User login
A Veteran Presenting With Leg Swelling, Dyspnea, and Proteinuria
*This article has been corrected to include a missing author.
Case Presentation. A 63-year-old male with well-controlled HIV (CD4 count 757, undetectable viral load), epilepsy, and hypertension presented to the VA Boston Healthcare System (VABHS) emergency department with 1 week of bilateral leg swelling and exertional shortness of breath. He reported having no fever, cough, chest pain, pain with inspiration and orthopnea. There was no personal or family history of pulmonary embolism. He reported weight gain but was unable to quantify how much. He also reported flare up of chronic knee pain, without swelling for which he had taken up to 4 tablets of naproxen daily for several weeks. His physical examination was notable for a heart rate of 105 beats per minute and bilateral pitting edema to his knees. Laboratory testing revealed a creatinine level of 2.5 mg/dL, which was increased from a baseline of 1.0 mg/dL (Table 1), and a urine protein-to-creatinine ratio of 7.8 mg/mg (Table 2). A renal ultrasound showed normal-sized kidneys without hydronephrosis or obstructing renal calculi. The patient was admitted for further workup of his dyspnea and acute kidney injury.
► Jonathan Li, MD, Chief Medical Resident, VABHS and Beth Israel Deaconess Medical Center (BIDMC). Dr. William, based on the degree of proteinuria and edema, a diagnosis of nephrotic syndrome was made. How is nephrotic syndrome defined, and how is it distinguished from glomerulonephritis?
► Jeffrey William, MD, Nephrologist, BIDMC, Assistant Professor of Medicine, Harvard Medical School. The pathophysiology of nephrotic disease and glomerulonephritis are quite distinct, resulting in symptoms and systemic manifestations that only slightly overlap. Glomerulonephritis is characterized by inflammation of the endothelial cells of the trilayered glomerular capillary, with a resulting active urine sediment with red blood cells, white blood cells, and casts. Nephrotic syndrome mostly affects the visceral epithelial cells of the glomerular capillary, commonly referred to as podocytes, and hence, the urine sediment in nephrotic disease is often inactive. Patients with nephrotic syndrome have nephrotic-range proteinuria (excretion of > 3.5 g per 24 h or a spot urine protein-creatinine ratio > 3.5 g in the steady state) and both hypoalbuminemia (< 3 g/dL) and peripheral edema. Lipiduria and hyperlipidemia are common findings in nephrotic syndrome but are not required for a clinical diagnosis.1 In contrast, glomerulonephritis is defined by a constellation of findings that include renal insufficiency (often indicated by an elevation in blood urea nitrogen and creatinine), hypertension, hematuria, and subnephrotic range proteinuria. In practice, patients may fulfill criteria of both nephrotic and nephritic syndromes, but the preponderance of clinical evidence often points one way or the other. In this case, nephrotic syndrome was diagnosed based on the urine protein-to-creatinine ratio of 7.8 mg/mg, hypoalbuminemia, and edema.
► Dr. Li. What would be your first-line workup for evaluation of the etiology of this patient’s nephrotic syndrome?
► Dr. William. Rather than memorizing a list of etiologies of nephrotic syndrome, it is essential to consider the pathophysiology of heavy proteinuria. Though the glomerular filtration barrier is extremely complex and defects in any component can cause proteinuria, disruption of the podocyte is often involved. Common disease processes that chiefly target the podocyte include minimal change disease, primary focal and segmental glomerulosclerosis (FSGS), and membranous nephropathy, all by differing mechanisms. Minimal change disease and idiopathic/primary FSGS are increasingly thought to be at differing points on a spectrum of the same disease.2 Secondary FSGS, on the other hand, is a progressive disease, commonly resulting from longstanding hypertension, diabetes mellitus, and obesity in adults. Membranous nephropathy can also be either primary or secondary. Primary membranous nephropathy is chiefly caused by a circulating IgG4 antibody to the podocyte membrane antigen PLA2R (M-type phospholipase A2 receptor), whereas secondary membranous nephropathy can be caused by a variety of systemic etiologies, including autoimmune disease (eg, systemic lupus erythematosus), certain malignancies, chronic infections (eg, hepatitis B and C), and many medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).3-5 Paraprotein deposition diseases can also cause glomerular damage leading to nephrotic-range proteinuria.
Given these potential diagnoses, a careful history should be taken to assess exposures and recent medication use. Urine sediment evaluation is essential in the evaluation of nephrotic syndrome to determine if there is an underlying nephritic process. Select serologies may be sent to look for autoimmune disease, such as systemic lupus erythematosus and common viral exposures like hepatitis B or C. Serum and urine protein electrophoreses would be appropriate initial tests of suspected paraprotein-related diseases. Other serologies, such as antineutrophil cytoplasmic antibodies or antiglomerular basement membrane antibodies, would not necessarily be indicated here given the lack of hematuria and presence of nephrotic-range proteinuria.
► Dr. Li. The initial evaluation was notable for an erythrocyte sedimentation rate > 120 (mm/h) and a weakly positive antinuclear antibody (ANA) titer of 1:40. The remainder of his initial workup did not reveal an etiology for his nephrotic syndrome (Table 3).
Dr. William, is there a role for starting urgent empiric steroids in nephrotic syndrome while workup is ongoing? If so, do the severity of proteinuria and/or symptoms play a role or is this determination based on something else?
► Dr. William. Edema is a primary symptom of nephrotic syndrome and can often be managed with diuretics alone. If a clear medication-mediated cause is suspected, discontinuation of this agent may result in spontaneous improvement without steroid treatment. However,in cases where an etiology is unclear and there are serious thrombotic complications requiring anticoagulation, and a renal biopsy is deemed to be too risky, then empiric steroid therapy may be necessary. Children with new-onset nephrotic syndrome are presumed to have minimal change disease, given its prevalence in this patient population, and are often given empiric steroids without obtaining a renal biopsy. However, in the adult population, a renal biopsy can typically be performed quickly and safely, with pathology results interpreted within days. In this patient, since a diagnosis was unclear and there was no contraindication to renal biopsy, a biopsy should be obtained before consideration of steroids.
► Dr. Li. Steroids were deferred in anticipation of renal biopsy, which showed stage I membranous nephropathy, suggestive of membranous lupus nephritis Class V. The deposits were strongly reactive for immunoglobuline G (IgG), IgA, and complement 1q (C1q), showed co-dominant staining for IgG1, IgG2, and IgG3, and were weakly positive for the PLA2 receptor. Focal intimal arteritis in a small interlobular vessel was seen.
Dr. William, the pathology returned suggestive of lupus nephritis. Does the overall clinical picture fit with lupus nephritis?
► Dr. William. Given the history and a rather low ANA, the diagnosis of lupus nephritis seems unlikely. The lack of IgG4 and PLA2R staining in the biopsy suggests that this membranous pattern on the biopsy is likely to be secondary to a systemic etiology, but further investigation should be pursued.
► Dr. Li. The patient was discharged after the biopsy with a planned outpatient nephrology follow-up to discuss results and treatment. He was prescribed an oral diuretic, and his symptoms improved. Several days after discharge, he developed blurry vision and was evaluated in the Ophthalmology clinic. On fundoscopy, he was found to have acute papillitis, a form of optic neuritis. As part of initial evaluation of infectious etiologies of papillitis, ophthalmology recommended testing for syphilis.
Dr. Strymish, when we are considering secondary syphilis, what is the recommended approach to diagnostic testing?
► Judith Strymish, MD, Infectious Diseases, BIDMC, Assistant Professor of Medicine, Harvard Medical School. The diagnosis of syphilis is usually made through serologic testing of blood specimens. Methods that detect the spirochete directly like dark-field smears are not readily available. Serologic tests include treponemal tests (eg, Treponema pallidum particle agglutination assay [TPPA]) and nontreponemal tests (eg, rapid plasma reagin [RPR]). One needs a confirmatory test because either test is associated with false positives. Either test can be done first. Most laboratories, including those at VABHS are now performing treponemal tests first as these have become more cost-effective.6 The TPPA treponemal test was found to have a lower false negative rate in primary syphilis compared with that of nontreponemal tests.7 Nontreponemal tests can be followed for response to therapy. If a patient has a history of treated syphilis, a nontreponemal test should be sent, since the treponemal test will remain positive for life.
If there is clinical concern for neurosyphilis, cerebrospinal fluid fluorescent (CSF) treponemal antibody needs to be sampled and sent for the nontreponemal venereal disease research laboratory (VDRL) test. The VDRL is highly specific for neurosyphilis but not as sensitive. Cerebrospinal fluid fluorescent treponemal antibody (CSF FTA) may also be sent; it is very sensitive but not very specific for neurosyphilis.
► Dr. Li. An RPR returned positive at 1:512 (was negative 14 months prior on a routine screening test), with positive reflex TPPA (Table 4). A diagnosis of secondary syphilis was made. Dr. Strymish, at this point, what additional testing and treatment is necessary?
► Dr. Strymish. With papillitis and a very high RPR, we need to assume that he has ophthalmic syphilis. This can occur in any stage of syphilis, but his eye findings and high RPR are consistent with secondary syphilis. Ophthalmic syphilis has been on the upswing, even more than is expected with recent increases in syphilis cases.8 Ophthalmic syphilis is considered a form of neurosyphilis. A lumbar puncture and treatment for neurosyphilis is recommended.9,10
► Dr. Li. A lumbar puncture was performed, and his CSF was VDRL positive. This confirmed a diagnosis of neurosyphilis (Table 4). The patient was treated for neurosyphilis with IV penicillin. The patient shared that he had episodes of unprotected oral sexual activity within the past year and approximately 1 year ago, he came in close contact (but no sexual activity) with a person who had a rash consistent with syphilis.Dr. William, syphilis would be a potential unifying diagnosis of his renal and ophthalmologic manifestations. Is syphilis known to cause membranous nephropathy?
► Dr. William. Though it is uncommon, the nephrotic syndrome is a well-described complication of secondary syphilis.11,12 Syphilis has been shown to cause nephrotic syndrome in a variety of ways. Case reports abound linking syphilis to minimal change disease and other glomerular diseases.13,14 A case report from 1993 shows a membranous pattern of glomerular disease similar to this case.15 As a form of secondary membranous nephropathy, the immunofluorescence pattern can demonstrate staining similar to the “full house” seen in lupus nephritis (IgA, IgM, and C1q, in addition to IgG and C3).16 This explains the initial interpretation of this patient’s biopsy, as lupus nephritis would be a much more common etiology of secondary membranous nephropathy than is acute syphilis with this immunofluorescence pattern. However, the data in this case are highly suggestive of a causal relationship between secondary syphilis and membranous nephropathy.
► Dr. Li. Dr. Strymish, how should this patient be screened for syphilis reinfection, and at what intervals would you recommend?
► Dr. Strymish. He will need follow-up testing to make sure that his syphilis is effectively treated. If CSF pleocytosis was present initially, a CSF examination should be repeated every 6 months until the cell count is normal. He will also need follow-up for normalization of his RPR. Persons with HIV infection and primary or secondary syphilis should be evaluated clinically and serologically for treatment failure at 3, 6, 9, 12, and 24 months after therapy according to US Centers for Disease Control and Prevention guidelines.9
His treponemal test for syphilis will likely stay positive for life. His RPR should decrease significantly with effective treatment. It makes sense to screen with RPR alone as long as he continues to have risk factors for acquiring syphilis. Routine syphilis testing is recommended for pregnant women, sexually active men who have sex with men, sexually active persons with HIV, and persons taking PrEP (pre-exposure prophylaxis) for HIV prevention. He should be screened at least yearly for syphilis.

► Dr. Li. Over the next several months, the patient’s creatinine normalized and his proteinuria resolved. His vision recovered, and he has had no further ophthalmologic complications.
Dr. William, what is his long-term renal prognosis? Do you expect that his acute episode of membranous nephropathy will have permanent effects on his renal function?
► Dr. William. His rapid response to therapy for neurosyphilis provides evidence for this etiology of his renal dysfunction and glomerulonephritis. His long-term prognosis is quite good if the syphilis is the only reason for him to have renal disease. The renal damage is often reversible in these cases. However, given his prior extensive NSAID exposure and history of hypertension, he may be at higher risk for chronic kidney disease than an otherwise healthy patient, especially after an episode of acute kidney injury. Therefore, his renal function should continue to be monitored as an outpatient.
Acknowledgments
The authors thank this veteran for sharing his story and allowing us to learn from this unusual case for the benefit of our future patients.
1. Rennke H, Denker BM. Renal Pathophysiology: The Essentials. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2014.
2. Maas RJ, Deegens JK, Smeets B, Moeller MJ, Wetzels JF. Minimal change disease and idiopathic FSGS: manifestations of the same disease. Nat Rev Nephrol. 2016;12(12):768-776.
3. Beck LH Jr, Bonegio RG, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361(1):11-21.
4. Rennke HG. Secondary membranoproliferative glomerulonephritis. Kidney Int. 1995;47(2):643-656.
5. Nawaz FA, Larsen CP, Troxell ML. Membranous nephropathy and nonsteroidal anti-inflammatory agents. Am J Kidney Dis. 2013;62(5):1012-1017.
6. Pillay A. Centers for Disease Control and Prevention Syphilis Summit—Diagnostics and laboratory issues. Sex Transm Dis. 2018;45(9S)(suppl 1):S13-S16.
7. Levett PN, Fonseca K, Tsang RS, et al. Canadian Public Health Laboratory Network laboratory guidelines for the use of serological tests (excluding point-of-care tests) for the diagnosis of syphilis in Canada. Can J Infect Dis Med Microbiol. 2015;26(suppl A):6A-12A.
8. Oliver SE, Aubin M, Atwell L, et al. Ocular syphilis—eight jurisdictions, United States, 2014-2015. MMWR Morb Mortal Wkly Rep. 2016;65(43):1185-1188.
9. Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recommendations and Reports 2015;64(RR3):1-137. [Erratum in MMWR Recomm Rep. 2015;64(33):924.]
10. US Centers for Disease Control and Prevention. Clinical advisory: ocular syphilis in the United States. https://www.cdc.gov/std/syphilis/clinicaladvisoryos2015.htm. Updated March 24, 2016. Accessed August 12, 2019.
11. Braunstein GD, Lewis EJ, Galvanek EG, Hamilton A, Bell WR. The nephrotic syndrome associated with secondary syphilis: an immune deposit disease. Am J Med. 1970;48:643-648.1.
12. Handoko ML, Duijvestein M, Scheepstra CG, de Fijter CW. Syphilis: a reversible cause of nephrotic syndrome. BMJ Case Rep. 2013;2013:pii:bcr2012008279
13. Krane NK, Espenan P, Walker PD, Bergman SM, Wallin JD. Renal disease and syphilis: a report of nephrotic syndrome with minimal change disease. Am J Kidney Dis. 1987;9(2):176-179.
14. Bhorade MS, Carag HB, Lee HJ, Potter EV, Dunea G. Nephropathy of secondary syphilis: a clinical and pathological spectrum. JAMA. 1971;216(7):1159-1166.
15. Hunte W, al-Ghraoui F, Cohen RJ. Secondary syphilis and the nephrotic syndrome. J Am Soc Nephrol. 1993;3(7):1351-1355.
16. Gamble CN, Reardan JB. Immunopathogenesis of syphilitic glomerulonephritis. Elution of antitreponemal antibody from glomerular immune-complex deposits. N Engl J Med. 1975;292(9):449-454.
*This article has been corrected to include a missing author.
Case Presentation. A 63-year-old male with well-controlled HIV (CD4 count 757, undetectable viral load), epilepsy, and hypertension presented to the VA Boston Healthcare System (VABHS) emergency department with 1 week of bilateral leg swelling and exertional shortness of breath. He reported having no fever, cough, chest pain, pain with inspiration and orthopnea. There was no personal or family history of pulmonary embolism. He reported weight gain but was unable to quantify how much. He also reported flare up of chronic knee pain, without swelling for which he had taken up to 4 tablets of naproxen daily for several weeks. His physical examination was notable for a heart rate of 105 beats per minute and bilateral pitting edema to his knees. Laboratory testing revealed a creatinine level of 2.5 mg/dL, which was increased from a baseline of 1.0 mg/dL (Table 1), and a urine protein-to-creatinine ratio of 7.8 mg/mg (Table 2). A renal ultrasound showed normal-sized kidneys without hydronephrosis or obstructing renal calculi. The patient was admitted for further workup of his dyspnea and acute kidney injury.
► Jonathan Li, MD, Chief Medical Resident, VABHS and Beth Israel Deaconess Medical Center (BIDMC). Dr. William, based on the degree of proteinuria and edema, a diagnosis of nephrotic syndrome was made. How is nephrotic syndrome defined, and how is it distinguished from glomerulonephritis?
► Jeffrey William, MD, Nephrologist, BIDMC, Assistant Professor of Medicine, Harvard Medical School. The pathophysiology of nephrotic disease and glomerulonephritis are quite distinct, resulting in symptoms and systemic manifestations that only slightly overlap. Glomerulonephritis is characterized by inflammation of the endothelial cells of the trilayered glomerular capillary, with a resulting active urine sediment with red blood cells, white blood cells, and casts. Nephrotic syndrome mostly affects the visceral epithelial cells of the glomerular capillary, commonly referred to as podocytes, and hence, the urine sediment in nephrotic disease is often inactive. Patients with nephrotic syndrome have nephrotic-range proteinuria (excretion of > 3.5 g per 24 h or a spot urine protein-creatinine ratio > 3.5 g in the steady state) and both hypoalbuminemia (< 3 g/dL) and peripheral edema. Lipiduria and hyperlipidemia are common findings in nephrotic syndrome but are not required for a clinical diagnosis.1 In contrast, glomerulonephritis is defined by a constellation of findings that include renal insufficiency (often indicated by an elevation in blood urea nitrogen and creatinine), hypertension, hematuria, and subnephrotic range proteinuria. In practice, patients may fulfill criteria of both nephrotic and nephritic syndromes, but the preponderance of clinical evidence often points one way or the other. In this case, nephrotic syndrome was diagnosed based on the urine protein-to-creatinine ratio of 7.8 mg/mg, hypoalbuminemia, and edema.
► Dr. Li. What would be your first-line workup for evaluation of the etiology of this patient’s nephrotic syndrome?
► Dr. William. Rather than memorizing a list of etiologies of nephrotic syndrome, it is essential to consider the pathophysiology of heavy proteinuria. Though the glomerular filtration barrier is extremely complex and defects in any component can cause proteinuria, disruption of the podocyte is often involved. Common disease processes that chiefly target the podocyte include minimal change disease, primary focal and segmental glomerulosclerosis (FSGS), and membranous nephropathy, all by differing mechanisms. Minimal change disease and idiopathic/primary FSGS are increasingly thought to be at differing points on a spectrum of the same disease.2 Secondary FSGS, on the other hand, is a progressive disease, commonly resulting from longstanding hypertension, diabetes mellitus, and obesity in adults. Membranous nephropathy can also be either primary or secondary. Primary membranous nephropathy is chiefly caused by a circulating IgG4 antibody to the podocyte membrane antigen PLA2R (M-type phospholipase A2 receptor), whereas secondary membranous nephropathy can be caused by a variety of systemic etiologies, including autoimmune disease (eg, systemic lupus erythematosus), certain malignancies, chronic infections (eg, hepatitis B and C), and many medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).3-5 Paraprotein deposition diseases can also cause glomerular damage leading to nephrotic-range proteinuria.
Given these potential diagnoses, a careful history should be taken to assess exposures and recent medication use. Urine sediment evaluation is essential in the evaluation of nephrotic syndrome to determine if there is an underlying nephritic process. Select serologies may be sent to look for autoimmune disease, such as systemic lupus erythematosus and common viral exposures like hepatitis B or C. Serum and urine protein electrophoreses would be appropriate initial tests of suspected paraprotein-related diseases. Other serologies, such as antineutrophil cytoplasmic antibodies or antiglomerular basement membrane antibodies, would not necessarily be indicated here given the lack of hematuria and presence of nephrotic-range proteinuria.
► Dr. Li. The initial evaluation was notable for an erythrocyte sedimentation rate > 120 (mm/h) and a weakly positive antinuclear antibody (ANA) titer of 1:40. The remainder of his initial workup did not reveal an etiology for his nephrotic syndrome (Table 3).
Dr. William, is there a role for starting urgent empiric steroids in nephrotic syndrome while workup is ongoing? If so, do the severity of proteinuria and/or symptoms play a role or is this determination based on something else?
► Dr. William. Edema is a primary symptom of nephrotic syndrome and can often be managed with diuretics alone. If a clear medication-mediated cause is suspected, discontinuation of this agent may result in spontaneous improvement without steroid treatment. However,in cases where an etiology is unclear and there are serious thrombotic complications requiring anticoagulation, and a renal biopsy is deemed to be too risky, then empiric steroid therapy may be necessary. Children with new-onset nephrotic syndrome are presumed to have minimal change disease, given its prevalence in this patient population, and are often given empiric steroids without obtaining a renal biopsy. However, in the adult population, a renal biopsy can typically be performed quickly and safely, with pathology results interpreted within days. In this patient, since a diagnosis was unclear and there was no contraindication to renal biopsy, a biopsy should be obtained before consideration of steroids.
► Dr. Li. Steroids were deferred in anticipation of renal biopsy, which showed stage I membranous nephropathy, suggestive of membranous lupus nephritis Class V. The deposits were strongly reactive for immunoglobuline G (IgG), IgA, and complement 1q (C1q), showed co-dominant staining for IgG1, IgG2, and IgG3, and were weakly positive for the PLA2 receptor. Focal intimal arteritis in a small interlobular vessel was seen.
Dr. William, the pathology returned suggestive of lupus nephritis. Does the overall clinical picture fit with lupus nephritis?
► Dr. William. Given the history and a rather low ANA, the diagnosis of lupus nephritis seems unlikely. The lack of IgG4 and PLA2R staining in the biopsy suggests that this membranous pattern on the biopsy is likely to be secondary to a systemic etiology, but further investigation should be pursued.
► Dr. Li. The patient was discharged after the biopsy with a planned outpatient nephrology follow-up to discuss results and treatment. He was prescribed an oral diuretic, and his symptoms improved. Several days after discharge, he developed blurry vision and was evaluated in the Ophthalmology clinic. On fundoscopy, he was found to have acute papillitis, a form of optic neuritis. As part of initial evaluation of infectious etiologies of papillitis, ophthalmology recommended testing for syphilis.
Dr. Strymish, when we are considering secondary syphilis, what is the recommended approach to diagnostic testing?
► Judith Strymish, MD, Infectious Diseases, BIDMC, Assistant Professor of Medicine, Harvard Medical School. The diagnosis of syphilis is usually made through serologic testing of blood specimens. Methods that detect the spirochete directly like dark-field smears are not readily available. Serologic tests include treponemal tests (eg, Treponema pallidum particle agglutination assay [TPPA]) and nontreponemal tests (eg, rapid plasma reagin [RPR]). One needs a confirmatory test because either test is associated with false positives. Either test can be done first. Most laboratories, including those at VABHS are now performing treponemal tests first as these have become more cost-effective.6 The TPPA treponemal test was found to have a lower false negative rate in primary syphilis compared with that of nontreponemal tests.7 Nontreponemal tests can be followed for response to therapy. If a patient has a history of treated syphilis, a nontreponemal test should be sent, since the treponemal test will remain positive for life.
If there is clinical concern for neurosyphilis, cerebrospinal fluid fluorescent (CSF) treponemal antibody needs to be sampled and sent for the nontreponemal venereal disease research laboratory (VDRL) test. The VDRL is highly specific for neurosyphilis but not as sensitive. Cerebrospinal fluid fluorescent treponemal antibody (CSF FTA) may also be sent; it is very sensitive but not very specific for neurosyphilis.
► Dr. Li. An RPR returned positive at 1:512 (was negative 14 months prior on a routine screening test), with positive reflex TPPA (Table 4). A diagnosis of secondary syphilis was made. Dr. Strymish, at this point, what additional testing and treatment is necessary?
► Dr. Strymish. With papillitis and a very high RPR, we need to assume that he has ophthalmic syphilis. This can occur in any stage of syphilis, but his eye findings and high RPR are consistent with secondary syphilis. Ophthalmic syphilis has been on the upswing, even more than is expected with recent increases in syphilis cases.8 Ophthalmic syphilis is considered a form of neurosyphilis. A lumbar puncture and treatment for neurosyphilis is recommended.9,10
► Dr. Li. A lumbar puncture was performed, and his CSF was VDRL positive. This confirmed a diagnosis of neurosyphilis (Table 4). The patient was treated for neurosyphilis with IV penicillin. The patient shared that he had episodes of unprotected oral sexual activity within the past year and approximately 1 year ago, he came in close contact (but no sexual activity) with a person who had a rash consistent with syphilis.Dr. William, syphilis would be a potential unifying diagnosis of his renal and ophthalmologic manifestations. Is syphilis known to cause membranous nephropathy?
► Dr. William. Though it is uncommon, the nephrotic syndrome is a well-described complication of secondary syphilis.11,12 Syphilis has been shown to cause nephrotic syndrome in a variety of ways. Case reports abound linking syphilis to minimal change disease and other glomerular diseases.13,14 A case report from 1993 shows a membranous pattern of glomerular disease similar to this case.15 As a form of secondary membranous nephropathy, the immunofluorescence pattern can demonstrate staining similar to the “full house” seen in lupus nephritis (IgA, IgM, and C1q, in addition to IgG and C3).16 This explains the initial interpretation of this patient’s biopsy, as lupus nephritis would be a much more common etiology of secondary membranous nephropathy than is acute syphilis with this immunofluorescence pattern. However, the data in this case are highly suggestive of a causal relationship between secondary syphilis and membranous nephropathy.
► Dr. Li. Dr. Strymish, how should this patient be screened for syphilis reinfection, and at what intervals would you recommend?
► Dr. Strymish. He will need follow-up testing to make sure that his syphilis is effectively treated. If CSF pleocytosis was present initially, a CSF examination should be repeated every 6 months until the cell count is normal. He will also need follow-up for normalization of his RPR. Persons with HIV infection and primary or secondary syphilis should be evaluated clinically and serologically for treatment failure at 3, 6, 9, 12, and 24 months after therapy according to US Centers for Disease Control and Prevention guidelines.9
His treponemal test for syphilis will likely stay positive for life. His RPR should decrease significantly with effective treatment. It makes sense to screen with RPR alone as long as he continues to have risk factors for acquiring syphilis. Routine syphilis testing is recommended for pregnant women, sexually active men who have sex with men, sexually active persons with HIV, and persons taking PrEP (pre-exposure prophylaxis) for HIV prevention. He should be screened at least yearly for syphilis.
► Dr. Li. Over the next several months, the patient’s creatinine normalized and his proteinuria resolved. His vision recovered, and he has had no further ophthalmologic complications.
Dr. William, what is his long-term renal prognosis? Do you expect that his acute episode of membranous nephropathy will have permanent effects on his renal function?
► Dr. William. His rapid response to therapy for neurosyphilis provides evidence for this etiology of his renal dysfunction and glomerulonephritis. His long-term prognosis is quite good if the syphilis is the only reason for him to have renal disease. The renal damage is often reversible in these cases. However, given his prior extensive NSAID exposure and history of hypertension, he may be at higher risk for chronic kidney disease than an otherwise healthy patient, especially after an episode of acute kidney injury. Therefore, his renal function should continue to be monitored as an outpatient.
Acknowledgments
The authors thank this veteran for sharing his story and allowing us to learn from this unusual case for the benefit of our future patients.
*This article has been corrected to include a missing author.
Case Presentation. A 63-year-old male with well-controlled HIV (CD4 count 757, undetectable viral load), epilepsy, and hypertension presented to the VA Boston Healthcare System (VABHS) emergency department with 1 week of bilateral leg swelling and exertional shortness of breath. He reported having no fever, cough, chest pain, pain with inspiration and orthopnea. There was no personal or family history of pulmonary embolism. He reported weight gain but was unable to quantify how much. He also reported flare up of chronic knee pain, without swelling for which he had taken up to 4 tablets of naproxen daily for several weeks. His physical examination was notable for a heart rate of 105 beats per minute and bilateral pitting edema to his knees. Laboratory testing revealed a creatinine level of 2.5 mg/dL, which was increased from a baseline of 1.0 mg/dL (Table 1), and a urine protein-to-creatinine ratio of 7.8 mg/mg (Table 2). A renal ultrasound showed normal-sized kidneys without hydronephrosis or obstructing renal calculi. The patient was admitted for further workup of his dyspnea and acute kidney injury.
► Jonathan Li, MD, Chief Medical Resident, VABHS and Beth Israel Deaconess Medical Center (BIDMC). Dr. William, based on the degree of proteinuria and edema, a diagnosis of nephrotic syndrome was made. How is nephrotic syndrome defined, and how is it distinguished from glomerulonephritis?
► Jeffrey William, MD, Nephrologist, BIDMC, Assistant Professor of Medicine, Harvard Medical School. The pathophysiology of nephrotic disease and glomerulonephritis are quite distinct, resulting in symptoms and systemic manifestations that only slightly overlap. Glomerulonephritis is characterized by inflammation of the endothelial cells of the trilayered glomerular capillary, with a resulting active urine sediment with red blood cells, white blood cells, and casts. Nephrotic syndrome mostly affects the visceral epithelial cells of the glomerular capillary, commonly referred to as podocytes, and hence, the urine sediment in nephrotic disease is often inactive. Patients with nephrotic syndrome have nephrotic-range proteinuria (excretion of > 3.5 g per 24 h or a spot urine protein-creatinine ratio > 3.5 g in the steady state) and both hypoalbuminemia (< 3 g/dL) and peripheral edema. Lipiduria and hyperlipidemia are common findings in nephrotic syndrome but are not required for a clinical diagnosis.1 In contrast, glomerulonephritis is defined by a constellation of findings that include renal insufficiency (often indicated by an elevation in blood urea nitrogen and creatinine), hypertension, hematuria, and subnephrotic range proteinuria. In practice, patients may fulfill criteria of both nephrotic and nephritic syndromes, but the preponderance of clinical evidence often points one way or the other. In this case, nephrotic syndrome was diagnosed based on the urine protein-to-creatinine ratio of 7.8 mg/mg, hypoalbuminemia, and edema.
► Dr. Li. What would be your first-line workup for evaluation of the etiology of this patient’s nephrotic syndrome?
► Dr. William. Rather than memorizing a list of etiologies of nephrotic syndrome, it is essential to consider the pathophysiology of heavy proteinuria. Though the glomerular filtration barrier is extremely complex and defects in any component can cause proteinuria, disruption of the podocyte is often involved. Common disease processes that chiefly target the podocyte include minimal change disease, primary focal and segmental glomerulosclerosis (FSGS), and membranous nephropathy, all by differing mechanisms. Minimal change disease and idiopathic/primary FSGS are increasingly thought to be at differing points on a spectrum of the same disease.2 Secondary FSGS, on the other hand, is a progressive disease, commonly resulting from longstanding hypertension, diabetes mellitus, and obesity in adults. Membranous nephropathy can also be either primary or secondary. Primary membranous nephropathy is chiefly caused by a circulating IgG4 antibody to the podocyte membrane antigen PLA2R (M-type phospholipase A2 receptor), whereas secondary membranous nephropathy can be caused by a variety of systemic etiologies, including autoimmune disease (eg, systemic lupus erythematosus), certain malignancies, chronic infections (eg, hepatitis B and C), and many medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).3-5 Paraprotein deposition diseases can also cause glomerular damage leading to nephrotic-range proteinuria.
Given these potential diagnoses, a careful history should be taken to assess exposures and recent medication use. Urine sediment evaluation is essential in the evaluation of nephrotic syndrome to determine if there is an underlying nephritic process. Select serologies may be sent to look for autoimmune disease, such as systemic lupus erythematosus and common viral exposures like hepatitis B or C. Serum and urine protein electrophoreses would be appropriate initial tests of suspected paraprotein-related diseases. Other serologies, such as antineutrophil cytoplasmic antibodies or antiglomerular basement membrane antibodies, would not necessarily be indicated here given the lack of hematuria and presence of nephrotic-range proteinuria.
► Dr. Li. The initial evaluation was notable for an erythrocyte sedimentation rate > 120 (mm/h) and a weakly positive antinuclear antibody (ANA) titer of 1:40. The remainder of his initial workup did not reveal an etiology for his nephrotic syndrome (Table 3).
Dr. William, is there a role for starting urgent empiric steroids in nephrotic syndrome while workup is ongoing? If so, do the severity of proteinuria and/or symptoms play a role or is this determination based on something else?
► Dr. William. Edema is a primary symptom of nephrotic syndrome and can often be managed with diuretics alone. If a clear medication-mediated cause is suspected, discontinuation of this agent may result in spontaneous improvement without steroid treatment. However,in cases where an etiology is unclear and there are serious thrombotic complications requiring anticoagulation, and a renal biopsy is deemed to be too risky, then empiric steroid therapy may be necessary. Children with new-onset nephrotic syndrome are presumed to have minimal change disease, given its prevalence in this patient population, and are often given empiric steroids without obtaining a renal biopsy. However, in the adult population, a renal biopsy can typically be performed quickly and safely, with pathology results interpreted within days. In this patient, since a diagnosis was unclear and there was no contraindication to renal biopsy, a biopsy should be obtained before consideration of steroids.
► Dr. Li. Steroids were deferred in anticipation of renal biopsy, which showed stage I membranous nephropathy, suggestive of membranous lupus nephritis Class V. The deposits were strongly reactive for immunoglobuline G (IgG), IgA, and complement 1q (C1q), showed co-dominant staining for IgG1, IgG2, and IgG3, and were weakly positive for the PLA2 receptor. Focal intimal arteritis in a small interlobular vessel was seen.
Dr. William, the pathology returned suggestive of lupus nephritis. Does the overall clinical picture fit with lupus nephritis?
► Dr. William. Given the history and a rather low ANA, the diagnosis of lupus nephritis seems unlikely. The lack of IgG4 and PLA2R staining in the biopsy suggests that this membranous pattern on the biopsy is likely to be secondary to a systemic etiology, but further investigation should be pursued.
► Dr. Li. The patient was discharged after the biopsy with a planned outpatient nephrology follow-up to discuss results and treatment. He was prescribed an oral diuretic, and his symptoms improved. Several days after discharge, he developed blurry vision and was evaluated in the Ophthalmology clinic. On fundoscopy, he was found to have acute papillitis, a form of optic neuritis. As part of initial evaluation of infectious etiologies of papillitis, ophthalmology recommended testing for syphilis.
Dr. Strymish, when we are considering secondary syphilis, what is the recommended approach to diagnostic testing?
► Judith Strymish, MD, Infectious Diseases, BIDMC, Assistant Professor of Medicine, Harvard Medical School. The diagnosis of syphilis is usually made through serologic testing of blood specimens. Methods that detect the spirochete directly like dark-field smears are not readily available. Serologic tests include treponemal tests (eg, Treponema pallidum particle agglutination assay [TPPA]) and nontreponemal tests (eg, rapid plasma reagin [RPR]). One needs a confirmatory test because either test is associated with false positives. Either test can be done first. Most laboratories, including those at VABHS are now performing treponemal tests first as these have become more cost-effective.6 The TPPA treponemal test was found to have a lower false negative rate in primary syphilis compared with that of nontreponemal tests.7 Nontreponemal tests can be followed for response to therapy. If a patient has a history of treated syphilis, a nontreponemal test should be sent, since the treponemal test will remain positive for life.
If there is clinical concern for neurosyphilis, cerebrospinal fluid fluorescent (CSF) treponemal antibody needs to be sampled and sent for the nontreponemal venereal disease research laboratory (VDRL) test. The VDRL is highly specific for neurosyphilis but not as sensitive. Cerebrospinal fluid fluorescent treponemal antibody (CSF FTA) may also be sent; it is very sensitive but not very specific for neurosyphilis.
► Dr. Li. An RPR returned positive at 1:512 (was negative 14 months prior on a routine screening test), with positive reflex TPPA (Table 4). A diagnosis of secondary syphilis was made. Dr. Strymish, at this point, what additional testing and treatment is necessary?
► Dr. Strymish. With papillitis and a very high RPR, we need to assume that he has ophthalmic syphilis. This can occur in any stage of syphilis, but his eye findings and high RPR are consistent with secondary syphilis. Ophthalmic syphilis has been on the upswing, even more than is expected with recent increases in syphilis cases.8 Ophthalmic syphilis is considered a form of neurosyphilis. A lumbar puncture and treatment for neurosyphilis is recommended.9,10
► Dr. Li. A lumbar puncture was performed, and his CSF was VDRL positive. This confirmed a diagnosis of neurosyphilis (Table 4). The patient was treated for neurosyphilis with IV penicillin. The patient shared that he had episodes of unprotected oral sexual activity within the past year and approximately 1 year ago, he came in close contact (but no sexual activity) with a person who had a rash consistent with syphilis.Dr. William, syphilis would be a potential unifying diagnosis of his renal and ophthalmologic manifestations. Is syphilis known to cause membranous nephropathy?
► Dr. William. Though it is uncommon, the nephrotic syndrome is a well-described complication of secondary syphilis.11,12 Syphilis has been shown to cause nephrotic syndrome in a variety of ways. Case reports abound linking syphilis to minimal change disease and other glomerular diseases.13,14 A case report from 1993 shows a membranous pattern of glomerular disease similar to this case.15 As a form of secondary membranous nephropathy, the immunofluorescence pattern can demonstrate staining similar to the “full house” seen in lupus nephritis (IgA, IgM, and C1q, in addition to IgG and C3).16 This explains the initial interpretation of this patient’s biopsy, as lupus nephritis would be a much more common etiology of secondary membranous nephropathy than is acute syphilis with this immunofluorescence pattern. However, the data in this case are highly suggestive of a causal relationship between secondary syphilis and membranous nephropathy.
► Dr. Li. Dr. Strymish, how should this patient be screened for syphilis reinfection, and at what intervals would you recommend?
► Dr. Strymish. He will need follow-up testing to make sure that his syphilis is effectively treated. If CSF pleocytosis was present initially, a CSF examination should be repeated every 6 months until the cell count is normal. He will also need follow-up for normalization of his RPR. Persons with HIV infection and primary or secondary syphilis should be evaluated clinically and serologically for treatment failure at 3, 6, 9, 12, and 24 months after therapy according to US Centers for Disease Control and Prevention guidelines.9
His treponemal test for syphilis will likely stay positive for life. His RPR should decrease significantly with effective treatment. It makes sense to screen with RPR alone as long as he continues to have risk factors for acquiring syphilis. Routine syphilis testing is recommended for pregnant women, sexually active men who have sex with men, sexually active persons with HIV, and persons taking PrEP (pre-exposure prophylaxis) for HIV prevention. He should be screened at least yearly for syphilis.
► Dr. Li. Over the next several months, the patient’s creatinine normalized and his proteinuria resolved. His vision recovered, and he has had no further ophthalmologic complications.
Dr. William, what is his long-term renal prognosis? Do you expect that his acute episode of membranous nephropathy will have permanent effects on his renal function?
► Dr. William. His rapid response to therapy for neurosyphilis provides evidence for this etiology of his renal dysfunction and glomerulonephritis. His long-term prognosis is quite good if the syphilis is the only reason for him to have renal disease. The renal damage is often reversible in these cases. However, given his prior extensive NSAID exposure and history of hypertension, he may be at higher risk for chronic kidney disease than an otherwise healthy patient, especially after an episode of acute kidney injury. Therefore, his renal function should continue to be monitored as an outpatient.
Acknowledgments
The authors thank this veteran for sharing his story and allowing us to learn from this unusual case for the benefit of our future patients.
1. Rennke H, Denker BM. Renal Pathophysiology: The Essentials. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2014.
2. Maas RJ, Deegens JK, Smeets B, Moeller MJ, Wetzels JF. Minimal change disease and idiopathic FSGS: manifestations of the same disease. Nat Rev Nephrol. 2016;12(12):768-776.
3. Beck LH Jr, Bonegio RG, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361(1):11-21.
4. Rennke HG. Secondary membranoproliferative glomerulonephritis. Kidney Int. 1995;47(2):643-656.
5. Nawaz FA, Larsen CP, Troxell ML. Membranous nephropathy and nonsteroidal anti-inflammatory agents. Am J Kidney Dis. 2013;62(5):1012-1017.
6. Pillay A. Centers for Disease Control and Prevention Syphilis Summit—Diagnostics and laboratory issues. Sex Transm Dis. 2018;45(9S)(suppl 1):S13-S16.
7. Levett PN, Fonseca K, Tsang RS, et al. Canadian Public Health Laboratory Network laboratory guidelines for the use of serological tests (excluding point-of-care tests) for the diagnosis of syphilis in Canada. Can J Infect Dis Med Microbiol. 2015;26(suppl A):6A-12A.
8. Oliver SE, Aubin M, Atwell L, et al. Ocular syphilis—eight jurisdictions, United States, 2014-2015. MMWR Morb Mortal Wkly Rep. 2016;65(43):1185-1188.
9. Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recommendations and Reports 2015;64(RR3):1-137. [Erratum in MMWR Recomm Rep. 2015;64(33):924.]
10. US Centers for Disease Control and Prevention. Clinical advisory: ocular syphilis in the United States. https://www.cdc.gov/std/syphilis/clinicaladvisoryos2015.htm. Updated March 24, 2016. Accessed August 12, 2019.
11. Braunstein GD, Lewis EJ, Galvanek EG, Hamilton A, Bell WR. The nephrotic syndrome associated with secondary syphilis: an immune deposit disease. Am J Med. 1970;48:643-648.1.
12. Handoko ML, Duijvestein M, Scheepstra CG, de Fijter CW. Syphilis: a reversible cause of nephrotic syndrome. BMJ Case Rep. 2013;2013:pii:bcr2012008279
13. Krane NK, Espenan P, Walker PD, Bergman SM, Wallin JD. Renal disease and syphilis: a report of nephrotic syndrome with minimal change disease. Am J Kidney Dis. 1987;9(2):176-179.
14. Bhorade MS, Carag HB, Lee HJ, Potter EV, Dunea G. Nephropathy of secondary syphilis: a clinical and pathological spectrum. JAMA. 1971;216(7):1159-1166.
15. Hunte W, al-Ghraoui F, Cohen RJ. Secondary syphilis and the nephrotic syndrome. J Am Soc Nephrol. 1993;3(7):1351-1355.
16. Gamble CN, Reardan JB. Immunopathogenesis of syphilitic glomerulonephritis. Elution of antitreponemal antibody from glomerular immune-complex deposits. N Engl J Med. 1975;292(9):449-454.
1. Rennke H, Denker BM. Renal Pathophysiology: The Essentials. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2014.
2. Maas RJ, Deegens JK, Smeets B, Moeller MJ, Wetzels JF. Minimal change disease and idiopathic FSGS: manifestations of the same disease. Nat Rev Nephrol. 2016;12(12):768-776.
3. Beck LH Jr, Bonegio RG, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361(1):11-21.
4. Rennke HG. Secondary membranoproliferative glomerulonephritis. Kidney Int. 1995;47(2):643-656.
5. Nawaz FA, Larsen CP, Troxell ML. Membranous nephropathy and nonsteroidal anti-inflammatory agents. Am J Kidney Dis. 2013;62(5):1012-1017.
6. Pillay A. Centers for Disease Control and Prevention Syphilis Summit—Diagnostics and laboratory issues. Sex Transm Dis. 2018;45(9S)(suppl 1):S13-S16.
7. Levett PN, Fonseca K, Tsang RS, et al. Canadian Public Health Laboratory Network laboratory guidelines for the use of serological tests (excluding point-of-care tests) for the diagnosis of syphilis in Canada. Can J Infect Dis Med Microbiol. 2015;26(suppl A):6A-12A.
8. Oliver SE, Aubin M, Atwell L, et al. Ocular syphilis—eight jurisdictions, United States, 2014-2015. MMWR Morb Mortal Wkly Rep. 2016;65(43):1185-1188.
9. Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recommendations and Reports 2015;64(RR3):1-137. [Erratum in MMWR Recomm Rep. 2015;64(33):924.]
10. US Centers for Disease Control and Prevention. Clinical advisory: ocular syphilis in the United States. https://www.cdc.gov/std/syphilis/clinicaladvisoryos2015.htm. Updated March 24, 2016. Accessed August 12, 2019.
11. Braunstein GD, Lewis EJ, Galvanek EG, Hamilton A, Bell WR. The nephrotic syndrome associated with secondary syphilis: an immune deposit disease. Am J Med. 1970;48:643-648.1.
12. Handoko ML, Duijvestein M, Scheepstra CG, de Fijter CW. Syphilis: a reversible cause of nephrotic syndrome. BMJ Case Rep. 2013;2013:pii:bcr2012008279
13. Krane NK, Espenan P, Walker PD, Bergman SM, Wallin JD. Renal disease and syphilis: a report of nephrotic syndrome with minimal change disease. Am J Kidney Dis. 1987;9(2):176-179.
14. Bhorade MS, Carag HB, Lee HJ, Potter EV, Dunea G. Nephropathy of secondary syphilis: a clinical and pathological spectrum. JAMA. 1971;216(7):1159-1166.
15. Hunte W, al-Ghraoui F, Cohen RJ. Secondary syphilis and the nephrotic syndrome. J Am Soc Nephrol. 1993;3(7):1351-1355.
16. Gamble CN, Reardan JB. Immunopathogenesis of syphilitic glomerulonephritis. Elution of antitreponemal antibody from glomerular immune-complex deposits. N Engl J Med. 1975;292(9):449-454.
Fatal Drug-Resistant Invasive Pulmonary Aspergillus fumigatus in a 56-Year-Old Immunosuppressed Man (FULL)
Historically, aspergillosis in patients with hematopoietic stem cell transplantation (HSCT) has carried a high mortality rate. However, recent data demonstrate a dramatic improvement in outcomes for patients with HSCT: 90-day survival increased from 22% before 2000 to 45% over the past 15 years.1 Improved outcomes coincide with changes in transplant immunosuppression practices, use of cross-sectional imaging for early disease identification, galactomannan screening, and the development of novel treatment options.
Voriconazole is an azole drug that blocks the synthesis of ergosterol, a vital component of the cellular membrane of fungi. Voriconazole was approved in 2002 after a clinical trial demonstrated an improvement in 50% of patients with invasive aspergillosis in the voriconazole arm vs 30% in the amphotericin B arm at 12 weeks.2 Amphotericin B is a polyene antifungal drug that binds with ergosterol, creating leaks in the cell membrane that lead to cellular demise. Voriconazole quickly became the first-line therapy for invasive aspergillosis and is recommended by both the Infectious Disease Society of American (IDSA) and the European Conference on Infections in Leukemia.3
Case Presentation
A 55-year-old man with high-risk chronic myelogenous leukemia (CML) underwent a 10 of 10 human leukocyte antigen allele and antigen-matched peripheral blood allogeneic HSCT with a myeloablative-conditioning regimen of busulfan and cyclophosphamide, along with prophylactic voriconazole, sulfamethoxazole/trimethoprim, and acyclovir. After successful engraftment (without significant neutropenia), his posttransplant course was complicated by grade 2 graft vs host disease (GVHD) of the skin, eyes, and liver, which responded well to steroids and tacrolimus. Voriconazole was continued for 5 months until immunosuppression was minimized (tacrolimus 1 mg twice daily). Two months later, the patient’s GVHD worsened, necessitating treatment at an outside hospital with high-dose prednisone (2 mg/kg/d) and cyclosporine (300 mg twice daily). Voriconazole prophylaxis was not reinitiated at that time.
One year later, at a routine follow-up appointment, the patient endorsed several weeks of malaise, weight loss, and nonproductive cough. The patient’s immunosuppression recently had been reduced to 1 mg/kg/d of prednisone and 100 mg of cyclosporine twice daily. A chest X-ray demonstrated multiple pulmonary nodules; follow-up chest computed tomography (CT) confirmed multiple nodular infiltrates with surrounding ground-glass opacities suspicious with a fungal infection (Figure 1). 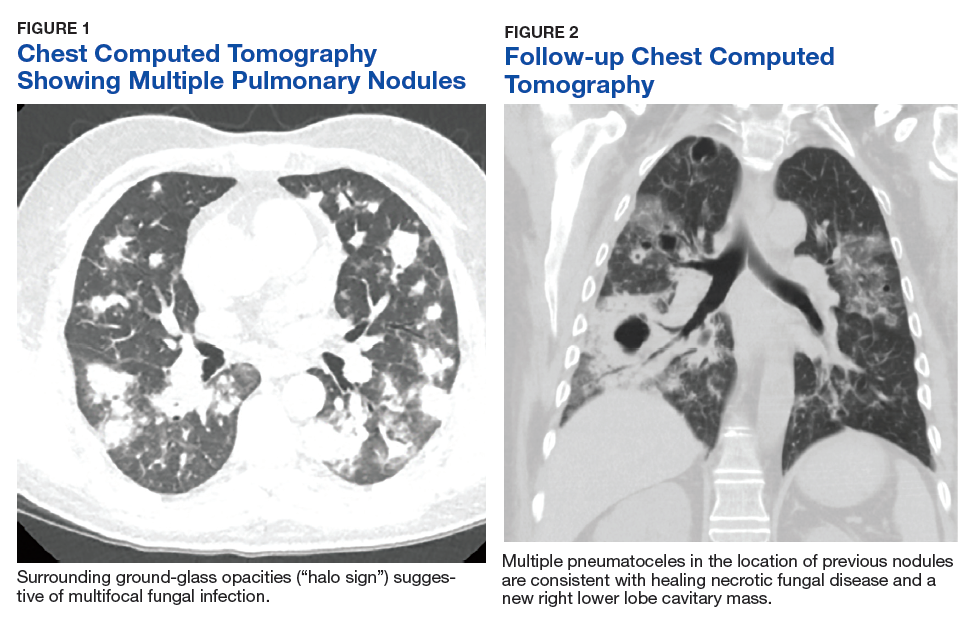
Treatment with oral voriconazole (300 mg twice daily) was initiated for probable pulmonary aspergillosis. Cyclosporine (150 mg twice daily) and prednisone (1 mg/kg/d) were continued throughout treatment out of concern for hepatic GVHD. The patient’s symptoms improved over the next 10 days, and follow-up chest imaging demonstrated improvement.
Two weeks after initiation of voriconazole treatment, the patient developed a new productive cough and dyspnea, associated with fevers and chills. Repeat imaging revealed right lower-lobe pneumonia. The serum voriconazole trough level was checked and was 3.1 mg/L, suggesting therapeutic dosing. The patient subsequently developed acute respiratory distress syndrome and required intubation and mechanical ventilation. Repeat BAL sampling demonstrated multidrug-resistant Escherichia coli, a BAL galactomannan level of 2.0 ODI, and negative fungal cultures. The patient’s hospital course was complicated by profound hypoxemia, requiring prone positioning and neuromuscular blockade. He was treated with meropenem and voriconazole. His immunosuppression was reduced, but he rapidly developed acute liver injury from hepatic GVHD that resolved after reinitiation of cyclosporine and prednisone at 0.75 mg/kg/d.
The patient improved over the next 3 weeks and was successfully extubated. Repeat chest CT imaging demonstrated numerous pneumatoceles in the location of previous nodules, consistent with healing necrotic fungal disease, and a new right lower-lobe cavitary mass (Figure 2). Two days after transferring out of the intensive care unit, the patient again developed hypoxemia and fevers to 39° C. Bronchoscopy with BAL of the right lower lobe revealed positive A fumigatus and Rhizopus sp polymerase chain reaction (PCR) assays, although fungal cultures were positive only for A fumigatus. Liposomal amphotericin B (5 mg/kg) was added to voriconazole therapy to treat mucormycosis and to provide a second active agent against A fumigatus.
Unfortunately, the patient’s clinical status continued to deteriorate with signs of progressive respiratory failure and infection despite empiric, broad-spectrum antibiotics and dual antifungal therapy. His serum voriconazole level continued to be therapeutic at 1.9 mg/L. The patient declined reintubation and invasive mechanical ventilation, and he ultimately transitioned to comfort measures and died with his family at the bedside.
Autopsy demonstrated widely disseminated Aspergillus infection as the cause of death, with evidence of myocardial, neural, and vascular invasion of A fumigatus (Figures 3 and 4). 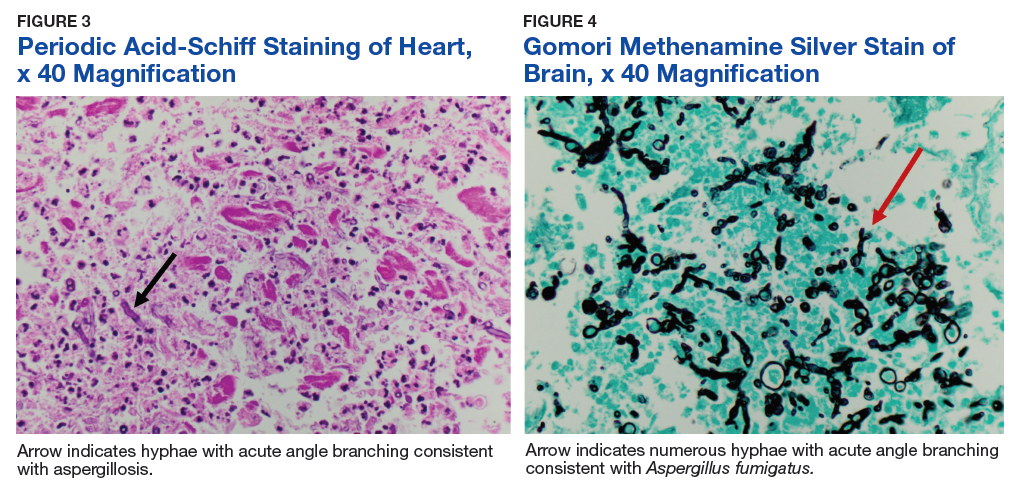
Discussion
This case of fatal, progressive, invasive, pulmonary aspergillosis demonstrates several important factors in the treatment of patients with this disease. Treatment failure usually relates to any of 4 possible factors: host immune status, severity or burden of disease, appropriate dosing of antifungal agents, and drug resistance. This patient’s immune system was heavily suppressed for a prolonged period. Attempts at reducing immunosuppression to the minimal required dosage to prevent a GVHD flare were unsuccessful and became an unmodifiable risk factor, a major contributor to his demise.
The risks of continuous high-dose immunosuppression in steroid-refractory GVHD is well understood and has been previously demonstrated to have up to 50% 4-year nonrelapse mortality, mainly due to overwhelming bacterial, viral, and fungal infections.4 All attempts should be made to cease or reduce immunosuppression in the setting of a severe infection, although this is sometimes impossible as in this case.
The patient’s disease burden was significant as evidenced by the bilateral, multifocal pulmonary nodules seen on chest imaging and the disseminated disease found at postmortem examination. His initial improvement in symptoms with voriconazole and the evolution of his images (with many of his initial pulmonary nodules becoming pneumatoceles) suggested a temporary positive immune response. The authors believe that the Rhizopus in his sputum represents noninvasive colonization of one of his pneumatoceles, because postmortem examination failed to reveal Rhizopus at any other location.
Voriconazole has excellent pulmonary and central nervous system penetration: In this patient serum levels were well within the therapeutic range. His peculiar drug resistance pattern (sensitivity to azoles and resistance to amphotericin) is unusual. Azole resistance in leukemia and patients with HSCT is more common than is amphotericin resistance, with current estimates of azole resistance close to 5%, ranging between 1% and 30%.5,6 Widespread use of antifungal prophylaxis with azoles likely selects for azole resistance.6
Despite this concern of azole resistance, current IDSA guidelines recommend against routine susceptibility testing of Aspergillus to azole therapy because of the current lack of consensus between the European Committee on Antibiotic Susceptibility Testing and Clinical and Laboratory Standards Institute on break points for resistance patterns.3,7 This is an area of emerging research, and proposed cut points for declaration of resistance do exist in the literature even if not globally agreed on.8
Combination antifungal therapy is an option for treatment in cases of possible drug resistance. Nonetheless, a recent randomized, double-blind, placebo-controlled, multicenter trial comparing voriconazole monotherapy with the combination of voriconazole and anidulafungin failed to demonstrate an overall mortality benefit in the primary analysis, although secondary analysis showed a mortality benefit with combination therapy in patients at highest risk for death.9
Despite the lack of unified standards with susceptibility testing, it may be reasonable to perform such tests in patients with demonstrating progressive disease. In this patient’s case, amphotericin B was added to treat the Rhizopus species found in his sputum, and while not the combination studied in the previously mentioned study, the drug should have provided an additional active agent for Aspergillus should this patient have had azole resistance.
Surprisingly, subsequent testing demonstrated the Aspergillus species to be resistant to amphotericin B. De novo amphotericin B-resistant A fumigates is extremely rare, with an expected incidence of 1% or less.10 The authors believe the patient may have demonstrated induction of amphotericin-B resistance through activation of fungal stress pathways by prior treatment with voriconazole. This has been demonstrated in vitro and should be considered should combination salvage therapy be required for the treatment of a refractory Aspergillus infection especially if patients have received prior treatment with voriconazole.11
Conclusion
This fatal case of invasive pulmonary aspergillosis illustrates the importance of considering the 4 main causes of treatment failure in an infection. Although the patient had a high burden of disease with a rare resistance pattern, he was treated with appropriate and well-dosed therapy. Ultimately, his unmodifiable immunosuppression was likely the driving factor leading to treatment failure and death. The indication for and number of bone marrow transplants continues to increase, thus exposure to and treatment of invasive fungal infections will increase accordingly. As such, providers should ensure that all causes of treatment failure are considered and addressed.
1. Upton A, Kirby KA, Carpenter P, Boeckh M, Marr KA. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin Infect Dis. 2007;44(4):531-540.
2. Herbrecht R, Denning DW, Patterson TF, et al; Invasive Fungal Infections Group of the European Organisation for Research and Treatment of Cancer and the Global Aspergillus Study Group. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408-415.
3. Patterson TF, Thompson GR III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Disease Society of America. Clin Infect Dis. 2016;63(4):e1-e60.
4. García-Cadenas I, Rivera I, Martino R, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transplant. 2017;52(1):107-113.
5. Vermeulen E, Maertens J, De Bel A, et al. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob Agents Chemother. 2015;59(8):4569-4576.
6. Wiederhold NP, Patterson TF. Emergence of azole resistance in Aspergillus. Semin Respir Crit Care Med. 2015;36(5):673-680.
7. Cuenca-Estrella M, Moore CB, Barchiesi F, et al; AFST Subcommittee of the European Committee on Antimicrobial Susceptibility Testing. Multicenter evaluation of the reproducibility of the proposed antifungal susceptibility testing method for fermentative yeasts of the Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST). Clin Microbiol Infect. 2003;9(6):467-474.
8. Pfaller MA, Diekema DJ, Ghannoum MA, et al; Clinical and Laboratory Standards Institute Antifungal Testing Subcommittee. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by Clinical and Laboratory Standards Institute for broth microdilution methods. J Clin Microbiol. 2009;47(10):3142-3146.
9. Marr KA, Schlamm HT, Herbrecht R, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2015;162(2):81-89.
10. Tashiro M, Izumikawa K, Minematsu A, et al. Antifungal susceptibilities of Aspergillus fumigatus clinical isolates obtained in Nagasaki, Japan. Antimicrob Agents Chemother. 2012;56(1):584-587.
11. Rajendran R, Mowat E, Jones B, Williams C, Ramage G. Prior in vitro exposure to voriconazole confers resistance to amphotericin B in Aspergillus fumigatus biofilms. Int J Antimicrob Agents. 2015;46(3):342-345.
Historically, aspergillosis in patients with hematopoietic stem cell transplantation (HSCT) has carried a high mortality rate. However, recent data demonstrate a dramatic improvement in outcomes for patients with HSCT: 90-day survival increased from 22% before 2000 to 45% over the past 15 years.1 Improved outcomes coincide with changes in transplant immunosuppression practices, use of cross-sectional imaging for early disease identification, galactomannan screening, and the development of novel treatment options.
Voriconazole is an azole drug that blocks the synthesis of ergosterol, a vital component of the cellular membrane of fungi. Voriconazole was approved in 2002 after a clinical trial demonstrated an improvement in 50% of patients with invasive aspergillosis in the voriconazole arm vs 30% in the amphotericin B arm at 12 weeks.2 Amphotericin B is a polyene antifungal drug that binds with ergosterol, creating leaks in the cell membrane that lead to cellular demise. Voriconazole quickly became the first-line therapy for invasive aspergillosis and is recommended by both the Infectious Disease Society of American (IDSA) and the European Conference on Infections in Leukemia.3
Case Presentation
A 55-year-old man with high-risk chronic myelogenous leukemia (CML) underwent a 10 of 10 human leukocyte antigen allele and antigen-matched peripheral blood allogeneic HSCT with a myeloablative-conditioning regimen of busulfan and cyclophosphamide, along with prophylactic voriconazole, sulfamethoxazole/trimethoprim, and acyclovir. After successful engraftment (without significant neutropenia), his posttransplant course was complicated by grade 2 graft vs host disease (GVHD) of the skin, eyes, and liver, which responded well to steroids and tacrolimus. Voriconazole was continued for 5 months until immunosuppression was minimized (tacrolimus 1 mg twice daily). Two months later, the patient’s GVHD worsened, necessitating treatment at an outside hospital with high-dose prednisone (2 mg/kg/d) and cyclosporine (300 mg twice daily). Voriconazole prophylaxis was not reinitiated at that time.
One year later, at a routine follow-up appointment, the patient endorsed several weeks of malaise, weight loss, and nonproductive cough. The patient’s immunosuppression recently had been reduced to 1 mg/kg/d of prednisone and 100 mg of cyclosporine twice daily. A chest X-ray demonstrated multiple pulmonary nodules; follow-up chest computed tomography (CT) confirmed multiple nodular infiltrates with surrounding ground-glass opacities suspicious with a fungal infection (Figure 1).
Treatment with oral voriconazole (300 mg twice daily) was initiated for probable pulmonary aspergillosis. Cyclosporine (150 mg twice daily) and prednisone (1 mg/kg/d) were continued throughout treatment out of concern for hepatic GVHD. The patient’s symptoms improved over the next 10 days, and follow-up chest imaging demonstrated improvement.
Two weeks after initiation of voriconazole treatment, the patient developed a new productive cough and dyspnea, associated with fevers and chills. Repeat imaging revealed right lower-lobe pneumonia. The serum voriconazole trough level was checked and was 3.1 mg/L, suggesting therapeutic dosing. The patient subsequently developed acute respiratory distress syndrome and required intubation and mechanical ventilation. Repeat BAL sampling demonstrated multidrug-resistant Escherichia coli, a BAL galactomannan level of 2.0 ODI, and negative fungal cultures. The patient’s hospital course was complicated by profound hypoxemia, requiring prone positioning and neuromuscular blockade. He was treated with meropenem and voriconazole. His immunosuppression was reduced, but he rapidly developed acute liver injury from hepatic GVHD that resolved after reinitiation of cyclosporine and prednisone at 0.75 mg/kg/d.
The patient improved over the next 3 weeks and was successfully extubated. Repeat chest CT imaging demonstrated numerous pneumatoceles in the location of previous nodules, consistent with healing necrotic fungal disease, and a new right lower-lobe cavitary mass (Figure 2). Two days after transferring out of the intensive care unit, the patient again developed hypoxemia and fevers to 39° C. Bronchoscopy with BAL of the right lower lobe revealed positive A fumigatus and Rhizopus sp polymerase chain reaction (PCR) assays, although fungal cultures were positive only for A fumigatus. Liposomal amphotericin B (5 mg/kg) was added to voriconazole therapy to treat mucormycosis and to provide a second active agent against A fumigatus.
Unfortunately, the patient’s clinical status continued to deteriorate with signs of progressive respiratory failure and infection despite empiric, broad-spectrum antibiotics and dual antifungal therapy. His serum voriconazole level continued to be therapeutic at 1.9 mg/L. The patient declined reintubation and invasive mechanical ventilation, and he ultimately transitioned to comfort measures and died with his family at the bedside.
Autopsy demonstrated widely disseminated Aspergillus infection as the cause of death, with evidence of myocardial, neural, and vascular invasion of A fumigatus (Figures 3 and 4).
Discussion
This case of fatal, progressive, invasive, pulmonary aspergillosis demonstrates several important factors in the treatment of patients with this disease. Treatment failure usually relates to any of 4 possible factors: host immune status, severity or burden of disease, appropriate dosing of antifungal agents, and drug resistance. This patient’s immune system was heavily suppressed for a prolonged period. Attempts at reducing immunosuppression to the minimal required dosage to prevent a GVHD flare were unsuccessful and became an unmodifiable risk factor, a major contributor to his demise.
The risks of continuous high-dose immunosuppression in steroid-refractory GVHD is well understood and has been previously demonstrated to have up to 50% 4-year nonrelapse mortality, mainly due to overwhelming bacterial, viral, and fungal infections.4 All attempts should be made to cease or reduce immunosuppression in the setting of a severe infection, although this is sometimes impossible as in this case.
The patient’s disease burden was significant as evidenced by the bilateral, multifocal pulmonary nodules seen on chest imaging and the disseminated disease found at postmortem examination. His initial improvement in symptoms with voriconazole and the evolution of his images (with many of his initial pulmonary nodules becoming pneumatoceles) suggested a temporary positive immune response. The authors believe that the Rhizopus in his sputum represents noninvasive colonization of one of his pneumatoceles, because postmortem examination failed to reveal Rhizopus at any other location.
Voriconazole has excellent pulmonary and central nervous system penetration: In this patient serum levels were well within the therapeutic range. His peculiar drug resistance pattern (sensitivity to azoles and resistance to amphotericin) is unusual. Azole resistance in leukemia and patients with HSCT is more common than is amphotericin resistance, with current estimates of azole resistance close to 5%, ranging between 1% and 30%.5,6 Widespread use of antifungal prophylaxis with azoles likely selects for azole resistance.6
Despite this concern of azole resistance, current IDSA guidelines recommend against routine susceptibility testing of Aspergillus to azole therapy because of the current lack of consensus between the European Committee on Antibiotic Susceptibility Testing and Clinical and Laboratory Standards Institute on break points for resistance patterns.3,7 This is an area of emerging research, and proposed cut points for declaration of resistance do exist in the literature even if not globally agreed on.8
Combination antifungal therapy is an option for treatment in cases of possible drug resistance. Nonetheless, a recent randomized, double-blind, placebo-controlled, multicenter trial comparing voriconazole monotherapy with the combination of voriconazole and anidulafungin failed to demonstrate an overall mortality benefit in the primary analysis, although secondary analysis showed a mortality benefit with combination therapy in patients at highest risk for death.9
Despite the lack of unified standards with susceptibility testing, it may be reasonable to perform such tests in patients with demonstrating progressive disease. In this patient’s case, amphotericin B was added to treat the Rhizopus species found in his sputum, and while not the combination studied in the previously mentioned study, the drug should have provided an additional active agent for Aspergillus should this patient have had azole resistance.
Surprisingly, subsequent testing demonstrated the Aspergillus species to be resistant to amphotericin B. De novo amphotericin B-resistant A fumigates is extremely rare, with an expected incidence of 1% or less.10 The authors believe the patient may have demonstrated induction of amphotericin-B resistance through activation of fungal stress pathways by prior treatment with voriconazole. This has been demonstrated in vitro and should be considered should combination salvage therapy be required for the treatment of a refractory Aspergillus infection especially if patients have received prior treatment with voriconazole.11
Conclusion
This fatal case of invasive pulmonary aspergillosis illustrates the importance of considering the 4 main causes of treatment failure in an infection. Although the patient had a high burden of disease with a rare resistance pattern, he was treated with appropriate and well-dosed therapy. Ultimately, his unmodifiable immunosuppression was likely the driving factor leading to treatment failure and death. The indication for and number of bone marrow transplants continues to increase, thus exposure to and treatment of invasive fungal infections will increase accordingly. As such, providers should ensure that all causes of treatment failure are considered and addressed.
Historically, aspergillosis in patients with hematopoietic stem cell transplantation (HSCT) has carried a high mortality rate. However, recent data demonstrate a dramatic improvement in outcomes for patients with HSCT: 90-day survival increased from 22% before 2000 to 45% over the past 15 years.1 Improved outcomes coincide with changes in transplant immunosuppression practices, use of cross-sectional imaging for early disease identification, galactomannan screening, and the development of novel treatment options.
Voriconazole is an azole drug that blocks the synthesis of ergosterol, a vital component of the cellular membrane of fungi. Voriconazole was approved in 2002 after a clinical trial demonstrated an improvement in 50% of patients with invasive aspergillosis in the voriconazole arm vs 30% in the amphotericin B arm at 12 weeks.2 Amphotericin B is a polyene antifungal drug that binds with ergosterol, creating leaks in the cell membrane that lead to cellular demise. Voriconazole quickly became the first-line therapy for invasive aspergillosis and is recommended by both the Infectious Disease Society of American (IDSA) and the European Conference on Infections in Leukemia.3
Case Presentation
A 55-year-old man with high-risk chronic myelogenous leukemia (CML) underwent a 10 of 10 human leukocyte antigen allele and antigen-matched peripheral blood allogeneic HSCT with a myeloablative-conditioning regimen of busulfan and cyclophosphamide, along with prophylactic voriconazole, sulfamethoxazole/trimethoprim, and acyclovir. After successful engraftment (without significant neutropenia), his posttransplant course was complicated by grade 2 graft vs host disease (GVHD) of the skin, eyes, and liver, which responded well to steroids and tacrolimus. Voriconazole was continued for 5 months until immunosuppression was minimized (tacrolimus 1 mg twice daily). Two months later, the patient’s GVHD worsened, necessitating treatment at an outside hospital with high-dose prednisone (2 mg/kg/d) and cyclosporine (300 mg twice daily). Voriconazole prophylaxis was not reinitiated at that time.
One year later, at a routine follow-up appointment, the patient endorsed several weeks of malaise, weight loss, and nonproductive cough. The patient’s immunosuppression recently had been reduced to 1 mg/kg/d of prednisone and 100 mg of cyclosporine twice daily. A chest X-ray demonstrated multiple pulmonary nodules; follow-up chest computed tomography (CT) confirmed multiple nodular infiltrates with surrounding ground-glass opacities suspicious with a fungal infection (Figure 1).
Treatment with oral voriconazole (300 mg twice daily) was initiated for probable pulmonary aspergillosis. Cyclosporine (150 mg twice daily) and prednisone (1 mg/kg/d) were continued throughout treatment out of concern for hepatic GVHD. The patient’s symptoms improved over the next 10 days, and follow-up chest imaging demonstrated improvement.
Two weeks after initiation of voriconazole treatment, the patient developed a new productive cough and dyspnea, associated with fevers and chills. Repeat imaging revealed right lower-lobe pneumonia. The serum voriconazole trough level was checked and was 3.1 mg/L, suggesting therapeutic dosing. The patient subsequently developed acute respiratory distress syndrome and required intubation and mechanical ventilation. Repeat BAL sampling demonstrated multidrug-resistant Escherichia coli, a BAL galactomannan level of 2.0 ODI, and negative fungal cultures. The patient’s hospital course was complicated by profound hypoxemia, requiring prone positioning and neuromuscular blockade. He was treated with meropenem and voriconazole. His immunosuppression was reduced, but he rapidly developed acute liver injury from hepatic GVHD that resolved after reinitiation of cyclosporine and prednisone at 0.75 mg/kg/d.
The patient improved over the next 3 weeks and was successfully extubated. Repeat chest CT imaging demonstrated numerous pneumatoceles in the location of previous nodules, consistent with healing necrotic fungal disease, and a new right lower-lobe cavitary mass (Figure 2). Two days after transferring out of the intensive care unit, the patient again developed hypoxemia and fevers to 39° C. Bronchoscopy with BAL of the right lower lobe revealed positive A fumigatus and Rhizopus sp polymerase chain reaction (PCR) assays, although fungal cultures were positive only for A fumigatus. Liposomal amphotericin B (5 mg/kg) was added to voriconazole therapy to treat mucormycosis and to provide a second active agent against A fumigatus.
Unfortunately, the patient’s clinical status continued to deteriorate with signs of progressive respiratory failure and infection despite empiric, broad-spectrum antibiotics and dual antifungal therapy. His serum voriconazole level continued to be therapeutic at 1.9 mg/L. The patient declined reintubation and invasive mechanical ventilation, and he ultimately transitioned to comfort measures and died with his family at the bedside.
Autopsy demonstrated widely disseminated Aspergillus infection as the cause of death, with evidence of myocardial, neural, and vascular invasion of A fumigatus (Figures 3 and 4).
Discussion
This case of fatal, progressive, invasive, pulmonary aspergillosis demonstrates several important factors in the treatment of patients with this disease. Treatment failure usually relates to any of 4 possible factors: host immune status, severity or burden of disease, appropriate dosing of antifungal agents, and drug resistance. This patient’s immune system was heavily suppressed for a prolonged period. Attempts at reducing immunosuppression to the minimal required dosage to prevent a GVHD flare were unsuccessful and became an unmodifiable risk factor, a major contributor to his demise.
The risks of continuous high-dose immunosuppression in steroid-refractory GVHD is well understood and has been previously demonstrated to have up to 50% 4-year nonrelapse mortality, mainly due to overwhelming bacterial, viral, and fungal infections.4 All attempts should be made to cease or reduce immunosuppression in the setting of a severe infection, although this is sometimes impossible as in this case.
The patient’s disease burden was significant as evidenced by the bilateral, multifocal pulmonary nodules seen on chest imaging and the disseminated disease found at postmortem examination. His initial improvement in symptoms with voriconazole and the evolution of his images (with many of his initial pulmonary nodules becoming pneumatoceles) suggested a temporary positive immune response. The authors believe that the Rhizopus in his sputum represents noninvasive colonization of one of his pneumatoceles, because postmortem examination failed to reveal Rhizopus at any other location.
Voriconazole has excellent pulmonary and central nervous system penetration: In this patient serum levels were well within the therapeutic range. His peculiar drug resistance pattern (sensitivity to azoles and resistance to amphotericin) is unusual. Azole resistance in leukemia and patients with HSCT is more common than is amphotericin resistance, with current estimates of azole resistance close to 5%, ranging between 1% and 30%.5,6 Widespread use of antifungal prophylaxis with azoles likely selects for azole resistance.6
Despite this concern of azole resistance, current IDSA guidelines recommend against routine susceptibility testing of Aspergillus to azole therapy because of the current lack of consensus between the European Committee on Antibiotic Susceptibility Testing and Clinical and Laboratory Standards Institute on break points for resistance patterns.3,7 This is an area of emerging research, and proposed cut points for declaration of resistance do exist in the literature even if not globally agreed on.8
Combination antifungal therapy is an option for treatment in cases of possible drug resistance. Nonetheless, a recent randomized, double-blind, placebo-controlled, multicenter trial comparing voriconazole monotherapy with the combination of voriconazole and anidulafungin failed to demonstrate an overall mortality benefit in the primary analysis, although secondary analysis showed a mortality benefit with combination therapy in patients at highest risk for death.9
Despite the lack of unified standards with susceptibility testing, it may be reasonable to perform such tests in patients with demonstrating progressive disease. In this patient’s case, amphotericin B was added to treat the Rhizopus species found in his sputum, and while not the combination studied in the previously mentioned study, the drug should have provided an additional active agent for Aspergillus should this patient have had azole resistance.
Surprisingly, subsequent testing demonstrated the Aspergillus species to be resistant to amphotericin B. De novo amphotericin B-resistant A fumigates is extremely rare, with an expected incidence of 1% or less.10 The authors believe the patient may have demonstrated induction of amphotericin-B resistance through activation of fungal stress pathways by prior treatment with voriconazole. This has been demonstrated in vitro and should be considered should combination salvage therapy be required for the treatment of a refractory Aspergillus infection especially if patients have received prior treatment with voriconazole.11
Conclusion
This fatal case of invasive pulmonary aspergillosis illustrates the importance of considering the 4 main causes of treatment failure in an infection. Although the patient had a high burden of disease with a rare resistance pattern, he was treated with appropriate and well-dosed therapy. Ultimately, his unmodifiable immunosuppression was likely the driving factor leading to treatment failure and death. The indication for and number of bone marrow transplants continues to increase, thus exposure to and treatment of invasive fungal infections will increase accordingly. As such, providers should ensure that all causes of treatment failure are considered and addressed.
1. Upton A, Kirby KA, Carpenter P, Boeckh M, Marr KA. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin Infect Dis. 2007;44(4):531-540.
2. Herbrecht R, Denning DW, Patterson TF, et al; Invasive Fungal Infections Group of the European Organisation for Research and Treatment of Cancer and the Global Aspergillus Study Group. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408-415.
3. Patterson TF, Thompson GR III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Disease Society of America. Clin Infect Dis. 2016;63(4):e1-e60.
4. García-Cadenas I, Rivera I, Martino R, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transplant. 2017;52(1):107-113.
5. Vermeulen E, Maertens J, De Bel A, et al. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob Agents Chemother. 2015;59(8):4569-4576.
6. Wiederhold NP, Patterson TF. Emergence of azole resistance in Aspergillus. Semin Respir Crit Care Med. 2015;36(5):673-680.
7. Cuenca-Estrella M, Moore CB, Barchiesi F, et al; AFST Subcommittee of the European Committee on Antimicrobial Susceptibility Testing. Multicenter evaluation of the reproducibility of the proposed antifungal susceptibility testing method for fermentative yeasts of the Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST). Clin Microbiol Infect. 2003;9(6):467-474.
8. Pfaller MA, Diekema DJ, Ghannoum MA, et al; Clinical and Laboratory Standards Institute Antifungal Testing Subcommittee. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by Clinical and Laboratory Standards Institute for broth microdilution methods. J Clin Microbiol. 2009;47(10):3142-3146.
9. Marr KA, Schlamm HT, Herbrecht R, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2015;162(2):81-89.
10. Tashiro M, Izumikawa K, Minematsu A, et al. Antifungal susceptibilities of Aspergillus fumigatus clinical isolates obtained in Nagasaki, Japan. Antimicrob Agents Chemother. 2012;56(1):584-587.
11. Rajendran R, Mowat E, Jones B, Williams C, Ramage G. Prior in vitro exposure to voriconazole confers resistance to amphotericin B in Aspergillus fumigatus biofilms. Int J Antimicrob Agents. 2015;46(3):342-345.
1. Upton A, Kirby KA, Carpenter P, Boeckh M, Marr KA. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin Infect Dis. 2007;44(4):531-540.
2. Herbrecht R, Denning DW, Patterson TF, et al; Invasive Fungal Infections Group of the European Organisation for Research and Treatment of Cancer and the Global Aspergillus Study Group. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408-415.
3. Patterson TF, Thompson GR III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Disease Society of America. Clin Infect Dis. 2016;63(4):e1-e60.
4. García-Cadenas I, Rivera I, Martino R, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transplant. 2017;52(1):107-113.
5. Vermeulen E, Maertens J, De Bel A, et al. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob Agents Chemother. 2015;59(8):4569-4576.
6. Wiederhold NP, Patterson TF. Emergence of azole resistance in Aspergillus. Semin Respir Crit Care Med. 2015;36(5):673-680.
7. Cuenca-Estrella M, Moore CB, Barchiesi F, et al; AFST Subcommittee of the European Committee on Antimicrobial Susceptibility Testing. Multicenter evaluation of the reproducibility of the proposed antifungal susceptibility testing method for fermentative yeasts of the Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST). Clin Microbiol Infect. 2003;9(6):467-474.
8. Pfaller MA, Diekema DJ, Ghannoum MA, et al; Clinical and Laboratory Standards Institute Antifungal Testing Subcommittee. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by Clinical and Laboratory Standards Institute for broth microdilution methods. J Clin Microbiol. 2009;47(10):3142-3146.
9. Marr KA, Schlamm HT, Herbrecht R, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2015;162(2):81-89.
10. Tashiro M, Izumikawa K, Minematsu A, et al. Antifungal susceptibilities of Aspergillus fumigatus clinical isolates obtained in Nagasaki, Japan. Antimicrob Agents Chemother. 2012;56(1):584-587.
11. Rajendran R, Mowat E, Jones B, Williams C, Ramage G. Prior in vitro exposure to voriconazole confers resistance to amphotericin B in Aspergillus fumigatus biofilms. Int J Antimicrob Agents. 2015;46(3):342-345.
Recurrence of Linear Basal Cell Carcinoma
Case Report
A 63-year-old man was evaluated in the Mohs clinic for a lesion on the right supraclavicular neck, which he described as a linear asymptomatic “birthmark” that had been present since childhood and stable for many years. It began to enlarge approximately 5 years prior, became increasingly red, and had occasional crusting. The lesion also gradually became more irritated with repeated mild trauma when he carried a backpack while hiking. On physical examination, a 10×2-cm, linear, pink plaque with an irregular border, translucent rolled edges, and central smooth atrophic skin was seen on the right supraclavicular neck (Figure). There was no visible epidermal nevus or nevus sebaceous in the area. A shave biopsy of the lesion confirmed the pathologic diagnosis of basal cell carcinoma, nodular type, along with the morphologic diagnosis of linear basal cell carcinoma (LBCC). The tumor was completely removed with standard excision using 5-mm margins.

Approximately 10 months after the original excision, the patient developed an irritated erosion that occasionally bled when his backpack rubbed against it. He returned to the clinic after the erosion failed to heal. Physical examination revealed a 1.4×0.7-cm, eroded, pink papule with large telangiectases at the superior pole of the excision scar. A shave biopsy confirmed the diagnosis of a recurrent infiltrative basal cell carcinoma. The tumor was then completely excised using Mohs micrographic surgery.
Comment
Linear basal cell carcinoma, first described by Lewis1 in 1985, is a rare morphologic variant of basal cell carcinoma. In 2011, Al-Niaimi and Lyon2 performed a comprehensive literature search on LBCC (1985-2008) and found only 39 cases (including 2 of their own) had been published since the pioneer case in 1985. It was determined that the most common sites affected were the periorbital area and neck (n=13 each [67%]), and the majority were histologically nodular (n=27 [69%]). Mohs micrographic surgery was the most common treatment method (n=23 [59%]), followed by primary excision (n=17 [44%]). A history of trauma, radiotherapy, or prior operation in association with the site of the LBCC was discovered in only 7 cases (18%).2 Although Peschen et al3 proposed that trauma—both physical and surgical—and radiotherapy may play a role in the development of LBCCs, the low incidence reported suggests that other factors may be involved. To determine if genetic factors were contributing to the development of LBCCs, Yamaguchi et al4 investigated the expression of p27 and PCTAIRE1, both known to contribute to tumorigenesis when mutated, as well as somatic gene mutations using deep sequencing in a case of LBCC; they found no associated genetic mutation.
Reported Cases of LBCC
According to a PubMed search of articles indexed for MEDLINE using the terms linear and basal cell carcinoma, 67 cases (including the current case) of LBCC have been published since 1985. The patient demographics, anatomic location, histologic subtype, treatment methods, and frequency of recurrence for all reported cases of LBCC are summarized in the Table.1-24 There were 36 women and 31 men, with an average age of 70 years (range, 40–92 years). The most commonly affected sites were the periocular region (n=27) and neck (n=18). Histologically, most LBCCs were nodular (n=35), with the next most common histologic subtype being infiltrative (n=20), which included the morphoeic, metatypical, and micronodular subtypes under the overarching infiltrative subtype. The most frequently chosen treatment option was primary excision (n=38 [57%]), followed by Mohs micrographic surgery (n=28 [42%]). Risk factors previously identified by Al-Niaimi and Lyon,2 including trauma, radiotherapy, or prior operation, were reported in 12 of 67 cases. Recurrence was reported in only 2 of 67 cases, 1 being the current case; however, an accurate recurrence rate could not be calculated due to lack of follow-up or short length of follow-up in most of the reported cases.
Presentation and Treatment
Currently, there are no set criteria for the diagnosis of LBCC, but it has been shown to follow a characteristic morphologic pattern, favoring extension in one direction leading to a length-to-width ratio that typically is at least 3 to 1.5 With most lesions presenting in the periocular region along relaxed skin tension lines, it has been speculated that these tumors expand along wrinkles.2 Pierard and Lapiere25 proposed that the preferential parallel orientation and a straightening of thin collagen bundles and elastic fibers within the reticular dermis combined with relaxed skin tension lines and muscle contraction perpendicular to these stromal parts may influence the growth of tumors preferentially in one direction, contributing to linearity of the lesion. In addition, the clinical appearance is not a reliable indicator of subclinical extension.2 Therefore, Lim et al6 recommended Mohs micrographic surgery as the best initial treatment of LBCCs.
Conclusion
Linear basal cell carcinoma should be considered a distinct morphologic variant of basal cell carcinoma. Although likely underreported, this variant is uncommon. It presents most often in the periocular and neck regions. The most common histologic subtypes are nodular and infiltrative. Because of the likelihood of subclinical spread, LBCC should be regarded as a high-risk subtype. As such, Mohs micrographic surgery or excision with complete circumferential peripheral and deep margin assessment is recommended as first-line treatment of LBCC.6
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1985;24:124-125.
- Al-Niaimi F, Lyon CC. Linear basal cell carcinoma: a distinct condition? Clin Exp Dermatol. 2011;36:231-234.
- Peschen M, Lo JS, Snow SN, et al. Linear basal cell carcinoma. Cutis. 1993;51:287-289.
- Yamaguchi Y, Yanagi T, Imafuku K, et al. A case of linear basal cell carcinoma: evaluation of proliferative activity by immunohistochemical staining of PCTAIRE1 and p27. J Eur Acad Dermatol Venereol. 2017;31:E359-E362.
- Mavirakis I, Malhotra R, Selva D, et al. Linear basal cell carcinoma: a distinct clinical entity. J Plast Reconstr Aesthet Surg. 2006;59:419-423.
- Lim KK, Randle HW, Roenigk RK, et al. Linear basal cell carcinoma: report of seventeen cases and review of the presentation and treatment. Dermatol Surg. 1999;25:63-67.
- Pardavila R, Rosón E, De la torre C, et al. Linear basal cell carcinoma. report of two cases [in Spanish]. Actas Dermosifiliogr. 2007;98:291.
- Shinsuke K, Hirohiko K, Yasuhiro T, et al. Linear basal cell carcinoma in an Asian patient. Open Ophthalmol J. 2007;1:20-22.
- Ning C, Chao S. Linear basal cell carcinoma of the scrotum. Dermatol Sinica. 2002;20:57-62.
- Chopra KF, Cohen PR. Linear basal cell carcinomas: report of multiple sequential tumors localized to a radiotherapy port and review of the literature. Tex Med. 1997;93:57-59.
- da Silva MO, Dadalt P, Santos OL, et al. Linear basal cell carcinoma. Int J Dermatol. 1995;34:488.
- Warthan TL, Lewis JE. Giant linear basal cell epithelioma. Int J Dermatol. 1994;33:284.
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1989;28:682-684.
- Alcántara-Reifs CM, Salido-Vallejo R, González-Menchen A, et al. Linear basal cell carcinoma: report of three cases with dermoscopic findings. Indian J Dermatol Venereol Leprol. 2016;82:708-711.
- Lee MS, Cho E, Lee JH, et al. Linearly curved, blackish macule on the wrist. Cutis. 2016;97:384, 406-407.
- Bajaj S, Sharma PK, Kar HK. Linear adamantinoid basal cell carcinoma in the axilla. Dermatol Online J. 2015;21. pii:13030/qt8k0713nb.
- Iga N, Sakurai K, Fujii H, et al. Linear basal cell carcinoma at the external genitalia. J Dermatol. 2014;41:275-276.
- Ichinokawa Y, Ohtuki A, Hattori M, et al. Linear basal cell carcinoma: a case report. Case Rep Dermatol. 2011;3:142-146.
- Becher GL, Affleck A, Fleming C, et al. Linear basal cell carcinoma occurs most commonly on the lower eyelid. Clin Exp Dermatol. 2011;36:311-312.
- Jellouli A, Triki S, Zghal M, et al. Linear basal cell carcinoma. Actas Dermosifiliogr. 2010;101:648-650.
- Takiyoshi N, Nakano H, Kaneko T, et al. A linear basal cell carcinoma undergoing spontaneous regression. Clin Exp Dermatol. 2009;34:E411-E413.
- Yoleri L, Ozden S, Kandiloglu A. A 46-year-old male with an ulcerated linear lesion on his neck. Ann Saudi Med. 2008;28:57-58.
- Palleschi GM, Corradini D, Bruscino N, et al. Linear basal cell carcinoma: clinical significance and better surgical approach. G Ital Dermatol Venereol. 2016;151:119-121.
- Rodriguez-Garijo N, Redondo P. Linear basal cell carcinoma of the lower eyelid: reconstruction with a musculocutaneous transposition flap. JAAD Case Rep. 2018;4:633-635.
- Pierard GE, Lapiere CM. Microanatomy of the dermis in relation to relaxed skin tension lines and Langer’s lines. Am J Dermatopathol. 1987;9:219-224.
Case Report
A 63-year-old man was evaluated in the Mohs clinic for a lesion on the right supraclavicular neck, which he described as a linear asymptomatic “birthmark” that had been present since childhood and stable for many years. It began to enlarge approximately 5 years prior, became increasingly red, and had occasional crusting. The lesion also gradually became more irritated with repeated mild trauma when he carried a backpack while hiking. On physical examination, a 10×2-cm, linear, pink plaque with an irregular border, translucent rolled edges, and central smooth atrophic skin was seen on the right supraclavicular neck (Figure). There was no visible epidermal nevus or nevus sebaceous in the area. A shave biopsy of the lesion confirmed the pathologic diagnosis of basal cell carcinoma, nodular type, along with the morphologic diagnosis of linear basal cell carcinoma (LBCC). The tumor was completely removed with standard excision using 5-mm margins.
Approximately 10 months after the original excision, the patient developed an irritated erosion that occasionally bled when his backpack rubbed against it. He returned to the clinic after the erosion failed to heal. Physical examination revealed a 1.4×0.7-cm, eroded, pink papule with large telangiectases at the superior pole of the excision scar. A shave biopsy confirmed the diagnosis of a recurrent infiltrative basal cell carcinoma. The tumor was then completely excised using Mohs micrographic surgery.
Comment
Linear basal cell carcinoma, first described by Lewis1 in 1985, is a rare morphologic variant of basal cell carcinoma. In 2011, Al-Niaimi and Lyon2 performed a comprehensive literature search on LBCC (1985-2008) and found only 39 cases (including 2 of their own) had been published since the pioneer case in 1985. It was determined that the most common sites affected were the periorbital area and neck (n=13 each [67%]), and the majority were histologically nodular (n=27 [69%]). Mohs micrographic surgery was the most common treatment method (n=23 [59%]), followed by primary excision (n=17 [44%]). A history of trauma, radiotherapy, or prior operation in association with the site of the LBCC was discovered in only 7 cases (18%).2 Although Peschen et al3 proposed that trauma—both physical and surgical—and radiotherapy may play a role in the development of LBCCs, the low incidence reported suggests that other factors may be involved. To determine if genetic factors were contributing to the development of LBCCs, Yamaguchi et al4 investigated the expression of p27 and PCTAIRE1, both known to contribute to tumorigenesis when mutated, as well as somatic gene mutations using deep sequencing in a case of LBCC; they found no associated genetic mutation.
Reported Cases of LBCC
According to a PubMed search of articles indexed for MEDLINE using the terms linear and basal cell carcinoma, 67 cases (including the current case) of LBCC have been published since 1985. The patient demographics, anatomic location, histologic subtype, treatment methods, and frequency of recurrence for all reported cases of LBCC are summarized in the Table.1-24 There were 36 women and 31 men, with an average age of 70 years (range, 40–92 years). The most commonly affected sites were the periocular region (n=27) and neck (n=18). Histologically, most LBCCs were nodular (n=35), with the next most common histologic subtype being infiltrative (n=20), which included the morphoeic, metatypical, and micronodular subtypes under the overarching infiltrative subtype. The most frequently chosen treatment option was primary excision (n=38 [57%]), followed by Mohs micrographic surgery (n=28 [42%]). Risk factors previously identified by Al-Niaimi and Lyon,2 including trauma, radiotherapy, or prior operation, were reported in 12 of 67 cases. Recurrence was reported in only 2 of 67 cases, 1 being the current case; however, an accurate recurrence rate could not be calculated due to lack of follow-up or short length of follow-up in most of the reported cases.
Presentation and Treatment
Currently, there are no set criteria for the diagnosis of LBCC, but it has been shown to follow a characteristic morphologic pattern, favoring extension in one direction leading to a length-to-width ratio that typically is at least 3 to 1.5 With most lesions presenting in the periocular region along relaxed skin tension lines, it has been speculated that these tumors expand along wrinkles.2 Pierard and Lapiere25 proposed that the preferential parallel orientation and a straightening of thin collagen bundles and elastic fibers within the reticular dermis combined with relaxed skin tension lines and muscle contraction perpendicular to these stromal parts may influence the growth of tumors preferentially in one direction, contributing to linearity of the lesion. In addition, the clinical appearance is not a reliable indicator of subclinical extension.2 Therefore, Lim et al6 recommended Mohs micrographic surgery as the best initial treatment of LBCCs.
Conclusion
Linear basal cell carcinoma should be considered a distinct morphologic variant of basal cell carcinoma. Although likely underreported, this variant is uncommon. It presents most often in the periocular and neck regions. The most common histologic subtypes are nodular and infiltrative. Because of the likelihood of subclinical spread, LBCC should be regarded as a high-risk subtype. As such, Mohs micrographic surgery or excision with complete circumferential peripheral and deep margin assessment is recommended as first-line treatment of LBCC.6
Case Report
A 63-year-old man was evaluated in the Mohs clinic for a lesion on the right supraclavicular neck, which he described as a linear asymptomatic “birthmark” that had been present since childhood and stable for many years. It began to enlarge approximately 5 years prior, became increasingly red, and had occasional crusting. The lesion also gradually became more irritated with repeated mild trauma when he carried a backpack while hiking. On physical examination, a 10×2-cm, linear, pink plaque with an irregular border, translucent rolled edges, and central smooth atrophic skin was seen on the right supraclavicular neck (Figure). There was no visible epidermal nevus or nevus sebaceous in the area. A shave biopsy of the lesion confirmed the pathologic diagnosis of basal cell carcinoma, nodular type, along with the morphologic diagnosis of linear basal cell carcinoma (LBCC). The tumor was completely removed with standard excision using 5-mm margins.
Approximately 10 months after the original excision, the patient developed an irritated erosion that occasionally bled when his backpack rubbed against it. He returned to the clinic after the erosion failed to heal. Physical examination revealed a 1.4×0.7-cm, eroded, pink papule with large telangiectases at the superior pole of the excision scar. A shave biopsy confirmed the diagnosis of a recurrent infiltrative basal cell carcinoma. The tumor was then completely excised using Mohs micrographic surgery.
Comment
Linear basal cell carcinoma, first described by Lewis1 in 1985, is a rare morphologic variant of basal cell carcinoma. In 2011, Al-Niaimi and Lyon2 performed a comprehensive literature search on LBCC (1985-2008) and found only 39 cases (including 2 of their own) had been published since the pioneer case in 1985. It was determined that the most common sites affected were the periorbital area and neck (n=13 each [67%]), and the majority were histologically nodular (n=27 [69%]). Mohs micrographic surgery was the most common treatment method (n=23 [59%]), followed by primary excision (n=17 [44%]). A history of trauma, radiotherapy, or prior operation in association with the site of the LBCC was discovered in only 7 cases (18%).2 Although Peschen et al3 proposed that trauma—both physical and surgical—and radiotherapy may play a role in the development of LBCCs, the low incidence reported suggests that other factors may be involved. To determine if genetic factors were contributing to the development of LBCCs, Yamaguchi et al4 investigated the expression of p27 and PCTAIRE1, both known to contribute to tumorigenesis when mutated, as well as somatic gene mutations using deep sequencing in a case of LBCC; they found no associated genetic mutation.
Reported Cases of LBCC
According to a PubMed search of articles indexed for MEDLINE using the terms linear and basal cell carcinoma, 67 cases (including the current case) of LBCC have been published since 1985. The patient demographics, anatomic location, histologic subtype, treatment methods, and frequency of recurrence for all reported cases of LBCC are summarized in the Table.1-24 There were 36 women and 31 men, with an average age of 70 years (range, 40–92 years). The most commonly affected sites were the periocular region (n=27) and neck (n=18). Histologically, most LBCCs were nodular (n=35), with the next most common histologic subtype being infiltrative (n=20), which included the morphoeic, metatypical, and micronodular subtypes under the overarching infiltrative subtype. The most frequently chosen treatment option was primary excision (n=38 [57%]), followed by Mohs micrographic surgery (n=28 [42%]). Risk factors previously identified by Al-Niaimi and Lyon,2 including trauma, radiotherapy, or prior operation, were reported in 12 of 67 cases. Recurrence was reported in only 2 of 67 cases, 1 being the current case; however, an accurate recurrence rate could not be calculated due to lack of follow-up or short length of follow-up in most of the reported cases.
Presentation and Treatment
Currently, there are no set criteria for the diagnosis of LBCC, but it has been shown to follow a characteristic morphologic pattern, favoring extension in one direction leading to a length-to-width ratio that typically is at least 3 to 1.5 With most lesions presenting in the periocular region along relaxed skin tension lines, it has been speculated that these tumors expand along wrinkles.2 Pierard and Lapiere25 proposed that the preferential parallel orientation and a straightening of thin collagen bundles and elastic fibers within the reticular dermis combined with relaxed skin tension lines and muscle contraction perpendicular to these stromal parts may influence the growth of tumors preferentially in one direction, contributing to linearity of the lesion. In addition, the clinical appearance is not a reliable indicator of subclinical extension.2 Therefore, Lim et al6 recommended Mohs micrographic surgery as the best initial treatment of LBCCs.
Conclusion
Linear basal cell carcinoma should be considered a distinct morphologic variant of basal cell carcinoma. Although likely underreported, this variant is uncommon. It presents most often in the periocular and neck regions. The most common histologic subtypes are nodular and infiltrative. Because of the likelihood of subclinical spread, LBCC should be regarded as a high-risk subtype. As such, Mohs micrographic surgery or excision with complete circumferential peripheral and deep margin assessment is recommended as first-line treatment of LBCC.6
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1985;24:124-125.
- Al-Niaimi F, Lyon CC. Linear basal cell carcinoma: a distinct condition? Clin Exp Dermatol. 2011;36:231-234.
- Peschen M, Lo JS, Snow SN, et al. Linear basal cell carcinoma. Cutis. 1993;51:287-289.
- Yamaguchi Y, Yanagi T, Imafuku K, et al. A case of linear basal cell carcinoma: evaluation of proliferative activity by immunohistochemical staining of PCTAIRE1 and p27. J Eur Acad Dermatol Venereol. 2017;31:E359-E362.
- Mavirakis I, Malhotra R, Selva D, et al. Linear basal cell carcinoma: a distinct clinical entity. J Plast Reconstr Aesthet Surg. 2006;59:419-423.
- Lim KK, Randle HW, Roenigk RK, et al. Linear basal cell carcinoma: report of seventeen cases and review of the presentation and treatment. Dermatol Surg. 1999;25:63-67.
- Pardavila R, Rosón E, De la torre C, et al. Linear basal cell carcinoma. report of two cases [in Spanish]. Actas Dermosifiliogr. 2007;98:291.
- Shinsuke K, Hirohiko K, Yasuhiro T, et al. Linear basal cell carcinoma in an Asian patient. Open Ophthalmol J. 2007;1:20-22.
- Ning C, Chao S. Linear basal cell carcinoma of the scrotum. Dermatol Sinica. 2002;20:57-62.
- Chopra KF, Cohen PR. Linear basal cell carcinomas: report of multiple sequential tumors localized to a radiotherapy port and review of the literature. Tex Med. 1997;93:57-59.
- da Silva MO, Dadalt P, Santos OL, et al. Linear basal cell carcinoma. Int J Dermatol. 1995;34:488.
- Warthan TL, Lewis JE. Giant linear basal cell epithelioma. Int J Dermatol. 1994;33:284.
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1989;28:682-684.
- Alcántara-Reifs CM, Salido-Vallejo R, González-Menchen A, et al. Linear basal cell carcinoma: report of three cases with dermoscopic findings. Indian J Dermatol Venereol Leprol. 2016;82:708-711.
- Lee MS, Cho E, Lee JH, et al. Linearly curved, blackish macule on the wrist. Cutis. 2016;97:384, 406-407.
- Bajaj S, Sharma PK, Kar HK. Linear adamantinoid basal cell carcinoma in the axilla. Dermatol Online J. 2015;21. pii:13030/qt8k0713nb.
- Iga N, Sakurai K, Fujii H, et al. Linear basal cell carcinoma at the external genitalia. J Dermatol. 2014;41:275-276.
- Ichinokawa Y, Ohtuki A, Hattori M, et al. Linear basal cell carcinoma: a case report. Case Rep Dermatol. 2011;3:142-146.
- Becher GL, Affleck A, Fleming C, et al. Linear basal cell carcinoma occurs most commonly on the lower eyelid. Clin Exp Dermatol. 2011;36:311-312.
- Jellouli A, Triki S, Zghal M, et al. Linear basal cell carcinoma. Actas Dermosifiliogr. 2010;101:648-650.
- Takiyoshi N, Nakano H, Kaneko T, et al. A linear basal cell carcinoma undergoing spontaneous regression. Clin Exp Dermatol. 2009;34:E411-E413.
- Yoleri L, Ozden S, Kandiloglu A. A 46-year-old male with an ulcerated linear lesion on his neck. Ann Saudi Med. 2008;28:57-58.
- Palleschi GM, Corradini D, Bruscino N, et al. Linear basal cell carcinoma: clinical significance and better surgical approach. G Ital Dermatol Venereol. 2016;151:119-121.
- Rodriguez-Garijo N, Redondo P. Linear basal cell carcinoma of the lower eyelid: reconstruction with a musculocutaneous transposition flap. JAAD Case Rep. 2018;4:633-635.
- Pierard GE, Lapiere CM. Microanatomy of the dermis in relation to relaxed skin tension lines and Langer’s lines. Am J Dermatopathol. 1987;9:219-224.
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1985;24:124-125.
- Al-Niaimi F, Lyon CC. Linear basal cell carcinoma: a distinct condition? Clin Exp Dermatol. 2011;36:231-234.
- Peschen M, Lo JS, Snow SN, et al. Linear basal cell carcinoma. Cutis. 1993;51:287-289.
- Yamaguchi Y, Yanagi T, Imafuku K, et al. A case of linear basal cell carcinoma: evaluation of proliferative activity by immunohistochemical staining of PCTAIRE1 and p27. J Eur Acad Dermatol Venereol. 2017;31:E359-E362.
- Mavirakis I, Malhotra R, Selva D, et al. Linear basal cell carcinoma: a distinct clinical entity. J Plast Reconstr Aesthet Surg. 2006;59:419-423.
- Lim KK, Randle HW, Roenigk RK, et al. Linear basal cell carcinoma: report of seventeen cases and review of the presentation and treatment. Dermatol Surg. 1999;25:63-67.
- Pardavila R, Rosón E, De la torre C, et al. Linear basal cell carcinoma. report of two cases [in Spanish]. Actas Dermosifiliogr. 2007;98:291.
- Shinsuke K, Hirohiko K, Yasuhiro T, et al. Linear basal cell carcinoma in an Asian patient. Open Ophthalmol J. 2007;1:20-22.
- Ning C, Chao S. Linear basal cell carcinoma of the scrotum. Dermatol Sinica. 2002;20:57-62.
- Chopra KF, Cohen PR. Linear basal cell carcinomas: report of multiple sequential tumors localized to a radiotherapy port and review of the literature. Tex Med. 1997;93:57-59.
- da Silva MO, Dadalt P, Santos OL, et al. Linear basal cell carcinoma. Int J Dermatol. 1995;34:488.
- Warthan TL, Lewis JE. Giant linear basal cell epithelioma. Int J Dermatol. 1994;33:284.
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1989;28:682-684.
- Alcántara-Reifs CM, Salido-Vallejo R, González-Menchen A, et al. Linear basal cell carcinoma: report of three cases with dermoscopic findings. Indian J Dermatol Venereol Leprol. 2016;82:708-711.
- Lee MS, Cho E, Lee JH, et al. Linearly curved, blackish macule on the wrist. Cutis. 2016;97:384, 406-407.
- Bajaj S, Sharma PK, Kar HK. Linear adamantinoid basal cell carcinoma in the axilla. Dermatol Online J. 2015;21. pii:13030/qt8k0713nb.
- Iga N, Sakurai K, Fujii H, et al. Linear basal cell carcinoma at the external genitalia. J Dermatol. 2014;41:275-276.
- Ichinokawa Y, Ohtuki A, Hattori M, et al. Linear basal cell carcinoma: a case report. Case Rep Dermatol. 2011;3:142-146.
- Becher GL, Affleck A, Fleming C, et al. Linear basal cell carcinoma occurs most commonly on the lower eyelid. Clin Exp Dermatol. 2011;36:311-312.
- Jellouli A, Triki S, Zghal M, et al. Linear basal cell carcinoma. Actas Dermosifiliogr. 2010;101:648-650.
- Takiyoshi N, Nakano H, Kaneko T, et al. A linear basal cell carcinoma undergoing spontaneous regression. Clin Exp Dermatol. 2009;34:E411-E413.
- Yoleri L, Ozden S, Kandiloglu A. A 46-year-old male with an ulcerated linear lesion on his neck. Ann Saudi Med. 2008;28:57-58.
- Palleschi GM, Corradini D, Bruscino N, et al. Linear basal cell carcinoma: clinical significance and better surgical approach. G Ital Dermatol Venereol. 2016;151:119-121.
- Rodriguez-Garijo N, Redondo P. Linear basal cell carcinoma of the lower eyelid: reconstruction with a musculocutaneous transposition flap. JAAD Case Rep. 2018;4:633-635.
- Pierard GE, Lapiere CM. Microanatomy of the dermis in relation to relaxed skin tension lines and Langer’s lines. Am J Dermatopathol. 1987;9:219-224.
Practice Points
- Linear basal cell carcinoma (LBCC) follows a characteristic morphologic pattern of a length-to-width ratio that typically is at least 3 to 1.
- Linear basal cell carcinomas most commonly present in the periocular region and on the neck along relaxed skin tension lines.
- Because of the likelihood of subclinical spread, LBCC should be regarded as a high-risk subtype of basal cell carcinoma.
- Mohs micrographic surgery or excision with complete circumferential peripheral and deep-margin assessment is recommended as first-line treatment.
Review of Radiologic Considerations in an Immunocompetent Patient With Primary Central Nervous System Lymphoma (FULL)
Central nervous system (CNS) lymphoma can be classified into 2 categories: primary CNS lymphoma (PCNSL), which includes disease limited to brain, eyes, spinal cord; and leptomeninges without coexisting or previous systemic lymphoma. Secondary CNS lymphoma (SCNSL) is essentially metastatic disease from a systemic primary site.1 The focus of this case presentation is PCNSL, with an emphasis on imaging characteristics and differential diagnosis.
The median age at diagnosis for PCNSL is 65 years, and the overall incidence has been decreasing since the mid-1990s, likely related to the increased use of highly-active antiretroviral therapy (HAART) in patients with AIDS.2,3 Although overall incidence has decreased, incidence in the elderly population has increased.4 Historically, PCNSL has been considered an AIDS-defining illness.5 These patients, among other immunocompromised patients, such as those on chronic immunosuppressive therapy, are at a higher risk for developing the malignancy.6
Clinical presentation varies because of the location of CNS involvement and may present with headache, mood or personality disturbances, or focal neurologic deficits. Seizures are less likely due to the tendency of PCNSL to spare gray matter. Initial workup generally includes a head computed tomography (CT) scan, as well as a contrast-enhanced magnetic resonance image (MRI), which may help direct clinicians to the appropriate diagnosis. However, there is significant overlap between the imaging characteristics of PCNSL and numerous other disease processes, including glioblastoma and demyelination. The imaging characteristics of PCNSL are considerably different depending on the patient’s immune status.7
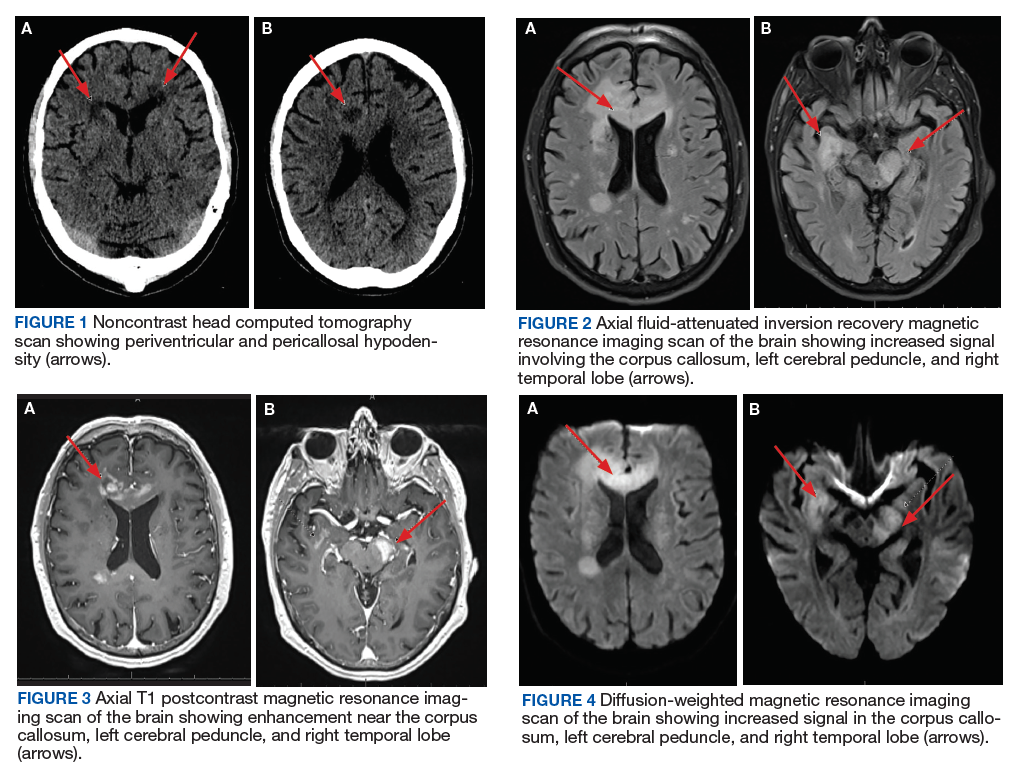
This case illustrates a rare presentation of PCNSL in an immunocompetent patient whose MRI characteristics were seemingly more consistent with those seen in patients with immunodeficiency. The main differential diagnoses and key imaging characteristics, which may help obtain accurate diagnosis, will be discussed.
Case Presentation
A 72-year-old male veteran presented with a 2-month history of subjective weakness in his upper and lower extremities progressing to multiple falls at home. He had no significant medical history other than a thymectomy at age 15 for an enlarged thymus, which per patient report, was benign. An initial laboratory test that included vitamin B12, folate, thyroid-stimulating hormone, complete blood cell count, and comprehensive metabolic panel, were unremarkable, with a white blood cell count of 8.5 K/uL. The initial neurologic evaluation did not show any focal neurologic deficits; however, during the initial hospital stay, the patient developed increasing lower extremity weakness on examination. A noncontrast CT head scan showed extensive nonspecific hypodensities within the periventricular white matter (Figure 1). A contrast-enhanced MRI showed enhancing lesions involving the corpus callosum, left cerebral peduncle, and right temporal lobe (Figures 2, 3, and 4). These lesions also exhibited significant restricted diffusion and a mild amount of surrounding vasogenic edema. The working diagnosis after the MRI included primary CNS lymphoma, multifocal glioblastoma, and tumefactive demyelinating disease. The patient was started on IV steroids and transferred for neurosurgical evaluation and biopsy at an outside hospital. The frontal lesion was biopsied, and the initial frozen section was consistent with lymphoma; a bone marrow biopsy was negative. The workup for immunodeficiency was unremarkable. Pathology revealed high-grade B-cell lymphoma, and the patient began a chemotherapy regimen.
Discussion
The workup of altered mental status, focal neurologic deficits, headaches, or other neurologic conditions often begins with a noncontrast CT scan. On CT, PCNSL generally appears isodense to hyperdense to gray matter, but appearance is variable. The often hyperdense appearance is attributable to the hypercellular nature of lymphoma. Many times, as in this case, CT may show only vague hypodensities, some of which may be associated with surrounding edema. This presentation is nonspecific and may be seen with advancing age due to changes of chronic microvascular ischemia as well as demyelination, other malignancies, and several other disease processes, both benign and malignant. After the initial CT scan, further workup requires evaluation with MRI. PCNSL exhibits restricted diffusion and variable signal intensity on T2-weighted imaging.
PCNSL is frequently centrally located within the periventricular white matter, often within the frontal lobe but can involve other lobes, the basal ganglia, brainstem, cerebellum, or less likely, the spinal canal.7 Contrary to primary CNS disease, secondary lymphoma within the CNS has been described classically as affecting a leptomeningeal (pia and arachnoid mater) distribution two-thirds of the time, with parenchymal involvement occurring in the other one-third of patients. A recent study by Malikova and colleagues found parenchymal involvement may be much more common than previously thought.1 Leptomeningeal spread of disease often involves the cranial nerves, subependymal regions, spinal cord, or spinal nerve roots. Dural involvement in primary or secondary lymphoma is rare.
PCNSL nearly always shows enhancement. Linear enhancement along perivascular spaces is highly characteristic of PCNSL. The typical appearance of PCNSL associated with immunodeficiency varies from that seen in an otherwise immunocompetent patient. Patients with immunodeficiency usually have multifocal involvement, central necrosis leading to a ring enhancement appearance, and have more propensity for spontaneous hemorrhage.7 Immunocompetent patients are less likely to present with multifocal disease and rarely show ring enhancement. Also, spontaneous hemorrhage is rare in immunocompetent patients. In our case, extensive multifocal involvement was present, whereas typically immunocompetent patients will present with a solitary homogeneously enhancing parenchymal mass.
The primary differential for PCNSL includes malignant glioma, tumefactive multiple sclerosis, metastatic disease, and in an immunocompromised patient, toxoplasmosis. The degree of associated vasogenic edema and mass effect is generally lower in PCNSL than that of malignant gliomas and metastasis. Also, PCNSL tends to spare the cerebral cortex.8
Classically, PCNSL, malignant gliomas, and demyelinating disease have been considered the main differential for lesions that cross midline and involve both cerebral hemispheres. Lymphoma generally exhibits more restricted diffusion than malignant gliomas and metastasis, attributable to the highly cellular nature of lymphoma.7 Tumefactive multiple sclerosis is associated with relatively minimal mass effect for lesion size and exhibits less restricted diffusion values when compared to high grade gliomas and PCNSL. One fairly specific finding for tumefactive demyelinating lesions is incomplete rim enhancement.9 Unfortunately, an MRI is not reliable in differentiating these entities, and biopsy is required for definitive diagnosis. Many advancing imaging modalities may help provide the correct diagnosis of PCNSL, including diffusion-weighted and apparent diffusion coefficient imaging, diffusion tensor imaging, MR spectroscopy and PET imaging.7
Conclusion
With the increasing use of HAART, the paradigm of PCNSL is shifting toward one predominantly affecting immunocompetent patients. PCNSL should be considered in any patient with multiple enhancing CNS lesions, regardless of immune status. Several key imaging characteristics may help differentiate PCNSL and other disease processes; however, at this time, biopsy is recommended for definitive diagnosis.
1. Malikova H, Burghardtova M, Koubska E, Mandys V, Kozak T, Weichet J. Secondary central nervous system lymphoma: spectrum of morphological MRI appearances. Neuropsychiatr Dis Treat. 2018;4:733-740.
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(suppl 5):v1-v49.
3. Diamond C, Taylor TH, Aboumrad T, Anton-Culver H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: incidence, presentation, treatment, and survival. Cancer. 2006;106(1):128-135.
4. O’Neill BP, Decker PA, Tieu C, Cerhan JR. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkins lymphoma. Am J Hematol. 2013;88(12):997-1000.
5. [no authors listed]. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(rr-17):1-19.
6. Maiuri F. Central nervous system lymphomas and immunodeficiency. Neurological Research. 1989;11(1):2-5.
7. Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR Am J Neuroradiol. 2010;32(6):984-992.
8. Gómez Roselló E, Quiles Granado AM, Laguillo Sala G, Gutiérrez S. Primary central nervous system lymphoma in immunocompetent patients: spectrum of findings and differential characteristics. Radiología. 2018;60(4):280-289.
9. Mabray MC, Cohen BA, Villanueva-Meyer JE, et al. Performance of Apparent Diffusion Coefficient Values and Conventional MRI Features in Differentiating Tumefactive Demyelinating Lesions From Primary Brain Neoplasms. American Journal of Roentgenology. 2015;205(5):1075-1085.
Central nervous system (CNS) lymphoma can be classified into 2 categories: primary CNS lymphoma (PCNSL), which includes disease limited to brain, eyes, spinal cord; and leptomeninges without coexisting or previous systemic lymphoma. Secondary CNS lymphoma (SCNSL) is essentially metastatic disease from a systemic primary site.1 The focus of this case presentation is PCNSL, with an emphasis on imaging characteristics and differential diagnosis.
The median age at diagnosis for PCNSL is 65 years, and the overall incidence has been decreasing since the mid-1990s, likely related to the increased use of highly-active antiretroviral therapy (HAART) in patients with AIDS.2,3 Although overall incidence has decreased, incidence in the elderly population has increased.4 Historically, PCNSL has been considered an AIDS-defining illness.5 These patients, among other immunocompromised patients, such as those on chronic immunosuppressive therapy, are at a higher risk for developing the malignancy.6
Clinical presentation varies because of the location of CNS involvement and may present with headache, mood or personality disturbances, or focal neurologic deficits. Seizures are less likely due to the tendency of PCNSL to spare gray matter. Initial workup generally includes a head computed tomography (CT) scan, as well as a contrast-enhanced magnetic resonance image (MRI), which may help direct clinicians to the appropriate diagnosis. However, there is significant overlap between the imaging characteristics of PCNSL and numerous other disease processes, including glioblastoma and demyelination. The imaging characteristics of PCNSL are considerably different depending on the patient’s immune status.7
This case illustrates a rare presentation of PCNSL in an immunocompetent patient whose MRI characteristics were seemingly more consistent with those seen in patients with immunodeficiency. The main differential diagnoses and key imaging characteristics, which may help obtain accurate diagnosis, will be discussed.
Case Presentation
A 72-year-old male veteran presented with a 2-month history of subjective weakness in his upper and lower extremities progressing to multiple falls at home. He had no significant medical history other than a thymectomy at age 15 for an enlarged thymus, which per patient report, was benign. An initial laboratory test that included vitamin B12, folate, thyroid-stimulating hormone, complete blood cell count, and comprehensive metabolic panel, were unremarkable, with a white blood cell count of 8.5 K/uL. The initial neurologic evaluation did not show any focal neurologic deficits; however, during the initial hospital stay, the patient developed increasing lower extremity weakness on examination. A noncontrast CT head scan showed extensive nonspecific hypodensities within the periventricular white matter (Figure 1). A contrast-enhanced MRI showed enhancing lesions involving the corpus callosum, left cerebral peduncle, and right temporal lobe (Figures 2, 3, and 4). These lesions also exhibited significant restricted diffusion and a mild amount of surrounding vasogenic edema. The working diagnosis after the MRI included primary CNS lymphoma, multifocal glioblastoma, and tumefactive demyelinating disease. The patient was started on IV steroids and transferred for neurosurgical evaluation and biopsy at an outside hospital. The frontal lesion was biopsied, and the initial frozen section was consistent with lymphoma; a bone marrow biopsy was negative. The workup for immunodeficiency was unremarkable. Pathology revealed high-grade B-cell lymphoma, and the patient began a chemotherapy regimen.
Discussion
The workup of altered mental status, focal neurologic deficits, headaches, or other neurologic conditions often begins with a noncontrast CT scan. On CT, PCNSL generally appears isodense to hyperdense to gray matter, but appearance is variable. The often hyperdense appearance is attributable to the hypercellular nature of lymphoma. Many times, as in this case, CT may show only vague hypodensities, some of which may be associated with surrounding edema. This presentation is nonspecific and may be seen with advancing age due to changes of chronic microvascular ischemia as well as demyelination, other malignancies, and several other disease processes, both benign and malignant. After the initial CT scan, further workup requires evaluation with MRI. PCNSL exhibits restricted diffusion and variable signal intensity on T2-weighted imaging.
PCNSL is frequently centrally located within the periventricular white matter, often within the frontal lobe but can involve other lobes, the basal ganglia, brainstem, cerebellum, or less likely, the spinal canal.7 Contrary to primary CNS disease, secondary lymphoma within the CNS has been described classically as affecting a leptomeningeal (pia and arachnoid mater) distribution two-thirds of the time, with parenchymal involvement occurring in the other one-third of patients. A recent study by Malikova and colleagues found parenchymal involvement may be much more common than previously thought.1 Leptomeningeal spread of disease often involves the cranial nerves, subependymal regions, spinal cord, or spinal nerve roots. Dural involvement in primary or secondary lymphoma is rare.
PCNSL nearly always shows enhancement. Linear enhancement along perivascular spaces is highly characteristic of PCNSL. The typical appearance of PCNSL associated with immunodeficiency varies from that seen in an otherwise immunocompetent patient. Patients with immunodeficiency usually have multifocal involvement, central necrosis leading to a ring enhancement appearance, and have more propensity for spontaneous hemorrhage.7 Immunocompetent patients are less likely to present with multifocal disease and rarely show ring enhancement. Also, spontaneous hemorrhage is rare in immunocompetent patients. In our case, extensive multifocal involvement was present, whereas typically immunocompetent patients will present with a solitary homogeneously enhancing parenchymal mass.
The primary differential for PCNSL includes malignant glioma, tumefactive multiple sclerosis, metastatic disease, and in an immunocompromised patient, toxoplasmosis. The degree of associated vasogenic edema and mass effect is generally lower in PCNSL than that of malignant gliomas and metastasis. Also, PCNSL tends to spare the cerebral cortex.8
Classically, PCNSL, malignant gliomas, and demyelinating disease have been considered the main differential for lesions that cross midline and involve both cerebral hemispheres. Lymphoma generally exhibits more restricted diffusion than malignant gliomas and metastasis, attributable to the highly cellular nature of lymphoma.7 Tumefactive multiple sclerosis is associated with relatively minimal mass effect for lesion size and exhibits less restricted diffusion values when compared to high grade gliomas and PCNSL. One fairly specific finding for tumefactive demyelinating lesions is incomplete rim enhancement.9 Unfortunately, an MRI is not reliable in differentiating these entities, and biopsy is required for definitive diagnosis. Many advancing imaging modalities may help provide the correct diagnosis of PCNSL, including diffusion-weighted and apparent diffusion coefficient imaging, diffusion tensor imaging, MR spectroscopy and PET imaging.7
Conclusion
With the increasing use of HAART, the paradigm of PCNSL is shifting toward one predominantly affecting immunocompetent patients. PCNSL should be considered in any patient with multiple enhancing CNS lesions, regardless of immune status. Several key imaging characteristics may help differentiate PCNSL and other disease processes; however, at this time, biopsy is recommended for definitive diagnosis.
Central nervous system (CNS) lymphoma can be classified into 2 categories: primary CNS lymphoma (PCNSL), which includes disease limited to brain, eyes, spinal cord; and leptomeninges without coexisting or previous systemic lymphoma. Secondary CNS lymphoma (SCNSL) is essentially metastatic disease from a systemic primary site.1 The focus of this case presentation is PCNSL, with an emphasis on imaging characteristics and differential diagnosis.
The median age at diagnosis for PCNSL is 65 years, and the overall incidence has been decreasing since the mid-1990s, likely related to the increased use of highly-active antiretroviral therapy (HAART) in patients with AIDS.2,3 Although overall incidence has decreased, incidence in the elderly population has increased.4 Historically, PCNSL has been considered an AIDS-defining illness.5 These patients, among other immunocompromised patients, such as those on chronic immunosuppressive therapy, are at a higher risk for developing the malignancy.6
Clinical presentation varies because of the location of CNS involvement and may present with headache, mood or personality disturbances, or focal neurologic deficits. Seizures are less likely due to the tendency of PCNSL to spare gray matter. Initial workup generally includes a head computed tomography (CT) scan, as well as a contrast-enhanced magnetic resonance image (MRI), which may help direct clinicians to the appropriate diagnosis. However, there is significant overlap between the imaging characteristics of PCNSL and numerous other disease processes, including glioblastoma and demyelination. The imaging characteristics of PCNSL are considerably different depending on the patient’s immune status.7
This case illustrates a rare presentation of PCNSL in an immunocompetent patient whose MRI characteristics were seemingly more consistent with those seen in patients with immunodeficiency. The main differential diagnoses and key imaging characteristics, which may help obtain accurate diagnosis, will be discussed.
Case Presentation
A 72-year-old male veteran presented with a 2-month history of subjective weakness in his upper and lower extremities progressing to multiple falls at home. He had no significant medical history other than a thymectomy at age 15 for an enlarged thymus, which per patient report, was benign. An initial laboratory test that included vitamin B12, folate, thyroid-stimulating hormone, complete blood cell count, and comprehensive metabolic panel, were unremarkable, with a white blood cell count of 8.5 K/uL. The initial neurologic evaluation did not show any focal neurologic deficits; however, during the initial hospital stay, the patient developed increasing lower extremity weakness on examination. A noncontrast CT head scan showed extensive nonspecific hypodensities within the periventricular white matter (Figure 1). A contrast-enhanced MRI showed enhancing lesions involving the corpus callosum, left cerebral peduncle, and right temporal lobe (Figures 2, 3, and 4). These lesions also exhibited significant restricted diffusion and a mild amount of surrounding vasogenic edema. The working diagnosis after the MRI included primary CNS lymphoma, multifocal glioblastoma, and tumefactive demyelinating disease. The patient was started on IV steroids and transferred for neurosurgical evaluation and biopsy at an outside hospital. The frontal lesion was biopsied, and the initial frozen section was consistent with lymphoma; a bone marrow biopsy was negative. The workup for immunodeficiency was unremarkable. Pathology revealed high-grade B-cell lymphoma, and the patient began a chemotherapy regimen.
Discussion
The workup of altered mental status, focal neurologic deficits, headaches, or other neurologic conditions often begins with a noncontrast CT scan. On CT, PCNSL generally appears isodense to hyperdense to gray matter, but appearance is variable. The often hyperdense appearance is attributable to the hypercellular nature of lymphoma. Many times, as in this case, CT may show only vague hypodensities, some of which may be associated with surrounding edema. This presentation is nonspecific and may be seen with advancing age due to changes of chronic microvascular ischemia as well as demyelination, other malignancies, and several other disease processes, both benign and malignant. After the initial CT scan, further workup requires evaluation with MRI. PCNSL exhibits restricted diffusion and variable signal intensity on T2-weighted imaging.
PCNSL is frequently centrally located within the periventricular white matter, often within the frontal lobe but can involve other lobes, the basal ganglia, brainstem, cerebellum, or less likely, the spinal canal.7 Contrary to primary CNS disease, secondary lymphoma within the CNS has been described classically as affecting a leptomeningeal (pia and arachnoid mater) distribution two-thirds of the time, with parenchymal involvement occurring in the other one-third of patients. A recent study by Malikova and colleagues found parenchymal involvement may be much more common than previously thought.1 Leptomeningeal spread of disease often involves the cranial nerves, subependymal regions, spinal cord, or spinal nerve roots. Dural involvement in primary or secondary lymphoma is rare.
PCNSL nearly always shows enhancement. Linear enhancement along perivascular spaces is highly characteristic of PCNSL. The typical appearance of PCNSL associated with immunodeficiency varies from that seen in an otherwise immunocompetent patient. Patients with immunodeficiency usually have multifocal involvement, central necrosis leading to a ring enhancement appearance, and have more propensity for spontaneous hemorrhage.7 Immunocompetent patients are less likely to present with multifocal disease and rarely show ring enhancement. Also, spontaneous hemorrhage is rare in immunocompetent patients. In our case, extensive multifocal involvement was present, whereas typically immunocompetent patients will present with a solitary homogeneously enhancing parenchymal mass.
The primary differential for PCNSL includes malignant glioma, tumefactive multiple sclerosis, metastatic disease, and in an immunocompromised patient, toxoplasmosis. The degree of associated vasogenic edema and mass effect is generally lower in PCNSL than that of malignant gliomas and metastasis. Also, PCNSL tends to spare the cerebral cortex.8
Classically, PCNSL, malignant gliomas, and demyelinating disease have been considered the main differential for lesions that cross midline and involve both cerebral hemispheres. Lymphoma generally exhibits more restricted diffusion than malignant gliomas and metastasis, attributable to the highly cellular nature of lymphoma.7 Tumefactive multiple sclerosis is associated with relatively minimal mass effect for lesion size and exhibits less restricted diffusion values when compared to high grade gliomas and PCNSL. One fairly specific finding for tumefactive demyelinating lesions is incomplete rim enhancement.9 Unfortunately, an MRI is not reliable in differentiating these entities, and biopsy is required for definitive diagnosis. Many advancing imaging modalities may help provide the correct diagnosis of PCNSL, including diffusion-weighted and apparent diffusion coefficient imaging, diffusion tensor imaging, MR spectroscopy and PET imaging.7
Conclusion
With the increasing use of HAART, the paradigm of PCNSL is shifting toward one predominantly affecting immunocompetent patients. PCNSL should be considered in any patient with multiple enhancing CNS lesions, regardless of immune status. Several key imaging characteristics may help differentiate PCNSL and other disease processes; however, at this time, biopsy is recommended for definitive diagnosis.
1. Malikova H, Burghardtova M, Koubska E, Mandys V, Kozak T, Weichet J. Secondary central nervous system lymphoma: spectrum of morphological MRI appearances. Neuropsychiatr Dis Treat. 2018;4:733-740.
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(suppl 5):v1-v49.
3. Diamond C, Taylor TH, Aboumrad T, Anton-Culver H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: incidence, presentation, treatment, and survival. Cancer. 2006;106(1):128-135.
4. O’Neill BP, Decker PA, Tieu C, Cerhan JR. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkins lymphoma. Am J Hematol. 2013;88(12):997-1000.
5. [no authors listed]. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(rr-17):1-19.
6. Maiuri F. Central nervous system lymphomas and immunodeficiency. Neurological Research. 1989;11(1):2-5.
7. Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR Am J Neuroradiol. 2010;32(6):984-992.
8. Gómez Roselló E, Quiles Granado AM, Laguillo Sala G, Gutiérrez S. Primary central nervous system lymphoma in immunocompetent patients: spectrum of findings and differential characteristics. Radiología. 2018;60(4):280-289.
9. Mabray MC, Cohen BA, Villanueva-Meyer JE, et al. Performance of Apparent Diffusion Coefficient Values and Conventional MRI Features in Differentiating Tumefactive Demyelinating Lesions From Primary Brain Neoplasms. American Journal of Roentgenology. 2015;205(5):1075-1085.
1. Malikova H, Burghardtova M, Koubska E, Mandys V, Kozak T, Weichet J. Secondary central nervous system lymphoma: spectrum of morphological MRI appearances. Neuropsychiatr Dis Treat. 2018;4:733-740.
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(suppl 5):v1-v49.
3. Diamond C, Taylor TH, Aboumrad T, Anton-Culver H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: incidence, presentation, treatment, and survival. Cancer. 2006;106(1):128-135.
4. O’Neill BP, Decker PA, Tieu C, Cerhan JR. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkins lymphoma. Am J Hematol. 2013;88(12):997-1000.
5. [no authors listed]. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(rr-17):1-19.
6. Maiuri F. Central nervous system lymphomas and immunodeficiency. Neurological Research. 1989;11(1):2-5.
7. Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR Am J Neuroradiol. 2010;32(6):984-992.
8. Gómez Roselló E, Quiles Granado AM, Laguillo Sala G, Gutiérrez S. Primary central nervous system lymphoma in immunocompetent patients: spectrum of findings and differential characteristics. Radiología. 2018;60(4):280-289.
9. Mabray MC, Cohen BA, Villanueva-Meyer JE, et al. Performance of Apparent Diffusion Coefficient Values and Conventional MRI Features in Differentiating Tumefactive Demyelinating Lesions From Primary Brain Neoplasms. American Journal of Roentgenology. 2015;205(5):1075-1085.
A Reticular Rash on the Leg
A 73-year-old male veteran with a history of ischemic stroke with left-sided deficits and edema, falls, poorly controlled hypertension, active tobacco use, obesity, and prediabetes was assessed on a routine visit by our home-based primary care team and found to have a new, unilateral, asymptomatic rash. He reported feeling no pain in the affected area or any significant increase in the baseline left lower extremity edema and weakness resulting from his stroke 2 years prior.
On the left lateral leg from mid-thigh to mid-calf, there was a nontender, flat, reticulated rash with pigmentary alteration ranging from light brown to dark brown (Figure).
On further questioning, the patient reported regular use of a space heater because his gas furnace had been destroyed in an earthquake more than 20 years before. He would place this heater close to his left leg when using the computer or while sleeping in his wheelchair.
- What is your diagnosis?
- How would you treat this patient?
Our Diagnosis
Erythema ab igne, also called hot water bottle rash, is a clinical diagnosis based on characteristic cutaneous findings and a clear history of chronic, moderate heat or infrared exposure.1 Although exposure to space heaters, open fire, radiators, hot water bottles, and heating pads are the classic causes, recently there have been reports of laptop computers, cell phones, infrared food lamps, automobile seat heaters, and heated recliners causing the same type of skin reaction.2
With chronic moderate heat or infrared exposure, the rash usually progresses over days to months. It begins as a mild, transient, reticulated, erythematous rash, which follows the pattern of the cutaneous venous plexus and resolves minutes to hours after removal of the offending source as vasodilation resolves. After months of continued exposure, the dermis around the affected vasculature eventually becomes hyperpigmented due to the deposition of melanin and sometimes hemosiderin.
The rash is usually asymptomatic but has been associated with pain, pruritis, and/or tingling. Once the diagnosis is made, treatment involves removal of the offending source. The discoloration may resolve over months to years, but permanent hyperpigmentation is not uncommon. There are a few case reports on treatment using Nd-Yag laser therapy, topical hydroquinone and tretinoin, 5-fluorouracil, and systemic mesoglycan with topical bioflavonoids.2-4
While the prognosis of erythema ab igne is excellent if detected early, failure to recognize this condition and remove the offending source can lead to sequalae, such as squamous cell carcinoma, poorly differentiated carcinoma, cutaneous marginal zone lymphoma, and Merkel cell carcinoma.5-8 Development of malignancy typically has a latency period of > 30 years. Patients should have periodic surveillance of their skin and any suspicious lesion in the involved area should be considered for biopsy.

Rashes may represent systemic or more localized pathology (Table). In contrast to erythema ab igne, the rash associated with a vasculitic process (autoimmune, drug-induced, or infectious) tends to be more generalized and bilateral but still follows the pattern of the cutaneous venous plexus. An example of this would be livedo reticularis. Although this rash is reticular, it is not hyperpigmented.9 A variant of livedo reticularis is cutis marmorata, which develops in response to cold exposure, particularly in infants or in the setting of hypothyroidism.Cutis marmorata is erythematous, blanchable, and reversible with rewarming. Unlike erythema ab igne, there is no hyperpigmentation and tends to be more diffuse.10
When evaluating a reticular rash, consider local and systemic etiologies. If more localized and hyperpigmented, ask about heat or infrared exposure. This may point to a diagnosis of erythema ab igne.
1. Page EH, Shear NH. Temperature-dependent skin disorders. J Am Acad Dermatol. 1988;18(5, pt 1):1003-1019.
2. Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162(1):77-78.
3. Kim HW, Kim EJ, Park HC, Ko JY, Ro YS, Kim JE. Erythema ab igne successfully treated with low fluenced 1,064-nm Q-switched Neodymium-Doped Yttrium Aluminum Garnet laser. J Cosmet Laser Ther. 2014;16(3):147-148.
4. Gianfaldoni S, Gianfaldoni R, Tchernev G, Lotti J, Wollina U, Lotti T. Erythema ab igne successfully treated with mesoglycan and bioflavonoids: a case-report. Open Access Maced J Med Sci. 2017;5(4):432-435.
5. Arrington JH 3rd, Lockman DS. Thermal keratoses and squamous cell carcinoma in situ associated with erythema ab igne. AMA Arch Derm. 1979;115(10):1226-1228.
6. Sigmon JR, Cantrell J, Teague D, Sangueza O, Sheehan DJ. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35(6):676-678
7. Wharton J, Roffwarg D, Miller J, Sheehan DJ. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62(6):1080-1081.
8. Jones CS. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124(1):110-113.
9. Sajjan VV, Lunge S, Swamy MB, Pandit AM. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6(5):315-321.
10. O’Connor NR, McLaughlin MR, Ham P. Newborn skin: part I. Common rashes. Am Fam Physician. 2008;77(1):47-52.
A 73-year-old male veteran with a history of ischemic stroke with left-sided deficits and edema, falls, poorly controlled hypertension, active tobacco use, obesity, and prediabetes was assessed on a routine visit by our home-based primary care team and found to have a new, unilateral, asymptomatic rash. He reported feeling no pain in the affected area or any significant increase in the baseline left lower extremity edema and weakness resulting from his stroke 2 years prior.
On the left lateral leg from mid-thigh to mid-calf, there was a nontender, flat, reticulated rash with pigmentary alteration ranging from light brown to dark brown (Figure).
On further questioning, the patient reported regular use of a space heater because his gas furnace had been destroyed in an earthquake more than 20 years before. He would place this heater close to his left leg when using the computer or while sleeping in his wheelchair.
- What is your diagnosis?
- How would you treat this patient?
Our Diagnosis
Erythema ab igne, also called hot water bottle rash, is a clinical diagnosis based on characteristic cutaneous findings and a clear history of chronic, moderate heat or infrared exposure.1 Although exposure to space heaters, open fire, radiators, hot water bottles, and heating pads are the classic causes, recently there have been reports of laptop computers, cell phones, infrared food lamps, automobile seat heaters, and heated recliners causing the same type of skin reaction.2
With chronic moderate heat or infrared exposure, the rash usually progresses over days to months. It begins as a mild, transient, reticulated, erythematous rash, which follows the pattern of the cutaneous venous plexus and resolves minutes to hours after removal of the offending source as vasodilation resolves. After months of continued exposure, the dermis around the affected vasculature eventually becomes hyperpigmented due to the deposition of melanin and sometimes hemosiderin.
The rash is usually asymptomatic but has been associated with pain, pruritis, and/or tingling. Once the diagnosis is made, treatment involves removal of the offending source. The discoloration may resolve over months to years, but permanent hyperpigmentation is not uncommon. There are a few case reports on treatment using Nd-Yag laser therapy, topical hydroquinone and tretinoin, 5-fluorouracil, and systemic mesoglycan with topical bioflavonoids.2-4
While the prognosis of erythema ab igne is excellent if detected early, failure to recognize this condition and remove the offending source can lead to sequalae, such as squamous cell carcinoma, poorly differentiated carcinoma, cutaneous marginal zone lymphoma, and Merkel cell carcinoma.5-8 Development of malignancy typically has a latency period of > 30 years. Patients should have periodic surveillance of their skin and any suspicious lesion in the involved area should be considered for biopsy.
Rashes may represent systemic or more localized pathology (Table). In contrast to erythema ab igne, the rash associated with a vasculitic process (autoimmune, drug-induced, or infectious) tends to be more generalized and bilateral but still follows the pattern of the cutaneous venous plexus. An example of this would be livedo reticularis. Although this rash is reticular, it is not hyperpigmented.9 A variant of livedo reticularis is cutis marmorata, which develops in response to cold exposure, particularly in infants or in the setting of hypothyroidism.Cutis marmorata is erythematous, blanchable, and reversible with rewarming. Unlike erythema ab igne, there is no hyperpigmentation and tends to be more diffuse.10
When evaluating a reticular rash, consider local and systemic etiologies. If more localized and hyperpigmented, ask about heat or infrared exposure. This may point to a diagnosis of erythema ab igne.
A 73-year-old male veteran with a history of ischemic stroke with left-sided deficits and edema, falls, poorly controlled hypertension, active tobacco use, obesity, and prediabetes was assessed on a routine visit by our home-based primary care team and found to have a new, unilateral, asymptomatic rash. He reported feeling no pain in the affected area or any significant increase in the baseline left lower extremity edema and weakness resulting from his stroke 2 years prior.
On the left lateral leg from mid-thigh to mid-calf, there was a nontender, flat, reticulated rash with pigmentary alteration ranging from light brown to dark brown (Figure).
On further questioning, the patient reported regular use of a space heater because his gas furnace had been destroyed in an earthquake more than 20 years before. He would place this heater close to his left leg when using the computer or while sleeping in his wheelchair.
- What is your diagnosis?
- How would you treat this patient?
Our Diagnosis
Erythema ab igne, also called hot water bottle rash, is a clinical diagnosis based on characteristic cutaneous findings and a clear history of chronic, moderate heat or infrared exposure.1 Although exposure to space heaters, open fire, radiators, hot water bottles, and heating pads are the classic causes, recently there have been reports of laptop computers, cell phones, infrared food lamps, automobile seat heaters, and heated recliners causing the same type of skin reaction.2
With chronic moderate heat or infrared exposure, the rash usually progresses over days to months. It begins as a mild, transient, reticulated, erythematous rash, which follows the pattern of the cutaneous venous plexus and resolves minutes to hours after removal of the offending source as vasodilation resolves. After months of continued exposure, the dermis around the affected vasculature eventually becomes hyperpigmented due to the deposition of melanin and sometimes hemosiderin.
The rash is usually asymptomatic but has been associated with pain, pruritis, and/or tingling. Once the diagnosis is made, treatment involves removal of the offending source. The discoloration may resolve over months to years, but permanent hyperpigmentation is not uncommon. There are a few case reports on treatment using Nd-Yag laser therapy, topical hydroquinone and tretinoin, 5-fluorouracil, and systemic mesoglycan with topical bioflavonoids.2-4
While the prognosis of erythema ab igne is excellent if detected early, failure to recognize this condition and remove the offending source can lead to sequalae, such as squamous cell carcinoma, poorly differentiated carcinoma, cutaneous marginal zone lymphoma, and Merkel cell carcinoma.5-8 Development of malignancy typically has a latency period of > 30 years. Patients should have periodic surveillance of their skin and any suspicious lesion in the involved area should be considered for biopsy.
Rashes may represent systemic or more localized pathology (Table). In contrast to erythema ab igne, the rash associated with a vasculitic process (autoimmune, drug-induced, or infectious) tends to be more generalized and bilateral but still follows the pattern of the cutaneous venous plexus. An example of this would be livedo reticularis. Although this rash is reticular, it is not hyperpigmented.9 A variant of livedo reticularis is cutis marmorata, which develops in response to cold exposure, particularly in infants or in the setting of hypothyroidism.Cutis marmorata is erythematous, blanchable, and reversible with rewarming. Unlike erythema ab igne, there is no hyperpigmentation and tends to be more diffuse.10
When evaluating a reticular rash, consider local and systemic etiologies. If more localized and hyperpigmented, ask about heat or infrared exposure. This may point to a diagnosis of erythema ab igne.
1. Page EH, Shear NH. Temperature-dependent skin disorders. J Am Acad Dermatol. 1988;18(5, pt 1):1003-1019.
2. Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162(1):77-78.
3. Kim HW, Kim EJ, Park HC, Ko JY, Ro YS, Kim JE. Erythema ab igne successfully treated with low fluenced 1,064-nm Q-switched Neodymium-Doped Yttrium Aluminum Garnet laser. J Cosmet Laser Ther. 2014;16(3):147-148.
4. Gianfaldoni S, Gianfaldoni R, Tchernev G, Lotti J, Wollina U, Lotti T. Erythema ab igne successfully treated with mesoglycan and bioflavonoids: a case-report. Open Access Maced J Med Sci. 2017;5(4):432-435.
5. Arrington JH 3rd, Lockman DS. Thermal keratoses and squamous cell carcinoma in situ associated with erythema ab igne. AMA Arch Derm. 1979;115(10):1226-1228.
6. Sigmon JR, Cantrell J, Teague D, Sangueza O, Sheehan DJ. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35(6):676-678
7. Wharton J, Roffwarg D, Miller J, Sheehan DJ. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62(6):1080-1081.
8. Jones CS. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124(1):110-113.
9. Sajjan VV, Lunge S, Swamy MB, Pandit AM. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6(5):315-321.
10. O’Connor NR, McLaughlin MR, Ham P. Newborn skin: part I. Common rashes. Am Fam Physician. 2008;77(1):47-52.
1. Page EH, Shear NH. Temperature-dependent skin disorders. J Am Acad Dermatol. 1988;18(5, pt 1):1003-1019.
2. Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162(1):77-78.
3. Kim HW, Kim EJ, Park HC, Ko JY, Ro YS, Kim JE. Erythema ab igne successfully treated with low fluenced 1,064-nm Q-switched Neodymium-Doped Yttrium Aluminum Garnet laser. J Cosmet Laser Ther. 2014;16(3):147-148.
4. Gianfaldoni S, Gianfaldoni R, Tchernev G, Lotti J, Wollina U, Lotti T. Erythema ab igne successfully treated with mesoglycan and bioflavonoids: a case-report. Open Access Maced J Med Sci. 2017;5(4):432-435.
5. Arrington JH 3rd, Lockman DS. Thermal keratoses and squamous cell carcinoma in situ associated with erythema ab igne. AMA Arch Derm. 1979;115(10):1226-1228.
6. Sigmon JR, Cantrell J, Teague D, Sangueza O, Sheehan DJ. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35(6):676-678
7. Wharton J, Roffwarg D, Miller J, Sheehan DJ. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62(6):1080-1081.
8. Jones CS. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124(1):110-113.
9. Sajjan VV, Lunge S, Swamy MB, Pandit AM. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6(5):315-321.
10. O’Connor NR, McLaughlin MR, Ham P. Newborn skin: part I. Common rashes. Am Fam Physician. 2008;77(1):47-52.
Shoulder Injury Related to Vaccine Administration: A Rare Reaction
Localized reactions and transient pain at the site of vaccine administration are frequent and well-described occurrences that are typically short-lived and mild in nature. The most common findings at the injection site are soreness, erythema, and edema.1 Although less common, generalized shoulder dysfunction after vaccine administration also has been reported. Bodor and colleagues described a peri-articular inflammatory response that led to shoulder pain and weakness.2 A single case report by Kuether and colleagues described atraumatic osteonecrosis of the humeral head after H1N1 vaccine administration in the deltoid.3 In 2010, shoulder injury related to vaccine administration (SIRVA) was described by Atanasoff and colleagues as the rapid onset of shoulder pain and dysfunction persisting as a complication of deltoid muscle vaccination in a case series of 13 patients.4 In our report, we present a case of an active-duty male eventually diagnosed with SIRVA after influenza vaccination and discuss factors that may prevent vaccine-related shoulder injuries.
Case Presentation
A 31-year-old active-duty male presented to the Allergy clinic for evaluation of persistent left shoulder pain and decreased range of motion (ROM) following influenza vaccination 4 months prior. He reported a history of chronic low back and right shoulder pain. Although the patient had a traumatic injury to his right shoulder, which was corrected with surgery, he had no surgeries on the left shoulder. He reported no prior pain or known trauma to his left shoulder. He had no personal or family history of atopy or vaccine reactions.
The patient weighed 91 kg and received an intramuscular (IM) quadrivalent influenza vaccine with a 25-gauge, 1-inch needle during a mass influenza immunization. He recalled that the site of vaccination was slightly more than 3 cm below the top of the shoulder in a region correlating to the left deltoid. The vaccine was administered while he was standing with his arm extended, adducted, and internally rotated. The patient experienced intense pain immediately after the vaccination and noted decreased ROM. Initially, he dismissed the pain and decreased ROM as routine but sought medical attention when there was no improvement after 3 weeks.
Six weeks after the onset of symptoms, a magnetic resonance image (MRI) revealed tendinopathy of the left distal subscapularis, infraspinatus, supraspinatus, and teres minor tendon. These findings were suggestive of a small partial thickness tear of the supraspinatus (Figure 1), possible calcific tendinopathy of the distal teres minor (Figure 2), and underlying humeral head edema (Figure 3). The patient was evaluated by Orthopedics and experienced no relief from ibuprofen, celecoxib, and a steroid/lidocaine intra-articular injection. Laboratory studies included an unremarkable complete blood count and erythrocyte sedimentation rate. He was diagnosed with SIRVA and continued in physical therapy with incomplete resolution of symptoms 6 months postvaccination.
Discussion
According to a 2018 report issued by the Centers for Disease Control and Prevention, local reactions following immunizations are seen in up to 80% of administered vaccine doses.1 While most of these reactions are mild, transient, cutaneous reactions, rarely these also may persist and impact quality of life significantly. SIRVA is one such process that can lead to persistent musculoskeletal dysfunction. SIRVA presents as shoulder pain and limited ROM that occurs after the administration of an injectable vaccine. In 2011, the Institute of Medicine determined that evidence supported a causal relationship between vaccine administration and deltoid bursitis.5
In 2017, SIRVA was included in the Vaccine Injury Compensation Program (VICP), a federal program that can provide compensation to individuals injured by certain vaccines.6 A diagnosis of SIRVA can be considered in patients who experience pain within 48 hours of vaccination, have no prior history of pain or dysfunction of the affected shoulder prior to vaccine administration, and have symptoms limited to the shoulder in which the vaccine was administered where no other abnormality is present to explain these symptoms (eg, brachial neuritis, other neuropathy). Currently, patients with back pain or musculoskeletal complaints that do not include the shoulder following deltoid vaccination do not meet the reporting criteria for SIRVA in the VICP.6
The exact prevalence or incidence of SIRVA is unknown. In a 2017 systematic review of the literature and the Spanish Pharmacovigilance System database, Martín Arias and colleagues found 45 cases of new onset, unilateral shoulder dysfunction without associated neuropathy or autoimmune conditions following vaccine administration. They noted a female to male predominance (71.1% vs 28.9%) with a mean age of 53.6 years (range 22-89 y). Most of the cases occurred following influenza vaccine (62%); pneumococcal vaccine was the next most common (13%).7 Shoulder injury also has been reported after tetanus-diphtheria toxoids, human papilloma virus, and hepatitis A virus vaccines.4,7 The review noted that all patients had onset of pain within the first week following vaccination with the majority (81%) having pain in the first 24 hours. Two cases found in the Spanish database had pain onset 2 months postvaccination.7 Atanasoff and colleagues found that 93% of patients had pain onset within 24 hours of vaccination with 54% reporting immediate pain.4
The Vaccine Adverse Event Reporting System (VAERS) tracks reports of shoulder dysfunction following certain vaccinations, but the system is unable to establish causality. According to VAERS reporting, between 2010 and 2016, there were 1006 possible reports of shoulder dysfunction following inactivated influenza vaccination (IIV) compared with an estimated 130 million doses of IIV given each influenza season in the US.8
Bodor and Montalvo postulated that vaccine antigen was being over penetrated into the synovial space of the shoulder, as the subdeltoid/subacromial bursa is located a mere 0.8 to 1.6 cm below the skin surface in patients with healthy body mass index.2 Atanasoff and colleagues expounded that antibodies from previous vaccination or natural infection may then form antigen-antibody complexes, creating prolonged local immune and inflammatory responses leading to bursitis or tendonitis.4 Martín Arias and colleagues hypothesized that improper injection technique, including wrong insertion angle, incorrect needle type/size, and failure to account for the patient’s physical characteristics were the most likely causes of SIRVA.7
Proper vaccine administration ensures that vaccinations are delivered in a safe and efficacious manner. Safe vaccination practices include the use of trained personnel who receive comprehensive, competency-based training regarding vaccine administration.1 Aspiration prior to an injection is a practice that has not been evaluated fully. Given that the 2 routinely recommended locations for IM vaccines (deltoid muscle in adults or vastus lateralis muscle in infants) lack large blood vessels, the practice of aspiration prior to an IM vaccine is not currently deemed necessary.1 Additional safe vaccine practices include the selection of appropriate needle length for muscle penetration and that anatomic landmarks determine the location of vaccination.1 Despite this, in a survey of 100 medical professionals, half could not name any structure at risk from improper deltoid vaccination technique.9
Cook and colleagues used anthropomorphic data to evaluate the potential for injury to the subdeltoid/subacromial bursa and/or the axillary nerve.10 Based on these data, they recommended safe IM vaccine administration can be assured by using the midpoint of the deltoid muscle located midway between the acromion and deltoid tuberosity with the arm abducted to 60°.10,11 In 46% of SIRVA cases described by Atanasoff and colleagues, patients reported that the vaccine was administered “too high.”4 The study also recommended that the clinician and the patient be in the seated position to ensure proper needle angle and location of administration.4 For most adults, a 1-inch needle is appropriate for vaccine administration in the deltoid; however, in females weighing < 70 kg and males < 75 kg, a 5/8-inch needle is recommended to avoid injury.7
Our 91-kg patient was appropriately administered his vaccine with a 1-inch needle. As he experienced immediate pain, it is unlikely that his symptoms were due to an immune-mediated process, as this would not be expected to occur immediately. Improper location of vaccine administration is a proposed mechanism of injury for our patient, though this cannot be confirmed by history alone. His prior history of traumatic injury to the opposite shoulder could represent a confounding factor as no prior imaging was available for the vaccine-affected shoulder. A preexisting shoulder abnormality or injury cannot be completely excluded, and it is possible that an underlying prior shoulder injury was aggravated postvaccination.
Evaluation and Treatment
There is no standardized approach for the evaluation of SIRVA to date. Awareness of SIRVA and a high index of suspicion are necessary to evaluate patients with shoulder concerns postvaccination. Laboratory evaluation should be considered to evaluate for other potential diagnoses (eg, infection, rheumatologic concerns). Routine X-rays are not helpful in cases of SIRVA. Ultrasound may be considered as it can show bursa abnormalities consistent with bursitis.2 MRI of the affected shoulder may provide improved diagnostic capability if SIRVA is suspected. MRI findings vary but include intraosseous edema, bursitis, tendonitis, and rotator cuff tears.4,12 Complete rotator cuff tears were found in 15% of cases reviewed by Atanasoff and colleagues.4 While there is no recommended timing for MRI, 63% of MRIs were performed within 3 months of symptom onset.4 As SIRVA is not a neurologic injury, nerve conduction, electromyographic studies, and neurologic evaluation or testing are expected to be normal.
Treatment of SIRVA and other vaccine-related shoulder injuries typically have involved pain management (eg, nonsteroidal anti-inflammatory agents), intra-articular steroid injections, and physical therapy, though some patients never experience complete resolution of symptoms.2,4,7 Both patients with vaccination-related shoulder dysfunction described by Bodor and colleagues improved after intra-articular triamcinolone injections, with up to 3 injections before complete resolution of pain in one patient.2 Orthopedics evaluation may need to be considered for persistent symptoms. According to Atanasoff and colleagues, most patients were symptomatic for at least 6 months, and complete recovery was seen in less than one-third of patients.4 Although the development of SIRVA is not a contraindication to future doses of the presumed causative vaccine, subsequent vaccination should include careful consideration of other administration sites if possible (eg, vastus lateralis may be used for IM injections in adults) (Figure 4).
Reporting
A diagnosis or concern for SIRVA also should be reported to the VAERS, the national database established in order to detect possible safety problems with US-licensed vaccines. VAERS reports can be submitted by anyone with concerns for vaccine adverse reactions, including patients, caregivers, and health care professionals at vaers.hhs.gov/reportevent.html. Additional information regarding VICP can be obtained at www.hrsa.gov/vaccine-compensation/index.html.
Military-Specific Issues
The military values readiness, which includes ensuring that active-duty members remain up-to-date on life-saving vaccinations. Immunization is of critical importance to mobility and success of the overall mission. Mobility processing lines where immunizations can be provided to multiple active-duty members can be a successful strategy for mass immunizations. Although the quick administration of immunizations maintains readiness and provides a medically necessary service, it also may increase the chances of incorrect vaccine placement in the deltoid, causing long-term shoulder immobility that may impact a service member’s retainability. The benefits of mobility processing lines can continue to outweigh the risks of immunization administration by ensuring proper staff training, seating both the administrator and recipient of vaccination, and selecting a proper needle length and site of administration specific to each recipient.
Conclusion
Correct administration of vaccines is of utmost importance in preventing SIRVA and other vaccine-related shoulder dysfunctions. Proper staff training and refresher training can help prevent vaccine-related shoulder injuries. Additionally, clinicians should be aware of this potential complication and maintain a high index of suspicion when evaluating patients with postvaccination shoulder complaints.
1. Centers for Disease Control and Prevention. Epidemiology and prevention of vaccine-preventable diseases. https://www.cdc.gov/vaccines/pubs/pinkbook/vac-admin.html. Published 2015. Accessed June 3, 2019.
2. Bodor M, Montalvo E. Vaccination-related shoulder dysfunction. Vaccine. 2007;25(4):585-587.
3. Kuether G, Dietrich B, Smith T, Peter C, Gruessner S. Atraumatic osteonecrosis of the humeral head after influenza A-(H1N1) v-2009 vaccination. Vaccine. 2011;29(40):6830-6833.
4. Atanasoff S, Ryan T, Lightfoot R, Johann-Liang R. Shoulder injury related to vaccine administration (SIRVA). Vaccine. 2010;28(51):8049-8052.
5. Institute of Medicine. Adverse effects of vaccines: evidence and causality. http://www.nationalacademies.org/hmd/~/media/Files/Report%20Files/2011/Adverse-Effects-of-Vaccines-Evidence-and-Causality/Vaccine-report-brief-FINAL.pdf. Published August 2011. Accessed June 3, 2019.
6. Health Resources and Services Administration, Health and Human Services Administration. National vaccine injury compensation program: revisions to the vaccine injury table. https://www.federalregister.gov/documents/2017/01/19/2017-00701/national-vaccine-injury-compensation-program-revisions-to-the-vaccine-injury-table. Published January 19, 2017. Accessed June 3, 2019.
7. Martín Arias LH, Sanz Fadrique R, Sáinz Gil M, Salgueiro-Vazquez ME. Risk of bursitis and other injuries and dysfunctions of the shoulder following vaccinations. Vaccine. 2017;35(37):4870-4876.
8. Centers for Disease Control and Prevention. Reports of shoulder dysfunction following inactivated influenza vaccine in the Vaccine Adverse Event Reporting System (VAERS), 2010-2016. https://stacks.cdc.gov/view/cdc/57624. Published January 4, 2018. Accessed June 3, 2019.
9. McGarvey MA, Hooper AC. The deltoid intramuscular injection site in the adult. Current practice among general practitioners and practice nurses. Ir Med J. 2005;98(4):105-107.
10. Cook IF. An evidence based protocol for the prevention of upper arm injury related to vaccine administration (UAIRVA). Hum Vaccin. 2011;7(8):845-848.
11. Cook IF. Best vaccination practice and medically attended injection site events following deltoid intramuscular injection. Hum Vaccin Immunother. 2015;11(5):1184-1191.
12. Okur G, Chaney KA, Lomasney LM. Magnetic resonance imaging of abnormal shoulder pain following influenza vaccination. Skeletal Radiol. 2014;43(9):1325-1331.
Localized reactions and transient pain at the site of vaccine administration are frequent and well-described occurrences that are typically short-lived and mild in nature. The most common findings at the injection site are soreness, erythema, and edema.1 Although less common, generalized shoulder dysfunction after vaccine administration also has been reported. Bodor and colleagues described a peri-articular inflammatory response that led to shoulder pain and weakness.2 A single case report by Kuether and colleagues described atraumatic osteonecrosis of the humeral head after H1N1 vaccine administration in the deltoid.3 In 2010, shoulder injury related to vaccine administration (SIRVA) was described by Atanasoff and colleagues as the rapid onset of shoulder pain and dysfunction persisting as a complication of deltoid muscle vaccination in a case series of 13 patients.4 In our report, we present a case of an active-duty male eventually diagnosed with SIRVA after influenza vaccination and discuss factors that may prevent vaccine-related shoulder injuries.
Case Presentation
A 31-year-old active-duty male presented to the Allergy clinic for evaluation of persistent left shoulder pain and decreased range of motion (ROM) following influenza vaccination 4 months prior. He reported a history of chronic low back and right shoulder pain. Although the patient had a traumatic injury to his right shoulder, which was corrected with surgery, he had no surgeries on the left shoulder. He reported no prior pain or known trauma to his left shoulder. He had no personal or family history of atopy or vaccine reactions.
The patient weighed 91 kg and received an intramuscular (IM) quadrivalent influenza vaccine with a 25-gauge, 1-inch needle during a mass influenza immunization. He recalled that the site of vaccination was slightly more than 3 cm below the top of the shoulder in a region correlating to the left deltoid. The vaccine was administered while he was standing with his arm extended, adducted, and internally rotated. The patient experienced intense pain immediately after the vaccination and noted decreased ROM. Initially, he dismissed the pain and decreased ROM as routine but sought medical attention when there was no improvement after 3 weeks.
Six weeks after the onset of symptoms, a magnetic resonance image (MRI) revealed tendinopathy of the left distal subscapularis, infraspinatus, supraspinatus, and teres minor tendon. These findings were suggestive of a small partial thickness tear of the supraspinatus (Figure 1), possible calcific tendinopathy of the distal teres minor (Figure 2), and underlying humeral head edema (Figure 3). The patient was evaluated by Orthopedics and experienced no relief from ibuprofen, celecoxib, and a steroid/lidocaine intra-articular injection. Laboratory studies included an unremarkable complete blood count and erythrocyte sedimentation rate. He was diagnosed with SIRVA and continued in physical therapy with incomplete resolution of symptoms 6 months postvaccination.
Discussion
According to a 2018 report issued by the Centers for Disease Control and Prevention, local reactions following immunizations are seen in up to 80% of administered vaccine doses.1 While most of these reactions are mild, transient, cutaneous reactions, rarely these also may persist and impact quality of life significantly. SIRVA is one such process that can lead to persistent musculoskeletal dysfunction. SIRVA presents as shoulder pain and limited ROM that occurs after the administration of an injectable vaccine. In 2011, the Institute of Medicine determined that evidence supported a causal relationship between vaccine administration and deltoid bursitis.5
In 2017, SIRVA was included in the Vaccine Injury Compensation Program (VICP), a federal program that can provide compensation to individuals injured by certain vaccines.6 A diagnosis of SIRVA can be considered in patients who experience pain within 48 hours of vaccination, have no prior history of pain or dysfunction of the affected shoulder prior to vaccine administration, and have symptoms limited to the shoulder in which the vaccine was administered where no other abnormality is present to explain these symptoms (eg, brachial neuritis, other neuropathy). Currently, patients with back pain or musculoskeletal complaints that do not include the shoulder following deltoid vaccination do not meet the reporting criteria for SIRVA in the VICP.6
The exact prevalence or incidence of SIRVA is unknown. In a 2017 systematic review of the literature and the Spanish Pharmacovigilance System database, Martín Arias and colleagues found 45 cases of new onset, unilateral shoulder dysfunction without associated neuropathy or autoimmune conditions following vaccine administration. They noted a female to male predominance (71.1% vs 28.9%) with a mean age of 53.6 years (range 22-89 y). Most of the cases occurred following influenza vaccine (62%); pneumococcal vaccine was the next most common (13%).7 Shoulder injury also has been reported after tetanus-diphtheria toxoids, human papilloma virus, and hepatitis A virus vaccines.4,7 The review noted that all patients had onset of pain within the first week following vaccination with the majority (81%) having pain in the first 24 hours. Two cases found in the Spanish database had pain onset 2 months postvaccination.7 Atanasoff and colleagues found that 93% of patients had pain onset within 24 hours of vaccination with 54% reporting immediate pain.4
The Vaccine Adverse Event Reporting System (VAERS) tracks reports of shoulder dysfunction following certain vaccinations, but the system is unable to establish causality. According to VAERS reporting, between 2010 and 2016, there were 1006 possible reports of shoulder dysfunction following inactivated influenza vaccination (IIV) compared with an estimated 130 million doses of IIV given each influenza season in the US.8
Bodor and Montalvo postulated that vaccine antigen was being over penetrated into the synovial space of the shoulder, as the subdeltoid/subacromial bursa is located a mere 0.8 to 1.6 cm below the skin surface in patients with healthy body mass index.2 Atanasoff and colleagues expounded that antibodies from previous vaccination or natural infection may then form antigen-antibody complexes, creating prolonged local immune and inflammatory responses leading to bursitis or tendonitis.4 Martín Arias and colleagues hypothesized that improper injection technique, including wrong insertion angle, incorrect needle type/size, and failure to account for the patient’s physical characteristics were the most likely causes of SIRVA.7
Proper vaccine administration ensures that vaccinations are delivered in a safe and efficacious manner. Safe vaccination practices include the use of trained personnel who receive comprehensive, competency-based training regarding vaccine administration.1 Aspiration prior to an injection is a practice that has not been evaluated fully. Given that the 2 routinely recommended locations for IM vaccines (deltoid muscle in adults or vastus lateralis muscle in infants) lack large blood vessels, the practice of aspiration prior to an IM vaccine is not currently deemed necessary.1 Additional safe vaccine practices include the selection of appropriate needle length for muscle penetration and that anatomic landmarks determine the location of vaccination.1 Despite this, in a survey of 100 medical professionals, half could not name any structure at risk from improper deltoid vaccination technique.9
Cook and colleagues used anthropomorphic data to evaluate the potential for injury to the subdeltoid/subacromial bursa and/or the axillary nerve.10 Based on these data, they recommended safe IM vaccine administration can be assured by using the midpoint of the deltoid muscle located midway between the acromion and deltoid tuberosity with the arm abducted to 60°.10,11 In 46% of SIRVA cases described by Atanasoff and colleagues, patients reported that the vaccine was administered “too high.”4 The study also recommended that the clinician and the patient be in the seated position to ensure proper needle angle and location of administration.4 For most adults, a 1-inch needle is appropriate for vaccine administration in the deltoid; however, in females weighing < 70 kg and males < 75 kg, a 5/8-inch needle is recommended to avoid injury.7
Our 91-kg patient was appropriately administered his vaccine with a 1-inch needle. As he experienced immediate pain, it is unlikely that his symptoms were due to an immune-mediated process, as this would not be expected to occur immediately. Improper location of vaccine administration is a proposed mechanism of injury for our patient, though this cannot be confirmed by history alone. His prior history of traumatic injury to the opposite shoulder could represent a confounding factor as no prior imaging was available for the vaccine-affected shoulder. A preexisting shoulder abnormality or injury cannot be completely excluded, and it is possible that an underlying prior shoulder injury was aggravated postvaccination.
Evaluation and Treatment
There is no standardized approach for the evaluation of SIRVA to date. Awareness of SIRVA and a high index of suspicion are necessary to evaluate patients with shoulder concerns postvaccination. Laboratory evaluation should be considered to evaluate for other potential diagnoses (eg, infection, rheumatologic concerns). Routine X-rays are not helpful in cases of SIRVA. Ultrasound may be considered as it can show bursa abnormalities consistent with bursitis.2 MRI of the affected shoulder may provide improved diagnostic capability if SIRVA is suspected. MRI findings vary but include intraosseous edema, bursitis, tendonitis, and rotator cuff tears.4,12 Complete rotator cuff tears were found in 15% of cases reviewed by Atanasoff and colleagues.4 While there is no recommended timing for MRI, 63% of MRIs were performed within 3 months of symptom onset.4 As SIRVA is not a neurologic injury, nerve conduction, electromyographic studies, and neurologic evaluation or testing are expected to be normal.
Treatment of SIRVA and other vaccine-related shoulder injuries typically have involved pain management (eg, nonsteroidal anti-inflammatory agents), intra-articular steroid injections, and physical therapy, though some patients never experience complete resolution of symptoms.2,4,7 Both patients with vaccination-related shoulder dysfunction described by Bodor and colleagues improved after intra-articular triamcinolone injections, with up to 3 injections before complete resolution of pain in one patient.2 Orthopedics evaluation may need to be considered for persistent symptoms. According to Atanasoff and colleagues, most patients were symptomatic for at least 6 months, and complete recovery was seen in less than one-third of patients.4 Although the development of SIRVA is not a contraindication to future doses of the presumed causative vaccine, subsequent vaccination should include careful consideration of other administration sites if possible (eg, vastus lateralis may be used for IM injections in adults) (Figure 4).
Reporting
A diagnosis or concern for SIRVA also should be reported to the VAERS, the national database established in order to detect possible safety problems with US-licensed vaccines. VAERS reports can be submitted by anyone with concerns for vaccine adverse reactions, including patients, caregivers, and health care professionals at vaers.hhs.gov/reportevent.html. Additional information regarding VICP can be obtained at www.hrsa.gov/vaccine-compensation/index.html.
Military-Specific Issues
The military values readiness, which includes ensuring that active-duty members remain up-to-date on life-saving vaccinations. Immunization is of critical importance to mobility and success of the overall mission. Mobility processing lines where immunizations can be provided to multiple active-duty members can be a successful strategy for mass immunizations. Although the quick administration of immunizations maintains readiness and provides a medically necessary service, it also may increase the chances of incorrect vaccine placement in the deltoid, causing long-term shoulder immobility that may impact a service member’s retainability. The benefits of mobility processing lines can continue to outweigh the risks of immunization administration by ensuring proper staff training, seating both the administrator and recipient of vaccination, and selecting a proper needle length and site of administration specific to each recipient.
Conclusion
Correct administration of vaccines is of utmost importance in preventing SIRVA and other vaccine-related shoulder dysfunctions. Proper staff training and refresher training can help prevent vaccine-related shoulder injuries. Additionally, clinicians should be aware of this potential complication and maintain a high index of suspicion when evaluating patients with postvaccination shoulder complaints.
Localized reactions and transient pain at the site of vaccine administration are frequent and well-described occurrences that are typically short-lived and mild in nature. The most common findings at the injection site are soreness, erythema, and edema.1 Although less common, generalized shoulder dysfunction after vaccine administration also has been reported. Bodor and colleagues described a peri-articular inflammatory response that led to shoulder pain and weakness.2 A single case report by Kuether and colleagues described atraumatic osteonecrosis of the humeral head after H1N1 vaccine administration in the deltoid.3 In 2010, shoulder injury related to vaccine administration (SIRVA) was described by Atanasoff and colleagues as the rapid onset of shoulder pain and dysfunction persisting as a complication of deltoid muscle vaccination in a case series of 13 patients.4 In our report, we present a case of an active-duty male eventually diagnosed with SIRVA after influenza vaccination and discuss factors that may prevent vaccine-related shoulder injuries.
Case Presentation
A 31-year-old active-duty male presented to the Allergy clinic for evaluation of persistent left shoulder pain and decreased range of motion (ROM) following influenza vaccination 4 months prior. He reported a history of chronic low back and right shoulder pain. Although the patient had a traumatic injury to his right shoulder, which was corrected with surgery, he had no surgeries on the left shoulder. He reported no prior pain or known trauma to his left shoulder. He had no personal or family history of atopy or vaccine reactions.
The patient weighed 91 kg and received an intramuscular (IM) quadrivalent influenza vaccine with a 25-gauge, 1-inch needle during a mass influenza immunization. He recalled that the site of vaccination was slightly more than 3 cm below the top of the shoulder in a region correlating to the left deltoid. The vaccine was administered while he was standing with his arm extended, adducted, and internally rotated. The patient experienced intense pain immediately after the vaccination and noted decreased ROM. Initially, he dismissed the pain and decreased ROM as routine but sought medical attention when there was no improvement after 3 weeks.
Six weeks after the onset of symptoms, a magnetic resonance image (MRI) revealed tendinopathy of the left distal subscapularis, infraspinatus, supraspinatus, and teres minor tendon. These findings were suggestive of a small partial thickness tear of the supraspinatus (Figure 1), possible calcific tendinopathy of the distal teres minor (Figure 2), and underlying humeral head edema (Figure 3). The patient was evaluated by Orthopedics and experienced no relief from ibuprofen, celecoxib, and a steroid/lidocaine intra-articular injection. Laboratory studies included an unremarkable complete blood count and erythrocyte sedimentation rate. He was diagnosed with SIRVA and continued in physical therapy with incomplete resolution of symptoms 6 months postvaccination.
Discussion
According to a 2018 report issued by the Centers for Disease Control and Prevention, local reactions following immunizations are seen in up to 80% of administered vaccine doses.1 While most of these reactions are mild, transient, cutaneous reactions, rarely these also may persist and impact quality of life significantly. SIRVA is one such process that can lead to persistent musculoskeletal dysfunction. SIRVA presents as shoulder pain and limited ROM that occurs after the administration of an injectable vaccine. In 2011, the Institute of Medicine determined that evidence supported a causal relationship between vaccine administration and deltoid bursitis.5
In 2017, SIRVA was included in the Vaccine Injury Compensation Program (VICP), a federal program that can provide compensation to individuals injured by certain vaccines.6 A diagnosis of SIRVA can be considered in patients who experience pain within 48 hours of vaccination, have no prior history of pain or dysfunction of the affected shoulder prior to vaccine administration, and have symptoms limited to the shoulder in which the vaccine was administered where no other abnormality is present to explain these symptoms (eg, brachial neuritis, other neuropathy). Currently, patients with back pain or musculoskeletal complaints that do not include the shoulder following deltoid vaccination do not meet the reporting criteria for SIRVA in the VICP.6
The exact prevalence or incidence of SIRVA is unknown. In a 2017 systematic review of the literature and the Spanish Pharmacovigilance System database, Martín Arias and colleagues found 45 cases of new onset, unilateral shoulder dysfunction without associated neuropathy or autoimmune conditions following vaccine administration. They noted a female to male predominance (71.1% vs 28.9%) with a mean age of 53.6 years (range 22-89 y). Most of the cases occurred following influenza vaccine (62%); pneumococcal vaccine was the next most common (13%).7 Shoulder injury also has been reported after tetanus-diphtheria toxoids, human papilloma virus, and hepatitis A virus vaccines.4,7 The review noted that all patients had onset of pain within the first week following vaccination with the majority (81%) having pain in the first 24 hours. Two cases found in the Spanish database had pain onset 2 months postvaccination.7 Atanasoff and colleagues found that 93% of patients had pain onset within 24 hours of vaccination with 54% reporting immediate pain.4
The Vaccine Adverse Event Reporting System (VAERS) tracks reports of shoulder dysfunction following certain vaccinations, but the system is unable to establish causality. According to VAERS reporting, between 2010 and 2016, there were 1006 possible reports of shoulder dysfunction following inactivated influenza vaccination (IIV) compared with an estimated 130 million doses of IIV given each influenza season in the US.8
Bodor and Montalvo postulated that vaccine antigen was being over penetrated into the synovial space of the shoulder, as the subdeltoid/subacromial bursa is located a mere 0.8 to 1.6 cm below the skin surface in patients with healthy body mass index.2 Atanasoff and colleagues expounded that antibodies from previous vaccination or natural infection may then form antigen-antibody complexes, creating prolonged local immune and inflammatory responses leading to bursitis or tendonitis.4 Martín Arias and colleagues hypothesized that improper injection technique, including wrong insertion angle, incorrect needle type/size, and failure to account for the patient’s physical characteristics were the most likely causes of SIRVA.7
Proper vaccine administration ensures that vaccinations are delivered in a safe and efficacious manner. Safe vaccination practices include the use of trained personnel who receive comprehensive, competency-based training regarding vaccine administration.1 Aspiration prior to an injection is a practice that has not been evaluated fully. Given that the 2 routinely recommended locations for IM vaccines (deltoid muscle in adults or vastus lateralis muscle in infants) lack large blood vessels, the practice of aspiration prior to an IM vaccine is not currently deemed necessary.1 Additional safe vaccine practices include the selection of appropriate needle length for muscle penetration and that anatomic landmarks determine the location of vaccination.1 Despite this, in a survey of 100 medical professionals, half could not name any structure at risk from improper deltoid vaccination technique.9
Cook and colleagues used anthropomorphic data to evaluate the potential for injury to the subdeltoid/subacromial bursa and/or the axillary nerve.10 Based on these data, they recommended safe IM vaccine administration can be assured by using the midpoint of the deltoid muscle located midway between the acromion and deltoid tuberosity with the arm abducted to 60°.10,11 In 46% of SIRVA cases described by Atanasoff and colleagues, patients reported that the vaccine was administered “too high.”4 The study also recommended that the clinician and the patient be in the seated position to ensure proper needle angle and location of administration.4 For most adults, a 1-inch needle is appropriate for vaccine administration in the deltoid; however, in females weighing < 70 kg and males < 75 kg, a 5/8-inch needle is recommended to avoid injury.7
Our 91-kg patient was appropriately administered his vaccine with a 1-inch needle. As he experienced immediate pain, it is unlikely that his symptoms were due to an immune-mediated process, as this would not be expected to occur immediately. Improper location of vaccine administration is a proposed mechanism of injury for our patient, though this cannot be confirmed by history alone. His prior history of traumatic injury to the opposite shoulder could represent a confounding factor as no prior imaging was available for the vaccine-affected shoulder. A preexisting shoulder abnormality or injury cannot be completely excluded, and it is possible that an underlying prior shoulder injury was aggravated postvaccination.
Evaluation and Treatment
There is no standardized approach for the evaluation of SIRVA to date. Awareness of SIRVA and a high index of suspicion are necessary to evaluate patients with shoulder concerns postvaccination. Laboratory evaluation should be considered to evaluate for other potential diagnoses (eg, infection, rheumatologic concerns). Routine X-rays are not helpful in cases of SIRVA. Ultrasound may be considered as it can show bursa abnormalities consistent with bursitis.2 MRI of the affected shoulder may provide improved diagnostic capability if SIRVA is suspected. MRI findings vary but include intraosseous edema, bursitis, tendonitis, and rotator cuff tears.4,12 Complete rotator cuff tears were found in 15% of cases reviewed by Atanasoff and colleagues.4 While there is no recommended timing for MRI, 63% of MRIs were performed within 3 months of symptom onset.4 As SIRVA is not a neurologic injury, nerve conduction, electromyographic studies, and neurologic evaluation or testing are expected to be normal.
Treatment of SIRVA and other vaccine-related shoulder injuries typically have involved pain management (eg, nonsteroidal anti-inflammatory agents), intra-articular steroid injections, and physical therapy, though some patients never experience complete resolution of symptoms.2,4,7 Both patients with vaccination-related shoulder dysfunction described by Bodor and colleagues improved after intra-articular triamcinolone injections, with up to 3 injections before complete resolution of pain in one patient.2 Orthopedics evaluation may need to be considered for persistent symptoms. According to Atanasoff and colleagues, most patients were symptomatic for at least 6 months, and complete recovery was seen in less than one-third of patients.4 Although the development of SIRVA is not a contraindication to future doses of the presumed causative vaccine, subsequent vaccination should include careful consideration of other administration sites if possible (eg, vastus lateralis may be used for IM injections in adults) (Figure 4).
Reporting
A diagnosis or concern for SIRVA also should be reported to the VAERS, the national database established in order to detect possible safety problems with US-licensed vaccines. VAERS reports can be submitted by anyone with concerns for vaccine adverse reactions, including patients, caregivers, and health care professionals at vaers.hhs.gov/reportevent.html. Additional information regarding VICP can be obtained at www.hrsa.gov/vaccine-compensation/index.html.
Military-Specific Issues
The military values readiness, which includes ensuring that active-duty members remain up-to-date on life-saving vaccinations. Immunization is of critical importance to mobility and success of the overall mission. Mobility processing lines where immunizations can be provided to multiple active-duty members can be a successful strategy for mass immunizations. Although the quick administration of immunizations maintains readiness and provides a medically necessary service, it also may increase the chances of incorrect vaccine placement in the deltoid, causing long-term shoulder immobility that may impact a service member’s retainability. The benefits of mobility processing lines can continue to outweigh the risks of immunization administration by ensuring proper staff training, seating both the administrator and recipient of vaccination, and selecting a proper needle length and site of administration specific to each recipient.
Conclusion
Correct administration of vaccines is of utmost importance in preventing SIRVA and other vaccine-related shoulder dysfunctions. Proper staff training and refresher training can help prevent vaccine-related shoulder injuries. Additionally, clinicians should be aware of this potential complication and maintain a high index of suspicion when evaluating patients with postvaccination shoulder complaints.
1. Centers for Disease Control and Prevention. Epidemiology and prevention of vaccine-preventable diseases. https://www.cdc.gov/vaccines/pubs/pinkbook/vac-admin.html. Published 2015. Accessed June 3, 2019.
2. Bodor M, Montalvo E. Vaccination-related shoulder dysfunction. Vaccine. 2007;25(4):585-587.
3. Kuether G, Dietrich B, Smith T, Peter C, Gruessner S. Atraumatic osteonecrosis of the humeral head after influenza A-(H1N1) v-2009 vaccination. Vaccine. 2011;29(40):6830-6833.
4. Atanasoff S, Ryan T, Lightfoot R, Johann-Liang R. Shoulder injury related to vaccine administration (SIRVA). Vaccine. 2010;28(51):8049-8052.
5. Institute of Medicine. Adverse effects of vaccines: evidence and causality. http://www.nationalacademies.org/hmd/~/media/Files/Report%20Files/2011/Adverse-Effects-of-Vaccines-Evidence-and-Causality/Vaccine-report-brief-FINAL.pdf. Published August 2011. Accessed June 3, 2019.
6. Health Resources and Services Administration, Health and Human Services Administration. National vaccine injury compensation program: revisions to the vaccine injury table. https://www.federalregister.gov/documents/2017/01/19/2017-00701/national-vaccine-injury-compensation-program-revisions-to-the-vaccine-injury-table. Published January 19, 2017. Accessed June 3, 2019.
7. Martín Arias LH, Sanz Fadrique R, Sáinz Gil M, Salgueiro-Vazquez ME. Risk of bursitis and other injuries and dysfunctions of the shoulder following vaccinations. Vaccine. 2017;35(37):4870-4876.
8. Centers for Disease Control and Prevention. Reports of shoulder dysfunction following inactivated influenza vaccine in the Vaccine Adverse Event Reporting System (VAERS), 2010-2016. https://stacks.cdc.gov/view/cdc/57624. Published January 4, 2018. Accessed June 3, 2019.
9. McGarvey MA, Hooper AC. The deltoid intramuscular injection site in the adult. Current practice among general practitioners and practice nurses. Ir Med J. 2005;98(4):105-107.
10. Cook IF. An evidence based protocol for the prevention of upper arm injury related to vaccine administration (UAIRVA). Hum Vaccin. 2011;7(8):845-848.
11. Cook IF. Best vaccination practice and medically attended injection site events following deltoid intramuscular injection. Hum Vaccin Immunother. 2015;11(5):1184-1191.
12. Okur G, Chaney KA, Lomasney LM. Magnetic resonance imaging of abnormal shoulder pain following influenza vaccination. Skeletal Radiol. 2014;43(9):1325-1331.
1. Centers for Disease Control and Prevention. Epidemiology and prevention of vaccine-preventable diseases. https://www.cdc.gov/vaccines/pubs/pinkbook/vac-admin.html. Published 2015. Accessed June 3, 2019.
2. Bodor M, Montalvo E. Vaccination-related shoulder dysfunction. Vaccine. 2007;25(4):585-587.
3. Kuether G, Dietrich B, Smith T, Peter C, Gruessner S. Atraumatic osteonecrosis of the humeral head after influenza A-(H1N1) v-2009 vaccination. Vaccine. 2011;29(40):6830-6833.
4. Atanasoff S, Ryan T, Lightfoot R, Johann-Liang R. Shoulder injury related to vaccine administration (SIRVA). Vaccine. 2010;28(51):8049-8052.
5. Institute of Medicine. Adverse effects of vaccines: evidence and causality. http://www.nationalacademies.org/hmd/~/media/Files/Report%20Files/2011/Adverse-Effects-of-Vaccines-Evidence-and-Causality/Vaccine-report-brief-FINAL.pdf. Published August 2011. Accessed June 3, 2019.
6. Health Resources and Services Administration, Health and Human Services Administration. National vaccine injury compensation program: revisions to the vaccine injury table. https://www.federalregister.gov/documents/2017/01/19/2017-00701/national-vaccine-injury-compensation-program-revisions-to-the-vaccine-injury-table. Published January 19, 2017. Accessed June 3, 2019.
7. Martín Arias LH, Sanz Fadrique R, Sáinz Gil M, Salgueiro-Vazquez ME. Risk of bursitis and other injuries and dysfunctions of the shoulder following vaccinations. Vaccine. 2017;35(37):4870-4876.
8. Centers for Disease Control and Prevention. Reports of shoulder dysfunction following inactivated influenza vaccine in the Vaccine Adverse Event Reporting System (VAERS), 2010-2016. https://stacks.cdc.gov/view/cdc/57624. Published January 4, 2018. Accessed June 3, 2019.
9. McGarvey MA, Hooper AC. The deltoid intramuscular injection site in the adult. Current practice among general practitioners and practice nurses. Ir Med J. 2005;98(4):105-107.
10. Cook IF. An evidence based protocol for the prevention of upper arm injury related to vaccine administration (UAIRVA). Hum Vaccin. 2011;7(8):845-848.
11. Cook IF. Best vaccination practice and medically attended injection site events following deltoid intramuscular injection. Hum Vaccin Immunother. 2015;11(5):1184-1191.
12. Okur G, Chaney KA, Lomasney LM. Magnetic resonance imaging of abnormal shoulder pain following influenza vaccination. Skeletal Radiol. 2014;43(9):1325-1331.
Using Optical Coherence Tomography in the Management of Postoperative Wound Leaks After Cataract Surgery
The term cataract is derived from the Latin word “catarractes,” which means “waterfall,” as the foamy white opacity of an advanced cataract can be likened to a tempestuous cascade. Cataract is the leading cause of preventable blindness worldwide.1,2 It is no surprise, therefore, that cataract surgery is the most frequently performed ophthalmic surgical procedure worldwide. Cataract surgeries may reach 30 million annual cases by 2020.3 Given the large number of surgeries being performed, postsurgical complications are not uncommon.
Early postoperative complications from lens exchange (cataract) surgery include increased intraocular pressure (IOP), corneal edema, and corneal wound leakage.4 Corneal wound leakage is not uncommon; one study showed that, in 100 cases, almost one-third of incisions leaked.5 A 2014 prospective study of 500 postcataract surgery eyes revealed that 48.8% had fluid egress.6 Early detection is important so that efforts to restore corneal integrity can immediately be implemented. If not caught early, patients are at risk for developing a cascade of sequelae, including endophthalmitis.
The majority of corneal wound leaks postphacoemulsification are self-limiting and self-sealing. Moderate wound leaks require treatment, as in the following case. Strategies to detect, image, and treat wound leaks are covered in this discussion.
Case Presentation
A 69-year-old male veteran presented with no complaints for a 1-day postoperative visit following right eye phacoemulsification cataract extraction. His best corrected visual acuity in the right eye was 20/40, and his pinhole visual acuity was 20/25+2. On slit-lamp examination, the temporally located main incision appeared well-adhered and was found to be Seidel negative; however, the inferior paracentesis wound was found to be Seidel positive, demonstrating a slow leak. Intraocular pressure (IOP) measured with tonopen was 9 mm Hg.
A bandage soft contact lens was placed on the eye. The patient was instructed not to rub or place any pressure on the eye and to avoid bending and heavy lifting. He was also instructed to continue his postoperative medications (prednisolone 1% every 2 hours and polymyxin B sulfate 4 times daily) in his right eye. A follow-up appointment was scheduled for the next day.

The patient presented for his postoperative day-2 visit with a best corrected visual acuity in the right eye of 20/20. He reported no visual problems, no eye pain, and mentioned that he had had a comfortable night sleep. A slit-lamp examination revealed trace diffuse injection in the operative eye, predominantly central Descemet membrane folds, 1+ stromal edema, and a Seidel negative main incision wound. However, the inferior paracentesis wound showed a moderate leak (Seidel positive), and the anterior chamber showed a 1+ cell and flare. Goldmann tonometry revealed an IOP of 5 mm Hg, indicating hypotony.

Anterior segment cube 512 x 128 optical coherence tomography (OCT) was obtained with the bandage contact lens (Figures 1 and 2), and then repeated with the bandage contact lens removed (Figures 3 and 4). OCT imaging confirmed epithelial and endothelial gaping, loss of coaptation, and a localized detachment of the Descemet membrane. The veteran was referred to his surgeon that same day, and 2 limbal vicryl sutures were placed. The patient was instructed to continue prednisolone 1% 4 times daily and polymyxin B sulfate every 2 hours; erythromycin ointment 3 times daily was added to his regimen.
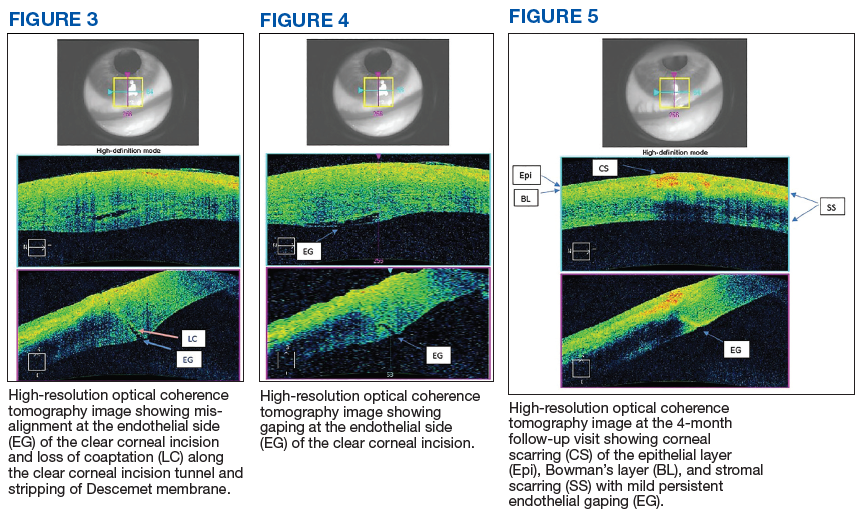
He was scheduled for a follow-up examination 1 week later. At that visit, the wound was no longer leaking and IOP had risen to a preoperative value of 17 mm Hg. The corneal sutures were removed at the 1-month postoperative examination and a follow-up was scheduled for 4 months later. An anterior segment OCT was obtained (Figure 5).
Discussion
In July 1967, Charles Kelman, MD, suggested using a dental ultrasonic tool, normally employed to clean teeth, to fragment the nucleus of the crystalline lens. Dr. Kelman’s first operation using phacoemulsification on a human eye took 3 hours.7 As the procedure for cataract removal has been refined, complication rates and surgical times have vastly improved.
Phacoemulsification is the most commonly performed outpatient surgery in the US; about 3 million cases are performed annually. Due to the high volume of cases, adverse events (AEs) are not uncommon. The incidence of complications following phacoemulsification is < 5%; the frequency of severe complications has been estimated at < 0.7%.8 Severe complications include endophthalmitis, suprachoroidal hemorrhage, and/or retinal detachment.9 Studies have shown a decline in rates of sight-threatening AEs from 1994 to 2006.9 A retrospective study of 45,082 veterans from 2005 to 2007 identified that a preoperative disease burden such as diabetes mellitus, chronic pulmonary disease, age-related macular degeneration, and diabetes with ophthalmic manifestations, was positively associated with a greater risk of cataract surgical complications.10
Complications
The level of a surgeon’s proficiency with phacoemulsification is directly correlated to the number of operations performed; there is a lower complication rate among more experienced surgeons, including those who work in high-volume settings.11,12 One study identified that the AE rate within 14 days of surgery was 0.8% for surgeons performing 50 to 250 cataract surgeries per year, but only 0.1% for those performing > 1000 cataract surgeries annually.12
Potential postoperative lens exchange complications include increased IOP, corneal wound leakage, corneal edema, bullous keratopathy, cystoid macular edema, retinal detachment, and endophthalmitis (Table 1). A corneal wound leak can provide a potential ingress for bacteria, putting the patient at risk for endophthalmitis, perhaps the most devastating complication following cataract surgery.

Endophthalmitis
Endophthalmitis has been reported to occur in .001% to .327% of patients during postoperative care.5,13-17 Early detection is important to maintain corneal integrity and prevent a cascade of detrimental ocular sequalae including the potential for endophthalmitis. According to Zaida and colleagues, endophthalmitis occurred in fewer than 1 of 1000 consecutive cases.14 A leaking clear corneal incision wound on the first day postoperatively has been associated with a 44-fold increased risk of endophthalmitis.13
Causes of endophthalmitis
In a retrospective case-controlled series of 57 patients with postcataract endophthalmitis, implantation of an intraocular lens with a resultant wound abnormality was thought to be the causative factor in 5%.17 Another source of endophthalmitis can be the intraocular lens (IOL), which may act as a vector for bacteria. By placing the IOL against the conjunctiva or exposing it to the theater air during surgery, bacteria can be introduced prior to implantation.17 Immunosuppressive treatment is the only patient antecedent factor that can be considered a predictor for endopthalmitis.17

The internal corneal seal is IOP dependent, and postoperative ocular hypotony may cause a seemingly watertight wound to leak. Taban and colleagues used anterior segment OCT to image numerous self-sealing incisions. They found that the corneal incision wound more tightly seals at higher IOPs. Additionally, more perpendicular (larger angle) incisions seal better at a lower IOP while less perpendicular (smaller angle) incisions seal better at a higher IOP (Figure 6).18
Incision Placement
Studies have shown that the main incision site is more clinically competent than is the side port incision site, as in our case study.19 Side-port incisions have a 1- or 2-plane architectural profile in contrast to the 3-plane profile typical of a main incision.19 Recent advances including the conversion to clear-corneal incisions of diminishing size, techniques used for wound construction, phacoemulsification machine design, and small-incision IOLs, should further reduce the prevalence and complications of wound compromise.20
Seidel Testing
Seidel testing is the most common method to evaluate corneal wound integrity and identify leaks. A drop of topical anesthetic is instilled in the eye and then a fluorescein strip (not fluorescein sodium and benoxinate hydrochloride ophthalmic solution, which may become less sterile since it has a multiuse container) is applied to the superior conjunctiva. The clinician then looks for evidence of fluid egress using the cobalt blue filter. The patient is instructed to blink once. Fluid egress appears as a black stream as the fluorescein dye becomes diluted by aqueous humor escaping the nonintact wound and the appearance of bright green dye surrounds the leak site. The term Seidel positive indicates a leak. An estimate should be made of the rate and volume of fluid exiting the wound.
Gonioscopy
Gonioscopy can be used to evaluate the postsurgical incision, more specifically for identification and management of internal incision wound gape. On gonioscopy, internal wound gape appears as an elongated oval opening resembling a fish mouth. If internal incision wound gape is identified gonioscopically before surgery is complete, the leak can be managed intraoperatively. The surgeon can irrigate along the length of the incision to remove cortical fragments or viscoelastic that may cause internal wound gaping. If unsuccessful, rapidly deepening the anterior chamber with balanced salt solution through the paracentesis incision may be employed. These methods may improve wound stability, reduce risk of postoperative hyphema, lower the incidence of endophthalmitis, and lessen the likelihood of late against-the-rule drift.21
Anterior Segment Optical Coherence Tomography
Instances when Seidel testing was negative despite actual wound gaping have been described.22,23 Anterior segment OCT is useful to evaluate incision architecture. A 2007 United Kingdom study investigated the corneal architecture in the immediate postoperative period following phacoemulsification using anterior segment OCT. This study showed the benefits of identifying architectural features such as epithelial gaping, endothelial gaping, stripping of Descemet membrane, and loss of coaptation. These features were found to be more common at low IOP and could represent a significant risk factor for endophthalmitis.24 Another study published by Behrens and colleagues indicated that a localized detachment of Descemet membrane may be more common than observed with slit-lamp (Figure 7). Corneal gaping, especially if along the entire length of the surgical wound, may lead to inadvertent bacterial access into the anterior chamber.25

Anterior segment OCT imaging was first described by Izatt and colleagues in 1994.26 Unlike posterior segment OCT, anterior segment OCT requires a greater depth of field and higher energy levels as images are commonly distorted by refraction at boundaries where the refractive index changes. Longer infrared wavelengths improve the penetration through tissues that scatter light, such as the sclera and limbus, which allows visualization, for example, of the iridocorneal angle.27,28

Two main scan patterns are used for anterior segment OCT: 512 x 128 cube scan (4-mm width x 4-mm length) and 5-line raster (3-mm length) with adjustable rotation and spacing. A recent software update allows measurement of corneal thickness, visualization of anterior chamber angle structures along with topographic analysis, anterior and posterior elevation maps of the cornea, and reliable pachymetric maps.29,30 The anterior segment cube acquires a series of 128 horizontal scan lines each composed of 512 A-scans. These high-definition scans acquire vertical and horizontal directions composed of 1024 A-scans each. This cube may be used to measure corneal thickness and visualize corneal architecture, creating a 3-D image of the data (Figure 8). The anterior segment 5-line raster scans through 5 parallel lines of equal length to view high-resolution images of the anterior chamber angle and cornea. Each line, fixed at 3-mm in length, is composed of 4096 A-scans.31 Anterior segment cube OCT allows identification of subtle variations in incision architecture at different locations across the width of the OCT image.
Bandage Soft Contact Lens
Upon reviewing the anterior segment OCT images of our patient with the bandage contact lens in place, it was evident that the adherent ocular bandage was protecting the incision. A tighter fitting bandage contact lens is ideal and adheres firmly to any area of epithelial damage and epithelial gaping to help seal the incision, protecting the wound and improving structural integrity. The bandage contact lens is gradually replaced by new cells via re-epithelialization; thus, it behaves as an adjunct to natural wound healing. A bandage contact lens also improves patient comfort.
It is hypothesized that a bandage contact lens improves the structural integrity of the incision site and helps prevent leaking, hypotony, and minor wound leaks. One study revealed a statistically significant lower IOP in nonbandage contact lens patients by an average of 6 mm Hg (mean [SD] 13.4 mm Hg [5.3]; range, 5 - 23 mm Hg) vs patients with a bandage contact lens (mean [SD] 19.4 mm Hg [5.9]; range, 11 - 29 mm Hg) in the immediate postoperative period.32 The authors suggested that the bandage contact lens may prevent microleaks, resulting in a higher IOP.
Aqueous Suppressants
Aqueous suppressants are a great option when IOP is abnormally elevated by decreasing the IOP and allowing the cornea to heal and self-seal.Effective aqueous suppressants are β blockers and carbonic anhydrase inhibitors.
After phacoemulsification ocular hypotony (< 6 mm Hg) occurs most commonly due to wound leakage or excessive intraocular inflammation. However, with the presence of corneal wound leakage and ocular hypotony, aqueous suppressants are not the best option.
Further Management of Wound Leaks
Management of a postoperative wound leak will vary based on severity. The majority of mild leaks are self-sealing. Anterior segment OCT helps the clinician to identify microleaks in an otherwise Seidel negative eye. If wound leakage is moderate with a formed anterior chamber, the use of a bandage contact lens is a good option, as can be the prescription of aqueous suppressants, depending on IOP.33
If the anterior chamber is flat, iris prolapse is apparent, or extremely low IOP exists, the patient needs to be referred to the surgeon. Current standard of care directs the surgeon to use sutures to further manage corneal wound leak. However, several studies have recognized the increased risk of suture-related complications, such as induced astigmatism, corneal opacities, incomplete wound closure, and corneal neovascularization.6,34-38 Other wound closure options include polyethylene glycol-based products, corneal welding, cyanoacrylate, or fibrin (Table 2).39 Traditionally nylon sutures have been used for clear corneal incision wound closure. However, tissue adhesives are gaining popularity as a substitute for sutures in wound closure.40

Cyanoacrylate
Numerous studies have been published on the efficacy of cyanoacrylate as a substitute for sutures, specifically in clear corneal incisions. AEs of cyanoacrylate include a transient foreign-body sensation and diffuse or focal bulbar conjunctival hyperemia.41,42 Shigemitsu and Majima found that fibrin and cyanoacrylate glue had tensile strength similar to sutures when used in cataract surgery.39 Polyethylene glycol-based products, also used in artificial tears and contact lens materials, may also help seal wound leaks. Another agent is ReSure (Ocular Therapeutix, Bedford, MA), an FDA-approved synthetic, polyethylene glycol hydrogel sealant that is 90% water after polymerization. ReSure has been shown to be safe and effective in sealing cataract surgical clear corneal incisions.6,43 ReSure takes about 20 seconds to prepare, and placement is aided by the use of a blue dye that dissipates within hours. This hydrogel will gradually slough off in the tears once the tissue has fully regenerated; there is no need to remove the sealant.44
Rossi and colleagues evaluated the efficacy of corneal welding to close wounds after cataract surgery. The technique involves laser-assisted closure of the corneal wound(s) by a diode laser that welds the stroma.45 Corneal welding takes seconds to achieve good closure without significant astigmatism or inflammation; however very careful application of the light absorbing dyes is required as they are toxic if allowed to enter the anterior chamber.45-47
Conclusion
Optometrists may be called to manage patients during both the preoperative and postoperative phases of cataract surgical care. Those who participate in postoperative care should carefully evaluate for the presence of wound leak or wound gape as a potential complication. The OCT may be employed to evaluate patients suspected of having these leaks or gapes. Proficiency in the interpretation of OCT results and more traditional evaluation methods allows for successful detection of wound leaks or gapes. The timely diagnosis and treatment of postoperative wound leaks allow for the best possible outcomes for cataract surgery patients.
1. Thylefors B, Négrel AD, Pararajasegaram R, Dadzie KY. Global data on blindness. Bull World Health Organ. 1995;73(1):115-121.
2. Flaxman SR, Bourne RRA, Resnikoff S, et al; Vision Loss Expert Group of the Global Burden of Disease Study. Global causes of blindness and distance vision impairment 1990-2020: a systematic review and meta-analysis. Lancet Glob Health. 2017;5(12):e1221-e1224.
3. Congdon N, Vingerling JR, Klein BE, et al; Eye Diseases Prevalence Research Group. Prevalence of cataract and pseudophakia/aphakia among adults in the United States. Arch Ophthalmol. 2004;122(4):487-494.
4. Kurt E, Mayalı H. Early post-operative complications in cataract surgery. In: Zaidi FH, ed. Cataract Surgery. IntechOpen; 2013. https://www.intechopen.com/books/cataract-surgery/post-operative-infections-associated-with-cataract-surgery. Accessed July 15, 2019.
5. Chee SP. Clear corneal incision leakage after phacoemulsification--detection using povidone iodine 5%. Int Ophthalmol. 2005;26(4-5):175-179.
6. Masket S, Hovanesian JA, Levenson J, et al. Hydrogel sealant versus sutures to prevent fluid egress after cataract surgery. J Cataract Refract Surg. 2014;40(12):2057-2066.
7. Kelman CD. Phaco-emulsification and aspiration: a new technique of cataract removal. A preliminary report. Am J Ophthalmol. 1967;64(1):23-35.
8. Powe NR, Schein OD, Gieser SC, et al. Synthesis of the literature on visual acuity and complications following cataract extraction with intraocular lens implantation. Cataract Patient Outcome Research Team [published correction appears in Arch Ophthalmol. 1994;112(7):889]. Arch Ophthalmol. 1994;112(2):239-252.
9. Stein JD, Grossman DS, Mundy KM, Sugar A, Sloan FA. Severe adverse events after cataract surgery among medicare beneficiaries. Ophthalmology. 2011;118(9):1716-1723.
10. Greenberg PB, Tseng VL, Wu WC, et al. Prevalence and predictors of ocular complications associated with cataract surgery in United States veterans. Ophthalmology. 2011;118(3):507-514.
11. Mangan MS, Atalay E, Anci C, Tuncer I, Bilqec MD. Comparison of different types of complications in the phacoemulsification surgery learning curve according to number of operations performed. Turk J Ophthalmol. 2016;46(1):7-10.
12. Bell CM, Hatch WV, Cernat G, Urbach DR. Surgeon volumes and selected patient outcomes in cataract surgery: a population-based analysis. Ophthalmology. 2007;114(3):405-410.
13. Wallin T, Parker J, Jin Y, Kefalopoulos G, Olson RJ. Cohort study of 27 cases of endophthalmitis at a single institution. J Cataract Refract Surg. 2005;31(4):735-741.
14. Zaidi FH, Corbett MC, Burton BJ, Bloom PA. Raising the benchmark for the 21st century--the 1000 cataract operations audit and survey: outcomes, consultant-supervised training and sourcing NHS choice. Br J Ophthalmol. 2007;91(6):731-736.
15. Nichamin LD, Chang DF, Johnson SH, et al; American Society of Cataract and Refractive Surgery Cataract Clinical Committee. ASCRS white paper: what is the association between clear corneal cataract incisions and postoperative endophthalmitis? J Cataract Refract Surg. 2006;32(9):1556-1559.
16. Packer M, Chang DF, Dewey SH, et al; ASCRS Cataract Clinical Committee. Prevention, diagnosis, and management of acute postoperative bacterial endophthalmitis. J Cataract Refract Surg. 2011;37(9):1699-1714.
17. Montan PG, Koranyi G, Setterquist HE, Stridh A, Philipson BT, Wiklund K. Endophthalmitis after cataract surgery: risk factors relating to technique and events of the operation and patient history: a retrospective case-control study. Ophthalmology. 1998;105(12):2171-2177.
18. Taban M, Rao B, Reznik J, Zhang J, Chen Z, McDonnell PJ. Dynamic morphology of sutureless cataract wounds—effect of incision angle and location. Surv Ophthalmol. 2004;49(suppl 2):S62-S72.
19. Chee SP, Ti SE, Lim L, Chan AS, Jap A. Anterior segment optical coherence tomography evaluation of the integrity of clear corneal incisions: a comparison between 2.2-mm and 2.65-mm main incisions. Am J Ophthalmol. 2010;149(5):768-776.e1.
20. Koch DD, Nacke RE, Wang L, Novak KD. Issues in wound management. In: Steinert R, ed. Cataract Surgery. 3rd ed. New York: Elsevier; 2009:581-588.
21. Gimbel HV, Sun R, DeBroff GM. Recognition and management of internal wound gape. J Cataract Refract Surg. 1995;21(2):121-124.
22. May WN, Castro-Combs J, Quinto GG, Kashiwabuchi R, Gower EW, Behrens A. Standardized Seidel test to evaluate different sutureless cataract incision configurations. J Cataract Refract Surg. 2010;36(6):1011-1017.
23. Kashiwabuchi FK, Khan YA, Rodrigues MW Jr, Wang J, McDonnell PJ, Daoud YJ. Seidel and India ink tests assessment of different clear cornea side-port incision configurations. Graefes Arch Clin Exp Ophthalmol. 2013;251(8):1961-1965.
24. Calladine D, Packard R. Clear corneal incision architecture in the immediate postoperative period evaluated using optical coherence tomography. J Cataract Refract Surg. 2007;33(8):1429-1435.
25. Behrens WJ, Stark KA, Pratzer, McDonnell PJ. Dynamics of small-incision clear cornea wounds after phacoemulsification surgery using optical coherence tomography in the early postoperative period. J Refractive Surgery. 2008;24(1):46-49.
26. Izatt JA, Hee MR, Swanson EA, et al. Micrometer-scale resolution imaging of the anterior eye in vivo with optical coherence tomography. Arch Ophthalmol. 1994;112(12):1584-1589.
27. Hurmeric V, Yoo SH, Mutlu FM. Optical coherence tomography in cornea and refractive surgery. Expert Rev Ophthalmol. 2012;7(3):241-250.
28. Schuman JS, Puliafito CA, Fujimoto JG, Duker JS. Optical Coherence Tomography of Ocular Diseases. 3rd ed. Thorofare, NJ: Slack Inc; 2013.
29. Salim S. The role of anterior segment optical coherence tomography in glaucoma. J Ophthalmol. 2012;2012:476801.
30. Kharousi NA, Wali UK, Azeem S. Current applications of optical coherence tomography in ophthalmology. In: Kawasaki M, ed. Optical Coherence Tomography. IntechOpen; 2013. https://www.intechopen.com/books/optical-coherence-tomography. Accessed July 31, 2019.
31. Rodrigues EB, Johanson M, Penha FM. Anterior segment tomography with the cirrus optical coherence tomography. J Ophthalmol. 2012;2012:806989.
32. Calladine D, Ward M, Packard R. Adherent ocular bandage for clear corneal incisions used in cataract surgery. J Cataract Refract Surg. 2010;36(11):1839-1848.
33. Haldar K, Saraff R. Closure technique for leaking wound resulting from thermal injury during phacoemulsification. J Cataract Refract Surg. 2014;40(9):1412-1414.
34. Zoghby JT, Cohen KL. Phacoemulsification-related corneal incision contracture. https://www.aao.org/eyenet/article/phacoemulsification-related-corneal-incision-contr. Published December 2012. Accessed June 16, 2019.
35. Bhatia SS. Ocular surface sealants and adhesives. Ocul Surf. 2006;4(3):146-154.
36. May WN, Castro-Combs J, Kashiwabuchi RT, et al. Bacterial-sized particle inflow through sutured clear corneal incisions in a laboratory human model. J Cataract Refract Surg. 2011;37(6):1140-1146.
37. Meskin SW, Ritterband DC, Shapiro DE, et al. Liquid bandage (2-octyl cyanoacrylate) as a temporary wound barrier in clear corneal cataract surgery. Ophthalmology. 2005;112(11):2015-2021.
38. Heaven CJ, Davison CR, Cockcroft PM. Bacterial contamination of nylon corneal sutures. Eye (Lond). 1995;9(pt 1):116-118.
39. Shigemitsu T, Majima Y. The utilization of a biological adhesive for wound treatment: comparison of suture, self-sealing sutureless and cyanoacrylate closure in the tensile strength test. Int Ophthalmol. 1996-1997;20:323-328.
40. Uy HS, Kenyon KR. Surgical outcomes after application of a liquid adhesive ocular bandage to clear corneal incisions during cataract surgery. J Cataract Refract Surg. 2013;39(11):1668-1674.
41. Meskin SW, Ritterband DC, Shapiro DE, et al. Liquid bandage (2-octyl cyanoacrylate) as a temporary wound barrier in clear corneal cataract surgery. Ophthalmology. 2005;112(11):2015-2021.
42. Tong AY, Gupta PK, Kim T. Wound closure and tissue adhesives in clear corneal incision cataract surgery. Curr Opin Ophthalmol. 2018;29(1):14-18.
43. US Food and Drug Administration. Summary of Safety and Effectiveness Data. Ophthalmic sealant: ReSure Sealant. https://www.accessdata.fda.gov/cdrh_docs/pdf13/P130004b.pdf. Published September 13, 2013. Accessed July 9, 2019.
44. About ReSure sealant. https://www.resuresealant.com/overview. Accessed July 31, 2019.
45. Menabuoni L, Pini R, Rossi F, Lenzetti I, Yoo SH, Parel JM. Laser-assisted corneal welding in cataract surgery: retrospective study. J Cataract Refract Surg. 2007;33(9):1608-1612.
46. Rasier R, Ozeren M, Artunay O, et al. Corneal tissue welding with infrared laser irradiation after clear corneal incision. Cornea. 2010;29(9):985-990.
47. Rossi F, Matteini P, Ratto F, Menabuoni L, Lenzetti I, Pini R. Laser tissue welding in ophthalmic surgery. J Biophotonics. 2008;1(4):331-342.
48. Taban M, Behrens A, Newcomb RL, et al. Acute endophthalmitis following cataract surgery: a systematic review of the literature. Arch Ophthalmol. 2005;123(5):613-620.
49. Taylor DM, Atlas BF, Romanchuk KG, Stern AL. Pseudophakic bullous keratopathy. Ophthalmology. 1983;90(1):19-24.
50. Lobo CL, Faria PM, Soares MA, Bernardes RC, Cunha-Vaz JG. Macular alterations after small-incision cataract surgery. J Cataract Refract Surg. 2004;30(4):752-760.
51. Flach AJ. The incidence, pathogenesis and treatment of cystoid macular edema following cataract surgery. Trans Am Ophthalmol Soc. 1998;96:557-634.
52. Wright PL, Wilkinson CP, Balyeat HD, Popham J, Reinke M. Angiographic cystoid macular edema after posterior chamber lens implantation. Arch Ophthalmol. 1988;106(6):740-744.
53. Kim SJ, Belair ML, Bressler NM, et al. A method of reporting macular edema after cataract surgery using optical coherence tomography. Retina. 2008;28(6):870-876.
54. Alio JL, Ruiz-Moreno JM, Shabayek MH, Lugo FL, Abd El Rahman AM. The risk of retinal detachment in high myopia after small incision coaxial phacoemulsification. Am J Ophthalmol. 2007;144(1):93-98.
55. Bhagwandien AC, Cheng YY, Wolfs RC, van Meurs JC, Luyten GP. Relationship between retinal detachment and biometry in 4262 cataractous eyes. Ophthalmology. 2006;113(4):643-649.
56. Boberg-Ans G, Henning V, Villumsen J, la Cour M. Longterm incidence of rhegmatogenous retinal detachment and survival in a defined population undergoing standardized phacoemulsification surgery. Acta Ophthalmol Scand. 2006;84(5):613-618.
57. Jakobsson G, Montan P, Zetterberg M, Stenevi U, Behndig A, Lundström M. Capsule complication during cataract surgery: retinal detachment after cataract surgery with capsule complication: Swedish Capsule Rupture Study Group report 4. J Cataract Refract Surg. 2009;35(10):1699-1705.
58. Neuhann IM, Neuhann TF, Heimann H, Schmickler S, Gerl RH, Foerster MH. Retinal detachment after phacoemulsification in high myopia: analysis of 2356 cases. J Cataract Refract Surg. 2008;34(10):1644-1657.
59. Russell M, Gaskin B, Russell D, Polkinghorne PJ. Pseudophakic retinal detachment after phacoemulsification cataract surgery: ten-year retrospective review. J Cataract Refract Surg. 2006;32(3):442-445.
60. Apple DJ, Solomon KD, Tetz MR, et al. Posterior capsule opacification. Surv Ophthalmol. 1992;37(2):73-116.
61. Wu S, Tong N, Pan L, et al. Retrospective analyses of potential risk factors for posterior capsule opacification after cataract surgery. J Ophthalmol. 2018;2018:9089285.
62. Clark A, Morlet N, Ng JQ, Preen DB, Semmens JB. Whole population trends in complications of cataract surgery over 22 years in Western Australia. Ophthalmology. 2011;118(6):1055-1061.
63. Adhikari S, Shrestha UD. Pediatric cataract surgery with hydrophilic acrylic intraocular lens implantation in Nepalese Children. Clin Ophthalmol. 2017;12:7-11.
64. Lee BJ, Smith SD, Jeng BH. Suture-related corneal infections after clear corneal cataract surgery. J Cataract Refract Surg. 2009;35(5):939-942.
65. May WN, Castro-Combs J, Kashiwabuchi RT, et al. Sutured clear corneal incision: wound apposition and permeability to bacterial-sized particles. Cornea. 2013;32(3):319-325.
66. Hillier RJ, Ajit RR, Kelly SP. Suture-related complications after cataract surgery: a patient safety issue. J Cataract Refract Surg. 2009;35(11):2035-2036.
67. Hovanesian JA, Karageozian VH. Watertight cataract incision closure using fibrin tissue adhesive. J Cataract Refract Surg. 2007;33(8):1461-1463.
The term cataract is derived from the Latin word “catarractes,” which means “waterfall,” as the foamy white opacity of an advanced cataract can be likened to a tempestuous cascade. Cataract is the leading cause of preventable blindness worldwide.1,2 It is no surprise, therefore, that cataract surgery is the most frequently performed ophthalmic surgical procedure worldwide. Cataract surgeries may reach 30 million annual cases by 2020.3 Given the large number of surgeries being performed, postsurgical complications are not uncommon.
Early postoperative complications from lens exchange (cataract) surgery include increased intraocular pressure (IOP), corneal edema, and corneal wound leakage.4 Corneal wound leakage is not uncommon; one study showed that, in 100 cases, almost one-third of incisions leaked.5 A 2014 prospective study of 500 postcataract surgery eyes revealed that 48.8% had fluid egress.6 Early detection is important so that efforts to restore corneal integrity can immediately be implemented. If not caught early, patients are at risk for developing a cascade of sequelae, including endophthalmitis.
The majority of corneal wound leaks postphacoemulsification are self-limiting and self-sealing. Moderate wound leaks require treatment, as in the following case. Strategies to detect, image, and treat wound leaks are covered in this discussion.
Case Presentation
A 69-year-old male veteran presented with no complaints for a 1-day postoperative visit following right eye phacoemulsification cataract extraction. His best corrected visual acuity in the right eye was 20/40, and his pinhole visual acuity was 20/25+2. On slit-lamp examination, the temporally located main incision appeared well-adhered and was found to be Seidel negative; however, the inferior paracentesis wound was found to be Seidel positive, demonstrating a slow leak. Intraocular pressure (IOP) measured with tonopen was 9 mm Hg.
A bandage soft contact lens was placed on the eye. The patient was instructed not to rub or place any pressure on the eye and to avoid bending and heavy lifting. He was also instructed to continue his postoperative medications (prednisolone 1% every 2 hours and polymyxin B sulfate 4 times daily) in his right eye. A follow-up appointment was scheduled for the next day.
The patient presented for his postoperative day-2 visit with a best corrected visual acuity in the right eye of 20/20. He reported no visual problems, no eye pain, and mentioned that he had had a comfortable night sleep. A slit-lamp examination revealed trace diffuse injection in the operative eye, predominantly central Descemet membrane folds, 1+ stromal edema, and a Seidel negative main incision wound. However, the inferior paracentesis wound showed a moderate leak (Seidel positive), and the anterior chamber showed a 1+ cell and flare. Goldmann tonometry revealed an IOP of 5 mm Hg, indicating hypotony.
Anterior segment cube 512 x 128 optical coherence tomography (OCT) was obtained with the bandage contact lens (Figures 1 and 2), and then repeated with the bandage contact lens removed (Figures 3 and 4). OCT imaging confirmed epithelial and endothelial gaping, loss of coaptation, and a localized detachment of the Descemet membrane. The veteran was referred to his surgeon that same day, and 2 limbal vicryl sutures were placed. The patient was instructed to continue prednisolone 1% 4 times daily and polymyxin B sulfate every 2 hours; erythromycin ointment 3 times daily was added to his regimen.
He was scheduled for a follow-up examination 1 week later. At that visit, the wound was no longer leaking and IOP had risen to a preoperative value of 17 mm Hg. The corneal sutures were removed at the 1-month postoperative examination and a follow-up was scheduled for 4 months later. An anterior segment OCT was obtained (Figure 5).
Discussion
In July 1967, Charles Kelman, MD, suggested using a dental ultrasonic tool, normally employed to clean teeth, to fragment the nucleus of the crystalline lens. Dr. Kelman’s first operation using phacoemulsification on a human eye took 3 hours.7 As the procedure for cataract removal has been refined, complication rates and surgical times have vastly improved.
Phacoemulsification is the most commonly performed outpatient surgery in the US; about 3 million cases are performed annually. Due to the high volume of cases, adverse events (AEs) are not uncommon. The incidence of complications following phacoemulsification is < 5%; the frequency of severe complications has been estimated at < 0.7%.8 Severe complications include endophthalmitis, suprachoroidal hemorrhage, and/or retinal detachment.9 Studies have shown a decline in rates of sight-threatening AEs from 1994 to 2006.9 A retrospective study of 45,082 veterans from 2005 to 2007 identified that a preoperative disease burden such as diabetes mellitus, chronic pulmonary disease, age-related macular degeneration, and diabetes with ophthalmic manifestations, was positively associated with a greater risk of cataract surgical complications.10
Complications
The level of a surgeon’s proficiency with phacoemulsification is directly correlated to the number of operations performed; there is a lower complication rate among more experienced surgeons, including those who work in high-volume settings.11,12 One study identified that the AE rate within 14 days of surgery was 0.8% for surgeons performing 50 to 250 cataract surgeries per year, but only 0.1% for those performing > 1000 cataract surgeries annually.12
Potential postoperative lens exchange complications include increased IOP, corneal wound leakage, corneal edema, bullous keratopathy, cystoid macular edema, retinal detachment, and endophthalmitis (Table 1). A corneal wound leak can provide a potential ingress for bacteria, putting the patient at risk for endophthalmitis, perhaps the most devastating complication following cataract surgery.
Endophthalmitis
Endophthalmitis has been reported to occur in .001% to .327% of patients during postoperative care.5,13-17 Early detection is important to maintain corneal integrity and prevent a cascade of detrimental ocular sequalae including the potential for endophthalmitis. According to Zaida and colleagues, endophthalmitis occurred in fewer than 1 of 1000 consecutive cases.14 A leaking clear corneal incision wound on the first day postoperatively has been associated with a 44-fold increased risk of endophthalmitis.13
Causes of endophthalmitis
In a retrospective case-controlled series of 57 patients with postcataract endophthalmitis, implantation of an intraocular lens with a resultant wound abnormality was thought to be the causative factor in 5%.17 Another source of endophthalmitis can be the intraocular lens (IOL), which may act as a vector for bacteria. By placing the IOL against the conjunctiva or exposing it to the theater air during surgery, bacteria can be introduced prior to implantation.17 Immunosuppressive treatment is the only patient antecedent factor that can be considered a predictor for endopthalmitis.17
The internal corneal seal is IOP dependent, and postoperative ocular hypotony may cause a seemingly watertight wound to leak. Taban and colleagues used anterior segment OCT to image numerous self-sealing incisions. They found that the corneal incision wound more tightly seals at higher IOPs. Additionally, more perpendicular (larger angle) incisions seal better at a lower IOP while less perpendicular (smaller angle) incisions seal better at a higher IOP (Figure 6).18
Incision Placement
Studies have shown that the main incision site is more clinically competent than is the side port incision site, as in our case study.19 Side-port incisions have a 1- or 2-plane architectural profile in contrast to the 3-plane profile typical of a main incision.19 Recent advances including the conversion to clear-corneal incisions of diminishing size, techniques used for wound construction, phacoemulsification machine design, and small-incision IOLs, should further reduce the prevalence and complications of wound compromise.20
Seidel Testing
Seidel testing is the most common method to evaluate corneal wound integrity and identify leaks. A drop of topical anesthetic is instilled in the eye and then a fluorescein strip (not fluorescein sodium and benoxinate hydrochloride ophthalmic solution, which may become less sterile since it has a multiuse container) is applied to the superior conjunctiva. The clinician then looks for evidence of fluid egress using the cobalt blue filter. The patient is instructed to blink once. Fluid egress appears as a black stream as the fluorescein dye becomes diluted by aqueous humor escaping the nonintact wound and the appearance of bright green dye surrounds the leak site. The term Seidel positive indicates a leak. An estimate should be made of the rate and volume of fluid exiting the wound.
Gonioscopy
Gonioscopy can be used to evaluate the postsurgical incision, more specifically for identification and management of internal incision wound gape. On gonioscopy, internal wound gape appears as an elongated oval opening resembling a fish mouth. If internal incision wound gape is identified gonioscopically before surgery is complete, the leak can be managed intraoperatively. The surgeon can irrigate along the length of the incision to remove cortical fragments or viscoelastic that may cause internal wound gaping. If unsuccessful, rapidly deepening the anterior chamber with balanced salt solution through the paracentesis incision may be employed. These methods may improve wound stability, reduce risk of postoperative hyphema, lower the incidence of endophthalmitis, and lessen the likelihood of late against-the-rule drift.21
Anterior Segment Optical Coherence Tomography
Instances when Seidel testing was negative despite actual wound gaping have been described.22,23 Anterior segment OCT is useful to evaluate incision architecture. A 2007 United Kingdom study investigated the corneal architecture in the immediate postoperative period following phacoemulsification using anterior segment OCT. This study showed the benefits of identifying architectural features such as epithelial gaping, endothelial gaping, stripping of Descemet membrane, and loss of coaptation. These features were found to be more common at low IOP and could represent a significant risk factor for endophthalmitis.24 Another study published by Behrens and colleagues indicated that a localized detachment of Descemet membrane may be more common than observed with slit-lamp (Figure 7). Corneal gaping, especially if along the entire length of the surgical wound, may lead to inadvertent bacterial access into the anterior chamber.25
Anterior segment OCT imaging was first described by Izatt and colleagues in 1994.26 Unlike posterior segment OCT, anterior segment OCT requires a greater depth of field and higher energy levels as images are commonly distorted by refraction at boundaries where the refractive index changes. Longer infrared wavelengths improve the penetration through tissues that scatter light, such as the sclera and limbus, which allows visualization, for example, of the iridocorneal angle.27,28
Two main scan patterns are used for anterior segment OCT: 512 x 128 cube scan (4-mm width x 4-mm length) and 5-line raster (3-mm length) with adjustable rotation and spacing. A recent software update allows measurement of corneal thickness, visualization of anterior chamber angle structures along with topographic analysis, anterior and posterior elevation maps of the cornea, and reliable pachymetric maps.29,30 The anterior segment cube acquires a series of 128 horizontal scan lines each composed of 512 A-scans. These high-definition scans acquire vertical and horizontal directions composed of 1024 A-scans each. This cube may be used to measure corneal thickness and visualize corneal architecture, creating a 3-D image of the data (Figure 8). The anterior segment 5-line raster scans through 5 parallel lines of equal length to view high-resolution images of the anterior chamber angle and cornea. Each line, fixed at 3-mm in length, is composed of 4096 A-scans.31 Anterior segment cube OCT allows identification of subtle variations in incision architecture at different locations across the width of the OCT image.
Bandage Soft Contact Lens
Upon reviewing the anterior segment OCT images of our patient with the bandage contact lens in place, it was evident that the adherent ocular bandage was protecting the incision. A tighter fitting bandage contact lens is ideal and adheres firmly to any area of epithelial damage and epithelial gaping to help seal the incision, protecting the wound and improving structural integrity. The bandage contact lens is gradually replaced by new cells via re-epithelialization; thus, it behaves as an adjunct to natural wound healing. A bandage contact lens also improves patient comfort.
It is hypothesized that a bandage contact lens improves the structural integrity of the incision site and helps prevent leaking, hypotony, and minor wound leaks. One study revealed a statistically significant lower IOP in nonbandage contact lens patients by an average of 6 mm Hg (mean [SD] 13.4 mm Hg [5.3]; range, 5 - 23 mm Hg) vs patients with a bandage contact lens (mean [SD] 19.4 mm Hg [5.9]; range, 11 - 29 mm Hg) in the immediate postoperative period.32 The authors suggested that the bandage contact lens may prevent microleaks, resulting in a higher IOP.
Aqueous Suppressants
Aqueous suppressants are a great option when IOP is abnormally elevated by decreasing the IOP and allowing the cornea to heal and self-seal.Effective aqueous suppressants are β blockers and carbonic anhydrase inhibitors.
After phacoemulsification ocular hypotony (< 6 mm Hg) occurs most commonly due to wound leakage or excessive intraocular inflammation. However, with the presence of corneal wound leakage and ocular hypotony, aqueous suppressants are not the best option.
Further Management of Wound Leaks
Management of a postoperative wound leak will vary based on severity. The majority of mild leaks are self-sealing. Anterior segment OCT helps the clinician to identify microleaks in an otherwise Seidel negative eye. If wound leakage is moderate with a formed anterior chamber, the use of a bandage contact lens is a good option, as can be the prescription of aqueous suppressants, depending on IOP.33
If the anterior chamber is flat, iris prolapse is apparent, or extremely low IOP exists, the patient needs to be referred to the surgeon. Current standard of care directs the surgeon to use sutures to further manage corneal wound leak. However, several studies have recognized the increased risk of suture-related complications, such as induced astigmatism, corneal opacities, incomplete wound closure, and corneal neovascularization.6,34-38 Other wound closure options include polyethylene glycol-based products, corneal welding, cyanoacrylate, or fibrin (Table 2).39 Traditionally nylon sutures have been used for clear corneal incision wound closure. However, tissue adhesives are gaining popularity as a substitute for sutures in wound closure.40
Cyanoacrylate
Numerous studies have been published on the efficacy of cyanoacrylate as a substitute for sutures, specifically in clear corneal incisions. AEs of cyanoacrylate include a transient foreign-body sensation and diffuse or focal bulbar conjunctival hyperemia.41,42 Shigemitsu and Majima found that fibrin and cyanoacrylate glue had tensile strength similar to sutures when used in cataract surgery.39 Polyethylene glycol-based products, also used in artificial tears and contact lens materials, may also help seal wound leaks. Another agent is ReSure (Ocular Therapeutix, Bedford, MA), an FDA-approved synthetic, polyethylene glycol hydrogel sealant that is 90% water after polymerization. ReSure has been shown to be safe and effective in sealing cataract surgical clear corneal incisions.6,43 ReSure takes about 20 seconds to prepare, and placement is aided by the use of a blue dye that dissipates within hours. This hydrogel will gradually slough off in the tears once the tissue has fully regenerated; there is no need to remove the sealant.44
Rossi and colleagues evaluated the efficacy of corneal welding to close wounds after cataract surgery. The technique involves laser-assisted closure of the corneal wound(s) by a diode laser that welds the stroma.45 Corneal welding takes seconds to achieve good closure without significant astigmatism or inflammation; however very careful application of the light absorbing dyes is required as they are toxic if allowed to enter the anterior chamber.45-47
Conclusion
Optometrists may be called to manage patients during both the preoperative and postoperative phases of cataract surgical care. Those who participate in postoperative care should carefully evaluate for the presence of wound leak or wound gape as a potential complication. The OCT may be employed to evaluate patients suspected of having these leaks or gapes. Proficiency in the interpretation of OCT results and more traditional evaluation methods allows for successful detection of wound leaks or gapes. The timely diagnosis and treatment of postoperative wound leaks allow for the best possible outcomes for cataract surgery patients.
The term cataract is derived from the Latin word “catarractes,” which means “waterfall,” as the foamy white opacity of an advanced cataract can be likened to a tempestuous cascade. Cataract is the leading cause of preventable blindness worldwide.1,2 It is no surprise, therefore, that cataract surgery is the most frequently performed ophthalmic surgical procedure worldwide. Cataract surgeries may reach 30 million annual cases by 2020.3 Given the large number of surgeries being performed, postsurgical complications are not uncommon.
Early postoperative complications from lens exchange (cataract) surgery include increased intraocular pressure (IOP), corneal edema, and corneal wound leakage.4 Corneal wound leakage is not uncommon; one study showed that, in 100 cases, almost one-third of incisions leaked.5 A 2014 prospective study of 500 postcataract surgery eyes revealed that 48.8% had fluid egress.6 Early detection is important so that efforts to restore corneal integrity can immediately be implemented. If not caught early, patients are at risk for developing a cascade of sequelae, including endophthalmitis.
The majority of corneal wound leaks postphacoemulsification are self-limiting and self-sealing. Moderate wound leaks require treatment, as in the following case. Strategies to detect, image, and treat wound leaks are covered in this discussion.
Case Presentation
A 69-year-old male veteran presented with no complaints for a 1-day postoperative visit following right eye phacoemulsification cataract extraction. His best corrected visual acuity in the right eye was 20/40, and his pinhole visual acuity was 20/25+2. On slit-lamp examination, the temporally located main incision appeared well-adhered and was found to be Seidel negative; however, the inferior paracentesis wound was found to be Seidel positive, demonstrating a slow leak. Intraocular pressure (IOP) measured with tonopen was 9 mm Hg.
A bandage soft contact lens was placed on the eye. The patient was instructed not to rub or place any pressure on the eye and to avoid bending and heavy lifting. He was also instructed to continue his postoperative medications (prednisolone 1% every 2 hours and polymyxin B sulfate 4 times daily) in his right eye. A follow-up appointment was scheduled for the next day.
The patient presented for his postoperative day-2 visit with a best corrected visual acuity in the right eye of 20/20. He reported no visual problems, no eye pain, and mentioned that he had had a comfortable night sleep. A slit-lamp examination revealed trace diffuse injection in the operative eye, predominantly central Descemet membrane folds, 1+ stromal edema, and a Seidel negative main incision wound. However, the inferior paracentesis wound showed a moderate leak (Seidel positive), and the anterior chamber showed a 1+ cell and flare. Goldmann tonometry revealed an IOP of 5 mm Hg, indicating hypotony.
Anterior segment cube 512 x 128 optical coherence tomography (OCT) was obtained with the bandage contact lens (Figures 1 and 2), and then repeated with the bandage contact lens removed (Figures 3 and 4). OCT imaging confirmed epithelial and endothelial gaping, loss of coaptation, and a localized detachment of the Descemet membrane. The veteran was referred to his surgeon that same day, and 2 limbal vicryl sutures were placed. The patient was instructed to continue prednisolone 1% 4 times daily and polymyxin B sulfate every 2 hours; erythromycin ointment 3 times daily was added to his regimen.
He was scheduled for a follow-up examination 1 week later. At that visit, the wound was no longer leaking and IOP had risen to a preoperative value of 17 mm Hg. The corneal sutures were removed at the 1-month postoperative examination and a follow-up was scheduled for 4 months later. An anterior segment OCT was obtained (Figure 5).
Discussion
In July 1967, Charles Kelman, MD, suggested using a dental ultrasonic tool, normally employed to clean teeth, to fragment the nucleus of the crystalline lens. Dr. Kelman’s first operation using phacoemulsification on a human eye took 3 hours.7 As the procedure for cataract removal has been refined, complication rates and surgical times have vastly improved.
Phacoemulsification is the most commonly performed outpatient surgery in the US; about 3 million cases are performed annually. Due to the high volume of cases, adverse events (AEs) are not uncommon. The incidence of complications following phacoemulsification is < 5%; the frequency of severe complications has been estimated at < 0.7%.8 Severe complications include endophthalmitis, suprachoroidal hemorrhage, and/or retinal detachment.9 Studies have shown a decline in rates of sight-threatening AEs from 1994 to 2006.9 A retrospective study of 45,082 veterans from 2005 to 2007 identified that a preoperative disease burden such as diabetes mellitus, chronic pulmonary disease, age-related macular degeneration, and diabetes with ophthalmic manifestations, was positively associated with a greater risk of cataract surgical complications.10
Complications
The level of a surgeon’s proficiency with phacoemulsification is directly correlated to the number of operations performed; there is a lower complication rate among more experienced surgeons, including those who work in high-volume settings.11,12 One study identified that the AE rate within 14 days of surgery was 0.8% for surgeons performing 50 to 250 cataract surgeries per year, but only 0.1% for those performing > 1000 cataract surgeries annually.12
Potential postoperative lens exchange complications include increased IOP, corneal wound leakage, corneal edema, bullous keratopathy, cystoid macular edema, retinal detachment, and endophthalmitis (Table 1). A corneal wound leak can provide a potential ingress for bacteria, putting the patient at risk for endophthalmitis, perhaps the most devastating complication following cataract surgery.
Endophthalmitis
Endophthalmitis has been reported to occur in .001% to .327% of patients during postoperative care.5,13-17 Early detection is important to maintain corneal integrity and prevent a cascade of detrimental ocular sequalae including the potential for endophthalmitis. According to Zaida and colleagues, endophthalmitis occurred in fewer than 1 of 1000 consecutive cases.14 A leaking clear corneal incision wound on the first day postoperatively has been associated with a 44-fold increased risk of endophthalmitis.13
Causes of endophthalmitis
In a retrospective case-controlled series of 57 patients with postcataract endophthalmitis, implantation of an intraocular lens with a resultant wound abnormality was thought to be the causative factor in 5%.17 Another source of endophthalmitis can be the intraocular lens (IOL), which may act as a vector for bacteria. By placing the IOL against the conjunctiva or exposing it to the theater air during surgery, bacteria can be introduced prior to implantation.17 Immunosuppressive treatment is the only patient antecedent factor that can be considered a predictor for endopthalmitis.17
The internal corneal seal is IOP dependent, and postoperative ocular hypotony may cause a seemingly watertight wound to leak. Taban and colleagues used anterior segment OCT to image numerous self-sealing incisions. They found that the corneal incision wound more tightly seals at higher IOPs. Additionally, more perpendicular (larger angle) incisions seal better at a lower IOP while less perpendicular (smaller angle) incisions seal better at a higher IOP (Figure 6).18
Incision Placement
Studies have shown that the main incision site is more clinically competent than is the side port incision site, as in our case study.19 Side-port incisions have a 1- or 2-plane architectural profile in contrast to the 3-plane profile typical of a main incision.19 Recent advances including the conversion to clear-corneal incisions of diminishing size, techniques used for wound construction, phacoemulsification machine design, and small-incision IOLs, should further reduce the prevalence and complications of wound compromise.20
Seidel Testing
Seidel testing is the most common method to evaluate corneal wound integrity and identify leaks. A drop of topical anesthetic is instilled in the eye and then a fluorescein strip (not fluorescein sodium and benoxinate hydrochloride ophthalmic solution, which may become less sterile since it has a multiuse container) is applied to the superior conjunctiva. The clinician then looks for evidence of fluid egress using the cobalt blue filter. The patient is instructed to blink once. Fluid egress appears as a black stream as the fluorescein dye becomes diluted by aqueous humor escaping the nonintact wound and the appearance of bright green dye surrounds the leak site. The term Seidel positive indicates a leak. An estimate should be made of the rate and volume of fluid exiting the wound.
Gonioscopy
Gonioscopy can be used to evaluate the postsurgical incision, more specifically for identification and management of internal incision wound gape. On gonioscopy, internal wound gape appears as an elongated oval opening resembling a fish mouth. If internal incision wound gape is identified gonioscopically before surgery is complete, the leak can be managed intraoperatively. The surgeon can irrigate along the length of the incision to remove cortical fragments or viscoelastic that may cause internal wound gaping. If unsuccessful, rapidly deepening the anterior chamber with balanced salt solution through the paracentesis incision may be employed. These methods may improve wound stability, reduce risk of postoperative hyphema, lower the incidence of endophthalmitis, and lessen the likelihood of late against-the-rule drift.21
Anterior Segment Optical Coherence Tomography
Instances when Seidel testing was negative despite actual wound gaping have been described.22,23 Anterior segment OCT is useful to evaluate incision architecture. A 2007 United Kingdom study investigated the corneal architecture in the immediate postoperative period following phacoemulsification using anterior segment OCT. This study showed the benefits of identifying architectural features such as epithelial gaping, endothelial gaping, stripping of Descemet membrane, and loss of coaptation. These features were found to be more common at low IOP and could represent a significant risk factor for endophthalmitis.24 Another study published by Behrens and colleagues indicated that a localized detachment of Descemet membrane may be more common than observed with slit-lamp (Figure 7). Corneal gaping, especially if along the entire length of the surgical wound, may lead to inadvertent bacterial access into the anterior chamber.25
Anterior segment OCT imaging was first described by Izatt and colleagues in 1994.26 Unlike posterior segment OCT, anterior segment OCT requires a greater depth of field and higher energy levels as images are commonly distorted by refraction at boundaries where the refractive index changes. Longer infrared wavelengths improve the penetration through tissues that scatter light, such as the sclera and limbus, which allows visualization, for example, of the iridocorneal angle.27,28
Two main scan patterns are used for anterior segment OCT: 512 x 128 cube scan (4-mm width x 4-mm length) and 5-line raster (3-mm length) with adjustable rotation and spacing. A recent software update allows measurement of corneal thickness, visualization of anterior chamber angle structures along with topographic analysis, anterior and posterior elevation maps of the cornea, and reliable pachymetric maps.29,30 The anterior segment cube acquires a series of 128 horizontal scan lines each composed of 512 A-scans. These high-definition scans acquire vertical and horizontal directions composed of 1024 A-scans each. This cube may be used to measure corneal thickness and visualize corneal architecture, creating a 3-D image of the data (Figure 8). The anterior segment 5-line raster scans through 5 parallel lines of equal length to view high-resolution images of the anterior chamber angle and cornea. Each line, fixed at 3-mm in length, is composed of 4096 A-scans.31 Anterior segment cube OCT allows identification of subtle variations in incision architecture at different locations across the width of the OCT image.
Bandage Soft Contact Lens
Upon reviewing the anterior segment OCT images of our patient with the bandage contact lens in place, it was evident that the adherent ocular bandage was protecting the incision. A tighter fitting bandage contact lens is ideal and adheres firmly to any area of epithelial damage and epithelial gaping to help seal the incision, protecting the wound and improving structural integrity. The bandage contact lens is gradually replaced by new cells via re-epithelialization; thus, it behaves as an adjunct to natural wound healing. A bandage contact lens also improves patient comfort.
It is hypothesized that a bandage contact lens improves the structural integrity of the incision site and helps prevent leaking, hypotony, and minor wound leaks. One study revealed a statistically significant lower IOP in nonbandage contact lens patients by an average of 6 mm Hg (mean [SD] 13.4 mm Hg [5.3]; range, 5 - 23 mm Hg) vs patients with a bandage contact lens (mean [SD] 19.4 mm Hg [5.9]; range, 11 - 29 mm Hg) in the immediate postoperative period.32 The authors suggested that the bandage contact lens may prevent microleaks, resulting in a higher IOP.
Aqueous Suppressants
Aqueous suppressants are a great option when IOP is abnormally elevated by decreasing the IOP and allowing the cornea to heal and self-seal.Effective aqueous suppressants are β blockers and carbonic anhydrase inhibitors.
After phacoemulsification ocular hypotony (< 6 mm Hg) occurs most commonly due to wound leakage or excessive intraocular inflammation. However, with the presence of corneal wound leakage and ocular hypotony, aqueous suppressants are not the best option.
Further Management of Wound Leaks
Management of a postoperative wound leak will vary based on severity. The majority of mild leaks are self-sealing. Anterior segment OCT helps the clinician to identify microleaks in an otherwise Seidel negative eye. If wound leakage is moderate with a formed anterior chamber, the use of a bandage contact lens is a good option, as can be the prescription of aqueous suppressants, depending on IOP.33
If the anterior chamber is flat, iris prolapse is apparent, or extremely low IOP exists, the patient needs to be referred to the surgeon. Current standard of care directs the surgeon to use sutures to further manage corneal wound leak. However, several studies have recognized the increased risk of suture-related complications, such as induced astigmatism, corneal opacities, incomplete wound closure, and corneal neovascularization.6,34-38 Other wound closure options include polyethylene glycol-based products, corneal welding, cyanoacrylate, or fibrin (Table 2).39 Traditionally nylon sutures have been used for clear corneal incision wound closure. However, tissue adhesives are gaining popularity as a substitute for sutures in wound closure.40
Cyanoacrylate
Numerous studies have been published on the efficacy of cyanoacrylate as a substitute for sutures, specifically in clear corneal incisions. AEs of cyanoacrylate include a transient foreign-body sensation and diffuse or focal bulbar conjunctival hyperemia.41,42 Shigemitsu and Majima found that fibrin and cyanoacrylate glue had tensile strength similar to sutures when used in cataract surgery.39 Polyethylene glycol-based products, also used in artificial tears and contact lens materials, may also help seal wound leaks. Another agent is ReSure (Ocular Therapeutix, Bedford, MA), an FDA-approved synthetic, polyethylene glycol hydrogel sealant that is 90% water after polymerization. ReSure has been shown to be safe and effective in sealing cataract surgical clear corneal incisions.6,43 ReSure takes about 20 seconds to prepare, and placement is aided by the use of a blue dye that dissipates within hours. This hydrogel will gradually slough off in the tears once the tissue has fully regenerated; there is no need to remove the sealant.44
Rossi and colleagues evaluated the efficacy of corneal welding to close wounds after cataract surgery. The technique involves laser-assisted closure of the corneal wound(s) by a diode laser that welds the stroma.45 Corneal welding takes seconds to achieve good closure without significant astigmatism or inflammation; however very careful application of the light absorbing dyes is required as they are toxic if allowed to enter the anterior chamber.45-47
Conclusion
Optometrists may be called to manage patients during both the preoperative and postoperative phases of cataract surgical care. Those who participate in postoperative care should carefully evaluate for the presence of wound leak or wound gape as a potential complication. The OCT may be employed to evaluate patients suspected of having these leaks or gapes. Proficiency in the interpretation of OCT results and more traditional evaluation methods allows for successful detection of wound leaks or gapes. The timely diagnosis and treatment of postoperative wound leaks allow for the best possible outcomes for cataract surgery patients.
1. Thylefors B, Négrel AD, Pararajasegaram R, Dadzie KY. Global data on blindness. Bull World Health Organ. 1995;73(1):115-121.
2. Flaxman SR, Bourne RRA, Resnikoff S, et al; Vision Loss Expert Group of the Global Burden of Disease Study. Global causes of blindness and distance vision impairment 1990-2020: a systematic review and meta-analysis. Lancet Glob Health. 2017;5(12):e1221-e1224.
3. Congdon N, Vingerling JR, Klein BE, et al; Eye Diseases Prevalence Research Group. Prevalence of cataract and pseudophakia/aphakia among adults in the United States. Arch Ophthalmol. 2004;122(4):487-494.
4. Kurt E, Mayalı H. Early post-operative complications in cataract surgery. In: Zaidi FH, ed. Cataract Surgery. IntechOpen; 2013. https://www.intechopen.com/books/cataract-surgery/post-operative-infections-associated-with-cataract-surgery. Accessed July 15, 2019.
5. Chee SP. Clear corneal incision leakage after phacoemulsification--detection using povidone iodine 5%. Int Ophthalmol. 2005;26(4-5):175-179.
6. Masket S, Hovanesian JA, Levenson J, et al. Hydrogel sealant versus sutures to prevent fluid egress after cataract surgery. J Cataract Refract Surg. 2014;40(12):2057-2066.
7. Kelman CD. Phaco-emulsification and aspiration: a new technique of cataract removal. A preliminary report. Am J Ophthalmol. 1967;64(1):23-35.
8. Powe NR, Schein OD, Gieser SC, et al. Synthesis of the literature on visual acuity and complications following cataract extraction with intraocular lens implantation. Cataract Patient Outcome Research Team [published correction appears in Arch Ophthalmol. 1994;112(7):889]. Arch Ophthalmol. 1994;112(2):239-252.
9. Stein JD, Grossman DS, Mundy KM, Sugar A, Sloan FA. Severe adverse events after cataract surgery among medicare beneficiaries. Ophthalmology. 2011;118(9):1716-1723.
10. Greenberg PB, Tseng VL, Wu WC, et al. Prevalence and predictors of ocular complications associated with cataract surgery in United States veterans. Ophthalmology. 2011;118(3):507-514.
11. Mangan MS, Atalay E, Anci C, Tuncer I, Bilqec MD. Comparison of different types of complications in the phacoemulsification surgery learning curve according to number of operations performed. Turk J Ophthalmol. 2016;46(1):7-10.
12. Bell CM, Hatch WV, Cernat G, Urbach DR. Surgeon volumes and selected patient outcomes in cataract surgery: a population-based analysis. Ophthalmology. 2007;114(3):405-410.
13. Wallin T, Parker J, Jin Y, Kefalopoulos G, Olson RJ. Cohort study of 27 cases of endophthalmitis at a single institution. J Cataract Refract Surg. 2005;31(4):735-741.
14. Zaidi FH, Corbett MC, Burton BJ, Bloom PA. Raising the benchmark for the 21st century--the 1000 cataract operations audit and survey: outcomes, consultant-supervised training and sourcing NHS choice. Br J Ophthalmol. 2007;91(6):731-736.
15. Nichamin LD, Chang DF, Johnson SH, et al; American Society of Cataract and Refractive Surgery Cataract Clinical Committee. ASCRS white paper: what is the association between clear corneal cataract incisions and postoperative endophthalmitis? J Cataract Refract Surg. 2006;32(9):1556-1559.
16. Packer M, Chang DF, Dewey SH, et al; ASCRS Cataract Clinical Committee. Prevention, diagnosis, and management of acute postoperative bacterial endophthalmitis. J Cataract Refract Surg. 2011;37(9):1699-1714.
17. Montan PG, Koranyi G, Setterquist HE, Stridh A, Philipson BT, Wiklund K. Endophthalmitis after cataract surgery: risk factors relating to technique and events of the operation and patient history: a retrospective case-control study. Ophthalmology. 1998;105(12):2171-2177.
18. Taban M, Rao B, Reznik J, Zhang J, Chen Z, McDonnell PJ. Dynamic morphology of sutureless cataract wounds—effect of incision angle and location. Surv Ophthalmol. 2004;49(suppl 2):S62-S72.
19. Chee SP, Ti SE, Lim L, Chan AS, Jap A. Anterior segment optical coherence tomography evaluation of the integrity of clear corneal incisions: a comparison between 2.2-mm and 2.65-mm main incisions. Am J Ophthalmol. 2010;149(5):768-776.e1.
20. Koch DD, Nacke RE, Wang L, Novak KD. Issues in wound management. In: Steinert R, ed. Cataract Surgery. 3rd ed. New York: Elsevier; 2009:581-588.
21. Gimbel HV, Sun R, DeBroff GM. Recognition and management of internal wound gape. J Cataract Refract Surg. 1995;21(2):121-124.
22. May WN, Castro-Combs J, Quinto GG, Kashiwabuchi R, Gower EW, Behrens A. Standardized Seidel test to evaluate different sutureless cataract incision configurations. J Cataract Refract Surg. 2010;36(6):1011-1017.
23. Kashiwabuchi FK, Khan YA, Rodrigues MW Jr, Wang J, McDonnell PJ, Daoud YJ. Seidel and India ink tests assessment of different clear cornea side-port incision configurations. Graefes Arch Clin Exp Ophthalmol. 2013;251(8):1961-1965.
24. Calladine D, Packard R. Clear corneal incision architecture in the immediate postoperative period evaluated using optical coherence tomography. J Cataract Refract Surg. 2007;33(8):1429-1435.
25. Behrens WJ, Stark KA, Pratzer, McDonnell PJ. Dynamics of small-incision clear cornea wounds after phacoemulsification surgery using optical coherence tomography in the early postoperative period. J Refractive Surgery. 2008;24(1):46-49.
26. Izatt JA, Hee MR, Swanson EA, et al. Micrometer-scale resolution imaging of the anterior eye in vivo with optical coherence tomography. Arch Ophthalmol. 1994;112(12):1584-1589.
27. Hurmeric V, Yoo SH, Mutlu FM. Optical coherence tomography in cornea and refractive surgery. Expert Rev Ophthalmol. 2012;7(3):241-250.
28. Schuman JS, Puliafito CA, Fujimoto JG, Duker JS. Optical Coherence Tomography of Ocular Diseases. 3rd ed. Thorofare, NJ: Slack Inc; 2013.
29. Salim S. The role of anterior segment optical coherence tomography in glaucoma. J Ophthalmol. 2012;2012:476801.
30. Kharousi NA, Wali UK, Azeem S. Current applications of optical coherence tomography in ophthalmology. In: Kawasaki M, ed. Optical Coherence Tomography. IntechOpen; 2013. https://www.intechopen.com/books/optical-coherence-tomography. Accessed July 31, 2019.
31. Rodrigues EB, Johanson M, Penha FM. Anterior segment tomography with the cirrus optical coherence tomography. J Ophthalmol. 2012;2012:806989.
32. Calladine D, Ward M, Packard R. Adherent ocular bandage for clear corneal incisions used in cataract surgery. J Cataract Refract Surg. 2010;36(11):1839-1848.
33. Haldar K, Saraff R. Closure technique for leaking wound resulting from thermal injury during phacoemulsification. J Cataract Refract Surg. 2014;40(9):1412-1414.
34. Zoghby JT, Cohen KL. Phacoemulsification-related corneal incision contracture. https://www.aao.org/eyenet/article/phacoemulsification-related-corneal-incision-contr. Published December 2012. Accessed June 16, 2019.
35. Bhatia SS. Ocular surface sealants and adhesives. Ocul Surf. 2006;4(3):146-154.
36. May WN, Castro-Combs J, Kashiwabuchi RT, et al. Bacterial-sized particle inflow through sutured clear corneal incisions in a laboratory human model. J Cataract Refract Surg. 2011;37(6):1140-1146.
37. Meskin SW, Ritterband DC, Shapiro DE, et al. Liquid bandage (2-octyl cyanoacrylate) as a temporary wound barrier in clear corneal cataract surgery. Ophthalmology. 2005;112(11):2015-2021.
38. Heaven CJ, Davison CR, Cockcroft PM. Bacterial contamination of nylon corneal sutures. Eye (Lond). 1995;9(pt 1):116-118.
39. Shigemitsu T, Majima Y. The utilization of a biological adhesive for wound treatment: comparison of suture, self-sealing sutureless and cyanoacrylate closure in the tensile strength test. Int Ophthalmol. 1996-1997;20:323-328.
40. Uy HS, Kenyon KR. Surgical outcomes after application of a liquid adhesive ocular bandage to clear corneal incisions during cataract surgery. J Cataract Refract Surg. 2013;39(11):1668-1674.
41. Meskin SW, Ritterband DC, Shapiro DE, et al. Liquid bandage (2-octyl cyanoacrylate) as a temporary wound barrier in clear corneal cataract surgery. Ophthalmology. 2005;112(11):2015-2021.
42. Tong AY, Gupta PK, Kim T. Wound closure and tissue adhesives in clear corneal incision cataract surgery. Curr Opin Ophthalmol. 2018;29(1):14-18.
43. US Food and Drug Administration. Summary of Safety and Effectiveness Data. Ophthalmic sealant: ReSure Sealant. https://www.accessdata.fda.gov/cdrh_docs/pdf13/P130004b.pdf. Published September 13, 2013. Accessed July 9, 2019.
44. About ReSure sealant. https://www.resuresealant.com/overview. Accessed July 31, 2019.
45. Menabuoni L, Pini R, Rossi F, Lenzetti I, Yoo SH, Parel JM. Laser-assisted corneal welding in cataract surgery: retrospective study. J Cataract Refract Surg. 2007;33(9):1608-1612.
46. Rasier R, Ozeren M, Artunay O, et al. Corneal tissue welding with infrared laser irradiation after clear corneal incision. Cornea. 2010;29(9):985-990.
47. Rossi F, Matteini P, Ratto F, Menabuoni L, Lenzetti I, Pini R. Laser tissue welding in ophthalmic surgery. J Biophotonics. 2008;1(4):331-342.
48. Taban M, Behrens A, Newcomb RL, et al. Acute endophthalmitis following cataract surgery: a systematic review of the literature. Arch Ophthalmol. 2005;123(5):613-620.
49. Taylor DM, Atlas BF, Romanchuk KG, Stern AL. Pseudophakic bullous keratopathy. Ophthalmology. 1983;90(1):19-24.
50. Lobo CL, Faria PM, Soares MA, Bernardes RC, Cunha-Vaz JG. Macular alterations after small-incision cataract surgery. J Cataract Refract Surg. 2004;30(4):752-760.
51. Flach AJ. The incidence, pathogenesis and treatment of cystoid macular edema following cataract surgery. Trans Am Ophthalmol Soc. 1998;96:557-634.
52. Wright PL, Wilkinson CP, Balyeat HD, Popham J, Reinke M. Angiographic cystoid macular edema after posterior chamber lens implantation. Arch Ophthalmol. 1988;106(6):740-744.
53. Kim SJ, Belair ML, Bressler NM, et al. A method of reporting macular edema after cataract surgery using optical coherence tomography. Retina. 2008;28(6):870-876.
54. Alio JL, Ruiz-Moreno JM, Shabayek MH, Lugo FL, Abd El Rahman AM. The risk of retinal detachment in high myopia after small incision coaxial phacoemulsification. Am J Ophthalmol. 2007;144(1):93-98.
55. Bhagwandien AC, Cheng YY, Wolfs RC, van Meurs JC, Luyten GP. Relationship between retinal detachment and biometry in 4262 cataractous eyes. Ophthalmology. 2006;113(4):643-649.
56. Boberg-Ans G, Henning V, Villumsen J, la Cour M. Longterm incidence of rhegmatogenous retinal detachment and survival in a defined population undergoing standardized phacoemulsification surgery. Acta Ophthalmol Scand. 2006;84(5):613-618.
57. Jakobsson G, Montan P, Zetterberg M, Stenevi U, Behndig A, Lundström M. Capsule complication during cataract surgery: retinal detachment after cataract surgery with capsule complication: Swedish Capsule Rupture Study Group report 4. J Cataract Refract Surg. 2009;35(10):1699-1705.
58. Neuhann IM, Neuhann TF, Heimann H, Schmickler S, Gerl RH, Foerster MH. Retinal detachment after phacoemulsification in high myopia: analysis of 2356 cases. J Cataract Refract Surg. 2008;34(10):1644-1657.
59. Russell M, Gaskin B, Russell D, Polkinghorne PJ. Pseudophakic retinal detachment after phacoemulsification cataract surgery: ten-year retrospective review. J Cataract Refract Surg. 2006;32(3):442-445.
60. Apple DJ, Solomon KD, Tetz MR, et al. Posterior capsule opacification. Surv Ophthalmol. 1992;37(2):73-116.
61. Wu S, Tong N, Pan L, et al. Retrospective analyses of potential risk factors for posterior capsule opacification after cataract surgery. J Ophthalmol. 2018;2018:9089285.
62. Clark A, Morlet N, Ng JQ, Preen DB, Semmens JB. Whole population trends in complications of cataract surgery over 22 years in Western Australia. Ophthalmology. 2011;118(6):1055-1061.
63. Adhikari S, Shrestha UD. Pediatric cataract surgery with hydrophilic acrylic intraocular lens implantation in Nepalese Children. Clin Ophthalmol. 2017;12:7-11.
64. Lee BJ, Smith SD, Jeng BH. Suture-related corneal infections after clear corneal cataract surgery. J Cataract Refract Surg. 2009;35(5):939-942.
65. May WN, Castro-Combs J, Kashiwabuchi RT, et al. Sutured clear corneal incision: wound apposition and permeability to bacterial-sized particles. Cornea. 2013;32(3):319-325.
66. Hillier RJ, Ajit RR, Kelly SP. Suture-related complications after cataract surgery: a patient safety issue. J Cataract Refract Surg. 2009;35(11):2035-2036.
67. Hovanesian JA, Karageozian VH. Watertight cataract incision closure using fibrin tissue adhesive. J Cataract Refract Surg. 2007;33(8):1461-1463.
1. Thylefors B, Négrel AD, Pararajasegaram R, Dadzie KY. Global data on blindness. Bull World Health Organ. 1995;73(1):115-121.
2. Flaxman SR, Bourne RRA, Resnikoff S, et al; Vision Loss Expert Group of the Global Burden of Disease Study. Global causes of blindness and distance vision impairment 1990-2020: a systematic review and meta-analysis. Lancet Glob Health. 2017;5(12):e1221-e1224.
3. Congdon N, Vingerling JR, Klein BE, et al; Eye Diseases Prevalence Research Group. Prevalence of cataract and pseudophakia/aphakia among adults in the United States. Arch Ophthalmol. 2004;122(4):487-494.
4. Kurt E, Mayalı H. Early post-operative complications in cataract surgery. In: Zaidi FH, ed. Cataract Surgery. IntechOpen; 2013. https://www.intechopen.com/books/cataract-surgery/post-operative-infections-associated-with-cataract-surgery. Accessed July 15, 2019.
5. Chee SP. Clear corneal incision leakage after phacoemulsification--detection using povidone iodine 5%. Int Ophthalmol. 2005;26(4-5):175-179.
6. Masket S, Hovanesian JA, Levenson J, et al. Hydrogel sealant versus sutures to prevent fluid egress after cataract surgery. J Cataract Refract Surg. 2014;40(12):2057-2066.
7. Kelman CD. Phaco-emulsification and aspiration: a new technique of cataract removal. A preliminary report. Am J Ophthalmol. 1967;64(1):23-35.
8. Powe NR, Schein OD, Gieser SC, et al. Synthesis of the literature on visual acuity and complications following cataract extraction with intraocular lens implantation. Cataract Patient Outcome Research Team [published correction appears in Arch Ophthalmol. 1994;112(7):889]. Arch Ophthalmol. 1994;112(2):239-252.
9. Stein JD, Grossman DS, Mundy KM, Sugar A, Sloan FA. Severe adverse events after cataract surgery among medicare beneficiaries. Ophthalmology. 2011;118(9):1716-1723.
10. Greenberg PB, Tseng VL, Wu WC, et al. Prevalence and predictors of ocular complications associated with cataract surgery in United States veterans. Ophthalmology. 2011;118(3):507-514.
11. Mangan MS, Atalay E, Anci C, Tuncer I, Bilqec MD. Comparison of different types of complications in the phacoemulsification surgery learning curve according to number of operations performed. Turk J Ophthalmol. 2016;46(1):7-10.
12. Bell CM, Hatch WV, Cernat G, Urbach DR. Surgeon volumes and selected patient outcomes in cataract surgery: a population-based analysis. Ophthalmology. 2007;114(3):405-410.
13. Wallin T, Parker J, Jin Y, Kefalopoulos G, Olson RJ. Cohort study of 27 cases of endophthalmitis at a single institution. J Cataract Refract Surg. 2005;31(4):735-741.
14. Zaidi FH, Corbett MC, Burton BJ, Bloom PA. Raising the benchmark for the 21st century--the 1000 cataract operations audit and survey: outcomes, consultant-supervised training and sourcing NHS choice. Br J Ophthalmol. 2007;91(6):731-736.
15. Nichamin LD, Chang DF, Johnson SH, et al; American Society of Cataract and Refractive Surgery Cataract Clinical Committee. ASCRS white paper: what is the association between clear corneal cataract incisions and postoperative endophthalmitis? J Cataract Refract Surg. 2006;32(9):1556-1559.
16. Packer M, Chang DF, Dewey SH, et al; ASCRS Cataract Clinical Committee. Prevention, diagnosis, and management of acute postoperative bacterial endophthalmitis. J Cataract Refract Surg. 2011;37(9):1699-1714.
17. Montan PG, Koranyi G, Setterquist HE, Stridh A, Philipson BT, Wiklund K. Endophthalmitis after cataract surgery: risk factors relating to technique and events of the operation and patient history: a retrospective case-control study. Ophthalmology. 1998;105(12):2171-2177.
18. Taban M, Rao B, Reznik J, Zhang J, Chen Z, McDonnell PJ. Dynamic morphology of sutureless cataract wounds—effect of incision angle and location. Surv Ophthalmol. 2004;49(suppl 2):S62-S72.
19. Chee SP, Ti SE, Lim L, Chan AS, Jap A. Anterior segment optical coherence tomography evaluation of the integrity of clear corneal incisions: a comparison between 2.2-mm and 2.65-mm main incisions. Am J Ophthalmol. 2010;149(5):768-776.e1.
20. Koch DD, Nacke RE, Wang L, Novak KD. Issues in wound management. In: Steinert R, ed. Cataract Surgery. 3rd ed. New York: Elsevier; 2009:581-588.
21. Gimbel HV, Sun R, DeBroff GM. Recognition and management of internal wound gape. J Cataract Refract Surg. 1995;21(2):121-124.
22. May WN, Castro-Combs J, Quinto GG, Kashiwabuchi R, Gower EW, Behrens A. Standardized Seidel test to evaluate different sutureless cataract incision configurations. J Cataract Refract Surg. 2010;36(6):1011-1017.
23. Kashiwabuchi FK, Khan YA, Rodrigues MW Jr, Wang J, McDonnell PJ, Daoud YJ. Seidel and India ink tests assessment of different clear cornea side-port incision configurations. Graefes Arch Clin Exp Ophthalmol. 2013;251(8):1961-1965.
24. Calladine D, Packard R. Clear corneal incision architecture in the immediate postoperative period evaluated using optical coherence tomography. J Cataract Refract Surg. 2007;33(8):1429-1435.
25. Behrens WJ, Stark KA, Pratzer, McDonnell PJ. Dynamics of small-incision clear cornea wounds after phacoemulsification surgery using optical coherence tomography in the early postoperative period. J Refractive Surgery. 2008;24(1):46-49.
26. Izatt JA, Hee MR, Swanson EA, et al. Micrometer-scale resolution imaging of the anterior eye in vivo with optical coherence tomography. Arch Ophthalmol. 1994;112(12):1584-1589.
27. Hurmeric V, Yoo SH, Mutlu FM. Optical coherence tomography in cornea and refractive surgery. Expert Rev Ophthalmol. 2012;7(3):241-250.
28. Schuman JS, Puliafito CA, Fujimoto JG, Duker JS. Optical Coherence Tomography of Ocular Diseases. 3rd ed. Thorofare, NJ: Slack Inc; 2013.
29. Salim S. The role of anterior segment optical coherence tomography in glaucoma. J Ophthalmol. 2012;2012:476801.
30. Kharousi NA, Wali UK, Azeem S. Current applications of optical coherence tomography in ophthalmology. In: Kawasaki M, ed. Optical Coherence Tomography. IntechOpen; 2013. https://www.intechopen.com/books/optical-coherence-tomography. Accessed July 31, 2019.
31. Rodrigues EB, Johanson M, Penha FM. Anterior segment tomography with the cirrus optical coherence tomography. J Ophthalmol. 2012;2012:806989.
32. Calladine D, Ward M, Packard R. Adherent ocular bandage for clear corneal incisions used in cataract surgery. J Cataract Refract Surg. 2010;36(11):1839-1848.
33. Haldar K, Saraff R. Closure technique for leaking wound resulting from thermal injury during phacoemulsification. J Cataract Refract Surg. 2014;40(9):1412-1414.
34. Zoghby JT, Cohen KL. Phacoemulsification-related corneal incision contracture. https://www.aao.org/eyenet/article/phacoemulsification-related-corneal-incision-contr. Published December 2012. Accessed June 16, 2019.
35. Bhatia SS. Ocular surface sealants and adhesives. Ocul Surf. 2006;4(3):146-154.
36. May WN, Castro-Combs J, Kashiwabuchi RT, et al. Bacterial-sized particle inflow through sutured clear corneal incisions in a laboratory human model. J Cataract Refract Surg. 2011;37(6):1140-1146.
37. Meskin SW, Ritterband DC, Shapiro DE, et al. Liquid bandage (2-octyl cyanoacrylate) as a temporary wound barrier in clear corneal cataract surgery. Ophthalmology. 2005;112(11):2015-2021.
38. Heaven CJ, Davison CR, Cockcroft PM. Bacterial contamination of nylon corneal sutures. Eye (Lond). 1995;9(pt 1):116-118.
39. Shigemitsu T, Majima Y. The utilization of a biological adhesive for wound treatment: comparison of suture, self-sealing sutureless and cyanoacrylate closure in the tensile strength test. Int Ophthalmol. 1996-1997;20:323-328.
40. Uy HS, Kenyon KR. Surgical outcomes after application of a liquid adhesive ocular bandage to clear corneal incisions during cataract surgery. J Cataract Refract Surg. 2013;39(11):1668-1674.
41. Meskin SW, Ritterband DC, Shapiro DE, et al. Liquid bandage (2-octyl cyanoacrylate) as a temporary wound barrier in clear corneal cataract surgery. Ophthalmology. 2005;112(11):2015-2021.
42. Tong AY, Gupta PK, Kim T. Wound closure and tissue adhesives in clear corneal incision cataract surgery. Curr Opin Ophthalmol. 2018;29(1):14-18.
43. US Food and Drug Administration. Summary of Safety and Effectiveness Data. Ophthalmic sealant: ReSure Sealant. https://www.accessdata.fda.gov/cdrh_docs/pdf13/P130004b.pdf. Published September 13, 2013. Accessed July 9, 2019.
44. About ReSure sealant. https://www.resuresealant.com/overview. Accessed July 31, 2019.
45. Menabuoni L, Pini R, Rossi F, Lenzetti I, Yoo SH, Parel JM. Laser-assisted corneal welding in cataract surgery: retrospective study. J Cataract Refract Surg. 2007;33(9):1608-1612.
46. Rasier R, Ozeren M, Artunay O, et al. Corneal tissue welding with infrared laser irradiation after clear corneal incision. Cornea. 2010;29(9):985-990.
47. Rossi F, Matteini P, Ratto F, Menabuoni L, Lenzetti I, Pini R. Laser tissue welding in ophthalmic surgery. J Biophotonics. 2008;1(4):331-342.
48. Taban M, Behrens A, Newcomb RL, et al. Acute endophthalmitis following cataract surgery: a systematic review of the literature. Arch Ophthalmol. 2005;123(5):613-620.
49. Taylor DM, Atlas BF, Romanchuk KG, Stern AL. Pseudophakic bullous keratopathy. Ophthalmology. 1983;90(1):19-24.
50. Lobo CL, Faria PM, Soares MA, Bernardes RC, Cunha-Vaz JG. Macular alterations after small-incision cataract surgery. J Cataract Refract Surg. 2004;30(4):752-760.
51. Flach AJ. The incidence, pathogenesis and treatment of cystoid macular edema following cataract surgery. Trans Am Ophthalmol Soc. 1998;96:557-634.
52. Wright PL, Wilkinson CP, Balyeat HD, Popham J, Reinke M. Angiographic cystoid macular edema after posterior chamber lens implantation. Arch Ophthalmol. 1988;106(6):740-744.
53. Kim SJ, Belair ML, Bressler NM, et al. A method of reporting macular edema after cataract surgery using optical coherence tomography. Retina. 2008;28(6):870-876.
54. Alio JL, Ruiz-Moreno JM, Shabayek MH, Lugo FL, Abd El Rahman AM. The risk of retinal detachment in high myopia after small incision coaxial phacoemulsification. Am J Ophthalmol. 2007;144(1):93-98.
55. Bhagwandien AC, Cheng YY, Wolfs RC, van Meurs JC, Luyten GP. Relationship between retinal detachment and biometry in 4262 cataractous eyes. Ophthalmology. 2006;113(4):643-649.
56. Boberg-Ans G, Henning V, Villumsen J, la Cour M. Longterm incidence of rhegmatogenous retinal detachment and survival in a defined population undergoing standardized phacoemulsification surgery. Acta Ophthalmol Scand. 2006;84(5):613-618.
57. Jakobsson G, Montan P, Zetterberg M, Stenevi U, Behndig A, Lundström M. Capsule complication during cataract surgery: retinal detachment after cataract surgery with capsule complication: Swedish Capsule Rupture Study Group report 4. J Cataract Refract Surg. 2009;35(10):1699-1705.
58. Neuhann IM, Neuhann TF, Heimann H, Schmickler S, Gerl RH, Foerster MH. Retinal detachment after phacoemulsification in high myopia: analysis of 2356 cases. J Cataract Refract Surg. 2008;34(10):1644-1657.
59. Russell M, Gaskin B, Russell D, Polkinghorne PJ. Pseudophakic retinal detachment after phacoemulsification cataract surgery: ten-year retrospective review. J Cataract Refract Surg. 2006;32(3):442-445.
60. Apple DJ, Solomon KD, Tetz MR, et al. Posterior capsule opacification. Surv Ophthalmol. 1992;37(2):73-116.
61. Wu S, Tong N, Pan L, et al. Retrospective analyses of potential risk factors for posterior capsule opacification after cataract surgery. J Ophthalmol. 2018;2018:9089285.
62. Clark A, Morlet N, Ng JQ, Preen DB, Semmens JB. Whole population trends in complications of cataract surgery over 22 years in Western Australia. Ophthalmology. 2011;118(6):1055-1061.
63. Adhikari S, Shrestha UD. Pediatric cataract surgery with hydrophilic acrylic intraocular lens implantation in Nepalese Children. Clin Ophthalmol. 2017;12:7-11.
64. Lee BJ, Smith SD, Jeng BH. Suture-related corneal infections after clear corneal cataract surgery. J Cataract Refract Surg. 2009;35(5):939-942.
65. May WN, Castro-Combs J, Kashiwabuchi RT, et al. Sutured clear corneal incision: wound apposition and permeability to bacterial-sized particles. Cornea. 2013;32(3):319-325.
66. Hillier RJ, Ajit RR, Kelly SP. Suture-related complications after cataract surgery: a patient safety issue. J Cataract Refract Surg. 2009;35(11):2035-2036.
67. Hovanesian JA, Karageozian VH. Watertight cataract incision closure using fibrin tissue adhesive. J Cataract Refract Surg. 2007;33(8):1461-1463.
Acute hearing loss, tinnitus, and fullness in the left ear • Weber test lateralized to the right ear • Positive Rinne test and normal tympanometry • Dx?
THE CASE
A healthy 48-year-old man presented to our otolaryngology clinic with a 2-hour history of hearing loss, tinnitus, and fullness in the left ear. He denied any vertigo, nausea, vomiting, otalgia, or otorrhea. He had noticed signs of a possible upper respiratory infection, including a sore throat and headache, the day before his symptoms started. His medical history was unremarkable. He denied any history of otologic surgery, trauma, or vision problems, and he was not taking any medications.
The patient was afebrile on physical examination with a heart rate of 48 beats/min and blood pressure of 117/68 mm Hg. A Weber test performed using a 512-Hz tuning fork lateralized to the right ear. A Rinne test showed air conduction was louder than bone conduction in the affected left ear—a normal finding. Tympanometry and otoscopic examination showed the bilateral tympanic membranes were normal.
THE DIAGNOSIS
Pure tone audiometry showed severe sensorineural hearing loss in the left ear and a poor speech discrimination score. The Weber test confirmed the hearing loss was sensorineural and not conductive, ruling out a middle ear effusion. Additionally, the normal tympanogram made conductive hearing loss from a middle ear effusion or tympanic membrane perforation unlikely. The positive Rinne test was consistent with a diagnosis of idiopathic sudden sensorineural hearing loss (SSNHL).
DISCUSSION
SSNHL is defined by hearing loss of more than 30 dB in at least 3 consecutive frequencies with acute onset of less than 72 hours.1,2 The most common symptoms include acute hearing loss, tinnitus, and fullness in the affected ear.1 The majority of cases of SSNHL are unilateral. The typical age of onset is in the fourth and fifth decades, occurring with equal distribution in both sexes.
Etiology.
Diagnosis. The initial evaluation should include an otoscopic examination, tuning fork tests, and pure tone audiometry.1-3 Weber and Rinne tests are essential when evaluating patients for unilateral hearing loss and determining the type of loss (ie, sensorineural vs conductive). The Weber test (ideally using a 512-Hz tuning fork) can detect either conductive or sensorineural hearing loss. In a normal Weber test, the patient should hear the vibration of the tuning fork equally in both ears. The tuning fork will be heard in both ears in conductive hearing loss but will only be heard in the unaffected hear if sensorineural hearing loss is present. So, for instance, if a patient has a perforation in the right tympanic membrane causing conductive hearing loss in the right hear, the tuning fork would be heard in both ears. If the patient has sensorineural hearing loss in the right ear, the tuning fork would only be heard in the left ear.
The Rinne test compares the perception of sound waves transmitted by air conduction vs bone conduction and serves as a rapid screen for conductive hearing loss.
Continue to: Magnetic resonance imaging...
Magnetic resonance imaging (MRI) of the brain and brainstem with gadolinium contrast can reveal vascular events (thrombotic or hemorrhagic), demyelinating disorders, or retrocochlear lesions such as vestibular schwannoma and is indicated in all cases of suspected SSNHL.4,5
Treatment and management. The current standard of care for treatment of idiopathic SSNHL is systemic steroids.1,2 Although the gold standard currently is oral prednisolone or methylprednisolone (1 mg/kg/d for 10 to 14 days with a taper,1,2 the evidence for this regimen stems from a single placebo-controlled trial (N = 67) that demonstrated greater improvement in the steroid group compared with the placebo group (61% vs 32%).6 A Cochrane review and other systematic analyses have not demonstrated clear efficacy of corticosteroid treatment for the management of idiopathic SSNHL.7,8
Because of the potential systemic adverse effects associated with oral corticosteroids, intratympanic (IT) corticosteroids have been advocated as an alternative treatment option. A prospective, randomized, noninferiority trial comparing the efficacy of oral vs IT corticosteroids for idiopathic SSNHL found IT corticosteroids to be noninferior to systemic treatment.9 IT treatment also has been advocated as a rescue therapy for patients who do not respond to systemic treatment.10
A combination of oral and IT corticosteroids was investigated in a retrospective study analyzing multiple treatment modalities.10 Researchers first compared 122 patients receiving one of 3 treatments: (1) IT corticosteroids, (2) oral corticosteroids, and (3) combination treatment (IT + oral corticosteroids). There was no difference in hearing recovery among any of the treatments. Fifty-eight patients who were refractory to initial treatment were then included in a second analysis in which they were divided into those who received additional IT corticosteroids (salvage treatment) vs no treatment (control). There was no difference in hearing recovery between the 2 groups. The authors concluded that IT corticosteroids were as effective as oral treatment and that salvage IT treatment did not add any benefit.10
The American Academy of Otolaryngology-Head and Neck Surgery (AAO-HNS) recently published guidelines on the diagnosis and management of SSNHL.11 The guidelines state that IT steroids should be considered in patients who cannot tolerate oral steroids, such as patients with diabetes. It is important to note, however, that the high cost of IT treatment (~$2000 for dexamethasone or methylprednisolone vs < $10 for oral prednisolone) is an issue that needs to be considered as health care costs continue to rise.
Continue to: Antivirals
Antivirals. Because an underlying viral etiology has been speculated as a potential cause of idiopathic SSNHL, antiviral agents such as valacyclovir or famciclovir also are potential treatment agents.12 Antiviral medications have minimal adverse effects and are relatively inexpensive, but the benefits have not yet been proven in randomized controlled trials,and they currently are not endorsed by the AAO-HNS in their guidelines for the management of SSNHL.11
Spontaneous recovery occurs in up to 40% of patients with idiopathic SSNHL. As many as 65% of those who experience recovery do so within 2 weeks of the onset of symptoms, regardless of treatment.1,2 Treatment beyond 2 weeks after onset of symptoms is unlikely to be of any benefit, although some otolaryngologists will treat for up to 6 weeks after the onset of hearing loss.
A substantial number of patients with SSNHL may not recover. Management of these patients begins with referral to an appropriate specialist to initiate counseling and lifestyle changes. Depending on the degree of hearing loss, audiologic rehabilitation may include use of a traditional or bone-anchored hearing aid or a frequency-modulation system.1,2,11 Tinnitus retraining therapy might be of benefit for patients with persistent tinnitus.11
Our patient. After a discussion of his treatment options, our patient decided on a combination of oral prednisolone (60 mg once daily for 9 days followed by a taper for 5 days) and intratympanic dexamethasone injections (1 mL [10 mg/mL] once weekly for 3 weeks).
The rationale for this approach was the minimal adverse effects associated with short-term (ie, days to 1–2 weeks) use of high-dose (ie, > 30 mg/d) corticosteroids. Although steroid therapy has been associated with adverse effects such as aseptic necrosis of the hip, these complications usually arise after longer periods (ie, months to years) of high-dose steroid therapy with a mean cumulative dose much higher than what was used in our patient.13
Continue to: Our patient...
Our patient noticed slight improvement within 48 hours of the initial onset of symptoms that continued for the next several weeks until full recovery was attained. An MRI performed 5 days after the onset of symptoms was negative for retrocochlear pathology.
THE TAKEAWAY
SSNHL is a medical emergency that requires prompt recognition and diagnosis. The steps in evaluating sudden hearing loss include: (1) appropriate history and physical examination (eg, otoscopic examination, tuning fork tests), (2) urgent audiometry to confirm hearing loss, (3) immediate referral to an otolaryngologist for further testing (eg, tympanometry, blood tests, MRI), and (4) initiation of treatment.
If a specific etiology is identified (eg, vestibular schwannoma), the patient should be referred to a specialist for appropriate treatment. If there is no identifiable cause (idiopathic SSNHL), the patient should be treated with oral and/or intratympanic steroids. Patients who do not recover following treatment should be offered audiologic rehabilitation.
CORRESPONDENCE
Sergio Huerta, MD, UT Southwestern Medical Center, 4500 S Lancaster Road #112L, Dallas, TX 75216; [email protected]
1. Schreiber BE, Agrup C, Haskard DO, et al. Sudden sensorineural hearing loss. Lancet. 2010;375:1203-1211.
2. Rauch SD. Clinical practice. Idiopathic sudden sensorineural hearing loss. N Engl J Med. 2008;359:833-840.
3. Paul BC, Roland JT Jr. An abnormal audiogram. JAMA. 2015;313:85-86.
4. Aarnisalo AA, Suoranta H, Ylikoski J. Magnetic resonance imaging findings in the auditory pathway of patients with sudden deafness. Otol Neurotol. 2004;25:245-249.
5. Cadoni G, Cianfoni A, Agostino S, et al. Magnetic resonance imaging findings in sudden sensorineural hearing loss. J Otolaryngol. 2006;35:310-316.
6. Wilson WR, Byl FM, Laird N. The efficacy of steroids in the treatment of idiopathic sudden hearing loss. A double-blind clinical study. Arch Otolaryngol. 1980;106:772-776.
7. Wei BPC, Stathopoulos D, O’Leary S. Steroids for idiopathic sudden sensorineural hearing loss. Cochrane Database Syst Rev. 2013. doi:10.1002/14651858.CD003998.pub3.
8. Conlin AE, Parnes LS. Treatment of sudden sensorineural hearing loss: II. a meta-analysis. Arch Otolaryngol Head Neck Surg. 2007;133:582-586.
9. Rauch SD, Halpin CF, Antonelli PJ, et al. Oral vs intratympanic corticosteroid therapy for idiopathic sudden sensorineural hearing loss: a randomized trial. JAMA. 2011;305:2071-2079.
10. Lee KH, Ryu SH, Lee HM, et al. Is intratympanic dexamethasone injection effective for the treatment of idiopathic sudden sensorineural hearing loss? J Audiol Otol. 2015;19:154-158.
11. Stachler RJ, Chandrasekhar SS, Archer SM, et al. Clinical practice guideline: sudden hearing loss. Otolaryngol Head Neck Surg. 2012;146(3 suppl):S1-S35.
12. Westerlaken BO, Stokroos RJ, Dhooge IJ, et al. Treatment of idiopathic sudden sensorineural hearing loss with antiviral therapy: a prospective, randomized, double-blind clinical trial. Ann Otol Rhinol Laryngol. 2003;112:993-1000.
13. Nowak DA, Yeung J. Steroid-induced osteonecrosis in dermatology: a review [published online March 30, 2015]. J Cutan Med Surg. 2015;19:358-360.
THE CASE
A healthy 48-year-old man presented to our otolaryngology clinic with a 2-hour history of hearing loss, tinnitus, and fullness in the left ear. He denied any vertigo, nausea, vomiting, otalgia, or otorrhea. He had noticed signs of a possible upper respiratory infection, including a sore throat and headache, the day before his symptoms started. His medical history was unremarkable. He denied any history of otologic surgery, trauma, or vision problems, and he was not taking any medications.
The patient was afebrile on physical examination with a heart rate of 48 beats/min and blood pressure of 117/68 mm Hg. A Weber test performed using a 512-Hz tuning fork lateralized to the right ear. A Rinne test showed air conduction was louder than bone conduction in the affected left ear—a normal finding. Tympanometry and otoscopic examination showed the bilateral tympanic membranes were normal.
THE DIAGNOSIS
Pure tone audiometry showed severe sensorineural hearing loss in the left ear and a poor speech discrimination score. The Weber test confirmed the hearing loss was sensorineural and not conductive, ruling out a middle ear effusion. Additionally, the normal tympanogram made conductive hearing loss from a middle ear effusion or tympanic membrane perforation unlikely. The positive Rinne test was consistent with a diagnosis of idiopathic sudden sensorineural hearing loss (SSNHL).
DISCUSSION
SSNHL is defined by hearing loss of more than 30 dB in at least 3 consecutive frequencies with acute onset of less than 72 hours.1,2 The most common symptoms include acute hearing loss, tinnitus, and fullness in the affected ear.1 The majority of cases of SSNHL are unilateral. The typical age of onset is in the fourth and fifth decades, occurring with equal distribution in both sexes.
Etiology.
Diagnosis. The initial evaluation should include an otoscopic examination, tuning fork tests, and pure tone audiometry.1-3 Weber and Rinne tests are essential when evaluating patients for unilateral hearing loss and determining the type of loss (ie, sensorineural vs conductive). The Weber test (ideally using a 512-Hz tuning fork) can detect either conductive or sensorineural hearing loss. In a normal Weber test, the patient should hear the vibration of the tuning fork equally in both ears. The tuning fork will be heard in both ears in conductive hearing loss but will only be heard in the unaffected hear if sensorineural hearing loss is present. So, for instance, if a patient has a perforation in the right tympanic membrane causing conductive hearing loss in the right hear, the tuning fork would be heard in both ears. If the patient has sensorineural hearing loss in the right ear, the tuning fork would only be heard in the left ear.
The Rinne test compares the perception of sound waves transmitted by air conduction vs bone conduction and serves as a rapid screen for conductive hearing loss.
Continue to: Magnetic resonance imaging...
Magnetic resonance imaging (MRI) of the brain and brainstem with gadolinium contrast can reveal vascular events (thrombotic or hemorrhagic), demyelinating disorders, or retrocochlear lesions such as vestibular schwannoma and is indicated in all cases of suspected SSNHL.4,5
Treatment and management. The current standard of care for treatment of idiopathic SSNHL is systemic steroids.1,2 Although the gold standard currently is oral prednisolone or methylprednisolone (1 mg/kg/d for 10 to 14 days with a taper,1,2 the evidence for this regimen stems from a single placebo-controlled trial (N = 67) that demonstrated greater improvement in the steroid group compared with the placebo group (61% vs 32%).6 A Cochrane review and other systematic analyses have not demonstrated clear efficacy of corticosteroid treatment for the management of idiopathic SSNHL.7,8
Because of the potential systemic adverse effects associated with oral corticosteroids, intratympanic (IT) corticosteroids have been advocated as an alternative treatment option. A prospective, randomized, noninferiority trial comparing the efficacy of oral vs IT corticosteroids for idiopathic SSNHL found IT corticosteroids to be noninferior to systemic treatment.9 IT treatment also has been advocated as a rescue therapy for patients who do not respond to systemic treatment.10
A combination of oral and IT corticosteroids was investigated in a retrospective study analyzing multiple treatment modalities.10 Researchers first compared 122 patients receiving one of 3 treatments: (1) IT corticosteroids, (2) oral corticosteroids, and (3) combination treatment (IT + oral corticosteroids). There was no difference in hearing recovery among any of the treatments. Fifty-eight patients who were refractory to initial treatment were then included in a second analysis in which they were divided into those who received additional IT corticosteroids (salvage treatment) vs no treatment (control). There was no difference in hearing recovery between the 2 groups. The authors concluded that IT corticosteroids were as effective as oral treatment and that salvage IT treatment did not add any benefit.10
The American Academy of Otolaryngology-Head and Neck Surgery (AAO-HNS) recently published guidelines on the diagnosis and management of SSNHL.11 The guidelines state that IT steroids should be considered in patients who cannot tolerate oral steroids, such as patients with diabetes. It is important to note, however, that the high cost of IT treatment (~$2000 for dexamethasone or methylprednisolone vs < $10 for oral prednisolone) is an issue that needs to be considered as health care costs continue to rise.
Continue to: Antivirals
Antivirals. Because an underlying viral etiology has been speculated as a potential cause of idiopathic SSNHL, antiviral agents such as valacyclovir or famciclovir also are potential treatment agents.12 Antiviral medications have minimal adverse effects and are relatively inexpensive, but the benefits have not yet been proven in randomized controlled trials,and they currently are not endorsed by the AAO-HNS in their guidelines for the management of SSNHL.11
Spontaneous recovery occurs in up to 40% of patients with idiopathic SSNHL. As many as 65% of those who experience recovery do so within 2 weeks of the onset of symptoms, regardless of treatment.1,2 Treatment beyond 2 weeks after onset of symptoms is unlikely to be of any benefit, although some otolaryngologists will treat for up to 6 weeks after the onset of hearing loss.
A substantial number of patients with SSNHL may not recover. Management of these patients begins with referral to an appropriate specialist to initiate counseling and lifestyle changes. Depending on the degree of hearing loss, audiologic rehabilitation may include use of a traditional or bone-anchored hearing aid or a frequency-modulation system.1,2,11 Tinnitus retraining therapy might be of benefit for patients with persistent tinnitus.11
Our patient. After a discussion of his treatment options, our patient decided on a combination of oral prednisolone (60 mg once daily for 9 days followed by a taper for 5 days) and intratympanic dexamethasone injections (1 mL [10 mg/mL] once weekly for 3 weeks).
The rationale for this approach was the minimal adverse effects associated with short-term (ie, days to 1–2 weeks) use of high-dose (ie, > 30 mg/d) corticosteroids. Although steroid therapy has been associated with adverse effects such as aseptic necrosis of the hip, these complications usually arise after longer periods (ie, months to years) of high-dose steroid therapy with a mean cumulative dose much higher than what was used in our patient.13
Continue to: Our patient...
Our patient noticed slight improvement within 48 hours of the initial onset of symptoms that continued for the next several weeks until full recovery was attained. An MRI performed 5 days after the onset of symptoms was negative for retrocochlear pathology.
THE TAKEAWAY
SSNHL is a medical emergency that requires prompt recognition and diagnosis. The steps in evaluating sudden hearing loss include: (1) appropriate history and physical examination (eg, otoscopic examination, tuning fork tests), (2) urgent audiometry to confirm hearing loss, (3) immediate referral to an otolaryngologist for further testing (eg, tympanometry, blood tests, MRI), and (4) initiation of treatment.
If a specific etiology is identified (eg, vestibular schwannoma), the patient should be referred to a specialist for appropriate treatment. If there is no identifiable cause (idiopathic SSNHL), the patient should be treated with oral and/or intratympanic steroids. Patients who do not recover following treatment should be offered audiologic rehabilitation.
CORRESPONDENCE
Sergio Huerta, MD, UT Southwestern Medical Center, 4500 S Lancaster Road #112L, Dallas, TX 75216; [email protected]
THE CASE
A healthy 48-year-old man presented to our otolaryngology clinic with a 2-hour history of hearing loss, tinnitus, and fullness in the left ear. He denied any vertigo, nausea, vomiting, otalgia, or otorrhea. He had noticed signs of a possible upper respiratory infection, including a sore throat and headache, the day before his symptoms started. His medical history was unremarkable. He denied any history of otologic surgery, trauma, or vision problems, and he was not taking any medications.
The patient was afebrile on physical examination with a heart rate of 48 beats/min and blood pressure of 117/68 mm Hg. A Weber test performed using a 512-Hz tuning fork lateralized to the right ear. A Rinne test showed air conduction was louder than bone conduction in the affected left ear—a normal finding. Tympanometry and otoscopic examination showed the bilateral tympanic membranes were normal.
THE DIAGNOSIS
Pure tone audiometry showed severe sensorineural hearing loss in the left ear and a poor speech discrimination score. The Weber test confirmed the hearing loss was sensorineural and not conductive, ruling out a middle ear effusion. Additionally, the normal tympanogram made conductive hearing loss from a middle ear effusion or tympanic membrane perforation unlikely. The positive Rinne test was consistent with a diagnosis of idiopathic sudden sensorineural hearing loss (SSNHL).
DISCUSSION
SSNHL is defined by hearing loss of more than 30 dB in at least 3 consecutive frequencies with acute onset of less than 72 hours.1,2 The most common symptoms include acute hearing loss, tinnitus, and fullness in the affected ear.1 The majority of cases of SSNHL are unilateral. The typical age of onset is in the fourth and fifth decades, occurring with equal distribution in both sexes.
Etiology.
Diagnosis. The initial evaluation should include an otoscopic examination, tuning fork tests, and pure tone audiometry.1-3 Weber and Rinne tests are essential when evaluating patients for unilateral hearing loss and determining the type of loss (ie, sensorineural vs conductive). The Weber test (ideally using a 512-Hz tuning fork) can detect either conductive or sensorineural hearing loss. In a normal Weber test, the patient should hear the vibration of the tuning fork equally in both ears. The tuning fork will be heard in both ears in conductive hearing loss but will only be heard in the unaffected hear if sensorineural hearing loss is present. So, for instance, if a patient has a perforation in the right tympanic membrane causing conductive hearing loss in the right hear, the tuning fork would be heard in both ears. If the patient has sensorineural hearing loss in the right ear, the tuning fork would only be heard in the left ear.
The Rinne test compares the perception of sound waves transmitted by air conduction vs bone conduction and serves as a rapid screen for conductive hearing loss.
Continue to: Magnetic resonance imaging...
Magnetic resonance imaging (MRI) of the brain and brainstem with gadolinium contrast can reveal vascular events (thrombotic or hemorrhagic), demyelinating disorders, or retrocochlear lesions such as vestibular schwannoma and is indicated in all cases of suspected SSNHL.4,5
Treatment and management. The current standard of care for treatment of idiopathic SSNHL is systemic steroids.1,2 Although the gold standard currently is oral prednisolone or methylprednisolone (1 mg/kg/d for 10 to 14 days with a taper,1,2 the evidence for this regimen stems from a single placebo-controlled trial (N = 67) that demonstrated greater improvement in the steroid group compared with the placebo group (61% vs 32%).6 A Cochrane review and other systematic analyses have not demonstrated clear efficacy of corticosteroid treatment for the management of idiopathic SSNHL.7,8
Because of the potential systemic adverse effects associated with oral corticosteroids, intratympanic (IT) corticosteroids have been advocated as an alternative treatment option. A prospective, randomized, noninferiority trial comparing the efficacy of oral vs IT corticosteroids for idiopathic SSNHL found IT corticosteroids to be noninferior to systemic treatment.9 IT treatment also has been advocated as a rescue therapy for patients who do not respond to systemic treatment.10
A combination of oral and IT corticosteroids was investigated in a retrospective study analyzing multiple treatment modalities.10 Researchers first compared 122 patients receiving one of 3 treatments: (1) IT corticosteroids, (2) oral corticosteroids, and (3) combination treatment (IT + oral corticosteroids). There was no difference in hearing recovery among any of the treatments. Fifty-eight patients who were refractory to initial treatment were then included in a second analysis in which they were divided into those who received additional IT corticosteroids (salvage treatment) vs no treatment (control). There was no difference in hearing recovery between the 2 groups. The authors concluded that IT corticosteroids were as effective as oral treatment and that salvage IT treatment did not add any benefit.10
The American Academy of Otolaryngology-Head and Neck Surgery (AAO-HNS) recently published guidelines on the diagnosis and management of SSNHL.11 The guidelines state that IT steroids should be considered in patients who cannot tolerate oral steroids, such as patients with diabetes. It is important to note, however, that the high cost of IT treatment (~$2000 for dexamethasone or methylprednisolone vs < $10 for oral prednisolone) is an issue that needs to be considered as health care costs continue to rise.
Continue to: Antivirals
Antivirals. Because an underlying viral etiology has been speculated as a potential cause of idiopathic SSNHL, antiviral agents such as valacyclovir or famciclovir also are potential treatment agents.12 Antiviral medications have minimal adverse effects and are relatively inexpensive, but the benefits have not yet been proven in randomized controlled trials,and they currently are not endorsed by the AAO-HNS in their guidelines for the management of SSNHL.11
Spontaneous recovery occurs in up to 40% of patients with idiopathic SSNHL. As many as 65% of those who experience recovery do so within 2 weeks of the onset of symptoms, regardless of treatment.1,2 Treatment beyond 2 weeks after onset of symptoms is unlikely to be of any benefit, although some otolaryngologists will treat for up to 6 weeks after the onset of hearing loss.
A substantial number of patients with SSNHL may not recover. Management of these patients begins with referral to an appropriate specialist to initiate counseling and lifestyle changes. Depending on the degree of hearing loss, audiologic rehabilitation may include use of a traditional or bone-anchored hearing aid or a frequency-modulation system.1,2,11 Tinnitus retraining therapy might be of benefit for patients with persistent tinnitus.11
Our patient. After a discussion of his treatment options, our patient decided on a combination of oral prednisolone (60 mg once daily for 9 days followed by a taper for 5 days) and intratympanic dexamethasone injections (1 mL [10 mg/mL] once weekly for 3 weeks).
The rationale for this approach was the minimal adverse effects associated with short-term (ie, days to 1–2 weeks) use of high-dose (ie, > 30 mg/d) corticosteroids. Although steroid therapy has been associated with adverse effects such as aseptic necrosis of the hip, these complications usually arise after longer periods (ie, months to years) of high-dose steroid therapy with a mean cumulative dose much higher than what was used in our patient.13
Continue to: Our patient...
Our patient noticed slight improvement within 48 hours of the initial onset of symptoms that continued for the next several weeks until full recovery was attained. An MRI performed 5 days after the onset of symptoms was negative for retrocochlear pathology.
THE TAKEAWAY
SSNHL is a medical emergency that requires prompt recognition and diagnosis. The steps in evaluating sudden hearing loss include: (1) appropriate history and physical examination (eg, otoscopic examination, tuning fork tests), (2) urgent audiometry to confirm hearing loss, (3) immediate referral to an otolaryngologist for further testing (eg, tympanometry, blood tests, MRI), and (4) initiation of treatment.
If a specific etiology is identified (eg, vestibular schwannoma), the patient should be referred to a specialist for appropriate treatment. If there is no identifiable cause (idiopathic SSNHL), the patient should be treated with oral and/or intratympanic steroids. Patients who do not recover following treatment should be offered audiologic rehabilitation.
CORRESPONDENCE
Sergio Huerta, MD, UT Southwestern Medical Center, 4500 S Lancaster Road #112L, Dallas, TX 75216; [email protected]
1. Schreiber BE, Agrup C, Haskard DO, et al. Sudden sensorineural hearing loss. Lancet. 2010;375:1203-1211.
2. Rauch SD. Clinical practice. Idiopathic sudden sensorineural hearing loss. N Engl J Med. 2008;359:833-840.
3. Paul BC, Roland JT Jr. An abnormal audiogram. JAMA. 2015;313:85-86.
4. Aarnisalo AA, Suoranta H, Ylikoski J. Magnetic resonance imaging findings in the auditory pathway of patients with sudden deafness. Otol Neurotol. 2004;25:245-249.
5. Cadoni G, Cianfoni A, Agostino S, et al. Magnetic resonance imaging findings in sudden sensorineural hearing loss. J Otolaryngol. 2006;35:310-316.
6. Wilson WR, Byl FM, Laird N. The efficacy of steroids in the treatment of idiopathic sudden hearing loss. A double-blind clinical study. Arch Otolaryngol. 1980;106:772-776.
7. Wei BPC, Stathopoulos D, O’Leary S. Steroids for idiopathic sudden sensorineural hearing loss. Cochrane Database Syst Rev. 2013. doi:10.1002/14651858.CD003998.pub3.
8. Conlin AE, Parnes LS. Treatment of sudden sensorineural hearing loss: II. a meta-analysis. Arch Otolaryngol Head Neck Surg. 2007;133:582-586.
9. Rauch SD, Halpin CF, Antonelli PJ, et al. Oral vs intratympanic corticosteroid therapy for idiopathic sudden sensorineural hearing loss: a randomized trial. JAMA. 2011;305:2071-2079.
10. Lee KH, Ryu SH, Lee HM, et al. Is intratympanic dexamethasone injection effective for the treatment of idiopathic sudden sensorineural hearing loss? J Audiol Otol. 2015;19:154-158.
11. Stachler RJ, Chandrasekhar SS, Archer SM, et al. Clinical practice guideline: sudden hearing loss. Otolaryngol Head Neck Surg. 2012;146(3 suppl):S1-S35.
12. Westerlaken BO, Stokroos RJ, Dhooge IJ, et al. Treatment of idiopathic sudden sensorineural hearing loss with antiviral therapy: a prospective, randomized, double-blind clinical trial. Ann Otol Rhinol Laryngol. 2003;112:993-1000.
13. Nowak DA, Yeung J. Steroid-induced osteonecrosis in dermatology: a review [published online March 30, 2015]. J Cutan Med Surg. 2015;19:358-360.
1. Schreiber BE, Agrup C, Haskard DO, et al. Sudden sensorineural hearing loss. Lancet. 2010;375:1203-1211.
2. Rauch SD. Clinical practice. Idiopathic sudden sensorineural hearing loss. N Engl J Med. 2008;359:833-840.
3. Paul BC, Roland JT Jr. An abnormal audiogram. JAMA. 2015;313:85-86.
4. Aarnisalo AA, Suoranta H, Ylikoski J. Magnetic resonance imaging findings in the auditory pathway of patients with sudden deafness. Otol Neurotol. 2004;25:245-249.
5. Cadoni G, Cianfoni A, Agostino S, et al. Magnetic resonance imaging findings in sudden sensorineural hearing loss. J Otolaryngol. 2006;35:310-316.
6. Wilson WR, Byl FM, Laird N. The efficacy of steroids in the treatment of idiopathic sudden hearing loss. A double-blind clinical study. Arch Otolaryngol. 1980;106:772-776.
7. Wei BPC, Stathopoulos D, O’Leary S. Steroids for idiopathic sudden sensorineural hearing loss. Cochrane Database Syst Rev. 2013. doi:10.1002/14651858.CD003998.pub3.
8. Conlin AE, Parnes LS. Treatment of sudden sensorineural hearing loss: II. a meta-analysis. Arch Otolaryngol Head Neck Surg. 2007;133:582-586.
9. Rauch SD, Halpin CF, Antonelli PJ, et al. Oral vs intratympanic corticosteroid therapy for idiopathic sudden sensorineural hearing loss: a randomized trial. JAMA. 2011;305:2071-2079.
10. Lee KH, Ryu SH, Lee HM, et al. Is intratympanic dexamethasone injection effective for the treatment of idiopathic sudden sensorineural hearing loss? J Audiol Otol. 2015;19:154-158.
11. Stachler RJ, Chandrasekhar SS, Archer SM, et al. Clinical practice guideline: sudden hearing loss. Otolaryngol Head Neck Surg. 2012;146(3 suppl):S1-S35.
12. Westerlaken BO, Stokroos RJ, Dhooge IJ, et al. Treatment of idiopathic sudden sensorineural hearing loss with antiviral therapy: a prospective, randomized, double-blind clinical trial. Ann Otol Rhinol Laryngol. 2003;112:993-1000.
13. Nowak DA, Yeung J. Steroid-induced osteonecrosis in dermatology: a review [published online March 30, 2015]. J Cutan Med Surg. 2015;19:358-360.

<i>Mycobacterium abscessus</i> Infection Following Home Dermabrasion
Case Report
A 32-year-old woman presented to the dermatology clinic with a tender lump overlying the right maxilla of 6 weeks’ duration. The lesion developed acutely 1 to 2 months after the patient began using an at-home microdermabrasion device, which she routinely cleaned with tap water. The physical examination was notable for a 1.5-cm, soft, superficially indurated plaque on the right cheek without associated lymphadenopathy (Figure).
A punch biopsy revealed underlying necrotic fat. Computed tomography of the neck showed 20-mm skin thickening overlying the right zygomatic arch, with minimal adjacent subcutaneous soft tissue stranding and reactive lymph nodes. Further histologic examination of the biopsy specimen revealed inflamed granulation tissue with granulomatous inflammation.
Acid-fast bacterial culture was positive. Subsequent speciation revealed the causal agent to be multidrug-resistant Mycobacterium abscessus. The patient was initially treated with trimethoprim-sulfamethoxazole, which was switched to a combination of doxycycline and levofloxacin a few days later after initial culture returned. The following week, after the specific microorganism was confirmed with specific sensitivity, treatment was changed to intravenous (IV) tigecycline and amikacin. This regimen was continued for 2 more months through a peripherally inserted central catheter, then discontinued after complete resolution of the skin lesion.
Comment
Mycobacterial Infection
Nontuberculous mycobacteria were not identified as human pathogens until the 1950s. They are known to cause skin disease, lymphadenitis, skeletal infection, pulmonary disease, and disseminated infection, with pulmonary disease being the most common clinical form overall.1Mycobacterium abscessus is a member of a more specific group known as rapidly growing nontuberculous mycobacteria, which also includes Mycobacterium fortuitum and Mycobacterium chelonae.2 Commonly found in water, soil, and dust, M abscessus causes skin and soft tissue infection after skin injury by inoculation, minor trauma, or surgery.2-4 An increased rate of infections recently has been attributed to an increase in cosmetic procedures such as tattooing, liposuction, mesotherapy, pedicures, and body piercing. Mycobacterial infections transmitted through acupuncture also have been documented.5,6
Causes of Skin and Soft Tissue Infections
Skin and soft tissue infections due to rapidly growing mycobacteria often are associated with systemic comorbidities that cause immunosuppression and with immunosuppressive medications.7 Our patient did not have a preexisting comorbidity and did not take any long-term medication. When multiple lesions have been reported, patients were more likely to either have a systemic comorbidity or be taking immunosuppressive medication compared to patients with a single lesion. A history of penetrating trauma or an invasive surgical procedure has been reported more often in patients with a single lesion.7
Our patient had a solitary lesion on the face; improper sterile technique while using an at-home microdermabrasion device was thought to be the cause of infection. Although generally considered a minimally abrasive treatment modality, microdermabrasion caused enough trauma to create a nidus of infection in our patient.
Presentation
Cutaneous infection from rapidly growing mycobacteria can manifest as a nonhealing ulceration, subcutaneous abscess, draining sinus, or subcutaneous fluctuant or firm nodules. Erythema may be found in association with ulcers or chronic drainage from a surgical wound.2,7
Histopathologic appearance varies, depending on the evolution of the disease and host immunologic status. Tuberculoid, palisading, and sarcoidlike granulomas; a diffuse infiltrate of histiocytic foamy cells; acute and chronic panniculitis; nonspecific chronic inflammation; cutaneous abscess; suppurative granuloma; and necrotizing folliculitis all can be seen.8 Immunosuppressed patients are less likely to form granulomas.6 Diagnosis often is delayed because acid-fast bacterial culture is not typically performed on skin biopsy specimens or surgical wound infections.7 Fortunately, a high index of suspicion in our patient’s case allowed for prompt diagnosis and expeditious management.
Management
Mycobacterium abscessus tends to be resistant to conventional antituberculous medications; overall, it is considered a highly drug-resistant pathogen that is difficult to treat.9,10 Treatment usually requires 3 to 6 months of therapy, with oral clarithromycin considered the first-line agent for localized infection.5 Because cases of clarithromycin resistance have been reported in patients with M chelonae infection, caution is warranted when deciding between monotherapy or combination therapy.7 Multidrug resistance often necessitates prolonged IV therapy. Amikacin is the mostly commonly used IV agent for M abscessus infection. Adverse effects of treatment are common, often leading to a change in or discontinuation of therapy.11
Our patient was initially given trimethoprim-sulfamethoxazole before being switched to doxycycline and levofloxacin prior to final results of susceptibility testing. Ultimately, due to the multidrug-resistant nature of M abscessus, clarithromycin was not a viable option. Therefore, the patient was administered tigecycline and amikacin through a peripherally inserted central catheter until symptoms fully resolved.
Surgery can be an important adjunctive measure for certain patients, especially those with a single lesion.7 Our patient did well with medical treatment alone.
Conclusion
Given the difficulty of treating skin and soft tissue infections caused by M abscessus and related mycobacteria, it is worth noting that these infections are increasingly caused by procedures generally considered to be minimally invasive. Microdermabrasion—performed at home in an unsterile environment and not by a trained medical professional—was the causal procedure in this case. An important consideration is whether clinicians can be comfortable with the use of these treatments at home or whether they should be advising patients against at-home treatments that have potentially serious complications.
- Lee WJ, Kang SM, Sung H, et al. Non-tuberculous mycobacterial infections of the skin: a retrospective study of 29 cases. J Dermatol. 2010;37:965-972.
- Fitzgerald DA, Smith AG, Lees A, et al. Cutaneous infection with Mycobacterium abscessus. Br J Dermatol. 1995;132:800-804.
- Moore M, Frerichs JB. An unusual acid-fast infection of the knee with subcutaneous, abscess-like lesions of the gluteal region; report of a case with a study of the organism, Mycobacterium abscessus, n. sp. J Invest Dermatol. 1953;20:133-169.
- Inman PM, Beck A, Brown AE, et al. Outbreak of injection abscesses due to Mycobacterium abscessus. Arch Dermatol. 1969;100:141-147.
- Ryu HJ, Kim WJ, Oh CH, et al. Iatrogenic Mycobacterium abscessus infection associated with acupuncture: clinical manifestations and its treatment. Int J Dermatol. 2005;44:846-850.
- Wentworth AB, Drage LA, Wengenack NL, et al. Increased incidence of cutaneous nontuberculous mycobacterial infection, 1980 to 2009: a population-based study. Mayo Clin Proc. 2013;88:38-45.
- Uslan DZ, Kowalski TJ, Wengenack NL, et al. Skin and soft tissue infections due to rapidly growing mycobacteria: comparison of clinical features, treatment, and susceptibility. Arch Dermatol. 2006;142:1287-1292.
- Bartralot R, Pujol RM, García-Patos V, et al. Cutaneous infections due to nontuberculous mycobacteria: histopathological review of 28 cases. comparative study between lesions observed in immunosuppressed patients and normal hosts. J Cutan Pathol. 2000;27:124-129.
- Morris-Jones R, Fletcher C, Morris-Jones S, et al. Mycobacterium abscessus: a cutaneous infection in a patient on renal replacement therapy. Clin Exp Dermatol. 2001;26:415-418.
- Jeong SH, Kim SY, Huh HJ, et al. Mycobacteriological characteristics and treatment outcomes in extrapulmonary Mycobacterium abscessus complex infections. Int J Infect Dis. 2017;60:49-56.
- Novosad SA, Beekmann SE, Polgreen PM, et al. Treatment of Mycobacterium abscessus infection. Emerg Infect Dis. 2016;22:511-514.
Case Report
A 32-year-old woman presented to the dermatology clinic with a tender lump overlying the right maxilla of 6 weeks’ duration. The lesion developed acutely 1 to 2 months after the patient began using an at-home microdermabrasion device, which she routinely cleaned with tap water. The physical examination was notable for a 1.5-cm, soft, superficially indurated plaque on the right cheek without associated lymphadenopathy (Figure).
A punch biopsy revealed underlying necrotic fat. Computed tomography of the neck showed 20-mm skin thickening overlying the right zygomatic arch, with minimal adjacent subcutaneous soft tissue stranding and reactive lymph nodes. Further histologic examination of the biopsy specimen revealed inflamed granulation tissue with granulomatous inflammation.
Acid-fast bacterial culture was positive. Subsequent speciation revealed the causal agent to be multidrug-resistant Mycobacterium abscessus. The patient was initially treated with trimethoprim-sulfamethoxazole, which was switched to a combination of doxycycline and levofloxacin a few days later after initial culture returned. The following week, after the specific microorganism was confirmed with specific sensitivity, treatment was changed to intravenous (IV) tigecycline and amikacin. This regimen was continued for 2 more months through a peripherally inserted central catheter, then discontinued after complete resolution of the skin lesion.
Comment
Mycobacterial Infection
Nontuberculous mycobacteria were not identified as human pathogens until the 1950s. They are known to cause skin disease, lymphadenitis, skeletal infection, pulmonary disease, and disseminated infection, with pulmonary disease being the most common clinical form overall.1Mycobacterium abscessus is a member of a more specific group known as rapidly growing nontuberculous mycobacteria, which also includes Mycobacterium fortuitum and Mycobacterium chelonae.2 Commonly found in water, soil, and dust, M abscessus causes skin and soft tissue infection after skin injury by inoculation, minor trauma, or surgery.2-4 An increased rate of infections recently has been attributed to an increase in cosmetic procedures such as tattooing, liposuction, mesotherapy, pedicures, and body piercing. Mycobacterial infections transmitted through acupuncture also have been documented.5,6
Causes of Skin and Soft Tissue Infections
Skin and soft tissue infections due to rapidly growing mycobacteria often are associated with systemic comorbidities that cause immunosuppression and with immunosuppressive medications.7 Our patient did not have a preexisting comorbidity and did not take any long-term medication. When multiple lesions have been reported, patients were more likely to either have a systemic comorbidity or be taking immunosuppressive medication compared to patients with a single lesion. A history of penetrating trauma or an invasive surgical procedure has been reported more often in patients with a single lesion.7
Our patient had a solitary lesion on the face; improper sterile technique while using an at-home microdermabrasion device was thought to be the cause of infection. Although generally considered a minimally abrasive treatment modality, microdermabrasion caused enough trauma to create a nidus of infection in our patient.
Presentation
Cutaneous infection from rapidly growing mycobacteria can manifest as a nonhealing ulceration, subcutaneous abscess, draining sinus, or subcutaneous fluctuant or firm nodules. Erythema may be found in association with ulcers or chronic drainage from a surgical wound.2,7
Histopathologic appearance varies, depending on the evolution of the disease and host immunologic status. Tuberculoid, palisading, and sarcoidlike granulomas; a diffuse infiltrate of histiocytic foamy cells; acute and chronic panniculitis; nonspecific chronic inflammation; cutaneous abscess; suppurative granuloma; and necrotizing folliculitis all can be seen.8 Immunosuppressed patients are less likely to form granulomas.6 Diagnosis often is delayed because acid-fast bacterial culture is not typically performed on skin biopsy specimens or surgical wound infections.7 Fortunately, a high index of suspicion in our patient’s case allowed for prompt diagnosis and expeditious management.
Management
Mycobacterium abscessus tends to be resistant to conventional antituberculous medications; overall, it is considered a highly drug-resistant pathogen that is difficult to treat.9,10 Treatment usually requires 3 to 6 months of therapy, with oral clarithromycin considered the first-line agent for localized infection.5 Because cases of clarithromycin resistance have been reported in patients with M chelonae infection, caution is warranted when deciding between monotherapy or combination therapy.7 Multidrug resistance often necessitates prolonged IV therapy. Amikacin is the mostly commonly used IV agent for M abscessus infection. Adverse effects of treatment are common, often leading to a change in or discontinuation of therapy.11
Our patient was initially given trimethoprim-sulfamethoxazole before being switched to doxycycline and levofloxacin prior to final results of susceptibility testing. Ultimately, due to the multidrug-resistant nature of M abscessus, clarithromycin was not a viable option. Therefore, the patient was administered tigecycline and amikacin through a peripherally inserted central catheter until symptoms fully resolved.
Surgery can be an important adjunctive measure for certain patients, especially those with a single lesion.7 Our patient did well with medical treatment alone.
Conclusion
Given the difficulty of treating skin and soft tissue infections caused by M abscessus and related mycobacteria, it is worth noting that these infections are increasingly caused by procedures generally considered to be minimally invasive. Microdermabrasion—performed at home in an unsterile environment and not by a trained medical professional—was the causal procedure in this case. An important consideration is whether clinicians can be comfortable with the use of these treatments at home or whether they should be advising patients against at-home treatments that have potentially serious complications.
Case Report
A 32-year-old woman presented to the dermatology clinic with a tender lump overlying the right maxilla of 6 weeks’ duration. The lesion developed acutely 1 to 2 months after the patient began using an at-home microdermabrasion device, which she routinely cleaned with tap water. The physical examination was notable for a 1.5-cm, soft, superficially indurated plaque on the right cheek without associated lymphadenopathy (Figure).
A punch biopsy revealed underlying necrotic fat. Computed tomography of the neck showed 20-mm skin thickening overlying the right zygomatic arch, with minimal adjacent subcutaneous soft tissue stranding and reactive lymph nodes. Further histologic examination of the biopsy specimen revealed inflamed granulation tissue with granulomatous inflammation.
Acid-fast bacterial culture was positive. Subsequent speciation revealed the causal agent to be multidrug-resistant Mycobacterium abscessus. The patient was initially treated with trimethoprim-sulfamethoxazole, which was switched to a combination of doxycycline and levofloxacin a few days later after initial culture returned. The following week, after the specific microorganism was confirmed with specific sensitivity, treatment was changed to intravenous (IV) tigecycline and amikacin. This regimen was continued for 2 more months through a peripherally inserted central catheter, then discontinued after complete resolution of the skin lesion.
Comment
Mycobacterial Infection
Nontuberculous mycobacteria were not identified as human pathogens until the 1950s. They are known to cause skin disease, lymphadenitis, skeletal infection, pulmonary disease, and disseminated infection, with pulmonary disease being the most common clinical form overall.1Mycobacterium abscessus is a member of a more specific group known as rapidly growing nontuberculous mycobacteria, which also includes Mycobacterium fortuitum and Mycobacterium chelonae.2 Commonly found in water, soil, and dust, M abscessus causes skin and soft tissue infection after skin injury by inoculation, minor trauma, or surgery.2-4 An increased rate of infections recently has been attributed to an increase in cosmetic procedures such as tattooing, liposuction, mesotherapy, pedicures, and body piercing. Mycobacterial infections transmitted through acupuncture also have been documented.5,6
Causes of Skin and Soft Tissue Infections
Skin and soft tissue infections due to rapidly growing mycobacteria often are associated with systemic comorbidities that cause immunosuppression and with immunosuppressive medications.7 Our patient did not have a preexisting comorbidity and did not take any long-term medication. When multiple lesions have been reported, patients were more likely to either have a systemic comorbidity or be taking immunosuppressive medication compared to patients with a single lesion. A history of penetrating trauma or an invasive surgical procedure has been reported more often in patients with a single lesion.7
Our patient had a solitary lesion on the face; improper sterile technique while using an at-home microdermabrasion device was thought to be the cause of infection. Although generally considered a minimally abrasive treatment modality, microdermabrasion caused enough trauma to create a nidus of infection in our patient.
Presentation
Cutaneous infection from rapidly growing mycobacteria can manifest as a nonhealing ulceration, subcutaneous abscess, draining sinus, or subcutaneous fluctuant or firm nodules. Erythema may be found in association with ulcers or chronic drainage from a surgical wound.2,7
Histopathologic appearance varies, depending on the evolution of the disease and host immunologic status. Tuberculoid, palisading, and sarcoidlike granulomas; a diffuse infiltrate of histiocytic foamy cells; acute and chronic panniculitis; nonspecific chronic inflammation; cutaneous abscess; suppurative granuloma; and necrotizing folliculitis all can be seen.8 Immunosuppressed patients are less likely to form granulomas.6 Diagnosis often is delayed because acid-fast bacterial culture is not typically performed on skin biopsy specimens or surgical wound infections.7 Fortunately, a high index of suspicion in our patient’s case allowed for prompt diagnosis and expeditious management.
Management
Mycobacterium abscessus tends to be resistant to conventional antituberculous medications; overall, it is considered a highly drug-resistant pathogen that is difficult to treat.9,10 Treatment usually requires 3 to 6 months of therapy, with oral clarithromycin considered the first-line agent for localized infection.5 Because cases of clarithromycin resistance have been reported in patients with M chelonae infection, caution is warranted when deciding between monotherapy or combination therapy.7 Multidrug resistance often necessitates prolonged IV therapy. Amikacin is the mostly commonly used IV agent for M abscessus infection. Adverse effects of treatment are common, often leading to a change in or discontinuation of therapy.11
Our patient was initially given trimethoprim-sulfamethoxazole before being switched to doxycycline and levofloxacin prior to final results of susceptibility testing. Ultimately, due to the multidrug-resistant nature of M abscessus, clarithromycin was not a viable option. Therefore, the patient was administered tigecycline and amikacin through a peripherally inserted central catheter until symptoms fully resolved.
Surgery can be an important adjunctive measure for certain patients, especially those with a single lesion.7 Our patient did well with medical treatment alone.
Conclusion
Given the difficulty of treating skin and soft tissue infections caused by M abscessus and related mycobacteria, it is worth noting that these infections are increasingly caused by procedures generally considered to be minimally invasive. Microdermabrasion—performed at home in an unsterile environment and not by a trained medical professional—was the causal procedure in this case. An important consideration is whether clinicians can be comfortable with the use of these treatments at home or whether they should be advising patients against at-home treatments that have potentially serious complications.
- Lee WJ, Kang SM, Sung H, et al. Non-tuberculous mycobacterial infections of the skin: a retrospective study of 29 cases. J Dermatol. 2010;37:965-972.
- Fitzgerald DA, Smith AG, Lees A, et al. Cutaneous infection with Mycobacterium abscessus. Br J Dermatol. 1995;132:800-804.
- Moore M, Frerichs JB. An unusual acid-fast infection of the knee with subcutaneous, abscess-like lesions of the gluteal region; report of a case with a study of the organism, Mycobacterium abscessus, n. sp. J Invest Dermatol. 1953;20:133-169.
- Inman PM, Beck A, Brown AE, et al. Outbreak of injection abscesses due to Mycobacterium abscessus. Arch Dermatol. 1969;100:141-147.
- Ryu HJ, Kim WJ, Oh CH, et al. Iatrogenic Mycobacterium abscessus infection associated with acupuncture: clinical manifestations and its treatment. Int J Dermatol. 2005;44:846-850.
- Wentworth AB, Drage LA, Wengenack NL, et al. Increased incidence of cutaneous nontuberculous mycobacterial infection, 1980 to 2009: a population-based study. Mayo Clin Proc. 2013;88:38-45.
- Uslan DZ, Kowalski TJ, Wengenack NL, et al. Skin and soft tissue infections due to rapidly growing mycobacteria: comparison of clinical features, treatment, and susceptibility. Arch Dermatol. 2006;142:1287-1292.
- Bartralot R, Pujol RM, García-Patos V, et al. Cutaneous infections due to nontuberculous mycobacteria: histopathological review of 28 cases. comparative study between lesions observed in immunosuppressed patients and normal hosts. J Cutan Pathol. 2000;27:124-129.
- Morris-Jones R, Fletcher C, Morris-Jones S, et al. Mycobacterium abscessus: a cutaneous infection in a patient on renal replacement therapy. Clin Exp Dermatol. 2001;26:415-418.
- Jeong SH, Kim SY, Huh HJ, et al. Mycobacteriological characteristics and treatment outcomes in extrapulmonary Mycobacterium abscessus complex infections. Int J Infect Dis. 2017;60:49-56.
- Novosad SA, Beekmann SE, Polgreen PM, et al. Treatment of Mycobacterium abscessus infection. Emerg Infect Dis. 2016;22:511-514.
- Lee WJ, Kang SM, Sung H, et al. Non-tuberculous mycobacterial infections of the skin: a retrospective study of 29 cases. J Dermatol. 2010;37:965-972.
- Fitzgerald DA, Smith AG, Lees A, et al. Cutaneous infection with Mycobacterium abscessus. Br J Dermatol. 1995;132:800-804.
- Moore M, Frerichs JB. An unusual acid-fast infection of the knee with subcutaneous, abscess-like lesions of the gluteal region; report of a case with a study of the organism, Mycobacterium abscessus, n. sp. J Invest Dermatol. 1953;20:133-169.
- Inman PM, Beck A, Brown AE, et al. Outbreak of injection abscesses due to Mycobacterium abscessus. Arch Dermatol. 1969;100:141-147.
- Ryu HJ, Kim WJ, Oh CH, et al. Iatrogenic Mycobacterium abscessus infection associated with acupuncture: clinical manifestations and its treatment. Int J Dermatol. 2005;44:846-850.
- Wentworth AB, Drage LA, Wengenack NL, et al. Increased incidence of cutaneous nontuberculous mycobacterial infection, 1980 to 2009: a population-based study. Mayo Clin Proc. 2013;88:38-45.
- Uslan DZ, Kowalski TJ, Wengenack NL, et al. Skin and soft tissue infections due to rapidly growing mycobacteria: comparison of clinical features, treatment, and susceptibility. Arch Dermatol. 2006;142:1287-1292.
- Bartralot R, Pujol RM, García-Patos V, et al. Cutaneous infections due to nontuberculous mycobacteria: histopathological review of 28 cases. comparative study between lesions observed in immunosuppressed patients and normal hosts. J Cutan Pathol. 2000;27:124-129.
- Morris-Jones R, Fletcher C, Morris-Jones S, et al. Mycobacterium abscessus: a cutaneous infection in a patient on renal replacement therapy. Clin Exp Dermatol. 2001;26:415-418.
- Jeong SH, Kim SY, Huh HJ, et al. Mycobacteriological characteristics and treatment outcomes in extrapulmonary Mycobacterium abscessus complex infections. Int J Infect Dis. 2017;60:49-56.
- Novosad SA, Beekmann SE, Polgreen PM, et al. Treatment of Mycobacterium abscessus infection. Emerg Infect Dis. 2016;22:511-514.
Practice Points
- Atypical mycobacteria are included in the differential for cutaneous abscesses.
- At-home cosmetic treatments often carry unrecognized risks for adverse events.
- Obtain culture prior to initiation of empiric antibiotics.