User login
Delayed Coronary Vasospasm in a Patient with Metastatic Gastric Cancer Receiving FOLFOX Therapy
A 40-year-old man with stage IV gastric adenocarcinoma was found to have coronary artery vasospasm in the setting of recent 5-fluorouracil administration.
Coronary artery vasospasm is a rare but well-known adverse effect of 5-fluorouracil (5-FU) that can be life threatening if unrecognized. Patients typically present with anginal chest pain and ST elevations on electrocardiogram (ECG) without atherosclerotic disease on coronary angiography. This phenomenon typically occurs during or shortly after infusion and resolves within hours to days after cessation of 5-FU.
In this report, we present an unusual case of coronary artery vasospasm that intermittently recurred for 25 days following 5-FU treatment in a 40-year-old male with stage IV gastric adenocarcinoma. We also review the literature on typical presentation and risk factors for 5-FU-induced coronary vasospasm, findings on coronary angiography, and management options.
5-FU is an IV administered antimetabolite chemotherapy commonly used to treat solid tumors, including gastrointestinal, pancreatic, breast, and head and neck tumors. 5-FU inhibits thymidylate synthase, which reduces levels of thymidine, a key pyrimidine nucleoside required for DNA replication within tumor cells.1 For several decades, 5-FU has remained one of the first-line drugs for colorectal cancer because it may be curative. It is the third most commonly used chemotherapy in the world and is included on the World Health Organization’s list of essential medicines.2
Cardiotoxicity occurs in 1.2 to 18% of patients who receive 5-FU therapy.3 Although there is variability in presentation for acute cardiotoxicity from 5-FU, including sudden death, angina pectoris, myocardial infarction, and ventricular arrhythmias, the mechanism most commonly implicated is coronary artery vasospasm.3 The direct observation of active coronary artery vasospasm during left heart catheterization is rare due its transient nature; however, several case studies have managed to demonstrate this.4,5 The pathophysiology of 5-FU-induced cardiotoxicity is unknown, but adverse effects on cardiac microvasculature, myocyte metabolism, platelet aggregation, and coronary vasoconstriction have all been proposed.3,6In the current case, we present a patient with stage IV gastric adenocarcinoma who complained of chest pain during hospitalization and was found to have coronary artery vasospasm in the setting of recent 5-FU administration. Following coronary angiography that showed a lack of atherosclerotic disease, the patient continued to experience episodes of chest pain with ST elevations on ECG that recurred despite cessation of 5-FU and repeated administration of vasodilatory medications.
Case Presentation
A male aged 40 years was admitted to the hospital for abdominal pain, with initial imaging concerning for partial small bowel obstruction. His history included recently diagnosed stage IV gastric adenocarcinoma complicated by peritoneal carcinomatosis status post initiation of infusional FOLFOX-4 (5-FU, leucovorin, and oxaliplatin) 11 days prior. The patient was treated for small bowel obstruction. However, several days after admission, he developed nonpleuritic, substernal chest pain unrelated to exertion and unrelieved by rest. The patient reported no known risk factors, family history, or personal history of coronary artery disease. Baseline echocardiography and ECG performed several months prior showed normal left ventricular function without ischemic findings.
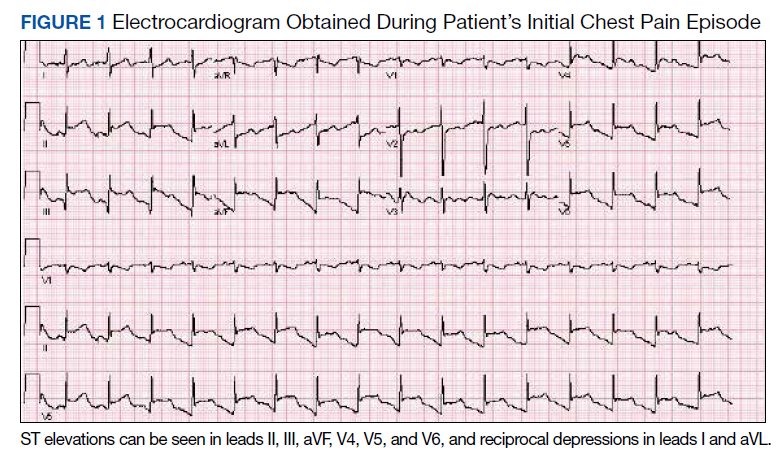
Physical examination at the time of chest pain revealed a heart rate of 140 beats/min. The remainder of his vital signs were within normal range. There were no murmurs, rubs, gallops, or additional heart sounds heard on cardiac auscultation. Chest pain was not reproducible to palpation or positional in nature. An ECG demonstrated dynamic inferolateral ST elevations with reciprocal changes in leads I and aVL (Figure 1). A bedside echocardiogram showed hypokinesis of the septal wall. Troponin-I returned below the detectable level.
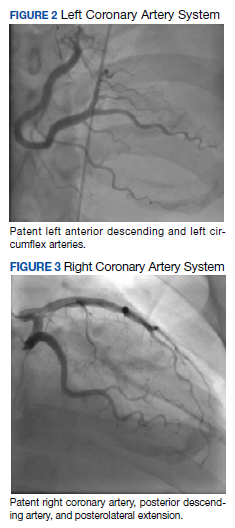
The patient was taken for emergent coronary catheterization, which demonstrated patent epicardial coronary arteries without atherosclerosis, a left ventricular ejection fraction of 60%, and a right dominant heart (Figures 2 and 3). Ventriculogram showed normal wall motion. Repeat troponin-I several hours after catheterization was again below detectable levels.
Given the patient’s acute onset of chest pain and inferolateral ST elevations seen on ECG, the working diagnosis prior to coronary catherization was acute coronary syndrome. The differential diagnosis included other causes of life-threatening chest pain, including pulmonary embolism, pneumonia, aortic dissection, myopericarditis, pericardial effusion, cardiac tamponade, or coronary artery vasospasm. Computed tomography (CT) angiography of the chest was not consistent with pulmonary embolism or other acute cardiopulmonary process. Based on findings from coronary angiography and recent exposure to 5-FU, as well as resolution followed by recurrence of chest pain and ECG changes over weeks, the most likely diagnosis after coronary catheterization was coronary artery vasospasm.
Treatment
Following catheterization, the patient returned to the medical intensive care unit, where he continued to report intermittent episodes of chest pain with ST elevations. In the following days, he was started on isosorbide mononitrate 150 mg daily and amlodipine 10 mg daily. Although these vasodilatory agents reduced the frequency of his chest pain episodes, intermittent chest pain associated with ST elevations on ECG continued even with maximal doses of isosorbide mononitrate and amlodipine. Administration of sublingual nitroglycerin during chest pain episodes effectively relieved his chest pain. Given the severity and frequency of the patient’s chest pain, the oncology consult team recommended foregoing further chemotherapeutic treatment with 5-FU.
Outcome
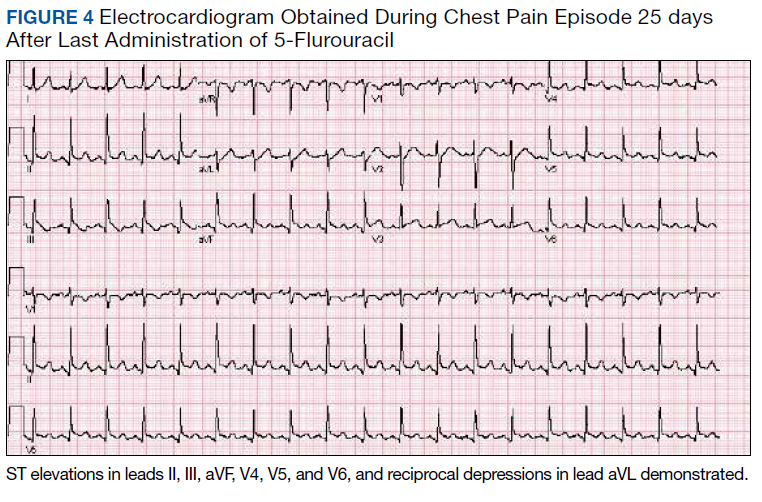
Despite holding 5-FU throughout the patient’s hospitalization and treating the patient with antianginal mediations, frequent chest pain episodes associated with ST elevations continued to recur until 25 days after his last treatment with 5-FU (Figure 4). The patient eventually expired during this hospital stay due to cancer-related complications.
Discussion
Coronary artery vasospasm is a well-known complication of 5-FU that can be life threatening if unrecognized.6-8 As seen in our case, patients typically present with anginal chest pain relieved with nitrates and ST elevations on ECG in the absence of occlusive macrovascular disease on coronary angiography.
A unique aspect of 5-FU is its variability in dose and frequency of administration across chemotherapeutic regimens. Particularly, 5-FU can be administered in daily intravenous bolus doses or as a continuous infusion for a protracted length of time. The spectrum of toxicity from 5-FU differs depending on the dose and frequency of administration. Bolus administration of 5-FU, for example, is thought to be associated with a higher rate of myelosuppression, while infusional administration of 5-FU is thought to be associated with a higher rate of cardiotoxicity and a higher tumor response rate.9
Most cases of coronary vasospasm occur either during infusion of 5-FU or within hours to days after completion. The median time of presentation for 5-FU-induced coronary artery vasospasm is about 12 hours postinfusion, while the most delayed presentation reported in the literature is 72 hours postinfusion.6,8 Delayed presentation of vasospasm may result from the release of potent vasoactive metabolites of 5-FU that accumulate over time; therefore, infusional administration may accentuate this effect.6,9 Remarkably, our patient’s chest pain episodes persisted for 25 days despite treatment with anti-anginal medications, highlighting the extent to which infusional 5-FU can produce a delay in adverse cardiotoxic effects and the importance of ongoing clinical vigilance after 5-FU exposure.
Vasospasm alone does not completely explain the spectrum of cardiac toxicity attributed to 5-FU administration. As in our case, coronary angiography during symptomatic episodes often fails to demonstrate coronary vasospasm.8 Additionally, ergonovine, an alkaloid agent used to assess coronary vasomotor function, failed to induce coronary vasospasm in some patients with suspected 5-FU-induced cardiac toxicity.10 The lack of vasospasm in some patients with 5-FU-induced cardiac toxicity suggests multiple independent effects of 5-FU on cardiac tissue that are poorly understood.
In the absence of obvious macrovascular effects, there also may be a deleterious effect of 5-FU on the coronary microvasculature that may result in coronary artery vasospasm. Though coronary microvasculature cannot be directly visualized, observation of slowed coronary blood velocity indicates a reduction in microvascular flow.8 Thus, the failure to observe epicardial coronary vasospasm in our patient does not preclude a vasospastic pathology.
The heterogeneous presentation of coronary artery vasospasm demands consideration of other disease processes such as atherosclerotic coronary artery disease, pericarditis, myopericarditis, primary arrythmias, and stress-induced cardiomyopathy, all of which have been described in association with 5-FU administration.8 A 12-lead ECG should be performed during a suspected attack. An ECG will typically demonstrate ST elevations corresponding to spasm of the involved vessel. Reciprocal ST depressions in the contralateral leads also may be seen. ECG may be useful in the acute setting to identify regional wall motion abnormalities or to rule out pericardial effusion as a cause. Cardiac biomarkers such as troponin-I, -C, and creatine kinase typically are less useful because they are often normal, even in known coronary artery vasospasm.11
Coronary angiography during an episode may show a localized region of vasospasm in an epicardial artery. Diffuse multivessel vasospasm does occur, and the location of vasospasm may change, but these events are rare. Under normal circumstances, provocative testing involving angiography with administration of acetylcholine, ergot agents, or hyperventilation can be performed. However, this type of investigation should be limited to specialized centers and should not be performed in the acute phase of the disease.12
Treatment of suspected coronary vasospasm in patients receiving 5-FU involves stopping the infusion and administering calcium channel blockers or oral nitrates to relieve anginal symptoms.13 5-FU-induced coronary artery vasospasm has a 90% rate of recurrence with subsequent infusions.8 If possible, alternate chemotherapy regimens should be considered once coronary artery vasospasm has been identified.14,15 If further 5-FU use is required, or if benefits are deemed to outweigh risks, infusions should be given in an inpatient setting with continuous cardiac monitoring.16
Calcium channel blockers and oral nitrates have been found to produce benefit in patients in acute settings; however, there is little evidence to attest to their effectiveness as prophylactic agents in those receiving 5-FU. Some reports demonstrate episodes where both calcium channel blockers and oral nitrates failed to prevent subsequent vasospasms.17 Although this was the case for our patient, short-acting sublingual nitroglycerin seemed to be effective in reducing the frequency of anginal symptoms.
Long-term outcomes have not been well investigated for patients with 5-FU-induced coronary vasospasm. However, many case reports show improvements in left ventricular function between 8 and 15 days after discontinuation of 5-FU.7,10 Although this would be a valuable topic for further research, the rarity of this phenomenon creates limitations.
Conclusions
5-FU is a first-line chemotherapy for gastrointestinal cancers that is generally well tolerated but may be associated with potentially life-threatening cardiotoxic effects, of which coronary artery vasospasm is the most common. Coronary artery vasospasm presents with anginal chest pain and ST elevations on ECG that can be indistinguishable from acute coronary syndrome. Diagnosis requires cardiac catheterization, which will reveal patent coronary arteries. Infusional administration of 5-FU may be more likely to produce late cardiotoxic effects and a longer period of persistent symptoms, necessitating close monitoring for days or even weeks from last administration of 5-FU. Coronary artery vasospasm should be treated with anti-anginal medications, though varying degrees of effectiveness can be seen; clinicians should remain vigilant for recurrent episodes of chest pain despite treatment.
1. Wacker A, Lersch C, Scherpinski U, Reindl L, Seyfarth M. High incidence of angina pectoris in patients treated with 5-fluorouracil. A planned surveillance study with 102 patients. Oncology. 2003;65(2):108-112. doi:10.1159/000072334
2. World Health Organization Model List of Essential Medicines, 21st List, 2019. Accessed April 14, 2021. https://apps.who.int/iris/rest/bitstreams/1237479/retrieve
3. Jensen SA, Sørensen JB. Risk factors and prevention of cardiotoxicity induced by 5-fluorouracil or capecitabine. Cancer Chemother Pharmacol. 2006;58(4):487-493. doi:10.1007/s00280-005-0178-1
4. Shoemaker LK, Arora U, Rocha Lima CM. 5-fluorouracil-induced coronary vasospasm. Cancer Control. 2004;11(1):46-49. doi:10.1177/107327480401100207
5. Luwaert RJ, Descamps O, Majois F, Chaudron JM, Beauduin M. Coronary artery spasm induced by 5-fluorouracil. Eur Heart J. 1991;12(3):468-470. doi:10.1093/oxfordjournals.eurheartj.a059919
6. Saif MW, Shah MM, Shah AR. Fluoropyrimidine-associated cardiotoxicity: revisited. Expert Opin Drug Saf. 2009;8(2):191-202. doi:10.1517/14740330902733961
7. Patel B, Kloner RA, Ensley J, Al-Sarraf M, Kish J, Wynne J. 5-Fluorouracil cardiotoxicity: left ventricular dysfunction and effect of coronary vasodilators. Am J Med Sci. 1987;294(4):238-243. doi:10.1097/00000441-198710000-00004
8. Sara JD, Kaur J, Khodadadi R, et al. 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol. 2018;10:1758835918780140. Published 2018 Jun 18. doi:10.1177/1758835918780140
9. Hansen RM, Ryan L, Anderson T, et al. Phase III study of bolus versus infusion fluorouracil with or without cisplatin in advanced colorectal cancer. J Natl Cancer Inst. 1996;88(10):668-674. doi:10.1093/jnci/88.10.668
10. Kim SM, Kwak CH, Lee B, et al. A case of severe coronary spasm associated with 5-fluorouracil chemotherapy. Korean J Intern Med. 2012;27(3):342-345. doi:10.3904/kjim.2012.27.3.342
11. Swarup S, Patibandla S, Grossman SA. Coronary Artery Vasospasm. StatPearls. Treasure Island (FL): StatPearls Publishing LLC.; 2021.
12. Beijk MA, Vlastra WV, Delewi R, et al. Myocardial infarction with non-obstructive coronary arteries: a focus on vasospastic angina. Neth Heart J. 2019;27(5):237-245. doi:10.1007/s12471-019-1232-7
13. Giza DE, Boccalandro F, Lopez-Mattei J, et al. Ischemic heart disease: special considerations in cardio-oncology. Curr Treat Options Cardiovasc Med. 2017;19(5):37. doi:10.1007/s11936-017-0535-5
14. Meydan N, Kundak I, Yavuzsen T, et al. Cardiotoxicity of de Gramont’s regimen: incidence, clinical characteristics and long-term follow-up. Jpn J Clin Oncol. 2005;35(5):265-270. doi:10.1093/jjco/hyi071
15. Senkus E, Jassem J. Cardiovascular effects of systemic cancer treatment. Cancer Treat Rev. 2011;37(4):300-311. doi:10.1016/j.ctrv.2010.11.001
16. Rezkalla S, Kloner RA, Ensley J, et al. Continuous ambulatory ECG monitoring during fluorouracil therapy: a prospective study. J Clin Oncol. 1989;7(4):509-514. doi:10.1200/JCO.1989.7.4.509
17. Akpek G, Hartshorn KL. Failure of oral nitrate and calcium channel blocker therapy to prevent 5-fluorouracil-related myocardial ischemia: a case report. Cancer Chemother Pharmacol. 1999;43(2):157-161. doi:10.1007/s002800050877
A 40-year-old man with stage IV gastric adenocarcinoma was found to have coronary artery vasospasm in the setting of recent 5-fluorouracil administration.
A 40-year-old man with stage IV gastric adenocarcinoma was found to have coronary artery vasospasm in the setting of recent 5-fluorouracil administration.
Coronary artery vasospasm is a rare but well-known adverse effect of 5-fluorouracil (5-FU) that can be life threatening if unrecognized. Patients typically present with anginal chest pain and ST elevations on electrocardiogram (ECG) without atherosclerotic disease on coronary angiography. This phenomenon typically occurs during or shortly after infusion and resolves within hours to days after cessation of 5-FU.
In this report, we present an unusual case of coronary artery vasospasm that intermittently recurred for 25 days following 5-FU treatment in a 40-year-old male with stage IV gastric adenocarcinoma. We also review the literature on typical presentation and risk factors for 5-FU-induced coronary vasospasm, findings on coronary angiography, and management options.
5-FU is an IV administered antimetabolite chemotherapy commonly used to treat solid tumors, including gastrointestinal, pancreatic, breast, and head and neck tumors. 5-FU inhibits thymidylate synthase, which reduces levels of thymidine, a key pyrimidine nucleoside required for DNA replication within tumor cells.1 For several decades, 5-FU has remained one of the first-line drugs for colorectal cancer because it may be curative. It is the third most commonly used chemotherapy in the world and is included on the World Health Organization’s list of essential medicines.2
Cardiotoxicity occurs in 1.2 to 18% of patients who receive 5-FU therapy.3 Although there is variability in presentation for acute cardiotoxicity from 5-FU, including sudden death, angina pectoris, myocardial infarction, and ventricular arrhythmias, the mechanism most commonly implicated is coronary artery vasospasm.3 The direct observation of active coronary artery vasospasm during left heart catheterization is rare due its transient nature; however, several case studies have managed to demonstrate this.4,5 The pathophysiology of 5-FU-induced cardiotoxicity is unknown, but adverse effects on cardiac microvasculature, myocyte metabolism, platelet aggregation, and coronary vasoconstriction have all been proposed.3,6In the current case, we present a patient with stage IV gastric adenocarcinoma who complained of chest pain during hospitalization and was found to have coronary artery vasospasm in the setting of recent 5-FU administration. Following coronary angiography that showed a lack of atherosclerotic disease, the patient continued to experience episodes of chest pain with ST elevations on ECG that recurred despite cessation of 5-FU and repeated administration of vasodilatory medications.
Case Presentation
A male aged 40 years was admitted to the hospital for abdominal pain, with initial imaging concerning for partial small bowel obstruction. His history included recently diagnosed stage IV gastric adenocarcinoma complicated by peritoneal carcinomatosis status post initiation of infusional FOLFOX-4 (5-FU, leucovorin, and oxaliplatin) 11 days prior. The patient was treated for small bowel obstruction. However, several days after admission, he developed nonpleuritic, substernal chest pain unrelated to exertion and unrelieved by rest. The patient reported no known risk factors, family history, or personal history of coronary artery disease. Baseline echocardiography and ECG performed several months prior showed normal left ventricular function without ischemic findings.
Physical examination at the time of chest pain revealed a heart rate of 140 beats/min. The remainder of his vital signs were within normal range. There were no murmurs, rubs, gallops, or additional heart sounds heard on cardiac auscultation. Chest pain was not reproducible to palpation or positional in nature. An ECG demonstrated dynamic inferolateral ST elevations with reciprocal changes in leads I and aVL (Figure 1). A bedside echocardiogram showed hypokinesis of the septal wall. Troponin-I returned below the detectable level.
The patient was taken for emergent coronary catheterization, which demonstrated patent epicardial coronary arteries without atherosclerosis, a left ventricular ejection fraction of 60%, and a right dominant heart (Figures 2 and 3). Ventriculogram showed normal wall motion. Repeat troponin-I several hours after catheterization was again below detectable levels.
Given the patient’s acute onset of chest pain and inferolateral ST elevations seen on ECG, the working diagnosis prior to coronary catherization was acute coronary syndrome. The differential diagnosis included other causes of life-threatening chest pain, including pulmonary embolism, pneumonia, aortic dissection, myopericarditis, pericardial effusion, cardiac tamponade, or coronary artery vasospasm. Computed tomography (CT) angiography of the chest was not consistent with pulmonary embolism or other acute cardiopulmonary process. Based on findings from coronary angiography and recent exposure to 5-FU, as well as resolution followed by recurrence of chest pain and ECG changes over weeks, the most likely diagnosis after coronary catheterization was coronary artery vasospasm.
Treatment
Following catheterization, the patient returned to the medical intensive care unit, where he continued to report intermittent episodes of chest pain with ST elevations. In the following days, he was started on isosorbide mononitrate 150 mg daily and amlodipine 10 mg daily. Although these vasodilatory agents reduced the frequency of his chest pain episodes, intermittent chest pain associated with ST elevations on ECG continued even with maximal doses of isosorbide mononitrate and amlodipine. Administration of sublingual nitroglycerin during chest pain episodes effectively relieved his chest pain. Given the severity and frequency of the patient’s chest pain, the oncology consult team recommended foregoing further chemotherapeutic treatment with 5-FU.
Outcome
Despite holding 5-FU throughout the patient’s hospitalization and treating the patient with antianginal mediations, frequent chest pain episodes associated with ST elevations continued to recur until 25 days after his last treatment with 5-FU (Figure 4). The patient eventually expired during this hospital stay due to cancer-related complications.
Discussion
Coronary artery vasospasm is a well-known complication of 5-FU that can be life threatening if unrecognized.6-8 As seen in our case, patients typically present with anginal chest pain relieved with nitrates and ST elevations on ECG in the absence of occlusive macrovascular disease on coronary angiography.
A unique aspect of 5-FU is its variability in dose and frequency of administration across chemotherapeutic regimens. Particularly, 5-FU can be administered in daily intravenous bolus doses or as a continuous infusion for a protracted length of time. The spectrum of toxicity from 5-FU differs depending on the dose and frequency of administration. Bolus administration of 5-FU, for example, is thought to be associated with a higher rate of myelosuppression, while infusional administration of 5-FU is thought to be associated with a higher rate of cardiotoxicity and a higher tumor response rate.9
Most cases of coronary vasospasm occur either during infusion of 5-FU or within hours to days after completion. The median time of presentation for 5-FU-induced coronary artery vasospasm is about 12 hours postinfusion, while the most delayed presentation reported in the literature is 72 hours postinfusion.6,8 Delayed presentation of vasospasm may result from the release of potent vasoactive metabolites of 5-FU that accumulate over time; therefore, infusional administration may accentuate this effect.6,9 Remarkably, our patient’s chest pain episodes persisted for 25 days despite treatment with anti-anginal medications, highlighting the extent to which infusional 5-FU can produce a delay in adverse cardiotoxic effects and the importance of ongoing clinical vigilance after 5-FU exposure.
Vasospasm alone does not completely explain the spectrum of cardiac toxicity attributed to 5-FU administration. As in our case, coronary angiography during symptomatic episodes often fails to demonstrate coronary vasospasm.8 Additionally, ergonovine, an alkaloid agent used to assess coronary vasomotor function, failed to induce coronary vasospasm in some patients with suspected 5-FU-induced cardiac toxicity.10 The lack of vasospasm in some patients with 5-FU-induced cardiac toxicity suggests multiple independent effects of 5-FU on cardiac tissue that are poorly understood.
In the absence of obvious macrovascular effects, there also may be a deleterious effect of 5-FU on the coronary microvasculature that may result in coronary artery vasospasm. Though coronary microvasculature cannot be directly visualized, observation of slowed coronary blood velocity indicates a reduction in microvascular flow.8 Thus, the failure to observe epicardial coronary vasospasm in our patient does not preclude a vasospastic pathology.
The heterogeneous presentation of coronary artery vasospasm demands consideration of other disease processes such as atherosclerotic coronary artery disease, pericarditis, myopericarditis, primary arrythmias, and stress-induced cardiomyopathy, all of which have been described in association with 5-FU administration.8 A 12-lead ECG should be performed during a suspected attack. An ECG will typically demonstrate ST elevations corresponding to spasm of the involved vessel. Reciprocal ST depressions in the contralateral leads also may be seen. ECG may be useful in the acute setting to identify regional wall motion abnormalities or to rule out pericardial effusion as a cause. Cardiac biomarkers such as troponin-I, -C, and creatine kinase typically are less useful because they are often normal, even in known coronary artery vasospasm.11
Coronary angiography during an episode may show a localized region of vasospasm in an epicardial artery. Diffuse multivessel vasospasm does occur, and the location of vasospasm may change, but these events are rare. Under normal circumstances, provocative testing involving angiography with administration of acetylcholine, ergot agents, or hyperventilation can be performed. However, this type of investigation should be limited to specialized centers and should not be performed in the acute phase of the disease.12
Treatment of suspected coronary vasospasm in patients receiving 5-FU involves stopping the infusion and administering calcium channel blockers or oral nitrates to relieve anginal symptoms.13 5-FU-induced coronary artery vasospasm has a 90% rate of recurrence with subsequent infusions.8 If possible, alternate chemotherapy regimens should be considered once coronary artery vasospasm has been identified.14,15 If further 5-FU use is required, or if benefits are deemed to outweigh risks, infusions should be given in an inpatient setting with continuous cardiac monitoring.16
Calcium channel blockers and oral nitrates have been found to produce benefit in patients in acute settings; however, there is little evidence to attest to their effectiveness as prophylactic agents in those receiving 5-FU. Some reports demonstrate episodes where both calcium channel blockers and oral nitrates failed to prevent subsequent vasospasms.17 Although this was the case for our patient, short-acting sublingual nitroglycerin seemed to be effective in reducing the frequency of anginal symptoms.
Long-term outcomes have not been well investigated for patients with 5-FU-induced coronary vasospasm. However, many case reports show improvements in left ventricular function between 8 and 15 days after discontinuation of 5-FU.7,10 Although this would be a valuable topic for further research, the rarity of this phenomenon creates limitations.
Conclusions
5-FU is a first-line chemotherapy for gastrointestinal cancers that is generally well tolerated but may be associated with potentially life-threatening cardiotoxic effects, of which coronary artery vasospasm is the most common. Coronary artery vasospasm presents with anginal chest pain and ST elevations on ECG that can be indistinguishable from acute coronary syndrome. Diagnosis requires cardiac catheterization, which will reveal patent coronary arteries. Infusional administration of 5-FU may be more likely to produce late cardiotoxic effects and a longer period of persistent symptoms, necessitating close monitoring for days or even weeks from last administration of 5-FU. Coronary artery vasospasm should be treated with anti-anginal medications, though varying degrees of effectiveness can be seen; clinicians should remain vigilant for recurrent episodes of chest pain despite treatment.
Coronary artery vasospasm is a rare but well-known adverse effect of 5-fluorouracil (5-FU) that can be life threatening if unrecognized. Patients typically present with anginal chest pain and ST elevations on electrocardiogram (ECG) without atherosclerotic disease on coronary angiography. This phenomenon typically occurs during or shortly after infusion and resolves within hours to days after cessation of 5-FU.
In this report, we present an unusual case of coronary artery vasospasm that intermittently recurred for 25 days following 5-FU treatment in a 40-year-old male with stage IV gastric adenocarcinoma. We also review the literature on typical presentation and risk factors for 5-FU-induced coronary vasospasm, findings on coronary angiography, and management options.
5-FU is an IV administered antimetabolite chemotherapy commonly used to treat solid tumors, including gastrointestinal, pancreatic, breast, and head and neck tumors. 5-FU inhibits thymidylate synthase, which reduces levels of thymidine, a key pyrimidine nucleoside required for DNA replication within tumor cells.1 For several decades, 5-FU has remained one of the first-line drugs for colorectal cancer because it may be curative. It is the third most commonly used chemotherapy in the world and is included on the World Health Organization’s list of essential medicines.2
Cardiotoxicity occurs in 1.2 to 18% of patients who receive 5-FU therapy.3 Although there is variability in presentation for acute cardiotoxicity from 5-FU, including sudden death, angina pectoris, myocardial infarction, and ventricular arrhythmias, the mechanism most commonly implicated is coronary artery vasospasm.3 The direct observation of active coronary artery vasospasm during left heart catheterization is rare due its transient nature; however, several case studies have managed to demonstrate this.4,5 The pathophysiology of 5-FU-induced cardiotoxicity is unknown, but adverse effects on cardiac microvasculature, myocyte metabolism, platelet aggregation, and coronary vasoconstriction have all been proposed.3,6In the current case, we present a patient with stage IV gastric adenocarcinoma who complained of chest pain during hospitalization and was found to have coronary artery vasospasm in the setting of recent 5-FU administration. Following coronary angiography that showed a lack of atherosclerotic disease, the patient continued to experience episodes of chest pain with ST elevations on ECG that recurred despite cessation of 5-FU and repeated administration of vasodilatory medications.
Case Presentation
A male aged 40 years was admitted to the hospital for abdominal pain, with initial imaging concerning for partial small bowel obstruction. His history included recently diagnosed stage IV gastric adenocarcinoma complicated by peritoneal carcinomatosis status post initiation of infusional FOLFOX-4 (5-FU, leucovorin, and oxaliplatin) 11 days prior. The patient was treated for small bowel obstruction. However, several days after admission, he developed nonpleuritic, substernal chest pain unrelated to exertion and unrelieved by rest. The patient reported no known risk factors, family history, or personal history of coronary artery disease. Baseline echocardiography and ECG performed several months prior showed normal left ventricular function without ischemic findings.
Physical examination at the time of chest pain revealed a heart rate of 140 beats/min. The remainder of his vital signs were within normal range. There were no murmurs, rubs, gallops, or additional heart sounds heard on cardiac auscultation. Chest pain was not reproducible to palpation or positional in nature. An ECG demonstrated dynamic inferolateral ST elevations with reciprocal changes in leads I and aVL (Figure 1). A bedside echocardiogram showed hypokinesis of the septal wall. Troponin-I returned below the detectable level.
The patient was taken for emergent coronary catheterization, which demonstrated patent epicardial coronary arteries without atherosclerosis, a left ventricular ejection fraction of 60%, and a right dominant heart (Figures 2 and 3). Ventriculogram showed normal wall motion. Repeat troponin-I several hours after catheterization was again below detectable levels.
Given the patient’s acute onset of chest pain and inferolateral ST elevations seen on ECG, the working diagnosis prior to coronary catherization was acute coronary syndrome. The differential diagnosis included other causes of life-threatening chest pain, including pulmonary embolism, pneumonia, aortic dissection, myopericarditis, pericardial effusion, cardiac tamponade, or coronary artery vasospasm. Computed tomography (CT) angiography of the chest was not consistent with pulmonary embolism or other acute cardiopulmonary process. Based on findings from coronary angiography and recent exposure to 5-FU, as well as resolution followed by recurrence of chest pain and ECG changes over weeks, the most likely diagnosis after coronary catheterization was coronary artery vasospasm.
Treatment
Following catheterization, the patient returned to the medical intensive care unit, where he continued to report intermittent episodes of chest pain with ST elevations. In the following days, he was started on isosorbide mononitrate 150 mg daily and amlodipine 10 mg daily. Although these vasodilatory agents reduced the frequency of his chest pain episodes, intermittent chest pain associated with ST elevations on ECG continued even with maximal doses of isosorbide mononitrate and amlodipine. Administration of sublingual nitroglycerin during chest pain episodes effectively relieved his chest pain. Given the severity and frequency of the patient’s chest pain, the oncology consult team recommended foregoing further chemotherapeutic treatment with 5-FU.
Outcome
Despite holding 5-FU throughout the patient’s hospitalization and treating the patient with antianginal mediations, frequent chest pain episodes associated with ST elevations continued to recur until 25 days after his last treatment with 5-FU (Figure 4). The patient eventually expired during this hospital stay due to cancer-related complications.
Discussion
Coronary artery vasospasm is a well-known complication of 5-FU that can be life threatening if unrecognized.6-8 As seen in our case, patients typically present with anginal chest pain relieved with nitrates and ST elevations on ECG in the absence of occlusive macrovascular disease on coronary angiography.
A unique aspect of 5-FU is its variability in dose and frequency of administration across chemotherapeutic regimens. Particularly, 5-FU can be administered in daily intravenous bolus doses or as a continuous infusion for a protracted length of time. The spectrum of toxicity from 5-FU differs depending on the dose and frequency of administration. Bolus administration of 5-FU, for example, is thought to be associated with a higher rate of myelosuppression, while infusional administration of 5-FU is thought to be associated with a higher rate of cardiotoxicity and a higher tumor response rate.9
Most cases of coronary vasospasm occur either during infusion of 5-FU or within hours to days after completion. The median time of presentation for 5-FU-induced coronary artery vasospasm is about 12 hours postinfusion, while the most delayed presentation reported in the literature is 72 hours postinfusion.6,8 Delayed presentation of vasospasm may result from the release of potent vasoactive metabolites of 5-FU that accumulate over time; therefore, infusional administration may accentuate this effect.6,9 Remarkably, our patient’s chest pain episodes persisted for 25 days despite treatment with anti-anginal medications, highlighting the extent to which infusional 5-FU can produce a delay in adverse cardiotoxic effects and the importance of ongoing clinical vigilance after 5-FU exposure.
Vasospasm alone does not completely explain the spectrum of cardiac toxicity attributed to 5-FU administration. As in our case, coronary angiography during symptomatic episodes often fails to demonstrate coronary vasospasm.8 Additionally, ergonovine, an alkaloid agent used to assess coronary vasomotor function, failed to induce coronary vasospasm in some patients with suspected 5-FU-induced cardiac toxicity.10 The lack of vasospasm in some patients with 5-FU-induced cardiac toxicity suggests multiple independent effects of 5-FU on cardiac tissue that are poorly understood.
In the absence of obvious macrovascular effects, there also may be a deleterious effect of 5-FU on the coronary microvasculature that may result in coronary artery vasospasm. Though coronary microvasculature cannot be directly visualized, observation of slowed coronary blood velocity indicates a reduction in microvascular flow.8 Thus, the failure to observe epicardial coronary vasospasm in our patient does not preclude a vasospastic pathology.
The heterogeneous presentation of coronary artery vasospasm demands consideration of other disease processes such as atherosclerotic coronary artery disease, pericarditis, myopericarditis, primary arrythmias, and stress-induced cardiomyopathy, all of which have been described in association with 5-FU administration.8 A 12-lead ECG should be performed during a suspected attack. An ECG will typically demonstrate ST elevations corresponding to spasm of the involved vessel. Reciprocal ST depressions in the contralateral leads also may be seen. ECG may be useful in the acute setting to identify regional wall motion abnormalities or to rule out pericardial effusion as a cause. Cardiac biomarkers such as troponin-I, -C, and creatine kinase typically are less useful because they are often normal, even in known coronary artery vasospasm.11
Coronary angiography during an episode may show a localized region of vasospasm in an epicardial artery. Diffuse multivessel vasospasm does occur, and the location of vasospasm may change, but these events are rare. Under normal circumstances, provocative testing involving angiography with administration of acetylcholine, ergot agents, or hyperventilation can be performed. However, this type of investigation should be limited to specialized centers and should not be performed in the acute phase of the disease.12
Treatment of suspected coronary vasospasm in patients receiving 5-FU involves stopping the infusion and administering calcium channel blockers or oral nitrates to relieve anginal symptoms.13 5-FU-induced coronary artery vasospasm has a 90% rate of recurrence with subsequent infusions.8 If possible, alternate chemotherapy regimens should be considered once coronary artery vasospasm has been identified.14,15 If further 5-FU use is required, or if benefits are deemed to outweigh risks, infusions should be given in an inpatient setting with continuous cardiac monitoring.16
Calcium channel blockers and oral nitrates have been found to produce benefit in patients in acute settings; however, there is little evidence to attest to their effectiveness as prophylactic agents in those receiving 5-FU. Some reports demonstrate episodes where both calcium channel blockers and oral nitrates failed to prevent subsequent vasospasms.17 Although this was the case for our patient, short-acting sublingual nitroglycerin seemed to be effective in reducing the frequency of anginal symptoms.
Long-term outcomes have not been well investigated for patients with 5-FU-induced coronary vasospasm. However, many case reports show improvements in left ventricular function between 8 and 15 days after discontinuation of 5-FU.7,10 Although this would be a valuable topic for further research, the rarity of this phenomenon creates limitations.
Conclusions
5-FU is a first-line chemotherapy for gastrointestinal cancers that is generally well tolerated but may be associated with potentially life-threatening cardiotoxic effects, of which coronary artery vasospasm is the most common. Coronary artery vasospasm presents with anginal chest pain and ST elevations on ECG that can be indistinguishable from acute coronary syndrome. Diagnosis requires cardiac catheterization, which will reveal patent coronary arteries. Infusional administration of 5-FU may be more likely to produce late cardiotoxic effects and a longer period of persistent symptoms, necessitating close monitoring for days or even weeks from last administration of 5-FU. Coronary artery vasospasm should be treated with anti-anginal medications, though varying degrees of effectiveness can be seen; clinicians should remain vigilant for recurrent episodes of chest pain despite treatment.
1. Wacker A, Lersch C, Scherpinski U, Reindl L, Seyfarth M. High incidence of angina pectoris in patients treated with 5-fluorouracil. A planned surveillance study with 102 patients. Oncology. 2003;65(2):108-112. doi:10.1159/000072334
2. World Health Organization Model List of Essential Medicines, 21st List, 2019. Accessed April 14, 2021. https://apps.who.int/iris/rest/bitstreams/1237479/retrieve
3. Jensen SA, Sørensen JB. Risk factors and prevention of cardiotoxicity induced by 5-fluorouracil or capecitabine. Cancer Chemother Pharmacol. 2006;58(4):487-493. doi:10.1007/s00280-005-0178-1
4. Shoemaker LK, Arora U, Rocha Lima CM. 5-fluorouracil-induced coronary vasospasm. Cancer Control. 2004;11(1):46-49. doi:10.1177/107327480401100207
5. Luwaert RJ, Descamps O, Majois F, Chaudron JM, Beauduin M. Coronary artery spasm induced by 5-fluorouracil. Eur Heart J. 1991;12(3):468-470. doi:10.1093/oxfordjournals.eurheartj.a059919
6. Saif MW, Shah MM, Shah AR. Fluoropyrimidine-associated cardiotoxicity: revisited. Expert Opin Drug Saf. 2009;8(2):191-202. doi:10.1517/14740330902733961
7. Patel B, Kloner RA, Ensley J, Al-Sarraf M, Kish J, Wynne J. 5-Fluorouracil cardiotoxicity: left ventricular dysfunction and effect of coronary vasodilators. Am J Med Sci. 1987;294(4):238-243. doi:10.1097/00000441-198710000-00004
8. Sara JD, Kaur J, Khodadadi R, et al. 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol. 2018;10:1758835918780140. Published 2018 Jun 18. doi:10.1177/1758835918780140
9. Hansen RM, Ryan L, Anderson T, et al. Phase III study of bolus versus infusion fluorouracil with or without cisplatin in advanced colorectal cancer. J Natl Cancer Inst. 1996;88(10):668-674. doi:10.1093/jnci/88.10.668
10. Kim SM, Kwak CH, Lee B, et al. A case of severe coronary spasm associated with 5-fluorouracil chemotherapy. Korean J Intern Med. 2012;27(3):342-345. doi:10.3904/kjim.2012.27.3.342
11. Swarup S, Patibandla S, Grossman SA. Coronary Artery Vasospasm. StatPearls. Treasure Island (FL): StatPearls Publishing LLC.; 2021.
12. Beijk MA, Vlastra WV, Delewi R, et al. Myocardial infarction with non-obstructive coronary arteries: a focus on vasospastic angina. Neth Heart J. 2019;27(5):237-245. doi:10.1007/s12471-019-1232-7
13. Giza DE, Boccalandro F, Lopez-Mattei J, et al. Ischemic heart disease: special considerations in cardio-oncology. Curr Treat Options Cardiovasc Med. 2017;19(5):37. doi:10.1007/s11936-017-0535-5
14. Meydan N, Kundak I, Yavuzsen T, et al. Cardiotoxicity of de Gramont’s regimen: incidence, clinical characteristics and long-term follow-up. Jpn J Clin Oncol. 2005;35(5):265-270. doi:10.1093/jjco/hyi071
15. Senkus E, Jassem J. Cardiovascular effects of systemic cancer treatment. Cancer Treat Rev. 2011;37(4):300-311. doi:10.1016/j.ctrv.2010.11.001
16. Rezkalla S, Kloner RA, Ensley J, et al. Continuous ambulatory ECG monitoring during fluorouracil therapy: a prospective study. J Clin Oncol. 1989;7(4):509-514. doi:10.1200/JCO.1989.7.4.509
17. Akpek G, Hartshorn KL. Failure of oral nitrate and calcium channel blocker therapy to prevent 5-fluorouracil-related myocardial ischemia: a case report. Cancer Chemother Pharmacol. 1999;43(2):157-161. doi:10.1007/s002800050877
1. Wacker A, Lersch C, Scherpinski U, Reindl L, Seyfarth M. High incidence of angina pectoris in patients treated with 5-fluorouracil. A planned surveillance study with 102 patients. Oncology. 2003;65(2):108-112. doi:10.1159/000072334
2. World Health Organization Model List of Essential Medicines, 21st List, 2019. Accessed April 14, 2021. https://apps.who.int/iris/rest/bitstreams/1237479/retrieve
3. Jensen SA, Sørensen JB. Risk factors and prevention of cardiotoxicity induced by 5-fluorouracil or capecitabine. Cancer Chemother Pharmacol. 2006;58(4):487-493. doi:10.1007/s00280-005-0178-1
4. Shoemaker LK, Arora U, Rocha Lima CM. 5-fluorouracil-induced coronary vasospasm. Cancer Control. 2004;11(1):46-49. doi:10.1177/107327480401100207
5. Luwaert RJ, Descamps O, Majois F, Chaudron JM, Beauduin M. Coronary artery spasm induced by 5-fluorouracil. Eur Heart J. 1991;12(3):468-470. doi:10.1093/oxfordjournals.eurheartj.a059919
6. Saif MW, Shah MM, Shah AR. Fluoropyrimidine-associated cardiotoxicity: revisited. Expert Opin Drug Saf. 2009;8(2):191-202. doi:10.1517/14740330902733961
7. Patel B, Kloner RA, Ensley J, Al-Sarraf M, Kish J, Wynne J. 5-Fluorouracil cardiotoxicity: left ventricular dysfunction and effect of coronary vasodilators. Am J Med Sci. 1987;294(4):238-243. doi:10.1097/00000441-198710000-00004
8. Sara JD, Kaur J, Khodadadi R, et al. 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol. 2018;10:1758835918780140. Published 2018 Jun 18. doi:10.1177/1758835918780140
9. Hansen RM, Ryan L, Anderson T, et al. Phase III study of bolus versus infusion fluorouracil with or without cisplatin in advanced colorectal cancer. J Natl Cancer Inst. 1996;88(10):668-674. doi:10.1093/jnci/88.10.668
10. Kim SM, Kwak CH, Lee B, et al. A case of severe coronary spasm associated with 5-fluorouracil chemotherapy. Korean J Intern Med. 2012;27(3):342-345. doi:10.3904/kjim.2012.27.3.342
11. Swarup S, Patibandla S, Grossman SA. Coronary Artery Vasospasm. StatPearls. Treasure Island (FL): StatPearls Publishing LLC.; 2021.
12. Beijk MA, Vlastra WV, Delewi R, et al. Myocardial infarction with non-obstructive coronary arteries: a focus on vasospastic angina. Neth Heart J. 2019;27(5):237-245. doi:10.1007/s12471-019-1232-7
13. Giza DE, Boccalandro F, Lopez-Mattei J, et al. Ischemic heart disease: special considerations in cardio-oncology. Curr Treat Options Cardiovasc Med. 2017;19(5):37. doi:10.1007/s11936-017-0535-5
14. Meydan N, Kundak I, Yavuzsen T, et al. Cardiotoxicity of de Gramont’s regimen: incidence, clinical characteristics and long-term follow-up. Jpn J Clin Oncol. 2005;35(5):265-270. doi:10.1093/jjco/hyi071
15. Senkus E, Jassem J. Cardiovascular effects of systemic cancer treatment. Cancer Treat Rev. 2011;37(4):300-311. doi:10.1016/j.ctrv.2010.11.001
16. Rezkalla S, Kloner RA, Ensley J, et al. Continuous ambulatory ECG monitoring during fluorouracil therapy: a prospective study. J Clin Oncol. 1989;7(4):509-514. doi:10.1200/JCO.1989.7.4.509
17. Akpek G, Hartshorn KL. Failure of oral nitrate and calcium channel blocker therapy to prevent 5-fluorouracil-related myocardial ischemia: a case report. Cancer Chemother Pharmacol. 1999;43(2):157-161. doi:10.1007/s002800050877
Beneath the Surface: Massive Retroperitoneal Liposarcoma Masquerading as Meralgia Paresthetica
In patients presenting with focal neurologic findings involving the lower extremities, a thorough abdominal examination should be considered an integral part of the full neurologic work up.
Meralgia paresthetica (MP) is a sensory mononeuropathy of the lateral femoral cutaneous nerve (LFCN), clinically characterized by numbness, pain, and paresthesias involving the anterolateral aspect of the thigh. Estimates of MP incidence are derived largely from observational studies and reported to be about 3.2 to 4.3 cases per 10,000 patient-years.1,2 Although typically arising during midlife and especially in the context of comorbid obesity, diabetes mellitus (DM), and excessive alcohol consumption, MP may occur at any age, and bears a slight predilection for males.2-4
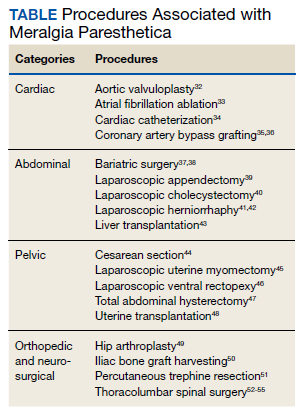
MP may be divided etiologically into iatrogenic and spontaneous subtypes.5 Iatrogenic cases generally are attributable to nerve injury in the setting of direct or indirect trauma (such as with patient malpositioning) arising in the context of multiple forms of procedural or surgical intervention (Table). Spontaneous MP is primarily thought to occur as a result of LFCN compression at the level of the inguinal ligament, wherein internal or external pressures may promote LFCN entrapment and resultant functional disruption (Figure 1).6,7
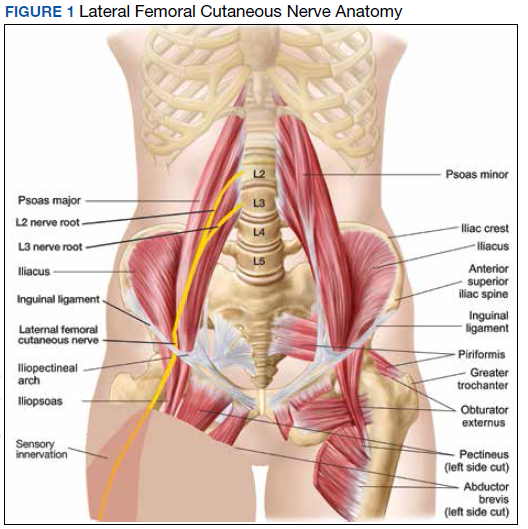
External forces, such as tight garments, wallets, or even elements of modern body armor, have been reported to provoke MP.8-11 Alternatively, states of increased intraabdominal pressure, such as obesity, ascites, and pregnancy may predispose to LFCN compression.2,12,13 Less commonly, lumbar radiculopathy, pelvic masses, and several forms of retroperitoneal pathology may present with clinical symptomatology indistinguishable from MP.14-17 Importantly, many of these represent must-not-miss diagnoses, and may be suggested via a focused history and physical examination.
Here, we present a case of MP secondary to a massive retroperitoneal sarcoma, ultimately drawing renewed attention to the known association of MP and retroperitoneal pathology, and therein highlighting the utility of a dedicated review of systems to identify red-flag features in patients who present with MP and a thorough abdominal examination in all patients presenting with focal neurologic deficits involving the lower extremities.
Case Presentation
A male Vietnam War veteran aged 69 years presented to a primary care clinic at West Roxbury Veterans Affairs Medical Center (WRVAMC) in Massachusetts with progressive right lower extremity numbness. Three months prior to this visit, he was evaluated in an urgent care clinic at WRVAMC for 6 months of numbness and increasingly painful nocturnal paresthesias involving the same extremity. A targeted physical examination at that visit revealed an obese male wearing tight suspenders, as well as focally diminished sensation to light touch involving the anterolateral aspect of the thigh, extending from just below the right hip to above the knee. Sensation in the medial thigh was spared. Strength and reflexes were normal in the bilateral lower extremities. An abdominal examination was not performed. He received a diagnosis of MP and counseled regarding weight loss, glycemic control, garment optimization, and conservative analgesia with as-needed nonsteroidal anti-inflammatory drugs. He was instructed to follow-up closely with his primary care physician for further monitoring.
During the current visit, the patient reported 2 atraumatic falls the prior 2 months, attributed to escalating right leg weakness. The patient reported that ascending stairs had become difficult, and he was unable to cross his right leg over his left while in a seated position. The territory of numbness expanded to his front and inner thigh. Although previously he was able to hike 4 miles, he now was unable to walk more than half of a mile without developing shortness of breath. He reported frequent urination without hematuria and a recent weight gain of 8 pounds despite early satiety.
His medical history included hypertension, hypercholesterolemia, truncal obesity, noninsulin dependent DM, coronary artery disease, atrial flutter, transient ischemic attack, and benign positional paroxysmal vertigo. He was exposed to Agent Orange during his service in Vietnam. Family history was notable for breast cancer (mother), lung cancer (father), and an unspecified form of lymphoma (brother). He had smoked approximately 2 packs of cigarettes daily for 15 years but quit 38 years prior. He reported consuming on average 3 alcohol-containing drinks per week and no illicit drug use. He was adherent with all medications, including furosemide 40 mg daily, losartan 25 mg daily, metoprolol succinate 50 mg daily, atorvastatin 80 mg daily, metformin 500 mg twice daily, and rivaroxaban 20 mg daily with dinner.
His vital signs included a blood pressure of 123/58 mmHg, a pulse of 74 beats per minute, a respiratory rate of 16 breaths per minute, and an oxygen saturation of 94% on ambient air. His temperature was recorded at 96.7°F, and his weight was 234 pounds with a body mass index (BMI) of 34. He was well groomed and in no acute distress. His cardiopulmonary examination was normal. Carotid, radial, and bilateral dorsalis pedis pulsations were 2+ bilaterally, and no jugular venous distension was observed at 30°. The abdomen was protuberant. Nonshifting dullness to percussion and firmness to palpation was observed throughout right upper and lower quadrants, with hyperactive bowel sounds primarily localized to the left upper and lower quadrants.
Neurologic examination revealed symmetric facies with normal phonation and diction. He was spontaneously moving all extremities, and his gait was normal. Sensation to light touch was severely diminished throughout the anterolateral and medial thigh, extending to the level of the knee, and otherwise reduced in a stocking-type pattern over the bilateral feet and toes. His right hip flexion, adduction, as well as internal and external rotation were focally diminished to 4- out of 5. Right knee extension was 4+ out of 5. Strength was otherwise 5 out of 5. The patient exhibited asymmetric Patellar reflexes—absent on the right and 2+ on the left. Achilles reflexes were absent bilaterally. Straight-leg raise test was negative bilaterally and did not clearly exacerbate his right leg numbness or paresthesias. There were no notable fasciculations. There was 2+ bilateral lower extremity pitting edema appreciated to the level of the midshin (right greater than left), without palpable cords or new skin lesions.
Upon referral to the neurology service, the patient underwent electromyography, which revealed complex repetitive discharges in the right tibialis anterior and pattern of reduced recruitment upon activation of the right vastus medialis, collectively suggestive of an L3-4 plexopathy. The patient was admitted for expedited workup.
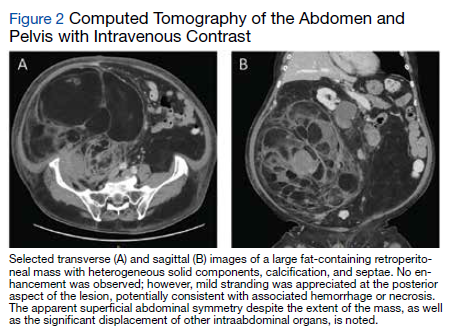
A complete blood count and metabolic panel that were taken in the emergency department were normal, save for a serum bicarbonate of 30 mEq/L. His hemoglobin A1c was 6.6%. Computed tomography (CT) of the abdomen and pelvis with IV contrast was obtained, and notable for a 30 cm fat-containing right-sided retroperitoneal mass with associated solid nodular components and calcification (Figure 2). No enhancement of the lesion was observed. There was significant associated mass effect, with superior displacement of the liver and right hemidiaphragm, as well as superomedial deflection of the right kidney, inferior vena cava, and other intraabdominal organs. Subsequent imaging with a CT of the chest, as well as magnetic resonance imaging of the brain, were without evidence of metastatic disease.
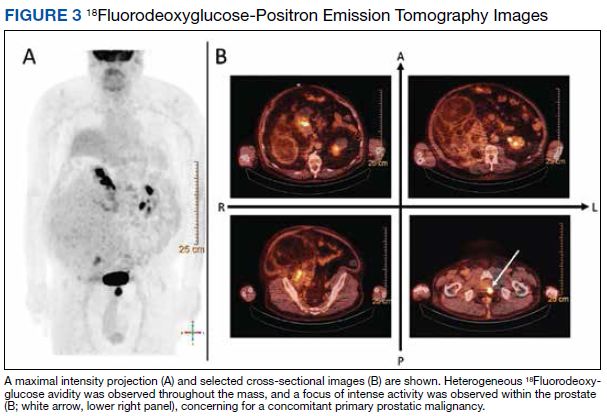
18Fluorodeoxyglucose-positron emission tomography (FDG-PET) was performed and demonstrated heterogeneous FDG avidity throughout the mass (SUVmax 5.9), as well as poor delineation of the boundary of the right psoas major, consistent with muscular invasion (Figure 3). The FDG-PET also revealed intense tracer uptake within the left prostate (SUVmax 26), concerning for a concomitant prostate malignancy.
To facilitate tissue diagnosis, the patient underwent a CT-guided biopsy of the retroperitoneal mass. Subsequent histopathologic analysis revealed a primarily well-differentiated spindle cell lesion with occasional adipocytic atypia, and a superimposed hypercellular element characterized by the presence of pleomorphic high-grade spindled cells. The neoplastic spindle cells were MDM2-positive by both immunohistochemistry and fluorescence in situ hybridization (FISH), and negative for pancytokeratin, smooth muscle myosin, and S100. The findings were collectively consistent with a dedifferentiated liposarcoma (DDLPS).
Given the focus of FDG avidity observed on the PET, the patient underwent a transrectal ultrasound-guided biopsy of the prostate, which yielded diagnosis of a concomitant high-risk (Gleason 4+4) prostate adenocarcinoma. A bone scan did not reveal evidence of osseous metastatic disease.
Outcome
The patient was treated with external beam radiotherapy (EBRT) delivered simultaneously to both the prostate and high-risk retroperitoneal margins of the DDLPS, as well as concurrent androgen deprivation therapy. Five months after completed radiotherapy, resection of the DDLPS was attempted. However, palliative tumor debulking was instead performed due to extensive locoregional invasion with involvement of the posterior peritoneum and ipsilateral quadratus, iliopsoas, and psoas muscles, as well as the adjacent lumbar nerve roots.
At present, the patient is undergoing surveillance imaging every 3 months to reevaluate his underlying disease burden, which has thus far been radiographically stable. Current management at the primary care level is focused on preserving quality of life, particularly maintaining mobility and functional independence.
Discussion
Although generally a benign entrapment neuropathy, MP bears well-established associations with multiple forms of must-not-miss pathology. Here, we present the case of a veteran in whom MP was the index presentation of a massive retroperitoneal liposarcoma, stressing the importance of a thorough history and physical examination in all patients presenting with MP. The case presented herein highlights many of the red-flag signs and symptoms that primary care physicians might encounter in patients with retroperitoneal pathology, including MP and MP-like syndromes (Figure 4).
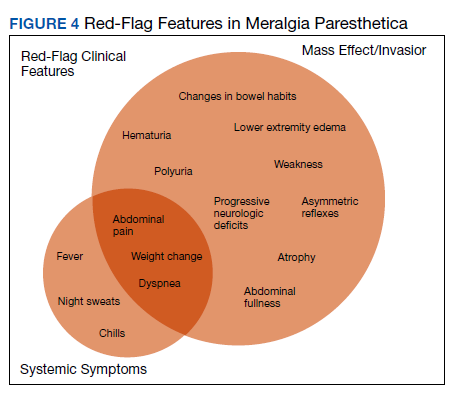
In this case, the pretest probability of a spontaneous and uncomplicated MP was high given the patient’s sex, age, body habitus, and DM; however, there important atypia that emerged as the case evolved, including: (1) the progressive course; (2) proximal right lower extremity weakness; (3) asymmetric patellar reflexes; and (4) numerous clinical stigmata of intraabdominal mass effect. The patient exhibited abnormalities on abdominal examination that suggested the presence of an underlying intraabdominal mass, providing key diagnostic insight into this case. Given the slowly progressive nature of liposarcomas, we feel the abnormalities appreciated on abdominal examination were likely apparent during the initial presentation.18
There are numerous cognitive biases that may explain why an abdominal examination was not prioritized during the initial presentation. Namely, the patient’s numerous risk factors for spontaneous MP, as detailed above, may have contributed to framing bias that limited consideration of alternative diagnoses. In addition, the patient’s physical examination likely contributed to search satisfaction, whereby alternative diagnoses were not further entertained after discovery of findings consistent with spontaneous MP.19 Finally, it remains conceivable that an abdominal examination was not prioritized as it is often perceived as being distinct from, rather than an integral part of, the neurologic examination.20 Given that numerous neurologic disorders may present with abdominal pathology, we feel a thorough abdominal examination should be considered part of the full neurologic examination, especially in cases presenting with focal neurologic findings involving the lower extremities.21
Collectively, this case alludes to the importance of close clinical follow-up, as well as adequate anticipatory patient guidance in cases of suspected MP. In most patients, the clinical course of spontaneous MP is benign and favorable, with up to 85% of patients experiencing resolution within 4 to 6 months of the initial presentation.22 Common conservative measures include weight loss, garment optimization, and nonsteroidal anti-inflammatory drugs as needed for analgesia. In refractory cases, procedural interventions such as with neurolysis or resection of the lateral femoral cutaneous nerve, may be required after the ruling out of alternative diagnoses.23,24
Importantly, in even prolonged and resistant cases of MP, patient discomfort remains localized to the territory of the LFCN. Additional lower motor neuron signs, such as an expanding territory of sensory involvement, muscle weakness, or diminished reflexes, should prompt additional testing for alternative diagnoses. In addition, clinical findings concerning for intraabdominal mass effect, many of which were observed in this case, should lead to further evaluation and expeditious cross-sectional imaging. Although this patient’s early satiety, polyuria, bilateral lower extremity edema, weight gain, and lumbar plexopathy each may be explained by direct compression, invasion, or displacement, his report of progressive exertional dyspnea merits further discussion.
Exertional dyspnea is an uncommon complication of soft tissue sarcoma, reported almost exclusively in cases with cardiac, mediastinal, or other thoracic involvement.25-28 In this case, there was no evidence of thoracic involvement, either through direct extension or metastasis. Instead, the patient’s exertional dyspnea may have been attributable to increased intraabdominal pressure leading to compromised diaphragm excursion and reduced pulmonary reserve. In addition, the radiographic findings also raise the possibility of a potential contribution from preload failure due to IVC compression. Overall, dyspnea is a concerning feature that may suggest advanced disease.
Despite the value of a thorough history and physical examination in patients with MP, major clinical guidelines from neurologic, neurosurgical, and orthopedic organizations do not formally address MP evaluation and management. Further, proposed clinical practice algorithms are inconsistent in their recommendations regarding the identification of red-flag features and ruling out of alternative diagnoses.22,29,30 To supplement the abdominal examination, it would be reasonable to perform a pelvic compression test (PCT) in patients presenting with suspected MP. The PCT is a highly sensitive and specific provocative maneuver shown to enable reliable differentiation between MP and lumbar radiculopathy, and is performed by placing downward force on the anterior superior iliac spine of the affected extremity for 45 seconds with the patient in the lateral recumbent position.31 As this maneuver is intended to force relaxation of the inguinal ligament, thereby relieving pressure on the LFCN, improvement in the patient’s symptoms with the PCT is consistent with MP.
Conclusions
1. van Slobbe AM, Bohnen AM, Bernsen RM, Koes BW, Bierma-Zeinstra SM. Incidence rates and determinants in meralgia paresthetica in general practice. J Neurol. 2004;251(3):294-297. doi:10.1007/s00415-004-0310-x
2. Parisi TJ, Mandrekar J, Dyck PJ, Klein CJ. Meralgia paresthetica: relation to obesity, advanced age, and diabetes mellitus. Neurology. 2011;77(16):1538-1542. doi:10.1212/WNL.0b013e318233b356
3. Ecker AD. Diagnosis of meralgia paresthetica. JAMA. 1985;253(7):976.
4. Massey EW, Pellock JM. Meralgia paraesthetica in a child. J Pediatr. 1978;93(2):325-326. doi:10.1016/s0022-3476(78)80566-6
5. Harney D, Patijn J. Meralgia paresthetica: diagnosis and management strategies. Pain Med. 2007;8(8):669-677. doi:10.1111/j.1526-4637.2006.00227.x
6. Berini SE, Spinner RJ, Jentoft ME, et al. Chronic meralgia paresthetica and neurectomy: a clinical pathologic study. Neurology. 2014;82(17):1551-1555. doi:10.1212/WNL.0000000000000367
7. Payne RA, Harbaugh K, Specht CS, Rizk E. Correlation of histopathology and clinical symptoms in meralgia paresthetica. Cureus. 2017;9(10):e1789. Published 2017 Oct 20. doi:10.7759/cureus.1789
8. Boyce JR. Meralgia paresthetica and tight trousers. JAMA. 1984;251(12):1553.
9. Orton D. Meralgia paresthetica from a wallet. JAMA. 1984;252(24):3368.
10. Fargo MV, Konitzer LN. Meralgia paresthetica due to body armor wear in U.S. soldiers serving in Iraq: a case report and review of the literature. Mil Med. 2007;172(6):663-665. doi:10.7205/milmed.172.6.663
11. Korkmaz N, Ozçakar L. Meralgia paresthetica in a policeman: the belt or the gun. Plast Reconstr Surg. 2004;114(4):1012-1013. doi:10.1097/01.prs.0000138706.86633.01
12. Gooding MS, Evangelista V, Pereira L. Carpal Tunnel Syndrome and Meralgia Paresthetica in Pregnancy. Obstet Gynecol Surv. 2020;75(2):121-126. doi:10.1097/OGX.0000000000000745
13. Pauwels A, Amarenco P, Chazouillères O, Pigot F, Calmus Y, Lévy VG. Une complication rare et méconnue de l’ascite: la méralgie paresthésique [Unusual and unknown complication of ascites: meralgia paresthetica]. Gastroenterol Clin Biol. 1990;14(3):295.
14. Braddom RL. L2 rather than L1 radiculopathy mimics meralgia paresthetica. Muscle Nerve. 2010;42(5):842. doi:10.1002/mus.21826
15. Suber DA, Massey EW. Pelvic mass presenting as meralgia paresthetica. Obstet Gynecol. 1979;53(2):257-258.
16. Flowers RS. Meralgia paresthetica. A clue to retroperitoneal malignant tumor. Am J Surg. 1968;116(1):89-92. doi:10.1016/0002-9610(68)90423-6
17. Yi TI, Yoon TH, Kim JS, Lee GE, Kim BR. Femoral neuropathy and meralgia paresthetica secondary to an iliacus hematoma. Ann Rehabil Med. 2012;36(2):273-277. doi:10.5535/arm.2012.36.2.273
18. Lee ATJ, Thway K, Huang PH, Jones RL. Clinical and molecular spectrum of liposarcoma. J Clin Oncol. 2018;36(2):151-159. doi:10.1200/JCO.2017.74.9598
19. O’Sullivan ED, Schofield SJ. Cognitive bias in clinical medicine. J R Coll Physicians Edinb. 2018;48(3):225-232. doi:10.4997/JRCPE.2018.306
20. Bickley, LS. Bates’ Guide to Physical Examination and History Taking. 12th Edition. Wolters Kluwer Health/Lippincott Williams and Wilkins; 2016.
21. Bhavsar AS, Verma S, Lamba R, Lall CG, Koenigsknecht V, Rajesh A. Abdominal manifestations of neurologic disorders. Radiographics. 2013;33(1):135-153. doi:10.1148/rg.331125097
22. Dureja GP, Gulaya V, Jayalakshmi TS, Mandal P. Management of meralgia paresthetica: a multimodality regimen. Anesth Analg. 1995;80(5):1060-1061. doi:10.1097/00000539-199505000-00043
23. Patijn J, Mekhail N, Hayek S, Lataster A, van Kleef M, Van Zundert J. Meralgia paresthetica. Pain Pract. 2011;11(3):302-308. doi:10.1111/j.1533-2500.2011.00458.x24. Ivins GK. Meralgia paresthetica, the elusive diagnosis: clinical experience with 14 adult patients. Ann Surg. 2000;232(2):281-286. doi:10.1097/00000658-200008000-00019
25. Munin MA, Goerner MS, Raggio I, et al. A rare cause of dyspnea: undifferentiated pleomorphic sarcoma in the left atrium. Cardiol Res. 2017;8(5):241-245. doi:10.14740/cr590w
26. Nguyen A, Awad WI. Cardiac sarcoma arising from malignant transformation of a preexisting atrial myxoma. Ann Thorac Surg. 2016;101(4):1571-1573. doi:10.1016/j.athoracsur.2015.05.129
27. Jiang S, Li J, Zeng Q, Liang J. Pulmonary artery intimal sarcoma misdiagnosed as pulmonary embolism: a case report. Oncol Lett. 2017;13(4):2713-2716. doi:10.3892/ol.2017.5775
28. Cojocaru A, Oliveira PJ, Pellecchia C. A pleural presentation of a rare soft tissue sarcoma. Am J Resp Crit Care Med. 2012;185:A5201. doi:10.1164/ajrccm-conference.2012.185.1_MeetingAbstracts.A5201
29. Grossman MG, Ducey SA, Nadler SS, Levy AS. Meralgia paresthetica: diagnosis and treatment. J Am Acad Orthop Surg. 2001;9(5):336-344. doi:10.5435/00124635-200109000-00007
30. Cheatham SW, Kolber MJ, Salamh PA. Meralgia paresthetica: a review of the literature. Int J Sports Phys Ther. 2013;8(6):883-893.
31. Nouraei SA, Anand B, Spink G, O’Neill KS. A novel approach to the diagnosis and management of meralgia paresthetica. Neurosurgery. 2007;60(4):696-700. doi:10.1227/01.NEU.0000255392.69914.F7
32. Antunes PE, Antunes MJ. Meralgia paresthetica after aortic valve surgery. J Heart Valve Dis. 1997;6(6):589-590.
33. Reddy YM, Singh D, Chikkam V, et al. Postprocedural neuropathy after atrial fibrillation ablation. J Interv Card Electrophysiol. 2013;36(3):279-285. doi:10.1007/s10840-012-9724-z
34. Butler R, Webster MW. Meralgia paresthetica: an unusual complication of cardiac catheterization via the femoral artery. Catheter Cardiovasc Interv. 2002;56(1):69-71. doi:10.1002/ccd.10149
35. Jellish WS, Oftadeh M. Peripheral nerve injury in cardiac surgery. J Cardiothorac Vasc Anesth. 2018;32(1):495-511. doi:10.1053/j.jvca.2017.08.030
36. Parsonnet V, Karasakalides A, Gielchinsky I, Hochberg M, Hussain SM. Meralgia paresthetica after coronary bypass surgery. J Thorac Cardiovasc Surg. 1991;101(2):219-221.
37. Macgregor AM, Thoburn EK. Meralgia paresthetica following bariatric surgery. Obes Surg. 1999;9(4):364-368. doi:10.1381/096089299765552945
38. Grace DM. Meralgia paresthetica after gastroplasty for morbid obesity. Can J Surg. 1987;30(1):64-65.
39. Polidori L, Magarelli M, Tramutoli R. Meralgia paresthetica as a complication of laparoscopic appendectomy. Surg Endosc. 2003;17(5):832. doi:10.1007/s00464-002-4279-1
40. Yamout B, Tayyim A, Farhat W. Meralgia paresthetica as a complication of laparoscopic cholecystectomy. Clin Neurol Neurosurg. 1994;96(2):143-144. doi:10.1016/0303-8467(94)90048-5
41. Broin EO, Horner C, Mealy K, et al. Meralgia paraesthetica following laparoscopic inguinal hernia repair. an anatomical analysis. Surg Endosc. 1995;9(1):76-78. doi:10.1007/BF00187893
42. Eubanks S, Newman L 3rd, Goehring L, et al. Meralgia paresthetica: a complication of laparoscopic herniorrhaphy. Surg Laparosc Endosc. 1993;3(5):381-385.
43. Atamaz F, Hepgüler S, Karasu Z, Kilic M. Meralgia paresthetica after liver transplantation: a case report. Transplant Proc. 2005;37(10):4424-4425. doi:10.1016/j.transproceed.2005.11.047
44. Chung KH, Lee JY, Ko TK, et al. Meralgia paresthetica affecting parturient women who underwent cesarean section -a case report-. Korean J Anesthesiol. 2010;59 Suppl(Suppl):S86-S89. doi:10.4097/kjae.2010.59.S.S86
45. Hutchins FL Jr, Huggins J, Delaney ML. Laparoscopic myomectomy-an unusual cause of meralgia paresthetica. J Am Assoc Gynecol Laparosc. 1998;5(3):309-311. doi:10.1016/s1074-3804(98)80039-x
46. Jones CD, Guiot L, Portelli M, Bullen T, Skaife P. Two interesting cases of meralgia paraesthetica. Pain Physician. 2017;20(6):E987-E989.
47. Peters G, Larner AJ. Meralgia paresthetica following gynecologic and obstetric surgery. Int J Gynaecol Obstet. 2006;95(1):42-43. doi:10.1016/j.ijgo.2006.05.025
48. Kvarnström N, Järvholm S, Johannesson L, Dahm-Kähler P, Olausson M, Brännström M. Live donors of the initial observational study of uterus transplantation-psychological and medical follow-up until 1 year after surgery in the 9 cases. Transplantation. 2017;101(3):664-670. doi:10.1097/TP.0000000000001567
49. Goulding K, Beaulé PE, Kim PR, Fazekas A. Incidence of lateral femoral cutaneous nerve neuropraxia after anterior approach hip arthroplasty. Clin Orthop Relat Res. 2010;468(9):2397-2404. doi:10.1007/s11999-010-1406-5
50. Yamamoto T, Nagira K, Kurosaka M. Meralgia paresthetica occurring 40 years after iliac bone graft harvesting: case report. Neurosurgery. 2001;49(6):1455-1457. doi:10.1097/00006123-200112000-00028
51. Roqueplan F, Porcher R, Hamzé B, et al. Long-term results of percutaneous resection and interstitial laser ablation of osteoid osteomas. Eur Radiol. 2010;20(1):209-217. doi:10.1007/s00330-009-1537-9
52. Gupta A, Muzumdar D, Ramani PS. Meralgia paraesthetica following lumbar spine surgery: a study in 110 consecutive surgically treated cases. Neurol India. 2004;52(1):64-66.
53. Yang SH, Wu CC, Chen PQ. Postoperative meralgia paresthetica after posterior spine surgery: incidence, risk factors, and clinical outcomes. Spine (Phila Pa 1976). 2005;30(18):E547-E550. doi:10.1097/01.brs.0000178821.14102.9d
54. Tejwani SG, Scaduto AA, Bowen RE. Transient meralgia paresthetica after pediatric posterior spine fusion. J Pediatr Orthop. 2006;26(4):530-533. doi:10.1097/01.bpo.0000217721.95480.9e
55. Peker S, Ay B, Sun I, Ozgen S, Pamir M. Meralgia paraesthetica: complications of prone position during lumbar disc surgery. Internet J Anesthesiol. 2003;8(1):24-29.
In patients presenting with focal neurologic findings involving the lower extremities, a thorough abdominal examination should be considered an integral part of the full neurologic work up.
In patients presenting with focal neurologic findings involving the lower extremities, a thorough abdominal examination should be considered an integral part of the full neurologic work up.
Meralgia paresthetica (MP) is a sensory mononeuropathy of the lateral femoral cutaneous nerve (LFCN), clinically characterized by numbness, pain, and paresthesias involving the anterolateral aspect of the thigh. Estimates of MP incidence are derived largely from observational studies and reported to be about 3.2 to 4.3 cases per 10,000 patient-years.1,2 Although typically arising during midlife and especially in the context of comorbid obesity, diabetes mellitus (DM), and excessive alcohol consumption, MP may occur at any age, and bears a slight predilection for males.2-4
MP may be divided etiologically into iatrogenic and spontaneous subtypes.5 Iatrogenic cases generally are attributable to nerve injury in the setting of direct or indirect trauma (such as with patient malpositioning) arising in the context of multiple forms of procedural or surgical intervention (Table). Spontaneous MP is primarily thought to occur as a result of LFCN compression at the level of the inguinal ligament, wherein internal or external pressures may promote LFCN entrapment and resultant functional disruption (Figure 1).6,7
External forces, such as tight garments, wallets, or even elements of modern body armor, have been reported to provoke MP.8-11 Alternatively, states of increased intraabdominal pressure, such as obesity, ascites, and pregnancy may predispose to LFCN compression.2,12,13 Less commonly, lumbar radiculopathy, pelvic masses, and several forms of retroperitoneal pathology may present with clinical symptomatology indistinguishable from MP.14-17 Importantly, many of these represent must-not-miss diagnoses, and may be suggested via a focused history and physical examination.
Here, we present a case of MP secondary to a massive retroperitoneal sarcoma, ultimately drawing renewed attention to the known association of MP and retroperitoneal pathology, and therein highlighting the utility of a dedicated review of systems to identify red-flag features in patients who present with MP and a thorough abdominal examination in all patients presenting with focal neurologic deficits involving the lower extremities.
Case Presentation
A male Vietnam War veteran aged 69 years presented to a primary care clinic at West Roxbury Veterans Affairs Medical Center (WRVAMC) in Massachusetts with progressive right lower extremity numbness. Three months prior to this visit, he was evaluated in an urgent care clinic at WRVAMC for 6 months of numbness and increasingly painful nocturnal paresthesias involving the same extremity. A targeted physical examination at that visit revealed an obese male wearing tight suspenders, as well as focally diminished sensation to light touch involving the anterolateral aspect of the thigh, extending from just below the right hip to above the knee. Sensation in the medial thigh was spared. Strength and reflexes were normal in the bilateral lower extremities. An abdominal examination was not performed. He received a diagnosis of MP and counseled regarding weight loss, glycemic control, garment optimization, and conservative analgesia with as-needed nonsteroidal anti-inflammatory drugs. He was instructed to follow-up closely with his primary care physician for further monitoring.
During the current visit, the patient reported 2 atraumatic falls the prior 2 months, attributed to escalating right leg weakness. The patient reported that ascending stairs had become difficult, and he was unable to cross his right leg over his left while in a seated position. The territory of numbness expanded to his front and inner thigh. Although previously he was able to hike 4 miles, he now was unable to walk more than half of a mile without developing shortness of breath. He reported frequent urination without hematuria and a recent weight gain of 8 pounds despite early satiety.
His medical history included hypertension, hypercholesterolemia, truncal obesity, noninsulin dependent DM, coronary artery disease, atrial flutter, transient ischemic attack, and benign positional paroxysmal vertigo. He was exposed to Agent Orange during his service in Vietnam. Family history was notable for breast cancer (mother), lung cancer (father), and an unspecified form of lymphoma (brother). He had smoked approximately 2 packs of cigarettes daily for 15 years but quit 38 years prior. He reported consuming on average 3 alcohol-containing drinks per week and no illicit drug use. He was adherent with all medications, including furosemide 40 mg daily, losartan 25 mg daily, metoprolol succinate 50 mg daily, atorvastatin 80 mg daily, metformin 500 mg twice daily, and rivaroxaban 20 mg daily with dinner.
His vital signs included a blood pressure of 123/58 mmHg, a pulse of 74 beats per minute, a respiratory rate of 16 breaths per minute, and an oxygen saturation of 94% on ambient air. His temperature was recorded at 96.7°F, and his weight was 234 pounds with a body mass index (BMI) of 34. He was well groomed and in no acute distress. His cardiopulmonary examination was normal. Carotid, radial, and bilateral dorsalis pedis pulsations were 2+ bilaterally, and no jugular venous distension was observed at 30°. The abdomen was protuberant. Nonshifting dullness to percussion and firmness to palpation was observed throughout right upper and lower quadrants, with hyperactive bowel sounds primarily localized to the left upper and lower quadrants.
Neurologic examination revealed symmetric facies with normal phonation and diction. He was spontaneously moving all extremities, and his gait was normal. Sensation to light touch was severely diminished throughout the anterolateral and medial thigh, extending to the level of the knee, and otherwise reduced in a stocking-type pattern over the bilateral feet and toes. His right hip flexion, adduction, as well as internal and external rotation were focally diminished to 4- out of 5. Right knee extension was 4+ out of 5. Strength was otherwise 5 out of 5. The patient exhibited asymmetric Patellar reflexes—absent on the right and 2+ on the left. Achilles reflexes were absent bilaterally. Straight-leg raise test was negative bilaterally and did not clearly exacerbate his right leg numbness or paresthesias. There were no notable fasciculations. There was 2+ bilateral lower extremity pitting edema appreciated to the level of the midshin (right greater than left), without palpable cords or new skin lesions.
Upon referral to the neurology service, the patient underwent electromyography, which revealed complex repetitive discharges in the right tibialis anterior and pattern of reduced recruitment upon activation of the right vastus medialis, collectively suggestive of an L3-4 plexopathy. The patient was admitted for expedited workup.
A complete blood count and metabolic panel that were taken in the emergency department were normal, save for a serum bicarbonate of 30 mEq/L. His hemoglobin A1c was 6.6%. Computed tomography (CT) of the abdomen and pelvis with IV contrast was obtained, and notable for a 30 cm fat-containing right-sided retroperitoneal mass with associated solid nodular components and calcification (Figure 2). No enhancement of the lesion was observed. There was significant associated mass effect, with superior displacement of the liver and right hemidiaphragm, as well as superomedial deflection of the right kidney, inferior vena cava, and other intraabdominal organs. Subsequent imaging with a CT of the chest, as well as magnetic resonance imaging of the brain, were without evidence of metastatic disease.
18Fluorodeoxyglucose-positron emission tomography (FDG-PET) was performed and demonstrated heterogeneous FDG avidity throughout the mass (SUVmax 5.9), as well as poor delineation of the boundary of the right psoas major, consistent with muscular invasion (Figure 3). The FDG-PET also revealed intense tracer uptake within the left prostate (SUVmax 26), concerning for a concomitant prostate malignancy.
To facilitate tissue diagnosis, the patient underwent a CT-guided biopsy of the retroperitoneal mass. Subsequent histopathologic analysis revealed a primarily well-differentiated spindle cell lesion with occasional adipocytic atypia, and a superimposed hypercellular element characterized by the presence of pleomorphic high-grade spindled cells. The neoplastic spindle cells were MDM2-positive by both immunohistochemistry and fluorescence in situ hybridization (FISH), and negative for pancytokeratin, smooth muscle myosin, and S100. The findings were collectively consistent with a dedifferentiated liposarcoma (DDLPS).
Given the focus of FDG avidity observed on the PET, the patient underwent a transrectal ultrasound-guided biopsy of the prostate, which yielded diagnosis of a concomitant high-risk (Gleason 4+4) prostate adenocarcinoma. A bone scan did not reveal evidence of osseous metastatic disease.
Outcome
The patient was treated with external beam radiotherapy (EBRT) delivered simultaneously to both the prostate and high-risk retroperitoneal margins of the DDLPS, as well as concurrent androgen deprivation therapy. Five months after completed radiotherapy, resection of the DDLPS was attempted. However, palliative tumor debulking was instead performed due to extensive locoregional invasion with involvement of the posterior peritoneum and ipsilateral quadratus, iliopsoas, and psoas muscles, as well as the adjacent lumbar nerve roots.
At present, the patient is undergoing surveillance imaging every 3 months to reevaluate his underlying disease burden, which has thus far been radiographically stable. Current management at the primary care level is focused on preserving quality of life, particularly maintaining mobility and functional independence.
Discussion
Although generally a benign entrapment neuropathy, MP bears well-established associations with multiple forms of must-not-miss pathology. Here, we present the case of a veteran in whom MP was the index presentation of a massive retroperitoneal liposarcoma, stressing the importance of a thorough history and physical examination in all patients presenting with MP. The case presented herein highlights many of the red-flag signs and symptoms that primary care physicians might encounter in patients with retroperitoneal pathology, including MP and MP-like syndromes (Figure 4).
In this case, the pretest probability of a spontaneous and uncomplicated MP was high given the patient’s sex, age, body habitus, and DM; however, there important atypia that emerged as the case evolved, including: (1) the progressive course; (2) proximal right lower extremity weakness; (3) asymmetric patellar reflexes; and (4) numerous clinical stigmata of intraabdominal mass effect. The patient exhibited abnormalities on abdominal examination that suggested the presence of an underlying intraabdominal mass, providing key diagnostic insight into this case. Given the slowly progressive nature of liposarcomas, we feel the abnormalities appreciated on abdominal examination were likely apparent during the initial presentation.18
There are numerous cognitive biases that may explain why an abdominal examination was not prioritized during the initial presentation. Namely, the patient’s numerous risk factors for spontaneous MP, as detailed above, may have contributed to framing bias that limited consideration of alternative diagnoses. In addition, the patient’s physical examination likely contributed to search satisfaction, whereby alternative diagnoses were not further entertained after discovery of findings consistent with spontaneous MP.19 Finally, it remains conceivable that an abdominal examination was not prioritized as it is often perceived as being distinct from, rather than an integral part of, the neurologic examination.20 Given that numerous neurologic disorders may present with abdominal pathology, we feel a thorough abdominal examination should be considered part of the full neurologic examination, especially in cases presenting with focal neurologic findings involving the lower extremities.21
Collectively, this case alludes to the importance of close clinical follow-up, as well as adequate anticipatory patient guidance in cases of suspected MP. In most patients, the clinical course of spontaneous MP is benign and favorable, with up to 85% of patients experiencing resolution within 4 to 6 months of the initial presentation.22 Common conservative measures include weight loss, garment optimization, and nonsteroidal anti-inflammatory drugs as needed for analgesia. In refractory cases, procedural interventions such as with neurolysis or resection of the lateral femoral cutaneous nerve, may be required after the ruling out of alternative diagnoses.23,24
Importantly, in even prolonged and resistant cases of MP, patient discomfort remains localized to the territory of the LFCN. Additional lower motor neuron signs, such as an expanding territory of sensory involvement, muscle weakness, or diminished reflexes, should prompt additional testing for alternative diagnoses. In addition, clinical findings concerning for intraabdominal mass effect, many of which were observed in this case, should lead to further evaluation and expeditious cross-sectional imaging. Although this patient’s early satiety, polyuria, bilateral lower extremity edema, weight gain, and lumbar plexopathy each may be explained by direct compression, invasion, or displacement, his report of progressive exertional dyspnea merits further discussion.
Exertional dyspnea is an uncommon complication of soft tissue sarcoma, reported almost exclusively in cases with cardiac, mediastinal, or other thoracic involvement.25-28 In this case, there was no evidence of thoracic involvement, either through direct extension or metastasis. Instead, the patient’s exertional dyspnea may have been attributable to increased intraabdominal pressure leading to compromised diaphragm excursion and reduced pulmonary reserve. In addition, the radiographic findings also raise the possibility of a potential contribution from preload failure due to IVC compression. Overall, dyspnea is a concerning feature that may suggest advanced disease.
Despite the value of a thorough history and physical examination in patients with MP, major clinical guidelines from neurologic, neurosurgical, and orthopedic organizations do not formally address MP evaluation and management. Further, proposed clinical practice algorithms are inconsistent in their recommendations regarding the identification of red-flag features and ruling out of alternative diagnoses.22,29,30 To supplement the abdominal examination, it would be reasonable to perform a pelvic compression test (PCT) in patients presenting with suspected MP. The PCT is a highly sensitive and specific provocative maneuver shown to enable reliable differentiation between MP and lumbar radiculopathy, and is performed by placing downward force on the anterior superior iliac spine of the affected extremity for 45 seconds with the patient in the lateral recumbent position.31 As this maneuver is intended to force relaxation of the inguinal ligament, thereby relieving pressure on the LFCN, improvement in the patient’s symptoms with the PCT is consistent with MP.
Conclusions
Meralgia paresthetica (MP) is a sensory mononeuropathy of the lateral femoral cutaneous nerve (LFCN), clinically characterized by numbness, pain, and paresthesias involving the anterolateral aspect of the thigh. Estimates of MP incidence are derived largely from observational studies and reported to be about 3.2 to 4.3 cases per 10,000 patient-years.1,2 Although typically arising during midlife and especially in the context of comorbid obesity, diabetes mellitus (DM), and excessive alcohol consumption, MP may occur at any age, and bears a slight predilection for males.2-4
MP may be divided etiologically into iatrogenic and spontaneous subtypes.5 Iatrogenic cases generally are attributable to nerve injury in the setting of direct or indirect trauma (such as with patient malpositioning) arising in the context of multiple forms of procedural or surgical intervention (Table). Spontaneous MP is primarily thought to occur as a result of LFCN compression at the level of the inguinal ligament, wherein internal or external pressures may promote LFCN entrapment and resultant functional disruption (Figure 1).6,7
External forces, such as tight garments, wallets, or even elements of modern body armor, have been reported to provoke MP.8-11 Alternatively, states of increased intraabdominal pressure, such as obesity, ascites, and pregnancy may predispose to LFCN compression.2,12,13 Less commonly, lumbar radiculopathy, pelvic masses, and several forms of retroperitoneal pathology may present with clinical symptomatology indistinguishable from MP.14-17 Importantly, many of these represent must-not-miss diagnoses, and may be suggested via a focused history and physical examination.
Here, we present a case of MP secondary to a massive retroperitoneal sarcoma, ultimately drawing renewed attention to the known association of MP and retroperitoneal pathology, and therein highlighting the utility of a dedicated review of systems to identify red-flag features in patients who present with MP and a thorough abdominal examination in all patients presenting with focal neurologic deficits involving the lower extremities.
Case Presentation
A male Vietnam War veteran aged 69 years presented to a primary care clinic at West Roxbury Veterans Affairs Medical Center (WRVAMC) in Massachusetts with progressive right lower extremity numbness. Three months prior to this visit, he was evaluated in an urgent care clinic at WRVAMC for 6 months of numbness and increasingly painful nocturnal paresthesias involving the same extremity. A targeted physical examination at that visit revealed an obese male wearing tight suspenders, as well as focally diminished sensation to light touch involving the anterolateral aspect of the thigh, extending from just below the right hip to above the knee. Sensation in the medial thigh was spared. Strength and reflexes were normal in the bilateral lower extremities. An abdominal examination was not performed. He received a diagnosis of MP and counseled regarding weight loss, glycemic control, garment optimization, and conservative analgesia with as-needed nonsteroidal anti-inflammatory drugs. He was instructed to follow-up closely with his primary care physician for further monitoring.
During the current visit, the patient reported 2 atraumatic falls the prior 2 months, attributed to escalating right leg weakness. The patient reported that ascending stairs had become difficult, and he was unable to cross his right leg over his left while in a seated position. The territory of numbness expanded to his front and inner thigh. Although previously he was able to hike 4 miles, he now was unable to walk more than half of a mile without developing shortness of breath. He reported frequent urination without hematuria and a recent weight gain of 8 pounds despite early satiety.
His medical history included hypertension, hypercholesterolemia, truncal obesity, noninsulin dependent DM, coronary artery disease, atrial flutter, transient ischemic attack, and benign positional paroxysmal vertigo. He was exposed to Agent Orange during his service in Vietnam. Family history was notable for breast cancer (mother), lung cancer (father), and an unspecified form of lymphoma (brother). He had smoked approximately 2 packs of cigarettes daily for 15 years but quit 38 years prior. He reported consuming on average 3 alcohol-containing drinks per week and no illicit drug use. He was adherent with all medications, including furosemide 40 mg daily, losartan 25 mg daily, metoprolol succinate 50 mg daily, atorvastatin 80 mg daily, metformin 500 mg twice daily, and rivaroxaban 20 mg daily with dinner.
His vital signs included a blood pressure of 123/58 mmHg, a pulse of 74 beats per minute, a respiratory rate of 16 breaths per minute, and an oxygen saturation of 94% on ambient air. His temperature was recorded at 96.7°F, and his weight was 234 pounds with a body mass index (BMI) of 34. He was well groomed and in no acute distress. His cardiopulmonary examination was normal. Carotid, radial, and bilateral dorsalis pedis pulsations were 2+ bilaterally, and no jugular venous distension was observed at 30°. The abdomen was protuberant. Nonshifting dullness to percussion and firmness to palpation was observed throughout right upper and lower quadrants, with hyperactive bowel sounds primarily localized to the left upper and lower quadrants.
Neurologic examination revealed symmetric facies with normal phonation and diction. He was spontaneously moving all extremities, and his gait was normal. Sensation to light touch was severely diminished throughout the anterolateral and medial thigh, extending to the level of the knee, and otherwise reduced in a stocking-type pattern over the bilateral feet and toes. His right hip flexion, adduction, as well as internal and external rotation were focally diminished to 4- out of 5. Right knee extension was 4+ out of 5. Strength was otherwise 5 out of 5. The patient exhibited asymmetric Patellar reflexes—absent on the right and 2+ on the left. Achilles reflexes were absent bilaterally. Straight-leg raise test was negative bilaterally and did not clearly exacerbate his right leg numbness or paresthesias. There were no notable fasciculations. There was 2+ bilateral lower extremity pitting edema appreciated to the level of the midshin (right greater than left), without palpable cords or new skin lesions.
Upon referral to the neurology service, the patient underwent electromyography, which revealed complex repetitive discharges in the right tibialis anterior and pattern of reduced recruitment upon activation of the right vastus medialis, collectively suggestive of an L3-4 plexopathy. The patient was admitted for expedited workup.
A complete blood count and metabolic panel that were taken in the emergency department were normal, save for a serum bicarbonate of 30 mEq/L. His hemoglobin A1c was 6.6%. Computed tomography (CT) of the abdomen and pelvis with IV contrast was obtained, and notable for a 30 cm fat-containing right-sided retroperitoneal mass with associated solid nodular components and calcification (Figure 2). No enhancement of the lesion was observed. There was significant associated mass effect, with superior displacement of the liver and right hemidiaphragm, as well as superomedial deflection of the right kidney, inferior vena cava, and other intraabdominal organs. Subsequent imaging with a CT of the chest, as well as magnetic resonance imaging of the brain, were without evidence of metastatic disease.
18Fluorodeoxyglucose-positron emission tomography (FDG-PET) was performed and demonstrated heterogeneous FDG avidity throughout the mass (SUVmax 5.9), as well as poor delineation of the boundary of the right psoas major, consistent with muscular invasion (Figure 3). The FDG-PET also revealed intense tracer uptake within the left prostate (SUVmax 26), concerning for a concomitant prostate malignancy.
To facilitate tissue diagnosis, the patient underwent a CT-guided biopsy of the retroperitoneal mass. Subsequent histopathologic analysis revealed a primarily well-differentiated spindle cell lesion with occasional adipocytic atypia, and a superimposed hypercellular element characterized by the presence of pleomorphic high-grade spindled cells. The neoplastic spindle cells were MDM2-positive by both immunohistochemistry and fluorescence in situ hybridization (FISH), and negative for pancytokeratin, smooth muscle myosin, and S100. The findings were collectively consistent with a dedifferentiated liposarcoma (DDLPS).
Given the focus of FDG avidity observed on the PET, the patient underwent a transrectal ultrasound-guided biopsy of the prostate, which yielded diagnosis of a concomitant high-risk (Gleason 4+4) prostate adenocarcinoma. A bone scan did not reveal evidence of osseous metastatic disease.
Outcome
The patient was treated with external beam radiotherapy (EBRT) delivered simultaneously to both the prostate and high-risk retroperitoneal margins of the DDLPS, as well as concurrent androgen deprivation therapy. Five months after completed radiotherapy, resection of the DDLPS was attempted. However, palliative tumor debulking was instead performed due to extensive locoregional invasion with involvement of the posterior peritoneum and ipsilateral quadratus, iliopsoas, and psoas muscles, as well as the adjacent lumbar nerve roots.
At present, the patient is undergoing surveillance imaging every 3 months to reevaluate his underlying disease burden, which has thus far been radiographically stable. Current management at the primary care level is focused on preserving quality of life, particularly maintaining mobility and functional independence.
Discussion
Although generally a benign entrapment neuropathy, MP bears well-established associations with multiple forms of must-not-miss pathology. Here, we present the case of a veteran in whom MP was the index presentation of a massive retroperitoneal liposarcoma, stressing the importance of a thorough history and physical examination in all patients presenting with MP. The case presented herein highlights many of the red-flag signs and symptoms that primary care physicians might encounter in patients with retroperitoneal pathology, including MP and MP-like syndromes (Figure 4).
In this case, the pretest probability of a spontaneous and uncomplicated MP was high given the patient’s sex, age, body habitus, and DM; however, there important atypia that emerged as the case evolved, including: (1) the progressive course; (2) proximal right lower extremity weakness; (3) asymmetric patellar reflexes; and (4) numerous clinical stigmata of intraabdominal mass effect. The patient exhibited abnormalities on abdominal examination that suggested the presence of an underlying intraabdominal mass, providing key diagnostic insight into this case. Given the slowly progressive nature of liposarcomas, we feel the abnormalities appreciated on abdominal examination were likely apparent during the initial presentation.18
There are numerous cognitive biases that may explain why an abdominal examination was not prioritized during the initial presentation. Namely, the patient’s numerous risk factors for spontaneous MP, as detailed above, may have contributed to framing bias that limited consideration of alternative diagnoses. In addition, the patient’s physical examination likely contributed to search satisfaction, whereby alternative diagnoses were not further entertained after discovery of findings consistent with spontaneous MP.19 Finally, it remains conceivable that an abdominal examination was not prioritized as it is often perceived as being distinct from, rather than an integral part of, the neurologic examination.20 Given that numerous neurologic disorders may present with abdominal pathology, we feel a thorough abdominal examination should be considered part of the full neurologic examination, especially in cases presenting with focal neurologic findings involving the lower extremities.21
Collectively, this case alludes to the importance of close clinical follow-up, as well as adequate anticipatory patient guidance in cases of suspected MP. In most patients, the clinical course of spontaneous MP is benign and favorable, with up to 85% of patients experiencing resolution within 4 to 6 months of the initial presentation.22 Common conservative measures include weight loss, garment optimization, and nonsteroidal anti-inflammatory drugs as needed for analgesia. In refractory cases, procedural interventions such as with neurolysis or resection of the lateral femoral cutaneous nerve, may be required after the ruling out of alternative diagnoses.23,24
Importantly, in even prolonged and resistant cases of MP, patient discomfort remains localized to the territory of the LFCN. Additional lower motor neuron signs, such as an expanding territory of sensory involvement, muscle weakness, or diminished reflexes, should prompt additional testing for alternative diagnoses. In addition, clinical findings concerning for intraabdominal mass effect, many of which were observed in this case, should lead to further evaluation and expeditious cross-sectional imaging. Although this patient’s early satiety, polyuria, bilateral lower extremity edema, weight gain, and lumbar plexopathy each may be explained by direct compression, invasion, or displacement, his report of progressive exertional dyspnea merits further discussion.
Exertional dyspnea is an uncommon complication of soft tissue sarcoma, reported almost exclusively in cases with cardiac, mediastinal, or other thoracic involvement.25-28 In this case, there was no evidence of thoracic involvement, either through direct extension or metastasis. Instead, the patient’s exertional dyspnea may have been attributable to increased intraabdominal pressure leading to compromised diaphragm excursion and reduced pulmonary reserve. In addition, the radiographic findings also raise the possibility of a potential contribution from preload failure due to IVC compression. Overall, dyspnea is a concerning feature that may suggest advanced disease.
Despite the value of a thorough history and physical examination in patients with MP, major clinical guidelines from neurologic, neurosurgical, and orthopedic organizations do not formally address MP evaluation and management. Further, proposed clinical practice algorithms are inconsistent in their recommendations regarding the identification of red-flag features and ruling out of alternative diagnoses.22,29,30 To supplement the abdominal examination, it would be reasonable to perform a pelvic compression test (PCT) in patients presenting with suspected MP. The PCT is a highly sensitive and specific provocative maneuver shown to enable reliable differentiation between MP and lumbar radiculopathy, and is performed by placing downward force on the anterior superior iliac spine of the affected extremity for 45 seconds with the patient in the lateral recumbent position.31 As this maneuver is intended to force relaxation of the inguinal ligament, thereby relieving pressure on the LFCN, improvement in the patient’s symptoms with the PCT is consistent with MP.
Conclusions
1. van Slobbe AM, Bohnen AM, Bernsen RM, Koes BW, Bierma-Zeinstra SM. Incidence rates and determinants in meralgia paresthetica in general practice. J Neurol. 2004;251(3):294-297. doi:10.1007/s00415-004-0310-x
2. Parisi TJ, Mandrekar J, Dyck PJ, Klein CJ. Meralgia paresthetica: relation to obesity, advanced age, and diabetes mellitus. Neurology. 2011;77(16):1538-1542. doi:10.1212/WNL.0b013e318233b356
3. Ecker AD. Diagnosis of meralgia paresthetica. JAMA. 1985;253(7):976.
4. Massey EW, Pellock JM. Meralgia paraesthetica in a child. J Pediatr. 1978;93(2):325-326. doi:10.1016/s0022-3476(78)80566-6
5. Harney D, Patijn J. Meralgia paresthetica: diagnosis and management strategies. Pain Med. 2007;8(8):669-677. doi:10.1111/j.1526-4637.2006.00227.x
6. Berini SE, Spinner RJ, Jentoft ME, et al. Chronic meralgia paresthetica and neurectomy: a clinical pathologic study. Neurology. 2014;82(17):1551-1555. doi:10.1212/WNL.0000000000000367
7. Payne RA, Harbaugh K, Specht CS, Rizk E. Correlation of histopathology and clinical symptoms in meralgia paresthetica. Cureus. 2017;9(10):e1789. Published 2017 Oct 20. doi:10.7759/cureus.1789
8. Boyce JR. Meralgia paresthetica and tight trousers. JAMA. 1984;251(12):1553.
9. Orton D. Meralgia paresthetica from a wallet. JAMA. 1984;252(24):3368.
10. Fargo MV, Konitzer LN. Meralgia paresthetica due to body armor wear in U.S. soldiers serving in Iraq: a case report and review of the literature. Mil Med. 2007;172(6):663-665. doi:10.7205/milmed.172.6.663
11. Korkmaz N, Ozçakar L. Meralgia paresthetica in a policeman: the belt or the gun. Plast Reconstr Surg. 2004;114(4):1012-1013. doi:10.1097/01.prs.0000138706.86633.01
12. Gooding MS, Evangelista V, Pereira L. Carpal Tunnel Syndrome and Meralgia Paresthetica in Pregnancy. Obstet Gynecol Surv. 2020;75(2):121-126. doi:10.1097/OGX.0000000000000745
13. Pauwels A, Amarenco P, Chazouillères O, Pigot F, Calmus Y, Lévy VG. Une complication rare et méconnue de l’ascite: la méralgie paresthésique [Unusual and unknown complication of ascites: meralgia paresthetica]. Gastroenterol Clin Biol. 1990;14(3):295.
14. Braddom RL. L2 rather than L1 radiculopathy mimics meralgia paresthetica. Muscle Nerve. 2010;42(5):842. doi:10.1002/mus.21826
15. Suber DA, Massey EW. Pelvic mass presenting as meralgia paresthetica. Obstet Gynecol. 1979;53(2):257-258.
16. Flowers RS. Meralgia paresthetica. A clue to retroperitoneal malignant tumor. Am J Surg. 1968;116(1):89-92. doi:10.1016/0002-9610(68)90423-6
17. Yi TI, Yoon TH, Kim JS, Lee GE, Kim BR. Femoral neuropathy and meralgia paresthetica secondary to an iliacus hematoma. Ann Rehabil Med. 2012;36(2):273-277. doi:10.5535/arm.2012.36.2.273
18. Lee ATJ, Thway K, Huang PH, Jones RL. Clinical and molecular spectrum of liposarcoma. J Clin Oncol. 2018;36(2):151-159. doi:10.1200/JCO.2017.74.9598
19. O’Sullivan ED, Schofield SJ. Cognitive bias in clinical medicine. J R Coll Physicians Edinb. 2018;48(3):225-232. doi:10.4997/JRCPE.2018.306
20. Bickley, LS. Bates’ Guide to Physical Examination and History Taking. 12th Edition. Wolters Kluwer Health/Lippincott Williams and Wilkins; 2016.
21. Bhavsar AS, Verma S, Lamba R, Lall CG, Koenigsknecht V, Rajesh A. Abdominal manifestations of neurologic disorders. Radiographics. 2013;33(1):135-153. doi:10.1148/rg.331125097
22. Dureja GP, Gulaya V, Jayalakshmi TS, Mandal P. Management of meralgia paresthetica: a multimodality regimen. Anesth Analg. 1995;80(5):1060-1061. doi:10.1097/00000539-199505000-00043
23. Patijn J, Mekhail N, Hayek S, Lataster A, van Kleef M, Van Zundert J. Meralgia paresthetica. Pain Pract. 2011;11(3):302-308. doi:10.1111/j.1533-2500.2011.00458.x24. Ivins GK. Meralgia paresthetica, the elusive diagnosis: clinical experience with 14 adult patients. Ann Surg. 2000;232(2):281-286. doi:10.1097/00000658-200008000-00019
25. Munin MA, Goerner MS, Raggio I, et al. A rare cause of dyspnea: undifferentiated pleomorphic sarcoma in the left atrium. Cardiol Res. 2017;8(5):241-245. doi:10.14740/cr590w
26. Nguyen A, Awad WI. Cardiac sarcoma arising from malignant transformation of a preexisting atrial myxoma. Ann Thorac Surg. 2016;101(4):1571-1573. doi:10.1016/j.athoracsur.2015.05.129
27. Jiang S, Li J, Zeng Q, Liang J. Pulmonary artery intimal sarcoma misdiagnosed as pulmonary embolism: a case report. Oncol Lett. 2017;13(4):2713-2716. doi:10.3892/ol.2017.5775
28. Cojocaru A, Oliveira PJ, Pellecchia C. A pleural presentation of a rare soft tissue sarcoma. Am J Resp Crit Care Med. 2012;185:A5201. doi:10.1164/ajrccm-conference.2012.185.1_MeetingAbstracts.A5201
29. Grossman MG, Ducey SA, Nadler SS, Levy AS. Meralgia paresthetica: diagnosis and treatment. J Am Acad Orthop Surg. 2001;9(5):336-344. doi:10.5435/00124635-200109000-00007
30. Cheatham SW, Kolber MJ, Salamh PA. Meralgia paresthetica: a review of the literature. Int J Sports Phys Ther. 2013;8(6):883-893.
31. Nouraei SA, Anand B, Spink G, O’Neill KS. A novel approach to the diagnosis and management of meralgia paresthetica. Neurosurgery. 2007;60(4):696-700. doi:10.1227/01.NEU.0000255392.69914.F7
32. Antunes PE, Antunes MJ. Meralgia paresthetica after aortic valve surgery. J Heart Valve Dis. 1997;6(6):589-590.
33. Reddy YM, Singh D, Chikkam V, et al. Postprocedural neuropathy after atrial fibrillation ablation. J Interv Card Electrophysiol. 2013;36(3):279-285. doi:10.1007/s10840-012-9724-z
34. Butler R, Webster MW. Meralgia paresthetica: an unusual complication of cardiac catheterization via the femoral artery. Catheter Cardiovasc Interv. 2002;56(1):69-71. doi:10.1002/ccd.10149
35. Jellish WS, Oftadeh M. Peripheral nerve injury in cardiac surgery. J Cardiothorac Vasc Anesth. 2018;32(1):495-511. doi:10.1053/j.jvca.2017.08.030
36. Parsonnet V, Karasakalides A, Gielchinsky I, Hochberg M, Hussain SM. Meralgia paresthetica after coronary bypass surgery. J Thorac Cardiovasc Surg. 1991;101(2):219-221.
37. Macgregor AM, Thoburn EK. Meralgia paresthetica following bariatric surgery. Obes Surg. 1999;9(4):364-368. doi:10.1381/096089299765552945
38. Grace DM. Meralgia paresthetica after gastroplasty for morbid obesity. Can J Surg. 1987;30(1):64-65.
39. Polidori L, Magarelli M, Tramutoli R. Meralgia paresthetica as a complication of laparoscopic appendectomy. Surg Endosc. 2003;17(5):832. doi:10.1007/s00464-002-4279-1
40. Yamout B, Tayyim A, Farhat W. Meralgia paresthetica as a complication of laparoscopic cholecystectomy. Clin Neurol Neurosurg. 1994;96(2):143-144. doi:10.1016/0303-8467(94)90048-5
41. Broin EO, Horner C, Mealy K, et al. Meralgia paraesthetica following laparoscopic inguinal hernia repair. an anatomical analysis. Surg Endosc. 1995;9(1):76-78. doi:10.1007/BF00187893
42. Eubanks S, Newman L 3rd, Goehring L, et al. Meralgia paresthetica: a complication of laparoscopic herniorrhaphy. Surg Laparosc Endosc. 1993;3(5):381-385.
43. Atamaz F, Hepgüler S, Karasu Z, Kilic M. Meralgia paresthetica after liver transplantation: a case report. Transplant Proc. 2005;37(10):4424-4425. doi:10.1016/j.transproceed.2005.11.047
44. Chung KH, Lee JY, Ko TK, et al. Meralgia paresthetica affecting parturient women who underwent cesarean section -a case report-. Korean J Anesthesiol. 2010;59 Suppl(Suppl):S86-S89. doi:10.4097/kjae.2010.59.S.S86
45. Hutchins FL Jr, Huggins J, Delaney ML. Laparoscopic myomectomy-an unusual cause of meralgia paresthetica. J Am Assoc Gynecol Laparosc. 1998;5(3):309-311. doi:10.1016/s1074-3804(98)80039-x
46. Jones CD, Guiot L, Portelli M, Bullen T, Skaife P. Two interesting cases of meralgia paraesthetica. Pain Physician. 2017;20(6):E987-E989.
47. Peters G, Larner AJ. Meralgia paresthetica following gynecologic and obstetric surgery. Int J Gynaecol Obstet. 2006;95(1):42-43. doi:10.1016/j.ijgo.2006.05.025
48. Kvarnström N, Järvholm S, Johannesson L, Dahm-Kähler P, Olausson M, Brännström M. Live donors of the initial observational study of uterus transplantation-psychological and medical follow-up until 1 year after surgery in the 9 cases. Transplantation. 2017;101(3):664-670. doi:10.1097/TP.0000000000001567
49. Goulding K, Beaulé PE, Kim PR, Fazekas A. Incidence of lateral femoral cutaneous nerve neuropraxia after anterior approach hip arthroplasty. Clin Orthop Relat Res. 2010;468(9):2397-2404. doi:10.1007/s11999-010-1406-5
50. Yamamoto T, Nagira K, Kurosaka M. Meralgia paresthetica occurring 40 years after iliac bone graft harvesting: case report. Neurosurgery. 2001;49(6):1455-1457. doi:10.1097/00006123-200112000-00028
51. Roqueplan F, Porcher R, Hamzé B, et al. Long-term results of percutaneous resection and interstitial laser ablation of osteoid osteomas. Eur Radiol. 2010;20(1):209-217. doi:10.1007/s00330-009-1537-9
52. Gupta A, Muzumdar D, Ramani PS. Meralgia paraesthetica following lumbar spine surgery: a study in 110 consecutive surgically treated cases. Neurol India. 2004;52(1):64-66.
53. Yang SH, Wu CC, Chen PQ. Postoperative meralgia paresthetica after posterior spine surgery: incidence, risk factors, and clinical outcomes. Spine (Phila Pa 1976). 2005;30(18):E547-E550. doi:10.1097/01.brs.0000178821.14102.9d
54. Tejwani SG, Scaduto AA, Bowen RE. Transient meralgia paresthetica after pediatric posterior spine fusion. J Pediatr Orthop. 2006;26(4):530-533. doi:10.1097/01.bpo.0000217721.95480.9e
55. Peker S, Ay B, Sun I, Ozgen S, Pamir M. Meralgia paraesthetica: complications of prone position during lumbar disc surgery. Internet J Anesthesiol. 2003;8(1):24-29.
1. van Slobbe AM, Bohnen AM, Bernsen RM, Koes BW, Bierma-Zeinstra SM. Incidence rates and determinants in meralgia paresthetica in general practice. J Neurol. 2004;251(3):294-297. doi:10.1007/s00415-004-0310-x
2. Parisi TJ, Mandrekar J, Dyck PJ, Klein CJ. Meralgia paresthetica: relation to obesity, advanced age, and diabetes mellitus. Neurology. 2011;77(16):1538-1542. doi:10.1212/WNL.0b013e318233b356
3. Ecker AD. Diagnosis of meralgia paresthetica. JAMA. 1985;253(7):976.
4. Massey EW, Pellock JM. Meralgia paraesthetica in a child. J Pediatr. 1978;93(2):325-326. doi:10.1016/s0022-3476(78)80566-6
5. Harney D, Patijn J. Meralgia paresthetica: diagnosis and management strategies. Pain Med. 2007;8(8):669-677. doi:10.1111/j.1526-4637.2006.00227.x
6. Berini SE, Spinner RJ, Jentoft ME, et al. Chronic meralgia paresthetica and neurectomy: a clinical pathologic study. Neurology. 2014;82(17):1551-1555. doi:10.1212/WNL.0000000000000367
7. Payne RA, Harbaugh K, Specht CS, Rizk E. Correlation of histopathology and clinical symptoms in meralgia paresthetica. Cureus. 2017;9(10):e1789. Published 2017 Oct 20. doi:10.7759/cureus.1789
8. Boyce JR. Meralgia paresthetica and tight trousers. JAMA. 1984;251(12):1553.
9. Orton D. Meralgia paresthetica from a wallet. JAMA. 1984;252(24):3368.
10. Fargo MV, Konitzer LN. Meralgia paresthetica due to body armor wear in U.S. soldiers serving in Iraq: a case report and review of the literature. Mil Med. 2007;172(6):663-665. doi:10.7205/milmed.172.6.663
11. Korkmaz N, Ozçakar L. Meralgia paresthetica in a policeman: the belt or the gun. Plast Reconstr Surg. 2004;114(4):1012-1013. doi:10.1097/01.prs.0000138706.86633.01
12. Gooding MS, Evangelista V, Pereira L. Carpal Tunnel Syndrome and Meralgia Paresthetica in Pregnancy. Obstet Gynecol Surv. 2020;75(2):121-126. doi:10.1097/OGX.0000000000000745
13. Pauwels A, Amarenco P, Chazouillères O, Pigot F, Calmus Y, Lévy VG. Une complication rare et méconnue de l’ascite: la méralgie paresthésique [Unusual and unknown complication of ascites: meralgia paresthetica]. Gastroenterol Clin Biol. 1990;14(3):295.
14. Braddom RL. L2 rather than L1 radiculopathy mimics meralgia paresthetica. Muscle Nerve. 2010;42(5):842. doi:10.1002/mus.21826
15. Suber DA, Massey EW. Pelvic mass presenting as meralgia paresthetica. Obstet Gynecol. 1979;53(2):257-258.
16. Flowers RS. Meralgia paresthetica. A clue to retroperitoneal malignant tumor. Am J Surg. 1968;116(1):89-92. doi:10.1016/0002-9610(68)90423-6
17. Yi TI, Yoon TH, Kim JS, Lee GE, Kim BR. Femoral neuropathy and meralgia paresthetica secondary to an iliacus hematoma. Ann Rehabil Med. 2012;36(2):273-277. doi:10.5535/arm.2012.36.2.273
18. Lee ATJ, Thway K, Huang PH, Jones RL. Clinical and molecular spectrum of liposarcoma. J Clin Oncol. 2018;36(2):151-159. doi:10.1200/JCO.2017.74.9598
19. O’Sullivan ED, Schofield SJ. Cognitive bias in clinical medicine. J R Coll Physicians Edinb. 2018;48(3):225-232. doi:10.4997/JRCPE.2018.306
20. Bickley, LS. Bates’ Guide to Physical Examination and History Taking. 12th Edition. Wolters Kluwer Health/Lippincott Williams and Wilkins; 2016.
21. Bhavsar AS, Verma S, Lamba R, Lall CG, Koenigsknecht V, Rajesh A. Abdominal manifestations of neurologic disorders. Radiographics. 2013;33(1):135-153. doi:10.1148/rg.331125097
22. Dureja GP, Gulaya V, Jayalakshmi TS, Mandal P. Management of meralgia paresthetica: a multimodality regimen. Anesth Analg. 1995;80(5):1060-1061. doi:10.1097/00000539-199505000-00043
23. Patijn J, Mekhail N, Hayek S, Lataster A, van Kleef M, Van Zundert J. Meralgia paresthetica. Pain Pract. 2011;11(3):302-308. doi:10.1111/j.1533-2500.2011.00458.x24. Ivins GK. Meralgia paresthetica, the elusive diagnosis: clinical experience with 14 adult patients. Ann Surg. 2000;232(2):281-286. doi:10.1097/00000658-200008000-00019
25. Munin MA, Goerner MS, Raggio I, et al. A rare cause of dyspnea: undifferentiated pleomorphic sarcoma in the left atrium. Cardiol Res. 2017;8(5):241-245. doi:10.14740/cr590w
26. Nguyen A, Awad WI. Cardiac sarcoma arising from malignant transformation of a preexisting atrial myxoma. Ann Thorac Surg. 2016;101(4):1571-1573. doi:10.1016/j.athoracsur.2015.05.129
27. Jiang S, Li J, Zeng Q, Liang J. Pulmonary artery intimal sarcoma misdiagnosed as pulmonary embolism: a case report. Oncol Lett. 2017;13(4):2713-2716. doi:10.3892/ol.2017.5775
28. Cojocaru A, Oliveira PJ, Pellecchia C. A pleural presentation of a rare soft tissue sarcoma. Am J Resp Crit Care Med. 2012;185:A5201. doi:10.1164/ajrccm-conference.2012.185.1_MeetingAbstracts.A5201
29. Grossman MG, Ducey SA, Nadler SS, Levy AS. Meralgia paresthetica: diagnosis and treatment. J Am Acad Orthop Surg. 2001;9(5):336-344. doi:10.5435/00124635-200109000-00007
30. Cheatham SW, Kolber MJ, Salamh PA. Meralgia paresthetica: a review of the literature. Int J Sports Phys Ther. 2013;8(6):883-893.
31. Nouraei SA, Anand B, Spink G, O’Neill KS. A novel approach to the diagnosis and management of meralgia paresthetica. Neurosurgery. 2007;60(4):696-700. doi:10.1227/01.NEU.0000255392.69914.F7
32. Antunes PE, Antunes MJ. Meralgia paresthetica after aortic valve surgery. J Heart Valve Dis. 1997;6(6):589-590.
33. Reddy YM, Singh D, Chikkam V, et al. Postprocedural neuropathy after atrial fibrillation ablation. J Interv Card Electrophysiol. 2013;36(3):279-285. doi:10.1007/s10840-012-9724-z
34. Butler R, Webster MW. Meralgia paresthetica: an unusual complication of cardiac catheterization via the femoral artery. Catheter Cardiovasc Interv. 2002;56(1):69-71. doi:10.1002/ccd.10149
35. Jellish WS, Oftadeh M. Peripheral nerve injury in cardiac surgery. J Cardiothorac Vasc Anesth. 2018;32(1):495-511. doi:10.1053/j.jvca.2017.08.030
36. Parsonnet V, Karasakalides A, Gielchinsky I, Hochberg M, Hussain SM. Meralgia paresthetica after coronary bypass surgery. J Thorac Cardiovasc Surg. 1991;101(2):219-221.
37. Macgregor AM, Thoburn EK. Meralgia paresthetica following bariatric surgery. Obes Surg. 1999;9(4):364-368. doi:10.1381/096089299765552945
38. Grace DM. Meralgia paresthetica after gastroplasty for morbid obesity. Can J Surg. 1987;30(1):64-65.
39. Polidori L, Magarelli M, Tramutoli R. Meralgia paresthetica as a complication of laparoscopic appendectomy. Surg Endosc. 2003;17(5):832. doi:10.1007/s00464-002-4279-1
40. Yamout B, Tayyim A, Farhat W. Meralgia paresthetica as a complication of laparoscopic cholecystectomy. Clin Neurol Neurosurg. 1994;96(2):143-144. doi:10.1016/0303-8467(94)90048-5
41. Broin EO, Horner C, Mealy K, et al. Meralgia paraesthetica following laparoscopic inguinal hernia repair. an anatomical analysis. Surg Endosc. 1995;9(1):76-78. doi:10.1007/BF00187893
42. Eubanks S, Newman L 3rd, Goehring L, et al. Meralgia paresthetica: a complication of laparoscopic herniorrhaphy. Surg Laparosc Endosc. 1993;3(5):381-385.
43. Atamaz F, Hepgüler S, Karasu Z, Kilic M. Meralgia paresthetica after liver transplantation: a case report. Transplant Proc. 2005;37(10):4424-4425. doi:10.1016/j.transproceed.2005.11.047
44. Chung KH, Lee JY, Ko TK, et al. Meralgia paresthetica affecting parturient women who underwent cesarean section -a case report-. Korean J Anesthesiol. 2010;59 Suppl(Suppl):S86-S89. doi:10.4097/kjae.2010.59.S.S86
45. Hutchins FL Jr, Huggins J, Delaney ML. Laparoscopic myomectomy-an unusual cause of meralgia paresthetica. J Am Assoc Gynecol Laparosc. 1998;5(3):309-311. doi:10.1016/s1074-3804(98)80039-x
46. Jones CD, Guiot L, Portelli M, Bullen T, Skaife P. Two interesting cases of meralgia paraesthetica. Pain Physician. 2017;20(6):E987-E989.
47. Peters G, Larner AJ. Meralgia paresthetica following gynecologic and obstetric surgery. Int J Gynaecol Obstet. 2006;95(1):42-43. doi:10.1016/j.ijgo.2006.05.025
48. Kvarnström N, Järvholm S, Johannesson L, Dahm-Kähler P, Olausson M, Brännström M. Live donors of the initial observational study of uterus transplantation-psychological and medical follow-up until 1 year after surgery in the 9 cases. Transplantation. 2017;101(3):664-670. doi:10.1097/TP.0000000000001567
49. Goulding K, Beaulé PE, Kim PR, Fazekas A. Incidence of lateral femoral cutaneous nerve neuropraxia after anterior approach hip arthroplasty. Clin Orthop Relat Res. 2010;468(9):2397-2404. doi:10.1007/s11999-010-1406-5
50. Yamamoto T, Nagira K, Kurosaka M. Meralgia paresthetica occurring 40 years after iliac bone graft harvesting: case report. Neurosurgery. 2001;49(6):1455-1457. doi:10.1097/00006123-200112000-00028
51. Roqueplan F, Porcher R, Hamzé B, et al. Long-term results of percutaneous resection and interstitial laser ablation of osteoid osteomas. Eur Radiol. 2010;20(1):209-217. doi:10.1007/s00330-009-1537-9
52. Gupta A, Muzumdar D, Ramani PS. Meralgia paraesthetica following lumbar spine surgery: a study in 110 consecutive surgically treated cases. Neurol India. 2004;52(1):64-66.
53. Yang SH, Wu CC, Chen PQ. Postoperative meralgia paresthetica after posterior spine surgery: incidence, risk factors, and clinical outcomes. Spine (Phila Pa 1976). 2005;30(18):E547-E550. doi:10.1097/01.brs.0000178821.14102.9d
54. Tejwani SG, Scaduto AA, Bowen RE. Transient meralgia paresthetica after pediatric posterior spine fusion. J Pediatr Orthop. 2006;26(4):530-533. doi:10.1097/01.bpo.0000217721.95480.9e
55. Peker S, Ay B, Sun I, Ozgen S, Pamir M. Meralgia paraesthetica: complications of prone position during lumbar disc surgery. Internet J Anesthesiol. 2003;8(1):24-29.
Photographic Confirmation of Biopsy Sites Saves Lives
Quality photographic documentation of lesions prior to biopsy can decrease the risk of wrong site surgery, improve patient care, and save lives.
Preventable errors by health care workers are widespread and cause significant morbidity and mortality. Wrong site surgery (WSS) is a preventable error that causes harm through both the direct insult of surgery and propagation of the untreated initial problem. WSS also can cause poor patient outcomes, low morale, malpractice claims, and increased costs to the health care system. The estimated median prevalence of WSS across all specialties is 9 events per 1,000,000 surgical procedures, and an institutional study of 112,500 surgical procedures reported 1 wrong-site event, which involved removing the incorrect skin lesion and not removing the intended lesion.1,2
Though the prevalence is low when examining all specialties together, dermatology is also susceptible to WSS.3 Watson and colleagues demonstrated that 31% of intervention errors were due to WSS and suggested that prebiopsy photography helps decrease errors.4 Thus, the American Academy of Dermatology has emphasized the importance of reducing WSS.5 A study by Nijhawan and colleagues found that 25% of patients receiving Mohs surgery at a private single cancer center could not identify their biopsy location because the duration between biopsy and surgery allowed biopsy sites to heal well, which made finding the lesion difficult.6
Risk factors for WSS include having multiple health care providers (HCPs) living remote from the surgery location involved in the case, being a traveling veteran, receiving care at multiple facilities inside and outside the US Department of Veterans Affairs (VA) system, mislabeling photographs or specimens, and photographs not taken at time of biopsy and too close with no frame of reference to assist in finding the correct site. The VA electronic health record (EHR) is not integrated with outside facility EHRs, and the Office of Community Care (OCC) at the VA is responsible for obtaining copies of outside records. If unsuccessful, the HCP and/or patient must provide the records. Frequently, records are not received or require multiple attempts to be obtained. This mostly affects veterans receiving care at multiple facilities inside and outside the VA system as the lack of or timely receipt of past health records could increase the risk for WSS.
To combat WSS, some institutions have implemented standardized protocols requiring photographic documentation of lesions before biopsy so that the surgeon can properly identify the correct site prior to operating.7 Fortunately, recent advances in technology have made it easier to provide photographic documentation of skin lesions. Highsmith and colleagues highlighted use of smartphones to avoid WSS in dermatology.7 Despite these advances, photographic documentation of lesions is not universal. A study by Rossy and colleagues found that less than half of patients referred for Mohs surgery had clear documentation of the biopsy site with photography, diagram, or measurements, and of those documented, only a small fraction used photographs.8
Photographic documentation is not currently required by the VA, increasing the risk of WSS. About 20% of the ~150 VA dermatology departments nationwide are associated with a dermatology residency program and have implemented photographic documentation of lesions before biopsy. The other 80% of departments may not be using photographic documentation. The following 3 cases experienced by the authors highlight instances of how quality photographic documentation of lesions prior to biopsy can improve patient care and save lives. Then, we propose a photographic documentation protocol for VA dermatology departments to follow based on the photographic standards outlined by the American Society for Dermatologic Surgery.9
Case 1 Presentation
A 36-year-old traveling veteran who relocates frequently and receives care at multiple VA medical centers (VAMCs) presented for excision of a melanoma. The patient had been managed at another VAMC where the lesion was biopsied in September 2016. He presented to the Orlando, Florida, VAMC dermatology clinic 5 months later with the photographs of his biopsy sites along with the biopsy reports. The patient had 6 biopsies labeled A through F. Lesion A at the right mid back was positive for melanoma (Figure 1), whereas lesion C on the mid lower back was not cancerous (Figure 2). On examination of the patient’s back, he had numerous moles and scars. The initial receiving HCP circled and photographed the scar presumed to be the melanoma on the mid lower back (Figure 3).
On the day of surgery, the surgeon routinely checked the biopsy report as well as the photograph from the patient’s most recent HCP visit. The surgeon noted that biopsy A (right mid back) on the pathology report had been identified as the melanoma; however, biopsy C (mid lower back) was circled and presumed to be the melanoma in the recent photograph by the receiving HCP—a nurse practitioner. The surgeon compared the initial photos from the referring VAMC with those from the receiving HCP and subsequently matched biopsy A (melanoma) with the correct location on the right mid back.
This discrepancy was explained to the patient with photographic confirmation, allowing for agreement on the correct site before the surgery. The pathology results of the surgical excision confirmed melanoma in the specimen and clear margins. Thus, the correct site was operated on.
Case 2 Presentation
A veteran aged 86 years with a medical history of a double transplant and long-term immunosuppression leading to numerous skin cancers was referred for surgical excision of a confirmed squamous cell carcinoma (SCC) on the left upper back. On the day of surgery, the biopsy site could not be identified clearly due to numerous preexisting scars (Figure 4). No photograph of the original biopsy site was available. The referring HCP was called to the bedside to assist in identifying the biopsy site but also was unable to clearly identify the site. This was explained to the patient. As 2-person confirmation was unsuccessful, conservative treatment was used with patient consent. The patient has since had subsequent close follow-up to monitor for recurrence, as SCC in transplant patients can display aggressive growth and potential for metastasis.
Case 3 Presentation
A veteran was referred for surgical excision of a nonmelanoma skin cancer. The biopsy was completed well in advance of the anticipated surgery day. On the day of surgery, the site could not be detected as it healed well after the biopsy. Although a clinical photograph was available, it was taken too close-up to find a frame of reference for identifying the location of the biopsy site. The referring HCP was called to the bedside to assist in identification of the biopsy site, but 2-person confirmation was unsuccessful. This was explained to the patient, and with his consent, the HCPs agreed on conservative treatment and close follow-up.
Discussion
To prevent and minimize poor outcomes associated with WSS, the health care team should routinely document the lesion location in detail before the biopsy. Many HCPs believe a preoperative photograph is the best method for documentation. As demonstrated in the third case presentation, photographs must be taken at a distance that includes nearby anatomic landmarks for reference. It is suggested that the providers obtain 2 images, one that is far enough to include landmarks, and one that is close enough to clearly differentiate the targeted lesion from others.10
Although high-resolution digital cameras are preferred, mobile phones also can be used if they provide quality images. As phones with built-in cameras are ubiquitous, they offer a quick and easy method of photographic documentation. St John and colleagues also presented the possibility of having patients keep pictures of the lesion on their phones, as this removes potential privacy concerns and facilitates easy transportation of information between HCPs.10 If it is discovered that a photograph was not taken at the time of biopsy, our practice contacts the patient and asks them to photograph and circle the biopsy site using their mobile phone or camera and bring it to the surgery appointment. We propose a VA protocol for photographic documentation of biopsy sites (Table).

HCPs who are not comfortable with technology may be hesitant to use photographic documentation using a smartphone or camera. Further, HCPs often face time constraints, and taking photographs and uploading them to the EHR could decrease patient contact time. Therefore, photographic documentation presents an opportunity for a team approach to patient-centered care: Nursing and other medical staff can assist with these duties and learn the proper photographic documentation of biopsy sites. Using phone or tablet applications that provide rapid photographic documentation and uploading to the EHR also would facilitate universal use of photographic documentation.
If a HCP is uncomfortable or unable to use photography to document lesions, alternative strategies for documenting lesions exist, including diagrams, anatomic landmarks, ultraviolet (UV) fluorescent tattoos, and patient identification of lesions.10 In the diagram method, a HCP marks the lesion location on a diagram of the body preferably with a short description of the lesion’s location and/or characteristics.11 The diagram should be uploaded into the EHR. There are other methods for documenting lesion location relative to anatomic landmarks. Triangulation involves documenting distance between the lesion and 3 distinct anatomic locations.10 UV fluorescent tattooing involves putting UV tattoo dye in the biopsy site and locating the dye using a Wood lamp at the time of surgery. The lamp was used in a single case report of a patient with recurrent basal cell carcinoma.12 Patient identification of lesions by phone applications that allow patients to track their lesion, a phone selfie of the biopsy site, or a direct account of a lesion can be used to confirm lesion location based on the other methods mentioned.10
Patients often are poorly adherent to instructions aimed at reducing the risk of WSS. In a study that asked patients undergoing elective foot or ankle surgery to mark the foot not being operated on, 41% of patients were either partially or nonadherent with this request.13 Educating patients on the importance of lesion self-identification has the potential to improve identification of biopsy location and prevent WSS. Nursing and medical staff can provide patient education while photographing the biopsy site including taking a photograph with the patient’s cell phone for their records.
Due to subsequent morbidity and mortality that can result from WSS, photographic confirmation of biopsy sites is a step that surgeons can take to ensure identification of the correct site prior to surgery. Case 1 provides an example of how photographs taken prior to biopsy can prevent WSS. In a disease such as melanoma, photographs are particularly important, as insufficient treatment can lead to fatal metastases. To increase quality of care, all available photographs should be reviewed, especially in cases where the pathology report does not match the clinical presentation.
If WSS occurs, HCPs may be hesitant to disclose their mistakes due to potential lawsuits, the possibility that disclosure may inadvertently harm the patient, and their relative inexperience in and training regarding disclosure skills.14 Surgeons who perform WSS may receive severe penalties from state licensing boards, including suspension of medical license. Financially, many insurers will not compensate providers for WSS. Also, many incidents of WSS result in a malpractice claim, with about 80% of those cases resulting in a malpractice award.15 However, it is important that HCPs are open with their patients regarding WSS.
As demonstrated in case presentations 2 and 3, having 2-person confirmation and patient confirmation before to surgery is important in preventing WSS for patients who have poor documentation of biopsy sites. In cases where agreement is not achieved, HCPs can consider several other options to help identify lesions. Dermabrasion and alcohol wipes are options.10 Dermabrasion uses friction to expose surgical sights that have healed, scarred, or been hidden by sun damage.10 Alcohol wipes remove surface scale and crust, creating a glisten with tangential lighting that highlights surface irregularities. Anesthesia injection prior to surgery creates a blister at the location of the cancer. This is because skin cancer weakens the attachments between keratinocytes, and as a result, the hydrostatic pressure from the anesthesia favorably blisters the malignancy location.10,16
Dermoscopy is another strategy shown to help identify scar margins.10,17 Under dermoscopy, a scar demonstrates a white-pink homogenous patch with underlying vessels, whereas basal cell carcinoma remnants include blue-gray ovoid nests and globules, telangiectasias, spoke wheel and leaflike structures.17 As a final option, HCPs can perform an additional biopsy of potential cancer locations to find the lesion again.10 If the lesions cannot be identified, HCPs should consider conservative measures or less invasive treatments with close and frequent follow-up.
Conclusions
The cases described here highlight how the lack of proper photographic documentation can prevent the use of curative surgical treatment. In order to reduce WSS and improve quality care, HCPs must continue to take steps and create safeguards to minimize risk. Proper documentation of lesions prior to biopsy provides an effective route to reduce incidence of WSS. If the biopsy site cannot be found, various strategies to properly identify the site can be employed. If WSS occurs, it is important that HCPs provide full disclosure to patients. With a growing emphasis on patient safety measures and advances in technology, HCPs are becoming increasingly cognizant about the most effective ways to optimize patient care, and it is anticipated that this will result in a decrease in morbidity and mortality.
1. Hempel S, Maggard-Gibbons M, Nguyen DK, et al. Wrong-site surgery, retained surgical items, and surgical fires: a systematic review of surgical never events. JAMA Surg. 2015;150(8):796-805. doi:10.1001/jamasurg.2015.0301
2. Knight N, Aucar J. Use of an anatomic marking form as an alternative to the Universal Protocol for Preventing Wrong Site, Wrong Procedure and Wrong Person Surgery. Am J Surg. 2010;200(6):803-809. doi:10.1016/j.amjsurg.2010.06.010
3. Elston DM, Stratman EJ, Miller SJ. Skin biopsy: biopsy issues in specific diseases [published correction appears in J Am Acad Dermatol. 2016 Oct;75(4):854]. J Am Acad Dermatol. 2016;74(1):1-18. doi:10.1016/j.jaad.2015.06.033
4. Watson AJ, Redbord K, Taylor JS, Shippy A, Kostecki J, Swerlick R. Medical error in dermatology practice: development of a classification system to drive priority setting in patient safety efforts. J Am Acad Dermatol. 2013;68(5):729-737. doi:10.1016/j.jaad.2012.10.058
5. Elston DM, Taylor JS, Coldiron B, et al. Patient safety: Part I. Patient safety and the dermatologist. J Am Acad Dermatol. 2009;61(2):179-191. doi:10.1016/j.jaad.2009.04.056
6. Nijhawan RI, Lee EH, Nehal KS. Biopsy site selfies--a quality improvement pilot study to assist with correct surgical site identification. Dermatol Surg. 2015;41(4):499-504. doi:10.1097/DSS.0000000000000305
7. Highsmith JT, Weinstein DA, Highsmith MJ, Etzkorn JR. BIOPSY 1-2-3 in dermatologic surgery: improving smartphone use to avoid wrong-site surgery. Technol Innov. 2016;18(2-3):203-206. doi:10.21300/18.2-3.2016.203
8. Rossy KM, Lawrence N. Difficulty with surgical site identification: what role does it play in dermatology? J Am Acad Dermatol. 2012;67(2):257-261. doi:10.1016/j.jaad.2012.02.034
9. American Society for Dermatologic Surgery. Photographic standards in dermatologic surgery poster. Accessed April 12, 2021. https://www.asds.net/medical-professionals/members-resources/product-details/productname/photographic-standards-poster
10. St John J, Walker J, Goldberg D, Maloney ME. Avoiding Medical Errors in Cutaneous Site Identification: A Best Practices Review. Dermatol Surg. 2016;42(4):477-484. doi:10.1097/DSS.0000000000000683
11. Alam M, Lee A, Ibrahimi OA, et al. A multistep approach to improving biopsy site identification in dermatology: physician, staff, and patient roles based on a Delphi consensus. JAMA Dermatol. 2014;150(5):550-558. doi:10.1001/jamadermatol.2013.9804
12. Chuang GS, Gilchrest BA. Ultraviolet-fluorescent tattoo location of cutaneous biopsy site. Dermatol Surg. 2012;38(3):479-483. doi:10.1111/j.1524-4725.2011.02238.x
13. DiGiovanni CW, Kang L, Manuel J. Patient compliance in avoiding wrong-site surgery. J Bone Joint Surg Am. 2003;85(5):815-819. doi:10.2106/00004623-200305000-00007
14. Gallagher TH. A 62-year-old woman with skin cancer who experienced wrong-site surgery: review of medical error. JAMA. 2009;302(6):669-677. doi:10.1001/jama.2009.1011
15. Mulloy DF, Hughes RG. Wrong-site surgery: a preventable medical error. In: Hughes RG, ed. Patient Safety and Quality: An Evidence-Based Handbook for Nurses. Agency for Healthcare Research and Quality (US); 2008:chap 36. Accessed April 23, 2021. https://www.ncbi.nlm.nih.gov/books/NBK2678
16. Zaiac M, Tongdee E, Porges L, Touloei K, Prodanovich S. Anesthetic blister induction to identify biopsy site prior to Mohs surgery. J Drugs Dermatol. 2015;14(5):446-447.
17. Jawed SI, Goldberg LH, Wang SQ. Dermoscopy to identify biopsy sites before Mohs surgery. Dermatol Surg. 2014;40(3):334-337. doi:10.1111/dsu.12422
Quality photographic documentation of lesions prior to biopsy can decrease the risk of wrong site surgery, improve patient care, and save lives.
Quality photographic documentation of lesions prior to biopsy can decrease the risk of wrong site surgery, improve patient care, and save lives.
Preventable errors by health care workers are widespread and cause significant morbidity and mortality. Wrong site surgery (WSS) is a preventable error that causes harm through both the direct insult of surgery and propagation of the untreated initial problem. WSS also can cause poor patient outcomes, low morale, malpractice claims, and increased costs to the health care system. The estimated median prevalence of WSS across all specialties is 9 events per 1,000,000 surgical procedures, and an institutional study of 112,500 surgical procedures reported 1 wrong-site event, which involved removing the incorrect skin lesion and not removing the intended lesion.1,2
Though the prevalence is low when examining all specialties together, dermatology is also susceptible to WSS.3 Watson and colleagues demonstrated that 31% of intervention errors were due to WSS and suggested that prebiopsy photography helps decrease errors.4 Thus, the American Academy of Dermatology has emphasized the importance of reducing WSS.5 A study by Nijhawan and colleagues found that 25% of patients receiving Mohs surgery at a private single cancer center could not identify their biopsy location because the duration between biopsy and surgery allowed biopsy sites to heal well, which made finding the lesion difficult.6
Risk factors for WSS include having multiple health care providers (HCPs) living remote from the surgery location involved in the case, being a traveling veteran, receiving care at multiple facilities inside and outside the US Department of Veterans Affairs (VA) system, mislabeling photographs or specimens, and photographs not taken at time of biopsy and too close with no frame of reference to assist in finding the correct site. The VA electronic health record (EHR) is not integrated with outside facility EHRs, and the Office of Community Care (OCC) at the VA is responsible for obtaining copies of outside records. If unsuccessful, the HCP and/or patient must provide the records. Frequently, records are not received or require multiple attempts to be obtained. This mostly affects veterans receiving care at multiple facilities inside and outside the VA system as the lack of or timely receipt of past health records could increase the risk for WSS.
To combat WSS, some institutions have implemented standardized protocols requiring photographic documentation of lesions before biopsy so that the surgeon can properly identify the correct site prior to operating.7 Fortunately, recent advances in technology have made it easier to provide photographic documentation of skin lesions. Highsmith and colleagues highlighted use of smartphones to avoid WSS in dermatology.7 Despite these advances, photographic documentation of lesions is not universal. A study by Rossy and colleagues found that less than half of patients referred for Mohs surgery had clear documentation of the biopsy site with photography, diagram, or measurements, and of those documented, only a small fraction used photographs.8
Photographic documentation is not currently required by the VA, increasing the risk of WSS. About 20% of the ~150 VA dermatology departments nationwide are associated with a dermatology residency program and have implemented photographic documentation of lesions before biopsy. The other 80% of departments may not be using photographic documentation. The following 3 cases experienced by the authors highlight instances of how quality photographic documentation of lesions prior to biopsy can improve patient care and save lives. Then, we propose a photographic documentation protocol for VA dermatology departments to follow based on the photographic standards outlined by the American Society for Dermatologic Surgery.9
Case 1 Presentation
A 36-year-old traveling veteran who relocates frequently and receives care at multiple VA medical centers (VAMCs) presented for excision of a melanoma. The patient had been managed at another VAMC where the lesion was biopsied in September 2016. He presented to the Orlando, Florida, VAMC dermatology clinic 5 months later with the photographs of his biopsy sites along with the biopsy reports. The patient had 6 biopsies labeled A through F. Lesion A at the right mid back was positive for melanoma (Figure 1), whereas lesion C on the mid lower back was not cancerous (Figure 2). On examination of the patient’s back, he had numerous moles and scars. The initial receiving HCP circled and photographed the scar presumed to be the melanoma on the mid lower back (Figure 3).
On the day of surgery, the surgeon routinely checked the biopsy report as well as the photograph from the patient’s most recent HCP visit. The surgeon noted that biopsy A (right mid back) on the pathology report had been identified as the melanoma; however, biopsy C (mid lower back) was circled and presumed to be the melanoma in the recent photograph by the receiving HCP—a nurse practitioner. The surgeon compared the initial photos from the referring VAMC with those from the receiving HCP and subsequently matched biopsy A (melanoma) with the correct location on the right mid back.
This discrepancy was explained to the patient with photographic confirmation, allowing for agreement on the correct site before the surgery. The pathology results of the surgical excision confirmed melanoma in the specimen and clear margins. Thus, the correct site was operated on.
Case 2 Presentation
A veteran aged 86 years with a medical history of a double transplant and long-term immunosuppression leading to numerous skin cancers was referred for surgical excision of a confirmed squamous cell carcinoma (SCC) on the left upper back. On the day of surgery, the biopsy site could not be identified clearly due to numerous preexisting scars (Figure 4). No photograph of the original biopsy site was available. The referring HCP was called to the bedside to assist in identifying the biopsy site but also was unable to clearly identify the site. This was explained to the patient. As 2-person confirmation was unsuccessful, conservative treatment was used with patient consent. The patient has since had subsequent close follow-up to monitor for recurrence, as SCC in transplant patients can display aggressive growth and potential for metastasis.
Case 3 Presentation
A veteran was referred for surgical excision of a nonmelanoma skin cancer. The biopsy was completed well in advance of the anticipated surgery day. On the day of surgery, the site could not be detected as it healed well after the biopsy. Although a clinical photograph was available, it was taken too close-up to find a frame of reference for identifying the location of the biopsy site. The referring HCP was called to the bedside to assist in identification of the biopsy site, but 2-person confirmation was unsuccessful. This was explained to the patient, and with his consent, the HCPs agreed on conservative treatment and close follow-up.
Discussion
To prevent and minimize poor outcomes associated with WSS, the health care team should routinely document the lesion location in detail before the biopsy. Many HCPs believe a preoperative photograph is the best method for documentation. As demonstrated in the third case presentation, photographs must be taken at a distance that includes nearby anatomic landmarks for reference. It is suggested that the providers obtain 2 images, one that is far enough to include landmarks, and one that is close enough to clearly differentiate the targeted lesion from others.10
Although high-resolution digital cameras are preferred, mobile phones also can be used if they provide quality images. As phones with built-in cameras are ubiquitous, they offer a quick and easy method of photographic documentation. St John and colleagues also presented the possibility of having patients keep pictures of the lesion on their phones, as this removes potential privacy concerns and facilitates easy transportation of information between HCPs.10 If it is discovered that a photograph was not taken at the time of biopsy, our practice contacts the patient and asks them to photograph and circle the biopsy site using their mobile phone or camera and bring it to the surgery appointment. We propose a VA protocol for photographic documentation of biopsy sites (Table).
HCPs who are not comfortable with technology may be hesitant to use photographic documentation using a smartphone or camera. Further, HCPs often face time constraints, and taking photographs and uploading them to the EHR could decrease patient contact time. Therefore, photographic documentation presents an opportunity for a team approach to patient-centered care: Nursing and other medical staff can assist with these duties and learn the proper photographic documentation of biopsy sites. Using phone or tablet applications that provide rapid photographic documentation and uploading to the EHR also would facilitate universal use of photographic documentation.
If a HCP is uncomfortable or unable to use photography to document lesions, alternative strategies for documenting lesions exist, including diagrams, anatomic landmarks, ultraviolet (UV) fluorescent tattoos, and patient identification of lesions.10 In the diagram method, a HCP marks the lesion location on a diagram of the body preferably with a short description of the lesion’s location and/or characteristics.11 The diagram should be uploaded into the EHR. There are other methods for documenting lesion location relative to anatomic landmarks. Triangulation involves documenting distance between the lesion and 3 distinct anatomic locations.10 UV fluorescent tattooing involves putting UV tattoo dye in the biopsy site and locating the dye using a Wood lamp at the time of surgery. The lamp was used in a single case report of a patient with recurrent basal cell carcinoma.12 Patient identification of lesions by phone applications that allow patients to track their lesion, a phone selfie of the biopsy site, or a direct account of a lesion can be used to confirm lesion location based on the other methods mentioned.10
Patients often are poorly adherent to instructions aimed at reducing the risk of WSS. In a study that asked patients undergoing elective foot or ankle surgery to mark the foot not being operated on, 41% of patients were either partially or nonadherent with this request.13 Educating patients on the importance of lesion self-identification has the potential to improve identification of biopsy location and prevent WSS. Nursing and medical staff can provide patient education while photographing the biopsy site including taking a photograph with the patient’s cell phone for their records.
Due to subsequent morbidity and mortality that can result from WSS, photographic confirmation of biopsy sites is a step that surgeons can take to ensure identification of the correct site prior to surgery. Case 1 provides an example of how photographs taken prior to biopsy can prevent WSS. In a disease such as melanoma, photographs are particularly important, as insufficient treatment can lead to fatal metastases. To increase quality of care, all available photographs should be reviewed, especially in cases where the pathology report does not match the clinical presentation.
If WSS occurs, HCPs may be hesitant to disclose their mistakes due to potential lawsuits, the possibility that disclosure may inadvertently harm the patient, and their relative inexperience in and training regarding disclosure skills.14 Surgeons who perform WSS may receive severe penalties from state licensing boards, including suspension of medical license. Financially, many insurers will not compensate providers for WSS. Also, many incidents of WSS result in a malpractice claim, with about 80% of those cases resulting in a malpractice award.15 However, it is important that HCPs are open with their patients regarding WSS.
As demonstrated in case presentations 2 and 3, having 2-person confirmation and patient confirmation before to surgery is important in preventing WSS for patients who have poor documentation of biopsy sites. In cases where agreement is not achieved, HCPs can consider several other options to help identify lesions. Dermabrasion and alcohol wipes are options.10 Dermabrasion uses friction to expose surgical sights that have healed, scarred, or been hidden by sun damage.10 Alcohol wipes remove surface scale and crust, creating a glisten with tangential lighting that highlights surface irregularities. Anesthesia injection prior to surgery creates a blister at the location of the cancer. This is because skin cancer weakens the attachments between keratinocytes, and as a result, the hydrostatic pressure from the anesthesia favorably blisters the malignancy location.10,16
Dermoscopy is another strategy shown to help identify scar margins.10,17 Under dermoscopy, a scar demonstrates a white-pink homogenous patch with underlying vessels, whereas basal cell carcinoma remnants include blue-gray ovoid nests and globules, telangiectasias, spoke wheel and leaflike structures.17 As a final option, HCPs can perform an additional biopsy of potential cancer locations to find the lesion again.10 If the lesions cannot be identified, HCPs should consider conservative measures or less invasive treatments with close and frequent follow-up.
Conclusions
The cases described here highlight how the lack of proper photographic documentation can prevent the use of curative surgical treatment. In order to reduce WSS and improve quality care, HCPs must continue to take steps and create safeguards to minimize risk. Proper documentation of lesions prior to biopsy provides an effective route to reduce incidence of WSS. If the biopsy site cannot be found, various strategies to properly identify the site can be employed. If WSS occurs, it is important that HCPs provide full disclosure to patients. With a growing emphasis on patient safety measures and advances in technology, HCPs are becoming increasingly cognizant about the most effective ways to optimize patient care, and it is anticipated that this will result in a decrease in morbidity and mortality.
Preventable errors by health care workers are widespread and cause significant morbidity and mortality. Wrong site surgery (WSS) is a preventable error that causes harm through both the direct insult of surgery and propagation of the untreated initial problem. WSS also can cause poor patient outcomes, low morale, malpractice claims, and increased costs to the health care system. The estimated median prevalence of WSS across all specialties is 9 events per 1,000,000 surgical procedures, and an institutional study of 112,500 surgical procedures reported 1 wrong-site event, which involved removing the incorrect skin lesion and not removing the intended lesion.1,2
Though the prevalence is low when examining all specialties together, dermatology is also susceptible to WSS.3 Watson and colleagues demonstrated that 31% of intervention errors were due to WSS and suggested that prebiopsy photography helps decrease errors.4 Thus, the American Academy of Dermatology has emphasized the importance of reducing WSS.5 A study by Nijhawan and colleagues found that 25% of patients receiving Mohs surgery at a private single cancer center could not identify their biopsy location because the duration between biopsy and surgery allowed biopsy sites to heal well, which made finding the lesion difficult.6
Risk factors for WSS include having multiple health care providers (HCPs) living remote from the surgery location involved in the case, being a traveling veteran, receiving care at multiple facilities inside and outside the US Department of Veterans Affairs (VA) system, mislabeling photographs or specimens, and photographs not taken at time of biopsy and too close with no frame of reference to assist in finding the correct site. The VA electronic health record (EHR) is not integrated with outside facility EHRs, and the Office of Community Care (OCC) at the VA is responsible for obtaining copies of outside records. If unsuccessful, the HCP and/or patient must provide the records. Frequently, records are not received or require multiple attempts to be obtained. This mostly affects veterans receiving care at multiple facilities inside and outside the VA system as the lack of or timely receipt of past health records could increase the risk for WSS.
To combat WSS, some institutions have implemented standardized protocols requiring photographic documentation of lesions before biopsy so that the surgeon can properly identify the correct site prior to operating.7 Fortunately, recent advances in technology have made it easier to provide photographic documentation of skin lesions. Highsmith and colleagues highlighted use of smartphones to avoid WSS in dermatology.7 Despite these advances, photographic documentation of lesions is not universal. A study by Rossy and colleagues found that less than half of patients referred for Mohs surgery had clear documentation of the biopsy site with photography, diagram, or measurements, and of those documented, only a small fraction used photographs.8
Photographic documentation is not currently required by the VA, increasing the risk of WSS. About 20% of the ~150 VA dermatology departments nationwide are associated with a dermatology residency program and have implemented photographic documentation of lesions before biopsy. The other 80% of departments may not be using photographic documentation. The following 3 cases experienced by the authors highlight instances of how quality photographic documentation of lesions prior to biopsy can improve patient care and save lives. Then, we propose a photographic documentation protocol for VA dermatology departments to follow based on the photographic standards outlined by the American Society for Dermatologic Surgery.9
Case 1 Presentation
A 36-year-old traveling veteran who relocates frequently and receives care at multiple VA medical centers (VAMCs) presented for excision of a melanoma. The patient had been managed at another VAMC where the lesion was biopsied in September 2016. He presented to the Orlando, Florida, VAMC dermatology clinic 5 months later with the photographs of his biopsy sites along with the biopsy reports. The patient had 6 biopsies labeled A through F. Lesion A at the right mid back was positive for melanoma (Figure 1), whereas lesion C on the mid lower back was not cancerous (Figure 2). On examination of the patient’s back, he had numerous moles and scars. The initial receiving HCP circled and photographed the scar presumed to be the melanoma on the mid lower back (Figure 3).
On the day of surgery, the surgeon routinely checked the biopsy report as well as the photograph from the patient’s most recent HCP visit. The surgeon noted that biopsy A (right mid back) on the pathology report had been identified as the melanoma; however, biopsy C (mid lower back) was circled and presumed to be the melanoma in the recent photograph by the receiving HCP—a nurse practitioner. The surgeon compared the initial photos from the referring VAMC with those from the receiving HCP and subsequently matched biopsy A (melanoma) with the correct location on the right mid back.
This discrepancy was explained to the patient with photographic confirmation, allowing for agreement on the correct site before the surgery. The pathology results of the surgical excision confirmed melanoma in the specimen and clear margins. Thus, the correct site was operated on.
Case 2 Presentation
A veteran aged 86 years with a medical history of a double transplant and long-term immunosuppression leading to numerous skin cancers was referred for surgical excision of a confirmed squamous cell carcinoma (SCC) on the left upper back. On the day of surgery, the biopsy site could not be identified clearly due to numerous preexisting scars (Figure 4). No photograph of the original biopsy site was available. The referring HCP was called to the bedside to assist in identifying the biopsy site but also was unable to clearly identify the site. This was explained to the patient. As 2-person confirmation was unsuccessful, conservative treatment was used with patient consent. The patient has since had subsequent close follow-up to monitor for recurrence, as SCC in transplant patients can display aggressive growth and potential for metastasis.
Case 3 Presentation
A veteran was referred for surgical excision of a nonmelanoma skin cancer. The biopsy was completed well in advance of the anticipated surgery day. On the day of surgery, the site could not be detected as it healed well after the biopsy. Although a clinical photograph was available, it was taken too close-up to find a frame of reference for identifying the location of the biopsy site. The referring HCP was called to the bedside to assist in identification of the biopsy site, but 2-person confirmation was unsuccessful. This was explained to the patient, and with his consent, the HCPs agreed on conservative treatment and close follow-up.
Discussion
To prevent and minimize poor outcomes associated with WSS, the health care team should routinely document the lesion location in detail before the biopsy. Many HCPs believe a preoperative photograph is the best method for documentation. As demonstrated in the third case presentation, photographs must be taken at a distance that includes nearby anatomic landmarks for reference. It is suggested that the providers obtain 2 images, one that is far enough to include landmarks, and one that is close enough to clearly differentiate the targeted lesion from others.10
Although high-resolution digital cameras are preferred, mobile phones also can be used if they provide quality images. As phones with built-in cameras are ubiquitous, they offer a quick and easy method of photographic documentation. St John and colleagues also presented the possibility of having patients keep pictures of the lesion on their phones, as this removes potential privacy concerns and facilitates easy transportation of information between HCPs.10 If it is discovered that a photograph was not taken at the time of biopsy, our practice contacts the patient and asks them to photograph and circle the biopsy site using their mobile phone or camera and bring it to the surgery appointment. We propose a VA protocol for photographic documentation of biopsy sites (Table).
HCPs who are not comfortable with technology may be hesitant to use photographic documentation using a smartphone or camera. Further, HCPs often face time constraints, and taking photographs and uploading them to the EHR could decrease patient contact time. Therefore, photographic documentation presents an opportunity for a team approach to patient-centered care: Nursing and other medical staff can assist with these duties and learn the proper photographic documentation of biopsy sites. Using phone or tablet applications that provide rapid photographic documentation and uploading to the EHR also would facilitate universal use of photographic documentation.
If a HCP is uncomfortable or unable to use photography to document lesions, alternative strategies for documenting lesions exist, including diagrams, anatomic landmarks, ultraviolet (UV) fluorescent tattoos, and patient identification of lesions.10 In the diagram method, a HCP marks the lesion location on a diagram of the body preferably with a short description of the lesion’s location and/or characteristics.11 The diagram should be uploaded into the EHR. There are other methods for documenting lesion location relative to anatomic landmarks. Triangulation involves documenting distance between the lesion and 3 distinct anatomic locations.10 UV fluorescent tattooing involves putting UV tattoo dye in the biopsy site and locating the dye using a Wood lamp at the time of surgery. The lamp was used in a single case report of a patient with recurrent basal cell carcinoma.12 Patient identification of lesions by phone applications that allow patients to track their lesion, a phone selfie of the biopsy site, or a direct account of a lesion can be used to confirm lesion location based on the other methods mentioned.10
Patients often are poorly adherent to instructions aimed at reducing the risk of WSS. In a study that asked patients undergoing elective foot or ankle surgery to mark the foot not being operated on, 41% of patients were either partially or nonadherent with this request.13 Educating patients on the importance of lesion self-identification has the potential to improve identification of biopsy location and prevent WSS. Nursing and medical staff can provide patient education while photographing the biopsy site including taking a photograph with the patient’s cell phone for their records.
Due to subsequent morbidity and mortality that can result from WSS, photographic confirmation of biopsy sites is a step that surgeons can take to ensure identification of the correct site prior to surgery. Case 1 provides an example of how photographs taken prior to biopsy can prevent WSS. In a disease such as melanoma, photographs are particularly important, as insufficient treatment can lead to fatal metastases. To increase quality of care, all available photographs should be reviewed, especially in cases where the pathology report does not match the clinical presentation.
If WSS occurs, HCPs may be hesitant to disclose their mistakes due to potential lawsuits, the possibility that disclosure may inadvertently harm the patient, and their relative inexperience in and training regarding disclosure skills.14 Surgeons who perform WSS may receive severe penalties from state licensing boards, including suspension of medical license. Financially, many insurers will not compensate providers for WSS. Also, many incidents of WSS result in a malpractice claim, with about 80% of those cases resulting in a malpractice award.15 However, it is important that HCPs are open with their patients regarding WSS.
As demonstrated in case presentations 2 and 3, having 2-person confirmation and patient confirmation before to surgery is important in preventing WSS for patients who have poor documentation of biopsy sites. In cases where agreement is not achieved, HCPs can consider several other options to help identify lesions. Dermabrasion and alcohol wipes are options.10 Dermabrasion uses friction to expose surgical sights that have healed, scarred, or been hidden by sun damage.10 Alcohol wipes remove surface scale and crust, creating a glisten with tangential lighting that highlights surface irregularities. Anesthesia injection prior to surgery creates a blister at the location of the cancer. This is because skin cancer weakens the attachments between keratinocytes, and as a result, the hydrostatic pressure from the anesthesia favorably blisters the malignancy location.10,16
Dermoscopy is another strategy shown to help identify scar margins.10,17 Under dermoscopy, a scar demonstrates a white-pink homogenous patch with underlying vessels, whereas basal cell carcinoma remnants include blue-gray ovoid nests and globules, telangiectasias, spoke wheel and leaflike structures.17 As a final option, HCPs can perform an additional biopsy of potential cancer locations to find the lesion again.10 If the lesions cannot be identified, HCPs should consider conservative measures or less invasive treatments with close and frequent follow-up.
Conclusions
The cases described here highlight how the lack of proper photographic documentation can prevent the use of curative surgical treatment. In order to reduce WSS and improve quality care, HCPs must continue to take steps and create safeguards to minimize risk. Proper documentation of lesions prior to biopsy provides an effective route to reduce incidence of WSS. If the biopsy site cannot be found, various strategies to properly identify the site can be employed. If WSS occurs, it is important that HCPs provide full disclosure to patients. With a growing emphasis on patient safety measures and advances in technology, HCPs are becoming increasingly cognizant about the most effective ways to optimize patient care, and it is anticipated that this will result in a decrease in morbidity and mortality.
1. Hempel S, Maggard-Gibbons M, Nguyen DK, et al. Wrong-site surgery, retained surgical items, and surgical fires: a systematic review of surgical never events. JAMA Surg. 2015;150(8):796-805. doi:10.1001/jamasurg.2015.0301
2. Knight N, Aucar J. Use of an anatomic marking form as an alternative to the Universal Protocol for Preventing Wrong Site, Wrong Procedure and Wrong Person Surgery. Am J Surg. 2010;200(6):803-809. doi:10.1016/j.amjsurg.2010.06.010
3. Elston DM, Stratman EJ, Miller SJ. Skin biopsy: biopsy issues in specific diseases [published correction appears in J Am Acad Dermatol. 2016 Oct;75(4):854]. J Am Acad Dermatol. 2016;74(1):1-18. doi:10.1016/j.jaad.2015.06.033
4. Watson AJ, Redbord K, Taylor JS, Shippy A, Kostecki J, Swerlick R. Medical error in dermatology practice: development of a classification system to drive priority setting in patient safety efforts. J Am Acad Dermatol. 2013;68(5):729-737. doi:10.1016/j.jaad.2012.10.058
5. Elston DM, Taylor JS, Coldiron B, et al. Patient safety: Part I. Patient safety and the dermatologist. J Am Acad Dermatol. 2009;61(2):179-191. doi:10.1016/j.jaad.2009.04.056
6. Nijhawan RI, Lee EH, Nehal KS. Biopsy site selfies--a quality improvement pilot study to assist with correct surgical site identification. Dermatol Surg. 2015;41(4):499-504. doi:10.1097/DSS.0000000000000305
7. Highsmith JT, Weinstein DA, Highsmith MJ, Etzkorn JR. BIOPSY 1-2-3 in dermatologic surgery: improving smartphone use to avoid wrong-site surgery. Technol Innov. 2016;18(2-3):203-206. doi:10.21300/18.2-3.2016.203
8. Rossy KM, Lawrence N. Difficulty with surgical site identification: what role does it play in dermatology? J Am Acad Dermatol. 2012;67(2):257-261. doi:10.1016/j.jaad.2012.02.034
9. American Society for Dermatologic Surgery. Photographic standards in dermatologic surgery poster. Accessed April 12, 2021. https://www.asds.net/medical-professionals/members-resources/product-details/productname/photographic-standards-poster
10. St John J, Walker J, Goldberg D, Maloney ME. Avoiding Medical Errors in Cutaneous Site Identification: A Best Practices Review. Dermatol Surg. 2016;42(4):477-484. doi:10.1097/DSS.0000000000000683
11. Alam M, Lee A, Ibrahimi OA, et al. A multistep approach to improving biopsy site identification in dermatology: physician, staff, and patient roles based on a Delphi consensus. JAMA Dermatol. 2014;150(5):550-558. doi:10.1001/jamadermatol.2013.9804
12. Chuang GS, Gilchrest BA. Ultraviolet-fluorescent tattoo location of cutaneous biopsy site. Dermatol Surg. 2012;38(3):479-483. doi:10.1111/j.1524-4725.2011.02238.x
13. DiGiovanni CW, Kang L, Manuel J. Patient compliance in avoiding wrong-site surgery. J Bone Joint Surg Am. 2003;85(5):815-819. doi:10.2106/00004623-200305000-00007
14. Gallagher TH. A 62-year-old woman with skin cancer who experienced wrong-site surgery: review of medical error. JAMA. 2009;302(6):669-677. doi:10.1001/jama.2009.1011
15. Mulloy DF, Hughes RG. Wrong-site surgery: a preventable medical error. In: Hughes RG, ed. Patient Safety and Quality: An Evidence-Based Handbook for Nurses. Agency for Healthcare Research and Quality (US); 2008:chap 36. Accessed April 23, 2021. https://www.ncbi.nlm.nih.gov/books/NBK2678
16. Zaiac M, Tongdee E, Porges L, Touloei K, Prodanovich S. Anesthetic blister induction to identify biopsy site prior to Mohs surgery. J Drugs Dermatol. 2015;14(5):446-447.
17. Jawed SI, Goldberg LH, Wang SQ. Dermoscopy to identify biopsy sites before Mohs surgery. Dermatol Surg. 2014;40(3):334-337. doi:10.1111/dsu.12422
1. Hempel S, Maggard-Gibbons M, Nguyen DK, et al. Wrong-site surgery, retained surgical items, and surgical fires: a systematic review of surgical never events. JAMA Surg. 2015;150(8):796-805. doi:10.1001/jamasurg.2015.0301
2. Knight N, Aucar J. Use of an anatomic marking form as an alternative to the Universal Protocol for Preventing Wrong Site, Wrong Procedure and Wrong Person Surgery. Am J Surg. 2010;200(6):803-809. doi:10.1016/j.amjsurg.2010.06.010
3. Elston DM, Stratman EJ, Miller SJ. Skin biopsy: biopsy issues in specific diseases [published correction appears in J Am Acad Dermatol. 2016 Oct;75(4):854]. J Am Acad Dermatol. 2016;74(1):1-18. doi:10.1016/j.jaad.2015.06.033
4. Watson AJ, Redbord K, Taylor JS, Shippy A, Kostecki J, Swerlick R. Medical error in dermatology practice: development of a classification system to drive priority setting in patient safety efforts. J Am Acad Dermatol. 2013;68(5):729-737. doi:10.1016/j.jaad.2012.10.058
5. Elston DM, Taylor JS, Coldiron B, et al. Patient safety: Part I. Patient safety and the dermatologist. J Am Acad Dermatol. 2009;61(2):179-191. doi:10.1016/j.jaad.2009.04.056
6. Nijhawan RI, Lee EH, Nehal KS. Biopsy site selfies--a quality improvement pilot study to assist with correct surgical site identification. Dermatol Surg. 2015;41(4):499-504. doi:10.1097/DSS.0000000000000305
7. Highsmith JT, Weinstein DA, Highsmith MJ, Etzkorn JR. BIOPSY 1-2-3 in dermatologic surgery: improving smartphone use to avoid wrong-site surgery. Technol Innov. 2016;18(2-3):203-206. doi:10.21300/18.2-3.2016.203
8. Rossy KM, Lawrence N. Difficulty with surgical site identification: what role does it play in dermatology? J Am Acad Dermatol. 2012;67(2):257-261. doi:10.1016/j.jaad.2012.02.034
9. American Society for Dermatologic Surgery. Photographic standards in dermatologic surgery poster. Accessed April 12, 2021. https://www.asds.net/medical-professionals/members-resources/product-details/productname/photographic-standards-poster
10. St John J, Walker J, Goldberg D, Maloney ME. Avoiding Medical Errors in Cutaneous Site Identification: A Best Practices Review. Dermatol Surg. 2016;42(4):477-484. doi:10.1097/DSS.0000000000000683
11. Alam M, Lee A, Ibrahimi OA, et al. A multistep approach to improving biopsy site identification in dermatology: physician, staff, and patient roles based on a Delphi consensus. JAMA Dermatol. 2014;150(5):550-558. doi:10.1001/jamadermatol.2013.9804
12. Chuang GS, Gilchrest BA. Ultraviolet-fluorescent tattoo location of cutaneous biopsy site. Dermatol Surg. 2012;38(3):479-483. doi:10.1111/j.1524-4725.2011.02238.x
13. DiGiovanni CW, Kang L, Manuel J. Patient compliance in avoiding wrong-site surgery. J Bone Joint Surg Am. 2003;85(5):815-819. doi:10.2106/00004623-200305000-00007
14. Gallagher TH. A 62-year-old woman with skin cancer who experienced wrong-site surgery: review of medical error. JAMA. 2009;302(6):669-677. doi:10.1001/jama.2009.1011
15. Mulloy DF, Hughes RG. Wrong-site surgery: a preventable medical error. In: Hughes RG, ed. Patient Safety and Quality: An Evidence-Based Handbook for Nurses. Agency for Healthcare Research and Quality (US); 2008:chap 36. Accessed April 23, 2021. https://www.ncbi.nlm.nih.gov/books/NBK2678
16. Zaiac M, Tongdee E, Porges L, Touloei K, Prodanovich S. Anesthetic blister induction to identify biopsy site prior to Mohs surgery. J Drugs Dermatol. 2015;14(5):446-447.
17. Jawed SI, Goldberg LH, Wang SQ. Dermoscopy to identify biopsy sites before Mohs surgery. Dermatol Surg. 2014;40(3):334-337. doi:10.1111/dsu.12422
Desmoplastic Melanoma Masquerading as Neurofibroma
Desmoplastic melanoma (DMM) is a rare variant of melanoma that presents major challenges to both clinicians and pathologists.1 Clinically, the lesions may appear as subtle bland papules, nodules, or plaques. They can be easily mistaken for benign growths, leading to a delayed diagnosis. Consequently, most DMMs at the time of diagnosis tend to be thick, with a mean Breslow depth ranging from 2.0 to 6.5 mm.2 Histopathologic evaluation has its difficulties. At scanning magnification, these tumors may show low cellularity, mimicking a benign proliferation. It is well recognized that S-100 and other tumor markers lack specificity for DMM, which can be positive in a range of neural tumors and other cell types.2 In some amelanotic tumors, DMM becomes virtually indistinguishable from benign peripheral sheath tumors such as neurofribroma.3
Desmoplastic melanoma is exceedingly uncommon in the United States, with an estimated annual incidence rate of 2.0 cases per million.2 Typical locations of presentation include sun-exposed skin, with the head and neck regions representing more than half of reported cases.2 Desmoplastic melanoma largely is a disease of fair-skinned patients, with 95.5% of cases in the United States occurring in white non-Hispanic individuals. Advancing age, male gender, and head and neck location are associated with an increased risk for DMM-specific death.2 It is important that new or changing lesions in the correct cohort and location are biopsied promptly. We present this case to highlight the ongoing challenges of diagnosing DMM both clinically and histologically and to review the salient features of this often benign-appearing tumor.
Case Report
A 51-year-old White man with a history of prostate cancer, a personal and family history of melanoma, and benign neurofibromas presented with a 6-mm, pink, well-demarcated, soft papule on the left lateral neck (Figure 1). The lesion had been stable for many years but began growing more rapidly 1 to 2 years prior to presentation. The lesion was asymptomatic, and he denied changes in color or texture. There also was no bleeding or ulceration. A review of systems was unremarkable. A shave biopsy of the lesion revealed a nodular spindle cell tumor in the dermis resembling a neurofibroma on low power (Figure 2). However, overlying the tumor was a confluent proliferation positive for MART-1 and S-100, which was consistent with a diagnosis of melanoma in situ (Figure 3). Higher-power evaluation of the dermal proliferation showed both bland and hyperchromatic spindled and epithelioid cells (Figure 4), with rare mitotic figures highlighted by PHH3, an uncommon finding in neurofibromas (Figure 5). The dermal spindle cells were positive for S-100 and p75 and negative for Melan-A. Epithelial membrane antigen highlighted a faint sheath surrounding the dermal component. Ki-67 revealed a mildly increased proliferative index in the dermal component. The diagnosis of DMM was made after outside dermatopathology consultation was in agreement. However, the possibility of a melanoma in situ growing in association with an underlying neurofibroma remained a diagnostic consideration histologically. The lesion was widely excised.
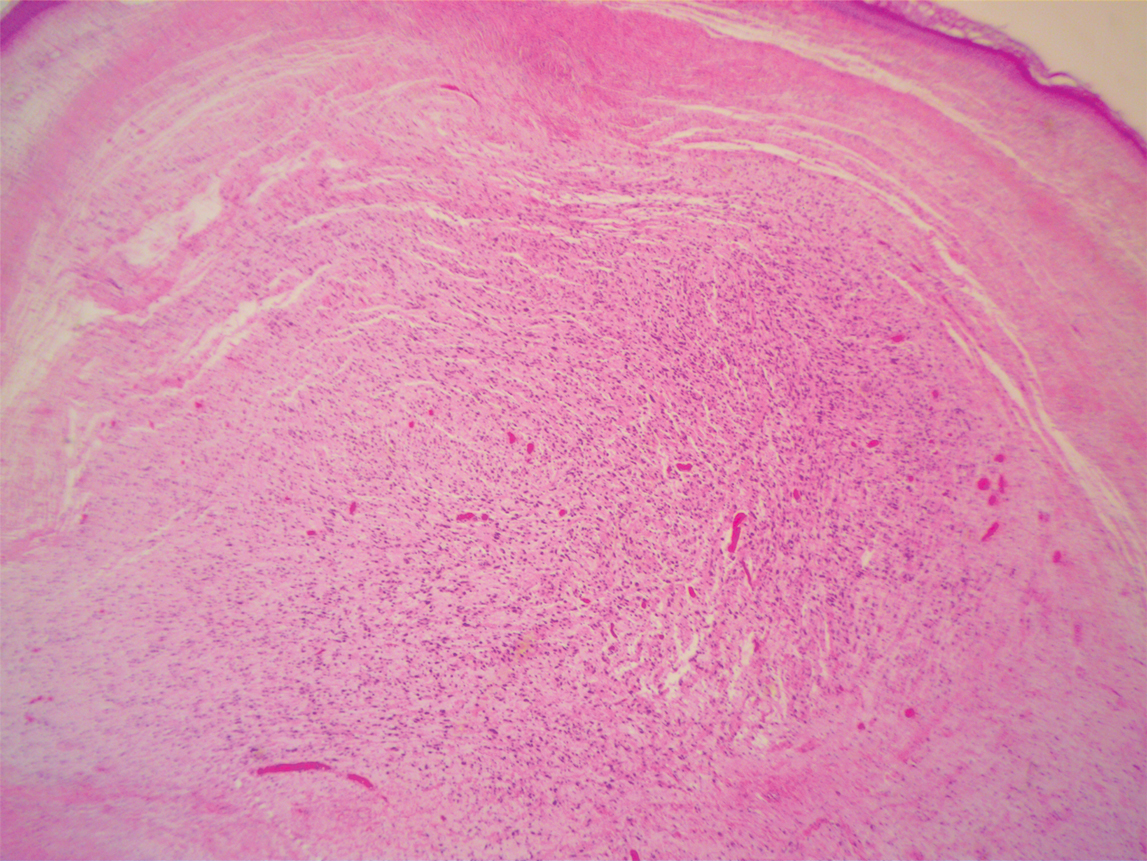
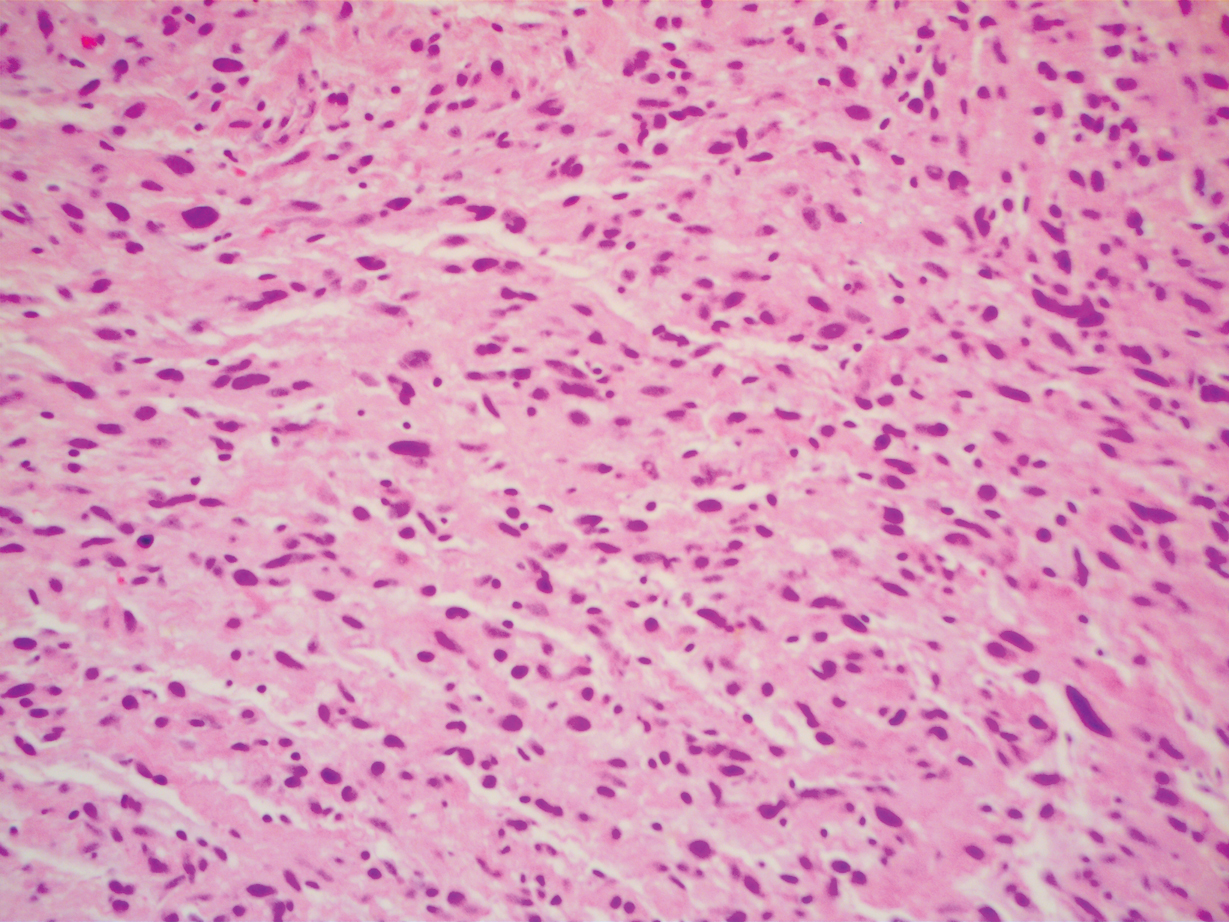
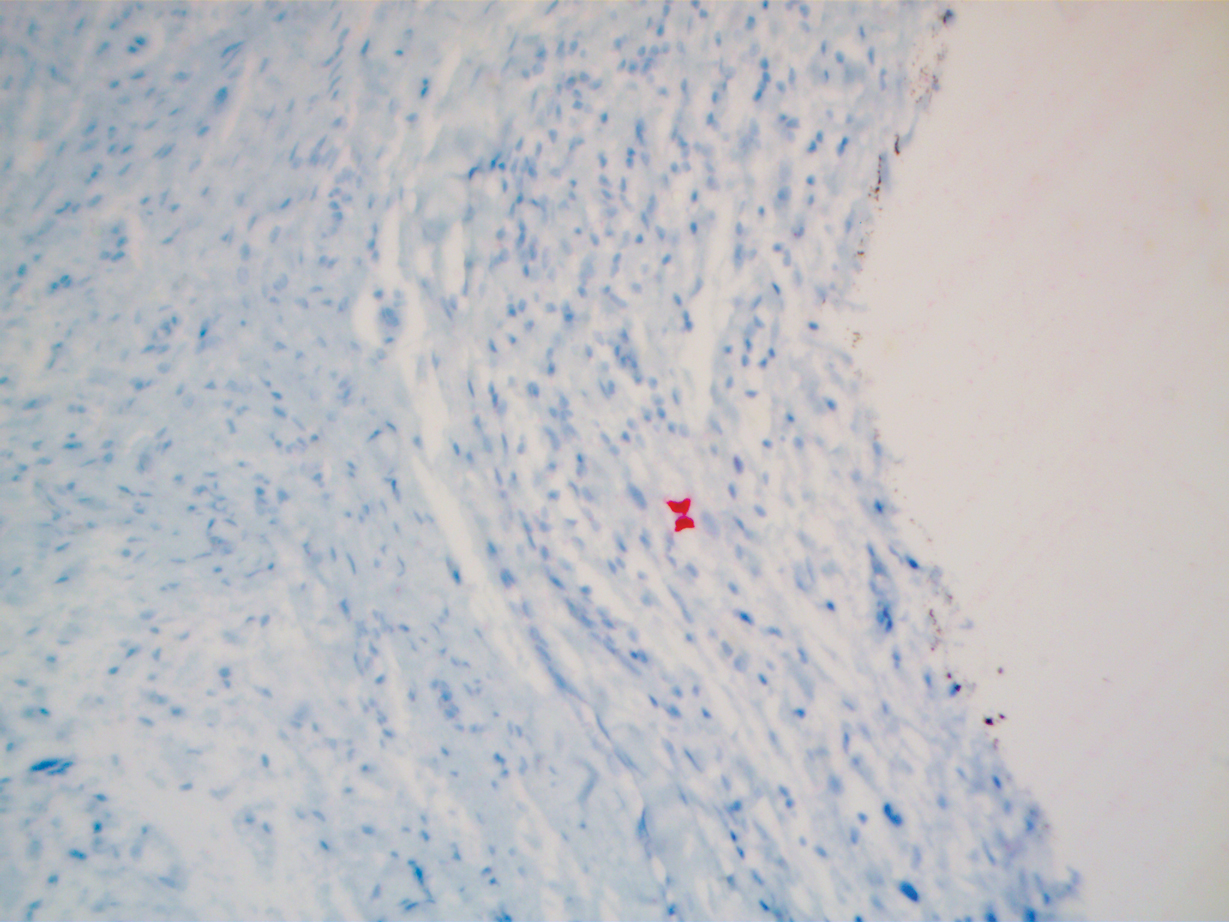
Comment
Differential for DMM
Early DMMs may not show sufficient cytologic atypia to permit obvious distinction from neurofibromas, which becomes problematic when encountering a spindle cell proliferation within severely sun-damaged skin, or even more so when an intraepidermal population of melanocytes is situated above a dermal population of slender, spindled, S-100–positive cells, as seen in our patient.4 For these challenging scenarios, Yeh and McCalmont4 have proposed evaluating for a CD34 “fingerprint” pattern. This pattern typically is widespread in neurofibroma but absent or limited in DMM, and it is a useful adjunct in the differential diagnosis when conventional immunohistochemistry has little contribution.
There are several case reports in the literature of DMM mimicking other benign or malignant proliferations. In 2012, Jou et al5 described a case of a 62-year-old White man who presented with an oral nodule consistent with fibrous inflammatory hyperplasia clinically. Incisional biopsy later confirmed the diagnosis of amelanotic DMM.5 Similar case reports have been described in which the diagnosis of DMM was later found to resemble a sarcoma and malignant peripheral nerve sheath tumor.6,7
Diagnosis of DMM
The prototypical DMM is an asymmetrical and deeply infiltrative spindle cell lesion in severely sun-damaged skin. By definition, the individual melanocytes are separated by connective tissue components, giving the tumor a paucicellular appearance.1 Although the low cellularity can give a deceptively bland scanning aspect, on high-power examination there usually are identifiable atypical spindled cells with enlarged, elongated, and hyperchromatic nuclei. S-100 typically is diffusely positive in DMM, though occasional cases show more limited staining.8 Other commonly used and more specific markers of melanocytic differentiation, including HMB45 and Melan-A, typically are negative in the paucicellular spindle cell components.9 Desmoplastic melanoma can be further categorized by the degree of fibrosis within a particular tumor. If fibrosis is prominent throughout the entire tumor, it is named pure DMM. On the other hand, fibrosis may only represent a portion of an otherwise nondesmoplastic melanoma, which is known as combined DMM.10
Conclusion
We present this case to highlight the ongoing challenges of diagnosing DMM both clinically and histologically. Although a bland-appearing lesion, key clinical features prompting a biopsy in our patient included recent growth of the lesion, a personal history of melanoma, the patient’s fair skin type, a history of heavy sun exposure, and the location of the lesion. According to Busam,11 an associated melanoma in situ component is identified in 80% to 85% of DMM cases. Detection of a melanoma in situ component associated with a malignant spindle cell tumor can help establish the diagnosis of DMM. In the absence of melanoma in situ, a strong diffuse immunoreactivity for S-100 and lack of epithelial markers support the diagnosis.11 After review of the literature, our case likely represents DMM as opposed to a melanoma in situ developing within a neurofibroma.
- Wood BA. Desmoplastic melanoma: recent advances and persisting challenges. Pathology. 2013;45:453-463.
- Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
- Machado I, Llombart B, Cruz J, et al. Desmoplastic melanoma may mimic a cutaneous peripheral nerve sheath tumor: report of 3 challenging cases. J Cutan Pathol. 2017;4:632-638.
- Yeh I, McCalmont, TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. J Cutan Pathol. 2011;38:625-630.
- Jou A, Miranda FV, Oliveira MG, et al. Oral desmoplastic melanoma mimicking inflammatory hyperplasia. Gerodontology. 2012;29:E1163-E1167.
- Ishikura H, Kojo T, Ichimura H, et al. Desmoplastic malignant melanoma of the uterine cervix: a rare primary malignancy in the uterus mimicking a sarcoma. Histopathology. 1998;33:93-94.
- Barnett SL, Wells MJ, Mickey B, et al. Perineural extension of cutaneous desmoplastic melanoma mimicking an intracranial malignant peripheral nerve sheath tumor. case report. J Neurosurg. 2011;115:273-277.
- Jain S, Allen PW. Desmoplastic malignant melanoma and its variants. a study of 45 cases. Am J Surg Pathol. 1989;13:358-373.
- Skelton HG, Maceira J, Smith KJ, et al. HMB45 negative spindle cell malignant melanoma. Am J Dermatopathol. 1997;19:580-584.
- George E, McClain SE, Slingluff CL, et al. Subclassification of desmoplastic melanoma: pure and mixed variants have significantly different capacities for lymph node metastasis. J Cutan Pathol. 2009;36:425-432.
- Busam KJ. Desmoplastic melanoma. Clin Lab Med. 2011;31:321-330.
Desmoplastic melanoma (DMM) is a rare variant of melanoma that presents major challenges to both clinicians and pathologists.1 Clinically, the lesions may appear as subtle bland papules, nodules, or plaques. They can be easily mistaken for benign growths, leading to a delayed diagnosis. Consequently, most DMMs at the time of diagnosis tend to be thick, with a mean Breslow depth ranging from 2.0 to 6.5 mm.2 Histopathologic evaluation has its difficulties. At scanning magnification, these tumors may show low cellularity, mimicking a benign proliferation. It is well recognized that S-100 and other tumor markers lack specificity for DMM, which can be positive in a range of neural tumors and other cell types.2 In some amelanotic tumors, DMM becomes virtually indistinguishable from benign peripheral sheath tumors such as neurofribroma.3
Desmoplastic melanoma is exceedingly uncommon in the United States, with an estimated annual incidence rate of 2.0 cases per million.2 Typical locations of presentation include sun-exposed skin, with the head and neck regions representing more than half of reported cases.2 Desmoplastic melanoma largely is a disease of fair-skinned patients, with 95.5% of cases in the United States occurring in white non-Hispanic individuals. Advancing age, male gender, and head and neck location are associated with an increased risk for DMM-specific death.2 It is important that new or changing lesions in the correct cohort and location are biopsied promptly. We present this case to highlight the ongoing challenges of diagnosing DMM both clinically and histologically and to review the salient features of this often benign-appearing tumor.
Case Report
A 51-year-old White man with a history of prostate cancer, a personal and family history of melanoma, and benign neurofibromas presented with a 6-mm, pink, well-demarcated, soft papule on the left lateral neck (Figure 1). The lesion had been stable for many years but began growing more rapidly 1 to 2 years prior to presentation. The lesion was asymptomatic, and he denied changes in color or texture. There also was no bleeding or ulceration. A review of systems was unremarkable. A shave biopsy of the lesion revealed a nodular spindle cell tumor in the dermis resembling a neurofibroma on low power (Figure 2). However, overlying the tumor was a confluent proliferation positive for MART-1 and S-100, which was consistent with a diagnosis of melanoma in situ (Figure 3). Higher-power evaluation of the dermal proliferation showed both bland and hyperchromatic spindled and epithelioid cells (Figure 4), with rare mitotic figures highlighted by PHH3, an uncommon finding in neurofibromas (Figure 5). The dermal spindle cells were positive for S-100 and p75 and negative for Melan-A. Epithelial membrane antigen highlighted a faint sheath surrounding the dermal component. Ki-67 revealed a mildly increased proliferative index in the dermal component. The diagnosis of DMM was made after outside dermatopathology consultation was in agreement. However, the possibility of a melanoma in situ growing in association with an underlying neurofibroma remained a diagnostic consideration histologically. The lesion was widely excised.
Comment
Differential for DMM
Early DMMs may not show sufficient cytologic atypia to permit obvious distinction from neurofibromas, which becomes problematic when encountering a spindle cell proliferation within severely sun-damaged skin, or even more so when an intraepidermal population of melanocytes is situated above a dermal population of slender, spindled, S-100–positive cells, as seen in our patient.4 For these challenging scenarios, Yeh and McCalmont4 have proposed evaluating for a CD34 “fingerprint” pattern. This pattern typically is widespread in neurofibroma but absent or limited in DMM, and it is a useful adjunct in the differential diagnosis when conventional immunohistochemistry has little contribution.
There are several case reports in the literature of DMM mimicking other benign or malignant proliferations. In 2012, Jou et al5 described a case of a 62-year-old White man who presented with an oral nodule consistent with fibrous inflammatory hyperplasia clinically. Incisional biopsy later confirmed the diagnosis of amelanotic DMM.5 Similar case reports have been described in which the diagnosis of DMM was later found to resemble a sarcoma and malignant peripheral nerve sheath tumor.6,7
Diagnosis of DMM
The prototypical DMM is an asymmetrical and deeply infiltrative spindle cell lesion in severely sun-damaged skin. By definition, the individual melanocytes are separated by connective tissue components, giving the tumor a paucicellular appearance.1 Although the low cellularity can give a deceptively bland scanning aspect, on high-power examination there usually are identifiable atypical spindled cells with enlarged, elongated, and hyperchromatic nuclei. S-100 typically is diffusely positive in DMM, though occasional cases show more limited staining.8 Other commonly used and more specific markers of melanocytic differentiation, including HMB45 and Melan-A, typically are negative in the paucicellular spindle cell components.9 Desmoplastic melanoma can be further categorized by the degree of fibrosis within a particular tumor. If fibrosis is prominent throughout the entire tumor, it is named pure DMM. On the other hand, fibrosis may only represent a portion of an otherwise nondesmoplastic melanoma, which is known as combined DMM.10
Conclusion
We present this case to highlight the ongoing challenges of diagnosing DMM both clinically and histologically. Although a bland-appearing lesion, key clinical features prompting a biopsy in our patient included recent growth of the lesion, a personal history of melanoma, the patient’s fair skin type, a history of heavy sun exposure, and the location of the lesion. According to Busam,11 an associated melanoma in situ component is identified in 80% to 85% of DMM cases. Detection of a melanoma in situ component associated with a malignant spindle cell tumor can help establish the diagnosis of DMM. In the absence of melanoma in situ, a strong diffuse immunoreactivity for S-100 and lack of epithelial markers support the diagnosis.11 After review of the literature, our case likely represents DMM as opposed to a melanoma in situ developing within a neurofibroma.
Desmoplastic melanoma (DMM) is a rare variant of melanoma that presents major challenges to both clinicians and pathologists.1 Clinically, the lesions may appear as subtle bland papules, nodules, or plaques. They can be easily mistaken for benign growths, leading to a delayed diagnosis. Consequently, most DMMs at the time of diagnosis tend to be thick, with a mean Breslow depth ranging from 2.0 to 6.5 mm.2 Histopathologic evaluation has its difficulties. At scanning magnification, these tumors may show low cellularity, mimicking a benign proliferation. It is well recognized that S-100 and other tumor markers lack specificity for DMM, which can be positive in a range of neural tumors and other cell types.2 In some amelanotic tumors, DMM becomes virtually indistinguishable from benign peripheral sheath tumors such as neurofribroma.3
Desmoplastic melanoma is exceedingly uncommon in the United States, with an estimated annual incidence rate of 2.0 cases per million.2 Typical locations of presentation include sun-exposed skin, with the head and neck regions representing more than half of reported cases.2 Desmoplastic melanoma largely is a disease of fair-skinned patients, with 95.5% of cases in the United States occurring in white non-Hispanic individuals. Advancing age, male gender, and head and neck location are associated with an increased risk for DMM-specific death.2 It is important that new or changing lesions in the correct cohort and location are biopsied promptly. We present this case to highlight the ongoing challenges of diagnosing DMM both clinically and histologically and to review the salient features of this often benign-appearing tumor.
Case Report
A 51-year-old White man with a history of prostate cancer, a personal and family history of melanoma, and benign neurofibromas presented with a 6-mm, pink, well-demarcated, soft papule on the left lateral neck (Figure 1). The lesion had been stable for many years but began growing more rapidly 1 to 2 years prior to presentation. The lesion was asymptomatic, and he denied changes in color or texture. There also was no bleeding or ulceration. A review of systems was unremarkable. A shave biopsy of the lesion revealed a nodular spindle cell tumor in the dermis resembling a neurofibroma on low power (Figure 2). However, overlying the tumor was a confluent proliferation positive for MART-1 and S-100, which was consistent with a diagnosis of melanoma in situ (Figure 3). Higher-power evaluation of the dermal proliferation showed both bland and hyperchromatic spindled and epithelioid cells (Figure 4), with rare mitotic figures highlighted by PHH3, an uncommon finding in neurofibromas (Figure 5). The dermal spindle cells were positive for S-100 and p75 and negative for Melan-A. Epithelial membrane antigen highlighted a faint sheath surrounding the dermal component. Ki-67 revealed a mildly increased proliferative index in the dermal component. The diagnosis of DMM was made after outside dermatopathology consultation was in agreement. However, the possibility of a melanoma in situ growing in association with an underlying neurofibroma remained a diagnostic consideration histologically. The lesion was widely excised.
Comment
Differential for DMM
Early DMMs may not show sufficient cytologic atypia to permit obvious distinction from neurofibromas, which becomes problematic when encountering a spindle cell proliferation within severely sun-damaged skin, or even more so when an intraepidermal population of melanocytes is situated above a dermal population of slender, spindled, S-100–positive cells, as seen in our patient.4 For these challenging scenarios, Yeh and McCalmont4 have proposed evaluating for a CD34 “fingerprint” pattern. This pattern typically is widespread in neurofibroma but absent or limited in DMM, and it is a useful adjunct in the differential diagnosis when conventional immunohistochemistry has little contribution.
There are several case reports in the literature of DMM mimicking other benign or malignant proliferations. In 2012, Jou et al5 described a case of a 62-year-old White man who presented with an oral nodule consistent with fibrous inflammatory hyperplasia clinically. Incisional biopsy later confirmed the diagnosis of amelanotic DMM.5 Similar case reports have been described in which the diagnosis of DMM was later found to resemble a sarcoma and malignant peripheral nerve sheath tumor.6,7
Diagnosis of DMM
The prototypical DMM is an asymmetrical and deeply infiltrative spindle cell lesion in severely sun-damaged skin. By definition, the individual melanocytes are separated by connective tissue components, giving the tumor a paucicellular appearance.1 Although the low cellularity can give a deceptively bland scanning aspect, on high-power examination there usually are identifiable atypical spindled cells with enlarged, elongated, and hyperchromatic nuclei. S-100 typically is diffusely positive in DMM, though occasional cases show more limited staining.8 Other commonly used and more specific markers of melanocytic differentiation, including HMB45 and Melan-A, typically are negative in the paucicellular spindle cell components.9 Desmoplastic melanoma can be further categorized by the degree of fibrosis within a particular tumor. If fibrosis is prominent throughout the entire tumor, it is named pure DMM. On the other hand, fibrosis may only represent a portion of an otherwise nondesmoplastic melanoma, which is known as combined DMM.10
Conclusion
We present this case to highlight the ongoing challenges of diagnosing DMM both clinically and histologically. Although a bland-appearing lesion, key clinical features prompting a biopsy in our patient included recent growth of the lesion, a personal history of melanoma, the patient’s fair skin type, a history of heavy sun exposure, and the location of the lesion. According to Busam,11 an associated melanoma in situ component is identified in 80% to 85% of DMM cases. Detection of a melanoma in situ component associated with a malignant spindle cell tumor can help establish the diagnosis of DMM. In the absence of melanoma in situ, a strong diffuse immunoreactivity for S-100 and lack of epithelial markers support the diagnosis.11 After review of the literature, our case likely represents DMM as opposed to a melanoma in situ developing within a neurofibroma.
- Wood BA. Desmoplastic melanoma: recent advances and persisting challenges. Pathology. 2013;45:453-463.
- Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
- Machado I, Llombart B, Cruz J, et al. Desmoplastic melanoma may mimic a cutaneous peripheral nerve sheath tumor: report of 3 challenging cases. J Cutan Pathol. 2017;4:632-638.
- Yeh I, McCalmont, TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. J Cutan Pathol. 2011;38:625-630.
- Jou A, Miranda FV, Oliveira MG, et al. Oral desmoplastic melanoma mimicking inflammatory hyperplasia. Gerodontology. 2012;29:E1163-E1167.
- Ishikura H, Kojo T, Ichimura H, et al. Desmoplastic malignant melanoma of the uterine cervix: a rare primary malignancy in the uterus mimicking a sarcoma. Histopathology. 1998;33:93-94.
- Barnett SL, Wells MJ, Mickey B, et al. Perineural extension of cutaneous desmoplastic melanoma mimicking an intracranial malignant peripheral nerve sheath tumor. case report. J Neurosurg. 2011;115:273-277.
- Jain S, Allen PW. Desmoplastic malignant melanoma and its variants. a study of 45 cases. Am J Surg Pathol. 1989;13:358-373.
- Skelton HG, Maceira J, Smith KJ, et al. HMB45 negative spindle cell malignant melanoma. Am J Dermatopathol. 1997;19:580-584.
- George E, McClain SE, Slingluff CL, et al. Subclassification of desmoplastic melanoma: pure and mixed variants have significantly different capacities for lymph node metastasis. J Cutan Pathol. 2009;36:425-432.
- Busam KJ. Desmoplastic melanoma. Clin Lab Med. 2011;31:321-330.
- Wood BA. Desmoplastic melanoma: recent advances and persisting challenges. Pathology. 2013;45:453-463.
- Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
- Machado I, Llombart B, Cruz J, et al. Desmoplastic melanoma may mimic a cutaneous peripheral nerve sheath tumor: report of 3 challenging cases. J Cutan Pathol. 2017;4:632-638.
- Yeh I, McCalmont, TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. J Cutan Pathol. 2011;38:625-630.
- Jou A, Miranda FV, Oliveira MG, et al. Oral desmoplastic melanoma mimicking inflammatory hyperplasia. Gerodontology. 2012;29:E1163-E1167.
- Ishikura H, Kojo T, Ichimura H, et al. Desmoplastic malignant melanoma of the uterine cervix: a rare primary malignancy in the uterus mimicking a sarcoma. Histopathology. 1998;33:93-94.
- Barnett SL, Wells MJ, Mickey B, et al. Perineural extension of cutaneous desmoplastic melanoma mimicking an intracranial malignant peripheral nerve sheath tumor. case report. J Neurosurg. 2011;115:273-277.
- Jain S, Allen PW. Desmoplastic malignant melanoma and its variants. a study of 45 cases. Am J Surg Pathol. 1989;13:358-373.
- Skelton HG, Maceira J, Smith KJ, et al. HMB45 negative spindle cell malignant melanoma. Am J Dermatopathol. 1997;19:580-584.
- George E, McClain SE, Slingluff CL, et al. Subclassification of desmoplastic melanoma: pure and mixed variants have significantly different capacities for lymph node metastasis. J Cutan Pathol. 2009;36:425-432.
- Busam KJ. Desmoplastic melanoma. Clin Lab Med. 2011;31:321-330.
Practice Points
- Desmoplastic melanoma remains a diagnostic challenge both clinically and histologically.
- New or changing lesions on sun-exposed sites of elderly patients with fair skin types should have a low threshold for biopsy.
- Consensus between more than one dermatopathologist is sometimes required to make the diagnosis histologically.
23-year-old woman • syncopal episode • sinus bradycardia • history of bipolar disorder • Dx?
THE CASE
A 23-year-old woman with past medical history of bipolar II disorder and a REM-specific seizure disorder that resolved at age 9 presented after a syncopal episode. The patient reported an initial sensation of lightheadedness while at work, which was followed by a syncopal episode with brief (1-2 min) loss of consciousness and a minor head injury.
She denied other prodromal symptoms including chest pain, shortness of breath, palpitations, and nausea. She also did not experience convulsions, urinary/bowel incontinence, or confusion upon regaining consciousness.
She denied previous syncopal episodes. However, she reported that, 2 weeks prior, there had been an event similar to that of her presenting complaint. During that episode, she experienced lightheadedness and a fall without loss of consciousness.
The patient had been prescribed a regimen of sertraline 100 mg/d and aripiprazole 10 mg/d to maintain mood stability. She had self-discontinued these medications about 8 months prior to presentation. A recent return of her depressive features had prompted a restart of this regimen 1 week before her first fall, without an initial taper upward.
While in the emergency department, she became bradycardic (heart rate, 38 beats/min) and hypotensive (blood pressure, 70/40 mm Hg). She subsequently became increasingly somnolent and had 1 episode of emesis. An electrocardiogram (EKG) revealed sinus bradycardia without other acute abnormalities (FIGURE).

Blood work including a basic metabolic panel, complete blood count, and cardiac enzymes were all within normal limits. Computed tomography of the head revealed no intracranial pathology. Her vitals were initially unresponsive to a fluid bolus but improved and stabilized after administration of intravenous atropine 0.5 mg.
Aripiprazole was held and sertraline was decreased to 75 mg on hospital Day 1, with close monitoring of her mood. Cardiology was consulted and followed the patient during her stay. The patient was monitored on telemetry for 3 days, exhibiting only sinus bradycardia with a stable heart rate of 45-55 beats/min. Systolic blood pressures were stable within 120 to 130 mm Hg. Transthoracic echocardiogram performed on hospital Day 2 was unremarkable, revealing a normal left ventricular ejection fraction of 65% and no wall motion abnormalities. She had no recurrence of the syncope or emesis.
Continue to: THE DIAGNOSIS
THE DIAGNOSIS
Given her benign cardiac work-up and symptom onset coinciding with the abrupt resumption of high doses of aripiprazole after an 8-month abstinence, the patient’s presentation was attributed to a rather uncommon adverse drug reaction to aripiprazole. This has only been described in a few case reports.
DISCUSSION
Aripiprazole (Abilify) is an atypical antipsychotic frequently used in the treatment of psychiatric conditions, including bipolar disorder and schizophrenia. While the specific therapeutic mechanism is unknown, it is believed that drug efficacy is related to partial agonism at dopamine D2, serotonin 5-HT1A, and serotonin 5-HT2A.1 As aripiprazole works on a variety of receptors involved in other physiologic processes, clinical adverse effects have been reported, most of which are associated with the adrenergic alpha1 receptors.1 These include cognitive impairment and seizures. Cardiovascular adverse effects of aripiprazole include orthostatic hypotension, cardiac arrhythmia, prolonged QT interval, and syncope.1-5
Selective serotonin reuptake inhibitors (SSRIs) such as sertraline (Zoloft) have also been shown to cause cardiac arrhythmia and syncope.6 Although sertraline may have contributed to the patient’s cardiac symptoms, it is more likely that the aripiprazole was the direct cause, as she remained asymptomatic while on a therapeutic dose of sertraline. Furthermore, aripiprazole is primarily metabolized though hepatic CYP2D6, which sertraline has been shown to inhibit.1,7 Therefore, the concomitant use of sertraline with no initial taper of either medication likely led to an increased effective dose of aripiprazole in our patient and subsequently to her presentation.
Few prior cases have identified aripiprazole as a cause of antipsychotic-associated bradycardic response.8 Based on the Adverse Drug Reaction Probability Scale, often referred to as the Naranjo Scale, we believe this to be a probable adverse response in our patient.9 Bradycardia followed a reasonable temporal sequence after aripiprazole use with a response previously described in the literature. Symptoms also improved after discontinuation of the drug and other etiologies of the bradycardia were ruled out.
Our patient was discharged with a 30-day cardiac event monitor and a scheduled appointment with Cardiology.
Continue to: THE TAKEAWAY
THE TAKEAWAY
As this case suggests, there may be an association between aripiprazole and symptomatic bradycardia. Therefore, family physicians should inquire about aripiprazole use in patients who present with cardiac symptoms and consider tapering this medication if other causes cannot be identified. Additionally, given the potential cardiac adverse effects of atypical antipsychotics, physicians may consider ordering baseline and follow-up EKGs to monitor for arrhythmias in patients prescribed aripiprazole. This may be especially prudent when an atypical antipsychotic is combined with an SSRI, as potential cardiac adverse effects may occur more frequently.
CORRESPONDENCE
Kyle Fletke, MD, Department of Family and Community Medicine, University of Maryland School of Medicine, 29 South Paca Street, Baltimore, MD 21201; [email protected]
1. Abilify [package insert]. Rockville, MD: Otsuka America Pharmaceutical, Inc; 2014.
2. Belemonte C, Ochoa D, Román M, et al. Evaluation of the relationship between pharmacokinetics and the safety of aripiprazole and its cardiovascular side effects in health volunteers. J Clin Psychopharmacol. 2016;36:608-614.
3. Torgovnic J, Sethi NK, Arsura E. Aripiprazole-induced orthostatic hypotension and cardiac arrhythmia. Psychiatry Clin Neurosci. 2008:62:485.
4. Pacher P, Kecskemeti V. Cardiovascular side effects of new antidepressants and antipsychotics: new drugs, old concerns? Curr Pharm Des. 2004;10:2463-2475.
5. Russo L, Rizzo A, Di Vincenzo A, et al. Aripiprazole overdose and transient 2:1 second degree atrioventricular block: only a coincidence? Curr Drug Saf. 2019;14:155-157.
6. Pacher P, Ungvari Z, Kecskemeti V, et al. Review of cardiovascular effects of fluoxetine, a selective serotonin reuptake inhibitor, compared to tricyclic antidepressants. Curr Med Chem. 1998;5:381-90.
7. Hemeryck A, Belpaire FM. Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update. Curr Drub Metab. 2002;3:13-37.
8. Snarr BS, Phan SV, Garner A, et al. Symptomatic bradycardia with oral aripiprazole and oral ziprasidone. Ann Pharmacother. 2010;44:760-763.
9. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
THE CASE
A 23-year-old woman with past medical history of bipolar II disorder and a REM-specific seizure disorder that resolved at age 9 presented after a syncopal episode. The patient reported an initial sensation of lightheadedness while at work, which was followed by a syncopal episode with brief (1-2 min) loss of consciousness and a minor head injury.
She denied other prodromal symptoms including chest pain, shortness of breath, palpitations, and nausea. She also did not experience convulsions, urinary/bowel incontinence, or confusion upon regaining consciousness.
She denied previous syncopal episodes. However, she reported that, 2 weeks prior, there had been an event similar to that of her presenting complaint. During that episode, she experienced lightheadedness and a fall without loss of consciousness.
The patient had been prescribed a regimen of sertraline 100 mg/d and aripiprazole 10 mg/d to maintain mood stability. She had self-discontinued these medications about 8 months prior to presentation. A recent return of her depressive features had prompted a restart of this regimen 1 week before her first fall, without an initial taper upward.
While in the emergency department, she became bradycardic (heart rate, 38 beats/min) and hypotensive (blood pressure, 70/40 mm Hg). She subsequently became increasingly somnolent and had 1 episode of emesis. An electrocardiogram (EKG) revealed sinus bradycardia without other acute abnormalities (FIGURE).
Blood work including a basic metabolic panel, complete blood count, and cardiac enzymes were all within normal limits. Computed tomography of the head revealed no intracranial pathology. Her vitals were initially unresponsive to a fluid bolus but improved and stabilized after administration of intravenous atropine 0.5 mg.
Aripiprazole was held and sertraline was decreased to 75 mg on hospital Day 1, with close monitoring of her mood. Cardiology was consulted and followed the patient during her stay. The patient was monitored on telemetry for 3 days, exhibiting only sinus bradycardia with a stable heart rate of 45-55 beats/min. Systolic blood pressures were stable within 120 to 130 mm Hg. Transthoracic echocardiogram performed on hospital Day 2 was unremarkable, revealing a normal left ventricular ejection fraction of 65% and no wall motion abnormalities. She had no recurrence of the syncope or emesis.
Continue to: THE DIAGNOSIS
THE DIAGNOSIS
Given her benign cardiac work-up and symptom onset coinciding with the abrupt resumption of high doses of aripiprazole after an 8-month abstinence, the patient’s presentation was attributed to a rather uncommon adverse drug reaction to aripiprazole. This has only been described in a few case reports.
DISCUSSION
Aripiprazole (Abilify) is an atypical antipsychotic frequently used in the treatment of psychiatric conditions, including bipolar disorder and schizophrenia. While the specific therapeutic mechanism is unknown, it is believed that drug efficacy is related to partial agonism at dopamine D2, serotonin 5-HT1A, and serotonin 5-HT2A.1 As aripiprazole works on a variety of receptors involved in other physiologic processes, clinical adverse effects have been reported, most of which are associated with the adrenergic alpha1 receptors.1 These include cognitive impairment and seizures. Cardiovascular adverse effects of aripiprazole include orthostatic hypotension, cardiac arrhythmia, prolonged QT interval, and syncope.1-5
Selective serotonin reuptake inhibitors (SSRIs) such as sertraline (Zoloft) have also been shown to cause cardiac arrhythmia and syncope.6 Although sertraline may have contributed to the patient’s cardiac symptoms, it is more likely that the aripiprazole was the direct cause, as she remained asymptomatic while on a therapeutic dose of sertraline. Furthermore, aripiprazole is primarily metabolized though hepatic CYP2D6, which sertraline has been shown to inhibit.1,7 Therefore, the concomitant use of sertraline with no initial taper of either medication likely led to an increased effective dose of aripiprazole in our patient and subsequently to her presentation.
Few prior cases have identified aripiprazole as a cause of antipsychotic-associated bradycardic response.8 Based on the Adverse Drug Reaction Probability Scale, often referred to as the Naranjo Scale, we believe this to be a probable adverse response in our patient.9 Bradycardia followed a reasonable temporal sequence after aripiprazole use with a response previously described in the literature. Symptoms also improved after discontinuation of the drug and other etiologies of the bradycardia were ruled out.
Our patient was discharged with a 30-day cardiac event monitor and a scheduled appointment with Cardiology.
Continue to: THE TAKEAWAY
THE TAKEAWAY
As this case suggests, there may be an association between aripiprazole and symptomatic bradycardia. Therefore, family physicians should inquire about aripiprazole use in patients who present with cardiac symptoms and consider tapering this medication if other causes cannot be identified. Additionally, given the potential cardiac adverse effects of atypical antipsychotics, physicians may consider ordering baseline and follow-up EKGs to monitor for arrhythmias in patients prescribed aripiprazole. This may be especially prudent when an atypical antipsychotic is combined with an SSRI, as potential cardiac adverse effects may occur more frequently.
CORRESPONDENCE
Kyle Fletke, MD, Department of Family and Community Medicine, University of Maryland School of Medicine, 29 South Paca Street, Baltimore, MD 21201; [email protected]
THE CASE
A 23-year-old woman with past medical history of bipolar II disorder and a REM-specific seizure disorder that resolved at age 9 presented after a syncopal episode. The patient reported an initial sensation of lightheadedness while at work, which was followed by a syncopal episode with brief (1-2 min) loss of consciousness and a minor head injury.
She denied other prodromal symptoms including chest pain, shortness of breath, palpitations, and nausea. She also did not experience convulsions, urinary/bowel incontinence, or confusion upon regaining consciousness.
She denied previous syncopal episodes. However, she reported that, 2 weeks prior, there had been an event similar to that of her presenting complaint. During that episode, she experienced lightheadedness and a fall without loss of consciousness.
The patient had been prescribed a regimen of sertraline 100 mg/d and aripiprazole 10 mg/d to maintain mood stability. She had self-discontinued these medications about 8 months prior to presentation. A recent return of her depressive features had prompted a restart of this regimen 1 week before her first fall, without an initial taper upward.
While in the emergency department, she became bradycardic (heart rate, 38 beats/min) and hypotensive (blood pressure, 70/40 mm Hg). She subsequently became increasingly somnolent and had 1 episode of emesis. An electrocardiogram (EKG) revealed sinus bradycardia without other acute abnormalities (FIGURE).
Blood work including a basic metabolic panel, complete blood count, and cardiac enzymes were all within normal limits. Computed tomography of the head revealed no intracranial pathology. Her vitals were initially unresponsive to a fluid bolus but improved and stabilized after administration of intravenous atropine 0.5 mg.
Aripiprazole was held and sertraline was decreased to 75 mg on hospital Day 1, with close monitoring of her mood. Cardiology was consulted and followed the patient during her stay. The patient was monitored on telemetry for 3 days, exhibiting only sinus bradycardia with a stable heart rate of 45-55 beats/min. Systolic blood pressures were stable within 120 to 130 mm Hg. Transthoracic echocardiogram performed on hospital Day 2 was unremarkable, revealing a normal left ventricular ejection fraction of 65% and no wall motion abnormalities. She had no recurrence of the syncope or emesis.
Continue to: THE DIAGNOSIS
THE DIAGNOSIS
Given her benign cardiac work-up and symptom onset coinciding with the abrupt resumption of high doses of aripiprazole after an 8-month abstinence, the patient’s presentation was attributed to a rather uncommon adverse drug reaction to aripiprazole. This has only been described in a few case reports.
DISCUSSION
Aripiprazole (Abilify) is an atypical antipsychotic frequently used in the treatment of psychiatric conditions, including bipolar disorder and schizophrenia. While the specific therapeutic mechanism is unknown, it is believed that drug efficacy is related to partial agonism at dopamine D2, serotonin 5-HT1A, and serotonin 5-HT2A.1 As aripiprazole works on a variety of receptors involved in other physiologic processes, clinical adverse effects have been reported, most of which are associated with the adrenergic alpha1 receptors.1 These include cognitive impairment and seizures. Cardiovascular adverse effects of aripiprazole include orthostatic hypotension, cardiac arrhythmia, prolonged QT interval, and syncope.1-5
Selective serotonin reuptake inhibitors (SSRIs) such as sertraline (Zoloft) have also been shown to cause cardiac arrhythmia and syncope.6 Although sertraline may have contributed to the patient’s cardiac symptoms, it is more likely that the aripiprazole was the direct cause, as she remained asymptomatic while on a therapeutic dose of sertraline. Furthermore, aripiprazole is primarily metabolized though hepatic CYP2D6, which sertraline has been shown to inhibit.1,7 Therefore, the concomitant use of sertraline with no initial taper of either medication likely led to an increased effective dose of aripiprazole in our patient and subsequently to her presentation.
Few prior cases have identified aripiprazole as a cause of antipsychotic-associated bradycardic response.8 Based on the Adverse Drug Reaction Probability Scale, often referred to as the Naranjo Scale, we believe this to be a probable adverse response in our patient.9 Bradycardia followed a reasonable temporal sequence after aripiprazole use with a response previously described in the literature. Symptoms also improved after discontinuation of the drug and other etiologies of the bradycardia were ruled out.
Our patient was discharged with a 30-day cardiac event monitor and a scheduled appointment with Cardiology.
Continue to: THE TAKEAWAY
THE TAKEAWAY
As this case suggests, there may be an association between aripiprazole and symptomatic bradycardia. Therefore, family physicians should inquire about aripiprazole use in patients who present with cardiac symptoms and consider tapering this medication if other causes cannot be identified. Additionally, given the potential cardiac adverse effects of atypical antipsychotics, physicians may consider ordering baseline and follow-up EKGs to monitor for arrhythmias in patients prescribed aripiprazole. This may be especially prudent when an atypical antipsychotic is combined with an SSRI, as potential cardiac adverse effects may occur more frequently.
CORRESPONDENCE
Kyle Fletke, MD, Department of Family and Community Medicine, University of Maryland School of Medicine, 29 South Paca Street, Baltimore, MD 21201; [email protected]
1. Abilify [package insert]. Rockville, MD: Otsuka America Pharmaceutical, Inc; 2014.
2. Belemonte C, Ochoa D, Román M, et al. Evaluation of the relationship between pharmacokinetics and the safety of aripiprazole and its cardiovascular side effects in health volunteers. J Clin Psychopharmacol. 2016;36:608-614.
3. Torgovnic J, Sethi NK, Arsura E. Aripiprazole-induced orthostatic hypotension and cardiac arrhythmia. Psychiatry Clin Neurosci. 2008:62:485.
4. Pacher P, Kecskemeti V. Cardiovascular side effects of new antidepressants and antipsychotics: new drugs, old concerns? Curr Pharm Des. 2004;10:2463-2475.
5. Russo L, Rizzo A, Di Vincenzo A, et al. Aripiprazole overdose and transient 2:1 second degree atrioventricular block: only a coincidence? Curr Drug Saf. 2019;14:155-157.
6. Pacher P, Ungvari Z, Kecskemeti V, et al. Review of cardiovascular effects of fluoxetine, a selective serotonin reuptake inhibitor, compared to tricyclic antidepressants. Curr Med Chem. 1998;5:381-90.
7. Hemeryck A, Belpaire FM. Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update. Curr Drub Metab. 2002;3:13-37.
8. Snarr BS, Phan SV, Garner A, et al. Symptomatic bradycardia with oral aripiprazole and oral ziprasidone. Ann Pharmacother. 2010;44:760-763.
9. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
1. Abilify [package insert]. Rockville, MD: Otsuka America Pharmaceutical, Inc; 2014.
2. Belemonte C, Ochoa D, Román M, et al. Evaluation of the relationship between pharmacokinetics and the safety of aripiprazole and its cardiovascular side effects in health volunteers. J Clin Psychopharmacol. 2016;36:608-614.
3. Torgovnic J, Sethi NK, Arsura E. Aripiprazole-induced orthostatic hypotension and cardiac arrhythmia. Psychiatry Clin Neurosci. 2008:62:485.
4. Pacher P, Kecskemeti V. Cardiovascular side effects of new antidepressants and antipsychotics: new drugs, old concerns? Curr Pharm Des. 2004;10:2463-2475.
5. Russo L, Rizzo A, Di Vincenzo A, et al. Aripiprazole overdose and transient 2:1 second degree atrioventricular block: only a coincidence? Curr Drug Saf. 2019;14:155-157.
6. Pacher P, Ungvari Z, Kecskemeti V, et al. Review of cardiovascular effects of fluoxetine, a selective serotonin reuptake inhibitor, compared to tricyclic antidepressants. Curr Med Chem. 1998;5:381-90.
7. Hemeryck A, Belpaire FM. Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update. Curr Drub Metab. 2002;3:13-37.
8. Snarr BS, Phan SV, Garner A, et al. Symptomatic bradycardia with oral aripiprazole and oral ziprasidone. Ann Pharmacother. 2010;44:760-763.
9. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.

The Natural History of a Patient With COVID-19 Pneumonia and Silent Hypoxemia
In less than a year, COVID-19 has infected nearly 100 million people worldwide and caused more than 2 million deaths and counting. Although the infection fatality rate is estimated to be 1% and the case fatality rate between 2% and 3%, COVID-19 has had a disproportionate effect on the older population and those with comorbidities. Some of these findings are mirrored in the US Department of Veterans Affairs (VA) population, which has seen a higher case fatality rate.1-4
As a respiratory tract infection, the most dreaded presentation is severe pneumonia with acute hypoxemia, which may rapidly deteriorate to acute respiratory distress syndrome (ARDS) and respiratory failure.5-7 This possibility has led to early intubation strategies aimed at preempting this rapid deterioration and minimizing viral exposure to health care workers. Intubation rates have varied widely with extremes of 6 to 88%.8,9
However, this early intubation strategy has waned as some of the rationale behind its endorsement has been called into question. Early intubation bypasses alternatives to intubation; high-flow nasal cannula oxygen, noninvasive ventilation, and awake proning are all effective maneuvers in the appropriate patient.10,11 The use of first-line high-flow nasal cannula oxygen and noninvasive ventilation has been widely reported. Reports of first-line use of high-flow nasal cannula oxygen has not demonstrated inferior outcomes, nor has the timing of intubation, suggesting a significant portion of patients could benefit from a trial of therapy and eventually avoid intubation.11-14 Other therapies, such as systemic corticosteroids, confer a mortality benefit in those patients with COVID-19 who require oxygen or mechanical ventilation, but their impact on the progression of respiratory failure and need for intubation are undetermined.
There also are reports of patients who report no signs of respiratory distress or dyspnea with their COVID-19 pneumonia despite profound hypoxemia or high oxygen requirements. Various terms, including silent hypoxemia or happy hypoxia, are descriptive of the demeanor of these patients, and treatment has invariably included oxygen.15,16 Nevertheless, low oxygen measurements have generally prompted higher levels of supplemental oxygen or more invasive therapies.
Treatment rendered may obscure the trajectory of response, which is important to understand to better position options for invasive therapies and other therapeutics. We recently encountered a patient with a course of illness that represented the natural history of COVID-19 pneumonia with low oxygen levels (referred to as hypoxemia for consistency) that highlighted several issues of management.
Case Presentation
A 62-year-old undomiciled woman with morbid obesity, prediabetes mellitus, long-standing schizophrenia, and bipolar disorder presented to our facility for evaluation of dry cough and need for tuberculosis clearance for admittance to a shelter. She appeared comfortable and was afebrile with blood pressure 111/74 mm Hg, heart rate 82 beats per minute. Her respiratory rate was 18 breaths per minute, but the pulse oximetry showed oxygen saturation of 70 to 75% on room air at rest. A chest X-ray showed bibasilar infiltrates (Figure 1), and a rapid COVID-19 nasopharyngeal polymerase chain reaction (PCR) test returned positive, confirmed by a second PCR test. Baseline inflammatory markers were elevated (Figure 2). In addition, the serum interleukin-6 also was elevated to 66.1 pg/mL (normal < 5.0), erythrocyte sedimentation rate elevated to 69 mm/h, but serum procalcitonin was essentially normal (0.22 ng/mL; normal < 20 ng/mL) as was the serum lactate (1.4 mmol/L).
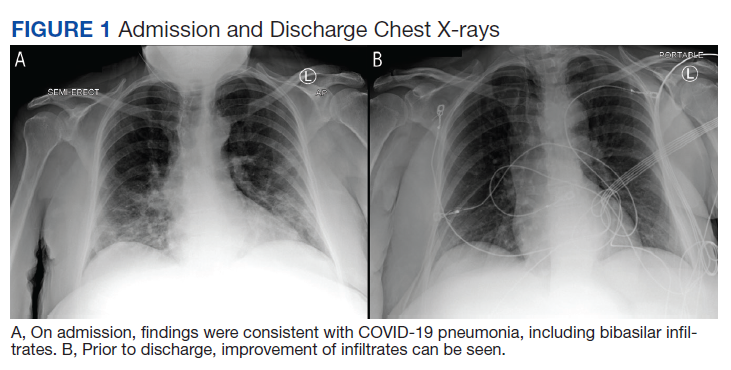
The patient was admitted to the intensive care unit (ICU) for close monitoring in anticipation of the possibility of decompensation based on her age, hypoxia, and elevated inflammatory markers.17 Besides a subsequent low-grade fever (100.4 oF) and lymphopenia (manual count 550/uL), she remained clinically unchanged. Throughout her hospitalization, she maintained a persistent psychotic delusion that she did not have COVID-19, refusing all medical interventions, including a peripheral IV line and supplemental oxygen for the entire duration. Extensive efforts to identify family or a surrogate decision maker were unsuccessful. After consultation with Psychiatry, Bio-Ethics, and hospital leadership, the patient was deemed to lack decision-making capacity regarding treatment or disposition and was placed on a psychiatric hold. However, since any interventions against her will would require sedation, IV access, and potentially increase the risk of nosocomial COVID-19 transmission, she was allowed to remain untreated and was closely monitored for symptoms of worsening respiratory failure.
Over the next 2 weeks, her hypoxemia, inflammatory markers, and the infiltrates on imaging resolved (Figure 2). The lowest daily awake room air pulse oximetry readings are reported, initially with consistent readings in the low 80% range, but on day 12, readings were > 90% and remained > 90% for the remainder of her hospitalization. Therefore, shortly after hospital day 12, she was clinically stable for discharge from acute care to a subacute facility, but this required documentation of the clearance of her viral infection. She refused to undergo a subsequent nasopharyngeal swab but allowed an oropharyngeal COVID-19 PCR swab, which was negative. She remained stable and unchanged for the remainder of her hospitalization, awaiting identification of a receiving facility and was able to be discharged to transitional housing on day 38.
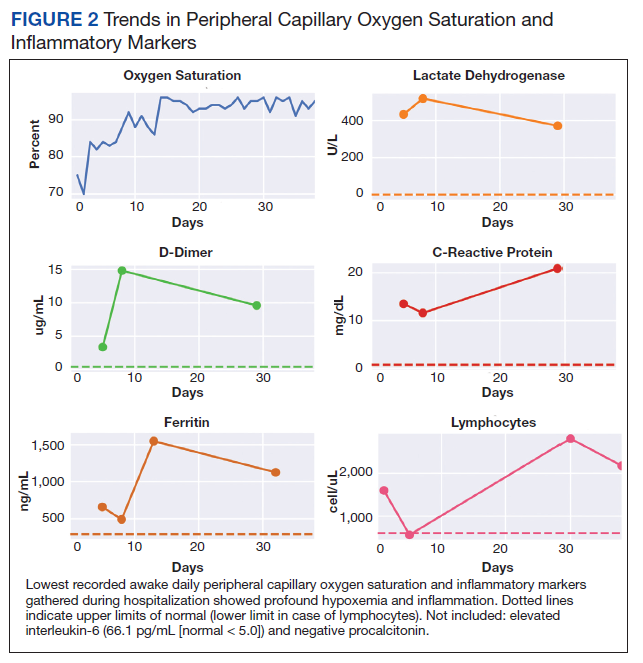
Discussion
The initial reports of COVID-19 pneumonia focused on ARDS and respiratory failure requiring mechanical ventilation with less emphasis on those with lower severity of illness. This was heightened by health care systems that were overwhelmed with large number of patients while faced with limited supplies and equipment. Given the risk to patients and providers of crash intubations, some recommended early intubation strategies.3 However, the natural history of COVID-19 pneumonia and the threshold for intubation of these patients remain poorly defined despite the creation of prognostic tools.17 This patient’s persistent hypoxemia and elevated inflammatory markers certainly met markers of disease associated with a high risk of progression.
The greatest concern would have been her level of hypoxemia. Acceptable thresholds of hypoxemia vary, but general consensus would classify pulse oximetry < 90% as hypoxemia and a threshold for administering supplemental oxygen. It is important to recognize how pulse oximetry readings translate to partial pressure of oxygen (PaO2) measurements (Table 1). Pulse oximetry readings of 90% corresponds to a PaO2 readings of 60 mm Hg in ideal conditions without the influence of acidosis, PaCO2, or temperature. While lower readings are of concern, these do not represent absolute indications for assisted ventilatory support as lower levels are well tolerated in a variety of conditions. A common example are patients with chronic obstructive pulmonary disease. Long-term mortality benefits of continuous supplemental oxygen are well established in specific populations, but the threshold for correction in the acute setting remains a case-by-case decision. This decision is complex and is based on more than an absolute number or the amount of oxygen required to achieve a threshold level of oxygenation.
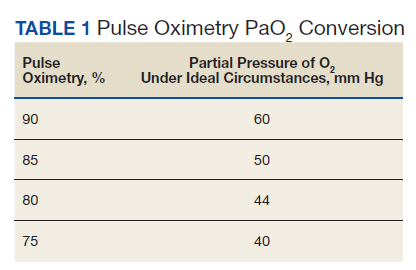
The PaO2/FIO2 (fraction of inspired oxygen) is a common measure used to address severity of disease and oxygen requirements. It also has been used to define the severity of ARDS, but the ratio is based on intubated and mechanically ventilated patients and may not translate well to those not on assisted ventilation. Treatment with supplemental oxygen also involves entrained air with associated imprecision in oxygen delivery.18 For this discussion, the patient’s admission PaO2/FIO2 on room air would have been between 190 and 260. Coupled with the bilateral infiltrates on imaging, there was justified concern for progression to severe ARDS. Her presentation would have met most of the epidemiologic criteria used in initial case finding for severe COVID-19 cases, including a blood oxygen saturation ≤ 93%, PaO2/FIO2 < 300 with infiltrates involving close to if not exceeding 50% of the lung.
With COVID-19 pneumonia, the pathologic injury to the alveoli resembles that of any viral pneumonia with recruitment of predominantly lymphocytic inflammatory cells that fill the alveoli, derangements in ventilation/perfusion mismatch as the core mechanism of hypoxemia with interstitial edema and shuntlike physiology developing at the extremes of involvement. In later stages, the histologic appearance is similar to ARDS, including hyaline membrane formation and thickened alveolar septa with perivascular lymphocytic-plasmocytic infiltration. In addition, there also are findings of organizing pneumonia with fibroblastic proliferation, thrombosis, and diffuse alveolar damage, a constellation of findings similar to that seen in the latter stages of ARDS.2
Although these histologic findings resemble ARDS, many patients with respiratory failure due to COVID-19 have a different physiologic profile compared with those with typical ARDS, with the most striking finding of lungs with low elastance or high compliance. From the critical care standpoint, this meant that the lungs were relatively easy to ventilate with lower peak airway and plateau pressures and low driving pressures. This condition suggested that there was relatively less lung that could be recruited with positive end expiratory pressure; therefore, a somewhat different entity from that associated with ARDS.19 These findings were often noted early in the course of respiratory failure, and although there is debate about whether this represents a different phenotype or timepoint in the spectrum of disease, it clearly represents a subset that is distinct from that which had been previously encountered.
On the other hand, the clinical features seen in those patients with COVID-19 pneumonia who progressed to advanced respiratory failure were essentially indistinguishable from those patients with traditional ARDS. Other explanations for this respiratory failure have included a disrupted vasoregulatory response to hypoxemia with failed hypoxic vasoconstriction, intravascular microthrombi, and impaired diffusion, all contributing to impaired gas exchange and hypoxemia.19-21 This can lead to shuntlike conditions that neither respond well to supplemental oxygen nor manifest the type of physiologic response seen with other causes of hypoxemia.
The severity of hypoxemia manifested by this patient may have elicited additional findings of respiratory distress, such as dyspnea and tachypnea. However, in patients with severe COVID-19 pneumonia, dyspnea was not a universal finding, reported in the 20 to 60% range of cohorts, higher in those with ARDS and mechanical ventilation, although some report near universal dyspnea in their series.1,4,8,22,23 Tachypnea is another symptom of interest. Using a threshold of > 24 breaths/min, tachypnea was noted in 16 to 29% of patients with a much greater proportion (63%) in nonsurvivors.6,24 Several explanations have been proposed for the discordance between the presence and severity of hypoxemia and lack of symptoms of dyspnea and tachypnea. It is important to recognize that misclassification of the severity of hypoxemia can occur due to technical issues and potential errors involving pulse oximetry measurement and shifts in the oxyhemoglobin dissociation curve. However, this is more pertinent for those with mild disease as the severity of hypoxemia in severe pneumonia is beyond what can be attributed to technical issues.
More important, the ventilatory response curve to hypoxemia may not be normal for some patients, blunted by as much as 50% in older patients, especially in those with diabetes mellitus.7,25,26 In addition, the ventilatory response varies widely even among normal individuals. This would translate to lower levels of minute ventilation (less tachypnea or respiratory effort) with hypoxemia. Hypocapnic hypoxemia also blunts the ventilatory response to hypoxemia. Subjects do not increase their minute ventilation if the PaCO2 remains low despite oxygen desaturation to < 70%, especially if PaCO2 < 30 mm Hg or alternatively, increases in minute ventilation are not seen until the PaCO2 exceeds 39 mm Hg.27 Both scenarios occur in those with COVID-19 pneumonia and provide another explanation for the absence of respiratory symptoms or signs of respiratory distress in some patients.
The observation of more compliant lungs may help in the understanding of the variable presentation of these patients. Compliant lungs do not require the increased pressure needed to achieve a specific tidal volume that, in turn, may increase the work of breathing. This may add to the explanation of seemingly paradoxical silent hypoxemia in those patients where the combination of a blunted ventilatory response, hypocapnia, shunt physiology, and normal respiratory system compliance is represented by the absence of increased breathing effort despite severe hypoxemia.
If not for the patient’s refusal of medical services, this patient quite possibly would have been intubated due to hypoxemia and health care providers’ concern for her risk of deterioration. Reported intubation and mechanical ventilation rates have varied widely from extremes of from < 5 to 88% in severely ill patients.9,22 About 75% will need oxygen, but many can be treated and recover without the need for intubation and mechanical ventilation.
As previously mentioned, options for treatment include standard and high-flow oxygen delivery, noninvasive ventilation, and awake prone ventilation. Their role in patient management has been recently outlined, and instead of an early intubation strategy, represents gradual escalation of support that may be sufficient to treat hypoxemia and avoid the need for intubation and mechanical ventilation (Table 2).
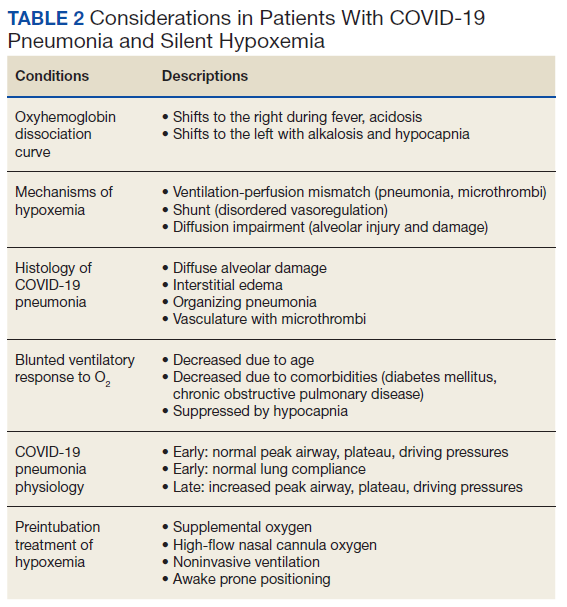
In addition, the patient’s hospital course was notable for the decline in known markers of active inflammation that mirrored the resolution of her hypoxemia and pneumonia. This included elevated lactate dehydrogenase, D-dimer, ferritin, and C-reactive protein with all but the latter rising and decreasing over 2 weeks. These findings provide additional information of the time for recovery and supports the use of these markers to monitor the course of pneumonia.
The patient declined all intervention, including oxygen, and recovered to her presumed prehospitalization condition. This experiment of nature due to unique circumstances may shed light on the natural time course of untreated hypoxemic COVID-19 pneumonia that has not previously been well appreciated. It is important to recognize that recovery occurred over 2 weeks. This is close to the observed and expected time for recovery that has been reported for those with severe COVID-19 pneumonia.
Conclusions
Since the emergence of the COVID-19, evidence has accumulated for the benefit of several adjunctive therapies in the treatment of this type of pneumonia, with corticosteroids providing a mortality benefit. Although unknown whether this patient’s experience can be generalized to others or whether it represents her unique response, this case provides another perspective for comparison of treatments and reinforces the need for prospective, randomized clinical trials to establish treatment efficacy. The exact nature of silent hypoxemia of COVID-19 remains incompletely understood; however, this case highlights the importance of treating the individual instead of clinical markers and provides a time course for recovery from pneumonia and severe hypoxemia that occurs without oxygen or any other treatment over about 2 weeks.
1. Ioannou GN, Locke E, Green P, et al. Risk factors for hospitalization, mechanical ventilation, or death among 10131 US veterans with SARS-CoV-2 infection. JAMA Netw Open. 2020;3(9):e2022310. doi:10.1001/jamanetworkopen.2020.22310
2. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA. 2020;324(8):782-793. doi:10.1001/jama.2020.12839
3. Alhazzani W, Moller MH, Arabi YM, et al. Surviving sepsis campaign: guidelines on the management of critically ill adults with coronavirus disease 2019 (COVID-19). Crit Care Med. 2020;48(6):e440-e469. doi:10.1097/CCM.0000000000004363
4. Ziehr DR, Alladina J, Petri CR, et al. Respiratory pathophysiology of mechanically ventilated patients with COVID-19: a cohort study. Am J Respir Crit Care Med. 2020;201(12):1560-1564. doi:10.1164/rccm.202004-1163LE
5. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020;323(13):1239-1242. doi:10.1001/jama.2020.2648
6. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054-1062. doi:10.1016/S01406736(20)30566-3
7. Tobin MJ, Laghi F, Jubran A. Why COVID-19 silent hypoxemia is baffling to physicians. Am J Respir Crit Care Med. 2020;202(3):356-360. doi:10.1164/rccm.202006-2157CP
8. Guan WJ, Ni ZY, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708-1720. doi:10.1056/NEJMoa2002032
9. Grasselli G, Zangrillo A, Zanella A, et al. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy Region, Italy. JAMA. 2020;323(16):1574-1581. doi:10.1001/jama.2020.5394
10. Raoof S, Nava S, Carpati C, Hill NS. High-flow, noninvasive ventilation and awake (nonintubation) proning in patients with coronavirus disease 2019 with respiratory failure. Chest. 2020;158(5):1992-2002. doi:10.1016/j.chest.2020.07.013
11. Ackermann M, Mentzer SJ, Jonigk D. Pulmonary vascular pathology in COVID-19. Reply. N Engl J Med. 2020;383(9):888-889. doi:10.1056/NEJMc2022068
12. McDonough G, Khaing P, Treacy T, McGrath C, Yoo EJ. The use of high-flow nasal oxygen in the ICU as a first-line therapy for acute hypoxemic respiratory failure secondary to coronavirus disease 2019. Crit Care Explor. 2020;2(10):e0257. doi:10.1097/CCE.0000000000000257
13. Hernandez-Romieu AC, Adelman MW, et al. Timing of intubation and mortality among critically ill coronavirus disease 2019 patients: a single-center cohort study. Crit Care Med. 2020;48(11):e1045-e1053. doi:10.1097/CCM.0000000000004600
14. Cummings MJ, Baldwin MR, Abrams D, et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: a prospective cohort study. Lancet. 2020;395(10239):1763-1770. doi:10.1016/S0140-6736(20)31189-2
15. Dhont S, Derom E, Van Braeckel E, Depuydt P, Lambrecht BN. The pathophysiology of ‘happy’ hypoxemia in COVID-19. Respir Res. 2020;21(1):198. doi:10.1186/s12931-020-01462-5
16. Wilkerson RG, Adler JD, Shah NG, Brown R. Silent hypoxia: a harbinger of clinical deterioration in patients with COVID-19. Am J Emerg Med. 2020;38(10):2243.e5-2243.e6. doi:10.1016/j.ajem.2020.05.044
17. Gong J, Ou J, Qiu X, et al. A tool for early prediction of severe coronavirus disease 2019 (COVID-19): a multicenter study using the risk nomogram in Wuhan and Guangdong, China. Clin Infect Dis. 2020;71(15):833-840. doi:10.1093/cid/ciaa443
18. Force ADT, Ranieri VM, Rubenfeld GD, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23):2526-2533. doi:10.1001/jama.2012.5669
19. Marini JJ, Gattinoni L. Management of COVID-19 respiratory distress. JAMA. 2020;323(22):2329-2330. doi:10.1001/jama.2020.6825
20. Schaller T, Hirschbuhl K, Burkhardt K, et al. Postmortem examination of patients with COVID-19. JAMA. 2020;323(24):2518-2520. doi:10.1001/jama.2020.8907
21. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383(2):120-128. doi:10.1056/NEJMoa2015432
22. Wu C, Chen X, Cai Y, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180(7):934-943. doi:10.1001/jamainternmed.2020.0994. Published correction appeared May 11, 2020. Errors in data and units of measure. doi:10.1001/jamainternmed.2020.1429
23. Yang J, Zheng Y, Gou X, et al. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int J Infect Dis. 2020;94:91-95. doi:10.1016/j.ijid.2020.03.017
24. Richardson S, Hirsch JS, Narasimhan M, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA. 2020;323(20):2052-2059. doi:10.1001/jama.2020.6775
25. Tobin MJ, Jubran A, Laghi F. Misconceptions of pathophysiology of happy hypoxemia and implications for management of COVID-19. Respir Res. 2020;21(1):249. doi:10.1186/s12931-020-01520-y
26. Bickler PE, Feiner JR, Lipnick MS, McKleroy W. “Silent” presentation of hypoxemia and cardiorespiratory compensation in COVID-19. Anesthesiology. 2020;134(2):262-269. doi:10.1097/ALN.0000000000003578
27. Jounieaux V, Parreira VF, Aubert G, Dury M, Delguste P, Rodenstein DO. Effects of hypocapnic hyperventilation on the response to hypoxia in normal subjects receiving intermittent positive-pressure ventilation. Chest. 2002;121(4):1141-1148. doi:10.1378/chest.121.4.1141
In less than a year, COVID-19 has infected nearly 100 million people worldwide and caused more than 2 million deaths and counting. Although the infection fatality rate is estimated to be 1% and the case fatality rate between 2% and 3%, COVID-19 has had a disproportionate effect on the older population and those with comorbidities. Some of these findings are mirrored in the US Department of Veterans Affairs (VA) population, which has seen a higher case fatality rate.1-4
As a respiratory tract infection, the most dreaded presentation is severe pneumonia with acute hypoxemia, which may rapidly deteriorate to acute respiratory distress syndrome (ARDS) and respiratory failure.5-7 This possibility has led to early intubation strategies aimed at preempting this rapid deterioration and minimizing viral exposure to health care workers. Intubation rates have varied widely with extremes of 6 to 88%.8,9
However, this early intubation strategy has waned as some of the rationale behind its endorsement has been called into question. Early intubation bypasses alternatives to intubation; high-flow nasal cannula oxygen, noninvasive ventilation, and awake proning are all effective maneuvers in the appropriate patient.10,11 The use of first-line high-flow nasal cannula oxygen and noninvasive ventilation has been widely reported. Reports of first-line use of high-flow nasal cannula oxygen has not demonstrated inferior outcomes, nor has the timing of intubation, suggesting a significant portion of patients could benefit from a trial of therapy and eventually avoid intubation.11-14 Other therapies, such as systemic corticosteroids, confer a mortality benefit in those patients with COVID-19 who require oxygen or mechanical ventilation, but their impact on the progression of respiratory failure and need for intubation are undetermined.
There also are reports of patients who report no signs of respiratory distress or dyspnea with their COVID-19 pneumonia despite profound hypoxemia or high oxygen requirements. Various terms, including silent hypoxemia or happy hypoxia, are descriptive of the demeanor of these patients, and treatment has invariably included oxygen.15,16 Nevertheless, low oxygen measurements have generally prompted higher levels of supplemental oxygen or more invasive therapies.
Treatment rendered may obscure the trajectory of response, which is important to understand to better position options for invasive therapies and other therapeutics. We recently encountered a patient with a course of illness that represented the natural history of COVID-19 pneumonia with low oxygen levels (referred to as hypoxemia for consistency) that highlighted several issues of management.
Case Presentation
A 62-year-old undomiciled woman with morbid obesity, prediabetes mellitus, long-standing schizophrenia, and bipolar disorder presented to our facility for evaluation of dry cough and need for tuberculosis clearance for admittance to a shelter. She appeared comfortable and was afebrile with blood pressure 111/74 mm Hg, heart rate 82 beats per minute. Her respiratory rate was 18 breaths per minute, but the pulse oximetry showed oxygen saturation of 70 to 75% on room air at rest. A chest X-ray showed bibasilar infiltrates (Figure 1), and a rapid COVID-19 nasopharyngeal polymerase chain reaction (PCR) test returned positive, confirmed by a second PCR test. Baseline inflammatory markers were elevated (Figure 2). In addition, the serum interleukin-6 also was elevated to 66.1 pg/mL (normal < 5.0), erythrocyte sedimentation rate elevated to 69 mm/h, but serum procalcitonin was essentially normal (0.22 ng/mL; normal < 20 ng/mL) as was the serum lactate (1.4 mmol/L).
The patient was admitted to the intensive care unit (ICU) for close monitoring in anticipation of the possibility of decompensation based on her age, hypoxia, and elevated inflammatory markers.17 Besides a subsequent low-grade fever (100.4 oF) and lymphopenia (manual count 550/uL), she remained clinically unchanged. Throughout her hospitalization, she maintained a persistent psychotic delusion that she did not have COVID-19, refusing all medical interventions, including a peripheral IV line and supplemental oxygen for the entire duration. Extensive efforts to identify family or a surrogate decision maker were unsuccessful. After consultation with Psychiatry, Bio-Ethics, and hospital leadership, the patient was deemed to lack decision-making capacity regarding treatment or disposition and was placed on a psychiatric hold. However, since any interventions against her will would require sedation, IV access, and potentially increase the risk of nosocomial COVID-19 transmission, she was allowed to remain untreated and was closely monitored for symptoms of worsening respiratory failure.
Over the next 2 weeks, her hypoxemia, inflammatory markers, and the infiltrates on imaging resolved (Figure 2). The lowest daily awake room air pulse oximetry readings are reported, initially with consistent readings in the low 80% range, but on day 12, readings were > 90% and remained > 90% for the remainder of her hospitalization. Therefore, shortly after hospital day 12, she was clinically stable for discharge from acute care to a subacute facility, but this required documentation of the clearance of her viral infection. She refused to undergo a subsequent nasopharyngeal swab but allowed an oropharyngeal COVID-19 PCR swab, which was negative. She remained stable and unchanged for the remainder of her hospitalization, awaiting identification of a receiving facility and was able to be discharged to transitional housing on day 38.
Discussion
The initial reports of COVID-19 pneumonia focused on ARDS and respiratory failure requiring mechanical ventilation with less emphasis on those with lower severity of illness. This was heightened by health care systems that were overwhelmed with large number of patients while faced with limited supplies and equipment. Given the risk to patients and providers of crash intubations, some recommended early intubation strategies.3 However, the natural history of COVID-19 pneumonia and the threshold for intubation of these patients remain poorly defined despite the creation of prognostic tools.17 This patient’s persistent hypoxemia and elevated inflammatory markers certainly met markers of disease associated with a high risk of progression.
The greatest concern would have been her level of hypoxemia. Acceptable thresholds of hypoxemia vary, but general consensus would classify pulse oximetry < 90% as hypoxemia and a threshold for administering supplemental oxygen. It is important to recognize how pulse oximetry readings translate to partial pressure of oxygen (PaO2) measurements (Table 1). Pulse oximetry readings of 90% corresponds to a PaO2 readings of 60 mm Hg in ideal conditions without the influence of acidosis, PaCO2, or temperature. While lower readings are of concern, these do not represent absolute indications for assisted ventilatory support as lower levels are well tolerated in a variety of conditions. A common example are patients with chronic obstructive pulmonary disease. Long-term mortality benefits of continuous supplemental oxygen are well established in specific populations, but the threshold for correction in the acute setting remains a case-by-case decision. This decision is complex and is based on more than an absolute number or the amount of oxygen required to achieve a threshold level of oxygenation.
The PaO2/FIO2 (fraction of inspired oxygen) is a common measure used to address severity of disease and oxygen requirements. It also has been used to define the severity of ARDS, but the ratio is based on intubated and mechanically ventilated patients and may not translate well to those not on assisted ventilation. Treatment with supplemental oxygen also involves entrained air with associated imprecision in oxygen delivery.18 For this discussion, the patient’s admission PaO2/FIO2 on room air would have been between 190 and 260. Coupled with the bilateral infiltrates on imaging, there was justified concern for progression to severe ARDS. Her presentation would have met most of the epidemiologic criteria used in initial case finding for severe COVID-19 cases, including a blood oxygen saturation ≤ 93%, PaO2/FIO2 < 300 with infiltrates involving close to if not exceeding 50% of the lung.
With COVID-19 pneumonia, the pathologic injury to the alveoli resembles that of any viral pneumonia with recruitment of predominantly lymphocytic inflammatory cells that fill the alveoli, derangements in ventilation/perfusion mismatch as the core mechanism of hypoxemia with interstitial edema and shuntlike physiology developing at the extremes of involvement. In later stages, the histologic appearance is similar to ARDS, including hyaline membrane formation and thickened alveolar septa with perivascular lymphocytic-plasmocytic infiltration. In addition, there also are findings of organizing pneumonia with fibroblastic proliferation, thrombosis, and diffuse alveolar damage, a constellation of findings similar to that seen in the latter stages of ARDS.2
Although these histologic findings resemble ARDS, many patients with respiratory failure due to COVID-19 have a different physiologic profile compared with those with typical ARDS, with the most striking finding of lungs with low elastance or high compliance. From the critical care standpoint, this meant that the lungs were relatively easy to ventilate with lower peak airway and plateau pressures and low driving pressures. This condition suggested that there was relatively less lung that could be recruited with positive end expiratory pressure; therefore, a somewhat different entity from that associated with ARDS.19 These findings were often noted early in the course of respiratory failure, and although there is debate about whether this represents a different phenotype or timepoint in the spectrum of disease, it clearly represents a subset that is distinct from that which had been previously encountered.
On the other hand, the clinical features seen in those patients with COVID-19 pneumonia who progressed to advanced respiratory failure were essentially indistinguishable from those patients with traditional ARDS. Other explanations for this respiratory failure have included a disrupted vasoregulatory response to hypoxemia with failed hypoxic vasoconstriction, intravascular microthrombi, and impaired diffusion, all contributing to impaired gas exchange and hypoxemia.19-21 This can lead to shuntlike conditions that neither respond well to supplemental oxygen nor manifest the type of physiologic response seen with other causes of hypoxemia.
The severity of hypoxemia manifested by this patient may have elicited additional findings of respiratory distress, such as dyspnea and tachypnea. However, in patients with severe COVID-19 pneumonia, dyspnea was not a universal finding, reported in the 20 to 60% range of cohorts, higher in those with ARDS and mechanical ventilation, although some report near universal dyspnea in their series.1,4,8,22,23 Tachypnea is another symptom of interest. Using a threshold of > 24 breaths/min, tachypnea was noted in 16 to 29% of patients with a much greater proportion (63%) in nonsurvivors.6,24 Several explanations have been proposed for the discordance between the presence and severity of hypoxemia and lack of symptoms of dyspnea and tachypnea. It is important to recognize that misclassification of the severity of hypoxemia can occur due to technical issues and potential errors involving pulse oximetry measurement and shifts in the oxyhemoglobin dissociation curve. However, this is more pertinent for those with mild disease as the severity of hypoxemia in severe pneumonia is beyond what can be attributed to technical issues.
More important, the ventilatory response curve to hypoxemia may not be normal for some patients, blunted by as much as 50% in older patients, especially in those with diabetes mellitus.7,25,26 In addition, the ventilatory response varies widely even among normal individuals. This would translate to lower levels of minute ventilation (less tachypnea or respiratory effort) with hypoxemia. Hypocapnic hypoxemia also blunts the ventilatory response to hypoxemia. Subjects do not increase their minute ventilation if the PaCO2 remains low despite oxygen desaturation to < 70%, especially if PaCO2 < 30 mm Hg or alternatively, increases in minute ventilation are not seen until the PaCO2 exceeds 39 mm Hg.27 Both scenarios occur in those with COVID-19 pneumonia and provide another explanation for the absence of respiratory symptoms or signs of respiratory distress in some patients.
The observation of more compliant lungs may help in the understanding of the variable presentation of these patients. Compliant lungs do not require the increased pressure needed to achieve a specific tidal volume that, in turn, may increase the work of breathing. This may add to the explanation of seemingly paradoxical silent hypoxemia in those patients where the combination of a blunted ventilatory response, hypocapnia, shunt physiology, and normal respiratory system compliance is represented by the absence of increased breathing effort despite severe hypoxemia.
If not for the patient’s refusal of medical services, this patient quite possibly would have been intubated due to hypoxemia and health care providers’ concern for her risk of deterioration. Reported intubation and mechanical ventilation rates have varied widely from extremes of from < 5 to 88% in severely ill patients.9,22 About 75% will need oxygen, but many can be treated and recover without the need for intubation and mechanical ventilation.
As previously mentioned, options for treatment include standard and high-flow oxygen delivery, noninvasive ventilation, and awake prone ventilation. Their role in patient management has been recently outlined, and instead of an early intubation strategy, represents gradual escalation of support that may be sufficient to treat hypoxemia and avoid the need for intubation and mechanical ventilation (Table 2).
In addition, the patient’s hospital course was notable for the decline in known markers of active inflammation that mirrored the resolution of her hypoxemia and pneumonia. This included elevated lactate dehydrogenase, D-dimer, ferritin, and C-reactive protein with all but the latter rising and decreasing over 2 weeks. These findings provide additional information of the time for recovery and supports the use of these markers to monitor the course of pneumonia.
The patient declined all intervention, including oxygen, and recovered to her presumed prehospitalization condition. This experiment of nature due to unique circumstances may shed light on the natural time course of untreated hypoxemic COVID-19 pneumonia that has not previously been well appreciated. It is important to recognize that recovery occurred over 2 weeks. This is close to the observed and expected time for recovery that has been reported for those with severe COVID-19 pneumonia.
Conclusions
Since the emergence of the COVID-19, evidence has accumulated for the benefit of several adjunctive therapies in the treatment of this type of pneumonia, with corticosteroids providing a mortality benefit. Although unknown whether this patient’s experience can be generalized to others or whether it represents her unique response, this case provides another perspective for comparison of treatments and reinforces the need for prospective, randomized clinical trials to establish treatment efficacy. The exact nature of silent hypoxemia of COVID-19 remains incompletely understood; however, this case highlights the importance of treating the individual instead of clinical markers and provides a time course for recovery from pneumonia and severe hypoxemia that occurs without oxygen or any other treatment over about 2 weeks.
In less than a year, COVID-19 has infected nearly 100 million people worldwide and caused more than 2 million deaths and counting. Although the infection fatality rate is estimated to be 1% and the case fatality rate between 2% and 3%, COVID-19 has had a disproportionate effect on the older population and those with comorbidities. Some of these findings are mirrored in the US Department of Veterans Affairs (VA) population, which has seen a higher case fatality rate.1-4
As a respiratory tract infection, the most dreaded presentation is severe pneumonia with acute hypoxemia, which may rapidly deteriorate to acute respiratory distress syndrome (ARDS) and respiratory failure.5-7 This possibility has led to early intubation strategies aimed at preempting this rapid deterioration and minimizing viral exposure to health care workers. Intubation rates have varied widely with extremes of 6 to 88%.8,9
However, this early intubation strategy has waned as some of the rationale behind its endorsement has been called into question. Early intubation bypasses alternatives to intubation; high-flow nasal cannula oxygen, noninvasive ventilation, and awake proning are all effective maneuvers in the appropriate patient.10,11 The use of first-line high-flow nasal cannula oxygen and noninvasive ventilation has been widely reported. Reports of first-line use of high-flow nasal cannula oxygen has not demonstrated inferior outcomes, nor has the timing of intubation, suggesting a significant portion of patients could benefit from a trial of therapy and eventually avoid intubation.11-14 Other therapies, such as systemic corticosteroids, confer a mortality benefit in those patients with COVID-19 who require oxygen or mechanical ventilation, but their impact on the progression of respiratory failure and need for intubation are undetermined.
There also are reports of patients who report no signs of respiratory distress or dyspnea with their COVID-19 pneumonia despite profound hypoxemia or high oxygen requirements. Various terms, including silent hypoxemia or happy hypoxia, are descriptive of the demeanor of these patients, and treatment has invariably included oxygen.15,16 Nevertheless, low oxygen measurements have generally prompted higher levels of supplemental oxygen or more invasive therapies.
Treatment rendered may obscure the trajectory of response, which is important to understand to better position options for invasive therapies and other therapeutics. We recently encountered a patient with a course of illness that represented the natural history of COVID-19 pneumonia with low oxygen levels (referred to as hypoxemia for consistency) that highlighted several issues of management.
Case Presentation
A 62-year-old undomiciled woman with morbid obesity, prediabetes mellitus, long-standing schizophrenia, and bipolar disorder presented to our facility for evaluation of dry cough and need for tuberculosis clearance for admittance to a shelter. She appeared comfortable and was afebrile with blood pressure 111/74 mm Hg, heart rate 82 beats per minute. Her respiratory rate was 18 breaths per minute, but the pulse oximetry showed oxygen saturation of 70 to 75% on room air at rest. A chest X-ray showed bibasilar infiltrates (Figure 1), and a rapid COVID-19 nasopharyngeal polymerase chain reaction (PCR) test returned positive, confirmed by a second PCR test. Baseline inflammatory markers were elevated (Figure 2). In addition, the serum interleukin-6 also was elevated to 66.1 pg/mL (normal < 5.0), erythrocyte sedimentation rate elevated to 69 mm/h, but serum procalcitonin was essentially normal (0.22 ng/mL; normal < 20 ng/mL) as was the serum lactate (1.4 mmol/L).
The patient was admitted to the intensive care unit (ICU) for close monitoring in anticipation of the possibility of decompensation based on her age, hypoxia, and elevated inflammatory markers.17 Besides a subsequent low-grade fever (100.4 oF) and lymphopenia (manual count 550/uL), she remained clinically unchanged. Throughout her hospitalization, she maintained a persistent psychotic delusion that she did not have COVID-19, refusing all medical interventions, including a peripheral IV line and supplemental oxygen for the entire duration. Extensive efforts to identify family or a surrogate decision maker were unsuccessful. After consultation with Psychiatry, Bio-Ethics, and hospital leadership, the patient was deemed to lack decision-making capacity regarding treatment or disposition and was placed on a psychiatric hold. However, since any interventions against her will would require sedation, IV access, and potentially increase the risk of nosocomial COVID-19 transmission, she was allowed to remain untreated and was closely monitored for symptoms of worsening respiratory failure.
Over the next 2 weeks, her hypoxemia, inflammatory markers, and the infiltrates on imaging resolved (Figure 2). The lowest daily awake room air pulse oximetry readings are reported, initially with consistent readings in the low 80% range, but on day 12, readings were > 90% and remained > 90% for the remainder of her hospitalization. Therefore, shortly after hospital day 12, she was clinically stable for discharge from acute care to a subacute facility, but this required documentation of the clearance of her viral infection. She refused to undergo a subsequent nasopharyngeal swab but allowed an oropharyngeal COVID-19 PCR swab, which was negative. She remained stable and unchanged for the remainder of her hospitalization, awaiting identification of a receiving facility and was able to be discharged to transitional housing on day 38.
Discussion
The initial reports of COVID-19 pneumonia focused on ARDS and respiratory failure requiring mechanical ventilation with less emphasis on those with lower severity of illness. This was heightened by health care systems that were overwhelmed with large number of patients while faced with limited supplies and equipment. Given the risk to patients and providers of crash intubations, some recommended early intubation strategies.3 However, the natural history of COVID-19 pneumonia and the threshold for intubation of these patients remain poorly defined despite the creation of prognostic tools.17 This patient’s persistent hypoxemia and elevated inflammatory markers certainly met markers of disease associated with a high risk of progression.
The greatest concern would have been her level of hypoxemia. Acceptable thresholds of hypoxemia vary, but general consensus would classify pulse oximetry < 90% as hypoxemia and a threshold for administering supplemental oxygen. It is important to recognize how pulse oximetry readings translate to partial pressure of oxygen (PaO2) measurements (Table 1). Pulse oximetry readings of 90% corresponds to a PaO2 readings of 60 mm Hg in ideal conditions without the influence of acidosis, PaCO2, or temperature. While lower readings are of concern, these do not represent absolute indications for assisted ventilatory support as lower levels are well tolerated in a variety of conditions. A common example are patients with chronic obstructive pulmonary disease. Long-term mortality benefits of continuous supplemental oxygen are well established in specific populations, but the threshold for correction in the acute setting remains a case-by-case decision. This decision is complex and is based on more than an absolute number or the amount of oxygen required to achieve a threshold level of oxygenation.
The PaO2/FIO2 (fraction of inspired oxygen) is a common measure used to address severity of disease and oxygen requirements. It also has been used to define the severity of ARDS, but the ratio is based on intubated and mechanically ventilated patients and may not translate well to those not on assisted ventilation. Treatment with supplemental oxygen also involves entrained air with associated imprecision in oxygen delivery.18 For this discussion, the patient’s admission PaO2/FIO2 on room air would have been between 190 and 260. Coupled with the bilateral infiltrates on imaging, there was justified concern for progression to severe ARDS. Her presentation would have met most of the epidemiologic criteria used in initial case finding for severe COVID-19 cases, including a blood oxygen saturation ≤ 93%, PaO2/FIO2 < 300 with infiltrates involving close to if not exceeding 50% of the lung.
With COVID-19 pneumonia, the pathologic injury to the alveoli resembles that of any viral pneumonia with recruitment of predominantly lymphocytic inflammatory cells that fill the alveoli, derangements in ventilation/perfusion mismatch as the core mechanism of hypoxemia with interstitial edema and shuntlike physiology developing at the extremes of involvement. In later stages, the histologic appearance is similar to ARDS, including hyaline membrane formation and thickened alveolar septa with perivascular lymphocytic-plasmocytic infiltration. In addition, there also are findings of organizing pneumonia with fibroblastic proliferation, thrombosis, and diffuse alveolar damage, a constellation of findings similar to that seen in the latter stages of ARDS.2
Although these histologic findings resemble ARDS, many patients with respiratory failure due to COVID-19 have a different physiologic profile compared with those with typical ARDS, with the most striking finding of lungs with low elastance or high compliance. From the critical care standpoint, this meant that the lungs were relatively easy to ventilate with lower peak airway and plateau pressures and low driving pressures. This condition suggested that there was relatively less lung that could be recruited with positive end expiratory pressure; therefore, a somewhat different entity from that associated with ARDS.19 These findings were often noted early in the course of respiratory failure, and although there is debate about whether this represents a different phenotype or timepoint in the spectrum of disease, it clearly represents a subset that is distinct from that which had been previously encountered.
On the other hand, the clinical features seen in those patients with COVID-19 pneumonia who progressed to advanced respiratory failure were essentially indistinguishable from those patients with traditional ARDS. Other explanations for this respiratory failure have included a disrupted vasoregulatory response to hypoxemia with failed hypoxic vasoconstriction, intravascular microthrombi, and impaired diffusion, all contributing to impaired gas exchange and hypoxemia.19-21 This can lead to shuntlike conditions that neither respond well to supplemental oxygen nor manifest the type of physiologic response seen with other causes of hypoxemia.
The severity of hypoxemia manifested by this patient may have elicited additional findings of respiratory distress, such as dyspnea and tachypnea. However, in patients with severe COVID-19 pneumonia, dyspnea was not a universal finding, reported in the 20 to 60% range of cohorts, higher in those with ARDS and mechanical ventilation, although some report near universal dyspnea in their series.1,4,8,22,23 Tachypnea is another symptom of interest. Using a threshold of > 24 breaths/min, tachypnea was noted in 16 to 29% of patients with a much greater proportion (63%) in nonsurvivors.6,24 Several explanations have been proposed for the discordance between the presence and severity of hypoxemia and lack of symptoms of dyspnea and tachypnea. It is important to recognize that misclassification of the severity of hypoxemia can occur due to technical issues and potential errors involving pulse oximetry measurement and shifts in the oxyhemoglobin dissociation curve. However, this is more pertinent for those with mild disease as the severity of hypoxemia in severe pneumonia is beyond what can be attributed to technical issues.
More important, the ventilatory response curve to hypoxemia may not be normal for some patients, blunted by as much as 50% in older patients, especially in those with diabetes mellitus.7,25,26 In addition, the ventilatory response varies widely even among normal individuals. This would translate to lower levels of minute ventilation (less tachypnea or respiratory effort) with hypoxemia. Hypocapnic hypoxemia also blunts the ventilatory response to hypoxemia. Subjects do not increase their minute ventilation if the PaCO2 remains low despite oxygen desaturation to < 70%, especially if PaCO2 < 30 mm Hg or alternatively, increases in minute ventilation are not seen until the PaCO2 exceeds 39 mm Hg.27 Both scenarios occur in those with COVID-19 pneumonia and provide another explanation for the absence of respiratory symptoms or signs of respiratory distress in some patients.
The observation of more compliant lungs may help in the understanding of the variable presentation of these patients. Compliant lungs do not require the increased pressure needed to achieve a specific tidal volume that, in turn, may increase the work of breathing. This may add to the explanation of seemingly paradoxical silent hypoxemia in those patients where the combination of a blunted ventilatory response, hypocapnia, shunt physiology, and normal respiratory system compliance is represented by the absence of increased breathing effort despite severe hypoxemia.
If not for the patient’s refusal of medical services, this patient quite possibly would have been intubated due to hypoxemia and health care providers’ concern for her risk of deterioration. Reported intubation and mechanical ventilation rates have varied widely from extremes of from < 5 to 88% in severely ill patients.9,22 About 75% will need oxygen, but many can be treated and recover without the need for intubation and mechanical ventilation.
As previously mentioned, options for treatment include standard and high-flow oxygen delivery, noninvasive ventilation, and awake prone ventilation. Their role in patient management has been recently outlined, and instead of an early intubation strategy, represents gradual escalation of support that may be sufficient to treat hypoxemia and avoid the need for intubation and mechanical ventilation (Table 2).
In addition, the patient’s hospital course was notable for the decline in known markers of active inflammation that mirrored the resolution of her hypoxemia and pneumonia. This included elevated lactate dehydrogenase, D-dimer, ferritin, and C-reactive protein with all but the latter rising and decreasing over 2 weeks. These findings provide additional information of the time for recovery and supports the use of these markers to monitor the course of pneumonia.
The patient declined all intervention, including oxygen, and recovered to her presumed prehospitalization condition. This experiment of nature due to unique circumstances may shed light on the natural time course of untreated hypoxemic COVID-19 pneumonia that has not previously been well appreciated. It is important to recognize that recovery occurred over 2 weeks. This is close to the observed and expected time for recovery that has been reported for those with severe COVID-19 pneumonia.
Conclusions
Since the emergence of the COVID-19, evidence has accumulated for the benefit of several adjunctive therapies in the treatment of this type of pneumonia, with corticosteroids providing a mortality benefit. Although unknown whether this patient’s experience can be generalized to others or whether it represents her unique response, this case provides another perspective for comparison of treatments and reinforces the need for prospective, randomized clinical trials to establish treatment efficacy. The exact nature of silent hypoxemia of COVID-19 remains incompletely understood; however, this case highlights the importance of treating the individual instead of clinical markers and provides a time course for recovery from pneumonia and severe hypoxemia that occurs without oxygen or any other treatment over about 2 weeks.
1. Ioannou GN, Locke E, Green P, et al. Risk factors for hospitalization, mechanical ventilation, or death among 10131 US veterans with SARS-CoV-2 infection. JAMA Netw Open. 2020;3(9):e2022310. doi:10.1001/jamanetworkopen.2020.22310
2. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA. 2020;324(8):782-793. doi:10.1001/jama.2020.12839
3. Alhazzani W, Moller MH, Arabi YM, et al. Surviving sepsis campaign: guidelines on the management of critically ill adults with coronavirus disease 2019 (COVID-19). Crit Care Med. 2020;48(6):e440-e469. doi:10.1097/CCM.0000000000004363
4. Ziehr DR, Alladina J, Petri CR, et al. Respiratory pathophysiology of mechanically ventilated patients with COVID-19: a cohort study. Am J Respir Crit Care Med. 2020;201(12):1560-1564. doi:10.1164/rccm.202004-1163LE
5. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020;323(13):1239-1242. doi:10.1001/jama.2020.2648
6. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054-1062. doi:10.1016/S01406736(20)30566-3
7. Tobin MJ, Laghi F, Jubran A. Why COVID-19 silent hypoxemia is baffling to physicians. Am J Respir Crit Care Med. 2020;202(3):356-360. doi:10.1164/rccm.202006-2157CP
8. Guan WJ, Ni ZY, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708-1720. doi:10.1056/NEJMoa2002032
9. Grasselli G, Zangrillo A, Zanella A, et al. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy Region, Italy. JAMA. 2020;323(16):1574-1581. doi:10.1001/jama.2020.5394
10. Raoof S, Nava S, Carpati C, Hill NS. High-flow, noninvasive ventilation and awake (nonintubation) proning in patients with coronavirus disease 2019 with respiratory failure. Chest. 2020;158(5):1992-2002. doi:10.1016/j.chest.2020.07.013
11. Ackermann M, Mentzer SJ, Jonigk D. Pulmonary vascular pathology in COVID-19. Reply. N Engl J Med. 2020;383(9):888-889. doi:10.1056/NEJMc2022068
12. McDonough G, Khaing P, Treacy T, McGrath C, Yoo EJ. The use of high-flow nasal oxygen in the ICU as a first-line therapy for acute hypoxemic respiratory failure secondary to coronavirus disease 2019. Crit Care Explor. 2020;2(10):e0257. doi:10.1097/CCE.0000000000000257
13. Hernandez-Romieu AC, Adelman MW, et al. Timing of intubation and mortality among critically ill coronavirus disease 2019 patients: a single-center cohort study. Crit Care Med. 2020;48(11):e1045-e1053. doi:10.1097/CCM.0000000000004600
14. Cummings MJ, Baldwin MR, Abrams D, et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: a prospective cohort study. Lancet. 2020;395(10239):1763-1770. doi:10.1016/S0140-6736(20)31189-2
15. Dhont S, Derom E, Van Braeckel E, Depuydt P, Lambrecht BN. The pathophysiology of ‘happy’ hypoxemia in COVID-19. Respir Res. 2020;21(1):198. doi:10.1186/s12931-020-01462-5
16. Wilkerson RG, Adler JD, Shah NG, Brown R. Silent hypoxia: a harbinger of clinical deterioration in patients with COVID-19. Am J Emerg Med. 2020;38(10):2243.e5-2243.e6. doi:10.1016/j.ajem.2020.05.044
17. Gong J, Ou J, Qiu X, et al. A tool for early prediction of severe coronavirus disease 2019 (COVID-19): a multicenter study using the risk nomogram in Wuhan and Guangdong, China. Clin Infect Dis. 2020;71(15):833-840. doi:10.1093/cid/ciaa443
18. Force ADT, Ranieri VM, Rubenfeld GD, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23):2526-2533. doi:10.1001/jama.2012.5669
19. Marini JJ, Gattinoni L. Management of COVID-19 respiratory distress. JAMA. 2020;323(22):2329-2330. doi:10.1001/jama.2020.6825
20. Schaller T, Hirschbuhl K, Burkhardt K, et al. Postmortem examination of patients with COVID-19. JAMA. 2020;323(24):2518-2520. doi:10.1001/jama.2020.8907
21. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383(2):120-128. doi:10.1056/NEJMoa2015432
22. Wu C, Chen X, Cai Y, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180(7):934-943. doi:10.1001/jamainternmed.2020.0994. Published correction appeared May 11, 2020. Errors in data and units of measure. doi:10.1001/jamainternmed.2020.1429
23. Yang J, Zheng Y, Gou X, et al. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int J Infect Dis. 2020;94:91-95. doi:10.1016/j.ijid.2020.03.017
24. Richardson S, Hirsch JS, Narasimhan M, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA. 2020;323(20):2052-2059. doi:10.1001/jama.2020.6775
25. Tobin MJ, Jubran A, Laghi F. Misconceptions of pathophysiology of happy hypoxemia and implications for management of COVID-19. Respir Res. 2020;21(1):249. doi:10.1186/s12931-020-01520-y
26. Bickler PE, Feiner JR, Lipnick MS, McKleroy W. “Silent” presentation of hypoxemia and cardiorespiratory compensation in COVID-19. Anesthesiology. 2020;134(2):262-269. doi:10.1097/ALN.0000000000003578
27. Jounieaux V, Parreira VF, Aubert G, Dury M, Delguste P, Rodenstein DO. Effects of hypocapnic hyperventilation on the response to hypoxia in normal subjects receiving intermittent positive-pressure ventilation. Chest. 2002;121(4):1141-1148. doi:10.1378/chest.121.4.1141
1. Ioannou GN, Locke E, Green P, et al. Risk factors for hospitalization, mechanical ventilation, or death among 10131 US veterans with SARS-CoV-2 infection. JAMA Netw Open. 2020;3(9):e2022310. doi:10.1001/jamanetworkopen.2020.22310
2. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA. 2020;324(8):782-793. doi:10.1001/jama.2020.12839
3. Alhazzani W, Moller MH, Arabi YM, et al. Surviving sepsis campaign: guidelines on the management of critically ill adults with coronavirus disease 2019 (COVID-19). Crit Care Med. 2020;48(6):e440-e469. doi:10.1097/CCM.0000000000004363
4. Ziehr DR, Alladina J, Petri CR, et al. Respiratory pathophysiology of mechanically ventilated patients with COVID-19: a cohort study. Am J Respir Crit Care Med. 2020;201(12):1560-1564. doi:10.1164/rccm.202004-1163LE
5. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020;323(13):1239-1242. doi:10.1001/jama.2020.2648
6. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054-1062. doi:10.1016/S01406736(20)30566-3
7. Tobin MJ, Laghi F, Jubran A. Why COVID-19 silent hypoxemia is baffling to physicians. Am J Respir Crit Care Med. 2020;202(3):356-360. doi:10.1164/rccm.202006-2157CP
8. Guan WJ, Ni ZY, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708-1720. doi:10.1056/NEJMoa2002032
9. Grasselli G, Zangrillo A, Zanella A, et al. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy Region, Italy. JAMA. 2020;323(16):1574-1581. doi:10.1001/jama.2020.5394
10. Raoof S, Nava S, Carpati C, Hill NS. High-flow, noninvasive ventilation and awake (nonintubation) proning in patients with coronavirus disease 2019 with respiratory failure. Chest. 2020;158(5):1992-2002. doi:10.1016/j.chest.2020.07.013
11. Ackermann M, Mentzer SJ, Jonigk D. Pulmonary vascular pathology in COVID-19. Reply. N Engl J Med. 2020;383(9):888-889. doi:10.1056/NEJMc2022068
12. McDonough G, Khaing P, Treacy T, McGrath C, Yoo EJ. The use of high-flow nasal oxygen in the ICU as a first-line therapy for acute hypoxemic respiratory failure secondary to coronavirus disease 2019. Crit Care Explor. 2020;2(10):e0257. doi:10.1097/CCE.0000000000000257
13. Hernandez-Romieu AC, Adelman MW, et al. Timing of intubation and mortality among critically ill coronavirus disease 2019 patients: a single-center cohort study. Crit Care Med. 2020;48(11):e1045-e1053. doi:10.1097/CCM.0000000000004600
14. Cummings MJ, Baldwin MR, Abrams D, et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: a prospective cohort study. Lancet. 2020;395(10239):1763-1770. doi:10.1016/S0140-6736(20)31189-2
15. Dhont S, Derom E, Van Braeckel E, Depuydt P, Lambrecht BN. The pathophysiology of ‘happy’ hypoxemia in COVID-19. Respir Res. 2020;21(1):198. doi:10.1186/s12931-020-01462-5
16. Wilkerson RG, Adler JD, Shah NG, Brown R. Silent hypoxia: a harbinger of clinical deterioration in patients with COVID-19. Am J Emerg Med. 2020;38(10):2243.e5-2243.e6. doi:10.1016/j.ajem.2020.05.044
17. Gong J, Ou J, Qiu X, et al. A tool for early prediction of severe coronavirus disease 2019 (COVID-19): a multicenter study using the risk nomogram in Wuhan and Guangdong, China. Clin Infect Dis. 2020;71(15):833-840. doi:10.1093/cid/ciaa443
18. Force ADT, Ranieri VM, Rubenfeld GD, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23):2526-2533. doi:10.1001/jama.2012.5669
19. Marini JJ, Gattinoni L. Management of COVID-19 respiratory distress. JAMA. 2020;323(22):2329-2330. doi:10.1001/jama.2020.6825
20. Schaller T, Hirschbuhl K, Burkhardt K, et al. Postmortem examination of patients with COVID-19. JAMA. 2020;323(24):2518-2520. doi:10.1001/jama.2020.8907
21. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383(2):120-128. doi:10.1056/NEJMoa2015432
22. Wu C, Chen X, Cai Y, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180(7):934-943. doi:10.1001/jamainternmed.2020.0994. Published correction appeared May 11, 2020. Errors in data and units of measure. doi:10.1001/jamainternmed.2020.1429
23. Yang J, Zheng Y, Gou X, et al. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int J Infect Dis. 2020;94:91-95. doi:10.1016/j.ijid.2020.03.017
24. Richardson S, Hirsch JS, Narasimhan M, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA. 2020;323(20):2052-2059. doi:10.1001/jama.2020.6775
25. Tobin MJ, Jubran A, Laghi F. Misconceptions of pathophysiology of happy hypoxemia and implications for management of COVID-19. Respir Res. 2020;21(1):249. doi:10.1186/s12931-020-01520-y
26. Bickler PE, Feiner JR, Lipnick MS, McKleroy W. “Silent” presentation of hypoxemia and cardiorespiratory compensation in COVID-19. Anesthesiology. 2020;134(2):262-269. doi:10.1097/ALN.0000000000003578
27. Jounieaux V, Parreira VF, Aubert G, Dury M, Delguste P, Rodenstein DO. Effects of hypocapnic hyperventilation on the response to hypoxia in normal subjects receiving intermittent positive-pressure ventilation. Chest. 2002;121(4):1141-1148. doi:10.1378/chest.121.4.1141
Correction of Dialysis-Induced Metabolic Alkalosis
Metabolic alkalosis, a disorder that causes elevations in serum bicarbonate and arterial pH, is a common metabolic abnormality found in nearly half of hospitalized patients but is rare in patients with end-stage renal disease (ESRD) on hemodialysis (HD) during the pretreatment state. The problem seems to arise due to a high rate of older patients with multiple comorbidities and malnutrition who are undergoing HD. Metabolic alkalosis is associated with increased morbidity and mortality. In this report, we present a case of metabolic alkalosis, describe an innovative approach to manage metabolic alkalosis in the dialysis population, and review the pathophysiology.
Case Presentation
A 63-year-old female with emphysema, diabetic nephropathy, and ESRD on regular HD for 2 months by a tunneled subclavian vein catheter was admitted with 2 weeks of orthopnea and leg swelling. The review of systems was negative for chest pain, cough, wheeze, or sputum production. She was a former smoker with no alcohol or drug misuse. The patient was taking carvedilol 25 mg daily, furosemide 20 mg twice daily, basal insulin premeal, lisinopril 40 mg daily, pantoprazole 40 mg daily, calcium carbonate 400 mg 3 times daily, ferrous sulphate 325 mg daily, and a vilanterol/tiotropium inhaler once daily. Her dialysate outpatient prescription included sodium 140 mEq/L, potassium 2 mEq/L, calcium 2.5 mEq/L, and bicarbonate 36 mEq/L. Our dialysis unit used NaturaLyte dry pack for bicarbonate dialysis.
The patient appeared tachypneic with 26 respirations/min, oxygen saturation of 89% on room air, which improved to 94% on a 2 L nasal cannula. Her heart rate was 89 beats/min, blood pressure was 129/72 mm Hg, and body mass index was 21.2. The physical examination revealed jugular venous distension, lung crackles, reduced air entry, and pedal edema. Muscle wasting was noted in the arms and thighs. The tunnel catheter did not appear infected.
The patient’s blood work showed sodium, 136 (reference, 132-140) mmol/L; potassium, 4.3 (reference, 3.5-5.0) mmol/L; chloride, 89 (reference, 98-111) mmol/L; total CO2, 36 (reference, 24-28) mEq/L; blood urea nitrogen, 21 (reference, 7-21) mg/dL; creatinine 3.4 (reference, 0.5-1.4) mg/dL; and albumin, 2.7 (reference, 3.7-5.0) mg/dL. Arterial gases showed pH, 7.56 (reference, 7.35-7.45), partial CO2, 47 (reference, 35-45) mm Hg; bicarbonate, 42 (reference, 22-26) mEq/L; partial O2, 54 (reference, 75 to 100) mm Hg. Brain natriuretic peptide was 2,800 (normal, < 100) pg/mL with a normal troponin. X-rays showed pulmonary congestion and bilateral pleural effusions that were transudative on fluid analysis. An echocardiogram showed ejection fraction of 20 to 25% with normal valves (baseline ejection fraction of 60%-65%). A coronary arteriogram revealed severe nonischemic cardiomyopathy.
Treatment
To reduce bicarbonate levels, 3 L of normal saline solution were infused prefilter during HD, and ultrafiltration (UF) of 4.5 L achieved a net UF of -1.5 L over 3.5 hours on lower dialysate bicarbonate (30 mEq/L). Good catheter flow was achieved with a blood flow rate of 350 mL/min and a dialysate flow of 700 mL/min. Venous blood gases and basic serum metabolic panels were obtained throughout the first HD session (Table 1). Improvement in pH from 7.5 to 7.43 and in total CO2 from 36 to 30 mEq/L were noted after the treatment. Subsequently, we used the same membrane (Optiflux F160NRe) for 2 consecutive daily treatments to remove excess fluid and prevent worsening alkalosis using the same minimal bicarbonate bath, but no further normal saline solution was given.
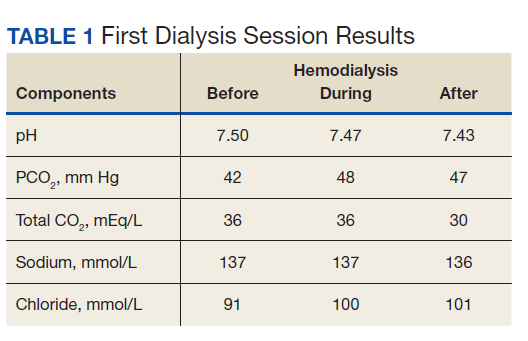
Outcome
Volume overload was controlled as needed with UF. The bicarbonate did not drop after the second HD session, suggesting low organic acid production in the intradialytic period. By shortening the duration of dialysis to 3 hours and improving nutritional intake, we achieved dry weight, and the patient was discharged home with a total CO2 of 25 mEq/L. Outpatient dialysis sessions were arranged to run at shorter duration (3 hours compared with 3.5 hours) and use low bicarbonate dialysate. The patient was admitted several times afterward for acute decompensated heart failure, but in all those admissions, her bicarbonate was in the normal-to-high range, between 23 and 30 mEq/L.
Discussion
Metabolic alkalosis is relatively rare in ESRD patients on HD. Particularly in the predialysis period, but with the growing number of older patients undergoing HD and the aggressive treatment of acidosis with relatively higher buffer concentrations; there has been an increase in the incidence of metabolic alkalosis in patients on HD. In the Fresenius Medical Care (FMC) prevalent HD patient study, predialysis bicarbonate levels have increased overtime from a mean (SD)22.9 (3.1) mEq/L in 2004 to a mean (SD) 24.1 (3.5) mEq/L in September 2011, with 25% of patients > 26.0 mEq/L compared with only 6% in 2004.1 The condition has been associated with cardiac arrhythmia, intradialytic hypocalcemia, hypokalemia, hypercapnia, hypoxia, accelerated hypertension, and seizure.2-4 Metabolic alkalosis may be associated with increased mortality.5-7 However, the effect dissipated after adjusting for inflammation and nutritional status.6
Our patient had primary metabolic alkalosis evident by her high pH of 7.56 and high total CO2 of 36 mEq/L. The serum total CO2 reflects the metabolic status more accurately than the blood gas bicarbonate, which is prone to calculation error by the Henderson-Hasselbalch equation. Her respiratory compensation for the metabolic alkalosis was appropriate, with an increase of arterial PaCO2 to 47 mm Hg (
In patients with ESRD on HD who have no residual urine output, causes of metabolic alkalosis are limited to loss of net acid or gain of alkali through the gastrointestinal tract; our patient had none of these. Similarly, all renal causes of metabolic alkalosis are not applicable to our patient, including mineralocorticoid excess and contraction alkalosis. In patients with preserved kidney function, loop diuretics can induce alkalosis through enhanced tubular absorption of HCO3. While acetazolamide can mitigate this scenario by blocking carbonic anhydrase in the luminal border of the collecting ducts resulting in excretion of bicarbonate in the urine, our patient had negligible urine output despite being on furosemide 20 mg twice daily, making this an unlikely cause.
Severe metabolic alkalosis in dialysis patients has been reported with cocaine use, pica ingestion, and citrate load as in plasma exchange, massive transfusions, and regional anticoagulation.2,8-11 Although calcium carbonate intake can contribute to alkalosis, her small daily dose of 1,200 mg contains approximately 12 mEq of carbonate, which is not a significant contributor to the alkalosis.
With all other causes excluded, the metabolic alkalosis in our patient is presumed to result from the bicarbonate-rich dialysate. Since the majority of patients with ESRD are acidotic before dialysis, the dialysate bicarbonate is set at a higher than normal physiologic level to bring the pH close to or even higher than normal after dialysis. The patient had been dialyzed with NaturaLyte as an outpatient, which was set at the dialysis unit default mode of 36 mEq/L. This form of alkalosis has been reported to peak immediately after treatment but in most patients returns to the predialysis acidotic state due to endogenous acid production.1,4,12 Normally, muscles play a significant role in buffering excess bicarbonate in patients with nonfunctioning kidneys; hence, malnutrition with muscle wasting tends to propagate and maintain alkalosis, as in our patient.
Managing alkalosis in patients on dialysis can be challenging and is often directed at identifying potential causes like overzealous bicarbonate dialysate and addressing comorbidities, especially malnutrition.6,7 Bicarbonate delivery can be set on dialysis machines as low as 20 mEq/L. However, the reliability of correcting serum bicarbonate by adjusting bicarbonate-based dialysis products is in question as these products deliver additional buffering capacity through mixing and metabolism of acetate, acetic acid, or citric acid (Table 2).
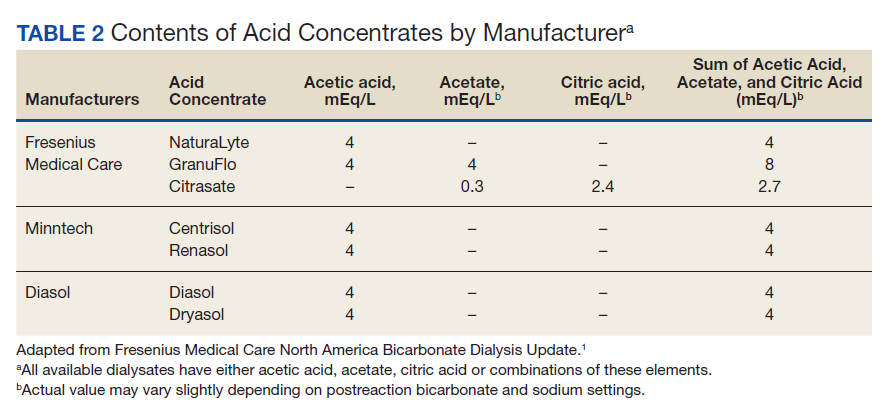
We infused a high volume of sodium chloride during dialysis to create hyperchloremic metabolic acidosis while removing the volume by UF, thereby eliminating more bicarbonate by convection. Normal saline has a pH of 5.5 and a chloride of 154 mmol/L. We have compensated for an inherent lack of flexibility in HD as it is currently practiced: dialysates are virtually all deliberately alkaline because most of the patients coming to HD have varying magnitudes of metabolic acidosis and acidemia. The dialysate concentrate that dilutes to a bicarbonate level of 30 mEq/L would have only a modest effect against this magnitude of metabolic alkalosis that this patient had at dialysis. We have compensated for this structural inadequacy of current HD by repairing the patient’s severe hypochloremic metabolic alkalosis by infusing a hyperchloremic sodium chloride solution and dialyzing off the excess sodium bicarbonate. This is the logical inverse of what usually happens in the severely acidotic patients seen prior to dialysis: dialyzing off an excess of normal saline and repairing the metabolic acidosis by transfer-in of sodium bicarbonate from the dialysate.
Fresenius Medical Care, which provides most HD machines and fluids in the United States, created charts to show the approximate degree that each contributes as additional buffer. That was in response to a class action lawsuit for metabolic alkalosis due to overdelivery of bicarbonate that resulted in alleged cardiac arrests in patients with HD.13 Their report cast doubt on the ability of a lower bicarbonate bath to correct metabolic alkalosis in a predictable fashion.1 We accordingly showed that normal saline delivery is a reliable option to promptly lower serum bicarbonate level. However, this is a temporary measure and long-term bicarbonate delivery during dialysis needs to be addressed.
Huber and Gennari demonstrated success in reducing severe alkalosis in patients with ESRD due to vomiting with the use of HCO3 bath of 30 mEq/L.14 In their report, the calculated bicarbonate dropped from 94 to 39 mEq/L; after 3 hours of HD, their patient also was receiving 2 L of an isotonic saline infusion daily. These observations suggest that lowering bicarbonate in the bath is effective in much more severe cases than ours, and even then, extra measures are needed to bring it down to desirable levels. In the early days, some health care providers used a specially prepared high-chloride (123 mEq/L) and low-acetate dialysate (18 mEq/L), which increased serum chloride and hydrogen ion concentrations and decreased the serum bicarbonate concentration compared with those in commercially available high-acetate dialysate (containing 37 mEq/L acetate and 104 mEq/L Cl).15 However, this method requires special preparation of dialysate. Oral potassium chloride also was used to correct metabolic alkalosis, but the risk of potassium overload precludes this approach in patients with ESRD.16
Likewise, adding oral sodium chloride risks causing volume overload, especially in patients with cardiomyopathy; it may increase thirst, resulting in interdialytic excess volume gains.17 In our patient, respiratory compensation took place by correcting pulmonary congestion by UF, and the gentle bicarbonate removal in addition to boosting chloride levels promptly improved the metabolic alkalosis.
Notably adequate volume control achieved by HD in persons with small muscle mass and severe cardiomyopathy can require longer treatment duration than required to achieve adequate clearance. Accordingly, more bicarbonate loading can take place, causing metabolic alkalosis. This problem is compounded by the potential overdelivery of bicarbonate than that entered by the physician’s order.1
Conclusions
Attention should be paid to detect elevated predialysis serum bicarbonate levels in ESRD patients on HD, especially those with values above 27 mmol/L due to higher mortality.6,7 Treatment of these patients is more challenging than for those who are acidotic predialysis, especially when alkalosis is compounded by malnutrition. Mitigation of this problem is achieved by using a lower bicarbonate bath and the shortest effective dialysis duration that achieves adequate clearance. Poor clearance also deleteriously affects patient nutrition and well-being. We have shown that normal saline solution infusion with concurrent removal by UF can correct pretreatment metabolic alkalosis when other measures are inadequate.
1. Fresenius Medical Care North America. Bicarbonate dialysis update. July 2012. Accessed May 14, 2018. http://www.renalweb.com/writings/alkalosis/FMC%20Jul%2025%202012.pdf
2. Rho M, Renda J. Pica presenting as metabolic alkalosis and seizure in a dialysis patient. Clin Nephrol. 2006;66(1):71-73. doi:10.5414/cnp66071
3. Bear R, Goldstein M, Phillipson E, et al. Effect of metabolic alkalosis on respiratory function in patients with chronic obstructive lung disease. Can Med Assoc J. 1977;117(8):900-903.
4. Javaheri S, Kazemi H. Metabolic alkalosis and hypoventilation in humans. Am Rev Respir Dis. 1987;136(4):1011-1016. doi:10.1164/ajrccm/136.4.1011
5. Yamamoto T, Shoji S, Yamakawa T, et al. Predialysis and postdialysis pH and bicarbonate and risk of all-cause and cardiovascular mortality in long-term hemodialysis patients. Am J Kidney Dis. 2015;66(3):469-478. doi:10.1053/j.ajkd.2015.04.014
6. Wu DY, Shinaberger CS, Regidor DL, McAllister CJ, Kopple JD, Kalantar-Zadeh K. Association between serum bicarbonate and death in hemodialysis patients: is it better to be acidotic or alkalotic? Clin J Am Soc Nephrol. 2006;1(1):70-78. doi:10.2215/CJN.00010505
7. Bommer J, Locatelli F, Satayathum S, et al. Association of predialysis serum bicarbonate levels with risk of mortality and hospitalization in the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am J Kidney Dis. 2004;44(4):661-671. doi:10.1053/j.ajkd.2004.06.008
8. Diskin CJ, Stokes TJ, Dansby LM, Radcliff L, Carter TB. Recurrent metabolic alkalosis and elevated troponins after crack cocaine use in a hemodialysis patient. Clin Exp Nephrol. 2006;10(2):156-158. doi:10.1007/s10157-006-0414-y
9. Ostermann ME, Girgis-Hanna Y, Nelson SR, Eastwood JB. Metabolic alkalosis in patients with renal failure. Nephrol Dial Transplant. 2003;18(11):2442-2448. doi:10.1093/ndt/gfg333
10. Rahilly GT, Berl T. Severe metabolic alkalosis caused by administration of plasma protein fraction in end-stage renal failure. N Engl J Med. 1979;301(15):824-826. doi:10.1056/NEJM197910113011506
11. Panesar M, Shah N, Vaqar S, et al. Changes in serum bicarbonate levels caused by acetate-containing bicarbonate-buffered hemodialysis solution: an observational prospective cohort study. Ther Apher Dial. 2017;21(2):157-165. doi:10.1111/1744-9987.12510
12. Noh U-S, Yi J-H, Han S-W, Kim H-J. Varying dialysate bicarbonate concentrations in maintenance hemodialysis patients affect post-dialysis alkalosis but not pre-dialysis acidosis. Electrolyte Blood Press. 2007;5(2):95-101. doi:10.5049/EBP.2007.5.2.95
13. Perriello B. Fresenius, plaintiffs ask for more time for $250m settlement in dialysate cases. Published March 4, 2016. Accessed May 14, 2018. https://www.massdevice.com/fresenius-askes-judge-time-250m-settlement-dialysate-cases
14. Huber L, Gennari FJ. Severe metabolic alkalosis in a hemodialysis patient. Am J Kidney Dis. 2011;58(1):144-149. doi:10.1053/j.ajkd.2011.03.016
15. Swartz RD, Rubin JE, Brown RS, Yager JM, Steinman TI, Frazier HS. Correction of postoperative metabolic alkalosis and renal failure by hemodialysis. Ann Intern Med. 1977;86(1):52-55. doi:10.7326/0003-4819-86-1-52
16. Rosen RA, Julian BA, Dubovsky EV, Galla JH, Luke RG. On the mechanism by which chloride corrects metabolic alkalosis in man. Am J Med. 1988;84(3, pt 1):449-458. doi:10.1016/0002-9343(88)90265-3
17. Hirakawa Y, Hanafusa N, Nangaku M. Correction of metabolic alkalosis and elevated calcium levels by sodium chloride in a hemodialysis patient with inadequate chloride intake. Ther Apher Dial. 2016;20(1):86-87. doi:10.1111/1744-9987.12335
Metabolic alkalosis, a disorder that causes elevations in serum bicarbonate and arterial pH, is a common metabolic abnormality found in nearly half of hospitalized patients but is rare in patients with end-stage renal disease (ESRD) on hemodialysis (HD) during the pretreatment state. The problem seems to arise due to a high rate of older patients with multiple comorbidities and malnutrition who are undergoing HD. Metabolic alkalosis is associated with increased morbidity and mortality. In this report, we present a case of metabolic alkalosis, describe an innovative approach to manage metabolic alkalosis in the dialysis population, and review the pathophysiology.
Case Presentation
A 63-year-old female with emphysema, diabetic nephropathy, and ESRD on regular HD for 2 months by a tunneled subclavian vein catheter was admitted with 2 weeks of orthopnea and leg swelling. The review of systems was negative for chest pain, cough, wheeze, or sputum production. She was a former smoker with no alcohol or drug misuse. The patient was taking carvedilol 25 mg daily, furosemide 20 mg twice daily, basal insulin premeal, lisinopril 40 mg daily, pantoprazole 40 mg daily, calcium carbonate 400 mg 3 times daily, ferrous sulphate 325 mg daily, and a vilanterol/tiotropium inhaler once daily. Her dialysate outpatient prescription included sodium 140 mEq/L, potassium 2 mEq/L, calcium 2.5 mEq/L, and bicarbonate 36 mEq/L. Our dialysis unit used NaturaLyte dry pack for bicarbonate dialysis.
The patient appeared tachypneic with 26 respirations/min, oxygen saturation of 89% on room air, which improved to 94% on a 2 L nasal cannula. Her heart rate was 89 beats/min, blood pressure was 129/72 mm Hg, and body mass index was 21.2. The physical examination revealed jugular venous distension, lung crackles, reduced air entry, and pedal edema. Muscle wasting was noted in the arms and thighs. The tunnel catheter did not appear infected.
The patient’s blood work showed sodium, 136 (reference, 132-140) mmol/L; potassium, 4.3 (reference, 3.5-5.0) mmol/L; chloride, 89 (reference, 98-111) mmol/L; total CO2, 36 (reference, 24-28) mEq/L; blood urea nitrogen, 21 (reference, 7-21) mg/dL; creatinine 3.4 (reference, 0.5-1.4) mg/dL; and albumin, 2.7 (reference, 3.7-5.0) mg/dL. Arterial gases showed pH, 7.56 (reference, 7.35-7.45), partial CO2, 47 (reference, 35-45) mm Hg; bicarbonate, 42 (reference, 22-26) mEq/L; partial O2, 54 (reference, 75 to 100) mm Hg. Brain natriuretic peptide was 2,800 (normal, < 100) pg/mL with a normal troponin. X-rays showed pulmonary congestion and bilateral pleural effusions that were transudative on fluid analysis. An echocardiogram showed ejection fraction of 20 to 25% with normal valves (baseline ejection fraction of 60%-65%). A coronary arteriogram revealed severe nonischemic cardiomyopathy.
Treatment
To reduce bicarbonate levels, 3 L of normal saline solution were infused prefilter during HD, and ultrafiltration (UF) of 4.5 L achieved a net UF of -1.5 L over 3.5 hours on lower dialysate bicarbonate (30 mEq/L). Good catheter flow was achieved with a blood flow rate of 350 mL/min and a dialysate flow of 700 mL/min. Venous blood gases and basic serum metabolic panels were obtained throughout the first HD session (Table 1). Improvement in pH from 7.5 to 7.43 and in total CO2 from 36 to 30 mEq/L were noted after the treatment. Subsequently, we used the same membrane (Optiflux F160NRe) for 2 consecutive daily treatments to remove excess fluid and prevent worsening alkalosis using the same minimal bicarbonate bath, but no further normal saline solution was given.
Outcome
Volume overload was controlled as needed with UF. The bicarbonate did not drop after the second HD session, suggesting low organic acid production in the intradialytic period. By shortening the duration of dialysis to 3 hours and improving nutritional intake, we achieved dry weight, and the patient was discharged home with a total CO2 of 25 mEq/L. Outpatient dialysis sessions were arranged to run at shorter duration (3 hours compared with 3.5 hours) and use low bicarbonate dialysate. The patient was admitted several times afterward for acute decompensated heart failure, but in all those admissions, her bicarbonate was in the normal-to-high range, between 23 and 30 mEq/L.
Discussion
Metabolic alkalosis is relatively rare in ESRD patients on HD. Particularly in the predialysis period, but with the growing number of older patients undergoing HD and the aggressive treatment of acidosis with relatively higher buffer concentrations; there has been an increase in the incidence of metabolic alkalosis in patients on HD. In the Fresenius Medical Care (FMC) prevalent HD patient study, predialysis bicarbonate levels have increased overtime from a mean (SD)22.9 (3.1) mEq/L in 2004 to a mean (SD) 24.1 (3.5) mEq/L in September 2011, with 25% of patients > 26.0 mEq/L compared with only 6% in 2004.1 The condition has been associated with cardiac arrhythmia, intradialytic hypocalcemia, hypokalemia, hypercapnia, hypoxia, accelerated hypertension, and seizure.2-4 Metabolic alkalosis may be associated with increased mortality.5-7 However, the effect dissipated after adjusting for inflammation and nutritional status.6
Our patient had primary metabolic alkalosis evident by her high pH of 7.56 and high total CO2 of 36 mEq/L. The serum total CO2 reflects the metabolic status more accurately than the blood gas bicarbonate, which is prone to calculation error by the Henderson-Hasselbalch equation. Her respiratory compensation for the metabolic alkalosis was appropriate, with an increase of arterial PaCO2 to 47 mm Hg (
In patients with ESRD on HD who have no residual urine output, causes of metabolic alkalosis are limited to loss of net acid or gain of alkali through the gastrointestinal tract; our patient had none of these. Similarly, all renal causes of metabolic alkalosis are not applicable to our patient, including mineralocorticoid excess and contraction alkalosis. In patients with preserved kidney function, loop diuretics can induce alkalosis through enhanced tubular absorption of HCO3. While acetazolamide can mitigate this scenario by blocking carbonic anhydrase in the luminal border of the collecting ducts resulting in excretion of bicarbonate in the urine, our patient had negligible urine output despite being on furosemide 20 mg twice daily, making this an unlikely cause.
Severe metabolic alkalosis in dialysis patients has been reported with cocaine use, pica ingestion, and citrate load as in plasma exchange, massive transfusions, and regional anticoagulation.2,8-11 Although calcium carbonate intake can contribute to alkalosis, her small daily dose of 1,200 mg contains approximately 12 mEq of carbonate, which is not a significant contributor to the alkalosis.
With all other causes excluded, the metabolic alkalosis in our patient is presumed to result from the bicarbonate-rich dialysate. Since the majority of patients with ESRD are acidotic before dialysis, the dialysate bicarbonate is set at a higher than normal physiologic level to bring the pH close to or even higher than normal after dialysis. The patient had been dialyzed with NaturaLyte as an outpatient, which was set at the dialysis unit default mode of 36 mEq/L. This form of alkalosis has been reported to peak immediately after treatment but in most patients returns to the predialysis acidotic state due to endogenous acid production.1,4,12 Normally, muscles play a significant role in buffering excess bicarbonate in patients with nonfunctioning kidneys; hence, malnutrition with muscle wasting tends to propagate and maintain alkalosis, as in our patient.
Managing alkalosis in patients on dialysis can be challenging and is often directed at identifying potential causes like overzealous bicarbonate dialysate and addressing comorbidities, especially malnutrition.6,7 Bicarbonate delivery can be set on dialysis machines as low as 20 mEq/L. However, the reliability of correcting serum bicarbonate by adjusting bicarbonate-based dialysis products is in question as these products deliver additional buffering capacity through mixing and metabolism of acetate, acetic acid, or citric acid (Table 2).
We infused a high volume of sodium chloride during dialysis to create hyperchloremic metabolic acidosis while removing the volume by UF, thereby eliminating more bicarbonate by convection. Normal saline has a pH of 5.5 and a chloride of 154 mmol/L. We have compensated for an inherent lack of flexibility in HD as it is currently practiced: dialysates are virtually all deliberately alkaline because most of the patients coming to HD have varying magnitudes of metabolic acidosis and acidemia. The dialysate concentrate that dilutes to a bicarbonate level of 30 mEq/L would have only a modest effect against this magnitude of metabolic alkalosis that this patient had at dialysis. We have compensated for this structural inadequacy of current HD by repairing the patient’s severe hypochloremic metabolic alkalosis by infusing a hyperchloremic sodium chloride solution and dialyzing off the excess sodium bicarbonate. This is the logical inverse of what usually happens in the severely acidotic patients seen prior to dialysis: dialyzing off an excess of normal saline and repairing the metabolic acidosis by transfer-in of sodium bicarbonate from the dialysate.
Fresenius Medical Care, which provides most HD machines and fluids in the United States, created charts to show the approximate degree that each contributes as additional buffer. That was in response to a class action lawsuit for metabolic alkalosis due to overdelivery of bicarbonate that resulted in alleged cardiac arrests in patients with HD.13 Their report cast doubt on the ability of a lower bicarbonate bath to correct metabolic alkalosis in a predictable fashion.1 We accordingly showed that normal saline delivery is a reliable option to promptly lower serum bicarbonate level. However, this is a temporary measure and long-term bicarbonate delivery during dialysis needs to be addressed.
Huber and Gennari demonstrated success in reducing severe alkalosis in patients with ESRD due to vomiting with the use of HCO3 bath of 30 mEq/L.14 In their report, the calculated bicarbonate dropped from 94 to 39 mEq/L; after 3 hours of HD, their patient also was receiving 2 L of an isotonic saline infusion daily. These observations suggest that lowering bicarbonate in the bath is effective in much more severe cases than ours, and even then, extra measures are needed to bring it down to desirable levels. In the early days, some health care providers used a specially prepared high-chloride (123 mEq/L) and low-acetate dialysate (18 mEq/L), which increased serum chloride and hydrogen ion concentrations and decreased the serum bicarbonate concentration compared with those in commercially available high-acetate dialysate (containing 37 mEq/L acetate and 104 mEq/L Cl).15 However, this method requires special preparation of dialysate. Oral potassium chloride also was used to correct metabolic alkalosis, but the risk of potassium overload precludes this approach in patients with ESRD.16
Likewise, adding oral sodium chloride risks causing volume overload, especially in patients with cardiomyopathy; it may increase thirst, resulting in interdialytic excess volume gains.17 In our patient, respiratory compensation took place by correcting pulmonary congestion by UF, and the gentle bicarbonate removal in addition to boosting chloride levels promptly improved the metabolic alkalosis.
Notably adequate volume control achieved by HD in persons with small muscle mass and severe cardiomyopathy can require longer treatment duration than required to achieve adequate clearance. Accordingly, more bicarbonate loading can take place, causing metabolic alkalosis. This problem is compounded by the potential overdelivery of bicarbonate than that entered by the physician’s order.1
Conclusions
Attention should be paid to detect elevated predialysis serum bicarbonate levels in ESRD patients on HD, especially those with values above 27 mmol/L due to higher mortality.6,7 Treatment of these patients is more challenging than for those who are acidotic predialysis, especially when alkalosis is compounded by malnutrition. Mitigation of this problem is achieved by using a lower bicarbonate bath and the shortest effective dialysis duration that achieves adequate clearance. Poor clearance also deleteriously affects patient nutrition and well-being. We have shown that normal saline solution infusion with concurrent removal by UF can correct pretreatment metabolic alkalosis when other measures are inadequate.
Metabolic alkalosis, a disorder that causes elevations in serum bicarbonate and arterial pH, is a common metabolic abnormality found in nearly half of hospitalized patients but is rare in patients with end-stage renal disease (ESRD) on hemodialysis (HD) during the pretreatment state. The problem seems to arise due to a high rate of older patients with multiple comorbidities and malnutrition who are undergoing HD. Metabolic alkalosis is associated with increased morbidity and mortality. In this report, we present a case of metabolic alkalosis, describe an innovative approach to manage metabolic alkalosis in the dialysis population, and review the pathophysiology.
Case Presentation
A 63-year-old female with emphysema, diabetic nephropathy, and ESRD on regular HD for 2 months by a tunneled subclavian vein catheter was admitted with 2 weeks of orthopnea and leg swelling. The review of systems was negative for chest pain, cough, wheeze, or sputum production. She was a former smoker with no alcohol or drug misuse. The patient was taking carvedilol 25 mg daily, furosemide 20 mg twice daily, basal insulin premeal, lisinopril 40 mg daily, pantoprazole 40 mg daily, calcium carbonate 400 mg 3 times daily, ferrous sulphate 325 mg daily, and a vilanterol/tiotropium inhaler once daily. Her dialysate outpatient prescription included sodium 140 mEq/L, potassium 2 mEq/L, calcium 2.5 mEq/L, and bicarbonate 36 mEq/L. Our dialysis unit used NaturaLyte dry pack for bicarbonate dialysis.
The patient appeared tachypneic with 26 respirations/min, oxygen saturation of 89% on room air, which improved to 94% on a 2 L nasal cannula. Her heart rate was 89 beats/min, blood pressure was 129/72 mm Hg, and body mass index was 21.2. The physical examination revealed jugular venous distension, lung crackles, reduced air entry, and pedal edema. Muscle wasting was noted in the arms and thighs. The tunnel catheter did not appear infected.
The patient’s blood work showed sodium, 136 (reference, 132-140) mmol/L; potassium, 4.3 (reference, 3.5-5.0) mmol/L; chloride, 89 (reference, 98-111) mmol/L; total CO2, 36 (reference, 24-28) mEq/L; blood urea nitrogen, 21 (reference, 7-21) mg/dL; creatinine 3.4 (reference, 0.5-1.4) mg/dL; and albumin, 2.7 (reference, 3.7-5.0) mg/dL. Arterial gases showed pH, 7.56 (reference, 7.35-7.45), partial CO2, 47 (reference, 35-45) mm Hg; bicarbonate, 42 (reference, 22-26) mEq/L; partial O2, 54 (reference, 75 to 100) mm Hg. Brain natriuretic peptide was 2,800 (normal, < 100) pg/mL with a normal troponin. X-rays showed pulmonary congestion and bilateral pleural effusions that were transudative on fluid analysis. An echocardiogram showed ejection fraction of 20 to 25% with normal valves (baseline ejection fraction of 60%-65%). A coronary arteriogram revealed severe nonischemic cardiomyopathy.
Treatment
To reduce bicarbonate levels, 3 L of normal saline solution were infused prefilter during HD, and ultrafiltration (UF) of 4.5 L achieved a net UF of -1.5 L over 3.5 hours on lower dialysate bicarbonate (30 mEq/L). Good catheter flow was achieved with a blood flow rate of 350 mL/min and a dialysate flow of 700 mL/min. Venous blood gases and basic serum metabolic panels were obtained throughout the first HD session (Table 1). Improvement in pH from 7.5 to 7.43 and in total CO2 from 36 to 30 mEq/L were noted after the treatment. Subsequently, we used the same membrane (Optiflux F160NRe) for 2 consecutive daily treatments to remove excess fluid and prevent worsening alkalosis using the same minimal bicarbonate bath, but no further normal saline solution was given.
Outcome
Volume overload was controlled as needed with UF. The bicarbonate did not drop after the second HD session, suggesting low organic acid production in the intradialytic period. By shortening the duration of dialysis to 3 hours and improving nutritional intake, we achieved dry weight, and the patient was discharged home with a total CO2 of 25 mEq/L. Outpatient dialysis sessions were arranged to run at shorter duration (3 hours compared with 3.5 hours) and use low bicarbonate dialysate. The patient was admitted several times afterward for acute decompensated heart failure, but in all those admissions, her bicarbonate was in the normal-to-high range, between 23 and 30 mEq/L.
Discussion
Metabolic alkalosis is relatively rare in ESRD patients on HD. Particularly in the predialysis period, but with the growing number of older patients undergoing HD and the aggressive treatment of acidosis with relatively higher buffer concentrations; there has been an increase in the incidence of metabolic alkalosis in patients on HD. In the Fresenius Medical Care (FMC) prevalent HD patient study, predialysis bicarbonate levels have increased overtime from a mean (SD)22.9 (3.1) mEq/L in 2004 to a mean (SD) 24.1 (3.5) mEq/L in September 2011, with 25% of patients > 26.0 mEq/L compared with only 6% in 2004.1 The condition has been associated with cardiac arrhythmia, intradialytic hypocalcemia, hypokalemia, hypercapnia, hypoxia, accelerated hypertension, and seizure.2-4 Metabolic alkalosis may be associated with increased mortality.5-7 However, the effect dissipated after adjusting for inflammation and nutritional status.6
Our patient had primary metabolic alkalosis evident by her high pH of 7.56 and high total CO2 of 36 mEq/L. The serum total CO2 reflects the metabolic status more accurately than the blood gas bicarbonate, which is prone to calculation error by the Henderson-Hasselbalch equation. Her respiratory compensation for the metabolic alkalosis was appropriate, with an increase of arterial PaCO2 to 47 mm Hg (
In patients with ESRD on HD who have no residual urine output, causes of metabolic alkalosis are limited to loss of net acid or gain of alkali through the gastrointestinal tract; our patient had none of these. Similarly, all renal causes of metabolic alkalosis are not applicable to our patient, including mineralocorticoid excess and contraction alkalosis. In patients with preserved kidney function, loop diuretics can induce alkalosis through enhanced tubular absorption of HCO3. While acetazolamide can mitigate this scenario by blocking carbonic anhydrase in the luminal border of the collecting ducts resulting in excretion of bicarbonate in the urine, our patient had negligible urine output despite being on furosemide 20 mg twice daily, making this an unlikely cause.
Severe metabolic alkalosis in dialysis patients has been reported with cocaine use, pica ingestion, and citrate load as in plasma exchange, massive transfusions, and regional anticoagulation.2,8-11 Although calcium carbonate intake can contribute to alkalosis, her small daily dose of 1,200 mg contains approximately 12 mEq of carbonate, which is not a significant contributor to the alkalosis.
With all other causes excluded, the metabolic alkalosis in our patient is presumed to result from the bicarbonate-rich dialysate. Since the majority of patients with ESRD are acidotic before dialysis, the dialysate bicarbonate is set at a higher than normal physiologic level to bring the pH close to or even higher than normal after dialysis. The patient had been dialyzed with NaturaLyte as an outpatient, which was set at the dialysis unit default mode of 36 mEq/L. This form of alkalosis has been reported to peak immediately after treatment but in most patients returns to the predialysis acidotic state due to endogenous acid production.1,4,12 Normally, muscles play a significant role in buffering excess bicarbonate in patients with nonfunctioning kidneys; hence, malnutrition with muscle wasting tends to propagate and maintain alkalosis, as in our patient.
Managing alkalosis in patients on dialysis can be challenging and is often directed at identifying potential causes like overzealous bicarbonate dialysate and addressing comorbidities, especially malnutrition.6,7 Bicarbonate delivery can be set on dialysis machines as low as 20 mEq/L. However, the reliability of correcting serum bicarbonate by adjusting bicarbonate-based dialysis products is in question as these products deliver additional buffering capacity through mixing and metabolism of acetate, acetic acid, or citric acid (Table 2).
We infused a high volume of sodium chloride during dialysis to create hyperchloremic metabolic acidosis while removing the volume by UF, thereby eliminating more bicarbonate by convection. Normal saline has a pH of 5.5 and a chloride of 154 mmol/L. We have compensated for an inherent lack of flexibility in HD as it is currently practiced: dialysates are virtually all deliberately alkaline because most of the patients coming to HD have varying magnitudes of metabolic acidosis and acidemia. The dialysate concentrate that dilutes to a bicarbonate level of 30 mEq/L would have only a modest effect against this magnitude of metabolic alkalosis that this patient had at dialysis. We have compensated for this structural inadequacy of current HD by repairing the patient’s severe hypochloremic metabolic alkalosis by infusing a hyperchloremic sodium chloride solution and dialyzing off the excess sodium bicarbonate. This is the logical inverse of what usually happens in the severely acidotic patients seen prior to dialysis: dialyzing off an excess of normal saline and repairing the metabolic acidosis by transfer-in of sodium bicarbonate from the dialysate.
Fresenius Medical Care, which provides most HD machines and fluids in the United States, created charts to show the approximate degree that each contributes as additional buffer. That was in response to a class action lawsuit for metabolic alkalosis due to overdelivery of bicarbonate that resulted in alleged cardiac arrests in patients with HD.13 Their report cast doubt on the ability of a lower bicarbonate bath to correct metabolic alkalosis in a predictable fashion.1 We accordingly showed that normal saline delivery is a reliable option to promptly lower serum bicarbonate level. However, this is a temporary measure and long-term bicarbonate delivery during dialysis needs to be addressed.
Huber and Gennari demonstrated success in reducing severe alkalosis in patients with ESRD due to vomiting with the use of HCO3 bath of 30 mEq/L.14 In their report, the calculated bicarbonate dropped from 94 to 39 mEq/L; after 3 hours of HD, their patient also was receiving 2 L of an isotonic saline infusion daily. These observations suggest that lowering bicarbonate in the bath is effective in much more severe cases than ours, and even then, extra measures are needed to bring it down to desirable levels. In the early days, some health care providers used a specially prepared high-chloride (123 mEq/L) and low-acetate dialysate (18 mEq/L), which increased serum chloride and hydrogen ion concentrations and decreased the serum bicarbonate concentration compared with those in commercially available high-acetate dialysate (containing 37 mEq/L acetate and 104 mEq/L Cl).15 However, this method requires special preparation of dialysate. Oral potassium chloride also was used to correct metabolic alkalosis, but the risk of potassium overload precludes this approach in patients with ESRD.16
Likewise, adding oral sodium chloride risks causing volume overload, especially in patients with cardiomyopathy; it may increase thirst, resulting in interdialytic excess volume gains.17 In our patient, respiratory compensation took place by correcting pulmonary congestion by UF, and the gentle bicarbonate removal in addition to boosting chloride levels promptly improved the metabolic alkalosis.
Notably adequate volume control achieved by HD in persons with small muscle mass and severe cardiomyopathy can require longer treatment duration than required to achieve adequate clearance. Accordingly, more bicarbonate loading can take place, causing metabolic alkalosis. This problem is compounded by the potential overdelivery of bicarbonate than that entered by the physician’s order.1
Conclusions
Attention should be paid to detect elevated predialysis serum bicarbonate levels in ESRD patients on HD, especially those with values above 27 mmol/L due to higher mortality.6,7 Treatment of these patients is more challenging than for those who are acidotic predialysis, especially when alkalosis is compounded by malnutrition. Mitigation of this problem is achieved by using a lower bicarbonate bath and the shortest effective dialysis duration that achieves adequate clearance. Poor clearance also deleteriously affects patient nutrition and well-being. We have shown that normal saline solution infusion with concurrent removal by UF can correct pretreatment metabolic alkalosis when other measures are inadequate.
1. Fresenius Medical Care North America. Bicarbonate dialysis update. July 2012. Accessed May 14, 2018. http://www.renalweb.com/writings/alkalosis/FMC%20Jul%2025%202012.pdf
2. Rho M, Renda J. Pica presenting as metabolic alkalosis and seizure in a dialysis patient. Clin Nephrol. 2006;66(1):71-73. doi:10.5414/cnp66071
3. Bear R, Goldstein M, Phillipson E, et al. Effect of metabolic alkalosis on respiratory function in patients with chronic obstructive lung disease. Can Med Assoc J. 1977;117(8):900-903.
4. Javaheri S, Kazemi H. Metabolic alkalosis and hypoventilation in humans. Am Rev Respir Dis. 1987;136(4):1011-1016. doi:10.1164/ajrccm/136.4.1011
5. Yamamoto T, Shoji S, Yamakawa T, et al. Predialysis and postdialysis pH and bicarbonate and risk of all-cause and cardiovascular mortality in long-term hemodialysis patients. Am J Kidney Dis. 2015;66(3):469-478. doi:10.1053/j.ajkd.2015.04.014
6. Wu DY, Shinaberger CS, Regidor DL, McAllister CJ, Kopple JD, Kalantar-Zadeh K. Association between serum bicarbonate and death in hemodialysis patients: is it better to be acidotic or alkalotic? Clin J Am Soc Nephrol. 2006;1(1):70-78. doi:10.2215/CJN.00010505
7. Bommer J, Locatelli F, Satayathum S, et al. Association of predialysis serum bicarbonate levels with risk of mortality and hospitalization in the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am J Kidney Dis. 2004;44(4):661-671. doi:10.1053/j.ajkd.2004.06.008
8. Diskin CJ, Stokes TJ, Dansby LM, Radcliff L, Carter TB. Recurrent metabolic alkalosis and elevated troponins after crack cocaine use in a hemodialysis patient. Clin Exp Nephrol. 2006;10(2):156-158. doi:10.1007/s10157-006-0414-y
9. Ostermann ME, Girgis-Hanna Y, Nelson SR, Eastwood JB. Metabolic alkalosis in patients with renal failure. Nephrol Dial Transplant. 2003;18(11):2442-2448. doi:10.1093/ndt/gfg333
10. Rahilly GT, Berl T. Severe metabolic alkalosis caused by administration of plasma protein fraction in end-stage renal failure. N Engl J Med. 1979;301(15):824-826. doi:10.1056/NEJM197910113011506
11. Panesar M, Shah N, Vaqar S, et al. Changes in serum bicarbonate levels caused by acetate-containing bicarbonate-buffered hemodialysis solution: an observational prospective cohort study. Ther Apher Dial. 2017;21(2):157-165. doi:10.1111/1744-9987.12510
12. Noh U-S, Yi J-H, Han S-W, Kim H-J. Varying dialysate bicarbonate concentrations in maintenance hemodialysis patients affect post-dialysis alkalosis but not pre-dialysis acidosis. Electrolyte Blood Press. 2007;5(2):95-101. doi:10.5049/EBP.2007.5.2.95
13. Perriello B. Fresenius, plaintiffs ask for more time for $250m settlement in dialysate cases. Published March 4, 2016. Accessed May 14, 2018. https://www.massdevice.com/fresenius-askes-judge-time-250m-settlement-dialysate-cases
14. Huber L, Gennari FJ. Severe metabolic alkalosis in a hemodialysis patient. Am J Kidney Dis. 2011;58(1):144-149. doi:10.1053/j.ajkd.2011.03.016
15. Swartz RD, Rubin JE, Brown RS, Yager JM, Steinman TI, Frazier HS. Correction of postoperative metabolic alkalosis and renal failure by hemodialysis. Ann Intern Med. 1977;86(1):52-55. doi:10.7326/0003-4819-86-1-52
16. Rosen RA, Julian BA, Dubovsky EV, Galla JH, Luke RG. On the mechanism by which chloride corrects metabolic alkalosis in man. Am J Med. 1988;84(3, pt 1):449-458. doi:10.1016/0002-9343(88)90265-3
17. Hirakawa Y, Hanafusa N, Nangaku M. Correction of metabolic alkalosis and elevated calcium levels by sodium chloride in a hemodialysis patient with inadequate chloride intake. Ther Apher Dial. 2016;20(1):86-87. doi:10.1111/1744-9987.12335
1. Fresenius Medical Care North America. Bicarbonate dialysis update. July 2012. Accessed May 14, 2018. http://www.renalweb.com/writings/alkalosis/FMC%20Jul%2025%202012.pdf
2. Rho M, Renda J. Pica presenting as metabolic alkalosis and seizure in a dialysis patient. Clin Nephrol. 2006;66(1):71-73. doi:10.5414/cnp66071
3. Bear R, Goldstein M, Phillipson E, et al. Effect of metabolic alkalosis on respiratory function in patients with chronic obstructive lung disease. Can Med Assoc J. 1977;117(8):900-903.
4. Javaheri S, Kazemi H. Metabolic alkalosis and hypoventilation in humans. Am Rev Respir Dis. 1987;136(4):1011-1016. doi:10.1164/ajrccm/136.4.1011
5. Yamamoto T, Shoji S, Yamakawa T, et al. Predialysis and postdialysis pH and bicarbonate and risk of all-cause and cardiovascular mortality in long-term hemodialysis patients. Am J Kidney Dis. 2015;66(3):469-478. doi:10.1053/j.ajkd.2015.04.014
6. Wu DY, Shinaberger CS, Regidor DL, McAllister CJ, Kopple JD, Kalantar-Zadeh K. Association between serum bicarbonate and death in hemodialysis patients: is it better to be acidotic or alkalotic? Clin J Am Soc Nephrol. 2006;1(1):70-78. doi:10.2215/CJN.00010505
7. Bommer J, Locatelli F, Satayathum S, et al. Association of predialysis serum bicarbonate levels with risk of mortality and hospitalization in the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am J Kidney Dis. 2004;44(4):661-671. doi:10.1053/j.ajkd.2004.06.008
8. Diskin CJ, Stokes TJ, Dansby LM, Radcliff L, Carter TB. Recurrent metabolic alkalosis and elevated troponins after crack cocaine use in a hemodialysis patient. Clin Exp Nephrol. 2006;10(2):156-158. doi:10.1007/s10157-006-0414-y
9. Ostermann ME, Girgis-Hanna Y, Nelson SR, Eastwood JB. Metabolic alkalosis in patients with renal failure. Nephrol Dial Transplant. 2003;18(11):2442-2448. doi:10.1093/ndt/gfg333
10. Rahilly GT, Berl T. Severe metabolic alkalosis caused by administration of plasma protein fraction in end-stage renal failure. N Engl J Med. 1979;301(15):824-826. doi:10.1056/NEJM197910113011506
11. Panesar M, Shah N, Vaqar S, et al. Changes in serum bicarbonate levels caused by acetate-containing bicarbonate-buffered hemodialysis solution: an observational prospective cohort study. Ther Apher Dial. 2017;21(2):157-165. doi:10.1111/1744-9987.12510
12. Noh U-S, Yi J-H, Han S-W, Kim H-J. Varying dialysate bicarbonate concentrations in maintenance hemodialysis patients affect post-dialysis alkalosis but not pre-dialysis acidosis. Electrolyte Blood Press. 2007;5(2):95-101. doi:10.5049/EBP.2007.5.2.95
13. Perriello B. Fresenius, plaintiffs ask for more time for $250m settlement in dialysate cases. Published March 4, 2016. Accessed May 14, 2018. https://www.massdevice.com/fresenius-askes-judge-time-250m-settlement-dialysate-cases
14. Huber L, Gennari FJ. Severe metabolic alkalosis in a hemodialysis patient. Am J Kidney Dis. 2011;58(1):144-149. doi:10.1053/j.ajkd.2011.03.016
15. Swartz RD, Rubin JE, Brown RS, Yager JM, Steinman TI, Frazier HS. Correction of postoperative metabolic alkalosis and renal failure by hemodialysis. Ann Intern Med. 1977;86(1):52-55. doi:10.7326/0003-4819-86-1-52
16. Rosen RA, Julian BA, Dubovsky EV, Galla JH, Luke RG. On the mechanism by which chloride corrects metabolic alkalosis in man. Am J Med. 1988;84(3, pt 1):449-458. doi:10.1016/0002-9343(88)90265-3
17. Hirakawa Y, Hanafusa N, Nangaku M. Correction of metabolic alkalosis and elevated calcium levels by sodium chloride in a hemodialysis patient with inadequate chloride intake. Ther Apher Dial. 2016;20(1):86-87. doi:10.1111/1744-9987.12335
37-year-old man • cough • increasing shortness of breath • pleuritic chest pain • Dx?
THE CASE
A 37-year-old man with a history of asthma, schizoaffective disorder, and tobacco use (36 packs per year) presented to the clinic after 5 days of worsening cough, reproducible left-sided chest pain, and increasing shortness of breath. He also experienced chills, fatigue, nausea, and vomiting but was afebrile. The patient had not travelled recently nor had direct contact with anyone sick. He also denied intravenous (IV) drug use, alcohol use, and bloody sputum. Recently, he had intentionally lost weight, as recommended by his psychiatrist.
Medication review revealed that he was taking many central-acting agents for schizoaffective disorder, including alprazolam, aripiprazole, desvenlafaxine, and quetiapine. Due to his intermittent asthma since childhood, he used an albuterol inhaler as needed, which currently offered only minimal relief. He denied any history of hospitalization or intubation for asthma.
During the clinic visit, his blood pressure was 90/60 mm Hg and his heart rate was normal. His pulse oximetry was 92% on room air. On physical examination, he had normal-appearing dentition. Auscultation revealed bilateral expiratory wheezes with decreased breath sounds at the left lower lobe.

A plain chest radiograph (CXR) performed in the clinic (FIGURE 1) showed a large, thick-walled cavitary lesion with an air-fluid level in the left lower lobe. The patient was directly admitted to the Family Medicine Inpatient Service. Computed tomography (CT) of the chest with contrast was ordered to rule out empyema or malignancy. The chest CT confirmed the previous findings while also revealing a surrounding satellite nodularity in the left lower lobe (FIGURE 2). QuantiFERON-TB Gold and HIV tests were both negative.

THE DIAGNOSIS
The patient was given a diagnosis of a lung abscess based on symptoms and imaging. An extensive smoking history, as well as multiple sedating medications, increased his likelihood of aspiration.
DISCUSSION
Lung abscess is the probable diagnosis in a patient with indolent infectious symptoms (cough, fever, night sweats) developing over days to weeks and a CXR finding of pulmonary opacity, often with an air-fluid level.1-4 A lung abscess is a circumscribed collection of pus in the lung parenchyma that develops as a result of microbial infection.4
Primary vs secondary abscess. Lung abscesses can be divided into 2 groups: primary and secondary abscesses. Primary abscesses (60%) occur without any other medical condition or in patients prone to aspiration.5 Secondary abscesses occur in the setting of a comorbid medical condition, such as lung disease, heart disease, bronchogenic neoplasm, or immunocompromised status.5
Continue to: With a primary lung abscess...
With a primary lung abscess, oropharyngeal contents are aspirated (generally while the patient is unconscious) and contain mixed flora.2 The aspirate typically migrates to the posterior segments of the upper lobes and to the superior segments of the lower lobes. These abscesses are usually singular and have an air-fluid level.1,2
Secondary lung abscesses occur in bronchial obstruction (by tumor, foreign body, or enlarged lymph nodes), with coexisting lung diseases (bronchiectasis, cystic fibrosis, infected pulmonary infarcts, lung contusion) or by direct spread (broncho-esophageal fistula, subphrenic abscess).6 Secondary abscesses are associated with a poorer prognosis, dependent on the patient’s general condition and underlying disease.7
What to rule out
The differential diagnosis of cavitary lung lesion includes tuberculosis, necrotizing pneumonia, bronchial carcinoma, pulmonary embolism, vasculitis (eg, Churg-Strauss syndrome), and localized pleural empyema.1,4 A CT scan is helpful to differentiate between a parenchymal lesion and pleural collection, which may not be as clear on CXR.1,4
Tuberculosis manifests with fatigue, weight loss, and night sweats; a chest CT will reveal a cavitating lesion (usually upper lobe) with a characteristic “rim sign” that includes caseous necrosis surrounded by a peripheral enhancing rim.8
Necrotizing pneumonia manifests as acute, fulminant infection. The most common causative organisms on sputum culture are Streptococcus pneumoniae, Staphylococcus aureus, Klebsiella pneumoniae, and Pseudomonas species. Plain radiography will reveal multiple cavities and often associated pleural effusion and empyema.9
Continue to: Excavating bronchogenic carcinomas
Excavating bronchogenic carcinomas differ from a lung abscess in that a patient with the latter is typically, but not always, febrile and has purulent sputum. On imaging, a bronchogenic carcinoma has a thicker and more irregular wall than a lung abscess.10
Treatment
When antibiotics first became available, penicillin was used to treat lung abscess.11 Then IV clindamycin became the drug of choice after 2 trials demonstrated its superiority to IV penicillin.12,13 More recently, clindamycin alone has fallen out of favor due to growing anaerobic resistance.14
Current therapy includes beta-lactam with beta-lactamase inhibitors.14 Lung abscesses are typically polymicrobial and thus carry different degrees of antibiotic resistance.15,16 If culture data are available, targeted therapy is preferred, especially for secondary abscesses.7 Antibiotic therapy is usually continued until a CXR reveals a small lesion or is clear, which may require several months of outpatient oral antibiotic therapy.4

Our patient was treated with IV clindamycin for 3 days in the hospital. Clindamycin was chosen due to his penicillin allergy and started empirically without any culture data. He was transitioned to oral clindamycin and completed a total 3-week course as his CXR continued to show improvement (FIGURE 3). He did not undergo bronchoscopy. A follow-up CXR showed resolution of lung abscess at 9 months. (FIGURE 4).

THE TAKEAWAY
All patients with lung abscesses should have sputum culture with gram stain done—ideally prior to starting antibiotics.3,4 Bronchoscopy should be considered for patients with atypical presentations or those who fail standard therapy, but may be used in other cases, as well.3
CORRESPONDENCE
Morteza Khodaee, MD, MPH, AFW Clinic, 3055 Roslyn Street, Denver, CO 80238; [email protected]
1. Hassan M, Asciak R, Rizk R, et al. Lung abscess or empyema? Taking a closer look. Thorax. 2018;73:887-889. https://doi. org/10.1136/thoraxjnl-2018-211604
2. Moreira J da SM, Camargo J de JP, Felicetti JC, et al. Lung abscess: analysis of 252 consecutive cases diagnosed between 1968 and 2004. J Bras Pneumol. 2006;32:136-43. https://doi.org/10.1590/ s1806-37132006000200009
3. Schiza S, Siafakas NM. Clinical presentation and management of empyema, lung abscess and pleural effusion. Curr Opin Pulm Med. 2006;12:205-211. https://doi.org/10.1097/01. mcp.0000219270.73180.8b
4. Yazbeck MF, Dahdel M, Kalra A, et al. Lung abscess: update on microbiology and management. Am J Ther. 2014;21:217-221. https://doi.org/10.1097/MJT.0b013e3182383c9b
5. Nicolini A, Cilloniz C, Senarega R, et al. Lung abscess due to Streptococcus pneumoniae: a case series and brief review of the literature. Pneumonol Alergol Pol. 2014;82:276-285. https://doi. org/10.5603/PiAP.2014.0033
6. Puligandla PS, Laberge J-M. Respiratory infections: pneumonia, lung abscess, and empyema. Semin Pediatr Surg. 2008;17:42-52. https://doi.org/10.1053/j.sempedsurg.2007.10.007
7. Marra A, Hillejan L, Ukena D. [Management of Lung Abscess]. Zentralbl Chir. 2015;140 (suppl 1):S47-S53. https://doi. org/10.1055/s-0035-1557883
THE CASE
A 37-year-old man with a history of asthma, schizoaffective disorder, and tobacco use (36 packs per year) presented to the clinic after 5 days of worsening cough, reproducible left-sided chest pain, and increasing shortness of breath. He also experienced chills, fatigue, nausea, and vomiting but was afebrile. The patient had not travelled recently nor had direct contact with anyone sick. He also denied intravenous (IV) drug use, alcohol use, and bloody sputum. Recently, he had intentionally lost weight, as recommended by his psychiatrist.
Medication review revealed that he was taking many central-acting agents for schizoaffective disorder, including alprazolam, aripiprazole, desvenlafaxine, and quetiapine. Due to his intermittent asthma since childhood, he used an albuterol inhaler as needed, which currently offered only minimal relief. He denied any history of hospitalization or intubation for asthma.
During the clinic visit, his blood pressure was 90/60 mm Hg and his heart rate was normal. His pulse oximetry was 92% on room air. On physical examination, he had normal-appearing dentition. Auscultation revealed bilateral expiratory wheezes with decreased breath sounds at the left lower lobe.
A plain chest radiograph (CXR) performed in the clinic (FIGURE 1) showed a large, thick-walled cavitary lesion with an air-fluid level in the left lower lobe. The patient was directly admitted to the Family Medicine Inpatient Service. Computed tomography (CT) of the chest with contrast was ordered to rule out empyema or malignancy. The chest CT confirmed the previous findings while also revealing a surrounding satellite nodularity in the left lower lobe (FIGURE 2). QuantiFERON-TB Gold and HIV tests were both negative.
THE DIAGNOSIS
The patient was given a diagnosis of a lung abscess based on symptoms and imaging. An extensive smoking history, as well as multiple sedating medications, increased his likelihood of aspiration.
DISCUSSION
Lung abscess is the probable diagnosis in a patient with indolent infectious symptoms (cough, fever, night sweats) developing over days to weeks and a CXR finding of pulmonary opacity, often with an air-fluid level.1-4 A lung abscess is a circumscribed collection of pus in the lung parenchyma that develops as a result of microbial infection.4
Primary vs secondary abscess. Lung abscesses can be divided into 2 groups: primary and secondary abscesses. Primary abscesses (60%) occur without any other medical condition or in patients prone to aspiration.5 Secondary abscesses occur in the setting of a comorbid medical condition, such as lung disease, heart disease, bronchogenic neoplasm, or immunocompromised status.5
Continue to: With a primary lung abscess...
With a primary lung abscess, oropharyngeal contents are aspirated (generally while the patient is unconscious) and contain mixed flora.2 The aspirate typically migrates to the posterior segments of the upper lobes and to the superior segments of the lower lobes. These abscesses are usually singular and have an air-fluid level.1,2
Secondary lung abscesses occur in bronchial obstruction (by tumor, foreign body, or enlarged lymph nodes), with coexisting lung diseases (bronchiectasis, cystic fibrosis, infected pulmonary infarcts, lung contusion) or by direct spread (broncho-esophageal fistula, subphrenic abscess).6 Secondary abscesses are associated with a poorer prognosis, dependent on the patient’s general condition and underlying disease.7
What to rule out
The differential diagnosis of cavitary lung lesion includes tuberculosis, necrotizing pneumonia, bronchial carcinoma, pulmonary embolism, vasculitis (eg, Churg-Strauss syndrome), and localized pleural empyema.1,4 A CT scan is helpful to differentiate between a parenchymal lesion and pleural collection, which may not be as clear on CXR.1,4
Tuberculosis manifests with fatigue, weight loss, and night sweats; a chest CT will reveal a cavitating lesion (usually upper lobe) with a characteristic “rim sign” that includes caseous necrosis surrounded by a peripheral enhancing rim.8
Necrotizing pneumonia manifests as acute, fulminant infection. The most common causative organisms on sputum culture are Streptococcus pneumoniae, Staphylococcus aureus, Klebsiella pneumoniae, and Pseudomonas species. Plain radiography will reveal multiple cavities and often associated pleural effusion and empyema.9
Continue to: Excavating bronchogenic carcinomas
Excavating bronchogenic carcinomas differ from a lung abscess in that a patient with the latter is typically, but not always, febrile and has purulent sputum. On imaging, a bronchogenic carcinoma has a thicker and more irregular wall than a lung abscess.10
Treatment
When antibiotics first became available, penicillin was used to treat lung abscess.11 Then IV clindamycin became the drug of choice after 2 trials demonstrated its superiority to IV penicillin.12,13 More recently, clindamycin alone has fallen out of favor due to growing anaerobic resistance.14
Current therapy includes beta-lactam with beta-lactamase inhibitors.14 Lung abscesses are typically polymicrobial and thus carry different degrees of antibiotic resistance.15,16 If culture data are available, targeted therapy is preferred, especially for secondary abscesses.7 Antibiotic therapy is usually continued until a CXR reveals a small lesion or is clear, which may require several months of outpatient oral antibiotic therapy.4
Our patient was treated with IV clindamycin for 3 days in the hospital. Clindamycin was chosen due to his penicillin allergy and started empirically without any culture data. He was transitioned to oral clindamycin and completed a total 3-week course as his CXR continued to show improvement (FIGURE 3). He did not undergo bronchoscopy. A follow-up CXR showed resolution of lung abscess at 9 months. (FIGURE 4).
THE TAKEAWAY
All patients with lung abscesses should have sputum culture with gram stain done—ideally prior to starting antibiotics.3,4 Bronchoscopy should be considered for patients with atypical presentations or those who fail standard therapy, but may be used in other cases, as well.3
CORRESPONDENCE
Morteza Khodaee, MD, MPH, AFW Clinic, 3055 Roslyn Street, Denver, CO 80238; [email protected]
THE CASE
A 37-year-old man with a history of asthma, schizoaffective disorder, and tobacco use (36 packs per year) presented to the clinic after 5 days of worsening cough, reproducible left-sided chest pain, and increasing shortness of breath. He also experienced chills, fatigue, nausea, and vomiting but was afebrile. The patient had not travelled recently nor had direct contact with anyone sick. He also denied intravenous (IV) drug use, alcohol use, and bloody sputum. Recently, he had intentionally lost weight, as recommended by his psychiatrist.
Medication review revealed that he was taking many central-acting agents for schizoaffective disorder, including alprazolam, aripiprazole, desvenlafaxine, and quetiapine. Due to his intermittent asthma since childhood, he used an albuterol inhaler as needed, which currently offered only minimal relief. He denied any history of hospitalization or intubation for asthma.
During the clinic visit, his blood pressure was 90/60 mm Hg and his heart rate was normal. His pulse oximetry was 92% on room air. On physical examination, he had normal-appearing dentition. Auscultation revealed bilateral expiratory wheezes with decreased breath sounds at the left lower lobe.
A plain chest radiograph (CXR) performed in the clinic (FIGURE 1) showed a large, thick-walled cavitary lesion with an air-fluid level in the left lower lobe. The patient was directly admitted to the Family Medicine Inpatient Service. Computed tomography (CT) of the chest with contrast was ordered to rule out empyema or malignancy. The chest CT confirmed the previous findings while also revealing a surrounding satellite nodularity in the left lower lobe (FIGURE 2). QuantiFERON-TB Gold and HIV tests were both negative.
THE DIAGNOSIS
The patient was given a diagnosis of a lung abscess based on symptoms and imaging. An extensive smoking history, as well as multiple sedating medications, increased his likelihood of aspiration.
DISCUSSION
Lung abscess is the probable diagnosis in a patient with indolent infectious symptoms (cough, fever, night sweats) developing over days to weeks and a CXR finding of pulmonary opacity, often with an air-fluid level.1-4 A lung abscess is a circumscribed collection of pus in the lung parenchyma that develops as a result of microbial infection.4
Primary vs secondary abscess. Lung abscesses can be divided into 2 groups: primary and secondary abscesses. Primary abscesses (60%) occur without any other medical condition or in patients prone to aspiration.5 Secondary abscesses occur in the setting of a comorbid medical condition, such as lung disease, heart disease, bronchogenic neoplasm, or immunocompromised status.5
Continue to: With a primary lung abscess...
With a primary lung abscess, oropharyngeal contents are aspirated (generally while the patient is unconscious) and contain mixed flora.2 The aspirate typically migrates to the posterior segments of the upper lobes and to the superior segments of the lower lobes. These abscesses are usually singular and have an air-fluid level.1,2
Secondary lung abscesses occur in bronchial obstruction (by tumor, foreign body, or enlarged lymph nodes), with coexisting lung diseases (bronchiectasis, cystic fibrosis, infected pulmonary infarcts, lung contusion) or by direct spread (broncho-esophageal fistula, subphrenic abscess).6 Secondary abscesses are associated with a poorer prognosis, dependent on the patient’s general condition and underlying disease.7
What to rule out
The differential diagnosis of cavitary lung lesion includes tuberculosis, necrotizing pneumonia, bronchial carcinoma, pulmonary embolism, vasculitis (eg, Churg-Strauss syndrome), and localized pleural empyema.1,4 A CT scan is helpful to differentiate between a parenchymal lesion and pleural collection, which may not be as clear on CXR.1,4
Tuberculosis manifests with fatigue, weight loss, and night sweats; a chest CT will reveal a cavitating lesion (usually upper lobe) with a characteristic “rim sign” that includes caseous necrosis surrounded by a peripheral enhancing rim.8
Necrotizing pneumonia manifests as acute, fulminant infection. The most common causative organisms on sputum culture are Streptococcus pneumoniae, Staphylococcus aureus, Klebsiella pneumoniae, and Pseudomonas species. Plain radiography will reveal multiple cavities and often associated pleural effusion and empyema.9
Continue to: Excavating bronchogenic carcinomas
Excavating bronchogenic carcinomas differ from a lung abscess in that a patient with the latter is typically, but not always, febrile and has purulent sputum. On imaging, a bronchogenic carcinoma has a thicker and more irregular wall than a lung abscess.10
Treatment
When antibiotics first became available, penicillin was used to treat lung abscess.11 Then IV clindamycin became the drug of choice after 2 trials demonstrated its superiority to IV penicillin.12,13 More recently, clindamycin alone has fallen out of favor due to growing anaerobic resistance.14
Current therapy includes beta-lactam with beta-lactamase inhibitors.14 Lung abscesses are typically polymicrobial and thus carry different degrees of antibiotic resistance.15,16 If culture data are available, targeted therapy is preferred, especially for secondary abscesses.7 Antibiotic therapy is usually continued until a CXR reveals a small lesion or is clear, which may require several months of outpatient oral antibiotic therapy.4
Our patient was treated with IV clindamycin for 3 days in the hospital. Clindamycin was chosen due to his penicillin allergy and started empirically without any culture data. He was transitioned to oral clindamycin and completed a total 3-week course as his CXR continued to show improvement (FIGURE 3). He did not undergo bronchoscopy. A follow-up CXR showed resolution of lung abscess at 9 months. (FIGURE 4).
THE TAKEAWAY
All patients with lung abscesses should have sputum culture with gram stain done—ideally prior to starting antibiotics.3,4 Bronchoscopy should be considered for patients with atypical presentations or those who fail standard therapy, but may be used in other cases, as well.3
CORRESPONDENCE
Morteza Khodaee, MD, MPH, AFW Clinic, 3055 Roslyn Street, Denver, CO 80238; [email protected]
1. Hassan M, Asciak R, Rizk R, et al. Lung abscess or empyema? Taking a closer look. Thorax. 2018;73:887-889. https://doi. org/10.1136/thoraxjnl-2018-211604
2. Moreira J da SM, Camargo J de JP, Felicetti JC, et al. Lung abscess: analysis of 252 consecutive cases diagnosed between 1968 and 2004. J Bras Pneumol. 2006;32:136-43. https://doi.org/10.1590/ s1806-37132006000200009
3. Schiza S, Siafakas NM. Clinical presentation and management of empyema, lung abscess and pleural effusion. Curr Opin Pulm Med. 2006;12:205-211. https://doi.org/10.1097/01. mcp.0000219270.73180.8b
4. Yazbeck MF, Dahdel M, Kalra A, et al. Lung abscess: update on microbiology and management. Am J Ther. 2014;21:217-221. https://doi.org/10.1097/MJT.0b013e3182383c9b
5. Nicolini A, Cilloniz C, Senarega R, et al. Lung abscess due to Streptococcus pneumoniae: a case series and brief review of the literature. Pneumonol Alergol Pol. 2014;82:276-285. https://doi. org/10.5603/PiAP.2014.0033
6. Puligandla PS, Laberge J-M. Respiratory infections: pneumonia, lung abscess, and empyema. Semin Pediatr Surg. 2008;17:42-52. https://doi.org/10.1053/j.sempedsurg.2007.10.007
7. Marra A, Hillejan L, Ukena D. [Management of Lung Abscess]. Zentralbl Chir. 2015;140 (suppl 1):S47-S53. https://doi. org/10.1055/s-0035-1557883
1. Hassan M, Asciak R, Rizk R, et al. Lung abscess or empyema? Taking a closer look. Thorax. 2018;73:887-889. https://doi. org/10.1136/thoraxjnl-2018-211604
2. Moreira J da SM, Camargo J de JP, Felicetti JC, et al. Lung abscess: analysis of 252 consecutive cases diagnosed between 1968 and 2004. J Bras Pneumol. 2006;32:136-43. https://doi.org/10.1590/ s1806-37132006000200009
3. Schiza S, Siafakas NM. Clinical presentation and management of empyema, lung abscess and pleural effusion. Curr Opin Pulm Med. 2006;12:205-211. https://doi.org/10.1097/01. mcp.0000219270.73180.8b
4. Yazbeck MF, Dahdel M, Kalra A, et al. Lung abscess: update on microbiology and management. Am J Ther. 2014;21:217-221. https://doi.org/10.1097/MJT.0b013e3182383c9b
5. Nicolini A, Cilloniz C, Senarega R, et al. Lung abscess due to Streptococcus pneumoniae: a case series and brief review of the literature. Pneumonol Alergol Pol. 2014;82:276-285. https://doi. org/10.5603/PiAP.2014.0033
6. Puligandla PS, Laberge J-M. Respiratory infections: pneumonia, lung abscess, and empyema. Semin Pediatr Surg. 2008;17:42-52. https://doi.org/10.1053/j.sempedsurg.2007.10.007
7. Marra A, Hillejan L, Ukena D. [Management of Lung Abscess]. Zentralbl Chir. 2015;140 (suppl 1):S47-S53. https://doi. org/10.1055/s-0035-1557883

Erethism Mercurialis and Reactions to Elemental Mercury
Evidence of human exposure to mercury dates as far back as the Egyptians in 1500
Mercury release in the environment primarily is a function of human activity, including coal-fired power plants, residential heating, and mining.9,10 Mercury from these sources is commonly found in the sediment of lakes and bays, where it is enzymatically converted to methylmercury by aquatic microorganisms; subsequent food chain biomagnification results in elevated mercury levels in apex predators. Substantial release of mercury into the environment also can be attributed to health care facilities from their use of thermometers containing 0.5 to 3 g of elemental mercury,11 blood pressure monitors, and medical waste incinerators.5
Mercury has been reported as the second most common cause of heavy metal poisoning after lead.12 Standards from the US Food and Drug Administration dictate that methylmercury levels in fish and wheat products must not exceed 1 ppm.13 Most plant and animal food sources contain methylmercury at levels between 0.0001 and 0.01 ppm; mercury concentrations are especially high in tuna, averaging 0.4 ppm, while larger predatory fish contain levels in excess of 1 ppm.14 The use of mercury-containing cosmetic products also presents a substantial exposure risk to consumers.5,10 In one study, 3.3% of skin-lightening creams and soaps purchased within the United States contained concentrations of mercury exceeding 1000 ppm.15
We describe a case of mercury toxicity resulting from intentional injection of liquid mercury into the right antecubital fossa in a suicide attempt.
Case Report
A 31-year-old woman presented to the family practice center for evaluation of a firm stained area on the skin of the right arm. She reported increasing anxiety, depression, tremors, irritability, and difficulty concentrating over the last 6 months. She denied headache and joint or muscle pain. Four years earlier, she had broken apart a thermometer and injected approximately 0.7 mL of its contents into the right arm in a suicide attempt. She intended to inject the thermometer’s contents directly into a vein, but the material instead entered the surrounding tissue. She denied notable pain or itching overlying the injection site. Her medications included aripiprazole and buspirone. She noted that she smoked half a pack of cigarettes per day and had a history of methamphetamine abuse. She was homeless and unemployed. Physical examination revealed an anxious tremulous woman with an erythematous to bluish gray, firm plaque on the right antecubital fossa (Figure 1). There were no notable tremors and no gait disturbance.
Her blood mercury level was greater than 100 µg/L and urine mercury was 477 µg/g (reference ranges, 1–8 μg/L and 4–5 μg/L, respectively). A radiograph of the right elbow area revealed scattered punctate foci of increased density within or overlying the anterolateral elbow soft tissues. She was diagnosed with mercury granuloma causing chronic mercury elevation. She underwent excision of the granuloma (Figure 2) with endovascular surgery via an elliptical incision. The patient was subsequently lost to follow-up.
Comment
Elemental mercury is a silver liquid at room temperature that spontaneously evaporates to form mercury vapor, an invisible, odorless, toxic gas. Accidental cutaneous exposure typically is safely managed by washing exposed skin with soap and water,16 though there is a potential risk for systemic absorption, especially when the skin is inflamed. When metallic mercury is subcutaneously injected, it is advised to promptly excise all subcutaneous areas containing mercury, regardless of any symptoms of systemic toxicity. Patients should subsequently be monitored for signs of both central nervous system (CNS) and renal deficits, undergo chelation therapy when systemic effects are apparent, and finally receive psychiatric consultation and treatment when necessary.17
Inorganic mercury compounds are formed when elemental mercury combines with sulfur or oxygen and often take the form of mercury salts, which appear as white crystals.16 These salts occur naturally in the environment and are used in pesticides, antiseptics, and skin-lightening creams and soaps.18
Methylmercury is a highly toxic, organic compound that is capable of crossing the placental and blood-brain barriers. It is the most common organic mercury compound found in the environment.16 Most humans have trace amounts of methylmercury in their bodies, typically as a result of consuming seafood.5
Exposure to mercury most commonly occurs through chronic consumption of methylmercury in seafood or acute inhalation of elemental mercury vapors.9 Iatrogenic cases of mercury exposure via injection also have been reported in the literature, including a case resulting in acute poisoning due to peritoneal lavage with mercury bichloride.19 Acute mercury-induced pulmonary damage typically resolves completely. However, there have been reported cases of exposure progressing to interstitial emphysema, pneumatocele, pneumothorax, pneumomediastinum, interstitial fibrosis, and chronic respiratory insufficiency, with examples of fatal acute respiratory distress syndrome being reported.5,16,20 Although individuals who inhale mercury vapors initially may be unaware of exposure due to little upper airway irritation, symptoms following an initial acute exposure may include ptyalism, a metallic taste, dysphagia, enteritis, diarrhea, nausea, renal damage, and CNS effects.16 Additionally, exposure may lead to confusion with signs and symptoms of metal fume fever, including shortness of breath, pleuritic chest pain, stomatitis, lethargy, and vomiting.20
Chronic exposure to mercury vapor can result in accumulation of mercury in the body, leading to neuropsychiatric, dermatologic, oropharyngeal, and renal manifestations. Sore throat, fever, headache, fatigue, dyspnea, chest pain, and pneumonitis are common.16 Typically, low-level exposure to elemental mercury does not lead to long-lasting health effects. However, individuals exposed to high-level elemental mercury vapors may require hospitalization. Treatment of acute mercury poisoning consists of removing the source of exposure, followed by cardiopulmonary support.16
Specific assays for mercury levels in blood and urine are useful to assess the level of exposure and risk to the patient. Blood mercury concentrations of 20 µg/L or below are considered within reference range; however, once blood and urine concentrations of mercury exceed 100 µg/L, clinical signs of acute mercury poisoning typically manifest.21 Chest radiographs can reveal pulmonary damage, while complete blood cell count, metabolic panel, and urinalysis can assess damage to other organs. Neuropsychiatric testing and nerve conduction studies may provide objective evidence of CNS toxicity. Assays for N-acetyl-β-D-glucosaminidase can provide an indication of early renal tubular dysfunction.16
Elemental mercury is not absorbed from the gastrointestinal tract, posing minimal risk for acute toxicity from ingestion. Generally, less than 10% of ingested inorganic mercury is absorbed from the gut, while elemental mercury is nonabsorbable.10 If an individual ingests a large amount of mercury, it may persist in the gastrointestinal tract for an extended period. Mercury is radiopaque, and abdominal radiographs should be obtained in all cases of ingestion.16
Mercury is toxic to the CNS and peripheral nervous system, resulting in erethism mercurialis, a constellation of neuropsychologic signs and symptoms including restlessness, irritability, insomnia, emotional lability, difficulty concentrating, and impaired memory. In severe cases, delirium and psychosis may develop. Other CNS effects include tremors, paresthesia, dysarthria, neuromuscular changes, headaches, polyneuropathy, and cerebellar ataxia, as well as ophthalmologic and audiologic impairment.5,16
Upon inhalation exposure, patients with respiratory concerns should be given oxygen. Bronchospasms are treated with bronchodilators; however, if multiple chemical exposures are suspected, bronchial-sensitizing agents may pose additional risks. Corticosteroids and antibiotics have been recommended for treatment of chemical pneumonitis, but their efficacy has not been substantiated.16
Skin reactions associated with skin contact to elemental mercury are rare. However, hives and dermatitis have been observed following accidental contact with inorganic mercury compounds.5 Manifestation in children chronically exposed to mercury includes a nonallergic hypersensitivity (acrodynia),5,17 which is characterized by pain and dusky pink discoloration in the hands and feet, most often seen in children chronically exposed to mercury absorbed from vapor inhalation or cutaneous exposure.16
Renal conditions associated with acute inhalation of elemental mercury vapor include proteinuria, nephrotic syndrome, temporary tubular dysfunction, acute tubular necrosis, and oliguric renal failure.16 Chronic exposure to inorganic mercury compounds also has been reported to cause renal damage.5 Chelation therapy should be performed for any symptomatic patient with a clear history of acute elemental mercury exposure.16 The most frequently used chelation agent in cases of acute inorganic mercury exposures is dimercaprol. In rare cases of mercury intoxication, hemodialysis is required in the treatment of renal failure and to expedite removal of dimercaprol-mercury complexes.16
Cardiovascular symptoms associated with acute inhalation of high levels of elemental mercury include tachycardia and hypertension.16 Increases in blood pressure, palpitations, and heart rate also have been observed in instances of acute elemental mercury exposure. Studies show that exposure to mercury increases both the risk for acute myocardial infarction as well as death from coronary heart and cardiovascular diseases.5
Conclusion
Mercury poisoning presents with varied neuropsychologic signs and symptoms. Our case provides insight into a unique route of exposure for mercury toxicity. In addition to the unusual presentation of a mercury granuloma, our case illustrates how surgical techniques can aid in removal of cutaneous reservoirs in the setting of percutaneous exposure.
- History of mercury. Government of Canada website. Modified April 26, 2010. Accessed March 11, 2021. https://www.canada.ca/en/environment-climate-change/services/pollutants/mercury-environment/about/history.html
- Dartmouth Toxic Metals Superfund Research Program website. Accessed March 11, 2021. https://sites.dartmouth.edu/toxmetal/
- Norn S, Permin H, Kruse E, et al. Mercury—a major agent in the history of medicine and alchemy [in Danish]. Dan Medicinhist Arbog. 2008;36:21-40.
- Waldron HA. Did the Mad Hatter have mercury poisoning? Br Med J (Clin Res Ed). 1983;287:1961.
- Poulin J, Gibb H. Mercury: assessing the environmental burden of disease at national and local levels. WHO Environmental Burden of Disease Series No. 16. World Health Organization; 2008.
- Charcot JM. Clinical lectures of the diseases of the nervous system. In: Kinnier Wilson SA. The Landmark Library of Neurology and Neurosurgery. Gryphon Editions; 1994:186.
- Kinnier Wilson SA. Neurology. In: Kinnier Wilson SA. The Landmark Library of Neurology and Neurosurgery. Gryphon Editions; 1994:739-740.
- Harada M. Minamata disease: methylmercury poisoning in Japan caused by environmental pollution. Crit Rev Toxicol. 1995;25:1-24.
- Mercury and health. World Health Organization website. Updated March 31, 2017. Accessed March 12, 2021. http://www.whoint/mediacentre/factsheets/fs361/en/
- Olson DA. Mercury toxicity. Updated November 5, 2018. Accessed March 12, 2021.http://emedicine.medscape.com/article/1175560-overview
- Mercury thermometers. Environmental Protection Agency website. Updated June 26, 2018. https://www.epa.gov/mercury/mercury-thermometers
- Jao-Tan C, Pope E. Cutaneous poisoning syndromes in children: a review. Curr Opin Pediatr. 2006;18:410-416.
- US Department of Health and Human Services: Public Health Service Agency for Toxic Substances and Disease Registry. Toxicological profile for mercury: regulations and advisories. Published March 1999. Accessed March 23, 2021. https://www.atsdr.cdc.gov/toxprofiles/tp46.pdf
- US Food and Drug Administration. Mercury levels in commercial fish and shellfish (1990-2012). Updated October 25, 2017. Accessed March 16, 2021. https://www.fda.gov/food/metals-and-your-food/mercury-levels-commercial-fish-and-shellfish-1990-2012
- Hamann CR, Boonchai W, Wen L, et al. Spectrometric analysis of mercury content in 549 skin-lightening products: is mercury toxicity a hidden global health hazard? J Am Acad Dermatol. 2014;70:281-287.e3.
- Mercury. Managing Hazardous Materials Incidents. Agency for Toxic Substances and Disease Registry website. Accessed March 16, 2021. https://www.atsdr.cdc.gov/MHMI/mmg46.pdf
- Krohn IT, Solof A, Mobini J, et al. Subcutaneous injection of metallic mercury. JAMA. 1980;243:548-549.
- Lai O, Parsi KK, Wu D, et al. Mercury toxicity presenting acrodynia and a papulovesicular eruption in a 5-year-old girl. Dermatol Online J. 2016;16;22:13030/qt6444r7nc.
- Dolianiti M, Tasiopoulou K, Kalostou A, et al. Mercury bichloride iatrogenic poisoning: a case report. J Clin Toxicol. 2016;6:2. doi:10.4172/2161-0495.1000290
- Broussard LA, Hammett-Stabler CA, Winecker RE, et al. The toxicology of mercury. Lab Med. 2002;33:614-625. doi:10.1309/5HY1-V3NE-2LFL-P9MT
- Byeong-Jin Y, Byoung-Gwon K, Man-Joong J, et al. Evaluation of mercury exposure levels, clinical diagnosis and treatment for mercury intoxication. Ann Occup Environ Med. 2016;28:5.
Evidence of human exposure to mercury dates as far back as the Egyptians in 1500
Mercury release in the environment primarily is a function of human activity, including coal-fired power plants, residential heating, and mining.9,10 Mercury from these sources is commonly found in the sediment of lakes and bays, where it is enzymatically converted to methylmercury by aquatic microorganisms; subsequent food chain biomagnification results in elevated mercury levels in apex predators. Substantial release of mercury into the environment also can be attributed to health care facilities from their use of thermometers containing 0.5 to 3 g of elemental mercury,11 blood pressure monitors, and medical waste incinerators.5
Mercury has been reported as the second most common cause of heavy metal poisoning after lead.12 Standards from the US Food and Drug Administration dictate that methylmercury levels in fish and wheat products must not exceed 1 ppm.13 Most plant and animal food sources contain methylmercury at levels between 0.0001 and 0.01 ppm; mercury concentrations are especially high in tuna, averaging 0.4 ppm, while larger predatory fish contain levels in excess of 1 ppm.14 The use of mercury-containing cosmetic products also presents a substantial exposure risk to consumers.5,10 In one study, 3.3% of skin-lightening creams and soaps purchased within the United States contained concentrations of mercury exceeding 1000 ppm.15
We describe a case of mercury toxicity resulting from intentional injection of liquid mercury into the right antecubital fossa in a suicide attempt.
Case Report
A 31-year-old woman presented to the family practice center for evaluation of a firm stained area on the skin of the right arm. She reported increasing anxiety, depression, tremors, irritability, and difficulty concentrating over the last 6 months. She denied headache and joint or muscle pain. Four years earlier, she had broken apart a thermometer and injected approximately 0.7 mL of its contents into the right arm in a suicide attempt. She intended to inject the thermometer’s contents directly into a vein, but the material instead entered the surrounding tissue. She denied notable pain or itching overlying the injection site. Her medications included aripiprazole and buspirone. She noted that she smoked half a pack of cigarettes per day and had a history of methamphetamine abuse. She was homeless and unemployed. Physical examination revealed an anxious tremulous woman with an erythematous to bluish gray, firm plaque on the right antecubital fossa (Figure 1). There were no notable tremors and no gait disturbance.
Her blood mercury level was greater than 100 µg/L and urine mercury was 477 µg/g (reference ranges, 1–8 μg/L and 4–5 μg/L, respectively). A radiograph of the right elbow area revealed scattered punctate foci of increased density within or overlying the anterolateral elbow soft tissues. She was diagnosed with mercury granuloma causing chronic mercury elevation. She underwent excision of the granuloma (Figure 2) with endovascular surgery via an elliptical incision. The patient was subsequently lost to follow-up.
Comment
Elemental mercury is a silver liquid at room temperature that spontaneously evaporates to form mercury vapor, an invisible, odorless, toxic gas. Accidental cutaneous exposure typically is safely managed by washing exposed skin with soap and water,16 though there is a potential risk for systemic absorption, especially when the skin is inflamed. When metallic mercury is subcutaneously injected, it is advised to promptly excise all subcutaneous areas containing mercury, regardless of any symptoms of systemic toxicity. Patients should subsequently be monitored for signs of both central nervous system (CNS) and renal deficits, undergo chelation therapy when systemic effects are apparent, and finally receive psychiatric consultation and treatment when necessary.17
Inorganic mercury compounds are formed when elemental mercury combines with sulfur or oxygen and often take the form of mercury salts, which appear as white crystals.16 These salts occur naturally in the environment and are used in pesticides, antiseptics, and skin-lightening creams and soaps.18
Methylmercury is a highly toxic, organic compound that is capable of crossing the placental and blood-brain barriers. It is the most common organic mercury compound found in the environment.16 Most humans have trace amounts of methylmercury in their bodies, typically as a result of consuming seafood.5
Exposure to mercury most commonly occurs through chronic consumption of methylmercury in seafood or acute inhalation of elemental mercury vapors.9 Iatrogenic cases of mercury exposure via injection also have been reported in the literature, including a case resulting in acute poisoning due to peritoneal lavage with mercury bichloride.19 Acute mercury-induced pulmonary damage typically resolves completely. However, there have been reported cases of exposure progressing to interstitial emphysema, pneumatocele, pneumothorax, pneumomediastinum, interstitial fibrosis, and chronic respiratory insufficiency, with examples of fatal acute respiratory distress syndrome being reported.5,16,20 Although individuals who inhale mercury vapors initially may be unaware of exposure due to little upper airway irritation, symptoms following an initial acute exposure may include ptyalism, a metallic taste, dysphagia, enteritis, diarrhea, nausea, renal damage, and CNS effects.16 Additionally, exposure may lead to confusion with signs and symptoms of metal fume fever, including shortness of breath, pleuritic chest pain, stomatitis, lethargy, and vomiting.20
Chronic exposure to mercury vapor can result in accumulation of mercury in the body, leading to neuropsychiatric, dermatologic, oropharyngeal, and renal manifestations. Sore throat, fever, headache, fatigue, dyspnea, chest pain, and pneumonitis are common.16 Typically, low-level exposure to elemental mercury does not lead to long-lasting health effects. However, individuals exposed to high-level elemental mercury vapors may require hospitalization. Treatment of acute mercury poisoning consists of removing the source of exposure, followed by cardiopulmonary support.16
Specific assays for mercury levels in blood and urine are useful to assess the level of exposure and risk to the patient. Blood mercury concentrations of 20 µg/L or below are considered within reference range; however, once blood and urine concentrations of mercury exceed 100 µg/L, clinical signs of acute mercury poisoning typically manifest.21 Chest radiographs can reveal pulmonary damage, while complete blood cell count, metabolic panel, and urinalysis can assess damage to other organs. Neuropsychiatric testing and nerve conduction studies may provide objective evidence of CNS toxicity. Assays for N-acetyl-β-D-glucosaminidase can provide an indication of early renal tubular dysfunction.16
Elemental mercury is not absorbed from the gastrointestinal tract, posing minimal risk for acute toxicity from ingestion. Generally, less than 10% of ingested inorganic mercury is absorbed from the gut, while elemental mercury is nonabsorbable.10 If an individual ingests a large amount of mercury, it may persist in the gastrointestinal tract for an extended period. Mercury is radiopaque, and abdominal radiographs should be obtained in all cases of ingestion.16
Mercury is toxic to the CNS and peripheral nervous system, resulting in erethism mercurialis, a constellation of neuropsychologic signs and symptoms including restlessness, irritability, insomnia, emotional lability, difficulty concentrating, and impaired memory. In severe cases, delirium and psychosis may develop. Other CNS effects include tremors, paresthesia, dysarthria, neuromuscular changes, headaches, polyneuropathy, and cerebellar ataxia, as well as ophthalmologic and audiologic impairment.5,16
Upon inhalation exposure, patients with respiratory concerns should be given oxygen. Bronchospasms are treated with bronchodilators; however, if multiple chemical exposures are suspected, bronchial-sensitizing agents may pose additional risks. Corticosteroids and antibiotics have been recommended for treatment of chemical pneumonitis, but their efficacy has not been substantiated.16
Skin reactions associated with skin contact to elemental mercury are rare. However, hives and dermatitis have been observed following accidental contact with inorganic mercury compounds.5 Manifestation in children chronically exposed to mercury includes a nonallergic hypersensitivity (acrodynia),5,17 which is characterized by pain and dusky pink discoloration in the hands and feet, most often seen in children chronically exposed to mercury absorbed from vapor inhalation or cutaneous exposure.16
Renal conditions associated with acute inhalation of elemental mercury vapor include proteinuria, nephrotic syndrome, temporary tubular dysfunction, acute tubular necrosis, and oliguric renal failure.16 Chronic exposure to inorganic mercury compounds also has been reported to cause renal damage.5 Chelation therapy should be performed for any symptomatic patient with a clear history of acute elemental mercury exposure.16 The most frequently used chelation agent in cases of acute inorganic mercury exposures is dimercaprol. In rare cases of mercury intoxication, hemodialysis is required in the treatment of renal failure and to expedite removal of dimercaprol-mercury complexes.16
Cardiovascular symptoms associated with acute inhalation of high levels of elemental mercury include tachycardia and hypertension.16 Increases in blood pressure, palpitations, and heart rate also have been observed in instances of acute elemental mercury exposure. Studies show that exposure to mercury increases both the risk for acute myocardial infarction as well as death from coronary heart and cardiovascular diseases.5
Conclusion
Mercury poisoning presents with varied neuropsychologic signs and symptoms. Our case provides insight into a unique route of exposure for mercury toxicity. In addition to the unusual presentation of a mercury granuloma, our case illustrates how surgical techniques can aid in removal of cutaneous reservoirs in the setting of percutaneous exposure.
Evidence of human exposure to mercury dates as far back as the Egyptians in 1500
Mercury release in the environment primarily is a function of human activity, including coal-fired power plants, residential heating, and mining.9,10 Mercury from these sources is commonly found in the sediment of lakes and bays, where it is enzymatically converted to methylmercury by aquatic microorganisms; subsequent food chain biomagnification results in elevated mercury levels in apex predators. Substantial release of mercury into the environment also can be attributed to health care facilities from their use of thermometers containing 0.5 to 3 g of elemental mercury,11 blood pressure monitors, and medical waste incinerators.5
Mercury has been reported as the second most common cause of heavy metal poisoning after lead.12 Standards from the US Food and Drug Administration dictate that methylmercury levels in fish and wheat products must not exceed 1 ppm.13 Most plant and animal food sources contain methylmercury at levels between 0.0001 and 0.01 ppm; mercury concentrations are especially high in tuna, averaging 0.4 ppm, while larger predatory fish contain levels in excess of 1 ppm.14 The use of mercury-containing cosmetic products also presents a substantial exposure risk to consumers.5,10 In one study, 3.3% of skin-lightening creams and soaps purchased within the United States contained concentrations of mercury exceeding 1000 ppm.15
We describe a case of mercury toxicity resulting from intentional injection of liquid mercury into the right antecubital fossa in a suicide attempt.
Case Report
A 31-year-old woman presented to the family practice center for evaluation of a firm stained area on the skin of the right arm. She reported increasing anxiety, depression, tremors, irritability, and difficulty concentrating over the last 6 months. She denied headache and joint or muscle pain. Four years earlier, she had broken apart a thermometer and injected approximately 0.7 mL of its contents into the right arm in a suicide attempt. She intended to inject the thermometer’s contents directly into a vein, but the material instead entered the surrounding tissue. She denied notable pain or itching overlying the injection site. Her medications included aripiprazole and buspirone. She noted that she smoked half a pack of cigarettes per day and had a history of methamphetamine abuse. She was homeless and unemployed. Physical examination revealed an anxious tremulous woman with an erythematous to bluish gray, firm plaque on the right antecubital fossa (Figure 1). There were no notable tremors and no gait disturbance.
Her blood mercury level was greater than 100 µg/L and urine mercury was 477 µg/g (reference ranges, 1–8 μg/L and 4–5 μg/L, respectively). A radiograph of the right elbow area revealed scattered punctate foci of increased density within or overlying the anterolateral elbow soft tissues. She was diagnosed with mercury granuloma causing chronic mercury elevation. She underwent excision of the granuloma (Figure 2) with endovascular surgery via an elliptical incision. The patient was subsequently lost to follow-up.
Comment
Elemental mercury is a silver liquid at room temperature that spontaneously evaporates to form mercury vapor, an invisible, odorless, toxic gas. Accidental cutaneous exposure typically is safely managed by washing exposed skin with soap and water,16 though there is a potential risk for systemic absorption, especially when the skin is inflamed. When metallic mercury is subcutaneously injected, it is advised to promptly excise all subcutaneous areas containing mercury, regardless of any symptoms of systemic toxicity. Patients should subsequently be monitored for signs of both central nervous system (CNS) and renal deficits, undergo chelation therapy when systemic effects are apparent, and finally receive psychiatric consultation and treatment when necessary.17
Inorganic mercury compounds are formed when elemental mercury combines with sulfur or oxygen and often take the form of mercury salts, which appear as white crystals.16 These salts occur naturally in the environment and are used in pesticides, antiseptics, and skin-lightening creams and soaps.18
Methylmercury is a highly toxic, organic compound that is capable of crossing the placental and blood-brain barriers. It is the most common organic mercury compound found in the environment.16 Most humans have trace amounts of methylmercury in their bodies, typically as a result of consuming seafood.5
Exposure to mercury most commonly occurs through chronic consumption of methylmercury in seafood or acute inhalation of elemental mercury vapors.9 Iatrogenic cases of mercury exposure via injection also have been reported in the literature, including a case resulting in acute poisoning due to peritoneal lavage with mercury bichloride.19 Acute mercury-induced pulmonary damage typically resolves completely. However, there have been reported cases of exposure progressing to interstitial emphysema, pneumatocele, pneumothorax, pneumomediastinum, interstitial fibrosis, and chronic respiratory insufficiency, with examples of fatal acute respiratory distress syndrome being reported.5,16,20 Although individuals who inhale mercury vapors initially may be unaware of exposure due to little upper airway irritation, symptoms following an initial acute exposure may include ptyalism, a metallic taste, dysphagia, enteritis, diarrhea, nausea, renal damage, and CNS effects.16 Additionally, exposure may lead to confusion with signs and symptoms of metal fume fever, including shortness of breath, pleuritic chest pain, stomatitis, lethargy, and vomiting.20
Chronic exposure to mercury vapor can result in accumulation of mercury in the body, leading to neuropsychiatric, dermatologic, oropharyngeal, and renal manifestations. Sore throat, fever, headache, fatigue, dyspnea, chest pain, and pneumonitis are common.16 Typically, low-level exposure to elemental mercury does not lead to long-lasting health effects. However, individuals exposed to high-level elemental mercury vapors may require hospitalization. Treatment of acute mercury poisoning consists of removing the source of exposure, followed by cardiopulmonary support.16
Specific assays for mercury levels in blood and urine are useful to assess the level of exposure and risk to the patient. Blood mercury concentrations of 20 µg/L or below are considered within reference range; however, once blood and urine concentrations of mercury exceed 100 µg/L, clinical signs of acute mercury poisoning typically manifest.21 Chest radiographs can reveal pulmonary damage, while complete blood cell count, metabolic panel, and urinalysis can assess damage to other organs. Neuropsychiatric testing and nerve conduction studies may provide objective evidence of CNS toxicity. Assays for N-acetyl-β-D-glucosaminidase can provide an indication of early renal tubular dysfunction.16
Elemental mercury is not absorbed from the gastrointestinal tract, posing minimal risk for acute toxicity from ingestion. Generally, less than 10% of ingested inorganic mercury is absorbed from the gut, while elemental mercury is nonabsorbable.10 If an individual ingests a large amount of mercury, it may persist in the gastrointestinal tract for an extended period. Mercury is radiopaque, and abdominal radiographs should be obtained in all cases of ingestion.16
Mercury is toxic to the CNS and peripheral nervous system, resulting in erethism mercurialis, a constellation of neuropsychologic signs and symptoms including restlessness, irritability, insomnia, emotional lability, difficulty concentrating, and impaired memory. In severe cases, delirium and psychosis may develop. Other CNS effects include tremors, paresthesia, dysarthria, neuromuscular changes, headaches, polyneuropathy, and cerebellar ataxia, as well as ophthalmologic and audiologic impairment.5,16
Upon inhalation exposure, patients with respiratory concerns should be given oxygen. Bronchospasms are treated with bronchodilators; however, if multiple chemical exposures are suspected, bronchial-sensitizing agents may pose additional risks. Corticosteroids and antibiotics have been recommended for treatment of chemical pneumonitis, but their efficacy has not been substantiated.16
Skin reactions associated with skin contact to elemental mercury are rare. However, hives and dermatitis have been observed following accidental contact with inorganic mercury compounds.5 Manifestation in children chronically exposed to mercury includes a nonallergic hypersensitivity (acrodynia),5,17 which is characterized by pain and dusky pink discoloration in the hands and feet, most often seen in children chronically exposed to mercury absorbed from vapor inhalation or cutaneous exposure.16
Renal conditions associated with acute inhalation of elemental mercury vapor include proteinuria, nephrotic syndrome, temporary tubular dysfunction, acute tubular necrosis, and oliguric renal failure.16 Chronic exposure to inorganic mercury compounds also has been reported to cause renal damage.5 Chelation therapy should be performed for any symptomatic patient with a clear history of acute elemental mercury exposure.16 The most frequently used chelation agent in cases of acute inorganic mercury exposures is dimercaprol. In rare cases of mercury intoxication, hemodialysis is required in the treatment of renal failure and to expedite removal of dimercaprol-mercury complexes.16
Cardiovascular symptoms associated with acute inhalation of high levels of elemental mercury include tachycardia and hypertension.16 Increases in blood pressure, palpitations, and heart rate also have been observed in instances of acute elemental mercury exposure. Studies show that exposure to mercury increases both the risk for acute myocardial infarction as well as death from coronary heart and cardiovascular diseases.5
Conclusion
Mercury poisoning presents with varied neuropsychologic signs and symptoms. Our case provides insight into a unique route of exposure for mercury toxicity. In addition to the unusual presentation of a mercury granuloma, our case illustrates how surgical techniques can aid in removal of cutaneous reservoirs in the setting of percutaneous exposure.
- History of mercury. Government of Canada website. Modified April 26, 2010. Accessed March 11, 2021. https://www.canada.ca/en/environment-climate-change/services/pollutants/mercury-environment/about/history.html
- Dartmouth Toxic Metals Superfund Research Program website. Accessed March 11, 2021. https://sites.dartmouth.edu/toxmetal/
- Norn S, Permin H, Kruse E, et al. Mercury—a major agent in the history of medicine and alchemy [in Danish]. Dan Medicinhist Arbog. 2008;36:21-40.
- Waldron HA. Did the Mad Hatter have mercury poisoning? Br Med J (Clin Res Ed). 1983;287:1961.
- Poulin J, Gibb H. Mercury: assessing the environmental burden of disease at national and local levels. WHO Environmental Burden of Disease Series No. 16. World Health Organization; 2008.
- Charcot JM. Clinical lectures of the diseases of the nervous system. In: Kinnier Wilson SA. The Landmark Library of Neurology and Neurosurgery. Gryphon Editions; 1994:186.
- Kinnier Wilson SA. Neurology. In: Kinnier Wilson SA. The Landmark Library of Neurology and Neurosurgery. Gryphon Editions; 1994:739-740.
- Harada M. Minamata disease: methylmercury poisoning in Japan caused by environmental pollution. Crit Rev Toxicol. 1995;25:1-24.
- Mercury and health. World Health Organization website. Updated March 31, 2017. Accessed March 12, 2021. http://www.whoint/mediacentre/factsheets/fs361/en/
- Olson DA. Mercury toxicity. Updated November 5, 2018. Accessed March 12, 2021.http://emedicine.medscape.com/article/1175560-overview
- Mercury thermometers. Environmental Protection Agency website. Updated June 26, 2018. https://www.epa.gov/mercury/mercury-thermometers
- Jao-Tan C, Pope E. Cutaneous poisoning syndromes in children: a review. Curr Opin Pediatr. 2006;18:410-416.
- US Department of Health and Human Services: Public Health Service Agency for Toxic Substances and Disease Registry. Toxicological profile for mercury: regulations and advisories. Published March 1999. Accessed March 23, 2021. https://www.atsdr.cdc.gov/toxprofiles/tp46.pdf
- US Food and Drug Administration. Mercury levels in commercial fish and shellfish (1990-2012). Updated October 25, 2017. Accessed March 16, 2021. https://www.fda.gov/food/metals-and-your-food/mercury-levels-commercial-fish-and-shellfish-1990-2012
- Hamann CR, Boonchai W, Wen L, et al. Spectrometric analysis of mercury content in 549 skin-lightening products: is mercury toxicity a hidden global health hazard? J Am Acad Dermatol. 2014;70:281-287.e3.
- Mercury. Managing Hazardous Materials Incidents. Agency for Toxic Substances and Disease Registry website. Accessed March 16, 2021. https://www.atsdr.cdc.gov/MHMI/mmg46.pdf
- Krohn IT, Solof A, Mobini J, et al. Subcutaneous injection of metallic mercury. JAMA. 1980;243:548-549.
- Lai O, Parsi KK, Wu D, et al. Mercury toxicity presenting acrodynia and a papulovesicular eruption in a 5-year-old girl. Dermatol Online J. 2016;16;22:13030/qt6444r7nc.
- Dolianiti M, Tasiopoulou K, Kalostou A, et al. Mercury bichloride iatrogenic poisoning: a case report. J Clin Toxicol. 2016;6:2. doi:10.4172/2161-0495.1000290
- Broussard LA, Hammett-Stabler CA, Winecker RE, et al. The toxicology of mercury. Lab Med. 2002;33:614-625. doi:10.1309/5HY1-V3NE-2LFL-P9MT
- Byeong-Jin Y, Byoung-Gwon K, Man-Joong J, et al. Evaluation of mercury exposure levels, clinical diagnosis and treatment for mercury intoxication. Ann Occup Environ Med. 2016;28:5.
- History of mercury. Government of Canada website. Modified April 26, 2010. Accessed March 11, 2021. https://www.canada.ca/en/environment-climate-change/services/pollutants/mercury-environment/about/history.html
- Dartmouth Toxic Metals Superfund Research Program website. Accessed March 11, 2021. https://sites.dartmouth.edu/toxmetal/
- Norn S, Permin H, Kruse E, et al. Mercury—a major agent in the history of medicine and alchemy [in Danish]. Dan Medicinhist Arbog. 2008;36:21-40.
- Waldron HA. Did the Mad Hatter have mercury poisoning? Br Med J (Clin Res Ed). 1983;287:1961.
- Poulin J, Gibb H. Mercury: assessing the environmental burden of disease at national and local levels. WHO Environmental Burden of Disease Series No. 16. World Health Organization; 2008.
- Charcot JM. Clinical lectures of the diseases of the nervous system. In: Kinnier Wilson SA. The Landmark Library of Neurology and Neurosurgery. Gryphon Editions; 1994:186.
- Kinnier Wilson SA. Neurology. In: Kinnier Wilson SA. The Landmark Library of Neurology and Neurosurgery. Gryphon Editions; 1994:739-740.
- Harada M. Minamata disease: methylmercury poisoning in Japan caused by environmental pollution. Crit Rev Toxicol. 1995;25:1-24.
- Mercury and health. World Health Organization website. Updated March 31, 2017. Accessed March 12, 2021. http://www.whoint/mediacentre/factsheets/fs361/en/
- Olson DA. Mercury toxicity. Updated November 5, 2018. Accessed March 12, 2021.http://emedicine.medscape.com/article/1175560-overview
- Mercury thermometers. Environmental Protection Agency website. Updated June 26, 2018. https://www.epa.gov/mercury/mercury-thermometers
- Jao-Tan C, Pope E. Cutaneous poisoning syndromes in children: a review. Curr Opin Pediatr. 2006;18:410-416.
- US Department of Health and Human Services: Public Health Service Agency for Toxic Substances and Disease Registry. Toxicological profile for mercury: regulations and advisories. Published March 1999. Accessed March 23, 2021. https://www.atsdr.cdc.gov/toxprofiles/tp46.pdf
- US Food and Drug Administration. Mercury levels in commercial fish and shellfish (1990-2012). Updated October 25, 2017. Accessed March 16, 2021. https://www.fda.gov/food/metals-and-your-food/mercury-levels-commercial-fish-and-shellfish-1990-2012
- Hamann CR, Boonchai W, Wen L, et al. Spectrometric analysis of mercury content in 549 skin-lightening products: is mercury toxicity a hidden global health hazard? J Am Acad Dermatol. 2014;70:281-287.e3.
- Mercury. Managing Hazardous Materials Incidents. Agency for Toxic Substances and Disease Registry website. Accessed March 16, 2021. https://www.atsdr.cdc.gov/MHMI/mmg46.pdf
- Krohn IT, Solof A, Mobini J, et al. Subcutaneous injection of metallic mercury. JAMA. 1980;243:548-549.
- Lai O, Parsi KK, Wu D, et al. Mercury toxicity presenting acrodynia and a papulovesicular eruption in a 5-year-old girl. Dermatol Online J. 2016;16;22:13030/qt6444r7nc.
- Dolianiti M, Tasiopoulou K, Kalostou A, et al. Mercury bichloride iatrogenic poisoning: a case report. J Clin Toxicol. 2016;6:2. doi:10.4172/2161-0495.1000290
- Broussard LA, Hammett-Stabler CA, Winecker RE, et al. The toxicology of mercury. Lab Med. 2002;33:614-625. doi:10.1309/5HY1-V3NE-2LFL-P9MT
- Byeong-Jin Y, Byoung-Gwon K, Man-Joong J, et al. Evaluation of mercury exposure levels, clinical diagnosis and treatment for mercury intoxication. Ann Occup Environ Med. 2016;28:5.
Practice Points
- Chronic mercury granulomas can present as firm, erythematous to bluish gray plaques.
- Accidental skin contact to elemental mercury may cause urticaria and dermatitis.
- Blood mercury concentrations below 20 11µg/L are considered within reference range; once blood and urine concentrations exceed 100 11µg/L, clinical signs of acute mercury poisoning typically manifest.
- Mercury is toxic to the central and peripheral nervous systems, resulting in erethism mercurialis, a constellation of neuropsychologic signs and symptoms including restlessness, irritability, insomnia, emotional lability, difficulty concentrating, and impaired memory.