User login
Biomarker testing for treatment of metastatic colorectal cancer: role of the pathologist in community practice
The past decade has been marked by significant advancements in the treatment of patients with metastatic colorectal cancer (mCRC), including the approval of novel biologic agents such as the angiogenesis inhibitors bevacizumab and afibercept and the epidermal growth factor receptor monoclonal antibodies (mAbs) cetuximab and panitumumab. Cetuximab was recently approved by the US Food and Drug Administration in combination with FOLFIRI (irinotecan, 5-fuorouracil, leucovorin) for the first-line treatment of patients with KRAS mutation-negative (wild-type) tumors as determined by an FDA-approved companion diagnostic. It was the first FDA approval in mCRC requiring use of a diagnostic test that is predictive of response prior to initiation of frontline therapy.
Click on the PDF icon at the top of this introduction to read the full article.
The past decade has been marked by significant advancements in the treatment of patients with metastatic colorectal cancer (mCRC), including the approval of novel biologic agents such as the angiogenesis inhibitors bevacizumab and afibercept and the epidermal growth factor receptor monoclonal antibodies (mAbs) cetuximab and panitumumab. Cetuximab was recently approved by the US Food and Drug Administration in combination with FOLFIRI (irinotecan, 5-fuorouracil, leucovorin) for the first-line treatment of patients with KRAS mutation-negative (wild-type) tumors as determined by an FDA-approved companion diagnostic. It was the first FDA approval in mCRC requiring use of a diagnostic test that is predictive of response prior to initiation of frontline therapy.
Click on the PDF icon at the top of this introduction to read the full article.
The past decade has been marked by significant advancements in the treatment of patients with metastatic colorectal cancer (mCRC), including the approval of novel biologic agents such as the angiogenesis inhibitors bevacizumab and afibercept and the epidermal growth factor receptor monoclonal antibodies (mAbs) cetuximab and panitumumab. Cetuximab was recently approved by the US Food and Drug Administration in combination with FOLFIRI (irinotecan, 5-fuorouracil, leucovorin) for the first-line treatment of patients with KRAS mutation-negative (wild-type) tumors as determined by an FDA-approved companion diagnostic. It was the first FDA approval in mCRC requiring use of a diagnostic test that is predictive of response prior to initiation of frontline therapy.
Click on the PDF icon at the top of this introduction to read the full article.
Current options and future directions in the systemic treatment of metastatic melanoma
Systemic treatment options for metastatic melanoma have historically been limited, with conventional cytotoxic chemotherapies demonstrating only modest benefit. Recent advances, however, have dramatically changed the treatment landscape and can be considered in 2 general categories: immunotherapeutic approaches that enhance antitumor immunity, and targeted therapeutic approaches that block oncogenic driver mutations. Immunotherapy with antibodies that block cytotoxic T-lymphocyte antigen 4 and programmed death-1 receptor can result in durable responses in a subset of patients. These treatments may be considered for patients irrespective of their mutational status, and ongoing research continues to investigate biomarkers associated with clinical outcomes. Side effects of these agents result from immune-mediated reactions involving various organ sites and can include: diarrhea, rash, hepatitis, and endocrinopathies.
Click on the PDF icon at the top of this introduction to read the full article.
Systemic treatment options for metastatic melanoma have historically been limited, with conventional cytotoxic chemotherapies demonstrating only modest benefit. Recent advances, however, have dramatically changed the treatment landscape and can be considered in 2 general categories: immunotherapeutic approaches that enhance antitumor immunity, and targeted therapeutic approaches that block oncogenic driver mutations. Immunotherapy with antibodies that block cytotoxic T-lymphocyte antigen 4 and programmed death-1 receptor can result in durable responses in a subset of patients. These treatments may be considered for patients irrespective of their mutational status, and ongoing research continues to investigate biomarkers associated with clinical outcomes. Side effects of these agents result from immune-mediated reactions involving various organ sites and can include: diarrhea, rash, hepatitis, and endocrinopathies.
Click on the PDF icon at the top of this introduction to read the full article.
Systemic treatment options for metastatic melanoma have historically been limited, with conventional cytotoxic chemotherapies demonstrating only modest benefit. Recent advances, however, have dramatically changed the treatment landscape and can be considered in 2 general categories: immunotherapeutic approaches that enhance antitumor immunity, and targeted therapeutic approaches that block oncogenic driver mutations. Immunotherapy with antibodies that block cytotoxic T-lymphocyte antigen 4 and programmed death-1 receptor can result in durable responses in a subset of patients. These treatments may be considered for patients irrespective of their mutational status, and ongoing research continues to investigate biomarkers associated with clinical outcomes. Side effects of these agents result from immune-mediated reactions involving various organ sites and can include: diarrhea, rash, hepatitis, and endocrinopathies.
Click on the PDF icon at the top of this introduction to read the full article.
Principles and Characteristics of an HMG
With the continuing growth of the specialty of hospital medicine, the capabilities and performance of hospital medicine groups (HMGs) varies significantly. There are few guidelines that HMGs can reference as tools to guide self‐improvement. To address this deficiency, the Society of Hospital Medicine (SHM) Board of Directors authorized a process to identify the key principles and characteristics of an effective HMG.
METHODS
Topic Development and Validation Prework
In providing direction to this effort, the SHM board felt that the principles and characteristics should be directed at both hospitals and hospitalists, addressing the full range of managerial, organizational, clinical, and quality activities necessary to achieve effectiveness. Furthermore, the board defined effectiveness as consisting of 2 components. First, the HMG must assure that the patients managed by hospitalists receive high‐quality care that is sensitive to their needs and preferences. Second, the HMG must understand that the central role of the hospitalist is to coordinate patient care and foster interdisciplinary communication across the care continuum to provide optimal patient outcomes.
The SHM board appointed an HMG Characteristics Workgroup consisting of individuals who have experience with a wide array of HMG models and who could offer expert opinions on the subject. The HMG Characteristics Workgroup felt it important to review the work of other organizations that develop and administer criteria, standards, and/or requirements for healthcare organizations. Examples cited were the American College of Surgeons[1]; The Joint Commission[2]; American Nurse Credentialing Center[3]; the National Committee for Quality Assurance[4]; the American Medical Group Association[5]; and the American Association of Critical‐Care Nurses.[6]
In March 2012 and April 2012, SHM staff reviewed the websites and published materials of these organizations. For each program, information was captured on the qualifications of applicants, history of the program, timing of administering the program, the nature of recognition granted, and the program's keys to success. The summary of these findings was shared with the workgroup.
Background research and the broad scope of characteristics to be addressed led to the workgroup's decision to develop the principles and characteristics using a consensus process, emphasizing expert opinion supplemented by feedback from a broad group of stakeholders.
Initial Draft
During April 2012 and May 2012, the HMG Characteristics Workgroup identified 3 domains for the key characteristics: (1) program structure and operations, (2) clinical care delivery, and (3) organizational performance improvement. Over the course of several meetings, the HMG Characteristics Workgroup developed an initial draft of 83 characteristics, grouped into 29 subgroups within the 3 domains.
From June 2012 to November 2012, this initial draft was reviewed by a broad cross section of the hospital medicine community including members of SHM's committees, a group of academic hospitalists, focus groups in 2 communities (Philadelphia and Boston), and the leaders of several regional and national hospitalist management companies. Quantitative and qualitative feedback was obtained.
In November 2012, the SHM Board of Directors held its annual leadership meeting, attended by approximately 25 national hospitalist thought leaders and chairpersons of SHM committees. At this meeting, a series of exercises were conducted in which these leaders of the hospital medicine movement, including the SHM board members, were each assigned individual characteristics and asked to review and edit them for clarity and appropriateness.
As a result of feedback at that meeting and subsequent discussion by the SHM board, the workgroup was asked to modify the characteristics in 3 ways. First, the list should be streamlined, reducing the number of characteristics. Second, the 3 domains should be eliminated, and a better organizing framework should be created. Third, additional context should be added to the list of characteristics.
Second Draft
During the period from November 2012 to December 2012, the HMG Characteristics Workgroup went through a 2‐step Delphi process to consolidate characteristics and/or eliminate characteristics that were redundant or unnecessary. In the first step, members of the workgroup rated each characteristic from 1 to 3. A rating of 1 meant not important; good quality, but not required for an effective HMG. A rating of 2 meant important; most effective HMGs will meet requirement. A rating of 3 meant highly important; mandatory for an effective HMG. In the second step, members of the workgroup received feedback on the scores for each characteristic and came to a consensus on which characteristics should be eliminated or merged with other characteristics.
As a result, the number of characteristics was reduced and consolidated from 83 to 47, and a new framing structure was defined, replacing the 3 domains with 10 organizing principles. Finally, a rationale for each characteristic was added, defending its inclusion in the list. In addition, consideration was given to including a section describing how an HMG could demonstrate that their organization met each characteristic. However, the workgroup and the board decided that these demonstration requirements should be vetted before they were published.
From January 2013 to June 2013, the revised key principles and characteristics were reviewed by selected chairpersons of SHM committees and by 2 focus groups of HMG leaders. These reviews were conducted at the SHM Annual Meeting. Finally, in June 2013, the Committee on Clinical Leadership of the American Hospital Association reviewed and commented on the draft of the principles and characteristics.
In addition, based on feedback received from the reviewers, the wording of many of the characteristics went through revisions to assure precision and clarity. Before submission to the Journal of Hospital Medicine, a professional editor was engaged to assure that the format and language of the characteristics were clear and consistent.
Final Approval
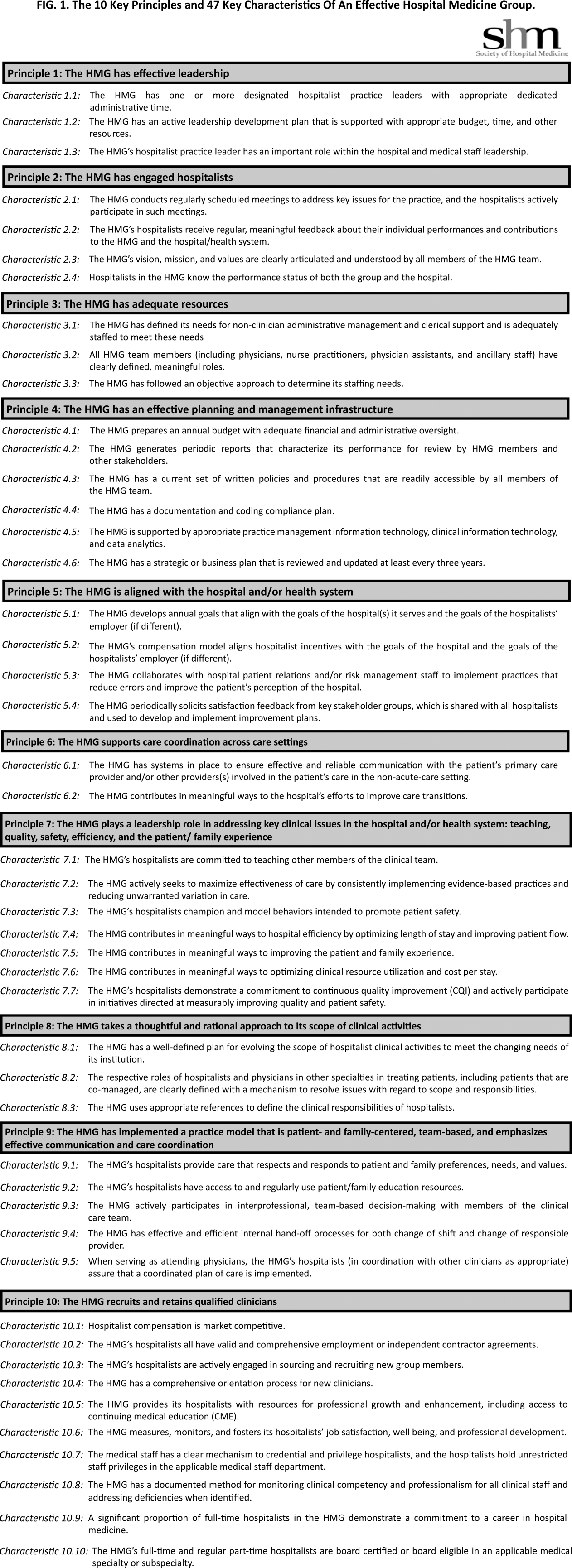
The final draft of the 10 principles and 47 characteristics was approved for publication at a meeting of the SHM Board of Directors in September 2013 (Figure 1).
RESULTS
A recurring issue that the workgroup addressed was the applicability of the characteristics from 1 practice setting to another. Confounding factors include the HMG's employment/organizational model (eg, hospital employed, academic, multispecialty group, private practice, and management company), its population served (eg, adult vs pediatric, more than 1 hospital), and the type of hospital served (eg, academic vs community, the hospital has more than 1 HMG). The workgroup has made an effort to assure that all 47 characteristics can be applied to every type of HMG.
In developing the 10 principles, the workgroup attempted to construct a list of the basic ingredients needed to build and sustain an effective HMG. These 10 principles stand on their own, independent of the 47 key characteristics, and include issues such as effective leadership, clinician engagement, adequate resources, management infrastructure, key hospitalist roles and responsibilities, alignment with the hospital, and the recruitment and retention of qualified hospitalists.
A more detailed version of the Key Principles and Characteristics of an Effective HMG is available in the online version of this article (see Supporting Information, Appendix, in the online version of this article). The online Appendix includes the rationales for each of the characteristics, guidance on how to provide feedback to the SHM on the framework, and the SHM's plan for further development of the key principles and characteristics.
DISCUSSION
To address the variability in capabilities and performance of HMGs, these principles and characteristics are designed to provide a framework for HMGs seeking to conduct self‐assessments and develop pathways for improvement.
Although there may be HMG arrangements that do not directly involve the hospital and its executive team, and therefore alternative approaches may make sense, for most HMGs hospitals are directly involved with the HMG as either an employer or a contractor. For that reason, the Key Principles and Characteristics of an Effective HMG is written for 2 audiences: the executive leadership of the hospital (most specifically the chief medical officer or a similar role) and the hospitalists in the HMG (most specifically the practice medical director). To address the key characteristics requires the active participation of both parties. For the hospital executives, the framework establishes expectations for the HMG. For the hospitalists, the framework provides guidance in the development of an improvement plan.
Hospital executives and hospitalists can use the key characteristics in a broad spectrum of ways. The easiest and least formalized approach would be to use the framework as the basis of an ongoing dialogue between the hospital leadership and the HMG. A more formal approach would be to use the framework to guide the planning and budgeting activities of the HMG. Finally, a hospital or health system can use the key principles and characteristics as a way to evaluate their affiliated HMG(s)for example, the HMG must address 80% of the 47 characteristics.
The Key Principles and Characteristics of an Effective HMG should be considered akin to the Core Competencies in Hospital Medicine previously published in the Journal of Hospital Medicine.[7] However, instead of focusing on the competencies of individual physicians, this framework focuses on the characteristics of hospitalist groups. Just as a physician or other healthcare provider is not expected to demonstrate competency for every element in the core competencies document, an HMG does not need to have all 47 characteristics to be effective. Effective hospitalists may have skills other than those listed in the Core Competencies in Hospital Medicine. Similarly, the 47 characteristics do not represent an exhaustive list of every desirable HMG attribute. In general, effective HMGs should possess most of the characteristics.
In applying the framework, the HMG should not simply attempt to evaluate each characteristic with a yes or no assessment. For HMGs responding yes, there may be a wide range of performancefrom meeting the bare minimum requirements to employing sophisticated, expansive measures to excel in the characteristic.
SHM encourages hospital leaders and HMG leaders to use these characteristics to perform an HMG self‐assessment and to develop a plan. The plan could address implementation of selected characteristics that are not currently being addressed by the HMG or the development of additional behaviors, tools, resources, and capabilities that more fully incorporate those characteristics for which the HMG meets only minimum requirements. In addition, the plan could address the impact that a larger organization (eg, health system, hospital, or employer) may have on a given characteristic.
As outlined above, the process used to develop the Key Principles and Characteristics of an Effective HMG was grounded in expert opinion and extensive review and feedback. HMGs that use the framework should recognize that others might have a different opinion. For example, characteristic 5.2 states, The HMG's compensation model aligns hospitalist incentives with the goals of the hospital and the goals of the hospitalist's employer (if different). There are likely to be experienced hospitalist leaders who believe that an effective HMG does not need to have an incentive compensation system. However, the consensus process employed to develop the key characteristics led to the conclusion that an effective HMG should have an incentive compensation system.
The publication of the Key Principles and Characteristics of an Effective HMG may lead to negative and/or unintended consequences. A self‐assessment by an HMG using this framework could require a significant level of effort on behalf of the HMG, whereas implementing remedial efforts to address the characteristics could require an investment of time and money that could take away from other important issues facing the HMG. Many HMGs may be held accountable for addressing these characteristics without the necessary financial support from their hospital or medical group. Finally, the publication of the document could create a backlash from members of the hospitalist community who do not think that the SHM should be in the business of defining what characterizes an effective HMG, rather that this definition should be left to the marketplace.
Despite these concerns, the leadership of the SHM expects that the publication of the Key Principles and Characteristics of an Effective HMG will lead to overall improvement in the capabilities and performance of HMGs.
CONCLUSIONS
The Key Principles and Characteristics of an Effective HMG have been designed to be aspirational, helping to raise the bar for the specialty of hospital medicine. These principles and characteristics could provide a framework for HMGs seeking to conduct self‐assessments, outlining a pathway for improvement, and better defining the central role of hospitalists in coordinating team‐based, patient‐centered care in the acute care setting.
Acknowledgments
Disclosures: Patrick Cawley, MD: none; Steven Deitelzweig, MD: none; Leslie Flores, MHA: provides consulting to hospital medicine groups; Joseph A. Miller, MS: none; John Nelson, MD: provides consulting to hospital medicine groups; Scott Rissmiller, MD: none; Laurence Wellikson, MD: none; Winthrop F. Whitcomb, MD: provides consulting to hospital medicine groups.
- American College of Surgeons. New verification site visit outcomes. Available at: http://www.facs.org/trauma/verifivisitoutcomes.html. Accessed September 3, 2013.
- Hospital accreditation standards 2012. Oakbrook Terrace, IL: The Joint Commission; 2012. Available at: Amazon.com: http://www.amazon.com/Hospital‐Accreditation‐Standards‐Joint‐Commission/dp/1599404257
- The magnet model: components and sources of evidence. Silver Spring, MD: American Nurse Credentialing Center; 2011. Available at: Amazon.com: http://www.amazon.com/Magnet‐Model‐Components‐Sources‐Evidence/dp/1935213229.
- Patient Centered Medical Home Standards and Guidelines. National Committee for Quality Assurance. Available at: https://inetshop01.pub.ncqa.org/Publications/deptCate.asp?dept_id=21(suppl 1):2–95.
With the continuing growth of the specialty of hospital medicine, the capabilities and performance of hospital medicine groups (HMGs) varies significantly. There are few guidelines that HMGs can reference as tools to guide self‐improvement. To address this deficiency, the Society of Hospital Medicine (SHM) Board of Directors authorized a process to identify the key principles and characteristics of an effective HMG.
METHODS
Topic Development and Validation Prework
In providing direction to this effort, the SHM board felt that the principles and characteristics should be directed at both hospitals and hospitalists, addressing the full range of managerial, organizational, clinical, and quality activities necessary to achieve effectiveness. Furthermore, the board defined effectiveness as consisting of 2 components. First, the HMG must assure that the patients managed by hospitalists receive high‐quality care that is sensitive to their needs and preferences. Second, the HMG must understand that the central role of the hospitalist is to coordinate patient care and foster interdisciplinary communication across the care continuum to provide optimal patient outcomes.
The SHM board appointed an HMG Characteristics Workgroup consisting of individuals who have experience with a wide array of HMG models and who could offer expert opinions on the subject. The HMG Characteristics Workgroup felt it important to review the work of other organizations that develop and administer criteria, standards, and/or requirements for healthcare organizations. Examples cited were the American College of Surgeons[1]; The Joint Commission[2]; American Nurse Credentialing Center[3]; the National Committee for Quality Assurance[4]; the American Medical Group Association[5]; and the American Association of Critical‐Care Nurses.[6]
In March 2012 and April 2012, SHM staff reviewed the websites and published materials of these organizations. For each program, information was captured on the qualifications of applicants, history of the program, timing of administering the program, the nature of recognition granted, and the program's keys to success. The summary of these findings was shared with the workgroup.
Background research and the broad scope of characteristics to be addressed led to the workgroup's decision to develop the principles and characteristics using a consensus process, emphasizing expert opinion supplemented by feedback from a broad group of stakeholders.
Initial Draft
During April 2012 and May 2012, the HMG Characteristics Workgroup identified 3 domains for the key characteristics: (1) program structure and operations, (2) clinical care delivery, and (3) organizational performance improvement. Over the course of several meetings, the HMG Characteristics Workgroup developed an initial draft of 83 characteristics, grouped into 29 subgroups within the 3 domains.
From June 2012 to November 2012, this initial draft was reviewed by a broad cross section of the hospital medicine community including members of SHM's committees, a group of academic hospitalists, focus groups in 2 communities (Philadelphia and Boston), and the leaders of several regional and national hospitalist management companies. Quantitative and qualitative feedback was obtained.
In November 2012, the SHM Board of Directors held its annual leadership meeting, attended by approximately 25 national hospitalist thought leaders and chairpersons of SHM committees. At this meeting, a series of exercises were conducted in which these leaders of the hospital medicine movement, including the SHM board members, were each assigned individual characteristics and asked to review and edit them for clarity and appropriateness.
As a result of feedback at that meeting and subsequent discussion by the SHM board, the workgroup was asked to modify the characteristics in 3 ways. First, the list should be streamlined, reducing the number of characteristics. Second, the 3 domains should be eliminated, and a better organizing framework should be created. Third, additional context should be added to the list of characteristics.
Second Draft
During the period from November 2012 to December 2012, the HMG Characteristics Workgroup went through a 2‐step Delphi process to consolidate characteristics and/or eliminate characteristics that were redundant or unnecessary. In the first step, members of the workgroup rated each characteristic from 1 to 3. A rating of 1 meant not important; good quality, but not required for an effective HMG. A rating of 2 meant important; most effective HMGs will meet requirement. A rating of 3 meant highly important; mandatory for an effective HMG. In the second step, members of the workgroup received feedback on the scores for each characteristic and came to a consensus on which characteristics should be eliminated or merged with other characteristics.
As a result, the number of characteristics was reduced and consolidated from 83 to 47, and a new framing structure was defined, replacing the 3 domains with 10 organizing principles. Finally, a rationale for each characteristic was added, defending its inclusion in the list. In addition, consideration was given to including a section describing how an HMG could demonstrate that their organization met each characteristic. However, the workgroup and the board decided that these demonstration requirements should be vetted before they were published.
From January 2013 to June 2013, the revised key principles and characteristics were reviewed by selected chairpersons of SHM committees and by 2 focus groups of HMG leaders. These reviews were conducted at the SHM Annual Meeting. Finally, in June 2013, the Committee on Clinical Leadership of the American Hospital Association reviewed and commented on the draft of the principles and characteristics.
In addition, based on feedback received from the reviewers, the wording of many of the characteristics went through revisions to assure precision and clarity. Before submission to the Journal of Hospital Medicine, a professional editor was engaged to assure that the format and language of the characteristics were clear and consistent.
Final Approval
The final draft of the 10 principles and 47 characteristics was approved for publication at a meeting of the SHM Board of Directors in September 2013 (Figure 1).
RESULTS
A recurring issue that the workgroup addressed was the applicability of the characteristics from 1 practice setting to another. Confounding factors include the HMG's employment/organizational model (eg, hospital employed, academic, multispecialty group, private practice, and management company), its population served (eg, adult vs pediatric, more than 1 hospital), and the type of hospital served (eg, academic vs community, the hospital has more than 1 HMG). The workgroup has made an effort to assure that all 47 characteristics can be applied to every type of HMG.
In developing the 10 principles, the workgroup attempted to construct a list of the basic ingredients needed to build and sustain an effective HMG. These 10 principles stand on their own, independent of the 47 key characteristics, and include issues such as effective leadership, clinician engagement, adequate resources, management infrastructure, key hospitalist roles and responsibilities, alignment with the hospital, and the recruitment and retention of qualified hospitalists.
A more detailed version of the Key Principles and Characteristics of an Effective HMG is available in the online version of this article (see Supporting Information, Appendix, in the online version of this article). The online Appendix includes the rationales for each of the characteristics, guidance on how to provide feedback to the SHM on the framework, and the SHM's plan for further development of the key principles and characteristics.
DISCUSSION
To address the variability in capabilities and performance of HMGs, these principles and characteristics are designed to provide a framework for HMGs seeking to conduct self‐assessments and develop pathways for improvement.
Although there may be HMG arrangements that do not directly involve the hospital and its executive team, and therefore alternative approaches may make sense, for most HMGs hospitals are directly involved with the HMG as either an employer or a contractor. For that reason, the Key Principles and Characteristics of an Effective HMG is written for 2 audiences: the executive leadership of the hospital (most specifically the chief medical officer or a similar role) and the hospitalists in the HMG (most specifically the practice medical director). To address the key characteristics requires the active participation of both parties. For the hospital executives, the framework establishes expectations for the HMG. For the hospitalists, the framework provides guidance in the development of an improvement plan.
Hospital executives and hospitalists can use the key characteristics in a broad spectrum of ways. The easiest and least formalized approach would be to use the framework as the basis of an ongoing dialogue between the hospital leadership and the HMG. A more formal approach would be to use the framework to guide the planning and budgeting activities of the HMG. Finally, a hospital or health system can use the key principles and characteristics as a way to evaluate their affiliated HMG(s)for example, the HMG must address 80% of the 47 characteristics.
The Key Principles and Characteristics of an Effective HMG should be considered akin to the Core Competencies in Hospital Medicine previously published in the Journal of Hospital Medicine.[7] However, instead of focusing on the competencies of individual physicians, this framework focuses on the characteristics of hospitalist groups. Just as a physician or other healthcare provider is not expected to demonstrate competency for every element in the core competencies document, an HMG does not need to have all 47 characteristics to be effective. Effective hospitalists may have skills other than those listed in the Core Competencies in Hospital Medicine. Similarly, the 47 characteristics do not represent an exhaustive list of every desirable HMG attribute. In general, effective HMGs should possess most of the characteristics.
In applying the framework, the HMG should not simply attempt to evaluate each characteristic with a yes or no assessment. For HMGs responding yes, there may be a wide range of performancefrom meeting the bare minimum requirements to employing sophisticated, expansive measures to excel in the characteristic.
SHM encourages hospital leaders and HMG leaders to use these characteristics to perform an HMG self‐assessment and to develop a plan. The plan could address implementation of selected characteristics that are not currently being addressed by the HMG or the development of additional behaviors, tools, resources, and capabilities that more fully incorporate those characteristics for which the HMG meets only minimum requirements. In addition, the plan could address the impact that a larger organization (eg, health system, hospital, or employer) may have on a given characteristic.
As outlined above, the process used to develop the Key Principles and Characteristics of an Effective HMG was grounded in expert opinion and extensive review and feedback. HMGs that use the framework should recognize that others might have a different opinion. For example, characteristic 5.2 states, The HMG's compensation model aligns hospitalist incentives with the goals of the hospital and the goals of the hospitalist's employer (if different). There are likely to be experienced hospitalist leaders who believe that an effective HMG does not need to have an incentive compensation system. However, the consensus process employed to develop the key characteristics led to the conclusion that an effective HMG should have an incentive compensation system.
The publication of the Key Principles and Characteristics of an Effective HMG may lead to negative and/or unintended consequences. A self‐assessment by an HMG using this framework could require a significant level of effort on behalf of the HMG, whereas implementing remedial efforts to address the characteristics could require an investment of time and money that could take away from other important issues facing the HMG. Many HMGs may be held accountable for addressing these characteristics without the necessary financial support from their hospital or medical group. Finally, the publication of the document could create a backlash from members of the hospitalist community who do not think that the SHM should be in the business of defining what characterizes an effective HMG, rather that this definition should be left to the marketplace.
Despite these concerns, the leadership of the SHM expects that the publication of the Key Principles and Characteristics of an Effective HMG will lead to overall improvement in the capabilities and performance of HMGs.
CONCLUSIONS
The Key Principles and Characteristics of an Effective HMG have been designed to be aspirational, helping to raise the bar for the specialty of hospital medicine. These principles and characteristics could provide a framework for HMGs seeking to conduct self‐assessments, outlining a pathway for improvement, and better defining the central role of hospitalists in coordinating team‐based, patient‐centered care in the acute care setting.
Acknowledgments
Disclosures: Patrick Cawley, MD: none; Steven Deitelzweig, MD: none; Leslie Flores, MHA: provides consulting to hospital medicine groups; Joseph A. Miller, MS: none; John Nelson, MD: provides consulting to hospital medicine groups; Scott Rissmiller, MD: none; Laurence Wellikson, MD: none; Winthrop F. Whitcomb, MD: provides consulting to hospital medicine groups.
With the continuing growth of the specialty of hospital medicine, the capabilities and performance of hospital medicine groups (HMGs) varies significantly. There are few guidelines that HMGs can reference as tools to guide self‐improvement. To address this deficiency, the Society of Hospital Medicine (SHM) Board of Directors authorized a process to identify the key principles and characteristics of an effective HMG.
METHODS
Topic Development and Validation Prework
In providing direction to this effort, the SHM board felt that the principles and characteristics should be directed at both hospitals and hospitalists, addressing the full range of managerial, organizational, clinical, and quality activities necessary to achieve effectiveness. Furthermore, the board defined effectiveness as consisting of 2 components. First, the HMG must assure that the patients managed by hospitalists receive high‐quality care that is sensitive to their needs and preferences. Second, the HMG must understand that the central role of the hospitalist is to coordinate patient care and foster interdisciplinary communication across the care continuum to provide optimal patient outcomes.
The SHM board appointed an HMG Characteristics Workgroup consisting of individuals who have experience with a wide array of HMG models and who could offer expert opinions on the subject. The HMG Characteristics Workgroup felt it important to review the work of other organizations that develop and administer criteria, standards, and/or requirements for healthcare organizations. Examples cited were the American College of Surgeons[1]; The Joint Commission[2]; American Nurse Credentialing Center[3]; the National Committee for Quality Assurance[4]; the American Medical Group Association[5]; and the American Association of Critical‐Care Nurses.[6]
In March 2012 and April 2012, SHM staff reviewed the websites and published materials of these organizations. For each program, information was captured on the qualifications of applicants, history of the program, timing of administering the program, the nature of recognition granted, and the program's keys to success. The summary of these findings was shared with the workgroup.
Background research and the broad scope of characteristics to be addressed led to the workgroup's decision to develop the principles and characteristics using a consensus process, emphasizing expert opinion supplemented by feedback from a broad group of stakeholders.
Initial Draft
During April 2012 and May 2012, the HMG Characteristics Workgroup identified 3 domains for the key characteristics: (1) program structure and operations, (2) clinical care delivery, and (3) organizational performance improvement. Over the course of several meetings, the HMG Characteristics Workgroup developed an initial draft of 83 characteristics, grouped into 29 subgroups within the 3 domains.
From June 2012 to November 2012, this initial draft was reviewed by a broad cross section of the hospital medicine community including members of SHM's committees, a group of academic hospitalists, focus groups in 2 communities (Philadelphia and Boston), and the leaders of several regional and national hospitalist management companies. Quantitative and qualitative feedback was obtained.
In November 2012, the SHM Board of Directors held its annual leadership meeting, attended by approximately 25 national hospitalist thought leaders and chairpersons of SHM committees. At this meeting, a series of exercises were conducted in which these leaders of the hospital medicine movement, including the SHM board members, were each assigned individual characteristics and asked to review and edit them for clarity and appropriateness.
As a result of feedback at that meeting and subsequent discussion by the SHM board, the workgroup was asked to modify the characteristics in 3 ways. First, the list should be streamlined, reducing the number of characteristics. Second, the 3 domains should be eliminated, and a better organizing framework should be created. Third, additional context should be added to the list of characteristics.
Second Draft
During the period from November 2012 to December 2012, the HMG Characteristics Workgroup went through a 2‐step Delphi process to consolidate characteristics and/or eliminate characteristics that were redundant or unnecessary. In the first step, members of the workgroup rated each characteristic from 1 to 3. A rating of 1 meant not important; good quality, but not required for an effective HMG. A rating of 2 meant important; most effective HMGs will meet requirement. A rating of 3 meant highly important; mandatory for an effective HMG. In the second step, members of the workgroup received feedback on the scores for each characteristic and came to a consensus on which characteristics should be eliminated or merged with other characteristics.
As a result, the number of characteristics was reduced and consolidated from 83 to 47, and a new framing structure was defined, replacing the 3 domains with 10 organizing principles. Finally, a rationale for each characteristic was added, defending its inclusion in the list. In addition, consideration was given to including a section describing how an HMG could demonstrate that their organization met each characteristic. However, the workgroup and the board decided that these demonstration requirements should be vetted before they were published.
From January 2013 to June 2013, the revised key principles and characteristics were reviewed by selected chairpersons of SHM committees and by 2 focus groups of HMG leaders. These reviews were conducted at the SHM Annual Meeting. Finally, in June 2013, the Committee on Clinical Leadership of the American Hospital Association reviewed and commented on the draft of the principles and characteristics.
In addition, based on feedback received from the reviewers, the wording of many of the characteristics went through revisions to assure precision and clarity. Before submission to the Journal of Hospital Medicine, a professional editor was engaged to assure that the format and language of the characteristics were clear and consistent.
Final Approval
The final draft of the 10 principles and 47 characteristics was approved for publication at a meeting of the SHM Board of Directors in September 2013 (Figure 1).
RESULTS
A recurring issue that the workgroup addressed was the applicability of the characteristics from 1 practice setting to another. Confounding factors include the HMG's employment/organizational model (eg, hospital employed, academic, multispecialty group, private practice, and management company), its population served (eg, adult vs pediatric, more than 1 hospital), and the type of hospital served (eg, academic vs community, the hospital has more than 1 HMG). The workgroup has made an effort to assure that all 47 characteristics can be applied to every type of HMG.
In developing the 10 principles, the workgroup attempted to construct a list of the basic ingredients needed to build and sustain an effective HMG. These 10 principles stand on their own, independent of the 47 key characteristics, and include issues such as effective leadership, clinician engagement, adequate resources, management infrastructure, key hospitalist roles and responsibilities, alignment with the hospital, and the recruitment and retention of qualified hospitalists.
A more detailed version of the Key Principles and Characteristics of an Effective HMG is available in the online version of this article (see Supporting Information, Appendix, in the online version of this article). The online Appendix includes the rationales for each of the characteristics, guidance on how to provide feedback to the SHM on the framework, and the SHM's plan for further development of the key principles and characteristics.
DISCUSSION
To address the variability in capabilities and performance of HMGs, these principles and characteristics are designed to provide a framework for HMGs seeking to conduct self‐assessments and develop pathways for improvement.
Although there may be HMG arrangements that do not directly involve the hospital and its executive team, and therefore alternative approaches may make sense, for most HMGs hospitals are directly involved with the HMG as either an employer or a contractor. For that reason, the Key Principles and Characteristics of an Effective HMG is written for 2 audiences: the executive leadership of the hospital (most specifically the chief medical officer or a similar role) and the hospitalists in the HMG (most specifically the practice medical director). To address the key characteristics requires the active participation of both parties. For the hospital executives, the framework establishes expectations for the HMG. For the hospitalists, the framework provides guidance in the development of an improvement plan.
Hospital executives and hospitalists can use the key characteristics in a broad spectrum of ways. The easiest and least formalized approach would be to use the framework as the basis of an ongoing dialogue between the hospital leadership and the HMG. A more formal approach would be to use the framework to guide the planning and budgeting activities of the HMG. Finally, a hospital or health system can use the key principles and characteristics as a way to evaluate their affiliated HMG(s)for example, the HMG must address 80% of the 47 characteristics.
The Key Principles and Characteristics of an Effective HMG should be considered akin to the Core Competencies in Hospital Medicine previously published in the Journal of Hospital Medicine.[7] However, instead of focusing on the competencies of individual physicians, this framework focuses on the characteristics of hospitalist groups. Just as a physician or other healthcare provider is not expected to demonstrate competency for every element in the core competencies document, an HMG does not need to have all 47 characteristics to be effective. Effective hospitalists may have skills other than those listed in the Core Competencies in Hospital Medicine. Similarly, the 47 characteristics do not represent an exhaustive list of every desirable HMG attribute. In general, effective HMGs should possess most of the characteristics.
In applying the framework, the HMG should not simply attempt to evaluate each characteristic with a yes or no assessment. For HMGs responding yes, there may be a wide range of performancefrom meeting the bare minimum requirements to employing sophisticated, expansive measures to excel in the characteristic.
SHM encourages hospital leaders and HMG leaders to use these characteristics to perform an HMG self‐assessment and to develop a plan. The plan could address implementation of selected characteristics that are not currently being addressed by the HMG or the development of additional behaviors, tools, resources, and capabilities that more fully incorporate those characteristics for which the HMG meets only minimum requirements. In addition, the plan could address the impact that a larger organization (eg, health system, hospital, or employer) may have on a given characteristic.
As outlined above, the process used to develop the Key Principles and Characteristics of an Effective HMG was grounded in expert opinion and extensive review and feedback. HMGs that use the framework should recognize that others might have a different opinion. For example, characteristic 5.2 states, The HMG's compensation model aligns hospitalist incentives with the goals of the hospital and the goals of the hospitalist's employer (if different). There are likely to be experienced hospitalist leaders who believe that an effective HMG does not need to have an incentive compensation system. However, the consensus process employed to develop the key characteristics led to the conclusion that an effective HMG should have an incentive compensation system.
The publication of the Key Principles and Characteristics of an Effective HMG may lead to negative and/or unintended consequences. A self‐assessment by an HMG using this framework could require a significant level of effort on behalf of the HMG, whereas implementing remedial efforts to address the characteristics could require an investment of time and money that could take away from other important issues facing the HMG. Many HMGs may be held accountable for addressing these characteristics without the necessary financial support from their hospital or medical group. Finally, the publication of the document could create a backlash from members of the hospitalist community who do not think that the SHM should be in the business of defining what characterizes an effective HMG, rather that this definition should be left to the marketplace.
Despite these concerns, the leadership of the SHM expects that the publication of the Key Principles and Characteristics of an Effective HMG will lead to overall improvement in the capabilities and performance of HMGs.
CONCLUSIONS
The Key Principles and Characteristics of an Effective HMG have been designed to be aspirational, helping to raise the bar for the specialty of hospital medicine. These principles and characteristics could provide a framework for HMGs seeking to conduct self‐assessments, outlining a pathway for improvement, and better defining the central role of hospitalists in coordinating team‐based, patient‐centered care in the acute care setting.
Acknowledgments
Disclosures: Patrick Cawley, MD: none; Steven Deitelzweig, MD: none; Leslie Flores, MHA: provides consulting to hospital medicine groups; Joseph A. Miller, MS: none; John Nelson, MD: provides consulting to hospital medicine groups; Scott Rissmiller, MD: none; Laurence Wellikson, MD: none; Winthrop F. Whitcomb, MD: provides consulting to hospital medicine groups.
- American College of Surgeons. New verification site visit outcomes. Available at: http://www.facs.org/trauma/verifivisitoutcomes.html. Accessed September 3, 2013.
- Hospital accreditation standards 2012. Oakbrook Terrace, IL: The Joint Commission; 2012. Available at: Amazon.com: http://www.amazon.com/Hospital‐Accreditation‐Standards‐Joint‐Commission/dp/1599404257
- The magnet model: components and sources of evidence. Silver Spring, MD: American Nurse Credentialing Center; 2011. Available at: Amazon.com: http://www.amazon.com/Magnet‐Model‐Components‐Sources‐Evidence/dp/1935213229.
- Patient Centered Medical Home Standards and Guidelines. National Committee for Quality Assurance. Available at: https://inetshop01.pub.ncqa.org/Publications/deptCate.asp?dept_id=21(suppl 1):2–95.
- American College of Surgeons. New verification site visit outcomes. Available at: http://www.facs.org/trauma/verifivisitoutcomes.html. Accessed September 3, 2013.
- Hospital accreditation standards 2012. Oakbrook Terrace, IL: The Joint Commission; 2012. Available at: Amazon.com: http://www.amazon.com/Hospital‐Accreditation‐Standards‐Joint‐Commission/dp/1599404257
- The magnet model: components and sources of evidence. Silver Spring, MD: American Nurse Credentialing Center; 2011. Available at: Amazon.com: http://www.amazon.com/Magnet‐Model‐Components‐Sources‐Evidence/dp/1935213229.
- Patient Centered Medical Home Standards and Guidelines. National Committee for Quality Assurance. Available at: https://inetshop01.pub.ncqa.org/Publications/deptCate.asp?dept_id=21(suppl 1):2–95.
von Willebrand Disease: Approach to Diagnosis and Management
von Willebrand disease (VWD) is an inherited bleeding disorder caused by deficient or defective plasma von Willebrand factor (VWF). VWF is an adhesive multimeric plasma glycoprotein that performs 2 major functions in hemostasis: it mediates platelet adhesion to injured subendothelium via glycoprotein 1bα (GPIbα), and it binds and stabilizes factor VIII (FVIII) in circulation, protecting it from proteolytic degradation by enzymes. The current VWD classification recognizes 3 types. In order to understand the role of the numerous laboratory investigations as well as the classification of VWD, it is important to review the structure and function of the VWF subunit. Bleeding symptoms reflect the defect in primary hemostasis: mucocutaneous bleeding and excessive bleeding after surgery or trauma. Treatment focuses on increasing VWF levels with desmopressin (1-deamino-8-D-arginine vasopressin, DDAVP) or clotting factor concentrates containing both VWF and FVIII (VWF/FVIII concentrate). Nonspecific treatment options include antifibrinolytic agents (tranexamic acid) and hormone therapy (oral contraceptive pill).
To read the full article in PDF:
von Willebrand disease (VWD) is an inherited bleeding disorder caused by deficient or defective plasma von Willebrand factor (VWF). VWF is an adhesive multimeric plasma glycoprotein that performs 2 major functions in hemostasis: it mediates platelet adhesion to injured subendothelium via glycoprotein 1bα (GPIbα), and it binds and stabilizes factor VIII (FVIII) in circulation, protecting it from proteolytic degradation by enzymes. The current VWD classification recognizes 3 types. In order to understand the role of the numerous laboratory investigations as well as the classification of VWD, it is important to review the structure and function of the VWF subunit. Bleeding symptoms reflect the defect in primary hemostasis: mucocutaneous bleeding and excessive bleeding after surgery or trauma. Treatment focuses on increasing VWF levels with desmopressin (1-deamino-8-D-arginine vasopressin, DDAVP) or clotting factor concentrates containing both VWF and FVIII (VWF/FVIII concentrate). Nonspecific treatment options include antifibrinolytic agents (tranexamic acid) and hormone therapy (oral contraceptive pill).
To read the full article in PDF:
von Willebrand disease (VWD) is an inherited bleeding disorder caused by deficient or defective plasma von Willebrand factor (VWF). VWF is an adhesive multimeric plasma glycoprotein that performs 2 major functions in hemostasis: it mediates platelet adhesion to injured subendothelium via glycoprotein 1bα (GPIbα), and it binds and stabilizes factor VIII (FVIII) in circulation, protecting it from proteolytic degradation by enzymes. The current VWD classification recognizes 3 types. In order to understand the role of the numerous laboratory investigations as well as the classification of VWD, it is important to review the structure and function of the VWF subunit. Bleeding symptoms reflect the defect in primary hemostasis: mucocutaneous bleeding and excessive bleeding after surgery or trauma. Treatment focuses on increasing VWF levels with desmopressin (1-deamino-8-D-arginine vasopressin, DDAVP) or clotting factor concentrates containing both VWF and FVIII (VWF/FVIII concentrate). Nonspecific treatment options include antifibrinolytic agents (tranexamic acid) and hormone therapy (oral contraceptive pill).
To read the full article in PDF:
Anticoagulation and antiplatelet therapy in acute coronary syndromes
Antiplatelet and anticoagulant drugs are a cornerstone of the medical treatment of acute coronary syndrome (ACS), reducing the rates of both morbidity and death.1–4 However, reductions in ischemic events with these drugs have uniformly been accompanied by increases in bleeding complications, which reduce the net benefit.5 Thus, clinical research has been exploring ways to maximize the benefit while minimizing the risk.
Here, we review the guidelines and evidence supporting the use of antiplatelet and anticoagulant drugs in ACS.
ACUTE CORONARY SYNDROMES WITH OR WITHOUT ST ELEVATION

A key distinction when treating ACS is whether the electrocardiogram shows ST-segment elevation. In cases of non-ST-elevation ACS (ie, unstable angina or non-ST-elevation myocardial infarction), a second key question is whether the initial strategy will be invasive (with angiography performed urgently) or conservative (with angiography performed later). In ST-elevation myocardial infarction, another distinction is how perfusion is to be restored, ie, with primary percutaneous coronary intervention or with thrombolysis. All these questions affect the choice of antiplatelet and anticoagulant therapy.
Figure 1 and Figure 2 summarize the guidelines of the American College of Cardiology Foundation and American Heart Association.1,2,6,7
ANTIPLATELET THERAPY
Aspirin for all

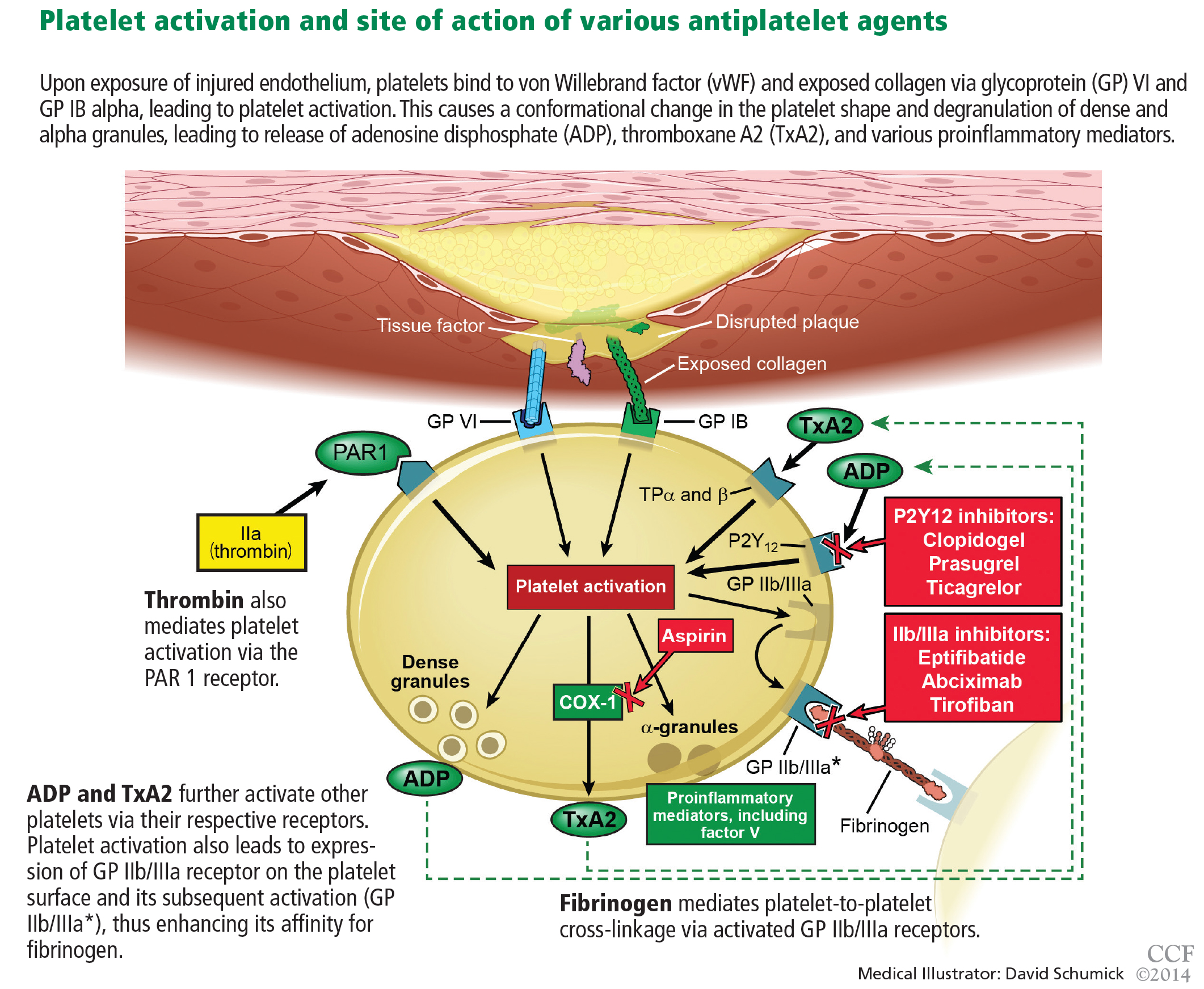
Aspirin irreversibly acetylates the enzyme cyclooxygenase-1, blocking intraplatelet formation of thromboxane A2 (Figure 3), a potent platelet aggregator and endothelial vasoconstrictor. Large clinical trials have confirmed that aspirin reduces morbidity and mortality rates by as much as 50% in patients with ACS.8
The ISIS-2 trial9 found that giving aspirin early in the emergency department significantly reduced the mortality rate.
The Antithrombotic Trialists’ Collaboration,10 in a meta-analysis of randomized controlled trials comparing different doses of aspirin in high-risk ACS patients, found no greater benefit for doses of aspirin higher than 162 mg per day when used long-term.
How to use. During an ACS, the patient should receive one dose of aspirin 325 mg (the standard high-dose pill in the United States). This dose should be chewed, as buccal absorption results in more rapid systemic effects.11
Thereafter, the patient should take 81 mg per day, continued indefinitely. The 81-mg dose also applies to patients who undergo a percutaneous coronary intervention with a drug-eluting stent.7 Previous recommendations called for higher doses, but studies have shown that higher doses pose a higher risk of bleeding without additional clinical benefit. The use of enteric-coated aspirin does not reduce this risk,12 and its delayed release may in fact cause aspirin “pseudoresistance.”13
The concurrent use of nonsteroidal anti-inflammatory drugs (NSAIDs) should be avoided, as NSAIDs reversibly bind to platelets, thus preventing aspirin from binding.14 As aspirin washes out of the body, NSAIDs may then become unbound from platelets, leaving platelets activated.
P2Y12 receptor inhibitors: Clopidogrel, prasugrel, ticagrelor
These agents bind to P2Y12 receptors on platelets to inhibit adenosine diphosphate-mediated platelet activation (Figure 3). Clopidogrel and prasugrel are irreversible prodrugs, whereas ticagrelor binds reversibly.
Clopidogrel, a prodrug
Clopidogrel has a half-life of 8 hours and a time to peak concentration of 4 hours. Eighty-five percent of a dose is inactivated by gut esterases. The remainder is metabolized primarily by the cytochrome P4502C19 enzyme system into its active metabolite.
How to use. The recommended dosage is a 600-mg bolus early in the course of ACS. This is associated with a lower rate of cardiovascular events than a 300-mg dose,2,15 although no trial has rigorously compared 300-mg vs 600-mg doses using major clinical end points. In patients presenting with ACS who cannot tolerate aspirin because of hypersensitivity or major gastrointestinal contraindication, clopidogrel is an alternative.1
The CURE trial16 randomized 12,526 patients with non-ST-elevation ACS to receive clopidogrel or placebo in addition to standard therapy. Clopidogrel was associated with a 20% lower rate of cardiovascular death, myocardial infarction, or stroke in both low- and high-risk patients regardless of whether an invasive or conservative strategy was pursued.

However, patients who underwent coronary artery bypass grafting (CABG) had a 53% higher risk of bleeding (an absolute risk of 3.3%) if they received clopidogrel within 5 days of the surgery. This has led to the practice in some centers of delaying giving clopidogrel until after the coronary anatomy has been defined. This deprives the patient of the anti-ischemic benefits conferred by giving clopidogrel early and remains a contentious issue, with most suggesting that the risk-benefit ratio still favors giving clopidogrel early, before angiography, unless there is a high likelihood that surgery will ultimately be required.17 Alternatively, one could consider using a shorter-acting intravenous glycoprotein IIb/IIIa inhibitor such as eptifibatide as a “bridge” until a definitive reperfusion strategy is chosen.
Effect of CYP2C19 variants. The CLOVIS-2 study18 assessed the effects of genetic variants on the clopidogrel concentration in 106 patients who had had a myocardial infarction. The study confirmed that patients who carry certain variants of the CYP2C19 gene attain lower plasma concentrations of clopidogrel after receiving this drug.19 This accounts for its delayed onset of action as well as its variability in response in patients who have reduced expression or inhibition of this enzyme system. Doubling the standard dose in patients who carry these variants does not appear to provide clinical benefit.20
Thus, the thought is emerging that one should consider using prasugrel or ticagrelor instead of clopidogrel in patients who have these polymorphisms, though this is yet to be backed by robust clinical evidence.
Possible interaction with proton pump inhibitors. Controversy exists about whether proton pump inhibitors inhibit clopidogrel’s action. Although the US Food and Drug Administration continues to warn against the concurrent use of omeprazole and clopidogrel,21 an analysis of the PLATO trial22 concluded that patients with ACS who were taking proton pump inhibitors were at higher risk of ischemic events regardless of whether they had been randomized to clopidogrel or ticagrelor (a drug that acts independently of the cytochrome P450 system). This observation suggests that patients on proton pump inhibitors are generally sicker and at higher risk of ischemic events regardless of the choice of antiplatelet therapy. The use of other gastroprotective agents did not appear to mitigate these risks.
Prasugrel: Faster metabolism to active drug
Prasugrel is an irreversible P2Y12 receptor antagonist (Figure 3) that is metabolized into its active metabolite faster and in a more predictable fashion than clopidogrel.23
The TRITON-TIMI 38 study24 included 13,608 ACS patients in whom an early invasive strategy was planned and who were pretreated with prasugrel or clopidogrel in addition to standard treatment. The rate of the primary efficacy end point of death, myocardial infarction, or stroke was 19% lower in the prasugrel group. In those who underwent percutaneous coronary intervention, the incidence of in-stent thrombosis was more than 50% lower in the prasugrel group regardless of whether bare metal stents or drug-eluting stents were used.
Greater platelet inhibition came at the price of a higher incidence of serious bleeding, particularly in the subgroups of patients who were over age 75, had a history of stroke or transient ischemic attack, or weighed less than 60 kg. Prasugrel is therefore contraindicated in patients with a history of transient ischemic attack or stroke. Some suggest that a 5-mg dose can be used with caution (rather than the usual 10-mg dose) in patients over age 75 years or those who have low body weight.
The TRILOGY-ACS trial25 compared prasugrel and clopidogrel in medically managed patients with high-risk non-ST-elevation ACS. It found no difference in the rates of the primary end points of cardiovascular death, myocardial infarction, or stroke at 1 year. In the prespecified subset of patients over age 75 years, the rate of bleeding end points was no higher with prasugrel 5 mg once daily than with clopidogrel.
Prasugrel’s half-life is 7 hours, and its peak antiplatelet effect is within 30 minutes after an oral dose, compared with 4 hours with clopidogrel. Therefore, if a patient with non-ST-elevation ACS is going to go to the catheterization laboratory soon, he or she should not receive prasugrel beforehand, and should receive it later only if the results of angiography indicate that CABG will not be needed urgently. This is an important consideration when using prasugrel, as the rate of surgery-related bleeding was four times higher than with clopidogrel. If possible, this drug should be withheld for at least 7 days before CABG.
Ticagrelor, a direct P2Y12 receptor inhibitor
Ticagrelor, a reversible direct inhibitor of the P2Y12 receptor, inhibits adenosine diphosphate-mediated activation and aggregation (Figure 3). It has a median time to peak concentration of 1.3 to 2 hours and a half-life of 9 hours.
The PLATO trial26 enrolled 18,624 patients with ACS who were given either ticagrelor or clopidogrel in addition to standard therapy. At 12 months, the composite primary end point of myocardial infarction, death, or stroke had occurred in 16% fewer patients receiving ticagrelor than in the clopidogrel group. Analyzed separately, there were 16% fewer myocardial infarctions, 21% fewer cardiovascular deaths, and 22% fewer deaths from any cause, regardless of whether an invasive or conservative strategy was used, and with or without prior clopidogrel use. Fewer cases of stent thrombosis occurred in the ticagrelor group, and the rate of major bleeding was the same.
In a prospectively defined subgroup analysis,27 ticagrelor was beneficial only in patients who received lower doses of aspirin (< 100 mg daily): the hazard ratio for the primary end point was 0.79 (95% confidence interval [CI] 0.71–0.88) in ticagrelor recipients who received low-dose aspirin and 1.45 (95% CI 1.01–2.09) in those who received high-dose aspirin.
Although this analysis is underpowered and controversial, the current evidence suggests that when used in combination with ticagrelor, the aspirin dose should be 81 mg.
Ticagrelor was also associated with a 19% higher incidence of non-CABG- or procedure-related major bleeding, more nonfatal and fatal intracranial bleeding, a higher incidence of dyspnea, and significantly more ventricular pauses.
Although ticagrelor carries no black-box warning about its use in patients with prior stroke or transient ischemic attack, the number of such patients in PLATO was small. Thus, caution should still be used in these patients.28
Ticagrelor should preferably be discontinued 5 days before CABG.
Glycoprotein IIb/IIIa inhibitors: Eptifibatide, tirofiban, abciximab
Glycoprotein IIb/IIIa inhibitors are intravenous agents that act by inhibiting fibrinogen-and von Willebrand factor-mediated platelet-to-platelet cross-linkage, the final pathway of platelet aggregation (Figure 3).
Use of these agents in ACS has been decreasing, as evidence supporting their use was largely established before the era of dual antiplatelet therapy.
A meta-analysis29 of 46,374 patients with non-ST-elevation ACS found that routinely adding a glycoprotein IIb/IIIa inhibitor “upstream” as a third agent in patients receiving dual antiplatelet therapy bought only a modest (11%) reduction in death or myocardial infarction at 30 days, at the price of a 23% increase in major bleeding and no decrease in the overall rate of death. Roughly 70% of the patients were receiving dual antiplatelet therapy before cardiac catheterization.
These agents can be considered in high-risk ACS patients, such as those with ST-segment changes or elevated troponin concentrations, and in diabetic patients, on the assumption that these patients likely have a high intracoronary thrombus burden and are at higher risk of microvascular embolization.6,30 They can also be considered at the time of primary percutaneous coronary intervention in selected patients receiving heparin.7
Eptifibatide
Eptifibatide is a small-molecule, short-acting glycoprotein IIb/IIIa inhibitor with a half-life of 2.5 hours. Its inhibition of platelet aggregation is reversible by stopping the drug infusion and is thought to be a result of dissociation of the drug from platelets.
The PURSUIT trial31 studied 10,948 patients presenting with non-ST-elevation ACS randomized to placebo, eptifibatide in a 180-μg/kg bolus followed by a 2.0-μg/kg/min infusion, or eptifibatide in a 180-μg/kg bolus followed by a 1.3-μg/kg/min infusion. Both eptifibatide groups had a 1.5% absolute reduction in the incidence of the primary end point of death or myocardial infarction, a benefit that was apparent at 96 hours and that persisted through 30 days. Bleeding was more common in the eptifibatide groups, but there was no increase in the rate of hemorrhagic stroke.
The ACUITY trial32 found that early use of eptifibatide or tirofiban had no effect on the primary outcome. (See the section below on bivalirudin for more information about the ACUITY trial.)
PARENTERAL ANTICOAGULANTS
Unfractionated heparin: A declining role
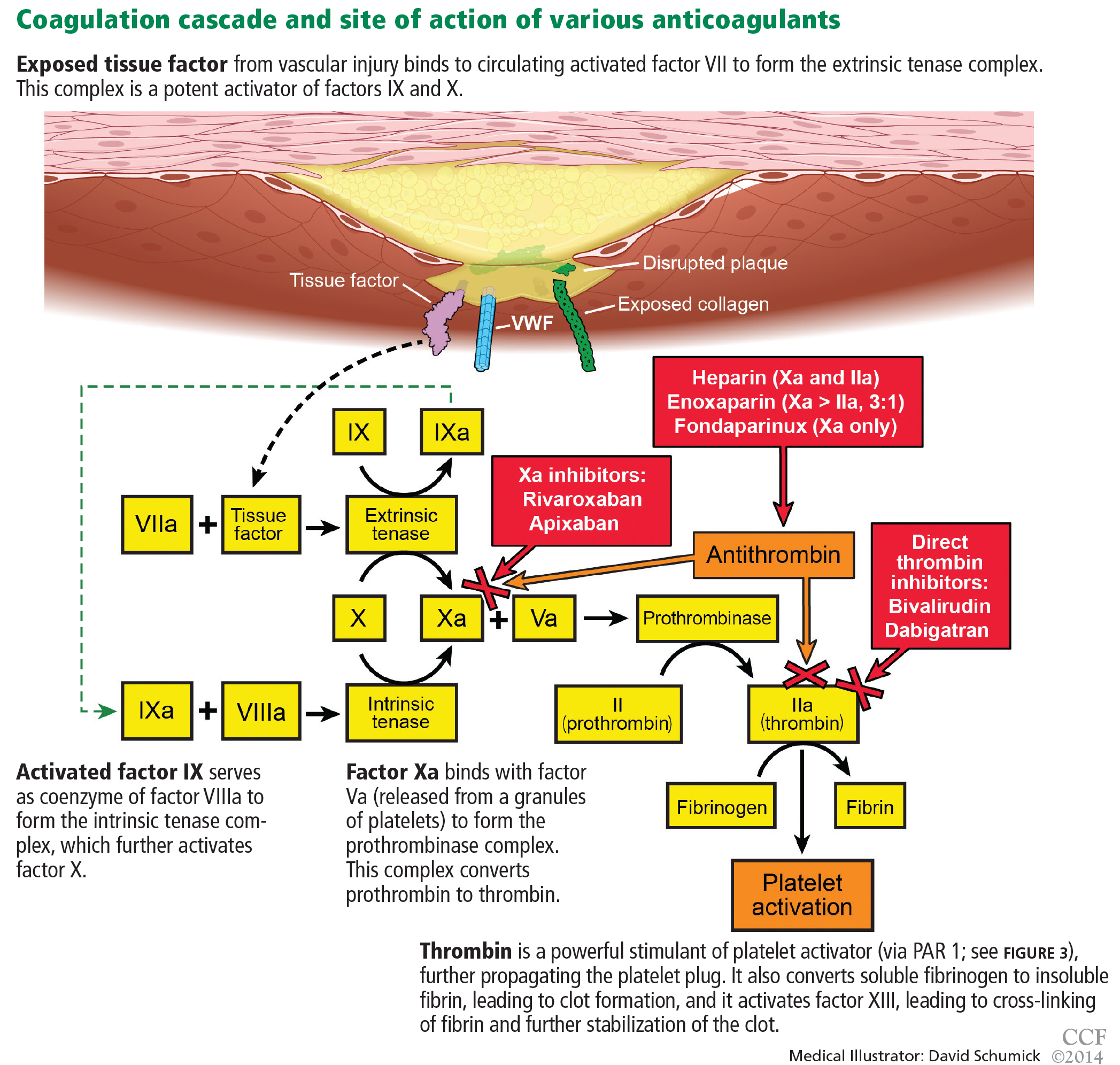
Heparin binds to antithrombin and induces a conformational change, causing rapid inhibition of factor IIa (thrombin), factor IXa, and factor Xa, thus preventing further thrombus propagation (Figure 4). An intravenous bolus of 60 units/kg produces a time to peak of 5 to 10 minutes and a half-life of 30 to 60 minutes.
Heparin can be reversed by giving protamine sulfate (1 mg per 100 units of heparin). For ACS, it is given in a bolus of 60 units/kg not exceeding 4,000 units, followed by an infusion of 12 units/kg/hour, with monitoring of the activated partial thromboplastin time every 6 hours with a goal value of 50 to 70 seconds or 1.5 to 2.5 times control.
Side effects include thrombocytopenia, heparin-induced thrombocytopenia (a distinct condition), and bleeding.
The use of unfractionated heparin was tested in ACS in the early 1990s. Oler et al33 performed a meta-analysis of six randomized trials and found a 33% lower rate of death in patients treated with heparin in addition to aspirin in ACS, as well less reported ischemic pain.
Advantages of unfractionated heparin are that it has stood the test of time, is inexpensive, and can be rapidly reversed. The disadvantages are that it can have serious side effects, including heparin-induced thrombocytopenia, and is more likely to cause bleeding than the newer intravenous anticoagulants discussed below. Thus, its position as the main anticoagulant in ACS is being challenged.
Bivalirudin, a direct thrombin inhibitor
Bivalirudin is a synthetic direct thrombin inhibitor of fluid-phase and clot-bound thrombin (Figure 4). It also inhibits platelets directly.
The ACUITY trial32 randomized 13,819 patients with moderate to high-risk ACS scheduled for invasive treatment into three treatment groups:
- Heparin (either unfractionated heparin or enoxaparin) plus a glycoprotein IIb/IIIa inhibitor (either eptifibatide, tirofiban, or abciximab)
- Bivalirudin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin alone.
The bivalirudin-alone treatment was as sociated with noninferior rates of composite ischemia end points and significantly lower rates of major bleeding, adding up to a significant reduction in the net clinical outcome end point. An important caveat is that bivalirudin’s noninferiority was mostly in the group of patients already receiving a thienopyridine before angiography and percutaneous coronary intervention (RR 0.97 vs 1.27, P = .054). There was less major, nonmajor, minor, CABG-related, and non-CABG-related bleeding as well as need for transfusion in the bivalirudin-alone group, making bivalirudin monotherapy an attractive option in ACS patients with or without ST-segment elevation undergoing a percutaneous coronary intervention.1,31
The ISAR-REACT trial34 later compared bivalirudin alone vs unfractionated heparin and abciximab in patients with non-ST-elevation myocardial infarction undergoing percutaneous coronary intervention pretreated with aspirin and clopidogrel. The composite rate of ischemia was similar in the two treatment groups, with significantly lower rates of bleeding in the bivalirudin group.
HORIZONS-AMI35 randomized 3,602 patients with ST-elevation myocardial infarction receiving aspirin and clopidogrel either to unfractionated heparin and a glycoprotein IIb/IIIa inhibitor or to bivalirudin. As in the ACUITY trial, there was no difference in ischemic end points and a 40% to 45% lower rate of major bleeding end points in the bivalirudin group, translating into an overall lower rate of death.
Enoxaparin, a low-molecular weight heparin
Enoxaparin is a low-molecular-weight heparin that inhibits factor IIa and factor Xa via antithrombin, roughly in a ratio of 1:3 (Figure 4). It has a time to peak effect of 10 minutes when given intravenously36 and 3 to 5 hours when given subcutaneously.37 Its half-life is 4.5 hours, but it is longer in patients with renal dysfunction, requiring dose adjustments in this population.
Its anticoagulant effect is partially reversible. If it is to be reversed between 0 and 8 hours after dosing, the recommended reversal regimen is 1 mg of protamine sulfate for every 1 mg of enoxaparin used. At 8 to 12 hours, it is 0.5 mg of protamine for every 1 mg of enoxaparin. After 12 hours, no protamine is required.
Compared with unfractionated heparin, enoxaparin has less plasma protein binding and a more consistent anticoagulant effect. Its high bioavailability also allows for subcutaneous dosing. Its greater anti-Xa activity inhibits thrombin generation more effectively, and it causes lower rates of thrombocytopenia and heparin-induced thrombocytopenia.
de Lemos et al38 found that, in ACS patients in whom an early conservative approach of medical management was planned, enoxaparin was more efficacious than unfractionated heparin and caused a similar rate of bleeding.
Murphy et al,39 in a meta-analysis of 12 trials in 49,088 ACS patients, also found that enoxaparin had a net clinical benefit compared with unfractionated heparin in reducing rates of myocardial infarction and death despite more bleeding.
The ESSENCE trial40 compared enoxaparin vs unfractionated heparin in 3,171 patients with ACS. It found fewer ischemic events with enoxaparin in the early phase, more minor bleeding, but no increase in major bleeding.
The SYNERGY trial,41 in 10,027 patients with high-risk non-ST-elevation ACS undergoing percutaneous coronary intervention, compared subcutaneous enoxaparin with intravenous heparin. Enoxaparin was found to be noninferior to heparin but caused more bleeding, including major bleeding, drops in hemoglobin, and intracranial hemorrhage.
The EXTRACT-TIMI 25 trial.42 In patients with ST-elevation myocardial infarction, enoxaparin has been shown to be beneficial both in patients treated with fibrinolysis and in those who underwent primary percutaneous coronary intervention. The EXTRACT-TIMI 25 trial randomized 20,749 patients to receive either enoxaparin (an intravenous bolus and maintenance subcutaneous dosing based on renal function) or intravenous heparin in addition to thrombolysis within 6 hours of the diagnosis of ST-elevation myocardial infarction. Although the enoxaparin group had more bleeding end points, they had fewer primary and secondary efficacy end points, translating into an overall net clinical benefit in favor of enoxaparin.
The ATOLL trial43 examined the use of enoxaparin (0.5 mg/kg intravenously) or unfractionated heparin in 910 patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention (via the radial artery in 66% to 69%). Although there was a trend towards benefit in terms of the primary end point of death, myocardial infarction complications, procedure failure, and major bleeding favoring enoxaparin, it was not statistically significant (95% CI 0.68–1.01, P = .06).
However, there was a 37% to 42% lower rate of the secondary end point of death, recurrent myocardial infarction or ACS, or urgent target-vessel revascularization in the enoxaparin group, with a 40% reduction in death from any cause, death from a cardiac cause, or shock. The safety profiles of the two drugs were similar, and the net clinical benefit significantly favored enoxaparin.
Fondaparinux, a factor Xa inhibitor
Fondaparinux is a synthetic pentasaccharide that indirectly inhibits factor Xa through the action of antithrombin (Figure 4). After a 2.5-mg subcutaneous dose, it has a time to peak concentration of 2 hours and a half-life of 17 to 21 hours.
The OASIS-5 trial44 compared fondaparinux and enoxaparin in 20,078 patients treated for non-ST-elevation ACS. Although the rates of death, myocardial infarction, and refractory ischemia at 9 days were similar for both drugs, the fondaparinux group had a significantly (almost 50%) lower rate of bleeding at 30 days, translating into significantly fewer deaths at 30 days. However, patients receiving fondaparinux who underwent percutaneous coronary intervention had a threefold higher rate of catheter-related thrombosis.
The OASIS-6 trial45 compared fondaparinux vs usual care (placebo in those in whom unfractionated heparin was not indicated or unfractionated heparin for up to 48 hours followed by placebo for up to 8 days) in 12,092 patients with ST-elevation myocardial infarction. There was a 1.5% absolute risk reduction in death and reinfarction without an increase in bleeding at 30 days, with trends persisting 6 months into the study. However, fondaparinux was not superior to heparin in the 3% of patients who underwent primary percutaneous coronary intervention. As in OASIS-5, there was more catheter-related thrombosis in the fondaparinux group.
Although the use of supplemental unfractionated heparin appears to have mitigated this risk, fondaparinux remains a less-than-ideal option in the era of primary percutaneous coronary intervention for ST-elevation myocardial infarction and has therefore found limited use in this group of patients. It should, however, be considered in patients for whom a conservative strategy is planned, especially if bleeding risk is deemed to be high.
ORAL ANTICOAGULANTS
Oral anticoagulants provide ischemic benefit in selected patients with ACS—at the price of a higher risk of significant bleeding.
Warfarin
Warfarin was investigated after myocardial infarction in the WARIS II,46 CARS,47 and CHAMP48 trials.
WARIS II46 looked at the use of aspirin alone, warfarin alone, and aspirin and warfarin in combination. The rates of the primary end points of stroke, nonfatal infarction, and death were lower in the warfarin group.
CARS47 found no difference in the rate of the primary end point of fatal infarction, nonfatal ischemic stroke, or cardiovascular death with aspirin vs warfarin plus aspirin.
CHAMP48 saw similar trends, ie, no difference in the rate of death, recurrent myocardial infarction, or stroke with warfarin plus aspirin vs aspirin alone.
All three studies showed increases in major bleeding with warfarin use.
Putting these trials into context, the significant net clinical benefit of dual antiplatelet therapy in the current era compared with the significant bleeding and questionable conflicting evidence supporting benefit with warfarin has limited its use in ACS patients.
Rivaroxaban, an oral factor Xa inhibitor
Rivaroxaban is a novel oral direct reversible factor Xa inhibitor.
The ATLAS ACS 2-TIMI 51 trial49 found rivaroxaban 2.5 mg or 5 mg to yield a significantly lower rate of the primary outcome of cardiovascular death, myocardial infarction, ischemic stroke, and in-stent thrombosis compared with placebo, but significantly more major non-CABG bleeding and intracranial hemorrhage.
The dose used in this trial was much lower than the dose used in trials investigating the role of this drug in stroke prophylaxis in atrial fibrillation.
Apixaban, an oral factor Xa inhibitor
Apixaban is another direct factor Xa inhibitor.
The APPRAISE-2 trial50 compared apixaban 5 mg twice daily vs placebo in ACS. There was no difference in the rate of cardiovascular death, myocardial infarction, or stroke, but there was significantly more bleeding in the apixaban group, prompting early termination of this study.
Dabigatran, an oral thrombin inhibitor
Dabigatran is an oral direct thrombin inhibitor.
The RE-DEEM trial51 compared four doses of dabigatran (50, 75, 110, and 150 mg twice daily) and placebo in ACS patients. The dabigatran groups had more major and minor bleeding, and the higher the dose, the higher the incidence of bleeding. In addition, the rates of ischemic end points were no lower with dabigatran, although this trial was not powered to show differences in clinical events.
REDUCING THE RISK OF BLEEDING
In the treatment of ACS, the benefits of restoring perfusion by preventing further propagation of thrombus and platelet aggregation come at a significant price of higher bleeding risk. This in turn increases the risk of death through various mechanisms, including shock, worsening ischemia, discontinuation of antiplatelet and anticoagulation therapy causing stent thrombosis, and anemia leading to transfusion, which propagates the underlying inflammatory milieu.52
Giugliano and Braunwald53 provide practical suggestions to reduce this risk, advising physicians to:
- Avoid inappropriately high dosing, particularly in patients with renal insufficiency
- Preferentially use agents that cause less bleeding (eg, bivalirudin, fondaparinux) without compromising anti-ischemic efficacy
- Minimize the concomitant use of other drugs that cause bleeding (eg, NSAIDs)
- Use drugs that protect against bleeding (eg, proton pump inhibitors) in patients at high risk
- Prevent access-site bleeding by using the radial artery, smaller sheaths, and appropriate sheath and closure device management. Indeed, the use of radial interventions in ACS has been shown to reduce access-site-related bleeding, even in patients at high risk.54
The reduction in bleeding risk may provide future trials the opportunity to increase antithrombotic efficacy of different agents with goals of reducing ischemic end points.
- Wright RS, Anderson JL, Adams CD, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2011 ACCF/AHA focused update incorporated into the ACC/AHA 2007 Guidelines for the Management of Patients with Unstable Angina/Non-ST-Elevation Myocardial Infarction. J Am Coll Cardiol 2011; 57:e215–e367.
- Jneid H, Anderson JL, Wright RS, et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2012; 60:645–681.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet 1996; 348:1329–1339.
- Cohen M, Adams PC, Parry G, et al. Combination antithrombotic therapy in unstable rest angina and non-Q-wave infarction in nonprior aspirin users. Primary end points analysis from the ATACS trial. Antithrombotic Therapy in Acute Coronary Syndromes Research Group. Circulation 1994; 89:81–88.
- Moscucci M, Fox KA, Cannon CP, et al. Predictors of major bleeding in acute coronary syndromes: the Global Registry of Acute Coronary Events (GRACE). Eur Heart J 2003; 24:1815–1823.
- Levine GN, Bates ER, Blankenship JC, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; Society for Cardiovascular Angiography and Interventions. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol 2011; 58:e44–e122.
- American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:485–510.
- Lewis HD, Davis JW, Archibald DG, et al. Protective effects of aspirin against acute myocardial infarction and death in men with unstable angina. Results of a Veterans Administration Cooperative Study. N Engl J Med 1983; 309:396–403.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Sweeny JM, Gorog DA, Fuster V. Antiplatelet drug ‘resistance’. Part 1: mechanisms and clinical measurements. Nat Rev Cardiol 2009; 6:273–282.
- Kelly JP, Kaufman DW, Jurgelon JM, Sheehan J, Koff RS, Shapiro S. Risk of aspirin-associated major upper-gastrointestinal bleeding with enteric-coated or buffered product. Lancet 1996; 348:1413–1416.
- Grosser T, Fries S, Lawson JA, Kapoor SC, Grant GR, FitzGerald GA. Drug resistance and pseudoresistance: an unintended consequence of enteric coating aspirin. Circulation 2013; 127:377–385.
- US Food and Drug Administration (FDA). Concomitant use of ibuprofen and aspirin: potential for attenuation of the anti-platelet effect of aspirin. http://www.fda.gov/downloads/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm161282.pdf. Accessed November 30, 2013.
- Patti G, Colonna G, Pasceri V, Pepe LL, Montinaro A, Di Sciascio G. Randomized trial of high loading dose of clopidogrel for reduction of periprocedural myocardial infarction in patients undergoing coronary intervention: results from the ARMYDA-2 (Antiplatelet therapy for Reduction of MYocardial Damage during Angioplasty) study. Circulation 2005; 111:2099–2106.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Bavry AA, Lincoff AM. Is clopidogrel cardiovascular medicine’s double-edged sword? Circulation 2006; 113:1638–1640.
- Collet JP, Hulot JS, Anzaha G, et al; CLOVIS-2 Investigators. High doses of clopidogrel to overcome genetic resistance: the randomized crossover CLOVIS-2 (Clopidogrel and Response Variability Investigation Study 2). JACC Cardiovasc Interv 2011; 4:392–402.
- Hulot JS, Collet JP, Cayla G, et al. CYP2C19 but not PON1 genetic variants influence clopidogrel pharmacokinetics, pharmacodynamics, and clinical efficacy in post-myocardial infarction patients. Circ Cardiovasc Interv 2011; 4:422–428.
- Cuisset T, Quilici J, Cohen W, et al. Usefulness of high clopidogrel maintenance dose according to CYP2C19 genotypes in clopidogrel low responders undergoing coronary stenting for non ST elevation acute coronary syndrome. Am J Cardiol 2011; 108:760–765.
- US Food and Drug Administration (FDA). FDA reminder to avoid concomitant use of Plavix (clopidogrel) and omeprazole. http://www.fda.gov/Drugs/DrugSafety/ucm231161.htm. Accessed November 30, 2013.
- Goodman SG, Clare R, Pieper KS, et al; Platelet Inhibition and Patient Outcomes Trial Investigators. Association of proton pump inhibitor use on cardiovascular outcomes with clopidogrel and ticagrelor: insights from the platelet inhibition and patient outcomes trial. Circulation 2012; 125:978–986.
- Solomon S, Vacek JL. Reducing cardiac ischemic events in patients with ACS: prasugrel versus clopidogrel. Commentary. Postgrad Med 2010; 122:198–200.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Roe MT, Armstrong PW, Fox KA, et al; TRILOGY ACS Investigators. Prasugrel versus clopidogrel for acute coronary syndromes without revascularization. N Engl J Med 2012; 367:1297–1309.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Mahaffey KW, Wojdyla DM, Carroll K, et al; PLATO Investigators. Ticagrelor compared with clopidogrel by geographic region in the Platelet Inhibition and Patient Outcomes (PLATO) trial. Circulation 2011; 124:544–554.
- Verheugt FW. Beware of novel antiplatelet therapy in acute coronary syndrome patients with previous stroke. Circulation 2012; 125:2821–2823.
- Tricoci P, Newby LK, Hasselblad V, et al. Upstream use of small-molecule glycoprotein iib/iiia inhibitors in patients with non-ST-segment elevation acute coronary syndromes: a systematic overview of randomized clinical trials. Circ Cardiovasc Qual Outcomes 2011; 4:448–458.
- Kastrati A, Mehilli J, Neumann FJ, et al; Intracoronary Stenting and Antithrombotic: Regimen Rapid Early Action for Coronary Treatment 2 (ISAR-REACT 2) Trial Investigators. Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: the ISAR-REACT 2 randomized trial. JAMA 2006; 295:1531–1538.
- Inhibition of platelet glycoprotein IIb/IIIa with eptifibatide in patients with acute coronary syndromes. The PURSUIT Trial Investigators. Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin Therapy. N Engl J Med 1998; 339:436–443.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Oler A, Whooley MA, Oler J, Grady D. Adding heparin to aspirin reduces the incidence of myocardial infarction and death in patients with unstable angina. A meta-analysis. JAMA 1996; 276:811–815.
- Kastrati A, Neumann FJ, Schulz S, et al; ISAR-REACT 4 Trial Investigators. Abciximab and heparin versus bivalirudin for non-ST-elevation myocardial infarction. N Engl J Med 2011; 365:1980–1989.
- Stone GW, Witzenbichler B, Guagliumi G, et al; HORIZONS-AMI Trial Investigators. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med 2008; 358:2218–2230.
- Aslam MS, Sundberg S, Sabri MN, Cooke D, Lakier JB. Pharmacokinetics of intravenous/subcutaneous enoxaparin in patients with acute coronary syndrome undergoing percutaneous coronary interventions. Catheter Cardiovasc Interv 2002; 57:187–190.
- Sanofi-Aventis US. Lovenox (enoxaparin sodium injection) product information. http://www.lovenox.com/hcp/clinical-data.aspx. Accessed December 1, 2013.
- de Lemos JA, Blazing MA, Wiviott SD, et al. Enoxaparin versus unfractionated heparin in patients treated with tirofiban, aspirin and an early conservative initial management strategy: results from the A phase of the A-to-Z trial. Eur Heart J 2004; 25:1688–1694.
- Murphy SA, Gibson CM, Morrow DA, et al. Efficacy and safety of the low-molecular weight heparin enoxaparin compared with unfractionated heparin across the acute coronary syndrome spectrum: a meta-analysis. Eur Heart J 2007; 28:2077–2086.
- Cohen M, Demers C, Gurfinkel EP, et al. A comparison of low-molecular-weight heparin with unfractionated heparin for unstable coronary artery disease. Efficacy and Safety of Subcutaneous Enoxaparin in Non-Q-Wave Coronary Events Study Group. N Engl J Med 1997; 337:447–452.
- Ferguson JJ, Califf RM, Antman EM, et al; SYNERGY Trial Investigators. Enoxaparin vs unfractionated heparin in high-risk patients with non-ST-segment elevation acute coronary syndromes managed with an intended early invasive strategy: primary results of the SYNERGY randomized trial. JAMA 2004; 292:45–54.
- Antman EM, Morrow DA, McCabe CH, et al; ExTRACT-TIMI 25 Investigators. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med 2006; 354:1477–1488.
- Montalescot G, Zeymer U, Silvain J, et al; ATOLL Investigators. Intravenous enoxaparin or unfractionated heparin in primary percutaneous coronary intervention for ST-elevation myocardial infarction: the international randomised open-label ATOLL trial. Lancet 2011; 378:693–703.
- Yusuf S, Mehta SR, Chrolavicius S, et al; Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Yusuf S, Mehta SR, Chrolavicius S, et al; OASIS-6 Trial Group. Effects of fondaparinux on mortality and reinfarction in patients with acute ST-segment elevation myocardial infarction: the OASIS-6 randomized trial. JAMA 2006; 295:1519–1530.
- Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002; 347:969–974.
- Coumadin Aspirin Reinfarction Study (CARS) Investigators. Randomised double-blind trial of fixed low-dose warfarin with aspirin after myocardial infarction. Lancet 1997; 350:389–396.
- Fiore LD, Ezekowitz MD, Brophy MT, Lu D, Sacco J, Peduzzi P; Combination Hemotherapy and Mortality Prevention (CHAMP) Study Group. Department of Veterans Affairs Cooperative Studies Program Clinical Trial comparing combined warfarin and aspirin with aspirin alone in survivors of acute myocardial infarction: primary results of the CHAMP study. Circulation 2002; 105:557–563.
- Mega JL, Braunwald E, Wiviott SD, et al; ATLAS ACS 2–TIMI 51 Investigators. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med 2012; 366:9–19.
- Alexander JH, Lopes RD, James S, et al; APPRAISE-2 Investigators. Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med 2011; 365:699–708.
- Oldgren J, Budaj A, Granger CB, et al; RE-DEEM Investigators. Dabigatran vs placebo in patients with acute coronary syndromes on dual antiplatelet therapy: a randomized, double-blind, phase II trial. Eur Heart J 2011; 32:2781–2789.
- Steg PG, Huber K, Andreotti F, et al. Bleeding in acute coronary syndromes and percutaneous coronary interventions: position paper by the Working Group on Thrombosis of the European Society of Cardiology. Eur Heart J 2011; 32:1854–1864.
- Giugliano RP, Braunwald E. The year in non-ST-segment elevation acute coronary syndrome. J Am Coll Cardiol 2012; 60:2127–039.
- Rao SV, Ou FS, Wang TY, et al. Trends in the prevalence and outcomes of radial and femoral approaches to percutaneous coronary intervention: a report from the National Cardiovascular Data Registry. JACC Cardiovasc Interv 2008; 1:379–386.
Antiplatelet and anticoagulant drugs are a cornerstone of the medical treatment of acute coronary syndrome (ACS), reducing the rates of both morbidity and death.1–4 However, reductions in ischemic events with these drugs have uniformly been accompanied by increases in bleeding complications, which reduce the net benefit.5 Thus, clinical research has been exploring ways to maximize the benefit while minimizing the risk.
Here, we review the guidelines and evidence supporting the use of antiplatelet and anticoagulant drugs in ACS.
ACUTE CORONARY SYNDROMES WITH OR WITHOUT ST ELEVATION
A key distinction when treating ACS is whether the electrocardiogram shows ST-segment elevation. In cases of non-ST-elevation ACS (ie, unstable angina or non-ST-elevation myocardial infarction), a second key question is whether the initial strategy will be invasive (with angiography performed urgently) or conservative (with angiography performed later). In ST-elevation myocardial infarction, another distinction is how perfusion is to be restored, ie, with primary percutaneous coronary intervention or with thrombolysis. All these questions affect the choice of antiplatelet and anticoagulant therapy.
Figure 1 and Figure 2 summarize the guidelines of the American College of Cardiology Foundation and American Heart Association.1,2,6,7
ANTIPLATELET THERAPY
Aspirin for all
Aspirin irreversibly acetylates the enzyme cyclooxygenase-1, blocking intraplatelet formation of thromboxane A2 (Figure 3), a potent platelet aggregator and endothelial vasoconstrictor. Large clinical trials have confirmed that aspirin reduces morbidity and mortality rates by as much as 50% in patients with ACS.8
The ISIS-2 trial9 found that giving aspirin early in the emergency department significantly reduced the mortality rate.
The Antithrombotic Trialists’ Collaboration,10 in a meta-analysis of randomized controlled trials comparing different doses of aspirin in high-risk ACS patients, found no greater benefit for doses of aspirin higher than 162 mg per day when used long-term.
How to use. During an ACS, the patient should receive one dose of aspirin 325 mg (the standard high-dose pill in the United States). This dose should be chewed, as buccal absorption results in more rapid systemic effects.11
Thereafter, the patient should take 81 mg per day, continued indefinitely. The 81-mg dose also applies to patients who undergo a percutaneous coronary intervention with a drug-eluting stent.7 Previous recommendations called for higher doses, but studies have shown that higher doses pose a higher risk of bleeding without additional clinical benefit. The use of enteric-coated aspirin does not reduce this risk,12 and its delayed release may in fact cause aspirin “pseudoresistance.”13
The concurrent use of nonsteroidal anti-inflammatory drugs (NSAIDs) should be avoided, as NSAIDs reversibly bind to platelets, thus preventing aspirin from binding.14 As aspirin washes out of the body, NSAIDs may then become unbound from platelets, leaving platelets activated.
P2Y12 receptor inhibitors: Clopidogrel, prasugrel, ticagrelor
These agents bind to P2Y12 receptors on platelets to inhibit adenosine diphosphate-mediated platelet activation (Figure 3). Clopidogrel and prasugrel are irreversible prodrugs, whereas ticagrelor binds reversibly.
Clopidogrel, a prodrug
Clopidogrel has a half-life of 8 hours and a time to peak concentration of 4 hours. Eighty-five percent of a dose is inactivated by gut esterases. The remainder is metabolized primarily by the cytochrome P4502C19 enzyme system into its active metabolite.
How to use. The recommended dosage is a 600-mg bolus early in the course of ACS. This is associated with a lower rate of cardiovascular events than a 300-mg dose,2,15 although no trial has rigorously compared 300-mg vs 600-mg doses using major clinical end points. In patients presenting with ACS who cannot tolerate aspirin because of hypersensitivity or major gastrointestinal contraindication, clopidogrel is an alternative.1
The CURE trial16 randomized 12,526 patients with non-ST-elevation ACS to receive clopidogrel or placebo in addition to standard therapy. Clopidogrel was associated with a 20% lower rate of cardiovascular death, myocardial infarction, or stroke in both low- and high-risk patients regardless of whether an invasive or conservative strategy was pursued.
However, patients who underwent coronary artery bypass grafting (CABG) had a 53% higher risk of bleeding (an absolute risk of 3.3%) if they received clopidogrel within 5 days of the surgery. This has led to the practice in some centers of delaying giving clopidogrel until after the coronary anatomy has been defined. This deprives the patient of the anti-ischemic benefits conferred by giving clopidogrel early and remains a contentious issue, with most suggesting that the risk-benefit ratio still favors giving clopidogrel early, before angiography, unless there is a high likelihood that surgery will ultimately be required.17 Alternatively, one could consider using a shorter-acting intravenous glycoprotein IIb/IIIa inhibitor such as eptifibatide as a “bridge” until a definitive reperfusion strategy is chosen.
Effect of CYP2C19 variants. The CLOVIS-2 study18 assessed the effects of genetic variants on the clopidogrel concentration in 106 patients who had had a myocardial infarction. The study confirmed that patients who carry certain variants of the CYP2C19 gene attain lower plasma concentrations of clopidogrel after receiving this drug.19 This accounts for its delayed onset of action as well as its variability in response in patients who have reduced expression or inhibition of this enzyme system. Doubling the standard dose in patients who carry these variants does not appear to provide clinical benefit.20
Thus, the thought is emerging that one should consider using prasugrel or ticagrelor instead of clopidogrel in patients who have these polymorphisms, though this is yet to be backed by robust clinical evidence.
Possible interaction with proton pump inhibitors. Controversy exists about whether proton pump inhibitors inhibit clopidogrel’s action. Although the US Food and Drug Administration continues to warn against the concurrent use of omeprazole and clopidogrel,21 an analysis of the PLATO trial22 concluded that patients with ACS who were taking proton pump inhibitors were at higher risk of ischemic events regardless of whether they had been randomized to clopidogrel or ticagrelor (a drug that acts independently of the cytochrome P450 system). This observation suggests that patients on proton pump inhibitors are generally sicker and at higher risk of ischemic events regardless of the choice of antiplatelet therapy. The use of other gastroprotective agents did not appear to mitigate these risks.
Prasugrel: Faster metabolism to active drug
Prasugrel is an irreversible P2Y12 receptor antagonist (Figure 3) that is metabolized into its active metabolite faster and in a more predictable fashion than clopidogrel.23
The TRITON-TIMI 38 study24 included 13,608 ACS patients in whom an early invasive strategy was planned and who were pretreated with prasugrel or clopidogrel in addition to standard treatment. The rate of the primary efficacy end point of death, myocardial infarction, or stroke was 19% lower in the prasugrel group. In those who underwent percutaneous coronary intervention, the incidence of in-stent thrombosis was more than 50% lower in the prasugrel group regardless of whether bare metal stents or drug-eluting stents were used.
Greater platelet inhibition came at the price of a higher incidence of serious bleeding, particularly in the subgroups of patients who were over age 75, had a history of stroke or transient ischemic attack, or weighed less than 60 kg. Prasugrel is therefore contraindicated in patients with a history of transient ischemic attack or stroke. Some suggest that a 5-mg dose can be used with caution (rather than the usual 10-mg dose) in patients over age 75 years or those who have low body weight.
The TRILOGY-ACS trial25 compared prasugrel and clopidogrel in medically managed patients with high-risk non-ST-elevation ACS. It found no difference in the rates of the primary end points of cardiovascular death, myocardial infarction, or stroke at 1 year. In the prespecified subset of patients over age 75 years, the rate of bleeding end points was no higher with prasugrel 5 mg once daily than with clopidogrel.
Prasugrel’s half-life is 7 hours, and its peak antiplatelet effect is within 30 minutes after an oral dose, compared with 4 hours with clopidogrel. Therefore, if a patient with non-ST-elevation ACS is going to go to the catheterization laboratory soon, he or she should not receive prasugrel beforehand, and should receive it later only if the results of angiography indicate that CABG will not be needed urgently. This is an important consideration when using prasugrel, as the rate of surgery-related bleeding was four times higher than with clopidogrel. If possible, this drug should be withheld for at least 7 days before CABG.
Ticagrelor, a direct P2Y12 receptor inhibitor
Ticagrelor, a reversible direct inhibitor of the P2Y12 receptor, inhibits adenosine diphosphate-mediated activation and aggregation (Figure 3). It has a median time to peak concentration of 1.3 to 2 hours and a half-life of 9 hours.
The PLATO trial26 enrolled 18,624 patients with ACS who were given either ticagrelor or clopidogrel in addition to standard therapy. At 12 months, the composite primary end point of myocardial infarction, death, or stroke had occurred in 16% fewer patients receiving ticagrelor than in the clopidogrel group. Analyzed separately, there were 16% fewer myocardial infarctions, 21% fewer cardiovascular deaths, and 22% fewer deaths from any cause, regardless of whether an invasive or conservative strategy was used, and with or without prior clopidogrel use. Fewer cases of stent thrombosis occurred in the ticagrelor group, and the rate of major bleeding was the same.
In a prospectively defined subgroup analysis,27 ticagrelor was beneficial only in patients who received lower doses of aspirin (< 100 mg daily): the hazard ratio for the primary end point was 0.79 (95% confidence interval [CI] 0.71–0.88) in ticagrelor recipients who received low-dose aspirin and 1.45 (95% CI 1.01–2.09) in those who received high-dose aspirin.
Although this analysis is underpowered and controversial, the current evidence suggests that when used in combination with ticagrelor, the aspirin dose should be 81 mg.
Ticagrelor was also associated with a 19% higher incidence of non-CABG- or procedure-related major bleeding, more nonfatal and fatal intracranial bleeding, a higher incidence of dyspnea, and significantly more ventricular pauses.
Although ticagrelor carries no black-box warning about its use in patients with prior stroke or transient ischemic attack, the number of such patients in PLATO was small. Thus, caution should still be used in these patients.28
Ticagrelor should preferably be discontinued 5 days before CABG.
Glycoprotein IIb/IIIa inhibitors: Eptifibatide, tirofiban, abciximab
Glycoprotein IIb/IIIa inhibitors are intravenous agents that act by inhibiting fibrinogen-and von Willebrand factor-mediated platelet-to-platelet cross-linkage, the final pathway of platelet aggregation (Figure 3).
Use of these agents in ACS has been decreasing, as evidence supporting their use was largely established before the era of dual antiplatelet therapy.
A meta-analysis29 of 46,374 patients with non-ST-elevation ACS found that routinely adding a glycoprotein IIb/IIIa inhibitor “upstream” as a third agent in patients receiving dual antiplatelet therapy bought only a modest (11%) reduction in death or myocardial infarction at 30 days, at the price of a 23% increase in major bleeding and no decrease in the overall rate of death. Roughly 70% of the patients were receiving dual antiplatelet therapy before cardiac catheterization.
These agents can be considered in high-risk ACS patients, such as those with ST-segment changes or elevated troponin concentrations, and in diabetic patients, on the assumption that these patients likely have a high intracoronary thrombus burden and are at higher risk of microvascular embolization.6,30 They can also be considered at the time of primary percutaneous coronary intervention in selected patients receiving heparin.7
Eptifibatide
Eptifibatide is a small-molecule, short-acting glycoprotein IIb/IIIa inhibitor with a half-life of 2.5 hours. Its inhibition of platelet aggregation is reversible by stopping the drug infusion and is thought to be a result of dissociation of the drug from platelets.
The PURSUIT trial31 studied 10,948 patients presenting with non-ST-elevation ACS randomized to placebo, eptifibatide in a 180-μg/kg bolus followed by a 2.0-μg/kg/min infusion, or eptifibatide in a 180-μg/kg bolus followed by a 1.3-μg/kg/min infusion. Both eptifibatide groups had a 1.5% absolute reduction in the incidence of the primary end point of death or myocardial infarction, a benefit that was apparent at 96 hours and that persisted through 30 days. Bleeding was more common in the eptifibatide groups, but there was no increase in the rate of hemorrhagic stroke.
The ACUITY trial32 found that early use of eptifibatide or tirofiban had no effect on the primary outcome. (See the section below on bivalirudin for more information about the ACUITY trial.)
PARENTERAL ANTICOAGULANTS
Unfractionated heparin: A declining role
Heparin binds to antithrombin and induces a conformational change, causing rapid inhibition of factor IIa (thrombin), factor IXa, and factor Xa, thus preventing further thrombus propagation (Figure 4). An intravenous bolus of 60 units/kg produces a time to peak of 5 to 10 minutes and a half-life of 30 to 60 minutes.
Heparin can be reversed by giving protamine sulfate (1 mg per 100 units of heparin). For ACS, it is given in a bolus of 60 units/kg not exceeding 4,000 units, followed by an infusion of 12 units/kg/hour, with monitoring of the activated partial thromboplastin time every 6 hours with a goal value of 50 to 70 seconds or 1.5 to 2.5 times control.
Side effects include thrombocytopenia, heparin-induced thrombocytopenia (a distinct condition), and bleeding.
The use of unfractionated heparin was tested in ACS in the early 1990s. Oler et al33 performed a meta-analysis of six randomized trials and found a 33% lower rate of death in patients treated with heparin in addition to aspirin in ACS, as well less reported ischemic pain.
Advantages of unfractionated heparin are that it has stood the test of time, is inexpensive, and can be rapidly reversed. The disadvantages are that it can have serious side effects, including heparin-induced thrombocytopenia, and is more likely to cause bleeding than the newer intravenous anticoagulants discussed below. Thus, its position as the main anticoagulant in ACS is being challenged.
Bivalirudin, a direct thrombin inhibitor
Bivalirudin is a synthetic direct thrombin inhibitor of fluid-phase and clot-bound thrombin (Figure 4). It also inhibits platelets directly.
The ACUITY trial32 randomized 13,819 patients with moderate to high-risk ACS scheduled for invasive treatment into three treatment groups:
- Heparin (either unfractionated heparin or enoxaparin) plus a glycoprotein IIb/IIIa inhibitor (either eptifibatide, tirofiban, or abciximab)
- Bivalirudin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin alone.
The bivalirudin-alone treatment was as sociated with noninferior rates of composite ischemia end points and significantly lower rates of major bleeding, adding up to a significant reduction in the net clinical outcome end point. An important caveat is that bivalirudin’s noninferiority was mostly in the group of patients already receiving a thienopyridine before angiography and percutaneous coronary intervention (RR 0.97 vs 1.27, P = .054). There was less major, nonmajor, minor, CABG-related, and non-CABG-related bleeding as well as need for transfusion in the bivalirudin-alone group, making bivalirudin monotherapy an attractive option in ACS patients with or without ST-segment elevation undergoing a percutaneous coronary intervention.1,31
The ISAR-REACT trial34 later compared bivalirudin alone vs unfractionated heparin and abciximab in patients with non-ST-elevation myocardial infarction undergoing percutaneous coronary intervention pretreated with aspirin and clopidogrel. The composite rate of ischemia was similar in the two treatment groups, with significantly lower rates of bleeding in the bivalirudin group.
HORIZONS-AMI35 randomized 3,602 patients with ST-elevation myocardial infarction receiving aspirin and clopidogrel either to unfractionated heparin and a glycoprotein IIb/IIIa inhibitor or to bivalirudin. As in the ACUITY trial, there was no difference in ischemic end points and a 40% to 45% lower rate of major bleeding end points in the bivalirudin group, translating into an overall lower rate of death.
Enoxaparin, a low-molecular weight heparin
Enoxaparin is a low-molecular-weight heparin that inhibits factor IIa and factor Xa via antithrombin, roughly in a ratio of 1:3 (Figure 4). It has a time to peak effect of 10 minutes when given intravenously36 and 3 to 5 hours when given subcutaneously.37 Its half-life is 4.5 hours, but it is longer in patients with renal dysfunction, requiring dose adjustments in this population.
Its anticoagulant effect is partially reversible. If it is to be reversed between 0 and 8 hours after dosing, the recommended reversal regimen is 1 mg of protamine sulfate for every 1 mg of enoxaparin used. At 8 to 12 hours, it is 0.5 mg of protamine for every 1 mg of enoxaparin. After 12 hours, no protamine is required.
Compared with unfractionated heparin, enoxaparin has less plasma protein binding and a more consistent anticoagulant effect. Its high bioavailability also allows for subcutaneous dosing. Its greater anti-Xa activity inhibits thrombin generation more effectively, and it causes lower rates of thrombocytopenia and heparin-induced thrombocytopenia.
de Lemos et al38 found that, in ACS patients in whom an early conservative approach of medical management was planned, enoxaparin was more efficacious than unfractionated heparin and caused a similar rate of bleeding.
Murphy et al,39 in a meta-analysis of 12 trials in 49,088 ACS patients, also found that enoxaparin had a net clinical benefit compared with unfractionated heparin in reducing rates of myocardial infarction and death despite more bleeding.
The ESSENCE trial40 compared enoxaparin vs unfractionated heparin in 3,171 patients with ACS. It found fewer ischemic events with enoxaparin in the early phase, more minor bleeding, but no increase in major bleeding.
The SYNERGY trial,41 in 10,027 patients with high-risk non-ST-elevation ACS undergoing percutaneous coronary intervention, compared subcutaneous enoxaparin with intravenous heparin. Enoxaparin was found to be noninferior to heparin but caused more bleeding, including major bleeding, drops in hemoglobin, and intracranial hemorrhage.
The EXTRACT-TIMI 25 trial.42 In patients with ST-elevation myocardial infarction, enoxaparin has been shown to be beneficial both in patients treated with fibrinolysis and in those who underwent primary percutaneous coronary intervention. The EXTRACT-TIMI 25 trial randomized 20,749 patients to receive either enoxaparin (an intravenous bolus and maintenance subcutaneous dosing based on renal function) or intravenous heparin in addition to thrombolysis within 6 hours of the diagnosis of ST-elevation myocardial infarction. Although the enoxaparin group had more bleeding end points, they had fewer primary and secondary efficacy end points, translating into an overall net clinical benefit in favor of enoxaparin.
The ATOLL trial43 examined the use of enoxaparin (0.5 mg/kg intravenously) or unfractionated heparin in 910 patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention (via the radial artery in 66% to 69%). Although there was a trend towards benefit in terms of the primary end point of death, myocardial infarction complications, procedure failure, and major bleeding favoring enoxaparin, it was not statistically significant (95% CI 0.68–1.01, P = .06).
However, there was a 37% to 42% lower rate of the secondary end point of death, recurrent myocardial infarction or ACS, or urgent target-vessel revascularization in the enoxaparin group, with a 40% reduction in death from any cause, death from a cardiac cause, or shock. The safety profiles of the two drugs were similar, and the net clinical benefit significantly favored enoxaparin.
Fondaparinux, a factor Xa inhibitor
Fondaparinux is a synthetic pentasaccharide that indirectly inhibits factor Xa through the action of antithrombin (Figure 4). After a 2.5-mg subcutaneous dose, it has a time to peak concentration of 2 hours and a half-life of 17 to 21 hours.
The OASIS-5 trial44 compared fondaparinux and enoxaparin in 20,078 patients treated for non-ST-elevation ACS. Although the rates of death, myocardial infarction, and refractory ischemia at 9 days were similar for both drugs, the fondaparinux group had a significantly (almost 50%) lower rate of bleeding at 30 days, translating into significantly fewer deaths at 30 days. However, patients receiving fondaparinux who underwent percutaneous coronary intervention had a threefold higher rate of catheter-related thrombosis.
The OASIS-6 trial45 compared fondaparinux vs usual care (placebo in those in whom unfractionated heparin was not indicated or unfractionated heparin for up to 48 hours followed by placebo for up to 8 days) in 12,092 patients with ST-elevation myocardial infarction. There was a 1.5% absolute risk reduction in death and reinfarction without an increase in bleeding at 30 days, with trends persisting 6 months into the study. However, fondaparinux was not superior to heparin in the 3% of patients who underwent primary percutaneous coronary intervention. As in OASIS-5, there was more catheter-related thrombosis in the fondaparinux group.
Although the use of supplemental unfractionated heparin appears to have mitigated this risk, fondaparinux remains a less-than-ideal option in the era of primary percutaneous coronary intervention for ST-elevation myocardial infarction and has therefore found limited use in this group of patients. It should, however, be considered in patients for whom a conservative strategy is planned, especially if bleeding risk is deemed to be high.
ORAL ANTICOAGULANTS
Oral anticoagulants provide ischemic benefit in selected patients with ACS—at the price of a higher risk of significant bleeding.
Warfarin
Warfarin was investigated after myocardial infarction in the WARIS II,46 CARS,47 and CHAMP48 trials.
WARIS II46 looked at the use of aspirin alone, warfarin alone, and aspirin and warfarin in combination. The rates of the primary end points of stroke, nonfatal infarction, and death were lower in the warfarin group.
CARS47 found no difference in the rate of the primary end point of fatal infarction, nonfatal ischemic stroke, or cardiovascular death with aspirin vs warfarin plus aspirin.
CHAMP48 saw similar trends, ie, no difference in the rate of death, recurrent myocardial infarction, or stroke with warfarin plus aspirin vs aspirin alone.
All three studies showed increases in major bleeding with warfarin use.
Putting these trials into context, the significant net clinical benefit of dual antiplatelet therapy in the current era compared with the significant bleeding and questionable conflicting evidence supporting benefit with warfarin has limited its use in ACS patients.
Rivaroxaban, an oral factor Xa inhibitor
Rivaroxaban is a novel oral direct reversible factor Xa inhibitor.
The ATLAS ACS 2-TIMI 51 trial49 found rivaroxaban 2.5 mg or 5 mg to yield a significantly lower rate of the primary outcome of cardiovascular death, myocardial infarction, ischemic stroke, and in-stent thrombosis compared with placebo, but significantly more major non-CABG bleeding and intracranial hemorrhage.
The dose used in this trial was much lower than the dose used in trials investigating the role of this drug in stroke prophylaxis in atrial fibrillation.
Apixaban, an oral factor Xa inhibitor
Apixaban is another direct factor Xa inhibitor.
The APPRAISE-2 trial50 compared apixaban 5 mg twice daily vs placebo in ACS. There was no difference in the rate of cardiovascular death, myocardial infarction, or stroke, but there was significantly more bleeding in the apixaban group, prompting early termination of this study.
Dabigatran, an oral thrombin inhibitor
Dabigatran is an oral direct thrombin inhibitor.
The RE-DEEM trial51 compared four doses of dabigatran (50, 75, 110, and 150 mg twice daily) and placebo in ACS patients. The dabigatran groups had more major and minor bleeding, and the higher the dose, the higher the incidence of bleeding. In addition, the rates of ischemic end points were no lower with dabigatran, although this trial was not powered to show differences in clinical events.
REDUCING THE RISK OF BLEEDING
In the treatment of ACS, the benefits of restoring perfusion by preventing further propagation of thrombus and platelet aggregation come at a significant price of higher bleeding risk. This in turn increases the risk of death through various mechanisms, including shock, worsening ischemia, discontinuation of antiplatelet and anticoagulation therapy causing stent thrombosis, and anemia leading to transfusion, which propagates the underlying inflammatory milieu.52
Giugliano and Braunwald53 provide practical suggestions to reduce this risk, advising physicians to:
- Avoid inappropriately high dosing, particularly in patients with renal insufficiency
- Preferentially use agents that cause less bleeding (eg, bivalirudin, fondaparinux) without compromising anti-ischemic efficacy
- Minimize the concomitant use of other drugs that cause bleeding (eg, NSAIDs)
- Use drugs that protect against bleeding (eg, proton pump inhibitors) in patients at high risk
- Prevent access-site bleeding by using the radial artery, smaller sheaths, and appropriate sheath and closure device management. Indeed, the use of radial interventions in ACS has been shown to reduce access-site-related bleeding, even in patients at high risk.54
The reduction in bleeding risk may provide future trials the opportunity to increase antithrombotic efficacy of different agents with goals of reducing ischemic end points.
Antiplatelet and anticoagulant drugs are a cornerstone of the medical treatment of acute coronary syndrome (ACS), reducing the rates of both morbidity and death.1–4 However, reductions in ischemic events with these drugs have uniformly been accompanied by increases in bleeding complications, which reduce the net benefit.5 Thus, clinical research has been exploring ways to maximize the benefit while minimizing the risk.
Here, we review the guidelines and evidence supporting the use of antiplatelet and anticoagulant drugs in ACS.
ACUTE CORONARY SYNDROMES WITH OR WITHOUT ST ELEVATION
A key distinction when treating ACS is whether the electrocardiogram shows ST-segment elevation. In cases of non-ST-elevation ACS (ie, unstable angina or non-ST-elevation myocardial infarction), a second key question is whether the initial strategy will be invasive (with angiography performed urgently) or conservative (with angiography performed later). In ST-elevation myocardial infarction, another distinction is how perfusion is to be restored, ie, with primary percutaneous coronary intervention or with thrombolysis. All these questions affect the choice of antiplatelet and anticoagulant therapy.
Figure 1 and Figure 2 summarize the guidelines of the American College of Cardiology Foundation and American Heart Association.1,2,6,7
ANTIPLATELET THERAPY
Aspirin for all
Aspirin irreversibly acetylates the enzyme cyclooxygenase-1, blocking intraplatelet formation of thromboxane A2 (Figure 3), a potent platelet aggregator and endothelial vasoconstrictor. Large clinical trials have confirmed that aspirin reduces morbidity and mortality rates by as much as 50% in patients with ACS.8
The ISIS-2 trial9 found that giving aspirin early in the emergency department significantly reduced the mortality rate.
The Antithrombotic Trialists’ Collaboration,10 in a meta-analysis of randomized controlled trials comparing different doses of aspirin in high-risk ACS patients, found no greater benefit for doses of aspirin higher than 162 mg per day when used long-term.
How to use. During an ACS, the patient should receive one dose of aspirin 325 mg (the standard high-dose pill in the United States). This dose should be chewed, as buccal absorption results in more rapid systemic effects.11
Thereafter, the patient should take 81 mg per day, continued indefinitely. The 81-mg dose also applies to patients who undergo a percutaneous coronary intervention with a drug-eluting stent.7 Previous recommendations called for higher doses, but studies have shown that higher doses pose a higher risk of bleeding without additional clinical benefit. The use of enteric-coated aspirin does not reduce this risk,12 and its delayed release may in fact cause aspirin “pseudoresistance.”13
The concurrent use of nonsteroidal anti-inflammatory drugs (NSAIDs) should be avoided, as NSAIDs reversibly bind to platelets, thus preventing aspirin from binding.14 As aspirin washes out of the body, NSAIDs may then become unbound from platelets, leaving platelets activated.
P2Y12 receptor inhibitors: Clopidogrel, prasugrel, ticagrelor
These agents bind to P2Y12 receptors on platelets to inhibit adenosine diphosphate-mediated platelet activation (Figure 3). Clopidogrel and prasugrel are irreversible prodrugs, whereas ticagrelor binds reversibly.
Clopidogrel, a prodrug
Clopidogrel has a half-life of 8 hours and a time to peak concentration of 4 hours. Eighty-five percent of a dose is inactivated by gut esterases. The remainder is metabolized primarily by the cytochrome P4502C19 enzyme system into its active metabolite.
How to use. The recommended dosage is a 600-mg bolus early in the course of ACS. This is associated with a lower rate of cardiovascular events than a 300-mg dose,2,15 although no trial has rigorously compared 300-mg vs 600-mg doses using major clinical end points. In patients presenting with ACS who cannot tolerate aspirin because of hypersensitivity or major gastrointestinal contraindication, clopidogrel is an alternative.1
The CURE trial16 randomized 12,526 patients with non-ST-elevation ACS to receive clopidogrel or placebo in addition to standard therapy. Clopidogrel was associated with a 20% lower rate of cardiovascular death, myocardial infarction, or stroke in both low- and high-risk patients regardless of whether an invasive or conservative strategy was pursued.
However, patients who underwent coronary artery bypass grafting (CABG) had a 53% higher risk of bleeding (an absolute risk of 3.3%) if they received clopidogrel within 5 days of the surgery. This has led to the practice in some centers of delaying giving clopidogrel until after the coronary anatomy has been defined. This deprives the patient of the anti-ischemic benefits conferred by giving clopidogrel early and remains a contentious issue, with most suggesting that the risk-benefit ratio still favors giving clopidogrel early, before angiography, unless there is a high likelihood that surgery will ultimately be required.17 Alternatively, one could consider using a shorter-acting intravenous glycoprotein IIb/IIIa inhibitor such as eptifibatide as a “bridge” until a definitive reperfusion strategy is chosen.
Effect of CYP2C19 variants. The CLOVIS-2 study18 assessed the effects of genetic variants on the clopidogrel concentration in 106 patients who had had a myocardial infarction. The study confirmed that patients who carry certain variants of the CYP2C19 gene attain lower plasma concentrations of clopidogrel after receiving this drug.19 This accounts for its delayed onset of action as well as its variability in response in patients who have reduced expression or inhibition of this enzyme system. Doubling the standard dose in patients who carry these variants does not appear to provide clinical benefit.20
Thus, the thought is emerging that one should consider using prasugrel or ticagrelor instead of clopidogrel in patients who have these polymorphisms, though this is yet to be backed by robust clinical evidence.
Possible interaction with proton pump inhibitors. Controversy exists about whether proton pump inhibitors inhibit clopidogrel’s action. Although the US Food and Drug Administration continues to warn against the concurrent use of omeprazole and clopidogrel,21 an analysis of the PLATO trial22 concluded that patients with ACS who were taking proton pump inhibitors were at higher risk of ischemic events regardless of whether they had been randomized to clopidogrel or ticagrelor (a drug that acts independently of the cytochrome P450 system). This observation suggests that patients on proton pump inhibitors are generally sicker and at higher risk of ischemic events regardless of the choice of antiplatelet therapy. The use of other gastroprotective agents did not appear to mitigate these risks.
Prasugrel: Faster metabolism to active drug
Prasugrel is an irreversible P2Y12 receptor antagonist (Figure 3) that is metabolized into its active metabolite faster and in a more predictable fashion than clopidogrel.23
The TRITON-TIMI 38 study24 included 13,608 ACS patients in whom an early invasive strategy was planned and who were pretreated with prasugrel or clopidogrel in addition to standard treatment. The rate of the primary efficacy end point of death, myocardial infarction, or stroke was 19% lower in the prasugrel group. In those who underwent percutaneous coronary intervention, the incidence of in-stent thrombosis was more than 50% lower in the prasugrel group regardless of whether bare metal stents or drug-eluting stents were used.
Greater platelet inhibition came at the price of a higher incidence of serious bleeding, particularly in the subgroups of patients who were over age 75, had a history of stroke or transient ischemic attack, or weighed less than 60 kg. Prasugrel is therefore contraindicated in patients with a history of transient ischemic attack or stroke. Some suggest that a 5-mg dose can be used with caution (rather than the usual 10-mg dose) in patients over age 75 years or those who have low body weight.
The TRILOGY-ACS trial25 compared prasugrel and clopidogrel in medically managed patients with high-risk non-ST-elevation ACS. It found no difference in the rates of the primary end points of cardiovascular death, myocardial infarction, or stroke at 1 year. In the prespecified subset of patients over age 75 years, the rate of bleeding end points was no higher with prasugrel 5 mg once daily than with clopidogrel.
Prasugrel’s half-life is 7 hours, and its peak antiplatelet effect is within 30 minutes after an oral dose, compared with 4 hours with clopidogrel. Therefore, if a patient with non-ST-elevation ACS is going to go to the catheterization laboratory soon, he or she should not receive prasugrel beforehand, and should receive it later only if the results of angiography indicate that CABG will not be needed urgently. This is an important consideration when using prasugrel, as the rate of surgery-related bleeding was four times higher than with clopidogrel. If possible, this drug should be withheld for at least 7 days before CABG.
Ticagrelor, a direct P2Y12 receptor inhibitor
Ticagrelor, a reversible direct inhibitor of the P2Y12 receptor, inhibits adenosine diphosphate-mediated activation and aggregation (Figure 3). It has a median time to peak concentration of 1.3 to 2 hours and a half-life of 9 hours.
The PLATO trial26 enrolled 18,624 patients with ACS who were given either ticagrelor or clopidogrel in addition to standard therapy. At 12 months, the composite primary end point of myocardial infarction, death, or stroke had occurred in 16% fewer patients receiving ticagrelor than in the clopidogrel group. Analyzed separately, there were 16% fewer myocardial infarctions, 21% fewer cardiovascular deaths, and 22% fewer deaths from any cause, regardless of whether an invasive or conservative strategy was used, and with or without prior clopidogrel use. Fewer cases of stent thrombosis occurred in the ticagrelor group, and the rate of major bleeding was the same.
In a prospectively defined subgroup analysis,27 ticagrelor was beneficial only in patients who received lower doses of aspirin (< 100 mg daily): the hazard ratio for the primary end point was 0.79 (95% confidence interval [CI] 0.71–0.88) in ticagrelor recipients who received low-dose aspirin and 1.45 (95% CI 1.01–2.09) in those who received high-dose aspirin.
Although this analysis is underpowered and controversial, the current evidence suggests that when used in combination with ticagrelor, the aspirin dose should be 81 mg.
Ticagrelor was also associated with a 19% higher incidence of non-CABG- or procedure-related major bleeding, more nonfatal and fatal intracranial bleeding, a higher incidence of dyspnea, and significantly more ventricular pauses.
Although ticagrelor carries no black-box warning about its use in patients with prior stroke or transient ischemic attack, the number of such patients in PLATO was small. Thus, caution should still be used in these patients.28
Ticagrelor should preferably be discontinued 5 days before CABG.
Glycoprotein IIb/IIIa inhibitors: Eptifibatide, tirofiban, abciximab
Glycoprotein IIb/IIIa inhibitors are intravenous agents that act by inhibiting fibrinogen-and von Willebrand factor-mediated platelet-to-platelet cross-linkage, the final pathway of platelet aggregation (Figure 3).
Use of these agents in ACS has been decreasing, as evidence supporting their use was largely established before the era of dual antiplatelet therapy.
A meta-analysis29 of 46,374 patients with non-ST-elevation ACS found that routinely adding a glycoprotein IIb/IIIa inhibitor “upstream” as a third agent in patients receiving dual antiplatelet therapy bought only a modest (11%) reduction in death or myocardial infarction at 30 days, at the price of a 23% increase in major bleeding and no decrease in the overall rate of death. Roughly 70% of the patients were receiving dual antiplatelet therapy before cardiac catheterization.
These agents can be considered in high-risk ACS patients, such as those with ST-segment changes or elevated troponin concentrations, and in diabetic patients, on the assumption that these patients likely have a high intracoronary thrombus burden and are at higher risk of microvascular embolization.6,30 They can also be considered at the time of primary percutaneous coronary intervention in selected patients receiving heparin.7
Eptifibatide
Eptifibatide is a small-molecule, short-acting glycoprotein IIb/IIIa inhibitor with a half-life of 2.5 hours. Its inhibition of platelet aggregation is reversible by stopping the drug infusion and is thought to be a result of dissociation of the drug from platelets.
The PURSUIT trial31 studied 10,948 patients presenting with non-ST-elevation ACS randomized to placebo, eptifibatide in a 180-μg/kg bolus followed by a 2.0-μg/kg/min infusion, or eptifibatide in a 180-μg/kg bolus followed by a 1.3-μg/kg/min infusion. Both eptifibatide groups had a 1.5% absolute reduction in the incidence of the primary end point of death or myocardial infarction, a benefit that was apparent at 96 hours and that persisted through 30 days. Bleeding was more common in the eptifibatide groups, but there was no increase in the rate of hemorrhagic stroke.
The ACUITY trial32 found that early use of eptifibatide or tirofiban had no effect on the primary outcome. (See the section below on bivalirudin for more information about the ACUITY trial.)
PARENTERAL ANTICOAGULANTS
Unfractionated heparin: A declining role
Heparin binds to antithrombin and induces a conformational change, causing rapid inhibition of factor IIa (thrombin), factor IXa, and factor Xa, thus preventing further thrombus propagation (Figure 4). An intravenous bolus of 60 units/kg produces a time to peak of 5 to 10 minutes and a half-life of 30 to 60 minutes.
Heparin can be reversed by giving protamine sulfate (1 mg per 100 units of heparin). For ACS, it is given in a bolus of 60 units/kg not exceeding 4,000 units, followed by an infusion of 12 units/kg/hour, with monitoring of the activated partial thromboplastin time every 6 hours with a goal value of 50 to 70 seconds or 1.5 to 2.5 times control.
Side effects include thrombocytopenia, heparin-induced thrombocytopenia (a distinct condition), and bleeding.
The use of unfractionated heparin was tested in ACS in the early 1990s. Oler et al33 performed a meta-analysis of six randomized trials and found a 33% lower rate of death in patients treated with heparin in addition to aspirin in ACS, as well less reported ischemic pain.
Advantages of unfractionated heparin are that it has stood the test of time, is inexpensive, and can be rapidly reversed. The disadvantages are that it can have serious side effects, including heparin-induced thrombocytopenia, and is more likely to cause bleeding than the newer intravenous anticoagulants discussed below. Thus, its position as the main anticoagulant in ACS is being challenged.
Bivalirudin, a direct thrombin inhibitor
Bivalirudin is a synthetic direct thrombin inhibitor of fluid-phase and clot-bound thrombin (Figure 4). It also inhibits platelets directly.
The ACUITY trial32 randomized 13,819 patients with moderate to high-risk ACS scheduled for invasive treatment into three treatment groups:
- Heparin (either unfractionated heparin or enoxaparin) plus a glycoprotein IIb/IIIa inhibitor (either eptifibatide, tirofiban, or abciximab)
- Bivalirudin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin alone.
The bivalirudin-alone treatment was as sociated with noninferior rates of composite ischemia end points and significantly lower rates of major bleeding, adding up to a significant reduction in the net clinical outcome end point. An important caveat is that bivalirudin’s noninferiority was mostly in the group of patients already receiving a thienopyridine before angiography and percutaneous coronary intervention (RR 0.97 vs 1.27, P = .054). There was less major, nonmajor, minor, CABG-related, and non-CABG-related bleeding as well as need for transfusion in the bivalirudin-alone group, making bivalirudin monotherapy an attractive option in ACS patients with or without ST-segment elevation undergoing a percutaneous coronary intervention.1,31
The ISAR-REACT trial34 later compared bivalirudin alone vs unfractionated heparin and abciximab in patients with non-ST-elevation myocardial infarction undergoing percutaneous coronary intervention pretreated with aspirin and clopidogrel. The composite rate of ischemia was similar in the two treatment groups, with significantly lower rates of bleeding in the bivalirudin group.
HORIZONS-AMI35 randomized 3,602 patients with ST-elevation myocardial infarction receiving aspirin and clopidogrel either to unfractionated heparin and a glycoprotein IIb/IIIa inhibitor or to bivalirudin. As in the ACUITY trial, there was no difference in ischemic end points and a 40% to 45% lower rate of major bleeding end points in the bivalirudin group, translating into an overall lower rate of death.
Enoxaparin, a low-molecular weight heparin
Enoxaparin is a low-molecular-weight heparin that inhibits factor IIa and factor Xa via antithrombin, roughly in a ratio of 1:3 (Figure 4). It has a time to peak effect of 10 minutes when given intravenously36 and 3 to 5 hours when given subcutaneously.37 Its half-life is 4.5 hours, but it is longer in patients with renal dysfunction, requiring dose adjustments in this population.
Its anticoagulant effect is partially reversible. If it is to be reversed between 0 and 8 hours after dosing, the recommended reversal regimen is 1 mg of protamine sulfate for every 1 mg of enoxaparin used. At 8 to 12 hours, it is 0.5 mg of protamine for every 1 mg of enoxaparin. After 12 hours, no protamine is required.
Compared with unfractionated heparin, enoxaparin has less plasma protein binding and a more consistent anticoagulant effect. Its high bioavailability also allows for subcutaneous dosing. Its greater anti-Xa activity inhibits thrombin generation more effectively, and it causes lower rates of thrombocytopenia and heparin-induced thrombocytopenia.
de Lemos et al38 found that, in ACS patients in whom an early conservative approach of medical management was planned, enoxaparin was more efficacious than unfractionated heparin and caused a similar rate of bleeding.
Murphy et al,39 in a meta-analysis of 12 trials in 49,088 ACS patients, also found that enoxaparin had a net clinical benefit compared with unfractionated heparin in reducing rates of myocardial infarction and death despite more bleeding.
The ESSENCE trial40 compared enoxaparin vs unfractionated heparin in 3,171 patients with ACS. It found fewer ischemic events with enoxaparin in the early phase, more minor bleeding, but no increase in major bleeding.
The SYNERGY trial,41 in 10,027 patients with high-risk non-ST-elevation ACS undergoing percutaneous coronary intervention, compared subcutaneous enoxaparin with intravenous heparin. Enoxaparin was found to be noninferior to heparin but caused more bleeding, including major bleeding, drops in hemoglobin, and intracranial hemorrhage.
The EXTRACT-TIMI 25 trial.42 In patients with ST-elevation myocardial infarction, enoxaparin has been shown to be beneficial both in patients treated with fibrinolysis and in those who underwent primary percutaneous coronary intervention. The EXTRACT-TIMI 25 trial randomized 20,749 patients to receive either enoxaparin (an intravenous bolus and maintenance subcutaneous dosing based on renal function) or intravenous heparin in addition to thrombolysis within 6 hours of the diagnosis of ST-elevation myocardial infarction. Although the enoxaparin group had more bleeding end points, they had fewer primary and secondary efficacy end points, translating into an overall net clinical benefit in favor of enoxaparin.
The ATOLL trial43 examined the use of enoxaparin (0.5 mg/kg intravenously) or unfractionated heparin in 910 patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention (via the radial artery in 66% to 69%). Although there was a trend towards benefit in terms of the primary end point of death, myocardial infarction complications, procedure failure, and major bleeding favoring enoxaparin, it was not statistically significant (95% CI 0.68–1.01, P = .06).
However, there was a 37% to 42% lower rate of the secondary end point of death, recurrent myocardial infarction or ACS, or urgent target-vessel revascularization in the enoxaparin group, with a 40% reduction in death from any cause, death from a cardiac cause, or shock. The safety profiles of the two drugs were similar, and the net clinical benefit significantly favored enoxaparin.
Fondaparinux, a factor Xa inhibitor
Fondaparinux is a synthetic pentasaccharide that indirectly inhibits factor Xa through the action of antithrombin (Figure 4). After a 2.5-mg subcutaneous dose, it has a time to peak concentration of 2 hours and a half-life of 17 to 21 hours.
The OASIS-5 trial44 compared fondaparinux and enoxaparin in 20,078 patients treated for non-ST-elevation ACS. Although the rates of death, myocardial infarction, and refractory ischemia at 9 days were similar for both drugs, the fondaparinux group had a significantly (almost 50%) lower rate of bleeding at 30 days, translating into significantly fewer deaths at 30 days. However, patients receiving fondaparinux who underwent percutaneous coronary intervention had a threefold higher rate of catheter-related thrombosis.
The OASIS-6 trial45 compared fondaparinux vs usual care (placebo in those in whom unfractionated heparin was not indicated or unfractionated heparin for up to 48 hours followed by placebo for up to 8 days) in 12,092 patients with ST-elevation myocardial infarction. There was a 1.5% absolute risk reduction in death and reinfarction without an increase in bleeding at 30 days, with trends persisting 6 months into the study. However, fondaparinux was not superior to heparin in the 3% of patients who underwent primary percutaneous coronary intervention. As in OASIS-5, there was more catheter-related thrombosis in the fondaparinux group.
Although the use of supplemental unfractionated heparin appears to have mitigated this risk, fondaparinux remains a less-than-ideal option in the era of primary percutaneous coronary intervention for ST-elevation myocardial infarction and has therefore found limited use in this group of patients. It should, however, be considered in patients for whom a conservative strategy is planned, especially if bleeding risk is deemed to be high.
ORAL ANTICOAGULANTS
Oral anticoagulants provide ischemic benefit in selected patients with ACS—at the price of a higher risk of significant bleeding.
Warfarin
Warfarin was investigated after myocardial infarction in the WARIS II,46 CARS,47 and CHAMP48 trials.
WARIS II46 looked at the use of aspirin alone, warfarin alone, and aspirin and warfarin in combination. The rates of the primary end points of stroke, nonfatal infarction, and death were lower in the warfarin group.
CARS47 found no difference in the rate of the primary end point of fatal infarction, nonfatal ischemic stroke, or cardiovascular death with aspirin vs warfarin plus aspirin.
CHAMP48 saw similar trends, ie, no difference in the rate of death, recurrent myocardial infarction, or stroke with warfarin plus aspirin vs aspirin alone.
All three studies showed increases in major bleeding with warfarin use.
Putting these trials into context, the significant net clinical benefit of dual antiplatelet therapy in the current era compared with the significant bleeding and questionable conflicting evidence supporting benefit with warfarin has limited its use in ACS patients.
Rivaroxaban, an oral factor Xa inhibitor
Rivaroxaban is a novel oral direct reversible factor Xa inhibitor.
The ATLAS ACS 2-TIMI 51 trial49 found rivaroxaban 2.5 mg or 5 mg to yield a significantly lower rate of the primary outcome of cardiovascular death, myocardial infarction, ischemic stroke, and in-stent thrombosis compared with placebo, but significantly more major non-CABG bleeding and intracranial hemorrhage.
The dose used in this trial was much lower than the dose used in trials investigating the role of this drug in stroke prophylaxis in atrial fibrillation.
Apixaban, an oral factor Xa inhibitor
Apixaban is another direct factor Xa inhibitor.
The APPRAISE-2 trial50 compared apixaban 5 mg twice daily vs placebo in ACS. There was no difference in the rate of cardiovascular death, myocardial infarction, or stroke, but there was significantly more bleeding in the apixaban group, prompting early termination of this study.
Dabigatran, an oral thrombin inhibitor
Dabigatran is an oral direct thrombin inhibitor.
The RE-DEEM trial51 compared four doses of dabigatran (50, 75, 110, and 150 mg twice daily) and placebo in ACS patients. The dabigatran groups had more major and minor bleeding, and the higher the dose, the higher the incidence of bleeding. In addition, the rates of ischemic end points were no lower with dabigatran, although this trial was not powered to show differences in clinical events.
REDUCING THE RISK OF BLEEDING
In the treatment of ACS, the benefits of restoring perfusion by preventing further propagation of thrombus and platelet aggregation come at a significant price of higher bleeding risk. This in turn increases the risk of death through various mechanisms, including shock, worsening ischemia, discontinuation of antiplatelet and anticoagulation therapy causing stent thrombosis, and anemia leading to transfusion, which propagates the underlying inflammatory milieu.52
Giugliano and Braunwald53 provide practical suggestions to reduce this risk, advising physicians to:
- Avoid inappropriately high dosing, particularly in patients with renal insufficiency
- Preferentially use agents that cause less bleeding (eg, bivalirudin, fondaparinux) without compromising anti-ischemic efficacy
- Minimize the concomitant use of other drugs that cause bleeding (eg, NSAIDs)
- Use drugs that protect against bleeding (eg, proton pump inhibitors) in patients at high risk
- Prevent access-site bleeding by using the radial artery, smaller sheaths, and appropriate sheath and closure device management. Indeed, the use of radial interventions in ACS has been shown to reduce access-site-related bleeding, even in patients at high risk.54
The reduction in bleeding risk may provide future trials the opportunity to increase antithrombotic efficacy of different agents with goals of reducing ischemic end points.
- Wright RS, Anderson JL, Adams CD, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2011 ACCF/AHA focused update incorporated into the ACC/AHA 2007 Guidelines for the Management of Patients with Unstable Angina/Non-ST-Elevation Myocardial Infarction. J Am Coll Cardiol 2011; 57:e215–e367.
- Jneid H, Anderson JL, Wright RS, et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2012; 60:645–681.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet 1996; 348:1329–1339.
- Cohen M, Adams PC, Parry G, et al. Combination antithrombotic therapy in unstable rest angina and non-Q-wave infarction in nonprior aspirin users. Primary end points analysis from the ATACS trial. Antithrombotic Therapy in Acute Coronary Syndromes Research Group. Circulation 1994; 89:81–88.
- Moscucci M, Fox KA, Cannon CP, et al. Predictors of major bleeding in acute coronary syndromes: the Global Registry of Acute Coronary Events (GRACE). Eur Heart J 2003; 24:1815–1823.
- Levine GN, Bates ER, Blankenship JC, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; Society for Cardiovascular Angiography and Interventions. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol 2011; 58:e44–e122.
- American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:485–510.
- Lewis HD, Davis JW, Archibald DG, et al. Protective effects of aspirin against acute myocardial infarction and death in men with unstable angina. Results of a Veterans Administration Cooperative Study. N Engl J Med 1983; 309:396–403.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Sweeny JM, Gorog DA, Fuster V. Antiplatelet drug ‘resistance’. Part 1: mechanisms and clinical measurements. Nat Rev Cardiol 2009; 6:273–282.
- Kelly JP, Kaufman DW, Jurgelon JM, Sheehan J, Koff RS, Shapiro S. Risk of aspirin-associated major upper-gastrointestinal bleeding with enteric-coated or buffered product. Lancet 1996; 348:1413–1416.
- Grosser T, Fries S, Lawson JA, Kapoor SC, Grant GR, FitzGerald GA. Drug resistance and pseudoresistance: an unintended consequence of enteric coating aspirin. Circulation 2013; 127:377–385.
- US Food and Drug Administration (FDA). Concomitant use of ibuprofen and aspirin: potential for attenuation of the anti-platelet effect of aspirin. http://www.fda.gov/downloads/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm161282.pdf. Accessed November 30, 2013.
- Patti G, Colonna G, Pasceri V, Pepe LL, Montinaro A, Di Sciascio G. Randomized trial of high loading dose of clopidogrel for reduction of periprocedural myocardial infarction in patients undergoing coronary intervention: results from the ARMYDA-2 (Antiplatelet therapy for Reduction of MYocardial Damage during Angioplasty) study. Circulation 2005; 111:2099–2106.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Bavry AA, Lincoff AM. Is clopidogrel cardiovascular medicine’s double-edged sword? Circulation 2006; 113:1638–1640.
- Collet JP, Hulot JS, Anzaha G, et al; CLOVIS-2 Investigators. High doses of clopidogrel to overcome genetic resistance: the randomized crossover CLOVIS-2 (Clopidogrel and Response Variability Investigation Study 2). JACC Cardiovasc Interv 2011; 4:392–402.
- Hulot JS, Collet JP, Cayla G, et al. CYP2C19 but not PON1 genetic variants influence clopidogrel pharmacokinetics, pharmacodynamics, and clinical efficacy in post-myocardial infarction patients. Circ Cardiovasc Interv 2011; 4:422–428.
- Cuisset T, Quilici J, Cohen W, et al. Usefulness of high clopidogrel maintenance dose according to CYP2C19 genotypes in clopidogrel low responders undergoing coronary stenting for non ST elevation acute coronary syndrome. Am J Cardiol 2011; 108:760–765.
- US Food and Drug Administration (FDA). FDA reminder to avoid concomitant use of Plavix (clopidogrel) and omeprazole. http://www.fda.gov/Drugs/DrugSafety/ucm231161.htm. Accessed November 30, 2013.
- Goodman SG, Clare R, Pieper KS, et al; Platelet Inhibition and Patient Outcomes Trial Investigators. Association of proton pump inhibitor use on cardiovascular outcomes with clopidogrel and ticagrelor: insights from the platelet inhibition and patient outcomes trial. Circulation 2012; 125:978–986.
- Solomon S, Vacek JL. Reducing cardiac ischemic events in patients with ACS: prasugrel versus clopidogrel. Commentary. Postgrad Med 2010; 122:198–200.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Roe MT, Armstrong PW, Fox KA, et al; TRILOGY ACS Investigators. Prasugrel versus clopidogrel for acute coronary syndromes without revascularization. N Engl J Med 2012; 367:1297–1309.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Mahaffey KW, Wojdyla DM, Carroll K, et al; PLATO Investigators. Ticagrelor compared with clopidogrel by geographic region in the Platelet Inhibition and Patient Outcomes (PLATO) trial. Circulation 2011; 124:544–554.
- Verheugt FW. Beware of novel antiplatelet therapy in acute coronary syndrome patients with previous stroke. Circulation 2012; 125:2821–2823.
- Tricoci P, Newby LK, Hasselblad V, et al. Upstream use of small-molecule glycoprotein iib/iiia inhibitors in patients with non-ST-segment elevation acute coronary syndromes: a systematic overview of randomized clinical trials. Circ Cardiovasc Qual Outcomes 2011; 4:448–458.
- Kastrati A, Mehilli J, Neumann FJ, et al; Intracoronary Stenting and Antithrombotic: Regimen Rapid Early Action for Coronary Treatment 2 (ISAR-REACT 2) Trial Investigators. Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: the ISAR-REACT 2 randomized trial. JAMA 2006; 295:1531–1538.
- Inhibition of platelet glycoprotein IIb/IIIa with eptifibatide in patients with acute coronary syndromes. The PURSUIT Trial Investigators. Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin Therapy. N Engl J Med 1998; 339:436–443.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Oler A, Whooley MA, Oler J, Grady D. Adding heparin to aspirin reduces the incidence of myocardial infarction and death in patients with unstable angina. A meta-analysis. JAMA 1996; 276:811–815.
- Kastrati A, Neumann FJ, Schulz S, et al; ISAR-REACT 4 Trial Investigators. Abciximab and heparin versus bivalirudin for non-ST-elevation myocardial infarction. N Engl J Med 2011; 365:1980–1989.
- Stone GW, Witzenbichler B, Guagliumi G, et al; HORIZONS-AMI Trial Investigators. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med 2008; 358:2218–2230.
- Aslam MS, Sundberg S, Sabri MN, Cooke D, Lakier JB. Pharmacokinetics of intravenous/subcutaneous enoxaparin in patients with acute coronary syndrome undergoing percutaneous coronary interventions. Catheter Cardiovasc Interv 2002; 57:187–190.
- Sanofi-Aventis US. Lovenox (enoxaparin sodium injection) product information. http://www.lovenox.com/hcp/clinical-data.aspx. Accessed December 1, 2013.
- de Lemos JA, Blazing MA, Wiviott SD, et al. Enoxaparin versus unfractionated heparin in patients treated with tirofiban, aspirin and an early conservative initial management strategy: results from the A phase of the A-to-Z trial. Eur Heart J 2004; 25:1688–1694.
- Murphy SA, Gibson CM, Morrow DA, et al. Efficacy and safety of the low-molecular weight heparin enoxaparin compared with unfractionated heparin across the acute coronary syndrome spectrum: a meta-analysis. Eur Heart J 2007; 28:2077–2086.
- Cohen M, Demers C, Gurfinkel EP, et al. A comparison of low-molecular-weight heparin with unfractionated heparin for unstable coronary artery disease. Efficacy and Safety of Subcutaneous Enoxaparin in Non-Q-Wave Coronary Events Study Group. N Engl J Med 1997; 337:447–452.
- Ferguson JJ, Califf RM, Antman EM, et al; SYNERGY Trial Investigators. Enoxaparin vs unfractionated heparin in high-risk patients with non-ST-segment elevation acute coronary syndromes managed with an intended early invasive strategy: primary results of the SYNERGY randomized trial. JAMA 2004; 292:45–54.
- Antman EM, Morrow DA, McCabe CH, et al; ExTRACT-TIMI 25 Investigators. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med 2006; 354:1477–1488.
- Montalescot G, Zeymer U, Silvain J, et al; ATOLL Investigators. Intravenous enoxaparin or unfractionated heparin in primary percutaneous coronary intervention for ST-elevation myocardial infarction: the international randomised open-label ATOLL trial. Lancet 2011; 378:693–703.
- Yusuf S, Mehta SR, Chrolavicius S, et al; Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Yusuf S, Mehta SR, Chrolavicius S, et al; OASIS-6 Trial Group. Effects of fondaparinux on mortality and reinfarction in patients with acute ST-segment elevation myocardial infarction: the OASIS-6 randomized trial. JAMA 2006; 295:1519–1530.
- Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002; 347:969–974.
- Coumadin Aspirin Reinfarction Study (CARS) Investigators. Randomised double-blind trial of fixed low-dose warfarin with aspirin after myocardial infarction. Lancet 1997; 350:389–396.
- Fiore LD, Ezekowitz MD, Brophy MT, Lu D, Sacco J, Peduzzi P; Combination Hemotherapy and Mortality Prevention (CHAMP) Study Group. Department of Veterans Affairs Cooperative Studies Program Clinical Trial comparing combined warfarin and aspirin with aspirin alone in survivors of acute myocardial infarction: primary results of the CHAMP study. Circulation 2002; 105:557–563.
- Mega JL, Braunwald E, Wiviott SD, et al; ATLAS ACS 2–TIMI 51 Investigators. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med 2012; 366:9–19.
- Alexander JH, Lopes RD, James S, et al; APPRAISE-2 Investigators. Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med 2011; 365:699–708.
- Oldgren J, Budaj A, Granger CB, et al; RE-DEEM Investigators. Dabigatran vs placebo in patients with acute coronary syndromes on dual antiplatelet therapy: a randomized, double-blind, phase II trial. Eur Heart J 2011; 32:2781–2789.
- Steg PG, Huber K, Andreotti F, et al. Bleeding in acute coronary syndromes and percutaneous coronary interventions: position paper by the Working Group on Thrombosis of the European Society of Cardiology. Eur Heart J 2011; 32:1854–1864.
- Giugliano RP, Braunwald E. The year in non-ST-segment elevation acute coronary syndrome. J Am Coll Cardiol 2012; 60:2127–039.
- Rao SV, Ou FS, Wang TY, et al. Trends in the prevalence and outcomes of radial and femoral approaches to percutaneous coronary intervention: a report from the National Cardiovascular Data Registry. JACC Cardiovasc Interv 2008; 1:379–386.
- Wright RS, Anderson JL, Adams CD, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2011 ACCF/AHA focused update incorporated into the ACC/AHA 2007 Guidelines for the Management of Patients with Unstable Angina/Non-ST-Elevation Myocardial Infarction. J Am Coll Cardiol 2011; 57:e215–e367.
- Jneid H, Anderson JL, Wright RS, et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2012; 60:645–681.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet 1996; 348:1329–1339.
- Cohen M, Adams PC, Parry G, et al. Combination antithrombotic therapy in unstable rest angina and non-Q-wave infarction in nonprior aspirin users. Primary end points analysis from the ATACS trial. Antithrombotic Therapy in Acute Coronary Syndromes Research Group. Circulation 1994; 89:81–88.
- Moscucci M, Fox KA, Cannon CP, et al. Predictors of major bleeding in acute coronary syndromes: the Global Registry of Acute Coronary Events (GRACE). Eur Heart J 2003; 24:1815–1823.
- Levine GN, Bates ER, Blankenship JC, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; Society for Cardiovascular Angiography and Interventions. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol 2011; 58:e44–e122.
- American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:485–510.
- Lewis HD, Davis JW, Archibald DG, et al. Protective effects of aspirin against acute myocardial infarction and death in men with unstable angina. Results of a Veterans Administration Cooperative Study. N Engl J Med 1983; 309:396–403.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Sweeny JM, Gorog DA, Fuster V. Antiplatelet drug ‘resistance’. Part 1: mechanisms and clinical measurements. Nat Rev Cardiol 2009; 6:273–282.
- Kelly JP, Kaufman DW, Jurgelon JM, Sheehan J, Koff RS, Shapiro S. Risk of aspirin-associated major upper-gastrointestinal bleeding with enteric-coated or buffered product. Lancet 1996; 348:1413–1416.
- Grosser T, Fries S, Lawson JA, Kapoor SC, Grant GR, FitzGerald GA. Drug resistance and pseudoresistance: an unintended consequence of enteric coating aspirin. Circulation 2013; 127:377–385.
- US Food and Drug Administration (FDA). Concomitant use of ibuprofen and aspirin: potential for attenuation of the anti-platelet effect of aspirin. http://www.fda.gov/downloads/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm161282.pdf. Accessed November 30, 2013.
- Patti G, Colonna G, Pasceri V, Pepe LL, Montinaro A, Di Sciascio G. Randomized trial of high loading dose of clopidogrel for reduction of periprocedural myocardial infarction in patients undergoing coronary intervention: results from the ARMYDA-2 (Antiplatelet therapy for Reduction of MYocardial Damage during Angioplasty) study. Circulation 2005; 111:2099–2106.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Bavry AA, Lincoff AM. Is clopidogrel cardiovascular medicine’s double-edged sword? Circulation 2006; 113:1638–1640.
- Collet JP, Hulot JS, Anzaha G, et al; CLOVIS-2 Investigators. High doses of clopidogrel to overcome genetic resistance: the randomized crossover CLOVIS-2 (Clopidogrel and Response Variability Investigation Study 2). JACC Cardiovasc Interv 2011; 4:392–402.
- Hulot JS, Collet JP, Cayla G, et al. CYP2C19 but not PON1 genetic variants influence clopidogrel pharmacokinetics, pharmacodynamics, and clinical efficacy in post-myocardial infarction patients. Circ Cardiovasc Interv 2011; 4:422–428.
- Cuisset T, Quilici J, Cohen W, et al. Usefulness of high clopidogrel maintenance dose according to CYP2C19 genotypes in clopidogrel low responders undergoing coronary stenting for non ST elevation acute coronary syndrome. Am J Cardiol 2011; 108:760–765.
- US Food and Drug Administration (FDA). FDA reminder to avoid concomitant use of Plavix (clopidogrel) and omeprazole. http://www.fda.gov/Drugs/DrugSafety/ucm231161.htm. Accessed November 30, 2013.
- Goodman SG, Clare R, Pieper KS, et al; Platelet Inhibition and Patient Outcomes Trial Investigators. Association of proton pump inhibitor use on cardiovascular outcomes with clopidogrel and ticagrelor: insights from the platelet inhibition and patient outcomes trial. Circulation 2012; 125:978–986.
- Solomon S, Vacek JL. Reducing cardiac ischemic events in patients with ACS: prasugrel versus clopidogrel. Commentary. Postgrad Med 2010; 122:198–200.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Roe MT, Armstrong PW, Fox KA, et al; TRILOGY ACS Investigators. Prasugrel versus clopidogrel for acute coronary syndromes without revascularization. N Engl J Med 2012; 367:1297–1309.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Mahaffey KW, Wojdyla DM, Carroll K, et al; PLATO Investigators. Ticagrelor compared with clopidogrel by geographic region in the Platelet Inhibition and Patient Outcomes (PLATO) trial. Circulation 2011; 124:544–554.
- Verheugt FW. Beware of novel antiplatelet therapy in acute coronary syndrome patients with previous stroke. Circulation 2012; 125:2821–2823.
- Tricoci P, Newby LK, Hasselblad V, et al. Upstream use of small-molecule glycoprotein iib/iiia inhibitors in patients with non-ST-segment elevation acute coronary syndromes: a systematic overview of randomized clinical trials. Circ Cardiovasc Qual Outcomes 2011; 4:448–458.
- Kastrati A, Mehilli J, Neumann FJ, et al; Intracoronary Stenting and Antithrombotic: Regimen Rapid Early Action for Coronary Treatment 2 (ISAR-REACT 2) Trial Investigators. Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: the ISAR-REACT 2 randomized trial. JAMA 2006; 295:1531–1538.
- Inhibition of platelet glycoprotein IIb/IIIa with eptifibatide in patients with acute coronary syndromes. The PURSUIT Trial Investigators. Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin Therapy. N Engl J Med 1998; 339:436–443.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Oler A, Whooley MA, Oler J, Grady D. Adding heparin to aspirin reduces the incidence of myocardial infarction and death in patients with unstable angina. A meta-analysis. JAMA 1996; 276:811–815.
- Kastrati A, Neumann FJ, Schulz S, et al; ISAR-REACT 4 Trial Investigators. Abciximab and heparin versus bivalirudin for non-ST-elevation myocardial infarction. N Engl J Med 2011; 365:1980–1989.
- Stone GW, Witzenbichler B, Guagliumi G, et al; HORIZONS-AMI Trial Investigators. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med 2008; 358:2218–2230.
- Aslam MS, Sundberg S, Sabri MN, Cooke D, Lakier JB. Pharmacokinetics of intravenous/subcutaneous enoxaparin in patients with acute coronary syndrome undergoing percutaneous coronary interventions. Catheter Cardiovasc Interv 2002; 57:187–190.
- Sanofi-Aventis US. Lovenox (enoxaparin sodium injection) product information. http://www.lovenox.com/hcp/clinical-data.aspx. Accessed December 1, 2013.
- de Lemos JA, Blazing MA, Wiviott SD, et al. Enoxaparin versus unfractionated heparin in patients treated with tirofiban, aspirin and an early conservative initial management strategy: results from the A phase of the A-to-Z trial. Eur Heart J 2004; 25:1688–1694.
- Murphy SA, Gibson CM, Morrow DA, et al. Efficacy and safety of the low-molecular weight heparin enoxaparin compared with unfractionated heparin across the acute coronary syndrome spectrum: a meta-analysis. Eur Heart J 2007; 28:2077–2086.
- Cohen M, Demers C, Gurfinkel EP, et al. A comparison of low-molecular-weight heparin with unfractionated heparin for unstable coronary artery disease. Efficacy and Safety of Subcutaneous Enoxaparin in Non-Q-Wave Coronary Events Study Group. N Engl J Med 1997; 337:447–452.
- Ferguson JJ, Califf RM, Antman EM, et al; SYNERGY Trial Investigators. Enoxaparin vs unfractionated heparin in high-risk patients with non-ST-segment elevation acute coronary syndromes managed with an intended early invasive strategy: primary results of the SYNERGY randomized trial. JAMA 2004; 292:45–54.
- Antman EM, Morrow DA, McCabe CH, et al; ExTRACT-TIMI 25 Investigators. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med 2006; 354:1477–1488.
- Montalescot G, Zeymer U, Silvain J, et al; ATOLL Investigators. Intravenous enoxaparin or unfractionated heparin in primary percutaneous coronary intervention for ST-elevation myocardial infarction: the international randomised open-label ATOLL trial. Lancet 2011; 378:693–703.
- Yusuf S, Mehta SR, Chrolavicius S, et al; Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Yusuf S, Mehta SR, Chrolavicius S, et al; OASIS-6 Trial Group. Effects of fondaparinux on mortality and reinfarction in patients with acute ST-segment elevation myocardial infarction: the OASIS-6 randomized trial. JAMA 2006; 295:1519–1530.
- Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002; 347:969–974.
- Coumadin Aspirin Reinfarction Study (CARS) Investigators. Randomised double-blind trial of fixed low-dose warfarin with aspirin after myocardial infarction. Lancet 1997; 350:389–396.
- Fiore LD, Ezekowitz MD, Brophy MT, Lu D, Sacco J, Peduzzi P; Combination Hemotherapy and Mortality Prevention (CHAMP) Study Group. Department of Veterans Affairs Cooperative Studies Program Clinical Trial comparing combined warfarin and aspirin with aspirin alone in survivors of acute myocardial infarction: primary results of the CHAMP study. Circulation 2002; 105:557–563.
- Mega JL, Braunwald E, Wiviott SD, et al; ATLAS ACS 2–TIMI 51 Investigators. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med 2012; 366:9–19.
- Alexander JH, Lopes RD, James S, et al; APPRAISE-2 Investigators. Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med 2011; 365:699–708.
- Oldgren J, Budaj A, Granger CB, et al; RE-DEEM Investigators. Dabigatran vs placebo in patients with acute coronary syndromes on dual antiplatelet therapy: a randomized, double-blind, phase II trial. Eur Heart J 2011; 32:2781–2789.
- Steg PG, Huber K, Andreotti F, et al. Bleeding in acute coronary syndromes and percutaneous coronary interventions: position paper by the Working Group on Thrombosis of the European Society of Cardiology. Eur Heart J 2011; 32:1854–1864.
- Giugliano RP, Braunwald E. The year in non-ST-segment elevation acute coronary syndrome. J Am Coll Cardiol 2012; 60:2127–039.
- Rao SV, Ou FS, Wang TY, et al. Trends in the prevalence and outcomes of radial and femoral approaches to percutaneous coronary intervention: a report from the National Cardiovascular Data Registry. JACC Cardiovasc Interv 2008; 1:379–386.
KEY POINTS
- Although antiplatelet and anticoagulant drugs reduce the risk of ischemic events, including coronary death, they also increase the risk of bleeding, reducing their net benefit. But the risk of bleeding can be managed.
- All patients experiencing an ACS should receive a single dose of aspirin 325 mg and should be instructed to chew it; this should be followed by 81 mg daily.
- Patients who are not expected to undergo coronary artery bypass grafting on an urgent basis should also receive clopidogrel, prasugrel, or ticagrelor.
- Glycoprotein IIb/IIIa inhibitors are being used less now than in the past.
- The use of unfractionated heparin is being challenged by newer parenteral anticoagulants, ie, bivalirudin, enoxaparin, and fondaparinux.
- The role of oral anticoagulants (warfarin, rivaroxaban, apixaban, and dabigatran) in ACS is uncertain.
Clinical update in sexually transmitted diseases 2014
With nearly 20 million new infections annually, sexually transmitted diseases (STDs) are very common in the United States.1,2 And with recent changes to the national health care landscape, including the passage of the Affordable Care Act and state budget cuts resulting in the closure of STD and human immunodeficiency virus (HIV) clinics, primary care providers can expect to encounter more patients with STDs.
For women and infants, STDs can have serious and long-term consequences, including infertility, facilitation of HIV infection, reproductive tract cancer, pelvic inflammatory disease, and poor perinatal outcomes.2 STDs cost the US health care system nearly $16 billion every year.3
STD prevention and control strategies traditionally include surveillance, screening, behavioral interventions, treatment, and partner management.4 This paper will review patient management by syndrome and provide guidance to clinicians to facilitate timely diagnosis and treatment, important components of any effective STD prevention strategy.
ANYONE CAN HAVE AN STD
STDs affect people of all races, ages, and sexual orientations. That said, some groups are at greater risk:
Adolescents and young adults. Persons ages 15 through 24 represent 25% of the sexually experienced population in the United States but account for nearly half of all incident STDs.1
Racial and ethnic minorities. STD disparities are one of the five greatest health disparities for African American communities.4

Men who have sex with men number approximately 2% to 4% of the US male population, yet account for approximately 70% of reported cases of primary and secondary syphilis and more than 50% of persons with HIV infection.3,5,6
HIGH-RISK BEHAVIOR AND SCREENING
A thorough sexual history will reveal behaviors that place a person at risk of infection. The US Preventive Services Task Force (USPSTF) defines high-risk sexual behavior as having multiple current partners, having a new partner, using condoms inconsistently, having sex while under the influence of alcohol or drugs, or exchanging sex for money or drugs.7
An effective strategy for obtaining a sexual history is the “five Ps”8:
- Partners (eg, Do you have sex with men, women, or both?)
- Prevention of pregnancy (eg, What are you doing to prevent pregnancy?)
- Protection from STDs (eg, What do you do to protect yourself from STDs?)
- Practices (eg, To understand your risks for STDs, I need to understand the kind of sex you have had recently.)
- Past history of STDs (eg, Have you ever had an STD?).8
The USPSTF and the US Centers for Disease Control and Prevention (CDC) recommend certain populations be screened for STDs.7,8
Everyone age 13 through 64 should be tested for HIV at least once, per CDC recommendation.8
Sexually active females up to age 24 should routinely be screened for chlamydia every year.7,8
Nonpregnant women at higher risk of infection should be screened for gonorrhea and syphilis.
Pregnant women, regardless of risk, should be screened for chlamydia, hepatitis B, HIV, and syphilis; pregnant women at higher risk of infection should also be screened for gonorrhea and hepatitis C.7,9
Men should be screened for HIV, and men at higher risk should also be screened for syphilis.8
Men who have sex with men should be screened at least annually for HIV and syphilis and undergo a test for urethral chlamydia and gonorrhea infection. Men who participate in receptive anal intercourse should be tested for rectal chlamydia and gonorrhea and, in those who participate in oral intercourse, for pharyngeal gonorrhea.8
PREVENTION
Vaccination against HPV, hepatitis A and B
Preexposure vaccination is one of the most effective ways to prevent human papillomavirus (HPV), hepatitis A, and hepatitis B infection.10
HPV vaccination. The Advisory Committee on Immunization Practices recommends routine HPV vaccination of female patients at age 11 or 12, or through age 26 if not previously vaccinated.11,12 Routine vaccination is also recommended for males at age 11 or 12 and through age 21, if not previously vaccinated.12 The upper age is extended through age 26 for men who have sex with men and for immunocompromised patients.12
Two HPV vaccines are available for females; one is quadrivalent and the other is bivalent. Both protect against two HPV types that cause cervical and other HPV-associated cancers.11 The quadrivalent vaccine also protects against the two types that cause 90% of genital warts.13 Only the quadrivalent vaccine is licensed for use in males.12
Hepatitis A and B vaccination. Hepatitis B vaccination is recommended for all unvaccinated, uninfected patients being evaluated for an STD.8,14
Vaccinating for hepatitis A and hepatitis B is important for men who have sex with men, who have a higher risk of acquiring and transmitting these infections.15
Other preventive practices
Male circumcision has been shown to reduce the risk of HIV infection, high-risk genital HPV infection, and genital herpes in heterosexual men.10,16,17
Male condoms, when used consistently and correctly, can reduce the risk of chlamydia, gonorrhea, and trichomoniasis.8 The risk of transmission of syphilis, genital HPV, and genital herpes can also be reduced by correctly and consistently using condoms when the infected area of exposure is covered.8
Microbiocides not recommended. Topical microbiocides do not have not enough evidence to recommend them for STD prevention. However, limited data suggest that tenofovir 1% vaginal gel may reduce the risk of acquiring genital herpes simplex virus type 2 (HSV-2) infection in women.18,19
GENITAL ULCERATIVE DISEASE: HERPES, SYPHILIS, OTHERS
The risk of acquiring HIV is two to five times higher if one is exposed to it when a genital ulcerative disease is present.20,21
In the United States, most cases of genital, anal, or perianal ulcers in sexually active persons are due to genital herpes or syphilis.8 Other causes include chancroid, granuloma inguinale, lymphogranuloma venereum, and noninfectious causes.
Although the frequency of these conditions varies by geographic area and demographic profile, herpes is the most prevalent of them.8 HSV-2 infection is one of the most prevalent STDs in the United States, with approximately 17% of all adolescents and adults infected,1,22 and a much higher prevalence in persons who use drugs.23
A thorough medical history and physical examination should be conducted. In addition, an accurate diagnosis requires specific tests: syphilis serology, dark field microscopy (if available), and culture or polymerase chain reaction (PCR) testing for herpes.8 A positive serologic test for HSV-1 or HSV-2 is enough to make the diagnosis in a patient whose symptoms suggest herpes, even if PCR and culture are negative.
GENITAL HERPES: MOSTLY ASYMPTOMATIC
Genital herpes is caused by HSV-1 or HSV-2. Of these, HSV-1 is on the rise, causing an increasing proportion of first episodes of genital herpes in some populations. It may now account for most new genital herpes infections in young women and in men who have sex with men.8,24
Most people infected with HSV-1 or HSV-2 have no symptoms or have subclinical disease. When symptoms do occur, one or more vesicles may appear on or around the genitals, rectum, or mouth. The average incubation period after exposure is 4 days (range 2–12).25 The first episode of genital herpes is often associated with systemic symptoms (eg, fever, headache, myalgia, and malaise) and local symptoms (eg, dysuria, vaginal or urethral discharge, and inguinal adenopathy).26,27
Genital herpes often recurs, especially during the first year. Recurrences are less frequent with HSV-1 than with HSV-2.
Diagnosing genital herpes requires laboratory testing
Diagnosing genital herpes by clinical signs and symptoms is both insensitive and nonspecific. Therefore, laboratory testing should be performed for patients who present with genital ulcerative disease.
Virologic tests. Viral culture and nucleic acid amplification methods, including PCR assays, are the preferred virologic tests for herpes.8 Although viral culture is widely available, its sensitivity depends on the stage of the lesion and rapidly declines as lesions begin to heal. PCR assays are more sensitive than viral culture, can be done in automated systems, and are increasingly being used in clinical settings.26 However, if the patient has no active lesions at the time of testing, failure to detect herpes by culture or PCR does not guarantee that the patient is not infected, as viral shedding is intermittent.
Serologic tests. Type-specific antibodies to HSV develop during the first several weeks following infection and persist indefinitely. Providers should specifically request serologic type-specific immunoglobulin G (IgG) assays. IgM testing for HSV should not be used, as IgM testing is not type-specific. Moreover, although some clinicians believe IgM is a good test for early infection because levels rise early and then decline, IgM may be positive during recurrent episodes.8
Both laboratory-based assays and point-of-care tests for HSV-2 are available. They are performed on capillary blood or serum and have sensitivities that range from 80% to 100% and specificities greater than 96%, compared with HSV-2 immunoblot and Western blot testing as the standard.28,29
False-negative results may be more frequent in the early stages of infection. HSV-2 antibody indicates anogenital infection, while HSV-1 antibody might also be due to orolabial infection, which is something to keep in mind in a patient without genital symptoms.8
Treatment can control herpes but not eradicate it
Several herpes vaccines have undergone clinical trials, but an effective one remains elusive.30,31
Antiviral therapy is used to control signs and symptoms of clinical disease but does not eradicate latent virus. For initial clinical episodes of genital herpes, the CDC recommends acyclovir, valacyclovir, or famciclovir for 7 to 10 days.8 In patients with established genital herpes, daily suppressive antiviral therapy can reduce recurrences, subclinical shedding, and the likelihood of transmission to partners; famciclovir is somewhat less efficacious for suppressing viral shedding.8
A diagnosis of herpes can carry considerable stigma, which can substantially interfere with a patient’s current and future relationships.25,32,33 Sex partners of patients with genital herpes may benefit from evaluation and counseling. Clinicians should appreciate the psychological impact of a genital herpes diagnosis and address these concerns by providing education, counseling, and support while encouraging patients to recognize that herpes is a manageable condition.34
SYPHILIS IS INCREASING IN MEN WHO HAVE SEX WITH MEN
Since 2001, rates of syphilis, a systemic disease caused by Treponema pallidum, have been increasing in men who have sex with men.3,35 As of 2011, this group accounts for approximately 72% of all cases of primary and secondary syphilis in the United States.3
Primary syphilis is characterized by a firm, painless chancre at the site of inoculation. The chancre lasts 3 to 6 weeks and heals regardless of treatment. However, if the infected person does not receive adequate treatment, the infection progresses to the secondary stage.26
Secondary syphilis typically starts with a nonpruritic rash, usually macular or papular, on the trunk and extremities, classically including the palms and soles. Other symptoms may include alopecia, lymphadenopathy, condylomata, and systemic symptoms.
Without treatment, the infection can progress to latent syphilis, which is further categorized as early (acquired during the preceding year), late latent, or of unknown duration.26
Practical diagnosis of syphilis relies on serologic testing
Definitive diagnosis of early syphilis requires dark field microscopy or PCR to detect T pallidum in lesion exudate or tissue.8 However, because there are no commercially available tests for T pallidum, serologic testing is the mainstay.
Two types of serologic tests must be performed to diagnose syphilis: a nontreponemal test and a treponemal test.8
Nontreponemal tests:
- The Venereal Disease Research Laboratory (VDRL) test
- The rapid plasma reagin (RPR) test.
Treponemal tests:
- T pallidum passive particle agglutination (TP-PA) assay
- Fluorescent treponemal antibody absorbed (FTA-ABS) test
- Enzyme immunoassay (EIA)
- Chemiluminescence immunoassay (CIA).
Using only one type of serologic test is in-sufficient for diagnosis because each type has limitations, including the possibility of false-positive results.
Nontreponemal test results may correlate with disease activity, and results should be reported quantitatively; a fourfold change in titer, equivalent to a change of two dilutions, is needed to demonstrate a clinically significant difference between two nontreponemal test results using the same serologic test.8
Which order of testing for syphilis is best?
The CDC recommends that nontreponemal tests be used to screen for syphilis and treponemal tests be used to confirm the diagnosis.36 This traditional algorithm performs well in identifying persons with active infection who require further evaluation and treatment, while minimizing false-positive results in low-prevalence populations.
However, some clinical laboratories have adopted the reverse sequence, using treponemal tests (usually an EIA or a CIA) to screen for syphilis, followed by a nontreponemal test to confirm active infection.36–38 This reverse-sequence testing may result in overdiagnosis and overtreatment of syphilis in some clinical settings.37 When reverse-sequence syphilis screening is used, the CDC recommends reflexively testing all sera that produce reactive EIA or CIA results with a quantitative nontreponemal test and reflexively testing sera with discordant results (ie, a reactive EIA or CIA and a nonreactive RPR or VDRL test) with a different treponemal test.36
Traditionally, the FTA-ABS test had been considered the gold standard treponemal test; however, it has lower specificity than other treponemal tests. Accordingly, TP-PA is the recommended confirmatory treponemal test.8,39 False-negative results, although rare, may occur for biological or technical reasons, such as the prozone phenomenon, resulting in a missed diagnosis. The prozone phenomenon occurs when the antibody titer is high and antigen binding sites are saturated, preventing the cross-linking reaction between antigens and antibodies. In this instance, when syphilis is suspected clinically and the RPR assay is nonreactive, the clinician can request RPR testing at dilutions of sera, ie, diluting the patient’s serum to bring the antibody concentration into the zone equivalence.40
Suspect neurosyphilis if neurologic symptoms arise
Central nervous system involvement can occur at any stage of syphilis. If clinical evidence of neurologic involvement (meningitis, stroke, cranial nerve dysfunction, or auditory or ophthalmic abnormalities) is observed in any patient with syphilis, regardless of stage, a cerebrospinal fluid examination should be performed.8 Laboratory testing can support the diagnosis of neurosyphilis; however, no single laboratory test can be used to diagnose it.
Cerebrospinal fluid abnormalities (ie, mononuclear pleocytosis, increased protein) are common in patients with early syphilis even in the absence of clinical neurologic findings. There is no firm evidence to support diverging from recommended treatment for early syphilis in these patients.35
Cerebrospinal fluid examination is recommended for all patients with serologic evidence of syphilis infection and neurologic symptoms and for patients who do not achieve an adequate serologic decline with stage-appropriate treatment.8
Penicillin is still the mainstay of syphilis treatment
Penicillin is still the mainstay of syphilis treatment.
- Patients with early syphilis (primary, secondary, or early latent) should receive a single intramuscular dose of benzathine penicillin G (2.4 million units).8,35
- Patients with late latent syphilis should receive three intramuscular doses of benzathine penicillin G (2.4 million units each) at 1-week intervals.
- Neurosyphilis should be treated with aqueous crystalline penicillin G 18 to 24 million units daily for 10 to 14 days.8
Doxycycline is the preferred alternative in nonpregnant patients who are allergic to penicillin. The dosage is 100 mg orally twice a day for 14 days (for primary, secondary, or early latent infections) or for 28 days (for late latent infections or those of unknown duration).35,41
Ceftriaxone (1–2 g daily) may be effective for treating early syphilis. However, data are limited, and the optimal dose and duration of therapy are not defined.8,42
Azithromycin in a single 2-g oral dose is effective for treating early syphilis. However, T pallidum chromosomal mutations associated with macrolide resistance are being detected.35,43 In view of this, azithromycin should not be used in men who have sex with men or in pregnant women.
Follow-up of syphilis patients and partners
Close clinical and serologic follow-up is strongly advised in persons who receive an alternate regimen to evaluate for treatment failure.8
Sex partners of patients with primary syphilis should be considered at risk and given treatment if they had sexual contact with the patient within 3 months plus the duration of symptoms, within 6 months plus the duration of symptoms for those with secondary syphilis, or 1 year for patients with early latent syphilis.8
Serologic and clinical evaluation should be repeated at 6, 12, and 24 months after treatment. HIV-infected patients should receive closer follow-up, ie, at 3, 6, 9, 12, and 24 months. The same quantitative nontreponemal serologic test should be used at each visit, with at least a fourfold decrease in titer representing an appropriate serologic decline.
Failure to achieve an appropriate serologic decline in 6 to 12 months may represent treatment failure.8 Optimal management in this instance is unclear; at a minimum, additional clinical and serologic follow-up should be performed.8 If additional follow-up cannot be ensured, retreatment (weekly intramuscular injections of benzathine penicillin G 2.4 million units for 3 weeks) is recommended. Cerebrospinal fluid examination can be considered to exclude unrecognized neurosyphilis.8
URETHRITIS: GONORRHEA, CHLAMYDIA TOP THE LIST
Symptoms of urethritis can include dysuria, discharge (purulent or mucopurulent), and urethral pruritus.26 However, asymptomatic infections are common.8
Several organisms are associated with infectious urethritis, including26:
- Neisseria gonorrhoeae
- Chlamydia trachomatis
- Mycoplasma genitalium
- Trichomonas vaginalis
- Ureaplasma urealyticum.
Diagnosing urethritis: Try to identify the agent
The clinician should attempt to obtain objective evidence of urethral inflammation. Urethral discharge should be examined with microscopy using Gram stain or methylene blue, or a first-void urine sample should be tested with microscopy and leukocyte esterase.26 If clinic-based diagnostic testing is not available, testing for N gonorrhoeae and C trachomatis using nucleic acid amplification can identify additional infections.8
During the clinic visit, either Gram-stain microscopy of a urethral swab or microscopic examination of a first-catch urine sample may identify the causative agent. If gram-negative intracellular diplococci are seen on urethral smear, gonorrhea infection is diagnosed. Nongonococcal urethritis is diagnosed when microscopy or urinalysis displays evidence of inflammation (positive leukocyte esterase or at least 10 white blood cells per high-power field) without gram-negative intracellular diplococci.8
Testing should be performed to determine the specific cause of urethritis, because both chlamydia and gonorrhea are reportable diseases. Nucleic acid amplification tests are the most sensitive tests for C trachomatis and N gonorrhoeae and can be performed on urethral swabs or urine.44 If clinic-based diagnostic tools are unavailable, patients should receive empiric treatment for chlamydia and gonorrhea.8
Treatment of urethritis, by organism
Urethral gonorrhea should be treated with dual therapy: ceftriaxone 250 mg in a single intramuscular dose and either azithromycin 1 g orally as a single dose or doxycycline 100 mg orally twice a day for 7 days. Oral cephalosporins are no longer recommended as first-line treatment of gonorrhea.45
Chlamydial urethritis is treated with azithromycin 1 g in a single oral dose or doxycycline 100 mg orally twice a day for 7 days. Although azithromycin provides the advantages of a single-dose regimen administered and directly observed by the provider, some evidence suggests that doxycycline may be more effective than azithromycin for symptomatic chlamydial urethritis.46
All sex partners within the preceding 60 days should be referred for evaluation, testing, and empiric treatment with a drug regimen effective against chlamydia (if nongonococcal urethritis or only C trachomatis was identified) and gonorrhea (if N gonorrhoeae was identified). When patients diagnosed with chlamydia or gonorrhea indicate that their partners are unlikely to seek evaluation, providers can offer patient-delivered partner therapy, a form of expedited partner therapy in which partners of infected persons are treated without previous medical evaluation. Providers should visit www.cdc.gov/std/ept for updated information for their individual jurisdiction, as expedited partner therapy is prohibited in some states. No studies have been published involving patient-delivered partner therapy for chlamydia or gonorrhea in men who have sex with men.8
Patients with recurrent or persistent urethritis can be retreated with the initial regimen if they did not comply with treatment or were reexposed to an untreated sex partner. However, persistent urethritis after doxycycline therapy may suggest the presence of doxycycline-resistant M genitalium or U urealyticum. T vaginalis may also cause urethritis in men. Diagnostic evaluation may include culture or nucleic acid amplification testing of a urethral swab or urine (ie, PCR [Amplicor] or transcription-mediated amplification).8
M genitalium or U urealyticum urethritis. Currently, no commercially available diagnostic test exists for M genitalium or U urealyticum, so clinicians must choose a treatment regimen on the basis of objective evidence of inflammation in the absence of an etiologic agent. If azithromycin was not given during the initial course, metronidazole 2 g orally or tinidazole 2 g orally in a single dose plus azithromycin 1 g orally should be considered.8
M genitalium is one of the most common pathogens in men with persistent urethritis, accounting for 15% to 25% of cases. Several studies have shown that moxifloxacin 400 mg orally daily for 7 days is effective against M genitalium.46–48 Therefore, men for whom an initial regimen of azithromycin fails should be retreated with moxifloxacin 400 mg orally once daily for 7 days.
If men require treatment with a new antibiotic regimen for persistent urethritis and a sexually transmitted agent is the suspected cause, all partners in the past 60 days before the initial diagnosis and any interim partners should be referred for evaluation and appropriate treatment.
CERVICITIS: CHLAMYDIA, GONORRHEA, OTHERS
Cervicitis is frequently asymptomatic, but signs on pelvic examination may include purulent or mucopurulent endocervical exudate and sustained endocervical bleeding easily induced by passage of a cotton swab through the cervical os.26
In most cases, the pathogen cannot be identified.49 When an organism is isolated, it is typically C trachomatis or N gonorrhoeae. Others that may cause cervicitis include the organisms responsible for bacterial vaginosis, T vaginalis, HSV, and possibly M genitalium.50
Diagnostic workup for cervicitis
Diagnostic workup for cervicitis should include microscopic evaluation of an endocervical specimen and testing for C trachomatis and N gonorrhoeae. A finding of leukorrhea (> 10 white blood cells per high-power field on microscopic examination of vaginal fluid) has been associated with chlamydial and gonococcal infection of the cervix.8 In the absence of inflammatory vaginitis, leukorrhea might be a sensitive indicator of cervical inflammation, with a high negative predictive value.
Nucleic acid amplification testing for C trachomatis and N gonorrhoeae can be performed on urine, endocervical, or vaginal swab specimens collected by the clinician or self-collected.51 The performance of C trachomatis nucleic acid amplification testing on patient-collected vaginal swab specimens has similar sensitivity and specificity to those performed on cervical and first-void urine samples.26,44,52
Women with cervicitis also should be evaluated for bacterial vaginosis and trichomoniasis, and if the organisms that cause these conditions are detected, treatment is advised. Microscopy has a low sensitivity (approximately 50%) for detecting T vaginalis; if the organism is not identified, further testing such as culture may be performed to exclude it as the pathogen.
Women with cervicitis should also be evaluated for clinical signs of pelvic inflammatory disease, including uterine, adnexal, and cervical motion tenderness, and fever.
Cervicitis can be treated presumptively
Women with cervicitis who should receive presumptive therapy include those at higher risk of chlamydial infection (ie, those with new or multiple sex partners, those age 25 or younger, and those who engage in unprotected intercourse, especially if follow-up cannot be ensured).8
Recommended therapy is either azithromycin 1 g orally in a single dose or doxycycline 100 mg orally twice a day for 7 days. The clinician should consider dual therapy to cover gonorrhea if the prevalence of gonorrhea is more than 5% (ie, in younger patients).8 If bacterial vaginosis or T vaginalis infection is diagnosed, these conditions should be treated at the time of clinical evaluation (see vaginitis section for more detail). For women in whom presumptive therapy is deferred, the results of diagnostic testing should guide appropriate treatment.
Repeat testing 3 to 6 months after treatment is recommended for all women diagnosed with chlamydia or gonorrhea,53 and all sex partners in the past 60 days should be referred for evaluation and receive treatment for the STDs for which the index patient received treatment. In states where it is allowed, patient-delivered partner therapy should be considered if the patient indicates her partner is unlikely to seek medical evaluation.
VAGINITIS: NOT JUST YEAST
Women with vaginitis may present with complaints of discharge, pruritus, or a bad vaginal odor. A careful medical history, including information on sexual behaviors and vaginal hygiene practices (ie, douching), should be conducted in addition to physical examination and diagnostic testing.
The most common conditions associated with vaginitis are bacterial vaginosis, trichomoniasis, and candidiasis. However, vulvovaginal candidiasis, most often caused by Candida albicans, is not transmitted sexually and will not be reviewed further here. Although bacterial vaginosis is associated with known risk factors for STDs (eg, new or multiple sex partners), the cause of the microbial alteration that precipitates it is not known.44 Co-infection with T vaginalis is extremely common.54
BACTERIAL VAGINOSIS: VERY COMMON
Bacterial vaginosis is the most common genital infection in reproductive-age women.55 It is a polymicrobial syndrome in which anaerobic bacteria (Prevotella and Mobiluncus species), Gardnerella vaginalis, Ureaplasma, and Mycoplasma replace the normal vaginal flora.
Diagnosis of bacterial vaginosis
Bacterial vaginosis can be diagnosed by clinical criteria or Gram staining. Clinical diagnosis requires three of the following clinical criteria proposed by Amsel et al56:
- Clue cells
- Vaginal fluid pH > 4.5
- Fishy odor before or after addition of 10% potassium hydroxide
- Thin, homogeneous, white discharge that smoothly coats the vaginal walls.
The clinical utility of PCR for diagnosing G vaginalis remains unclear, and culture is not recommended because it has low specificity. Gram staining can be used to determine the concentration of lactobacilli, small gram-negative or variable rods (G vaginalis and anaerobic rods), and curved gram-negative rods (ie, Mobiluncus species).
Treatment of bacterial vaginosis: Metronidazole or clindamycin
Treatment is recommended to relieve vaginal symptoms, with the potential benefit of reducing the risk of acquiring chlamydia, gonorrhea, and HIV and other viral STDs.8
Recommended treatment is with metronidazole 500 mg orally twice a day for 7 days, metronidazole gel 0.75% vaginal suppository once a day for 5 days, or clindamycin cream 2% vaginal suppository once a day for 7 days.44 A meta-analysis of several trials found that clindamycin and metronidazole have equivalent effectiveness for eradicating bacterial vaginosis symptoms.57 Accordingly, providers can consider patient preference, co-infections, and possible side effects when selecting a regimen. Alternative regimens include tinidazole or clindamycin orally or in vaginal ovules.
Women should be advised to refrain from sexual intercourse during treatment. Routine treatment of male or female sexual partners is not warranted.
Bacterial vaginosis commonly recurs, and limited data exist regarding optimal management of recurrences. Using a different treatment regimen may be an option in patients who have a recurrence; however, re-treatment with the same topical regimen is an acceptable approach for treating recurrent bacterial vaginosis during the early stages of infection.58 One study suggests that metronidazole for 7 days, followed by intravaginal boric acid for 21 days, and then, for those in remission, suppressive metronidazole gel for 16 weeks may be another option.59 For women with multiple recurrences, metronidazole gel twice weekly for 4 to 6 months has been shown to reduce recurrences, although its benefit may not persist after it is stopped. The therapeutic role for probiotics remains unclear.60
TRICHOMONIASIS: TREAT PARTNERS
Although most women with trichomoniasis have few or no symptoms, some have vaginal discharge that may be diffuse, malodorous, and yellow-greenish, and some have vulvar irritation.
Diagnosis of trichomoniasis: Microscopy is first-line but insensitive
The most common method for diagnosing T vaginalis infection remains microscopic evaluation of wet preparations of genital secretions, because of its convenience and relatively low cost. This may demonstrate the motile, flagellated protozoa T vaginalis and many white blood cells. Slides of vaginal fluid specimens should be examined immediately after collection to maximize performance. Unfortunately, the sensitivity of wet preparation is 44% to 80% in vaginal specimens.61
Culture is still considered the gold standard for diagnosing trichomoniasis and, if available, should be performed when direct microscopy is unrevealing.
Point-of-care diagnostic tests for T vaginalis infection include the OSOM Trichomonas Rapid Test (Sekisui Diagnostics, San Diego, CA), which is an immunochromatographic capillary flow dipstick test, and the Affirm VPIII (Becton, Dickinson and Company, Franklin Lakes, NJ), a nucleic acid probe-hydridization test that identifies T vaginalis, G vaginalis, and C albicans.44,62 Liquid-based Pap tests may demonstrate T vaginalis, although they should not be performed exclusively for this purpose. Among women, nucleic acid amplification tests may detect a prevalence three to five times higher than indicated by wet mount microscopy. The APTIMA Trichomonas vaginalis assay (Hologic Gen-Probe, San Diego, CA) was the most sensitive test for trichomonas detection in this study.63
Extragenital testing with nucleic acid amplification tests is not recommended for T vaginalis, as it remains unclear if the rectum can serve as a reservoir for infection, and T vaginalis has not been found to infect oral sites.8
Treatment of trichomoniasis: Metronidazole or tinidazole
Nitroimidazoles, ie, metronidazole and tinidazole, are the only class of drugs available to treat trichomoniasis. The recommended regimen is metronidazole or tinidazole 2 g orally in a single dose. Studies suggest that tinidazole may be superior to metronidazole, with higher cure rates due to its longer half-life and higher tissue concentrations.64
Low-level metronidazole resistance is estimated to occur in 2% to 5% of trichomoniasis infections65; high-level resistance occurs rarely. If a single dose of metronidazole 2 g fails to cure the infection and reinfection is ruled out, the patient should be treated with metronidazole 500 mg orally twice a day for 7 days. If this regimen is not effective, providers can consider tinidazole or metronidazole 2 g orally for 7 days.44,62,64 Consultation and susceptibility testing for T vaginalis is available from the CDC if these alternative regimens are ineffective.
T vaginalis infection has a high rate of transmission to sexual partners,66 and all partners should be treated. Male sexual partners should be treated with metronidazole 500 mg twice a day orally for 7 days, tinidazole 2 g orally in a single dose, or tinidazole 500 mg twice a day for 7 days. Patient-delivered partner therapy may have a role in partner management for trichomoniasis.67
PROCTITIS: SUSPECT LYMPHOGRANULOMA VENEREUM
Acute proctitis in men and women who practice receptive anal intercourse is usually sexually acquired. The most common causative organisms are N gonorrhoeae, C trachomatis (serotypes associated with or not associated with lymphogranuloma venereum), and HSV; T pallidum is less common.68 Co-infections are not uncommon in this setting.69
Symptoms of proctitis may include anal discharge, rectal ulcers and bleeding, anorectal pain, tenesmus, and constipation. Patients with lymphogranuloma venereum may also present with tender, fluctuant inguinal or femoral lymphadenopathy (buboes), or herpetiform genital ulcers or papules.
Diagnosis of proctitis
Clinical evaluation should include digital rectal examination and anoscopy (if possible) to look for abnormalities such as ulcerations, hemorrhoids, anal fissures, condylomas, strictures, exudate, and bleeding.
Appropriate diagnostic testing includes Gram staining and culture of discharge, herpes viral culture or PCR, and nucleic acid amplification testing for chlamydia and gonorrhea (in laboratories with Clinical Laboratory Improvement Amendments validation).8
Nucleic acid amplification tests detect C trachomatis serotypes L1–L3, responsible for lymphogranuloma venereum, and non-lymphogranuloma venereum serotypes A–K, but cannot distinguish between the two, whereas PCR-based genotyping can.8,26 Although this distinction is important to ensure appropriate evaluation and management of sex partners, empiric treatment for lymphogranuloma venereum (doxycycline 100 mg orally twice a day for 21 days) should be provided to patients at high risk, including men who have sex with men and who have anorectal chlamydia, HIV infection, or bloody discharge and perianal or mucosal ulcers.8
Treatment of proctitis
Patients with painful perianal or mucosal ulceration should receive presumptive treatment for lymphogranuloma venereum and HSV while awaiting results of diagnostic testing. If rectal discharge is detected or Gram staining of anorectal secretions detects polymorphonuclear leukocytes, treatment should include ceftriaxone 250 mg intramuscularly and doxycycline 100 mg orally twice a day for 7 days.8 Additional testing for syphilis and HIV should also be performed.
All sexual partners should be evaluated for any disease diagnosed in the index patient.
- Satterwhite CL, Torrone E, Meites E, et al. Sexually transmitted infections among US women and men: prevalence and incidence estimates, 2008. Sex Transm Dis 2013; 40:187–193.
- Institute of Medicine (US); Committee on Prevention and Control of Sexually Transmitted Diseases. The hidden epidemic: confronting sexually transmitted diseases. Washington, DC: National Academy Press; 1997.
- Centers for Disease Control and Prevention (CDC). 2011 Sexually Transmitted Disease Surveillance. http://www.cdc.gov/std/stats11/toc.htm. Accessed January 10, 2014.
- Barrow RY, Newman LM, Douglas JM. Taking positive steps to address STD disparities for African-American communities. Sex Transm Dis 2008; 35(suppl 12):S1–S3.
- Mitchell JW, Petroll AE. Patterns of HIV and sexually transmitted infection testing among men who have sex with men couples in the United States. Sex Transm Dis 2012; 39:871–876.
- Centers for Disease Control and Prevention (CDC). HIV testing among men who have sex with men—21 cities, United States, 2008. MMWR Morb Mortal Wkly Rep 2011; 60:694–699.
- Meyers D, Wolff T, Gregory K, et al., USPSTF. USPSTF recommendations for STI screening. Am Fam Physician 2008; 77:819–824.
- Workowski KA, Berman S; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm Rep 2010; 59:1–110.
- Summaries for patients. Screening for chlamydial infection: recommendations from the US Preventive Services Task Force. Ann Intern Med 2007; 147:I44.
- Marrazzo JM, Cates W. Interventions to prevent sexually transmitted infections, including HIV infection. Clin Infect Dis 2011; 53(suppl 3):S64–S78.
- Markowitz LE, Dunne EF, Saraiya M, Lawson HW, Chesson H, Unger ER; Centers for Disease Control and Prevention (CDC); Advisory Committee on Immunization Practices (ACIP). Quadrivalent Human Papillomavirus Vaccine: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2007; 56:1–24.
- Centers for Disease Control and Prevention (CDC). Recommendations on the use of quadrivalent human papillomavirus vaccine in males—Advisory Committee on Immunization Practices (ACIP), 2011. MMWR Morb Mortal Wkly Rep 2011; 60:1705–1708.
- Paavonen J, Jenkins D, Bosch FX, et al; HPV PATRICIA study group. Efficacy of a prophylactic adjuvanted bivalent L1 virus-like-particle vaccine against infection with human papillomavirus types 16 and 18 in young women: an interim analysis of a phase III double-blind, randomised controlled trial. Lancet 2007; 369:2161–2170.
- Mast EE, Weinbaum CM, Fiore AE, et al; Advisory Committee on Immunization Practices (ACIP); Centers for Disease Control and Prevention (CDC). A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) Part II: immunization of adults. MMWR Recomm Rep 2006; 55:1–33.
- Mayer KH. Sexually transmitted diseases in men who have sex with men. Clin Infect Dis 2011; 53(suppl 3):S79–S83.
- Tobian AA, Serwadda D, Quinn TC, et al. Male circumcision for the prevention of HSV-2 and HPV infections and syphilis. N Engl J Med 2009; 360:1298–1309.
- Auvert B, Sobngwi-Tambekou J, Cutler E, et al. Effect of male circumcision on the prevalence of high-risk human papillomavirus in young men: results of a randomized controlled trial conducted in Orange Farm, South Africa. J Infect Dis 2009; 199:14–19.
- Obiero J, Mwethera PG, Wiysonge CS. Topical microbicides for prevention of sexually transmitted infections. Cochrane Database Syst Rev 2012; 6:CD007961.
- Abdool Karim Q, Abdool Karim SS, Frohlich JA, et al; CAPRISA 004 Trial Group. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science 2010; 329:1168–1174.
- HIV prevention through early detection and treatment of other sexually transmitted diseases—United States. Recommendations of the Advisory Committee for HIV and STD prevention. MMWR Recomm Rep 1998; 47:1–24.
- Fleming DT, Wasserheit JN. From epidemiological synergy to public health policy and practice: the contribution of other sexually transmitted diseases to sexual transmission of HIV infection. Sex Transm Infect 1999; 75:3–17.
- Centers for Disease Control and Prevention (CDC). Seroprevalence of herpes simplex virus type 2 among persons aged 14–49 years—United States, 2005–2008. MMWR Morb Mortal Wkly Rep 2010; 59:456–459.
- Semaan S, Leinhos M, Neumann MS. Public health strategies for prevention and control of HSV-2 in persons who use drugs in the United States. Drug Alcohol Depend 2013; 131:182–197.
- Bernstein DI, Bellamy AR, Hook EW, et al. Epidemiology, clinical presentation, and antibody response to primary infection with herpes simplex virus type 1 and type 2 in young women. Clin Infect Dis 2013; 56:344–351.
- Kimberlin DW, Rouse DJ. Clinical practice. Genital herpes. N Engl J Med 2004; 350:1970–1977.
- Sexually Transmitted Diseases. 4th ed. New York, NY: McGraw-Hill Professional Publishing; 2007.
- Johnston C, Magaret A, Selke S, Remington M, Corey L, Wald A. Herpes simplex virus viremia during primary genital infection. J Infect Dis 2008; 198:31–34.
- Laderman EI, Whitworth E, Dumaual E, et al. Rapid, sensitive, and specific lateral-flow immunochromatographic point-of-care device for detection of herpes simplex virus type 2-specific immunoglobulin G antibodies in serum and whole blood. Clin Vaccine Immunol 2008; 15:159–163.
- Philip SS, Ahrens K, Shayevich C, et al. Evaluation of a new point-of-care serologic assay for herpes simplex virus type 2 infection. Clin Infect Dis 2008; 47:e79–e82.
- Belshe RB, Leone PA, Bernstein DI, et al; Herpevac Trial for Women. Efficacy results of a trial of a herpes simplex vaccine. N Engl J Med 2012; 366:34–43.
- Stanberry LR, Spruance SL, Cunningham AL, et al; GlaxoSmithKline Herpes Vaccine Efficacy Study Group. Glycoprotein-D-adjuvant vaccine to prevent genital herpes. N Engl J Med 2002; 347:1652–1661.
- Mark H, Gilbert L, Nanda J. Psychosocial well-being and quality of life among women newly diagnosed with genital herpes. J Obstet Gynecol Neonatal Nurs 2009; 38:320–326.
- Gilbert LK, Omisore F. Common questions about herpes: analysis of chat-room transcripts. Herpes 2009; 15:57–61.
- Alexander L, Naisbett B. Patient and physician partnerships in managing genital herpes. J Infect Dis 2002; 186(suppl 1):S57–S65.
- Ghanem KG, Workowski KA. Management of adult syphilis. Clin Infect Dis 2011; 53(suppl 3):S110–S128.
- Centers for Disease Control and Prevention (CDC). Discordant results from reverse sequence syphilis screening—five laboratories, United States, 2006–2010. MMWR Morb Mortal Wkly Rep 2011; 60:133–137.
- Centers for Disease Control and Prevention (CDC). Syphilis testing algorithms using treponemal tests for initial screening—four laboratories, New York City, 2005–2006. MMWR Morb Mortal Wkly Rep 2008; 57:872–875.
- Seña AC, White BL, Sparling PF. Novel Treponema pallidum serologic tests: a paradigm shift in syphilis screening for the 21st century. Clin Infect Dis 2010; 51:700–708.
- Marangoni A, Sambri V, Storni E, D’Antuono A, Negosanti M, Cevenini R. Treponema pallidum surface immunofluorescence assay for serologic diagnosis of syphilis. Clin Diagn Lab Immunol 2000; 7:417–421.
- Post JJ, Khor C, Furner V, Smith DE, Whybin LR, Robertson PW. Case report and evaluation of the frequency of the prozone phenomenon in syphilis serology—an infrequent but important laboratory phenomenon. Sex Health 2012; 9:488–490.
- Ghanem KG, Erbelding EJ, Cheng WW, Rompalo AM. Doxycycline compared with benzathine penicillin for the treatment of early syphilis. Clin Infect Dis 2006; 42:e45–e49.
- Hook EW, Roddy RE, Handsfield HH. Ceftriaxone therapy for incubating and early syphilis. J Infect Dis 1988; 158:881–884.
- A2058G Prevalence Workgroup. Prevalence of the 23S rRNA A2058G point mutation and molecular subtypes in Treponema pallidum in the United States, 2007 to 2009. Sex Transm Dis 2012; 39:794–798.
- Workowski KA, Berman SM. Centers for Disease Control and Prevention Sexually Transmitted Disease Treatment Guidelines. Clin Infect Dis 2011; 53(suppl 3):S59–S63.
- Kirkcaldy RD, Bolan GA, Wasserheit JN. Cephalosporin-resistant gonorrhea in North America. JAMA 2013; 309:185–187.
- Seña AC, Lensing S, Rompalo A, et al. Chlamydia trachomatis, Mycoplasma genitalium, and Trichomonas vaginalis infections in men with nongonococcal urethritis: predictors and persistence after therapy. J Infect Dis 2012; 206:357–365.
- Manhart LE, Broad JM, Golden MR. Mycoplasma genitalium: should we treat and how? Clin Infect Dis 2011; 53(suppl 3):S129–S142.
- Jernberg E, Moghaddam A, Moi H. Azithromycin and moxifloxacin for microbiological cure of Mycoplasma genitalium infection: an open study. Int J STD AIDS 2008; 19:676–679.
- Taylor SN, Lensing S, Schwebke J, et al. Prevalence and treatment outcome of cervicitis of unknown etiology. Sex Transm Dis 2013; 40:379–385.
- Lusk MJ, Konecny P. Cervicitis: a review. Curr Opin Infect Dis 2008; 21:49–55.
- Geisler WM. Diagnosis and management of uncomplicated Chlamydia trachomatis infections in adolescents and adults: summary of evidence reviewed for the 2010 Centers for Disease Control and Prevention Sexually Transmitted Diseases Treatment Guidelines. Clin Infect Dis 2011; 53(suppl 3):S92–S98.
- Schachter J, Chernesky MA, Willis DE, et al. Vaginal swabs are the specimens of choice when screening for Chlamydia trachomatis and Neisseria gonorrhoeae: results from a multicenter evaluation of the APTIMA assays for both infections. Sex Transm Dis 2005; 32:725–728.
- Hosenfeld CB, Workowski KA, Berman S, et al. Repeat infection with Chlamydia and gonorrhea among females: a systematic review of the literature. Sex Transm Dis 2009; 36:478–489.
- Sobel JD, Subramanian C, Foxman B, Fairfax M, Gygax SE. Mixed vaginitis-more than coinfection and with therapeutic implications. Curr Infect Dis Rep 2013; 15:104–108.
- Taylor BD, Darville T, Haggerty CL. Does bacterial vaginosis cause pelvic inflammatory disease? Sex Transm Dis 2013; 40:117–122.
- Amsel R, Totten PA, Spiegel CA, Chen KC, Eschenbach D, Holmes KK. Nonspecific vaginitis. Diagnostic criteria and microbial and epidemiologic associations. Am J Med 1983; 74:14–22.
- Oduyebo OO, Anorlu RI, Ogunsola FT. The effects of antimicrobial therapy on bacterial vaginosis in non-pregnant women. Cochrane Database Syst Rev 2009; ( 3):CD006055.
- Bunge KE, Beigi RH, Meyn LA, Hillier SL. The efficacy of retreatment with the same medication for early treatment failure of bacterial vaginosis. Sex Transm Dis 2009; 36:711–713.
- Reichman O, Akins R, Sobel JD. Boric acid addition to suppressive antimicrobial therapy for recurrent bacterial vaginosis. Sex Transm Dis 2009; 36:732–734.
- Senok AC, Verstraelen H, Temmerman M, Botta GA. Probiotics for the treatment of bacterial vaginosis. Cochrane Database Syst Rev 2009; (4):CD006289.
- Nye MB, Schwebke JR, Body BA. Comparison of APTIMA Trichomonas vaginalis transcription-mediated amplification to wet mount microscopy, culture, and polymerase chain reaction for diagnosis of trichomoniasis in men and women. Am J Obstet Gynecol 2009; 200:188.e1–188.e7.
- Bachmann LH, Hobbs MM, Seña AC, et al. Trichomonas vaginalis genital infections: progress and challenges. Clin Infect Dis 2011; 53(suppl 3):S160–S172.
- Schwebke JR, Hobbs MM, Taylor SN, et al. Molecular testing for Trichomonas vaginalis in women: results from a prospective US clinical trial. J Clin Microbiol 2011; 49:4106–4111.
- Coleman JS, Gaydos CA, Witter F. Trichomonas vaginalis vaginitis in obstetrics and gynecology practice: new concepts and controversies. Obstet Gynecol Surv 2013; 68:43–50.
- Kirkcaldy RD, Augostini P, Asbel LE, et al. Trichomonas vaginalis antimicrobial drug resistance in 6 US cities, STD Surveillance Network, 2009–2010. Emerg Infect Dis 2012; 18:939–943.
- Hoots BE, Peterman TA, Torrone EA, Weinstock H, Meites E, Bolan GA. A Trich-y question: should Trichomonas vaginalis infection be reportable? Sex Transm Dis 2013; 40:113–116.
- Schwebke JR, Desmond RA. A randomized controlled trial of partner notification methods for prevention of trichomoniasis in women. Sex Transm Dis 2010; 37:392–396.
- Studemeister A. Cytomegalovirus proctitis: a rare and disregarded sexually transmitted disease. Sex Transm Dis 2011; 38:876–878.
- Hoentjen F, Rubin DT. Infectious proctitis: when to suspect it is not inflammatory bowel disease. Dig Dis Sci 2012; 57:269–273.
With nearly 20 million new infections annually, sexually transmitted diseases (STDs) are very common in the United States.1,2 And with recent changes to the national health care landscape, including the passage of the Affordable Care Act and state budget cuts resulting in the closure of STD and human immunodeficiency virus (HIV) clinics, primary care providers can expect to encounter more patients with STDs.
For women and infants, STDs can have serious and long-term consequences, including infertility, facilitation of HIV infection, reproductive tract cancer, pelvic inflammatory disease, and poor perinatal outcomes.2 STDs cost the US health care system nearly $16 billion every year.3
STD prevention and control strategies traditionally include surveillance, screening, behavioral interventions, treatment, and partner management.4 This paper will review patient management by syndrome and provide guidance to clinicians to facilitate timely diagnosis and treatment, important components of any effective STD prevention strategy.
ANYONE CAN HAVE AN STD
STDs affect people of all races, ages, and sexual orientations. That said, some groups are at greater risk:
Adolescents and young adults. Persons ages 15 through 24 represent 25% of the sexually experienced population in the United States but account for nearly half of all incident STDs.1
Racial and ethnic minorities. STD disparities are one of the five greatest health disparities for African American communities.4
Men who have sex with men number approximately 2% to 4% of the US male population, yet account for approximately 70% of reported cases of primary and secondary syphilis and more than 50% of persons with HIV infection.3,5,6
HIGH-RISK BEHAVIOR AND SCREENING
A thorough sexual history will reveal behaviors that place a person at risk of infection. The US Preventive Services Task Force (USPSTF) defines high-risk sexual behavior as having multiple current partners, having a new partner, using condoms inconsistently, having sex while under the influence of alcohol or drugs, or exchanging sex for money or drugs.7
An effective strategy for obtaining a sexual history is the “five Ps”8:
- Partners (eg, Do you have sex with men, women, or both?)
- Prevention of pregnancy (eg, What are you doing to prevent pregnancy?)
- Protection from STDs (eg, What do you do to protect yourself from STDs?)
- Practices (eg, To understand your risks for STDs, I need to understand the kind of sex you have had recently.)
- Past history of STDs (eg, Have you ever had an STD?).8
The USPSTF and the US Centers for Disease Control and Prevention (CDC) recommend certain populations be screened for STDs.7,8
Everyone age 13 through 64 should be tested for HIV at least once, per CDC recommendation.8
Sexually active females up to age 24 should routinely be screened for chlamydia every year.7,8
Nonpregnant women at higher risk of infection should be screened for gonorrhea and syphilis.
Pregnant women, regardless of risk, should be screened for chlamydia, hepatitis B, HIV, and syphilis; pregnant women at higher risk of infection should also be screened for gonorrhea and hepatitis C.7,9
Men should be screened for HIV, and men at higher risk should also be screened for syphilis.8
Men who have sex with men should be screened at least annually for HIV and syphilis and undergo a test for urethral chlamydia and gonorrhea infection. Men who participate in receptive anal intercourse should be tested for rectal chlamydia and gonorrhea and, in those who participate in oral intercourse, for pharyngeal gonorrhea.8
PREVENTION
Vaccination against HPV, hepatitis A and B
Preexposure vaccination is one of the most effective ways to prevent human papillomavirus (HPV), hepatitis A, and hepatitis B infection.10
HPV vaccination. The Advisory Committee on Immunization Practices recommends routine HPV vaccination of female patients at age 11 or 12, or through age 26 if not previously vaccinated.11,12 Routine vaccination is also recommended for males at age 11 or 12 and through age 21, if not previously vaccinated.12 The upper age is extended through age 26 for men who have sex with men and for immunocompromised patients.12
Two HPV vaccines are available for females; one is quadrivalent and the other is bivalent. Both protect against two HPV types that cause cervical and other HPV-associated cancers.11 The quadrivalent vaccine also protects against the two types that cause 90% of genital warts.13 Only the quadrivalent vaccine is licensed for use in males.12
Hepatitis A and B vaccination. Hepatitis B vaccination is recommended for all unvaccinated, uninfected patients being evaluated for an STD.8,14
Vaccinating for hepatitis A and hepatitis B is important for men who have sex with men, who have a higher risk of acquiring and transmitting these infections.15
Other preventive practices
Male circumcision has been shown to reduce the risk of HIV infection, high-risk genital HPV infection, and genital herpes in heterosexual men.10,16,17
Male condoms, when used consistently and correctly, can reduce the risk of chlamydia, gonorrhea, and trichomoniasis.8 The risk of transmission of syphilis, genital HPV, and genital herpes can also be reduced by correctly and consistently using condoms when the infected area of exposure is covered.8
Microbiocides not recommended. Topical microbiocides do not have not enough evidence to recommend them for STD prevention. However, limited data suggest that tenofovir 1% vaginal gel may reduce the risk of acquiring genital herpes simplex virus type 2 (HSV-2) infection in women.18,19
GENITAL ULCERATIVE DISEASE: HERPES, SYPHILIS, OTHERS
The risk of acquiring HIV is two to five times higher if one is exposed to it when a genital ulcerative disease is present.20,21
In the United States, most cases of genital, anal, or perianal ulcers in sexually active persons are due to genital herpes or syphilis.8 Other causes include chancroid, granuloma inguinale, lymphogranuloma venereum, and noninfectious causes.
Although the frequency of these conditions varies by geographic area and demographic profile, herpes is the most prevalent of them.8 HSV-2 infection is one of the most prevalent STDs in the United States, with approximately 17% of all adolescents and adults infected,1,22 and a much higher prevalence in persons who use drugs.23
A thorough medical history and physical examination should be conducted. In addition, an accurate diagnosis requires specific tests: syphilis serology, dark field microscopy (if available), and culture or polymerase chain reaction (PCR) testing for herpes.8 A positive serologic test for HSV-1 or HSV-2 is enough to make the diagnosis in a patient whose symptoms suggest herpes, even if PCR and culture are negative.
GENITAL HERPES: MOSTLY ASYMPTOMATIC
Genital herpes is caused by HSV-1 or HSV-2. Of these, HSV-1 is on the rise, causing an increasing proportion of first episodes of genital herpes in some populations. It may now account for most new genital herpes infections in young women and in men who have sex with men.8,24
Most people infected with HSV-1 or HSV-2 have no symptoms or have subclinical disease. When symptoms do occur, one or more vesicles may appear on or around the genitals, rectum, or mouth. The average incubation period after exposure is 4 days (range 2–12).25 The first episode of genital herpes is often associated with systemic symptoms (eg, fever, headache, myalgia, and malaise) and local symptoms (eg, dysuria, vaginal or urethral discharge, and inguinal adenopathy).26,27
Genital herpes often recurs, especially during the first year. Recurrences are less frequent with HSV-1 than with HSV-2.
Diagnosing genital herpes requires laboratory testing
Diagnosing genital herpes by clinical signs and symptoms is both insensitive and nonspecific. Therefore, laboratory testing should be performed for patients who present with genital ulcerative disease.
Virologic tests. Viral culture and nucleic acid amplification methods, including PCR assays, are the preferred virologic tests for herpes.8 Although viral culture is widely available, its sensitivity depends on the stage of the lesion and rapidly declines as lesions begin to heal. PCR assays are more sensitive than viral culture, can be done in automated systems, and are increasingly being used in clinical settings.26 However, if the patient has no active lesions at the time of testing, failure to detect herpes by culture or PCR does not guarantee that the patient is not infected, as viral shedding is intermittent.
Serologic tests. Type-specific antibodies to HSV develop during the first several weeks following infection and persist indefinitely. Providers should specifically request serologic type-specific immunoglobulin G (IgG) assays. IgM testing for HSV should not be used, as IgM testing is not type-specific. Moreover, although some clinicians believe IgM is a good test for early infection because levels rise early and then decline, IgM may be positive during recurrent episodes.8
Both laboratory-based assays and point-of-care tests for HSV-2 are available. They are performed on capillary blood or serum and have sensitivities that range from 80% to 100% and specificities greater than 96%, compared with HSV-2 immunoblot and Western blot testing as the standard.28,29
False-negative results may be more frequent in the early stages of infection. HSV-2 antibody indicates anogenital infection, while HSV-1 antibody might also be due to orolabial infection, which is something to keep in mind in a patient without genital symptoms.8
Treatment can control herpes but not eradicate it
Several herpes vaccines have undergone clinical trials, but an effective one remains elusive.30,31
Antiviral therapy is used to control signs and symptoms of clinical disease but does not eradicate latent virus. For initial clinical episodes of genital herpes, the CDC recommends acyclovir, valacyclovir, or famciclovir for 7 to 10 days.8 In patients with established genital herpes, daily suppressive antiviral therapy can reduce recurrences, subclinical shedding, and the likelihood of transmission to partners; famciclovir is somewhat less efficacious for suppressing viral shedding.8
A diagnosis of herpes can carry considerable stigma, which can substantially interfere with a patient’s current and future relationships.25,32,33 Sex partners of patients with genital herpes may benefit from evaluation and counseling. Clinicians should appreciate the psychological impact of a genital herpes diagnosis and address these concerns by providing education, counseling, and support while encouraging patients to recognize that herpes is a manageable condition.34
SYPHILIS IS INCREASING IN MEN WHO HAVE SEX WITH MEN
Since 2001, rates of syphilis, a systemic disease caused by Treponema pallidum, have been increasing in men who have sex with men.3,35 As of 2011, this group accounts for approximately 72% of all cases of primary and secondary syphilis in the United States.3
Primary syphilis is characterized by a firm, painless chancre at the site of inoculation. The chancre lasts 3 to 6 weeks and heals regardless of treatment. However, if the infected person does not receive adequate treatment, the infection progresses to the secondary stage.26
Secondary syphilis typically starts with a nonpruritic rash, usually macular or papular, on the trunk and extremities, classically including the palms and soles. Other symptoms may include alopecia, lymphadenopathy, condylomata, and systemic symptoms.
Without treatment, the infection can progress to latent syphilis, which is further categorized as early (acquired during the preceding year), late latent, or of unknown duration.26
Practical diagnosis of syphilis relies on serologic testing
Definitive diagnosis of early syphilis requires dark field microscopy or PCR to detect T pallidum in lesion exudate or tissue.8 However, because there are no commercially available tests for T pallidum, serologic testing is the mainstay.
Two types of serologic tests must be performed to diagnose syphilis: a nontreponemal test and a treponemal test.8
Nontreponemal tests:
- The Venereal Disease Research Laboratory (VDRL) test
- The rapid plasma reagin (RPR) test.
Treponemal tests:
- T pallidum passive particle agglutination (TP-PA) assay
- Fluorescent treponemal antibody absorbed (FTA-ABS) test
- Enzyme immunoassay (EIA)
- Chemiluminescence immunoassay (CIA).
Using only one type of serologic test is in-sufficient for diagnosis because each type has limitations, including the possibility of false-positive results.
Nontreponemal test results may correlate with disease activity, and results should be reported quantitatively; a fourfold change in titer, equivalent to a change of two dilutions, is needed to demonstrate a clinically significant difference between two nontreponemal test results using the same serologic test.8
Which order of testing for syphilis is best?
The CDC recommends that nontreponemal tests be used to screen for syphilis and treponemal tests be used to confirm the diagnosis.36 This traditional algorithm performs well in identifying persons with active infection who require further evaluation and treatment, while minimizing false-positive results in low-prevalence populations.
However, some clinical laboratories have adopted the reverse sequence, using treponemal tests (usually an EIA or a CIA) to screen for syphilis, followed by a nontreponemal test to confirm active infection.36–38 This reverse-sequence testing may result in overdiagnosis and overtreatment of syphilis in some clinical settings.37 When reverse-sequence syphilis screening is used, the CDC recommends reflexively testing all sera that produce reactive EIA or CIA results with a quantitative nontreponemal test and reflexively testing sera with discordant results (ie, a reactive EIA or CIA and a nonreactive RPR or VDRL test) with a different treponemal test.36
Traditionally, the FTA-ABS test had been considered the gold standard treponemal test; however, it has lower specificity than other treponemal tests. Accordingly, TP-PA is the recommended confirmatory treponemal test.8,39 False-negative results, although rare, may occur for biological or technical reasons, such as the prozone phenomenon, resulting in a missed diagnosis. The prozone phenomenon occurs when the antibody titer is high and antigen binding sites are saturated, preventing the cross-linking reaction between antigens and antibodies. In this instance, when syphilis is suspected clinically and the RPR assay is nonreactive, the clinician can request RPR testing at dilutions of sera, ie, diluting the patient’s serum to bring the antibody concentration into the zone equivalence.40
Suspect neurosyphilis if neurologic symptoms arise
Central nervous system involvement can occur at any stage of syphilis. If clinical evidence of neurologic involvement (meningitis, stroke, cranial nerve dysfunction, or auditory or ophthalmic abnormalities) is observed in any patient with syphilis, regardless of stage, a cerebrospinal fluid examination should be performed.8 Laboratory testing can support the diagnosis of neurosyphilis; however, no single laboratory test can be used to diagnose it.
Cerebrospinal fluid abnormalities (ie, mononuclear pleocytosis, increased protein) are common in patients with early syphilis even in the absence of clinical neurologic findings. There is no firm evidence to support diverging from recommended treatment for early syphilis in these patients.35
Cerebrospinal fluid examination is recommended for all patients with serologic evidence of syphilis infection and neurologic symptoms and for patients who do not achieve an adequate serologic decline with stage-appropriate treatment.8
Penicillin is still the mainstay of syphilis treatment
Penicillin is still the mainstay of syphilis treatment.
- Patients with early syphilis (primary, secondary, or early latent) should receive a single intramuscular dose of benzathine penicillin G (2.4 million units).8,35
- Patients with late latent syphilis should receive three intramuscular doses of benzathine penicillin G (2.4 million units each) at 1-week intervals.
- Neurosyphilis should be treated with aqueous crystalline penicillin G 18 to 24 million units daily for 10 to 14 days.8
Doxycycline is the preferred alternative in nonpregnant patients who are allergic to penicillin. The dosage is 100 mg orally twice a day for 14 days (for primary, secondary, or early latent infections) or for 28 days (for late latent infections or those of unknown duration).35,41
Ceftriaxone (1–2 g daily) may be effective for treating early syphilis. However, data are limited, and the optimal dose and duration of therapy are not defined.8,42
Azithromycin in a single 2-g oral dose is effective for treating early syphilis. However, T pallidum chromosomal mutations associated with macrolide resistance are being detected.35,43 In view of this, azithromycin should not be used in men who have sex with men or in pregnant women.
Follow-up of syphilis patients and partners
Close clinical and serologic follow-up is strongly advised in persons who receive an alternate regimen to evaluate for treatment failure.8
Sex partners of patients with primary syphilis should be considered at risk and given treatment if they had sexual contact with the patient within 3 months plus the duration of symptoms, within 6 months plus the duration of symptoms for those with secondary syphilis, or 1 year for patients with early latent syphilis.8
Serologic and clinical evaluation should be repeated at 6, 12, and 24 months after treatment. HIV-infected patients should receive closer follow-up, ie, at 3, 6, 9, 12, and 24 months. The same quantitative nontreponemal serologic test should be used at each visit, with at least a fourfold decrease in titer representing an appropriate serologic decline.
Failure to achieve an appropriate serologic decline in 6 to 12 months may represent treatment failure.8 Optimal management in this instance is unclear; at a minimum, additional clinical and serologic follow-up should be performed.8 If additional follow-up cannot be ensured, retreatment (weekly intramuscular injections of benzathine penicillin G 2.4 million units for 3 weeks) is recommended. Cerebrospinal fluid examination can be considered to exclude unrecognized neurosyphilis.8
URETHRITIS: GONORRHEA, CHLAMYDIA TOP THE LIST
Symptoms of urethritis can include dysuria, discharge (purulent or mucopurulent), and urethral pruritus.26 However, asymptomatic infections are common.8
Several organisms are associated with infectious urethritis, including26:
- Neisseria gonorrhoeae
- Chlamydia trachomatis
- Mycoplasma genitalium
- Trichomonas vaginalis
- Ureaplasma urealyticum.
Diagnosing urethritis: Try to identify the agent
The clinician should attempt to obtain objective evidence of urethral inflammation. Urethral discharge should be examined with microscopy using Gram stain or methylene blue, or a first-void urine sample should be tested with microscopy and leukocyte esterase.26 If clinic-based diagnostic testing is not available, testing for N gonorrhoeae and C trachomatis using nucleic acid amplification can identify additional infections.8
During the clinic visit, either Gram-stain microscopy of a urethral swab or microscopic examination of a first-catch urine sample may identify the causative agent. If gram-negative intracellular diplococci are seen on urethral smear, gonorrhea infection is diagnosed. Nongonococcal urethritis is diagnosed when microscopy or urinalysis displays evidence of inflammation (positive leukocyte esterase or at least 10 white blood cells per high-power field) without gram-negative intracellular diplococci.8
Testing should be performed to determine the specific cause of urethritis, because both chlamydia and gonorrhea are reportable diseases. Nucleic acid amplification tests are the most sensitive tests for C trachomatis and N gonorrhoeae and can be performed on urethral swabs or urine.44 If clinic-based diagnostic tools are unavailable, patients should receive empiric treatment for chlamydia and gonorrhea.8
Treatment of urethritis, by organism
Urethral gonorrhea should be treated with dual therapy: ceftriaxone 250 mg in a single intramuscular dose and either azithromycin 1 g orally as a single dose or doxycycline 100 mg orally twice a day for 7 days. Oral cephalosporins are no longer recommended as first-line treatment of gonorrhea.45
Chlamydial urethritis is treated with azithromycin 1 g in a single oral dose or doxycycline 100 mg orally twice a day for 7 days. Although azithromycin provides the advantages of a single-dose regimen administered and directly observed by the provider, some evidence suggests that doxycycline may be more effective than azithromycin for symptomatic chlamydial urethritis.46
All sex partners within the preceding 60 days should be referred for evaluation, testing, and empiric treatment with a drug regimen effective against chlamydia (if nongonococcal urethritis or only C trachomatis was identified) and gonorrhea (if N gonorrhoeae was identified). When patients diagnosed with chlamydia or gonorrhea indicate that their partners are unlikely to seek evaluation, providers can offer patient-delivered partner therapy, a form of expedited partner therapy in which partners of infected persons are treated without previous medical evaluation. Providers should visit www.cdc.gov/std/ept for updated information for their individual jurisdiction, as expedited partner therapy is prohibited in some states. No studies have been published involving patient-delivered partner therapy for chlamydia or gonorrhea in men who have sex with men.8
Patients with recurrent or persistent urethritis can be retreated with the initial regimen if they did not comply with treatment or were reexposed to an untreated sex partner. However, persistent urethritis after doxycycline therapy may suggest the presence of doxycycline-resistant M genitalium or U urealyticum. T vaginalis may also cause urethritis in men. Diagnostic evaluation may include culture or nucleic acid amplification testing of a urethral swab or urine (ie, PCR [Amplicor] or transcription-mediated amplification).8
M genitalium or U urealyticum urethritis. Currently, no commercially available diagnostic test exists for M genitalium or U urealyticum, so clinicians must choose a treatment regimen on the basis of objective evidence of inflammation in the absence of an etiologic agent. If azithromycin was not given during the initial course, metronidazole 2 g orally or tinidazole 2 g orally in a single dose plus azithromycin 1 g orally should be considered.8
M genitalium is one of the most common pathogens in men with persistent urethritis, accounting for 15% to 25% of cases. Several studies have shown that moxifloxacin 400 mg orally daily for 7 days is effective against M genitalium.46–48 Therefore, men for whom an initial regimen of azithromycin fails should be retreated with moxifloxacin 400 mg orally once daily for 7 days.
If men require treatment with a new antibiotic regimen for persistent urethritis and a sexually transmitted agent is the suspected cause, all partners in the past 60 days before the initial diagnosis and any interim partners should be referred for evaluation and appropriate treatment.
CERVICITIS: CHLAMYDIA, GONORRHEA, OTHERS
Cervicitis is frequently asymptomatic, but signs on pelvic examination may include purulent or mucopurulent endocervical exudate and sustained endocervical bleeding easily induced by passage of a cotton swab through the cervical os.26
In most cases, the pathogen cannot be identified.49 When an organism is isolated, it is typically C trachomatis or N gonorrhoeae. Others that may cause cervicitis include the organisms responsible for bacterial vaginosis, T vaginalis, HSV, and possibly M genitalium.50
Diagnostic workup for cervicitis
Diagnostic workup for cervicitis should include microscopic evaluation of an endocervical specimen and testing for C trachomatis and N gonorrhoeae. A finding of leukorrhea (> 10 white blood cells per high-power field on microscopic examination of vaginal fluid) has been associated with chlamydial and gonococcal infection of the cervix.8 In the absence of inflammatory vaginitis, leukorrhea might be a sensitive indicator of cervical inflammation, with a high negative predictive value.
Nucleic acid amplification testing for C trachomatis and N gonorrhoeae can be performed on urine, endocervical, or vaginal swab specimens collected by the clinician or self-collected.51 The performance of C trachomatis nucleic acid amplification testing on patient-collected vaginal swab specimens has similar sensitivity and specificity to those performed on cervical and first-void urine samples.26,44,52
Women with cervicitis also should be evaluated for bacterial vaginosis and trichomoniasis, and if the organisms that cause these conditions are detected, treatment is advised. Microscopy has a low sensitivity (approximately 50%) for detecting T vaginalis; if the organism is not identified, further testing such as culture may be performed to exclude it as the pathogen.
Women with cervicitis should also be evaluated for clinical signs of pelvic inflammatory disease, including uterine, adnexal, and cervical motion tenderness, and fever.
Cervicitis can be treated presumptively
Women with cervicitis who should receive presumptive therapy include those at higher risk of chlamydial infection (ie, those with new or multiple sex partners, those age 25 or younger, and those who engage in unprotected intercourse, especially if follow-up cannot be ensured).8
Recommended therapy is either azithromycin 1 g orally in a single dose or doxycycline 100 mg orally twice a day for 7 days. The clinician should consider dual therapy to cover gonorrhea if the prevalence of gonorrhea is more than 5% (ie, in younger patients).8 If bacterial vaginosis or T vaginalis infection is diagnosed, these conditions should be treated at the time of clinical evaluation (see vaginitis section for more detail). For women in whom presumptive therapy is deferred, the results of diagnostic testing should guide appropriate treatment.
Repeat testing 3 to 6 months after treatment is recommended for all women diagnosed with chlamydia or gonorrhea,53 and all sex partners in the past 60 days should be referred for evaluation and receive treatment for the STDs for which the index patient received treatment. In states where it is allowed, patient-delivered partner therapy should be considered if the patient indicates her partner is unlikely to seek medical evaluation.
VAGINITIS: NOT JUST YEAST
Women with vaginitis may present with complaints of discharge, pruritus, or a bad vaginal odor. A careful medical history, including information on sexual behaviors and vaginal hygiene practices (ie, douching), should be conducted in addition to physical examination and diagnostic testing.
The most common conditions associated with vaginitis are bacterial vaginosis, trichomoniasis, and candidiasis. However, vulvovaginal candidiasis, most often caused by Candida albicans, is not transmitted sexually and will not be reviewed further here. Although bacterial vaginosis is associated with known risk factors for STDs (eg, new or multiple sex partners), the cause of the microbial alteration that precipitates it is not known.44 Co-infection with T vaginalis is extremely common.54
BACTERIAL VAGINOSIS: VERY COMMON
Bacterial vaginosis is the most common genital infection in reproductive-age women.55 It is a polymicrobial syndrome in which anaerobic bacteria (Prevotella and Mobiluncus species), Gardnerella vaginalis, Ureaplasma, and Mycoplasma replace the normal vaginal flora.
Diagnosis of bacterial vaginosis
Bacterial vaginosis can be diagnosed by clinical criteria or Gram staining. Clinical diagnosis requires three of the following clinical criteria proposed by Amsel et al56:
- Clue cells
- Vaginal fluid pH > 4.5
- Fishy odor before or after addition of 10% potassium hydroxide
- Thin, homogeneous, white discharge that smoothly coats the vaginal walls.
The clinical utility of PCR for diagnosing G vaginalis remains unclear, and culture is not recommended because it has low specificity. Gram staining can be used to determine the concentration of lactobacilli, small gram-negative or variable rods (G vaginalis and anaerobic rods), and curved gram-negative rods (ie, Mobiluncus species).
Treatment of bacterial vaginosis: Metronidazole or clindamycin
Treatment is recommended to relieve vaginal symptoms, with the potential benefit of reducing the risk of acquiring chlamydia, gonorrhea, and HIV and other viral STDs.8
Recommended treatment is with metronidazole 500 mg orally twice a day for 7 days, metronidazole gel 0.75% vaginal suppository once a day for 5 days, or clindamycin cream 2% vaginal suppository once a day for 7 days.44 A meta-analysis of several trials found that clindamycin and metronidazole have equivalent effectiveness for eradicating bacterial vaginosis symptoms.57 Accordingly, providers can consider patient preference, co-infections, and possible side effects when selecting a regimen. Alternative regimens include tinidazole or clindamycin orally or in vaginal ovules.
Women should be advised to refrain from sexual intercourse during treatment. Routine treatment of male or female sexual partners is not warranted.
Bacterial vaginosis commonly recurs, and limited data exist regarding optimal management of recurrences. Using a different treatment regimen may be an option in patients who have a recurrence; however, re-treatment with the same topical regimen is an acceptable approach for treating recurrent bacterial vaginosis during the early stages of infection.58 One study suggests that metronidazole for 7 days, followed by intravaginal boric acid for 21 days, and then, for those in remission, suppressive metronidazole gel for 16 weeks may be another option.59 For women with multiple recurrences, metronidazole gel twice weekly for 4 to 6 months has been shown to reduce recurrences, although its benefit may not persist after it is stopped. The therapeutic role for probiotics remains unclear.60
TRICHOMONIASIS: TREAT PARTNERS
Although most women with trichomoniasis have few or no symptoms, some have vaginal discharge that may be diffuse, malodorous, and yellow-greenish, and some have vulvar irritation.
Diagnosis of trichomoniasis: Microscopy is first-line but insensitive
The most common method for diagnosing T vaginalis infection remains microscopic evaluation of wet preparations of genital secretions, because of its convenience and relatively low cost. This may demonstrate the motile, flagellated protozoa T vaginalis and many white blood cells. Slides of vaginal fluid specimens should be examined immediately after collection to maximize performance. Unfortunately, the sensitivity of wet preparation is 44% to 80% in vaginal specimens.61
Culture is still considered the gold standard for diagnosing trichomoniasis and, if available, should be performed when direct microscopy is unrevealing.
Point-of-care diagnostic tests for T vaginalis infection include the OSOM Trichomonas Rapid Test (Sekisui Diagnostics, San Diego, CA), which is an immunochromatographic capillary flow dipstick test, and the Affirm VPIII (Becton, Dickinson and Company, Franklin Lakes, NJ), a nucleic acid probe-hydridization test that identifies T vaginalis, G vaginalis, and C albicans.44,62 Liquid-based Pap tests may demonstrate T vaginalis, although they should not be performed exclusively for this purpose. Among women, nucleic acid amplification tests may detect a prevalence three to five times higher than indicated by wet mount microscopy. The APTIMA Trichomonas vaginalis assay (Hologic Gen-Probe, San Diego, CA) was the most sensitive test for trichomonas detection in this study.63
Extragenital testing with nucleic acid amplification tests is not recommended for T vaginalis, as it remains unclear if the rectum can serve as a reservoir for infection, and T vaginalis has not been found to infect oral sites.8
Treatment of trichomoniasis: Metronidazole or tinidazole
Nitroimidazoles, ie, metronidazole and tinidazole, are the only class of drugs available to treat trichomoniasis. The recommended regimen is metronidazole or tinidazole 2 g orally in a single dose. Studies suggest that tinidazole may be superior to metronidazole, with higher cure rates due to its longer half-life and higher tissue concentrations.64
Low-level metronidazole resistance is estimated to occur in 2% to 5% of trichomoniasis infections65; high-level resistance occurs rarely. If a single dose of metronidazole 2 g fails to cure the infection and reinfection is ruled out, the patient should be treated with metronidazole 500 mg orally twice a day for 7 days. If this regimen is not effective, providers can consider tinidazole or metronidazole 2 g orally for 7 days.44,62,64 Consultation and susceptibility testing for T vaginalis is available from the CDC if these alternative regimens are ineffective.
T vaginalis infection has a high rate of transmission to sexual partners,66 and all partners should be treated. Male sexual partners should be treated with metronidazole 500 mg twice a day orally for 7 days, tinidazole 2 g orally in a single dose, or tinidazole 500 mg twice a day for 7 days. Patient-delivered partner therapy may have a role in partner management for trichomoniasis.67
PROCTITIS: SUSPECT LYMPHOGRANULOMA VENEREUM
Acute proctitis in men and women who practice receptive anal intercourse is usually sexually acquired. The most common causative organisms are N gonorrhoeae, C trachomatis (serotypes associated with or not associated with lymphogranuloma venereum), and HSV; T pallidum is less common.68 Co-infections are not uncommon in this setting.69
Symptoms of proctitis may include anal discharge, rectal ulcers and bleeding, anorectal pain, tenesmus, and constipation. Patients with lymphogranuloma venereum may also present with tender, fluctuant inguinal or femoral lymphadenopathy (buboes), or herpetiform genital ulcers or papules.
Diagnosis of proctitis
Clinical evaluation should include digital rectal examination and anoscopy (if possible) to look for abnormalities such as ulcerations, hemorrhoids, anal fissures, condylomas, strictures, exudate, and bleeding.
Appropriate diagnostic testing includes Gram staining and culture of discharge, herpes viral culture or PCR, and nucleic acid amplification testing for chlamydia and gonorrhea (in laboratories with Clinical Laboratory Improvement Amendments validation).8
Nucleic acid amplification tests detect C trachomatis serotypes L1–L3, responsible for lymphogranuloma venereum, and non-lymphogranuloma venereum serotypes A–K, but cannot distinguish between the two, whereas PCR-based genotyping can.8,26 Although this distinction is important to ensure appropriate evaluation and management of sex partners, empiric treatment for lymphogranuloma venereum (doxycycline 100 mg orally twice a day for 21 days) should be provided to patients at high risk, including men who have sex with men and who have anorectal chlamydia, HIV infection, or bloody discharge and perianal or mucosal ulcers.8
Treatment of proctitis
Patients with painful perianal or mucosal ulceration should receive presumptive treatment for lymphogranuloma venereum and HSV while awaiting results of diagnostic testing. If rectal discharge is detected or Gram staining of anorectal secretions detects polymorphonuclear leukocytes, treatment should include ceftriaxone 250 mg intramuscularly and doxycycline 100 mg orally twice a day for 7 days.8 Additional testing for syphilis and HIV should also be performed.
All sexual partners should be evaluated for any disease diagnosed in the index patient.
With nearly 20 million new infections annually, sexually transmitted diseases (STDs) are very common in the United States.1,2 And with recent changes to the national health care landscape, including the passage of the Affordable Care Act and state budget cuts resulting in the closure of STD and human immunodeficiency virus (HIV) clinics, primary care providers can expect to encounter more patients with STDs.
For women and infants, STDs can have serious and long-term consequences, including infertility, facilitation of HIV infection, reproductive tract cancer, pelvic inflammatory disease, and poor perinatal outcomes.2 STDs cost the US health care system nearly $16 billion every year.3
STD prevention and control strategies traditionally include surveillance, screening, behavioral interventions, treatment, and partner management.4 This paper will review patient management by syndrome and provide guidance to clinicians to facilitate timely diagnosis and treatment, important components of any effective STD prevention strategy.
ANYONE CAN HAVE AN STD
STDs affect people of all races, ages, and sexual orientations. That said, some groups are at greater risk:
Adolescents and young adults. Persons ages 15 through 24 represent 25% of the sexually experienced population in the United States but account for nearly half of all incident STDs.1
Racial and ethnic minorities. STD disparities are one of the five greatest health disparities for African American communities.4
Men who have sex with men number approximately 2% to 4% of the US male population, yet account for approximately 70% of reported cases of primary and secondary syphilis and more than 50% of persons with HIV infection.3,5,6
HIGH-RISK BEHAVIOR AND SCREENING
A thorough sexual history will reveal behaviors that place a person at risk of infection. The US Preventive Services Task Force (USPSTF) defines high-risk sexual behavior as having multiple current partners, having a new partner, using condoms inconsistently, having sex while under the influence of alcohol or drugs, or exchanging sex for money or drugs.7
An effective strategy for obtaining a sexual history is the “five Ps”8:
- Partners (eg, Do you have sex with men, women, or both?)
- Prevention of pregnancy (eg, What are you doing to prevent pregnancy?)
- Protection from STDs (eg, What do you do to protect yourself from STDs?)
- Practices (eg, To understand your risks for STDs, I need to understand the kind of sex you have had recently.)
- Past history of STDs (eg, Have you ever had an STD?).8
The USPSTF and the US Centers for Disease Control and Prevention (CDC) recommend certain populations be screened for STDs.7,8
Everyone age 13 through 64 should be tested for HIV at least once, per CDC recommendation.8
Sexually active females up to age 24 should routinely be screened for chlamydia every year.7,8
Nonpregnant women at higher risk of infection should be screened for gonorrhea and syphilis.
Pregnant women, regardless of risk, should be screened for chlamydia, hepatitis B, HIV, and syphilis; pregnant women at higher risk of infection should also be screened for gonorrhea and hepatitis C.7,9
Men should be screened for HIV, and men at higher risk should also be screened for syphilis.8
Men who have sex with men should be screened at least annually for HIV and syphilis and undergo a test for urethral chlamydia and gonorrhea infection. Men who participate in receptive anal intercourse should be tested for rectal chlamydia and gonorrhea and, in those who participate in oral intercourse, for pharyngeal gonorrhea.8
PREVENTION
Vaccination against HPV, hepatitis A and B
Preexposure vaccination is one of the most effective ways to prevent human papillomavirus (HPV), hepatitis A, and hepatitis B infection.10
HPV vaccination. The Advisory Committee on Immunization Practices recommends routine HPV vaccination of female patients at age 11 or 12, or through age 26 if not previously vaccinated.11,12 Routine vaccination is also recommended for males at age 11 or 12 and through age 21, if not previously vaccinated.12 The upper age is extended through age 26 for men who have sex with men and for immunocompromised patients.12
Two HPV vaccines are available for females; one is quadrivalent and the other is bivalent. Both protect against two HPV types that cause cervical and other HPV-associated cancers.11 The quadrivalent vaccine also protects against the two types that cause 90% of genital warts.13 Only the quadrivalent vaccine is licensed for use in males.12
Hepatitis A and B vaccination. Hepatitis B vaccination is recommended for all unvaccinated, uninfected patients being evaluated for an STD.8,14
Vaccinating for hepatitis A and hepatitis B is important for men who have sex with men, who have a higher risk of acquiring and transmitting these infections.15
Other preventive practices
Male circumcision has been shown to reduce the risk of HIV infection, high-risk genital HPV infection, and genital herpes in heterosexual men.10,16,17
Male condoms, when used consistently and correctly, can reduce the risk of chlamydia, gonorrhea, and trichomoniasis.8 The risk of transmission of syphilis, genital HPV, and genital herpes can also be reduced by correctly and consistently using condoms when the infected area of exposure is covered.8
Microbiocides not recommended. Topical microbiocides do not have not enough evidence to recommend them for STD prevention. However, limited data suggest that tenofovir 1% vaginal gel may reduce the risk of acquiring genital herpes simplex virus type 2 (HSV-2) infection in women.18,19
GENITAL ULCERATIVE DISEASE: HERPES, SYPHILIS, OTHERS
The risk of acquiring HIV is two to five times higher if one is exposed to it when a genital ulcerative disease is present.20,21
In the United States, most cases of genital, anal, or perianal ulcers in sexually active persons are due to genital herpes or syphilis.8 Other causes include chancroid, granuloma inguinale, lymphogranuloma venereum, and noninfectious causes.
Although the frequency of these conditions varies by geographic area and demographic profile, herpes is the most prevalent of them.8 HSV-2 infection is one of the most prevalent STDs in the United States, with approximately 17% of all adolescents and adults infected,1,22 and a much higher prevalence in persons who use drugs.23
A thorough medical history and physical examination should be conducted. In addition, an accurate diagnosis requires specific tests: syphilis serology, dark field microscopy (if available), and culture or polymerase chain reaction (PCR) testing for herpes.8 A positive serologic test for HSV-1 or HSV-2 is enough to make the diagnosis in a patient whose symptoms suggest herpes, even if PCR and culture are negative.
GENITAL HERPES: MOSTLY ASYMPTOMATIC
Genital herpes is caused by HSV-1 or HSV-2. Of these, HSV-1 is on the rise, causing an increasing proportion of first episodes of genital herpes in some populations. It may now account for most new genital herpes infections in young women and in men who have sex with men.8,24
Most people infected with HSV-1 or HSV-2 have no symptoms or have subclinical disease. When symptoms do occur, one or more vesicles may appear on or around the genitals, rectum, or mouth. The average incubation period after exposure is 4 days (range 2–12).25 The first episode of genital herpes is often associated with systemic symptoms (eg, fever, headache, myalgia, and malaise) and local symptoms (eg, dysuria, vaginal or urethral discharge, and inguinal adenopathy).26,27
Genital herpes often recurs, especially during the first year. Recurrences are less frequent with HSV-1 than with HSV-2.
Diagnosing genital herpes requires laboratory testing
Diagnosing genital herpes by clinical signs and symptoms is both insensitive and nonspecific. Therefore, laboratory testing should be performed for patients who present with genital ulcerative disease.
Virologic tests. Viral culture and nucleic acid amplification methods, including PCR assays, are the preferred virologic tests for herpes.8 Although viral culture is widely available, its sensitivity depends on the stage of the lesion and rapidly declines as lesions begin to heal. PCR assays are more sensitive than viral culture, can be done in automated systems, and are increasingly being used in clinical settings.26 However, if the patient has no active lesions at the time of testing, failure to detect herpes by culture or PCR does not guarantee that the patient is not infected, as viral shedding is intermittent.
Serologic tests. Type-specific antibodies to HSV develop during the first several weeks following infection and persist indefinitely. Providers should specifically request serologic type-specific immunoglobulin G (IgG) assays. IgM testing for HSV should not be used, as IgM testing is not type-specific. Moreover, although some clinicians believe IgM is a good test for early infection because levels rise early and then decline, IgM may be positive during recurrent episodes.8
Both laboratory-based assays and point-of-care tests for HSV-2 are available. They are performed on capillary blood or serum and have sensitivities that range from 80% to 100% and specificities greater than 96%, compared with HSV-2 immunoblot and Western blot testing as the standard.28,29
False-negative results may be more frequent in the early stages of infection. HSV-2 antibody indicates anogenital infection, while HSV-1 antibody might also be due to orolabial infection, which is something to keep in mind in a patient without genital symptoms.8
Treatment can control herpes but not eradicate it
Several herpes vaccines have undergone clinical trials, but an effective one remains elusive.30,31
Antiviral therapy is used to control signs and symptoms of clinical disease but does not eradicate latent virus. For initial clinical episodes of genital herpes, the CDC recommends acyclovir, valacyclovir, or famciclovir for 7 to 10 days.8 In patients with established genital herpes, daily suppressive antiviral therapy can reduce recurrences, subclinical shedding, and the likelihood of transmission to partners; famciclovir is somewhat less efficacious for suppressing viral shedding.8
A diagnosis of herpes can carry considerable stigma, which can substantially interfere with a patient’s current and future relationships.25,32,33 Sex partners of patients with genital herpes may benefit from evaluation and counseling. Clinicians should appreciate the psychological impact of a genital herpes diagnosis and address these concerns by providing education, counseling, and support while encouraging patients to recognize that herpes is a manageable condition.34
SYPHILIS IS INCREASING IN MEN WHO HAVE SEX WITH MEN
Since 2001, rates of syphilis, a systemic disease caused by Treponema pallidum, have been increasing in men who have sex with men.3,35 As of 2011, this group accounts for approximately 72% of all cases of primary and secondary syphilis in the United States.3
Primary syphilis is characterized by a firm, painless chancre at the site of inoculation. The chancre lasts 3 to 6 weeks and heals regardless of treatment. However, if the infected person does not receive adequate treatment, the infection progresses to the secondary stage.26
Secondary syphilis typically starts with a nonpruritic rash, usually macular or papular, on the trunk and extremities, classically including the palms and soles. Other symptoms may include alopecia, lymphadenopathy, condylomata, and systemic symptoms.
Without treatment, the infection can progress to latent syphilis, which is further categorized as early (acquired during the preceding year), late latent, or of unknown duration.26
Practical diagnosis of syphilis relies on serologic testing
Definitive diagnosis of early syphilis requires dark field microscopy or PCR to detect T pallidum in lesion exudate or tissue.8 However, because there are no commercially available tests for T pallidum, serologic testing is the mainstay.
Two types of serologic tests must be performed to diagnose syphilis: a nontreponemal test and a treponemal test.8
Nontreponemal tests:
- The Venereal Disease Research Laboratory (VDRL) test
- The rapid plasma reagin (RPR) test.
Treponemal tests:
- T pallidum passive particle agglutination (TP-PA) assay
- Fluorescent treponemal antibody absorbed (FTA-ABS) test
- Enzyme immunoassay (EIA)
- Chemiluminescence immunoassay (CIA).
Using only one type of serologic test is in-sufficient for diagnosis because each type has limitations, including the possibility of false-positive results.
Nontreponemal test results may correlate with disease activity, and results should be reported quantitatively; a fourfold change in titer, equivalent to a change of two dilutions, is needed to demonstrate a clinically significant difference between two nontreponemal test results using the same serologic test.8
Which order of testing for syphilis is best?
The CDC recommends that nontreponemal tests be used to screen for syphilis and treponemal tests be used to confirm the diagnosis.36 This traditional algorithm performs well in identifying persons with active infection who require further evaluation and treatment, while minimizing false-positive results in low-prevalence populations.
However, some clinical laboratories have adopted the reverse sequence, using treponemal tests (usually an EIA or a CIA) to screen for syphilis, followed by a nontreponemal test to confirm active infection.36–38 This reverse-sequence testing may result in overdiagnosis and overtreatment of syphilis in some clinical settings.37 When reverse-sequence syphilis screening is used, the CDC recommends reflexively testing all sera that produce reactive EIA or CIA results with a quantitative nontreponemal test and reflexively testing sera with discordant results (ie, a reactive EIA or CIA and a nonreactive RPR or VDRL test) with a different treponemal test.36
Traditionally, the FTA-ABS test had been considered the gold standard treponemal test; however, it has lower specificity than other treponemal tests. Accordingly, TP-PA is the recommended confirmatory treponemal test.8,39 False-negative results, although rare, may occur for biological or technical reasons, such as the prozone phenomenon, resulting in a missed diagnosis. The prozone phenomenon occurs when the antibody titer is high and antigen binding sites are saturated, preventing the cross-linking reaction between antigens and antibodies. In this instance, when syphilis is suspected clinically and the RPR assay is nonreactive, the clinician can request RPR testing at dilutions of sera, ie, diluting the patient’s serum to bring the antibody concentration into the zone equivalence.40
Suspect neurosyphilis if neurologic symptoms arise
Central nervous system involvement can occur at any stage of syphilis. If clinical evidence of neurologic involvement (meningitis, stroke, cranial nerve dysfunction, or auditory or ophthalmic abnormalities) is observed in any patient with syphilis, regardless of stage, a cerebrospinal fluid examination should be performed.8 Laboratory testing can support the diagnosis of neurosyphilis; however, no single laboratory test can be used to diagnose it.
Cerebrospinal fluid abnormalities (ie, mononuclear pleocytosis, increased protein) are common in patients with early syphilis even in the absence of clinical neurologic findings. There is no firm evidence to support diverging from recommended treatment for early syphilis in these patients.35
Cerebrospinal fluid examination is recommended for all patients with serologic evidence of syphilis infection and neurologic symptoms and for patients who do not achieve an adequate serologic decline with stage-appropriate treatment.8
Penicillin is still the mainstay of syphilis treatment
Penicillin is still the mainstay of syphilis treatment.
- Patients with early syphilis (primary, secondary, or early latent) should receive a single intramuscular dose of benzathine penicillin G (2.4 million units).8,35
- Patients with late latent syphilis should receive three intramuscular doses of benzathine penicillin G (2.4 million units each) at 1-week intervals.
- Neurosyphilis should be treated with aqueous crystalline penicillin G 18 to 24 million units daily for 10 to 14 days.8
Doxycycline is the preferred alternative in nonpregnant patients who are allergic to penicillin. The dosage is 100 mg orally twice a day for 14 days (for primary, secondary, or early latent infections) or for 28 days (for late latent infections or those of unknown duration).35,41
Ceftriaxone (1–2 g daily) may be effective for treating early syphilis. However, data are limited, and the optimal dose and duration of therapy are not defined.8,42
Azithromycin in a single 2-g oral dose is effective for treating early syphilis. However, T pallidum chromosomal mutations associated with macrolide resistance are being detected.35,43 In view of this, azithromycin should not be used in men who have sex with men or in pregnant women.
Follow-up of syphilis patients and partners
Close clinical and serologic follow-up is strongly advised in persons who receive an alternate regimen to evaluate for treatment failure.8
Sex partners of patients with primary syphilis should be considered at risk and given treatment if they had sexual contact with the patient within 3 months plus the duration of symptoms, within 6 months plus the duration of symptoms for those with secondary syphilis, or 1 year for patients with early latent syphilis.8
Serologic and clinical evaluation should be repeated at 6, 12, and 24 months after treatment. HIV-infected patients should receive closer follow-up, ie, at 3, 6, 9, 12, and 24 months. The same quantitative nontreponemal serologic test should be used at each visit, with at least a fourfold decrease in titer representing an appropriate serologic decline.
Failure to achieve an appropriate serologic decline in 6 to 12 months may represent treatment failure.8 Optimal management in this instance is unclear; at a minimum, additional clinical and serologic follow-up should be performed.8 If additional follow-up cannot be ensured, retreatment (weekly intramuscular injections of benzathine penicillin G 2.4 million units for 3 weeks) is recommended. Cerebrospinal fluid examination can be considered to exclude unrecognized neurosyphilis.8
URETHRITIS: GONORRHEA, CHLAMYDIA TOP THE LIST
Symptoms of urethritis can include dysuria, discharge (purulent or mucopurulent), and urethral pruritus.26 However, asymptomatic infections are common.8
Several organisms are associated with infectious urethritis, including26:
- Neisseria gonorrhoeae
- Chlamydia trachomatis
- Mycoplasma genitalium
- Trichomonas vaginalis
- Ureaplasma urealyticum.
Diagnosing urethritis: Try to identify the agent
The clinician should attempt to obtain objective evidence of urethral inflammation. Urethral discharge should be examined with microscopy using Gram stain or methylene blue, or a first-void urine sample should be tested with microscopy and leukocyte esterase.26 If clinic-based diagnostic testing is not available, testing for N gonorrhoeae and C trachomatis using nucleic acid amplification can identify additional infections.8
During the clinic visit, either Gram-stain microscopy of a urethral swab or microscopic examination of a first-catch urine sample may identify the causative agent. If gram-negative intracellular diplococci are seen on urethral smear, gonorrhea infection is diagnosed. Nongonococcal urethritis is diagnosed when microscopy or urinalysis displays evidence of inflammation (positive leukocyte esterase or at least 10 white blood cells per high-power field) without gram-negative intracellular diplococci.8
Testing should be performed to determine the specific cause of urethritis, because both chlamydia and gonorrhea are reportable diseases. Nucleic acid amplification tests are the most sensitive tests for C trachomatis and N gonorrhoeae and can be performed on urethral swabs or urine.44 If clinic-based diagnostic tools are unavailable, patients should receive empiric treatment for chlamydia and gonorrhea.8
Treatment of urethritis, by organism
Urethral gonorrhea should be treated with dual therapy: ceftriaxone 250 mg in a single intramuscular dose and either azithromycin 1 g orally as a single dose or doxycycline 100 mg orally twice a day for 7 days. Oral cephalosporins are no longer recommended as first-line treatment of gonorrhea.45
Chlamydial urethritis is treated with azithromycin 1 g in a single oral dose or doxycycline 100 mg orally twice a day for 7 days. Although azithromycin provides the advantages of a single-dose regimen administered and directly observed by the provider, some evidence suggests that doxycycline may be more effective than azithromycin for symptomatic chlamydial urethritis.46
All sex partners within the preceding 60 days should be referred for evaluation, testing, and empiric treatment with a drug regimen effective against chlamydia (if nongonococcal urethritis or only C trachomatis was identified) and gonorrhea (if N gonorrhoeae was identified). When patients diagnosed with chlamydia or gonorrhea indicate that their partners are unlikely to seek evaluation, providers can offer patient-delivered partner therapy, a form of expedited partner therapy in which partners of infected persons are treated without previous medical evaluation. Providers should visit www.cdc.gov/std/ept for updated information for their individual jurisdiction, as expedited partner therapy is prohibited in some states. No studies have been published involving patient-delivered partner therapy for chlamydia or gonorrhea in men who have sex with men.8
Patients with recurrent or persistent urethritis can be retreated with the initial regimen if they did not comply with treatment or were reexposed to an untreated sex partner. However, persistent urethritis after doxycycline therapy may suggest the presence of doxycycline-resistant M genitalium or U urealyticum. T vaginalis may also cause urethritis in men. Diagnostic evaluation may include culture or nucleic acid amplification testing of a urethral swab or urine (ie, PCR [Amplicor] or transcription-mediated amplification).8
M genitalium or U urealyticum urethritis. Currently, no commercially available diagnostic test exists for M genitalium or U urealyticum, so clinicians must choose a treatment regimen on the basis of objective evidence of inflammation in the absence of an etiologic agent. If azithromycin was not given during the initial course, metronidazole 2 g orally or tinidazole 2 g orally in a single dose plus azithromycin 1 g orally should be considered.8
M genitalium is one of the most common pathogens in men with persistent urethritis, accounting for 15% to 25% of cases. Several studies have shown that moxifloxacin 400 mg orally daily for 7 days is effective against M genitalium.46–48 Therefore, men for whom an initial regimen of azithromycin fails should be retreated with moxifloxacin 400 mg orally once daily for 7 days.
If men require treatment with a new antibiotic regimen for persistent urethritis and a sexually transmitted agent is the suspected cause, all partners in the past 60 days before the initial diagnosis and any interim partners should be referred for evaluation and appropriate treatment.
CERVICITIS: CHLAMYDIA, GONORRHEA, OTHERS
Cervicitis is frequently asymptomatic, but signs on pelvic examination may include purulent or mucopurulent endocervical exudate and sustained endocervical bleeding easily induced by passage of a cotton swab through the cervical os.26
In most cases, the pathogen cannot be identified.49 When an organism is isolated, it is typically C trachomatis or N gonorrhoeae. Others that may cause cervicitis include the organisms responsible for bacterial vaginosis, T vaginalis, HSV, and possibly M genitalium.50
Diagnostic workup for cervicitis
Diagnostic workup for cervicitis should include microscopic evaluation of an endocervical specimen and testing for C trachomatis and N gonorrhoeae. A finding of leukorrhea (> 10 white blood cells per high-power field on microscopic examination of vaginal fluid) has been associated with chlamydial and gonococcal infection of the cervix.8 In the absence of inflammatory vaginitis, leukorrhea might be a sensitive indicator of cervical inflammation, with a high negative predictive value.
Nucleic acid amplification testing for C trachomatis and N gonorrhoeae can be performed on urine, endocervical, or vaginal swab specimens collected by the clinician or self-collected.51 The performance of C trachomatis nucleic acid amplification testing on patient-collected vaginal swab specimens has similar sensitivity and specificity to those performed on cervical and first-void urine samples.26,44,52
Women with cervicitis also should be evaluated for bacterial vaginosis and trichomoniasis, and if the organisms that cause these conditions are detected, treatment is advised. Microscopy has a low sensitivity (approximately 50%) for detecting T vaginalis; if the organism is not identified, further testing such as culture may be performed to exclude it as the pathogen.
Women with cervicitis should also be evaluated for clinical signs of pelvic inflammatory disease, including uterine, adnexal, and cervical motion tenderness, and fever.
Cervicitis can be treated presumptively
Women with cervicitis who should receive presumptive therapy include those at higher risk of chlamydial infection (ie, those with new or multiple sex partners, those age 25 or younger, and those who engage in unprotected intercourse, especially if follow-up cannot be ensured).8
Recommended therapy is either azithromycin 1 g orally in a single dose or doxycycline 100 mg orally twice a day for 7 days. The clinician should consider dual therapy to cover gonorrhea if the prevalence of gonorrhea is more than 5% (ie, in younger patients).8 If bacterial vaginosis or T vaginalis infection is diagnosed, these conditions should be treated at the time of clinical evaluation (see vaginitis section for more detail). For women in whom presumptive therapy is deferred, the results of diagnostic testing should guide appropriate treatment.
Repeat testing 3 to 6 months after treatment is recommended for all women diagnosed with chlamydia or gonorrhea,53 and all sex partners in the past 60 days should be referred for evaluation and receive treatment for the STDs for which the index patient received treatment. In states where it is allowed, patient-delivered partner therapy should be considered if the patient indicates her partner is unlikely to seek medical evaluation.
VAGINITIS: NOT JUST YEAST
Women with vaginitis may present with complaints of discharge, pruritus, or a bad vaginal odor. A careful medical history, including information on sexual behaviors and vaginal hygiene practices (ie, douching), should be conducted in addition to physical examination and diagnostic testing.
The most common conditions associated with vaginitis are bacterial vaginosis, trichomoniasis, and candidiasis. However, vulvovaginal candidiasis, most often caused by Candida albicans, is not transmitted sexually and will not be reviewed further here. Although bacterial vaginosis is associated with known risk factors for STDs (eg, new or multiple sex partners), the cause of the microbial alteration that precipitates it is not known.44 Co-infection with T vaginalis is extremely common.54
BACTERIAL VAGINOSIS: VERY COMMON
Bacterial vaginosis is the most common genital infection in reproductive-age women.55 It is a polymicrobial syndrome in which anaerobic bacteria (Prevotella and Mobiluncus species), Gardnerella vaginalis, Ureaplasma, and Mycoplasma replace the normal vaginal flora.
Diagnosis of bacterial vaginosis
Bacterial vaginosis can be diagnosed by clinical criteria or Gram staining. Clinical diagnosis requires three of the following clinical criteria proposed by Amsel et al56:
- Clue cells
- Vaginal fluid pH > 4.5
- Fishy odor before or after addition of 10% potassium hydroxide
- Thin, homogeneous, white discharge that smoothly coats the vaginal walls.
The clinical utility of PCR for diagnosing G vaginalis remains unclear, and culture is not recommended because it has low specificity. Gram staining can be used to determine the concentration of lactobacilli, small gram-negative or variable rods (G vaginalis and anaerobic rods), and curved gram-negative rods (ie, Mobiluncus species).
Treatment of bacterial vaginosis: Metronidazole or clindamycin
Treatment is recommended to relieve vaginal symptoms, with the potential benefit of reducing the risk of acquiring chlamydia, gonorrhea, and HIV and other viral STDs.8
Recommended treatment is with metronidazole 500 mg orally twice a day for 7 days, metronidazole gel 0.75% vaginal suppository once a day for 5 days, or clindamycin cream 2% vaginal suppository once a day for 7 days.44 A meta-analysis of several trials found that clindamycin and metronidazole have equivalent effectiveness for eradicating bacterial vaginosis symptoms.57 Accordingly, providers can consider patient preference, co-infections, and possible side effects when selecting a regimen. Alternative regimens include tinidazole or clindamycin orally or in vaginal ovules.
Women should be advised to refrain from sexual intercourse during treatment. Routine treatment of male or female sexual partners is not warranted.
Bacterial vaginosis commonly recurs, and limited data exist regarding optimal management of recurrences. Using a different treatment regimen may be an option in patients who have a recurrence; however, re-treatment with the same topical regimen is an acceptable approach for treating recurrent bacterial vaginosis during the early stages of infection.58 One study suggests that metronidazole for 7 days, followed by intravaginal boric acid for 21 days, and then, for those in remission, suppressive metronidazole gel for 16 weeks may be another option.59 For women with multiple recurrences, metronidazole gel twice weekly for 4 to 6 months has been shown to reduce recurrences, although its benefit may not persist after it is stopped. The therapeutic role for probiotics remains unclear.60
TRICHOMONIASIS: TREAT PARTNERS
Although most women with trichomoniasis have few or no symptoms, some have vaginal discharge that may be diffuse, malodorous, and yellow-greenish, and some have vulvar irritation.
Diagnosis of trichomoniasis: Microscopy is first-line but insensitive
The most common method for diagnosing T vaginalis infection remains microscopic evaluation of wet preparations of genital secretions, because of its convenience and relatively low cost. This may demonstrate the motile, flagellated protozoa T vaginalis and many white blood cells. Slides of vaginal fluid specimens should be examined immediately after collection to maximize performance. Unfortunately, the sensitivity of wet preparation is 44% to 80% in vaginal specimens.61
Culture is still considered the gold standard for diagnosing trichomoniasis and, if available, should be performed when direct microscopy is unrevealing.
Point-of-care diagnostic tests for T vaginalis infection include the OSOM Trichomonas Rapid Test (Sekisui Diagnostics, San Diego, CA), which is an immunochromatographic capillary flow dipstick test, and the Affirm VPIII (Becton, Dickinson and Company, Franklin Lakes, NJ), a nucleic acid probe-hydridization test that identifies T vaginalis, G vaginalis, and C albicans.44,62 Liquid-based Pap tests may demonstrate T vaginalis, although they should not be performed exclusively for this purpose. Among women, nucleic acid amplification tests may detect a prevalence three to five times higher than indicated by wet mount microscopy. The APTIMA Trichomonas vaginalis assay (Hologic Gen-Probe, San Diego, CA) was the most sensitive test for trichomonas detection in this study.63
Extragenital testing with nucleic acid amplification tests is not recommended for T vaginalis, as it remains unclear if the rectum can serve as a reservoir for infection, and T vaginalis has not been found to infect oral sites.8
Treatment of trichomoniasis: Metronidazole or tinidazole
Nitroimidazoles, ie, metronidazole and tinidazole, are the only class of drugs available to treat trichomoniasis. The recommended regimen is metronidazole or tinidazole 2 g orally in a single dose. Studies suggest that tinidazole may be superior to metronidazole, with higher cure rates due to its longer half-life and higher tissue concentrations.64
Low-level metronidazole resistance is estimated to occur in 2% to 5% of trichomoniasis infections65; high-level resistance occurs rarely. If a single dose of metronidazole 2 g fails to cure the infection and reinfection is ruled out, the patient should be treated with metronidazole 500 mg orally twice a day for 7 days. If this regimen is not effective, providers can consider tinidazole or metronidazole 2 g orally for 7 days.44,62,64 Consultation and susceptibility testing for T vaginalis is available from the CDC if these alternative regimens are ineffective.
T vaginalis infection has a high rate of transmission to sexual partners,66 and all partners should be treated. Male sexual partners should be treated with metronidazole 500 mg twice a day orally for 7 days, tinidazole 2 g orally in a single dose, or tinidazole 500 mg twice a day for 7 days. Patient-delivered partner therapy may have a role in partner management for trichomoniasis.67
PROCTITIS: SUSPECT LYMPHOGRANULOMA VENEREUM
Acute proctitis in men and women who practice receptive anal intercourse is usually sexually acquired. The most common causative organisms are N gonorrhoeae, C trachomatis (serotypes associated with or not associated with lymphogranuloma venereum), and HSV; T pallidum is less common.68 Co-infections are not uncommon in this setting.69
Symptoms of proctitis may include anal discharge, rectal ulcers and bleeding, anorectal pain, tenesmus, and constipation. Patients with lymphogranuloma venereum may also present with tender, fluctuant inguinal or femoral lymphadenopathy (buboes), or herpetiform genital ulcers or papules.
Diagnosis of proctitis
Clinical evaluation should include digital rectal examination and anoscopy (if possible) to look for abnormalities such as ulcerations, hemorrhoids, anal fissures, condylomas, strictures, exudate, and bleeding.
Appropriate diagnostic testing includes Gram staining and culture of discharge, herpes viral culture or PCR, and nucleic acid amplification testing for chlamydia and gonorrhea (in laboratories with Clinical Laboratory Improvement Amendments validation).8
Nucleic acid amplification tests detect C trachomatis serotypes L1–L3, responsible for lymphogranuloma venereum, and non-lymphogranuloma venereum serotypes A–K, but cannot distinguish between the two, whereas PCR-based genotyping can.8,26 Although this distinction is important to ensure appropriate evaluation and management of sex partners, empiric treatment for lymphogranuloma venereum (doxycycline 100 mg orally twice a day for 21 days) should be provided to patients at high risk, including men who have sex with men and who have anorectal chlamydia, HIV infection, or bloody discharge and perianal or mucosal ulcers.8
Treatment of proctitis
Patients with painful perianal or mucosal ulceration should receive presumptive treatment for lymphogranuloma venereum and HSV while awaiting results of diagnostic testing. If rectal discharge is detected or Gram staining of anorectal secretions detects polymorphonuclear leukocytes, treatment should include ceftriaxone 250 mg intramuscularly and doxycycline 100 mg orally twice a day for 7 days.8 Additional testing for syphilis and HIV should also be performed.
All sexual partners should be evaluated for any disease diagnosed in the index patient.
- Satterwhite CL, Torrone E, Meites E, et al. Sexually transmitted infections among US women and men: prevalence and incidence estimates, 2008. Sex Transm Dis 2013; 40:187–193.
- Institute of Medicine (US); Committee on Prevention and Control of Sexually Transmitted Diseases. The hidden epidemic: confronting sexually transmitted diseases. Washington, DC: National Academy Press; 1997.
- Centers for Disease Control and Prevention (CDC). 2011 Sexually Transmitted Disease Surveillance. http://www.cdc.gov/std/stats11/toc.htm. Accessed January 10, 2014.
- Barrow RY, Newman LM, Douglas JM. Taking positive steps to address STD disparities for African-American communities. Sex Transm Dis 2008; 35(suppl 12):S1–S3.
- Mitchell JW, Petroll AE. Patterns of HIV and sexually transmitted infection testing among men who have sex with men couples in the United States. Sex Transm Dis 2012; 39:871–876.
- Centers for Disease Control and Prevention (CDC). HIV testing among men who have sex with men—21 cities, United States, 2008. MMWR Morb Mortal Wkly Rep 2011; 60:694–699.
- Meyers D, Wolff T, Gregory K, et al., USPSTF. USPSTF recommendations for STI screening. Am Fam Physician 2008; 77:819–824.
- Workowski KA, Berman S; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm Rep 2010; 59:1–110.
- Summaries for patients. Screening for chlamydial infection: recommendations from the US Preventive Services Task Force. Ann Intern Med 2007; 147:I44.
- Marrazzo JM, Cates W. Interventions to prevent sexually transmitted infections, including HIV infection. Clin Infect Dis 2011; 53(suppl 3):S64–S78.
- Markowitz LE, Dunne EF, Saraiya M, Lawson HW, Chesson H, Unger ER; Centers for Disease Control and Prevention (CDC); Advisory Committee on Immunization Practices (ACIP). Quadrivalent Human Papillomavirus Vaccine: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2007; 56:1–24.
- Centers for Disease Control and Prevention (CDC). Recommendations on the use of quadrivalent human papillomavirus vaccine in males—Advisory Committee on Immunization Practices (ACIP), 2011. MMWR Morb Mortal Wkly Rep 2011; 60:1705–1708.
- Paavonen J, Jenkins D, Bosch FX, et al; HPV PATRICIA study group. Efficacy of a prophylactic adjuvanted bivalent L1 virus-like-particle vaccine against infection with human papillomavirus types 16 and 18 in young women: an interim analysis of a phase III double-blind, randomised controlled trial. Lancet 2007; 369:2161–2170.
- Mast EE, Weinbaum CM, Fiore AE, et al; Advisory Committee on Immunization Practices (ACIP); Centers for Disease Control and Prevention (CDC). A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) Part II: immunization of adults. MMWR Recomm Rep 2006; 55:1–33.
- Mayer KH. Sexually transmitted diseases in men who have sex with men. Clin Infect Dis 2011; 53(suppl 3):S79–S83.
- Tobian AA, Serwadda D, Quinn TC, et al. Male circumcision for the prevention of HSV-2 and HPV infections and syphilis. N Engl J Med 2009; 360:1298–1309.
- Auvert B, Sobngwi-Tambekou J, Cutler E, et al. Effect of male circumcision on the prevalence of high-risk human papillomavirus in young men: results of a randomized controlled trial conducted in Orange Farm, South Africa. J Infect Dis 2009; 199:14–19.
- Obiero J, Mwethera PG, Wiysonge CS. Topical microbicides for prevention of sexually transmitted infections. Cochrane Database Syst Rev 2012; 6:CD007961.
- Abdool Karim Q, Abdool Karim SS, Frohlich JA, et al; CAPRISA 004 Trial Group. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science 2010; 329:1168–1174.
- HIV prevention through early detection and treatment of other sexually transmitted diseases—United States. Recommendations of the Advisory Committee for HIV and STD prevention. MMWR Recomm Rep 1998; 47:1–24.
- Fleming DT, Wasserheit JN. From epidemiological synergy to public health policy and practice: the contribution of other sexually transmitted diseases to sexual transmission of HIV infection. Sex Transm Infect 1999; 75:3–17.
- Centers for Disease Control and Prevention (CDC). Seroprevalence of herpes simplex virus type 2 among persons aged 14–49 years—United States, 2005–2008. MMWR Morb Mortal Wkly Rep 2010; 59:456–459.
- Semaan S, Leinhos M, Neumann MS. Public health strategies for prevention and control of HSV-2 in persons who use drugs in the United States. Drug Alcohol Depend 2013; 131:182–197.
- Bernstein DI, Bellamy AR, Hook EW, et al. Epidemiology, clinical presentation, and antibody response to primary infection with herpes simplex virus type 1 and type 2 in young women. Clin Infect Dis 2013; 56:344–351.
- Kimberlin DW, Rouse DJ. Clinical practice. Genital herpes. N Engl J Med 2004; 350:1970–1977.
- Sexually Transmitted Diseases. 4th ed. New York, NY: McGraw-Hill Professional Publishing; 2007.
- Johnston C, Magaret A, Selke S, Remington M, Corey L, Wald A. Herpes simplex virus viremia during primary genital infection. J Infect Dis 2008; 198:31–34.
- Laderman EI, Whitworth E, Dumaual E, et al. Rapid, sensitive, and specific lateral-flow immunochromatographic point-of-care device for detection of herpes simplex virus type 2-specific immunoglobulin G antibodies in serum and whole blood. Clin Vaccine Immunol 2008; 15:159–163.
- Philip SS, Ahrens K, Shayevich C, et al. Evaluation of a new point-of-care serologic assay for herpes simplex virus type 2 infection. Clin Infect Dis 2008; 47:e79–e82.
- Belshe RB, Leone PA, Bernstein DI, et al; Herpevac Trial for Women. Efficacy results of a trial of a herpes simplex vaccine. N Engl J Med 2012; 366:34–43.
- Stanberry LR, Spruance SL, Cunningham AL, et al; GlaxoSmithKline Herpes Vaccine Efficacy Study Group. Glycoprotein-D-adjuvant vaccine to prevent genital herpes. N Engl J Med 2002; 347:1652–1661.
- Mark H, Gilbert L, Nanda J. Psychosocial well-being and quality of life among women newly diagnosed with genital herpes. J Obstet Gynecol Neonatal Nurs 2009; 38:320–326.
- Gilbert LK, Omisore F. Common questions about herpes: analysis of chat-room transcripts. Herpes 2009; 15:57–61.
- Alexander L, Naisbett B. Patient and physician partnerships in managing genital herpes. J Infect Dis 2002; 186(suppl 1):S57–S65.
- Ghanem KG, Workowski KA. Management of adult syphilis. Clin Infect Dis 2011; 53(suppl 3):S110–S128.
- Centers for Disease Control and Prevention (CDC). Discordant results from reverse sequence syphilis screening—five laboratories, United States, 2006–2010. MMWR Morb Mortal Wkly Rep 2011; 60:133–137.
- Centers for Disease Control and Prevention (CDC). Syphilis testing algorithms using treponemal tests for initial screening—four laboratories, New York City, 2005–2006. MMWR Morb Mortal Wkly Rep 2008; 57:872–875.
- Seña AC, White BL, Sparling PF. Novel Treponema pallidum serologic tests: a paradigm shift in syphilis screening for the 21st century. Clin Infect Dis 2010; 51:700–708.
- Marangoni A, Sambri V, Storni E, D’Antuono A, Negosanti M, Cevenini R. Treponema pallidum surface immunofluorescence assay for serologic diagnosis of syphilis. Clin Diagn Lab Immunol 2000; 7:417–421.
- Post JJ, Khor C, Furner V, Smith DE, Whybin LR, Robertson PW. Case report and evaluation of the frequency of the prozone phenomenon in syphilis serology—an infrequent but important laboratory phenomenon. Sex Health 2012; 9:488–490.
- Ghanem KG, Erbelding EJ, Cheng WW, Rompalo AM. Doxycycline compared with benzathine penicillin for the treatment of early syphilis. Clin Infect Dis 2006; 42:e45–e49.
- Hook EW, Roddy RE, Handsfield HH. Ceftriaxone therapy for incubating and early syphilis. J Infect Dis 1988; 158:881–884.
- A2058G Prevalence Workgroup. Prevalence of the 23S rRNA A2058G point mutation and molecular subtypes in Treponema pallidum in the United States, 2007 to 2009. Sex Transm Dis 2012; 39:794–798.
- Workowski KA, Berman SM. Centers for Disease Control and Prevention Sexually Transmitted Disease Treatment Guidelines. Clin Infect Dis 2011; 53(suppl 3):S59–S63.
- Kirkcaldy RD, Bolan GA, Wasserheit JN. Cephalosporin-resistant gonorrhea in North America. JAMA 2013; 309:185–187.
- Seña AC, Lensing S, Rompalo A, et al. Chlamydia trachomatis, Mycoplasma genitalium, and Trichomonas vaginalis infections in men with nongonococcal urethritis: predictors and persistence after therapy. J Infect Dis 2012; 206:357–365.
- Manhart LE, Broad JM, Golden MR. Mycoplasma genitalium: should we treat and how? Clin Infect Dis 2011; 53(suppl 3):S129–S142.
- Jernberg E, Moghaddam A, Moi H. Azithromycin and moxifloxacin for microbiological cure of Mycoplasma genitalium infection: an open study. Int J STD AIDS 2008; 19:676–679.
- Taylor SN, Lensing S, Schwebke J, et al. Prevalence and treatment outcome of cervicitis of unknown etiology. Sex Transm Dis 2013; 40:379–385.
- Lusk MJ, Konecny P. Cervicitis: a review. Curr Opin Infect Dis 2008; 21:49–55.
- Geisler WM. Diagnosis and management of uncomplicated Chlamydia trachomatis infections in adolescents and adults: summary of evidence reviewed for the 2010 Centers for Disease Control and Prevention Sexually Transmitted Diseases Treatment Guidelines. Clin Infect Dis 2011; 53(suppl 3):S92–S98.
- Schachter J, Chernesky MA, Willis DE, et al. Vaginal swabs are the specimens of choice when screening for Chlamydia trachomatis and Neisseria gonorrhoeae: results from a multicenter evaluation of the APTIMA assays for both infections. Sex Transm Dis 2005; 32:725–728.
- Hosenfeld CB, Workowski KA, Berman S, et al. Repeat infection with Chlamydia and gonorrhea among females: a systematic review of the literature. Sex Transm Dis 2009; 36:478–489.
- Sobel JD, Subramanian C, Foxman B, Fairfax M, Gygax SE. Mixed vaginitis-more than coinfection and with therapeutic implications. Curr Infect Dis Rep 2013; 15:104–108.
- Taylor BD, Darville T, Haggerty CL. Does bacterial vaginosis cause pelvic inflammatory disease? Sex Transm Dis 2013; 40:117–122.
- Amsel R, Totten PA, Spiegel CA, Chen KC, Eschenbach D, Holmes KK. Nonspecific vaginitis. Diagnostic criteria and microbial and epidemiologic associations. Am J Med 1983; 74:14–22.
- Oduyebo OO, Anorlu RI, Ogunsola FT. The effects of antimicrobial therapy on bacterial vaginosis in non-pregnant women. Cochrane Database Syst Rev 2009; ( 3):CD006055.
- Bunge KE, Beigi RH, Meyn LA, Hillier SL. The efficacy of retreatment with the same medication for early treatment failure of bacterial vaginosis. Sex Transm Dis 2009; 36:711–713.
- Reichman O, Akins R, Sobel JD. Boric acid addition to suppressive antimicrobial therapy for recurrent bacterial vaginosis. Sex Transm Dis 2009; 36:732–734.
- Senok AC, Verstraelen H, Temmerman M, Botta GA. Probiotics for the treatment of bacterial vaginosis. Cochrane Database Syst Rev 2009; (4):CD006289.
- Nye MB, Schwebke JR, Body BA. Comparison of APTIMA Trichomonas vaginalis transcription-mediated amplification to wet mount microscopy, culture, and polymerase chain reaction for diagnosis of trichomoniasis in men and women. Am J Obstet Gynecol 2009; 200:188.e1–188.e7.
- Bachmann LH, Hobbs MM, Seña AC, et al. Trichomonas vaginalis genital infections: progress and challenges. Clin Infect Dis 2011; 53(suppl 3):S160–S172.
- Schwebke JR, Hobbs MM, Taylor SN, et al. Molecular testing for Trichomonas vaginalis in women: results from a prospective US clinical trial. J Clin Microbiol 2011; 49:4106–4111.
- Coleman JS, Gaydos CA, Witter F. Trichomonas vaginalis vaginitis in obstetrics and gynecology practice: new concepts and controversies. Obstet Gynecol Surv 2013; 68:43–50.
- Kirkcaldy RD, Augostini P, Asbel LE, et al. Trichomonas vaginalis antimicrobial drug resistance in 6 US cities, STD Surveillance Network, 2009–2010. Emerg Infect Dis 2012; 18:939–943.
- Hoots BE, Peterman TA, Torrone EA, Weinstock H, Meites E, Bolan GA. A Trich-y question: should Trichomonas vaginalis infection be reportable? Sex Transm Dis 2013; 40:113–116.
- Schwebke JR, Desmond RA. A randomized controlled trial of partner notification methods for prevention of trichomoniasis in women. Sex Transm Dis 2010; 37:392–396.
- Studemeister A. Cytomegalovirus proctitis: a rare and disregarded sexually transmitted disease. Sex Transm Dis 2011; 38:876–878.
- Hoentjen F, Rubin DT. Infectious proctitis: when to suspect it is not inflammatory bowel disease. Dig Dis Sci 2012; 57:269–273.
- Satterwhite CL, Torrone E, Meites E, et al. Sexually transmitted infections among US women and men: prevalence and incidence estimates, 2008. Sex Transm Dis 2013; 40:187–193.
- Institute of Medicine (US); Committee on Prevention and Control of Sexually Transmitted Diseases. The hidden epidemic: confronting sexually transmitted diseases. Washington, DC: National Academy Press; 1997.
- Centers for Disease Control and Prevention (CDC). 2011 Sexually Transmitted Disease Surveillance. http://www.cdc.gov/std/stats11/toc.htm. Accessed January 10, 2014.
- Barrow RY, Newman LM, Douglas JM. Taking positive steps to address STD disparities for African-American communities. Sex Transm Dis 2008; 35(suppl 12):S1–S3.
- Mitchell JW, Petroll AE. Patterns of HIV and sexually transmitted infection testing among men who have sex with men couples in the United States. Sex Transm Dis 2012; 39:871–876.
- Centers for Disease Control and Prevention (CDC). HIV testing among men who have sex with men—21 cities, United States, 2008. MMWR Morb Mortal Wkly Rep 2011; 60:694–699.
- Meyers D, Wolff T, Gregory K, et al., USPSTF. USPSTF recommendations for STI screening. Am Fam Physician 2008; 77:819–824.
- Workowski KA, Berman S; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm Rep 2010; 59:1–110.
- Summaries for patients. Screening for chlamydial infection: recommendations from the US Preventive Services Task Force. Ann Intern Med 2007; 147:I44.
- Marrazzo JM, Cates W. Interventions to prevent sexually transmitted infections, including HIV infection. Clin Infect Dis 2011; 53(suppl 3):S64–S78.
- Markowitz LE, Dunne EF, Saraiya M, Lawson HW, Chesson H, Unger ER; Centers for Disease Control and Prevention (CDC); Advisory Committee on Immunization Practices (ACIP). Quadrivalent Human Papillomavirus Vaccine: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2007; 56:1–24.
- Centers for Disease Control and Prevention (CDC). Recommendations on the use of quadrivalent human papillomavirus vaccine in males—Advisory Committee on Immunization Practices (ACIP), 2011. MMWR Morb Mortal Wkly Rep 2011; 60:1705–1708.
- Paavonen J, Jenkins D, Bosch FX, et al; HPV PATRICIA study group. Efficacy of a prophylactic adjuvanted bivalent L1 virus-like-particle vaccine against infection with human papillomavirus types 16 and 18 in young women: an interim analysis of a phase III double-blind, randomised controlled trial. Lancet 2007; 369:2161–2170.
- Mast EE, Weinbaum CM, Fiore AE, et al; Advisory Committee on Immunization Practices (ACIP); Centers for Disease Control and Prevention (CDC). A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) Part II: immunization of adults. MMWR Recomm Rep 2006; 55:1–33.
- Mayer KH. Sexually transmitted diseases in men who have sex with men. Clin Infect Dis 2011; 53(suppl 3):S79–S83.
- Tobian AA, Serwadda D, Quinn TC, et al. Male circumcision for the prevention of HSV-2 and HPV infections and syphilis. N Engl J Med 2009; 360:1298–1309.
- Auvert B, Sobngwi-Tambekou J, Cutler E, et al. Effect of male circumcision on the prevalence of high-risk human papillomavirus in young men: results of a randomized controlled trial conducted in Orange Farm, South Africa. J Infect Dis 2009; 199:14–19.
- Obiero J, Mwethera PG, Wiysonge CS. Topical microbicides for prevention of sexually transmitted infections. Cochrane Database Syst Rev 2012; 6:CD007961.
- Abdool Karim Q, Abdool Karim SS, Frohlich JA, et al; CAPRISA 004 Trial Group. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science 2010; 329:1168–1174.
- HIV prevention through early detection and treatment of other sexually transmitted diseases—United States. Recommendations of the Advisory Committee for HIV and STD prevention. MMWR Recomm Rep 1998; 47:1–24.
- Fleming DT, Wasserheit JN. From epidemiological synergy to public health policy and practice: the contribution of other sexually transmitted diseases to sexual transmission of HIV infection. Sex Transm Infect 1999; 75:3–17.
- Centers for Disease Control and Prevention (CDC). Seroprevalence of herpes simplex virus type 2 among persons aged 14–49 years—United States, 2005–2008. MMWR Morb Mortal Wkly Rep 2010; 59:456–459.
- Semaan S, Leinhos M, Neumann MS. Public health strategies for prevention and control of HSV-2 in persons who use drugs in the United States. Drug Alcohol Depend 2013; 131:182–197.
- Bernstein DI, Bellamy AR, Hook EW, et al. Epidemiology, clinical presentation, and antibody response to primary infection with herpes simplex virus type 1 and type 2 in young women. Clin Infect Dis 2013; 56:344–351.
- Kimberlin DW, Rouse DJ. Clinical practice. Genital herpes. N Engl J Med 2004; 350:1970–1977.
- Sexually Transmitted Diseases. 4th ed. New York, NY: McGraw-Hill Professional Publishing; 2007.
- Johnston C, Magaret A, Selke S, Remington M, Corey L, Wald A. Herpes simplex virus viremia during primary genital infection. J Infect Dis 2008; 198:31–34.
- Laderman EI, Whitworth E, Dumaual E, et al. Rapid, sensitive, and specific lateral-flow immunochromatographic point-of-care device for detection of herpes simplex virus type 2-specific immunoglobulin G antibodies in serum and whole blood. Clin Vaccine Immunol 2008; 15:159–163.
- Philip SS, Ahrens K, Shayevich C, et al. Evaluation of a new point-of-care serologic assay for herpes simplex virus type 2 infection. Clin Infect Dis 2008; 47:e79–e82.
- Belshe RB, Leone PA, Bernstein DI, et al; Herpevac Trial for Women. Efficacy results of a trial of a herpes simplex vaccine. N Engl J Med 2012; 366:34–43.
- Stanberry LR, Spruance SL, Cunningham AL, et al; GlaxoSmithKline Herpes Vaccine Efficacy Study Group. Glycoprotein-D-adjuvant vaccine to prevent genital herpes. N Engl J Med 2002; 347:1652–1661.
- Mark H, Gilbert L, Nanda J. Psychosocial well-being and quality of life among women newly diagnosed with genital herpes. J Obstet Gynecol Neonatal Nurs 2009; 38:320–326.
- Gilbert LK, Omisore F. Common questions about herpes: analysis of chat-room transcripts. Herpes 2009; 15:57–61.
- Alexander L, Naisbett B. Patient and physician partnerships in managing genital herpes. J Infect Dis 2002; 186(suppl 1):S57–S65.
- Ghanem KG, Workowski KA. Management of adult syphilis. Clin Infect Dis 2011; 53(suppl 3):S110–S128.
- Centers for Disease Control and Prevention (CDC). Discordant results from reverse sequence syphilis screening—five laboratories, United States, 2006–2010. MMWR Morb Mortal Wkly Rep 2011; 60:133–137.
- Centers for Disease Control and Prevention (CDC). Syphilis testing algorithms using treponemal tests for initial screening—four laboratories, New York City, 2005–2006. MMWR Morb Mortal Wkly Rep 2008; 57:872–875.
- Seña AC, White BL, Sparling PF. Novel Treponema pallidum serologic tests: a paradigm shift in syphilis screening for the 21st century. Clin Infect Dis 2010; 51:700–708.
- Marangoni A, Sambri V, Storni E, D’Antuono A, Negosanti M, Cevenini R. Treponema pallidum surface immunofluorescence assay for serologic diagnosis of syphilis. Clin Diagn Lab Immunol 2000; 7:417–421.
- Post JJ, Khor C, Furner V, Smith DE, Whybin LR, Robertson PW. Case report and evaluation of the frequency of the prozone phenomenon in syphilis serology—an infrequent but important laboratory phenomenon. Sex Health 2012; 9:488–490.
- Ghanem KG, Erbelding EJ, Cheng WW, Rompalo AM. Doxycycline compared with benzathine penicillin for the treatment of early syphilis. Clin Infect Dis 2006; 42:e45–e49.
- Hook EW, Roddy RE, Handsfield HH. Ceftriaxone therapy for incubating and early syphilis. J Infect Dis 1988; 158:881–884.
- A2058G Prevalence Workgroup. Prevalence of the 23S rRNA A2058G point mutation and molecular subtypes in Treponema pallidum in the United States, 2007 to 2009. Sex Transm Dis 2012; 39:794–798.
- Workowski KA, Berman SM. Centers for Disease Control and Prevention Sexually Transmitted Disease Treatment Guidelines. Clin Infect Dis 2011; 53(suppl 3):S59–S63.
- Kirkcaldy RD, Bolan GA, Wasserheit JN. Cephalosporin-resistant gonorrhea in North America. JAMA 2013; 309:185–187.
- Seña AC, Lensing S, Rompalo A, et al. Chlamydia trachomatis, Mycoplasma genitalium, and Trichomonas vaginalis infections in men with nongonococcal urethritis: predictors and persistence after therapy. J Infect Dis 2012; 206:357–365.
- Manhart LE, Broad JM, Golden MR. Mycoplasma genitalium: should we treat and how? Clin Infect Dis 2011; 53(suppl 3):S129–S142.
- Jernberg E, Moghaddam A, Moi H. Azithromycin and moxifloxacin for microbiological cure of Mycoplasma genitalium infection: an open study. Int J STD AIDS 2008; 19:676–679.
- Taylor SN, Lensing S, Schwebke J, et al. Prevalence and treatment outcome of cervicitis of unknown etiology. Sex Transm Dis 2013; 40:379–385.
- Lusk MJ, Konecny P. Cervicitis: a review. Curr Opin Infect Dis 2008; 21:49–55.
- Geisler WM. Diagnosis and management of uncomplicated Chlamydia trachomatis infections in adolescents and adults: summary of evidence reviewed for the 2010 Centers for Disease Control and Prevention Sexually Transmitted Diseases Treatment Guidelines. Clin Infect Dis 2011; 53(suppl 3):S92–S98.
- Schachter J, Chernesky MA, Willis DE, et al. Vaginal swabs are the specimens of choice when screening for Chlamydia trachomatis and Neisseria gonorrhoeae: results from a multicenter evaluation of the APTIMA assays for both infections. Sex Transm Dis 2005; 32:725–728.
- Hosenfeld CB, Workowski KA, Berman S, et al. Repeat infection with Chlamydia and gonorrhea among females: a systematic review of the literature. Sex Transm Dis 2009; 36:478–489.
- Sobel JD, Subramanian C, Foxman B, Fairfax M, Gygax SE. Mixed vaginitis-more than coinfection and with therapeutic implications. Curr Infect Dis Rep 2013; 15:104–108.
- Taylor BD, Darville T, Haggerty CL. Does bacterial vaginosis cause pelvic inflammatory disease? Sex Transm Dis 2013; 40:117–122.
- Amsel R, Totten PA, Spiegel CA, Chen KC, Eschenbach D, Holmes KK. Nonspecific vaginitis. Diagnostic criteria and microbial and epidemiologic associations. Am J Med 1983; 74:14–22.
- Oduyebo OO, Anorlu RI, Ogunsola FT. The effects of antimicrobial therapy on bacterial vaginosis in non-pregnant women. Cochrane Database Syst Rev 2009; ( 3):CD006055.
- Bunge KE, Beigi RH, Meyn LA, Hillier SL. The efficacy of retreatment with the same medication for early treatment failure of bacterial vaginosis. Sex Transm Dis 2009; 36:711–713.
- Reichman O, Akins R, Sobel JD. Boric acid addition to suppressive antimicrobial therapy for recurrent bacterial vaginosis. Sex Transm Dis 2009; 36:732–734.
- Senok AC, Verstraelen H, Temmerman M, Botta GA. Probiotics for the treatment of bacterial vaginosis. Cochrane Database Syst Rev 2009; (4):CD006289.
- Nye MB, Schwebke JR, Body BA. Comparison of APTIMA Trichomonas vaginalis transcription-mediated amplification to wet mount microscopy, culture, and polymerase chain reaction for diagnosis of trichomoniasis in men and women. Am J Obstet Gynecol 2009; 200:188.e1–188.e7.
- Bachmann LH, Hobbs MM, Seña AC, et al. Trichomonas vaginalis genital infections: progress and challenges. Clin Infect Dis 2011; 53(suppl 3):S160–S172.
- Schwebke JR, Hobbs MM, Taylor SN, et al. Molecular testing for Trichomonas vaginalis in women: results from a prospective US clinical trial. J Clin Microbiol 2011; 49:4106–4111.
- Coleman JS, Gaydos CA, Witter F. Trichomonas vaginalis vaginitis in obstetrics and gynecology practice: new concepts and controversies. Obstet Gynecol Surv 2013; 68:43–50.
- Kirkcaldy RD, Augostini P, Asbel LE, et al. Trichomonas vaginalis antimicrobial drug resistance in 6 US cities, STD Surveillance Network, 2009–2010. Emerg Infect Dis 2012; 18:939–943.
- Hoots BE, Peterman TA, Torrone EA, Weinstock H, Meites E, Bolan GA. A Trich-y question: should Trichomonas vaginalis infection be reportable? Sex Transm Dis 2013; 40:113–116.
- Schwebke JR, Desmond RA. A randomized controlled trial of partner notification methods for prevention of trichomoniasis in women. Sex Transm Dis 2010; 37:392–396.
- Studemeister A. Cytomegalovirus proctitis: a rare and disregarded sexually transmitted disease. Sex Transm Dis 2011; 38:876–878.
- Hoentjen F, Rubin DT. Infectious proctitis: when to suspect it is not inflammatory bowel disease. Dig Dis Sci 2012; 57:269–273.
KEY POINTS
- Anyone can have an STD, although the prevalence is higher in some groups, such as younger sexually active people, certain racial and ethnic minorities, men who have sex with men, and people who engage in risky sexual behavior.
- Preexposure vaccination is one of the most effective ways to prevent human papillomavirus, hepatitis A virus, and hepatitis B virus infections.
- The risk of acquiring human immunodeficiency virus is two to five times higher if the patient has a genital ulcerative disease such as syphilis or herpes at the time of exposure.
- Chlamydia trachomatis and Neisseria gonorrhoeae are major players in urethritis, cervicitis, and proctitis.
- The most common conditions associated with vaginitis include bacterial vaginosis, trichomoniasis, and candidiasis.
Managing risks of TNF inhibitors: An update for the internist
Biologic agents such as those that block tumor necrosis factor (TNF) alpha have revolutionized the treatment of autoimmune diseases like rheumatoid arthritis and inflammatory bowel disease, dramatically improving disease control and quality of life. In addition, they have made true disease remission possible in some cases.
However, as with any new therapy, a variety of side effects must be considered.
WHAT ARE BIOLOGIC AGENTS?

Biologic agents are genetically engineered drugs manufactured or synthesized in vitro from molecules such as proteins, genes, and antibodies present in living organisms. By targeting specific molecular components of the inflammatory cascade, including cytokines, these drugs alter certain aspects of the body’s inflammatory response in autoimmune diseases. Because TNF inhibitors are the most widely used biologic agents in the United States, this review will focus on them.
TNF INHIBITORS
TNF inhibitors suppress the inflammatory cascade by inactivating TNF alpha, a cytokine that promotes inflammation in the intestine, synovial tissue, and other sites.1,2
Several TNF inhibitors are available. Etanercept and infliximab were the first two to receive US Food and Drug Administration (FDA) approval, and they are extensively used. Etanercept, a soluble TNF receptor given subcutaneously, was approved for treating rheumatoid arthritis in 1998. Infliximab, a chimeric monoclonal antibody (75% human and 25% mouse protein sequence) given intravenously, received FDA approval for treating Crohn disease in 1998 and for rheumatoid arthritis in 1999. Other anti-TNF agents with varying properties are also available.
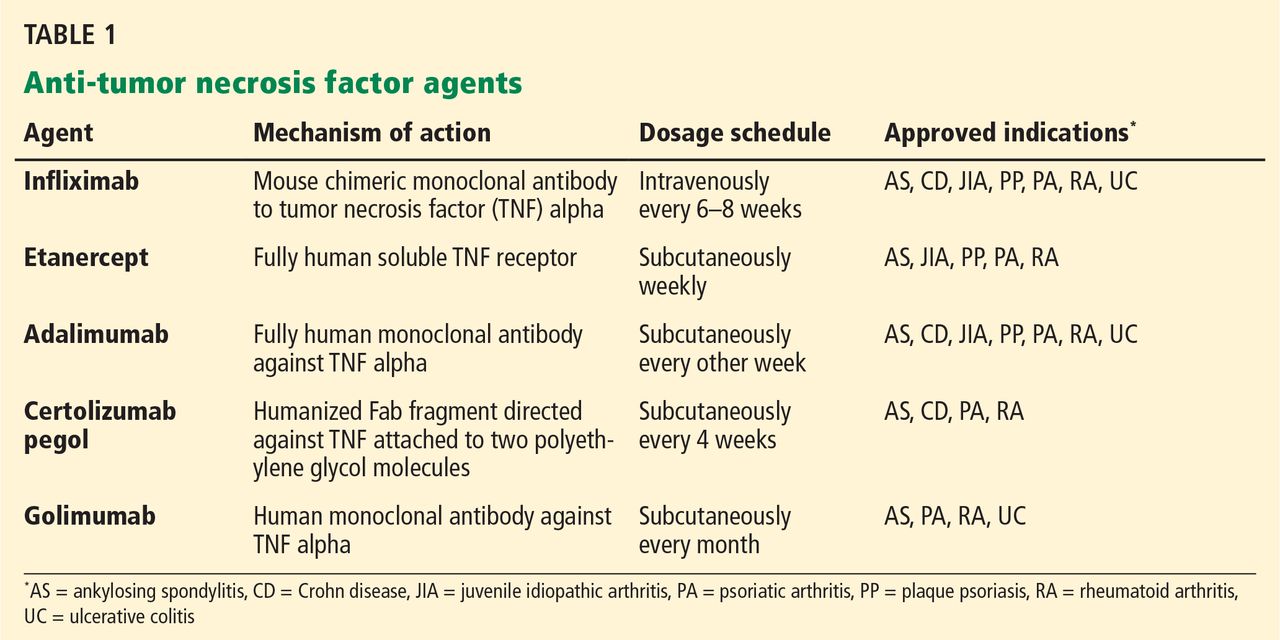
Table 1 lists anti-TNF agents approved for treating rheumatoid arthritis and inflammatory bowel disease. These are chronic, relapsing diseases that significantly and dramatically reduce quality of life, especially when they are poorly controlled.3,4 Untreated disease has been associated with malignancy, infection, pregnancy loss, and malnutrition.5–8 TNF inhibitors are aimed at patients who have moderate to severe disease or for whom previous treatments have failed, to help them achieve and maintain steroid-free remission. However, use of these agents is tempered by the risk of potentially serious side effects.
Special thought should also be given to the direct costs of the drugs (up to $30,000 per year, not counting the cost of their administration) and to the indirect costs such as time away from work to receive treatment. These are major considerations in some cases, and patients should be selected carefully for treatment with these drugs.
BEFORE STARTING THERAPY
Before starting anti-TNF therapy, several steps can reduce the risk of serious adverse events.
Take a focused history
The clinical history should include inquiries about previous bacterial, fungal, and tuberculosis infections or exposure; diabetes; and other immunocompromised states that increase the risk of acquiring potentially life-threatening infections.
Details of particular geographic areas of residence, occupational exposures, and social history should be sought. These include history of incarceration (which may put patients at risk of tuberculosis) and residence in the Ohio River valley or in the southwestern or midwestern United States (which may increase the risk of histoplasmosis, coccidioidomycosis, and other fungal infections).
Bring vaccinations up to date
Age-appropriate vaccinations should be discussed and given, ideally before starting therapy. These include influenza vaccine every year and tetanus boosters every 10 years for all and, as appropriate, varicella, human papillomavirus, and pneumococcal vaccinations. The US Centers for Disease Control and Prevention recommend an additional dose of the pneumococcal vaccine if more than 5 years have elapsed since the first one, and many clinicians opt to give it every 5 years.
In general, live-attenuated vaccines, including the intranasal influenza vaccine, are contraindicated in patients taking biologic agents.9 For patients at high risk of exposure or infection, it may be reasonable to hold the biologic agent for a period of time, vaccinate, and resume the biologic agent a month later.
Recent data suggest that the varicella zoster vaccine may be safely given to older patients with immune-mediated diseases such as rheumatoid arthritis and inflammatory bowel disease taking biologic agents.10 New guidelines from the American College of Rheumatology recommend age-appropriate vaccines for rheumatoid arthritis patients age 60 and older before biologic treatments are started. Case-by-case discussion with the subspecialist and the patient is recommended.
Screen for chronic infections
Tuberculosis screening with a purified protein derivative test or an interferon-gamma-release (Quantiferon) assay followed by chest radiography in patients with a positive test is mandatory before giving a TNF inhibitor.
Hepatitis B virus status should be determined before starting anti-TNF therapy.11
Hepatitis B vaccination has been recommended for patients with inflammatory bowel disease, but no clear recommendation exists for patients with rheumatic disease. Patients with inflammatory bowel disease tend to have low rates of response to hepatitis B vaccination12,13; possible reasons include their lack of an appropriate innate immune response to infectious agents, malnutrition, surgery, older age, and immunosuppressive drugs.14 An accelerated vaccination protocol with recombinant hepatitis B vaccine (Energix-B) in a double dose at 0, 1, and 2 months has been shown to improve response rates.15
Whenever possible, it may be better to vaccinate patients before starting immunosuppressive therapy, and to check postvaccination titers to ensure adequate response.
Perform an examination
A full physical examination with special attention to skin rashes should be performed. This may serve as a baseline to assist early detection of new rashes associated with anti-TNF therapy.

A baseline complete blood cell count and complete metabolic panel should be routinely obtained before starting therapy (and thereafter at the discretion of the physician). In conjunction with follow-up tests, they can help detect an unexpected decrease in white blood cell count or abnormal results on the liver panel.16 These baseline and follow-up tests are generally performed by the subspecialist, and the results are shared with the primary care physician.
Table 2 summarizes key information to be sought before starting a patient on a TNF inhibitor.
ADVERSE EFFECTS OF ANTI-TNF DRUGS
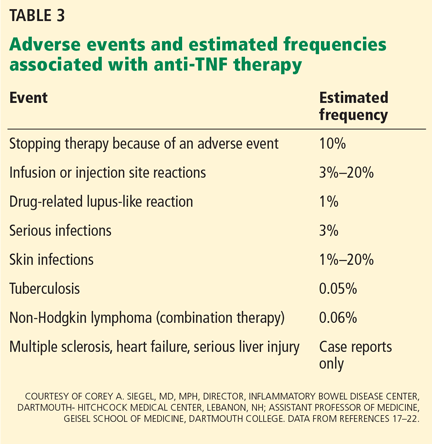
Infusion reactions, infections, cardiac arrhythmias, demyelinating disorders, skin infections, and malignancies have been reported with anti-TNF therapy. The relative frequencies of these adverse events are summarized in Table 3.17–22
NONINFECTIOUS COMPLICATIONS OF TNF INHIBITORS
Injection site reactions
When anti-TNF agents are given subcutaneously, injection site reactions are common (occurring in up to 40% of patients) and are considered minor.11 Reactions, including significant pain, typically occur within the first few months of therapy. They can last 2 to 5 days but rarely warrant stopping therapy. Treatment with ice and an antihistamine is almost always sufficient to control symptoms.
Infusion reactions with infliximab
Infliximab can cause both acute and delayed infusion reactions. Acute reactions can occur up to 24 hours after infusion but usually appear within 10 minutes of administration and are handled by the infusion suite staff. They range from the severe immunoglobulin E-mediated type I reaction, manifesting with hypotension, bronchospasm, and urticaria, to the milder anaphylactoid-type reaction, which constitutes the majority.23–25
While most primary care physicians will not encounter an acute reaction, family doctors and emergency room physicians may encounter delayed reactions, which can develop 1 to 14 days after infusion. These reactions usually resemble serum sickness and present with joint pain, fatigue, myalgia, and fever. But, unlike classic serum sickness, these reactions are generally not associated with a rash.
With nonspecific symptoms, the diagnosis may be easy to overlook. However, establishing this diagnosis is important because repeat therapy may result in a more severe reaction upon reexposure to the drug.
Once diagnosed, these reactions can be treated symptomatically with a combination of acetaminophen and diphenhydramine after discussion between the primary care physician and subspecialist.23,24
Autoimmune syndromes
Several studies have reported a small percentage of patients treated with anti-TNF agents who develop paradoxical autoimmune conditions. These range from asymptomatic immunologic alterations, including the formation of antinuclear antibodies and antibodies to double-stranded DNA, to life-threatening systemic autoimmune diseases.26,27
Autoimmune diseases associated with anti-TNF treatment include a lupus-like syndrome, vasculitides, and psoriatic skin lesions. These syndromes warrant stopping the inciting drug and, on occasion, giving corticosteroids. Most cases arise between 1 month and 1 year of starting treatment, and almost 75% resolve completely after the anti-TNF therapy is stopped.26
Interestingly, anti-TNF agents are approved for treating psoriasis and psoriatic arthritis, but psoriasis has paradoxically developed in patients being treated with these drugs for other autoimmune diseases. The FDA has reviewed 69 cases of new-onset psoriasis with anti-TNF therapy, including 17 pustular and 15 palmoplantar cases. The 12 most severe cases resulted in hospitalization, and symptoms resolved in most after treatment cessation.28
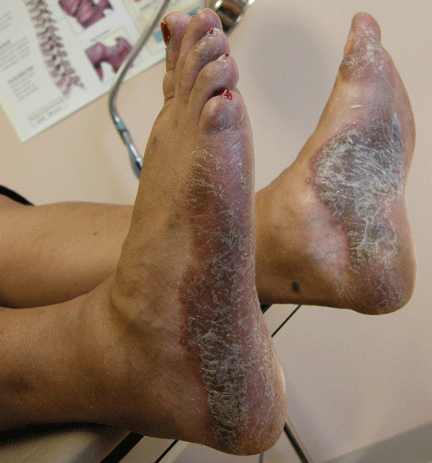
Fiorino et al29 counted 18 reported cases of psoriasis induced by anti-TNF therapy in patients with inflammatory bowel disease and concluded that it is rare. Harrison et al30 reported similar findings in patients with rheumatoid arthritis, with an increased incidence rate of 1.04 per 1,000 person-years. New-onset psoriasis was most common in patients treated with adalimumab. An example of the rash is seen in Figure 1.
CARDIOVASCULAR SIDE EFFECTS
Cardiovascular side effects of anti-TNF agents range from nonspecific and asymptomatic arrhythmias to worsening of heart failure.
Circulating levels of TNF are increased in patients with heart failure, and studies have evaluated the effects of TNF inhibition with infliximab on cardiac function and overall survival.31,32 The combined risk of death from any cause or hospitalization from heart failure was significantly higher in the infliximab groups, and the effects persisted for up to 5 months after stopping therapy.
Other studies22,33 have evaluated the effects of infliximab and etanercept on cardiac function and overall survival. Results showed possible exacerbation of heart failure with etanercept and increased risk of death with infliximab in patients with New York Heart Association (NYHA) class III or IV heart failure and left ventricular ejection fractions less than 35%.
Case reports have also described patients with worsening or new-onset heart failure on TNF inhibitors, including patients younger than 50 years and without identifiable cardiovascular risk factors.
Data analyses31,34,35 from large clinical registries have reported no significant increase in heart failure attributable to TNF inhibitors. However, we have concerns about the methodology of these analyses.
Currently, anti-TNF therapy is contraindicated in patients with NYHA class III or IV heart failure. Data are inconclusive for patients with class I or II heart failure. Baseline echocardiography and cardiology consultation can be considered, with close monitoring and avoidance of high doses of TNF inhibitors. If heart failure develops in a patient on anti-TNF therapy, the drug should be discontinued and the patient should be evaluated further.36
DEMYELINATING DISEASE, INCLUDING MULTIPLE SCLEROSIS
Anti-TNF agents have been associated with the onset or exacerbation of clinical symptoms and radiographic evidence of central nervous system demyelinating disorders, including multiple sclerosis.37–40 Mohan et al38 identified 19 cases of demyelinating events occurring after administration of anti-TNF agents in early 2001. In most cases, symptoms improved or resolved after therapy was stopped.
Optic neuritis,41,42 bilateral optic neuropathy,43 and aseptic meningitis44 have also been reported, but these have occurred only rarely.
How common are these effects? Postmarketing surveillance in patients with rheumatoid arthritis yields an estimated incidence of demyelinating disorders of 1 per 1,000 patient-years with adalimumab therapy.45 Complicating the assessment is an observed slight increase in risk of demyelinating conditions associated with inflammatory bowel disease.
Symptoms that should heighten the physician’s suspicion of this adverse effect include confusion, paresthesias, and ataxia. Patients on anti-TNF therapy who develop new visual symptoms should be checked for painless visual loss as a sign of early demyelinating disease.
Although data that conclusively link anti-TNF agents to multiple sclerosis are lacking, these drugs should not be initiated in patients who have a history of demyelinating disease, and treatment should be stopped promptly if the diagnosis is suspected.
MALIGNANCY
Whether anti-TNF therapy is directly linked to development of malignancies is difficult to determine. There are many confounding factors, including the risk of malignancy in underlying inflammatory disease and the concomitant use of other medications such as thiopurines, which have a known association with lymphoma.46
The incidence of lymphoma is twice as high in rheumatoid arthritis patients as in the general population.47 The risk is higher in those with more aggressive joint disease—the subset of rheumatoid arthritis patients who are more likely to be given anti-TNF agents.5
In patients with inflammatory bowel disease, the risk of cholangiocarcinoma is four times higher, and the risk of small-bowel adenocarcinoma is 16 to 18 times higher, but no increased risk of lymphoma has been identified in this population.48,49
Data from six clinical trials of infliximab, including a long-term study of its safety in Crohn disease, suggest it poses no increase in overall risk of malignancy.50–52 Similar results have been reported in patients with other rheumatic diseases.8 Information on this topic is constantly evolving, and studies range from case series to clinical trials to large patient registries.
The decision to use a TNF inhibitor should be based on the patient’s clinical picture and risk factors. Discussion of the risks and benefits of therapy with the patient should be clearly documented.
Non-Hodgkin lymphoma
Evidence about the risk of lymphoma with anti-TNF use is mixed, as up to two-thirds of patients on anti-TNF therapy have received concomitant nonbiologic immunosuppressive medications, making it difficult to determine the true risk from the biologic agents alone.53 Current evidence both supports9,53–56 and refutes7,55,57–60 the idea that anti-TNF agents increase lymphoma risk.
In patients with inflammatory bowel disease, several population-based studies have not shown a clear increase in lymphoma risk with anti-TNF use.56,59,60 Pedersen et al,61 in a meta-analysis of eight studies, confirmed these findings by showing no overall lymphoma risk in patients with inflammatory bowel disease.
However, a Canadian population-based study found a statistically significant increase in non-Hodgkin lymphoma in males with Crohn disease, with an incidence ratio of 3.63 (95% confidence interval [CI] 1.53–8.62).61 Additionally, Siegel et al62 found a significantly higher risk (6.1 cases per 10,000 patients) in patients treated with anti-TNF agents and thiopurines than in the general population (1.9 cases per 10,000 people). Although the difference was statistically significant, the overall risk is still very low.
Patients with rheumatoid arthritis seem to have a risk of lymphoma two to three times higher than in the general population. However, large population-based studies have not shown a statistically significant increase in the risk of lymphoma with anti-TNF therapy.63
Hepatosplenic T-cell lymphoma is a rare subtype of peripheral T-cell non-Hodgkin lymphoma; 25 cases have been reported in patients receiving anti-TNF therapy.64 Although the risk is extremely low (< 0.05%), physicians must carefully consider the risks and benefits of combination therapy, especially in young male patients with inflammatory bowel disease, since death is the usual outcome of this disease.65–67
Skin cancers
Wolfe and Michaud8 evaluated malignancy risk in rheumatoid arthritis patients being treated with biologic agents, including TNF inhibitors, using a large longitudinal database. These data were compared with those of the US Surveillance, Epidemiology, and End-Results (SEER) national database. No increase in the overall cancer rate was seen in rheumatoid arthritis patients (standardized incidence ratio [SIR] 1.0, 95% CI 1.0–1.1).
However, melanoma was more common in rheumatoid arthritis patients compared with SEER rates (SIR 1.7, 95% CI 1.3–2.3).8 In addition, biologic therapy was associated with a higher (but not statistically significant) risk of melanoma (odds ratio [OR] 2.3, 95% CI 0.9–5.4) and a higher risk of nonmelanoma skin cancer (OR 1.5, 95% CI 1.2–1.8), but not of other types of cancer.8
INFECTION
Patients on anti-TNF therapy are at a higher risk of infection, ranging from minor to life-threatening bacterial infections, and including the reactivation of granulomatous and fungal infections. More importantly, these agents are similar to steroids in blunting signs of infection, which may delay diagnosis and treatment.
The management of infection in patients on anti-TNF medications varies from case to case. In general, patients with a minor infection that does not require hospitalization or intravenous antibiotics can continue the biologic therapy while taking oral antibiotics. TNF inhibitors must be held in the event of a major infection.
Consultation with an infectious disease specialist is recommended, especially in complex cases.
Bacterial infections
An increased risk of minor bacterial infections such as urinary tract and respiratory infections has been well documented in several randomized control trials of anti-TNF agents, though other studies have shown no such increase in risk.33,51–59
The threshold for using antibiotics for a suspected bacterial infection is somewhat shifted in favor of treatment in patients on anti-TNF therapy. The reason is twofold: as previously noted, infections may be worse than they appear, because anti-TNF drugs can mask the signs and symptoms of a serious infection, and in patients on these drugs, an untreated bacterial infection may rapidly become life-threatening.
In general, broad-spectrum antibiotics are not warranted unless the source of infection is unclear or the patient is in danger of hemodynamic compromise.
Opportunistic infections
The association of anti-TNF agents with opportunistic infections could be viewed as an extension of their normal and intended therapeutic activity as potent immunosuppressive agents.68 Rheumatoid arthritis and inflammatory bowel disease are usually associated with conditions and situations that predispose patients to opportunistic infections, such as decreased immune response, malnutrition or malabsorption, surgeries, and concomitant immunosuppressive medications.7 Combination therapy with other immunosuppressive drugs and older age appear to markedly increase the risk of opportunistic infections, including mycobacterial and fungal infections, in patients with inflammatory bowel disease.7
Overall, opportunistic infections represent a measurable risk of anti-TNF therapy, and awareness and vigilance are important, especially in areas where opportunistic infections such as histoplasmosis and coccidiomycosis are endemic.50 Furthermore, physicians must be aware of the higher risk of opportunistic infections when multiple immunosuppressive drugs are used concurrently.
Granulomatous infections such as tuberculosis
Anti-TNF agents increase the risk of de novo granulomatous infections and of reactivating such infections. Granuloma formation and intracellular destruction of mycobacteria depend on TNF. TNF is important in maintaining the anatomic integrity of granulomas where these organisms have been sequestered, and blocking TNF leads to breakdown of granulomas and release of virulent organisms.69,70
TNF inhibitors increase the risk of reactivation of latent tuberculosis infection. The risk is greater with infliximab and adalimumab than with etanercept,71,72 and it has been described with certolizumab.74 Study results are varied thus far but show a risk of tuberculosis reactivation five to 30 times higher than in the general population, with tremendous variability in risk depending on background rates of previous exposure.
The absence of typical tuberculosis symptoms further complicates care in these cases. Fever, weight loss, and night sweats tend to be TNF-mediated and are therefore masked by anti-TNF agents, leading to atypical presentations. In addition, active tuberculosis infection associated with TNF inhibitors is more likely to involve extrapulmonary sites such as the skin and musculoskeletal system and to be disseminated at presentation.
A paradoxical worsening of tuberculosis symptoms may also be seen in patients with latent tuberculosis reactivation, especially after discontinuing anti-TNF therapy. This is thought to result from an immune reconstitution inflammatory syndrome.
The pretreatment evaluation should include a history of risk factors, a physical examination, and either a tuberculin skin test or an interferon-gamma-release assay. Interferon-gamma-release assays are particularly helpful in patients who have received bacille Calmette-Guérin vaccination. In patients who test positive or have been exposed, tuberculosis treatment should begin 4 weeks before starting anti-TNF therapy, though the optimal timing of antituberculosis agents is still controversial.74–77
If tuberculosis develops in a patient on anti-TNF therapy, he or she should receive antituberculosis drugs. Anti-TNF therapy should be stopped and should be resumed after 2 months only if no other treatment option is available.75
Invasive opportunistic fungal infections
Invasive opportunistic fungal infections have been reported with anti-TNF therapy, including histoplasmosis and coccidioidomycosis.78–80 Most patients who had histoplasmosis were treated with other immunosuppressive therapies and resided in or were raised near the Ohio or Mississippi River valleys, where this disease is endemic. Similarly, most cases of coccidioidomycosis were in endemic areas of Arizona, California, and Nevada, and patients on concomitant immunosuppressive therapy.78
Currently, there is no evidence to recommend obtaining Histoplasma capsulatum or Coccidioides immitis serologies before initiating anti-TNF therapy in patients in endemic areas.81 However, patients must be instructed to seek medical attention quickly for pulmonary or febrile illnesses.
Viral hepatitis infections
The data on hepatitis B and hepatitis C in patients on biologic therapies are mostly limited to case reports.
Hepatitis B. A small prospective study from Spain followed the liver biochemistry tests and hepatitis B status of 80 patients with Crohn disease treated with infliximab. Of three patients who were chronic hepatitis B carriers before starting infliximab, two experienced reactivation of hepatitis B after discontinuing infliximab, and one ultimately died. The third patient was treated with lamivudine concurrently with infliximab without clinical or biochemical changes during or after therapy.82
Similar findings were observed in two patients with rheumatoid arthritis on treatment with infliximab. One of the patients required liver transplantation, and both were treated with lamivudine, resulting in normalization of liver function test results.83,84
Recent reviews indicate that despite these findings, hepatitis B reactivation after anti-TNF withdrawal may not be common.85 There are limited data on hepatitis B reactivation and associated liver dysfunction in patients with inflammatory bowel disease treated with immunosuppressants. In a retrospective multicenter trial by Loras et al86 in patients with inflammatory bowel disease who had viral hepatitis, 36% of patients positive for hepatitis B surface antigen developed liver dysfunction, and six patients developed liver failure. In that study, treatment with more than two immunosuppressants was an independent predictor of hepatitis B reactivation.
Prolonged immunosuppression (longer than 3 months) has also been identified as an independent predictor of liver dysfunction (OR 3.06; 95% 95% CI 1.02–9.16).87
The European Association for the Study of the Liver and the American Association for the Study of Liver Diseases recommend starting lamivudine before chemotherapy or immunomodulator or immunosuppressive therapy in hepatitis B virus carriers and continuing preventive treatment for at least 6 months after stopping immunomodulating drugs. Lamivudine at a dose of 100 mg/day may reduce the risk of reactivation of hepatitis B.88–90 Tenofovir and entecavir may be useful alternatives in patients with hepatitis B who have never received nucleoside analogues. Hepatitis B reactivation did not occur in any of the 16 patients who received preventive entecavir treatment while receiving immunosuppressive treatments.89,91
In patients receiving immunosuppressive therapy, hepatitis B reactivation is associated with significant morbidity and mortality. Although risk factors for reactivation of hepatitis B virus infection have been identified, we recommend preventive treatment for all carriers positive for hepatitis B surface antigen. This should be done regardless of the number, type, and dosage of immunosuppressants and regardless of hepatitis B virus DNA levels.90
The frequency of hepatitis B and hepatitis C infection in patients with Crohn disease has been reported to be as high as 24%. The high incidence is thought to be secondary to multiple blood transfusions and surgeries.92 The use of biologic agents, including anti-TNF agents, in chronic hepatitis B virus- or hepatitis C virus-infected patients can lead to enhanced viral replication and hepatitis exacerbation.
Although active viral replication can occur during treatment with biologic agents, reactivation or exacerbation can also occur after the anti-TNF agent is stopped.82 This finding has prompted the recommendation that all candidates for biologic therapy be tested for hepatitis B immunization status, followed by immunization in nonimmune patients before starting anti-TNF therapy.93,94
Hepatitis C. There are no guidelines that adequately address the use of anti-TNF agents in patients with chronic hepatitis C infection.
Several small retrospective studies in rheumatoid arthritis patients with hepatitis C have shown that TNF inhibitors can be safely used without worsening liver function tests or changing the viral load.95–98 This is reassuring and provides the subspecialist with another treatment option, as other therapies such as disease-modifying antirheumatic drugs and steroids are known to aggravate viral hepatitis and increase the risk of viremia.96
Although small retrospective studies and one large randomized double-blind placebo-controlled trial have shown TNF inhibitors to be relatively safe in rheumatoid arthritis patients with hepatitis C, their use in these patients should be considered only with caution if they have evidence of hepatic synthetic dysfunction (eg, hypoalbuminemia, thrombocytopenia, increased international normalized ratio). The American College of Rheumatology recommends avoiding TNF inhibitors in Child-Pugh classes B and C.99
PREGNANCY
Physicians caring for patients with rheumatoid arthritis and inflammatory bowel disease must be aware of how these diseases affect fecundity and fertility and how the medications can affect conception and pregnancy. Many patients have difficulty conceiving while their autoimmune disease is active, and better disease control may improve fecundity and result in unanticipated pregnancy. Patients should be advised of the need for contraception if pregnancy is ill-advised or undesired.
Many patients seek advice about teratogenicity before conceiving, or seek guidance about rheumatoid arthritis and inflammatory bowel disease treatment while pregnant.
Several studies have reported a higher risk of adverse pregnancy outcomes in patients with rheumatoid arthritis and inflammatory bowel disease than in the general population.99–105 In inflammatory bowel disease, the odds of a premature delivery or having a low-birth-weight child are twice as high as in the normal population.100,103 Higher rates of cesarean delivery and stillbirth have also been reported. The main predisposing factor appears to be the disease activity at the time of conception, as active disease seems to be linked to adverse pregnancy outcomes.
Treatment with anti-TNF agents may rapidly achieve and maintain remission, raising the question of the safety of continued anti-TNF use during pregnancy. The FDA classifies anti-TNF agents as category B drugs, as animal studies have not demonstrated fetal risk, and no well-controlled prospective study has yet been conducted with pregnant women.
In an observational study, Schnitzler et al105 assessed the outcomes of 42 pregnancies in 35 patients with inflammatory bowel disease receiving either infliximab or adalimumab during pregnancy, compared with 56 pregnancies in 45 healthy patients without inflammatory bowel disease. There was no statistical difference in abortion rates between patients receiving anti-TNF agents and healthy women without inflammatory bowel disease (21% vs 14%, P = .4234). There was also no significant difference observed in birth weight, birth length, or cranial circumference of the children between the two groups. However, pregnancies with direct exposure to anti-TNF agents resulted in a higher frequency of premature delivery (25% vs 6%, P = .023).
Similar results were noted from the Crohn’s Therapy, Resource, Evaluation, and Assessment Tool, or TREAT, registry, as well as from a large systematic review by Vinet et al106 and case reports of rheumatoid arthritis and inflammatory bowel disease in women exposed to anti-TNF agents during pregnancy.
In addition, results from a recent systematic review of 38 studies of anti-TNF use and fetal risk, with a total of 437 women (189 on infliximab, 230 on adalimumab, 18 on certolizumab pegol), showed similar results.107 In pregnancies exposed to anti-TNF agents, the rates of congenital abnormalities (3.4%), fetal deaths (8.5%), and preterm births (2.7%) were similar to those in the general population.
For patients in disease remission on TNF inhibitors, it is reasonable to continue these agents during pregnancy after careful discussion with the patient. Fetal safety and infant immunization response after delivery are the primary concerns in these cases.
Both infliximab and adalimumab cross the placenta and remain detectable in the baby’s circulation 4 months (for adalimumab) to 6 months (for infliximab) after delivery. It is currently recommended that infliximab be stopped at 32 weeks of gestation for the remainder of the pregnancy and that adalimumab be stopped at 34 to 36 weeks, given a planned 40-week gestation.
Certolizumab does not cross the placenta in significant amounts and should be continued throughout pregnancy; drug levels in infants were shown to be less than 2 μg/mL, even when dosed the week of delivery.108–112
TAKE-HOME POINTS
- All health care providers of patients with rheumatoid arthritis and inflammatory bowel disease should be familiar with anti-TNF agents used in treating these diseases.
- The benefits of controlling the disease far outweigh the risks of therapy when used appropriately.
- Care of these patients should be multidisciplinary, with clear communication between primary care physician and specialist.
- Patient education and monitoring combined with prompt communication between primary care physician and specialist are key.
- Wijbrandts CA, Dijkgraaf MG, Kraan MC, et al. The clinical response to infliximab in rheumatoid arthritis is in part dependent on pretreatment tumour necrosis factor alpha expression in the synovium. Ann Rheum Dis 2008; 67:1139–1144.
- Sfikakis PP. The first decade of biologic TNF antagonists in clinical practice: lessons learned, unresolved issues and future directions. Curr Dir Autoimmun 2010; 11:180–210.
- Casellas F, Alcalá MJ, Prieto L, Miró JR, Malagelada JR. Assessment of the influence of disease activity on the quality of life of patients with inflammatory bowel disease using a short questionnaire. Am J Gastroenterol 2004; 99:457–461.
- Casellas F, López-Vivancos J, Badia X, Vilaseca J, Malagelada JR. Influence of inflammatory bowel disease on different dimensions of quality of life. Eur J Gastroenterol Hepatol 2001; 13:567–572.
- Baecklund E, Iliadou A, Askling J, et al. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum 2006; 54:692–701.
- Franklin J, Lunt M, Bunn D, Symmons D, Silman A. Incidence of lymphoma in a large primary care derived cohort of cases of inflammatory polyarthritis. Ann Rheum Dis 2006; 65:617–622.
- Toruner M, Loftus EV, Harmsen WS, et al. Risk factors for opportunistic infections in patients with inflammatory bowel disease. Gastroenterology 2008; 134:929–936.
- Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum 2007; 56:2886–2895.
- Prevention of pneumococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 1997; 46:1–24.
- Zhang J, Xie F, Delzell E, et al. Association between vaccination for herpes zoster and risk of herpes zoster infection among older patients with selected immune-mediated diseases. JAMA 2012; 308:43–49.
- Moreland LW, Baumgartner SW, Schiff MH, et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med 1997; 337:141–147.
- Altunöz ME, Senates E, Yesil A, Calhan T, Ovünç AO. Patients with inflammatory bowel disease have a lower response rate to HBV vaccination compared to controls. Dig Dis Sci 2012; 57:1039–1044.
- Gisbert JP, Villagrasa JR, Rodríguez-Nogueiras A, Chaparro M. Efficacy of hepatitis B vaccination and revaccination and factors impacting on response in patients with inflammatory bowel disease. Am J Gastroenterol 2012; 107:1460–1466.
- Carrera E, Manzano R, Garrido E. Efficacy of the vaccination in inflammatory bowel disease. World J Gastroenterol 2013; 19:1349–1353.
- Gisbert JP, Menchén L, García-Sánchez V, Marín I, Villagrasa JR, Chaparro M. Comparison of the effectiveness of two protocols for vaccination (standard and double dosage) against hepatitis B virus in patients with inflammatory bowel disease. Aliment Pharmacol Ther 2012; 35:1379–1385.
- Ferkolj I. How to improve the safety of biologic therapy in Crohn’s disease. J Physiol Pharmacol 2009; 60(suppl 7):67–70.
- Siegel CA. The risks of biologic therapy for inflammatory bowel disease. In:Bernstein ED, editor. The Inflammatory Bowel Disease Yearbook, volume 6. London, UK: Remedica, 2010:89–108.
- Remicade (infliximab) Package Insert. Horsham, PA: Janssen Biotech, Inc; 2013. http://www.remicade.com/shared/product/remicade/prescribing-information.pdf. Accessed January 2, 2014.
- Vermeire S, Noman M, Van Assche G, et al. Autoimmunity associated with anti-tumor necrosis factor alpha treatment in Crohn’s disease: a prospective cohort study. Gastroenterology 2003; 125:32–39.
- Cush JJ. Biological drug use: US perspectives on indications and monitoring. Ann Rheum Dis 2005; 64(suppl 4):iv18–iv23.
- TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 1999; 53:457–465.
- Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT; Anti-TNF Therapy Against Congestive Heart Failure Investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003; 107:3133–3140.
- Cheifetz A, Mayer L. Monoclonal antibodies, immunogenicity, and associated infusion reactions. Mt Sinai J Med 2005; 72:250–256.
- Cheifetz A, Smedley M, Martin S, et al. The incidence and management of infusion reactions to infliximab: a large center experience. Am J Gastroenterol 2003; 98:1315–1324.
- Vultaggio A, Matucci A, Nencini F, et al. Anti-infliximab IgE and non-IgE antibodies and induction of infusion-related severe anaphylactic reactions. Allergy 2010; 65:657–661.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore) 2007; 86:242–251.
- Stallmach A, Hagel S, Bruns T. Adverse effects of biologics used for treating IBD. Best Pract Res Clin Gastroenterol 2010; 24:167–182.
- Haagsma CJ, Blom HJ, van Riel PL, et al. Influence of sulphasalazine, methotrexate, and the combination of both on plasma homocysteine concentrations in patients with rheumatoid arthritis. Ann Rheum Dis 1999; 58:79–84.
- Fiorino G, Allez M, Malesci A, Danese S. Review article: anti TNF-alpha induced psoriasis in patients with inflammatory bowel disease. Aliment Pharmacol Ther 2009; 29:921–927.
- Harrison MJ, Dixon WG, Watson KD, et al; British Society for Rheumatology Biologics Register Control Centre Consortium; BSRBR. Rates of new-onset psoriasis in patients with rheumatoid arthritis receiving anti-tumour necrosis factor alpha therapy: results from the British Society for Rheumatology Biologics Register. Ann Rheum Dis 2009; 68:209–215.
- Marchesoni A, Zaccara E, Gorla R, et al. TNF-alpha antagonist survival rate in a cohort of rheumatoid arthritis patients observed under conditions of standard clinical practice. Ann N Y Acad Sci 2009; 1173:837–846.
- Tomas L, Lazurova I, Pundova L, et al. Acute and long-term effect of infliximab on humoral and echocardiographic parameters in patients with chronic inflammatory diseases. Clin Rheumatol 2013 32:61–66.
- Senel S, Cobankara V, Taskoylu O, et al. The safety and efficacy of etanercept on cardiac functions and lipid profile in patients with active rheumatoid arthritis. J Investig Med 2012; 60:62–65.
- Al-Aly Z, Pan H, Zeringue A, et al. Tumor necrosis factor-a blockade, cardiovascular outcomes, and survival in rheumatoid arthritis. Transl Res 2011; 157:10–18.
- Listing J, Strangfeld A, Kekow J, et al. Does tumor necrosis factor alpha inhibition promote or prevent heart failure in patients with rheumatoid arthritis? Arthritis Rheum 2008; 58:667–677.
- Wolfe F, Michaud K. Heart failure in rheumatoid arthritis: rates, predictors, and the effect of anti-tumor necrosis factor therapy. Am J Med 2004; 116:305–311.
- Enayati PJ, Papadakis KA. Association of anti-tumor necrosis factor therapy with the development of multiple sclerosis. J Clin Gastroenterol 2005; 39:303–306.
- Mohan N, Edwards ET, Cupps TR, et al. Demyelination occurring during anti-tumor necrosis factor alpha therapy for inflammatory arthritides. Arthritis Rheum 2001; 44:2862–2869.
- Sands BE, Anderson FH, Bernstein CN, et al. Infliximab maintenance therapy for fistulizing Crohn’s disease. N Engl J Med 2004; 350:876–885.
- Thomas CW, Weinshenker BG, Sandborn WJ. Demyelination during anti-tumor necrosis factor alpha therapy with infliximab for Crohn’s disease. Inflamm Bowel Dis 2004; 10:28–31.
- Foroozan R, Buono LM, Sergott RC, Savino PJ. Retrobulbar optic neuritis associated with infliximab. Arch Ophthalmol 2002; 120:985–987.
- Strong BY, Erny BC, Herzenberg H, Razzeca KJ. Retrobulbar optic neuritis associated with infliximab in a patient with Crohn disease. Ann Intern Med 2004; 140:W34.
- ten Tusscher MP, Jacobs PJ, Busch MJ, de Graaf L, Diemont WL. Bilateral anterior toxic optic neuropathy and the use of infliximab. BMJ 2003; 326:579.
- Hegde N, Gayomali C, Rich MW. Infliximab-induced headache and infliximab-induced meningitis: two ends of the same spectrum? South Med J 2005; 98:564–566.
- Schiff MH, Burmester GR, Kent JD, et al. Safety analyses of adalimumab (HUMIRA) in global clinical trials and US postmarketing surveillance of patients with rheumatoid arthritis. Ann Rheum Dis 2006; 65:889–894.
- Beaugerie L, Brousse N, Bouvier AM, et al; CESAME Study Group. Lymphoproliferative disorders in patients receiving thiopurines for inflammatory bowel disease: a prospective observational cohort study. Lancet 2009; 374:1617–1625.
- Ekström K, Hjalgrim H, Brandt L, et al. Risk of malignant lymphomas in patients with rheumatoid arthritis and in their first-degree relatives. Arthritis Rheum 2003; 48:963–970.
- Lakatos PL, Lakatos L. Risk for colorectal cancer in ulcerative colitis: changes, causes and management strategies. World J Gastroenterol 2008; 14:3937–3947.
- Persson PG, Karlén P, Bernell O, et al. Crohn’s disease and cancer: a population-based cohort study. Gastroenterology 1994; 107:1675–1679.
- de Silva S, Devlin S, Panaccione R. Optimizing the safety of biologic therapy for IBD. Nat Rev Gastroenterol Hepatol 2010; 7:93–101.
- Lichtenstein GR, Feagan BG, Cohen RD, et al. Serious infections and mortality in association with therapies for Crohn’s disease: TREAT registry. Clin Gastroenterol Hepatol 2006; 4:621–630.
- Peyrin-Biroulet L, Deltenre P, de Suray N, Branche J, Sandborn WJ, Colombel JF. Efficacy and safety of tumor necrosis factor antagonists in Crohn’s disease: meta-analysis of placebo-controlled trials. Clin Gastroenterol Hepatol 2008; 6:644–653.
- Hanauer SB, Feagan BG, Lichtenstein GR, et al; ACCENT I Study Group. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet 2002; 359:1541–1549.
- Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: the CHARM trial. Gastroenterology 2007; 132:52–65.
- Sandborn WJ, Feagan BG, Stoinov S, et al; PRECISE 1 Study Investigators. Certolizumab pegol for the treatment of Crohn’s disease. N Engl J Med 2007; 357:228–238.
- Sandborn WJ, Hanauer SB, Rutgeerts P, et al. Adalimumab for maintenance treatment of Crohn’s disease: results of the CLASSIC II trial. Gut 2007; 56:1232–1239.
- Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: the CLASSIC-I trial. Gastroenterology 2006; 130:323–333.
- Rutgeerts P, D’Haens G, Targan S, et al. Efficacy and safety of retreatment with anti-tumor necrosis factor antibody (infliximab) to maintain remission in Crohn’s disease. Gastroenterology 1999; 117:761–769.
- Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med 2005; 353:2462–2476.
- Pedersen N, Duricova D, Elkjaer M, Gamborg M, Munkholm P, Jess T. Risk of extra-intestinal cancer in inflammatory bowel disease: meta-analysis of population-based cohort studies. Am J Gastroenterol 2010; 105:1480–1487.
- Bernstein CN, Blanchard JF, Kliewer E, Wajda A. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer 2001; 91:854–862.
- Siegel CA, Marden SM, Persing SN, Larson RJ, Sands BE. Risk of lymphoma associated with combination anti-tumor necrosis factor and immunomodulator therapy for the treatment of Crohn’s disease: a meta-analysis. Clin Gastroenterol Hepatol 2009; 7:874–881.
- Wolfe F, Michaud K. Lympyhoma in rheumatoid arthritis. The effect of methotrexate and anti-tumor necrosis factor therapy in 18,572 patients. Arthritis Rheum 2004; 50:1740–1751.
- Parakkal D, Sifuentes H, Semer R, Ehrenpreis ED. Hepatosplenic T-cell lymphoma in patients receiving TNF-a inhibitor therapy: expanding the groups at risk. Eur J Gastroenterol Hepatol 2011; 23:1150–1156.
- Rosh JR, Gross T, Mamula P, Griffiths A, Hyams J. Hepatosplenic T-cell lymphoma in adolescents and young adults with Crohn’s disease: a cautionary tale? Inflamm Bowel Dis 2007; 13:1024–1030.
- Shale M, Kanfer E, Panaccione R, Ghosh S. Hepatosplenic T cell lymphoma in inflammatory bowel disease. Gut 2008; 57:1639–1641.
- Thai A, Prindiville T. Hepatosplenic T-cell lymphoma and inflammatory bowel disease. J Crohns Colitis 2010; 4:511–522.
- Viget N, Vernier-Massouille G, Salmon-Ceron D, Yazdanpanah Y, Colombel JF. Opportunistic infections in patients with inflammatory bowel disease: prevention and diagnosis. Gut 2008; 57:549–558.
- Bekker LG, Freeman S, Murray PJ, Ryffel B, Kaplan G. TNF-alpha controls intracellular mycobacterial growth by both inducible nitric oxide synthase-dependent and inducible nitric oxide synthase-independent pathways. J Immunol 2001; 166:6728–6734.
- Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol 2002; 168:4620–4627.
- Gómez-Reino JJ, Carmona L, Valverde VR, Mola EM, Montero MDBIOBADASER Group. Treatment of rheumatoid arthritis with tumor necrosis factor inhibitors may predispose to significant increase in tuberculosis risk: a multicenter active-surveillance report. Arthritis Rheum 2003; 48:2122–2127.
- Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med 2001; 345:1098–1104.
- Smolen J, Landewé RB, Mease P, et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis 2009; 68:797–804.
- Demkow U, Broniarek-Samson B, Filewska M, et al. Prevalence of latent tuberculosis infection in health care workers in Poland assessed by interferon-gamma whole blood and tuberculin skin tests. J Physiol Pharmacol 2008; 59(suppl 6):209–217.
- Pache I, Rogler G, Felley C. TNF-alpha blockers in inflammatory bowel diseases: practical consensus recommendations and a user’s guide. Swiss Med Wkly 2009; 139:278–287.
- Rahier JF, Ben-Horin S, Chowers Y, et al; European Crohn’s and Colitis Organisation (ECCO). European evidence-based consensus on the prevention, diagnosis and management of opportunistic infections in inflammatory bowel disease. J Crohns Colitis 2009; 3:47–91.
- Rahier JF, Yazdanpanah Y, Colombel JF, Travis S. The European (ECCO) consensus on infection in IBD: what does it change for the clinician? Gut 2009; 58:1313–1315.
- Bergstrom L, Yocum DE, Ampel NM, et al. Increased risk of coccidioidomycosis in patients treated with tumor necrosis factor alpha antagonists. Arthritis Rheum 2004; 50:1959–1966.
- Lee JH, Slifman NR, Gershon SK, et al. Life-threatening histoplasmosis complicating immunotherapy with tumor necrosis factor alpha antagonists infliximab and etanercept. Arthritis Rheum 2002; 46:2565–2570.
- Wood KL, Hage CA, Knox KS, et al. Histoplasmosis after treatment with anti-tumor necrosis factor-alpha therapy. Am J Respir Crit Care Med 2003; 167:1279–1282.
- Reddy JG, Loftus EV. Safety of infliximab and other biologic agents in the inflammatory bowel diseases. Gastroenterol Clin North Am 2006; 35:837–855.
- Esteve M, Saro C, González-Huix F, Suarez F, Forné M, Viver JM. Chronic hepatitis B reactivation following infliximab therapy in Crohn’s disease patients: need for primary prophylaxis. Gut 2004; 53:1363–1365.
- Michel M, Duvoux C, Hezode C, Cherqui D. Fulminant hepatitis after infliximab in a patient with hepatitis B virus treated for an adult onset Still’s disease. J Rheumatol 2003; 30:1624–1625.
- Ostuni P, Botsios C, Punzi L, Sfriso P, Todesco S. Hepatitis B reactivation in a chronic hepatitis B surface antigen carrier with rheumatoid arthritis treated with infliximab and low dose methotrexate. Ann Rheum Dis 2003; 62:686–687.
- Pérez-Alvarez R, Díaz-Lagares C, García-Hernández F, et al; BIOGEAS Study Group. Hepatitis B virus (HBV) reactivation in patients receiving tumor necrosis factor (TNF)-targeted therapy: analysis of 257 cases. Medicine (Baltimore) 2011; 90:359–371.
- Loras C, Gisbert JP, Mínguez M, et al; REPENTINA study; GETECCU (Grupo Español de Enfermedades de Crohn y Colitis Ulcerosa) Group. Liver dysfunction related to hepatitis B and C in patients with inflammatory bowel disease treated with immunosuppressive therapy. Gut 2010; 59:1340–1346.
- Park SH, Yang SK, Lim YS, et al. Clinical courses of chronic hepatitis B virus infection and inflammatory bowel disease in patients with both diseases. Inflamm Bowel Dis 2012; 18:2004–2010.
- Loomba R, Rowley A, Wesley R, et al. Systematic review: the effect of preventive lamivudine on hepatitis B reactivation during chemotherapy. Ann Intern Med 2008; 148:519–528.
- Lok AS, McMahon BJ. Chronic hepatitis B: update 2009. Hepatology 2009; 50:661–662.
- European Association For The Study Of The Liver. EASL Clinical Practice Guidelines: management of chronic hepatitis B. J Hepatol 2009; 50:227–242.
- Watanabe M, Shibuya A, Takada J, et al. Entecavir is an optional agent to prevent hepatitis B virus (HBV) reactivation: a review of 16 patients. Eur J Intern Med 2010; 21:333–337.
- Biancone L, Pavia M, Del Vecchio Blanco G, et al; Italian Group for the Study of the Colon and Rectum (GISC). Hepatitis B and C virus infection in Crohn’s disease. Inflamm Bowel Dis 2001; 7:287–294.
- Melmed GY. Vaccination strategies for patients with inflammatory bowel disease on immunomodulators and biologics. Inflamm Bowel Dis 2009; 15:1410–1416.
- Melmed GY, Ippoliti AF, Papadakis KA, et al. Patients with inflammatory bowel disease are at risk for vaccine-preventable illnesses. Am J Gastroenterol 2006; 101:1834–1840.
- Ferri C, Ferraccioli G, Ferrari D, et al. Safety of anti-tumor necrosis factor-alpha therapy in patients with rheumatoid arthritis and chronic hepatitis C virus infection. J Rheumatol 2008; 35:1944–1949.
- Mok MY, Ng WL, Yuen MF, Wong RW, Lau CS. Safety of disease modifying anti-rheumatic agents in rheumatoid arthritis patients with chronic viral hepatitis. Clin Exp Rheumatol 2000; 18:363–368.
- Vassilopoulos D, Calabrese LH. Risks of immunosuppressive therapies including biologic agents in patients with rheumatic diseases and coexisting chronic viral infections. Curr Opin Rheumatol 2007; 19:619–625.
- Vassilopoulos D, Apostolopoulou A, Hadziyannis E, et al. Long-term safety of anti-TNF treatment in patients with rheumatic diseases and chronic or resolved hepatitis B virus infection. Ann Rheum Dis 2010; 69:1352–1355.
- Saag KG, Teng GG, Patkar NM, et al; American College of Rheumatology. American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Rheum 2008; 59:762–784.
- Cornish J, Tan E, Teare J, et al. A meta-analysis on the influence of inflammatory bowel disease on pregnancy. Gut 2007; 56:830–837.
- Dominitz JA, Young JC, Boyko EJ. Outcomes of infants born to mothers with inflammatory bowel disease: a population-based cohort study. Am J Gastroenterol 2002; 97:641–648.
- Kornfeld D, Cnattingius S, Ekbom A. Pregnancy outcomes in women with inflammatory bowel disease—a population-based cohort study. Am J Obstet Gynecol 1997; 177:942–946.
- Mahadevan U, Sandborn WJ, Li DK, Hakimian S, Kane S, Corley DA. Pregnancy outcomes in women with inflammatory bowel disease: a large community-based study from Northern California. Gastroenterology 2007; 133:1106–1112.
- Nguyen GC, Boudreau H, Harris ML, Maxwell CV. Outcomes of obstetric hospitalizations among women with inflammatory bowel disease in the United States. Clin Gastroenterol Hepatol 2009; 7:329–334.
- Schnitzler F, Fidder H, Ferrante M, et al. Outcome of pregnancy in women with inflammatory bowel disease treated with antitumor necrosis factor therapy. Inflamm Bowel Dis 2011; 17:1846–1854.
- Vinet E, Pineau C, Gordon C, Clarke AE, Bernatsky S. Biologic therapy and pregnancy outcomes in women with rheumatic diseases. Arthritis Rheum 2009; 61:587–592.
- Guidi L, Pugliese D, Armuzzi A. Update on the management of inflammatory bowel disease: specific role of adalimumab. Clin Exp Gastroenterol 2011; 4:163–172.
- Mahadevan U. Continuing immunomodulators and biologic medications in pregnant IBD patients – pro. IInflamm Bowel Dis 2007; 13:1439–1440.
- Mahadevan U. Gastrointestinal medications in pregnancy. Best Pract Res Clin Gastroenterol 2007; 21:849–877.
- Mahadevan U, Cucchiara S, Hyams JS, et al. The London Position Statement of the World Congress of Gastroenterology on Biological Therapy for IBD with the European Crohn’s and Colitis Organisation: pregnancy and pediatrics. Am J Gastroenterol 2011; 106:214–223.
- Mahadevan U, Kane S. Use of infliximab in pregnancy. Am J Gastroenterol 2010; 105:219–220.
- Vasiliauskas EA, Church JA, Silverman N, Barry M, Targan SR, Dubinsky MC. Case report: evidence for transplacental transfer of maternally administered infliximab to the newborn. Clin Gastroenterol Hepatol 2006; 4:1255–1258.
Biologic agents such as those that block tumor necrosis factor (TNF) alpha have revolutionized the treatment of autoimmune diseases like rheumatoid arthritis and inflammatory bowel disease, dramatically improving disease control and quality of life. In addition, they have made true disease remission possible in some cases.
However, as with any new therapy, a variety of side effects must be considered.
WHAT ARE BIOLOGIC AGENTS?
Biologic agents are genetically engineered drugs manufactured or synthesized in vitro from molecules such as proteins, genes, and antibodies present in living organisms. By targeting specific molecular components of the inflammatory cascade, including cytokines, these drugs alter certain aspects of the body’s inflammatory response in autoimmune diseases. Because TNF inhibitors are the most widely used biologic agents in the United States, this review will focus on them.
TNF INHIBITORS
TNF inhibitors suppress the inflammatory cascade by inactivating TNF alpha, a cytokine that promotes inflammation in the intestine, synovial tissue, and other sites.1,2
Several TNF inhibitors are available. Etanercept and infliximab were the first two to receive US Food and Drug Administration (FDA) approval, and they are extensively used. Etanercept, a soluble TNF receptor given subcutaneously, was approved for treating rheumatoid arthritis in 1998. Infliximab, a chimeric monoclonal antibody (75% human and 25% mouse protein sequence) given intravenously, received FDA approval for treating Crohn disease in 1998 and for rheumatoid arthritis in 1999. Other anti-TNF agents with varying properties are also available.
Table 1 lists anti-TNF agents approved for treating rheumatoid arthritis and inflammatory bowel disease. These are chronic, relapsing diseases that significantly and dramatically reduce quality of life, especially when they are poorly controlled.3,4 Untreated disease has been associated with malignancy, infection, pregnancy loss, and malnutrition.5–8 TNF inhibitors are aimed at patients who have moderate to severe disease or for whom previous treatments have failed, to help them achieve and maintain steroid-free remission. However, use of these agents is tempered by the risk of potentially serious side effects.
Special thought should also be given to the direct costs of the drugs (up to $30,000 per year, not counting the cost of their administration) and to the indirect costs such as time away from work to receive treatment. These are major considerations in some cases, and patients should be selected carefully for treatment with these drugs.
BEFORE STARTING THERAPY
Before starting anti-TNF therapy, several steps can reduce the risk of serious adverse events.
Take a focused history
The clinical history should include inquiries about previous bacterial, fungal, and tuberculosis infections or exposure; diabetes; and other immunocompromised states that increase the risk of acquiring potentially life-threatening infections.
Details of particular geographic areas of residence, occupational exposures, and social history should be sought. These include history of incarceration (which may put patients at risk of tuberculosis) and residence in the Ohio River valley or in the southwestern or midwestern United States (which may increase the risk of histoplasmosis, coccidioidomycosis, and other fungal infections).
Bring vaccinations up to date
Age-appropriate vaccinations should be discussed and given, ideally before starting therapy. These include influenza vaccine every year and tetanus boosters every 10 years for all and, as appropriate, varicella, human papillomavirus, and pneumococcal vaccinations. The US Centers for Disease Control and Prevention recommend an additional dose of the pneumococcal vaccine if more than 5 years have elapsed since the first one, and many clinicians opt to give it every 5 years.
In general, live-attenuated vaccines, including the intranasal influenza vaccine, are contraindicated in patients taking biologic agents.9 For patients at high risk of exposure or infection, it may be reasonable to hold the biologic agent for a period of time, vaccinate, and resume the biologic agent a month later.
Recent data suggest that the varicella zoster vaccine may be safely given to older patients with immune-mediated diseases such as rheumatoid arthritis and inflammatory bowel disease taking biologic agents.10 New guidelines from the American College of Rheumatology recommend age-appropriate vaccines for rheumatoid arthritis patients age 60 and older before biologic treatments are started. Case-by-case discussion with the subspecialist and the patient is recommended.
Screen for chronic infections
Tuberculosis screening with a purified protein derivative test or an interferon-gamma-release (Quantiferon) assay followed by chest radiography in patients with a positive test is mandatory before giving a TNF inhibitor.
Hepatitis B virus status should be determined before starting anti-TNF therapy.11
Hepatitis B vaccination has been recommended for patients with inflammatory bowel disease, but no clear recommendation exists for patients with rheumatic disease. Patients with inflammatory bowel disease tend to have low rates of response to hepatitis B vaccination12,13; possible reasons include their lack of an appropriate innate immune response to infectious agents, malnutrition, surgery, older age, and immunosuppressive drugs.14 An accelerated vaccination protocol with recombinant hepatitis B vaccine (Energix-B) in a double dose at 0, 1, and 2 months has been shown to improve response rates.15
Whenever possible, it may be better to vaccinate patients before starting immunosuppressive therapy, and to check postvaccination titers to ensure adequate response.
Perform an examination
A full physical examination with special attention to skin rashes should be performed. This may serve as a baseline to assist early detection of new rashes associated with anti-TNF therapy.
A baseline complete blood cell count and complete metabolic panel should be routinely obtained before starting therapy (and thereafter at the discretion of the physician). In conjunction with follow-up tests, they can help detect an unexpected decrease in white blood cell count or abnormal results on the liver panel.16 These baseline and follow-up tests are generally performed by the subspecialist, and the results are shared with the primary care physician.
Table 2 summarizes key information to be sought before starting a patient on a TNF inhibitor.
ADVERSE EFFECTS OF ANTI-TNF DRUGS
Infusion reactions, infections, cardiac arrhythmias, demyelinating disorders, skin infections, and malignancies have been reported with anti-TNF therapy. The relative frequencies of these adverse events are summarized in Table 3.17–22
NONINFECTIOUS COMPLICATIONS OF TNF INHIBITORS
Injection site reactions
When anti-TNF agents are given subcutaneously, injection site reactions are common (occurring in up to 40% of patients) and are considered minor.11 Reactions, including significant pain, typically occur within the first few months of therapy. They can last 2 to 5 days but rarely warrant stopping therapy. Treatment with ice and an antihistamine is almost always sufficient to control symptoms.
Infusion reactions with infliximab
Infliximab can cause both acute and delayed infusion reactions. Acute reactions can occur up to 24 hours after infusion but usually appear within 10 minutes of administration and are handled by the infusion suite staff. They range from the severe immunoglobulin E-mediated type I reaction, manifesting with hypotension, bronchospasm, and urticaria, to the milder anaphylactoid-type reaction, which constitutes the majority.23–25
While most primary care physicians will not encounter an acute reaction, family doctors and emergency room physicians may encounter delayed reactions, which can develop 1 to 14 days after infusion. These reactions usually resemble serum sickness and present with joint pain, fatigue, myalgia, and fever. But, unlike classic serum sickness, these reactions are generally not associated with a rash.
With nonspecific symptoms, the diagnosis may be easy to overlook. However, establishing this diagnosis is important because repeat therapy may result in a more severe reaction upon reexposure to the drug.
Once diagnosed, these reactions can be treated symptomatically with a combination of acetaminophen and diphenhydramine after discussion between the primary care physician and subspecialist.23,24
Autoimmune syndromes
Several studies have reported a small percentage of patients treated with anti-TNF agents who develop paradoxical autoimmune conditions. These range from asymptomatic immunologic alterations, including the formation of antinuclear antibodies and antibodies to double-stranded DNA, to life-threatening systemic autoimmune diseases.26,27
Autoimmune diseases associated with anti-TNF treatment include a lupus-like syndrome, vasculitides, and psoriatic skin lesions. These syndromes warrant stopping the inciting drug and, on occasion, giving corticosteroids. Most cases arise between 1 month and 1 year of starting treatment, and almost 75% resolve completely after the anti-TNF therapy is stopped.26
Interestingly, anti-TNF agents are approved for treating psoriasis and psoriatic arthritis, but psoriasis has paradoxically developed in patients being treated with these drugs for other autoimmune diseases. The FDA has reviewed 69 cases of new-onset psoriasis with anti-TNF therapy, including 17 pustular and 15 palmoplantar cases. The 12 most severe cases resulted in hospitalization, and symptoms resolved in most after treatment cessation.28
Fiorino et al29 counted 18 reported cases of psoriasis induced by anti-TNF therapy in patients with inflammatory bowel disease and concluded that it is rare. Harrison et al30 reported similar findings in patients with rheumatoid arthritis, with an increased incidence rate of 1.04 per 1,000 person-years. New-onset psoriasis was most common in patients treated with adalimumab. An example of the rash is seen in Figure 1.
CARDIOVASCULAR SIDE EFFECTS
Cardiovascular side effects of anti-TNF agents range from nonspecific and asymptomatic arrhythmias to worsening of heart failure.
Circulating levels of TNF are increased in patients with heart failure, and studies have evaluated the effects of TNF inhibition with infliximab on cardiac function and overall survival.31,32 The combined risk of death from any cause or hospitalization from heart failure was significantly higher in the infliximab groups, and the effects persisted for up to 5 months after stopping therapy.
Other studies22,33 have evaluated the effects of infliximab and etanercept on cardiac function and overall survival. Results showed possible exacerbation of heart failure with etanercept and increased risk of death with infliximab in patients with New York Heart Association (NYHA) class III or IV heart failure and left ventricular ejection fractions less than 35%.
Case reports have also described patients with worsening or new-onset heart failure on TNF inhibitors, including patients younger than 50 years and without identifiable cardiovascular risk factors.
Data analyses31,34,35 from large clinical registries have reported no significant increase in heart failure attributable to TNF inhibitors. However, we have concerns about the methodology of these analyses.
Currently, anti-TNF therapy is contraindicated in patients with NYHA class III or IV heart failure. Data are inconclusive for patients with class I or II heart failure. Baseline echocardiography and cardiology consultation can be considered, with close monitoring and avoidance of high doses of TNF inhibitors. If heart failure develops in a patient on anti-TNF therapy, the drug should be discontinued and the patient should be evaluated further.36
DEMYELINATING DISEASE, INCLUDING MULTIPLE SCLEROSIS
Anti-TNF agents have been associated with the onset or exacerbation of clinical symptoms and radiographic evidence of central nervous system demyelinating disorders, including multiple sclerosis.37–40 Mohan et al38 identified 19 cases of demyelinating events occurring after administration of anti-TNF agents in early 2001. In most cases, symptoms improved or resolved after therapy was stopped.
Optic neuritis,41,42 bilateral optic neuropathy,43 and aseptic meningitis44 have also been reported, but these have occurred only rarely.
How common are these effects? Postmarketing surveillance in patients with rheumatoid arthritis yields an estimated incidence of demyelinating disorders of 1 per 1,000 patient-years with adalimumab therapy.45 Complicating the assessment is an observed slight increase in risk of demyelinating conditions associated with inflammatory bowel disease.
Symptoms that should heighten the physician’s suspicion of this adverse effect include confusion, paresthesias, and ataxia. Patients on anti-TNF therapy who develop new visual symptoms should be checked for painless visual loss as a sign of early demyelinating disease.
Although data that conclusively link anti-TNF agents to multiple sclerosis are lacking, these drugs should not be initiated in patients who have a history of demyelinating disease, and treatment should be stopped promptly if the diagnosis is suspected.
MALIGNANCY
Whether anti-TNF therapy is directly linked to development of malignancies is difficult to determine. There are many confounding factors, including the risk of malignancy in underlying inflammatory disease and the concomitant use of other medications such as thiopurines, which have a known association with lymphoma.46
The incidence of lymphoma is twice as high in rheumatoid arthritis patients as in the general population.47 The risk is higher in those with more aggressive joint disease—the subset of rheumatoid arthritis patients who are more likely to be given anti-TNF agents.5
In patients with inflammatory bowel disease, the risk of cholangiocarcinoma is four times higher, and the risk of small-bowel adenocarcinoma is 16 to 18 times higher, but no increased risk of lymphoma has been identified in this population.48,49
Data from six clinical trials of infliximab, including a long-term study of its safety in Crohn disease, suggest it poses no increase in overall risk of malignancy.50–52 Similar results have been reported in patients with other rheumatic diseases.8 Information on this topic is constantly evolving, and studies range from case series to clinical trials to large patient registries.
The decision to use a TNF inhibitor should be based on the patient’s clinical picture and risk factors. Discussion of the risks and benefits of therapy with the patient should be clearly documented.
Non-Hodgkin lymphoma
Evidence about the risk of lymphoma with anti-TNF use is mixed, as up to two-thirds of patients on anti-TNF therapy have received concomitant nonbiologic immunosuppressive medications, making it difficult to determine the true risk from the biologic agents alone.53 Current evidence both supports9,53–56 and refutes7,55,57–60 the idea that anti-TNF agents increase lymphoma risk.
In patients with inflammatory bowel disease, several population-based studies have not shown a clear increase in lymphoma risk with anti-TNF use.56,59,60 Pedersen et al,61 in a meta-analysis of eight studies, confirmed these findings by showing no overall lymphoma risk in patients with inflammatory bowel disease.
However, a Canadian population-based study found a statistically significant increase in non-Hodgkin lymphoma in males with Crohn disease, with an incidence ratio of 3.63 (95% confidence interval [CI] 1.53–8.62).61 Additionally, Siegel et al62 found a significantly higher risk (6.1 cases per 10,000 patients) in patients treated with anti-TNF agents and thiopurines than in the general population (1.9 cases per 10,000 people). Although the difference was statistically significant, the overall risk is still very low.
Patients with rheumatoid arthritis seem to have a risk of lymphoma two to three times higher than in the general population. However, large population-based studies have not shown a statistically significant increase in the risk of lymphoma with anti-TNF therapy.63
Hepatosplenic T-cell lymphoma is a rare subtype of peripheral T-cell non-Hodgkin lymphoma; 25 cases have been reported in patients receiving anti-TNF therapy.64 Although the risk is extremely low (< 0.05%), physicians must carefully consider the risks and benefits of combination therapy, especially in young male patients with inflammatory bowel disease, since death is the usual outcome of this disease.65–67
Skin cancers
Wolfe and Michaud8 evaluated malignancy risk in rheumatoid arthritis patients being treated with biologic agents, including TNF inhibitors, using a large longitudinal database. These data were compared with those of the US Surveillance, Epidemiology, and End-Results (SEER) national database. No increase in the overall cancer rate was seen in rheumatoid arthritis patients (standardized incidence ratio [SIR] 1.0, 95% CI 1.0–1.1).
However, melanoma was more common in rheumatoid arthritis patients compared with SEER rates (SIR 1.7, 95% CI 1.3–2.3).8 In addition, biologic therapy was associated with a higher (but not statistically significant) risk of melanoma (odds ratio [OR] 2.3, 95% CI 0.9–5.4) and a higher risk of nonmelanoma skin cancer (OR 1.5, 95% CI 1.2–1.8), but not of other types of cancer.8
INFECTION
Patients on anti-TNF therapy are at a higher risk of infection, ranging from minor to life-threatening bacterial infections, and including the reactivation of granulomatous and fungal infections. More importantly, these agents are similar to steroids in blunting signs of infection, which may delay diagnosis and treatment.
The management of infection in patients on anti-TNF medications varies from case to case. In general, patients with a minor infection that does not require hospitalization or intravenous antibiotics can continue the biologic therapy while taking oral antibiotics. TNF inhibitors must be held in the event of a major infection.
Consultation with an infectious disease specialist is recommended, especially in complex cases.
Bacterial infections
An increased risk of minor bacterial infections such as urinary tract and respiratory infections has been well documented in several randomized control trials of anti-TNF agents, though other studies have shown no such increase in risk.33,51–59
The threshold for using antibiotics for a suspected bacterial infection is somewhat shifted in favor of treatment in patients on anti-TNF therapy. The reason is twofold: as previously noted, infections may be worse than they appear, because anti-TNF drugs can mask the signs and symptoms of a serious infection, and in patients on these drugs, an untreated bacterial infection may rapidly become life-threatening.
In general, broad-spectrum antibiotics are not warranted unless the source of infection is unclear or the patient is in danger of hemodynamic compromise.
Opportunistic infections
The association of anti-TNF agents with opportunistic infections could be viewed as an extension of their normal and intended therapeutic activity as potent immunosuppressive agents.68 Rheumatoid arthritis and inflammatory bowel disease are usually associated with conditions and situations that predispose patients to opportunistic infections, such as decreased immune response, malnutrition or malabsorption, surgeries, and concomitant immunosuppressive medications.7 Combination therapy with other immunosuppressive drugs and older age appear to markedly increase the risk of opportunistic infections, including mycobacterial and fungal infections, in patients with inflammatory bowel disease.7
Overall, opportunistic infections represent a measurable risk of anti-TNF therapy, and awareness and vigilance are important, especially in areas where opportunistic infections such as histoplasmosis and coccidiomycosis are endemic.50 Furthermore, physicians must be aware of the higher risk of opportunistic infections when multiple immunosuppressive drugs are used concurrently.
Granulomatous infections such as tuberculosis
Anti-TNF agents increase the risk of de novo granulomatous infections and of reactivating such infections. Granuloma formation and intracellular destruction of mycobacteria depend on TNF. TNF is important in maintaining the anatomic integrity of granulomas where these organisms have been sequestered, and blocking TNF leads to breakdown of granulomas and release of virulent organisms.69,70
TNF inhibitors increase the risk of reactivation of latent tuberculosis infection. The risk is greater with infliximab and adalimumab than with etanercept,71,72 and it has been described with certolizumab.74 Study results are varied thus far but show a risk of tuberculosis reactivation five to 30 times higher than in the general population, with tremendous variability in risk depending on background rates of previous exposure.
The absence of typical tuberculosis symptoms further complicates care in these cases. Fever, weight loss, and night sweats tend to be TNF-mediated and are therefore masked by anti-TNF agents, leading to atypical presentations. In addition, active tuberculosis infection associated with TNF inhibitors is more likely to involve extrapulmonary sites such as the skin and musculoskeletal system and to be disseminated at presentation.
A paradoxical worsening of tuberculosis symptoms may also be seen in patients with latent tuberculosis reactivation, especially after discontinuing anti-TNF therapy. This is thought to result from an immune reconstitution inflammatory syndrome.
The pretreatment evaluation should include a history of risk factors, a physical examination, and either a tuberculin skin test or an interferon-gamma-release assay. Interferon-gamma-release assays are particularly helpful in patients who have received bacille Calmette-Guérin vaccination. In patients who test positive or have been exposed, tuberculosis treatment should begin 4 weeks before starting anti-TNF therapy, though the optimal timing of antituberculosis agents is still controversial.74–77
If tuberculosis develops in a patient on anti-TNF therapy, he or she should receive antituberculosis drugs. Anti-TNF therapy should be stopped and should be resumed after 2 months only if no other treatment option is available.75
Invasive opportunistic fungal infections
Invasive opportunistic fungal infections have been reported with anti-TNF therapy, including histoplasmosis and coccidioidomycosis.78–80 Most patients who had histoplasmosis were treated with other immunosuppressive therapies and resided in or were raised near the Ohio or Mississippi River valleys, where this disease is endemic. Similarly, most cases of coccidioidomycosis were in endemic areas of Arizona, California, and Nevada, and patients on concomitant immunosuppressive therapy.78
Currently, there is no evidence to recommend obtaining Histoplasma capsulatum or Coccidioides immitis serologies before initiating anti-TNF therapy in patients in endemic areas.81 However, patients must be instructed to seek medical attention quickly for pulmonary or febrile illnesses.
Viral hepatitis infections
The data on hepatitis B and hepatitis C in patients on biologic therapies are mostly limited to case reports.
Hepatitis B. A small prospective study from Spain followed the liver biochemistry tests and hepatitis B status of 80 patients with Crohn disease treated with infliximab. Of three patients who were chronic hepatitis B carriers before starting infliximab, two experienced reactivation of hepatitis B after discontinuing infliximab, and one ultimately died. The third patient was treated with lamivudine concurrently with infliximab without clinical or biochemical changes during or after therapy.82
Similar findings were observed in two patients with rheumatoid arthritis on treatment with infliximab. One of the patients required liver transplantation, and both were treated with lamivudine, resulting in normalization of liver function test results.83,84
Recent reviews indicate that despite these findings, hepatitis B reactivation after anti-TNF withdrawal may not be common.85 There are limited data on hepatitis B reactivation and associated liver dysfunction in patients with inflammatory bowel disease treated with immunosuppressants. In a retrospective multicenter trial by Loras et al86 in patients with inflammatory bowel disease who had viral hepatitis, 36% of patients positive for hepatitis B surface antigen developed liver dysfunction, and six patients developed liver failure. In that study, treatment with more than two immunosuppressants was an independent predictor of hepatitis B reactivation.
Prolonged immunosuppression (longer than 3 months) has also been identified as an independent predictor of liver dysfunction (OR 3.06; 95% 95% CI 1.02–9.16).87
The European Association for the Study of the Liver and the American Association for the Study of Liver Diseases recommend starting lamivudine before chemotherapy or immunomodulator or immunosuppressive therapy in hepatitis B virus carriers and continuing preventive treatment for at least 6 months after stopping immunomodulating drugs. Lamivudine at a dose of 100 mg/day may reduce the risk of reactivation of hepatitis B.88–90 Tenofovir and entecavir may be useful alternatives in patients with hepatitis B who have never received nucleoside analogues. Hepatitis B reactivation did not occur in any of the 16 patients who received preventive entecavir treatment while receiving immunosuppressive treatments.89,91
In patients receiving immunosuppressive therapy, hepatitis B reactivation is associated with significant morbidity and mortality. Although risk factors for reactivation of hepatitis B virus infection have been identified, we recommend preventive treatment for all carriers positive for hepatitis B surface antigen. This should be done regardless of the number, type, and dosage of immunosuppressants and regardless of hepatitis B virus DNA levels.90
The frequency of hepatitis B and hepatitis C infection in patients with Crohn disease has been reported to be as high as 24%. The high incidence is thought to be secondary to multiple blood transfusions and surgeries.92 The use of biologic agents, including anti-TNF agents, in chronic hepatitis B virus- or hepatitis C virus-infected patients can lead to enhanced viral replication and hepatitis exacerbation.
Although active viral replication can occur during treatment with biologic agents, reactivation or exacerbation can also occur after the anti-TNF agent is stopped.82 This finding has prompted the recommendation that all candidates for biologic therapy be tested for hepatitis B immunization status, followed by immunization in nonimmune patients before starting anti-TNF therapy.93,94
Hepatitis C. There are no guidelines that adequately address the use of anti-TNF agents in patients with chronic hepatitis C infection.
Several small retrospective studies in rheumatoid arthritis patients with hepatitis C have shown that TNF inhibitors can be safely used without worsening liver function tests or changing the viral load.95–98 This is reassuring and provides the subspecialist with another treatment option, as other therapies such as disease-modifying antirheumatic drugs and steroids are known to aggravate viral hepatitis and increase the risk of viremia.96
Although small retrospective studies and one large randomized double-blind placebo-controlled trial have shown TNF inhibitors to be relatively safe in rheumatoid arthritis patients with hepatitis C, their use in these patients should be considered only with caution if they have evidence of hepatic synthetic dysfunction (eg, hypoalbuminemia, thrombocytopenia, increased international normalized ratio). The American College of Rheumatology recommends avoiding TNF inhibitors in Child-Pugh classes B and C.99
PREGNANCY
Physicians caring for patients with rheumatoid arthritis and inflammatory bowel disease must be aware of how these diseases affect fecundity and fertility and how the medications can affect conception and pregnancy. Many patients have difficulty conceiving while their autoimmune disease is active, and better disease control may improve fecundity and result in unanticipated pregnancy. Patients should be advised of the need for contraception if pregnancy is ill-advised or undesired.
Many patients seek advice about teratogenicity before conceiving, or seek guidance about rheumatoid arthritis and inflammatory bowel disease treatment while pregnant.
Several studies have reported a higher risk of adverse pregnancy outcomes in patients with rheumatoid arthritis and inflammatory bowel disease than in the general population.99–105 In inflammatory bowel disease, the odds of a premature delivery or having a low-birth-weight child are twice as high as in the normal population.100,103 Higher rates of cesarean delivery and stillbirth have also been reported. The main predisposing factor appears to be the disease activity at the time of conception, as active disease seems to be linked to adverse pregnancy outcomes.
Treatment with anti-TNF agents may rapidly achieve and maintain remission, raising the question of the safety of continued anti-TNF use during pregnancy. The FDA classifies anti-TNF agents as category B drugs, as animal studies have not demonstrated fetal risk, and no well-controlled prospective study has yet been conducted with pregnant women.
In an observational study, Schnitzler et al105 assessed the outcomes of 42 pregnancies in 35 patients with inflammatory bowel disease receiving either infliximab or adalimumab during pregnancy, compared with 56 pregnancies in 45 healthy patients without inflammatory bowel disease. There was no statistical difference in abortion rates between patients receiving anti-TNF agents and healthy women without inflammatory bowel disease (21% vs 14%, P = .4234). There was also no significant difference observed in birth weight, birth length, or cranial circumference of the children between the two groups. However, pregnancies with direct exposure to anti-TNF agents resulted in a higher frequency of premature delivery (25% vs 6%, P = .023).
Similar results were noted from the Crohn’s Therapy, Resource, Evaluation, and Assessment Tool, or TREAT, registry, as well as from a large systematic review by Vinet et al106 and case reports of rheumatoid arthritis and inflammatory bowel disease in women exposed to anti-TNF agents during pregnancy.
In addition, results from a recent systematic review of 38 studies of anti-TNF use and fetal risk, with a total of 437 women (189 on infliximab, 230 on adalimumab, 18 on certolizumab pegol), showed similar results.107 In pregnancies exposed to anti-TNF agents, the rates of congenital abnormalities (3.4%), fetal deaths (8.5%), and preterm births (2.7%) were similar to those in the general population.
For patients in disease remission on TNF inhibitors, it is reasonable to continue these agents during pregnancy after careful discussion with the patient. Fetal safety and infant immunization response after delivery are the primary concerns in these cases.
Both infliximab and adalimumab cross the placenta and remain detectable in the baby’s circulation 4 months (for adalimumab) to 6 months (for infliximab) after delivery. It is currently recommended that infliximab be stopped at 32 weeks of gestation for the remainder of the pregnancy and that adalimumab be stopped at 34 to 36 weeks, given a planned 40-week gestation.
Certolizumab does not cross the placenta in significant amounts and should be continued throughout pregnancy; drug levels in infants were shown to be less than 2 μg/mL, even when dosed the week of delivery.108–112
TAKE-HOME POINTS
- All health care providers of patients with rheumatoid arthritis and inflammatory bowel disease should be familiar with anti-TNF agents used in treating these diseases.
- The benefits of controlling the disease far outweigh the risks of therapy when used appropriately.
- Care of these patients should be multidisciplinary, with clear communication between primary care physician and specialist.
- Patient education and monitoring combined with prompt communication between primary care physician and specialist are key.
Biologic agents such as those that block tumor necrosis factor (TNF) alpha have revolutionized the treatment of autoimmune diseases like rheumatoid arthritis and inflammatory bowel disease, dramatically improving disease control and quality of life. In addition, they have made true disease remission possible in some cases.
However, as with any new therapy, a variety of side effects must be considered.
WHAT ARE BIOLOGIC AGENTS?
Biologic agents are genetically engineered drugs manufactured or synthesized in vitro from molecules such as proteins, genes, and antibodies present in living organisms. By targeting specific molecular components of the inflammatory cascade, including cytokines, these drugs alter certain aspects of the body’s inflammatory response in autoimmune diseases. Because TNF inhibitors are the most widely used biologic agents in the United States, this review will focus on them.
TNF INHIBITORS
TNF inhibitors suppress the inflammatory cascade by inactivating TNF alpha, a cytokine that promotes inflammation in the intestine, synovial tissue, and other sites.1,2
Several TNF inhibitors are available. Etanercept and infliximab were the first two to receive US Food and Drug Administration (FDA) approval, and they are extensively used. Etanercept, a soluble TNF receptor given subcutaneously, was approved for treating rheumatoid arthritis in 1998. Infliximab, a chimeric monoclonal antibody (75% human and 25% mouse protein sequence) given intravenously, received FDA approval for treating Crohn disease in 1998 and for rheumatoid arthritis in 1999. Other anti-TNF agents with varying properties are also available.
Table 1 lists anti-TNF agents approved for treating rheumatoid arthritis and inflammatory bowel disease. These are chronic, relapsing diseases that significantly and dramatically reduce quality of life, especially when they are poorly controlled.3,4 Untreated disease has been associated with malignancy, infection, pregnancy loss, and malnutrition.5–8 TNF inhibitors are aimed at patients who have moderate to severe disease or for whom previous treatments have failed, to help them achieve and maintain steroid-free remission. However, use of these agents is tempered by the risk of potentially serious side effects.
Special thought should also be given to the direct costs of the drugs (up to $30,000 per year, not counting the cost of their administration) and to the indirect costs such as time away from work to receive treatment. These are major considerations in some cases, and patients should be selected carefully for treatment with these drugs.
BEFORE STARTING THERAPY
Before starting anti-TNF therapy, several steps can reduce the risk of serious adverse events.
Take a focused history
The clinical history should include inquiries about previous bacterial, fungal, and tuberculosis infections or exposure; diabetes; and other immunocompromised states that increase the risk of acquiring potentially life-threatening infections.
Details of particular geographic areas of residence, occupational exposures, and social history should be sought. These include history of incarceration (which may put patients at risk of tuberculosis) and residence in the Ohio River valley or in the southwestern or midwestern United States (which may increase the risk of histoplasmosis, coccidioidomycosis, and other fungal infections).
Bring vaccinations up to date
Age-appropriate vaccinations should be discussed and given, ideally before starting therapy. These include influenza vaccine every year and tetanus boosters every 10 years for all and, as appropriate, varicella, human papillomavirus, and pneumococcal vaccinations. The US Centers for Disease Control and Prevention recommend an additional dose of the pneumococcal vaccine if more than 5 years have elapsed since the first one, and many clinicians opt to give it every 5 years.
In general, live-attenuated vaccines, including the intranasal influenza vaccine, are contraindicated in patients taking biologic agents.9 For patients at high risk of exposure or infection, it may be reasonable to hold the biologic agent for a period of time, vaccinate, and resume the biologic agent a month later.
Recent data suggest that the varicella zoster vaccine may be safely given to older patients with immune-mediated diseases such as rheumatoid arthritis and inflammatory bowel disease taking biologic agents.10 New guidelines from the American College of Rheumatology recommend age-appropriate vaccines for rheumatoid arthritis patients age 60 and older before biologic treatments are started. Case-by-case discussion with the subspecialist and the patient is recommended.
Screen for chronic infections
Tuberculosis screening with a purified protein derivative test or an interferon-gamma-release (Quantiferon) assay followed by chest radiography in patients with a positive test is mandatory before giving a TNF inhibitor.
Hepatitis B virus status should be determined before starting anti-TNF therapy.11
Hepatitis B vaccination has been recommended for patients with inflammatory bowel disease, but no clear recommendation exists for patients with rheumatic disease. Patients with inflammatory bowel disease tend to have low rates of response to hepatitis B vaccination12,13; possible reasons include their lack of an appropriate innate immune response to infectious agents, malnutrition, surgery, older age, and immunosuppressive drugs.14 An accelerated vaccination protocol with recombinant hepatitis B vaccine (Energix-B) in a double dose at 0, 1, and 2 months has been shown to improve response rates.15
Whenever possible, it may be better to vaccinate patients before starting immunosuppressive therapy, and to check postvaccination titers to ensure adequate response.
Perform an examination
A full physical examination with special attention to skin rashes should be performed. This may serve as a baseline to assist early detection of new rashes associated with anti-TNF therapy.
A baseline complete blood cell count and complete metabolic panel should be routinely obtained before starting therapy (and thereafter at the discretion of the physician). In conjunction with follow-up tests, they can help detect an unexpected decrease in white blood cell count or abnormal results on the liver panel.16 These baseline and follow-up tests are generally performed by the subspecialist, and the results are shared with the primary care physician.
Table 2 summarizes key information to be sought before starting a patient on a TNF inhibitor.
ADVERSE EFFECTS OF ANTI-TNF DRUGS
Infusion reactions, infections, cardiac arrhythmias, demyelinating disorders, skin infections, and malignancies have been reported with anti-TNF therapy. The relative frequencies of these adverse events are summarized in Table 3.17–22
NONINFECTIOUS COMPLICATIONS OF TNF INHIBITORS
Injection site reactions
When anti-TNF agents are given subcutaneously, injection site reactions are common (occurring in up to 40% of patients) and are considered minor.11 Reactions, including significant pain, typically occur within the first few months of therapy. They can last 2 to 5 days but rarely warrant stopping therapy. Treatment with ice and an antihistamine is almost always sufficient to control symptoms.
Infusion reactions with infliximab
Infliximab can cause both acute and delayed infusion reactions. Acute reactions can occur up to 24 hours after infusion but usually appear within 10 minutes of administration and are handled by the infusion suite staff. They range from the severe immunoglobulin E-mediated type I reaction, manifesting with hypotension, bronchospasm, and urticaria, to the milder anaphylactoid-type reaction, which constitutes the majority.23–25
While most primary care physicians will not encounter an acute reaction, family doctors and emergency room physicians may encounter delayed reactions, which can develop 1 to 14 days after infusion. These reactions usually resemble serum sickness and present with joint pain, fatigue, myalgia, and fever. But, unlike classic serum sickness, these reactions are generally not associated with a rash.
With nonspecific symptoms, the diagnosis may be easy to overlook. However, establishing this diagnosis is important because repeat therapy may result in a more severe reaction upon reexposure to the drug.
Once diagnosed, these reactions can be treated symptomatically with a combination of acetaminophen and diphenhydramine after discussion between the primary care physician and subspecialist.23,24
Autoimmune syndromes
Several studies have reported a small percentage of patients treated with anti-TNF agents who develop paradoxical autoimmune conditions. These range from asymptomatic immunologic alterations, including the formation of antinuclear antibodies and antibodies to double-stranded DNA, to life-threatening systemic autoimmune diseases.26,27
Autoimmune diseases associated with anti-TNF treatment include a lupus-like syndrome, vasculitides, and psoriatic skin lesions. These syndromes warrant stopping the inciting drug and, on occasion, giving corticosteroids. Most cases arise between 1 month and 1 year of starting treatment, and almost 75% resolve completely after the anti-TNF therapy is stopped.26
Interestingly, anti-TNF agents are approved for treating psoriasis and psoriatic arthritis, but psoriasis has paradoxically developed in patients being treated with these drugs for other autoimmune diseases. The FDA has reviewed 69 cases of new-onset psoriasis with anti-TNF therapy, including 17 pustular and 15 palmoplantar cases. The 12 most severe cases resulted in hospitalization, and symptoms resolved in most after treatment cessation.28
Fiorino et al29 counted 18 reported cases of psoriasis induced by anti-TNF therapy in patients with inflammatory bowel disease and concluded that it is rare. Harrison et al30 reported similar findings in patients with rheumatoid arthritis, with an increased incidence rate of 1.04 per 1,000 person-years. New-onset psoriasis was most common in patients treated with adalimumab. An example of the rash is seen in Figure 1.
CARDIOVASCULAR SIDE EFFECTS
Cardiovascular side effects of anti-TNF agents range from nonspecific and asymptomatic arrhythmias to worsening of heart failure.
Circulating levels of TNF are increased in patients with heart failure, and studies have evaluated the effects of TNF inhibition with infliximab on cardiac function and overall survival.31,32 The combined risk of death from any cause or hospitalization from heart failure was significantly higher in the infliximab groups, and the effects persisted for up to 5 months after stopping therapy.
Other studies22,33 have evaluated the effects of infliximab and etanercept on cardiac function and overall survival. Results showed possible exacerbation of heart failure with etanercept and increased risk of death with infliximab in patients with New York Heart Association (NYHA) class III or IV heart failure and left ventricular ejection fractions less than 35%.
Case reports have also described patients with worsening or new-onset heart failure on TNF inhibitors, including patients younger than 50 years and without identifiable cardiovascular risk factors.
Data analyses31,34,35 from large clinical registries have reported no significant increase in heart failure attributable to TNF inhibitors. However, we have concerns about the methodology of these analyses.
Currently, anti-TNF therapy is contraindicated in patients with NYHA class III or IV heart failure. Data are inconclusive for patients with class I or II heart failure. Baseline echocardiography and cardiology consultation can be considered, with close monitoring and avoidance of high doses of TNF inhibitors. If heart failure develops in a patient on anti-TNF therapy, the drug should be discontinued and the patient should be evaluated further.36
DEMYELINATING DISEASE, INCLUDING MULTIPLE SCLEROSIS
Anti-TNF agents have been associated with the onset or exacerbation of clinical symptoms and radiographic evidence of central nervous system demyelinating disorders, including multiple sclerosis.37–40 Mohan et al38 identified 19 cases of demyelinating events occurring after administration of anti-TNF agents in early 2001. In most cases, symptoms improved or resolved after therapy was stopped.
Optic neuritis,41,42 bilateral optic neuropathy,43 and aseptic meningitis44 have also been reported, but these have occurred only rarely.
How common are these effects? Postmarketing surveillance in patients with rheumatoid arthritis yields an estimated incidence of demyelinating disorders of 1 per 1,000 patient-years with adalimumab therapy.45 Complicating the assessment is an observed slight increase in risk of demyelinating conditions associated with inflammatory bowel disease.
Symptoms that should heighten the physician’s suspicion of this adverse effect include confusion, paresthesias, and ataxia. Patients on anti-TNF therapy who develop new visual symptoms should be checked for painless visual loss as a sign of early demyelinating disease.
Although data that conclusively link anti-TNF agents to multiple sclerosis are lacking, these drugs should not be initiated in patients who have a history of demyelinating disease, and treatment should be stopped promptly if the diagnosis is suspected.
MALIGNANCY
Whether anti-TNF therapy is directly linked to development of malignancies is difficult to determine. There are many confounding factors, including the risk of malignancy in underlying inflammatory disease and the concomitant use of other medications such as thiopurines, which have a known association with lymphoma.46
The incidence of lymphoma is twice as high in rheumatoid arthritis patients as in the general population.47 The risk is higher in those with more aggressive joint disease—the subset of rheumatoid arthritis patients who are more likely to be given anti-TNF agents.5
In patients with inflammatory bowel disease, the risk of cholangiocarcinoma is four times higher, and the risk of small-bowel adenocarcinoma is 16 to 18 times higher, but no increased risk of lymphoma has been identified in this population.48,49
Data from six clinical trials of infliximab, including a long-term study of its safety in Crohn disease, suggest it poses no increase in overall risk of malignancy.50–52 Similar results have been reported in patients with other rheumatic diseases.8 Information on this topic is constantly evolving, and studies range from case series to clinical trials to large patient registries.
The decision to use a TNF inhibitor should be based on the patient’s clinical picture and risk factors. Discussion of the risks and benefits of therapy with the patient should be clearly documented.
Non-Hodgkin lymphoma
Evidence about the risk of lymphoma with anti-TNF use is mixed, as up to two-thirds of patients on anti-TNF therapy have received concomitant nonbiologic immunosuppressive medications, making it difficult to determine the true risk from the biologic agents alone.53 Current evidence both supports9,53–56 and refutes7,55,57–60 the idea that anti-TNF agents increase lymphoma risk.
In patients with inflammatory bowel disease, several population-based studies have not shown a clear increase in lymphoma risk with anti-TNF use.56,59,60 Pedersen et al,61 in a meta-analysis of eight studies, confirmed these findings by showing no overall lymphoma risk in patients with inflammatory bowel disease.
However, a Canadian population-based study found a statistically significant increase in non-Hodgkin lymphoma in males with Crohn disease, with an incidence ratio of 3.63 (95% confidence interval [CI] 1.53–8.62).61 Additionally, Siegel et al62 found a significantly higher risk (6.1 cases per 10,000 patients) in patients treated with anti-TNF agents and thiopurines than in the general population (1.9 cases per 10,000 people). Although the difference was statistically significant, the overall risk is still very low.
Patients with rheumatoid arthritis seem to have a risk of lymphoma two to three times higher than in the general population. However, large population-based studies have not shown a statistically significant increase in the risk of lymphoma with anti-TNF therapy.63
Hepatosplenic T-cell lymphoma is a rare subtype of peripheral T-cell non-Hodgkin lymphoma; 25 cases have been reported in patients receiving anti-TNF therapy.64 Although the risk is extremely low (< 0.05%), physicians must carefully consider the risks and benefits of combination therapy, especially in young male patients with inflammatory bowel disease, since death is the usual outcome of this disease.65–67
Skin cancers
Wolfe and Michaud8 evaluated malignancy risk in rheumatoid arthritis patients being treated with biologic agents, including TNF inhibitors, using a large longitudinal database. These data were compared with those of the US Surveillance, Epidemiology, and End-Results (SEER) national database. No increase in the overall cancer rate was seen in rheumatoid arthritis patients (standardized incidence ratio [SIR] 1.0, 95% CI 1.0–1.1).
However, melanoma was more common in rheumatoid arthritis patients compared with SEER rates (SIR 1.7, 95% CI 1.3–2.3).8 In addition, biologic therapy was associated with a higher (but not statistically significant) risk of melanoma (odds ratio [OR] 2.3, 95% CI 0.9–5.4) and a higher risk of nonmelanoma skin cancer (OR 1.5, 95% CI 1.2–1.8), but not of other types of cancer.8
INFECTION
Patients on anti-TNF therapy are at a higher risk of infection, ranging from minor to life-threatening bacterial infections, and including the reactivation of granulomatous and fungal infections. More importantly, these agents are similar to steroids in blunting signs of infection, which may delay diagnosis and treatment.
The management of infection in patients on anti-TNF medications varies from case to case. In general, patients with a minor infection that does not require hospitalization or intravenous antibiotics can continue the biologic therapy while taking oral antibiotics. TNF inhibitors must be held in the event of a major infection.
Consultation with an infectious disease specialist is recommended, especially in complex cases.
Bacterial infections
An increased risk of minor bacterial infections such as urinary tract and respiratory infections has been well documented in several randomized control trials of anti-TNF agents, though other studies have shown no such increase in risk.33,51–59
The threshold for using antibiotics for a suspected bacterial infection is somewhat shifted in favor of treatment in patients on anti-TNF therapy. The reason is twofold: as previously noted, infections may be worse than they appear, because anti-TNF drugs can mask the signs and symptoms of a serious infection, and in patients on these drugs, an untreated bacterial infection may rapidly become life-threatening.
In general, broad-spectrum antibiotics are not warranted unless the source of infection is unclear or the patient is in danger of hemodynamic compromise.
Opportunistic infections
The association of anti-TNF agents with opportunistic infections could be viewed as an extension of their normal and intended therapeutic activity as potent immunosuppressive agents.68 Rheumatoid arthritis and inflammatory bowel disease are usually associated with conditions and situations that predispose patients to opportunistic infections, such as decreased immune response, malnutrition or malabsorption, surgeries, and concomitant immunosuppressive medications.7 Combination therapy with other immunosuppressive drugs and older age appear to markedly increase the risk of opportunistic infections, including mycobacterial and fungal infections, in patients with inflammatory bowel disease.7
Overall, opportunistic infections represent a measurable risk of anti-TNF therapy, and awareness and vigilance are important, especially in areas where opportunistic infections such as histoplasmosis and coccidiomycosis are endemic.50 Furthermore, physicians must be aware of the higher risk of opportunistic infections when multiple immunosuppressive drugs are used concurrently.
Granulomatous infections such as tuberculosis
Anti-TNF agents increase the risk of de novo granulomatous infections and of reactivating such infections. Granuloma formation and intracellular destruction of mycobacteria depend on TNF. TNF is important in maintaining the anatomic integrity of granulomas where these organisms have been sequestered, and blocking TNF leads to breakdown of granulomas and release of virulent organisms.69,70
TNF inhibitors increase the risk of reactivation of latent tuberculosis infection. The risk is greater with infliximab and adalimumab than with etanercept,71,72 and it has been described with certolizumab.74 Study results are varied thus far but show a risk of tuberculosis reactivation five to 30 times higher than in the general population, with tremendous variability in risk depending on background rates of previous exposure.
The absence of typical tuberculosis symptoms further complicates care in these cases. Fever, weight loss, and night sweats tend to be TNF-mediated and are therefore masked by anti-TNF agents, leading to atypical presentations. In addition, active tuberculosis infection associated with TNF inhibitors is more likely to involve extrapulmonary sites such as the skin and musculoskeletal system and to be disseminated at presentation.
A paradoxical worsening of tuberculosis symptoms may also be seen in patients with latent tuberculosis reactivation, especially after discontinuing anti-TNF therapy. This is thought to result from an immune reconstitution inflammatory syndrome.
The pretreatment evaluation should include a history of risk factors, a physical examination, and either a tuberculin skin test or an interferon-gamma-release assay. Interferon-gamma-release assays are particularly helpful in patients who have received bacille Calmette-Guérin vaccination. In patients who test positive or have been exposed, tuberculosis treatment should begin 4 weeks before starting anti-TNF therapy, though the optimal timing of antituberculosis agents is still controversial.74–77
If tuberculosis develops in a patient on anti-TNF therapy, he or she should receive antituberculosis drugs. Anti-TNF therapy should be stopped and should be resumed after 2 months only if no other treatment option is available.75
Invasive opportunistic fungal infections
Invasive opportunistic fungal infections have been reported with anti-TNF therapy, including histoplasmosis and coccidioidomycosis.78–80 Most patients who had histoplasmosis were treated with other immunosuppressive therapies and resided in or were raised near the Ohio or Mississippi River valleys, where this disease is endemic. Similarly, most cases of coccidioidomycosis were in endemic areas of Arizona, California, and Nevada, and patients on concomitant immunosuppressive therapy.78
Currently, there is no evidence to recommend obtaining Histoplasma capsulatum or Coccidioides immitis serologies before initiating anti-TNF therapy in patients in endemic areas.81 However, patients must be instructed to seek medical attention quickly for pulmonary or febrile illnesses.
Viral hepatitis infections
The data on hepatitis B and hepatitis C in patients on biologic therapies are mostly limited to case reports.
Hepatitis B. A small prospective study from Spain followed the liver biochemistry tests and hepatitis B status of 80 patients with Crohn disease treated with infliximab. Of three patients who were chronic hepatitis B carriers before starting infliximab, two experienced reactivation of hepatitis B after discontinuing infliximab, and one ultimately died. The third patient was treated with lamivudine concurrently with infliximab without clinical or biochemical changes during or after therapy.82
Similar findings were observed in two patients with rheumatoid arthritis on treatment with infliximab. One of the patients required liver transplantation, and both were treated with lamivudine, resulting in normalization of liver function test results.83,84
Recent reviews indicate that despite these findings, hepatitis B reactivation after anti-TNF withdrawal may not be common.85 There are limited data on hepatitis B reactivation and associated liver dysfunction in patients with inflammatory bowel disease treated with immunosuppressants. In a retrospective multicenter trial by Loras et al86 in patients with inflammatory bowel disease who had viral hepatitis, 36% of patients positive for hepatitis B surface antigen developed liver dysfunction, and six patients developed liver failure. In that study, treatment with more than two immunosuppressants was an independent predictor of hepatitis B reactivation.
Prolonged immunosuppression (longer than 3 months) has also been identified as an independent predictor of liver dysfunction (OR 3.06; 95% 95% CI 1.02–9.16).87
The European Association for the Study of the Liver and the American Association for the Study of Liver Diseases recommend starting lamivudine before chemotherapy or immunomodulator or immunosuppressive therapy in hepatitis B virus carriers and continuing preventive treatment for at least 6 months after stopping immunomodulating drugs. Lamivudine at a dose of 100 mg/day may reduce the risk of reactivation of hepatitis B.88–90 Tenofovir and entecavir may be useful alternatives in patients with hepatitis B who have never received nucleoside analogues. Hepatitis B reactivation did not occur in any of the 16 patients who received preventive entecavir treatment while receiving immunosuppressive treatments.89,91
In patients receiving immunosuppressive therapy, hepatitis B reactivation is associated with significant morbidity and mortality. Although risk factors for reactivation of hepatitis B virus infection have been identified, we recommend preventive treatment for all carriers positive for hepatitis B surface antigen. This should be done regardless of the number, type, and dosage of immunosuppressants and regardless of hepatitis B virus DNA levels.90
The frequency of hepatitis B and hepatitis C infection in patients with Crohn disease has been reported to be as high as 24%. The high incidence is thought to be secondary to multiple blood transfusions and surgeries.92 The use of biologic agents, including anti-TNF agents, in chronic hepatitis B virus- or hepatitis C virus-infected patients can lead to enhanced viral replication and hepatitis exacerbation.
Although active viral replication can occur during treatment with biologic agents, reactivation or exacerbation can also occur after the anti-TNF agent is stopped.82 This finding has prompted the recommendation that all candidates for biologic therapy be tested for hepatitis B immunization status, followed by immunization in nonimmune patients before starting anti-TNF therapy.93,94
Hepatitis C. There are no guidelines that adequately address the use of anti-TNF agents in patients with chronic hepatitis C infection.
Several small retrospective studies in rheumatoid arthritis patients with hepatitis C have shown that TNF inhibitors can be safely used without worsening liver function tests or changing the viral load.95–98 This is reassuring and provides the subspecialist with another treatment option, as other therapies such as disease-modifying antirheumatic drugs and steroids are known to aggravate viral hepatitis and increase the risk of viremia.96
Although small retrospective studies and one large randomized double-blind placebo-controlled trial have shown TNF inhibitors to be relatively safe in rheumatoid arthritis patients with hepatitis C, their use in these patients should be considered only with caution if they have evidence of hepatic synthetic dysfunction (eg, hypoalbuminemia, thrombocytopenia, increased international normalized ratio). The American College of Rheumatology recommends avoiding TNF inhibitors in Child-Pugh classes B and C.99
PREGNANCY
Physicians caring for patients with rheumatoid arthritis and inflammatory bowel disease must be aware of how these diseases affect fecundity and fertility and how the medications can affect conception and pregnancy. Many patients have difficulty conceiving while their autoimmune disease is active, and better disease control may improve fecundity and result in unanticipated pregnancy. Patients should be advised of the need for contraception if pregnancy is ill-advised or undesired.
Many patients seek advice about teratogenicity before conceiving, or seek guidance about rheumatoid arthritis and inflammatory bowel disease treatment while pregnant.
Several studies have reported a higher risk of adverse pregnancy outcomes in patients with rheumatoid arthritis and inflammatory bowel disease than in the general population.99–105 In inflammatory bowel disease, the odds of a premature delivery or having a low-birth-weight child are twice as high as in the normal population.100,103 Higher rates of cesarean delivery and stillbirth have also been reported. The main predisposing factor appears to be the disease activity at the time of conception, as active disease seems to be linked to adverse pregnancy outcomes.
Treatment with anti-TNF agents may rapidly achieve and maintain remission, raising the question of the safety of continued anti-TNF use during pregnancy. The FDA classifies anti-TNF agents as category B drugs, as animal studies have not demonstrated fetal risk, and no well-controlled prospective study has yet been conducted with pregnant women.
In an observational study, Schnitzler et al105 assessed the outcomes of 42 pregnancies in 35 patients with inflammatory bowel disease receiving either infliximab or adalimumab during pregnancy, compared with 56 pregnancies in 45 healthy patients without inflammatory bowel disease. There was no statistical difference in abortion rates between patients receiving anti-TNF agents and healthy women without inflammatory bowel disease (21% vs 14%, P = .4234). There was also no significant difference observed in birth weight, birth length, or cranial circumference of the children between the two groups. However, pregnancies with direct exposure to anti-TNF agents resulted in a higher frequency of premature delivery (25% vs 6%, P = .023).
Similar results were noted from the Crohn’s Therapy, Resource, Evaluation, and Assessment Tool, or TREAT, registry, as well as from a large systematic review by Vinet et al106 and case reports of rheumatoid arthritis and inflammatory bowel disease in women exposed to anti-TNF agents during pregnancy.
In addition, results from a recent systematic review of 38 studies of anti-TNF use and fetal risk, with a total of 437 women (189 on infliximab, 230 on adalimumab, 18 on certolizumab pegol), showed similar results.107 In pregnancies exposed to anti-TNF agents, the rates of congenital abnormalities (3.4%), fetal deaths (8.5%), and preterm births (2.7%) were similar to those in the general population.
For patients in disease remission on TNF inhibitors, it is reasonable to continue these agents during pregnancy after careful discussion with the patient. Fetal safety and infant immunization response after delivery are the primary concerns in these cases.
Both infliximab and adalimumab cross the placenta and remain detectable in the baby’s circulation 4 months (for adalimumab) to 6 months (for infliximab) after delivery. It is currently recommended that infliximab be stopped at 32 weeks of gestation for the remainder of the pregnancy and that adalimumab be stopped at 34 to 36 weeks, given a planned 40-week gestation.
Certolizumab does not cross the placenta in significant amounts and should be continued throughout pregnancy; drug levels in infants were shown to be less than 2 μg/mL, even when dosed the week of delivery.108–112
TAKE-HOME POINTS
- All health care providers of patients with rheumatoid arthritis and inflammatory bowel disease should be familiar with anti-TNF agents used in treating these diseases.
- The benefits of controlling the disease far outweigh the risks of therapy when used appropriately.
- Care of these patients should be multidisciplinary, with clear communication between primary care physician and specialist.
- Patient education and monitoring combined with prompt communication between primary care physician and specialist are key.
- Wijbrandts CA, Dijkgraaf MG, Kraan MC, et al. The clinical response to infliximab in rheumatoid arthritis is in part dependent on pretreatment tumour necrosis factor alpha expression in the synovium. Ann Rheum Dis 2008; 67:1139–1144.
- Sfikakis PP. The first decade of biologic TNF antagonists in clinical practice: lessons learned, unresolved issues and future directions. Curr Dir Autoimmun 2010; 11:180–210.
- Casellas F, Alcalá MJ, Prieto L, Miró JR, Malagelada JR. Assessment of the influence of disease activity on the quality of life of patients with inflammatory bowel disease using a short questionnaire. Am J Gastroenterol 2004; 99:457–461.
- Casellas F, López-Vivancos J, Badia X, Vilaseca J, Malagelada JR. Influence of inflammatory bowel disease on different dimensions of quality of life. Eur J Gastroenterol Hepatol 2001; 13:567–572.
- Baecklund E, Iliadou A, Askling J, et al. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum 2006; 54:692–701.
- Franklin J, Lunt M, Bunn D, Symmons D, Silman A. Incidence of lymphoma in a large primary care derived cohort of cases of inflammatory polyarthritis. Ann Rheum Dis 2006; 65:617–622.
- Toruner M, Loftus EV, Harmsen WS, et al. Risk factors for opportunistic infections in patients with inflammatory bowel disease. Gastroenterology 2008; 134:929–936.
- Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum 2007; 56:2886–2895.
- Prevention of pneumococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 1997; 46:1–24.
- Zhang J, Xie F, Delzell E, et al. Association between vaccination for herpes zoster and risk of herpes zoster infection among older patients with selected immune-mediated diseases. JAMA 2012; 308:43–49.
- Moreland LW, Baumgartner SW, Schiff MH, et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med 1997; 337:141–147.
- Altunöz ME, Senates E, Yesil A, Calhan T, Ovünç AO. Patients with inflammatory bowel disease have a lower response rate to HBV vaccination compared to controls. Dig Dis Sci 2012; 57:1039–1044.
- Gisbert JP, Villagrasa JR, Rodríguez-Nogueiras A, Chaparro M. Efficacy of hepatitis B vaccination and revaccination and factors impacting on response in patients with inflammatory bowel disease. Am J Gastroenterol 2012; 107:1460–1466.
- Carrera E, Manzano R, Garrido E. Efficacy of the vaccination in inflammatory bowel disease. World J Gastroenterol 2013; 19:1349–1353.
- Gisbert JP, Menchén L, García-Sánchez V, Marín I, Villagrasa JR, Chaparro M. Comparison of the effectiveness of two protocols for vaccination (standard and double dosage) against hepatitis B virus in patients with inflammatory bowel disease. Aliment Pharmacol Ther 2012; 35:1379–1385.
- Ferkolj I. How to improve the safety of biologic therapy in Crohn’s disease. J Physiol Pharmacol 2009; 60(suppl 7):67–70.
- Siegel CA. The risks of biologic therapy for inflammatory bowel disease. In:Bernstein ED, editor. The Inflammatory Bowel Disease Yearbook, volume 6. London, UK: Remedica, 2010:89–108.
- Remicade (infliximab) Package Insert. Horsham, PA: Janssen Biotech, Inc; 2013. http://www.remicade.com/shared/product/remicade/prescribing-information.pdf. Accessed January 2, 2014.
- Vermeire S, Noman M, Van Assche G, et al. Autoimmunity associated with anti-tumor necrosis factor alpha treatment in Crohn’s disease: a prospective cohort study. Gastroenterology 2003; 125:32–39.
- Cush JJ. Biological drug use: US perspectives on indications and monitoring. Ann Rheum Dis 2005; 64(suppl 4):iv18–iv23.
- TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 1999; 53:457–465.
- Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT; Anti-TNF Therapy Against Congestive Heart Failure Investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003; 107:3133–3140.
- Cheifetz A, Mayer L. Monoclonal antibodies, immunogenicity, and associated infusion reactions. Mt Sinai J Med 2005; 72:250–256.
- Cheifetz A, Smedley M, Martin S, et al. The incidence and management of infusion reactions to infliximab: a large center experience. Am J Gastroenterol 2003; 98:1315–1324.
- Vultaggio A, Matucci A, Nencini F, et al. Anti-infliximab IgE and non-IgE antibodies and induction of infusion-related severe anaphylactic reactions. Allergy 2010; 65:657–661.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore) 2007; 86:242–251.
- Stallmach A, Hagel S, Bruns T. Adverse effects of biologics used for treating IBD. Best Pract Res Clin Gastroenterol 2010; 24:167–182.
- Haagsma CJ, Blom HJ, van Riel PL, et al. Influence of sulphasalazine, methotrexate, and the combination of both on plasma homocysteine concentrations in patients with rheumatoid arthritis. Ann Rheum Dis 1999; 58:79–84.
- Fiorino G, Allez M, Malesci A, Danese S. Review article: anti TNF-alpha induced psoriasis in patients with inflammatory bowel disease. Aliment Pharmacol Ther 2009; 29:921–927.
- Harrison MJ, Dixon WG, Watson KD, et al; British Society for Rheumatology Biologics Register Control Centre Consortium; BSRBR. Rates of new-onset psoriasis in patients with rheumatoid arthritis receiving anti-tumour necrosis factor alpha therapy: results from the British Society for Rheumatology Biologics Register. Ann Rheum Dis 2009; 68:209–215.
- Marchesoni A, Zaccara E, Gorla R, et al. TNF-alpha antagonist survival rate in a cohort of rheumatoid arthritis patients observed under conditions of standard clinical practice. Ann N Y Acad Sci 2009; 1173:837–846.
- Tomas L, Lazurova I, Pundova L, et al. Acute and long-term effect of infliximab on humoral and echocardiographic parameters in patients with chronic inflammatory diseases. Clin Rheumatol 2013 32:61–66.
- Senel S, Cobankara V, Taskoylu O, et al. The safety and efficacy of etanercept on cardiac functions and lipid profile in patients with active rheumatoid arthritis. J Investig Med 2012; 60:62–65.
- Al-Aly Z, Pan H, Zeringue A, et al. Tumor necrosis factor-a blockade, cardiovascular outcomes, and survival in rheumatoid arthritis. Transl Res 2011; 157:10–18.
- Listing J, Strangfeld A, Kekow J, et al. Does tumor necrosis factor alpha inhibition promote or prevent heart failure in patients with rheumatoid arthritis? Arthritis Rheum 2008; 58:667–677.
- Wolfe F, Michaud K. Heart failure in rheumatoid arthritis: rates, predictors, and the effect of anti-tumor necrosis factor therapy. Am J Med 2004; 116:305–311.
- Enayati PJ, Papadakis KA. Association of anti-tumor necrosis factor therapy with the development of multiple sclerosis. J Clin Gastroenterol 2005; 39:303–306.
- Mohan N, Edwards ET, Cupps TR, et al. Demyelination occurring during anti-tumor necrosis factor alpha therapy for inflammatory arthritides. Arthritis Rheum 2001; 44:2862–2869.
- Sands BE, Anderson FH, Bernstein CN, et al. Infliximab maintenance therapy for fistulizing Crohn’s disease. N Engl J Med 2004; 350:876–885.
- Thomas CW, Weinshenker BG, Sandborn WJ. Demyelination during anti-tumor necrosis factor alpha therapy with infliximab for Crohn’s disease. Inflamm Bowel Dis 2004; 10:28–31.
- Foroozan R, Buono LM, Sergott RC, Savino PJ. Retrobulbar optic neuritis associated with infliximab. Arch Ophthalmol 2002; 120:985–987.
- Strong BY, Erny BC, Herzenberg H, Razzeca KJ. Retrobulbar optic neuritis associated with infliximab in a patient with Crohn disease. Ann Intern Med 2004; 140:W34.
- ten Tusscher MP, Jacobs PJ, Busch MJ, de Graaf L, Diemont WL. Bilateral anterior toxic optic neuropathy and the use of infliximab. BMJ 2003; 326:579.
- Hegde N, Gayomali C, Rich MW. Infliximab-induced headache and infliximab-induced meningitis: two ends of the same spectrum? South Med J 2005; 98:564–566.
- Schiff MH, Burmester GR, Kent JD, et al. Safety analyses of adalimumab (HUMIRA) in global clinical trials and US postmarketing surveillance of patients with rheumatoid arthritis. Ann Rheum Dis 2006; 65:889–894.
- Beaugerie L, Brousse N, Bouvier AM, et al; CESAME Study Group. Lymphoproliferative disorders in patients receiving thiopurines for inflammatory bowel disease: a prospective observational cohort study. Lancet 2009; 374:1617–1625.
- Ekström K, Hjalgrim H, Brandt L, et al. Risk of malignant lymphomas in patients with rheumatoid arthritis and in their first-degree relatives. Arthritis Rheum 2003; 48:963–970.
- Lakatos PL, Lakatos L. Risk for colorectal cancer in ulcerative colitis: changes, causes and management strategies. World J Gastroenterol 2008; 14:3937–3947.
- Persson PG, Karlén P, Bernell O, et al. Crohn’s disease and cancer: a population-based cohort study. Gastroenterology 1994; 107:1675–1679.
- de Silva S, Devlin S, Panaccione R. Optimizing the safety of biologic therapy for IBD. Nat Rev Gastroenterol Hepatol 2010; 7:93–101.
- Lichtenstein GR, Feagan BG, Cohen RD, et al. Serious infections and mortality in association with therapies for Crohn’s disease: TREAT registry. Clin Gastroenterol Hepatol 2006; 4:621–630.
- Peyrin-Biroulet L, Deltenre P, de Suray N, Branche J, Sandborn WJ, Colombel JF. Efficacy and safety of tumor necrosis factor antagonists in Crohn’s disease: meta-analysis of placebo-controlled trials. Clin Gastroenterol Hepatol 2008; 6:644–653.
- Hanauer SB, Feagan BG, Lichtenstein GR, et al; ACCENT I Study Group. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet 2002; 359:1541–1549.
- Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: the CHARM trial. Gastroenterology 2007; 132:52–65.
- Sandborn WJ, Feagan BG, Stoinov S, et al; PRECISE 1 Study Investigators. Certolizumab pegol for the treatment of Crohn’s disease. N Engl J Med 2007; 357:228–238.
- Sandborn WJ, Hanauer SB, Rutgeerts P, et al. Adalimumab for maintenance treatment of Crohn’s disease: results of the CLASSIC II trial. Gut 2007; 56:1232–1239.
- Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: the CLASSIC-I trial. Gastroenterology 2006; 130:323–333.
- Rutgeerts P, D’Haens G, Targan S, et al. Efficacy and safety of retreatment with anti-tumor necrosis factor antibody (infliximab) to maintain remission in Crohn’s disease. Gastroenterology 1999; 117:761–769.
- Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med 2005; 353:2462–2476.
- Pedersen N, Duricova D, Elkjaer M, Gamborg M, Munkholm P, Jess T. Risk of extra-intestinal cancer in inflammatory bowel disease: meta-analysis of population-based cohort studies. Am J Gastroenterol 2010; 105:1480–1487.
- Bernstein CN, Blanchard JF, Kliewer E, Wajda A. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer 2001; 91:854–862.
- Siegel CA, Marden SM, Persing SN, Larson RJ, Sands BE. Risk of lymphoma associated with combination anti-tumor necrosis factor and immunomodulator therapy for the treatment of Crohn’s disease: a meta-analysis. Clin Gastroenterol Hepatol 2009; 7:874–881.
- Wolfe F, Michaud K. Lympyhoma in rheumatoid arthritis. The effect of methotrexate and anti-tumor necrosis factor therapy in 18,572 patients. Arthritis Rheum 2004; 50:1740–1751.
- Parakkal D, Sifuentes H, Semer R, Ehrenpreis ED. Hepatosplenic T-cell lymphoma in patients receiving TNF-a inhibitor therapy: expanding the groups at risk. Eur J Gastroenterol Hepatol 2011; 23:1150–1156.
- Rosh JR, Gross T, Mamula P, Griffiths A, Hyams J. Hepatosplenic T-cell lymphoma in adolescents and young adults with Crohn’s disease: a cautionary tale? Inflamm Bowel Dis 2007; 13:1024–1030.
- Shale M, Kanfer E, Panaccione R, Ghosh S. Hepatosplenic T cell lymphoma in inflammatory bowel disease. Gut 2008; 57:1639–1641.
- Thai A, Prindiville T. Hepatosplenic T-cell lymphoma and inflammatory bowel disease. J Crohns Colitis 2010; 4:511–522.
- Viget N, Vernier-Massouille G, Salmon-Ceron D, Yazdanpanah Y, Colombel JF. Opportunistic infections in patients with inflammatory bowel disease: prevention and diagnosis. Gut 2008; 57:549–558.
- Bekker LG, Freeman S, Murray PJ, Ryffel B, Kaplan G. TNF-alpha controls intracellular mycobacterial growth by both inducible nitric oxide synthase-dependent and inducible nitric oxide synthase-independent pathways. J Immunol 2001; 166:6728–6734.
- Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol 2002; 168:4620–4627.
- Gómez-Reino JJ, Carmona L, Valverde VR, Mola EM, Montero MDBIOBADASER Group. Treatment of rheumatoid arthritis with tumor necrosis factor inhibitors may predispose to significant increase in tuberculosis risk: a multicenter active-surveillance report. Arthritis Rheum 2003; 48:2122–2127.
- Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med 2001; 345:1098–1104.
- Smolen J, Landewé RB, Mease P, et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis 2009; 68:797–804.
- Demkow U, Broniarek-Samson B, Filewska M, et al. Prevalence of latent tuberculosis infection in health care workers in Poland assessed by interferon-gamma whole blood and tuberculin skin tests. J Physiol Pharmacol 2008; 59(suppl 6):209–217.
- Pache I, Rogler G, Felley C. TNF-alpha blockers in inflammatory bowel diseases: practical consensus recommendations and a user’s guide. Swiss Med Wkly 2009; 139:278–287.
- Rahier JF, Ben-Horin S, Chowers Y, et al; European Crohn’s and Colitis Organisation (ECCO). European evidence-based consensus on the prevention, diagnosis and management of opportunistic infections in inflammatory bowel disease. J Crohns Colitis 2009; 3:47–91.
- Rahier JF, Yazdanpanah Y, Colombel JF, Travis S. The European (ECCO) consensus on infection in IBD: what does it change for the clinician? Gut 2009; 58:1313–1315.
- Bergstrom L, Yocum DE, Ampel NM, et al. Increased risk of coccidioidomycosis in patients treated with tumor necrosis factor alpha antagonists. Arthritis Rheum 2004; 50:1959–1966.
- Lee JH, Slifman NR, Gershon SK, et al. Life-threatening histoplasmosis complicating immunotherapy with tumor necrosis factor alpha antagonists infliximab and etanercept. Arthritis Rheum 2002; 46:2565–2570.
- Wood KL, Hage CA, Knox KS, et al. Histoplasmosis after treatment with anti-tumor necrosis factor-alpha therapy. Am J Respir Crit Care Med 2003; 167:1279–1282.
- Reddy JG, Loftus EV. Safety of infliximab and other biologic agents in the inflammatory bowel diseases. Gastroenterol Clin North Am 2006; 35:837–855.
- Esteve M, Saro C, González-Huix F, Suarez F, Forné M, Viver JM. Chronic hepatitis B reactivation following infliximab therapy in Crohn’s disease patients: need for primary prophylaxis. Gut 2004; 53:1363–1365.
- Michel M, Duvoux C, Hezode C, Cherqui D. Fulminant hepatitis after infliximab in a patient with hepatitis B virus treated for an adult onset Still’s disease. J Rheumatol 2003; 30:1624–1625.
- Ostuni P, Botsios C, Punzi L, Sfriso P, Todesco S. Hepatitis B reactivation in a chronic hepatitis B surface antigen carrier with rheumatoid arthritis treated with infliximab and low dose methotrexate. Ann Rheum Dis 2003; 62:686–687.
- Pérez-Alvarez R, Díaz-Lagares C, García-Hernández F, et al; BIOGEAS Study Group. Hepatitis B virus (HBV) reactivation in patients receiving tumor necrosis factor (TNF)-targeted therapy: analysis of 257 cases. Medicine (Baltimore) 2011; 90:359–371.
- Loras C, Gisbert JP, Mínguez M, et al; REPENTINA study; GETECCU (Grupo Español de Enfermedades de Crohn y Colitis Ulcerosa) Group. Liver dysfunction related to hepatitis B and C in patients with inflammatory bowel disease treated with immunosuppressive therapy. Gut 2010; 59:1340–1346.
- Park SH, Yang SK, Lim YS, et al. Clinical courses of chronic hepatitis B virus infection and inflammatory bowel disease in patients with both diseases. Inflamm Bowel Dis 2012; 18:2004–2010.
- Loomba R, Rowley A, Wesley R, et al. Systematic review: the effect of preventive lamivudine on hepatitis B reactivation during chemotherapy. Ann Intern Med 2008; 148:519–528.
- Lok AS, McMahon BJ. Chronic hepatitis B: update 2009. Hepatology 2009; 50:661–662.
- European Association For The Study Of The Liver. EASL Clinical Practice Guidelines: management of chronic hepatitis B. J Hepatol 2009; 50:227–242.
- Watanabe M, Shibuya A, Takada J, et al. Entecavir is an optional agent to prevent hepatitis B virus (HBV) reactivation: a review of 16 patients. Eur J Intern Med 2010; 21:333–337.
- Biancone L, Pavia M, Del Vecchio Blanco G, et al; Italian Group for the Study of the Colon and Rectum (GISC). Hepatitis B and C virus infection in Crohn’s disease. Inflamm Bowel Dis 2001; 7:287–294.
- Melmed GY. Vaccination strategies for patients with inflammatory bowel disease on immunomodulators and biologics. Inflamm Bowel Dis 2009; 15:1410–1416.
- Melmed GY, Ippoliti AF, Papadakis KA, et al. Patients with inflammatory bowel disease are at risk for vaccine-preventable illnesses. Am J Gastroenterol 2006; 101:1834–1840.
- Ferri C, Ferraccioli G, Ferrari D, et al. Safety of anti-tumor necrosis factor-alpha therapy in patients with rheumatoid arthritis and chronic hepatitis C virus infection. J Rheumatol 2008; 35:1944–1949.
- Mok MY, Ng WL, Yuen MF, Wong RW, Lau CS. Safety of disease modifying anti-rheumatic agents in rheumatoid arthritis patients with chronic viral hepatitis. Clin Exp Rheumatol 2000; 18:363–368.
- Vassilopoulos D, Calabrese LH. Risks of immunosuppressive therapies including biologic agents in patients with rheumatic diseases and coexisting chronic viral infections. Curr Opin Rheumatol 2007; 19:619–625.
- Vassilopoulos D, Apostolopoulou A, Hadziyannis E, et al. Long-term safety of anti-TNF treatment in patients with rheumatic diseases and chronic or resolved hepatitis B virus infection. Ann Rheum Dis 2010; 69:1352–1355.
- Saag KG, Teng GG, Patkar NM, et al; American College of Rheumatology. American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Rheum 2008; 59:762–784.
- Cornish J, Tan E, Teare J, et al. A meta-analysis on the influence of inflammatory bowel disease on pregnancy. Gut 2007; 56:830–837.
- Dominitz JA, Young JC, Boyko EJ. Outcomes of infants born to mothers with inflammatory bowel disease: a population-based cohort study. Am J Gastroenterol 2002; 97:641–648.
- Kornfeld D, Cnattingius S, Ekbom A. Pregnancy outcomes in women with inflammatory bowel disease—a population-based cohort study. Am J Obstet Gynecol 1997; 177:942–946.
- Mahadevan U, Sandborn WJ, Li DK, Hakimian S, Kane S, Corley DA. Pregnancy outcomes in women with inflammatory bowel disease: a large community-based study from Northern California. Gastroenterology 2007; 133:1106–1112.
- Nguyen GC, Boudreau H, Harris ML, Maxwell CV. Outcomes of obstetric hospitalizations among women with inflammatory bowel disease in the United States. Clin Gastroenterol Hepatol 2009; 7:329–334.
- Schnitzler F, Fidder H, Ferrante M, et al. Outcome of pregnancy in women with inflammatory bowel disease treated with antitumor necrosis factor therapy. Inflamm Bowel Dis 2011; 17:1846–1854.
- Vinet E, Pineau C, Gordon C, Clarke AE, Bernatsky S. Biologic therapy and pregnancy outcomes in women with rheumatic diseases. Arthritis Rheum 2009; 61:587–592.
- Guidi L, Pugliese D, Armuzzi A. Update on the management of inflammatory bowel disease: specific role of adalimumab. Clin Exp Gastroenterol 2011; 4:163–172.
- Mahadevan U. Continuing immunomodulators and biologic medications in pregnant IBD patients – pro. IInflamm Bowel Dis 2007; 13:1439–1440.
- Mahadevan U. Gastrointestinal medications in pregnancy. Best Pract Res Clin Gastroenterol 2007; 21:849–877.
- Mahadevan U, Cucchiara S, Hyams JS, et al. The London Position Statement of the World Congress of Gastroenterology on Biological Therapy for IBD with the European Crohn’s and Colitis Organisation: pregnancy and pediatrics. Am J Gastroenterol 2011; 106:214–223.
- Mahadevan U, Kane S. Use of infliximab in pregnancy. Am J Gastroenterol 2010; 105:219–220.
- Vasiliauskas EA, Church JA, Silverman N, Barry M, Targan SR, Dubinsky MC. Case report: evidence for transplacental transfer of maternally administered infliximab to the newborn. Clin Gastroenterol Hepatol 2006; 4:1255–1258.
- Wijbrandts CA, Dijkgraaf MG, Kraan MC, et al. The clinical response to infliximab in rheumatoid arthritis is in part dependent on pretreatment tumour necrosis factor alpha expression in the synovium. Ann Rheum Dis 2008; 67:1139–1144.
- Sfikakis PP. The first decade of biologic TNF antagonists in clinical practice: lessons learned, unresolved issues and future directions. Curr Dir Autoimmun 2010; 11:180–210.
- Casellas F, Alcalá MJ, Prieto L, Miró JR, Malagelada JR. Assessment of the influence of disease activity on the quality of life of patients with inflammatory bowel disease using a short questionnaire. Am J Gastroenterol 2004; 99:457–461.
- Casellas F, López-Vivancos J, Badia X, Vilaseca J, Malagelada JR. Influence of inflammatory bowel disease on different dimensions of quality of life. Eur J Gastroenterol Hepatol 2001; 13:567–572.
- Baecklund E, Iliadou A, Askling J, et al. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum 2006; 54:692–701.
- Franklin J, Lunt M, Bunn D, Symmons D, Silman A. Incidence of lymphoma in a large primary care derived cohort of cases of inflammatory polyarthritis. Ann Rheum Dis 2006; 65:617–622.
- Toruner M, Loftus EV, Harmsen WS, et al. Risk factors for opportunistic infections in patients with inflammatory bowel disease. Gastroenterology 2008; 134:929–936.
- Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum 2007; 56:2886–2895.
- Prevention of pneumococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 1997; 46:1–24.
- Zhang J, Xie F, Delzell E, et al. Association between vaccination for herpes zoster and risk of herpes zoster infection among older patients with selected immune-mediated diseases. JAMA 2012; 308:43–49.
- Moreland LW, Baumgartner SW, Schiff MH, et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med 1997; 337:141–147.
- Altunöz ME, Senates E, Yesil A, Calhan T, Ovünç AO. Patients with inflammatory bowel disease have a lower response rate to HBV vaccination compared to controls. Dig Dis Sci 2012; 57:1039–1044.
- Gisbert JP, Villagrasa JR, Rodríguez-Nogueiras A, Chaparro M. Efficacy of hepatitis B vaccination and revaccination and factors impacting on response in patients with inflammatory bowel disease. Am J Gastroenterol 2012; 107:1460–1466.
- Carrera E, Manzano R, Garrido E. Efficacy of the vaccination in inflammatory bowel disease. World J Gastroenterol 2013; 19:1349–1353.
- Gisbert JP, Menchén L, García-Sánchez V, Marín I, Villagrasa JR, Chaparro M. Comparison of the effectiveness of two protocols for vaccination (standard and double dosage) against hepatitis B virus in patients with inflammatory bowel disease. Aliment Pharmacol Ther 2012; 35:1379–1385.
- Ferkolj I. How to improve the safety of biologic therapy in Crohn’s disease. J Physiol Pharmacol 2009; 60(suppl 7):67–70.
- Siegel CA. The risks of biologic therapy for inflammatory bowel disease. In:Bernstein ED, editor. The Inflammatory Bowel Disease Yearbook, volume 6. London, UK: Remedica, 2010:89–108.
- Remicade (infliximab) Package Insert. Horsham, PA: Janssen Biotech, Inc; 2013. http://www.remicade.com/shared/product/remicade/prescribing-information.pdf. Accessed January 2, 2014.
- Vermeire S, Noman M, Van Assche G, et al. Autoimmunity associated with anti-tumor necrosis factor alpha treatment in Crohn’s disease: a prospective cohort study. Gastroenterology 2003; 125:32–39.
- Cush JJ. Biological drug use: US perspectives on indications and monitoring. Ann Rheum Dis 2005; 64(suppl 4):iv18–iv23.
- TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 1999; 53:457–465.
- Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT; Anti-TNF Therapy Against Congestive Heart Failure Investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003; 107:3133–3140.
- Cheifetz A, Mayer L. Monoclonal antibodies, immunogenicity, and associated infusion reactions. Mt Sinai J Med 2005; 72:250–256.
- Cheifetz A, Smedley M, Martin S, et al. The incidence and management of infusion reactions to infliximab: a large center experience. Am J Gastroenterol 2003; 98:1315–1324.
- Vultaggio A, Matucci A, Nencini F, et al. Anti-infliximab IgE and non-IgE antibodies and induction of infusion-related severe anaphylactic reactions. Allergy 2010; 65:657–661.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore) 2007; 86:242–251.
- Stallmach A, Hagel S, Bruns T. Adverse effects of biologics used for treating IBD. Best Pract Res Clin Gastroenterol 2010; 24:167–182.
- Haagsma CJ, Blom HJ, van Riel PL, et al. Influence of sulphasalazine, methotrexate, and the combination of both on plasma homocysteine concentrations in patients with rheumatoid arthritis. Ann Rheum Dis 1999; 58:79–84.
- Fiorino G, Allez M, Malesci A, Danese S. Review article: anti TNF-alpha induced psoriasis in patients with inflammatory bowel disease. Aliment Pharmacol Ther 2009; 29:921–927.
- Harrison MJ, Dixon WG, Watson KD, et al; British Society for Rheumatology Biologics Register Control Centre Consortium; BSRBR. Rates of new-onset psoriasis in patients with rheumatoid arthritis receiving anti-tumour necrosis factor alpha therapy: results from the British Society for Rheumatology Biologics Register. Ann Rheum Dis 2009; 68:209–215.
- Marchesoni A, Zaccara E, Gorla R, et al. TNF-alpha antagonist survival rate in a cohort of rheumatoid arthritis patients observed under conditions of standard clinical practice. Ann N Y Acad Sci 2009; 1173:837–846.
- Tomas L, Lazurova I, Pundova L, et al. Acute and long-term effect of infliximab on humoral and echocardiographic parameters in patients with chronic inflammatory diseases. Clin Rheumatol 2013 32:61–66.
- Senel S, Cobankara V, Taskoylu O, et al. The safety and efficacy of etanercept on cardiac functions and lipid profile in patients with active rheumatoid arthritis. J Investig Med 2012; 60:62–65.
- Al-Aly Z, Pan H, Zeringue A, et al. Tumor necrosis factor-a blockade, cardiovascular outcomes, and survival in rheumatoid arthritis. Transl Res 2011; 157:10–18.
- Listing J, Strangfeld A, Kekow J, et al. Does tumor necrosis factor alpha inhibition promote or prevent heart failure in patients with rheumatoid arthritis? Arthritis Rheum 2008; 58:667–677.
- Wolfe F, Michaud K. Heart failure in rheumatoid arthritis: rates, predictors, and the effect of anti-tumor necrosis factor therapy. Am J Med 2004; 116:305–311.
- Enayati PJ, Papadakis KA. Association of anti-tumor necrosis factor therapy with the development of multiple sclerosis. J Clin Gastroenterol 2005; 39:303–306.
- Mohan N, Edwards ET, Cupps TR, et al. Demyelination occurring during anti-tumor necrosis factor alpha therapy for inflammatory arthritides. Arthritis Rheum 2001; 44:2862–2869.
- Sands BE, Anderson FH, Bernstein CN, et al. Infliximab maintenance therapy for fistulizing Crohn’s disease. N Engl J Med 2004; 350:876–885.
- Thomas CW, Weinshenker BG, Sandborn WJ. Demyelination during anti-tumor necrosis factor alpha therapy with infliximab for Crohn’s disease. Inflamm Bowel Dis 2004; 10:28–31.
- Foroozan R, Buono LM, Sergott RC, Savino PJ. Retrobulbar optic neuritis associated with infliximab. Arch Ophthalmol 2002; 120:985–987.
- Strong BY, Erny BC, Herzenberg H, Razzeca KJ. Retrobulbar optic neuritis associated with infliximab in a patient with Crohn disease. Ann Intern Med 2004; 140:W34.
- ten Tusscher MP, Jacobs PJ, Busch MJ, de Graaf L, Diemont WL. Bilateral anterior toxic optic neuropathy and the use of infliximab. BMJ 2003; 326:579.
- Hegde N, Gayomali C, Rich MW. Infliximab-induced headache and infliximab-induced meningitis: two ends of the same spectrum? South Med J 2005; 98:564–566.
- Schiff MH, Burmester GR, Kent JD, et al. Safety analyses of adalimumab (HUMIRA) in global clinical trials and US postmarketing surveillance of patients with rheumatoid arthritis. Ann Rheum Dis 2006; 65:889–894.
- Beaugerie L, Brousse N, Bouvier AM, et al; CESAME Study Group. Lymphoproliferative disorders in patients receiving thiopurines for inflammatory bowel disease: a prospective observational cohort study. Lancet 2009; 374:1617–1625.
- Ekström K, Hjalgrim H, Brandt L, et al. Risk of malignant lymphomas in patients with rheumatoid arthritis and in their first-degree relatives. Arthritis Rheum 2003; 48:963–970.
- Lakatos PL, Lakatos L. Risk for colorectal cancer in ulcerative colitis: changes, causes and management strategies. World J Gastroenterol 2008; 14:3937–3947.
- Persson PG, Karlén P, Bernell O, et al. Crohn’s disease and cancer: a population-based cohort study. Gastroenterology 1994; 107:1675–1679.
- de Silva S, Devlin S, Panaccione R. Optimizing the safety of biologic therapy for IBD. Nat Rev Gastroenterol Hepatol 2010; 7:93–101.
- Lichtenstein GR, Feagan BG, Cohen RD, et al. Serious infections and mortality in association with therapies for Crohn’s disease: TREAT registry. Clin Gastroenterol Hepatol 2006; 4:621–630.
- Peyrin-Biroulet L, Deltenre P, de Suray N, Branche J, Sandborn WJ, Colombel JF. Efficacy and safety of tumor necrosis factor antagonists in Crohn’s disease: meta-analysis of placebo-controlled trials. Clin Gastroenterol Hepatol 2008; 6:644–653.
- Hanauer SB, Feagan BG, Lichtenstein GR, et al; ACCENT I Study Group. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet 2002; 359:1541–1549.
- Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: the CHARM trial. Gastroenterology 2007; 132:52–65.
- Sandborn WJ, Feagan BG, Stoinov S, et al; PRECISE 1 Study Investigators. Certolizumab pegol for the treatment of Crohn’s disease. N Engl J Med 2007; 357:228–238.
- Sandborn WJ, Hanauer SB, Rutgeerts P, et al. Adalimumab for maintenance treatment of Crohn’s disease: results of the CLASSIC II trial. Gut 2007; 56:1232–1239.
- Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: the CLASSIC-I trial. Gastroenterology 2006; 130:323–333.
- Rutgeerts P, D’Haens G, Targan S, et al. Efficacy and safety of retreatment with anti-tumor necrosis factor antibody (infliximab) to maintain remission in Crohn’s disease. Gastroenterology 1999; 117:761–769.
- Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med 2005; 353:2462–2476.
- Pedersen N, Duricova D, Elkjaer M, Gamborg M, Munkholm P, Jess T. Risk of extra-intestinal cancer in inflammatory bowel disease: meta-analysis of population-based cohort studies. Am J Gastroenterol 2010; 105:1480–1487.
- Bernstein CN, Blanchard JF, Kliewer E, Wajda A. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer 2001; 91:854–862.
- Siegel CA, Marden SM, Persing SN, Larson RJ, Sands BE. Risk of lymphoma associated with combination anti-tumor necrosis factor and immunomodulator therapy for the treatment of Crohn’s disease: a meta-analysis. Clin Gastroenterol Hepatol 2009; 7:874–881.
- Wolfe F, Michaud K. Lympyhoma in rheumatoid arthritis. The effect of methotrexate and anti-tumor necrosis factor therapy in 18,572 patients. Arthritis Rheum 2004; 50:1740–1751.
- Parakkal D, Sifuentes H, Semer R, Ehrenpreis ED. Hepatosplenic T-cell lymphoma in patients receiving TNF-a inhibitor therapy: expanding the groups at risk. Eur J Gastroenterol Hepatol 2011; 23:1150–1156.
- Rosh JR, Gross T, Mamula P, Griffiths A, Hyams J. Hepatosplenic T-cell lymphoma in adolescents and young adults with Crohn’s disease: a cautionary tale? Inflamm Bowel Dis 2007; 13:1024–1030.
- Shale M, Kanfer E, Panaccione R, Ghosh S. Hepatosplenic T cell lymphoma in inflammatory bowel disease. Gut 2008; 57:1639–1641.
- Thai A, Prindiville T. Hepatosplenic T-cell lymphoma and inflammatory bowel disease. J Crohns Colitis 2010; 4:511–522.
- Viget N, Vernier-Massouille G, Salmon-Ceron D, Yazdanpanah Y, Colombel JF. Opportunistic infections in patients with inflammatory bowel disease: prevention and diagnosis. Gut 2008; 57:549–558.
- Bekker LG, Freeman S, Murray PJ, Ryffel B, Kaplan G. TNF-alpha controls intracellular mycobacterial growth by both inducible nitric oxide synthase-dependent and inducible nitric oxide synthase-independent pathways. J Immunol 2001; 166:6728–6734.
- Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol 2002; 168:4620–4627.
- Gómez-Reino JJ, Carmona L, Valverde VR, Mola EM, Montero MDBIOBADASER Group. Treatment of rheumatoid arthritis with tumor necrosis factor inhibitors may predispose to significant increase in tuberculosis risk: a multicenter active-surveillance report. Arthritis Rheum 2003; 48:2122–2127.
- Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med 2001; 345:1098–1104.
- Smolen J, Landewé RB, Mease P, et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis 2009; 68:797–804.
- Demkow U, Broniarek-Samson B, Filewska M, et al. Prevalence of latent tuberculosis infection in health care workers in Poland assessed by interferon-gamma whole blood and tuberculin skin tests. J Physiol Pharmacol 2008; 59(suppl 6):209–217.
- Pache I, Rogler G, Felley C. TNF-alpha blockers in inflammatory bowel diseases: practical consensus recommendations and a user’s guide. Swiss Med Wkly 2009; 139:278–287.
- Rahier JF, Ben-Horin S, Chowers Y, et al; European Crohn’s and Colitis Organisation (ECCO). European evidence-based consensus on the prevention, diagnosis and management of opportunistic infections in inflammatory bowel disease. J Crohns Colitis 2009; 3:47–91.
- Rahier JF, Yazdanpanah Y, Colombel JF, Travis S. The European (ECCO) consensus on infection in IBD: what does it change for the clinician? Gut 2009; 58:1313–1315.
- Bergstrom L, Yocum DE, Ampel NM, et al. Increased risk of coccidioidomycosis in patients treated with tumor necrosis factor alpha antagonists. Arthritis Rheum 2004; 50:1959–1966.
- Lee JH, Slifman NR, Gershon SK, et al. Life-threatening histoplasmosis complicating immunotherapy with tumor necrosis factor alpha antagonists infliximab and etanercept. Arthritis Rheum 2002; 46:2565–2570.
- Wood KL, Hage CA, Knox KS, et al. Histoplasmosis after treatment with anti-tumor necrosis factor-alpha therapy. Am J Respir Crit Care Med 2003; 167:1279–1282.
- Reddy JG, Loftus EV. Safety of infliximab and other biologic agents in the inflammatory bowel diseases. Gastroenterol Clin North Am 2006; 35:837–855.
- Esteve M, Saro C, González-Huix F, Suarez F, Forné M, Viver JM. Chronic hepatitis B reactivation following infliximab therapy in Crohn’s disease patients: need for primary prophylaxis. Gut 2004; 53:1363–1365.
- Michel M, Duvoux C, Hezode C, Cherqui D. Fulminant hepatitis after infliximab in a patient with hepatitis B virus treated for an adult onset Still’s disease. J Rheumatol 2003; 30:1624–1625.
- Ostuni P, Botsios C, Punzi L, Sfriso P, Todesco S. Hepatitis B reactivation in a chronic hepatitis B surface antigen carrier with rheumatoid arthritis treated with infliximab and low dose methotrexate. Ann Rheum Dis 2003; 62:686–687.
- Pérez-Alvarez R, Díaz-Lagares C, García-Hernández F, et al; BIOGEAS Study Group. Hepatitis B virus (HBV) reactivation in patients receiving tumor necrosis factor (TNF)-targeted therapy: analysis of 257 cases. Medicine (Baltimore) 2011; 90:359–371.
- Loras C, Gisbert JP, Mínguez M, et al; REPENTINA study; GETECCU (Grupo Español de Enfermedades de Crohn y Colitis Ulcerosa) Group. Liver dysfunction related to hepatitis B and C in patients with inflammatory bowel disease treated with immunosuppressive therapy. Gut 2010; 59:1340–1346.
- Park SH, Yang SK, Lim YS, et al. Clinical courses of chronic hepatitis B virus infection and inflammatory bowel disease in patients with both diseases. Inflamm Bowel Dis 2012; 18:2004–2010.
- Loomba R, Rowley A, Wesley R, et al. Systematic review: the effect of preventive lamivudine on hepatitis B reactivation during chemotherapy. Ann Intern Med 2008; 148:519–528.
- Lok AS, McMahon BJ. Chronic hepatitis B: update 2009. Hepatology 2009; 50:661–662.
- European Association For The Study Of The Liver. EASL Clinical Practice Guidelines: management of chronic hepatitis B. J Hepatol 2009; 50:227–242.
- Watanabe M, Shibuya A, Takada J, et al. Entecavir is an optional agent to prevent hepatitis B virus (HBV) reactivation: a review of 16 patients. Eur J Intern Med 2010; 21:333–337.
- Biancone L, Pavia M, Del Vecchio Blanco G, et al; Italian Group for the Study of the Colon and Rectum (GISC). Hepatitis B and C virus infection in Crohn’s disease. Inflamm Bowel Dis 2001; 7:287–294.
- Melmed GY. Vaccination strategies for patients with inflammatory bowel disease on immunomodulators and biologics. Inflamm Bowel Dis 2009; 15:1410–1416.
- Melmed GY, Ippoliti AF, Papadakis KA, et al. Patients with inflammatory bowel disease are at risk for vaccine-preventable illnesses. Am J Gastroenterol 2006; 101:1834–1840.
- Ferri C, Ferraccioli G, Ferrari D, et al. Safety of anti-tumor necrosis factor-alpha therapy in patients with rheumatoid arthritis and chronic hepatitis C virus infection. J Rheumatol 2008; 35:1944–1949.
- Mok MY, Ng WL, Yuen MF, Wong RW, Lau CS. Safety of disease modifying anti-rheumatic agents in rheumatoid arthritis patients with chronic viral hepatitis. Clin Exp Rheumatol 2000; 18:363–368.
- Vassilopoulos D, Calabrese LH. Risks of immunosuppressive therapies including biologic agents in patients with rheumatic diseases and coexisting chronic viral infections. Curr Opin Rheumatol 2007; 19:619–625.
- Vassilopoulos D, Apostolopoulou A, Hadziyannis E, et al. Long-term safety of anti-TNF treatment in patients with rheumatic diseases and chronic or resolved hepatitis B virus infection. Ann Rheum Dis 2010; 69:1352–1355.
- Saag KG, Teng GG, Patkar NM, et al; American College of Rheumatology. American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Rheum 2008; 59:762–784.
- Cornish J, Tan E, Teare J, et al. A meta-analysis on the influence of inflammatory bowel disease on pregnancy. Gut 2007; 56:830–837.
- Dominitz JA, Young JC, Boyko EJ. Outcomes of infants born to mothers with inflammatory bowel disease: a population-based cohort study. Am J Gastroenterol 2002; 97:641–648.
- Kornfeld D, Cnattingius S, Ekbom A. Pregnancy outcomes in women with inflammatory bowel disease—a population-based cohort study. Am J Obstet Gynecol 1997; 177:942–946.
- Mahadevan U, Sandborn WJ, Li DK, Hakimian S, Kane S, Corley DA. Pregnancy outcomes in women with inflammatory bowel disease: a large community-based study from Northern California. Gastroenterology 2007; 133:1106–1112.
- Nguyen GC, Boudreau H, Harris ML, Maxwell CV. Outcomes of obstetric hospitalizations among women with inflammatory bowel disease in the United States. Clin Gastroenterol Hepatol 2009; 7:329–334.
- Schnitzler F, Fidder H, Ferrante M, et al. Outcome of pregnancy in women with inflammatory bowel disease treated with antitumor necrosis factor therapy. Inflamm Bowel Dis 2011; 17:1846–1854.
- Vinet E, Pineau C, Gordon C, Clarke AE, Bernatsky S. Biologic therapy and pregnancy outcomes in women with rheumatic diseases. Arthritis Rheum 2009; 61:587–592.
- Guidi L, Pugliese D, Armuzzi A. Update on the management of inflammatory bowel disease: specific role of adalimumab. Clin Exp Gastroenterol 2011; 4:163–172.
- Mahadevan U. Continuing immunomodulators and biologic medications in pregnant IBD patients – pro. IInflamm Bowel Dis 2007; 13:1439–1440.
- Mahadevan U. Gastrointestinal medications in pregnancy. Best Pract Res Clin Gastroenterol 2007; 21:849–877.
- Mahadevan U, Cucchiara S, Hyams JS, et al. The London Position Statement of the World Congress of Gastroenterology on Biological Therapy for IBD with the European Crohn’s and Colitis Organisation: pregnancy and pediatrics. Am J Gastroenterol 2011; 106:214–223.
- Mahadevan U, Kane S. Use of infliximab in pregnancy. Am J Gastroenterol 2010; 105:219–220.
- Vasiliauskas EA, Church JA, Silverman N, Barry M, Targan SR, Dubinsky MC. Case report: evidence for transplacental transfer of maternally administered infliximab to the newborn. Clin Gastroenterol Hepatol 2006; 4:1255–1258.
KEY POINTS
- Over the past 10 years, TNF inhibitors have substantially altered the management of autoimmune diseases such as rheumatoid arthritis and inflammatory bowel disease.
- Safety concerns include risks of infection, reactivation of latent infection (eg, fungal infection, granulomatous infection), malignancy, and autoimmune and neurologic effects.
- Before treating, take a complete history, including exposure to latent infections and geographic considerations, and bring patients’ immunizations up to date.
- Regular clinical and laboratory monitoring during treatment helps optimize therapy and minimize the risk of adverse effects.
- Physicians must be aware of atypical presentations of infection and understand how their treatment may differ in patients on biologic therapy.
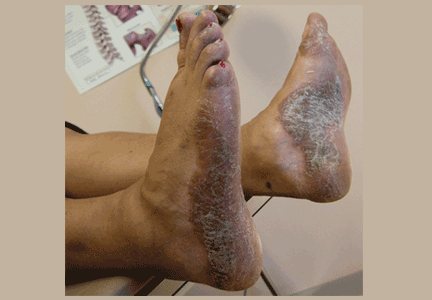
Using the Internet in your practice. Part 1: Why social media are important and how to get started
Let’s rewind to the year 2000, the dawning of a new millennium. It was then that many physicians decided the time was ripe to establish a Web presence. It wasn’t that difficult, after all: Just take the practice’s three-color, trifold brochure and convert it into a Web-site template. A teenager could do it—and many did, sometimes guided by a college student in computer sciences.
These early implementers were confident that they could cruise into the 21st Century with this new technology. They had no idea how much the Internet would change…or how fast…but their basic impulse was a wise one, to harness the power of the Internet for the good of their patients and their practices.
In this four-part series, we focus on the rapidly expanding utilization of the Internet for health-related purposes. In Part 1, we focus on why it’s important to address the Web, particularly social media, and we zoom in on creating a blog for your practice. In Part 2, our focus will be the “big three”: Facebook, Twitter, and YouTube. We will take up search engine optimization and online reputation management in Parts 3 and 4, respectively.
WHY IS THE INTERNET IMPORTANT?
It isn’t uncommon for patients to arrive in their doctor’s office with a stack of pages downloaded from the Internet that describe their disease state or tests they are about to undergo. Many patients also are beginning to expect to interact with their physicians through Web sites, blogs, and Facebook and Twitter accounts.
Related Article: Why (and how) you should encourage your patients' search for health information on the Web Jennifer Gunter, MD (December 2011)
In fact, so much of health care is moving online that many physicians assume that everybody uses the Internet. The most recent data from the Pew Internet & American Life Project indicate that, in the United States, one in three adults have gone online to find out more about a medical condition, and 59% of all adults use the Internet to search for health information (TABLE 1).1,2 Eight in 10 people who regularly use the Internet look online for health information, making it the third most popular online pursuit tracked by the Pew project, after reading and sending email and using a search engine.
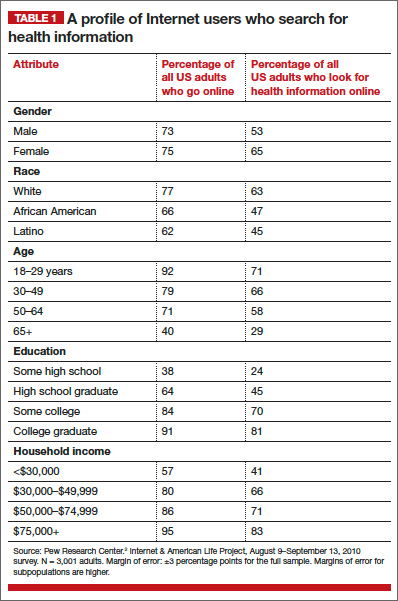
What types of health information do US adults look for online? Most people (66%) who use the Web to search for health information look for information on a specific disease or medical problem (see TABLE 2 for a list of other common health topics).3
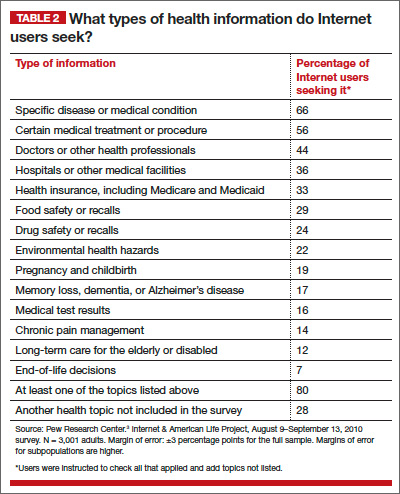
The Pew Research Center also found that some demographic groups are more likely than others to seek health information online. They include:
- adults who have provided unpaid care to a parent, child, friend, or other loved one in the past 12 months
- women
- white adults
- adults aged 18 to 49 years
- adults with at least some college education
- adults in higher-income households.1
Check out the QUICK POLL on the OBG Management home page. To give your answer and see how other physicians have responded, Click Here.
WHAT ARE SOCIAL MEDIA AND WHY DO WE NEED THEM?
Social media encompass Web sites and other online communication applications used for social networking. Three of the most widely used media are Facebook, Twitter, and YouTube.
When someone once asked hockey great Wayne Gretzky about his sport strategy, he replied: “I don’t skate to where the puck is or where the puck has been; I skate to where the puck is going to be.” Social media are where the puck (ie, our patients) are going to be today and tomorrow.
If we review other media launches, we discover that it took nearly 40 years for radio to attract 50 million listeners, and 13 years for television to reach 50 million viewers. But it took only 4 years for the Internet to achieve 50 million users. Facebook alone reached 100 million users in just 9 months!
Just a decade ago, the Mayo Clinic relied on standard marketing techniques using radio, TV, and print media to attract new patients. Today, the Mayo Clinic makes use of Facebook, Twitter, YouTube, podcasts, and blogging. The Mayo Clinic even has developed a Center for Social Media to focus on the use of social media for its centers in Rochester, Minnesota; Jacksonville, Florida; and Phoenix, Arizona. If something is good for the Mayo Clinic, it has to be OK for the rest of us.
Social media also make it possible for smaller practices to compete with much larger practices that have huge marketing budgets. With very little expense, small practices—even solo practices—can develop a social media presence that can rival those of larger competitors.

HOW TO GET STARTED
There are four major social media programs to consider: Facebook, Twitter, YouTube, and blogging. We suggest that ObGyns who are ready to develop a social media presence begin with blogging, the focus of this article. We will cover Facebook, Twitter, and YouTube in Part 2 of this series.
Blogging is the easiest way to enter the world of social media. It’s free, can be accomplished reasonably quickly, and allows you to communicate with existing patients and attract new patients to your practice.
What is a blog? A blog is a Web site that is maintained with regular entries (posts) that invite comments from readers. Blogging allows feedback from people who visit your site and offers you the opportunity to respond to their comments. This creates a dialogue between you, your existing patients, and potential patients that is hard to achieve on an ordinary Web site.
The only expense for a blog is the cost of your time. There are several sites that will host your blog:
- WordPress.com offers free traffic stats, anti-spam features, search engine optimization, and more. Its platform is used by many popular blogs, including Forbes, Flickr, and CNN.
- Blogger.com (powered by Google) offers a user-friendly interface and smooth integration with the blogger’s Google account
- Blog.com provides the same basic features as other blog-hosting platforms, including free templates, but it charges a fee to keep ads off your site
- MovableType.com is a high-end hosting platform that charges a fee for its use
- LiveJournal.com provides its basic service at no charge but, like Blog.com, charges a fee to keep ads off your site.
We prefer WordPress.com because it was recommended in The Social Media Bible. WordPress.com offers tutorials that help you create a blog, enter content, and publish your material. You can access them at http://learn.wordpress.com.
We suggest that you develop your blog by incorporating a “hook” or other enticement to capture readers’ attention, keep your message relevant to their lives, and link the blog to your Web site so readers can find solutions to their medical problems.
Social media experts agree that regular posting is the key to success, particularly in regard to blogging. Commit to posting at least weekly. Visitors are more likely to return to your blog when they can count on regular updates.
Related Article: To blog or not to blog? What's the answer for you and your practice? Jennifer Gunter, MD (August 2011)
How to tell your story
One way to start your post is by offering a startling statistic or analogy. For example, if you are writing about breast cancer, you might begin by observing that more than 1,000 women under age 40 died of the disease in 2013—or that only lung cancer causes more cancer deaths in women.
Humor is another way to engage readers. We have found that people are attracted to funny anecdotes and stories. For example, when Dr. Baum is writing about erectile dysfunction, he might tell a story about arriving at a hotel and finding only 32 cents in his pocket to tip the bellman. When he offered the young bellman a copy of his new book, Impotence: It’s Reversible, the bellman replied, “Dr. Baum, if it’s all right with you, I’d just like to have the 32 cents.” In a blog post about this exchange, Dr. Baum might explain that the article is intended to give readers a little more than 32 cents’ worth of information about erectile dysfunction. The post would carry on from there.
Another option is to relate a compelling story about a recent patient (without using her name) that describes how you identified a problem, made a diagnosis, and resolved the patient’s complaint.
At the end of each blog post, we recommend that you invite readers to submit open-ended questions and comments. This motivates them to respond and starts a dialogue between your practice and potential new patients. Also include a call to action, preferably with a link from your blog to your Web site, inviting readers to visit your site or contact your practice to become a patient.
Most comments on your blog are likely to be positive, or to consist of requests for clarification or specific information. And most blog-hosting platforms allow you to review comments before they are published to your blog site. Any unnecessarily harsh or abusive comments can simply be rejected.
Once you have created a blog and begun to post regularly, we recommend that you check traffic to the site using the built-in analytics available through most hosting platforms. The traffic stats give you information on the number of visitors you have, how long they are spending at your blog, and how many are connecting to your main Web site. You can use this valuable information to identify what is working and tweak your blog posts accordingly.
Catchy titles make a difference
Strive to create titles that will capture the attention of your readers. People often decide whether or not to read a blog post on the basis of its title alone. Think of an effective title as a billboard. Drivers are speeding down the highway and have only 3 or 4 seconds to read the billboard and decide whether they will visit the restaurant, buy the product, or call for more information. The same holds true for titles on your blogs.
For example, Dr. Baum once titled a blog post “Urinary incontinence: Diagnosis and treatment.” It drew few readers. When he changed the title to “Urinary incontinence: You don’t have to depend on Depends,” nearly 1,000 readers commented on the post. Same article, different title.
Four pillars of a successful practice: 2. Attract new patients Neil H. Baum, MD (Four-part series, May 2013)
Pay attention to your practice Web site
We mentioned getting visitors from your blog site to your practice’s Web site. Once they arrive, two strategies are vital:
- visitor navigation
- patient-conversion systems.
Visitor navigation. The visitor comes to your Web site to get information that provides a solution to her problem. Once she lands on your site, you have less than 10 seconds to engage her; otherwise, she’ll leave instantly with the click of the mouse. Make it easy for her to find what she is looking for. For example, are the procedures and treatments you offer listed prominently so that the visitor can see them immediately and click on the link she wants? How about adding an icon, at the top right on every page, that says: “Schedule an appointment” or “Schedule a consultation.” The words you use (and their placement) are critically important if you want the visitor to become a patient!
Related Article: My #1 strategy for retaining patients Neil H. Baum, MD (Audiocast, March 2013)
Patient-conversion systems. Many Web sites are designed by people other than marketers. Even many Web-design companies focus on the look of the site rather than its main purpose: to convert visitors to patients.
If you want to get a Web-site visitor to schedule an appointment, your phone number should be clearly visible (along with the “Schedule an appointment” icon) on every page above the fold. “Above the fold” simply means that the visitor does not have to scroll down the page to see it. Believe it or not, many fancy Web sites fail to put these items in plain view!
And because you want to position yourself as a trusted authority in your field, patient testimonials are an important feature to include on your home page. And keep the information simple—stay away from technical jargon that the visitor will not understand.
Capture the visitor’s email address and use an auto-responder to follow up. You can get the visitor’s email address by offering something of value, such as a complimentary medical guide to a common condition. Once you have her email address, you have a way to stay in touch with the prospective patient and build a relationship of trust and confidence in your ability to solve her problem.
BOTTOM LINE: SOCIAL MEDIA ARE WORTH THE EFFORT
Social media marketing is a tool that most medical practices will be considering in the near future. A blog is a social media tool that can educate and inform existing patients and attract new ones to your practice. It is inexpensive, effective, and well worth the time and effort required to create a presence.
WE WANT TO HEAR FROM YOU!
Drop us a line and let us know what you think about current articles, which topics you'd like to see covered in future issues, and what challenges you face in daily practice. Tell us what you think by emailing us at: [email protected]
- Fox S, Duggan M. Health Online 2013: Summary of Findings. Pew Internet & American Life Project. http://www.pewinternet.org/Reports/2013/Health-online/Summary-of-Findings.aspx. Published January 15, 2013. Accessed January 9, 2014.
- Fox S. Health Topics: Health Information is a Popular Pursuit Online. Pew Internet & American Life Project. http://www.pewinternet.org/Reports/2011/HealthTopics/Part-1.aspx. Published February 1, 2011. Accessed January 9, 2014.
- Fox S. Health Topics: Eight in Ten Adult Internet Users Look for Information Online. Pew Internet & American Life Project. http://www.pewinternet.org/Reports/2011/Social-Life-of-Health-Info/Part-2/Section-1.aspx. Published May 12, 2011. Accessed January 9, 2014.

Neil H. Baum, MD, practices urology in New Orleans, Louisiana. He is Associate Clinical Professor of Urology at Tulane Medical School and Louisiana State University School of Medicine, both in New Orleans. He is also on the medical staff at Touro Infirmary in New Orleans, and East Jefferson General Hospital in Metairie, Louisiana. And he is the author of Marketing Your Clinical Practice: Ethically, Effectively, Economically (4th edition, 2009; Jones & Bartlett).

Ron Romano is CEO of Instant Marketing Systems in Toronto, Ontario.
The authors report no financial relationships relevant to this article.
Neil H. Baum, MD, practices urology in New Orleans, Louisiana. He is Associate Clinical Professor of Urology at Tulane Medical School and Louisiana State University School of Medicine, both in New Orleans. He is also on the medical staff at Touro Infirmary in New Orleans, and East Jefferson General Hospital in Metairie, Louisiana. And he is the author of Marketing Your Clinical Practice: Ethically, Effectively, Economically (4th edition, 2009; Jones & Bartlett).
Ron Romano is CEO of Instant Marketing Systems in Toronto, Ontario.
The authors report no financial relationships relevant to this article.
Neil H. Baum, MD, practices urology in New Orleans, Louisiana. He is Associate Clinical Professor of Urology at Tulane Medical School and Louisiana State University School of Medicine, both in New Orleans. He is also on the medical staff at Touro Infirmary in New Orleans, and East Jefferson General Hospital in Metairie, Louisiana. And he is the author of Marketing Your Clinical Practice: Ethically, Effectively, Economically (4th edition, 2009; Jones & Bartlett).
Ron Romano is CEO of Instant Marketing Systems in Toronto, Ontario.
The authors report no financial relationships relevant to this article.
Let’s rewind to the year 2000, the dawning of a new millennium. It was then that many physicians decided the time was ripe to establish a Web presence. It wasn’t that difficult, after all: Just take the practice’s three-color, trifold brochure and convert it into a Web-site template. A teenager could do it—and many did, sometimes guided by a college student in computer sciences.
These early implementers were confident that they could cruise into the 21st Century with this new technology. They had no idea how much the Internet would change…or how fast…but their basic impulse was a wise one, to harness the power of the Internet for the good of their patients and their practices.
In this four-part series, we focus on the rapidly expanding utilization of the Internet for health-related purposes. In Part 1, we focus on why it’s important to address the Web, particularly social media, and we zoom in on creating a blog for your practice. In Part 2, our focus will be the “big three”: Facebook, Twitter, and YouTube. We will take up search engine optimization and online reputation management in Parts 3 and 4, respectively.
WHY IS THE INTERNET IMPORTANT?
It isn’t uncommon for patients to arrive in their doctor’s office with a stack of pages downloaded from the Internet that describe their disease state or tests they are about to undergo. Many patients also are beginning to expect to interact with their physicians through Web sites, blogs, and Facebook and Twitter accounts.
Related Article: Why (and how) you should encourage your patients' search for health information on the Web Jennifer Gunter, MD (December 2011)
In fact, so much of health care is moving online that many physicians assume that everybody uses the Internet. The most recent data from the Pew Internet & American Life Project indicate that, in the United States, one in three adults have gone online to find out more about a medical condition, and 59% of all adults use the Internet to search for health information (TABLE 1).1,2 Eight in 10 people who regularly use the Internet look online for health information, making it the third most popular online pursuit tracked by the Pew project, after reading and sending email and using a search engine.
What types of health information do US adults look for online? Most people (66%) who use the Web to search for health information look for information on a specific disease or medical problem (see TABLE 2 for a list of other common health topics).3
The Pew Research Center also found that some demographic groups are more likely than others to seek health information online. They include:
- adults who have provided unpaid care to a parent, child, friend, or other loved one in the past 12 months
- women
- white adults
- adults aged 18 to 49 years
- adults with at least some college education
- adults in higher-income households.1
Check out the QUICK POLL on the OBG Management home page. To give your answer and see how other physicians have responded, Click Here.
WHAT ARE SOCIAL MEDIA AND WHY DO WE NEED THEM?
Social media encompass Web sites and other online communication applications used for social networking. Three of the most widely used media are Facebook, Twitter, and YouTube.
When someone once asked hockey great Wayne Gretzky about his sport strategy, he replied: “I don’t skate to where the puck is or where the puck has been; I skate to where the puck is going to be.” Social media are where the puck (ie, our patients) are going to be today and tomorrow.
If we review other media launches, we discover that it took nearly 40 years for radio to attract 50 million listeners, and 13 years for television to reach 50 million viewers. But it took only 4 years for the Internet to achieve 50 million users. Facebook alone reached 100 million users in just 9 months!
Just a decade ago, the Mayo Clinic relied on standard marketing techniques using radio, TV, and print media to attract new patients. Today, the Mayo Clinic makes use of Facebook, Twitter, YouTube, podcasts, and blogging. The Mayo Clinic even has developed a Center for Social Media to focus on the use of social media for its centers in Rochester, Minnesota; Jacksonville, Florida; and Phoenix, Arizona. If something is good for the Mayo Clinic, it has to be OK for the rest of us.
Social media also make it possible for smaller practices to compete with much larger practices that have huge marketing budgets. With very little expense, small practices—even solo practices—can develop a social media presence that can rival those of larger competitors.
HOW TO GET STARTED
There are four major social media programs to consider: Facebook, Twitter, YouTube, and blogging. We suggest that ObGyns who are ready to develop a social media presence begin with blogging, the focus of this article. We will cover Facebook, Twitter, and YouTube in Part 2 of this series.
Blogging is the easiest way to enter the world of social media. It’s free, can be accomplished reasonably quickly, and allows you to communicate with existing patients and attract new patients to your practice.
What is a blog? A blog is a Web site that is maintained with regular entries (posts) that invite comments from readers. Blogging allows feedback from people who visit your site and offers you the opportunity to respond to their comments. This creates a dialogue between you, your existing patients, and potential patients that is hard to achieve on an ordinary Web site.
The only expense for a blog is the cost of your time. There are several sites that will host your blog:
- WordPress.com offers free traffic stats, anti-spam features, search engine optimization, and more. Its platform is used by many popular blogs, including Forbes, Flickr, and CNN.
- Blogger.com (powered by Google) offers a user-friendly interface and smooth integration with the blogger’s Google account
- Blog.com provides the same basic features as other blog-hosting platforms, including free templates, but it charges a fee to keep ads off your site
- MovableType.com is a high-end hosting platform that charges a fee for its use
- LiveJournal.com provides its basic service at no charge but, like Blog.com, charges a fee to keep ads off your site.
We prefer WordPress.com because it was recommended in The Social Media Bible. WordPress.com offers tutorials that help you create a blog, enter content, and publish your material. You can access them at http://learn.wordpress.com.
We suggest that you develop your blog by incorporating a “hook” or other enticement to capture readers’ attention, keep your message relevant to their lives, and link the blog to your Web site so readers can find solutions to their medical problems.
Social media experts agree that regular posting is the key to success, particularly in regard to blogging. Commit to posting at least weekly. Visitors are more likely to return to your blog when they can count on regular updates.
Related Article: To blog or not to blog? What's the answer for you and your practice? Jennifer Gunter, MD (August 2011)
How to tell your story
One way to start your post is by offering a startling statistic or analogy. For example, if you are writing about breast cancer, you might begin by observing that more than 1,000 women under age 40 died of the disease in 2013—or that only lung cancer causes more cancer deaths in women.
Humor is another way to engage readers. We have found that people are attracted to funny anecdotes and stories. For example, when Dr. Baum is writing about erectile dysfunction, he might tell a story about arriving at a hotel and finding only 32 cents in his pocket to tip the bellman. When he offered the young bellman a copy of his new book, Impotence: It’s Reversible, the bellman replied, “Dr. Baum, if it’s all right with you, I’d just like to have the 32 cents.” In a blog post about this exchange, Dr. Baum might explain that the article is intended to give readers a little more than 32 cents’ worth of information about erectile dysfunction. The post would carry on from there.
Another option is to relate a compelling story about a recent patient (without using her name) that describes how you identified a problem, made a diagnosis, and resolved the patient’s complaint.
At the end of each blog post, we recommend that you invite readers to submit open-ended questions and comments. This motivates them to respond and starts a dialogue between your practice and potential new patients. Also include a call to action, preferably with a link from your blog to your Web site, inviting readers to visit your site or contact your practice to become a patient.
Most comments on your blog are likely to be positive, or to consist of requests for clarification or specific information. And most blog-hosting platforms allow you to review comments before they are published to your blog site. Any unnecessarily harsh or abusive comments can simply be rejected.
Once you have created a blog and begun to post regularly, we recommend that you check traffic to the site using the built-in analytics available through most hosting platforms. The traffic stats give you information on the number of visitors you have, how long they are spending at your blog, and how many are connecting to your main Web site. You can use this valuable information to identify what is working and tweak your blog posts accordingly.
Catchy titles make a difference
Strive to create titles that will capture the attention of your readers. People often decide whether or not to read a blog post on the basis of its title alone. Think of an effective title as a billboard. Drivers are speeding down the highway and have only 3 or 4 seconds to read the billboard and decide whether they will visit the restaurant, buy the product, or call for more information. The same holds true for titles on your blogs.
For example, Dr. Baum once titled a blog post “Urinary incontinence: Diagnosis and treatment.” It drew few readers. When he changed the title to “Urinary incontinence: You don’t have to depend on Depends,” nearly 1,000 readers commented on the post. Same article, different title.
Four pillars of a successful practice: 2. Attract new patients Neil H. Baum, MD (Four-part series, May 2013)
Pay attention to your practice Web site
We mentioned getting visitors from your blog site to your practice’s Web site. Once they arrive, two strategies are vital:
- visitor navigation
- patient-conversion systems.
Visitor navigation. The visitor comes to your Web site to get information that provides a solution to her problem. Once she lands on your site, you have less than 10 seconds to engage her; otherwise, she’ll leave instantly with the click of the mouse. Make it easy for her to find what she is looking for. For example, are the procedures and treatments you offer listed prominently so that the visitor can see them immediately and click on the link she wants? How about adding an icon, at the top right on every page, that says: “Schedule an appointment” or “Schedule a consultation.” The words you use (and their placement) are critically important if you want the visitor to become a patient!
Related Article: My #1 strategy for retaining patients Neil H. Baum, MD (Audiocast, March 2013)
Patient-conversion systems. Many Web sites are designed by people other than marketers. Even many Web-design companies focus on the look of the site rather than its main purpose: to convert visitors to patients.
If you want to get a Web-site visitor to schedule an appointment, your phone number should be clearly visible (along with the “Schedule an appointment” icon) on every page above the fold. “Above the fold” simply means that the visitor does not have to scroll down the page to see it. Believe it or not, many fancy Web sites fail to put these items in plain view!
And because you want to position yourself as a trusted authority in your field, patient testimonials are an important feature to include on your home page. And keep the information simple—stay away from technical jargon that the visitor will not understand.
Capture the visitor’s email address and use an auto-responder to follow up. You can get the visitor’s email address by offering something of value, such as a complimentary medical guide to a common condition. Once you have her email address, you have a way to stay in touch with the prospective patient and build a relationship of trust and confidence in your ability to solve her problem.
BOTTOM LINE: SOCIAL MEDIA ARE WORTH THE EFFORT
Social media marketing is a tool that most medical practices will be considering in the near future. A blog is a social media tool that can educate and inform existing patients and attract new ones to your practice. It is inexpensive, effective, and well worth the time and effort required to create a presence.
WE WANT TO HEAR FROM YOU!
Drop us a line and let us know what you think about current articles, which topics you'd like to see covered in future issues, and what challenges you face in daily practice. Tell us what you think by emailing us at: [email protected]
Let’s rewind to the year 2000, the dawning of a new millennium. It was then that many physicians decided the time was ripe to establish a Web presence. It wasn’t that difficult, after all: Just take the practice’s three-color, trifold brochure and convert it into a Web-site template. A teenager could do it—and many did, sometimes guided by a college student in computer sciences.
These early implementers were confident that they could cruise into the 21st Century with this new technology. They had no idea how much the Internet would change…or how fast…but their basic impulse was a wise one, to harness the power of the Internet for the good of their patients and their practices.
In this four-part series, we focus on the rapidly expanding utilization of the Internet for health-related purposes. In Part 1, we focus on why it’s important to address the Web, particularly social media, and we zoom in on creating a blog for your practice. In Part 2, our focus will be the “big three”: Facebook, Twitter, and YouTube. We will take up search engine optimization and online reputation management in Parts 3 and 4, respectively.
WHY IS THE INTERNET IMPORTANT?
It isn’t uncommon for patients to arrive in their doctor’s office with a stack of pages downloaded from the Internet that describe their disease state or tests they are about to undergo. Many patients also are beginning to expect to interact with their physicians through Web sites, blogs, and Facebook and Twitter accounts.
Related Article: Why (and how) you should encourage your patients' search for health information on the Web Jennifer Gunter, MD (December 2011)
In fact, so much of health care is moving online that many physicians assume that everybody uses the Internet. The most recent data from the Pew Internet & American Life Project indicate that, in the United States, one in three adults have gone online to find out more about a medical condition, and 59% of all adults use the Internet to search for health information (TABLE 1).1,2 Eight in 10 people who regularly use the Internet look online for health information, making it the third most popular online pursuit tracked by the Pew project, after reading and sending email and using a search engine.
What types of health information do US adults look for online? Most people (66%) who use the Web to search for health information look for information on a specific disease or medical problem (see TABLE 2 for a list of other common health topics).3
The Pew Research Center also found that some demographic groups are more likely than others to seek health information online. They include:
- adults who have provided unpaid care to a parent, child, friend, or other loved one in the past 12 months
- women
- white adults
- adults aged 18 to 49 years
- adults with at least some college education
- adults in higher-income households.1
Check out the QUICK POLL on the OBG Management home page. To give your answer and see how other physicians have responded, Click Here.
WHAT ARE SOCIAL MEDIA AND WHY DO WE NEED THEM?
Social media encompass Web sites and other online communication applications used for social networking. Three of the most widely used media are Facebook, Twitter, and YouTube.
When someone once asked hockey great Wayne Gretzky about his sport strategy, he replied: “I don’t skate to where the puck is or where the puck has been; I skate to where the puck is going to be.” Social media are where the puck (ie, our patients) are going to be today and tomorrow.
If we review other media launches, we discover that it took nearly 40 years for radio to attract 50 million listeners, and 13 years for television to reach 50 million viewers. But it took only 4 years for the Internet to achieve 50 million users. Facebook alone reached 100 million users in just 9 months!
Just a decade ago, the Mayo Clinic relied on standard marketing techniques using radio, TV, and print media to attract new patients. Today, the Mayo Clinic makes use of Facebook, Twitter, YouTube, podcasts, and blogging. The Mayo Clinic even has developed a Center for Social Media to focus on the use of social media for its centers in Rochester, Minnesota; Jacksonville, Florida; and Phoenix, Arizona. If something is good for the Mayo Clinic, it has to be OK for the rest of us.
Social media also make it possible for smaller practices to compete with much larger practices that have huge marketing budgets. With very little expense, small practices—even solo practices—can develop a social media presence that can rival those of larger competitors.
HOW TO GET STARTED
There are four major social media programs to consider: Facebook, Twitter, YouTube, and blogging. We suggest that ObGyns who are ready to develop a social media presence begin with blogging, the focus of this article. We will cover Facebook, Twitter, and YouTube in Part 2 of this series.
Blogging is the easiest way to enter the world of social media. It’s free, can be accomplished reasonably quickly, and allows you to communicate with existing patients and attract new patients to your practice.
What is a blog? A blog is a Web site that is maintained with regular entries (posts) that invite comments from readers. Blogging allows feedback from people who visit your site and offers you the opportunity to respond to their comments. This creates a dialogue between you, your existing patients, and potential patients that is hard to achieve on an ordinary Web site.
The only expense for a blog is the cost of your time. There are several sites that will host your blog:
- WordPress.com offers free traffic stats, anti-spam features, search engine optimization, and more. Its platform is used by many popular blogs, including Forbes, Flickr, and CNN.
- Blogger.com (powered by Google) offers a user-friendly interface and smooth integration with the blogger’s Google account
- Blog.com provides the same basic features as other blog-hosting platforms, including free templates, but it charges a fee to keep ads off your site
- MovableType.com is a high-end hosting platform that charges a fee for its use
- LiveJournal.com provides its basic service at no charge but, like Blog.com, charges a fee to keep ads off your site.
We prefer WordPress.com because it was recommended in The Social Media Bible. WordPress.com offers tutorials that help you create a blog, enter content, and publish your material. You can access them at http://learn.wordpress.com.
We suggest that you develop your blog by incorporating a “hook” or other enticement to capture readers’ attention, keep your message relevant to their lives, and link the blog to your Web site so readers can find solutions to their medical problems.
Social media experts agree that regular posting is the key to success, particularly in regard to blogging. Commit to posting at least weekly. Visitors are more likely to return to your blog when they can count on regular updates.
Related Article: To blog or not to blog? What's the answer for you and your practice? Jennifer Gunter, MD (August 2011)
How to tell your story
One way to start your post is by offering a startling statistic or analogy. For example, if you are writing about breast cancer, you might begin by observing that more than 1,000 women under age 40 died of the disease in 2013—or that only lung cancer causes more cancer deaths in women.
Humor is another way to engage readers. We have found that people are attracted to funny anecdotes and stories. For example, when Dr. Baum is writing about erectile dysfunction, he might tell a story about arriving at a hotel and finding only 32 cents in his pocket to tip the bellman. When he offered the young bellman a copy of his new book, Impotence: It’s Reversible, the bellman replied, “Dr. Baum, if it’s all right with you, I’d just like to have the 32 cents.” In a blog post about this exchange, Dr. Baum might explain that the article is intended to give readers a little more than 32 cents’ worth of information about erectile dysfunction. The post would carry on from there.
Another option is to relate a compelling story about a recent patient (without using her name) that describes how you identified a problem, made a diagnosis, and resolved the patient’s complaint.
At the end of each blog post, we recommend that you invite readers to submit open-ended questions and comments. This motivates them to respond and starts a dialogue between your practice and potential new patients. Also include a call to action, preferably with a link from your blog to your Web site, inviting readers to visit your site or contact your practice to become a patient.
Most comments on your blog are likely to be positive, or to consist of requests for clarification or specific information. And most blog-hosting platforms allow you to review comments before they are published to your blog site. Any unnecessarily harsh or abusive comments can simply be rejected.
Once you have created a blog and begun to post regularly, we recommend that you check traffic to the site using the built-in analytics available through most hosting platforms. The traffic stats give you information on the number of visitors you have, how long they are spending at your blog, and how many are connecting to your main Web site. You can use this valuable information to identify what is working and tweak your blog posts accordingly.
Catchy titles make a difference
Strive to create titles that will capture the attention of your readers. People often decide whether or not to read a blog post on the basis of its title alone. Think of an effective title as a billboard. Drivers are speeding down the highway and have only 3 or 4 seconds to read the billboard and decide whether they will visit the restaurant, buy the product, or call for more information. The same holds true for titles on your blogs.
For example, Dr. Baum once titled a blog post “Urinary incontinence: Diagnosis and treatment.” It drew few readers. When he changed the title to “Urinary incontinence: You don’t have to depend on Depends,” nearly 1,000 readers commented on the post. Same article, different title.
Four pillars of a successful practice: 2. Attract new patients Neil H. Baum, MD (Four-part series, May 2013)
Pay attention to your practice Web site
We mentioned getting visitors from your blog site to your practice’s Web site. Once they arrive, two strategies are vital:
- visitor navigation
- patient-conversion systems.
Visitor navigation. The visitor comes to your Web site to get information that provides a solution to her problem. Once she lands on your site, you have less than 10 seconds to engage her; otherwise, she’ll leave instantly with the click of the mouse. Make it easy for her to find what she is looking for. For example, are the procedures and treatments you offer listed prominently so that the visitor can see them immediately and click on the link she wants? How about adding an icon, at the top right on every page, that says: “Schedule an appointment” or “Schedule a consultation.” The words you use (and their placement) are critically important if you want the visitor to become a patient!
Related Article: My #1 strategy for retaining patients Neil H. Baum, MD (Audiocast, March 2013)
Patient-conversion systems. Many Web sites are designed by people other than marketers. Even many Web-design companies focus on the look of the site rather than its main purpose: to convert visitors to patients.
If you want to get a Web-site visitor to schedule an appointment, your phone number should be clearly visible (along with the “Schedule an appointment” icon) on every page above the fold. “Above the fold” simply means that the visitor does not have to scroll down the page to see it. Believe it or not, many fancy Web sites fail to put these items in plain view!
And because you want to position yourself as a trusted authority in your field, patient testimonials are an important feature to include on your home page. And keep the information simple—stay away from technical jargon that the visitor will not understand.
Capture the visitor’s email address and use an auto-responder to follow up. You can get the visitor’s email address by offering something of value, such as a complimentary medical guide to a common condition. Once you have her email address, you have a way to stay in touch with the prospective patient and build a relationship of trust and confidence in your ability to solve her problem.
BOTTOM LINE: SOCIAL MEDIA ARE WORTH THE EFFORT
Social media marketing is a tool that most medical practices will be considering in the near future. A blog is a social media tool that can educate and inform existing patients and attract new ones to your practice. It is inexpensive, effective, and well worth the time and effort required to create a presence.
WE WANT TO HEAR FROM YOU!
Drop us a line and let us know what you think about current articles, which topics you'd like to see covered in future issues, and what challenges you face in daily practice. Tell us what you think by emailing us at: [email protected]
- Fox S, Duggan M. Health Online 2013: Summary of Findings. Pew Internet & American Life Project. http://www.pewinternet.org/Reports/2013/Health-online/Summary-of-Findings.aspx. Published January 15, 2013. Accessed January 9, 2014.
- Fox S. Health Topics: Health Information is a Popular Pursuit Online. Pew Internet & American Life Project. http://www.pewinternet.org/Reports/2011/HealthTopics/Part-1.aspx. Published February 1, 2011. Accessed January 9, 2014.
- Fox S. Health Topics: Eight in Ten Adult Internet Users Look for Information Online. Pew Internet & American Life Project. http://www.pewinternet.org/Reports/2011/Social-Life-of-Health-Info/Part-2/Section-1.aspx. Published May 12, 2011. Accessed January 9, 2014.
- Fox S, Duggan M. Health Online 2013: Summary of Findings. Pew Internet & American Life Project. http://www.pewinternet.org/Reports/2013/Health-online/Summary-of-Findings.aspx. Published January 15, 2013. Accessed January 9, 2014.
- Fox S. Health Topics: Health Information is a Popular Pursuit Online. Pew Internet & American Life Project. http://www.pewinternet.org/Reports/2011/HealthTopics/Part-1.aspx. Published February 1, 2011. Accessed January 9, 2014.
- Fox S. Health Topics: Eight in Ten Adult Internet Users Look for Information Online. Pew Internet & American Life Project. http://www.pewinternet.org/Reports/2011/Social-Life-of-Health-Info/Part-2/Section-1.aspx. Published May 12, 2011. Accessed January 9, 2014.
THE SERIES: USING THE INTERNET IN YOUR PRACTICE
Part 2: Generating new patients using social media (April 2014)
Part 3: Search engine optimization
Part 4: Online reputation management
(Look for Parts 2 through 4 in 2014)

Obesity in the elderly: More complicated than you think
Should older obese people try to lose weight? Such a simple question is more complicated than one would think.
At issue is whether obesity is harmful in older people, and whether treating it will reduce their health risks. True, obesity is an independent risk factor for cardiovascular disease and is associated with many comorbidities, including type 2 diabetes mellitus, hyperlipidemia, heart failure, and hypertension.1 An independent association also exists between obesity and all-cause mortality.2 However, there is also evidence suggesting that obesity in this age group is associated with a lower, not higher, risk of death—a finding termed the obesity paradox.3 And for that matter, what exactly constitutes obesity in elderly people, who naturally undergo changes in body composition as they age?
This article examines the literature on these controversial issues, including changes in body composition with age, the definition of obesity in older adults, the obesity paradox, and treatment of obesity in older adults.
AMERICANS ARE GETTING OLDER—AND BIGGER
Americans are living longer than ever before; life expectancy has reached a new high of 77.8 years.4,5 According to the US Census Bureau,6 about 27 million people in the United States are over age 70, and this number is expected to nearly double by 2030.

Meanwhile, the prevalence of obesity, defined as a body mass index (BMI) of 30 kg/m2 or higher, has increased in the last 25 years in all age groups in the United States, including those age 65 and older.7,8 These two trends add up to an increase in the number of obese older people. In 2000, 22.9% of people age 60 to 69 and 15.5% of those over age 70 and older were obese.9 This amounted to a 56% increase in the former group and a 36% increase in the latter group in the interval since 1991.5,9
BUT WHAT CONSTITUTES ‘OBESITY’?
Obesity is the excess accumulation of body fat, leading to a higher risk of medical illness and premature death. But measuring it is not as simple as one might think.
The body mass index can mislead
The BMI, ie, weight in kilograms divided by the square of the height in meters, correlates fairly well with body fat stores and is generally used to classify medical risk.
However, the BMI can classify some older people as overweight (BMI 30.0–34.9 kg/m2) or obese (BMI ≥ 35.0 kg/m2) who actually do not have an excess of body fat—and can fail to classify others as overweight or obese who do. For example, if a person loses height as a result of vertebral compression fractures, his or her BMI would become higher, even with no change in weight or body fat. Conversely, changes in body composition with age, including loss of muscle and an increase in fat, may not be reflected in the BMI, even if the person really does have too much body fat.10
This second limitation of the BMI is important when estimating risk in older adults, who have a particular fat distribution. Visceral, subcutaneous, intramuscular, and intrahepatic fat increase with age, and they are all risk factors for insulin resistance and type 2 diabetes mellitus.11 And in older people, having too much visceral fat is more prevalent than the BMI might predict.10
Percent body fat awaits investigation
Percent body fat is another way to assess body fat. Defined as the total weight of fat divided by total weight, it is measured in various ways.
Dual-energy x-ray absorptiometry, computed tomography, and magnetic resonance imaging can measure percent body fat, and they can differentiate visceral from subcutaneous fat (which is less metabolically active). Unfortunately, most of these tests are used for this purpose only in research, and they are relatively expensive.
Commercially available bioelectrical impedance devices send a weak electric current through the body and measure the resistance, and using this information and four other factors (height, weight, age, and sex), they calculate percent body fat. This method is fast, easy, painless, and cheap. A disadvantage is that the handheld devices measure body composition of the upper body only. Because the lower body is excluded, they do not give an accurate measurement of body fat of the abdomen and hips. Also, they cannot differentiate visceral from subcutaneous fat.
Bioelectrical impedance devices work well in healthy individuals with stable water balance. The values are only an estimate of fat-free mass, and therefore this method is not the gold standard for assessing body fat. Bioelectrical impedance is better at tracking body composition in an individual over time than at diagnosing obesity.
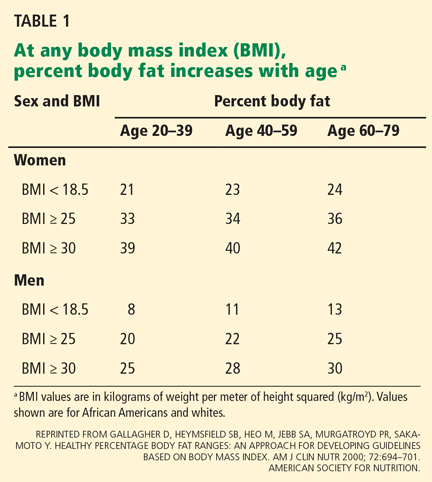
Percent body fat can vary by sex and race. Asians, for example, have higher percent body fats at lower BMIs, particularly when younger.12 Also, Gallagher et al12 found that percent body fat increased with age at every given BMI in both men and women (Table 1).
The traditional universal cutoffs for defining obesity by percent body fat are 25% in men and 35% in women. However, research has indicated that cutoffs of 20% to 25% in men and 30% to 38% in women may better identify those at risk of metabolic disease.13 Guidelines and evidence-based cutoffs for percent body fat must await further investigation.
Waist circumference is useful
In older adults, obesity can be diagnosed by a measurement such as waist circumference, which correlates highly with total fat and intra-abdominal fat.14 It is very cost-effective, simple, and useful for the office assessment of adiposity.
The measurement should be made halfway between the iliac crest and the lower anterior ribs, with the patient standing, and at the end of expiration.
The traditional standard for waist circumference is less than 89 cm (35 inches) for women and 102 cm (40 inches) for men. However, opinion differs, and different reference ranges exist depending on ethnicity. Additionally, because stature and body composition change with age, concerns have been raised about misclassification of the health risks related to obesity in older adults using the current standard.15,16
The waist circumference is as good as or even better than the BMI as a measure of excess adiposity in older adults.16–18 This is in part because of the age-dependent height decrease in older adults.15,19 (Recall that the BMI is calculated using the height squared as the denominator; as a result, the BMI would give a higher reading and thus an overestimate of total body fat.) Conversely, we can underestimate the amount of adiposity because of decreases in abdominal muscle tone.17
Cutoffs for waist circumference should be age-specific.16
Investigators in the Netherlands15,16 prospectively took 4,996 measurements in 2,232 people with a mean age of 70, from 1992 through 2006. They concluded that the best cutoffs for predicting the health risks of obesity in the elderly were 109 cm (43 inches) in men and 98 cm (39 inches) in women.
A group of researchers has proposed that the cutoffs be shifted upward in older adults, with new values for those age 70 and over.20 The Health Survey for England aimed to describe the patterns and trends in waist circumference and abdominal obesity and overweight in people age 70 through 89, comparing both the standard and the new cutoffs. Optimal cutoffs recommended for abdominal obesity for patients age 70 and older were 100 to 106 cm in men and 99 cm in women.20 Estimates of the prevalence of abdominal obesity are much lower using the new cutoffs.
SARCOPENIA: LOSS OF MUSCLE WITH AGE
With age comes sarcopenia—the progressive loss of muscle mass, primarily skeletal muscle, resulting in a decrease in strength and power.21 The process begins as early as the 20s or 30s.22 It is distinct from wasting (involuntary weight loss from inadequate intake), seen in starvation.21
Sarcopenia is defined as an appendicular skeletal muscle mass index (the appendicular skeletal mass divided by the square of the height in meters) of less than 2 standard deviations below a young adult reference, and a percentage of body fat over the 60th percentile for the individual’s sex and age.23,24 Estimates of its prevalence vary, but it is common and it increases with age.14,20
Sarcopenic obesity: Less muscle, more fat
Progressive loss of skeletal muscle with age, along with an increase and redistribution of body fat, is known as sarcopenic obesity.25 It is associated with higher morbidity and mortality rates as well as a decline in functional strength, which leads to frailty.23 This loss of muscle mass may go unnoticed in an older person until he or she begins to lose physical function.
As noted, in an older person with sarcopenic obesity, the BMI may mislead because of the high percentage of fat and the low lean mass.26
Why we change with age
This change in body composition with age is a result of several factors. Illness or inactivity can lead to loss of muscle, while body fat is preserved.17 The combination of reduced physical activity, a lower resting metabolic rate, and an unchanged intake of food can increase the likelihood of sarcopenia.27 Also possibly contributing are hormonal changes, including reduced production of growth hormone and testosterone and decreased responsiveness to thyroid hormone and leptin.28
Moreover, the interaction of several factors can lead to a vicious circle of progressive loss of muscle and increase in fat. As people age, their physical activity tends to decrease, resulting in muscle loss. As muscle mass decreases, the amount of available insulin-responsive tissue is reduced, resulting in insulin resistance, which in turn promotes the metabolic syndrome and an increase in fat. With more fat, people produce more of the adipokines tumor necrosis factor alpha and interleukin 6, which further promote insulin resistance.
Other changes contribute to a decrease in muscle quality and performance, including an increase in intramuscular and intrahepatic fat, which is associated with insulin resistance.11 The increases in adipose stores occur mostly in intra-abdominal fat rather than in subcutaneous fat.
ADVERSE EFFECTS OF OBESITY
A number of comorbidities arise with obesity, regardless of age.19
The diseases most strongly associated with obesity are the metabolic syndrome and type 2 diabetes mellitus.17 Studies have shown that in older adults, obesity as measured by waist circumference is associated with hyperglycemia and dyslipidemia.29,30
Metabolic abnormalities may ensue in obese older people through complex mechanisms involving an age-related decline in sex hormones. For example, late-onset hypogonadism in men, which is more common in those who are obese, is related to the metabolic syndrome.29
These mechanisms are also complex in women. Because estrogens can be produced in adipose tissue, obese postmenopausal women have higher concentrations of estrogens than their lean counterparts, and this may lead to metabolic abnormalities.31 (On the other hand, higher estrogen levels in obese menopausal women can protect against osteoporosis by increasing bone mass.)
Older people who weigh more and have more adipose tissue, especially those who became obese at a young age, have a greater risk of osteoarthritis of the knee,32,33 which when combined with obesity can cause disability and physical impairment.19 And cardiovascular risk factors,18,33 hypertension,34 and certain cancers35 are more common in older people with higher waist circumference.
THE OBESITY PARADOX
In general, obesity in younger adults has been shown to shorten life expectancy. This risk of death is often associated with obesity-related health problems.
In older people, the effect of obesity is much more complex.36 The optimal weight in terms of survival increases with age. More interesting is the finding that although the risk of cardiovascular disease is higher in overweight or obese older adults, studies also suggest that in this age group, being overweight or obese is paradoxically associated with lower mortality rates from these diseases.26 This phenomenon is called the obesity paradox.37
For those over age 75, the relative risk of death from all causes and from cardiovascular disease has been found to decrease with increasing BMI.25 The relationship between BMI and death from all causes in older adults may actually be a U-shaped curve, meaning that the risk of death rises at both extremes of BMI values.26
Possible explanations for the paradox
Several hypotheses have been proposed to explain the change in the relationship between BMI and the risk of death that occurs with aging.
The BMI is an imperfect measure of obesity. The obesity paradox may be an artifact of using the BMI to measure obesity in older adults.17 As described above, sarcopenic obesity must be considered in those over age 65 because the BMI does not differentiate between fat and muscle. Older adults tend to have a higher proportion of body and visceral fat that is distributed differently, making the waist circumference or waist-hip ratio a more appropriate measure of obesity in this group.38 Janssen et al39 found that in people age 65 and older, after controlling for waist circumference, higher BMI values were associated with lower death rates; after controlling for BMI, waist circumference was associated with a higher risk of death.
The survival effect suggests that people who are susceptible to the negative effects of obesity die sooner,40 and those who survive until old age may be resistant to the effects of obesity.41 If true, the survival effect would explain why the death rate seems to be unaffected by BMI in the older population.
Unhealthy weight loss. Smoking and diseases such as cancer that can cause early death may also induce weight loss, further complicating the relationship between BMI and death.19 After age 80, the association between BMI and the risk of death is weak because those with a low BMI include not only those who have always been lean and physically active, but also those who lost weight through chronic ill health or smoking.17
Further study needed. Thus, a number of confounding variables may muddy the association between obesity and death in older adults. Obesity should not be misinterpreted as being harmless or beneficial in older adults. Stevens et al36 found that a greater BMI was associated with a higher rate of death from all causes and from cardiovascular disease in men and women up to age 75, but that the relative risk of death associated with a greater BMI decreased with age.
Optimal BMI targets in older people have yet to be validated in a large prospective trial. However, multiple studies have examined the relationship between BMI and all-cause mortality in older adults and have identified a BMI of 24 to 35 as “ideal” and associated with the lowest risk of death, with a lower range for men and a higher range for women.42,43 The topic has been reviewed by Oreopoulos et al.26 More research is needed to evaluate this relationship.
THE BENEFIT OF WEIGHT LOSS IN OLDER ADULTS IS CONTROVERSIAL
In younger obese people, weight loss brings a multitude of benefits by reducing the risk of complications arising from obesity. However, in older adults, the effects of weight loss remain controversial, and evidence to guide treatment is limited.44,45 The few trials that have been published have typically focused on cardiovascular risk factors rather than physical function.45
In a 1-year trial, 107 people age 65 or older were randomized to a control group, to weight management, to exercise, or to weight management plus exercise. The combination of weight loss and exercise yielded the greatest improvement in physical function.46
Intentional vs unintentional weight loss
Intentional weight loss is altogether different from unintentional weight loss.
In most cases, weight loss in older adults is unintentional and may indicate underlying disease and impending death.17 For example, older men who lose weight unintentionally have significantly greater rates of smoking, disability, cancer, and respiratory disease and less obesity and physical activity than those who lose weight intentionally.47
Studies have shown an increase in life expectancy in older patients with type 2 diabetes mellitus who lost weight intentionally.48,49 In fact, moderate weight loss—just 5% to 10%—has been shown to improve cardiovascular risk factors,44 osteoarthritis, and type 2 diabetes.50
Bales and Buhr44 performed a systematic review of 16 studies that had lasted at least 6 months. Patients were age 60 or older with a minimum baseline BMI of 27 kg/m2 who intentionally lost at least 3% of body weight or 2 kg. Levels of the inflammatory markers C-reactive protein, tumor necrosis factor alpha, and interleukin 6 declined with weight, along with blood pressure, fasting glucose, waist circumference, and low-density lipoprotein cholesterol. On the downside, bone mineral density and lean body mass also declined slightly. The best way to avoid losing lean body mass and to preserve bone density during weight loss is to include a program of resistance-training exercises.
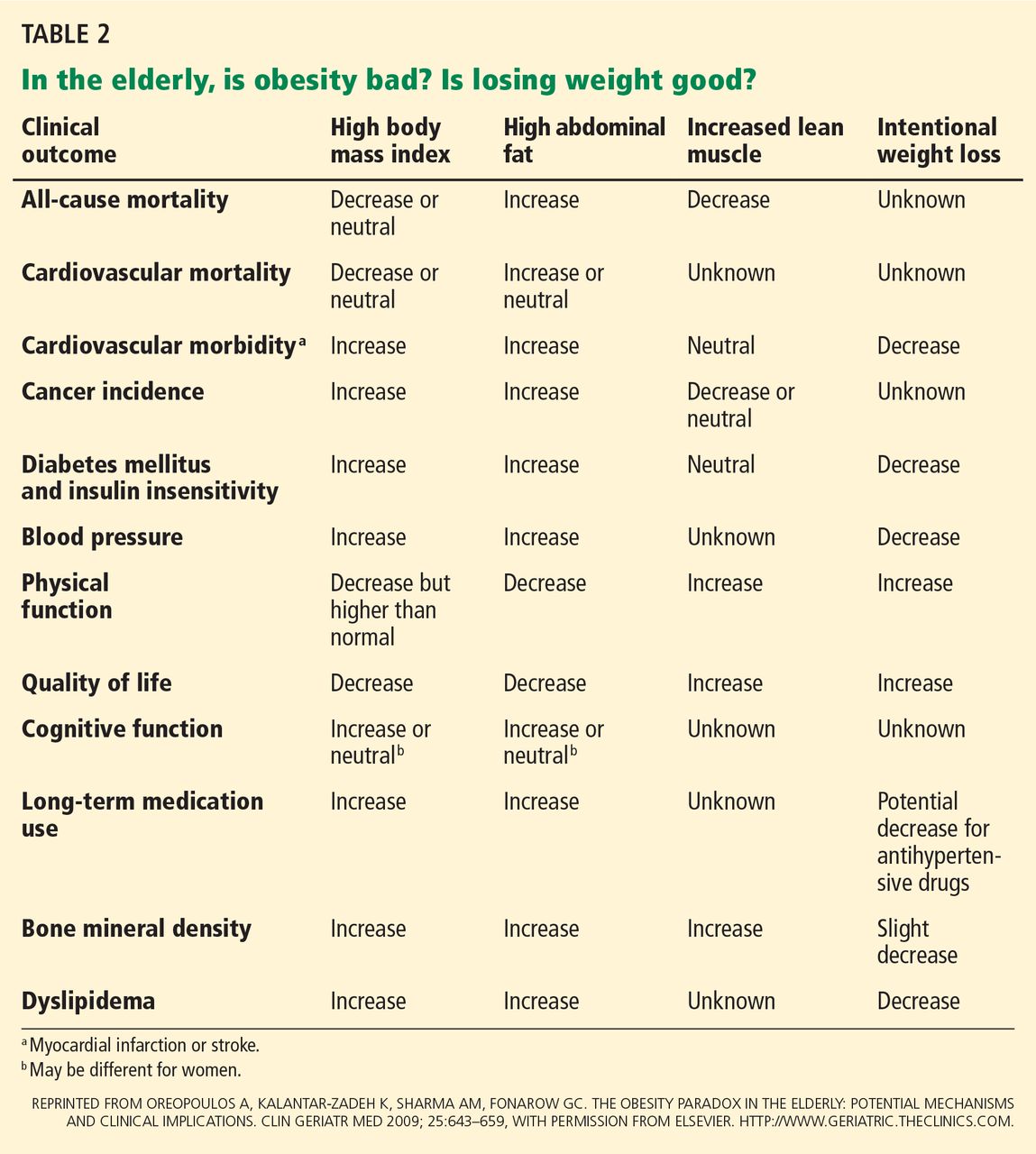
No clinical trial has evaluated the effects of intentional weight loss on death rates in older obese people.25 As a result, evidence-based recommendations cannot be made. Rather, advice on weight loss must be individualized, with special emphasis on the patient’s weight history and medical comorbidities.44
Oreopoulos et al26 summarized the possible effects of BMI, abdominal fat, lean body mass, and intentional weight loss on morbidity and mortality outcomes in older adults (Table 2).
TREATMENT GUIDELINES AND RECOMMENDATIONS
Many of the methods of weight management in older adults are the same as in young and middle-aged adults.51 Recommendations for all age groups include lifestyle changes, increased activity, dietary changes, drug therapy, and bariatric surgery.
Whether there should be separate guidelines for older adults is controversial. In view of the obesity paradox, physicians have been reluctant to recommend weight loss in elderly patients. Caution is advised in recommending weight loss solely on the basis of body weight, as studies have shown that the weight associated with maximal survival increases with age. Because of age-related changes in body composition and reduced energy requirements and expenditure, recommendations for the young and middle-aged should not be applied directly to older adults.
In this group, especially those who have survived into old age with good health and an intact functional status, one could argue that significant caloric restriction should not be recommended. In these people, the goal is often to maintain weight and incorporate a daily exercise program rather than to aggressively lose weight. Adding resistance training can improve physical function, which can improve quality of life. There is less emphasis on cardiovascular risk, but both outcomes apply for both age groups.52
Intentional weight loss should be recommended to high-risk older adults, including those with cardiovascular disease, type 2 diabetes mellitus, and metabolic syndrome, because the absolute risk of death and morbidity is higher in this group. Most health benefits can be achieved with modest weight loss.53 Potential benefits include prevention of cognitive impairment, protection from bone fractures, an increase in antioxidant defense, a reserve of fat and energy stores, and an increase in longevity.26
Treatment differs from that in the younger population primarily because of the importance of preventing loss of muscle with intentional weight loss. People of all ages who lose weight intentionally lose fat and, to a lesser extent, skeletal muscle. Older patients have already lost muscle mass, but further changes in body composition, especially a further reduction in muscle mass, can be limited by consuming about 1.0 g/kg of high-quality protein in the diet and by engaging in resistance training and weight training.52
Improving quality of life and physical function are important goals. Information is emerging about when obesity needs to be managed in older adults. There is also evidence to support dietary and exercise therapy.54 Weight-loss options include lifestyle interventions, pharmacotherapy, and bariatric surgery.
Lifestyle interventions: Diet and exercise
The goal is to induce an energy deficit by reducing energy intake, increasing energy expenditure, or both—by 500 to 1,000 calories a day. This generally leads to a loss of 1 to 2 lb per week, and possibly up to 10% of weight in 6 months. Loss of about 10 to 20 lb with diet and exercise can translate to a relatively large reduction in visceral fat, with subsequent improvement in metabolic abnormalities.
A regular exercise program is important for improving overall physical function, which can slow progression to frailty. Adding aerobic, endurance, and resistance training helps preserve fat-free mass, which otherwise tends to diminish during active weight loss.55–57
The exercise program should begin at the outset of the weight-loss effort to help maintain weight loss and to prevent weight regain.58 Exercise is not essential for reaching the targeted weight loss, but starting early is important to reduce the loss of lean muscle that is usually already seen in the older population.
Several studies indicate that diet and exercise are just as effective in middle-aged and older people (over age 60) as in the younger population.58–60 Older people in the Diabetes Prevention Program were more compliant with lifestyle interventions and lost more weight than younger participants49: 60% of the older group met the 7% weight-loss goal at the end of 24 weeks, compared with 43% of those under age 45. At 3 years, the numbers were 63% vs 27%.
In a small randomized controlled trial,61 fat mass decreased by 6.6 kg in 17 people assigned to a program of diet and exercise, compared with a gain of 1.7 kg in a control group of 10 patients. Fat-free mass decreased by about 1 kg in both groups. The authors concluded that diet plus exercise (resistance training and strength training in this trial) could ameliorate frailty in obese older adults.
If exercise is appropriate, a physician should write a prescription for it, especially for resistance training, strengthening, flexibility, and stretching. This is important for patients with sarcopenic obesity and for those at high risk of chronic bone loss. The 2007 American College of Sports Medicine guidelines recommend muscle-strengthening activity of 8 to 10 exercises involving the major muscle groups, 10 to 15 repetitions at least twice a week. Flexibility and balance exercises should be included for those at risk of falls.62
Pharmacotherapy
At present, there are two general classes of weight-loss drugs: appetite suppressants and drugs that interfere with nutrient absorption.
Appetite suppressants include the sympathomimetics, which stimulate the release of dopamine and norepinephrine, resulting in increased satiety. Data—and therefore, recommendations—on their use in the elderly are very scarce, as most randomized controlled trials included only a small number of older people. A meta-analysis of drug therapy to treat obesity noted that the study population ranged in age from 34 to 54.63
The only approved drug currently available for use in older adults is orlistat, which blocks absorption of dietary fat by binding to intestinal lipase. A randomized controlled trial found the weight loss with orlistat to be comparable in older and younger adults.64,65
Review medications than can cause weight gain
When assessing older adults, always review the drugs they are taking. Those known to cause weight gain include certain of the following:
- Antiepileptics (eg, gabapentin)
- Antipsychotics (eg, olanzapine)
- Antidepressants (eg, tricyclics)
- Antihyperglycemic drugs (eg, sulfonylureas, thiazolidinediones)
- Beta-blockers
- Steroids.
If medically appropriate, a weight-neutral drug should be substituted for one suspected of causing weight gain. If a different physician (eg, a specialist) prescribed the original drug, he or she should be notified or consulted about any change.
Bariatric surgery
Bariatric surgery is the most effective weight-loss option, and more older patients are undergoing it than in the past. Dorman et al66 showed that the number of patients age 65 or older undergoing bariatric surgery increased from the year 2005 (when they accounted for 2%) to 2009 (when they accounted for 4.8%).
However, very few studies have provided information on the safety and effectiveness of bariatric surgery in older people. Several reports concluded that rates of perioperative morbidity and mortality are higher in older patients.67–69 Surgery resulted in marked weight loss and improvement in obesity-related complications and physical disability in older patients, although by a lower rate than in younger patients.
Varela et al70 examined the outcomes of bariatric surgery in a database from the University Health System Consortium Centers between 1999 and 2005. Patients over age 60 accounted for 1,339 (2.7%) of all bariatric operations performed. Compared with young and middle-aged patients, older patients had more comorbidities, longer hospital stays, and more complications, in addition to a higher in-hospital mortality rate. When risk-adjusted, the observed-to-expected mortality ratio for the older group was 0.9, compared with 0.7 in the young and middle-aged cohort.
Willkomm et al71 found an apparently higher operative risk profile in those over age 65 (n = 100) than in younger patients (n = 1,374), with higher rates of sleep apnea, diabetes, and hypertension. However, the operative outcomes were similar in the two groups in terms of operative time, length of stay, and 30-day readmission rates. The authors concluded that patients over age 65 had excellent outcomes compared with younger patients, suggesting that older age is not a risk factor for complications or death with bariatric surgery.
The American College of Surgeons National Surgical Quality Improvement Program evaluated the outcomes of 48,378 adults with a BMI greater than or equal to 35 kg/m2 who underwent bariatric surgery between 2005 and 2009.66 During this time, the number of patients age 65 and older seeking bariatric surgery increased from 1.5% to 4%. A total of 1,449 patients were in this age range. Thirty-day mortality rates did not differ significantly by age group and were less than 1% for all age ranges. Being age 65 or older was a significant predictor of prolonged length of stay but not of major adverse events. Significant predictors of major adverse events were a BMI greater than or equal to 55 kg/m2, cardiac comorbidities, a severe American Society of Anesthesiologists score, albumin levels lower than 3 g/dL, and creatinine levels greater than 1.5 mg/dL.
The most up-to-date study of the outcomes of bariatric surgery in patients over age 70 was a retrospective review at a single institution from 2007 to 2008 of 42 patients who underwent bariatric surgery.72 Twenty-two patients had laparoscopic gastric banding, 12 had laparoscopic sleeve gastrectomy, and 8 underwent laparoscopic Roux-en-Y gastric bypass. No patient died, complications occurred in 9 patients, and the rates of postoperative use of medications for hypertension, hyperlipidemia, diabetes, and osteoarthritis were reduced by about half. With the increasing number of patients seeking bariatric surgery, especially those over age 70, further prospective studies will determine if the outcomes are statistically significant.
If bariatric surgery is considered
The outcomes, complications, and mortality rates associated with bariatric surgery have been shown to be acceptable for adults age 65 and older. Perioperative risk assessment in the older obese patient seeking bariatric surgery is paramount to ensure that the benefits of the procedure justify any associated risks to the patient. Consequently, patients over age 65 should not be excluded out of hand: the patient’s individual risk of major adverse events must be identified beforehand.
If the patient is at risk, efforts should be made to reduce the risk to an acceptable level, including cardiac risk stratification, optimization of drug therapy, and discussions with the bariatric surgeon to plan on a less-invasive laparoscopic procedure. Otherwise, older obese patients can safely proceed with conventional bariatric surgery, which will help them achieve durable weight loss, improve quality of life, and reduce associated comorbidities.
The aforementioned studies of bariatric surgery are retrospective, include small numbers of patients, and lack long-term follow-up. The issues of long-term safety and the risk of death and morbidity in the aging population will require randomized controlled trials to answer these important questions.
At our hospital, we have seen an increase in the number of patients referred for a possible additional procedure (revision) to correct a problem from a previous bariatric surgery. The problems arising from the previous surgery can lead to weight gain or to excessive weight loss and malnutrition. To date, our institution has no policy on when to consider a revisional procedure in an older patient. All patients, including older ones, are assessed for the procedure on a case-by-case basis.
- Bray GA, Macdiarmid J. The epidemic of obesity. West J Med 2000; 172:78–79.
- Calle EE, Thun MJ, Petrelli JM, Rodriguez C, Heath CW. Body-mass index and mortality in a prospective cohort of US adults. N Engl J Med 1999; 341:1097–1105.
- Kalantar-Zadeh K, Horwich TB, Oreopoulos A, et al. Risk factor paradox in wasting diseases. Curr Opin Clin Nutr Metab Care 2007; 10:433–442.
- Mokdad AH, Marks JS, Stroup DF, Gerberding JL. Actual causes of death in the United States, 2000. JAMA 2004; 291:1238–1245.
- Arias E, Rostron BL, Tejada-Vera B. United States life tables, 2005. National vital statistics reports; vol 58no 10. Hyattsville, MD: National Center for Health Statistics. 2010.
- US Census Bureau International Database (IDB). Population projections of the US by age, sex, race, Hispanic origin, population division. http://www.census.gov/ipc/www/idb/country.php. Accessed September 13, 2013.
- Hedley AA, Ogden CL, Johnson CL, Carroll MD, Curtin LR, Flegal KM. Prevalence of overweight and obesity among US children, adolescents, and adults, 1999–2002. JAMA 2004; 291:2847–2850.
- Kuczmarski RJ, Flegal KM, Campbell SM, Johnson CL. Increasing prevalence of overweight among US adults. The National Health and Nutrition Examination Surveys, 1960 to 1991. JAMA 1994; 272:205–211.
- Mokdad AH, Serdula MK, Dietz WH, Bowman BA, Marks JS, Koplan JP. The spread of the obesity epidemic in the United States, 1991–1998. JAMA 1999; 282:1519–1522.
- Horani MH, Mooradian AD. Management of obesity in the elderly: special considerations. Treat Endocrinol 2002; 1:387–398.
- Beaufrère B, Morio B. Fat and protein redistribution with aging: metabolic considerations. Eur J Clin Nutr 2000; 54(suppl 3):S48–S53.
- Gallagher D, Heymsfield SB, Heo M, Jebb SA, Murgatroyd PR, Sakamoto Y. Healthy percentage body fat ranges: an approach for developing guidelines based on body mass index. Am J Clin Nutr 2000; 72:694–701.
- Snitker S. Use of body fatness cutoff points (author reply). Mayo Clin Proc 2010; 85:1057; author reply 1057–1058.
- Baumgartner RN, Koehler KM, Gallagher D, et al. Epidemiology of sarcopenia among the elderly in New Mexico. Am J Epidemiol 1998; 147:755–763. Erratum in Am J Epidemiol 1999; 149:1161.
- Visscher TL, Seidell JC, Molarius A, van der Kuip D, Hofman A, Witteman JC. A comparison of body mass index, waist-hip ratio and waist circumference as predictors of all-cause mortality among the elderly: the Rotterdam study. Int J Obes Relat Metab Disord 2001; 25:1730–1735.
- Molarius A, Seidell JC, Visscher TL, Hofman A. Misclassification of high-risk older subjects using waist action levels established for young and middle-aged adults—results from the Rotterdam Study. J Am Geriatr Soc 2000; 48:1638–1645.
- Han TS, Tajar A, Lean ME. Obesity and weight management in the elderly. Br Med Bull 2011; 97:169–196.
- Turcato E, Bosello O, Di Francesco V, et al. Waist circumference and abdominal sagittal diameter as surrogates of body fat distribution in the elderly: their relation with cardiovascular risk factors. Int J Obes Relat Metab Disord 2000; 24:1005–1010.
- Zamboni M, Mazzali G, Zoico E, et al. Health consequences of obesity in the elderly: a review of four unresolved questions. Int J Obes (Lond) 2005; 29:1011–1029.
- Heim N, Snijder MB, Heymans MW, Deeg DJ, Seidell JC, Visser M. Optimal cutoff values for high-risk waist circumference in older adults based on related health outcomes. Am J Epidemiol 2011; 174:479–489.
- Roubenoff R, Castaneda C. Sarcopenia—understanding the dynamics of aging muscle. JAMA 2001; 286:1230–1231.
- Schutz Y, Kyle UU, Pichard C. Fat-free mass index and fat mass index percentiles in Caucasians aged 18-98 y. Int J Obes Relat Metab Disord 2002; 26:953–960.
- Baumgartner RN, Wayne SJ, Waters DL, Janssen I, Gallagher D, Morley JE. Sarcopenic obesity predicts instrumental activities of daily living disability in the elderly. Obes Res 2004; 12:1995–2004.
- Morley JE, Baumgartner RN, Roubenoff R, Mayer J, Nair KS. Sarcopenia. J Lab Clin Med 2001; 137:231–243.
- Roubenoff R. Sarcopenic obesity: the confluence of two epidemics. Obes Res 2004; 12:887–888.
- Oreopoulos A, Kalantar-Zadeh K, Sharma AM, Fonarow GC. The obesity paradox in the elderly: potential mechanisms and clinical implications. Clin Geriatr Med 2009; 25:643–659.
- Elia M, Ritz P, Stubbs RJ. Total energy expenditure in the elderly. Eur J Clin Nutr 2000; 54(suppl 3):S92–S103.
- Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988; 37:1595–1607.
- Corona G, Mannucci E, Forti G, Maggi M. Hypogonadism, ED, metabolic syndrome and obesity: a pathological link supporting cardiovascular diseases. Int J Androl 2009; 32:587–598.
- Haarbo J, Hassager C, Riis BJ, Christiansen C. Relation of body fat distribution to serum lipids and lipoproteins in elderly women. Atherosclerosis 1989; 80:57–62.
- Cignarella A, Kratz M, Bolego C. Emerging role of estrogen in the control of cardiometabolic disease. Trends Pharmacol Sci 2010; 31:183–189.
- Felson DT, Anderson JJ, Naimark A, Walker AM, Meenan RF. Obesity and knee osteoarthritis. The Framingham Study. Ann Intern Med 1988; 109:18–24.
- Gelber AC, Hochberg MC, Mead LA, Wang NY, Wigley FM, Klag MJ. Body mass index in young men and the risk of subsequent knee and hip osteoarthritis. Am J Med 1999; 107:542–548.
- Iwao S, Iwao N, Muller DC, Elahi D, Shimokata H, Andres R. Effect of aging on the relationship between multiple risk factors and waist circumference. J Am Geriatr Soc 2000; 48:788–794.
- Folsom AR, Kushi LH, Anderson KE, et al. Associations of general and abdominal obesity with multiple health outcomes in older women: the Iowa Women’s Health Study. Arch Intern Med 2000; 160:2117–2128.
- Stevens J, Cai J, Pamuk ER, Williamson DF, Thun MJ, Wood JL. The effect of age on the association between body-mass index and mortality. N Engl J Med 1998; 338:1–7.
- Kalantar-Zadeh K, Horwich TB, Oreopoulos A, et al. Risk factor paradox in wasting diseases. Curr Opin Clin Nutr Metab Care 2007; 10:433–442.
- Zamboni M, Armellini F, Harris T, et al. Effects of age on body fat distribution and cardiovascular risk factors in women. Am J Clin Nutr 1997; 66:111–115.
- Janssen I, Katzmarzyk PT, Ross R. Body mass index is inversely related to mortality in older people after adjustment for waist circumference. J Am Geriatr Soc 2005; 53:2112–2118.
- Inelmen EM, Sergi G, Coin A, Miotto F, Peruzza S, Enzi G. Can obesity be a risk factor in elderly people? Obes Rev 2003; 4:147–155.
- Elia M. Obesity in the elderly. Obes Res 2001; 9(suppl 4):244S–248S.
- Losonczy KG, Harris TB, Cornoni-Huntley J, et al. Does weight loss from middle age to old age explain the inverse weight mortality relation in old age? Am J Epidemiol 1995; 141:312–321.
- Corrada MM, Kawas CH, Mozaffar F, Paganini-Hill A. Association of body mass index and weight change with all-cause mortality in the elderly. Am J Epidemiol 2006; 163:938–949.
- Bales CW, Buhr G. Is obesity bad for older persons? A systematic review of the pros and cons of weight reduction in later life. J Am Med Dir Assoc 2008; 9:302–312.
- Witham MD, Avenell A. Interventions to achieve long-term weight loss in obese older people: a systematic review and meta-analysis. Age Ageing 2010; 39:176–184.
- Villareal DT, Chode S, Parimi N, et al. Weight loss, exercise, or both and physical function in obese older adults. N Engl J Med 2011; 364:1218–1229.
- Wannamethee SG, Shaper AG, Whincup PH, Walker M. Characteristics of older men who lose weight intentionally or unintentionally. Am J Epidemiol 2000; 151:667–675.
- Lean ME, Powrie JK, Anderson AS, Garthwaite PH. Obesity, weight loss and prognosis in type 2 diabetes. Diabet Med 1990; 7:228–233.
- Williamson DF, Thompson TJ, Thun M, Flanders D, Pamuk E, Byers T. Intentional weight loss and mortality among overweight individuals with diabetes. Diabetes Care 2000; 23:1499–1504.
- Hamman RF, Wing RR, Edelstein SL, et al. Effect of weight loss with lifestyle intervention on risk of diabetes. Diabetes Care 2006; 29:2102-2107.
- National Heart, Lung, and Blood Institute in cooperation with The National Institute of Diabetes and Digestive and Kidney Diseases. Clinical guidelines on the identification, evaluation and treatment of the overweight and obesity in adults, the evidence report. NIH Publication number 98-4803 http://www.nhlbi.nih.gov/guidelines/obesity/ob_gdlns.pdf. Accessed September 13, 2013.
- Villareal DT, Apovian CM, Kushner RF, Klein S; American Society for Nutrition; NAASO, The Obesity Society. Obesity in older adults: technical review and position statement of the American Society for Nutrition and NAASO, The Obesity Society. Am J Clin Nutr 2005; 82:923–934.
- Williamson DF, Pamuk E, Thun M, Flanders D, Byers T, Heath C. Prospective study of intentional weight loss and mortality in never-smoking overweight US white women aged 40-64 years. Am J Epidemiol 1995; 141:1128–1141.
- McTigue KM, Hess R, Ziouras J. Obesity in older adults: a systematic review of the evidence for diagnosis and treatment. Obesity (Silver Spring). 2006; 14:1485–1497.
- Ryan AS, Pratley RE, Elahi D, Goldberg AP. Resistive training increases fat-free mass and maintains RMR despite weight loss in postmenopausal women. J Appl Physiol 1995; 79:818–823.
- Pavlou KN, Krey S, Steffee WP. Exercise as an adjunct to weight loss and maintenance in moderately obese subjects. Am J Clin Nutr 1989; 49(suppl 5):1115–1123.
- Kraemer WJ, Volek JS, Clark KL, et al. Influence of exercise training on physiological and performance changes with weight loss in men. Med Sci Sports Exerc 1999; 31:1320–1329.
- Wing RR, Hill JO. Successful weight loss maintenance. Annu Rev Nutr 2001; 21:323–341.
- Banks M, Klein S, Sinacore D, Siener C, Villareal DT. Effects of weight loss and exercise on frailty in obese elderly subjects. J Am Geriatr Soc 2005; 53:S16.
- Messier SP, Loeser RF, Miller GD, et al. Exercise and dietary weight loss in overweight and obese older adults with knee osteoarthritis: the Arthritis, Diet, and Activity Promotion Trial. Arthritis Rheum 2004; 50:1501–1510.
- Villareal DT, Banks M, Sinacore DR, Siener C, Klein S. Effect of weight loss and exercise on frailty in obese older adults. Arch Intern Med 2006; 166:860–866.
- Nelson ME, Rejeski WJ, Blair SN, et al; American College of Sports Medicine; American Heart Association. Physical activity and public health in older adults: recommendation from the American College of Sports Medicine and the American Heart Association. Circulation 2007; 116:1094–1105.
- Li Z, Maglione M, Tu W, et al. Meta-analysis: pharmacologic treatment of obesity. Ann Intern Med 2005; 142:532–546.
- Segal KR, Lucas C, Boldrin M, Hauptman J. Weight loss efficacy of orlistat in obese elderly adults (abstract). Obes Res 1999; 7(suppl):26S.
- Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med 2000; 9:160–167.
- Dorman RB, Abraham AA, Al-Refaie WB, Parsons HM, Ikramuddin S, Habermann EB. Bariatric surgery outcomes in the elderly: an ACS NSQIP study. J Gastrointest Surg 2012; 16:35–44.
- Sugerman HJ, DeMaria EJ, Kellum JM, Sugerman EL, Meador JG, Wolfe LG. Effects of bariatric surgery in older patients. Ann Surg 2004; 240:243–247.
- St. Peter SD, Craft RO, Tiede JL, Swain JM. Impact of advanced age on weight loss and health benefits after laparoscopic gastric bypass. Arch Surg 2005; 140:165–168.
- Sosa JL, Pombo H, Pallavicini H, Ruiz-Rodriguez M. Laparoscopic gastric bypass beyond age 60. Obes Surg 2004; 14:1398–1401.
- Varela JE, Wilson SE, Nguyen NT. Outcomes of bariatric surgery in the elderly. Am Surg 2006; 72:865–869.
- Willkomm CM, Fisher TL, Barnes GS, Kennedy CI, Kuhn JA. Surgical weight loss >65 years old: is it worth the risk? Surg Obes Relat Dis 2010; 6:491–496.
- Heiat A, Vaccarino V, Krumholz HM. An evidence-based assessment of federal guidelines for overweight and obesity as they apply to elderly persons. Arch Intern Med 2001; 161:1194–1203.
Should older obese people try to lose weight? Such a simple question is more complicated than one would think.
At issue is whether obesity is harmful in older people, and whether treating it will reduce their health risks. True, obesity is an independent risk factor for cardiovascular disease and is associated with many comorbidities, including type 2 diabetes mellitus, hyperlipidemia, heart failure, and hypertension.1 An independent association also exists between obesity and all-cause mortality.2 However, there is also evidence suggesting that obesity in this age group is associated with a lower, not higher, risk of death—a finding termed the obesity paradox.3 And for that matter, what exactly constitutes obesity in elderly people, who naturally undergo changes in body composition as they age?
This article examines the literature on these controversial issues, including changes in body composition with age, the definition of obesity in older adults, the obesity paradox, and treatment of obesity in older adults.
AMERICANS ARE GETTING OLDER—AND BIGGER
Americans are living longer than ever before; life expectancy has reached a new high of 77.8 years.4,5 According to the US Census Bureau,6 about 27 million people in the United States are over age 70, and this number is expected to nearly double by 2030.
Meanwhile, the prevalence of obesity, defined as a body mass index (BMI) of 30 kg/m2 or higher, has increased in the last 25 years in all age groups in the United States, including those age 65 and older.7,8 These two trends add up to an increase in the number of obese older people. In 2000, 22.9% of people age 60 to 69 and 15.5% of those over age 70 and older were obese.9 This amounted to a 56% increase in the former group and a 36% increase in the latter group in the interval since 1991.5,9
BUT WHAT CONSTITUTES ‘OBESITY’?
Obesity is the excess accumulation of body fat, leading to a higher risk of medical illness and premature death. But measuring it is not as simple as one might think.
The body mass index can mislead
The BMI, ie, weight in kilograms divided by the square of the height in meters, correlates fairly well with body fat stores and is generally used to classify medical risk.
However, the BMI can classify some older people as overweight (BMI 30.0–34.9 kg/m2) or obese (BMI ≥ 35.0 kg/m2) who actually do not have an excess of body fat—and can fail to classify others as overweight or obese who do. For example, if a person loses height as a result of vertebral compression fractures, his or her BMI would become higher, even with no change in weight or body fat. Conversely, changes in body composition with age, including loss of muscle and an increase in fat, may not be reflected in the BMI, even if the person really does have too much body fat.10
This second limitation of the BMI is important when estimating risk in older adults, who have a particular fat distribution. Visceral, subcutaneous, intramuscular, and intrahepatic fat increase with age, and they are all risk factors for insulin resistance and type 2 diabetes mellitus.11 And in older people, having too much visceral fat is more prevalent than the BMI might predict.10
Percent body fat awaits investigation
Percent body fat is another way to assess body fat. Defined as the total weight of fat divided by total weight, it is measured in various ways.
Dual-energy x-ray absorptiometry, computed tomography, and magnetic resonance imaging can measure percent body fat, and they can differentiate visceral from subcutaneous fat (which is less metabolically active). Unfortunately, most of these tests are used for this purpose only in research, and they are relatively expensive.
Commercially available bioelectrical impedance devices send a weak electric current through the body and measure the resistance, and using this information and four other factors (height, weight, age, and sex), they calculate percent body fat. This method is fast, easy, painless, and cheap. A disadvantage is that the handheld devices measure body composition of the upper body only. Because the lower body is excluded, they do not give an accurate measurement of body fat of the abdomen and hips. Also, they cannot differentiate visceral from subcutaneous fat.
Bioelectrical impedance devices work well in healthy individuals with stable water balance. The values are only an estimate of fat-free mass, and therefore this method is not the gold standard for assessing body fat. Bioelectrical impedance is better at tracking body composition in an individual over time than at diagnosing obesity.
Percent body fat can vary by sex and race. Asians, for example, have higher percent body fats at lower BMIs, particularly when younger.12 Also, Gallagher et al12 found that percent body fat increased with age at every given BMI in both men and women (Table 1).
The traditional universal cutoffs for defining obesity by percent body fat are 25% in men and 35% in women. However, research has indicated that cutoffs of 20% to 25% in men and 30% to 38% in women may better identify those at risk of metabolic disease.13 Guidelines and evidence-based cutoffs for percent body fat must await further investigation.
Waist circumference is useful
In older adults, obesity can be diagnosed by a measurement such as waist circumference, which correlates highly with total fat and intra-abdominal fat.14 It is very cost-effective, simple, and useful for the office assessment of adiposity.
The measurement should be made halfway between the iliac crest and the lower anterior ribs, with the patient standing, and at the end of expiration.
The traditional standard for waist circumference is less than 89 cm (35 inches) for women and 102 cm (40 inches) for men. However, opinion differs, and different reference ranges exist depending on ethnicity. Additionally, because stature and body composition change with age, concerns have been raised about misclassification of the health risks related to obesity in older adults using the current standard.15,16
The waist circumference is as good as or even better than the BMI as a measure of excess adiposity in older adults.16–18 This is in part because of the age-dependent height decrease in older adults.15,19 (Recall that the BMI is calculated using the height squared as the denominator; as a result, the BMI would give a higher reading and thus an overestimate of total body fat.) Conversely, we can underestimate the amount of adiposity because of decreases in abdominal muscle tone.17
Cutoffs for waist circumference should be age-specific.16
Investigators in the Netherlands15,16 prospectively took 4,996 measurements in 2,232 people with a mean age of 70, from 1992 through 2006. They concluded that the best cutoffs for predicting the health risks of obesity in the elderly were 109 cm (43 inches) in men and 98 cm (39 inches) in women.
A group of researchers has proposed that the cutoffs be shifted upward in older adults, with new values for those age 70 and over.20 The Health Survey for England aimed to describe the patterns and trends in waist circumference and abdominal obesity and overweight in people age 70 through 89, comparing both the standard and the new cutoffs. Optimal cutoffs recommended for abdominal obesity for patients age 70 and older were 100 to 106 cm in men and 99 cm in women.20 Estimates of the prevalence of abdominal obesity are much lower using the new cutoffs.
SARCOPENIA: LOSS OF MUSCLE WITH AGE
With age comes sarcopenia—the progressive loss of muscle mass, primarily skeletal muscle, resulting in a decrease in strength and power.21 The process begins as early as the 20s or 30s.22 It is distinct from wasting (involuntary weight loss from inadequate intake), seen in starvation.21
Sarcopenia is defined as an appendicular skeletal muscle mass index (the appendicular skeletal mass divided by the square of the height in meters) of less than 2 standard deviations below a young adult reference, and a percentage of body fat over the 60th percentile for the individual’s sex and age.23,24 Estimates of its prevalence vary, but it is common and it increases with age.14,20
Sarcopenic obesity: Less muscle, more fat
Progressive loss of skeletal muscle with age, along with an increase and redistribution of body fat, is known as sarcopenic obesity.25 It is associated with higher morbidity and mortality rates as well as a decline in functional strength, which leads to frailty.23 This loss of muscle mass may go unnoticed in an older person until he or she begins to lose physical function.
As noted, in an older person with sarcopenic obesity, the BMI may mislead because of the high percentage of fat and the low lean mass.26
Why we change with age
This change in body composition with age is a result of several factors. Illness or inactivity can lead to loss of muscle, while body fat is preserved.17 The combination of reduced physical activity, a lower resting metabolic rate, and an unchanged intake of food can increase the likelihood of sarcopenia.27 Also possibly contributing are hormonal changes, including reduced production of growth hormone and testosterone and decreased responsiveness to thyroid hormone and leptin.28
Moreover, the interaction of several factors can lead to a vicious circle of progressive loss of muscle and increase in fat. As people age, their physical activity tends to decrease, resulting in muscle loss. As muscle mass decreases, the amount of available insulin-responsive tissue is reduced, resulting in insulin resistance, which in turn promotes the metabolic syndrome and an increase in fat. With more fat, people produce more of the adipokines tumor necrosis factor alpha and interleukin 6, which further promote insulin resistance.
Other changes contribute to a decrease in muscle quality and performance, including an increase in intramuscular and intrahepatic fat, which is associated with insulin resistance.11 The increases in adipose stores occur mostly in intra-abdominal fat rather than in subcutaneous fat.
ADVERSE EFFECTS OF OBESITY
A number of comorbidities arise with obesity, regardless of age.19
The diseases most strongly associated with obesity are the metabolic syndrome and type 2 diabetes mellitus.17 Studies have shown that in older adults, obesity as measured by waist circumference is associated with hyperglycemia and dyslipidemia.29,30
Metabolic abnormalities may ensue in obese older people through complex mechanisms involving an age-related decline in sex hormones. For example, late-onset hypogonadism in men, which is more common in those who are obese, is related to the metabolic syndrome.29
These mechanisms are also complex in women. Because estrogens can be produced in adipose tissue, obese postmenopausal women have higher concentrations of estrogens than their lean counterparts, and this may lead to metabolic abnormalities.31 (On the other hand, higher estrogen levels in obese menopausal women can protect against osteoporosis by increasing bone mass.)
Older people who weigh more and have more adipose tissue, especially those who became obese at a young age, have a greater risk of osteoarthritis of the knee,32,33 which when combined with obesity can cause disability and physical impairment.19 And cardiovascular risk factors,18,33 hypertension,34 and certain cancers35 are more common in older people with higher waist circumference.
THE OBESITY PARADOX
In general, obesity in younger adults has been shown to shorten life expectancy. This risk of death is often associated with obesity-related health problems.
In older people, the effect of obesity is much more complex.36 The optimal weight in terms of survival increases with age. More interesting is the finding that although the risk of cardiovascular disease is higher in overweight or obese older adults, studies also suggest that in this age group, being overweight or obese is paradoxically associated with lower mortality rates from these diseases.26 This phenomenon is called the obesity paradox.37
For those over age 75, the relative risk of death from all causes and from cardiovascular disease has been found to decrease with increasing BMI.25 The relationship between BMI and death from all causes in older adults may actually be a U-shaped curve, meaning that the risk of death rises at both extremes of BMI values.26
Possible explanations for the paradox
Several hypotheses have been proposed to explain the change in the relationship between BMI and the risk of death that occurs with aging.
The BMI is an imperfect measure of obesity. The obesity paradox may be an artifact of using the BMI to measure obesity in older adults.17 As described above, sarcopenic obesity must be considered in those over age 65 because the BMI does not differentiate between fat and muscle. Older adults tend to have a higher proportion of body and visceral fat that is distributed differently, making the waist circumference or waist-hip ratio a more appropriate measure of obesity in this group.38 Janssen et al39 found that in people age 65 and older, after controlling for waist circumference, higher BMI values were associated with lower death rates; after controlling for BMI, waist circumference was associated with a higher risk of death.
The survival effect suggests that people who are susceptible to the negative effects of obesity die sooner,40 and those who survive until old age may be resistant to the effects of obesity.41 If true, the survival effect would explain why the death rate seems to be unaffected by BMI in the older population.
Unhealthy weight loss. Smoking and diseases such as cancer that can cause early death may also induce weight loss, further complicating the relationship between BMI and death.19 After age 80, the association between BMI and the risk of death is weak because those with a low BMI include not only those who have always been lean and physically active, but also those who lost weight through chronic ill health or smoking.17
Further study needed. Thus, a number of confounding variables may muddy the association between obesity and death in older adults. Obesity should not be misinterpreted as being harmless or beneficial in older adults. Stevens et al36 found that a greater BMI was associated with a higher rate of death from all causes and from cardiovascular disease in men and women up to age 75, but that the relative risk of death associated with a greater BMI decreased with age.
Optimal BMI targets in older people have yet to be validated in a large prospective trial. However, multiple studies have examined the relationship between BMI and all-cause mortality in older adults and have identified a BMI of 24 to 35 as “ideal” and associated with the lowest risk of death, with a lower range for men and a higher range for women.42,43 The topic has been reviewed by Oreopoulos et al.26 More research is needed to evaluate this relationship.
THE BENEFIT OF WEIGHT LOSS IN OLDER ADULTS IS CONTROVERSIAL
In younger obese people, weight loss brings a multitude of benefits by reducing the risk of complications arising from obesity. However, in older adults, the effects of weight loss remain controversial, and evidence to guide treatment is limited.44,45 The few trials that have been published have typically focused on cardiovascular risk factors rather than physical function.45
In a 1-year trial, 107 people age 65 or older were randomized to a control group, to weight management, to exercise, or to weight management plus exercise. The combination of weight loss and exercise yielded the greatest improvement in physical function.46
Intentional vs unintentional weight loss
Intentional weight loss is altogether different from unintentional weight loss.
In most cases, weight loss in older adults is unintentional and may indicate underlying disease and impending death.17 For example, older men who lose weight unintentionally have significantly greater rates of smoking, disability, cancer, and respiratory disease and less obesity and physical activity than those who lose weight intentionally.47
Studies have shown an increase in life expectancy in older patients with type 2 diabetes mellitus who lost weight intentionally.48,49 In fact, moderate weight loss—just 5% to 10%—has been shown to improve cardiovascular risk factors,44 osteoarthritis, and type 2 diabetes.50
Bales and Buhr44 performed a systematic review of 16 studies that had lasted at least 6 months. Patients were age 60 or older with a minimum baseline BMI of 27 kg/m2 who intentionally lost at least 3% of body weight or 2 kg. Levels of the inflammatory markers C-reactive protein, tumor necrosis factor alpha, and interleukin 6 declined with weight, along with blood pressure, fasting glucose, waist circumference, and low-density lipoprotein cholesterol. On the downside, bone mineral density and lean body mass also declined slightly. The best way to avoid losing lean body mass and to preserve bone density during weight loss is to include a program of resistance-training exercises.
No clinical trial has evaluated the effects of intentional weight loss on death rates in older obese people.25 As a result, evidence-based recommendations cannot be made. Rather, advice on weight loss must be individualized, with special emphasis on the patient’s weight history and medical comorbidities.44
Oreopoulos et al26 summarized the possible effects of BMI, abdominal fat, lean body mass, and intentional weight loss on morbidity and mortality outcomes in older adults (Table 2).
TREATMENT GUIDELINES AND RECOMMENDATIONS
Many of the methods of weight management in older adults are the same as in young and middle-aged adults.51 Recommendations for all age groups include lifestyle changes, increased activity, dietary changes, drug therapy, and bariatric surgery.
Whether there should be separate guidelines for older adults is controversial. In view of the obesity paradox, physicians have been reluctant to recommend weight loss in elderly patients. Caution is advised in recommending weight loss solely on the basis of body weight, as studies have shown that the weight associated with maximal survival increases with age. Because of age-related changes in body composition and reduced energy requirements and expenditure, recommendations for the young and middle-aged should not be applied directly to older adults.
In this group, especially those who have survived into old age with good health and an intact functional status, one could argue that significant caloric restriction should not be recommended. In these people, the goal is often to maintain weight and incorporate a daily exercise program rather than to aggressively lose weight. Adding resistance training can improve physical function, which can improve quality of life. There is less emphasis on cardiovascular risk, but both outcomes apply for both age groups.52
Intentional weight loss should be recommended to high-risk older adults, including those with cardiovascular disease, type 2 diabetes mellitus, and metabolic syndrome, because the absolute risk of death and morbidity is higher in this group. Most health benefits can be achieved with modest weight loss.53 Potential benefits include prevention of cognitive impairment, protection from bone fractures, an increase in antioxidant defense, a reserve of fat and energy stores, and an increase in longevity.26
Treatment differs from that in the younger population primarily because of the importance of preventing loss of muscle with intentional weight loss. People of all ages who lose weight intentionally lose fat and, to a lesser extent, skeletal muscle. Older patients have already lost muscle mass, but further changes in body composition, especially a further reduction in muscle mass, can be limited by consuming about 1.0 g/kg of high-quality protein in the diet and by engaging in resistance training and weight training.52
Improving quality of life and physical function are important goals. Information is emerging about when obesity needs to be managed in older adults. There is also evidence to support dietary and exercise therapy.54 Weight-loss options include lifestyle interventions, pharmacotherapy, and bariatric surgery.
Lifestyle interventions: Diet and exercise
The goal is to induce an energy deficit by reducing energy intake, increasing energy expenditure, or both—by 500 to 1,000 calories a day. This generally leads to a loss of 1 to 2 lb per week, and possibly up to 10% of weight in 6 months. Loss of about 10 to 20 lb with diet and exercise can translate to a relatively large reduction in visceral fat, with subsequent improvement in metabolic abnormalities.
A regular exercise program is important for improving overall physical function, which can slow progression to frailty. Adding aerobic, endurance, and resistance training helps preserve fat-free mass, which otherwise tends to diminish during active weight loss.55–57
The exercise program should begin at the outset of the weight-loss effort to help maintain weight loss and to prevent weight regain.58 Exercise is not essential for reaching the targeted weight loss, but starting early is important to reduce the loss of lean muscle that is usually already seen in the older population.
Several studies indicate that diet and exercise are just as effective in middle-aged and older people (over age 60) as in the younger population.58–60 Older people in the Diabetes Prevention Program were more compliant with lifestyle interventions and lost more weight than younger participants49: 60% of the older group met the 7% weight-loss goal at the end of 24 weeks, compared with 43% of those under age 45. At 3 years, the numbers were 63% vs 27%.
In a small randomized controlled trial,61 fat mass decreased by 6.6 kg in 17 people assigned to a program of diet and exercise, compared with a gain of 1.7 kg in a control group of 10 patients. Fat-free mass decreased by about 1 kg in both groups. The authors concluded that diet plus exercise (resistance training and strength training in this trial) could ameliorate frailty in obese older adults.
If exercise is appropriate, a physician should write a prescription for it, especially for resistance training, strengthening, flexibility, and stretching. This is important for patients with sarcopenic obesity and for those at high risk of chronic bone loss. The 2007 American College of Sports Medicine guidelines recommend muscle-strengthening activity of 8 to 10 exercises involving the major muscle groups, 10 to 15 repetitions at least twice a week. Flexibility and balance exercises should be included for those at risk of falls.62
Pharmacotherapy
At present, there are two general classes of weight-loss drugs: appetite suppressants and drugs that interfere with nutrient absorption.
Appetite suppressants include the sympathomimetics, which stimulate the release of dopamine and norepinephrine, resulting in increased satiety. Data—and therefore, recommendations—on their use in the elderly are very scarce, as most randomized controlled trials included only a small number of older people. A meta-analysis of drug therapy to treat obesity noted that the study population ranged in age from 34 to 54.63
The only approved drug currently available for use in older adults is orlistat, which blocks absorption of dietary fat by binding to intestinal lipase. A randomized controlled trial found the weight loss with orlistat to be comparable in older and younger adults.64,65
Review medications than can cause weight gain
When assessing older adults, always review the drugs they are taking. Those known to cause weight gain include certain of the following:
- Antiepileptics (eg, gabapentin)
- Antipsychotics (eg, olanzapine)
- Antidepressants (eg, tricyclics)
- Antihyperglycemic drugs (eg, sulfonylureas, thiazolidinediones)
- Beta-blockers
- Steroids.
If medically appropriate, a weight-neutral drug should be substituted for one suspected of causing weight gain. If a different physician (eg, a specialist) prescribed the original drug, he or she should be notified or consulted about any change.
Bariatric surgery
Bariatric surgery is the most effective weight-loss option, and more older patients are undergoing it than in the past. Dorman et al66 showed that the number of patients age 65 or older undergoing bariatric surgery increased from the year 2005 (when they accounted for 2%) to 2009 (when they accounted for 4.8%).
However, very few studies have provided information on the safety and effectiveness of bariatric surgery in older people. Several reports concluded that rates of perioperative morbidity and mortality are higher in older patients.67–69 Surgery resulted in marked weight loss and improvement in obesity-related complications and physical disability in older patients, although by a lower rate than in younger patients.
Varela et al70 examined the outcomes of bariatric surgery in a database from the University Health System Consortium Centers between 1999 and 2005. Patients over age 60 accounted for 1,339 (2.7%) of all bariatric operations performed. Compared with young and middle-aged patients, older patients had more comorbidities, longer hospital stays, and more complications, in addition to a higher in-hospital mortality rate. When risk-adjusted, the observed-to-expected mortality ratio for the older group was 0.9, compared with 0.7 in the young and middle-aged cohort.
Willkomm et al71 found an apparently higher operative risk profile in those over age 65 (n = 100) than in younger patients (n = 1,374), with higher rates of sleep apnea, diabetes, and hypertension. However, the operative outcomes were similar in the two groups in terms of operative time, length of stay, and 30-day readmission rates. The authors concluded that patients over age 65 had excellent outcomes compared with younger patients, suggesting that older age is not a risk factor for complications or death with bariatric surgery.
The American College of Surgeons National Surgical Quality Improvement Program evaluated the outcomes of 48,378 adults with a BMI greater than or equal to 35 kg/m2 who underwent bariatric surgery between 2005 and 2009.66 During this time, the number of patients age 65 and older seeking bariatric surgery increased from 1.5% to 4%. A total of 1,449 patients were in this age range. Thirty-day mortality rates did not differ significantly by age group and were less than 1% for all age ranges. Being age 65 or older was a significant predictor of prolonged length of stay but not of major adverse events. Significant predictors of major adverse events were a BMI greater than or equal to 55 kg/m2, cardiac comorbidities, a severe American Society of Anesthesiologists score, albumin levels lower than 3 g/dL, and creatinine levels greater than 1.5 mg/dL.
The most up-to-date study of the outcomes of bariatric surgery in patients over age 70 was a retrospective review at a single institution from 2007 to 2008 of 42 patients who underwent bariatric surgery.72 Twenty-two patients had laparoscopic gastric banding, 12 had laparoscopic sleeve gastrectomy, and 8 underwent laparoscopic Roux-en-Y gastric bypass. No patient died, complications occurred in 9 patients, and the rates of postoperative use of medications for hypertension, hyperlipidemia, diabetes, and osteoarthritis were reduced by about half. With the increasing number of patients seeking bariatric surgery, especially those over age 70, further prospective studies will determine if the outcomes are statistically significant.
If bariatric surgery is considered
The outcomes, complications, and mortality rates associated with bariatric surgery have been shown to be acceptable for adults age 65 and older. Perioperative risk assessment in the older obese patient seeking bariatric surgery is paramount to ensure that the benefits of the procedure justify any associated risks to the patient. Consequently, patients over age 65 should not be excluded out of hand: the patient’s individual risk of major adverse events must be identified beforehand.
If the patient is at risk, efforts should be made to reduce the risk to an acceptable level, including cardiac risk stratification, optimization of drug therapy, and discussions with the bariatric surgeon to plan on a less-invasive laparoscopic procedure. Otherwise, older obese patients can safely proceed with conventional bariatric surgery, which will help them achieve durable weight loss, improve quality of life, and reduce associated comorbidities.
The aforementioned studies of bariatric surgery are retrospective, include small numbers of patients, and lack long-term follow-up. The issues of long-term safety and the risk of death and morbidity in the aging population will require randomized controlled trials to answer these important questions.
At our hospital, we have seen an increase in the number of patients referred for a possible additional procedure (revision) to correct a problem from a previous bariatric surgery. The problems arising from the previous surgery can lead to weight gain or to excessive weight loss and malnutrition. To date, our institution has no policy on when to consider a revisional procedure in an older patient. All patients, including older ones, are assessed for the procedure on a case-by-case basis.
Should older obese people try to lose weight? Such a simple question is more complicated than one would think.
At issue is whether obesity is harmful in older people, and whether treating it will reduce their health risks. True, obesity is an independent risk factor for cardiovascular disease and is associated with many comorbidities, including type 2 diabetes mellitus, hyperlipidemia, heart failure, and hypertension.1 An independent association also exists between obesity and all-cause mortality.2 However, there is also evidence suggesting that obesity in this age group is associated with a lower, not higher, risk of death—a finding termed the obesity paradox.3 And for that matter, what exactly constitutes obesity in elderly people, who naturally undergo changes in body composition as they age?
This article examines the literature on these controversial issues, including changes in body composition with age, the definition of obesity in older adults, the obesity paradox, and treatment of obesity in older adults.
AMERICANS ARE GETTING OLDER—AND BIGGER
Americans are living longer than ever before; life expectancy has reached a new high of 77.8 years.4,5 According to the US Census Bureau,6 about 27 million people in the United States are over age 70, and this number is expected to nearly double by 2030.
Meanwhile, the prevalence of obesity, defined as a body mass index (BMI) of 30 kg/m2 or higher, has increased in the last 25 years in all age groups in the United States, including those age 65 and older.7,8 These two trends add up to an increase in the number of obese older people. In 2000, 22.9% of people age 60 to 69 and 15.5% of those over age 70 and older were obese.9 This amounted to a 56% increase in the former group and a 36% increase in the latter group in the interval since 1991.5,9
BUT WHAT CONSTITUTES ‘OBESITY’?
Obesity is the excess accumulation of body fat, leading to a higher risk of medical illness and premature death. But measuring it is not as simple as one might think.
The body mass index can mislead
The BMI, ie, weight in kilograms divided by the square of the height in meters, correlates fairly well with body fat stores and is generally used to classify medical risk.
However, the BMI can classify some older people as overweight (BMI 30.0–34.9 kg/m2) or obese (BMI ≥ 35.0 kg/m2) who actually do not have an excess of body fat—and can fail to classify others as overweight or obese who do. For example, if a person loses height as a result of vertebral compression fractures, his or her BMI would become higher, even with no change in weight or body fat. Conversely, changes in body composition with age, including loss of muscle and an increase in fat, may not be reflected in the BMI, even if the person really does have too much body fat.10
This second limitation of the BMI is important when estimating risk in older adults, who have a particular fat distribution. Visceral, subcutaneous, intramuscular, and intrahepatic fat increase with age, and they are all risk factors for insulin resistance and type 2 diabetes mellitus.11 And in older people, having too much visceral fat is more prevalent than the BMI might predict.10
Percent body fat awaits investigation
Percent body fat is another way to assess body fat. Defined as the total weight of fat divided by total weight, it is measured in various ways.
Dual-energy x-ray absorptiometry, computed tomography, and magnetic resonance imaging can measure percent body fat, and they can differentiate visceral from subcutaneous fat (which is less metabolically active). Unfortunately, most of these tests are used for this purpose only in research, and they are relatively expensive.
Commercially available bioelectrical impedance devices send a weak electric current through the body and measure the resistance, and using this information and four other factors (height, weight, age, and sex), they calculate percent body fat. This method is fast, easy, painless, and cheap. A disadvantage is that the handheld devices measure body composition of the upper body only. Because the lower body is excluded, they do not give an accurate measurement of body fat of the abdomen and hips. Also, they cannot differentiate visceral from subcutaneous fat.
Bioelectrical impedance devices work well in healthy individuals with stable water balance. The values are only an estimate of fat-free mass, and therefore this method is not the gold standard for assessing body fat. Bioelectrical impedance is better at tracking body composition in an individual over time than at diagnosing obesity.
Percent body fat can vary by sex and race. Asians, for example, have higher percent body fats at lower BMIs, particularly when younger.12 Also, Gallagher et al12 found that percent body fat increased with age at every given BMI in both men and women (Table 1).
The traditional universal cutoffs for defining obesity by percent body fat are 25% in men and 35% in women. However, research has indicated that cutoffs of 20% to 25% in men and 30% to 38% in women may better identify those at risk of metabolic disease.13 Guidelines and evidence-based cutoffs for percent body fat must await further investigation.
Waist circumference is useful
In older adults, obesity can be diagnosed by a measurement such as waist circumference, which correlates highly with total fat and intra-abdominal fat.14 It is very cost-effective, simple, and useful for the office assessment of adiposity.
The measurement should be made halfway between the iliac crest and the lower anterior ribs, with the patient standing, and at the end of expiration.
The traditional standard for waist circumference is less than 89 cm (35 inches) for women and 102 cm (40 inches) for men. However, opinion differs, and different reference ranges exist depending on ethnicity. Additionally, because stature and body composition change with age, concerns have been raised about misclassification of the health risks related to obesity in older adults using the current standard.15,16
The waist circumference is as good as or even better than the BMI as a measure of excess adiposity in older adults.16–18 This is in part because of the age-dependent height decrease in older adults.15,19 (Recall that the BMI is calculated using the height squared as the denominator; as a result, the BMI would give a higher reading and thus an overestimate of total body fat.) Conversely, we can underestimate the amount of adiposity because of decreases in abdominal muscle tone.17
Cutoffs for waist circumference should be age-specific.16
Investigators in the Netherlands15,16 prospectively took 4,996 measurements in 2,232 people with a mean age of 70, from 1992 through 2006. They concluded that the best cutoffs for predicting the health risks of obesity in the elderly were 109 cm (43 inches) in men and 98 cm (39 inches) in women.
A group of researchers has proposed that the cutoffs be shifted upward in older adults, with new values for those age 70 and over.20 The Health Survey for England aimed to describe the patterns and trends in waist circumference and abdominal obesity and overweight in people age 70 through 89, comparing both the standard and the new cutoffs. Optimal cutoffs recommended for abdominal obesity for patients age 70 and older were 100 to 106 cm in men and 99 cm in women.20 Estimates of the prevalence of abdominal obesity are much lower using the new cutoffs.
SARCOPENIA: LOSS OF MUSCLE WITH AGE
With age comes sarcopenia—the progressive loss of muscle mass, primarily skeletal muscle, resulting in a decrease in strength and power.21 The process begins as early as the 20s or 30s.22 It is distinct from wasting (involuntary weight loss from inadequate intake), seen in starvation.21
Sarcopenia is defined as an appendicular skeletal muscle mass index (the appendicular skeletal mass divided by the square of the height in meters) of less than 2 standard deviations below a young adult reference, and a percentage of body fat over the 60th percentile for the individual’s sex and age.23,24 Estimates of its prevalence vary, but it is common and it increases with age.14,20
Sarcopenic obesity: Less muscle, more fat
Progressive loss of skeletal muscle with age, along with an increase and redistribution of body fat, is known as sarcopenic obesity.25 It is associated with higher morbidity and mortality rates as well as a decline in functional strength, which leads to frailty.23 This loss of muscle mass may go unnoticed in an older person until he or she begins to lose physical function.
As noted, in an older person with sarcopenic obesity, the BMI may mislead because of the high percentage of fat and the low lean mass.26
Why we change with age
This change in body composition with age is a result of several factors. Illness or inactivity can lead to loss of muscle, while body fat is preserved.17 The combination of reduced physical activity, a lower resting metabolic rate, and an unchanged intake of food can increase the likelihood of sarcopenia.27 Also possibly contributing are hormonal changes, including reduced production of growth hormone and testosterone and decreased responsiveness to thyroid hormone and leptin.28
Moreover, the interaction of several factors can lead to a vicious circle of progressive loss of muscle and increase in fat. As people age, their physical activity tends to decrease, resulting in muscle loss. As muscle mass decreases, the amount of available insulin-responsive tissue is reduced, resulting in insulin resistance, which in turn promotes the metabolic syndrome and an increase in fat. With more fat, people produce more of the adipokines tumor necrosis factor alpha and interleukin 6, which further promote insulin resistance.
Other changes contribute to a decrease in muscle quality and performance, including an increase in intramuscular and intrahepatic fat, which is associated with insulin resistance.11 The increases in adipose stores occur mostly in intra-abdominal fat rather than in subcutaneous fat.
ADVERSE EFFECTS OF OBESITY
A number of comorbidities arise with obesity, regardless of age.19
The diseases most strongly associated with obesity are the metabolic syndrome and type 2 diabetes mellitus.17 Studies have shown that in older adults, obesity as measured by waist circumference is associated with hyperglycemia and dyslipidemia.29,30
Metabolic abnormalities may ensue in obese older people through complex mechanisms involving an age-related decline in sex hormones. For example, late-onset hypogonadism in men, which is more common in those who are obese, is related to the metabolic syndrome.29
These mechanisms are also complex in women. Because estrogens can be produced in adipose tissue, obese postmenopausal women have higher concentrations of estrogens than their lean counterparts, and this may lead to metabolic abnormalities.31 (On the other hand, higher estrogen levels in obese menopausal women can protect against osteoporosis by increasing bone mass.)
Older people who weigh more and have more adipose tissue, especially those who became obese at a young age, have a greater risk of osteoarthritis of the knee,32,33 which when combined with obesity can cause disability and physical impairment.19 And cardiovascular risk factors,18,33 hypertension,34 and certain cancers35 are more common in older people with higher waist circumference.
THE OBESITY PARADOX
In general, obesity in younger adults has been shown to shorten life expectancy. This risk of death is often associated with obesity-related health problems.
In older people, the effect of obesity is much more complex.36 The optimal weight in terms of survival increases with age. More interesting is the finding that although the risk of cardiovascular disease is higher in overweight or obese older adults, studies also suggest that in this age group, being overweight or obese is paradoxically associated with lower mortality rates from these diseases.26 This phenomenon is called the obesity paradox.37
For those over age 75, the relative risk of death from all causes and from cardiovascular disease has been found to decrease with increasing BMI.25 The relationship between BMI and death from all causes in older adults may actually be a U-shaped curve, meaning that the risk of death rises at both extremes of BMI values.26
Possible explanations for the paradox
Several hypotheses have been proposed to explain the change in the relationship between BMI and the risk of death that occurs with aging.
The BMI is an imperfect measure of obesity. The obesity paradox may be an artifact of using the BMI to measure obesity in older adults.17 As described above, sarcopenic obesity must be considered in those over age 65 because the BMI does not differentiate between fat and muscle. Older adults tend to have a higher proportion of body and visceral fat that is distributed differently, making the waist circumference or waist-hip ratio a more appropriate measure of obesity in this group.38 Janssen et al39 found that in people age 65 and older, after controlling for waist circumference, higher BMI values were associated with lower death rates; after controlling for BMI, waist circumference was associated with a higher risk of death.
The survival effect suggests that people who are susceptible to the negative effects of obesity die sooner,40 and those who survive until old age may be resistant to the effects of obesity.41 If true, the survival effect would explain why the death rate seems to be unaffected by BMI in the older population.
Unhealthy weight loss. Smoking and diseases such as cancer that can cause early death may also induce weight loss, further complicating the relationship between BMI and death.19 After age 80, the association between BMI and the risk of death is weak because those with a low BMI include not only those who have always been lean and physically active, but also those who lost weight through chronic ill health or smoking.17
Further study needed. Thus, a number of confounding variables may muddy the association between obesity and death in older adults. Obesity should not be misinterpreted as being harmless or beneficial in older adults. Stevens et al36 found that a greater BMI was associated with a higher rate of death from all causes and from cardiovascular disease in men and women up to age 75, but that the relative risk of death associated with a greater BMI decreased with age.
Optimal BMI targets in older people have yet to be validated in a large prospective trial. However, multiple studies have examined the relationship between BMI and all-cause mortality in older adults and have identified a BMI of 24 to 35 as “ideal” and associated with the lowest risk of death, with a lower range for men and a higher range for women.42,43 The topic has been reviewed by Oreopoulos et al.26 More research is needed to evaluate this relationship.
THE BENEFIT OF WEIGHT LOSS IN OLDER ADULTS IS CONTROVERSIAL
In younger obese people, weight loss brings a multitude of benefits by reducing the risk of complications arising from obesity. However, in older adults, the effects of weight loss remain controversial, and evidence to guide treatment is limited.44,45 The few trials that have been published have typically focused on cardiovascular risk factors rather than physical function.45
In a 1-year trial, 107 people age 65 or older were randomized to a control group, to weight management, to exercise, or to weight management plus exercise. The combination of weight loss and exercise yielded the greatest improvement in physical function.46
Intentional vs unintentional weight loss
Intentional weight loss is altogether different from unintentional weight loss.
In most cases, weight loss in older adults is unintentional and may indicate underlying disease and impending death.17 For example, older men who lose weight unintentionally have significantly greater rates of smoking, disability, cancer, and respiratory disease and less obesity and physical activity than those who lose weight intentionally.47
Studies have shown an increase in life expectancy in older patients with type 2 diabetes mellitus who lost weight intentionally.48,49 In fact, moderate weight loss—just 5% to 10%—has been shown to improve cardiovascular risk factors,44 osteoarthritis, and type 2 diabetes.50
Bales and Buhr44 performed a systematic review of 16 studies that had lasted at least 6 months. Patients were age 60 or older with a minimum baseline BMI of 27 kg/m2 who intentionally lost at least 3% of body weight or 2 kg. Levels of the inflammatory markers C-reactive protein, tumor necrosis factor alpha, and interleukin 6 declined with weight, along with blood pressure, fasting glucose, waist circumference, and low-density lipoprotein cholesterol. On the downside, bone mineral density and lean body mass also declined slightly. The best way to avoid losing lean body mass and to preserve bone density during weight loss is to include a program of resistance-training exercises.
No clinical trial has evaluated the effects of intentional weight loss on death rates in older obese people.25 As a result, evidence-based recommendations cannot be made. Rather, advice on weight loss must be individualized, with special emphasis on the patient’s weight history and medical comorbidities.44
Oreopoulos et al26 summarized the possible effects of BMI, abdominal fat, lean body mass, and intentional weight loss on morbidity and mortality outcomes in older adults (Table 2).
TREATMENT GUIDELINES AND RECOMMENDATIONS
Many of the methods of weight management in older adults are the same as in young and middle-aged adults.51 Recommendations for all age groups include lifestyle changes, increased activity, dietary changes, drug therapy, and bariatric surgery.
Whether there should be separate guidelines for older adults is controversial. In view of the obesity paradox, physicians have been reluctant to recommend weight loss in elderly patients. Caution is advised in recommending weight loss solely on the basis of body weight, as studies have shown that the weight associated with maximal survival increases with age. Because of age-related changes in body composition and reduced energy requirements and expenditure, recommendations for the young and middle-aged should not be applied directly to older adults.
In this group, especially those who have survived into old age with good health and an intact functional status, one could argue that significant caloric restriction should not be recommended. In these people, the goal is often to maintain weight and incorporate a daily exercise program rather than to aggressively lose weight. Adding resistance training can improve physical function, which can improve quality of life. There is less emphasis on cardiovascular risk, but both outcomes apply for both age groups.52
Intentional weight loss should be recommended to high-risk older adults, including those with cardiovascular disease, type 2 diabetes mellitus, and metabolic syndrome, because the absolute risk of death and morbidity is higher in this group. Most health benefits can be achieved with modest weight loss.53 Potential benefits include prevention of cognitive impairment, protection from bone fractures, an increase in antioxidant defense, a reserve of fat and energy stores, and an increase in longevity.26
Treatment differs from that in the younger population primarily because of the importance of preventing loss of muscle with intentional weight loss. People of all ages who lose weight intentionally lose fat and, to a lesser extent, skeletal muscle. Older patients have already lost muscle mass, but further changes in body composition, especially a further reduction in muscle mass, can be limited by consuming about 1.0 g/kg of high-quality protein in the diet and by engaging in resistance training and weight training.52
Improving quality of life and physical function are important goals. Information is emerging about when obesity needs to be managed in older adults. There is also evidence to support dietary and exercise therapy.54 Weight-loss options include lifestyle interventions, pharmacotherapy, and bariatric surgery.
Lifestyle interventions: Diet and exercise
The goal is to induce an energy deficit by reducing energy intake, increasing energy expenditure, or both—by 500 to 1,000 calories a day. This generally leads to a loss of 1 to 2 lb per week, and possibly up to 10% of weight in 6 months. Loss of about 10 to 20 lb with diet and exercise can translate to a relatively large reduction in visceral fat, with subsequent improvement in metabolic abnormalities.
A regular exercise program is important for improving overall physical function, which can slow progression to frailty. Adding aerobic, endurance, and resistance training helps preserve fat-free mass, which otherwise tends to diminish during active weight loss.55–57
The exercise program should begin at the outset of the weight-loss effort to help maintain weight loss and to prevent weight regain.58 Exercise is not essential for reaching the targeted weight loss, but starting early is important to reduce the loss of lean muscle that is usually already seen in the older population.
Several studies indicate that diet and exercise are just as effective in middle-aged and older people (over age 60) as in the younger population.58–60 Older people in the Diabetes Prevention Program were more compliant with lifestyle interventions and lost more weight than younger participants49: 60% of the older group met the 7% weight-loss goal at the end of 24 weeks, compared with 43% of those under age 45. At 3 years, the numbers were 63% vs 27%.
In a small randomized controlled trial,61 fat mass decreased by 6.6 kg in 17 people assigned to a program of diet and exercise, compared with a gain of 1.7 kg in a control group of 10 patients. Fat-free mass decreased by about 1 kg in both groups. The authors concluded that diet plus exercise (resistance training and strength training in this trial) could ameliorate frailty in obese older adults.
If exercise is appropriate, a physician should write a prescription for it, especially for resistance training, strengthening, flexibility, and stretching. This is important for patients with sarcopenic obesity and for those at high risk of chronic bone loss. The 2007 American College of Sports Medicine guidelines recommend muscle-strengthening activity of 8 to 10 exercises involving the major muscle groups, 10 to 15 repetitions at least twice a week. Flexibility and balance exercises should be included for those at risk of falls.62
Pharmacotherapy
At present, there are two general classes of weight-loss drugs: appetite suppressants and drugs that interfere with nutrient absorption.
Appetite suppressants include the sympathomimetics, which stimulate the release of dopamine and norepinephrine, resulting in increased satiety. Data—and therefore, recommendations—on their use in the elderly are very scarce, as most randomized controlled trials included only a small number of older people. A meta-analysis of drug therapy to treat obesity noted that the study population ranged in age from 34 to 54.63
The only approved drug currently available for use in older adults is orlistat, which blocks absorption of dietary fat by binding to intestinal lipase. A randomized controlled trial found the weight loss with orlistat to be comparable in older and younger adults.64,65
Review medications than can cause weight gain
When assessing older adults, always review the drugs they are taking. Those known to cause weight gain include certain of the following:
- Antiepileptics (eg, gabapentin)
- Antipsychotics (eg, olanzapine)
- Antidepressants (eg, tricyclics)
- Antihyperglycemic drugs (eg, sulfonylureas, thiazolidinediones)
- Beta-blockers
- Steroids.
If medically appropriate, a weight-neutral drug should be substituted for one suspected of causing weight gain. If a different physician (eg, a specialist) prescribed the original drug, he or she should be notified or consulted about any change.
Bariatric surgery
Bariatric surgery is the most effective weight-loss option, and more older patients are undergoing it than in the past. Dorman et al66 showed that the number of patients age 65 or older undergoing bariatric surgery increased from the year 2005 (when they accounted for 2%) to 2009 (when they accounted for 4.8%).
However, very few studies have provided information on the safety and effectiveness of bariatric surgery in older people. Several reports concluded that rates of perioperative morbidity and mortality are higher in older patients.67–69 Surgery resulted in marked weight loss and improvement in obesity-related complications and physical disability in older patients, although by a lower rate than in younger patients.
Varela et al70 examined the outcomes of bariatric surgery in a database from the University Health System Consortium Centers between 1999 and 2005. Patients over age 60 accounted for 1,339 (2.7%) of all bariatric operations performed. Compared with young and middle-aged patients, older patients had more comorbidities, longer hospital stays, and more complications, in addition to a higher in-hospital mortality rate. When risk-adjusted, the observed-to-expected mortality ratio for the older group was 0.9, compared with 0.7 in the young and middle-aged cohort.
Willkomm et al71 found an apparently higher operative risk profile in those over age 65 (n = 100) than in younger patients (n = 1,374), with higher rates of sleep apnea, diabetes, and hypertension. However, the operative outcomes were similar in the two groups in terms of operative time, length of stay, and 30-day readmission rates. The authors concluded that patients over age 65 had excellent outcomes compared with younger patients, suggesting that older age is not a risk factor for complications or death with bariatric surgery.
The American College of Surgeons National Surgical Quality Improvement Program evaluated the outcomes of 48,378 adults with a BMI greater than or equal to 35 kg/m2 who underwent bariatric surgery between 2005 and 2009.66 During this time, the number of patients age 65 and older seeking bariatric surgery increased from 1.5% to 4%. A total of 1,449 patients were in this age range. Thirty-day mortality rates did not differ significantly by age group and were less than 1% for all age ranges. Being age 65 or older was a significant predictor of prolonged length of stay but not of major adverse events. Significant predictors of major adverse events were a BMI greater than or equal to 55 kg/m2, cardiac comorbidities, a severe American Society of Anesthesiologists score, albumin levels lower than 3 g/dL, and creatinine levels greater than 1.5 mg/dL.
The most up-to-date study of the outcomes of bariatric surgery in patients over age 70 was a retrospective review at a single institution from 2007 to 2008 of 42 patients who underwent bariatric surgery.72 Twenty-two patients had laparoscopic gastric banding, 12 had laparoscopic sleeve gastrectomy, and 8 underwent laparoscopic Roux-en-Y gastric bypass. No patient died, complications occurred in 9 patients, and the rates of postoperative use of medications for hypertension, hyperlipidemia, diabetes, and osteoarthritis were reduced by about half. With the increasing number of patients seeking bariatric surgery, especially those over age 70, further prospective studies will determine if the outcomes are statistically significant.
If bariatric surgery is considered
The outcomes, complications, and mortality rates associated with bariatric surgery have been shown to be acceptable for adults age 65 and older. Perioperative risk assessment in the older obese patient seeking bariatric surgery is paramount to ensure that the benefits of the procedure justify any associated risks to the patient. Consequently, patients over age 65 should not be excluded out of hand: the patient’s individual risk of major adverse events must be identified beforehand.
If the patient is at risk, efforts should be made to reduce the risk to an acceptable level, including cardiac risk stratification, optimization of drug therapy, and discussions with the bariatric surgeon to plan on a less-invasive laparoscopic procedure. Otherwise, older obese patients can safely proceed with conventional bariatric surgery, which will help them achieve durable weight loss, improve quality of life, and reduce associated comorbidities.
The aforementioned studies of bariatric surgery are retrospective, include small numbers of patients, and lack long-term follow-up. The issues of long-term safety and the risk of death and morbidity in the aging population will require randomized controlled trials to answer these important questions.
At our hospital, we have seen an increase in the number of patients referred for a possible additional procedure (revision) to correct a problem from a previous bariatric surgery. The problems arising from the previous surgery can lead to weight gain or to excessive weight loss and malnutrition. To date, our institution has no policy on when to consider a revisional procedure in an older patient. All patients, including older ones, are assessed for the procedure on a case-by-case basis.
- Bray GA, Macdiarmid J. The epidemic of obesity. West J Med 2000; 172:78–79.
- Calle EE, Thun MJ, Petrelli JM, Rodriguez C, Heath CW. Body-mass index and mortality in a prospective cohort of US adults. N Engl J Med 1999; 341:1097–1105.
- Kalantar-Zadeh K, Horwich TB, Oreopoulos A, et al. Risk factor paradox in wasting diseases. Curr Opin Clin Nutr Metab Care 2007; 10:433–442.
- Mokdad AH, Marks JS, Stroup DF, Gerberding JL. Actual causes of death in the United States, 2000. JAMA 2004; 291:1238–1245.
- Arias E, Rostron BL, Tejada-Vera B. United States life tables, 2005. National vital statistics reports; vol 58no 10. Hyattsville, MD: National Center for Health Statistics. 2010.
- US Census Bureau International Database (IDB). Population projections of the US by age, sex, race, Hispanic origin, population division. http://www.census.gov/ipc/www/idb/country.php. Accessed September 13, 2013.
- Hedley AA, Ogden CL, Johnson CL, Carroll MD, Curtin LR, Flegal KM. Prevalence of overweight and obesity among US children, adolescents, and adults, 1999–2002. JAMA 2004; 291:2847–2850.
- Kuczmarski RJ, Flegal KM, Campbell SM, Johnson CL. Increasing prevalence of overweight among US adults. The National Health and Nutrition Examination Surveys, 1960 to 1991. JAMA 1994; 272:205–211.
- Mokdad AH, Serdula MK, Dietz WH, Bowman BA, Marks JS, Koplan JP. The spread of the obesity epidemic in the United States, 1991–1998. JAMA 1999; 282:1519–1522.
- Horani MH, Mooradian AD. Management of obesity in the elderly: special considerations. Treat Endocrinol 2002; 1:387–398.
- Beaufrère B, Morio B. Fat and protein redistribution with aging: metabolic considerations. Eur J Clin Nutr 2000; 54(suppl 3):S48–S53.
- Gallagher D, Heymsfield SB, Heo M, Jebb SA, Murgatroyd PR, Sakamoto Y. Healthy percentage body fat ranges: an approach for developing guidelines based on body mass index. Am J Clin Nutr 2000; 72:694–701.
- Snitker S. Use of body fatness cutoff points (author reply). Mayo Clin Proc 2010; 85:1057; author reply 1057–1058.
- Baumgartner RN, Koehler KM, Gallagher D, et al. Epidemiology of sarcopenia among the elderly in New Mexico. Am J Epidemiol 1998; 147:755–763. Erratum in Am J Epidemiol 1999; 149:1161.
- Visscher TL, Seidell JC, Molarius A, van der Kuip D, Hofman A, Witteman JC. A comparison of body mass index, waist-hip ratio and waist circumference as predictors of all-cause mortality among the elderly: the Rotterdam study. Int J Obes Relat Metab Disord 2001; 25:1730–1735.
- Molarius A, Seidell JC, Visscher TL, Hofman A. Misclassification of high-risk older subjects using waist action levels established for young and middle-aged adults—results from the Rotterdam Study. J Am Geriatr Soc 2000; 48:1638–1645.
- Han TS, Tajar A, Lean ME. Obesity and weight management in the elderly. Br Med Bull 2011; 97:169–196.
- Turcato E, Bosello O, Di Francesco V, et al. Waist circumference and abdominal sagittal diameter as surrogates of body fat distribution in the elderly: their relation with cardiovascular risk factors. Int J Obes Relat Metab Disord 2000; 24:1005–1010.
- Zamboni M, Mazzali G, Zoico E, et al. Health consequences of obesity in the elderly: a review of four unresolved questions. Int J Obes (Lond) 2005; 29:1011–1029.
- Heim N, Snijder MB, Heymans MW, Deeg DJ, Seidell JC, Visser M. Optimal cutoff values for high-risk waist circumference in older adults based on related health outcomes. Am J Epidemiol 2011; 174:479–489.
- Roubenoff R, Castaneda C. Sarcopenia—understanding the dynamics of aging muscle. JAMA 2001; 286:1230–1231.
- Schutz Y, Kyle UU, Pichard C. Fat-free mass index and fat mass index percentiles in Caucasians aged 18-98 y. Int J Obes Relat Metab Disord 2002; 26:953–960.
- Baumgartner RN, Wayne SJ, Waters DL, Janssen I, Gallagher D, Morley JE. Sarcopenic obesity predicts instrumental activities of daily living disability in the elderly. Obes Res 2004; 12:1995–2004.
- Morley JE, Baumgartner RN, Roubenoff R, Mayer J, Nair KS. Sarcopenia. J Lab Clin Med 2001; 137:231–243.
- Roubenoff R. Sarcopenic obesity: the confluence of two epidemics. Obes Res 2004; 12:887–888.
- Oreopoulos A, Kalantar-Zadeh K, Sharma AM, Fonarow GC. The obesity paradox in the elderly: potential mechanisms and clinical implications. Clin Geriatr Med 2009; 25:643–659.
- Elia M, Ritz P, Stubbs RJ. Total energy expenditure in the elderly. Eur J Clin Nutr 2000; 54(suppl 3):S92–S103.
- Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988; 37:1595–1607.
- Corona G, Mannucci E, Forti G, Maggi M. Hypogonadism, ED, metabolic syndrome and obesity: a pathological link supporting cardiovascular diseases. Int J Androl 2009; 32:587–598.
- Haarbo J, Hassager C, Riis BJ, Christiansen C. Relation of body fat distribution to serum lipids and lipoproteins in elderly women. Atherosclerosis 1989; 80:57–62.
- Cignarella A, Kratz M, Bolego C. Emerging role of estrogen in the control of cardiometabolic disease. Trends Pharmacol Sci 2010; 31:183–189.
- Felson DT, Anderson JJ, Naimark A, Walker AM, Meenan RF. Obesity and knee osteoarthritis. The Framingham Study. Ann Intern Med 1988; 109:18–24.
- Gelber AC, Hochberg MC, Mead LA, Wang NY, Wigley FM, Klag MJ. Body mass index in young men and the risk of subsequent knee and hip osteoarthritis. Am J Med 1999; 107:542–548.
- Iwao S, Iwao N, Muller DC, Elahi D, Shimokata H, Andres R. Effect of aging on the relationship between multiple risk factors and waist circumference. J Am Geriatr Soc 2000; 48:788–794.
- Folsom AR, Kushi LH, Anderson KE, et al. Associations of general and abdominal obesity with multiple health outcomes in older women: the Iowa Women’s Health Study. Arch Intern Med 2000; 160:2117–2128.
- Stevens J, Cai J, Pamuk ER, Williamson DF, Thun MJ, Wood JL. The effect of age on the association between body-mass index and mortality. N Engl J Med 1998; 338:1–7.
- Kalantar-Zadeh K, Horwich TB, Oreopoulos A, et al. Risk factor paradox in wasting diseases. Curr Opin Clin Nutr Metab Care 2007; 10:433–442.
- Zamboni M, Armellini F, Harris T, et al. Effects of age on body fat distribution and cardiovascular risk factors in women. Am J Clin Nutr 1997; 66:111–115.
- Janssen I, Katzmarzyk PT, Ross R. Body mass index is inversely related to mortality in older people after adjustment for waist circumference. J Am Geriatr Soc 2005; 53:2112–2118.
- Inelmen EM, Sergi G, Coin A, Miotto F, Peruzza S, Enzi G. Can obesity be a risk factor in elderly people? Obes Rev 2003; 4:147–155.
- Elia M. Obesity in the elderly. Obes Res 2001; 9(suppl 4):244S–248S.
- Losonczy KG, Harris TB, Cornoni-Huntley J, et al. Does weight loss from middle age to old age explain the inverse weight mortality relation in old age? Am J Epidemiol 1995; 141:312–321.
- Corrada MM, Kawas CH, Mozaffar F, Paganini-Hill A. Association of body mass index and weight change with all-cause mortality in the elderly. Am J Epidemiol 2006; 163:938–949.
- Bales CW, Buhr G. Is obesity bad for older persons? A systematic review of the pros and cons of weight reduction in later life. J Am Med Dir Assoc 2008; 9:302–312.
- Witham MD, Avenell A. Interventions to achieve long-term weight loss in obese older people: a systematic review and meta-analysis. Age Ageing 2010; 39:176–184.
- Villareal DT, Chode S, Parimi N, et al. Weight loss, exercise, or both and physical function in obese older adults. N Engl J Med 2011; 364:1218–1229.
- Wannamethee SG, Shaper AG, Whincup PH, Walker M. Characteristics of older men who lose weight intentionally or unintentionally. Am J Epidemiol 2000; 151:667–675.
- Lean ME, Powrie JK, Anderson AS, Garthwaite PH. Obesity, weight loss and prognosis in type 2 diabetes. Diabet Med 1990; 7:228–233.
- Williamson DF, Thompson TJ, Thun M, Flanders D, Pamuk E, Byers T. Intentional weight loss and mortality among overweight individuals with diabetes. Diabetes Care 2000; 23:1499–1504.
- Hamman RF, Wing RR, Edelstein SL, et al. Effect of weight loss with lifestyle intervention on risk of diabetes. Diabetes Care 2006; 29:2102-2107.
- National Heart, Lung, and Blood Institute in cooperation with The National Institute of Diabetes and Digestive and Kidney Diseases. Clinical guidelines on the identification, evaluation and treatment of the overweight and obesity in adults, the evidence report. NIH Publication number 98-4803 http://www.nhlbi.nih.gov/guidelines/obesity/ob_gdlns.pdf. Accessed September 13, 2013.
- Villareal DT, Apovian CM, Kushner RF, Klein S; American Society for Nutrition; NAASO, The Obesity Society. Obesity in older adults: technical review and position statement of the American Society for Nutrition and NAASO, The Obesity Society. Am J Clin Nutr 2005; 82:923–934.
- Williamson DF, Pamuk E, Thun M, Flanders D, Byers T, Heath C. Prospective study of intentional weight loss and mortality in never-smoking overweight US white women aged 40-64 years. Am J Epidemiol 1995; 141:1128–1141.
- McTigue KM, Hess R, Ziouras J. Obesity in older adults: a systematic review of the evidence for diagnosis and treatment. Obesity (Silver Spring). 2006; 14:1485–1497.
- Ryan AS, Pratley RE, Elahi D, Goldberg AP. Resistive training increases fat-free mass and maintains RMR despite weight loss in postmenopausal women. J Appl Physiol 1995; 79:818–823.
- Pavlou KN, Krey S, Steffee WP. Exercise as an adjunct to weight loss and maintenance in moderately obese subjects. Am J Clin Nutr 1989; 49(suppl 5):1115–1123.
- Kraemer WJ, Volek JS, Clark KL, et al. Influence of exercise training on physiological and performance changes with weight loss in men. Med Sci Sports Exerc 1999; 31:1320–1329.
- Wing RR, Hill JO. Successful weight loss maintenance. Annu Rev Nutr 2001; 21:323–341.
- Banks M, Klein S, Sinacore D, Siener C, Villareal DT. Effects of weight loss and exercise on frailty in obese elderly subjects. J Am Geriatr Soc 2005; 53:S16.
- Messier SP, Loeser RF, Miller GD, et al. Exercise and dietary weight loss in overweight and obese older adults with knee osteoarthritis: the Arthritis, Diet, and Activity Promotion Trial. Arthritis Rheum 2004; 50:1501–1510.
- Villareal DT, Banks M, Sinacore DR, Siener C, Klein S. Effect of weight loss and exercise on frailty in obese older adults. Arch Intern Med 2006; 166:860–866.
- Nelson ME, Rejeski WJ, Blair SN, et al; American College of Sports Medicine; American Heart Association. Physical activity and public health in older adults: recommendation from the American College of Sports Medicine and the American Heart Association. Circulation 2007; 116:1094–1105.
- Li Z, Maglione M, Tu W, et al. Meta-analysis: pharmacologic treatment of obesity. Ann Intern Med 2005; 142:532–546.
- Segal KR, Lucas C, Boldrin M, Hauptman J. Weight loss efficacy of orlistat in obese elderly adults (abstract). Obes Res 1999; 7(suppl):26S.
- Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med 2000; 9:160–167.
- Dorman RB, Abraham AA, Al-Refaie WB, Parsons HM, Ikramuddin S, Habermann EB. Bariatric surgery outcomes in the elderly: an ACS NSQIP study. J Gastrointest Surg 2012; 16:35–44.
- Sugerman HJ, DeMaria EJ, Kellum JM, Sugerman EL, Meador JG, Wolfe LG. Effects of bariatric surgery in older patients. Ann Surg 2004; 240:243–247.
- St. Peter SD, Craft RO, Tiede JL, Swain JM. Impact of advanced age on weight loss and health benefits after laparoscopic gastric bypass. Arch Surg 2005; 140:165–168.
- Sosa JL, Pombo H, Pallavicini H, Ruiz-Rodriguez M. Laparoscopic gastric bypass beyond age 60. Obes Surg 2004; 14:1398–1401.
- Varela JE, Wilson SE, Nguyen NT. Outcomes of bariatric surgery in the elderly. Am Surg 2006; 72:865–869.
- Willkomm CM, Fisher TL, Barnes GS, Kennedy CI, Kuhn JA. Surgical weight loss >65 years old: is it worth the risk? Surg Obes Relat Dis 2010; 6:491–496.
- Heiat A, Vaccarino V, Krumholz HM. An evidence-based assessment of federal guidelines for overweight and obesity as they apply to elderly persons. Arch Intern Med 2001; 161:1194–1203.
- Bray GA, Macdiarmid J. The epidemic of obesity. West J Med 2000; 172:78–79.
- Calle EE, Thun MJ, Petrelli JM, Rodriguez C, Heath CW. Body-mass index and mortality in a prospective cohort of US adults. N Engl J Med 1999; 341:1097–1105.
- Kalantar-Zadeh K, Horwich TB, Oreopoulos A, et al. Risk factor paradox in wasting diseases. Curr Opin Clin Nutr Metab Care 2007; 10:433–442.
- Mokdad AH, Marks JS, Stroup DF, Gerberding JL. Actual causes of death in the United States, 2000. JAMA 2004; 291:1238–1245.
- Arias E, Rostron BL, Tejada-Vera B. United States life tables, 2005. National vital statistics reports; vol 58no 10. Hyattsville, MD: National Center for Health Statistics. 2010.
- US Census Bureau International Database (IDB). Population projections of the US by age, sex, race, Hispanic origin, population division. http://www.census.gov/ipc/www/idb/country.php. Accessed September 13, 2013.
- Hedley AA, Ogden CL, Johnson CL, Carroll MD, Curtin LR, Flegal KM. Prevalence of overweight and obesity among US children, adolescents, and adults, 1999–2002. JAMA 2004; 291:2847–2850.
- Kuczmarski RJ, Flegal KM, Campbell SM, Johnson CL. Increasing prevalence of overweight among US adults. The National Health and Nutrition Examination Surveys, 1960 to 1991. JAMA 1994; 272:205–211.
- Mokdad AH, Serdula MK, Dietz WH, Bowman BA, Marks JS, Koplan JP. The spread of the obesity epidemic in the United States, 1991–1998. JAMA 1999; 282:1519–1522.
- Horani MH, Mooradian AD. Management of obesity in the elderly: special considerations. Treat Endocrinol 2002; 1:387–398.
- Beaufrère B, Morio B. Fat and protein redistribution with aging: metabolic considerations. Eur J Clin Nutr 2000; 54(suppl 3):S48–S53.
- Gallagher D, Heymsfield SB, Heo M, Jebb SA, Murgatroyd PR, Sakamoto Y. Healthy percentage body fat ranges: an approach for developing guidelines based on body mass index. Am J Clin Nutr 2000; 72:694–701.
- Snitker S. Use of body fatness cutoff points (author reply). Mayo Clin Proc 2010; 85:1057; author reply 1057–1058.
- Baumgartner RN, Koehler KM, Gallagher D, et al. Epidemiology of sarcopenia among the elderly in New Mexico. Am J Epidemiol 1998; 147:755–763. Erratum in Am J Epidemiol 1999; 149:1161.
- Visscher TL, Seidell JC, Molarius A, van der Kuip D, Hofman A, Witteman JC. A comparison of body mass index, waist-hip ratio and waist circumference as predictors of all-cause mortality among the elderly: the Rotterdam study. Int J Obes Relat Metab Disord 2001; 25:1730–1735.
- Molarius A, Seidell JC, Visscher TL, Hofman A. Misclassification of high-risk older subjects using waist action levels established for young and middle-aged adults—results from the Rotterdam Study. J Am Geriatr Soc 2000; 48:1638–1645.
- Han TS, Tajar A, Lean ME. Obesity and weight management in the elderly. Br Med Bull 2011; 97:169–196.
- Turcato E, Bosello O, Di Francesco V, et al. Waist circumference and abdominal sagittal diameter as surrogates of body fat distribution in the elderly: their relation with cardiovascular risk factors. Int J Obes Relat Metab Disord 2000; 24:1005–1010.
- Zamboni M, Mazzali G, Zoico E, et al. Health consequences of obesity in the elderly: a review of four unresolved questions. Int J Obes (Lond) 2005; 29:1011–1029.
- Heim N, Snijder MB, Heymans MW, Deeg DJ, Seidell JC, Visser M. Optimal cutoff values for high-risk waist circumference in older adults based on related health outcomes. Am J Epidemiol 2011; 174:479–489.
- Roubenoff R, Castaneda C. Sarcopenia—understanding the dynamics of aging muscle. JAMA 2001; 286:1230–1231.
- Schutz Y, Kyle UU, Pichard C. Fat-free mass index and fat mass index percentiles in Caucasians aged 18-98 y. Int J Obes Relat Metab Disord 2002; 26:953–960.
- Baumgartner RN, Wayne SJ, Waters DL, Janssen I, Gallagher D, Morley JE. Sarcopenic obesity predicts instrumental activities of daily living disability in the elderly. Obes Res 2004; 12:1995–2004.
- Morley JE, Baumgartner RN, Roubenoff R, Mayer J, Nair KS. Sarcopenia. J Lab Clin Med 2001; 137:231–243.
- Roubenoff R. Sarcopenic obesity: the confluence of two epidemics. Obes Res 2004; 12:887–888.
- Oreopoulos A, Kalantar-Zadeh K, Sharma AM, Fonarow GC. The obesity paradox in the elderly: potential mechanisms and clinical implications. Clin Geriatr Med 2009; 25:643–659.
- Elia M, Ritz P, Stubbs RJ. Total energy expenditure in the elderly. Eur J Clin Nutr 2000; 54(suppl 3):S92–S103.
- Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988; 37:1595–1607.
- Corona G, Mannucci E, Forti G, Maggi M. Hypogonadism, ED, metabolic syndrome and obesity: a pathological link supporting cardiovascular diseases. Int J Androl 2009; 32:587–598.
- Haarbo J, Hassager C, Riis BJ, Christiansen C. Relation of body fat distribution to serum lipids and lipoproteins in elderly women. Atherosclerosis 1989; 80:57–62.
- Cignarella A, Kratz M, Bolego C. Emerging role of estrogen in the control of cardiometabolic disease. Trends Pharmacol Sci 2010; 31:183–189.
- Felson DT, Anderson JJ, Naimark A, Walker AM, Meenan RF. Obesity and knee osteoarthritis. The Framingham Study. Ann Intern Med 1988; 109:18–24.
- Gelber AC, Hochberg MC, Mead LA, Wang NY, Wigley FM, Klag MJ. Body mass index in young men and the risk of subsequent knee and hip osteoarthritis. Am J Med 1999; 107:542–548.
- Iwao S, Iwao N, Muller DC, Elahi D, Shimokata H, Andres R. Effect of aging on the relationship between multiple risk factors and waist circumference. J Am Geriatr Soc 2000; 48:788–794.
- Folsom AR, Kushi LH, Anderson KE, et al. Associations of general and abdominal obesity with multiple health outcomes in older women: the Iowa Women’s Health Study. Arch Intern Med 2000; 160:2117–2128.
- Stevens J, Cai J, Pamuk ER, Williamson DF, Thun MJ, Wood JL. The effect of age on the association between body-mass index and mortality. N Engl J Med 1998; 338:1–7.
- Kalantar-Zadeh K, Horwich TB, Oreopoulos A, et al. Risk factor paradox in wasting diseases. Curr Opin Clin Nutr Metab Care 2007; 10:433–442.
- Zamboni M, Armellini F, Harris T, et al. Effects of age on body fat distribution and cardiovascular risk factors in women. Am J Clin Nutr 1997; 66:111–115.
- Janssen I, Katzmarzyk PT, Ross R. Body mass index is inversely related to mortality in older people after adjustment for waist circumference. J Am Geriatr Soc 2005; 53:2112–2118.
- Inelmen EM, Sergi G, Coin A, Miotto F, Peruzza S, Enzi G. Can obesity be a risk factor in elderly people? Obes Rev 2003; 4:147–155.
- Elia M. Obesity in the elderly. Obes Res 2001; 9(suppl 4):244S–248S.
- Losonczy KG, Harris TB, Cornoni-Huntley J, et al. Does weight loss from middle age to old age explain the inverse weight mortality relation in old age? Am J Epidemiol 1995; 141:312–321.
- Corrada MM, Kawas CH, Mozaffar F, Paganini-Hill A. Association of body mass index and weight change with all-cause mortality in the elderly. Am J Epidemiol 2006; 163:938–949.
- Bales CW, Buhr G. Is obesity bad for older persons? A systematic review of the pros and cons of weight reduction in later life. J Am Med Dir Assoc 2008; 9:302–312.
- Witham MD, Avenell A. Interventions to achieve long-term weight loss in obese older people: a systematic review and meta-analysis. Age Ageing 2010; 39:176–184.
- Villareal DT, Chode S, Parimi N, et al. Weight loss, exercise, or both and physical function in obese older adults. N Engl J Med 2011; 364:1218–1229.
- Wannamethee SG, Shaper AG, Whincup PH, Walker M. Characteristics of older men who lose weight intentionally or unintentionally. Am J Epidemiol 2000; 151:667–675.
- Lean ME, Powrie JK, Anderson AS, Garthwaite PH. Obesity, weight loss and prognosis in type 2 diabetes. Diabet Med 1990; 7:228–233.
- Williamson DF, Thompson TJ, Thun M, Flanders D, Pamuk E, Byers T. Intentional weight loss and mortality among overweight individuals with diabetes. Diabetes Care 2000; 23:1499–1504.
- Hamman RF, Wing RR, Edelstein SL, et al. Effect of weight loss with lifestyle intervention on risk of diabetes. Diabetes Care 2006; 29:2102-2107.
- National Heart, Lung, and Blood Institute in cooperation with The National Institute of Diabetes and Digestive and Kidney Diseases. Clinical guidelines on the identification, evaluation and treatment of the overweight and obesity in adults, the evidence report. NIH Publication number 98-4803 http://www.nhlbi.nih.gov/guidelines/obesity/ob_gdlns.pdf. Accessed September 13, 2013.
- Villareal DT, Apovian CM, Kushner RF, Klein S; American Society for Nutrition; NAASO, The Obesity Society. Obesity in older adults: technical review and position statement of the American Society for Nutrition and NAASO, The Obesity Society. Am J Clin Nutr 2005; 82:923–934.
- Williamson DF, Pamuk E, Thun M, Flanders D, Byers T, Heath C. Prospective study of intentional weight loss and mortality in never-smoking overweight US white women aged 40-64 years. Am J Epidemiol 1995; 141:1128–1141.
- McTigue KM, Hess R, Ziouras J. Obesity in older adults: a systematic review of the evidence for diagnosis and treatment. Obesity (Silver Spring). 2006; 14:1485–1497.
- Ryan AS, Pratley RE, Elahi D, Goldberg AP. Resistive training increases fat-free mass and maintains RMR despite weight loss in postmenopausal women. J Appl Physiol 1995; 79:818–823.
- Pavlou KN, Krey S, Steffee WP. Exercise as an adjunct to weight loss and maintenance in moderately obese subjects. Am J Clin Nutr 1989; 49(suppl 5):1115–1123.
- Kraemer WJ, Volek JS, Clark KL, et al. Influence of exercise training on physiological and performance changes with weight loss in men. Med Sci Sports Exerc 1999; 31:1320–1329.
- Wing RR, Hill JO. Successful weight loss maintenance. Annu Rev Nutr 2001; 21:323–341.
- Banks M, Klein S, Sinacore D, Siener C, Villareal DT. Effects of weight loss and exercise on frailty in obese elderly subjects. J Am Geriatr Soc 2005; 53:S16.
- Messier SP, Loeser RF, Miller GD, et al. Exercise and dietary weight loss in overweight and obese older adults with knee osteoarthritis: the Arthritis, Diet, and Activity Promotion Trial. Arthritis Rheum 2004; 50:1501–1510.
- Villareal DT, Banks M, Sinacore DR, Siener C, Klein S. Effect of weight loss and exercise on frailty in obese older adults. Arch Intern Med 2006; 166:860–866.
- Nelson ME, Rejeski WJ, Blair SN, et al; American College of Sports Medicine; American Heart Association. Physical activity and public health in older adults: recommendation from the American College of Sports Medicine and the American Heart Association. Circulation 2007; 116:1094–1105.
- Li Z, Maglione M, Tu W, et al. Meta-analysis: pharmacologic treatment of obesity. Ann Intern Med 2005; 142:532–546.
- Segal KR, Lucas C, Boldrin M, Hauptman J. Weight loss efficacy of orlistat in obese elderly adults (abstract). Obes Res 1999; 7(suppl):26S.
- Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med 2000; 9:160–167.
- Dorman RB, Abraham AA, Al-Refaie WB, Parsons HM, Ikramuddin S, Habermann EB. Bariatric surgery outcomes in the elderly: an ACS NSQIP study. J Gastrointest Surg 2012; 16:35–44.
- Sugerman HJ, DeMaria EJ, Kellum JM, Sugerman EL, Meador JG, Wolfe LG. Effects of bariatric surgery in older patients. Ann Surg 2004; 240:243–247.
- St. Peter SD, Craft RO, Tiede JL, Swain JM. Impact of advanced age on weight loss and health benefits after laparoscopic gastric bypass. Arch Surg 2005; 140:165–168.
- Sosa JL, Pombo H, Pallavicini H, Ruiz-Rodriguez M. Laparoscopic gastric bypass beyond age 60. Obes Surg 2004; 14:1398–1401.
- Varela JE, Wilson SE, Nguyen NT. Outcomes of bariatric surgery in the elderly. Am Surg 2006; 72:865–869.
- Willkomm CM, Fisher TL, Barnes GS, Kennedy CI, Kuhn JA. Surgical weight loss >65 years old: is it worth the risk? Surg Obes Relat Dis 2010; 6:491–496.
- Heiat A, Vaccarino V, Krumholz HM. An evidence-based assessment of federal guidelines for overweight and obesity as they apply to elderly persons. Arch Intern Med 2001; 161:1194–1203.
KEY POINTS
- In older patients, the waist circumference may be more appropriate than the body mass index as a measure of adiposity.
- Data suggest that being moderately overweight may offer a survival advantage in older people, but a body mass index of 30 kg/m2 or higher continues to be associated with many health risks in this age group.
- In obese patients, intensive lifestyle interventions with an emphasis on exercise and strength training can optimize their overall health and quality of life.
- Weight-loss recommendations in older obese patients should take into account the benefits and risks of lifestyle interventions, drug therapy, and bariatric surgery.
Albuminuria: When urine predicts kidney and cardiovascular disease
“One can obtain considerable information concerning the general health by examining the urine.”
—Hippocrates (460?–355? BCE)
Chronic kidney disease is a notable public health concern because it is an important risk factor for end-stage renal disease, cardiovascular disease, and death. Its prevalence1 exceeds 10% and is considerably higher in high-risk groups, such as those with diabetes or hypertension, which are growing in the United States.
While high levels of total protein in the urine have always been recognized as pathologic, a growing body of evidence links excretion of the protein albumin to adverse cardiovascular outcomes, and most international guidelines now recommend measuring albumin specifically. Albuminuria is a predictor of declining renal function and is independently associated with adverse cardiovascular outcomes. Thus, clinicians need to detect it early, manage it effectively, and reduce concurrent risk factors for cardiovascular disease.
Therefore, this review will focus on albuminuria. However, because the traditional standard for urinary protein measurement was total protein, and because a few guidelines still recommend measuring total protein rather than albumin, we will also briefly discuss total urinary protein.
MOST URINARY PROTEIN IS ALBUMIN
Most of the protein in the urine is albumin filtered from the plasma. Less than half of the rest is derived from the distal renal tubules (uromodulin or Tamm-Horsfall mucoprotein), 2 and urine also contains a small and varying proportion of immunoglobulins, low-molecular-weight proteins, and light chains.
Normal healthy people lose less than 30 mg of albumin in the urine per day. In greater amounts, albumin is the major urinary protein in most kidney diseases. Other proteins in urine can be specific markers of less-common illnesses such as plasma cell dyscrasia, glomerulopathy, and renal tubular disease.
MEASURING PROTEINURIA AND ALBUMINURIA
Albumin is not a homogeneous molecule in urine. It undergoes changes to its molecular configuration in the presence of certain ions, peptides, hormones, and drugs, and as a result of proteolytic fragmentation both in the plasma and in renal tubules.3 Consequently, measuring urinary albumin involves a trade-off between convenience and accuracy.
A 24-hour timed urine sample has long been the gold standard for measuring albuminuria, but the collection is cumbersome and time-consuming, and the test is prone to laboratory error.
Dipstick measurements are more convenient and are better at detecting albumin than other proteins in urine, but they have low sensitivity and high interobserver variation.3–5
The albumin-to-creatinine ratio (ACR). As the quantity of protein in the urine changes with time of day, exertion, stress level, and posture, spot-checking of urine samples is not as good as timed collection. However, a simultaneous measurement of creatinine in a spot urine sample adjusts for protein concentration, which can vary with a person’s hydration status. The ACR so obtained is consistent with the 24-hour timed collection (the gold standard) and is the recommended method of assessing albuminuria.3 An early morning urine sample is favored, as it avoids orthostatic variations and varies less in the same individual.
In a study in the general population comparing the ACR in a random sample and in an early morning sample, only 44% of those who had an ACR of 30 mg/g or higher in the random sample had one this high in the early morning sample.6 However, getting an early morning sample is not always feasible in clinical practice. If you are going to measure albuminuria, the Kidney Disease Outcomes and Quality Initiative7 suggests checking the ACR in a random sample and then, if the test is positive, following up and confirming it within 3 months with an early morning sample.

Also, since creatinine excretion differs with race, diet, and muscle mass, if the 24-hour creatinine excretion is not close to 1 g, the ACR will give an erroneous estimate of the 24-hour excretion rate.3
Table 1 compares the various methods of measuring protein in the urine.3,5,8,9 Of note, methods of measuring albumin and total protein vary considerably in their precision and accuracy, making it impossible to reliably translate values from one to the other.5
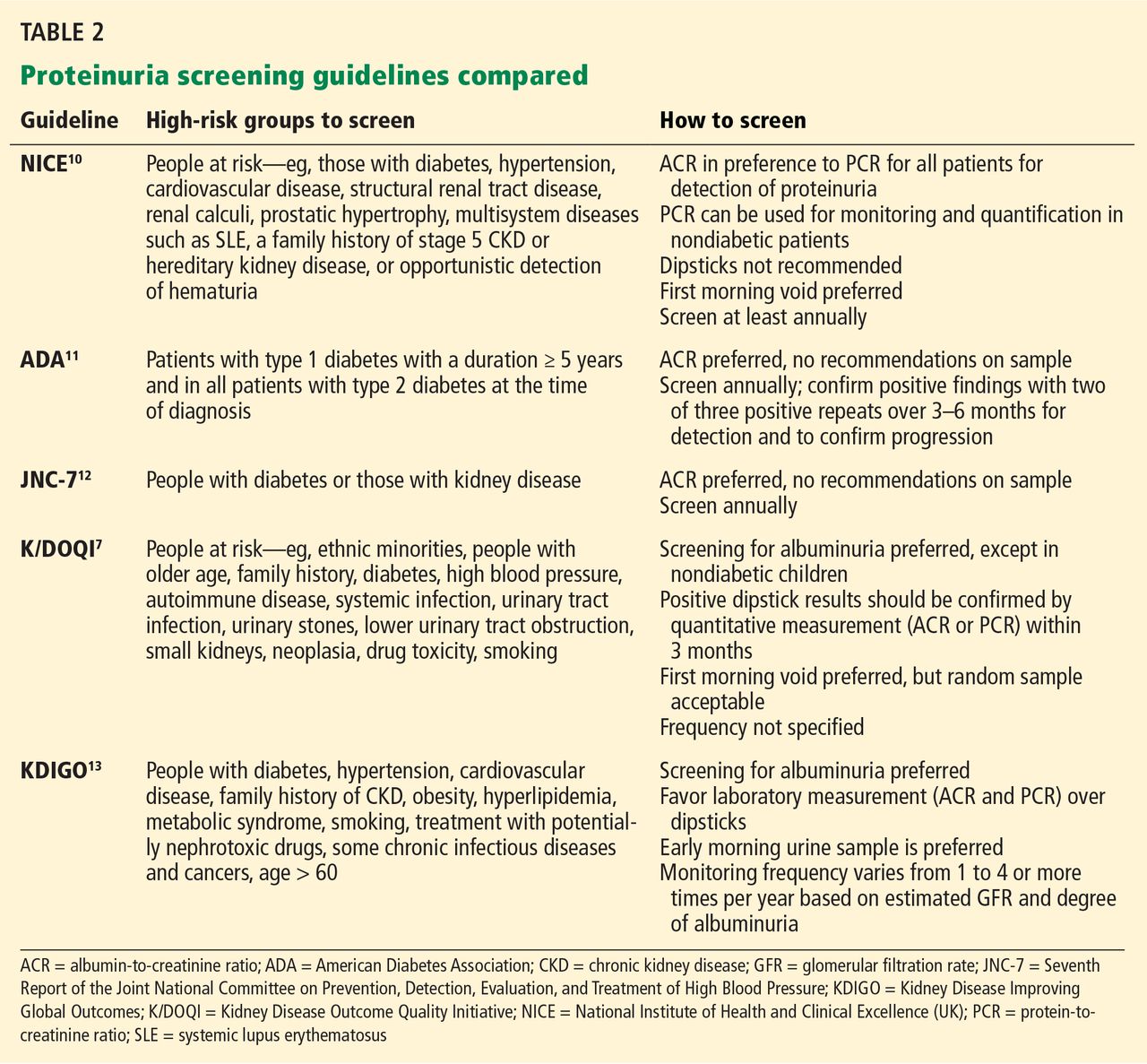
National and international guidelines (Table 2)7,10–13 agree that albuminuria should be tested in diabetic patients, as it is a surrogate marker for early diabetic nephropathy.3,13 Most guidelines also recommend measuring albuminuria by a urine ACR test as the preferred measure, even in people without diabetes.
Also, no single cutoff is universally accepted for distinguishing pathologic albuminuria from physiologic albuminuria, nor is there a universally accepted unit of measure.14 Because approximately 1 g of creatinine is lost in the urine per day, the ACR has the convenient property of numerically matching the albumin excretory rate expressed in milligrams per 24 hours. The other commonly used unit is milligrams of albumin per millimole of creatinine; 30 mg/g is roughly equal to 3 mg/mmol.
The term microalbuminuria was traditionally used to refer to albumin excretion of 30 to 299 mg per 24 hours, and macroalbuminuria to 300 mg or more per 24 hours. However, as there is no pathophysiologic basis to these thresholds (see outcomes data below), the current Kidney Disease Improving Global Outcomes (KDIGO) guidelines do not recommend using these terms.13,15
RENAL COMPLICATIONS OF ALBUMINURIA
A failure of the glomerular filtration barrier or of proximal tubular reabsorption accounts for most cases of pathologic albuminuria.16 Processes affecting the glomerular filtration of albumin include endothelial cell dysfunction and abnormalities with the glomerular basement membrane, podocytes, or the slit diaphragms among the podocytic processes.17
Ultrafiltrated albumin has been directly implicated in tubulointerstitial damage and glomerulosclerosis through diverse pathways. In the proximal tubule, albumin up-regulates interleukin 8 (a chemoattractant for lymphocytes and neutrophils), induces synthesis of endothelin 1 (which stimulates renal cell proliferation, extracellular matrix production, and monocyte attraction), and causes apoptosis of tubular cells.18 In response to albumin, proximal tubular cells also stimulate interstitial fibroblasts via paracrine release of transforming growth factor beta, either directly or by activating complement or macrophages.18,19
Studies linking albuminuria to kidney disease
Albuminuria has traditionally been associated with diabetes mellitus as a predictor of overt diabetic nephropathy,20,21 although in type 1 diabetes, established albuminuria can spontaneously regress.22
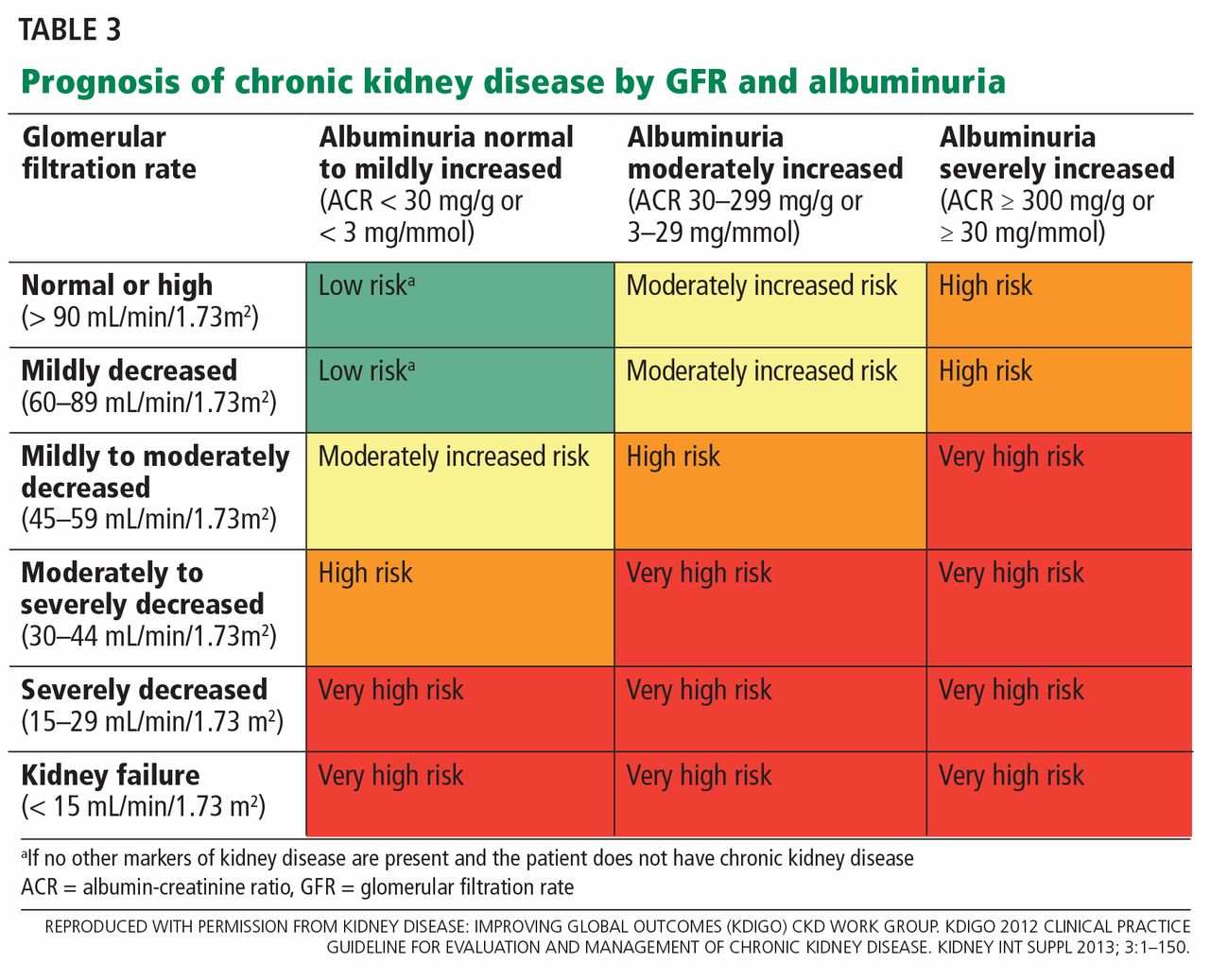
Albuminuria is also a strong predictor of progression in chronic kidney disease.23 In fact, in the last decade, albuminuria has become an independent criterion in the definition of chronic kidney disease; excretion of more than 30 mg of albumin per day, sustained for at least 3 months, qualifies as chronic kidney disease, with independent prognostic implications (Table 3).13
Astor et al,24 in a meta-analysis of 13 studies with more than 21,000 patients with chronic kidney disease, found that the risk of end-stage renal disease was three times higher in those with albuminuria.
Gansevoort et al,23 in a meta-analysis of nine studies with nearly 850,000 participants from the general population, found that the risk of end-stage renal disease increased continuously as albumin excretion increased. They also found that as albuminuria increased, so did the risk of progression of chronic kidney disease and the incidence of acute kidney injury.
Hemmelgarn et al,25 in a large pooled cohort study with more than 1.5 million participants from the general population, showed that increasing albuminuria was associated with a decline in the estimated glomerular filtration rate (GFR) and with progression to end-stage renal disease across all strata of baseline renal function. For example, in persons with an estimated GFR of 60 mL/min/1.73 m2
- 1 per 1,000 person-years for those with no proteinuria
- 2.8 per 1,000 person-years for those with mild proteinuria (trace or 1+ by dipstick or ACR 30–300 mg/g)
- 13.4 per 1,000 person-years for those with heavy proteinuria (2+ or ACR > 300 mg/g).
Rates of progression to end-stage renal disease were:
- 0.03 per 1,000 person-years with no proteinuria
- 0.05 per 1,000 person-years with mild proteinuria
- 1 per 1,000 person-years with heavy proteinuria.25
CARDIOVASCULAR CONSEQUENCES OF ALBUMINURIA
The exact pathophysiologic link between albuminuria and cardiovascular disease is unknown, but several mechanisms have been proposed.
One is that generalized endothelial dysfunction causes both albuminuria and cardiovascular disease.26 Endothelium-derived nitric oxide has vasodilator, antiplatelet, antiproliferative, antiadhesive, permeability-decreasing, and anti-inflammatory properties. Impaired endothelial synthesis of nitric oxide has been independently associated with both microalbuminuria and diabetes.27
Low levels of heparan sulfate (which has antithrombogenic effects and decreases vessel permeability) in the glycocalyx lining vessel walls may also account for albuminuria and for the other cardiovascular effects.28–30 These changes may be the consequence of chronic low-grade inflammation, which precedes the occurrence and progression of both albuminuria and atherothrombotic disease. The resulting abnormalities in the endothelial glycocalyx could lead to increased glomerular permeability to albumin and may also be implicated in the pathogenesis of atherosclerosis.26
In an atherosclerotic aorta and coronary arteries, the endothelial dysfunction may cause increased leakage of cholesterol and glycated end-products into the myocardium, resulting in increasing wall stiffness and left ventricular mass. A similar atherosclerotic process may account for coronary artery microthrombi, resulting in subendocardial ischemia that could lead to systolic and diastolic heart dysfunction.31
Studies linking albuminuria to heart disease
There is convincing evidence that albuminuria is associated with cardiovascular disease. An ACR between 30 and 300 mg/g was independently associated with myocardial infarction and ischemia.32 People with albuminuria have more than twice the risk of severe coronary artery disease, and albuminuria is also associated with increased intimal thickening in the carotid arteries.33,34 An ACR in the same range has been associated with increased incidence and progression of coronary artery calcification.35 Higher brachial-ankle pulse-wave velocity has also been demonstrated with albuminuria in a dose-dependent fashion.36,37
An ACR of 30 to 300 mg/g has been linked to left ventricular hypertrophy independently of other risk factors,38 and functionally with diastolic dysfunction and abnormal midwall shortening.39 In a study of a subgroup of patients with diabetes from a population-based cohort of Native American patients (the Strong Heart Study),39 the prevalence of diastolic dysfunction was:
- 16% in those with no albuminuria
- 26% in those with an ACR of 30 to 300 mg/g
- 31% in those with an ACR greater than 300 mg/g.
The association persisted even after controlling for age, sex, hypertension, and other covariates.
Those pathologic associations have been directly linked to clinical outcomes. For patients with heart failure (New York Heart Association class II–IV), a study found that albuminuria (an ACR > 30 mg/g) conferred a 41% higher risk of admission for heart failure, and an ACR greater than 300 mg/g was associated with an 88% higher risk.40
In an analysis of a prospective cohort from the general population with albuminuria and a low prevalence of renal dysfunction (the Prevention of Renal and Vascular Endstage Disease study),41 albuminuria was associated with a modest increase in the incidence of the composite end point of myocardial infarction, stroke, ischemic heart disease, revascularization procedures, and all-cause mortality per doubling of the urine albumin excretion (hazard ratio 1.08, range 1.04 –1.12).
The relationship to cardiovascular outcomes extends below traditional lower-limit thresholds of albuminuria (corresponding to an ACR > 30 mg/g). A subgroup of patients from the Framingham Offspring Study without prevalent cardiovascular disease, hypertension, diabetes, or kidney disease, and thus with a low to intermediate probability of cardiovascular events, were found to have thresholds for albuminuria as low as 5.3 mg/g in men and 10.8 mg/g in women to discriminate between incident coronary artery disease, heart failure, cerebrovascular disease, other peripheral vascular disease, or death.42
In a meta-analysis including more than 1 million patients in the general population, increasing albuminuria was associated with an increase in deaths from all causes in a continuous manner, with no threshold effect.43 In patients with an ACR of 30 mg/g, the hazard ratio for death was 1.63, increasing to 2.22 for those with more than 300 mg/g compared with those with no albuminuria. A similar increase in the risk of myocardial infarction, heart failure, stroke, or sudden cardiac death was noted with higher ACR.43
Important prospective cohort studies and meta-analyses related to albuminuria and kidney and cardiovascular disease and death are summarized in the eTable.23,39–50
THE CASE FOR TREATING ALBUMINURIA
Reduced progression of renal disease
Treating patients who have proteinuric chronic kidney disease with an angiotensin-converting enzyme (ACE) inhibitor or an angiotensin receptor blocker (ARB) can reduce the risk of progression of renal failure. However, it is unclear whether this benefit is the result of treating concomitant risk factors independent of the reduction in albuminuria, and there is no consistent treatment effect in improving renal outcomes across studies.
Fink et al,51 in a meta-analysis, found that chronic kidney disease patients with diabetes, hypertension, and macroalbuminuria had a 40% lower risk of progression to end-stage renal disease if they received an ACE inhibitor (relative risk [RR] 0.60, 95% confidence interval [CI] 0.43–0.83). In the same meta-analysis, ARBs also reduced the risk of progression to end-stage renal disease (RR 0.77, 95% CI 0.66–0.90).
Jafar et al,52 in an analysis of pooled patient-level data including only nondiabetic patients on ACE inhibitor therapy (n = 1,860), found that the risk of progression of renal failure, defined as a doubling of serum creatinine or end-stage renal disease, was reduced (RR 0.70, 95% CI 0.55–0.88). Patients with higher levels of albuminuria showed the most benefit, but the effect was not conclusive for albuminuria below 500 mg/day at baseline.
Maione et al,53 in a meta-analysis that included patients with albuminuria who were treated with ACE inhibitors vs placebo (n = 8,231), found a similar reduction in risk of:
- Progression to end-stage renal disease (RR 0.67, 95% CI 0.54–0.84)
- Doubling of serum creatinine (RR 0.62, 95% CI 0.46–0.84)
- Progression of albuminuria (RR 0.49, 95% CI 0.36–0.65)
- Normalization of pathologic albuminuria (as defined by the trialists in the individual studies) (RR 2.99, 95% CI 1.82–4.91).
Similar results were obtained for patients with albuminuria who were treated with ARBs.53
ONTARGET.54 In contrast, in the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial, the combination of an ACE inhibitor and an ARB showed no benefit in reducing the progression of renal failure, and in those patients with chronic kidney disease there was a higher risk of a doubling of serum creatinine or of the development of end-stage renal disease and hyperkalemia.
Also, in a pooled analysis of the ONTARGET and Telmisartan Randomized Assessment Study in ACE Intolerant Subjects With Cardiovascular Disease (TRANSCEND) trials, a 50% reduction in baseline albuminuria was associated with reduced progression of renal failure in those with a baseline ACR less than 10 mg/g.55
Improved cardiovascular outcomes
There is also evidence of better cardiovascular outcomes with treatment of albuminuria. Again, it is uncertain whether this is a result of treating risk factors other than albuminuria with ACE inhibitors or ARBs, and there is no parallel benefit demonstrated across all studies.
LIFE.47,48 In the Losartan Intervention for Endpoint Reduction in Hypertension trial, survival analyses suggested a decrease in risk of cardiovascular adverse events as the degree of proteinuria improved with ARB therapy.
Maione et al,53 in a meta-analysis including 8,231 patients with albuminuria and at least one other risk factor, found a significant reduction in the rate of nonfatal cardiovascular outcomes (angina, myocardial infarction, revascularization, stroke, transient ischemic attack, or heart failure) with ACE inhibitors vs placebo (RR 0.88, CI 0.82–0.94) and also in 3,888 patients treated with ARBs vs placebo (RR 0.77, CI 0.61–0.98). However, the meta-analysis did not show that ACE inhibitor or ARB therapy reduced rate of cardiovascular or all-cause mortality.
Fink et al,51 in their meta-analysis of 18 trials of ACE inhibitors and four trials of ARBs, also found no evidence that ACE inhibitor or ARB therapy reduced cardiovascular mortality rates.38
The ONTARGET trial evaluated the combination of an ACE inhibitor and ARB therapy in patients with diabetes or preexisting peripheral vascular disease. Reductions in the rate of cardiovascular disease or death were not observed, and in those with chronic kidney disease, there was a higher risk of doubling of serum creatinine or development of end-stage renal disease and adverse events of hyperkalemia.56 And although an increase in baseline albuminuria was associated with worse cardiovascular outcomes, its reduction in the ONTARGET and TRANSCEND trials did not demonstrate better outcomes when the baseline ACR was greater than 10 mg/g.55
WHO SHOULD BE TESTED?
The benefit of adding albuminuria to conventional cardiovascular risk stratification such as Framingham risk scoring is not conclusive. However, today’s clinician may view albuminuria as a biomarker for renal and cardiovascular disease, as albuminuria might be a surrogate marker for endothelial dysfunction in the glomerular capillaries or other vital vascular beds.
High-risk populations and chronic kidney disease patients
Nearly all the current guidelines recommend annual screening for albuminuria in patients with diabetes and hypertension (Table 2).7,10–13 Other high-risk populations include people with cardiovascular disease, a family history of end-stage renal disease, and metabolic syndrome. Additionally, chronic kidney disease patients whose estimated GFR defines them as being in stage 3 or higher (ie, GFR < 60 mL/min/1.73m2), regardless of other comorbidities, should be tested for albuminuria, as it is an important risk predictor.
Most experts prefer that albuminuria be measured by urine ACR in a first morning voided sample, though this is not the only option.
Screening the general population
Given that albuminuria has been shown to be such an important prognosticator for patients at high risk and those with chronic kidney disease, the question arises whether screening for albuminuria in the asymptomatic low-risk general population would foster earlier detection and therefore enable earlier intervention and result in improved outcomes. However, a systematic review done for the United States Preventive Services Task Force and for an American College of Physicians clinical practice guideline did not find robust evidence to support this.51
OUR RECOMMENDATIONS
Who should be tested?
- Patients with chronic kidney disease stage 3, 4, or 5 (GFR < 60 mL/min/1.73m2) who are not on dialysis
- Patients who are considered at higher risk of adverse outcomes, such as those with diabetes, hypertension, a family history of end-stage renal disease, or cardiovascular disease. Testing is useful for recognizing increased renal and cardiovascular risk and may lead clinicians to prescribe or titrate a renin-angiotensin system antagonist, a statin, or both, or to modify other cardiovascular risk factors.
- Not recommended: routine screening in the general population who are asymptomatic or are considered at low risk.
Which test should be used?
Based on current evidence and most guidelines, we recommend the urine ACR test as the screening test for people with diabetes and others deemed to be at high risk.
What should be done about albuminuria?
- Controlling blood pressure is important, and though there is debate about the target blood pressure, an individualized plan should be developed with the patient based on age, comorbidities, and goals of care.
- An ACE inhibitor or ARB, if not contraindicated, is recommended for patients with diabetes whose ACR is greater than 30 mg/g and for patients with chronic kidney disease and an ACR greater than 300 mg/g.
- Current evidence does not support the combined use of an ACE inhibitor and an ARB, as proof of benefit is lacking and the risk of adverse events is higher.
- Refer patients with high or unexplained albuminuria to a nephrologist or clinic specializing in chronic kidney disease.
- Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA 2007; 298:2038–2047.
- Hoyer JR, Seiler MW. Pathophysiology of Tamm-Horsfall protein. Kidney Int 1979; 16:279–289.
- Viswanathan G, Upadhyay A. Assessment of proteinuria. Adv Chronic Kidney Dis 2011; 18:243–248.
- Guh JY. Proteinuria versus albuminuria in chronic kidney disease. Nephrology (Carlton) 2010; 15(suppl 2):53–56.
- Lamb EJ, MacKenzie F, Stevens PE. How should proteinuria be detected and measured? Ann Clin Biochem 2009; 46:205–217.
- Saydah SH, Pavkov ME, Zhang C, et al. Albuminuria prevalence in first morning void compared with previous random urine from adults in the National Health and Nutrition Examination Survey, 2009-2010. Clin Chem 2013; 59:675–683.
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39(suppl 1):S1–S266.
- Younes N, Cleary PA, Steffes MW, et al; DCCT/EDIC Research Group. Comparison of urinary albumin-creatinine ratio and albumin excretion rate in the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study. Clin J Am Soc Nephrol 2010; 5:1235–1242.
- Brinkman JW, de Zeeuw D, Duker JJ, et al. Falsely low urinary albumin concentrations after prolonged frozen storage of urine samples. Clin Chem 2005; 51:2181–2183.
- National Collaborating Centre for Chronic Conditions (UK). Chronic Kidney Disease: National Clinical Guideline for Early Identification and Management in Adults in Primary and Secondary Care. London: Royal College of Physicians (UK) 2008.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Chobanian AV, Bakris GL, Black HR, et al; Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- Kidney Disease Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl 2013; 3:1–150.
- Johnson DW. Global proteinuria guidelines: are we nearly there yet? Clin Biochem Rev 2011; 32:89–95.
- Ruggenenti P, Remuzzi G. Time to abandon microalbuminuria? Kidney Int 2006; 70:1214–1222.
- Glassock RJ. Is the presence of microalbuminuria a relevant marker of kidney disease? Curr Hypertens Rep 2010; 12:364–368.
- Zhang A, Huang S. Progress in pathogenesis of proteinuria. Int J Nephrol 2012; 2012:314251.
- Abbate M, Zoja C, Remuzzi G. How does proteinuria cause progressive renal damage? J Am Soc Nephrol 2006; 17:2974–2984.
- Karalliedde J, Viberti G. Proteinuria in diabetes: bystander or pathway to cardiorenal disease? J Am Soc Nephrol 2010; 21:2020–2027.
- Svendsen PA, Oxenbøll B, Christiansen JS. Microalbuminuria in diabetic patients—a longitudinal study. Acta Endocrinol Suppl (Copenh) 1981; 242:53–54.
- Viberti GC, Hill RD, Jarrett RJ, Argyropoulos A, Mahmud U, Keen H. Microalbuminuria as a predictor of clinical nephropathy in insulin-dependent diabetes mellitus. Lancet 1982; 1:1430–1432.
- Perkins BA, Ficociello LH, Silva KH, Finkelstein DM, Warram JH, Krolewski AS. Regression of microalbuminuria in type 1 diabetes. N Engl J Med 2003; 348:2285–2293.
- Gansevoort RT, Matsushita K, van der Velde M, et al; Chronic Kidney Disease Prognosis Consortium. Lower estimated GFR and higher albuminuria are associated with adverse kidney outcomes. A collaborative meta-analysis of general and high-risk population cohorts. Kidney Int 2011; 80:93–104.
- Astor BC, Matsushita K, Gansevoort RT, et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with mortality and end-stage renal disease. A collaborative meta-analysis of kidney disease population cohorts. Kidney Int 2011; 79:1331–1340.
- Hemmelgarn BR, Manns BJ, Lloyd A, et al; Alberta Kidney Disease Network. Relation between kidney function, proteinuria, and adverse outcomes. JAMA 2010; 303:423–429.
- Stehouwer CD, Smulders YM. Microalbuminuria and risk for cardiovascular disease: analysis of potential mechanisms. J Am Soc Nephrol 2006; 17:2106–2111.
- Stehouwer CD, Henry RM, Dekker JM, Nijpels G, Heine RJ, Bouter LM. Microalbuminuria is associated with impaired brachial artery, flow-mediated vasodilation in elderly individuals without and with diabetes: further evidence for a link between microalbuminuria and endothelial dysfunction—the Hoorn Study. Kidney Int Suppl 2004; 92:S42–S44.
- Wasty F, Alavi MZ, Moore S. Distribution of glycosaminoglycans in the intima of human aortas: changes in atherosclerosis and diabetes mellitus. Diabetologia 1993; 36:316–322.
- Ylä-Herttuala S, Sumuvuori H, Karkola K, Möttönen M, Nikkari T. Glycosaminoglycans in normal and atherosclerotic human coronary arteries. Lab Invest 1986; 54:402–407.
- Deckert T, Feldt-Rasmussen B, Borch-Johnsen K, Jensen T, Kofoed-Enevoldsen A. Albuminuria reflects widespread vascular damage. The Steno hypothesis. Diabetologia 1989; 32:219–226.
- van Hoeven KH, Factor SM. A comparison of the pathological spectrum of hypertensive, diabetic, and hypertensive-diabetic heart disease. Circulation 1990; 82:848–855.
- Diercks GF, van Boven AJ, Hillege HL, et al. Microalbuminuria is independently associated with ischaemic electrocardiographic abnormalities in a large non-diabetic population. The PREVEND (Prevention of REnal and Vascular ENdstage Disease) study. Eur Heart J 2000; 21:1922–1927.
- Bigazzi R, Bianchi S, Nenci R, Baldari D, Baldari G, Campese VM. Increased thickness of the carotid artery in patients with essential hypertension and microalbuminuria. J Hum Hypertens 1995; 9:827–833.
- Tuttle KR, Puhlman ME, Cooney SK, Short R. Urinary albumin and insulin as predictors of coronary artery disease: an angiographic study. Am J Kidney Dis 1999; 34:918–925.
- DeFilippis AP, Kramer HJ, Katz R, et al. Association between coronary artery calcification progression and microalbuminuria: the MESA study. JACC Cardiovasc Imaging 2010; 3:595–604.
- Liu CS, Pi-Sunyer FX, Li CI, et al. Albuminuria is strongly associated with arterial stiffness, especially in diabetic or hypertensive subjects—a population-based study (Taichung Community Health Study, TCHS). Atherosclerosis 2010; 211:315–321.
- Upadhyay A, Hwang SJ, Mitchell GF, et al. Arterial stiffness in mild-to-moderate CKD. J Am Soc Nephrol 2009; 20:2044–2053.
- Pontremoli R, Sofia A, Ravera M, et al. Prevalence and clinical correlates of microalbuminuria in essential hypertension: the MAGIC Study. Microalbuminuria: a Genoa Investigation on Complications. Hypertension 1997; 30:1135–1143.
- Liu JE, Robbins DC, Palmieri V, et al. Association of albuminuria with systolic and diastolic left ventricular dysfunction in type 2 diabetes: the Strong Heart Study. J Am Coll Cardiol 2003; 41:2022–2028.
- Jackson CE, Solomon SD, Gerstein HC, et al; CHARM Investigators and Committees. Albuminuria in chronic heart failure: prevalence and prognostic importance. Lancet 2009; 374:543–550.
- Smink PA, Lambers Heerspink HJ, Gansevoort RT, et al. Albuminuria, estimated GFR, traditional risk factors, and incident cardiovascular disease: the PREVEND (Prevention of Renal and Vascular Endstage Disease) study. Am J Kidney Dis 2012; 60:804–811.
- Arnlöv J, Evans JC, Meigs JB, et al. Low-grade albuminuria and incidence of cardiovascular disease events in nonhypertensive and nondiabetic individuals: the Framingham Heart Study. Circulation 2005; 112:969–975.
- Chronic Kidney Disease Prognosis Consortium; Matsushita K, van der Velde M, Astor BC, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet 2010; 375:2073–2081.
- van der Velde M, Matsushita K, Coresh J, et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with all-cause and cardiovascular mortality. A collaborative meta-analysis of high-risk population cohorts. Kidney Int 2011; 79:1341–1352.
- Ruggenenti P, Porrini E, Motterlini N, et al; BENEDICT Study Investigators. Measurable urinary albumin predicts cardiovascular risk among normoalbuminuric patients with type 2 diabetes. J Am Soc Nephrol 2012; 23:1717–1724.
- Hallan S, Astor B, Romundstad S, Aasarød K, Kvenild K, Coresh J. Association of kidney function and albuminuria with cardiovascular mortality in older vs younger individuals: the HUNT II Study. Arch Intern Med 2007; 167:2490–2496.
- Ibsen H, Wachtell K, Olsen MH, et al. Albuminuria and cardiovascular risk in hypertensive patients with left ventricular hypertrophy: the LIFE Study. Kidney Int Suppl 2004; 92:S56–S58.
- Olsen MH, Wachtell K, Bella JN, et al. Albuminuria predicts cardiovascular events independently of left ventricular mass in hypertension: a LIFE substudy. J Hum Hypertens 2004; 18:453–459.
- Klausen K, Borch-Johnsen K, Feldt-Rasmussen B, et al. Very low levels of microalbuminuria are associated with increased risk of coronary heart disease and death independently of renal function, hypertension, and diabetes. Circulation 2004; 110:32–35.
- Gerstein HC, Mann JF, Yi Q, et al; HOPE Study Investigators. Albuminuria and risk of cardiovascular events, death, and heart failure in diabetic and nondiabetic individuals. JAMA 2001; 286:421–426.
- Fink HA, Ishani A, Taylor BC, et al. Screening for, monitoring, and treatment of chronic kidney disease stages 1 to 3: a systematic review for the US Preventive Services Task Force and for an American College of Physicians Clinical Practice Guideline. Ann Intern Med 2012; 156:570–581.
- Jafar TH, Schmid CH, Landa M, et al. Angiotensin-converting enzyme inhibitors and progression of nondiabetic renal disease. A meta-analysis of patient-level data. Ann Intern Med 2001; 135:73–87.
- Maione A, Navaneethan SD, Graziano G, et al. Angiotensin-converting enzyme inhibitors, angiotensin receptor blockers and combined therapy in patients with micro- and macroalbuminuria and other cardiovascular risk factors: a systematic review of randomized controlled trials. Nephrol Dial Transplant 2011; 26:2827–2847.
- Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Schmieder RE, Mann JF, Schumacher H, et al; ONTARGET Investigators. Changes in albuminuria predict mortality and morbidity in patients with vascular disease. J Am Soc Nephrol 2011; 22:1353–1364.
- Tobe SW, Clase CM, Gao P, et al; ONTARGET and TRANSCEND Investigators. Cardiovascular and renal outcomes with telmisartan, ramipril, or both in people at high renal risk: results from the ONTARGET and TRANSCEND studies. Circulation 2011; 123:1098–1107.
“One can obtain considerable information concerning the general health by examining the urine.”
—Hippocrates (460?–355? BCE)
Chronic kidney disease is a notable public health concern because it is an important risk factor for end-stage renal disease, cardiovascular disease, and death. Its prevalence1 exceeds 10% and is considerably higher in high-risk groups, such as those with diabetes or hypertension, which are growing in the United States.
While high levels of total protein in the urine have always been recognized as pathologic, a growing body of evidence links excretion of the protein albumin to adverse cardiovascular outcomes, and most international guidelines now recommend measuring albumin specifically. Albuminuria is a predictor of declining renal function and is independently associated with adverse cardiovascular outcomes. Thus, clinicians need to detect it early, manage it effectively, and reduce concurrent risk factors for cardiovascular disease.
Therefore, this review will focus on albuminuria. However, because the traditional standard for urinary protein measurement was total protein, and because a few guidelines still recommend measuring total protein rather than albumin, we will also briefly discuss total urinary protein.
MOST URINARY PROTEIN IS ALBUMIN
Most of the protein in the urine is albumin filtered from the plasma. Less than half of the rest is derived from the distal renal tubules (uromodulin or Tamm-Horsfall mucoprotein), 2 and urine also contains a small and varying proportion of immunoglobulins, low-molecular-weight proteins, and light chains.
Normal healthy people lose less than 30 mg of albumin in the urine per day. In greater amounts, albumin is the major urinary protein in most kidney diseases. Other proteins in urine can be specific markers of less-common illnesses such as plasma cell dyscrasia, glomerulopathy, and renal tubular disease.
MEASURING PROTEINURIA AND ALBUMINURIA
Albumin is not a homogeneous molecule in urine. It undergoes changes to its molecular configuration in the presence of certain ions, peptides, hormones, and drugs, and as a result of proteolytic fragmentation both in the plasma and in renal tubules.3 Consequently, measuring urinary albumin involves a trade-off between convenience and accuracy.
A 24-hour timed urine sample has long been the gold standard for measuring albuminuria, but the collection is cumbersome and time-consuming, and the test is prone to laboratory error.
Dipstick measurements are more convenient and are better at detecting albumin than other proteins in urine, but they have low sensitivity and high interobserver variation.3–5
The albumin-to-creatinine ratio (ACR). As the quantity of protein in the urine changes with time of day, exertion, stress level, and posture, spot-checking of urine samples is not as good as timed collection. However, a simultaneous measurement of creatinine in a spot urine sample adjusts for protein concentration, which can vary with a person’s hydration status. The ACR so obtained is consistent with the 24-hour timed collection (the gold standard) and is the recommended method of assessing albuminuria.3 An early morning urine sample is favored, as it avoids orthostatic variations and varies less in the same individual.
In a study in the general population comparing the ACR in a random sample and in an early morning sample, only 44% of those who had an ACR of 30 mg/g or higher in the random sample had one this high in the early morning sample.6 However, getting an early morning sample is not always feasible in clinical practice. If you are going to measure albuminuria, the Kidney Disease Outcomes and Quality Initiative7 suggests checking the ACR in a random sample and then, if the test is positive, following up and confirming it within 3 months with an early morning sample.
Also, since creatinine excretion differs with race, diet, and muscle mass, if the 24-hour creatinine excretion is not close to 1 g, the ACR will give an erroneous estimate of the 24-hour excretion rate.3
Table 1 compares the various methods of measuring protein in the urine.3,5,8,9 Of note, methods of measuring albumin and total protein vary considerably in their precision and accuracy, making it impossible to reliably translate values from one to the other.5
National and international guidelines (Table 2)7,10–13 agree that albuminuria should be tested in diabetic patients, as it is a surrogate marker for early diabetic nephropathy.3,13 Most guidelines also recommend measuring albuminuria by a urine ACR test as the preferred measure, even in people without diabetes.
Also, no single cutoff is universally accepted for distinguishing pathologic albuminuria from physiologic albuminuria, nor is there a universally accepted unit of measure.14 Because approximately 1 g of creatinine is lost in the urine per day, the ACR has the convenient property of numerically matching the albumin excretory rate expressed in milligrams per 24 hours. The other commonly used unit is milligrams of albumin per millimole of creatinine; 30 mg/g is roughly equal to 3 mg/mmol.
The term microalbuminuria was traditionally used to refer to albumin excretion of 30 to 299 mg per 24 hours, and macroalbuminuria to 300 mg or more per 24 hours. However, as there is no pathophysiologic basis to these thresholds (see outcomes data below), the current Kidney Disease Improving Global Outcomes (KDIGO) guidelines do not recommend using these terms.13,15
RENAL COMPLICATIONS OF ALBUMINURIA
A failure of the glomerular filtration barrier or of proximal tubular reabsorption accounts for most cases of pathologic albuminuria.16 Processes affecting the glomerular filtration of albumin include endothelial cell dysfunction and abnormalities with the glomerular basement membrane, podocytes, or the slit diaphragms among the podocytic processes.17
Ultrafiltrated albumin has been directly implicated in tubulointerstitial damage and glomerulosclerosis through diverse pathways. In the proximal tubule, albumin up-regulates interleukin 8 (a chemoattractant for lymphocytes and neutrophils), induces synthesis of endothelin 1 (which stimulates renal cell proliferation, extracellular matrix production, and monocyte attraction), and causes apoptosis of tubular cells.18 In response to albumin, proximal tubular cells also stimulate interstitial fibroblasts via paracrine release of transforming growth factor beta, either directly or by activating complement or macrophages.18,19
Studies linking albuminuria to kidney disease
Albuminuria has traditionally been associated with diabetes mellitus as a predictor of overt diabetic nephropathy,20,21 although in type 1 diabetes, established albuminuria can spontaneously regress.22
Albuminuria is also a strong predictor of progression in chronic kidney disease.23 In fact, in the last decade, albuminuria has become an independent criterion in the definition of chronic kidney disease; excretion of more than 30 mg of albumin per day, sustained for at least 3 months, qualifies as chronic kidney disease, with independent prognostic implications (Table 3).13
Astor et al,24 in a meta-analysis of 13 studies with more than 21,000 patients with chronic kidney disease, found that the risk of end-stage renal disease was three times higher in those with albuminuria.
Gansevoort et al,23 in a meta-analysis of nine studies with nearly 850,000 participants from the general population, found that the risk of end-stage renal disease increased continuously as albumin excretion increased. They also found that as albuminuria increased, so did the risk of progression of chronic kidney disease and the incidence of acute kidney injury.
Hemmelgarn et al,25 in a large pooled cohort study with more than 1.5 million participants from the general population, showed that increasing albuminuria was associated with a decline in the estimated glomerular filtration rate (GFR) and with progression to end-stage renal disease across all strata of baseline renal function. For example, in persons with an estimated GFR of 60 mL/min/1.73 m2
- 1 per 1,000 person-years for those with no proteinuria
- 2.8 per 1,000 person-years for those with mild proteinuria (trace or 1+ by dipstick or ACR 30–300 mg/g)
- 13.4 per 1,000 person-years for those with heavy proteinuria (2+ or ACR > 300 mg/g).
Rates of progression to end-stage renal disease were:
- 0.03 per 1,000 person-years with no proteinuria
- 0.05 per 1,000 person-years with mild proteinuria
- 1 per 1,000 person-years with heavy proteinuria.25
CARDIOVASCULAR CONSEQUENCES OF ALBUMINURIA
The exact pathophysiologic link between albuminuria and cardiovascular disease is unknown, but several mechanisms have been proposed.
One is that generalized endothelial dysfunction causes both albuminuria and cardiovascular disease.26 Endothelium-derived nitric oxide has vasodilator, antiplatelet, antiproliferative, antiadhesive, permeability-decreasing, and anti-inflammatory properties. Impaired endothelial synthesis of nitric oxide has been independently associated with both microalbuminuria and diabetes.27
Low levels of heparan sulfate (which has antithrombogenic effects and decreases vessel permeability) in the glycocalyx lining vessel walls may also account for albuminuria and for the other cardiovascular effects.28–30 These changes may be the consequence of chronic low-grade inflammation, which precedes the occurrence and progression of both albuminuria and atherothrombotic disease. The resulting abnormalities in the endothelial glycocalyx could lead to increased glomerular permeability to albumin and may also be implicated in the pathogenesis of atherosclerosis.26
In an atherosclerotic aorta and coronary arteries, the endothelial dysfunction may cause increased leakage of cholesterol and glycated end-products into the myocardium, resulting in increasing wall stiffness and left ventricular mass. A similar atherosclerotic process may account for coronary artery microthrombi, resulting in subendocardial ischemia that could lead to systolic and diastolic heart dysfunction.31
Studies linking albuminuria to heart disease
There is convincing evidence that albuminuria is associated with cardiovascular disease. An ACR between 30 and 300 mg/g was independently associated with myocardial infarction and ischemia.32 People with albuminuria have more than twice the risk of severe coronary artery disease, and albuminuria is also associated with increased intimal thickening in the carotid arteries.33,34 An ACR in the same range has been associated with increased incidence and progression of coronary artery calcification.35 Higher brachial-ankle pulse-wave velocity has also been demonstrated with albuminuria in a dose-dependent fashion.36,37
An ACR of 30 to 300 mg/g has been linked to left ventricular hypertrophy independently of other risk factors,38 and functionally with diastolic dysfunction and abnormal midwall shortening.39 In a study of a subgroup of patients with diabetes from a population-based cohort of Native American patients (the Strong Heart Study),39 the prevalence of diastolic dysfunction was:
- 16% in those with no albuminuria
- 26% in those with an ACR of 30 to 300 mg/g
- 31% in those with an ACR greater than 300 mg/g.
The association persisted even after controlling for age, sex, hypertension, and other covariates.
Those pathologic associations have been directly linked to clinical outcomes. For patients with heart failure (New York Heart Association class II–IV), a study found that albuminuria (an ACR > 30 mg/g) conferred a 41% higher risk of admission for heart failure, and an ACR greater than 300 mg/g was associated with an 88% higher risk.40
In an analysis of a prospective cohort from the general population with albuminuria and a low prevalence of renal dysfunction (the Prevention of Renal and Vascular Endstage Disease study),41 albuminuria was associated with a modest increase in the incidence of the composite end point of myocardial infarction, stroke, ischemic heart disease, revascularization procedures, and all-cause mortality per doubling of the urine albumin excretion (hazard ratio 1.08, range 1.04 –1.12).
The relationship to cardiovascular outcomes extends below traditional lower-limit thresholds of albuminuria (corresponding to an ACR > 30 mg/g). A subgroup of patients from the Framingham Offspring Study without prevalent cardiovascular disease, hypertension, diabetes, or kidney disease, and thus with a low to intermediate probability of cardiovascular events, were found to have thresholds for albuminuria as low as 5.3 mg/g in men and 10.8 mg/g in women to discriminate between incident coronary artery disease, heart failure, cerebrovascular disease, other peripheral vascular disease, or death.42
In a meta-analysis including more than 1 million patients in the general population, increasing albuminuria was associated with an increase in deaths from all causes in a continuous manner, with no threshold effect.43 In patients with an ACR of 30 mg/g, the hazard ratio for death was 1.63, increasing to 2.22 for those with more than 300 mg/g compared with those with no albuminuria. A similar increase in the risk of myocardial infarction, heart failure, stroke, or sudden cardiac death was noted with higher ACR.43
Important prospective cohort studies and meta-analyses related to albuminuria and kidney and cardiovascular disease and death are summarized in the eTable.23,39–50
THE CASE FOR TREATING ALBUMINURIA
Reduced progression of renal disease
Treating patients who have proteinuric chronic kidney disease with an angiotensin-converting enzyme (ACE) inhibitor or an angiotensin receptor blocker (ARB) can reduce the risk of progression of renal failure. However, it is unclear whether this benefit is the result of treating concomitant risk factors independent of the reduction in albuminuria, and there is no consistent treatment effect in improving renal outcomes across studies.
Fink et al,51 in a meta-analysis, found that chronic kidney disease patients with diabetes, hypertension, and macroalbuminuria had a 40% lower risk of progression to end-stage renal disease if they received an ACE inhibitor (relative risk [RR] 0.60, 95% confidence interval [CI] 0.43–0.83). In the same meta-analysis, ARBs also reduced the risk of progression to end-stage renal disease (RR 0.77, 95% CI 0.66–0.90).
Jafar et al,52 in an analysis of pooled patient-level data including only nondiabetic patients on ACE inhibitor therapy (n = 1,860), found that the risk of progression of renal failure, defined as a doubling of serum creatinine or end-stage renal disease, was reduced (RR 0.70, 95% CI 0.55–0.88). Patients with higher levels of albuminuria showed the most benefit, but the effect was not conclusive for albuminuria below 500 mg/day at baseline.
Maione et al,53 in a meta-analysis that included patients with albuminuria who were treated with ACE inhibitors vs placebo (n = 8,231), found a similar reduction in risk of:
- Progression to end-stage renal disease (RR 0.67, 95% CI 0.54–0.84)
- Doubling of serum creatinine (RR 0.62, 95% CI 0.46–0.84)
- Progression of albuminuria (RR 0.49, 95% CI 0.36–0.65)
- Normalization of pathologic albuminuria (as defined by the trialists in the individual studies) (RR 2.99, 95% CI 1.82–4.91).
Similar results were obtained for patients with albuminuria who were treated with ARBs.53
ONTARGET.54 In contrast, in the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial, the combination of an ACE inhibitor and an ARB showed no benefit in reducing the progression of renal failure, and in those patients with chronic kidney disease there was a higher risk of a doubling of serum creatinine or of the development of end-stage renal disease and hyperkalemia.
Also, in a pooled analysis of the ONTARGET and Telmisartan Randomized Assessment Study in ACE Intolerant Subjects With Cardiovascular Disease (TRANSCEND) trials, a 50% reduction in baseline albuminuria was associated with reduced progression of renal failure in those with a baseline ACR less than 10 mg/g.55
Improved cardiovascular outcomes
There is also evidence of better cardiovascular outcomes with treatment of albuminuria. Again, it is uncertain whether this is a result of treating risk factors other than albuminuria with ACE inhibitors or ARBs, and there is no parallel benefit demonstrated across all studies.
LIFE.47,48 In the Losartan Intervention for Endpoint Reduction in Hypertension trial, survival analyses suggested a decrease in risk of cardiovascular adverse events as the degree of proteinuria improved with ARB therapy.
Maione et al,53 in a meta-analysis including 8,231 patients with albuminuria and at least one other risk factor, found a significant reduction in the rate of nonfatal cardiovascular outcomes (angina, myocardial infarction, revascularization, stroke, transient ischemic attack, or heart failure) with ACE inhibitors vs placebo (RR 0.88, CI 0.82–0.94) and also in 3,888 patients treated with ARBs vs placebo (RR 0.77, CI 0.61–0.98). However, the meta-analysis did not show that ACE inhibitor or ARB therapy reduced rate of cardiovascular or all-cause mortality.
Fink et al,51 in their meta-analysis of 18 trials of ACE inhibitors and four trials of ARBs, also found no evidence that ACE inhibitor or ARB therapy reduced cardiovascular mortality rates.38
The ONTARGET trial evaluated the combination of an ACE inhibitor and ARB therapy in patients with diabetes or preexisting peripheral vascular disease. Reductions in the rate of cardiovascular disease or death were not observed, and in those with chronic kidney disease, there was a higher risk of doubling of serum creatinine or development of end-stage renal disease and adverse events of hyperkalemia.56 And although an increase in baseline albuminuria was associated with worse cardiovascular outcomes, its reduction in the ONTARGET and TRANSCEND trials did not demonstrate better outcomes when the baseline ACR was greater than 10 mg/g.55
WHO SHOULD BE TESTED?
The benefit of adding albuminuria to conventional cardiovascular risk stratification such as Framingham risk scoring is not conclusive. However, today’s clinician may view albuminuria as a biomarker for renal and cardiovascular disease, as albuminuria might be a surrogate marker for endothelial dysfunction in the glomerular capillaries or other vital vascular beds.
High-risk populations and chronic kidney disease patients
Nearly all the current guidelines recommend annual screening for albuminuria in patients with diabetes and hypertension (Table 2).7,10–13 Other high-risk populations include people with cardiovascular disease, a family history of end-stage renal disease, and metabolic syndrome. Additionally, chronic kidney disease patients whose estimated GFR defines them as being in stage 3 or higher (ie, GFR < 60 mL/min/1.73m2), regardless of other comorbidities, should be tested for albuminuria, as it is an important risk predictor.
Most experts prefer that albuminuria be measured by urine ACR in a first morning voided sample, though this is not the only option.
Screening the general population
Given that albuminuria has been shown to be such an important prognosticator for patients at high risk and those with chronic kidney disease, the question arises whether screening for albuminuria in the asymptomatic low-risk general population would foster earlier detection and therefore enable earlier intervention and result in improved outcomes. However, a systematic review done for the United States Preventive Services Task Force and for an American College of Physicians clinical practice guideline did not find robust evidence to support this.51
OUR RECOMMENDATIONS
Who should be tested?
- Patients with chronic kidney disease stage 3, 4, or 5 (GFR < 60 mL/min/1.73m2) who are not on dialysis
- Patients who are considered at higher risk of adverse outcomes, such as those with diabetes, hypertension, a family history of end-stage renal disease, or cardiovascular disease. Testing is useful for recognizing increased renal and cardiovascular risk and may lead clinicians to prescribe or titrate a renin-angiotensin system antagonist, a statin, or both, or to modify other cardiovascular risk factors.
- Not recommended: routine screening in the general population who are asymptomatic or are considered at low risk.
Which test should be used?
Based on current evidence and most guidelines, we recommend the urine ACR test as the screening test for people with diabetes and others deemed to be at high risk.
What should be done about albuminuria?
- Controlling blood pressure is important, and though there is debate about the target blood pressure, an individualized plan should be developed with the patient based on age, comorbidities, and goals of care.
- An ACE inhibitor or ARB, if not contraindicated, is recommended for patients with diabetes whose ACR is greater than 30 mg/g and for patients with chronic kidney disease and an ACR greater than 300 mg/g.
- Current evidence does not support the combined use of an ACE inhibitor and an ARB, as proof of benefit is lacking and the risk of adverse events is higher.
- Refer patients with high or unexplained albuminuria to a nephrologist or clinic specializing in chronic kidney disease.
“One can obtain considerable information concerning the general health by examining the urine.”
—Hippocrates (460?–355? BCE)
Chronic kidney disease is a notable public health concern because it is an important risk factor for end-stage renal disease, cardiovascular disease, and death. Its prevalence1 exceeds 10% and is considerably higher in high-risk groups, such as those with diabetes or hypertension, which are growing in the United States.
While high levels of total protein in the urine have always been recognized as pathologic, a growing body of evidence links excretion of the protein albumin to adverse cardiovascular outcomes, and most international guidelines now recommend measuring albumin specifically. Albuminuria is a predictor of declining renal function and is independently associated with adverse cardiovascular outcomes. Thus, clinicians need to detect it early, manage it effectively, and reduce concurrent risk factors for cardiovascular disease.
Therefore, this review will focus on albuminuria. However, because the traditional standard for urinary protein measurement was total protein, and because a few guidelines still recommend measuring total protein rather than albumin, we will also briefly discuss total urinary protein.
MOST URINARY PROTEIN IS ALBUMIN
Most of the protein in the urine is albumin filtered from the plasma. Less than half of the rest is derived from the distal renal tubules (uromodulin or Tamm-Horsfall mucoprotein), 2 and urine also contains a small and varying proportion of immunoglobulins, low-molecular-weight proteins, and light chains.
Normal healthy people lose less than 30 mg of albumin in the urine per day. In greater amounts, albumin is the major urinary protein in most kidney diseases. Other proteins in urine can be specific markers of less-common illnesses such as plasma cell dyscrasia, glomerulopathy, and renal tubular disease.
MEASURING PROTEINURIA AND ALBUMINURIA
Albumin is not a homogeneous molecule in urine. It undergoes changes to its molecular configuration in the presence of certain ions, peptides, hormones, and drugs, and as a result of proteolytic fragmentation both in the plasma and in renal tubules.3 Consequently, measuring urinary albumin involves a trade-off between convenience and accuracy.
A 24-hour timed urine sample has long been the gold standard for measuring albuminuria, but the collection is cumbersome and time-consuming, and the test is prone to laboratory error.
Dipstick measurements are more convenient and are better at detecting albumin than other proteins in urine, but they have low sensitivity and high interobserver variation.3–5
The albumin-to-creatinine ratio (ACR). As the quantity of protein in the urine changes with time of day, exertion, stress level, and posture, spot-checking of urine samples is not as good as timed collection. However, a simultaneous measurement of creatinine in a spot urine sample adjusts for protein concentration, which can vary with a person’s hydration status. The ACR so obtained is consistent with the 24-hour timed collection (the gold standard) and is the recommended method of assessing albuminuria.3 An early morning urine sample is favored, as it avoids orthostatic variations and varies less in the same individual.
In a study in the general population comparing the ACR in a random sample and in an early morning sample, only 44% of those who had an ACR of 30 mg/g or higher in the random sample had one this high in the early morning sample.6 However, getting an early morning sample is not always feasible in clinical practice. If you are going to measure albuminuria, the Kidney Disease Outcomes and Quality Initiative7 suggests checking the ACR in a random sample and then, if the test is positive, following up and confirming it within 3 months with an early morning sample.
Also, since creatinine excretion differs with race, diet, and muscle mass, if the 24-hour creatinine excretion is not close to 1 g, the ACR will give an erroneous estimate of the 24-hour excretion rate.3
Table 1 compares the various methods of measuring protein in the urine.3,5,8,9 Of note, methods of measuring albumin and total protein vary considerably in their precision and accuracy, making it impossible to reliably translate values from one to the other.5
National and international guidelines (Table 2)7,10–13 agree that albuminuria should be tested in diabetic patients, as it is a surrogate marker for early diabetic nephropathy.3,13 Most guidelines also recommend measuring albuminuria by a urine ACR test as the preferred measure, even in people without diabetes.
Also, no single cutoff is universally accepted for distinguishing pathologic albuminuria from physiologic albuminuria, nor is there a universally accepted unit of measure.14 Because approximately 1 g of creatinine is lost in the urine per day, the ACR has the convenient property of numerically matching the albumin excretory rate expressed in milligrams per 24 hours. The other commonly used unit is milligrams of albumin per millimole of creatinine; 30 mg/g is roughly equal to 3 mg/mmol.
The term microalbuminuria was traditionally used to refer to albumin excretion of 30 to 299 mg per 24 hours, and macroalbuminuria to 300 mg or more per 24 hours. However, as there is no pathophysiologic basis to these thresholds (see outcomes data below), the current Kidney Disease Improving Global Outcomes (KDIGO) guidelines do not recommend using these terms.13,15
RENAL COMPLICATIONS OF ALBUMINURIA
A failure of the glomerular filtration barrier or of proximal tubular reabsorption accounts for most cases of pathologic albuminuria.16 Processes affecting the glomerular filtration of albumin include endothelial cell dysfunction and abnormalities with the glomerular basement membrane, podocytes, or the slit diaphragms among the podocytic processes.17
Ultrafiltrated albumin has been directly implicated in tubulointerstitial damage and glomerulosclerosis through diverse pathways. In the proximal tubule, albumin up-regulates interleukin 8 (a chemoattractant for lymphocytes and neutrophils), induces synthesis of endothelin 1 (which stimulates renal cell proliferation, extracellular matrix production, and monocyte attraction), and causes apoptosis of tubular cells.18 In response to albumin, proximal tubular cells also stimulate interstitial fibroblasts via paracrine release of transforming growth factor beta, either directly or by activating complement or macrophages.18,19
Studies linking albuminuria to kidney disease
Albuminuria has traditionally been associated with diabetes mellitus as a predictor of overt diabetic nephropathy,20,21 although in type 1 diabetes, established albuminuria can spontaneously regress.22
Albuminuria is also a strong predictor of progression in chronic kidney disease.23 In fact, in the last decade, albuminuria has become an independent criterion in the definition of chronic kidney disease; excretion of more than 30 mg of albumin per day, sustained for at least 3 months, qualifies as chronic kidney disease, with independent prognostic implications (Table 3).13
Astor et al,24 in a meta-analysis of 13 studies with more than 21,000 patients with chronic kidney disease, found that the risk of end-stage renal disease was three times higher in those with albuminuria.
Gansevoort et al,23 in a meta-analysis of nine studies with nearly 850,000 participants from the general population, found that the risk of end-stage renal disease increased continuously as albumin excretion increased. They also found that as albuminuria increased, so did the risk of progression of chronic kidney disease and the incidence of acute kidney injury.
Hemmelgarn et al,25 in a large pooled cohort study with more than 1.5 million participants from the general population, showed that increasing albuminuria was associated with a decline in the estimated glomerular filtration rate (GFR) and with progression to end-stage renal disease across all strata of baseline renal function. For example, in persons with an estimated GFR of 60 mL/min/1.73 m2
- 1 per 1,000 person-years for those with no proteinuria
- 2.8 per 1,000 person-years for those with mild proteinuria (trace or 1+ by dipstick or ACR 30–300 mg/g)
- 13.4 per 1,000 person-years for those with heavy proteinuria (2+ or ACR > 300 mg/g).
Rates of progression to end-stage renal disease were:
- 0.03 per 1,000 person-years with no proteinuria
- 0.05 per 1,000 person-years with mild proteinuria
- 1 per 1,000 person-years with heavy proteinuria.25
CARDIOVASCULAR CONSEQUENCES OF ALBUMINURIA
The exact pathophysiologic link between albuminuria and cardiovascular disease is unknown, but several mechanisms have been proposed.
One is that generalized endothelial dysfunction causes both albuminuria and cardiovascular disease.26 Endothelium-derived nitric oxide has vasodilator, antiplatelet, antiproliferative, antiadhesive, permeability-decreasing, and anti-inflammatory properties. Impaired endothelial synthesis of nitric oxide has been independently associated with both microalbuminuria and diabetes.27
Low levels of heparan sulfate (which has antithrombogenic effects and decreases vessel permeability) in the glycocalyx lining vessel walls may also account for albuminuria and for the other cardiovascular effects.28–30 These changes may be the consequence of chronic low-grade inflammation, which precedes the occurrence and progression of both albuminuria and atherothrombotic disease. The resulting abnormalities in the endothelial glycocalyx could lead to increased glomerular permeability to albumin and may also be implicated in the pathogenesis of atherosclerosis.26
In an atherosclerotic aorta and coronary arteries, the endothelial dysfunction may cause increased leakage of cholesterol and glycated end-products into the myocardium, resulting in increasing wall stiffness and left ventricular mass. A similar atherosclerotic process may account for coronary artery microthrombi, resulting in subendocardial ischemia that could lead to systolic and diastolic heart dysfunction.31
Studies linking albuminuria to heart disease
There is convincing evidence that albuminuria is associated with cardiovascular disease. An ACR between 30 and 300 mg/g was independently associated with myocardial infarction and ischemia.32 People with albuminuria have more than twice the risk of severe coronary artery disease, and albuminuria is also associated with increased intimal thickening in the carotid arteries.33,34 An ACR in the same range has been associated with increased incidence and progression of coronary artery calcification.35 Higher brachial-ankle pulse-wave velocity has also been demonstrated with albuminuria in a dose-dependent fashion.36,37
An ACR of 30 to 300 mg/g has been linked to left ventricular hypertrophy independently of other risk factors,38 and functionally with diastolic dysfunction and abnormal midwall shortening.39 In a study of a subgroup of patients with diabetes from a population-based cohort of Native American patients (the Strong Heart Study),39 the prevalence of diastolic dysfunction was:
- 16% in those with no albuminuria
- 26% in those with an ACR of 30 to 300 mg/g
- 31% in those with an ACR greater than 300 mg/g.
The association persisted even after controlling for age, sex, hypertension, and other covariates.
Those pathologic associations have been directly linked to clinical outcomes. For patients with heart failure (New York Heart Association class II–IV), a study found that albuminuria (an ACR > 30 mg/g) conferred a 41% higher risk of admission for heart failure, and an ACR greater than 300 mg/g was associated with an 88% higher risk.40
In an analysis of a prospective cohort from the general population with albuminuria and a low prevalence of renal dysfunction (the Prevention of Renal and Vascular Endstage Disease study),41 albuminuria was associated with a modest increase in the incidence of the composite end point of myocardial infarction, stroke, ischemic heart disease, revascularization procedures, and all-cause mortality per doubling of the urine albumin excretion (hazard ratio 1.08, range 1.04 –1.12).
The relationship to cardiovascular outcomes extends below traditional lower-limit thresholds of albuminuria (corresponding to an ACR > 30 mg/g). A subgroup of patients from the Framingham Offspring Study without prevalent cardiovascular disease, hypertension, diabetes, or kidney disease, and thus with a low to intermediate probability of cardiovascular events, were found to have thresholds for albuminuria as low as 5.3 mg/g in men and 10.8 mg/g in women to discriminate between incident coronary artery disease, heart failure, cerebrovascular disease, other peripheral vascular disease, or death.42
In a meta-analysis including more than 1 million patients in the general population, increasing albuminuria was associated with an increase in deaths from all causes in a continuous manner, with no threshold effect.43 In patients with an ACR of 30 mg/g, the hazard ratio for death was 1.63, increasing to 2.22 for those with more than 300 mg/g compared with those with no albuminuria. A similar increase in the risk of myocardial infarction, heart failure, stroke, or sudden cardiac death was noted with higher ACR.43
Important prospective cohort studies and meta-analyses related to albuminuria and kidney and cardiovascular disease and death are summarized in the eTable.23,39–50
THE CASE FOR TREATING ALBUMINURIA
Reduced progression of renal disease
Treating patients who have proteinuric chronic kidney disease with an angiotensin-converting enzyme (ACE) inhibitor or an angiotensin receptor blocker (ARB) can reduce the risk of progression of renal failure. However, it is unclear whether this benefit is the result of treating concomitant risk factors independent of the reduction in albuminuria, and there is no consistent treatment effect in improving renal outcomes across studies.
Fink et al,51 in a meta-analysis, found that chronic kidney disease patients with diabetes, hypertension, and macroalbuminuria had a 40% lower risk of progression to end-stage renal disease if they received an ACE inhibitor (relative risk [RR] 0.60, 95% confidence interval [CI] 0.43–0.83). In the same meta-analysis, ARBs also reduced the risk of progression to end-stage renal disease (RR 0.77, 95% CI 0.66–0.90).
Jafar et al,52 in an analysis of pooled patient-level data including only nondiabetic patients on ACE inhibitor therapy (n = 1,860), found that the risk of progression of renal failure, defined as a doubling of serum creatinine or end-stage renal disease, was reduced (RR 0.70, 95% CI 0.55–0.88). Patients with higher levels of albuminuria showed the most benefit, but the effect was not conclusive for albuminuria below 500 mg/day at baseline.
Maione et al,53 in a meta-analysis that included patients with albuminuria who were treated with ACE inhibitors vs placebo (n = 8,231), found a similar reduction in risk of:
- Progression to end-stage renal disease (RR 0.67, 95% CI 0.54–0.84)
- Doubling of serum creatinine (RR 0.62, 95% CI 0.46–0.84)
- Progression of albuminuria (RR 0.49, 95% CI 0.36–0.65)
- Normalization of pathologic albuminuria (as defined by the trialists in the individual studies) (RR 2.99, 95% CI 1.82–4.91).
Similar results were obtained for patients with albuminuria who were treated with ARBs.53
ONTARGET.54 In contrast, in the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial, the combination of an ACE inhibitor and an ARB showed no benefit in reducing the progression of renal failure, and in those patients with chronic kidney disease there was a higher risk of a doubling of serum creatinine or of the development of end-stage renal disease and hyperkalemia.
Also, in a pooled analysis of the ONTARGET and Telmisartan Randomized Assessment Study in ACE Intolerant Subjects With Cardiovascular Disease (TRANSCEND) trials, a 50% reduction in baseline albuminuria was associated with reduced progression of renal failure in those with a baseline ACR less than 10 mg/g.55
Improved cardiovascular outcomes
There is also evidence of better cardiovascular outcomes with treatment of albuminuria. Again, it is uncertain whether this is a result of treating risk factors other than albuminuria with ACE inhibitors or ARBs, and there is no parallel benefit demonstrated across all studies.
LIFE.47,48 In the Losartan Intervention for Endpoint Reduction in Hypertension trial, survival analyses suggested a decrease in risk of cardiovascular adverse events as the degree of proteinuria improved with ARB therapy.
Maione et al,53 in a meta-analysis including 8,231 patients with albuminuria and at least one other risk factor, found a significant reduction in the rate of nonfatal cardiovascular outcomes (angina, myocardial infarction, revascularization, stroke, transient ischemic attack, or heart failure) with ACE inhibitors vs placebo (RR 0.88, CI 0.82–0.94) and also in 3,888 patients treated with ARBs vs placebo (RR 0.77, CI 0.61–0.98). However, the meta-analysis did not show that ACE inhibitor or ARB therapy reduced rate of cardiovascular or all-cause mortality.
Fink et al,51 in their meta-analysis of 18 trials of ACE inhibitors and four trials of ARBs, also found no evidence that ACE inhibitor or ARB therapy reduced cardiovascular mortality rates.38
The ONTARGET trial evaluated the combination of an ACE inhibitor and ARB therapy in patients with diabetes or preexisting peripheral vascular disease. Reductions in the rate of cardiovascular disease or death were not observed, and in those with chronic kidney disease, there was a higher risk of doubling of serum creatinine or development of end-stage renal disease and adverse events of hyperkalemia.56 And although an increase in baseline albuminuria was associated with worse cardiovascular outcomes, its reduction in the ONTARGET and TRANSCEND trials did not demonstrate better outcomes when the baseline ACR was greater than 10 mg/g.55
WHO SHOULD BE TESTED?
The benefit of adding albuminuria to conventional cardiovascular risk stratification such as Framingham risk scoring is not conclusive. However, today’s clinician may view albuminuria as a biomarker for renal and cardiovascular disease, as albuminuria might be a surrogate marker for endothelial dysfunction in the glomerular capillaries or other vital vascular beds.
High-risk populations and chronic kidney disease patients
Nearly all the current guidelines recommend annual screening for albuminuria in patients with diabetes and hypertension (Table 2).7,10–13 Other high-risk populations include people with cardiovascular disease, a family history of end-stage renal disease, and metabolic syndrome. Additionally, chronic kidney disease patients whose estimated GFR defines them as being in stage 3 or higher (ie, GFR < 60 mL/min/1.73m2), regardless of other comorbidities, should be tested for albuminuria, as it is an important risk predictor.
Most experts prefer that albuminuria be measured by urine ACR in a first morning voided sample, though this is not the only option.
Screening the general population
Given that albuminuria has been shown to be such an important prognosticator for patients at high risk and those with chronic kidney disease, the question arises whether screening for albuminuria in the asymptomatic low-risk general population would foster earlier detection and therefore enable earlier intervention and result in improved outcomes. However, a systematic review done for the United States Preventive Services Task Force and for an American College of Physicians clinical practice guideline did not find robust evidence to support this.51
OUR RECOMMENDATIONS
Who should be tested?
- Patients with chronic kidney disease stage 3, 4, or 5 (GFR < 60 mL/min/1.73m2) who are not on dialysis
- Patients who are considered at higher risk of adverse outcomes, such as those with diabetes, hypertension, a family history of end-stage renal disease, or cardiovascular disease. Testing is useful for recognizing increased renal and cardiovascular risk and may lead clinicians to prescribe or titrate a renin-angiotensin system antagonist, a statin, or both, or to modify other cardiovascular risk factors.
- Not recommended: routine screening in the general population who are asymptomatic or are considered at low risk.
Which test should be used?
Based on current evidence and most guidelines, we recommend the urine ACR test as the screening test for people with diabetes and others deemed to be at high risk.
What should be done about albuminuria?
- Controlling blood pressure is important, and though there is debate about the target blood pressure, an individualized plan should be developed with the patient based on age, comorbidities, and goals of care.
- An ACE inhibitor or ARB, if not contraindicated, is recommended for patients with diabetes whose ACR is greater than 30 mg/g and for patients with chronic kidney disease and an ACR greater than 300 mg/g.
- Current evidence does not support the combined use of an ACE inhibitor and an ARB, as proof of benefit is lacking and the risk of adverse events is higher.
- Refer patients with high or unexplained albuminuria to a nephrologist or clinic specializing in chronic kidney disease.
- Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA 2007; 298:2038–2047.
- Hoyer JR, Seiler MW. Pathophysiology of Tamm-Horsfall protein. Kidney Int 1979; 16:279–289.
- Viswanathan G, Upadhyay A. Assessment of proteinuria. Adv Chronic Kidney Dis 2011; 18:243–248.
- Guh JY. Proteinuria versus albuminuria in chronic kidney disease. Nephrology (Carlton) 2010; 15(suppl 2):53–56.
- Lamb EJ, MacKenzie F, Stevens PE. How should proteinuria be detected and measured? Ann Clin Biochem 2009; 46:205–217.
- Saydah SH, Pavkov ME, Zhang C, et al. Albuminuria prevalence in first morning void compared with previous random urine from adults in the National Health and Nutrition Examination Survey, 2009-2010. Clin Chem 2013; 59:675–683.
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39(suppl 1):S1–S266.
- Younes N, Cleary PA, Steffes MW, et al; DCCT/EDIC Research Group. Comparison of urinary albumin-creatinine ratio and albumin excretion rate in the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study. Clin J Am Soc Nephrol 2010; 5:1235–1242.
- Brinkman JW, de Zeeuw D, Duker JJ, et al. Falsely low urinary albumin concentrations after prolonged frozen storage of urine samples. Clin Chem 2005; 51:2181–2183.
- National Collaborating Centre for Chronic Conditions (UK). Chronic Kidney Disease: National Clinical Guideline for Early Identification and Management in Adults in Primary and Secondary Care. London: Royal College of Physicians (UK) 2008.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Chobanian AV, Bakris GL, Black HR, et al; Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- Kidney Disease Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl 2013; 3:1–150.
- Johnson DW. Global proteinuria guidelines: are we nearly there yet? Clin Biochem Rev 2011; 32:89–95.
- Ruggenenti P, Remuzzi G. Time to abandon microalbuminuria? Kidney Int 2006; 70:1214–1222.
- Glassock RJ. Is the presence of microalbuminuria a relevant marker of kidney disease? Curr Hypertens Rep 2010; 12:364–368.
- Zhang A, Huang S. Progress in pathogenesis of proteinuria. Int J Nephrol 2012; 2012:314251.
- Abbate M, Zoja C, Remuzzi G. How does proteinuria cause progressive renal damage? J Am Soc Nephrol 2006; 17:2974–2984.
- Karalliedde J, Viberti G. Proteinuria in diabetes: bystander or pathway to cardiorenal disease? J Am Soc Nephrol 2010; 21:2020–2027.
- Svendsen PA, Oxenbøll B, Christiansen JS. Microalbuminuria in diabetic patients—a longitudinal study. Acta Endocrinol Suppl (Copenh) 1981; 242:53–54.
- Viberti GC, Hill RD, Jarrett RJ, Argyropoulos A, Mahmud U, Keen H. Microalbuminuria as a predictor of clinical nephropathy in insulin-dependent diabetes mellitus. Lancet 1982; 1:1430–1432.
- Perkins BA, Ficociello LH, Silva KH, Finkelstein DM, Warram JH, Krolewski AS. Regression of microalbuminuria in type 1 diabetes. N Engl J Med 2003; 348:2285–2293.
- Gansevoort RT, Matsushita K, van der Velde M, et al; Chronic Kidney Disease Prognosis Consortium. Lower estimated GFR and higher albuminuria are associated with adverse kidney outcomes. A collaborative meta-analysis of general and high-risk population cohorts. Kidney Int 2011; 80:93–104.
- Astor BC, Matsushita K, Gansevoort RT, et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with mortality and end-stage renal disease. A collaborative meta-analysis of kidney disease population cohorts. Kidney Int 2011; 79:1331–1340.
- Hemmelgarn BR, Manns BJ, Lloyd A, et al; Alberta Kidney Disease Network. Relation between kidney function, proteinuria, and adverse outcomes. JAMA 2010; 303:423–429.
- Stehouwer CD, Smulders YM. Microalbuminuria and risk for cardiovascular disease: analysis of potential mechanisms. J Am Soc Nephrol 2006; 17:2106–2111.
- Stehouwer CD, Henry RM, Dekker JM, Nijpels G, Heine RJ, Bouter LM. Microalbuminuria is associated with impaired brachial artery, flow-mediated vasodilation in elderly individuals without and with diabetes: further evidence for a link between microalbuminuria and endothelial dysfunction—the Hoorn Study. Kidney Int Suppl 2004; 92:S42–S44.
- Wasty F, Alavi MZ, Moore S. Distribution of glycosaminoglycans in the intima of human aortas: changes in atherosclerosis and diabetes mellitus. Diabetologia 1993; 36:316–322.
- Ylä-Herttuala S, Sumuvuori H, Karkola K, Möttönen M, Nikkari T. Glycosaminoglycans in normal and atherosclerotic human coronary arteries. Lab Invest 1986; 54:402–407.
- Deckert T, Feldt-Rasmussen B, Borch-Johnsen K, Jensen T, Kofoed-Enevoldsen A. Albuminuria reflects widespread vascular damage. The Steno hypothesis. Diabetologia 1989; 32:219–226.
- van Hoeven KH, Factor SM. A comparison of the pathological spectrum of hypertensive, diabetic, and hypertensive-diabetic heart disease. Circulation 1990; 82:848–855.
- Diercks GF, van Boven AJ, Hillege HL, et al. Microalbuminuria is independently associated with ischaemic electrocardiographic abnormalities in a large non-diabetic population. The PREVEND (Prevention of REnal and Vascular ENdstage Disease) study. Eur Heart J 2000; 21:1922–1927.
- Bigazzi R, Bianchi S, Nenci R, Baldari D, Baldari G, Campese VM. Increased thickness of the carotid artery in patients with essential hypertension and microalbuminuria. J Hum Hypertens 1995; 9:827–833.
- Tuttle KR, Puhlman ME, Cooney SK, Short R. Urinary albumin and insulin as predictors of coronary artery disease: an angiographic study. Am J Kidney Dis 1999; 34:918–925.
- DeFilippis AP, Kramer HJ, Katz R, et al. Association between coronary artery calcification progression and microalbuminuria: the MESA study. JACC Cardiovasc Imaging 2010; 3:595–604.
- Liu CS, Pi-Sunyer FX, Li CI, et al. Albuminuria is strongly associated with arterial stiffness, especially in diabetic or hypertensive subjects—a population-based study (Taichung Community Health Study, TCHS). Atherosclerosis 2010; 211:315–321.
- Upadhyay A, Hwang SJ, Mitchell GF, et al. Arterial stiffness in mild-to-moderate CKD. J Am Soc Nephrol 2009; 20:2044–2053.
- Pontremoli R, Sofia A, Ravera M, et al. Prevalence and clinical correlates of microalbuminuria in essential hypertension: the MAGIC Study. Microalbuminuria: a Genoa Investigation on Complications. Hypertension 1997; 30:1135–1143.
- Liu JE, Robbins DC, Palmieri V, et al. Association of albuminuria with systolic and diastolic left ventricular dysfunction in type 2 diabetes: the Strong Heart Study. J Am Coll Cardiol 2003; 41:2022–2028.
- Jackson CE, Solomon SD, Gerstein HC, et al; CHARM Investigators and Committees. Albuminuria in chronic heart failure: prevalence and prognostic importance. Lancet 2009; 374:543–550.
- Smink PA, Lambers Heerspink HJ, Gansevoort RT, et al. Albuminuria, estimated GFR, traditional risk factors, and incident cardiovascular disease: the PREVEND (Prevention of Renal and Vascular Endstage Disease) study. Am J Kidney Dis 2012; 60:804–811.
- Arnlöv J, Evans JC, Meigs JB, et al. Low-grade albuminuria and incidence of cardiovascular disease events in nonhypertensive and nondiabetic individuals: the Framingham Heart Study. Circulation 2005; 112:969–975.
- Chronic Kidney Disease Prognosis Consortium; Matsushita K, van der Velde M, Astor BC, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet 2010; 375:2073–2081.
- van der Velde M, Matsushita K, Coresh J, et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with all-cause and cardiovascular mortality. A collaborative meta-analysis of high-risk population cohorts. Kidney Int 2011; 79:1341–1352.
- Ruggenenti P, Porrini E, Motterlini N, et al; BENEDICT Study Investigators. Measurable urinary albumin predicts cardiovascular risk among normoalbuminuric patients with type 2 diabetes. J Am Soc Nephrol 2012; 23:1717–1724.
- Hallan S, Astor B, Romundstad S, Aasarød K, Kvenild K, Coresh J. Association of kidney function and albuminuria with cardiovascular mortality in older vs younger individuals: the HUNT II Study. Arch Intern Med 2007; 167:2490–2496.
- Ibsen H, Wachtell K, Olsen MH, et al. Albuminuria and cardiovascular risk in hypertensive patients with left ventricular hypertrophy: the LIFE Study. Kidney Int Suppl 2004; 92:S56–S58.
- Olsen MH, Wachtell K, Bella JN, et al. Albuminuria predicts cardiovascular events independently of left ventricular mass in hypertension: a LIFE substudy. J Hum Hypertens 2004; 18:453–459.
- Klausen K, Borch-Johnsen K, Feldt-Rasmussen B, et al. Very low levels of microalbuminuria are associated with increased risk of coronary heart disease and death independently of renal function, hypertension, and diabetes. Circulation 2004; 110:32–35.
- Gerstein HC, Mann JF, Yi Q, et al; HOPE Study Investigators. Albuminuria and risk of cardiovascular events, death, and heart failure in diabetic and nondiabetic individuals. JAMA 2001; 286:421–426.
- Fink HA, Ishani A, Taylor BC, et al. Screening for, monitoring, and treatment of chronic kidney disease stages 1 to 3: a systematic review for the US Preventive Services Task Force and for an American College of Physicians Clinical Practice Guideline. Ann Intern Med 2012; 156:570–581.
- Jafar TH, Schmid CH, Landa M, et al. Angiotensin-converting enzyme inhibitors and progression of nondiabetic renal disease. A meta-analysis of patient-level data. Ann Intern Med 2001; 135:73–87.
- Maione A, Navaneethan SD, Graziano G, et al. Angiotensin-converting enzyme inhibitors, angiotensin receptor blockers and combined therapy in patients with micro- and macroalbuminuria and other cardiovascular risk factors: a systematic review of randomized controlled trials. Nephrol Dial Transplant 2011; 26:2827–2847.
- Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Schmieder RE, Mann JF, Schumacher H, et al; ONTARGET Investigators. Changes in albuminuria predict mortality and morbidity in patients with vascular disease. J Am Soc Nephrol 2011; 22:1353–1364.
- Tobe SW, Clase CM, Gao P, et al; ONTARGET and TRANSCEND Investigators. Cardiovascular and renal outcomes with telmisartan, ramipril, or both in people at high renal risk: results from the ONTARGET and TRANSCEND studies. Circulation 2011; 123:1098–1107.
- Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA 2007; 298:2038–2047.
- Hoyer JR, Seiler MW. Pathophysiology of Tamm-Horsfall protein. Kidney Int 1979; 16:279–289.
- Viswanathan G, Upadhyay A. Assessment of proteinuria. Adv Chronic Kidney Dis 2011; 18:243–248.
- Guh JY. Proteinuria versus albuminuria in chronic kidney disease. Nephrology (Carlton) 2010; 15(suppl 2):53–56.
- Lamb EJ, MacKenzie F, Stevens PE. How should proteinuria be detected and measured? Ann Clin Biochem 2009; 46:205–217.
- Saydah SH, Pavkov ME, Zhang C, et al. Albuminuria prevalence in first morning void compared with previous random urine from adults in the National Health and Nutrition Examination Survey, 2009-2010. Clin Chem 2013; 59:675–683.
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39(suppl 1):S1–S266.
- Younes N, Cleary PA, Steffes MW, et al; DCCT/EDIC Research Group. Comparison of urinary albumin-creatinine ratio and albumin excretion rate in the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study. Clin J Am Soc Nephrol 2010; 5:1235–1242.
- Brinkman JW, de Zeeuw D, Duker JJ, et al. Falsely low urinary albumin concentrations after prolonged frozen storage of urine samples. Clin Chem 2005; 51:2181–2183.
- National Collaborating Centre for Chronic Conditions (UK). Chronic Kidney Disease: National Clinical Guideline for Early Identification and Management in Adults in Primary and Secondary Care. London: Royal College of Physicians (UK) 2008.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Chobanian AV, Bakris GL, Black HR, et al; Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- Kidney Disease Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl 2013; 3:1–150.
- Johnson DW. Global proteinuria guidelines: are we nearly there yet? Clin Biochem Rev 2011; 32:89–95.
- Ruggenenti P, Remuzzi G. Time to abandon microalbuminuria? Kidney Int 2006; 70:1214–1222.
- Glassock RJ. Is the presence of microalbuminuria a relevant marker of kidney disease? Curr Hypertens Rep 2010; 12:364–368.
- Zhang A, Huang S. Progress in pathogenesis of proteinuria. Int J Nephrol 2012; 2012:314251.
- Abbate M, Zoja C, Remuzzi G. How does proteinuria cause progressive renal damage? J Am Soc Nephrol 2006; 17:2974–2984.
- Karalliedde J, Viberti G. Proteinuria in diabetes: bystander or pathway to cardiorenal disease? J Am Soc Nephrol 2010; 21:2020–2027.
- Svendsen PA, Oxenbøll B, Christiansen JS. Microalbuminuria in diabetic patients—a longitudinal study. Acta Endocrinol Suppl (Copenh) 1981; 242:53–54.
- Viberti GC, Hill RD, Jarrett RJ, Argyropoulos A, Mahmud U, Keen H. Microalbuminuria as a predictor of clinical nephropathy in insulin-dependent diabetes mellitus. Lancet 1982; 1:1430–1432.
- Perkins BA, Ficociello LH, Silva KH, Finkelstein DM, Warram JH, Krolewski AS. Regression of microalbuminuria in type 1 diabetes. N Engl J Med 2003; 348:2285–2293.
- Gansevoort RT, Matsushita K, van der Velde M, et al; Chronic Kidney Disease Prognosis Consortium. Lower estimated GFR and higher albuminuria are associated with adverse kidney outcomes. A collaborative meta-analysis of general and high-risk population cohorts. Kidney Int 2011; 80:93–104.
- Astor BC, Matsushita K, Gansevoort RT, et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with mortality and end-stage renal disease. A collaborative meta-analysis of kidney disease population cohorts. Kidney Int 2011; 79:1331–1340.
- Hemmelgarn BR, Manns BJ, Lloyd A, et al; Alberta Kidney Disease Network. Relation between kidney function, proteinuria, and adverse outcomes. JAMA 2010; 303:423–429.
- Stehouwer CD, Smulders YM. Microalbuminuria and risk for cardiovascular disease: analysis of potential mechanisms. J Am Soc Nephrol 2006; 17:2106–2111.
- Stehouwer CD, Henry RM, Dekker JM, Nijpels G, Heine RJ, Bouter LM. Microalbuminuria is associated with impaired brachial artery, flow-mediated vasodilation in elderly individuals without and with diabetes: further evidence for a link between microalbuminuria and endothelial dysfunction—the Hoorn Study. Kidney Int Suppl 2004; 92:S42–S44.
- Wasty F, Alavi MZ, Moore S. Distribution of glycosaminoglycans in the intima of human aortas: changes in atherosclerosis and diabetes mellitus. Diabetologia 1993; 36:316–322.
- Ylä-Herttuala S, Sumuvuori H, Karkola K, Möttönen M, Nikkari T. Glycosaminoglycans in normal and atherosclerotic human coronary arteries. Lab Invest 1986; 54:402–407.
- Deckert T, Feldt-Rasmussen B, Borch-Johnsen K, Jensen T, Kofoed-Enevoldsen A. Albuminuria reflects widespread vascular damage. The Steno hypothesis. Diabetologia 1989; 32:219–226.
- van Hoeven KH, Factor SM. A comparison of the pathological spectrum of hypertensive, diabetic, and hypertensive-diabetic heart disease. Circulation 1990; 82:848–855.
- Diercks GF, van Boven AJ, Hillege HL, et al. Microalbuminuria is independently associated with ischaemic electrocardiographic abnormalities in a large non-diabetic population. The PREVEND (Prevention of REnal and Vascular ENdstage Disease) study. Eur Heart J 2000; 21:1922–1927.
- Bigazzi R, Bianchi S, Nenci R, Baldari D, Baldari G, Campese VM. Increased thickness of the carotid artery in patients with essential hypertension and microalbuminuria. J Hum Hypertens 1995; 9:827–833.
- Tuttle KR, Puhlman ME, Cooney SK, Short R. Urinary albumin and insulin as predictors of coronary artery disease: an angiographic study. Am J Kidney Dis 1999; 34:918–925.
- DeFilippis AP, Kramer HJ, Katz R, et al. Association between coronary artery calcification progression and microalbuminuria: the MESA study. JACC Cardiovasc Imaging 2010; 3:595–604.
- Liu CS, Pi-Sunyer FX, Li CI, et al. Albuminuria is strongly associated with arterial stiffness, especially in diabetic or hypertensive subjects—a population-based study (Taichung Community Health Study, TCHS). Atherosclerosis 2010; 211:315–321.
- Upadhyay A, Hwang SJ, Mitchell GF, et al. Arterial stiffness in mild-to-moderate CKD. J Am Soc Nephrol 2009; 20:2044–2053.
- Pontremoli R, Sofia A, Ravera M, et al. Prevalence and clinical correlates of microalbuminuria in essential hypertension: the MAGIC Study. Microalbuminuria: a Genoa Investigation on Complications. Hypertension 1997; 30:1135–1143.
- Liu JE, Robbins DC, Palmieri V, et al. Association of albuminuria with systolic and diastolic left ventricular dysfunction in type 2 diabetes: the Strong Heart Study. J Am Coll Cardiol 2003; 41:2022–2028.
- Jackson CE, Solomon SD, Gerstein HC, et al; CHARM Investigators and Committees. Albuminuria in chronic heart failure: prevalence and prognostic importance. Lancet 2009; 374:543–550.
- Smink PA, Lambers Heerspink HJ, Gansevoort RT, et al. Albuminuria, estimated GFR, traditional risk factors, and incident cardiovascular disease: the PREVEND (Prevention of Renal and Vascular Endstage Disease) study. Am J Kidney Dis 2012; 60:804–811.
- Arnlöv J, Evans JC, Meigs JB, et al. Low-grade albuminuria and incidence of cardiovascular disease events in nonhypertensive and nondiabetic individuals: the Framingham Heart Study. Circulation 2005; 112:969–975.
- Chronic Kidney Disease Prognosis Consortium; Matsushita K, van der Velde M, Astor BC, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet 2010; 375:2073–2081.
- van der Velde M, Matsushita K, Coresh J, et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with all-cause and cardiovascular mortality. A collaborative meta-analysis of high-risk population cohorts. Kidney Int 2011; 79:1341–1352.
- Ruggenenti P, Porrini E, Motterlini N, et al; BENEDICT Study Investigators. Measurable urinary albumin predicts cardiovascular risk among normoalbuminuric patients with type 2 diabetes. J Am Soc Nephrol 2012; 23:1717–1724.
- Hallan S, Astor B, Romundstad S, Aasarød K, Kvenild K, Coresh J. Association of kidney function and albuminuria with cardiovascular mortality in older vs younger individuals: the HUNT II Study. Arch Intern Med 2007; 167:2490–2496.
- Ibsen H, Wachtell K, Olsen MH, et al. Albuminuria and cardiovascular risk in hypertensive patients with left ventricular hypertrophy: the LIFE Study. Kidney Int Suppl 2004; 92:S56–S58.
- Olsen MH, Wachtell K, Bella JN, et al. Albuminuria predicts cardiovascular events independently of left ventricular mass in hypertension: a LIFE substudy. J Hum Hypertens 2004; 18:453–459.
- Klausen K, Borch-Johnsen K, Feldt-Rasmussen B, et al. Very low levels of microalbuminuria are associated with increased risk of coronary heart disease and death independently of renal function, hypertension, and diabetes. Circulation 2004; 110:32–35.
- Gerstein HC, Mann JF, Yi Q, et al; HOPE Study Investigators. Albuminuria and risk of cardiovascular events, death, and heart failure in diabetic and nondiabetic individuals. JAMA 2001; 286:421–426.
- Fink HA, Ishani A, Taylor BC, et al. Screening for, monitoring, and treatment of chronic kidney disease stages 1 to 3: a systematic review for the US Preventive Services Task Force and for an American College of Physicians Clinical Practice Guideline. Ann Intern Med 2012; 156:570–581.
- Jafar TH, Schmid CH, Landa M, et al. Angiotensin-converting enzyme inhibitors and progression of nondiabetic renal disease. A meta-analysis of patient-level data. Ann Intern Med 2001; 135:73–87.
- Maione A, Navaneethan SD, Graziano G, et al. Angiotensin-converting enzyme inhibitors, angiotensin receptor blockers and combined therapy in patients with micro- and macroalbuminuria and other cardiovascular risk factors: a systematic review of randomized controlled trials. Nephrol Dial Transplant 2011; 26:2827–2847.
- Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Schmieder RE, Mann JF, Schumacher H, et al; ONTARGET Investigators. Changes in albuminuria predict mortality and morbidity in patients with vascular disease. J Am Soc Nephrol 2011; 22:1353–1364.
- Tobe SW, Clase CM, Gao P, et al; ONTARGET and TRANSCEND Investigators. Cardiovascular and renal outcomes with telmisartan, ramipril, or both in people at high renal risk: results from the ONTARGET and TRANSCEND studies. Circulation 2011; 123:1098–1107.
KEY POINTS
- Albuminuria is best measured by the albumin-to-creatinine ratio.
- In several studies, albuminuria has been independently associated with a higher risk of death, cardiovascular events, heart failure, stroke, and progression of chronic kidney disease.
- Despite strong evidence linking albuminuria to adverse outcomes, evidence is limited in favor of routinely screening for it in the general population.
- Evaluating and managing albuminuria require understanding the limits of its clinical measures, controlling other risk factors for progression of renal disease, managing it medically, and referring to a specialist in certain situations.