User login
Management of Rodenticide Poisoning Associated with Synthetic Cannabinoids
Between March 7, 2018, and May 9, 2018, at least 164 people in Illinois were sickened by synthetic cannabinoids laced with rodenticides. The Illinois Department of Public Health has reported 4 deaths connected with the use of synthetic cannabinoids (sold under names such as Spice, K2, Legal Weed, etc).1 Synthetic cannabinoids are mind-altering chemicals that are sprayed on dried plant material and often sold at convenience stores. Some users have reported smoking these substances because they are generally not detected by standard urine toxicology tests.
Recreational use of synthetic cannabinoids can lead to serious and, at times, deadly complications. Chemicals found in rat poison have contaminated batches of synthetic cannabinoids, leading to coagulopathy and severe bleeding. Affected patients have reported hemoptysis, hematuria, severe epistaxis, bleeding gums, conjunctival hemorrhages, and gastrointestinal bleeding. The following case is of a patient who presented to an emergency department (ED) with severe coagulopathy and cardiotoxicity after using an adulterated synthetic cannabinoid product.
Case Presentation
A 65-year-old man presented to the ED reporting hematochezia, hematuria, and hemoptysis. He reported that these symptoms began about 1 day after he had smoked a synthetic cannabinoid called K2. The patient stated that some of his friends who used the same product were experiencing similar symptoms. He reported mild generalized abdominal pain but reported no chest pain, dyspnea, headache, fevers, chills, or dysuria.
The patient’s past medical history included hypertension, dyslipidemia, chronic lower back pain, and vitamin D deficiency. His past surgical history was notable for an exploratory laparotomy after a stab wound to the abdomen. The patient reported taking the following medications: morphine SA 30 mg bid, meloxicam 15 mg daily, amitriptyline 100 mg qhs, amlodipine 5 mg daily, hydrocodone/acetaminophen 5/325 mg q12h prn, atorvastatin 20 mg qhs, omeprazole 20 mg qam, senna 187 mg daily prn, psyllium 1 packet dissolved in water daily prn, and cholecalciferol 1,000 IU daily.
The patient’s temperature was 98o F, blood pressure, 144/80 mm Hg; pulse, 131 beats per minute; respiratory rate, 18 breaths per minute; and O2 saturation, 98% (ambient air). A physical examination revealed no acute distress; he was coughing up blood; clear lungs; heart sounds were tachycardic and irregularly irregular; soft, nondistended, mild generalized tenderness in the abdomen with no guarding and no rebound. The pertinent laboratory tests were international normalized ratio (INR), > 20; prothrombin time, > 150 seconds; prothrombin thromboplastin time, 157 seconds; hemoglobin, 13.3 g/dL; platelet count, 195 k/uL; white blood count, 11.3 k/uL; creatinine, 0.57mg/dL; potassium, 3.8 mmol/L, D-dimertest, 0.87 ug/mL fibrinogen equivalent units; fibrinogen level, 624 mg/dL; troponin, < 0.04 ng/mL; lactic acid, 1.3 mmol/L; total bilirubin, 0.8 mg/dL; alanine aminotransferase, 22 U/L, aspartate aminotransferase, 22 U/L; alkaline phosphatase, 89 U/L; urinalysis with > 50 red blood cells/high power field; large blood, negative leukocyte esterase, negative nitrite. The patient’s urine toxicology was negative for cannabinoids, methadone, amphetamines, cocaine, and benzodiazepines; but was positive for opiates. An anticoagulant poisoning panel also was ordered.
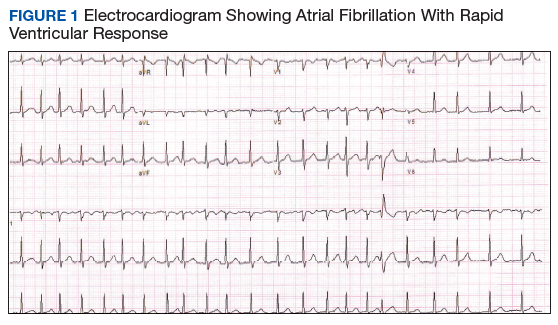
An electrocardiogram (ECG) and imaging studies were ordered. The ECG showed atrial fibrillation (AF) with rapid ventricular response (Figure 1). A chest X-ray indicated bibasilar consolidations that were worse on the right side. A noncontrast computed tomography (CT) of the head did not show intracranial bleeding. An abdomen/pelvis CT showed bilateral diffuse patchy peribronchovascular ground-glass opacities in the lung bases that could represent pulmonary hemorrhage, but no peritoneal or retroperitoneal bleeding.
Treatment
In the ED, the case was discussed with the Illinois Poison Control Center. The patient was diagnosed with coagulopathy likely due to anticoagulant poisoning. He was immediately treated with 10 mg of IV vitamin K, a fixed dose of 2,000 units of 4-factor prothrombin complex concentrate, and 4 units of fresh frozen plasma. His INR improved to 1.42 within several hours. He received 5 mg of IV metoprolol for uncontrolled AF and was admitted to the intensive care unit (ICU) for further care.
In the ICU the patient was started on oral vitamin K 50 mg tid for ongoing treatment of coagulopathy due to concern for possible rodenticide poisoning associated with very long half-life. This dose was then decreased to 50 mg bid. He was given IV fluid resuscitation with normal saline and started on rate control for AF with oral metoprolol. His heart rate improved. An echocardiogram showed new cardiomyopathy with an ejection fraction of 25% to 30%. Given basilar infiltrates and 1 episode of low-grade fever, he was started on ceftriaxone for possible community-acquired pneumonia. The patient was started on cholestyramine to help with washout of the possible rodenticide. No endoscopic interventions were performed.
The patient was transferred to an inpatient telemetry floor 24 hours after admission to the ICU once his tachycardia and bleeding improved. He did not require transfusion of packed red blood cells. In the ICU his INR had ranged between 1.62 and 2.46 (down from > 20 in the ED). His hemoglobin dropped from 13.3 g/dL on admission to 12 g/dL on transfer from the ICU, before stabilizing around 11 g/dL on the floor. The patient’s heart rate required better control, so metoprolol was increased to a total daily dose of 200 mg on the telemetry floor. Oral digoxin was then added after a digoxin load for additional rate control, as the patient remained tachycardic. Twice a day the patient continued to take 50 mg vitamin K. Cholestyramine and ceftriaxone were initially continued, but when the INR started increasing again, the cholestyramine was stopped to allow for an increase to more frequent 3-times daily vitamin 50 mg K administration (cholestyramine can interfere with vitamin K absorption). According to the toxicology service, there was only weak evidence to support use of cholestyramine in this setting.
Given his ongoing mild hemoptysis, the patient received first 1 unit, and then another 4 units of FFP when the INR increased to 3.96 despite oral vitamin K. After FFP, the INR decreased to 1.93 and subsequently to 1.52. A CT of the chest showed patchy ground-glass densities throughout the lungs, predominantly at the lung bases and to a lesser extent in the upper lobes. The findings were felt to represent pulmonary hemorrhage given the patient’s history of hemoptysis (Figure 2). 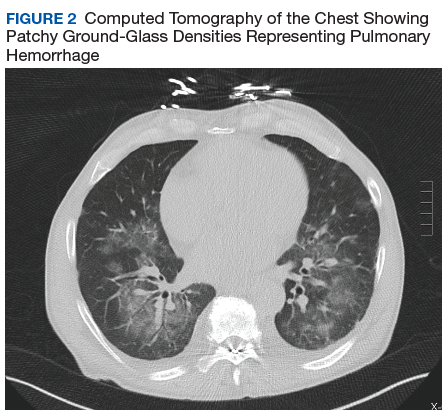
The patient’s heart rate control improved, and he remained hemodynamically stable. A thyroid function test was within normal limits. Lisinopril was added to metoprolol and digoxin given his newly diagnosed cardiomyopathy. The patient was observed for a total of 4 days on the inpatient floor and discharged after his INR stabilized around 1.5 on twice daily 50 mg vitamin K. The patient’s hematuria and hematochezia completely resolved, and hemoptysis was much improved at the time of discharge. His hemoglobin remained stable. The anticoagulant poisoning panel came back positive for
The patient has remained in AF at all follow-up visits. The INR normalized by 6 weeks after hospital discharge, and the dose of vitamin K slowly was tapered with close monitoring of the INR. Vitamin K was tapered for about 6 months after his initial presentation, and the patient was started on a direct oral anticoagulant (DOAC) for anticoagulation when the INR remained stable off vitamin K. He subsequently underwent a transesophageal echocardiogram followed by an attempt at direct current (DC) cardioversion; however, he did not remain in sinus rhythm, and is being continued on anticoagulation and rate control for his AF.
Discussion
Users generally smoke synthetic cannabinoids, which produce cannabis-like effects. However, atypical intoxication effects with worse complications often occur.2 These products typically contain dried shredded plant material that is soaked in or sprayed with several synthetic cannabinoids, varying in dosage and combination.3 Synthetic cannabinoids have been associated with serious adverse effects (AEs), including drowsiness, light-headedness, and fast or irregular heartbeat.4 More severe clinical features such as psychosis, delirium, cardiotoxicity, seizures, rhabdomyolysis, acute kidney injury, hyperthermia, myocardial ischemia, ischemic strokes, and death have also been noted.4
It is not known how some batches of synthetic cannabinoids came to be contaminated with rat poison or how commonly such an adulteration is found across the country. Several different guidelines provide pathways for the treatment of acute bleeding in the setting of coagulopathy due to vitamin K antagonists.5,6 Each guideline divides the indications for reversal into either severity of bleeding or the criticality of the bleeding based on location.5,6 All guidelines recommend the use of vitamin K (either oral or IV) followed by FFP or 4-factor prothrombin complex concentrate (PCC) for more severe bleeding.5,6 However, recommendations regarding the use of PCC vary in dosing for vitamin K antagonists (in contrast to treatment of coagulopathy due to DOACs). Recent studies and guidelines suggest that fixed-dose (rather than weight-based dose) PCC is effective for the reversal of coagulopathy due to vitamin K antagonists.6,7 Using fixed rather than weight-based dosing decreases cost and may decrease the possibility of thrombotic AEs.7 In this patient, a fixed-dose of 2,000 units of PCC was given based on data that were extrapolated from warfarin reversal using PCC.7
The vitamin K antagonists that adulterated this patient’s synthetic cannabinoid were difenacoum and brodifacoum, which are 4-hydroxycoumarin derivatives. These are second-generation long-acting anticoagulant rodenticides (LAARs) that are about 100 times more potent than warfarin.8 As the name implies, LAARs have a longer duration of action in the body of any organism that ingests the poison, which is due to the highly lipophilic groups that have been added to the warfarin molecule to combat resistance in rodents.9
As a result of the deposition in the tissues, there have been reports of the duration of action of brodifacoum ranging from 51 days to 9 months after ingestion, with the latter caused by an intentional overdose in a human.9-12 Reports suggest that coagulopathy is not likely to occur when the serum brodifacoum concentration is < 10 ng/mL.13,14 Animal models show difenacoum has a tissue half-life of about 62 days.15 Reports of difenacoum poisoning in humans have shown variable lengths of treatment, ranging from 30 to 47 days.16-18 The length of treatment for either brodifacoum or difenacoum will depend on the amount of poison exposure.
The long duration of action and treatment duration may lead to problems with drug procurement, especially in the early phase of treatment in which IV vitamin K is used. The supply of IV vitamin K recently has been limited for at least some manufacturers. According to the American Society of Health System Pharmacists Current Drug Shortage List, the increased demand is thought to be due to increased use of synthetic inhaled cannabinoids laced with anticoagulant.19 IV vitamin K products are available from suppliers such as Amphastar (Rancho Cucamonga, CA) and Hospira (Lake Forest, IL).
The American College of Chest Physicians recommends IV vitamin K administration in patients with major bleeding secondary to vitamin K antagonists.20 The oral route is thought to be more effective than a subcutaneous route in the treatment of nonbleeding patients with rodenticide-associated coagulopathy. Due to erratic and unpredictable absorption, the subcutaneous route of administration has fallen out of favor. Oral vitamin K products were not affected by the recent shortage. However, large doses of oral vitamin K can be costly. Due to the long half-life of LAAR, many patients are discharged with a prescription for oral vitamin K. Although vitamin K is found in most over-the-counter (OTC) multivitamins, the strength is insufficient. Most OTC formulations are ≤ 100 μg, whereas the prescription strength is 5 mg, but patients being treated for rodenticide poisoning require much larger doses.
Commercial insurance carriers and Medicare Part D usually do not cover vitamins and minerals unless it is for a medically accepted indication or is an indication supported by citation in either the American Hospital Formulary System, United States Pharmacopeia drug information book, or an electronic information resource that is supported by evidence such as Micromedex.21 For a patient without insurance coverage being treated with high-dose vitamin K therapy for rodenticide poisoning outside of a federal health care system, the cost could be as high as $500 to $1,000 per day, depending on the dose of vitamin K needed to maintain an acceptable INR.
Conclusion
In addition to bleeding as a result of coagulopathy, this patient presented with new onset of AF with rapid ventricular response and a newly diagnosed cardiomyopathy. Although the patient had other cardiovascular risk factors, such as hypertension, dyslipidemia, and a remote history of cocaine use, it is likely that the use of the synthetic cannabinoids contributed to the development and/or worsening of this arrhythmia and cardiomyopathy. The patient remained in AF 6 weeks after hospital discharge with a controlled ventricular rate on metoprolol and digoxin. An interval echocardiogram 6 weeks after hospital discharge showed a recovered ejection fraction. In cases of tachycardia-induced cardiomyopathy, the ejection fraction often recovers with control of the tachycardia. The patient was weaned off vitamin K about 6 months after his initial presentation and started on a DOAC for anticoagulation. He subsequently underwent a transesophageal echocardiogram followed by an attempt at DC cardioversion; however, he did not remain in sinus rhythm and is being continued on anticoagulation and rate control for his AF.
Although unclear how synthetic cannabinoids became adulterated with a potent vitamin K antagonist, health care practitioners should consider this if a patient presents with unexplained coagulopathy and widespread bleeding. Fixed-dose PCC should be considered as an alternative to weight-based dosing in these cases. Physicians and pharmacy personnel should anticipate a need for long-term high doses of vitamin K in order to begin work early to obtain sufficient supplies to treat presenting patients.
1. Illinois Department of Public Health. Synthetic cannabinoids. http://dph.illinois.gov/topics-services/prevention-wellness/medical-cannabis/synthetic-cannabinoids. Updated May 30, 2018. Accessed April 8, 2019.
2. Tournebize J, Gibaja V, Kahn JP. Acute effects of synthetic cannabinoids: update 2015. Subst Abus. 2017;38(3):344-366.
3. United Nations Office on Drugs and Crime. Global SMART update. https://www.unodc.org/documents/scientific/Global_SMART_Update_13_web.pdf. Published March 2015. Accessed April 8, 2019.
4. Adams AJ, Banister SD, Irizarry L, Trecki J, Schwartz M, Gerona R, “Zombie” outbreak caused by the synthetic cannabinoid AMB-FUBINACA in New York. N Engl J Med. 2017;376(3):235-242.
5. Tomaselli GF, Mahaffey KW, Cuker A, et al. 2017 ACC expert consensus decision pathway on management of bleeding in patients on oral anticoagulants: a report of the American College of Cardiology Task Force on Expert Consensus Decision Pathways. J Am Coll Cardiol. 2017;70(24):3042-3067.
6. Cushman M, Lim W, Zakai NA. 2011 Clinical Practice guide on anticoagulant dosing and management of anticoagulant-associated bleeding complications in adults. http://www.hematology.org/Clinicians/Guidelines-Quality/Quick-Ref/525.aspx. Published 2011. Accessed April 8, 2019.
7. Klein L, Peters J, Miner J, Gorlin J. Evaluation of fixed dose 4-factor prothrombin complex concentrate for emergent warfarin reversal. Am J Emerg Med. 2015;33(9):1213-1218.
8. Bachmann KA, Sullivan TJ. Dispositional and pharmacodynamic characteristics of brodifacoum in warfarin-sensitive rats. Pharmacology. 1983;27(5):281-288.
9. Lipton RA, Klass EM. Human ingestion of ‘superwarfarin’ rodenticide resulting in a prolonged anticoagulant effect. JAMA. 1984;252(21):3004-3005.
10. Chong LL, Chau WK, Ho CH. A case of ‘superwarfarin’ poisoning. Scand J Haematol. 1986;36(3):314-331.
11. Jones EC, Growe GH, Naiman SC. Prolonged anticoagulation in rat poisoning. JAMA. 1984;252(21):3005-3007.
12. Babcock J, Hartman K, Pedersen A, Murphy M, Alving B. Rodenticide-induced coagulopathy in a young child. A case of Munchausen syndrome by proxy. Am J Pediatr Hematol Oncol. 1993;15(1):126-130.
13. Hollinger BR, Pastoor TP. Case management and plasma half-life in a case of brodifacoum poisoning. Arch Intern Med. 1993;153(16):1925-1928.
14. Bruno GR, Howland MA, McMeeking A, Hoffman RS. Long-acting anticoagulant overdose: brodifacoum kinetics and optimal vitamin K dosing. Ann Emerg Med. 2000;36(3):262-267.
15. Vandenbrouke V, Bousquet-Meloua A, De Backer P, Croubels S. Pharmacokinetics of eight anticoagulant rodenticides in mice after single oral administration. J Vet Pharmacol Ther. 2008;31(5):437-445.
16. Barlow AM, Gay AL, Park BK. Difenacoum (Neosorexa) poisoning. Br Med J (Clin Res Ed). 1982;285(6341):541.
17. Katona B, Wason S. Superwarfarin poisoning. J Emerg Med. 1989;7(6):627-631.
18. Butcher GP, Shearer MJ, MacNicoll AD, Kelly MJ, Ind PW. Difenacoum poisoning as a cause of haematuria. Hum Exp Toxicol. 1992;11(6):553-554.
19. American Society of Health System Pharmacists. Current drug shortages. Vitamin K (phytonadione) injection. https://www.ashp.org/drug-shortages/current-shortages/Drug-Shortage-Detail.aspx?id=100. Updated July 5, 2018. Accessed April 8, 2019.
20. Holbrook A, Schulman S, Witt DM, et al. Evidence-based management of anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(suppl 2):e152S-e184S.
21. Centers for Medicare and Medicaid Services. Part D Excluded Drugs. https://www.medicareadvocacy.org/old-site/News/Archives/PartD_ExcludedDrugsByState.htm. Accessed on August 23, 2018.
Between March 7, 2018, and May 9, 2018, at least 164 people in Illinois were sickened by synthetic cannabinoids laced with rodenticides. The Illinois Department of Public Health has reported 4 deaths connected with the use of synthetic cannabinoids (sold under names such as Spice, K2, Legal Weed, etc).1 Synthetic cannabinoids are mind-altering chemicals that are sprayed on dried plant material and often sold at convenience stores. Some users have reported smoking these substances because they are generally not detected by standard urine toxicology tests.
Recreational use of synthetic cannabinoids can lead to serious and, at times, deadly complications. Chemicals found in rat poison have contaminated batches of synthetic cannabinoids, leading to coagulopathy and severe bleeding. Affected patients have reported hemoptysis, hematuria, severe epistaxis, bleeding gums, conjunctival hemorrhages, and gastrointestinal bleeding. The following case is of a patient who presented to an emergency department (ED) with severe coagulopathy and cardiotoxicity after using an adulterated synthetic cannabinoid product.
Case Presentation
A 65-year-old man presented to the ED reporting hematochezia, hematuria, and hemoptysis. He reported that these symptoms began about 1 day after he had smoked a synthetic cannabinoid called K2. The patient stated that some of his friends who used the same product were experiencing similar symptoms. He reported mild generalized abdominal pain but reported no chest pain, dyspnea, headache, fevers, chills, or dysuria.
The patient’s past medical history included hypertension, dyslipidemia, chronic lower back pain, and vitamin D deficiency. His past surgical history was notable for an exploratory laparotomy after a stab wound to the abdomen. The patient reported taking the following medications: morphine SA 30 mg bid, meloxicam 15 mg daily, amitriptyline 100 mg qhs, amlodipine 5 mg daily, hydrocodone/acetaminophen 5/325 mg q12h prn, atorvastatin 20 mg qhs, omeprazole 20 mg qam, senna 187 mg daily prn, psyllium 1 packet dissolved in water daily prn, and cholecalciferol 1,000 IU daily.
The patient’s temperature was 98o F, blood pressure, 144/80 mm Hg; pulse, 131 beats per minute; respiratory rate, 18 breaths per minute; and O2 saturation, 98% (ambient air). A physical examination revealed no acute distress; he was coughing up blood; clear lungs; heart sounds were tachycardic and irregularly irregular; soft, nondistended, mild generalized tenderness in the abdomen with no guarding and no rebound. The pertinent laboratory tests were international normalized ratio (INR), > 20; prothrombin time, > 150 seconds; prothrombin thromboplastin time, 157 seconds; hemoglobin, 13.3 g/dL; platelet count, 195 k/uL; white blood count, 11.3 k/uL; creatinine, 0.57mg/dL; potassium, 3.8 mmol/L, D-dimertest, 0.87 ug/mL fibrinogen equivalent units; fibrinogen level, 624 mg/dL; troponin, < 0.04 ng/mL; lactic acid, 1.3 mmol/L; total bilirubin, 0.8 mg/dL; alanine aminotransferase, 22 U/L, aspartate aminotransferase, 22 U/L; alkaline phosphatase, 89 U/L; urinalysis with > 50 red blood cells/high power field; large blood, negative leukocyte esterase, negative nitrite. The patient’s urine toxicology was negative for cannabinoids, methadone, amphetamines, cocaine, and benzodiazepines; but was positive for opiates. An anticoagulant poisoning panel also was ordered.
An electrocardiogram (ECG) and imaging studies were ordered. The ECG showed atrial fibrillation (AF) with rapid ventricular response (Figure 1). A chest X-ray indicated bibasilar consolidations that were worse on the right side. A noncontrast computed tomography (CT) of the head did not show intracranial bleeding. An abdomen/pelvis CT showed bilateral diffuse patchy peribronchovascular ground-glass opacities in the lung bases that could represent pulmonary hemorrhage, but no peritoneal or retroperitoneal bleeding.
Treatment
In the ED, the case was discussed with the Illinois Poison Control Center. The patient was diagnosed with coagulopathy likely due to anticoagulant poisoning. He was immediately treated with 10 mg of IV vitamin K, a fixed dose of 2,000 units of 4-factor prothrombin complex concentrate, and 4 units of fresh frozen plasma. His INR improved to 1.42 within several hours. He received 5 mg of IV metoprolol for uncontrolled AF and was admitted to the intensive care unit (ICU) for further care.
In the ICU the patient was started on oral vitamin K 50 mg tid for ongoing treatment of coagulopathy due to concern for possible rodenticide poisoning associated with very long half-life. This dose was then decreased to 50 mg bid. He was given IV fluid resuscitation with normal saline and started on rate control for AF with oral metoprolol. His heart rate improved. An echocardiogram showed new cardiomyopathy with an ejection fraction of 25% to 30%. Given basilar infiltrates and 1 episode of low-grade fever, he was started on ceftriaxone for possible community-acquired pneumonia. The patient was started on cholestyramine to help with washout of the possible rodenticide. No endoscopic interventions were performed.
The patient was transferred to an inpatient telemetry floor 24 hours after admission to the ICU once his tachycardia and bleeding improved. He did not require transfusion of packed red blood cells. In the ICU his INR had ranged between 1.62 and 2.46 (down from > 20 in the ED). His hemoglobin dropped from 13.3 g/dL on admission to 12 g/dL on transfer from the ICU, before stabilizing around 11 g/dL on the floor. The patient’s heart rate required better control, so metoprolol was increased to a total daily dose of 200 mg on the telemetry floor. Oral digoxin was then added after a digoxin load for additional rate control, as the patient remained tachycardic. Twice a day the patient continued to take 50 mg vitamin K. Cholestyramine and ceftriaxone were initially continued, but when the INR started increasing again, the cholestyramine was stopped to allow for an increase to more frequent 3-times daily vitamin 50 mg K administration (cholestyramine can interfere with vitamin K absorption). According to the toxicology service, there was only weak evidence to support use of cholestyramine in this setting.
Given his ongoing mild hemoptysis, the patient received first 1 unit, and then another 4 units of FFP when the INR increased to 3.96 despite oral vitamin K. After FFP, the INR decreased to 1.93 and subsequently to 1.52. A CT of the chest showed patchy ground-glass densities throughout the lungs, predominantly at the lung bases and to a lesser extent in the upper lobes. The findings were felt to represent pulmonary hemorrhage given the patient’s history of hemoptysis (Figure 2).
The patient’s heart rate control improved, and he remained hemodynamically stable. A thyroid function test was within normal limits. Lisinopril was added to metoprolol and digoxin given his newly diagnosed cardiomyopathy. The patient was observed for a total of 4 days on the inpatient floor and discharged after his INR stabilized around 1.5 on twice daily 50 mg vitamin K. The patient’s hematuria and hematochezia completely resolved, and hemoptysis was much improved at the time of discharge. His hemoglobin remained stable. The anticoagulant poisoning panel came back positive for
The patient has remained in AF at all follow-up visits. The INR normalized by 6 weeks after hospital discharge, and the dose of vitamin K slowly was tapered with close monitoring of the INR. Vitamin K was tapered for about 6 months after his initial presentation, and the patient was started on a direct oral anticoagulant (DOAC) for anticoagulation when the INR remained stable off vitamin K. He subsequently underwent a transesophageal echocardiogram followed by an attempt at direct current (DC) cardioversion; however, he did not remain in sinus rhythm, and is being continued on anticoagulation and rate control for his AF.
Discussion
Users generally smoke synthetic cannabinoids, which produce cannabis-like effects. However, atypical intoxication effects with worse complications often occur.2 These products typically contain dried shredded plant material that is soaked in or sprayed with several synthetic cannabinoids, varying in dosage and combination.3 Synthetic cannabinoids have been associated with serious adverse effects (AEs), including drowsiness, light-headedness, and fast or irregular heartbeat.4 More severe clinical features such as psychosis, delirium, cardiotoxicity, seizures, rhabdomyolysis, acute kidney injury, hyperthermia, myocardial ischemia, ischemic strokes, and death have also been noted.4
It is not known how some batches of synthetic cannabinoids came to be contaminated with rat poison or how commonly such an adulteration is found across the country. Several different guidelines provide pathways for the treatment of acute bleeding in the setting of coagulopathy due to vitamin K antagonists.5,6 Each guideline divides the indications for reversal into either severity of bleeding or the criticality of the bleeding based on location.5,6 All guidelines recommend the use of vitamin K (either oral or IV) followed by FFP or 4-factor prothrombin complex concentrate (PCC) for more severe bleeding.5,6 However, recommendations regarding the use of PCC vary in dosing for vitamin K antagonists (in contrast to treatment of coagulopathy due to DOACs). Recent studies and guidelines suggest that fixed-dose (rather than weight-based dose) PCC is effective for the reversal of coagulopathy due to vitamin K antagonists.6,7 Using fixed rather than weight-based dosing decreases cost and may decrease the possibility of thrombotic AEs.7 In this patient, a fixed-dose of 2,000 units of PCC was given based on data that were extrapolated from warfarin reversal using PCC.7
The vitamin K antagonists that adulterated this patient’s synthetic cannabinoid were difenacoum and brodifacoum, which are 4-hydroxycoumarin derivatives. These are second-generation long-acting anticoagulant rodenticides (LAARs) that are about 100 times more potent than warfarin.8 As the name implies, LAARs have a longer duration of action in the body of any organism that ingests the poison, which is due to the highly lipophilic groups that have been added to the warfarin molecule to combat resistance in rodents.9
As a result of the deposition in the tissues, there have been reports of the duration of action of brodifacoum ranging from 51 days to 9 months after ingestion, with the latter caused by an intentional overdose in a human.9-12 Reports suggest that coagulopathy is not likely to occur when the serum brodifacoum concentration is < 10 ng/mL.13,14 Animal models show difenacoum has a tissue half-life of about 62 days.15 Reports of difenacoum poisoning in humans have shown variable lengths of treatment, ranging from 30 to 47 days.16-18 The length of treatment for either brodifacoum or difenacoum will depend on the amount of poison exposure.
The long duration of action and treatment duration may lead to problems with drug procurement, especially in the early phase of treatment in which IV vitamin K is used. The supply of IV vitamin K recently has been limited for at least some manufacturers. According to the American Society of Health System Pharmacists Current Drug Shortage List, the increased demand is thought to be due to increased use of synthetic inhaled cannabinoids laced with anticoagulant.19 IV vitamin K products are available from suppliers such as Amphastar (Rancho Cucamonga, CA) and Hospira (Lake Forest, IL).
The American College of Chest Physicians recommends IV vitamin K administration in patients with major bleeding secondary to vitamin K antagonists.20 The oral route is thought to be more effective than a subcutaneous route in the treatment of nonbleeding patients with rodenticide-associated coagulopathy. Due to erratic and unpredictable absorption, the subcutaneous route of administration has fallen out of favor. Oral vitamin K products were not affected by the recent shortage. However, large doses of oral vitamin K can be costly. Due to the long half-life of LAAR, many patients are discharged with a prescription for oral vitamin K. Although vitamin K is found in most over-the-counter (OTC) multivitamins, the strength is insufficient. Most OTC formulations are ≤ 100 μg, whereas the prescription strength is 5 mg, but patients being treated for rodenticide poisoning require much larger doses.
Commercial insurance carriers and Medicare Part D usually do not cover vitamins and minerals unless it is for a medically accepted indication or is an indication supported by citation in either the American Hospital Formulary System, United States Pharmacopeia drug information book, or an electronic information resource that is supported by evidence such as Micromedex.21 For a patient without insurance coverage being treated with high-dose vitamin K therapy for rodenticide poisoning outside of a federal health care system, the cost could be as high as $500 to $1,000 per day, depending on the dose of vitamin K needed to maintain an acceptable INR.
Conclusion
In addition to bleeding as a result of coagulopathy, this patient presented with new onset of AF with rapid ventricular response and a newly diagnosed cardiomyopathy. Although the patient had other cardiovascular risk factors, such as hypertension, dyslipidemia, and a remote history of cocaine use, it is likely that the use of the synthetic cannabinoids contributed to the development and/or worsening of this arrhythmia and cardiomyopathy. The patient remained in AF 6 weeks after hospital discharge with a controlled ventricular rate on metoprolol and digoxin. An interval echocardiogram 6 weeks after hospital discharge showed a recovered ejection fraction. In cases of tachycardia-induced cardiomyopathy, the ejection fraction often recovers with control of the tachycardia. The patient was weaned off vitamin K about 6 months after his initial presentation and started on a DOAC for anticoagulation. He subsequently underwent a transesophageal echocardiogram followed by an attempt at DC cardioversion; however, he did not remain in sinus rhythm and is being continued on anticoagulation and rate control for his AF.
Although unclear how synthetic cannabinoids became adulterated with a potent vitamin K antagonist, health care practitioners should consider this if a patient presents with unexplained coagulopathy and widespread bleeding. Fixed-dose PCC should be considered as an alternative to weight-based dosing in these cases. Physicians and pharmacy personnel should anticipate a need for long-term high doses of vitamin K in order to begin work early to obtain sufficient supplies to treat presenting patients.
Between March 7, 2018, and May 9, 2018, at least 164 people in Illinois were sickened by synthetic cannabinoids laced with rodenticides. The Illinois Department of Public Health has reported 4 deaths connected with the use of synthetic cannabinoids (sold under names such as Spice, K2, Legal Weed, etc).1 Synthetic cannabinoids are mind-altering chemicals that are sprayed on dried plant material and often sold at convenience stores. Some users have reported smoking these substances because they are generally not detected by standard urine toxicology tests.
Recreational use of synthetic cannabinoids can lead to serious and, at times, deadly complications. Chemicals found in rat poison have contaminated batches of synthetic cannabinoids, leading to coagulopathy and severe bleeding. Affected patients have reported hemoptysis, hematuria, severe epistaxis, bleeding gums, conjunctival hemorrhages, and gastrointestinal bleeding. The following case is of a patient who presented to an emergency department (ED) with severe coagulopathy and cardiotoxicity after using an adulterated synthetic cannabinoid product.
Case Presentation
A 65-year-old man presented to the ED reporting hematochezia, hematuria, and hemoptysis. He reported that these symptoms began about 1 day after he had smoked a synthetic cannabinoid called K2. The patient stated that some of his friends who used the same product were experiencing similar symptoms. He reported mild generalized abdominal pain but reported no chest pain, dyspnea, headache, fevers, chills, or dysuria.
The patient’s past medical history included hypertension, dyslipidemia, chronic lower back pain, and vitamin D deficiency. His past surgical history was notable for an exploratory laparotomy after a stab wound to the abdomen. The patient reported taking the following medications: morphine SA 30 mg bid, meloxicam 15 mg daily, amitriptyline 100 mg qhs, amlodipine 5 mg daily, hydrocodone/acetaminophen 5/325 mg q12h prn, atorvastatin 20 mg qhs, omeprazole 20 mg qam, senna 187 mg daily prn, psyllium 1 packet dissolved in water daily prn, and cholecalciferol 1,000 IU daily.
The patient’s temperature was 98o F, blood pressure, 144/80 mm Hg; pulse, 131 beats per minute; respiratory rate, 18 breaths per minute; and O2 saturation, 98% (ambient air). A physical examination revealed no acute distress; he was coughing up blood; clear lungs; heart sounds were tachycardic and irregularly irregular; soft, nondistended, mild generalized tenderness in the abdomen with no guarding and no rebound. The pertinent laboratory tests were international normalized ratio (INR), > 20; prothrombin time, > 150 seconds; prothrombin thromboplastin time, 157 seconds; hemoglobin, 13.3 g/dL; platelet count, 195 k/uL; white blood count, 11.3 k/uL; creatinine, 0.57mg/dL; potassium, 3.8 mmol/L, D-dimertest, 0.87 ug/mL fibrinogen equivalent units; fibrinogen level, 624 mg/dL; troponin, < 0.04 ng/mL; lactic acid, 1.3 mmol/L; total bilirubin, 0.8 mg/dL; alanine aminotransferase, 22 U/L, aspartate aminotransferase, 22 U/L; alkaline phosphatase, 89 U/L; urinalysis with > 50 red blood cells/high power field; large blood, negative leukocyte esterase, negative nitrite. The patient’s urine toxicology was negative for cannabinoids, methadone, amphetamines, cocaine, and benzodiazepines; but was positive for opiates. An anticoagulant poisoning panel also was ordered.
An electrocardiogram (ECG) and imaging studies were ordered. The ECG showed atrial fibrillation (AF) with rapid ventricular response (Figure 1). A chest X-ray indicated bibasilar consolidations that were worse on the right side. A noncontrast computed tomography (CT) of the head did not show intracranial bleeding. An abdomen/pelvis CT showed bilateral diffuse patchy peribronchovascular ground-glass opacities in the lung bases that could represent pulmonary hemorrhage, but no peritoneal or retroperitoneal bleeding.
Treatment
In the ED, the case was discussed with the Illinois Poison Control Center. The patient was diagnosed with coagulopathy likely due to anticoagulant poisoning. He was immediately treated with 10 mg of IV vitamin K, a fixed dose of 2,000 units of 4-factor prothrombin complex concentrate, and 4 units of fresh frozen plasma. His INR improved to 1.42 within several hours. He received 5 mg of IV metoprolol for uncontrolled AF and was admitted to the intensive care unit (ICU) for further care.
In the ICU the patient was started on oral vitamin K 50 mg tid for ongoing treatment of coagulopathy due to concern for possible rodenticide poisoning associated with very long half-life. This dose was then decreased to 50 mg bid. He was given IV fluid resuscitation with normal saline and started on rate control for AF with oral metoprolol. His heart rate improved. An echocardiogram showed new cardiomyopathy with an ejection fraction of 25% to 30%. Given basilar infiltrates and 1 episode of low-grade fever, he was started on ceftriaxone for possible community-acquired pneumonia. The patient was started on cholestyramine to help with washout of the possible rodenticide. No endoscopic interventions were performed.
The patient was transferred to an inpatient telemetry floor 24 hours after admission to the ICU once his tachycardia and bleeding improved. He did not require transfusion of packed red blood cells. In the ICU his INR had ranged between 1.62 and 2.46 (down from > 20 in the ED). His hemoglobin dropped from 13.3 g/dL on admission to 12 g/dL on transfer from the ICU, before stabilizing around 11 g/dL on the floor. The patient’s heart rate required better control, so metoprolol was increased to a total daily dose of 200 mg on the telemetry floor. Oral digoxin was then added after a digoxin load for additional rate control, as the patient remained tachycardic. Twice a day the patient continued to take 50 mg vitamin K. Cholestyramine and ceftriaxone were initially continued, but when the INR started increasing again, the cholestyramine was stopped to allow for an increase to more frequent 3-times daily vitamin 50 mg K administration (cholestyramine can interfere with vitamin K absorption). According to the toxicology service, there was only weak evidence to support use of cholestyramine in this setting.
Given his ongoing mild hemoptysis, the patient received first 1 unit, and then another 4 units of FFP when the INR increased to 3.96 despite oral vitamin K. After FFP, the INR decreased to 1.93 and subsequently to 1.52. A CT of the chest showed patchy ground-glass densities throughout the lungs, predominantly at the lung bases and to a lesser extent in the upper lobes. The findings were felt to represent pulmonary hemorrhage given the patient’s history of hemoptysis (Figure 2).
The patient’s heart rate control improved, and he remained hemodynamically stable. A thyroid function test was within normal limits. Lisinopril was added to metoprolol and digoxin given his newly diagnosed cardiomyopathy. The patient was observed for a total of 4 days on the inpatient floor and discharged after his INR stabilized around 1.5 on twice daily 50 mg vitamin K. The patient’s hematuria and hematochezia completely resolved, and hemoptysis was much improved at the time of discharge. His hemoglobin remained stable. The anticoagulant poisoning panel came back positive for
The patient has remained in AF at all follow-up visits. The INR normalized by 6 weeks after hospital discharge, and the dose of vitamin K slowly was tapered with close monitoring of the INR. Vitamin K was tapered for about 6 months after his initial presentation, and the patient was started on a direct oral anticoagulant (DOAC) for anticoagulation when the INR remained stable off vitamin K. He subsequently underwent a transesophageal echocardiogram followed by an attempt at direct current (DC) cardioversion; however, he did not remain in sinus rhythm, and is being continued on anticoagulation and rate control for his AF.
Discussion
Users generally smoke synthetic cannabinoids, which produce cannabis-like effects. However, atypical intoxication effects with worse complications often occur.2 These products typically contain dried shredded plant material that is soaked in or sprayed with several synthetic cannabinoids, varying in dosage and combination.3 Synthetic cannabinoids have been associated with serious adverse effects (AEs), including drowsiness, light-headedness, and fast or irregular heartbeat.4 More severe clinical features such as psychosis, delirium, cardiotoxicity, seizures, rhabdomyolysis, acute kidney injury, hyperthermia, myocardial ischemia, ischemic strokes, and death have also been noted.4
It is not known how some batches of synthetic cannabinoids came to be contaminated with rat poison or how commonly such an adulteration is found across the country. Several different guidelines provide pathways for the treatment of acute bleeding in the setting of coagulopathy due to vitamin K antagonists.5,6 Each guideline divides the indications for reversal into either severity of bleeding or the criticality of the bleeding based on location.5,6 All guidelines recommend the use of vitamin K (either oral or IV) followed by FFP or 4-factor prothrombin complex concentrate (PCC) for more severe bleeding.5,6 However, recommendations regarding the use of PCC vary in dosing for vitamin K antagonists (in contrast to treatment of coagulopathy due to DOACs). Recent studies and guidelines suggest that fixed-dose (rather than weight-based dose) PCC is effective for the reversal of coagulopathy due to vitamin K antagonists.6,7 Using fixed rather than weight-based dosing decreases cost and may decrease the possibility of thrombotic AEs.7 In this patient, a fixed-dose of 2,000 units of PCC was given based on data that were extrapolated from warfarin reversal using PCC.7
The vitamin K antagonists that adulterated this patient’s synthetic cannabinoid were difenacoum and brodifacoum, which are 4-hydroxycoumarin derivatives. These are second-generation long-acting anticoagulant rodenticides (LAARs) that are about 100 times more potent than warfarin.8 As the name implies, LAARs have a longer duration of action in the body of any organism that ingests the poison, which is due to the highly lipophilic groups that have been added to the warfarin molecule to combat resistance in rodents.9
As a result of the deposition in the tissues, there have been reports of the duration of action of brodifacoum ranging from 51 days to 9 months after ingestion, with the latter caused by an intentional overdose in a human.9-12 Reports suggest that coagulopathy is not likely to occur when the serum brodifacoum concentration is < 10 ng/mL.13,14 Animal models show difenacoum has a tissue half-life of about 62 days.15 Reports of difenacoum poisoning in humans have shown variable lengths of treatment, ranging from 30 to 47 days.16-18 The length of treatment for either brodifacoum or difenacoum will depend on the amount of poison exposure.
The long duration of action and treatment duration may lead to problems with drug procurement, especially in the early phase of treatment in which IV vitamin K is used. The supply of IV vitamin K recently has been limited for at least some manufacturers. According to the American Society of Health System Pharmacists Current Drug Shortage List, the increased demand is thought to be due to increased use of synthetic inhaled cannabinoids laced with anticoagulant.19 IV vitamin K products are available from suppliers such as Amphastar (Rancho Cucamonga, CA) and Hospira (Lake Forest, IL).
The American College of Chest Physicians recommends IV vitamin K administration in patients with major bleeding secondary to vitamin K antagonists.20 The oral route is thought to be more effective than a subcutaneous route in the treatment of nonbleeding patients with rodenticide-associated coagulopathy. Due to erratic and unpredictable absorption, the subcutaneous route of administration has fallen out of favor. Oral vitamin K products were not affected by the recent shortage. However, large doses of oral vitamin K can be costly. Due to the long half-life of LAAR, many patients are discharged with a prescription for oral vitamin K. Although vitamin K is found in most over-the-counter (OTC) multivitamins, the strength is insufficient. Most OTC formulations are ≤ 100 μg, whereas the prescription strength is 5 mg, but patients being treated for rodenticide poisoning require much larger doses.
Commercial insurance carriers and Medicare Part D usually do not cover vitamins and minerals unless it is for a medically accepted indication or is an indication supported by citation in either the American Hospital Formulary System, United States Pharmacopeia drug information book, or an electronic information resource that is supported by evidence such as Micromedex.21 For a patient without insurance coverage being treated with high-dose vitamin K therapy for rodenticide poisoning outside of a federal health care system, the cost could be as high as $500 to $1,000 per day, depending on the dose of vitamin K needed to maintain an acceptable INR.
Conclusion
In addition to bleeding as a result of coagulopathy, this patient presented with new onset of AF with rapid ventricular response and a newly diagnosed cardiomyopathy. Although the patient had other cardiovascular risk factors, such as hypertension, dyslipidemia, and a remote history of cocaine use, it is likely that the use of the synthetic cannabinoids contributed to the development and/or worsening of this arrhythmia and cardiomyopathy. The patient remained in AF 6 weeks after hospital discharge with a controlled ventricular rate on metoprolol and digoxin. An interval echocardiogram 6 weeks after hospital discharge showed a recovered ejection fraction. In cases of tachycardia-induced cardiomyopathy, the ejection fraction often recovers with control of the tachycardia. The patient was weaned off vitamin K about 6 months after his initial presentation and started on a DOAC for anticoagulation. He subsequently underwent a transesophageal echocardiogram followed by an attempt at DC cardioversion; however, he did not remain in sinus rhythm and is being continued on anticoagulation and rate control for his AF.
Although unclear how synthetic cannabinoids became adulterated with a potent vitamin K antagonist, health care practitioners should consider this if a patient presents with unexplained coagulopathy and widespread bleeding. Fixed-dose PCC should be considered as an alternative to weight-based dosing in these cases. Physicians and pharmacy personnel should anticipate a need for long-term high doses of vitamin K in order to begin work early to obtain sufficient supplies to treat presenting patients.
1. Illinois Department of Public Health. Synthetic cannabinoids. http://dph.illinois.gov/topics-services/prevention-wellness/medical-cannabis/synthetic-cannabinoids. Updated May 30, 2018. Accessed April 8, 2019.
2. Tournebize J, Gibaja V, Kahn JP. Acute effects of synthetic cannabinoids: update 2015. Subst Abus. 2017;38(3):344-366.
3. United Nations Office on Drugs and Crime. Global SMART update. https://www.unodc.org/documents/scientific/Global_SMART_Update_13_web.pdf. Published March 2015. Accessed April 8, 2019.
4. Adams AJ, Banister SD, Irizarry L, Trecki J, Schwartz M, Gerona R, “Zombie” outbreak caused by the synthetic cannabinoid AMB-FUBINACA in New York. N Engl J Med. 2017;376(3):235-242.
5. Tomaselli GF, Mahaffey KW, Cuker A, et al. 2017 ACC expert consensus decision pathway on management of bleeding in patients on oral anticoagulants: a report of the American College of Cardiology Task Force on Expert Consensus Decision Pathways. J Am Coll Cardiol. 2017;70(24):3042-3067.
6. Cushman M, Lim W, Zakai NA. 2011 Clinical Practice guide on anticoagulant dosing and management of anticoagulant-associated bleeding complications in adults. http://www.hematology.org/Clinicians/Guidelines-Quality/Quick-Ref/525.aspx. Published 2011. Accessed April 8, 2019.
7. Klein L, Peters J, Miner J, Gorlin J. Evaluation of fixed dose 4-factor prothrombin complex concentrate for emergent warfarin reversal. Am J Emerg Med. 2015;33(9):1213-1218.
8. Bachmann KA, Sullivan TJ. Dispositional and pharmacodynamic characteristics of brodifacoum in warfarin-sensitive rats. Pharmacology. 1983;27(5):281-288.
9. Lipton RA, Klass EM. Human ingestion of ‘superwarfarin’ rodenticide resulting in a prolonged anticoagulant effect. JAMA. 1984;252(21):3004-3005.
10. Chong LL, Chau WK, Ho CH. A case of ‘superwarfarin’ poisoning. Scand J Haematol. 1986;36(3):314-331.
11. Jones EC, Growe GH, Naiman SC. Prolonged anticoagulation in rat poisoning. JAMA. 1984;252(21):3005-3007.
12. Babcock J, Hartman K, Pedersen A, Murphy M, Alving B. Rodenticide-induced coagulopathy in a young child. A case of Munchausen syndrome by proxy. Am J Pediatr Hematol Oncol. 1993;15(1):126-130.
13. Hollinger BR, Pastoor TP. Case management and plasma half-life in a case of brodifacoum poisoning. Arch Intern Med. 1993;153(16):1925-1928.
14. Bruno GR, Howland MA, McMeeking A, Hoffman RS. Long-acting anticoagulant overdose: brodifacoum kinetics and optimal vitamin K dosing. Ann Emerg Med. 2000;36(3):262-267.
15. Vandenbrouke V, Bousquet-Meloua A, De Backer P, Croubels S. Pharmacokinetics of eight anticoagulant rodenticides in mice after single oral administration. J Vet Pharmacol Ther. 2008;31(5):437-445.
16. Barlow AM, Gay AL, Park BK. Difenacoum (Neosorexa) poisoning. Br Med J (Clin Res Ed). 1982;285(6341):541.
17. Katona B, Wason S. Superwarfarin poisoning. J Emerg Med. 1989;7(6):627-631.
18. Butcher GP, Shearer MJ, MacNicoll AD, Kelly MJ, Ind PW. Difenacoum poisoning as a cause of haematuria. Hum Exp Toxicol. 1992;11(6):553-554.
19. American Society of Health System Pharmacists. Current drug shortages. Vitamin K (phytonadione) injection. https://www.ashp.org/drug-shortages/current-shortages/Drug-Shortage-Detail.aspx?id=100. Updated July 5, 2018. Accessed April 8, 2019.
20. Holbrook A, Schulman S, Witt DM, et al. Evidence-based management of anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(suppl 2):e152S-e184S.
21. Centers for Medicare and Medicaid Services. Part D Excluded Drugs. https://www.medicareadvocacy.org/old-site/News/Archives/PartD_ExcludedDrugsByState.htm. Accessed on August 23, 2018.
1. Illinois Department of Public Health. Synthetic cannabinoids. http://dph.illinois.gov/topics-services/prevention-wellness/medical-cannabis/synthetic-cannabinoids. Updated May 30, 2018. Accessed April 8, 2019.
2. Tournebize J, Gibaja V, Kahn JP. Acute effects of synthetic cannabinoids: update 2015. Subst Abus. 2017;38(3):344-366.
3. United Nations Office on Drugs and Crime. Global SMART update. https://www.unodc.org/documents/scientific/Global_SMART_Update_13_web.pdf. Published March 2015. Accessed April 8, 2019.
4. Adams AJ, Banister SD, Irizarry L, Trecki J, Schwartz M, Gerona R, “Zombie” outbreak caused by the synthetic cannabinoid AMB-FUBINACA in New York. N Engl J Med. 2017;376(3):235-242.
5. Tomaselli GF, Mahaffey KW, Cuker A, et al. 2017 ACC expert consensus decision pathway on management of bleeding in patients on oral anticoagulants: a report of the American College of Cardiology Task Force on Expert Consensus Decision Pathways. J Am Coll Cardiol. 2017;70(24):3042-3067.
6. Cushman M, Lim W, Zakai NA. 2011 Clinical Practice guide on anticoagulant dosing and management of anticoagulant-associated bleeding complications in adults. http://www.hematology.org/Clinicians/Guidelines-Quality/Quick-Ref/525.aspx. Published 2011. Accessed April 8, 2019.
7. Klein L, Peters J, Miner J, Gorlin J. Evaluation of fixed dose 4-factor prothrombin complex concentrate for emergent warfarin reversal. Am J Emerg Med. 2015;33(9):1213-1218.
8. Bachmann KA, Sullivan TJ. Dispositional and pharmacodynamic characteristics of brodifacoum in warfarin-sensitive rats. Pharmacology. 1983;27(5):281-288.
9. Lipton RA, Klass EM. Human ingestion of ‘superwarfarin’ rodenticide resulting in a prolonged anticoagulant effect. JAMA. 1984;252(21):3004-3005.
10. Chong LL, Chau WK, Ho CH. A case of ‘superwarfarin’ poisoning. Scand J Haematol. 1986;36(3):314-331.
11. Jones EC, Growe GH, Naiman SC. Prolonged anticoagulation in rat poisoning. JAMA. 1984;252(21):3005-3007.
12. Babcock J, Hartman K, Pedersen A, Murphy M, Alving B. Rodenticide-induced coagulopathy in a young child. A case of Munchausen syndrome by proxy. Am J Pediatr Hematol Oncol. 1993;15(1):126-130.
13. Hollinger BR, Pastoor TP. Case management and plasma half-life in a case of brodifacoum poisoning. Arch Intern Med. 1993;153(16):1925-1928.
14. Bruno GR, Howland MA, McMeeking A, Hoffman RS. Long-acting anticoagulant overdose: brodifacoum kinetics and optimal vitamin K dosing. Ann Emerg Med. 2000;36(3):262-267.
15. Vandenbrouke V, Bousquet-Meloua A, De Backer P, Croubels S. Pharmacokinetics of eight anticoagulant rodenticides in mice after single oral administration. J Vet Pharmacol Ther. 2008;31(5):437-445.
16. Barlow AM, Gay AL, Park BK. Difenacoum (Neosorexa) poisoning. Br Med J (Clin Res Ed). 1982;285(6341):541.
17. Katona B, Wason S. Superwarfarin poisoning. J Emerg Med. 1989;7(6):627-631.
18. Butcher GP, Shearer MJ, MacNicoll AD, Kelly MJ, Ind PW. Difenacoum poisoning as a cause of haematuria. Hum Exp Toxicol. 1992;11(6):553-554.
19. American Society of Health System Pharmacists. Current drug shortages. Vitamin K (phytonadione) injection. https://www.ashp.org/drug-shortages/current-shortages/Drug-Shortage-Detail.aspx?id=100. Updated July 5, 2018. Accessed April 8, 2019.
20. Holbrook A, Schulman S, Witt DM, et al. Evidence-based management of anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(suppl 2):e152S-e184S.
21. Centers for Medicare and Medicaid Services. Part D Excluded Drugs. https://www.medicareadvocacy.org/old-site/News/Archives/PartD_ExcludedDrugsByState.htm. Accessed on August 23, 2018.
Basal Cell Carcinoma Masquerading as a Dermoid Cyst and Bursitis of the Knee
Basal cell carcinoma (BCC) is the most frequently diagnosed skin cancer in the United States. It develops most often on sun-exposed skin, including the face and neck. Although BCCs are slow-growing tumors that rarely metastasize, they can cause notable local destruction with disfigurement if neglected or inadequately treated. Basal cell carcinoma arising on the legs is relatively uncommon.1,2 We present an interesting case of delayed diagnosis of BCC on the left knee due to earlier misdiagnoses of a dermoid cyst and bursitis.
Case Report
A 67-year-old man with no history of skin cancer presented with a painful growing tumor on the left knee of approximately 2 years’ duration. The patient’s primary care physician as well as a general surgeon initially diagnosed it as a dermoid cyst and bursitis. The nodule failed to respond to conservative therapy with nonsteroidal anti-inflammatory drugs and continued to grow until it began to ulcerate. Concerned about the possibility of septic arthritis, the patient’s primary care physician referred him to the emergency department. He was subsequently sent to the dermatology clinic.
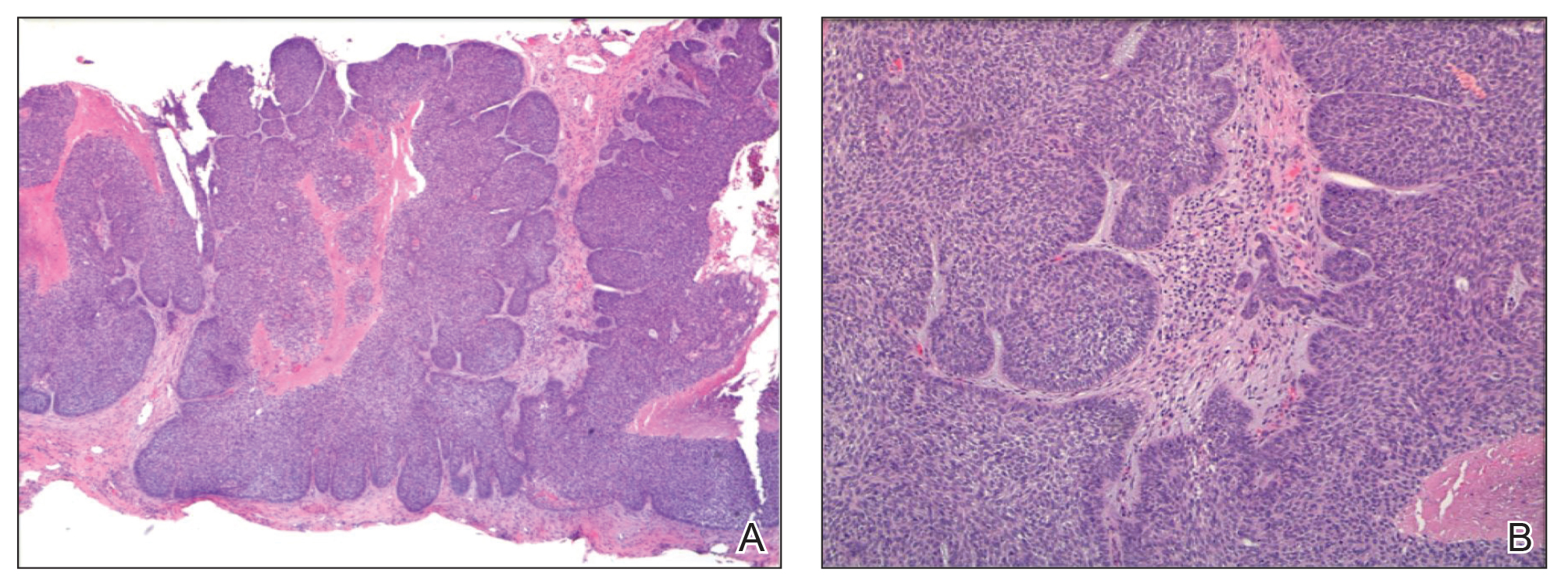
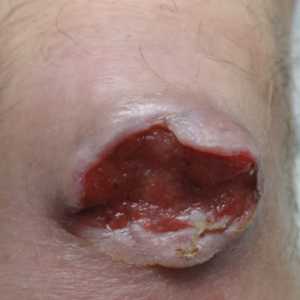
On examination by dermatology, a 6.3×4.4-cm, tender, mobile, ulcerated nodule was noted on the left knee (Figure 1A). No popliteal or inguinal lymph nodes were palpable. Basal cell carcinoma, squamous cell carcinoma, or atypical infection (eg, Leishmania, deep fungal, mycobacterial) was suspected clinically. The patient underwent a diagnostic skin biopsy; hematoxylin and eosin–stained sections revealed lobular proliferation of basaloid cells with peripheral palisading and central tumoral necrosis, consistent with primary BCC (Figure 2).
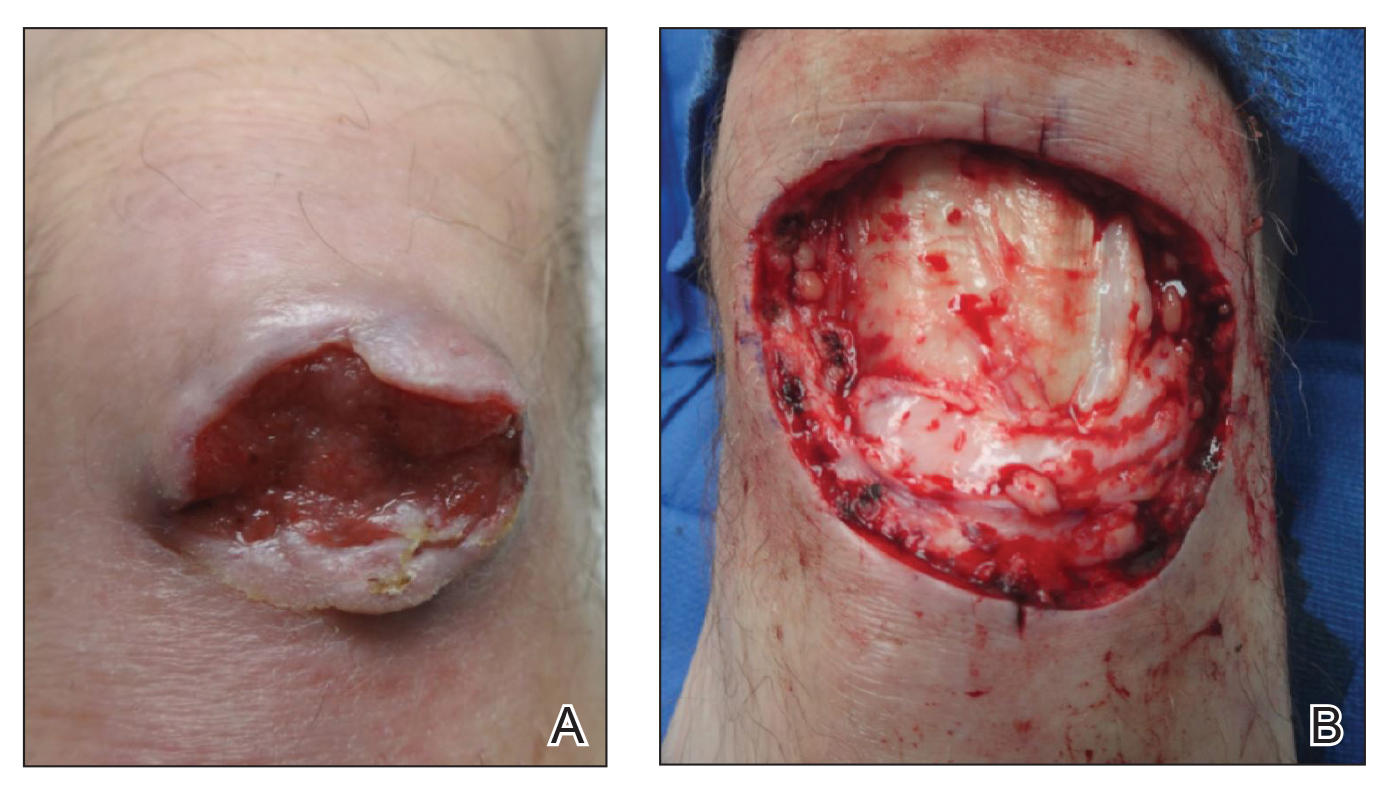
Given the size of the tumor, the patient was referred for Mohs micrographic surgery and eventual reconstruction by a plastic surgeon. The tumor was cleared after 2 stages of Mohs surgery, with a final wound size of 7.7×5.4 cm (Figure 1B). Plastic surgery later performed a gastrocnemius muscle flap with a split-thickness skin graft (175 cm2) to repair the wound.
Comment
Exposure to UV radiation is the primary causative agent of most BCCs, accounting for the preferential distribution of these tumors on sun-exposed areas of the body. Approximately 80% of BCCs are located on the head and neck, 10% occur on the trunk, and only 8% are found on the lower extremities.1
Giant BCC, the finding in this case, is defined by the American Joint Committee on Cancer as a tumor larger than 5 cm in diameter. Fewer than 1% of all BCCs achieve this size; they appear more commonly on the back where they can go unnoticed.2 Neglect and inadequate treatment of the primary tumor are the most important contributing factors to the size of giant BCCs. Giant BCCs also have more aggressive biologic behavior, with an increased risk for local invasion and metastasis.3 In this case, the lesion was larger than 5 cm in diameter and occurred on the lower extremity rather than on the trunk.
This case is unusual because delayed diagnosis of BCC was the result of misdiagnoses of a dermoid cyst and bursitis, with a diagnostic skin biopsy demonstrating BCC almost 2 years later. It should be emphasized that early diagnosis and treatment could prevent tumor expansion. Physicians should have a high degree of suspicion for BCC, especially when a dermoid cyst and knee bursitis fail to respond to conservative management.
- Pearson G, King LE, Boyd AS. Basal cell carcinoma of the lower extremities. Int J Dermatol. 1999;38:852-854.
- Arnaiz J, Gallardo E, Piedra T, et al. Giant basal cell carcinoma on the lower leg: MRI findings. J Plast Reconstr Aesthet Surg. 2007;60:1167-1168.
- Randle HW. Giant basal cell carcinoma [letter]. Int J Dermatol. 1996;35:222-223.
Basal cell carcinoma (BCC) is the most frequently diagnosed skin cancer in the United States. It develops most often on sun-exposed skin, including the face and neck. Although BCCs are slow-growing tumors that rarely metastasize, they can cause notable local destruction with disfigurement if neglected or inadequately treated. Basal cell carcinoma arising on the legs is relatively uncommon.1,2 We present an interesting case of delayed diagnosis of BCC on the left knee due to earlier misdiagnoses of a dermoid cyst and bursitis.
Case Report
A 67-year-old man with no history of skin cancer presented with a painful growing tumor on the left knee of approximately 2 years’ duration. The patient’s primary care physician as well as a general surgeon initially diagnosed it as a dermoid cyst and bursitis. The nodule failed to respond to conservative therapy with nonsteroidal anti-inflammatory drugs and continued to grow until it began to ulcerate. Concerned about the possibility of septic arthritis, the patient’s primary care physician referred him to the emergency department. He was subsequently sent to the dermatology clinic.
On examination by dermatology, a 6.3×4.4-cm, tender, mobile, ulcerated nodule was noted on the left knee (Figure 1A). No popliteal or inguinal lymph nodes were palpable. Basal cell carcinoma, squamous cell carcinoma, or atypical infection (eg, Leishmania, deep fungal, mycobacterial) was suspected clinically. The patient underwent a diagnostic skin biopsy; hematoxylin and eosin–stained sections revealed lobular proliferation of basaloid cells with peripheral palisading and central tumoral necrosis, consistent with primary BCC (Figure 2).
Given the size of the tumor, the patient was referred for Mohs micrographic surgery and eventual reconstruction by a plastic surgeon. The tumor was cleared after 2 stages of Mohs surgery, with a final wound size of 7.7×5.4 cm (Figure 1B). Plastic surgery later performed a gastrocnemius muscle flap with a split-thickness skin graft (175 cm2) to repair the wound.
Comment
Exposure to UV radiation is the primary causative agent of most BCCs, accounting for the preferential distribution of these tumors on sun-exposed areas of the body. Approximately 80% of BCCs are located on the head and neck, 10% occur on the trunk, and only 8% are found on the lower extremities.1
Giant BCC, the finding in this case, is defined by the American Joint Committee on Cancer as a tumor larger than 5 cm in diameter. Fewer than 1% of all BCCs achieve this size; they appear more commonly on the back where they can go unnoticed.2 Neglect and inadequate treatment of the primary tumor are the most important contributing factors to the size of giant BCCs. Giant BCCs also have more aggressive biologic behavior, with an increased risk for local invasion and metastasis.3 In this case, the lesion was larger than 5 cm in diameter and occurred on the lower extremity rather than on the trunk.
This case is unusual because delayed diagnosis of BCC was the result of misdiagnoses of a dermoid cyst and bursitis, with a diagnostic skin biopsy demonstrating BCC almost 2 years later. It should be emphasized that early diagnosis and treatment could prevent tumor expansion. Physicians should have a high degree of suspicion for BCC, especially when a dermoid cyst and knee bursitis fail to respond to conservative management.
Basal cell carcinoma (BCC) is the most frequently diagnosed skin cancer in the United States. It develops most often on sun-exposed skin, including the face and neck. Although BCCs are slow-growing tumors that rarely metastasize, they can cause notable local destruction with disfigurement if neglected or inadequately treated. Basal cell carcinoma arising on the legs is relatively uncommon.1,2 We present an interesting case of delayed diagnosis of BCC on the left knee due to earlier misdiagnoses of a dermoid cyst and bursitis.
Case Report
A 67-year-old man with no history of skin cancer presented with a painful growing tumor on the left knee of approximately 2 years’ duration. The patient’s primary care physician as well as a general surgeon initially diagnosed it as a dermoid cyst and bursitis. The nodule failed to respond to conservative therapy with nonsteroidal anti-inflammatory drugs and continued to grow until it began to ulcerate. Concerned about the possibility of septic arthritis, the patient’s primary care physician referred him to the emergency department. He was subsequently sent to the dermatology clinic.
On examination by dermatology, a 6.3×4.4-cm, tender, mobile, ulcerated nodule was noted on the left knee (Figure 1A). No popliteal or inguinal lymph nodes were palpable. Basal cell carcinoma, squamous cell carcinoma, or atypical infection (eg, Leishmania, deep fungal, mycobacterial) was suspected clinically. The patient underwent a diagnostic skin biopsy; hematoxylin and eosin–stained sections revealed lobular proliferation of basaloid cells with peripheral palisading and central tumoral necrosis, consistent with primary BCC (Figure 2).
Given the size of the tumor, the patient was referred for Mohs micrographic surgery and eventual reconstruction by a plastic surgeon. The tumor was cleared after 2 stages of Mohs surgery, with a final wound size of 7.7×5.4 cm (Figure 1B). Plastic surgery later performed a gastrocnemius muscle flap with a split-thickness skin graft (175 cm2) to repair the wound.
Comment
Exposure to UV radiation is the primary causative agent of most BCCs, accounting for the preferential distribution of these tumors on sun-exposed areas of the body. Approximately 80% of BCCs are located on the head and neck, 10% occur on the trunk, and only 8% are found on the lower extremities.1
Giant BCC, the finding in this case, is defined by the American Joint Committee on Cancer as a tumor larger than 5 cm in diameter. Fewer than 1% of all BCCs achieve this size; they appear more commonly on the back where they can go unnoticed.2 Neglect and inadequate treatment of the primary tumor are the most important contributing factors to the size of giant BCCs. Giant BCCs also have more aggressive biologic behavior, with an increased risk for local invasion and metastasis.3 In this case, the lesion was larger than 5 cm in diameter and occurred on the lower extremity rather than on the trunk.
This case is unusual because delayed diagnosis of BCC was the result of misdiagnoses of a dermoid cyst and bursitis, with a diagnostic skin biopsy demonstrating BCC almost 2 years later. It should be emphasized that early diagnosis and treatment could prevent tumor expansion. Physicians should have a high degree of suspicion for BCC, especially when a dermoid cyst and knee bursitis fail to respond to conservative management.
- Pearson G, King LE, Boyd AS. Basal cell carcinoma of the lower extremities. Int J Dermatol. 1999;38:852-854.
- Arnaiz J, Gallardo E, Piedra T, et al. Giant basal cell carcinoma on the lower leg: MRI findings. J Plast Reconstr Aesthet Surg. 2007;60:1167-1168.
- Randle HW. Giant basal cell carcinoma [letter]. Int J Dermatol. 1996;35:222-223.
- Pearson G, King LE, Boyd AS. Basal cell carcinoma of the lower extremities. Int J Dermatol. 1999;38:852-854.
- Arnaiz J, Gallardo E, Piedra T, et al. Giant basal cell carcinoma on the lower leg: MRI findings. J Plast Reconstr Aesthet Surg. 2007;60:1167-1168.
- Randle HW. Giant basal cell carcinoma [letter]. Int J Dermatol. 1996;35:222-223.
Practice Points
- This case highlights an unusual presentation of basal cell carcinoma masquerading as bursitis.
- Clinicians should be aware of confirmation bias, especially when multiple physicians and specialists are involved in a case.
- When the initial clinical impression is not corroborated by objective data or the condition is not responding to conventional therapy, it is important for clinicians to revisit the possibility of an inaccurate diagnosis.
Squamous Cell Carcinoma With Perineural Involvement in Nevus Sebaceus
First reported in 1895, nevus sebaceus (NS) is a con genital papillomatous hamartoma most commonly found on the scalp and face. 1 Lesions typically are yellow-orange plaques and often are hairless. Nevus sebaceus is most prominent in the few first months after birth and again at puberty during development of the sebaceous glands. Development of epithelial hyperplasia, cysts, verrucas, and benign or malignant tumors has been reported. 1 The most common benign tumors are syringocystadenoma papilliferum and trichoblastoma. Cases of malignancy are rare, and basal cell carcinoma is the predominant form (approximately 2% of cases). Squamous cell carcinoma (SCC) and adnexal carcinoma are reported at even lower rates. 1 Malignant transformation occurring during childhood is extremely uncommon. According to a PubMed search of articles indexed for MEDLINE using the terms nevus sebaceous, malignancy, and squamous cell carcinoma and narrowing the results to children, there have been only 4 prior reports of SCC developing within an NS in a child. 2-5 We report a case of SCC arising in an NS in a 13-year-old adolescent girl with perineural invasion.
Case Report
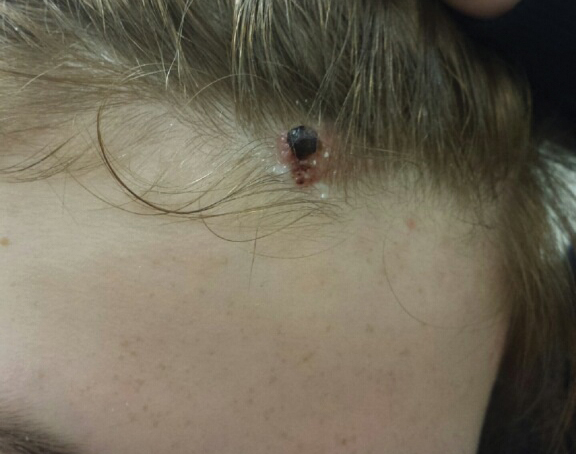
A 13-year-old fair-skinned adolescent girl presented with a hairless 2×2.5-cm yellow plaque at the hairline on the anterior central scalp. The plaque had been present since birth and had progressively developed a superiorly located 3×5-mm erythematous verrucous nodule (Figure 1) with an approximate height of 6 mm over the last year. The nodule was subjected to regular trauma and bled with minimal insult. The patient appeared otherwise healthy, with no history of skin cancer or other chronic medical conditions. There was no evidence of lymphadenopathy on examination, and no other skin abnormalities were noted. There was no reported family history of skin cancer or chronic skin conditions suggestive of increased risk for cancer or other pathologic dermatoses. Differential diagnoses for the plaque and nodule complex included verruca, Spitz nevus, or secondary neoplasm within NS.
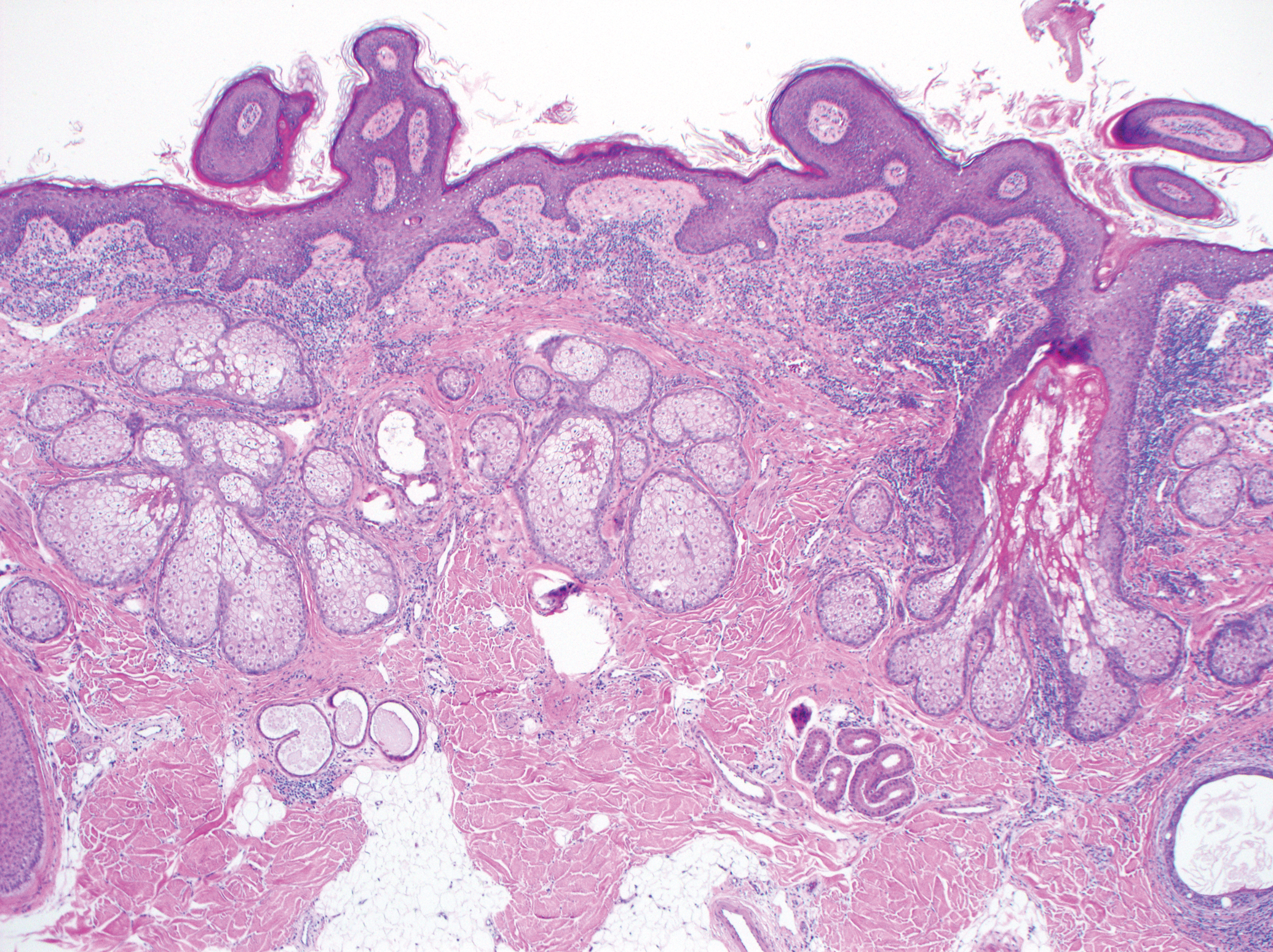
Excision was conducted under local anesthesia without complication. An elliptical section of skin measuring 0.8×2.5 cm was excised to a depth of 3 mm. The resulting wound was closed using a complex linear repair. The section was placed in formalin specimen transport medium and sent to Walter Reed National Military Medical Center (Bethesda, Maryland). Microscopic examination of the specimen revealed features typical for NS, including mild verrucous epidermal hyperplasia, sebaceous gland hyperplasia, presence of apocrine glands, and hamartomatous follicular proliferations (Figure 2). An even more papillomatous epidermal proliferation that was comprised of atypical squamous cells was present within the lesion. Similar atypical squamous cells infiltrated the superficial dermis in nests, cords, and single cells (Figure 3A). One focus showed perineural invasion with a small superficial nerve fiber surrounded by SCC (Figure 3B). The tumor was completely excised, with negative surgical margins extending approximately 2 mm. Adjuvant radiation therapy and further specialized Mohs micrographic excision were not performed because of the clear histologic appearance of the carcinoma and strong evidence of complete excision.
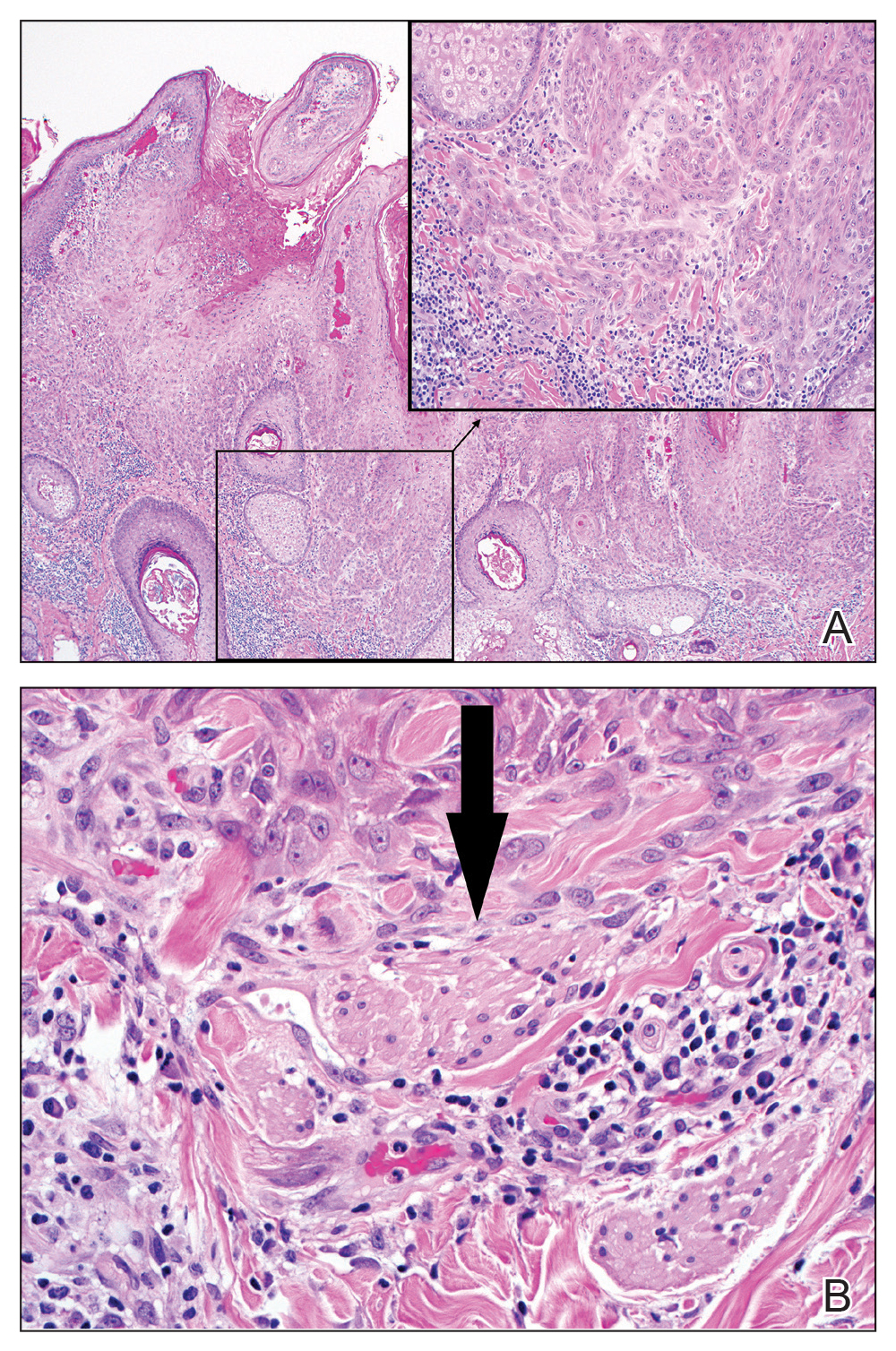
At 2-week follow-up, the surgical scar on the anterior central forehead was well healed without evidence of SCC recurrence. On physical examination there was neither lymphadenopathy nor signs of neurologic deficit, except for superficial cutaneous hypoesthesia in the immediate area surrounding the healed site. Following discussion with the patient and her parents, it was decided that the patient would obtain baseline laboratory tests, chest radiography, and abdominal ultrasonography, and she would undergo serial follow-up examinations every 3 months for the next 2 years. Annual follow-up was recommended after 2 years, with the caveat to return sooner if recurrence or symptoms were to arise.
Comment
Historically, there has been variability in the histopathologic interpretation of SCC in NS in the literature. Retrospective analysis of the histologic evidence of SCC in the 2 earliest possible cases of pediatric SCC in NS have been questioned due to the lack of clinical data presented and the possibility that the diagnosis of SCC was inaccurate.6 Our case was histopathologically interpreted as superficially invasive, well-differentiated SCC arising within an NS; therefore, we classified this case as SCC and took every precaution to ensure the lesion was completely excised, given the potentially invasive nature of SCC.
Our case is unique because it represents SCC in NS with histologic evidence of perineural involvement. Perineural invasion is a major route of tumor spread in SCC and may result in increased occurrence of regional lymph node spread and distant metastases, with path of least resistance or neural cell adhesion as possible spreading methods.7-9 However, there is a notable amount of prognostic variability based on tumor type, the nerve involved, and degree of involvement.9 It is common for cutaneous SCC to occur with invasion of small intradermal nerves, but a poor outcome is less likely in asymptomatic patients who have perineural involvement that was incidentally discovered on histologic examination.10
In our patient, the entire tumor was completely removed with local excision. Recurrence of the SCC or future symptoms of deep neural invasion were not anticipated given the postoperative evidence of clear margins in the excised skin and subdermal structures as well as the lack of preoperative and postoperative symptoms. Close clinical follow-up was warranted to monitor for early signs of recurrence or neural involvement. We have confidence that the planned follow-up regimen in our patient will reveal any early signs of new occurrence or recurrence.
In the case of recurrence, Mohs micrographic surgery would likely be indicated. We elected not to treat with adjuvant radiotherapy because its benefit in cutaneous SCC with perineural invasion is debatable based on the lack of randomized controlled clinical evidence.10,11 The patient obtained postoperative baseline complete blood cell count with differential, posterior/anterior and lateral chest radiographs, as well as abdominal ultrasonography. Each returned negative findings of hematologic or distant organ metastases, with subsequent follow-up visits also negative for any new concerning findings.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2, pt 1):263-268.
- Aguayo R, Pallares J, Cassanova JM, et al. Squamous cell carcinoma developing in Jadassohn’s sebaceous nevus: case report and review of the literature. Dermatol Surg. 2010;36:1763-1768.
- Taher M, Feibleman C, Bennet R. Squamous cell carcinoma arising in a nevus sebaceous of Jadassohn in a 9-year-old girl: treatment using Mohs micrographic surgery with literature review. Dermatol Surg. 2010;36:1203-1208.
- Hidvegi NC, Kangesu L, Wolfe KQ. Squamous cell carcinoma complicating naevus sebaceous of Jadassohn in a child. Br J Plast Surg. 2003;56:50-52.
- Belhadjali H, Moussa A, Yahia S, et al. Simultaneous occurrence of squamous cell carcinomas within a nevus sebaceous of Jadassohn in an 11-year-old girl. Pediatr Dermatol. 2009;26:236-237.
- Wilson-Jones EW, Heyl T. Naevus sebaceus: a report of 140 cases with special regard to the development of secondary malignant tumors. Br J Dermatol. 1970;82:99-117.
- Ballantyne AJ, McCarten AB, Ibanez ML. The extension of cancer of the head and neck through perineural peripheral nerves. Am J Surg. 1963;106:651-667.
- Goepfert H, Dichtel WJ, Medina JE, et al. Perineural invasion in squamous cell skin carcinoma of the head and neck. Am J Surg. 1984;148:542-547.
- Feasel AM, Brown TJ, Bogle MA, et al. Perineural invasion of cutaneous malignancies. Dermatol Surg. 2001;27:531-542.
- Cottel WI. Perineural invasion by squamous cell carcinoma. J Dermatol Surg Oncol. 1982;8:589-600.
- Mendenhall WM, Parsons JT, Mendenhall NP, et al. Carcinoma of the skin of the head and neck with perineural invasion. Head Neck. 1989;11:301-308.
First reported in 1895, nevus sebaceus (NS) is a con genital papillomatous hamartoma most commonly found on the scalp and face. 1 Lesions typically are yellow-orange plaques and often are hairless. Nevus sebaceus is most prominent in the few first months after birth and again at puberty during development of the sebaceous glands. Development of epithelial hyperplasia, cysts, verrucas, and benign or malignant tumors has been reported. 1 The most common benign tumors are syringocystadenoma papilliferum and trichoblastoma. Cases of malignancy are rare, and basal cell carcinoma is the predominant form (approximately 2% of cases). Squamous cell carcinoma (SCC) and adnexal carcinoma are reported at even lower rates. 1 Malignant transformation occurring during childhood is extremely uncommon. According to a PubMed search of articles indexed for MEDLINE using the terms nevus sebaceous, malignancy, and squamous cell carcinoma and narrowing the results to children, there have been only 4 prior reports of SCC developing within an NS in a child. 2-5 We report a case of SCC arising in an NS in a 13-year-old adolescent girl with perineural invasion.
Case Report
A 13-year-old fair-skinned adolescent girl presented with a hairless 2×2.5-cm yellow plaque at the hairline on the anterior central scalp. The plaque had been present since birth and had progressively developed a superiorly located 3×5-mm erythematous verrucous nodule (Figure 1) with an approximate height of 6 mm over the last year. The nodule was subjected to regular trauma and bled with minimal insult. The patient appeared otherwise healthy, with no history of skin cancer or other chronic medical conditions. There was no evidence of lymphadenopathy on examination, and no other skin abnormalities were noted. There was no reported family history of skin cancer or chronic skin conditions suggestive of increased risk for cancer or other pathologic dermatoses. Differential diagnoses for the plaque and nodule complex included verruca, Spitz nevus, or secondary neoplasm within NS.
Excision was conducted under local anesthesia without complication. An elliptical section of skin measuring 0.8×2.5 cm was excised to a depth of 3 mm. The resulting wound was closed using a complex linear repair. The section was placed in formalin specimen transport medium and sent to Walter Reed National Military Medical Center (Bethesda, Maryland). Microscopic examination of the specimen revealed features typical for NS, including mild verrucous epidermal hyperplasia, sebaceous gland hyperplasia, presence of apocrine glands, and hamartomatous follicular proliferations (Figure 2). An even more papillomatous epidermal proliferation that was comprised of atypical squamous cells was present within the lesion. Similar atypical squamous cells infiltrated the superficial dermis in nests, cords, and single cells (Figure 3A). One focus showed perineural invasion with a small superficial nerve fiber surrounded by SCC (Figure 3B). The tumor was completely excised, with negative surgical margins extending approximately 2 mm. Adjuvant radiation therapy and further specialized Mohs micrographic excision were not performed because of the clear histologic appearance of the carcinoma and strong evidence of complete excision.
At 2-week follow-up, the surgical scar on the anterior central forehead was well healed without evidence of SCC recurrence. On physical examination there was neither lymphadenopathy nor signs of neurologic deficit, except for superficial cutaneous hypoesthesia in the immediate area surrounding the healed site. Following discussion with the patient and her parents, it was decided that the patient would obtain baseline laboratory tests, chest radiography, and abdominal ultrasonography, and she would undergo serial follow-up examinations every 3 months for the next 2 years. Annual follow-up was recommended after 2 years, with the caveat to return sooner if recurrence or symptoms were to arise.
Comment
Historically, there has been variability in the histopathologic interpretation of SCC in NS in the literature. Retrospective analysis of the histologic evidence of SCC in the 2 earliest possible cases of pediatric SCC in NS have been questioned due to the lack of clinical data presented and the possibility that the diagnosis of SCC was inaccurate.6 Our case was histopathologically interpreted as superficially invasive, well-differentiated SCC arising within an NS; therefore, we classified this case as SCC and took every precaution to ensure the lesion was completely excised, given the potentially invasive nature of SCC.
Our case is unique because it represents SCC in NS with histologic evidence of perineural involvement. Perineural invasion is a major route of tumor spread in SCC and may result in increased occurrence of regional lymph node spread and distant metastases, with path of least resistance or neural cell adhesion as possible spreading methods.7-9 However, there is a notable amount of prognostic variability based on tumor type, the nerve involved, and degree of involvement.9 It is common for cutaneous SCC to occur with invasion of small intradermal nerves, but a poor outcome is less likely in asymptomatic patients who have perineural involvement that was incidentally discovered on histologic examination.10
In our patient, the entire tumor was completely removed with local excision. Recurrence of the SCC or future symptoms of deep neural invasion were not anticipated given the postoperative evidence of clear margins in the excised skin and subdermal structures as well as the lack of preoperative and postoperative symptoms. Close clinical follow-up was warranted to monitor for early signs of recurrence or neural involvement. We have confidence that the planned follow-up regimen in our patient will reveal any early signs of new occurrence or recurrence.
In the case of recurrence, Mohs micrographic surgery would likely be indicated. We elected not to treat with adjuvant radiotherapy because its benefit in cutaneous SCC with perineural invasion is debatable based on the lack of randomized controlled clinical evidence.10,11 The patient obtained postoperative baseline complete blood cell count with differential, posterior/anterior and lateral chest radiographs, as well as abdominal ultrasonography. Each returned negative findings of hematologic or distant organ metastases, with subsequent follow-up visits also negative for any new concerning findings.
First reported in 1895, nevus sebaceus (NS) is a con genital papillomatous hamartoma most commonly found on the scalp and face. 1 Lesions typically are yellow-orange plaques and often are hairless. Nevus sebaceus is most prominent in the few first months after birth and again at puberty during development of the sebaceous glands. Development of epithelial hyperplasia, cysts, verrucas, and benign or malignant tumors has been reported. 1 The most common benign tumors are syringocystadenoma papilliferum and trichoblastoma. Cases of malignancy are rare, and basal cell carcinoma is the predominant form (approximately 2% of cases). Squamous cell carcinoma (SCC) and adnexal carcinoma are reported at even lower rates. 1 Malignant transformation occurring during childhood is extremely uncommon. According to a PubMed search of articles indexed for MEDLINE using the terms nevus sebaceous, malignancy, and squamous cell carcinoma and narrowing the results to children, there have been only 4 prior reports of SCC developing within an NS in a child. 2-5 We report a case of SCC arising in an NS in a 13-year-old adolescent girl with perineural invasion.
Case Report
A 13-year-old fair-skinned adolescent girl presented with a hairless 2×2.5-cm yellow plaque at the hairline on the anterior central scalp. The plaque had been present since birth and had progressively developed a superiorly located 3×5-mm erythematous verrucous nodule (Figure 1) with an approximate height of 6 mm over the last year. The nodule was subjected to regular trauma and bled with minimal insult. The patient appeared otherwise healthy, with no history of skin cancer or other chronic medical conditions. There was no evidence of lymphadenopathy on examination, and no other skin abnormalities were noted. There was no reported family history of skin cancer or chronic skin conditions suggestive of increased risk for cancer or other pathologic dermatoses. Differential diagnoses for the plaque and nodule complex included verruca, Spitz nevus, or secondary neoplasm within NS.
Excision was conducted under local anesthesia without complication. An elliptical section of skin measuring 0.8×2.5 cm was excised to a depth of 3 mm. The resulting wound was closed using a complex linear repair. The section was placed in formalin specimen transport medium and sent to Walter Reed National Military Medical Center (Bethesda, Maryland). Microscopic examination of the specimen revealed features typical for NS, including mild verrucous epidermal hyperplasia, sebaceous gland hyperplasia, presence of apocrine glands, and hamartomatous follicular proliferations (Figure 2). An even more papillomatous epidermal proliferation that was comprised of atypical squamous cells was present within the lesion. Similar atypical squamous cells infiltrated the superficial dermis in nests, cords, and single cells (Figure 3A). One focus showed perineural invasion with a small superficial nerve fiber surrounded by SCC (Figure 3B). The tumor was completely excised, with negative surgical margins extending approximately 2 mm. Adjuvant radiation therapy and further specialized Mohs micrographic excision were not performed because of the clear histologic appearance of the carcinoma and strong evidence of complete excision.
At 2-week follow-up, the surgical scar on the anterior central forehead was well healed without evidence of SCC recurrence. On physical examination there was neither lymphadenopathy nor signs of neurologic deficit, except for superficial cutaneous hypoesthesia in the immediate area surrounding the healed site. Following discussion with the patient and her parents, it was decided that the patient would obtain baseline laboratory tests, chest radiography, and abdominal ultrasonography, and she would undergo serial follow-up examinations every 3 months for the next 2 years. Annual follow-up was recommended after 2 years, with the caveat to return sooner if recurrence or symptoms were to arise.
Comment
Historically, there has been variability in the histopathologic interpretation of SCC in NS in the literature. Retrospective analysis of the histologic evidence of SCC in the 2 earliest possible cases of pediatric SCC in NS have been questioned due to the lack of clinical data presented and the possibility that the diagnosis of SCC was inaccurate.6 Our case was histopathologically interpreted as superficially invasive, well-differentiated SCC arising within an NS; therefore, we classified this case as SCC and took every precaution to ensure the lesion was completely excised, given the potentially invasive nature of SCC.
Our case is unique because it represents SCC in NS with histologic evidence of perineural involvement. Perineural invasion is a major route of tumor spread in SCC and may result in increased occurrence of regional lymph node spread and distant metastases, with path of least resistance or neural cell adhesion as possible spreading methods.7-9 However, there is a notable amount of prognostic variability based on tumor type, the nerve involved, and degree of involvement.9 It is common for cutaneous SCC to occur with invasion of small intradermal nerves, but a poor outcome is less likely in asymptomatic patients who have perineural involvement that was incidentally discovered on histologic examination.10
In our patient, the entire tumor was completely removed with local excision. Recurrence of the SCC or future symptoms of deep neural invasion were not anticipated given the postoperative evidence of clear margins in the excised skin and subdermal structures as well as the lack of preoperative and postoperative symptoms. Close clinical follow-up was warranted to monitor for early signs of recurrence or neural involvement. We have confidence that the planned follow-up regimen in our patient will reveal any early signs of new occurrence or recurrence.
In the case of recurrence, Mohs micrographic surgery would likely be indicated. We elected not to treat with adjuvant radiotherapy because its benefit in cutaneous SCC with perineural invasion is debatable based on the lack of randomized controlled clinical evidence.10,11 The patient obtained postoperative baseline complete blood cell count with differential, posterior/anterior and lateral chest radiographs, as well as abdominal ultrasonography. Each returned negative findings of hematologic or distant organ metastases, with subsequent follow-up visits also negative for any new concerning findings.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2, pt 1):263-268.
- Aguayo R, Pallares J, Cassanova JM, et al. Squamous cell carcinoma developing in Jadassohn’s sebaceous nevus: case report and review of the literature. Dermatol Surg. 2010;36:1763-1768.
- Taher M, Feibleman C, Bennet R. Squamous cell carcinoma arising in a nevus sebaceous of Jadassohn in a 9-year-old girl: treatment using Mohs micrographic surgery with literature review. Dermatol Surg. 2010;36:1203-1208.
- Hidvegi NC, Kangesu L, Wolfe KQ. Squamous cell carcinoma complicating naevus sebaceous of Jadassohn in a child. Br J Plast Surg. 2003;56:50-52.
- Belhadjali H, Moussa A, Yahia S, et al. Simultaneous occurrence of squamous cell carcinomas within a nevus sebaceous of Jadassohn in an 11-year-old girl. Pediatr Dermatol. 2009;26:236-237.
- Wilson-Jones EW, Heyl T. Naevus sebaceus: a report of 140 cases with special regard to the development of secondary malignant tumors. Br J Dermatol. 1970;82:99-117.
- Ballantyne AJ, McCarten AB, Ibanez ML. The extension of cancer of the head and neck through perineural peripheral nerves. Am J Surg. 1963;106:651-667.
- Goepfert H, Dichtel WJ, Medina JE, et al. Perineural invasion in squamous cell skin carcinoma of the head and neck. Am J Surg. 1984;148:542-547.
- Feasel AM, Brown TJ, Bogle MA, et al. Perineural invasion of cutaneous malignancies. Dermatol Surg. 2001;27:531-542.
- Cottel WI. Perineural invasion by squamous cell carcinoma. J Dermatol Surg Oncol. 1982;8:589-600.
- Mendenhall WM, Parsons JT, Mendenhall NP, et al. Carcinoma of the skin of the head and neck with perineural invasion. Head Neck. 1989;11:301-308.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2, pt 1):263-268.
- Aguayo R, Pallares J, Cassanova JM, et al. Squamous cell carcinoma developing in Jadassohn’s sebaceous nevus: case report and review of the literature. Dermatol Surg. 2010;36:1763-1768.
- Taher M, Feibleman C, Bennet R. Squamous cell carcinoma arising in a nevus sebaceous of Jadassohn in a 9-year-old girl: treatment using Mohs micrographic surgery with literature review. Dermatol Surg. 2010;36:1203-1208.
- Hidvegi NC, Kangesu L, Wolfe KQ. Squamous cell carcinoma complicating naevus sebaceous of Jadassohn in a child. Br J Plast Surg. 2003;56:50-52.
- Belhadjali H, Moussa A, Yahia S, et al. Simultaneous occurrence of squamous cell carcinomas within a nevus sebaceous of Jadassohn in an 11-year-old girl. Pediatr Dermatol. 2009;26:236-237.
- Wilson-Jones EW, Heyl T. Naevus sebaceus: a report of 140 cases with special regard to the development of secondary malignant tumors. Br J Dermatol. 1970;82:99-117.
- Ballantyne AJ, McCarten AB, Ibanez ML. The extension of cancer of the head and neck through perineural peripheral nerves. Am J Surg. 1963;106:651-667.
- Goepfert H, Dichtel WJ, Medina JE, et al. Perineural invasion in squamous cell skin carcinoma of the head and neck. Am J Surg. 1984;148:542-547.
- Feasel AM, Brown TJ, Bogle MA, et al. Perineural invasion of cutaneous malignancies. Dermatol Surg. 2001;27:531-542.
- Cottel WI. Perineural invasion by squamous cell carcinoma. J Dermatol Surg Oncol. 1982;8:589-600.
- Mendenhall WM, Parsons JT, Mendenhall NP, et al. Carcinoma of the skin of the head and neck with perineural invasion. Head Neck. 1989;11:301-308.
Practice Points
- Nevus sebaceus (NS) is frequently found on the scalp and may increase in size during puberty.
- Commonly found additional neoplasms within NS include trichoblastoma and syringocystadenoma papilliferum. Malignancies are possible but rare.
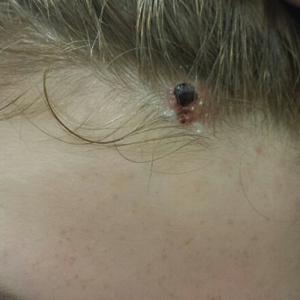
Acute Encephalopathy Following Hyperbaric Oxygen Therapy in a Patient on Metronidazole
Altered mental status (AMS) is a common presentation to the emergency department (ED) for older patients and is often due to underlying drug-associated adverse effects (AEs), medical or psychiatric illness, or neurologic disease. EDs often have protocols for diagnosing and managing AMS to assess the underlying etiology. A formal assessment with a full history and physical examination is paramount to diagnosing the cause of AMS.
Oral metronidazole is a commonly used antibiotic for anaerobic bacterial infections and Clostridium difficile-associated diarrhea and colitis.1Metronidazole produces cytotoxic intermediates that cause DNA strand breakage and destabilization, resulting in bactericidal activity in host cells.2Common AEs include gastrointestinal symptoms such as nausea, vomiting, and diarrhea; less common AEs can involve the nervous system and include seizures, peripheral neuropathy, dizziness, ataxia, and encephalopathy.3,4A pattern of magnetic resonance image (MRI) abnormalities typically located at the cerebellar dentate nucleus midbrain, dorsal pons, medulla, and splenium of the corpus callosum have been associated with metronidazole usage.5
Hyperbaric oxygen therapy (HBOT) is a treatment modality used as the primary therapy for decompression sickness, arterial gas embolism, and carbon monoxide poisoning. HBOT is used as adjuvant therapy for osteonecrosis caused by radiation or bisphosphonate use.6,7 HBOT increases the partial pressure of oxygen in plasma and increases the amount of oxygen delivered to tissues throughout the body.8Hyperoxia, defined as an elevated partial pressure of oxygen leading to excess oxygenation to tissues and organs, increases production of reactive oxygen and nitrogen species, which are signaling factors in a variety of pathways that stimulate angiogenesis.8 AEs of HBOT include barotrauma-related injuries and oxygen toxicity, such as respiratory distress or central nervous system (CNS) symptoms.9 Severe CNS AEs occur in 1% to 2% of patients undergoing therapy and manifest as generalized tonic-clonic seizures, typically in patients with preexisting neurologic disorders, brain injury, or lowered seizure threshold.7,8,10 There have been no documented incidences of HBOT inducing acute encephalopathy.
Case Presentation
A 63-year-old male smoker with no history of alcohol use presented to the ED with an acute onset of lightheadedness, confusion, and poor coordination following his second HBOT for radiation-induced osteonecrosis of the mandible. The patient reported chronic, slowly progressive pain and numbness of the feet that began 4 years earlier. He noted marked worsening of pain and difficulty standing and walking 3 to 4 months prior to presentation.
Ten years prior, the patient was diagnosed with cancer of the right tonsil. A tonsillectomy with wide margins was performed, followed by 35 rounds of radiation treatment and 2 rounds of chemotherapy with cisplatin.
In May 2017, the patient presented with a lump in the right cheek that was diagnosed as osteonecrosis of the mandible. An oral surgeon prescribed metronidazole 500 mg qid and amoxicillin 500 mg tid. The patient was adherent until presentation in November 2017. Following lack of improvement of the osteonecrosis from antibiotic therapy, oral surgery was planned, and the patient was referred for HBOT with a planned 20 HBOT preoperative treatments and 10 postoperative treatments.
Following his first 2-hour HBOT treatment on November 13, 2017, the patient complained of light-headedness, confusion, and incoordination. While driving on a familiar route to his home, he collided with a tree that was 6 feet from the curb. The patient attempted to drive another vehicle later that day, resulting in a second motor vehicle accident. There was no significant injury reported in either accident.
His partner described the patient’s episode of disorientation lasting 6 to 8 hours, during which he “looked drunk” and was unable to sit in a chair without falling. The following morning, the patient had improved mental status but had not returned to baseline. His second HBOT treatment took place that day, and again, the patient acutely experienced light-headedness and confusion following completion. Therapy was suspended, and the patient was referred to the ED for further evaluation. Mild facial asymmetry without weakness, decreased sensation from toes to knees bilaterally, and absent Achilles reflexes bilaterally were found on neurologic examination. He exhibited past-pointing on finger-to-nose testing bilaterally. He was able to ambulate independently, but he could not perform tandem gait.
An MRI of the brain showed abnormal T2 hyperintensity found bilaterally at the dentate nuclei and inferior colliculi. The splenium of the corpus callosum also showed mild involvement with hyperintense lesions. Laboratory tests of the patient’s complete blood count; comprehensive metabolic panel; vitamins B1, B6, B12; and folic acid levels had no notable abnormalities and were within normal limits.
Metronidazole and HBOT therapy were discontinued, and all of the patient’s symptoms resolved within 2 weeks. A repeat examination and MRI performed 1 month later showed resolution of all the patient’s clinical findings and MRI abnormalities. HBOT was resumed without the recurrence of previously described symptoms.
Discussion
This patient’s encephalopathic symptoms correlate temporally with the onset of HBOT. There is no medical literature suggesting a relationship between HBOT and encephalopathic symptoms with MRI abnormalities, and in fact, some studies suggest HBOT as a treatment for hypoxic-ischemic encephalopathy in neonates.11 This led us to believe that the HBOT may have exacerbated some underlying condition, evidenced by the specific MRI findings of T2 fluid-attenuated inversion recovery (FLAIR) hyperintensities in the dentate nuclei and inferior colliculi (Figures 1 and 2). 
Differential diagnoses for T2 hyperintense lesions in the dentate nuclei include metronidazole toxicity, acute Wernicke encephalopathy (WE), and methyl bromide intoxication. Diseases that would have presented in infancy with similar MRI findings (Canavan disease, maple-syrup urine disease, and glutaric aciduria type 1) were not considered plausible.12-14 
Despite his denial of alcohol use, the patient was at risk for malnutrition secondary to his mandibular lesion and difficulty eating. Clinically, he presented with episodes of confusion and ataxia, consistent with 2 of the classic triad of symptoms of WE (no ocular abnormalities noted on exam). Typical MRI findings in WE include signal intensity alterations (including T2 hyperintensities) in the medial thalami, mammillary bodies, collicular bodies, and periaqueductal and periventricular regions.14,15 Atypical MRI findings in WE include symmetric signal intensity changes in the cerebellum, dentate nuclei, caudate nuclei, red nuclei, cranial nerve nuclei, and splenium.14 Of note, atypical MRI findings were more common in patients without alcohol use disorders and WE, and typical MRI findings were more common in patients with alcohol use disorders.14 However, this patient’s report of no alcohol use and the serum thiamine level being within normal limits (173 nmol/L; range 78-185 nmol/L) made acute WE less likely than metronidonazale-induced encephalopathy (MIE).
The most common neurologic AE of metronidazole is distal symmetric sensory polyneuropathy, which also can have motor or autonomic features.16,17 While our patient had a history of peripheral neuropathy, he noted marked worsening of foot pain 3 months after initiating metronidazole therapy. A potential mechanism involves metronidazole or its cytotoxic intermediates binding neuronal ribonucleic acids, thus inhibiting protein synthesis and resulting in degenerative neuronal changes and reversible axonal swelling (as opposed to the DNA interference attributed to the drug’s mechanism of bactericidal action).18 Neuropathies may result from prolonged high-dose metronidazole therapy (cumulative dose > 42 g),3 but they also have been seen in short-term use of high dosages.17
CNS AEs are much rarer and are thought to be associated with metronidazole’s ability to cross the blood-brain barrier. These patients present as a toxic encephalopathy with cerebellar dysfunction (dysarthria, ataxia) as the most common presentation, followed by AMS and seizures.4 Our patient presented with acute confusion and ataxia. Animal studies suggest that γ-aminobutyric acid (GABA) receptor modulation in the cerebellar and vestibular systems may contribute to this neurotoxicity, but no definitive mechanism of injury has been found.19
On MRI, MIE most commonly presents with hyperintense lesions in the bilateral cerebellar dentate nucleus on T2-weighted and FLAIR images.5,20 The midbrain, dorsal pons, medulla, and corpus callosum also can show increased signal intensity.5 This AE does not seem to be dose- or duration-dependent, and most cases report complete or partial resolution of symptoms following discontinuation of the drug, though this is not absolute.4,13,21 The patient’s MRI findings were highly consistent with MIE (Figure 2).
Conclusion
This patient’s highly specific MRI findings, neurologic examination consistent with confusion, ataxia, length-dependent sensory neuropathy, and 360-g cumulative dose of metronidazole over the previous 6 months suggest he experienced MIE. The mechanism of how HBOT precipitated the patient’s altered mental status, incoordination, and worsening of peripheral neuropathy is unknown. Although encephalopathy with MRI abnormalities as described is not a reported AE of HBOT, it may be unrecognized. It is possible that without HBOT the patient would have remained asymptomatic apart from his peripheral neuropathy.
We propose HBOT may exacerbate or increase the risk of a patient developing MIE. Our patient was able to safely resume HBOT after metronidazole was discontinued, suggesting that the combination was the causation for the development of encephalopathy. We do not believe any similar cases have been reported.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541.
2. Edwards DI. The action of metronidazole on DNA. J Antimicrob Chemother. 1977;3(1):43-48.
3. Goolsby TA, Jakeman B, Gaynes RP. Clinical relevance of metronidazole and peripheral neuropathy: a systematic review of the literature. Int J Antimicrob Agents. 2018;51(3):319-325.
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systematic review. Clin Neuropharmacol. 2011;34(6):241-247.
5. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658.
6. Ceponis P, Keilman C, Guerry C, Freiberger JJ. Hyperbaric oxygen therapy and osteonecrosis. Oral Dis. 2017;23(2):141-151.
7. Leach R, Rees P, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317(7166):1140-1143.
8. Thom SR. Hyperbaric oxygen–its mechanisms and efficacy. Plastic Reconstr Surg. 2011;127(suppl 1):131S-141S.
9. Plafki C, Peters P, Almeling M, Welslau W, Busch R. Complications and side effects of hyperbaric oxygen therapy. Aviation Space Environ Med. 2000;71(2):119-124.
10. Hadanny A, Meir O, Bechor Y, Fishlev G, Bergan J, Efrati S. Seizures during hyperbaric oxygen therapy: retrospective analysis of 62,614 treatment sessions. Undersea Hyperb Med. 2016;43(1):21-28.
11. Liu Z, Xiong T, Meads C. Clinical effectiveness of treatment with hyperbaric oxygen for neonatal hypoxic-ischaemic encephalopathy: systematic review of Chinese literature. BMJ. 2006;333(7564):374.
12. Bond KM, Brinjikji W, Eckel LJ, Kallmes DF, McDonald RJ, Carr CM. Dentate update: imaging features of entities that affect the dentate nucleus. AJNR Am J Neuroradiol. 2017;38(8):1467-1474.
13. Agarwal A, Kanekar S, Sabat S, Thamburaj K. Metronidazole-induced cerebellar toxicity. Neurol Int. 2016;8(1):6365.
14. Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. AJR Am J Roentgenol. 2009;192(2):501-508.
15. Jung YC, Chanraud S, Sullivan EV. Neuroimaging of Wernicke’s encephalopathy and Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):170-180.
16. Hobson-Webb LD, Roach ES, Donofrio PD. Metronidazole: newly recognized cause of autonomic neuropathy. J Child Neurol. 2006;21(5):429-431.
17. Nath Chaurasia R. Rapid onset metronidazole induced sensory neuropathy: case series and review of literature. Int J Neurorehabilitation. 2015;02:152.
18. Bradley WG, Karlsson IJ, Rassol CG. Metronidazole neuropathy. Br Med J. 1977;2(6087):610-611.
19. Evans J, Levesque D, Knowles K, Longshore R, Plummer S. Diazepam as a treatment for metronidazole toxicosis in dogs: a retrospective study of 21 cases. J Vet Intern Med. 2003;17(3):304-310.
20. Farmakiotis D, Zeluff B. Images in clinical medicine. Metronidazole-associated encephalopathy. N Engl J Med. 2016;374(15):1465.
21. Hobbs K, Stern-Nezer S, Buckwalter MS, Fischbein N, Finley Caulfield A. Metronidazole-induced encephalopathy: not always a reversible situation. Neurocrit Care. 2015;22(3):429-436.
Altered mental status (AMS) is a common presentation to the emergency department (ED) for older patients and is often due to underlying drug-associated adverse effects (AEs), medical or psychiatric illness, or neurologic disease. EDs often have protocols for diagnosing and managing AMS to assess the underlying etiology. A formal assessment with a full history and physical examination is paramount to diagnosing the cause of AMS.
Oral metronidazole is a commonly used antibiotic for anaerobic bacterial infections and Clostridium difficile-associated diarrhea and colitis.1Metronidazole produces cytotoxic intermediates that cause DNA strand breakage and destabilization, resulting in bactericidal activity in host cells.2Common AEs include gastrointestinal symptoms such as nausea, vomiting, and diarrhea; less common AEs can involve the nervous system and include seizures, peripheral neuropathy, dizziness, ataxia, and encephalopathy.3,4A pattern of magnetic resonance image (MRI) abnormalities typically located at the cerebellar dentate nucleus midbrain, dorsal pons, medulla, and splenium of the corpus callosum have been associated with metronidazole usage.5
Hyperbaric oxygen therapy (HBOT) is a treatment modality used as the primary therapy for decompression sickness, arterial gas embolism, and carbon monoxide poisoning. HBOT is used as adjuvant therapy for osteonecrosis caused by radiation or bisphosphonate use.6,7 HBOT increases the partial pressure of oxygen in plasma and increases the amount of oxygen delivered to tissues throughout the body.8Hyperoxia, defined as an elevated partial pressure of oxygen leading to excess oxygenation to tissues and organs, increases production of reactive oxygen and nitrogen species, which are signaling factors in a variety of pathways that stimulate angiogenesis.8 AEs of HBOT include barotrauma-related injuries and oxygen toxicity, such as respiratory distress or central nervous system (CNS) symptoms.9 Severe CNS AEs occur in 1% to 2% of patients undergoing therapy and manifest as generalized tonic-clonic seizures, typically in patients with preexisting neurologic disorders, brain injury, or lowered seizure threshold.7,8,10 There have been no documented incidences of HBOT inducing acute encephalopathy.
Case Presentation
A 63-year-old male smoker with no history of alcohol use presented to the ED with an acute onset of lightheadedness, confusion, and poor coordination following his second HBOT for radiation-induced osteonecrosis of the mandible. The patient reported chronic, slowly progressive pain and numbness of the feet that began 4 years earlier. He noted marked worsening of pain and difficulty standing and walking 3 to 4 months prior to presentation.
Ten years prior, the patient was diagnosed with cancer of the right tonsil. A tonsillectomy with wide margins was performed, followed by 35 rounds of radiation treatment and 2 rounds of chemotherapy with cisplatin.
In May 2017, the patient presented with a lump in the right cheek that was diagnosed as osteonecrosis of the mandible. An oral surgeon prescribed metronidazole 500 mg qid and amoxicillin 500 mg tid. The patient was adherent until presentation in November 2017. Following lack of improvement of the osteonecrosis from antibiotic therapy, oral surgery was planned, and the patient was referred for HBOT with a planned 20 HBOT preoperative treatments and 10 postoperative treatments.
Following his first 2-hour HBOT treatment on November 13, 2017, the patient complained of light-headedness, confusion, and incoordination. While driving on a familiar route to his home, he collided with a tree that was 6 feet from the curb. The patient attempted to drive another vehicle later that day, resulting in a second motor vehicle accident. There was no significant injury reported in either accident.
His partner described the patient’s episode of disorientation lasting 6 to 8 hours, during which he “looked drunk” and was unable to sit in a chair without falling. The following morning, the patient had improved mental status but had not returned to baseline. His second HBOT treatment took place that day, and again, the patient acutely experienced light-headedness and confusion following completion. Therapy was suspended, and the patient was referred to the ED for further evaluation. Mild facial asymmetry without weakness, decreased sensation from toes to knees bilaterally, and absent Achilles reflexes bilaterally were found on neurologic examination. He exhibited past-pointing on finger-to-nose testing bilaterally. He was able to ambulate independently, but he could not perform tandem gait.
An MRI of the brain showed abnormal T2 hyperintensity found bilaterally at the dentate nuclei and inferior colliculi. The splenium of the corpus callosum also showed mild involvement with hyperintense lesions. Laboratory tests of the patient’s complete blood count; comprehensive metabolic panel; vitamins B1, B6, B12; and folic acid levels had no notable abnormalities and were within normal limits.
Metronidazole and HBOT therapy were discontinued, and all of the patient’s symptoms resolved within 2 weeks. A repeat examination and MRI performed 1 month later showed resolution of all the patient’s clinical findings and MRI abnormalities. HBOT was resumed without the recurrence of previously described symptoms.
Discussion
This patient’s encephalopathic symptoms correlate temporally with the onset of HBOT. There is no medical literature suggesting a relationship between HBOT and encephalopathic symptoms with MRI abnormalities, and in fact, some studies suggest HBOT as a treatment for hypoxic-ischemic encephalopathy in neonates.11 This led us to believe that the HBOT may have exacerbated some underlying condition, evidenced by the specific MRI findings of T2 fluid-attenuated inversion recovery (FLAIR) hyperintensities in the dentate nuclei and inferior colliculi (Figures 1 and 2).
Differential diagnoses for T2 hyperintense lesions in the dentate nuclei include metronidazole toxicity, acute Wernicke encephalopathy (WE), and methyl bromide intoxication. Diseases that would have presented in infancy with similar MRI findings (Canavan disease, maple-syrup urine disease, and glutaric aciduria type 1) were not considered plausible.12-14
Despite his denial of alcohol use, the patient was at risk for malnutrition secondary to his mandibular lesion and difficulty eating. Clinically, he presented with episodes of confusion and ataxia, consistent with 2 of the classic triad of symptoms of WE (no ocular abnormalities noted on exam). Typical MRI findings in WE include signal intensity alterations (including T2 hyperintensities) in the medial thalami, mammillary bodies, collicular bodies, and periaqueductal and periventricular regions.14,15 Atypical MRI findings in WE include symmetric signal intensity changes in the cerebellum, dentate nuclei, caudate nuclei, red nuclei, cranial nerve nuclei, and splenium.14 Of note, atypical MRI findings were more common in patients without alcohol use disorders and WE, and typical MRI findings were more common in patients with alcohol use disorders.14 However, this patient’s report of no alcohol use and the serum thiamine level being within normal limits (173 nmol/L; range 78-185 nmol/L) made acute WE less likely than metronidonazale-induced encephalopathy (MIE).
The most common neurologic AE of metronidazole is distal symmetric sensory polyneuropathy, which also can have motor or autonomic features.16,17 While our patient had a history of peripheral neuropathy, he noted marked worsening of foot pain 3 months after initiating metronidazole therapy. A potential mechanism involves metronidazole or its cytotoxic intermediates binding neuronal ribonucleic acids, thus inhibiting protein synthesis and resulting in degenerative neuronal changes and reversible axonal swelling (as opposed to the DNA interference attributed to the drug’s mechanism of bactericidal action).18 Neuropathies may result from prolonged high-dose metronidazole therapy (cumulative dose > 42 g),3 but they also have been seen in short-term use of high dosages.17
CNS AEs are much rarer and are thought to be associated with metronidazole’s ability to cross the blood-brain barrier. These patients present as a toxic encephalopathy with cerebellar dysfunction (dysarthria, ataxia) as the most common presentation, followed by AMS and seizures.4 Our patient presented with acute confusion and ataxia. Animal studies suggest that γ-aminobutyric acid (GABA) receptor modulation in the cerebellar and vestibular systems may contribute to this neurotoxicity, but no definitive mechanism of injury has been found.19
On MRI, MIE most commonly presents with hyperintense lesions in the bilateral cerebellar dentate nucleus on T2-weighted and FLAIR images.5,20 The midbrain, dorsal pons, medulla, and corpus callosum also can show increased signal intensity.5 This AE does not seem to be dose- or duration-dependent, and most cases report complete or partial resolution of symptoms following discontinuation of the drug, though this is not absolute.4,13,21 The patient’s MRI findings were highly consistent with MIE (Figure 2).
Conclusion
This patient’s highly specific MRI findings, neurologic examination consistent with confusion, ataxia, length-dependent sensory neuropathy, and 360-g cumulative dose of metronidazole over the previous 6 months suggest he experienced MIE. The mechanism of how HBOT precipitated the patient’s altered mental status, incoordination, and worsening of peripheral neuropathy is unknown. Although encephalopathy with MRI abnormalities as described is not a reported AE of HBOT, it may be unrecognized. It is possible that without HBOT the patient would have remained asymptomatic apart from his peripheral neuropathy.
We propose HBOT may exacerbate or increase the risk of a patient developing MIE. Our patient was able to safely resume HBOT after metronidazole was discontinued, suggesting that the combination was the causation for the development of encephalopathy. We do not believe any similar cases have been reported.
Altered mental status (AMS) is a common presentation to the emergency department (ED) for older patients and is often due to underlying drug-associated adverse effects (AEs), medical or psychiatric illness, or neurologic disease. EDs often have protocols for diagnosing and managing AMS to assess the underlying etiology. A formal assessment with a full history and physical examination is paramount to diagnosing the cause of AMS.
Oral metronidazole is a commonly used antibiotic for anaerobic bacterial infections and Clostridium difficile-associated diarrhea and colitis.1Metronidazole produces cytotoxic intermediates that cause DNA strand breakage and destabilization, resulting in bactericidal activity in host cells.2Common AEs include gastrointestinal symptoms such as nausea, vomiting, and diarrhea; less common AEs can involve the nervous system and include seizures, peripheral neuropathy, dizziness, ataxia, and encephalopathy.3,4A pattern of magnetic resonance image (MRI) abnormalities typically located at the cerebellar dentate nucleus midbrain, dorsal pons, medulla, and splenium of the corpus callosum have been associated with metronidazole usage.5
Hyperbaric oxygen therapy (HBOT) is a treatment modality used as the primary therapy for decompression sickness, arterial gas embolism, and carbon monoxide poisoning. HBOT is used as adjuvant therapy for osteonecrosis caused by radiation or bisphosphonate use.6,7 HBOT increases the partial pressure of oxygen in plasma and increases the amount of oxygen delivered to tissues throughout the body.8Hyperoxia, defined as an elevated partial pressure of oxygen leading to excess oxygenation to tissues and organs, increases production of reactive oxygen and nitrogen species, which are signaling factors in a variety of pathways that stimulate angiogenesis.8 AEs of HBOT include barotrauma-related injuries and oxygen toxicity, such as respiratory distress or central nervous system (CNS) symptoms.9 Severe CNS AEs occur in 1% to 2% of patients undergoing therapy and manifest as generalized tonic-clonic seizures, typically in patients with preexisting neurologic disorders, brain injury, or lowered seizure threshold.7,8,10 There have been no documented incidences of HBOT inducing acute encephalopathy.
Case Presentation
A 63-year-old male smoker with no history of alcohol use presented to the ED with an acute onset of lightheadedness, confusion, and poor coordination following his second HBOT for radiation-induced osteonecrosis of the mandible. The patient reported chronic, slowly progressive pain and numbness of the feet that began 4 years earlier. He noted marked worsening of pain and difficulty standing and walking 3 to 4 months prior to presentation.
Ten years prior, the patient was diagnosed with cancer of the right tonsil. A tonsillectomy with wide margins was performed, followed by 35 rounds of radiation treatment and 2 rounds of chemotherapy with cisplatin.
In May 2017, the patient presented with a lump in the right cheek that was diagnosed as osteonecrosis of the mandible. An oral surgeon prescribed metronidazole 500 mg qid and amoxicillin 500 mg tid. The patient was adherent until presentation in November 2017. Following lack of improvement of the osteonecrosis from antibiotic therapy, oral surgery was planned, and the patient was referred for HBOT with a planned 20 HBOT preoperative treatments and 10 postoperative treatments.
Following his first 2-hour HBOT treatment on November 13, 2017, the patient complained of light-headedness, confusion, and incoordination. While driving on a familiar route to his home, he collided with a tree that was 6 feet from the curb. The patient attempted to drive another vehicle later that day, resulting in a second motor vehicle accident. There was no significant injury reported in either accident.
His partner described the patient’s episode of disorientation lasting 6 to 8 hours, during which he “looked drunk” and was unable to sit in a chair without falling. The following morning, the patient had improved mental status but had not returned to baseline. His second HBOT treatment took place that day, and again, the patient acutely experienced light-headedness and confusion following completion. Therapy was suspended, and the patient was referred to the ED for further evaluation. Mild facial asymmetry without weakness, decreased sensation from toes to knees bilaterally, and absent Achilles reflexes bilaterally were found on neurologic examination. He exhibited past-pointing on finger-to-nose testing bilaterally. He was able to ambulate independently, but he could not perform tandem gait.
An MRI of the brain showed abnormal T2 hyperintensity found bilaterally at the dentate nuclei and inferior colliculi. The splenium of the corpus callosum also showed mild involvement with hyperintense lesions. Laboratory tests of the patient’s complete blood count; comprehensive metabolic panel; vitamins B1, B6, B12; and folic acid levels had no notable abnormalities and were within normal limits.
Metronidazole and HBOT therapy were discontinued, and all of the patient’s symptoms resolved within 2 weeks. A repeat examination and MRI performed 1 month later showed resolution of all the patient’s clinical findings and MRI abnormalities. HBOT was resumed without the recurrence of previously described symptoms.
Discussion
This patient’s encephalopathic symptoms correlate temporally with the onset of HBOT. There is no medical literature suggesting a relationship between HBOT and encephalopathic symptoms with MRI abnormalities, and in fact, some studies suggest HBOT as a treatment for hypoxic-ischemic encephalopathy in neonates.11 This led us to believe that the HBOT may have exacerbated some underlying condition, evidenced by the specific MRI findings of T2 fluid-attenuated inversion recovery (FLAIR) hyperintensities in the dentate nuclei and inferior colliculi (Figures 1 and 2).
Differential diagnoses for T2 hyperintense lesions in the dentate nuclei include metronidazole toxicity, acute Wernicke encephalopathy (WE), and methyl bromide intoxication. Diseases that would have presented in infancy with similar MRI findings (Canavan disease, maple-syrup urine disease, and glutaric aciduria type 1) were not considered plausible.12-14
Despite his denial of alcohol use, the patient was at risk for malnutrition secondary to his mandibular lesion and difficulty eating. Clinically, he presented with episodes of confusion and ataxia, consistent with 2 of the classic triad of symptoms of WE (no ocular abnormalities noted on exam). Typical MRI findings in WE include signal intensity alterations (including T2 hyperintensities) in the medial thalami, mammillary bodies, collicular bodies, and periaqueductal and periventricular regions.14,15 Atypical MRI findings in WE include symmetric signal intensity changes in the cerebellum, dentate nuclei, caudate nuclei, red nuclei, cranial nerve nuclei, and splenium.14 Of note, atypical MRI findings were more common in patients without alcohol use disorders and WE, and typical MRI findings were more common in patients with alcohol use disorders.14 However, this patient’s report of no alcohol use and the serum thiamine level being within normal limits (173 nmol/L; range 78-185 nmol/L) made acute WE less likely than metronidonazale-induced encephalopathy (MIE).
The most common neurologic AE of metronidazole is distal symmetric sensory polyneuropathy, which also can have motor or autonomic features.16,17 While our patient had a history of peripheral neuropathy, he noted marked worsening of foot pain 3 months after initiating metronidazole therapy. A potential mechanism involves metronidazole or its cytotoxic intermediates binding neuronal ribonucleic acids, thus inhibiting protein synthesis and resulting in degenerative neuronal changes and reversible axonal swelling (as opposed to the DNA interference attributed to the drug’s mechanism of bactericidal action).18 Neuropathies may result from prolonged high-dose metronidazole therapy (cumulative dose > 42 g),3 but they also have been seen in short-term use of high dosages.17
CNS AEs are much rarer and are thought to be associated with metronidazole’s ability to cross the blood-brain barrier. These patients present as a toxic encephalopathy with cerebellar dysfunction (dysarthria, ataxia) as the most common presentation, followed by AMS and seizures.4 Our patient presented with acute confusion and ataxia. Animal studies suggest that γ-aminobutyric acid (GABA) receptor modulation in the cerebellar and vestibular systems may contribute to this neurotoxicity, but no definitive mechanism of injury has been found.19
On MRI, MIE most commonly presents with hyperintense lesions in the bilateral cerebellar dentate nucleus on T2-weighted and FLAIR images.5,20 The midbrain, dorsal pons, medulla, and corpus callosum also can show increased signal intensity.5 This AE does not seem to be dose- or duration-dependent, and most cases report complete or partial resolution of symptoms following discontinuation of the drug, though this is not absolute.4,13,21 The patient’s MRI findings were highly consistent with MIE (Figure 2).
Conclusion
This patient’s highly specific MRI findings, neurologic examination consistent with confusion, ataxia, length-dependent sensory neuropathy, and 360-g cumulative dose of metronidazole over the previous 6 months suggest he experienced MIE. The mechanism of how HBOT precipitated the patient’s altered mental status, incoordination, and worsening of peripheral neuropathy is unknown. Although encephalopathy with MRI abnormalities as described is not a reported AE of HBOT, it may be unrecognized. It is possible that without HBOT the patient would have remained asymptomatic apart from his peripheral neuropathy.
We propose HBOT may exacerbate or increase the risk of a patient developing MIE. Our patient was able to safely resume HBOT after metronidazole was discontinued, suggesting that the combination was the causation for the development of encephalopathy. We do not believe any similar cases have been reported.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541.
2. Edwards DI. The action of metronidazole on DNA. J Antimicrob Chemother. 1977;3(1):43-48.
3. Goolsby TA, Jakeman B, Gaynes RP. Clinical relevance of metronidazole and peripheral neuropathy: a systematic review of the literature. Int J Antimicrob Agents. 2018;51(3):319-325.
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systematic review. Clin Neuropharmacol. 2011;34(6):241-247.
5. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658.
6. Ceponis P, Keilman C, Guerry C, Freiberger JJ. Hyperbaric oxygen therapy and osteonecrosis. Oral Dis. 2017;23(2):141-151.
7. Leach R, Rees P, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317(7166):1140-1143.
8. Thom SR. Hyperbaric oxygen–its mechanisms and efficacy. Plastic Reconstr Surg. 2011;127(suppl 1):131S-141S.
9. Plafki C, Peters P, Almeling M, Welslau W, Busch R. Complications and side effects of hyperbaric oxygen therapy. Aviation Space Environ Med. 2000;71(2):119-124.
10. Hadanny A, Meir O, Bechor Y, Fishlev G, Bergan J, Efrati S. Seizures during hyperbaric oxygen therapy: retrospective analysis of 62,614 treatment sessions. Undersea Hyperb Med. 2016;43(1):21-28.
11. Liu Z, Xiong T, Meads C. Clinical effectiveness of treatment with hyperbaric oxygen for neonatal hypoxic-ischaemic encephalopathy: systematic review of Chinese literature. BMJ. 2006;333(7564):374.
12. Bond KM, Brinjikji W, Eckel LJ, Kallmes DF, McDonald RJ, Carr CM. Dentate update: imaging features of entities that affect the dentate nucleus. AJNR Am J Neuroradiol. 2017;38(8):1467-1474.
13. Agarwal A, Kanekar S, Sabat S, Thamburaj K. Metronidazole-induced cerebellar toxicity. Neurol Int. 2016;8(1):6365.
14. Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. AJR Am J Roentgenol. 2009;192(2):501-508.
15. Jung YC, Chanraud S, Sullivan EV. Neuroimaging of Wernicke’s encephalopathy and Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):170-180.
16. Hobson-Webb LD, Roach ES, Donofrio PD. Metronidazole: newly recognized cause of autonomic neuropathy. J Child Neurol. 2006;21(5):429-431.
17. Nath Chaurasia R. Rapid onset metronidazole induced sensory neuropathy: case series and review of literature. Int J Neurorehabilitation. 2015;02:152.
18. Bradley WG, Karlsson IJ, Rassol CG. Metronidazole neuropathy. Br Med J. 1977;2(6087):610-611.
19. Evans J, Levesque D, Knowles K, Longshore R, Plummer S. Diazepam as a treatment for metronidazole toxicosis in dogs: a retrospective study of 21 cases. J Vet Intern Med. 2003;17(3):304-310.
20. Farmakiotis D, Zeluff B. Images in clinical medicine. Metronidazole-associated encephalopathy. N Engl J Med. 2016;374(15):1465.
21. Hobbs K, Stern-Nezer S, Buckwalter MS, Fischbein N, Finley Caulfield A. Metronidazole-induced encephalopathy: not always a reversible situation. Neurocrit Care. 2015;22(3):429-436.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541.
2. Edwards DI. The action of metronidazole on DNA. J Antimicrob Chemother. 1977;3(1):43-48.
3. Goolsby TA, Jakeman B, Gaynes RP. Clinical relevance of metronidazole and peripheral neuropathy: a systematic review of the literature. Int J Antimicrob Agents. 2018;51(3):319-325.
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systematic review. Clin Neuropharmacol. 2011;34(6):241-247.
5. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658.
6. Ceponis P, Keilman C, Guerry C, Freiberger JJ. Hyperbaric oxygen therapy and osteonecrosis. Oral Dis. 2017;23(2):141-151.
7. Leach R, Rees P, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317(7166):1140-1143.
8. Thom SR. Hyperbaric oxygen–its mechanisms and efficacy. Plastic Reconstr Surg. 2011;127(suppl 1):131S-141S.
9. Plafki C, Peters P, Almeling M, Welslau W, Busch R. Complications and side effects of hyperbaric oxygen therapy. Aviation Space Environ Med. 2000;71(2):119-124.
10. Hadanny A, Meir O, Bechor Y, Fishlev G, Bergan J, Efrati S. Seizures during hyperbaric oxygen therapy: retrospective analysis of 62,614 treatment sessions. Undersea Hyperb Med. 2016;43(1):21-28.
11. Liu Z, Xiong T, Meads C. Clinical effectiveness of treatment with hyperbaric oxygen for neonatal hypoxic-ischaemic encephalopathy: systematic review of Chinese literature. BMJ. 2006;333(7564):374.
12. Bond KM, Brinjikji W, Eckel LJ, Kallmes DF, McDonald RJ, Carr CM. Dentate update: imaging features of entities that affect the dentate nucleus. AJNR Am J Neuroradiol. 2017;38(8):1467-1474.
13. Agarwal A, Kanekar S, Sabat S, Thamburaj K. Metronidazole-induced cerebellar toxicity. Neurol Int. 2016;8(1):6365.
14. Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. AJR Am J Roentgenol. 2009;192(2):501-508.
15. Jung YC, Chanraud S, Sullivan EV. Neuroimaging of Wernicke’s encephalopathy and Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):170-180.
16. Hobson-Webb LD, Roach ES, Donofrio PD. Metronidazole: newly recognized cause of autonomic neuropathy. J Child Neurol. 2006;21(5):429-431.
17. Nath Chaurasia R. Rapid onset metronidazole induced sensory neuropathy: case series and review of literature. Int J Neurorehabilitation. 2015;02:152.
18. Bradley WG, Karlsson IJ, Rassol CG. Metronidazole neuropathy. Br Med J. 1977;2(6087):610-611.
19. Evans J, Levesque D, Knowles K, Longshore R, Plummer S. Diazepam as a treatment for metronidazole toxicosis in dogs: a retrospective study of 21 cases. J Vet Intern Med. 2003;17(3):304-310.
20. Farmakiotis D, Zeluff B. Images in clinical medicine. Metronidazole-associated encephalopathy. N Engl J Med. 2016;374(15):1465.
21. Hobbs K, Stern-Nezer S, Buckwalter MS, Fischbein N, Finley Caulfield A. Metronidazole-induced encephalopathy: not always a reversible situation. Neurocrit Care. 2015;22(3):429-436.
Fingernail Abnormalities After a Systemic Illness
A 45-year-old African American woman presented with painless fingernail detachment and cracks on her fingernails that had developed over the previous month. Her medical history was notable for an episode of Stevens-Johnson syndrome 2 months prior that required treatment with prednisone, IV immunoglobulin, etanercept, acetaminophen, and diphenhydramine.
A physical examination revealed multiple fingernails on both hands that exhibited 4 mm of proximal painless nail detachment with cream-colored discoloration, friability, and horizontal splitting (Figure). New, healthy nail was visible beneath the affected areas. Toenails were not affected.
- What is your diagnosis?
- How would you treat this patient?
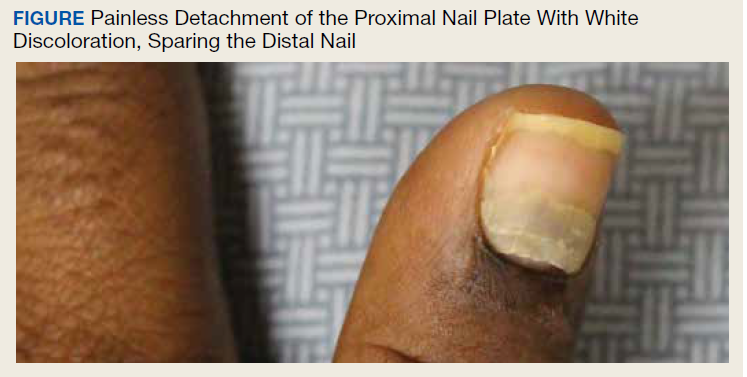
Diagnosis
Based on the timing and characteristics of her nail detachment, the patient was diagnosed with onychomadesis, which is defined as painless detachment of the proximal nail plate from the nail matrix and nail bed after at least 40 days from an initial insult. Air beneath the detached nail plate causes a characteristic creamy-white discoloration. The severity of onychomadesis ranges from transverse furrows that affect a single nail without shedding, known as Beau lines, to multiple nails that are completely shed.1,2 Nail plate shedding is typical because the nail matrix, the site of stem cells and the most proximal portion of the nail apparatus, is damaged and transiently arrested.
Various etiologies can halt nail plate production abruptly within the matrix. These typically manifest ≥ 40 days after the initial insult (the length of time for a fingernail to emerge from the proximal nail fold).2 The annual incidence of these etiologies ranges from approximately 1 per 1 million people for Stevens-Johnson syndrome, a rare cause of onychomadesis, to 1 per 10 people for onychomycosis, one of the more common causes of onychomadesis.3 The Table compares the characteristics of the diagnoses that are most commonly associated with nail detachment and discoloration. 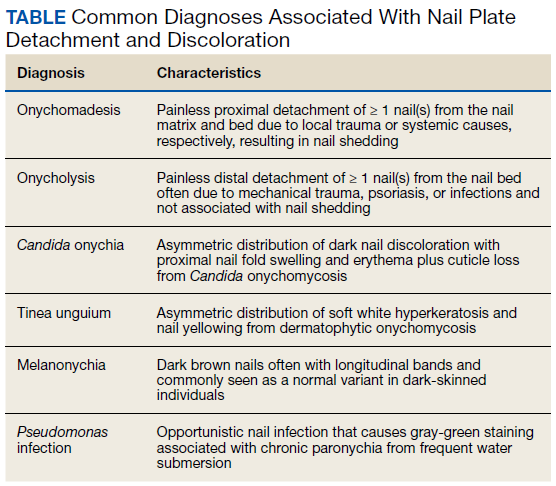
When a single nail is affected, the etiology of onychomadesis usually is primary and local, including mechanical nail trauma and fungal nail infections (onychomycosis).1,2 Candida onychia is onychomycosis caused by Candida species typically Candida albicans, which result in localized nail darkening, chronic inflammation of the paronychial skin, and cuticle loss. The infection favors immunocompromised people; coinfections are common, and onychomadesis or onycholysis can occur. Unlike onychomadesis, onycholysis is defined by painless detachment of the distal nail plate from the nail bed, but nail shedding typically does not occur because the nail matrix is spared. The preferred treatment for Candida onychia is oral itraconazole, and guided screenings for immunodeficiencies and endocrinopathies, especially diabetes mellitus, should be completed.3,4
Tinea unguium is another form of onychomycosis, but it is caused by dermatophytes, typically Trichophyton rubrum or Trichophyton mentagrophytes, which produce white and yellow nail discoloration followed by distal to proximal nail thickening and softening. Infection usually begins in toenails and demonstrates variable involvement in each nail as well as asymmetric distribution among digits.3 This condition also may eventuate in onychomadesis or onycholysis. Debridement followed by oral terbinafine is the treatment of choice.4
Two other causes of localized nail discoloration with or without nail detachment include melanonychia and nail bed infection by Pseudomonas aeruginosa (P aeruginosa). Melanonychia can be linear or diffuse brown discoloration of 1 or more nails caused by melanin deposition. Either pattern is a common finding in dark-skinned people, especially by age 50 years, but melanocyte hyperplasia should be excluded in all individuals along with drug adverse effects, exogenous pigments, infections, and systemic diseases.3,5 P aeruginosa produces pyocyanin, the green pigment responsible for the discoloration seen in this opportunistic infection often localized to a single nail. Prior maceration of the nail apparatus by repeated water submersion is common among affected individuals. Avoidance of submerging fingernails in liquids followed by nail debridement and oral antipseudomonal antibiotics is the preferred treatment course.3
The etiology is usually secondary and systemic when multiple nails demonstrate onychomadesis, but the exact pathophysiology is poorly understood. One of the most studied infectious etiologies of onychomadesis is hand-foot-and-mouth disease (HFMD), which typically affects children aged < 10 years. Parents often will recall their child being ill 1 to 2 months prior to the nail findings. Scarlet fever and varicella also can result in onychomadesis. Although not common systemic causes, Stevens-Johnson syndrome and toxic epidermal necrolysis can trigger onychomadesis of multiple nails that usually resolves in several months, but other nail deformities often persist.2,6 Onycholysis also can accompany this finding.7 Autoimmune etiologies of onychomadesis include alopecia areata and pemphigus vulgaris. Inciting medications that are toxic to the nail matrix include chemotherapy agents, valproic acid, carbamazepine, lithium, and azithromycin. Rare congenital disorders and birth trauma also can present with onychomadesis of multiple nails during infancy.2
Systemic etiologies typically affect fingernails more than toenails because of the faster growth rate of fingernails. Once the source of onychomadesis is controlled or eradicated, complete regrowth of fingernails can take from 4 to 6 months. Toenails can take twice as long and older age increases all regrowth periods.5
Our patient was treated with analgesics until her mucosal surfaces fully healed, and topical emollients and keratolytics were used to soften eschars from previous blisters and prevent further scar formation. Her affected fingernails shed and regrew after 6 months without additional interventions.
Conclusion
Although Stevens-Johnson syndrome is a rare cause of onychomadesis, and the pathophysiology of this sequela is poorly understood, this case illustrates a common nail abnormality with multiple potential etiologies that are discerned by an accurate history and thorough exam. In the absence of decorative nail polish, nails can be easily examined to provide helpful clues for past injuries or underlying diseases. An understanding of nail growth mechanics and associated terminology reveals the diagnostic and therapeutic implications of proximal vs distal nail detachment, the hue of nail discoloration, as well as single vs multiple affected nails.
Onychomadesis in single nails should prompt questions about nail trauma or risk factors for fungal infections. Depending on the etiology, manual activities need to be adjusted, or antifungals need to be initiated while investigating for an immunocompromised state. Onychomadesis in multiple nails in children should raise suspicion for HFMD or even birth trauma and congenital disorders. Multiple affected nails in adults should prompt guided questions for autoimmune diseases and inciting medications. For onycholysis, trauma, psoriasis, or certain infections should be the target. Green nails are easily recognized and treated with a defined regiment, whereas dark nails should be examined closely to differentiate Candida onychia from melanonychia. Whether from a rare cause in an adult to a common illness in a child, primary care providers have sufficient expertise to diagnose and treat various nail disorders and reassure worried patients and parents with an understanding of nail regrowth.
1. Salgado F, Handler MZ, Schwartz RA. Shedding light on onychomadesis. Cutis. 2017;99(1):33-36.
2. Hardin J, Haber RM. Oncyhomadesis: literature review. Br J Dermatol. 2015;172(3):592-596.
3. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology. 5th ed. New York, NY: McGraw-Hill; 2005.
4. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Saunders; 2012.
5. Shemer A, Daniel CR III. Common nail disorders. Clin Dermatol. 2013;31(5):578-586.
6. Acharya S, Balachandran C. Onychomadesis in Stevens-Johnson syndrome. Indian J Dermatol Venereol Leprol. 1996;62(4):264-265.
7. Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: part II. Prognosis, sequelae, diagnosis, differential diagnosis, prevention, and treatment. J Am Acad Dermatol. 2013;69(2):187.e1-e16.
A 45-year-old African American woman presented with painless fingernail detachment and cracks on her fingernails that had developed over the previous month. Her medical history was notable for an episode of Stevens-Johnson syndrome 2 months prior that required treatment with prednisone, IV immunoglobulin, etanercept, acetaminophen, and diphenhydramine.
A physical examination revealed multiple fingernails on both hands that exhibited 4 mm of proximal painless nail detachment with cream-colored discoloration, friability, and horizontal splitting (Figure). New, healthy nail was visible beneath the affected areas. Toenails were not affected.
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
Based on the timing and characteristics of her nail detachment, the patient was diagnosed with onychomadesis, which is defined as painless detachment of the proximal nail plate from the nail matrix and nail bed after at least 40 days from an initial insult. Air beneath the detached nail plate causes a characteristic creamy-white discoloration. The severity of onychomadesis ranges from transverse furrows that affect a single nail without shedding, known as Beau lines, to multiple nails that are completely shed.1,2 Nail plate shedding is typical because the nail matrix, the site of stem cells and the most proximal portion of the nail apparatus, is damaged and transiently arrested.
Various etiologies can halt nail plate production abruptly within the matrix. These typically manifest ≥ 40 days after the initial insult (the length of time for a fingernail to emerge from the proximal nail fold).2 The annual incidence of these etiologies ranges from approximately 1 per 1 million people for Stevens-Johnson syndrome, a rare cause of onychomadesis, to 1 per 10 people for onychomycosis, one of the more common causes of onychomadesis.3 The Table compares the characteristics of the diagnoses that are most commonly associated with nail detachment and discoloration.
When a single nail is affected, the etiology of onychomadesis usually is primary and local, including mechanical nail trauma and fungal nail infections (onychomycosis).1,2 Candida onychia is onychomycosis caused by Candida species typically Candida albicans, which result in localized nail darkening, chronic inflammation of the paronychial skin, and cuticle loss. The infection favors immunocompromised people; coinfections are common, and onychomadesis or onycholysis can occur. Unlike onychomadesis, onycholysis is defined by painless detachment of the distal nail plate from the nail bed, but nail shedding typically does not occur because the nail matrix is spared. The preferred treatment for Candida onychia is oral itraconazole, and guided screenings for immunodeficiencies and endocrinopathies, especially diabetes mellitus, should be completed.3,4
Tinea unguium is another form of onychomycosis, but it is caused by dermatophytes, typically Trichophyton rubrum or Trichophyton mentagrophytes, which produce white and yellow nail discoloration followed by distal to proximal nail thickening and softening. Infection usually begins in toenails and demonstrates variable involvement in each nail as well as asymmetric distribution among digits.3 This condition also may eventuate in onychomadesis or onycholysis. Debridement followed by oral terbinafine is the treatment of choice.4
Two other causes of localized nail discoloration with or without nail detachment include melanonychia and nail bed infection by Pseudomonas aeruginosa (P aeruginosa). Melanonychia can be linear or diffuse brown discoloration of 1 or more nails caused by melanin deposition. Either pattern is a common finding in dark-skinned people, especially by age 50 years, but melanocyte hyperplasia should be excluded in all individuals along with drug adverse effects, exogenous pigments, infections, and systemic diseases.3,5 P aeruginosa produces pyocyanin, the green pigment responsible for the discoloration seen in this opportunistic infection often localized to a single nail. Prior maceration of the nail apparatus by repeated water submersion is common among affected individuals. Avoidance of submerging fingernails in liquids followed by nail debridement and oral antipseudomonal antibiotics is the preferred treatment course.3
The etiology is usually secondary and systemic when multiple nails demonstrate onychomadesis, but the exact pathophysiology is poorly understood. One of the most studied infectious etiologies of onychomadesis is hand-foot-and-mouth disease (HFMD), which typically affects children aged < 10 years. Parents often will recall their child being ill 1 to 2 months prior to the nail findings. Scarlet fever and varicella also can result in onychomadesis. Although not common systemic causes, Stevens-Johnson syndrome and toxic epidermal necrolysis can trigger onychomadesis of multiple nails that usually resolves in several months, but other nail deformities often persist.2,6 Onycholysis also can accompany this finding.7 Autoimmune etiologies of onychomadesis include alopecia areata and pemphigus vulgaris. Inciting medications that are toxic to the nail matrix include chemotherapy agents, valproic acid, carbamazepine, lithium, and azithromycin. Rare congenital disorders and birth trauma also can present with onychomadesis of multiple nails during infancy.2
Systemic etiologies typically affect fingernails more than toenails because of the faster growth rate of fingernails. Once the source of onychomadesis is controlled or eradicated, complete regrowth of fingernails can take from 4 to 6 months. Toenails can take twice as long and older age increases all regrowth periods.5
Our patient was treated with analgesics until her mucosal surfaces fully healed, and topical emollients and keratolytics were used to soften eschars from previous blisters and prevent further scar formation. Her affected fingernails shed and regrew after 6 months without additional interventions.
Conclusion
Although Stevens-Johnson syndrome is a rare cause of onychomadesis, and the pathophysiology of this sequela is poorly understood, this case illustrates a common nail abnormality with multiple potential etiologies that are discerned by an accurate history and thorough exam. In the absence of decorative nail polish, nails can be easily examined to provide helpful clues for past injuries or underlying diseases. An understanding of nail growth mechanics and associated terminology reveals the diagnostic and therapeutic implications of proximal vs distal nail detachment, the hue of nail discoloration, as well as single vs multiple affected nails.
Onychomadesis in single nails should prompt questions about nail trauma or risk factors for fungal infections. Depending on the etiology, manual activities need to be adjusted, or antifungals need to be initiated while investigating for an immunocompromised state. Onychomadesis in multiple nails in children should raise suspicion for HFMD or even birth trauma and congenital disorders. Multiple affected nails in adults should prompt guided questions for autoimmune diseases and inciting medications. For onycholysis, trauma, psoriasis, or certain infections should be the target. Green nails are easily recognized and treated with a defined regiment, whereas dark nails should be examined closely to differentiate Candida onychia from melanonychia. Whether from a rare cause in an adult to a common illness in a child, primary care providers have sufficient expertise to diagnose and treat various nail disorders and reassure worried patients and parents with an understanding of nail regrowth.
A 45-year-old African American woman presented with painless fingernail detachment and cracks on her fingernails that had developed over the previous month. Her medical history was notable for an episode of Stevens-Johnson syndrome 2 months prior that required treatment with prednisone, IV immunoglobulin, etanercept, acetaminophen, and diphenhydramine.
A physical examination revealed multiple fingernails on both hands that exhibited 4 mm of proximal painless nail detachment with cream-colored discoloration, friability, and horizontal splitting (Figure). New, healthy nail was visible beneath the affected areas. Toenails were not affected.
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
Based on the timing and characteristics of her nail detachment, the patient was diagnosed with onychomadesis, which is defined as painless detachment of the proximal nail plate from the nail matrix and nail bed after at least 40 days from an initial insult. Air beneath the detached nail plate causes a characteristic creamy-white discoloration. The severity of onychomadesis ranges from transverse furrows that affect a single nail without shedding, known as Beau lines, to multiple nails that are completely shed.1,2 Nail plate shedding is typical because the nail matrix, the site of stem cells and the most proximal portion of the nail apparatus, is damaged and transiently arrested.
Various etiologies can halt nail plate production abruptly within the matrix. These typically manifest ≥ 40 days after the initial insult (the length of time for a fingernail to emerge from the proximal nail fold).2 The annual incidence of these etiologies ranges from approximately 1 per 1 million people for Stevens-Johnson syndrome, a rare cause of onychomadesis, to 1 per 10 people for onychomycosis, one of the more common causes of onychomadesis.3 The Table compares the characteristics of the diagnoses that are most commonly associated with nail detachment and discoloration.
When a single nail is affected, the etiology of onychomadesis usually is primary and local, including mechanical nail trauma and fungal nail infections (onychomycosis).1,2 Candida onychia is onychomycosis caused by Candida species typically Candida albicans, which result in localized nail darkening, chronic inflammation of the paronychial skin, and cuticle loss. The infection favors immunocompromised people; coinfections are common, and onychomadesis or onycholysis can occur. Unlike onychomadesis, onycholysis is defined by painless detachment of the distal nail plate from the nail bed, but nail shedding typically does not occur because the nail matrix is spared. The preferred treatment for Candida onychia is oral itraconazole, and guided screenings for immunodeficiencies and endocrinopathies, especially diabetes mellitus, should be completed.3,4
Tinea unguium is another form of onychomycosis, but it is caused by dermatophytes, typically Trichophyton rubrum or Trichophyton mentagrophytes, which produce white and yellow nail discoloration followed by distal to proximal nail thickening and softening. Infection usually begins in toenails and demonstrates variable involvement in each nail as well as asymmetric distribution among digits.3 This condition also may eventuate in onychomadesis or onycholysis. Debridement followed by oral terbinafine is the treatment of choice.4
Two other causes of localized nail discoloration with or without nail detachment include melanonychia and nail bed infection by Pseudomonas aeruginosa (P aeruginosa). Melanonychia can be linear or diffuse brown discoloration of 1 or more nails caused by melanin deposition. Either pattern is a common finding in dark-skinned people, especially by age 50 years, but melanocyte hyperplasia should be excluded in all individuals along with drug adverse effects, exogenous pigments, infections, and systemic diseases.3,5 P aeruginosa produces pyocyanin, the green pigment responsible for the discoloration seen in this opportunistic infection often localized to a single nail. Prior maceration of the nail apparatus by repeated water submersion is common among affected individuals. Avoidance of submerging fingernails in liquids followed by nail debridement and oral antipseudomonal antibiotics is the preferred treatment course.3
The etiology is usually secondary and systemic when multiple nails demonstrate onychomadesis, but the exact pathophysiology is poorly understood. One of the most studied infectious etiologies of onychomadesis is hand-foot-and-mouth disease (HFMD), which typically affects children aged < 10 years. Parents often will recall their child being ill 1 to 2 months prior to the nail findings. Scarlet fever and varicella also can result in onychomadesis. Although not common systemic causes, Stevens-Johnson syndrome and toxic epidermal necrolysis can trigger onychomadesis of multiple nails that usually resolves in several months, but other nail deformities often persist.2,6 Onycholysis also can accompany this finding.7 Autoimmune etiologies of onychomadesis include alopecia areata and pemphigus vulgaris. Inciting medications that are toxic to the nail matrix include chemotherapy agents, valproic acid, carbamazepine, lithium, and azithromycin. Rare congenital disorders and birth trauma also can present with onychomadesis of multiple nails during infancy.2
Systemic etiologies typically affect fingernails more than toenails because of the faster growth rate of fingernails. Once the source of onychomadesis is controlled or eradicated, complete regrowth of fingernails can take from 4 to 6 months. Toenails can take twice as long and older age increases all regrowth periods.5
Our patient was treated with analgesics until her mucosal surfaces fully healed, and topical emollients and keratolytics were used to soften eschars from previous blisters and prevent further scar formation. Her affected fingernails shed and regrew after 6 months without additional interventions.
Conclusion
Although Stevens-Johnson syndrome is a rare cause of onychomadesis, and the pathophysiology of this sequela is poorly understood, this case illustrates a common nail abnormality with multiple potential etiologies that are discerned by an accurate history and thorough exam. In the absence of decorative nail polish, nails can be easily examined to provide helpful clues for past injuries or underlying diseases. An understanding of nail growth mechanics and associated terminology reveals the diagnostic and therapeutic implications of proximal vs distal nail detachment, the hue of nail discoloration, as well as single vs multiple affected nails.
Onychomadesis in single nails should prompt questions about nail trauma or risk factors for fungal infections. Depending on the etiology, manual activities need to be adjusted, or antifungals need to be initiated while investigating for an immunocompromised state. Onychomadesis in multiple nails in children should raise suspicion for HFMD or even birth trauma and congenital disorders. Multiple affected nails in adults should prompt guided questions for autoimmune diseases and inciting medications. For onycholysis, trauma, psoriasis, or certain infections should be the target. Green nails are easily recognized and treated with a defined regiment, whereas dark nails should be examined closely to differentiate Candida onychia from melanonychia. Whether from a rare cause in an adult to a common illness in a child, primary care providers have sufficient expertise to diagnose and treat various nail disorders and reassure worried patients and parents with an understanding of nail regrowth.
1. Salgado F, Handler MZ, Schwartz RA. Shedding light on onychomadesis. Cutis. 2017;99(1):33-36.
2. Hardin J, Haber RM. Oncyhomadesis: literature review. Br J Dermatol. 2015;172(3):592-596.
3. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology. 5th ed. New York, NY: McGraw-Hill; 2005.
4. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Saunders; 2012.
5. Shemer A, Daniel CR III. Common nail disorders. Clin Dermatol. 2013;31(5):578-586.
6. Acharya S, Balachandran C. Onychomadesis in Stevens-Johnson syndrome. Indian J Dermatol Venereol Leprol. 1996;62(4):264-265.
7. Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: part II. Prognosis, sequelae, diagnosis, differential diagnosis, prevention, and treatment. J Am Acad Dermatol. 2013;69(2):187.e1-e16.
1. Salgado F, Handler MZ, Schwartz RA. Shedding light on onychomadesis. Cutis. 2017;99(1):33-36.
2. Hardin J, Haber RM. Oncyhomadesis: literature review. Br J Dermatol. 2015;172(3):592-596.
3. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology. 5th ed. New York, NY: McGraw-Hill; 2005.
4. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Saunders; 2012.
5. Shemer A, Daniel CR III. Common nail disorders. Clin Dermatol. 2013;31(5):578-586.
6. Acharya S, Balachandran C. Onychomadesis in Stevens-Johnson syndrome. Indian J Dermatol Venereol Leprol. 1996;62(4):264-265.
7. Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: part II. Prognosis, sequelae, diagnosis, differential diagnosis, prevention, and treatment. J Am Acad Dermatol. 2013;69(2):187.e1-e16.
Daily headaches • associated nausea • obesity • Dx?
THE CASE
A 22-year-old woman presented to our office complaining of headaches that started 6 weeks earlier. Initially the headache was throbbing, nonpositional, infrequent, and intermittent, lasting 15 to 45 minutes, often starting in the neck and migrating towards the right frontotemporal region. During the week prior to presentation, the headaches became daily and constant, with brief periods of relief after the patient took ibuprofen 400 mg 4 times a day as needed. The patient reported associated nausea, a sensation of pressure changes in the ears, and intermittent dimming of vision in the right eye (sometimes independent of headache). The patient denied photophobia and phonophobia. Her only medication was an oral contraceptive pill (OCP). She had no prior history of headaches.
Physical examination showed a blood pressure of 148/66 mm Hg, body mass index of 44.38, muscle tenderness in the neck and upper back, and no focal neurological findings. Funduscopic examination was unsuccessful. A working diagnosis of atypical migraine was made, but because of unilateral visual disturbance the patient was referred to Ophthalmology for further evaluation. The following day, ophthalmological consultation found bilateral papilledema and the patient was admitted to our hospitalist service via the Emergency Department. She subsequently was referred to inpatient Neurology.
THE DIAGNOSIS
Magnetic resonance imaging (MRI) of the brain and orbits with and without contrast was unremarkable. Magnetic resonance venography (MRV) with contrast of the brain showed possible stenosis at the junction of the transverse and sigmoid sinuses but no mass lesion nor venous sinus thrombosis. Lumbar puncture (LP) revealed an opening pressure of 650 mm H20 (reference range, 60–250 mm H2O).1 A diagnosis of idiopathic intracranial hypertension (IIH) was made.

DISCUSSION
IIH, previously known as pseudotumor cerebri and benign intracranial hypertension, is defined by signs and symptoms of elevated intracranial pressure (ICP) without obvious cause on neuroimaging (TABLE 12-5). It is well documented that IIH is consequential and can result in vision loss and intractable chronic headaches.5,6 Older terms such as pseudotumor cerebri and benign intracranial hypertension are therefore no longer recommended because they are considered misleading and not reflective of the severity of potential injury caused by the condition3,4,6 IIH is considered a diagnosis of exclusion requiring certain criteria to be met (TABLE 22). Although the etiology of IIH is unclear, associations have been made between IIH and various medications and conditions2-5,7 (TABLE 33,5).

Classically, IIH affects women who are obese and of childbearing age, but studies have shown that this condition also can affect men and children—albeit less frequently.3,5-7 The incidence of IIH in the general population is between 0.03 to 2.36/100,000 people per year, but in women, the incidence is 0.65 to 4.65/100,000 per year.6 Furthermore, females who are obese have an incidence of 2.7 to 19.3/100,000 per year.6

Headache is the most common symptom of IIH. Unfortunately, the differential diagnosis of headache is vast; thus, a careful history is needed to narrow the field3,5-7 (TABLE 42). Associated symptoms of transient visual changes, pulsatile tinnitus, neck and back pain, nausea, vomiting, photo/phonophobia, and findings of abducens nerve palsy or papilledema—while nonspecific— should raise suspicion for elevated ICP and IIH, especially in women who are obese.2-8 Once IIH is suspected, an urgent diagnosis and treatment is necessary to prevent permanent vision loss.3,4,6

Headache with findings of papilledema warrants neuroimaging, preferably with MRI, to rule out intracranial mass and hydrocephalus.1,2,5 MRV also is recommended to assess for intracranial venous thrombosis, an alternate cause for papilledema and increased ICP.1,2,4,5
Continue to: Recently, a classification of IIH...
Recently, a classification of IIH without papilledema has been acknowledged by the International Headache Society.2,8 Specific MRI findings have been suggested to help make this diagnosis5,9 (TABLE 55).

TREATMENT FOR IIH CAN BE MEDICAL OR SURGICAL
Medications associated with IIH should be discontinued.7 The first-line medication for IIH is acetazolamide, a carbonic anhydrase inhibitor that works in the choroid plexus to decrease cerebrospinal fluid (CSF) production and thus, lower ICP.3,6 An adult dose of 1 to 2 g/day3,4,6 is tolerated well, but can be increased to 4 g/day,10 if necessary. Weight loss via diet and exercise or bariatric surgery has been shown to be effective in patients who are obese and have been given a diagnosis of IIH.3,4
Topiramate also has been suggested as a treatment option, based on its usefulness in weight loss and because of its action as a weak carbonic anhydrase inhibitor.3,6 Also, LP has therapeutic merit—although relief is only short-term.3,6 Patients who fail medical therapy and have intractable headache or progressive visual loss appear to benefit from optic nerve sheath fenestration.3,7,8
Our patient experienced notable improvement in her headache after LP. Her OCP was discontinued, a diuretic regimen started, and weight loss counseling was provided. Prior to discharge, the patient was seen by a neuro-ophthalmologist for perimetry, a visual field test that assesses for acute vision loss and establishes a baseline for follow-up monitoring of vision.7
THE TAKEAWAY
Headache is a common condition that may be challenging to correctly diagnose. A thorough history and neurological examination, including fundoscopy, are essential in the evaluation of headache and suspected IIH. In the primary care setting, limited time, lack of mydriatic agents, suboptimal lighting, and practitioner inexperience may pose challenges for funduscopic examination. Ophthalmoscopes incorporating new technology to expand and magnify the examiner’s field of view may facilitate this exam.11 A global rise in the prevalence of obesity underscores a need for primary care providers to be compulsive about their clinical evaluation when symptoms suspicious of IIH are present. Lastly, if IIH cannot be ruled out confidently, recommend a prompt evaluation by an ophthalmologist.
CORRESPONDENCE
Aarti Paltoo, MD, MSc, CCFP, Peel Village Medical Center, 28 Rambler Drive, Brampton, Ontario L6W 1E2 Canada; [email protected]
1. Lee SC, Lueck CJ. Cerebrospinal fluid pressure in adults. J Neuroophthalmol. 2014;34:278-283.
2. International Headache Society. Idiopathic intracranial hypertension. The International Classification of Headache Disorders. 2nd ed. Oxford, UK: Blackwell Publishing; 2003:1-232.
3. Biousse V, Bruce BB, Newman NJ. Update on the pathophysiology and management of idiopathic intracranial hypertension. J Neurol Neurosurg Psychiatry. 2012;83:488-494.
4. Mollan SP, Markey KA, Benzimra JD, et al. A practical approach to diagnosis, assessment and management of idiopathic intracranial hypertension. Pract Neurol. 2014;14:380-390.
5. Friedman DI, Liu GT, Digre KB. Revised diagnostic criteria for the pseudotumor cerebri syndrome in adults and children. Neurology. 2013;81:1159-1165.
6. Julayanont P, Karukote A, Ruthirago D, et al. Idiopathic intracranial hypertension: ongoing clinical challenges and future prospects. J Pain Res. 2016;9:87-99.
7. Friedman DI, Digre KB. Headache medicine meets neuro-ophthalmology: exam techniques and challenging cases. Headache. 2013;53:703-716.
8. Digre KB, Nakamoto BK, Warner JE, et al. A comparison of idiopathic intracranaial hypertension with and without papilledema. Headache. 2009;49:185-193.
9. Digre KB. Imaging characteristics of IIH: are they reliable? Cephalagia. 2013;33:1067-1069.
10. Horton J. Acetazolamide for pseudotumor cerebri--evidence from the NORDIC trial. JAMA. 2014;311:1618-1619.
11. Petrushkin H, Barsam A, Mavrakakis M, et al. Optic disc assessment in the emergency department: a comparative study between the PanOptic and direct ophthalmoscopes. Emerg Med J. 2012;29:1007-1008.
THE CASE
A 22-year-old woman presented to our office complaining of headaches that started 6 weeks earlier. Initially the headache was throbbing, nonpositional, infrequent, and intermittent, lasting 15 to 45 minutes, often starting in the neck and migrating towards the right frontotemporal region. During the week prior to presentation, the headaches became daily and constant, with brief periods of relief after the patient took ibuprofen 400 mg 4 times a day as needed. The patient reported associated nausea, a sensation of pressure changes in the ears, and intermittent dimming of vision in the right eye (sometimes independent of headache). The patient denied photophobia and phonophobia. Her only medication was an oral contraceptive pill (OCP). She had no prior history of headaches.
Physical examination showed a blood pressure of 148/66 mm Hg, body mass index of 44.38, muscle tenderness in the neck and upper back, and no focal neurological findings. Funduscopic examination was unsuccessful. A working diagnosis of atypical migraine was made, but because of unilateral visual disturbance the patient was referred to Ophthalmology for further evaluation. The following day, ophthalmological consultation found bilateral papilledema and the patient was admitted to our hospitalist service via the Emergency Department. She subsequently was referred to inpatient Neurology.
THE DIAGNOSIS
Magnetic resonance imaging (MRI) of the brain and orbits with and without contrast was unremarkable. Magnetic resonance venography (MRV) with contrast of the brain showed possible stenosis at the junction of the transverse and sigmoid sinuses but no mass lesion nor venous sinus thrombosis. Lumbar puncture (LP) revealed an opening pressure of 650 mm H20 (reference range, 60–250 mm H2O).1 A diagnosis of idiopathic intracranial hypertension (IIH) was made.
DISCUSSION
IIH, previously known as pseudotumor cerebri and benign intracranial hypertension, is defined by signs and symptoms of elevated intracranial pressure (ICP) without obvious cause on neuroimaging (TABLE 12-5). It is well documented that IIH is consequential and can result in vision loss and intractable chronic headaches.5,6 Older terms such as pseudotumor cerebri and benign intracranial hypertension are therefore no longer recommended because they are considered misleading and not reflective of the severity of potential injury caused by the condition3,4,6 IIH is considered a diagnosis of exclusion requiring certain criteria to be met (TABLE 22). Although the etiology of IIH is unclear, associations have been made between IIH and various medications and conditions2-5,7 (TABLE 33,5).
Classically, IIH affects women who are obese and of childbearing age, but studies have shown that this condition also can affect men and children—albeit less frequently.3,5-7 The incidence of IIH in the general population is between 0.03 to 2.36/100,000 people per year, but in women, the incidence is 0.65 to 4.65/100,000 per year.6 Furthermore, females who are obese have an incidence of 2.7 to 19.3/100,000 per year.6
Headache is the most common symptom of IIH. Unfortunately, the differential diagnosis of headache is vast; thus, a careful history is needed to narrow the field3,5-7 (TABLE 42). Associated symptoms of transient visual changes, pulsatile tinnitus, neck and back pain, nausea, vomiting, photo/phonophobia, and findings of abducens nerve palsy or papilledema—while nonspecific— should raise suspicion for elevated ICP and IIH, especially in women who are obese.2-8 Once IIH is suspected, an urgent diagnosis and treatment is necessary to prevent permanent vision loss.3,4,6
Headache with findings of papilledema warrants neuroimaging, preferably with MRI, to rule out intracranial mass and hydrocephalus.1,2,5 MRV also is recommended to assess for intracranial venous thrombosis, an alternate cause for papilledema and increased ICP.1,2,4,5
Continue to: Recently, a classification of IIH...
Recently, a classification of IIH without papilledema has been acknowledged by the International Headache Society.2,8 Specific MRI findings have been suggested to help make this diagnosis5,9 (TABLE 55).
TREATMENT FOR IIH CAN BE MEDICAL OR SURGICAL
Medications associated with IIH should be discontinued.7 The first-line medication for IIH is acetazolamide, a carbonic anhydrase inhibitor that works in the choroid plexus to decrease cerebrospinal fluid (CSF) production and thus, lower ICP.3,6 An adult dose of 1 to 2 g/day3,4,6 is tolerated well, but can be increased to 4 g/day,10 if necessary. Weight loss via diet and exercise or bariatric surgery has been shown to be effective in patients who are obese and have been given a diagnosis of IIH.3,4
Topiramate also has been suggested as a treatment option, based on its usefulness in weight loss and because of its action as a weak carbonic anhydrase inhibitor.3,6 Also, LP has therapeutic merit—although relief is only short-term.3,6 Patients who fail medical therapy and have intractable headache or progressive visual loss appear to benefit from optic nerve sheath fenestration.3,7,8
Our patient experienced notable improvement in her headache after LP. Her OCP was discontinued, a diuretic regimen started, and weight loss counseling was provided. Prior to discharge, the patient was seen by a neuro-ophthalmologist for perimetry, a visual field test that assesses for acute vision loss and establishes a baseline for follow-up monitoring of vision.7
THE TAKEAWAY
Headache is a common condition that may be challenging to correctly diagnose. A thorough history and neurological examination, including fundoscopy, are essential in the evaluation of headache and suspected IIH. In the primary care setting, limited time, lack of mydriatic agents, suboptimal lighting, and practitioner inexperience may pose challenges for funduscopic examination. Ophthalmoscopes incorporating new technology to expand and magnify the examiner’s field of view may facilitate this exam.11 A global rise in the prevalence of obesity underscores a need for primary care providers to be compulsive about their clinical evaluation when symptoms suspicious of IIH are present. Lastly, if IIH cannot be ruled out confidently, recommend a prompt evaluation by an ophthalmologist.
CORRESPONDENCE
Aarti Paltoo, MD, MSc, CCFP, Peel Village Medical Center, 28 Rambler Drive, Brampton, Ontario L6W 1E2 Canada; [email protected]
THE CASE
A 22-year-old woman presented to our office complaining of headaches that started 6 weeks earlier. Initially the headache was throbbing, nonpositional, infrequent, and intermittent, lasting 15 to 45 minutes, often starting in the neck and migrating towards the right frontotemporal region. During the week prior to presentation, the headaches became daily and constant, with brief periods of relief after the patient took ibuprofen 400 mg 4 times a day as needed. The patient reported associated nausea, a sensation of pressure changes in the ears, and intermittent dimming of vision in the right eye (sometimes independent of headache). The patient denied photophobia and phonophobia. Her only medication was an oral contraceptive pill (OCP). She had no prior history of headaches.
Physical examination showed a blood pressure of 148/66 mm Hg, body mass index of 44.38, muscle tenderness in the neck and upper back, and no focal neurological findings. Funduscopic examination was unsuccessful. A working diagnosis of atypical migraine was made, but because of unilateral visual disturbance the patient was referred to Ophthalmology for further evaluation. The following day, ophthalmological consultation found bilateral papilledema and the patient was admitted to our hospitalist service via the Emergency Department. She subsequently was referred to inpatient Neurology.
THE DIAGNOSIS
Magnetic resonance imaging (MRI) of the brain and orbits with and without contrast was unremarkable. Magnetic resonance venography (MRV) with contrast of the brain showed possible stenosis at the junction of the transverse and sigmoid sinuses but no mass lesion nor venous sinus thrombosis. Lumbar puncture (LP) revealed an opening pressure of 650 mm H20 (reference range, 60–250 mm H2O).1 A diagnosis of idiopathic intracranial hypertension (IIH) was made.
DISCUSSION
IIH, previously known as pseudotumor cerebri and benign intracranial hypertension, is defined by signs and symptoms of elevated intracranial pressure (ICP) without obvious cause on neuroimaging (TABLE 12-5). It is well documented that IIH is consequential and can result in vision loss and intractable chronic headaches.5,6 Older terms such as pseudotumor cerebri and benign intracranial hypertension are therefore no longer recommended because they are considered misleading and not reflective of the severity of potential injury caused by the condition3,4,6 IIH is considered a diagnosis of exclusion requiring certain criteria to be met (TABLE 22). Although the etiology of IIH is unclear, associations have been made between IIH and various medications and conditions2-5,7 (TABLE 33,5).
Classically, IIH affects women who are obese and of childbearing age, but studies have shown that this condition also can affect men and children—albeit less frequently.3,5-7 The incidence of IIH in the general population is between 0.03 to 2.36/100,000 people per year, but in women, the incidence is 0.65 to 4.65/100,000 per year.6 Furthermore, females who are obese have an incidence of 2.7 to 19.3/100,000 per year.6
Headache is the most common symptom of IIH. Unfortunately, the differential diagnosis of headache is vast; thus, a careful history is needed to narrow the field3,5-7 (TABLE 42). Associated symptoms of transient visual changes, pulsatile tinnitus, neck and back pain, nausea, vomiting, photo/phonophobia, and findings of abducens nerve palsy or papilledema—while nonspecific— should raise suspicion for elevated ICP and IIH, especially in women who are obese.2-8 Once IIH is suspected, an urgent diagnosis and treatment is necessary to prevent permanent vision loss.3,4,6
Headache with findings of papilledema warrants neuroimaging, preferably with MRI, to rule out intracranial mass and hydrocephalus.1,2,5 MRV also is recommended to assess for intracranial venous thrombosis, an alternate cause for papilledema and increased ICP.1,2,4,5
Continue to: Recently, a classification of IIH...
Recently, a classification of IIH without papilledema has been acknowledged by the International Headache Society.2,8 Specific MRI findings have been suggested to help make this diagnosis5,9 (TABLE 55).
TREATMENT FOR IIH CAN BE MEDICAL OR SURGICAL
Medications associated with IIH should be discontinued.7 The first-line medication for IIH is acetazolamide, a carbonic anhydrase inhibitor that works in the choroid plexus to decrease cerebrospinal fluid (CSF) production and thus, lower ICP.3,6 An adult dose of 1 to 2 g/day3,4,6 is tolerated well, but can be increased to 4 g/day,10 if necessary. Weight loss via diet and exercise or bariatric surgery has been shown to be effective in patients who are obese and have been given a diagnosis of IIH.3,4
Topiramate also has been suggested as a treatment option, based on its usefulness in weight loss and because of its action as a weak carbonic anhydrase inhibitor.3,6 Also, LP has therapeutic merit—although relief is only short-term.3,6 Patients who fail medical therapy and have intractable headache or progressive visual loss appear to benefit from optic nerve sheath fenestration.3,7,8
Our patient experienced notable improvement in her headache after LP. Her OCP was discontinued, a diuretic regimen started, and weight loss counseling was provided. Prior to discharge, the patient was seen by a neuro-ophthalmologist for perimetry, a visual field test that assesses for acute vision loss and establishes a baseline for follow-up monitoring of vision.7
THE TAKEAWAY
Headache is a common condition that may be challenging to correctly diagnose. A thorough history and neurological examination, including fundoscopy, are essential in the evaluation of headache and suspected IIH. In the primary care setting, limited time, lack of mydriatic agents, suboptimal lighting, and practitioner inexperience may pose challenges for funduscopic examination. Ophthalmoscopes incorporating new technology to expand and magnify the examiner’s field of view may facilitate this exam.11 A global rise in the prevalence of obesity underscores a need for primary care providers to be compulsive about their clinical evaluation when symptoms suspicious of IIH are present. Lastly, if IIH cannot be ruled out confidently, recommend a prompt evaluation by an ophthalmologist.
CORRESPONDENCE
Aarti Paltoo, MD, MSc, CCFP, Peel Village Medical Center, 28 Rambler Drive, Brampton, Ontario L6W 1E2 Canada; [email protected]
1. Lee SC, Lueck CJ. Cerebrospinal fluid pressure in adults. J Neuroophthalmol. 2014;34:278-283.
2. International Headache Society. Idiopathic intracranial hypertension. The International Classification of Headache Disorders. 2nd ed. Oxford, UK: Blackwell Publishing; 2003:1-232.
3. Biousse V, Bruce BB, Newman NJ. Update on the pathophysiology and management of idiopathic intracranial hypertension. J Neurol Neurosurg Psychiatry. 2012;83:488-494.
4. Mollan SP, Markey KA, Benzimra JD, et al. A practical approach to diagnosis, assessment and management of idiopathic intracranial hypertension. Pract Neurol. 2014;14:380-390.
5. Friedman DI, Liu GT, Digre KB. Revised diagnostic criteria for the pseudotumor cerebri syndrome in adults and children. Neurology. 2013;81:1159-1165.
6. Julayanont P, Karukote A, Ruthirago D, et al. Idiopathic intracranial hypertension: ongoing clinical challenges and future prospects. J Pain Res. 2016;9:87-99.
7. Friedman DI, Digre KB. Headache medicine meets neuro-ophthalmology: exam techniques and challenging cases. Headache. 2013;53:703-716.
8. Digre KB, Nakamoto BK, Warner JE, et al. A comparison of idiopathic intracranaial hypertension with and without papilledema. Headache. 2009;49:185-193.
9. Digre KB. Imaging characteristics of IIH: are they reliable? Cephalagia. 2013;33:1067-1069.
10. Horton J. Acetazolamide for pseudotumor cerebri--evidence from the NORDIC trial. JAMA. 2014;311:1618-1619.
11. Petrushkin H, Barsam A, Mavrakakis M, et al. Optic disc assessment in the emergency department: a comparative study between the PanOptic and direct ophthalmoscopes. Emerg Med J. 2012;29:1007-1008.
1. Lee SC, Lueck CJ. Cerebrospinal fluid pressure in adults. J Neuroophthalmol. 2014;34:278-283.
2. International Headache Society. Idiopathic intracranial hypertension. The International Classification of Headache Disorders. 2nd ed. Oxford, UK: Blackwell Publishing; 2003:1-232.
3. Biousse V, Bruce BB, Newman NJ. Update on the pathophysiology and management of idiopathic intracranial hypertension. J Neurol Neurosurg Psychiatry. 2012;83:488-494.
4. Mollan SP, Markey KA, Benzimra JD, et al. A practical approach to diagnosis, assessment and management of idiopathic intracranial hypertension. Pract Neurol. 2014;14:380-390.
5. Friedman DI, Liu GT, Digre KB. Revised diagnostic criteria for the pseudotumor cerebri syndrome in adults and children. Neurology. 2013;81:1159-1165.
6. Julayanont P, Karukote A, Ruthirago D, et al. Idiopathic intracranial hypertension: ongoing clinical challenges and future prospects. J Pain Res. 2016;9:87-99.
7. Friedman DI, Digre KB. Headache medicine meets neuro-ophthalmology: exam techniques and challenging cases. Headache. 2013;53:703-716.
8. Digre KB, Nakamoto BK, Warner JE, et al. A comparison of idiopathic intracranaial hypertension with and without papilledema. Headache. 2009;49:185-193.
9. Digre KB. Imaging characteristics of IIH: are they reliable? Cephalagia. 2013;33:1067-1069.
10. Horton J. Acetazolamide for pseudotumor cerebri--evidence from the NORDIC trial. JAMA. 2014;311:1618-1619.
11. Petrushkin H, Barsam A, Mavrakakis M, et al. Optic disc assessment in the emergency department: a comparative study between the PanOptic and direct ophthalmoscopes. Emerg Med J. 2012;29:1007-1008.

Subacute polyarticular arthralgias • swelling of the ankles and right knee • recent travel to the Dominican Republic • Dx?
THE CASE
A 78-year-old woman with a history of anxiety and hypertension presented to our family medicine residency practice in Massachusetts with subacute polyarticular arthralgias that had been present for 2 months. She complained of pain and swelling of both ankles and the right knee. She noted that her symptoms had started on a recent trip to the Dominican Republic, where she developed generalized joint pain and a fever that lasted 1 to 2 weeks and subsequently resolved with the lingering polyarthralgias. She denied any rash, constitutional symptoms, photosensitivity, headaches, photophobia, or history of tick bite. Physical examination revealed normal vital signs, notable warmth and swelling of the bilateral ankles that was worse on the right side, and swelling of the right knee with effusion—but no tenderness—to palpation.
THE DIAGNOSIS
The patient’s labwork revealed a white blood cell count of 5900/mcL (reference range, 4500–11,000/mcL), hemoglobin count of 12.5 g/dL (reference range, 14–17.5 g/dL), and a platelet count of 230×103/mcL. Electrolytes and renal function were normal. She had an elevated erythrocyte sedimentation rate of 34 mm/h (reference range, 0–20 mm/h) and a positive antinuclear antibody (ANA) test, but no titer was reported. Anti-chikungunya IgG and IgM antibodies were positive on enzyme-linked immunosorbent assay (ELISA) serologic testing.
DISCUSSION
Chikungunya is an infectious disease that is relatively rare in the United States. Chikungunya was rarely identified in American travelers prior to 2006, but incidence increased over the next decade. In 2014, a total of 2811 cases were reported.1 Chikungunya is an RNA arbovirus that is transmitted by Aedes aegypti and Aedes albopictus mosquitoes and is endemic to West Africa. Within the last 2 decades, there has been an increasing number of outbreaks in India, Asia, Europe, and the Americas, where the highest incidence is in South America, followed by Central America. In the United States, almost all reported cases of chikungunya infection have been in travelers returning from endemic areas.2 The first 2 known cases of local transmission in the United States were reported in Florida in July 2014.3 Local transmission of chikungunya is significant in that it represents the possibility of a local reservoir for sustained transmission.
Disease presentation. Patients will initially complain of a high fever and severe distal polyarthralgias that usually are symmetric. The most common symptoms are polyarthralgias (87%–98% of patients), myalgias (46%–59%), and a maculopapular rash
The term chikungunya is derived from a Kimakonde (central Bantu) word meaning “that which bends up” because of the arthralgia caused by the disease. Fever usually lasts 3 to 7 days; polyarthralgia begins shortly after the onset of fever.4 Frank arthritis also may be present. Infection often exacerbates a previously damaged or diseased joint. Acute symptoms usually persist for 1 to 2 weeks, but arthralgias and arthritis can persist for months to years following resolution of the acute disease.6 In one study of 47 patients with acute chikungunya in Marseilles, France, the number of patients who were symptomatic declined from 88% to 86%, 48%, and 4% at 1, 3, 6, and 15 months, respectively.7
The differential diagnosis includes tropical infectious diseases (dengue, chikungunya, Zika, and leptospirosis) in patients who have recently traveled to the tropics and who complain of subacute polyarticular arthralgias or arthritis; locally acquired infections associated with arthralgia/arthritis such as Lyme disease and other tick-borne diseases and rickettsial infections; parvovirus B19 and other postinfectious arthritides; and rheumatologic conditions such as systemic lupus.
Clinical differentiation among dengue, chikungunya, and Zika may be difficult, although persistent frank arthritis is much more common in chikungunya than in dengue or Zika. Furthermore, conjunctivitis is present in Zika but is absent in chikungunya. Chikungunya also is more likely to cause high fever, severe arthralgia, arthritis, rash, and lymphopenia than Zika or dengue. Dengue is more likely to cause lymphopenia and hemorrhagic consequences than is chikungunya or Zika.8
Continue to: In our patient...
In our patient, dengue titers were not obtained because the duration of symptoms was thought to be more consistent with chikungunya, but testing for dengue also would have been appropriate.
The most common test for diagnosing acute chikungunya is ELISA serologic testing for IgM antibodies, which develop toward the end of the first week of infection; earlier in that first week, serum testing for viral RNA may be performed by polymerase chain reaction.
Treatment is largely supportive
Treatment of acute chikungunya is largely supportive and includes anti-inflammatory agents. To our knowledge, no antiviral agents have been shown to be effective. Postacute or chronic symptoms may require treatment with glucocorticoids or other immunomodulatory medications. A 2017 literature review of treatments for chikungunya-associated rheumatic disorders showed evidence that chloroquine was more effective than placebo for chronic pain relief. Also, adding a disease-modifying antirheumatic agent in combination with chloroquine was more effective for controlling pain and reducing disability than hydroxychloroquine monotherapy.10
Our patient was treated with ibuprofen only and experienced resolution of joint symptoms several months after the initial presentation. A repeat ANA test 12 months later was negative.
A 2009 review of the medical literature revealed a single case report of chikungunya associated with positive ANA.8 Although a positive ANA may be associated with acute viral infections, significantly elevated ANA levels typically are associated with autoimmunity. Resolution of the patient’s serum ANA 1 year later suggested that the positive ANA was not secondary to a pre-existing rheumatologic condition but rather a consequence of her body’s response to the chikungunya infection itself. Our case raises the hypothesis that, at least in some cases, chikungunya somehow stimulates a temporary autoimmune response, which may help explain why immunomodulatory medications can be effective treatment options.
Continue to: THE TAKEAWAY
THE TAKEAWAY
Chikungunya is increasingly common in tropical and subtropical regions. Family physicians practicing in the United States should become familiar with the common patterns of presentation of viruses such as chikungunya, dengue, and Zika. Obtaining a travel history for patients presenting with arthritis improves the differential diagnosis and may even reveal the cause of the condition.
CORRESPONDENCE
Jeremy Golding, MD, 279 Lincoln Street, Worcester, MA 01605; [email protected]
1. Chikungunya virus. Centers for Disease Control and Prevention website. https://www.cdc.gov/chikungunya/geo/united-states.html. Reviewed December 17, 2018. Accessed March 5, 2019.
2. Pan American Health Organization. Preparedness and response for chikungunya virus: introduction into the Americas. https://www.paho.org/hq/dmdocuments/2012/CHIKV-English.pdf. Published 2011. Accessed March 5, 2019.
3. First chikungunya case acquired in the United States reported in Florida [press release]. Atlanta, GA: Centers for Disease Control and Prevention; July 17, 2014. http://www.cdc.gov/media/releases/2014/p0717-chikungunya.html. Accessed March 5, 2019.
4. Taubitz W, Cramer JP, Kapaun A, et al. Chikungunya fever in travelers: clinical presentation and course [published online May 23, 2007]. Clin Infect Dis. 2007;45:e1-e4.
5. Thiberville SD, Moyen N, Dupuis-Maguiraga L, et al. Chikungunya fever: epidemiology, clinical syndrome, pathogenesis and therapy. Antiviral Res. 2013;99:345-370.
6. Burt FJ, Rolph MS, Rulli NE, et al. Chikungunya: a re-emerging virus. Lancet. 2012;379:662-671.
7. Simon F, Parola P, Grandadam M, et al. Chikungunya infection: an emerging rheumatism among travelers returned from Indian Ocean islands. Report of 47 cases. Medicine (Baltimore). 2007;86:123-137.
8. Chikungunya virus. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/chikungunya/hc/clinicalevaluation.html. Reviewed December 17, 2018. Accessed March 5, 2019.
9. Petersen LR, Jamieson DJ, Powers AM, et al. Zika virus. N Engl J Med. 2016;374:1552-1563.
10. Martí-Carvajal A, Ramon-Pardo P, Javelle E, et al. Interventions for treating patients with chikungunya virus infection-related rheumatic and musculoskeletal disorders: a systematic review. PLoS One. 2017;12:e0179028.
THE CASE
A 78-year-old woman with a history of anxiety and hypertension presented to our family medicine residency practice in Massachusetts with subacute polyarticular arthralgias that had been present for 2 months. She complained of pain and swelling of both ankles and the right knee. She noted that her symptoms had started on a recent trip to the Dominican Republic, where she developed generalized joint pain and a fever that lasted 1 to 2 weeks and subsequently resolved with the lingering polyarthralgias. She denied any rash, constitutional symptoms, photosensitivity, headaches, photophobia, or history of tick bite. Physical examination revealed normal vital signs, notable warmth and swelling of the bilateral ankles that was worse on the right side, and swelling of the right knee with effusion—but no tenderness—to palpation.
THE DIAGNOSIS
The patient’s labwork revealed a white blood cell count of 5900/mcL (reference range, 4500–11,000/mcL), hemoglobin count of 12.5 g/dL (reference range, 14–17.5 g/dL), and a platelet count of 230×103/mcL. Electrolytes and renal function were normal. She had an elevated erythrocyte sedimentation rate of 34 mm/h (reference range, 0–20 mm/h) and a positive antinuclear antibody (ANA) test, but no titer was reported. Anti-chikungunya IgG and IgM antibodies were positive on enzyme-linked immunosorbent assay (ELISA) serologic testing.
DISCUSSION
Chikungunya is an infectious disease that is relatively rare in the United States. Chikungunya was rarely identified in American travelers prior to 2006, but incidence increased over the next decade. In 2014, a total of 2811 cases were reported.1 Chikungunya is an RNA arbovirus that is transmitted by Aedes aegypti and Aedes albopictus mosquitoes and is endemic to West Africa. Within the last 2 decades, there has been an increasing number of outbreaks in India, Asia, Europe, and the Americas, where the highest incidence is in South America, followed by Central America. In the United States, almost all reported cases of chikungunya infection have been in travelers returning from endemic areas.2 The first 2 known cases of local transmission in the United States were reported in Florida in July 2014.3 Local transmission of chikungunya is significant in that it represents the possibility of a local reservoir for sustained transmission.
Disease presentation. Patients will initially complain of a high fever and severe distal polyarthralgias that usually are symmetric. The most common symptoms are polyarthralgias (87%–98% of patients), myalgias (46%–59%), and a maculopapular rash
The term chikungunya is derived from a Kimakonde (central Bantu) word meaning “that which bends up” because of the arthralgia caused by the disease. Fever usually lasts 3 to 7 days; polyarthralgia begins shortly after the onset of fever.4 Frank arthritis also may be present. Infection often exacerbates a previously damaged or diseased joint. Acute symptoms usually persist for 1 to 2 weeks, but arthralgias and arthritis can persist for months to years following resolution of the acute disease.6 In one study of 47 patients with acute chikungunya in Marseilles, France, the number of patients who were symptomatic declined from 88% to 86%, 48%, and 4% at 1, 3, 6, and 15 months, respectively.7
The differential diagnosis includes tropical infectious diseases (dengue, chikungunya, Zika, and leptospirosis) in patients who have recently traveled to the tropics and who complain of subacute polyarticular arthralgias or arthritis; locally acquired infections associated with arthralgia/arthritis such as Lyme disease and other tick-borne diseases and rickettsial infections; parvovirus B19 and other postinfectious arthritides; and rheumatologic conditions such as systemic lupus.
Clinical differentiation among dengue, chikungunya, and Zika may be difficult, although persistent frank arthritis is much more common in chikungunya than in dengue or Zika. Furthermore, conjunctivitis is present in Zika but is absent in chikungunya. Chikungunya also is more likely to cause high fever, severe arthralgia, arthritis, rash, and lymphopenia than Zika or dengue. Dengue is more likely to cause lymphopenia and hemorrhagic consequences than is chikungunya or Zika.8
Continue to: In our patient...
In our patient, dengue titers were not obtained because the duration of symptoms was thought to be more consistent with chikungunya, but testing for dengue also would have been appropriate.
The most common test for diagnosing acute chikungunya is ELISA serologic testing for IgM antibodies, which develop toward the end of the first week of infection; earlier in that first week, serum testing for viral RNA may be performed by polymerase chain reaction.
Treatment is largely supportive
Treatment of acute chikungunya is largely supportive and includes anti-inflammatory agents. To our knowledge, no antiviral agents have been shown to be effective. Postacute or chronic symptoms may require treatment with glucocorticoids or other immunomodulatory medications. A 2017 literature review of treatments for chikungunya-associated rheumatic disorders showed evidence that chloroquine was more effective than placebo for chronic pain relief. Also, adding a disease-modifying antirheumatic agent in combination with chloroquine was more effective for controlling pain and reducing disability than hydroxychloroquine monotherapy.10
Our patient was treated with ibuprofen only and experienced resolution of joint symptoms several months after the initial presentation. A repeat ANA test 12 months later was negative.
A 2009 review of the medical literature revealed a single case report of chikungunya associated with positive ANA.8 Although a positive ANA may be associated with acute viral infections, significantly elevated ANA levels typically are associated with autoimmunity. Resolution of the patient’s serum ANA 1 year later suggested that the positive ANA was not secondary to a pre-existing rheumatologic condition but rather a consequence of her body’s response to the chikungunya infection itself. Our case raises the hypothesis that, at least in some cases, chikungunya somehow stimulates a temporary autoimmune response, which may help explain why immunomodulatory medications can be effective treatment options.
Continue to: THE TAKEAWAY
THE TAKEAWAY
Chikungunya is increasingly common in tropical and subtropical regions. Family physicians practicing in the United States should become familiar with the common patterns of presentation of viruses such as chikungunya, dengue, and Zika. Obtaining a travel history for patients presenting with arthritis improves the differential diagnosis and may even reveal the cause of the condition.
CORRESPONDENCE
Jeremy Golding, MD, 279 Lincoln Street, Worcester, MA 01605; [email protected]
THE CASE
A 78-year-old woman with a history of anxiety and hypertension presented to our family medicine residency practice in Massachusetts with subacute polyarticular arthralgias that had been present for 2 months. She complained of pain and swelling of both ankles and the right knee. She noted that her symptoms had started on a recent trip to the Dominican Republic, where she developed generalized joint pain and a fever that lasted 1 to 2 weeks and subsequently resolved with the lingering polyarthralgias. She denied any rash, constitutional symptoms, photosensitivity, headaches, photophobia, or history of tick bite. Physical examination revealed normal vital signs, notable warmth and swelling of the bilateral ankles that was worse on the right side, and swelling of the right knee with effusion—but no tenderness—to palpation.
THE DIAGNOSIS
The patient’s labwork revealed a white blood cell count of 5900/mcL (reference range, 4500–11,000/mcL), hemoglobin count of 12.5 g/dL (reference range, 14–17.5 g/dL), and a platelet count of 230×103/mcL. Electrolytes and renal function were normal. She had an elevated erythrocyte sedimentation rate of 34 mm/h (reference range, 0–20 mm/h) and a positive antinuclear antibody (ANA) test, but no titer was reported. Anti-chikungunya IgG and IgM antibodies were positive on enzyme-linked immunosorbent assay (ELISA) serologic testing.
DISCUSSION
Chikungunya is an infectious disease that is relatively rare in the United States. Chikungunya was rarely identified in American travelers prior to 2006, but incidence increased over the next decade. In 2014, a total of 2811 cases were reported.1 Chikungunya is an RNA arbovirus that is transmitted by Aedes aegypti and Aedes albopictus mosquitoes and is endemic to West Africa. Within the last 2 decades, there has been an increasing number of outbreaks in India, Asia, Europe, and the Americas, where the highest incidence is in South America, followed by Central America. In the United States, almost all reported cases of chikungunya infection have been in travelers returning from endemic areas.2 The first 2 known cases of local transmission in the United States were reported in Florida in July 2014.3 Local transmission of chikungunya is significant in that it represents the possibility of a local reservoir for sustained transmission.
Disease presentation. Patients will initially complain of a high fever and severe distal polyarthralgias that usually are symmetric. The most common symptoms are polyarthralgias (87%–98% of patients), myalgias (46%–59%), and a maculopapular rash
The term chikungunya is derived from a Kimakonde (central Bantu) word meaning “that which bends up” because of the arthralgia caused by the disease. Fever usually lasts 3 to 7 days; polyarthralgia begins shortly after the onset of fever.4 Frank arthritis also may be present. Infection often exacerbates a previously damaged or diseased joint. Acute symptoms usually persist for 1 to 2 weeks, but arthralgias and arthritis can persist for months to years following resolution of the acute disease.6 In one study of 47 patients with acute chikungunya in Marseilles, France, the number of patients who were symptomatic declined from 88% to 86%, 48%, and 4% at 1, 3, 6, and 15 months, respectively.7
The differential diagnosis includes tropical infectious diseases (dengue, chikungunya, Zika, and leptospirosis) in patients who have recently traveled to the tropics and who complain of subacute polyarticular arthralgias or arthritis; locally acquired infections associated with arthralgia/arthritis such as Lyme disease and other tick-borne diseases and rickettsial infections; parvovirus B19 and other postinfectious arthritides; and rheumatologic conditions such as systemic lupus.
Clinical differentiation among dengue, chikungunya, and Zika may be difficult, although persistent frank arthritis is much more common in chikungunya than in dengue or Zika. Furthermore, conjunctivitis is present in Zika but is absent in chikungunya. Chikungunya also is more likely to cause high fever, severe arthralgia, arthritis, rash, and lymphopenia than Zika or dengue. Dengue is more likely to cause lymphopenia and hemorrhagic consequences than is chikungunya or Zika.8
Continue to: In our patient...
In our patient, dengue titers were not obtained because the duration of symptoms was thought to be more consistent with chikungunya, but testing for dengue also would have been appropriate.
The most common test for diagnosing acute chikungunya is ELISA serologic testing for IgM antibodies, which develop toward the end of the first week of infection; earlier in that first week, serum testing for viral RNA may be performed by polymerase chain reaction.
Treatment is largely supportive
Treatment of acute chikungunya is largely supportive and includes anti-inflammatory agents. To our knowledge, no antiviral agents have been shown to be effective. Postacute or chronic symptoms may require treatment with glucocorticoids or other immunomodulatory medications. A 2017 literature review of treatments for chikungunya-associated rheumatic disorders showed evidence that chloroquine was more effective than placebo for chronic pain relief. Also, adding a disease-modifying antirheumatic agent in combination with chloroquine was more effective for controlling pain and reducing disability than hydroxychloroquine monotherapy.10
Our patient was treated with ibuprofen only and experienced resolution of joint symptoms several months after the initial presentation. A repeat ANA test 12 months later was negative.
A 2009 review of the medical literature revealed a single case report of chikungunya associated with positive ANA.8 Although a positive ANA may be associated with acute viral infections, significantly elevated ANA levels typically are associated with autoimmunity. Resolution of the patient’s serum ANA 1 year later suggested that the positive ANA was not secondary to a pre-existing rheumatologic condition but rather a consequence of her body’s response to the chikungunya infection itself. Our case raises the hypothesis that, at least in some cases, chikungunya somehow stimulates a temporary autoimmune response, which may help explain why immunomodulatory medications can be effective treatment options.
Continue to: THE TAKEAWAY
THE TAKEAWAY
Chikungunya is increasingly common in tropical and subtropical regions. Family physicians practicing in the United States should become familiar with the common patterns of presentation of viruses such as chikungunya, dengue, and Zika. Obtaining a travel history for patients presenting with arthritis improves the differential diagnosis and may even reveal the cause of the condition.
CORRESPONDENCE
Jeremy Golding, MD, 279 Lincoln Street, Worcester, MA 01605; [email protected]
1. Chikungunya virus. Centers for Disease Control and Prevention website. https://www.cdc.gov/chikungunya/geo/united-states.html. Reviewed December 17, 2018. Accessed March 5, 2019.
2. Pan American Health Organization. Preparedness and response for chikungunya virus: introduction into the Americas. https://www.paho.org/hq/dmdocuments/2012/CHIKV-English.pdf. Published 2011. Accessed March 5, 2019.
3. First chikungunya case acquired in the United States reported in Florida [press release]. Atlanta, GA: Centers for Disease Control and Prevention; July 17, 2014. http://www.cdc.gov/media/releases/2014/p0717-chikungunya.html. Accessed March 5, 2019.
4. Taubitz W, Cramer JP, Kapaun A, et al. Chikungunya fever in travelers: clinical presentation and course [published online May 23, 2007]. Clin Infect Dis. 2007;45:e1-e4.
5. Thiberville SD, Moyen N, Dupuis-Maguiraga L, et al. Chikungunya fever: epidemiology, clinical syndrome, pathogenesis and therapy. Antiviral Res. 2013;99:345-370.
6. Burt FJ, Rolph MS, Rulli NE, et al. Chikungunya: a re-emerging virus. Lancet. 2012;379:662-671.
7. Simon F, Parola P, Grandadam M, et al. Chikungunya infection: an emerging rheumatism among travelers returned from Indian Ocean islands. Report of 47 cases. Medicine (Baltimore). 2007;86:123-137.
8. Chikungunya virus. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/chikungunya/hc/clinicalevaluation.html. Reviewed December 17, 2018. Accessed March 5, 2019.
9. Petersen LR, Jamieson DJ, Powers AM, et al. Zika virus. N Engl J Med. 2016;374:1552-1563.
10. Martí-Carvajal A, Ramon-Pardo P, Javelle E, et al. Interventions for treating patients with chikungunya virus infection-related rheumatic and musculoskeletal disorders: a systematic review. PLoS One. 2017;12:e0179028.
1. Chikungunya virus. Centers for Disease Control and Prevention website. https://www.cdc.gov/chikungunya/geo/united-states.html. Reviewed December 17, 2018. Accessed March 5, 2019.
2. Pan American Health Organization. Preparedness and response for chikungunya virus: introduction into the Americas. https://www.paho.org/hq/dmdocuments/2012/CHIKV-English.pdf. Published 2011. Accessed March 5, 2019.
3. First chikungunya case acquired in the United States reported in Florida [press release]. Atlanta, GA: Centers for Disease Control and Prevention; July 17, 2014. http://www.cdc.gov/media/releases/2014/p0717-chikungunya.html. Accessed March 5, 2019.
4. Taubitz W, Cramer JP, Kapaun A, et al. Chikungunya fever in travelers: clinical presentation and course [published online May 23, 2007]. Clin Infect Dis. 2007;45:e1-e4.
5. Thiberville SD, Moyen N, Dupuis-Maguiraga L, et al. Chikungunya fever: epidemiology, clinical syndrome, pathogenesis and therapy. Antiviral Res. 2013;99:345-370.
6. Burt FJ, Rolph MS, Rulli NE, et al. Chikungunya: a re-emerging virus. Lancet. 2012;379:662-671.
7. Simon F, Parola P, Grandadam M, et al. Chikungunya infection: an emerging rheumatism among travelers returned from Indian Ocean islands. Report of 47 cases. Medicine (Baltimore). 2007;86:123-137.
8. Chikungunya virus. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/chikungunya/hc/clinicalevaluation.html. Reviewed December 17, 2018. Accessed March 5, 2019.
9. Petersen LR, Jamieson DJ, Powers AM, et al. Zika virus. N Engl J Med. 2016;374:1552-1563.
10. Martí-Carvajal A, Ramon-Pardo P, Javelle E, et al. Interventions for treating patients with chikungunya virus infection-related rheumatic and musculoskeletal disorders: a systematic review. PLoS One. 2017;12:e0179028.

Nivolumab-Induced Lichen Planus Pemphigoides
Nivolumab, an immune checkpoint modulator, acts by binding to the programmed cell death 1 (PD-1) receptor on T cells, which blocks the inhibition of T cells. Nivolumab ultimately leads to stimulation of the T-cell response1 and overcomes evasive adaptations of certain cancers. Cutaneous adverse events (AEs) have been reported in approximately 20% to 40% of patients treated with the anti–PD-1 class of drugs, including nivolumab.2-4 The most common cutaneous AEs include pruritus; vitiligo; and various forms of rash, such as lichenoid dermatitis, psoriasiform eruptions, and bullous pemphigoid.1-3,5-7 We report a patient with non–small cell lung cancer being treated with nivolumab who developed a bullous lichenoid eruption consistent with the diagnosis of lichen planus pemphigoides (LPP).
Case Report
An 87-year-old woman presented with a pruritic rash on the trunk and extremities of 3 weeks’ duration. Her medical history included stage IV non–small cell lung cancer, congestive heart failure, coronary artery disease, chronic kidney disease, and hypertension. Her long-term medications were ipratropium-albuterol, alendronate, amlodipine, aspirin, carvedilol, colesevelam, probiotic granules, and bumetanide. She was previously treated with carboplatin and docetaxel, which were discontinued secondary to fatigue, diarrhea, poor appetite, loss of taste, and a nonspecific rash. Six months later (approximately 3 months prior to the onset of cutaneous symptoms), she was started on nivolumab monotherapy every 14 days for a total of 9 infusions.
At the current presentation, physical examination revealed erythematous crusted erosions on the trunk and extremities and 1 flaccid bulla on the back. A punch biopsy revealed lichenoid dermatitis. The patient returned 2 weeks later with worsening of cutaneous manifestations, including more blisters and erosions. Figure 1 shows the clinical appearance of the eruption on the patient’s leg. At this time, additional biopsies revealed a subepidermal bullous lichenoid eruption with eosinophils (Figure 2). Direct immunofluorescence (DIF) was negative; however, indirect immunofluorescence (IIF) revealed weak linear staining for IgG antibodies along the basement membrane zone on monkey esophagus substrate. Examination of salt-split skin was noncontributory. The patient improved with a 2-week oral prednisone taper (starting at 40 mg daily). The dose was decreased incrementally over the course of 2 weeks from 40 mg to 20 mg to 0 mg. Because of the presumed grade 3 (severe) cutaneous drug eruption linked to nivolumab and further discussion with the medical oncology team, the patient decided to cease therapy. Since cessation of therapy, she has been seen twice for follow-up. At 2-month follow-up, she presented with drastic improvement of the eruption, and at 1 year she has continued to forego any further treatment for the stable and nonprogressing malignancy.
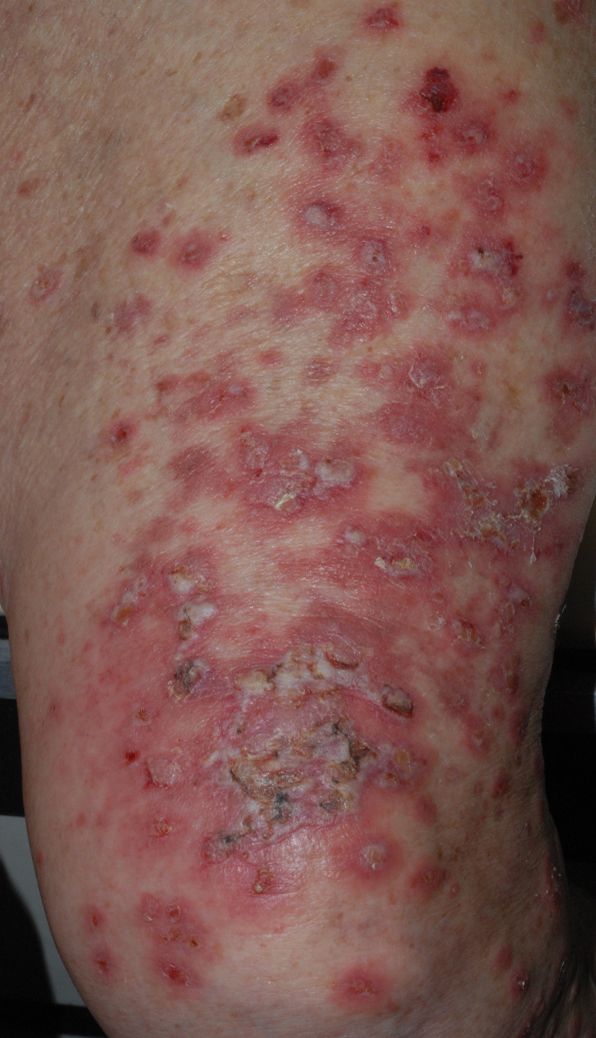
Widespread coalescent lesions with crusted and hemorrhagic bullae were present on the thigh and knee.
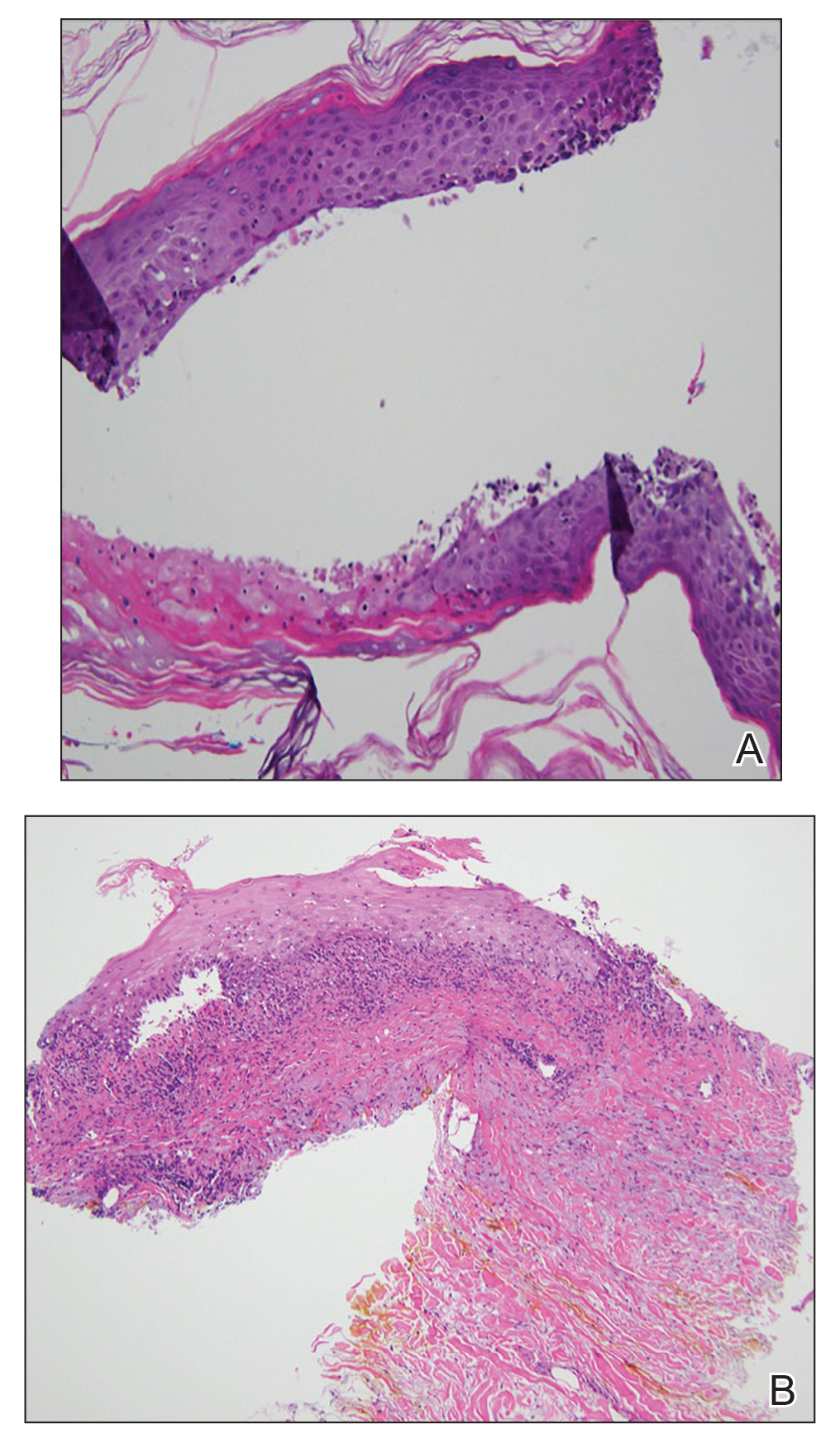
Comment
Immunotherapy
The interaction between the PD-1 receptor and its ligands, programmed death ligand 1 (PD-L1) and programmed death ligand 2, is an immune checkpoint.8,9 Under normal physiologic conditions, this checkpoint serves to prevent autoimmunity.10 When the PD-1 receptor is left unbound, T cells are more inclined to mount an immune response. If the receptor is ligand bound, the response of T cells is suppressed via mechanisms such as anergy or apoptosis.8 Tumor cells are known to produce PD-L1 as an adaptive resistance mechanism to evade immunity.8 Nivolumab is a human monoclonal antibody that targets the PD-1 receptor, thereby preventing the interaction with its ligand and allowing for unsuppressed activity of T cells.10 This therapy ultimately blocks the tumor’s local immune suppression mechanisms, which allows T cells to recognize cancer antigens.10
Adverse Events
Dermatologic AEs are among the most common with nivolumab treatment. In a pooled retrospective analysis of melanoma patients, Weber et al9 found that 34% of 576 patients experience cutaneous any-grade AEs associated with nivolumab treatment, most commonly pruritus. It has been well documented that anti–PD-1 therapy AEs of the skin as well as other organ systems have a delayed onset of at least 1 month.9 The average time of onset for bullous eruptions associated with anti–PD-1 therapy has been reported to be approximately 12 weeks, with a range of 7 to 16.1 weeks.11 Our patient had a bullous eruption with an onset of 12 weeks following initiation of treatment.
Although lichenoid reactions appear to be relatively common AEs of anti–PD-1 therapy,2,5,6 only a small number of cases of bullous pemphigoid eruptions have been reported.7 It has been hypothesized that blockade of the PD-1/PD-L1 pathway increases production of hemidesmosomal protein BP180 autoantibody, which is involved in the pathogenesis of LPP.7 Bullous eruptions have not been reported in the use of anticytotoxic T-lymphocyte–associated protein 4 agents, which could indicate that such eruptions are specific to the anti–PD-1 class of drugs.7
Diagnosis
Our patient represents a rare drug reaction involving both lichenoid and bullous components. Our differential diagnosis included drug-induced bullous lichen planus (BLP) and drug-induced LPP. Differentiation of these diagnoses can be difficult. In fact, in 2017 Fujii et al12 found reason to reprise the hypothesis that BLP is a transitional step toward LPP. The histologic evaluation of LPP differs depending on the type of lesion biopsied and can be indistinguishable from BLP as well as bullous pemphigoid. Therefore, clinical history and immunofluorescence should be used to make a diagnosis. Lichen planus pemphigoides typically will have linear IgG deposition along the basement membrane zone on both DIF and IIF, findings that will be negative in patients with BLP.13 Direct immunofluorescence findings in BLP include shaggy deposits of fibrin along the basement membrane zone. In this patient, DIF was negative, which may have been caused by variability among lesions in LPP, but IIF was positive. Given the clinicopathologic correlation, the diagnosis of LPP was made. Further studies, such as immunoblot and enzyme-linked immunosorbent assay, also can be used to aid diagnosis.
A similar presentation has been documented in a patient with metastatic melanoma.14 The diagnosis in this patient was LPP induced by pembrolizumab, which is another agent within the anti–PD-1 class. The Naranjo probability scale scored our patient’s eruption as a possible adverse drug reaction.15 Thus, other etiologies, such as a paraneoplastic process, cannot be completely ruled out. However, our patient has not had recurrence after 1 year, and the timing of the eruption appeared to be related to drug therapy, making alternative etiologies less likely.
Management
Cessation of nivolumab therapy and a short course of oral corticosteroid therapy led to marked improvement of symptoms. Given the emergent treatment of our patient, the resolution of her symptoms cannot be solely attributed to the cessation of nivolumab or to treatment with prednisone. Oral rather than topical corticosteroids were chosen because of the severity of the eruption. Topical corticosteroids and oral antihistamines can provide relief in less severe cases of bullous reactions to anti–PD-1 therapy.7,11 This regimen also has proven to be effective in lichenoid dermatitis induced by anti–PD-1.2
Conclusion
We hope this case report will contribute to the growing body of evidence regarding recognition and management of unique reactions to cancer immunotherapies.
- Macdonald JB, Macdonald B, Golitz LE, et al. Cutaneous adverse effects of targeted therapies: part II: inhibitors of intracellular molecular signaling pathways. J Am Acad Dermatol. 2015;72:221-236; quiz 237-238.
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25.
- Abdel-Rahman O, El Halawani H, Fouad M. Risk of cutaneous toxicities in patients with solid tumors treated with immune checkpoint inhibitors: a meta-analysis. Future Oncol. 2015;11:2471-2484.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-2454.
- Hwang SJ, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort [published online January 12, 2016]. J Am Acad Dermatol. 2016;74:455-461.e1.
- Sibaud V, Meyer N, Lamant L, et al. Dermatologic complications of anti-PD-1/PD-L1 immune checkpoint antibodies. Curr Opin Oncol. 2016;28:254-263.
- Naidoo J, Schindler K, Querfeld C, et al. Autoimmune bullous skin disorders with immune checkpoint inhibitors targeting PD-1 and PD-L1. Cancer Immunol Res. 2016;4:383-389.
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4.
- Weber JS, Hodi FS, Wolchok JD, et al. Safety profile of nivolumab monotherapy: a pooled analysis of patients with advanced melanoma. J Clin Oncol. 2017;35:785-792.
- Mamalis A, Garcha M, Jagdeo J. Targeting the PD-1 pathway: a promising future for the treatment of melanoma. Arch Dermatol Res. 2014;306:511-519.
- Jour G, Glitza IC, Ellis RM, et al. Autoimmune dermatologic toxicities from immune checkpoint blockade with anti-PD-1 antibody therapy: a report on bullous skin eruptions. J Cutan Pathol. 2016;43:688-696.
- Fujii M, Takahashi I, Honma M, et al. Bullous lichen planus accompanied by elevation of serum anti-BP180 autoantibody: a possible transitional mechanism to lichen planus pemphigoides. J Dermatol. 2017;44:E124-E125.
- Arbache ST, Nogueira TG, Delgado L, et al. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-889.
- Schmidgen MI, Butsch F, Schadmand-Fischer S, et al. Pembrolizumab-induced lichen planus pemphigoides in a patient with metastatic melanoma. J Dtsch Dermatol Ges. 2017;15:742-745.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
Nivolumab, an immune checkpoint modulator, acts by binding to the programmed cell death 1 (PD-1) receptor on T cells, which blocks the inhibition of T cells. Nivolumab ultimately leads to stimulation of the T-cell response1 and overcomes evasive adaptations of certain cancers. Cutaneous adverse events (AEs) have been reported in approximately 20% to 40% of patients treated with the anti–PD-1 class of drugs, including nivolumab.2-4 The most common cutaneous AEs include pruritus; vitiligo; and various forms of rash, such as lichenoid dermatitis, psoriasiform eruptions, and bullous pemphigoid.1-3,5-7 We report a patient with non–small cell lung cancer being treated with nivolumab who developed a bullous lichenoid eruption consistent with the diagnosis of lichen planus pemphigoides (LPP).
Case Report
An 87-year-old woman presented with a pruritic rash on the trunk and extremities of 3 weeks’ duration. Her medical history included stage IV non–small cell lung cancer, congestive heart failure, coronary artery disease, chronic kidney disease, and hypertension. Her long-term medications were ipratropium-albuterol, alendronate, amlodipine, aspirin, carvedilol, colesevelam, probiotic granules, and bumetanide. She was previously treated with carboplatin and docetaxel, which were discontinued secondary to fatigue, diarrhea, poor appetite, loss of taste, and a nonspecific rash. Six months later (approximately 3 months prior to the onset of cutaneous symptoms), she was started on nivolumab monotherapy every 14 days for a total of 9 infusions.
At the current presentation, physical examination revealed erythematous crusted erosions on the trunk and extremities and 1 flaccid bulla on the back. A punch biopsy revealed lichenoid dermatitis. The patient returned 2 weeks later with worsening of cutaneous manifestations, including more blisters and erosions. Figure 1 shows the clinical appearance of the eruption on the patient’s leg. At this time, additional biopsies revealed a subepidermal bullous lichenoid eruption with eosinophils (Figure 2). Direct immunofluorescence (DIF) was negative; however, indirect immunofluorescence (IIF) revealed weak linear staining for IgG antibodies along the basement membrane zone on monkey esophagus substrate. Examination of salt-split skin was noncontributory. The patient improved with a 2-week oral prednisone taper (starting at 40 mg daily). The dose was decreased incrementally over the course of 2 weeks from 40 mg to 20 mg to 0 mg. Because of the presumed grade 3 (severe) cutaneous drug eruption linked to nivolumab and further discussion with the medical oncology team, the patient decided to cease therapy. Since cessation of therapy, she has been seen twice for follow-up. At 2-month follow-up, she presented with drastic improvement of the eruption, and at 1 year she has continued to forego any further treatment for the stable and nonprogressing malignancy.
Widespread coalescent lesions with crusted and hemorrhagic bullae were present on the thigh and knee.
Comment
Immunotherapy
The interaction between the PD-1 receptor and its ligands, programmed death ligand 1 (PD-L1) and programmed death ligand 2, is an immune checkpoint.8,9 Under normal physiologic conditions, this checkpoint serves to prevent autoimmunity.10 When the PD-1 receptor is left unbound, T cells are more inclined to mount an immune response. If the receptor is ligand bound, the response of T cells is suppressed via mechanisms such as anergy or apoptosis.8 Tumor cells are known to produce PD-L1 as an adaptive resistance mechanism to evade immunity.8 Nivolumab is a human monoclonal antibody that targets the PD-1 receptor, thereby preventing the interaction with its ligand and allowing for unsuppressed activity of T cells.10 This therapy ultimately blocks the tumor’s local immune suppression mechanisms, which allows T cells to recognize cancer antigens.10
Adverse Events
Dermatologic AEs are among the most common with nivolumab treatment. In a pooled retrospective analysis of melanoma patients, Weber et al9 found that 34% of 576 patients experience cutaneous any-grade AEs associated with nivolumab treatment, most commonly pruritus. It has been well documented that anti–PD-1 therapy AEs of the skin as well as other organ systems have a delayed onset of at least 1 month.9 The average time of onset for bullous eruptions associated with anti–PD-1 therapy has been reported to be approximately 12 weeks, with a range of 7 to 16.1 weeks.11 Our patient had a bullous eruption with an onset of 12 weeks following initiation of treatment.
Although lichenoid reactions appear to be relatively common AEs of anti–PD-1 therapy,2,5,6 only a small number of cases of bullous pemphigoid eruptions have been reported.7 It has been hypothesized that blockade of the PD-1/PD-L1 pathway increases production of hemidesmosomal protein BP180 autoantibody, which is involved in the pathogenesis of LPP.7 Bullous eruptions have not been reported in the use of anticytotoxic T-lymphocyte–associated protein 4 agents, which could indicate that such eruptions are specific to the anti–PD-1 class of drugs.7
Diagnosis
Our patient represents a rare drug reaction involving both lichenoid and bullous components. Our differential diagnosis included drug-induced bullous lichen planus (BLP) and drug-induced LPP. Differentiation of these diagnoses can be difficult. In fact, in 2017 Fujii et al12 found reason to reprise the hypothesis that BLP is a transitional step toward LPP. The histologic evaluation of LPP differs depending on the type of lesion biopsied and can be indistinguishable from BLP as well as bullous pemphigoid. Therefore, clinical history and immunofluorescence should be used to make a diagnosis. Lichen planus pemphigoides typically will have linear IgG deposition along the basement membrane zone on both DIF and IIF, findings that will be negative in patients with BLP.13 Direct immunofluorescence findings in BLP include shaggy deposits of fibrin along the basement membrane zone. In this patient, DIF was negative, which may have been caused by variability among lesions in LPP, but IIF was positive. Given the clinicopathologic correlation, the diagnosis of LPP was made. Further studies, such as immunoblot and enzyme-linked immunosorbent assay, also can be used to aid diagnosis.
A similar presentation has been documented in a patient with metastatic melanoma.14 The diagnosis in this patient was LPP induced by pembrolizumab, which is another agent within the anti–PD-1 class. The Naranjo probability scale scored our patient’s eruption as a possible adverse drug reaction.15 Thus, other etiologies, such as a paraneoplastic process, cannot be completely ruled out. However, our patient has not had recurrence after 1 year, and the timing of the eruption appeared to be related to drug therapy, making alternative etiologies less likely.
Management
Cessation of nivolumab therapy and a short course of oral corticosteroid therapy led to marked improvement of symptoms. Given the emergent treatment of our patient, the resolution of her symptoms cannot be solely attributed to the cessation of nivolumab or to treatment with prednisone. Oral rather than topical corticosteroids were chosen because of the severity of the eruption. Topical corticosteroids and oral antihistamines can provide relief in less severe cases of bullous reactions to anti–PD-1 therapy.7,11 This regimen also has proven to be effective in lichenoid dermatitis induced by anti–PD-1.2
Conclusion
We hope this case report will contribute to the growing body of evidence regarding recognition and management of unique reactions to cancer immunotherapies.
Nivolumab, an immune checkpoint modulator, acts by binding to the programmed cell death 1 (PD-1) receptor on T cells, which blocks the inhibition of T cells. Nivolumab ultimately leads to stimulation of the T-cell response1 and overcomes evasive adaptations of certain cancers. Cutaneous adverse events (AEs) have been reported in approximately 20% to 40% of patients treated with the anti–PD-1 class of drugs, including nivolumab.2-4 The most common cutaneous AEs include pruritus; vitiligo; and various forms of rash, such as lichenoid dermatitis, psoriasiform eruptions, and bullous pemphigoid.1-3,5-7 We report a patient with non–small cell lung cancer being treated with nivolumab who developed a bullous lichenoid eruption consistent with the diagnosis of lichen planus pemphigoides (LPP).
Case Report
An 87-year-old woman presented with a pruritic rash on the trunk and extremities of 3 weeks’ duration. Her medical history included stage IV non–small cell lung cancer, congestive heart failure, coronary artery disease, chronic kidney disease, and hypertension. Her long-term medications were ipratropium-albuterol, alendronate, amlodipine, aspirin, carvedilol, colesevelam, probiotic granules, and bumetanide. She was previously treated with carboplatin and docetaxel, which were discontinued secondary to fatigue, diarrhea, poor appetite, loss of taste, and a nonspecific rash. Six months later (approximately 3 months prior to the onset of cutaneous symptoms), she was started on nivolumab monotherapy every 14 days for a total of 9 infusions.
At the current presentation, physical examination revealed erythematous crusted erosions on the trunk and extremities and 1 flaccid bulla on the back. A punch biopsy revealed lichenoid dermatitis. The patient returned 2 weeks later with worsening of cutaneous manifestations, including more blisters and erosions. Figure 1 shows the clinical appearance of the eruption on the patient’s leg. At this time, additional biopsies revealed a subepidermal bullous lichenoid eruption with eosinophils (Figure 2). Direct immunofluorescence (DIF) was negative; however, indirect immunofluorescence (IIF) revealed weak linear staining for IgG antibodies along the basement membrane zone on monkey esophagus substrate. Examination of salt-split skin was noncontributory. The patient improved with a 2-week oral prednisone taper (starting at 40 mg daily). The dose was decreased incrementally over the course of 2 weeks from 40 mg to 20 mg to 0 mg. Because of the presumed grade 3 (severe) cutaneous drug eruption linked to nivolumab and further discussion with the medical oncology team, the patient decided to cease therapy. Since cessation of therapy, she has been seen twice for follow-up. At 2-month follow-up, she presented with drastic improvement of the eruption, and at 1 year she has continued to forego any further treatment for the stable and nonprogressing malignancy.
Widespread coalescent lesions with crusted and hemorrhagic bullae were present on the thigh and knee.
Comment
Immunotherapy
The interaction between the PD-1 receptor and its ligands, programmed death ligand 1 (PD-L1) and programmed death ligand 2, is an immune checkpoint.8,9 Under normal physiologic conditions, this checkpoint serves to prevent autoimmunity.10 When the PD-1 receptor is left unbound, T cells are more inclined to mount an immune response. If the receptor is ligand bound, the response of T cells is suppressed via mechanisms such as anergy or apoptosis.8 Tumor cells are known to produce PD-L1 as an adaptive resistance mechanism to evade immunity.8 Nivolumab is a human monoclonal antibody that targets the PD-1 receptor, thereby preventing the interaction with its ligand and allowing for unsuppressed activity of T cells.10 This therapy ultimately blocks the tumor’s local immune suppression mechanisms, which allows T cells to recognize cancer antigens.10
Adverse Events
Dermatologic AEs are among the most common with nivolumab treatment. In a pooled retrospective analysis of melanoma patients, Weber et al9 found that 34% of 576 patients experience cutaneous any-grade AEs associated with nivolumab treatment, most commonly pruritus. It has been well documented that anti–PD-1 therapy AEs of the skin as well as other organ systems have a delayed onset of at least 1 month.9 The average time of onset for bullous eruptions associated with anti–PD-1 therapy has been reported to be approximately 12 weeks, with a range of 7 to 16.1 weeks.11 Our patient had a bullous eruption with an onset of 12 weeks following initiation of treatment.
Although lichenoid reactions appear to be relatively common AEs of anti–PD-1 therapy,2,5,6 only a small number of cases of bullous pemphigoid eruptions have been reported.7 It has been hypothesized that blockade of the PD-1/PD-L1 pathway increases production of hemidesmosomal protein BP180 autoantibody, which is involved in the pathogenesis of LPP.7 Bullous eruptions have not been reported in the use of anticytotoxic T-lymphocyte–associated protein 4 agents, which could indicate that such eruptions are specific to the anti–PD-1 class of drugs.7
Diagnosis
Our patient represents a rare drug reaction involving both lichenoid and bullous components. Our differential diagnosis included drug-induced bullous lichen planus (BLP) and drug-induced LPP. Differentiation of these diagnoses can be difficult. In fact, in 2017 Fujii et al12 found reason to reprise the hypothesis that BLP is a transitional step toward LPP. The histologic evaluation of LPP differs depending on the type of lesion biopsied and can be indistinguishable from BLP as well as bullous pemphigoid. Therefore, clinical history and immunofluorescence should be used to make a diagnosis. Lichen planus pemphigoides typically will have linear IgG deposition along the basement membrane zone on both DIF and IIF, findings that will be negative in patients with BLP.13 Direct immunofluorescence findings in BLP include shaggy deposits of fibrin along the basement membrane zone. In this patient, DIF was negative, which may have been caused by variability among lesions in LPP, but IIF was positive. Given the clinicopathologic correlation, the diagnosis of LPP was made. Further studies, such as immunoblot and enzyme-linked immunosorbent assay, also can be used to aid diagnosis.
A similar presentation has been documented in a patient with metastatic melanoma.14 The diagnosis in this patient was LPP induced by pembrolizumab, which is another agent within the anti–PD-1 class. The Naranjo probability scale scored our patient’s eruption as a possible adverse drug reaction.15 Thus, other etiologies, such as a paraneoplastic process, cannot be completely ruled out. However, our patient has not had recurrence after 1 year, and the timing of the eruption appeared to be related to drug therapy, making alternative etiologies less likely.
Management
Cessation of nivolumab therapy and a short course of oral corticosteroid therapy led to marked improvement of symptoms. Given the emergent treatment of our patient, the resolution of her symptoms cannot be solely attributed to the cessation of nivolumab or to treatment with prednisone. Oral rather than topical corticosteroids were chosen because of the severity of the eruption. Topical corticosteroids and oral antihistamines can provide relief in less severe cases of bullous reactions to anti–PD-1 therapy.7,11 This regimen also has proven to be effective in lichenoid dermatitis induced by anti–PD-1.2
Conclusion
We hope this case report will contribute to the growing body of evidence regarding recognition and management of unique reactions to cancer immunotherapies.
- Macdonald JB, Macdonald B, Golitz LE, et al. Cutaneous adverse effects of targeted therapies: part II: inhibitors of intracellular molecular signaling pathways. J Am Acad Dermatol. 2015;72:221-236; quiz 237-238.
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25.
- Abdel-Rahman O, El Halawani H, Fouad M. Risk of cutaneous toxicities in patients with solid tumors treated with immune checkpoint inhibitors: a meta-analysis. Future Oncol. 2015;11:2471-2484.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-2454.
- Hwang SJ, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort [published online January 12, 2016]. J Am Acad Dermatol. 2016;74:455-461.e1.
- Sibaud V, Meyer N, Lamant L, et al. Dermatologic complications of anti-PD-1/PD-L1 immune checkpoint antibodies. Curr Opin Oncol. 2016;28:254-263.
- Naidoo J, Schindler K, Querfeld C, et al. Autoimmune bullous skin disorders with immune checkpoint inhibitors targeting PD-1 and PD-L1. Cancer Immunol Res. 2016;4:383-389.
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4.
- Weber JS, Hodi FS, Wolchok JD, et al. Safety profile of nivolumab monotherapy: a pooled analysis of patients with advanced melanoma. J Clin Oncol. 2017;35:785-792.
- Mamalis A, Garcha M, Jagdeo J. Targeting the PD-1 pathway: a promising future for the treatment of melanoma. Arch Dermatol Res. 2014;306:511-519.
- Jour G, Glitza IC, Ellis RM, et al. Autoimmune dermatologic toxicities from immune checkpoint blockade with anti-PD-1 antibody therapy: a report on bullous skin eruptions. J Cutan Pathol. 2016;43:688-696.
- Fujii M, Takahashi I, Honma M, et al. Bullous lichen planus accompanied by elevation of serum anti-BP180 autoantibody: a possible transitional mechanism to lichen planus pemphigoides. J Dermatol. 2017;44:E124-E125.
- Arbache ST, Nogueira TG, Delgado L, et al. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-889.
- Schmidgen MI, Butsch F, Schadmand-Fischer S, et al. Pembrolizumab-induced lichen planus pemphigoides in a patient with metastatic melanoma. J Dtsch Dermatol Ges. 2017;15:742-745.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Macdonald JB, Macdonald B, Golitz LE, et al. Cutaneous adverse effects of targeted therapies: part II: inhibitors of intracellular molecular signaling pathways. J Am Acad Dermatol. 2015;72:221-236; quiz 237-238.
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25.
- Abdel-Rahman O, El Halawani H, Fouad M. Risk of cutaneous toxicities in patients with solid tumors treated with immune checkpoint inhibitors: a meta-analysis. Future Oncol. 2015;11:2471-2484.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-2454.
- Hwang SJ, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort [published online January 12, 2016]. J Am Acad Dermatol. 2016;74:455-461.e1.
- Sibaud V, Meyer N, Lamant L, et al. Dermatologic complications of anti-PD-1/PD-L1 immune checkpoint antibodies. Curr Opin Oncol. 2016;28:254-263.
- Naidoo J, Schindler K, Querfeld C, et al. Autoimmune bullous skin disorders with immune checkpoint inhibitors targeting PD-1 and PD-L1. Cancer Immunol Res. 2016;4:383-389.
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4.
- Weber JS, Hodi FS, Wolchok JD, et al. Safety profile of nivolumab monotherapy: a pooled analysis of patients with advanced melanoma. J Clin Oncol. 2017;35:785-792.
- Mamalis A, Garcha M, Jagdeo J. Targeting the PD-1 pathway: a promising future for the treatment of melanoma. Arch Dermatol Res. 2014;306:511-519.
- Jour G, Glitza IC, Ellis RM, et al. Autoimmune dermatologic toxicities from immune checkpoint blockade with anti-PD-1 antibody therapy: a report on bullous skin eruptions. J Cutan Pathol. 2016;43:688-696.
- Fujii M, Takahashi I, Honma M, et al. Bullous lichen planus accompanied by elevation of serum anti-BP180 autoantibody: a possible transitional mechanism to lichen planus pemphigoides. J Dermatol. 2017;44:E124-E125.
- Arbache ST, Nogueira TG, Delgado L, et al. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-889.
- Schmidgen MI, Butsch F, Schadmand-Fischer S, et al. Pembrolizumab-induced lichen planus pemphigoides in a patient with metastatic melanoma. J Dtsch Dermatol Ges. 2017;15:742-745.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
Practice Points
- Dermatologists should be aware that lichen planus pemphigoides is within the spectrum of toxicity for patients treated with nivolumab.
- Bullous eruptions related to anti–programmed cell death 1 agents tend to appear 4 months after initiation of therapy.
- A severe cutaneous toxicity of a checkpoint inhibitor should be managed using oral corticosteroids with consideration of withdrawing the offending agent.
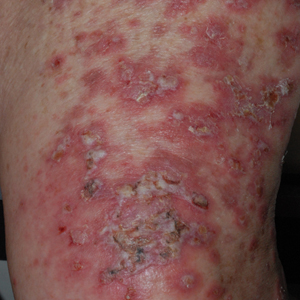
Relapsing Polychondritis in Human Immunodeficiency Virus
Relapsing polychondritis (RP) is a recurrent inflammatory condition involving primarily cartilaginous structures. The disease, first described as a clinical entity in 1960 by Pearson et al,1 is rare with an estimated incidence of 3.5 cases per 1 million individuals.2 The pathogenesis of RP is widely accepted as being autoimmune in nature, largely due to the identification of circulating autoantibodies seen in the sera of patients with similar clinical pictures.3
Although in most patients the primary process involves inflammation of cartilage, a subset of patients experience involvement of noncartilaginous sites.4 The degree of systemic involvement varies from none to notable, affecting the cardiovascular and respiratory systems and potentially leading to life-threatening complications, including cardiac valve compromise and airway collapse. Relapsing polychondritis is considered to be a progressive disease with the ultimate potential to be life-threatening.5
Human immunodeficiency virus (HIV) infection leads to a profound state of immune dysregulation, affecting innate, adaptive, and natural killer components of the immune system.6 There is variability in the development of autoimmune disease in HIV patients depending on the stage of infection. The frequency of rheumatologic disease in HIV patients might be as high as 60%.6 Relapsing polychondritis is rare in patients with HIV.7-9 Of 4 reported cases, 2 patients had other coexisting autoimmune disease—sarcoidosis and Behçet disease.8,9
Case Report
A 36-year-old man presented to the clinic with a concern of recurrent ear pain and swelling of approximately 2 years’ duration. Onset was sudden without inciting event. Symptoms initially involved the right ear with eventual progression to both ears. Additional symptoms included an auditory perception of underwater submersion, intermittent vertigo, and 3 episodes of throat closure sensation.
The patient’s medical history was notable for asthma; gastritis; depression; and HIV infection, which was diagnosed 4 years earlier and adequately managed with highly active antiretroviral therapy. His family history was notable for systemic lupus erythematosus in his mother, maternal aunt, and maternal cousin.
At presentation, the patient’s CD4 count was 799 cells/mm3 with an undetectable viral load. Medications included abacavir-dolutegravir-lamivudine, hydroxyzine, meclizine, mometasone, and quetiapine. Physical examination showed erythema, swelling, and tenderness of the left and right auricles with sparing of the earlobe that was more noticeable on the left ear (Figure 1). Bacterial culture from the external auditory meatus was positive for methicillin-resistant Staphylococcus aureus. Biopsy revealed chronic inflammatory perichondritis with mild to moderate fibrosis and chronic lymphocytic inflammation at the dermal cartilaginous junction (Figure 2). A direct immunofluorescent biopsy was unremarkable, but subsequent type II collagen antibodies were positive (35.5 endotoxin units/mL [reference range, <20 endotoxin units/mL]).
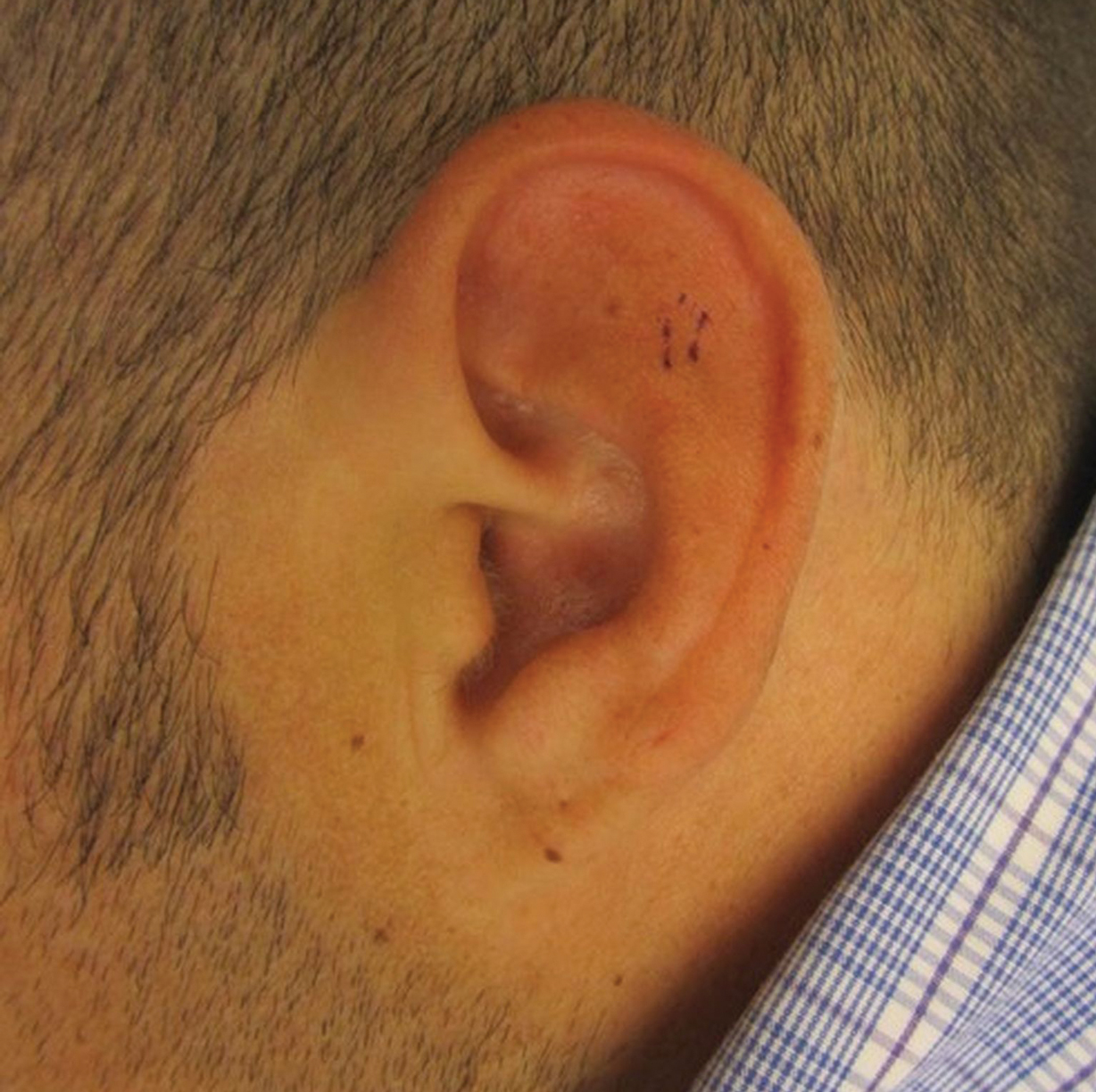
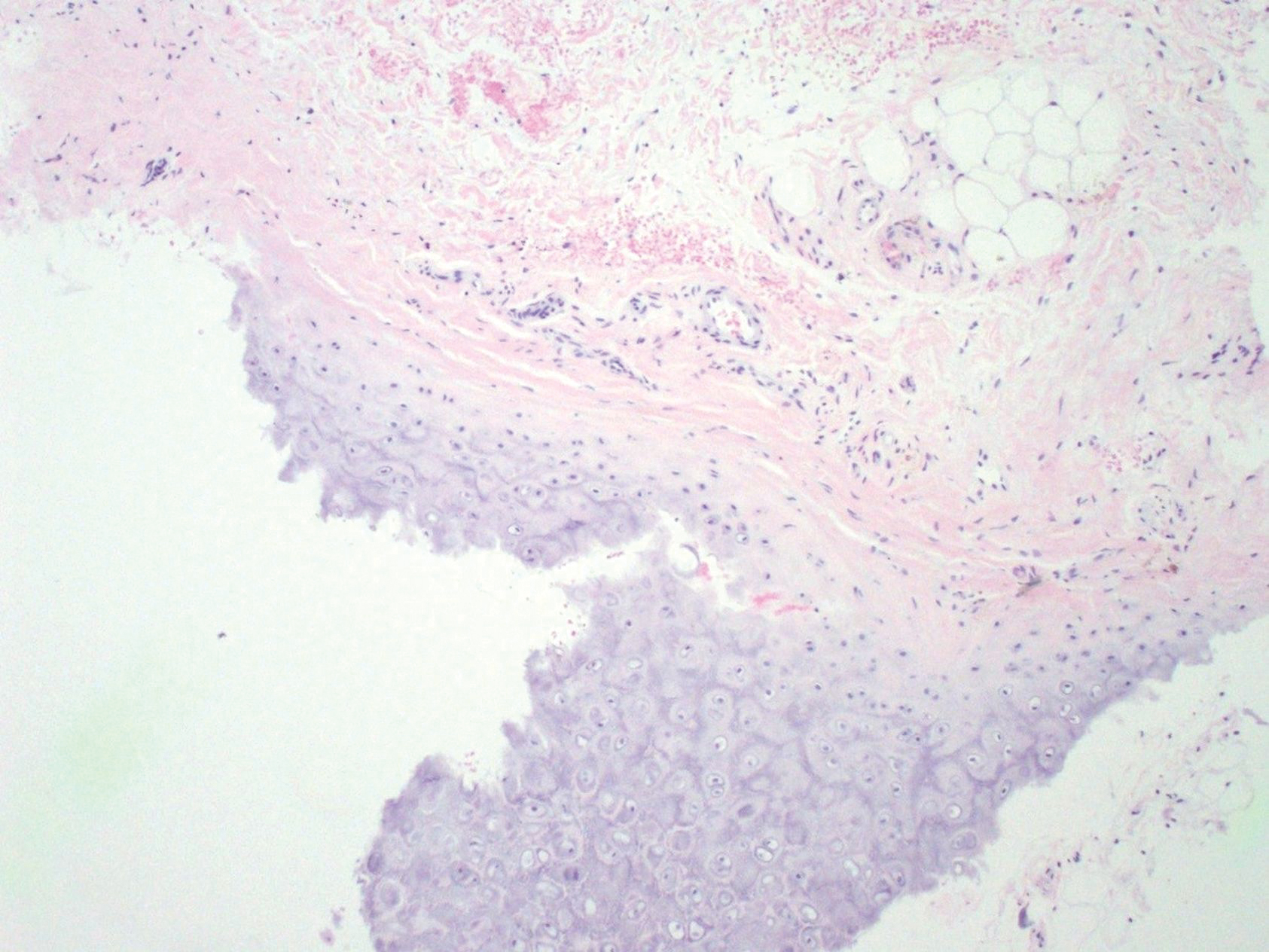
Comment
Relapsing polychondritis is an uncommon progressive disease characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures.4 The most consistent clinical features of RP are ear inflammation that involves the auricle and spares the lobe, nasal chondritis, and arthralgia.10 Laryngotracheal compromise may occur from tracheal cartilage inflammation. The involvement of these specific structures is due to commonality between their component collagens.5 Although any organ system can be affected, as many as 50% of patients have respiratory tract involvement, which may affect any portion of the respiratory tree.11 If involving the larynx, this inflammation can lead to severe edema warranting intubation. Cardiovascular involvement is present in 24% to 52% of patients,10 which most commonly manifests as valvular impairment affecting the aortic valve more frequently than the mitral valve.5
Pathogenesis
Although the etiology of RP remains undetermined, multiple hypotheses have been proposed. One is that a certain subset of patients is predisposed to autoimmunity, and a secondary triggering event in the form of infection, malignancy, or medication catalyzes development of RP. A second hypothesis is that mechanical trauma to cartilage exposes the immune system to certain antigens that would have otherwise remained hidden, prompting autosensitization.12,13
Regardless of cause, an autoimmune pathogenesis is favored based on the following observations: RP is frequently associated with other autoimmune diseases in the same patient, glucocorticosteroids and other immunosuppressive therapies are effective for treatment, and histopathologic findings include an infiltrate of CD4+ T lymphocytes with detection of immunoglobulins and plasma cells in different lesions.5 The detection of autoantibodies against collagen in the serum of patients with RP further supports an autoimmune pathogenesis.3 The earliest identified autoantibodies in patients with RP were against type II collagen. Subsequent studies have identified autoantibodies against type IV and type XI collagens as well as other cartilage-related proteins such as matrilin 114 and cartilage oligomeric matrix proteins.15 Although circulating antibodies to type II collagen are present in a variable number of patients with the disease (30%–70%), levels likely correlate with disease activity and are highest at times of acute inflammation.3 Additionally, titers of type II collagen antibodies have been shown to decrease upon institution of immunosuppressive therapy.16
Although a humoral response dominates the picture of RP, there also is an associated T cell–mediated response.13 Histopathologically, biopsy of an active lesion of auricular cartilage shows a mixed inflammatory infiltrate composed primarily of lymphocytes, with variable numbers of polymorphonuclear cells, monocytes, and plasma cells. Loss of basophilia of the cartilage matrix can be observed, thought to be the result of proteoglycan depletion.13 Later, lesions classically display apoptosis of chondrocytes, focal calcification, or fibrosis.5
Diagnosis
Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge. Many sets of diagnostic criteria have been proposed. The most referenced remains the original criteria described by McAdam et al.17 In 2012, the Relapsing Polychondritis Disease Activity Index modified criteria set forth by Michet et al18 and might serve as the standard for diagnosis going forward.19
McAdam et al17 proposed that 3 of 6 clinical features are necessary for diagnosis: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation, respiratory tract chondritis, and audiovestibular damage. Michet et al18 proposed that 1 of 2 conditions are necessary for diagnosis of RP: (1) proven inflammation in 2 of 3 of the auricular, nasal, or laryngotracheal cartilages; or (2) proven inflammation in 1 of 3 of the auricular, nasal, or laryngotracheal cartilages, plus 2 other signs, including ocular inflammation, vestibular dysfunction, seronegative inflammatory arthritis, and hearing loss.
These criteria were proposed originally in 197617 and modified in 1986.18 No further updates have been offered since then. As such, serologic findings, such as antibodies against type II collagen, are not included in the diagnostic criteria. Additionally, these antibodies are not specific for RP and can be seen in other conditions such as rheumatoid arthritis.20
More recently, imaging analysis has been employed in conjunction with clinical and serologic data to diagnose the disease and evaluate its severity. The use of imaging modalities for these purposes is most beneficial in patients with notable disease and respiratory involvement.21
Although the clinical picture is typified by the classic findings described above, the clinician must be aware of more subtle clues to diagnosis,11 which is of particular importance to the dermatologist because 35% of patients with RP alone will have skin manifestations that can precede onset of chondritis.10 Most commonly, dermatologic manifestations are nonspecific and can include nodules on the limbs, purpura, and urticarial lesions.22 Individual case reports have noted the coexistence of RP with erythema multiforme,18 erythema annulare centrifugum,23 pyoderma gangrenosum,24 and panniculitis,18 among other disorders.
Treatment
Standardized guidelines for treatment do not exist. Treatments should be chosen based on severity of disease. Mild disease, presenting with recurrent chondritis and arthritis without evidence of systemic involvement, can be treated with nonsteroidal anti-inflammatory drugs, dapsone, or colchicine. Refractory disease often requires high-dose systemic corticosteroids.5
Severe systemic involvement leads to increased mortality and warrants more aggressive treatment.22 Commonly used agents include the immunosuppressants cyclophosphamide, cyclosporine, and methotrexate. Tumor necrosis factor α inhibitors have been the most widely utilized immunomodulatory agent for treatment of RP.25,26 Abatacept and rituximab also have been used with variable efficacy in patients with severe disease. Recently, the IL-6 receptor blocker tocilizumab has been used with some success.27
Prognosis
The prognosis for patients with RP largely depends on the severity of disease and degree of internal involvement. With improved management, largely due to awareness and recognition of disease, the survival rate among RP patients has increased from 55% at 10 years to 94% at the end of 8 years.18 The main cause of death in RP patients is airway complications related to laryngotracheal involvement.10 The second most common cause of death is cardiovascular complications in which valvular disease predominates.5
Concomitant Illness
Thirty-five percent of RP patients have coexisting autoimmune disease, the most common being antineutrophil cytoplasmic antibody–associated vasculitis.5,28 Although this association with autoimmune disease is well described, reports of RP occurring in other states of immune dysfunction are sparse. One case of RP has been reported in a child with common variable immunodeficiency thought to be related to underlying abnormal immune regulation and immunodeficiency.29 Relapsing polychondritis has been described in 4 patients with HIV, 2 of whom had concomitant autoimmune disease.7-9
Human immunodeficiency virus infection is a well-established cause of immune dysregulation and has variable association with autoimmunity. This variability depends largely on the stage of infection. When divided into stages, autoimmune diseases develop predominantly in stage I, during acute infection with an intact immune system; in stage III, with immunosuppression, a low CD4 count, and development of AIDS; and in stage IV, when the immune system is restored with the institution of highly active antiretroviral therapy.6 The interplay between HIV infection and development of autoimmune disease is complex, and pathogenesis remains speculative.
Conclusion
Our patient represents a case of RP in an HIV-positive patient. Additionally, our patient had no other identifiable autoimmune conditions but did have a strong family history of them. It is important for providers to be aware of the potential for development of RP as well as other autoimmune disease in the setting of HIV infection. The implications of a missed diagnosis could be dire because the disease course of RP is progressive and has the potential to decrease survival.
- Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med. 1960;263:51-58.
- Kent PD, Michet CJ Jr, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56-61.
- Ebringer R, Rook G, Swana GT, et al. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473-479.
- Sharma A, Law AD, Bambery P, et al. Relapsing polychondritis: clinical presentations, disease activity and outcomes. Orphanet J Rare Dis. 2014;9:198.
- Vitale A, Sota J, Rigante D, et al. Relapsing polychondritis: an update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives. Curr Rheumatol Rep. 2016;18:3.
- Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
- Dolev JC, Maurer TA, Reddy SG, et al. Relapsing polychondritis in HIV-infected patients: a report of two cases. J Am Acad Dermatol. 2004;51:1023-1025.
- Zandman-Goddard G, Peeva E, Barland P. Combined autoimmune disease in a patient with AIDS. Clin Rheumatol. 2002;21:70-72.
- Belzunegui J, Cancio J, Pego JM, et al. Relapsing polychondritis and Behc¸et’s syndrome in a patient with HIV infection. Ann Rheum Dis. 1995;54:780.
- Sharma A, Gnanapandithan K, Sharma K, et al. Relapsing polychondritis: a review. Clin Rheumatol. 2013;32:1575-1583.
- Cantarini L, Vitale A, Brizi MG, et al. Diagnosis and classification of relapsing polychondritis. J Autoimmun. 2014;48-49:53-59.
- Cañas CA, Bonilla Abadía F. Local cartilage trauma as a pathogenic factor in autoimmunity (one hypothesis based on patients with relapsing polychondritis triggered by cartilage trauma). Autoimmune Dis. 2012;2012:453698.
- Ouchi N, Uzuki M, Kamataki A, et al. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. 2011;38:730-737.
- Buckner JH, Wu JJ, Reife RA, et al. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939-943.
- Kempta Lekpa F, Piette JC, Bastuji-Garin S, et al. Serum cartilage oligomeric matrix protein (COMP) is a marker of disease activity in relapsing polychondritis. Clin Exp Rheumatol. 2010;28:553-555.
- Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203-1207.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet CJ, McKenna CH, Luthra HS, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Arnaud L, Devilliers H, Peng SL, et al. The Relapsing Polychondritis Disease Activity Index: development of a disease activity score for relapsing polychondritis. Autoimmun Rev. 2012;12:204-209.
- Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin Immunopathol. 2003;25:3-18.
- Thaiss WM, Nikolaou K, Spengler W, et al. Imaging diagnosis in relapsing polychondritis and correlation with clinical and serological data. Skeletal Radiol. 2015;5:339-346.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Francès C, el Rassi R, Laporte JL, et al. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). 2001;80:173-179.
- Chopra R, Chaudhary N, Kay J. Relapsing polychondritis. Rheum Dis Clin North Am. 2013;39:263-276.
- Moulis G, Sailler L, Pugnet G, et al. Biologics in relapsing polychondritis: a case series. Clin Exp Rheumatol. 2013;31:937-939.
- Henes CJ, Xenitidis T, Horger M. Tocilizumab for refractory relapsing polychondritis—long-term response monitoring by magnetic resonance imaging. Joint Bone Spine. 2016;83:365-366.
- Weinberger A, Myers AR. Relapsing polychondritis associated with cutaneous vasculitis. Arch Dermatol. 1979;115:980-981.
- Karaca NE, Aksu G, Yildiz B, et al. Relapsing polychondritis in a child with common variable immunodeficiency. Int J Dermatol. 2009;48:525-528.
Relapsing polychondritis (RP) is a recurrent inflammatory condition involving primarily cartilaginous structures. The disease, first described as a clinical entity in 1960 by Pearson et al,1 is rare with an estimated incidence of 3.5 cases per 1 million individuals.2 The pathogenesis of RP is widely accepted as being autoimmune in nature, largely due to the identification of circulating autoantibodies seen in the sera of patients with similar clinical pictures.3
Although in most patients the primary process involves inflammation of cartilage, a subset of patients experience involvement of noncartilaginous sites.4 The degree of systemic involvement varies from none to notable, affecting the cardiovascular and respiratory systems and potentially leading to life-threatening complications, including cardiac valve compromise and airway collapse. Relapsing polychondritis is considered to be a progressive disease with the ultimate potential to be life-threatening.5
Human immunodeficiency virus (HIV) infection leads to a profound state of immune dysregulation, affecting innate, adaptive, and natural killer components of the immune system.6 There is variability in the development of autoimmune disease in HIV patients depending on the stage of infection. The frequency of rheumatologic disease in HIV patients might be as high as 60%.6 Relapsing polychondritis is rare in patients with HIV.7-9 Of 4 reported cases, 2 patients had other coexisting autoimmune disease—sarcoidosis and Behçet disease.8,9
Case Report
A 36-year-old man presented to the clinic with a concern of recurrent ear pain and swelling of approximately 2 years’ duration. Onset was sudden without inciting event. Symptoms initially involved the right ear with eventual progression to both ears. Additional symptoms included an auditory perception of underwater submersion, intermittent vertigo, and 3 episodes of throat closure sensation.
The patient’s medical history was notable for asthma; gastritis; depression; and HIV infection, which was diagnosed 4 years earlier and adequately managed with highly active antiretroviral therapy. His family history was notable for systemic lupus erythematosus in his mother, maternal aunt, and maternal cousin.
At presentation, the patient’s CD4 count was 799 cells/mm3 with an undetectable viral load. Medications included abacavir-dolutegravir-lamivudine, hydroxyzine, meclizine, mometasone, and quetiapine. Physical examination showed erythema, swelling, and tenderness of the left and right auricles with sparing of the earlobe that was more noticeable on the left ear (Figure 1). Bacterial culture from the external auditory meatus was positive for methicillin-resistant Staphylococcus aureus. Biopsy revealed chronic inflammatory perichondritis with mild to moderate fibrosis and chronic lymphocytic inflammation at the dermal cartilaginous junction (Figure 2). A direct immunofluorescent biopsy was unremarkable, but subsequent type II collagen antibodies were positive (35.5 endotoxin units/mL [reference range, <20 endotoxin units/mL]).
Comment
Relapsing polychondritis is an uncommon progressive disease characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures.4 The most consistent clinical features of RP are ear inflammation that involves the auricle and spares the lobe, nasal chondritis, and arthralgia.10 Laryngotracheal compromise may occur from tracheal cartilage inflammation. The involvement of these specific structures is due to commonality between their component collagens.5 Although any organ system can be affected, as many as 50% of patients have respiratory tract involvement, which may affect any portion of the respiratory tree.11 If involving the larynx, this inflammation can lead to severe edema warranting intubation. Cardiovascular involvement is present in 24% to 52% of patients,10 which most commonly manifests as valvular impairment affecting the aortic valve more frequently than the mitral valve.5
Pathogenesis
Although the etiology of RP remains undetermined, multiple hypotheses have been proposed. One is that a certain subset of patients is predisposed to autoimmunity, and a secondary triggering event in the form of infection, malignancy, or medication catalyzes development of RP. A second hypothesis is that mechanical trauma to cartilage exposes the immune system to certain antigens that would have otherwise remained hidden, prompting autosensitization.12,13
Regardless of cause, an autoimmune pathogenesis is favored based on the following observations: RP is frequently associated with other autoimmune diseases in the same patient, glucocorticosteroids and other immunosuppressive therapies are effective for treatment, and histopathologic findings include an infiltrate of CD4+ T lymphocytes with detection of immunoglobulins and plasma cells in different lesions.5 The detection of autoantibodies against collagen in the serum of patients with RP further supports an autoimmune pathogenesis.3 The earliest identified autoantibodies in patients with RP were against type II collagen. Subsequent studies have identified autoantibodies against type IV and type XI collagens as well as other cartilage-related proteins such as matrilin 114 and cartilage oligomeric matrix proteins.15 Although circulating antibodies to type II collagen are present in a variable number of patients with the disease (30%–70%), levels likely correlate with disease activity and are highest at times of acute inflammation.3 Additionally, titers of type II collagen antibodies have been shown to decrease upon institution of immunosuppressive therapy.16
Although a humoral response dominates the picture of RP, there also is an associated T cell–mediated response.13 Histopathologically, biopsy of an active lesion of auricular cartilage shows a mixed inflammatory infiltrate composed primarily of lymphocytes, with variable numbers of polymorphonuclear cells, monocytes, and plasma cells. Loss of basophilia of the cartilage matrix can be observed, thought to be the result of proteoglycan depletion.13 Later, lesions classically display apoptosis of chondrocytes, focal calcification, or fibrosis.5
Diagnosis
Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge. Many sets of diagnostic criteria have been proposed. The most referenced remains the original criteria described by McAdam et al.17 In 2012, the Relapsing Polychondritis Disease Activity Index modified criteria set forth by Michet et al18 and might serve as the standard for diagnosis going forward.19
McAdam et al17 proposed that 3 of 6 clinical features are necessary for diagnosis: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation, respiratory tract chondritis, and audiovestibular damage. Michet et al18 proposed that 1 of 2 conditions are necessary for diagnosis of RP: (1) proven inflammation in 2 of 3 of the auricular, nasal, or laryngotracheal cartilages; or (2) proven inflammation in 1 of 3 of the auricular, nasal, or laryngotracheal cartilages, plus 2 other signs, including ocular inflammation, vestibular dysfunction, seronegative inflammatory arthritis, and hearing loss.
These criteria were proposed originally in 197617 and modified in 1986.18 No further updates have been offered since then. As such, serologic findings, such as antibodies against type II collagen, are not included in the diagnostic criteria. Additionally, these antibodies are not specific for RP and can be seen in other conditions such as rheumatoid arthritis.20
More recently, imaging analysis has been employed in conjunction with clinical and serologic data to diagnose the disease and evaluate its severity. The use of imaging modalities for these purposes is most beneficial in patients with notable disease and respiratory involvement.21
Although the clinical picture is typified by the classic findings described above, the clinician must be aware of more subtle clues to diagnosis,11 which is of particular importance to the dermatologist because 35% of patients with RP alone will have skin manifestations that can precede onset of chondritis.10 Most commonly, dermatologic manifestations are nonspecific and can include nodules on the limbs, purpura, and urticarial lesions.22 Individual case reports have noted the coexistence of RP with erythema multiforme,18 erythema annulare centrifugum,23 pyoderma gangrenosum,24 and panniculitis,18 among other disorders.
Treatment
Standardized guidelines for treatment do not exist. Treatments should be chosen based on severity of disease. Mild disease, presenting with recurrent chondritis and arthritis without evidence of systemic involvement, can be treated with nonsteroidal anti-inflammatory drugs, dapsone, or colchicine. Refractory disease often requires high-dose systemic corticosteroids.5
Severe systemic involvement leads to increased mortality and warrants more aggressive treatment.22 Commonly used agents include the immunosuppressants cyclophosphamide, cyclosporine, and methotrexate. Tumor necrosis factor α inhibitors have been the most widely utilized immunomodulatory agent for treatment of RP.25,26 Abatacept and rituximab also have been used with variable efficacy in patients with severe disease. Recently, the IL-6 receptor blocker tocilizumab has been used with some success.27
Prognosis
The prognosis for patients with RP largely depends on the severity of disease and degree of internal involvement. With improved management, largely due to awareness and recognition of disease, the survival rate among RP patients has increased from 55% at 10 years to 94% at the end of 8 years.18 The main cause of death in RP patients is airway complications related to laryngotracheal involvement.10 The second most common cause of death is cardiovascular complications in which valvular disease predominates.5
Concomitant Illness
Thirty-five percent of RP patients have coexisting autoimmune disease, the most common being antineutrophil cytoplasmic antibody–associated vasculitis.5,28 Although this association with autoimmune disease is well described, reports of RP occurring in other states of immune dysfunction are sparse. One case of RP has been reported in a child with common variable immunodeficiency thought to be related to underlying abnormal immune regulation and immunodeficiency.29 Relapsing polychondritis has been described in 4 patients with HIV, 2 of whom had concomitant autoimmune disease.7-9
Human immunodeficiency virus infection is a well-established cause of immune dysregulation and has variable association with autoimmunity. This variability depends largely on the stage of infection. When divided into stages, autoimmune diseases develop predominantly in stage I, during acute infection with an intact immune system; in stage III, with immunosuppression, a low CD4 count, and development of AIDS; and in stage IV, when the immune system is restored with the institution of highly active antiretroviral therapy.6 The interplay between HIV infection and development of autoimmune disease is complex, and pathogenesis remains speculative.
Conclusion
Our patient represents a case of RP in an HIV-positive patient. Additionally, our patient had no other identifiable autoimmune conditions but did have a strong family history of them. It is important for providers to be aware of the potential for development of RP as well as other autoimmune disease in the setting of HIV infection. The implications of a missed diagnosis could be dire because the disease course of RP is progressive and has the potential to decrease survival.
Relapsing polychondritis (RP) is a recurrent inflammatory condition involving primarily cartilaginous structures. The disease, first described as a clinical entity in 1960 by Pearson et al,1 is rare with an estimated incidence of 3.5 cases per 1 million individuals.2 The pathogenesis of RP is widely accepted as being autoimmune in nature, largely due to the identification of circulating autoantibodies seen in the sera of patients with similar clinical pictures.3
Although in most patients the primary process involves inflammation of cartilage, a subset of patients experience involvement of noncartilaginous sites.4 The degree of systemic involvement varies from none to notable, affecting the cardiovascular and respiratory systems and potentially leading to life-threatening complications, including cardiac valve compromise and airway collapse. Relapsing polychondritis is considered to be a progressive disease with the ultimate potential to be life-threatening.5
Human immunodeficiency virus (HIV) infection leads to a profound state of immune dysregulation, affecting innate, adaptive, and natural killer components of the immune system.6 There is variability in the development of autoimmune disease in HIV patients depending on the stage of infection. The frequency of rheumatologic disease in HIV patients might be as high as 60%.6 Relapsing polychondritis is rare in patients with HIV.7-9 Of 4 reported cases, 2 patients had other coexisting autoimmune disease—sarcoidosis and Behçet disease.8,9
Case Report
A 36-year-old man presented to the clinic with a concern of recurrent ear pain and swelling of approximately 2 years’ duration. Onset was sudden without inciting event. Symptoms initially involved the right ear with eventual progression to both ears. Additional symptoms included an auditory perception of underwater submersion, intermittent vertigo, and 3 episodes of throat closure sensation.
The patient’s medical history was notable for asthma; gastritis; depression; and HIV infection, which was diagnosed 4 years earlier and adequately managed with highly active antiretroviral therapy. His family history was notable for systemic lupus erythematosus in his mother, maternal aunt, and maternal cousin.
At presentation, the patient’s CD4 count was 799 cells/mm3 with an undetectable viral load. Medications included abacavir-dolutegravir-lamivudine, hydroxyzine, meclizine, mometasone, and quetiapine. Physical examination showed erythema, swelling, and tenderness of the left and right auricles with sparing of the earlobe that was more noticeable on the left ear (Figure 1). Bacterial culture from the external auditory meatus was positive for methicillin-resistant Staphylococcus aureus. Biopsy revealed chronic inflammatory perichondritis with mild to moderate fibrosis and chronic lymphocytic inflammation at the dermal cartilaginous junction (Figure 2). A direct immunofluorescent biopsy was unremarkable, but subsequent type II collagen antibodies were positive (35.5 endotoxin units/mL [reference range, <20 endotoxin units/mL]).
Comment
Relapsing polychondritis is an uncommon progressive disease characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures.4 The most consistent clinical features of RP are ear inflammation that involves the auricle and spares the lobe, nasal chondritis, and arthralgia.10 Laryngotracheal compromise may occur from tracheal cartilage inflammation. The involvement of these specific structures is due to commonality between their component collagens.5 Although any organ system can be affected, as many as 50% of patients have respiratory tract involvement, which may affect any portion of the respiratory tree.11 If involving the larynx, this inflammation can lead to severe edema warranting intubation. Cardiovascular involvement is present in 24% to 52% of patients,10 which most commonly manifests as valvular impairment affecting the aortic valve more frequently than the mitral valve.5
Pathogenesis
Although the etiology of RP remains undetermined, multiple hypotheses have been proposed. One is that a certain subset of patients is predisposed to autoimmunity, and a secondary triggering event in the form of infection, malignancy, or medication catalyzes development of RP. A second hypothesis is that mechanical trauma to cartilage exposes the immune system to certain antigens that would have otherwise remained hidden, prompting autosensitization.12,13
Regardless of cause, an autoimmune pathogenesis is favored based on the following observations: RP is frequently associated with other autoimmune diseases in the same patient, glucocorticosteroids and other immunosuppressive therapies are effective for treatment, and histopathologic findings include an infiltrate of CD4+ T lymphocytes with detection of immunoglobulins and plasma cells in different lesions.5 The detection of autoantibodies against collagen in the serum of patients with RP further supports an autoimmune pathogenesis.3 The earliest identified autoantibodies in patients with RP were against type II collagen. Subsequent studies have identified autoantibodies against type IV and type XI collagens as well as other cartilage-related proteins such as matrilin 114 and cartilage oligomeric matrix proteins.15 Although circulating antibodies to type II collagen are present in a variable number of patients with the disease (30%–70%), levels likely correlate with disease activity and are highest at times of acute inflammation.3 Additionally, titers of type II collagen antibodies have been shown to decrease upon institution of immunosuppressive therapy.16
Although a humoral response dominates the picture of RP, there also is an associated T cell–mediated response.13 Histopathologically, biopsy of an active lesion of auricular cartilage shows a mixed inflammatory infiltrate composed primarily of lymphocytes, with variable numbers of polymorphonuclear cells, monocytes, and plasma cells. Loss of basophilia of the cartilage matrix can be observed, thought to be the result of proteoglycan depletion.13 Later, lesions classically display apoptosis of chondrocytes, focal calcification, or fibrosis.5
Diagnosis
Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge. Many sets of diagnostic criteria have been proposed. The most referenced remains the original criteria described by McAdam et al.17 In 2012, the Relapsing Polychondritis Disease Activity Index modified criteria set forth by Michet et al18 and might serve as the standard for diagnosis going forward.19
McAdam et al17 proposed that 3 of 6 clinical features are necessary for diagnosis: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation, respiratory tract chondritis, and audiovestibular damage. Michet et al18 proposed that 1 of 2 conditions are necessary for diagnosis of RP: (1) proven inflammation in 2 of 3 of the auricular, nasal, or laryngotracheal cartilages; or (2) proven inflammation in 1 of 3 of the auricular, nasal, or laryngotracheal cartilages, plus 2 other signs, including ocular inflammation, vestibular dysfunction, seronegative inflammatory arthritis, and hearing loss.
These criteria were proposed originally in 197617 and modified in 1986.18 No further updates have been offered since then. As such, serologic findings, such as antibodies against type II collagen, are not included in the diagnostic criteria. Additionally, these antibodies are not specific for RP and can be seen in other conditions such as rheumatoid arthritis.20
More recently, imaging analysis has been employed in conjunction with clinical and serologic data to diagnose the disease and evaluate its severity. The use of imaging modalities for these purposes is most beneficial in patients with notable disease and respiratory involvement.21
Although the clinical picture is typified by the classic findings described above, the clinician must be aware of more subtle clues to diagnosis,11 which is of particular importance to the dermatologist because 35% of patients with RP alone will have skin manifestations that can precede onset of chondritis.10 Most commonly, dermatologic manifestations are nonspecific and can include nodules on the limbs, purpura, and urticarial lesions.22 Individual case reports have noted the coexistence of RP with erythema multiforme,18 erythema annulare centrifugum,23 pyoderma gangrenosum,24 and panniculitis,18 among other disorders.
Treatment
Standardized guidelines for treatment do not exist. Treatments should be chosen based on severity of disease. Mild disease, presenting with recurrent chondritis and arthritis without evidence of systemic involvement, can be treated with nonsteroidal anti-inflammatory drugs, dapsone, or colchicine. Refractory disease often requires high-dose systemic corticosteroids.5
Severe systemic involvement leads to increased mortality and warrants more aggressive treatment.22 Commonly used agents include the immunosuppressants cyclophosphamide, cyclosporine, and methotrexate. Tumor necrosis factor α inhibitors have been the most widely utilized immunomodulatory agent for treatment of RP.25,26 Abatacept and rituximab also have been used with variable efficacy in patients with severe disease. Recently, the IL-6 receptor blocker tocilizumab has been used with some success.27
Prognosis
The prognosis for patients with RP largely depends on the severity of disease and degree of internal involvement. With improved management, largely due to awareness and recognition of disease, the survival rate among RP patients has increased from 55% at 10 years to 94% at the end of 8 years.18 The main cause of death in RP patients is airway complications related to laryngotracheal involvement.10 The second most common cause of death is cardiovascular complications in which valvular disease predominates.5
Concomitant Illness
Thirty-five percent of RP patients have coexisting autoimmune disease, the most common being antineutrophil cytoplasmic antibody–associated vasculitis.5,28 Although this association with autoimmune disease is well described, reports of RP occurring in other states of immune dysfunction are sparse. One case of RP has been reported in a child with common variable immunodeficiency thought to be related to underlying abnormal immune regulation and immunodeficiency.29 Relapsing polychondritis has been described in 4 patients with HIV, 2 of whom had concomitant autoimmune disease.7-9
Human immunodeficiency virus infection is a well-established cause of immune dysregulation and has variable association with autoimmunity. This variability depends largely on the stage of infection. When divided into stages, autoimmune diseases develop predominantly in stage I, during acute infection with an intact immune system; in stage III, with immunosuppression, a low CD4 count, and development of AIDS; and in stage IV, when the immune system is restored with the institution of highly active antiretroviral therapy.6 The interplay between HIV infection and development of autoimmune disease is complex, and pathogenesis remains speculative.
Conclusion
Our patient represents a case of RP in an HIV-positive patient. Additionally, our patient had no other identifiable autoimmune conditions but did have a strong family history of them. It is important for providers to be aware of the potential for development of RP as well as other autoimmune disease in the setting of HIV infection. The implications of a missed diagnosis could be dire because the disease course of RP is progressive and has the potential to decrease survival.
- Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med. 1960;263:51-58.
- Kent PD, Michet CJ Jr, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56-61.
- Ebringer R, Rook G, Swana GT, et al. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473-479.
- Sharma A, Law AD, Bambery P, et al. Relapsing polychondritis: clinical presentations, disease activity and outcomes. Orphanet J Rare Dis. 2014;9:198.
- Vitale A, Sota J, Rigante D, et al. Relapsing polychondritis: an update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives. Curr Rheumatol Rep. 2016;18:3.
- Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
- Dolev JC, Maurer TA, Reddy SG, et al. Relapsing polychondritis in HIV-infected patients: a report of two cases. J Am Acad Dermatol. 2004;51:1023-1025.
- Zandman-Goddard G, Peeva E, Barland P. Combined autoimmune disease in a patient with AIDS. Clin Rheumatol. 2002;21:70-72.
- Belzunegui J, Cancio J, Pego JM, et al. Relapsing polychondritis and Behc¸et’s syndrome in a patient with HIV infection. Ann Rheum Dis. 1995;54:780.
- Sharma A, Gnanapandithan K, Sharma K, et al. Relapsing polychondritis: a review. Clin Rheumatol. 2013;32:1575-1583.
- Cantarini L, Vitale A, Brizi MG, et al. Diagnosis and classification of relapsing polychondritis. J Autoimmun. 2014;48-49:53-59.
- Cañas CA, Bonilla Abadía F. Local cartilage trauma as a pathogenic factor in autoimmunity (one hypothesis based on patients with relapsing polychondritis triggered by cartilage trauma). Autoimmune Dis. 2012;2012:453698.
- Ouchi N, Uzuki M, Kamataki A, et al. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. 2011;38:730-737.
- Buckner JH, Wu JJ, Reife RA, et al. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939-943.
- Kempta Lekpa F, Piette JC, Bastuji-Garin S, et al. Serum cartilage oligomeric matrix protein (COMP) is a marker of disease activity in relapsing polychondritis. Clin Exp Rheumatol. 2010;28:553-555.
- Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203-1207.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet CJ, McKenna CH, Luthra HS, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Arnaud L, Devilliers H, Peng SL, et al. The Relapsing Polychondritis Disease Activity Index: development of a disease activity score for relapsing polychondritis. Autoimmun Rev. 2012;12:204-209.
- Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin Immunopathol. 2003;25:3-18.
- Thaiss WM, Nikolaou K, Spengler W, et al. Imaging diagnosis in relapsing polychondritis and correlation with clinical and serological data. Skeletal Radiol. 2015;5:339-346.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Francès C, el Rassi R, Laporte JL, et al. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). 2001;80:173-179.
- Chopra R, Chaudhary N, Kay J. Relapsing polychondritis. Rheum Dis Clin North Am. 2013;39:263-276.
- Moulis G, Sailler L, Pugnet G, et al. Biologics in relapsing polychondritis: a case series. Clin Exp Rheumatol. 2013;31:937-939.
- Henes CJ, Xenitidis T, Horger M. Tocilizumab for refractory relapsing polychondritis—long-term response monitoring by magnetic resonance imaging. Joint Bone Spine. 2016;83:365-366.
- Weinberger A, Myers AR. Relapsing polychondritis associated with cutaneous vasculitis. Arch Dermatol. 1979;115:980-981.
- Karaca NE, Aksu G, Yildiz B, et al. Relapsing polychondritis in a child with common variable immunodeficiency. Int J Dermatol. 2009;48:525-528.
- Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med. 1960;263:51-58.
- Kent PD, Michet CJ Jr, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56-61.
- Ebringer R, Rook G, Swana GT, et al. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473-479.
- Sharma A, Law AD, Bambery P, et al. Relapsing polychondritis: clinical presentations, disease activity and outcomes. Orphanet J Rare Dis. 2014;9:198.
- Vitale A, Sota J, Rigante D, et al. Relapsing polychondritis: an update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives. Curr Rheumatol Rep. 2016;18:3.
- Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
- Dolev JC, Maurer TA, Reddy SG, et al. Relapsing polychondritis in HIV-infected patients: a report of two cases. J Am Acad Dermatol. 2004;51:1023-1025.
- Zandman-Goddard G, Peeva E, Barland P. Combined autoimmune disease in a patient with AIDS. Clin Rheumatol. 2002;21:70-72.
- Belzunegui J, Cancio J, Pego JM, et al. Relapsing polychondritis and Behc¸et’s syndrome in a patient with HIV infection. Ann Rheum Dis. 1995;54:780.
- Sharma A, Gnanapandithan K, Sharma K, et al. Relapsing polychondritis: a review. Clin Rheumatol. 2013;32:1575-1583.
- Cantarini L, Vitale A, Brizi MG, et al. Diagnosis and classification of relapsing polychondritis. J Autoimmun. 2014;48-49:53-59.
- Cañas CA, Bonilla Abadía F. Local cartilage trauma as a pathogenic factor in autoimmunity (one hypothesis based on patients with relapsing polychondritis triggered by cartilage trauma). Autoimmune Dis. 2012;2012:453698.
- Ouchi N, Uzuki M, Kamataki A, et al. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. 2011;38:730-737.
- Buckner JH, Wu JJ, Reife RA, et al. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939-943.
- Kempta Lekpa F, Piette JC, Bastuji-Garin S, et al. Serum cartilage oligomeric matrix protein (COMP) is a marker of disease activity in relapsing polychondritis. Clin Exp Rheumatol. 2010;28:553-555.
- Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203-1207.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet CJ, McKenna CH, Luthra HS, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Arnaud L, Devilliers H, Peng SL, et al. The Relapsing Polychondritis Disease Activity Index: development of a disease activity score for relapsing polychondritis. Autoimmun Rev. 2012;12:204-209.
- Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin Immunopathol. 2003;25:3-18.
- Thaiss WM, Nikolaou K, Spengler W, et al. Imaging diagnosis in relapsing polychondritis and correlation with clinical and serological data. Skeletal Radiol. 2015;5:339-346.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Francès C, el Rassi R, Laporte JL, et al. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). 2001;80:173-179.
- Chopra R, Chaudhary N, Kay J. Relapsing polychondritis. Rheum Dis Clin North Am. 2013;39:263-276.
- Moulis G, Sailler L, Pugnet G, et al. Biologics in relapsing polychondritis: a case series. Clin Exp Rheumatol. 2013;31:937-939.
- Henes CJ, Xenitidis T, Horger M. Tocilizumab for refractory relapsing polychondritis—long-term response monitoring by magnetic resonance imaging. Joint Bone Spine. 2016;83:365-366.
- Weinberger A, Myers AR. Relapsing polychondritis associated with cutaneous vasculitis. Arch Dermatol. 1979;115:980-981.
- Karaca NE, Aksu G, Yildiz B, et al. Relapsing polychondritis in a child with common variable immunodeficiency. Int J Dermatol. 2009;48:525-528.
Practice Points
- Relapsing polychondritis (RP) is characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures, most often manifesting as ear inflammation that involves the auricle but spares the lobe, nasal chondritis, and arthralgia.
- Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge.
- One-third of RP patients have coexisting autoimmune disease.
- Treatment of RP depends on severity of disease.
- Dermatologists must be aware of the potential for development of RP in the setting of human immunodeficiency virus infection; a missed diagnosis of this progressive disease has the potential to be life-threatening.
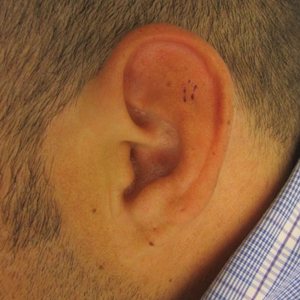
Cutaneous Metastasis of Endometrial Carcinoma: An Unusual and Dramatic Presentation
Case Report
A 62-year-old woman presented with multiple large friable tumors of the abdominal panniculus. The patient also reported an unintentional 75-lb weight loss over the last 9 months as well as vaginal bleeding and fecal discharge from the vagina of 2 weeks’ duration. The patient had a surgical and medical history of a robotic-assisted hysterectomy and bilateral salpingo-oophorectomy performed 4 years prior to presentation. Final surgical pathology showed complex atypical endometrial hyperplasia with no adenocarcinoma identified.
Physical examination revealed multiple large, friable, exophytic tumors of the left side of the lower abdominal panniculus within close vicinity of the patient’s abdominal hysterectomy scars (Figure 1). The largest lesion measured approximately 6 cm in length. Laboratory values were elevated for carcinoembryonic antigen (5.9 ng/mL [reference range, <3.0 ng/mL]) and cancer antigen 125 (202 U/mL [reference range, <35 U/mL]). Computed tomography of the abdomen and pelvis revealed diffuse metastatic disease.
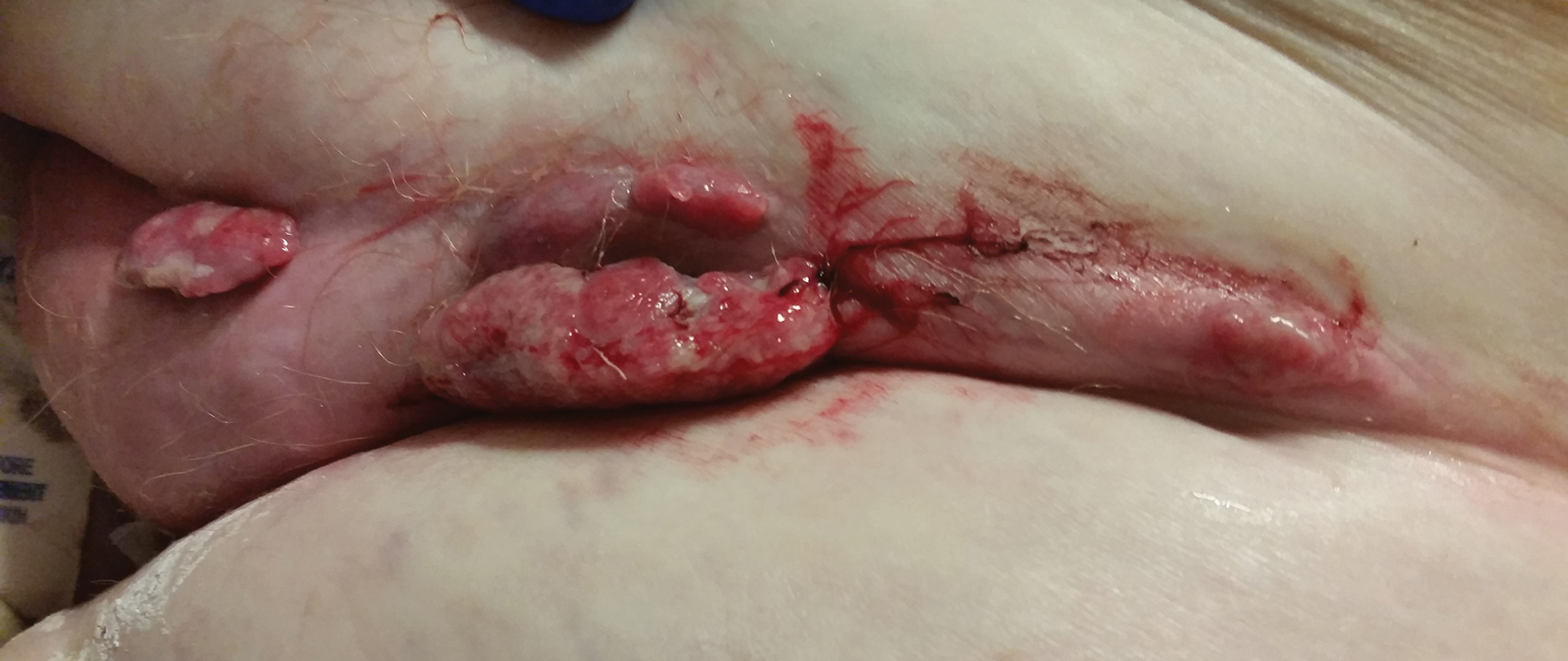
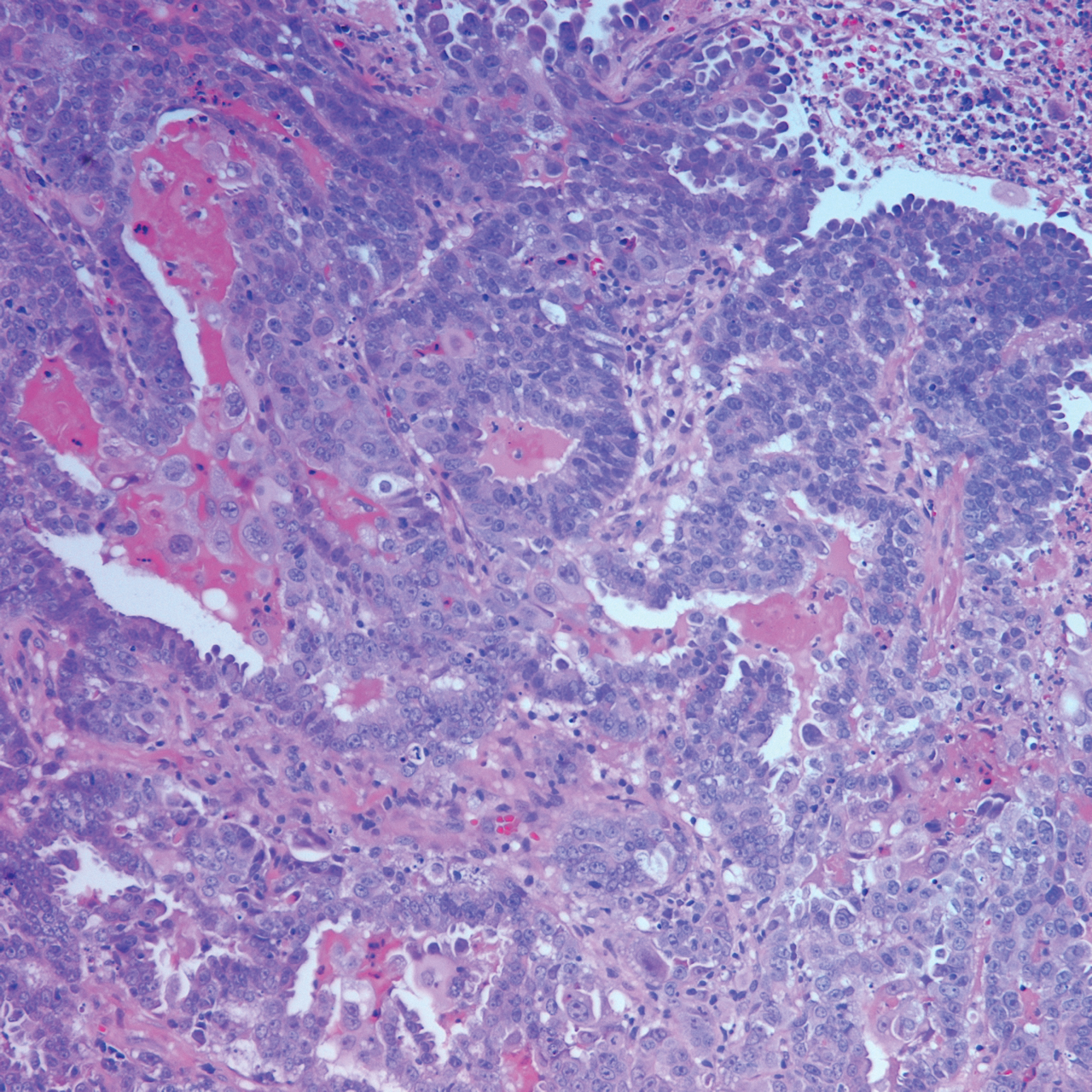
Comment
Incidence and Pathogenesis
Endometrial carcinoma is the most common gynecologic malignancy in the United States, but it rarely progresses to disseminated disease because of routine gynecologic examinations and the low threshold for surgical intervention. Cutaneous metastases represent one of the rarest presentations of disseminated disease, occurring in only 0.8% of those diagnosed with endometrial carcinoma.1 Cutaneous metastases occur almost exclusively in women older than 50 years and typically appear several months to years after hysterectomy. Although the exact pathogenesis is unknown, it is theorized that small foci of malignant cells may be seeded during surgery, leading to visceral and cutaneous involvement.
Clinical Presentation
Lesions vary morphologically, most commonly presenting as nonspecific, painless, hemorrhagic nodules. Lesions typically present in areas of direct local extension; prior radiotherapy; or areas of initial surgery, as was the case with our patient.2 Approximately 20 cases of umbilical involvement (Sister Mary Joseph nodule) have been reported in the literature. These cases are thought to occur from direct local spread of disease from the peritoneum.3 Hematogenous and lymphatic spread to distant sites such as the scalp and mandible also have been reported. More than 50% of patients will have underlying visceral metastatic disease at the time of diagnosis.3
Histopathologic Findings
Histopathology varies with the morphology of the underlying primary tumor, with endometrioid adenocarcinoma being the most common form associated with cutaneous metastasis, as was the case with our patient.4 Histology is characterized by dermal proliferation of atypical glandular epithelium with diffuse hemorrhage. Staining typically is positive for CK7 and negative for CK20 and CDX2.5 Histopathology and immunohistochemical staining are not specific for diagnosis and must be correlated with clinical history.
Management and Prognosis
Similar to cutaneous metastasis in other internal malignancies, prognosis is poor, as widespread dissemination of the underlying malignancy typically is present. Mean life expectancy is 4 to 12 months.6 Treatment is primarily palliative, as chemotherapy and radiotherapy are largely ineffective.
Conclusion
Our patient represents a dramatic form of cutaneous extension of a common disease. Dermatologists often are consulted because of the nonspecific nature of the lesions and must be conscious of this entity. As with other cutaneous metastases, a thorough medical and surgical history in conjunction with histopathology are necessary for an accurate diagnosis.
- Atallah D, el Kassis N, Lutfallah F, et al. Cutaneous metastasis in endometrial cancer: once in a blue moon—case report. World J Surg Oncol. 2014;12:86.
- Temkin SM, Hellman M, Lee YC, et al. Surgical resection of vulvar metastases of endometrial cancer: a presentation of two cases. J Low Genit Tract Dis. 2007;11:118-121.
- Kushner DM, Lurain JR, Fu TS, et al. Endometrial adenocarcinoma metastatic to the scalp: case report and literature review. Gynecol Oncol. 1997;65:530-533.
- El M’rabet FZ, Hottinger A, George AC. Cutaneous metastasis of endometrial carcinoma: a case report and literature review. J Clin Gynecol Obstet. 2012;1:19-23.
- Stonard CM, Manek S. Cutaneous metastasis from an endometrial carcinoma: a case history and review of the literature. Histopathology. 2003;43:201-203
- Damewood MD, Rosenshein NB, Grumbine FC, et al. Cutaneous metastasis of endometrial carcinoma. Cancer. 1980;46:1471-1477.
Case Report
A 62-year-old woman presented with multiple large friable tumors of the abdominal panniculus. The patient also reported an unintentional 75-lb weight loss over the last 9 months as well as vaginal bleeding and fecal discharge from the vagina of 2 weeks’ duration. The patient had a surgical and medical history of a robotic-assisted hysterectomy and bilateral salpingo-oophorectomy performed 4 years prior to presentation. Final surgical pathology showed complex atypical endometrial hyperplasia with no adenocarcinoma identified.
Physical examination revealed multiple large, friable, exophytic tumors of the left side of the lower abdominal panniculus within close vicinity of the patient’s abdominal hysterectomy scars (Figure 1). The largest lesion measured approximately 6 cm in length. Laboratory values were elevated for carcinoembryonic antigen (5.9 ng/mL [reference range, <3.0 ng/mL]) and cancer antigen 125 (202 U/mL [reference range, <35 U/mL]). Computed tomography of the abdomen and pelvis revealed diffuse metastatic disease.
Comment
Incidence and Pathogenesis
Endometrial carcinoma is the most common gynecologic malignancy in the United States, but it rarely progresses to disseminated disease because of routine gynecologic examinations and the low threshold for surgical intervention. Cutaneous metastases represent one of the rarest presentations of disseminated disease, occurring in only 0.8% of those diagnosed with endometrial carcinoma.1 Cutaneous metastases occur almost exclusively in women older than 50 years and typically appear several months to years after hysterectomy. Although the exact pathogenesis is unknown, it is theorized that small foci of malignant cells may be seeded during surgery, leading to visceral and cutaneous involvement.
Clinical Presentation
Lesions vary morphologically, most commonly presenting as nonspecific, painless, hemorrhagic nodules. Lesions typically present in areas of direct local extension; prior radiotherapy; or areas of initial surgery, as was the case with our patient.2 Approximately 20 cases of umbilical involvement (Sister Mary Joseph nodule) have been reported in the literature. These cases are thought to occur from direct local spread of disease from the peritoneum.3 Hematogenous and lymphatic spread to distant sites such as the scalp and mandible also have been reported. More than 50% of patients will have underlying visceral metastatic disease at the time of diagnosis.3
Histopathologic Findings
Histopathology varies with the morphology of the underlying primary tumor, with endometrioid adenocarcinoma being the most common form associated with cutaneous metastasis, as was the case with our patient.4 Histology is characterized by dermal proliferation of atypical glandular epithelium with diffuse hemorrhage. Staining typically is positive for CK7 and negative for CK20 and CDX2.5 Histopathology and immunohistochemical staining are not specific for diagnosis and must be correlated with clinical history.
Management and Prognosis
Similar to cutaneous metastasis in other internal malignancies, prognosis is poor, as widespread dissemination of the underlying malignancy typically is present. Mean life expectancy is 4 to 12 months.6 Treatment is primarily palliative, as chemotherapy and radiotherapy are largely ineffective.
Conclusion
Our patient represents a dramatic form of cutaneous extension of a common disease. Dermatologists often are consulted because of the nonspecific nature of the lesions and must be conscious of this entity. As with other cutaneous metastases, a thorough medical and surgical history in conjunction with histopathology are necessary for an accurate diagnosis.
Case Report
A 62-year-old woman presented with multiple large friable tumors of the abdominal panniculus. The patient also reported an unintentional 75-lb weight loss over the last 9 months as well as vaginal bleeding and fecal discharge from the vagina of 2 weeks’ duration. The patient had a surgical and medical history of a robotic-assisted hysterectomy and bilateral salpingo-oophorectomy performed 4 years prior to presentation. Final surgical pathology showed complex atypical endometrial hyperplasia with no adenocarcinoma identified.
Physical examination revealed multiple large, friable, exophytic tumors of the left side of the lower abdominal panniculus within close vicinity of the patient’s abdominal hysterectomy scars (Figure 1). The largest lesion measured approximately 6 cm in length. Laboratory values were elevated for carcinoembryonic antigen (5.9 ng/mL [reference range, <3.0 ng/mL]) and cancer antigen 125 (202 U/mL [reference range, <35 U/mL]). Computed tomography of the abdomen and pelvis revealed diffuse metastatic disease.
Comment
Incidence and Pathogenesis
Endometrial carcinoma is the most common gynecologic malignancy in the United States, but it rarely progresses to disseminated disease because of routine gynecologic examinations and the low threshold for surgical intervention. Cutaneous metastases represent one of the rarest presentations of disseminated disease, occurring in only 0.8% of those diagnosed with endometrial carcinoma.1 Cutaneous metastases occur almost exclusively in women older than 50 years and typically appear several months to years after hysterectomy. Although the exact pathogenesis is unknown, it is theorized that small foci of malignant cells may be seeded during surgery, leading to visceral and cutaneous involvement.
Clinical Presentation
Lesions vary morphologically, most commonly presenting as nonspecific, painless, hemorrhagic nodules. Lesions typically present in areas of direct local extension; prior radiotherapy; or areas of initial surgery, as was the case with our patient.2 Approximately 20 cases of umbilical involvement (Sister Mary Joseph nodule) have been reported in the literature. These cases are thought to occur from direct local spread of disease from the peritoneum.3 Hematogenous and lymphatic spread to distant sites such as the scalp and mandible also have been reported. More than 50% of patients will have underlying visceral metastatic disease at the time of diagnosis.3
Histopathologic Findings
Histopathology varies with the morphology of the underlying primary tumor, with endometrioid adenocarcinoma being the most common form associated with cutaneous metastasis, as was the case with our patient.4 Histology is characterized by dermal proliferation of atypical glandular epithelium with diffuse hemorrhage. Staining typically is positive for CK7 and negative for CK20 and CDX2.5 Histopathology and immunohistochemical staining are not specific for diagnosis and must be correlated with clinical history.
Management and Prognosis
Similar to cutaneous metastasis in other internal malignancies, prognosis is poor, as widespread dissemination of the underlying malignancy typically is present. Mean life expectancy is 4 to 12 months.6 Treatment is primarily palliative, as chemotherapy and radiotherapy are largely ineffective.
Conclusion
Our patient represents a dramatic form of cutaneous extension of a common disease. Dermatologists often are consulted because of the nonspecific nature of the lesions and must be conscious of this entity. As with other cutaneous metastases, a thorough medical and surgical history in conjunction with histopathology are necessary for an accurate diagnosis.
- Atallah D, el Kassis N, Lutfallah F, et al. Cutaneous metastasis in endometrial cancer: once in a blue moon—case report. World J Surg Oncol. 2014;12:86.
- Temkin SM, Hellman M, Lee YC, et al. Surgical resection of vulvar metastases of endometrial cancer: a presentation of two cases. J Low Genit Tract Dis. 2007;11:118-121.
- Kushner DM, Lurain JR, Fu TS, et al. Endometrial adenocarcinoma metastatic to the scalp: case report and literature review. Gynecol Oncol. 1997;65:530-533.
- El M’rabet FZ, Hottinger A, George AC. Cutaneous metastasis of endometrial carcinoma: a case report and literature review. J Clin Gynecol Obstet. 2012;1:19-23.
- Stonard CM, Manek S. Cutaneous metastasis from an endometrial carcinoma: a case history and review of the literature. Histopathology. 2003;43:201-203
- Damewood MD, Rosenshein NB, Grumbine FC, et al. Cutaneous metastasis of endometrial carcinoma. Cancer. 1980;46:1471-1477.
- Atallah D, el Kassis N, Lutfallah F, et al. Cutaneous metastasis in endometrial cancer: once in a blue moon—case report. World J Surg Oncol. 2014;12:86.
- Temkin SM, Hellman M, Lee YC, et al. Surgical resection of vulvar metastases of endometrial cancer: a presentation of two cases. J Low Genit Tract Dis. 2007;11:118-121.
- Kushner DM, Lurain JR, Fu TS, et al. Endometrial adenocarcinoma metastatic to the scalp: case report and literature review. Gynecol Oncol. 1997;65:530-533.
- El M’rabet FZ, Hottinger A, George AC. Cutaneous metastasis of endometrial carcinoma: a case report and literature review. J Clin Gynecol Obstet. 2012;1:19-23.
- Stonard CM, Manek S. Cutaneous metastasis from an endometrial carcinoma: a case history and review of the literature. Histopathology. 2003;43:201-203
- Damewood MD, Rosenshein NB, Grumbine FC, et al. Cutaneous metastasis of endometrial carcinoma. Cancer. 1980;46:1471-1477.
Practice Points
- Cutaneous metastases of endometrial carcinoma are extremely rare and typically present in areas of direct local spread.
- As with other cutaneous metastases, lesions often are nonspecific, making history and histopathology essential for diagnosis.