User login
Psoriasiform Drug Eruption Secondary to Sorafenib: Case Series and Review of the Literature
The expanded use of targeted anticancer agents such as sorafenib has revealed a growing spectrum of adverse cutaneous eruptions. We describe 3 patients with sorafenib-induced psoriasiform dermatitis and review the literature of only 10 other similar reported cases based on a search of PubMed, Web of Science, and American Society of Clinical Oncology abstracts using the terms psoriasis or psoriasiform dermatitis and sorafenib.1-10 We seek to increase awareness of this particular drug eruption in response to sorafenib and to describe potential effective treatment options, especially when sorafenib cannot be discontinued.
Case Reports
Patient 1
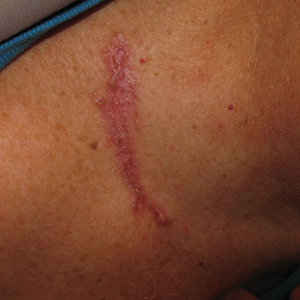
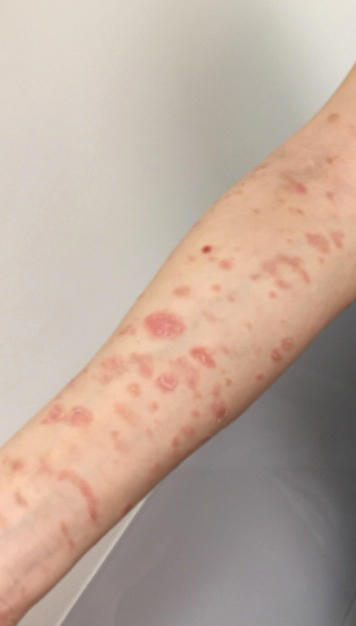
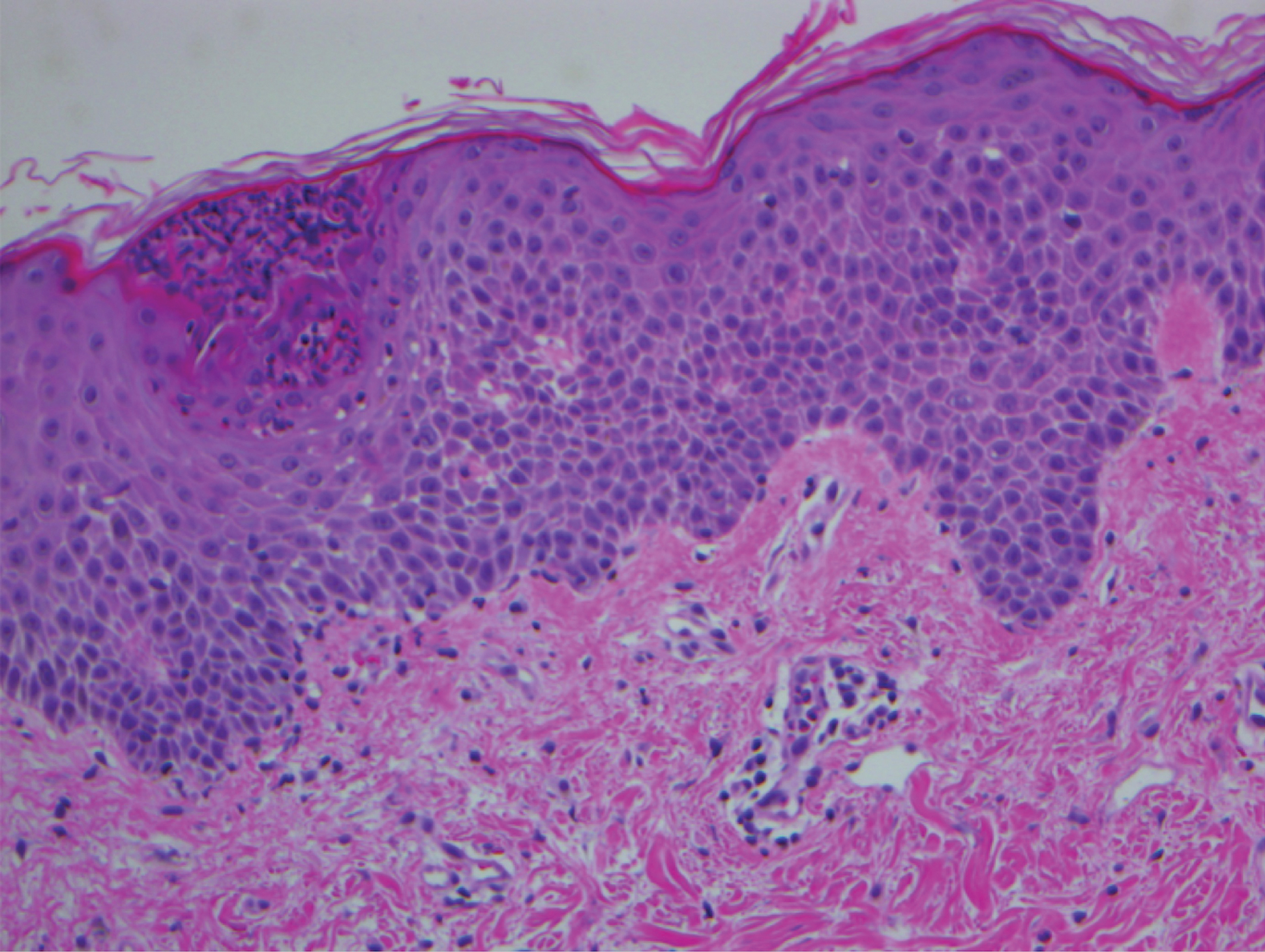
A 68-year-old man with chronic hepatitis B infection and hepatocellular carcinoma (HCC) was started on sorafenib 400 mg daily. After 2 months of treatment, he developed painful hyperkeratotic lesions on the bilateral palms and soles with formation of calluses and superficial blisters on an erythematous base that was consistent with hand-foot skin reaction (HFSR). He also had numerous erythematous thin papules and plaques with adherent white scale and yellow crust on the bilateral thighs, lower legs, forearms, dorsal hands, abdomen, back, and buttocks (Figure 1). He had no personal or family history of psoriasis, and blood tests were unremarkable. Histologic analysis of punch biopsies from the buttocks and right leg revealed focal parakeratosis with neutrophils and serous crust, acanthosis, mild spongiosis, and lymphocytes at the dermoepidermal junction and surrounding dermal vessels, consistent with psoriasiform dermatitis (Figure 2). Sorafenib was discontinued and the eruption began to resolve within a week. A lower dose of sorafenib (200 mg daily) was attempted and the psoriasiform eruption recurred.
Patient 2
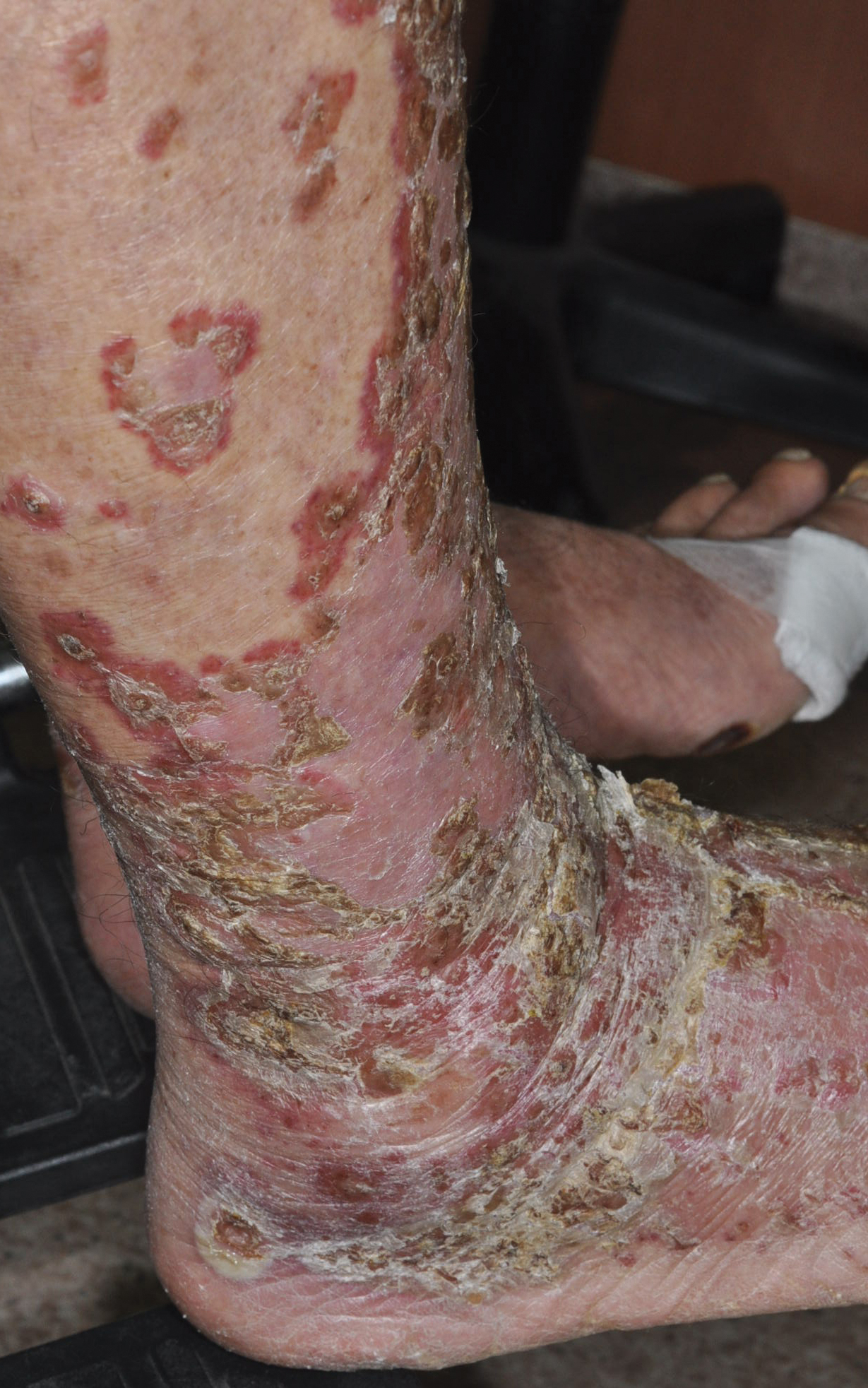
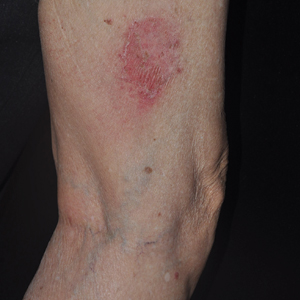
An 82-year-old man with chronic hepatitis B infection and HCC with lung metastasis was treated with sorafenib 400 mg daily. One week after treatment, he developed painful, thick, erythematous lesions on acral surfaces, consistent with HFSR. The sorafenib dose was decreased to 200 mg daily and HFSR resolved. Four months later, he developed well-demarcated, erythematous, scaly plaques with peripheral pustules on the right thigh (Figure 3) and right shin. He had no personal or family history of psoriasis, and blood tests were unremarkable. Samples from the pustules were taken for bacterial culture and fungal stain, but both were negative. Histologic analysis of a punch biopsy from the right thigh revealed necrotic parakeratosis, spongiform pustules, mild acanthosis, and a perivascular lymphocytic infiltrate with many neutrophils in the dermis. These findings suggested a diagnosis of pustular psoriasis, pustular drug eruption, or acute generalized exanthematous pustulosis. Treatment was initiated with mometasone cream. The patient subsequently developed hemoptysis and ascites from sorafenib. Sorafenib was discontinued and his skin eruption gradually resolved.
Patient 3
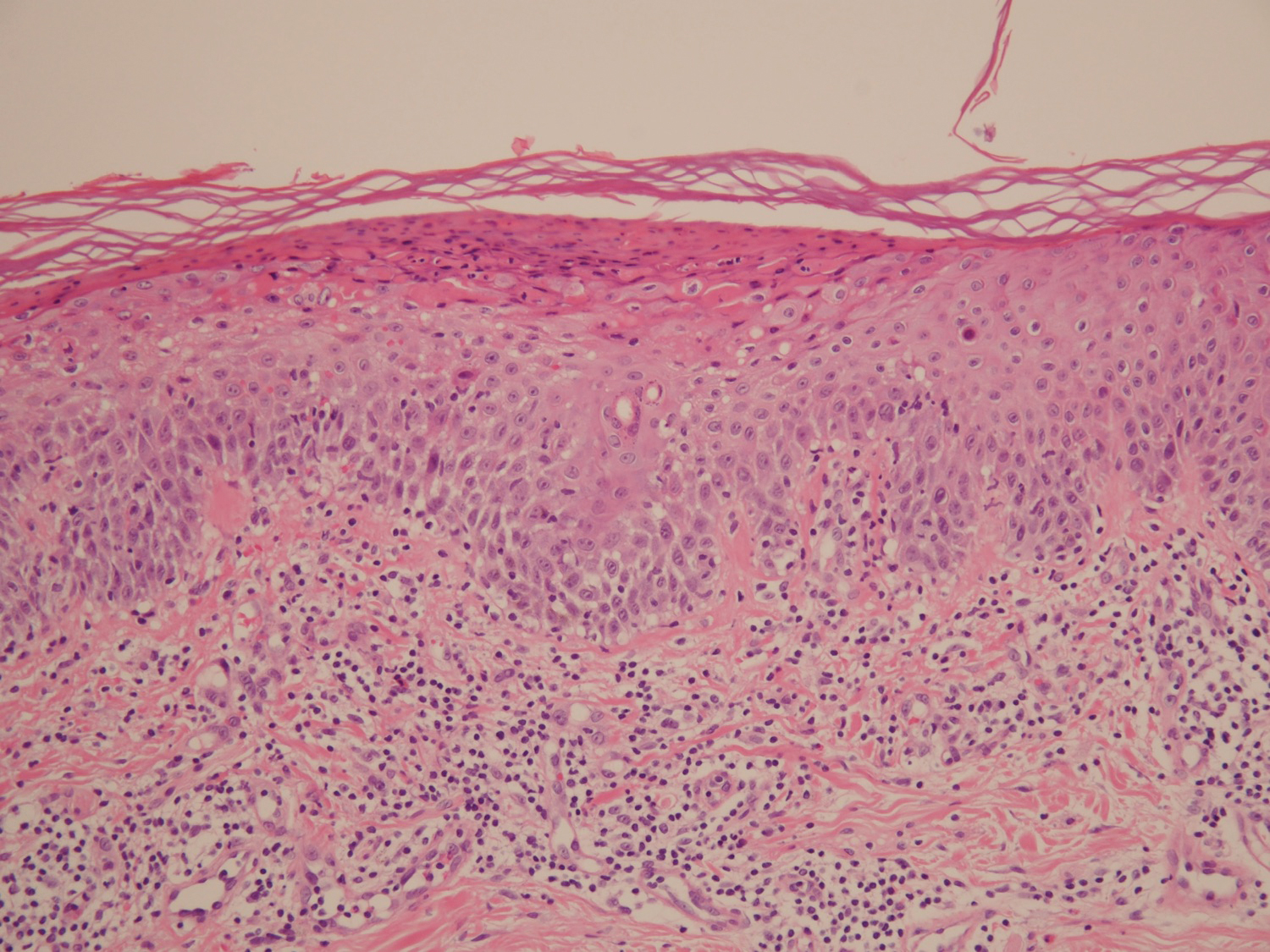
A 45-year-old woman with history of acute myeloid leukemia (AML) was started on sorafenib 200 mg twice daily as part of a clinical pilot study to maintain remission following an allogeneic bone marrow transplant. Four months after beginning sorafenib, the patient developed multiple well-defined, erythematous, thin papules and plaques with overlying flaky white scale on the bilateral upper extremities and trunk and scattered on the bilateral upper thighs (Figure 4) along with abdominal pain. Her other medical history, physical findings, and laboratory results were unremarkable, and there was no personal or family history of psoriasis. Her oncologist suspected that the eruption and symptoms were due to sorafenib and reduced the dose to 200 mg daily. Histologic analysis of a punch biopsy specimen revealed subcorneal neutrophilic collections with mild spongiosis and mild perivascular inflammatory infiltrate composed of lymphocytes and neutrophils (Figure 5). Direct immunofluorescence was negative for antibody or complement deposition. A bone marrow biopsy was negative for AML recurrence. The patient was continued on sorafenib to prevent AML recurrence, and she was started on triamcinolone cream 0.1% twice daily. Two weeks later, the eruption worsened and the patient was started on oral hydroxyzine for pruritus and narrowband UVB (NB-UVB) phototherapy 3 times a week. After 9 applications of NB-UVB phototherapy, there was complete resolution of the eruption.
Comment
Sorafenib is an oral tyrosine kinase inhibitor that blocks tumor cell proliferation and angiogenesis due to its activity against vascular endothelial growth factor (VEGF) receptor, platelet-derived growth factor receptor, stem cell growth factor receptor, and rapidly accelerated fibrosarcoma kinases.11 It is primarily used for the treatment of solid tumors, such as advanced renal cell carcinoma, unresectable HCC, and thyroid carcinoma, and more recently has been expanded for treatment of AML due to potential inhibition of FMS-like tyrosine kinase 3 receptor. Although dermatologic toxicity is a common adverse event during treatment with sorafenib,11 reports of psoriasiform drug eruptions are rare.
Review of Cases
Based on our literature search, there are 10 previously reported cases of psoriasiform drug eruption secondary to sorafenib. Of the 13 total cases (including the 3 patients in this report), 7 patients had a history of psoriasis; most were middle-aged men; and the treatment with sorafenib was for solid tumors, primarily HCC with the exception of patient 3 from the current report who was treated for AML (Table). In all cases, the dose of sorafenib ranged from 200 to 800 mg daily. In 5 cases, HFSR preceded (as with patient 2 in the current report) or presented concurrently (as with patient 1 in the current report) with the onset of psoriasiform rash.1,3,5
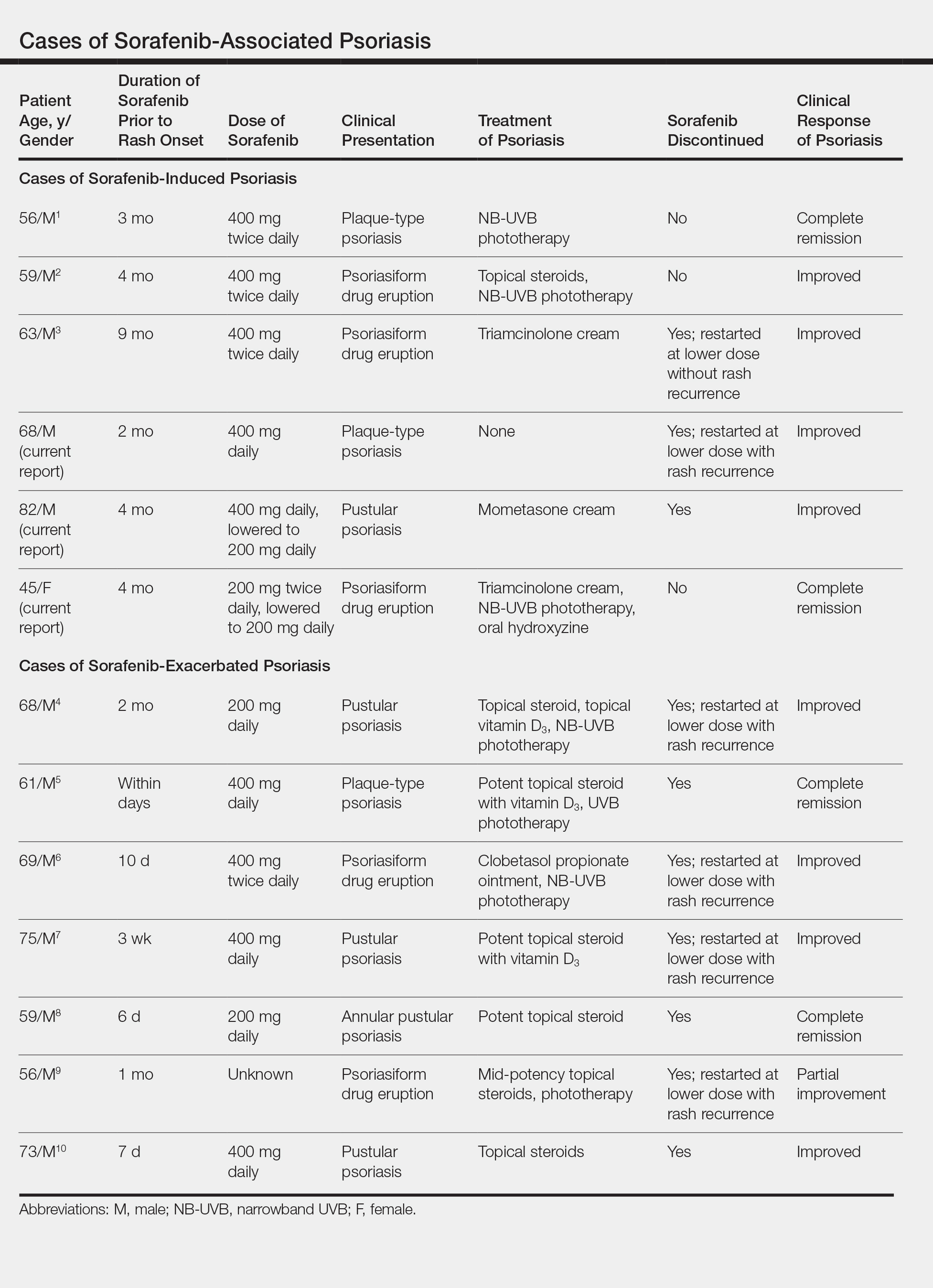
Of the 13 total cases, patients with a history of psoriasis generally developed the eruption in a shorter period of time after starting sorafenib (eg, days to 2 months) compared to those without a history of psoriasis (eg, 2 to 9 months)(Table), suggesting that patients with preexisting psoriasis more rapidly developed the drug eruption than patients without a history. In these patients with a history of psoriasis, all had long-standing mild to moderate stable plaque psoriasis, with the exception of 1 case in which the type of psoriasis was not described (Table).7 The presentation of the drug eruption following sorafenib varied from psoriasiform drug eruption (5 patients, including patient 3),2,3,6,9 pustular psoriasis (5 patients, including patient 2),4,7,8,10 and plaque psoriasis (3 patients, including patient 1).1,5 Interestingly, 5 of 6 patients with a history of plaque psoriasis presented with pustular psoriasis or psoriasiform drug eruption after treatment with sorafenib.4-6,8-10 These results suggest a causal relationship between sorafenib and exacerbation of preexisting psoriasis.
In the 13 total cases, treatments included mid- to high-potency topical steroids (10 cases), UVB or NB-UVB phototherapy (7 cases), and discontinuation of sorafenib (10 cases)(Table). All of these treatments led to improvement of the eruption with the exception of 1 case in which hand involvement was recalcitrant to therapy.9 Of the 10 cases in which sorafenib was discontinued, rechallenge at a lower dose was performed in 6 cases (including patient 1)3,4,6,7,9 with recurrence of psoriasiform rash seen in 5 cases (including patient 1)(Table).4,6,7,9 These data strongly implicate sorafenib as the direct cause of these psoriasiform eruptions. In the 3 cases in which sorafenib was not discontinued (including patient 3), there was notable improvement of the eruption with NB-UVB phototherapy.1,2
Vascular endothelial growth factor is overexpressed on psoriatic keratinocytes, contributes to epidermal hyperplasia, and induces angiogenesis in the dermis.12 The development of psoriasiform eruptions in patients treated with sorafenib seems paradoxical, as this drug has been considered as potential therapy for psoriasis due to its ability to block VEGF receptor signaling. Indeed, an improvement of psoriasis has been reported in 1 case of a patient treated with sorafenib13 and in multiple patients with psoriasis treated with other VEGF antagonists (eg, bevacizumab).14 The underlying mechanisms by which sorafenib induced or exacerbated psoriasis are not entirely clear. Palmoplantar hyperkeratosis, keratosis pilaris–like eruption, multiple cysts, eruptive keratoacanthomas, and squamous cell carcinoma have been described in patients treated with sorafenib, supporting the hypothesis that treatment with sorafenib alters keratinocyte proliferation and differentiation.15 In addition, B-Raf inhibitors such as imatinib are known to induce or exacerbate psoriasiform dermatitis.16 The activity of sorafenib resulting in psoriasis may be specific to RAF kinase inhibition, as there are no reports in the literature that describe psoriasiform dermatitis with agents that preferentially block other sorafenib targets such as VEGF receptor, stem cell growth factor receptor, or platelet-derived growth factor receptor. Future studies are needed to fully elucidate the underlying mechanisms by which sorafenib induces or exacerbates psoriasiform dermatitis and whether the severity of the drug eruption correlates with the antitumor efficacy of sorafenib.
Conclusion
Although psoriasiform drug eruptions secondary to sorafenib are not life-threatening, they impact quality of life with associated pain, pruritus, infection, and limitation of daily activities. Dose reduction or discontinuation of sorafenib resulted in resolution of the psoriasiform dermatitis; however, as demonstrated in 3 cases (including patient 3),1,2 psoriasiform dermatitis can be managed while maintaining the patient on sorafenib so that treatment of the malignancy is not compromised.
- Hung CT, Chiang CP, Wu BY. Sorafenib-induced psoriasis and hand-foot skin reaction responded dramatically to systemic narrowband ultraviolet B phototherapy. J Dermatol. 2012;39:1076-1077.
- González-López M, Yáñez S, Val-Bernal JF, et al. Psoriasiform skin eruption associated with sorafenib therapy. Indian J Dermatol Venereol Leprol. 2011;77:614-615.
- Diamantis ML, Chon SY. Sorafenib-induced psoriasiform eruption in a patient with metastatic thyroid carcinoma. J Drugs Dermatol. 2010;9:169-171.
- Hsu MC, Chen CC. Psoriasis flare-ups following sorafenib therapy: a rare case. Dermatologica Sin. 2016;34:148-150.
- Yiu ZZ, Ali FR, Griffiths CE. Paradoxical exacerbation of chronic plaque psoriasis by sorafenib. Clin Exp Dermatol. 2016;41:407-409.
- I˙lknur T, Akarsu S, Çarsanbali S, et al. Sorafenib-associated psoriasiform eruption in a patient with hepatocellular carcinoma. J Drugs Dermatol. 2014;13:899-900.
- Maki N, Komine M, Takatsuka Y, et al. Pustular eruption induced by sorafenib in a case of psoriasis vulgaris. J Dermatol. 2013;40:299-300.
- Du-Thanh A, Girard C, Pageaux GP, et al. Sorafenib-induced annular pustular psoriasis (Milian-Katchoura type). Eur J Dermatol. 2013;23:900-901.
- Laquer V, Saedi N, Dann F, et al. Sorafenib-associated psoriasiform skin changes. Cutis. 2010;85:301-302.
- Ohashi T, Yamamoto T. Exacerbation of psoriasis with pustulation by sorafenib in a patient with metastatic hepatocellular carcinoma. Indian J Dermatol. 2019;64:75-77.
- Chu D, Lacouture ME, Fillos T, et al. Risk of hand-foot skin reaction with sorafenib: a systematic review and meta-analysis. Acta Oncol (Madr). 2008;47:176-186.
- Canavese M, Altruda F, Ruzicka T, et al. Vascular endothelial growth factor (VEGF) in the pathogenesis of psoriasis--a possible target for novel therapies? J Dermatol Sci. 2010;58:171-176.
- Fournier C, Tisman G. Sorafenib-associated remission of psoriasis in hypernephroma: case report. Dermatol Online J. 2010;16:17.
- Akman A, Yilmaz E, Mutlu H, et al. Complete remission of psoriasis following bevacizumab therapy for colon cancer. Clin Exp Dermatol. 2009;34:E202-E204.
- Kong HH, Turner ML. Array of cutaneous adverse effects associated with sorafenib. J Am Acad Dermatol. 2009;61:360-361.
- Atalay F, Kızılkılıç E, Ada RS. Imatinib-induced psoriasis. Turk J Haematol. 2013;30:216-218.
The expanded use of targeted anticancer agents such as sorafenib has revealed a growing spectrum of adverse cutaneous eruptions. We describe 3 patients with sorafenib-induced psoriasiform dermatitis and review the literature of only 10 other similar reported cases based on a search of PubMed, Web of Science, and American Society of Clinical Oncology abstracts using the terms psoriasis or psoriasiform dermatitis and sorafenib.1-10 We seek to increase awareness of this particular drug eruption in response to sorafenib and to describe potential effective treatment options, especially when sorafenib cannot be discontinued.
Case Reports
Patient 1
A 68-year-old man with chronic hepatitis B infection and hepatocellular carcinoma (HCC) was started on sorafenib 400 mg daily. After 2 months of treatment, he developed painful hyperkeratotic lesions on the bilateral palms and soles with formation of calluses and superficial blisters on an erythematous base that was consistent with hand-foot skin reaction (HFSR). He also had numerous erythematous thin papules and plaques with adherent white scale and yellow crust on the bilateral thighs, lower legs, forearms, dorsal hands, abdomen, back, and buttocks (Figure 1). He had no personal or family history of psoriasis, and blood tests were unremarkable. Histologic analysis of punch biopsies from the buttocks and right leg revealed focal parakeratosis with neutrophils and serous crust, acanthosis, mild spongiosis, and lymphocytes at the dermoepidermal junction and surrounding dermal vessels, consistent with psoriasiform dermatitis (Figure 2). Sorafenib was discontinued and the eruption began to resolve within a week. A lower dose of sorafenib (200 mg daily) was attempted and the psoriasiform eruption recurred.
Patient 2
An 82-year-old man with chronic hepatitis B infection and HCC with lung metastasis was treated with sorafenib 400 mg daily. One week after treatment, he developed painful, thick, erythematous lesions on acral surfaces, consistent with HFSR. The sorafenib dose was decreased to 200 mg daily and HFSR resolved. Four months later, he developed well-demarcated, erythematous, scaly plaques with peripheral pustules on the right thigh (Figure 3) and right shin. He had no personal or family history of psoriasis, and blood tests were unremarkable. Samples from the pustules were taken for bacterial culture and fungal stain, but both were negative. Histologic analysis of a punch biopsy from the right thigh revealed necrotic parakeratosis, spongiform pustules, mild acanthosis, and a perivascular lymphocytic infiltrate with many neutrophils in the dermis. These findings suggested a diagnosis of pustular psoriasis, pustular drug eruption, or acute generalized exanthematous pustulosis. Treatment was initiated with mometasone cream. The patient subsequently developed hemoptysis and ascites from sorafenib. Sorafenib was discontinued and his skin eruption gradually resolved.
Patient 3
A 45-year-old woman with history of acute myeloid leukemia (AML) was started on sorafenib 200 mg twice daily as part of a clinical pilot study to maintain remission following an allogeneic bone marrow transplant. Four months after beginning sorafenib, the patient developed multiple well-defined, erythematous, thin papules and plaques with overlying flaky white scale on the bilateral upper extremities and trunk and scattered on the bilateral upper thighs (Figure 4) along with abdominal pain. Her other medical history, physical findings, and laboratory results were unremarkable, and there was no personal or family history of psoriasis. Her oncologist suspected that the eruption and symptoms were due to sorafenib and reduced the dose to 200 mg daily. Histologic analysis of a punch biopsy specimen revealed subcorneal neutrophilic collections with mild spongiosis and mild perivascular inflammatory infiltrate composed of lymphocytes and neutrophils (Figure 5). Direct immunofluorescence was negative for antibody or complement deposition. A bone marrow biopsy was negative for AML recurrence. The patient was continued on sorafenib to prevent AML recurrence, and she was started on triamcinolone cream 0.1% twice daily. Two weeks later, the eruption worsened and the patient was started on oral hydroxyzine for pruritus and narrowband UVB (NB-UVB) phototherapy 3 times a week. After 9 applications of NB-UVB phototherapy, there was complete resolution of the eruption.
Comment
Sorafenib is an oral tyrosine kinase inhibitor that blocks tumor cell proliferation and angiogenesis due to its activity against vascular endothelial growth factor (VEGF) receptor, platelet-derived growth factor receptor, stem cell growth factor receptor, and rapidly accelerated fibrosarcoma kinases.11 It is primarily used for the treatment of solid tumors, such as advanced renal cell carcinoma, unresectable HCC, and thyroid carcinoma, and more recently has been expanded for treatment of AML due to potential inhibition of FMS-like tyrosine kinase 3 receptor. Although dermatologic toxicity is a common adverse event during treatment with sorafenib,11 reports of psoriasiform drug eruptions are rare.
Review of Cases
Based on our literature search, there are 10 previously reported cases of psoriasiform drug eruption secondary to sorafenib. Of the 13 total cases (including the 3 patients in this report), 7 patients had a history of psoriasis; most were middle-aged men; and the treatment with sorafenib was for solid tumors, primarily HCC with the exception of patient 3 from the current report who was treated for AML (Table). In all cases, the dose of sorafenib ranged from 200 to 800 mg daily. In 5 cases, HFSR preceded (as with patient 2 in the current report) or presented concurrently (as with patient 1 in the current report) with the onset of psoriasiform rash.1,3,5
Of the 13 total cases, patients with a history of psoriasis generally developed the eruption in a shorter period of time after starting sorafenib (eg, days to 2 months) compared to those without a history of psoriasis (eg, 2 to 9 months)(Table), suggesting that patients with preexisting psoriasis more rapidly developed the drug eruption than patients without a history. In these patients with a history of psoriasis, all had long-standing mild to moderate stable plaque psoriasis, with the exception of 1 case in which the type of psoriasis was not described (Table).7 The presentation of the drug eruption following sorafenib varied from psoriasiform drug eruption (5 patients, including patient 3),2,3,6,9 pustular psoriasis (5 patients, including patient 2),4,7,8,10 and plaque psoriasis (3 patients, including patient 1).1,5 Interestingly, 5 of 6 patients with a history of plaque psoriasis presented with pustular psoriasis or psoriasiform drug eruption after treatment with sorafenib.4-6,8-10 These results suggest a causal relationship between sorafenib and exacerbation of preexisting psoriasis.
In the 13 total cases, treatments included mid- to high-potency topical steroids (10 cases), UVB or NB-UVB phototherapy (7 cases), and discontinuation of sorafenib (10 cases)(Table). All of these treatments led to improvement of the eruption with the exception of 1 case in which hand involvement was recalcitrant to therapy.9 Of the 10 cases in which sorafenib was discontinued, rechallenge at a lower dose was performed in 6 cases (including patient 1)3,4,6,7,9 with recurrence of psoriasiform rash seen in 5 cases (including patient 1)(Table).4,6,7,9 These data strongly implicate sorafenib as the direct cause of these psoriasiform eruptions. In the 3 cases in which sorafenib was not discontinued (including patient 3), there was notable improvement of the eruption with NB-UVB phototherapy.1,2
Vascular endothelial growth factor is overexpressed on psoriatic keratinocytes, contributes to epidermal hyperplasia, and induces angiogenesis in the dermis.12 The development of psoriasiform eruptions in patients treated with sorafenib seems paradoxical, as this drug has been considered as potential therapy for psoriasis due to its ability to block VEGF receptor signaling. Indeed, an improvement of psoriasis has been reported in 1 case of a patient treated with sorafenib13 and in multiple patients with psoriasis treated with other VEGF antagonists (eg, bevacizumab).14 The underlying mechanisms by which sorafenib induced or exacerbated psoriasis are not entirely clear. Palmoplantar hyperkeratosis, keratosis pilaris–like eruption, multiple cysts, eruptive keratoacanthomas, and squamous cell carcinoma have been described in patients treated with sorafenib, supporting the hypothesis that treatment with sorafenib alters keratinocyte proliferation and differentiation.15 In addition, B-Raf inhibitors such as imatinib are known to induce or exacerbate psoriasiform dermatitis.16 The activity of sorafenib resulting in psoriasis may be specific to RAF kinase inhibition, as there are no reports in the literature that describe psoriasiform dermatitis with agents that preferentially block other sorafenib targets such as VEGF receptor, stem cell growth factor receptor, or platelet-derived growth factor receptor. Future studies are needed to fully elucidate the underlying mechanisms by which sorafenib induces or exacerbates psoriasiform dermatitis and whether the severity of the drug eruption correlates with the antitumor efficacy of sorafenib.
Conclusion
Although psoriasiform drug eruptions secondary to sorafenib are not life-threatening, they impact quality of life with associated pain, pruritus, infection, and limitation of daily activities. Dose reduction or discontinuation of sorafenib resulted in resolution of the psoriasiform dermatitis; however, as demonstrated in 3 cases (including patient 3),1,2 psoriasiform dermatitis can be managed while maintaining the patient on sorafenib so that treatment of the malignancy is not compromised.
The expanded use of targeted anticancer agents such as sorafenib has revealed a growing spectrum of adverse cutaneous eruptions. We describe 3 patients with sorafenib-induced psoriasiform dermatitis and review the literature of only 10 other similar reported cases based on a search of PubMed, Web of Science, and American Society of Clinical Oncology abstracts using the terms psoriasis or psoriasiform dermatitis and sorafenib.1-10 We seek to increase awareness of this particular drug eruption in response to sorafenib and to describe potential effective treatment options, especially when sorafenib cannot be discontinued.
Case Reports
Patient 1
A 68-year-old man with chronic hepatitis B infection and hepatocellular carcinoma (HCC) was started on sorafenib 400 mg daily. After 2 months of treatment, he developed painful hyperkeratotic lesions on the bilateral palms and soles with formation of calluses and superficial blisters on an erythematous base that was consistent with hand-foot skin reaction (HFSR). He also had numerous erythematous thin papules and plaques with adherent white scale and yellow crust on the bilateral thighs, lower legs, forearms, dorsal hands, abdomen, back, and buttocks (Figure 1). He had no personal or family history of psoriasis, and blood tests were unremarkable. Histologic analysis of punch biopsies from the buttocks and right leg revealed focal parakeratosis with neutrophils and serous crust, acanthosis, mild spongiosis, and lymphocytes at the dermoepidermal junction and surrounding dermal vessels, consistent with psoriasiform dermatitis (Figure 2). Sorafenib was discontinued and the eruption began to resolve within a week. A lower dose of sorafenib (200 mg daily) was attempted and the psoriasiform eruption recurred.
Patient 2
An 82-year-old man with chronic hepatitis B infection and HCC with lung metastasis was treated with sorafenib 400 mg daily. One week after treatment, he developed painful, thick, erythematous lesions on acral surfaces, consistent with HFSR. The sorafenib dose was decreased to 200 mg daily and HFSR resolved. Four months later, he developed well-demarcated, erythematous, scaly plaques with peripheral pustules on the right thigh (Figure 3) and right shin. He had no personal or family history of psoriasis, and blood tests were unremarkable. Samples from the pustules were taken for bacterial culture and fungal stain, but both were negative. Histologic analysis of a punch biopsy from the right thigh revealed necrotic parakeratosis, spongiform pustules, mild acanthosis, and a perivascular lymphocytic infiltrate with many neutrophils in the dermis. These findings suggested a diagnosis of pustular psoriasis, pustular drug eruption, or acute generalized exanthematous pustulosis. Treatment was initiated with mometasone cream. The patient subsequently developed hemoptysis and ascites from sorafenib. Sorafenib was discontinued and his skin eruption gradually resolved.
Patient 3
A 45-year-old woman with history of acute myeloid leukemia (AML) was started on sorafenib 200 mg twice daily as part of a clinical pilot study to maintain remission following an allogeneic bone marrow transplant. Four months after beginning sorafenib, the patient developed multiple well-defined, erythematous, thin papules and plaques with overlying flaky white scale on the bilateral upper extremities and trunk and scattered on the bilateral upper thighs (Figure 4) along with abdominal pain. Her other medical history, physical findings, and laboratory results were unremarkable, and there was no personal or family history of psoriasis. Her oncologist suspected that the eruption and symptoms were due to sorafenib and reduced the dose to 200 mg daily. Histologic analysis of a punch biopsy specimen revealed subcorneal neutrophilic collections with mild spongiosis and mild perivascular inflammatory infiltrate composed of lymphocytes and neutrophils (Figure 5). Direct immunofluorescence was negative for antibody or complement deposition. A bone marrow biopsy was negative for AML recurrence. The patient was continued on sorafenib to prevent AML recurrence, and she was started on triamcinolone cream 0.1% twice daily. Two weeks later, the eruption worsened and the patient was started on oral hydroxyzine for pruritus and narrowband UVB (NB-UVB) phototherapy 3 times a week. After 9 applications of NB-UVB phototherapy, there was complete resolution of the eruption.
Comment
Sorafenib is an oral tyrosine kinase inhibitor that blocks tumor cell proliferation and angiogenesis due to its activity against vascular endothelial growth factor (VEGF) receptor, platelet-derived growth factor receptor, stem cell growth factor receptor, and rapidly accelerated fibrosarcoma kinases.11 It is primarily used for the treatment of solid tumors, such as advanced renal cell carcinoma, unresectable HCC, and thyroid carcinoma, and more recently has been expanded for treatment of AML due to potential inhibition of FMS-like tyrosine kinase 3 receptor. Although dermatologic toxicity is a common adverse event during treatment with sorafenib,11 reports of psoriasiform drug eruptions are rare.
Review of Cases
Based on our literature search, there are 10 previously reported cases of psoriasiform drug eruption secondary to sorafenib. Of the 13 total cases (including the 3 patients in this report), 7 patients had a history of psoriasis; most were middle-aged men; and the treatment with sorafenib was for solid tumors, primarily HCC with the exception of patient 3 from the current report who was treated for AML (Table). In all cases, the dose of sorafenib ranged from 200 to 800 mg daily. In 5 cases, HFSR preceded (as with patient 2 in the current report) or presented concurrently (as with patient 1 in the current report) with the onset of psoriasiform rash.1,3,5
Of the 13 total cases, patients with a history of psoriasis generally developed the eruption in a shorter period of time after starting sorafenib (eg, days to 2 months) compared to those without a history of psoriasis (eg, 2 to 9 months)(Table), suggesting that patients with preexisting psoriasis more rapidly developed the drug eruption than patients without a history. In these patients with a history of psoriasis, all had long-standing mild to moderate stable plaque psoriasis, with the exception of 1 case in which the type of psoriasis was not described (Table).7 The presentation of the drug eruption following sorafenib varied from psoriasiform drug eruption (5 patients, including patient 3),2,3,6,9 pustular psoriasis (5 patients, including patient 2),4,7,8,10 and plaque psoriasis (3 patients, including patient 1).1,5 Interestingly, 5 of 6 patients with a history of plaque psoriasis presented with pustular psoriasis or psoriasiform drug eruption after treatment with sorafenib.4-6,8-10 These results suggest a causal relationship between sorafenib and exacerbation of preexisting psoriasis.
In the 13 total cases, treatments included mid- to high-potency topical steroids (10 cases), UVB or NB-UVB phototherapy (7 cases), and discontinuation of sorafenib (10 cases)(Table). All of these treatments led to improvement of the eruption with the exception of 1 case in which hand involvement was recalcitrant to therapy.9 Of the 10 cases in which sorafenib was discontinued, rechallenge at a lower dose was performed in 6 cases (including patient 1)3,4,6,7,9 with recurrence of psoriasiform rash seen in 5 cases (including patient 1)(Table).4,6,7,9 These data strongly implicate sorafenib as the direct cause of these psoriasiform eruptions. In the 3 cases in which sorafenib was not discontinued (including patient 3), there was notable improvement of the eruption with NB-UVB phototherapy.1,2
Vascular endothelial growth factor is overexpressed on psoriatic keratinocytes, contributes to epidermal hyperplasia, and induces angiogenesis in the dermis.12 The development of psoriasiform eruptions in patients treated with sorafenib seems paradoxical, as this drug has been considered as potential therapy for psoriasis due to its ability to block VEGF receptor signaling. Indeed, an improvement of psoriasis has been reported in 1 case of a patient treated with sorafenib13 and in multiple patients with psoriasis treated with other VEGF antagonists (eg, bevacizumab).14 The underlying mechanisms by which sorafenib induced or exacerbated psoriasis are not entirely clear. Palmoplantar hyperkeratosis, keratosis pilaris–like eruption, multiple cysts, eruptive keratoacanthomas, and squamous cell carcinoma have been described in patients treated with sorafenib, supporting the hypothesis that treatment with sorafenib alters keratinocyte proliferation and differentiation.15 In addition, B-Raf inhibitors such as imatinib are known to induce or exacerbate psoriasiform dermatitis.16 The activity of sorafenib resulting in psoriasis may be specific to RAF kinase inhibition, as there are no reports in the literature that describe psoriasiform dermatitis with agents that preferentially block other sorafenib targets such as VEGF receptor, stem cell growth factor receptor, or platelet-derived growth factor receptor. Future studies are needed to fully elucidate the underlying mechanisms by which sorafenib induces or exacerbates psoriasiform dermatitis and whether the severity of the drug eruption correlates with the antitumor efficacy of sorafenib.
Conclusion
Although psoriasiform drug eruptions secondary to sorafenib are not life-threatening, they impact quality of life with associated pain, pruritus, infection, and limitation of daily activities. Dose reduction or discontinuation of sorafenib resulted in resolution of the psoriasiform dermatitis; however, as demonstrated in 3 cases (including patient 3),1,2 psoriasiform dermatitis can be managed while maintaining the patient on sorafenib so that treatment of the malignancy is not compromised.
- Hung CT, Chiang CP, Wu BY. Sorafenib-induced psoriasis and hand-foot skin reaction responded dramatically to systemic narrowband ultraviolet B phototherapy. J Dermatol. 2012;39:1076-1077.
- González-López M, Yáñez S, Val-Bernal JF, et al. Psoriasiform skin eruption associated with sorafenib therapy. Indian J Dermatol Venereol Leprol. 2011;77:614-615.
- Diamantis ML, Chon SY. Sorafenib-induced psoriasiform eruption in a patient with metastatic thyroid carcinoma. J Drugs Dermatol. 2010;9:169-171.
- Hsu MC, Chen CC. Psoriasis flare-ups following sorafenib therapy: a rare case. Dermatologica Sin. 2016;34:148-150.
- Yiu ZZ, Ali FR, Griffiths CE. Paradoxical exacerbation of chronic plaque psoriasis by sorafenib. Clin Exp Dermatol. 2016;41:407-409.
- I˙lknur T, Akarsu S, Çarsanbali S, et al. Sorafenib-associated psoriasiform eruption in a patient with hepatocellular carcinoma. J Drugs Dermatol. 2014;13:899-900.
- Maki N, Komine M, Takatsuka Y, et al. Pustular eruption induced by sorafenib in a case of psoriasis vulgaris. J Dermatol. 2013;40:299-300.
- Du-Thanh A, Girard C, Pageaux GP, et al. Sorafenib-induced annular pustular psoriasis (Milian-Katchoura type). Eur J Dermatol. 2013;23:900-901.
- Laquer V, Saedi N, Dann F, et al. Sorafenib-associated psoriasiform skin changes. Cutis. 2010;85:301-302.
- Ohashi T, Yamamoto T. Exacerbation of psoriasis with pustulation by sorafenib in a patient with metastatic hepatocellular carcinoma. Indian J Dermatol. 2019;64:75-77.
- Chu D, Lacouture ME, Fillos T, et al. Risk of hand-foot skin reaction with sorafenib: a systematic review and meta-analysis. Acta Oncol (Madr). 2008;47:176-186.
- Canavese M, Altruda F, Ruzicka T, et al. Vascular endothelial growth factor (VEGF) in the pathogenesis of psoriasis--a possible target for novel therapies? J Dermatol Sci. 2010;58:171-176.
- Fournier C, Tisman G. Sorafenib-associated remission of psoriasis in hypernephroma: case report. Dermatol Online J. 2010;16:17.
- Akman A, Yilmaz E, Mutlu H, et al. Complete remission of psoriasis following bevacizumab therapy for colon cancer. Clin Exp Dermatol. 2009;34:E202-E204.
- Kong HH, Turner ML. Array of cutaneous adverse effects associated with sorafenib. J Am Acad Dermatol. 2009;61:360-361.
- Atalay F, Kızılkılıç E, Ada RS. Imatinib-induced psoriasis. Turk J Haematol. 2013;30:216-218.
- Hung CT, Chiang CP, Wu BY. Sorafenib-induced psoriasis and hand-foot skin reaction responded dramatically to systemic narrowband ultraviolet B phototherapy. J Dermatol. 2012;39:1076-1077.
- González-López M, Yáñez S, Val-Bernal JF, et al. Psoriasiform skin eruption associated with sorafenib therapy. Indian J Dermatol Venereol Leprol. 2011;77:614-615.
- Diamantis ML, Chon SY. Sorafenib-induced psoriasiform eruption in a patient with metastatic thyroid carcinoma. J Drugs Dermatol. 2010;9:169-171.
- Hsu MC, Chen CC. Psoriasis flare-ups following sorafenib therapy: a rare case. Dermatologica Sin. 2016;34:148-150.
- Yiu ZZ, Ali FR, Griffiths CE. Paradoxical exacerbation of chronic plaque psoriasis by sorafenib. Clin Exp Dermatol. 2016;41:407-409.
- I˙lknur T, Akarsu S, Çarsanbali S, et al. Sorafenib-associated psoriasiform eruption in a patient with hepatocellular carcinoma. J Drugs Dermatol. 2014;13:899-900.
- Maki N, Komine M, Takatsuka Y, et al. Pustular eruption induced by sorafenib in a case of psoriasis vulgaris. J Dermatol. 2013;40:299-300.
- Du-Thanh A, Girard C, Pageaux GP, et al. Sorafenib-induced annular pustular psoriasis (Milian-Katchoura type). Eur J Dermatol. 2013;23:900-901.
- Laquer V, Saedi N, Dann F, et al. Sorafenib-associated psoriasiform skin changes. Cutis. 2010;85:301-302.
- Ohashi T, Yamamoto T. Exacerbation of psoriasis with pustulation by sorafenib in a patient with metastatic hepatocellular carcinoma. Indian J Dermatol. 2019;64:75-77.
- Chu D, Lacouture ME, Fillos T, et al. Risk of hand-foot skin reaction with sorafenib: a systematic review and meta-analysis. Acta Oncol (Madr). 2008;47:176-186.
- Canavese M, Altruda F, Ruzicka T, et al. Vascular endothelial growth factor (VEGF) in the pathogenesis of psoriasis--a possible target for novel therapies? J Dermatol Sci. 2010;58:171-176.
- Fournier C, Tisman G. Sorafenib-associated remission of psoriasis in hypernephroma: case report. Dermatol Online J. 2010;16:17.
- Akman A, Yilmaz E, Mutlu H, et al. Complete remission of psoriasis following bevacizumab therapy for colon cancer. Clin Exp Dermatol. 2009;34:E202-E204.
- Kong HH, Turner ML. Array of cutaneous adverse effects associated with sorafenib. J Am Acad Dermatol. 2009;61:360-361.
- Atalay F, Kızılkılıç E, Ada RS. Imatinib-induced psoriasis. Turk J Haematol. 2013;30:216-218.
Practice Points
- The use of targeted anticancer agents continues to expand. With this expansion, the number and type of cutaneous adverse events continues to increase.
- Although sorafenib is known to cause various dermatologic side effects, there are few reports of psoriasiform dermatitis.
- Increased awareness of sorafenib-induced psoriasiform dermatitis and its management is vital to prevent discontinuation of potentially life-saving anticancer therapy.
Significant clinical response induced by vismodegib in advanced sarcoma: Hedgehog pathway inhibition
Spindle cell sarcomas are part of a rare, heterogeneous family of connective tissue tumors. These tumors are primarily treated with surgery and have a high risk of recurrence and distant metastasis with elevated mortality rates.1 Other than the evidence for first-line therapy with doxorubicin in advanced soft tissue sarcoma, little evidence exists to point to an optimal second-line therapy. This is due to the diversity of soft tissue sarcomas, which encompass approximately 70 different histologic subtypes that can each respond differently to treatment.2 As such, newer strategies, including immunotherapy and targeted molecular drugs, are being developed.
Quiescent in most adult tissues, the Hedgehog signaling pathway, when inappropriately activated, has been implicated in the development of multiple types of cancers, including basal cell, breast, prostate, hepatocellular, pancreatic, and brain cancer.3 The Hedgehog signaling pathway is an important regulator of cell growth and differentiation in early development, but when inappropriately activated can lead to cell proliferation and increased angiogenic factors, decreased apoptosis, and breakdown of tight junctions promoting cancer growth and metastasis.4 Recent data reveal that the Hedgehog pathway plays a specific role in activation of satellite cells, proliferation of myoblasts, and differentiation of skeletal muscle.5 Activation of this embryonic pathway has been implicated in embryonal rhabdoymyosarcoma, osteosarcoma, and chondrosarcoma.5-7
This pathway has recently been recognized as a therapeutic target, with the development of vismodegib, a targeted Hedgehog pathway inhibitor. This novel agent is in active use for treatment of advanced basal cell carcinoma and is currently undergoing trials for various other malignancies. Recently, a phase 2a basket study, called MyPathway, evaluated the use of targeted therapies in 35 different advanced refractory solid tumors harboring specific molecular alterations. Out of 21 patients with mutations in the Hedgehog pathway, 3 had a partial response to vismodegib—one had an unknown primary tumor, another a squamous skin cancer, and the third a salivary gland cancer.8 Vismodegib (GDC-0449) was also evaluated in a phase 2 multicenter clinical trial in patients with progressive advanced chondrosarcoma.7 Although the study did not meet its primary endpoint, the proportion of patients with non-progressive disease was 25.6% at 6 months. Investigators observed that the benefit occurred in the subset of patients with overexpression of the Hedgehog ligand. Genomic studies for mutations in SMO and PTCH genes were available for only 28 and 26 patients, respectively, of the 45 patients enrolled on the trial. While there were no mutations identified, expression data revealed that overexpression of the Hedgehog ligand was present in 65% of cases tested (13 out of 20 patients). In patients with stable disease at 6 months, all had overexpression of the Hedgehog ligand.7 These studies point to the potential use of vismodegib in both bone and soft tissue sarcomas, and more specifically, to the importance of genomic testing in these cases.
Case Presentation and Summary
This report describes the novel use of vismodegib, an oral Hedgehog signaling pathway inhibitor, in the treatment of a patient with metastatic soft tissue sarcoma.
An 18-year-old female with no particular previous illnesses was initially diagnosed with superficial soft tissue sarcoma overlying the right hip in 2013. Due to the complexity of pathology, a second opinion was requested and revealed atypical cellular spindle and epithelioid cells, morphologically and immunohistochemically suggestive of spindle cell sarcoma, not otherwise specified. She underwent negative-margin resection in January 2014. Her course was complicated by two recurrences in the right inguinal lymph nodes in July 2014 and July 2015. She was treated with lymph node dissection in 2014, followed by numerous right lymph node dissections and adjuvant radiation in 2015.
A routine computerized tomography (CT) scan of the thorax-abdomen and pelvis in August 2016 revealed recurrence of disease, with multiple lung nodules as well as metastases in the retroperitoneum. She received 6 cycles of gemcitabine and docetaxel with stability of disease. The patient was then started on a PI3K inhibitor as part of a clinical trial, as genotypic analysis of the tumor revealed an activating mutation of the PI3K gene. The patient’s course was complicated by acute obstructive renal failure requiring a double J stent for right-sided hydronephrosis.
Repeat imaging revealed disease progression, and the patient was then switched to liposomal doxorubicin alone for 4 months and then in combination with olaratumab. She received the combined treatment for a total of 3 months, which was then stopped when she was found to have new peritoneal implants and worsening ascites. At this time, tissue was sent for FoundationOne® next generation sequencing (NGS)-based genomic testing, and the patient received one dose of nivolumab.
In January 2018, 2 days after receiving her first dose of nivolumab, the patient required admission for worsening abdominal pain secondary to progression of her disease (FIGURE 1). She was found to have acute kidney injury on top of chronic kidney disease due to hydronephrosis requiring a left-sided double J stent. She also had transaminitis resulting from a common bile duct stricture treated with a biliary stent and worsening ascites requiring regular paracentesis. This was all in the context of new or growing metastatic implants.
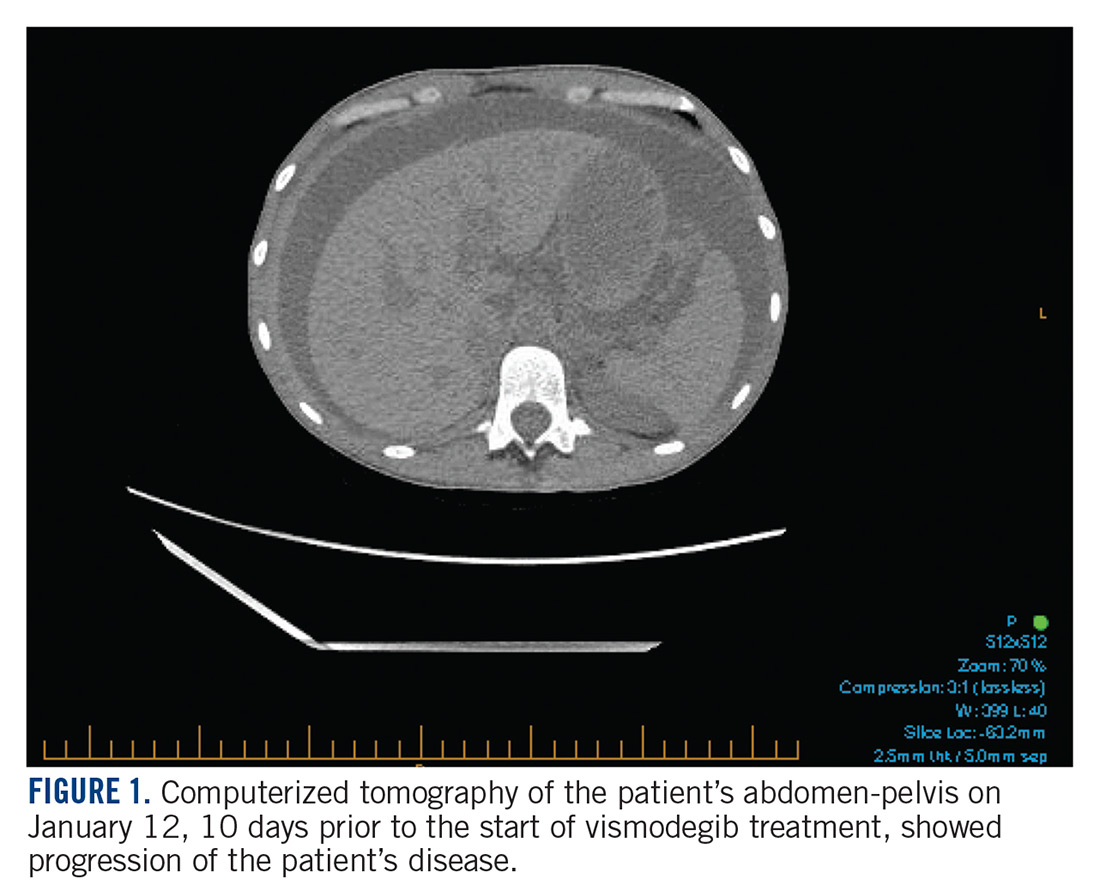
At this time, the result of the FoundationOne genomic testing revealed PTCH1 loss of exons 1-24 and CDKN2A/B loss. Mutation of tumor suppressor gene PTCH1 leads to Hedgehog pathway activation and therefore the patient was started on vismodegib on January 22, 2018. She was discharged from the hospital in stable condition a day later, on January 23.
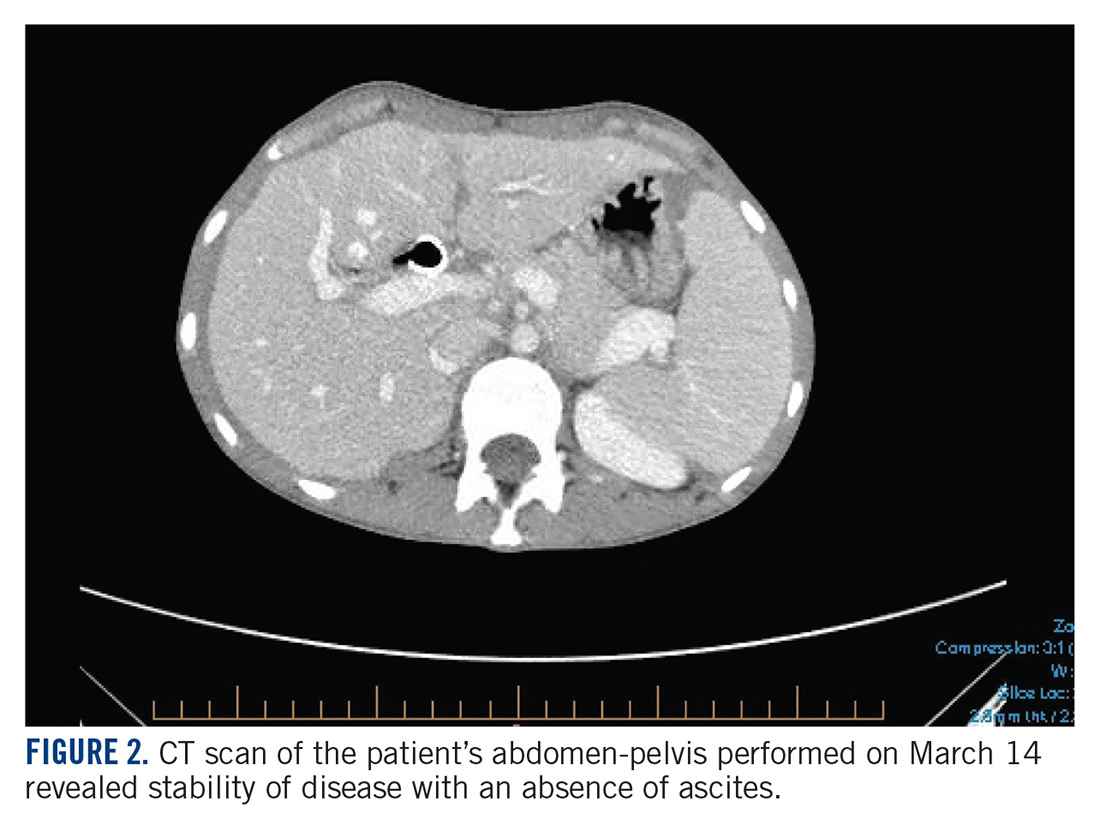
The patient’s clinical status subsequently improved, with significant reduction in her chronic abdominal pain and very minimal side effects. Clinically, the patient’s acute kidney injury resolved (from a creatinine of 272 μmol/L at discharge to 85 μmol/L after a week of treatment) and her liver enzymes normalized (from an alkaline phosphatase of 301 U/L to 83 U/L, and alanine transaminase of 111 U/L to 38 U/L). CT scan of her chest and abdomen, which was performed 1 month post treatment, revealed stability of disease with absence of ascites (FIGURE 2). The patient continued to have a good response to treatment for 6 months, with no recurrence of pain or ascites.
Six months later, in July 2018, the patient developed increasing pain and a CT scan revealed worsening of abdominopelvic carcinomatosis. In this context, vismodegib was discontinued on July 17. In the next 5 months, she went on to receive carboplatin and paclitaxel, gemcitabine, and nivolumab consecutively with no response. She was admitted to hospital on December 30 for a pain crisis. She passed away on January 9, 2019, from fecal peritonitis.
Discussion
To the best of our knowledge, this is the first patient with metastatic sarcoma to receive vismodegib, a Hedgehog signaling pathway inhibitor. She achieved an excellent clinical response with progression- free disease for approximately 6 months after starting treatment.
There is no current standard second- line treatment for metastatic soft tissue sarcoma. The choice of systemic therapy is histology-driven and therefore treatment is individualized for each patient. The future of oncology is heading towards an even more personalized approach with molecular profiling. Our case report highlights the relevance of genomic testing and targeted therapies, especially in cases of diverse clinical and biological disease behavior.
Molecular targeting is even more necessary in patients with advanced cancer who have failed multiple lines of treatment. As in our study, these patients can obtain a significant response with meaningful improvement in their quality of life. Future research is currently focusing on identifying new molecular targets in patients with advanced refractory cancers. Further studies will need to be done to determine whether these molecular targeting agents, such as vismodegib, lead to significant outcome changes in these patients.
1. Collini P, Sorensen PHB, Patel S, et al. Sarcomas with spindle cell morphology. Semin Oncol. 2009;36(4):324-337.
2. Frezza AM, Stacchiotti S, Gronchi A. Systemic treatment in advanced soft tissue sarcoma: what is standard, what is new. BMC Med. 2017;15(1):109.
3. Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properties and tumor microenvironment. Mol Cancer. 2016;15:24.
4. Abidi A. Hedgehog signaling pathway: a novel target for cancer therapy: vismodegib, a promising therapeutic option in treatment of basal cell carcinomas. Indian J Pharmacol. 2014;46(1): 3-12.
5. Belyea B, Kephart JG, Blum J, Kirsch DG, Linardic CM. Embryonic signaling pathways and rhabdomyosarcoma: contributions to cancer development and opportunities for therapeutic targeting. Sarcoma. 2012;2012:13.
6. Yao Z, Han L, Chen Y, et al. Hedgehog signalling in the tumourigenesis and metastasis of osteosarcoma, and its potential value in the clinical therapy of osteosarcoma. Cell Death Dis. 2018;9(6):701.
7. Italiano A, Le Cesne A, Bellera C, et al. GDC- 0449 in patients with advanced chondrosarcomas: a French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann Oncol. 2013;24(11):2922-2926.
8. Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from MyPathway, an open-label, phase IIa multiple basket study. J Clin Oncol. 2018;36(6): 536-542.
Spindle cell sarcomas are part of a rare, heterogeneous family of connective tissue tumors. These tumors are primarily treated with surgery and have a high risk of recurrence and distant metastasis with elevated mortality rates.1 Other than the evidence for first-line therapy with doxorubicin in advanced soft tissue sarcoma, little evidence exists to point to an optimal second-line therapy. This is due to the diversity of soft tissue sarcomas, which encompass approximately 70 different histologic subtypes that can each respond differently to treatment.2 As such, newer strategies, including immunotherapy and targeted molecular drugs, are being developed.
Quiescent in most adult tissues, the Hedgehog signaling pathway, when inappropriately activated, has been implicated in the development of multiple types of cancers, including basal cell, breast, prostate, hepatocellular, pancreatic, and brain cancer.3 The Hedgehog signaling pathway is an important regulator of cell growth and differentiation in early development, but when inappropriately activated can lead to cell proliferation and increased angiogenic factors, decreased apoptosis, and breakdown of tight junctions promoting cancer growth and metastasis.4 Recent data reveal that the Hedgehog pathway plays a specific role in activation of satellite cells, proliferation of myoblasts, and differentiation of skeletal muscle.5 Activation of this embryonic pathway has been implicated in embryonal rhabdoymyosarcoma, osteosarcoma, and chondrosarcoma.5-7
This pathway has recently been recognized as a therapeutic target, with the development of vismodegib, a targeted Hedgehog pathway inhibitor. This novel agent is in active use for treatment of advanced basal cell carcinoma and is currently undergoing trials for various other malignancies. Recently, a phase 2a basket study, called MyPathway, evaluated the use of targeted therapies in 35 different advanced refractory solid tumors harboring specific molecular alterations. Out of 21 patients with mutations in the Hedgehog pathway, 3 had a partial response to vismodegib—one had an unknown primary tumor, another a squamous skin cancer, and the third a salivary gland cancer.8 Vismodegib (GDC-0449) was also evaluated in a phase 2 multicenter clinical trial in patients with progressive advanced chondrosarcoma.7 Although the study did not meet its primary endpoint, the proportion of patients with non-progressive disease was 25.6% at 6 months. Investigators observed that the benefit occurred in the subset of patients with overexpression of the Hedgehog ligand. Genomic studies for mutations in SMO and PTCH genes were available for only 28 and 26 patients, respectively, of the 45 patients enrolled on the trial. While there were no mutations identified, expression data revealed that overexpression of the Hedgehog ligand was present in 65% of cases tested (13 out of 20 patients). In patients with stable disease at 6 months, all had overexpression of the Hedgehog ligand.7 These studies point to the potential use of vismodegib in both bone and soft tissue sarcomas, and more specifically, to the importance of genomic testing in these cases.
Case Presentation and Summary
This report describes the novel use of vismodegib, an oral Hedgehog signaling pathway inhibitor, in the treatment of a patient with metastatic soft tissue sarcoma.
An 18-year-old female with no particular previous illnesses was initially diagnosed with superficial soft tissue sarcoma overlying the right hip in 2013. Due to the complexity of pathology, a second opinion was requested and revealed atypical cellular spindle and epithelioid cells, morphologically and immunohistochemically suggestive of spindle cell sarcoma, not otherwise specified. She underwent negative-margin resection in January 2014. Her course was complicated by two recurrences in the right inguinal lymph nodes in July 2014 and July 2015. She was treated with lymph node dissection in 2014, followed by numerous right lymph node dissections and adjuvant radiation in 2015.
A routine computerized tomography (CT) scan of the thorax-abdomen and pelvis in August 2016 revealed recurrence of disease, with multiple lung nodules as well as metastases in the retroperitoneum. She received 6 cycles of gemcitabine and docetaxel with stability of disease. The patient was then started on a PI3K inhibitor as part of a clinical trial, as genotypic analysis of the tumor revealed an activating mutation of the PI3K gene. The patient’s course was complicated by acute obstructive renal failure requiring a double J stent for right-sided hydronephrosis.
Repeat imaging revealed disease progression, and the patient was then switched to liposomal doxorubicin alone for 4 months and then in combination with olaratumab. She received the combined treatment for a total of 3 months, which was then stopped when she was found to have new peritoneal implants and worsening ascites. At this time, tissue was sent for FoundationOne® next generation sequencing (NGS)-based genomic testing, and the patient received one dose of nivolumab.
In January 2018, 2 days after receiving her first dose of nivolumab, the patient required admission for worsening abdominal pain secondary to progression of her disease (FIGURE 1). She was found to have acute kidney injury on top of chronic kidney disease due to hydronephrosis requiring a left-sided double J stent. She also had transaminitis resulting from a common bile duct stricture treated with a biliary stent and worsening ascites requiring regular paracentesis. This was all in the context of new or growing metastatic implants.
At this time, the result of the FoundationOne genomic testing revealed PTCH1 loss of exons 1-24 and CDKN2A/B loss. Mutation of tumor suppressor gene PTCH1 leads to Hedgehog pathway activation and therefore the patient was started on vismodegib on January 22, 2018. She was discharged from the hospital in stable condition a day later, on January 23.
The patient’s clinical status subsequently improved, with significant reduction in her chronic abdominal pain and very minimal side effects. Clinically, the patient’s acute kidney injury resolved (from a creatinine of 272 μmol/L at discharge to 85 μmol/L after a week of treatment) and her liver enzymes normalized (from an alkaline phosphatase of 301 U/L to 83 U/L, and alanine transaminase of 111 U/L to 38 U/L). CT scan of her chest and abdomen, which was performed 1 month post treatment, revealed stability of disease with absence of ascites (FIGURE 2). The patient continued to have a good response to treatment for 6 months, with no recurrence of pain or ascites.
Six months later, in July 2018, the patient developed increasing pain and a CT scan revealed worsening of abdominopelvic carcinomatosis. In this context, vismodegib was discontinued on July 17. In the next 5 months, she went on to receive carboplatin and paclitaxel, gemcitabine, and nivolumab consecutively with no response. She was admitted to hospital on December 30 for a pain crisis. She passed away on January 9, 2019, from fecal peritonitis.
Discussion
To the best of our knowledge, this is the first patient with metastatic sarcoma to receive vismodegib, a Hedgehog signaling pathway inhibitor. She achieved an excellent clinical response with progression- free disease for approximately 6 months after starting treatment.
There is no current standard second- line treatment for metastatic soft tissue sarcoma. The choice of systemic therapy is histology-driven and therefore treatment is individualized for each patient. The future of oncology is heading towards an even more personalized approach with molecular profiling. Our case report highlights the relevance of genomic testing and targeted therapies, especially in cases of diverse clinical and biological disease behavior.
Molecular targeting is even more necessary in patients with advanced cancer who have failed multiple lines of treatment. As in our study, these patients can obtain a significant response with meaningful improvement in their quality of life. Future research is currently focusing on identifying new molecular targets in patients with advanced refractory cancers. Further studies will need to be done to determine whether these molecular targeting agents, such as vismodegib, lead to significant outcome changes in these patients.
Spindle cell sarcomas are part of a rare, heterogeneous family of connective tissue tumors. These tumors are primarily treated with surgery and have a high risk of recurrence and distant metastasis with elevated mortality rates.1 Other than the evidence for first-line therapy with doxorubicin in advanced soft tissue sarcoma, little evidence exists to point to an optimal second-line therapy. This is due to the diversity of soft tissue sarcomas, which encompass approximately 70 different histologic subtypes that can each respond differently to treatment.2 As such, newer strategies, including immunotherapy and targeted molecular drugs, are being developed.
Quiescent in most adult tissues, the Hedgehog signaling pathway, when inappropriately activated, has been implicated in the development of multiple types of cancers, including basal cell, breast, prostate, hepatocellular, pancreatic, and brain cancer.3 The Hedgehog signaling pathway is an important regulator of cell growth and differentiation in early development, but when inappropriately activated can lead to cell proliferation and increased angiogenic factors, decreased apoptosis, and breakdown of tight junctions promoting cancer growth and metastasis.4 Recent data reveal that the Hedgehog pathway plays a specific role in activation of satellite cells, proliferation of myoblasts, and differentiation of skeletal muscle.5 Activation of this embryonic pathway has been implicated in embryonal rhabdoymyosarcoma, osteosarcoma, and chondrosarcoma.5-7
This pathway has recently been recognized as a therapeutic target, with the development of vismodegib, a targeted Hedgehog pathway inhibitor. This novel agent is in active use for treatment of advanced basal cell carcinoma and is currently undergoing trials for various other malignancies. Recently, a phase 2a basket study, called MyPathway, evaluated the use of targeted therapies in 35 different advanced refractory solid tumors harboring specific molecular alterations. Out of 21 patients with mutations in the Hedgehog pathway, 3 had a partial response to vismodegib—one had an unknown primary tumor, another a squamous skin cancer, and the third a salivary gland cancer.8 Vismodegib (GDC-0449) was also evaluated in a phase 2 multicenter clinical trial in patients with progressive advanced chondrosarcoma.7 Although the study did not meet its primary endpoint, the proportion of patients with non-progressive disease was 25.6% at 6 months. Investigators observed that the benefit occurred in the subset of patients with overexpression of the Hedgehog ligand. Genomic studies for mutations in SMO and PTCH genes were available for only 28 and 26 patients, respectively, of the 45 patients enrolled on the trial. While there were no mutations identified, expression data revealed that overexpression of the Hedgehog ligand was present in 65% of cases tested (13 out of 20 patients). In patients with stable disease at 6 months, all had overexpression of the Hedgehog ligand.7 These studies point to the potential use of vismodegib in both bone and soft tissue sarcomas, and more specifically, to the importance of genomic testing in these cases.
Case Presentation and Summary
This report describes the novel use of vismodegib, an oral Hedgehog signaling pathway inhibitor, in the treatment of a patient with metastatic soft tissue sarcoma.
An 18-year-old female with no particular previous illnesses was initially diagnosed with superficial soft tissue sarcoma overlying the right hip in 2013. Due to the complexity of pathology, a second opinion was requested and revealed atypical cellular spindle and epithelioid cells, morphologically and immunohistochemically suggestive of spindle cell sarcoma, not otherwise specified. She underwent negative-margin resection in January 2014. Her course was complicated by two recurrences in the right inguinal lymph nodes in July 2014 and July 2015. She was treated with lymph node dissection in 2014, followed by numerous right lymph node dissections and adjuvant radiation in 2015.
A routine computerized tomography (CT) scan of the thorax-abdomen and pelvis in August 2016 revealed recurrence of disease, with multiple lung nodules as well as metastases in the retroperitoneum. She received 6 cycles of gemcitabine and docetaxel with stability of disease. The patient was then started on a PI3K inhibitor as part of a clinical trial, as genotypic analysis of the tumor revealed an activating mutation of the PI3K gene. The patient’s course was complicated by acute obstructive renal failure requiring a double J stent for right-sided hydronephrosis.
Repeat imaging revealed disease progression, and the patient was then switched to liposomal doxorubicin alone for 4 months and then in combination with olaratumab. She received the combined treatment for a total of 3 months, which was then stopped when she was found to have new peritoneal implants and worsening ascites. At this time, tissue was sent for FoundationOne® next generation sequencing (NGS)-based genomic testing, and the patient received one dose of nivolumab.
In January 2018, 2 days after receiving her first dose of nivolumab, the patient required admission for worsening abdominal pain secondary to progression of her disease (FIGURE 1). She was found to have acute kidney injury on top of chronic kidney disease due to hydronephrosis requiring a left-sided double J stent. She also had transaminitis resulting from a common bile duct stricture treated with a biliary stent and worsening ascites requiring regular paracentesis. This was all in the context of new or growing metastatic implants.
At this time, the result of the FoundationOne genomic testing revealed PTCH1 loss of exons 1-24 and CDKN2A/B loss. Mutation of tumor suppressor gene PTCH1 leads to Hedgehog pathway activation and therefore the patient was started on vismodegib on January 22, 2018. She was discharged from the hospital in stable condition a day later, on January 23.
The patient’s clinical status subsequently improved, with significant reduction in her chronic abdominal pain and very minimal side effects. Clinically, the patient’s acute kidney injury resolved (from a creatinine of 272 μmol/L at discharge to 85 μmol/L after a week of treatment) and her liver enzymes normalized (from an alkaline phosphatase of 301 U/L to 83 U/L, and alanine transaminase of 111 U/L to 38 U/L). CT scan of her chest and abdomen, which was performed 1 month post treatment, revealed stability of disease with absence of ascites (FIGURE 2). The patient continued to have a good response to treatment for 6 months, with no recurrence of pain or ascites.
Six months later, in July 2018, the patient developed increasing pain and a CT scan revealed worsening of abdominopelvic carcinomatosis. In this context, vismodegib was discontinued on July 17. In the next 5 months, she went on to receive carboplatin and paclitaxel, gemcitabine, and nivolumab consecutively with no response. She was admitted to hospital on December 30 for a pain crisis. She passed away on January 9, 2019, from fecal peritonitis.
Discussion
To the best of our knowledge, this is the first patient with metastatic sarcoma to receive vismodegib, a Hedgehog signaling pathway inhibitor. She achieved an excellent clinical response with progression- free disease for approximately 6 months after starting treatment.
There is no current standard second- line treatment for metastatic soft tissue sarcoma. The choice of systemic therapy is histology-driven and therefore treatment is individualized for each patient. The future of oncology is heading towards an even more personalized approach with molecular profiling. Our case report highlights the relevance of genomic testing and targeted therapies, especially in cases of diverse clinical and biological disease behavior.
Molecular targeting is even more necessary in patients with advanced cancer who have failed multiple lines of treatment. As in our study, these patients can obtain a significant response with meaningful improvement in their quality of life. Future research is currently focusing on identifying new molecular targets in patients with advanced refractory cancers. Further studies will need to be done to determine whether these molecular targeting agents, such as vismodegib, lead to significant outcome changes in these patients.
1. Collini P, Sorensen PHB, Patel S, et al. Sarcomas with spindle cell morphology. Semin Oncol. 2009;36(4):324-337.
2. Frezza AM, Stacchiotti S, Gronchi A. Systemic treatment in advanced soft tissue sarcoma: what is standard, what is new. BMC Med. 2017;15(1):109.
3. Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properties and tumor microenvironment. Mol Cancer. 2016;15:24.
4. Abidi A. Hedgehog signaling pathway: a novel target for cancer therapy: vismodegib, a promising therapeutic option in treatment of basal cell carcinomas. Indian J Pharmacol. 2014;46(1): 3-12.
5. Belyea B, Kephart JG, Blum J, Kirsch DG, Linardic CM. Embryonic signaling pathways and rhabdomyosarcoma: contributions to cancer development and opportunities for therapeutic targeting. Sarcoma. 2012;2012:13.
6. Yao Z, Han L, Chen Y, et al. Hedgehog signalling in the tumourigenesis and metastasis of osteosarcoma, and its potential value in the clinical therapy of osteosarcoma. Cell Death Dis. 2018;9(6):701.
7. Italiano A, Le Cesne A, Bellera C, et al. GDC- 0449 in patients with advanced chondrosarcomas: a French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann Oncol. 2013;24(11):2922-2926.
8. Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from MyPathway, an open-label, phase IIa multiple basket study. J Clin Oncol. 2018;36(6): 536-542.
1. Collini P, Sorensen PHB, Patel S, et al. Sarcomas with spindle cell morphology. Semin Oncol. 2009;36(4):324-337.
2. Frezza AM, Stacchiotti S, Gronchi A. Systemic treatment in advanced soft tissue sarcoma: what is standard, what is new. BMC Med. 2017;15(1):109.
3. Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properties and tumor microenvironment. Mol Cancer. 2016;15:24.
4. Abidi A. Hedgehog signaling pathway: a novel target for cancer therapy: vismodegib, a promising therapeutic option in treatment of basal cell carcinomas. Indian J Pharmacol. 2014;46(1): 3-12.
5. Belyea B, Kephart JG, Blum J, Kirsch DG, Linardic CM. Embryonic signaling pathways and rhabdomyosarcoma: contributions to cancer development and opportunities for therapeutic targeting. Sarcoma. 2012;2012:13.
6. Yao Z, Han L, Chen Y, et al. Hedgehog signalling in the tumourigenesis and metastasis of osteosarcoma, and its potential value in the clinical therapy of osteosarcoma. Cell Death Dis. 2018;9(6):701.
7. Italiano A, Le Cesne A, Bellera C, et al. GDC- 0449 in patients with advanced chondrosarcomas: a French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann Oncol. 2013;24(11):2922-2926.
8. Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from MyPathway, an open-label, phase IIa multiple basket study. J Clin Oncol. 2018;36(6): 536-542.
Red patches on the tongue with white borders • history of geographic tongue • incompletely treated celiac disease • Dx?
THE CASE
A 49-year-old woman presented to our clinic with concerns about the changing appearance of her tongue over the past 2 to 3 weeks. She had been given a diagnosis of celiac disease by her gastroenterologist approximately 5 years earlier. At the time of that diagnosis, she had smooth patches on the surface of her tongue with missing papillae and slightly raised borders. (This gave her tongue a map-like appearance, consistent with geographic tongue [GT].) The patient’s symptoms improved after she started a gluten-free diet, but she reported occasional noncompliance over the past year.
At the current presentation, the patient noted that new lesions on the tongue had started as diffuse shiny red patches surrounded by clearly delineated white borders, ultimately progressing to structural changes. She denied any burning of the tongue or other oral symptoms but reported feelings of anxiety, a “foggy mind,” and diffuse arthralgia for the past several weeks. The patient’s list of medications included vitamin D and magnesium supplements, a multivitamin, and probiotics.
On physical examination, her tongue showed areas of shiny erythematous mucosa and deep central grooves with small radiating furrows giving a wrinkled appearance (FIGURE). A review of systems revealed nonspecific abdominal pain including bloating, cramping, and gas for the previous few months. An examination of her throat and oral cavity was unremarkable, and the remainder of the physical examination was normal.

THE DIAGNOSIS
A diagnosis of fissured tongue (FT) was suspected based on the clinical appearance of the patient’s tongue. Laboratory studies including a complete blood count; antinuclear antibody test; rheumatoid factor test; anticyclic citrullinated peptide test; a comprehensive metabolic panel; and thyroid-stimulating hormone, 25-hydroxyvitamin D, and vitamin B₁₂ level tests were performed based on her symptoms and current medications to rule out any other potential diagnoses. All laboratory results were normal, and a tissue transglutaminase IgA test was not repeated because it was positive when previously tested by the gastroenterologist at the time of her celiac disease diagnosis. A diagnosis of FT due to incompletely treated celiac disease was confirmed.
DISCUSSION
Clinical presentation. FT commonly presents in association with GT,1,2 with some cases of GT naturally progressing to FT.3,4 In most cases, FT is asymptomatic unless debris becomes entrapped in the fissures. Rarely, patients may complain of a burning sensation on the tongue. The clinical appearance of the tongue includes deep grooves with possible malodor or halitosis along with discoloration if trapping of debris and subsequent inflammation occurs.1
Etiology. FT has been linked to celiac disease; systemic conditions such as arthritis, iron deficiency, depression, anxiety, and neuropathy; and poor oral hygiene. Genetics also may play a role, as some cases of FT may be inherited. Getting to the source requires a careful history to uncover signs and symptoms (that may not have been reported until now) and to determine if other family members also have FT. A careful examination of the oral cavity, with an eye toward the patient’s oral hygiene, is also instructive (TABLE).5-8 In general, FT is believed to be a normal tongue variant in less than 10% of the general population.5,6 Additionally, local factors such as ill-fitting prosthesis, infection, parafunctional habits, allergic reaction, xerostomia, and galvanism have been implicated in the etiology of FT.5

In our patient, progression of GT to FT was caused by incompletely treated celiac disease. Both FT and GT may represent different reaction patterns caused by the same hematologic and immunologic diseases.3 In fact, the appearance of the tongue may aid in the diagnosis of celiac disease, which has been observed in 15% of patients with GT.7 Fissured tongue also may indicate an inability of the gastrointestinal mucosa to absorb nutrients; therefore, close nutrition monitoring is recommended.9
Continue to: Other oral and dental manifestations...
Other oral and dental manifestations of celiac disease include enamel defects, delayed tooth eruption, recurrent aphthous ulcers, cheilosis, oral lichen planus, and atrophic glossitis.10 Our patient also reported anxiety, “foggy mind,” diffuse arthralgia, and abdominal pain, which are symptoms of uncontrolled celiac disease. There is no known etiology of tongue manifestations in patients with incompletely treated celiac disease.
Treatment. FT generally does not require specific therapy other than the treatment of the underlying inflammatory condition. It is important to maintain proper oral and dental care, such as brushing the top surface of the tongue to clean and remove food debris. Bacteria and plaque can collect in the fissures, leading to bad breath and an increased potential for tooth decay.
Our patient was referred to a dietitian to assist with adherence to the gluten-free diet. At follow-up 3 months later, the appearance of her tongue had improved and fewer fissures were visible. The majority of her other symptoms also had resolved.
THE TAKEAWAY
FT may be a normal variant of the tongue in some patients or may be associated with poor oral hygiene. Additionally, FT often is associated with an underlying medical or inherited condition and may serve as a marker for an untreated or partially treated condition such as celiac disease, as was the case with our patient. When other signs or symptoms of systemic disease are present, further laboratory and endoscopic workup is necessary to rule out other causes and to diagnose celiac disease, if present.
As FT has been reported to be a natural progression from GT, the appearance of FT may indicate partial treatment of the underlying disease process and therefore more intensive therapy and follow-up would be needed. In this case, more intensive dietary guidance was provided with subsequent improvement of symptoms.
CORRESPONDENCE
Peter J. Carek, MD, MS, Department of Community Health and Family Medicine, College of Medicine, University of Florida, P.O. Box 100237, Gainesville, FL 32610-0237; [email protected]
1. Reamy BV, Cerby R, Bunt CW. Common tongue conditions in primary care. Am Fam Physician. 2010;81:627-634.
2. Yarom N, Cantony U, Gorsky M. Prevalence of fissured tongue, geographic tongue and median rhomboid glossitis among Israeli adults of different ethnic origins. Dermatology. 2004;209:88-94.
3. Dafar A, Cevik-Aras H, Robledo-Sierra J, et al. Factors associated with geographic tongue and fissured tongue. Acta Odontol Scad. 2016;74:210-216.
4. Hume WJ. Geographic stomatitis: a critical review. J Dent. 1975;3:25-43.
5. Sudarshan R, Sree Vijayabala G, Samata Y, et al. Newer classification system for fissured tongue: an epidemiological approach. J Tropical Med. doi:10.1155/2015/262079.
6. Mangold AR, Torgerson RR, Rogers RS. Diseases of the tongue. Clin Dermatol. 2016;34:458-469.
7. Cigic L, Galic T, Kero D, et al. The prevalence of celiac disease in patients with geographic tongue. J Oral Pathol Med. 2016;45:791-796.
8. Zargari O. The prevalence and significance of fissured tongue and geographical tongue in psoriatic patients. Clin Exp Dermatology. 2006;31:192-195.
9. Kullaa-Mikkonen A, Penttila I, Kotilainen R, et al. Haematological and immunological features of patients with fissured tongue syndrome. Br J Oral Maxillofac Surg. 1987;25:481-487.
10. Rashid M, Zarkadas M, Anca A, et al. Oral manifestations of celiac disease: a clinical guide for dentists. J Can Dent Assoc. 2011;77:b39.
THE CASE
A 49-year-old woman presented to our clinic with concerns about the changing appearance of her tongue over the past 2 to 3 weeks. She had been given a diagnosis of celiac disease by her gastroenterologist approximately 5 years earlier. At the time of that diagnosis, she had smooth patches on the surface of her tongue with missing papillae and slightly raised borders. (This gave her tongue a map-like appearance, consistent with geographic tongue [GT].) The patient’s symptoms improved after she started a gluten-free diet, but she reported occasional noncompliance over the past year.
At the current presentation, the patient noted that new lesions on the tongue had started as diffuse shiny red patches surrounded by clearly delineated white borders, ultimately progressing to structural changes. She denied any burning of the tongue or other oral symptoms but reported feelings of anxiety, a “foggy mind,” and diffuse arthralgia for the past several weeks. The patient’s list of medications included vitamin D and magnesium supplements, a multivitamin, and probiotics.
On physical examination, her tongue showed areas of shiny erythematous mucosa and deep central grooves with small radiating furrows giving a wrinkled appearance (FIGURE). A review of systems revealed nonspecific abdominal pain including bloating, cramping, and gas for the previous few months. An examination of her throat and oral cavity was unremarkable, and the remainder of the physical examination was normal.
THE DIAGNOSIS
A diagnosis of fissured tongue (FT) was suspected based on the clinical appearance of the patient’s tongue. Laboratory studies including a complete blood count; antinuclear antibody test; rheumatoid factor test; anticyclic citrullinated peptide test; a comprehensive metabolic panel; and thyroid-stimulating hormone, 25-hydroxyvitamin D, and vitamin B₁₂ level tests were performed based on her symptoms and current medications to rule out any other potential diagnoses. All laboratory results were normal, and a tissue transglutaminase IgA test was not repeated because it was positive when previously tested by the gastroenterologist at the time of her celiac disease diagnosis. A diagnosis of FT due to incompletely treated celiac disease was confirmed.
DISCUSSION
Clinical presentation. FT commonly presents in association with GT,1,2 with some cases of GT naturally progressing to FT.3,4 In most cases, FT is asymptomatic unless debris becomes entrapped in the fissures. Rarely, patients may complain of a burning sensation on the tongue. The clinical appearance of the tongue includes deep grooves with possible malodor or halitosis along with discoloration if trapping of debris and subsequent inflammation occurs.1
Etiology. FT has been linked to celiac disease; systemic conditions such as arthritis, iron deficiency, depression, anxiety, and neuropathy; and poor oral hygiene. Genetics also may play a role, as some cases of FT may be inherited. Getting to the source requires a careful history to uncover signs and symptoms (that may not have been reported until now) and to determine if other family members also have FT. A careful examination of the oral cavity, with an eye toward the patient’s oral hygiene, is also instructive (TABLE).5-8 In general, FT is believed to be a normal tongue variant in less than 10% of the general population.5,6 Additionally, local factors such as ill-fitting prosthesis, infection, parafunctional habits, allergic reaction, xerostomia, and galvanism have been implicated in the etiology of FT.5
In our patient, progression of GT to FT was caused by incompletely treated celiac disease. Both FT and GT may represent different reaction patterns caused by the same hematologic and immunologic diseases.3 In fact, the appearance of the tongue may aid in the diagnosis of celiac disease, which has been observed in 15% of patients with GT.7 Fissured tongue also may indicate an inability of the gastrointestinal mucosa to absorb nutrients; therefore, close nutrition monitoring is recommended.9
Continue to: Other oral and dental manifestations...
Other oral and dental manifestations of celiac disease include enamel defects, delayed tooth eruption, recurrent aphthous ulcers, cheilosis, oral lichen planus, and atrophic glossitis.10 Our patient also reported anxiety, “foggy mind,” diffuse arthralgia, and abdominal pain, which are symptoms of uncontrolled celiac disease. There is no known etiology of tongue manifestations in patients with incompletely treated celiac disease.
Treatment. FT generally does not require specific therapy other than the treatment of the underlying inflammatory condition. It is important to maintain proper oral and dental care, such as brushing the top surface of the tongue to clean and remove food debris. Bacteria and plaque can collect in the fissures, leading to bad breath and an increased potential for tooth decay.
Our patient was referred to a dietitian to assist with adherence to the gluten-free diet. At follow-up 3 months later, the appearance of her tongue had improved and fewer fissures were visible. The majority of her other symptoms also had resolved.
THE TAKEAWAY
FT may be a normal variant of the tongue in some patients or may be associated with poor oral hygiene. Additionally, FT often is associated with an underlying medical or inherited condition and may serve as a marker for an untreated or partially treated condition such as celiac disease, as was the case with our patient. When other signs or symptoms of systemic disease are present, further laboratory and endoscopic workup is necessary to rule out other causes and to diagnose celiac disease, if present.
As FT has been reported to be a natural progression from GT, the appearance of FT may indicate partial treatment of the underlying disease process and therefore more intensive therapy and follow-up would be needed. In this case, more intensive dietary guidance was provided with subsequent improvement of symptoms.
CORRESPONDENCE
Peter J. Carek, MD, MS, Department of Community Health and Family Medicine, College of Medicine, University of Florida, P.O. Box 100237, Gainesville, FL 32610-0237; [email protected]
THE CASE
A 49-year-old woman presented to our clinic with concerns about the changing appearance of her tongue over the past 2 to 3 weeks. She had been given a diagnosis of celiac disease by her gastroenterologist approximately 5 years earlier. At the time of that diagnosis, she had smooth patches on the surface of her tongue with missing papillae and slightly raised borders. (This gave her tongue a map-like appearance, consistent with geographic tongue [GT].) The patient’s symptoms improved after she started a gluten-free diet, but she reported occasional noncompliance over the past year.
At the current presentation, the patient noted that new lesions on the tongue had started as diffuse shiny red patches surrounded by clearly delineated white borders, ultimately progressing to structural changes. She denied any burning of the tongue or other oral symptoms but reported feelings of anxiety, a “foggy mind,” and diffuse arthralgia for the past several weeks. The patient’s list of medications included vitamin D and magnesium supplements, a multivitamin, and probiotics.
On physical examination, her tongue showed areas of shiny erythematous mucosa and deep central grooves with small radiating furrows giving a wrinkled appearance (FIGURE). A review of systems revealed nonspecific abdominal pain including bloating, cramping, and gas for the previous few months. An examination of her throat and oral cavity was unremarkable, and the remainder of the physical examination was normal.
THE DIAGNOSIS
A diagnosis of fissured tongue (FT) was suspected based on the clinical appearance of the patient’s tongue. Laboratory studies including a complete blood count; antinuclear antibody test; rheumatoid factor test; anticyclic citrullinated peptide test; a comprehensive metabolic panel; and thyroid-stimulating hormone, 25-hydroxyvitamin D, and vitamin B₁₂ level tests were performed based on her symptoms and current medications to rule out any other potential diagnoses. All laboratory results were normal, and a tissue transglutaminase IgA test was not repeated because it was positive when previously tested by the gastroenterologist at the time of her celiac disease diagnosis. A diagnosis of FT due to incompletely treated celiac disease was confirmed.
DISCUSSION
Clinical presentation. FT commonly presents in association with GT,1,2 with some cases of GT naturally progressing to FT.3,4 In most cases, FT is asymptomatic unless debris becomes entrapped in the fissures. Rarely, patients may complain of a burning sensation on the tongue. The clinical appearance of the tongue includes deep grooves with possible malodor or halitosis along with discoloration if trapping of debris and subsequent inflammation occurs.1
Etiology. FT has been linked to celiac disease; systemic conditions such as arthritis, iron deficiency, depression, anxiety, and neuropathy; and poor oral hygiene. Genetics also may play a role, as some cases of FT may be inherited. Getting to the source requires a careful history to uncover signs and symptoms (that may not have been reported until now) and to determine if other family members also have FT. A careful examination of the oral cavity, with an eye toward the patient’s oral hygiene, is also instructive (TABLE).5-8 In general, FT is believed to be a normal tongue variant in less than 10% of the general population.5,6 Additionally, local factors such as ill-fitting prosthesis, infection, parafunctional habits, allergic reaction, xerostomia, and galvanism have been implicated in the etiology of FT.5
In our patient, progression of GT to FT was caused by incompletely treated celiac disease. Both FT and GT may represent different reaction patterns caused by the same hematologic and immunologic diseases.3 In fact, the appearance of the tongue may aid in the diagnosis of celiac disease, which has been observed in 15% of patients with GT.7 Fissured tongue also may indicate an inability of the gastrointestinal mucosa to absorb nutrients; therefore, close nutrition monitoring is recommended.9
Continue to: Other oral and dental manifestations...
Other oral and dental manifestations of celiac disease include enamel defects, delayed tooth eruption, recurrent aphthous ulcers, cheilosis, oral lichen planus, and atrophic glossitis.10 Our patient also reported anxiety, “foggy mind,” diffuse arthralgia, and abdominal pain, which are symptoms of uncontrolled celiac disease. There is no known etiology of tongue manifestations in patients with incompletely treated celiac disease.
Treatment. FT generally does not require specific therapy other than the treatment of the underlying inflammatory condition. It is important to maintain proper oral and dental care, such as brushing the top surface of the tongue to clean and remove food debris. Bacteria and plaque can collect in the fissures, leading to bad breath and an increased potential for tooth decay.
Our patient was referred to a dietitian to assist with adherence to the gluten-free diet. At follow-up 3 months later, the appearance of her tongue had improved and fewer fissures were visible. The majority of her other symptoms also had resolved.
THE TAKEAWAY
FT may be a normal variant of the tongue in some patients or may be associated with poor oral hygiene. Additionally, FT often is associated with an underlying medical or inherited condition and may serve as a marker for an untreated or partially treated condition such as celiac disease, as was the case with our patient. When other signs or symptoms of systemic disease are present, further laboratory and endoscopic workup is necessary to rule out other causes and to diagnose celiac disease, if present.
As FT has been reported to be a natural progression from GT, the appearance of FT may indicate partial treatment of the underlying disease process and therefore more intensive therapy and follow-up would be needed. In this case, more intensive dietary guidance was provided with subsequent improvement of symptoms.
CORRESPONDENCE
Peter J. Carek, MD, MS, Department of Community Health and Family Medicine, College of Medicine, University of Florida, P.O. Box 100237, Gainesville, FL 32610-0237; [email protected]
1. Reamy BV, Cerby R, Bunt CW. Common tongue conditions in primary care. Am Fam Physician. 2010;81:627-634.
2. Yarom N, Cantony U, Gorsky M. Prevalence of fissured tongue, geographic tongue and median rhomboid glossitis among Israeli adults of different ethnic origins. Dermatology. 2004;209:88-94.
3. Dafar A, Cevik-Aras H, Robledo-Sierra J, et al. Factors associated with geographic tongue and fissured tongue. Acta Odontol Scad. 2016;74:210-216.
4. Hume WJ. Geographic stomatitis: a critical review. J Dent. 1975;3:25-43.
5. Sudarshan R, Sree Vijayabala G, Samata Y, et al. Newer classification system for fissured tongue: an epidemiological approach. J Tropical Med. doi:10.1155/2015/262079.
6. Mangold AR, Torgerson RR, Rogers RS. Diseases of the tongue. Clin Dermatol. 2016;34:458-469.
7. Cigic L, Galic T, Kero D, et al. The prevalence of celiac disease in patients with geographic tongue. J Oral Pathol Med. 2016;45:791-796.
8. Zargari O. The prevalence and significance of fissured tongue and geographical tongue in psoriatic patients. Clin Exp Dermatology. 2006;31:192-195.
9. Kullaa-Mikkonen A, Penttila I, Kotilainen R, et al. Haematological and immunological features of patients with fissured tongue syndrome. Br J Oral Maxillofac Surg. 1987;25:481-487.
10. Rashid M, Zarkadas M, Anca A, et al. Oral manifestations of celiac disease: a clinical guide for dentists. J Can Dent Assoc. 2011;77:b39.
1. Reamy BV, Cerby R, Bunt CW. Common tongue conditions in primary care. Am Fam Physician. 2010;81:627-634.
2. Yarom N, Cantony U, Gorsky M. Prevalence of fissured tongue, geographic tongue and median rhomboid glossitis among Israeli adults of different ethnic origins. Dermatology. 2004;209:88-94.
3. Dafar A, Cevik-Aras H, Robledo-Sierra J, et al. Factors associated with geographic tongue and fissured tongue. Acta Odontol Scad. 2016;74:210-216.
4. Hume WJ. Geographic stomatitis: a critical review. J Dent. 1975;3:25-43.
5. Sudarshan R, Sree Vijayabala G, Samata Y, et al. Newer classification system for fissured tongue: an epidemiological approach. J Tropical Med. doi:10.1155/2015/262079.
6. Mangold AR, Torgerson RR, Rogers RS. Diseases of the tongue. Clin Dermatol. 2016;34:458-469.
7. Cigic L, Galic T, Kero D, et al. The prevalence of celiac disease in patients with geographic tongue. J Oral Pathol Med. 2016;45:791-796.
8. Zargari O. The prevalence and significance of fissured tongue and geographical tongue in psoriatic patients. Clin Exp Dermatology. 2006;31:192-195.
9. Kullaa-Mikkonen A, Penttila I, Kotilainen R, et al. Haematological and immunological features of patients with fissured tongue syndrome. Br J Oral Maxillofac Surg. 1987;25:481-487.
10. Rashid M, Zarkadas M, Anca A, et al. Oral manifestations of celiac disease: a clinical guide for dentists. J Can Dent Assoc. 2011;77:b39.

8-year-old boy • palpable purpura on the legs with arthralgia • absence of coagulopathy • upper respiratory infection • Dx?
THE CASE
An 8-year-old boy presented to his family physician (FP) with pharyngitis, nasal drainage, and a dry cough of 3 days’ duration. He denied any fever, chills, vomiting, or diarrhea. He had no sick contacts or prior history of streptococcal pharyngitis, but a rapid strep test was positive. No throat culture was performed at this time. The patient was started on amoxicillin 250 mg 3 times daily for 10 days.
On Day 7 of symptoms, the patient presented to the emergency department with elbow and knee pain, as well as mild swelling and purpura of his legs of 3 days’ duration. He was normotensive and reported no abdominal pain. A laboratory workup, including a complete blood cell count and differential, prothrombin time, partial thromboplastin time, comprehensive metabolic panel, creatinine kinase test, urinalysis, and chest radiograph, was normal, but his erythrocyte sedimentation rate (ESR) was mildly elevated at 22 mm/h (reference range, 0–20 mm/h). The patient was discharged on acetaminophen 15 mg/kg every 4 hours as needed for pain.
THE DIAGNOSIS
Based on the distinctive palpable purpura on the legs, arthralgia, upper respiratory infection, and lack of thrombocytopenia and coagulopathy, a presumptive diagnosis of Henoch-Schönlein purpura (HSP) was made.
On Day 9 of symptoms, the patient returned to his FP’s office because the arthralgia persisted in his ankles, knees, and hips. He had developed lower back pain, but the pharyngitis and upper respiratory symptoms had resolved. On physical examination, he was normotensive with a normal abdominal exam. The patient reported that it hurt to move his wrists, hands, elbows, shoulders, knees, and ankles. He also had mild swelling in his left wrist, hand, and ankle. The paraspinal muscles in the lower thoracic and lumbar back were mildly tender to palpation. A complete metabolic panel and urinalysis were normal. Dermatologic examination revealed discrete purpuric lesions ranging from 1 to 8 mm in diameter on the child’s shins, thighs, and buttocks. Urinalysis, blood urea nitrogen, and creatinine kinase were normal. His ESR remained mildly elevated at 24 mm/h. Since there was no evidence of glomerulonephritis, ibuprofen 10 mg/kg every 8 hours as needed was added for pain management.
The child was brought back to his FP on Day 18 for a scheduled follow-up visit. The parents reported that his arthralgia was improved during the day, but by the evening, his knees and ankles hurt so much that they had to carry him to the bathroom. On physical examination, he still had palpable purpura of the legs. There was no swelling, but his joints were still tender to palpation. His parents were reminded to give him ibuprofen after school to control evening pain. Over the next 2 weeks, the patient showed gradual improvement, and by Day 33 the rash and all of the associated symptoms had resolved.
DISCUSSION
Clinical presentation. HSP is an IgA immune complex vasculitis in which abnormal glycosylation of IgA creates large immune complexes that are deposited in the walls of the skin capillaries and arterioles. The primary clinical finding in HSP is a distinctive nonthrombocytopenic purpuric rash that is not associated with coagulopathy and is characterized by reddish purple macules that progress to palpable purpura with petechiae (

A preceding upper respiratory infection has been found in 37% of patients,1 and in patients with renal complications, 20% to 50% have been found to have a group A Streptococcus infection.2 Other associations include food allergies, cold exposure, insect bites, and drug allergies.
Continue to: HSP vasculitis causes...
HSP vasculitis causes abdominal pain in 50% to 75% of patients due to proximal small-bowel submucosal hemorrhage and bowel wall edema.3 In children with HSP, 20% to 55% have been shown to develop renal disease,4 which can range in severity from microscopic hematuria to nephrotic syndrome.3 To ensure prompt treatment of renal manifestations, renal function should be monitored regularly via blood pressure and urinalysis during the course of HSP and after resolution. Renal disease associated with HSP can be acute or chronic.
This case was different because our patient did not exhibit all elements of the classic tetrad of HSP, which includes the characteristic rash, abdominal pain, renal involvement, and arthralgia.
Incidence. HSP is more common in children than adults, with average annual incidence rates of 20/100,000 and 70/100,000 in children in the United States and Asia, respectively.5 While 90% of HSP cases occur in children < 10 years, the peak incidence is at 6 years of age.6 Complications from HSP are more common in adults than in children.7 Caucasian and Asian populations have a 3- to 4-times higher prevalence of HSP than black populations. The male-to-female ratio is 2 to 1.6
The diagnosis of HSP is usually made clinically, based on the distinctive rash, which typically is symmetrical, involving the buttocks, lower legs, elbows, and/or knees. HSP also can be confirmed via skin biopsy and/or direct immunofluorescence, which can identify the presence of IgA in the vessel walls.
The presence of 3 or more of the following criteria also suggests HSP: palpable purpura, bowel angina, gastrointestinal (GI) bleeding, hematuria, ≤ 20 years of age at onset, and no medications prior to presentation of symptoms (87% of cases correctly classified). Fewer than 3 of these factors favor hypersensitivity vasculitis (74% of cases correctly classified).8
Continue to: The differential diagnosis
The differential diagnosis for HSP includes polyarteritis nodosa, a vasculitis with a different characteristic rash; acute abdomen, distinguished by the absence of purpura or arthralgia; meningococcemia, in which fever and meningeal signs may occur; hypersensitivity vasculitis, which arises due to prior exposure to medications or food allergens; and thrombocytopenic purpura, which is characterized by low platelet count.9
Treatment focuses on pain management
In the absence of renal disease, HSP commonly is treated with naproxen for pain management (dosage for children < 2 years of age: 5-7 mg/kg orally every 8-12 hours; dosage for children ≥ 2 years of age, adolescents, and adults: 10-20 mg/kg/d divided into 2 doses; maximum adolescent and adult dose is 1500 mg/d for 3 days followed by a maximum of 1000 mg/d thereafter).
For patients of all ages with severe pain and those with GI effects limiting oral intake of medication, use oral prednisone (1-2 mg/kg/d [maximum dose, 60-80 mg/d]) or intravenous methylprednisolone (0.8-1.6 mg/kg/d [maximum dose, 64 mg/d). Glucocorticoids may then be tapered slowly over 4 to 8 weeks to avoid rebound since they help with inflammation but do not shorten the course of disease. Steroids can ease GI and joint symptoms in HSP but will not improve the rash.
THE TAKEAWAY
The classic tetrad of HSP includes the characteristic rash, abdominal pain, renal involvement, and arthralgia. Diagnosis usually is made clinically, but skin biopsy and direct immunofluorescence can confirm small vessel vasculitis with IgA deposits. More severe manifestations of HSP such as renal disease, hemorrhage, severe anemia, signs of intestinal obstruction, or peritonitis require rapid subspecialty referral.
CORRESPONDENCE
Rachel Bramson, MD, Department of Primary Care, Baylor Scott and White Health, University Clinic, 1700 University Drive, College Station, TX 77840; [email protected]
1. Rigante D, Castellazzi L, Bosco A, et al. Is there a crossroad between infections, genetics, and Henoch-Schönlein purpura? Autoimmun Rev. 2013;12:1016-1021.
2. LaConti JJ, Donet JA, Cho-Vega JH, et al. Henoch-Schönlein Purpura with adalimumab therapy for ulcerative colitis: a case report and review of the literature [published online July 27, 2016]. Case Rep Rheumatol. 2016;2016:2812980.
3. Trnka P. Henoch-Schönlein purpura in children. J Paediatr Child Health. 2013;49:995-1003.
4. Audemard-Verger A, Pillebout E, Guillevin L, et al. IgA vasculitis (Henoch-Shönlein purpura) in adults: diagnostic and therapeutic aspects. Autoimmun Rev. 2015;14:579-585.
5. Chen J, Mao J. Henoch-Schönlein purpura nephritis in children: incidence, pathogenesis and management. World J Pediatr. 2015;11:29-34.
6. Michel B, Hunder G, Bloch D, et al. Hypersensitivity vasculitis and Henoch-Schönlein purpura: a comparison between the 2 disorders. J Rheumatol. 1992;19:721-728.
7. Reamy BV, Williams PM, Lindsay TJ. Henoch-Schönlein purpura. Am Fam Physician. 2009;80:697-704.
8. Yang YH, Yu HH, Chiang BL. The diagnosis and classification of Henoch-Schönlein purpura: an updated review. Autoimmun Rev. 2014;13:355-358.
9. Floege J, Feehally J. Treatment of IgA nephropathy and Henoch-Schönlein nephritis. Nat Rev Nephrol. 2013;9:320-327.
THE CASE
An 8-year-old boy presented to his family physician (FP) with pharyngitis, nasal drainage, and a dry cough of 3 days’ duration. He denied any fever, chills, vomiting, or diarrhea. He had no sick contacts or prior history of streptococcal pharyngitis, but a rapid strep test was positive. No throat culture was performed at this time. The patient was started on amoxicillin 250 mg 3 times daily for 10 days.
On Day 7 of symptoms, the patient presented to the emergency department with elbow and knee pain, as well as mild swelling and purpura of his legs of 3 days’ duration. He was normotensive and reported no abdominal pain. A laboratory workup, including a complete blood cell count and differential, prothrombin time, partial thromboplastin time, comprehensive metabolic panel, creatinine kinase test, urinalysis, and chest radiograph, was normal, but his erythrocyte sedimentation rate (ESR) was mildly elevated at 22 mm/h (reference range, 0–20 mm/h). The patient was discharged on acetaminophen 15 mg/kg every 4 hours as needed for pain.
THE DIAGNOSIS
Based on the distinctive palpable purpura on the legs, arthralgia, upper respiratory infection, and lack of thrombocytopenia and coagulopathy, a presumptive diagnosis of Henoch-Schönlein purpura (HSP) was made.
On Day 9 of symptoms, the patient returned to his FP’s office because the arthralgia persisted in his ankles, knees, and hips. He had developed lower back pain, but the pharyngitis and upper respiratory symptoms had resolved. On physical examination, he was normotensive with a normal abdominal exam. The patient reported that it hurt to move his wrists, hands, elbows, shoulders, knees, and ankles. He also had mild swelling in his left wrist, hand, and ankle. The paraspinal muscles in the lower thoracic and lumbar back were mildly tender to palpation. A complete metabolic panel and urinalysis were normal. Dermatologic examination revealed discrete purpuric lesions ranging from 1 to 8 mm in diameter on the child’s shins, thighs, and buttocks. Urinalysis, blood urea nitrogen, and creatinine kinase were normal. His ESR remained mildly elevated at 24 mm/h. Since there was no evidence of glomerulonephritis, ibuprofen 10 mg/kg every 8 hours as needed was added for pain management.
The child was brought back to his FP on Day 18 for a scheduled follow-up visit. The parents reported that his arthralgia was improved during the day, but by the evening, his knees and ankles hurt so much that they had to carry him to the bathroom. On physical examination, he still had palpable purpura of the legs. There was no swelling, but his joints were still tender to palpation. His parents were reminded to give him ibuprofen after school to control evening pain. Over the next 2 weeks, the patient showed gradual improvement, and by Day 33 the rash and all of the associated symptoms had resolved.
DISCUSSION
Clinical presentation. HSP is an IgA immune complex vasculitis in which abnormal glycosylation of IgA creates large immune complexes that are deposited in the walls of the skin capillaries and arterioles. The primary clinical finding in HSP is a distinctive nonthrombocytopenic purpuric rash that is not associated with coagulopathy and is characterized by reddish purple macules that progress to palpable purpura with petechiae (
A preceding upper respiratory infection has been found in 37% of patients,1 and in patients with renal complications, 20% to 50% have been found to have a group A Streptococcus infection.2 Other associations include food allergies, cold exposure, insect bites, and drug allergies.
Continue to: HSP vasculitis causes...
HSP vasculitis causes abdominal pain in 50% to 75% of patients due to proximal small-bowel submucosal hemorrhage and bowel wall edema.3 In children with HSP, 20% to 55% have been shown to develop renal disease,4 which can range in severity from microscopic hematuria to nephrotic syndrome.3 To ensure prompt treatment of renal manifestations, renal function should be monitored regularly via blood pressure and urinalysis during the course of HSP and after resolution. Renal disease associated with HSP can be acute or chronic.
This case was different because our patient did not exhibit all elements of the classic tetrad of HSP, which includes the characteristic rash, abdominal pain, renal involvement, and arthralgia.
Incidence. HSP is more common in children than adults, with average annual incidence rates of 20/100,000 and 70/100,000 in children in the United States and Asia, respectively.5 While 90% of HSP cases occur in children < 10 years, the peak incidence is at 6 years of age.6 Complications from HSP are more common in adults than in children.7 Caucasian and Asian populations have a 3- to 4-times higher prevalence of HSP than black populations. The male-to-female ratio is 2 to 1.6
The diagnosis of HSP is usually made clinically, based on the distinctive rash, which typically is symmetrical, involving the buttocks, lower legs, elbows, and/or knees. HSP also can be confirmed via skin biopsy and/or direct immunofluorescence, which can identify the presence of IgA in the vessel walls.
The presence of 3 or more of the following criteria also suggests HSP: palpable purpura, bowel angina, gastrointestinal (GI) bleeding, hematuria, ≤ 20 years of age at onset, and no medications prior to presentation of symptoms (87% of cases correctly classified). Fewer than 3 of these factors favor hypersensitivity vasculitis (74% of cases correctly classified).8
Continue to: The differential diagnosis
The differential diagnosis for HSP includes polyarteritis nodosa, a vasculitis with a different characteristic rash; acute abdomen, distinguished by the absence of purpura or arthralgia; meningococcemia, in which fever and meningeal signs may occur; hypersensitivity vasculitis, which arises due to prior exposure to medications or food allergens; and thrombocytopenic purpura, which is characterized by low platelet count.9
Treatment focuses on pain management
In the absence of renal disease, HSP commonly is treated with naproxen for pain management (dosage for children < 2 years of age: 5-7 mg/kg orally every 8-12 hours; dosage for children ≥ 2 years of age, adolescents, and adults: 10-20 mg/kg/d divided into 2 doses; maximum adolescent and adult dose is 1500 mg/d for 3 days followed by a maximum of 1000 mg/d thereafter).
For patients of all ages with severe pain and those with GI effects limiting oral intake of medication, use oral prednisone (1-2 mg/kg/d [maximum dose, 60-80 mg/d]) or intravenous methylprednisolone (0.8-1.6 mg/kg/d [maximum dose, 64 mg/d). Glucocorticoids may then be tapered slowly over 4 to 8 weeks to avoid rebound since they help with inflammation but do not shorten the course of disease. Steroids can ease GI and joint symptoms in HSP but will not improve the rash.
THE TAKEAWAY
The classic tetrad of HSP includes the characteristic rash, abdominal pain, renal involvement, and arthralgia. Diagnosis usually is made clinically, but skin biopsy and direct immunofluorescence can confirm small vessel vasculitis with IgA deposits. More severe manifestations of HSP such as renal disease, hemorrhage, severe anemia, signs of intestinal obstruction, or peritonitis require rapid subspecialty referral.
CORRESPONDENCE
Rachel Bramson, MD, Department of Primary Care, Baylor Scott and White Health, University Clinic, 1700 University Drive, College Station, TX 77840; [email protected]
THE CASE
An 8-year-old boy presented to his family physician (FP) with pharyngitis, nasal drainage, and a dry cough of 3 days’ duration. He denied any fever, chills, vomiting, or diarrhea. He had no sick contacts or prior history of streptococcal pharyngitis, but a rapid strep test was positive. No throat culture was performed at this time. The patient was started on amoxicillin 250 mg 3 times daily for 10 days.
On Day 7 of symptoms, the patient presented to the emergency department with elbow and knee pain, as well as mild swelling and purpura of his legs of 3 days’ duration. He was normotensive and reported no abdominal pain. A laboratory workup, including a complete blood cell count and differential, prothrombin time, partial thromboplastin time, comprehensive metabolic panel, creatinine kinase test, urinalysis, and chest radiograph, was normal, but his erythrocyte sedimentation rate (ESR) was mildly elevated at 22 mm/h (reference range, 0–20 mm/h). The patient was discharged on acetaminophen 15 mg/kg every 4 hours as needed for pain.
THE DIAGNOSIS
Based on the distinctive palpable purpura on the legs, arthralgia, upper respiratory infection, and lack of thrombocytopenia and coagulopathy, a presumptive diagnosis of Henoch-Schönlein purpura (HSP) was made.
On Day 9 of symptoms, the patient returned to his FP’s office because the arthralgia persisted in his ankles, knees, and hips. He had developed lower back pain, but the pharyngitis and upper respiratory symptoms had resolved. On physical examination, he was normotensive with a normal abdominal exam. The patient reported that it hurt to move his wrists, hands, elbows, shoulders, knees, and ankles. He also had mild swelling in his left wrist, hand, and ankle. The paraspinal muscles in the lower thoracic and lumbar back were mildly tender to palpation. A complete metabolic panel and urinalysis were normal. Dermatologic examination revealed discrete purpuric lesions ranging from 1 to 8 mm in diameter on the child’s shins, thighs, and buttocks. Urinalysis, blood urea nitrogen, and creatinine kinase were normal. His ESR remained mildly elevated at 24 mm/h. Since there was no evidence of glomerulonephritis, ibuprofen 10 mg/kg every 8 hours as needed was added for pain management.
The child was brought back to his FP on Day 18 for a scheduled follow-up visit. The parents reported that his arthralgia was improved during the day, but by the evening, his knees and ankles hurt so much that they had to carry him to the bathroom. On physical examination, he still had palpable purpura of the legs. There was no swelling, but his joints were still tender to palpation. His parents were reminded to give him ibuprofen after school to control evening pain. Over the next 2 weeks, the patient showed gradual improvement, and by Day 33 the rash and all of the associated symptoms had resolved.
DISCUSSION
Clinical presentation. HSP is an IgA immune complex vasculitis in which abnormal glycosylation of IgA creates large immune complexes that are deposited in the walls of the skin capillaries and arterioles. The primary clinical finding in HSP is a distinctive nonthrombocytopenic purpuric rash that is not associated with coagulopathy and is characterized by reddish purple macules that progress to palpable purpura with petechiae (
A preceding upper respiratory infection has been found in 37% of patients,1 and in patients with renal complications, 20% to 50% have been found to have a group A Streptococcus infection.2 Other associations include food allergies, cold exposure, insect bites, and drug allergies.
Continue to: HSP vasculitis causes...
HSP vasculitis causes abdominal pain in 50% to 75% of patients due to proximal small-bowel submucosal hemorrhage and bowel wall edema.3 In children with HSP, 20% to 55% have been shown to develop renal disease,4 which can range in severity from microscopic hematuria to nephrotic syndrome.3 To ensure prompt treatment of renal manifestations, renal function should be monitored regularly via blood pressure and urinalysis during the course of HSP and after resolution. Renal disease associated with HSP can be acute or chronic.
This case was different because our patient did not exhibit all elements of the classic tetrad of HSP, which includes the characteristic rash, abdominal pain, renal involvement, and arthralgia.
Incidence. HSP is more common in children than adults, with average annual incidence rates of 20/100,000 and 70/100,000 in children in the United States and Asia, respectively.5 While 90% of HSP cases occur in children < 10 years, the peak incidence is at 6 years of age.6 Complications from HSP are more common in adults than in children.7 Caucasian and Asian populations have a 3- to 4-times higher prevalence of HSP than black populations. The male-to-female ratio is 2 to 1.6
The diagnosis of HSP is usually made clinically, based on the distinctive rash, which typically is symmetrical, involving the buttocks, lower legs, elbows, and/or knees. HSP also can be confirmed via skin biopsy and/or direct immunofluorescence, which can identify the presence of IgA in the vessel walls.
The presence of 3 or more of the following criteria also suggests HSP: palpable purpura, bowel angina, gastrointestinal (GI) bleeding, hematuria, ≤ 20 years of age at onset, and no medications prior to presentation of symptoms (87% of cases correctly classified). Fewer than 3 of these factors favor hypersensitivity vasculitis (74% of cases correctly classified).8
Continue to: The differential diagnosis
The differential diagnosis for HSP includes polyarteritis nodosa, a vasculitis with a different characteristic rash; acute abdomen, distinguished by the absence of purpura or arthralgia; meningococcemia, in which fever and meningeal signs may occur; hypersensitivity vasculitis, which arises due to prior exposure to medications or food allergens; and thrombocytopenic purpura, which is characterized by low platelet count.9
Treatment focuses on pain management
In the absence of renal disease, HSP commonly is treated with naproxen for pain management (dosage for children < 2 years of age: 5-7 mg/kg orally every 8-12 hours; dosage for children ≥ 2 years of age, adolescents, and adults: 10-20 mg/kg/d divided into 2 doses; maximum adolescent and adult dose is 1500 mg/d for 3 days followed by a maximum of 1000 mg/d thereafter).
For patients of all ages with severe pain and those with GI effects limiting oral intake of medication, use oral prednisone (1-2 mg/kg/d [maximum dose, 60-80 mg/d]) or intravenous methylprednisolone (0.8-1.6 mg/kg/d [maximum dose, 64 mg/d). Glucocorticoids may then be tapered slowly over 4 to 8 weeks to avoid rebound since they help with inflammation but do not shorten the course of disease. Steroids can ease GI and joint symptoms in HSP but will not improve the rash.
THE TAKEAWAY
The classic tetrad of HSP includes the characteristic rash, abdominal pain, renal involvement, and arthralgia. Diagnosis usually is made clinically, but skin biopsy and direct immunofluorescence can confirm small vessel vasculitis with IgA deposits. More severe manifestations of HSP such as renal disease, hemorrhage, severe anemia, signs of intestinal obstruction, or peritonitis require rapid subspecialty referral.
CORRESPONDENCE
Rachel Bramson, MD, Department of Primary Care, Baylor Scott and White Health, University Clinic, 1700 University Drive, College Station, TX 77840; [email protected]
1. Rigante D, Castellazzi L, Bosco A, et al. Is there a crossroad between infections, genetics, and Henoch-Schönlein purpura? Autoimmun Rev. 2013;12:1016-1021.
2. LaConti JJ, Donet JA, Cho-Vega JH, et al. Henoch-Schönlein Purpura with adalimumab therapy for ulcerative colitis: a case report and review of the literature [published online July 27, 2016]. Case Rep Rheumatol. 2016;2016:2812980.
3. Trnka P. Henoch-Schönlein purpura in children. J Paediatr Child Health. 2013;49:995-1003.
4. Audemard-Verger A, Pillebout E, Guillevin L, et al. IgA vasculitis (Henoch-Shönlein purpura) in adults: diagnostic and therapeutic aspects. Autoimmun Rev. 2015;14:579-585.
5. Chen J, Mao J. Henoch-Schönlein purpura nephritis in children: incidence, pathogenesis and management. World J Pediatr. 2015;11:29-34.
6. Michel B, Hunder G, Bloch D, et al. Hypersensitivity vasculitis and Henoch-Schönlein purpura: a comparison between the 2 disorders. J Rheumatol. 1992;19:721-728.
7. Reamy BV, Williams PM, Lindsay TJ. Henoch-Schönlein purpura. Am Fam Physician. 2009;80:697-704.
8. Yang YH, Yu HH, Chiang BL. The diagnosis and classification of Henoch-Schönlein purpura: an updated review. Autoimmun Rev. 2014;13:355-358.
9. Floege J, Feehally J. Treatment of IgA nephropathy and Henoch-Schönlein nephritis. Nat Rev Nephrol. 2013;9:320-327.
1. Rigante D, Castellazzi L, Bosco A, et al. Is there a crossroad between infections, genetics, and Henoch-Schönlein purpura? Autoimmun Rev. 2013;12:1016-1021.
2. LaConti JJ, Donet JA, Cho-Vega JH, et al. Henoch-Schönlein Purpura with adalimumab therapy for ulcerative colitis: a case report and review of the literature [published online July 27, 2016]. Case Rep Rheumatol. 2016;2016:2812980.
3. Trnka P. Henoch-Schönlein purpura in children. J Paediatr Child Health. 2013;49:995-1003.
4. Audemard-Verger A, Pillebout E, Guillevin L, et al. IgA vasculitis (Henoch-Shönlein purpura) in adults: diagnostic and therapeutic aspects. Autoimmun Rev. 2015;14:579-585.
5. Chen J, Mao J. Henoch-Schönlein purpura nephritis in children: incidence, pathogenesis and management. World J Pediatr. 2015;11:29-34.
6. Michel B, Hunder G, Bloch D, et al. Hypersensitivity vasculitis and Henoch-Schönlein purpura: a comparison between the 2 disorders. J Rheumatol. 1992;19:721-728.
7. Reamy BV, Williams PM, Lindsay TJ. Henoch-Schönlein purpura. Am Fam Physician. 2009;80:697-704.
8. Yang YH, Yu HH, Chiang BL. The diagnosis and classification of Henoch-Schönlein purpura: an updated review. Autoimmun Rev. 2014;13:355-358.
9. Floege J, Feehally J. Treatment of IgA nephropathy and Henoch-Schönlein nephritis. Nat Rev Nephrol. 2013;9:320-327.

Allergic Contact Dermatitis From Sorbitans in Beer and Bread
Sorbitan sesquioleate (SSO), sorbitan monooleate (SMO), and related compounds are increasingly recognized contact allergens. Sorbitan sesquioleate and SMO are nonionic emulsifying agents derived from sorbitol.1
Sorbitan sesquioleate, SMO, and other sorbitol derivatives are used as emulsifiers and dispersing agents in cosmetics, topical medications, topical emollients, produce, and other commercial products. Related compounds also are found in foods such as apples, berries, cherries, and sucrose-free cakes and cookies.1 We present a case of allergic contact dermatitis (ACD) with positive patch testing to sorbitans and clinical correlation with beer and bread exposure.
Case Report
A 62-year-old man presented with a persistent pruritic rash of 6 months’ duration. Erythematous eczematous papules and plaques were observed on the face, neck, chest, abdomen, back, and upper and lower extremities, affecting approximately 60% of the body surface area. His current list of medications was reviewed and included a multivitamin, fish oil, and vitamin C. A punch biopsy revealed spongiotic dermatitis with eosinophils. Patch testing using the North American Contact Dermatitis Group Standard Series with supplemental allergens found in toiletries revealed a positive reaction to SSO and SMO that was persistent at 48 and 96 hours. Notably, patch testing for sodium benzoate, nickel, potassium dichromate, and balsam of Peru were negative. Investigation into the personal care products the patient used identified the presence of sorbitol solution in Vanicream bar soap and Vanicream moisturizing cream (Pharmaceutical Specialties Inc). These products were started after the development of the rash and were discontinued after positive patch testing, but the patient continued to experience the eruption with no improvement.
Retrospectively, the patient was able to correlate exacerbations with drinking beer and eating sandwiches. He habitually ate a sandwich on the same type of bread every single day and enjoyed the same brand of beer 2 to 4 times per week without much variation. To limit allergens, the patient gave up the daily sandwich and avoided bread altogether, noting remarkable clinical improvement over a few weeks. Later, he described even more improvement while on a trip where he did not have access to his usual beer. The eruption recurred when he returned home and excessively indulged in his favorite beer. He also noted recurrence with exposure to certain breads. No new lesions developed with avoidance of beer and bread, and he had less than 1% body surface area involvement at 2-month follow-up and 0% involvement at 1 year. For educational purposes, follow-up patch testing was performed using Vanicream sorbitol solution and the specific beer and bread the patient consumed. The Vanicream solution was obtained from the manufacturer. The beer was placed directly onto a test disc. The bread was moistened with a drop of saline and then placed directly onto a test disc. All were negative at 48 and 96 hours.
Comment
Sorbitol Ingredients
We report a case of systemic ACD with a positive patch test to sorbitans that was exacerbated with consumption of beer and bread and resolved with avoidance of these products. Although it was determined that the patient used personal care products containing a sorbitol solution, discontinuation did not result in clinical improvement. Sorbitol, sorbitans, and sorbitol derivatives are not commonly reported in the ingredient lists of foods such as beer and bread. Both beer and bread are created with the addition of yeast cultures, for fermentation in beer and for leavening in bread. Sorbitol is used as an osmotic stabilizer in the preparation of yeast strains2 and also is a by-product of fermentation by certain bacteria3 found in beer. Additionally, review of commercially available preparations of baker’s and brewer’s yeasts, such as Fleischmann’s and Red Star, list sorbitan monostearate in the ingredients.4-7 We propose that trace amounts are present in the yeast preparations for brewing and baking.
In this case, the offending beer and bread were locally made products (Abita Beer, Covington, Louisiana; Leidenheimer Bread, New Orleans, Louisiana). Both companies were unable to share their yeast sources, limiting our ability to confirm the use of sorbitol in their preparation. We hypothesize that if sorbitol is commonly used in yeast culture preparation and can be a by-product of fermentation, then it is present in trace amounts in many beers and breads and is not specific to these two products.
Contact Allergy
There are few prior reports of ACD due to beer. A case series in 1969 described 4 patients with positive patch testing to ethanol and alcohol by-products and clinical resolution with avoidance of alcohol.8 Another case from 1985 described ACD to beer where patch testing was positive to the beer itself.9 Other published cases of cutaneous reactions to beer demonstrated immediate-type hypersensitivity resulting from both ingestion and skin contact, which is thought to be caused by IgE antibodies to malt and barley proteins.10,11
It is important to distinguish between systemic ACD and oral allergy syndrome (OAS). Although the defining features and criteria for diagnosing OAS have not been officially established, OAS is an IgE-mediated immune reaction commonly described as itching, tingling, or swelling, usually confined to the oral cavity after recent consumption of foods such as raw fruits, vegetables, and nuts.12 Oral allergy syndrome is treated with antihistamines and avoidance of known food allergens. In comparison, ACD is a type IV hypersensitivity, delayed cell-mediated reaction, commonly presenting with widespread rash.
Occupational contact dermatitis is common in bakers and food handlers and is more often irritant than allergic. Several relevant allergens have been identified in these groups13,14 and do not include sorbitans; our patient tested positive to both SSO and SMO. Sorbitan sesquioleate and SMO have been increasingly recognized as contact allergens over the last several years, both as standalone allergens and as potential cross-reactors.1 Sorbitan sesquioleate, SMO, and other sorbitol derivatives are found in cosmetics, topical and oral medications, topical emollients, produce, and other commercial products, including but not limited to topical clindamycin, topical metronidazole, topical ketoconazole, tazarotene cream 0.05% and 0.1%, toothpastes, acetaminophen maximum strength liquid, apples, berries, and sucrose-free cakes and cookies.1,15,16
In 2014, a study evaluated 12 oral antihistamines as potential sources for systemic contact allergens; 55% of these 12 oral antihistamine preparations included at least 1 of 10 allergen groups specifically identified. The sorbitans and sorbitol derivatives group ranked highest among the group of allergens found listed in these oral medications.17
Most patients found to have a contact allergy to the products containing SSO, SMO, or sorbitol derivatives reported notable improvement with discontinuation and change to sorbitol-free product use.1,18 It should be noted that SSO is added as an emulsifier to many of the fragrances used for patch testing. A positive patch test to fragrance mix without concomitant sorbitan testing may incorrectly diagnose the allergen.19
Patients with atopic dermatitis, particularly those with a filaggrin mutation, are at increased risk for ACD to sorbitans due to a compromised skin barrier and frequent use of topical steroids. In one study, 75% of patients (n=12) with a positive patch test to SSO were using a topical steroid emulsified with sorbitol or sorbitan derivatives.19
Conclusion
Sorbitan sesquioleate and SMO are increasingly relevant contact allergens. Sorbitol and related substances have been identified in numerous products and may be present in yeast-fermented and leavened goods. When patch testing is positive to SSO and SMO, the dermatologist should inquire about dietary habits with specific attention to beer and bread, in addition to inventorying other dietary preferences, prescription and over-the-counter medications, and personal care products. We suggest dietary considerations only if topical exposures have been eliminated and the rash has not improved.
- Asarch A, Scheinman PL. Sorbitan sesquioleate: an emerging contact allergen. Dermatitis. 2008;19:339-341.
- Lundblad V, Struhl K. Yeast. In: Adelman K, Ausubel F, Brent R, et al. Current Protocols in Molecular Biology, Supplement 64. New York, NY: John Wiley & Sons, Inc; 2008:13.0.1-13.0.4. https://onlinelibrary.wiley.com. Accessed August 19, 2019.
- Spitaels F, Wieme A, Balzarini T, et al. Gluconobacter cerevisiae sp. nov., isolated from the brewery environment. Int J Sys Evol Microbiol. 2014;64(pt 4):1134-1141.
- Fleischmann’s, n.d. Product Label for Rapid Rise Instant Yeast. Memphis, TN. 2017.
- Fleischmann’s, n.d. Product Label for Active Dry Yeast. Memphis, TN. 2017.
- Red Star, n.d. Product Label for Quick-Rise. Milwaukee, WI. 2017.
- Red Star, n.d. Product Label for Platinum Superior Baking Yeast. Milwaukee, WI. 2017.
- Fregert S, Groth O, Hjorth N, et al. Alcohol dermatitis. Acta Derm Venereol. 1969;49:493-497.
- Clarke P. Contact dermatitis due to beer. Med J Aust. 1985;143:92.
- Koelemij I, Van Zuuren EJ. Contact urticaria from beer. Clin Exp Dermatol. 2014;39:395-407.
- Santucci B, Cristaudo A, Cannistraci C, et al. Urticaria from beer in 3 patients. Contact Dermatitis. 1996;34:368.
- Kohn JB. What is oral allergy syndrome? J Acad Nutr Diet. 2017;117:988.
- Vincenzi C, Stinchi C, Ricci C, et al. Contact dermatitis due to an emulsifying agent in a baker. Contact Dermatitis. 1995;32:57.
- Nethercott JR, Holness DL. Occupational dermatitis in food handlers and bakers. J Am Acad Dermatol. 1989;21:485-490.
- Pereira F, Cunha H, Dias M. Contact dermatitis due to emulsifiers. Contact Dermatitis. 1997;36:114.
- Gao Z, Maurousset L, Lemoine R, et al. Cloning, expression, and characterization of sorbitol transporters from developing sour cherry fruit and leaf sink tissues. Plant Physiol. 2003;131:1566-1575.
- McEnery-Stonelake M, Silvestri DL. Contact allergens in oral antihistamines. Dermatitis. 2014;25:83-88.
- Asarch A, Scheinman PL. Sorbitan sesquioleate, a common emulsifier in topical steroids, is an important contact allergen. Dermatitis. 2008;19:323-327.
- Hald M, Menné T, Johansen JD, et al. Allergic contact dermatitis caused by sorbitan sesquioleate imitating severe glove dermatitis in a patient with filaggrin mutation. Contact Dermatitis. 2013;69:311-322.
Sorbitan sesquioleate (SSO), sorbitan monooleate (SMO), and related compounds are increasingly recognized contact allergens. Sorbitan sesquioleate and SMO are nonionic emulsifying agents derived from sorbitol.1
Sorbitan sesquioleate, SMO, and other sorbitol derivatives are used as emulsifiers and dispersing agents in cosmetics, topical medications, topical emollients, produce, and other commercial products. Related compounds also are found in foods such as apples, berries, cherries, and sucrose-free cakes and cookies.1 We present a case of allergic contact dermatitis (ACD) with positive patch testing to sorbitans and clinical correlation with beer and bread exposure.
Case Report
A 62-year-old man presented with a persistent pruritic rash of 6 months’ duration. Erythematous eczematous papules and plaques were observed on the face, neck, chest, abdomen, back, and upper and lower extremities, affecting approximately 60% of the body surface area. His current list of medications was reviewed and included a multivitamin, fish oil, and vitamin C. A punch biopsy revealed spongiotic dermatitis with eosinophils. Patch testing using the North American Contact Dermatitis Group Standard Series with supplemental allergens found in toiletries revealed a positive reaction to SSO and SMO that was persistent at 48 and 96 hours. Notably, patch testing for sodium benzoate, nickel, potassium dichromate, and balsam of Peru were negative. Investigation into the personal care products the patient used identified the presence of sorbitol solution in Vanicream bar soap and Vanicream moisturizing cream (Pharmaceutical Specialties Inc). These products were started after the development of the rash and were discontinued after positive patch testing, but the patient continued to experience the eruption with no improvement.
Retrospectively, the patient was able to correlate exacerbations with drinking beer and eating sandwiches. He habitually ate a sandwich on the same type of bread every single day and enjoyed the same brand of beer 2 to 4 times per week without much variation. To limit allergens, the patient gave up the daily sandwich and avoided bread altogether, noting remarkable clinical improvement over a few weeks. Later, he described even more improvement while on a trip where he did not have access to his usual beer. The eruption recurred when he returned home and excessively indulged in his favorite beer. He also noted recurrence with exposure to certain breads. No new lesions developed with avoidance of beer and bread, and he had less than 1% body surface area involvement at 2-month follow-up and 0% involvement at 1 year. For educational purposes, follow-up patch testing was performed using Vanicream sorbitol solution and the specific beer and bread the patient consumed. The Vanicream solution was obtained from the manufacturer. The beer was placed directly onto a test disc. The bread was moistened with a drop of saline and then placed directly onto a test disc. All were negative at 48 and 96 hours.
Comment
Sorbitol Ingredients
We report a case of systemic ACD with a positive patch test to sorbitans that was exacerbated with consumption of beer and bread and resolved with avoidance of these products. Although it was determined that the patient used personal care products containing a sorbitol solution, discontinuation did not result in clinical improvement. Sorbitol, sorbitans, and sorbitol derivatives are not commonly reported in the ingredient lists of foods such as beer and bread. Both beer and bread are created with the addition of yeast cultures, for fermentation in beer and for leavening in bread. Sorbitol is used as an osmotic stabilizer in the preparation of yeast strains2 and also is a by-product of fermentation by certain bacteria3 found in beer. Additionally, review of commercially available preparations of baker’s and brewer’s yeasts, such as Fleischmann’s and Red Star, list sorbitan monostearate in the ingredients.4-7 We propose that trace amounts are present in the yeast preparations for brewing and baking.
In this case, the offending beer and bread were locally made products (Abita Beer, Covington, Louisiana; Leidenheimer Bread, New Orleans, Louisiana). Both companies were unable to share their yeast sources, limiting our ability to confirm the use of sorbitol in their preparation. We hypothesize that if sorbitol is commonly used in yeast culture preparation and can be a by-product of fermentation, then it is present in trace amounts in many beers and breads and is not specific to these two products.
Contact Allergy
There are few prior reports of ACD due to beer. A case series in 1969 described 4 patients with positive patch testing to ethanol and alcohol by-products and clinical resolution with avoidance of alcohol.8 Another case from 1985 described ACD to beer where patch testing was positive to the beer itself.9 Other published cases of cutaneous reactions to beer demonstrated immediate-type hypersensitivity resulting from both ingestion and skin contact, which is thought to be caused by IgE antibodies to malt and barley proteins.10,11
It is important to distinguish between systemic ACD and oral allergy syndrome (OAS). Although the defining features and criteria for diagnosing OAS have not been officially established, OAS is an IgE-mediated immune reaction commonly described as itching, tingling, or swelling, usually confined to the oral cavity after recent consumption of foods such as raw fruits, vegetables, and nuts.12 Oral allergy syndrome is treated with antihistamines and avoidance of known food allergens. In comparison, ACD is a type IV hypersensitivity, delayed cell-mediated reaction, commonly presenting with widespread rash.
Occupational contact dermatitis is common in bakers and food handlers and is more often irritant than allergic. Several relevant allergens have been identified in these groups13,14 and do not include sorbitans; our patient tested positive to both SSO and SMO. Sorbitan sesquioleate and SMO have been increasingly recognized as contact allergens over the last several years, both as standalone allergens and as potential cross-reactors.1 Sorbitan sesquioleate, SMO, and other sorbitol derivatives are found in cosmetics, topical and oral medications, topical emollients, produce, and other commercial products, including but not limited to topical clindamycin, topical metronidazole, topical ketoconazole, tazarotene cream 0.05% and 0.1%, toothpastes, acetaminophen maximum strength liquid, apples, berries, and sucrose-free cakes and cookies.1,15,16
In 2014, a study evaluated 12 oral antihistamines as potential sources for systemic contact allergens; 55% of these 12 oral antihistamine preparations included at least 1 of 10 allergen groups specifically identified. The sorbitans and sorbitol derivatives group ranked highest among the group of allergens found listed in these oral medications.17
Most patients found to have a contact allergy to the products containing SSO, SMO, or sorbitol derivatives reported notable improvement with discontinuation and change to sorbitol-free product use.1,18 It should be noted that SSO is added as an emulsifier to many of the fragrances used for patch testing. A positive patch test to fragrance mix without concomitant sorbitan testing may incorrectly diagnose the allergen.19
Patients with atopic dermatitis, particularly those with a filaggrin mutation, are at increased risk for ACD to sorbitans due to a compromised skin barrier and frequent use of topical steroids. In one study, 75% of patients (n=12) with a positive patch test to SSO were using a topical steroid emulsified with sorbitol or sorbitan derivatives.19
Conclusion
Sorbitan sesquioleate and SMO are increasingly relevant contact allergens. Sorbitol and related substances have been identified in numerous products and may be present in yeast-fermented and leavened goods. When patch testing is positive to SSO and SMO, the dermatologist should inquire about dietary habits with specific attention to beer and bread, in addition to inventorying other dietary preferences, prescription and over-the-counter medications, and personal care products. We suggest dietary considerations only if topical exposures have been eliminated and the rash has not improved.
Sorbitan sesquioleate (SSO), sorbitan monooleate (SMO), and related compounds are increasingly recognized contact allergens. Sorbitan sesquioleate and SMO are nonionic emulsifying agents derived from sorbitol.1
Sorbitan sesquioleate, SMO, and other sorbitol derivatives are used as emulsifiers and dispersing agents in cosmetics, topical medications, topical emollients, produce, and other commercial products. Related compounds also are found in foods such as apples, berries, cherries, and sucrose-free cakes and cookies.1 We present a case of allergic contact dermatitis (ACD) with positive patch testing to sorbitans and clinical correlation with beer and bread exposure.
Case Report
A 62-year-old man presented with a persistent pruritic rash of 6 months’ duration. Erythematous eczematous papules and plaques were observed on the face, neck, chest, abdomen, back, and upper and lower extremities, affecting approximately 60% of the body surface area. His current list of medications was reviewed and included a multivitamin, fish oil, and vitamin C. A punch biopsy revealed spongiotic dermatitis with eosinophils. Patch testing using the North American Contact Dermatitis Group Standard Series with supplemental allergens found in toiletries revealed a positive reaction to SSO and SMO that was persistent at 48 and 96 hours. Notably, patch testing for sodium benzoate, nickel, potassium dichromate, and balsam of Peru were negative. Investigation into the personal care products the patient used identified the presence of sorbitol solution in Vanicream bar soap and Vanicream moisturizing cream (Pharmaceutical Specialties Inc). These products were started after the development of the rash and were discontinued after positive patch testing, but the patient continued to experience the eruption with no improvement.
Retrospectively, the patient was able to correlate exacerbations with drinking beer and eating sandwiches. He habitually ate a sandwich on the same type of bread every single day and enjoyed the same brand of beer 2 to 4 times per week without much variation. To limit allergens, the patient gave up the daily sandwich and avoided bread altogether, noting remarkable clinical improvement over a few weeks. Later, he described even more improvement while on a trip where he did not have access to his usual beer. The eruption recurred when he returned home and excessively indulged in his favorite beer. He also noted recurrence with exposure to certain breads. No new lesions developed with avoidance of beer and bread, and he had less than 1% body surface area involvement at 2-month follow-up and 0% involvement at 1 year. For educational purposes, follow-up patch testing was performed using Vanicream sorbitol solution and the specific beer and bread the patient consumed. The Vanicream solution was obtained from the manufacturer. The beer was placed directly onto a test disc. The bread was moistened with a drop of saline and then placed directly onto a test disc. All were negative at 48 and 96 hours.
Comment
Sorbitol Ingredients
We report a case of systemic ACD with a positive patch test to sorbitans that was exacerbated with consumption of beer and bread and resolved with avoidance of these products. Although it was determined that the patient used personal care products containing a sorbitol solution, discontinuation did not result in clinical improvement. Sorbitol, sorbitans, and sorbitol derivatives are not commonly reported in the ingredient lists of foods such as beer and bread. Both beer and bread are created with the addition of yeast cultures, for fermentation in beer and for leavening in bread. Sorbitol is used as an osmotic stabilizer in the preparation of yeast strains2 and also is a by-product of fermentation by certain bacteria3 found in beer. Additionally, review of commercially available preparations of baker’s and brewer’s yeasts, such as Fleischmann’s and Red Star, list sorbitan monostearate in the ingredients.4-7 We propose that trace amounts are present in the yeast preparations for brewing and baking.
In this case, the offending beer and bread were locally made products (Abita Beer, Covington, Louisiana; Leidenheimer Bread, New Orleans, Louisiana). Both companies were unable to share their yeast sources, limiting our ability to confirm the use of sorbitol in their preparation. We hypothesize that if sorbitol is commonly used in yeast culture preparation and can be a by-product of fermentation, then it is present in trace amounts in many beers and breads and is not specific to these two products.
Contact Allergy
There are few prior reports of ACD due to beer. A case series in 1969 described 4 patients with positive patch testing to ethanol and alcohol by-products and clinical resolution with avoidance of alcohol.8 Another case from 1985 described ACD to beer where patch testing was positive to the beer itself.9 Other published cases of cutaneous reactions to beer demonstrated immediate-type hypersensitivity resulting from both ingestion and skin contact, which is thought to be caused by IgE antibodies to malt and barley proteins.10,11
It is important to distinguish between systemic ACD and oral allergy syndrome (OAS). Although the defining features and criteria for diagnosing OAS have not been officially established, OAS is an IgE-mediated immune reaction commonly described as itching, tingling, or swelling, usually confined to the oral cavity after recent consumption of foods such as raw fruits, vegetables, and nuts.12 Oral allergy syndrome is treated with antihistamines and avoidance of known food allergens. In comparison, ACD is a type IV hypersensitivity, delayed cell-mediated reaction, commonly presenting with widespread rash.
Occupational contact dermatitis is common in bakers and food handlers and is more often irritant than allergic. Several relevant allergens have been identified in these groups13,14 and do not include sorbitans; our patient tested positive to both SSO and SMO. Sorbitan sesquioleate and SMO have been increasingly recognized as contact allergens over the last several years, both as standalone allergens and as potential cross-reactors.1 Sorbitan sesquioleate, SMO, and other sorbitol derivatives are found in cosmetics, topical and oral medications, topical emollients, produce, and other commercial products, including but not limited to topical clindamycin, topical metronidazole, topical ketoconazole, tazarotene cream 0.05% and 0.1%, toothpastes, acetaminophen maximum strength liquid, apples, berries, and sucrose-free cakes and cookies.1,15,16
In 2014, a study evaluated 12 oral antihistamines as potential sources for systemic contact allergens; 55% of these 12 oral antihistamine preparations included at least 1 of 10 allergen groups specifically identified. The sorbitans and sorbitol derivatives group ranked highest among the group of allergens found listed in these oral medications.17
Most patients found to have a contact allergy to the products containing SSO, SMO, or sorbitol derivatives reported notable improvement with discontinuation and change to sorbitol-free product use.1,18 It should be noted that SSO is added as an emulsifier to many of the fragrances used for patch testing. A positive patch test to fragrance mix without concomitant sorbitan testing may incorrectly diagnose the allergen.19
Patients with atopic dermatitis, particularly those with a filaggrin mutation, are at increased risk for ACD to sorbitans due to a compromised skin barrier and frequent use of topical steroids. In one study, 75% of patients (n=12) with a positive patch test to SSO were using a topical steroid emulsified with sorbitol or sorbitan derivatives.19
Conclusion
Sorbitan sesquioleate and SMO are increasingly relevant contact allergens. Sorbitol and related substances have been identified in numerous products and may be present in yeast-fermented and leavened goods. When patch testing is positive to SSO and SMO, the dermatologist should inquire about dietary habits with specific attention to beer and bread, in addition to inventorying other dietary preferences, prescription and over-the-counter medications, and personal care products. We suggest dietary considerations only if topical exposures have been eliminated and the rash has not improved.
- Asarch A, Scheinman PL. Sorbitan sesquioleate: an emerging contact allergen. Dermatitis. 2008;19:339-341.
- Lundblad V, Struhl K. Yeast. In: Adelman K, Ausubel F, Brent R, et al. Current Protocols in Molecular Biology, Supplement 64. New York, NY: John Wiley & Sons, Inc; 2008:13.0.1-13.0.4. https://onlinelibrary.wiley.com. Accessed August 19, 2019.
- Spitaels F, Wieme A, Balzarini T, et al. Gluconobacter cerevisiae sp. nov., isolated from the brewery environment. Int J Sys Evol Microbiol. 2014;64(pt 4):1134-1141.
- Fleischmann’s, n.d. Product Label for Rapid Rise Instant Yeast. Memphis, TN. 2017.
- Fleischmann’s, n.d. Product Label for Active Dry Yeast. Memphis, TN. 2017.
- Red Star, n.d. Product Label for Quick-Rise. Milwaukee, WI. 2017.
- Red Star, n.d. Product Label for Platinum Superior Baking Yeast. Milwaukee, WI. 2017.
- Fregert S, Groth O, Hjorth N, et al. Alcohol dermatitis. Acta Derm Venereol. 1969;49:493-497.
- Clarke P. Contact dermatitis due to beer. Med J Aust. 1985;143:92.
- Koelemij I, Van Zuuren EJ. Contact urticaria from beer. Clin Exp Dermatol. 2014;39:395-407.
- Santucci B, Cristaudo A, Cannistraci C, et al. Urticaria from beer in 3 patients. Contact Dermatitis. 1996;34:368.
- Kohn JB. What is oral allergy syndrome? J Acad Nutr Diet. 2017;117:988.
- Vincenzi C, Stinchi C, Ricci C, et al. Contact dermatitis due to an emulsifying agent in a baker. Contact Dermatitis. 1995;32:57.
- Nethercott JR, Holness DL. Occupational dermatitis in food handlers and bakers. J Am Acad Dermatol. 1989;21:485-490.
- Pereira F, Cunha H, Dias M. Contact dermatitis due to emulsifiers. Contact Dermatitis. 1997;36:114.
- Gao Z, Maurousset L, Lemoine R, et al. Cloning, expression, and characterization of sorbitol transporters from developing sour cherry fruit and leaf sink tissues. Plant Physiol. 2003;131:1566-1575.
- McEnery-Stonelake M, Silvestri DL. Contact allergens in oral antihistamines. Dermatitis. 2014;25:83-88.
- Asarch A, Scheinman PL. Sorbitan sesquioleate, a common emulsifier in topical steroids, is an important contact allergen. Dermatitis. 2008;19:323-327.
- Hald M, Menné T, Johansen JD, et al. Allergic contact dermatitis caused by sorbitan sesquioleate imitating severe glove dermatitis in a patient with filaggrin mutation. Contact Dermatitis. 2013;69:311-322.
- Asarch A, Scheinman PL. Sorbitan sesquioleate: an emerging contact allergen. Dermatitis. 2008;19:339-341.
- Lundblad V, Struhl K. Yeast. In: Adelman K, Ausubel F, Brent R, et al. Current Protocols in Molecular Biology, Supplement 64. New York, NY: John Wiley & Sons, Inc; 2008:13.0.1-13.0.4. https://onlinelibrary.wiley.com. Accessed August 19, 2019.
- Spitaels F, Wieme A, Balzarini T, et al. Gluconobacter cerevisiae sp. nov., isolated from the brewery environment. Int J Sys Evol Microbiol. 2014;64(pt 4):1134-1141.
- Fleischmann’s, n.d. Product Label for Rapid Rise Instant Yeast. Memphis, TN. 2017.
- Fleischmann’s, n.d. Product Label for Active Dry Yeast. Memphis, TN. 2017.
- Red Star, n.d. Product Label for Quick-Rise. Milwaukee, WI. 2017.
- Red Star, n.d. Product Label for Platinum Superior Baking Yeast. Milwaukee, WI. 2017.
- Fregert S, Groth O, Hjorth N, et al. Alcohol dermatitis. Acta Derm Venereol. 1969;49:493-497.
- Clarke P. Contact dermatitis due to beer. Med J Aust. 1985;143:92.
- Koelemij I, Van Zuuren EJ. Contact urticaria from beer. Clin Exp Dermatol. 2014;39:395-407.
- Santucci B, Cristaudo A, Cannistraci C, et al. Urticaria from beer in 3 patients. Contact Dermatitis. 1996;34:368.
- Kohn JB. What is oral allergy syndrome? J Acad Nutr Diet. 2017;117:988.
- Vincenzi C, Stinchi C, Ricci C, et al. Contact dermatitis due to an emulsifying agent in a baker. Contact Dermatitis. 1995;32:57.
- Nethercott JR, Holness DL. Occupational dermatitis in food handlers and bakers. J Am Acad Dermatol. 1989;21:485-490.
- Pereira F, Cunha H, Dias M. Contact dermatitis due to emulsifiers. Contact Dermatitis. 1997;36:114.
- Gao Z, Maurousset L, Lemoine R, et al. Cloning, expression, and characterization of sorbitol transporters from developing sour cherry fruit and leaf sink tissues. Plant Physiol. 2003;131:1566-1575.
- McEnery-Stonelake M, Silvestri DL. Contact allergens in oral antihistamines. Dermatitis. 2014;25:83-88.
- Asarch A, Scheinman PL. Sorbitan sesquioleate, a common emulsifier in topical steroids, is an important contact allergen. Dermatitis. 2008;19:323-327.
- Hald M, Menné T, Johansen JD, et al. Allergic contact dermatitis caused by sorbitan sesquioleate imitating severe glove dermatitis in a patient with filaggrin mutation. Contact Dermatitis. 2013;69:311-322.
Practice Points
- Sorbitan sesquioleate (SSO) and sorbitan monooleate (SMO) are increasingly relevant contact allergens that may be present in yeast-fermented and leavened products.
- When patch testing is positive to SSO and SMO, the dermatologist should inquire about dietary habits with specific attention to beer and bread.
- Consider elimination of beer, bread, and other leavened products when rash persists after avoidance of topical exposures.
Disseminated Invasive Candidiasis in an Immunocompetent Host
Candida albicans (C albicans) is a normal commensal in the human gastrointestinal (GI) tract. In addition to localized infections in healthy human beings, dissemination with fatal outcome can occur in immunocompromised individuals.1
Invasive candidiasis (IC) due to C albicans is the most common nosocomial mycosis in the world and has 2 forms, candidemia and deep-seated tissue candidiasis, which can lead to multisystem organ failure.2 The deep-seated form may originate from nonhematogenous routes, such as introduction through a peritoneal catheter or ascending infection from cystitis.2 In addition, about 50% of primary candidemia cases lead to secondary deep-seated candidiasis; however, only about 40% of these cases show positive blood cultures. Since the window of opportunity for a positive culture is narrow, active candidemia may be missed.3,4
Once developed, the prognosis for IC is grim: Mortality is 40% regardless of therapy.2 IC typically occurs in immunocompromised hosts; IC in immunocompetent persons has rarely been reported.5,6 It is challenging to diagnose IC in the immunocompetent patients as 50% to 70% of the general population is naturally colonized by this organism, and when found, it is assumed to be mostly innocuous. Neutrophil-driven cell-mediated immunity associated with IL-1 and IL-17 response prevent fungal growth and dissemination, protecting the immunocompetent host.7
We report on a patient who showed no neutropenia or leukocytopenia but developed disseminated candidiasis. This report is one of the rare cases of full-blown disseminated candidiasis with lesions related to C albicans found in almost all of the important organs.
Case Presentation
A 67-year-old male patient with a history of hypertension, peripheral vascular disease, daily heavy alcohol consumption, and a 50-pack-year history of smoking developed gangrene of the left fifth toe. He underwent vascular surgery consultation with an aortogram/left lower extremity angiography that showed occlusion of the left external iliac artery as well as the left common femoral artery. It was decided to improve inflow in the common iliac artery by placing a bare metal stent and subsequent balloon dilatation before a right to left femoral to femoral artery bypass. The patient tolerated the procedure well and was discharged home.
Two days later, the patient was admitted to a US Department of Veterans Affairs (VA) complexity level 1a hospital with weakness and worsening pain in the left lower extremities. Examination revealed chronic ischemic changes in the feet bilaterally and evidence of dry gangrene in the left fifth toe requiring femoral bypass surgery. But poor nutritional status and cardiac status prevented pursuing a permanent solution.
Following completion of a stress echocardiogram, the patient developed shock with systolic blood pressure of 60 mm Hg, and atrial fibrillation (AF) with rapid ventricular rate (RVR). He was initially treated with IV fluid supplementation, vasopressor therapy, synchronized cardioversion, and IV amiodarone/anticoagulation therapy, due to his persistent AF with RVR. The patient was transferred to a tertiary care center for persistent hypothermia and received treatment with warm saline. After initial recovery with warm saline resuscitation, he had a prolonged, complicated hospital course in which he developed progressive respiratory failure requiring intubation and critical care support. He developed a right internal jugular deep venous thrombosis, heparin-induced thrombocytopenia, lower GI bleeding requiring emergent embolization by interventional radiology, inferior vena cava filter placement, renal failure requiring dialysis, small bowel obstruction secondary to right lower quadrant phlegmon and perforation requiring small bowel resection and end ileostomy. His antibiotic regimen included therapy with vancomycin and piperacillin-tazobactam.
He eventually recovered and was extubated and subsequently transferred back to the VA hospital where cefepime was initiated because of suspicion of a urinary tract infection and septicemia (urine cultures eventually grew C albicans). Over the subsequent 3 days, the patient’s renal output and hyperkalemia worsened, he also developed increased anion gap metabolic acidosis and was intubated again and placed on full mechanical ventilatory support. His blood cultures were negative, and sputum cultures revealed normal respiratory flora and 1+ C albicans. Infectious diseases consultation recommended an abdominal ultrasound, which revealed nonspecific findings. The antibiotic regimen was changed to daptomycin and piperacillin-tazobactam. A follow-up chest X-ray revealed a developing right lower lobe pneumonia and hilar prominence suggestive of lymphadenopathy. The patient’s clinical condition deteriorated, and he subsequently developed cardiac arrest; resuscitation was not successful and he expired.
Outcome and Follow-up
An autopsy disclosed the cause of death to be bilateral candida pneumonia, part of a disseminated (invasive) candidiasis, in a patient rendered vulnerable to such infection by peripheral vascular disease and renal insufficiency. Purulent inflammation was noted at the site of disarticulation of the left foot and confluent consolidation of the lower lobes of both lungs as well as focal consolidation of the middle lobe of the right lung. Examination of histologic sections, with staining both by routine method (hematoxylin and eosin) and the Grocott-Gömöri methenamine silver method for fungus, disclosed fungal forms (yeast and filamentous) in most tissues, including the lungs (Figure 1 A and B) and kidneys (Figure 1 C and D). The pulmonary sections in addition to massive inflammation showed macrophages with engulfed yeast (Figure 2 A) and a lymphatic channel, stuffed with yeast in an alveolar septum (Figure 2 B). These findings confirmed the antemortem presence of the fungus and the body’s response to it. Inflammation was noted around glomeruli overgrown by candida (Figure 1 C and D); fungi also were seen in capsular regions (not depicted). C albicans was present in the myocardium (Figure 1 E and F), brain, thyroid, and adrenal glands (Figure 3); the only organ without C albicans was the liver, either because invasion was truly absent here or because sampling had not managed to retrieve it.
Paraffin-embedded blocks of lung tissue, sent to the University of Washington Molecular Diagnosis Microbiology Laboratory for broad-range polymerase chain reaction (PCR) identification, were positive for C albicans after extraction of gDNA and conduction of PCR using internal transcribed spacer 1 and 2 specific primers.
Discussion
IC is rare among immunocompetent individuals, but C albicans can evolve into a fatal disseminated infection. We report an atypical case of IC, with profound pulmonary infection in a patient who died 1 month after hospitalization for lower extremity pain.
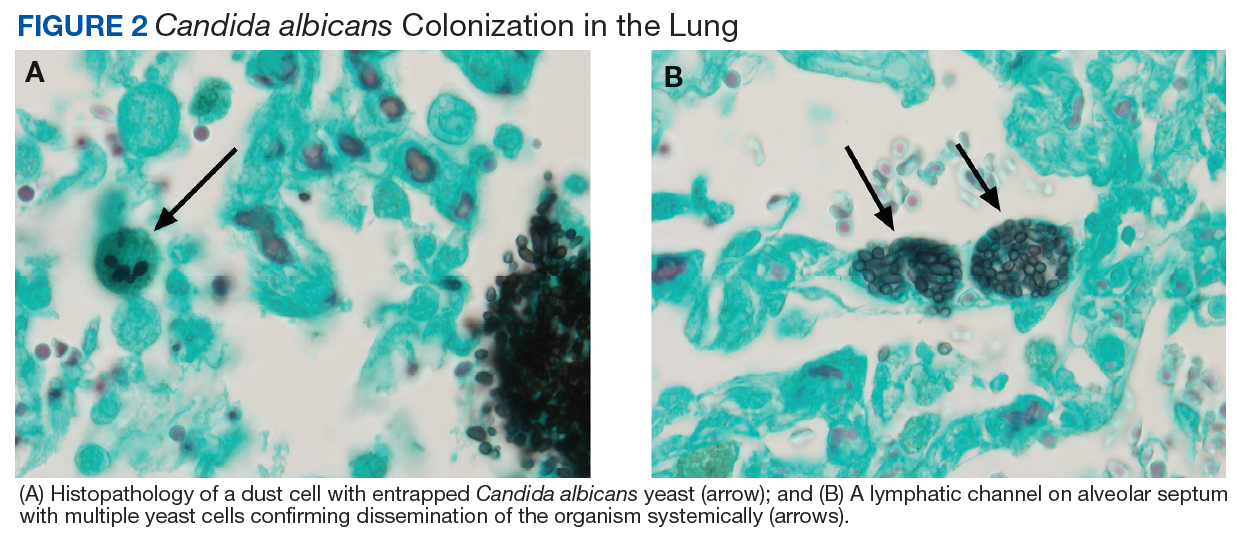
Cell-mediated immunity involving neutrophils and macrophages plays a major role in protection against candidiasis, while cytokines and chemokines involve regulating balanced immunity.1,2 A series of recent studies show that alcohol impairs neutrophil-mediated killing and phagocytic-mediated uptake of a pathogen in this process.8,9 As the patient chronically misused alcohol, his immune system may have experienced a subclinical immunosuppression, which would have become clinically relevant once C albicans was introduced systemically. Recent studies of bacterial pathogenesis and alcoholism strongly support this hypothesis.10,11
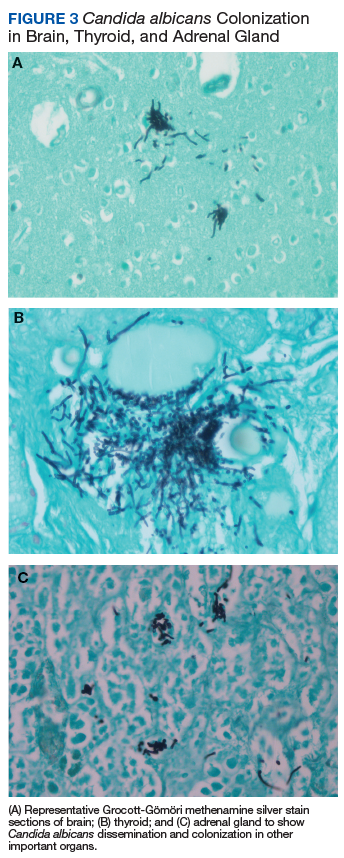
Most patients with the unusual diagnosis of candida pneumonia have had a background of malignancy or immunosuppressive factors (eg, administration of corticosteroids).12 In a series of 20 cases, 14 had sputum cultures positive for the organism, 6 had positive urine cultures, and 6 had positive blood cultures. Chest radiographs usually showed confluent bronchopneumonia. Five patients were diagnosed antemortem and treated with amphotericin B, but none survived.13 In the literature a positive blood culture or demonstration of yeast within pulmonary histiocytes has been considered proof of the pathogenicity of the fungus, as opposed to noninvasive colonization of the airways, a common occurrence in patients receiving mechanical ventilation.2
As previously discussed, blood cultures are often negative with invasive candidiasis, as the window of opportunity is short and may be missed. As shown in murine models, it is easy to miss a narrow window of candidemia, leading to false-negative blood cultures in clinical practice.14,15 Mouse model studies also have found that the window of candidemia is very short in disseminated candidiasis as a lethal IV dose of C albicans disappeared from blood within 48 hours of postinoculation.15 The biomarker of serum procalcitonin is a great diagnostic resource for the elimination of a likely bacterial sepsis, and conversely, the early suspicion of a fungemia, as serum procalcitonin would typically be elevated in a bacterial but not a fungal septicemia.16 The average cost per test is only about $30, and we recommend testing for serum procalcitonin as well as monitoring of serum lactate levels in cases of nonresponding septicemia.
The C albicans in this case may have been introduced hematogenously from the amputation site or through an ascending cystitis, or possibly have been derived from commensal flora in the GI tract. The iron supplementation provided to the patient may have promoted the growth and virulence of the candida; studies have shown that the kidneys assimilate increased levels of iron during disseminated candidiasis thus providing a more favorable site for colonization.17The presence of C albicans in a single collection of sputum or urine does not ordinarily indicate infection in an immunocompetent individual. Estimation of serum procalcitonin, a biomarker for bacterial infection and sepsis, might be useful if negative, for turning attention to a nonbacterial (such as, candida) source as the causative agent.18
Conclusion
C albicans can rarely cause disseminated disease in nonimmunocompromised critically ill patients. Low serum procalcitonin levels in a septic patient might indicate nonbacterial cause such as candidiasis. Even with disseminated candidiasis, blood cultures may remain negative.
1. Navarathna DH, Stein EV, Lessey-Morillon EC, Nayak D, Martin-Manso G, Roberts DD. CD47 promotes protective innate and adaptive immunity in a mouse model of disseminated candidiasis. PLoS One. 2015;10(5):e0128220.
2. Kullberg BJ, Arendrup MC. Invasive candidiasis. N Engl J Med. 2015;373(15):1445-1456.
3. Clancy CJ, Nguyen MH. Diagnosing invasive candidiasis. J Clin Microbiol. 2018;56(5):e01909-e01917.
4. Ericson EL, Klingspor L, Ullberg M, Ozenci V. Clinical comparison of the Bactec Mycosis IC/F, BacT/Alert FA, and BacT/Alert FN blood culture vials for the detection of candidemia. Diagn Microbiol Infect Dis. 2012;73(2):153-156.
5. Baum GL. The significance of Candida albicans in human sputum. N Engl J Med. 1960;263:70-73.
6. el-Ebiary M, Torres A, Fàbregas N, et al. Significance of the isolation of Candida species from respiratory samples in critically ill, non-neutropenic patients. An immediate postmortem histologic study. Am J Respir Crit Care Med. 1997;156(2, pt 1):583-590.
7. Altmeier S, Toska A, Sparber F, Teijeira A, Halin C, LeibundGut-Landmann S. IL-1 coordinates the neutrophil response to C. albicans in the oral mucosa. PLoS Pathog. 2016;12(9):e1005882.
8. Karavitis J, Kovacs EJ. Macrophage phagocytosis: effects of environmental pollutants, alcohol, cigarette smoke, and other external factors. J Leukoc Biol. 2011;90(6):1065-1078.
9. Chiu C-H, Wang Y-C, Yeh K-M, Lin J-C, Siu LK, Chang F-Y. Influence of ethanol concentration in the phagocytic function of neutrophils against Klebsiella pneumoniae isolates in an experimental model. J Microbiol Immunol Infect. 2018;51(1):64-69.
10. Khocht A, Schleifer S, Janal M, Keller S. Neutrophil function and periodontitis in alcohol-dependent males without medical disorders. J Int Acad Periodontol. 2013;15(3):68-74.
11. Gandhi JA, Ekhar VV, Asplund MB, et al. Alcohol enhances Acinetobacter baumannii-associated pneumonia and systemic dissemination by impairing neutrophil antimicrobial activity in a murine model of infection. PLoS One. 2014;9(4):e95707.
12. Mohsenifar Z, Chopra SK, Johnson BL, Simmons DH. Candida pneumonia: experience with 20 patients. West J Med. 1979;131(3):196-200.
13. Jones JM. Laboratory diagnosis of invasive candidiasis. Clin Microbiol Rev. 1990;3(1):32-45.
14. Clancy CJ, Nguyen MH. Finding the “missing 50%” of invasive candidiasis: how nonculture diagnostics will improve understanding of disease spectrum and transform patient care. Clin Infect Dis. 2013;56(9):1284-1292.
15. Kappe R, Mu¨ ller J. Rapid clearance of Candida albicans mannan antigens by liver and spleen in contrast to prolonged circulation of Cryptococcus neoformans antigens. J Clin Microbiol. 1991;29(8):1665-1669.
16. Balk RA, Kadri SS, Cao Z, Robinson SB, Lipkin C, Bozzette SA. Effect of procalcitonin testing on health-care utilization and costs in critically ill patients in the United States. Chest. 2017;151(1):23-33.
17. Potrykus J, Stead D, Maccallum DM, et al. Fungal iron availability during deep seated candidiasis is defined by a complex interplay involving systemic and local events. PLoS Pathog. 2013;9(10):e1003676.
18. Soni NJ, Samson DJ, Galaydick JL, Vats V, Pitrak DL, Aronson N. Procalcitonin-Guided Antibiotic Therapy. Rockville, MD: Agency for Healthcare Research and Quality (US); 2012.
Candida albicans (C albicans) is a normal commensal in the human gastrointestinal (GI) tract. In addition to localized infections in healthy human beings, dissemination with fatal outcome can occur in immunocompromised individuals.1
Invasive candidiasis (IC) due to C albicans is the most common nosocomial mycosis in the world and has 2 forms, candidemia and deep-seated tissue candidiasis, which can lead to multisystem organ failure.2 The deep-seated form may originate from nonhematogenous routes, such as introduction through a peritoneal catheter or ascending infection from cystitis.2 In addition, about 50% of primary candidemia cases lead to secondary deep-seated candidiasis; however, only about 40% of these cases show positive blood cultures. Since the window of opportunity for a positive culture is narrow, active candidemia may be missed.3,4
Once developed, the prognosis for IC is grim: Mortality is 40% regardless of therapy.2 IC typically occurs in immunocompromised hosts; IC in immunocompetent persons has rarely been reported.5,6 It is challenging to diagnose IC in the immunocompetent patients as 50% to 70% of the general population is naturally colonized by this organism, and when found, it is assumed to be mostly innocuous. Neutrophil-driven cell-mediated immunity associated with IL-1 and IL-17 response prevent fungal growth and dissemination, protecting the immunocompetent host.7
We report on a patient who showed no neutropenia or leukocytopenia but developed disseminated candidiasis. This report is one of the rare cases of full-blown disseminated candidiasis with lesions related to C albicans found in almost all of the important organs.
Case Presentation
A 67-year-old male patient with a history of hypertension, peripheral vascular disease, daily heavy alcohol consumption, and a 50-pack-year history of smoking developed gangrene of the left fifth toe. He underwent vascular surgery consultation with an aortogram/left lower extremity angiography that showed occlusion of the left external iliac artery as well as the left common femoral artery. It was decided to improve inflow in the common iliac artery by placing a bare metal stent and subsequent balloon dilatation before a right to left femoral to femoral artery bypass. The patient tolerated the procedure well and was discharged home.
Two days later, the patient was admitted to a US Department of Veterans Affairs (VA) complexity level 1a hospital with weakness and worsening pain in the left lower extremities. Examination revealed chronic ischemic changes in the feet bilaterally and evidence of dry gangrene in the left fifth toe requiring femoral bypass surgery. But poor nutritional status and cardiac status prevented pursuing a permanent solution.
Following completion of a stress echocardiogram, the patient developed shock with systolic blood pressure of 60 mm Hg, and atrial fibrillation (AF) with rapid ventricular rate (RVR). He was initially treated with IV fluid supplementation, vasopressor therapy, synchronized cardioversion, and IV amiodarone/anticoagulation therapy, due to his persistent AF with RVR. The patient was transferred to a tertiary care center for persistent hypothermia and received treatment with warm saline. After initial recovery with warm saline resuscitation, he had a prolonged, complicated hospital course in which he developed progressive respiratory failure requiring intubation and critical care support. He developed a right internal jugular deep venous thrombosis, heparin-induced thrombocytopenia, lower GI bleeding requiring emergent embolization by interventional radiology, inferior vena cava filter placement, renal failure requiring dialysis, small bowel obstruction secondary to right lower quadrant phlegmon and perforation requiring small bowel resection and end ileostomy. His antibiotic regimen included therapy with vancomycin and piperacillin-tazobactam.
He eventually recovered and was extubated and subsequently transferred back to the VA hospital where cefepime was initiated because of suspicion of a urinary tract infection and septicemia (urine cultures eventually grew C albicans). Over the subsequent 3 days, the patient’s renal output and hyperkalemia worsened, he also developed increased anion gap metabolic acidosis and was intubated again and placed on full mechanical ventilatory support. His blood cultures were negative, and sputum cultures revealed normal respiratory flora and 1+ C albicans. Infectious diseases consultation recommended an abdominal ultrasound, which revealed nonspecific findings. The antibiotic regimen was changed to daptomycin and piperacillin-tazobactam. A follow-up chest X-ray revealed a developing right lower lobe pneumonia and hilar prominence suggestive of lymphadenopathy. The patient’s clinical condition deteriorated, and he subsequently developed cardiac arrest; resuscitation was not successful and he expired.
Outcome and Follow-up
An autopsy disclosed the cause of death to be bilateral candida pneumonia, part of a disseminated (invasive) candidiasis, in a patient rendered vulnerable to such infection by peripheral vascular disease and renal insufficiency. Purulent inflammation was noted at the site of disarticulation of the left foot and confluent consolidation of the lower lobes of both lungs as well as focal consolidation of the middle lobe of the right lung. Examination of histologic sections, with staining both by routine method (hematoxylin and eosin) and the Grocott-Gömöri methenamine silver method for fungus, disclosed fungal forms (yeast and filamentous) in most tissues, including the lungs (Figure 1 A and B) and kidneys (Figure 1 C and D). The pulmonary sections in addition to massive inflammation showed macrophages with engulfed yeast (Figure 2 A) and a lymphatic channel, stuffed with yeast in an alveolar septum (Figure 2 B). These findings confirmed the antemortem presence of the fungus and the body’s response to it. Inflammation was noted around glomeruli overgrown by candida (Figure 1 C and D); fungi also were seen in capsular regions (not depicted). C albicans was present in the myocardium (Figure 1 E and F), brain, thyroid, and adrenal glands (Figure 3); the only organ without C albicans was the liver, either because invasion was truly absent here or because sampling had not managed to retrieve it.
Paraffin-embedded blocks of lung tissue, sent to the University of Washington Molecular Diagnosis Microbiology Laboratory for broad-range polymerase chain reaction (PCR) identification, were positive for C albicans after extraction of gDNA and conduction of PCR using internal transcribed spacer 1 and 2 specific primers.
Discussion
IC is rare among immunocompetent individuals, but C albicans can evolve into a fatal disseminated infection. We report an atypical case of IC, with profound pulmonary infection in a patient who died 1 month after hospitalization for lower extremity pain.
Cell-mediated immunity involving neutrophils and macrophages plays a major role in protection against candidiasis, while cytokines and chemokines involve regulating balanced immunity.1,2 A series of recent studies show that alcohol impairs neutrophil-mediated killing and phagocytic-mediated uptake of a pathogen in this process.8,9 As the patient chronically misused alcohol, his immune system may have experienced a subclinical immunosuppression, which would have become clinically relevant once C albicans was introduced systemically. Recent studies of bacterial pathogenesis and alcoholism strongly support this hypothesis.10,11
Most patients with the unusual diagnosis of candida pneumonia have had a background of malignancy or immunosuppressive factors (eg, administration of corticosteroids).12 In a series of 20 cases, 14 had sputum cultures positive for the organism, 6 had positive urine cultures, and 6 had positive blood cultures. Chest radiographs usually showed confluent bronchopneumonia. Five patients were diagnosed antemortem and treated with amphotericin B, but none survived.13 In the literature a positive blood culture or demonstration of yeast within pulmonary histiocytes has been considered proof of the pathogenicity of the fungus, as opposed to noninvasive colonization of the airways, a common occurrence in patients receiving mechanical ventilation.2
As previously discussed, blood cultures are often negative with invasive candidiasis, as the window of opportunity is short and may be missed. As shown in murine models, it is easy to miss a narrow window of candidemia, leading to false-negative blood cultures in clinical practice.14,15 Mouse model studies also have found that the window of candidemia is very short in disseminated candidiasis as a lethal IV dose of C albicans disappeared from blood within 48 hours of postinoculation.15 The biomarker of serum procalcitonin is a great diagnostic resource for the elimination of a likely bacterial sepsis, and conversely, the early suspicion of a fungemia, as serum procalcitonin would typically be elevated in a bacterial but not a fungal septicemia.16 The average cost per test is only about $30, and we recommend testing for serum procalcitonin as well as monitoring of serum lactate levels in cases of nonresponding septicemia.
The C albicans in this case may have been introduced hematogenously from the amputation site or through an ascending cystitis, or possibly have been derived from commensal flora in the GI tract. The iron supplementation provided to the patient may have promoted the growth and virulence of the candida; studies have shown that the kidneys assimilate increased levels of iron during disseminated candidiasis thus providing a more favorable site for colonization.17The presence of C albicans in a single collection of sputum or urine does not ordinarily indicate infection in an immunocompetent individual. Estimation of serum procalcitonin, a biomarker for bacterial infection and sepsis, might be useful if negative, for turning attention to a nonbacterial (such as, candida) source as the causative agent.18
Conclusion
C albicans can rarely cause disseminated disease in nonimmunocompromised critically ill patients. Low serum procalcitonin levels in a septic patient might indicate nonbacterial cause such as candidiasis. Even with disseminated candidiasis, blood cultures may remain negative.
Candida albicans (C albicans) is a normal commensal in the human gastrointestinal (GI) tract. In addition to localized infections in healthy human beings, dissemination with fatal outcome can occur in immunocompromised individuals.1
Invasive candidiasis (IC) due to C albicans is the most common nosocomial mycosis in the world and has 2 forms, candidemia and deep-seated tissue candidiasis, which can lead to multisystem organ failure.2 The deep-seated form may originate from nonhematogenous routes, such as introduction through a peritoneal catheter or ascending infection from cystitis.2 In addition, about 50% of primary candidemia cases lead to secondary deep-seated candidiasis; however, only about 40% of these cases show positive blood cultures. Since the window of opportunity for a positive culture is narrow, active candidemia may be missed.3,4
Once developed, the prognosis for IC is grim: Mortality is 40% regardless of therapy.2 IC typically occurs in immunocompromised hosts; IC in immunocompetent persons has rarely been reported.5,6 It is challenging to diagnose IC in the immunocompetent patients as 50% to 70% of the general population is naturally colonized by this organism, and when found, it is assumed to be mostly innocuous. Neutrophil-driven cell-mediated immunity associated with IL-1 and IL-17 response prevent fungal growth and dissemination, protecting the immunocompetent host.7
We report on a patient who showed no neutropenia or leukocytopenia but developed disseminated candidiasis. This report is one of the rare cases of full-blown disseminated candidiasis with lesions related to C albicans found in almost all of the important organs.
Case Presentation
A 67-year-old male patient with a history of hypertension, peripheral vascular disease, daily heavy alcohol consumption, and a 50-pack-year history of smoking developed gangrene of the left fifth toe. He underwent vascular surgery consultation with an aortogram/left lower extremity angiography that showed occlusion of the left external iliac artery as well as the left common femoral artery. It was decided to improve inflow in the common iliac artery by placing a bare metal stent and subsequent balloon dilatation before a right to left femoral to femoral artery bypass. The patient tolerated the procedure well and was discharged home.
Two days later, the patient was admitted to a US Department of Veterans Affairs (VA) complexity level 1a hospital with weakness and worsening pain in the left lower extremities. Examination revealed chronic ischemic changes in the feet bilaterally and evidence of dry gangrene in the left fifth toe requiring femoral bypass surgery. But poor nutritional status and cardiac status prevented pursuing a permanent solution.
Following completion of a stress echocardiogram, the patient developed shock with systolic blood pressure of 60 mm Hg, and atrial fibrillation (AF) with rapid ventricular rate (RVR). He was initially treated with IV fluid supplementation, vasopressor therapy, synchronized cardioversion, and IV amiodarone/anticoagulation therapy, due to his persistent AF with RVR. The patient was transferred to a tertiary care center for persistent hypothermia and received treatment with warm saline. After initial recovery with warm saline resuscitation, he had a prolonged, complicated hospital course in which he developed progressive respiratory failure requiring intubation and critical care support. He developed a right internal jugular deep venous thrombosis, heparin-induced thrombocytopenia, lower GI bleeding requiring emergent embolization by interventional radiology, inferior vena cava filter placement, renal failure requiring dialysis, small bowel obstruction secondary to right lower quadrant phlegmon and perforation requiring small bowel resection and end ileostomy. His antibiotic regimen included therapy with vancomycin and piperacillin-tazobactam.
He eventually recovered and was extubated and subsequently transferred back to the VA hospital where cefepime was initiated because of suspicion of a urinary tract infection and septicemia (urine cultures eventually grew C albicans). Over the subsequent 3 days, the patient’s renal output and hyperkalemia worsened, he also developed increased anion gap metabolic acidosis and was intubated again and placed on full mechanical ventilatory support. His blood cultures were negative, and sputum cultures revealed normal respiratory flora and 1+ C albicans. Infectious diseases consultation recommended an abdominal ultrasound, which revealed nonspecific findings. The antibiotic regimen was changed to daptomycin and piperacillin-tazobactam. A follow-up chest X-ray revealed a developing right lower lobe pneumonia and hilar prominence suggestive of lymphadenopathy. The patient’s clinical condition deteriorated, and he subsequently developed cardiac arrest; resuscitation was not successful and he expired.
Outcome and Follow-up
An autopsy disclosed the cause of death to be bilateral candida pneumonia, part of a disseminated (invasive) candidiasis, in a patient rendered vulnerable to such infection by peripheral vascular disease and renal insufficiency. Purulent inflammation was noted at the site of disarticulation of the left foot and confluent consolidation of the lower lobes of both lungs as well as focal consolidation of the middle lobe of the right lung. Examination of histologic sections, with staining both by routine method (hematoxylin and eosin) and the Grocott-Gömöri methenamine silver method for fungus, disclosed fungal forms (yeast and filamentous) in most tissues, including the lungs (Figure 1 A and B) and kidneys (Figure 1 C and D). The pulmonary sections in addition to massive inflammation showed macrophages with engulfed yeast (Figure 2 A) and a lymphatic channel, stuffed with yeast in an alveolar septum (Figure 2 B). These findings confirmed the antemortem presence of the fungus and the body’s response to it. Inflammation was noted around glomeruli overgrown by candida (Figure 1 C and D); fungi also were seen in capsular regions (not depicted). C albicans was present in the myocardium (Figure 1 E and F), brain, thyroid, and adrenal glands (Figure 3); the only organ without C albicans was the liver, either because invasion was truly absent here or because sampling had not managed to retrieve it.
Paraffin-embedded blocks of lung tissue, sent to the University of Washington Molecular Diagnosis Microbiology Laboratory for broad-range polymerase chain reaction (PCR) identification, were positive for C albicans after extraction of gDNA and conduction of PCR using internal transcribed spacer 1 and 2 specific primers.
Discussion
IC is rare among immunocompetent individuals, but C albicans can evolve into a fatal disseminated infection. We report an atypical case of IC, with profound pulmonary infection in a patient who died 1 month after hospitalization for lower extremity pain.
Cell-mediated immunity involving neutrophils and macrophages plays a major role in protection against candidiasis, while cytokines and chemokines involve regulating balanced immunity.1,2 A series of recent studies show that alcohol impairs neutrophil-mediated killing and phagocytic-mediated uptake of a pathogen in this process.8,9 As the patient chronically misused alcohol, his immune system may have experienced a subclinical immunosuppression, which would have become clinically relevant once C albicans was introduced systemically. Recent studies of bacterial pathogenesis and alcoholism strongly support this hypothesis.10,11
Most patients with the unusual diagnosis of candida pneumonia have had a background of malignancy or immunosuppressive factors (eg, administration of corticosteroids).12 In a series of 20 cases, 14 had sputum cultures positive for the organism, 6 had positive urine cultures, and 6 had positive blood cultures. Chest radiographs usually showed confluent bronchopneumonia. Five patients were diagnosed antemortem and treated with amphotericin B, but none survived.13 In the literature a positive blood culture or demonstration of yeast within pulmonary histiocytes has been considered proof of the pathogenicity of the fungus, as opposed to noninvasive colonization of the airways, a common occurrence in patients receiving mechanical ventilation.2
As previously discussed, blood cultures are often negative with invasive candidiasis, as the window of opportunity is short and may be missed. As shown in murine models, it is easy to miss a narrow window of candidemia, leading to false-negative blood cultures in clinical practice.14,15 Mouse model studies also have found that the window of candidemia is very short in disseminated candidiasis as a lethal IV dose of C albicans disappeared from blood within 48 hours of postinoculation.15 The biomarker of serum procalcitonin is a great diagnostic resource for the elimination of a likely bacterial sepsis, and conversely, the early suspicion of a fungemia, as serum procalcitonin would typically be elevated in a bacterial but not a fungal septicemia.16 The average cost per test is only about $30, and we recommend testing for serum procalcitonin as well as monitoring of serum lactate levels in cases of nonresponding septicemia.
The C albicans in this case may have been introduced hematogenously from the amputation site or through an ascending cystitis, or possibly have been derived from commensal flora in the GI tract. The iron supplementation provided to the patient may have promoted the growth and virulence of the candida; studies have shown that the kidneys assimilate increased levels of iron during disseminated candidiasis thus providing a more favorable site for colonization.17The presence of C albicans in a single collection of sputum or urine does not ordinarily indicate infection in an immunocompetent individual. Estimation of serum procalcitonin, a biomarker for bacterial infection and sepsis, might be useful if negative, for turning attention to a nonbacterial (such as, candida) source as the causative agent.18
Conclusion
C albicans can rarely cause disseminated disease in nonimmunocompromised critically ill patients. Low serum procalcitonin levels in a septic patient might indicate nonbacterial cause such as candidiasis. Even with disseminated candidiasis, blood cultures may remain negative.
1. Navarathna DH, Stein EV, Lessey-Morillon EC, Nayak D, Martin-Manso G, Roberts DD. CD47 promotes protective innate and adaptive immunity in a mouse model of disseminated candidiasis. PLoS One. 2015;10(5):e0128220.
2. Kullberg BJ, Arendrup MC. Invasive candidiasis. N Engl J Med. 2015;373(15):1445-1456.
3. Clancy CJ, Nguyen MH. Diagnosing invasive candidiasis. J Clin Microbiol. 2018;56(5):e01909-e01917.
4. Ericson EL, Klingspor L, Ullberg M, Ozenci V. Clinical comparison of the Bactec Mycosis IC/F, BacT/Alert FA, and BacT/Alert FN blood culture vials for the detection of candidemia. Diagn Microbiol Infect Dis. 2012;73(2):153-156.
5. Baum GL. The significance of Candida albicans in human sputum. N Engl J Med. 1960;263:70-73.
6. el-Ebiary M, Torres A, Fàbregas N, et al. Significance of the isolation of Candida species from respiratory samples in critically ill, non-neutropenic patients. An immediate postmortem histologic study. Am J Respir Crit Care Med. 1997;156(2, pt 1):583-590.
7. Altmeier S, Toska A, Sparber F, Teijeira A, Halin C, LeibundGut-Landmann S. IL-1 coordinates the neutrophil response to C. albicans in the oral mucosa. PLoS Pathog. 2016;12(9):e1005882.
8. Karavitis J, Kovacs EJ. Macrophage phagocytosis: effects of environmental pollutants, alcohol, cigarette smoke, and other external factors. J Leukoc Biol. 2011;90(6):1065-1078.
9. Chiu C-H, Wang Y-C, Yeh K-M, Lin J-C, Siu LK, Chang F-Y. Influence of ethanol concentration in the phagocytic function of neutrophils against Klebsiella pneumoniae isolates in an experimental model. J Microbiol Immunol Infect. 2018;51(1):64-69.
10. Khocht A, Schleifer S, Janal M, Keller S. Neutrophil function and periodontitis in alcohol-dependent males without medical disorders. J Int Acad Periodontol. 2013;15(3):68-74.
11. Gandhi JA, Ekhar VV, Asplund MB, et al. Alcohol enhances Acinetobacter baumannii-associated pneumonia and systemic dissemination by impairing neutrophil antimicrobial activity in a murine model of infection. PLoS One. 2014;9(4):e95707.
12. Mohsenifar Z, Chopra SK, Johnson BL, Simmons DH. Candida pneumonia: experience with 20 patients. West J Med. 1979;131(3):196-200.
13. Jones JM. Laboratory diagnosis of invasive candidiasis. Clin Microbiol Rev. 1990;3(1):32-45.
14. Clancy CJ, Nguyen MH. Finding the “missing 50%” of invasive candidiasis: how nonculture diagnostics will improve understanding of disease spectrum and transform patient care. Clin Infect Dis. 2013;56(9):1284-1292.
15. Kappe R, Mu¨ ller J. Rapid clearance of Candida albicans mannan antigens by liver and spleen in contrast to prolonged circulation of Cryptococcus neoformans antigens. J Clin Microbiol. 1991;29(8):1665-1669.
16. Balk RA, Kadri SS, Cao Z, Robinson SB, Lipkin C, Bozzette SA. Effect of procalcitonin testing on health-care utilization and costs in critically ill patients in the United States. Chest. 2017;151(1):23-33.
17. Potrykus J, Stead D, Maccallum DM, et al. Fungal iron availability during deep seated candidiasis is defined by a complex interplay involving systemic and local events. PLoS Pathog. 2013;9(10):e1003676.
18. Soni NJ, Samson DJ, Galaydick JL, Vats V, Pitrak DL, Aronson N. Procalcitonin-Guided Antibiotic Therapy. Rockville, MD: Agency for Healthcare Research and Quality (US); 2012.
1. Navarathna DH, Stein EV, Lessey-Morillon EC, Nayak D, Martin-Manso G, Roberts DD. CD47 promotes protective innate and adaptive immunity in a mouse model of disseminated candidiasis. PLoS One. 2015;10(5):e0128220.
2. Kullberg BJ, Arendrup MC. Invasive candidiasis. N Engl J Med. 2015;373(15):1445-1456.
3. Clancy CJ, Nguyen MH. Diagnosing invasive candidiasis. J Clin Microbiol. 2018;56(5):e01909-e01917.
4. Ericson EL, Klingspor L, Ullberg M, Ozenci V. Clinical comparison of the Bactec Mycosis IC/F, BacT/Alert FA, and BacT/Alert FN blood culture vials for the detection of candidemia. Diagn Microbiol Infect Dis. 2012;73(2):153-156.
5. Baum GL. The significance of Candida albicans in human sputum. N Engl J Med. 1960;263:70-73.
6. el-Ebiary M, Torres A, Fàbregas N, et al. Significance of the isolation of Candida species from respiratory samples in critically ill, non-neutropenic patients. An immediate postmortem histologic study. Am J Respir Crit Care Med. 1997;156(2, pt 1):583-590.
7. Altmeier S, Toska A, Sparber F, Teijeira A, Halin C, LeibundGut-Landmann S. IL-1 coordinates the neutrophil response to C. albicans in the oral mucosa. PLoS Pathog. 2016;12(9):e1005882.
8. Karavitis J, Kovacs EJ. Macrophage phagocytosis: effects of environmental pollutants, alcohol, cigarette smoke, and other external factors. J Leukoc Biol. 2011;90(6):1065-1078.
9. Chiu C-H, Wang Y-C, Yeh K-M, Lin J-C, Siu LK, Chang F-Y. Influence of ethanol concentration in the phagocytic function of neutrophils against Klebsiella pneumoniae isolates in an experimental model. J Microbiol Immunol Infect. 2018;51(1):64-69.
10. Khocht A, Schleifer S, Janal M, Keller S. Neutrophil function and periodontitis in alcohol-dependent males without medical disorders. J Int Acad Periodontol. 2013;15(3):68-74.
11. Gandhi JA, Ekhar VV, Asplund MB, et al. Alcohol enhances Acinetobacter baumannii-associated pneumonia and systemic dissemination by impairing neutrophil antimicrobial activity in a murine model of infection. PLoS One. 2014;9(4):e95707.
12. Mohsenifar Z, Chopra SK, Johnson BL, Simmons DH. Candida pneumonia: experience with 20 patients. West J Med. 1979;131(3):196-200.
13. Jones JM. Laboratory diagnosis of invasive candidiasis. Clin Microbiol Rev. 1990;3(1):32-45.
14. Clancy CJ, Nguyen MH. Finding the “missing 50%” of invasive candidiasis: how nonculture diagnostics will improve understanding of disease spectrum and transform patient care. Clin Infect Dis. 2013;56(9):1284-1292.
15. Kappe R, Mu¨ ller J. Rapid clearance of Candida albicans mannan antigens by liver and spleen in contrast to prolonged circulation of Cryptococcus neoformans antigens. J Clin Microbiol. 1991;29(8):1665-1669.
16. Balk RA, Kadri SS, Cao Z, Robinson SB, Lipkin C, Bozzette SA. Effect of procalcitonin testing on health-care utilization and costs in critically ill patients in the United States. Chest. 2017;151(1):23-33.
17. Potrykus J, Stead D, Maccallum DM, et al. Fungal iron availability during deep seated candidiasis is defined by a complex interplay involving systemic and local events. PLoS Pathog. 2013;9(10):e1003676.
18. Soni NJ, Samson DJ, Galaydick JL, Vats V, Pitrak DL, Aronson N. Procalcitonin-Guided Antibiotic Therapy. Rockville, MD: Agency for Healthcare Research and Quality (US); 2012.
A Veteran Presenting With Leg Swelling, Dyspnea, and Proteinuria
*This article has been corrected to include a missing author.
Case Presentation. A 63-year-old male with well-controlled HIV (CD4 count 757, undetectable viral load), epilepsy, and hypertension presented to the VA Boston Healthcare System (VABHS) emergency department with 1 week of bilateral leg swelling and exertional shortness of breath. He reported having no fever, cough, chest pain, pain with inspiration and orthopnea. There was no personal or family history of pulmonary embolism. He reported weight gain but was unable to quantify how much. He also reported flare up of chronic knee pain, without swelling for which he had taken up to 4 tablets of naproxen daily for several weeks. His physical examination was notable for a heart rate of 105 beats per minute and bilateral pitting edema to his knees. Laboratory testing revealed a creatinine level of 2.5 mg/dL, which was increased from a baseline of 1.0 mg/dL (Table 1), and a urine protein-to-creatinine ratio of 7.8 mg/mg (Table 2). A renal ultrasound showed normal-sized kidneys without hydronephrosis or obstructing renal calculi. The patient was admitted for further workup of his dyspnea and acute kidney injury.
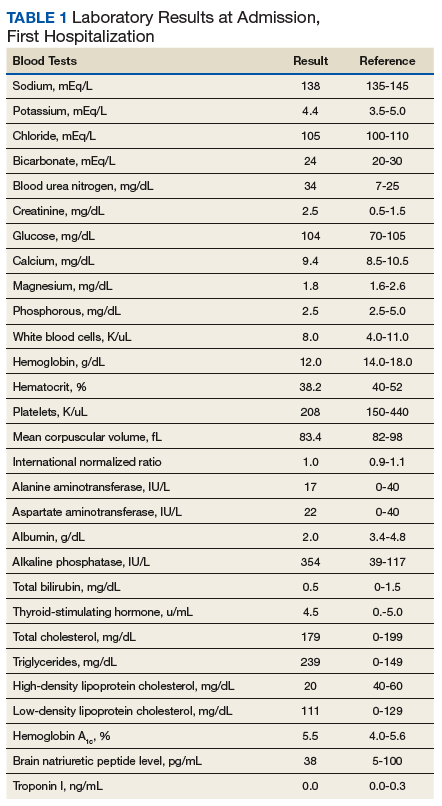
► Jonathan Li, MD, Chief Medical Resident, VABHS and Beth Israel Deaconess Medical Center (BIDMC). Dr. William, based on the degree of proteinuria and edema, a diagnosis of nephrotic syndrome was made. How is nephrotic syndrome defined, and how is it distinguished from glomerulonephritis?
► Jeffrey William, MD, Nephrologist, BIDMC, Assistant Professor of Medicine, Harvard Medical School. The pathophysiology of nephrotic disease and glomerulonephritis are quite distinct, resulting in symptoms and systemic manifestations that only slightly overlap. Glomerulonephritis is characterized by inflammation of the endothelial cells of the trilayered glomerular capillary, with a resulting active urine sediment with red blood cells, white blood cells, and casts. Nephrotic syndrome mostly affects the visceral epithelial cells of the glomerular capillary, commonly referred to as podocytes, and hence, the urine sediment in nephrotic disease is often inactive. Patients with nephrotic syndrome have nephrotic-range proteinuria (excretion of > 3.5 g per 24 h or a spot urine protein-creatinine ratio > 3.5 g in the steady state) and both hypoalbuminemia (< 3 g/dL) and peripheral edema. Lipiduria and hyperlipidemia are common findings in nephrotic syndrome but are not required for a clinical diagnosis.1 In contrast, glomerulonephritis is defined by a constellation of findings that include renal insufficiency (often indicated by an elevation in blood urea nitrogen and creatinine), hypertension, hematuria, and subnephrotic range proteinuria. In practice, patients may fulfill criteria of both nephrotic and nephritic syndromes, but the preponderance of clinical evidence often points one way or the other. In this case, nephrotic syndrome was diagnosed based on the urine protein-to-creatinine ratio of 7.8 mg/mg, hypoalbuminemia, and edema.
► Dr. Li. What would be your first-line workup for evaluation of the etiology of this patient’s nephrotic syndrome?
► Dr. William. Rather than memorizing a list of etiologies of nephrotic syndrome, it is essential to consider the pathophysiology of heavy proteinuria. Though the glomerular filtration barrier is extremely complex and defects in any component can cause proteinuria, disruption of the podocyte is often involved. Common disease processes that chiefly target the podocyte include minimal change disease, primary focal and segmental glomerulosclerosis (FSGS), and membranous nephropathy, all by differing mechanisms. Minimal change disease and idiopathic/primary FSGS are increasingly thought to be at differing points on a spectrum of the same disease.2 Secondary FSGS, on the other hand, is a progressive disease, commonly resulting from longstanding hypertension, diabetes mellitus, and obesity in adults. Membranous nephropathy can also be either primary or secondary. Primary membranous nephropathy is chiefly caused by a circulating IgG4 antibody to the podocyte membrane antigen PLA2R (M-type phospholipase A2 receptor), whereas secondary membranous nephropathy can be caused by a variety of systemic etiologies, including autoimmune disease (eg, systemic lupus erythematosus), certain malignancies, chronic infections (eg, hepatitis B and C), and many medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).3-5 Paraprotein deposition diseases can also cause glomerular damage leading to nephrotic-range proteinuria.
Given these potential diagnoses, a careful history should be taken to assess exposures and recent medication use. Urine sediment evaluation is essential in the evaluation of nephrotic syndrome to determine if there is an underlying nephritic process. Select serologies may be sent to look for autoimmune disease, such as systemic lupus erythematosus and common viral exposures like hepatitis B or C. Serum and urine protein electrophoreses would be appropriate initial tests of suspected paraprotein-related diseases. Other serologies, such as antineutrophil cytoplasmic antibodies or antiglomerular basement membrane antibodies, would not necessarily be indicated here given the lack of hematuria and presence of nephrotic-range proteinuria.
► Dr. Li. The initial evaluation was notable for an erythrocyte sedimentation rate > 120 (mm/h) and a weakly positive antinuclear antibody (ANA) titer of 1:40. The remainder of his initial workup did not reveal an etiology for his nephrotic syndrome (Table 3).
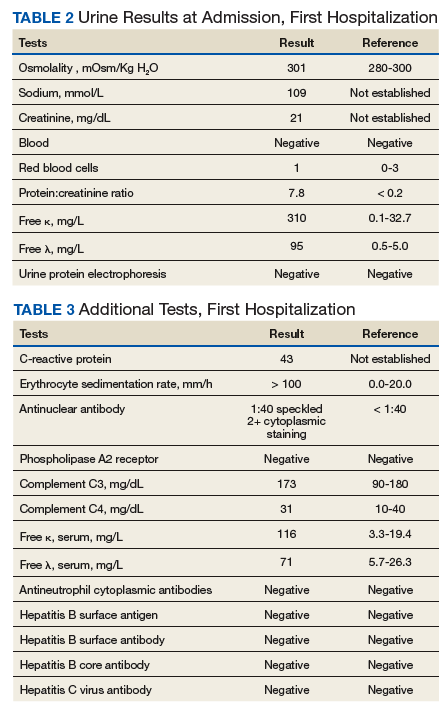
Dr. William, is there a role for starting urgent empiric steroids in nephrotic syndrome while workup is ongoing? If so, do the severity of proteinuria and/or symptoms play a role or is this determination based on something else?
► Dr. William. Edema is a primary symptom of nephrotic syndrome and can often be managed with diuretics alone. If a clear medication-mediated cause is suspected, discontinuation of this agent may result in spontaneous improvement without steroid treatment. However,in cases where an etiology is unclear and there are serious thrombotic complications requiring anticoagulation, and a renal biopsy is deemed to be too risky, then empiric steroid therapy may be necessary. Children with new-onset nephrotic syndrome are presumed to have minimal change disease, given its prevalence in this patient population, and are often given empiric steroids without obtaining a renal biopsy. However, in the adult population, a renal biopsy can typically be performed quickly and safely, with pathology results interpreted within days. In this patient, since a diagnosis was unclear and there was no contraindication to renal biopsy, a biopsy should be obtained before consideration of steroids.
► Dr. Li. Steroids were deferred in anticipation of renal biopsy, which showed stage I membranous nephropathy, suggestive of membranous lupus nephritis Class V. The deposits were strongly reactive for immunoglobuline G (IgG), IgA, and complement 1q (C1q), showed co-dominant staining for IgG1, IgG2, and IgG3, and were weakly positive for the PLA2 receptor. Focal intimal arteritis in a small interlobular vessel was seen.
Dr. William, the pathology returned suggestive of lupus nephritis. Does the overall clinical picture fit with lupus nephritis?
► Dr. William. Given the history and a rather low ANA, the diagnosis of lupus nephritis seems unlikely. The lack of IgG4 and PLA2R staining in the biopsy suggests that this membranous pattern on the biopsy is likely to be secondary to a systemic etiology, but further investigation should be pursued.
► Dr. Li. The patient was discharged after the biopsy with a planned outpatient nephrology follow-up to discuss results and treatment. He was prescribed an oral diuretic, and his symptoms improved. Several days after discharge, he developed blurry vision and was evaluated in the Ophthalmology clinic. On fundoscopy, he was found to have acute papillitis, a form of optic neuritis. As part of initial evaluation of infectious etiologies of papillitis, ophthalmology recommended testing for syphilis.
Dr. Strymish, when we are considering secondary syphilis, what is the recommended approach to diagnostic testing?
► Judith Strymish, MD, Infectious Diseases, BIDMC, Assistant Professor of Medicine, Harvard Medical School. The diagnosis of syphilis is usually made through serologic testing of blood specimens. Methods that detect the spirochete directly like dark-field smears are not readily available. Serologic tests include treponemal tests (eg, Treponema pallidum particle agglutination assay [TPPA]) and nontreponemal tests (eg, rapid plasma reagin [RPR]). One needs a confirmatory test because either test is associated with false positives. Either test can be done first. Most laboratories, including those at VABHS are now performing treponemal tests first as these have become more cost-effective.6 The TPPA treponemal test was found to have a lower false negative rate in primary syphilis compared with that of nontreponemal tests.7 Nontreponemal tests can be followed for response to therapy. If a patient has a history of treated syphilis, a nontreponemal test should be sent, since the treponemal test will remain positive for life.
If there is clinical concern for neurosyphilis, cerebrospinal fluid fluorescent (CSF) treponemal antibody needs to be sampled and sent for the nontreponemal venereal disease research laboratory (VDRL) test. The VDRL is highly specific for neurosyphilis but not as sensitive. Cerebrospinal fluid fluorescent treponemal antibody (CSF FTA) may also be sent; it is very sensitive but not very specific for neurosyphilis.
► Dr. Li. An RPR returned positive at 1:512 (was negative 14 months prior on a routine screening test), with positive reflex TPPA (Table 4). A diagnosis of secondary syphilis was made. Dr. Strymish, at this point, what additional testing and treatment is necessary?
► Dr. Strymish. With papillitis and a very high RPR, we need to assume that he has ophthalmic syphilis. This can occur in any stage of syphilis, but his eye findings and high RPR are consistent with secondary syphilis. Ophthalmic syphilis has been on the upswing, even more than is expected with recent increases in syphilis cases.8 Ophthalmic syphilis is considered a form of neurosyphilis. A lumbar puncture and treatment for neurosyphilis is recommended.9,10
► Dr. Li. A lumbar puncture was performed, and his CSF was VDRL positive. This confirmed a diagnosis of neurosyphilis (Table 4). The patient was treated for neurosyphilis with IV penicillin. The patient shared that he had episodes of unprotected oral sexual activity within the past year and approximately 1 year ago, he came in close contact (but no sexual activity) with a person who had a rash consistent with syphilis.Dr. William, syphilis would be a potential unifying diagnosis of his renal and ophthalmologic manifestations. Is syphilis known to cause membranous nephropathy?
► Dr. William. Though it is uncommon, the nephrotic syndrome is a well-described complication of secondary syphilis.11,12 Syphilis has been shown to cause nephrotic syndrome in a variety of ways. Case reports abound linking syphilis to minimal change disease and other glomerular diseases.13,14 A case report from 1993 shows a membranous pattern of glomerular disease similar to this case.15 As a form of secondary membranous nephropathy, the immunofluorescence pattern can demonstrate staining similar to the “full house” seen in lupus nephritis (IgA, IgM, and C1q, in addition to IgG and C3).16 This explains the initial interpretation of this patient’s biopsy, as lupus nephritis would be a much more common etiology of secondary membranous nephropathy than is acute syphilis with this immunofluorescence pattern. However, the data in this case are highly suggestive of a causal relationship between secondary syphilis and membranous nephropathy.
► Dr. Li. Dr. Strymish, how should this patient be screened for syphilis reinfection, and at what intervals would you recommend?
► Dr. Strymish. He will need follow-up testing to make sure that his syphilis is effectively treated. If CSF pleocytosis was present initially, a CSF examination should be repeated every 6 months until the cell count is normal. He will also need follow-up for normalization of his RPR. Persons with HIV infection and primary or secondary syphilis should be evaluated clinically and serologically for treatment failure at 3, 6, 9, 12, and 24 months after therapy according to US Centers for Disease Control and Prevention guidelines.9
His treponemal test for syphilis will likely stay positive for life. His RPR should decrease significantly with effective treatment. It makes sense to screen with RPR alone as long as he continues to have risk factors for acquiring syphilis. Routine syphilis testing is recommended for pregnant women, sexually active men who have sex with men, sexually active persons with HIV, and persons taking PrEP (pre-exposure prophylaxis) for HIV prevention. He should be screened at least yearly for syphilis.

► Dr. Li. Over the next several months, the patient’s creatinine normalized and his proteinuria resolved. His vision recovered, and he has had no further ophthalmologic complications.
Dr. William, what is his long-term renal prognosis? Do you expect that his acute episode of membranous nephropathy will have permanent effects on his renal function?
► Dr. William. His rapid response to therapy for neurosyphilis provides evidence for this etiology of his renal dysfunction and glomerulonephritis. His long-term prognosis is quite good if the syphilis is the only reason for him to have renal disease. The renal damage is often reversible in these cases. However, given his prior extensive NSAID exposure and history of hypertension, he may be at higher risk for chronic kidney disease than an otherwise healthy patient, especially after an episode of acute kidney injury. Therefore, his renal function should continue to be monitored as an outpatient.
Acknowledgments
The authors thank this veteran for sharing his story and allowing us to learn from this unusual case for the benefit of our future patients.
1. Rennke H, Denker BM. Renal Pathophysiology: The Essentials. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2014.
2. Maas RJ, Deegens JK, Smeets B, Moeller MJ, Wetzels JF. Minimal change disease and idiopathic FSGS: manifestations of the same disease. Nat Rev Nephrol. 2016;12(12):768-776.
3. Beck LH Jr, Bonegio RG, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361(1):11-21.
4. Rennke HG. Secondary membranoproliferative glomerulonephritis. Kidney Int. 1995;47(2):643-656.
5. Nawaz FA, Larsen CP, Troxell ML. Membranous nephropathy and nonsteroidal anti-inflammatory agents. Am J Kidney Dis. 2013;62(5):1012-1017.
6. Pillay A. Centers for Disease Control and Prevention Syphilis Summit—Diagnostics and laboratory issues. Sex Transm Dis. 2018;45(9S)(suppl 1):S13-S16.
7. Levett PN, Fonseca K, Tsang RS, et al. Canadian Public Health Laboratory Network laboratory guidelines for the use of serological tests (excluding point-of-care tests) for the diagnosis of syphilis in Canada. Can J Infect Dis Med Microbiol. 2015;26(suppl A):6A-12A.
8. Oliver SE, Aubin M, Atwell L, et al. Ocular syphilis—eight jurisdictions, United States, 2014-2015. MMWR Morb Mortal Wkly Rep. 2016;65(43):1185-1188.
9. Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recommendations and Reports 2015;64(RR3):1-137. [Erratum in MMWR Recomm Rep. 2015;64(33):924.]
10. US Centers for Disease Control and Prevention. Clinical advisory: ocular syphilis in the United States. https://www.cdc.gov/std/syphilis/clinicaladvisoryos2015.htm. Updated March 24, 2016. Accessed August 12, 2019.
11. Braunstein GD, Lewis EJ, Galvanek EG, Hamilton A, Bell WR. The nephrotic syndrome associated with secondary syphilis: an immune deposit disease. Am J Med. 1970;48:643-648.1.
12. Handoko ML, Duijvestein M, Scheepstra CG, de Fijter CW. Syphilis: a reversible cause of nephrotic syndrome. BMJ Case Rep. 2013;2013:pii:bcr2012008279
13. Krane NK, Espenan P, Walker PD, Bergman SM, Wallin JD. Renal disease and syphilis: a report of nephrotic syndrome with minimal change disease. Am J Kidney Dis. 1987;9(2):176-179.
14. Bhorade MS, Carag HB, Lee HJ, Potter EV, Dunea G. Nephropathy of secondary syphilis: a clinical and pathological spectrum. JAMA. 1971;216(7):1159-1166.
15. Hunte W, al-Ghraoui F, Cohen RJ. Secondary syphilis and the nephrotic syndrome. J Am Soc Nephrol. 1993;3(7):1351-1355.
16. Gamble CN, Reardan JB. Immunopathogenesis of syphilitic glomerulonephritis. Elution of antitreponemal antibody from glomerular immune-complex deposits. N Engl J Med. 1975;292(9):449-454.
*This article has been corrected to include a missing author.
Case Presentation. A 63-year-old male with well-controlled HIV (CD4 count 757, undetectable viral load), epilepsy, and hypertension presented to the VA Boston Healthcare System (VABHS) emergency department with 1 week of bilateral leg swelling and exertional shortness of breath. He reported having no fever, cough, chest pain, pain with inspiration and orthopnea. There was no personal or family history of pulmonary embolism. He reported weight gain but was unable to quantify how much. He also reported flare up of chronic knee pain, without swelling for which he had taken up to 4 tablets of naproxen daily for several weeks. His physical examination was notable for a heart rate of 105 beats per minute and bilateral pitting edema to his knees. Laboratory testing revealed a creatinine level of 2.5 mg/dL, which was increased from a baseline of 1.0 mg/dL (Table 1), and a urine protein-to-creatinine ratio of 7.8 mg/mg (Table 2). A renal ultrasound showed normal-sized kidneys without hydronephrosis or obstructing renal calculi. The patient was admitted for further workup of his dyspnea and acute kidney injury.
► Jonathan Li, MD, Chief Medical Resident, VABHS and Beth Israel Deaconess Medical Center (BIDMC). Dr. William, based on the degree of proteinuria and edema, a diagnosis of nephrotic syndrome was made. How is nephrotic syndrome defined, and how is it distinguished from glomerulonephritis?
► Jeffrey William, MD, Nephrologist, BIDMC, Assistant Professor of Medicine, Harvard Medical School. The pathophysiology of nephrotic disease and glomerulonephritis are quite distinct, resulting in symptoms and systemic manifestations that only slightly overlap. Glomerulonephritis is characterized by inflammation of the endothelial cells of the trilayered glomerular capillary, with a resulting active urine sediment with red blood cells, white blood cells, and casts. Nephrotic syndrome mostly affects the visceral epithelial cells of the glomerular capillary, commonly referred to as podocytes, and hence, the urine sediment in nephrotic disease is often inactive. Patients with nephrotic syndrome have nephrotic-range proteinuria (excretion of > 3.5 g per 24 h or a spot urine protein-creatinine ratio > 3.5 g in the steady state) and both hypoalbuminemia (< 3 g/dL) and peripheral edema. Lipiduria and hyperlipidemia are common findings in nephrotic syndrome but are not required for a clinical diagnosis.1 In contrast, glomerulonephritis is defined by a constellation of findings that include renal insufficiency (often indicated by an elevation in blood urea nitrogen and creatinine), hypertension, hematuria, and subnephrotic range proteinuria. In practice, patients may fulfill criteria of both nephrotic and nephritic syndromes, but the preponderance of clinical evidence often points one way or the other. In this case, nephrotic syndrome was diagnosed based on the urine protein-to-creatinine ratio of 7.8 mg/mg, hypoalbuminemia, and edema.
► Dr. Li. What would be your first-line workup for evaluation of the etiology of this patient’s nephrotic syndrome?
► Dr. William. Rather than memorizing a list of etiologies of nephrotic syndrome, it is essential to consider the pathophysiology of heavy proteinuria. Though the glomerular filtration barrier is extremely complex and defects in any component can cause proteinuria, disruption of the podocyte is often involved. Common disease processes that chiefly target the podocyte include minimal change disease, primary focal and segmental glomerulosclerosis (FSGS), and membranous nephropathy, all by differing mechanisms. Minimal change disease and idiopathic/primary FSGS are increasingly thought to be at differing points on a spectrum of the same disease.2 Secondary FSGS, on the other hand, is a progressive disease, commonly resulting from longstanding hypertension, diabetes mellitus, and obesity in adults. Membranous nephropathy can also be either primary or secondary. Primary membranous nephropathy is chiefly caused by a circulating IgG4 antibody to the podocyte membrane antigen PLA2R (M-type phospholipase A2 receptor), whereas secondary membranous nephropathy can be caused by a variety of systemic etiologies, including autoimmune disease (eg, systemic lupus erythematosus), certain malignancies, chronic infections (eg, hepatitis B and C), and many medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).3-5 Paraprotein deposition diseases can also cause glomerular damage leading to nephrotic-range proteinuria.
Given these potential diagnoses, a careful history should be taken to assess exposures and recent medication use. Urine sediment evaluation is essential in the evaluation of nephrotic syndrome to determine if there is an underlying nephritic process. Select serologies may be sent to look for autoimmune disease, such as systemic lupus erythematosus and common viral exposures like hepatitis B or C. Serum and urine protein electrophoreses would be appropriate initial tests of suspected paraprotein-related diseases. Other serologies, such as antineutrophil cytoplasmic antibodies or antiglomerular basement membrane antibodies, would not necessarily be indicated here given the lack of hematuria and presence of nephrotic-range proteinuria.
► Dr. Li. The initial evaluation was notable for an erythrocyte sedimentation rate > 120 (mm/h) and a weakly positive antinuclear antibody (ANA) titer of 1:40. The remainder of his initial workup did not reveal an etiology for his nephrotic syndrome (Table 3).
Dr. William, is there a role for starting urgent empiric steroids in nephrotic syndrome while workup is ongoing? If so, do the severity of proteinuria and/or symptoms play a role or is this determination based on something else?
► Dr. William. Edema is a primary symptom of nephrotic syndrome and can often be managed with diuretics alone. If a clear medication-mediated cause is suspected, discontinuation of this agent may result in spontaneous improvement without steroid treatment. However,in cases where an etiology is unclear and there are serious thrombotic complications requiring anticoagulation, and a renal biopsy is deemed to be too risky, then empiric steroid therapy may be necessary. Children with new-onset nephrotic syndrome are presumed to have minimal change disease, given its prevalence in this patient population, and are often given empiric steroids without obtaining a renal biopsy. However, in the adult population, a renal biopsy can typically be performed quickly and safely, with pathology results interpreted within days. In this patient, since a diagnosis was unclear and there was no contraindication to renal biopsy, a biopsy should be obtained before consideration of steroids.
► Dr. Li. Steroids were deferred in anticipation of renal biopsy, which showed stage I membranous nephropathy, suggestive of membranous lupus nephritis Class V. The deposits were strongly reactive for immunoglobuline G (IgG), IgA, and complement 1q (C1q), showed co-dominant staining for IgG1, IgG2, and IgG3, and were weakly positive for the PLA2 receptor. Focal intimal arteritis in a small interlobular vessel was seen.
Dr. William, the pathology returned suggestive of lupus nephritis. Does the overall clinical picture fit with lupus nephritis?
► Dr. William. Given the history and a rather low ANA, the diagnosis of lupus nephritis seems unlikely. The lack of IgG4 and PLA2R staining in the biopsy suggests that this membranous pattern on the biopsy is likely to be secondary to a systemic etiology, but further investigation should be pursued.
► Dr. Li. The patient was discharged after the biopsy with a planned outpatient nephrology follow-up to discuss results and treatment. He was prescribed an oral diuretic, and his symptoms improved. Several days after discharge, he developed blurry vision and was evaluated in the Ophthalmology clinic. On fundoscopy, he was found to have acute papillitis, a form of optic neuritis. As part of initial evaluation of infectious etiologies of papillitis, ophthalmology recommended testing for syphilis.
Dr. Strymish, when we are considering secondary syphilis, what is the recommended approach to diagnostic testing?
► Judith Strymish, MD, Infectious Diseases, BIDMC, Assistant Professor of Medicine, Harvard Medical School. The diagnosis of syphilis is usually made through serologic testing of blood specimens. Methods that detect the spirochete directly like dark-field smears are not readily available. Serologic tests include treponemal tests (eg, Treponema pallidum particle agglutination assay [TPPA]) and nontreponemal tests (eg, rapid plasma reagin [RPR]). One needs a confirmatory test because either test is associated with false positives. Either test can be done first. Most laboratories, including those at VABHS are now performing treponemal tests first as these have become more cost-effective.6 The TPPA treponemal test was found to have a lower false negative rate in primary syphilis compared with that of nontreponemal tests.7 Nontreponemal tests can be followed for response to therapy. If a patient has a history of treated syphilis, a nontreponemal test should be sent, since the treponemal test will remain positive for life.
If there is clinical concern for neurosyphilis, cerebrospinal fluid fluorescent (CSF) treponemal antibody needs to be sampled and sent for the nontreponemal venereal disease research laboratory (VDRL) test. The VDRL is highly specific for neurosyphilis but not as sensitive. Cerebrospinal fluid fluorescent treponemal antibody (CSF FTA) may also be sent; it is very sensitive but not very specific for neurosyphilis.
► Dr. Li. An RPR returned positive at 1:512 (was negative 14 months prior on a routine screening test), with positive reflex TPPA (Table 4). A diagnosis of secondary syphilis was made. Dr. Strymish, at this point, what additional testing and treatment is necessary?
► Dr. Strymish. With papillitis and a very high RPR, we need to assume that he has ophthalmic syphilis. This can occur in any stage of syphilis, but his eye findings and high RPR are consistent with secondary syphilis. Ophthalmic syphilis has been on the upswing, even more than is expected with recent increases in syphilis cases.8 Ophthalmic syphilis is considered a form of neurosyphilis. A lumbar puncture and treatment for neurosyphilis is recommended.9,10
► Dr. Li. A lumbar puncture was performed, and his CSF was VDRL positive. This confirmed a diagnosis of neurosyphilis (Table 4). The patient was treated for neurosyphilis with IV penicillin. The patient shared that he had episodes of unprotected oral sexual activity within the past year and approximately 1 year ago, he came in close contact (but no sexual activity) with a person who had a rash consistent with syphilis.Dr. William, syphilis would be a potential unifying diagnosis of his renal and ophthalmologic manifestations. Is syphilis known to cause membranous nephropathy?
► Dr. William. Though it is uncommon, the nephrotic syndrome is a well-described complication of secondary syphilis.11,12 Syphilis has been shown to cause nephrotic syndrome in a variety of ways. Case reports abound linking syphilis to minimal change disease and other glomerular diseases.13,14 A case report from 1993 shows a membranous pattern of glomerular disease similar to this case.15 As a form of secondary membranous nephropathy, the immunofluorescence pattern can demonstrate staining similar to the “full house” seen in lupus nephritis (IgA, IgM, and C1q, in addition to IgG and C3).16 This explains the initial interpretation of this patient’s biopsy, as lupus nephritis would be a much more common etiology of secondary membranous nephropathy than is acute syphilis with this immunofluorescence pattern. However, the data in this case are highly suggestive of a causal relationship between secondary syphilis and membranous nephropathy.
► Dr. Li. Dr. Strymish, how should this patient be screened for syphilis reinfection, and at what intervals would you recommend?
► Dr. Strymish. He will need follow-up testing to make sure that his syphilis is effectively treated. If CSF pleocytosis was present initially, a CSF examination should be repeated every 6 months until the cell count is normal. He will also need follow-up for normalization of his RPR. Persons with HIV infection and primary or secondary syphilis should be evaluated clinically and serologically for treatment failure at 3, 6, 9, 12, and 24 months after therapy according to US Centers for Disease Control and Prevention guidelines.9
His treponemal test for syphilis will likely stay positive for life. His RPR should decrease significantly with effective treatment. It makes sense to screen with RPR alone as long as he continues to have risk factors for acquiring syphilis. Routine syphilis testing is recommended for pregnant women, sexually active men who have sex with men, sexually active persons with HIV, and persons taking PrEP (pre-exposure prophylaxis) for HIV prevention. He should be screened at least yearly for syphilis.
► Dr. Li. Over the next several months, the patient’s creatinine normalized and his proteinuria resolved. His vision recovered, and he has had no further ophthalmologic complications.
Dr. William, what is his long-term renal prognosis? Do you expect that his acute episode of membranous nephropathy will have permanent effects on his renal function?
► Dr. William. His rapid response to therapy for neurosyphilis provides evidence for this etiology of his renal dysfunction and glomerulonephritis. His long-term prognosis is quite good if the syphilis is the only reason for him to have renal disease. The renal damage is often reversible in these cases. However, given his prior extensive NSAID exposure and history of hypertension, he may be at higher risk for chronic kidney disease than an otherwise healthy patient, especially after an episode of acute kidney injury. Therefore, his renal function should continue to be monitored as an outpatient.
Acknowledgments
The authors thank this veteran for sharing his story and allowing us to learn from this unusual case for the benefit of our future patients.
*This article has been corrected to include a missing author.
Case Presentation. A 63-year-old male with well-controlled HIV (CD4 count 757, undetectable viral load), epilepsy, and hypertension presented to the VA Boston Healthcare System (VABHS) emergency department with 1 week of bilateral leg swelling and exertional shortness of breath. He reported having no fever, cough, chest pain, pain with inspiration and orthopnea. There was no personal or family history of pulmonary embolism. He reported weight gain but was unable to quantify how much. He also reported flare up of chronic knee pain, without swelling for which he had taken up to 4 tablets of naproxen daily for several weeks. His physical examination was notable for a heart rate of 105 beats per minute and bilateral pitting edema to his knees. Laboratory testing revealed a creatinine level of 2.5 mg/dL, which was increased from a baseline of 1.0 mg/dL (Table 1), and a urine protein-to-creatinine ratio of 7.8 mg/mg (Table 2). A renal ultrasound showed normal-sized kidneys without hydronephrosis or obstructing renal calculi. The patient was admitted for further workup of his dyspnea and acute kidney injury.
► Jonathan Li, MD, Chief Medical Resident, VABHS and Beth Israel Deaconess Medical Center (BIDMC). Dr. William, based on the degree of proteinuria and edema, a diagnosis of nephrotic syndrome was made. How is nephrotic syndrome defined, and how is it distinguished from glomerulonephritis?
► Jeffrey William, MD, Nephrologist, BIDMC, Assistant Professor of Medicine, Harvard Medical School. The pathophysiology of nephrotic disease and glomerulonephritis are quite distinct, resulting in symptoms and systemic manifestations that only slightly overlap. Glomerulonephritis is characterized by inflammation of the endothelial cells of the trilayered glomerular capillary, with a resulting active urine sediment with red blood cells, white blood cells, and casts. Nephrotic syndrome mostly affects the visceral epithelial cells of the glomerular capillary, commonly referred to as podocytes, and hence, the urine sediment in nephrotic disease is often inactive. Patients with nephrotic syndrome have nephrotic-range proteinuria (excretion of > 3.5 g per 24 h or a spot urine protein-creatinine ratio > 3.5 g in the steady state) and both hypoalbuminemia (< 3 g/dL) and peripheral edema. Lipiduria and hyperlipidemia are common findings in nephrotic syndrome but are not required for a clinical diagnosis.1 In contrast, glomerulonephritis is defined by a constellation of findings that include renal insufficiency (often indicated by an elevation in blood urea nitrogen and creatinine), hypertension, hematuria, and subnephrotic range proteinuria. In practice, patients may fulfill criteria of both nephrotic and nephritic syndromes, but the preponderance of clinical evidence often points one way or the other. In this case, nephrotic syndrome was diagnosed based on the urine protein-to-creatinine ratio of 7.8 mg/mg, hypoalbuminemia, and edema.
► Dr. Li. What would be your first-line workup for evaluation of the etiology of this patient’s nephrotic syndrome?
► Dr. William. Rather than memorizing a list of etiologies of nephrotic syndrome, it is essential to consider the pathophysiology of heavy proteinuria. Though the glomerular filtration barrier is extremely complex and defects in any component can cause proteinuria, disruption of the podocyte is often involved. Common disease processes that chiefly target the podocyte include minimal change disease, primary focal and segmental glomerulosclerosis (FSGS), and membranous nephropathy, all by differing mechanisms. Minimal change disease and idiopathic/primary FSGS are increasingly thought to be at differing points on a spectrum of the same disease.2 Secondary FSGS, on the other hand, is a progressive disease, commonly resulting from longstanding hypertension, diabetes mellitus, and obesity in adults. Membranous nephropathy can also be either primary or secondary. Primary membranous nephropathy is chiefly caused by a circulating IgG4 antibody to the podocyte membrane antigen PLA2R (M-type phospholipase A2 receptor), whereas secondary membranous nephropathy can be caused by a variety of systemic etiologies, including autoimmune disease (eg, systemic lupus erythematosus), certain malignancies, chronic infections (eg, hepatitis B and C), and many medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).3-5 Paraprotein deposition diseases can also cause glomerular damage leading to nephrotic-range proteinuria.
Given these potential diagnoses, a careful history should be taken to assess exposures and recent medication use. Urine sediment evaluation is essential in the evaluation of nephrotic syndrome to determine if there is an underlying nephritic process. Select serologies may be sent to look for autoimmune disease, such as systemic lupus erythematosus and common viral exposures like hepatitis B or C. Serum and urine protein electrophoreses would be appropriate initial tests of suspected paraprotein-related diseases. Other serologies, such as antineutrophil cytoplasmic antibodies or antiglomerular basement membrane antibodies, would not necessarily be indicated here given the lack of hematuria and presence of nephrotic-range proteinuria.
► Dr. Li. The initial evaluation was notable for an erythrocyte sedimentation rate > 120 (mm/h) and a weakly positive antinuclear antibody (ANA) titer of 1:40. The remainder of his initial workup did not reveal an etiology for his nephrotic syndrome (Table 3).
Dr. William, is there a role for starting urgent empiric steroids in nephrotic syndrome while workup is ongoing? If so, do the severity of proteinuria and/or symptoms play a role or is this determination based on something else?
► Dr. William. Edema is a primary symptom of nephrotic syndrome and can often be managed with diuretics alone. If a clear medication-mediated cause is suspected, discontinuation of this agent may result in spontaneous improvement without steroid treatment. However,in cases where an etiology is unclear and there are serious thrombotic complications requiring anticoagulation, and a renal biopsy is deemed to be too risky, then empiric steroid therapy may be necessary. Children with new-onset nephrotic syndrome are presumed to have minimal change disease, given its prevalence in this patient population, and are often given empiric steroids without obtaining a renal biopsy. However, in the adult population, a renal biopsy can typically be performed quickly and safely, with pathology results interpreted within days. In this patient, since a diagnosis was unclear and there was no contraindication to renal biopsy, a biopsy should be obtained before consideration of steroids.
► Dr. Li. Steroids were deferred in anticipation of renal biopsy, which showed stage I membranous nephropathy, suggestive of membranous lupus nephritis Class V. The deposits were strongly reactive for immunoglobuline G (IgG), IgA, and complement 1q (C1q), showed co-dominant staining for IgG1, IgG2, and IgG3, and were weakly positive for the PLA2 receptor. Focal intimal arteritis in a small interlobular vessel was seen.
Dr. William, the pathology returned suggestive of lupus nephritis. Does the overall clinical picture fit with lupus nephritis?
► Dr. William. Given the history and a rather low ANA, the diagnosis of lupus nephritis seems unlikely. The lack of IgG4 and PLA2R staining in the biopsy suggests that this membranous pattern on the biopsy is likely to be secondary to a systemic etiology, but further investigation should be pursued.
► Dr. Li. The patient was discharged after the biopsy with a planned outpatient nephrology follow-up to discuss results and treatment. He was prescribed an oral diuretic, and his symptoms improved. Several days after discharge, he developed blurry vision and was evaluated in the Ophthalmology clinic. On fundoscopy, he was found to have acute papillitis, a form of optic neuritis. As part of initial evaluation of infectious etiologies of papillitis, ophthalmology recommended testing for syphilis.
Dr. Strymish, when we are considering secondary syphilis, what is the recommended approach to diagnostic testing?
► Judith Strymish, MD, Infectious Diseases, BIDMC, Assistant Professor of Medicine, Harvard Medical School. The diagnosis of syphilis is usually made through serologic testing of blood specimens. Methods that detect the spirochete directly like dark-field smears are not readily available. Serologic tests include treponemal tests (eg, Treponema pallidum particle agglutination assay [TPPA]) and nontreponemal tests (eg, rapid plasma reagin [RPR]). One needs a confirmatory test because either test is associated with false positives. Either test can be done first. Most laboratories, including those at VABHS are now performing treponemal tests first as these have become more cost-effective.6 The TPPA treponemal test was found to have a lower false negative rate in primary syphilis compared with that of nontreponemal tests.7 Nontreponemal tests can be followed for response to therapy. If a patient has a history of treated syphilis, a nontreponemal test should be sent, since the treponemal test will remain positive for life.
If there is clinical concern for neurosyphilis, cerebrospinal fluid fluorescent (CSF) treponemal antibody needs to be sampled and sent for the nontreponemal venereal disease research laboratory (VDRL) test. The VDRL is highly specific for neurosyphilis but not as sensitive. Cerebrospinal fluid fluorescent treponemal antibody (CSF FTA) may also be sent; it is very sensitive but not very specific for neurosyphilis.
► Dr. Li. An RPR returned positive at 1:512 (was negative 14 months prior on a routine screening test), with positive reflex TPPA (Table 4). A diagnosis of secondary syphilis was made. Dr. Strymish, at this point, what additional testing and treatment is necessary?
► Dr. Strymish. With papillitis and a very high RPR, we need to assume that he has ophthalmic syphilis. This can occur in any stage of syphilis, but his eye findings and high RPR are consistent with secondary syphilis. Ophthalmic syphilis has been on the upswing, even more than is expected with recent increases in syphilis cases.8 Ophthalmic syphilis is considered a form of neurosyphilis. A lumbar puncture and treatment for neurosyphilis is recommended.9,10
► Dr. Li. A lumbar puncture was performed, and his CSF was VDRL positive. This confirmed a diagnosis of neurosyphilis (Table 4). The patient was treated for neurosyphilis with IV penicillin. The patient shared that he had episodes of unprotected oral sexual activity within the past year and approximately 1 year ago, he came in close contact (but no sexual activity) with a person who had a rash consistent with syphilis.Dr. William, syphilis would be a potential unifying diagnosis of his renal and ophthalmologic manifestations. Is syphilis known to cause membranous nephropathy?
► Dr. William. Though it is uncommon, the nephrotic syndrome is a well-described complication of secondary syphilis.11,12 Syphilis has been shown to cause nephrotic syndrome in a variety of ways. Case reports abound linking syphilis to minimal change disease and other glomerular diseases.13,14 A case report from 1993 shows a membranous pattern of glomerular disease similar to this case.15 As a form of secondary membranous nephropathy, the immunofluorescence pattern can demonstrate staining similar to the “full house” seen in lupus nephritis (IgA, IgM, and C1q, in addition to IgG and C3).16 This explains the initial interpretation of this patient’s biopsy, as lupus nephritis would be a much more common etiology of secondary membranous nephropathy than is acute syphilis with this immunofluorescence pattern. However, the data in this case are highly suggestive of a causal relationship between secondary syphilis and membranous nephropathy.
► Dr. Li. Dr. Strymish, how should this patient be screened for syphilis reinfection, and at what intervals would you recommend?
► Dr. Strymish. He will need follow-up testing to make sure that his syphilis is effectively treated. If CSF pleocytosis was present initially, a CSF examination should be repeated every 6 months until the cell count is normal. He will also need follow-up for normalization of his RPR. Persons with HIV infection and primary or secondary syphilis should be evaluated clinically and serologically for treatment failure at 3, 6, 9, 12, and 24 months after therapy according to US Centers for Disease Control and Prevention guidelines.9
His treponemal test for syphilis will likely stay positive for life. His RPR should decrease significantly with effective treatment. It makes sense to screen with RPR alone as long as he continues to have risk factors for acquiring syphilis. Routine syphilis testing is recommended for pregnant women, sexually active men who have sex with men, sexually active persons with HIV, and persons taking PrEP (pre-exposure prophylaxis) for HIV prevention. He should be screened at least yearly for syphilis.
► Dr. Li. Over the next several months, the patient’s creatinine normalized and his proteinuria resolved. His vision recovered, and he has had no further ophthalmologic complications.
Dr. William, what is his long-term renal prognosis? Do you expect that his acute episode of membranous nephropathy will have permanent effects on his renal function?
► Dr. William. His rapid response to therapy for neurosyphilis provides evidence for this etiology of his renal dysfunction and glomerulonephritis. His long-term prognosis is quite good if the syphilis is the only reason for him to have renal disease. The renal damage is often reversible in these cases. However, given his prior extensive NSAID exposure and history of hypertension, he may be at higher risk for chronic kidney disease than an otherwise healthy patient, especially after an episode of acute kidney injury. Therefore, his renal function should continue to be monitored as an outpatient.
Acknowledgments
The authors thank this veteran for sharing his story and allowing us to learn from this unusual case for the benefit of our future patients.
1. Rennke H, Denker BM. Renal Pathophysiology: The Essentials. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2014.
2. Maas RJ, Deegens JK, Smeets B, Moeller MJ, Wetzels JF. Minimal change disease and idiopathic FSGS: manifestations of the same disease. Nat Rev Nephrol. 2016;12(12):768-776.
3. Beck LH Jr, Bonegio RG, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361(1):11-21.
4. Rennke HG. Secondary membranoproliferative glomerulonephritis. Kidney Int. 1995;47(2):643-656.
5. Nawaz FA, Larsen CP, Troxell ML. Membranous nephropathy and nonsteroidal anti-inflammatory agents. Am J Kidney Dis. 2013;62(5):1012-1017.
6. Pillay A. Centers for Disease Control and Prevention Syphilis Summit—Diagnostics and laboratory issues. Sex Transm Dis. 2018;45(9S)(suppl 1):S13-S16.
7. Levett PN, Fonseca K, Tsang RS, et al. Canadian Public Health Laboratory Network laboratory guidelines for the use of serological tests (excluding point-of-care tests) for the diagnosis of syphilis in Canada. Can J Infect Dis Med Microbiol. 2015;26(suppl A):6A-12A.
8. Oliver SE, Aubin M, Atwell L, et al. Ocular syphilis—eight jurisdictions, United States, 2014-2015. MMWR Morb Mortal Wkly Rep. 2016;65(43):1185-1188.
9. Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recommendations and Reports 2015;64(RR3):1-137. [Erratum in MMWR Recomm Rep. 2015;64(33):924.]
10. US Centers for Disease Control and Prevention. Clinical advisory: ocular syphilis in the United States. https://www.cdc.gov/std/syphilis/clinicaladvisoryos2015.htm. Updated March 24, 2016. Accessed August 12, 2019.
11. Braunstein GD, Lewis EJ, Galvanek EG, Hamilton A, Bell WR. The nephrotic syndrome associated with secondary syphilis: an immune deposit disease. Am J Med. 1970;48:643-648.1.
12. Handoko ML, Duijvestein M, Scheepstra CG, de Fijter CW. Syphilis: a reversible cause of nephrotic syndrome. BMJ Case Rep. 2013;2013:pii:bcr2012008279
13. Krane NK, Espenan P, Walker PD, Bergman SM, Wallin JD. Renal disease and syphilis: a report of nephrotic syndrome with minimal change disease. Am J Kidney Dis. 1987;9(2):176-179.
14. Bhorade MS, Carag HB, Lee HJ, Potter EV, Dunea G. Nephropathy of secondary syphilis: a clinical and pathological spectrum. JAMA. 1971;216(7):1159-1166.
15. Hunte W, al-Ghraoui F, Cohen RJ. Secondary syphilis and the nephrotic syndrome. J Am Soc Nephrol. 1993;3(7):1351-1355.
16. Gamble CN, Reardan JB. Immunopathogenesis of syphilitic glomerulonephritis. Elution of antitreponemal antibody from glomerular immune-complex deposits. N Engl J Med. 1975;292(9):449-454.
1. Rennke H, Denker BM. Renal Pathophysiology: The Essentials. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2014.
2. Maas RJ, Deegens JK, Smeets B, Moeller MJ, Wetzels JF. Minimal change disease and idiopathic FSGS: manifestations of the same disease. Nat Rev Nephrol. 2016;12(12):768-776.
3. Beck LH Jr, Bonegio RG, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361(1):11-21.
4. Rennke HG. Secondary membranoproliferative glomerulonephritis. Kidney Int. 1995;47(2):643-656.
5. Nawaz FA, Larsen CP, Troxell ML. Membranous nephropathy and nonsteroidal anti-inflammatory agents. Am J Kidney Dis. 2013;62(5):1012-1017.
6. Pillay A. Centers for Disease Control and Prevention Syphilis Summit—Diagnostics and laboratory issues. Sex Transm Dis. 2018;45(9S)(suppl 1):S13-S16.
7. Levett PN, Fonseca K, Tsang RS, et al. Canadian Public Health Laboratory Network laboratory guidelines for the use of serological tests (excluding point-of-care tests) for the diagnosis of syphilis in Canada. Can J Infect Dis Med Microbiol. 2015;26(suppl A):6A-12A.
8. Oliver SE, Aubin M, Atwell L, et al. Ocular syphilis—eight jurisdictions, United States, 2014-2015. MMWR Morb Mortal Wkly Rep. 2016;65(43):1185-1188.
9. Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recommendations and Reports 2015;64(RR3):1-137. [Erratum in MMWR Recomm Rep. 2015;64(33):924.]
10. US Centers for Disease Control and Prevention. Clinical advisory: ocular syphilis in the United States. https://www.cdc.gov/std/syphilis/clinicaladvisoryos2015.htm. Updated March 24, 2016. Accessed August 12, 2019.
11. Braunstein GD, Lewis EJ, Galvanek EG, Hamilton A, Bell WR. The nephrotic syndrome associated with secondary syphilis: an immune deposit disease. Am J Med. 1970;48:643-648.1.
12. Handoko ML, Duijvestein M, Scheepstra CG, de Fijter CW. Syphilis: a reversible cause of nephrotic syndrome. BMJ Case Rep. 2013;2013:pii:bcr2012008279
13. Krane NK, Espenan P, Walker PD, Bergman SM, Wallin JD. Renal disease and syphilis: a report of nephrotic syndrome with minimal change disease. Am J Kidney Dis. 1987;9(2):176-179.
14. Bhorade MS, Carag HB, Lee HJ, Potter EV, Dunea G. Nephropathy of secondary syphilis: a clinical and pathological spectrum. JAMA. 1971;216(7):1159-1166.
15. Hunte W, al-Ghraoui F, Cohen RJ. Secondary syphilis and the nephrotic syndrome. J Am Soc Nephrol. 1993;3(7):1351-1355.
16. Gamble CN, Reardan JB. Immunopathogenesis of syphilitic glomerulonephritis. Elution of antitreponemal antibody from glomerular immune-complex deposits. N Engl J Med. 1975;292(9):449-454.
Fatal Drug-Resistant Invasive Pulmonary Aspergillus fumigatus in a 56-Year-Old Immunosuppressed Man (FULL)
Historically, aspergillosis in patients with hematopoietic stem cell transplantation (HSCT) has carried a high mortality rate. However, recent data demonstrate a dramatic improvement in outcomes for patients with HSCT: 90-day survival increased from 22% before 2000 to 45% over the past 15 years.1 Improved outcomes coincide with changes in transplant immunosuppression practices, use of cross-sectional imaging for early disease identification, galactomannan screening, and the development of novel treatment options.
Voriconazole is an azole drug that blocks the synthesis of ergosterol, a vital component of the cellular membrane of fungi. Voriconazole was approved in 2002 after a clinical trial demonstrated an improvement in 50% of patients with invasive aspergillosis in the voriconazole arm vs 30% in the amphotericin B arm at 12 weeks.2 Amphotericin B is a polyene antifungal drug that binds with ergosterol, creating leaks in the cell membrane that lead to cellular demise. Voriconazole quickly became the first-line therapy for invasive aspergillosis and is recommended by both the Infectious Disease Society of American (IDSA) and the European Conference on Infections in Leukemia.3
Case Presentation
A 55-year-old man with high-risk chronic myelogenous leukemia (CML) underwent a 10 of 10 human leukocyte antigen allele and antigen-matched peripheral blood allogeneic HSCT with a myeloablative-conditioning regimen of busulfan and cyclophosphamide, along with prophylactic voriconazole, sulfamethoxazole/trimethoprim, and acyclovir. After successful engraftment (without significant neutropenia), his posttransplant course was complicated by grade 2 graft vs host disease (GVHD) of the skin, eyes, and liver, which responded well to steroids and tacrolimus. Voriconazole was continued for 5 months until immunosuppression was minimized (tacrolimus 1 mg twice daily). Two months later, the patient’s GVHD worsened, necessitating treatment at an outside hospital with high-dose prednisone (2 mg/kg/d) and cyclosporine (300 mg twice daily). Voriconazole prophylaxis was not reinitiated at that time.
One year later, at a routine follow-up appointment, the patient endorsed several weeks of malaise, weight loss, and nonproductive cough. The patient’s immunosuppression recently had been reduced to 1 mg/kg/d of prednisone and 100 mg of cyclosporine twice daily. A chest X-ray demonstrated multiple pulmonary nodules; follow-up chest computed tomography (CT) confirmed multiple nodular infiltrates with surrounding ground-glass opacities suspicious with a fungal infection (Figure 1). 
Treatment with oral voriconazole (300 mg twice daily) was initiated for probable pulmonary aspergillosis. Cyclosporine (150 mg twice daily) and prednisone (1 mg/kg/d) were continued throughout treatment out of concern for hepatic GVHD. The patient’s symptoms improved over the next 10 days, and follow-up chest imaging demonstrated improvement.
Two weeks after initiation of voriconazole treatment, the patient developed a new productive cough and dyspnea, associated with fevers and chills. Repeat imaging revealed right lower-lobe pneumonia. The serum voriconazole trough level was checked and was 3.1 mg/L, suggesting therapeutic dosing. The patient subsequently developed acute respiratory distress syndrome and required intubation and mechanical ventilation. Repeat BAL sampling demonstrated multidrug-resistant Escherichia coli, a BAL galactomannan level of 2.0 ODI, and negative fungal cultures. The patient’s hospital course was complicated by profound hypoxemia, requiring prone positioning and neuromuscular blockade. He was treated with meropenem and voriconazole. His immunosuppression was reduced, but he rapidly developed acute liver injury from hepatic GVHD that resolved after reinitiation of cyclosporine and prednisone at 0.75 mg/kg/d.
The patient improved over the next 3 weeks and was successfully extubated. Repeat chest CT imaging demonstrated numerous pneumatoceles in the location of previous nodules, consistent with healing necrotic fungal disease, and a new right lower-lobe cavitary mass (Figure 2). Two days after transferring out of the intensive care unit, the patient again developed hypoxemia and fevers to 39° C. Bronchoscopy with BAL of the right lower lobe revealed positive A fumigatus and Rhizopus sp polymerase chain reaction (PCR) assays, although fungal cultures were positive only for A fumigatus. Liposomal amphotericin B (5 mg/kg) was added to voriconazole therapy to treat mucormycosis and to provide a second active agent against A fumigatus.
Unfortunately, the patient’s clinical status continued to deteriorate with signs of progressive respiratory failure and infection despite empiric, broad-spectrum antibiotics and dual antifungal therapy. His serum voriconazole level continued to be therapeutic at 1.9 mg/L. The patient declined reintubation and invasive mechanical ventilation, and he ultimately transitioned to comfort measures and died with his family at the bedside.
Autopsy demonstrated widely disseminated Aspergillus infection as the cause of death, with evidence of myocardial, neural, and vascular invasion of A fumigatus (Figures 3 and 4). 
Discussion
This case of fatal, progressive, invasive, pulmonary aspergillosis demonstrates several important factors in the treatment of patients with this disease. Treatment failure usually relates to any of 4 possible factors: host immune status, severity or burden of disease, appropriate dosing of antifungal agents, and drug resistance. This patient’s immune system was heavily suppressed for a prolonged period. Attempts at reducing immunosuppression to the minimal required dosage to prevent a GVHD flare were unsuccessful and became an unmodifiable risk factor, a major contributor to his demise.
The risks of continuous high-dose immunosuppression in steroid-refractory GVHD is well understood and has been previously demonstrated to have up to 50% 4-year nonrelapse mortality, mainly due to overwhelming bacterial, viral, and fungal infections.4 All attempts should be made to cease or reduce immunosuppression in the setting of a severe infection, although this is sometimes impossible as in this case.
The patient’s disease burden was significant as evidenced by the bilateral, multifocal pulmonary nodules seen on chest imaging and the disseminated disease found at postmortem examination. His initial improvement in symptoms with voriconazole and the evolution of his images (with many of his initial pulmonary nodules becoming pneumatoceles) suggested a temporary positive immune response. The authors believe that the Rhizopus in his sputum represents noninvasive colonization of one of his pneumatoceles, because postmortem examination failed to reveal Rhizopus at any other location.
Voriconazole has excellent pulmonary and central nervous system penetration: In this patient serum levels were well within the therapeutic range. His peculiar drug resistance pattern (sensitivity to azoles and resistance to amphotericin) is unusual. Azole resistance in leukemia and patients with HSCT is more common than is amphotericin resistance, with current estimates of azole resistance close to 5%, ranging between 1% and 30%.5,6 Widespread use of antifungal prophylaxis with azoles likely selects for azole resistance.6
Despite this concern of azole resistance, current IDSA guidelines recommend against routine susceptibility testing of Aspergillus to azole therapy because of the current lack of consensus between the European Committee on Antibiotic Susceptibility Testing and Clinical and Laboratory Standards Institute on break points for resistance patterns.3,7 This is an area of emerging research, and proposed cut points for declaration of resistance do exist in the literature even if not globally agreed on.8
Combination antifungal therapy is an option for treatment in cases of possible drug resistance. Nonetheless, a recent randomized, double-blind, placebo-controlled, multicenter trial comparing voriconazole monotherapy with the combination of voriconazole and anidulafungin failed to demonstrate an overall mortality benefit in the primary analysis, although secondary analysis showed a mortality benefit with combination therapy in patients at highest risk for death.9
Despite the lack of unified standards with susceptibility testing, it may be reasonable to perform such tests in patients with demonstrating progressive disease. In this patient’s case, amphotericin B was added to treat the Rhizopus species found in his sputum, and while not the combination studied in the previously mentioned study, the drug should have provided an additional active agent for Aspergillus should this patient have had azole resistance.
Surprisingly, subsequent testing demonstrated the Aspergillus species to be resistant to amphotericin B. De novo amphotericin B-resistant A fumigates is extremely rare, with an expected incidence of 1% or less.10 The authors believe the patient may have demonstrated induction of amphotericin-B resistance through activation of fungal stress pathways by prior treatment with voriconazole. This has been demonstrated in vitro and should be considered should combination salvage therapy be required for the treatment of a refractory Aspergillus infection especially if patients have received prior treatment with voriconazole.11
Conclusion
This fatal case of invasive pulmonary aspergillosis illustrates the importance of considering the 4 main causes of treatment failure in an infection. Although the patient had a high burden of disease with a rare resistance pattern, he was treated with appropriate and well-dosed therapy. Ultimately, his unmodifiable immunosuppression was likely the driving factor leading to treatment failure and death. The indication for and number of bone marrow transplants continues to increase, thus exposure to and treatment of invasive fungal infections will increase accordingly. As such, providers should ensure that all causes of treatment failure are considered and addressed.
1. Upton A, Kirby KA, Carpenter P, Boeckh M, Marr KA. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin Infect Dis. 2007;44(4):531-540.
2. Herbrecht R, Denning DW, Patterson TF, et al; Invasive Fungal Infections Group of the European Organisation for Research and Treatment of Cancer and the Global Aspergillus Study Group. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408-415.
3. Patterson TF, Thompson GR III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Disease Society of America. Clin Infect Dis. 2016;63(4):e1-e60.
4. García-Cadenas I, Rivera I, Martino R, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transplant. 2017;52(1):107-113.
5. Vermeulen E, Maertens J, De Bel A, et al. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob Agents Chemother. 2015;59(8):4569-4576.
6. Wiederhold NP, Patterson TF. Emergence of azole resistance in Aspergillus. Semin Respir Crit Care Med. 2015;36(5):673-680.
7. Cuenca-Estrella M, Moore CB, Barchiesi F, et al; AFST Subcommittee of the European Committee on Antimicrobial Susceptibility Testing. Multicenter evaluation of the reproducibility of the proposed antifungal susceptibility testing method for fermentative yeasts of the Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST). Clin Microbiol Infect. 2003;9(6):467-474.
8. Pfaller MA, Diekema DJ, Ghannoum MA, et al; Clinical and Laboratory Standards Institute Antifungal Testing Subcommittee. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by Clinical and Laboratory Standards Institute for broth microdilution methods. J Clin Microbiol. 2009;47(10):3142-3146.
9. Marr KA, Schlamm HT, Herbrecht R, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2015;162(2):81-89.
10. Tashiro M, Izumikawa K, Minematsu A, et al. Antifungal susceptibilities of Aspergillus fumigatus clinical isolates obtained in Nagasaki, Japan. Antimicrob Agents Chemother. 2012;56(1):584-587.
11. Rajendran R, Mowat E, Jones B, Williams C, Ramage G. Prior in vitro exposure to voriconazole confers resistance to amphotericin B in Aspergillus fumigatus biofilms. Int J Antimicrob Agents. 2015;46(3):342-345.
Historically, aspergillosis in patients with hematopoietic stem cell transplantation (HSCT) has carried a high mortality rate. However, recent data demonstrate a dramatic improvement in outcomes for patients with HSCT: 90-day survival increased from 22% before 2000 to 45% over the past 15 years.1 Improved outcomes coincide with changes in transplant immunosuppression practices, use of cross-sectional imaging for early disease identification, galactomannan screening, and the development of novel treatment options.
Voriconazole is an azole drug that blocks the synthesis of ergosterol, a vital component of the cellular membrane of fungi. Voriconazole was approved in 2002 after a clinical trial demonstrated an improvement in 50% of patients with invasive aspergillosis in the voriconazole arm vs 30% in the amphotericin B arm at 12 weeks.2 Amphotericin B is a polyene antifungal drug that binds with ergosterol, creating leaks in the cell membrane that lead to cellular demise. Voriconazole quickly became the first-line therapy for invasive aspergillosis and is recommended by both the Infectious Disease Society of American (IDSA) and the European Conference on Infections in Leukemia.3
Case Presentation
A 55-year-old man with high-risk chronic myelogenous leukemia (CML) underwent a 10 of 10 human leukocyte antigen allele and antigen-matched peripheral blood allogeneic HSCT with a myeloablative-conditioning regimen of busulfan and cyclophosphamide, along with prophylactic voriconazole, sulfamethoxazole/trimethoprim, and acyclovir. After successful engraftment (without significant neutropenia), his posttransplant course was complicated by grade 2 graft vs host disease (GVHD) of the skin, eyes, and liver, which responded well to steroids and tacrolimus. Voriconazole was continued for 5 months until immunosuppression was minimized (tacrolimus 1 mg twice daily). Two months later, the patient’s GVHD worsened, necessitating treatment at an outside hospital with high-dose prednisone (2 mg/kg/d) and cyclosporine (300 mg twice daily). Voriconazole prophylaxis was not reinitiated at that time.
One year later, at a routine follow-up appointment, the patient endorsed several weeks of malaise, weight loss, and nonproductive cough. The patient’s immunosuppression recently had been reduced to 1 mg/kg/d of prednisone and 100 mg of cyclosporine twice daily. A chest X-ray demonstrated multiple pulmonary nodules; follow-up chest computed tomography (CT) confirmed multiple nodular infiltrates with surrounding ground-glass opacities suspicious with a fungal infection (Figure 1).
Treatment with oral voriconazole (300 mg twice daily) was initiated for probable pulmonary aspergillosis. Cyclosporine (150 mg twice daily) and prednisone (1 mg/kg/d) were continued throughout treatment out of concern for hepatic GVHD. The patient’s symptoms improved over the next 10 days, and follow-up chest imaging demonstrated improvement.
Two weeks after initiation of voriconazole treatment, the patient developed a new productive cough and dyspnea, associated with fevers and chills. Repeat imaging revealed right lower-lobe pneumonia. The serum voriconazole trough level was checked and was 3.1 mg/L, suggesting therapeutic dosing. The patient subsequently developed acute respiratory distress syndrome and required intubation and mechanical ventilation. Repeat BAL sampling demonstrated multidrug-resistant Escherichia coli, a BAL galactomannan level of 2.0 ODI, and negative fungal cultures. The patient’s hospital course was complicated by profound hypoxemia, requiring prone positioning and neuromuscular blockade. He was treated with meropenem and voriconazole. His immunosuppression was reduced, but he rapidly developed acute liver injury from hepatic GVHD that resolved after reinitiation of cyclosporine and prednisone at 0.75 mg/kg/d.
The patient improved over the next 3 weeks and was successfully extubated. Repeat chest CT imaging demonstrated numerous pneumatoceles in the location of previous nodules, consistent with healing necrotic fungal disease, and a new right lower-lobe cavitary mass (Figure 2). Two days after transferring out of the intensive care unit, the patient again developed hypoxemia and fevers to 39° C. Bronchoscopy with BAL of the right lower lobe revealed positive A fumigatus and Rhizopus sp polymerase chain reaction (PCR) assays, although fungal cultures were positive only for A fumigatus. Liposomal amphotericin B (5 mg/kg) was added to voriconazole therapy to treat mucormycosis and to provide a second active agent against A fumigatus.
Unfortunately, the patient’s clinical status continued to deteriorate with signs of progressive respiratory failure and infection despite empiric, broad-spectrum antibiotics and dual antifungal therapy. His serum voriconazole level continued to be therapeutic at 1.9 mg/L. The patient declined reintubation and invasive mechanical ventilation, and he ultimately transitioned to comfort measures and died with his family at the bedside.
Autopsy demonstrated widely disseminated Aspergillus infection as the cause of death, with evidence of myocardial, neural, and vascular invasion of A fumigatus (Figures 3 and 4).
Discussion
This case of fatal, progressive, invasive, pulmonary aspergillosis demonstrates several important factors in the treatment of patients with this disease. Treatment failure usually relates to any of 4 possible factors: host immune status, severity or burden of disease, appropriate dosing of antifungal agents, and drug resistance. This patient’s immune system was heavily suppressed for a prolonged period. Attempts at reducing immunosuppression to the minimal required dosage to prevent a GVHD flare were unsuccessful and became an unmodifiable risk factor, a major contributor to his demise.
The risks of continuous high-dose immunosuppression in steroid-refractory GVHD is well understood and has been previously demonstrated to have up to 50% 4-year nonrelapse mortality, mainly due to overwhelming bacterial, viral, and fungal infections.4 All attempts should be made to cease or reduce immunosuppression in the setting of a severe infection, although this is sometimes impossible as in this case.
The patient’s disease burden was significant as evidenced by the bilateral, multifocal pulmonary nodules seen on chest imaging and the disseminated disease found at postmortem examination. His initial improvement in symptoms with voriconazole and the evolution of his images (with many of his initial pulmonary nodules becoming pneumatoceles) suggested a temporary positive immune response. The authors believe that the Rhizopus in his sputum represents noninvasive colonization of one of his pneumatoceles, because postmortem examination failed to reveal Rhizopus at any other location.
Voriconazole has excellent pulmonary and central nervous system penetration: In this patient serum levels were well within the therapeutic range. His peculiar drug resistance pattern (sensitivity to azoles and resistance to amphotericin) is unusual. Azole resistance in leukemia and patients with HSCT is more common than is amphotericin resistance, with current estimates of azole resistance close to 5%, ranging between 1% and 30%.5,6 Widespread use of antifungal prophylaxis with azoles likely selects for azole resistance.6
Despite this concern of azole resistance, current IDSA guidelines recommend against routine susceptibility testing of Aspergillus to azole therapy because of the current lack of consensus between the European Committee on Antibiotic Susceptibility Testing and Clinical and Laboratory Standards Institute on break points for resistance patterns.3,7 This is an area of emerging research, and proposed cut points for declaration of resistance do exist in the literature even if not globally agreed on.8
Combination antifungal therapy is an option for treatment in cases of possible drug resistance. Nonetheless, a recent randomized, double-blind, placebo-controlled, multicenter trial comparing voriconazole monotherapy with the combination of voriconazole and anidulafungin failed to demonstrate an overall mortality benefit in the primary analysis, although secondary analysis showed a mortality benefit with combination therapy in patients at highest risk for death.9
Despite the lack of unified standards with susceptibility testing, it may be reasonable to perform such tests in patients with demonstrating progressive disease. In this patient’s case, amphotericin B was added to treat the Rhizopus species found in his sputum, and while not the combination studied in the previously mentioned study, the drug should have provided an additional active agent for Aspergillus should this patient have had azole resistance.
Surprisingly, subsequent testing demonstrated the Aspergillus species to be resistant to amphotericin B. De novo amphotericin B-resistant A fumigates is extremely rare, with an expected incidence of 1% or less.10 The authors believe the patient may have demonstrated induction of amphotericin-B resistance through activation of fungal stress pathways by prior treatment with voriconazole. This has been demonstrated in vitro and should be considered should combination salvage therapy be required for the treatment of a refractory Aspergillus infection especially if patients have received prior treatment with voriconazole.11
Conclusion
This fatal case of invasive pulmonary aspergillosis illustrates the importance of considering the 4 main causes of treatment failure in an infection. Although the patient had a high burden of disease with a rare resistance pattern, he was treated with appropriate and well-dosed therapy. Ultimately, his unmodifiable immunosuppression was likely the driving factor leading to treatment failure and death. The indication for and number of bone marrow transplants continues to increase, thus exposure to and treatment of invasive fungal infections will increase accordingly. As such, providers should ensure that all causes of treatment failure are considered and addressed.
Historically, aspergillosis in patients with hematopoietic stem cell transplantation (HSCT) has carried a high mortality rate. However, recent data demonstrate a dramatic improvement in outcomes for patients with HSCT: 90-day survival increased from 22% before 2000 to 45% over the past 15 years.1 Improved outcomes coincide with changes in transplant immunosuppression practices, use of cross-sectional imaging for early disease identification, galactomannan screening, and the development of novel treatment options.
Voriconazole is an azole drug that blocks the synthesis of ergosterol, a vital component of the cellular membrane of fungi. Voriconazole was approved in 2002 after a clinical trial demonstrated an improvement in 50% of patients with invasive aspergillosis in the voriconazole arm vs 30% in the amphotericin B arm at 12 weeks.2 Amphotericin B is a polyene antifungal drug that binds with ergosterol, creating leaks in the cell membrane that lead to cellular demise. Voriconazole quickly became the first-line therapy for invasive aspergillosis and is recommended by both the Infectious Disease Society of American (IDSA) and the European Conference on Infections in Leukemia.3
Case Presentation
A 55-year-old man with high-risk chronic myelogenous leukemia (CML) underwent a 10 of 10 human leukocyte antigen allele and antigen-matched peripheral blood allogeneic HSCT with a myeloablative-conditioning regimen of busulfan and cyclophosphamide, along with prophylactic voriconazole, sulfamethoxazole/trimethoprim, and acyclovir. After successful engraftment (without significant neutropenia), his posttransplant course was complicated by grade 2 graft vs host disease (GVHD) of the skin, eyes, and liver, which responded well to steroids and tacrolimus. Voriconazole was continued for 5 months until immunosuppression was minimized (tacrolimus 1 mg twice daily). Two months later, the patient’s GVHD worsened, necessitating treatment at an outside hospital with high-dose prednisone (2 mg/kg/d) and cyclosporine (300 mg twice daily). Voriconazole prophylaxis was not reinitiated at that time.
One year later, at a routine follow-up appointment, the patient endorsed several weeks of malaise, weight loss, and nonproductive cough. The patient’s immunosuppression recently had been reduced to 1 mg/kg/d of prednisone and 100 mg of cyclosporine twice daily. A chest X-ray demonstrated multiple pulmonary nodules; follow-up chest computed tomography (CT) confirmed multiple nodular infiltrates with surrounding ground-glass opacities suspicious with a fungal infection (Figure 1).
Treatment with oral voriconazole (300 mg twice daily) was initiated for probable pulmonary aspergillosis. Cyclosporine (150 mg twice daily) and prednisone (1 mg/kg/d) were continued throughout treatment out of concern for hepatic GVHD. The patient’s symptoms improved over the next 10 days, and follow-up chest imaging demonstrated improvement.
Two weeks after initiation of voriconazole treatment, the patient developed a new productive cough and dyspnea, associated with fevers and chills. Repeat imaging revealed right lower-lobe pneumonia. The serum voriconazole trough level was checked and was 3.1 mg/L, suggesting therapeutic dosing. The patient subsequently developed acute respiratory distress syndrome and required intubation and mechanical ventilation. Repeat BAL sampling demonstrated multidrug-resistant Escherichia coli, a BAL galactomannan level of 2.0 ODI, and negative fungal cultures. The patient’s hospital course was complicated by profound hypoxemia, requiring prone positioning and neuromuscular blockade. He was treated with meropenem and voriconazole. His immunosuppression was reduced, but he rapidly developed acute liver injury from hepatic GVHD that resolved after reinitiation of cyclosporine and prednisone at 0.75 mg/kg/d.
The patient improved over the next 3 weeks and was successfully extubated. Repeat chest CT imaging demonstrated numerous pneumatoceles in the location of previous nodules, consistent with healing necrotic fungal disease, and a new right lower-lobe cavitary mass (Figure 2). Two days after transferring out of the intensive care unit, the patient again developed hypoxemia and fevers to 39° C. Bronchoscopy with BAL of the right lower lobe revealed positive A fumigatus and Rhizopus sp polymerase chain reaction (PCR) assays, although fungal cultures were positive only for A fumigatus. Liposomal amphotericin B (5 mg/kg) was added to voriconazole therapy to treat mucormycosis and to provide a second active agent against A fumigatus.
Unfortunately, the patient’s clinical status continued to deteriorate with signs of progressive respiratory failure and infection despite empiric, broad-spectrum antibiotics and dual antifungal therapy. His serum voriconazole level continued to be therapeutic at 1.9 mg/L. The patient declined reintubation and invasive mechanical ventilation, and he ultimately transitioned to comfort measures and died with his family at the bedside.
Autopsy demonstrated widely disseminated Aspergillus infection as the cause of death, with evidence of myocardial, neural, and vascular invasion of A fumigatus (Figures 3 and 4).
Discussion
This case of fatal, progressive, invasive, pulmonary aspergillosis demonstrates several important factors in the treatment of patients with this disease. Treatment failure usually relates to any of 4 possible factors: host immune status, severity or burden of disease, appropriate dosing of antifungal agents, and drug resistance. This patient’s immune system was heavily suppressed for a prolonged period. Attempts at reducing immunosuppression to the minimal required dosage to prevent a GVHD flare were unsuccessful and became an unmodifiable risk factor, a major contributor to his demise.
The risks of continuous high-dose immunosuppression in steroid-refractory GVHD is well understood and has been previously demonstrated to have up to 50% 4-year nonrelapse mortality, mainly due to overwhelming bacterial, viral, and fungal infections.4 All attempts should be made to cease or reduce immunosuppression in the setting of a severe infection, although this is sometimes impossible as in this case.
The patient’s disease burden was significant as evidenced by the bilateral, multifocal pulmonary nodules seen on chest imaging and the disseminated disease found at postmortem examination. His initial improvement in symptoms with voriconazole and the evolution of his images (with many of his initial pulmonary nodules becoming pneumatoceles) suggested a temporary positive immune response. The authors believe that the Rhizopus in his sputum represents noninvasive colonization of one of his pneumatoceles, because postmortem examination failed to reveal Rhizopus at any other location.
Voriconazole has excellent pulmonary and central nervous system penetration: In this patient serum levels were well within the therapeutic range. His peculiar drug resistance pattern (sensitivity to azoles and resistance to amphotericin) is unusual. Azole resistance in leukemia and patients with HSCT is more common than is amphotericin resistance, with current estimates of azole resistance close to 5%, ranging between 1% and 30%.5,6 Widespread use of antifungal prophylaxis with azoles likely selects for azole resistance.6
Despite this concern of azole resistance, current IDSA guidelines recommend against routine susceptibility testing of Aspergillus to azole therapy because of the current lack of consensus between the European Committee on Antibiotic Susceptibility Testing and Clinical and Laboratory Standards Institute on break points for resistance patterns.3,7 This is an area of emerging research, and proposed cut points for declaration of resistance do exist in the literature even if not globally agreed on.8
Combination antifungal therapy is an option for treatment in cases of possible drug resistance. Nonetheless, a recent randomized, double-blind, placebo-controlled, multicenter trial comparing voriconazole monotherapy with the combination of voriconazole and anidulafungin failed to demonstrate an overall mortality benefit in the primary analysis, although secondary analysis showed a mortality benefit with combination therapy in patients at highest risk for death.9
Despite the lack of unified standards with susceptibility testing, it may be reasonable to perform such tests in patients with demonstrating progressive disease. In this patient’s case, amphotericin B was added to treat the Rhizopus species found in his sputum, and while not the combination studied in the previously mentioned study, the drug should have provided an additional active agent for Aspergillus should this patient have had azole resistance.
Surprisingly, subsequent testing demonstrated the Aspergillus species to be resistant to amphotericin B. De novo amphotericin B-resistant A fumigates is extremely rare, with an expected incidence of 1% or less.10 The authors believe the patient may have demonstrated induction of amphotericin-B resistance through activation of fungal stress pathways by prior treatment with voriconazole. This has been demonstrated in vitro and should be considered should combination salvage therapy be required for the treatment of a refractory Aspergillus infection especially if patients have received prior treatment with voriconazole.11
Conclusion
This fatal case of invasive pulmonary aspergillosis illustrates the importance of considering the 4 main causes of treatment failure in an infection. Although the patient had a high burden of disease with a rare resistance pattern, he was treated with appropriate and well-dosed therapy. Ultimately, his unmodifiable immunosuppression was likely the driving factor leading to treatment failure and death. The indication for and number of bone marrow transplants continues to increase, thus exposure to and treatment of invasive fungal infections will increase accordingly. As such, providers should ensure that all causes of treatment failure are considered and addressed.
1. Upton A, Kirby KA, Carpenter P, Boeckh M, Marr KA. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin Infect Dis. 2007;44(4):531-540.
2. Herbrecht R, Denning DW, Patterson TF, et al; Invasive Fungal Infections Group of the European Organisation for Research and Treatment of Cancer and the Global Aspergillus Study Group. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408-415.
3. Patterson TF, Thompson GR III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Disease Society of America. Clin Infect Dis. 2016;63(4):e1-e60.
4. García-Cadenas I, Rivera I, Martino R, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transplant. 2017;52(1):107-113.
5. Vermeulen E, Maertens J, De Bel A, et al. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob Agents Chemother. 2015;59(8):4569-4576.
6. Wiederhold NP, Patterson TF. Emergence of azole resistance in Aspergillus. Semin Respir Crit Care Med. 2015;36(5):673-680.
7. Cuenca-Estrella M, Moore CB, Barchiesi F, et al; AFST Subcommittee of the European Committee on Antimicrobial Susceptibility Testing. Multicenter evaluation of the reproducibility of the proposed antifungal susceptibility testing method for fermentative yeasts of the Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST). Clin Microbiol Infect. 2003;9(6):467-474.
8. Pfaller MA, Diekema DJ, Ghannoum MA, et al; Clinical and Laboratory Standards Institute Antifungal Testing Subcommittee. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by Clinical and Laboratory Standards Institute for broth microdilution methods. J Clin Microbiol. 2009;47(10):3142-3146.
9. Marr KA, Schlamm HT, Herbrecht R, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2015;162(2):81-89.
10. Tashiro M, Izumikawa K, Minematsu A, et al. Antifungal susceptibilities of Aspergillus fumigatus clinical isolates obtained in Nagasaki, Japan. Antimicrob Agents Chemother. 2012;56(1):584-587.
11. Rajendran R, Mowat E, Jones B, Williams C, Ramage G. Prior in vitro exposure to voriconazole confers resistance to amphotericin B in Aspergillus fumigatus biofilms. Int J Antimicrob Agents. 2015;46(3):342-345.
1. Upton A, Kirby KA, Carpenter P, Boeckh M, Marr KA. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin Infect Dis. 2007;44(4):531-540.
2. Herbrecht R, Denning DW, Patterson TF, et al; Invasive Fungal Infections Group of the European Organisation for Research and Treatment of Cancer and the Global Aspergillus Study Group. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408-415.
3. Patterson TF, Thompson GR III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Disease Society of America. Clin Infect Dis. 2016;63(4):e1-e60.
4. García-Cadenas I, Rivera I, Martino R, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transplant. 2017;52(1):107-113.
5. Vermeulen E, Maertens J, De Bel A, et al. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob Agents Chemother. 2015;59(8):4569-4576.
6. Wiederhold NP, Patterson TF. Emergence of azole resistance in Aspergillus. Semin Respir Crit Care Med. 2015;36(5):673-680.
7. Cuenca-Estrella M, Moore CB, Barchiesi F, et al; AFST Subcommittee of the European Committee on Antimicrobial Susceptibility Testing. Multicenter evaluation of the reproducibility of the proposed antifungal susceptibility testing method for fermentative yeasts of the Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST). Clin Microbiol Infect. 2003;9(6):467-474.
8. Pfaller MA, Diekema DJ, Ghannoum MA, et al; Clinical and Laboratory Standards Institute Antifungal Testing Subcommittee. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by Clinical and Laboratory Standards Institute for broth microdilution methods. J Clin Microbiol. 2009;47(10):3142-3146.
9. Marr KA, Schlamm HT, Herbrecht R, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2015;162(2):81-89.
10. Tashiro M, Izumikawa K, Minematsu A, et al. Antifungal susceptibilities of Aspergillus fumigatus clinical isolates obtained in Nagasaki, Japan. Antimicrob Agents Chemother. 2012;56(1):584-587.
11. Rajendran R, Mowat E, Jones B, Williams C, Ramage G. Prior in vitro exposure to voriconazole confers resistance to amphotericin B in Aspergillus fumigatus biofilms. Int J Antimicrob Agents. 2015;46(3):342-345.
Recurrence of Linear Basal Cell Carcinoma
Case Report
A 63-year-old man was evaluated in the Mohs clinic for a lesion on the right supraclavicular neck, which he described as a linear asymptomatic “birthmark” that had been present since childhood and stable for many years. It began to enlarge approximately 5 years prior, became increasingly red, and had occasional crusting. The lesion also gradually became more irritated with repeated mild trauma when he carried a backpack while hiking. On physical examination, a 10×2-cm, linear, pink plaque with an irregular border, translucent rolled edges, and central smooth atrophic skin was seen on the right supraclavicular neck (Figure). There was no visible epidermal nevus or nevus sebaceous in the area. A shave biopsy of the lesion confirmed the pathologic diagnosis of basal cell carcinoma, nodular type, along with the morphologic diagnosis of linear basal cell carcinoma (LBCC). The tumor was completely removed with standard excision using 5-mm margins.

Approximately 10 months after the original excision, the patient developed an irritated erosion that occasionally bled when his backpack rubbed against it. He returned to the clinic after the erosion failed to heal. Physical examination revealed a 1.4×0.7-cm, eroded, pink papule with large telangiectases at the superior pole of the excision scar. A shave biopsy confirmed the diagnosis of a recurrent infiltrative basal cell carcinoma. The tumor was then completely excised using Mohs micrographic surgery.
Comment
Linear basal cell carcinoma, first described by Lewis1 in 1985, is a rare morphologic variant of basal cell carcinoma. In 2011, Al-Niaimi and Lyon2 performed a comprehensive literature search on LBCC (1985-2008) and found only 39 cases (including 2 of their own) had been published since the pioneer case in 1985. It was determined that the most common sites affected were the periorbital area and neck (n=13 each [67%]), and the majority were histologically nodular (n=27 [69%]). Mohs micrographic surgery was the most common treatment method (n=23 [59%]), followed by primary excision (n=17 [44%]). A history of trauma, radiotherapy, or prior operation in association with the site of the LBCC was discovered in only 7 cases (18%).2 Although Peschen et al3 proposed that trauma—both physical and surgical—and radiotherapy may play a role in the development of LBCCs, the low incidence reported suggests that other factors may be involved. To determine if genetic factors were contributing to the development of LBCCs, Yamaguchi et al4 investigated the expression of p27 and PCTAIRE1, both known to contribute to tumorigenesis when mutated, as well as somatic gene mutations using deep sequencing in a case of LBCC; they found no associated genetic mutation.
Reported Cases of LBCC
According to a PubMed search of articles indexed for MEDLINE using the terms linear and basal cell carcinoma, 67 cases (including the current case) of LBCC have been published since 1985. The patient demographics, anatomic location, histologic subtype, treatment methods, and frequency of recurrence for all reported cases of LBCC are summarized in the Table.1-24 There were 36 women and 31 men, with an average age of 70 years (range, 40–92 years). The most commonly affected sites were the periocular region (n=27) and neck (n=18). Histologically, most LBCCs were nodular (n=35), with the next most common histologic subtype being infiltrative (n=20), which included the morphoeic, metatypical, and micronodular subtypes under the overarching infiltrative subtype. The most frequently chosen treatment option was primary excision (n=38 [57%]), followed by Mohs micrographic surgery (n=28 [42%]). Risk factors previously identified by Al-Niaimi and Lyon,2 including trauma, radiotherapy, or prior operation, were reported in 12 of 67 cases. Recurrence was reported in only 2 of 67 cases, 1 being the current case; however, an accurate recurrence rate could not be calculated due to lack of follow-up or short length of follow-up in most of the reported cases.
Presentation and Treatment
Currently, there are no set criteria for the diagnosis of LBCC, but it has been shown to follow a characteristic morphologic pattern, favoring extension in one direction leading to a length-to-width ratio that typically is at least 3 to 1.5 With most lesions presenting in the periocular region along relaxed skin tension lines, it has been speculated that these tumors expand along wrinkles.2 Pierard and Lapiere25 proposed that the preferential parallel orientation and a straightening of thin collagen bundles and elastic fibers within the reticular dermis combined with relaxed skin tension lines and muscle contraction perpendicular to these stromal parts may influence the growth of tumors preferentially in one direction, contributing to linearity of the lesion. In addition, the clinical appearance is not a reliable indicator of subclinical extension.2 Therefore, Lim et al6 recommended Mohs micrographic surgery as the best initial treatment of LBCCs.
Conclusion
Linear basal cell carcinoma should be considered a distinct morphologic variant of basal cell carcinoma. Although likely underreported, this variant is uncommon. It presents most often in the periocular and neck regions. The most common histologic subtypes are nodular and infiltrative. Because of the likelihood of subclinical spread, LBCC should be regarded as a high-risk subtype. As such, Mohs micrographic surgery or excision with complete circumferential peripheral and deep margin assessment is recommended as first-line treatment of LBCC.6
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1985;24:124-125.
- Al-Niaimi F, Lyon CC. Linear basal cell carcinoma: a distinct condition? Clin Exp Dermatol. 2011;36:231-234.
- Peschen M, Lo JS, Snow SN, et al. Linear basal cell carcinoma. Cutis. 1993;51:287-289.
- Yamaguchi Y, Yanagi T, Imafuku K, et al. A case of linear basal cell carcinoma: evaluation of proliferative activity by immunohistochemical staining of PCTAIRE1 and p27. J Eur Acad Dermatol Venereol. 2017;31:E359-E362.
- Mavirakis I, Malhotra R, Selva D, et al. Linear basal cell carcinoma: a distinct clinical entity. J Plast Reconstr Aesthet Surg. 2006;59:419-423.
- Lim KK, Randle HW, Roenigk RK, et al. Linear basal cell carcinoma: report of seventeen cases and review of the presentation and treatment. Dermatol Surg. 1999;25:63-67.
- Pardavila R, Rosón E, De la torre C, et al. Linear basal cell carcinoma. report of two cases [in Spanish]. Actas Dermosifiliogr. 2007;98:291.
- Shinsuke K, Hirohiko K, Yasuhiro T, et al. Linear basal cell carcinoma in an Asian patient. Open Ophthalmol J. 2007;1:20-22.
- Ning C, Chao S. Linear basal cell carcinoma of the scrotum. Dermatol Sinica. 2002;20:57-62.
- Chopra KF, Cohen PR. Linear basal cell carcinomas: report of multiple sequential tumors localized to a radiotherapy port and review of the literature. Tex Med. 1997;93:57-59.
- da Silva MO, Dadalt P, Santos OL, et al. Linear basal cell carcinoma. Int J Dermatol. 1995;34:488.
- Warthan TL, Lewis JE. Giant linear basal cell epithelioma. Int J Dermatol. 1994;33:284.
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1989;28:682-684.
- Alcántara-Reifs CM, Salido-Vallejo R, González-Menchen A, et al. Linear basal cell carcinoma: report of three cases with dermoscopic findings. Indian J Dermatol Venereol Leprol. 2016;82:708-711.
- Lee MS, Cho E, Lee JH, et al. Linearly curved, blackish macule on the wrist. Cutis. 2016;97:384, 406-407.
- Bajaj S, Sharma PK, Kar HK. Linear adamantinoid basal cell carcinoma in the axilla. Dermatol Online J. 2015;21. pii:13030/qt8k0713nb.
- Iga N, Sakurai K, Fujii H, et al. Linear basal cell carcinoma at the external genitalia. J Dermatol. 2014;41:275-276.
- Ichinokawa Y, Ohtuki A, Hattori M, et al. Linear basal cell carcinoma: a case report. Case Rep Dermatol. 2011;3:142-146.
- Becher GL, Affleck A, Fleming C, et al. Linear basal cell carcinoma occurs most commonly on the lower eyelid. Clin Exp Dermatol. 2011;36:311-312.
- Jellouli A, Triki S, Zghal M, et al. Linear basal cell carcinoma. Actas Dermosifiliogr. 2010;101:648-650.
- Takiyoshi N, Nakano H, Kaneko T, et al. A linear basal cell carcinoma undergoing spontaneous regression. Clin Exp Dermatol. 2009;34:E411-E413.
- Yoleri L, Ozden S, Kandiloglu A. A 46-year-old male with an ulcerated linear lesion on his neck. Ann Saudi Med. 2008;28:57-58.
- Palleschi GM, Corradini D, Bruscino N, et al. Linear basal cell carcinoma: clinical significance and better surgical approach. G Ital Dermatol Venereol. 2016;151:119-121.
- Rodriguez-Garijo N, Redondo P. Linear basal cell carcinoma of the lower eyelid: reconstruction with a musculocutaneous transposition flap. JAAD Case Rep. 2018;4:633-635.
- Pierard GE, Lapiere CM. Microanatomy of the dermis in relation to relaxed skin tension lines and Langer’s lines. Am J Dermatopathol. 1987;9:219-224.
Case Report
A 63-year-old man was evaluated in the Mohs clinic for a lesion on the right supraclavicular neck, which he described as a linear asymptomatic “birthmark” that had been present since childhood and stable for many years. It began to enlarge approximately 5 years prior, became increasingly red, and had occasional crusting. The lesion also gradually became more irritated with repeated mild trauma when he carried a backpack while hiking. On physical examination, a 10×2-cm, linear, pink plaque with an irregular border, translucent rolled edges, and central smooth atrophic skin was seen on the right supraclavicular neck (Figure). There was no visible epidermal nevus or nevus sebaceous in the area. A shave biopsy of the lesion confirmed the pathologic diagnosis of basal cell carcinoma, nodular type, along with the morphologic diagnosis of linear basal cell carcinoma (LBCC). The tumor was completely removed with standard excision using 5-mm margins.
Approximately 10 months after the original excision, the patient developed an irritated erosion that occasionally bled when his backpack rubbed against it. He returned to the clinic after the erosion failed to heal. Physical examination revealed a 1.4×0.7-cm, eroded, pink papule with large telangiectases at the superior pole of the excision scar. A shave biopsy confirmed the diagnosis of a recurrent infiltrative basal cell carcinoma. The tumor was then completely excised using Mohs micrographic surgery.
Comment
Linear basal cell carcinoma, first described by Lewis1 in 1985, is a rare morphologic variant of basal cell carcinoma. In 2011, Al-Niaimi and Lyon2 performed a comprehensive literature search on LBCC (1985-2008) and found only 39 cases (including 2 of their own) had been published since the pioneer case in 1985. It was determined that the most common sites affected were the periorbital area and neck (n=13 each [67%]), and the majority were histologically nodular (n=27 [69%]). Mohs micrographic surgery was the most common treatment method (n=23 [59%]), followed by primary excision (n=17 [44%]). A history of trauma, radiotherapy, or prior operation in association with the site of the LBCC was discovered in only 7 cases (18%).2 Although Peschen et al3 proposed that trauma—both physical and surgical—and radiotherapy may play a role in the development of LBCCs, the low incidence reported suggests that other factors may be involved. To determine if genetic factors were contributing to the development of LBCCs, Yamaguchi et al4 investigated the expression of p27 and PCTAIRE1, both known to contribute to tumorigenesis when mutated, as well as somatic gene mutations using deep sequencing in a case of LBCC; they found no associated genetic mutation.
Reported Cases of LBCC
According to a PubMed search of articles indexed for MEDLINE using the terms linear and basal cell carcinoma, 67 cases (including the current case) of LBCC have been published since 1985. The patient demographics, anatomic location, histologic subtype, treatment methods, and frequency of recurrence for all reported cases of LBCC are summarized in the Table.1-24 There were 36 women and 31 men, with an average age of 70 years (range, 40–92 years). The most commonly affected sites were the periocular region (n=27) and neck (n=18). Histologically, most LBCCs were nodular (n=35), with the next most common histologic subtype being infiltrative (n=20), which included the morphoeic, metatypical, and micronodular subtypes under the overarching infiltrative subtype. The most frequently chosen treatment option was primary excision (n=38 [57%]), followed by Mohs micrographic surgery (n=28 [42%]). Risk factors previously identified by Al-Niaimi and Lyon,2 including trauma, radiotherapy, or prior operation, were reported in 12 of 67 cases. Recurrence was reported in only 2 of 67 cases, 1 being the current case; however, an accurate recurrence rate could not be calculated due to lack of follow-up or short length of follow-up in most of the reported cases.
Presentation and Treatment
Currently, there are no set criteria for the diagnosis of LBCC, but it has been shown to follow a characteristic morphologic pattern, favoring extension in one direction leading to a length-to-width ratio that typically is at least 3 to 1.5 With most lesions presenting in the periocular region along relaxed skin tension lines, it has been speculated that these tumors expand along wrinkles.2 Pierard and Lapiere25 proposed that the preferential parallel orientation and a straightening of thin collagen bundles and elastic fibers within the reticular dermis combined with relaxed skin tension lines and muscle contraction perpendicular to these stromal parts may influence the growth of tumors preferentially in one direction, contributing to linearity of the lesion. In addition, the clinical appearance is not a reliable indicator of subclinical extension.2 Therefore, Lim et al6 recommended Mohs micrographic surgery as the best initial treatment of LBCCs.
Conclusion
Linear basal cell carcinoma should be considered a distinct morphologic variant of basal cell carcinoma. Although likely underreported, this variant is uncommon. It presents most often in the periocular and neck regions. The most common histologic subtypes are nodular and infiltrative. Because of the likelihood of subclinical spread, LBCC should be regarded as a high-risk subtype. As such, Mohs micrographic surgery or excision with complete circumferential peripheral and deep margin assessment is recommended as first-line treatment of LBCC.6
Case Report
A 63-year-old man was evaluated in the Mohs clinic for a lesion on the right supraclavicular neck, which he described as a linear asymptomatic “birthmark” that had been present since childhood and stable for many years. It began to enlarge approximately 5 years prior, became increasingly red, and had occasional crusting. The lesion also gradually became more irritated with repeated mild trauma when he carried a backpack while hiking. On physical examination, a 10×2-cm, linear, pink plaque with an irregular border, translucent rolled edges, and central smooth atrophic skin was seen on the right supraclavicular neck (Figure). There was no visible epidermal nevus or nevus sebaceous in the area. A shave biopsy of the lesion confirmed the pathologic diagnosis of basal cell carcinoma, nodular type, along with the morphologic diagnosis of linear basal cell carcinoma (LBCC). The tumor was completely removed with standard excision using 5-mm margins.
Approximately 10 months after the original excision, the patient developed an irritated erosion that occasionally bled when his backpack rubbed against it. He returned to the clinic after the erosion failed to heal. Physical examination revealed a 1.4×0.7-cm, eroded, pink papule with large telangiectases at the superior pole of the excision scar. A shave biopsy confirmed the diagnosis of a recurrent infiltrative basal cell carcinoma. The tumor was then completely excised using Mohs micrographic surgery.
Comment
Linear basal cell carcinoma, first described by Lewis1 in 1985, is a rare morphologic variant of basal cell carcinoma. In 2011, Al-Niaimi and Lyon2 performed a comprehensive literature search on LBCC (1985-2008) and found only 39 cases (including 2 of their own) had been published since the pioneer case in 1985. It was determined that the most common sites affected were the periorbital area and neck (n=13 each [67%]), and the majority were histologically nodular (n=27 [69%]). Mohs micrographic surgery was the most common treatment method (n=23 [59%]), followed by primary excision (n=17 [44%]). A history of trauma, radiotherapy, or prior operation in association with the site of the LBCC was discovered in only 7 cases (18%).2 Although Peschen et al3 proposed that trauma—both physical and surgical—and radiotherapy may play a role in the development of LBCCs, the low incidence reported suggests that other factors may be involved. To determine if genetic factors were contributing to the development of LBCCs, Yamaguchi et al4 investigated the expression of p27 and PCTAIRE1, both known to contribute to tumorigenesis when mutated, as well as somatic gene mutations using deep sequencing in a case of LBCC; they found no associated genetic mutation.
Reported Cases of LBCC
According to a PubMed search of articles indexed for MEDLINE using the terms linear and basal cell carcinoma, 67 cases (including the current case) of LBCC have been published since 1985. The patient demographics, anatomic location, histologic subtype, treatment methods, and frequency of recurrence for all reported cases of LBCC are summarized in the Table.1-24 There were 36 women and 31 men, with an average age of 70 years (range, 40–92 years). The most commonly affected sites were the periocular region (n=27) and neck (n=18). Histologically, most LBCCs were nodular (n=35), with the next most common histologic subtype being infiltrative (n=20), which included the morphoeic, metatypical, and micronodular subtypes under the overarching infiltrative subtype. The most frequently chosen treatment option was primary excision (n=38 [57%]), followed by Mohs micrographic surgery (n=28 [42%]). Risk factors previously identified by Al-Niaimi and Lyon,2 including trauma, radiotherapy, or prior operation, were reported in 12 of 67 cases. Recurrence was reported in only 2 of 67 cases, 1 being the current case; however, an accurate recurrence rate could not be calculated due to lack of follow-up or short length of follow-up in most of the reported cases.
Presentation and Treatment
Currently, there are no set criteria for the diagnosis of LBCC, but it has been shown to follow a characteristic morphologic pattern, favoring extension in one direction leading to a length-to-width ratio that typically is at least 3 to 1.5 With most lesions presenting in the periocular region along relaxed skin tension lines, it has been speculated that these tumors expand along wrinkles.2 Pierard and Lapiere25 proposed that the preferential parallel orientation and a straightening of thin collagen bundles and elastic fibers within the reticular dermis combined with relaxed skin tension lines and muscle contraction perpendicular to these stromal parts may influence the growth of tumors preferentially in one direction, contributing to linearity of the lesion. In addition, the clinical appearance is not a reliable indicator of subclinical extension.2 Therefore, Lim et al6 recommended Mohs micrographic surgery as the best initial treatment of LBCCs.
Conclusion
Linear basal cell carcinoma should be considered a distinct morphologic variant of basal cell carcinoma. Although likely underreported, this variant is uncommon. It presents most often in the periocular and neck regions. The most common histologic subtypes are nodular and infiltrative. Because of the likelihood of subclinical spread, LBCC should be regarded as a high-risk subtype. As such, Mohs micrographic surgery or excision with complete circumferential peripheral and deep margin assessment is recommended as first-line treatment of LBCC.6
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1985;24:124-125.
- Al-Niaimi F, Lyon CC. Linear basal cell carcinoma: a distinct condition? Clin Exp Dermatol. 2011;36:231-234.
- Peschen M, Lo JS, Snow SN, et al. Linear basal cell carcinoma. Cutis. 1993;51:287-289.
- Yamaguchi Y, Yanagi T, Imafuku K, et al. A case of linear basal cell carcinoma: evaluation of proliferative activity by immunohistochemical staining of PCTAIRE1 and p27. J Eur Acad Dermatol Venereol. 2017;31:E359-E362.
- Mavirakis I, Malhotra R, Selva D, et al. Linear basal cell carcinoma: a distinct clinical entity. J Plast Reconstr Aesthet Surg. 2006;59:419-423.
- Lim KK, Randle HW, Roenigk RK, et al. Linear basal cell carcinoma: report of seventeen cases and review of the presentation and treatment. Dermatol Surg. 1999;25:63-67.
- Pardavila R, Rosón E, De la torre C, et al. Linear basal cell carcinoma. report of two cases [in Spanish]. Actas Dermosifiliogr. 2007;98:291.
- Shinsuke K, Hirohiko K, Yasuhiro T, et al. Linear basal cell carcinoma in an Asian patient. Open Ophthalmol J. 2007;1:20-22.
- Ning C, Chao S. Linear basal cell carcinoma of the scrotum. Dermatol Sinica. 2002;20:57-62.
- Chopra KF, Cohen PR. Linear basal cell carcinomas: report of multiple sequential tumors localized to a radiotherapy port and review of the literature. Tex Med. 1997;93:57-59.
- da Silva MO, Dadalt P, Santos OL, et al. Linear basal cell carcinoma. Int J Dermatol. 1995;34:488.
- Warthan TL, Lewis JE. Giant linear basal cell epithelioma. Int J Dermatol. 1994;33:284.
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1989;28:682-684.
- Alcántara-Reifs CM, Salido-Vallejo R, González-Menchen A, et al. Linear basal cell carcinoma: report of three cases with dermoscopic findings. Indian J Dermatol Venereol Leprol. 2016;82:708-711.
- Lee MS, Cho E, Lee JH, et al. Linearly curved, blackish macule on the wrist. Cutis. 2016;97:384, 406-407.
- Bajaj S, Sharma PK, Kar HK. Linear adamantinoid basal cell carcinoma in the axilla. Dermatol Online J. 2015;21. pii:13030/qt8k0713nb.
- Iga N, Sakurai K, Fujii H, et al. Linear basal cell carcinoma at the external genitalia. J Dermatol. 2014;41:275-276.
- Ichinokawa Y, Ohtuki A, Hattori M, et al. Linear basal cell carcinoma: a case report. Case Rep Dermatol. 2011;3:142-146.
- Becher GL, Affleck A, Fleming C, et al. Linear basal cell carcinoma occurs most commonly on the lower eyelid. Clin Exp Dermatol. 2011;36:311-312.
- Jellouli A, Triki S, Zghal M, et al. Linear basal cell carcinoma. Actas Dermosifiliogr. 2010;101:648-650.
- Takiyoshi N, Nakano H, Kaneko T, et al. A linear basal cell carcinoma undergoing spontaneous regression. Clin Exp Dermatol. 2009;34:E411-E413.
- Yoleri L, Ozden S, Kandiloglu A. A 46-year-old male with an ulcerated linear lesion on his neck. Ann Saudi Med. 2008;28:57-58.
- Palleschi GM, Corradini D, Bruscino N, et al. Linear basal cell carcinoma: clinical significance and better surgical approach. G Ital Dermatol Venereol. 2016;151:119-121.
- Rodriguez-Garijo N, Redondo P. Linear basal cell carcinoma of the lower eyelid: reconstruction with a musculocutaneous transposition flap. JAAD Case Rep. 2018;4:633-635.
- Pierard GE, Lapiere CM. Microanatomy of the dermis in relation to relaxed skin tension lines and Langer’s lines. Am J Dermatopathol. 1987;9:219-224.
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1985;24:124-125.
- Al-Niaimi F, Lyon CC. Linear basal cell carcinoma: a distinct condition? Clin Exp Dermatol. 2011;36:231-234.
- Peschen M, Lo JS, Snow SN, et al. Linear basal cell carcinoma. Cutis. 1993;51:287-289.
- Yamaguchi Y, Yanagi T, Imafuku K, et al. A case of linear basal cell carcinoma: evaluation of proliferative activity by immunohistochemical staining of PCTAIRE1 and p27. J Eur Acad Dermatol Venereol. 2017;31:E359-E362.
- Mavirakis I, Malhotra R, Selva D, et al. Linear basal cell carcinoma: a distinct clinical entity. J Plast Reconstr Aesthet Surg. 2006;59:419-423.
- Lim KK, Randle HW, Roenigk RK, et al. Linear basal cell carcinoma: report of seventeen cases and review of the presentation and treatment. Dermatol Surg. 1999;25:63-67.
- Pardavila R, Rosón E, De la torre C, et al. Linear basal cell carcinoma. report of two cases [in Spanish]. Actas Dermosifiliogr. 2007;98:291.
- Shinsuke K, Hirohiko K, Yasuhiro T, et al. Linear basal cell carcinoma in an Asian patient. Open Ophthalmol J. 2007;1:20-22.
- Ning C, Chao S. Linear basal cell carcinoma of the scrotum. Dermatol Sinica. 2002;20:57-62.
- Chopra KF, Cohen PR. Linear basal cell carcinomas: report of multiple sequential tumors localized to a radiotherapy port and review of the literature. Tex Med. 1997;93:57-59.
- da Silva MO, Dadalt P, Santos OL, et al. Linear basal cell carcinoma. Int J Dermatol. 1995;34:488.
- Warthan TL, Lewis JE. Giant linear basal cell epithelioma. Int J Dermatol. 1994;33:284.
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1989;28:682-684.
- Alcántara-Reifs CM, Salido-Vallejo R, González-Menchen A, et al. Linear basal cell carcinoma: report of three cases with dermoscopic findings. Indian J Dermatol Venereol Leprol. 2016;82:708-711.
- Lee MS, Cho E, Lee JH, et al. Linearly curved, blackish macule on the wrist. Cutis. 2016;97:384, 406-407.
- Bajaj S, Sharma PK, Kar HK. Linear adamantinoid basal cell carcinoma in the axilla. Dermatol Online J. 2015;21. pii:13030/qt8k0713nb.
- Iga N, Sakurai K, Fujii H, et al. Linear basal cell carcinoma at the external genitalia. J Dermatol. 2014;41:275-276.
- Ichinokawa Y, Ohtuki A, Hattori M, et al. Linear basal cell carcinoma: a case report. Case Rep Dermatol. 2011;3:142-146.
- Becher GL, Affleck A, Fleming C, et al. Linear basal cell carcinoma occurs most commonly on the lower eyelid. Clin Exp Dermatol. 2011;36:311-312.
- Jellouli A, Triki S, Zghal M, et al. Linear basal cell carcinoma. Actas Dermosifiliogr. 2010;101:648-650.
- Takiyoshi N, Nakano H, Kaneko T, et al. A linear basal cell carcinoma undergoing spontaneous regression. Clin Exp Dermatol. 2009;34:E411-E413.
- Yoleri L, Ozden S, Kandiloglu A. A 46-year-old male with an ulcerated linear lesion on his neck. Ann Saudi Med. 2008;28:57-58.
- Palleschi GM, Corradini D, Bruscino N, et al. Linear basal cell carcinoma: clinical significance and better surgical approach. G Ital Dermatol Venereol. 2016;151:119-121.
- Rodriguez-Garijo N, Redondo P. Linear basal cell carcinoma of the lower eyelid: reconstruction with a musculocutaneous transposition flap. JAAD Case Rep. 2018;4:633-635.
- Pierard GE, Lapiere CM. Microanatomy of the dermis in relation to relaxed skin tension lines and Langer’s lines. Am J Dermatopathol. 1987;9:219-224.
Practice Points
- Linear basal cell carcinoma (LBCC) follows a characteristic morphologic pattern of a length-to-width ratio that typically is at least 3 to 1.
- Linear basal cell carcinomas most commonly present in the periocular region and on the neck along relaxed skin tension lines.
- Because of the likelihood of subclinical spread, LBCC should be regarded as a high-risk subtype of basal cell carcinoma.
- Mohs micrographic surgery or excision with complete circumferential peripheral and deep-margin assessment is recommended as first-line treatment.