User login
Clinically Impressive Tophaceous Gout With Significant Bony Destruction
Gout is an in inflammatory condition that is generally characterized by red, hot, swollen, and painful joints. The disease is often associated with increased serum uric acid levels; which are considered elevated when they are > 6 mg/dL in women and > 7 mg/dL in men. When gout affects joints, the subchondral bone may be involved, leading to destructive, painful changes. This article presents the case of a patient diagnosed with tophaceous gout of the left second toe with bony erosive changes and calcified nodules noted on magnetic resonance images (MRI).
Case Presentation
A 70-year-old white male presented to the podiatry clinic for a left second-toe mass that was diagnosed as tophaceous gout after being seen by his primary care physician. The patient reported that the mass had slowly grown over the past 10 years. At presentation, he had a 0.2-cm ulcer on the dorsal aspect of the left second-toe mass. The patient stated that the ulcer had recently appeared with some exudate; however, there was no active drainage of material. The patient had a 20-year history of gout that was untreated with dietary modifications or medication. The patient also stated that although the left second-toe mass did not cause any pain on rest, it did cause pain with shoe gear and during ambulation. A community-based podiatrist had recommended amputation of the second toe and as a result the patient was seeking a second opinion at the US Department of Veterans Affairs (VA) Lebanon VA Medical Center (VAMC) in Pennsylvania. The patient had not had acute gouty attacks during the past 10 years.
The patient’s medical history was significant for uncontrolled gout, hyperlipidemia, coronary artery disease with a 4-vessel coronary artery bypass grafting, impaired fasting glucose, prostate cancer that was in remission, alcohol misuse (currently limited to ≤ 2 drinks per night), and 30-year history of cigarette smoking (quit 2 months prior to visit).1,2
At his first visit to the clinic, an examination revealed distinct evidence of bulging of the soft tissues of the second toe of the left foot with a dry sinus tract that was not malodorous (Figure 1). The left second toe was erythematous and edematous. A local increase in skin temperature was present on the second toe of the left foot compared with that of the contralateral foot and other toes. The dorsalis pedis and tibialis posterior pulses were easily palpated, and the capillary return was within normal limits. Palpation of the left second-toe plantar elicited mild tenderness. Crepitation was not present at the left second metatarsophalangeal joint (MPJ) nor at the interphalangeal joint. There was restricted range of motion at the left second MPJ compared with that of the right foot and no motion at the proximal interphalangeal joint. The movement at the left second metatarsophalangeal elicited tenderness. The mass on the left second toe was firm, nonpulsatile, oval-shaped, with a white pigmented consistency that measured 2 cm x 2.5 cm.
There were no deficits present on the neurologic examination, which was noncontributory. There also was no gross evidence of motor weakness. His initial temporal temperature was 98.2° F. The initial laboratory findings were uric acid, 9.5 mg/dL; fasting glucose, 117 g/dL; estimated glomerular filtration rate, 55 mL/min/1.73 m2; erythrocyte sedimentation rate, 6.5 mm/h; and white blood count, 6.6 K/uL.3,4-6
Diagnostic imaging included X-rays of the patient’s feet and a MRI of the left foot. The X-rays showed diffusely osteopenic bones with severe soft tissue swelling surrounding the second proximal interphalangeal joint. Also present was moderate soft tissue swelling at the level of the first metatarsophalangeal joint accompanied by extensive erosions at both of these joints, most pronounced at the second proximal interphalangeal joint. Also, there was narrowing at the first MPJ and the first interphalangeal joint. Erosive changes at the tarsometatarsal articulations and small lucencies within the navicular/midfoot joint were suggestive of additional gouty erosions. A small-to-moderate posterior calcaneal enthesophyte was present as well as a tiny calcaneal enthesophyte (Figure 2).
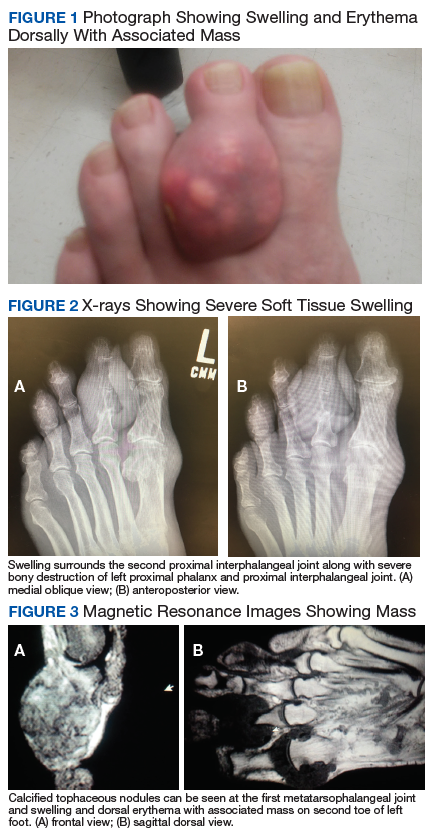
A MRI showed a destructive soft tissue mass, resulting in overhanging edges, with foci of calcifications centered about the proximal interphalangeal joint of the second toe, which is consistent with a calcified tophaceous gout nodule. The widest dimension of the mass measured 3.2 cm. There also was a less prominent calcified tophaceous gout nodule at the first MPJ. There were additional small punched-out lesions involving the bases of the first through fourth metatarsi and at the distal aspect of the first cuneiform in keeping with gouty arthropathy (Figure 3).4,7-10
The initial treatment plan presented to the patient was to amputate the left second toe. But the patient decided against amputation. Treatment guidelines for allopurinol are to titrate in 100-mg increments every 2 weeks until the serum uric acid levels are consistently < 6, tophi resolve, and the patient should be free of gout attacks.11 We initiated uric acid-lowering therapy with allopurinol at 50 mg/d for 7 days, increasing to 100 mg/d for 7 days, then to 200 mg/d for 10 days. The patient’s serum uric acid level was checked at 200 mg/d. Our patient could not tolerate the allopurinol and decided to discontinue treatment. After 1 year he started having severe pain and returned to have the toe amputated. The patient healed uneventfully.
Discussion
Tophaceous gout is characterized by collections of solid urate accompanied by chronic inflammatory and often destructive changes in the surrounding tissue brought on by periods of increased uric acid levels. Due to the patient’s 20-year history of untreated tophaceous gout, we saw the extent of bony and soft tissue destruction that this pathology created. This patient’s uric acid laboratory value of 9.5 mg/dL was well above the normal reference values of 2.6 to 7.2 mg/dL. The X-rays performed suggested that there was not only bony destruction, but also deformity.
The destruction to the surrounding soft tissues noted as advanced nonhealing wounds formed to the area of the tophi. The size of the second digit also was impressive, causing displacement of the other digits. As stated in the literature, tophaceous gout is usually painless as was the case in our patient. It is the combination of the relatively painless nature of this pathology accompanied by no treatment over many years that led to the patient’s level of deformity and tissue destruction.
Conclusion
We describe a common presentation of bone involvement secondary to significant tophaceous gout in the absence osteomyelitis. The goal of treatment was to maintain a functional foot free of major deformity, pain, or associated risk factors that could lead to a more significant surgical procedure, such as a proximal amputation.11 Given the destructive nature of this pathology, it is important to educate the patient, perform regular examinations, and start medications early to control uric acid levels. These measures will improve the patient’s prognosis and avoid severe sequelae.
1. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63(10):3136-3141.
2. Roddy E, Choi HK. Epidemiology of gout. Rheum Dis Clin North Am. 2014;40(2):155-175.
3. Choi H. Epidemiology of crystal arthropathy. Rheum Dis Clin North Am. 2006;32(2):255-273.
4. Nakayama DA, Barthelemy C, Carrera G, Lightfoot RW Jr, Wortmann RL. Tophaceous gout: a clinical and radiographic assessment. Arthritis Rheum. 1984;27(4):468-471.
5. Dalbeth N, Haskard DO. Pathophysiology of crystal-induced arthritis. In: Wortmann RL, Schumacher HR Jr, Becker MA, Ryan LM, eds. Crystal-induced Arthropathies. New York: Taylor & Francis; 2006.
6. Dalbeth N, Pool B, Gamble GD, et al. Cellular characterization of the gouty tophus: a quantitative analysis. Arthritis Rheum. 2010;62(5):1549-1556.
7. Hsu CY, Shih TT, Huang KM, Chen PQ, Sheu JJ, Li YW. Tophaceous gout of the spine: MR imaging features. Clin Radiol. 2002;57(10):919-925.
8. Schumacher HR Jr, Becker MA, Edwards NL, et al. Magnetic resonance imaging in the quantitative assessment of gouty tophi. Int J Clin Pract. 2006;60(4):408-414.
9. McQueen FM, Doyle A, Dalbeth N. Imaging in the crystal arthropathies. Rheum Dis Clin North Am. 2014;40(2):231-249.
10. Choi HK, Al-Arfaj AM, Eftekhari A, et al. Dual energy computed tomography in tophaceous gout. Ann Rheum Dis. 2009;68(10):1609-1612.
11. Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64(10):1431-1446.
Gout is an in inflammatory condition that is generally characterized by red, hot, swollen, and painful joints. The disease is often associated with increased serum uric acid levels; which are considered elevated when they are > 6 mg/dL in women and > 7 mg/dL in men. When gout affects joints, the subchondral bone may be involved, leading to destructive, painful changes. This article presents the case of a patient diagnosed with tophaceous gout of the left second toe with bony erosive changes and calcified nodules noted on magnetic resonance images (MRI).
Case Presentation
A 70-year-old white male presented to the podiatry clinic for a left second-toe mass that was diagnosed as tophaceous gout after being seen by his primary care physician. The patient reported that the mass had slowly grown over the past 10 years. At presentation, he had a 0.2-cm ulcer on the dorsal aspect of the left second-toe mass. The patient stated that the ulcer had recently appeared with some exudate; however, there was no active drainage of material. The patient had a 20-year history of gout that was untreated with dietary modifications or medication. The patient also stated that although the left second-toe mass did not cause any pain on rest, it did cause pain with shoe gear and during ambulation. A community-based podiatrist had recommended amputation of the second toe and as a result the patient was seeking a second opinion at the US Department of Veterans Affairs (VA) Lebanon VA Medical Center (VAMC) in Pennsylvania. The patient had not had acute gouty attacks during the past 10 years.
The patient’s medical history was significant for uncontrolled gout, hyperlipidemia, coronary artery disease with a 4-vessel coronary artery bypass grafting, impaired fasting glucose, prostate cancer that was in remission, alcohol misuse (currently limited to ≤ 2 drinks per night), and 30-year history of cigarette smoking (quit 2 months prior to visit).1,2
At his first visit to the clinic, an examination revealed distinct evidence of bulging of the soft tissues of the second toe of the left foot with a dry sinus tract that was not malodorous (Figure 1). The left second toe was erythematous and edematous. A local increase in skin temperature was present on the second toe of the left foot compared with that of the contralateral foot and other toes. The dorsalis pedis and tibialis posterior pulses were easily palpated, and the capillary return was within normal limits. Palpation of the left second-toe plantar elicited mild tenderness. Crepitation was not present at the left second metatarsophalangeal joint (MPJ) nor at the interphalangeal joint. There was restricted range of motion at the left second MPJ compared with that of the right foot and no motion at the proximal interphalangeal joint. The movement at the left second metatarsophalangeal elicited tenderness. The mass on the left second toe was firm, nonpulsatile, oval-shaped, with a white pigmented consistency that measured 2 cm x 2.5 cm.
There were no deficits present on the neurologic examination, which was noncontributory. There also was no gross evidence of motor weakness. His initial temporal temperature was 98.2° F. The initial laboratory findings were uric acid, 9.5 mg/dL; fasting glucose, 117 g/dL; estimated glomerular filtration rate, 55 mL/min/1.73 m2; erythrocyte sedimentation rate, 6.5 mm/h; and white blood count, 6.6 K/uL.3,4-6
Diagnostic imaging included X-rays of the patient’s feet and a MRI of the left foot. The X-rays showed diffusely osteopenic bones with severe soft tissue swelling surrounding the second proximal interphalangeal joint. Also present was moderate soft tissue swelling at the level of the first metatarsophalangeal joint accompanied by extensive erosions at both of these joints, most pronounced at the second proximal interphalangeal joint. Also, there was narrowing at the first MPJ and the first interphalangeal joint. Erosive changes at the tarsometatarsal articulations and small lucencies within the navicular/midfoot joint were suggestive of additional gouty erosions. A small-to-moderate posterior calcaneal enthesophyte was present as well as a tiny calcaneal enthesophyte (Figure 2).
A MRI showed a destructive soft tissue mass, resulting in overhanging edges, with foci of calcifications centered about the proximal interphalangeal joint of the second toe, which is consistent with a calcified tophaceous gout nodule. The widest dimension of the mass measured 3.2 cm. There also was a less prominent calcified tophaceous gout nodule at the first MPJ. There were additional small punched-out lesions involving the bases of the first through fourth metatarsi and at the distal aspect of the first cuneiform in keeping with gouty arthropathy (Figure 3).4,7-10
The initial treatment plan presented to the patient was to amputate the left second toe. But the patient decided against amputation. Treatment guidelines for allopurinol are to titrate in 100-mg increments every 2 weeks until the serum uric acid levels are consistently < 6, tophi resolve, and the patient should be free of gout attacks.11 We initiated uric acid-lowering therapy with allopurinol at 50 mg/d for 7 days, increasing to 100 mg/d for 7 days, then to 200 mg/d for 10 days. The patient’s serum uric acid level was checked at 200 mg/d. Our patient could not tolerate the allopurinol and decided to discontinue treatment. After 1 year he started having severe pain and returned to have the toe amputated. The patient healed uneventfully.
Discussion
Tophaceous gout is characterized by collections of solid urate accompanied by chronic inflammatory and often destructive changes in the surrounding tissue brought on by periods of increased uric acid levels. Due to the patient’s 20-year history of untreated tophaceous gout, we saw the extent of bony and soft tissue destruction that this pathology created. This patient’s uric acid laboratory value of 9.5 mg/dL was well above the normal reference values of 2.6 to 7.2 mg/dL. The X-rays performed suggested that there was not only bony destruction, but also deformity.
The destruction to the surrounding soft tissues noted as advanced nonhealing wounds formed to the area of the tophi. The size of the second digit also was impressive, causing displacement of the other digits. As stated in the literature, tophaceous gout is usually painless as was the case in our patient. It is the combination of the relatively painless nature of this pathology accompanied by no treatment over many years that led to the patient’s level of deformity and tissue destruction.
Conclusion
We describe a common presentation of bone involvement secondary to significant tophaceous gout in the absence osteomyelitis. The goal of treatment was to maintain a functional foot free of major deformity, pain, or associated risk factors that could lead to a more significant surgical procedure, such as a proximal amputation.11 Given the destructive nature of this pathology, it is important to educate the patient, perform regular examinations, and start medications early to control uric acid levels. These measures will improve the patient’s prognosis and avoid severe sequelae.
Gout is an in inflammatory condition that is generally characterized by red, hot, swollen, and painful joints. The disease is often associated with increased serum uric acid levels; which are considered elevated when they are > 6 mg/dL in women and > 7 mg/dL in men. When gout affects joints, the subchondral bone may be involved, leading to destructive, painful changes. This article presents the case of a patient diagnosed with tophaceous gout of the left second toe with bony erosive changes and calcified nodules noted on magnetic resonance images (MRI).
Case Presentation
A 70-year-old white male presented to the podiatry clinic for a left second-toe mass that was diagnosed as tophaceous gout after being seen by his primary care physician. The patient reported that the mass had slowly grown over the past 10 years. At presentation, he had a 0.2-cm ulcer on the dorsal aspect of the left second-toe mass. The patient stated that the ulcer had recently appeared with some exudate; however, there was no active drainage of material. The patient had a 20-year history of gout that was untreated with dietary modifications or medication. The patient also stated that although the left second-toe mass did not cause any pain on rest, it did cause pain with shoe gear and during ambulation. A community-based podiatrist had recommended amputation of the second toe and as a result the patient was seeking a second opinion at the US Department of Veterans Affairs (VA) Lebanon VA Medical Center (VAMC) in Pennsylvania. The patient had not had acute gouty attacks during the past 10 years.
The patient’s medical history was significant for uncontrolled gout, hyperlipidemia, coronary artery disease with a 4-vessel coronary artery bypass grafting, impaired fasting glucose, prostate cancer that was in remission, alcohol misuse (currently limited to ≤ 2 drinks per night), and 30-year history of cigarette smoking (quit 2 months prior to visit).1,2
At his first visit to the clinic, an examination revealed distinct evidence of bulging of the soft tissues of the second toe of the left foot with a dry sinus tract that was not malodorous (Figure 1). The left second toe was erythematous and edematous. A local increase in skin temperature was present on the second toe of the left foot compared with that of the contralateral foot and other toes. The dorsalis pedis and tibialis posterior pulses were easily palpated, and the capillary return was within normal limits. Palpation of the left second-toe plantar elicited mild tenderness. Crepitation was not present at the left second metatarsophalangeal joint (MPJ) nor at the interphalangeal joint. There was restricted range of motion at the left second MPJ compared with that of the right foot and no motion at the proximal interphalangeal joint. The movement at the left second metatarsophalangeal elicited tenderness. The mass on the left second toe was firm, nonpulsatile, oval-shaped, with a white pigmented consistency that measured 2 cm x 2.5 cm.
There were no deficits present on the neurologic examination, which was noncontributory. There also was no gross evidence of motor weakness. His initial temporal temperature was 98.2° F. The initial laboratory findings were uric acid, 9.5 mg/dL; fasting glucose, 117 g/dL; estimated glomerular filtration rate, 55 mL/min/1.73 m2; erythrocyte sedimentation rate, 6.5 mm/h; and white blood count, 6.6 K/uL.3,4-6
Diagnostic imaging included X-rays of the patient’s feet and a MRI of the left foot. The X-rays showed diffusely osteopenic bones with severe soft tissue swelling surrounding the second proximal interphalangeal joint. Also present was moderate soft tissue swelling at the level of the first metatarsophalangeal joint accompanied by extensive erosions at both of these joints, most pronounced at the second proximal interphalangeal joint. Also, there was narrowing at the first MPJ and the first interphalangeal joint. Erosive changes at the tarsometatarsal articulations and small lucencies within the navicular/midfoot joint were suggestive of additional gouty erosions. A small-to-moderate posterior calcaneal enthesophyte was present as well as a tiny calcaneal enthesophyte (Figure 2).
A MRI showed a destructive soft tissue mass, resulting in overhanging edges, with foci of calcifications centered about the proximal interphalangeal joint of the second toe, which is consistent with a calcified tophaceous gout nodule. The widest dimension of the mass measured 3.2 cm. There also was a less prominent calcified tophaceous gout nodule at the first MPJ. There were additional small punched-out lesions involving the bases of the first through fourth metatarsi and at the distal aspect of the first cuneiform in keeping with gouty arthropathy (Figure 3).4,7-10
The initial treatment plan presented to the patient was to amputate the left second toe. But the patient decided against amputation. Treatment guidelines for allopurinol are to titrate in 100-mg increments every 2 weeks until the serum uric acid levels are consistently < 6, tophi resolve, and the patient should be free of gout attacks.11 We initiated uric acid-lowering therapy with allopurinol at 50 mg/d for 7 days, increasing to 100 mg/d for 7 days, then to 200 mg/d for 10 days. The patient’s serum uric acid level was checked at 200 mg/d. Our patient could not tolerate the allopurinol and decided to discontinue treatment. After 1 year he started having severe pain and returned to have the toe amputated. The patient healed uneventfully.
Discussion
Tophaceous gout is characterized by collections of solid urate accompanied by chronic inflammatory and often destructive changes in the surrounding tissue brought on by periods of increased uric acid levels. Due to the patient’s 20-year history of untreated tophaceous gout, we saw the extent of bony and soft tissue destruction that this pathology created. This patient’s uric acid laboratory value of 9.5 mg/dL was well above the normal reference values of 2.6 to 7.2 mg/dL. The X-rays performed suggested that there was not only bony destruction, but also deformity.
The destruction to the surrounding soft tissues noted as advanced nonhealing wounds formed to the area of the tophi. The size of the second digit also was impressive, causing displacement of the other digits. As stated in the literature, tophaceous gout is usually painless as was the case in our patient. It is the combination of the relatively painless nature of this pathology accompanied by no treatment over many years that led to the patient’s level of deformity and tissue destruction.
Conclusion
We describe a common presentation of bone involvement secondary to significant tophaceous gout in the absence osteomyelitis. The goal of treatment was to maintain a functional foot free of major deformity, pain, or associated risk factors that could lead to a more significant surgical procedure, such as a proximal amputation.11 Given the destructive nature of this pathology, it is important to educate the patient, perform regular examinations, and start medications early to control uric acid levels. These measures will improve the patient’s prognosis and avoid severe sequelae.
1. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63(10):3136-3141.
2. Roddy E, Choi HK. Epidemiology of gout. Rheum Dis Clin North Am. 2014;40(2):155-175.
3. Choi H. Epidemiology of crystal arthropathy. Rheum Dis Clin North Am. 2006;32(2):255-273.
4. Nakayama DA, Barthelemy C, Carrera G, Lightfoot RW Jr, Wortmann RL. Tophaceous gout: a clinical and radiographic assessment. Arthritis Rheum. 1984;27(4):468-471.
5. Dalbeth N, Haskard DO. Pathophysiology of crystal-induced arthritis. In: Wortmann RL, Schumacher HR Jr, Becker MA, Ryan LM, eds. Crystal-induced Arthropathies. New York: Taylor & Francis; 2006.
6. Dalbeth N, Pool B, Gamble GD, et al. Cellular characterization of the gouty tophus: a quantitative analysis. Arthritis Rheum. 2010;62(5):1549-1556.
7. Hsu CY, Shih TT, Huang KM, Chen PQ, Sheu JJ, Li YW. Tophaceous gout of the spine: MR imaging features. Clin Radiol. 2002;57(10):919-925.
8. Schumacher HR Jr, Becker MA, Edwards NL, et al. Magnetic resonance imaging in the quantitative assessment of gouty tophi. Int J Clin Pract. 2006;60(4):408-414.
9. McQueen FM, Doyle A, Dalbeth N. Imaging in the crystal arthropathies. Rheum Dis Clin North Am. 2014;40(2):231-249.
10. Choi HK, Al-Arfaj AM, Eftekhari A, et al. Dual energy computed tomography in tophaceous gout. Ann Rheum Dis. 2009;68(10):1609-1612.
11. Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64(10):1431-1446.
1. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63(10):3136-3141.
2. Roddy E, Choi HK. Epidemiology of gout. Rheum Dis Clin North Am. 2014;40(2):155-175.
3. Choi H. Epidemiology of crystal arthropathy. Rheum Dis Clin North Am. 2006;32(2):255-273.
4. Nakayama DA, Barthelemy C, Carrera G, Lightfoot RW Jr, Wortmann RL. Tophaceous gout: a clinical and radiographic assessment. Arthritis Rheum. 1984;27(4):468-471.
5. Dalbeth N, Haskard DO. Pathophysiology of crystal-induced arthritis. In: Wortmann RL, Schumacher HR Jr, Becker MA, Ryan LM, eds. Crystal-induced Arthropathies. New York: Taylor & Francis; 2006.
6. Dalbeth N, Pool B, Gamble GD, et al. Cellular characterization of the gouty tophus: a quantitative analysis. Arthritis Rheum. 2010;62(5):1549-1556.
7. Hsu CY, Shih TT, Huang KM, Chen PQ, Sheu JJ, Li YW. Tophaceous gout of the spine: MR imaging features. Clin Radiol. 2002;57(10):919-925.
8. Schumacher HR Jr, Becker MA, Edwards NL, et al. Magnetic resonance imaging in the quantitative assessment of gouty tophi. Int J Clin Pract. 2006;60(4):408-414.
9. McQueen FM, Doyle A, Dalbeth N. Imaging in the crystal arthropathies. Rheum Dis Clin North Am. 2014;40(2):231-249.
10. Choi HK, Al-Arfaj AM, Eftekhari A, et al. Dual energy computed tomography in tophaceous gout. Ann Rheum Dis. 2009;68(10):1609-1612.
11. Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64(10):1431-1446.
Partners in Oncology Care: Coordinated Follicular Lymphoma Management (FULL)
Four case examples illustrate the important role of multidisciplinary medical care for the optimal long-term care of patients with follicular lymphoma.
Patients benefit from multidisciplinary care that coordinates management of complex medical problems. Traditionally, multidisciplinary cancer care involves oncology specialty providers in fields that include medical oncology, radiation oncology, and surgical oncology. Multidisciplinary cancer care intends to improve patient outcomes by bringing together different health care providers (HCPs) who are involved in the treatment of patients with cancer. Because new therapies are more effective and allow patients with cancer to live longer, adverse effects (AEs) are more likely to impact patients’ well-being, both while receiving treatment and long after it has completed. Thus, this population may benefit from an expanded approach to multidisciplinary care that includes input from specialty and primary care providers (PCPs), clinical pharmacy specialists (CPS), physical and occupational therapists, and patient navigators and educators.
We present 4 hypothetical cases, based on actual patients, that illustrate opportunities where multidisciplinary care coordination may improve patient experiences. These cases draw on current quality initiatives from the National Cancer Institute Community Cancer Centers Program, which has focused on improving the quality of multidisciplinary cancer care at selected community centers, and the Veterans Health Administration (VHA) patient-aligned care team (PACT) model, which brings together different health professionals to optimize primary care coordination.1,2 In addition, the National Committee for Quality Assurance has introduced an educational initiative to facilitate implementation of an oncologic medical home.3 This initiative stresses increased multidisciplinary communication, patient-centered care delivery, and reduced fragmentation of care for this population. Despite these guidelines and experiences from other medical specialties, models for integrated cancer care have not been implemented in a prospective fashion within the VHA.
In this article, we focus on opportunities to take collaborative care approaches for the treatment of patients with follicular lymphoma (FL): a common, incurable, and often indolent B-cell non-Hodgkin lymphoma.4 FL was selected because these patients may be treated numerous times and long-term sequalae can accumulate throughout their cancer continuum (a series of health events encompassing cancer screening, diagnosis, treatment, survivorship, relapse, and death).5 HCPs in distinct roles can assist patients with cancer in optimizing their health outcomes and overall wellbeing.6
Case Example 1
A 70-year-old male was diagnosed with stage IV FL. Because of his advanced disease, he began therapy with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone). Prednisone was administered at 100 mg daily on the first 5 days of each 21-day cycle. On day 4 of the first treatment cycle, the patient notified his oncologist that he had been very thirsty and his random blood sugar values on 2 different days were 283 mg/dL and 312 mg/dL. A laboratory review revealed his hemoglobin A1c (HbA1c) 7 months prior was 5.6%.
Discussion
The high-dose prednisone component of this and other lymphoma therapy regimens can worsen diabetes mellitus (DM) control and/or worsen prediabetes. Patient characteristics that increase the risk of developing glucocorticoid-induced DM after CHOP chemotherapy include age ≥ 60 years, HbA1c > 6.1%, and body mass index > 30.7 This patient did not have DM prior to the FL therapy initiation, but afterwards he met diagnostic criteria for DM. For completeness, other causes for elevated blood glucose should be ruled out (ie, infection, laboratory error, etc.). An oncologist often will triage acute hyperglycemia, treating immediately with IV fluids and/or insulin. Thereafter, ongoing chronic disease management for DM may be best managed by PCPs, certified DM educators, and registered dieticians.
Several programs involving multidisciplinary DM care, comprised of physicians, advanced practice providers, nurses, certified DM educators, and/or pharmacists have been shown to improve HbA1c, cardiovascular outcomes, and all-cause mortality, while reducing health care costs.8 In addition, patient navigators can assist patients with coordinating visits to disease-state specialists and identifying further educational needs. For example, in 1 program, nonclinical peer navigators were shown to improve the number of appointments attended and reduce HbA1c in a population of patients with DM who were primarily minority, urban, and of low socioeconomic status.9 Thus, integrating DM care shows potential to improve outcomes for patients with lymphoma who develop glucocorticoid
Case Example 2
A 75-year-old male was diagnosed with FL. He was treated initially with bendamustine and rituximab. He required reinitiation of therapy 20 months later when he developed lymphadenopathy, fatigue, and night sweats and began treatment with oral idelalisib, a second-line therapy. Later, the patient presented to his PCP for a routine visit, and on medication reconciliation review, the patient reported regular use of trimethoprim-sulfamethoxazole.
Discussion
Upon consultation with the CPS and the patient’s oncologist, the PCP confirmed trimethoprim-sulfamethoxazole should be continued during therapy and for about 6 months following completion of therapy. Trimethoprim-sulfamethoxazole is used for prophylaxis against Pneumocystis jirovecii (formerly Pneumocystis carinii). While use of prophylactic therapy is not necessary for all patients with FL, idelalisib impairs the function of circulating lymphoid B-cells and thus has been associated with an increased risk of serious infection.10 A CPS can provide insight that maximizes medication adherence and efficacy while minimizing food-drug, drug-drug interactions, and AEs. CPS have been shown to: improve adherence to oral therapies, increase prospective monitoring required for safe therapy dose selection, and document assessment of chemotherapy-related AEs.11,12 Thus, multidisciplinary, integrated care is an important component of providing quality oncology care.
Case Example 3
A 60-year-old female presented to her PCP with a 2-week history of shortness of breath and leg swelling. She was treated for FL 4 years previously with 6 cycles of R-CHOP. She reported no chest pain and did not have a prior history of hypertension, DM, or heart disease. On physical exam, she had elevated jugular venous pressure to jaw at 45°, bilateral pulmonary rales, and 2+ pitting pretibial edema. Laboratory tests that included complete blood count, basic chemistries, and thyroid stimulating hormone were unremarkable, though brain natriuretic peptide (BNP) was elevated at 425 pg/mL.
As this patient’s laboratory results and physical examination suggested new-onset congestive heart failure, the PCP obtained an echocardiogram, which demonstrated an ejection fraction of 35% and global hypokinesis. Because the patient was symptomatic, she was admitted to the hospital to begin guideline-directed medical therapy (GDMT) including IV diuresis.
Discussion
Given the absence of significant risk factors and prior history of coronary artery disease, the most probable cause for this patient’s cardiomyopathy is doxorubicin. Doxorubicin is an anthracycline chemotherapy that can cause nonischemic, dilated cardiomyopathy, particularly when cumulative doses > 400 mg/m2 are administered, or when combined with chest radiation.13 This patient benefited from GDMT for reduced ejection-fraction heart failure (HFrEF). Studies have demonstrated positive outcomes when HFrEF patients are cared for by a multidisciplinary team who focus of volume management as well as uptitration of therapies to target doses.14
Case Example 4
An 80-year-old female was diagnosed with stage III FL but did not require immediate therapy. After developing discomfort due to enlarging lymphadenopathy, she initiated therapy with rituximab, cyclophosphamide, vincristine, and prednisone (R-CVP). She presented to her oncologist for consideration of her fifth cycle of R-CVP and reported a burning sensation on the soles of her feet and numbness in her fingertips and toes. On examination, her pulses were intact and there were no signs of infection, reduced blood flow, or edema. The patient demonstrated decreased sensation on monofilament testing. She had no history of DM and a recent HbA1c test was 4.9% An evaluation for other causes of neuropathy, such as hypothyroidism and vitamin B12 deficiency was negative. Thus, vincristine therapy was identified as the most likely etiology for her peripheral neuropathy. The oncologist decided to proceed with cycle 5 of chemotherapy but reduced the dose of vincristine by 50%.
Discussion
Vincristine is a microtubule inhibitor used in many chemotherapy regimens and may cause reversible or permanent neuropathy, including autonomic (constipation), sensory (stocking-glove distribution), or motor (foot-drop).15 A nerve conduction study may be indicated as part of the diagnostic evaluation. Treatment for painful sensory neuropathy may include pharmacologic therapy (such as gabapentin, pregabalin, capsaicin cream).16 Podiatrists can provide foot care and may provide shoes and inserts if appropriate. Physical therapists may assist with safety and mobility evaluations and can provide therapeutic exercises and assistive devices that improve function and quality of life.17
Conclusion
As cancer becomes more curable and more manageable, patients with cancer and survivors no longer rely exclusively on their oncologists for medical care. This is increasingly prevalent for patients with incurable but indolent cancers that may be present for years to decades, as acute and cumulative toxicities may complicate existing comorbidities. Thus, in this era of increasingly complex cancer therapies, multidisciplinary medical care that involves PCPs, specialists, and allied medical professionals, is essential for providing care that optimizes health and fully addresses patients’ needs.
1. Friedman EL, Chawla N, Morris PT, et al. Assessing the development of multidisciplinary care: experience of the National Cancer Institute community cancer centers program. J Oncol Pract. 2015;11(1):e36-e43.
2. Peterson K, Helfand M, Humphrey L, Christensen V, Carson S. Evidence brief: effectiveness of intensive primary care programs. https://www.hsrd.research.va.gov/publications/esp/Intensive-Primary-Care-Supplement.pdf. Published February 2013. Accessed April 5, 2019.
3. National Committee for Quality Assurance. Oncology medical home recognition. https://www.ncqa.org/programs/health-care-providers-practices/oncology-medical-home. Accessed April 5, 2019.
4. Kahl BS, Yang DT. Follicular lymphoma: evolving therapeutic strategies. Blood. 2016;127(17):2055-2063.
5. Dulaney C, Wallace AS, Everett AS, Dover L, McDonald A, Kropp L. Defining health across the cancer continuum. Cureus. 2017;9(2):e1029.
6. Hopkins J, Mumber MP. Patient navigation through the cancer care continuum: an overview. J Oncol Pract. 2009;5(4):150-152.
7. Lee SY, Kurita N, Yokoyama Y, et al. Glucocorticoid-induced diabetes mellitus in patients with lymphoma treated with CHOP chemotherapy. Support Care Cancer. 2014;22(5):1385-1390.
8. McGill M, Blonde L, Juliana CN, et al; Global Partnership for Effective Diabetes Management. The interdisciplinary team in type 2 diabetes management: challenges and best practice solutions from real-world scenarios. J Clin Transl Endocrinol. 2017;7:21-27.
9. Horný M, Glover W, Gupte G, Saraswat A, Vimalananda V, Rosenzweig J. Patient navigation to improve diabetes outpatient care at a safety-net hospital: a retrospective cohort study. BMC Health Serv Res. 2017;17(1):759.
10. Reinwald M, Silva JT, Mueller NJ, et al. ESCMID Study Group for Infections in Compromised Hosts (ESGICH) Consensus Document on the safety of targeted and biological therapies: an infectious diseases perspective (Intracellular signaling pathways: tyrosine kinase and mTOR inhibitors). Clin Microbiol Infect. 2018;24(suppl 2):S53-S70.
11. Holle LM, Boehnke Michaud L. Oncology pharmacists in health care delivery: vital members of the cancer care team. J. Oncol. Pract. 2014;10(3):e142-e145.
12. Morgan KP, Muluneh B, Dean AM, Amerine LB. Impact of an integrated oral chemotherapy program on patient adherence. J Oncol Pharm Pract. 2018;24(5):332-336.
13. Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97(11):2869-2879.
14. Feltner C, Jones CD, Cené CW, et al. Transitional care interventions to prevent readmissions for persons with heart failure: a systematic review and meta-analysis. Ann Intern Med. 2014;160(11):774-784.
15. Mora E, Smith EM, Donohoe C, Hertz DL. Vincristine-induced peripheral neuropathy in pediatric cancer patients. Am J Cancer Res. 2016;6(11):2416-2430.
16. Hershman DL, Lacchetti C, Dworkin RH, et al; American Society of Clinical Oncology. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2014;32(18):1941–1967
17. Duregon F, Vendramin B, Bullo V, et al. Effects of exercise on cancer patients suffering chemotherapy-induced peripheral neuropathy undergoing treatment: a systematic review. Crit Rev Oncol Hematol. 2018;121:90-100.
Four case examples illustrate the important role of multidisciplinary medical care for the optimal long-term care of patients with follicular lymphoma.
Four case examples illustrate the important role of multidisciplinary medical care for the optimal long-term care of patients with follicular lymphoma.
Patients benefit from multidisciplinary care that coordinates management of complex medical problems. Traditionally, multidisciplinary cancer care involves oncology specialty providers in fields that include medical oncology, radiation oncology, and surgical oncology. Multidisciplinary cancer care intends to improve patient outcomes by bringing together different health care providers (HCPs) who are involved in the treatment of patients with cancer. Because new therapies are more effective and allow patients with cancer to live longer, adverse effects (AEs) are more likely to impact patients’ well-being, both while receiving treatment and long after it has completed. Thus, this population may benefit from an expanded approach to multidisciplinary care that includes input from specialty and primary care providers (PCPs), clinical pharmacy specialists (CPS), physical and occupational therapists, and patient navigators and educators.
We present 4 hypothetical cases, based on actual patients, that illustrate opportunities where multidisciplinary care coordination may improve patient experiences. These cases draw on current quality initiatives from the National Cancer Institute Community Cancer Centers Program, which has focused on improving the quality of multidisciplinary cancer care at selected community centers, and the Veterans Health Administration (VHA) patient-aligned care team (PACT) model, which brings together different health professionals to optimize primary care coordination.1,2 In addition, the National Committee for Quality Assurance has introduced an educational initiative to facilitate implementation of an oncologic medical home.3 This initiative stresses increased multidisciplinary communication, patient-centered care delivery, and reduced fragmentation of care for this population. Despite these guidelines and experiences from other medical specialties, models for integrated cancer care have not been implemented in a prospective fashion within the VHA.
In this article, we focus on opportunities to take collaborative care approaches for the treatment of patients with follicular lymphoma (FL): a common, incurable, and often indolent B-cell non-Hodgkin lymphoma.4 FL was selected because these patients may be treated numerous times and long-term sequalae can accumulate throughout their cancer continuum (a series of health events encompassing cancer screening, diagnosis, treatment, survivorship, relapse, and death).5 HCPs in distinct roles can assist patients with cancer in optimizing their health outcomes and overall wellbeing.6
Case Example 1
A 70-year-old male was diagnosed with stage IV FL. Because of his advanced disease, he began therapy with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone). Prednisone was administered at 100 mg daily on the first 5 days of each 21-day cycle. On day 4 of the first treatment cycle, the patient notified his oncologist that he had been very thirsty and his random blood sugar values on 2 different days were 283 mg/dL and 312 mg/dL. A laboratory review revealed his hemoglobin A1c (HbA1c) 7 months prior was 5.6%.
Discussion
The high-dose prednisone component of this and other lymphoma therapy regimens can worsen diabetes mellitus (DM) control and/or worsen prediabetes. Patient characteristics that increase the risk of developing glucocorticoid-induced DM after CHOP chemotherapy include age ≥ 60 years, HbA1c > 6.1%, and body mass index > 30.7 This patient did not have DM prior to the FL therapy initiation, but afterwards he met diagnostic criteria for DM. For completeness, other causes for elevated blood glucose should be ruled out (ie, infection, laboratory error, etc.). An oncologist often will triage acute hyperglycemia, treating immediately with IV fluids and/or insulin. Thereafter, ongoing chronic disease management for DM may be best managed by PCPs, certified DM educators, and registered dieticians.
Several programs involving multidisciplinary DM care, comprised of physicians, advanced practice providers, nurses, certified DM educators, and/or pharmacists have been shown to improve HbA1c, cardiovascular outcomes, and all-cause mortality, while reducing health care costs.8 In addition, patient navigators can assist patients with coordinating visits to disease-state specialists and identifying further educational needs. For example, in 1 program, nonclinical peer navigators were shown to improve the number of appointments attended and reduce HbA1c in a population of patients with DM who were primarily minority, urban, and of low socioeconomic status.9 Thus, integrating DM care shows potential to improve outcomes for patients with lymphoma who develop glucocorticoid
Case Example 2
A 75-year-old male was diagnosed with FL. He was treated initially with bendamustine and rituximab. He required reinitiation of therapy 20 months later when he developed lymphadenopathy, fatigue, and night sweats and began treatment with oral idelalisib, a second-line therapy. Later, the patient presented to his PCP for a routine visit, and on medication reconciliation review, the patient reported regular use of trimethoprim-sulfamethoxazole.
Discussion
Upon consultation with the CPS and the patient’s oncologist, the PCP confirmed trimethoprim-sulfamethoxazole should be continued during therapy and for about 6 months following completion of therapy. Trimethoprim-sulfamethoxazole is used for prophylaxis against Pneumocystis jirovecii (formerly Pneumocystis carinii). While use of prophylactic therapy is not necessary for all patients with FL, idelalisib impairs the function of circulating lymphoid B-cells and thus has been associated with an increased risk of serious infection.10 A CPS can provide insight that maximizes medication adherence and efficacy while minimizing food-drug, drug-drug interactions, and AEs. CPS have been shown to: improve adherence to oral therapies, increase prospective monitoring required for safe therapy dose selection, and document assessment of chemotherapy-related AEs.11,12 Thus, multidisciplinary, integrated care is an important component of providing quality oncology care.
Case Example 3
A 60-year-old female presented to her PCP with a 2-week history of shortness of breath and leg swelling. She was treated for FL 4 years previously with 6 cycles of R-CHOP. She reported no chest pain and did not have a prior history of hypertension, DM, or heart disease. On physical exam, she had elevated jugular venous pressure to jaw at 45°, bilateral pulmonary rales, and 2+ pitting pretibial edema. Laboratory tests that included complete blood count, basic chemistries, and thyroid stimulating hormone were unremarkable, though brain natriuretic peptide (BNP) was elevated at 425 pg/mL.
As this patient’s laboratory results and physical examination suggested new-onset congestive heart failure, the PCP obtained an echocardiogram, which demonstrated an ejection fraction of 35% and global hypokinesis. Because the patient was symptomatic, she was admitted to the hospital to begin guideline-directed medical therapy (GDMT) including IV diuresis.
Discussion
Given the absence of significant risk factors and prior history of coronary artery disease, the most probable cause for this patient’s cardiomyopathy is doxorubicin. Doxorubicin is an anthracycline chemotherapy that can cause nonischemic, dilated cardiomyopathy, particularly when cumulative doses > 400 mg/m2 are administered, or when combined with chest radiation.13 This patient benefited from GDMT for reduced ejection-fraction heart failure (HFrEF). Studies have demonstrated positive outcomes when HFrEF patients are cared for by a multidisciplinary team who focus of volume management as well as uptitration of therapies to target doses.14
Case Example 4
An 80-year-old female was diagnosed with stage III FL but did not require immediate therapy. After developing discomfort due to enlarging lymphadenopathy, she initiated therapy with rituximab, cyclophosphamide, vincristine, and prednisone (R-CVP). She presented to her oncologist for consideration of her fifth cycle of R-CVP and reported a burning sensation on the soles of her feet and numbness in her fingertips and toes. On examination, her pulses were intact and there were no signs of infection, reduced blood flow, or edema. The patient demonstrated decreased sensation on monofilament testing. She had no history of DM and a recent HbA1c test was 4.9% An evaluation for other causes of neuropathy, such as hypothyroidism and vitamin B12 deficiency was negative. Thus, vincristine therapy was identified as the most likely etiology for her peripheral neuropathy. The oncologist decided to proceed with cycle 5 of chemotherapy but reduced the dose of vincristine by 50%.
Discussion
Vincristine is a microtubule inhibitor used in many chemotherapy regimens and may cause reversible or permanent neuropathy, including autonomic (constipation), sensory (stocking-glove distribution), or motor (foot-drop).15 A nerve conduction study may be indicated as part of the diagnostic evaluation. Treatment for painful sensory neuropathy may include pharmacologic therapy (such as gabapentin, pregabalin, capsaicin cream).16 Podiatrists can provide foot care and may provide shoes and inserts if appropriate. Physical therapists may assist with safety and mobility evaluations and can provide therapeutic exercises and assistive devices that improve function and quality of life.17
Conclusion
As cancer becomes more curable and more manageable, patients with cancer and survivors no longer rely exclusively on their oncologists for medical care. This is increasingly prevalent for patients with incurable but indolent cancers that may be present for years to decades, as acute and cumulative toxicities may complicate existing comorbidities. Thus, in this era of increasingly complex cancer therapies, multidisciplinary medical care that involves PCPs, specialists, and allied medical professionals, is essential for providing care that optimizes health and fully addresses patients’ needs.
Patients benefit from multidisciplinary care that coordinates management of complex medical problems. Traditionally, multidisciplinary cancer care involves oncology specialty providers in fields that include medical oncology, radiation oncology, and surgical oncology. Multidisciplinary cancer care intends to improve patient outcomes by bringing together different health care providers (HCPs) who are involved in the treatment of patients with cancer. Because new therapies are more effective and allow patients with cancer to live longer, adverse effects (AEs) are more likely to impact patients’ well-being, both while receiving treatment and long after it has completed. Thus, this population may benefit from an expanded approach to multidisciplinary care that includes input from specialty and primary care providers (PCPs), clinical pharmacy specialists (CPS), physical and occupational therapists, and patient navigators and educators.
We present 4 hypothetical cases, based on actual patients, that illustrate opportunities where multidisciplinary care coordination may improve patient experiences. These cases draw on current quality initiatives from the National Cancer Institute Community Cancer Centers Program, which has focused on improving the quality of multidisciplinary cancer care at selected community centers, and the Veterans Health Administration (VHA) patient-aligned care team (PACT) model, which brings together different health professionals to optimize primary care coordination.1,2 In addition, the National Committee for Quality Assurance has introduced an educational initiative to facilitate implementation of an oncologic medical home.3 This initiative stresses increased multidisciplinary communication, patient-centered care delivery, and reduced fragmentation of care for this population. Despite these guidelines and experiences from other medical specialties, models for integrated cancer care have not been implemented in a prospective fashion within the VHA.
In this article, we focus on opportunities to take collaborative care approaches for the treatment of patients with follicular lymphoma (FL): a common, incurable, and often indolent B-cell non-Hodgkin lymphoma.4 FL was selected because these patients may be treated numerous times and long-term sequalae can accumulate throughout their cancer continuum (a series of health events encompassing cancer screening, diagnosis, treatment, survivorship, relapse, and death).5 HCPs in distinct roles can assist patients with cancer in optimizing their health outcomes and overall wellbeing.6
Case Example 1
A 70-year-old male was diagnosed with stage IV FL. Because of his advanced disease, he began therapy with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone). Prednisone was administered at 100 mg daily on the first 5 days of each 21-day cycle. On day 4 of the first treatment cycle, the patient notified his oncologist that he had been very thirsty and his random blood sugar values on 2 different days were 283 mg/dL and 312 mg/dL. A laboratory review revealed his hemoglobin A1c (HbA1c) 7 months prior was 5.6%.
Discussion
The high-dose prednisone component of this and other lymphoma therapy regimens can worsen diabetes mellitus (DM) control and/or worsen prediabetes. Patient characteristics that increase the risk of developing glucocorticoid-induced DM after CHOP chemotherapy include age ≥ 60 years, HbA1c > 6.1%, and body mass index > 30.7 This patient did not have DM prior to the FL therapy initiation, but afterwards he met diagnostic criteria for DM. For completeness, other causes for elevated blood glucose should be ruled out (ie, infection, laboratory error, etc.). An oncologist often will triage acute hyperglycemia, treating immediately with IV fluids and/or insulin. Thereafter, ongoing chronic disease management for DM may be best managed by PCPs, certified DM educators, and registered dieticians.
Several programs involving multidisciplinary DM care, comprised of physicians, advanced practice providers, nurses, certified DM educators, and/or pharmacists have been shown to improve HbA1c, cardiovascular outcomes, and all-cause mortality, while reducing health care costs.8 In addition, patient navigators can assist patients with coordinating visits to disease-state specialists and identifying further educational needs. For example, in 1 program, nonclinical peer navigators were shown to improve the number of appointments attended and reduce HbA1c in a population of patients with DM who were primarily minority, urban, and of low socioeconomic status.9 Thus, integrating DM care shows potential to improve outcomes for patients with lymphoma who develop glucocorticoid
Case Example 2
A 75-year-old male was diagnosed with FL. He was treated initially with bendamustine and rituximab. He required reinitiation of therapy 20 months later when he developed lymphadenopathy, fatigue, and night sweats and began treatment with oral idelalisib, a second-line therapy. Later, the patient presented to his PCP for a routine visit, and on medication reconciliation review, the patient reported regular use of trimethoprim-sulfamethoxazole.
Discussion
Upon consultation with the CPS and the patient’s oncologist, the PCP confirmed trimethoprim-sulfamethoxazole should be continued during therapy and for about 6 months following completion of therapy. Trimethoprim-sulfamethoxazole is used for prophylaxis against Pneumocystis jirovecii (formerly Pneumocystis carinii). While use of prophylactic therapy is not necessary for all patients with FL, idelalisib impairs the function of circulating lymphoid B-cells and thus has been associated with an increased risk of serious infection.10 A CPS can provide insight that maximizes medication adherence and efficacy while minimizing food-drug, drug-drug interactions, and AEs. CPS have been shown to: improve adherence to oral therapies, increase prospective monitoring required for safe therapy dose selection, and document assessment of chemotherapy-related AEs.11,12 Thus, multidisciplinary, integrated care is an important component of providing quality oncology care.
Case Example 3
A 60-year-old female presented to her PCP with a 2-week history of shortness of breath and leg swelling. She was treated for FL 4 years previously with 6 cycles of R-CHOP. She reported no chest pain and did not have a prior history of hypertension, DM, or heart disease. On physical exam, she had elevated jugular venous pressure to jaw at 45°, bilateral pulmonary rales, and 2+ pitting pretibial edema. Laboratory tests that included complete blood count, basic chemistries, and thyroid stimulating hormone were unremarkable, though brain natriuretic peptide (BNP) was elevated at 425 pg/mL.
As this patient’s laboratory results and physical examination suggested new-onset congestive heart failure, the PCP obtained an echocardiogram, which demonstrated an ejection fraction of 35% and global hypokinesis. Because the patient was symptomatic, she was admitted to the hospital to begin guideline-directed medical therapy (GDMT) including IV diuresis.
Discussion
Given the absence of significant risk factors and prior history of coronary artery disease, the most probable cause for this patient’s cardiomyopathy is doxorubicin. Doxorubicin is an anthracycline chemotherapy that can cause nonischemic, dilated cardiomyopathy, particularly when cumulative doses > 400 mg/m2 are administered, or when combined with chest radiation.13 This patient benefited from GDMT for reduced ejection-fraction heart failure (HFrEF). Studies have demonstrated positive outcomes when HFrEF patients are cared for by a multidisciplinary team who focus of volume management as well as uptitration of therapies to target doses.14
Case Example 4
An 80-year-old female was diagnosed with stage III FL but did not require immediate therapy. After developing discomfort due to enlarging lymphadenopathy, she initiated therapy with rituximab, cyclophosphamide, vincristine, and prednisone (R-CVP). She presented to her oncologist for consideration of her fifth cycle of R-CVP and reported a burning sensation on the soles of her feet and numbness in her fingertips and toes. On examination, her pulses were intact and there were no signs of infection, reduced blood flow, or edema. The patient demonstrated decreased sensation on monofilament testing. She had no history of DM and a recent HbA1c test was 4.9% An evaluation for other causes of neuropathy, such as hypothyroidism and vitamin B12 deficiency was negative. Thus, vincristine therapy was identified as the most likely etiology for her peripheral neuropathy. The oncologist decided to proceed with cycle 5 of chemotherapy but reduced the dose of vincristine by 50%.
Discussion
Vincristine is a microtubule inhibitor used in many chemotherapy regimens and may cause reversible or permanent neuropathy, including autonomic (constipation), sensory (stocking-glove distribution), or motor (foot-drop).15 A nerve conduction study may be indicated as part of the diagnostic evaluation. Treatment for painful sensory neuropathy may include pharmacologic therapy (such as gabapentin, pregabalin, capsaicin cream).16 Podiatrists can provide foot care and may provide shoes and inserts if appropriate. Physical therapists may assist with safety and mobility evaluations and can provide therapeutic exercises and assistive devices that improve function and quality of life.17
Conclusion
As cancer becomes more curable and more manageable, patients with cancer and survivors no longer rely exclusively on their oncologists for medical care. This is increasingly prevalent for patients with incurable but indolent cancers that may be present for years to decades, as acute and cumulative toxicities may complicate existing comorbidities. Thus, in this era of increasingly complex cancer therapies, multidisciplinary medical care that involves PCPs, specialists, and allied medical professionals, is essential for providing care that optimizes health and fully addresses patients’ needs.
1. Friedman EL, Chawla N, Morris PT, et al. Assessing the development of multidisciplinary care: experience of the National Cancer Institute community cancer centers program. J Oncol Pract. 2015;11(1):e36-e43.
2. Peterson K, Helfand M, Humphrey L, Christensen V, Carson S. Evidence brief: effectiveness of intensive primary care programs. https://www.hsrd.research.va.gov/publications/esp/Intensive-Primary-Care-Supplement.pdf. Published February 2013. Accessed April 5, 2019.
3. National Committee for Quality Assurance. Oncology medical home recognition. https://www.ncqa.org/programs/health-care-providers-practices/oncology-medical-home. Accessed April 5, 2019.
4. Kahl BS, Yang DT. Follicular lymphoma: evolving therapeutic strategies. Blood. 2016;127(17):2055-2063.
5. Dulaney C, Wallace AS, Everett AS, Dover L, McDonald A, Kropp L. Defining health across the cancer continuum. Cureus. 2017;9(2):e1029.
6. Hopkins J, Mumber MP. Patient navigation through the cancer care continuum: an overview. J Oncol Pract. 2009;5(4):150-152.
7. Lee SY, Kurita N, Yokoyama Y, et al. Glucocorticoid-induced diabetes mellitus in patients with lymphoma treated with CHOP chemotherapy. Support Care Cancer. 2014;22(5):1385-1390.
8. McGill M, Blonde L, Juliana CN, et al; Global Partnership for Effective Diabetes Management. The interdisciplinary team in type 2 diabetes management: challenges and best practice solutions from real-world scenarios. J Clin Transl Endocrinol. 2017;7:21-27.
9. Horný M, Glover W, Gupte G, Saraswat A, Vimalananda V, Rosenzweig J. Patient navigation to improve diabetes outpatient care at a safety-net hospital: a retrospective cohort study. BMC Health Serv Res. 2017;17(1):759.
10. Reinwald M, Silva JT, Mueller NJ, et al. ESCMID Study Group for Infections in Compromised Hosts (ESGICH) Consensus Document on the safety of targeted and biological therapies: an infectious diseases perspective (Intracellular signaling pathways: tyrosine kinase and mTOR inhibitors). Clin Microbiol Infect. 2018;24(suppl 2):S53-S70.
11. Holle LM, Boehnke Michaud L. Oncology pharmacists in health care delivery: vital members of the cancer care team. J. Oncol. Pract. 2014;10(3):e142-e145.
12. Morgan KP, Muluneh B, Dean AM, Amerine LB. Impact of an integrated oral chemotherapy program on patient adherence. J Oncol Pharm Pract. 2018;24(5):332-336.
13. Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97(11):2869-2879.
14. Feltner C, Jones CD, Cené CW, et al. Transitional care interventions to prevent readmissions for persons with heart failure: a systematic review and meta-analysis. Ann Intern Med. 2014;160(11):774-784.
15. Mora E, Smith EM, Donohoe C, Hertz DL. Vincristine-induced peripheral neuropathy in pediatric cancer patients. Am J Cancer Res. 2016;6(11):2416-2430.
16. Hershman DL, Lacchetti C, Dworkin RH, et al; American Society of Clinical Oncology. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2014;32(18):1941–1967
17. Duregon F, Vendramin B, Bullo V, et al. Effects of exercise on cancer patients suffering chemotherapy-induced peripheral neuropathy undergoing treatment: a systematic review. Crit Rev Oncol Hematol. 2018;121:90-100.
1. Friedman EL, Chawla N, Morris PT, et al. Assessing the development of multidisciplinary care: experience of the National Cancer Institute community cancer centers program. J Oncol Pract. 2015;11(1):e36-e43.
2. Peterson K, Helfand M, Humphrey L, Christensen V, Carson S. Evidence brief: effectiveness of intensive primary care programs. https://www.hsrd.research.va.gov/publications/esp/Intensive-Primary-Care-Supplement.pdf. Published February 2013. Accessed April 5, 2019.
3. National Committee for Quality Assurance. Oncology medical home recognition. https://www.ncqa.org/programs/health-care-providers-practices/oncology-medical-home. Accessed April 5, 2019.
4. Kahl BS, Yang DT. Follicular lymphoma: evolving therapeutic strategies. Blood. 2016;127(17):2055-2063.
5. Dulaney C, Wallace AS, Everett AS, Dover L, McDonald A, Kropp L. Defining health across the cancer continuum. Cureus. 2017;9(2):e1029.
6. Hopkins J, Mumber MP. Patient navigation through the cancer care continuum: an overview. J Oncol Pract. 2009;5(4):150-152.
7. Lee SY, Kurita N, Yokoyama Y, et al. Glucocorticoid-induced diabetes mellitus in patients with lymphoma treated with CHOP chemotherapy. Support Care Cancer. 2014;22(5):1385-1390.
8. McGill M, Blonde L, Juliana CN, et al; Global Partnership for Effective Diabetes Management. The interdisciplinary team in type 2 diabetes management: challenges and best practice solutions from real-world scenarios. J Clin Transl Endocrinol. 2017;7:21-27.
9. Horný M, Glover W, Gupte G, Saraswat A, Vimalananda V, Rosenzweig J. Patient navigation to improve diabetes outpatient care at a safety-net hospital: a retrospective cohort study. BMC Health Serv Res. 2017;17(1):759.
10. Reinwald M, Silva JT, Mueller NJ, et al. ESCMID Study Group for Infections in Compromised Hosts (ESGICH) Consensus Document on the safety of targeted and biological therapies: an infectious diseases perspective (Intracellular signaling pathways: tyrosine kinase and mTOR inhibitors). Clin Microbiol Infect. 2018;24(suppl 2):S53-S70.
11. Holle LM, Boehnke Michaud L. Oncology pharmacists in health care delivery: vital members of the cancer care team. J. Oncol. Pract. 2014;10(3):e142-e145.
12. Morgan KP, Muluneh B, Dean AM, Amerine LB. Impact of an integrated oral chemotherapy program on patient adherence. J Oncol Pharm Pract. 2018;24(5):332-336.
13. Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97(11):2869-2879.
14. Feltner C, Jones CD, Cené CW, et al. Transitional care interventions to prevent readmissions for persons with heart failure: a systematic review and meta-analysis. Ann Intern Med. 2014;160(11):774-784.
15. Mora E, Smith EM, Donohoe C, Hertz DL. Vincristine-induced peripheral neuropathy in pediatric cancer patients. Am J Cancer Res. 2016;6(11):2416-2430.
16. Hershman DL, Lacchetti C, Dworkin RH, et al; American Society of Clinical Oncology. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2014;32(18):1941–1967
17. Duregon F, Vendramin B, Bullo V, et al. Effects of exercise on cancer patients suffering chemotherapy-induced peripheral neuropathy undergoing treatment: a systematic review. Crit Rev Oncol Hematol. 2018;121:90-100.
Multiple Atypical Vascular Lesions Following Breast-Conserving Surgery and Radiation
Atypical vascular lesions (AVLs) are rare flesh-colored, erythematous, or violaceous macules, patches, papules, or plaques that may occur following adjuvant radiation in breast cancer patients who have undergone conservative lumpectomy.1,2 They range in size from 1 mm to 6 cm and are most often confined to the radiation field. Presentation occurs 1 to 20 years following radiation, though the lesions most often present within 5 years.1,2 Although generally considered benign, 2 of 29 cases of AVLs progressed to angiosarcoma over a 5-year follow-up period in a retrospective clinicopathologic study.1
Atypical vascular lesions show considerable histologic and clinical overlap with radiation-induced angiosarcomas (RIAs), making differentiation between the two challenging.3,4 Mentzel et al5 compared benign, atypical, and malignant postradiation vascular lesions with nonradiation-associated angiosarcomas and found that RIAs were highly variable histopathologically, ranging from well differentiated to poorly differentiated, with atypia ranging from mild to severe. Radiation-induced angiosarcomas could be distinguished from AVLs and nonradiation-associated angiosarcomas by their oncogene amplification and protein expression profiles. Most strikingly, they found amplification of the MYC oncogene by fluorescence in situ hybridization in the nucleus of almost all the RIA cells, which was not seen in AVLs or nonradiation-associated angiosarcomas. Similarly, they found positive nuclear staining for MYC protein by immunohistochemistry in the nucleus of almost all cases of RIA but not in AVL or nonradiation-associated angiosarcomas, making MYC staining a useful diagnostic marker.5 In contrast, a study by Patton et al1 concluded that AVLs demonstrate morphologic patterns and clinical outcomes that suggest they are precursors of angiosarcoma rather than just markers of risk.
Atypical vascular lesions and RIAs usually follow a total radiation dose of 40 to 50 Gy, but RIAs typically are diagnosed later (approximately 10 years following exposure).6,7 Although RIAs are rare, they are known to be aggressive and often high grade, with a median survival of less than 5 years.6,7 Survival is poor even with radical surgical treatment.8 We present a patient with at least 29 AVLs following breast-conserving surgery and radiation and suggest the need for increased awareness of the elevated risk for RIA in patients with numerous benign AVLs.
Case Report
A 43-year-old woman with a history of breast cancer who underwent breast-conserving lumpectomy and adjuvant radiation presented to dermatology upon referral from surgical oncology for multiple lesions on the right breast (Figure 1). Seven years prior to presentation she was diagnosed with grade 3 poorly differentiated invasive ductal carcinoma with lobular features in the right breast that was positive for human epidermal growth factor receptor 2 but negative for estrogen or progesterone receptors. She was given neoadjuvant treatment with trastuzumab, docetaxel, and carboplatin prior to conservation lumpectomy with adjuvant radiation. She received a total dose of 50.4 Gy in 28 fractions of 1.8 Gy each over 1 month, with a final boost of 10 Gy in 5 fractions of 2 Gy, each with local skin irritation as the only concern posttreatment.

She initially presented to dermatology approximately 3 years after radiotherapy (5 years prior to current presentation) with lesions on the breast that had been present for 6 to 9 months. Physical examination showed 2 firm, painless, 4- to 5-mm papules on the right upper breast. The patient was reassured that the lesions were not suspicious for malignancy; however, 3 years later she presented to surgical oncology with 8 bluish papules or macules (all approximately 4 mm in diameter) on the right breast. These lesions were biopsied and examined by 2 institutions. Pathology of the initial punch biopsy favored a diagnosis of AVLs, though the possibility of RIA could not be ruled out without a complete excisional biopsy. Two excisional biopsies a month later were again consistent with AVLs. In all cases, the lesions were negative for MYC protein. The patient was again reassured but referred to dermatology for a second opinion.
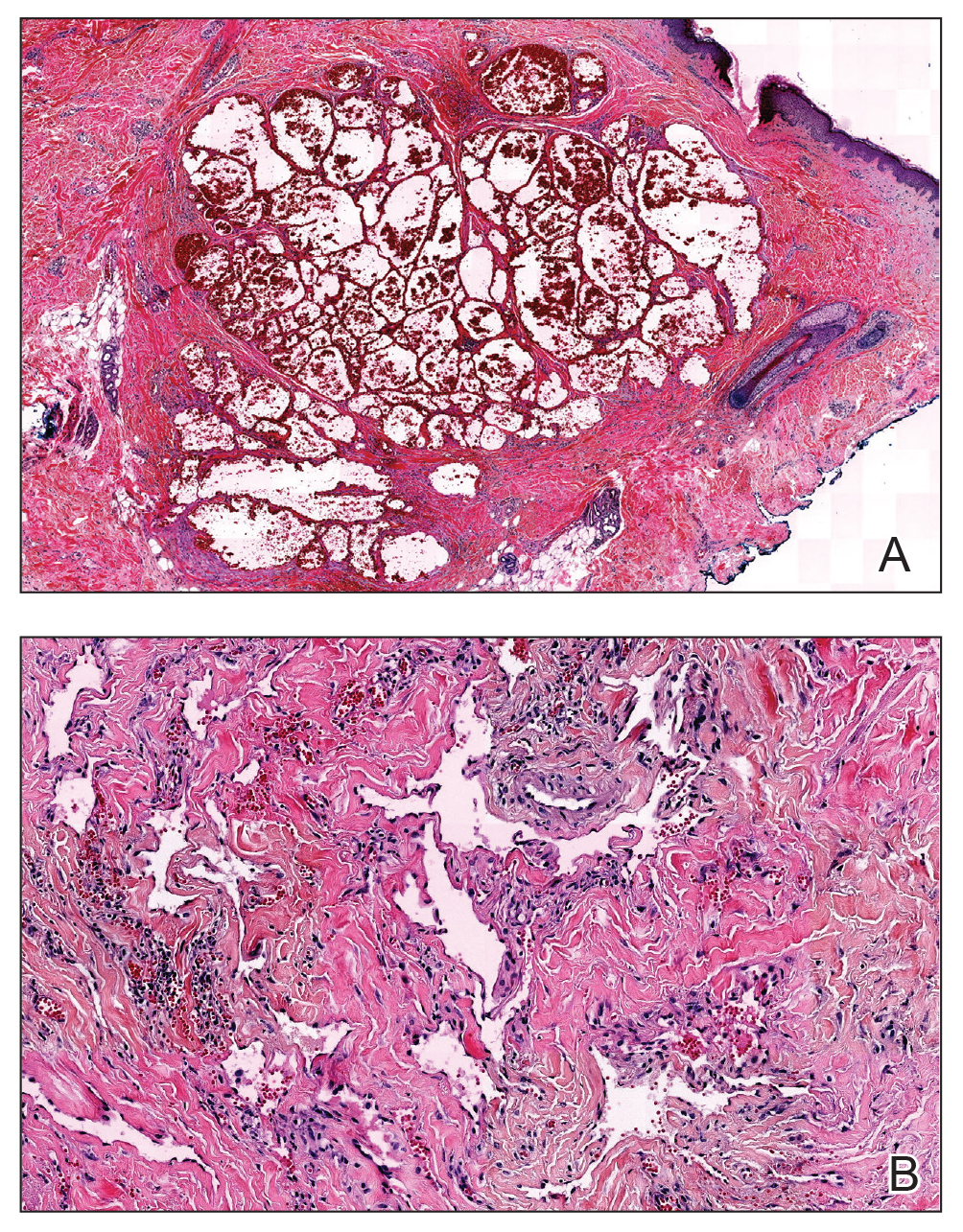
At the current presentation, physical examination showed at least 29 subcutaneous nodules on the right breast ranging in color from pink to deep blue to flesh colored with others more superficially hyperpigmented, possibly secondary to prior biopsy, and measuring 2 to 8 mm in diameter. Histopathologic examination of the biopsy specimens showed a vascular proliferation extending from the dermis into the subcutaneous tissue comprised of dilated and cavernous vascular channels lined by a single layer of endothelial cells with minimal cytologic atypia (Figure 2). There were focal areas of anastomosing slitlike vascular spaces dissecting dermal collagen. No features of malignancy, such as nuclear crowding, multilayering, or increased mitotic activity, were evident. Immunohistochemical studies for MYC protein were negative. The overall morphologic features and immunoprofile were felt to be most consistent with postradiation AVLs.
At the time, surgical oncology felt that the risk of radical mastectomy outweighed the risk of angiosarcoma due to the absence of frank angiosarcoma and the patient’s notable comorbidities, including diabetes mellitus, cerebrovascular disease, peripheral vascular disease, and smoking; however, after reviewing the literature and considering the difficulty of following such a large number of lesions, the dermatology team brought the patient’s case to the multidisciplinary cutaneous tumor board at the University of Massachusetts (Worcester, Massachusetts). In consensus, the tumor board recommended radical mastectomy despite the comorbidities, given her young age and the potential risk for malignant transformation of any one of the numerous AVLs to angiosarcoma.
Postmastectomy pathology showed multiple scattered foci of AVLs ranging from 1.5 to 4 mm in the dermis, similar to those seen on prior biopsies, with no frank evidence of RIA. At 3-year follow-up, the patient has had no recurrence of AVLs or findings suggestive of RIA. There were no reported complications.
Comment
Conservative breast cancer surgery and radiotherapy are becoming more prevalent for breast cancer treatment, thus the number of patients likely to present with AVLs has increased. These patients are at risk for transformation to RIAs.6 It is important for clinicians to be aware of the diagnosis of both AVLs and RIAs and their management given their more frequent presentation. In most cases, one or a few AVLs are present, and excision is the treatment of choice. In a retrospective study by Brenn and Fletcher3 examining 16 patients with AVLs and 26 patients with RIA, the majority of cases of AVL had a single lesion and the maximum number of AVLs was 4. One patient in their study had 30 AVLs (each 3–4 mm in diameter), and she was diagnosed with RIA.3 Our patient—with at least 29 identifiable AVL lesions—was felt to be at considerable risk for developing RIA, as the only other case reported with this many AVLs developed RIA.1 Given the large number of lesions, it was neither feasible to excise each one individually nor monitor all of them for malignant transformation.
Our case demonstrates the important role dermatologists may play in orchestrating care by a multispecialty team including oncology, radiation oncology, surgery, and plastic surgery. In our patient, a close examination of the literature by the dermatology team led to recognition of the potentially elevated risk for malignant transformation. The dermatology team also brought the case for review at the tumor board.
Although future studies are required to determine the relationship between AVL burden and the risk for progression to RIA, it is clear that a multidisciplinary approach and careful consideration of the current literature can prevent unnecessary morbidity and mortality for patients with this increasingly common problem.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol. 2008;32:943-950.
- Mandrell J, Mehta S, McClure S. Atypical vascular lesion of the breast. J Am Acad Dermatol. 2010;63:337-340.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Losch A, Chilek KD, Zirwas MJ. Post-radiation atypical vascular proliferation mimicking angiosarcoma eight months following breast-conserving therapy for breast carcinoma. J Clin Aesthet Dermatol. 2011;4:47-48.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological, immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Tahir M, Hendry P, Baird L, et al. Radiation induced angiosarcoma a sequela of radiotherapy for breast cancer following conservative surgery. Int Semin Surg Oncol. 2006;3:26.
- Hillenbrand T, Menge F, Hohenberger P, et al. Primary and secondary angiosarcomas: a comparative single-center analysis. Clin Sarcoma Res. 2015;5:14.
- Seinen JM, Styring E, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
Atypical vascular lesions (AVLs) are rare flesh-colored, erythematous, or violaceous macules, patches, papules, or plaques that may occur following adjuvant radiation in breast cancer patients who have undergone conservative lumpectomy.1,2 They range in size from 1 mm to 6 cm and are most often confined to the radiation field. Presentation occurs 1 to 20 years following radiation, though the lesions most often present within 5 years.1,2 Although generally considered benign, 2 of 29 cases of AVLs progressed to angiosarcoma over a 5-year follow-up period in a retrospective clinicopathologic study.1
Atypical vascular lesions show considerable histologic and clinical overlap with radiation-induced angiosarcomas (RIAs), making differentiation between the two challenging.3,4 Mentzel et al5 compared benign, atypical, and malignant postradiation vascular lesions with nonradiation-associated angiosarcomas and found that RIAs were highly variable histopathologically, ranging from well differentiated to poorly differentiated, with atypia ranging from mild to severe. Radiation-induced angiosarcomas could be distinguished from AVLs and nonradiation-associated angiosarcomas by their oncogene amplification and protein expression profiles. Most strikingly, they found amplification of the MYC oncogene by fluorescence in situ hybridization in the nucleus of almost all the RIA cells, which was not seen in AVLs or nonradiation-associated angiosarcomas. Similarly, they found positive nuclear staining for MYC protein by immunohistochemistry in the nucleus of almost all cases of RIA but not in AVL or nonradiation-associated angiosarcomas, making MYC staining a useful diagnostic marker.5 In contrast, a study by Patton et al1 concluded that AVLs demonstrate morphologic patterns and clinical outcomes that suggest they are precursors of angiosarcoma rather than just markers of risk.
Atypical vascular lesions and RIAs usually follow a total radiation dose of 40 to 50 Gy, but RIAs typically are diagnosed later (approximately 10 years following exposure).6,7 Although RIAs are rare, they are known to be aggressive and often high grade, with a median survival of less than 5 years.6,7 Survival is poor even with radical surgical treatment.8 We present a patient with at least 29 AVLs following breast-conserving surgery and radiation and suggest the need for increased awareness of the elevated risk for RIA in patients with numerous benign AVLs.
Case Report
A 43-year-old woman with a history of breast cancer who underwent breast-conserving lumpectomy and adjuvant radiation presented to dermatology upon referral from surgical oncology for multiple lesions on the right breast (Figure 1). Seven years prior to presentation she was diagnosed with grade 3 poorly differentiated invasive ductal carcinoma with lobular features in the right breast that was positive for human epidermal growth factor receptor 2 but negative for estrogen or progesterone receptors. She was given neoadjuvant treatment with trastuzumab, docetaxel, and carboplatin prior to conservation lumpectomy with adjuvant radiation. She received a total dose of 50.4 Gy in 28 fractions of 1.8 Gy each over 1 month, with a final boost of 10 Gy in 5 fractions of 2 Gy, each with local skin irritation as the only concern posttreatment.
She initially presented to dermatology approximately 3 years after radiotherapy (5 years prior to current presentation) with lesions on the breast that had been present for 6 to 9 months. Physical examination showed 2 firm, painless, 4- to 5-mm papules on the right upper breast. The patient was reassured that the lesions were not suspicious for malignancy; however, 3 years later she presented to surgical oncology with 8 bluish papules or macules (all approximately 4 mm in diameter) on the right breast. These lesions were biopsied and examined by 2 institutions. Pathology of the initial punch biopsy favored a diagnosis of AVLs, though the possibility of RIA could not be ruled out without a complete excisional biopsy. Two excisional biopsies a month later were again consistent with AVLs. In all cases, the lesions were negative for MYC protein. The patient was again reassured but referred to dermatology for a second opinion.
At the current presentation, physical examination showed at least 29 subcutaneous nodules on the right breast ranging in color from pink to deep blue to flesh colored with others more superficially hyperpigmented, possibly secondary to prior biopsy, and measuring 2 to 8 mm in diameter. Histopathologic examination of the biopsy specimens showed a vascular proliferation extending from the dermis into the subcutaneous tissue comprised of dilated and cavernous vascular channels lined by a single layer of endothelial cells with minimal cytologic atypia (Figure 2). There were focal areas of anastomosing slitlike vascular spaces dissecting dermal collagen. No features of malignancy, such as nuclear crowding, multilayering, or increased mitotic activity, were evident. Immunohistochemical studies for MYC protein were negative. The overall morphologic features and immunoprofile were felt to be most consistent with postradiation AVLs.
At the time, surgical oncology felt that the risk of radical mastectomy outweighed the risk of angiosarcoma due to the absence of frank angiosarcoma and the patient’s notable comorbidities, including diabetes mellitus, cerebrovascular disease, peripheral vascular disease, and smoking; however, after reviewing the literature and considering the difficulty of following such a large number of lesions, the dermatology team brought the patient’s case to the multidisciplinary cutaneous tumor board at the University of Massachusetts (Worcester, Massachusetts). In consensus, the tumor board recommended radical mastectomy despite the comorbidities, given her young age and the potential risk for malignant transformation of any one of the numerous AVLs to angiosarcoma.
Postmastectomy pathology showed multiple scattered foci of AVLs ranging from 1.5 to 4 mm in the dermis, similar to those seen on prior biopsies, with no frank evidence of RIA. At 3-year follow-up, the patient has had no recurrence of AVLs or findings suggestive of RIA. There were no reported complications.
Comment
Conservative breast cancer surgery and radiotherapy are becoming more prevalent for breast cancer treatment, thus the number of patients likely to present with AVLs has increased. These patients are at risk for transformation to RIAs.6 It is important for clinicians to be aware of the diagnosis of both AVLs and RIAs and their management given their more frequent presentation. In most cases, one or a few AVLs are present, and excision is the treatment of choice. In a retrospective study by Brenn and Fletcher3 examining 16 patients with AVLs and 26 patients with RIA, the majority of cases of AVL had a single lesion and the maximum number of AVLs was 4. One patient in their study had 30 AVLs (each 3–4 mm in diameter), and she was diagnosed with RIA.3 Our patient—with at least 29 identifiable AVL lesions—was felt to be at considerable risk for developing RIA, as the only other case reported with this many AVLs developed RIA.1 Given the large number of lesions, it was neither feasible to excise each one individually nor monitor all of them for malignant transformation.
Our case demonstrates the important role dermatologists may play in orchestrating care by a multispecialty team including oncology, radiation oncology, surgery, and plastic surgery. In our patient, a close examination of the literature by the dermatology team led to recognition of the potentially elevated risk for malignant transformation. The dermatology team also brought the case for review at the tumor board.
Although future studies are required to determine the relationship between AVL burden and the risk for progression to RIA, it is clear that a multidisciplinary approach and careful consideration of the current literature can prevent unnecessary morbidity and mortality for patients with this increasingly common problem.
Atypical vascular lesions (AVLs) are rare flesh-colored, erythematous, or violaceous macules, patches, papules, or plaques that may occur following adjuvant radiation in breast cancer patients who have undergone conservative lumpectomy.1,2 They range in size from 1 mm to 6 cm and are most often confined to the radiation field. Presentation occurs 1 to 20 years following radiation, though the lesions most often present within 5 years.1,2 Although generally considered benign, 2 of 29 cases of AVLs progressed to angiosarcoma over a 5-year follow-up period in a retrospective clinicopathologic study.1
Atypical vascular lesions show considerable histologic and clinical overlap with radiation-induced angiosarcomas (RIAs), making differentiation between the two challenging.3,4 Mentzel et al5 compared benign, atypical, and malignant postradiation vascular lesions with nonradiation-associated angiosarcomas and found that RIAs were highly variable histopathologically, ranging from well differentiated to poorly differentiated, with atypia ranging from mild to severe. Radiation-induced angiosarcomas could be distinguished from AVLs and nonradiation-associated angiosarcomas by their oncogene amplification and protein expression profiles. Most strikingly, they found amplification of the MYC oncogene by fluorescence in situ hybridization in the nucleus of almost all the RIA cells, which was not seen in AVLs or nonradiation-associated angiosarcomas. Similarly, they found positive nuclear staining for MYC protein by immunohistochemistry in the nucleus of almost all cases of RIA but not in AVL or nonradiation-associated angiosarcomas, making MYC staining a useful diagnostic marker.5 In contrast, a study by Patton et al1 concluded that AVLs demonstrate morphologic patterns and clinical outcomes that suggest they are precursors of angiosarcoma rather than just markers of risk.
Atypical vascular lesions and RIAs usually follow a total radiation dose of 40 to 50 Gy, but RIAs typically are diagnosed later (approximately 10 years following exposure).6,7 Although RIAs are rare, they are known to be aggressive and often high grade, with a median survival of less than 5 years.6,7 Survival is poor even with radical surgical treatment.8 We present a patient with at least 29 AVLs following breast-conserving surgery and radiation and suggest the need for increased awareness of the elevated risk for RIA in patients with numerous benign AVLs.
Case Report
A 43-year-old woman with a history of breast cancer who underwent breast-conserving lumpectomy and adjuvant radiation presented to dermatology upon referral from surgical oncology for multiple lesions on the right breast (Figure 1). Seven years prior to presentation she was diagnosed with grade 3 poorly differentiated invasive ductal carcinoma with lobular features in the right breast that was positive for human epidermal growth factor receptor 2 but negative for estrogen or progesterone receptors. She was given neoadjuvant treatment with trastuzumab, docetaxel, and carboplatin prior to conservation lumpectomy with adjuvant radiation. She received a total dose of 50.4 Gy in 28 fractions of 1.8 Gy each over 1 month, with a final boost of 10 Gy in 5 fractions of 2 Gy, each with local skin irritation as the only concern posttreatment.
She initially presented to dermatology approximately 3 years after radiotherapy (5 years prior to current presentation) with lesions on the breast that had been present for 6 to 9 months. Physical examination showed 2 firm, painless, 4- to 5-mm papules on the right upper breast. The patient was reassured that the lesions were not suspicious for malignancy; however, 3 years later she presented to surgical oncology with 8 bluish papules or macules (all approximately 4 mm in diameter) on the right breast. These lesions were biopsied and examined by 2 institutions. Pathology of the initial punch biopsy favored a diagnosis of AVLs, though the possibility of RIA could not be ruled out without a complete excisional biopsy. Two excisional biopsies a month later were again consistent with AVLs. In all cases, the lesions were negative for MYC protein. The patient was again reassured but referred to dermatology for a second opinion.
At the current presentation, physical examination showed at least 29 subcutaneous nodules on the right breast ranging in color from pink to deep blue to flesh colored with others more superficially hyperpigmented, possibly secondary to prior biopsy, and measuring 2 to 8 mm in diameter. Histopathologic examination of the biopsy specimens showed a vascular proliferation extending from the dermis into the subcutaneous tissue comprised of dilated and cavernous vascular channels lined by a single layer of endothelial cells with minimal cytologic atypia (Figure 2). There were focal areas of anastomosing slitlike vascular spaces dissecting dermal collagen. No features of malignancy, such as nuclear crowding, multilayering, or increased mitotic activity, were evident. Immunohistochemical studies for MYC protein were negative. The overall morphologic features and immunoprofile were felt to be most consistent with postradiation AVLs.
At the time, surgical oncology felt that the risk of radical mastectomy outweighed the risk of angiosarcoma due to the absence of frank angiosarcoma and the patient’s notable comorbidities, including diabetes mellitus, cerebrovascular disease, peripheral vascular disease, and smoking; however, after reviewing the literature and considering the difficulty of following such a large number of lesions, the dermatology team brought the patient’s case to the multidisciplinary cutaneous tumor board at the University of Massachusetts (Worcester, Massachusetts). In consensus, the tumor board recommended radical mastectomy despite the comorbidities, given her young age and the potential risk for malignant transformation of any one of the numerous AVLs to angiosarcoma.
Postmastectomy pathology showed multiple scattered foci of AVLs ranging from 1.5 to 4 mm in the dermis, similar to those seen on prior biopsies, with no frank evidence of RIA. At 3-year follow-up, the patient has had no recurrence of AVLs or findings suggestive of RIA. There were no reported complications.
Comment
Conservative breast cancer surgery and radiotherapy are becoming more prevalent for breast cancer treatment, thus the number of patients likely to present with AVLs has increased. These patients are at risk for transformation to RIAs.6 It is important for clinicians to be aware of the diagnosis of both AVLs and RIAs and their management given their more frequent presentation. In most cases, one or a few AVLs are present, and excision is the treatment of choice. In a retrospective study by Brenn and Fletcher3 examining 16 patients with AVLs and 26 patients with RIA, the majority of cases of AVL had a single lesion and the maximum number of AVLs was 4. One patient in their study had 30 AVLs (each 3–4 mm in diameter), and she was diagnosed with RIA.3 Our patient—with at least 29 identifiable AVL lesions—was felt to be at considerable risk for developing RIA, as the only other case reported with this many AVLs developed RIA.1 Given the large number of lesions, it was neither feasible to excise each one individually nor monitor all of them for malignant transformation.
Our case demonstrates the important role dermatologists may play in orchestrating care by a multispecialty team including oncology, radiation oncology, surgery, and plastic surgery. In our patient, a close examination of the literature by the dermatology team led to recognition of the potentially elevated risk for malignant transformation. The dermatology team also brought the case for review at the tumor board.
Although future studies are required to determine the relationship between AVL burden and the risk for progression to RIA, it is clear that a multidisciplinary approach and careful consideration of the current literature can prevent unnecessary morbidity and mortality for patients with this increasingly common problem.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol. 2008;32:943-950.
- Mandrell J, Mehta S, McClure S. Atypical vascular lesion of the breast. J Am Acad Dermatol. 2010;63:337-340.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Losch A, Chilek KD, Zirwas MJ. Post-radiation atypical vascular proliferation mimicking angiosarcoma eight months following breast-conserving therapy for breast carcinoma. J Clin Aesthet Dermatol. 2011;4:47-48.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological, immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Tahir M, Hendry P, Baird L, et al. Radiation induced angiosarcoma a sequela of radiotherapy for breast cancer following conservative surgery. Int Semin Surg Oncol. 2006;3:26.
- Hillenbrand T, Menge F, Hohenberger P, et al. Primary and secondary angiosarcomas: a comparative single-center analysis. Clin Sarcoma Res. 2015;5:14.
- Seinen JM, Styring E, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol. 2008;32:943-950.
- Mandrell J, Mehta S, McClure S. Atypical vascular lesion of the breast. J Am Acad Dermatol. 2010;63:337-340.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Losch A, Chilek KD, Zirwas MJ. Post-radiation atypical vascular proliferation mimicking angiosarcoma eight months following breast-conserving therapy for breast carcinoma. J Clin Aesthet Dermatol. 2011;4:47-48.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological, immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Tahir M, Hendry P, Baird L, et al. Radiation induced angiosarcoma a sequela of radiotherapy for breast cancer following conservative surgery. Int Semin Surg Oncol. 2006;3:26.
- Hillenbrand T, Menge F, Hohenberger P, et al. Primary and secondary angiosarcomas: a comparative single-center analysis. Clin Sarcoma Res. 2015;5:14.
- Seinen JM, Styring E, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
Practice Points
- Atypical vascular lesions (AVLs) of the breast have been reported in breast cancer patients following radiation treatment.
- Conservative breast cancer surgery and radiotherapy are becoming more prevalent for breast cancer treatment, thus the number of patients likely to present with AVLs has increased.
- Differentiation between AVLs and radiation-induced angiosarcomas (RIAs) can be challenging due to considerable histologic and clinical overlap; therefore, it is important for clinicians to be aware of the diagnosis and management of both AVLs and RIAs.
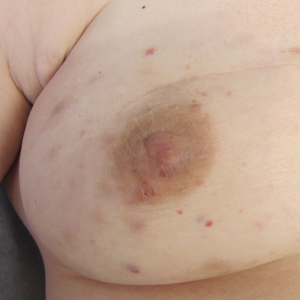
Wolf in sheep’s clothing: metatarsal osteosarcoma
Metatarsal bones are an unusual subsite for small bone involvement in osteosarcomas. This subgroup is often misdiagnosed and hence associated with significant treatment delays. The standard treatment of metatarsal osteosarcomas remains the same as for those treated at other sites, namely neoadjuvant chemotherapy followed by surgery and adjuvant chemotherapy. Limb salvage surgery or metatarsectomy in the foot is often a challenge owing to the poor compartmentalization of the disease. We hereby describe the case of a young girl with a metatarsal osteosarcoma who was managed with neoadjuvant chemotherapy and limb salvage surgery.
Introduction
Osteosarcomas are the most common primary malignant bone tumor in children and adolescents. Although predominantly occurring in pediatric and adolescent age groups, bimodal distribution (with a second incidence peak occurring in the sixth and seventh decades) is not uncommon.1 Osteosarcomas of the foot and small bones represent a rare and distinct clinical entity. This must have been a well-known observation for years that led to Watson-Jones stating, “Sarcoma of this [metatarsal] bone has not yet been reported in thousands of years in any country.”2 The incidence of osteosarcomas of the foot is estimated to be from 0.2% to 2%.3
These tumors, owing to their rarity, often lead to diagnostic dilemmas and hence treatment delays.4 They are usually mistaken for inflammatory conditions and often treated with—but not limited to—curettages and drainage procedures.5 The following case of osteosarcoma of the metatarsal bone in a young girl highlights the importance of having a high index of clinical suspicion prior to treatment.
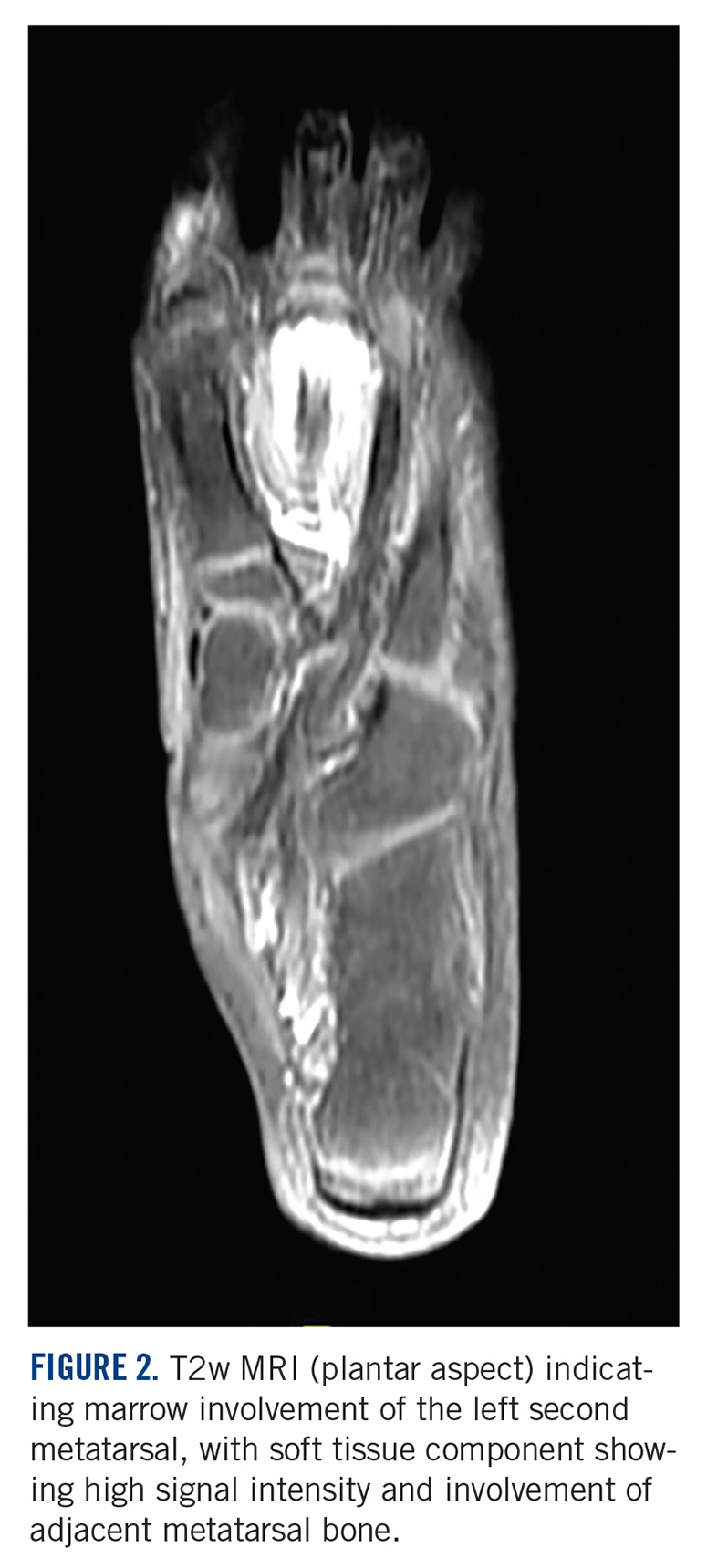
Case Presentation and Summary
A 10-year-old girl visited our outpatient clinic with a painful progressive swelling on the dorsum of the left foot of 2 months’ duration. There was no history of antecedent trauma or fever. Physical examination revealed a bony hard swelling measuring around 5 x 6 cm on the dorsum of the left foot around the region of the second metatarsal. There was no regional lymphadenopathy or distal neurovascular deficit. She was evaluated with a plain radiograph that demonstrated a lytic lesion in the left second metatarsal associated with cortical destruction and periosteal reaction (Figure 1). A subsequent magnetic resonance image (MRI) revealed a bony lesion destroying part of the left second metatarsal with cortical destruction and marrow involvement and affecting the soft tissue around the adjacent third metatarsal (Figure 2). Needle biopsy showed chondroblastic osteosarcoma. Computed tomography (CT) of the thorax and bone scan were both negative for distant metastases.
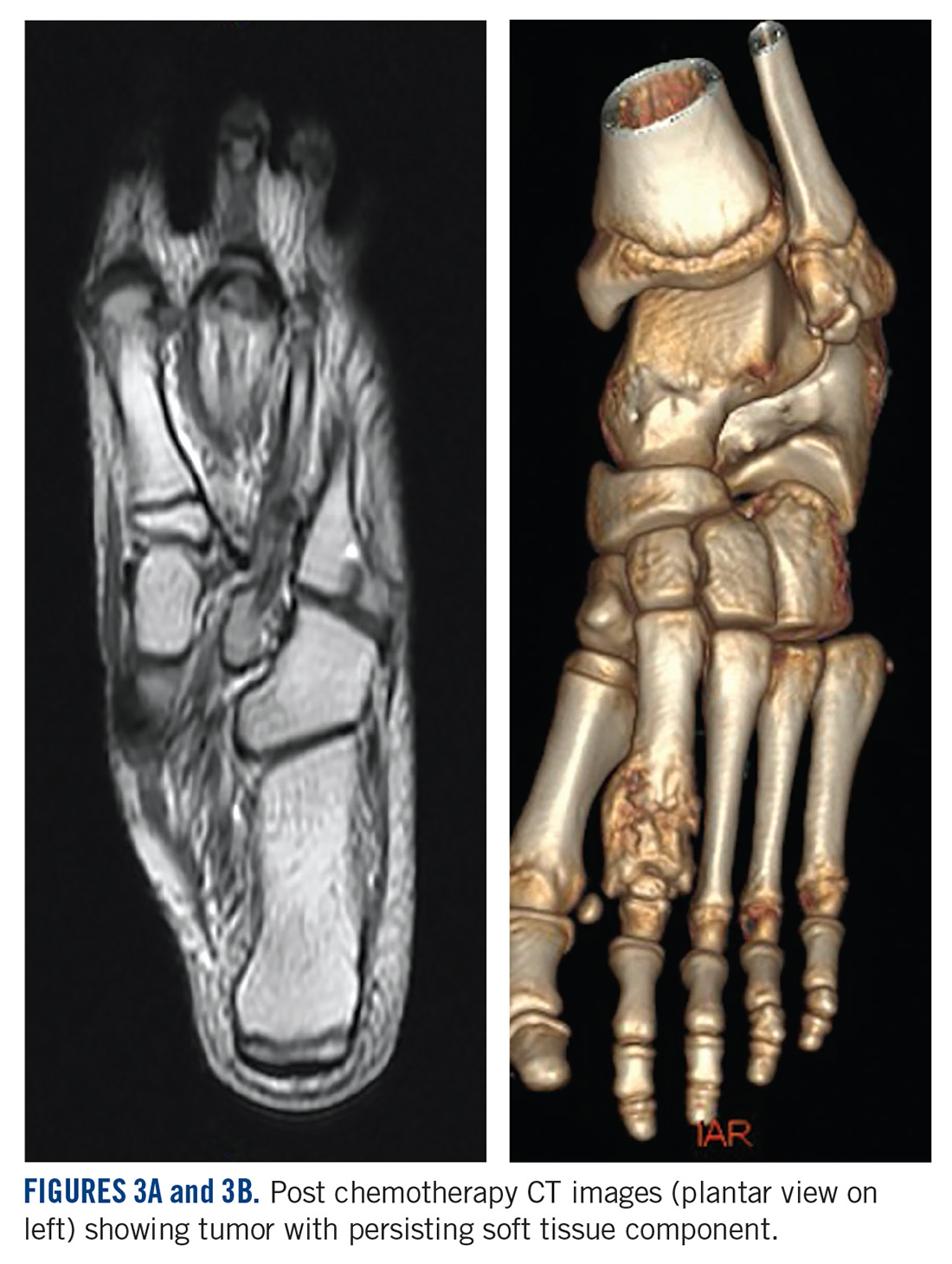
She received 3 cycles of a MAP (highdose methotrexate, doxorubicin, and cisplatin) regimen as neoadjuvant chemotherapy. Response assessment scans showed partial response (Figures 3A and B). We performed a wide excision of the second and third metatarsal with reconstruction using a segment of non-vascularized fibular graft as rigid fixation (Figure 4). The postoperative period was uneventful. She was able to begin partial weight bearing on the fourth postoperative day and her sutures were removed on the twelfth postoperative day. She received adjuvant chemotherapy following surgery. The final histopathology report showed residual disease with Huvos grade III response (>90% necrosis) with all margins negative for malignancy (Figure 5). At present, the child is disease-free at 5 months of treatment completion and is undergoing regular follow-up visits.
Discussion
Metatarsal involvement amongst smallbone osteosarcomas is uncommon.3 There are about 32 cases of osteosarcomas reported in the literature from 1940 to 2018 involving the metatarsal bones (Table 1). According to a review article from the Mayo Clinic, the most common bone of the foot involved is the calcaneum.6 While the incidence of osteosarcomas of the foot as a whole is around 0.2% to 2%,3 metatarsal involvement is documented in 0.5% of these patients.7 However, a recent study depicted metatarsal involvement in 33% of all osteosarcomas of the foot.8
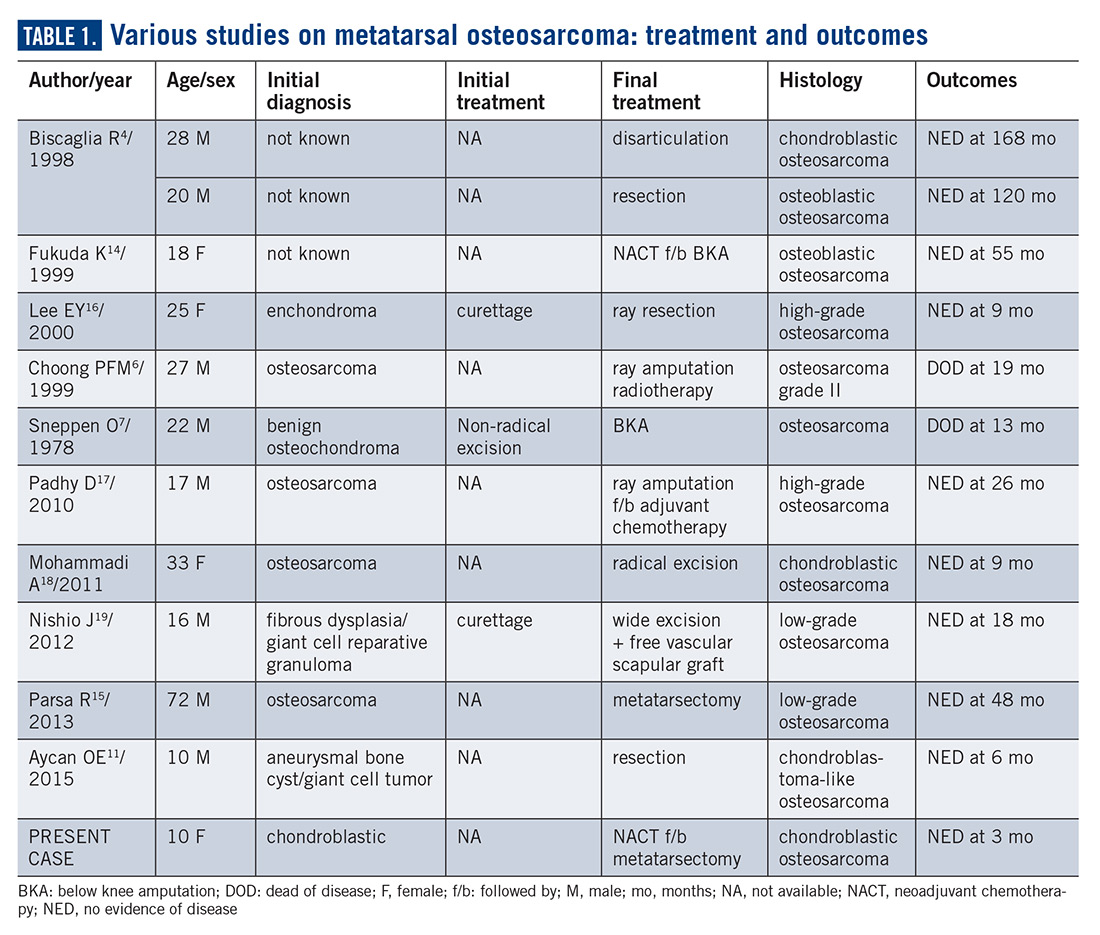
Osteosarcomas at conventional sites tend to have a bimodal age distribution with respect to disease affliction.9 Metatarsal osteosarcomas, however, are more common in an older age group.4,10 Our patient is probably the second youngest reported case of metatarsal osteosarcoma in the literature.11
Biscaglia et al propounded that osteosarcomas of the metatarsal were a distinct subgroup due to the rarity of occurrence, anatomical location, and prognosis.4 This often led to misdiagnosis and subsequent inadequate or inappropriate surgery. In six out of the ten cases (60%) described in Table 1, an incorrect pretreatment diagnosis was made that led to treatment delay. None, except one patient, received neoadjuvant chemotherapy, which is currently the standard of care. The average duration from symptom onset to diagnosis was found to be 2 years.4 However, in our case, the duration of symptoms was approximately 2 months.
Surgery for metatarsal osteosarcomas can be challenging, as the compartments of the foot are narrow spaces with poor demarcation. Limb salvage surgery in the form of metatarsectomy needs proper preoperative planning and execution. Neoadjuvant chemotherapy will serve to downstage the tumor within the fascial barriers of the metatarsal compartment.It has also been postulated that osteosarcoma of the foot may have a better prognosis and survival compared to other osteosarcoma subsites.10 This can be extrapolated from the fact that the majority are found to be low grade, and despite a long delay in treatment, there was no rapid increase in size and/or metastatic spread. However, tumor grade remains an important factor affecting survival— patients with higher grade tumors have worse survival.8
A number of differentials, including benign tumors, are to be kept in mind when diagnosing and treating such patients (Table 2). The most common benign tumors affecting the metatarsal are giant cell tumors (GCT) followed by chondromyxoid fibroma. Osteosarcomas and Ewing sarcomas constitute the malignant tumors.12 Occasionally, infections like osteomyelitis of the small bones may mimic malignancy. The absence of an extensive soft tissue component and/or calcifications with the presence of bony changes (like sequestrum) favors a diagnosis of infection/osteomyelitis. In addition, clinical findings like fever, skin redness, and presence of a painful swelling (especially after onset of fever) point to an inflammatory pathology rather than malignancy. Stress fractures rarely simulate tumors. MRI showing marrow and soft tissue edema with a visible fracture line points to the diagnosis.

A plane radiograph showing cortical bone destruction with a soft tissue component and calcification should be considered suspicious and must be thoroughly evaluated prior to surgical treatment.13 In a young patient such as ours, the important differentials that need to be considered include Ewing sarcoma, chronic osteomyelitis, and eosinophilic granuloma, which can radiologically mimic osteosarcoma at this location.
Conclusions
Osteosarcoma of the metatarsal is rare. Our case remains unique as it reports the second youngest patient in the literature. Erroneous or delayed diagnosis resulting in inadequate tumor excision and limb loss (amputation) often occurs in a majority of the cases. Proper pretreatment radiological imaging becomes imperative, and when clinical suspicion is high, a needle biopsy must follow in those cases. Early diagnosis with administration of neoadjuvant chemotherapy may allow us to perform limb salvage surgery or wide excision in these cases.
Acknowledgement
We would like to thank Dr. Sithara Aravind, Associate Professor, Department of Pathology, Malabar Cancer Center, for the photomicrographs.
1. Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3-13.
2. Watson-Jones R. Fractures and Joint Injuries. Vol. I, 4th ed. Edinburgh and London: E & S Livingstone Ltd.1960:347.
3. Wu KK. Osteogenic sarcoma of the tarsal navicular bone. J Foot Surg. 1989;28(4):363-369.
4. Biscaglia R, Gasbarrini A, Böhling T, Bacchini P, Bertoni F, Picci P. Osteosarcoma of the bones of the foot: an easily misdiagnosed malignant tumour. Mayo Clin Proc. 1998;73(9):842-847.
5. Kundu ZS, Gupta V, Sangwan SS, Rana P. Curettage of benign bone tumors and tumor like lesions: A retrospective analysis. Indian J Orthop. 2013;47(3):295-301.
6. Choong PFM, Qureshil AA, Sim FH, Unni KK. Osteosarcoma of the foot. A review of 52 patients at the Mayo Clinic. Acta Orthop Scand. 1999;70(4):361-364.
7. Sneppen O, Dissing I, Heerfordt J, Schiödt T. Osteosarcoma of the metatarsal bones: Review of the literature and report of a case. Acta Orthop Scand. 1978;49(2):220-223.
8. Anninga JK, Picci P, Fiocco M, et al. Osteosarcoma of the hands and feet: a distinct clinico-pathological subgroup. Virchows Arch. 2013;462(1):109-120.
9. Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, Epidemiology and End Results Program. Cancer.
2009;115(7):1531-1543.
10. Wang CW, Chen CY, Yang RS. Talar osteosarcoma treated with limb sparing surgery. J Bone Joint Surg Am. 2011;93:e22.
11. Aycan OE, Vanel D, Righi A, Arikan Y, Manfrini M. Chondroblastoma-like osteosarcoma:
a case report and review. Skeletal Radiol. 2015;44(6):869-873.
12. Jarkiewicz-Kochman E, Gołebiowski M, Swiatkowski J, Pacholec E, Rajewski R. Tumours of the metatarsus. Ortop Traumatol Rehabil. 2007;9(3):319-330.
13. Schatz J, Soper J, McCormack S, Healy M, Deady L, Brown W. Imaging of tumours in the ankle and foot. Top Magn Reson Imaging. 2010;21(1):37-50.
14. Fukuda K, Ushigome S, Nikaidou T, Asanuma K, Masui F. Osteosarcoma of the metatarsal. Skeletal Radiol. 1999;28(5):294-297.
15. Parsa R, Marcus M, Orlando R, Parsa C. Low-grade central osteosarcoma of the second metatarsal in a 72 year old male. Internet J Orthop Surg. 2013;21(2): 1-8.
16. Lee EY, Seeger LL, Nelson SD, Eckardt JJ. Primary osteosarcoma of a metatarsal bone. Skeletal Radiol. 2000;29(8):474-476.
17. Padhy D, Madhuri V, Pulimood SA, Danda S, Walter NM, Wang LL. Metatarsal osteosarcoma in Rothmund-Thomson syndrome: a case report. J Bone Joint
Surg Am. 2010;92(3):726-730.
18. Mohammadi A, Porghasem J, Noroozinia F, Ilkhanizadeh B, Ghasemi-Rad M, Khenari S. Periosteal osteosarcoma of the fifth metatarsal: A rare pedal tumor. J Foot Ankle Surg. 2011;50(5):620-622.
19. Nishio J, Iwasaki H, Takagi S, et al. Low-grade central osteosarcoma of the metatarsal bone: A clinicopathological, immunohistochemical, cytogenetic and molecular cytogenetic analysis. Anticancer Res. 2012;32(12):5429-5435.
Metatarsal bones are an unusual subsite for small bone involvement in osteosarcomas. This subgroup is often misdiagnosed and hence associated with significant treatment delays. The standard treatment of metatarsal osteosarcomas remains the same as for those treated at other sites, namely neoadjuvant chemotherapy followed by surgery and adjuvant chemotherapy. Limb salvage surgery or metatarsectomy in the foot is often a challenge owing to the poor compartmentalization of the disease. We hereby describe the case of a young girl with a metatarsal osteosarcoma who was managed with neoadjuvant chemotherapy and limb salvage surgery.
Introduction
Osteosarcomas are the most common primary malignant bone tumor in children and adolescents. Although predominantly occurring in pediatric and adolescent age groups, bimodal distribution (with a second incidence peak occurring in the sixth and seventh decades) is not uncommon.1 Osteosarcomas of the foot and small bones represent a rare and distinct clinical entity. This must have been a well-known observation for years that led to Watson-Jones stating, “Sarcoma of this [metatarsal] bone has not yet been reported in thousands of years in any country.”2 The incidence of osteosarcomas of the foot is estimated to be from 0.2% to 2%.3
These tumors, owing to their rarity, often lead to diagnostic dilemmas and hence treatment delays.4 They are usually mistaken for inflammatory conditions and often treated with—but not limited to—curettages and drainage procedures.5 The following case of osteosarcoma of the metatarsal bone in a young girl highlights the importance of having a high index of clinical suspicion prior to treatment.
Case Presentation and Summary
A 10-year-old girl visited our outpatient clinic with a painful progressive swelling on the dorsum of the left foot of 2 months’ duration. There was no history of antecedent trauma or fever. Physical examination revealed a bony hard swelling measuring around 5 x 6 cm on the dorsum of the left foot around the region of the second metatarsal. There was no regional lymphadenopathy or distal neurovascular deficit. She was evaluated with a plain radiograph that demonstrated a lytic lesion in the left second metatarsal associated with cortical destruction and periosteal reaction (Figure 1). A subsequent magnetic resonance image (MRI) revealed a bony lesion destroying part of the left second metatarsal with cortical destruction and marrow involvement and affecting the soft tissue around the adjacent third metatarsal (Figure 2). Needle biopsy showed chondroblastic osteosarcoma. Computed tomography (CT) of the thorax and bone scan were both negative for distant metastases.
She received 3 cycles of a MAP (highdose methotrexate, doxorubicin, and cisplatin) regimen as neoadjuvant chemotherapy. Response assessment scans showed partial response (Figures 3A and B). We performed a wide excision of the second and third metatarsal with reconstruction using a segment of non-vascularized fibular graft as rigid fixation (Figure 4). The postoperative period was uneventful. She was able to begin partial weight bearing on the fourth postoperative day and her sutures were removed on the twelfth postoperative day. She received adjuvant chemotherapy following surgery. The final histopathology report showed residual disease with Huvos grade III response (>90% necrosis) with all margins negative for malignancy (Figure 5). At present, the child is disease-free at 5 months of treatment completion and is undergoing regular follow-up visits.
Discussion
Metatarsal involvement amongst smallbone osteosarcomas is uncommon.3 There are about 32 cases of osteosarcomas reported in the literature from 1940 to 2018 involving the metatarsal bones (Table 1). According to a review article from the Mayo Clinic, the most common bone of the foot involved is the calcaneum.6 While the incidence of osteosarcomas of the foot as a whole is around 0.2% to 2%,3 metatarsal involvement is documented in 0.5% of these patients.7 However, a recent study depicted metatarsal involvement in 33% of all osteosarcomas of the foot.8
Osteosarcomas at conventional sites tend to have a bimodal age distribution with respect to disease affliction.9 Metatarsal osteosarcomas, however, are more common in an older age group.4,10 Our patient is probably the second youngest reported case of metatarsal osteosarcoma in the literature.11
Biscaglia et al propounded that osteosarcomas of the metatarsal were a distinct subgroup due to the rarity of occurrence, anatomical location, and prognosis.4 This often led to misdiagnosis and subsequent inadequate or inappropriate surgery. In six out of the ten cases (60%) described in Table 1, an incorrect pretreatment diagnosis was made that led to treatment delay. None, except one patient, received neoadjuvant chemotherapy, which is currently the standard of care. The average duration from symptom onset to diagnosis was found to be 2 years.4 However, in our case, the duration of symptoms was approximately 2 months.
Surgery for metatarsal osteosarcomas can be challenging, as the compartments of the foot are narrow spaces with poor demarcation. Limb salvage surgery in the form of metatarsectomy needs proper preoperative planning and execution. Neoadjuvant chemotherapy will serve to downstage the tumor within the fascial barriers of the metatarsal compartment.It has also been postulated that osteosarcoma of the foot may have a better prognosis and survival compared to other osteosarcoma subsites.10 This can be extrapolated from the fact that the majority are found to be low grade, and despite a long delay in treatment, there was no rapid increase in size and/or metastatic spread. However, tumor grade remains an important factor affecting survival— patients with higher grade tumors have worse survival.8
A number of differentials, including benign tumors, are to be kept in mind when diagnosing and treating such patients (Table 2). The most common benign tumors affecting the metatarsal are giant cell tumors (GCT) followed by chondromyxoid fibroma. Osteosarcomas and Ewing sarcomas constitute the malignant tumors.12 Occasionally, infections like osteomyelitis of the small bones may mimic malignancy. The absence of an extensive soft tissue component and/or calcifications with the presence of bony changes (like sequestrum) favors a diagnosis of infection/osteomyelitis. In addition, clinical findings like fever, skin redness, and presence of a painful swelling (especially after onset of fever) point to an inflammatory pathology rather than malignancy. Stress fractures rarely simulate tumors. MRI showing marrow and soft tissue edema with a visible fracture line points to the diagnosis.
A plane radiograph showing cortical bone destruction with a soft tissue component and calcification should be considered suspicious and must be thoroughly evaluated prior to surgical treatment.13 In a young patient such as ours, the important differentials that need to be considered include Ewing sarcoma, chronic osteomyelitis, and eosinophilic granuloma, which can radiologically mimic osteosarcoma at this location.
Conclusions
Osteosarcoma of the metatarsal is rare. Our case remains unique as it reports the second youngest patient in the literature. Erroneous or delayed diagnosis resulting in inadequate tumor excision and limb loss (amputation) often occurs in a majority of the cases. Proper pretreatment radiological imaging becomes imperative, and when clinical suspicion is high, a needle biopsy must follow in those cases. Early diagnosis with administration of neoadjuvant chemotherapy may allow us to perform limb salvage surgery or wide excision in these cases.
Acknowledgement
We would like to thank Dr. Sithara Aravind, Associate Professor, Department of Pathology, Malabar Cancer Center, for the photomicrographs.
Metatarsal bones are an unusual subsite for small bone involvement in osteosarcomas. This subgroup is often misdiagnosed and hence associated with significant treatment delays. The standard treatment of metatarsal osteosarcomas remains the same as for those treated at other sites, namely neoadjuvant chemotherapy followed by surgery and adjuvant chemotherapy. Limb salvage surgery or metatarsectomy in the foot is often a challenge owing to the poor compartmentalization of the disease. We hereby describe the case of a young girl with a metatarsal osteosarcoma who was managed with neoadjuvant chemotherapy and limb salvage surgery.
Introduction
Osteosarcomas are the most common primary malignant bone tumor in children and adolescents. Although predominantly occurring in pediatric and adolescent age groups, bimodal distribution (with a second incidence peak occurring in the sixth and seventh decades) is not uncommon.1 Osteosarcomas of the foot and small bones represent a rare and distinct clinical entity. This must have been a well-known observation for years that led to Watson-Jones stating, “Sarcoma of this [metatarsal] bone has not yet been reported in thousands of years in any country.”2 The incidence of osteosarcomas of the foot is estimated to be from 0.2% to 2%.3
These tumors, owing to their rarity, often lead to diagnostic dilemmas and hence treatment delays.4 They are usually mistaken for inflammatory conditions and often treated with—but not limited to—curettages and drainage procedures.5 The following case of osteosarcoma of the metatarsal bone in a young girl highlights the importance of having a high index of clinical suspicion prior to treatment.
Case Presentation and Summary
A 10-year-old girl visited our outpatient clinic with a painful progressive swelling on the dorsum of the left foot of 2 months’ duration. There was no history of antecedent trauma or fever. Physical examination revealed a bony hard swelling measuring around 5 x 6 cm on the dorsum of the left foot around the region of the second metatarsal. There was no regional lymphadenopathy or distal neurovascular deficit. She was evaluated with a plain radiograph that demonstrated a lytic lesion in the left second metatarsal associated with cortical destruction and periosteal reaction (Figure 1). A subsequent magnetic resonance image (MRI) revealed a bony lesion destroying part of the left second metatarsal with cortical destruction and marrow involvement and affecting the soft tissue around the adjacent third metatarsal (Figure 2). Needle biopsy showed chondroblastic osteosarcoma. Computed tomography (CT) of the thorax and bone scan were both negative for distant metastases.
She received 3 cycles of a MAP (highdose methotrexate, doxorubicin, and cisplatin) regimen as neoadjuvant chemotherapy. Response assessment scans showed partial response (Figures 3A and B). We performed a wide excision of the second and third metatarsal with reconstruction using a segment of non-vascularized fibular graft as rigid fixation (Figure 4). The postoperative period was uneventful. She was able to begin partial weight bearing on the fourth postoperative day and her sutures were removed on the twelfth postoperative day. She received adjuvant chemotherapy following surgery. The final histopathology report showed residual disease with Huvos grade III response (>90% necrosis) with all margins negative for malignancy (Figure 5). At present, the child is disease-free at 5 months of treatment completion and is undergoing regular follow-up visits.
Discussion
Metatarsal involvement amongst smallbone osteosarcomas is uncommon.3 There are about 32 cases of osteosarcomas reported in the literature from 1940 to 2018 involving the metatarsal bones (Table 1). According to a review article from the Mayo Clinic, the most common bone of the foot involved is the calcaneum.6 While the incidence of osteosarcomas of the foot as a whole is around 0.2% to 2%,3 metatarsal involvement is documented in 0.5% of these patients.7 However, a recent study depicted metatarsal involvement in 33% of all osteosarcomas of the foot.8
Osteosarcomas at conventional sites tend to have a bimodal age distribution with respect to disease affliction.9 Metatarsal osteosarcomas, however, are more common in an older age group.4,10 Our patient is probably the second youngest reported case of metatarsal osteosarcoma in the literature.11
Biscaglia et al propounded that osteosarcomas of the metatarsal were a distinct subgroup due to the rarity of occurrence, anatomical location, and prognosis.4 This often led to misdiagnosis and subsequent inadequate or inappropriate surgery. In six out of the ten cases (60%) described in Table 1, an incorrect pretreatment diagnosis was made that led to treatment delay. None, except one patient, received neoadjuvant chemotherapy, which is currently the standard of care. The average duration from symptom onset to diagnosis was found to be 2 years.4 However, in our case, the duration of symptoms was approximately 2 months.
Surgery for metatarsal osteosarcomas can be challenging, as the compartments of the foot are narrow spaces with poor demarcation. Limb salvage surgery in the form of metatarsectomy needs proper preoperative planning and execution. Neoadjuvant chemotherapy will serve to downstage the tumor within the fascial barriers of the metatarsal compartment.It has also been postulated that osteosarcoma of the foot may have a better prognosis and survival compared to other osteosarcoma subsites.10 This can be extrapolated from the fact that the majority are found to be low grade, and despite a long delay in treatment, there was no rapid increase in size and/or metastatic spread. However, tumor grade remains an important factor affecting survival— patients with higher grade tumors have worse survival.8
A number of differentials, including benign tumors, are to be kept in mind when diagnosing and treating such patients (Table 2). The most common benign tumors affecting the metatarsal are giant cell tumors (GCT) followed by chondromyxoid fibroma. Osteosarcomas and Ewing sarcomas constitute the malignant tumors.12 Occasionally, infections like osteomyelitis of the small bones may mimic malignancy. The absence of an extensive soft tissue component and/or calcifications with the presence of bony changes (like sequestrum) favors a diagnosis of infection/osteomyelitis. In addition, clinical findings like fever, skin redness, and presence of a painful swelling (especially after onset of fever) point to an inflammatory pathology rather than malignancy. Stress fractures rarely simulate tumors. MRI showing marrow and soft tissue edema with a visible fracture line points to the diagnosis.
A plane radiograph showing cortical bone destruction with a soft tissue component and calcification should be considered suspicious and must be thoroughly evaluated prior to surgical treatment.13 In a young patient such as ours, the important differentials that need to be considered include Ewing sarcoma, chronic osteomyelitis, and eosinophilic granuloma, which can radiologically mimic osteosarcoma at this location.
Conclusions
Osteosarcoma of the metatarsal is rare. Our case remains unique as it reports the second youngest patient in the literature. Erroneous or delayed diagnosis resulting in inadequate tumor excision and limb loss (amputation) often occurs in a majority of the cases. Proper pretreatment radiological imaging becomes imperative, and when clinical suspicion is high, a needle biopsy must follow in those cases. Early diagnosis with administration of neoadjuvant chemotherapy may allow us to perform limb salvage surgery or wide excision in these cases.
Acknowledgement
We would like to thank Dr. Sithara Aravind, Associate Professor, Department of Pathology, Malabar Cancer Center, for the photomicrographs.
1. Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3-13.
2. Watson-Jones R. Fractures and Joint Injuries. Vol. I, 4th ed. Edinburgh and London: E & S Livingstone Ltd.1960:347.
3. Wu KK. Osteogenic sarcoma of the tarsal navicular bone. J Foot Surg. 1989;28(4):363-369.
4. Biscaglia R, Gasbarrini A, Böhling T, Bacchini P, Bertoni F, Picci P. Osteosarcoma of the bones of the foot: an easily misdiagnosed malignant tumour. Mayo Clin Proc. 1998;73(9):842-847.
5. Kundu ZS, Gupta V, Sangwan SS, Rana P. Curettage of benign bone tumors and tumor like lesions: A retrospective analysis. Indian J Orthop. 2013;47(3):295-301.
6. Choong PFM, Qureshil AA, Sim FH, Unni KK. Osteosarcoma of the foot. A review of 52 patients at the Mayo Clinic. Acta Orthop Scand. 1999;70(4):361-364.
7. Sneppen O, Dissing I, Heerfordt J, Schiödt T. Osteosarcoma of the metatarsal bones: Review of the literature and report of a case. Acta Orthop Scand. 1978;49(2):220-223.
8. Anninga JK, Picci P, Fiocco M, et al. Osteosarcoma of the hands and feet: a distinct clinico-pathological subgroup. Virchows Arch. 2013;462(1):109-120.
9. Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, Epidemiology and End Results Program. Cancer.
2009;115(7):1531-1543.
10. Wang CW, Chen CY, Yang RS. Talar osteosarcoma treated with limb sparing surgery. J Bone Joint Surg Am. 2011;93:e22.
11. Aycan OE, Vanel D, Righi A, Arikan Y, Manfrini M. Chondroblastoma-like osteosarcoma:
a case report and review. Skeletal Radiol. 2015;44(6):869-873.
12. Jarkiewicz-Kochman E, Gołebiowski M, Swiatkowski J, Pacholec E, Rajewski R. Tumours of the metatarsus. Ortop Traumatol Rehabil. 2007;9(3):319-330.
13. Schatz J, Soper J, McCormack S, Healy M, Deady L, Brown W. Imaging of tumours in the ankle and foot. Top Magn Reson Imaging. 2010;21(1):37-50.
14. Fukuda K, Ushigome S, Nikaidou T, Asanuma K, Masui F. Osteosarcoma of the metatarsal. Skeletal Radiol. 1999;28(5):294-297.
15. Parsa R, Marcus M, Orlando R, Parsa C. Low-grade central osteosarcoma of the second metatarsal in a 72 year old male. Internet J Orthop Surg. 2013;21(2): 1-8.
16. Lee EY, Seeger LL, Nelson SD, Eckardt JJ. Primary osteosarcoma of a metatarsal bone. Skeletal Radiol. 2000;29(8):474-476.
17. Padhy D, Madhuri V, Pulimood SA, Danda S, Walter NM, Wang LL. Metatarsal osteosarcoma in Rothmund-Thomson syndrome: a case report. J Bone Joint
Surg Am. 2010;92(3):726-730.
18. Mohammadi A, Porghasem J, Noroozinia F, Ilkhanizadeh B, Ghasemi-Rad M, Khenari S. Periosteal osteosarcoma of the fifth metatarsal: A rare pedal tumor. J Foot Ankle Surg. 2011;50(5):620-622.
19. Nishio J, Iwasaki H, Takagi S, et al. Low-grade central osteosarcoma of the metatarsal bone: A clinicopathological, immunohistochemical, cytogenetic and molecular cytogenetic analysis. Anticancer Res. 2012;32(12):5429-5435.
1. Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3-13.
2. Watson-Jones R. Fractures and Joint Injuries. Vol. I, 4th ed. Edinburgh and London: E & S Livingstone Ltd.1960:347.
3. Wu KK. Osteogenic sarcoma of the tarsal navicular bone. J Foot Surg. 1989;28(4):363-369.
4. Biscaglia R, Gasbarrini A, Böhling T, Bacchini P, Bertoni F, Picci P. Osteosarcoma of the bones of the foot: an easily misdiagnosed malignant tumour. Mayo Clin Proc. 1998;73(9):842-847.
5. Kundu ZS, Gupta V, Sangwan SS, Rana P. Curettage of benign bone tumors and tumor like lesions: A retrospective analysis. Indian J Orthop. 2013;47(3):295-301.
6. Choong PFM, Qureshil AA, Sim FH, Unni KK. Osteosarcoma of the foot. A review of 52 patients at the Mayo Clinic. Acta Orthop Scand. 1999;70(4):361-364.
7. Sneppen O, Dissing I, Heerfordt J, Schiödt T. Osteosarcoma of the metatarsal bones: Review of the literature and report of a case. Acta Orthop Scand. 1978;49(2):220-223.
8. Anninga JK, Picci P, Fiocco M, et al. Osteosarcoma of the hands and feet: a distinct clinico-pathological subgroup. Virchows Arch. 2013;462(1):109-120.
9. Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, Epidemiology and End Results Program. Cancer.
2009;115(7):1531-1543.
10. Wang CW, Chen CY, Yang RS. Talar osteosarcoma treated with limb sparing surgery. J Bone Joint Surg Am. 2011;93:e22.
11. Aycan OE, Vanel D, Righi A, Arikan Y, Manfrini M. Chondroblastoma-like osteosarcoma:
a case report and review. Skeletal Radiol. 2015;44(6):869-873.
12. Jarkiewicz-Kochman E, Gołebiowski M, Swiatkowski J, Pacholec E, Rajewski R. Tumours of the metatarsus. Ortop Traumatol Rehabil. 2007;9(3):319-330.
13. Schatz J, Soper J, McCormack S, Healy M, Deady L, Brown W. Imaging of tumours in the ankle and foot. Top Magn Reson Imaging. 2010;21(1):37-50.
14. Fukuda K, Ushigome S, Nikaidou T, Asanuma K, Masui F. Osteosarcoma of the metatarsal. Skeletal Radiol. 1999;28(5):294-297.
15. Parsa R, Marcus M, Orlando R, Parsa C. Low-grade central osteosarcoma of the second metatarsal in a 72 year old male. Internet J Orthop Surg. 2013;21(2): 1-8.
16. Lee EY, Seeger LL, Nelson SD, Eckardt JJ. Primary osteosarcoma of a metatarsal bone. Skeletal Radiol. 2000;29(8):474-476.
17. Padhy D, Madhuri V, Pulimood SA, Danda S, Walter NM, Wang LL. Metatarsal osteosarcoma in Rothmund-Thomson syndrome: a case report. J Bone Joint
Surg Am. 2010;92(3):726-730.
18. Mohammadi A, Porghasem J, Noroozinia F, Ilkhanizadeh B, Ghasemi-Rad M, Khenari S. Periosteal osteosarcoma of the fifth metatarsal: A rare pedal tumor. J Foot Ankle Surg. 2011;50(5):620-622.
19. Nishio J, Iwasaki H, Takagi S, et al. Low-grade central osteosarcoma of the metatarsal bone: A clinicopathological, immunohistochemical, cytogenetic and molecular cytogenetic analysis. Anticancer Res. 2012;32(12):5429-5435.
An unusual presentation of low-grade clavicle osteosarcoma: a case report and literature review
Osteosarcoma (OS) is a rare disease with approximately 800- 900 newly diagnosed cases each year in the United States. Of those, the majority occur about the knee. The distal femur is the most common site, followed by the proximal tibia, with the proximal humerus being a distant third. OS of the clavicle has been reported, with the earliest case report dating from 1975.1 Since then, additional case reports of high-grade OS of the clavicle have been published.2,3 We describe the case of a 16-year-old female who presented with a mass on her right medial clavicle, which was confirmed to be a low-grade central OS.
Case Presentation
The patient is a 16-year-old female who presented to the Emergency Department (ED) for evaluation of a mass on her right clavicle, after being evaluated by her primary care physician (PCP). She noted an enlarging mass over the previous 2 months but stated that it had been asymptomatic until 4 days prior to presentation to her PCP, at which time she had developed tenderness to palpation and pain with range of motion of the right arm. X-rays were obtained at the PCP’s office and she was referred to the ED for further evaluation. She denied constitutional symptoms.
At the ED visit, she was noted to have an area of erythema and tenderness over the medial aspect of the right clavicle with increased bony prominence. A chest x-ray demonstrated medial clavicle enlargement with periosteal reaction and sclerosis (Figure 1).
MRI demonstrated a 6-cm x 3.8-cm x 4.1-cm mass arising from the right medial clavicle with cortical destruction and concomitant displacement of the right subclavian and brachiocephalic veins (Figure 2). A CT-guided biopsy was performed 1 week later and demonstrated low-grade OS. The pathologist was concerned about the possibility of sampling error and the presence of a higher-grade component, as low-grade OS of the clavicle had not been reported.
The patient was evaluated by a pediatric hematologist/oncologist 2 weeks later after having obtained the biopsy and a PET/CT scan. At that time, the PET/CT showed an FDG-avid mass at the clavicle without evidence of pulmonary metastatic disease (Figure 3). She was subsequently evaluated by orthopedic oncology, at which time a discussion was had regarding further treatment. There was essentially no literature to guide the surgical and medical teams, as low-grade clavicular OS is unknown. Based on the evidence of localized, low-grade disease, the determination was made to proceedwith surgical resection. In the event that high-grade disease was identified at the time of final pathological evaluation, the pediatric hematology/oncology team felt that administering all of the patient’s chemotherapy postoperatively would be acceptable and not affect her long-term prognosis. CT and CT angiogram were obtained for further operative planning (Figure 4).

Given the intimacy of the mass to the subclavian vessels, she was also seen preoperatively by pediatric general and cardiothoracic surgeons. The plan was formulated to have them in the operating room for mobilization of the subclavian vessels and in the event that a sternotomy was required for proximal control of the vessels. Following this visit, the case was discussed at the multidisciplinary pediatric tumor board and the consensus was to proceed with surgical resection.
Surgical Technique
General endotracheal anesthesia was administered without complication. The patient was positioned supine with a soft bump under her shoulders to place her neck in slight extension and thus facilitate access to the clavicle and great vessels. A 14-cm oblique incision was made over the subcutaneous clavicle extending to the contralateral sternoclavicular joint. Dissection was carried down to the fascia and the biopsy site was excised with the skin paddle. Dissection was carried through the sternocleidomastoid superiorly and the pectoralis major inferiorly, to 8 cm lateral from the right sternoclavicular joint. The clavicle was osteotomized well lateral of the palpable tumor and a marrow margin was sent for frozen section, which was found to be negative.
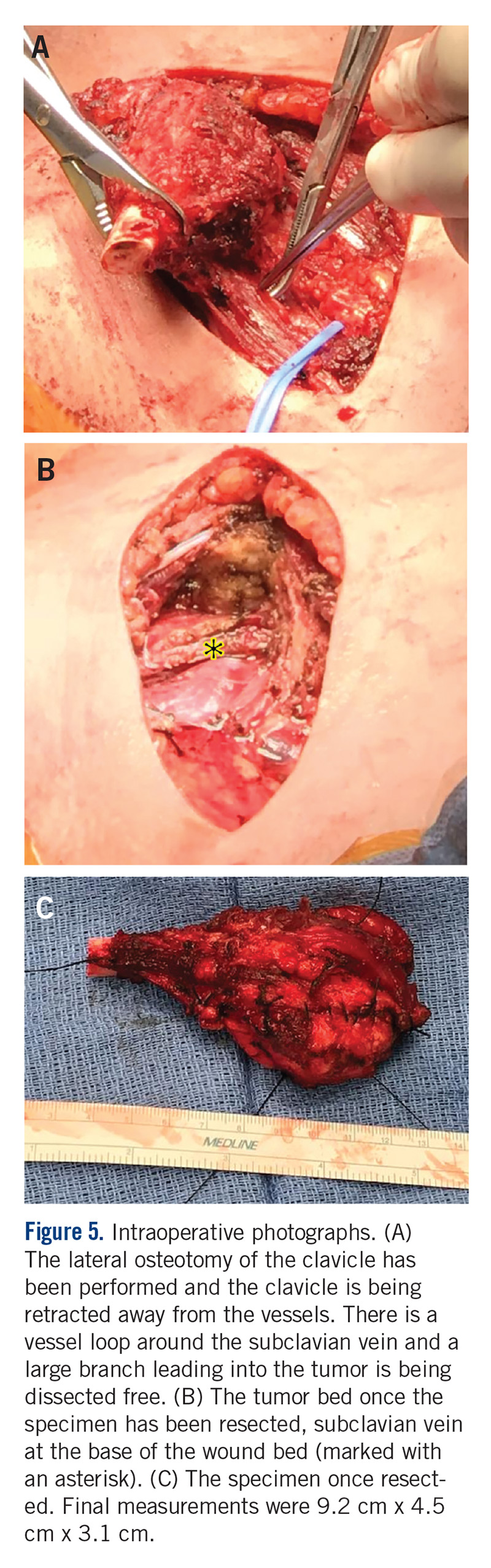
Dissection was continued circumferentially. Assistance from pediatric general and cardiothoracic surgery was required at the inferior aspect of the mass to assist with exposure and control of the subclavian vein (Figure 5A). A large branch of the subclavian vein near its junction with the internal jugular vein was found to be involved with the tumor and thus required suture ligation. The subclavian vein was noted to be intimate with the mass and somewhat friable. With the vein mobilized, a cuff of normal tissue was obtained inferiorly and superiorly to the mass. Medially, the sternoclavicular joint was disarticulated (Figure 5B). At this point, the specimen was delivered from the operative field and tagged in the usual fashion (Figure 5C). A medial soft tissue margin from the sternal side of the sternoclavicular joint was also sent and found to be negative for tumor. The wound was closed in layered fashion over a ¼” Penrose drain. A soft dressing was placed, and the patient was successfully extubated and transferred to the post-anesthesia care unit in stable condition.
Postoperative Course
The patient was found to be neurologically and vascularly intact on postoperative exam and was discharged on postoperative day 1.
She was seen 14 days postoperatively and was doing well at that time, with full range of motion of the shoulder, elbow, wrist, and hand. Final pathology confirmed a low-grade OS with extraosseous extension. All margins were negative except the medial (sternoclavicular joint) margin and the inferior margin adjacent to the subclavian vein. The intraoperative frozen section from the medial margin was negative for tumor.
The pediatric hematology/oncology team determined that, as no high-grade areas were identified, chemotherapy should be deferred. The positive margins were also discussed with the patient and her family specifically regarding further possible treatments. The findings from the pathology were discussed in a multidisciplinary tumor board and it was felt that, given the low-grade nature of the lesion as well as the high morbidity and risk of mortality with further surgery, additional surgery would be potentially more harmful than helpful. Additionally, low-grade OS is extremely resistant to radiotherapy. The plan remains to monitor her for local recurrence as well as metastases with serial imaging.
Discussion
The clavicle is one of the first bones in the body to ossify but one of the last to have final physeal closure. Its unique characteristics have led to various descriptions, such as a “short tubular bone” versus a “flat bone.”4,5 Of note are its paucity of a true intramedullary space and scanty red marrow, which make it an unlikely site for a primarily intramedullary- based neoplasm to arise.4 However, it has also been noted that malignant lesions are more common in the clavicle than benign lesions, and special attention should be paid to aggressiveappearing lesions in the clavicle.
Radiographs can be misleading as well. Prior studies have demonstrated that low-grade central OS can be readily misdiagnosed as fibrous dysplasia, desmoplastic fibroma, nonossifying fibroma, osteoblastoma, and aneurysmal bone cyst.6 Findings found in low-grade OS can include evidence of cortical interruption, local soft tissue mass development, intramedullary involvement, cortical destruction, and poor margination; however, low-grade OS is typically sclerotic and highly trabeculated. Cross-sectional imaging can help differentiate between OS and other more benign pathologies and should be considered in the clavicle where biopsy may be perilous.5
The difficulty of clavicular biopsy has been reported. Not only does clavicular anatomy make biopsy hazardous, but also the potential for sampling error does exist. In a case report of one patient with a highgrade lesion, fine needle aspiration biopsy was initially diagnosed as an aneurysmal bone cyst but was ultimately found to be osteosarcoma.2 Histology of low-grade lesions usually demonstrates minimal cytological atypia, rare mitotic activity, and variable osteoid production.5 Lower mitotic indices typically make wide resection curative for these patients, without the need for chemotherapy.
In this case, wide resection was carried out with the subclavian vein as the posterior-inferior margin and the sternoclavicular joint as the medial margin. Though the intra-operative medial margin was clear of disease, final pathology demonstrated focal (microscopic) involvement of the posterior and medial margins. A study of soft tissue sarcoma evaluated positive margins and concluded that the imperative of preservation of vital structures supersedes the need for negative margins.7,8 The rate of metastasis and overall survival was similar to surgical resections with positive margins. In the case of our patient, further resection would have carried significant morbidity and possibly mortality, including sacrifice of the major vessels to the arm below and entering into the sternum and thoracic cavity. The likely disability as well as the hazards of surgery were deemed to be too great to justify further excision. Frequent cross-sectional imaging will be necessary to evaluate the presence of recurrent or metastatic disease. To our knowledge, this is the first documented case of low-grade clavicle OS. This report demonstrates the need for multidisciplinary sarcoma care at a center of excellence, particularly in instances of unusual diagnoses.
1. Zinghi G. Osteosarcoma of the clavicle (description of a case) [in Italian]. Chir Organi Mov. 1975;62(6):671-674.
2. Cundy WJ, Carter C, Dhatrak D, Clayer M. Primary osteosarcoma of the clavicle and the perils of bone biopsy. BMJ Case Rep. 2015;2015:bcr2014208859.
3. Greenspan A, Unni KK, Mann J. Case report 804: Chondroblastic osteosarcoma grade 3 of the left clavicle. Skeletal Radiol. 1993;22(6):469-471.
4. Rossi B, Fabbriciani C, Chalidis BE, Visci F, Maccauro G. Primary malignant clavicular tumours: a clinicopathological analysis of six cases and evaluation of surgical management. Arch Orthop Trauma Surg. 2011;131(7):935-939.
5. Andresen KJ, Sundaram M, Unni KK, Sim FH. Imaging features of low-grade central osteosarcoma of the long bones and pelvis. Skeletal Radiol. 2004;33(7):373-379.
6. Malhas AM, Sumathi VP, James SL, et al. Low-grade central osteosarcoma: A difficult condition to diagnose. Sarcoma. 2012; 2012:764796.
7. O’Donnell PW, Griffin AM, Eward WC, et al. The effect of the setting of a positive surgical margin in soft tissue sarcoma. Cancer. 2014;120(18):2866-2875.
8. Kawaguchi N, Ahmed AR, Matsumoto S, Manabe J, Matsushita Y. The concept of curative margin in surgery for bone and soft tissue sarcoma. Clin Orthop Relat Res. 2004;419:165-172.
Osteosarcoma (OS) is a rare disease with approximately 800- 900 newly diagnosed cases each year in the United States. Of those, the majority occur about the knee. The distal femur is the most common site, followed by the proximal tibia, with the proximal humerus being a distant third. OS of the clavicle has been reported, with the earliest case report dating from 1975.1 Since then, additional case reports of high-grade OS of the clavicle have been published.2,3 We describe the case of a 16-year-old female who presented with a mass on her right medial clavicle, which was confirmed to be a low-grade central OS.
Case Presentation
The patient is a 16-year-old female who presented to the Emergency Department (ED) for evaluation of a mass on her right clavicle, after being evaluated by her primary care physician (PCP). She noted an enlarging mass over the previous 2 months but stated that it had been asymptomatic until 4 days prior to presentation to her PCP, at which time she had developed tenderness to palpation and pain with range of motion of the right arm. X-rays were obtained at the PCP’s office and she was referred to the ED for further evaluation. She denied constitutional symptoms.
At the ED visit, she was noted to have an area of erythema and tenderness over the medial aspect of the right clavicle with increased bony prominence. A chest x-ray demonstrated medial clavicle enlargement with periosteal reaction and sclerosis (Figure 1).
MRI demonstrated a 6-cm x 3.8-cm x 4.1-cm mass arising from the right medial clavicle with cortical destruction and concomitant displacement of the right subclavian and brachiocephalic veins (Figure 2). A CT-guided biopsy was performed 1 week later and demonstrated low-grade OS. The pathologist was concerned about the possibility of sampling error and the presence of a higher-grade component, as low-grade OS of the clavicle had not been reported.
The patient was evaluated by a pediatric hematologist/oncologist 2 weeks later after having obtained the biopsy and a PET/CT scan. At that time, the PET/CT showed an FDG-avid mass at the clavicle without evidence of pulmonary metastatic disease (Figure 3). She was subsequently evaluated by orthopedic oncology, at which time a discussion was had regarding further treatment. There was essentially no literature to guide the surgical and medical teams, as low-grade clavicular OS is unknown. Based on the evidence of localized, low-grade disease, the determination was made to proceedwith surgical resection. In the event that high-grade disease was identified at the time of final pathological evaluation, the pediatric hematology/oncology team felt that administering all of the patient’s chemotherapy postoperatively would be acceptable and not affect her long-term prognosis. CT and CT angiogram were obtained for further operative planning (Figure 4).
Given the intimacy of the mass to the subclavian vessels, she was also seen preoperatively by pediatric general and cardiothoracic surgeons. The plan was formulated to have them in the operating room for mobilization of the subclavian vessels and in the event that a sternotomy was required for proximal control of the vessels. Following this visit, the case was discussed at the multidisciplinary pediatric tumor board and the consensus was to proceed with surgical resection.
Surgical Technique
General endotracheal anesthesia was administered without complication. The patient was positioned supine with a soft bump under her shoulders to place her neck in slight extension and thus facilitate access to the clavicle and great vessels. A 14-cm oblique incision was made over the subcutaneous clavicle extending to the contralateral sternoclavicular joint. Dissection was carried down to the fascia and the biopsy site was excised with the skin paddle. Dissection was carried through the sternocleidomastoid superiorly and the pectoralis major inferiorly, to 8 cm lateral from the right sternoclavicular joint. The clavicle was osteotomized well lateral of the palpable tumor and a marrow margin was sent for frozen section, which was found to be negative.
Dissection was continued circumferentially. Assistance from pediatric general and cardiothoracic surgery was required at the inferior aspect of the mass to assist with exposure and control of the subclavian vein (Figure 5A). A large branch of the subclavian vein near its junction with the internal jugular vein was found to be involved with the tumor and thus required suture ligation. The subclavian vein was noted to be intimate with the mass and somewhat friable. With the vein mobilized, a cuff of normal tissue was obtained inferiorly and superiorly to the mass. Medially, the sternoclavicular joint was disarticulated (Figure 5B). At this point, the specimen was delivered from the operative field and tagged in the usual fashion (Figure 5C). A medial soft tissue margin from the sternal side of the sternoclavicular joint was also sent and found to be negative for tumor. The wound was closed in layered fashion over a ¼” Penrose drain. A soft dressing was placed, and the patient was successfully extubated and transferred to the post-anesthesia care unit in stable condition.
Postoperative Course
The patient was found to be neurologically and vascularly intact on postoperative exam and was discharged on postoperative day 1.
She was seen 14 days postoperatively and was doing well at that time, with full range of motion of the shoulder, elbow, wrist, and hand. Final pathology confirmed a low-grade OS with extraosseous extension. All margins were negative except the medial (sternoclavicular joint) margin and the inferior margin adjacent to the subclavian vein. The intraoperative frozen section from the medial margin was negative for tumor.
The pediatric hematology/oncology team determined that, as no high-grade areas were identified, chemotherapy should be deferred. The positive margins were also discussed with the patient and her family specifically regarding further possible treatments. The findings from the pathology were discussed in a multidisciplinary tumor board and it was felt that, given the low-grade nature of the lesion as well as the high morbidity and risk of mortality with further surgery, additional surgery would be potentially more harmful than helpful. Additionally, low-grade OS is extremely resistant to radiotherapy. The plan remains to monitor her for local recurrence as well as metastases with serial imaging.
Discussion
The clavicle is one of the first bones in the body to ossify but one of the last to have final physeal closure. Its unique characteristics have led to various descriptions, such as a “short tubular bone” versus a “flat bone.”4,5 Of note are its paucity of a true intramedullary space and scanty red marrow, which make it an unlikely site for a primarily intramedullary- based neoplasm to arise.4 However, it has also been noted that malignant lesions are more common in the clavicle than benign lesions, and special attention should be paid to aggressiveappearing lesions in the clavicle.
Radiographs can be misleading as well. Prior studies have demonstrated that low-grade central OS can be readily misdiagnosed as fibrous dysplasia, desmoplastic fibroma, nonossifying fibroma, osteoblastoma, and aneurysmal bone cyst.6 Findings found in low-grade OS can include evidence of cortical interruption, local soft tissue mass development, intramedullary involvement, cortical destruction, and poor margination; however, low-grade OS is typically sclerotic and highly trabeculated. Cross-sectional imaging can help differentiate between OS and other more benign pathologies and should be considered in the clavicle where biopsy may be perilous.5
The difficulty of clavicular biopsy has been reported. Not only does clavicular anatomy make biopsy hazardous, but also the potential for sampling error does exist. In a case report of one patient with a highgrade lesion, fine needle aspiration biopsy was initially diagnosed as an aneurysmal bone cyst but was ultimately found to be osteosarcoma.2 Histology of low-grade lesions usually demonstrates minimal cytological atypia, rare mitotic activity, and variable osteoid production.5 Lower mitotic indices typically make wide resection curative for these patients, without the need for chemotherapy.
In this case, wide resection was carried out with the subclavian vein as the posterior-inferior margin and the sternoclavicular joint as the medial margin. Though the intra-operative medial margin was clear of disease, final pathology demonstrated focal (microscopic) involvement of the posterior and medial margins. A study of soft tissue sarcoma evaluated positive margins and concluded that the imperative of preservation of vital structures supersedes the need for negative margins.7,8 The rate of metastasis and overall survival was similar to surgical resections with positive margins. In the case of our patient, further resection would have carried significant morbidity and possibly mortality, including sacrifice of the major vessels to the arm below and entering into the sternum and thoracic cavity. The likely disability as well as the hazards of surgery were deemed to be too great to justify further excision. Frequent cross-sectional imaging will be necessary to evaluate the presence of recurrent or metastatic disease. To our knowledge, this is the first documented case of low-grade clavicle OS. This report demonstrates the need for multidisciplinary sarcoma care at a center of excellence, particularly in instances of unusual diagnoses.
Osteosarcoma (OS) is a rare disease with approximately 800- 900 newly diagnosed cases each year in the United States. Of those, the majority occur about the knee. The distal femur is the most common site, followed by the proximal tibia, with the proximal humerus being a distant third. OS of the clavicle has been reported, with the earliest case report dating from 1975.1 Since then, additional case reports of high-grade OS of the clavicle have been published.2,3 We describe the case of a 16-year-old female who presented with a mass on her right medial clavicle, which was confirmed to be a low-grade central OS.
Case Presentation
The patient is a 16-year-old female who presented to the Emergency Department (ED) for evaluation of a mass on her right clavicle, after being evaluated by her primary care physician (PCP). She noted an enlarging mass over the previous 2 months but stated that it had been asymptomatic until 4 days prior to presentation to her PCP, at which time she had developed tenderness to palpation and pain with range of motion of the right arm. X-rays were obtained at the PCP’s office and she was referred to the ED for further evaluation. She denied constitutional symptoms.
At the ED visit, she was noted to have an area of erythema and tenderness over the medial aspect of the right clavicle with increased bony prominence. A chest x-ray demonstrated medial clavicle enlargement with periosteal reaction and sclerosis (Figure 1).
MRI demonstrated a 6-cm x 3.8-cm x 4.1-cm mass arising from the right medial clavicle with cortical destruction and concomitant displacement of the right subclavian and brachiocephalic veins (Figure 2). A CT-guided biopsy was performed 1 week later and demonstrated low-grade OS. The pathologist was concerned about the possibility of sampling error and the presence of a higher-grade component, as low-grade OS of the clavicle had not been reported.
The patient was evaluated by a pediatric hematologist/oncologist 2 weeks later after having obtained the biopsy and a PET/CT scan. At that time, the PET/CT showed an FDG-avid mass at the clavicle without evidence of pulmonary metastatic disease (Figure 3). She was subsequently evaluated by orthopedic oncology, at which time a discussion was had regarding further treatment. There was essentially no literature to guide the surgical and medical teams, as low-grade clavicular OS is unknown. Based on the evidence of localized, low-grade disease, the determination was made to proceedwith surgical resection. In the event that high-grade disease was identified at the time of final pathological evaluation, the pediatric hematology/oncology team felt that administering all of the patient’s chemotherapy postoperatively would be acceptable and not affect her long-term prognosis. CT and CT angiogram were obtained for further operative planning (Figure 4).
Given the intimacy of the mass to the subclavian vessels, she was also seen preoperatively by pediatric general and cardiothoracic surgeons. The plan was formulated to have them in the operating room for mobilization of the subclavian vessels and in the event that a sternotomy was required for proximal control of the vessels. Following this visit, the case was discussed at the multidisciplinary pediatric tumor board and the consensus was to proceed with surgical resection.
Surgical Technique
General endotracheal anesthesia was administered without complication. The patient was positioned supine with a soft bump under her shoulders to place her neck in slight extension and thus facilitate access to the clavicle and great vessels. A 14-cm oblique incision was made over the subcutaneous clavicle extending to the contralateral sternoclavicular joint. Dissection was carried down to the fascia and the biopsy site was excised with the skin paddle. Dissection was carried through the sternocleidomastoid superiorly and the pectoralis major inferiorly, to 8 cm lateral from the right sternoclavicular joint. The clavicle was osteotomized well lateral of the palpable tumor and a marrow margin was sent for frozen section, which was found to be negative.
Dissection was continued circumferentially. Assistance from pediatric general and cardiothoracic surgery was required at the inferior aspect of the mass to assist with exposure and control of the subclavian vein (Figure 5A). A large branch of the subclavian vein near its junction with the internal jugular vein was found to be involved with the tumor and thus required suture ligation. The subclavian vein was noted to be intimate with the mass and somewhat friable. With the vein mobilized, a cuff of normal tissue was obtained inferiorly and superiorly to the mass. Medially, the sternoclavicular joint was disarticulated (Figure 5B). At this point, the specimen was delivered from the operative field and tagged in the usual fashion (Figure 5C). A medial soft tissue margin from the sternal side of the sternoclavicular joint was also sent and found to be negative for tumor. The wound was closed in layered fashion over a ¼” Penrose drain. A soft dressing was placed, and the patient was successfully extubated and transferred to the post-anesthesia care unit in stable condition.
Postoperative Course
The patient was found to be neurologically and vascularly intact on postoperative exam and was discharged on postoperative day 1.
She was seen 14 days postoperatively and was doing well at that time, with full range of motion of the shoulder, elbow, wrist, and hand. Final pathology confirmed a low-grade OS with extraosseous extension. All margins were negative except the medial (sternoclavicular joint) margin and the inferior margin adjacent to the subclavian vein. The intraoperative frozen section from the medial margin was negative for tumor.
The pediatric hematology/oncology team determined that, as no high-grade areas were identified, chemotherapy should be deferred. The positive margins were also discussed with the patient and her family specifically regarding further possible treatments. The findings from the pathology were discussed in a multidisciplinary tumor board and it was felt that, given the low-grade nature of the lesion as well as the high morbidity and risk of mortality with further surgery, additional surgery would be potentially more harmful than helpful. Additionally, low-grade OS is extremely resistant to radiotherapy. The plan remains to monitor her for local recurrence as well as metastases with serial imaging.
Discussion
The clavicle is one of the first bones in the body to ossify but one of the last to have final physeal closure. Its unique characteristics have led to various descriptions, such as a “short tubular bone” versus a “flat bone.”4,5 Of note are its paucity of a true intramedullary space and scanty red marrow, which make it an unlikely site for a primarily intramedullary- based neoplasm to arise.4 However, it has also been noted that malignant lesions are more common in the clavicle than benign lesions, and special attention should be paid to aggressiveappearing lesions in the clavicle.
Radiographs can be misleading as well. Prior studies have demonstrated that low-grade central OS can be readily misdiagnosed as fibrous dysplasia, desmoplastic fibroma, nonossifying fibroma, osteoblastoma, and aneurysmal bone cyst.6 Findings found in low-grade OS can include evidence of cortical interruption, local soft tissue mass development, intramedullary involvement, cortical destruction, and poor margination; however, low-grade OS is typically sclerotic and highly trabeculated. Cross-sectional imaging can help differentiate between OS and other more benign pathologies and should be considered in the clavicle where biopsy may be perilous.5
The difficulty of clavicular biopsy has been reported. Not only does clavicular anatomy make biopsy hazardous, but also the potential for sampling error does exist. In a case report of one patient with a highgrade lesion, fine needle aspiration biopsy was initially diagnosed as an aneurysmal bone cyst but was ultimately found to be osteosarcoma.2 Histology of low-grade lesions usually demonstrates minimal cytological atypia, rare mitotic activity, and variable osteoid production.5 Lower mitotic indices typically make wide resection curative for these patients, without the need for chemotherapy.
In this case, wide resection was carried out with the subclavian vein as the posterior-inferior margin and the sternoclavicular joint as the medial margin. Though the intra-operative medial margin was clear of disease, final pathology demonstrated focal (microscopic) involvement of the posterior and medial margins. A study of soft tissue sarcoma evaluated positive margins and concluded that the imperative of preservation of vital structures supersedes the need for negative margins.7,8 The rate of metastasis and overall survival was similar to surgical resections with positive margins. In the case of our patient, further resection would have carried significant morbidity and possibly mortality, including sacrifice of the major vessels to the arm below and entering into the sternum and thoracic cavity. The likely disability as well as the hazards of surgery were deemed to be too great to justify further excision. Frequent cross-sectional imaging will be necessary to evaluate the presence of recurrent or metastatic disease. To our knowledge, this is the first documented case of low-grade clavicle OS. This report demonstrates the need for multidisciplinary sarcoma care at a center of excellence, particularly in instances of unusual diagnoses.
1. Zinghi G. Osteosarcoma of the clavicle (description of a case) [in Italian]. Chir Organi Mov. 1975;62(6):671-674.
2. Cundy WJ, Carter C, Dhatrak D, Clayer M. Primary osteosarcoma of the clavicle and the perils of bone biopsy. BMJ Case Rep. 2015;2015:bcr2014208859.
3. Greenspan A, Unni KK, Mann J. Case report 804: Chondroblastic osteosarcoma grade 3 of the left clavicle. Skeletal Radiol. 1993;22(6):469-471.
4. Rossi B, Fabbriciani C, Chalidis BE, Visci F, Maccauro G. Primary malignant clavicular tumours: a clinicopathological analysis of six cases and evaluation of surgical management. Arch Orthop Trauma Surg. 2011;131(7):935-939.
5. Andresen KJ, Sundaram M, Unni KK, Sim FH. Imaging features of low-grade central osteosarcoma of the long bones and pelvis. Skeletal Radiol. 2004;33(7):373-379.
6. Malhas AM, Sumathi VP, James SL, et al. Low-grade central osteosarcoma: A difficult condition to diagnose. Sarcoma. 2012; 2012:764796.
7. O’Donnell PW, Griffin AM, Eward WC, et al. The effect of the setting of a positive surgical margin in soft tissue sarcoma. Cancer. 2014;120(18):2866-2875.
8. Kawaguchi N, Ahmed AR, Matsumoto S, Manabe J, Matsushita Y. The concept of curative margin in surgery for bone and soft tissue sarcoma. Clin Orthop Relat Res. 2004;419:165-172.
1. Zinghi G. Osteosarcoma of the clavicle (description of a case) [in Italian]. Chir Organi Mov. 1975;62(6):671-674.
2. Cundy WJ, Carter C, Dhatrak D, Clayer M. Primary osteosarcoma of the clavicle and the perils of bone biopsy. BMJ Case Rep. 2015;2015:bcr2014208859.
3. Greenspan A, Unni KK, Mann J. Case report 804: Chondroblastic osteosarcoma grade 3 of the left clavicle. Skeletal Radiol. 1993;22(6):469-471.
4. Rossi B, Fabbriciani C, Chalidis BE, Visci F, Maccauro G. Primary malignant clavicular tumours: a clinicopathological analysis of six cases and evaluation of surgical management. Arch Orthop Trauma Surg. 2011;131(7):935-939.
5. Andresen KJ, Sundaram M, Unni KK, Sim FH. Imaging features of low-grade central osteosarcoma of the long bones and pelvis. Skeletal Radiol. 2004;33(7):373-379.
6. Malhas AM, Sumathi VP, James SL, et al. Low-grade central osteosarcoma: A difficult condition to diagnose. Sarcoma. 2012; 2012:764796.
7. O’Donnell PW, Griffin AM, Eward WC, et al. The effect of the setting of a positive surgical margin in soft tissue sarcoma. Cancer. 2014;120(18):2866-2875.
8. Kawaguchi N, Ahmed AR, Matsumoto S, Manabe J, Matsushita Y. The concept of curative margin in surgery for bone and soft tissue sarcoma. Clin Orthop Relat Res. 2004;419:165-172.
Bullous Systemic Lupus Erythematosus Successfully Treated With Rituximab
Bullous systemic lupus erythematosus (BSLE) is a rare cutaneous presentation of systemic lupus erythematosus (SLE).1 Although 59% to 85% of SLE patients develop skin-related symptoms, fewer than 5% of SLE patients develop BSLE.1-3 This acquired autoimmune bullous disease, characterized by subepidermal bullae with a neutrophilic infiltrate on histopathology, is precipitated by autoantibodies to type VII collagen. Bullae can appear on both cutaneous and mucosal surfaces but tend to favor the trunk, upper extremities, neck, face, and vermilion border.3
Our case of an 18-year-old black woman with BSLE was originally reported in 2011.4 We update the case to illustrate the heterogeneous presentation of BSLE in a single patient and to expand on the role of rituximab in this disease.
Case Report
An 18-year-old black woman presented with a vesicular eruption of 3 weeks’ duration that started on the trunk and buttocks and progressed to involve the face, oral mucosa, and posterior auricular area. The vesicular eruption was accompanied by fatigue, arthralgia, and myalgia.
Physical examination revealed multiple tense, fluid-filled vesicles, measuring roughly 2 to 3 mm in diameter, over the cheeks, chin, postauricular area, vermilion border, oral mucosa, and left side of the neck and shoulder. Resolved lesions on the trunk and buttocks were marked by superficial crust and postinflammatory hyperpigmentation. Scarring was absent.
Laboratory analysis demonstrated hemolytic anemia with a positive direct antiglobulin test, hypocomplementemia, and an elevated erythrocyte sedimentation rate. Antinuclear antibody testing was positive (titer, 1:640).
Biopsies were taken from the left cheek for hematoxylin and eosin (H&E) staining and direct immunofluorescence (DIF), which revealed subepidermal clefting, few neutrophils, and notable mucin deposition. Direct immunofluorescence showed a broad deposition of IgG, IgA, and IgM, as well as C3 in a ribbonlike pattern at the dermoepidermal junction.
A diagnosis of SLE with BSLE was made. The patient initially was treated with prednisone, hydroxychloroquine, mycophenolate mofetil, and intravenous immunoglobulin, but the cutaneous disease persisted. The bullous eruption resolved with 2 infusions of rituximab (1000 mg) spaced 2 weeks apart.
The patient was in remission on 5 mg of prednisone for 2 years following the initial course of rituximab. However, she developed a flare of SLE, with fatigue, arthralgia, hypocomplementemia, and recurrence of BSLE with tense bullae on the face and lips. The flare resolved with prednisone and a single infusion of rituximab (1000 mg). She was then maintained on hydroxychloroquine (200 mg/d).
Three years later (5 years after the initial presentation), the patient presented with pruritic erythematous papulovesicles on the bilateral extensor elbows and right knee (Figure 1). The clinical appearance suggested dermatitis herpetiformis (DH).
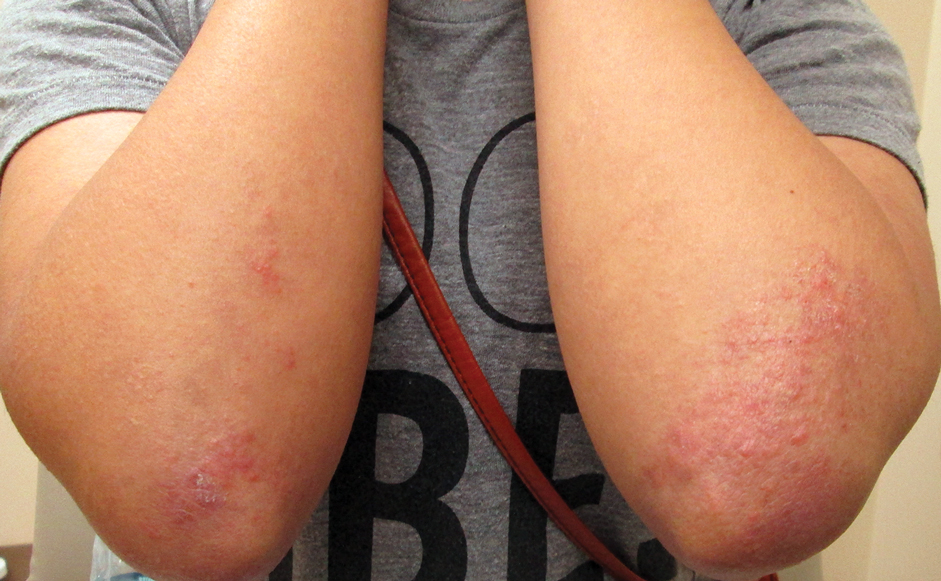
Punch biopsies were obtained from the right elbow for H&E and DIF testing; the H&E-stained specimen showed lichenoid dermatitis with prominent dermal mucin, consistent with cutaneous lupus erythematosus. Direct immunofluorescence showed prominent linear IgG, linear IgA, and granular IgM along the basement membrane, which were identical to DIF findings of the original eruption.
Further laboratory testing revealed hypocomplementemia, anemia of chronic disease (hemoglobin, 8.4 g/dL [reference range, 14.0–17.5 g/dL]), and an elevated erythrocyte sedimentation rate. Given the clinical appearance of the vesicles, DIF findings, and the corresponding SLE flare, a diagnosis of BSLE was made. Because of the systemic symptoms, skin findings, and laboratory results, azathioprine was started. The cutaneous symptoms were treated and resolved with the addition of triamcinolone ointment 0.1% twice daily.
Six months later, the patient presented to our facility with fatigue, arthralgia, and numerous erythematous papules coalescing into a large plaque on the left upper arm (Figure 2). Biopsy showed interface dermatitis with numerous neutrophils and early vesiculation, consistent with BSLE (Figure 3). She underwent another course of 2 infusions of rituximab (1000 mg) administered 2 weeks apart, with resolution of cutaneous and systemic disease.
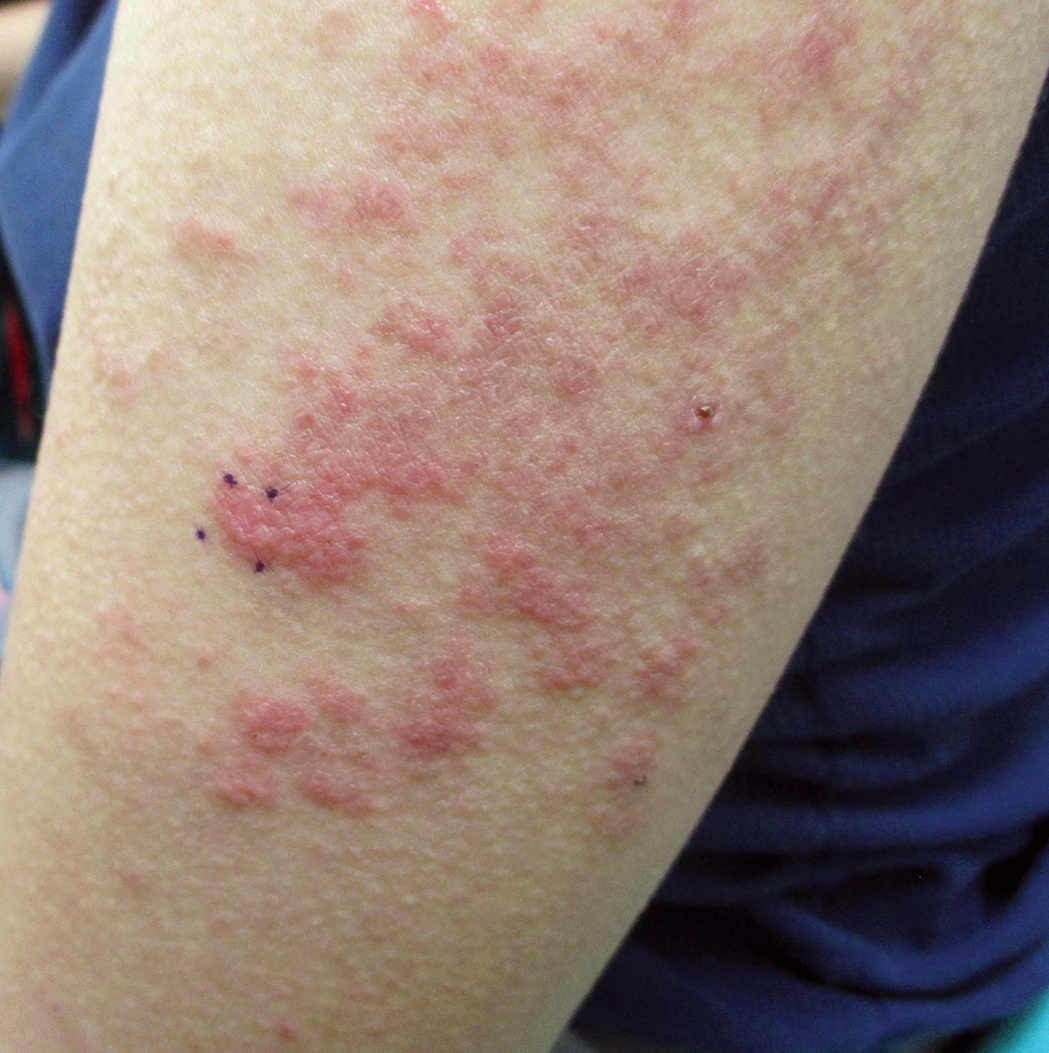
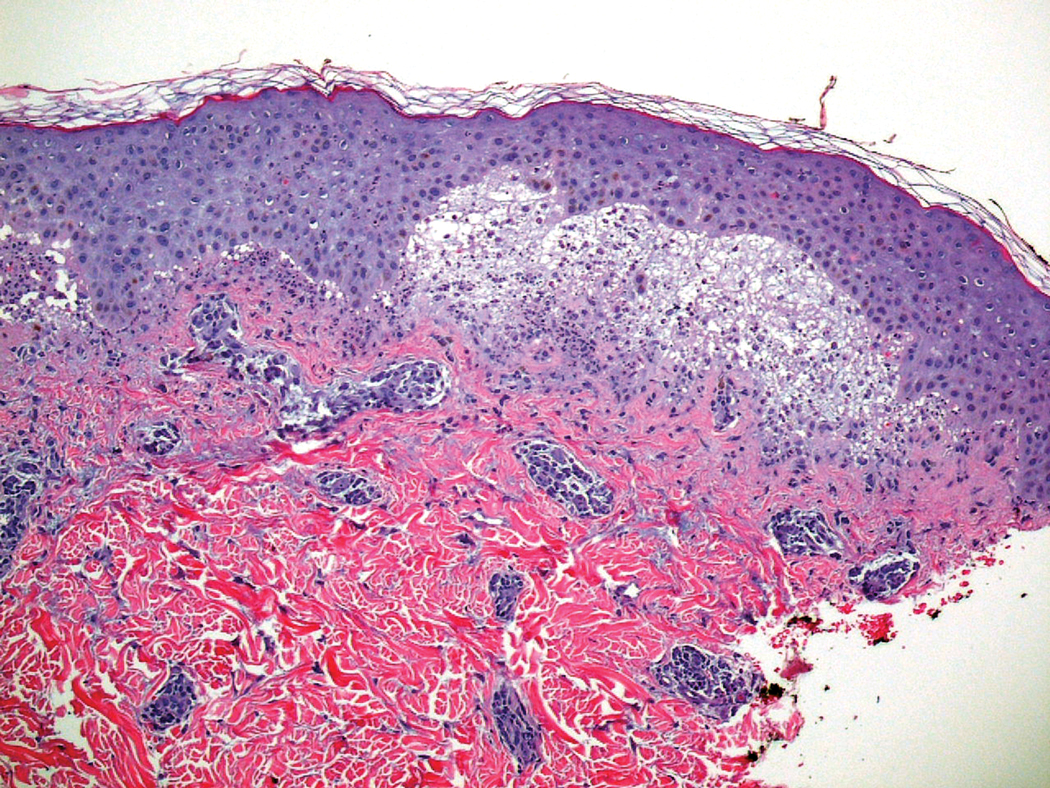
Comment
Diagnosis of BSLE
Bullous systemic lupus erythematosus is a rare cutaneous complication of SLE. It typically affects young black women in the second to fourth decades of life.1 It is a heterogeneous disorder with several clinical variants reported in the literature, and it can be mistaken for bullous pemphigoid, epidermolysis bullosa acquisita (EBA), linear IgA bullous dermatosis, and DH.1-3 Despite its varying clinical phenotypes, BSLE is associated with autoantibodies to the EBA antigen, type VII collagen.1
Current diagnostic criteria for BSLE, revised in 1995,5 include the following: (1) a diagnosis of SLE, based on criteria outlined by the American College of Rheumatology6; (2) vesicles or bullae, or both, involving but not limited to sun-exposed skin; (3) histopathologic features similar to DH; (4) DIF with IgG or IgM, or both, and IgA at the basement membrane zone; and (5) indirect immunofluorescence testing for circulating autoantibodies against the basement membrane zone, using the salt-split skin technique.
Clinical Presentation of BSLE
The classic phenotype associated with BSLE is similar to our patient’s original eruption, with tense bullae favoring the upper trunk and healing without scarring. The extensor surfaces typically are spared. Another presentation of BSLE is an EBA-like phenotype, with bullae on acral and extensor surfaces that heal with scarring. The EBA-like phenotype usually is more difficult to control. Lesions appearing clinically similar to DH have been reported, either as DH associated with SLE (later postulated to have been BSLE) or as herpetiform BSLE.1,4,7-10
Histopathology of BSLE
The typical histologic appearance of BSLE is similar to DH or linear IgA bullous dermatosis, with a predominantly neutrophilic inflammatory infiltrate in the upper dermis and a subepidermal split. Direct immunofluorescence shows broad deposition of IgG along the basement membrane zone (93% of cases; 60% of which are linear and 40% are granular), with approximately 70% of cases showing positive IgA or IgM, or both, at the basement membrane zone. Indirect immunofluorescence performed on 1 M NaCl salt-split skin showed staining on the dermal side of the split, similar to EBA.11
Treatment Options
Rapid clinical response has been reported with dapsone, usually in combination with other immunosuppresants.1,2 A subset of patients does not respond to dapsone, however, as was the case in our patient who tried dapsone early in the disease course but was not effective. Other therapies including azathioprine, cyclophosphamide, mycophenolate mofetil, and antimalarials have been used with some success.3
Rituximab, an anti-CD20 monoclonal antibody, has been used off label to treat BSLE cases that are resistant to dapsone, corticosteroids, and other immunosuppressants.12 Rituximab functions by depleting CD20+ B cells, thus altering the production of autoantibodies and, in the case of BSLE, reducing the concentration of circulating anti–type VII collagen antibodies. Rituximab was approved by the US Food and Drug Administration in 1997 for the treatment of non–Hodgkin lymphoma and later for chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis (Wegener granulomatosis), and microscopic polyangiitis.12 Off-label administration of rituximab to treat autoimmune bullous dermatoses has been increasing, and the drug is now approved by the US Food and Drug Administration to treat pemphigus vulgaris (as of June 2018).13
In 2011, Alsanafi et al12 reported successful treatment of BSLE with rituximab in a 61-year-old black woman who had rapid clearance of skin lesions. Our patient had rapid resolution of cutaneous disease with rituximab after the second infusion in a 2-infusion regimen. Interestingly, rituximab is the only agent that has reliably resulted in resolution of our patient’s cutaneous and systemic disease during multiple episodes.
There is little information in the literature regarding the duration of response to rituximab in BSLE or its use in subsequent flares. Our patient relapsed at 2 years and again 3 years later (5 years after the initial presentation). The original cutaneous outbreak and subsequent relapse had classic clinical and histological findings for BSLE; however, the third cutaneous relapse was more similar to DH, given its distribution and appearance. However, the histopathologic findings were the same at the third relapse as they were at the initial presentation and not reflective of DH. We propose that our patient’s prior treatment with rituximab and ongoing immunosuppression at presentation contributed to the more atypical cutaneous findings observed late in the disease course.
Conclusion
We report this case to highlight the heterogeneity of BSLE, even in a single patient, and to report the time course of treatment with rituximab. Although BSLE is considered a rare cutaneous complication of SLE, it is important to note that BSLE also can present as the initial manifestation of SLE.7 As such, BSLE should always be included in the differential diagnosis for a patient presenting with a bullous eruption and symptoms that suggest SLE.
This case also illustrates the repeated use of rituximab for the treatment of BSLE over a 5-year period and justifies the need for larger population-based studies to demonstrate the efficacy of rituximab in BSLE.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Camisa C. Vesiculobullous systemic lupus erythematosus. a report of four cases. J Am Acad Dermatol. 1988;18(1, pt 1):93-100.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Burke KR, Green BP, Meyerle J. Bullous lupus in an 18-year-old. Pediatr Dermatol. 2011;28:483.
- Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132:921-928.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheumat. 1997;40:1725.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Moncada B. Dermatitis herpetiformis in association with systemic lupus erythematosus. Arch Dermatol. 1974;109:723-725.
- Davies MG, Marks R, Waddington E. Simultaneous systemic lupus erythematosus and dermatitis herpetiformis. Arch Dermatol. 1976;112:1292-1294.
- Burrows N, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Alsanafi S, Kovarik C, Mermelstein AL, et al. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142-144.
- Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-708.
Bullous systemic lupus erythematosus (BSLE) is a rare cutaneous presentation of systemic lupus erythematosus (SLE).1 Although 59% to 85% of SLE patients develop skin-related symptoms, fewer than 5% of SLE patients develop BSLE.1-3 This acquired autoimmune bullous disease, characterized by subepidermal bullae with a neutrophilic infiltrate on histopathology, is precipitated by autoantibodies to type VII collagen. Bullae can appear on both cutaneous and mucosal surfaces but tend to favor the trunk, upper extremities, neck, face, and vermilion border.3
Our case of an 18-year-old black woman with BSLE was originally reported in 2011.4 We update the case to illustrate the heterogeneous presentation of BSLE in a single patient and to expand on the role of rituximab in this disease.
Case Report
An 18-year-old black woman presented with a vesicular eruption of 3 weeks’ duration that started on the trunk and buttocks and progressed to involve the face, oral mucosa, and posterior auricular area. The vesicular eruption was accompanied by fatigue, arthralgia, and myalgia.
Physical examination revealed multiple tense, fluid-filled vesicles, measuring roughly 2 to 3 mm in diameter, over the cheeks, chin, postauricular area, vermilion border, oral mucosa, and left side of the neck and shoulder. Resolved lesions on the trunk and buttocks were marked by superficial crust and postinflammatory hyperpigmentation. Scarring was absent.
Laboratory analysis demonstrated hemolytic anemia with a positive direct antiglobulin test, hypocomplementemia, and an elevated erythrocyte sedimentation rate. Antinuclear antibody testing was positive (titer, 1:640).
Biopsies were taken from the left cheek for hematoxylin and eosin (H&E) staining and direct immunofluorescence (DIF), which revealed subepidermal clefting, few neutrophils, and notable mucin deposition. Direct immunofluorescence showed a broad deposition of IgG, IgA, and IgM, as well as C3 in a ribbonlike pattern at the dermoepidermal junction.
A diagnosis of SLE with BSLE was made. The patient initially was treated with prednisone, hydroxychloroquine, mycophenolate mofetil, and intravenous immunoglobulin, but the cutaneous disease persisted. The bullous eruption resolved with 2 infusions of rituximab (1000 mg) spaced 2 weeks apart.
The patient was in remission on 5 mg of prednisone for 2 years following the initial course of rituximab. However, she developed a flare of SLE, with fatigue, arthralgia, hypocomplementemia, and recurrence of BSLE with tense bullae on the face and lips. The flare resolved with prednisone and a single infusion of rituximab (1000 mg). She was then maintained on hydroxychloroquine (200 mg/d).
Three years later (5 years after the initial presentation), the patient presented with pruritic erythematous papulovesicles on the bilateral extensor elbows and right knee (Figure 1). The clinical appearance suggested dermatitis herpetiformis (DH).
Punch biopsies were obtained from the right elbow for H&E and DIF testing; the H&E-stained specimen showed lichenoid dermatitis with prominent dermal mucin, consistent with cutaneous lupus erythematosus. Direct immunofluorescence showed prominent linear IgG, linear IgA, and granular IgM along the basement membrane, which were identical to DIF findings of the original eruption.
Further laboratory testing revealed hypocomplementemia, anemia of chronic disease (hemoglobin, 8.4 g/dL [reference range, 14.0–17.5 g/dL]), and an elevated erythrocyte sedimentation rate. Given the clinical appearance of the vesicles, DIF findings, and the corresponding SLE flare, a diagnosis of BSLE was made. Because of the systemic symptoms, skin findings, and laboratory results, azathioprine was started. The cutaneous symptoms were treated and resolved with the addition of triamcinolone ointment 0.1% twice daily.
Six months later, the patient presented to our facility with fatigue, arthralgia, and numerous erythematous papules coalescing into a large plaque on the left upper arm (Figure 2). Biopsy showed interface dermatitis with numerous neutrophils and early vesiculation, consistent with BSLE (Figure 3). She underwent another course of 2 infusions of rituximab (1000 mg) administered 2 weeks apart, with resolution of cutaneous and systemic disease.
Comment
Diagnosis of BSLE
Bullous systemic lupus erythematosus is a rare cutaneous complication of SLE. It typically affects young black women in the second to fourth decades of life.1 It is a heterogeneous disorder with several clinical variants reported in the literature, and it can be mistaken for bullous pemphigoid, epidermolysis bullosa acquisita (EBA), linear IgA bullous dermatosis, and DH.1-3 Despite its varying clinical phenotypes, BSLE is associated with autoantibodies to the EBA antigen, type VII collagen.1
Current diagnostic criteria for BSLE, revised in 1995,5 include the following: (1) a diagnosis of SLE, based on criteria outlined by the American College of Rheumatology6; (2) vesicles or bullae, or both, involving but not limited to sun-exposed skin; (3) histopathologic features similar to DH; (4) DIF with IgG or IgM, or both, and IgA at the basement membrane zone; and (5) indirect immunofluorescence testing for circulating autoantibodies against the basement membrane zone, using the salt-split skin technique.
Clinical Presentation of BSLE
The classic phenotype associated with BSLE is similar to our patient’s original eruption, with tense bullae favoring the upper trunk and healing without scarring. The extensor surfaces typically are spared. Another presentation of BSLE is an EBA-like phenotype, with bullae on acral and extensor surfaces that heal with scarring. The EBA-like phenotype usually is more difficult to control. Lesions appearing clinically similar to DH have been reported, either as DH associated with SLE (later postulated to have been BSLE) or as herpetiform BSLE.1,4,7-10
Histopathology of BSLE
The typical histologic appearance of BSLE is similar to DH or linear IgA bullous dermatosis, with a predominantly neutrophilic inflammatory infiltrate in the upper dermis and a subepidermal split. Direct immunofluorescence shows broad deposition of IgG along the basement membrane zone (93% of cases; 60% of which are linear and 40% are granular), with approximately 70% of cases showing positive IgA or IgM, or both, at the basement membrane zone. Indirect immunofluorescence performed on 1 M NaCl salt-split skin showed staining on the dermal side of the split, similar to EBA.11
Treatment Options
Rapid clinical response has been reported with dapsone, usually in combination with other immunosuppresants.1,2 A subset of patients does not respond to dapsone, however, as was the case in our patient who tried dapsone early in the disease course but was not effective. Other therapies including azathioprine, cyclophosphamide, mycophenolate mofetil, and antimalarials have been used with some success.3
Rituximab, an anti-CD20 monoclonal antibody, has been used off label to treat BSLE cases that are resistant to dapsone, corticosteroids, and other immunosuppressants.12 Rituximab functions by depleting CD20+ B cells, thus altering the production of autoantibodies and, in the case of BSLE, reducing the concentration of circulating anti–type VII collagen antibodies. Rituximab was approved by the US Food and Drug Administration in 1997 for the treatment of non–Hodgkin lymphoma and later for chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis (Wegener granulomatosis), and microscopic polyangiitis.12 Off-label administration of rituximab to treat autoimmune bullous dermatoses has been increasing, and the drug is now approved by the US Food and Drug Administration to treat pemphigus vulgaris (as of June 2018).13
In 2011, Alsanafi et al12 reported successful treatment of BSLE with rituximab in a 61-year-old black woman who had rapid clearance of skin lesions. Our patient had rapid resolution of cutaneous disease with rituximab after the second infusion in a 2-infusion regimen. Interestingly, rituximab is the only agent that has reliably resulted in resolution of our patient’s cutaneous and systemic disease during multiple episodes.
There is little information in the literature regarding the duration of response to rituximab in BSLE or its use in subsequent flares. Our patient relapsed at 2 years and again 3 years later (5 years after the initial presentation). The original cutaneous outbreak and subsequent relapse had classic clinical and histological findings for BSLE; however, the third cutaneous relapse was more similar to DH, given its distribution and appearance. However, the histopathologic findings were the same at the third relapse as they were at the initial presentation and not reflective of DH. We propose that our patient’s prior treatment with rituximab and ongoing immunosuppression at presentation contributed to the more atypical cutaneous findings observed late in the disease course.
Conclusion
We report this case to highlight the heterogeneity of BSLE, even in a single patient, and to report the time course of treatment with rituximab. Although BSLE is considered a rare cutaneous complication of SLE, it is important to note that BSLE also can present as the initial manifestation of SLE.7 As such, BSLE should always be included in the differential diagnosis for a patient presenting with a bullous eruption and symptoms that suggest SLE.
This case also illustrates the repeated use of rituximab for the treatment of BSLE over a 5-year period and justifies the need for larger population-based studies to demonstrate the efficacy of rituximab in BSLE.
Bullous systemic lupus erythematosus (BSLE) is a rare cutaneous presentation of systemic lupus erythematosus (SLE).1 Although 59% to 85% of SLE patients develop skin-related symptoms, fewer than 5% of SLE patients develop BSLE.1-3 This acquired autoimmune bullous disease, characterized by subepidermal bullae with a neutrophilic infiltrate on histopathology, is precipitated by autoantibodies to type VII collagen. Bullae can appear on both cutaneous and mucosal surfaces but tend to favor the trunk, upper extremities, neck, face, and vermilion border.3
Our case of an 18-year-old black woman with BSLE was originally reported in 2011.4 We update the case to illustrate the heterogeneous presentation of BSLE in a single patient and to expand on the role of rituximab in this disease.
Case Report
An 18-year-old black woman presented with a vesicular eruption of 3 weeks’ duration that started on the trunk and buttocks and progressed to involve the face, oral mucosa, and posterior auricular area. The vesicular eruption was accompanied by fatigue, arthralgia, and myalgia.
Physical examination revealed multiple tense, fluid-filled vesicles, measuring roughly 2 to 3 mm in diameter, over the cheeks, chin, postauricular area, vermilion border, oral mucosa, and left side of the neck and shoulder. Resolved lesions on the trunk and buttocks were marked by superficial crust and postinflammatory hyperpigmentation. Scarring was absent.
Laboratory analysis demonstrated hemolytic anemia with a positive direct antiglobulin test, hypocomplementemia, and an elevated erythrocyte sedimentation rate. Antinuclear antibody testing was positive (titer, 1:640).
Biopsies were taken from the left cheek for hematoxylin and eosin (H&E) staining and direct immunofluorescence (DIF), which revealed subepidermal clefting, few neutrophils, and notable mucin deposition. Direct immunofluorescence showed a broad deposition of IgG, IgA, and IgM, as well as C3 in a ribbonlike pattern at the dermoepidermal junction.
A diagnosis of SLE with BSLE was made. The patient initially was treated with prednisone, hydroxychloroquine, mycophenolate mofetil, and intravenous immunoglobulin, but the cutaneous disease persisted. The bullous eruption resolved with 2 infusions of rituximab (1000 mg) spaced 2 weeks apart.
The patient was in remission on 5 mg of prednisone for 2 years following the initial course of rituximab. However, she developed a flare of SLE, with fatigue, arthralgia, hypocomplementemia, and recurrence of BSLE with tense bullae on the face and lips. The flare resolved with prednisone and a single infusion of rituximab (1000 mg). She was then maintained on hydroxychloroquine (200 mg/d).
Three years later (5 years after the initial presentation), the patient presented with pruritic erythematous papulovesicles on the bilateral extensor elbows and right knee (Figure 1). The clinical appearance suggested dermatitis herpetiformis (DH).
Punch biopsies were obtained from the right elbow for H&E and DIF testing; the H&E-stained specimen showed lichenoid dermatitis with prominent dermal mucin, consistent with cutaneous lupus erythematosus. Direct immunofluorescence showed prominent linear IgG, linear IgA, and granular IgM along the basement membrane, which were identical to DIF findings of the original eruption.
Further laboratory testing revealed hypocomplementemia, anemia of chronic disease (hemoglobin, 8.4 g/dL [reference range, 14.0–17.5 g/dL]), and an elevated erythrocyte sedimentation rate. Given the clinical appearance of the vesicles, DIF findings, and the corresponding SLE flare, a diagnosis of BSLE was made. Because of the systemic symptoms, skin findings, and laboratory results, azathioprine was started. The cutaneous symptoms were treated and resolved with the addition of triamcinolone ointment 0.1% twice daily.
Six months later, the patient presented to our facility with fatigue, arthralgia, and numerous erythematous papules coalescing into a large plaque on the left upper arm (Figure 2). Biopsy showed interface dermatitis with numerous neutrophils and early vesiculation, consistent with BSLE (Figure 3). She underwent another course of 2 infusions of rituximab (1000 mg) administered 2 weeks apart, with resolution of cutaneous and systemic disease.
Comment
Diagnosis of BSLE
Bullous systemic lupus erythematosus is a rare cutaneous complication of SLE. It typically affects young black women in the second to fourth decades of life.1 It is a heterogeneous disorder with several clinical variants reported in the literature, and it can be mistaken for bullous pemphigoid, epidermolysis bullosa acquisita (EBA), linear IgA bullous dermatosis, and DH.1-3 Despite its varying clinical phenotypes, BSLE is associated with autoantibodies to the EBA antigen, type VII collagen.1
Current diagnostic criteria for BSLE, revised in 1995,5 include the following: (1) a diagnosis of SLE, based on criteria outlined by the American College of Rheumatology6; (2) vesicles or bullae, or both, involving but not limited to sun-exposed skin; (3) histopathologic features similar to DH; (4) DIF with IgG or IgM, or both, and IgA at the basement membrane zone; and (5) indirect immunofluorescence testing for circulating autoantibodies against the basement membrane zone, using the salt-split skin technique.
Clinical Presentation of BSLE
The classic phenotype associated with BSLE is similar to our patient’s original eruption, with tense bullae favoring the upper trunk and healing without scarring. The extensor surfaces typically are spared. Another presentation of BSLE is an EBA-like phenotype, with bullae on acral and extensor surfaces that heal with scarring. The EBA-like phenotype usually is more difficult to control. Lesions appearing clinically similar to DH have been reported, either as DH associated with SLE (later postulated to have been BSLE) or as herpetiform BSLE.1,4,7-10
Histopathology of BSLE
The typical histologic appearance of BSLE is similar to DH or linear IgA bullous dermatosis, with a predominantly neutrophilic inflammatory infiltrate in the upper dermis and a subepidermal split. Direct immunofluorescence shows broad deposition of IgG along the basement membrane zone (93% of cases; 60% of which are linear and 40% are granular), with approximately 70% of cases showing positive IgA or IgM, or both, at the basement membrane zone. Indirect immunofluorescence performed on 1 M NaCl salt-split skin showed staining on the dermal side of the split, similar to EBA.11
Treatment Options
Rapid clinical response has been reported with dapsone, usually in combination with other immunosuppresants.1,2 A subset of patients does not respond to dapsone, however, as was the case in our patient who tried dapsone early in the disease course but was not effective. Other therapies including azathioprine, cyclophosphamide, mycophenolate mofetil, and antimalarials have been used with some success.3
Rituximab, an anti-CD20 monoclonal antibody, has been used off label to treat BSLE cases that are resistant to dapsone, corticosteroids, and other immunosuppressants.12 Rituximab functions by depleting CD20+ B cells, thus altering the production of autoantibodies and, in the case of BSLE, reducing the concentration of circulating anti–type VII collagen antibodies. Rituximab was approved by the US Food and Drug Administration in 1997 for the treatment of non–Hodgkin lymphoma and later for chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis (Wegener granulomatosis), and microscopic polyangiitis.12 Off-label administration of rituximab to treat autoimmune bullous dermatoses has been increasing, and the drug is now approved by the US Food and Drug Administration to treat pemphigus vulgaris (as of June 2018).13
In 2011, Alsanafi et al12 reported successful treatment of BSLE with rituximab in a 61-year-old black woman who had rapid clearance of skin lesions. Our patient had rapid resolution of cutaneous disease with rituximab after the second infusion in a 2-infusion regimen. Interestingly, rituximab is the only agent that has reliably resulted in resolution of our patient’s cutaneous and systemic disease during multiple episodes.
There is little information in the literature regarding the duration of response to rituximab in BSLE or its use in subsequent flares. Our patient relapsed at 2 years and again 3 years later (5 years after the initial presentation). The original cutaneous outbreak and subsequent relapse had classic clinical and histological findings for BSLE; however, the third cutaneous relapse was more similar to DH, given its distribution and appearance. However, the histopathologic findings were the same at the third relapse as they were at the initial presentation and not reflective of DH. We propose that our patient’s prior treatment with rituximab and ongoing immunosuppression at presentation contributed to the more atypical cutaneous findings observed late in the disease course.
Conclusion
We report this case to highlight the heterogeneity of BSLE, even in a single patient, and to report the time course of treatment with rituximab. Although BSLE is considered a rare cutaneous complication of SLE, it is important to note that BSLE also can present as the initial manifestation of SLE.7 As such, BSLE should always be included in the differential diagnosis for a patient presenting with a bullous eruption and symptoms that suggest SLE.
This case also illustrates the repeated use of rituximab for the treatment of BSLE over a 5-year period and justifies the need for larger population-based studies to demonstrate the efficacy of rituximab in BSLE.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Camisa C. Vesiculobullous systemic lupus erythematosus. a report of four cases. J Am Acad Dermatol. 1988;18(1, pt 1):93-100.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Burke KR, Green BP, Meyerle J. Bullous lupus in an 18-year-old. Pediatr Dermatol. 2011;28:483.
- Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132:921-928.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheumat. 1997;40:1725.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Moncada B. Dermatitis herpetiformis in association with systemic lupus erythematosus. Arch Dermatol. 1974;109:723-725.
- Davies MG, Marks R, Waddington E. Simultaneous systemic lupus erythematosus and dermatitis herpetiformis. Arch Dermatol. 1976;112:1292-1294.
- Burrows N, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Alsanafi S, Kovarik C, Mermelstein AL, et al. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142-144.
- Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-708.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Camisa C. Vesiculobullous systemic lupus erythematosus. a report of four cases. J Am Acad Dermatol. 1988;18(1, pt 1):93-100.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Burke KR, Green BP, Meyerle J. Bullous lupus in an 18-year-old. Pediatr Dermatol. 2011;28:483.
- Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132:921-928.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheumat. 1997;40:1725.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Moncada B. Dermatitis herpetiformis in association with systemic lupus erythematosus. Arch Dermatol. 1974;109:723-725.
- Davies MG, Marks R, Waddington E. Simultaneous systemic lupus erythematosus and dermatitis herpetiformis. Arch Dermatol. 1976;112:1292-1294.
- Burrows N, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Alsanafi S, Kovarik C, Mermelstein AL, et al. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142-144.
- Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-708.
Practice Points
- Bullous systemic lupus erythematosus (BSLE) can present with a waxing and waning course punctuated by flares.
- Different clinical presentations can occur over the disease course.
- Rituximab is a viable treatment option in BSLE.
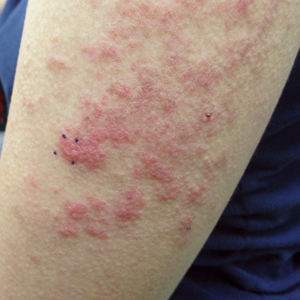
Acute Graft-vs-host Disease Following Liver Transplantation
Acute graft-vs-host disease (GVHD) is a T-cell mediated immunogenic response in which T lymphocytes from a donor regard host tissue as foreign and attack it in the setting of immunosuppression.1 The most common cause of acute GVHD is allogeneic stem cell transplantation, with solid-organ transplantation being a much less common cause.2 The incidence of acute GVHD following orthotopic liver transplantation (OLT) is 0.1%, as reported by the United Network for Organ Sharing, compared to an incidence of 40% to 60% in hematopoietic stem cell transplant recipients.3,4
Early recognition and treatment of acute GVHD following liver transplantation is imperative, as the mortality rate is 85% to 90%.2 We present a case of acute GVHD in a liver transplantation patient, with a focus on diagnostic criteria and comparison to acute GVHD following hematopoietic stem cell transplantation.
Case Report
A 68-year-old woman with a history of hepatitis C virus infection, hepatocellular carcinoma, and OLT 1 month prior presented to the hospital with fever and abdominal cellulitis in close proximity to the surgical site of 1 week’s duration. The patient was started on vancomycin and cefepime; pan cultures were performed.
At 10 days of hospitalization, the patient developed a pruritic, nontender, erythematous rash on the abdomen, with extension onto the chest and legs. The rash was associated with low-grade fever but not with diarrhea. Physical examination was notable for a few erythematous macules and scattered papules over the neck and chest and a large erythematous plaque with multiple ecchymoses over the lower abdomen (Figure 1A). Erythematous macules and papules coalescing into plaques were present on the lower back (Figure 1B) and proximal thighs. Oral, ocular, and genital lesions were absent.
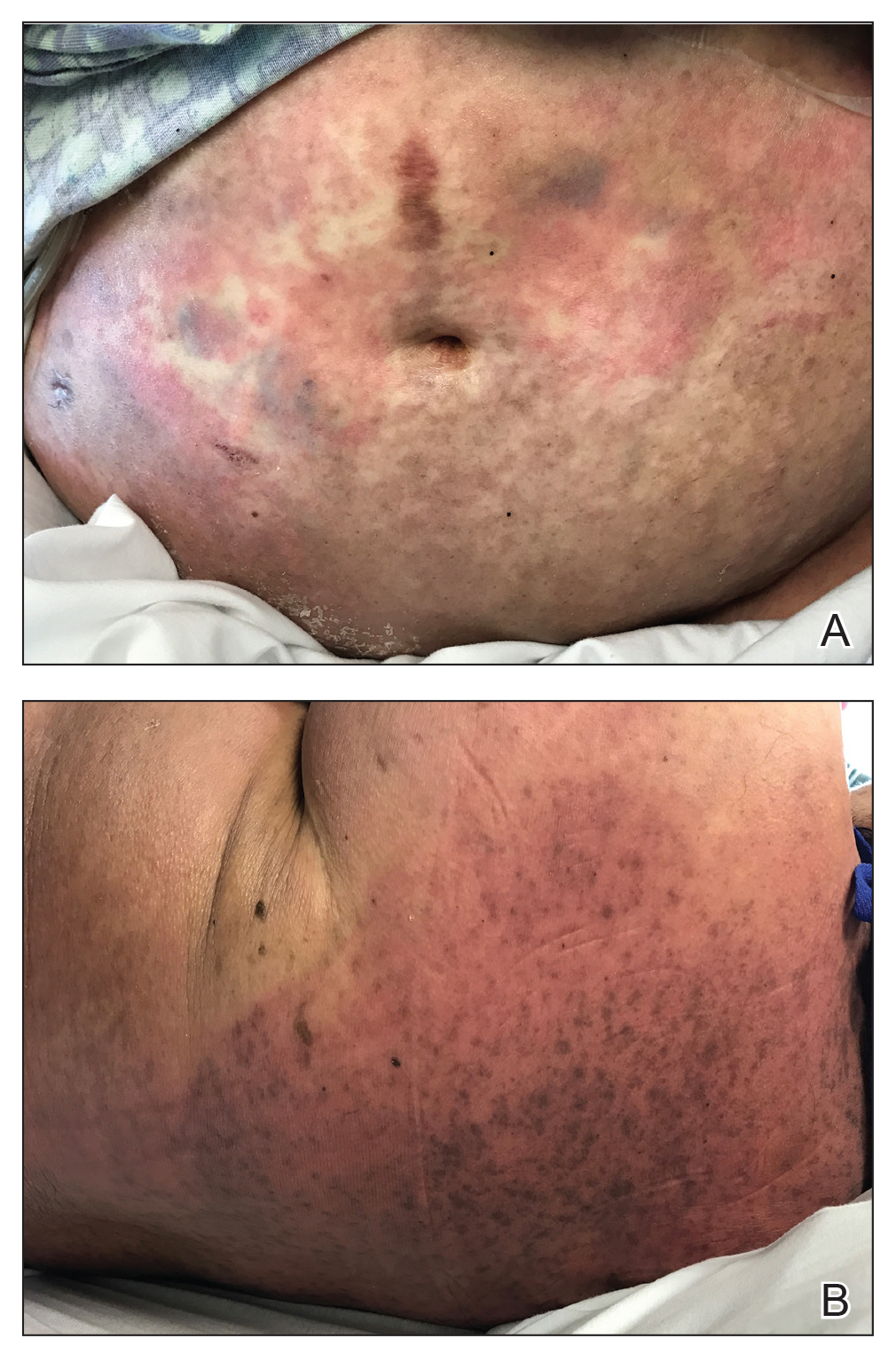
The differential diagnosis included drug reaction, viral infection, and acute GVHD. A skin biopsy was performed from the left side of the chest. Cefepime and vancomycin were discontinued; triamcinolone ointment 0.1% twice daily and antihistamines as needed for itching were started.
Over a 2-day period, the rash progressed to diffuse erythematous papules over the chest (Figure 2A) and bilateral arms (Figure 2B) including the palms. The patient also developed erythematous papules over the jawline and forehead as well as confluent erythematous plaques over the back with extension of the rash to involve the legs. She also had erythema and swelling bilaterally over the ears. She reported diarrhea. The low-grade fever resolved.
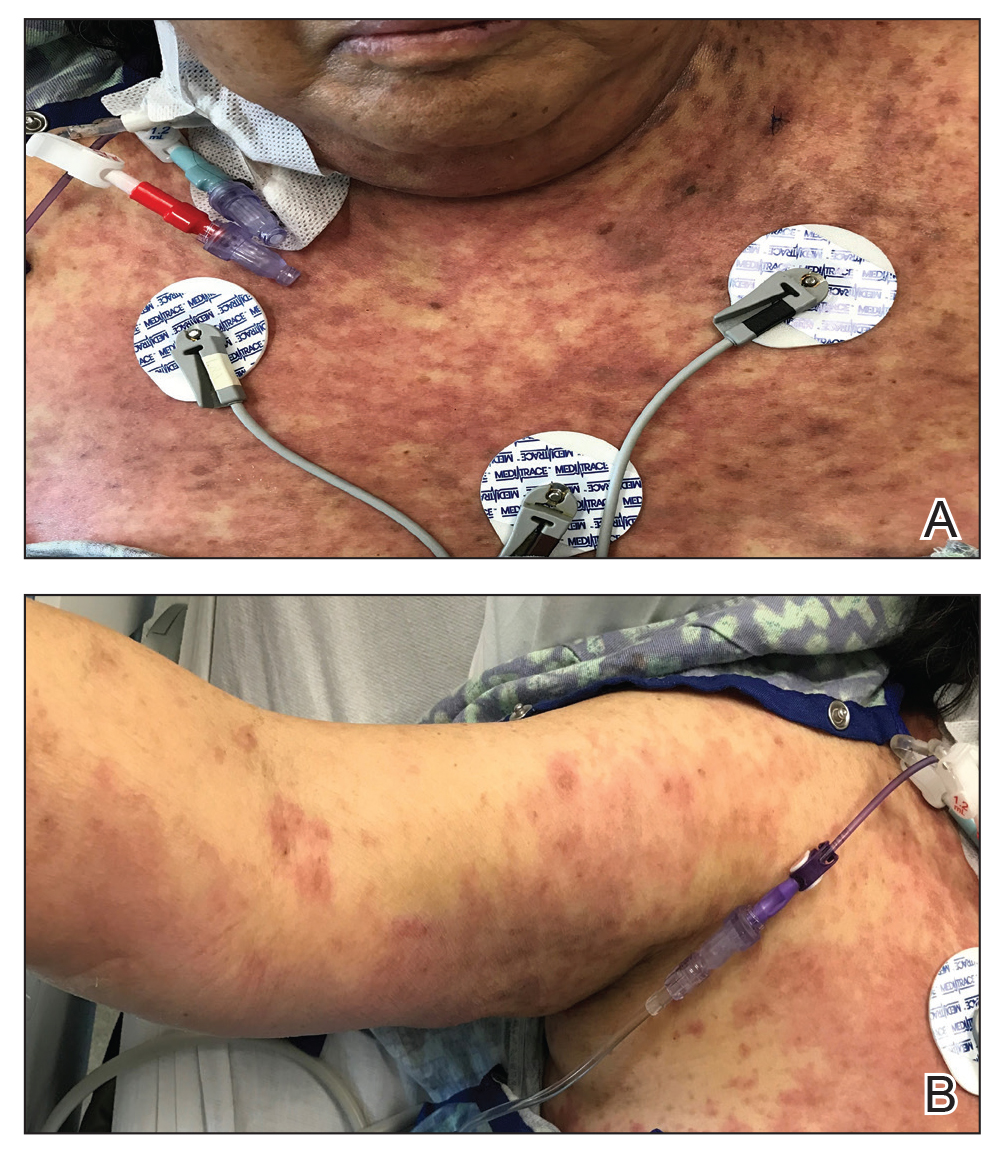
Laboratory review showed new-onset pancytopenia, normal liver function, and an elevated creatinine level of 2.3 mg/dL (reference range, 0.6–1.2 mg/dL), consistent with the patient’s baseline of stage 3 chronic kidney disease. Polymerase chain reaction analysis for cytomegalovirus was negative. Histology revealed vacuolar interface dermatitis with apoptotic keratinocytes, consistent with grade I GVHD (Figure 3). Duodenal biopsy revealed rare patchy glands with increased apoptosis, compatible with grade I GVHD.
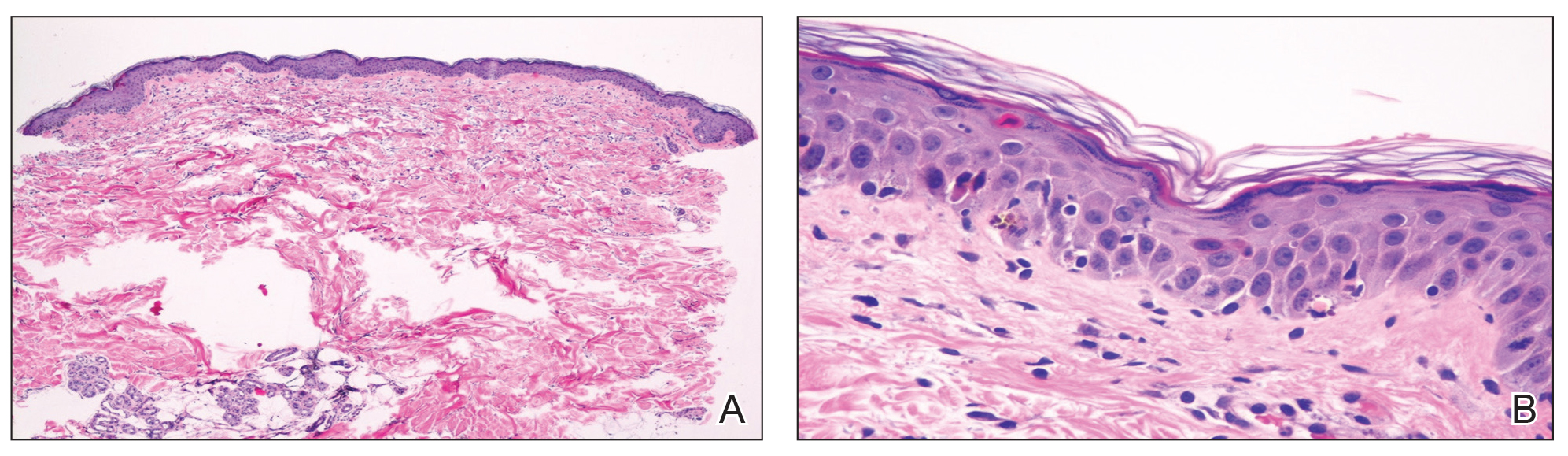
The patient was started on intravenous methylprednisolone 1 mg/kg for 3 days, then transitioned to an oral steroid taper, with improvement of the rash and other systemic symptoms.
Comment
GVHD Subtypes
The 2 types of GVHD are humoral and cellular.5 The humoral type results from ABO blood type incompatibility between donor and recipient and causes mild hemolytic anemia and fever. The cellular type is directed against major histocompatibility complexes and is associated with high morbidity and mortality.
Presentation of GVHD
Acute GVHD following OLT usually occurs 3 to 5 weeks after transplantation,6 as in our patient. Symptoms include rash, fever, pancytopenia, and diarrhea.2 Skin is the most commonly involved organ in acute GVHD; rash is the earliest manifestation.1 The rash can be asymptomatic or associated with pain and pruritus. Initial cutaneous manifestations include palmar erythema and erythematous to violaceous discoloration of the face and ears. A diffuse maculopapular rash can develop, involving the face, abdomen, and trunk. The rash may progress to formation of bullae or skin sloughing, resembling Stevens-Johnson syndrome or toxic epidermal necrolysis.1 The skin manifestation of acute GVHD following OLT is similar to hematopoietic stem cell transplantation (Table).7,8
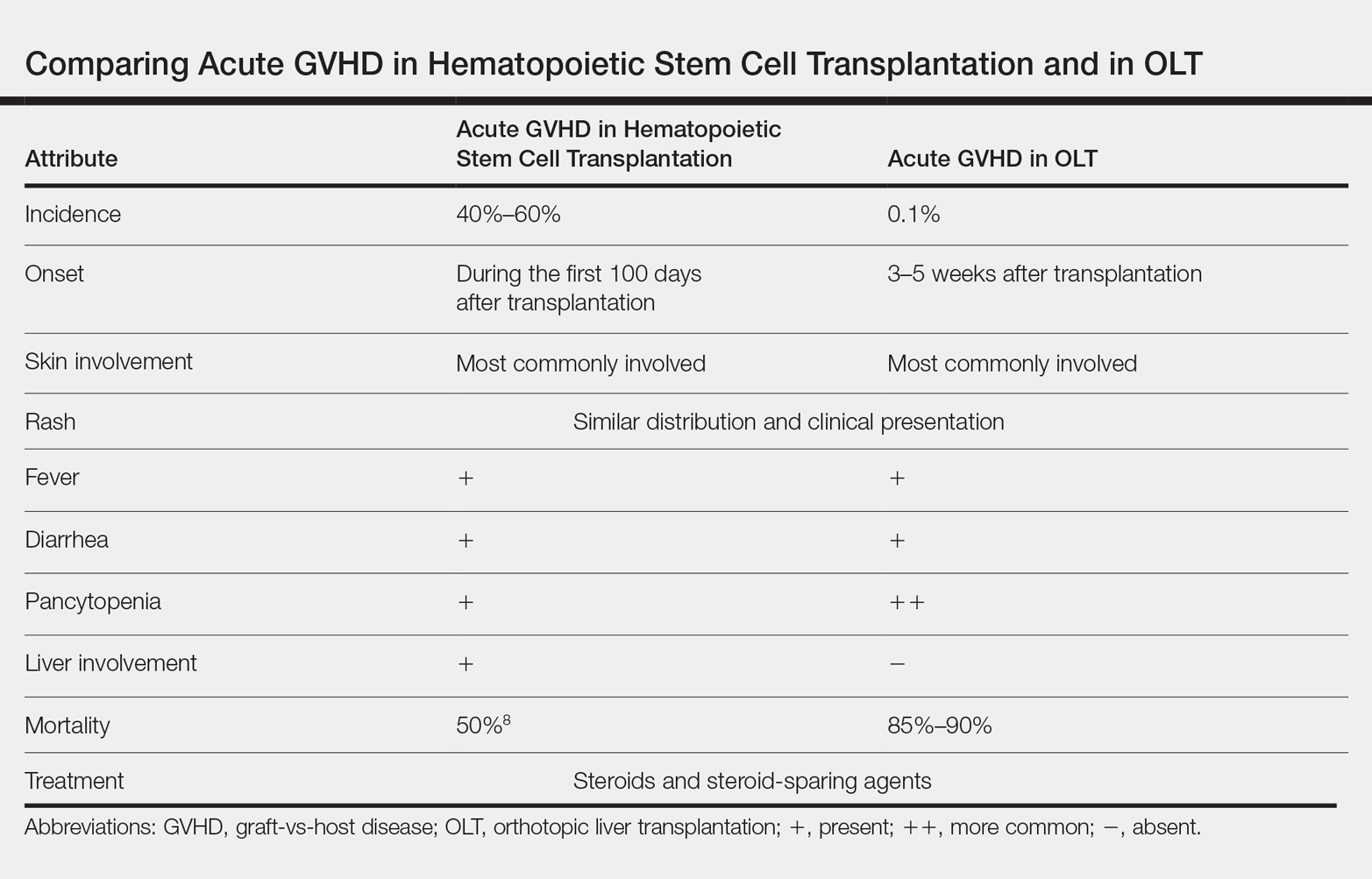
Pancytopenia is a common manifestation of GVHD following liver transplantation and is rarely seen following hematopoietic stem cell transplantation.7 Donor lymphocytes engraft and proliferate in the bone marrow, attacking recipient hematopoietic stem cells. It is important to note that more common causes of cytopenia following liver transplantation, including infection and drug-induced bone marrow suppression, should be ruled out before diagnosing acute GVHD.6
Acute GVHD can affect the gastrointestinal tract, causing diarrhea; however, other infectious and medication-induced causes of diarrhea also should be considered.6 In contrast to hematopoietic stem cell transplantation, in which the liver is usually involved,1 the liver is spared in acute GVHD following liver transplantation.5
Diagnosis of GVHD
The diagnosis of acute GVHD following liver transplantation can be challenging because the clinical manifestations can be caused by a drug reaction or viral infection, such as cytomegalovirus infection.2 Patients who are older than 50 years and glucose intolerant are at a higher risk of acute GVHD following OLT. The combination of younger donor age and the presence of an HLA class I match also increases the risk of acute GVHD.6 The diagnosis of acute GVHD is confirmed with biopsy of the skin or gastrointestinal tract.
Morbidity and Mortality of GVHD
Because of the high morbidity and mortality associated with acute GVHD following liver transplantation, early diagnosis and treatment are crucial.5 Death in patients with acute GVHD following OLT is mainly attributable to sepsis, multiorgan failure, and gastrointestinal tract bleeding.6 It remains unclear whether this high mortality is associated with delayed diagnosis due to nonspecific signs of acute GVHD following OLT or to the lack of appropriate treatment guidelines.6
Treatment Options
Because of the low incidence of acute GVHD following OLT, most treatment modalities are extrapolated from the literature on acute GVHD following stem cell transplantation.5 The most commonly used therapies include high-dose systemic steroids and anti–thymocyte globulin that attacks activated donor T cells.6 Other treatment modalities, including anti–tumor necrosis factor agents and antibodies to CD20, have been reported to be effective in steroid-refractory GVHD.2 The major drawback of systemic steroids is an increase in the risk for sepsis and infection; therefore, these patients should be diligently screened for infection and covered with antibiotics and antifungals. Extracorporeal photopheresis is another treatment modality that does not cause generalized immunosuppression but is not well studied in the setting of acute GVHD following OLT.6
Prevention
Acute GVHD following OLT can be prevented by eliminating donor T lymphocytes from the liver before transplantation. However, because the incidence of acute GVHD following OLT is very low, this approach is not routinely taken.2
Conclusion
Acute GVHD following liver transplantation is a rare complication; however, it has high mortality, necessitating further research regarding treatment and prevention. Early recognition and treatment of this condition can improve outcomes. Dermatologists should be familiar with the skin manifestations of acute GVHD following liver transplantation due to the rising number of cases of solid-organ transplantation.
- Hu SW, Cotliar J. Acute graft-versus-host disease following hematopoietic stem-cell transplantation. Dermatol Ther. 2011;24:411-423.
- Akbulut S, Yilmaz M, Yilmaz S. Graft-versus-host disease after liver transplantation: a comprehensive literature review. World J Gastroenterol. 2012;18:5240-5248.
- Taylor AL, Gibbs P, Bradley JA. Acute graft versus host disease following liver transplantation: the enemy within. Am J Transplant. 2004;4:466-474.
- Jagasia M, Arora M, Flowers ME, et al. Risk factor for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119:296-307.
- Kang WH, Hwang S, Song GW, et al. Acute graft-vs-host disease after liver transplantation: experience at a high-volume liver transplantation center in Korea. Transplant Proc. 2016;48:3368-3372.
- Murali AR, Chandra S, Stewart Z, et al. Graft versus host disease after liver transplantation in adults: a case series, review of literature, and an approach to management. Transplantation. 2016;100:2661-2670.
- Chaib E, Silva FD, Figueira ER, et al. Graft-versus-host disease after liver transplantation. Clinics (Sao Paulo). 2011;66:1115-1118.
- Barton-Burke M, Dwinell DM, Kafkas L, et al. Graft-versus-host disease: a complex long-term side effect of hematopoietic stem cell transplant. Oncology (Williston Park). 2008;22(11 Suppl Nurse Ed):31-45.
Acute graft-vs-host disease (GVHD) is a T-cell mediated immunogenic response in which T lymphocytes from a donor regard host tissue as foreign and attack it in the setting of immunosuppression.1 The most common cause of acute GVHD is allogeneic stem cell transplantation, with solid-organ transplantation being a much less common cause.2 The incidence of acute GVHD following orthotopic liver transplantation (OLT) is 0.1%, as reported by the United Network for Organ Sharing, compared to an incidence of 40% to 60% in hematopoietic stem cell transplant recipients.3,4
Early recognition and treatment of acute GVHD following liver transplantation is imperative, as the mortality rate is 85% to 90%.2 We present a case of acute GVHD in a liver transplantation patient, with a focus on diagnostic criteria and comparison to acute GVHD following hematopoietic stem cell transplantation.
Case Report
A 68-year-old woman with a history of hepatitis C virus infection, hepatocellular carcinoma, and OLT 1 month prior presented to the hospital with fever and abdominal cellulitis in close proximity to the surgical site of 1 week’s duration. The patient was started on vancomycin and cefepime; pan cultures were performed.
At 10 days of hospitalization, the patient developed a pruritic, nontender, erythematous rash on the abdomen, with extension onto the chest and legs. The rash was associated with low-grade fever but not with diarrhea. Physical examination was notable for a few erythematous macules and scattered papules over the neck and chest and a large erythematous plaque with multiple ecchymoses over the lower abdomen (Figure 1A). Erythematous macules and papules coalescing into plaques were present on the lower back (Figure 1B) and proximal thighs. Oral, ocular, and genital lesions were absent.
The differential diagnosis included drug reaction, viral infection, and acute GVHD. A skin biopsy was performed from the left side of the chest. Cefepime and vancomycin were discontinued; triamcinolone ointment 0.1% twice daily and antihistamines as needed for itching were started.
Over a 2-day period, the rash progressed to diffuse erythematous papules over the chest (Figure 2A) and bilateral arms (Figure 2B) including the palms. The patient also developed erythematous papules over the jawline and forehead as well as confluent erythematous plaques over the back with extension of the rash to involve the legs. She also had erythema and swelling bilaterally over the ears. She reported diarrhea. The low-grade fever resolved.
Laboratory review showed new-onset pancytopenia, normal liver function, and an elevated creatinine level of 2.3 mg/dL (reference range, 0.6–1.2 mg/dL), consistent with the patient’s baseline of stage 3 chronic kidney disease. Polymerase chain reaction analysis for cytomegalovirus was negative. Histology revealed vacuolar interface dermatitis with apoptotic keratinocytes, consistent with grade I GVHD (Figure 3). Duodenal biopsy revealed rare patchy glands with increased apoptosis, compatible with grade I GVHD.
The patient was started on intravenous methylprednisolone 1 mg/kg for 3 days, then transitioned to an oral steroid taper, with improvement of the rash and other systemic symptoms.
Comment
GVHD Subtypes
The 2 types of GVHD are humoral and cellular.5 The humoral type results from ABO blood type incompatibility between donor and recipient and causes mild hemolytic anemia and fever. The cellular type is directed against major histocompatibility complexes and is associated with high morbidity and mortality.
Presentation of GVHD
Acute GVHD following OLT usually occurs 3 to 5 weeks after transplantation,6 as in our patient. Symptoms include rash, fever, pancytopenia, and diarrhea.2 Skin is the most commonly involved organ in acute GVHD; rash is the earliest manifestation.1 The rash can be asymptomatic or associated with pain and pruritus. Initial cutaneous manifestations include palmar erythema and erythematous to violaceous discoloration of the face and ears. A diffuse maculopapular rash can develop, involving the face, abdomen, and trunk. The rash may progress to formation of bullae or skin sloughing, resembling Stevens-Johnson syndrome or toxic epidermal necrolysis.1 The skin manifestation of acute GVHD following OLT is similar to hematopoietic stem cell transplantation (Table).7,8
Pancytopenia is a common manifestation of GVHD following liver transplantation and is rarely seen following hematopoietic stem cell transplantation.7 Donor lymphocytes engraft and proliferate in the bone marrow, attacking recipient hematopoietic stem cells. It is important to note that more common causes of cytopenia following liver transplantation, including infection and drug-induced bone marrow suppression, should be ruled out before diagnosing acute GVHD.6
Acute GVHD can affect the gastrointestinal tract, causing diarrhea; however, other infectious and medication-induced causes of diarrhea also should be considered.6 In contrast to hematopoietic stem cell transplantation, in which the liver is usually involved,1 the liver is spared in acute GVHD following liver transplantation.5
Diagnosis of GVHD
The diagnosis of acute GVHD following liver transplantation can be challenging because the clinical manifestations can be caused by a drug reaction or viral infection, such as cytomegalovirus infection.2 Patients who are older than 50 years and glucose intolerant are at a higher risk of acute GVHD following OLT. The combination of younger donor age and the presence of an HLA class I match also increases the risk of acute GVHD.6 The diagnosis of acute GVHD is confirmed with biopsy of the skin or gastrointestinal tract.
Morbidity and Mortality of GVHD
Because of the high morbidity and mortality associated with acute GVHD following liver transplantation, early diagnosis and treatment are crucial.5 Death in patients with acute GVHD following OLT is mainly attributable to sepsis, multiorgan failure, and gastrointestinal tract bleeding.6 It remains unclear whether this high mortality is associated with delayed diagnosis due to nonspecific signs of acute GVHD following OLT or to the lack of appropriate treatment guidelines.6
Treatment Options
Because of the low incidence of acute GVHD following OLT, most treatment modalities are extrapolated from the literature on acute GVHD following stem cell transplantation.5 The most commonly used therapies include high-dose systemic steroids and anti–thymocyte globulin that attacks activated donor T cells.6 Other treatment modalities, including anti–tumor necrosis factor agents and antibodies to CD20, have been reported to be effective in steroid-refractory GVHD.2 The major drawback of systemic steroids is an increase in the risk for sepsis and infection; therefore, these patients should be diligently screened for infection and covered with antibiotics and antifungals. Extracorporeal photopheresis is another treatment modality that does not cause generalized immunosuppression but is not well studied in the setting of acute GVHD following OLT.6
Prevention
Acute GVHD following OLT can be prevented by eliminating donor T lymphocytes from the liver before transplantation. However, because the incidence of acute GVHD following OLT is very low, this approach is not routinely taken.2
Conclusion
Acute GVHD following liver transplantation is a rare complication; however, it has high mortality, necessitating further research regarding treatment and prevention. Early recognition and treatment of this condition can improve outcomes. Dermatologists should be familiar with the skin manifestations of acute GVHD following liver transplantation due to the rising number of cases of solid-organ transplantation.
Acute graft-vs-host disease (GVHD) is a T-cell mediated immunogenic response in which T lymphocytes from a donor regard host tissue as foreign and attack it in the setting of immunosuppression.1 The most common cause of acute GVHD is allogeneic stem cell transplantation, with solid-organ transplantation being a much less common cause.2 The incidence of acute GVHD following orthotopic liver transplantation (OLT) is 0.1%, as reported by the United Network for Organ Sharing, compared to an incidence of 40% to 60% in hematopoietic stem cell transplant recipients.3,4
Early recognition and treatment of acute GVHD following liver transplantation is imperative, as the mortality rate is 85% to 90%.2 We present a case of acute GVHD in a liver transplantation patient, with a focus on diagnostic criteria and comparison to acute GVHD following hematopoietic stem cell transplantation.
Case Report
A 68-year-old woman with a history of hepatitis C virus infection, hepatocellular carcinoma, and OLT 1 month prior presented to the hospital with fever and abdominal cellulitis in close proximity to the surgical site of 1 week’s duration. The patient was started on vancomycin and cefepime; pan cultures were performed.
At 10 days of hospitalization, the patient developed a pruritic, nontender, erythematous rash on the abdomen, with extension onto the chest and legs. The rash was associated with low-grade fever but not with diarrhea. Physical examination was notable for a few erythematous macules and scattered papules over the neck and chest and a large erythematous plaque with multiple ecchymoses over the lower abdomen (Figure 1A). Erythematous macules and papules coalescing into plaques were present on the lower back (Figure 1B) and proximal thighs. Oral, ocular, and genital lesions were absent.
The differential diagnosis included drug reaction, viral infection, and acute GVHD. A skin biopsy was performed from the left side of the chest. Cefepime and vancomycin were discontinued; triamcinolone ointment 0.1% twice daily and antihistamines as needed for itching were started.
Over a 2-day period, the rash progressed to diffuse erythematous papules over the chest (Figure 2A) and bilateral arms (Figure 2B) including the palms. The patient also developed erythematous papules over the jawline and forehead as well as confluent erythematous plaques over the back with extension of the rash to involve the legs. She also had erythema and swelling bilaterally over the ears. She reported diarrhea. The low-grade fever resolved.
Laboratory review showed new-onset pancytopenia, normal liver function, and an elevated creatinine level of 2.3 mg/dL (reference range, 0.6–1.2 mg/dL), consistent with the patient’s baseline of stage 3 chronic kidney disease. Polymerase chain reaction analysis for cytomegalovirus was negative. Histology revealed vacuolar interface dermatitis with apoptotic keratinocytes, consistent with grade I GVHD (Figure 3). Duodenal biopsy revealed rare patchy glands with increased apoptosis, compatible with grade I GVHD.
The patient was started on intravenous methylprednisolone 1 mg/kg for 3 days, then transitioned to an oral steroid taper, with improvement of the rash and other systemic symptoms.
Comment
GVHD Subtypes
The 2 types of GVHD are humoral and cellular.5 The humoral type results from ABO blood type incompatibility between donor and recipient and causes mild hemolytic anemia and fever. The cellular type is directed against major histocompatibility complexes and is associated with high morbidity and mortality.
Presentation of GVHD
Acute GVHD following OLT usually occurs 3 to 5 weeks after transplantation,6 as in our patient. Symptoms include rash, fever, pancytopenia, and diarrhea.2 Skin is the most commonly involved organ in acute GVHD; rash is the earliest manifestation.1 The rash can be asymptomatic or associated with pain and pruritus. Initial cutaneous manifestations include palmar erythema and erythematous to violaceous discoloration of the face and ears. A diffuse maculopapular rash can develop, involving the face, abdomen, and trunk. The rash may progress to formation of bullae or skin sloughing, resembling Stevens-Johnson syndrome or toxic epidermal necrolysis.1 The skin manifestation of acute GVHD following OLT is similar to hematopoietic stem cell transplantation (Table).7,8
Pancytopenia is a common manifestation of GVHD following liver transplantation and is rarely seen following hematopoietic stem cell transplantation.7 Donor lymphocytes engraft and proliferate in the bone marrow, attacking recipient hematopoietic stem cells. It is important to note that more common causes of cytopenia following liver transplantation, including infection and drug-induced bone marrow suppression, should be ruled out before diagnosing acute GVHD.6
Acute GVHD can affect the gastrointestinal tract, causing diarrhea; however, other infectious and medication-induced causes of diarrhea also should be considered.6 In contrast to hematopoietic stem cell transplantation, in which the liver is usually involved,1 the liver is spared in acute GVHD following liver transplantation.5
Diagnosis of GVHD
The diagnosis of acute GVHD following liver transplantation can be challenging because the clinical manifestations can be caused by a drug reaction or viral infection, such as cytomegalovirus infection.2 Patients who are older than 50 years and glucose intolerant are at a higher risk of acute GVHD following OLT. The combination of younger donor age and the presence of an HLA class I match also increases the risk of acute GVHD.6 The diagnosis of acute GVHD is confirmed with biopsy of the skin or gastrointestinal tract.
Morbidity and Mortality of GVHD
Because of the high morbidity and mortality associated with acute GVHD following liver transplantation, early diagnosis and treatment are crucial.5 Death in patients with acute GVHD following OLT is mainly attributable to sepsis, multiorgan failure, and gastrointestinal tract bleeding.6 It remains unclear whether this high mortality is associated with delayed diagnosis due to nonspecific signs of acute GVHD following OLT or to the lack of appropriate treatment guidelines.6
Treatment Options
Because of the low incidence of acute GVHD following OLT, most treatment modalities are extrapolated from the literature on acute GVHD following stem cell transplantation.5 The most commonly used therapies include high-dose systemic steroids and anti–thymocyte globulin that attacks activated donor T cells.6 Other treatment modalities, including anti–tumor necrosis factor agents and antibodies to CD20, have been reported to be effective in steroid-refractory GVHD.2 The major drawback of systemic steroids is an increase in the risk for sepsis and infection; therefore, these patients should be diligently screened for infection and covered with antibiotics and antifungals. Extracorporeal photopheresis is another treatment modality that does not cause generalized immunosuppression but is not well studied in the setting of acute GVHD following OLT.6
Prevention
Acute GVHD following OLT can be prevented by eliminating donor T lymphocytes from the liver before transplantation. However, because the incidence of acute GVHD following OLT is very low, this approach is not routinely taken.2
Conclusion
Acute GVHD following liver transplantation is a rare complication; however, it has high mortality, necessitating further research regarding treatment and prevention. Early recognition and treatment of this condition can improve outcomes. Dermatologists should be familiar with the skin manifestations of acute GVHD following liver transplantation due to the rising number of cases of solid-organ transplantation.
- Hu SW, Cotliar J. Acute graft-versus-host disease following hematopoietic stem-cell transplantation. Dermatol Ther. 2011;24:411-423.
- Akbulut S, Yilmaz M, Yilmaz S. Graft-versus-host disease after liver transplantation: a comprehensive literature review. World J Gastroenterol. 2012;18:5240-5248.
- Taylor AL, Gibbs P, Bradley JA. Acute graft versus host disease following liver transplantation: the enemy within. Am J Transplant. 2004;4:466-474.
- Jagasia M, Arora M, Flowers ME, et al. Risk factor for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119:296-307.
- Kang WH, Hwang S, Song GW, et al. Acute graft-vs-host disease after liver transplantation: experience at a high-volume liver transplantation center in Korea. Transplant Proc. 2016;48:3368-3372.
- Murali AR, Chandra S, Stewart Z, et al. Graft versus host disease after liver transplantation in adults: a case series, review of literature, and an approach to management. Transplantation. 2016;100:2661-2670.
- Chaib E, Silva FD, Figueira ER, et al. Graft-versus-host disease after liver transplantation. Clinics (Sao Paulo). 2011;66:1115-1118.
- Barton-Burke M, Dwinell DM, Kafkas L, et al. Graft-versus-host disease: a complex long-term side effect of hematopoietic stem cell transplant. Oncology (Williston Park). 2008;22(11 Suppl Nurse Ed):31-45.
- Hu SW, Cotliar J. Acute graft-versus-host disease following hematopoietic stem-cell transplantation. Dermatol Ther. 2011;24:411-423.
- Akbulut S, Yilmaz M, Yilmaz S. Graft-versus-host disease after liver transplantation: a comprehensive literature review. World J Gastroenterol. 2012;18:5240-5248.
- Taylor AL, Gibbs P, Bradley JA. Acute graft versus host disease following liver transplantation: the enemy within. Am J Transplant. 2004;4:466-474.
- Jagasia M, Arora M, Flowers ME, et al. Risk factor for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119:296-307.
- Kang WH, Hwang S, Song GW, et al. Acute graft-vs-host disease after liver transplantation: experience at a high-volume liver transplantation center in Korea. Transplant Proc. 2016;48:3368-3372.
- Murali AR, Chandra S, Stewart Z, et al. Graft versus host disease after liver transplantation in adults: a case series, review of literature, and an approach to management. Transplantation. 2016;100:2661-2670.
- Chaib E, Silva FD, Figueira ER, et al. Graft-versus-host disease after liver transplantation. Clinics (Sao Paulo). 2011;66:1115-1118.
- Barton-Burke M, Dwinell DM, Kafkas L, et al. Graft-versus-host disease: a complex long-term side effect of hematopoietic stem cell transplant. Oncology (Williston Park). 2008;22(11 Suppl Nurse Ed):31-45.
Practice Points
- Acute graft-vs-host disease (GVHD) is a T cell–mediated reaction in which donor T lymphocytes attack host tissue in the setting of immunosuppression.
- Acute GVHD is more common in allogeneic stem cell transplantation but can occur in the setting of solid organ transplantation.
- Symptoms of acute GVHD include rash with or without pruritus, fever, pancytopenia, and diarrhea.
- Early recognition and treatment with systemic steroids can improve mortality.
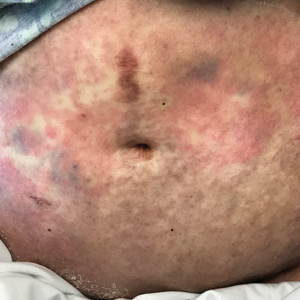
4-year-old girl • genital discomfort and dysuria • clitoral hood swelling • Blood blister on the labia minora • Dx?
THE CASE
A 4-year-old girl presented to her pediatrician with genital discomfort and dysuria of 6 months’ duration. The patient’s mother said that 3 days earlier, she noticed a tear near the child’s clitoris and a scab on the labia minora that the mother attributed to minor trauma from scratching. The pediatrician was concerned about genital trauma from sexual abuse and referred the patient to the emergency department, where a report with child protective services (CPS) was filed. The mother reported that the patient and her 8-year-old sibling spent 3 to 4 hours a day with a babysitter, who was always supervised, and the parents had no concerns about possible sexual abuse.
Physical examination by our institution’s Child Protection Team revealed clitoral hood swelling with subepithelial hemorrhages, a blood blister on the right labia minora, a fissure and subepithelial hemorrhages on the posterior fourchette, and a thin depigmented figure-of-eight lesion around the vulva and anus.
THE DIAGNOSIS
Since the clinical findings were consistent with prepubertal lichen sclerosus (LS), the CPS case was closed and the patient was referred to Pediatric Gynecology. Treatment with high-potency topical steroids was initiated with clobetasol ointment 0.05% twice daily for 2 weeks, then once daily for 2 weeks. She was then switched to triamcinolone ointment 0.01% twice daily for 2 weeks, then once daily for 2 weeks. These treatments were enough to stop the LS flare and decrease the anogenital itching.
DISCUSSION
Lichen sclerosus is a chronic inflammatory skin disease that primarily presents in the anogenital region; however, extragenital lesions on the upper extremities, thighs, and breasts have been reported in 15% to 20% of patients.1 Lichen sclerosus most commonly affects females as a result of low estrogen and may occur during puberty or following menopause, but it also is seen in males.1,2 The estimated prevalence of LS in prepubertal girls is 1 in 900.3 The effects of increased estrogen exposure on LS during puberty are not entirely clear. Lichen sclerosus previously was thought to improve with puberty, since it is not as common in women of reproductive age; however, studies have shown persistent symptoms after menarche in some patients.4-6
The pathogenesis of LS is multifactorial, likely with an autoimmune component, as it often is associated with other autoimmune findings such as thyroiditis, alopecia, pernicious anemia, and vitiligo.2 Diagnosis of prepubertal LS usually is made based on a review of the patient’s history and clinical examination. Presenting symptoms may include pruritus, skin irritation, vulvar pain, dysuria, bleeding from excoriations, fissures, and constipation.1,3,7
On physical examination, LS can present on the anogenital skin as smooth white spots or wrinkled, blotchy, atrophic patches. The skin around the vaginal opening and anus is thin and often is described as resembling parchment or cigarette paper in a figure-of-eight pattern (FIGURE 1A). Vulvar and anal fissures and subepithelial hemorrhages with the appearance of blood blisters also can be found (FIGURE 1B).8 Affected areas are fragile and susceptible to minor trauma, which may result in bruising or bleeding (FIGURE 1C).

Over time, scarring can occur and may result in disruption of the anogenital architecture—specifically loss of the labia minora, narrowing of the introitus, and burying of the clitoris.1,2 These changes can be similar to the scarring seen in postmenopausal women with LS.
Continue to: The differential diagnosis...
The differential diagnosis for prepubertal LS includes vitiligo, lichen planus, lichen simplex chronicus, psoriasis, eczema, vulvovaginitis, contact dermatitis, and trauma.2,7 On average, it takes 1 to 2 years after onset of symptoms before a correct diagnosis of prepubertal LS is made, and trauma and/or sexual abuse often are first suspected.7,9 For clinicians who are unfamiliar with prepubertal LS, the clinical findings of anogenital bruising and bleeding understandably may be suggestive of abuse. It is important to note that diagnosis of LS does not preclude the possibility of sexual abuse; in some cases, LS can be triggered or exacerbated by anogenital trauma, known as the Koebner phenomenon.2
Treatment. After the diagnosis of prepubertal LS is established, the goals of treatment are to provide symptom relief and prevent scarring of the external genitalia. To our knowledge, there have been no randomized controlled trials for treatment of LS in prepubertal girls. In general, acute symptoms are treated with high-potency topical steroids, such as clobetasol propionate or betamethasone valerate, and treatment regimens are variable.7
LS has an unpredictable clinical course and there often are recurrences that require repeat courses of topical steroids.9 Since concurrent bacterial infection is common,10 genital cultures should be obtained prior to initiation of topical steroids if an infection is suspected.
Topical calcineurin inhibitors have been used successfully, but proof of their effectiveness is limited to case reports in the literature.7 Surgical treatment of LS typically is reserved for complications associated with symptomatic adhesions that are refractory to medical management.7,11 Vulvar hygiene is paramount to symptom control, and topical emollients can be used to manage minor irritation.7,8 In our patient, clobetasol and triamcinolone ointments were enough to stop the LS flare and decrease the anogenital itching.
THE TAKEAWAY
Although LS has very characteristic skin findings, the diagnosis continues to be challenging for physicians who are unfamiliar with this condition. Failure to recognize prepubertal LS not only delays diagnosis and treatment but also may lead to repeated genital examinations and investigation by CPS for suspected sexual abuse. As with any genital complaint in a prepubertal girl, diagnosis of LS should not preclude appropriate screening for sexual abuse. Although providers should be vigilant about potential sexual abuse, familiarity with skin conditions that mimic genital trauma is essential.
CORRESPONDENCE
Monica Rosen, MD, L4000 Women’s Hospital, 1500 E Medical Center Drive, SPC 5276 Ann Arbor, MI 48109; [email protected]
1. Powell JJ, Wojnarowska F. Lichen sclerosus. Lancet. 1999;353:1777-1783.
2. Murphy R. Lichen sclerosus. Dermatol Clin. 2010;28:707-715.
3. Powell J, Wojnarowska F. Childhood vulvar lichen sclerosus: an increasingly common problem. J Am Acad Dermatol. 2001;44:803-806.
4. Powell J, Wojnarowska F. Childhood vulvar lichen sclerosus. The course after puberty. J Reprod Med. 2002;47:706-709.
5. Smith SD, Fischer G. Childhood onset vulvar lichen sclerosus does not resolve at puberty: a prospective case series. Pediatr Dermatol. 2009;26:725-729.
6. Focseneanu MA, Gupta M, Squires KC, et al. The course of lichen sclerosus diagnosed prior to puberty. J Pediatr Adolesc Gynecol. 2013;26:153-155.
7. Bercaw-Pratt JL, Boardman LA, Simms-Cendan JS. Clinical recommendation: pediatric lichen sclerosus. J Pediatr Adolesc Gynecol. 2014;27:111-116.
8. Jenny C, Kirby P, Fuquay D. Genital lichen sclerosus mistaken for child sexual abuse. Pediatrics. 1989;83:597-599.
9. Dendrinos ML, Quint EH. Lichen sclerosus in children and adolescents. Curr Opin Obstet Gynecol. 2013;25:370-374.
10. Lagerstedt M, Karvinen K, Joki-Erkkila M, et al. Childhood lichen sclerosus—a challenge for clinicians. Pediatr Dermatol. 2013;30:444-450.
11. Gurumurthy M, Morah N, Gioffre G, et al. The surgical management of complications of vulval lichen sclerosus. Eur J Obstet Gynecol Reprod Biol. 2012;162:79-82.
THE CASE
A 4-year-old girl presented to her pediatrician with genital discomfort and dysuria of 6 months’ duration. The patient’s mother said that 3 days earlier, she noticed a tear near the child’s clitoris and a scab on the labia minora that the mother attributed to minor trauma from scratching. The pediatrician was concerned about genital trauma from sexual abuse and referred the patient to the emergency department, where a report with child protective services (CPS) was filed. The mother reported that the patient and her 8-year-old sibling spent 3 to 4 hours a day with a babysitter, who was always supervised, and the parents had no concerns about possible sexual abuse.
Physical examination by our institution’s Child Protection Team revealed clitoral hood swelling with subepithelial hemorrhages, a blood blister on the right labia minora, a fissure and subepithelial hemorrhages on the posterior fourchette, and a thin depigmented figure-of-eight lesion around the vulva and anus.
THE DIAGNOSIS
Since the clinical findings were consistent with prepubertal lichen sclerosus (LS), the CPS case was closed and the patient was referred to Pediatric Gynecology. Treatment with high-potency topical steroids was initiated with clobetasol ointment 0.05% twice daily for 2 weeks, then once daily for 2 weeks. She was then switched to triamcinolone ointment 0.01% twice daily for 2 weeks, then once daily for 2 weeks. These treatments were enough to stop the LS flare and decrease the anogenital itching.
DISCUSSION
Lichen sclerosus is a chronic inflammatory skin disease that primarily presents in the anogenital region; however, extragenital lesions on the upper extremities, thighs, and breasts have been reported in 15% to 20% of patients.1 Lichen sclerosus most commonly affects females as a result of low estrogen and may occur during puberty or following menopause, but it also is seen in males.1,2 The estimated prevalence of LS in prepubertal girls is 1 in 900.3 The effects of increased estrogen exposure on LS during puberty are not entirely clear. Lichen sclerosus previously was thought to improve with puberty, since it is not as common in women of reproductive age; however, studies have shown persistent symptoms after menarche in some patients.4-6
The pathogenesis of LS is multifactorial, likely with an autoimmune component, as it often is associated with other autoimmune findings such as thyroiditis, alopecia, pernicious anemia, and vitiligo.2 Diagnosis of prepubertal LS usually is made based on a review of the patient’s history and clinical examination. Presenting symptoms may include pruritus, skin irritation, vulvar pain, dysuria, bleeding from excoriations, fissures, and constipation.1,3,7
On physical examination, LS can present on the anogenital skin as smooth white spots or wrinkled, blotchy, atrophic patches. The skin around the vaginal opening and anus is thin and often is described as resembling parchment or cigarette paper in a figure-of-eight pattern (FIGURE 1A). Vulvar and anal fissures and subepithelial hemorrhages with the appearance of blood blisters also can be found (FIGURE 1B).8 Affected areas are fragile and susceptible to minor trauma, which may result in bruising or bleeding (FIGURE 1C).
Over time, scarring can occur and may result in disruption of the anogenital architecture—specifically loss of the labia minora, narrowing of the introitus, and burying of the clitoris.1,2 These changes can be similar to the scarring seen in postmenopausal women with LS.
Continue to: The differential diagnosis...
The differential diagnosis for prepubertal LS includes vitiligo, lichen planus, lichen simplex chronicus, psoriasis, eczema, vulvovaginitis, contact dermatitis, and trauma.2,7 On average, it takes 1 to 2 years after onset of symptoms before a correct diagnosis of prepubertal LS is made, and trauma and/or sexual abuse often are first suspected.7,9 For clinicians who are unfamiliar with prepubertal LS, the clinical findings of anogenital bruising and bleeding understandably may be suggestive of abuse. It is important to note that diagnosis of LS does not preclude the possibility of sexual abuse; in some cases, LS can be triggered or exacerbated by anogenital trauma, known as the Koebner phenomenon.2
Treatment. After the diagnosis of prepubertal LS is established, the goals of treatment are to provide symptom relief and prevent scarring of the external genitalia. To our knowledge, there have been no randomized controlled trials for treatment of LS in prepubertal girls. In general, acute symptoms are treated with high-potency topical steroids, such as clobetasol propionate or betamethasone valerate, and treatment regimens are variable.7
LS has an unpredictable clinical course and there often are recurrences that require repeat courses of topical steroids.9 Since concurrent bacterial infection is common,10 genital cultures should be obtained prior to initiation of topical steroids if an infection is suspected.
Topical calcineurin inhibitors have been used successfully, but proof of their effectiveness is limited to case reports in the literature.7 Surgical treatment of LS typically is reserved for complications associated with symptomatic adhesions that are refractory to medical management.7,11 Vulvar hygiene is paramount to symptom control, and topical emollients can be used to manage minor irritation.7,8 In our patient, clobetasol and triamcinolone ointments were enough to stop the LS flare and decrease the anogenital itching.
THE TAKEAWAY
Although LS has very characteristic skin findings, the diagnosis continues to be challenging for physicians who are unfamiliar with this condition. Failure to recognize prepubertal LS not only delays diagnosis and treatment but also may lead to repeated genital examinations and investigation by CPS for suspected sexual abuse. As with any genital complaint in a prepubertal girl, diagnosis of LS should not preclude appropriate screening for sexual abuse. Although providers should be vigilant about potential sexual abuse, familiarity with skin conditions that mimic genital trauma is essential.
CORRESPONDENCE
Monica Rosen, MD, L4000 Women’s Hospital, 1500 E Medical Center Drive, SPC 5276 Ann Arbor, MI 48109; [email protected]
THE CASE
A 4-year-old girl presented to her pediatrician with genital discomfort and dysuria of 6 months’ duration. The patient’s mother said that 3 days earlier, she noticed a tear near the child’s clitoris and a scab on the labia minora that the mother attributed to minor trauma from scratching. The pediatrician was concerned about genital trauma from sexual abuse and referred the patient to the emergency department, where a report with child protective services (CPS) was filed. The mother reported that the patient and her 8-year-old sibling spent 3 to 4 hours a day with a babysitter, who was always supervised, and the parents had no concerns about possible sexual abuse.
Physical examination by our institution’s Child Protection Team revealed clitoral hood swelling with subepithelial hemorrhages, a blood blister on the right labia minora, a fissure and subepithelial hemorrhages on the posterior fourchette, and a thin depigmented figure-of-eight lesion around the vulva and anus.
THE DIAGNOSIS
Since the clinical findings were consistent with prepubertal lichen sclerosus (LS), the CPS case was closed and the patient was referred to Pediatric Gynecology. Treatment with high-potency topical steroids was initiated with clobetasol ointment 0.05% twice daily for 2 weeks, then once daily for 2 weeks. She was then switched to triamcinolone ointment 0.01% twice daily for 2 weeks, then once daily for 2 weeks. These treatments were enough to stop the LS flare and decrease the anogenital itching.
DISCUSSION
Lichen sclerosus is a chronic inflammatory skin disease that primarily presents in the anogenital region; however, extragenital lesions on the upper extremities, thighs, and breasts have been reported in 15% to 20% of patients.1 Lichen sclerosus most commonly affects females as a result of low estrogen and may occur during puberty or following menopause, but it also is seen in males.1,2 The estimated prevalence of LS in prepubertal girls is 1 in 900.3 The effects of increased estrogen exposure on LS during puberty are not entirely clear. Lichen sclerosus previously was thought to improve with puberty, since it is not as common in women of reproductive age; however, studies have shown persistent symptoms after menarche in some patients.4-6
The pathogenesis of LS is multifactorial, likely with an autoimmune component, as it often is associated with other autoimmune findings such as thyroiditis, alopecia, pernicious anemia, and vitiligo.2 Diagnosis of prepubertal LS usually is made based on a review of the patient’s history and clinical examination. Presenting symptoms may include pruritus, skin irritation, vulvar pain, dysuria, bleeding from excoriations, fissures, and constipation.1,3,7
On physical examination, LS can present on the anogenital skin as smooth white spots or wrinkled, blotchy, atrophic patches. The skin around the vaginal opening and anus is thin and often is described as resembling parchment or cigarette paper in a figure-of-eight pattern (FIGURE 1A). Vulvar and anal fissures and subepithelial hemorrhages with the appearance of blood blisters also can be found (FIGURE 1B).8 Affected areas are fragile and susceptible to minor trauma, which may result in bruising or bleeding (FIGURE 1C).
Over time, scarring can occur and may result in disruption of the anogenital architecture—specifically loss of the labia minora, narrowing of the introitus, and burying of the clitoris.1,2 These changes can be similar to the scarring seen in postmenopausal women with LS.
Continue to: The differential diagnosis...
The differential diagnosis for prepubertal LS includes vitiligo, lichen planus, lichen simplex chronicus, psoriasis, eczema, vulvovaginitis, contact dermatitis, and trauma.2,7 On average, it takes 1 to 2 years after onset of symptoms before a correct diagnosis of prepubertal LS is made, and trauma and/or sexual abuse often are first suspected.7,9 For clinicians who are unfamiliar with prepubertal LS, the clinical findings of anogenital bruising and bleeding understandably may be suggestive of abuse. It is important to note that diagnosis of LS does not preclude the possibility of sexual abuse; in some cases, LS can be triggered or exacerbated by anogenital trauma, known as the Koebner phenomenon.2
Treatment. After the diagnosis of prepubertal LS is established, the goals of treatment are to provide symptom relief and prevent scarring of the external genitalia. To our knowledge, there have been no randomized controlled trials for treatment of LS in prepubertal girls. In general, acute symptoms are treated with high-potency topical steroids, such as clobetasol propionate or betamethasone valerate, and treatment regimens are variable.7
LS has an unpredictable clinical course and there often are recurrences that require repeat courses of topical steroids.9 Since concurrent bacterial infection is common,10 genital cultures should be obtained prior to initiation of topical steroids if an infection is suspected.
Topical calcineurin inhibitors have been used successfully, but proof of their effectiveness is limited to case reports in the literature.7 Surgical treatment of LS typically is reserved for complications associated with symptomatic adhesions that are refractory to medical management.7,11 Vulvar hygiene is paramount to symptom control, and topical emollients can be used to manage minor irritation.7,8 In our patient, clobetasol and triamcinolone ointments were enough to stop the LS flare and decrease the anogenital itching.
THE TAKEAWAY
Although LS has very characteristic skin findings, the diagnosis continues to be challenging for physicians who are unfamiliar with this condition. Failure to recognize prepubertal LS not only delays diagnosis and treatment but also may lead to repeated genital examinations and investigation by CPS for suspected sexual abuse. As with any genital complaint in a prepubertal girl, diagnosis of LS should not preclude appropriate screening for sexual abuse. Although providers should be vigilant about potential sexual abuse, familiarity with skin conditions that mimic genital trauma is essential.
CORRESPONDENCE
Monica Rosen, MD, L4000 Women’s Hospital, 1500 E Medical Center Drive, SPC 5276 Ann Arbor, MI 48109; [email protected]
1. Powell JJ, Wojnarowska F. Lichen sclerosus. Lancet. 1999;353:1777-1783.
2. Murphy R. Lichen sclerosus. Dermatol Clin. 2010;28:707-715.
3. Powell J, Wojnarowska F. Childhood vulvar lichen sclerosus: an increasingly common problem. J Am Acad Dermatol. 2001;44:803-806.
4. Powell J, Wojnarowska F. Childhood vulvar lichen sclerosus. The course after puberty. J Reprod Med. 2002;47:706-709.
5. Smith SD, Fischer G. Childhood onset vulvar lichen sclerosus does not resolve at puberty: a prospective case series. Pediatr Dermatol. 2009;26:725-729.
6. Focseneanu MA, Gupta M, Squires KC, et al. The course of lichen sclerosus diagnosed prior to puberty. J Pediatr Adolesc Gynecol. 2013;26:153-155.
7. Bercaw-Pratt JL, Boardman LA, Simms-Cendan JS. Clinical recommendation: pediatric lichen sclerosus. J Pediatr Adolesc Gynecol. 2014;27:111-116.
8. Jenny C, Kirby P, Fuquay D. Genital lichen sclerosus mistaken for child sexual abuse. Pediatrics. 1989;83:597-599.
9. Dendrinos ML, Quint EH. Lichen sclerosus in children and adolescents. Curr Opin Obstet Gynecol. 2013;25:370-374.
10. Lagerstedt M, Karvinen K, Joki-Erkkila M, et al. Childhood lichen sclerosus—a challenge for clinicians. Pediatr Dermatol. 2013;30:444-450.
11. Gurumurthy M, Morah N, Gioffre G, et al. The surgical management of complications of vulval lichen sclerosus. Eur J Obstet Gynecol Reprod Biol. 2012;162:79-82.
1. Powell JJ, Wojnarowska F. Lichen sclerosus. Lancet. 1999;353:1777-1783.
2. Murphy R. Lichen sclerosus. Dermatol Clin. 2010;28:707-715.
3. Powell J, Wojnarowska F. Childhood vulvar lichen sclerosus: an increasingly common problem. J Am Acad Dermatol. 2001;44:803-806.
4. Powell J, Wojnarowska F. Childhood vulvar lichen sclerosus. The course after puberty. J Reprod Med. 2002;47:706-709.
5. Smith SD, Fischer G. Childhood onset vulvar lichen sclerosus does not resolve at puberty: a prospective case series. Pediatr Dermatol. 2009;26:725-729.
6. Focseneanu MA, Gupta M, Squires KC, et al. The course of lichen sclerosus diagnosed prior to puberty. J Pediatr Adolesc Gynecol. 2013;26:153-155.
7. Bercaw-Pratt JL, Boardman LA, Simms-Cendan JS. Clinical recommendation: pediatric lichen sclerosus. J Pediatr Adolesc Gynecol. 2014;27:111-116.
8. Jenny C, Kirby P, Fuquay D. Genital lichen sclerosus mistaken for child sexual abuse. Pediatrics. 1989;83:597-599.
9. Dendrinos ML, Quint EH. Lichen sclerosus in children and adolescents. Curr Opin Obstet Gynecol. 2013;25:370-374.
10. Lagerstedt M, Karvinen K, Joki-Erkkila M, et al. Childhood lichen sclerosus—a challenge for clinicians. Pediatr Dermatol. 2013;30:444-450.
11. Gurumurthy M, Morah N, Gioffre G, et al. The surgical management of complications of vulval lichen sclerosus. Eur J Obstet Gynecol Reprod Biol. 2012;162:79-82.

Would you be able to recognize the signs and symptoms of this particular drug overdose?
CASE 1
Two days after reviving her boyfriend with naloxone, a woman and her 30-year-old boyfriend presented to our family medicine clinic. They explained that he had injected heroin and shortly thereafter he stopped breathing and his lips turned blue. The patient’s girlfriend did not call emergency medical services (EMS) at the time because she was afraid of getting arrested due to past incarceration for possession of illegal drugs. Instead, she revived him with naloxone that she found in his bag.
Both the patient and his girlfriend were scared and surprised by his “terrible reaction,” as he had previously purchased heroin from the same dealer and used the same dose without similar effects. However, the patient did note that the drug he purchased this time had a bright white tinge, when normally the drug was light yellow.
On physical examination, the patient’s heart rate and blood pressure were normal. There were needle track marks on both forearms, elbows, and upper arms. A laboratory workup obtained during this visit revealed anemia and a normal basic metabolic panel. A hepatitis C virus antibody test was positive, and a hepatic function panel revealed elevated transaminase levels. Urine toxicology was positive for opioids and negative for other substances.
CASE 2
A 58-year-old man with a history of chronic hepatitis C, polysubstance abuse, and schizophrenia was transported to the emergency department by EMS after his family found him unresponsive in his bedroom. The patient had agonal breathing when EMS arrived, so they administered naloxone (4 mg intranasal and 4 mg intravenous). His breathing improved, but his mental status did not. He was still obtunded upon arrival in the emergency department and vomited 4 tan-colored patches. The patient was tachycardic (heart rate, 108 beats/min), hypertensive (blood pressure, 189/95 mm Hg), and had rapid shallow breathing (respiratory rate, 38 breaths/min). He was intubated for airway protection, at which time 2 more tan-colored patches were removed from his pharynx.
Laboratory evaluation revealed an acute kidney injury with a high anion metabolic acidosis. A hepatic function panel showed elevated transaminase levels. Plasma acetaminophen and salicylate levels were normal. A computed tomography head scan was normal. Urine toxicology was negative for opioids but was positive for cocaine and benzodiazepines.
THE DIAGNOSIS
Opioid overdose caused the acute respiratory depression in both cases. In Case 1, the patient unknowingly overdosed on heroin laced with fentanyl, known as China White, which likely caused the drug’s bright white tinge. In Case 2, the patient’s overdose was the result of oral ingestion of fentanyl patches. (Limited urine toxicology was negative for opiates because fentanyl is a fully synthetic opioid that shows up only with a specific or extended assay. More on this in a bit.)
DISCUSSION
The fatal drug overdose epidemic in the United States is growing. From 2000 to 2014, the mortality rate from drug overdose increased by 137%, including a 200% increase in the rate of overdose deaths related to opioids (ie, pain medications, heroin).1 Between 2013 and 2014, the age-adjusted mortality rate related to methadone, a synthetic opioid, remained unchanged; however, age-adjusted mortality rates related to natural and semisynthetic opioid pain medications, heroin, and synthetic opioids other than methadone (eg, fentanyl) increased by 9%, 26%, and 80%, respectively. In 2014, a sharp increase in overdose deaths related to synthetic opioids other than methadone coincided with law enforcement reports of increased availability of illegal fentanyl; however, the toxicology panel used by coroners and medical examiners at that time could not distinguish between illegal and prescription fentanyl.1
Continue to: Among 70,237 drug overdose deaths...
Among 70,237 drug overdose deaths in the United States in 2017, 47,600 (67.8%) involved an opioid. From 2013 to 2017, drug overdose death rates increased in 35 of 50 states and the District of Columbia, and significant increases in death rates involving synthetic opioids occurred in 15 out of 20 states, likely driven by illicitly manufactured fentanyl.2
Fentanyl-laced heroin: More common, but not new
In October 1991, 3-methylfentanyl was identified in 16 fatal drug overdoses in Allegheny County, Pennsylvania, contributing to a 4-fold increase in overdose deaths compared to the previous year. Fentanyl mixed with heroin and other drugs is commonly found in the Midwest, Northeast, and Southern regions of the United States; in 2014, more than 80% of fentanyl confiscations occurred in 10 states within these regions, with the highest incidence occurring in Ohio.3
When combined with fentanyl, heroin becomes 50 to 100 times more potent, resulting in a subjective high with exaggerated central nervous system depression manifesting as lethargy, miosis, and respiratory depression.4 Most drug users are unaware and unable to identify when heroin is laced with fentanyl, which may contribute to the rise in deaths from unintentional drug overdose.1,5,6
Oral abuse of fentanyl patches can be fatal
Outcomes from oral abuse of fentanyl patches have ranged from transient overdose symptoms, such as lethargy and respiratory depression, to death.7-9 When administered in a medical setting, transbuccal fentanyl has a bioavailability of 50% to 65% across the buccal membrane. Nearly 20% of the drug escapes hepatic first pass metabolism when fentanyl patches are ingested orally and enters the systemic circulation, resulting in severe overdose and potentially death. Prolonged chewing and sucking on fentanyl patches increases the contact time with the buccal membrane, resulting in increased systemic absorption compared to oral ingestion without chewing/sucking.7-9
Urine toxicology screening detects compounds based on a chemical assay for drugs—generally codeine, morphine, and their metabolites. Because fentanyl is a fully synthetic opioid, its structure is not
Continue to: Survival of fentanyl overdose depends on naloxone availability
Survival of fentanyl overdose depends on naloxone availability
Naloxone is a safe and effective antidote to an opioid overdose. It comes in 3 preparations, including intramuscular and subcutaneous injections and an intranasal spray.12 Concerns that naloxone will harm patients with opioid dependence are unfounded. Naloxone can induce symptoms of opioid withdrawal, such as yawning, lacrimation, piloerection, diaphoresis, myalgia, vomiting, and diarrhea. While these withdrawal symptoms are unpleasant, they are not life threatening.12 Due to its high potency, large doses of naloxone (ie, 4–16 mg) are required to reverse the effects of a fentanyl overdose.13 Intranasal naloxone hydrochloride 4 mg delivered in a single spray is preferred due to the ease of administration. Repeat doses may be necessary if respiratory depression continues or recurs prior to the arrival of emergency medical services. Increasing the availability of naloxone to first responders has the potential to save many lives.6
THE TAKEAWAY
Fentanyl is a major contributor to the growing drug overdose crisis in the United States. When laced with heroin or consumed orally in the form of transdermal patches, fentanyl becomes more potent and is increasingly fatal. It’s crucial that primary care physicians be able to identify and educate at-risk patients about the fatal consequences of fentanyl overdose and coordinate care to help get them into an appropriate rehabilitation program.
In order to quickly recognize the signs of fentanyl-related overdose, it’s important to be alert for this possibility. At the bedside, the most easily recognized abnormality associated with fentanyl or other opioid overdose is a decline in respiratory rate culminating in apnea.10 A respiratory rate of 12 breaths/min or less in a patient who is not in physiologic sleep strongly suggests acute opioid intoxication, particularly when accompanied by miosis or stupor. Other signs include bradycardia, hypotension, and seizures from anoxia.10
Apart from the severity of symptoms, it is hard to clinically distinguish fentanyl overdose from other opiate overdose incidents. Given the degree to which illegal opiates are contaminated with fentanyl in the United States,3 it is appropriate to screen for fentanyl with extended panel urine toxicology testing in patients with suspected opioid overdose.
CORRESPONDENCE
Jaividhya Dasarathy, MD, 2500 MetroHealth Medical Center, Cleveland, OH 44109; [email protected]
1. Rudd RA, Aleshire N, Zibbell JE, et al. Increases in drug and opioid overdose deaths—United States, 2000–2014. MMWR Morb Mortal Wkly Rep. 2016;64:1378-1382.
2. Scholl L, Seth P, Kariisa M, et al. Drug and opioid-involved overdose deaths—United States, 2013–2017. MMWR Morb Mortal Wkly Rep. 2019;67:1419-1427.
3. Hibbs J, Perper J, Winek CL. An outbreak of designer drug-related deaths in Pennsylvania. JAMA. 1991;265:1011-1013.
4. Increases in fentanyl drug confiscations and fentanyl-related overdose fatalities. Centers for Disease Control and Prevention Web site. https://emergency.cdc.gov/han/han00384.asp. Published October 26, 2015. Accessed May 3, 2019.
5. Fentanyl. Centers for Disease Control and Prevention Web site. https://www.cdc.gov/drugoverdose/opioids/fentanyl.html. Updated December 19, 2018. Accessed May 3, 2019.
6. Peterson AB, Gladden RM, Delcher C, et al. Increases in fentanyl-related overdose deaths—Florida and Ohio, 2013–2015. MMWR Morb Mortal Wkly Rep. 2016;65:844-849.
7. Streisand JB, Varvel JR, Stanski DR, et al. Absorption and bioavailability of oral transmucosal fentanyl citrate. Anesthesiology. 1991;75:223-229.
8. Kharasch ED, Whittington D, Hoffer C. Influence of hepatic and intestinal cytochrome P4503A activity on the acute disposition and effects of oral transmucosal fentanyl citrate. Anesthesiology. 2004;101:729-737.
9. Woodall KL, Martin TL, McLellan BA. Oral abuse of fentanyl patches (Duragesic): seven case reports. J Forensic Sci. 2008;53:222-225.
10. Moeller KE, Lee KC, Kissack JC. Urine drug screening: practical guide for clinicians. Mayo Clin Proc. 2008;83:66-76.
11. Appropriate Use of Drug Testing in Clinical Addiction Medicine. American Society of Addiction Medicine Web site. https://www.asam.org/docs/default-source/quality-science/appropriate_use_of_drug_testing_in_clinical-1-(7).pdf?sfvrsn=2. Published April 5, 2017. Accessed May 30, 2019.
12. Boyer EW. Management of opioid analgesic overdose. N Engl J Med. 2012;367:146-155.
13. Drugs@FDA: FDA approved drug products. US Food and Drug Administration Web site. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=208411. Accessed May 22, 2019.
CASE 1
Two days after reviving her boyfriend with naloxone, a woman and her 30-year-old boyfriend presented to our family medicine clinic. They explained that he had injected heroin and shortly thereafter he stopped breathing and his lips turned blue. The patient’s girlfriend did not call emergency medical services (EMS) at the time because she was afraid of getting arrested due to past incarceration for possession of illegal drugs. Instead, she revived him with naloxone that she found in his bag.
Both the patient and his girlfriend were scared and surprised by his “terrible reaction,” as he had previously purchased heroin from the same dealer and used the same dose without similar effects. However, the patient did note that the drug he purchased this time had a bright white tinge, when normally the drug was light yellow.
On physical examination, the patient’s heart rate and blood pressure were normal. There were needle track marks on both forearms, elbows, and upper arms. A laboratory workup obtained during this visit revealed anemia and a normal basic metabolic panel. A hepatitis C virus antibody test was positive, and a hepatic function panel revealed elevated transaminase levels. Urine toxicology was positive for opioids and negative for other substances.
CASE 2
A 58-year-old man with a history of chronic hepatitis C, polysubstance abuse, and schizophrenia was transported to the emergency department by EMS after his family found him unresponsive in his bedroom. The patient had agonal breathing when EMS arrived, so they administered naloxone (4 mg intranasal and 4 mg intravenous). His breathing improved, but his mental status did not. He was still obtunded upon arrival in the emergency department and vomited 4 tan-colored patches. The patient was tachycardic (heart rate, 108 beats/min), hypertensive (blood pressure, 189/95 mm Hg), and had rapid shallow breathing (respiratory rate, 38 breaths/min). He was intubated for airway protection, at which time 2 more tan-colored patches were removed from his pharynx.
Laboratory evaluation revealed an acute kidney injury with a high anion metabolic acidosis. A hepatic function panel showed elevated transaminase levels. Plasma acetaminophen and salicylate levels were normal. A computed tomography head scan was normal. Urine toxicology was negative for opioids but was positive for cocaine and benzodiazepines.
THE DIAGNOSIS
Opioid overdose caused the acute respiratory depression in both cases. In Case 1, the patient unknowingly overdosed on heroin laced with fentanyl, known as China White, which likely caused the drug’s bright white tinge. In Case 2, the patient’s overdose was the result of oral ingestion of fentanyl patches. (Limited urine toxicology was negative for opiates because fentanyl is a fully synthetic opioid that shows up only with a specific or extended assay. More on this in a bit.)
DISCUSSION
The fatal drug overdose epidemic in the United States is growing. From 2000 to 2014, the mortality rate from drug overdose increased by 137%, including a 200% increase in the rate of overdose deaths related to opioids (ie, pain medications, heroin).1 Between 2013 and 2014, the age-adjusted mortality rate related to methadone, a synthetic opioid, remained unchanged; however, age-adjusted mortality rates related to natural and semisynthetic opioid pain medications, heroin, and synthetic opioids other than methadone (eg, fentanyl) increased by 9%, 26%, and 80%, respectively. In 2014, a sharp increase in overdose deaths related to synthetic opioids other than methadone coincided with law enforcement reports of increased availability of illegal fentanyl; however, the toxicology panel used by coroners and medical examiners at that time could not distinguish between illegal and prescription fentanyl.1
Continue to: Among 70,237 drug overdose deaths...
Among 70,237 drug overdose deaths in the United States in 2017, 47,600 (67.8%) involved an opioid. From 2013 to 2017, drug overdose death rates increased in 35 of 50 states and the District of Columbia, and significant increases in death rates involving synthetic opioids occurred in 15 out of 20 states, likely driven by illicitly manufactured fentanyl.2
Fentanyl-laced heroin: More common, but not new
In October 1991, 3-methylfentanyl was identified in 16 fatal drug overdoses in Allegheny County, Pennsylvania, contributing to a 4-fold increase in overdose deaths compared to the previous year. Fentanyl mixed with heroin and other drugs is commonly found in the Midwest, Northeast, and Southern regions of the United States; in 2014, more than 80% of fentanyl confiscations occurred in 10 states within these regions, with the highest incidence occurring in Ohio.3
When combined with fentanyl, heroin becomes 50 to 100 times more potent, resulting in a subjective high with exaggerated central nervous system depression manifesting as lethargy, miosis, and respiratory depression.4 Most drug users are unaware and unable to identify when heroin is laced with fentanyl, which may contribute to the rise in deaths from unintentional drug overdose.1,5,6
Oral abuse of fentanyl patches can be fatal
Outcomes from oral abuse of fentanyl patches have ranged from transient overdose symptoms, such as lethargy and respiratory depression, to death.7-9 When administered in a medical setting, transbuccal fentanyl has a bioavailability of 50% to 65% across the buccal membrane. Nearly 20% of the drug escapes hepatic first pass metabolism when fentanyl patches are ingested orally and enters the systemic circulation, resulting in severe overdose and potentially death. Prolonged chewing and sucking on fentanyl patches increases the contact time with the buccal membrane, resulting in increased systemic absorption compared to oral ingestion without chewing/sucking.7-9
Urine toxicology screening detects compounds based on a chemical assay for drugs—generally codeine, morphine, and their metabolites. Because fentanyl is a fully synthetic opioid, its structure is not
Continue to: Survival of fentanyl overdose depends on naloxone availability
Survival of fentanyl overdose depends on naloxone availability
Naloxone is a safe and effective antidote to an opioid overdose. It comes in 3 preparations, including intramuscular and subcutaneous injections and an intranasal spray.12 Concerns that naloxone will harm patients with opioid dependence are unfounded. Naloxone can induce symptoms of opioid withdrawal, such as yawning, lacrimation, piloerection, diaphoresis, myalgia, vomiting, and diarrhea. While these withdrawal symptoms are unpleasant, they are not life threatening.12 Due to its high potency, large doses of naloxone (ie, 4–16 mg) are required to reverse the effects of a fentanyl overdose.13 Intranasal naloxone hydrochloride 4 mg delivered in a single spray is preferred due to the ease of administration. Repeat doses may be necessary if respiratory depression continues or recurs prior to the arrival of emergency medical services. Increasing the availability of naloxone to first responders has the potential to save many lives.6
THE TAKEAWAY
Fentanyl is a major contributor to the growing drug overdose crisis in the United States. When laced with heroin or consumed orally in the form of transdermal patches, fentanyl becomes more potent and is increasingly fatal. It’s crucial that primary care physicians be able to identify and educate at-risk patients about the fatal consequences of fentanyl overdose and coordinate care to help get them into an appropriate rehabilitation program.
In order to quickly recognize the signs of fentanyl-related overdose, it’s important to be alert for this possibility. At the bedside, the most easily recognized abnormality associated with fentanyl or other opioid overdose is a decline in respiratory rate culminating in apnea.10 A respiratory rate of 12 breaths/min or less in a patient who is not in physiologic sleep strongly suggests acute opioid intoxication, particularly when accompanied by miosis or stupor. Other signs include bradycardia, hypotension, and seizures from anoxia.10
Apart from the severity of symptoms, it is hard to clinically distinguish fentanyl overdose from other opiate overdose incidents. Given the degree to which illegal opiates are contaminated with fentanyl in the United States,3 it is appropriate to screen for fentanyl with extended panel urine toxicology testing in patients with suspected opioid overdose.
CORRESPONDENCE
Jaividhya Dasarathy, MD, 2500 MetroHealth Medical Center, Cleveland, OH 44109; [email protected]
CASE 1
Two days after reviving her boyfriend with naloxone, a woman and her 30-year-old boyfriend presented to our family medicine clinic. They explained that he had injected heroin and shortly thereafter he stopped breathing and his lips turned blue. The patient’s girlfriend did not call emergency medical services (EMS) at the time because she was afraid of getting arrested due to past incarceration for possession of illegal drugs. Instead, she revived him with naloxone that she found in his bag.
Both the patient and his girlfriend were scared and surprised by his “terrible reaction,” as he had previously purchased heroin from the same dealer and used the same dose without similar effects. However, the patient did note that the drug he purchased this time had a bright white tinge, when normally the drug was light yellow.
On physical examination, the patient’s heart rate and blood pressure were normal. There were needle track marks on both forearms, elbows, and upper arms. A laboratory workup obtained during this visit revealed anemia and a normal basic metabolic panel. A hepatitis C virus antibody test was positive, and a hepatic function panel revealed elevated transaminase levels. Urine toxicology was positive for opioids and negative for other substances.
CASE 2
A 58-year-old man with a history of chronic hepatitis C, polysubstance abuse, and schizophrenia was transported to the emergency department by EMS after his family found him unresponsive in his bedroom. The patient had agonal breathing when EMS arrived, so they administered naloxone (4 mg intranasal and 4 mg intravenous). His breathing improved, but his mental status did not. He was still obtunded upon arrival in the emergency department and vomited 4 tan-colored patches. The patient was tachycardic (heart rate, 108 beats/min), hypertensive (blood pressure, 189/95 mm Hg), and had rapid shallow breathing (respiratory rate, 38 breaths/min). He was intubated for airway protection, at which time 2 more tan-colored patches were removed from his pharynx.
Laboratory evaluation revealed an acute kidney injury with a high anion metabolic acidosis. A hepatic function panel showed elevated transaminase levels. Plasma acetaminophen and salicylate levels were normal. A computed tomography head scan was normal. Urine toxicology was negative for opioids but was positive for cocaine and benzodiazepines.
THE DIAGNOSIS
Opioid overdose caused the acute respiratory depression in both cases. In Case 1, the patient unknowingly overdosed on heroin laced with fentanyl, known as China White, which likely caused the drug’s bright white tinge. In Case 2, the patient’s overdose was the result of oral ingestion of fentanyl patches. (Limited urine toxicology was negative for opiates because fentanyl is a fully synthetic opioid that shows up only with a specific or extended assay. More on this in a bit.)
DISCUSSION
The fatal drug overdose epidemic in the United States is growing. From 2000 to 2014, the mortality rate from drug overdose increased by 137%, including a 200% increase in the rate of overdose deaths related to opioids (ie, pain medications, heroin).1 Between 2013 and 2014, the age-adjusted mortality rate related to methadone, a synthetic opioid, remained unchanged; however, age-adjusted mortality rates related to natural and semisynthetic opioid pain medications, heroin, and synthetic opioids other than methadone (eg, fentanyl) increased by 9%, 26%, and 80%, respectively. In 2014, a sharp increase in overdose deaths related to synthetic opioids other than methadone coincided with law enforcement reports of increased availability of illegal fentanyl; however, the toxicology panel used by coroners and medical examiners at that time could not distinguish between illegal and prescription fentanyl.1
Continue to: Among 70,237 drug overdose deaths...
Among 70,237 drug overdose deaths in the United States in 2017, 47,600 (67.8%) involved an opioid. From 2013 to 2017, drug overdose death rates increased in 35 of 50 states and the District of Columbia, and significant increases in death rates involving synthetic opioids occurred in 15 out of 20 states, likely driven by illicitly manufactured fentanyl.2
Fentanyl-laced heroin: More common, but not new
In October 1991, 3-methylfentanyl was identified in 16 fatal drug overdoses in Allegheny County, Pennsylvania, contributing to a 4-fold increase in overdose deaths compared to the previous year. Fentanyl mixed with heroin and other drugs is commonly found in the Midwest, Northeast, and Southern regions of the United States; in 2014, more than 80% of fentanyl confiscations occurred in 10 states within these regions, with the highest incidence occurring in Ohio.3
When combined with fentanyl, heroin becomes 50 to 100 times more potent, resulting in a subjective high with exaggerated central nervous system depression manifesting as lethargy, miosis, and respiratory depression.4 Most drug users are unaware and unable to identify when heroin is laced with fentanyl, which may contribute to the rise in deaths from unintentional drug overdose.1,5,6
Oral abuse of fentanyl patches can be fatal
Outcomes from oral abuse of fentanyl patches have ranged from transient overdose symptoms, such as lethargy and respiratory depression, to death.7-9 When administered in a medical setting, transbuccal fentanyl has a bioavailability of 50% to 65% across the buccal membrane. Nearly 20% of the drug escapes hepatic first pass metabolism when fentanyl patches are ingested orally and enters the systemic circulation, resulting in severe overdose and potentially death. Prolonged chewing and sucking on fentanyl patches increases the contact time with the buccal membrane, resulting in increased systemic absorption compared to oral ingestion without chewing/sucking.7-9
Urine toxicology screening detects compounds based on a chemical assay for drugs—generally codeine, morphine, and their metabolites. Because fentanyl is a fully synthetic opioid, its structure is not
Continue to: Survival of fentanyl overdose depends on naloxone availability
Survival of fentanyl overdose depends on naloxone availability
Naloxone is a safe and effective antidote to an opioid overdose. It comes in 3 preparations, including intramuscular and subcutaneous injections and an intranasal spray.12 Concerns that naloxone will harm patients with opioid dependence are unfounded. Naloxone can induce symptoms of opioid withdrawal, such as yawning, lacrimation, piloerection, diaphoresis, myalgia, vomiting, and diarrhea. While these withdrawal symptoms are unpleasant, they are not life threatening.12 Due to its high potency, large doses of naloxone (ie, 4–16 mg) are required to reverse the effects of a fentanyl overdose.13 Intranasal naloxone hydrochloride 4 mg delivered in a single spray is preferred due to the ease of administration. Repeat doses may be necessary if respiratory depression continues or recurs prior to the arrival of emergency medical services. Increasing the availability of naloxone to first responders has the potential to save many lives.6
THE TAKEAWAY
Fentanyl is a major contributor to the growing drug overdose crisis in the United States. When laced with heroin or consumed orally in the form of transdermal patches, fentanyl becomes more potent and is increasingly fatal. It’s crucial that primary care physicians be able to identify and educate at-risk patients about the fatal consequences of fentanyl overdose and coordinate care to help get them into an appropriate rehabilitation program.
In order to quickly recognize the signs of fentanyl-related overdose, it’s important to be alert for this possibility. At the bedside, the most easily recognized abnormality associated with fentanyl or other opioid overdose is a decline in respiratory rate culminating in apnea.10 A respiratory rate of 12 breaths/min or less in a patient who is not in physiologic sleep strongly suggests acute opioid intoxication, particularly when accompanied by miosis or stupor. Other signs include bradycardia, hypotension, and seizures from anoxia.10
Apart from the severity of symptoms, it is hard to clinically distinguish fentanyl overdose from other opiate overdose incidents. Given the degree to which illegal opiates are contaminated with fentanyl in the United States,3 it is appropriate to screen for fentanyl with extended panel urine toxicology testing in patients with suspected opioid overdose.
CORRESPONDENCE
Jaividhya Dasarathy, MD, 2500 MetroHealth Medical Center, Cleveland, OH 44109; [email protected]
1. Rudd RA, Aleshire N, Zibbell JE, et al. Increases in drug and opioid overdose deaths—United States, 2000–2014. MMWR Morb Mortal Wkly Rep. 2016;64:1378-1382.
2. Scholl L, Seth P, Kariisa M, et al. Drug and opioid-involved overdose deaths—United States, 2013–2017. MMWR Morb Mortal Wkly Rep. 2019;67:1419-1427.
3. Hibbs J, Perper J, Winek CL. An outbreak of designer drug-related deaths in Pennsylvania. JAMA. 1991;265:1011-1013.
4. Increases in fentanyl drug confiscations and fentanyl-related overdose fatalities. Centers for Disease Control and Prevention Web site. https://emergency.cdc.gov/han/han00384.asp. Published October 26, 2015. Accessed May 3, 2019.
5. Fentanyl. Centers for Disease Control and Prevention Web site. https://www.cdc.gov/drugoverdose/opioids/fentanyl.html. Updated December 19, 2018. Accessed May 3, 2019.
6. Peterson AB, Gladden RM, Delcher C, et al. Increases in fentanyl-related overdose deaths—Florida and Ohio, 2013–2015. MMWR Morb Mortal Wkly Rep. 2016;65:844-849.
7. Streisand JB, Varvel JR, Stanski DR, et al. Absorption and bioavailability of oral transmucosal fentanyl citrate. Anesthesiology. 1991;75:223-229.
8. Kharasch ED, Whittington D, Hoffer C. Influence of hepatic and intestinal cytochrome P4503A activity on the acute disposition and effects of oral transmucosal fentanyl citrate. Anesthesiology. 2004;101:729-737.
9. Woodall KL, Martin TL, McLellan BA. Oral abuse of fentanyl patches (Duragesic): seven case reports. J Forensic Sci. 2008;53:222-225.
10. Moeller KE, Lee KC, Kissack JC. Urine drug screening: practical guide for clinicians. Mayo Clin Proc. 2008;83:66-76.
11. Appropriate Use of Drug Testing in Clinical Addiction Medicine. American Society of Addiction Medicine Web site. https://www.asam.org/docs/default-source/quality-science/appropriate_use_of_drug_testing_in_clinical-1-(7).pdf?sfvrsn=2. Published April 5, 2017. Accessed May 30, 2019.
12. Boyer EW. Management of opioid analgesic overdose. N Engl J Med. 2012;367:146-155.
13. Drugs@FDA: FDA approved drug products. US Food and Drug Administration Web site. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=208411. Accessed May 22, 2019.
1. Rudd RA, Aleshire N, Zibbell JE, et al. Increases in drug and opioid overdose deaths—United States, 2000–2014. MMWR Morb Mortal Wkly Rep. 2016;64:1378-1382.
2. Scholl L, Seth P, Kariisa M, et al. Drug and opioid-involved overdose deaths—United States, 2013–2017. MMWR Morb Mortal Wkly Rep. 2019;67:1419-1427.
3. Hibbs J, Perper J, Winek CL. An outbreak of designer drug-related deaths in Pennsylvania. JAMA. 1991;265:1011-1013.
4. Increases in fentanyl drug confiscations and fentanyl-related overdose fatalities. Centers for Disease Control and Prevention Web site. https://emergency.cdc.gov/han/han00384.asp. Published October 26, 2015. Accessed May 3, 2019.
5. Fentanyl. Centers for Disease Control and Prevention Web site. https://www.cdc.gov/drugoverdose/opioids/fentanyl.html. Updated December 19, 2018. Accessed May 3, 2019.
6. Peterson AB, Gladden RM, Delcher C, et al. Increases in fentanyl-related overdose deaths—Florida and Ohio, 2013–2015. MMWR Morb Mortal Wkly Rep. 2016;65:844-849.
7. Streisand JB, Varvel JR, Stanski DR, et al. Absorption and bioavailability of oral transmucosal fentanyl citrate. Anesthesiology. 1991;75:223-229.
8. Kharasch ED, Whittington D, Hoffer C. Influence of hepatic and intestinal cytochrome P4503A activity on the acute disposition and effects of oral transmucosal fentanyl citrate. Anesthesiology. 2004;101:729-737.
9. Woodall KL, Martin TL, McLellan BA. Oral abuse of fentanyl patches (Duragesic): seven case reports. J Forensic Sci. 2008;53:222-225.
10. Moeller KE, Lee KC, Kissack JC. Urine drug screening: practical guide for clinicians. Mayo Clin Proc. 2008;83:66-76.
11. Appropriate Use of Drug Testing in Clinical Addiction Medicine. American Society of Addiction Medicine Web site. https://www.asam.org/docs/default-source/quality-science/appropriate_use_of_drug_testing_in_clinical-1-(7).pdf?sfvrsn=2. Published April 5, 2017. Accessed May 30, 2019.
12. Boyer EW. Management of opioid analgesic overdose. N Engl J Med. 2012;367:146-155.
13. Drugs@FDA: FDA approved drug products. US Food and Drug Administration Web site. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=208411. Accessed May 22, 2019.
