carrier.
4. Complete the patient’s medical record and describe the facts as they occurred. Date the records accurately with clarification on notes entered after the suicide. Avoid drawing conclusions. Do not apologize for, or justify, your treatment decisions.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Alexander DA, Klein S, Gray NM, et al. Suicide by patients: questionnaire study of its effect on consultant psychiatrists. BMJ. 2000;320(7249):1571-1574.
2. Courtenay KP, Stephens JP. The experience of patient suicide among trainees in psychiatry. The Psychiatrist. 2001;25:51-52.
3. Chemtob CM, Hamada RS, Bauer G, et al. Patients’ suicides: frequency and impact on psychiatrists. Am J Psychiatry. 1988;145(2):224-228.
4. Kaye NS, Soreff SM. The psychiatrist’s role, responses, and responsibilities when a patient commits suicide. Am J Psychiatry. 1991;148(6):739-743.
1. Alexander DA, Klein S, Gray NM, et al. Suicide by patients: questionnaire study of its effect on consultant psychiatrists. BMJ. 2000;320(7249):1571-1574.
2. Courtenay KP, Stephens JP. The experience of patient suicide among trainees in psychiatry. The Psychiatrist. 2001;25:51-52.
3. Chemtob CM, Hamada RS, Bauer G, et al. Patients’ suicides: frequency and impact on psychiatrists. Am J Psychiatry. 1988;145(2):224-228.
4. Kaye NS, Soreff SM. The psychiatrist’s role, responses, and responsibilities when a patient commits suicide. Am J Psychiatry. 1991;148(6):739-743.
Vortioxetine for major depressive disorder
Vortioxetine is FDA-approved to treat major depressive disorder (MDD) (Table 1), having shown efficacy in relieving depressive symptoms in clinical trials.1 Vortioxetine’s mechanism of action enhances CNS serotonergic activity through inhibiting serotonin (5-HT) reuptake, agonizing the 5-HT1A receptor, partially agonizing the 5-HT1B receptor, and antagonizing the 5-HT3, 5-HT1D, and 5-HT7 receptors.
Clinical implications
It is hypothesized that depression is a heterogeneous disease caused by dysregulation of serotonin, norepinephrine, and dopamine, subsequently producing mood and neurovegetative symptoms of depression. Preclinical, in vivo studies indicate that vortioxetine enhances levels of serotonin, norepinephrine, dopamine, acetylcholine, and histamine in specific areas of the brain with the ability to improve depressive symptoms. Vortioxetine’s multimodal activity can be a useful alternative to other serotonergic antidepressants for some patients who are partial responders or non-responders to other treatment options. In addition, vortioxetine appears to have minimal effect on weight2 and sexual function—the latter being dose-dependent.3
How does it work?
Vortioxetine differs from other antidepressants in its multimodal activity (ie, affecting G-protein mode receptors, ion channel mode receptors, and neurotransmitter transporters). It inhibits the serotonin transporter (Ki = 1.6 nM), causing subsequent inhibition of serotonin reuptake into presynaptic neurons as well as selectively acting on the other subtypes of serotonergic receptors; however, activity on the norepinephrine transporter (Ki = 113 nM) and dopamine transporter (Ki > 1000 nM) is minimal. It is believed that mood-regulating effects of vortioxetine are caused by inhibition of serotonin reuptake, prolonged availability of serotonin to the postsynaptic neurons, its agonist activity on the 5-HT1A receptor (Ki = 15 nM), and partial agonist activity on the 5-HT1B receptor (Ki = 33 nM). Vortioxetine has strong affinity for the 5-HT3 receptor (Ki = 3.7 nM), which plays a role in modulation of centrally mediated nausea and vomiting. Positron emission tomography studies in humans determined that the occupancy of 5-HT transporter was 50% at 5 mg/d, 65% at 10 mg/d, and 80% at 20 mg/d.1,4 Human studies did not show that vortioxetine causes QTc prolongation.
Pharmacokinetics
Therapeutic activity of vortioxetine is thought to be due to the parent drug. It has a half-life of approximately 66 hours, and achieves steady state in 13.5 to 19 days. Bioavailability of vortioxetine is 75%; absorption does not depend on food; and 98% of drug is bound on plasma proteins.
Vortioxetine has linear pharmacokinetics, with maximum plasma concentration 7 to 11 hours after ingestion. The medication is metabolized primarily by oxidation through cytochrome P (CYP) 450: CYP2D6 (primary), CYP 3A4/5, CYP 2C19, CYP 2C9, CYP2A6, CYP2C8, and CYP2B6 with subsequent glucuronidation. This predisposes vortioxetine to potential pharmacokinetic drug-drug interaction warranting dose adjustment consideration when vortioxetine is coadministered with compounds inhibiting CYP2D6 or inducing CYP3A4 for ≥14 days, or for patients identified as poor 2D6 metabolizers.
In addition, coadministration of vortioxetine with serotonergic medications such as triptans, other antidepressants, and tramadol can cause potentially life-threatening serotonin syndrome, characterized by mental status changes, autonomic instability, neuromuscular aberrations, and GI symptoms. Concomitant use of vortioxetine and a nonsteroidal anti-inflammatory drug, aspirin, or warfarin can result in abnormal bleeding. Coadministration of vortioxetine with another highly protein-bound drug may increase or decrease the free concentration of either drug depending on the binding affinity of the drug for the protein.
Efficacy
Vortioxetine reduced depressive symptoms in 6 positive, 6- to 8-week, double-blind, placebo controlled and randomized studies and 1 maintenance study.1 Subjects were adults (Studies 1 to 5) and geriatric patients from age 64 to 88 who had ≥1 depressive episode before age 60 (Study 6). All met DSM-IV-TR criteria for MDD. Subjects with cognitive impairment scoring <24 on the Mini-Mental Status Examination and children were excluded. Depending on the study, response to the treatment was primarily measured on the Montgomery-Åsberg Depression Rating Scale (MADRS) or Hamilton Depression Rating Scale (HAM-D).
See Table 2 for a description of the positive studies, including dosages. In all studies, vortioxetine was superior to placebo at least one dosage for treating depression. In the 6- to 8-week placebo-controlled studies, an effect of vortioxetine based on the primary efficacy measure was generally observed starting at Week 2; that effect increased in subsequent weeks with the full antidepressant effect of vortioxetine generally not seen until study Week 4 or later.1
The maintenance treatment study included 639 patients who met DSM-IV-TR criteria for MDD. This study lasted for as long as 64 weeks. The first 12-week period was open-label, during which patients were treated with vortioxetine, 5 mg/d or 10 mg/d, with a possibility to adjust the dosage in the first 8 weeks. By the end of Week 12, 396 subjects achieved remission (MADRS <10), 75% of whom were taking vortioxetine, 10 mg/d. These patients were then randomly assigned to placebo or the dosage of vortioxetine to which they had responded, and continued the study for as long as 64 weeks. Time to relapse (MADRS total score ≥22) or an insufficient therapeutic response (as judged by the investigator) was the primary efficacy outcome, and demonstrated that vortioxetine was superior to placebo.
Tolerability
The tolerability of vortioxetine is comparable with other serotonergic antidepressants. In pooled analysis of pre-marketing studies, 5% to 8% of patients receiving vortioxetine (5 to 20 mg/d) discontinued treatment because of adverse effects (AEs), compared with 4% in the placebo group. Nausea was the most commonly reported AE leading to discontinuation and appeared to be dose dependent.
AEs, such as nausea, constipation, and vomiting, most commonly occurred in the first week of treatment, with a median duration of 2 weeks.5 In the 6- to 8-week trials, the most common AEs were nausea, constipation, and vomiting. In longer trials (24 to 64 weeks), the most common AE was nausea.
In 6- to 8-week placebo-controlled studies, vortioxetine was not associated with any clinically significant effect on vital signs or laboratory values in hematology, urinalysis, or serum chemistry (except sodium). Hyponatremia, the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH), has occurred. The risk of developing SIADH and resultant hyponatremia is greater in geriatric patients and patients taking a diuretic.
Abruptly discontinuing vortioxetine can cause transient withdrawal symptoms, including headache and muscle tension, especially at a higher dosage (15 to 20 mg/d). Gradual tapering can reduce withdrawal symptoms.
Specific clinical issues
All antidepressants have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation and behaviors during the first months of treatment and with dosage changes.
Vortioxetine is categorized as pregnancy category C. Newborns exposed to a selective serotonin reuptake inhibitor (SSRI) in pregnancy may have an increased risk of persistent pulmonary hypertension during the neonatal period. When taken during the third trimester of pregnancy, SSRIs and serotonin-norepinephrine reuptake inhibitors can cause serious neonatal complications, including respiratory distress, cyanosis, apnea, and seizures, which may require longer hospitalization, respiratory support, or tube feeding for the infant. Consider risks and benefits of third-trimester use of an antidepressant. It is not known if vortioxetine is present in human breast milk.
Clinical studies on vortioxetine in pediatric patients have not been conducted.
No dosage adjustment is recommended on the basis of age for geriatric patients. No dose adjustment of vortioxetine is necessary on the basis of race, sex, ethnicity, renal function, or mild to moderate hepatic impairment. See Table 3 for practice points when prescribing vortioxetine. See Table 4 for contraindications to vortioxetine.


Dosing
The recommended starting dosage is 10 mg, administered orally once daily without regard to meals. Dosage should then be increased to 20 mg/d, as clinically warranted and tolerated. Consider a dosage decrease to 5 mg/d in patients who do not tolerate higher dosages or require drug adjustment because of drug-drug interaction or poor 2D6 metabolizer status.
Bottom Line
FDA-approved for major depressive disorder in adults, vortioxetine reduced depressive symptoms in 6 positive, double-blind, placebo-controlled, and randomized studies. The multimodal activity of vortioxeine can be a useful alternative to serotonergic antidepressants for some patients who are partial responders or nonresponders. Tolerability is comparable with other serotonergic antidepressants.
Related Resources
- Alam MY, Jacobsen PL, Chen Y, et al. Safety, tolerability, and efficacy of vortioxetine (Lu AA21004) in major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2014; 29(1):36-44.
- Mahableshwarkar AR, Jacobsen PL, Chen Y. A randomized, double-blind trial of 2.5 mg and 5 mg vortioxetine (Lu AA21004) versus placebo for 8 weeks in adults with major depressive disorder. Curr Med Res Opin. 2013;29(3):217-226.
Drug Brand Names
Linezolid • Zyvox Vortioxetine • Brintellix
Methylene blue • Urolene Blue Warfarin • Coumadin
Tramadol • Ultram
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Vortioxetine [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc.; 2013.
2. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71(10):1259-1272.
3. Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a comprehensive review and meta-analysis. J Clin Psychopharmacol. 2009; 29(3):259-266.
4. Chen G, Lee R, Højer A, et al. Pharmacokinetic drug interactions involving vortioxetine (LU AA 21004), a multimodal antidepressant. Clin Drug Invetig. 2013; 33(10):727-736.
5. Citrome L. Vortioxetine for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Prac. 2014;68(1):60-82.
Vortioxetine is FDA-approved to treat major depressive disorder (MDD) (Table 1), having shown efficacy in relieving depressive symptoms in clinical trials.1 Vortioxetine’s mechanism of action enhances CNS serotonergic activity through inhibiting serotonin (5-HT) reuptake, agonizing the 5-HT1A receptor, partially agonizing the 5-HT1B receptor, and antagonizing the 5-HT3, 5-HT1D, and 5-HT7 receptors.
Clinical implications
It is hypothesized that depression is a heterogeneous disease caused by dysregulation of serotonin, norepinephrine, and dopamine, subsequently producing mood and neurovegetative symptoms of depression. Preclinical, in vivo studies indicate that vortioxetine enhances levels of serotonin, norepinephrine, dopamine, acetylcholine, and histamine in specific areas of the brain with the ability to improve depressive symptoms. Vortioxetine’s multimodal activity can be a useful alternative to other serotonergic antidepressants for some patients who are partial responders or non-responders to other treatment options. In addition, vortioxetine appears to have minimal effect on weight2 and sexual function—the latter being dose-dependent.3
How does it work?
Vortioxetine differs from other antidepressants in its multimodal activity (ie, affecting G-protein mode receptors, ion channel mode receptors, and neurotransmitter transporters). It inhibits the serotonin transporter (Ki = 1.6 nM), causing subsequent inhibition of serotonin reuptake into presynaptic neurons as well as selectively acting on the other subtypes of serotonergic receptors; however, activity on the norepinephrine transporter (Ki = 113 nM) and dopamine transporter (Ki > 1000 nM) is minimal. It is believed that mood-regulating effects of vortioxetine are caused by inhibition of serotonin reuptake, prolonged availability of serotonin to the postsynaptic neurons, its agonist activity on the 5-HT1A receptor (Ki = 15 nM), and partial agonist activity on the 5-HT1B receptor (Ki = 33 nM). Vortioxetine has strong affinity for the 5-HT3 receptor (Ki = 3.7 nM), which plays a role in modulation of centrally mediated nausea and vomiting. Positron emission tomography studies in humans determined that the occupancy of 5-HT transporter was 50% at 5 mg/d, 65% at 10 mg/d, and 80% at 20 mg/d.1,4 Human studies did not show that vortioxetine causes QTc prolongation.
Pharmacokinetics
Therapeutic activity of vortioxetine is thought to be due to the parent drug. It has a half-life of approximately 66 hours, and achieves steady state in 13.5 to 19 days. Bioavailability of vortioxetine is 75%; absorption does not depend on food; and 98% of drug is bound on plasma proteins.
Vortioxetine has linear pharmacokinetics, with maximum plasma concentration 7 to 11 hours after ingestion. The medication is metabolized primarily by oxidation through cytochrome P (CYP) 450: CYP2D6 (primary), CYP 3A4/5, CYP 2C19, CYP 2C9, CYP2A6, CYP2C8, and CYP2B6 with subsequent glucuronidation. This predisposes vortioxetine to potential pharmacokinetic drug-drug interaction warranting dose adjustment consideration when vortioxetine is coadministered with compounds inhibiting CYP2D6 or inducing CYP3A4 for ≥14 days, or for patients identified as poor 2D6 metabolizers.
In addition, coadministration of vortioxetine with serotonergic medications such as triptans, other antidepressants, and tramadol can cause potentially life-threatening serotonin syndrome, characterized by mental status changes, autonomic instability, neuromuscular aberrations, and GI symptoms. Concomitant use of vortioxetine and a nonsteroidal anti-inflammatory drug, aspirin, or warfarin can result in abnormal bleeding. Coadministration of vortioxetine with another highly protein-bound drug may increase or decrease the free concentration of either drug depending on the binding affinity of the drug for the protein.
Efficacy
Vortioxetine reduced depressive symptoms in 6 positive, 6- to 8-week, double-blind, placebo controlled and randomized studies and 1 maintenance study.1 Subjects were adults (Studies 1 to 5) and geriatric patients from age 64 to 88 who had ≥1 depressive episode before age 60 (Study 6). All met DSM-IV-TR criteria for MDD. Subjects with cognitive impairment scoring <24 on the Mini-Mental Status Examination and children were excluded. Depending on the study, response to the treatment was primarily measured on the Montgomery-Åsberg Depression Rating Scale (MADRS) or Hamilton Depression Rating Scale (HAM-D).
See Table 2 for a description of the positive studies, including dosages. In all studies, vortioxetine was superior to placebo at least one dosage for treating depression. In the 6- to 8-week placebo-controlled studies, an effect of vortioxetine based on the primary efficacy measure was generally observed starting at Week 2; that effect increased in subsequent weeks with the full antidepressant effect of vortioxetine generally not seen until study Week 4 or later.1
The maintenance treatment study included 639 patients who met DSM-IV-TR criteria for MDD. This study lasted for as long as 64 weeks. The first 12-week period was open-label, during which patients were treated with vortioxetine, 5 mg/d or 10 mg/d, with a possibility to adjust the dosage in the first 8 weeks. By the end of Week 12, 396 subjects achieved remission (MADRS <10), 75% of whom were taking vortioxetine, 10 mg/d. These patients were then randomly assigned to placebo or the dosage of vortioxetine to which they had responded, and continued the study for as long as 64 weeks. Time to relapse (MADRS total score ≥22) or an insufficient therapeutic response (as judged by the investigator) was the primary efficacy outcome, and demonstrated that vortioxetine was superior to placebo.
Tolerability
The tolerability of vortioxetine is comparable with other serotonergic antidepressants. In pooled analysis of pre-marketing studies, 5% to 8% of patients receiving vortioxetine (5 to 20 mg/d) discontinued treatment because of adverse effects (AEs), compared with 4% in the placebo group. Nausea was the most commonly reported AE leading to discontinuation and appeared to be dose dependent.
AEs, such as nausea, constipation, and vomiting, most commonly occurred in the first week of treatment, with a median duration of 2 weeks.5 In the 6- to 8-week trials, the most common AEs were nausea, constipation, and vomiting. In longer trials (24 to 64 weeks), the most common AE was nausea.
In 6- to 8-week placebo-controlled studies, vortioxetine was not associated with any clinically significant effect on vital signs or laboratory values in hematology, urinalysis, or serum chemistry (except sodium). Hyponatremia, the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH), has occurred. The risk of developing SIADH and resultant hyponatremia is greater in geriatric patients and patients taking a diuretic.
Abruptly discontinuing vortioxetine can cause transient withdrawal symptoms, including headache and muscle tension, especially at a higher dosage (15 to 20 mg/d). Gradual tapering can reduce withdrawal symptoms.
Specific clinical issues
All antidepressants have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation and behaviors during the first months of treatment and with dosage changes.
Vortioxetine is categorized as pregnancy category C. Newborns exposed to a selective serotonin reuptake inhibitor (SSRI) in pregnancy may have an increased risk of persistent pulmonary hypertension during the neonatal period. When taken during the third trimester of pregnancy, SSRIs and serotonin-norepinephrine reuptake inhibitors can cause serious neonatal complications, including respiratory distress, cyanosis, apnea, and seizures, which may require longer hospitalization, respiratory support, or tube feeding for the infant. Consider risks and benefits of third-trimester use of an antidepressant. It is not known if vortioxetine is present in human breast milk.
Clinical studies on vortioxetine in pediatric patients have not been conducted.
No dosage adjustment is recommended on the basis of age for geriatric patients. No dose adjustment of vortioxetine is necessary on the basis of race, sex, ethnicity, renal function, or mild to moderate hepatic impairment. See Table 3 for practice points when prescribing vortioxetine. See Table 4 for contraindications to vortioxetine.
Dosing
The recommended starting dosage is 10 mg, administered orally once daily without regard to meals. Dosage should then be increased to 20 mg/d, as clinically warranted and tolerated. Consider a dosage decrease to 5 mg/d in patients who do not tolerate higher dosages or require drug adjustment because of drug-drug interaction or poor 2D6 metabolizer status.
Bottom Line
FDA-approved for major depressive disorder in adults, vortioxetine reduced depressive symptoms in 6 positive, double-blind, placebo-controlled, and randomized studies. The multimodal activity of vortioxeine can be a useful alternative to serotonergic antidepressants for some patients who are partial responders or nonresponders. Tolerability is comparable with other serotonergic antidepressants.
Related Resources
- Alam MY, Jacobsen PL, Chen Y, et al. Safety, tolerability, and efficacy of vortioxetine (Lu AA21004) in major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2014; 29(1):36-44.
- Mahableshwarkar AR, Jacobsen PL, Chen Y. A randomized, double-blind trial of 2.5 mg and 5 mg vortioxetine (Lu AA21004) versus placebo for 8 weeks in adults with major depressive disorder. Curr Med Res Opin. 2013;29(3):217-226.
Drug Brand Names
Linezolid • Zyvox Vortioxetine • Brintellix
Methylene blue • Urolene Blue Warfarin • Coumadin
Tramadol • Ultram
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Vortioxetine is FDA-approved to treat major depressive disorder (MDD) (Table 1), having shown efficacy in relieving depressive symptoms in clinical trials.1 Vortioxetine’s mechanism of action enhances CNS serotonergic activity through inhibiting serotonin (5-HT) reuptake, agonizing the 5-HT1A receptor, partially agonizing the 5-HT1B receptor, and antagonizing the 5-HT3, 5-HT1D, and 5-HT7 receptors.
Clinical implications
It is hypothesized that depression is a heterogeneous disease caused by dysregulation of serotonin, norepinephrine, and dopamine, subsequently producing mood and neurovegetative symptoms of depression. Preclinical, in vivo studies indicate that vortioxetine enhances levels of serotonin, norepinephrine, dopamine, acetylcholine, and histamine in specific areas of the brain with the ability to improve depressive symptoms. Vortioxetine’s multimodal activity can be a useful alternative to other serotonergic antidepressants for some patients who are partial responders or non-responders to other treatment options. In addition, vortioxetine appears to have minimal effect on weight2 and sexual function—the latter being dose-dependent.3
How does it work?
Vortioxetine differs from other antidepressants in its multimodal activity (ie, affecting G-protein mode receptors, ion channel mode receptors, and neurotransmitter transporters). It inhibits the serotonin transporter (Ki = 1.6 nM), causing subsequent inhibition of serotonin reuptake into presynaptic neurons as well as selectively acting on the other subtypes of serotonergic receptors; however, activity on the norepinephrine transporter (Ki = 113 nM) and dopamine transporter (Ki > 1000 nM) is minimal. It is believed that mood-regulating effects of vortioxetine are caused by inhibition of serotonin reuptake, prolonged availability of serotonin to the postsynaptic neurons, its agonist activity on the 5-HT1A receptor (Ki = 15 nM), and partial agonist activity on the 5-HT1B receptor (Ki = 33 nM). Vortioxetine has strong affinity for the 5-HT3 receptor (Ki = 3.7 nM), which plays a role in modulation of centrally mediated nausea and vomiting. Positron emission tomography studies in humans determined that the occupancy of 5-HT transporter was 50% at 5 mg/d, 65% at 10 mg/d, and 80% at 20 mg/d.1,4 Human studies did not show that vortioxetine causes QTc prolongation.
Pharmacokinetics
Therapeutic activity of vortioxetine is thought to be due to the parent drug. It has a half-life of approximately 66 hours, and achieves steady state in 13.5 to 19 days. Bioavailability of vortioxetine is 75%; absorption does not depend on food; and 98% of drug is bound on plasma proteins.
Vortioxetine has linear pharmacokinetics, with maximum plasma concentration 7 to 11 hours after ingestion. The medication is metabolized primarily by oxidation through cytochrome P (CYP) 450: CYP2D6 (primary), CYP 3A4/5, CYP 2C19, CYP 2C9, CYP2A6, CYP2C8, and CYP2B6 with subsequent glucuronidation. This predisposes vortioxetine to potential pharmacokinetic drug-drug interaction warranting dose adjustment consideration when vortioxetine is coadministered with compounds inhibiting CYP2D6 or inducing CYP3A4 for ≥14 days, or for patients identified as poor 2D6 metabolizers.
In addition, coadministration of vortioxetine with serotonergic medications such as triptans, other antidepressants, and tramadol can cause potentially life-threatening serotonin syndrome, characterized by mental status changes, autonomic instability, neuromuscular aberrations, and GI symptoms. Concomitant use of vortioxetine and a nonsteroidal anti-inflammatory drug, aspirin, or warfarin can result in abnormal bleeding. Coadministration of vortioxetine with another highly protein-bound drug may increase or decrease the free concentration of either drug depending on the binding affinity of the drug for the protein.
Efficacy
Vortioxetine reduced depressive symptoms in 6 positive, 6- to 8-week, double-blind, placebo controlled and randomized studies and 1 maintenance study.1 Subjects were adults (Studies 1 to 5) and geriatric patients from age 64 to 88 who had ≥1 depressive episode before age 60 (Study 6). All met DSM-IV-TR criteria for MDD. Subjects with cognitive impairment scoring <24 on the Mini-Mental Status Examination and children were excluded. Depending on the study, response to the treatment was primarily measured on the Montgomery-Åsberg Depression Rating Scale (MADRS) or Hamilton Depression Rating Scale (HAM-D).
See Table 2 for a description of the positive studies, including dosages. In all studies, vortioxetine was superior to placebo at least one dosage for treating depression. In the 6- to 8-week placebo-controlled studies, an effect of vortioxetine based on the primary efficacy measure was generally observed starting at Week 2; that effect increased in subsequent weeks with the full antidepressant effect of vortioxetine generally not seen until study Week 4 or later.1
The maintenance treatment study included 639 patients who met DSM-IV-TR criteria for MDD. This study lasted for as long as 64 weeks. The first 12-week period was open-label, during which patients were treated with vortioxetine, 5 mg/d or 10 mg/d, with a possibility to adjust the dosage in the first 8 weeks. By the end of Week 12, 396 subjects achieved remission (MADRS <10), 75% of whom were taking vortioxetine, 10 mg/d. These patients were then randomly assigned to placebo or the dosage of vortioxetine to which they had responded, and continued the study for as long as 64 weeks. Time to relapse (MADRS total score ≥22) or an insufficient therapeutic response (as judged by the investigator) was the primary efficacy outcome, and demonstrated that vortioxetine was superior to placebo.
Tolerability
The tolerability of vortioxetine is comparable with other serotonergic antidepressants. In pooled analysis of pre-marketing studies, 5% to 8% of patients receiving vortioxetine (5 to 20 mg/d) discontinued treatment because of adverse effects (AEs), compared with 4% in the placebo group. Nausea was the most commonly reported AE leading to discontinuation and appeared to be dose dependent.
AEs, such as nausea, constipation, and vomiting, most commonly occurred in the first week of treatment, with a median duration of 2 weeks.5 In the 6- to 8-week trials, the most common AEs were nausea, constipation, and vomiting. In longer trials (24 to 64 weeks), the most common AE was nausea.
In 6- to 8-week placebo-controlled studies, vortioxetine was not associated with any clinically significant effect on vital signs or laboratory values in hematology, urinalysis, or serum chemistry (except sodium). Hyponatremia, the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH), has occurred. The risk of developing SIADH and resultant hyponatremia is greater in geriatric patients and patients taking a diuretic.
Abruptly discontinuing vortioxetine can cause transient withdrawal symptoms, including headache and muscle tension, especially at a higher dosage (15 to 20 mg/d). Gradual tapering can reduce withdrawal symptoms.
Specific clinical issues
All antidepressants have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation and behaviors during the first months of treatment and with dosage changes.
Vortioxetine is categorized as pregnancy category C. Newborns exposed to a selective serotonin reuptake inhibitor (SSRI) in pregnancy may have an increased risk of persistent pulmonary hypertension during the neonatal period. When taken during the third trimester of pregnancy, SSRIs and serotonin-norepinephrine reuptake inhibitors can cause serious neonatal complications, including respiratory distress, cyanosis, apnea, and seizures, which may require longer hospitalization, respiratory support, or tube feeding for the infant. Consider risks and benefits of third-trimester use of an antidepressant. It is not known if vortioxetine is present in human breast milk.
Clinical studies on vortioxetine in pediatric patients have not been conducted.
No dosage adjustment is recommended on the basis of age for geriatric patients. No dose adjustment of vortioxetine is necessary on the basis of race, sex, ethnicity, renal function, or mild to moderate hepatic impairment. See Table 3 for practice points when prescribing vortioxetine. See Table 4 for contraindications to vortioxetine.
Dosing
The recommended starting dosage is 10 mg, administered orally once daily without regard to meals. Dosage should then be increased to 20 mg/d, as clinically warranted and tolerated. Consider a dosage decrease to 5 mg/d in patients who do not tolerate higher dosages or require drug adjustment because of drug-drug interaction or poor 2D6 metabolizer status.
Bottom Line
FDA-approved for major depressive disorder in adults, vortioxetine reduced depressive symptoms in 6 positive, double-blind, placebo-controlled, and randomized studies. The multimodal activity of vortioxeine can be a useful alternative to serotonergic antidepressants for some patients who are partial responders or nonresponders. Tolerability is comparable with other serotonergic antidepressants.
Related Resources
- Alam MY, Jacobsen PL, Chen Y, et al. Safety, tolerability, and efficacy of vortioxetine (Lu AA21004) in major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2014; 29(1):36-44.
- Mahableshwarkar AR, Jacobsen PL, Chen Y. A randomized, double-blind trial of 2.5 mg and 5 mg vortioxetine (Lu AA21004) versus placebo for 8 weeks in adults with major depressive disorder. Curr Med Res Opin. 2013;29(3):217-226.
Drug Brand Names
Linezolid • Zyvox Vortioxetine • Brintellix
Methylene blue • Urolene Blue Warfarin • Coumadin
Tramadol • Ultram
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Vortioxetine [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc.; 2013.
2. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71(10):1259-1272.
3. Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a comprehensive review and meta-analysis. J Clin Psychopharmacol. 2009; 29(3):259-266.
4. Chen G, Lee R, Højer A, et al. Pharmacokinetic drug interactions involving vortioxetine (LU AA 21004), a multimodal antidepressant. Clin Drug Invetig. 2013; 33(10):727-736.
5. Citrome L. Vortioxetine for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Prac. 2014;68(1):60-82.
1. Vortioxetine [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc.; 2013.
2. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71(10):1259-1272.
3. Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a comprehensive review and meta-analysis. J Clin Psychopharmacol. 2009; 29(3):259-266.
4. Chen G, Lee R, Højer A, et al. Pharmacokinetic drug interactions involving vortioxetine (LU AA 21004), a multimodal antidepressant. Clin Drug Invetig. 2013; 33(10):727-736.
5. Citrome L. Vortioxetine for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Prac. 2014;68(1):60-82.
Awakening to the dangers of obstructive sleep apnea
Estimates are that 50 to 70 million Americans suffer from a chronic disorder of sleep and wakefulness, hindering daily functioning and affecting health.1 Psychiatric illness is common among people who have a sleep disorder. The relationship between psychiatric illness and sleep disorders is bidirectional: People with mental illness often have sleep complaints, and a primary sleep disorder often results in neuropsychiatric complications.
What is obstructive sleep apnea?
The most common type of sleep-disordered breathing, obstructive sleep apnea (OSA) is characterized by frequent cessations of breathing during sleep because of an obstruction of the upper airway. The obstruction occurs secondary to inadequate motor tone of the tongue or airway dilator muscles, or both.1 In addition, many people with OSA have central apneic episodes, in which breathing stops temporarily without airway blockage or respiratory effort.2
The prevalence of OSA is growing as obesity in the United States increases. Risk factors for OSA include obesity, a craniofacial abnormality, an upper-airway abnormality, heredity, smoking, and nasal congestion. OSA plays a role in causing and exacerbating medical illness in people with severe and persistent mental illness, contributing to a significantly shortened life span. Attending to the general health of people who suffer from severe mental illness—including effective treatment of illnesses such as OSA—is crucial.3
Clinical features of OSA
OSA is characterized by hypopnea (a decrease in breathing during sleep) or apnea (an actual pause in breathing). Pauses in breathing during sleep of at least 10 seconds, with obstruction of oronasal airflow despite continuous chest and abdominal movements, are referred to as obstructive apneas. These pauses are associated with a decrease in oxygen saturation or arousal from sleep, or both.1
Primary features of OSA include sleep fragmentation accompanied by nocturnal hypoxemia and hypercapnia, with resulting excessive daytime sleepiness, mood problems, and poor neurocognitive performance (Table 1). OSA often causes potentially serious organ system dysfunction, including adverse cardiovascular and metabolic effects. Studies have suggested that executive dysfunction can be a feature of OSA, which is thought to be related to prefrontal lobe dysfunction caused by intermittent hypoxia. All of these conditions can contribute significantly to decreased quality of life.1

The prevalence of OSA in the general population is approximately 20% when the condition is defined as an apnea-hypopnea index >5 events an hour. The index is the number of apnea and hypopnea episodes that occur during 1 hour of sleep.4
OSA and psychiatric illness
Psychiatric disorders often are comorbid with OSA. These include depression, anxiety, bipolar disorder, schizophrenia, posttraumatic stress disorder (PTSD), panic disorder, and substance use disorder.
Depression. Several studies have documented that OSA and depressive disorder often are comorbid. Many symptoms are common to both, including fatigue, daytime sleepiness, poor concentration, irritability, and weight gain (Figure), although some core symptoms of depression (eg, sadness, anhedonia, guilt, and agitation) are clearly distinguishable from symptoms of OSA. The current recommendation is that a mood disorder should be considered secondary to OSA, and treated accordingly.5
Anxiety. OSA also has been linked to anxiety and nocturnal panic attacks. Frequent awakening due to choking from breathing cessation might play a role in the development of anxiety in patients with OSA, although the association is unproven. Studies have shown a correlation between anxiety disorders and excessive daytime sleepiness, one of the core symptoms of OSA.6 OSA is highly prevalent among combat veterans who have PTSD and complain of being overly vigilant at night; experiencing nightmares and frequent awakening; and having non-restorative sleep.7 Anecdotal reports suggest an association between OSA and bipolar disorder: namely, that continuous positive airway pressure (CPAP) treatment (see “How is OSA treated?,” below) might switch depressed patients to mania.8
Schizophrenia. A strong association exists between OSA and schizophrenia. In a study,9 an OSA diagnosis was made 6 times more often in patients with schizophrenia than in patients with other psychiatric illnesses. Obesity, male sex, and chronic antipsychotic administration were risk factors for OSA in patients with
schizophrenia.9 OSA might be underdiagnosed in patients with schizophrenia because excessive daytime sleepiness, the most common daytime symptom of OSA, can be misattributed as a negative symptom of the disease or a side effect of pharmacotherapy.
OSA and medical illness
OSA can be comorbid with several medical conditions (Table 2). Sleep research in the past 15 years has demonstrated that chronic sleep deprivation has multiple untoward health consequences apart from excessive daytime sleepiness.10 Recent research suggests that chronic sleep loss (<7 hours a night), including sleep loss secondary to OSA, has wide-ranging effects on the cardiovascular, endocrine, immune, and nervous systems, including:
• obesity (adults and children)
• diabetes mellitus and impaired glucose tolerance
• cardiovascular disease and hypertension.

Obesity is one of the primary and more modifiable risk factors for OSA (Box). Studies suggest that reducing the severity of obesity would likely benefit people with a sleep disorder, and that treating sleep deprivation and sleep disorders might benefit persons with obesity.12 Chronic sleep loss can have a deleterious influence on appetite regulation through effects on 2 hormones, leptin and ghrelin, that play a major role in appetite regulation. Chronic sleep loss causes and perpetuates obesity through its interplay with these, and other, hormones.12

Diabetes. The link between obesity and diabetes is well-established, as is the long-term morbidity and mortality of these 2 diseases.13 Evidence shows that OSA is associated with impaired glucose tolerance and an increased risk of diabetes.14
Cardiovascular disease. OSA has a strong association with cardiovascular disease, including systemic hypertension, possibly myocardial infarction, congestive heart failure, and stroke.15 Institution of appropriate treatment for OSA including CPAP can minimize or reverse many of these effects.16
Making an OSA diagnosis
A diagnostic polysomnogram (PSG), or sleep study, is the standard test when OSA is suspected. It is performed most often at an attended sleep laboratory. Typically, a PSG measures several physiologic measures, including, but not limited to:
• airflow through mouth and nose
• stages of sleep (by means of electroencephalography channels)
• thoracic and abdominal movements (to assess effort of breathing)
• muscle activity of the chin
• oxyhemoglobin saturation (to monitor variability in oxygen saturation [SaO2] during OSA events).
Portable diagnostic instruments can provide reliable information when a patient cannot be studied in a laboratory. Assessments available on portable instruments include cardiopulmonary monitoring of respiration only; PSG; and peripheral arterial tonometry, which measures autonomic manifestations of respiratory obstructive events.17,18
The severity of OSA is established by the apnea/hypopnea index, which measures the number of apneas and hypopneas per hour of sleep.
How is OSA treated?
CPAP is still the gold standard for treating OSA. CPAP provides a pneumatic splint for the upper airway by administering positive pressure through a nasal or oronasal mask. CPAP distinctly improves daytime sleepiness.19,20
Pressure is determined initially by titration during PSG, although a number of automated CPAP machines are available in which pressure is adjusted based on the machine’s response to airflow obstruction. Advantages of using PSG to titrate CPAP are direct observation to control mask leak and the ability to observe the effects of body position and sleep stage and clearly distinguish periods of sleep from wakefulness.
Regrettably, adherence to a nightly regimen of CPAP is less than ideal for several reasons, including claustrophobia, interface failure, and other motivational variables. Some patients who experience claustrophobia can use desensitization techniques; others are, ultimately, unable to use the mask.
Oral appliances. A patient who has mild or moderate OSA but who cannot use the CPAP mask might be a good candidate for an oral appliance. These appliances, which hold the mandible in an advanced position during the night, can be effective in such cases.
CPAP autotitration changes the treatment pressure based on feedback from such patient measures as airflow and airway resistance. Autotitrating devices might have a role in beginning treatment in patients with OSA by means of a portable sleep study, in which CPAP titration is not performed. In addition, autotitrating offers the possibility of changing pressure over time—such as with changes in position during the night or over the longer term in response to weight loss or gain.
Surgery. In patients who are unable to use CPAP, surgery might be indicated to relieve an anatomical obstruction, such as adenotonsillar hypertrophy or other type of mass lesion.
Sleep positioning. A patient who demonstrates OSA exclusively while sleeping supine might benefit from being trained to sleep on either side only or arranging pillows so that he can only sleep on his side.
In conclusion
OSA is common and easily treatable. It coexists with, and exacerbates, medical and psychiatric illness. Treating OSA concomitantly with comorbid medical and psychiatric illness is essential to achieve full symptom remission and prevent associated long-term consequences of both medical and psychiatric illness.
BOTTOM LINE
Obstructive sleep apnea (OSA) and psychiatric illness, especially depression, often co-exist. Screen depressed patients—especially those with risk factors for OSA, such as obesity, smoking, and an upper-airway abnormality—for a sleep disorder. This is especially important if a patient complains of daytime somnolence, fatigue, cognitive problems, poor concentration, or weight gain. For optimal results, treat comorbid psychiatric illness and OSA concurrently; the same is true for other sleep disorders.
Related Resources
- Babson KA, Del Re AC, Bonn-Miller MO, et al. The comorbidity of sleep apnea and mood, anxiety, and substance use disorders among obese military veterans within the Veterans Health Administration. J Clin Sleep Med. 2013; 9(12):1253-1258.
- Karkoulias K, Lykouras D, Sampsonas F, et al. The impact of obstructive sleep apnea syndrome severity on physical performance and mental health. The use of SF-36 questionnaire in sleep apnea. Eur Rev Med Pharmacol Sci. 2013;17(4):531-536.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Dr. Muhammad Awais Aftab, psychiatry resident at Hamad Medical Corporation, Doha, Qatar, and Umair Amin, final year MBBS student at King Edward Medical University, Lahore, Pakistan, assisted with development of the manuscript of this article.
1. Institute of Medicine. Sleep disorders and sleep deprivation: an unmet public health problem. Washington, DC: The National Academies Press; 2006:20.
2. Badr MS. Central sleep apnea. Prim Care. 2005;32(2):361-374.
3. Freedland KE, Carney RM, Hayano J, et al. Effect of obstructive sleep apnea on response to cognitive behavior therapy for depression after an acute myocardial infarction. J Psychosom Res. 2012;72(4):276-281.
4. Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008;5(2):136-143.
5. El-Sherbini AM, Bediwy AS, El-Mitwalli A. Association between obstructive sleep apnea (OSA) and depression and the effect of continuous positive airway pressure (CPAP) treatment. Neuropsychiatr Dis Treat. 2011;7:715-721.
6. Hasler G, Buysse DJ, Gamma A, et al. Excessive daytime sleepiness in young adults: a 20-year prospective community study. J Clin Psychiatry. 2005;66(4):521-529.
7. Yesavage JA, Kinoshita LM, Kimball T, et al. Sleep-disordered breathing in Vietnam veterans with posttraumatic stress disorder. Am J Geriatr Psychiatry. 2012;20(3):199-204.
8. Plante D, Winkelman J. Sleep disturbance in bipolar disorder: therapeutic implications. Am J Psychiatry. 2008; 165(7):830-843.
9. Winkelman J. Schizophrenia, obesity, and obstructive sleep apnea. J Clin Psychiatry. 2001;62(1):8-11.
10. Partinen M, Hublin C. Epidemiology of sleep disorders. Philadelphia, PA: Elsevier Saunders; 2005.
11. Valderas JM, Starfield B, Sibbald B, et al. Defining comorbidity: implications for understanding health and health services. Ann Fam Med. 2009;7(4):357-363.
12. Romero-Corral A, Caples SM, Lopez-Jimenez F, et al. Interactions between obesity and obstructive sleep apnea: implications for treatment. Chest. 2010;137(3):711-719.
13. Villareal DT, Apovian CM, Kushner RF, et al. Obesity in older adults: technical review and position statement of the American Society for Nutrition and NAASO, The Obesity Society. Obes Res. 2005;13(11):1849-1863.
14. Pamidi S, Aronsohn RS, Tasali E. Obstructive sleep apnea: role in the risk and severity of diabetes. Best Pract Res Clin Endocrinol Metab. 2010;24(5):703-715.
15. Malhotra A, Loscalzo J. Sleep and cardiovascular disease: an overview. Prog Cardiovasc Dis. 2009;51(4):279-284.
16. Bradley TD, Floras JS. Obstructive sleep apnoea and its cardiovascular consequences. Lancet. 2009;373(9657):82-93.
17. Chesson A, Berry R, Pack A. Practice parameters for the use of portable monitoring devices in the investigation of suspected obstructive sleep apnea in adults. Sleep. 2003; 26(7):907-913.
18. Pittman S, Ayas N, MacDonald M, et al. Using a wrist-worn device based on peripheral arterial tonometry to diagnose obstructive sleep apnea: in-laboratory and ambulatory validation. Sleep. 2004;27(1):923-933.
19. Ballester E, Badia J, Hernandez L, et al. Evidence of the effectiveness of continuous positive airway pressure in the treatment of sleep apnea/hypopnea syndrome. Am J Respir Crit Care Med. 1999;159:495-501.
20. Jenkinson D, Davies J, Mullins R, et al. Comparison of therapeutic and subtherapeutic nasal continuous positive airway pressure for obstructive sleep apnoea: a randomised prospective parallel trial. Lancet. 1999;353:2100-2105.
Estimates are that 50 to 70 million Americans suffer from a chronic disorder of sleep and wakefulness, hindering daily functioning and affecting health.1 Psychiatric illness is common among people who have a sleep disorder. The relationship between psychiatric illness and sleep disorders is bidirectional: People with mental illness often have sleep complaints, and a primary sleep disorder often results in neuropsychiatric complications.
What is obstructive sleep apnea?
The most common type of sleep-disordered breathing, obstructive sleep apnea (OSA) is characterized by frequent cessations of breathing during sleep because of an obstruction of the upper airway. The obstruction occurs secondary to inadequate motor tone of the tongue or airway dilator muscles, or both.1 In addition, many people with OSA have central apneic episodes, in which breathing stops temporarily without airway blockage or respiratory effort.2
The prevalence of OSA is growing as obesity in the United States increases. Risk factors for OSA include obesity, a craniofacial abnormality, an upper-airway abnormality, heredity, smoking, and nasal congestion. OSA plays a role in causing and exacerbating medical illness in people with severe and persistent mental illness, contributing to a significantly shortened life span. Attending to the general health of people who suffer from severe mental illness—including effective treatment of illnesses such as OSA—is crucial.3
Clinical features of OSA
OSA is characterized by hypopnea (a decrease in breathing during sleep) or apnea (an actual pause in breathing). Pauses in breathing during sleep of at least 10 seconds, with obstruction of oronasal airflow despite continuous chest and abdominal movements, are referred to as obstructive apneas. These pauses are associated with a decrease in oxygen saturation or arousal from sleep, or both.1
Primary features of OSA include sleep fragmentation accompanied by nocturnal hypoxemia and hypercapnia, with resulting excessive daytime sleepiness, mood problems, and poor neurocognitive performance (Table 1). OSA often causes potentially serious organ system dysfunction, including adverse cardiovascular and metabolic effects. Studies have suggested that executive dysfunction can be a feature of OSA, which is thought to be related to prefrontal lobe dysfunction caused by intermittent hypoxia. All of these conditions can contribute significantly to decreased quality of life.1
The prevalence of OSA in the general population is approximately 20% when the condition is defined as an apnea-hypopnea index >5 events an hour. The index is the number of apnea and hypopnea episodes that occur during 1 hour of sleep.4
OSA and psychiatric illness
Psychiatric disorders often are comorbid with OSA. These include depression, anxiety, bipolar disorder, schizophrenia, posttraumatic stress disorder (PTSD), panic disorder, and substance use disorder.
Depression. Several studies have documented that OSA and depressive disorder often are comorbid. Many symptoms are common to both, including fatigue, daytime sleepiness, poor concentration, irritability, and weight gain (Figure), although some core symptoms of depression (eg, sadness, anhedonia, guilt, and agitation) are clearly distinguishable from symptoms of OSA. The current recommendation is that a mood disorder should be considered secondary to OSA, and treated accordingly.5
Anxiety. OSA also has been linked to anxiety and nocturnal panic attacks. Frequent awakening due to choking from breathing cessation might play a role in the development of anxiety in patients with OSA, although the association is unproven. Studies have shown a correlation between anxiety disorders and excessive daytime sleepiness, one of the core symptoms of OSA.6 OSA is highly prevalent among combat veterans who have PTSD and complain of being overly vigilant at night; experiencing nightmares and frequent awakening; and having non-restorative sleep.7 Anecdotal reports suggest an association between OSA and bipolar disorder: namely, that continuous positive airway pressure (CPAP) treatment (see “How is OSA treated?,” below) might switch depressed patients to mania.8
Schizophrenia. A strong association exists between OSA and schizophrenia. In a study,9 an OSA diagnosis was made 6 times more often in patients with schizophrenia than in patients with other psychiatric illnesses. Obesity, male sex, and chronic antipsychotic administration were risk factors for OSA in patients with
schizophrenia.9 OSA might be underdiagnosed in patients with schizophrenia because excessive daytime sleepiness, the most common daytime symptom of OSA, can be misattributed as a negative symptom of the disease or a side effect of pharmacotherapy.
OSA and medical illness
OSA can be comorbid with several medical conditions (Table 2). Sleep research in the past 15 years has demonstrated that chronic sleep deprivation has multiple untoward health consequences apart from excessive daytime sleepiness.10 Recent research suggests that chronic sleep loss (<7 hours a night), including sleep loss secondary to OSA, has wide-ranging effects on the cardiovascular, endocrine, immune, and nervous systems, including:
• obesity (adults and children)
• diabetes mellitus and impaired glucose tolerance
• cardiovascular disease and hypertension.
Obesity is one of the primary and more modifiable risk factors for OSA (Box). Studies suggest that reducing the severity of obesity would likely benefit people with a sleep disorder, and that treating sleep deprivation and sleep disorders might benefit persons with obesity.12 Chronic sleep loss can have a deleterious influence on appetite regulation through effects on 2 hormones, leptin and ghrelin, that play a major role in appetite regulation. Chronic sleep loss causes and perpetuates obesity through its interplay with these, and other, hormones.12
Diabetes. The link between obesity and diabetes is well-established, as is the long-term morbidity and mortality of these 2 diseases.13 Evidence shows that OSA is associated with impaired glucose tolerance and an increased risk of diabetes.14
Cardiovascular disease. OSA has a strong association with cardiovascular disease, including systemic hypertension, possibly myocardial infarction, congestive heart failure, and stroke.15 Institution of appropriate treatment for OSA including CPAP can minimize or reverse many of these effects.16
Making an OSA diagnosis
A diagnostic polysomnogram (PSG), or sleep study, is the standard test when OSA is suspected. It is performed most often at an attended sleep laboratory. Typically, a PSG measures several physiologic measures, including, but not limited to:
• airflow through mouth and nose
• stages of sleep (by means of electroencephalography channels)
• thoracic and abdominal movements (to assess effort of breathing)
• muscle activity of the chin
• oxyhemoglobin saturation (to monitor variability in oxygen saturation [SaO2] during OSA events).
Portable diagnostic instruments can provide reliable information when a patient cannot be studied in a laboratory. Assessments available on portable instruments include cardiopulmonary monitoring of respiration only; PSG; and peripheral arterial tonometry, which measures autonomic manifestations of respiratory obstructive events.17,18
The severity of OSA is established by the apnea/hypopnea index, which measures the number of apneas and hypopneas per hour of sleep.
How is OSA treated?
CPAP is still the gold standard for treating OSA. CPAP provides a pneumatic splint for the upper airway by administering positive pressure through a nasal or oronasal mask. CPAP distinctly improves daytime sleepiness.19,20
Pressure is determined initially by titration during PSG, although a number of automated CPAP machines are available in which pressure is adjusted based on the machine’s response to airflow obstruction. Advantages of using PSG to titrate CPAP are direct observation to control mask leak and the ability to observe the effects of body position and sleep stage and clearly distinguish periods of sleep from wakefulness.
Regrettably, adherence to a nightly regimen of CPAP is less than ideal for several reasons, including claustrophobia, interface failure, and other motivational variables. Some patients who experience claustrophobia can use desensitization techniques; others are, ultimately, unable to use the mask.
Oral appliances. A patient who has mild or moderate OSA but who cannot use the CPAP mask might be a good candidate for an oral appliance. These appliances, which hold the mandible in an advanced position during the night, can be effective in such cases.
CPAP autotitration changes the treatment pressure based on feedback from such patient measures as airflow and airway resistance. Autotitrating devices might have a role in beginning treatment in patients with OSA by means of a portable sleep study, in which CPAP titration is not performed. In addition, autotitrating offers the possibility of changing pressure over time—such as with changes in position during the night or over the longer term in response to weight loss or gain.
Surgery. In patients who are unable to use CPAP, surgery might be indicated to relieve an anatomical obstruction, such as adenotonsillar hypertrophy or other type of mass lesion.
Sleep positioning. A patient who demonstrates OSA exclusively while sleeping supine might benefit from being trained to sleep on either side only or arranging pillows so that he can only sleep on his side.
In conclusion
OSA is common and easily treatable. It coexists with, and exacerbates, medical and psychiatric illness. Treating OSA concomitantly with comorbid medical and psychiatric illness is essential to achieve full symptom remission and prevent associated long-term consequences of both medical and psychiatric illness.
BOTTOM LINE
Obstructive sleep apnea (OSA) and psychiatric illness, especially depression, often co-exist. Screen depressed patients—especially those with risk factors for OSA, such as obesity, smoking, and an upper-airway abnormality—for a sleep disorder. This is especially important if a patient complains of daytime somnolence, fatigue, cognitive problems, poor concentration, or weight gain. For optimal results, treat comorbid psychiatric illness and OSA concurrently; the same is true for other sleep disorders.
Related Resources
- Babson KA, Del Re AC, Bonn-Miller MO, et al. The comorbidity of sleep apnea and mood, anxiety, and substance use disorders among obese military veterans within the Veterans Health Administration. J Clin Sleep Med. 2013; 9(12):1253-1258.
- Karkoulias K, Lykouras D, Sampsonas F, et al. The impact of obstructive sleep apnea syndrome severity on physical performance and mental health. The use of SF-36 questionnaire in sleep apnea. Eur Rev Med Pharmacol Sci. 2013;17(4):531-536.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Dr. Muhammad Awais Aftab, psychiatry resident at Hamad Medical Corporation, Doha, Qatar, and Umair Amin, final year MBBS student at King Edward Medical University, Lahore, Pakistan, assisted with development of the manuscript of this article.
Estimates are that 50 to 70 million Americans suffer from a chronic disorder of sleep and wakefulness, hindering daily functioning and affecting health.1 Psychiatric illness is common among people who have a sleep disorder. The relationship between psychiatric illness and sleep disorders is bidirectional: People with mental illness often have sleep complaints, and a primary sleep disorder often results in neuropsychiatric complications.
What is obstructive sleep apnea?
The most common type of sleep-disordered breathing, obstructive sleep apnea (OSA) is characterized by frequent cessations of breathing during sleep because of an obstruction of the upper airway. The obstruction occurs secondary to inadequate motor tone of the tongue or airway dilator muscles, or both.1 In addition, many people with OSA have central apneic episodes, in which breathing stops temporarily without airway blockage or respiratory effort.2
The prevalence of OSA is growing as obesity in the United States increases. Risk factors for OSA include obesity, a craniofacial abnormality, an upper-airway abnormality, heredity, smoking, and nasal congestion. OSA plays a role in causing and exacerbating medical illness in people with severe and persistent mental illness, contributing to a significantly shortened life span. Attending to the general health of people who suffer from severe mental illness—including effective treatment of illnesses such as OSA—is crucial.3
Clinical features of OSA
OSA is characterized by hypopnea (a decrease in breathing during sleep) or apnea (an actual pause in breathing). Pauses in breathing during sleep of at least 10 seconds, with obstruction of oronasal airflow despite continuous chest and abdominal movements, are referred to as obstructive apneas. These pauses are associated with a decrease in oxygen saturation or arousal from sleep, or both.1
Primary features of OSA include sleep fragmentation accompanied by nocturnal hypoxemia and hypercapnia, with resulting excessive daytime sleepiness, mood problems, and poor neurocognitive performance (Table 1). OSA often causes potentially serious organ system dysfunction, including adverse cardiovascular and metabolic effects. Studies have suggested that executive dysfunction can be a feature of OSA, which is thought to be related to prefrontal lobe dysfunction caused by intermittent hypoxia. All of these conditions can contribute significantly to decreased quality of life.1
The prevalence of OSA in the general population is approximately 20% when the condition is defined as an apnea-hypopnea index >5 events an hour. The index is the number of apnea and hypopnea episodes that occur during 1 hour of sleep.4
OSA and psychiatric illness
Psychiatric disorders often are comorbid with OSA. These include depression, anxiety, bipolar disorder, schizophrenia, posttraumatic stress disorder (PTSD), panic disorder, and substance use disorder.
Depression. Several studies have documented that OSA and depressive disorder often are comorbid. Many symptoms are common to both, including fatigue, daytime sleepiness, poor concentration, irritability, and weight gain (Figure), although some core symptoms of depression (eg, sadness, anhedonia, guilt, and agitation) are clearly distinguishable from symptoms of OSA. The current recommendation is that a mood disorder should be considered secondary to OSA, and treated accordingly.5
Anxiety. OSA also has been linked to anxiety and nocturnal panic attacks. Frequent awakening due to choking from breathing cessation might play a role in the development of anxiety in patients with OSA, although the association is unproven. Studies have shown a correlation between anxiety disorders and excessive daytime sleepiness, one of the core symptoms of OSA.6 OSA is highly prevalent among combat veterans who have PTSD and complain of being overly vigilant at night; experiencing nightmares and frequent awakening; and having non-restorative sleep.7 Anecdotal reports suggest an association between OSA and bipolar disorder: namely, that continuous positive airway pressure (CPAP) treatment (see “How is OSA treated?,” below) might switch depressed patients to mania.8
Schizophrenia. A strong association exists between OSA and schizophrenia. In a study,9 an OSA diagnosis was made 6 times more often in patients with schizophrenia than in patients with other psychiatric illnesses. Obesity, male sex, and chronic antipsychotic administration were risk factors for OSA in patients with
schizophrenia.9 OSA might be underdiagnosed in patients with schizophrenia because excessive daytime sleepiness, the most common daytime symptom of OSA, can be misattributed as a negative symptom of the disease or a side effect of pharmacotherapy.
OSA and medical illness
OSA can be comorbid with several medical conditions (Table 2). Sleep research in the past 15 years has demonstrated that chronic sleep deprivation has multiple untoward health consequences apart from excessive daytime sleepiness.10 Recent research suggests that chronic sleep loss (<7 hours a night), including sleep loss secondary to OSA, has wide-ranging effects on the cardiovascular, endocrine, immune, and nervous systems, including:
• obesity (adults and children)
• diabetes mellitus and impaired glucose tolerance
• cardiovascular disease and hypertension.
Obesity is one of the primary and more modifiable risk factors for OSA (Box). Studies suggest that reducing the severity of obesity would likely benefit people with a sleep disorder, and that treating sleep deprivation and sleep disorders might benefit persons with obesity.12 Chronic sleep loss can have a deleterious influence on appetite regulation through effects on 2 hormones, leptin and ghrelin, that play a major role in appetite regulation. Chronic sleep loss causes and perpetuates obesity through its interplay with these, and other, hormones.12
Diabetes. The link between obesity and diabetes is well-established, as is the long-term morbidity and mortality of these 2 diseases.13 Evidence shows that OSA is associated with impaired glucose tolerance and an increased risk of diabetes.14
Cardiovascular disease. OSA has a strong association with cardiovascular disease, including systemic hypertension, possibly myocardial infarction, congestive heart failure, and stroke.15 Institution of appropriate treatment for OSA including CPAP can minimize or reverse many of these effects.16
Making an OSA diagnosis
A diagnostic polysomnogram (PSG), or sleep study, is the standard test when OSA is suspected. It is performed most often at an attended sleep laboratory. Typically, a PSG measures several physiologic measures, including, but not limited to:
• airflow through mouth and nose
• stages of sleep (by means of electroencephalography channels)
• thoracic and abdominal movements (to assess effort of breathing)
• muscle activity of the chin
• oxyhemoglobin saturation (to monitor variability in oxygen saturation [SaO2] during OSA events).
Portable diagnostic instruments can provide reliable information when a patient cannot be studied in a laboratory. Assessments available on portable instruments include cardiopulmonary monitoring of respiration only; PSG; and peripheral arterial tonometry, which measures autonomic manifestations of respiratory obstructive events.17,18
The severity of OSA is established by the apnea/hypopnea index, which measures the number of apneas and hypopneas per hour of sleep.
How is OSA treated?
CPAP is still the gold standard for treating OSA. CPAP provides a pneumatic splint for the upper airway by administering positive pressure through a nasal or oronasal mask. CPAP distinctly improves daytime sleepiness.19,20
Pressure is determined initially by titration during PSG, although a number of automated CPAP machines are available in which pressure is adjusted based on the machine’s response to airflow obstruction. Advantages of using PSG to titrate CPAP are direct observation to control mask leak and the ability to observe the effects of body position and sleep stage and clearly distinguish periods of sleep from wakefulness.
Regrettably, adherence to a nightly regimen of CPAP is less than ideal for several reasons, including claustrophobia, interface failure, and other motivational variables. Some patients who experience claustrophobia can use desensitization techniques; others are, ultimately, unable to use the mask.
Oral appliances. A patient who has mild or moderate OSA but who cannot use the CPAP mask might be a good candidate for an oral appliance. These appliances, which hold the mandible in an advanced position during the night, can be effective in such cases.
CPAP autotitration changes the treatment pressure based on feedback from such patient measures as airflow and airway resistance. Autotitrating devices might have a role in beginning treatment in patients with OSA by means of a portable sleep study, in which CPAP titration is not performed. In addition, autotitrating offers the possibility of changing pressure over time—such as with changes in position during the night or over the longer term in response to weight loss or gain.
Surgery. In patients who are unable to use CPAP, surgery might be indicated to relieve an anatomical obstruction, such as adenotonsillar hypertrophy or other type of mass lesion.
Sleep positioning. A patient who demonstrates OSA exclusively while sleeping supine might benefit from being trained to sleep on either side only or arranging pillows so that he can only sleep on his side.
In conclusion
OSA is common and easily treatable. It coexists with, and exacerbates, medical and psychiatric illness. Treating OSA concomitantly with comorbid medical and psychiatric illness is essential to achieve full symptom remission and prevent associated long-term consequences of both medical and psychiatric illness.
BOTTOM LINE
Obstructive sleep apnea (OSA) and psychiatric illness, especially depression, often co-exist. Screen depressed patients—especially those with risk factors for OSA, such as obesity, smoking, and an upper-airway abnormality—for a sleep disorder. This is especially important if a patient complains of daytime somnolence, fatigue, cognitive problems, poor concentration, or weight gain. For optimal results, treat comorbid psychiatric illness and OSA concurrently; the same is true for other sleep disorders.
Related Resources
- Babson KA, Del Re AC, Bonn-Miller MO, et al. The comorbidity of sleep apnea and mood, anxiety, and substance use disorders among obese military veterans within the Veterans Health Administration. J Clin Sleep Med. 2013; 9(12):1253-1258.
- Karkoulias K, Lykouras D, Sampsonas F, et al. The impact of obstructive sleep apnea syndrome severity on physical performance and mental health. The use of SF-36 questionnaire in sleep apnea. Eur Rev Med Pharmacol Sci. 2013;17(4):531-536.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Dr. Muhammad Awais Aftab, psychiatry resident at Hamad Medical Corporation, Doha, Qatar, and Umair Amin, final year MBBS student at King Edward Medical University, Lahore, Pakistan, assisted with development of the manuscript of this article.
1. Institute of Medicine. Sleep disorders and sleep deprivation: an unmet public health problem. Washington, DC: The National Academies Press; 2006:20.
2. Badr MS. Central sleep apnea. Prim Care. 2005;32(2):361-374.
3. Freedland KE, Carney RM, Hayano J, et al. Effect of obstructive sleep apnea on response to cognitive behavior therapy for depression after an acute myocardial infarction. J Psychosom Res. 2012;72(4):276-281.
4. Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008;5(2):136-143.
5. El-Sherbini AM, Bediwy AS, El-Mitwalli A. Association between obstructive sleep apnea (OSA) and depression and the effect of continuous positive airway pressure (CPAP) treatment. Neuropsychiatr Dis Treat. 2011;7:715-721.
6. Hasler G, Buysse DJ, Gamma A, et al. Excessive daytime sleepiness in young adults: a 20-year prospective community study. J Clin Psychiatry. 2005;66(4):521-529.
7. Yesavage JA, Kinoshita LM, Kimball T, et al. Sleep-disordered breathing in Vietnam veterans with posttraumatic stress disorder. Am J Geriatr Psychiatry. 2012;20(3):199-204.
8. Plante D, Winkelman J. Sleep disturbance in bipolar disorder: therapeutic implications. Am J Psychiatry. 2008; 165(7):830-843.
9. Winkelman J. Schizophrenia, obesity, and obstructive sleep apnea. J Clin Psychiatry. 2001;62(1):8-11.
10. Partinen M, Hublin C. Epidemiology of sleep disorders. Philadelphia, PA: Elsevier Saunders; 2005.
11. Valderas JM, Starfield B, Sibbald B, et al. Defining comorbidity: implications for understanding health and health services. Ann Fam Med. 2009;7(4):357-363.
12. Romero-Corral A, Caples SM, Lopez-Jimenez F, et al. Interactions between obesity and obstructive sleep apnea: implications for treatment. Chest. 2010;137(3):711-719.
13. Villareal DT, Apovian CM, Kushner RF, et al. Obesity in older adults: technical review and position statement of the American Society for Nutrition and NAASO, The Obesity Society. Obes Res. 2005;13(11):1849-1863.
14. Pamidi S, Aronsohn RS, Tasali E. Obstructive sleep apnea: role in the risk and severity of diabetes. Best Pract Res Clin Endocrinol Metab. 2010;24(5):703-715.
15. Malhotra A, Loscalzo J. Sleep and cardiovascular disease: an overview. Prog Cardiovasc Dis. 2009;51(4):279-284.
16. Bradley TD, Floras JS. Obstructive sleep apnoea and its cardiovascular consequences. Lancet. 2009;373(9657):82-93.
17. Chesson A, Berry R, Pack A. Practice parameters for the use of portable monitoring devices in the investigation of suspected obstructive sleep apnea in adults. Sleep. 2003; 26(7):907-913.
18. Pittman S, Ayas N, MacDonald M, et al. Using a wrist-worn device based on peripheral arterial tonometry to diagnose obstructive sleep apnea: in-laboratory and ambulatory validation. Sleep. 2004;27(1):923-933.
19. Ballester E, Badia J, Hernandez L, et al. Evidence of the effectiveness of continuous positive airway pressure in the treatment of sleep apnea/hypopnea syndrome. Am J Respir Crit Care Med. 1999;159:495-501.
20. Jenkinson D, Davies J, Mullins R, et al. Comparison of therapeutic and subtherapeutic nasal continuous positive airway pressure for obstructive sleep apnoea: a randomised prospective parallel trial. Lancet. 1999;353:2100-2105.
1. Institute of Medicine. Sleep disorders and sleep deprivation: an unmet public health problem. Washington, DC: The National Academies Press; 2006:20.
2. Badr MS. Central sleep apnea. Prim Care. 2005;32(2):361-374.
3. Freedland KE, Carney RM, Hayano J, et al. Effect of obstructive sleep apnea on response to cognitive behavior therapy for depression after an acute myocardial infarction. J Psychosom Res. 2012;72(4):276-281.
4. Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008;5(2):136-143.
5. El-Sherbini AM, Bediwy AS, El-Mitwalli A. Association between obstructive sleep apnea (OSA) and depression and the effect of continuous positive airway pressure (CPAP) treatment. Neuropsychiatr Dis Treat. 2011;7:715-721.
6. Hasler G, Buysse DJ, Gamma A, et al. Excessive daytime sleepiness in young adults: a 20-year prospective community study. J Clin Psychiatry. 2005;66(4):521-529.
7. Yesavage JA, Kinoshita LM, Kimball T, et al. Sleep-disordered breathing in Vietnam veterans with posttraumatic stress disorder. Am J Geriatr Psychiatry. 2012;20(3):199-204.
8. Plante D, Winkelman J. Sleep disturbance in bipolar disorder: therapeutic implications. Am J Psychiatry. 2008; 165(7):830-843.
9. Winkelman J. Schizophrenia, obesity, and obstructive sleep apnea. J Clin Psychiatry. 2001;62(1):8-11.
10. Partinen M, Hublin C. Epidemiology of sleep disorders. Philadelphia, PA: Elsevier Saunders; 2005.
11. Valderas JM, Starfield B, Sibbald B, et al. Defining comorbidity: implications for understanding health and health services. Ann Fam Med. 2009;7(4):357-363.
12. Romero-Corral A, Caples SM, Lopez-Jimenez F, et al. Interactions between obesity and obstructive sleep apnea: implications for treatment. Chest. 2010;137(3):711-719.
13. Villareal DT, Apovian CM, Kushner RF, et al. Obesity in older adults: technical review and position statement of the American Society for Nutrition and NAASO, The Obesity Society. Obes Res. 2005;13(11):1849-1863.
14. Pamidi S, Aronsohn RS, Tasali E. Obstructive sleep apnea: role in the risk and severity of diabetes. Best Pract Res Clin Endocrinol Metab. 2010;24(5):703-715.
15. Malhotra A, Loscalzo J. Sleep and cardiovascular disease: an overview. Prog Cardiovasc Dis. 2009;51(4):279-284.
16. Bradley TD, Floras JS. Obstructive sleep apnoea and its cardiovascular consequences. Lancet. 2009;373(9657):82-93.
17. Chesson A, Berry R, Pack A. Practice parameters for the use of portable monitoring devices in the investigation of suspected obstructive sleep apnea in adults. Sleep. 2003; 26(7):907-913.
18. Pittman S, Ayas N, MacDonald M, et al. Using a wrist-worn device based on peripheral arterial tonometry to diagnose obstructive sleep apnea: in-laboratory and ambulatory validation. Sleep. 2004;27(1):923-933.
19. Ballester E, Badia J, Hernandez L, et al. Evidence of the effectiveness of continuous positive airway pressure in the treatment of sleep apnea/hypopnea syndrome. Am J Respir Crit Care Med. 1999;159:495-501.
20. Jenkinson D, Davies J, Mullins R, et al. Comparison of therapeutic and subtherapeutic nasal continuous positive airway pressure for obstructive sleep apnoea: a randomised prospective parallel trial. Lancet. 1999;353:2100-2105.

Dissecting melancholia with evidence-based biomarker tools
For more than 50 years, depression has been studied, and understood, as a deficiency of specific neurotransmitters in the brain—namely dopamine, norepinephrine, and serotonin. Treatments for depression have been engineered to increase the release, or block the degradation, of these neurotransmitters within the synaptic cleft. Although a large body of evidence supports involvement of dopamine, norepinephrine, and serotonin in the pathophysiology of depression, the observation that pharmacotherapy is able to induce remission only in <50% of patients1 has prompted researchers to look beyond neurotransmitters for an understanding of depressive disorders (Table 1).

Today, theories of depression focus more on differences in neuron density in various regions of the brain; the effect of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of proinflammatory mediators evoked during the stress response (Box,2,3). These theories should not be viewed as separate entities because they are highly interconnected. Integrating them provides for a more expansive understanding of the pathophysiology of depression and biomarkers that are involved (Table 2).


In this article, we:
- integrate the large body of evidence supporting the contribution of the above variables to the onset and persistence of depression
- propose a possible risk stratification model
- explore possibilities for treatment.
The stress response: How does it affect the brain?
Stress initiates a cascade of events in the brain and peripheral systems that enable an organism to cope with, and adapt to, new and challenging situations. That is why physiologic and behavioral responses to stress generally are considered beneficial to survival.
When stress is maintained for a long period, both brain and body are harmed because target cells undergo prolonged exposure to physiologic stress mediators. For example, Woolley and Gould4 exposed rats to varying durations of glucocorticoids and observed that treating animals with corticosterone injection for 21 days induced neuronal atrophy in the hippocampus and prefrontal cortex and increased release of proinflammatory cytokines from astrocytes within the limbic system. Stressful experiences are believed to be closely associated with development of psychological alterations and, thus, neuropsychiatric disorders.5 To go further: Chronic stress is believed to be the leading cause of depression.
When the brain perceives an external threat, the stress response is called into action. The amygdala, part of the primitive limbic system, is the primary area of the brain responsible for triggering the stress response,6 signaling the hypothalamus to release corticotropin-releasing hormone (CRH) to the anterior pituitary gland, which, in turn releases adrenocorticotropic hormone to the adrenal glands (Figure 1).7 The adrenal glands are responsible for releasing glucocorticoids, which, because of their lipophilic nature, can cross the blood-brain barrier and are found in higher levels in the cerebrospinal fluid (CSF) of depressed persons.7
Once in the brain, glucocorticoids can be irreversibly degraded in the cytosol by the enzyme 11-β hydroxysteroid dehydrogenase type 2, a potential target for treating depression, or can bind to the glucocorticoid receptor (GR). Results of a research study of the role of cortisol in suppression of proinflammatory cytokine signaling activity in rainbow trout hepatocytes suggest a negative feedback loop for GR gene regulation during stress.8
Because this auto-regulation is a crucial step in the physiological stress response, the idea of the GR as an important biomarker in depression has gained popularity. In humans, when the GR binds to glucocorticoids that are released from the adrenal cortex during the stress response, the activated GR-cortisol complex represses expression of proinflammatory proteins in astrocytes and microglial cells and in all cells in the periphery before they are transcribed into proteins.9 The GR also has been shown to modulate neurogenesis.8 Repeated stress that persists over a long period leads to GR resistance, thereby reducing inhibition of production of proinflammatory cytokines.
Exposure to stress for >21 days leads to overactivity of the HPA axis and GR resistance,10 which decreases suppression of proinflammatory cytokines. There is evidence that proinflammatory cytokines, tumor necrosis factor-α, and interleukin-6 further induce GR receptor resistance by preventing the cortisol-GR receptor complex from entering cell nuclei and decreasing binding to DNA within the nuclei.11 Dexamethasone, a GR agonist, has been implicated in research studies for potential re-regulation of the HPA axis in depressed persons.12
Nerve cell death in the hippocampus
Studies showing reduced hippocampal volume in unipolar depression and a correlation between the number of episodes and a consequence of untreated depression and studies suggesting that treatment can stop or reduce shrinkage,13 and recent findings of rapid neurogenesis in hippocampi in response to ketamine, brings our focus to hippocampus in depression.
The greatest density of GRs is found in the hippocampus, which is closely associated with the limbic system.7 Therefore, the hippocampus is sensitive to increases in glucocorticoids in the brain and plays a crucial role in regulation of the HPA axis.
Evidence shows that in chronic stress exposure (≥21 days), nerve cells in the hippocampus begin to atrophy and can no longer provide negative feedback inhibition to the hypothalamus, causing HPA axis dysregulation and uncontrolled release of glucocorticoids into the bloodstream and CSF.2 In patients with Cushing syndrome, who produce abnormally high levels of glucocorticoid, the incidence of depression is as high as 50%.14 Similarly, patients treated with glucocorticoids such as prednisone often experience psychiatric symptoms, the most common being depression. Gould found that partial adrenalectomy increased hippocampal neurogenesis in rat brains, indicating the beneficial effect of stress hormone antagonism.4 CRH antagonists are being looked at as a promising and less invasive treatment option for depression.
Focus has been diverted to the role of the hippocampus in depression because of its ability to regenerate throughout adulthood, leading potentially to a re-regulation of the HPA axis and subsiding of the stress response, which is universally believed to be the primary precipitating factor in depression onset. Rats require 10 to 21 days of rest to recover from the effects of chronic (21 days) administration of glucocorticoids.15 If this proves to be a directly proportional relationship, then rats would need an estimated 120 days to recover from 6 months of constant glucocorticoid exposure. Considering that the same is true for humans, current depression treatment programs, which average 6 weeks, are not long enough for adequate recovery.
Antidepressants such as selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and tricyclics stimulate neurogenesis in the hippocampus via increases in brain-derived neurotrophic factor (BDNF), suggesting that these neurotransmitters play an important role depression.16
Repetitive transcranial magnetic stimulation (rTMS), a noninvasive neuromodulation therapy approved to treat major depression, delivers brief magnetic pulses to the limbic structures. Treatment facilitates focal stimulation, rapidly applying electrical charges to the cortical neurons. TMS targets prefrontal circuits of the brain that are underactive during depressive episodes. Recent animal studies have suggested that bromodeoxyuridine (BrdU)-positive cells (newborn cells) are increased significantly in the dentate gyrus, in turn suggesting that hippocampal neurogenesis might be involved in the antidepressant effects of chronic rTMS.17 Although the underlying therapeutic mechanisms of rTMS treatment of depression remain unclear, it appears that hippocampal neurogenesis might be required to produce the effects of antidepressant treatments, including drugs and electroconvulsive therapy.17
Selective ‘shunting’ of energy occurs during the stress response
Hormones released from the adrenal glands during stress divert glucose to exercising muscles and the brain’s limbic system, which are involved in the fight-or-flight response.18 However, metabolic functions and areas of the brain that are not involved in the stress response, such as the cerebral cortex and hippocampus, are deprived of energy as a consequence of this innate selective shunting (Figure 2).19
Positron-emission tomography (PET) scanning of the resting brain shows that components of the cerebral cortex (prefrontal cortex, hippocampus, striatum) and areas connecting the cerebral cortex to the limbic system exhibit the most energy consumption in the brain during rest (Figure 3).20 PET studies also show that neuronal connections within these energy-demanding areas atrophy more rapidly than in any other area of the brain when their energy supply is reduced or cut off.6
When the supply of oxygen and glucose to certain areas of the brain is reduced—such as in traumatic brain injury or stroke—the excitatory neurotransmitter glutamate accumulates in extracellular fluid and causes nerve-cell death.21 When a conditioned stimulus is presented during fear acquisition, functional magnetic resonance imaging (fMRI) studies of fear-conditioning have consistently reported, in the prefrontal cortex:
- a decrease in the blood oxygen level-dependent signal, below resting baseline
- a reduction in blood flow (Figure 4).22
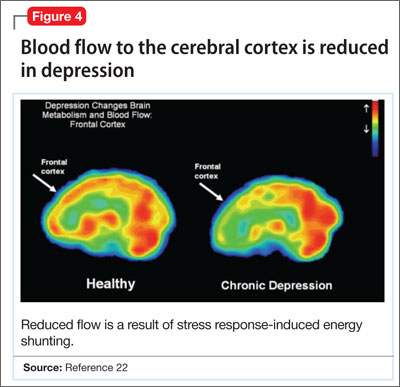
This discovery adds to evidence that demonstrates a decrease in gray-matter density in the frontal lobes as a result of glutaminergic toxicity (Figure 5).

Activation of L-glutamate, believed to play a significant role in depression and other neuropsychiatric disorders, triggers calcium-dependent intracellular responses that “excite cells to death,” so to speak—thereby causing nerve-cell apoptosis and a reduction in synaptic connections between different areas of the brain responsible for learning and memory.23 Malfunction of these synaptic connections is thought to be partially responsible for depression and other psychiatric disorders.
Excessive activation of N-methyl-d-asparate (NMDA) receptors is thought to be the underlying mechanism that leads to neuronal cell death in glutaminergic toxicity. Therefore, NMDA receptor proteins have become a target in treating neurodegenerative psychiatric illnesses. There is more than one type of NMDA receptor; some of them are excitatory, others are inhibitory. Four compounds have presented as therapeutic candidates for inhibition of NMDA receptor functioning and treatment of depression: those that inhibit glutamate binding, those that block the ion channel, and those that inhibit receptor binding to the terminal regulatory domain.24
Regrettably, these chemical compounds are not receptor-selective, but small structural modifications of these NMDA receptors have been found and lead to significant changes in potency and selectivity. This should serve as a unique starting point for developing highly specific NMDA receptor modulator agents for a variety of neuropsychiatric and neurological conditions. GLYX-13, a derivative of ketamine (an NMDA receptor antagonist), has been implicated for use in treating depression. It has been tested on 2 large phase-II study groups.25
Neuronal circuitry of depression is altered by prolonged stress
Symptoms of depression can be explained by the anatomical circuit shown in Figure 6.15,20 Impaired concentration, diminished ability to process new information, and decline in memory function are associated with decreased nerve density in the hippocampus, which plays a key role in learning, memory, and encoding of emotionally relevant data into memory.26 The hippocampus interacts with the amygdala to provide input about the context in which stimuli occur.
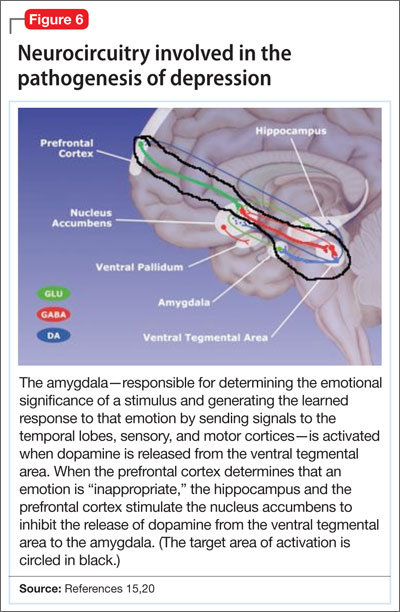
Depressed people often demonstrate impulsivity and have difficulty controlling expression of emotions—traits that are attributed to increased neuronal density in the amygdala and insula, which has been illustrated in PET scans and voxel-based morphometry in depressed patients.27 These brain areas are implicated in subjective emotional experience, processing of emotional reactions, and impulsive decision-making. The amygdala is normally highly regulated by the prefrontal cortex, which uses rational judgment to interpret stimuli and regulate the expression of emotion.
A study involving a facial expression processing task demonstrated reduced connectivity between the amygdala and prefrontal cortex and increased functional connectivity among the amygdala, hippocampus, and caudate-putamen in depressed patients.24 And in a study that measured white matter conduction in various brain areas in depressed patients, the greatest reduction was found in areas connecting the limbic system to the prefrontal cortex and hippocampus—believed to be caused by stress response-induced ischemic glutaminergic neuroapoptosis.21 Such neuroapoptosis might lead to irrational interpretation of stimuli, unchecked expression of emotion, and impulsive thoughts and behavior that are often present in depression and other mood disorders.
Deep brain stimulation (DBS), in which electrodes are implanted in the brain, has proved effective at increasing synaptic connections between the prefrontal cortex and the limbic system when electrodes are placed appropriately.28 Patients with refractory depression who are treated with DBS show increased gray-matter density and functional activity in the prefrontal cortex, hippocampus, and fronto-limbic connections.29 DBS also increases neurotransmission of dopamine, serotonin, and norepinephrine within the fronto-limbic circuitry.30
Identifying risk factors for depression
Genetic risk factors. Forty percent of patients with depression have a first-degree relative with depression, suggesting a strong genetic component.10 Inherited differences in hippocampal volume, synaptic connections between the prefrontal cortex and amygdala, γ-aminobutyric acid (GABA)/glutamate balance, BDNF neurotransmitter receptors, and anatomic positioning of the limbic system in relation to other brain structures might account for the heritability of psychiatric disorders such as depression.
Evidence has been consistent that hippocampal volume is diminished in the brain of depressed persons. However, there is no prospective cohort study to determine whether people who have lower gray-matter hippocampal density or volume, or both, before depression onset develop symptoms later in life. There also is no study to determine the percentage of people who have lower-than-average hippocampal gray-matter density or volume and who have a first-degree relative with depression. Such studies would yield valuable information about anatomic variables that increase the risk of depression.
It has been proposed that low GABA function is an inherited biomarker for depression. Bjork and co-workers found a lower plasma level of GABA in depressed subjects and in their first-degree relatives, confirming that GABAergic tone might be under genetic control.11 Genetic loci studies in mice have linked depressive-like behavior to GABAergic loci on chromosomes 8 and 11, encoding alpha 1, alpha 6, and gamma subunits of GABAA receptors.23
A recent study in humans showed that severe, treatment-resistant depression with anxiety was linked to a mutation in the B1 subunit of the GABAA receptor. Positive genetic associations were found between polymorphism in human GABAA receptor subunit genes.11
GABA metabolizing enzymes also can be considered biological modifiers of depression. For example:
- GABA uptake and metabolism is controlled by the enzyme glutamic acid decarboxylase (GAD); depression has been found to be associated with a polymorphism in the GAD67 gene encoding an isoform of GAD.11
- GABA transaminase (GABA-T) is another key enzyme in GABA turnover.31 It catabolizes GABA.
We can conclude that, to a high degree, depression depends on GABA production and metabolism.
A variant in the human BDNF gene, in which valine is substituted for methionine in position 66 of the pro-domain of the BDNF protein, is associated with
- a decrease in the production of BDNF
- increased susceptibility to neuropsychiatric disorders, including depression, anxiety disorder, and bipolar disorder (Figure 7).32
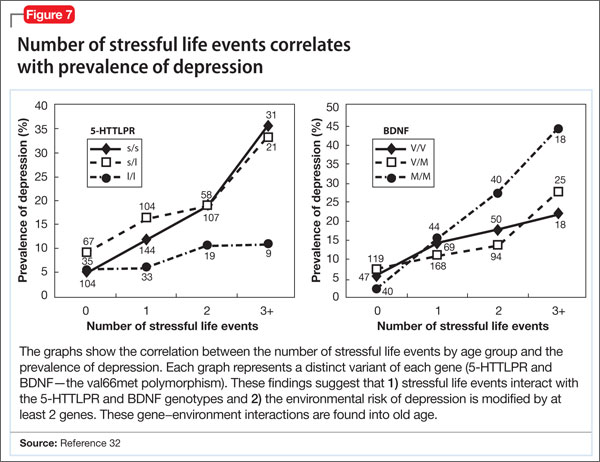
People with the MM allele have been found to have a small hippocampal neuronal density and poor hippocampus-dependent memory function in neuroimaging studies.23 They also displayed diminished ventromedial prefrontal cortex volume and presented with aversive memory extinction deficit (ie, “holding grudges”).
Another neurotrophic factor, vascular endothelial growth factor (VEGF), is a survival factor for endothelial cells and neurons and a modulator of synaptic transmission. Understanding the molecular and cellular specificity of antidepressant-induced VEGF will be critical to determine its potential as a therapeutic target in depression.33 Delineating the relationship between VEGF and depression has, ultimately, the potential to shed light on the still elusive neural mechanisms that underlie the pathophysiology of depression and the mechanisms by which antidepressants exert their effects.34
Genetic polymorphisms in monoamine receptors (5-HT2A), transporters (SERTPR, 5-HTTLPR, STin2, rs25531, SLC6A4), and regulatory enzymes should not be overlooked.35 There is reproducible evidence that variability in these polymorphisms are associated with variability in:
- vulnerability to depression
- the response to treatment with existing antidepressant medications.1
Most studies that look at changes in neuronal circuitry focus on the integrity of synaptic connections between the frontal cortex and limbic system; few of them have closely examined the importance of the anatomic proximity of the 2 regions. It might be that having an amygdala that is relatively closer to the frontal cortex and the hippocampus reduces a person’s risk of depression, and vice versa. This association needs to be investigated further with imaging studies.
Environmental risk factors. The brain is thought to be plastic until age 30.5 Plasticity diminishes with age after age 7—except for the hippocampus, which can regenerate throughout life.36 Early life experiences play an important role in forming synaptic connections between the frontal cortex and the limbic system, through a process known as fear conditioning.
Children learn early in life which stimuli are to be perceived as threatening or aversive and how to respond to best preserves their safety and internal sense of well-being. Those who grow up in a hostile environment learn to perceive more stimuli as threatening than children who grow up in a nurturing environment.32 It is possible that the amygdala is larger in children who grow up in less-than-ideal circumstances because this region is constantly being recruited—at the expense of the more rational frontal cortex.
Evidence suggests that these conditions reduce hippocampal neurogenesis37:
- increasing age
- substance abuse (opiates and methamphetamines)
- inadequate housing
- minimal physical activity
- little opportunity for social stimulation
- minimal learning experience.
Bottom Line
Depression has been understood as a neurotransmitter deficiency in the brain; treatments were engineered to increase release, or block degradation, of those neurotransmitters. Novel theories—all interconnected—of the neuroanatomical pathophysiology of depression focus more on differences in neuron density in the brain; effects of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of pro-inflammatory mediators evoked during the stress response.
Related Resources
- Fuchs E. Neurogenesis in the adult brain: is there an association with mental disorders? Eur Arch Psychiatry Clin Neurosci. 2007;257(5):247-249.
- Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004; 161(11):1957-1966.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
Anita Rao, second-year medical student, Stritch School of Medicine, Loyola University, Chicago, Illinois, assisted in the preparation of this manuscript.
1. Eley TC, Sugden K, Corsico A, et al. Gene-environment interaction analysis of serotonin system markers with adolescent depression. Mol Psychiatry. 2004;9(10):908-915.
2. Haber SN, Rauch SL. Neurocircuitry: a window into the networks underlying neuropsychiatric disease. Neuropsychopharmacology. 2010;35(1):1-3.
3. Frodl T, Bokde AL, Scheuerecker J, et al. Functional connectivity bias of the orbitofrontal cortex in drug-free patients with major depression. Biol Psychiatry. 2010; 67(2):161-167.
4. Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531(1-2): 225-231.
5. Heim C, Nemeroff CB. The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol Psychiatry. 1999;46(11):1509-1522.
6. Isgor C, Kabbaj M, Akil H, et al. Delayed effects of chronic variable stress during peripubertal-juvenile period on hippocampal morphology and on cognitive and stress axis functions in rats. Hippocampus. 2004;14(5):636-648.
7. De Kloet ER, Vreugdenhil E, Oitzl MS, et al. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269-301.
8. Philip AM, Kim SD, Vijayan MM. Cortisol modulates the expression of cytokines and suppressors of cytokine signaling (SOCS) in rainbow trout hepatocytes. Dev Comp Immunol. 2012;38(2):360-367.
9. Coplan JD, Lydiard RB. Brain circuits in panic disorder. Biol Psychiatry. 1998;44(12):1264-1276.
10. Anisman H, Merali Z. Cytokines, stress and depressive illness: brain-immune interactions. Ann Med. 2003;35(1):2-11.
11. Crowley JJ, Lucki I. Opportunities to discover genes regulating depression and antidepressant response from rodent behavioral genetics. Curr Pharm Des. 2005;11(2):157-169.
12. Covington HE 3rd, Vialou V, Nestler EJ. From synapse to nucleus: novel targets for treating depression. Neuropharmacology. 2010;58(4-5):683-693.
13. Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161(11):1957-1966.
14. Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5(12):917-930.
15. Hartley CA, Phelps EA. Changing fear: the neurocircuitry of emotion regulation. Neuropsychopharmacology. 2010;35(1): 136-146.
16. Kim DK, Lim SW, Lee S, et al. Serotonin transporter gene polymorphism and antidepressant response. Neuroreport. 2000;11(1):215-219.
17. Ueyama E, Ukai S, Ogawa A, et al, Chronic repetitive transcranial magnetic stimulation increases hippocampal neurogenesis in rats. Psychiatry Clin Neurosci. 2011; 65(1):77-81.
18. Irwin W, Anderle MJ, Abercrombie HC, et al. Amygdalar interhemispheric functional connectivity differs between the non-depressed and depressed human brain. Neuroimage. 2004;21(2):674-686.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007; 87(3):873-904.
20. Gusnard DA, Raichle ME, Raichle ME. Searching for a baseline: functional imaging and the resting human brain. Nat Rev Neurosci. 2001;2(10):685-694.
21. Hulsebosch CE, Hains BC, Crown ED, et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60(1):202-213.
22. Gottfried JA, Dolan RJ. Human orbitofrontal cortex mediates extinction learning while accessing conditioned representations of value. Nat Neurosci. 2004;7(10):1144-1152.
23 Arnone D, McKie S, Elliott R, et al. State-dependent changes in hippocampal grey matter in depression. Mol Psychiatry. 2012;1(8):1359-4184.
24. Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11(8):1169-1180.
25. Maeng S, Zarate CA Jr. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9(6):467-474.
26. Vaidya VA, Fernandes K, Jha S. Regulation of adult hippocampal neurogenesis: relevance to depression. Expert Rev Neurother. 2007;7(7):853-864.
27. Lisiecka DM, Carballedo A, Fagan AJ, et al. Altered inhibition of negative emotions in subjects at family risk of major depressive disorder. J Psychiatr Res. 2012;46(2):181-188.
28. Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651-660.
29. Levkovitz Y, Harel EV, Roth Y, et al. Deep transcranial magnetic stimulation over the prefrontal cortex: evaluation of antidepressant and cognitive effects in depressive patients. Brain Stimul. 2009;2(4):188-200.
30. Schlaepfer TE, Lieb K. Deep brain stimulation for treatment of refractory depression. Lancet. 2005;366(9495):1420-1422.
31. Astrup, J. Energy-requiring cell functions in the ischemic brain. Their critical supply and possible inhibition in protective therapy. J Neurosurg. 1982;56(4):482-497.
32. Fletcher JM. Childhood mistreatment and adolescent and young adult depression. Soc Sci Med. 2009;68(5):799-806.
33. Warner-Schmidt JL, Duman R. VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol. 2008;8(1):14-19.
34. Clark-Raymond A, Halaris A. VEGF and depression: a comprehensive assessment of clinical data. J Psychiatr Res. 2013;47(8):1080-1087.
35. Alonso R, Griebel G, Pavone G, et al. Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry. 2004;9(3):278-286.
36. Thomas RM, Peterson DA. A neurogenic theory of depression gains momentum. Mol Interv. 2003;3(8):441-444.
37. Jacobs BL. Adult brain neurogenesis and depression. Brain Behav Immun. 2002;16(5):602-609.
For more than 50 years, depression has been studied, and understood, as a deficiency of specific neurotransmitters in the brain—namely dopamine, norepinephrine, and serotonin. Treatments for depression have been engineered to increase the release, or block the degradation, of these neurotransmitters within the synaptic cleft. Although a large body of evidence supports involvement of dopamine, norepinephrine, and serotonin in the pathophysiology of depression, the observation that pharmacotherapy is able to induce remission only in <50% of patients1 has prompted researchers to look beyond neurotransmitters for an understanding of depressive disorders (Table 1).
Today, theories of depression focus more on differences in neuron density in various regions of the brain; the effect of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of proinflammatory mediators evoked during the stress response (Box,2,3). These theories should not be viewed as separate entities because they are highly interconnected. Integrating them provides for a more expansive understanding of the pathophysiology of depression and biomarkers that are involved (Table 2).
In this article, we:
- integrate the large body of evidence supporting the contribution of the above variables to the onset and persistence of depression
- propose a possible risk stratification model
- explore possibilities for treatment.
The stress response: How does it affect the brain?
Stress initiates a cascade of events in the brain and peripheral systems that enable an organism to cope with, and adapt to, new and challenging situations. That is why physiologic and behavioral responses to stress generally are considered beneficial to survival.
When stress is maintained for a long period, both brain and body are harmed because target cells undergo prolonged exposure to physiologic stress mediators. For example, Woolley and Gould4 exposed rats to varying durations of glucocorticoids and observed that treating animals with corticosterone injection for 21 days induced neuronal atrophy in the hippocampus and prefrontal cortex and increased release of proinflammatory cytokines from astrocytes within the limbic system. Stressful experiences are believed to be closely associated with development of psychological alterations and, thus, neuropsychiatric disorders.5 To go further: Chronic stress is believed to be the leading cause of depression.
When the brain perceives an external threat, the stress response is called into action. The amygdala, part of the primitive limbic system, is the primary area of the brain responsible for triggering the stress response,6 signaling the hypothalamus to release corticotropin-releasing hormone (CRH) to the anterior pituitary gland, which, in turn releases adrenocorticotropic hormone to the adrenal glands (Figure 1).7 The adrenal glands are responsible for releasing glucocorticoids, which, because of their lipophilic nature, can cross the blood-brain barrier and are found in higher levels in the cerebrospinal fluid (CSF) of depressed persons.7
Once in the brain, glucocorticoids can be irreversibly degraded in the cytosol by the enzyme 11-β hydroxysteroid dehydrogenase type 2, a potential target for treating depression, or can bind to the glucocorticoid receptor (GR). Results of a research study of the role of cortisol in suppression of proinflammatory cytokine signaling activity in rainbow trout hepatocytes suggest a negative feedback loop for GR gene regulation during stress.8
Because this auto-regulation is a crucial step in the physiological stress response, the idea of the GR as an important biomarker in depression has gained popularity. In humans, when the GR binds to glucocorticoids that are released from the adrenal cortex during the stress response, the activated GR-cortisol complex represses expression of proinflammatory proteins in astrocytes and microglial cells and in all cells in the periphery before they are transcribed into proteins.9 The GR also has been shown to modulate neurogenesis.8 Repeated stress that persists over a long period leads to GR resistance, thereby reducing inhibition of production of proinflammatory cytokines.
Exposure to stress for >21 days leads to overactivity of the HPA axis and GR resistance,10 which decreases suppression of proinflammatory cytokines. There is evidence that proinflammatory cytokines, tumor necrosis factor-α, and interleukin-6 further induce GR receptor resistance by preventing the cortisol-GR receptor complex from entering cell nuclei and decreasing binding to DNA within the nuclei.11 Dexamethasone, a GR agonist, has been implicated in research studies for potential re-regulation of the HPA axis in depressed persons.12
Nerve cell death in the hippocampus
Studies showing reduced hippocampal volume in unipolar depression and a correlation between the number of episodes and a consequence of untreated depression and studies suggesting that treatment can stop or reduce shrinkage,13 and recent findings of rapid neurogenesis in hippocampi in response to ketamine, brings our focus to hippocampus in depression.
The greatest density of GRs is found in the hippocampus, which is closely associated with the limbic system.7 Therefore, the hippocampus is sensitive to increases in glucocorticoids in the brain and plays a crucial role in regulation of the HPA axis.
Evidence shows that in chronic stress exposure (≥21 days), nerve cells in the hippocampus begin to atrophy and can no longer provide negative feedback inhibition to the hypothalamus, causing HPA axis dysregulation and uncontrolled release of glucocorticoids into the bloodstream and CSF.2 In patients with Cushing syndrome, who produce abnormally high levels of glucocorticoid, the incidence of depression is as high as 50%.14 Similarly, patients treated with glucocorticoids such as prednisone often experience psychiatric symptoms, the most common being depression. Gould found that partial adrenalectomy increased hippocampal neurogenesis in rat brains, indicating the beneficial effect of stress hormone antagonism.4 CRH antagonists are being looked at as a promising and less invasive treatment option for depression.
Focus has been diverted to the role of the hippocampus in depression because of its ability to regenerate throughout adulthood, leading potentially to a re-regulation of the HPA axis and subsiding of the stress response, which is universally believed to be the primary precipitating factor in depression onset. Rats require 10 to 21 days of rest to recover from the effects of chronic (21 days) administration of glucocorticoids.15 If this proves to be a directly proportional relationship, then rats would need an estimated 120 days to recover from 6 months of constant glucocorticoid exposure. Considering that the same is true for humans, current depression treatment programs, which average 6 weeks, are not long enough for adequate recovery.
Antidepressants such as selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and tricyclics stimulate neurogenesis in the hippocampus via increases in brain-derived neurotrophic factor (BDNF), suggesting that these neurotransmitters play an important role depression.16
Repetitive transcranial magnetic stimulation (rTMS), a noninvasive neuromodulation therapy approved to treat major depression, delivers brief magnetic pulses to the limbic structures. Treatment facilitates focal stimulation, rapidly applying electrical charges to the cortical neurons. TMS targets prefrontal circuits of the brain that are underactive during depressive episodes. Recent animal studies have suggested that bromodeoxyuridine (BrdU)-positive cells (newborn cells) are increased significantly in the dentate gyrus, in turn suggesting that hippocampal neurogenesis might be involved in the antidepressant effects of chronic rTMS.17 Although the underlying therapeutic mechanisms of rTMS treatment of depression remain unclear, it appears that hippocampal neurogenesis might be required to produce the effects of antidepressant treatments, including drugs and electroconvulsive therapy.17
Selective ‘shunting’ of energy occurs during the stress response
Hormones released from the adrenal glands during stress divert glucose to exercising muscles and the brain’s limbic system, which are involved in the fight-or-flight response.18 However, metabolic functions and areas of the brain that are not involved in the stress response, such as the cerebral cortex and hippocampus, are deprived of energy as a consequence of this innate selective shunting (Figure 2).19
Positron-emission tomography (PET) scanning of the resting brain shows that components of the cerebral cortex (prefrontal cortex, hippocampus, striatum) and areas connecting the cerebral cortex to the limbic system exhibit the most energy consumption in the brain during rest (Figure 3).20 PET studies also show that neuronal connections within these energy-demanding areas atrophy more rapidly than in any other area of the brain when their energy supply is reduced or cut off.6
When the supply of oxygen and glucose to certain areas of the brain is reduced—such as in traumatic brain injury or stroke—the excitatory neurotransmitter glutamate accumulates in extracellular fluid and causes nerve-cell death.21 When a conditioned stimulus is presented during fear acquisition, functional magnetic resonance imaging (fMRI) studies of fear-conditioning have consistently reported, in the prefrontal cortex:
- a decrease in the blood oxygen level-dependent signal, below resting baseline
- a reduction in blood flow (Figure 4).22
This discovery adds to evidence that demonstrates a decrease in gray-matter density in the frontal lobes as a result of glutaminergic toxicity (Figure 5).
Activation of L-glutamate, believed to play a significant role in depression and other neuropsychiatric disorders, triggers calcium-dependent intracellular responses that “excite cells to death,” so to speak—thereby causing nerve-cell apoptosis and a reduction in synaptic connections between different areas of the brain responsible for learning and memory.23 Malfunction of these synaptic connections is thought to be partially responsible for depression and other psychiatric disorders.
Excessive activation of N-methyl-d-asparate (NMDA) receptors is thought to be the underlying mechanism that leads to neuronal cell death in glutaminergic toxicity. Therefore, NMDA receptor proteins have become a target in treating neurodegenerative psychiatric illnesses. There is more than one type of NMDA receptor; some of them are excitatory, others are inhibitory. Four compounds have presented as therapeutic candidates for inhibition of NMDA receptor functioning and treatment of depression: those that inhibit glutamate binding, those that block the ion channel, and those that inhibit receptor binding to the terminal regulatory domain.24
Regrettably, these chemical compounds are not receptor-selective, but small structural modifications of these NMDA receptors have been found and lead to significant changes in potency and selectivity. This should serve as a unique starting point for developing highly specific NMDA receptor modulator agents for a variety of neuropsychiatric and neurological conditions. GLYX-13, a derivative of ketamine (an NMDA receptor antagonist), has been implicated for use in treating depression. It has been tested on 2 large phase-II study groups.25
Neuronal circuitry of depression is altered by prolonged stress
Symptoms of depression can be explained by the anatomical circuit shown in Figure 6.15,20 Impaired concentration, diminished ability to process new information, and decline in memory function are associated with decreased nerve density in the hippocampus, which plays a key role in learning, memory, and encoding of emotionally relevant data into memory.26 The hippocampus interacts with the amygdala to provide input about the context in which stimuli occur.
Depressed people often demonstrate impulsivity and have difficulty controlling expression of emotions—traits that are attributed to increased neuronal density in the amygdala and insula, which has been illustrated in PET scans and voxel-based morphometry in depressed patients.27 These brain areas are implicated in subjective emotional experience, processing of emotional reactions, and impulsive decision-making. The amygdala is normally highly regulated by the prefrontal cortex, which uses rational judgment to interpret stimuli and regulate the expression of emotion.
A study involving a facial expression processing task demonstrated reduced connectivity between the amygdala and prefrontal cortex and increased functional connectivity among the amygdala, hippocampus, and caudate-putamen in depressed patients.24 And in a study that measured white matter conduction in various brain areas in depressed patients, the greatest reduction was found in areas connecting the limbic system to the prefrontal cortex and hippocampus—believed to be caused by stress response-induced ischemic glutaminergic neuroapoptosis.21 Such neuroapoptosis might lead to irrational interpretation of stimuli, unchecked expression of emotion, and impulsive thoughts and behavior that are often present in depression and other mood disorders.
Deep brain stimulation (DBS), in which electrodes are implanted in the brain, has proved effective at increasing synaptic connections between the prefrontal cortex and the limbic system when electrodes are placed appropriately.28 Patients with refractory depression who are treated with DBS show increased gray-matter density and functional activity in the prefrontal cortex, hippocampus, and fronto-limbic connections.29 DBS also increases neurotransmission of dopamine, serotonin, and norepinephrine within the fronto-limbic circuitry.30
Identifying risk factors for depression
Genetic risk factors. Forty percent of patients with depression have a first-degree relative with depression, suggesting a strong genetic component.10 Inherited differences in hippocampal volume, synaptic connections between the prefrontal cortex and amygdala, γ-aminobutyric acid (GABA)/glutamate balance, BDNF neurotransmitter receptors, and anatomic positioning of the limbic system in relation to other brain structures might account for the heritability of psychiatric disorders such as depression.
Evidence has been consistent that hippocampal volume is diminished in the brain of depressed persons. However, there is no prospective cohort study to determine whether people who have lower gray-matter hippocampal density or volume, or both, before depression onset develop symptoms later in life. There also is no study to determine the percentage of people who have lower-than-average hippocampal gray-matter density or volume and who have a first-degree relative with depression. Such studies would yield valuable information about anatomic variables that increase the risk of depression.
It has been proposed that low GABA function is an inherited biomarker for depression. Bjork and co-workers found a lower plasma level of GABA in depressed subjects and in their first-degree relatives, confirming that GABAergic tone might be under genetic control.11 Genetic loci studies in mice have linked depressive-like behavior to GABAergic loci on chromosomes 8 and 11, encoding alpha 1, alpha 6, and gamma subunits of GABAA receptors.23
A recent study in humans showed that severe, treatment-resistant depression with anxiety was linked to a mutation in the B1 subunit of the GABAA receptor. Positive genetic associations were found between polymorphism in human GABAA receptor subunit genes.11
GABA metabolizing enzymes also can be considered biological modifiers of depression. For example:
- GABA uptake and metabolism is controlled by the enzyme glutamic acid decarboxylase (GAD); depression has been found to be associated with a polymorphism in the GAD67 gene encoding an isoform of GAD.11
- GABA transaminase (GABA-T) is another key enzyme in GABA turnover.31 It catabolizes GABA.
We can conclude that, to a high degree, depression depends on GABA production and metabolism.
A variant in the human BDNF gene, in which valine is substituted for methionine in position 66 of the pro-domain of the BDNF protein, is associated with
- a decrease in the production of BDNF
- increased susceptibility to neuropsychiatric disorders, including depression, anxiety disorder, and bipolar disorder (Figure 7).32
People with the MM allele have been found to have a small hippocampal neuronal density and poor hippocampus-dependent memory function in neuroimaging studies.23 They also displayed diminished ventromedial prefrontal cortex volume and presented with aversive memory extinction deficit (ie, “holding grudges”).
Another neurotrophic factor, vascular endothelial growth factor (VEGF), is a survival factor for endothelial cells and neurons and a modulator of synaptic transmission. Understanding the molecular and cellular specificity of antidepressant-induced VEGF will be critical to determine its potential as a therapeutic target in depression.33 Delineating the relationship between VEGF and depression has, ultimately, the potential to shed light on the still elusive neural mechanisms that underlie the pathophysiology of depression and the mechanisms by which antidepressants exert their effects.34
Genetic polymorphisms in monoamine receptors (5-HT2A), transporters (SERTPR, 5-HTTLPR, STin2, rs25531, SLC6A4), and regulatory enzymes should not be overlooked.35 There is reproducible evidence that variability in these polymorphisms are associated with variability in:
- vulnerability to depression
- the response to treatment with existing antidepressant medications.1
Most studies that look at changes in neuronal circuitry focus on the integrity of synaptic connections between the frontal cortex and limbic system; few of them have closely examined the importance of the anatomic proximity of the 2 regions. It might be that having an amygdala that is relatively closer to the frontal cortex and the hippocampus reduces a person’s risk of depression, and vice versa. This association needs to be investigated further with imaging studies.
Environmental risk factors. The brain is thought to be plastic until age 30.5 Plasticity diminishes with age after age 7—except for the hippocampus, which can regenerate throughout life.36 Early life experiences play an important role in forming synaptic connections between the frontal cortex and the limbic system, through a process known as fear conditioning.
Children learn early in life which stimuli are to be perceived as threatening or aversive and how to respond to best preserves their safety and internal sense of well-being. Those who grow up in a hostile environment learn to perceive more stimuli as threatening than children who grow up in a nurturing environment.32 It is possible that the amygdala is larger in children who grow up in less-than-ideal circumstances because this region is constantly being recruited—at the expense of the more rational frontal cortex.
Evidence suggests that these conditions reduce hippocampal neurogenesis37:
- increasing age
- substance abuse (opiates and methamphetamines)
- inadequate housing
- minimal physical activity
- little opportunity for social stimulation
- minimal learning experience.
Bottom Line
Depression has been understood as a neurotransmitter deficiency in the brain; treatments were engineered to increase release, or block degradation, of those neurotransmitters. Novel theories—all interconnected—of the neuroanatomical pathophysiology of depression focus more on differences in neuron density in the brain; effects of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of pro-inflammatory mediators evoked during the stress response.
Related Resources
- Fuchs E. Neurogenesis in the adult brain: is there an association with mental disorders? Eur Arch Psychiatry Clin Neurosci. 2007;257(5):247-249.
- Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004; 161(11):1957-1966.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
Anita Rao, second-year medical student, Stritch School of Medicine, Loyola University, Chicago, Illinois, assisted in the preparation of this manuscript.
For more than 50 years, depression has been studied, and understood, as a deficiency of specific neurotransmitters in the brain—namely dopamine, norepinephrine, and serotonin. Treatments for depression have been engineered to increase the release, or block the degradation, of these neurotransmitters within the synaptic cleft. Although a large body of evidence supports involvement of dopamine, norepinephrine, and serotonin in the pathophysiology of depression, the observation that pharmacotherapy is able to induce remission only in <50% of patients1 has prompted researchers to look beyond neurotransmitters for an understanding of depressive disorders (Table 1).
Today, theories of depression focus more on differences in neuron density in various regions of the brain; the effect of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of proinflammatory mediators evoked during the stress response (Box,2,3). These theories should not be viewed as separate entities because they are highly interconnected. Integrating them provides for a more expansive understanding of the pathophysiology of depression and biomarkers that are involved (Table 2).
In this article, we:
- integrate the large body of evidence supporting the contribution of the above variables to the onset and persistence of depression
- propose a possible risk stratification model
- explore possibilities for treatment.
The stress response: How does it affect the brain?
Stress initiates a cascade of events in the brain and peripheral systems that enable an organism to cope with, and adapt to, new and challenging situations. That is why physiologic and behavioral responses to stress generally are considered beneficial to survival.
When stress is maintained for a long period, both brain and body are harmed because target cells undergo prolonged exposure to physiologic stress mediators. For example, Woolley and Gould4 exposed rats to varying durations of glucocorticoids and observed that treating animals with corticosterone injection for 21 days induced neuronal atrophy in the hippocampus and prefrontal cortex and increased release of proinflammatory cytokines from astrocytes within the limbic system. Stressful experiences are believed to be closely associated with development of psychological alterations and, thus, neuropsychiatric disorders.5 To go further: Chronic stress is believed to be the leading cause of depression.
When the brain perceives an external threat, the stress response is called into action. The amygdala, part of the primitive limbic system, is the primary area of the brain responsible for triggering the stress response,6 signaling the hypothalamus to release corticotropin-releasing hormone (CRH) to the anterior pituitary gland, which, in turn releases adrenocorticotropic hormone to the adrenal glands (Figure 1).7 The adrenal glands are responsible for releasing glucocorticoids, which, because of their lipophilic nature, can cross the blood-brain barrier and are found in higher levels in the cerebrospinal fluid (CSF) of depressed persons.7
Once in the brain, glucocorticoids can be irreversibly degraded in the cytosol by the enzyme 11-β hydroxysteroid dehydrogenase type 2, a potential target for treating depression, or can bind to the glucocorticoid receptor (GR). Results of a research study of the role of cortisol in suppression of proinflammatory cytokine signaling activity in rainbow trout hepatocytes suggest a negative feedback loop for GR gene regulation during stress.8
Because this auto-regulation is a crucial step in the physiological stress response, the idea of the GR as an important biomarker in depression has gained popularity. In humans, when the GR binds to glucocorticoids that are released from the adrenal cortex during the stress response, the activated GR-cortisol complex represses expression of proinflammatory proteins in astrocytes and microglial cells and in all cells in the periphery before they are transcribed into proteins.9 The GR also has been shown to modulate neurogenesis.8 Repeated stress that persists over a long period leads to GR resistance, thereby reducing inhibition of production of proinflammatory cytokines.
Exposure to stress for >21 days leads to overactivity of the HPA axis and GR resistance,10 which decreases suppression of proinflammatory cytokines. There is evidence that proinflammatory cytokines, tumor necrosis factor-α, and interleukin-6 further induce GR receptor resistance by preventing the cortisol-GR receptor complex from entering cell nuclei and decreasing binding to DNA within the nuclei.11 Dexamethasone, a GR agonist, has been implicated in research studies for potential re-regulation of the HPA axis in depressed persons.12
Nerve cell death in the hippocampus
Studies showing reduced hippocampal volume in unipolar depression and a correlation between the number of episodes and a consequence of untreated depression and studies suggesting that treatment can stop or reduce shrinkage,13 and recent findings of rapid neurogenesis in hippocampi in response to ketamine, brings our focus to hippocampus in depression.
The greatest density of GRs is found in the hippocampus, which is closely associated with the limbic system.7 Therefore, the hippocampus is sensitive to increases in glucocorticoids in the brain and plays a crucial role in regulation of the HPA axis.
Evidence shows that in chronic stress exposure (≥21 days), nerve cells in the hippocampus begin to atrophy and can no longer provide negative feedback inhibition to the hypothalamus, causing HPA axis dysregulation and uncontrolled release of glucocorticoids into the bloodstream and CSF.2 In patients with Cushing syndrome, who produce abnormally high levels of glucocorticoid, the incidence of depression is as high as 50%.14 Similarly, patients treated with glucocorticoids such as prednisone often experience psychiatric symptoms, the most common being depression. Gould found that partial adrenalectomy increased hippocampal neurogenesis in rat brains, indicating the beneficial effect of stress hormone antagonism.4 CRH antagonists are being looked at as a promising and less invasive treatment option for depression.
Focus has been diverted to the role of the hippocampus in depression because of its ability to regenerate throughout adulthood, leading potentially to a re-regulation of the HPA axis and subsiding of the stress response, which is universally believed to be the primary precipitating factor in depression onset. Rats require 10 to 21 days of rest to recover from the effects of chronic (21 days) administration of glucocorticoids.15 If this proves to be a directly proportional relationship, then rats would need an estimated 120 days to recover from 6 months of constant glucocorticoid exposure. Considering that the same is true for humans, current depression treatment programs, which average 6 weeks, are not long enough for adequate recovery.
Antidepressants such as selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and tricyclics stimulate neurogenesis in the hippocampus via increases in brain-derived neurotrophic factor (BDNF), suggesting that these neurotransmitters play an important role depression.16
Repetitive transcranial magnetic stimulation (rTMS), a noninvasive neuromodulation therapy approved to treat major depression, delivers brief magnetic pulses to the limbic structures. Treatment facilitates focal stimulation, rapidly applying electrical charges to the cortical neurons. TMS targets prefrontal circuits of the brain that are underactive during depressive episodes. Recent animal studies have suggested that bromodeoxyuridine (BrdU)-positive cells (newborn cells) are increased significantly in the dentate gyrus, in turn suggesting that hippocampal neurogenesis might be involved in the antidepressant effects of chronic rTMS.17 Although the underlying therapeutic mechanisms of rTMS treatment of depression remain unclear, it appears that hippocampal neurogenesis might be required to produce the effects of antidepressant treatments, including drugs and electroconvulsive therapy.17
Selective ‘shunting’ of energy occurs during the stress response
Hormones released from the adrenal glands during stress divert glucose to exercising muscles and the brain’s limbic system, which are involved in the fight-or-flight response.18 However, metabolic functions and areas of the brain that are not involved in the stress response, such as the cerebral cortex and hippocampus, are deprived of energy as a consequence of this innate selective shunting (Figure 2).19
Positron-emission tomography (PET) scanning of the resting brain shows that components of the cerebral cortex (prefrontal cortex, hippocampus, striatum) and areas connecting the cerebral cortex to the limbic system exhibit the most energy consumption in the brain during rest (Figure 3).20 PET studies also show that neuronal connections within these energy-demanding areas atrophy more rapidly than in any other area of the brain when their energy supply is reduced or cut off.6
When the supply of oxygen and glucose to certain areas of the brain is reduced—such as in traumatic brain injury or stroke—the excitatory neurotransmitter glutamate accumulates in extracellular fluid and causes nerve-cell death.21 When a conditioned stimulus is presented during fear acquisition, functional magnetic resonance imaging (fMRI) studies of fear-conditioning have consistently reported, in the prefrontal cortex:
- a decrease in the blood oxygen level-dependent signal, below resting baseline
- a reduction in blood flow (Figure 4).22
This discovery adds to evidence that demonstrates a decrease in gray-matter density in the frontal lobes as a result of glutaminergic toxicity (Figure 5).
Activation of L-glutamate, believed to play a significant role in depression and other neuropsychiatric disorders, triggers calcium-dependent intracellular responses that “excite cells to death,” so to speak—thereby causing nerve-cell apoptosis and a reduction in synaptic connections between different areas of the brain responsible for learning and memory.23 Malfunction of these synaptic connections is thought to be partially responsible for depression and other psychiatric disorders.
Excessive activation of N-methyl-d-asparate (NMDA) receptors is thought to be the underlying mechanism that leads to neuronal cell death in glutaminergic toxicity. Therefore, NMDA receptor proteins have become a target in treating neurodegenerative psychiatric illnesses. There is more than one type of NMDA receptor; some of them are excitatory, others are inhibitory. Four compounds have presented as therapeutic candidates for inhibition of NMDA receptor functioning and treatment of depression: those that inhibit glutamate binding, those that block the ion channel, and those that inhibit receptor binding to the terminal regulatory domain.24
Regrettably, these chemical compounds are not receptor-selective, but small structural modifications of these NMDA receptors have been found and lead to significant changes in potency and selectivity. This should serve as a unique starting point for developing highly specific NMDA receptor modulator agents for a variety of neuropsychiatric and neurological conditions. GLYX-13, a derivative of ketamine (an NMDA receptor antagonist), has been implicated for use in treating depression. It has been tested on 2 large phase-II study groups.25
Neuronal circuitry of depression is altered by prolonged stress
Symptoms of depression can be explained by the anatomical circuit shown in Figure 6.15,20 Impaired concentration, diminished ability to process new information, and decline in memory function are associated with decreased nerve density in the hippocampus, which plays a key role in learning, memory, and encoding of emotionally relevant data into memory.26 The hippocampus interacts with the amygdala to provide input about the context in which stimuli occur.
Depressed people often demonstrate impulsivity and have difficulty controlling expression of emotions—traits that are attributed to increased neuronal density in the amygdala and insula, which has been illustrated in PET scans and voxel-based morphometry in depressed patients.27 These brain areas are implicated in subjective emotional experience, processing of emotional reactions, and impulsive decision-making. The amygdala is normally highly regulated by the prefrontal cortex, which uses rational judgment to interpret stimuli and regulate the expression of emotion.
A study involving a facial expression processing task demonstrated reduced connectivity between the amygdala and prefrontal cortex and increased functional connectivity among the amygdala, hippocampus, and caudate-putamen in depressed patients.24 And in a study that measured white matter conduction in various brain areas in depressed patients, the greatest reduction was found in areas connecting the limbic system to the prefrontal cortex and hippocampus—believed to be caused by stress response-induced ischemic glutaminergic neuroapoptosis.21 Such neuroapoptosis might lead to irrational interpretation of stimuli, unchecked expression of emotion, and impulsive thoughts and behavior that are often present in depression and other mood disorders.
Deep brain stimulation (DBS), in which electrodes are implanted in the brain, has proved effective at increasing synaptic connections between the prefrontal cortex and the limbic system when electrodes are placed appropriately.28 Patients with refractory depression who are treated with DBS show increased gray-matter density and functional activity in the prefrontal cortex, hippocampus, and fronto-limbic connections.29 DBS also increases neurotransmission of dopamine, serotonin, and norepinephrine within the fronto-limbic circuitry.30
Identifying risk factors for depression
Genetic risk factors. Forty percent of patients with depression have a first-degree relative with depression, suggesting a strong genetic component.10 Inherited differences in hippocampal volume, synaptic connections between the prefrontal cortex and amygdala, γ-aminobutyric acid (GABA)/glutamate balance, BDNF neurotransmitter receptors, and anatomic positioning of the limbic system in relation to other brain structures might account for the heritability of psychiatric disorders such as depression.
Evidence has been consistent that hippocampal volume is diminished in the brain of depressed persons. However, there is no prospective cohort study to determine whether people who have lower gray-matter hippocampal density or volume, or both, before depression onset develop symptoms later in life. There also is no study to determine the percentage of people who have lower-than-average hippocampal gray-matter density or volume and who have a first-degree relative with depression. Such studies would yield valuable information about anatomic variables that increase the risk of depression.
It has been proposed that low GABA function is an inherited biomarker for depression. Bjork and co-workers found a lower plasma level of GABA in depressed subjects and in their first-degree relatives, confirming that GABAergic tone might be under genetic control.11 Genetic loci studies in mice have linked depressive-like behavior to GABAergic loci on chromosomes 8 and 11, encoding alpha 1, alpha 6, and gamma subunits of GABAA receptors.23
A recent study in humans showed that severe, treatment-resistant depression with anxiety was linked to a mutation in the B1 subunit of the GABAA receptor. Positive genetic associations were found between polymorphism in human GABAA receptor subunit genes.11
GABA metabolizing enzymes also can be considered biological modifiers of depression. For example:
- GABA uptake and metabolism is controlled by the enzyme glutamic acid decarboxylase (GAD); depression has been found to be associated with a polymorphism in the GAD67 gene encoding an isoform of GAD.11
- GABA transaminase (GABA-T) is another key enzyme in GABA turnover.31 It catabolizes GABA.
We can conclude that, to a high degree, depression depends on GABA production and metabolism.
A variant in the human BDNF gene, in which valine is substituted for methionine in position 66 of the pro-domain of the BDNF protein, is associated with
- a decrease in the production of BDNF
- increased susceptibility to neuropsychiatric disorders, including depression, anxiety disorder, and bipolar disorder (Figure 7).32
People with the MM allele have been found to have a small hippocampal neuronal density and poor hippocampus-dependent memory function in neuroimaging studies.23 They also displayed diminished ventromedial prefrontal cortex volume and presented with aversive memory extinction deficit (ie, “holding grudges”).
Another neurotrophic factor, vascular endothelial growth factor (VEGF), is a survival factor for endothelial cells and neurons and a modulator of synaptic transmission. Understanding the molecular and cellular specificity of antidepressant-induced VEGF will be critical to determine its potential as a therapeutic target in depression.33 Delineating the relationship between VEGF and depression has, ultimately, the potential to shed light on the still elusive neural mechanisms that underlie the pathophysiology of depression and the mechanisms by which antidepressants exert their effects.34
Genetic polymorphisms in monoamine receptors (5-HT2A), transporters (SERTPR, 5-HTTLPR, STin2, rs25531, SLC6A4), and regulatory enzymes should not be overlooked.35 There is reproducible evidence that variability in these polymorphisms are associated with variability in:
- vulnerability to depression
- the response to treatment with existing antidepressant medications.1
Most studies that look at changes in neuronal circuitry focus on the integrity of synaptic connections between the frontal cortex and limbic system; few of them have closely examined the importance of the anatomic proximity of the 2 regions. It might be that having an amygdala that is relatively closer to the frontal cortex and the hippocampus reduces a person’s risk of depression, and vice versa. This association needs to be investigated further with imaging studies.
Environmental risk factors. The brain is thought to be plastic until age 30.5 Plasticity diminishes with age after age 7—except for the hippocampus, which can regenerate throughout life.36 Early life experiences play an important role in forming synaptic connections between the frontal cortex and the limbic system, through a process known as fear conditioning.
Children learn early in life which stimuli are to be perceived as threatening or aversive and how to respond to best preserves their safety and internal sense of well-being. Those who grow up in a hostile environment learn to perceive more stimuli as threatening than children who grow up in a nurturing environment.32 It is possible that the amygdala is larger in children who grow up in less-than-ideal circumstances because this region is constantly being recruited—at the expense of the more rational frontal cortex.
Evidence suggests that these conditions reduce hippocampal neurogenesis37:
- increasing age
- substance abuse (opiates and methamphetamines)
- inadequate housing
- minimal physical activity
- little opportunity for social stimulation
- minimal learning experience.
Bottom Line
Depression has been understood as a neurotransmitter deficiency in the brain; treatments were engineered to increase release, or block degradation, of those neurotransmitters. Novel theories—all interconnected—of the neuroanatomical pathophysiology of depression focus more on differences in neuron density in the brain; effects of stress on neurogenesis and neuronal cell apoptosis; alterations in feedback pathways connecting the pre-frontal cortex to the limbic system; and the role of pro-inflammatory mediators evoked during the stress response.
Related Resources
- Fuchs E. Neurogenesis in the adult brain: is there an association with mental disorders? Eur Arch Psychiatry Clin Neurosci. 2007;257(5):247-249.
- Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004; 161(11):1957-1966.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
Anita Rao, second-year medical student, Stritch School of Medicine, Loyola University, Chicago, Illinois, assisted in the preparation of this manuscript.
1. Eley TC, Sugden K, Corsico A, et al. Gene-environment interaction analysis of serotonin system markers with adolescent depression. Mol Psychiatry. 2004;9(10):908-915.
2. Haber SN, Rauch SL. Neurocircuitry: a window into the networks underlying neuropsychiatric disease. Neuropsychopharmacology. 2010;35(1):1-3.
3. Frodl T, Bokde AL, Scheuerecker J, et al. Functional connectivity bias of the orbitofrontal cortex in drug-free patients with major depression. Biol Psychiatry. 2010; 67(2):161-167.
4. Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531(1-2): 225-231.
5. Heim C, Nemeroff CB. The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol Psychiatry. 1999;46(11):1509-1522.
6. Isgor C, Kabbaj M, Akil H, et al. Delayed effects of chronic variable stress during peripubertal-juvenile period on hippocampal morphology and on cognitive and stress axis functions in rats. Hippocampus. 2004;14(5):636-648.
7. De Kloet ER, Vreugdenhil E, Oitzl MS, et al. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269-301.
8. Philip AM, Kim SD, Vijayan MM. Cortisol modulates the expression of cytokines and suppressors of cytokine signaling (SOCS) in rainbow trout hepatocytes. Dev Comp Immunol. 2012;38(2):360-367.
9. Coplan JD, Lydiard RB. Brain circuits in panic disorder. Biol Psychiatry. 1998;44(12):1264-1276.
10. Anisman H, Merali Z. Cytokines, stress and depressive illness: brain-immune interactions. Ann Med. 2003;35(1):2-11.
11. Crowley JJ, Lucki I. Opportunities to discover genes regulating depression and antidepressant response from rodent behavioral genetics. Curr Pharm Des. 2005;11(2):157-169.
12. Covington HE 3rd, Vialou V, Nestler EJ. From synapse to nucleus: novel targets for treating depression. Neuropharmacology. 2010;58(4-5):683-693.
13. Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161(11):1957-1966.
14. Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5(12):917-930.
15. Hartley CA, Phelps EA. Changing fear: the neurocircuitry of emotion regulation. Neuropsychopharmacology. 2010;35(1): 136-146.
16. Kim DK, Lim SW, Lee S, et al. Serotonin transporter gene polymorphism and antidepressant response. Neuroreport. 2000;11(1):215-219.
17. Ueyama E, Ukai S, Ogawa A, et al, Chronic repetitive transcranial magnetic stimulation increases hippocampal neurogenesis in rats. Psychiatry Clin Neurosci. 2011; 65(1):77-81.
18. Irwin W, Anderle MJ, Abercrombie HC, et al. Amygdalar interhemispheric functional connectivity differs between the non-depressed and depressed human brain. Neuroimage. 2004;21(2):674-686.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007; 87(3):873-904.
20. Gusnard DA, Raichle ME, Raichle ME. Searching for a baseline: functional imaging and the resting human brain. Nat Rev Neurosci. 2001;2(10):685-694.
21. Hulsebosch CE, Hains BC, Crown ED, et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60(1):202-213.
22. Gottfried JA, Dolan RJ. Human orbitofrontal cortex mediates extinction learning while accessing conditioned representations of value. Nat Neurosci. 2004;7(10):1144-1152.
23 Arnone D, McKie S, Elliott R, et al. State-dependent changes in hippocampal grey matter in depression. Mol Psychiatry. 2012;1(8):1359-4184.
24. Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11(8):1169-1180.
25. Maeng S, Zarate CA Jr. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9(6):467-474.
26. Vaidya VA, Fernandes K, Jha S. Regulation of adult hippocampal neurogenesis: relevance to depression. Expert Rev Neurother. 2007;7(7):853-864.
27. Lisiecka DM, Carballedo A, Fagan AJ, et al. Altered inhibition of negative emotions in subjects at family risk of major depressive disorder. J Psychiatr Res. 2012;46(2):181-188.
28. Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651-660.
29. Levkovitz Y, Harel EV, Roth Y, et al. Deep transcranial magnetic stimulation over the prefrontal cortex: evaluation of antidepressant and cognitive effects in depressive patients. Brain Stimul. 2009;2(4):188-200.
30. Schlaepfer TE, Lieb K. Deep brain stimulation for treatment of refractory depression. Lancet. 2005;366(9495):1420-1422.
31. Astrup, J. Energy-requiring cell functions in the ischemic brain. Their critical supply and possible inhibition in protective therapy. J Neurosurg. 1982;56(4):482-497.
32. Fletcher JM. Childhood mistreatment and adolescent and young adult depression. Soc Sci Med. 2009;68(5):799-806.
33. Warner-Schmidt JL, Duman R. VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol. 2008;8(1):14-19.
34. Clark-Raymond A, Halaris A. VEGF and depression: a comprehensive assessment of clinical data. J Psychiatr Res. 2013;47(8):1080-1087.
35. Alonso R, Griebel G, Pavone G, et al. Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry. 2004;9(3):278-286.
36. Thomas RM, Peterson DA. A neurogenic theory of depression gains momentum. Mol Interv. 2003;3(8):441-444.
37. Jacobs BL. Adult brain neurogenesis and depression. Brain Behav Immun. 2002;16(5):602-609.
1. Eley TC, Sugden K, Corsico A, et al. Gene-environment interaction analysis of serotonin system markers with adolescent depression. Mol Psychiatry. 2004;9(10):908-915.
2. Haber SN, Rauch SL. Neurocircuitry: a window into the networks underlying neuropsychiatric disease. Neuropsychopharmacology. 2010;35(1):1-3.
3. Frodl T, Bokde AL, Scheuerecker J, et al. Functional connectivity bias of the orbitofrontal cortex in drug-free patients with major depression. Biol Psychiatry. 2010; 67(2):161-167.
4. Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531(1-2): 225-231.
5. Heim C, Nemeroff CB. The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol Psychiatry. 1999;46(11):1509-1522.
6. Isgor C, Kabbaj M, Akil H, et al. Delayed effects of chronic variable stress during peripubertal-juvenile period on hippocampal morphology and on cognitive and stress axis functions in rats. Hippocampus. 2004;14(5):636-648.
7. De Kloet ER, Vreugdenhil E, Oitzl MS, et al. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269-301.
8. Philip AM, Kim SD, Vijayan MM. Cortisol modulates the expression of cytokines and suppressors of cytokine signaling (SOCS) in rainbow trout hepatocytes. Dev Comp Immunol. 2012;38(2):360-367.
9. Coplan JD, Lydiard RB. Brain circuits in panic disorder. Biol Psychiatry. 1998;44(12):1264-1276.
10. Anisman H, Merali Z. Cytokines, stress and depressive illness: brain-immune interactions. Ann Med. 2003;35(1):2-11.
11. Crowley JJ, Lucki I. Opportunities to discover genes regulating depression and antidepressant response from rodent behavioral genetics. Curr Pharm Des. 2005;11(2):157-169.
12. Covington HE 3rd, Vialou V, Nestler EJ. From synapse to nucleus: novel targets for treating depression. Neuropharmacology. 2010;58(4-5):683-693.
13. Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161(11):1957-1966.
14. Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5(12):917-930.
15. Hartley CA, Phelps EA. Changing fear: the neurocircuitry of emotion regulation. Neuropsychopharmacology. 2010;35(1): 136-146.
16. Kim DK, Lim SW, Lee S, et al. Serotonin transporter gene polymorphism and antidepressant response. Neuroreport. 2000;11(1):215-219.
17. Ueyama E, Ukai S, Ogawa A, et al, Chronic repetitive transcranial magnetic stimulation increases hippocampal neurogenesis in rats. Psychiatry Clin Neurosci. 2011; 65(1):77-81.
18. Irwin W, Anderle MJ, Abercrombie HC, et al. Amygdalar interhemispheric functional connectivity differs between the non-depressed and depressed human brain. Neuroimage. 2004;21(2):674-686.
19. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007; 87(3):873-904.
20. Gusnard DA, Raichle ME, Raichle ME. Searching for a baseline: functional imaging and the resting human brain. Nat Rev Neurosci. 2001;2(10):685-694.
21. Hulsebosch CE, Hains BC, Crown ED, et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60(1):202-213.
22. Gottfried JA, Dolan RJ. Human orbitofrontal cortex mediates extinction learning while accessing conditioned representations of value. Nat Neurosci. 2004;7(10):1144-1152.
23 Arnone D, McKie S, Elliott R, et al. State-dependent changes in hippocampal grey matter in depression. Mol Psychiatry. 2012;1(8):1359-4184.
24. Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11(8):1169-1180.
25. Maeng S, Zarate CA Jr. The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9(6):467-474.
26. Vaidya VA, Fernandes K, Jha S. Regulation of adult hippocampal neurogenesis: relevance to depression. Expert Rev Neurother. 2007;7(7):853-864.
27. Lisiecka DM, Carballedo A, Fagan AJ, et al. Altered inhibition of negative emotions in subjects at family risk of major depressive disorder. J Psychiatr Res. 2012;46(2):181-188.
28. Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651-660.
29. Levkovitz Y, Harel EV, Roth Y, et al. Deep transcranial magnetic stimulation over the prefrontal cortex: evaluation of antidepressant and cognitive effects in depressive patients. Brain Stimul. 2009;2(4):188-200.
30. Schlaepfer TE, Lieb K. Deep brain stimulation for treatment of refractory depression. Lancet. 2005;366(9495):1420-1422.
31. Astrup, J. Energy-requiring cell functions in the ischemic brain. Their critical supply and possible inhibition in protective therapy. J Neurosurg. 1982;56(4):482-497.
32. Fletcher JM. Childhood mistreatment and adolescent and young adult depression. Soc Sci Med. 2009;68(5):799-806.
33. Warner-Schmidt JL, Duman R. VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol. 2008;8(1):14-19.
34. Clark-Raymond A, Halaris A. VEGF and depression: a comprehensive assessment of clinical data. J Psychiatr Res. 2013;47(8):1080-1087.
35. Alonso R, Griebel G, Pavone G, et al. Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry. 2004;9(3):278-286.
36. Thomas RM, Peterson DA. A neurogenic theory of depression gains momentum. Mol Interv. 2003;3(8):441-444.
37. Jacobs BL. Adult brain neurogenesis and depression. Brain Behav Immun. 2002;16(5):602-609.

Recovery on higher ground: Spirituality in the treatment of substance abuse
Mr. W, age 45, is a divorced Army veteran living on the street who has entered alcohol treatment for the sixth time. He has never stayed sober for longer than 1 month after each of his previous treatment episodes.
On a typical day, Mr. W drinks 40 oz of beer, a pint of vodka, and other alcoholic beverages when available. Although he has used drugs, he reports that he did so only to augment the effects of alcohol. Before entering rehab, Mr. W worked for a television network for 16 years and was promoted to associate vice president. He lost that job as a result of drinking.
Mr. W comes from a large Irish Catholic family and has sustained an active religious faith, going to Mass 2 or 3 times a month. Throughout the interview, he appears introspective and describes frequent periods of “going inside” of himself to “rehash” things. He states that he has never been satisfied with his spiritual life and has been unable to “quiet the hunger inside.”
He describes his benders as “mini-retreats” and comments that focusing on the condensation droplets on a glass of beer is nearly a “sacramental experience” for him. He states: “My drinking is a spiritual thing for me. I believe that every time I drink I am on a spiritual search. I believe this with all my heart. I have this emptiness inside of me and alcohol would temporarily fill the enormous hole in my insides. Just for a short period of time, I would feel at peace and connected to others, and maybe even to God.”
Mr. W observes that “Every time I relapse it’s because I’m going through a spiritual withdrawal. Booze filled the void inside of me and now the void is back again. Physically and psychologically I’m fine. I’m just empty inside and when I can’t stand it any longer, I drink again.”
Few patients can so directly articulate the role they feel that spirituality plays in their substance use disorder. It is important for clinicians to be aware of the dynamics of spirituality and religion in the cause, maintenance, and treatment of substance misuse problems.
In this article, we discuss how spirituality can be assessed and suggest ethical and clinical practice concerns that we believe may support treatment of substance use disorder. We do not advocate incorporating spiritual interventions into clinical practice for patients who are uncomfortable with doing so, nor do we feel that consideration of and respect for patients’ spirituality precludes evidence-based pharmaceutical and behavioral treatment strategies. We believe, however, that addressing these issues can enhance treatment adherence in select patients.
Defining spirituality
Although religion and spirituality are related concepts, they differ.
- Religion has been defined as “an organized system of beliefs, practices, rituals, and symbols through which ones’ relationship to God or others is nurtured and exercised.”1
- Spirituality is more complex and multifaceted. Reflecting his extensive review of articles on spirituality and addiction, Cook2 proposed the following definition:
Spirituality is a distinctive, potentially creative, and universal dimension of
human experience arising within inner subjective awareness of individuals and within communities, social groups, and traditions. It may be experienced as relationship with that which is intimately “inner,” immanent, and personal, within the self and others, and/or as relationship with that which is wholly “other,” transcendent and beyond the self. It is experienced as being of fundamental or ultimate importance and is therefore concerned with matters of meaning and purpose in life, truth, and values.
In some religions, any use of alcohol or drugs is forbidden; in most religions, abuse of these substances violates norms. Those who misuse a substance also might be alienated from their religious and social support community. People struggling with addiction might feel they are compromising their spiritual values directly through the action itself and indirectly because of the harm their substance misuse causes to close friends and family. Misuse of substances also might be an attempt to “fill the void,” as Mr. W described it, of a spiritual longing or a consequence of doubts about meaning, purpose in life, and God.
Perhaps it isn’t surprising that, among psychiatric disorders, substance abuse problems seem to be most associated with spiritual intervention—especially Alcoholics Anonymous (AA), Narcotics Anonymous (NA), and Cocaine Anonymous (CA). Many of the 12 steps contain references to God and spirituality. Although much of the evidence supporting the effectiveness of the spiritual components of AA/NA/CA is correlational,3,4 the resonance that many persons who are recovering from substance abuse find in the 12-step model suggests that spiritual issues are relevant to understanding patients’ viewpoints and for planning treatment.
Religion and spirituality in risk and recovery
Although alcohol and drugs can have positive religious significance (eg, wine in the Christian Eucharist or in the Hindu practice of Ayurvedic medicine; peyote in some indigenous American religious rituals), substance use and abuse are less prevalent among persons who identify themselves as highly religious or spiritual.5 Studies indicate that alcohol use and abuse are less prevalent among persons who identify as Jewish, Muslim, or conservative Protestant, compared with those who are Catholic or liberal Protestant.6
Research suggests that consideration and accommodation of religious and spiritual practices in the recovery process is effective7-9—and preferred—by many patients.10 Psychiatrists do not need to and, in many cases, should not deliver overtly spiritual treatments to patients recovering from substance abuse. Spirituality and religion are complex issues largely outside the expertise of psychiatry; it would be naïve to consider spirituality and religion as universally beneficial elements for all patients recovering from substance abuse. Instead, psychiatrists should be equipped to:
- assess for religion and spirituality in patients
- be aware of, and supportive of, resources for integrating spirituality into treatment (eg, clergy and hospital chaplains, local AA/NA/CA groups, community religious organizations, spirituality groups).
Assessment
Post and colleagues11 note: “When patients feel that their spiritual needs are neglected in standard clinical environments, many of them may be driven away from effective medical treatment.” This is of particular concern when working with persons who have a substance use disorder—among whom, regrettably, only a minority avail themselves of professional care. You should carefully gauge the importance of these dimensions in how patients understand their disorder and the components of treatment they think their recovery process should involve.
Asking about the religious and spiritual aspects of patients’ lives shows respect for their views and facilitates a therapeutic alliance by recognizing their autonomy in treatment. Pargament and Lomax12 state: “Religion speaks to highly sensitive issues that lie at the core of the individual’s identity, commitments, values and world view. Patients are unlikely to engage in a conversation about the deepest side of themselves unless their psychiatrist demonstrates an openness to, interest in, and appreciation of the patient’s religiousness.” This exploration process can suggest natural environmental support systems available to complement recovery efforts and can indicate whether consultation with a clergyperson knowledgeable about and sensitive to their particular religious tradition is appropriate.
Assessment of spirituality and religious practice usually should occur during the initial clinical interview. Examples of revealing interview questions are listed in Box 1.

The Table13-20 lists well-researched psychometric measures that can provide clues to aspects of your patient’s spirituality. These scales yield quantitative results, yet it may be more useful to review with the patient his (her) responses to the items and to pursue issues further based on the responses.
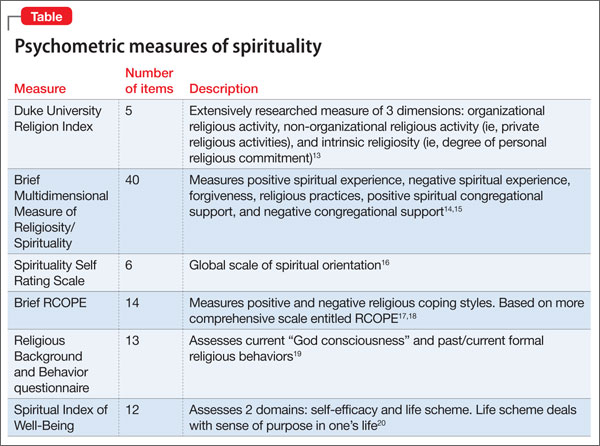
In discussing responses to questions about their spirituality and religious practices, some patients might ask about your views and whether you agree with their views. You could respond with a direct, concise answer, but refocusing the discussion on why the topic of spirituality is important to understanding one’s life and choices might be more therapeutic. This also might be a good time to remind patients that the treatment plan should reflect their view of the role of religion and spirituality in their life.
Of course, some patients are disinclined to discuss spirituality or religion, or prefer that it not be considered in treatment. This is clearly a matter of patient choice, but be aware that the patient may change his (her) mind as the recovery process continues and that the topic can be revisited if desired.
Recommendations for practice
Give careful consideration to ethical and clinical practice issues related to spiritual components in the recovery process. Plante21 and Meador and Koenig1 addressed several relevant ethical principles in considering spirituality in, respectively, psychological and psychiatric practice. Delaney and co-workers22 also presented similar principles, specifically within the context of substance use treatment. These can be summarized as a series of recommendations:
- Although psychiatrists as a group typically have a lower rate of conventional spirituality and religious practice than many of their patients,23-25 it is important not to ignore the patient’s perspective and show respect for the patient’s spiritual needs. However, if the psychiatrist has strong personal religious beliefs, he (she) must carefully guard against proselytizing or exerting undue, unintentional influence.
- Given the variety of religious traditions in the United States, understanding their particular features, as with other issues of cultural diversity, requires competence and sensitivity. Your work requires study and consultation with knowledgeable peers and experts in various faith traditions. Clergy and clinically trained chaplains, in particular, can conduct more comprehensive spiritual assessments that can yield additional treatment-relevant information.
- Maintaining professional boundaries is critical when dealing with religious and spiritual aspects of the patient’s life and thinking. Keep in mind issues of transference and the authoritative influence of the psychiatrist. The patient should understand the difference between services offered by a mental health professional and those given by a spiritual counselor or member of the clergy. Spiritual and religious issues, such as addressing concerns over guilt and sin and a relationship with God, should be referred to an appropriate clergyperson.
- Spiritual interventions introduced into the therapeutic process always should derive from the patient’s perspective and value system; they should not be imposed from an external source.
- Referral to AA often will help alcohol-dependent patients. The heart of AA’s philosophy is that addiction should be seen as a spiritual problem and that genuine recovery requires a profound spiritual awakening (Box 2). AA, as well as interventions inspired by it (eg, NA), are based on peer support, are readily available, and free. Although there is a dearth of controlled research demonstrating the efficacy of AA compared with other interventions, many recovering alcoholics credit these 12-step programs with their having maintained sobriety and adopting a positive lifestyle.

Recent research has identified specific components of AA participation that seem to be helpful.26 These include activities that are spiritual in nature and other generally active components of substance abuse care. Patient preference should be respected when encouraging AA involvement. For patients who are uncomfortable with AA—especially with its emphasis on spirituality—alternative peer support groups are available, such as SMART Recovery.27,28 If the patient adopts AA’s philosophy, it might be helpful for you to employ the language of AA and its constructs when talking with him (her).
Useful strategies on how therapists can encourage AA participation and integrate mutual help groups into treatment planning are described by Nowinski.25 Some AA members believe that use of medication is antithetical to the recovery process, but this is not the position of AA29; using FDA-approved medications, such as naltrexone and acamprosate, is evidence-based and often should be a part of the treatment regimen for alcohol dependence.
Bottom Line
Awareness of, and sensitivity to, the religious and spiritual characteristics of patients with substance use disorder can enhance clinical rapport, inform development of individualized treatment plans, and suggest strategies, such as professional consultation, that might increase the prospects for successful treatment.
Related Resources
- Galanter M. The concept of spirituality in relation to addiction recovery and general psychiatry. Recent Dev Alcohol. 2008;18:125-140.
- Monod S, Brennan M, Rochat E, et al. Instruments measuring spirituality in clinical research: a systematic review. J Gen Intern Med. 2011;26(11):1345-1357.
Drug Brand Names
Acamprosate • Campral
Naltrexone • ReVia, Vivitrol
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Featured Audio
John P. Allen, PhD, MPA, discusses whether spirituality plays a different role in treating substance use disorder than it might in treating other psychiatric illnesses. Dr. Allen works at the Department of Veterans Affairs, Mid-Atlantic Mental Illness Research, Education and Clinical Center, Division of Addictions Research and Treatment, Department of Psychiatry and Behavioral Sciences, Duke University School of Medicine, Durham, North Carolina.
1. Meador KG, Koenig HG. Spirituality and religion in psychiatry practice: parameters and implications. Psychiatr Ann. 2000;30(8):549-555.
2. Cook CC. Addiction and spirituality. Addiction. 2004;99(5):539-551.
3. Kelly J, Stout RL, Magill M, et al. Spirituality in recovery: a lagged mediational analysis of alcoholics anonymous’ principal theoretical mechanism of behavior change. Alcohol Clin Exp Res. 2011;35(3):454-463.
4. Zemore SE. A role for spiritual change in the benefits of 12-step involvement. Alcohol Clin Exp Res. 2007;31(10 suppl):76s-79s.
5. Kendler KS, Liu XQ, Gardner CO, et al. Dimensions of religiosity and their relationship to lifetime psychiatric and substance use disorders. Am J Psychiatry. 2003;160(3):496-503.
6. Koenig HG, King D, Carson VB. Handbook of religion and health, 2nd ed. New York, NY: Oxford University Press; 2012.
7. Carter TM. The effects of spiritual practices on recovery from substance abuse. J Psychiatr Ment Health Nurs. 1998; 5(5):409-413.
8. Conner BT, Anglin MD, Annon J, et al. Effect of religiosity and spirituality on drug treatment outcomes. J Behav Health Serv Res. 2009;36(2):189-198.
9. Robinson EA, Krentzman AR, Webb JR, et al. Six-month changes in spirituality and religiousness in alcoholics predict drinking outcomes at nine months. J Stud Alcohol Drugs. 2011;72(4):660-668.
10. Heinz AJ, Disney ER, Epstein DH, et al. A focus-group study on spirituality and substance-user treatment. Subst Use Misuse. 2010;45(1-2):134-153.
11. Post SG, Puchalski CM, Larson DB. Physicians and patient spirituality: professional boundaries, competency, and ethics. Ann Intern Med. 2000;132(7):578-583.
12. Pargament KI, Lomax JW. Understanding and addressing religion among people with mental illness. World Psychiatry. 2013;12(1):26-32.
13. Koenig HG, Büssing A. The Duke University Religion Index (DUREL): a five-item measure for use in epidemiological studies. Religions. 2010;1(1):78-85.
14. Brief Multidimensional Measure of Religiousness/Spirituality: 1999. http://www.primarycarecore.org/PDF/137.pdf. Accessed January 9, 2014.
15. Johnstone B, Yoon DP, Franklin KL, et al. Re-conceptualizing the factor structure of the Brief Multidimensional Measure of Religiousness/Spirituality. J Relig Health. 2009;48(2):146-163.
16. Galanter M, Dermatis H, Bunt G, et al. Assessment of spirituality and its relevance to addiction treatment. J Subst Abuse Treat. 2007;33(3):257-264.
17. Pargament K, Feuille M, Burdzy D. The Brief RCOPE: current psychometric status of a short measure of religious coping. Religions. 2011;2(1):51-76.
18. Afterdeployment.org. Spirituality assessment. http://www.afterdeployment.org/sites/default/files/pdfs/assessment-tools/spirituality-assessment.pdf. Accessed January 9, 2014.
19. Connors GJ, Tonigan JS, Miller WR. A measure of religious background and behavior for use in behavior change research. Psychol Addict Behav. 1996;10(2):90-96.
20. Daaleman TP, Frey BB. The Spirituality Index of Well-Being: a new Instrument for health-related quality-of-life research. Ann Fam Med. 2004;2(5):499-503.
21. Plante TG. Integrating spirituality and psychotherapy: ethical issues and principles to consider. J Clin Psychol. 2007;63(9):891-902.
22. Delaney HD, Forcehimes AA, Campbell WP, et al. Integrating spirituality into alcohol treatment. J Clin Psychol. 2009;65(2):185-198.
23. Shafranske EP. Religious involvement and professional practices of psychiatrists and other mental health professionals. Psychiatr Ann. 2000;30(8):525-532.
24. Curlin FA, Lantos JD, Roach CJ, et al. Religious characteristics of U.S. physicians: a national survey. Gen Intern Med. 2005;20(7):629-634.
25. Curlin FA, Odell SV, Lawrence RE, et al. The relationship between psychiatry and religion among U.S. physicians. Psychiatr Serv. 2007;58(9):1193-1198.
26. Kelly JF, Hoeppner B, Stout RL, et al. Determining the relative importance of the mechanisms of behavior change within Alcoholics Anonymous: a multiple mediator analysis. Addiction. 2012;107(2):289-299.
27. Nowinski J. Self-help groups for addictions. In: McCrady BS, Epstein EE, eds. Addictions. New York, NY: Oxford University Press; 1999:328-346.
28. SMART recovery: self-management and recovery training. http://www.smartrecovery.org. Accessed January 9, 2014.
29. Alcoholics Anonymous World Services. The AA member—medication & other drugs. 2nd ed. New York, NY: Alcoholics Anonymous World Services; 2011.
Mr. W, age 45, is a divorced Army veteran living on the street who has entered alcohol treatment for the sixth time. He has never stayed sober for longer than 1 month after each of his previous treatment episodes.
On a typical day, Mr. W drinks 40 oz of beer, a pint of vodka, and other alcoholic beverages when available. Although he has used drugs, he reports that he did so only to augment the effects of alcohol. Before entering rehab, Mr. W worked for a television network for 16 years and was promoted to associate vice president. He lost that job as a result of drinking.
Mr. W comes from a large Irish Catholic family and has sustained an active religious faith, going to Mass 2 or 3 times a month. Throughout the interview, he appears introspective and describes frequent periods of “going inside” of himself to “rehash” things. He states that he has never been satisfied with his spiritual life and has been unable to “quiet the hunger inside.”
He describes his benders as “mini-retreats” and comments that focusing on the condensation droplets on a glass of beer is nearly a “sacramental experience” for him. He states: “My drinking is a spiritual thing for me. I believe that every time I drink I am on a spiritual search. I believe this with all my heart. I have this emptiness inside of me and alcohol would temporarily fill the enormous hole in my insides. Just for a short period of time, I would feel at peace and connected to others, and maybe even to God.”
Mr. W observes that “Every time I relapse it’s because I’m going through a spiritual withdrawal. Booze filled the void inside of me and now the void is back again. Physically and psychologically I’m fine. I’m just empty inside and when I can’t stand it any longer, I drink again.”
Few patients can so directly articulate the role they feel that spirituality plays in their substance use disorder. It is important for clinicians to be aware of the dynamics of spirituality and religion in the cause, maintenance, and treatment of substance misuse problems.
In this article, we discuss how spirituality can be assessed and suggest ethical and clinical practice concerns that we believe may support treatment of substance use disorder. We do not advocate incorporating spiritual interventions into clinical practice for patients who are uncomfortable with doing so, nor do we feel that consideration of and respect for patients’ spirituality precludes evidence-based pharmaceutical and behavioral treatment strategies. We believe, however, that addressing these issues can enhance treatment adherence in select patients.
Defining spirituality
Although religion and spirituality are related concepts, they differ.
- Religion has been defined as “an organized system of beliefs, practices, rituals, and symbols through which ones’ relationship to God or others is nurtured and exercised.”1
- Spirituality is more complex and multifaceted. Reflecting his extensive review of articles on spirituality and addiction, Cook2 proposed the following definition:
Spirituality is a distinctive, potentially creative, and universal dimension of
human experience arising within inner subjective awareness of individuals and within communities, social groups, and traditions. It may be experienced as relationship with that which is intimately “inner,” immanent, and personal, within the self and others, and/or as relationship with that which is wholly “other,” transcendent and beyond the self. It is experienced as being of fundamental or ultimate importance and is therefore concerned with matters of meaning and purpose in life, truth, and values.
In some religions, any use of alcohol or drugs is forbidden; in most religions, abuse of these substances violates norms. Those who misuse a substance also might be alienated from their religious and social support community. People struggling with addiction might feel they are compromising their spiritual values directly through the action itself and indirectly because of the harm their substance misuse causes to close friends and family. Misuse of substances also might be an attempt to “fill the void,” as Mr. W described it, of a spiritual longing or a consequence of doubts about meaning, purpose in life, and God.
Perhaps it isn’t surprising that, among psychiatric disorders, substance abuse problems seem to be most associated with spiritual intervention—especially Alcoholics Anonymous (AA), Narcotics Anonymous (NA), and Cocaine Anonymous (CA). Many of the 12 steps contain references to God and spirituality. Although much of the evidence supporting the effectiveness of the spiritual components of AA/NA/CA is correlational,3,4 the resonance that many persons who are recovering from substance abuse find in the 12-step model suggests that spiritual issues are relevant to understanding patients’ viewpoints and for planning treatment.
Religion and spirituality in risk and recovery
Although alcohol and drugs can have positive religious significance (eg, wine in the Christian Eucharist or in the Hindu practice of Ayurvedic medicine; peyote in some indigenous American religious rituals), substance use and abuse are less prevalent among persons who identify themselves as highly religious or spiritual.5 Studies indicate that alcohol use and abuse are less prevalent among persons who identify as Jewish, Muslim, or conservative Protestant, compared with those who are Catholic or liberal Protestant.6
Research suggests that consideration and accommodation of religious and spiritual practices in the recovery process is effective7-9—and preferred—by many patients.10 Psychiatrists do not need to and, in many cases, should not deliver overtly spiritual treatments to patients recovering from substance abuse. Spirituality and religion are complex issues largely outside the expertise of psychiatry; it would be naïve to consider spirituality and religion as universally beneficial elements for all patients recovering from substance abuse. Instead, psychiatrists should be equipped to:
- assess for religion and spirituality in patients
- be aware of, and supportive of, resources for integrating spirituality into treatment (eg, clergy and hospital chaplains, local AA/NA/CA groups, community religious organizations, spirituality groups).
Assessment
Post and colleagues11 note: “When patients feel that their spiritual needs are neglected in standard clinical environments, many of them may be driven away from effective medical treatment.” This is of particular concern when working with persons who have a substance use disorder—among whom, regrettably, only a minority avail themselves of professional care. You should carefully gauge the importance of these dimensions in how patients understand their disorder and the components of treatment they think their recovery process should involve.
Asking about the religious and spiritual aspects of patients’ lives shows respect for their views and facilitates a therapeutic alliance by recognizing their autonomy in treatment. Pargament and Lomax12 state: “Religion speaks to highly sensitive issues that lie at the core of the individual’s identity, commitments, values and world view. Patients are unlikely to engage in a conversation about the deepest side of themselves unless their psychiatrist demonstrates an openness to, interest in, and appreciation of the patient’s religiousness.” This exploration process can suggest natural environmental support systems available to complement recovery efforts and can indicate whether consultation with a clergyperson knowledgeable about and sensitive to their particular religious tradition is appropriate.
Assessment of spirituality and religious practice usually should occur during the initial clinical interview. Examples of revealing interview questions are listed in Box 1.
The Table13-20 lists well-researched psychometric measures that can provide clues to aspects of your patient’s spirituality. These scales yield quantitative results, yet it may be more useful to review with the patient his (her) responses to the items and to pursue issues further based on the responses.
In discussing responses to questions about their spirituality and religious practices, some patients might ask about your views and whether you agree with their views. You could respond with a direct, concise answer, but refocusing the discussion on why the topic of spirituality is important to understanding one’s life and choices might be more therapeutic. This also might be a good time to remind patients that the treatment plan should reflect their view of the role of religion and spirituality in their life.
Of course, some patients are disinclined to discuss spirituality or religion, or prefer that it not be considered in treatment. This is clearly a matter of patient choice, but be aware that the patient may change his (her) mind as the recovery process continues and that the topic can be revisited if desired.
Recommendations for practice
Give careful consideration to ethical and clinical practice issues related to spiritual components in the recovery process. Plante21 and Meador and Koenig1 addressed several relevant ethical principles in considering spirituality in, respectively, psychological and psychiatric practice. Delaney and co-workers22 also presented similar principles, specifically within the context of substance use treatment. These can be summarized as a series of recommendations:
- Although psychiatrists as a group typically have a lower rate of conventional spirituality and religious practice than many of their patients,23-25 it is important not to ignore the patient’s perspective and show respect for the patient’s spiritual needs. However, if the psychiatrist has strong personal religious beliefs, he (she) must carefully guard against proselytizing or exerting undue, unintentional influence.
- Given the variety of religious traditions in the United States, understanding their particular features, as with other issues of cultural diversity, requires competence and sensitivity. Your work requires study and consultation with knowledgeable peers and experts in various faith traditions. Clergy and clinically trained chaplains, in particular, can conduct more comprehensive spiritual assessments that can yield additional treatment-relevant information.
- Maintaining professional boundaries is critical when dealing with religious and spiritual aspects of the patient’s life and thinking. Keep in mind issues of transference and the authoritative influence of the psychiatrist. The patient should understand the difference between services offered by a mental health professional and those given by a spiritual counselor or member of the clergy. Spiritual and religious issues, such as addressing concerns over guilt and sin and a relationship with God, should be referred to an appropriate clergyperson.
- Spiritual interventions introduced into the therapeutic process always should derive from the patient’s perspective and value system; they should not be imposed from an external source.
- Referral to AA often will help alcohol-dependent patients. The heart of AA’s philosophy is that addiction should be seen as a spiritual problem and that genuine recovery requires a profound spiritual awakening (Box 2). AA, as well as interventions inspired by it (eg, NA), are based on peer support, are readily available, and free. Although there is a dearth of controlled research demonstrating the efficacy of AA compared with other interventions, many recovering alcoholics credit these 12-step programs with their having maintained sobriety and adopting a positive lifestyle.
Recent research has identified specific components of AA participation that seem to be helpful.26 These include activities that are spiritual in nature and other generally active components of substance abuse care. Patient preference should be respected when encouraging AA involvement. For patients who are uncomfortable with AA—especially with its emphasis on spirituality—alternative peer support groups are available, such as SMART Recovery.27,28 If the patient adopts AA’s philosophy, it might be helpful for you to employ the language of AA and its constructs when talking with him (her).
Useful strategies on how therapists can encourage AA participation and integrate mutual help groups into treatment planning are described by Nowinski.25 Some AA members believe that use of medication is antithetical to the recovery process, but this is not the position of AA29; using FDA-approved medications, such as naltrexone and acamprosate, is evidence-based and often should be a part of the treatment regimen for alcohol dependence.
Bottom Line
Awareness of, and sensitivity to, the religious and spiritual characteristics of patients with substance use disorder can enhance clinical rapport, inform development of individualized treatment plans, and suggest strategies, such as professional consultation, that might increase the prospects for successful treatment.
Related Resources
- Galanter M. The concept of spirituality in relation to addiction recovery and general psychiatry. Recent Dev Alcohol. 2008;18:125-140.
- Monod S, Brennan M, Rochat E, et al. Instruments measuring spirituality in clinical research: a systematic review. J Gen Intern Med. 2011;26(11):1345-1357.
Drug Brand Names
Acamprosate • Campral
Naltrexone • ReVia, Vivitrol
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Featured Audio
John P. Allen, PhD, MPA, discusses whether spirituality plays a different role in treating substance use disorder than it might in treating other psychiatric illnesses. Dr. Allen works at the Department of Veterans Affairs, Mid-Atlantic Mental Illness Research, Education and Clinical Center, Division of Addictions Research and Treatment, Department of Psychiatry and Behavioral Sciences, Duke University School of Medicine, Durham, North Carolina.
Mr. W, age 45, is a divorced Army veteran living on the street who has entered alcohol treatment for the sixth time. He has never stayed sober for longer than 1 month after each of his previous treatment episodes.
On a typical day, Mr. W drinks 40 oz of beer, a pint of vodka, and other alcoholic beverages when available. Although he has used drugs, he reports that he did so only to augment the effects of alcohol. Before entering rehab, Mr. W worked for a television network for 16 years and was promoted to associate vice president. He lost that job as a result of drinking.
Mr. W comes from a large Irish Catholic family and has sustained an active religious faith, going to Mass 2 or 3 times a month. Throughout the interview, he appears introspective and describes frequent periods of “going inside” of himself to “rehash” things. He states that he has never been satisfied with his spiritual life and has been unable to “quiet the hunger inside.”
He describes his benders as “mini-retreats” and comments that focusing on the condensation droplets on a glass of beer is nearly a “sacramental experience” for him. He states: “My drinking is a spiritual thing for me. I believe that every time I drink I am on a spiritual search. I believe this with all my heart. I have this emptiness inside of me and alcohol would temporarily fill the enormous hole in my insides. Just for a short period of time, I would feel at peace and connected to others, and maybe even to God.”
Mr. W observes that “Every time I relapse it’s because I’m going through a spiritual withdrawal. Booze filled the void inside of me and now the void is back again. Physically and psychologically I’m fine. I’m just empty inside and when I can’t stand it any longer, I drink again.”
Few patients can so directly articulate the role they feel that spirituality plays in their substance use disorder. It is important for clinicians to be aware of the dynamics of spirituality and religion in the cause, maintenance, and treatment of substance misuse problems.
In this article, we discuss how spirituality can be assessed and suggest ethical and clinical practice concerns that we believe may support treatment of substance use disorder. We do not advocate incorporating spiritual interventions into clinical practice for patients who are uncomfortable with doing so, nor do we feel that consideration of and respect for patients’ spirituality precludes evidence-based pharmaceutical and behavioral treatment strategies. We believe, however, that addressing these issues can enhance treatment adherence in select patients.
Defining spirituality
Although religion and spirituality are related concepts, they differ.
- Religion has been defined as “an organized system of beliefs, practices, rituals, and symbols through which ones’ relationship to God or others is nurtured and exercised.”1
- Spirituality is more complex and multifaceted. Reflecting his extensive review of articles on spirituality and addiction, Cook2 proposed the following definition:
Spirituality is a distinctive, potentially creative, and universal dimension of
human experience arising within inner subjective awareness of individuals and within communities, social groups, and traditions. It may be experienced as relationship with that which is intimately “inner,” immanent, and personal, within the self and others, and/or as relationship with that which is wholly “other,” transcendent and beyond the self. It is experienced as being of fundamental or ultimate importance and is therefore concerned with matters of meaning and purpose in life, truth, and values.
In some religions, any use of alcohol or drugs is forbidden; in most religions, abuse of these substances violates norms. Those who misuse a substance also might be alienated from their religious and social support community. People struggling with addiction might feel they are compromising their spiritual values directly through the action itself and indirectly because of the harm their substance misuse causes to close friends and family. Misuse of substances also might be an attempt to “fill the void,” as Mr. W described it, of a spiritual longing or a consequence of doubts about meaning, purpose in life, and God.
Perhaps it isn’t surprising that, among psychiatric disorders, substance abuse problems seem to be most associated with spiritual intervention—especially Alcoholics Anonymous (AA), Narcotics Anonymous (NA), and Cocaine Anonymous (CA). Many of the 12 steps contain references to God and spirituality. Although much of the evidence supporting the effectiveness of the spiritual components of AA/NA/CA is correlational,3,4 the resonance that many persons who are recovering from substance abuse find in the 12-step model suggests that spiritual issues are relevant to understanding patients’ viewpoints and for planning treatment.
Religion and spirituality in risk and recovery
Although alcohol and drugs can have positive religious significance (eg, wine in the Christian Eucharist or in the Hindu practice of Ayurvedic medicine; peyote in some indigenous American religious rituals), substance use and abuse are less prevalent among persons who identify themselves as highly religious or spiritual.5 Studies indicate that alcohol use and abuse are less prevalent among persons who identify as Jewish, Muslim, or conservative Protestant, compared with those who are Catholic or liberal Protestant.6
Research suggests that consideration and accommodation of religious and spiritual practices in the recovery process is effective7-9—and preferred—by many patients.10 Psychiatrists do not need to and, in many cases, should not deliver overtly spiritual treatments to patients recovering from substance abuse. Spirituality and religion are complex issues largely outside the expertise of psychiatry; it would be naïve to consider spirituality and religion as universally beneficial elements for all patients recovering from substance abuse. Instead, psychiatrists should be equipped to:
- assess for religion and spirituality in patients
- be aware of, and supportive of, resources for integrating spirituality into treatment (eg, clergy and hospital chaplains, local AA/NA/CA groups, community religious organizations, spirituality groups).
Assessment
Post and colleagues11 note: “When patients feel that their spiritual needs are neglected in standard clinical environments, many of them may be driven away from effective medical treatment.” This is of particular concern when working with persons who have a substance use disorder—among whom, regrettably, only a minority avail themselves of professional care. You should carefully gauge the importance of these dimensions in how patients understand their disorder and the components of treatment they think their recovery process should involve.
Asking about the religious and spiritual aspects of patients’ lives shows respect for their views and facilitates a therapeutic alliance by recognizing their autonomy in treatment. Pargament and Lomax12 state: “Religion speaks to highly sensitive issues that lie at the core of the individual’s identity, commitments, values and world view. Patients are unlikely to engage in a conversation about the deepest side of themselves unless their psychiatrist demonstrates an openness to, interest in, and appreciation of the patient’s religiousness.” This exploration process can suggest natural environmental support systems available to complement recovery efforts and can indicate whether consultation with a clergyperson knowledgeable about and sensitive to their particular religious tradition is appropriate.
Assessment of spirituality and religious practice usually should occur during the initial clinical interview. Examples of revealing interview questions are listed in Box 1.
The Table13-20 lists well-researched psychometric measures that can provide clues to aspects of your patient’s spirituality. These scales yield quantitative results, yet it may be more useful to review with the patient his (her) responses to the items and to pursue issues further based on the responses.
In discussing responses to questions about their spirituality and religious practices, some patients might ask about your views and whether you agree with their views. You could respond with a direct, concise answer, but refocusing the discussion on why the topic of spirituality is important to understanding one’s life and choices might be more therapeutic. This also might be a good time to remind patients that the treatment plan should reflect their view of the role of religion and spirituality in their life.
Of course, some patients are disinclined to discuss spirituality or religion, or prefer that it not be considered in treatment. This is clearly a matter of patient choice, but be aware that the patient may change his (her) mind as the recovery process continues and that the topic can be revisited if desired.
Recommendations for practice
Give careful consideration to ethical and clinical practice issues related to spiritual components in the recovery process. Plante21 and Meador and Koenig1 addressed several relevant ethical principles in considering spirituality in, respectively, psychological and psychiatric practice. Delaney and co-workers22 also presented similar principles, specifically within the context of substance use treatment. These can be summarized as a series of recommendations:
- Although psychiatrists as a group typically have a lower rate of conventional spirituality and religious practice than many of their patients,23-25 it is important not to ignore the patient’s perspective and show respect for the patient’s spiritual needs. However, if the psychiatrist has strong personal religious beliefs, he (she) must carefully guard against proselytizing or exerting undue, unintentional influence.
- Given the variety of religious traditions in the United States, understanding their particular features, as with other issues of cultural diversity, requires competence and sensitivity. Your work requires study and consultation with knowledgeable peers and experts in various faith traditions. Clergy and clinically trained chaplains, in particular, can conduct more comprehensive spiritual assessments that can yield additional treatment-relevant information.
- Maintaining professional boundaries is critical when dealing with religious and spiritual aspects of the patient’s life and thinking. Keep in mind issues of transference and the authoritative influence of the psychiatrist. The patient should understand the difference between services offered by a mental health professional and those given by a spiritual counselor or member of the clergy. Spiritual and religious issues, such as addressing concerns over guilt and sin and a relationship with God, should be referred to an appropriate clergyperson.
- Spiritual interventions introduced into the therapeutic process always should derive from the patient’s perspective and value system; they should not be imposed from an external source.
- Referral to AA often will help alcohol-dependent patients. The heart of AA’s philosophy is that addiction should be seen as a spiritual problem and that genuine recovery requires a profound spiritual awakening (Box 2). AA, as well as interventions inspired by it (eg, NA), are based on peer support, are readily available, and free. Although there is a dearth of controlled research demonstrating the efficacy of AA compared with other interventions, many recovering alcoholics credit these 12-step programs with their having maintained sobriety and adopting a positive lifestyle.
Recent research has identified specific components of AA participation that seem to be helpful.26 These include activities that are spiritual in nature and other generally active components of substance abuse care. Patient preference should be respected when encouraging AA involvement. For patients who are uncomfortable with AA—especially with its emphasis on spirituality—alternative peer support groups are available, such as SMART Recovery.27,28 If the patient adopts AA’s philosophy, it might be helpful for you to employ the language of AA and its constructs when talking with him (her).
Useful strategies on how therapists can encourage AA participation and integrate mutual help groups into treatment planning are described by Nowinski.25 Some AA members believe that use of medication is antithetical to the recovery process, but this is not the position of AA29; using FDA-approved medications, such as naltrexone and acamprosate, is evidence-based and often should be a part of the treatment regimen for alcohol dependence.
Bottom Line
Awareness of, and sensitivity to, the religious and spiritual characteristics of patients with substance use disorder can enhance clinical rapport, inform development of individualized treatment plans, and suggest strategies, such as professional consultation, that might increase the prospects for successful treatment.
Related Resources
- Galanter M. The concept of spirituality in relation to addiction recovery and general psychiatry. Recent Dev Alcohol. 2008;18:125-140.
- Monod S, Brennan M, Rochat E, et al. Instruments measuring spirituality in clinical research: a systematic review. J Gen Intern Med. 2011;26(11):1345-1357.
Drug Brand Names
Acamprosate • Campral
Naltrexone • ReVia, Vivitrol
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Featured Audio
John P. Allen, PhD, MPA, discusses whether spirituality plays a different role in treating substance use disorder than it might in treating other psychiatric illnesses. Dr. Allen works at the Department of Veterans Affairs, Mid-Atlantic Mental Illness Research, Education and Clinical Center, Division of Addictions Research and Treatment, Department of Psychiatry and Behavioral Sciences, Duke University School of Medicine, Durham, North Carolina.
1. Meador KG, Koenig HG. Spirituality and religion in psychiatry practice: parameters and implications. Psychiatr Ann. 2000;30(8):549-555.
2. Cook CC. Addiction and spirituality. Addiction. 2004;99(5):539-551.
3. Kelly J, Stout RL, Magill M, et al. Spirituality in recovery: a lagged mediational analysis of alcoholics anonymous’ principal theoretical mechanism of behavior change. Alcohol Clin Exp Res. 2011;35(3):454-463.
4. Zemore SE. A role for spiritual change in the benefits of 12-step involvement. Alcohol Clin Exp Res. 2007;31(10 suppl):76s-79s.
5. Kendler KS, Liu XQ, Gardner CO, et al. Dimensions of religiosity and their relationship to lifetime psychiatric and substance use disorders. Am J Psychiatry. 2003;160(3):496-503.
6. Koenig HG, King D, Carson VB. Handbook of religion and health, 2nd ed. New York, NY: Oxford University Press; 2012.
7. Carter TM. The effects of spiritual practices on recovery from substance abuse. J Psychiatr Ment Health Nurs. 1998; 5(5):409-413.
8. Conner BT, Anglin MD, Annon J, et al. Effect of religiosity and spirituality on drug treatment outcomes. J Behav Health Serv Res. 2009;36(2):189-198.
9. Robinson EA, Krentzman AR, Webb JR, et al. Six-month changes in spirituality and religiousness in alcoholics predict drinking outcomes at nine months. J Stud Alcohol Drugs. 2011;72(4):660-668.
10. Heinz AJ, Disney ER, Epstein DH, et al. A focus-group study on spirituality and substance-user treatment. Subst Use Misuse. 2010;45(1-2):134-153.
11. Post SG, Puchalski CM, Larson DB. Physicians and patient spirituality: professional boundaries, competency, and ethics. Ann Intern Med. 2000;132(7):578-583.
12. Pargament KI, Lomax JW. Understanding and addressing religion among people with mental illness. World Psychiatry. 2013;12(1):26-32.
13. Koenig HG, Büssing A. The Duke University Religion Index (DUREL): a five-item measure for use in epidemiological studies. Religions. 2010;1(1):78-85.
14. Brief Multidimensional Measure of Religiousness/Spirituality: 1999. http://www.primarycarecore.org/PDF/137.pdf. Accessed January 9, 2014.
15. Johnstone B, Yoon DP, Franklin KL, et al. Re-conceptualizing the factor structure of the Brief Multidimensional Measure of Religiousness/Spirituality. J Relig Health. 2009;48(2):146-163.
16. Galanter M, Dermatis H, Bunt G, et al. Assessment of spirituality and its relevance to addiction treatment. J Subst Abuse Treat. 2007;33(3):257-264.
17. Pargament K, Feuille M, Burdzy D. The Brief RCOPE: current psychometric status of a short measure of religious coping. Religions. 2011;2(1):51-76.
18. Afterdeployment.org. Spirituality assessment. http://www.afterdeployment.org/sites/default/files/pdfs/assessment-tools/spirituality-assessment.pdf. Accessed January 9, 2014.
19. Connors GJ, Tonigan JS, Miller WR. A measure of religious background and behavior for use in behavior change research. Psychol Addict Behav. 1996;10(2):90-96.
20. Daaleman TP, Frey BB. The Spirituality Index of Well-Being: a new Instrument for health-related quality-of-life research. Ann Fam Med. 2004;2(5):499-503.
21. Plante TG. Integrating spirituality and psychotherapy: ethical issues and principles to consider. J Clin Psychol. 2007;63(9):891-902.
22. Delaney HD, Forcehimes AA, Campbell WP, et al. Integrating spirituality into alcohol treatment. J Clin Psychol. 2009;65(2):185-198.
23. Shafranske EP. Religious involvement and professional practices of psychiatrists and other mental health professionals. Psychiatr Ann. 2000;30(8):525-532.
24. Curlin FA, Lantos JD, Roach CJ, et al. Religious characteristics of U.S. physicians: a national survey. Gen Intern Med. 2005;20(7):629-634.
25. Curlin FA, Odell SV, Lawrence RE, et al. The relationship between psychiatry and religion among U.S. physicians. Psychiatr Serv. 2007;58(9):1193-1198.
26. Kelly JF, Hoeppner B, Stout RL, et al. Determining the relative importance of the mechanisms of behavior change within Alcoholics Anonymous: a multiple mediator analysis. Addiction. 2012;107(2):289-299.
27. Nowinski J. Self-help groups for addictions. In: McCrady BS, Epstein EE, eds. Addictions. New York, NY: Oxford University Press; 1999:328-346.
28. SMART recovery: self-management and recovery training. http://www.smartrecovery.org. Accessed January 9, 2014.
29. Alcoholics Anonymous World Services. The AA member—medication & other drugs. 2nd ed. New York, NY: Alcoholics Anonymous World Services; 2011.
1. Meador KG, Koenig HG. Spirituality and religion in psychiatry practice: parameters and implications. Psychiatr Ann. 2000;30(8):549-555.
2. Cook CC. Addiction and spirituality. Addiction. 2004;99(5):539-551.
3. Kelly J, Stout RL, Magill M, et al. Spirituality in recovery: a lagged mediational analysis of alcoholics anonymous’ principal theoretical mechanism of behavior change. Alcohol Clin Exp Res. 2011;35(3):454-463.
4. Zemore SE. A role for spiritual change in the benefits of 12-step involvement. Alcohol Clin Exp Res. 2007;31(10 suppl):76s-79s.
5. Kendler KS, Liu XQ, Gardner CO, et al. Dimensions of religiosity and their relationship to lifetime psychiatric and substance use disorders. Am J Psychiatry. 2003;160(3):496-503.
6. Koenig HG, King D, Carson VB. Handbook of religion and health, 2nd ed. New York, NY: Oxford University Press; 2012.
7. Carter TM. The effects of spiritual practices on recovery from substance abuse. J Psychiatr Ment Health Nurs. 1998; 5(5):409-413.
8. Conner BT, Anglin MD, Annon J, et al. Effect of religiosity and spirituality on drug treatment outcomes. J Behav Health Serv Res. 2009;36(2):189-198.
9. Robinson EA, Krentzman AR, Webb JR, et al. Six-month changes in spirituality and religiousness in alcoholics predict drinking outcomes at nine months. J Stud Alcohol Drugs. 2011;72(4):660-668.
10. Heinz AJ, Disney ER, Epstein DH, et al. A focus-group study on spirituality and substance-user treatment. Subst Use Misuse. 2010;45(1-2):134-153.
11. Post SG, Puchalski CM, Larson DB. Physicians and patient spirituality: professional boundaries, competency, and ethics. Ann Intern Med. 2000;132(7):578-583.
12. Pargament KI, Lomax JW. Understanding and addressing religion among people with mental illness. World Psychiatry. 2013;12(1):26-32.
13. Koenig HG, Büssing A. The Duke University Religion Index (DUREL): a five-item measure for use in epidemiological studies. Religions. 2010;1(1):78-85.
14. Brief Multidimensional Measure of Religiousness/Spirituality: 1999. http://www.primarycarecore.org/PDF/137.pdf. Accessed January 9, 2014.
15. Johnstone B, Yoon DP, Franklin KL, et al. Re-conceptualizing the factor structure of the Brief Multidimensional Measure of Religiousness/Spirituality. J Relig Health. 2009;48(2):146-163.
16. Galanter M, Dermatis H, Bunt G, et al. Assessment of spirituality and its relevance to addiction treatment. J Subst Abuse Treat. 2007;33(3):257-264.
17. Pargament K, Feuille M, Burdzy D. The Brief RCOPE: current psychometric status of a short measure of religious coping. Religions. 2011;2(1):51-76.
18. Afterdeployment.org. Spirituality assessment. http://www.afterdeployment.org/sites/default/files/pdfs/assessment-tools/spirituality-assessment.pdf. Accessed January 9, 2014.
19. Connors GJ, Tonigan JS, Miller WR. A measure of religious background and behavior for use in behavior change research. Psychol Addict Behav. 1996;10(2):90-96.
20. Daaleman TP, Frey BB. The Spirituality Index of Well-Being: a new Instrument for health-related quality-of-life research. Ann Fam Med. 2004;2(5):499-503.
21. Plante TG. Integrating spirituality and psychotherapy: ethical issues and principles to consider. J Clin Psychol. 2007;63(9):891-902.
22. Delaney HD, Forcehimes AA, Campbell WP, et al. Integrating spirituality into alcohol treatment. J Clin Psychol. 2009;65(2):185-198.
23. Shafranske EP. Religious involvement and professional practices of psychiatrists and other mental health professionals. Psychiatr Ann. 2000;30(8):525-532.
24. Curlin FA, Lantos JD, Roach CJ, et al. Religious characteristics of U.S. physicians: a national survey. Gen Intern Med. 2005;20(7):629-634.
25. Curlin FA, Odell SV, Lawrence RE, et al. The relationship between psychiatry and religion among U.S. physicians. Psychiatr Serv. 2007;58(9):1193-1198.
26. Kelly JF, Hoeppner B, Stout RL, et al. Determining the relative importance of the mechanisms of behavior change within Alcoholics Anonymous: a multiple mediator analysis. Addiction. 2012;107(2):289-299.
27. Nowinski J. Self-help groups for addictions. In: McCrady BS, Epstein EE, eds. Addictions. New York, NY: Oxford University Press; 1999:328-346.
28. SMART recovery: self-management and recovery training. http://www.smartrecovery.org. Accessed January 9, 2014.
29. Alcoholics Anonymous World Services. The AA member—medication & other drugs. 2nd ed. New York, NY: Alcoholics Anonymous World Services; 2011.

Apply your psychiatric skills to managing rheumatoid arthritis
Joint disease is the most common cause of disability and the source of considerable psychological distress. In the United States, 50 million adults complain of joint pain; in 2007, 1.5 million people suffered from rheumatoid arthritis (RA). A chronic inflammatory autoimmune disease of joints, RA can involve almost all organs.1
The link to mental illness
Mental illness in RA patients often is underdiagnosed and undertreated. These missed opportunities contribute to poor compliance with medical therapy, suboptimal therapeutic response, greater disability, and diminished quality of life.2
Limited mobility, chronic pain, sleep disturbance, fatigue, and immunological factors predispose RA patients to depression and anxiety.3 The proinflammatory cytokines, tumor necrosis factor-α (TNF-α), interleukin 1 (IL-1), IL-6, and interferon-g have a role in inducing affective symptoms. There also is a relationship between an elevated IL-17 level and anxiety.
Research substantiates a relationship between RA and depression.3 The prevalence of affective illness is approximately 6% among the general population, and 13% to 30% among RA patients.4 In arthritic populations, 52% exhibit depression and anxiety; joint discomfort contributes to insomnia in 25% to 42% of cases.4
Arthritic pain persists despite suppressed inflammation, which suggests involvement of the CNS.5 Increased levels of IL-6 and TNF-α can cause insomnia and affect pain perception.6 Decreased conditioned pain modulation, a lower pain threshold, and pressure pain intolerance lead to increased pain awareness and heightened discomfort.
How can you help your patient who has RA?
Because the focus of care in RA is on the disease’s physical attributes, psychiatric symptoms sometimes receive less attention.7 And because arthritic symptoms overlap with anorexia, weight loss, fatigue, pain, and insomnia, affective illness can go unrecognized.
Depression rating scales can overestimate affective illness, but a history and follow-up questionnaire can facilitate an accurate diagnosis of depression and help determine the need for, and type of, intervention.
Selective serotonin reuptake inhibitors (SSRIs) are considered first‐line treatment of depression associated with RA.7 Although SSRIs for RA can be administered to the maximum recommended dosage, titration is advised in accordance with patient response and tolerance.
Tricyclic antidepressants are not as well tolerated in RA, especially in older patients; however, they have more of an analgesic effect, even at lower dosages.
Joint disease activity and mood are associated with sleep disturbance, and vice versa.5 Insomnia calls for patient education about sleep hygiene, avoiding caffeine and other stimulants, and an individualized appraisal of options for pharmacotherapy.
Alleviating RA pain is important for psychosocial health.8 Although the medical team’s emphasis should be on controlling inflammation to minimize joint damage and pain, be sure to address your RA patients’ mood symptoms to improve the quality of their life.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Centers for Disease Control and Prevention. Arthritis-related statistics. http://www.cdc.gov/arthritis/data_statistics/arthritis_related_stats.htm. Updated August 1, 2011. Accessed January 4, 2013.
2. Shih M, Hootman JM, Strine TW, et al. Serious psychological distress in U.S. adults with arthritis. J Gen Intern Med. 2006;21(11):1160-1166.
3. Sato E, Nishimura K, Nakajima A, et al. Major depressive disorder in patients with rheumatoid arthritis. Mod Rheumatol. 2013;23(2):237-244.
4. Wolfe F, Michaud K, Li T. Sleep disturbance in patients with rheumatoid arthritis: evaluation by medical outcomes study and visual analog sleep scales. J Rheumatol. 2006;33(10):1942-1951.
5. Fragiadaki K, Tektonidou MG, Konsta M, et al. Sleep disturbances and interleukin 6 receptor inhibition in rheumatoid arthritis. J Rheumatol. 2012;39(1):60-62.
6. Lee YC, Lu B, Edwards RR, et al. The role of sleep problems in central pain processing in rheumatoid arthritis. Arthritis Rheum. 2013;65(1):59-68.
7. Dickens C, Creed F. The burden of depression in patients with rheumatoid arthritis. Rheumatology (Oxford). 2001; 40(12):1327-1330.
8. Courvoisier DS, Agoritsas T, Glauser J, et al. Pain as an important predictor of psychosocial health in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2012;64(2):190-196.
Joint disease is the most common cause of disability and the source of considerable psychological distress. In the United States, 50 million adults complain of joint pain; in 2007, 1.5 million people suffered from rheumatoid arthritis (RA). A chronic inflammatory autoimmune disease of joints, RA can involve almost all organs.1
The link to mental illness
Mental illness in RA patients often is underdiagnosed and undertreated. These missed opportunities contribute to poor compliance with medical therapy, suboptimal therapeutic response, greater disability, and diminished quality of life.2
Limited mobility, chronic pain, sleep disturbance, fatigue, and immunological factors predispose RA patients to depression and anxiety.3 The proinflammatory cytokines, tumor necrosis factor-α (TNF-α), interleukin 1 (IL-1), IL-6, and interferon-g have a role in inducing affective symptoms. There also is a relationship between an elevated IL-17 level and anxiety.
Research substantiates a relationship between RA and depression.3 The prevalence of affective illness is approximately 6% among the general population, and 13% to 30% among RA patients.4 In arthritic populations, 52% exhibit depression and anxiety; joint discomfort contributes to insomnia in 25% to 42% of cases.4
Arthritic pain persists despite suppressed inflammation, which suggests involvement of the CNS.5 Increased levels of IL-6 and TNF-α can cause insomnia and affect pain perception.6 Decreased conditioned pain modulation, a lower pain threshold, and pressure pain intolerance lead to increased pain awareness and heightened discomfort.
How can you help your patient who has RA?
Because the focus of care in RA is on the disease’s physical attributes, psychiatric symptoms sometimes receive less attention.7 And because arthritic symptoms overlap with anorexia, weight loss, fatigue, pain, and insomnia, affective illness can go unrecognized.
Depression rating scales can overestimate affective illness, but a history and follow-up questionnaire can facilitate an accurate diagnosis of depression and help determine the need for, and type of, intervention.
Selective serotonin reuptake inhibitors (SSRIs) are considered first‐line treatment of depression associated with RA.7 Although SSRIs for RA can be administered to the maximum recommended dosage, titration is advised in accordance with patient response and tolerance.
Tricyclic antidepressants are not as well tolerated in RA, especially in older patients; however, they have more of an analgesic effect, even at lower dosages.
Joint disease activity and mood are associated with sleep disturbance, and vice versa.5 Insomnia calls for patient education about sleep hygiene, avoiding caffeine and other stimulants, and an individualized appraisal of options for pharmacotherapy.
Alleviating RA pain is important for psychosocial health.8 Although the medical team’s emphasis should be on controlling inflammation to minimize joint damage and pain, be sure to address your RA patients’ mood symptoms to improve the quality of their life.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Joint disease is the most common cause of disability and the source of considerable psychological distress. In the United States, 50 million adults complain of joint pain; in 2007, 1.5 million people suffered from rheumatoid arthritis (RA). A chronic inflammatory autoimmune disease of joints, RA can involve almost all organs.1
The link to mental illness
Mental illness in RA patients often is underdiagnosed and undertreated. These missed opportunities contribute to poor compliance with medical therapy, suboptimal therapeutic response, greater disability, and diminished quality of life.2
Limited mobility, chronic pain, sleep disturbance, fatigue, and immunological factors predispose RA patients to depression and anxiety.3 The proinflammatory cytokines, tumor necrosis factor-α (TNF-α), interleukin 1 (IL-1), IL-6, and interferon-g have a role in inducing affective symptoms. There also is a relationship between an elevated IL-17 level and anxiety.
Research substantiates a relationship between RA and depression.3 The prevalence of affective illness is approximately 6% among the general population, and 13% to 30% among RA patients.4 In arthritic populations, 52% exhibit depression and anxiety; joint discomfort contributes to insomnia in 25% to 42% of cases.4
Arthritic pain persists despite suppressed inflammation, which suggests involvement of the CNS.5 Increased levels of IL-6 and TNF-α can cause insomnia and affect pain perception.6 Decreased conditioned pain modulation, a lower pain threshold, and pressure pain intolerance lead to increased pain awareness and heightened discomfort.
How can you help your patient who has RA?
Because the focus of care in RA is on the disease’s physical attributes, psychiatric symptoms sometimes receive less attention.7 And because arthritic symptoms overlap with anorexia, weight loss, fatigue, pain, and insomnia, affective illness can go unrecognized.
Depression rating scales can overestimate affective illness, but a history and follow-up questionnaire can facilitate an accurate diagnosis of depression and help determine the need for, and type of, intervention.
Selective serotonin reuptake inhibitors (SSRIs) are considered first‐line treatment of depression associated with RA.7 Although SSRIs for RA can be administered to the maximum recommended dosage, titration is advised in accordance with patient response and tolerance.
Tricyclic antidepressants are not as well tolerated in RA, especially in older patients; however, they have more of an analgesic effect, even at lower dosages.
Joint disease activity and mood are associated with sleep disturbance, and vice versa.5 Insomnia calls for patient education about sleep hygiene, avoiding caffeine and other stimulants, and an individualized appraisal of options for pharmacotherapy.
Alleviating RA pain is important for psychosocial health.8 Although the medical team’s emphasis should be on controlling inflammation to minimize joint damage and pain, be sure to address your RA patients’ mood symptoms to improve the quality of their life.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Centers for Disease Control and Prevention. Arthritis-related statistics. http://www.cdc.gov/arthritis/data_statistics/arthritis_related_stats.htm. Updated August 1, 2011. Accessed January 4, 2013.
2. Shih M, Hootman JM, Strine TW, et al. Serious psychological distress in U.S. adults with arthritis. J Gen Intern Med. 2006;21(11):1160-1166.
3. Sato E, Nishimura K, Nakajima A, et al. Major depressive disorder in patients with rheumatoid arthritis. Mod Rheumatol. 2013;23(2):237-244.
4. Wolfe F, Michaud K, Li T. Sleep disturbance in patients with rheumatoid arthritis: evaluation by medical outcomes study and visual analog sleep scales. J Rheumatol. 2006;33(10):1942-1951.
5. Fragiadaki K, Tektonidou MG, Konsta M, et al. Sleep disturbances and interleukin 6 receptor inhibition in rheumatoid arthritis. J Rheumatol. 2012;39(1):60-62.
6. Lee YC, Lu B, Edwards RR, et al. The role of sleep problems in central pain processing in rheumatoid arthritis. Arthritis Rheum. 2013;65(1):59-68.
7. Dickens C, Creed F. The burden of depression in patients with rheumatoid arthritis. Rheumatology (Oxford). 2001; 40(12):1327-1330.
8. Courvoisier DS, Agoritsas T, Glauser J, et al. Pain as an important predictor of psychosocial health in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2012;64(2):190-196.
1. Centers for Disease Control and Prevention. Arthritis-related statistics. http://www.cdc.gov/arthritis/data_statistics/arthritis_related_stats.htm. Updated August 1, 2011. Accessed January 4, 2013.
2. Shih M, Hootman JM, Strine TW, et al. Serious psychological distress in U.S. adults with arthritis. J Gen Intern Med. 2006;21(11):1160-1166.
3. Sato E, Nishimura K, Nakajima A, et al. Major depressive disorder in patients with rheumatoid arthritis. Mod Rheumatol. 2013;23(2):237-244.
4. Wolfe F, Michaud K, Li T. Sleep disturbance in patients with rheumatoid arthritis: evaluation by medical outcomes study and visual analog sleep scales. J Rheumatol. 2006;33(10):1942-1951.
5. Fragiadaki K, Tektonidou MG, Konsta M, et al. Sleep disturbances and interleukin 6 receptor inhibition in rheumatoid arthritis. J Rheumatol. 2012;39(1):60-62.
6. Lee YC, Lu B, Edwards RR, et al. The role of sleep problems in central pain processing in rheumatoid arthritis. Arthritis Rheum. 2013;65(1):59-68.
7. Dickens C, Creed F. The burden of depression in patients with rheumatoid arthritis. Rheumatology (Oxford). 2001; 40(12):1327-1330.
8. Courvoisier DS, Agoritsas T, Glauser J, et al. Pain as an important predictor of psychosocial health in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2012;64(2):190-196.