User login
Acroangiodermatitis of Mali and Stewart-Bluefarb Syndrome
Case Reports
Patient 1
A 56-year-old white man with a history of hypertension, hyperlipidemia, sleep apnea, bilateral knee replacement, and cataract removal presented to the emergency department with a worsening rash on the left posterior medial leg of 6 months’ duration. He reported associated redness and tenderness with the plaques as well as increased swelling and firmness of the leg. He was admitted to the hospital where the infectious disease team treated him with cefazolin for presumed cellulitis. His condition did not improve, and another course of cefazolin was started in addition to oral fluconazole and clotrimazole–betamethasone dipropionate lotion for a possible fungal cause. Again, treatment provided no improvement.
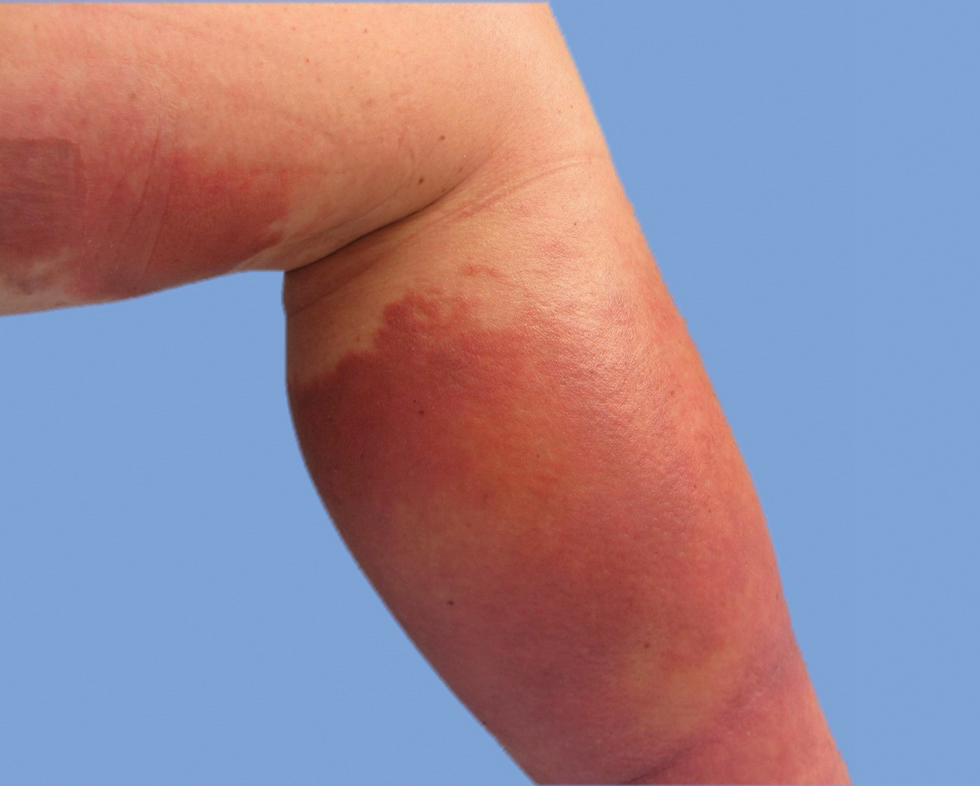
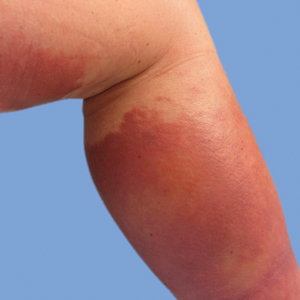
He was then evaluated by dermatology. On physical examination, the patient had edema, warmth, and induration of the left lower leg. There also was an annular and serpiginous indurated plaque with minimal scale on the left lower leg (Figure 1). A firm, dark red to purple plaque on the left medial thigh with mild scale was present. There also was scaling of the right plantar foot.
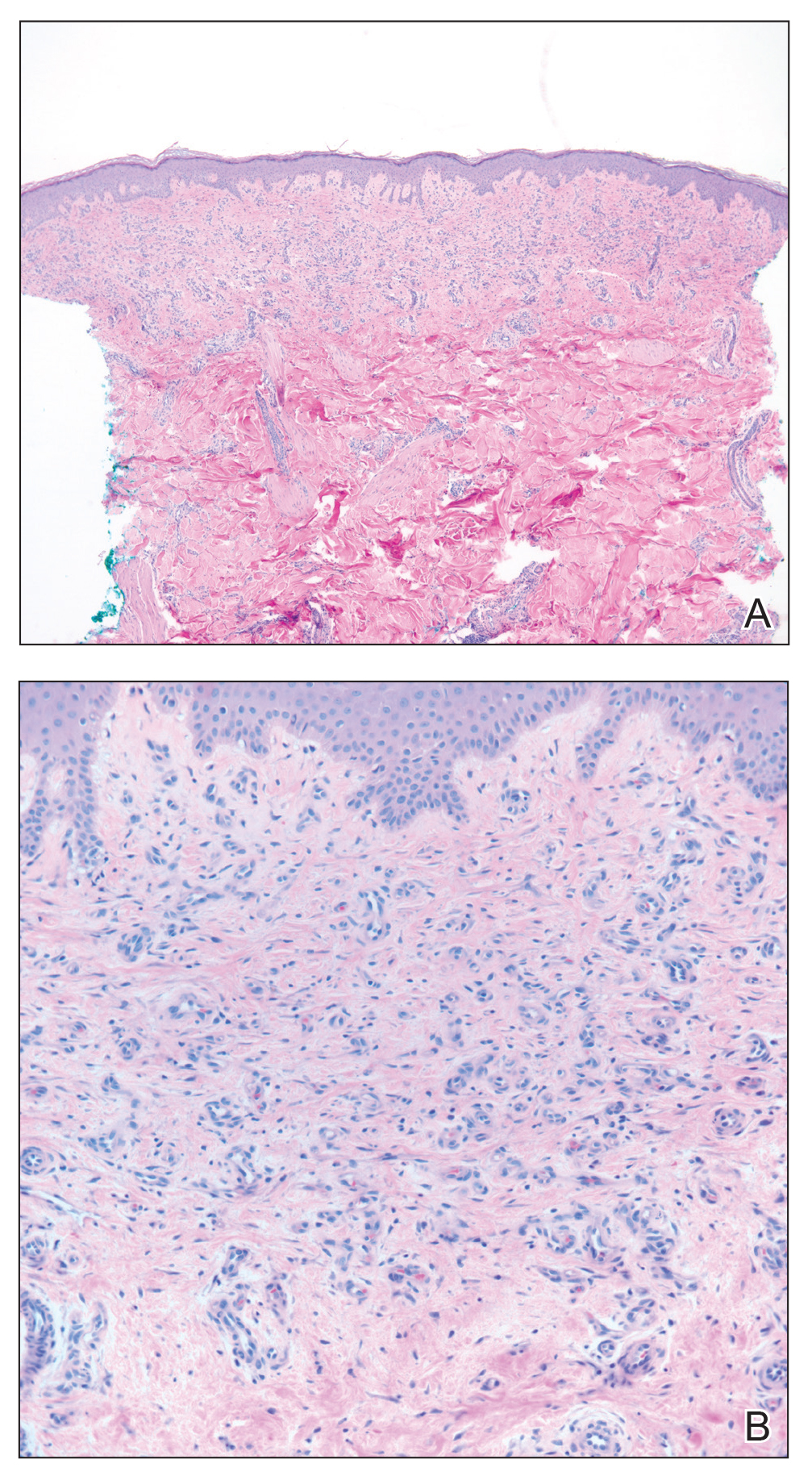
Skin biopsy revealed a dermal capillary proliferation with a scattering of inflammatory cells including eosinophils as well as dermal fibrosis (Figure 2). Periodic acid–Schiff and human herpesvirus 8 (HHV-8) immunostains were negative. Considering the degree and depth of vascular proliferation, Mali-type acroangiodermatitis (AAD) was the favored diagnosis.
Patient 2
A 72-year-old white man presented with a firm asymptomatic growth on the left dorsal forearm of 3 months’ duration. It was located near the site of a prior squamous cell carcinoma that was excised 1 year prior to presentation. The patient had no treatment or biopsy of the presenting lesion. His medical and surgical history included polycystic kidney disease and renal transplantation 4 years prior to presentation. He also had an arteriovenous fistula of the left arm. His other chronic diseases included chronic obstructive lung disease, congestive heart failure, hypertension, type 2 diabetes mellitus, and obstructive sleep apnea.
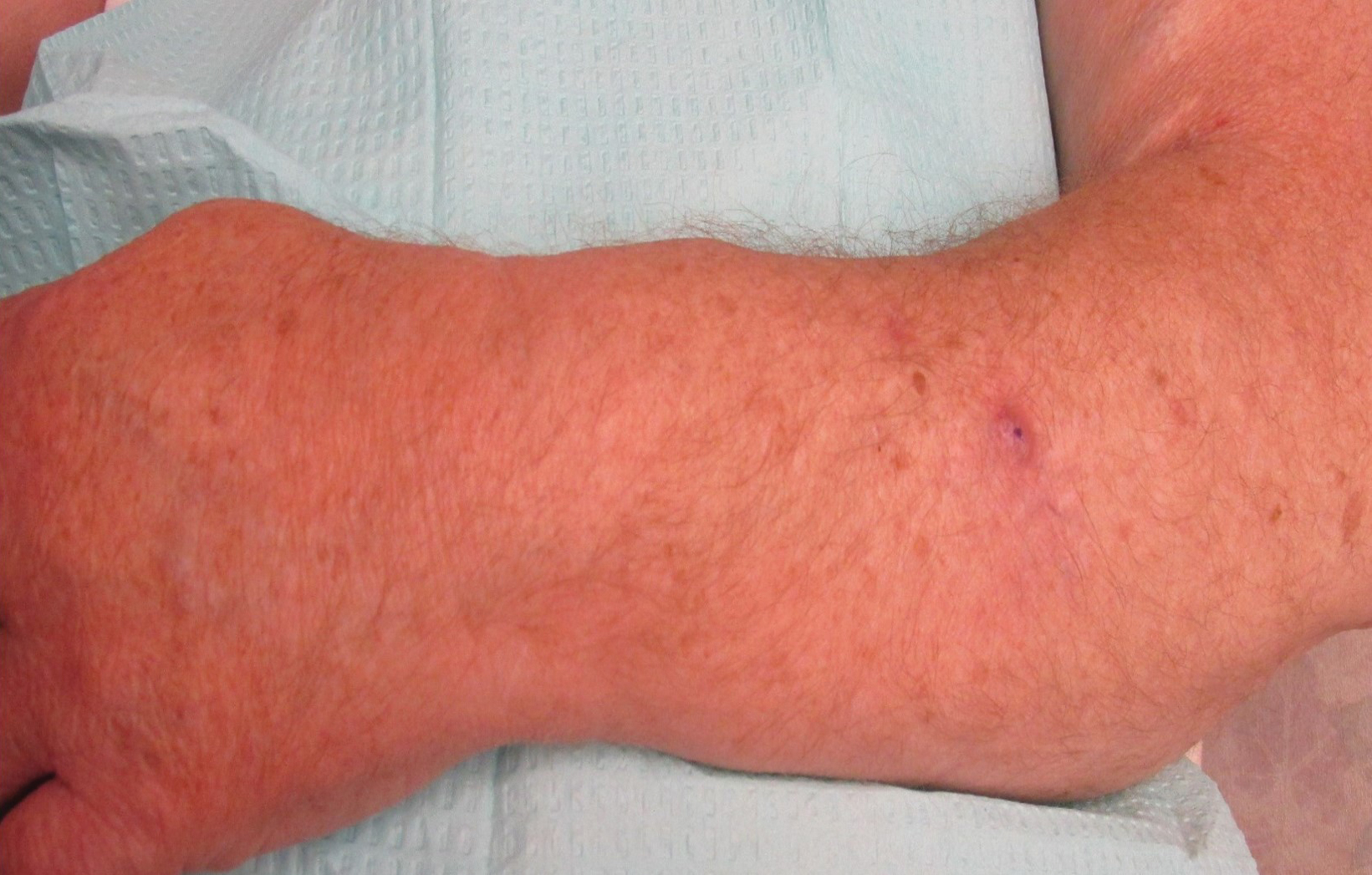
On physical examination, the patient had a 1-cm violaceous nodule on the extensor surface of the left mid forearm. An arteriovenous fistula was present proximal to the lesion on the left arm (Figure 3).
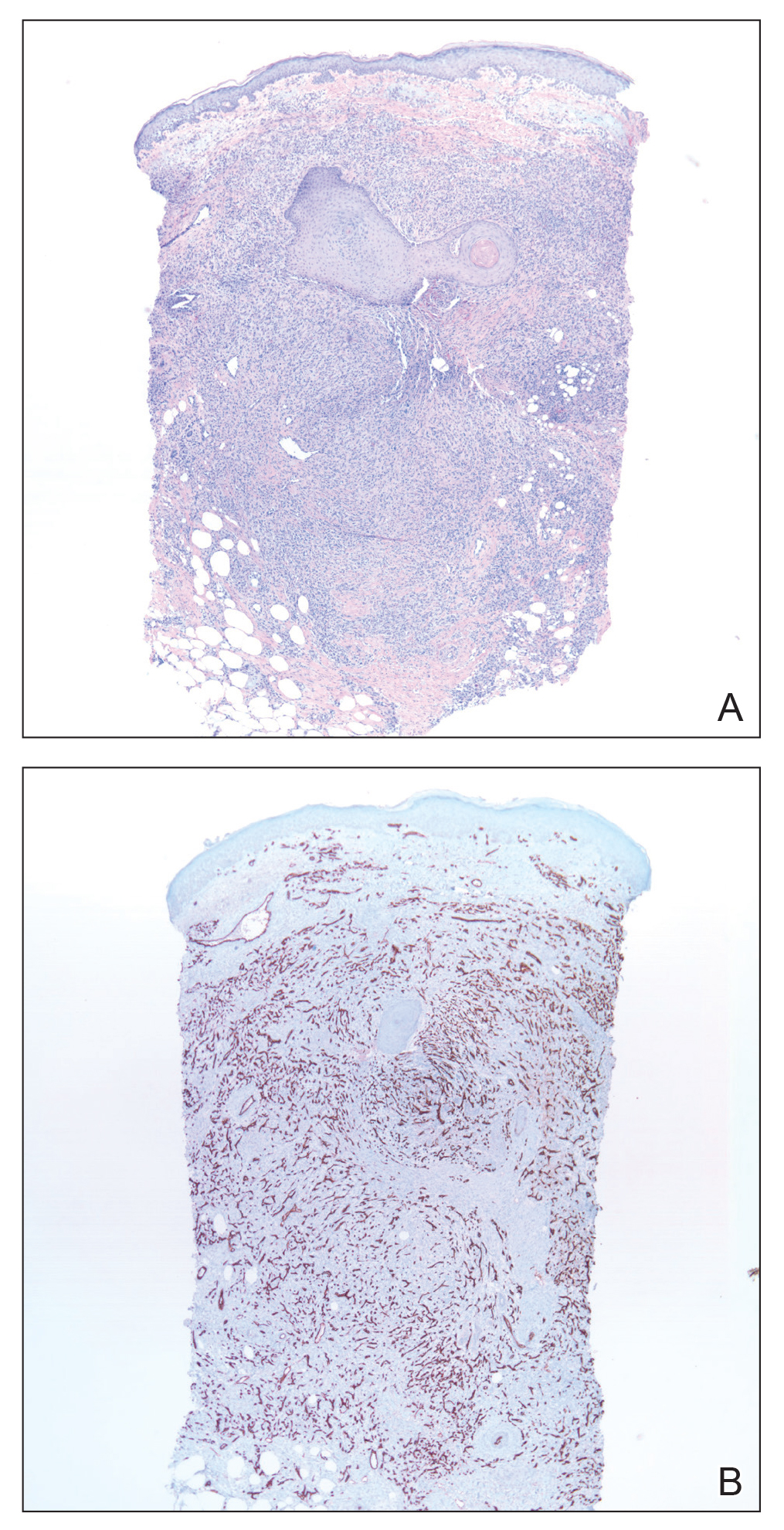
Skin biopsy revealed a tightly packed proliferation of small vascular channels that tested negative for HHV-8, tumor protein p63, and cytokeratin 5/6. Erythrocytes were noted in the lumen of some of these vessels. Neutrophils were scattered and clustered throughout the specimen (Figure 4A). Blood vessels were highlighted with CD34 (Figure 4B). Grocott-Gomori methenamine-silver stain was negative for infectious agents. These findings favored AAD secondary to an arteriovenous malformation, consistent with Stewart-Bluefarb syndrome (SBS).
Comment
Presentation of AAD
Acroangiodermatitis is a rare chronic inflammatory skin process involving a reactive proliferation of capillaries and fibrosis of the skin that resembles Kaposi sarcoma both clinically and histopathologically. The condition has been reported in patients with chronic venous insufficiency,1 congenital arteriovenous malformation,2 acquired iatrogenic arteriovenous fistula,3 paralyzed extremity,4 suction socket lower limb prosthesis (amputees),5 and minor trauma.6-8 The lesions of AAD tend to be circumscribed, slowly evolving, red-violaceous (or brown or dusky) macules, papules, or plaques that may become verrucous or develop into painful ulcerations. They generally occur on the distal dorsal aspects of the lower legs and feet.110
Variants of AAD
Mali et al9 first reported cutaneous manifestations resembling Kaposi sarcoma in 18 patients with chronic venous insufficiency in 1965. Two years later, Bluefarb and Adams10 described kaposiform skin lesions in one patient with a congenital arteriovenous malformation without chronic venous insufficiency. It was not until 1974, however, that Earhart et al11 proposed the term pseudo-Kaposi sarcoma.10,11 Based on these findings, AAD is described as 2 variants: Mali type and SBS.
Mali-type AAD is more common and typically occurs in elderly men. It classically presents bilaterally on the lower extremities in association with severe chronic venous insufficiency.5 Skin lesions usually occur on the medial aspect of the lower legs (as in patient 1), dorsum of the heel, hallux, or second toe.12
The etiology of Mali-type AAD is poorly understood. The leading theory is that the condition involves reduced perfusion due to chronic edema, resulting in neovascularization, fibroblast proliferation, hypertrophy, and inflammatory skin changes. When AAD occurs in the setting of a suction socket prosthesis, the negative pressure of the stump-socket environment is thought to alter local circulation, leading to proliferation of small blood vessels.5,13
Stewart-Bluefarb syndrome usually involves a single extremity in young adults with congenital arteriovenous malformations, amputees, and individuals with hemiplegia or iatrogenic arteriovenous fistulae (as in patient 2).1 It was once thought to occur secondary to Klippel-Trenaunay-Weber syndrome; however, SBS rarely is accompanied by limb hypertrophy.9 Pathogenesis is thought to involve an angiogenic response to a high perfusion rate and high oxygen saturation, which leads to fibroblast proliferation and reactive endothelial hyperplasia.1,14
Diagnosis and Differential Diagnosis
Prompt identification of an underlying arteriovenous anomaly is critical, given the sequelae of high-flow shunts, which may result in skin ulceration, limb length discrepancy, cortical thinning of bone with regional osteoporosis, and congestive heart failure.1,5 Duplex ultrasonography is the first-line diagnostic modality because it is noninvasive and widely available. The key doppler feature of an arteriovenous malformation is low resistance and high diastolic pulsatile flow,1 which should be confirmed with magnetic resonance angiography or computed tomography angiography if present on ultrasonography.
The differential diagnosis of AAD includes Kaposi sarcoma, reactive angioendotheliomatosis, diffuse dermal angiomatosis, intravascular histiocytosis, glomeruloid angioendotheliomatosis, and angiopericytomatosis.15,16 These entities present as multiple erythematous, violaceous, purpuric patches and plaques generally on the extremities but can have a widely varied distribution. Some lesions evolve to necrosis or ulceration. Histopathologic analysis is useful to differentiate these entities.
Histopathology
The histopathologic features of AAD can be nonspecific; clinicopathologic correlation often is necessary to establish the diagnosis. Features include a proliferation of small thick-walled vessels, often in a lobular arrangement, in an edematous papillary dermis. Small thrombi may be observed. There may be increased fibroblasts; plump endothelial cells; a superficial mixed infiltrate comprised of lymphocytes, histiocytes, and eosinophils; and deposition of hemosiderin.2,5 These characteristics overlap with features of Kaposi sarcoma; AAD, however, lacks slitlike vascular spaces, perivascular CD34+ expression, and nuclear atypia. A negative HHV-8 stain will assist in ruling out Kaposi sarcoma.1,17
Management
Treatment reports are anecdotal. The goal is to correct underlying venous hypertension. Conservative measures with compression garments, intermittent pneumatic compression, and limb elevation are first line.18 Oral antibiotics and local wound care with topical emollients and corticosteroids have been shown to be effective treatments.19-21
Oral erythromycin 500 mg 4 times daily for 3 weeks and clobetasol propionate cream 0.05% healed a lower extremity ulcer in a patient with Mali-type AAD.21 In another patient, conservative treatment of Mali-type AAD failed, but rapid improvement of 2 lower extremity ulcers resulted after 3 weeks of oral dapsone 50 mg twice daily.22
Conclusion
Acroangiodermatitis is a rare entity that is characterized by erythematous violaceous papules and plaques of the extremities, commonly in the setting of chronic venous insufficiency or an arteriovenous shunt. Histopathologic analysis shows proliferation of capillaries with fibrosis, extravasation of erythrocytes, and deposition of hemosiderin without the spindle cells and slitlike vascular spaces characteristic of Kaposi sarcoma. Detection of an underlying arteriovenous malformation is essential, as the disease can have local and systemic consequences, such as skin ulceration and congestive heart failure.1 Treatment options are conservative, directed toward local wound care, compression, and management of complications, such as ulceration and infection, as well as obliterating any underlying arteriovenous malformation.
- Parsi K, O’Connor AA, Bester L. Stewart-Bluefarb syndrome: report of five cases and a review of literature. Phlebology. 2015;30:505-514.
- Larralde M, Gonzalez V, Marietti R, et al. Pseudo-Kaposi sarcoma with arteriovenous malformation. Pediatr Dermatol. 2001;18:325-327.
- Nakanishi G, Tachibana T, Soga H, et al. Pseudo-Kaposi’s sarcoma of the hand associated with acquired iatrogenic arteriovenous fistula. Indian J Dermatol. 2014;59:415-416.
- Landthaler M, Langehenke H, Holzmann H, et al. Mali’s acroangiodermatitis (pseudo-Kaposi) in paralyzed legs. Hautarzt. 1988;39:304-307.
- Trindade F, Requena L. Pseudo-Kaposi’s sarcoma because of suction socket lower limb prosthesis. J Cutan Pathol. 2009;36:482-485.
- Yu-Lu W, Tao Q, Hong-Zhong J, et al. Non-tender pedal plaques and nodules: pseudo-Kaposi’s sarcoma (Stewart-Bluefarb type) induced by trauma. J Dtsch Dermatol Ges. 2015;13:927-930.
- Del-Río E, Aguilar A, Ambrojo P, et al. Pseudo-Kaposi sarcoma induced by minor trauma in a patient with Klippel-Trenaunay-Weber syndrome. Clin Exp Dermatol. 1993;18:151-153.
- Archie M, Khademi S, Aungst D, et al. A rare case of acroangiodermatitis associated with a congenital arteriovenous malformation (Stewart-Bluefarb Syndrome) in a young veteran: case report and review of the literature. Ann Vasc Surg. 2015;29:1448.e5-1448.e10.
- Mali JW, Kuiper JP, Hamers AA. Acro-angiodermatitis of the foot. Arch Dermatol. 1965;92:515-518.
- Bluefarb SM, Adams LA. Arteriovenous malformation with angiodermatitis. stasis dermatitis simulating Kaposi’s disease. Arch Dermatol. 1967;96:176-181.
- Earhart RN, Aeling JA, Nuss DD, et al. Pseudo-Kaposi sarcoma. A patient with arteriovenous malformation and skin lesions simulating Kaposi sarcoma. Arch Dermatol. 1974;110:907-910.
- Lugovic´ L, Pusic´ J, Situm M, et al. Acroangiodermatitis (pseudo-Kaposi sarcoma): three case reports. Acta Dermatovenerol Croat. 2007;15:152-157.
- Horiguchi Y, Takahashi K, Tanizaki H, et al. Case of bilateral acroangiodermatitis due to symmetrical arteriovenous fistulas of the soles. J Dermatol. 2015;42:989-991.
- Dog˘an S, Boztepe G, Karaduman A. Pseudo-Kaposi sarcoma: a challenging vascular phenomenon. Dermatol Online J. 2007;13:22.
- Mazloom SE, Stallings A, Kyei A. Differentiating intralymphatic histiocytosis, intravascular histiocytosis, and subtypes of reactive angioendotheliomatosis: review of clinical and histologic features of all cases reported to date. Am J Dermatopathol. 2017;39:33-39.
- Rongioletti F, Rebora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
- Kanitakis J, Narvaez D, Claudy A. Expression of the CD34 antigen distinguishes Kaposi’s sarcoma from pseudo-Kaposi’s sarcoma (acroangiodermatitis). Br J Dermatol. 1996;134:44-46.
- Pires A, Depairon M, Ricci C, et al. Effect of compression therapy on a pseudo-Kaposi sarcoma. Dermatology. 1999;198:439-441.
- Hayek S, Atiyeh B, Zgheib E. Stewart-Bluefarb syndrome: review of the literature and case report of chronic ulcer treatment with heparan sulphate (Cacipliq20®). Int Wound J. 2015;12:169-172.
- Varyani N, Thukral A, Kumar N, et al. Nonhealing ulcer: acroangiodermatitis of Mali. Case Rep Dermatol Med. 2011;2011:909383.
- Mehta AA, Pereira RR, Nayak C, et al. Acroangiodermatitis of Mali: a rare vascular phenomenon. Indian J Dermatol Venereol Leprol. 2010;76:553-556.
- Rashkovsky I, Gilead L, Schamroth J, et al. Acro-angiodermatitis: review of the literature and report of a case. Acta Derm Venereol. 1995;75:475-478.
Case Reports
Patient 1
A 56-year-old white man with a history of hypertension, hyperlipidemia, sleep apnea, bilateral knee replacement, and cataract removal presented to the emergency department with a worsening rash on the left posterior medial leg of 6 months’ duration. He reported associated redness and tenderness with the plaques as well as increased swelling and firmness of the leg. He was admitted to the hospital where the infectious disease team treated him with cefazolin for presumed cellulitis. His condition did not improve, and another course of cefazolin was started in addition to oral fluconazole and clotrimazole–betamethasone dipropionate lotion for a possible fungal cause. Again, treatment provided no improvement.
He was then evaluated by dermatology. On physical examination, the patient had edema, warmth, and induration of the left lower leg. There also was an annular and serpiginous indurated plaque with minimal scale on the left lower leg (Figure 1). A firm, dark red to purple plaque on the left medial thigh with mild scale was present. There also was scaling of the right plantar foot.
Skin biopsy revealed a dermal capillary proliferation with a scattering of inflammatory cells including eosinophils as well as dermal fibrosis (Figure 2). Periodic acid–Schiff and human herpesvirus 8 (HHV-8) immunostains were negative. Considering the degree and depth of vascular proliferation, Mali-type acroangiodermatitis (AAD) was the favored diagnosis.
Patient 2
A 72-year-old white man presented with a firm asymptomatic growth on the left dorsal forearm of 3 months’ duration. It was located near the site of a prior squamous cell carcinoma that was excised 1 year prior to presentation. The patient had no treatment or biopsy of the presenting lesion. His medical and surgical history included polycystic kidney disease and renal transplantation 4 years prior to presentation. He also had an arteriovenous fistula of the left arm. His other chronic diseases included chronic obstructive lung disease, congestive heart failure, hypertension, type 2 diabetes mellitus, and obstructive sleep apnea.
On physical examination, the patient had a 1-cm violaceous nodule on the extensor surface of the left mid forearm. An arteriovenous fistula was present proximal to the lesion on the left arm (Figure 3).
Skin biopsy revealed a tightly packed proliferation of small vascular channels that tested negative for HHV-8, tumor protein p63, and cytokeratin 5/6. Erythrocytes were noted in the lumen of some of these vessels. Neutrophils were scattered and clustered throughout the specimen (Figure 4A). Blood vessels were highlighted with CD34 (Figure 4B). Grocott-Gomori methenamine-silver stain was negative for infectious agents. These findings favored AAD secondary to an arteriovenous malformation, consistent with Stewart-Bluefarb syndrome (SBS).
Comment
Presentation of AAD
Acroangiodermatitis is a rare chronic inflammatory skin process involving a reactive proliferation of capillaries and fibrosis of the skin that resembles Kaposi sarcoma both clinically and histopathologically. The condition has been reported in patients with chronic venous insufficiency,1 congenital arteriovenous malformation,2 acquired iatrogenic arteriovenous fistula,3 paralyzed extremity,4 suction socket lower limb prosthesis (amputees),5 and minor trauma.6-8 The lesions of AAD tend to be circumscribed, slowly evolving, red-violaceous (or brown or dusky) macules, papules, or plaques that may become verrucous or develop into painful ulcerations. They generally occur on the distal dorsal aspects of the lower legs and feet.110
Variants of AAD
Mali et al9 first reported cutaneous manifestations resembling Kaposi sarcoma in 18 patients with chronic venous insufficiency in 1965. Two years later, Bluefarb and Adams10 described kaposiform skin lesions in one patient with a congenital arteriovenous malformation without chronic venous insufficiency. It was not until 1974, however, that Earhart et al11 proposed the term pseudo-Kaposi sarcoma.10,11 Based on these findings, AAD is described as 2 variants: Mali type and SBS.
Mali-type AAD is more common and typically occurs in elderly men. It classically presents bilaterally on the lower extremities in association with severe chronic venous insufficiency.5 Skin lesions usually occur on the medial aspect of the lower legs (as in patient 1), dorsum of the heel, hallux, or second toe.12
The etiology of Mali-type AAD is poorly understood. The leading theory is that the condition involves reduced perfusion due to chronic edema, resulting in neovascularization, fibroblast proliferation, hypertrophy, and inflammatory skin changes. When AAD occurs in the setting of a suction socket prosthesis, the negative pressure of the stump-socket environment is thought to alter local circulation, leading to proliferation of small blood vessels.5,13
Stewart-Bluefarb syndrome usually involves a single extremity in young adults with congenital arteriovenous malformations, amputees, and individuals with hemiplegia or iatrogenic arteriovenous fistulae (as in patient 2).1 It was once thought to occur secondary to Klippel-Trenaunay-Weber syndrome; however, SBS rarely is accompanied by limb hypertrophy.9 Pathogenesis is thought to involve an angiogenic response to a high perfusion rate and high oxygen saturation, which leads to fibroblast proliferation and reactive endothelial hyperplasia.1,14
Diagnosis and Differential Diagnosis
Prompt identification of an underlying arteriovenous anomaly is critical, given the sequelae of high-flow shunts, which may result in skin ulceration, limb length discrepancy, cortical thinning of bone with regional osteoporosis, and congestive heart failure.1,5 Duplex ultrasonography is the first-line diagnostic modality because it is noninvasive and widely available. The key doppler feature of an arteriovenous malformation is low resistance and high diastolic pulsatile flow,1 which should be confirmed with magnetic resonance angiography or computed tomography angiography if present on ultrasonography.
The differential diagnosis of AAD includes Kaposi sarcoma, reactive angioendotheliomatosis, diffuse dermal angiomatosis, intravascular histiocytosis, glomeruloid angioendotheliomatosis, and angiopericytomatosis.15,16 These entities present as multiple erythematous, violaceous, purpuric patches and plaques generally on the extremities but can have a widely varied distribution. Some lesions evolve to necrosis or ulceration. Histopathologic analysis is useful to differentiate these entities.
Histopathology
The histopathologic features of AAD can be nonspecific; clinicopathologic correlation often is necessary to establish the diagnosis. Features include a proliferation of small thick-walled vessels, often in a lobular arrangement, in an edematous papillary dermis. Small thrombi may be observed. There may be increased fibroblasts; plump endothelial cells; a superficial mixed infiltrate comprised of lymphocytes, histiocytes, and eosinophils; and deposition of hemosiderin.2,5 These characteristics overlap with features of Kaposi sarcoma; AAD, however, lacks slitlike vascular spaces, perivascular CD34+ expression, and nuclear atypia. A negative HHV-8 stain will assist in ruling out Kaposi sarcoma.1,17
Management
Treatment reports are anecdotal. The goal is to correct underlying venous hypertension. Conservative measures with compression garments, intermittent pneumatic compression, and limb elevation are first line.18 Oral antibiotics and local wound care with topical emollients and corticosteroids have been shown to be effective treatments.19-21
Oral erythromycin 500 mg 4 times daily for 3 weeks and clobetasol propionate cream 0.05% healed a lower extremity ulcer in a patient with Mali-type AAD.21 In another patient, conservative treatment of Mali-type AAD failed, but rapid improvement of 2 lower extremity ulcers resulted after 3 weeks of oral dapsone 50 mg twice daily.22
Conclusion
Acroangiodermatitis is a rare entity that is characterized by erythematous violaceous papules and plaques of the extremities, commonly in the setting of chronic venous insufficiency or an arteriovenous shunt. Histopathologic analysis shows proliferation of capillaries with fibrosis, extravasation of erythrocytes, and deposition of hemosiderin without the spindle cells and slitlike vascular spaces characteristic of Kaposi sarcoma. Detection of an underlying arteriovenous malformation is essential, as the disease can have local and systemic consequences, such as skin ulceration and congestive heart failure.1 Treatment options are conservative, directed toward local wound care, compression, and management of complications, such as ulceration and infection, as well as obliterating any underlying arteriovenous malformation.
Case Reports
Patient 1
A 56-year-old white man with a history of hypertension, hyperlipidemia, sleep apnea, bilateral knee replacement, and cataract removal presented to the emergency department with a worsening rash on the left posterior medial leg of 6 months’ duration. He reported associated redness and tenderness with the plaques as well as increased swelling and firmness of the leg. He was admitted to the hospital where the infectious disease team treated him with cefazolin for presumed cellulitis. His condition did not improve, and another course of cefazolin was started in addition to oral fluconazole and clotrimazole–betamethasone dipropionate lotion for a possible fungal cause. Again, treatment provided no improvement.
He was then evaluated by dermatology. On physical examination, the patient had edema, warmth, and induration of the left lower leg. There also was an annular and serpiginous indurated plaque with minimal scale on the left lower leg (Figure 1). A firm, dark red to purple plaque on the left medial thigh with mild scale was present. There also was scaling of the right plantar foot.
Skin biopsy revealed a dermal capillary proliferation with a scattering of inflammatory cells including eosinophils as well as dermal fibrosis (Figure 2). Periodic acid–Schiff and human herpesvirus 8 (HHV-8) immunostains were negative. Considering the degree and depth of vascular proliferation, Mali-type acroangiodermatitis (AAD) was the favored diagnosis.
Patient 2
A 72-year-old white man presented with a firm asymptomatic growth on the left dorsal forearm of 3 months’ duration. It was located near the site of a prior squamous cell carcinoma that was excised 1 year prior to presentation. The patient had no treatment or biopsy of the presenting lesion. His medical and surgical history included polycystic kidney disease and renal transplantation 4 years prior to presentation. He also had an arteriovenous fistula of the left arm. His other chronic diseases included chronic obstructive lung disease, congestive heart failure, hypertension, type 2 diabetes mellitus, and obstructive sleep apnea.
On physical examination, the patient had a 1-cm violaceous nodule on the extensor surface of the left mid forearm. An arteriovenous fistula was present proximal to the lesion on the left arm (Figure 3).
Skin biopsy revealed a tightly packed proliferation of small vascular channels that tested negative for HHV-8, tumor protein p63, and cytokeratin 5/6. Erythrocytes were noted in the lumen of some of these vessels. Neutrophils were scattered and clustered throughout the specimen (Figure 4A). Blood vessels were highlighted with CD34 (Figure 4B). Grocott-Gomori methenamine-silver stain was negative for infectious agents. These findings favored AAD secondary to an arteriovenous malformation, consistent with Stewart-Bluefarb syndrome (SBS).
Comment
Presentation of AAD
Acroangiodermatitis is a rare chronic inflammatory skin process involving a reactive proliferation of capillaries and fibrosis of the skin that resembles Kaposi sarcoma both clinically and histopathologically. The condition has been reported in patients with chronic venous insufficiency,1 congenital arteriovenous malformation,2 acquired iatrogenic arteriovenous fistula,3 paralyzed extremity,4 suction socket lower limb prosthesis (amputees),5 and minor trauma.6-8 The lesions of AAD tend to be circumscribed, slowly evolving, red-violaceous (or brown or dusky) macules, papules, or plaques that may become verrucous or develop into painful ulcerations. They generally occur on the distal dorsal aspects of the lower legs and feet.110
Variants of AAD
Mali et al9 first reported cutaneous manifestations resembling Kaposi sarcoma in 18 patients with chronic venous insufficiency in 1965. Two years later, Bluefarb and Adams10 described kaposiform skin lesions in one patient with a congenital arteriovenous malformation without chronic venous insufficiency. It was not until 1974, however, that Earhart et al11 proposed the term pseudo-Kaposi sarcoma.10,11 Based on these findings, AAD is described as 2 variants: Mali type and SBS.
Mali-type AAD is more common and typically occurs in elderly men. It classically presents bilaterally on the lower extremities in association with severe chronic venous insufficiency.5 Skin lesions usually occur on the medial aspect of the lower legs (as in patient 1), dorsum of the heel, hallux, or second toe.12
The etiology of Mali-type AAD is poorly understood. The leading theory is that the condition involves reduced perfusion due to chronic edema, resulting in neovascularization, fibroblast proliferation, hypertrophy, and inflammatory skin changes. When AAD occurs in the setting of a suction socket prosthesis, the negative pressure of the stump-socket environment is thought to alter local circulation, leading to proliferation of small blood vessels.5,13
Stewart-Bluefarb syndrome usually involves a single extremity in young adults with congenital arteriovenous malformations, amputees, and individuals with hemiplegia or iatrogenic arteriovenous fistulae (as in patient 2).1 It was once thought to occur secondary to Klippel-Trenaunay-Weber syndrome; however, SBS rarely is accompanied by limb hypertrophy.9 Pathogenesis is thought to involve an angiogenic response to a high perfusion rate and high oxygen saturation, which leads to fibroblast proliferation and reactive endothelial hyperplasia.1,14
Diagnosis and Differential Diagnosis
Prompt identification of an underlying arteriovenous anomaly is critical, given the sequelae of high-flow shunts, which may result in skin ulceration, limb length discrepancy, cortical thinning of bone with regional osteoporosis, and congestive heart failure.1,5 Duplex ultrasonography is the first-line diagnostic modality because it is noninvasive and widely available. The key doppler feature of an arteriovenous malformation is low resistance and high diastolic pulsatile flow,1 which should be confirmed with magnetic resonance angiography or computed tomography angiography if present on ultrasonography.
The differential diagnosis of AAD includes Kaposi sarcoma, reactive angioendotheliomatosis, diffuse dermal angiomatosis, intravascular histiocytosis, glomeruloid angioendotheliomatosis, and angiopericytomatosis.15,16 These entities present as multiple erythematous, violaceous, purpuric patches and plaques generally on the extremities but can have a widely varied distribution. Some lesions evolve to necrosis or ulceration. Histopathologic analysis is useful to differentiate these entities.
Histopathology
The histopathologic features of AAD can be nonspecific; clinicopathologic correlation often is necessary to establish the diagnosis. Features include a proliferation of small thick-walled vessels, often in a lobular arrangement, in an edematous papillary dermis. Small thrombi may be observed. There may be increased fibroblasts; plump endothelial cells; a superficial mixed infiltrate comprised of lymphocytes, histiocytes, and eosinophils; and deposition of hemosiderin.2,5 These characteristics overlap with features of Kaposi sarcoma; AAD, however, lacks slitlike vascular spaces, perivascular CD34+ expression, and nuclear atypia. A negative HHV-8 stain will assist in ruling out Kaposi sarcoma.1,17
Management
Treatment reports are anecdotal. The goal is to correct underlying venous hypertension. Conservative measures with compression garments, intermittent pneumatic compression, and limb elevation are first line.18 Oral antibiotics and local wound care with topical emollients and corticosteroids have been shown to be effective treatments.19-21
Oral erythromycin 500 mg 4 times daily for 3 weeks and clobetasol propionate cream 0.05% healed a lower extremity ulcer in a patient with Mali-type AAD.21 In another patient, conservative treatment of Mali-type AAD failed, but rapid improvement of 2 lower extremity ulcers resulted after 3 weeks of oral dapsone 50 mg twice daily.22
Conclusion
Acroangiodermatitis is a rare entity that is characterized by erythematous violaceous papules and plaques of the extremities, commonly in the setting of chronic venous insufficiency or an arteriovenous shunt. Histopathologic analysis shows proliferation of capillaries with fibrosis, extravasation of erythrocytes, and deposition of hemosiderin without the spindle cells and slitlike vascular spaces characteristic of Kaposi sarcoma. Detection of an underlying arteriovenous malformation is essential, as the disease can have local and systemic consequences, such as skin ulceration and congestive heart failure.1 Treatment options are conservative, directed toward local wound care, compression, and management of complications, such as ulceration and infection, as well as obliterating any underlying arteriovenous malformation.
- Parsi K, O’Connor AA, Bester L. Stewart-Bluefarb syndrome: report of five cases and a review of literature. Phlebology. 2015;30:505-514.
- Larralde M, Gonzalez V, Marietti R, et al. Pseudo-Kaposi sarcoma with arteriovenous malformation. Pediatr Dermatol. 2001;18:325-327.
- Nakanishi G, Tachibana T, Soga H, et al. Pseudo-Kaposi’s sarcoma of the hand associated with acquired iatrogenic arteriovenous fistula. Indian J Dermatol. 2014;59:415-416.
- Landthaler M, Langehenke H, Holzmann H, et al. Mali’s acroangiodermatitis (pseudo-Kaposi) in paralyzed legs. Hautarzt. 1988;39:304-307.
- Trindade F, Requena L. Pseudo-Kaposi’s sarcoma because of suction socket lower limb prosthesis. J Cutan Pathol. 2009;36:482-485.
- Yu-Lu W, Tao Q, Hong-Zhong J, et al. Non-tender pedal plaques and nodules: pseudo-Kaposi’s sarcoma (Stewart-Bluefarb type) induced by trauma. J Dtsch Dermatol Ges. 2015;13:927-930.
- Del-Río E, Aguilar A, Ambrojo P, et al. Pseudo-Kaposi sarcoma induced by minor trauma in a patient with Klippel-Trenaunay-Weber syndrome. Clin Exp Dermatol. 1993;18:151-153.
- Archie M, Khademi S, Aungst D, et al. A rare case of acroangiodermatitis associated with a congenital arteriovenous malformation (Stewart-Bluefarb Syndrome) in a young veteran: case report and review of the literature. Ann Vasc Surg. 2015;29:1448.e5-1448.e10.
- Mali JW, Kuiper JP, Hamers AA. Acro-angiodermatitis of the foot. Arch Dermatol. 1965;92:515-518.
- Bluefarb SM, Adams LA. Arteriovenous malformation with angiodermatitis. stasis dermatitis simulating Kaposi’s disease. Arch Dermatol. 1967;96:176-181.
- Earhart RN, Aeling JA, Nuss DD, et al. Pseudo-Kaposi sarcoma. A patient with arteriovenous malformation and skin lesions simulating Kaposi sarcoma. Arch Dermatol. 1974;110:907-910.
- Lugovic´ L, Pusic´ J, Situm M, et al. Acroangiodermatitis (pseudo-Kaposi sarcoma): three case reports. Acta Dermatovenerol Croat. 2007;15:152-157.
- Horiguchi Y, Takahashi K, Tanizaki H, et al. Case of bilateral acroangiodermatitis due to symmetrical arteriovenous fistulas of the soles. J Dermatol. 2015;42:989-991.
- Dog˘an S, Boztepe G, Karaduman A. Pseudo-Kaposi sarcoma: a challenging vascular phenomenon. Dermatol Online J. 2007;13:22.
- Mazloom SE, Stallings A, Kyei A. Differentiating intralymphatic histiocytosis, intravascular histiocytosis, and subtypes of reactive angioendotheliomatosis: review of clinical and histologic features of all cases reported to date. Am J Dermatopathol. 2017;39:33-39.
- Rongioletti F, Rebora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
- Kanitakis J, Narvaez D, Claudy A. Expression of the CD34 antigen distinguishes Kaposi’s sarcoma from pseudo-Kaposi’s sarcoma (acroangiodermatitis). Br J Dermatol. 1996;134:44-46.
- Pires A, Depairon M, Ricci C, et al. Effect of compression therapy on a pseudo-Kaposi sarcoma. Dermatology. 1999;198:439-441.
- Hayek S, Atiyeh B, Zgheib E. Stewart-Bluefarb syndrome: review of the literature and case report of chronic ulcer treatment with heparan sulphate (Cacipliq20®). Int Wound J. 2015;12:169-172.
- Varyani N, Thukral A, Kumar N, et al. Nonhealing ulcer: acroangiodermatitis of Mali. Case Rep Dermatol Med. 2011;2011:909383.
- Mehta AA, Pereira RR, Nayak C, et al. Acroangiodermatitis of Mali: a rare vascular phenomenon. Indian J Dermatol Venereol Leprol. 2010;76:553-556.
- Rashkovsky I, Gilead L, Schamroth J, et al. Acro-angiodermatitis: review of the literature and report of a case. Acta Derm Venereol. 1995;75:475-478.
- Parsi K, O’Connor AA, Bester L. Stewart-Bluefarb syndrome: report of five cases and a review of literature. Phlebology. 2015;30:505-514.
- Larralde M, Gonzalez V, Marietti R, et al. Pseudo-Kaposi sarcoma with arteriovenous malformation. Pediatr Dermatol. 2001;18:325-327.
- Nakanishi G, Tachibana T, Soga H, et al. Pseudo-Kaposi’s sarcoma of the hand associated with acquired iatrogenic arteriovenous fistula. Indian J Dermatol. 2014;59:415-416.
- Landthaler M, Langehenke H, Holzmann H, et al. Mali’s acroangiodermatitis (pseudo-Kaposi) in paralyzed legs. Hautarzt. 1988;39:304-307.
- Trindade F, Requena L. Pseudo-Kaposi’s sarcoma because of suction socket lower limb prosthesis. J Cutan Pathol. 2009;36:482-485.
- Yu-Lu W, Tao Q, Hong-Zhong J, et al. Non-tender pedal plaques and nodules: pseudo-Kaposi’s sarcoma (Stewart-Bluefarb type) induced by trauma. J Dtsch Dermatol Ges. 2015;13:927-930.
- Del-Río E, Aguilar A, Ambrojo P, et al. Pseudo-Kaposi sarcoma induced by minor trauma in a patient with Klippel-Trenaunay-Weber syndrome. Clin Exp Dermatol. 1993;18:151-153.
- Archie M, Khademi S, Aungst D, et al. A rare case of acroangiodermatitis associated with a congenital arteriovenous malformation (Stewart-Bluefarb Syndrome) in a young veteran: case report and review of the literature. Ann Vasc Surg. 2015;29:1448.e5-1448.e10.
- Mali JW, Kuiper JP, Hamers AA. Acro-angiodermatitis of the foot. Arch Dermatol. 1965;92:515-518.
- Bluefarb SM, Adams LA. Arteriovenous malformation with angiodermatitis. stasis dermatitis simulating Kaposi’s disease. Arch Dermatol. 1967;96:176-181.
- Earhart RN, Aeling JA, Nuss DD, et al. Pseudo-Kaposi sarcoma. A patient with arteriovenous malformation and skin lesions simulating Kaposi sarcoma. Arch Dermatol. 1974;110:907-910.
- Lugovic´ L, Pusic´ J, Situm M, et al. Acroangiodermatitis (pseudo-Kaposi sarcoma): three case reports. Acta Dermatovenerol Croat. 2007;15:152-157.
- Horiguchi Y, Takahashi K, Tanizaki H, et al. Case of bilateral acroangiodermatitis due to symmetrical arteriovenous fistulas of the soles. J Dermatol. 2015;42:989-991.
- Dog˘an S, Boztepe G, Karaduman A. Pseudo-Kaposi sarcoma: a challenging vascular phenomenon. Dermatol Online J. 2007;13:22.
- Mazloom SE, Stallings A, Kyei A. Differentiating intralymphatic histiocytosis, intravascular histiocytosis, and subtypes of reactive angioendotheliomatosis: review of clinical and histologic features of all cases reported to date. Am J Dermatopathol. 2017;39:33-39.
- Rongioletti F, Rebora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
- Kanitakis J, Narvaez D, Claudy A. Expression of the CD34 antigen distinguishes Kaposi’s sarcoma from pseudo-Kaposi’s sarcoma (acroangiodermatitis). Br J Dermatol. 1996;134:44-46.
- Pires A, Depairon M, Ricci C, et al. Effect of compression therapy on a pseudo-Kaposi sarcoma. Dermatology. 1999;198:439-441.
- Hayek S, Atiyeh B, Zgheib E. Stewart-Bluefarb syndrome: review of the literature and case report of chronic ulcer treatment with heparan sulphate (Cacipliq20®). Int Wound J. 2015;12:169-172.
- Varyani N, Thukral A, Kumar N, et al. Nonhealing ulcer: acroangiodermatitis of Mali. Case Rep Dermatol Med. 2011;2011:909383.
- Mehta AA, Pereira RR, Nayak C, et al. Acroangiodermatitis of Mali: a rare vascular phenomenon. Indian J Dermatol Venereol Leprol. 2010;76:553-556.
- Rashkovsky I, Gilead L, Schamroth J, et al. Acro-angiodermatitis: review of the literature and report of a case. Acta Derm Venereol. 1995;75:475-478.
Practice Points
- Acroangiodermatitis (AAD) may mimic Kaposi sarcoma clinically and histopathologically. A human herpesvirus 8 stain is helpful to differentiate these two entities.
- Diagnosis of AAD should prompt investigation of an underlying arteriovenous malformation, as the disease may have systemic consequences such as congestive heart failure.
Heparin-Induced Bullous Hemorrhagic Dermatosis Confined to the Oral Mucosa
Heparin is a naturally occurring anticoagulant and is commonly used to treat or prevent venous thrombosis or the extension of thrombosis.
Adverse effects of heparin administration include bleeding, injection-site pain, and thrombocytopenia. Heparin-induced thrombocytopenia (HIT) is a serious side effect wherein antibodies are formed against platelet antigens and predispose the patient to venous and arterial thrombosis.
Bullous hemorrhagic dermatosis is a poorly understood idiosyncratic drug reaction characterized by tense, blood-filled blisters that arise following the administration of subcutaneous low-molecular-weight heparin or intravenous unfractionated heparin (UFH). First reported in 2006 by Perrinaud et al
Case Report
An 84-year-old man was admitted to the cardiology service with severe substernal chest pain. An electrocardiogram did not show any ST-segment elevations; however, he had elevated troponin T levels. He had a medical history of coronary artery disease complicated by myocardial infarction (MI), as well as ischemic cardiomyopathy, hypertension, hyperlipidemia, ischemic stroke, and pulmonary embolism for which he was on long-term anticoagulation for years with warfarin, aspirin, and clopidogrel. The patient was diagnosed with a non–ST-segment elevation MI. Accordingly, the patient’s warfarin was discontinued, and he was administered a bolus and continuous infusion of UFH. He also was continued on aspirin and clopidogrel. Within 6 hours of initiation of UFH, the patient noted multiple discrete swollen lesions in the mouth. Dermatology consultation and biopsy of the lesions were deferred due to acute management of the patient’s MI.
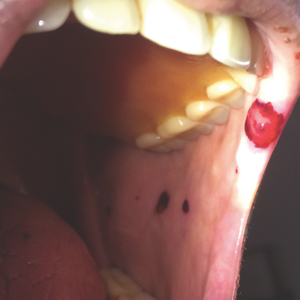
Physical examination revealed a moist oral mucosa with 7 slightly raised, hemorrhagic bullae ranging from 2 to 7 mm in diameter (Figure, A and B). One oral lesion was tense and had become denuded prior to evaluation. Laboratory testing included a normal platelet count (160,000/µL), a nearly therapeutic international normalized ratio (1.9), and a partial thromboplastin time that was initially normal (27 seconds) prior to admission and development of the oral lesions but found to be elevated (176 seconds) after admission and initial UFH bolus.
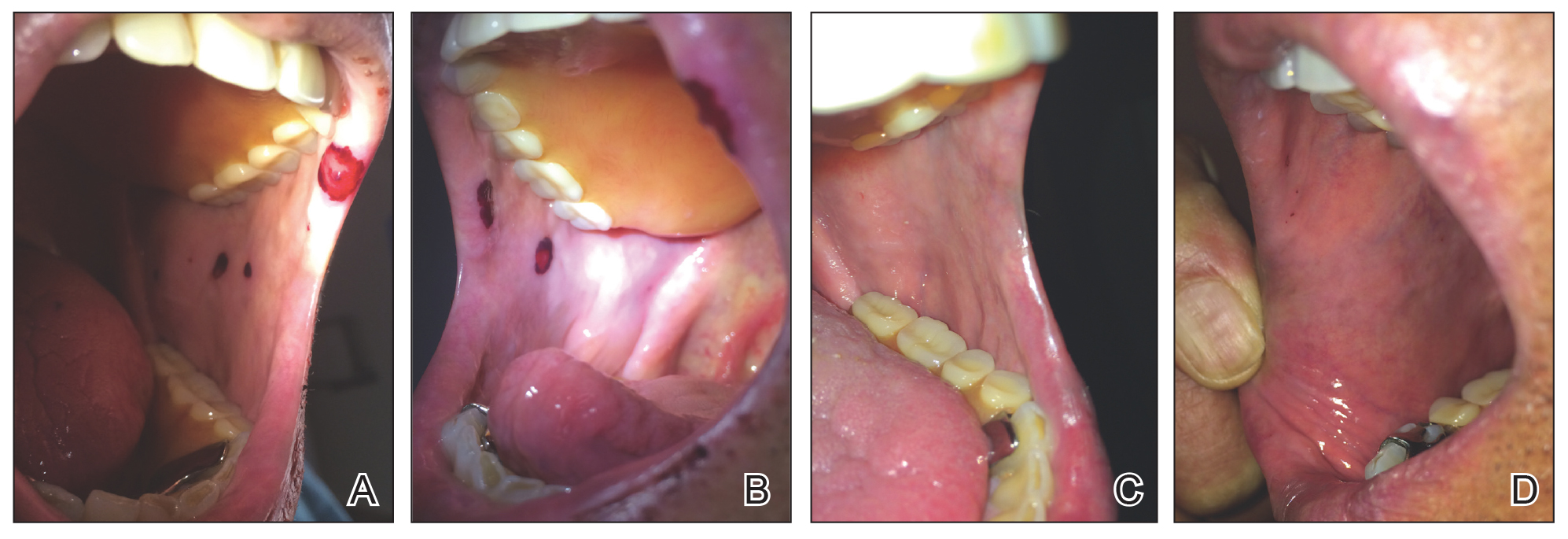
Upon further questioning, the patient revealed a history of similar oral lesions 1 year prior, following exposure to subcutaneous enoxaparin. At that time, formal evaluation by dermatology was deferred due to the rapid resolution of the blisters. Despite these new oral lesions, the patient was continued on a heparin drip for the next 48 hours because of the mortality benefit of heparin in non–ST-segment elevation MI. The patient was discharged from the hospital on a regimen of aspirin, warfarin, and clopidogrel. At 2-week follow-up, the oral lesions had resolved (Figure, C and D).
Comment
Heparin-Induced Skin Lesions
The 2 most common types of heparin-induced skin lesions are delayed-type hypersensitivity reactions and immune-mediated HIT. A 2009 Canadian study found that the overwhelming majority of heparin-induced skin lesions are due to delayed-type hypersensitivity reactions.
Types of HIT
Heparin-induced thrombocytopenia is one of the most serious adverse reactions to heparin administration. There are 2 subtypes of HIT, which differ in their clinical significance and pathophysiology.
Type II HIT is an immune-mediated response caused by the formation of IgG autoantibodies against the heparin–platelet factor 4 complex. Antibody formation and thrombocytopenia typically occur after 4 to 10 days of heparin exposure, and there can be devastating arterial and venous thrombotic complications.
Diagnosis of HIT
Heparin-induced thrombocytopenia should be suspected in patients with a lowered platelet count, particularly if the decrease is more than 50% from baseline, and in patients who develop stroke, MI, pulmonary embolism, or deep vein thrombosis while on heparin. Heparin-induced thrombocytopenia was not observed in our patient, as his platelet count remained stable between 160,000 and 164,000/µL throughout his hospital stay and he did not develop any evidence of thrombosis.
Differential Diagnosis
Our patient’s lesions appeared morphologically similar to
Bullous pemphigoid also was considered given the presence of tense bullae in an elderly patient. However, the rapid and spontaneous resolution of these lesions with complete lack of skin involvement made this diagnosis less likely.12
Heparin-Induced Bullous Hemorrhagic Dermatosis
Because our patient described a similar reaction while taking enoxaparin in the past, this case represents an idiosyncratic drug reaction, possibly from antibodies to a heparin-antigen complex. Heparin-induced bullous hemorrhagic dermatosis is a rarely reported condition with the majority of lesions presenting on the extremities.
Conclusion
We describe a rare side effect of heparin therapy characterized by discrete blisters on the oral mucosa. However, familiarity with the spectrum of reactions to heparin allowed the patient to continue heparin therapy despite this side effect, as the eruption was not life-threatening and the benefit of continuing heparin outweighed this adverse effect.
- Gómez-Outes A, Suárez-Gea ML, Calvo-Rojas G, et al. Discovery of anticoagulant drugs: a historical perspective. Curr Drug Discov Technol. 2012;9:83-104.
- Noti C, Seeberger PH. Chemical approaches to define the structure-activity relationship of heparin-like glycosaminoglycans. Chem Biol. 2005;12:731-756.
- Bakchoul T. An update on heparin-induced thrombocytopenia: diagnosis and management. Expert Opin Drug Saf. 2016;15:787-797.
- Schindewolf M, Schwaner S, Wolter M, et al. Incidence and causes of heparin-induced skin lesions. Can Med Assoc J. 2009;181:477-481.
- Perrinaud A, Jacobi D, Machet MC, et al. Bullous hemorrhagic dermatosis occurring at sites distant from subcutaneous injections of heparin: three cases. J Am Acad Dermatol. 2006;54(2 suppl):S5-S7.
- Naveen KN, Rai V. Bullous hemorrhagic dermatosis: a case report. Indian J Dermatol. 2014;59:423.
- Choudhry S, Fishman PM, Hernandez C. Heparin-induced bullous hemorrhagic dermatosis. Cutis. 2013;91:93-98.
- Villanueva CA, Nájera L, Espinosa P, et al. Bullous hemorrhagic dermatosis at distant sites: a report of 2 new cases due to enoxaparin injection and a review of the literature. Actas Dermosifiliogr. 2012;103:816-819.
- Ahmed I, Majeed A, Powell R. Heparin induced thrombocytopenia: diagnosis and management update. Postgrad Med J. 2007;83:575-582.
- Horie N, Kawano R, Inaba J, et al. Angina bullosa hemorrhagica of the soft palate: a clinical study of 16 cases. J Oral Sci. 2008;50:33-36.
- Rai S, Kaur M, Goel S. Angina bullosa hemorrhagica: report of 2 cases. Indian J Dermatol. 2012;57:503.
- Lawson W. Bullous oral lesions: clues to identifying—and managing—the cause. Consultant. 2013;53:168-176.
Heparin is a naturally occurring anticoagulant and is commonly used to treat or prevent venous thrombosis or the extension of thrombosis.
Adverse effects of heparin administration include bleeding, injection-site pain, and thrombocytopenia. Heparin-induced thrombocytopenia (HIT) is a serious side effect wherein antibodies are formed against platelet antigens and predispose the patient to venous and arterial thrombosis.
Bullous hemorrhagic dermatosis is a poorly understood idiosyncratic drug reaction characterized by tense, blood-filled blisters that arise following the administration of subcutaneous low-molecular-weight heparin or intravenous unfractionated heparin (UFH). First reported in 2006 by Perrinaud et al
Case Report
An 84-year-old man was admitted to the cardiology service with severe substernal chest pain. An electrocardiogram did not show any ST-segment elevations; however, he had elevated troponin T levels. He had a medical history of coronary artery disease complicated by myocardial infarction (MI), as well as ischemic cardiomyopathy, hypertension, hyperlipidemia, ischemic stroke, and pulmonary embolism for which he was on long-term anticoagulation for years with warfarin, aspirin, and clopidogrel. The patient was diagnosed with a non–ST-segment elevation MI. Accordingly, the patient’s warfarin was discontinued, and he was administered a bolus and continuous infusion of UFH. He also was continued on aspirin and clopidogrel. Within 6 hours of initiation of UFH, the patient noted multiple discrete swollen lesions in the mouth. Dermatology consultation and biopsy of the lesions were deferred due to acute management of the patient’s MI.
Physical examination revealed a moist oral mucosa with 7 slightly raised, hemorrhagic bullae ranging from 2 to 7 mm in diameter (Figure, A and B). One oral lesion was tense and had become denuded prior to evaluation. Laboratory testing included a normal platelet count (160,000/µL), a nearly therapeutic international normalized ratio (1.9), and a partial thromboplastin time that was initially normal (27 seconds) prior to admission and development of the oral lesions but found to be elevated (176 seconds) after admission and initial UFH bolus.
Upon further questioning, the patient revealed a history of similar oral lesions 1 year prior, following exposure to subcutaneous enoxaparin. At that time, formal evaluation by dermatology was deferred due to the rapid resolution of the blisters. Despite these new oral lesions, the patient was continued on a heparin drip for the next 48 hours because of the mortality benefit of heparin in non–ST-segment elevation MI. The patient was discharged from the hospital on a regimen of aspirin, warfarin, and clopidogrel. At 2-week follow-up, the oral lesions had resolved (Figure, C and D).
Comment
Heparin-Induced Skin Lesions
The 2 most common types of heparin-induced skin lesions are delayed-type hypersensitivity reactions and immune-mediated HIT. A 2009 Canadian study found that the overwhelming majority of heparin-induced skin lesions are due to delayed-type hypersensitivity reactions.
Types of HIT
Heparin-induced thrombocytopenia is one of the most serious adverse reactions to heparin administration. There are 2 subtypes of HIT, which differ in their clinical significance and pathophysiology.
Type II HIT is an immune-mediated response caused by the formation of IgG autoantibodies against the heparin–platelet factor 4 complex. Antibody formation and thrombocytopenia typically occur after 4 to 10 days of heparin exposure, and there can be devastating arterial and venous thrombotic complications.
Diagnosis of HIT
Heparin-induced thrombocytopenia should be suspected in patients with a lowered platelet count, particularly if the decrease is more than 50% from baseline, and in patients who develop stroke, MI, pulmonary embolism, or deep vein thrombosis while on heparin. Heparin-induced thrombocytopenia was not observed in our patient, as his platelet count remained stable between 160,000 and 164,000/µL throughout his hospital stay and he did not develop any evidence of thrombosis.
Differential Diagnosis
Our patient’s lesions appeared morphologically similar to
Bullous pemphigoid also was considered given the presence of tense bullae in an elderly patient. However, the rapid and spontaneous resolution of these lesions with complete lack of skin involvement made this diagnosis less likely.12
Heparin-Induced Bullous Hemorrhagic Dermatosis
Because our patient described a similar reaction while taking enoxaparin in the past, this case represents an idiosyncratic drug reaction, possibly from antibodies to a heparin-antigen complex. Heparin-induced bullous hemorrhagic dermatosis is a rarely reported condition with the majority of lesions presenting on the extremities.
Conclusion
We describe a rare side effect of heparin therapy characterized by discrete blisters on the oral mucosa. However, familiarity with the spectrum of reactions to heparin allowed the patient to continue heparin therapy despite this side effect, as the eruption was not life-threatening and the benefit of continuing heparin outweighed this adverse effect.
Heparin is a naturally occurring anticoagulant and is commonly used to treat or prevent venous thrombosis or the extension of thrombosis.
Adverse effects of heparin administration include bleeding, injection-site pain, and thrombocytopenia. Heparin-induced thrombocytopenia (HIT) is a serious side effect wherein antibodies are formed against platelet antigens and predispose the patient to venous and arterial thrombosis.
Bullous hemorrhagic dermatosis is a poorly understood idiosyncratic drug reaction characterized by tense, blood-filled blisters that arise following the administration of subcutaneous low-molecular-weight heparin or intravenous unfractionated heparin (UFH). First reported in 2006 by Perrinaud et al
Case Report
An 84-year-old man was admitted to the cardiology service with severe substernal chest pain. An electrocardiogram did not show any ST-segment elevations; however, he had elevated troponin T levels. He had a medical history of coronary artery disease complicated by myocardial infarction (MI), as well as ischemic cardiomyopathy, hypertension, hyperlipidemia, ischemic stroke, and pulmonary embolism for which he was on long-term anticoagulation for years with warfarin, aspirin, and clopidogrel. The patient was diagnosed with a non–ST-segment elevation MI. Accordingly, the patient’s warfarin was discontinued, and he was administered a bolus and continuous infusion of UFH. He also was continued on aspirin and clopidogrel. Within 6 hours of initiation of UFH, the patient noted multiple discrete swollen lesions in the mouth. Dermatology consultation and biopsy of the lesions were deferred due to acute management of the patient’s MI.
Physical examination revealed a moist oral mucosa with 7 slightly raised, hemorrhagic bullae ranging from 2 to 7 mm in diameter (Figure, A and B). One oral lesion was tense and had become denuded prior to evaluation. Laboratory testing included a normal platelet count (160,000/µL), a nearly therapeutic international normalized ratio (1.9), and a partial thromboplastin time that was initially normal (27 seconds) prior to admission and development of the oral lesions but found to be elevated (176 seconds) after admission and initial UFH bolus.
Upon further questioning, the patient revealed a history of similar oral lesions 1 year prior, following exposure to subcutaneous enoxaparin. At that time, formal evaluation by dermatology was deferred due to the rapid resolution of the blisters. Despite these new oral lesions, the patient was continued on a heparin drip for the next 48 hours because of the mortality benefit of heparin in non–ST-segment elevation MI. The patient was discharged from the hospital on a regimen of aspirin, warfarin, and clopidogrel. At 2-week follow-up, the oral lesions had resolved (Figure, C and D).
Comment
Heparin-Induced Skin Lesions
The 2 most common types of heparin-induced skin lesions are delayed-type hypersensitivity reactions and immune-mediated HIT. A 2009 Canadian study found that the overwhelming majority of heparin-induced skin lesions are due to delayed-type hypersensitivity reactions.
Types of HIT
Heparin-induced thrombocytopenia is one of the most serious adverse reactions to heparin administration. There are 2 subtypes of HIT, which differ in their clinical significance and pathophysiology.
Type II HIT is an immune-mediated response caused by the formation of IgG autoantibodies against the heparin–platelet factor 4 complex. Antibody formation and thrombocytopenia typically occur after 4 to 10 days of heparin exposure, and there can be devastating arterial and venous thrombotic complications.
Diagnosis of HIT
Heparin-induced thrombocytopenia should be suspected in patients with a lowered platelet count, particularly if the decrease is more than 50% from baseline, and in patients who develop stroke, MI, pulmonary embolism, or deep vein thrombosis while on heparin. Heparin-induced thrombocytopenia was not observed in our patient, as his platelet count remained stable between 160,000 and 164,000/µL throughout his hospital stay and he did not develop any evidence of thrombosis.
Differential Diagnosis
Our patient’s lesions appeared morphologically similar to
Bullous pemphigoid also was considered given the presence of tense bullae in an elderly patient. However, the rapid and spontaneous resolution of these lesions with complete lack of skin involvement made this diagnosis less likely.12
Heparin-Induced Bullous Hemorrhagic Dermatosis
Because our patient described a similar reaction while taking enoxaparin in the past, this case represents an idiosyncratic drug reaction, possibly from antibodies to a heparin-antigen complex. Heparin-induced bullous hemorrhagic dermatosis is a rarely reported condition with the majority of lesions presenting on the extremities.
Conclusion
We describe a rare side effect of heparin therapy characterized by discrete blisters on the oral mucosa. However, familiarity with the spectrum of reactions to heparin allowed the patient to continue heparin therapy despite this side effect, as the eruption was not life-threatening and the benefit of continuing heparin outweighed this adverse effect.
- Gómez-Outes A, Suárez-Gea ML, Calvo-Rojas G, et al. Discovery of anticoagulant drugs: a historical perspective. Curr Drug Discov Technol. 2012;9:83-104.
- Noti C, Seeberger PH. Chemical approaches to define the structure-activity relationship of heparin-like glycosaminoglycans. Chem Biol. 2005;12:731-756.
- Bakchoul T. An update on heparin-induced thrombocytopenia: diagnosis and management. Expert Opin Drug Saf. 2016;15:787-797.
- Schindewolf M, Schwaner S, Wolter M, et al. Incidence and causes of heparin-induced skin lesions. Can Med Assoc J. 2009;181:477-481.
- Perrinaud A, Jacobi D, Machet MC, et al. Bullous hemorrhagic dermatosis occurring at sites distant from subcutaneous injections of heparin: three cases. J Am Acad Dermatol. 2006;54(2 suppl):S5-S7.
- Naveen KN, Rai V. Bullous hemorrhagic dermatosis: a case report. Indian J Dermatol. 2014;59:423.
- Choudhry S, Fishman PM, Hernandez C. Heparin-induced bullous hemorrhagic dermatosis. Cutis. 2013;91:93-98.
- Villanueva CA, Nájera L, Espinosa P, et al. Bullous hemorrhagic dermatosis at distant sites: a report of 2 new cases due to enoxaparin injection and a review of the literature. Actas Dermosifiliogr. 2012;103:816-819.
- Ahmed I, Majeed A, Powell R. Heparin induced thrombocytopenia: diagnosis and management update. Postgrad Med J. 2007;83:575-582.
- Horie N, Kawano R, Inaba J, et al. Angina bullosa hemorrhagica of the soft palate: a clinical study of 16 cases. J Oral Sci. 2008;50:33-36.
- Rai S, Kaur M, Goel S. Angina bullosa hemorrhagica: report of 2 cases. Indian J Dermatol. 2012;57:503.
- Lawson W. Bullous oral lesions: clues to identifying—and managing—the cause. Consultant. 2013;53:168-176.
- Gómez-Outes A, Suárez-Gea ML, Calvo-Rojas G, et al. Discovery of anticoagulant drugs: a historical perspective. Curr Drug Discov Technol. 2012;9:83-104.
- Noti C, Seeberger PH. Chemical approaches to define the structure-activity relationship of heparin-like glycosaminoglycans. Chem Biol. 2005;12:731-756.
- Bakchoul T. An update on heparin-induced thrombocytopenia: diagnosis and management. Expert Opin Drug Saf. 2016;15:787-797.
- Schindewolf M, Schwaner S, Wolter M, et al. Incidence and causes of heparin-induced skin lesions. Can Med Assoc J. 2009;181:477-481.
- Perrinaud A, Jacobi D, Machet MC, et al. Bullous hemorrhagic dermatosis occurring at sites distant from subcutaneous injections of heparin: three cases. J Am Acad Dermatol. 2006;54(2 suppl):S5-S7.
- Naveen KN, Rai V. Bullous hemorrhagic dermatosis: a case report. Indian J Dermatol. 2014;59:423.
- Choudhry S, Fishman PM, Hernandez C. Heparin-induced bullous hemorrhagic dermatosis. Cutis. 2013;91:93-98.
- Villanueva CA, Nájera L, Espinosa P, et al. Bullous hemorrhagic dermatosis at distant sites: a report of 2 new cases due to enoxaparin injection and a review of the literature. Actas Dermosifiliogr. 2012;103:816-819.
- Ahmed I, Majeed A, Powell R. Heparin induced thrombocytopenia: diagnosis and management update. Postgrad Med J. 2007;83:575-582.
- Horie N, Kawano R, Inaba J, et al. Angina bullosa hemorrhagica of the soft palate: a clinical study of 16 cases. J Oral Sci. 2008;50:33-36.
- Rai S, Kaur M, Goel S. Angina bullosa hemorrhagica: report of 2 cases. Indian J Dermatol. 2012;57:503.
- Lawson W. Bullous oral lesions: clues to identifying—and managing—the cause. Consultant. 2013;53:168-176.
Practice Points
- It is important for physicians to recognize the clinical appearance of cutaneous adverse reactions to heparin, including bullous hemorrhagic dermatosis.
- Heparin-induced bullous hemorrhagic dermatosis tends to self-resolve, even with continuation of unfractionated heparin.
Erythema Gyratum Repens–like Eruption in Sézary Syndrome: Evidence for the Role of a Dermatophyte
Case Report
A 65-year-old woman presented with stage IVA2 mycosis fungoides (MF)(T4N3M0B2)/Sézary syndrome (SS). A peripheral blood count contained 6000 Sézary cells with cerebriform nuclei, a CD2+/−CD3+CD4+CD5+/−CD7+CD8−CD26−immunophenotype, and a highly abnormal CD4 to CD8 ratio (70:1). Positron emission tomography and computed tomography demonstrated hypermetabolic subcutaneous nodules in the base of the neck and generalized lymphadenopathy. Lymph node biopsy showed involvement by T-cell lymphoma and dominant T-cell receptor γ clonality by polymerase chain reaction.
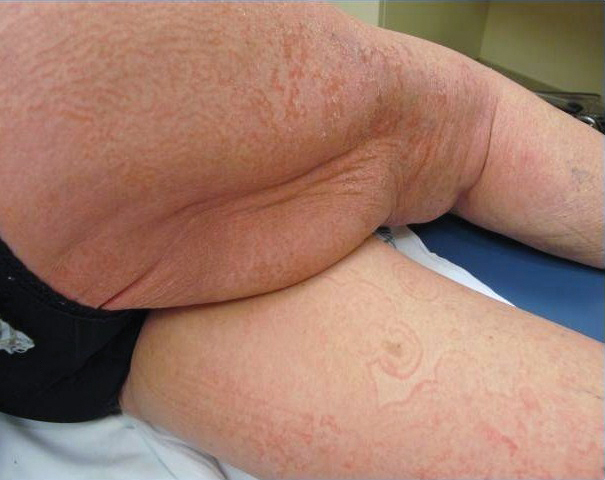
On initial presentation to the Cutaneous Lymphoma Clinic at the University of Wisconsin-Madison, the patient was erythrodermic. She also was noted to have undulating wavy bands and concentric annular, ringlike, thin, erythematous plaques with trailing scale, giving a wood grain, zebra hide–like appearance involving the buttocks, abdomen, and lower extremities (Figure 1). Lesions were markedly pruritic and were advancing rapidly. A diagnosis of erythema gyratum repens (EGR)–like eruption was made.
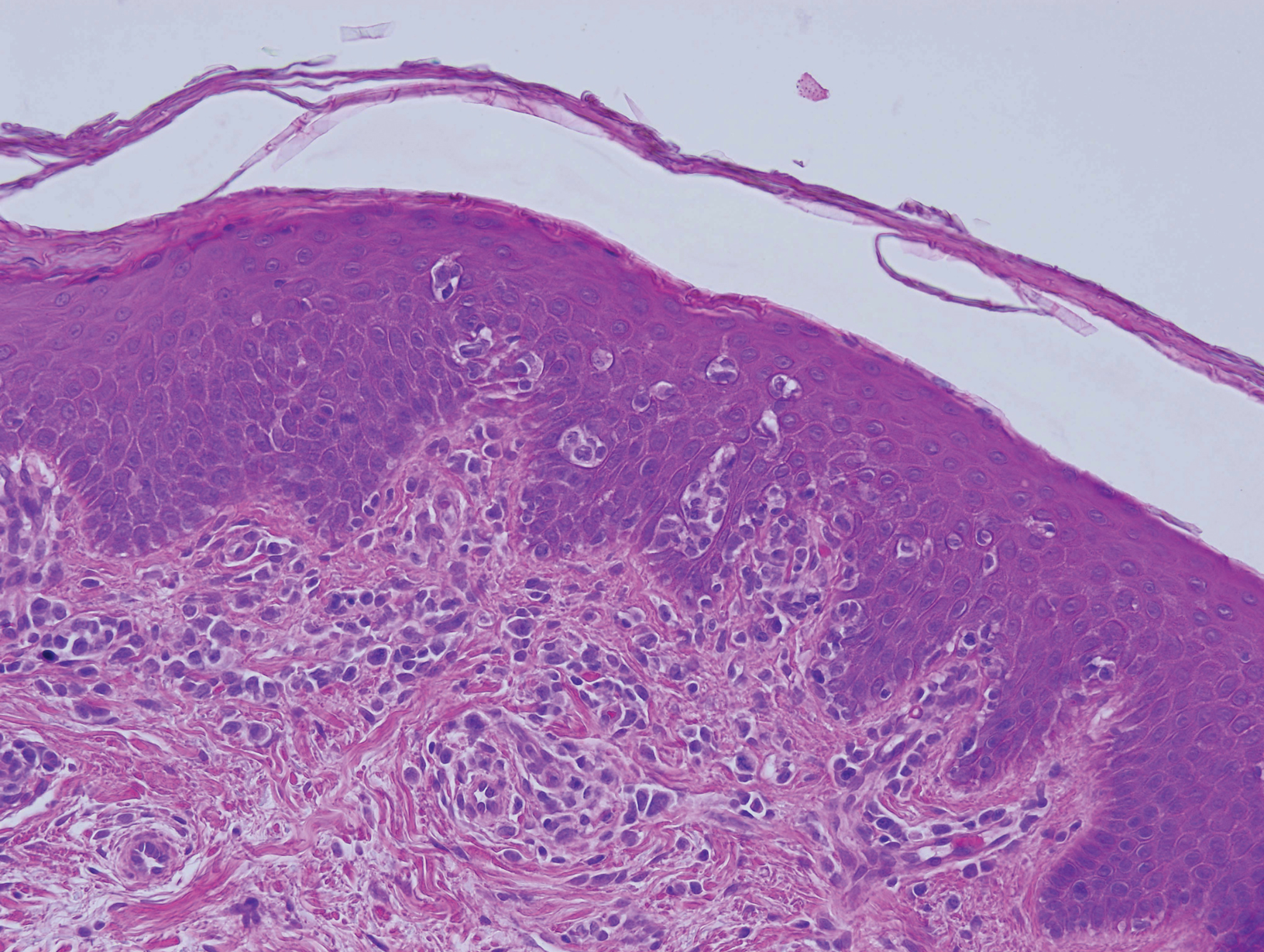
Biopsy of an EGR-like area on the leg showed a superficial perivascular and somewhat lichenoid lymphoid infiltrate (Figure 2). Lymphocytes were lined up along the basal layer, occasionally forming nests within the epidermis. Nearly all mononuclear cells in the epidermis and dermis exhibited positive CD3 and CD4 staining, with only scattered CD8 cells. These features were compatible with cutaneous involvement in SS. A concurrent biopsy from diffusely erythrodermic forearm skin, which lacked EGR-like morphology, showed similar histopathologic and immunophenotypic features.
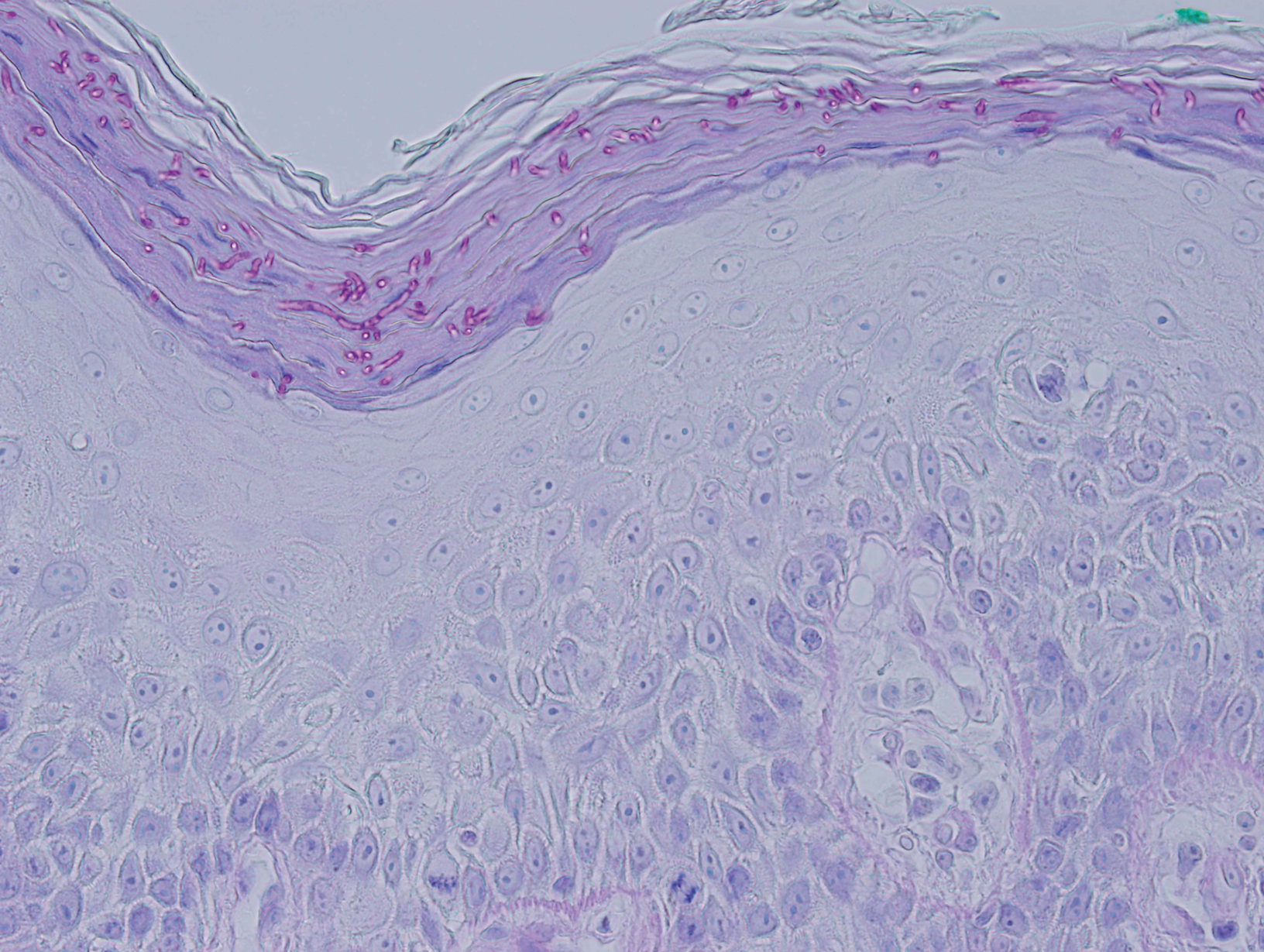
Periodic acid–Schiff (PAS) with diastase stain revealed numerous septate hyphae within the stratum corneum in both skin biopsy specimens (Figure 3). Fungal culture of EGR-like lesions was positive for a nonsporulating filamentous fungus, identified as Trichophyton rubrum by DNA sequencing.
A diagnosis of EGR-like eruption secondary to tinea corporis in SS was made. The possibility of tinea incognito also was considered to explain the presence of dermatophytes in the biopsy from skin that exhibited only erythroderma clinically; however, the patient did not have a history of corticosteroid use.
Interferon alfa-2b and methotrexate therapy was initiated. Additionally, oral terbinafine (250 mg/d) was initiated for 14 days, resulting in complete resolution of the EGR-like eruption; nevertheless, diffuse erythema remained. Subsequently, within 3 months of treatment, the cutaneous T-cell lymphoma (CTCL) improved with continued interferon alfa-2b and methotrexate. Erythroderma became minimal; the circulating Sézary cell count decreased by 50%. The patient ultimately had multiple relapses in erythroderma and progression of SS. Erythema gyratum repens–like lesions recurred on multiple occasions, with a temporary response to repeat courses of oral terbinafine.
Comment
Defining True EGR vs EGR-like Eruption
Sézary syndrome represents the leukemic stage of CTCL, which is defined by the triad of erythroderma; generalized lymphadenopathy; and neoplastic T cells in the skin, lymph nodes, and peripheral blood. It is well known that CTCL can mimic multiple benign and malignant dermatoses. One rare presentation of CTCL is an EGR-like eruption.
Erythema gyratum repens presents as rapidly advancing, erythematous, concentric bands that can be figurate, gyrate, or annular, with a fine trailing edge of scale (wood grain pattern). The diagnosis is based on the characteristic clinical pattern of EGR and by ruling out other mimicking conditions with biopsy.1 Patients with the characteristic clinical pattern but with an alternate underlying dermatosis are described as having an EGR-like eruption rather than true EGR.
True EGR is most often but not always associated with underlying malignancy. Biopsy of true EGR eruptions show nonspecific histopathologic features, with perivascular superficial mononuclear dermatitis, occasional mild spongiosis, and focal parakeratosis; specific features of an alternate dermatosis are lacking.2 In addition to CTCL, EGR-like eruptions have been described in a number of diseases, including systemic lupus erythematosus, erythema annulare centrifugum, bullous dermatosis, erythrokeratodermia variabilis, urticarial vasculitis, leukocytoclastic vasculitis, and neutrophilic dermatoses.
Prior Reports of EGR-like Eruption in Association With MF
According to a PubMed search of articles indexed for MEDLINE using the terms erythema gyratum repens in mycosis fungoides, mycosis fungoides with tinea, and concentric wood grain erythema, there have been 6 other cases of an EGR-like eruption in association with MF (Table). Poonawalla et al3 first described an EGR-like eruption (utilizing the term tinea pseudoimbricata) in a 55-year-old man with stage IB MF (T2N0M0B0). The patient had a preceding history of tinea pedis and tinea corporis that preceded the diagnosis of MF. At the time of MF diagnosis, the patient presented with extensive concentric, gyrate, wood grain, annular lesions. His MF was resistant to topical mechlorethamine, psoralen plus UVA, and oral bexarotene. The body surface area involvement decreased from 60% to less than 1% after institution of oral and topical antifungal therapy. It was postulated that the widespread dermatophytosis that preceded the development of MF may have been the persistent antigen leading to his disease. Preceding the diagnosis of MF, skin scrapings were floridly positive for dermatophyte hyphae. Fungal cultures from the affected areas of skin grew T rubrum.3
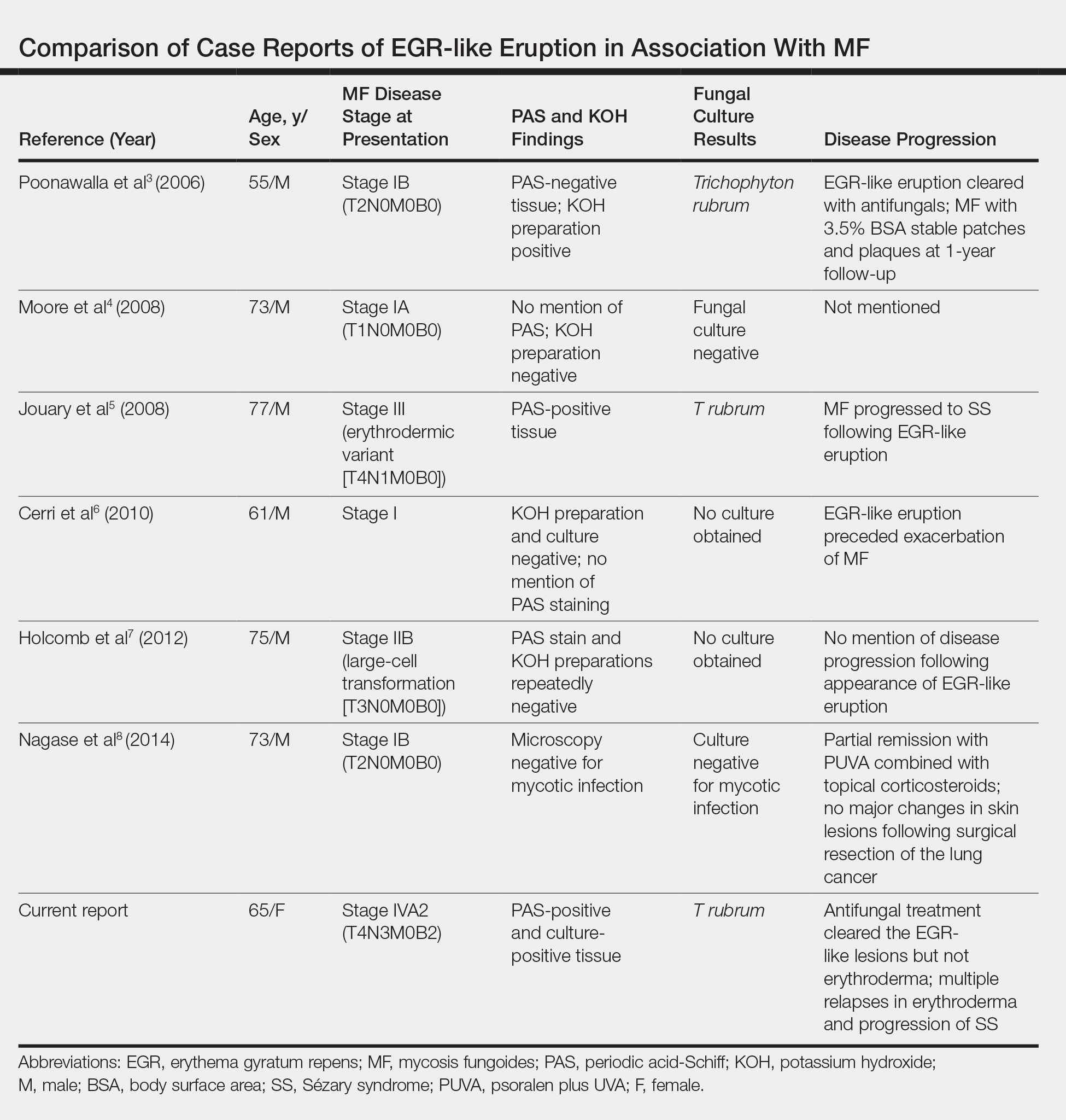
Moore et al4 described an EGR-like eruption on the trunk of a 73-year-old man with stage IA MF (T1N0M0B0). Biopsy was consistent with MF, but no fungal organisms were seen. Potassium hydroxide preparation and fungal cultures of the lesions also were negative for organisms. The patient was successfully treated with topical betamethasone.4Jouary et al5 described an EGR-like eruption in a 77-year-old man with stage III erythrodermic MF (T4N1M0B0). Biopsy showed mycelia on PAS stain. Subsequent culture isolated T rubrum. Terbinafine (250 mg/d) and ketoconazole cream 2% daily were initiated and the patient’s EGR-like rash quickly cleared, while MF progressed to SS.5
Cerri et al6 later described a case of EGR-like eruption in a 61-year-old man with stage I MF and an EGR-like eruption. Microscopic examination of potassium hydroxide (KOH) preparations and fungal culture of the lesions failed to demonstrate mycotic infection. There was no mention of PAS stain of skin biopsy specimens. In this case, the authors mentioned that EGR-like lesions preceded exacerbation of MF and questioned the prognostic significance of the EGR-like eruption in relation to MF.6
Holcomb et al7 reported the next case of a 75-year-old man with stage IIB MF (T3N0M0B0) with CD25+ and CD30+ large cell transformation who presented with an EGR-like eruption. In this case, PAS stain and KOH preparations were repeatedly negative for mycotic infection. Disease progression was not mentioned following the appearance of the EGR-like eruption.7
Nagase et al8 most recently described a case of a 73-year-old Japanese man with stage IB (T2N0M0B0) CD4−CD8− MF and lung cancer who developed a cutaneous eruption mimicking EGR. Microscopy and culture excluded the presence of a mycotic infection. The patient achieved partial remission with photochemotherapy (psoralen plus UVA) combined with topical corticosteroids. No major changes in the patient’s skin lesions were noted following surgical resection of the lung cancer.8
Dermatophyte Infection
It is known that conventional tinea corporis can occur in the setting of CTCL. However, EGR-like eruptions in CTCL can be distinguished from standard tinea corporis by the classic morphology of EGR and clinical history of rapid migration of these characteristic lesions.
Tinea imbricata is known to have a clinical appearance that is similar to EGR, but the infection is caused by Tinea concentricum, which is limited to southwest Polynesia, Melanesia, Southeast Asia, India, and Central America. Although T rubrum was the dermatophyte isolated by Poonawalla et al,3 Jouary et al,5 and in our case, whether T rubrum infection in the setting of CTCL has any impact on prognosis needs further study.
Our case of an EGR-like eruption presented in a patient with SS and tinea corporis. Biopsy specimens showed CTCL and concomitant dermatophytic infection that was confirmed with PAS stain and identified as T rubrum. Interestingly, our patient’s EGR-like eruption cleared with oral terbinafine therapy, consistent with findings described by Poonawalla et al3 and Jouary et al5 in which treatment of the dermatophytic infection led to resolution of the EGR-like eruption, suggesting a causative role.
However, testing for dermatophytes was negative in the other reported cases of EGR-like eruptions in patients with MF, despite screening for the presence of fungal microorganisms using KOH preparation, PAS staining, or fungal culture, or a combination of these methods,3-8 which raises the question: Do the cases reported without dermatophytic infection represent false-negative test results, or can the distinct clinical appearance of EGR indeed be seen in patients with CTCL who lack superimposed dermatophytosis? In 3 prior reported cases of EGR-like eruptions in MF, the eruption was preceded by immunosuppressive therapy.5-7
Further investigation is needed to correlate the role of dermatophytic infection in EGR-like eruptions. Our case and the Jouary et al5 case reported dermatophyte-positive EGR-like eruptions in MF and SS detected with histopathologic analysis and PAS stain. This low-cost screening method should be considered in future cases. If the test result is dermatophyte positive, a 14-day course of oral terbinafine (250 mg/d) might induce resolution of the EGR-like eruption.
Conclusion
The role of dermatophyte-induced EGR or EGR-like eruptions in other settings also warrants further investigation to shed light on this poorly understood yet striking dermatologic condition. Our patient showed both MF and dermatophytes in skin biopsy results, regardless of whether those sites showed erythroderma or EGR-like features clinically. On 3 occasions, antifungal treatment cleared the EGR-like lesions and associated pruritus but not erythroderma. Therefore, it appears that the mere presence of dermatophytes was necessary but not sufficient to produce the EGR-like lesions observed in our case.
- Rongioletti F, Fausti V, Parodi A. Erythema gyratum repens is not an obligate paraneoplastic disease: a systematic review of the literature and personal experience. J Eur Acad Dermatol Venereol. 2012;28:112-115.
- Albers SE, Fenske NA, Glass LF. Erythema gyratum repens: direct immunofluorescence microscopic findings. J Am Acad Dermatol. 1993;29:493-494.
- Poonawalla T, Chen W, Duvic M. Mycosis fungoides with tinea pseudoimbricata owing to Trichophyton rubrum infection. J Cutan Med Surg. 2006;10:52-56.
- Moore E, McFarlane R, Olerud J. Concentric wood grain erythema on the trunk. Arch Dermatol. 2008;144:673-678.
- Jouary T, Lalanne N, Stanislas S, et al. Erythema gyratum repens-like eruption in mycosis fungoides: is dermatophyte superinfection underdiagnosed in cutaneous T-cell lymphomas? J Eur Acad Dermatol Venereol. 2008;22:1276-1278.
- Cerri A, Vezzoli P, Serini SM, et al. Mycosis fungoides mimicking erythema gyratum repens: an additional variant? Eur J Dermatol. 2010;20:540-541.
- Holcomb M, Duvic M, Cutlan J. Erythema gyratum repens-like eruptions with large cell transformation in a patient with mycosis fungoides. Int J Dermatol. 2012;51:1231-1233.
- Nagase K, Shirai R, Okawa T, et al. CD4/CD8 double-negative mycosis fungoides mimicking erythema gyratum repens in a patient with underlying lung cancer. Acta Derm Venereol. 2014;94:89-90.
Case Report
A 65-year-old woman presented with stage IVA2 mycosis fungoides (MF)(T4N3M0B2)/Sézary syndrome (SS). A peripheral blood count contained 6000 Sézary cells with cerebriform nuclei, a CD2+/−CD3+CD4+CD5+/−CD7+CD8−CD26−immunophenotype, and a highly abnormal CD4 to CD8 ratio (70:1). Positron emission tomography and computed tomography demonstrated hypermetabolic subcutaneous nodules in the base of the neck and generalized lymphadenopathy. Lymph node biopsy showed involvement by T-cell lymphoma and dominant T-cell receptor γ clonality by polymerase chain reaction.
On initial presentation to the Cutaneous Lymphoma Clinic at the University of Wisconsin-Madison, the patient was erythrodermic. She also was noted to have undulating wavy bands and concentric annular, ringlike, thin, erythematous plaques with trailing scale, giving a wood grain, zebra hide–like appearance involving the buttocks, abdomen, and lower extremities (Figure 1). Lesions were markedly pruritic and were advancing rapidly. A diagnosis of erythema gyratum repens (EGR)–like eruption was made.
Biopsy of an EGR-like area on the leg showed a superficial perivascular and somewhat lichenoid lymphoid infiltrate (Figure 2). Lymphocytes were lined up along the basal layer, occasionally forming nests within the epidermis. Nearly all mononuclear cells in the epidermis and dermis exhibited positive CD3 and CD4 staining, with only scattered CD8 cells. These features were compatible with cutaneous involvement in SS. A concurrent biopsy from diffusely erythrodermic forearm skin, which lacked EGR-like morphology, showed similar histopathologic and immunophenotypic features.
Periodic acid–Schiff (PAS) with diastase stain revealed numerous septate hyphae within the stratum corneum in both skin biopsy specimens (Figure 3). Fungal culture of EGR-like lesions was positive for a nonsporulating filamentous fungus, identified as Trichophyton rubrum by DNA sequencing.
A diagnosis of EGR-like eruption secondary to tinea corporis in SS was made. The possibility of tinea incognito also was considered to explain the presence of dermatophytes in the biopsy from skin that exhibited only erythroderma clinically; however, the patient did not have a history of corticosteroid use.
Interferon alfa-2b and methotrexate therapy was initiated. Additionally, oral terbinafine (250 mg/d) was initiated for 14 days, resulting in complete resolution of the EGR-like eruption; nevertheless, diffuse erythema remained. Subsequently, within 3 months of treatment, the cutaneous T-cell lymphoma (CTCL) improved with continued interferon alfa-2b and methotrexate. Erythroderma became minimal; the circulating Sézary cell count decreased by 50%. The patient ultimately had multiple relapses in erythroderma and progression of SS. Erythema gyratum repens–like lesions recurred on multiple occasions, with a temporary response to repeat courses of oral terbinafine.
Comment
Defining True EGR vs EGR-like Eruption
Sézary syndrome represents the leukemic stage of CTCL, which is defined by the triad of erythroderma; generalized lymphadenopathy; and neoplastic T cells in the skin, lymph nodes, and peripheral blood. It is well known that CTCL can mimic multiple benign and malignant dermatoses. One rare presentation of CTCL is an EGR-like eruption.
Erythema gyratum repens presents as rapidly advancing, erythematous, concentric bands that can be figurate, gyrate, or annular, with a fine trailing edge of scale (wood grain pattern). The diagnosis is based on the characteristic clinical pattern of EGR and by ruling out other mimicking conditions with biopsy.1 Patients with the characteristic clinical pattern but with an alternate underlying dermatosis are described as having an EGR-like eruption rather than true EGR.
True EGR is most often but not always associated with underlying malignancy. Biopsy of true EGR eruptions show nonspecific histopathologic features, with perivascular superficial mononuclear dermatitis, occasional mild spongiosis, and focal parakeratosis; specific features of an alternate dermatosis are lacking.2 In addition to CTCL, EGR-like eruptions have been described in a number of diseases, including systemic lupus erythematosus, erythema annulare centrifugum, bullous dermatosis, erythrokeratodermia variabilis, urticarial vasculitis, leukocytoclastic vasculitis, and neutrophilic dermatoses.
Prior Reports of EGR-like Eruption in Association With MF
According to a PubMed search of articles indexed for MEDLINE using the terms erythema gyratum repens in mycosis fungoides, mycosis fungoides with tinea, and concentric wood grain erythema, there have been 6 other cases of an EGR-like eruption in association with MF (Table). Poonawalla et al3 first described an EGR-like eruption (utilizing the term tinea pseudoimbricata) in a 55-year-old man with stage IB MF (T2N0M0B0). The patient had a preceding history of tinea pedis and tinea corporis that preceded the diagnosis of MF. At the time of MF diagnosis, the patient presented with extensive concentric, gyrate, wood grain, annular lesions. His MF was resistant to topical mechlorethamine, psoralen plus UVA, and oral bexarotene. The body surface area involvement decreased from 60% to less than 1% after institution of oral and topical antifungal therapy. It was postulated that the widespread dermatophytosis that preceded the development of MF may have been the persistent antigen leading to his disease. Preceding the diagnosis of MF, skin scrapings were floridly positive for dermatophyte hyphae. Fungal cultures from the affected areas of skin grew T rubrum.3
Moore et al4 described an EGR-like eruption on the trunk of a 73-year-old man with stage IA MF (T1N0M0B0). Biopsy was consistent with MF, but no fungal organisms were seen. Potassium hydroxide preparation and fungal cultures of the lesions also were negative for organisms. The patient was successfully treated with topical betamethasone.4Jouary et al5 described an EGR-like eruption in a 77-year-old man with stage III erythrodermic MF (T4N1M0B0). Biopsy showed mycelia on PAS stain. Subsequent culture isolated T rubrum. Terbinafine (250 mg/d) and ketoconazole cream 2% daily were initiated and the patient’s EGR-like rash quickly cleared, while MF progressed to SS.5
Cerri et al6 later described a case of EGR-like eruption in a 61-year-old man with stage I MF and an EGR-like eruption. Microscopic examination of potassium hydroxide (KOH) preparations and fungal culture of the lesions failed to demonstrate mycotic infection. There was no mention of PAS stain of skin biopsy specimens. In this case, the authors mentioned that EGR-like lesions preceded exacerbation of MF and questioned the prognostic significance of the EGR-like eruption in relation to MF.6
Holcomb et al7 reported the next case of a 75-year-old man with stage IIB MF (T3N0M0B0) with CD25+ and CD30+ large cell transformation who presented with an EGR-like eruption. In this case, PAS stain and KOH preparations were repeatedly negative for mycotic infection. Disease progression was not mentioned following the appearance of the EGR-like eruption.7
Nagase et al8 most recently described a case of a 73-year-old Japanese man with stage IB (T2N0M0B0) CD4−CD8− MF and lung cancer who developed a cutaneous eruption mimicking EGR. Microscopy and culture excluded the presence of a mycotic infection. The patient achieved partial remission with photochemotherapy (psoralen plus UVA) combined with topical corticosteroids. No major changes in the patient’s skin lesions were noted following surgical resection of the lung cancer.8
Dermatophyte Infection
It is known that conventional tinea corporis can occur in the setting of CTCL. However, EGR-like eruptions in CTCL can be distinguished from standard tinea corporis by the classic morphology of EGR and clinical history of rapid migration of these characteristic lesions.
Tinea imbricata is known to have a clinical appearance that is similar to EGR, but the infection is caused by Tinea concentricum, which is limited to southwest Polynesia, Melanesia, Southeast Asia, India, and Central America. Although T rubrum was the dermatophyte isolated by Poonawalla et al,3 Jouary et al,5 and in our case, whether T rubrum infection in the setting of CTCL has any impact on prognosis needs further study.
Our case of an EGR-like eruption presented in a patient with SS and tinea corporis. Biopsy specimens showed CTCL and concomitant dermatophytic infection that was confirmed with PAS stain and identified as T rubrum. Interestingly, our patient’s EGR-like eruption cleared with oral terbinafine therapy, consistent with findings described by Poonawalla et al3 and Jouary et al5 in which treatment of the dermatophytic infection led to resolution of the EGR-like eruption, suggesting a causative role.
However, testing for dermatophytes was negative in the other reported cases of EGR-like eruptions in patients with MF, despite screening for the presence of fungal microorganisms using KOH preparation, PAS staining, or fungal culture, or a combination of these methods,3-8 which raises the question: Do the cases reported without dermatophytic infection represent false-negative test results, or can the distinct clinical appearance of EGR indeed be seen in patients with CTCL who lack superimposed dermatophytosis? In 3 prior reported cases of EGR-like eruptions in MF, the eruption was preceded by immunosuppressive therapy.5-7
Further investigation is needed to correlate the role of dermatophytic infection in EGR-like eruptions. Our case and the Jouary et al5 case reported dermatophyte-positive EGR-like eruptions in MF and SS detected with histopathologic analysis and PAS stain. This low-cost screening method should be considered in future cases. If the test result is dermatophyte positive, a 14-day course of oral terbinafine (250 mg/d) might induce resolution of the EGR-like eruption.
Conclusion
The role of dermatophyte-induced EGR or EGR-like eruptions in other settings also warrants further investigation to shed light on this poorly understood yet striking dermatologic condition. Our patient showed both MF and dermatophytes in skin biopsy results, regardless of whether those sites showed erythroderma or EGR-like features clinically. On 3 occasions, antifungal treatment cleared the EGR-like lesions and associated pruritus but not erythroderma. Therefore, it appears that the mere presence of dermatophytes was necessary but not sufficient to produce the EGR-like lesions observed in our case.
Case Report
A 65-year-old woman presented with stage IVA2 mycosis fungoides (MF)(T4N3M0B2)/Sézary syndrome (SS). A peripheral blood count contained 6000 Sézary cells with cerebriform nuclei, a CD2+/−CD3+CD4+CD5+/−CD7+CD8−CD26−immunophenotype, and a highly abnormal CD4 to CD8 ratio (70:1). Positron emission tomography and computed tomography demonstrated hypermetabolic subcutaneous nodules in the base of the neck and generalized lymphadenopathy. Lymph node biopsy showed involvement by T-cell lymphoma and dominant T-cell receptor γ clonality by polymerase chain reaction.
On initial presentation to the Cutaneous Lymphoma Clinic at the University of Wisconsin-Madison, the patient was erythrodermic. She also was noted to have undulating wavy bands and concentric annular, ringlike, thin, erythematous plaques with trailing scale, giving a wood grain, zebra hide–like appearance involving the buttocks, abdomen, and lower extremities (Figure 1). Lesions were markedly pruritic and were advancing rapidly. A diagnosis of erythema gyratum repens (EGR)–like eruption was made.
Biopsy of an EGR-like area on the leg showed a superficial perivascular and somewhat lichenoid lymphoid infiltrate (Figure 2). Lymphocytes were lined up along the basal layer, occasionally forming nests within the epidermis. Nearly all mononuclear cells in the epidermis and dermis exhibited positive CD3 and CD4 staining, with only scattered CD8 cells. These features were compatible with cutaneous involvement in SS. A concurrent biopsy from diffusely erythrodermic forearm skin, which lacked EGR-like morphology, showed similar histopathologic and immunophenotypic features.
Periodic acid–Schiff (PAS) with diastase stain revealed numerous septate hyphae within the stratum corneum in both skin biopsy specimens (Figure 3). Fungal culture of EGR-like lesions was positive for a nonsporulating filamentous fungus, identified as Trichophyton rubrum by DNA sequencing.
A diagnosis of EGR-like eruption secondary to tinea corporis in SS was made. The possibility of tinea incognito also was considered to explain the presence of dermatophytes in the biopsy from skin that exhibited only erythroderma clinically; however, the patient did not have a history of corticosteroid use.
Interferon alfa-2b and methotrexate therapy was initiated. Additionally, oral terbinafine (250 mg/d) was initiated for 14 days, resulting in complete resolution of the EGR-like eruption; nevertheless, diffuse erythema remained. Subsequently, within 3 months of treatment, the cutaneous T-cell lymphoma (CTCL) improved with continued interferon alfa-2b and methotrexate. Erythroderma became minimal; the circulating Sézary cell count decreased by 50%. The patient ultimately had multiple relapses in erythroderma and progression of SS. Erythema gyratum repens–like lesions recurred on multiple occasions, with a temporary response to repeat courses of oral terbinafine.
Comment
Defining True EGR vs EGR-like Eruption
Sézary syndrome represents the leukemic stage of CTCL, which is defined by the triad of erythroderma; generalized lymphadenopathy; and neoplastic T cells in the skin, lymph nodes, and peripheral blood. It is well known that CTCL can mimic multiple benign and malignant dermatoses. One rare presentation of CTCL is an EGR-like eruption.
Erythema gyratum repens presents as rapidly advancing, erythematous, concentric bands that can be figurate, gyrate, or annular, with a fine trailing edge of scale (wood grain pattern). The diagnosis is based on the characteristic clinical pattern of EGR and by ruling out other mimicking conditions with biopsy.1 Patients with the characteristic clinical pattern but with an alternate underlying dermatosis are described as having an EGR-like eruption rather than true EGR.
True EGR is most often but not always associated with underlying malignancy. Biopsy of true EGR eruptions show nonspecific histopathologic features, with perivascular superficial mononuclear dermatitis, occasional mild spongiosis, and focal parakeratosis; specific features of an alternate dermatosis are lacking.2 In addition to CTCL, EGR-like eruptions have been described in a number of diseases, including systemic lupus erythematosus, erythema annulare centrifugum, bullous dermatosis, erythrokeratodermia variabilis, urticarial vasculitis, leukocytoclastic vasculitis, and neutrophilic dermatoses.
Prior Reports of EGR-like Eruption in Association With MF
According to a PubMed search of articles indexed for MEDLINE using the terms erythema gyratum repens in mycosis fungoides, mycosis fungoides with tinea, and concentric wood grain erythema, there have been 6 other cases of an EGR-like eruption in association with MF (Table). Poonawalla et al3 first described an EGR-like eruption (utilizing the term tinea pseudoimbricata) in a 55-year-old man with stage IB MF (T2N0M0B0). The patient had a preceding history of tinea pedis and tinea corporis that preceded the diagnosis of MF. At the time of MF diagnosis, the patient presented with extensive concentric, gyrate, wood grain, annular lesions. His MF was resistant to topical mechlorethamine, psoralen plus UVA, and oral bexarotene. The body surface area involvement decreased from 60% to less than 1% after institution of oral and topical antifungal therapy. It was postulated that the widespread dermatophytosis that preceded the development of MF may have been the persistent antigen leading to his disease. Preceding the diagnosis of MF, skin scrapings were floridly positive for dermatophyte hyphae. Fungal cultures from the affected areas of skin grew T rubrum.3
Moore et al4 described an EGR-like eruption on the trunk of a 73-year-old man with stage IA MF (T1N0M0B0). Biopsy was consistent with MF, but no fungal organisms were seen. Potassium hydroxide preparation and fungal cultures of the lesions also were negative for organisms. The patient was successfully treated with topical betamethasone.4Jouary et al5 described an EGR-like eruption in a 77-year-old man with stage III erythrodermic MF (T4N1M0B0). Biopsy showed mycelia on PAS stain. Subsequent culture isolated T rubrum. Terbinafine (250 mg/d) and ketoconazole cream 2% daily were initiated and the patient’s EGR-like rash quickly cleared, while MF progressed to SS.5
Cerri et al6 later described a case of EGR-like eruption in a 61-year-old man with stage I MF and an EGR-like eruption. Microscopic examination of potassium hydroxide (KOH) preparations and fungal culture of the lesions failed to demonstrate mycotic infection. There was no mention of PAS stain of skin biopsy specimens. In this case, the authors mentioned that EGR-like lesions preceded exacerbation of MF and questioned the prognostic significance of the EGR-like eruption in relation to MF.6
Holcomb et al7 reported the next case of a 75-year-old man with stage IIB MF (T3N0M0B0) with CD25+ and CD30+ large cell transformation who presented with an EGR-like eruption. In this case, PAS stain and KOH preparations were repeatedly negative for mycotic infection. Disease progression was not mentioned following the appearance of the EGR-like eruption.7
Nagase et al8 most recently described a case of a 73-year-old Japanese man with stage IB (T2N0M0B0) CD4−CD8− MF and lung cancer who developed a cutaneous eruption mimicking EGR. Microscopy and culture excluded the presence of a mycotic infection. The patient achieved partial remission with photochemotherapy (psoralen plus UVA) combined with topical corticosteroids. No major changes in the patient’s skin lesions were noted following surgical resection of the lung cancer.8
Dermatophyte Infection
It is known that conventional tinea corporis can occur in the setting of CTCL. However, EGR-like eruptions in CTCL can be distinguished from standard tinea corporis by the classic morphology of EGR and clinical history of rapid migration of these characteristic lesions.
Tinea imbricata is known to have a clinical appearance that is similar to EGR, but the infection is caused by Tinea concentricum, which is limited to southwest Polynesia, Melanesia, Southeast Asia, India, and Central America. Although T rubrum was the dermatophyte isolated by Poonawalla et al,3 Jouary et al,5 and in our case, whether T rubrum infection in the setting of CTCL has any impact on prognosis needs further study.
Our case of an EGR-like eruption presented in a patient with SS and tinea corporis. Biopsy specimens showed CTCL and concomitant dermatophytic infection that was confirmed with PAS stain and identified as T rubrum. Interestingly, our patient’s EGR-like eruption cleared with oral terbinafine therapy, consistent with findings described by Poonawalla et al3 and Jouary et al5 in which treatment of the dermatophytic infection led to resolution of the EGR-like eruption, suggesting a causative role.
However, testing for dermatophytes was negative in the other reported cases of EGR-like eruptions in patients with MF, despite screening for the presence of fungal microorganisms using KOH preparation, PAS staining, or fungal culture, or a combination of these methods,3-8 which raises the question: Do the cases reported without dermatophytic infection represent false-negative test results, or can the distinct clinical appearance of EGR indeed be seen in patients with CTCL who lack superimposed dermatophytosis? In 3 prior reported cases of EGR-like eruptions in MF, the eruption was preceded by immunosuppressive therapy.5-7
Further investigation is needed to correlate the role of dermatophytic infection in EGR-like eruptions. Our case and the Jouary et al5 case reported dermatophyte-positive EGR-like eruptions in MF and SS detected with histopathologic analysis and PAS stain. This low-cost screening method should be considered in future cases. If the test result is dermatophyte positive, a 14-day course of oral terbinafine (250 mg/d) might induce resolution of the EGR-like eruption.
Conclusion
The role of dermatophyte-induced EGR or EGR-like eruptions in other settings also warrants further investigation to shed light on this poorly understood yet striking dermatologic condition. Our patient showed both MF and dermatophytes in skin biopsy results, regardless of whether those sites showed erythroderma or EGR-like features clinically. On 3 occasions, antifungal treatment cleared the EGR-like lesions and associated pruritus but not erythroderma. Therefore, it appears that the mere presence of dermatophytes was necessary but not sufficient to produce the EGR-like lesions observed in our case.
- Rongioletti F, Fausti V, Parodi A. Erythema gyratum repens is not an obligate paraneoplastic disease: a systematic review of the literature and personal experience. J Eur Acad Dermatol Venereol. 2012;28:112-115.
- Albers SE, Fenske NA, Glass LF. Erythema gyratum repens: direct immunofluorescence microscopic findings. J Am Acad Dermatol. 1993;29:493-494.
- Poonawalla T, Chen W, Duvic M. Mycosis fungoides with tinea pseudoimbricata owing to Trichophyton rubrum infection. J Cutan Med Surg. 2006;10:52-56.
- Moore E, McFarlane R, Olerud J. Concentric wood grain erythema on the trunk. Arch Dermatol. 2008;144:673-678.
- Jouary T, Lalanne N, Stanislas S, et al. Erythema gyratum repens-like eruption in mycosis fungoides: is dermatophyte superinfection underdiagnosed in cutaneous T-cell lymphomas? J Eur Acad Dermatol Venereol. 2008;22:1276-1278.
- Cerri A, Vezzoli P, Serini SM, et al. Mycosis fungoides mimicking erythema gyratum repens: an additional variant? Eur J Dermatol. 2010;20:540-541.
- Holcomb M, Duvic M, Cutlan J. Erythema gyratum repens-like eruptions with large cell transformation in a patient with mycosis fungoides. Int J Dermatol. 2012;51:1231-1233.
- Nagase K, Shirai R, Okawa T, et al. CD4/CD8 double-negative mycosis fungoides mimicking erythema gyratum repens in a patient with underlying lung cancer. Acta Derm Venereol. 2014;94:89-90.
- Rongioletti F, Fausti V, Parodi A. Erythema gyratum repens is not an obligate paraneoplastic disease: a systematic review of the literature and personal experience. J Eur Acad Dermatol Venereol. 2012;28:112-115.
- Albers SE, Fenske NA, Glass LF. Erythema gyratum repens: direct immunofluorescence microscopic findings. J Am Acad Dermatol. 1993;29:493-494.
- Poonawalla T, Chen W, Duvic M. Mycosis fungoides with tinea pseudoimbricata owing to Trichophyton rubrum infection. J Cutan Med Surg. 2006;10:52-56.
- Moore E, McFarlane R, Olerud J. Concentric wood grain erythema on the trunk. Arch Dermatol. 2008;144:673-678.
- Jouary T, Lalanne N, Stanislas S, et al. Erythema gyratum repens-like eruption in mycosis fungoides: is dermatophyte superinfection underdiagnosed in cutaneous T-cell lymphomas? J Eur Acad Dermatol Venereol. 2008;22:1276-1278.
- Cerri A, Vezzoli P, Serini SM, et al. Mycosis fungoides mimicking erythema gyratum repens: an additional variant? Eur J Dermatol. 2010;20:540-541.
- Holcomb M, Duvic M, Cutlan J. Erythema gyratum repens-like eruptions with large cell transformation in a patient with mycosis fungoides. Int J Dermatol. 2012;51:1231-1233.
- Nagase K, Shirai R, Okawa T, et al. CD4/CD8 double-negative mycosis fungoides mimicking erythema gyratum repens in a patient with underlying lung cancer. Acta Derm Venereol. 2014;94:89-90.
Practice Points
- Erythema gyratum repens (EGR) presents as rapidly advancing, erythematous, concentric bands that can be figurate, gyrate, or annular, with fine trailing scale.
- Although EGR typically is associated with underlying malignancy, it is not an obligate paraneoplastic syndrome. There are numerous cases that are not associated with underlying neoplasms.
- An EGR-like eruption may be observed in Sézary syndrome, and an overlying superficial dermatophyte infection may play a role.
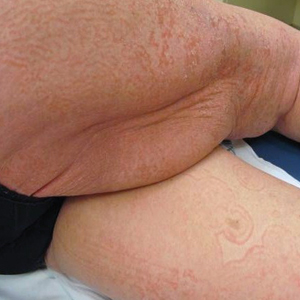
Ocular Chemical Burns in the Dermatology Office: A Practical Approach to Managing Safety Precautions
Many dermatologic procedures are performed on the face, such as skin biopsies, surgical excisions, and cosmetic procedures, which can increase the risk for accidental ocular injuries.1,2 Ocular chemical burns have been reported to account for approximately 3% to 20% of ocular injuries3,4 and are one of the few ocular emergencies dermatologists may encounter in practice. Given the potentially severe consequences of permanent vision changes or loss, it is important to take precautionary steps in preventing chemical exposures and know how to appropriately manage ophthalmic emergencies when they occur.1,5-8 In this article, we describe a patient with a transient ocular chemical injury from exposure to aluminum chloride hexahydrate that completely resolved with immediate care. We also offer practical guidance for the general dermatologist in the acute management of acidic chemical burns to the eye, highlighting immediate copious irrigation as the most important step in preventing severe permanent damage. Given that aluminum chloride hexahydrate is an acidic solution, we focus predominantly on the approach to acidic chemical exposures to the eye.
Case Report
A 61-year-old woman was seen in the dermatology outpatient clinic for a shave biopsy on the left cheek followed by aluminum chloride application for hemostasis. Following the biopsy, the patient stated she felt the sensation that something had dripped into the left eye and she felt a burning pain. There was a 30- to 60-second delay in irrigation of the eye, as it was at first unclear what had occurred. The patient reported an increased burning sensation, and at that point she was instructed to begin flushing the eye with tap water from the examination room sink for 15 to 20 minutes; she wanted to stop irrigation after a few minutes, and convincing her to continue thorough irrigation was somewhat challenging. It was determined that aluminum chloride hexahydrate had dripped from an oversaturated cotton swab in transit from the tray to the biopsy site.
The patient was urgently directed to the ophthalmology clinic and evaluated by an ophthalmologist within 1 to 2 hours of chemical exposure. Visual acuity of the affected left eye was noted to be 20/30 -2 with correctional glasses, and slit lamp examination revealed moderate injection of the conjunctiva and sclera, and at least 3 punctate epithelial erosions and punctate staining of the inferior aspects of the cornea, consistent with a chemical injury. The remaining ocular examination was normal for both eyes. She was diagnosed with keratitis of the left eye from chemical exposure to aluminum chloride and was prescribed loteprednol etabonate ophthalmic suspension 0.5% and tobramycin ophthalmic solution 0.3% to be applied to the left eye 4 times daily, with follow-up 4 days later.
At follow-up, the patient denied any pain, though she was not using the prescribed eye drops consistently. On examination, the patient showed improvement in visual acuity to 20/20 -2 and complete resolution of the keratitis, with slit lamp examination showing clear conjunctiva, sclera, and cornea. Given complete resolution, the eye drops were discontinued.
Comment
Factors Contributing to Ocular Chemical Injuries
Chemical burns to the eyes during cosmetic or surgical procedures are one of the few acute ocular emergencies dermatologists may encounter in practice. If not managed properly, the eye may be permanently damaged. Therefore, dermatologists must be confident in the initial management of ocular chemical burns (Table 1; Figure).

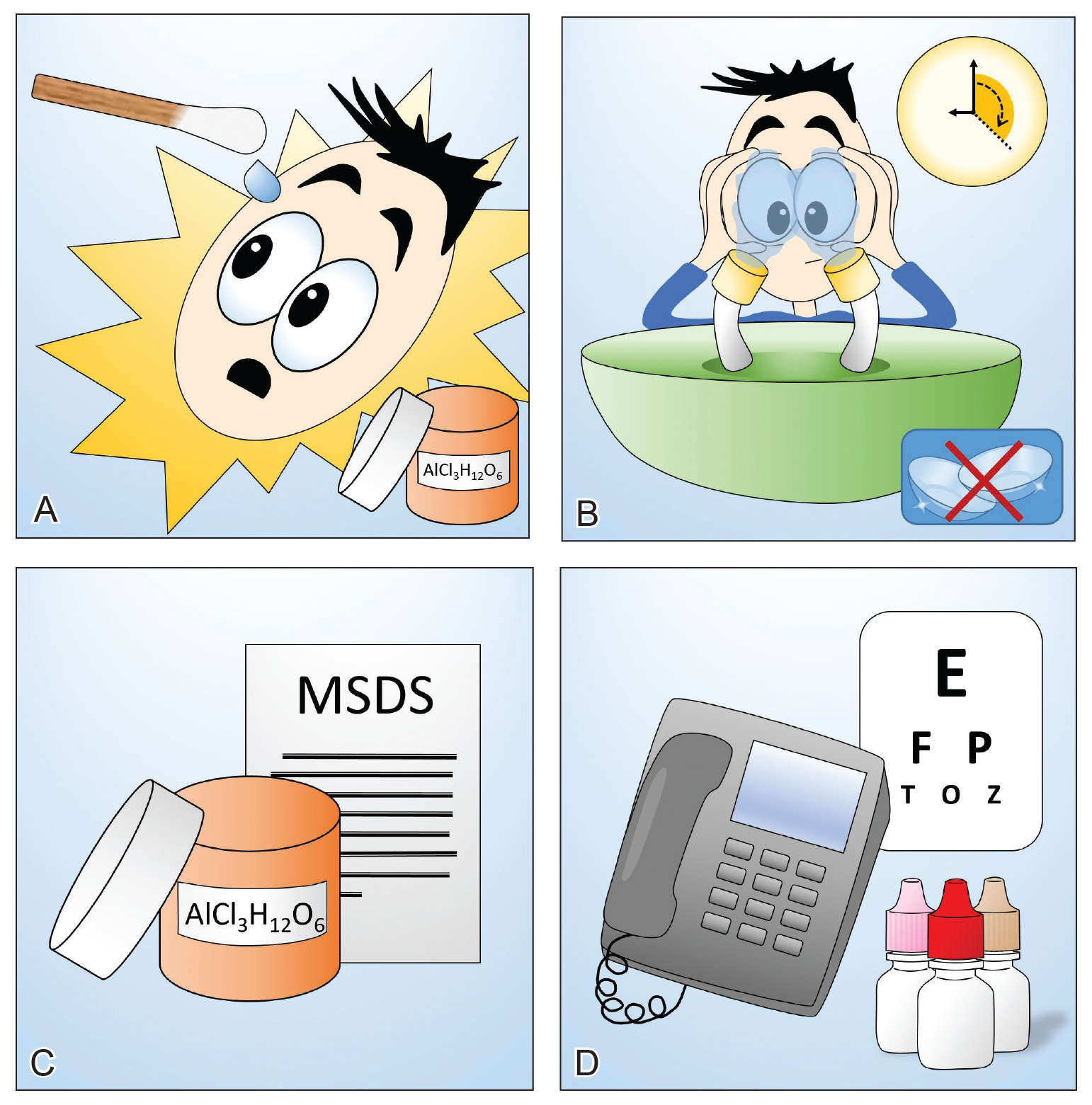
obtain the material safety data sheet. D, Refer the patient urgently to ophthalmology for a visual acuity test and treatment. Images courtesy of Deborah J. Moon, MD (Los Angeles, California).
Mechanism of Ocular Chemical Burns
The extent of injury is predominantly determined by 2 factors: (1) the chemical properties of the substance, and (2) the length of exposure.5,9,10 Potential chemical exposures and their reported ocular effects are listed in Table 2.11-21 Alkaline chemical burns often have the gravest outcome, as they can rapidly penetrate into the internal ocular structures, potentially leading to cataracts and glaucoma.9 Hydroxyl ions, often found in alkaline chemicals, are capable of rapidly denaturing the corneal matrix and triggering release of proteolytic enzymes through a series of inflammatory responses. Conversely, ocular damage from most acidic chemicals often is limited to the more superficial structures, such as the cornea and conjunctiva, given that acids may cause corneal proteins to coagulate, thus forming a barrier that slows further penetration into deeper structures.9 Nonetheless, corneal damage can still have a devastating impact on visual acuity, as the cornea provides 65% to 75% of the eye’s total focusing power.22 For both alkaline and acidic chemicals, immediate profuse irrigation is most critical in determining the clinical course.23-26 To provide perspective, potent alkaline chemicals may penetrate into the anterior chamber of the eye within 15 seconds,9 and delayed initiation of irrigation by even 5 to 15 minutes may lead to irreversible intraocular damage.27
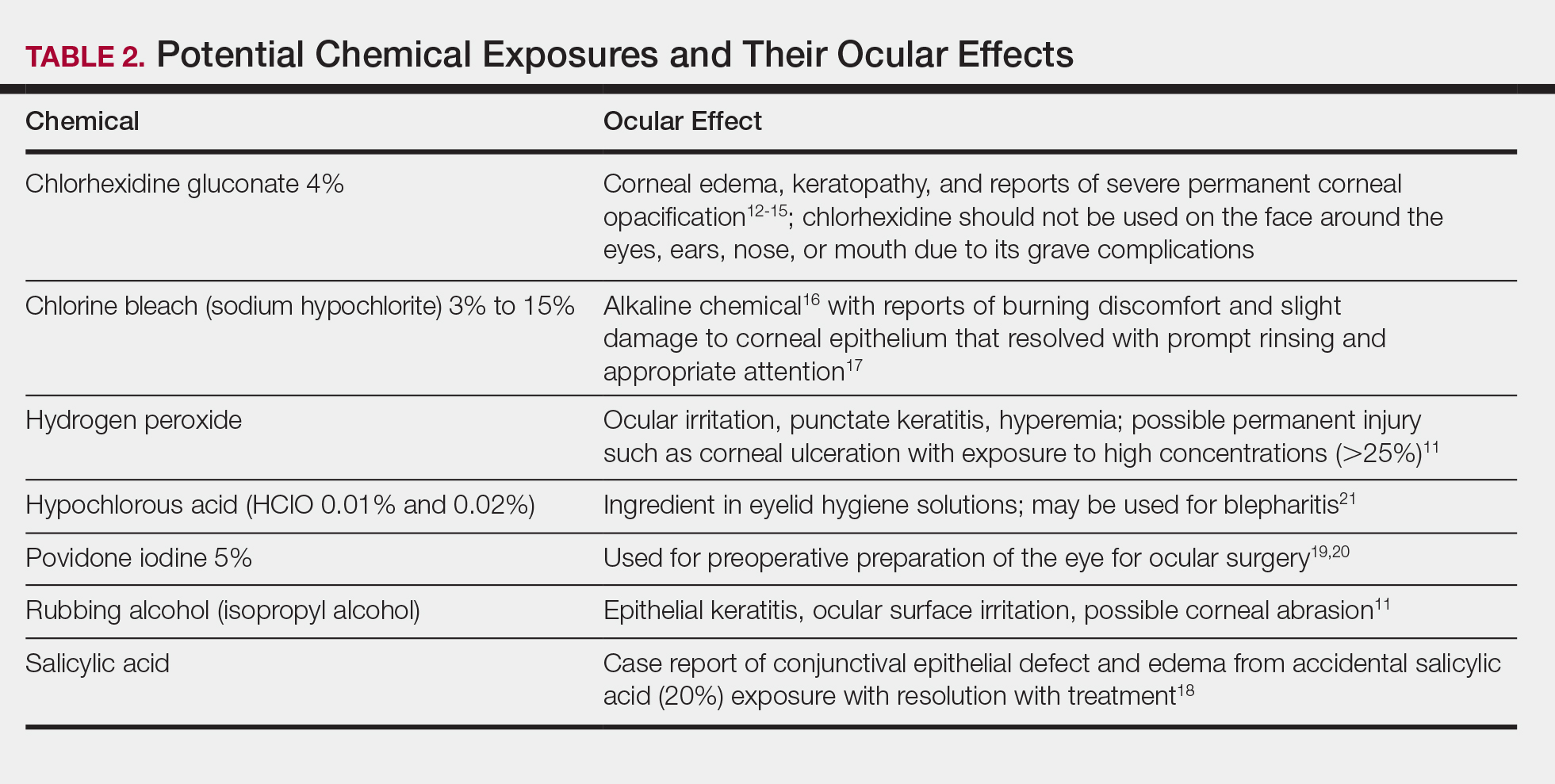
Symptoms of Ocular Chemical Exposure
Signs and symptoms associated with ocular chemical exposures include erythema, pain, tearing, photosensitivity, eyelid swelling, foreign body sensation, changes in vision, and corneal clouding.3,5,9,28 Specifically, aluminum chloride hexahydrate, a hemostatic agent commonly used by dermatologists, has potentially caused eye irritation and conjunctivitis, according to its material safety data sheet,29 as well as blepharospasms, transient disturbances in corneal epithelium, and a persistent faint nebula in the corneal stroma.30 Similar antiperspirants also showed damaging effects to bovine lenses, ocular irritation, and subjective reports of burning and watery eyes.31-33
Immediate Management
If potential chemical exposure to the eye is suspected either by the health care provider or patient, immediately irrigate the affected eye(s) for at least 15 to 30 minutes (longer for alkaline burns) with at least 1 to 2 L of irrigation fluid until the pH is between 7 and 7.2.3-5,9,27,34,35 Irrigation fluids reported to be used include normal saline, Ringer lactate solution, normal saline with sodium bicarbonate, and balanced salt solution.5 If no solutions are readily available, immediate irrigation with tap water is sufficient for diluting and washing away the chemical and has been reported to have better clinical outcomes than delaying irrigation.5,24-26 Studies have shown that prolonged irrigation corresponded with reduced severity, shortened healing time, shorter in-hospital treatment duration, and quicker return to work.5,26
If an eye wash station is not available, the patient can gently flush the eye under a sink faucet set to a gentle stream of lukewarm water.6,7 The health care provider also may manually irrigate the eye. Necessary equipment includes a large syringe or clean eyecup, irrigating fluid, local anesthetic drops for comfort, a towel to soak up excessive fluid, and a bowl or kidney dish to collect the irrigated fluid.34 Providers should first wash their hands. If necessary, anesthetic eye drops may be added for comfort. Lay a towel over the patient’s neck and shoulders and position the patient at a comfortable angle. Place a bowl adjacent to the patient’s cheek to collect the irrigating fluid and have the patient tilt his/her head such that the irrigated fluid would flow into the bowl. Pour a steady stream of the irrigating fluid over the eye from a height of no more than 5 cm.6,7,34
During irrigation, ensure that the patient’s eye(s) is wide open and that all ocular surfaces, including the area underneath the eyelids, are thoroughly washed; everting the eyelids may be beneficial. Ask the patient to move his/her eye(s) in all directions while irrigating. If available, place a litmus strip in the conjunctival fornix to ensure that the goal pH of 7 to 7.2 is reached.9 The pH should be rechecked every 15 to 30 minutes to ensure there has been no change, as hidden crystalized chemical particles may continue to elute chemicals, causing further injury.3 Contact lenses, if present, should be removed as soon as practical, as lenses can trap chemicals; however, immediate initiation of irrigation should not be delayed8 (Table 1).
Identify and verify the chemical suspected to have been exposed to the patient’s eye. The material safety data sheet, which may often be found online if a hard copy is not available, may provide valuable information for the ophthalmologist.36 After thorough irrigation, refer the patient urgently to ophthalmology or the emergency department for prompt evaluation. The emergency department is frequently equipped with polymethylmethacrylate scleral lenses, also called Morgan Lens, which consist of a plastic lens connected via tubing to a bag of irrigation fluid (eg, Ringer lactate solution), allowing for prolonged continuous irrigation of the conjunctiva and cornea. The ophthalmologist will conduct a visual acuity test and complete a thorough eye examination to assess the extent of ischemic injury to the conjunctiva or sclera and damage to the corneal epithelium and internal ocular structures.9
Generally, topical antibiotics, artificial tears, and topical steroids may be provided to patients with mild injury with close follow-up.9,37 For higher-grade injuries, broad-spectrum topical antibiotics, oral antibiotics, topical corticosteroids, vitamin C, and surgical treatments may be additionally recommended (Table 3). Long-term follow-up may be recommended by the ophthalmologist to monitor for potential late complications, such as glaucoma from damage to the trabecular meshwork, corneal abnormalities and limbal stem cell deficiency, symblepharon formation, or eyelid abnormalities.9
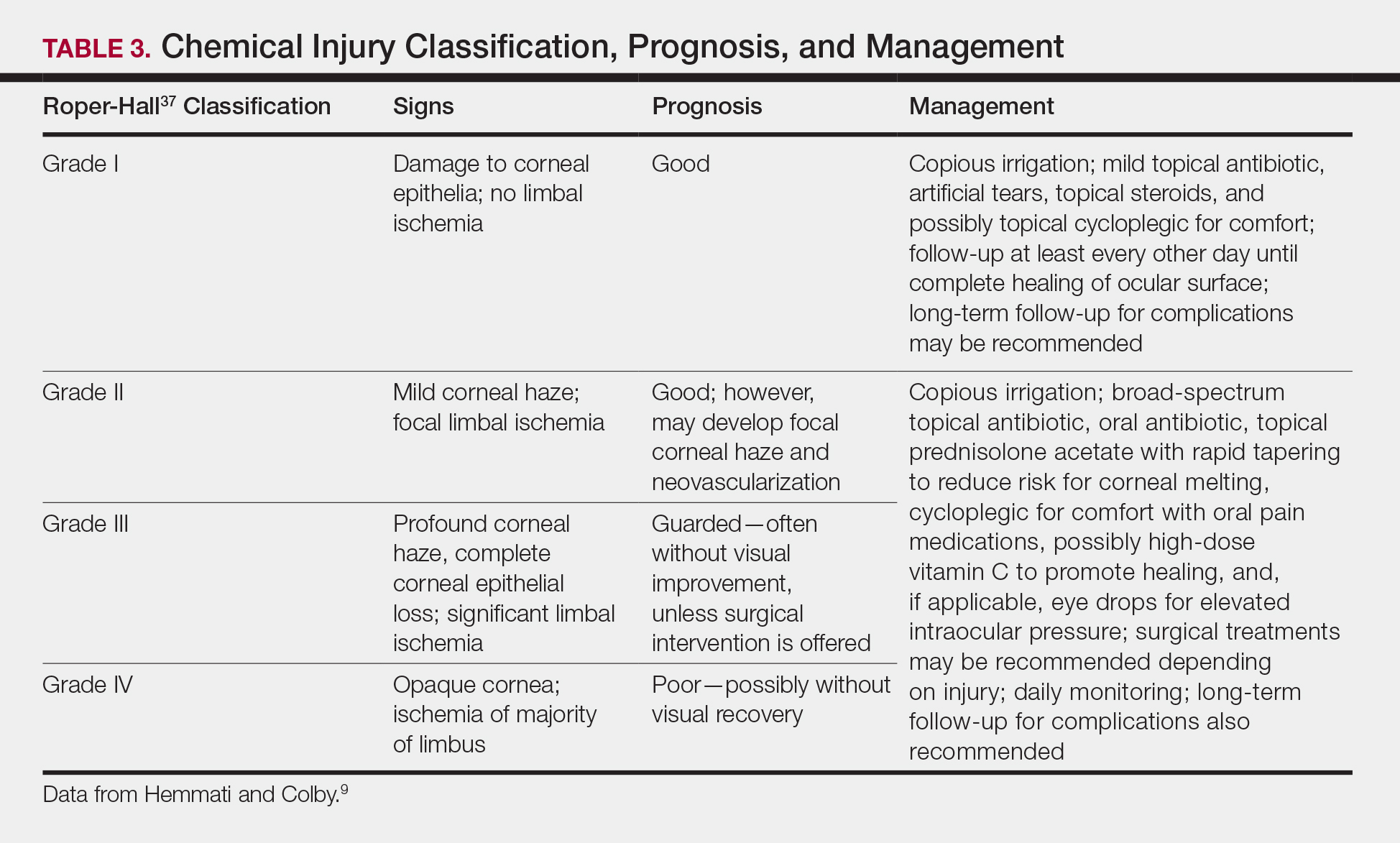
Conclusion
We report a case of a transient chemical burn to the eye secondary to exposure to aluminum chloride hexahydrate. Complete resolution of the injury was achieved with prompt irrigation and urgent medical management by ophthalmology. This case emphasizes the potential for ocular emergencies in the dermatology setting and highlights the steps for appropriate management should a chemical burn to the eye occur. We emphasize the importance of immediate profuse irrigation for 15 to 30 minutes and urgent evaluation by an ophthalmologist. Dermatologists should be cognizant of potential hazards to the eye during facial procedures and always take proper precautions to decrease the risk for ocular injuries.
- Ricci LH, Navajas SV, Carneiro PR, et al. Ocular adverse effects after facial cosmetic procedures: a review of case reports. J Cosmet Dermatol. 2015;14:145-151.
- Boonsiri M, Marks KC, Ditre CM. Benzocaine/lidocaine/tetracainecream: report of corneal damage and review. J Clin Aesthet Dermatol. 2016;9:48-50.
- Gelston CD. Common eye emergencies. Am Fam Physician. 2013;88:515-519.
- Sharma N, Kaur M, Agarwal T, et al. Treatment of acute ocular chemical burns. Surv Ophthalmol. 2018;63:214-235.
- Chau JP, Lee DT, Lo SH. A systematic review of methods of eye irrigation for adults and children with ocular chemical burns. Worldviews Evid Based Nurs. 2012;9:129-138.
- Sears W, Sears M, Sears R, et al. The Portable Pediatrician: Everything You Need to Know About Your Child’s Health. New York, NY: Little, Brown and Company; 2011.
- Kuckelkorn R, Schrage N, Keller G, et al. Emergency treatment of chemical and thermal eye burns. Acta Ophthalmol Scand. 2002;80:4-10.
- Schulte PA, Ahlers HW, Jackson LL, et al. Contact Lens Use in a Chemical Environment. Cincinnati, OH: National Institute for Occupational Safety and Health, US Department of Health and Human Services; 2005. NIOSH publication 2005-139.
- Hemmati HD, Colby KA. Treating acute chemical injuries of the cornea. Eyenet. October 2012. https://www.aao.org/eyenet/article/treating-acute-chemical-injuries-of-cornea. Accessed May 28, 2019.
- Schrage NF, Langefeld S, Zschocke J, et al. Eye burns: an emergency and continuing problem. Burns. 2000;26:689-699.
- Gattey D. Chemical-induced ocular side effects. In: Fraunfelder FT, Fraunfelder FW, Chambers WA, eds. Clinical Ocular Toxicology. Edinburgh, Scotland: W.B. Saunders; 2008:289-306.
- Apt L, Isenberg SJ. Hibiclens keratitis. Am J Ophthalmol. 1987;104:670-671.
- Tabor E, Bostwick DC, Evans C. Corneal damage due to eye contact with chlorhexidine gluconate. JAMA. 1989;261:557-558.
- Galor A, Jeng BH, Lowder CY. A curious case of corneal edema. Eyenet. January 2007. https://www.aao.org/eyenet/article/curious-case-of-corneal-edema. Accessed May 28, 2019.
- Hamed LM, Ellis FD, Boudreault G, et al. Hibiclens keratitis. Am J Ophthalmol. 1987;104:50-56.
- Haring R, Sheffield ID, Channa R, et al. Epidemiologic trends of chemical ocular burns in the United States. JAMA Ophthalmol. 2016;134:1119-1124.
- Racioppi F, Daskaleros PA, Besbelli N, et al. Household bleaches based on sodium hypochlorite: review of acute toxicology and poison control center experience. Food Chem Toxicol. 1994;32:845-861.
- Shazly TA. Ocular acid burn due to 20% concentrated salicylic acid. Cutan Ocul Toxicol. 2011;30:84-86.
- Speaker MG, Menikoff JA. Prophylaxis of endophthalmitis with topical povidone-iodine. Ophthalmology. 1991;98:1769-1775.
- Apt L, Isenberg S, Yoshimori R, et al. Chemical preparation of the eye in ophthalmic surgery: III. effect of povidone-iodine on the conjunctiva. Arch Ophthalmol. 1984;102:728-729.
- Stroman DW, Mintun K, Epstein AB, et al. Reduction in bacterial load using hypochlorous acid hygiene solution on ocular skin. Clin Ophthalmol. 2017;11:707-714.
- Paul M, Sieving A. Facts about the cornea and corneal disease. National Eye Institute, National Institutes of Health website. https://nei.nih.gov/health/cornealdisease. Accessed May 20, 2019.
- Khaw P, Shah P, Elkington A. Injury to the eye. BMJ. 2004;328:36-38.
- Duffy B. Managing chemical eye injuries: Bernice Duffy says initial management of potentially devastating chemical eye injuries by emergency nurses can affect patients’ future prognosis as much as subsequent ophthalmic treatment. Emerg Nurse. 2008;16:25-30.
- Burns F, Paterson C. Prompt irrigation of chemical eye injuries may avert severe damage. Occup Health Saf. 1989;58:33-36.
- Ikeda N, Hayasaka S, Hayasaka Y, et al. Alkali burns of the eye: effect of immediate copious irrigation with tap water on their severity. Ophthalmologica. 2006;220:225-228.
- Eslani M, Baradaran-Rafii A, Movahedan A, et al. The ocular surface chemical burns. J Ophthalmol. 2014;2014:196827.
- Pokhrel PK, Loftus SA. Ocular emergencies. Am Fam Physician. 2007;76:829-836.
- Drysol. MSDS No. BLVCL; Glendale, CA: Person & Covey Inc; March 9, 1991. http://msdsreport.com/msds/blvcl. Accessed May 20, 2019.
- Grant WM, Schuman JS. Toxicology of the Eye: Effects on the Eyes and Visual System From Chemicals, Drugs, Metals and Minerals, Plants, Toxins and Venoms: Also Systemic Side Effects From Eye Medications. Vol 1. Springfield, IL: Charles C. Thomas Publisher; 1993.
- Wong W, Sivak JG, Moran KL. Optical response of the cultured bovine lens; testing opaque or partially transparent semi-solid/solid common consumer hygiene products. Toxicol In Vitro. 2003;17:785-790.
- Donahue DA, Kaufman LE, Avalos J, et al. Survey of ocular irritation predictive capacity using chorioallantoic membrane vascular assay (CAMVA) and bovine corneal opacity and permeability (BCOP) test historical data for 319 personal care products over fourteen years. Toxicol In Vitro. 2011;25:563-572.
- Groot AC, Nater JP, Lender R, et al. Adverse effects of cosmetics and toiletries: a retrospective study in the general population. Int J Cosmet Sci. 1987;9:255-259.
- Stevens S. Ophthalmic practice. Community Eye Health. 2005;18:109-110.
- Hoyt KS, Haley RJ. Innovations in advanced practice: assessment and management of eye emergencies. Adv Emerg Nurs J. 2005;27:101-117.
- LaDou J, Harrison RJ, eds. CURRENT Diagnosis & Treatment: Occupational & Environmental Medicine. 5th ed. New York, NY: McGraw-Hill Education; 2013.
- Roper-Hall M. Thermal and chemical burns. Trans Ophthalmol Soc U K. 1965;85:631-653.
Many dermatologic procedures are performed on the face, such as skin biopsies, surgical excisions, and cosmetic procedures, which can increase the risk for accidental ocular injuries.1,2 Ocular chemical burns have been reported to account for approximately 3% to 20% of ocular injuries3,4 and are one of the few ocular emergencies dermatologists may encounter in practice. Given the potentially severe consequences of permanent vision changes or loss, it is important to take precautionary steps in preventing chemical exposures and know how to appropriately manage ophthalmic emergencies when they occur.1,5-8 In this article, we describe a patient with a transient ocular chemical injury from exposure to aluminum chloride hexahydrate that completely resolved with immediate care. We also offer practical guidance for the general dermatologist in the acute management of acidic chemical burns to the eye, highlighting immediate copious irrigation as the most important step in preventing severe permanent damage. Given that aluminum chloride hexahydrate is an acidic solution, we focus predominantly on the approach to acidic chemical exposures to the eye.
Case Report
A 61-year-old woman was seen in the dermatology outpatient clinic for a shave biopsy on the left cheek followed by aluminum chloride application for hemostasis. Following the biopsy, the patient stated she felt the sensation that something had dripped into the left eye and she felt a burning pain. There was a 30- to 60-second delay in irrigation of the eye, as it was at first unclear what had occurred. The patient reported an increased burning sensation, and at that point she was instructed to begin flushing the eye with tap water from the examination room sink for 15 to 20 minutes; she wanted to stop irrigation after a few minutes, and convincing her to continue thorough irrigation was somewhat challenging. It was determined that aluminum chloride hexahydrate had dripped from an oversaturated cotton swab in transit from the tray to the biopsy site.
The patient was urgently directed to the ophthalmology clinic and evaluated by an ophthalmologist within 1 to 2 hours of chemical exposure. Visual acuity of the affected left eye was noted to be 20/30 -2 with correctional glasses, and slit lamp examination revealed moderate injection of the conjunctiva and sclera, and at least 3 punctate epithelial erosions and punctate staining of the inferior aspects of the cornea, consistent with a chemical injury. The remaining ocular examination was normal for both eyes. She was diagnosed with keratitis of the left eye from chemical exposure to aluminum chloride and was prescribed loteprednol etabonate ophthalmic suspension 0.5% and tobramycin ophthalmic solution 0.3% to be applied to the left eye 4 times daily, with follow-up 4 days later.
At follow-up, the patient denied any pain, though she was not using the prescribed eye drops consistently. On examination, the patient showed improvement in visual acuity to 20/20 -2 and complete resolution of the keratitis, with slit lamp examination showing clear conjunctiva, sclera, and cornea. Given complete resolution, the eye drops were discontinued.
Comment
Factors Contributing to Ocular Chemical Injuries
Chemical burns to the eyes during cosmetic or surgical procedures are one of the few acute ocular emergencies dermatologists may encounter in practice. If not managed properly, the eye may be permanently damaged. Therefore, dermatologists must be confident in the initial management of ocular chemical burns (Table 1; Figure).
obtain the material safety data sheet. D, Refer the patient urgently to ophthalmology for a visual acuity test and treatment. Images courtesy of Deborah J. Moon, MD (Los Angeles, California).
Mechanism of Ocular Chemical Burns
The extent of injury is predominantly determined by 2 factors: (1) the chemical properties of the substance, and (2) the length of exposure.5,9,10 Potential chemical exposures and their reported ocular effects are listed in Table 2.11-21 Alkaline chemical burns often have the gravest outcome, as they can rapidly penetrate into the internal ocular structures, potentially leading to cataracts and glaucoma.9 Hydroxyl ions, often found in alkaline chemicals, are capable of rapidly denaturing the corneal matrix and triggering release of proteolytic enzymes through a series of inflammatory responses. Conversely, ocular damage from most acidic chemicals often is limited to the more superficial structures, such as the cornea and conjunctiva, given that acids may cause corneal proteins to coagulate, thus forming a barrier that slows further penetration into deeper structures.9 Nonetheless, corneal damage can still have a devastating impact on visual acuity, as the cornea provides 65% to 75% of the eye’s total focusing power.22 For both alkaline and acidic chemicals, immediate profuse irrigation is most critical in determining the clinical course.23-26 To provide perspective, potent alkaline chemicals may penetrate into the anterior chamber of the eye within 15 seconds,9 and delayed initiation of irrigation by even 5 to 15 minutes may lead to irreversible intraocular damage.27
Symptoms of Ocular Chemical Exposure
Signs and symptoms associated with ocular chemical exposures include erythema, pain, tearing, photosensitivity, eyelid swelling, foreign body sensation, changes in vision, and corneal clouding.3,5,9,28 Specifically, aluminum chloride hexahydrate, a hemostatic agent commonly used by dermatologists, has potentially caused eye irritation and conjunctivitis, according to its material safety data sheet,29 as well as blepharospasms, transient disturbances in corneal epithelium, and a persistent faint nebula in the corneal stroma.30 Similar antiperspirants also showed damaging effects to bovine lenses, ocular irritation, and subjective reports of burning and watery eyes.31-33
Immediate Management
If potential chemical exposure to the eye is suspected either by the health care provider or patient, immediately irrigate the affected eye(s) for at least 15 to 30 minutes (longer for alkaline burns) with at least 1 to 2 L of irrigation fluid until the pH is between 7 and 7.2.3-5,9,27,34,35 Irrigation fluids reported to be used include normal saline, Ringer lactate solution, normal saline with sodium bicarbonate, and balanced salt solution.5 If no solutions are readily available, immediate irrigation with tap water is sufficient for diluting and washing away the chemical and has been reported to have better clinical outcomes than delaying irrigation.5,24-26 Studies have shown that prolonged irrigation corresponded with reduced severity, shortened healing time, shorter in-hospital treatment duration, and quicker return to work.5,26
If an eye wash station is not available, the patient can gently flush the eye under a sink faucet set to a gentle stream of lukewarm water.6,7 The health care provider also may manually irrigate the eye. Necessary equipment includes a large syringe or clean eyecup, irrigating fluid, local anesthetic drops for comfort, a towel to soak up excessive fluid, and a bowl or kidney dish to collect the irrigated fluid.34 Providers should first wash their hands. If necessary, anesthetic eye drops may be added for comfort. Lay a towel over the patient’s neck and shoulders and position the patient at a comfortable angle. Place a bowl adjacent to the patient’s cheek to collect the irrigating fluid and have the patient tilt his/her head such that the irrigated fluid would flow into the bowl. Pour a steady stream of the irrigating fluid over the eye from a height of no more than 5 cm.6,7,34
During irrigation, ensure that the patient’s eye(s) is wide open and that all ocular surfaces, including the area underneath the eyelids, are thoroughly washed; everting the eyelids may be beneficial. Ask the patient to move his/her eye(s) in all directions while irrigating. If available, place a litmus strip in the conjunctival fornix to ensure that the goal pH of 7 to 7.2 is reached.9 The pH should be rechecked every 15 to 30 minutes to ensure there has been no change, as hidden crystalized chemical particles may continue to elute chemicals, causing further injury.3 Contact lenses, if present, should be removed as soon as practical, as lenses can trap chemicals; however, immediate initiation of irrigation should not be delayed8 (Table 1).
Identify and verify the chemical suspected to have been exposed to the patient’s eye. The material safety data sheet, which may often be found online if a hard copy is not available, may provide valuable information for the ophthalmologist.36 After thorough irrigation, refer the patient urgently to ophthalmology or the emergency department for prompt evaluation. The emergency department is frequently equipped with polymethylmethacrylate scleral lenses, also called Morgan Lens, which consist of a plastic lens connected via tubing to a bag of irrigation fluid (eg, Ringer lactate solution), allowing for prolonged continuous irrigation of the conjunctiva and cornea. The ophthalmologist will conduct a visual acuity test and complete a thorough eye examination to assess the extent of ischemic injury to the conjunctiva or sclera and damage to the corneal epithelium and internal ocular structures.9
Generally, topical antibiotics, artificial tears, and topical steroids may be provided to patients with mild injury with close follow-up.9,37 For higher-grade injuries, broad-spectrum topical antibiotics, oral antibiotics, topical corticosteroids, vitamin C, and surgical treatments may be additionally recommended (Table 3). Long-term follow-up may be recommended by the ophthalmologist to monitor for potential late complications, such as glaucoma from damage to the trabecular meshwork, corneal abnormalities and limbal stem cell deficiency, symblepharon formation, or eyelid abnormalities.9
Conclusion
We report a case of a transient chemical burn to the eye secondary to exposure to aluminum chloride hexahydrate. Complete resolution of the injury was achieved with prompt irrigation and urgent medical management by ophthalmology. This case emphasizes the potential for ocular emergencies in the dermatology setting and highlights the steps for appropriate management should a chemical burn to the eye occur. We emphasize the importance of immediate profuse irrigation for 15 to 30 minutes and urgent evaluation by an ophthalmologist. Dermatologists should be cognizant of potential hazards to the eye during facial procedures and always take proper precautions to decrease the risk for ocular injuries.
Many dermatologic procedures are performed on the face, such as skin biopsies, surgical excisions, and cosmetic procedures, which can increase the risk for accidental ocular injuries.1,2 Ocular chemical burns have been reported to account for approximately 3% to 20% of ocular injuries3,4 and are one of the few ocular emergencies dermatologists may encounter in practice. Given the potentially severe consequences of permanent vision changes or loss, it is important to take precautionary steps in preventing chemical exposures and know how to appropriately manage ophthalmic emergencies when they occur.1,5-8 In this article, we describe a patient with a transient ocular chemical injury from exposure to aluminum chloride hexahydrate that completely resolved with immediate care. We also offer practical guidance for the general dermatologist in the acute management of acidic chemical burns to the eye, highlighting immediate copious irrigation as the most important step in preventing severe permanent damage. Given that aluminum chloride hexahydrate is an acidic solution, we focus predominantly on the approach to acidic chemical exposures to the eye.
Case Report
A 61-year-old woman was seen in the dermatology outpatient clinic for a shave biopsy on the left cheek followed by aluminum chloride application for hemostasis. Following the biopsy, the patient stated she felt the sensation that something had dripped into the left eye and she felt a burning pain. There was a 30- to 60-second delay in irrigation of the eye, as it was at first unclear what had occurred. The patient reported an increased burning sensation, and at that point she was instructed to begin flushing the eye with tap water from the examination room sink for 15 to 20 minutes; she wanted to stop irrigation after a few minutes, and convincing her to continue thorough irrigation was somewhat challenging. It was determined that aluminum chloride hexahydrate had dripped from an oversaturated cotton swab in transit from the tray to the biopsy site.
The patient was urgently directed to the ophthalmology clinic and evaluated by an ophthalmologist within 1 to 2 hours of chemical exposure. Visual acuity of the affected left eye was noted to be 20/30 -2 with correctional glasses, and slit lamp examination revealed moderate injection of the conjunctiva and sclera, and at least 3 punctate epithelial erosions and punctate staining of the inferior aspects of the cornea, consistent with a chemical injury. The remaining ocular examination was normal for both eyes. She was diagnosed with keratitis of the left eye from chemical exposure to aluminum chloride and was prescribed loteprednol etabonate ophthalmic suspension 0.5% and tobramycin ophthalmic solution 0.3% to be applied to the left eye 4 times daily, with follow-up 4 days later.
At follow-up, the patient denied any pain, though she was not using the prescribed eye drops consistently. On examination, the patient showed improvement in visual acuity to 20/20 -2 and complete resolution of the keratitis, with slit lamp examination showing clear conjunctiva, sclera, and cornea. Given complete resolution, the eye drops were discontinued.
Comment
Factors Contributing to Ocular Chemical Injuries
Chemical burns to the eyes during cosmetic or surgical procedures are one of the few acute ocular emergencies dermatologists may encounter in practice. If not managed properly, the eye may be permanently damaged. Therefore, dermatologists must be confident in the initial management of ocular chemical burns (Table 1; Figure).
obtain the material safety data sheet. D, Refer the patient urgently to ophthalmology for a visual acuity test and treatment. Images courtesy of Deborah J. Moon, MD (Los Angeles, California).
Mechanism of Ocular Chemical Burns
The extent of injury is predominantly determined by 2 factors: (1) the chemical properties of the substance, and (2) the length of exposure.5,9,10 Potential chemical exposures and their reported ocular effects are listed in Table 2.11-21 Alkaline chemical burns often have the gravest outcome, as they can rapidly penetrate into the internal ocular structures, potentially leading to cataracts and glaucoma.9 Hydroxyl ions, often found in alkaline chemicals, are capable of rapidly denaturing the corneal matrix and triggering release of proteolytic enzymes through a series of inflammatory responses. Conversely, ocular damage from most acidic chemicals often is limited to the more superficial structures, such as the cornea and conjunctiva, given that acids may cause corneal proteins to coagulate, thus forming a barrier that slows further penetration into deeper structures.9 Nonetheless, corneal damage can still have a devastating impact on visual acuity, as the cornea provides 65% to 75% of the eye’s total focusing power.22 For both alkaline and acidic chemicals, immediate profuse irrigation is most critical in determining the clinical course.23-26 To provide perspective, potent alkaline chemicals may penetrate into the anterior chamber of the eye within 15 seconds,9 and delayed initiation of irrigation by even 5 to 15 minutes may lead to irreversible intraocular damage.27
Symptoms of Ocular Chemical Exposure
Signs and symptoms associated with ocular chemical exposures include erythema, pain, tearing, photosensitivity, eyelid swelling, foreign body sensation, changes in vision, and corneal clouding.3,5,9,28 Specifically, aluminum chloride hexahydrate, a hemostatic agent commonly used by dermatologists, has potentially caused eye irritation and conjunctivitis, according to its material safety data sheet,29 as well as blepharospasms, transient disturbances in corneal epithelium, and a persistent faint nebula in the corneal stroma.30 Similar antiperspirants also showed damaging effects to bovine lenses, ocular irritation, and subjective reports of burning and watery eyes.31-33
Immediate Management
If potential chemical exposure to the eye is suspected either by the health care provider or patient, immediately irrigate the affected eye(s) for at least 15 to 30 minutes (longer for alkaline burns) with at least 1 to 2 L of irrigation fluid until the pH is between 7 and 7.2.3-5,9,27,34,35 Irrigation fluids reported to be used include normal saline, Ringer lactate solution, normal saline with sodium bicarbonate, and balanced salt solution.5 If no solutions are readily available, immediate irrigation with tap water is sufficient for diluting and washing away the chemical and has been reported to have better clinical outcomes than delaying irrigation.5,24-26 Studies have shown that prolonged irrigation corresponded with reduced severity, shortened healing time, shorter in-hospital treatment duration, and quicker return to work.5,26
If an eye wash station is not available, the patient can gently flush the eye under a sink faucet set to a gentle stream of lukewarm water.6,7 The health care provider also may manually irrigate the eye. Necessary equipment includes a large syringe or clean eyecup, irrigating fluid, local anesthetic drops for comfort, a towel to soak up excessive fluid, and a bowl or kidney dish to collect the irrigated fluid.34 Providers should first wash their hands. If necessary, anesthetic eye drops may be added for comfort. Lay a towel over the patient’s neck and shoulders and position the patient at a comfortable angle. Place a bowl adjacent to the patient’s cheek to collect the irrigating fluid and have the patient tilt his/her head such that the irrigated fluid would flow into the bowl. Pour a steady stream of the irrigating fluid over the eye from a height of no more than 5 cm.6,7,34
During irrigation, ensure that the patient’s eye(s) is wide open and that all ocular surfaces, including the area underneath the eyelids, are thoroughly washed; everting the eyelids may be beneficial. Ask the patient to move his/her eye(s) in all directions while irrigating. If available, place a litmus strip in the conjunctival fornix to ensure that the goal pH of 7 to 7.2 is reached.9 The pH should be rechecked every 15 to 30 minutes to ensure there has been no change, as hidden crystalized chemical particles may continue to elute chemicals, causing further injury.3 Contact lenses, if present, should be removed as soon as practical, as lenses can trap chemicals; however, immediate initiation of irrigation should not be delayed8 (Table 1).
Identify and verify the chemical suspected to have been exposed to the patient’s eye. The material safety data sheet, which may often be found online if a hard copy is not available, may provide valuable information for the ophthalmologist.36 After thorough irrigation, refer the patient urgently to ophthalmology or the emergency department for prompt evaluation. The emergency department is frequently equipped with polymethylmethacrylate scleral lenses, also called Morgan Lens, which consist of a plastic lens connected via tubing to a bag of irrigation fluid (eg, Ringer lactate solution), allowing for prolonged continuous irrigation of the conjunctiva and cornea. The ophthalmologist will conduct a visual acuity test and complete a thorough eye examination to assess the extent of ischemic injury to the conjunctiva or sclera and damage to the corneal epithelium and internal ocular structures.9
Generally, topical antibiotics, artificial tears, and topical steroids may be provided to patients with mild injury with close follow-up.9,37 For higher-grade injuries, broad-spectrum topical antibiotics, oral antibiotics, topical corticosteroids, vitamin C, and surgical treatments may be additionally recommended (Table 3). Long-term follow-up may be recommended by the ophthalmologist to monitor for potential late complications, such as glaucoma from damage to the trabecular meshwork, corneal abnormalities and limbal stem cell deficiency, symblepharon formation, or eyelid abnormalities.9
Conclusion
We report a case of a transient chemical burn to the eye secondary to exposure to aluminum chloride hexahydrate. Complete resolution of the injury was achieved with prompt irrigation and urgent medical management by ophthalmology. This case emphasizes the potential for ocular emergencies in the dermatology setting and highlights the steps for appropriate management should a chemical burn to the eye occur. We emphasize the importance of immediate profuse irrigation for 15 to 30 minutes and urgent evaluation by an ophthalmologist. Dermatologists should be cognizant of potential hazards to the eye during facial procedures and always take proper precautions to decrease the risk for ocular injuries.
- Ricci LH, Navajas SV, Carneiro PR, et al. Ocular adverse effects after facial cosmetic procedures: a review of case reports. J Cosmet Dermatol. 2015;14:145-151.
- Boonsiri M, Marks KC, Ditre CM. Benzocaine/lidocaine/tetracainecream: report of corneal damage and review. J Clin Aesthet Dermatol. 2016;9:48-50.
- Gelston CD. Common eye emergencies. Am Fam Physician. 2013;88:515-519.
- Sharma N, Kaur M, Agarwal T, et al. Treatment of acute ocular chemical burns. Surv Ophthalmol. 2018;63:214-235.
- Chau JP, Lee DT, Lo SH. A systematic review of methods of eye irrigation for adults and children with ocular chemical burns. Worldviews Evid Based Nurs. 2012;9:129-138.
- Sears W, Sears M, Sears R, et al. The Portable Pediatrician: Everything You Need to Know About Your Child’s Health. New York, NY: Little, Brown and Company; 2011.
- Kuckelkorn R, Schrage N, Keller G, et al. Emergency treatment of chemical and thermal eye burns. Acta Ophthalmol Scand. 2002;80:4-10.
- Schulte PA, Ahlers HW, Jackson LL, et al. Contact Lens Use in a Chemical Environment. Cincinnati, OH: National Institute for Occupational Safety and Health, US Department of Health and Human Services; 2005. NIOSH publication 2005-139.
- Hemmati HD, Colby KA. Treating acute chemical injuries of the cornea. Eyenet. October 2012. https://www.aao.org/eyenet/article/treating-acute-chemical-injuries-of-cornea. Accessed May 28, 2019.
- Schrage NF, Langefeld S, Zschocke J, et al. Eye burns: an emergency and continuing problem. Burns. 2000;26:689-699.
- Gattey D. Chemical-induced ocular side effects. In: Fraunfelder FT, Fraunfelder FW, Chambers WA, eds. Clinical Ocular Toxicology. Edinburgh, Scotland: W.B. Saunders; 2008:289-306.
- Apt L, Isenberg SJ. Hibiclens keratitis. Am J Ophthalmol. 1987;104:670-671.
- Tabor E, Bostwick DC, Evans C. Corneal damage due to eye contact with chlorhexidine gluconate. JAMA. 1989;261:557-558.
- Galor A, Jeng BH, Lowder CY. A curious case of corneal edema. Eyenet. January 2007. https://www.aao.org/eyenet/article/curious-case-of-corneal-edema. Accessed May 28, 2019.
- Hamed LM, Ellis FD, Boudreault G, et al. Hibiclens keratitis. Am J Ophthalmol. 1987;104:50-56.
- Haring R, Sheffield ID, Channa R, et al. Epidemiologic trends of chemical ocular burns in the United States. JAMA Ophthalmol. 2016;134:1119-1124.
- Racioppi F, Daskaleros PA, Besbelli N, et al. Household bleaches based on sodium hypochlorite: review of acute toxicology and poison control center experience. Food Chem Toxicol. 1994;32:845-861.
- Shazly TA. Ocular acid burn due to 20% concentrated salicylic acid. Cutan Ocul Toxicol. 2011;30:84-86.
- Speaker MG, Menikoff JA. Prophylaxis of endophthalmitis with topical povidone-iodine. Ophthalmology. 1991;98:1769-1775.
- Apt L, Isenberg S, Yoshimori R, et al. Chemical preparation of the eye in ophthalmic surgery: III. effect of povidone-iodine on the conjunctiva. Arch Ophthalmol. 1984;102:728-729.
- Stroman DW, Mintun K, Epstein AB, et al. Reduction in bacterial load using hypochlorous acid hygiene solution on ocular skin. Clin Ophthalmol. 2017;11:707-714.
- Paul M, Sieving A. Facts about the cornea and corneal disease. National Eye Institute, National Institutes of Health website. https://nei.nih.gov/health/cornealdisease. Accessed May 20, 2019.
- Khaw P, Shah P, Elkington A. Injury to the eye. BMJ. 2004;328:36-38.
- Duffy B. Managing chemical eye injuries: Bernice Duffy says initial management of potentially devastating chemical eye injuries by emergency nurses can affect patients’ future prognosis as much as subsequent ophthalmic treatment. Emerg Nurse. 2008;16:25-30.
- Burns F, Paterson C. Prompt irrigation of chemical eye injuries may avert severe damage. Occup Health Saf. 1989;58:33-36.
- Ikeda N, Hayasaka S, Hayasaka Y, et al. Alkali burns of the eye: effect of immediate copious irrigation with tap water on their severity. Ophthalmologica. 2006;220:225-228.
- Eslani M, Baradaran-Rafii A, Movahedan A, et al. The ocular surface chemical burns. J Ophthalmol. 2014;2014:196827.
- Pokhrel PK, Loftus SA. Ocular emergencies. Am Fam Physician. 2007;76:829-836.
- Drysol. MSDS No. BLVCL; Glendale, CA: Person & Covey Inc; March 9, 1991. http://msdsreport.com/msds/blvcl. Accessed May 20, 2019.
- Grant WM, Schuman JS. Toxicology of the Eye: Effects on the Eyes and Visual System From Chemicals, Drugs, Metals and Minerals, Plants, Toxins and Venoms: Also Systemic Side Effects From Eye Medications. Vol 1. Springfield, IL: Charles C. Thomas Publisher; 1993.
- Wong W, Sivak JG, Moran KL. Optical response of the cultured bovine lens; testing opaque or partially transparent semi-solid/solid common consumer hygiene products. Toxicol In Vitro. 2003;17:785-790.
- Donahue DA, Kaufman LE, Avalos J, et al. Survey of ocular irritation predictive capacity using chorioallantoic membrane vascular assay (CAMVA) and bovine corneal opacity and permeability (BCOP) test historical data for 319 personal care products over fourteen years. Toxicol In Vitro. 2011;25:563-572.
- Groot AC, Nater JP, Lender R, et al. Adverse effects of cosmetics and toiletries: a retrospective study in the general population. Int J Cosmet Sci. 1987;9:255-259.
- Stevens S. Ophthalmic practice. Community Eye Health. 2005;18:109-110.
- Hoyt KS, Haley RJ. Innovations in advanced practice: assessment and management of eye emergencies. Adv Emerg Nurs J. 2005;27:101-117.
- LaDou J, Harrison RJ, eds. CURRENT Diagnosis & Treatment: Occupational & Environmental Medicine. 5th ed. New York, NY: McGraw-Hill Education; 2013.
- Roper-Hall M. Thermal and chemical burns. Trans Ophthalmol Soc U K. 1965;85:631-653.
- Ricci LH, Navajas SV, Carneiro PR, et al. Ocular adverse effects after facial cosmetic procedures: a review of case reports. J Cosmet Dermatol. 2015;14:145-151.
- Boonsiri M, Marks KC, Ditre CM. Benzocaine/lidocaine/tetracainecream: report of corneal damage and review. J Clin Aesthet Dermatol. 2016;9:48-50.
- Gelston CD. Common eye emergencies. Am Fam Physician. 2013;88:515-519.
- Sharma N, Kaur M, Agarwal T, et al. Treatment of acute ocular chemical burns. Surv Ophthalmol. 2018;63:214-235.
- Chau JP, Lee DT, Lo SH. A systematic review of methods of eye irrigation for adults and children with ocular chemical burns. Worldviews Evid Based Nurs. 2012;9:129-138.
- Sears W, Sears M, Sears R, et al. The Portable Pediatrician: Everything You Need to Know About Your Child’s Health. New York, NY: Little, Brown and Company; 2011.
- Kuckelkorn R, Schrage N, Keller G, et al. Emergency treatment of chemical and thermal eye burns. Acta Ophthalmol Scand. 2002;80:4-10.
- Schulte PA, Ahlers HW, Jackson LL, et al. Contact Lens Use in a Chemical Environment. Cincinnati, OH: National Institute for Occupational Safety and Health, US Department of Health and Human Services; 2005. NIOSH publication 2005-139.
- Hemmati HD, Colby KA. Treating acute chemical injuries of the cornea. Eyenet. October 2012. https://www.aao.org/eyenet/article/treating-acute-chemical-injuries-of-cornea. Accessed May 28, 2019.
- Schrage NF, Langefeld S, Zschocke J, et al. Eye burns: an emergency and continuing problem. Burns. 2000;26:689-699.
- Gattey D. Chemical-induced ocular side effects. In: Fraunfelder FT, Fraunfelder FW, Chambers WA, eds. Clinical Ocular Toxicology. Edinburgh, Scotland: W.B. Saunders; 2008:289-306.
- Apt L, Isenberg SJ. Hibiclens keratitis. Am J Ophthalmol. 1987;104:670-671.
- Tabor E, Bostwick DC, Evans C. Corneal damage due to eye contact with chlorhexidine gluconate. JAMA. 1989;261:557-558.
- Galor A, Jeng BH, Lowder CY. A curious case of corneal edema. Eyenet. January 2007. https://www.aao.org/eyenet/article/curious-case-of-corneal-edema. Accessed May 28, 2019.
- Hamed LM, Ellis FD, Boudreault G, et al. Hibiclens keratitis. Am J Ophthalmol. 1987;104:50-56.
- Haring R, Sheffield ID, Channa R, et al. Epidemiologic trends of chemical ocular burns in the United States. JAMA Ophthalmol. 2016;134:1119-1124.
- Racioppi F, Daskaleros PA, Besbelli N, et al. Household bleaches based on sodium hypochlorite: review of acute toxicology and poison control center experience. Food Chem Toxicol. 1994;32:845-861.
- Shazly TA. Ocular acid burn due to 20% concentrated salicylic acid. Cutan Ocul Toxicol. 2011;30:84-86.
- Speaker MG, Menikoff JA. Prophylaxis of endophthalmitis with topical povidone-iodine. Ophthalmology. 1991;98:1769-1775.
- Apt L, Isenberg S, Yoshimori R, et al. Chemical preparation of the eye in ophthalmic surgery: III. effect of povidone-iodine on the conjunctiva. Arch Ophthalmol. 1984;102:728-729.
- Stroman DW, Mintun K, Epstein AB, et al. Reduction in bacterial load using hypochlorous acid hygiene solution on ocular skin. Clin Ophthalmol. 2017;11:707-714.
- Paul M, Sieving A. Facts about the cornea and corneal disease. National Eye Institute, National Institutes of Health website. https://nei.nih.gov/health/cornealdisease. Accessed May 20, 2019.
- Khaw P, Shah P, Elkington A. Injury to the eye. BMJ. 2004;328:36-38.
- Duffy B. Managing chemical eye injuries: Bernice Duffy says initial management of potentially devastating chemical eye injuries by emergency nurses can affect patients’ future prognosis as much as subsequent ophthalmic treatment. Emerg Nurse. 2008;16:25-30.
- Burns F, Paterson C. Prompt irrigation of chemical eye injuries may avert severe damage. Occup Health Saf. 1989;58:33-36.
- Ikeda N, Hayasaka S, Hayasaka Y, et al. Alkali burns of the eye: effect of immediate copious irrigation with tap water on their severity. Ophthalmologica. 2006;220:225-228.
- Eslani M, Baradaran-Rafii A, Movahedan A, et al. The ocular surface chemical burns. J Ophthalmol. 2014;2014:196827.
- Pokhrel PK, Loftus SA. Ocular emergencies. Am Fam Physician. 2007;76:829-836.
- Drysol. MSDS No. BLVCL; Glendale, CA: Person & Covey Inc; March 9, 1991. http://msdsreport.com/msds/blvcl. Accessed May 20, 2019.
- Grant WM, Schuman JS. Toxicology of the Eye: Effects on the Eyes and Visual System From Chemicals, Drugs, Metals and Minerals, Plants, Toxins and Venoms: Also Systemic Side Effects From Eye Medications. Vol 1. Springfield, IL: Charles C. Thomas Publisher; 1993.
- Wong W, Sivak JG, Moran KL. Optical response of the cultured bovine lens; testing opaque or partially transparent semi-solid/solid common consumer hygiene products. Toxicol In Vitro. 2003;17:785-790.
- Donahue DA, Kaufman LE, Avalos J, et al. Survey of ocular irritation predictive capacity using chorioallantoic membrane vascular assay (CAMVA) and bovine corneal opacity and permeability (BCOP) test historical data for 319 personal care products over fourteen years. Toxicol In Vitro. 2011;25:563-572.
- Groot AC, Nater JP, Lender R, et al. Adverse effects of cosmetics and toiletries: a retrospective study in the general population. Int J Cosmet Sci. 1987;9:255-259.
- Stevens S. Ophthalmic practice. Community Eye Health. 2005;18:109-110.
- Hoyt KS, Haley RJ. Innovations in advanced practice: assessment and management of eye emergencies. Adv Emerg Nurs J. 2005;27:101-117.
- LaDou J, Harrison RJ, eds. CURRENT Diagnosis & Treatment: Occupational & Environmental Medicine. 5th ed. New York, NY: McGraw-Hill Education; 2013.
- Roper-Hall M. Thermal and chemical burns. Trans Ophthalmol Soc U K. 1965;85:631-653.
Practice Points
- Dermatologists should be cognizant of potential hazards to the eyes during facial procedures and always take proper precautions to decrease the risk for ocular injuries.
- If a patient’s eye(s) becomes exposed to a chemical during a dermatologic procedure, immediate copious irrigation for at least 15 to 30 minutes (longer for alkaline burns) is crucial, followed by prompt evaluation by an ophthalmologist.
- The patient should be instructed to manually hold open the eye and move the eyeball in all directions to achieve the most effective irrigation of the chemical.
- If the patient is wearing contact lenses, they should be removed promptly, but do not delay the irrigation to do so. Lenses should be removed once irrigation is underway.

Acquired Digital Fibrokeratoma Presenting as a Painless Nodule on the Right Fifth Fingernail
Case Report
A 53-year-old woman presented for an initial visit to the dermatology clinic for a growth under the right fifth fingernail of 1 year’s duration. She had no history of trauma to the digit or pain or bleeding. She self-treated with over-the-counter wart remover for several months without improvement. She reported no other skin concerns. She had a medical history of rheumatoid arthritis (RA) and basal cell carcinoma of the nose; she was taking methotrexate and adalimumab for the RA. She had a family history of melanoma in her father.
On physical examination, a firm nontender nodule was noted on the distal nail bed of the right fifth fingernail with onycholysis; the nail plate was otherwise intact (Figure 1). All other nails were normal. A plain radiograph of the involved digit showed no bony abnormality. Excisional biopsy of the nodule was performed and analyzed by histopathology (Figure 2). The biopsy specimen showed a benign epidermis that was acanthotic and surmounted by hyperkeratotic scale. The dermis was fibrotic with collagen bundles assuming a vertical orientation to the long axis of the epidermis, typical of a fibrokeratoma. There were no atypical features in the dermal component or epidermis (Figure 2). These findings were consistent with the diagnosis of acquired digital fibrokeratoma (ADF). The patient tolerated excisional biopsy well and had no evidence of recurrence 4 months following excision.
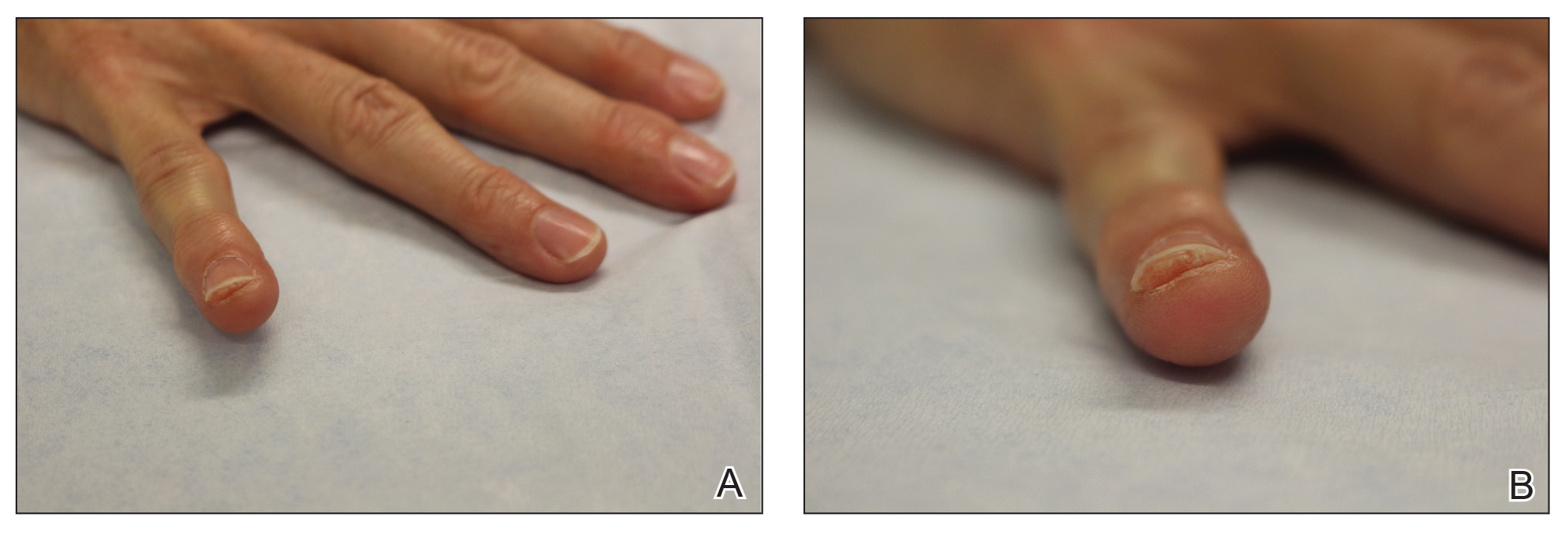
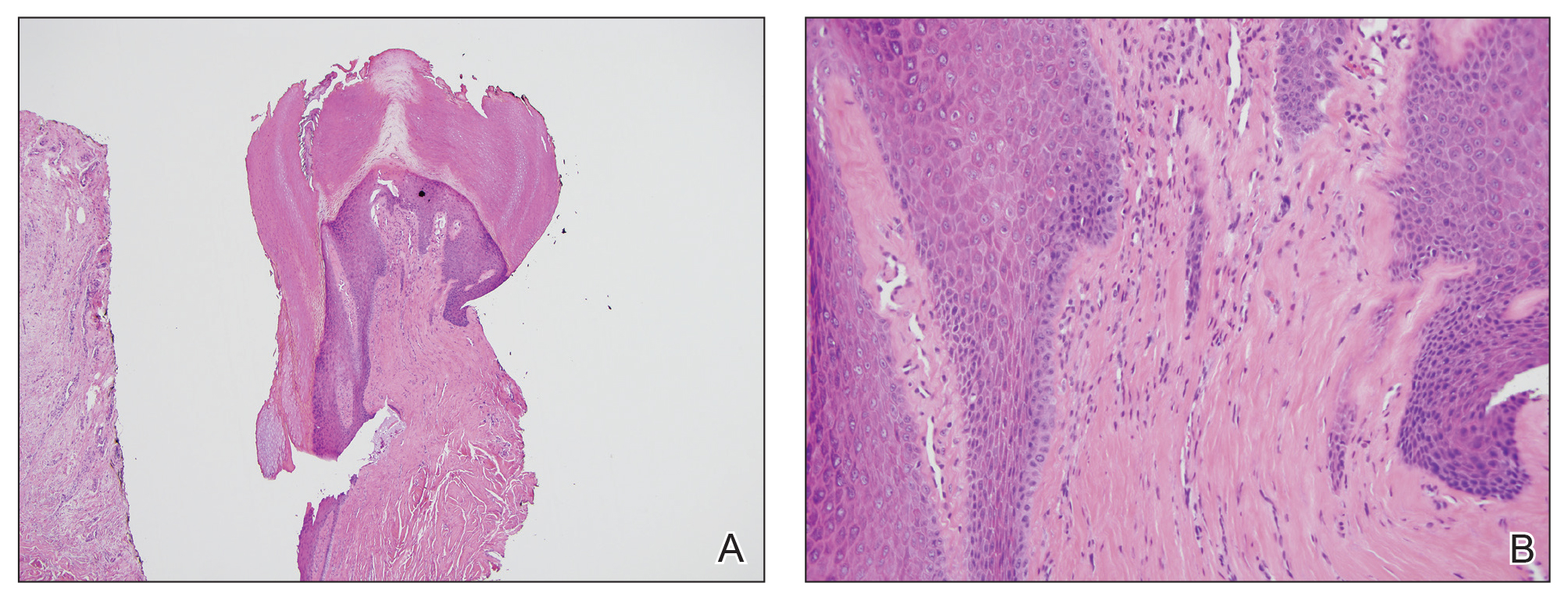
Comment
History and Clinical Presentation
First described by Bart et al1 in 1968, ADF is a rare benign fibrous tumor localized to the nail bed or periungual area.1 Typically, it presents as a solitary flesh-colored papule measuring 3 to 5 mm in diameter. It can be keratotic with a surrounding collarette of elevated skin. Acquired digital fibrokeratoma usually is localized to the digits of the hands or feet; when presenting subungually, it is more commonly found arising from the proximal matrix or nail bed of the great toe. Observed nail changes include longitudinal grooves, trachyonychia, subungual hyperkeratosis, and onycholysis.2 The affected nail can be painful, depending on the size and location of the tumor.
Acquired digital fibrokeratoma is more commonly found in middle-aged men; however, it has been reported among patients of various ages and in both sexes.1,3 In a study of 20 cases, the average duration before presenting for medical advice was 28 months.2 Acquired digital fibrokeratoma arises sporadically; some patients report prior local trauma. Lesions typically do not self-resolve.
Diagnosis
The diagnosis of ADF is made using a combination of clinical and histopathological findings. Dermoscopy is helpful and may show homogenous white or milky white structures, likely representing hyperkeratosis, proliferation of capillaries, and an increase in collagen bundles with a surrounding collarette of scale.4,5 Histopathology shows acanthosis and hyperkeratosis of the epidermis. Collagen bundles assume a characteristic vertical orientation to the long axis of the epidermis.
Two other histomorphologic subtypes, less common than the type I variant, are the type II variant, in which the number of fibroblasts is increased and the number of elastic fibers is decreased, and the type III variant, in which the stroma are edematous and cell poor. There is an even greater reduction in elastic tissue content in the type III variant than in the type I variant. There is evidence that type II ADFs exhibit more hyperkeratosis clinically than the other 2 subtypes, but from a practical perspective, this subclassification is not conducted in routine practice because it does not have clinical significance.5
Differential Diagnosis
The clinical differential diagnosis of ADF is broad and includes squamous cell carcinoma, onychomatricoma, onychopapilloma, verruca vulgaris, supernumerary digit, neurofibroma, cellular digital fibroma, and Koenen tumor (periungual fibroma). Almost all of these entities are easily differentiated from ADF on biopsy. A fibrokeratoma does not exhibit the atypia seen in squamous cell carcinoma. The multiple fibroepithelial projections and nail plate perforations characteristic of onychomatricoma are not observed in ADF. Onychopapilloma shows acanthosis and papillomatosis, similar to ADF; however, onychopapilloma lacks the characteristic vertical orientation of collagen in ADF. Verruca vulgaris classically shows koilocytosis, dilated blood vessels in papillae, and hypergranulosis. A supernumerary digit clinically lacks a collarette of scale and often presents in a bilateral fashion on the lateral fifth digits in children; histopathologically, a supernumerary digit is distinct from an ADF in that nerve bundles are abundant in the dermis, defining a form of amputation neuroma. Neurofibroma exhibits a spindle cell proliferation that assumes a patternless disposition in the dermis, accompanied by mucin, mast cells, and delicate collagen. The defining cell populace has a typical serpiginous nuclear outline that is characteristic of a Schwann cell. Cellular digital fibroma can present similar to ADF; it is considered by some to be a mucin-poor variant of superficial acral fibromyxoma. Its morphology is distinct: a proliferation of bland-appearing spindled cells exhibiting a storiform or fascicular growth pattern and CD34 positivity.
The differential diagnosis to consider when ADF is suspected is a Koenen tumor, which resembles a fibrokeratoma clinically and also is localized to the digits. Koenen tumors can be differentiated from fibrokeratoma by its association with tuberous sclerosis; a multiple, rather than solitary, presentation; a distinctive clove-shaped gross appearance; and an appearance on histopathology of stellate-shaped fibroblasts with occasional giant cells. Despite these important differences, Koenen tumor does exhibit a striking morphologic similarity to ADF, given that the vertical orientation of collagen bundles in Koenen tumor is virtually identical to ADF.6
Management
There are no known associations between ADF and medication use, including methotrexate and adalimumab, which our patient was taking; additionally, no association with RA or other systemic disorder has been reported.2 The preferred treatment of ADF is complete excision to the basal attachment of the tumor; recurrence is uncommon. Alternative therapies include destructive methods, such as cryotherapy, CO2 laser ablation, and electrodesiccation.2
- Bart RS, Andrade R, Kopf AW, et al. Acquired digital fibrokeratomas. Arch Dermatol. 1968;2:120-129.
- Hwang S, Kim M, Cho BK, et al. Clinical characteristics of acquired ungual fibrokeratoma. Indian J Dermatol Venereol Leprol. 2017;83:337-343.
- Yu D, Morgan RF. Acquired digital fibrokeratoma: a case report. Ann Plast Surg. 2015;74:304-305.
- Ehara Y, Yoshida Y, Ishizu S, et al. Acquired subungual fibrokeratoma. J Dermatol. 2017;44:e140-e141.
- Rubegni P, Poggiali S, Lamberti A, et al. Dermoscopy of acquired digital fibrokeratoma. Australas J Dermatol. 2012:53:47-48.
- Kint A, Baran R, De Keyser H. Acquired (digital) fibrokeratoma. J Am Acad Dermatol. 1985;12:816-821.
Case Report
A 53-year-old woman presented for an initial visit to the dermatology clinic for a growth under the right fifth fingernail of 1 year’s duration. She had no history of trauma to the digit or pain or bleeding. She self-treated with over-the-counter wart remover for several months without improvement. She reported no other skin concerns. She had a medical history of rheumatoid arthritis (RA) and basal cell carcinoma of the nose; she was taking methotrexate and adalimumab for the RA. She had a family history of melanoma in her father.
On physical examination, a firm nontender nodule was noted on the distal nail bed of the right fifth fingernail with onycholysis; the nail plate was otherwise intact (Figure 1). All other nails were normal. A plain radiograph of the involved digit showed no bony abnormality. Excisional biopsy of the nodule was performed and analyzed by histopathology (Figure 2). The biopsy specimen showed a benign epidermis that was acanthotic and surmounted by hyperkeratotic scale. The dermis was fibrotic with collagen bundles assuming a vertical orientation to the long axis of the epidermis, typical of a fibrokeratoma. There were no atypical features in the dermal component or epidermis (Figure 2). These findings were consistent with the diagnosis of acquired digital fibrokeratoma (ADF). The patient tolerated excisional biopsy well and had no evidence of recurrence 4 months following excision.
Comment
History and Clinical Presentation
First described by Bart et al1 in 1968, ADF is a rare benign fibrous tumor localized to the nail bed or periungual area.1 Typically, it presents as a solitary flesh-colored papule measuring 3 to 5 mm in diameter. It can be keratotic with a surrounding collarette of elevated skin. Acquired digital fibrokeratoma usually is localized to the digits of the hands or feet; when presenting subungually, it is more commonly found arising from the proximal matrix or nail bed of the great toe. Observed nail changes include longitudinal grooves, trachyonychia, subungual hyperkeratosis, and onycholysis.2 The affected nail can be painful, depending on the size and location of the tumor.
Acquired digital fibrokeratoma is more commonly found in middle-aged men; however, it has been reported among patients of various ages and in both sexes.1,3 In a study of 20 cases, the average duration before presenting for medical advice was 28 months.2 Acquired digital fibrokeratoma arises sporadically; some patients report prior local trauma. Lesions typically do not self-resolve.
Diagnosis
The diagnosis of ADF is made using a combination of clinical and histopathological findings. Dermoscopy is helpful and may show homogenous white or milky white structures, likely representing hyperkeratosis, proliferation of capillaries, and an increase in collagen bundles with a surrounding collarette of scale.4,5 Histopathology shows acanthosis and hyperkeratosis of the epidermis. Collagen bundles assume a characteristic vertical orientation to the long axis of the epidermis.
Two other histomorphologic subtypes, less common than the type I variant, are the type II variant, in which the number of fibroblasts is increased and the number of elastic fibers is decreased, and the type III variant, in which the stroma are edematous and cell poor. There is an even greater reduction in elastic tissue content in the type III variant than in the type I variant. There is evidence that type II ADFs exhibit more hyperkeratosis clinically than the other 2 subtypes, but from a practical perspective, this subclassification is not conducted in routine practice because it does not have clinical significance.5
Differential Diagnosis
The clinical differential diagnosis of ADF is broad and includes squamous cell carcinoma, onychomatricoma, onychopapilloma, verruca vulgaris, supernumerary digit, neurofibroma, cellular digital fibroma, and Koenen tumor (periungual fibroma). Almost all of these entities are easily differentiated from ADF on biopsy. A fibrokeratoma does not exhibit the atypia seen in squamous cell carcinoma. The multiple fibroepithelial projections and nail plate perforations characteristic of onychomatricoma are not observed in ADF. Onychopapilloma shows acanthosis and papillomatosis, similar to ADF; however, onychopapilloma lacks the characteristic vertical orientation of collagen in ADF. Verruca vulgaris classically shows koilocytosis, dilated blood vessels in papillae, and hypergranulosis. A supernumerary digit clinically lacks a collarette of scale and often presents in a bilateral fashion on the lateral fifth digits in children; histopathologically, a supernumerary digit is distinct from an ADF in that nerve bundles are abundant in the dermis, defining a form of amputation neuroma. Neurofibroma exhibits a spindle cell proliferation that assumes a patternless disposition in the dermis, accompanied by mucin, mast cells, and delicate collagen. The defining cell populace has a typical serpiginous nuclear outline that is characteristic of a Schwann cell. Cellular digital fibroma can present similar to ADF; it is considered by some to be a mucin-poor variant of superficial acral fibromyxoma. Its morphology is distinct: a proliferation of bland-appearing spindled cells exhibiting a storiform or fascicular growth pattern and CD34 positivity.
The differential diagnosis to consider when ADF is suspected is a Koenen tumor, which resembles a fibrokeratoma clinically and also is localized to the digits. Koenen tumors can be differentiated from fibrokeratoma by its association with tuberous sclerosis; a multiple, rather than solitary, presentation; a distinctive clove-shaped gross appearance; and an appearance on histopathology of stellate-shaped fibroblasts with occasional giant cells. Despite these important differences, Koenen tumor does exhibit a striking morphologic similarity to ADF, given that the vertical orientation of collagen bundles in Koenen tumor is virtually identical to ADF.6
Management
There are no known associations between ADF and medication use, including methotrexate and adalimumab, which our patient was taking; additionally, no association with RA or other systemic disorder has been reported.2 The preferred treatment of ADF is complete excision to the basal attachment of the tumor; recurrence is uncommon. Alternative therapies include destructive methods, such as cryotherapy, CO2 laser ablation, and electrodesiccation.2
Case Report
A 53-year-old woman presented for an initial visit to the dermatology clinic for a growth under the right fifth fingernail of 1 year’s duration. She had no history of trauma to the digit or pain or bleeding. She self-treated with over-the-counter wart remover for several months without improvement. She reported no other skin concerns. She had a medical history of rheumatoid arthritis (RA) and basal cell carcinoma of the nose; she was taking methotrexate and adalimumab for the RA. She had a family history of melanoma in her father.
On physical examination, a firm nontender nodule was noted on the distal nail bed of the right fifth fingernail with onycholysis; the nail plate was otherwise intact (Figure 1). All other nails were normal. A plain radiograph of the involved digit showed no bony abnormality. Excisional biopsy of the nodule was performed and analyzed by histopathology (Figure 2). The biopsy specimen showed a benign epidermis that was acanthotic and surmounted by hyperkeratotic scale. The dermis was fibrotic with collagen bundles assuming a vertical orientation to the long axis of the epidermis, typical of a fibrokeratoma. There were no atypical features in the dermal component or epidermis (Figure 2). These findings were consistent with the diagnosis of acquired digital fibrokeratoma (ADF). The patient tolerated excisional biopsy well and had no evidence of recurrence 4 months following excision.
Comment
History and Clinical Presentation
First described by Bart et al1 in 1968, ADF is a rare benign fibrous tumor localized to the nail bed or periungual area.1 Typically, it presents as a solitary flesh-colored papule measuring 3 to 5 mm in diameter. It can be keratotic with a surrounding collarette of elevated skin. Acquired digital fibrokeratoma usually is localized to the digits of the hands or feet; when presenting subungually, it is more commonly found arising from the proximal matrix or nail bed of the great toe. Observed nail changes include longitudinal grooves, trachyonychia, subungual hyperkeratosis, and onycholysis.2 The affected nail can be painful, depending on the size and location of the tumor.
Acquired digital fibrokeratoma is more commonly found in middle-aged men; however, it has been reported among patients of various ages and in both sexes.1,3 In a study of 20 cases, the average duration before presenting for medical advice was 28 months.2 Acquired digital fibrokeratoma arises sporadically; some patients report prior local trauma. Lesions typically do not self-resolve.
Diagnosis
The diagnosis of ADF is made using a combination of clinical and histopathological findings. Dermoscopy is helpful and may show homogenous white or milky white structures, likely representing hyperkeratosis, proliferation of capillaries, and an increase in collagen bundles with a surrounding collarette of scale.4,5 Histopathology shows acanthosis and hyperkeratosis of the epidermis. Collagen bundles assume a characteristic vertical orientation to the long axis of the epidermis.
Two other histomorphologic subtypes, less common than the type I variant, are the type II variant, in which the number of fibroblasts is increased and the number of elastic fibers is decreased, and the type III variant, in which the stroma are edematous and cell poor. There is an even greater reduction in elastic tissue content in the type III variant than in the type I variant. There is evidence that type II ADFs exhibit more hyperkeratosis clinically than the other 2 subtypes, but from a practical perspective, this subclassification is not conducted in routine practice because it does not have clinical significance.5
Differential Diagnosis
The clinical differential diagnosis of ADF is broad and includes squamous cell carcinoma, onychomatricoma, onychopapilloma, verruca vulgaris, supernumerary digit, neurofibroma, cellular digital fibroma, and Koenen tumor (periungual fibroma). Almost all of these entities are easily differentiated from ADF on biopsy. A fibrokeratoma does not exhibit the atypia seen in squamous cell carcinoma. The multiple fibroepithelial projections and nail plate perforations characteristic of onychomatricoma are not observed in ADF. Onychopapilloma shows acanthosis and papillomatosis, similar to ADF; however, onychopapilloma lacks the characteristic vertical orientation of collagen in ADF. Verruca vulgaris classically shows koilocytosis, dilated blood vessels in papillae, and hypergranulosis. A supernumerary digit clinically lacks a collarette of scale and often presents in a bilateral fashion on the lateral fifth digits in children; histopathologically, a supernumerary digit is distinct from an ADF in that nerve bundles are abundant in the dermis, defining a form of amputation neuroma. Neurofibroma exhibits a spindle cell proliferation that assumes a patternless disposition in the dermis, accompanied by mucin, mast cells, and delicate collagen. The defining cell populace has a typical serpiginous nuclear outline that is characteristic of a Schwann cell. Cellular digital fibroma can present similar to ADF; it is considered by some to be a mucin-poor variant of superficial acral fibromyxoma. Its morphology is distinct: a proliferation of bland-appearing spindled cells exhibiting a storiform or fascicular growth pattern and CD34 positivity.
The differential diagnosis to consider when ADF is suspected is a Koenen tumor, which resembles a fibrokeratoma clinically and also is localized to the digits. Koenen tumors can be differentiated from fibrokeratoma by its association with tuberous sclerosis; a multiple, rather than solitary, presentation; a distinctive clove-shaped gross appearance; and an appearance on histopathology of stellate-shaped fibroblasts with occasional giant cells. Despite these important differences, Koenen tumor does exhibit a striking morphologic similarity to ADF, given that the vertical orientation of collagen bundles in Koenen tumor is virtually identical to ADF.6
Management
There are no known associations between ADF and medication use, including methotrexate and adalimumab, which our patient was taking; additionally, no association with RA or other systemic disorder has been reported.2 The preferred treatment of ADF is complete excision to the basal attachment of the tumor; recurrence is uncommon. Alternative therapies include destructive methods, such as cryotherapy, CO2 laser ablation, and electrodesiccation.2
- Bart RS, Andrade R, Kopf AW, et al. Acquired digital fibrokeratomas. Arch Dermatol. 1968;2:120-129.
- Hwang S, Kim M, Cho BK, et al. Clinical characteristics of acquired ungual fibrokeratoma. Indian J Dermatol Venereol Leprol. 2017;83:337-343.
- Yu D, Morgan RF. Acquired digital fibrokeratoma: a case report. Ann Plast Surg. 2015;74:304-305.
- Ehara Y, Yoshida Y, Ishizu S, et al. Acquired subungual fibrokeratoma. J Dermatol. 2017;44:e140-e141.
- Rubegni P, Poggiali S, Lamberti A, et al. Dermoscopy of acquired digital fibrokeratoma. Australas J Dermatol. 2012:53:47-48.
- Kint A, Baran R, De Keyser H. Acquired (digital) fibrokeratoma. J Am Acad Dermatol. 1985;12:816-821.
- Bart RS, Andrade R, Kopf AW, et al. Acquired digital fibrokeratomas. Arch Dermatol. 1968;2:120-129.
- Hwang S, Kim M, Cho BK, et al. Clinical characteristics of acquired ungual fibrokeratoma. Indian J Dermatol Venereol Leprol. 2017;83:337-343.
- Yu D, Morgan RF. Acquired digital fibrokeratoma: a case report. Ann Plast Surg. 2015;74:304-305.
- Ehara Y, Yoshida Y, Ishizu S, et al. Acquired subungual fibrokeratoma. J Dermatol. 2017;44:e140-e141.
- Rubegni P, Poggiali S, Lamberti A, et al. Dermoscopy of acquired digital fibrokeratoma. Australas J Dermatol. 2012:53:47-48.
- Kint A, Baran R, De Keyser H. Acquired (digital) fibrokeratoma. J Am Acad Dermatol. 1985;12:816-821.
Practice Points
- Acquired digital fibrokeratoma is a benign tumor of the nail bed and periungual area.
- Histopathology shows epidermal acanthosis and hyperkeratosis, and collagen bundles are arranged in a vertical orientation to the long axis of the epidermis.
- Acquired digital fibrokeratoma should be considered in the differential diagnosis of flesh-colored papules on the nail unit associated with longitudinal grooves, trachyonychia, subungual hyperkeratosis, and onycholysis.
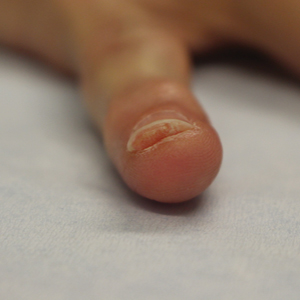
Melanocytic Matrical Carcinoma in a Solid-Organ Transplant Recipient
To the Editor:
A 68-year-old white man presented with a firm, gradually enlarging, mildly tender, grayish black papule with central ulceration on the left dorsal wrist of 4 months’ duration (Figure 1). His relevant medical history included multiple basal cell carcinomas (BCCs) and squamous cell carcinomas, as well as a single-lung transplant 2 years prior, for which he was on chronic immunosuppressive therapy with azathioprine, everolimus, tacrolimus, and prednisone. The clinical differential diagnosis included pigmented BCC, malignant melanoma, and ulcerated squamous cell carcinoma.
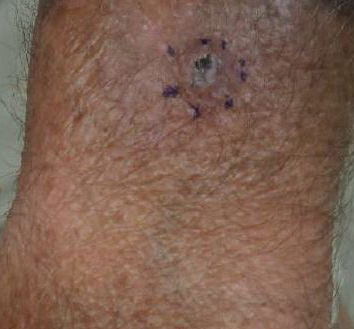
Histologic examination of the lesion (Figure 2) demonstrated irregular nodules of basaloid tumor cells with rounded nuclei, visible nucleoli, and scant cytoplasm involving the dermis. The tumor produced abrupt matrical-type keratinization, forming ghost cells. The lesion also contained frequent mitotic figures, apoptotic cells, focal areas of necrosis, and abundant melanin pigment. Admixed throughout the lesion were pigmented and dendritic melanocytic cells. The overlying epidermis was focally ulcerated with an adjacent localized connection between the tumor and the epidermis. Keratinocyte atypia was found in the surrounding epidermis, which contained melanophages, solar elastosis, and scattered chronic inflammatory cells. An immunohistochemical study (Figure 3) for tyrosinase demonstrated abundant admixed melanocytic cells. β-Catenin expression was shown in both nuclear and cytoplasmic distributions, and there was focal labeling on BerEP4 staining.
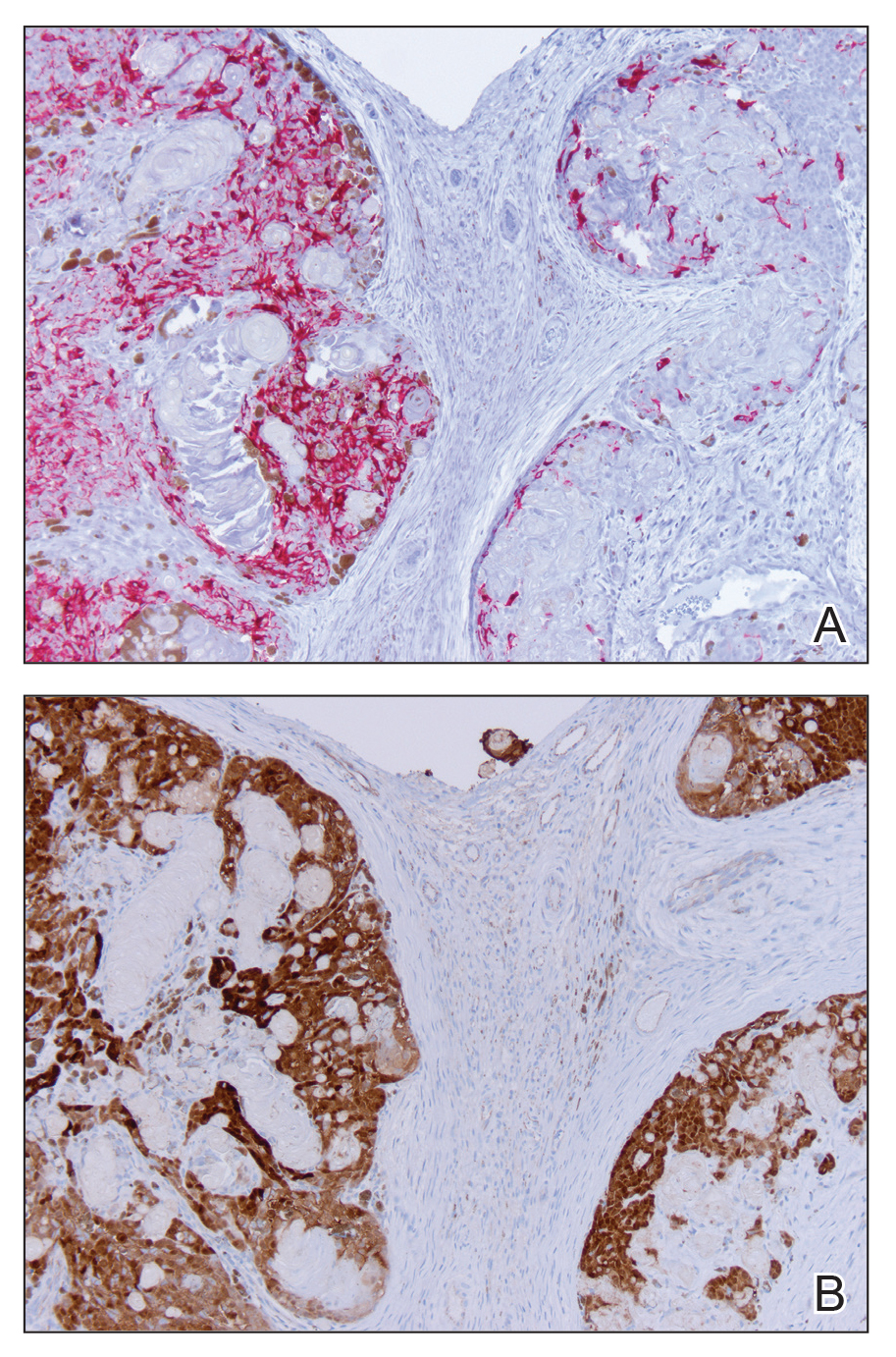
The lesion was subsequently treated with wide local excision. The patient has not had recurrence to date.
Melanocytic matricoma (MM), a rare adnexal tumor, was first described in 1999 by Carlson et al.1 A PubMed search of articles indexed for MEDLINE using the terms melanocytic and matricoma yielded 24 reported cases in the English-language literature.1-17 It consists of an admixed population of basaloid matrical and supramatrical cells, ghost cells, and dendritic melanocytes in a well-circumscribed dermal nodule, typically without epidermal or adnexal connection. In comparison to the more commonly described pilomatricoma, which can be uncommonly pigmented, MM typically has only focal areas of ghost cells and lacks cystic architecture.1,9,10,18 A granulomatous reaction to keratinaceous debris is variably present.1,9,10 Histologically, the scattered dendritic melanocytes are classically benign, but cases demonstrating melanocyte atypia have been reported.10,13 Melanocytic matricoma appears most commonly as a black or gray papule on sun-damaged skin in older men and tends not to recur following complete excision; thus, MM is considered to be a clinically benign neoplasm. Given the demographics and distribution of the lesions, exposure to UV radiation is thought to play a contributory role in the pathogenesis.2,10,19 Melanocytic matricoma is believed to recapitulate the hair follicle in the anagen phase, where there is close interplay between matrical keratinocytes and melanocytes prior to cessation of melanogenesis during the catagen phase.5,6,8,20,21 Evidence demonstrating highly conserved β-catenin and downstream lymphoid enhancer binding factor 1 (LEF1) expression, as well as pleckstrin homology-like domain, family A, member 1 (PHLDA1) expression (as a marker for follicular stem cells), points to constitutive activity in the Wnt signaling pathway in follicular stem cells of the bulge area as a major agent of tumorigenesis.12
Melanocytic matrical carcinoma, also known as malignant MM or matrical carcinoma with melanocytic hyperplasia, may be considered the malignant counterpart to MM.22 A PubMed search of articles indexed for MEDLINE using the terms melanocytic matrical carcinoma, malignant melanocytic matricoma, and matrical carcinoma with melanocytic hyperplasia, with review of references to identify additional citations, yielded 13 reported cases of MMC in the English-language literature (Table).19,22-30 As with MM, MMC is a biphasic tumor with basaloid matrical and supramatrical cells; focal areas of ghost cells; and admixed, banal-appearing dendritic melanocytes. However, the basaloid component also demonstrates nuclear atypia, mitoses, occasional ulceration, and variably poor circumscription. Clinically these lesions can mimic pigmented BCC, malignant melanoma, or other malignant adnexal tumors.25 Their natural history is unknown due to few reported cases, but they can be correlated with matrical carcinomas, which were first described by Weedon et al31 in 1980. A summary of more than 130 cases of matrical carcinomas in the English-language literature found that MMCs have high rates of local recurrence and metastasize in approximately 13% of cases. Wide local excision demonstrated lower rates of recurrence than simple excision (23% vs 83%), but there were insufficient cases to determine the incidence following Mohs micrographic surgery.32 Melanocytic matrical carcinomas also demonstrate mutations in the β-catenin pathway,pointing to a similar pathogenesis as their benign counterparts or perhaps direct malignant transformation.25,33,34
A subset of MMCs are combined cutaneous tumors (CCTs) consisting of epithelial neoplasms in close association with malignant melanocytes. Two of the more common variants include dermal squamomelanocytic tumors, a term first used by Pool et al,35 and malignant basomelanocytic tumors, as named by Erickson et al,36 but trichoblastomelanomas and other types have been documented.37 Although CCTs typically occur in the same patient populations as MMCs, namely elderly white men with chronically sun-damaged skin,they exhibit several important distinctions.37-39 By definition, CCTs have a malignant melanocytic component, whereas melanocytes are nonneoplastic in MMCs. The pathogenesis may differ as well. Various mechanisms for the close association of epithelial tumors and melanoma have been proposed, including field cancerization, tumor collision, tumor-tumor metastases, tumor colonization, and others, though CCTs likely arise through combinations of these processes depending upon their subtype.37-39 Paracrine signaling may play an important role in the pathogenesis of both tumors.5,6,8,38 As with MMCs, the prognosis of CCTs is limited by relatively few reported cases. Despite advanced Breslow depths in many cases, these tumors display more indolent behavior suggestive of melanoma in situ rather than invasive melanoma, perhaps due to dependence upon epithelial paracrine factors.37,39-42
Solid-organ transplant recipients have higher rates of more aggressive malignancies, of which skin cancer is the most common.43-49 Squamous cell carcinoma of the skin accounts for 95% of cutaneous malignancies in this population and occurs at approximately 65 times the rate of the general population.50 The risk of other skin cancers also is increased, though less dramatically, including BCC (10-fold increased risk) and melanoma (2- to 8-fold increased risk).46,50-53 The cause likely is multifactorial, including older age, history of skin cancer pretransplant, more than 5 years posttransplant, male sex, and incrementally as Fitzpatrick skin type decreases from VI to I.54-56 Immunosuppressive therapy also plays a role in tumorigenesis. Azathioprine metabolites have specifically been implicated in UVA radiation–induced promutagenic oxidative damage to DNA.57 Other studies have found no significant differences in the type of immunosuppressant used but instead have correlated rates of skin cancer to overall immunosuppression.48,55,58 Lung transplant recipients in particular demonstrate high rates of cutaneous malignancy, likely due in part to the necessity of more potent immunosuppressive regimens. Nearly one-third of patients develop a cutaneous malignancy by 5 years and nearly half by 10 years posttransplant.55
We report a rare case of MMC in a solid-organ transplant recipient. We hypothesize that the combination of UV radiation exposure–induced photodamage acquired pretransplant in addition to an aggressive immunosuppressive regimen with azathioprine and other agents posttransplant contributed to the development of this patient’s rare malignancy. Although rare, these tumors should remain in the differential diagnosis of clinicians and pathologists caring for this unique patient population.
- Carlson JA, Healy K, Slominski A, et al. Melanocytic matricoma: a report of two cases of a new entity. Am J Dermatopathol. 1999;21:344-349.
- Rizzardi C, Brollo A, Colonna A, et al. A tumor with composite pilo-folliculosebaceous differentiation harboring a recently described new entity—melanocytic matricoma. Am J Dermatopathol. 2002;24:493-497.
- Williams CM, Bozner P, Oliveri CV, et al. Melanocytic matricoma: case confirmation of a recently described entity. J Cutan Pathol. 2003;30:275-278.
- Horenstein MG, Kahn AG. Pathologic quiz case: a 69-year-old man with a brown-black facial papule. melanocytic matricoma. Arch Pathol Lab Med. 2004;128:e163-e164.
- Soler AP, Burchette JL, Bellet JS, et al. Cell adhesion protein expression in melanocytic matricoma. J Cutan Pathol. 2007;34:456-460.
- Islam MN, Bhattacharyya I, Proper SA, et al. Melanocytic matricoma: a distinctive clinicopathologic entity. Dermatol Surg. 2007;33:857-863.
- Monteagudo B, Requena L, Used-Aznar MM, et al. Melanocytic matricoma. Actas Dermosifiliogr. 2008;99:573-582.
- Cartaginese F, Sidoni A. Melanocytic matricoma. report of a further case with clinicopathological and immunohistochemical findings, differential diagnosis and review of the literature. Histol Histopathol. 2010;25:713-717.
- Tallon B, Cerroni L. Where pigmented pilomatricoma and melanocytic matricoma collide. Am J Dermatopathol. 2010;32:769-773.
- Zussman J, Sheth S, Ra SH, et al. Melanocytic matricoma with melanocytic atypia: report of a unique case and review of the literature. Am J Dermatopathol. 2011;33:508-512.
- Tanboon J, Manonukul J, Pattanaprichakul P. Melanocytic matricoma: two cases of a rare entity in women. J Cutan Pathol. 2014;41:775-782.
- Battistella M, Carlson JA, Oslo A, et al. Skin tumors with matrical differentiation: lessons from hair keratins, beta-catenin and PHLDA-1 expression. J Cutan Pathol. 2014;41:427-436.
- Barrado-Solis N, Moles-Poveda P, Roca-Estelles MJ, et al. Melanocytic matricoma with melanocytic atypia: report of a new case [published online February 11, 2015]. J Eur Acad Dermatol Venereol. 2016;30:859-860.
- Pagliarello C, Stanganelli I, Ricci R, et al. A pinkish-blue exophytic nodule on the arm of an elderly man: a quiz. melanocytic matricoma. Acta Derm Venereol. 2017;97:1261-1262.
- Winslow CY, Camacho I, Nousari CH. Melanocytic matricoma with consumption of the epidermis: an atypical histologic attribute or a malignant variant? Am J Dermatopathol. 2017;39:907-909.
- Sangiorgio V, Moneghini L, Tosi D, et al. A case of melanocytic matricoma with prominent mitotic activity and melanocytic hyperplasia. Int J Dermatol. 2018;57:e78-e81.
- Song J, Lu S, Wu Z. An unusual case of melanocytic matricoma in a young pregnant woman. Australas J Dermatol. 2019;60:140-141.
- Ishida M, Okabe H. Pigmented pilomatricoma: an underrecognized variant. Int J Clin Exp Pathol. 2013;6:1890-1893.
- Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol. 2008;30:174-177.
- Slominski A, Paus R. Melanogenesis is coupled to murine anagen: toward new concepts for the role of melanocytes and the regulation of melanogenesis in hair growth. J Invest Dermatol. 1993;101:90S-97S.
- De Berker D, Higgins CA, Jahada C, et al. Biology of hair and nails. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:1075-1092.
- Monteagudo C, Fernandez-Figueras MT, San Juan J, et al. Matrical carcinoma with prominent melanocytc hyperplasia (malignant melanocytic matricoma?). Am J Dermatopathol. 2003;25:485-489.
- Sloan JB, Sueki H, Jaworsky C. Pigmented malignant pilomatrixoma: report of a case and review of the literature. J Cutan Pathol. 1992;19:240-246.
- Hardisson D, Linares MD, Cuevas-Santos J, et al. Pilomatrix carcinoma: a clinicopathologic study of six cases and review of the literature. Am J Dermatopathol. 2001;23:394-401.
- Soler AP, Kindel SE, McCloskey G, et al. Cell-cell adhesion proteins in melanocytic pilomatrix carcinoma. Rare Tumors. 2010;2:e43-e45.
- Ardakani NM, Palmer DL, Wood BA. Malignant melanocytic matricoma: a report of 2 cases and review of the literature. Am J Dermatopathol. 2016;38:33-38.
- Villada G, Romagosa R, Miteva M, et al. Matrical carcinoma with melanocytic proliferation and prominent squamoid whorls. Am J Dermatopathol. 2016;38:e11-e14.
- Ji C, Zhang Y, Heller P, et al. Melanocytic matrical carcinoma mimicking melanoma. Am J Dermatopathol. 2017;39:903-906.
- Nielson CB, Vincek V. Malignant melanocytic matricoma and criteria for malignancy. Open J Pathol. 2018;8:94-100.
- Lehmer L, Carly SK, de Feraudy S. Matrical carcinoma with melanocytic hyperplasia mimicking nodular melanoma in an elderly Mexican male. J Cutan Pathol. 2019;46:442-446.
- Weedon D, Bell J, Mayze J. Matrical carcinoma of the skin. J Cutan Pathol. 1980;7:39-42.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Lazar AJ, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32:148-157.
- Hassanein AM, Glanz SM. Beta-catenin expression in benign and malignant pilomatrix neoplasms. Br J Dermatol. 2004;150:511-516.
- Pool SE, Manieei F, Clark WH Jr, et al. Dermal squamo-melanocytic tumor: a unique biphenotypic neoplasm of uncertain biological potential. Hum Pathol. 1999;30:525-529.
- Erickson LA, Myers JL, Mihm MC, et al. Malignant basomelanocytic tumor manifesting as metastatic melanoma. Am J Surg Pathol. 2004;28:1393-1396.
- Amin SM, Cooper C, Yelamos O, et al. Combined cutaneous tumors with a melanoma component: a clinical, histologic, and molecular study. J Am Acad Dermatol. 2015;73:451-460.
- Miteva M, Herschthal D, Ricotti C, et al. A rare case of a cutaneous squamomelanocytic tumor: revisiting the histogenesis of combined neoplasms. Am J Dermatopathol. 2009;31:599-603.
- Satter EK, Metcalf J, Lountzis N, et al. Tumors composed of malignant epithelial and melanocytic populations: a case series and review of the literature. J Cutan Pathol. 2009;36:211-219.
- Pouryazdanparast P, Yu L, Johnson T, et al. An unusual squamo-melanocytic tumor of uncertain biologic behavior: a variant of melanoma? Am J Dermatopathol. 2009;31:457-461.
- Burkhalter A, White W. Malignant melanoma in situ colonizing basal cell carcinoma: a simulator of invasive melanoma. Am J Dermatopathol. 1997;19:303-307.
- Papa G, Grandi G, Pascone M. Collision tumor of malignant skin cancers: a case of melanoma in basal cell carcinoma. Pathol Res Pract. 2006;202:691-694.
- Miao Y, Everly JJ, Gross TG, et al. De novo cancers arising in organ transplant recipients are associated with adverse outcomes compared with the general population. Transplantation. 2009;87:1347-1359.
- Bouwes Bavinck JN, Hardie DR, Green A, et al. The risk of skin cancer in renal transplant recipients in Queensland, Australia. a follow-up study. Transplantation. 1996;61:715-721.
- Berg D, Otley CC. Skin cancer in organ transplant recipients: epidemiology, pathogenesis, and management. J Am Acad Dermatol. 2002;47:1-17.
- Zwald FO, Brown M. Skin cancer in solid organ transplant recipients: advances in therapy and management: part I. epidemiology of skin cancer in solid organ transplant recipients. J Am Acad Dermatol. 2011;65:253-261.
- Zwald FO, Brown M. Skin cancer in solid organ transplant recipients: advances in therapy and management: part II. management of skin cancer in solid organ transplant recipients. J Am Acad Dermatol. 2011;65:263-273.
- DePry JL, Reed KB, Cook-Harris RH, et al. Iatrogenic immunosuppression and cutaneous malignancy. Clin Dermatol. 2011;29:602-613.
- Tessari G, Girolomoni G. Nonmelanoma skin cancer in solid organ transplant recipients: update on epidemiology, risk factors, and management. Dermatol Surg. 2012;38:1622-1630.
- Jensen P, Hansen S, Møller B, et al. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol. 1999;40:177-186.
- Kasiske BL, Snyder JJ, Gilbertson DT, et al. Cancer after kidney transplantation in the United States. Am J Transplant. 2004;4:905-913.
- Hollenbeak CS, Todd MM, Billingsley EM, et al. Increased incidence of melanoma in renal transplantation recipients. Cancer. 2005;104:1962-1967.
- Le Mire L, Hollowood K, Gray D, et al. Melanomas in renal transplant recipients. Br J Dermatol. 2006;154:472-477.
- Gogia R, Binstock M, Hirose R, et al. Fitzpatrick skin phototype is an independent predictor of squamous cell carcinoma risk after solid organ transplantation. J Am Acad Dermatol. 2013;68:585-591.
- Rashtak S, Dierkhising RA, Kremers WK, et al. Incidence and risk factors for skin cancer following lung transplantation. J Am Acad Dermatol. 2015;72:92-98.
- Ruiz DE, Luzuriaga AM, Hsieh C. Yearly burden of skin cancer in non-Caucasian and Caucasian solid-organ transplant recipients. J Clin Aesthet Dermatol. 2015;8:16-19.
- Perrett CM, Walker SL, O’Donovan P, et al. Azathioprine treatment photosensitizes human skin to ultraviolet A radiation. Br J Dermatol. 2008;159:198-204.
- Abou Ayache R, Thierry A, Bridoux F, et al. Long-term maintenance of calcineurin inhibitor monotherapy reduces the risk for squamous cell carcinomas after kidney transplantation compared with bi- or tritherapy. Transplant Proc. 2007;39:2592-2594.
To the Editor:
A 68-year-old white man presented with a firm, gradually enlarging, mildly tender, grayish black papule with central ulceration on the left dorsal wrist of 4 months’ duration (Figure 1). His relevant medical history included multiple basal cell carcinomas (BCCs) and squamous cell carcinomas, as well as a single-lung transplant 2 years prior, for which he was on chronic immunosuppressive therapy with azathioprine, everolimus, tacrolimus, and prednisone. The clinical differential diagnosis included pigmented BCC, malignant melanoma, and ulcerated squamous cell carcinoma.
Histologic examination of the lesion (Figure 2) demonstrated irregular nodules of basaloid tumor cells with rounded nuclei, visible nucleoli, and scant cytoplasm involving the dermis. The tumor produced abrupt matrical-type keratinization, forming ghost cells. The lesion also contained frequent mitotic figures, apoptotic cells, focal areas of necrosis, and abundant melanin pigment. Admixed throughout the lesion were pigmented and dendritic melanocytic cells. The overlying epidermis was focally ulcerated with an adjacent localized connection between the tumor and the epidermis. Keratinocyte atypia was found in the surrounding epidermis, which contained melanophages, solar elastosis, and scattered chronic inflammatory cells. An immunohistochemical study (Figure 3) for tyrosinase demonstrated abundant admixed melanocytic cells. β-Catenin expression was shown in both nuclear and cytoplasmic distributions, and there was focal labeling on BerEP4 staining.
The lesion was subsequently treated with wide local excision. The patient has not had recurrence to date.
Melanocytic matricoma (MM), a rare adnexal tumor, was first described in 1999 by Carlson et al.1 A PubMed search of articles indexed for MEDLINE using the terms melanocytic and matricoma yielded 24 reported cases in the English-language literature.1-17 It consists of an admixed population of basaloid matrical and supramatrical cells, ghost cells, and dendritic melanocytes in a well-circumscribed dermal nodule, typically without epidermal or adnexal connection. In comparison to the more commonly described pilomatricoma, which can be uncommonly pigmented, MM typically has only focal areas of ghost cells and lacks cystic architecture.1,9,10,18 A granulomatous reaction to keratinaceous debris is variably present.1,9,10 Histologically, the scattered dendritic melanocytes are classically benign, but cases demonstrating melanocyte atypia have been reported.10,13 Melanocytic matricoma appears most commonly as a black or gray papule on sun-damaged skin in older men and tends not to recur following complete excision; thus, MM is considered to be a clinically benign neoplasm. Given the demographics and distribution of the lesions, exposure to UV radiation is thought to play a contributory role in the pathogenesis.2,10,19 Melanocytic matricoma is believed to recapitulate the hair follicle in the anagen phase, where there is close interplay between matrical keratinocytes and melanocytes prior to cessation of melanogenesis during the catagen phase.5,6,8,20,21 Evidence demonstrating highly conserved β-catenin and downstream lymphoid enhancer binding factor 1 (LEF1) expression, as well as pleckstrin homology-like domain, family A, member 1 (PHLDA1) expression (as a marker for follicular stem cells), points to constitutive activity in the Wnt signaling pathway in follicular stem cells of the bulge area as a major agent of tumorigenesis.12
Melanocytic matrical carcinoma, also known as malignant MM or matrical carcinoma with melanocytic hyperplasia, may be considered the malignant counterpart to MM.22 A PubMed search of articles indexed for MEDLINE using the terms melanocytic matrical carcinoma, malignant melanocytic matricoma, and matrical carcinoma with melanocytic hyperplasia, with review of references to identify additional citations, yielded 13 reported cases of MMC in the English-language literature (Table).19,22-30 As with MM, MMC is a biphasic tumor with basaloid matrical and supramatrical cells; focal areas of ghost cells; and admixed, banal-appearing dendritic melanocytes. However, the basaloid component also demonstrates nuclear atypia, mitoses, occasional ulceration, and variably poor circumscription. Clinically these lesions can mimic pigmented BCC, malignant melanoma, or other malignant adnexal tumors.25 Their natural history is unknown due to few reported cases, but they can be correlated with matrical carcinomas, which were first described by Weedon et al31 in 1980. A summary of more than 130 cases of matrical carcinomas in the English-language literature found that MMCs have high rates of local recurrence and metastasize in approximately 13% of cases. Wide local excision demonstrated lower rates of recurrence than simple excision (23% vs 83%), but there were insufficient cases to determine the incidence following Mohs micrographic surgery.32 Melanocytic matrical carcinomas also demonstrate mutations in the β-catenin pathway,pointing to a similar pathogenesis as their benign counterparts or perhaps direct malignant transformation.25,33,34
A subset of MMCs are combined cutaneous tumors (CCTs) consisting of epithelial neoplasms in close association with malignant melanocytes. Two of the more common variants include dermal squamomelanocytic tumors, a term first used by Pool et al,35 and malignant basomelanocytic tumors, as named by Erickson et al,36 but trichoblastomelanomas and other types have been documented.37 Although CCTs typically occur in the same patient populations as MMCs, namely elderly white men with chronically sun-damaged skin,they exhibit several important distinctions.37-39 By definition, CCTs have a malignant melanocytic component, whereas melanocytes are nonneoplastic in MMCs. The pathogenesis may differ as well. Various mechanisms for the close association of epithelial tumors and melanoma have been proposed, including field cancerization, tumor collision, tumor-tumor metastases, tumor colonization, and others, though CCTs likely arise through combinations of these processes depending upon their subtype.37-39 Paracrine signaling may play an important role in the pathogenesis of both tumors.5,6,8,38 As with MMCs, the prognosis of CCTs is limited by relatively few reported cases. Despite advanced Breslow depths in many cases, these tumors display more indolent behavior suggestive of melanoma in situ rather than invasive melanoma, perhaps due to dependence upon epithelial paracrine factors.37,39-42
Solid-organ transplant recipients have higher rates of more aggressive malignancies, of which skin cancer is the most common.43-49 Squamous cell carcinoma of the skin accounts for 95% of cutaneous malignancies in this population and occurs at approximately 65 times the rate of the general population.50 The risk of other skin cancers also is increased, though less dramatically, including BCC (10-fold increased risk) and melanoma (2- to 8-fold increased risk).46,50-53 The cause likely is multifactorial, including older age, history of skin cancer pretransplant, more than 5 years posttransplant, male sex, and incrementally as Fitzpatrick skin type decreases from VI to I.54-56 Immunosuppressive therapy also plays a role in tumorigenesis. Azathioprine metabolites have specifically been implicated in UVA radiation–induced promutagenic oxidative damage to DNA.57 Other studies have found no significant differences in the type of immunosuppressant used but instead have correlated rates of skin cancer to overall immunosuppression.48,55,58 Lung transplant recipients in particular demonstrate high rates of cutaneous malignancy, likely due in part to the necessity of more potent immunosuppressive regimens. Nearly one-third of patients develop a cutaneous malignancy by 5 years and nearly half by 10 years posttransplant.55
We report a rare case of MMC in a solid-organ transplant recipient. We hypothesize that the combination of UV radiation exposure–induced photodamage acquired pretransplant in addition to an aggressive immunosuppressive regimen with azathioprine and other agents posttransplant contributed to the development of this patient’s rare malignancy. Although rare, these tumors should remain in the differential diagnosis of clinicians and pathologists caring for this unique patient population.
To the Editor:
A 68-year-old white man presented with a firm, gradually enlarging, mildly tender, grayish black papule with central ulceration on the left dorsal wrist of 4 months’ duration (Figure 1). His relevant medical history included multiple basal cell carcinomas (BCCs) and squamous cell carcinomas, as well as a single-lung transplant 2 years prior, for which he was on chronic immunosuppressive therapy with azathioprine, everolimus, tacrolimus, and prednisone. The clinical differential diagnosis included pigmented BCC, malignant melanoma, and ulcerated squamous cell carcinoma.
Histologic examination of the lesion (Figure 2) demonstrated irregular nodules of basaloid tumor cells with rounded nuclei, visible nucleoli, and scant cytoplasm involving the dermis. The tumor produced abrupt matrical-type keratinization, forming ghost cells. The lesion also contained frequent mitotic figures, apoptotic cells, focal areas of necrosis, and abundant melanin pigment. Admixed throughout the lesion were pigmented and dendritic melanocytic cells. The overlying epidermis was focally ulcerated with an adjacent localized connection between the tumor and the epidermis. Keratinocyte atypia was found in the surrounding epidermis, which contained melanophages, solar elastosis, and scattered chronic inflammatory cells. An immunohistochemical study (Figure 3) for tyrosinase demonstrated abundant admixed melanocytic cells. β-Catenin expression was shown in both nuclear and cytoplasmic distributions, and there was focal labeling on BerEP4 staining.
The lesion was subsequently treated with wide local excision. The patient has not had recurrence to date.
Melanocytic matricoma (MM), a rare adnexal tumor, was first described in 1999 by Carlson et al.1 A PubMed search of articles indexed for MEDLINE using the terms melanocytic and matricoma yielded 24 reported cases in the English-language literature.1-17 It consists of an admixed population of basaloid matrical and supramatrical cells, ghost cells, and dendritic melanocytes in a well-circumscribed dermal nodule, typically without epidermal or adnexal connection. In comparison to the more commonly described pilomatricoma, which can be uncommonly pigmented, MM typically has only focal areas of ghost cells and lacks cystic architecture.1,9,10,18 A granulomatous reaction to keratinaceous debris is variably present.1,9,10 Histologically, the scattered dendritic melanocytes are classically benign, but cases demonstrating melanocyte atypia have been reported.10,13 Melanocytic matricoma appears most commonly as a black or gray papule on sun-damaged skin in older men and tends not to recur following complete excision; thus, MM is considered to be a clinically benign neoplasm. Given the demographics and distribution of the lesions, exposure to UV radiation is thought to play a contributory role in the pathogenesis.2,10,19 Melanocytic matricoma is believed to recapitulate the hair follicle in the anagen phase, where there is close interplay between matrical keratinocytes and melanocytes prior to cessation of melanogenesis during the catagen phase.5,6,8,20,21 Evidence demonstrating highly conserved β-catenin and downstream lymphoid enhancer binding factor 1 (LEF1) expression, as well as pleckstrin homology-like domain, family A, member 1 (PHLDA1) expression (as a marker for follicular stem cells), points to constitutive activity in the Wnt signaling pathway in follicular stem cells of the bulge area as a major agent of tumorigenesis.12
Melanocytic matrical carcinoma, also known as malignant MM or matrical carcinoma with melanocytic hyperplasia, may be considered the malignant counterpart to MM.22 A PubMed search of articles indexed for MEDLINE using the terms melanocytic matrical carcinoma, malignant melanocytic matricoma, and matrical carcinoma with melanocytic hyperplasia, with review of references to identify additional citations, yielded 13 reported cases of MMC in the English-language literature (Table).19,22-30 As with MM, MMC is a biphasic tumor with basaloid matrical and supramatrical cells; focal areas of ghost cells; and admixed, banal-appearing dendritic melanocytes. However, the basaloid component also demonstrates nuclear atypia, mitoses, occasional ulceration, and variably poor circumscription. Clinically these lesions can mimic pigmented BCC, malignant melanoma, or other malignant adnexal tumors.25 Their natural history is unknown due to few reported cases, but they can be correlated with matrical carcinomas, which were first described by Weedon et al31 in 1980. A summary of more than 130 cases of matrical carcinomas in the English-language literature found that MMCs have high rates of local recurrence and metastasize in approximately 13% of cases. Wide local excision demonstrated lower rates of recurrence than simple excision (23% vs 83%), but there were insufficient cases to determine the incidence following Mohs micrographic surgery.32 Melanocytic matrical carcinomas also demonstrate mutations in the β-catenin pathway,pointing to a similar pathogenesis as their benign counterparts or perhaps direct malignant transformation.25,33,34
A subset of MMCs are combined cutaneous tumors (CCTs) consisting of epithelial neoplasms in close association with malignant melanocytes. Two of the more common variants include dermal squamomelanocytic tumors, a term first used by Pool et al,35 and malignant basomelanocytic tumors, as named by Erickson et al,36 but trichoblastomelanomas and other types have been documented.37 Although CCTs typically occur in the same patient populations as MMCs, namely elderly white men with chronically sun-damaged skin,they exhibit several important distinctions.37-39 By definition, CCTs have a malignant melanocytic component, whereas melanocytes are nonneoplastic in MMCs. The pathogenesis may differ as well. Various mechanisms for the close association of epithelial tumors and melanoma have been proposed, including field cancerization, tumor collision, tumor-tumor metastases, tumor colonization, and others, though CCTs likely arise through combinations of these processes depending upon their subtype.37-39 Paracrine signaling may play an important role in the pathogenesis of both tumors.5,6,8,38 As with MMCs, the prognosis of CCTs is limited by relatively few reported cases. Despite advanced Breslow depths in many cases, these tumors display more indolent behavior suggestive of melanoma in situ rather than invasive melanoma, perhaps due to dependence upon epithelial paracrine factors.37,39-42
Solid-organ transplant recipients have higher rates of more aggressive malignancies, of which skin cancer is the most common.43-49 Squamous cell carcinoma of the skin accounts for 95% of cutaneous malignancies in this population and occurs at approximately 65 times the rate of the general population.50 The risk of other skin cancers also is increased, though less dramatically, including BCC (10-fold increased risk) and melanoma (2- to 8-fold increased risk).46,50-53 The cause likely is multifactorial, including older age, history of skin cancer pretransplant, more than 5 years posttransplant, male sex, and incrementally as Fitzpatrick skin type decreases from VI to I.54-56 Immunosuppressive therapy also plays a role in tumorigenesis. Azathioprine metabolites have specifically been implicated in UVA radiation–induced promutagenic oxidative damage to DNA.57 Other studies have found no significant differences in the type of immunosuppressant used but instead have correlated rates of skin cancer to overall immunosuppression.48,55,58 Lung transplant recipients in particular demonstrate high rates of cutaneous malignancy, likely due in part to the necessity of more potent immunosuppressive regimens. Nearly one-third of patients develop a cutaneous malignancy by 5 years and nearly half by 10 years posttransplant.55
We report a rare case of MMC in a solid-organ transplant recipient. We hypothesize that the combination of UV radiation exposure–induced photodamage acquired pretransplant in addition to an aggressive immunosuppressive regimen with azathioprine and other agents posttransplant contributed to the development of this patient’s rare malignancy. Although rare, these tumors should remain in the differential diagnosis of clinicians and pathologists caring for this unique patient population.
- Carlson JA, Healy K, Slominski A, et al. Melanocytic matricoma: a report of two cases of a new entity. Am J Dermatopathol. 1999;21:344-349.
- Rizzardi C, Brollo A, Colonna A, et al. A tumor with composite pilo-folliculosebaceous differentiation harboring a recently described new entity—melanocytic matricoma. Am J Dermatopathol. 2002;24:493-497.
- Williams CM, Bozner P, Oliveri CV, et al. Melanocytic matricoma: case confirmation of a recently described entity. J Cutan Pathol. 2003;30:275-278.
- Horenstein MG, Kahn AG. Pathologic quiz case: a 69-year-old man with a brown-black facial papule. melanocytic matricoma. Arch Pathol Lab Med. 2004;128:e163-e164.
- Soler AP, Burchette JL, Bellet JS, et al. Cell adhesion protein expression in melanocytic matricoma. J Cutan Pathol. 2007;34:456-460.
- Islam MN, Bhattacharyya I, Proper SA, et al. Melanocytic matricoma: a distinctive clinicopathologic entity. Dermatol Surg. 2007;33:857-863.
- Monteagudo B, Requena L, Used-Aznar MM, et al. Melanocytic matricoma. Actas Dermosifiliogr. 2008;99:573-582.
- Cartaginese F, Sidoni A. Melanocytic matricoma. report of a further case with clinicopathological and immunohistochemical findings, differential diagnosis and review of the literature. Histol Histopathol. 2010;25:713-717.
- Tallon B, Cerroni L. Where pigmented pilomatricoma and melanocytic matricoma collide. Am J Dermatopathol. 2010;32:769-773.
- Zussman J, Sheth S, Ra SH, et al. Melanocytic matricoma with melanocytic atypia: report of a unique case and review of the literature. Am J Dermatopathol. 2011;33:508-512.
- Tanboon J, Manonukul J, Pattanaprichakul P. Melanocytic matricoma: two cases of a rare entity in women. J Cutan Pathol. 2014;41:775-782.
- Battistella M, Carlson JA, Oslo A, et al. Skin tumors with matrical differentiation: lessons from hair keratins, beta-catenin and PHLDA-1 expression. J Cutan Pathol. 2014;41:427-436.
- Barrado-Solis N, Moles-Poveda P, Roca-Estelles MJ, et al. Melanocytic matricoma with melanocytic atypia: report of a new case [published online February 11, 2015]. J Eur Acad Dermatol Venereol. 2016;30:859-860.
- Pagliarello C, Stanganelli I, Ricci R, et al. A pinkish-blue exophytic nodule on the arm of an elderly man: a quiz. melanocytic matricoma. Acta Derm Venereol. 2017;97:1261-1262.
- Winslow CY, Camacho I, Nousari CH. Melanocytic matricoma with consumption of the epidermis: an atypical histologic attribute or a malignant variant? Am J Dermatopathol. 2017;39:907-909.
- Sangiorgio V, Moneghini L, Tosi D, et al. A case of melanocytic matricoma with prominent mitotic activity and melanocytic hyperplasia. Int J Dermatol. 2018;57:e78-e81.
- Song J, Lu S, Wu Z. An unusual case of melanocytic matricoma in a young pregnant woman. Australas J Dermatol. 2019;60:140-141.
- Ishida M, Okabe H. Pigmented pilomatricoma: an underrecognized variant. Int J Clin Exp Pathol. 2013;6:1890-1893.
- Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol. 2008;30:174-177.
- Slominski A, Paus R. Melanogenesis is coupled to murine anagen: toward new concepts for the role of melanocytes and the regulation of melanogenesis in hair growth. J Invest Dermatol. 1993;101:90S-97S.
- De Berker D, Higgins CA, Jahada C, et al. Biology of hair and nails. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:1075-1092.
- Monteagudo C, Fernandez-Figueras MT, San Juan J, et al. Matrical carcinoma with prominent melanocytc hyperplasia (malignant melanocytic matricoma?). Am J Dermatopathol. 2003;25:485-489.
- Sloan JB, Sueki H, Jaworsky C. Pigmented malignant pilomatrixoma: report of a case and review of the literature. J Cutan Pathol. 1992;19:240-246.
- Hardisson D, Linares MD, Cuevas-Santos J, et al. Pilomatrix carcinoma: a clinicopathologic study of six cases and review of the literature. Am J Dermatopathol. 2001;23:394-401.
- Soler AP, Kindel SE, McCloskey G, et al. Cell-cell adhesion proteins in melanocytic pilomatrix carcinoma. Rare Tumors. 2010;2:e43-e45.
- Ardakani NM, Palmer DL, Wood BA. Malignant melanocytic matricoma: a report of 2 cases and review of the literature. Am J Dermatopathol. 2016;38:33-38.
- Villada G, Romagosa R, Miteva M, et al. Matrical carcinoma with melanocytic proliferation and prominent squamoid whorls. Am J Dermatopathol. 2016;38:e11-e14.
- Ji C, Zhang Y, Heller P, et al. Melanocytic matrical carcinoma mimicking melanoma. Am J Dermatopathol. 2017;39:903-906.
- Nielson CB, Vincek V. Malignant melanocytic matricoma and criteria for malignancy. Open J Pathol. 2018;8:94-100.
- Lehmer L, Carly SK, de Feraudy S. Matrical carcinoma with melanocytic hyperplasia mimicking nodular melanoma in an elderly Mexican male. J Cutan Pathol. 2019;46:442-446.
- Weedon D, Bell J, Mayze J. Matrical carcinoma of the skin. J Cutan Pathol. 1980;7:39-42.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Lazar AJ, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32:148-157.
- Hassanein AM, Glanz SM. Beta-catenin expression in benign and malignant pilomatrix neoplasms. Br J Dermatol. 2004;150:511-516.
- Pool SE, Manieei F, Clark WH Jr, et al. Dermal squamo-melanocytic tumor: a unique biphenotypic neoplasm of uncertain biological potential. Hum Pathol. 1999;30:525-529.
- Erickson LA, Myers JL, Mihm MC, et al. Malignant basomelanocytic tumor manifesting as metastatic melanoma. Am J Surg Pathol. 2004;28:1393-1396.
- Amin SM, Cooper C, Yelamos O, et al. Combined cutaneous tumors with a melanoma component: a clinical, histologic, and molecular study. J Am Acad Dermatol. 2015;73:451-460.
- Miteva M, Herschthal D, Ricotti C, et al. A rare case of a cutaneous squamomelanocytic tumor: revisiting the histogenesis of combined neoplasms. Am J Dermatopathol. 2009;31:599-603.
- Satter EK, Metcalf J, Lountzis N, et al. Tumors composed of malignant epithelial and melanocytic populations: a case series and review of the literature. J Cutan Pathol. 2009;36:211-219.
- Pouryazdanparast P, Yu L, Johnson T, et al. An unusual squamo-melanocytic tumor of uncertain biologic behavior: a variant of melanoma? Am J Dermatopathol. 2009;31:457-461.
- Burkhalter A, White W. Malignant melanoma in situ colonizing basal cell carcinoma: a simulator of invasive melanoma. Am J Dermatopathol. 1997;19:303-307.
- Papa G, Grandi G, Pascone M. Collision tumor of malignant skin cancers: a case of melanoma in basal cell carcinoma. Pathol Res Pract. 2006;202:691-694.
- Miao Y, Everly JJ, Gross TG, et al. De novo cancers arising in organ transplant recipients are associated with adverse outcomes compared with the general population. Transplantation. 2009;87:1347-1359.
- Bouwes Bavinck JN, Hardie DR, Green A, et al. The risk of skin cancer in renal transplant recipients in Queensland, Australia. a follow-up study. Transplantation. 1996;61:715-721.
- Berg D, Otley CC. Skin cancer in organ transplant recipients: epidemiology, pathogenesis, and management. J Am Acad Dermatol. 2002;47:1-17.
- Zwald FO, Brown M. Skin cancer in solid organ transplant recipients: advances in therapy and management: part I. epidemiology of skin cancer in solid organ transplant recipients. J Am Acad Dermatol. 2011;65:253-261.
- Zwald FO, Brown M. Skin cancer in solid organ transplant recipients: advances in therapy and management: part II. management of skin cancer in solid organ transplant recipients. J Am Acad Dermatol. 2011;65:263-273.
- DePry JL, Reed KB, Cook-Harris RH, et al. Iatrogenic immunosuppression and cutaneous malignancy. Clin Dermatol. 2011;29:602-613.
- Tessari G, Girolomoni G. Nonmelanoma skin cancer in solid organ transplant recipients: update on epidemiology, risk factors, and management. Dermatol Surg. 2012;38:1622-1630.
- Jensen P, Hansen S, Møller B, et al. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol. 1999;40:177-186.
- Kasiske BL, Snyder JJ, Gilbertson DT, et al. Cancer after kidney transplantation in the United States. Am J Transplant. 2004;4:905-913.
- Hollenbeak CS, Todd MM, Billingsley EM, et al. Increased incidence of melanoma in renal transplantation recipients. Cancer. 2005;104:1962-1967.
- Le Mire L, Hollowood K, Gray D, et al. Melanomas in renal transplant recipients. Br J Dermatol. 2006;154:472-477.
- Gogia R, Binstock M, Hirose R, et al. Fitzpatrick skin phototype is an independent predictor of squamous cell carcinoma risk after solid organ transplantation. J Am Acad Dermatol. 2013;68:585-591.
- Rashtak S, Dierkhising RA, Kremers WK, et al. Incidence and risk factors for skin cancer following lung transplantation. J Am Acad Dermatol. 2015;72:92-98.
- Ruiz DE, Luzuriaga AM, Hsieh C. Yearly burden of skin cancer in non-Caucasian and Caucasian solid-organ transplant recipients. J Clin Aesthet Dermatol. 2015;8:16-19.
- Perrett CM, Walker SL, O’Donovan P, et al. Azathioprine treatment photosensitizes human skin to ultraviolet A radiation. Br J Dermatol. 2008;159:198-204.
- Abou Ayache R, Thierry A, Bridoux F, et al. Long-term maintenance of calcineurin inhibitor monotherapy reduces the risk for squamous cell carcinomas after kidney transplantation compared with bi- or tritherapy. Transplant Proc. 2007;39:2592-2594.
- Carlson JA, Healy K, Slominski A, et al. Melanocytic matricoma: a report of two cases of a new entity. Am J Dermatopathol. 1999;21:344-349.
- Rizzardi C, Brollo A, Colonna A, et al. A tumor with composite pilo-folliculosebaceous differentiation harboring a recently described new entity—melanocytic matricoma. Am J Dermatopathol. 2002;24:493-497.
- Williams CM, Bozner P, Oliveri CV, et al. Melanocytic matricoma: case confirmation of a recently described entity. J Cutan Pathol. 2003;30:275-278.
- Horenstein MG, Kahn AG. Pathologic quiz case: a 69-year-old man with a brown-black facial papule. melanocytic matricoma. Arch Pathol Lab Med. 2004;128:e163-e164.
- Soler AP, Burchette JL, Bellet JS, et al. Cell adhesion protein expression in melanocytic matricoma. J Cutan Pathol. 2007;34:456-460.
- Islam MN, Bhattacharyya I, Proper SA, et al. Melanocytic matricoma: a distinctive clinicopathologic entity. Dermatol Surg. 2007;33:857-863.
- Monteagudo B, Requena L, Used-Aznar MM, et al. Melanocytic matricoma. Actas Dermosifiliogr. 2008;99:573-582.
- Cartaginese F, Sidoni A. Melanocytic matricoma. report of a further case with clinicopathological and immunohistochemical findings, differential diagnosis and review of the literature. Histol Histopathol. 2010;25:713-717.
- Tallon B, Cerroni L. Where pigmented pilomatricoma and melanocytic matricoma collide. Am J Dermatopathol. 2010;32:769-773.
- Zussman J, Sheth S, Ra SH, et al. Melanocytic matricoma with melanocytic atypia: report of a unique case and review of the literature. Am J Dermatopathol. 2011;33:508-512.
- Tanboon J, Manonukul J, Pattanaprichakul P. Melanocytic matricoma: two cases of a rare entity in women. J Cutan Pathol. 2014;41:775-782.
- Battistella M, Carlson JA, Oslo A, et al. Skin tumors with matrical differentiation: lessons from hair keratins, beta-catenin and PHLDA-1 expression. J Cutan Pathol. 2014;41:427-436.
- Barrado-Solis N, Moles-Poveda P, Roca-Estelles MJ, et al. Melanocytic matricoma with melanocytic atypia: report of a new case [published online February 11, 2015]. J Eur Acad Dermatol Venereol. 2016;30:859-860.
- Pagliarello C, Stanganelli I, Ricci R, et al. A pinkish-blue exophytic nodule on the arm of an elderly man: a quiz. melanocytic matricoma. Acta Derm Venereol. 2017;97:1261-1262.
- Winslow CY, Camacho I, Nousari CH. Melanocytic matricoma with consumption of the epidermis: an atypical histologic attribute or a malignant variant? Am J Dermatopathol. 2017;39:907-909.
- Sangiorgio V, Moneghini L, Tosi D, et al. A case of melanocytic matricoma with prominent mitotic activity and melanocytic hyperplasia. Int J Dermatol. 2018;57:e78-e81.
- Song J, Lu S, Wu Z. An unusual case of melanocytic matricoma in a young pregnant woman. Australas J Dermatol. 2019;60:140-141.
- Ishida M, Okabe H. Pigmented pilomatricoma: an underrecognized variant. Int J Clin Exp Pathol. 2013;6:1890-1893.
- Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol. 2008;30:174-177.
- Slominski A, Paus R. Melanogenesis is coupled to murine anagen: toward new concepts for the role of melanocytes and the regulation of melanogenesis in hair growth. J Invest Dermatol. 1993;101:90S-97S.
- De Berker D, Higgins CA, Jahada C, et al. Biology of hair and nails. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:1075-1092.
- Monteagudo C, Fernandez-Figueras MT, San Juan J, et al. Matrical carcinoma with prominent melanocytc hyperplasia (malignant melanocytic matricoma?). Am J Dermatopathol. 2003;25:485-489.
- Sloan JB, Sueki H, Jaworsky C. Pigmented malignant pilomatrixoma: report of a case and review of the literature. J Cutan Pathol. 1992;19:240-246.
- Hardisson D, Linares MD, Cuevas-Santos J, et al. Pilomatrix carcinoma: a clinicopathologic study of six cases and review of the literature. Am J Dermatopathol. 2001;23:394-401.
- Soler AP, Kindel SE, McCloskey G, et al. Cell-cell adhesion proteins in melanocytic pilomatrix carcinoma. Rare Tumors. 2010;2:e43-e45.
- Ardakani NM, Palmer DL, Wood BA. Malignant melanocytic matricoma: a report of 2 cases and review of the literature. Am J Dermatopathol. 2016;38:33-38.
- Villada G, Romagosa R, Miteva M, et al. Matrical carcinoma with melanocytic proliferation and prominent squamoid whorls. Am J Dermatopathol. 2016;38:e11-e14.
- Ji C, Zhang Y, Heller P, et al. Melanocytic matrical carcinoma mimicking melanoma. Am J Dermatopathol. 2017;39:903-906.
- Nielson CB, Vincek V. Malignant melanocytic matricoma and criteria for malignancy. Open J Pathol. 2018;8:94-100.
- Lehmer L, Carly SK, de Feraudy S. Matrical carcinoma with melanocytic hyperplasia mimicking nodular melanoma in an elderly Mexican male. J Cutan Pathol. 2019;46:442-446.
- Weedon D, Bell J, Mayze J. Matrical carcinoma of the skin. J Cutan Pathol. 1980;7:39-42.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Lazar AJ, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32:148-157.
- Hassanein AM, Glanz SM. Beta-catenin expression in benign and malignant pilomatrix neoplasms. Br J Dermatol. 2004;150:511-516.
- Pool SE, Manieei F, Clark WH Jr, et al. Dermal squamo-melanocytic tumor: a unique biphenotypic neoplasm of uncertain biological potential. Hum Pathol. 1999;30:525-529.
- Erickson LA, Myers JL, Mihm MC, et al. Malignant basomelanocytic tumor manifesting as metastatic melanoma. Am J Surg Pathol. 2004;28:1393-1396.
- Amin SM, Cooper C, Yelamos O, et al. Combined cutaneous tumors with a melanoma component: a clinical, histologic, and molecular study. J Am Acad Dermatol. 2015;73:451-460.
- Miteva M, Herschthal D, Ricotti C, et al. A rare case of a cutaneous squamomelanocytic tumor: revisiting the histogenesis of combined neoplasms. Am J Dermatopathol. 2009;31:599-603.
- Satter EK, Metcalf J, Lountzis N, et al. Tumors composed of malignant epithelial and melanocytic populations: a case series and review of the literature. J Cutan Pathol. 2009;36:211-219.
- Pouryazdanparast P, Yu L, Johnson T, et al. An unusual squamo-melanocytic tumor of uncertain biologic behavior: a variant of melanoma? Am J Dermatopathol. 2009;31:457-461.
- Burkhalter A, White W. Malignant melanoma in situ colonizing basal cell carcinoma: a simulator of invasive melanoma. Am J Dermatopathol. 1997;19:303-307.
- Papa G, Grandi G, Pascone M. Collision tumor of malignant skin cancers: a case of melanoma in basal cell carcinoma. Pathol Res Pract. 2006;202:691-694.
- Miao Y, Everly JJ, Gross TG, et al. De novo cancers arising in organ transplant recipients are associated with adverse outcomes compared with the general population. Transplantation. 2009;87:1347-1359.
- Bouwes Bavinck JN, Hardie DR, Green A, et al. The risk of skin cancer in renal transplant recipients in Queensland, Australia. a follow-up study. Transplantation. 1996;61:715-721.
- Berg D, Otley CC. Skin cancer in organ transplant recipients: epidemiology, pathogenesis, and management. J Am Acad Dermatol. 2002;47:1-17.
- Zwald FO, Brown M. Skin cancer in solid organ transplant recipients: advances in therapy and management: part I. epidemiology of skin cancer in solid organ transplant recipients. J Am Acad Dermatol. 2011;65:253-261.
- Zwald FO, Brown M. Skin cancer in solid organ transplant recipients: advances in therapy and management: part II. management of skin cancer in solid organ transplant recipients. J Am Acad Dermatol. 2011;65:263-273.
- DePry JL, Reed KB, Cook-Harris RH, et al. Iatrogenic immunosuppression and cutaneous malignancy. Clin Dermatol. 2011;29:602-613.
- Tessari G, Girolomoni G. Nonmelanoma skin cancer in solid organ transplant recipients: update on epidemiology, risk factors, and management. Dermatol Surg. 2012;38:1622-1630.
- Jensen P, Hansen S, Møller B, et al. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol. 1999;40:177-186.
- Kasiske BL, Snyder JJ, Gilbertson DT, et al. Cancer after kidney transplantation in the United States. Am J Transplant. 2004;4:905-913.
- Hollenbeak CS, Todd MM, Billingsley EM, et al. Increased incidence of melanoma in renal transplantation recipients. Cancer. 2005;104:1962-1967.
- Le Mire L, Hollowood K, Gray D, et al. Melanomas in renal transplant recipients. Br J Dermatol. 2006;154:472-477.
- Gogia R, Binstock M, Hirose R, et al. Fitzpatrick skin phototype is an independent predictor of squamous cell carcinoma risk after solid organ transplantation. J Am Acad Dermatol. 2013;68:585-591.
- Rashtak S, Dierkhising RA, Kremers WK, et al. Incidence and risk factors for skin cancer following lung transplantation. J Am Acad Dermatol. 2015;72:92-98.
- Ruiz DE, Luzuriaga AM, Hsieh C. Yearly burden of skin cancer in non-Caucasian and Caucasian solid-organ transplant recipients. J Clin Aesthet Dermatol. 2015;8:16-19.
- Perrett CM, Walker SL, O’Donovan P, et al. Azathioprine treatment photosensitizes human skin to ultraviolet A radiation. Br J Dermatol. 2008;159:198-204.
- Abou Ayache R, Thierry A, Bridoux F, et al. Long-term maintenance of calcineurin inhibitor monotherapy reduces the risk for squamous cell carcinomas after kidney transplantation compared with bi- or tritherapy. Transplant Proc. 2007;39:2592-2594.
Practice Points
- Melanocytic matrical carcinoma (MMC) is an extremely rare adnexal malignancy that can present as a hyperpigmented papule with or without ulceration.
- Histologically, the lesion resembles a matrical carcinoma with admixed, banal-appearing dendritic melanocytes.
- Solid-organ transplant recipients are at an increased risk of cutaneous malignancies, including rare cancers such as MMC, and these neoplasms should remain in the clinician’s differential diagnosis.
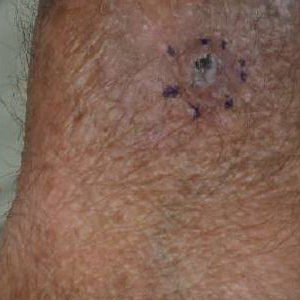
Vandetanib Photoinduced Cutaneous Toxicities
Vandetanib is a once-daily oral multikinase inhibitor that targets the rearranged during transfection (RET) tyrosine kinase, vascular endothelial growth factor receptor, and epidermal growth factor receptor. It has shown efficacy at doses of 300 mg daily in the treatment of progressive medullary thyroid cancer and has shown promise in non–small cell lung cancer and breast cancer. Vandetanib’s toxicity profile includes QT prolongation, diarrhea, and rash.1-3 Cutaneous involvement has been described in the literature as a photodistributed drug reaction with both erythema multiforme (EM) and Stevens-Johnson syndrome (SJS)–like eruptions, phototoxicity, and photoallergy (Table).4-12 Photoinduction is the common thread, but various mechanisms have been proposed, including drug deposition within the dermis and direct toxicity to keratinocytes; however, an understanding of the varied presentation is lacking.
We present 3 cases of vandetanib photoinduced cutaneous toxicities and review the literature on this novel kinase inhibitor. This discussion highlights the spectrum of photosensitivity reactions to vandetanib among patients with varying histologic and clinical presentations.
Case Reports
Patient 1A
74-year-old woman with a history of recurrent metastatic squamous cell carcinoma of the cervix and Fitzpatrick skin type III presented with erythematous, well-demarcated, photodistributed, eczematous papules that were coalescing into plaques on the scalp, hands, and face. The rash appeared sharply demarcated at the wrists bilaterally and principally involved the dorsal sun-exposed areas of her hands (Figure 1). The rash also involved the face and the V of the neck with sharp demarcation. Two weeks prior to onset, she initiated a phase 1 trial of oral vandetanib 100 mg twice daily and oral everolimus 5 mg daily. She did not recall practicing sun protection or experiencing increased sun exposure after starting that trial. The patient demonstrated symptom improvement with desonide cream, hydrocortisone cream 2.5%, and over-the-counter analgesic cream while continuing with the study drugs. However, she developed new, warm, painful papules on the hands and face. Phototesting and biopsy were not performed, and the etiology of the photosensitivity was unknown.
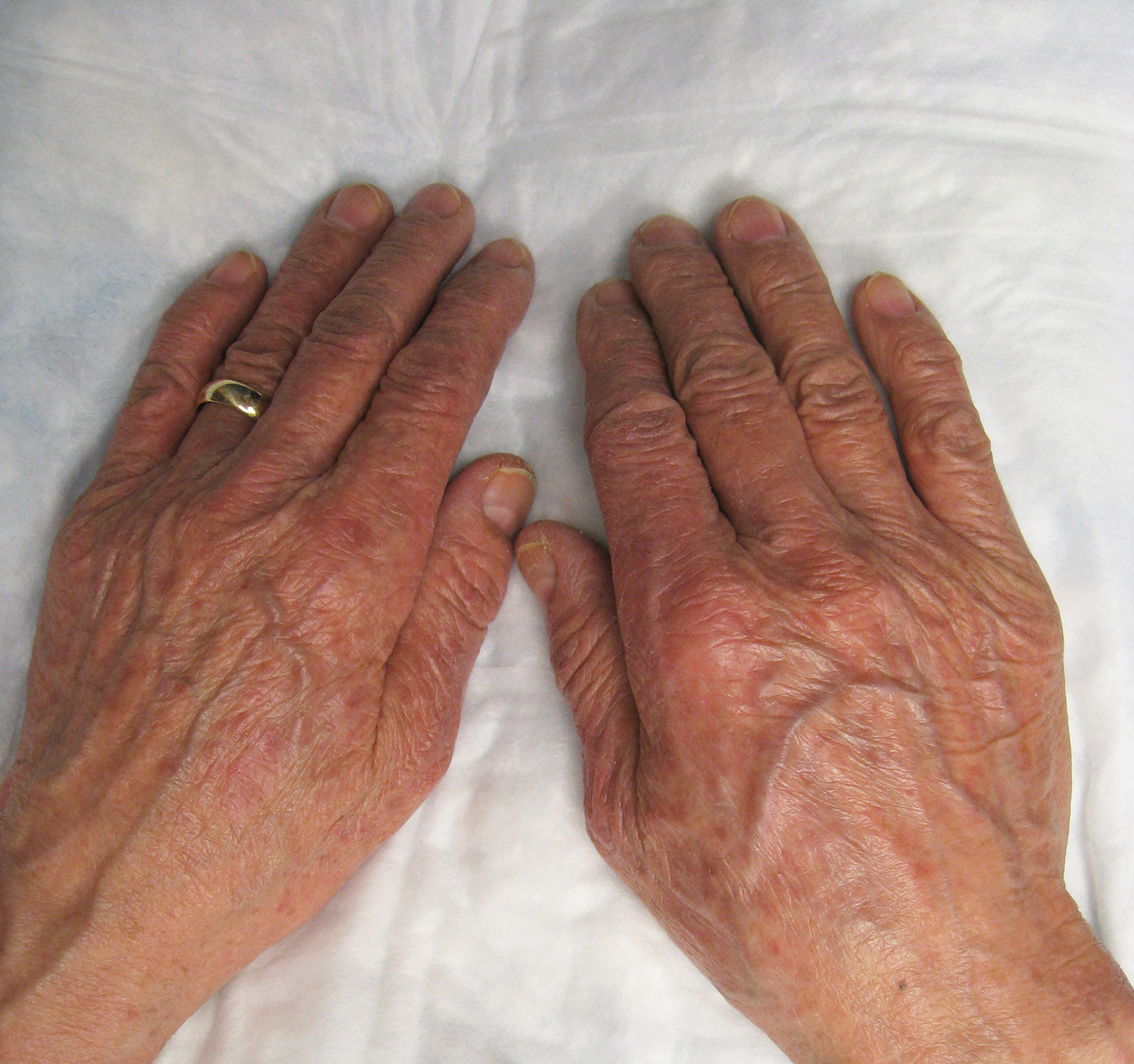
The patient was counseled about regular sun protection and was prescribed triamcinolone cream 0.1% for the arms and hydrocortisone cream 2.5% for the affected facial areas. Therapy with vandetanib and everolimus was continued without dose reduction or further cutaneous eruptions.
Patient 2
A 54-year-old man with a history of progressive medullary thyroid carcinoma and Fitzpatrick skin type II presented with erythematous, well-demarcated, photodistributed, edematous plaques and bullae of the head and neck, bilateral dorsal hands, and bilateral palms of 2 weeks’ duration. The rash spared the upper back and chest with a well-demarcated border (Figure 2A). There were ulcerations and erosions at the base of the neck and the dorsal hands (Figure 2B). He also had conjunctivitis but uninvolved oral and genital mucosae.
Two weeks before the rash appeared, oral vandetanib 300 mg daily was initiated. The patient initially noted some dry skin, which progressed to an eruption involving the face and neck and later the hands with palmar blistering and desquamation. Medication cessation for 1 month led to moderate improvement of the rash on the face and neck. He had not been practicing sun protection but did wear a baseball cap when outside. The patient did not recall an incidence of increased sun exposure. He underwent a skin biopsy of the right dorsal hand, which revealed interface dermatitis with dyskeratosis and subepidermal and intraepidermal bullae (Figure 3). The biopsy findings were most consistent with a phototoxic eruption. Phototesting was not performed.
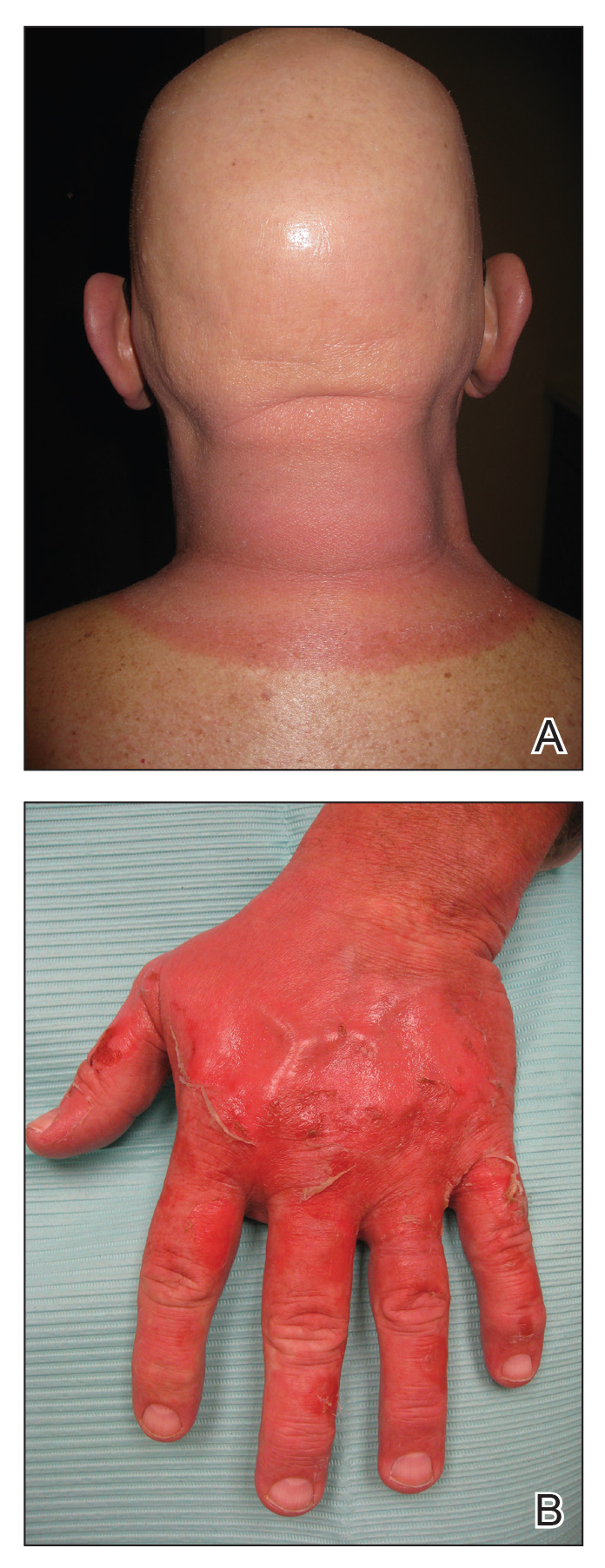

The patient then initiated sun-protective measures, a prednisone taper, and high-potency steroid ointments. As he tapered his prednisone, he noted continued improvement in the rash. His disease progressed, however, and he did not restart vandetanib.
Patient 3
A 73-year-old man with a history of metastatic lung carcinoma and Fitzpatrick skin type II presented with a rash on the scalp, face, and arms of 2.5 weeks’ duration. There was sharp demarcation at the edges of sun-exposed skin, and no bullae were noted (Figure 4). Prior to presentation, the patient started a 4-week phase 1 trial with vandetanib 300 mg daily and everolimus 10 mg daily. He did not recall any episodes of increased sun exposure. A punch biopsy of the arm showed an interface dermatitis suggestive of a phototoxic reaction. Phototesting was not performed to further clarify if there was a diminished minimal erythema dose with UVA or UVB radiation. Both drugs were discontinued, strict photoprotection was practiced, and triamcinolone cream 0.1% was initiated with resolution of rash. Vandetanib and everolimus were resumed at initial doses with strict photoprotection, and the rash has not recurred.

Comment
Adverse Events Associated With Vandetanib
Vandetanib is a novel multikinase inhibitor that targets RET tyrosine kinase, vascular endothelial growth factor receptor, and epidermal growth factor receptor.1,2 It currently is approved by the US Food and Drug Administration for the treatment of progressive medullary thyroid cancer and is being used in clinical trials for non–small cell lung cancer, glioma, advanced biliary tract cancer, breast cancer, and other advanced solid malignancies. Frequently reported adverse events (AEs) include QT prolongation, diarrhea, and rash.1-3 In a large phase 3 trial, 45% of patients had a rash; of these, 4% were grade 3 and above.3 The most common reasons for dose decrease or cessation were diarrhea and rash (1% and 1.3%, respectively).13 Outside of a trial setting, 75% (45/60) of patients in one French study reported a cutaneous AE, with photosensitivity noted in 22% (13/60). Thus, cutaneous reactions tend to be a common occurrence for patients on this drug, requiring diligent dermatologic examinations.14 In one meta-analysis comprising 9 studies with a total of 2961 patients, the incidence of all-grade rash was 46.1% (95% CI, 40.6%-51.8%), and it was concluded that vandetanib has the highest association of all-grade rash among the anti–vascular endothelial growth factor tyrosine kinase inhibitors. In this meta-analysis, the specific diagnosis of AEs was not further classified.15 In another cohort of vandetanib-treated patients, as many as 37% (28/63) of patients had photosensitivity, with no clarification of the etiology.16
Photoallergic vs Phototoxic Reactions
Photosensitivity reactions are cutaneous reactions that occur from UV light exposure, typically in conjunction with a photosensitizing agent. Photosensitivity reactions can be further classified into phototoxic and photoallergic reactions, which can be distinguished by histopathologic evaluation and history. Although phototoxic reactions will cause keratinocyte necrosis similar to a sunburn, photoallergic reactions will cause epidermal spongiosis similar to allergic contact dermatitis or eczema. Also, phototoxic reactions appear within 1 to 2 days of UV exposure and often are painful, whereas photoallergic reactions can be delayed for 2 to 3 weeks and usually are pruritic. Photosensitivity reactions related to vandetanib have been reported and are summarized in the Table.4-12
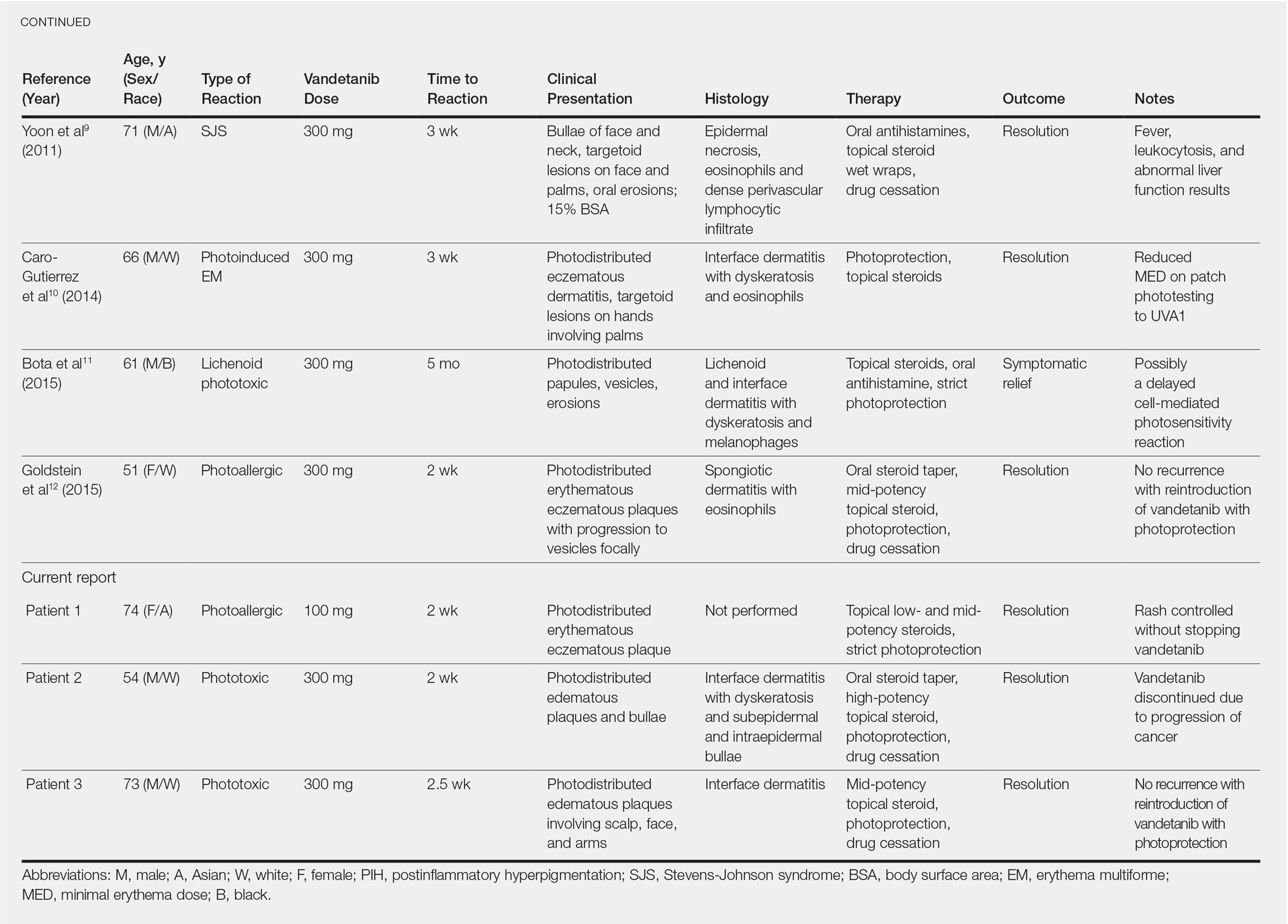
Although reported cutaneous reactions to vandetanib thus far in the literature were reported as photoinduced reactions, there have been isolated case reports of other eruptions including cutaneous pigmentation5 and one case of SJS.9 According to a PubMed search of articles indexed for MEDLINE using the terms vandetanib and rash, we found that there are a variety of clinical findings, but most of the reported photosensitivity cases were phototoxic. Fava et al7 and Goldstein et al12 both reported 1 photoallergic reaction each, plus patient 1 in our case series was noted to have a photoallergic reaction. Phototoxic reactions were reported in 4 patients (including our patient 2) who had dyskeratotic keratinocytes and vacuolar degeneration of the basal layer on histopathology.4,8 Fava et al7 described a lichenoid infiltrate with spongiosis consistent with a photoallergic reaction, but Chang et al4 and Bota et al11 described a lichenoid infiltrate with dyskeratotic cells. Also, Giacchero et al16 described a photosensitivity reaction in 28 of 63 patients. Although only 6 patients had biopsies performed, the range of photosensitivity reactions was demonstrated with lichenoid, dyskeratotic, and spongiotic reactions. However, the cases were not further defined as photoallergic or phototoxic.16 Vandetanib also has been associated with cutaneous blue pigmentation after likely phototoxic reactions. Pigment changes occurred after photosensitivity, but the clinical presentation of photosensitivity was not further characterized.5,16
Classic Drug Eruptions
Two patients were described as having classic drug eruptions—EM10 and SJS9—in photodistributed locations. Histologically, these entities are identical to phototoxic reactions, resulting in epidermal necrosis and an interface dermatitis, but the presence of targetoid lesions on the palms prompted the diagnosis of photodistributed EM and SJS in both cases.9,10 Unique to the SJS case was oral involvement.9
Distinguishing between a phototoxic reaction and photodistributed EM or SJS may be inconsequential if both can be prevented with photoprotection. Rechallenging patients with vandetanib while practicing photoprotection would help to clarify the mechanism, though this course is not always practical.
Mechanism of Action
As seen in our case series, cutaneous reactions occurred only on sun-exposed surfaces, and patients presented with sharp cutoff points that spared non–sun-exposed areas. Although clinically organized as a subtype of photosensitivity, the phototoxicity mechanism of action is considered a direct toxic effect on keratinocytes, which explains the histopathologic finding of dyskeratotic cells and the clinical spectrum of sunburn reaction, phototoxic EM, and SJS. UVA1 induces 2 photoproducts of vandetanib via a UVA1-mediated debromination process,17 but these photoproducts are not responsible for epidermal dyskeratosis.18 It was subsequently demonstrated that keratinocyte death was induced by apoptosis through photoinduced DNA cleavage and the formation of an aryl radical, which can induce further DNA damage.18 Caro-Gutierrez et al10 demonstrated a lowered minimal erythema dose in their patient with vandetanib-induced phototoxic EM.
Conversely, photoallergic reactions are considered immune-mediated delayed-type hypersensitivity reactions.4,7,11 Although the mechanism of a photoallergic reaction remains unclear, it is possible that vandetanib or a metabolite (in susceptible patients) induces an immune-mediated delayed-type hypersensitivity reaction with repeated exposure to the compound, which may explain the varied timing of photoallergic onset, including the events featured in the Bota et al11 case that occurred several months after drug initiation.
Conclusion
Considering the high prevalence of cutaneous AEs, especially varied photosensitivity reactions, these cases emphasize the importance of sun protection to help prevent dose reduction or drug cessation among patients taking vandetanib therapy.
- Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62:7284-7290.
- Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645-4655.
- Wells SA Jr, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134-141.
- Chang CH, Chang JW, Hui CY, et al. Severe photosensitivity reaction to vandetanib. J Clin Oncol. 2009;27:E114-E115.
- Kong HH, Fine HA, Stern JB, et al. Cutaneous pigmentation after photosensitivity induced by vandetanib therapy. Arch Dermatol. 2009;145:923-925.
- Brooks S, Linehan WM, Srinivasan R, et al. Successful laser treatment of vandetanib-associated cutaneous pigmentation. Arch Dermatol. 2011;147:364-365.
- Fava P, Quaglino P, Fierro MT, et al. Therapeutic hotline. a rare vandetanib-induced photo-allergic drug eruption. Dermatol Ther. 2010;23:553-555.
- Son YM, Roh JY, Cho EK, et al. Photosensitivity reactions to vandetanib: redevelopment after sequential treatment with docetaxel. Ann Dermatol. 2011;23(suppl 3):S314-S318.
- Yoon J, Oh CW, Kim CY. Stevens-Johnson syndrome induced by vandetanib. Ann Dermatol. 2011;23(suppl 3):S343-S345.
- Caro-Gutierrez D, Floristan Muruzabal MU, Gomez de la Fuente E, et al. Photo-induced erythema multiforme associated with vandetanib administration. J Am Acad Dermatol. 2014;71:E142-E144.11.
- Bota J, Harvey V, Ferguson C, et al. A rare case of late-onset lichenoid photodermatitis after vandetanib therapy. JAAD Case Rep. 2015;1:141-143.
- Goldstein J, Patel AB, Curry JL, et al. Photoallergic reaction in a patient receiving vandetanib for metastatic follicular thyroid carcinoma: a case report. BMC Dermatol. 2015;15:2.
- Thornton K, Kim G, Maher VE, et al. Vandetanib for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease: US Food and Drug Administration drug approval summary. Clin Cancer Res. 2012;18:3722-3730.
- Chougnet CN, Borget I, Leboulleux S, et al. Vandetanib for the treatment of advanced medullary thyroid cancer outside a clinical trial: results from a French cohort. Thyroid. 2015;25:386-391.
- Rosen AC, Wu S, Damse A, et al. Risk of rash in cancer patients treated with vandetanib: systematic review and meta-analysis. J Clin Endocrinol Metab. 2012;97:1125-1133.
- Giacchero D, Ramacciotti C, Arnault JP, et al. A new spectrum of skin toxic effects associated with the multikinase inhibitor vandetanib. Arch Dermatol. 2012;148:1418-1420.
- Dall’acqua S, Vedaldi D, Salvador A. Isolation and structure elucidation of the main UV-A photoproducts of vandetanib. J Pharm Biomed Anal. 2013;84:196-200.
- Salvador A, Vedaldi D, Brun P, et al. Vandetanib-induced phototoxicity in human keratinocytes NCTC-2544. Toxicol In Vitro. 2014;28:803-811.
Vandetanib is a once-daily oral multikinase inhibitor that targets the rearranged during transfection (RET) tyrosine kinase, vascular endothelial growth factor receptor, and epidermal growth factor receptor. It has shown efficacy at doses of 300 mg daily in the treatment of progressive medullary thyroid cancer and has shown promise in non–small cell lung cancer and breast cancer. Vandetanib’s toxicity profile includes QT prolongation, diarrhea, and rash.1-3 Cutaneous involvement has been described in the literature as a photodistributed drug reaction with both erythema multiforme (EM) and Stevens-Johnson syndrome (SJS)–like eruptions, phototoxicity, and photoallergy (Table).4-12 Photoinduction is the common thread, but various mechanisms have been proposed, including drug deposition within the dermis and direct toxicity to keratinocytes; however, an understanding of the varied presentation is lacking.
We present 3 cases of vandetanib photoinduced cutaneous toxicities and review the literature on this novel kinase inhibitor. This discussion highlights the spectrum of photosensitivity reactions to vandetanib among patients with varying histologic and clinical presentations.
Case Reports
Patient 1A
74-year-old woman with a history of recurrent metastatic squamous cell carcinoma of the cervix and Fitzpatrick skin type III presented with erythematous, well-demarcated, photodistributed, eczematous papules that were coalescing into plaques on the scalp, hands, and face. The rash appeared sharply demarcated at the wrists bilaterally and principally involved the dorsal sun-exposed areas of her hands (Figure 1). The rash also involved the face and the V of the neck with sharp demarcation. Two weeks prior to onset, she initiated a phase 1 trial of oral vandetanib 100 mg twice daily and oral everolimus 5 mg daily. She did not recall practicing sun protection or experiencing increased sun exposure after starting that trial. The patient demonstrated symptom improvement with desonide cream, hydrocortisone cream 2.5%, and over-the-counter analgesic cream while continuing with the study drugs. However, she developed new, warm, painful papules on the hands and face. Phototesting and biopsy were not performed, and the etiology of the photosensitivity was unknown.
The patient was counseled about regular sun protection and was prescribed triamcinolone cream 0.1% for the arms and hydrocortisone cream 2.5% for the affected facial areas. Therapy with vandetanib and everolimus was continued without dose reduction or further cutaneous eruptions.
Patient 2
A 54-year-old man with a history of progressive medullary thyroid carcinoma and Fitzpatrick skin type II presented with erythematous, well-demarcated, photodistributed, edematous plaques and bullae of the head and neck, bilateral dorsal hands, and bilateral palms of 2 weeks’ duration. The rash spared the upper back and chest with a well-demarcated border (Figure 2A). There were ulcerations and erosions at the base of the neck and the dorsal hands (Figure 2B). He also had conjunctivitis but uninvolved oral and genital mucosae.
Two weeks before the rash appeared, oral vandetanib 300 mg daily was initiated. The patient initially noted some dry skin, which progressed to an eruption involving the face and neck and later the hands with palmar blistering and desquamation. Medication cessation for 1 month led to moderate improvement of the rash on the face and neck. He had not been practicing sun protection but did wear a baseball cap when outside. The patient did not recall an incidence of increased sun exposure. He underwent a skin biopsy of the right dorsal hand, which revealed interface dermatitis with dyskeratosis and subepidermal and intraepidermal bullae (Figure 3). The biopsy findings were most consistent with a phototoxic eruption. Phototesting was not performed.
The patient then initiated sun-protective measures, a prednisone taper, and high-potency steroid ointments. As he tapered his prednisone, he noted continued improvement in the rash. His disease progressed, however, and he did not restart vandetanib.
Patient 3
A 73-year-old man with a history of metastatic lung carcinoma and Fitzpatrick skin type II presented with a rash on the scalp, face, and arms of 2.5 weeks’ duration. There was sharp demarcation at the edges of sun-exposed skin, and no bullae were noted (Figure 4). Prior to presentation, the patient started a 4-week phase 1 trial with vandetanib 300 mg daily and everolimus 10 mg daily. He did not recall any episodes of increased sun exposure. A punch biopsy of the arm showed an interface dermatitis suggestive of a phototoxic reaction. Phototesting was not performed to further clarify if there was a diminished minimal erythema dose with UVA or UVB radiation. Both drugs were discontinued, strict photoprotection was practiced, and triamcinolone cream 0.1% was initiated with resolution of rash. Vandetanib and everolimus were resumed at initial doses with strict photoprotection, and the rash has not recurred.
Comment
Adverse Events Associated With Vandetanib
Vandetanib is a novel multikinase inhibitor that targets RET tyrosine kinase, vascular endothelial growth factor receptor, and epidermal growth factor receptor.1,2 It currently is approved by the US Food and Drug Administration for the treatment of progressive medullary thyroid cancer and is being used in clinical trials for non–small cell lung cancer, glioma, advanced biliary tract cancer, breast cancer, and other advanced solid malignancies. Frequently reported adverse events (AEs) include QT prolongation, diarrhea, and rash.1-3 In a large phase 3 trial, 45% of patients had a rash; of these, 4% were grade 3 and above.3 The most common reasons for dose decrease or cessation were diarrhea and rash (1% and 1.3%, respectively).13 Outside of a trial setting, 75% (45/60) of patients in one French study reported a cutaneous AE, with photosensitivity noted in 22% (13/60). Thus, cutaneous reactions tend to be a common occurrence for patients on this drug, requiring diligent dermatologic examinations.14 In one meta-analysis comprising 9 studies with a total of 2961 patients, the incidence of all-grade rash was 46.1% (95% CI, 40.6%-51.8%), and it was concluded that vandetanib has the highest association of all-grade rash among the anti–vascular endothelial growth factor tyrosine kinase inhibitors. In this meta-analysis, the specific diagnosis of AEs was not further classified.15 In another cohort of vandetanib-treated patients, as many as 37% (28/63) of patients had photosensitivity, with no clarification of the etiology.16
Photoallergic vs Phototoxic Reactions
Photosensitivity reactions are cutaneous reactions that occur from UV light exposure, typically in conjunction with a photosensitizing agent. Photosensitivity reactions can be further classified into phototoxic and photoallergic reactions, which can be distinguished by histopathologic evaluation and history. Although phototoxic reactions will cause keratinocyte necrosis similar to a sunburn, photoallergic reactions will cause epidermal spongiosis similar to allergic contact dermatitis or eczema. Also, phototoxic reactions appear within 1 to 2 days of UV exposure and often are painful, whereas photoallergic reactions can be delayed for 2 to 3 weeks and usually are pruritic. Photosensitivity reactions related to vandetanib have been reported and are summarized in the Table.4-12
Although reported cutaneous reactions to vandetanib thus far in the literature were reported as photoinduced reactions, there have been isolated case reports of other eruptions including cutaneous pigmentation5 and one case of SJS.9 According to a PubMed search of articles indexed for MEDLINE using the terms vandetanib and rash, we found that there are a variety of clinical findings, but most of the reported photosensitivity cases were phototoxic. Fava et al7 and Goldstein et al12 both reported 1 photoallergic reaction each, plus patient 1 in our case series was noted to have a photoallergic reaction. Phototoxic reactions were reported in 4 patients (including our patient 2) who had dyskeratotic keratinocytes and vacuolar degeneration of the basal layer on histopathology.4,8 Fava et al7 described a lichenoid infiltrate with spongiosis consistent with a photoallergic reaction, but Chang et al4 and Bota et al11 described a lichenoid infiltrate with dyskeratotic cells. Also, Giacchero et al16 described a photosensitivity reaction in 28 of 63 patients. Although only 6 patients had biopsies performed, the range of photosensitivity reactions was demonstrated with lichenoid, dyskeratotic, and spongiotic reactions. However, the cases were not further defined as photoallergic or phototoxic.16 Vandetanib also has been associated with cutaneous blue pigmentation after likely phototoxic reactions. Pigment changes occurred after photosensitivity, but the clinical presentation of photosensitivity was not further characterized.5,16
Classic Drug Eruptions
Two patients were described as having classic drug eruptions—EM10 and SJS9—in photodistributed locations. Histologically, these entities are identical to phototoxic reactions, resulting in epidermal necrosis and an interface dermatitis, but the presence of targetoid lesions on the palms prompted the diagnosis of photodistributed EM and SJS in both cases.9,10 Unique to the SJS case was oral involvement.9
Distinguishing between a phototoxic reaction and photodistributed EM or SJS may be inconsequential if both can be prevented with photoprotection. Rechallenging patients with vandetanib while practicing photoprotection would help to clarify the mechanism, though this course is not always practical.
Mechanism of Action
As seen in our case series, cutaneous reactions occurred only on sun-exposed surfaces, and patients presented with sharp cutoff points that spared non–sun-exposed areas. Although clinically organized as a subtype of photosensitivity, the phototoxicity mechanism of action is considered a direct toxic effect on keratinocytes, which explains the histopathologic finding of dyskeratotic cells and the clinical spectrum of sunburn reaction, phototoxic EM, and SJS. UVA1 induces 2 photoproducts of vandetanib via a UVA1-mediated debromination process,17 but these photoproducts are not responsible for epidermal dyskeratosis.18 It was subsequently demonstrated that keratinocyte death was induced by apoptosis through photoinduced DNA cleavage and the formation of an aryl radical, which can induce further DNA damage.18 Caro-Gutierrez et al10 demonstrated a lowered minimal erythema dose in their patient with vandetanib-induced phototoxic EM.
Conversely, photoallergic reactions are considered immune-mediated delayed-type hypersensitivity reactions.4,7,11 Although the mechanism of a photoallergic reaction remains unclear, it is possible that vandetanib or a metabolite (in susceptible patients) induces an immune-mediated delayed-type hypersensitivity reaction with repeated exposure to the compound, which may explain the varied timing of photoallergic onset, including the events featured in the Bota et al11 case that occurred several months after drug initiation.
Conclusion
Considering the high prevalence of cutaneous AEs, especially varied photosensitivity reactions, these cases emphasize the importance of sun protection to help prevent dose reduction or drug cessation among patients taking vandetanib therapy.
Vandetanib is a once-daily oral multikinase inhibitor that targets the rearranged during transfection (RET) tyrosine kinase, vascular endothelial growth factor receptor, and epidermal growth factor receptor. It has shown efficacy at doses of 300 mg daily in the treatment of progressive medullary thyroid cancer and has shown promise in non–small cell lung cancer and breast cancer. Vandetanib’s toxicity profile includes QT prolongation, diarrhea, and rash.1-3 Cutaneous involvement has been described in the literature as a photodistributed drug reaction with both erythema multiforme (EM) and Stevens-Johnson syndrome (SJS)–like eruptions, phototoxicity, and photoallergy (Table).4-12 Photoinduction is the common thread, but various mechanisms have been proposed, including drug deposition within the dermis and direct toxicity to keratinocytes; however, an understanding of the varied presentation is lacking.
We present 3 cases of vandetanib photoinduced cutaneous toxicities and review the literature on this novel kinase inhibitor. This discussion highlights the spectrum of photosensitivity reactions to vandetanib among patients with varying histologic and clinical presentations.
Case Reports
Patient 1A
74-year-old woman with a history of recurrent metastatic squamous cell carcinoma of the cervix and Fitzpatrick skin type III presented with erythematous, well-demarcated, photodistributed, eczematous papules that were coalescing into plaques on the scalp, hands, and face. The rash appeared sharply demarcated at the wrists bilaterally and principally involved the dorsal sun-exposed areas of her hands (Figure 1). The rash also involved the face and the V of the neck with sharp demarcation. Two weeks prior to onset, she initiated a phase 1 trial of oral vandetanib 100 mg twice daily and oral everolimus 5 mg daily. She did not recall practicing sun protection or experiencing increased sun exposure after starting that trial. The patient demonstrated symptom improvement with desonide cream, hydrocortisone cream 2.5%, and over-the-counter analgesic cream while continuing with the study drugs. However, she developed new, warm, painful papules on the hands and face. Phototesting and biopsy were not performed, and the etiology of the photosensitivity was unknown.
The patient was counseled about regular sun protection and was prescribed triamcinolone cream 0.1% for the arms and hydrocortisone cream 2.5% for the affected facial areas. Therapy with vandetanib and everolimus was continued without dose reduction or further cutaneous eruptions.
Patient 2
A 54-year-old man with a history of progressive medullary thyroid carcinoma and Fitzpatrick skin type II presented with erythematous, well-demarcated, photodistributed, edematous plaques and bullae of the head and neck, bilateral dorsal hands, and bilateral palms of 2 weeks’ duration. The rash spared the upper back and chest with a well-demarcated border (Figure 2A). There were ulcerations and erosions at the base of the neck and the dorsal hands (Figure 2B). He also had conjunctivitis but uninvolved oral and genital mucosae.
Two weeks before the rash appeared, oral vandetanib 300 mg daily was initiated. The patient initially noted some dry skin, which progressed to an eruption involving the face and neck and later the hands with palmar blistering and desquamation. Medication cessation for 1 month led to moderate improvement of the rash on the face and neck. He had not been practicing sun protection but did wear a baseball cap when outside. The patient did not recall an incidence of increased sun exposure. He underwent a skin biopsy of the right dorsal hand, which revealed interface dermatitis with dyskeratosis and subepidermal and intraepidermal bullae (Figure 3). The biopsy findings were most consistent with a phototoxic eruption. Phototesting was not performed.
The patient then initiated sun-protective measures, a prednisone taper, and high-potency steroid ointments. As he tapered his prednisone, he noted continued improvement in the rash. His disease progressed, however, and he did not restart vandetanib.
Patient 3
A 73-year-old man with a history of metastatic lung carcinoma and Fitzpatrick skin type II presented with a rash on the scalp, face, and arms of 2.5 weeks’ duration. There was sharp demarcation at the edges of sun-exposed skin, and no bullae were noted (Figure 4). Prior to presentation, the patient started a 4-week phase 1 trial with vandetanib 300 mg daily and everolimus 10 mg daily. He did not recall any episodes of increased sun exposure. A punch biopsy of the arm showed an interface dermatitis suggestive of a phototoxic reaction. Phototesting was not performed to further clarify if there was a diminished minimal erythema dose with UVA or UVB radiation. Both drugs were discontinued, strict photoprotection was practiced, and triamcinolone cream 0.1% was initiated with resolution of rash. Vandetanib and everolimus were resumed at initial doses with strict photoprotection, and the rash has not recurred.
Comment
Adverse Events Associated With Vandetanib
Vandetanib is a novel multikinase inhibitor that targets RET tyrosine kinase, vascular endothelial growth factor receptor, and epidermal growth factor receptor.1,2 It currently is approved by the US Food and Drug Administration for the treatment of progressive medullary thyroid cancer and is being used in clinical trials for non–small cell lung cancer, glioma, advanced biliary tract cancer, breast cancer, and other advanced solid malignancies. Frequently reported adverse events (AEs) include QT prolongation, diarrhea, and rash.1-3 In a large phase 3 trial, 45% of patients had a rash; of these, 4% were grade 3 and above.3 The most common reasons for dose decrease or cessation were diarrhea and rash (1% and 1.3%, respectively).13 Outside of a trial setting, 75% (45/60) of patients in one French study reported a cutaneous AE, with photosensitivity noted in 22% (13/60). Thus, cutaneous reactions tend to be a common occurrence for patients on this drug, requiring diligent dermatologic examinations.14 In one meta-analysis comprising 9 studies with a total of 2961 patients, the incidence of all-grade rash was 46.1% (95% CI, 40.6%-51.8%), and it was concluded that vandetanib has the highest association of all-grade rash among the anti–vascular endothelial growth factor tyrosine kinase inhibitors. In this meta-analysis, the specific diagnosis of AEs was not further classified.15 In another cohort of vandetanib-treated patients, as many as 37% (28/63) of patients had photosensitivity, with no clarification of the etiology.16
Photoallergic vs Phototoxic Reactions
Photosensitivity reactions are cutaneous reactions that occur from UV light exposure, typically in conjunction with a photosensitizing agent. Photosensitivity reactions can be further classified into phototoxic and photoallergic reactions, which can be distinguished by histopathologic evaluation and history. Although phototoxic reactions will cause keratinocyte necrosis similar to a sunburn, photoallergic reactions will cause epidermal spongiosis similar to allergic contact dermatitis or eczema. Also, phototoxic reactions appear within 1 to 2 days of UV exposure and often are painful, whereas photoallergic reactions can be delayed for 2 to 3 weeks and usually are pruritic. Photosensitivity reactions related to vandetanib have been reported and are summarized in the Table.4-12
Although reported cutaneous reactions to vandetanib thus far in the literature were reported as photoinduced reactions, there have been isolated case reports of other eruptions including cutaneous pigmentation5 and one case of SJS.9 According to a PubMed search of articles indexed for MEDLINE using the terms vandetanib and rash, we found that there are a variety of clinical findings, but most of the reported photosensitivity cases were phototoxic. Fava et al7 and Goldstein et al12 both reported 1 photoallergic reaction each, plus patient 1 in our case series was noted to have a photoallergic reaction. Phototoxic reactions were reported in 4 patients (including our patient 2) who had dyskeratotic keratinocytes and vacuolar degeneration of the basal layer on histopathology.4,8 Fava et al7 described a lichenoid infiltrate with spongiosis consistent with a photoallergic reaction, but Chang et al4 and Bota et al11 described a lichenoid infiltrate with dyskeratotic cells. Also, Giacchero et al16 described a photosensitivity reaction in 28 of 63 patients. Although only 6 patients had biopsies performed, the range of photosensitivity reactions was demonstrated with lichenoid, dyskeratotic, and spongiotic reactions. However, the cases were not further defined as photoallergic or phototoxic.16 Vandetanib also has been associated with cutaneous blue pigmentation after likely phototoxic reactions. Pigment changes occurred after photosensitivity, but the clinical presentation of photosensitivity was not further characterized.5,16
Classic Drug Eruptions
Two patients were described as having classic drug eruptions—EM10 and SJS9—in photodistributed locations. Histologically, these entities are identical to phototoxic reactions, resulting in epidermal necrosis and an interface dermatitis, but the presence of targetoid lesions on the palms prompted the diagnosis of photodistributed EM and SJS in both cases.9,10 Unique to the SJS case was oral involvement.9
Distinguishing between a phototoxic reaction and photodistributed EM or SJS may be inconsequential if both can be prevented with photoprotection. Rechallenging patients with vandetanib while practicing photoprotection would help to clarify the mechanism, though this course is not always practical.
Mechanism of Action
As seen in our case series, cutaneous reactions occurred only on sun-exposed surfaces, and patients presented with sharp cutoff points that spared non–sun-exposed areas. Although clinically organized as a subtype of photosensitivity, the phototoxicity mechanism of action is considered a direct toxic effect on keratinocytes, which explains the histopathologic finding of dyskeratotic cells and the clinical spectrum of sunburn reaction, phototoxic EM, and SJS. UVA1 induces 2 photoproducts of vandetanib via a UVA1-mediated debromination process,17 but these photoproducts are not responsible for epidermal dyskeratosis.18 It was subsequently demonstrated that keratinocyte death was induced by apoptosis through photoinduced DNA cleavage and the formation of an aryl radical, which can induce further DNA damage.18 Caro-Gutierrez et al10 demonstrated a lowered minimal erythema dose in their patient with vandetanib-induced phototoxic EM.
Conversely, photoallergic reactions are considered immune-mediated delayed-type hypersensitivity reactions.4,7,11 Although the mechanism of a photoallergic reaction remains unclear, it is possible that vandetanib or a metabolite (in susceptible patients) induces an immune-mediated delayed-type hypersensitivity reaction with repeated exposure to the compound, which may explain the varied timing of photoallergic onset, including the events featured in the Bota et al11 case that occurred several months after drug initiation.
Conclusion
Considering the high prevalence of cutaneous AEs, especially varied photosensitivity reactions, these cases emphasize the importance of sun protection to help prevent dose reduction or drug cessation among patients taking vandetanib therapy.
- Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62:7284-7290.
- Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645-4655.
- Wells SA Jr, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134-141.
- Chang CH, Chang JW, Hui CY, et al. Severe photosensitivity reaction to vandetanib. J Clin Oncol. 2009;27:E114-E115.
- Kong HH, Fine HA, Stern JB, et al. Cutaneous pigmentation after photosensitivity induced by vandetanib therapy. Arch Dermatol. 2009;145:923-925.
- Brooks S, Linehan WM, Srinivasan R, et al. Successful laser treatment of vandetanib-associated cutaneous pigmentation. Arch Dermatol. 2011;147:364-365.
- Fava P, Quaglino P, Fierro MT, et al. Therapeutic hotline. a rare vandetanib-induced photo-allergic drug eruption. Dermatol Ther. 2010;23:553-555.
- Son YM, Roh JY, Cho EK, et al. Photosensitivity reactions to vandetanib: redevelopment after sequential treatment with docetaxel. Ann Dermatol. 2011;23(suppl 3):S314-S318.
- Yoon J, Oh CW, Kim CY. Stevens-Johnson syndrome induced by vandetanib. Ann Dermatol. 2011;23(suppl 3):S343-S345.
- Caro-Gutierrez D, Floristan Muruzabal MU, Gomez de la Fuente E, et al. Photo-induced erythema multiforme associated with vandetanib administration. J Am Acad Dermatol. 2014;71:E142-E144.11.
- Bota J, Harvey V, Ferguson C, et al. A rare case of late-onset lichenoid photodermatitis after vandetanib therapy. JAAD Case Rep. 2015;1:141-143.
- Goldstein J, Patel AB, Curry JL, et al. Photoallergic reaction in a patient receiving vandetanib for metastatic follicular thyroid carcinoma: a case report. BMC Dermatol. 2015;15:2.
- Thornton K, Kim G, Maher VE, et al. Vandetanib for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease: US Food and Drug Administration drug approval summary. Clin Cancer Res. 2012;18:3722-3730.
- Chougnet CN, Borget I, Leboulleux S, et al. Vandetanib for the treatment of advanced medullary thyroid cancer outside a clinical trial: results from a French cohort. Thyroid. 2015;25:386-391.
- Rosen AC, Wu S, Damse A, et al. Risk of rash in cancer patients treated with vandetanib: systematic review and meta-analysis. J Clin Endocrinol Metab. 2012;97:1125-1133.
- Giacchero D, Ramacciotti C, Arnault JP, et al. A new spectrum of skin toxic effects associated with the multikinase inhibitor vandetanib. Arch Dermatol. 2012;148:1418-1420.
- Dall’acqua S, Vedaldi D, Salvador A. Isolation and structure elucidation of the main UV-A photoproducts of vandetanib. J Pharm Biomed Anal. 2013;84:196-200.
- Salvador A, Vedaldi D, Brun P, et al. Vandetanib-induced phototoxicity in human keratinocytes NCTC-2544. Toxicol In Vitro. 2014;28:803-811.
- Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62:7284-7290.
- Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645-4655.
- Wells SA Jr, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134-141.
- Chang CH, Chang JW, Hui CY, et al. Severe photosensitivity reaction to vandetanib. J Clin Oncol. 2009;27:E114-E115.
- Kong HH, Fine HA, Stern JB, et al. Cutaneous pigmentation after photosensitivity induced by vandetanib therapy. Arch Dermatol. 2009;145:923-925.
- Brooks S, Linehan WM, Srinivasan R, et al. Successful laser treatment of vandetanib-associated cutaneous pigmentation. Arch Dermatol. 2011;147:364-365.
- Fava P, Quaglino P, Fierro MT, et al. Therapeutic hotline. a rare vandetanib-induced photo-allergic drug eruption. Dermatol Ther. 2010;23:553-555.
- Son YM, Roh JY, Cho EK, et al. Photosensitivity reactions to vandetanib: redevelopment after sequential treatment with docetaxel. Ann Dermatol. 2011;23(suppl 3):S314-S318.
- Yoon J, Oh CW, Kim CY. Stevens-Johnson syndrome induced by vandetanib. Ann Dermatol. 2011;23(suppl 3):S343-S345.
- Caro-Gutierrez D, Floristan Muruzabal MU, Gomez de la Fuente E, et al. Photo-induced erythema multiforme associated with vandetanib administration. J Am Acad Dermatol. 2014;71:E142-E144.11.
- Bota J, Harvey V, Ferguson C, et al. A rare case of late-onset lichenoid photodermatitis after vandetanib therapy. JAAD Case Rep. 2015;1:141-143.
- Goldstein J, Patel AB, Curry JL, et al. Photoallergic reaction in a patient receiving vandetanib for metastatic follicular thyroid carcinoma: a case report. BMC Dermatol. 2015;15:2.
- Thornton K, Kim G, Maher VE, et al. Vandetanib for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease: US Food and Drug Administration drug approval summary. Clin Cancer Res. 2012;18:3722-3730.
- Chougnet CN, Borget I, Leboulleux S, et al. Vandetanib for the treatment of advanced medullary thyroid cancer outside a clinical trial: results from a French cohort. Thyroid. 2015;25:386-391.
- Rosen AC, Wu S, Damse A, et al. Risk of rash in cancer patients treated with vandetanib: systematic review and meta-analysis. J Clin Endocrinol Metab. 2012;97:1125-1133.
- Giacchero D, Ramacciotti C, Arnault JP, et al. A new spectrum of skin toxic effects associated with the multikinase inhibitor vandetanib. Arch Dermatol. 2012;148:1418-1420.
- Dall’acqua S, Vedaldi D, Salvador A. Isolation and structure elucidation of the main UV-A photoproducts of vandetanib. J Pharm Biomed Anal. 2013;84:196-200.
- Salvador A, Vedaldi D, Brun P, et al. Vandetanib-induced phototoxicity in human keratinocytes NCTC-2544. Toxicol In Vitro. 2014;28:803-811.
Practice Points
- Vandetanib is a US Food and Drug Administration– approved once-daily oral multikinase inhibitor for patients with progressive medullary thyroid cancer with a high incidence of cutaneous toxicities including phototoxicity. Early recognition of such cutaneous toxicities leads to early intervention and may allow greater compliance with treatment.
- The most common toxicity is phototoxicity. Diligent interventions include photoprotection such as sunscreen, sun-protective clothing, and avoiding peak hours of sun exposure.
- Topical steroids as well as bland emollients are the mainstay of therapy for symptomatic lesions.
- Extensive cutaneous involvement may include blistering, pain, and pruritus and necessitate dose reduction or even drug cessation.
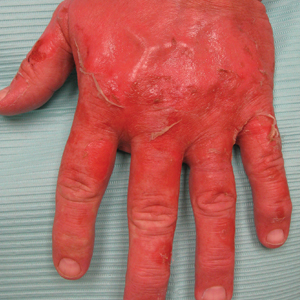
Lambert-Eaton Myasthenic Syndrome and Merkel Cell Carcinoma
Merkel cell carcinoma (MCC) is an aggressive neuroendocrine malignancy of the skin that is thought to arise from neural crest cells. It has an estimated annual incidence of 0.6 per 100,000 individuals, typically occurs in the elderly population, and is most common in white males.1 The tumor presents as a rapidly growing, violaceous nodule in sun-exposed areas of the skin; early in the course, it can be mistaken for a benign entity such as an epidermal cyst.2 Merkel cell carcinoma has a propensity to spread to regional lymph nodes, and in some cases, it occurs in the absence of skin findings.3 Histologically, MCC is nearly indistinguishable from small cell lung carcinoma (SCLC).4 The overall prognosis for patients with MCC is poor and largely dependent on the stage at diagnosis. Patients with regional and distant metastases have a 5-year survival rate of 26% to 42% and 18%, respectively.3
Lambert-Eaton myasthenic syndrome (LEMS) is a paraneoplastic or autoimmune disorder of the neuromuscular junction that is found in 3% of cases of SCLC.4 Reported cases of LEMS in patients with MCC are exceedingly rare.5-8 We provide a full report and longitudinal clinical follow-up of a case that was briefly discussed by Simmons et al,8 and we review the literature regarding paraneoplastic syndromes associated with MCC and other extrapulmonary small cell carcinomas (EPSCCs).
Case Report
A 63-year-old man was evaluated in the neurology clinic due to difficulty walking, climbing stairs, and performing push-ups over the last month. Prior to the onset of symptoms, he was otherwise healthy, walking 3 miles daily; however, at presentation he required use of a cane. Leg weakness worsened as the day progressed. In addition, he reported constipation, urinary urgency, dry mouth, mild dysphagia, reduced sensation below the knees, and a nasal quality in his speech. He had no ptosis, diplopia, dysarthria, muscle cramps, myalgia, or facial weakness. He denied fevers, chills, and night sweats but did admit to an unintentional 10- to 15-lb weight loss over the preceding few months.
The neurologic examination revealed mild proximal upper extremity weakness in the bilateral shoulder abductors, infraspinatus, hip extensors, and hip flexors (Medical Research Council muscle scale grade 4). All deep tendon reflexes, except the Achilles reflex, were present. Despite subjective sensory concerns, objective examination of all sensory modalities was normal. Cranial nerve examination was normal, except for a slight nasal quality to his voice.
A qualitative assay was positive for the presence of P/Q-type voltage-gated calcium channel (VGCC) antibodies. Other laboratory studies were within reference range, including acetylcholine-receptor antibodies (blocking, binding, and modulating) and muscle-specific kinase antibodies.
Lumbar and cervical spine magnetic resonance imaging revealed multilevel neuroforaminal stenosis without spinal canal stenosis or myelopathy. Computed tomography (CT) of the chest was notable for 2 pathologically enlarged lymph nodes in the left axilla and no evidence of primary pulmonary malignancy. Nerve-conduction studies (NCSs) in conjunction with other clinical findings were consistent with the diagnosis of LEMS.
Ultrasound-guided biopsy of the enlarged axillary lymph nodes demonstrated sheets and nests of small round blue tumor cells with minimal cytoplasm, high mitotic rate, and foci of necrosis (Figure 1). The tumor cells were positive for pancytokeratin (Lu-5) and cytokeratin (CK) 20 in a perinuclear dotlike pattern (Figure 2), as well as for the neuroendocrine markers synaptophysin (Figure 3), chromogranin A, and CD56. The tumor cells showed no immunoreactivity for CK7, thyroid transcription factor 1, CD3, CD5, or CD20. Flow cytometry demonstrated low cellularity, low specimen viability, and no evidence of an abnormal B-cell population. These findings were consistent with the diagnosis of MCC.
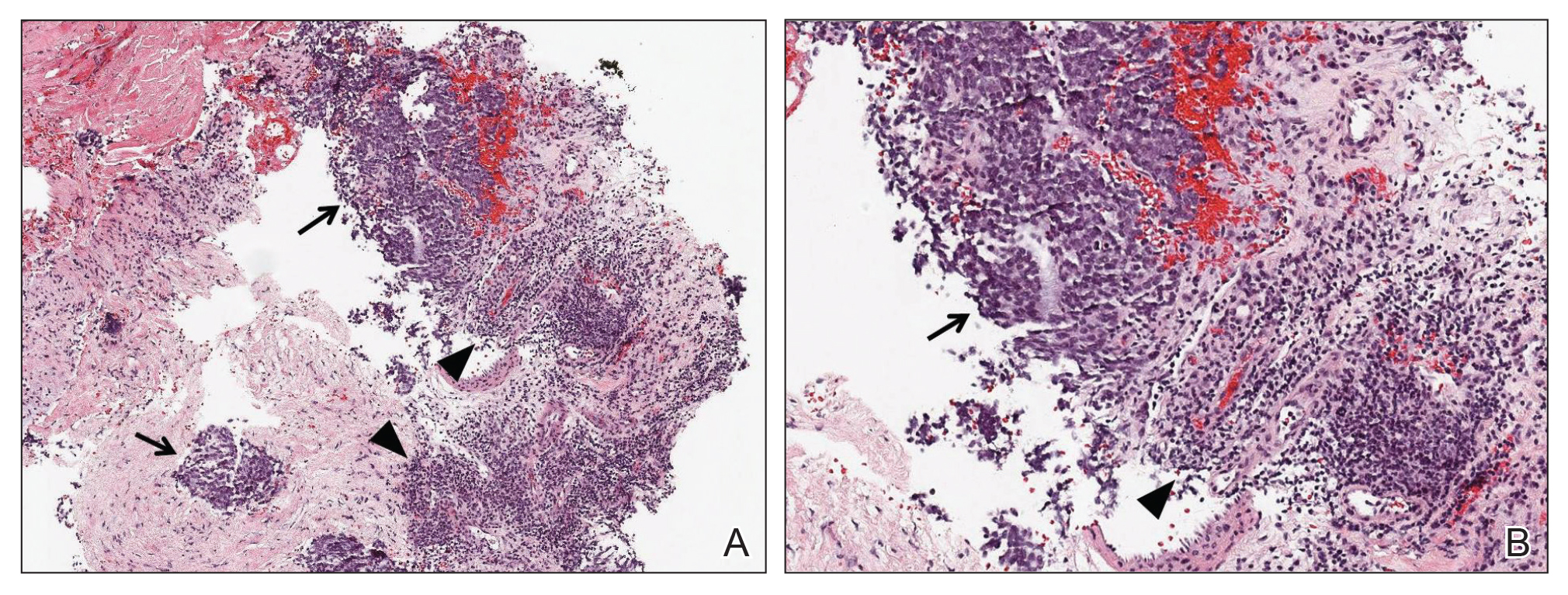
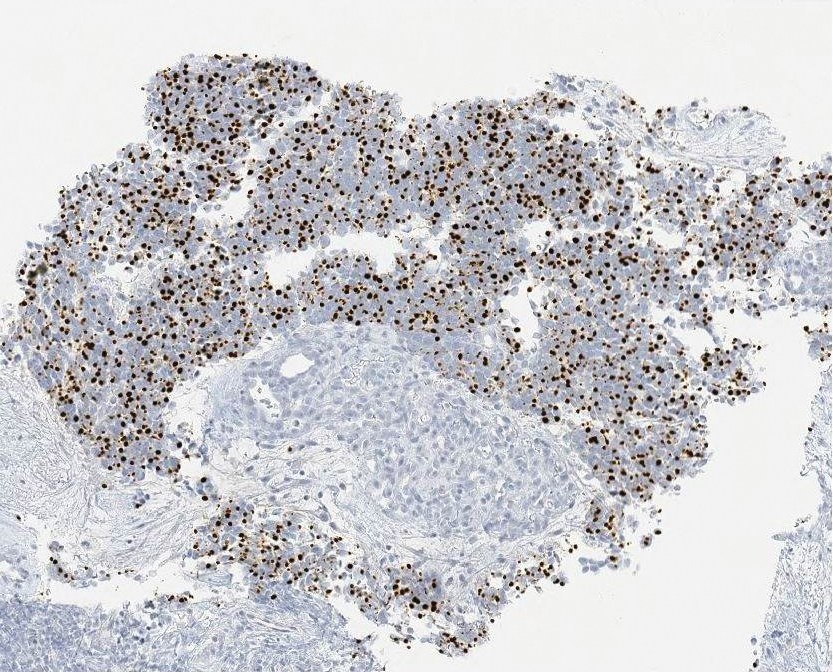
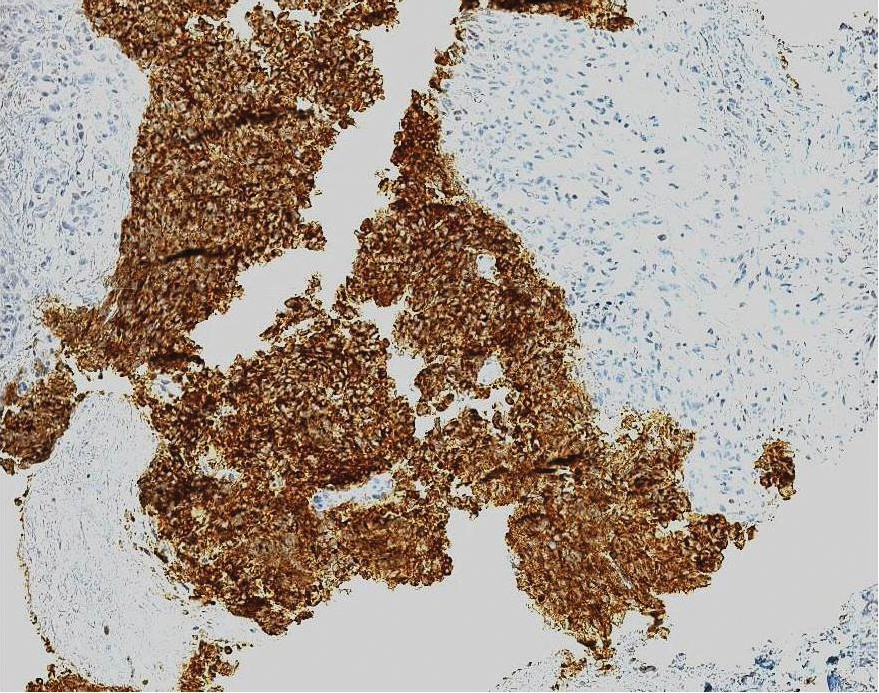
The patient underwent surgical excision of the involved lymph nodes. Four weeks after surgery, he reported dramatic improvement in strength, with complete resolution of the nasal speech, dysphagia, dry mouth, urinary retention, and constipation. Two months after surgery, his strength had normalized, except for slight persistent weakness in the bilateral shoulder abductors, trace weakness in the hip flexors, and a slight Trendelenburg gait. He was able to rise from a chair without using his arms and no longer required a cane for ambulation.
The patient underwent adjuvant radiation therapy after 2-month surgical follow-up with 5000-cGy radiation treatment to the left axillary region. Six months following primary definitive surgery and 4 months following adjuvant radiation therapy, he reported a 95% subjective return of physical strength. The patient was able to return to near-baseline physical activity. He continued to deny symptoms of dry mouth, incontinence, or constipation. Objectively, he had no focal neurologic deficits or weakness; no evidence of new skin lesions or lymphadenopathy was noted.
Comment
MCC vs SCLC
Merkel cell carcinoma is classified as a type of EPSCC. The histologic appearance of MCC is indistinguishable from SCLC. Both tumors are composed of uniform sheets of small round cells with a high nucleus to cytoplasm ratio, and both can express neuroendocrine markers, such as neuron-specific enolase, chromogranin A, and synaptophysin.9 Immunohistochemical positivity for CK20 and neurofilaments in combination with negative staining for thyroid transcription factor 1 and CK7 effectively differentiate MCC from SCLC.9 In addition, MCC often displays CK20 positivity in a perinuclear dotlike or punctate pattern, which is characteristic of this tumor.3,9,10 Negative immunohistochemical markers for B cells (CD20) and T cells (CD3) are important in excluding lymphoma.
LEMS Diagnosis
Lambert-Eaton myasthenic syndrome is a paraneoplastic or autoimmune disorder involving the neuromuscular junction. Autoantibodies to VGCC impair calcium influx into the presynaptic terminal, resulting in marked reduction of acetylcholine release into the synaptic cleft. The reduction in acetylcholine activity impairs production of muscle fiber action potentials, resulting in clinical weakness. The diagnosis of LEMS rests on clinical presentation, positive serology, and confirmatory neurophysiologic testing by NCS. Clinically, patients present with proximal weakness, hyporeflexia or areflexia, and autonomic dysfunction. Antibodies to P/Q-type VGCCs are found in 85% to 90% of cases of LEMS and are thought to play a direct causative role in the development of weakness.11 The finding of postexercise facilitation on motor NCS is the neurophysiologic hallmark and is highly specific for the diagnosis.
Approximately 50% to 60% of patients who present with LEMS have an underlying tumor, the vast majority of which are SCLC.11 There are a few reports of LEMS associated with other malignancies, including lymphoma; thymoma; neuroblastoma; and carcinoma of the breast, stomach, prostate, bladder, kidney, and gallbladder.12 Patients with nontumor or autoimmune LEMS tend to be younger, and there is no male predominance, as there is in paraneoplastic LEMS.13 Given the risk of underlying malignancy in LEMS, Titulaer et al14 proposed a screening protocol for patients presenting with LEMS, recommending initial primary screening using CT of the chest. If the CT scan is negative, total-body fludeoxyglucose positron emission tomography should be performed to assess for fludeoxyglucose avid lesions. If both initial studies are negative, routine follow-up with CT of the chest at 6-month intervals for a minimum of 2 to 4 years after the initial diagnosis of LEMS was recommended. An exception to this protocol was suggested to allow consideration to stop screening after the first 6-month follow-up chest CT for patients younger than 45 years who have never smoked and who have an HLA 8.1 haplotype for which nontumor LEMS would be a more probable diagnosis.14
In addition to a screening protocol, a validated prediction tool, the Dutch-English LEMS Tumor Association prediction score, was developed. It uses common signs and symptoms of LEMS and risk factors for SCLC to help guide the need for further screening.15
Paraneoplastic Syndromes Associated With MCC
Other paraneoplastic syndromes have been reported in association with MCC. A patient with brainstem encephalitis associated with MCC was reported in a trial of a novel immunotherapy for paraneoplastic neurologic syndromes.16,17 A syndrome of inappropriate antidiuretic hormone (SIADH) secretion was reported in a patient with N-type calcium channel antibodies.18 Two cases of paraneoplastic cerebellar degeneration have been reported; the first was associated with a novel 70-kD antibody,19 and the second was associated with the P/Q-type VGCC antibody.20 Anti-Hu antibodies have been found in a handful of reports of neurologic deterioration in patients with MCC. Hocar et al21 reported a severe necrotizing myopathy; Greenlee et al22 described a syndrome of progressive sensorimotor and autonomic neuropathy with encephalopathy; and Lopez et al23 described a constellation of vision changes, gait imbalance, and proximal weakness. Support for a pathophysiologic connection among these 3 cases is suggested by the finding of Hu antigen expression by MCC in 2 studies.24,25 Because MCC can present with occult lymph node involvement in the absence of primary cutaneous findings,3 there are more cases of paraneoplastic neurologic syndromes that were not recognized.
Extrapulmonary small cell carcinomas such as MCC are morphologically indistinguishable from their pulmonary counterparts and have been reported in most anatomic regions of the body, including gynecologic organs (eg, ovaries, cervix), genitourinary organs (eg, bladder, prostate), the gastrointestinal tract (eg, esophagus), skin (eg, MCC), and the head and neck region. Extrapulmonary small cell carcinoma is a rare entity, with the most common form found in the gynecologic tract, representing only 2% of gynecologic malignancies.26
Paraneoplastic syndromes of EPSCC are rare given the paucity of the malignancy. Several case reports discuss findings of SIADH in EPSCC of the cervix, as well as hypercalcemia, polyneuropathy, Cushing syndrome, limbic encephalitis, and peripheral neuropathy in EPSCC of the prostate.27,28 In contrast, SCLC has long been associated with paraneoplastic syndromes. Numerous case reports have been published describing SCLC-associated paraneoplastic syndromes to include hypercalcemia, Cushing syndrome, SIADH, vasoactive peptide production, cerebellar degeneration, limbic encephalitis, visceral plexopathy, autonomic dysfunction, and LEMS.29 As more cases of EPSCC with paraneoplastic syndromes are identified and reported, we might gain a better understanding of this interesting phenomenon.
Conclusion
Merkel cell carcinoma is an aggressive neuroendocrine malignancy associated with paraneoplastic neurologic syndromes, including LEMS. A thorough search for an underlying malignancy is highly recommended in patients with diagnosed LEMS without clear cause. Early identification and treatment of the primary tumor can lead to improvement of neurologic symptoms.
We present a case of LEMS with no clearly identifiable cause on presentation with later diagnosis of metastatic MCC of unknown primary origin. After surgical excision of affected lymph nodes and adjuvant radiation therapy, the patient had near-complete resolution of LEMS symptoms at 6-month follow-up, without additional findings of lymphadenopathy or skin lesions. Although this patient is not undergoing routine surveillance imaging to monitor for recurrence of MCC, a chest CT or positron emission tomography–CT for secondary screening would be considered if the patient experienced clinical symptoms consistent with LEMS.
In cases of LEMS without pulmonary malignancy, we recommend considering MCC in the differential diagnosis during the workup of an underlying malignancy
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2010;37:20-27.
- Senchenkov A, Moran SL. Merkel cell carcinoma: diagnosis, management, and outcomes. Plast Reconstr Surg. 2013;131:E771-E778.
- Han SY, North JP, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
- Vernino S. Paraneoplastic disorders affecting the neuromuscular junction or anterior horn cell. CONTINUUM Lifelong Learning in Neurology. 2009;15:132-146.
- Eggers SD, Salomao DR, Dinapoli RP, et al. Paraneoplastic and metastatic neurologic complications of Merkel cell carcinoma. Mayo Clin Proc. 2001;76:327-330.
- Siau RT, Morris A, Karoo RO. Surgery results in complete cure of Lambert-Eaton myasthenic syndrome in a patient with metastatic Merkel cell carcinoma. J Plast Reconstr Aesthet Surg. 2014;67:e162-e164.
- Bombelli F, Lispi L, Calabrò F, et al. Lambert-Eaton myasthenic syndrome associated to Merkel cell carcinoma: report of a case. Neurol Sci. 2015;36:1491-1492.
- Simmons DB, Duginski TM, McClean JC, et al. Lambert-eaton myasthenic syndrome and merkel cell carcinoma. Muscle Nerve. 2015;53:325-326.
- Bobos M, Hytiroglou P, Kostopoulos I, et al. Immunohistochemical distinction between Merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol. 2006;28:99-104.
- Jensen K, Kohler S, Rouse RV. Cytokeratin staining in Merkel cell carcinoma: an immunohistochemical study of cytokeratins 5/6, 7, 17, and 20. Appl Immunohistochem Mol Morphol. 2000;8:310-315.
- Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10:1098-1107.
- Sanders DB. Lambert-Eaton myasthenic syndrome. In: Daroff R, Aminoff MJ, eds. Encyclopedia of the Neurological Sciences. Vol 2. New York, NY: Elsevier; 2009:819-822.
- Wirtz PW, Smallegange TM, Wintzen AR, et al. Differences in clinical features between the Lambert-Eaton myasthenic syndrome with and without cancer: an analysis of 227 published cases. Clin Neurol Neurosurg. 2002;104:359-363.
- Titulaer MJ, Wirtz PW, Willems LN, et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008;26:4276-4281.
- Titulaer MJ, Maddison P, Sont JK, et al. Clinical Dutch-English Lambert-Eaton Myasthenic Syndrome (LEMS) Tumor Association prediction score accurately predicts small-cell lung cancer in the LEMS. J Clin Oncol. 2011;7:902-908.
- Cher LM, Hochberg FH, Teruya J, et al. Therapy for paraneoplastic neurologic syndromes in six patients with protein A column immunoadsorption. Cancer. 1995;75:1678-1683.
- Batchelor TT, Platten M, Hochberg FH. Immunoadsorption therapy for paraneoplastic syndromes. J Neurooncol. 1998;40:131-136.
- Blondin NA, Vortmeyer AO, Harel NY. Paraneoplastic syndrome of inappropriate antidiuretic hormone mimicking limbic encephalitis. Arch Neurol. 2011;68:1591-1594.
- Balegno S, Ceroni M, Corato M, et al. Antibodies to cerebellar nerve fibres in two patients with paraneoplastic cerebellar ataxia. Anticancer Res. 2005;25:3211-3214.
- Zhang C, Emery L, Lancaster E. Paraneoplastic cerebellar degeneration associated with noncutaneous Merkel cell carcinoma. Neurol Neuroimmunol Neuroinflamm. 2014;1:e17.
- Hocar O, Poszepczynska-Guigné E, Faye O, et al. Severe necrotizing myopathy subsequent to Merkel cell carcinoma. Ann Dermatol Venereol. 2011;138:130-134.
- Greenlee JE, Steffens JD, Clawson SA, et al. Anti-Hu antibodies in Merkel cell carcinoma. Ann Neurol. 2002;52:111-115.
- Lopez MC, Pericay C, Agustí M, et al. Merkel cell carcinoma associated with a paraneoplastic neurologic syndrome. Histopathology. 2004;44:628-629.
- Dalmau J, Furneaux HM, Cordon-Cardo C, et al. The expression of the Hu (paraneoplastic encephalomyelitis/sensory neuronopathy) antigen in human normal and tumor tissues. Am J Pathol. 1992;141:881-886.
- Gultekin SH, Rosai J, Demopoulos A, et al. Hu immunolabeling as a marker of neural and neuroendocrine differentiation in normal and neoplastic human tissues: assessment using a recombinant anti-Hu Fab fragment. Int J Surg Pathol. 2000;8:109-117.
- Zheng X, Liu D, Fallon JT, et al. Distinct genetic alterations in small cell carcinoma from different anatomic sites. Exp Hematol Oncol. 2015;4:2.
- Kim D, Yun H, Lee Y, et al. Small cell neuroendocrine carcinoma of the uterine cervix presenting with syndrome of inappropriate antidiuretic hormone secretion. Obstet Gynecol Sci. 2013;56:420-425.
- Venkatesh PK, Motwani B, Sherman N, et al. Metastatic pure small-cell carcinoma of prostate. Am J Med Sci. 2004;328:286-289.
- Kaltsas G, Androulakis II, de Herder WW, et al. Paraneoplastic syndromes secondary to neuroendocrine tumours. Endocr Relat Cancer. 2010;17:R173-R193.
Merkel cell carcinoma (MCC) is an aggressive neuroendocrine malignancy of the skin that is thought to arise from neural crest cells. It has an estimated annual incidence of 0.6 per 100,000 individuals, typically occurs in the elderly population, and is most common in white males.1 The tumor presents as a rapidly growing, violaceous nodule in sun-exposed areas of the skin; early in the course, it can be mistaken for a benign entity such as an epidermal cyst.2 Merkel cell carcinoma has a propensity to spread to regional lymph nodes, and in some cases, it occurs in the absence of skin findings.3 Histologically, MCC is nearly indistinguishable from small cell lung carcinoma (SCLC).4 The overall prognosis for patients with MCC is poor and largely dependent on the stage at diagnosis. Patients with regional and distant metastases have a 5-year survival rate of 26% to 42% and 18%, respectively.3
Lambert-Eaton myasthenic syndrome (LEMS) is a paraneoplastic or autoimmune disorder of the neuromuscular junction that is found in 3% of cases of SCLC.4 Reported cases of LEMS in patients with MCC are exceedingly rare.5-8 We provide a full report and longitudinal clinical follow-up of a case that was briefly discussed by Simmons et al,8 and we review the literature regarding paraneoplastic syndromes associated with MCC and other extrapulmonary small cell carcinomas (EPSCCs).
Case Report
A 63-year-old man was evaluated in the neurology clinic due to difficulty walking, climbing stairs, and performing push-ups over the last month. Prior to the onset of symptoms, he was otherwise healthy, walking 3 miles daily; however, at presentation he required use of a cane. Leg weakness worsened as the day progressed. In addition, he reported constipation, urinary urgency, dry mouth, mild dysphagia, reduced sensation below the knees, and a nasal quality in his speech. He had no ptosis, diplopia, dysarthria, muscle cramps, myalgia, or facial weakness. He denied fevers, chills, and night sweats but did admit to an unintentional 10- to 15-lb weight loss over the preceding few months.
The neurologic examination revealed mild proximal upper extremity weakness in the bilateral shoulder abductors, infraspinatus, hip extensors, and hip flexors (Medical Research Council muscle scale grade 4). All deep tendon reflexes, except the Achilles reflex, were present. Despite subjective sensory concerns, objective examination of all sensory modalities was normal. Cranial nerve examination was normal, except for a slight nasal quality to his voice.
A qualitative assay was positive for the presence of P/Q-type voltage-gated calcium channel (VGCC) antibodies. Other laboratory studies were within reference range, including acetylcholine-receptor antibodies (blocking, binding, and modulating) and muscle-specific kinase antibodies.
Lumbar and cervical spine magnetic resonance imaging revealed multilevel neuroforaminal stenosis without spinal canal stenosis or myelopathy. Computed tomography (CT) of the chest was notable for 2 pathologically enlarged lymph nodes in the left axilla and no evidence of primary pulmonary malignancy. Nerve-conduction studies (NCSs) in conjunction with other clinical findings were consistent with the diagnosis of LEMS.
Ultrasound-guided biopsy of the enlarged axillary lymph nodes demonstrated sheets and nests of small round blue tumor cells with minimal cytoplasm, high mitotic rate, and foci of necrosis (Figure 1). The tumor cells were positive for pancytokeratin (Lu-5) and cytokeratin (CK) 20 in a perinuclear dotlike pattern (Figure 2), as well as for the neuroendocrine markers synaptophysin (Figure 3), chromogranin A, and CD56. The tumor cells showed no immunoreactivity for CK7, thyroid transcription factor 1, CD3, CD5, or CD20. Flow cytometry demonstrated low cellularity, low specimen viability, and no evidence of an abnormal B-cell population. These findings were consistent with the diagnosis of MCC.
The patient underwent surgical excision of the involved lymph nodes. Four weeks after surgery, he reported dramatic improvement in strength, with complete resolution of the nasal speech, dysphagia, dry mouth, urinary retention, and constipation. Two months after surgery, his strength had normalized, except for slight persistent weakness in the bilateral shoulder abductors, trace weakness in the hip flexors, and a slight Trendelenburg gait. He was able to rise from a chair without using his arms and no longer required a cane for ambulation.
The patient underwent adjuvant radiation therapy after 2-month surgical follow-up with 5000-cGy radiation treatment to the left axillary region. Six months following primary definitive surgery and 4 months following adjuvant radiation therapy, he reported a 95% subjective return of physical strength. The patient was able to return to near-baseline physical activity. He continued to deny symptoms of dry mouth, incontinence, or constipation. Objectively, he had no focal neurologic deficits or weakness; no evidence of new skin lesions or lymphadenopathy was noted.
Comment
MCC vs SCLC
Merkel cell carcinoma is classified as a type of EPSCC. The histologic appearance of MCC is indistinguishable from SCLC. Both tumors are composed of uniform sheets of small round cells with a high nucleus to cytoplasm ratio, and both can express neuroendocrine markers, such as neuron-specific enolase, chromogranin A, and synaptophysin.9 Immunohistochemical positivity for CK20 and neurofilaments in combination with negative staining for thyroid transcription factor 1 and CK7 effectively differentiate MCC from SCLC.9 In addition, MCC often displays CK20 positivity in a perinuclear dotlike or punctate pattern, which is characteristic of this tumor.3,9,10 Negative immunohistochemical markers for B cells (CD20) and T cells (CD3) are important in excluding lymphoma.
LEMS Diagnosis
Lambert-Eaton myasthenic syndrome is a paraneoplastic or autoimmune disorder involving the neuromuscular junction. Autoantibodies to VGCC impair calcium influx into the presynaptic terminal, resulting in marked reduction of acetylcholine release into the synaptic cleft. The reduction in acetylcholine activity impairs production of muscle fiber action potentials, resulting in clinical weakness. The diagnosis of LEMS rests on clinical presentation, positive serology, and confirmatory neurophysiologic testing by NCS. Clinically, patients present with proximal weakness, hyporeflexia or areflexia, and autonomic dysfunction. Antibodies to P/Q-type VGCCs are found in 85% to 90% of cases of LEMS and are thought to play a direct causative role in the development of weakness.11 The finding of postexercise facilitation on motor NCS is the neurophysiologic hallmark and is highly specific for the diagnosis.
Approximately 50% to 60% of patients who present with LEMS have an underlying tumor, the vast majority of which are SCLC.11 There are a few reports of LEMS associated with other malignancies, including lymphoma; thymoma; neuroblastoma; and carcinoma of the breast, stomach, prostate, bladder, kidney, and gallbladder.12 Patients with nontumor or autoimmune LEMS tend to be younger, and there is no male predominance, as there is in paraneoplastic LEMS.13 Given the risk of underlying malignancy in LEMS, Titulaer et al14 proposed a screening protocol for patients presenting with LEMS, recommending initial primary screening using CT of the chest. If the CT scan is negative, total-body fludeoxyglucose positron emission tomography should be performed to assess for fludeoxyglucose avid lesions. If both initial studies are negative, routine follow-up with CT of the chest at 6-month intervals for a minimum of 2 to 4 years after the initial diagnosis of LEMS was recommended. An exception to this protocol was suggested to allow consideration to stop screening after the first 6-month follow-up chest CT for patients younger than 45 years who have never smoked and who have an HLA 8.1 haplotype for which nontumor LEMS would be a more probable diagnosis.14
In addition to a screening protocol, a validated prediction tool, the Dutch-English LEMS Tumor Association prediction score, was developed. It uses common signs and symptoms of LEMS and risk factors for SCLC to help guide the need for further screening.15
Paraneoplastic Syndromes Associated With MCC
Other paraneoplastic syndromes have been reported in association with MCC. A patient with brainstem encephalitis associated with MCC was reported in a trial of a novel immunotherapy for paraneoplastic neurologic syndromes.16,17 A syndrome of inappropriate antidiuretic hormone (SIADH) secretion was reported in a patient with N-type calcium channel antibodies.18 Two cases of paraneoplastic cerebellar degeneration have been reported; the first was associated with a novel 70-kD antibody,19 and the second was associated with the P/Q-type VGCC antibody.20 Anti-Hu antibodies have been found in a handful of reports of neurologic deterioration in patients with MCC. Hocar et al21 reported a severe necrotizing myopathy; Greenlee et al22 described a syndrome of progressive sensorimotor and autonomic neuropathy with encephalopathy; and Lopez et al23 described a constellation of vision changes, gait imbalance, and proximal weakness. Support for a pathophysiologic connection among these 3 cases is suggested by the finding of Hu antigen expression by MCC in 2 studies.24,25 Because MCC can present with occult lymph node involvement in the absence of primary cutaneous findings,3 there are more cases of paraneoplastic neurologic syndromes that were not recognized.
Extrapulmonary small cell carcinomas such as MCC are morphologically indistinguishable from their pulmonary counterparts and have been reported in most anatomic regions of the body, including gynecologic organs (eg, ovaries, cervix), genitourinary organs (eg, bladder, prostate), the gastrointestinal tract (eg, esophagus), skin (eg, MCC), and the head and neck region. Extrapulmonary small cell carcinoma is a rare entity, with the most common form found in the gynecologic tract, representing only 2% of gynecologic malignancies.26
Paraneoplastic syndromes of EPSCC are rare given the paucity of the malignancy. Several case reports discuss findings of SIADH in EPSCC of the cervix, as well as hypercalcemia, polyneuropathy, Cushing syndrome, limbic encephalitis, and peripheral neuropathy in EPSCC of the prostate.27,28 In contrast, SCLC has long been associated with paraneoplastic syndromes. Numerous case reports have been published describing SCLC-associated paraneoplastic syndromes to include hypercalcemia, Cushing syndrome, SIADH, vasoactive peptide production, cerebellar degeneration, limbic encephalitis, visceral plexopathy, autonomic dysfunction, and LEMS.29 As more cases of EPSCC with paraneoplastic syndromes are identified and reported, we might gain a better understanding of this interesting phenomenon.
Conclusion
Merkel cell carcinoma is an aggressive neuroendocrine malignancy associated with paraneoplastic neurologic syndromes, including LEMS. A thorough search for an underlying malignancy is highly recommended in patients with diagnosed LEMS without clear cause. Early identification and treatment of the primary tumor can lead to improvement of neurologic symptoms.
We present a case of LEMS with no clearly identifiable cause on presentation with later diagnosis of metastatic MCC of unknown primary origin. After surgical excision of affected lymph nodes and adjuvant radiation therapy, the patient had near-complete resolution of LEMS symptoms at 6-month follow-up, without additional findings of lymphadenopathy or skin lesions. Although this patient is not undergoing routine surveillance imaging to monitor for recurrence of MCC, a chest CT or positron emission tomography–CT for secondary screening would be considered if the patient experienced clinical symptoms consistent with LEMS.
In cases of LEMS without pulmonary malignancy, we recommend considering MCC in the differential diagnosis during the workup of an underlying malignancy
Merkel cell carcinoma (MCC) is an aggressive neuroendocrine malignancy of the skin that is thought to arise from neural crest cells. It has an estimated annual incidence of 0.6 per 100,000 individuals, typically occurs in the elderly population, and is most common in white males.1 The tumor presents as a rapidly growing, violaceous nodule in sun-exposed areas of the skin; early in the course, it can be mistaken for a benign entity such as an epidermal cyst.2 Merkel cell carcinoma has a propensity to spread to regional lymph nodes, and in some cases, it occurs in the absence of skin findings.3 Histologically, MCC is nearly indistinguishable from small cell lung carcinoma (SCLC).4 The overall prognosis for patients with MCC is poor and largely dependent on the stage at diagnosis. Patients with regional and distant metastases have a 5-year survival rate of 26% to 42% and 18%, respectively.3
Lambert-Eaton myasthenic syndrome (LEMS) is a paraneoplastic or autoimmune disorder of the neuromuscular junction that is found in 3% of cases of SCLC.4 Reported cases of LEMS in patients with MCC are exceedingly rare.5-8 We provide a full report and longitudinal clinical follow-up of a case that was briefly discussed by Simmons et al,8 and we review the literature regarding paraneoplastic syndromes associated with MCC and other extrapulmonary small cell carcinomas (EPSCCs).
Case Report
A 63-year-old man was evaluated in the neurology clinic due to difficulty walking, climbing stairs, and performing push-ups over the last month. Prior to the onset of symptoms, he was otherwise healthy, walking 3 miles daily; however, at presentation he required use of a cane. Leg weakness worsened as the day progressed. In addition, he reported constipation, urinary urgency, dry mouth, mild dysphagia, reduced sensation below the knees, and a nasal quality in his speech. He had no ptosis, diplopia, dysarthria, muscle cramps, myalgia, or facial weakness. He denied fevers, chills, and night sweats but did admit to an unintentional 10- to 15-lb weight loss over the preceding few months.
The neurologic examination revealed mild proximal upper extremity weakness in the bilateral shoulder abductors, infraspinatus, hip extensors, and hip flexors (Medical Research Council muscle scale grade 4). All deep tendon reflexes, except the Achilles reflex, were present. Despite subjective sensory concerns, objective examination of all sensory modalities was normal. Cranial nerve examination was normal, except for a slight nasal quality to his voice.
A qualitative assay was positive for the presence of P/Q-type voltage-gated calcium channel (VGCC) antibodies. Other laboratory studies were within reference range, including acetylcholine-receptor antibodies (blocking, binding, and modulating) and muscle-specific kinase antibodies.
Lumbar and cervical spine magnetic resonance imaging revealed multilevel neuroforaminal stenosis without spinal canal stenosis or myelopathy. Computed tomography (CT) of the chest was notable for 2 pathologically enlarged lymph nodes in the left axilla and no evidence of primary pulmonary malignancy. Nerve-conduction studies (NCSs) in conjunction with other clinical findings were consistent with the diagnosis of LEMS.
Ultrasound-guided biopsy of the enlarged axillary lymph nodes demonstrated sheets and nests of small round blue tumor cells with minimal cytoplasm, high mitotic rate, and foci of necrosis (Figure 1). The tumor cells were positive for pancytokeratin (Lu-5) and cytokeratin (CK) 20 in a perinuclear dotlike pattern (Figure 2), as well as for the neuroendocrine markers synaptophysin (Figure 3), chromogranin A, and CD56. The tumor cells showed no immunoreactivity for CK7, thyroid transcription factor 1, CD3, CD5, or CD20. Flow cytometry demonstrated low cellularity, low specimen viability, and no evidence of an abnormal B-cell population. These findings were consistent with the diagnosis of MCC.
The patient underwent surgical excision of the involved lymph nodes. Four weeks after surgery, he reported dramatic improvement in strength, with complete resolution of the nasal speech, dysphagia, dry mouth, urinary retention, and constipation. Two months after surgery, his strength had normalized, except for slight persistent weakness in the bilateral shoulder abductors, trace weakness in the hip flexors, and a slight Trendelenburg gait. He was able to rise from a chair without using his arms and no longer required a cane for ambulation.
The patient underwent adjuvant radiation therapy after 2-month surgical follow-up with 5000-cGy radiation treatment to the left axillary region. Six months following primary definitive surgery and 4 months following adjuvant radiation therapy, he reported a 95% subjective return of physical strength. The patient was able to return to near-baseline physical activity. He continued to deny symptoms of dry mouth, incontinence, or constipation. Objectively, he had no focal neurologic deficits or weakness; no evidence of new skin lesions or lymphadenopathy was noted.
Comment
MCC vs SCLC
Merkel cell carcinoma is classified as a type of EPSCC. The histologic appearance of MCC is indistinguishable from SCLC. Both tumors are composed of uniform sheets of small round cells with a high nucleus to cytoplasm ratio, and both can express neuroendocrine markers, such as neuron-specific enolase, chromogranin A, and synaptophysin.9 Immunohistochemical positivity for CK20 and neurofilaments in combination with negative staining for thyroid transcription factor 1 and CK7 effectively differentiate MCC from SCLC.9 In addition, MCC often displays CK20 positivity in a perinuclear dotlike or punctate pattern, which is characteristic of this tumor.3,9,10 Negative immunohistochemical markers for B cells (CD20) and T cells (CD3) are important in excluding lymphoma.
LEMS Diagnosis
Lambert-Eaton myasthenic syndrome is a paraneoplastic or autoimmune disorder involving the neuromuscular junction. Autoantibodies to VGCC impair calcium influx into the presynaptic terminal, resulting in marked reduction of acetylcholine release into the synaptic cleft. The reduction in acetylcholine activity impairs production of muscle fiber action potentials, resulting in clinical weakness. The diagnosis of LEMS rests on clinical presentation, positive serology, and confirmatory neurophysiologic testing by NCS. Clinically, patients present with proximal weakness, hyporeflexia or areflexia, and autonomic dysfunction. Antibodies to P/Q-type VGCCs are found in 85% to 90% of cases of LEMS and are thought to play a direct causative role in the development of weakness.11 The finding of postexercise facilitation on motor NCS is the neurophysiologic hallmark and is highly specific for the diagnosis.
Approximately 50% to 60% of patients who present with LEMS have an underlying tumor, the vast majority of which are SCLC.11 There are a few reports of LEMS associated with other malignancies, including lymphoma; thymoma; neuroblastoma; and carcinoma of the breast, stomach, prostate, bladder, kidney, and gallbladder.12 Patients with nontumor or autoimmune LEMS tend to be younger, and there is no male predominance, as there is in paraneoplastic LEMS.13 Given the risk of underlying malignancy in LEMS, Titulaer et al14 proposed a screening protocol for patients presenting with LEMS, recommending initial primary screening using CT of the chest. If the CT scan is negative, total-body fludeoxyglucose positron emission tomography should be performed to assess for fludeoxyglucose avid lesions. If both initial studies are negative, routine follow-up with CT of the chest at 6-month intervals for a minimum of 2 to 4 years after the initial diagnosis of LEMS was recommended. An exception to this protocol was suggested to allow consideration to stop screening after the first 6-month follow-up chest CT for patients younger than 45 years who have never smoked and who have an HLA 8.1 haplotype for which nontumor LEMS would be a more probable diagnosis.14
In addition to a screening protocol, a validated prediction tool, the Dutch-English LEMS Tumor Association prediction score, was developed. It uses common signs and symptoms of LEMS and risk factors for SCLC to help guide the need for further screening.15
Paraneoplastic Syndromes Associated With MCC
Other paraneoplastic syndromes have been reported in association with MCC. A patient with brainstem encephalitis associated with MCC was reported in a trial of a novel immunotherapy for paraneoplastic neurologic syndromes.16,17 A syndrome of inappropriate antidiuretic hormone (SIADH) secretion was reported in a patient with N-type calcium channel antibodies.18 Two cases of paraneoplastic cerebellar degeneration have been reported; the first was associated with a novel 70-kD antibody,19 and the second was associated with the P/Q-type VGCC antibody.20 Anti-Hu antibodies have been found in a handful of reports of neurologic deterioration in patients with MCC. Hocar et al21 reported a severe necrotizing myopathy; Greenlee et al22 described a syndrome of progressive sensorimotor and autonomic neuropathy with encephalopathy; and Lopez et al23 described a constellation of vision changes, gait imbalance, and proximal weakness. Support for a pathophysiologic connection among these 3 cases is suggested by the finding of Hu antigen expression by MCC in 2 studies.24,25 Because MCC can present with occult lymph node involvement in the absence of primary cutaneous findings,3 there are more cases of paraneoplastic neurologic syndromes that were not recognized.
Extrapulmonary small cell carcinomas such as MCC are morphologically indistinguishable from their pulmonary counterparts and have been reported in most anatomic regions of the body, including gynecologic organs (eg, ovaries, cervix), genitourinary organs (eg, bladder, prostate), the gastrointestinal tract (eg, esophagus), skin (eg, MCC), and the head and neck region. Extrapulmonary small cell carcinoma is a rare entity, with the most common form found in the gynecologic tract, representing only 2% of gynecologic malignancies.26
Paraneoplastic syndromes of EPSCC are rare given the paucity of the malignancy. Several case reports discuss findings of SIADH in EPSCC of the cervix, as well as hypercalcemia, polyneuropathy, Cushing syndrome, limbic encephalitis, and peripheral neuropathy in EPSCC of the prostate.27,28 In contrast, SCLC has long been associated with paraneoplastic syndromes. Numerous case reports have been published describing SCLC-associated paraneoplastic syndromes to include hypercalcemia, Cushing syndrome, SIADH, vasoactive peptide production, cerebellar degeneration, limbic encephalitis, visceral plexopathy, autonomic dysfunction, and LEMS.29 As more cases of EPSCC with paraneoplastic syndromes are identified and reported, we might gain a better understanding of this interesting phenomenon.
Conclusion
Merkel cell carcinoma is an aggressive neuroendocrine malignancy associated with paraneoplastic neurologic syndromes, including LEMS. A thorough search for an underlying malignancy is highly recommended in patients with diagnosed LEMS without clear cause. Early identification and treatment of the primary tumor can lead to improvement of neurologic symptoms.
We present a case of LEMS with no clearly identifiable cause on presentation with later diagnosis of metastatic MCC of unknown primary origin. After surgical excision of affected lymph nodes and adjuvant radiation therapy, the patient had near-complete resolution of LEMS symptoms at 6-month follow-up, without additional findings of lymphadenopathy or skin lesions. Although this patient is not undergoing routine surveillance imaging to monitor for recurrence of MCC, a chest CT or positron emission tomography–CT for secondary screening would be considered if the patient experienced clinical symptoms consistent with LEMS.
In cases of LEMS without pulmonary malignancy, we recommend considering MCC in the differential diagnosis during the workup of an underlying malignancy
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2010;37:20-27.
- Senchenkov A, Moran SL. Merkel cell carcinoma: diagnosis, management, and outcomes. Plast Reconstr Surg. 2013;131:E771-E778.
- Han SY, North JP, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
- Vernino S. Paraneoplastic disorders affecting the neuromuscular junction or anterior horn cell. CONTINUUM Lifelong Learning in Neurology. 2009;15:132-146.
- Eggers SD, Salomao DR, Dinapoli RP, et al. Paraneoplastic and metastatic neurologic complications of Merkel cell carcinoma. Mayo Clin Proc. 2001;76:327-330.
- Siau RT, Morris A, Karoo RO. Surgery results in complete cure of Lambert-Eaton myasthenic syndrome in a patient with metastatic Merkel cell carcinoma. J Plast Reconstr Aesthet Surg. 2014;67:e162-e164.
- Bombelli F, Lispi L, Calabrò F, et al. Lambert-Eaton myasthenic syndrome associated to Merkel cell carcinoma: report of a case. Neurol Sci. 2015;36:1491-1492.
- Simmons DB, Duginski TM, McClean JC, et al. Lambert-eaton myasthenic syndrome and merkel cell carcinoma. Muscle Nerve. 2015;53:325-326.
- Bobos M, Hytiroglou P, Kostopoulos I, et al. Immunohistochemical distinction between Merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol. 2006;28:99-104.
- Jensen K, Kohler S, Rouse RV. Cytokeratin staining in Merkel cell carcinoma: an immunohistochemical study of cytokeratins 5/6, 7, 17, and 20. Appl Immunohistochem Mol Morphol. 2000;8:310-315.
- Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10:1098-1107.
- Sanders DB. Lambert-Eaton myasthenic syndrome. In: Daroff R, Aminoff MJ, eds. Encyclopedia of the Neurological Sciences. Vol 2. New York, NY: Elsevier; 2009:819-822.
- Wirtz PW, Smallegange TM, Wintzen AR, et al. Differences in clinical features between the Lambert-Eaton myasthenic syndrome with and without cancer: an analysis of 227 published cases. Clin Neurol Neurosurg. 2002;104:359-363.
- Titulaer MJ, Wirtz PW, Willems LN, et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008;26:4276-4281.
- Titulaer MJ, Maddison P, Sont JK, et al. Clinical Dutch-English Lambert-Eaton Myasthenic Syndrome (LEMS) Tumor Association prediction score accurately predicts small-cell lung cancer in the LEMS. J Clin Oncol. 2011;7:902-908.
- Cher LM, Hochberg FH, Teruya J, et al. Therapy for paraneoplastic neurologic syndromes in six patients with protein A column immunoadsorption. Cancer. 1995;75:1678-1683.
- Batchelor TT, Platten M, Hochberg FH. Immunoadsorption therapy for paraneoplastic syndromes. J Neurooncol. 1998;40:131-136.
- Blondin NA, Vortmeyer AO, Harel NY. Paraneoplastic syndrome of inappropriate antidiuretic hormone mimicking limbic encephalitis. Arch Neurol. 2011;68:1591-1594.
- Balegno S, Ceroni M, Corato M, et al. Antibodies to cerebellar nerve fibres in two patients with paraneoplastic cerebellar ataxia. Anticancer Res. 2005;25:3211-3214.
- Zhang C, Emery L, Lancaster E. Paraneoplastic cerebellar degeneration associated with noncutaneous Merkel cell carcinoma. Neurol Neuroimmunol Neuroinflamm. 2014;1:e17.
- Hocar O, Poszepczynska-Guigné E, Faye O, et al. Severe necrotizing myopathy subsequent to Merkel cell carcinoma. Ann Dermatol Venereol. 2011;138:130-134.
- Greenlee JE, Steffens JD, Clawson SA, et al. Anti-Hu antibodies in Merkel cell carcinoma. Ann Neurol. 2002;52:111-115.
- Lopez MC, Pericay C, Agustí M, et al. Merkel cell carcinoma associated with a paraneoplastic neurologic syndrome. Histopathology. 2004;44:628-629.
- Dalmau J, Furneaux HM, Cordon-Cardo C, et al. The expression of the Hu (paraneoplastic encephalomyelitis/sensory neuronopathy) antigen in human normal and tumor tissues. Am J Pathol. 1992;141:881-886.
- Gultekin SH, Rosai J, Demopoulos A, et al. Hu immunolabeling as a marker of neural and neuroendocrine differentiation in normal and neoplastic human tissues: assessment using a recombinant anti-Hu Fab fragment. Int J Surg Pathol. 2000;8:109-117.
- Zheng X, Liu D, Fallon JT, et al. Distinct genetic alterations in small cell carcinoma from different anatomic sites. Exp Hematol Oncol. 2015;4:2.
- Kim D, Yun H, Lee Y, et al. Small cell neuroendocrine carcinoma of the uterine cervix presenting with syndrome of inappropriate antidiuretic hormone secretion. Obstet Gynecol Sci. 2013;56:420-425.
- Venkatesh PK, Motwani B, Sherman N, et al. Metastatic pure small-cell carcinoma of prostate. Am J Med Sci. 2004;328:286-289.
- Kaltsas G, Androulakis II, de Herder WW, et al. Paraneoplastic syndromes secondary to neuroendocrine tumours. Endocr Relat Cancer. 2010;17:R173-R193.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2010;37:20-27.
- Senchenkov A, Moran SL. Merkel cell carcinoma: diagnosis, management, and outcomes. Plast Reconstr Surg. 2013;131:E771-E778.
- Han SY, North JP, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
- Vernino S. Paraneoplastic disorders affecting the neuromuscular junction or anterior horn cell. CONTINUUM Lifelong Learning in Neurology. 2009;15:132-146.
- Eggers SD, Salomao DR, Dinapoli RP, et al. Paraneoplastic and metastatic neurologic complications of Merkel cell carcinoma. Mayo Clin Proc. 2001;76:327-330.
- Siau RT, Morris A, Karoo RO. Surgery results in complete cure of Lambert-Eaton myasthenic syndrome in a patient with metastatic Merkel cell carcinoma. J Plast Reconstr Aesthet Surg. 2014;67:e162-e164.
- Bombelli F, Lispi L, Calabrò F, et al. Lambert-Eaton myasthenic syndrome associated to Merkel cell carcinoma: report of a case. Neurol Sci. 2015;36:1491-1492.
- Simmons DB, Duginski TM, McClean JC, et al. Lambert-eaton myasthenic syndrome and merkel cell carcinoma. Muscle Nerve. 2015;53:325-326.
- Bobos M, Hytiroglou P, Kostopoulos I, et al. Immunohistochemical distinction between Merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol. 2006;28:99-104.
- Jensen K, Kohler S, Rouse RV. Cytokeratin staining in Merkel cell carcinoma: an immunohistochemical study of cytokeratins 5/6, 7, 17, and 20. Appl Immunohistochem Mol Morphol. 2000;8:310-315.
- Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10:1098-1107.
- Sanders DB. Lambert-Eaton myasthenic syndrome. In: Daroff R, Aminoff MJ, eds. Encyclopedia of the Neurological Sciences. Vol 2. New York, NY: Elsevier; 2009:819-822.
- Wirtz PW, Smallegange TM, Wintzen AR, et al. Differences in clinical features between the Lambert-Eaton myasthenic syndrome with and without cancer: an analysis of 227 published cases. Clin Neurol Neurosurg. 2002;104:359-363.
- Titulaer MJ, Wirtz PW, Willems LN, et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008;26:4276-4281.
- Titulaer MJ, Maddison P, Sont JK, et al. Clinical Dutch-English Lambert-Eaton Myasthenic Syndrome (LEMS) Tumor Association prediction score accurately predicts small-cell lung cancer in the LEMS. J Clin Oncol. 2011;7:902-908.
- Cher LM, Hochberg FH, Teruya J, et al. Therapy for paraneoplastic neurologic syndromes in six patients with protein A column immunoadsorption. Cancer. 1995;75:1678-1683.
- Batchelor TT, Platten M, Hochberg FH. Immunoadsorption therapy for paraneoplastic syndromes. J Neurooncol. 1998;40:131-136.
- Blondin NA, Vortmeyer AO, Harel NY. Paraneoplastic syndrome of inappropriate antidiuretic hormone mimicking limbic encephalitis. Arch Neurol. 2011;68:1591-1594.
- Balegno S, Ceroni M, Corato M, et al. Antibodies to cerebellar nerve fibres in two patients with paraneoplastic cerebellar ataxia. Anticancer Res. 2005;25:3211-3214.
- Zhang C, Emery L, Lancaster E. Paraneoplastic cerebellar degeneration associated with noncutaneous Merkel cell carcinoma. Neurol Neuroimmunol Neuroinflamm. 2014;1:e17.
- Hocar O, Poszepczynska-Guigné E, Faye O, et al. Severe necrotizing myopathy subsequent to Merkel cell carcinoma. Ann Dermatol Venereol. 2011;138:130-134.
- Greenlee JE, Steffens JD, Clawson SA, et al. Anti-Hu antibodies in Merkel cell carcinoma. Ann Neurol. 2002;52:111-115.
- Lopez MC, Pericay C, Agustí M, et al. Merkel cell carcinoma associated with a paraneoplastic neurologic syndrome. Histopathology. 2004;44:628-629.
- Dalmau J, Furneaux HM, Cordon-Cardo C, et al. The expression of the Hu (paraneoplastic encephalomyelitis/sensory neuronopathy) antigen in human normal and tumor tissues. Am J Pathol. 1992;141:881-886.
- Gultekin SH, Rosai J, Demopoulos A, et al. Hu immunolabeling as a marker of neural and neuroendocrine differentiation in normal and neoplastic human tissues: assessment using a recombinant anti-Hu Fab fragment. Int J Surg Pathol. 2000;8:109-117.
- Zheng X, Liu D, Fallon JT, et al. Distinct genetic alterations in small cell carcinoma from different anatomic sites. Exp Hematol Oncol. 2015;4:2.
- Kim D, Yun H, Lee Y, et al. Small cell neuroendocrine carcinoma of the uterine cervix presenting with syndrome of inappropriate antidiuretic hormone secretion. Obstet Gynecol Sci. 2013;56:420-425.
- Venkatesh PK, Motwani B, Sherman N, et al. Metastatic pure small-cell carcinoma of prostate. Am J Med Sci. 2004;328:286-289.
- Kaltsas G, Androulakis II, de Herder WW, et al. Paraneoplastic syndromes secondary to neuroendocrine tumours. Endocr Relat Cancer. 2010;17:R173-R193.
Practice Points
- Approximately 50% to 60% of patients with Lambert-Eaton myasthenic syndrome (LEMS) have an underlying tumor, most commonly small cell lung carcinoma.
- A thorough search for an underlying malignancy is highly recommended in patients with diagnosed LEMS without clear cause; to this end, a screening protocol comprising computed tomography and total-body fludeoxyglucose positron emission tomography has been established.
- Because Merkel cell carcinoma (MCC) can present as occult lymph node involvement with primary cutaneous findings absent, it is recommended that MCC be considered in the differential diagnosis of an underlying malignancy in a LEMS patient.
- Early identification and treatment of the primary tumor can lead to improvement of neurologic symptoms.
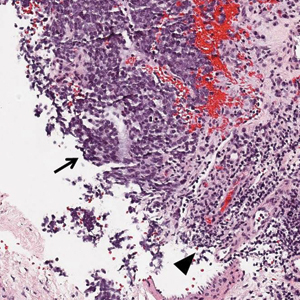
Hyperextension of the bilateral knees in a 1-day-old neonate • no knee fractures or dislocation on x-ray • Dx?
THE CASE
A 29-year-old G7P2315 woman gave birth to a girl at 37 weeks via spontaneous vaginal delivery. APGAR scores were 9 and 9. Birth weight was 2760 g. Cardiovascular and pulmonary examinations were normal (heart rate, 154 beats/min; respiratory rate, 52 breaths/min). Following delivery, the neonate appeared healthy, had a lusty cry, and had no visible craniofacial or cutaneous abnormalities; however, the bilateral knees were hyperextended to 90° to 110° (FIGURE 1A).

The mother had started prenatal care at 7 weeks with 10 total visits to her family physician (JD) throughout the pregnancy. Routine laboratory screening and prenatal ultrasounds (including an anatomy scan) were normal. She had a history of 3 preterm deliveries at 35 weeks, 36 weeks, and 36 weeks, respectively, and had been on progesterone shots once weekly starting at 18 weeks during the current pregnancy. She had no history of infections or recent travel. Her family history was remarkable for a sister who gave birth to a child with
THE DIAGNOSIS
The neonate tolerated passive flexion of the knees to a neutral position. Hip examination demonstrated appropriate range of movement with negative Ortolani and Barlow tests. The infant’s feet aligned correctly, with toes in the front and heels in the back, and an x-ray of the bilateral knees showed no fractures or dislocation.
Based on the clinical examination and x-ray findings, we made a diagnosis of congenital genu recurvatum. A pediatric orthopedics consultation was obtained, and the knees were placed in short leg splints in comfortable flexion to neutral on Day 1 of life. She was discharged the next day.
DISCUSSION
Congenital genu recurvatum, also known as congenital dislocation of the knee, is a rare condition involving abnormal hyperextension of the unilateral or bilateral knees with limited flexion.1 Reports in the literature are limited, but there seems to be a female predominance among known cases of congenital genu recurvatum.2 The clinical presentation varies. Finding may be isolated to the knee(s) but also can present in association with other congenital abnormalities, such as developmental dysplasia of the hip, clubfoot, and hindfoot and forefoot deformities.3,4
Diagnosis is made clinically with radiographic imaging
Diagnosis of congenital genu recurvatum is made clinically and can be confirmed via radiographic imaging of the knees.5 Clinical diagnosis requires assessment of the degree of hyperextension and palpation of the femoral condyles, which become more prominent as the severity of the hyperextension increases.6 X-rays help assess if a true dislocation or subluxation of the tibia on the femur has occurred. Based on the clinical and radiographic findings, congenital genu recurvatum typically is classified according to 3 levels of severity: grade 1 classification only involves hyperextension of the knees without dislocation or subluxation, grade 2 involves the same characteristic hyperextension along with anterior subluxation of the tibia on the femur, and grade 3 includes hyperextension with true dislocation of the tibia on the femur.1 Grades 1 and 2 on this spectrum technically are diagnosed as congenital genu recurvatum while grade 3 is diagnosed as a congenital dislocation of the knee,7 although the 2 terms are used interchangeably in the literature. We classified our case as a grade 1 congenital genu recurvatum based on the clinical and radiographic findings.
Congenital knee hyperextension has intrinsic and extrinsic causes
Hyperextension of the knees at birth may be caused by various intrinsic or extrinsic factors. Intrinsic causes may include breech position, lack of intrauterine space, trauma to the mother, quadriceps contracture or fibrosis, absence of the suprapatellar pouch, deficient or hypoplastic anterior cruciate ligament, pathological tissues, arthrogryposis, or genetic disorders such as Larsen syndrome or achondroplasia.6
Continue to: Extrinsic causes...
Extrinsic causes may include traumatic dislocation during the birthing process3 or intrauterine pressure leading to malposition of the joints. When intrauterine pressure is combined with reduced intrauterine space, this phenomenon is known as packaging disorder.6 Entanglement of the umbilical cord around the legs of the fetus during development may be another potential factor.1
The exact etiology in our patient was unknown, but we determined the cause was extrinsic based on the lack of other genetic abnormalities. We initially considered a possible connection between our patient’s diagnosis and her family history of thrombocytopenia absent radius syndrome, but it was later determined that both were isolated cases and the limb abnormalities were coincidental.
Treatment options and outcomes for extrinsic and intrinsic etiologies depend on the severity of the hyperextension and any associated abnormalities, as well as the time in which therapy is initiated.1 Reduction of the hyperextension within 24 hours of birth has been associated with excellent outcomes.8 Regardless of the cause, all cases of congenital genu recurvatum should first be treated conservatively. Evidence has suggested that conservative therapy involving early gentle manipulation of the knee combined with serial splinting and casting should be the first line of treatment.6 If initial treatment attempts fail or in cases occurring later in life, surgical interventions (eg, quadriceps release procedures such as percutaneous quadriceps recession or V-Y quadricepsplasty, proximal tibial closing-wedge, anterior displacement osteotomy) likely is warranted.6,9
Our patient. At 1 week of life, our patient’s short leg splints were replaced with long leg splints with a maximal flexion of 20° to 30° (FIGURE 1B). Weekly follow-ups with serial casting were initiated in the pediatric orthopedics clinic. At 3 weeks of life, the patient’s knee flexion had improved and the splints were removed (FIGURE 1C). Upon clinical examination, the bilateral knees were extended to a neutral position, and both could be actively and passively flexed to 90°. The patient was referred to Physical Therapy to perform range of movement exercises on the knees.

At 8 weeks of life, the bilateral legs were in full extension, and knee flexion was up to 130°. Physical therapy for knee range of movement exercise was continued on a weekly basis until 6 months of life, then twice monthly until the patient was 1 year old. Ultimately, the hyperextension was corrected, and the patient started walking at around 16 months of age. Her prognosis is good, and she will be able to participate in low-impact sports, after consulting with her orthopedist.

Continue to: THE TAKEAWAY
THE TAKEAWAY
Congenital genu recurvatum is a rare condition that presents with abnormal hyperextension of the knee(s) with limited flexion. Early diagnosis and assessment of the severity of the hyperextension is crucial in determining the type of intervention to pursue. Conservative management entails serial casting and splinting to increase knee flexion. If conservative management fails or if the diagnosis is made later in life, surgical options often are pursued.
CORRESPONDENCE
Jaividhya Dasarathy, MD, FAAFP, 2500 MetroHealth Medical Drive, Cleveland, OH 44109; [email protected]
1. Donaire AR, Sethuram S, Kitsos E, et al. Congenital bilateral knee hyperextension in a well-newborn infant. Res J Clin Pediatr. 2017;1. https://www.scitechnol.com/peer-review/congenital-bilateral-knee-hyperextension-in-a-wellnewborn-infant-V63Y.php?article_id=5940. Accessed April 2, 2019.
2. Osakwe GO, Asuquo EJ, Abang EI, et al. Congenital knee dislocation: challenges in management in a low resource center. Journal of dental and medical sciences. 2016;15:78-82.
3. Katz MP, Grogono BJ, Soper KC. The etiology and treatment of congenital dislocation of the knee. J Bone Joint Surg Br. 1967;49:112-20.
4. Elmada M, Ceylan H, Erdil M, et al. Congenital dislocation of knee. Eur J Med. 2013;10:164-166.
5. Abdelaziz TH, Samir S. Congenital dislocation of the knee: a protocol for management based on degree of knee flexion. J Child Orthop. 2011;5:143-149.
6. Tiwari M, Sharma N. Unilateral congenital knee and hip dislocation with bilateral clubfoot—a rare packaging disorder. J Orthop Case Rep. 2013;3:21-24.
7. Ahmadi B, Shahriaree H, Silver CM. Severe congenital genu recurvatum. case report. J Bone Joint Surg Am. 1979;61:622-623.
8. Cheng CC, Ko JY. Early reduction for congenital dislocation of the knee within twenty-four hours of birth. Chang Gung Med J. 2010;33:266-273.
9. Youssef AO. Limited open quadriceps release for treatment of congenital dislocation of the knee. J Pediatric Orthop. 2017;37:192-198.
THE CASE
A 29-year-old G7P2315 woman gave birth to a girl at 37 weeks via spontaneous vaginal delivery. APGAR scores were 9 and 9. Birth weight was 2760 g. Cardiovascular and pulmonary examinations were normal (heart rate, 154 beats/min; respiratory rate, 52 breaths/min). Following delivery, the neonate appeared healthy, had a lusty cry, and had no visible craniofacial or cutaneous abnormalities; however, the bilateral knees were hyperextended to 90° to 110° (FIGURE 1A).
The mother had started prenatal care at 7 weeks with 10 total visits to her family physician (JD) throughout the pregnancy. Routine laboratory screening and prenatal ultrasounds (including an anatomy scan) were normal. She had a history of 3 preterm deliveries at 35 weeks, 36 weeks, and 36 weeks, respectively, and had been on progesterone shots once weekly starting at 18 weeks during the current pregnancy. She had no history of infections or recent travel. Her family history was remarkable for a sister who gave birth to a child with
THE DIAGNOSIS
The neonate tolerated passive flexion of the knees to a neutral position. Hip examination demonstrated appropriate range of movement with negative Ortolani and Barlow tests. The infant’s feet aligned correctly, with toes in the front and heels in the back, and an x-ray of the bilateral knees showed no fractures or dislocation.
Based on the clinical examination and x-ray findings, we made a diagnosis of congenital genu recurvatum. A pediatric orthopedics consultation was obtained, and the knees were placed in short leg splints in comfortable flexion to neutral on Day 1 of life. She was discharged the next day.
DISCUSSION
Congenital genu recurvatum, also known as congenital dislocation of the knee, is a rare condition involving abnormal hyperextension of the unilateral or bilateral knees with limited flexion.1 Reports in the literature are limited, but there seems to be a female predominance among known cases of congenital genu recurvatum.2 The clinical presentation varies. Finding may be isolated to the knee(s) but also can present in association with other congenital abnormalities, such as developmental dysplasia of the hip, clubfoot, and hindfoot and forefoot deformities.3,4
Diagnosis is made clinically with radiographic imaging
Diagnosis of congenital genu recurvatum is made clinically and can be confirmed via radiographic imaging of the knees.5 Clinical diagnosis requires assessment of the degree of hyperextension and palpation of the femoral condyles, which become more prominent as the severity of the hyperextension increases.6 X-rays help assess if a true dislocation or subluxation of the tibia on the femur has occurred. Based on the clinical and radiographic findings, congenital genu recurvatum typically is classified according to 3 levels of severity: grade 1 classification only involves hyperextension of the knees without dislocation or subluxation, grade 2 involves the same characteristic hyperextension along with anterior subluxation of the tibia on the femur, and grade 3 includes hyperextension with true dislocation of the tibia on the femur.1 Grades 1 and 2 on this spectrum technically are diagnosed as congenital genu recurvatum while grade 3 is diagnosed as a congenital dislocation of the knee,7 although the 2 terms are used interchangeably in the literature. We classified our case as a grade 1 congenital genu recurvatum based on the clinical and radiographic findings.
Congenital knee hyperextension has intrinsic and extrinsic causes
Hyperextension of the knees at birth may be caused by various intrinsic or extrinsic factors. Intrinsic causes may include breech position, lack of intrauterine space, trauma to the mother, quadriceps contracture or fibrosis, absence of the suprapatellar pouch, deficient or hypoplastic anterior cruciate ligament, pathological tissues, arthrogryposis, or genetic disorders such as Larsen syndrome or achondroplasia.6
Continue to: Extrinsic causes...
Extrinsic causes may include traumatic dislocation during the birthing process3 or intrauterine pressure leading to malposition of the joints. When intrauterine pressure is combined with reduced intrauterine space, this phenomenon is known as packaging disorder.6 Entanglement of the umbilical cord around the legs of the fetus during development may be another potential factor.1
The exact etiology in our patient was unknown, but we determined the cause was extrinsic based on the lack of other genetic abnormalities. We initially considered a possible connection between our patient’s diagnosis and her family history of thrombocytopenia absent radius syndrome, but it was later determined that both were isolated cases and the limb abnormalities were coincidental.
Treatment options and outcomes for extrinsic and intrinsic etiologies depend on the severity of the hyperextension and any associated abnormalities, as well as the time in which therapy is initiated.1 Reduction of the hyperextension within 24 hours of birth has been associated with excellent outcomes.8 Regardless of the cause, all cases of congenital genu recurvatum should first be treated conservatively. Evidence has suggested that conservative therapy involving early gentle manipulation of the knee combined with serial splinting and casting should be the first line of treatment.6 If initial treatment attempts fail or in cases occurring later in life, surgical interventions (eg, quadriceps release procedures such as percutaneous quadriceps recession or V-Y quadricepsplasty, proximal tibial closing-wedge, anterior displacement osteotomy) likely is warranted.6,9
Our patient. At 1 week of life, our patient’s short leg splints were replaced with long leg splints with a maximal flexion of 20° to 30° (FIGURE 1B). Weekly follow-ups with serial casting were initiated in the pediatric orthopedics clinic. At 3 weeks of life, the patient’s knee flexion had improved and the splints were removed (FIGURE 1C). Upon clinical examination, the bilateral knees were extended to a neutral position, and both could be actively and passively flexed to 90°. The patient was referred to Physical Therapy to perform range of movement exercises on the knees.
At 8 weeks of life, the bilateral legs were in full extension, and knee flexion was up to 130°. Physical therapy for knee range of movement exercise was continued on a weekly basis until 6 months of life, then twice monthly until the patient was 1 year old. Ultimately, the hyperextension was corrected, and the patient started walking at around 16 months of age. Her prognosis is good, and she will be able to participate in low-impact sports, after consulting with her orthopedist.
Continue to: THE TAKEAWAY
THE TAKEAWAY
Congenital genu recurvatum is a rare condition that presents with abnormal hyperextension of the knee(s) with limited flexion. Early diagnosis and assessment of the severity of the hyperextension is crucial in determining the type of intervention to pursue. Conservative management entails serial casting and splinting to increase knee flexion. If conservative management fails or if the diagnosis is made later in life, surgical options often are pursued.
CORRESPONDENCE
Jaividhya Dasarathy, MD, FAAFP, 2500 MetroHealth Medical Drive, Cleveland, OH 44109; [email protected]
THE CASE
A 29-year-old G7P2315 woman gave birth to a girl at 37 weeks via spontaneous vaginal delivery. APGAR scores were 9 and 9. Birth weight was 2760 g. Cardiovascular and pulmonary examinations were normal (heart rate, 154 beats/min; respiratory rate, 52 breaths/min). Following delivery, the neonate appeared healthy, had a lusty cry, and had no visible craniofacial or cutaneous abnormalities; however, the bilateral knees were hyperextended to 90° to 110° (FIGURE 1A).
The mother had started prenatal care at 7 weeks with 10 total visits to her family physician (JD) throughout the pregnancy. Routine laboratory screening and prenatal ultrasounds (including an anatomy scan) were normal. She had a history of 3 preterm deliveries at 35 weeks, 36 weeks, and 36 weeks, respectively, and had been on progesterone shots once weekly starting at 18 weeks during the current pregnancy. She had no history of infections or recent travel. Her family history was remarkable for a sister who gave birth to a child with
THE DIAGNOSIS
The neonate tolerated passive flexion of the knees to a neutral position. Hip examination demonstrated appropriate range of movement with negative Ortolani and Barlow tests. The infant’s feet aligned correctly, with toes in the front and heels in the back, and an x-ray of the bilateral knees showed no fractures or dislocation.
Based on the clinical examination and x-ray findings, we made a diagnosis of congenital genu recurvatum. A pediatric orthopedics consultation was obtained, and the knees were placed in short leg splints in comfortable flexion to neutral on Day 1 of life. She was discharged the next day.
DISCUSSION
Congenital genu recurvatum, also known as congenital dislocation of the knee, is a rare condition involving abnormal hyperextension of the unilateral or bilateral knees with limited flexion.1 Reports in the literature are limited, but there seems to be a female predominance among known cases of congenital genu recurvatum.2 The clinical presentation varies. Finding may be isolated to the knee(s) but also can present in association with other congenital abnormalities, such as developmental dysplasia of the hip, clubfoot, and hindfoot and forefoot deformities.3,4
Diagnosis is made clinically with radiographic imaging
Diagnosis of congenital genu recurvatum is made clinically and can be confirmed via radiographic imaging of the knees.5 Clinical diagnosis requires assessment of the degree of hyperextension and palpation of the femoral condyles, which become more prominent as the severity of the hyperextension increases.6 X-rays help assess if a true dislocation or subluxation of the tibia on the femur has occurred. Based on the clinical and radiographic findings, congenital genu recurvatum typically is classified according to 3 levels of severity: grade 1 classification only involves hyperextension of the knees without dislocation or subluxation, grade 2 involves the same characteristic hyperextension along with anterior subluxation of the tibia on the femur, and grade 3 includes hyperextension with true dislocation of the tibia on the femur.1 Grades 1 and 2 on this spectrum technically are diagnosed as congenital genu recurvatum while grade 3 is diagnosed as a congenital dislocation of the knee,7 although the 2 terms are used interchangeably in the literature. We classified our case as a grade 1 congenital genu recurvatum based on the clinical and radiographic findings.
Congenital knee hyperextension has intrinsic and extrinsic causes
Hyperextension of the knees at birth may be caused by various intrinsic or extrinsic factors. Intrinsic causes may include breech position, lack of intrauterine space, trauma to the mother, quadriceps contracture or fibrosis, absence of the suprapatellar pouch, deficient or hypoplastic anterior cruciate ligament, pathological tissues, arthrogryposis, or genetic disorders such as Larsen syndrome or achondroplasia.6
Continue to: Extrinsic causes...
Extrinsic causes may include traumatic dislocation during the birthing process3 or intrauterine pressure leading to malposition of the joints. When intrauterine pressure is combined with reduced intrauterine space, this phenomenon is known as packaging disorder.6 Entanglement of the umbilical cord around the legs of the fetus during development may be another potential factor.1
The exact etiology in our patient was unknown, but we determined the cause was extrinsic based on the lack of other genetic abnormalities. We initially considered a possible connection between our patient’s diagnosis and her family history of thrombocytopenia absent radius syndrome, but it was later determined that both were isolated cases and the limb abnormalities were coincidental.
Treatment options and outcomes for extrinsic and intrinsic etiologies depend on the severity of the hyperextension and any associated abnormalities, as well as the time in which therapy is initiated.1 Reduction of the hyperextension within 24 hours of birth has been associated with excellent outcomes.8 Regardless of the cause, all cases of congenital genu recurvatum should first be treated conservatively. Evidence has suggested that conservative therapy involving early gentle manipulation of the knee combined with serial splinting and casting should be the first line of treatment.6 If initial treatment attempts fail or in cases occurring later in life, surgical interventions (eg, quadriceps release procedures such as percutaneous quadriceps recession or V-Y quadricepsplasty, proximal tibial closing-wedge, anterior displacement osteotomy) likely is warranted.6,9
Our patient. At 1 week of life, our patient’s short leg splints were replaced with long leg splints with a maximal flexion of 20° to 30° (FIGURE 1B). Weekly follow-ups with serial casting were initiated in the pediatric orthopedics clinic. At 3 weeks of life, the patient’s knee flexion had improved and the splints were removed (FIGURE 1C). Upon clinical examination, the bilateral knees were extended to a neutral position, and both could be actively and passively flexed to 90°. The patient was referred to Physical Therapy to perform range of movement exercises on the knees.
At 8 weeks of life, the bilateral legs were in full extension, and knee flexion was up to 130°. Physical therapy for knee range of movement exercise was continued on a weekly basis until 6 months of life, then twice monthly until the patient was 1 year old. Ultimately, the hyperextension was corrected, and the patient started walking at around 16 months of age. Her prognosis is good, and she will be able to participate in low-impact sports, after consulting with her orthopedist.
Continue to: THE TAKEAWAY
THE TAKEAWAY
Congenital genu recurvatum is a rare condition that presents with abnormal hyperextension of the knee(s) with limited flexion. Early diagnosis and assessment of the severity of the hyperextension is crucial in determining the type of intervention to pursue. Conservative management entails serial casting and splinting to increase knee flexion. If conservative management fails or if the diagnosis is made later in life, surgical options often are pursued.
CORRESPONDENCE
Jaividhya Dasarathy, MD, FAAFP, 2500 MetroHealth Medical Drive, Cleveland, OH 44109; [email protected]
1. Donaire AR, Sethuram S, Kitsos E, et al. Congenital bilateral knee hyperextension in a well-newborn infant. Res J Clin Pediatr. 2017;1. https://www.scitechnol.com/peer-review/congenital-bilateral-knee-hyperextension-in-a-wellnewborn-infant-V63Y.php?article_id=5940. Accessed April 2, 2019.
2. Osakwe GO, Asuquo EJ, Abang EI, et al. Congenital knee dislocation: challenges in management in a low resource center. Journal of dental and medical sciences. 2016;15:78-82.
3. Katz MP, Grogono BJ, Soper KC. The etiology and treatment of congenital dislocation of the knee. J Bone Joint Surg Br. 1967;49:112-20.
4. Elmada M, Ceylan H, Erdil M, et al. Congenital dislocation of knee. Eur J Med. 2013;10:164-166.
5. Abdelaziz TH, Samir S. Congenital dislocation of the knee: a protocol for management based on degree of knee flexion. J Child Orthop. 2011;5:143-149.
6. Tiwari M, Sharma N. Unilateral congenital knee and hip dislocation with bilateral clubfoot—a rare packaging disorder. J Orthop Case Rep. 2013;3:21-24.
7. Ahmadi B, Shahriaree H, Silver CM. Severe congenital genu recurvatum. case report. J Bone Joint Surg Am. 1979;61:622-623.
8. Cheng CC, Ko JY. Early reduction for congenital dislocation of the knee within twenty-four hours of birth. Chang Gung Med J. 2010;33:266-273.
9. Youssef AO. Limited open quadriceps release for treatment of congenital dislocation of the knee. J Pediatric Orthop. 2017;37:192-198.
1. Donaire AR, Sethuram S, Kitsos E, et al. Congenital bilateral knee hyperextension in a well-newborn infant. Res J Clin Pediatr. 2017;1. https://www.scitechnol.com/peer-review/congenital-bilateral-knee-hyperextension-in-a-wellnewborn-infant-V63Y.php?article_id=5940. Accessed April 2, 2019.
2. Osakwe GO, Asuquo EJ, Abang EI, et al. Congenital knee dislocation: challenges in management in a low resource center. Journal of dental and medical sciences. 2016;15:78-82.
3. Katz MP, Grogono BJ, Soper KC. The etiology and treatment of congenital dislocation of the knee. J Bone Joint Surg Br. 1967;49:112-20.
4. Elmada M, Ceylan H, Erdil M, et al. Congenital dislocation of knee. Eur J Med. 2013;10:164-166.
5. Abdelaziz TH, Samir S. Congenital dislocation of the knee: a protocol for management based on degree of knee flexion. J Child Orthop. 2011;5:143-149.
6. Tiwari M, Sharma N. Unilateral congenital knee and hip dislocation with bilateral clubfoot—a rare packaging disorder. J Orthop Case Rep. 2013;3:21-24.
7. Ahmadi B, Shahriaree H, Silver CM. Severe congenital genu recurvatum. case report. J Bone Joint Surg Am. 1979;61:622-623.
8. Cheng CC, Ko JY. Early reduction for congenital dislocation of the knee within twenty-four hours of birth. Chang Gung Med J. 2010;33:266-273.
9. Youssef AO. Limited open quadriceps release for treatment of congenital dislocation of the knee. J Pediatric Orthop. 2017;37:192-198.

Failure to thrive in a 6-day-old neonate • intermittent retractions with inspiratory stridor • Dx?
THE CASE
A primiparous mother gave birth to a girl at 38 and 4/7 weeks via uncomplicated vaginal delivery. Prenatal labs were normal. Neonatal physical examination was normal and the child’s birth weight was in the 33rd percentile. APGAR scores were 8 and 9. The neonate was afebrile during hospitalization, with a heart rate of 120 to 150 beats/min and a respiratory rate of 30 to 48 breaths/min. Her preductal and postductal oxygen saturations were 100% and 98%, respectively. She was discharged on Day 2 of life, having lost only 3% of her birth weight.
The patient was seen in clinic on Day 6 of life for a well-child exam and was in the 17th percentile for weight. At another visit for a well-child exam on Day 14 of life, she had not fully regained her birth weight. At both visits, the mother reported no issues with breastfeeding and said she was supplementing with formula. The patient was seen again for follow-up on Days 16 and 21 of life and demonstrated no weight gain despite close follow-up with the Special Supplemental Nutrition Program for Women, Infants, and Children (WIC), which determined the newborn had some breastfeeding issues but seemed to be consuming adequate calories. However, WIC assessments revealed that during feeding, the child was expending too many calories and had nasal congestion. The patient was admitted to the hospital on Day 21 of life with a diagnosis of failure to thrive (FTT), at which point she was in the 12th percentile for weight.
THE DIAGNOSIS
Shortly after the infant was admitted, she showed signs of respiratory distress. On physical examination, the on-call resident noted intermittent retractions with inspiratory stridor, and the patient demonstrated intermittent severe oxygen desaturations into the 70s. She also was sucking her pacifier furiously, which appeared to provide some relief from the respiratory distress. The child’s parents noted that she had demonstrated intermittent periods of respiratory distress since shortly after birth that seemed to be increasing in frequency.
Upon careful examination, the on-call resident identified a cystic lesion at the base of the child’s tongue. The otolaryngologist on call was brought in for an urgent consultation but was unable to visualize the lesion on physical examination and did not recommend further intervention at that time. The patient continued to demonstrate respiratory distress with hypoxia and was transferred to the pediatric intensive care unit for close monitoring.
The next morning a second otolaryngology consultation was requested. A computed tomography scan of the neck demonstrated a 1.5-cm cystic-appearing mass at the base of the tongue that was obstructing the patient’s airway. Direct flexible bronchoscopy confirmed the radiographic findings. The patient underwent immediate surgical resection of the lesion using a laser. A clear and milky gray cystic fluid exuded from the cyst when the lesion was pierced. The otolaryngologist visualized a widely patent airway following excision of the lesion (FIGURE).

Pathology results revealed no evidence of malignancy. The final diagnosis was a simple base-of-tongue cyst.
DISCUSSION
Failure to thrive is common in neonates and occurs most often due to inadequate caloric intake; however, it also can be caused by systemic disease associated with inadequate gastrointestinal absorption or increased caloric expenditure, such as congenital heart disease, renal disease (eg, renal tubular acidosis), chronic pulmonary disease (eg, cystic fibrosis), laryngomalacia, malignancy, immunodeficiency, or thyroid disease.1
Continue to: Respiratory distress
Respiratory distress in neonates also is common but tends to occur shortly after birth.2 Conditions associated with respiratory distress in neonates include transient tachypnea of the newborn, respiratory distress syndrome, pneumothorax, persistent pulmonary hypertension of the newborn, pneumonia, and meconium aspiration syndrome.2 Interestingly, there are additional reports in the literature of FTT and respiratory distress in neonat
Base-of-tongue cysts are rare in infants. Fewer than 50 cases were reported prior to 2011, with many being described as asymptomatic nonpainful lesions.6 Given the anatomic location of base-of-tongue cysts, the differential diagnosis should also include mucoceles, thyroglossal duct cysts, dermoid cysts, epidermoid cysts, vallecular cysts, hemangiomas, cystic hygromas, lymphangiomas, thyroid remnant cysts, teratomas, and hamartomas.4,7,8 When tongue cysts are initially discovered, inspiratory stridor, FTT, swallowing deficits, oxygen desaturation, respiratory failure, and/or acute life-threatening events have been reported.6,9,10
One important clinical observation made in our case was the use of an external apparatus to relieve the neonate’s respiratory distress. During physical examination, the on-call resident noted the patient was furiously sucking her pacifier, which seemed to reduce the respiratory difficulty and desaturations. It is known that non-nutritive sucking (NNS) can provide provisions for stress relief, improve oxygenation, and provide proprioceptive positioning of key anatomical structures within the oral cavity.11 Without the use of an external apparatus like a pacifier during restful states, neonates may develop vacuum-glossoptosis syndrome, in which the dorsum of the tongue and the soft palate adhere to the posterior pharyngeal wall and obstruct the airway.12 Our patient may have used the pacifier as an NNS task to move the tongue forward and break the glossoptosis-pharyngeal seal by sucking hard and fast during periods of respiratory distress, which reduced the potential for a vacuum-glossoptosis phenomenon that was likely created by the cyst during restful states.
Our patient was seen in clinic for follow-up after surgery on Day 35 of life. She was thriving and her weight was in the 24th percentile. She was seen again on Day 67 of life for a well-child exam and was in the 43rd percentile for weight.
THE TAKEAWAY
There is a sizeable list of possible diagnoses to consider when a neonate presents with FTT and respiratory distress. It is important to consider mechanical obstruction as a possible diagnosis and one which, if identified early, may be lifesaving. Our case demonstrates a proposed mechanism by which a mechanical obstruction such as a base-of-tongue cyst can cause the vacuum-glossoptosis syndrome; it also highlights NNS as a potential means of overcoming this phenomenon.
CORRESPONDENCE
Benjamin P. Hansen, MD, Renown Medical Group, 4796 Caughlin Pkwy, Ste 108, Reno, NV 89519; [email protected]
1. Larson-Nath C, Biank VF. Clinical review of failure to thrive in pediatric patients. Pediatr Ann. 2016;45:e46-e49.
2. Edwards MO, Kotecha SJ, Kotecha S. Respiratory distress of the term newborn infant. Paediatr Respir Rev. 2013;14:29-37.
3. Brennan T, Rastatter JC. Multilevel airway obstruction including rare tongue base mass presenting as severe croup in an infant. Int J Pediatr Otorhinolaryngol. 2013;77:128-129.
4. Gutiérrez JP, Berkowitz RG, Robertson CF. Vallecular cysts in newborns and young infants. Pediatr Pulmonol. 1999;27:282-285.
5. Wong KS, Huang YH, Wu CT. A vanishing tongue-base cyst. Turk J Pediatr. 2007;49:451-452.
6. Aubin A, Lescanne E, Pondaven S, et al. Stridor and lingual thyroglossal duct cyst in a newborn. Eur Ann Otorhinolaryngol Head Neck Dis. 2011;128:321-323.
7. Hur JH, Byun JS, Kim JK, et al. Mucocele in the base of the tongue mimicking a thyroglossal duct cyst: a very rare location. Iran J Radiol. 2016;13:4-7.
8. Tárrega ER, Rojas SF, Portero RG, et al. Prenatal ultrasound diagnosis of a cyst of the oral cavity: an unusual case of thyroglossal duct cyst located on the tongue base [published online January 21, 2016]. 2016;2016:7816306.
9. Parelkar SV, Patel JL, Sanghvi BV, et al. An unusual presentation of vallecular cyst with near fatal respiratory distress and management using conventional laparoscopic instruments. J Surg Tech Case Rep. 2012;4:118-120.
10. Sands NB, Anand SM, Manoukian JJ. Series of congenital vallecular cysts: a rare yet potentially fatal cause of upper airway obstruction and failure to thrive in the newborn. J Otolaryngol Head Neck Surg. 2009;38:6-10.
11. Pinelli J, Symington A. Non-nutritive sucking for promoting physiologic stability and nutrition in preterm infants. Cochrane Database Syst Rev 2005. 2010;4:CD001071.
12. Cozzi F, Albani R, Cardi E. A common pathophysiology for sudden cot death and sleep apnoea. “the vacuum-glossoptosis syndrome.” Med Hypotheses. 1979;5:329-338.
THE CASE
A primiparous mother gave birth to a girl at 38 and 4/7 weeks via uncomplicated vaginal delivery. Prenatal labs were normal. Neonatal physical examination was normal and the child’s birth weight was in the 33rd percentile. APGAR scores were 8 and 9. The neonate was afebrile during hospitalization, with a heart rate of 120 to 150 beats/min and a respiratory rate of 30 to 48 breaths/min. Her preductal and postductal oxygen saturations were 100% and 98%, respectively. She was discharged on Day 2 of life, having lost only 3% of her birth weight.
The patient was seen in clinic on Day 6 of life for a well-child exam and was in the 17th percentile for weight. At another visit for a well-child exam on Day 14 of life, she had not fully regained her birth weight. At both visits, the mother reported no issues with breastfeeding and said she was supplementing with formula. The patient was seen again for follow-up on Days 16 and 21 of life and demonstrated no weight gain despite close follow-up with the Special Supplemental Nutrition Program for Women, Infants, and Children (WIC), which determined the newborn had some breastfeeding issues but seemed to be consuming adequate calories. However, WIC assessments revealed that during feeding, the child was expending too many calories and had nasal congestion. The patient was admitted to the hospital on Day 21 of life with a diagnosis of failure to thrive (FTT), at which point she was in the 12th percentile for weight.
THE DIAGNOSIS
Shortly after the infant was admitted, she showed signs of respiratory distress. On physical examination, the on-call resident noted intermittent retractions with inspiratory stridor, and the patient demonstrated intermittent severe oxygen desaturations into the 70s. She also was sucking her pacifier furiously, which appeared to provide some relief from the respiratory distress. The child’s parents noted that she had demonstrated intermittent periods of respiratory distress since shortly after birth that seemed to be increasing in frequency.
Upon careful examination, the on-call resident identified a cystic lesion at the base of the child’s tongue. The otolaryngologist on call was brought in for an urgent consultation but was unable to visualize the lesion on physical examination and did not recommend further intervention at that time. The patient continued to demonstrate respiratory distress with hypoxia and was transferred to the pediatric intensive care unit for close monitoring.
The next morning a second otolaryngology consultation was requested. A computed tomography scan of the neck demonstrated a 1.5-cm cystic-appearing mass at the base of the tongue that was obstructing the patient’s airway. Direct flexible bronchoscopy confirmed the radiographic findings. The patient underwent immediate surgical resection of the lesion using a laser. A clear and milky gray cystic fluid exuded from the cyst when the lesion was pierced. The otolaryngologist visualized a widely patent airway following excision of the lesion (FIGURE).
Pathology results revealed no evidence of malignancy. The final diagnosis was a simple base-of-tongue cyst.
DISCUSSION
Failure to thrive is common in neonates and occurs most often due to inadequate caloric intake; however, it also can be caused by systemic disease associated with inadequate gastrointestinal absorption or increased caloric expenditure, such as congenital heart disease, renal disease (eg, renal tubular acidosis), chronic pulmonary disease (eg, cystic fibrosis), laryngomalacia, malignancy, immunodeficiency, or thyroid disease.1
Continue to: Respiratory distress
Respiratory distress in neonates also is common but tends to occur shortly after birth.2 Conditions associated with respiratory distress in neonates include transient tachypnea of the newborn, respiratory distress syndrome, pneumothorax, persistent pulmonary hypertension of the newborn, pneumonia, and meconium aspiration syndrome.2 Interestingly, there are additional reports in the literature of FTT and respiratory distress in neonat
Base-of-tongue cysts are rare in infants. Fewer than 50 cases were reported prior to 2011, with many being described as asymptomatic nonpainful lesions.6 Given the anatomic location of base-of-tongue cysts, the differential diagnosis should also include mucoceles, thyroglossal duct cysts, dermoid cysts, epidermoid cysts, vallecular cysts, hemangiomas, cystic hygromas, lymphangiomas, thyroid remnant cysts, teratomas, and hamartomas.4,7,8 When tongue cysts are initially discovered, inspiratory stridor, FTT, swallowing deficits, oxygen desaturation, respiratory failure, and/or acute life-threatening events have been reported.6,9,10
One important clinical observation made in our case was the use of an external apparatus to relieve the neonate’s respiratory distress. During physical examination, the on-call resident noted the patient was furiously sucking her pacifier, which seemed to reduce the respiratory difficulty and desaturations. It is known that non-nutritive sucking (NNS) can provide provisions for stress relief, improve oxygenation, and provide proprioceptive positioning of key anatomical structures within the oral cavity.11 Without the use of an external apparatus like a pacifier during restful states, neonates may develop vacuum-glossoptosis syndrome, in which the dorsum of the tongue and the soft palate adhere to the posterior pharyngeal wall and obstruct the airway.12 Our patient may have used the pacifier as an NNS task to move the tongue forward and break the glossoptosis-pharyngeal seal by sucking hard and fast during periods of respiratory distress, which reduced the potential for a vacuum-glossoptosis phenomenon that was likely created by the cyst during restful states.
Our patient was seen in clinic for follow-up after surgery on Day 35 of life. She was thriving and her weight was in the 24th percentile. She was seen again on Day 67 of life for a well-child exam and was in the 43rd percentile for weight.
THE TAKEAWAY
There is a sizeable list of possible diagnoses to consider when a neonate presents with FTT and respiratory distress. It is important to consider mechanical obstruction as a possible diagnosis and one which, if identified early, may be lifesaving. Our case demonstrates a proposed mechanism by which a mechanical obstruction such as a base-of-tongue cyst can cause the vacuum-glossoptosis syndrome; it also highlights NNS as a potential means of overcoming this phenomenon.
CORRESPONDENCE
Benjamin P. Hansen, MD, Renown Medical Group, 4796 Caughlin Pkwy, Ste 108, Reno, NV 89519; [email protected]
THE CASE
A primiparous mother gave birth to a girl at 38 and 4/7 weeks via uncomplicated vaginal delivery. Prenatal labs were normal. Neonatal physical examination was normal and the child’s birth weight was in the 33rd percentile. APGAR scores were 8 and 9. The neonate was afebrile during hospitalization, with a heart rate of 120 to 150 beats/min and a respiratory rate of 30 to 48 breaths/min. Her preductal and postductal oxygen saturations were 100% and 98%, respectively. She was discharged on Day 2 of life, having lost only 3% of her birth weight.
The patient was seen in clinic on Day 6 of life for a well-child exam and was in the 17th percentile for weight. At another visit for a well-child exam on Day 14 of life, she had not fully regained her birth weight. At both visits, the mother reported no issues with breastfeeding and said she was supplementing with formula. The patient was seen again for follow-up on Days 16 and 21 of life and demonstrated no weight gain despite close follow-up with the Special Supplemental Nutrition Program for Women, Infants, and Children (WIC), which determined the newborn had some breastfeeding issues but seemed to be consuming adequate calories. However, WIC assessments revealed that during feeding, the child was expending too many calories and had nasal congestion. The patient was admitted to the hospital on Day 21 of life with a diagnosis of failure to thrive (FTT), at which point she was in the 12th percentile for weight.
THE DIAGNOSIS
Shortly after the infant was admitted, she showed signs of respiratory distress. On physical examination, the on-call resident noted intermittent retractions with inspiratory stridor, and the patient demonstrated intermittent severe oxygen desaturations into the 70s. She also was sucking her pacifier furiously, which appeared to provide some relief from the respiratory distress. The child’s parents noted that she had demonstrated intermittent periods of respiratory distress since shortly after birth that seemed to be increasing in frequency.
Upon careful examination, the on-call resident identified a cystic lesion at the base of the child’s tongue. The otolaryngologist on call was brought in for an urgent consultation but was unable to visualize the lesion on physical examination and did not recommend further intervention at that time. The patient continued to demonstrate respiratory distress with hypoxia and was transferred to the pediatric intensive care unit for close monitoring.
The next morning a second otolaryngology consultation was requested. A computed tomography scan of the neck demonstrated a 1.5-cm cystic-appearing mass at the base of the tongue that was obstructing the patient’s airway. Direct flexible bronchoscopy confirmed the radiographic findings. The patient underwent immediate surgical resection of the lesion using a laser. A clear and milky gray cystic fluid exuded from the cyst when the lesion was pierced. The otolaryngologist visualized a widely patent airway following excision of the lesion (FIGURE).
Pathology results revealed no evidence of malignancy. The final diagnosis was a simple base-of-tongue cyst.
DISCUSSION
Failure to thrive is common in neonates and occurs most often due to inadequate caloric intake; however, it also can be caused by systemic disease associated with inadequate gastrointestinal absorption or increased caloric expenditure, such as congenital heart disease, renal disease (eg, renal tubular acidosis), chronic pulmonary disease (eg, cystic fibrosis), laryngomalacia, malignancy, immunodeficiency, or thyroid disease.1
Continue to: Respiratory distress
Respiratory distress in neonates also is common but tends to occur shortly after birth.2 Conditions associated with respiratory distress in neonates include transient tachypnea of the newborn, respiratory distress syndrome, pneumothorax, persistent pulmonary hypertension of the newborn, pneumonia, and meconium aspiration syndrome.2 Interestingly, there are additional reports in the literature of FTT and respiratory distress in neonat
Base-of-tongue cysts are rare in infants. Fewer than 50 cases were reported prior to 2011, with many being described as asymptomatic nonpainful lesions.6 Given the anatomic location of base-of-tongue cysts, the differential diagnosis should also include mucoceles, thyroglossal duct cysts, dermoid cysts, epidermoid cysts, vallecular cysts, hemangiomas, cystic hygromas, lymphangiomas, thyroid remnant cysts, teratomas, and hamartomas.4,7,8 When tongue cysts are initially discovered, inspiratory stridor, FTT, swallowing deficits, oxygen desaturation, respiratory failure, and/or acute life-threatening events have been reported.6,9,10
One important clinical observation made in our case was the use of an external apparatus to relieve the neonate’s respiratory distress. During physical examination, the on-call resident noted the patient was furiously sucking her pacifier, which seemed to reduce the respiratory difficulty and desaturations. It is known that non-nutritive sucking (NNS) can provide provisions for stress relief, improve oxygenation, and provide proprioceptive positioning of key anatomical structures within the oral cavity.11 Without the use of an external apparatus like a pacifier during restful states, neonates may develop vacuum-glossoptosis syndrome, in which the dorsum of the tongue and the soft palate adhere to the posterior pharyngeal wall and obstruct the airway.12 Our patient may have used the pacifier as an NNS task to move the tongue forward and break the glossoptosis-pharyngeal seal by sucking hard and fast during periods of respiratory distress, which reduced the potential for a vacuum-glossoptosis phenomenon that was likely created by the cyst during restful states.
Our patient was seen in clinic for follow-up after surgery on Day 35 of life. She was thriving and her weight was in the 24th percentile. She was seen again on Day 67 of life for a well-child exam and was in the 43rd percentile for weight.
THE TAKEAWAY
There is a sizeable list of possible diagnoses to consider when a neonate presents with FTT and respiratory distress. It is important to consider mechanical obstruction as a possible diagnosis and one which, if identified early, may be lifesaving. Our case demonstrates a proposed mechanism by which a mechanical obstruction such as a base-of-tongue cyst can cause the vacuum-glossoptosis syndrome; it also highlights NNS as a potential means of overcoming this phenomenon.
CORRESPONDENCE
Benjamin P. Hansen, MD, Renown Medical Group, 4796 Caughlin Pkwy, Ste 108, Reno, NV 89519; [email protected]
1. Larson-Nath C, Biank VF. Clinical review of failure to thrive in pediatric patients. Pediatr Ann. 2016;45:e46-e49.
2. Edwards MO, Kotecha SJ, Kotecha S. Respiratory distress of the term newborn infant. Paediatr Respir Rev. 2013;14:29-37.
3. Brennan T, Rastatter JC. Multilevel airway obstruction including rare tongue base mass presenting as severe croup in an infant. Int J Pediatr Otorhinolaryngol. 2013;77:128-129.
4. Gutiérrez JP, Berkowitz RG, Robertson CF. Vallecular cysts in newborns and young infants. Pediatr Pulmonol. 1999;27:282-285.
5. Wong KS, Huang YH, Wu CT. A vanishing tongue-base cyst. Turk J Pediatr. 2007;49:451-452.
6. Aubin A, Lescanne E, Pondaven S, et al. Stridor and lingual thyroglossal duct cyst in a newborn. Eur Ann Otorhinolaryngol Head Neck Dis. 2011;128:321-323.
7. Hur JH, Byun JS, Kim JK, et al. Mucocele in the base of the tongue mimicking a thyroglossal duct cyst: a very rare location. Iran J Radiol. 2016;13:4-7.
8. Tárrega ER, Rojas SF, Portero RG, et al. Prenatal ultrasound diagnosis of a cyst of the oral cavity: an unusual case of thyroglossal duct cyst located on the tongue base [published online January 21, 2016]. 2016;2016:7816306.
9. Parelkar SV, Patel JL, Sanghvi BV, et al. An unusual presentation of vallecular cyst with near fatal respiratory distress and management using conventional laparoscopic instruments. J Surg Tech Case Rep. 2012;4:118-120.
10. Sands NB, Anand SM, Manoukian JJ. Series of congenital vallecular cysts: a rare yet potentially fatal cause of upper airway obstruction and failure to thrive in the newborn. J Otolaryngol Head Neck Surg. 2009;38:6-10.
11. Pinelli J, Symington A. Non-nutritive sucking for promoting physiologic stability and nutrition in preterm infants. Cochrane Database Syst Rev 2005. 2010;4:CD001071.
12. Cozzi F, Albani R, Cardi E. A common pathophysiology for sudden cot death and sleep apnoea. “the vacuum-glossoptosis syndrome.” Med Hypotheses. 1979;5:329-338.
1. Larson-Nath C, Biank VF. Clinical review of failure to thrive in pediatric patients. Pediatr Ann. 2016;45:e46-e49.
2. Edwards MO, Kotecha SJ, Kotecha S. Respiratory distress of the term newborn infant. Paediatr Respir Rev. 2013;14:29-37.
3. Brennan T, Rastatter JC. Multilevel airway obstruction including rare tongue base mass presenting as severe croup in an infant. Int J Pediatr Otorhinolaryngol. 2013;77:128-129.
4. Gutiérrez JP, Berkowitz RG, Robertson CF. Vallecular cysts in newborns and young infants. Pediatr Pulmonol. 1999;27:282-285.
5. Wong KS, Huang YH, Wu CT. A vanishing tongue-base cyst. Turk J Pediatr. 2007;49:451-452.
6. Aubin A, Lescanne E, Pondaven S, et al. Stridor and lingual thyroglossal duct cyst in a newborn. Eur Ann Otorhinolaryngol Head Neck Dis. 2011;128:321-323.
7. Hur JH, Byun JS, Kim JK, et al. Mucocele in the base of the tongue mimicking a thyroglossal duct cyst: a very rare location. Iran J Radiol. 2016;13:4-7.
8. Tárrega ER, Rojas SF, Portero RG, et al. Prenatal ultrasound diagnosis of a cyst of the oral cavity: an unusual case of thyroglossal duct cyst located on the tongue base [published online January 21, 2016]. 2016;2016:7816306.
9. Parelkar SV, Patel JL, Sanghvi BV, et al. An unusual presentation of vallecular cyst with near fatal respiratory distress and management using conventional laparoscopic instruments. J Surg Tech Case Rep. 2012;4:118-120.
10. Sands NB, Anand SM, Manoukian JJ. Series of congenital vallecular cysts: a rare yet potentially fatal cause of upper airway obstruction and failure to thrive in the newborn. J Otolaryngol Head Neck Surg. 2009;38:6-10.
11. Pinelli J, Symington A. Non-nutritive sucking for promoting physiologic stability and nutrition in preterm infants. Cochrane Database Syst Rev 2005. 2010;4:CD001071.
12. Cozzi F, Albani R, Cardi E. A common pathophysiology for sudden cot death and sleep apnoea. “the vacuum-glossoptosis syndrome.” Med Hypotheses. 1979;5:329-338.
