User login
Spontaneously Regressing Primary Nodular Melanoma of the Glans Penis
To the Editor:
Primary malignant melanoma (PMM) of the penis is rare, comprising 1% of melanomas overall and less than 4% of malignancies in the male genitourinary tract.1 However, regression of PMM is not rare. Melanoma is 6 times more likely to undergo regression compared to other malignancies.2 Approximately 10% to 35% of cutaneous PMMs undergo partial regression, but only 42 cases of completely regressed cutaneous PMMs have been reported,3,4 which may be due to underreporting of completely regressed cutaneous PMMs, as they often are clinically inconspicuous. Additionally, completely regressed cutaneous PMMs may be incorrectly reported as metastatic melanoma of unknown primary.5 Clinical characteristics of regression include pink coloration and a lightening or whitening of baseline lesional color. Dermatoscopic features of regression include white areas, blue areas, or vascular structures that translate microscopically to dermal fibrosis, melanophages, and telangiectases.5 We report a case of complete clinical regression of a nodular, mucosal, penile PMM with no evidence of metastatic disease.
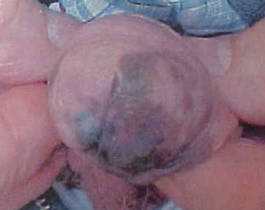
An 86-year-old man presented with a progressively enlarging, pigmented lesion on the glans penis of 2 years’ duration. His medical history was notable for retinal detachment, macular degeneration, lumbar stenosis, and seizures postneurosurgery for a subdural hematoma. Physical examination revealed a healthy man with a mottled, black-brown, macular, and nodular lesion with irregular margins and irregular shape on the glans penis (Figure 1). No other similar skin lesions or lymphadenopathy were detectable. A lesional deep shave biopsy obtained at presentation demonstrated a nodular-type malignant melanoma with a Breslow thickness of approximately 3.5 mm (Figure 2).
|
| Figure 1. Mottled, black-brown, macular, and nodular lesion with irregular margins and irregular shape on the glans penis. |
|
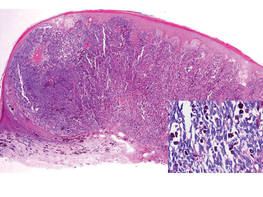
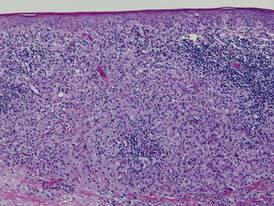
| Figure 2. Histologic section showed a nodular melanocytic proliferation with a dense sheetlike collection of melanocytes, predominantly in the dermis (H&E, original magnification ×20). Prominent cytologic atypia and multiple mitotic figures were consistent with melanoma (inset)(H&E, original magnification ×400). |
|
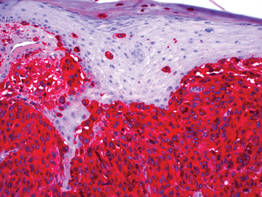
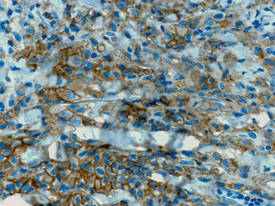
| Figure 3. High-power view of HMB-45 stain showed strong and diffuse staining, including several pagetoid intraepidermal melanocytes (original magnification ×200). |
|
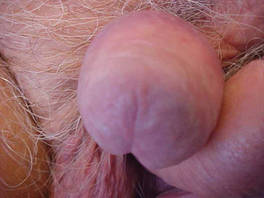
| Figure 4. Skin examination 8 years following the initial primary malignant melanoma diagnosis showed no clinical evidence of recurrent or metastatic melanoma and almost complete loss of pigmentation at the prior melanoma site. |
Histologic examination showed nodular nests of malignant melanocytes that were dispersed along the dermal-epidermal junction and coalesced into sheets within the dermis. Numerous dermal mitoses were present. The tumor was strongly and diffusely positive with Melan-A and HMB-45, which also highlighted scattered pagetoid intraepidermal cells (Figure 3). These findings were diagnostic of PMM of the mucocutaneous glans penis. The tumor was nonulcerated and invaded to a Breslow thickness of approximately 3.5 mm, corresponding to American Joint Committee on Cancer stage IIA (T3aN0M0) with an expected 5-year survival rate of 79%.6
He was referred to the urology department and was offered cystoscopy, urethrography, and phallectomy, which he refused. He also refused a trial of imiquimod. Computed tomography (CT) scans of his brain, chest, abdomen, and pelvis were negative for metastatic disease. Following the initial melanoma diagnosis, he had yearly dermatologic evaluations consisting of total-body skin and lymph node (LN) examinations. At 87 years of age (1 year following the initial diagnosis), the melanoma became dramatically smaller. At 88 years of age (2 years after diagnosis), the melanoma had near-complete clinical resolution. At 89 years of age, the patient reported asymmetric hearing loss. A cranial magnetic resonance imaging study showed no evidence of metastases.
At 92 years of age (6 years after the initial diagnosis), the patient reported bilateral leg pain. A CT scan of the lumbar spine showed no evidence of metastasis. He also reported abdominal pain. A CT scan of the abdomen and pelvis revealed an ileocecal mass. Biopsy of the ileocecal mass showed moderately differentiated invasive adenocarcinoma and no evidence of metastatic melanoma. The adenocarcinoma was resected and he continues to do well. Skin and LN examination 8 years after the initial diagnosis showed no clinical evidence of recurrent penile mucosal melanoma or metastatic melanoma (Figure 4). The PMM appeared to have clinically regressed spontaneously. He refused repeat skin biopsy and additional imaging studies.
The criteria for complete melanoma regression were initially described in 19657 and revised in 2005.2 Although our patient demonstrated complete clinical regression of his PMM, he did not meet the revised criteria for complete regression because there was no histopathologic confirmation of regression or of the absence of melanoma as well as no lymphatic involvement. It is extremely difficult to quantify the percentage of PMMs that completely regress. A case of a completely regressed untreated PMM with no metastatic disease 4 years after diagnosis has been reported. This case involved a nonulcerated melanoma with a Breslow thickness of 0.7 mm (American Joint Committee on Cancer stage IA).4 The prognosis of penile mucosal PMM is comparable to that of cutaneous PMM with a similar Breslow thickness.1
The prognostic significance of melanoma regression is controversial. Regression may be mediated by host immunity, apoptosis, and/or antiangiogenesis. The lymphocytic infiltrate in regressive melanomas consists of cytotoxic T cells with selective antitumor activity, which induces HLA class I–restricted melanoma lysis.8 Lymph node migration may result in T-lymphocyte priming and induction of antitumor immunity.9 Therefore, regression may indicate risk for sentinel LN metastasis.
It is possible that complete regression of melanoma does not truly exist, and late recurrence due to cancer dormancy is inevitable. Late recurrence is defined as first metastasis 10 years after complete removal of the PMM.10 Our patient has only been followed for 8 years, so this possibility cannot be entirely excluded.
1. van Geel AN, den Bakker MA, Kirkels W, et al. Prognosis of primary mucosal penile melanoma: a series of 19 Dutch patients and 47 patients from the literature. Urology. 2007;70:143-147.
2. High WA, Stewart D, Wilbers CR, et al. Completely regressed primary cutaneous malignant melanoma with nodal and/or visceral metastases: a report of 5 cases and assessment of the literature and diagnostic criteria. J Am Acad Dermatol. 2005;53:89-100.
3. Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
4. Muniesa C, Ferreres JR, Moreno A, et al. Completely regressed primary cutaneous malignant melanoma with metastases [published online ahead of print June 23, 2008]. J Eur Acad Dermatol Venereol. 2009;23:327-328.
5. Bories N, Dalle S, Debarbieux S, et al. Dermoscopy of fully regressive cutaneous melanoma [published online ahead of print March 13, 2008]. Br J Dermatol. 2008;158:1224-1229.
6. Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification [published online ahead of print November 16, 2009]. J Clin Oncol. 2009;27:6199-6206.
7. Smith JL Jr, Stehlin JS Jr. Spontaneous regression of primary malignant melanomas with regional metastasis. Cancer. 1965;18:1399-1415.
8. Bottger D, Dowden RV, Kay PP. Complete spontaneous regression of cutaneous primary malignant melanoma. Plast Reconstr Surg. 1992;89:548-553.
9. Shaw HM, McCarthy SW, McCarthy WH, et al. Thin regressing malignant melanoma: significance of concurrent regional lymph node metastases. Histopathology. 1989;15:257-265.
10. Hansel G, Schönlebe J, Haroske G, et al. Late recurrence (10 years or more) of malignant melanoma in south-east Germany (Saxony). a single-centre analysis of 1881 patients with a follow-up of 10 years or more [published online ahead of print January 11, 2010]. J Eur Acad Dermatol Venereol. 2010;24:833-836.
To the Editor:
Primary malignant melanoma (PMM) of the penis is rare, comprising 1% of melanomas overall and less than 4% of malignancies in the male genitourinary tract.1 However, regression of PMM is not rare. Melanoma is 6 times more likely to undergo regression compared to other malignancies.2 Approximately 10% to 35% of cutaneous PMMs undergo partial regression, but only 42 cases of completely regressed cutaneous PMMs have been reported,3,4 which may be due to underreporting of completely regressed cutaneous PMMs, as they often are clinically inconspicuous. Additionally, completely regressed cutaneous PMMs may be incorrectly reported as metastatic melanoma of unknown primary.5 Clinical characteristics of regression include pink coloration and a lightening or whitening of baseline lesional color. Dermatoscopic features of regression include white areas, blue areas, or vascular structures that translate microscopically to dermal fibrosis, melanophages, and telangiectases.5 We report a case of complete clinical regression of a nodular, mucosal, penile PMM with no evidence of metastatic disease.
An 86-year-old man presented with a progressively enlarging, pigmented lesion on the glans penis of 2 years’ duration. His medical history was notable for retinal detachment, macular degeneration, lumbar stenosis, and seizures postneurosurgery for a subdural hematoma. Physical examination revealed a healthy man with a mottled, black-brown, macular, and nodular lesion with irregular margins and irregular shape on the glans penis (Figure 1). No other similar skin lesions or lymphadenopathy were detectable. A lesional deep shave biopsy obtained at presentation demonstrated a nodular-type malignant melanoma with a Breslow thickness of approximately 3.5 mm (Figure 2).
|
|
| Figure 1. Mottled, black-brown, macular, and nodular lesion with irregular margins and irregular shape on the glans penis. |
|
|
| Figure 2. Histologic section showed a nodular melanocytic proliferation with a dense sheetlike collection of melanocytes, predominantly in the dermis (H&E, original magnification ×20). Prominent cytologic atypia and multiple mitotic figures were consistent with melanoma (inset)(H&E, original magnification ×400). |
|
|
| Figure 3. High-power view of HMB-45 stain showed strong and diffuse staining, including several pagetoid intraepidermal melanocytes (original magnification ×200). |
|
|
| Figure 4. Skin examination 8 years following the initial primary malignant melanoma diagnosis showed no clinical evidence of recurrent or metastatic melanoma and almost complete loss of pigmentation at the prior melanoma site. |
Histologic examination showed nodular nests of malignant melanocytes that were dispersed along the dermal-epidermal junction and coalesced into sheets within the dermis. Numerous dermal mitoses were present. The tumor was strongly and diffusely positive with Melan-A and HMB-45, which also highlighted scattered pagetoid intraepidermal cells (Figure 3). These findings were diagnostic of PMM of the mucocutaneous glans penis. The tumor was nonulcerated and invaded to a Breslow thickness of approximately 3.5 mm, corresponding to American Joint Committee on Cancer stage IIA (T3aN0M0) with an expected 5-year survival rate of 79%.6
He was referred to the urology department and was offered cystoscopy, urethrography, and phallectomy, which he refused. He also refused a trial of imiquimod. Computed tomography (CT) scans of his brain, chest, abdomen, and pelvis were negative for metastatic disease. Following the initial melanoma diagnosis, he had yearly dermatologic evaluations consisting of total-body skin and lymph node (LN) examinations. At 87 years of age (1 year following the initial diagnosis), the melanoma became dramatically smaller. At 88 years of age (2 years after diagnosis), the melanoma had near-complete clinical resolution. At 89 years of age, the patient reported asymmetric hearing loss. A cranial magnetic resonance imaging study showed no evidence of metastases.
At 92 years of age (6 years after the initial diagnosis), the patient reported bilateral leg pain. A CT scan of the lumbar spine showed no evidence of metastasis. He also reported abdominal pain. A CT scan of the abdomen and pelvis revealed an ileocecal mass. Biopsy of the ileocecal mass showed moderately differentiated invasive adenocarcinoma and no evidence of metastatic melanoma. The adenocarcinoma was resected and he continues to do well. Skin and LN examination 8 years after the initial diagnosis showed no clinical evidence of recurrent penile mucosal melanoma or metastatic melanoma (Figure 4). The PMM appeared to have clinically regressed spontaneously. He refused repeat skin biopsy and additional imaging studies.
The criteria for complete melanoma regression were initially described in 19657 and revised in 2005.2 Although our patient demonstrated complete clinical regression of his PMM, he did not meet the revised criteria for complete regression because there was no histopathologic confirmation of regression or of the absence of melanoma as well as no lymphatic involvement. It is extremely difficult to quantify the percentage of PMMs that completely regress. A case of a completely regressed untreated PMM with no metastatic disease 4 years after diagnosis has been reported. This case involved a nonulcerated melanoma with a Breslow thickness of 0.7 mm (American Joint Committee on Cancer stage IA).4 The prognosis of penile mucosal PMM is comparable to that of cutaneous PMM with a similar Breslow thickness.1
The prognostic significance of melanoma regression is controversial. Regression may be mediated by host immunity, apoptosis, and/or antiangiogenesis. The lymphocytic infiltrate in regressive melanomas consists of cytotoxic T cells with selective antitumor activity, which induces HLA class I–restricted melanoma lysis.8 Lymph node migration may result in T-lymphocyte priming and induction of antitumor immunity.9 Therefore, regression may indicate risk for sentinel LN metastasis.
It is possible that complete regression of melanoma does not truly exist, and late recurrence due to cancer dormancy is inevitable. Late recurrence is defined as first metastasis 10 years after complete removal of the PMM.10 Our patient has only been followed for 8 years, so this possibility cannot be entirely excluded.
To the Editor:
Primary malignant melanoma (PMM) of the penis is rare, comprising 1% of melanomas overall and less than 4% of malignancies in the male genitourinary tract.1 However, regression of PMM is not rare. Melanoma is 6 times more likely to undergo regression compared to other malignancies.2 Approximately 10% to 35% of cutaneous PMMs undergo partial regression, but only 42 cases of completely regressed cutaneous PMMs have been reported,3,4 which may be due to underreporting of completely regressed cutaneous PMMs, as they often are clinically inconspicuous. Additionally, completely regressed cutaneous PMMs may be incorrectly reported as metastatic melanoma of unknown primary.5 Clinical characteristics of regression include pink coloration and a lightening or whitening of baseline lesional color. Dermatoscopic features of regression include white areas, blue areas, or vascular structures that translate microscopically to dermal fibrosis, melanophages, and telangiectases.5 We report a case of complete clinical regression of a nodular, mucosal, penile PMM with no evidence of metastatic disease.
An 86-year-old man presented with a progressively enlarging, pigmented lesion on the glans penis of 2 years’ duration. His medical history was notable for retinal detachment, macular degeneration, lumbar stenosis, and seizures postneurosurgery for a subdural hematoma. Physical examination revealed a healthy man with a mottled, black-brown, macular, and nodular lesion with irregular margins and irregular shape on the glans penis (Figure 1). No other similar skin lesions or lymphadenopathy were detectable. A lesional deep shave biopsy obtained at presentation demonstrated a nodular-type malignant melanoma with a Breslow thickness of approximately 3.5 mm (Figure 2).
|
|
| Figure 1. Mottled, black-brown, macular, and nodular lesion with irregular margins and irregular shape on the glans penis. |
|
|
| Figure 2. Histologic section showed a nodular melanocytic proliferation with a dense sheetlike collection of melanocytes, predominantly in the dermis (H&E, original magnification ×20). Prominent cytologic atypia and multiple mitotic figures were consistent with melanoma (inset)(H&E, original magnification ×400). |
|
|
| Figure 3. High-power view of HMB-45 stain showed strong and diffuse staining, including several pagetoid intraepidermal melanocytes (original magnification ×200). |
|
|
| Figure 4. Skin examination 8 years following the initial primary malignant melanoma diagnosis showed no clinical evidence of recurrent or metastatic melanoma and almost complete loss of pigmentation at the prior melanoma site. |
Histologic examination showed nodular nests of malignant melanocytes that were dispersed along the dermal-epidermal junction and coalesced into sheets within the dermis. Numerous dermal mitoses were present. The tumor was strongly and diffusely positive with Melan-A and HMB-45, which also highlighted scattered pagetoid intraepidermal cells (Figure 3). These findings were diagnostic of PMM of the mucocutaneous glans penis. The tumor was nonulcerated and invaded to a Breslow thickness of approximately 3.5 mm, corresponding to American Joint Committee on Cancer stage IIA (T3aN0M0) with an expected 5-year survival rate of 79%.6
He was referred to the urology department and was offered cystoscopy, urethrography, and phallectomy, which he refused. He also refused a trial of imiquimod. Computed tomography (CT) scans of his brain, chest, abdomen, and pelvis were negative for metastatic disease. Following the initial melanoma diagnosis, he had yearly dermatologic evaluations consisting of total-body skin and lymph node (LN) examinations. At 87 years of age (1 year following the initial diagnosis), the melanoma became dramatically smaller. At 88 years of age (2 years after diagnosis), the melanoma had near-complete clinical resolution. At 89 years of age, the patient reported asymmetric hearing loss. A cranial magnetic resonance imaging study showed no evidence of metastases.
At 92 years of age (6 years after the initial diagnosis), the patient reported bilateral leg pain. A CT scan of the lumbar spine showed no evidence of metastasis. He also reported abdominal pain. A CT scan of the abdomen and pelvis revealed an ileocecal mass. Biopsy of the ileocecal mass showed moderately differentiated invasive adenocarcinoma and no evidence of metastatic melanoma. The adenocarcinoma was resected and he continues to do well. Skin and LN examination 8 years after the initial diagnosis showed no clinical evidence of recurrent penile mucosal melanoma or metastatic melanoma (Figure 4). The PMM appeared to have clinically regressed spontaneously. He refused repeat skin biopsy and additional imaging studies.
The criteria for complete melanoma regression were initially described in 19657 and revised in 2005.2 Although our patient demonstrated complete clinical regression of his PMM, he did not meet the revised criteria for complete regression because there was no histopathologic confirmation of regression or of the absence of melanoma as well as no lymphatic involvement. It is extremely difficult to quantify the percentage of PMMs that completely regress. A case of a completely regressed untreated PMM with no metastatic disease 4 years after diagnosis has been reported. This case involved a nonulcerated melanoma with a Breslow thickness of 0.7 mm (American Joint Committee on Cancer stage IA).4 The prognosis of penile mucosal PMM is comparable to that of cutaneous PMM with a similar Breslow thickness.1
The prognostic significance of melanoma regression is controversial. Regression may be mediated by host immunity, apoptosis, and/or antiangiogenesis. The lymphocytic infiltrate in regressive melanomas consists of cytotoxic T cells with selective antitumor activity, which induces HLA class I–restricted melanoma lysis.8 Lymph node migration may result in T-lymphocyte priming and induction of antitumor immunity.9 Therefore, regression may indicate risk for sentinel LN metastasis.
It is possible that complete regression of melanoma does not truly exist, and late recurrence due to cancer dormancy is inevitable. Late recurrence is defined as first metastasis 10 years after complete removal of the PMM.10 Our patient has only been followed for 8 years, so this possibility cannot be entirely excluded.
1. van Geel AN, den Bakker MA, Kirkels W, et al. Prognosis of primary mucosal penile melanoma: a series of 19 Dutch patients and 47 patients from the literature. Urology. 2007;70:143-147.
2. High WA, Stewart D, Wilbers CR, et al. Completely regressed primary cutaneous malignant melanoma with nodal and/or visceral metastases: a report of 5 cases and assessment of the literature and diagnostic criteria. J Am Acad Dermatol. 2005;53:89-100.
3. Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
4. Muniesa C, Ferreres JR, Moreno A, et al. Completely regressed primary cutaneous malignant melanoma with metastases [published online ahead of print June 23, 2008]. J Eur Acad Dermatol Venereol. 2009;23:327-328.
5. Bories N, Dalle S, Debarbieux S, et al. Dermoscopy of fully regressive cutaneous melanoma [published online ahead of print March 13, 2008]. Br J Dermatol. 2008;158:1224-1229.
6. Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification [published online ahead of print November 16, 2009]. J Clin Oncol. 2009;27:6199-6206.
7. Smith JL Jr, Stehlin JS Jr. Spontaneous regression of primary malignant melanomas with regional metastasis. Cancer. 1965;18:1399-1415.
8. Bottger D, Dowden RV, Kay PP. Complete spontaneous regression of cutaneous primary malignant melanoma. Plast Reconstr Surg. 1992;89:548-553.
9. Shaw HM, McCarthy SW, McCarthy WH, et al. Thin regressing malignant melanoma: significance of concurrent regional lymph node metastases. Histopathology. 1989;15:257-265.
10. Hansel G, Schönlebe J, Haroske G, et al. Late recurrence (10 years or more) of malignant melanoma in south-east Germany (Saxony). a single-centre analysis of 1881 patients with a follow-up of 10 years or more [published online ahead of print January 11, 2010]. J Eur Acad Dermatol Venereol. 2010;24:833-836.
1. van Geel AN, den Bakker MA, Kirkels W, et al. Prognosis of primary mucosal penile melanoma: a series of 19 Dutch patients and 47 patients from the literature. Urology. 2007;70:143-147.
2. High WA, Stewart D, Wilbers CR, et al. Completely regressed primary cutaneous malignant melanoma with nodal and/or visceral metastases: a report of 5 cases and assessment of the literature and diagnostic criteria. J Am Acad Dermatol. 2005;53:89-100.
3. Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
4. Muniesa C, Ferreres JR, Moreno A, et al. Completely regressed primary cutaneous malignant melanoma with metastases [published online ahead of print June 23, 2008]. J Eur Acad Dermatol Venereol. 2009;23:327-328.
5. Bories N, Dalle S, Debarbieux S, et al. Dermoscopy of fully regressive cutaneous melanoma [published online ahead of print March 13, 2008]. Br J Dermatol. 2008;158:1224-1229.
6. Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification [published online ahead of print November 16, 2009]. J Clin Oncol. 2009;27:6199-6206.
7. Smith JL Jr, Stehlin JS Jr. Spontaneous regression of primary malignant melanomas with regional metastasis. Cancer. 1965;18:1399-1415.
8. Bottger D, Dowden RV, Kay PP. Complete spontaneous regression of cutaneous primary malignant melanoma. Plast Reconstr Surg. 1992;89:548-553.
9. Shaw HM, McCarthy SW, McCarthy WH, et al. Thin regressing malignant melanoma: significance of concurrent regional lymph node metastases. Histopathology. 1989;15:257-265.
10. Hansel G, Schönlebe J, Haroske G, et al. Late recurrence (10 years or more) of malignant melanoma in south-east Germany (Saxony). a single-centre analysis of 1881 patients with a follow-up of 10 years or more [published online ahead of print January 11, 2010]. J Eur Acad Dermatol Venereol. 2010;24:833-836.
Adult-Type Langerhans Cell Histiocytosis: Minimal Treatment for Maximal Results
To the Editor:
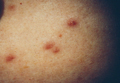
A 78-year-old man presented with erythematous circular skin papules that were widely scattered over the trunk. He denied recent contact with ill individuals and denied any systemic symptoms indicating internal involvement or malignancy leading to possible paraneoplastic presentation. Physical examination showed erythematous, circular, slightly elevated plaques of varying sizes scattered over the trunk (Figure 1) and right axilla.

|
| Figure 1. Nontender, erythematous, brown nodules scattered over the trunk. |
|
| Figure 2. Light microscopy revealed Langerhans cells filling the superficial dermis, abutting the epidermis, and extending into the deep dermis with surrounding inflammatory infiltrates (CD1a, original magnification ×100). |
|
| Figure 3. Langerhans cells appeared strongly positive for CD1a (original magnification ×400). |
Biopsies of lesions were taken and stained with immunoperoxidase. On light microscopy there was a reticular and papillary dermal dense infiltrate of cells with indented nuclei (Figure 2). At higher magnification, cells appeared strongly positive for CD1a (Figure 3) and S-100 protein, which was histologically consistent with adult-type Langerhans cell histiocytosis (ALCH).
Computed tomography of the head, chest, abdomen, and pelvis were ordered to rule out spread of ALCH to other organ sites. Results were clear of evidence of systemic spread. Additionally, a complete blood cell count and comprehensive metabolic panel were within reference range.
He was started on topical tacrolimus; however, most of the lesions resolved on their own. As a result, tacrolimus was discontinued due to its propensity to cause skin irritation and lack of change in disease progression. At 3-month follow-up, he was prescribed triamcinolone acetonide cream 0.1% for minor outbreaks. After 2 years, he was completely clear of all skin signs of ALCH.
Adult-type Langerhans cell histiocytosis is characterized as a group of disorders associated with abnormal spread and proliferation of dendritic cells of the epidermis. The disease primarily affects children aged 1 to 4 years. It is estimated that only 1 to 2 cases of ALCH per million occur.1 The pathophysiology of ALCH is unknown; it is speculated that it may be associated with a reactive inflammatory process triggered by proliferation of Langerhans-type dendritic cells. It is possible that the release of multiple cytokines by dendritic cells and T cells in ALCH lesions leads to erythematous eruptions and can contribute to spontaneous remission of the disorder.2 Various cases of ALCH have reported high serum levels of IL-17 and IL-10 proinflammatory cytokines, supporting the theory of an inflammatory etiology of ALCH.3
Comparative genomic hybridization with loss of heterozygosity of pulmonary lesions has provided further evidence to suggest that chromosomal aberrations also may contribute to the pathophysiology of ALCH.4 One study evaluated 14 cases of pulmonary ALCH for loss of heterozygosity and found allelic loss of 1p, 1q, 3p, 5p, 17p, and 22q.5 In addition, allelic loss of 1 or more tumor suppressor genes was identified in 19 of 24 specimens, suggesting a neoplastic type of pathology through uncontrolled cellular proliferation.6
Lesions of ALCH can be broad but typically present as red-brown maculopapular lesions with petechiae that erupt over the trunk, axilla, and perivulvar or retroauricular regions.7 The papules may unify to form an erythematous, weeping, or crusted eruption that appears similar to seborrheic dermatitis. Typically the lesions remit on their own; however, lesions can recur with the same or decreased severity as the primary eruption. Complications have been noted with lesions, particularly secondary infection and ulceration.7
Systemic involvement has been noted in adults, particularly in the lungs. Patients typically present with chronic cough, dyspnea, and chest pain with evidence of a solitary nodular lesion on radiologic testing. In addition, bone involvement has been noted as eosinophilic granulomas that can produce osteolytic lesions that lead to spontaneous fractures. Use of corticosteroids and immunosuppressive agents, as opposed to just observation, is warranted in cases of systemic involvement, according to the National Cancer Institute.7
Exact treatment modalities have not yet been elucidated due to the ambiguity of pathogenesis. In addition, ALCH is known to remit and relapse in patients, which increases the difficulty in evaluating the efficacy of particular treatments. Trials conducted by the Histiocyte Society have shown that treatment regimens should be tailored to disease severity. Epidermal involvement of ALCH typically responds to corticosteroid creams, whereas patients with systemic involvement respond well to strong chemotherapeutic agents such as vincristine and prednisone with mercaptopurine.8 However, as demonstrated in our case, lesions may remit on their own and use of corticosteroids and immunosuppressive agents may lead to further detriment without treating disease progression.
Because of a low prevalence among adults, ALCH is difficult to recognize and diagnose, and the uncertainty of the pathogenesis of ALCH limits treatment alternatives. Further study into proper treatment modalities is warranted given that the remitting and relapsing course of the disease and cosmetic quandaries are detrimental to patient well-being. Our case illustrates that it is appropriate to simply monitor lesions for cases limited to cutaneous involvement. Systemic agents may be used when there are signs of organ involvement outside the skin, but providers must proceed to do so with caution.
1. Baumgartner I, von Hochstetter A, Baumert B, et al. Langerhans’-cell histiocytosis in adults. Med Pediatr Oncol. 1997;28:9-14.
2. Egeler RM, Favara BE, van Meurs M, et al. Differential in situ cytokine profiles of Langerhans-like cells and T cells in Langerhans cell histiocytosis: abundant expression of cytokines relevant to disease and treatment. Blood. 1999;94:4195-4201.
3. da Costa CE, Szuhai K, van Eijk R, et al. No genomic aberrations in Langerhans cell histiocytosis as assessed by diverse molecular technologies. Genes Chromosomes Cancer. 2009;48:239-249.
4. Murakami I, Gogusev J, Fournet JC, et al. Detection of molecular cytogenetic aberrations in Langerhans cell histiocytosis of bone. Hum Pathol. 2002;33:555-560.
5. Dacic S, Trusky C, Bakker A, et al. Genotypic analysis of pulmonary Langerhans cell histiocytosis. Hum Pathol. 2003;34:1345-1349.
6. Chikwava KR, Hunt JL, Mantha GS, et al. Analysis of loss of heterozygosity in single-system and multisystem Langerhans’ cell histiocytosis. Pediatr Dev Pathol. 2007;10:18-24.
7. Langerhans cell histiocytosis treatment. National Cancer Institute Web site. http://www.cancer.gov/cancertopics/pdq/treatment/lchistio/HealthProfessional/page5. Updated June 4, 2014. Accessed August 27, 2014.
8. Weitzman S, Wayne AS, Arceci R, et al. Nucleoside analogues in the therapy of Langerhans cell histiocytosis: a survey of members of the histiocyte society and review of the literature. Med Pediatr Oncol. 1999;33:476-481.
To the Editor:
A 78-year-old man presented with erythematous circular skin papules that were widely scattered over the trunk. He denied recent contact with ill individuals and denied any systemic symptoms indicating internal involvement or malignancy leading to possible paraneoplastic presentation. Physical examination showed erythematous, circular, slightly elevated plaques of varying sizes scattered over the trunk (Figure 1) and right axilla.
|
|
| Figure 1. Nontender, erythematous, brown nodules scattered over the trunk. |
|
|
| Figure 2. Light microscopy revealed Langerhans cells filling the superficial dermis, abutting the epidermis, and extending into the deep dermis with surrounding inflammatory infiltrates (CD1a, original magnification ×100). |
|
|
| Figure 3. Langerhans cells appeared strongly positive for CD1a (original magnification ×400). |
Biopsies of lesions were taken and stained with immunoperoxidase. On light microscopy there was a reticular and papillary dermal dense infiltrate of cells with indented nuclei (Figure 2). At higher magnification, cells appeared strongly positive for CD1a (Figure 3) and S-100 protein, which was histologically consistent with adult-type Langerhans cell histiocytosis (ALCH).
Computed tomography of the head, chest, abdomen, and pelvis were ordered to rule out spread of ALCH to other organ sites. Results were clear of evidence of systemic spread. Additionally, a complete blood cell count and comprehensive metabolic panel were within reference range.
He was started on topical tacrolimus; however, most of the lesions resolved on their own. As a result, tacrolimus was discontinued due to its propensity to cause skin irritation and lack of change in disease progression. At 3-month follow-up, he was prescribed triamcinolone acetonide cream 0.1% for minor outbreaks. After 2 years, he was completely clear of all skin signs of ALCH.
Adult-type Langerhans cell histiocytosis is characterized as a group of disorders associated with abnormal spread and proliferation of dendritic cells of the epidermis. The disease primarily affects children aged 1 to 4 years. It is estimated that only 1 to 2 cases of ALCH per million occur.1 The pathophysiology of ALCH is unknown; it is speculated that it may be associated with a reactive inflammatory process triggered by proliferation of Langerhans-type dendritic cells. It is possible that the release of multiple cytokines by dendritic cells and T cells in ALCH lesions leads to erythematous eruptions and can contribute to spontaneous remission of the disorder.2 Various cases of ALCH have reported high serum levels of IL-17 and IL-10 proinflammatory cytokines, supporting the theory of an inflammatory etiology of ALCH.3
Comparative genomic hybridization with loss of heterozygosity of pulmonary lesions has provided further evidence to suggest that chromosomal aberrations also may contribute to the pathophysiology of ALCH.4 One study evaluated 14 cases of pulmonary ALCH for loss of heterozygosity and found allelic loss of 1p, 1q, 3p, 5p, 17p, and 22q.5 In addition, allelic loss of 1 or more tumor suppressor genes was identified in 19 of 24 specimens, suggesting a neoplastic type of pathology through uncontrolled cellular proliferation.6
Lesions of ALCH can be broad but typically present as red-brown maculopapular lesions with petechiae that erupt over the trunk, axilla, and perivulvar or retroauricular regions.7 The papules may unify to form an erythematous, weeping, or crusted eruption that appears similar to seborrheic dermatitis. Typically the lesions remit on their own; however, lesions can recur with the same or decreased severity as the primary eruption. Complications have been noted with lesions, particularly secondary infection and ulceration.7
Systemic involvement has been noted in adults, particularly in the lungs. Patients typically present with chronic cough, dyspnea, and chest pain with evidence of a solitary nodular lesion on radiologic testing. In addition, bone involvement has been noted as eosinophilic granulomas that can produce osteolytic lesions that lead to spontaneous fractures. Use of corticosteroids and immunosuppressive agents, as opposed to just observation, is warranted in cases of systemic involvement, according to the National Cancer Institute.7
Exact treatment modalities have not yet been elucidated due to the ambiguity of pathogenesis. In addition, ALCH is known to remit and relapse in patients, which increases the difficulty in evaluating the efficacy of particular treatments. Trials conducted by the Histiocyte Society have shown that treatment regimens should be tailored to disease severity. Epidermal involvement of ALCH typically responds to corticosteroid creams, whereas patients with systemic involvement respond well to strong chemotherapeutic agents such as vincristine and prednisone with mercaptopurine.8 However, as demonstrated in our case, lesions may remit on their own and use of corticosteroids and immunosuppressive agents may lead to further detriment without treating disease progression.
Because of a low prevalence among adults, ALCH is difficult to recognize and diagnose, and the uncertainty of the pathogenesis of ALCH limits treatment alternatives. Further study into proper treatment modalities is warranted given that the remitting and relapsing course of the disease and cosmetic quandaries are detrimental to patient well-being. Our case illustrates that it is appropriate to simply monitor lesions for cases limited to cutaneous involvement. Systemic agents may be used when there are signs of organ involvement outside the skin, but providers must proceed to do so with caution.
To the Editor:
A 78-year-old man presented with erythematous circular skin papules that were widely scattered over the trunk. He denied recent contact with ill individuals and denied any systemic symptoms indicating internal involvement or malignancy leading to possible paraneoplastic presentation. Physical examination showed erythematous, circular, slightly elevated plaques of varying sizes scattered over the trunk (Figure 1) and right axilla.
|
|
| Figure 1. Nontender, erythematous, brown nodules scattered over the trunk. |
|
|
| Figure 2. Light microscopy revealed Langerhans cells filling the superficial dermis, abutting the epidermis, and extending into the deep dermis with surrounding inflammatory infiltrates (CD1a, original magnification ×100). |
|
|
| Figure 3. Langerhans cells appeared strongly positive for CD1a (original magnification ×400). |
Biopsies of lesions were taken and stained with immunoperoxidase. On light microscopy there was a reticular and papillary dermal dense infiltrate of cells with indented nuclei (Figure 2). At higher magnification, cells appeared strongly positive for CD1a (Figure 3) and S-100 protein, which was histologically consistent with adult-type Langerhans cell histiocytosis (ALCH).
Computed tomography of the head, chest, abdomen, and pelvis were ordered to rule out spread of ALCH to other organ sites. Results were clear of evidence of systemic spread. Additionally, a complete blood cell count and comprehensive metabolic panel were within reference range.
He was started on topical tacrolimus; however, most of the lesions resolved on their own. As a result, tacrolimus was discontinued due to its propensity to cause skin irritation and lack of change in disease progression. At 3-month follow-up, he was prescribed triamcinolone acetonide cream 0.1% for minor outbreaks. After 2 years, he was completely clear of all skin signs of ALCH.
Adult-type Langerhans cell histiocytosis is characterized as a group of disorders associated with abnormal spread and proliferation of dendritic cells of the epidermis. The disease primarily affects children aged 1 to 4 years. It is estimated that only 1 to 2 cases of ALCH per million occur.1 The pathophysiology of ALCH is unknown; it is speculated that it may be associated with a reactive inflammatory process triggered by proliferation of Langerhans-type dendritic cells. It is possible that the release of multiple cytokines by dendritic cells and T cells in ALCH lesions leads to erythematous eruptions and can contribute to spontaneous remission of the disorder.2 Various cases of ALCH have reported high serum levels of IL-17 and IL-10 proinflammatory cytokines, supporting the theory of an inflammatory etiology of ALCH.3
Comparative genomic hybridization with loss of heterozygosity of pulmonary lesions has provided further evidence to suggest that chromosomal aberrations also may contribute to the pathophysiology of ALCH.4 One study evaluated 14 cases of pulmonary ALCH for loss of heterozygosity and found allelic loss of 1p, 1q, 3p, 5p, 17p, and 22q.5 In addition, allelic loss of 1 or more tumor suppressor genes was identified in 19 of 24 specimens, suggesting a neoplastic type of pathology through uncontrolled cellular proliferation.6
Lesions of ALCH can be broad but typically present as red-brown maculopapular lesions with petechiae that erupt over the trunk, axilla, and perivulvar or retroauricular regions.7 The papules may unify to form an erythematous, weeping, or crusted eruption that appears similar to seborrheic dermatitis. Typically the lesions remit on their own; however, lesions can recur with the same or decreased severity as the primary eruption. Complications have been noted with lesions, particularly secondary infection and ulceration.7
Systemic involvement has been noted in adults, particularly in the lungs. Patients typically present with chronic cough, dyspnea, and chest pain with evidence of a solitary nodular lesion on radiologic testing. In addition, bone involvement has been noted as eosinophilic granulomas that can produce osteolytic lesions that lead to spontaneous fractures. Use of corticosteroids and immunosuppressive agents, as opposed to just observation, is warranted in cases of systemic involvement, according to the National Cancer Institute.7
Exact treatment modalities have not yet been elucidated due to the ambiguity of pathogenesis. In addition, ALCH is known to remit and relapse in patients, which increases the difficulty in evaluating the efficacy of particular treatments. Trials conducted by the Histiocyte Society have shown that treatment regimens should be tailored to disease severity. Epidermal involvement of ALCH typically responds to corticosteroid creams, whereas patients with systemic involvement respond well to strong chemotherapeutic agents such as vincristine and prednisone with mercaptopurine.8 However, as demonstrated in our case, lesions may remit on their own and use of corticosteroids and immunosuppressive agents may lead to further detriment without treating disease progression.
Because of a low prevalence among adults, ALCH is difficult to recognize and diagnose, and the uncertainty of the pathogenesis of ALCH limits treatment alternatives. Further study into proper treatment modalities is warranted given that the remitting and relapsing course of the disease and cosmetic quandaries are detrimental to patient well-being. Our case illustrates that it is appropriate to simply monitor lesions for cases limited to cutaneous involvement. Systemic agents may be used when there are signs of organ involvement outside the skin, but providers must proceed to do so with caution.
1. Baumgartner I, von Hochstetter A, Baumert B, et al. Langerhans’-cell histiocytosis in adults. Med Pediatr Oncol. 1997;28:9-14.
2. Egeler RM, Favara BE, van Meurs M, et al. Differential in situ cytokine profiles of Langerhans-like cells and T cells in Langerhans cell histiocytosis: abundant expression of cytokines relevant to disease and treatment. Blood. 1999;94:4195-4201.
3. da Costa CE, Szuhai K, van Eijk R, et al. No genomic aberrations in Langerhans cell histiocytosis as assessed by diverse molecular technologies. Genes Chromosomes Cancer. 2009;48:239-249.
4. Murakami I, Gogusev J, Fournet JC, et al. Detection of molecular cytogenetic aberrations in Langerhans cell histiocytosis of bone. Hum Pathol. 2002;33:555-560.
5. Dacic S, Trusky C, Bakker A, et al. Genotypic analysis of pulmonary Langerhans cell histiocytosis. Hum Pathol. 2003;34:1345-1349.
6. Chikwava KR, Hunt JL, Mantha GS, et al. Analysis of loss of heterozygosity in single-system and multisystem Langerhans’ cell histiocytosis. Pediatr Dev Pathol. 2007;10:18-24.
7. Langerhans cell histiocytosis treatment. National Cancer Institute Web site. http://www.cancer.gov/cancertopics/pdq/treatment/lchistio/HealthProfessional/page5. Updated June 4, 2014. Accessed August 27, 2014.
8. Weitzman S, Wayne AS, Arceci R, et al. Nucleoside analogues in the therapy of Langerhans cell histiocytosis: a survey of members of the histiocyte society and review of the literature. Med Pediatr Oncol. 1999;33:476-481.
1. Baumgartner I, von Hochstetter A, Baumert B, et al. Langerhans’-cell histiocytosis in adults. Med Pediatr Oncol. 1997;28:9-14.
2. Egeler RM, Favara BE, van Meurs M, et al. Differential in situ cytokine profiles of Langerhans-like cells and T cells in Langerhans cell histiocytosis: abundant expression of cytokines relevant to disease and treatment. Blood. 1999;94:4195-4201.
3. da Costa CE, Szuhai K, van Eijk R, et al. No genomic aberrations in Langerhans cell histiocytosis as assessed by diverse molecular technologies. Genes Chromosomes Cancer. 2009;48:239-249.
4. Murakami I, Gogusev J, Fournet JC, et al. Detection of molecular cytogenetic aberrations in Langerhans cell histiocytosis of bone. Hum Pathol. 2002;33:555-560.
5. Dacic S, Trusky C, Bakker A, et al. Genotypic analysis of pulmonary Langerhans cell histiocytosis. Hum Pathol. 2003;34:1345-1349.
6. Chikwava KR, Hunt JL, Mantha GS, et al. Analysis of loss of heterozygosity in single-system and multisystem Langerhans’ cell histiocytosis. Pediatr Dev Pathol. 2007;10:18-24.
7. Langerhans cell histiocytosis treatment. National Cancer Institute Web site. http://www.cancer.gov/cancertopics/pdq/treatment/lchistio/HealthProfessional/page5. Updated June 4, 2014. Accessed August 27, 2014.
8. Weitzman S, Wayne AS, Arceci R, et al. Nucleoside analogues in the therapy of Langerhans cell histiocytosis: a survey of members of the histiocyte society and review of the literature. Med Pediatr Oncol. 1999;33:476-481.
Successful Treatment of Schnitzler Syndrome With Canakinumab
To the Editor:
Schnitzler syndrome occurs with a triad of chronic urticaria, recurring fevers, and monoclonal gammopathy. It was recognized as a clinical entity in 1972; now nearly 200 patients are reported in the medical literature.1-3 Flulike symptoms, arthralgia, bone pain, lymphadenopathy, and hepatosplenomegaly also are clinical findings.4,5 The erythrocyte sedimentation rate (ESR) often is markedly elevated, as are other acute phase reactants. Leukocytosis with neutrophilia and IgM and IgG monoclonal gammopathies have been described.4
Schnitzler syndrome shares many clinical characteristics with a subset of autoinflammatory disorders referred to as cryopyrin-associated periodic syndromes (CAPS), which includes familial cold autoinflammatory syndrome and Muckle-Wellssyndrome. These syndromes are associated with mutations in the cold-induced autoinflammatory syndrome 1 gene, CIAS1, which encodes the NALP3 inflammasome, leading to overproduction of IL-1β.5 A gain-of-function mutation in CIAS1 has been described in a patient with Schnitzler syndrome.6
Treatment of urticaria and constitutional symptoms associated with Schnitzler syndrome is challenging. Antihistamines are ineffective, though high-dose systemic glucocorticosteroids control most of the clinical manifestations. Methotrexate sodium, cyclosporine, and tumor necrosis factor antagonists are utilized as glucocorticosteroid-sparing agents. Anakinra, an IL-1 receptor monoclonal antibody that is approved for use in CAPS, has been reported to induce complete resolution of Schnitzler syndrome when administered daily; however, it is not approved by the US Food and Drug Administration for this disorder.7 Canakinumab, an IL-1β monoclonal antibody that is dosed every 8 weeks, was approved by the US Food and Drug Administration in 2009 for the treatment of CAPS. Given the similar clinical characteristics and genetic mutations found in CAPS and Schnitzler syndrome, canakinumab may be an effective treatment of both disorders. We report successful treatment with this monoclonal antibody in 2 patients with Schnitzler syndrome.
A 63-year-old man reported having night sweats and fatigue but had no arthralgia or arthritis. He had a 1-year history of severe urticaria and recurrent fevers (temperature, up to 38.4°C) and he also had type 1 diabetes mellitus, hypothyroidism, and celiac disease. Physical examination revealed an elevated temperature (38.4°C) and generalized urticaria but no evidence of hepatosplenomegaly, adenopathy, or arthritis. Leukocytosis was revealed (white blood cell count, 12,400/μL [reference range, 4500–11,000/μL]) with neutrophilia (88.5% [reference range, 56%]), elevated ESR (81 mm/h [reference range, 0–20 mm/h]), and IgM κ monoclonal gammopathy (0.37 g/L [reference range, 0.4–2.3 g/L]). Clinical examination as well as laboratory and imaging studies did not show evidence of malignancy or autoimmune disease. A skin biopsy identified neutrophilic urticaria without vasculitis. Prednisone 20 mg daily controlled the urticaria and fever, but symptoms recurred within days of glucocorticosteroid withdrawal.
A 47-year-old woman presented with a 7-year history of severe urticaria, fever (temperature, 38.9°C), myalgia, and arthralgia. She had a medical history of allergic rhinitis, gastroesophageal reflux disease, chronic pain syndrome, and depression. Physical examination revealed generalized urticaria with cervical and axillary lymphadenopathy 1 to 2 cm in size but no hepatosplenomegaly or arthritis. Prior evaluations for fever of unknown origin as well as autoimmune and malignant disorders were negative. Skin biopsies reported neutrophilic urticaria without vasculitis, and a lymph node biopsy from the left axilla revealed neutrophilic inflammation. A white blood cell count of 17,800/μL with 61.6% neutrophils, elevated C-reactive protein (153.4 mg/L [reference range, 0.08–3.1 mg/L]) and ESR (90 mm/h), and an IgG λ monoclonal gammopathy were present. She was previously treated with etanercept, methotrexate sodium, golimumab, and adalimumab, with only a partial response. For more than 5 years, prednisone 20 to 50 mg daily was necessary to control her symptoms. Cyclosporine 200 mg twice daily was added as a corticosteroid-sparing drug with partial response.
Both patients were diagnosed with Schnitzler syndrome and were started on canakinumab 150 mg administered subcutaneously in the upper arm every 8 weeks. Resolution of the urticaria and fevers occurred within 2 weeks, and all other medications for the treatment of Schnitzler syndrome were withdrawn without recurrence of symptoms after 3 years. The neutrophil count and acute phase reactants returned within reference range in each patient, but the monoclonal gammopathies remained unchanged. Patient 2 noted worsening of arthralgia after initiation of canakinumab, but long-term corticosteroid withdrawal was considered the cause. Patient 1 has been able to increase the interval of dosing to every 3 to 4 months without recurrence of symptoms. Patient 2 has not tolerated similar changes in dosing interval.
Canakinumab given at 8-week intervals was a safe and effective treatment of Schnitzler syndrome in this open trial of 2 patients. Anakinra also induces remission, but daily dosing is required. Cost may be a notable factor in the choice of therapy, as canakinumab costs substantially more per year than anakinra. Further investigation is required to determine if treatment with canakinumab will result in long-term remission and if less-frequent dosing will provide continued efficacy.
1. Schnitzler L. Lésions urticariennes chroniques permanentes (érythème pétaloïde?). Cas cliniques. nº 46 B. Journee Dermatologique d’Angers. October 1972.
2. Schnitzler L, Schubert B, Boasson M, et al. Urticaire chronique, lésions osseuses, macroglobulinémie IgM: maladie de Waldenstrӧm. Bull Soc Fr Dermatol Syphiligr. 1974;81:363.
3. Simon A, Asli B, Braun-Falco M, et al. Schnitzler’s syndrome: diagnosis, treatment, and follow-up. Allergy. 2013;68:562-568.
4. de Koning HD, Bodar EJ, van der Meer JW, et al. Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment [published online ahead of print June 21, 2007]. Semin Arthritis Rheum. 2007;37:137-148.
5. Lipsker D, Veran Y, Grunenberger F, et al. The Schnitzler syndrome. four new cases and review of the literature. Medicine (Baltimore). 2001;80:37-44.
6. Loock J, Lamprecht P, Timmann C, et al. Genetic predisposition (NLRP3 V198M mutation) for IL-1-mediated inflammation in a patient with Schnitzler syndrome. J Allergy Clin Immunol. 2010;125:500-502.
7. Ryan JG, de Koning HD, Beck LA, et al. IL-1 blockade in Schnitzler syndrome: ex vivo findings correlate with clinical remission [published online ahead of print October 22, 2007]. J Allergy Clin Immunol. 2008;121:260-262.
To the Editor:
Schnitzler syndrome occurs with a triad of chronic urticaria, recurring fevers, and monoclonal gammopathy. It was recognized as a clinical entity in 1972; now nearly 200 patients are reported in the medical literature.1-3 Flulike symptoms, arthralgia, bone pain, lymphadenopathy, and hepatosplenomegaly also are clinical findings.4,5 The erythrocyte sedimentation rate (ESR) often is markedly elevated, as are other acute phase reactants. Leukocytosis with neutrophilia and IgM and IgG monoclonal gammopathies have been described.4
Schnitzler syndrome shares many clinical characteristics with a subset of autoinflammatory disorders referred to as cryopyrin-associated periodic syndromes (CAPS), which includes familial cold autoinflammatory syndrome and Muckle-Wellssyndrome. These syndromes are associated with mutations in the cold-induced autoinflammatory syndrome 1 gene, CIAS1, which encodes the NALP3 inflammasome, leading to overproduction of IL-1β.5 A gain-of-function mutation in CIAS1 has been described in a patient with Schnitzler syndrome.6
Treatment of urticaria and constitutional symptoms associated with Schnitzler syndrome is challenging. Antihistamines are ineffective, though high-dose systemic glucocorticosteroids control most of the clinical manifestations. Methotrexate sodium, cyclosporine, and tumor necrosis factor antagonists are utilized as glucocorticosteroid-sparing agents. Anakinra, an IL-1 receptor monoclonal antibody that is approved for use in CAPS, has been reported to induce complete resolution of Schnitzler syndrome when administered daily; however, it is not approved by the US Food and Drug Administration for this disorder.7 Canakinumab, an IL-1β monoclonal antibody that is dosed every 8 weeks, was approved by the US Food and Drug Administration in 2009 for the treatment of CAPS. Given the similar clinical characteristics and genetic mutations found in CAPS and Schnitzler syndrome, canakinumab may be an effective treatment of both disorders. We report successful treatment with this monoclonal antibody in 2 patients with Schnitzler syndrome.
A 63-year-old man reported having night sweats and fatigue but had no arthralgia or arthritis. He had a 1-year history of severe urticaria and recurrent fevers (temperature, up to 38.4°C) and he also had type 1 diabetes mellitus, hypothyroidism, and celiac disease. Physical examination revealed an elevated temperature (38.4°C) and generalized urticaria but no evidence of hepatosplenomegaly, adenopathy, or arthritis. Leukocytosis was revealed (white blood cell count, 12,400/μL [reference range, 4500–11,000/μL]) with neutrophilia (88.5% [reference range, 56%]), elevated ESR (81 mm/h [reference range, 0–20 mm/h]), and IgM κ monoclonal gammopathy (0.37 g/L [reference range, 0.4–2.3 g/L]). Clinical examination as well as laboratory and imaging studies did not show evidence of malignancy or autoimmune disease. A skin biopsy identified neutrophilic urticaria without vasculitis. Prednisone 20 mg daily controlled the urticaria and fever, but symptoms recurred within days of glucocorticosteroid withdrawal.
A 47-year-old woman presented with a 7-year history of severe urticaria, fever (temperature, 38.9°C), myalgia, and arthralgia. She had a medical history of allergic rhinitis, gastroesophageal reflux disease, chronic pain syndrome, and depression. Physical examination revealed generalized urticaria with cervical and axillary lymphadenopathy 1 to 2 cm in size but no hepatosplenomegaly or arthritis. Prior evaluations for fever of unknown origin as well as autoimmune and malignant disorders were negative. Skin biopsies reported neutrophilic urticaria without vasculitis, and a lymph node biopsy from the left axilla revealed neutrophilic inflammation. A white blood cell count of 17,800/μL with 61.6% neutrophils, elevated C-reactive protein (153.4 mg/L [reference range, 0.08–3.1 mg/L]) and ESR (90 mm/h), and an IgG λ monoclonal gammopathy were present. She was previously treated with etanercept, methotrexate sodium, golimumab, and adalimumab, with only a partial response. For more than 5 years, prednisone 20 to 50 mg daily was necessary to control her symptoms. Cyclosporine 200 mg twice daily was added as a corticosteroid-sparing drug with partial response.
Both patients were diagnosed with Schnitzler syndrome and were started on canakinumab 150 mg administered subcutaneously in the upper arm every 8 weeks. Resolution of the urticaria and fevers occurred within 2 weeks, and all other medications for the treatment of Schnitzler syndrome were withdrawn without recurrence of symptoms after 3 years. The neutrophil count and acute phase reactants returned within reference range in each patient, but the monoclonal gammopathies remained unchanged. Patient 2 noted worsening of arthralgia after initiation of canakinumab, but long-term corticosteroid withdrawal was considered the cause. Patient 1 has been able to increase the interval of dosing to every 3 to 4 months without recurrence of symptoms. Patient 2 has not tolerated similar changes in dosing interval.
Canakinumab given at 8-week intervals was a safe and effective treatment of Schnitzler syndrome in this open trial of 2 patients. Anakinra also induces remission, but daily dosing is required. Cost may be a notable factor in the choice of therapy, as canakinumab costs substantially more per year than anakinra. Further investigation is required to determine if treatment with canakinumab will result in long-term remission and if less-frequent dosing will provide continued efficacy.
To the Editor:
Schnitzler syndrome occurs with a triad of chronic urticaria, recurring fevers, and monoclonal gammopathy. It was recognized as a clinical entity in 1972; now nearly 200 patients are reported in the medical literature.1-3 Flulike symptoms, arthralgia, bone pain, lymphadenopathy, and hepatosplenomegaly also are clinical findings.4,5 The erythrocyte sedimentation rate (ESR) often is markedly elevated, as are other acute phase reactants. Leukocytosis with neutrophilia and IgM and IgG monoclonal gammopathies have been described.4
Schnitzler syndrome shares many clinical characteristics with a subset of autoinflammatory disorders referred to as cryopyrin-associated periodic syndromes (CAPS), which includes familial cold autoinflammatory syndrome and Muckle-Wellssyndrome. These syndromes are associated with mutations in the cold-induced autoinflammatory syndrome 1 gene, CIAS1, which encodes the NALP3 inflammasome, leading to overproduction of IL-1β.5 A gain-of-function mutation in CIAS1 has been described in a patient with Schnitzler syndrome.6
Treatment of urticaria and constitutional symptoms associated with Schnitzler syndrome is challenging. Antihistamines are ineffective, though high-dose systemic glucocorticosteroids control most of the clinical manifestations. Methotrexate sodium, cyclosporine, and tumor necrosis factor antagonists are utilized as glucocorticosteroid-sparing agents. Anakinra, an IL-1 receptor monoclonal antibody that is approved for use in CAPS, has been reported to induce complete resolution of Schnitzler syndrome when administered daily; however, it is not approved by the US Food and Drug Administration for this disorder.7 Canakinumab, an IL-1β monoclonal antibody that is dosed every 8 weeks, was approved by the US Food and Drug Administration in 2009 for the treatment of CAPS. Given the similar clinical characteristics and genetic mutations found in CAPS and Schnitzler syndrome, canakinumab may be an effective treatment of both disorders. We report successful treatment with this monoclonal antibody in 2 patients with Schnitzler syndrome.
A 63-year-old man reported having night sweats and fatigue but had no arthralgia or arthritis. He had a 1-year history of severe urticaria and recurrent fevers (temperature, up to 38.4°C) and he also had type 1 diabetes mellitus, hypothyroidism, and celiac disease. Physical examination revealed an elevated temperature (38.4°C) and generalized urticaria but no evidence of hepatosplenomegaly, adenopathy, or arthritis. Leukocytosis was revealed (white blood cell count, 12,400/μL [reference range, 4500–11,000/μL]) with neutrophilia (88.5% [reference range, 56%]), elevated ESR (81 mm/h [reference range, 0–20 mm/h]), and IgM κ monoclonal gammopathy (0.37 g/L [reference range, 0.4–2.3 g/L]). Clinical examination as well as laboratory and imaging studies did not show evidence of malignancy or autoimmune disease. A skin biopsy identified neutrophilic urticaria without vasculitis. Prednisone 20 mg daily controlled the urticaria and fever, but symptoms recurred within days of glucocorticosteroid withdrawal.
A 47-year-old woman presented with a 7-year history of severe urticaria, fever (temperature, 38.9°C), myalgia, and arthralgia. She had a medical history of allergic rhinitis, gastroesophageal reflux disease, chronic pain syndrome, and depression. Physical examination revealed generalized urticaria with cervical and axillary lymphadenopathy 1 to 2 cm in size but no hepatosplenomegaly or arthritis. Prior evaluations for fever of unknown origin as well as autoimmune and malignant disorders were negative. Skin biopsies reported neutrophilic urticaria without vasculitis, and a lymph node biopsy from the left axilla revealed neutrophilic inflammation. A white blood cell count of 17,800/μL with 61.6% neutrophils, elevated C-reactive protein (153.4 mg/L [reference range, 0.08–3.1 mg/L]) and ESR (90 mm/h), and an IgG λ monoclonal gammopathy were present. She was previously treated with etanercept, methotrexate sodium, golimumab, and adalimumab, with only a partial response. For more than 5 years, prednisone 20 to 50 mg daily was necessary to control her symptoms. Cyclosporine 200 mg twice daily was added as a corticosteroid-sparing drug with partial response.
Both patients were diagnosed with Schnitzler syndrome and were started on canakinumab 150 mg administered subcutaneously in the upper arm every 8 weeks. Resolution of the urticaria and fevers occurred within 2 weeks, and all other medications for the treatment of Schnitzler syndrome were withdrawn without recurrence of symptoms after 3 years. The neutrophil count and acute phase reactants returned within reference range in each patient, but the monoclonal gammopathies remained unchanged. Patient 2 noted worsening of arthralgia after initiation of canakinumab, but long-term corticosteroid withdrawal was considered the cause. Patient 1 has been able to increase the interval of dosing to every 3 to 4 months without recurrence of symptoms. Patient 2 has not tolerated similar changes in dosing interval.
Canakinumab given at 8-week intervals was a safe and effective treatment of Schnitzler syndrome in this open trial of 2 patients. Anakinra also induces remission, but daily dosing is required. Cost may be a notable factor in the choice of therapy, as canakinumab costs substantially more per year than anakinra. Further investigation is required to determine if treatment with canakinumab will result in long-term remission and if less-frequent dosing will provide continued efficacy.
1. Schnitzler L. Lésions urticariennes chroniques permanentes (érythème pétaloïde?). Cas cliniques. nº 46 B. Journee Dermatologique d’Angers. October 1972.
2. Schnitzler L, Schubert B, Boasson M, et al. Urticaire chronique, lésions osseuses, macroglobulinémie IgM: maladie de Waldenstrӧm. Bull Soc Fr Dermatol Syphiligr. 1974;81:363.
3. Simon A, Asli B, Braun-Falco M, et al. Schnitzler’s syndrome: diagnosis, treatment, and follow-up. Allergy. 2013;68:562-568.
4. de Koning HD, Bodar EJ, van der Meer JW, et al. Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment [published online ahead of print June 21, 2007]. Semin Arthritis Rheum. 2007;37:137-148.
5. Lipsker D, Veran Y, Grunenberger F, et al. The Schnitzler syndrome. four new cases and review of the literature. Medicine (Baltimore). 2001;80:37-44.
6. Loock J, Lamprecht P, Timmann C, et al. Genetic predisposition (NLRP3 V198M mutation) for IL-1-mediated inflammation in a patient with Schnitzler syndrome. J Allergy Clin Immunol. 2010;125:500-502.
7. Ryan JG, de Koning HD, Beck LA, et al. IL-1 blockade in Schnitzler syndrome: ex vivo findings correlate with clinical remission [published online ahead of print October 22, 2007]. J Allergy Clin Immunol. 2008;121:260-262.
1. Schnitzler L. Lésions urticariennes chroniques permanentes (érythème pétaloïde?). Cas cliniques. nº 46 B. Journee Dermatologique d’Angers. October 1972.
2. Schnitzler L, Schubert B, Boasson M, et al. Urticaire chronique, lésions osseuses, macroglobulinémie IgM: maladie de Waldenstrӧm. Bull Soc Fr Dermatol Syphiligr. 1974;81:363.
3. Simon A, Asli B, Braun-Falco M, et al. Schnitzler’s syndrome: diagnosis, treatment, and follow-up. Allergy. 2013;68:562-568.
4. de Koning HD, Bodar EJ, van der Meer JW, et al. Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment [published online ahead of print June 21, 2007]. Semin Arthritis Rheum. 2007;37:137-148.
5. Lipsker D, Veran Y, Grunenberger F, et al. The Schnitzler syndrome. four new cases and review of the literature. Medicine (Baltimore). 2001;80:37-44.
6. Loock J, Lamprecht P, Timmann C, et al. Genetic predisposition (NLRP3 V198M mutation) for IL-1-mediated inflammation in a patient with Schnitzler syndrome. J Allergy Clin Immunol. 2010;125:500-502.
7. Ryan JG, de Koning HD, Beck LA, et al. IL-1 blockade in Schnitzler syndrome: ex vivo findings correlate with clinical remission [published online ahead of print October 22, 2007]. J Allergy Clin Immunol. 2008;121:260-262.
Fungal Melanonychia Caused by Trichophyton rubrum and the Value of Dermoscopy
To the Editor:
Longitudinal melanonychia encompasses a broad spectrum of diseases and often is a complex diagnostic problem. Differential diagnoses include ethnic-type nail pigmentation, which is more frequently seen in darker-skinned individuals; drug-induced pigmentation; subungual hemorrhage; fungal or bacterial infection; nevus; and melanoma.1,2 Fungal melan-onychia is an uncommon presentation of onychomycosis. Dermoscopy can assist in the evaluation of nail pigmentation caused by fungi to avoid unnecessary nail biopsies.
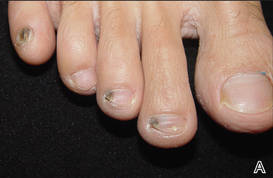
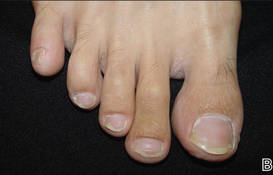
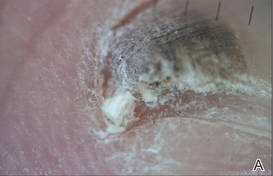
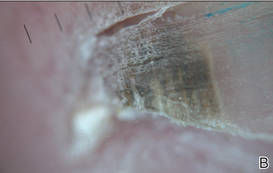
A 39-year-old man visited the dermatology clinic with a concern for melanoma because of blackish pigmentation of the toenails of 1 month’s duration. He denied history of trauma and was not taking any medications. On physical examination the second and third toenails revealed a 2-mm longitudinal band of black pigment on the lateral side; the fifth toenail showed diffuse black pigment (Figure 1A). The nail plates were thickened. Dermoscopy revealed prominent subungual hyperkeratosis, a homogeneous brown-black band with wide yellow streaks that were wider in the distal ends, and some focal reddish hue. No visible melanin inclusions were observed (Figure 2). These findings were suggestive of fungal infection. Cultures from the diseased nail grew a fungus identified as Trichophyton rubrum. The patient was treated with itraconazole 200 mg daily for 3 months. Clinical cure with disappearance of pigment was obtained at 5-month follow-up (Figure 1B).
|
|
| Figure 1. Blackish discoloration of the right second, third, and fifth toenails (A). Resolution of pigmentation and subungual hyperkeratosis was achieved at 5-month follow-up after treatment (B). |
|
|
| Figure 2. Top view (A) and front view (B) of a homogeneous brown-black band with subungual hyperkeratosis and wide intervening yellow streaks. Each ruler mark denotes 1 mm. |
Our patient illustrates the value of dermoscopy in evaluating melanonychia. The pigmentation of adult-onset melanonychia involving multiple fingers can be divided into nonmelanocytic or melanocytic origin. Causes of the former include subungual hematoma, fungal or bacterial infection, and exogenous pigmentation. The nonmelanocytic pigment often is homogeneously distributed without melanin inclusions under the dermoscope.2
On the contrary, melanin inclusions can be detected as fine granules in pigmentation of melanocytic origin, either from focal melanocytic activation or melanocyte proliferation. Causes of focal melanocytic activation include ethnic-type nail hyperpigmentation; inflammatory nail diseases; or drug-, radiation-, and friction-induced hyperpigmentation. The characteristic dermoscopic features are thin longitudinal gray lines with regular thickness and spacing in a grayish background.1-3
Melanocyte proliferation can result in a nevus or melanoma of the nail apparatus. Both share dermoscopic features of brown-black longitudinal lines in a brown background. However, the longitudinal lines in melanoma are irregular in coloration, spacing, thickness, and parallelism, in contrast with the regular pattern of a nevus.1-4 Although patients often are concerned about melanoma, involvement of multiple fingers at the same time is less likely.
In our patient, the homogeneous deep brown color without melanin inclusions favored a nonmel-anocytic origin. The distally wider pigmentation suggested fungal infection because most ungual infections extend from the distal to the proximal part of the nail.5,6 The focal reddish hue may be related with traumatic hemorrhage from subungual hyperkeratosis.
Cases of fungal melanonychia are being reported at an increasing rate. Some fungal strains are capable of synthesizing melanin, which is associated with virulence and acts as a fungal armor against toxic insults.5 In T rubrum, the melanoid variant, the diffusible black pigment infiltrates the nail plate and attributes to the black nail clinically.4 The most frequently isolated fungi in fungal melanonychia are T rubrum and Scytalidium dimidiatum6; however, Candida species,7,8 dematiaceous fungus,9 and other dermatophytes such as Trichophyton soudanense10 have been reported to be the cause.5
Our patient presented with fungal melanonychia due to T rubrum with dermoscopic features. The prominent subungual hyperkeratosis, distally wider homogeneous brown-black pigmented band, and wide yellow streaks with focal reddish hue all suggested fungal melanonychia. The diagnosis was further confirmed by a good response to antifungal agents.
1. Ronger S, Touzet S, Ligeron C, et al. Dermoscopic examination of nail pigmentation. Arch Dermatol. 2002;138:1327-1333.
2. Braun RP, Baran R, Le Gal FA, et al. Diagnosis and management of nail pigmentations [published online ahead of print February 22, 2007]. J Am Acad Dermatol. 2007;56:835-847.
3. Koga H, Saida T, Uhara H. Key point in dermoscopic differentiation between early nail apparatus melanoma and benign longitudinal melanonychia. J Dermatol. 2011;38:45-52.
4. Phan A, Dalle S, Touzet S, et al. Dermoscopic features of acral lentiginous melanoma in a large series of 110 cases in a white population [published online ahead of print November 18, 2009]. Br J Dermatol. 2010;162:765-771.
5. Finch J, Arenas R, Baran R. Fungal melanonychia [published online ahead of print January 17, 2012]. J Am Acad Dermatol. 2012;66:830-841.
6. Lee SW, Kim YC, Kim DK, et al. Fungal melanonychia. J Dermatol. 2004;31:904-909.
7. Parlak AH, Goksugur N, Karabay O. A case of melanonychia due to Candida albicans. Clin Exp Dermatol. 2006;31:398-400.
8. Gautret P, Rodier MH, Kauffmann-Lacroix C, et al. Case report and review. onychomycosis due to Candida parapsilosis. Mycoses. 2000;43:433-435.
9. Barua P, Barua S, Borkakoty B, et al. Onychomycosis by Scytalidium dimidiatum in green tea leaf pluckers: report of two cases [published online ahead of print July 20, 2007]. Mycopathologia. 2007;164:193-195.
10. Ricci C, Monod M, Baudraz-Rosselet F. Onychomycosis due to Trichophyton soudanense in Switzerland. Dermatology. 1998;197:297-298.
To the Editor:
Longitudinal melanonychia encompasses a broad spectrum of diseases and often is a complex diagnostic problem. Differential diagnoses include ethnic-type nail pigmentation, which is more frequently seen in darker-skinned individuals; drug-induced pigmentation; subungual hemorrhage; fungal or bacterial infection; nevus; and melanoma.1,2 Fungal melan-onychia is an uncommon presentation of onychomycosis. Dermoscopy can assist in the evaluation of nail pigmentation caused by fungi to avoid unnecessary nail biopsies.
A 39-year-old man visited the dermatology clinic with a concern for melanoma because of blackish pigmentation of the toenails of 1 month’s duration. He denied history of trauma and was not taking any medications. On physical examination the second and third toenails revealed a 2-mm longitudinal band of black pigment on the lateral side; the fifth toenail showed diffuse black pigment (Figure 1A). The nail plates were thickened. Dermoscopy revealed prominent subungual hyperkeratosis, a homogeneous brown-black band with wide yellow streaks that were wider in the distal ends, and some focal reddish hue. No visible melanin inclusions were observed (Figure 2). These findings were suggestive of fungal infection. Cultures from the diseased nail grew a fungus identified as Trichophyton rubrum. The patient was treated with itraconazole 200 mg daily for 3 months. Clinical cure with disappearance of pigment was obtained at 5-month follow-up (Figure 1B).
|
|
|
|
| Figure 1. Blackish discoloration of the right second, third, and fifth toenails (A). Resolution of pigmentation and subungual hyperkeratosis was achieved at 5-month follow-up after treatment (B). |
|
|
|
|
| Figure 2. Top view (A) and front view (B) of a homogeneous brown-black band with subungual hyperkeratosis and wide intervening yellow streaks. Each ruler mark denotes 1 mm. |
Our patient illustrates the value of dermoscopy in evaluating melanonychia. The pigmentation of adult-onset melanonychia involving multiple fingers can be divided into nonmelanocytic or melanocytic origin. Causes of the former include subungual hematoma, fungal or bacterial infection, and exogenous pigmentation. The nonmelanocytic pigment often is homogeneously distributed without melanin inclusions under the dermoscope.2
On the contrary, melanin inclusions can be detected as fine granules in pigmentation of melanocytic origin, either from focal melanocytic activation or melanocyte proliferation. Causes of focal melanocytic activation include ethnic-type nail hyperpigmentation; inflammatory nail diseases; or drug-, radiation-, and friction-induced hyperpigmentation. The characteristic dermoscopic features are thin longitudinal gray lines with regular thickness and spacing in a grayish background.1-3
Melanocyte proliferation can result in a nevus or melanoma of the nail apparatus. Both share dermoscopic features of brown-black longitudinal lines in a brown background. However, the longitudinal lines in melanoma are irregular in coloration, spacing, thickness, and parallelism, in contrast with the regular pattern of a nevus.1-4 Although patients often are concerned about melanoma, involvement of multiple fingers at the same time is less likely.
In our patient, the homogeneous deep brown color without melanin inclusions favored a nonmel-anocytic origin. The distally wider pigmentation suggested fungal infection because most ungual infections extend from the distal to the proximal part of the nail.5,6 The focal reddish hue may be related with traumatic hemorrhage from subungual hyperkeratosis.
Cases of fungal melanonychia are being reported at an increasing rate. Some fungal strains are capable of synthesizing melanin, which is associated with virulence and acts as a fungal armor against toxic insults.5 In T rubrum, the melanoid variant, the diffusible black pigment infiltrates the nail plate and attributes to the black nail clinically.4 The most frequently isolated fungi in fungal melanonychia are T rubrum and Scytalidium dimidiatum6; however, Candida species,7,8 dematiaceous fungus,9 and other dermatophytes such as Trichophyton soudanense10 have been reported to be the cause.5
Our patient presented with fungal melanonychia due to T rubrum with dermoscopic features. The prominent subungual hyperkeratosis, distally wider homogeneous brown-black pigmented band, and wide yellow streaks with focal reddish hue all suggested fungal melanonychia. The diagnosis was further confirmed by a good response to antifungal agents.
To the Editor:
Longitudinal melanonychia encompasses a broad spectrum of diseases and often is a complex diagnostic problem. Differential diagnoses include ethnic-type nail pigmentation, which is more frequently seen in darker-skinned individuals; drug-induced pigmentation; subungual hemorrhage; fungal or bacterial infection; nevus; and melanoma.1,2 Fungal melan-onychia is an uncommon presentation of onychomycosis. Dermoscopy can assist in the evaluation of nail pigmentation caused by fungi to avoid unnecessary nail biopsies.
A 39-year-old man visited the dermatology clinic with a concern for melanoma because of blackish pigmentation of the toenails of 1 month’s duration. He denied history of trauma and was not taking any medications. On physical examination the second and third toenails revealed a 2-mm longitudinal band of black pigment on the lateral side; the fifth toenail showed diffuse black pigment (Figure 1A). The nail plates were thickened. Dermoscopy revealed prominent subungual hyperkeratosis, a homogeneous brown-black band with wide yellow streaks that were wider in the distal ends, and some focal reddish hue. No visible melanin inclusions were observed (Figure 2). These findings were suggestive of fungal infection. Cultures from the diseased nail grew a fungus identified as Trichophyton rubrum. The patient was treated with itraconazole 200 mg daily for 3 months. Clinical cure with disappearance of pigment was obtained at 5-month follow-up (Figure 1B).
|
|
|
|
| Figure 1. Blackish discoloration of the right second, third, and fifth toenails (A). Resolution of pigmentation and subungual hyperkeratosis was achieved at 5-month follow-up after treatment (B). |
|
|
|
|
| Figure 2. Top view (A) and front view (B) of a homogeneous brown-black band with subungual hyperkeratosis and wide intervening yellow streaks. Each ruler mark denotes 1 mm. |
Our patient illustrates the value of dermoscopy in evaluating melanonychia. The pigmentation of adult-onset melanonychia involving multiple fingers can be divided into nonmelanocytic or melanocytic origin. Causes of the former include subungual hematoma, fungal or bacterial infection, and exogenous pigmentation. The nonmelanocytic pigment often is homogeneously distributed without melanin inclusions under the dermoscope.2
On the contrary, melanin inclusions can be detected as fine granules in pigmentation of melanocytic origin, either from focal melanocytic activation or melanocyte proliferation. Causes of focal melanocytic activation include ethnic-type nail hyperpigmentation; inflammatory nail diseases; or drug-, radiation-, and friction-induced hyperpigmentation. The characteristic dermoscopic features are thin longitudinal gray lines with regular thickness and spacing in a grayish background.1-3
Melanocyte proliferation can result in a nevus or melanoma of the nail apparatus. Both share dermoscopic features of brown-black longitudinal lines in a brown background. However, the longitudinal lines in melanoma are irregular in coloration, spacing, thickness, and parallelism, in contrast with the regular pattern of a nevus.1-4 Although patients often are concerned about melanoma, involvement of multiple fingers at the same time is less likely.
In our patient, the homogeneous deep brown color without melanin inclusions favored a nonmel-anocytic origin. The distally wider pigmentation suggested fungal infection because most ungual infections extend from the distal to the proximal part of the nail.5,6 The focal reddish hue may be related with traumatic hemorrhage from subungual hyperkeratosis.
Cases of fungal melanonychia are being reported at an increasing rate. Some fungal strains are capable of synthesizing melanin, which is associated with virulence and acts as a fungal armor against toxic insults.5 In T rubrum, the melanoid variant, the diffusible black pigment infiltrates the nail plate and attributes to the black nail clinically.4 The most frequently isolated fungi in fungal melanonychia are T rubrum and Scytalidium dimidiatum6; however, Candida species,7,8 dematiaceous fungus,9 and other dermatophytes such as Trichophyton soudanense10 have been reported to be the cause.5
Our patient presented with fungal melanonychia due to T rubrum with dermoscopic features. The prominent subungual hyperkeratosis, distally wider homogeneous brown-black pigmented band, and wide yellow streaks with focal reddish hue all suggested fungal melanonychia. The diagnosis was further confirmed by a good response to antifungal agents.
1. Ronger S, Touzet S, Ligeron C, et al. Dermoscopic examination of nail pigmentation. Arch Dermatol. 2002;138:1327-1333.
2. Braun RP, Baran R, Le Gal FA, et al. Diagnosis and management of nail pigmentations [published online ahead of print February 22, 2007]. J Am Acad Dermatol. 2007;56:835-847.
3. Koga H, Saida T, Uhara H. Key point in dermoscopic differentiation between early nail apparatus melanoma and benign longitudinal melanonychia. J Dermatol. 2011;38:45-52.
4. Phan A, Dalle S, Touzet S, et al. Dermoscopic features of acral lentiginous melanoma in a large series of 110 cases in a white population [published online ahead of print November 18, 2009]. Br J Dermatol. 2010;162:765-771.
5. Finch J, Arenas R, Baran R. Fungal melanonychia [published online ahead of print January 17, 2012]. J Am Acad Dermatol. 2012;66:830-841.
6. Lee SW, Kim YC, Kim DK, et al. Fungal melanonychia. J Dermatol. 2004;31:904-909.
7. Parlak AH, Goksugur N, Karabay O. A case of melanonychia due to Candida albicans. Clin Exp Dermatol. 2006;31:398-400.
8. Gautret P, Rodier MH, Kauffmann-Lacroix C, et al. Case report and review. onychomycosis due to Candida parapsilosis. Mycoses. 2000;43:433-435.
9. Barua P, Barua S, Borkakoty B, et al. Onychomycosis by Scytalidium dimidiatum in green tea leaf pluckers: report of two cases [published online ahead of print July 20, 2007]. Mycopathologia. 2007;164:193-195.
10. Ricci C, Monod M, Baudraz-Rosselet F. Onychomycosis due to Trichophyton soudanense in Switzerland. Dermatology. 1998;197:297-298.
1. Ronger S, Touzet S, Ligeron C, et al. Dermoscopic examination of nail pigmentation. Arch Dermatol. 2002;138:1327-1333.
2. Braun RP, Baran R, Le Gal FA, et al. Diagnosis and management of nail pigmentations [published online ahead of print February 22, 2007]. J Am Acad Dermatol. 2007;56:835-847.
3. Koga H, Saida T, Uhara H. Key point in dermoscopic differentiation between early nail apparatus melanoma and benign longitudinal melanonychia. J Dermatol. 2011;38:45-52.
4. Phan A, Dalle S, Touzet S, et al. Dermoscopic features of acral lentiginous melanoma in a large series of 110 cases in a white population [published online ahead of print November 18, 2009]. Br J Dermatol. 2010;162:765-771.
5. Finch J, Arenas R, Baran R. Fungal melanonychia [published online ahead of print January 17, 2012]. J Am Acad Dermatol. 2012;66:830-841.
6. Lee SW, Kim YC, Kim DK, et al. Fungal melanonychia. J Dermatol. 2004;31:904-909.
7. Parlak AH, Goksugur N, Karabay O. A case of melanonychia due to Candida albicans. Clin Exp Dermatol. 2006;31:398-400.
8. Gautret P, Rodier MH, Kauffmann-Lacroix C, et al. Case report and review. onychomycosis due to Candida parapsilosis. Mycoses. 2000;43:433-435.
9. Barua P, Barua S, Borkakoty B, et al. Onychomycosis by Scytalidium dimidiatum in green tea leaf pluckers: report of two cases [published online ahead of print July 20, 2007]. Mycopathologia. 2007;164:193-195.
10. Ricci C, Monod M, Baudraz-Rosselet F. Onychomycosis due to Trichophyton soudanense in Switzerland. Dermatology. 1998;197:297-298.
Narrow-Toed Shoes and the Toe-to-Toe Sign
To the Editor:
Macro- or microtrauma to nails can cause and/or exacerbate chronic diseases such as ingrown nails, onycholysis, onychauxis, onychogryposis, and hallux valgus (bunions). This trauma also can break the anatomic barrier of the hyponychium, thereby creating a portal for dermatophytes and other organisms to penetrate the nail apparatus.1
For many years, one author (C.R.D.) has used the following demonstration to communicate to patients how improper shoe fit may cause toenail trauma. The patient’s shoe is flipped 180° and placed toe-to-toe with the patient’s foot (Figure). Most patients can comprehend the relationship between their shoe fit and their toenail disease when they see this demonstration. We have termed it toe-to-toe sign.
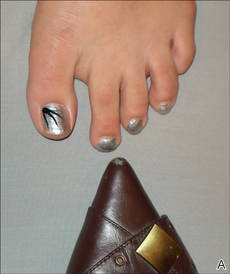
|
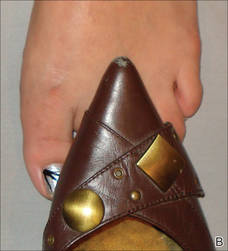
|
| The foot and shoe are toe-to-toe, demonstrating the narrow toe box in comparison to the actual toes (A). This comparison is accentuated when the shoe is placed over the foot with toes splayed from standing (B). |
Narrow toe box is the usual culprit. The toe-to-toe sign best emphasizes this relationship. This sign serves as a powerful tool when demonstrating how much the foot widens when bearing full weight, and how forces of ambulation and foot strike can damage the nails with narrow-toed footwear. This trauma is compounded with high-heeled shoes that force the toes forward. However, proper shoe fit may compete with idealized shoe style. It is not until patients realize the relationship between improper shoe fit, foot strike, and toe/toenail trauma that they can make long-term decisions that favorably impact their toes and toenails.
Reference
1. Daniel CR 3rd, Jellinek NJ. The pedal fungus reservoir. Arch Dermatol. 2006;142:1344-1346.
To the Editor:
Macro- or microtrauma to nails can cause and/or exacerbate chronic diseases such as ingrown nails, onycholysis, onychauxis, onychogryposis, and hallux valgus (bunions). This trauma also can break the anatomic barrier of the hyponychium, thereby creating a portal for dermatophytes and other organisms to penetrate the nail apparatus.1
For many years, one author (C.R.D.) has used the following demonstration to communicate to patients how improper shoe fit may cause toenail trauma. The patient’s shoe is flipped 180° and placed toe-to-toe with the patient’s foot (Figure). Most patients can comprehend the relationship between their shoe fit and their toenail disease when they see this demonstration. We have termed it toe-to-toe sign.
|
|
|
|
| The foot and shoe are toe-to-toe, demonstrating the narrow toe box in comparison to the actual toes (A). This comparison is accentuated when the shoe is placed over the foot with toes splayed from standing (B). |
Narrow toe box is the usual culprit. The toe-to-toe sign best emphasizes this relationship. This sign serves as a powerful tool when demonstrating how much the foot widens when bearing full weight, and how forces of ambulation and foot strike can damage the nails with narrow-toed footwear. This trauma is compounded with high-heeled shoes that force the toes forward. However, proper shoe fit may compete with idealized shoe style. It is not until patients realize the relationship between improper shoe fit, foot strike, and toe/toenail trauma that they can make long-term decisions that favorably impact their toes and toenails.
To the Editor:
Macro- or microtrauma to nails can cause and/or exacerbate chronic diseases such as ingrown nails, onycholysis, onychauxis, onychogryposis, and hallux valgus (bunions). This trauma also can break the anatomic barrier of the hyponychium, thereby creating a portal for dermatophytes and other organisms to penetrate the nail apparatus.1
For many years, one author (C.R.D.) has used the following demonstration to communicate to patients how improper shoe fit may cause toenail trauma. The patient’s shoe is flipped 180° and placed toe-to-toe with the patient’s foot (Figure). Most patients can comprehend the relationship between their shoe fit and their toenail disease when they see this demonstration. We have termed it toe-to-toe sign.
|
|
|
|
| The foot and shoe are toe-to-toe, demonstrating the narrow toe box in comparison to the actual toes (A). This comparison is accentuated when the shoe is placed over the foot with toes splayed from standing (B). |
Narrow toe box is the usual culprit. The toe-to-toe sign best emphasizes this relationship. This sign serves as a powerful tool when demonstrating how much the foot widens when bearing full weight, and how forces of ambulation and foot strike can damage the nails with narrow-toed footwear. This trauma is compounded with high-heeled shoes that force the toes forward. However, proper shoe fit may compete with idealized shoe style. It is not until patients realize the relationship between improper shoe fit, foot strike, and toe/toenail trauma that they can make long-term decisions that favorably impact their toes and toenails.
Reference
1. Daniel CR 3rd, Jellinek NJ. The pedal fungus reservoir. Arch Dermatol. 2006;142:1344-1346.
Reference
1. Daniel CR 3rd, Jellinek NJ. The pedal fungus reservoir. Arch Dermatol. 2006;142:1344-1346.

Plaques: A Rare Presentation of Acrokeratoelastoidosis
To the Editor:
Acrokeratoelastoidosis (AKE) is a rare disease first described by Costa1 in 1953. Typically it is only a cosmetic nuisance in the majority of patients and presents as asymptomatic, small, firm, flesh-colored to yellowish, round to polygonal papules with occasional keratosis or umbilication on the radial and ulnar margins of the hands and/or feet.1-3 In some cases, the lesions occur on the anterior aspects of the wrists, fingers, or lower legs.1 The lesions are always bilaterally distributed. Acrokeratoelastoidosis is a chronic skin disorder that commonly presents during childhood or adolescence, but presentation in adulthood also has been described.3 Histologically, AKE always shows hyperkeratosis, acanthosis, decrease of elastic tissue, and elastorrhexis of remaining elastic fibers. Plaque-type lesions are rare. We describe a patient who presented with plaques on the radial and ulnar margins of the hands.
A 36-year-old Chinese woman presented with asymptomatic, small, firm papules of 6 months’ duration that initially developed on the hands and gradually increased in number, coalescing into plaques. The feet were spared. She had no medical history of hyperhidrosis, chronic trauma, friction, or excessive sun exposure, and no family history of similar symptoms. No prior therapy had been attempted.
Physical examination showed nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules with wavy edges that were symmetrically distributed on the radial and ulnar margins of the hands; some papules had coalesced into plaques (Figure 1). A biopsy specimen taken from a plaque on the hypothenar eminence of the right hand revealed focal hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation with hematoxylin and eosin stain (Figure 2A). Aldehyde fuchsin staining showed fragmented and rarefied elastic fibers in the reticular dermis (Figure 2B). The patient was diagnosed with AKE. Oral tretinoin 10 mg twice daily was initiated and resulted in an evident response after 2 weeks of treatment. However, the patient stopped taking the medication because of pruritus and dry skin and the lesions then reappeared.

|
| Figure 1. Nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules distributed on the radial margin of the hand; some of papules coalesced into plaques. |
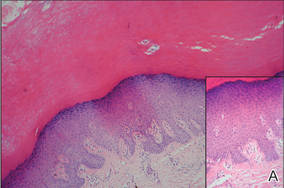
|
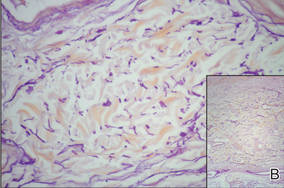
|
| Figure 2. Histopathology revealed hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation (A) (H&E, original magnification ×250 [inset, original magnification ×400]), as well as fragmented elastic fibers in the reticular dermis (B) (Aldehyde fuchsin, original magnification ×400 [inset, original magnification ×100]). |
Acrokeratoelastoidosis is a rare keratotic disorder. It seems to have no racial or ethnic predilection and occurs more frequently in women.4,5 It also is rare in China, with few cases reported, all women.5 The reason for the gender predilection in China remains unknown. The course is chronic, but it may rapidly progress during pregnancy.6
The pathogenesis of AKE is still unresolved.2,3 Although many cases are sporadic,5 it appears to be inherited in an autosomal-dominant fashion, most likely related to chromosome 2.7 Typically, AKE presents as papules that are discrete and bilaterally distributed in the palmoplantar margins,2,3 but some of the papules in our patient coalesced into plaques, which is unique. The histologic hallmarks indicated that the lesions were AKE.
The differential diagnosis of AKE includes hereditary papulotranslucent acrokeratoderma, focal acral hyperkeratosis, and keratoelastoidosis marginalis.8 Hereditary papulotranslucent acrokeratoderma also is inherited in an autosomal-dominant fashion and shares similar acral, translucent, keratotic papules with AKE, but there is no chronic inflammatory cell infiltrate, degeneration of collagenous fibers, or fragmentation of elastic fibers. The clinical appearance of focal acral hyperkeratosis is similar to AKE, but no changes are revealed in the elastic tissue.9 Because AKE, focal acral hyperkeratosis, and hereditary papulotranslucent acrokeratoderma have similar lesions and overlapping histologic changes, they may be considered variants of the same entity.4 Keratoelastoidosis marginalis, also called degenerative collagenous plaques of the hand, mainly affects white individuals aged 40 to 60 years with a history of prolonged sun exposure. Papules often are distributed over the junction of the dorsal and palmar skin and less often on the ulnar sides of the hands. The clinical lesions are similar to those in our patient, but histopathology of keratoelastoidosis marginalis shows amorphous, basophilic, elastotic masses and thickened, fragmented, calcified elastic fibers in the upper and mid dermis.
Therapies including liquid nitrogen, topical salicylic acid, methotrexate, dapsone, tar, cryotherapy, systemic prednisone, retinoic acid, clobetasone cream,5 and erbium:YAG laser10 have been applied. Thus far, no optimal treatment has been recommended and no tendency of spontaneous resolution has been previously reported in the literature. Our patient responded to tretinoin, but the lesions recurred after withdrawal of the medication; therefore, tretinoin may not be an optimal treatment option. Because the lesions are limited to the skin and AKE is only considered a cosmetic problem with a good prognosis, we recommend a wait-and-watch approach.
Acknowledgment—We thank Rashmi Sarkar, MD, New Delhi, India, for her assistance.
1. Costa OG. Akrokerato-elastoidosis; a hitherto undescribed skin disease. Dermatologica. 1953;107:164-168.
2. Bogle MA, Huang LY, Tschen JA. Acrokeratoelastoidosis. J Am Acad Dermatol. 2002;47:448-451.
3. Highet AS, Rook A, Anderson JR. Acrokeratoelastoidosis. Br J Dermatol. 1982;106:337-344.
4. Abulafia J, Vignale RA. Degenerative collagenous
plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
5. Luo DQ, Zhang B, Huang YB, et al. Papules on a young woman’s hands and feet. Clin Exp Dermatol. 2010;35:451-452.
6. Nelson-Adesokan P, Mallory SB, Lombardi C, et al. Acrokeratoelastoidosis of Costa [published correction appears in Int J Dermatol. 1996;35:380]. Int J Dermatol. 1995;34:431-433.
7. Greiner J, Krüger J, Palden L, et al. A linkage study of acrokeratoelastoidosis. possible mapping to chromosome 2. Hum Genet. 1983;63:222-227.
8. Hu W, Cook TF, Vicki GJ, et al. Acrokeratoelastoidosis. Pediatr Dermatol. 2002;19:320-322.
9. Dowd PM, Harman RR, Black MM. Focal acral hyperkeratosis. Br J Dermatol. 1983;109:97-103.
10. Erbil AH, Sezer E, Koç E, et al. Acrokeratoelastoidosis treated with the erbium:YAG laser [published online ahead of print November 3, 2007]. Clin Exp Dermatol. 2008;33:30-31.
To the Editor:
Acrokeratoelastoidosis (AKE) is a rare disease first described by Costa1 in 1953. Typically it is only a cosmetic nuisance in the majority of patients and presents as asymptomatic, small, firm, flesh-colored to yellowish, round to polygonal papules with occasional keratosis or umbilication on the radial and ulnar margins of the hands and/or feet.1-3 In some cases, the lesions occur on the anterior aspects of the wrists, fingers, or lower legs.1 The lesions are always bilaterally distributed. Acrokeratoelastoidosis is a chronic skin disorder that commonly presents during childhood or adolescence, but presentation in adulthood also has been described.3 Histologically, AKE always shows hyperkeratosis, acanthosis, decrease of elastic tissue, and elastorrhexis of remaining elastic fibers. Plaque-type lesions are rare. We describe a patient who presented with plaques on the radial and ulnar margins of the hands.
A 36-year-old Chinese woman presented with asymptomatic, small, firm papules of 6 months’ duration that initially developed on the hands and gradually increased in number, coalescing into plaques. The feet were spared. She had no medical history of hyperhidrosis, chronic trauma, friction, or excessive sun exposure, and no family history of similar symptoms. No prior therapy had been attempted.
Physical examination showed nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules with wavy edges that were symmetrically distributed on the radial and ulnar margins of the hands; some papules had coalesced into plaques (Figure 1). A biopsy specimen taken from a plaque on the hypothenar eminence of the right hand revealed focal hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation with hematoxylin and eosin stain (Figure 2A). Aldehyde fuchsin staining showed fragmented and rarefied elastic fibers in the reticular dermis (Figure 2B). The patient was diagnosed with AKE. Oral tretinoin 10 mg twice daily was initiated and resulted in an evident response after 2 weeks of treatment. However, the patient stopped taking the medication because of pruritus and dry skin and the lesions then reappeared.
|
|
| Figure 1. Nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules distributed on the radial margin of the hand; some of papules coalesced into plaques. |
|
|
|
|
| Figure 2. Histopathology revealed hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation (A) (H&E, original magnification ×250 [inset, original magnification ×400]), as well as fragmented elastic fibers in the reticular dermis (B) (Aldehyde fuchsin, original magnification ×400 [inset, original magnification ×100]). |
Acrokeratoelastoidosis is a rare keratotic disorder. It seems to have no racial or ethnic predilection and occurs more frequently in women.4,5 It also is rare in China, with few cases reported, all women.5 The reason for the gender predilection in China remains unknown. The course is chronic, but it may rapidly progress during pregnancy.6
The pathogenesis of AKE is still unresolved.2,3 Although many cases are sporadic,5 it appears to be inherited in an autosomal-dominant fashion, most likely related to chromosome 2.7 Typically, AKE presents as papules that are discrete and bilaterally distributed in the palmoplantar margins,2,3 but some of the papules in our patient coalesced into plaques, which is unique. The histologic hallmarks indicated that the lesions were AKE.
The differential diagnosis of AKE includes hereditary papulotranslucent acrokeratoderma, focal acral hyperkeratosis, and keratoelastoidosis marginalis.8 Hereditary papulotranslucent acrokeratoderma also is inherited in an autosomal-dominant fashion and shares similar acral, translucent, keratotic papules with AKE, but there is no chronic inflammatory cell infiltrate, degeneration of collagenous fibers, or fragmentation of elastic fibers. The clinical appearance of focal acral hyperkeratosis is similar to AKE, but no changes are revealed in the elastic tissue.9 Because AKE, focal acral hyperkeratosis, and hereditary papulotranslucent acrokeratoderma have similar lesions and overlapping histologic changes, they may be considered variants of the same entity.4 Keratoelastoidosis marginalis, also called degenerative collagenous plaques of the hand, mainly affects white individuals aged 40 to 60 years with a history of prolonged sun exposure. Papules often are distributed over the junction of the dorsal and palmar skin and less often on the ulnar sides of the hands. The clinical lesions are similar to those in our patient, but histopathology of keratoelastoidosis marginalis shows amorphous, basophilic, elastotic masses and thickened, fragmented, calcified elastic fibers in the upper and mid dermis.
Therapies including liquid nitrogen, topical salicylic acid, methotrexate, dapsone, tar, cryotherapy, systemic prednisone, retinoic acid, clobetasone cream,5 and erbium:YAG laser10 have been applied. Thus far, no optimal treatment has been recommended and no tendency of spontaneous resolution has been previously reported in the literature. Our patient responded to tretinoin, but the lesions recurred after withdrawal of the medication; therefore, tretinoin may not be an optimal treatment option. Because the lesions are limited to the skin and AKE is only considered a cosmetic problem with a good prognosis, we recommend a wait-and-watch approach.
Acknowledgment—We thank Rashmi Sarkar, MD, New Delhi, India, for her assistance.
To the Editor:
Acrokeratoelastoidosis (AKE) is a rare disease first described by Costa1 in 1953. Typically it is only a cosmetic nuisance in the majority of patients and presents as asymptomatic, small, firm, flesh-colored to yellowish, round to polygonal papules with occasional keratosis or umbilication on the radial and ulnar margins of the hands and/or feet.1-3 In some cases, the lesions occur on the anterior aspects of the wrists, fingers, or lower legs.1 The lesions are always bilaterally distributed. Acrokeratoelastoidosis is a chronic skin disorder that commonly presents during childhood or adolescence, but presentation in adulthood also has been described.3 Histologically, AKE always shows hyperkeratosis, acanthosis, decrease of elastic tissue, and elastorrhexis of remaining elastic fibers. Plaque-type lesions are rare. We describe a patient who presented with plaques on the radial and ulnar margins of the hands.
A 36-year-old Chinese woman presented with asymptomatic, small, firm papules of 6 months’ duration that initially developed on the hands and gradually increased in number, coalescing into plaques. The feet were spared. She had no medical history of hyperhidrosis, chronic trauma, friction, or excessive sun exposure, and no family history of similar symptoms. No prior therapy had been attempted.
Physical examination showed nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules with wavy edges that were symmetrically distributed on the radial and ulnar margins of the hands; some papules had coalesced into plaques (Figure 1). A biopsy specimen taken from a plaque on the hypothenar eminence of the right hand revealed focal hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation with hematoxylin and eosin stain (Figure 2A). Aldehyde fuchsin staining showed fragmented and rarefied elastic fibers in the reticular dermis (Figure 2B). The patient was diagnosed with AKE. Oral tretinoin 10 mg twice daily was initiated and resulted in an evident response after 2 weeks of treatment. However, the patient stopped taking the medication because of pruritus and dry skin and the lesions then reappeared.
|
|
| Figure 1. Nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules distributed on the radial margin of the hand; some of papules coalesced into plaques. |
|
|
|
|
| Figure 2. Histopathology revealed hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation (A) (H&E, original magnification ×250 [inset, original magnification ×400]), as well as fragmented elastic fibers in the reticular dermis (B) (Aldehyde fuchsin, original magnification ×400 [inset, original magnification ×100]). |
Acrokeratoelastoidosis is a rare keratotic disorder. It seems to have no racial or ethnic predilection and occurs more frequently in women.4,5 It also is rare in China, with few cases reported, all women.5 The reason for the gender predilection in China remains unknown. The course is chronic, but it may rapidly progress during pregnancy.6
The pathogenesis of AKE is still unresolved.2,3 Although many cases are sporadic,5 it appears to be inherited in an autosomal-dominant fashion, most likely related to chromosome 2.7 Typically, AKE presents as papules that are discrete and bilaterally distributed in the palmoplantar margins,2,3 but some of the papules in our patient coalesced into plaques, which is unique. The histologic hallmarks indicated that the lesions were AKE.
The differential diagnosis of AKE includes hereditary papulotranslucent acrokeratoderma, focal acral hyperkeratosis, and keratoelastoidosis marginalis.8 Hereditary papulotranslucent acrokeratoderma also is inherited in an autosomal-dominant fashion and shares similar acral, translucent, keratotic papules with AKE, but there is no chronic inflammatory cell infiltrate, degeneration of collagenous fibers, or fragmentation of elastic fibers. The clinical appearance of focal acral hyperkeratosis is similar to AKE, but no changes are revealed in the elastic tissue.9 Because AKE, focal acral hyperkeratosis, and hereditary papulotranslucent acrokeratoderma have similar lesions and overlapping histologic changes, they may be considered variants of the same entity.4 Keratoelastoidosis marginalis, also called degenerative collagenous plaques of the hand, mainly affects white individuals aged 40 to 60 years with a history of prolonged sun exposure. Papules often are distributed over the junction of the dorsal and palmar skin and less often on the ulnar sides of the hands. The clinical lesions are similar to those in our patient, but histopathology of keratoelastoidosis marginalis shows amorphous, basophilic, elastotic masses and thickened, fragmented, calcified elastic fibers in the upper and mid dermis.
Therapies including liquid nitrogen, topical salicylic acid, methotrexate, dapsone, tar, cryotherapy, systemic prednisone, retinoic acid, clobetasone cream,5 and erbium:YAG laser10 have been applied. Thus far, no optimal treatment has been recommended and no tendency of spontaneous resolution has been previously reported in the literature. Our patient responded to tretinoin, but the lesions recurred after withdrawal of the medication; therefore, tretinoin may not be an optimal treatment option. Because the lesions are limited to the skin and AKE is only considered a cosmetic problem with a good prognosis, we recommend a wait-and-watch approach.
Acknowledgment—We thank Rashmi Sarkar, MD, New Delhi, India, for her assistance.
1. Costa OG. Akrokerato-elastoidosis; a hitherto undescribed skin disease. Dermatologica. 1953;107:164-168.
2. Bogle MA, Huang LY, Tschen JA. Acrokeratoelastoidosis. J Am Acad Dermatol. 2002;47:448-451.
3. Highet AS, Rook A, Anderson JR. Acrokeratoelastoidosis. Br J Dermatol. 1982;106:337-344.
4. Abulafia J, Vignale RA. Degenerative collagenous
plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
5. Luo DQ, Zhang B, Huang YB, et al. Papules on a young woman’s hands and feet. Clin Exp Dermatol. 2010;35:451-452.
6. Nelson-Adesokan P, Mallory SB, Lombardi C, et al. Acrokeratoelastoidosis of Costa [published correction appears in Int J Dermatol. 1996;35:380]. Int J Dermatol. 1995;34:431-433.
7. Greiner J, Krüger J, Palden L, et al. A linkage study of acrokeratoelastoidosis. possible mapping to chromosome 2. Hum Genet. 1983;63:222-227.
8. Hu W, Cook TF, Vicki GJ, et al. Acrokeratoelastoidosis. Pediatr Dermatol. 2002;19:320-322.
9. Dowd PM, Harman RR, Black MM. Focal acral hyperkeratosis. Br J Dermatol. 1983;109:97-103.
10. Erbil AH, Sezer E, Koç E, et al. Acrokeratoelastoidosis treated with the erbium:YAG laser [published online ahead of print November 3, 2007]. Clin Exp Dermatol. 2008;33:30-31.
1. Costa OG. Akrokerato-elastoidosis; a hitherto undescribed skin disease. Dermatologica. 1953;107:164-168.
2. Bogle MA, Huang LY, Tschen JA. Acrokeratoelastoidosis. J Am Acad Dermatol. 2002;47:448-451.
3. Highet AS, Rook A, Anderson JR. Acrokeratoelastoidosis. Br J Dermatol. 1982;106:337-344.
4. Abulafia J, Vignale RA. Degenerative collagenous
plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
5. Luo DQ, Zhang B, Huang YB, et al. Papules on a young woman’s hands and feet. Clin Exp Dermatol. 2010;35:451-452.
6. Nelson-Adesokan P, Mallory SB, Lombardi C, et al. Acrokeratoelastoidosis of Costa [published correction appears in Int J Dermatol. 1996;35:380]. Int J Dermatol. 1995;34:431-433.
7. Greiner J, Krüger J, Palden L, et al. A linkage study of acrokeratoelastoidosis. possible mapping to chromosome 2. Hum Genet. 1983;63:222-227.
8. Hu W, Cook TF, Vicki GJ, et al. Acrokeratoelastoidosis. Pediatr Dermatol. 2002;19:320-322.
9. Dowd PM, Harman RR, Black MM. Focal acral hyperkeratosis. Br J Dermatol. 1983;109:97-103.
10. Erbil AH, Sezer E, Koç E, et al. Acrokeratoelastoidosis treated with the erbium:YAG laser [published online ahead of print November 3, 2007]. Clin Exp Dermatol. 2008;33:30-31.

Primary Mucinous Carcinoma of the Skin
To the Editor:
A 41-year-old man presented to our dermatology clinic with a recurrent asymptomatic nodule on the right cheek that had gradually increased in size over 1 year. The patient underwent laser excision at an outside facility 1 year after the first presentation. Pathology reports and tissue cultures from the excision were not available. The lesion recurred in the same location 6 months following excision. The patient reported no history of pain, fever, cough, weight loss, or loss of appetite, and he denied any trauma or radiation to the affected area. Dermatologic examination revealed a 24×18-mm, slightly elevated, dome-shaped, translucent, pink-colored tumor with telangiectasia on the right cheek (Figure 1). There was no tenderness or pruritus. Clinical examination and extensive radiographic studies revealed no primary disease. A complete blood cell count, biochemical tests, and serous tumor markers were within reference range. The lesion was resected with a 2-mm margin. The margins were free of tumor cells. Histopathology showed a circumscribed tumor with large amounts of mucin compartmentalized by fibrous septa and scattered floating islands of tumor cells in the dermis. Small-sized glands were organized in some areas of the tumor cells (Figure 2). Epithelial tumor cell islands contained uniform oval nuclei and focally vacuolated cytoplasm. The tumor cells were positive for cytokeratin 7 (CK7) (Figure 3) and negative for cytokeratin 20 (CK20), carcinoembryonic antigen, homeobox transcription factor (CDX2), villin, mucus-associated peptides of the thyroid transcription factor (TTF1), and prostate-specific antigen, all indicating a diagnosis of primary mucinous carcinoma of the skin (PMCS). No local recurrence, regional lymph node involvement, or distant metastasis was observed after resection.

|
| Figure 1. A 24×18-mm, slightly elevated, dome-shaped, translucent, pink-colored tumor with telangiectasia on the right cheek. |
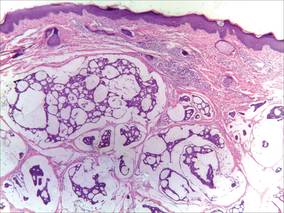
|
| Figure 2. Histopathology showed a circumscribed tumor with large amounts of mucin compartmentalized by fibrous septa that contained scattered floating islands of tumor cells in the dermis (H&E, original magnification ×40). |
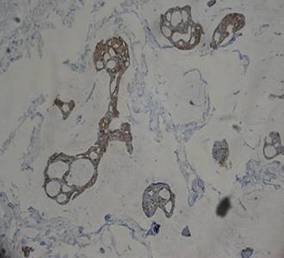
|
| Figure 3. The tumor cells were positive for cytokeratin 7 (original magnification ×400). |
Primary mucinous carcinoma of the skin is a rare malignant adnexal tumor. It was first described by Lennox et al1 in 1952 and was later given its name by Mendoza and Helwig2 through summarizing clinical and histochemical characteristics based on the observation of 14 cases. A PubMed search of articles indexed for MEDLINE using the search terms adenocarcinoma, mucinous (Medical Subject Headings); mucinous adenocarcinoma (title/abstract); colloid carcinoma* (title/abstract); skin neoplasms (Medical Subject Headings); skin neoplasm (title/abstract); skin cancer* (title/abstract), and primary (title/abstract) revealed more than 200 reported cases of PMCS. It usually is located in the head and neck region, with the eyelids being the most common site of presentation, but rare presentations in the gluteal region, axillae, hemithoraxes, vulva, arms, and legs also have been reported.3-6 Primary mucinous carcinoma of the skin mainly affects elderly patients (age range, 61–80 years), with men being affected more often than women. It often presents as a painless, solitary, nontender, sometimes ulcerated, translucent, white or reddish nodule. The tumors generally grow slowly with high rates of local recurrence and rare chances of distant metastasis.7 Most metastases involve the regional lymph nodes and lungs, but rare cases of bone marrow and parotid gland metastases also have been reported.8 Because PMCS is an indolent tumor, which may be mistaken for a benign tumor, it is not always histologically examined on initial presentation.
Primary mucinous carcinoma of the skin is characterized histopathologically by floating epithelial cell nests in mucinous lakes. It is unknown if this neoplasm shows eccrine- or apocrine-type differentiation. S-100 protein seems to indicate eccrine rather than apocrine differentiation.9 Misdiagnosis of PMCS is common, as it has an uncharacteristic gross appearance and may microscopically resemble cutaneous metastasis from a mucinous carcinoma of the breast, gastrointestinal tract, lungs, ovaries, or prostate. It is important to distinguish between metastatic tumors and PMCS because PMCS generally is more benign and has a better prognosis. A variety of stains have been reported as helpful in the diagnosis of PMCS, including CK7 and CK20, D2-40, p63, and others. In some reports, labeling for CK7 and CK20 was helpful in differentiating colon carcinoma from breast or skin malignancies but did not differentiate primary skin lesions from metastasis of breast malignancies to the skin.10-13 Paradela et al5 proposed D2-40 as a reliable immunostain to detect lymphatic invasion in node-negative primary tumors. Kalebi and Hale11 and Ivan et al12 highlighted the importance of p63 in excluding metastasis to the skin, particularly from the gastrointestinal tract, pancreaticobiliary tree, and lungs.
Even though these factors may help in establishing the primary site of the tumor, a final diagnosis only can be drawn when the patient has been subjected to a thorough investigation. In our patient, clinical workup did not reveal other possible primary sites in other organs or evidence of metastasis from the skin lesion to other sites. Based on the histologic findings, the diagnosis of PMCS was made.
The recommended treatment of PMCS varies from standard excision to wide local excision including dissection of the regional lymph nodes. Alam et al14 reported that Mohs micrographic surgery may be appropriate for first-line therapy because the procedure gives complete margin control and spares tissue. Immunohistochemistry should be used as an adjunct to routine hematoxylin and eosin staining to aid in ensuring negative margins.15 Considering a high potential for recurrence following surgical excision, it is important to detect hormone receptors in this tumor because patients may be treated using antiestrogenic drugs such as tamoxifen. Moreover, recurrent or metastatic PMCS is both resistant to radiotherapy and unresponsive to chemotherapy.3 Annual follow-up is recommended due to the potential for recurrence or metastasis.
1. Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin, with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-890.
2. Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
3. Breiting L, Dahlstrøm K, Christensen L, et al. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2007;29:595-596.
4. Krishnamurthy J, Saba F, Sunila. Primary mucinous carcinoma of the skin: a rare tumor in the gluteal region. Indian J Pathol Microbiol. 2009;52:225-227.
5. Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
6. Laco J, Simáková E, Svobodová J, et al. Recurrent mucinous carcinoma of skin mimicking primary mucinous carcinoma of parotid gland: a diagnostic pitfall. Cesk Patol. 2009;45:79-82.
7. Breiting L, Christensen L, Dahlstrøm K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
8. Ajithkumar TV, Nileena N, Abraham EK, et al. Bone marrow relapse in primary mucinous carcinoma of skin. Am J Clin Oncol. 1999;22:303-304.
9. Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
10. Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin [published online ahead of print September 24, 2009]. J Cutan Pathol. 2010;37:411-415.
11. Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
12. Ivan D, Nash JW, Prieto VG, et al. Use of p63 expression in distinguishing primary and metastatic cutaneous adnexal neoplasms from metastatic adenocarcinoma to skin. J Cutan Pathol. 2007;34:474-480.
13. Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
14. Alam M, Trela R, Kim N, et al. Treatment of primary mucinous carcinoma of the skin: meta-analysis of 189 cases. J Invest Dermatol. 2009;129:382.
15. Stranahan D, Cherpelis BS, Glass LF, et al. Immunohistochemical stains in Mohs surgery: a review [published online ahead of print April 16, 2009]. Dermatol Surg. 2009;35:1023-1034.
To the Editor:
A 41-year-old man presented to our dermatology clinic with a recurrent asymptomatic nodule on the right cheek that had gradually increased in size over 1 year. The patient underwent laser excision at an outside facility 1 year after the first presentation. Pathology reports and tissue cultures from the excision were not available. The lesion recurred in the same location 6 months following excision. The patient reported no history of pain, fever, cough, weight loss, or loss of appetite, and he denied any trauma or radiation to the affected area. Dermatologic examination revealed a 24×18-mm, slightly elevated, dome-shaped, translucent, pink-colored tumor with telangiectasia on the right cheek (Figure 1). There was no tenderness or pruritus. Clinical examination and extensive radiographic studies revealed no primary disease. A complete blood cell count, biochemical tests, and serous tumor markers were within reference range. The lesion was resected with a 2-mm margin. The margins were free of tumor cells. Histopathology showed a circumscribed tumor with large amounts of mucin compartmentalized by fibrous septa and scattered floating islands of tumor cells in the dermis. Small-sized glands were organized in some areas of the tumor cells (Figure 2). Epithelial tumor cell islands contained uniform oval nuclei and focally vacuolated cytoplasm. The tumor cells were positive for cytokeratin 7 (CK7) (Figure 3) and negative for cytokeratin 20 (CK20), carcinoembryonic antigen, homeobox transcription factor (CDX2), villin, mucus-associated peptides of the thyroid transcription factor (TTF1), and prostate-specific antigen, all indicating a diagnosis of primary mucinous carcinoma of the skin (PMCS). No local recurrence, regional lymph node involvement, or distant metastasis was observed after resection.
|
|
| Figure 1. A 24×18-mm, slightly elevated, dome-shaped, translucent, pink-colored tumor with telangiectasia on the right cheek. |
|
|
| Figure 2. Histopathology showed a circumscribed tumor with large amounts of mucin compartmentalized by fibrous septa that contained scattered floating islands of tumor cells in the dermis (H&E, original magnification ×40). |
|
|
| Figure 3. The tumor cells were positive for cytokeratin 7 (original magnification ×400). |
Primary mucinous carcinoma of the skin is a rare malignant adnexal tumor. It was first described by Lennox et al1 in 1952 and was later given its name by Mendoza and Helwig2 through summarizing clinical and histochemical characteristics based on the observation of 14 cases. A PubMed search of articles indexed for MEDLINE using the search terms adenocarcinoma, mucinous (Medical Subject Headings); mucinous adenocarcinoma (title/abstract); colloid carcinoma* (title/abstract); skin neoplasms (Medical Subject Headings); skin neoplasm (title/abstract); skin cancer* (title/abstract), and primary (title/abstract) revealed more than 200 reported cases of PMCS. It usually is located in the head and neck region, with the eyelids being the most common site of presentation, but rare presentations in the gluteal region, axillae, hemithoraxes, vulva, arms, and legs also have been reported.3-6 Primary mucinous carcinoma of the skin mainly affects elderly patients (age range, 61–80 years), with men being affected more often than women. It often presents as a painless, solitary, nontender, sometimes ulcerated, translucent, white or reddish nodule. The tumors generally grow slowly with high rates of local recurrence and rare chances of distant metastasis.7 Most metastases involve the regional lymph nodes and lungs, but rare cases of bone marrow and parotid gland metastases also have been reported.8 Because PMCS is an indolent tumor, which may be mistaken for a benign tumor, it is not always histologically examined on initial presentation.
Primary mucinous carcinoma of the skin is characterized histopathologically by floating epithelial cell nests in mucinous lakes. It is unknown if this neoplasm shows eccrine- or apocrine-type differentiation. S-100 protein seems to indicate eccrine rather than apocrine differentiation.9 Misdiagnosis of PMCS is common, as it has an uncharacteristic gross appearance and may microscopically resemble cutaneous metastasis from a mucinous carcinoma of the breast, gastrointestinal tract, lungs, ovaries, or prostate. It is important to distinguish between metastatic tumors and PMCS because PMCS generally is more benign and has a better prognosis. A variety of stains have been reported as helpful in the diagnosis of PMCS, including CK7 and CK20, D2-40, p63, and others. In some reports, labeling for CK7 and CK20 was helpful in differentiating colon carcinoma from breast or skin malignancies but did not differentiate primary skin lesions from metastasis of breast malignancies to the skin.10-13 Paradela et al5 proposed D2-40 as a reliable immunostain to detect lymphatic invasion in node-negative primary tumors. Kalebi and Hale11 and Ivan et al12 highlighted the importance of p63 in excluding metastasis to the skin, particularly from the gastrointestinal tract, pancreaticobiliary tree, and lungs.
Even though these factors may help in establishing the primary site of the tumor, a final diagnosis only can be drawn when the patient has been subjected to a thorough investigation. In our patient, clinical workup did not reveal other possible primary sites in other organs or evidence of metastasis from the skin lesion to other sites. Based on the histologic findings, the diagnosis of PMCS was made.
The recommended treatment of PMCS varies from standard excision to wide local excision including dissection of the regional lymph nodes. Alam et al14 reported that Mohs micrographic surgery may be appropriate for first-line therapy because the procedure gives complete margin control and spares tissue. Immunohistochemistry should be used as an adjunct to routine hematoxylin and eosin staining to aid in ensuring negative margins.15 Considering a high potential for recurrence following surgical excision, it is important to detect hormone receptors in this tumor because patients may be treated using antiestrogenic drugs such as tamoxifen. Moreover, recurrent or metastatic PMCS is both resistant to radiotherapy and unresponsive to chemotherapy.3 Annual follow-up is recommended due to the potential for recurrence or metastasis.
To the Editor:
A 41-year-old man presented to our dermatology clinic with a recurrent asymptomatic nodule on the right cheek that had gradually increased in size over 1 year. The patient underwent laser excision at an outside facility 1 year after the first presentation. Pathology reports and tissue cultures from the excision were not available. The lesion recurred in the same location 6 months following excision. The patient reported no history of pain, fever, cough, weight loss, or loss of appetite, and he denied any trauma or radiation to the affected area. Dermatologic examination revealed a 24×18-mm, slightly elevated, dome-shaped, translucent, pink-colored tumor with telangiectasia on the right cheek (Figure 1). There was no tenderness or pruritus. Clinical examination and extensive radiographic studies revealed no primary disease. A complete blood cell count, biochemical tests, and serous tumor markers were within reference range. The lesion was resected with a 2-mm margin. The margins were free of tumor cells. Histopathology showed a circumscribed tumor with large amounts of mucin compartmentalized by fibrous septa and scattered floating islands of tumor cells in the dermis. Small-sized glands were organized in some areas of the tumor cells (Figure 2). Epithelial tumor cell islands contained uniform oval nuclei and focally vacuolated cytoplasm. The tumor cells were positive for cytokeratin 7 (CK7) (Figure 3) and negative for cytokeratin 20 (CK20), carcinoembryonic antigen, homeobox transcription factor (CDX2), villin, mucus-associated peptides of the thyroid transcription factor (TTF1), and prostate-specific antigen, all indicating a diagnosis of primary mucinous carcinoma of the skin (PMCS). No local recurrence, regional lymph node involvement, or distant metastasis was observed after resection.
|
|
| Figure 1. A 24×18-mm, slightly elevated, dome-shaped, translucent, pink-colored tumor with telangiectasia on the right cheek. |
|
|
| Figure 2. Histopathology showed a circumscribed tumor with large amounts of mucin compartmentalized by fibrous septa that contained scattered floating islands of tumor cells in the dermis (H&E, original magnification ×40). |
|
|
| Figure 3. The tumor cells were positive for cytokeratin 7 (original magnification ×400). |
Primary mucinous carcinoma of the skin is a rare malignant adnexal tumor. It was first described by Lennox et al1 in 1952 and was later given its name by Mendoza and Helwig2 through summarizing clinical and histochemical characteristics based on the observation of 14 cases. A PubMed search of articles indexed for MEDLINE using the search terms adenocarcinoma, mucinous (Medical Subject Headings); mucinous adenocarcinoma (title/abstract); colloid carcinoma* (title/abstract); skin neoplasms (Medical Subject Headings); skin neoplasm (title/abstract); skin cancer* (title/abstract), and primary (title/abstract) revealed more than 200 reported cases of PMCS. It usually is located in the head and neck region, with the eyelids being the most common site of presentation, but rare presentations in the gluteal region, axillae, hemithoraxes, vulva, arms, and legs also have been reported.3-6 Primary mucinous carcinoma of the skin mainly affects elderly patients (age range, 61–80 years), with men being affected more often than women. It often presents as a painless, solitary, nontender, sometimes ulcerated, translucent, white or reddish nodule. The tumors generally grow slowly with high rates of local recurrence and rare chances of distant metastasis.7 Most metastases involve the regional lymph nodes and lungs, but rare cases of bone marrow and parotid gland metastases also have been reported.8 Because PMCS is an indolent tumor, which may be mistaken for a benign tumor, it is not always histologically examined on initial presentation.
Primary mucinous carcinoma of the skin is characterized histopathologically by floating epithelial cell nests in mucinous lakes. It is unknown if this neoplasm shows eccrine- or apocrine-type differentiation. S-100 protein seems to indicate eccrine rather than apocrine differentiation.9 Misdiagnosis of PMCS is common, as it has an uncharacteristic gross appearance and may microscopically resemble cutaneous metastasis from a mucinous carcinoma of the breast, gastrointestinal tract, lungs, ovaries, or prostate. It is important to distinguish between metastatic tumors and PMCS because PMCS generally is more benign and has a better prognosis. A variety of stains have been reported as helpful in the diagnosis of PMCS, including CK7 and CK20, D2-40, p63, and others. In some reports, labeling for CK7 and CK20 was helpful in differentiating colon carcinoma from breast or skin malignancies but did not differentiate primary skin lesions from metastasis of breast malignancies to the skin.10-13 Paradela et al5 proposed D2-40 as a reliable immunostain to detect lymphatic invasion in node-negative primary tumors. Kalebi and Hale11 and Ivan et al12 highlighted the importance of p63 in excluding metastasis to the skin, particularly from the gastrointestinal tract, pancreaticobiliary tree, and lungs.
Even though these factors may help in establishing the primary site of the tumor, a final diagnosis only can be drawn when the patient has been subjected to a thorough investigation. In our patient, clinical workup did not reveal other possible primary sites in other organs or evidence of metastasis from the skin lesion to other sites. Based on the histologic findings, the diagnosis of PMCS was made.
The recommended treatment of PMCS varies from standard excision to wide local excision including dissection of the regional lymph nodes. Alam et al14 reported that Mohs micrographic surgery may be appropriate for first-line therapy because the procedure gives complete margin control and spares tissue. Immunohistochemistry should be used as an adjunct to routine hematoxylin and eosin staining to aid in ensuring negative margins.15 Considering a high potential for recurrence following surgical excision, it is important to detect hormone receptors in this tumor because patients may be treated using antiestrogenic drugs such as tamoxifen. Moreover, recurrent or metastatic PMCS is both resistant to radiotherapy and unresponsive to chemotherapy.3 Annual follow-up is recommended due to the potential for recurrence or metastasis.
1. Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin, with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-890.
2. Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
3. Breiting L, Dahlstrøm K, Christensen L, et al. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2007;29:595-596.
4. Krishnamurthy J, Saba F, Sunila. Primary mucinous carcinoma of the skin: a rare tumor in the gluteal region. Indian J Pathol Microbiol. 2009;52:225-227.
5. Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
6. Laco J, Simáková E, Svobodová J, et al. Recurrent mucinous carcinoma of skin mimicking primary mucinous carcinoma of parotid gland: a diagnostic pitfall. Cesk Patol. 2009;45:79-82.
7. Breiting L, Christensen L, Dahlstrøm K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
8. Ajithkumar TV, Nileena N, Abraham EK, et al. Bone marrow relapse in primary mucinous carcinoma of skin. Am J Clin Oncol. 1999;22:303-304.
9. Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
10. Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin [published online ahead of print September 24, 2009]. J Cutan Pathol. 2010;37:411-415.
11. Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
12. Ivan D, Nash JW, Prieto VG, et al. Use of p63 expression in distinguishing primary and metastatic cutaneous adnexal neoplasms from metastatic adenocarcinoma to skin. J Cutan Pathol. 2007;34:474-480.
13. Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
14. Alam M, Trela R, Kim N, et al. Treatment of primary mucinous carcinoma of the skin: meta-analysis of 189 cases. J Invest Dermatol. 2009;129:382.
15. Stranahan D, Cherpelis BS, Glass LF, et al. Immunohistochemical stains in Mohs surgery: a review [published online ahead of print April 16, 2009]. Dermatol Surg. 2009;35:1023-1034.
1. Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin, with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-890.
2. Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
3. Breiting L, Dahlstrøm K, Christensen L, et al. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2007;29:595-596.
4. Krishnamurthy J, Saba F, Sunila. Primary mucinous carcinoma of the skin: a rare tumor in the gluteal region. Indian J Pathol Microbiol. 2009;52:225-227.
5. Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
6. Laco J, Simáková E, Svobodová J, et al. Recurrent mucinous carcinoma of skin mimicking primary mucinous carcinoma of parotid gland: a diagnostic pitfall. Cesk Patol. 2009;45:79-82.
7. Breiting L, Christensen L, Dahlstrøm K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
8. Ajithkumar TV, Nileena N, Abraham EK, et al. Bone marrow relapse in primary mucinous carcinoma of skin. Am J Clin Oncol. 1999;22:303-304.
9. Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
10. Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin [published online ahead of print September 24, 2009]. J Cutan Pathol. 2010;37:411-415.
11. Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
12. Ivan D, Nash JW, Prieto VG, et al. Use of p63 expression in distinguishing primary and metastatic cutaneous adnexal neoplasms from metastatic adenocarcinoma to skin. J Cutan Pathol. 2007;34:474-480.
13. Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
14. Alam M, Trela R, Kim N, et al. Treatment of primary mucinous carcinoma of the skin: meta-analysis of 189 cases. J Invest Dermatol. 2009;129:382.
15. Stranahan D, Cherpelis BS, Glass LF, et al. Immunohistochemical stains in Mohs surgery: a review [published online ahead of print April 16, 2009]. Dermatol Surg. 2009;35:1023-1034.
Large Plaque-Type Benign Cephalic Histiocytosis Showing Rapid Aggravation Following Vaccination
To the Editor:
Benign cephalic histiocytosis (BCH) is a rare benign dermatosis in which self-healing papular eruptions develop. This condition is classified as non-X histiocytosis and behaves as a benign histiocytic proliferation. Since the first report by Gianotti et al1 in 1971, the etiology and pathophysiology of this condition have not been elucidated. The clinical and histologic features of BCH overlap with juvenile xanthogranuloma (JXG) and generalized eruptive histiocytosis.2 Rodriguez-Jurado et al3 reported that BCH showed clinical evolution to JXG with varicella-zoster infection, suggesting that BCH is a part of a spectrum of diseases that non–Langerhans cell histiocytosis encompasses. We present a case of plaque-type BCH in an infant.
An 11-month-old male infant presented with asymptomatic, ill-defined, infiltrated plaques on the cheeks of 1 month’s duration (Figure 1). Similar red plaques were present on both ears, the left forearm, and the right middle finger. One month following the initial onset as tiny red papules, the lesions had become larger and more prominent after an episode of fever (temperature, 39°C) and immunization against rubella virus and Haemophilus influenzae type b. No therapeutic effect was observed with the application of a topical steroid at the time of presentation. A skin biopsy specimen was taken from a flat plaque on the right cheek. Histopathologic findings revealed dense cell infiltrates predominated by histiocytic cells with some vacuolization in the papillary and upper reticular dermis (Figure 2). Neither foamy histiocytic cells nor giant cells were seen. Immunohistochemical analysis revealed that the cells were CD68+ (Figure 3) and negative for S-100. Following 2 months of observation without any treatment, the plaques on the ears, right forearm, and right middle finger had begun to spontaneously subside. Overall, the remaining lesions showed complete remission by 21 months of age.
|
| Figure 1. Asymptomatic red, infiltrated, flat plaques on the left cheek. |
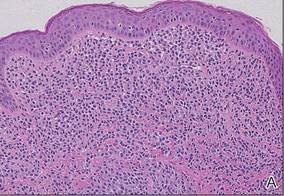
|
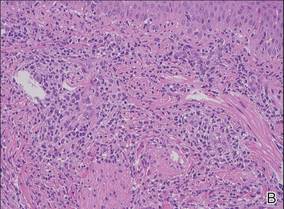
|
| Figure 2. A biopsy revealed an infiltrate of histiocytes with vacuolization of cells in the papillary and upper reticular dermis (A and B) (both H&E, original magnifications ×50 and ×200). |
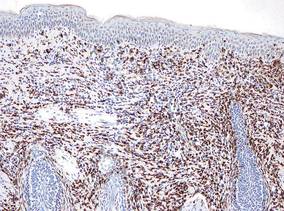
|
| Figure 3. Immunohistochemistry revealed histiocytic infiltrate cells that were CD68+ (original magnification ×50). |
Benign cephalic histiocytosis commonly presents as smooth, round, or flat-topped papules with a diameter of 1 to 8 mm and color ranging from tan-yellow to brown-red.2 The etiology of BCH is unknown. Plaque-type BCH represented by this patient is a rare feature, suggesting sarcoidosis, perniosis, or Sjögren syndrome because the lesions are comprised of infiltrated erythematous plaques rather than papules. Rodriguez-Jurado et al3 described a transitional form of BCH to JXG in a 2-year-old girl with facial plaques after an episode of varicella-zoster virus infection. Vasconcelos et al4 also indicated that JXG is associated with cytomegalovirus infection. These findings support the notion that BCH and non–Langerhans cell histiocytosis are on the same spectrum of diseases, representing a reactive histiocytic process to an infectious agent.
The difference in clinical manifestation of each non–Langerhans cell histiocytosis subgroup may reflect the degree or stage of infection. T lymphocytes may release different cytokines and activate macrophage migration when sensitized by a virus or vaccine. As a result, BCH may show various clinical features even when confirmed by histological analysis. Our patient might have had an early stage of BCH that would transform into JXG.
1. Gianotti F, Caputo R, Ermacora E. Singular “infantile histiocytosis with cells with intracytoplasmic vermiform particles” [in French]. Bull Soc Fr Dermatol Syphiligr. 1971;78:232-233.
2. Jih DM, Salcedo SL, Jaworsky C. Benign cephalic histiocytosis: a case report and review. J Am Acad Dermatol. 2002;47:908-913.
3. Rodriguez-Jurado R, Duran-McKinster C, Ruiz-Maldonado R. Benign cephalic histiocytosis progressing into juvenile xanthogranuloma: a non-Langerhans cell histiocytosis transforming under the influence of a virus? Am J Dermatopathol. 2000;22:70-74.
4. Vasconcelos FO, Oliveira LA, Naves MD, et al. Juvenile xanthogranuloma: case report with immunohistochemical identification of early and late cytomegalovirus antigens. J Oral Sci. 2001;43:21-25.
To the Editor:
Benign cephalic histiocytosis (BCH) is a rare benign dermatosis in which self-healing papular eruptions develop. This condition is classified as non-X histiocytosis and behaves as a benign histiocytic proliferation. Since the first report by Gianotti et al1 in 1971, the etiology and pathophysiology of this condition have not been elucidated. The clinical and histologic features of BCH overlap with juvenile xanthogranuloma (JXG) and generalized eruptive histiocytosis.2 Rodriguez-Jurado et al3 reported that BCH showed clinical evolution to JXG with varicella-zoster infection, suggesting that BCH is a part of a spectrum of diseases that non–Langerhans cell histiocytosis encompasses. We present a case of plaque-type BCH in an infant.
An 11-month-old male infant presented with asymptomatic, ill-defined, infiltrated plaques on the cheeks of 1 month’s duration (Figure 1). Similar red plaques were present on both ears, the left forearm, and the right middle finger. One month following the initial onset as tiny red papules, the lesions had become larger and more prominent after an episode of fever (temperature, 39°C) and immunization against rubella virus and Haemophilus influenzae type b. No therapeutic effect was observed with the application of a topical steroid at the time of presentation. A skin biopsy specimen was taken from a flat plaque on the right cheek. Histopathologic findings revealed dense cell infiltrates predominated by histiocytic cells with some vacuolization in the papillary and upper reticular dermis (Figure 2). Neither foamy histiocytic cells nor giant cells were seen. Immunohistochemical analysis revealed that the cells were CD68+ (Figure 3) and negative for S-100. Following 2 months of observation without any treatment, the plaques on the ears, right forearm, and right middle finger had begun to spontaneously subside. Overall, the remaining lesions showed complete remission by 21 months of age.
|
|
| Figure 1. Asymptomatic red, infiltrated, flat plaques on the left cheek. |
|
|
|
|
| Figure 2. A biopsy revealed an infiltrate of histiocytes with vacuolization of cells in the papillary and upper reticular dermis (A and B) (both H&E, original magnifications ×50 and ×200). |
|
|
| Figure 3. Immunohistochemistry revealed histiocytic infiltrate cells that were CD68+ (original magnification ×50). |
Benign cephalic histiocytosis commonly presents as smooth, round, or flat-topped papules with a diameter of 1 to 8 mm and color ranging from tan-yellow to brown-red.2 The etiology of BCH is unknown. Plaque-type BCH represented by this patient is a rare feature, suggesting sarcoidosis, perniosis, or Sjögren syndrome because the lesions are comprised of infiltrated erythematous plaques rather than papules. Rodriguez-Jurado et al3 described a transitional form of BCH to JXG in a 2-year-old girl with facial plaques after an episode of varicella-zoster virus infection. Vasconcelos et al4 also indicated that JXG is associated with cytomegalovirus infection. These findings support the notion that BCH and non–Langerhans cell histiocytosis are on the same spectrum of diseases, representing a reactive histiocytic process to an infectious agent.
The difference in clinical manifestation of each non–Langerhans cell histiocytosis subgroup may reflect the degree or stage of infection. T lymphocytes may release different cytokines and activate macrophage migration when sensitized by a virus or vaccine. As a result, BCH may show various clinical features even when confirmed by histological analysis. Our patient might have had an early stage of BCH that would transform into JXG.
To the Editor:
Benign cephalic histiocytosis (BCH) is a rare benign dermatosis in which self-healing papular eruptions develop. This condition is classified as non-X histiocytosis and behaves as a benign histiocytic proliferation. Since the first report by Gianotti et al1 in 1971, the etiology and pathophysiology of this condition have not been elucidated. The clinical and histologic features of BCH overlap with juvenile xanthogranuloma (JXG) and generalized eruptive histiocytosis.2 Rodriguez-Jurado et al3 reported that BCH showed clinical evolution to JXG with varicella-zoster infection, suggesting that BCH is a part of a spectrum of diseases that non–Langerhans cell histiocytosis encompasses. We present a case of plaque-type BCH in an infant.
An 11-month-old male infant presented with asymptomatic, ill-defined, infiltrated plaques on the cheeks of 1 month’s duration (Figure 1). Similar red plaques were present on both ears, the left forearm, and the right middle finger. One month following the initial onset as tiny red papules, the lesions had become larger and more prominent after an episode of fever (temperature, 39°C) and immunization against rubella virus and Haemophilus influenzae type b. No therapeutic effect was observed with the application of a topical steroid at the time of presentation. A skin biopsy specimen was taken from a flat plaque on the right cheek. Histopathologic findings revealed dense cell infiltrates predominated by histiocytic cells with some vacuolization in the papillary and upper reticular dermis (Figure 2). Neither foamy histiocytic cells nor giant cells were seen. Immunohistochemical analysis revealed that the cells were CD68+ (Figure 3) and negative for S-100. Following 2 months of observation without any treatment, the plaques on the ears, right forearm, and right middle finger had begun to spontaneously subside. Overall, the remaining lesions showed complete remission by 21 months of age.
|
|
| Figure 1. Asymptomatic red, infiltrated, flat plaques on the left cheek. |
|
|
|
|
| Figure 2. A biopsy revealed an infiltrate of histiocytes with vacuolization of cells in the papillary and upper reticular dermis (A and B) (both H&E, original magnifications ×50 and ×200). |
|
|
| Figure 3. Immunohistochemistry revealed histiocytic infiltrate cells that were CD68+ (original magnification ×50). |
Benign cephalic histiocytosis commonly presents as smooth, round, or flat-topped papules with a diameter of 1 to 8 mm and color ranging from tan-yellow to brown-red.2 The etiology of BCH is unknown. Plaque-type BCH represented by this patient is a rare feature, suggesting sarcoidosis, perniosis, or Sjögren syndrome because the lesions are comprised of infiltrated erythematous plaques rather than papules. Rodriguez-Jurado et al3 described a transitional form of BCH to JXG in a 2-year-old girl with facial plaques after an episode of varicella-zoster virus infection. Vasconcelos et al4 also indicated that JXG is associated with cytomegalovirus infection. These findings support the notion that BCH and non–Langerhans cell histiocytosis are on the same spectrum of diseases, representing a reactive histiocytic process to an infectious agent.
The difference in clinical manifestation of each non–Langerhans cell histiocytosis subgroup may reflect the degree or stage of infection. T lymphocytes may release different cytokines and activate macrophage migration when sensitized by a virus or vaccine. As a result, BCH may show various clinical features even when confirmed by histological analysis. Our patient might have had an early stage of BCH that would transform into JXG.
1. Gianotti F, Caputo R, Ermacora E. Singular “infantile histiocytosis with cells with intracytoplasmic vermiform particles” [in French]. Bull Soc Fr Dermatol Syphiligr. 1971;78:232-233.
2. Jih DM, Salcedo SL, Jaworsky C. Benign cephalic histiocytosis: a case report and review. J Am Acad Dermatol. 2002;47:908-913.
3. Rodriguez-Jurado R, Duran-McKinster C, Ruiz-Maldonado R. Benign cephalic histiocytosis progressing into juvenile xanthogranuloma: a non-Langerhans cell histiocytosis transforming under the influence of a virus? Am J Dermatopathol. 2000;22:70-74.
4. Vasconcelos FO, Oliveira LA, Naves MD, et al. Juvenile xanthogranuloma: case report with immunohistochemical identification of early and late cytomegalovirus antigens. J Oral Sci. 2001;43:21-25.
1. Gianotti F, Caputo R, Ermacora E. Singular “infantile histiocytosis with cells with intracytoplasmic vermiform particles” [in French]. Bull Soc Fr Dermatol Syphiligr. 1971;78:232-233.
2. Jih DM, Salcedo SL, Jaworsky C. Benign cephalic histiocytosis: a case report and review. J Am Acad Dermatol. 2002;47:908-913.
3. Rodriguez-Jurado R, Duran-McKinster C, Ruiz-Maldonado R. Benign cephalic histiocytosis progressing into juvenile xanthogranuloma: a non-Langerhans cell histiocytosis transforming under the influence of a virus? Am J Dermatopathol. 2000;22:70-74.
4. Vasconcelos FO, Oliveira LA, Naves MD, et al. Juvenile xanthogranuloma: case report with immunohistochemical identification of early and late cytomegalovirus antigens. J Oral Sci. 2001;43:21-25.
Pedunculated Scrotal Nodule in an Elderly Male: A Rare Presentation of Trichoblastoma
To the Editor:
A 78-year-old man with no relevant dermatologic history presented for a routine skin examination. He had a large pedunculated nodule on the right scrotum that had been stable in size for more than 40 years. He reported the lesion had been diagnosed as a skin tag by his urologist 20 years prior and had remained asymptomatic in nature. On examination, a 2.5×1-cm, pedunculated, firm, rubbery nodule protruded from the anterior aspect of the right scrotal skin (Figure 1). Prominent telangiectases were noted on the surface of the lesion along with an irregular pigmented macule on the tip. There was no inguinal lymphadenopathy. Given the pigmentation at the tip of the lesion, he was advised to have it completely excised to rule out melanocytic atypia. The lesion subsequently was excised under local anesthesia and the resulting defect was primarily closed. A group of basaloid lobules within the dermis with scant stroma was noted (Figure 2). There was no epidermal connection and retraction artifact was absent. The epidermis appeared normal, except for basilar pigmentation corresponding to the pigmented macule at the tip of the lesion. Higher magnification of one of the basaloid islands demonstrated a papillary mesenchymal body (Figure 3). Immunohistochemistry with cytokeratin 20 showed positive staining of Merkel cells colonizing the basaloid islands (Figure 4). A diagnosis of trichoblastoma was rendered. Two nodular basal cell carcinomas (BCCs) were seen on initial presentation, but he remained free of recurrence of the scrotal tumor at 2-year follow-up.
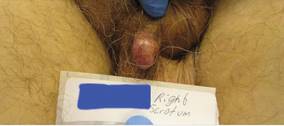
|
Figure 1. Firm, pedunculated, flesh-colored nodule on the scrotum. |
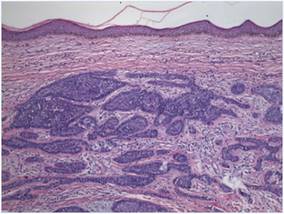
|
Figure 2. Basaloid tumor islands within the dermis with no epidermal connection (H&E, original magnification ×20). |
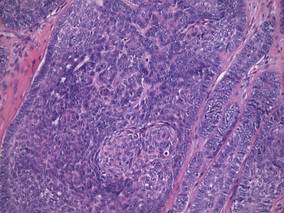
|
Figure 3. Papillary mesenchymal body adjacent to several dermal basaloid islands (H&E, original magnification ×40). |
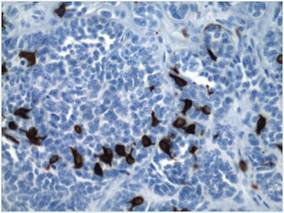
|
Figure 4. Cytokeratin 20 immunohistochemical staining indicated Merkel cells within the basaloid islands (original magnification ×40). |
Trichoblastoma is a rare benign tumor with differentiation toward the primitive hair follicle. The lesion typically presents as a slow-growing, solitary, well-circumscribed nodule that typically measures up to 3 cm and is predominantly located on the head and neck with a predilection for the scalp. Rarely the lesion will appear on the trunk, proximal extremities, or perianal and genital regions.1 Trichoblastomas show no gender predilection and predominantly affect adults between the fifth and seventh decades of life. In children it is the most common neoplasm arising in preexisting nevus sebaceous (Jadassohn nevus).2 Several clinical variants of trichoblastoma have been described, including giant trichoblastoma, pigmented trichoblastoma, and subcutaneous trichoblastoma, among others.3
Histologically, these tumors are characterized by well-circumscribed basaloid islands in the dermis with peripheral palisading closely resembling BCCs and trichoepitheliomas. Unlike BCCs, retraction artifact in trichoblastoma is a rare finding, while papillary mesenchymal bodies and an increased number of Merkel cells characterize both trichoblastomas and trichoepitheliomas, as in our patient.4 Other histologic features include lack of epidermal connection and location in the deeper dermis and/or subcutis. There also are reported cases with sebaceous, pigmented, rippled patterns and clear cell differentiation on histology.3,5
The genetic basis for trichoblastoma remains unclear. Although these primitive hair follicle tumors share histologic features with BCC and trichoepithelioma, which have both demonstrated PTCH (patched) mutations, no classic mutations in the PTCH gene were seen in 11 cases of trichoblastoma.6 In contrast, there have been reports of trichoblastoma occurring in Brooke-Spiegler syndrome and a case of a somatic CYLD (cylindromatosis) frameshift mutation in a trichoblastoma in a patient with Brooke-Spiegler syndrome7; however, the significance of CYLD mutation in thepathogenesis of trichoblastoma has not been fully elucidated.8
Trichoblastomas generally behave in an indolent asymptomatic manner, with malignant transformation being a rare occurrence.9 However, complete excision of these tumors is recommended given the potential for continued slow growth if incompletely removed. In the case of our patient, the lesion was completely excised at the outset. We plan to clinically monitor for evidence of disease recurrence as part of an annual full-body examination and have noted no evidence of recurrence in more than 2 years.
1. Brenn T, McKee PH. Tumors of the hair follicle. In: McKee PH, Calonje E, Granter SR, eds. Pathology of the Skin With Clinical Correlations. 3rd ed. London, England: Elsevier Mosby; 2005:1552-1557.
2. Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceous of Jadassohn: a clinicopathologic study of series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
3. Kim DW, Lee JH, Kim I. Giant melanotrichoblastoma. Am J Dermatopathol. 2011;33:e37-e40.
4. Schulz T, Hartschuh W. Merkel cells are absent in basal cell carcinomas but frequently found in trichoblastomas. an immunohistochemical study. J Cutan Pathol. 1997;24:14-24.
5. Swick BL, Baum CL, Walling HW. Rippled-patterntrichoblastoma with apocrine differentiation arising in a nevus sebaceus: report of a case and review of the literature. J Cutan Pathol. 2009;36:1200-1205.
6. Hafner C, Schmiemann V, Ruetten A, et al. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas [published online ahead of print June 26, 2007]. Hum Pathol. 2007;38:1496-1500.
7. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27-33.
8. Kazakov DV, Schaller J, Vanecek T, et al. Brooke-Spiegler syndrome: report of a case with a novel mutation in the CYLD gene and different types of somatic mutations in benign and malignant tumors [published online ahead of print February 4, 2010]. J Cutan Pathol. 2010;37:886-890.
9. Schulz T, Proske S, Hartschuh W, et al. High-grade trichoblastic carcinoma arising in trichoblastoma: a rare adnexal neoplasm often showing metastatic spread. Am J Dermatopathol. 2005;27:9-16
To the Editor:
A 78-year-old man with no relevant dermatologic history presented for a routine skin examination. He had a large pedunculated nodule on the right scrotum that had been stable in size for more than 40 years. He reported the lesion had been diagnosed as a skin tag by his urologist 20 years prior and had remained asymptomatic in nature. On examination, a 2.5×1-cm, pedunculated, firm, rubbery nodule protruded from the anterior aspect of the right scrotal skin (Figure 1). Prominent telangiectases were noted on the surface of the lesion along with an irregular pigmented macule on the tip. There was no inguinal lymphadenopathy. Given the pigmentation at the tip of the lesion, he was advised to have it completely excised to rule out melanocytic atypia. The lesion subsequently was excised under local anesthesia and the resulting defect was primarily closed. A group of basaloid lobules within the dermis with scant stroma was noted (Figure 2). There was no epidermal connection and retraction artifact was absent. The epidermis appeared normal, except for basilar pigmentation corresponding to the pigmented macule at the tip of the lesion. Higher magnification of one of the basaloid islands demonstrated a papillary mesenchymal body (Figure 3). Immunohistochemistry with cytokeratin 20 showed positive staining of Merkel cells colonizing the basaloid islands (Figure 4). A diagnosis of trichoblastoma was rendered. Two nodular basal cell carcinomas (BCCs) were seen on initial presentation, but he remained free of recurrence of the scrotal tumor at 2-year follow-up.
|
|
Figure 1. Firm, pedunculated, flesh-colored nodule on the scrotum. |
|
|
Figure 2. Basaloid tumor islands within the dermis with no epidermal connection (H&E, original magnification ×20). |
|
|
Figure 3. Papillary mesenchymal body adjacent to several dermal basaloid islands (H&E, original magnification ×40). |
|
|
Figure 4. Cytokeratin 20 immunohistochemical staining indicated Merkel cells within the basaloid islands (original magnification ×40). |
Trichoblastoma is a rare benign tumor with differentiation toward the primitive hair follicle. The lesion typically presents as a slow-growing, solitary, well-circumscribed nodule that typically measures up to 3 cm and is predominantly located on the head and neck with a predilection for the scalp. Rarely the lesion will appear on the trunk, proximal extremities, or perianal and genital regions.1 Trichoblastomas show no gender predilection and predominantly affect adults between the fifth and seventh decades of life. In children it is the most common neoplasm arising in preexisting nevus sebaceous (Jadassohn nevus).2 Several clinical variants of trichoblastoma have been described, including giant trichoblastoma, pigmented trichoblastoma, and subcutaneous trichoblastoma, among others.3
Histologically, these tumors are characterized by well-circumscribed basaloid islands in the dermis with peripheral palisading closely resembling BCCs and trichoepitheliomas. Unlike BCCs, retraction artifact in trichoblastoma is a rare finding, while papillary mesenchymal bodies and an increased number of Merkel cells characterize both trichoblastomas and trichoepitheliomas, as in our patient.4 Other histologic features include lack of epidermal connection and location in the deeper dermis and/or subcutis. There also are reported cases with sebaceous, pigmented, rippled patterns and clear cell differentiation on histology.3,5
The genetic basis for trichoblastoma remains unclear. Although these primitive hair follicle tumors share histologic features with BCC and trichoepithelioma, which have both demonstrated PTCH (patched) mutations, no classic mutations in the PTCH gene were seen in 11 cases of trichoblastoma.6 In contrast, there have been reports of trichoblastoma occurring in Brooke-Spiegler syndrome and a case of a somatic CYLD (cylindromatosis) frameshift mutation in a trichoblastoma in a patient with Brooke-Spiegler syndrome7; however, the significance of CYLD mutation in thepathogenesis of trichoblastoma has not been fully elucidated.8
Trichoblastomas generally behave in an indolent asymptomatic manner, with malignant transformation being a rare occurrence.9 However, complete excision of these tumors is recommended given the potential for continued slow growth if incompletely removed. In the case of our patient, the lesion was completely excised at the outset. We plan to clinically monitor for evidence of disease recurrence as part of an annual full-body examination and have noted no evidence of recurrence in more than 2 years.
To the Editor:
A 78-year-old man with no relevant dermatologic history presented for a routine skin examination. He had a large pedunculated nodule on the right scrotum that had been stable in size for more than 40 years. He reported the lesion had been diagnosed as a skin tag by his urologist 20 years prior and had remained asymptomatic in nature. On examination, a 2.5×1-cm, pedunculated, firm, rubbery nodule protruded from the anterior aspect of the right scrotal skin (Figure 1). Prominent telangiectases were noted on the surface of the lesion along with an irregular pigmented macule on the tip. There was no inguinal lymphadenopathy. Given the pigmentation at the tip of the lesion, he was advised to have it completely excised to rule out melanocytic atypia. The lesion subsequently was excised under local anesthesia and the resulting defect was primarily closed. A group of basaloid lobules within the dermis with scant stroma was noted (Figure 2). There was no epidermal connection and retraction artifact was absent. The epidermis appeared normal, except for basilar pigmentation corresponding to the pigmented macule at the tip of the lesion. Higher magnification of one of the basaloid islands demonstrated a papillary mesenchymal body (Figure 3). Immunohistochemistry with cytokeratin 20 showed positive staining of Merkel cells colonizing the basaloid islands (Figure 4). A diagnosis of trichoblastoma was rendered. Two nodular basal cell carcinomas (BCCs) were seen on initial presentation, but he remained free of recurrence of the scrotal tumor at 2-year follow-up.
|
|
Figure 1. Firm, pedunculated, flesh-colored nodule on the scrotum. |
|
|
Figure 2. Basaloid tumor islands within the dermis with no epidermal connection (H&E, original magnification ×20). |
|
|
Figure 3. Papillary mesenchymal body adjacent to several dermal basaloid islands (H&E, original magnification ×40). |
|
|
Figure 4. Cytokeratin 20 immunohistochemical staining indicated Merkel cells within the basaloid islands (original magnification ×40). |
Trichoblastoma is a rare benign tumor with differentiation toward the primitive hair follicle. The lesion typically presents as a slow-growing, solitary, well-circumscribed nodule that typically measures up to 3 cm and is predominantly located on the head and neck with a predilection for the scalp. Rarely the lesion will appear on the trunk, proximal extremities, or perianal and genital regions.1 Trichoblastomas show no gender predilection and predominantly affect adults between the fifth and seventh decades of life. In children it is the most common neoplasm arising in preexisting nevus sebaceous (Jadassohn nevus).2 Several clinical variants of trichoblastoma have been described, including giant trichoblastoma, pigmented trichoblastoma, and subcutaneous trichoblastoma, among others.3
Histologically, these tumors are characterized by well-circumscribed basaloid islands in the dermis with peripheral palisading closely resembling BCCs and trichoepitheliomas. Unlike BCCs, retraction artifact in trichoblastoma is a rare finding, while papillary mesenchymal bodies and an increased number of Merkel cells characterize both trichoblastomas and trichoepitheliomas, as in our patient.4 Other histologic features include lack of epidermal connection and location in the deeper dermis and/or subcutis. There also are reported cases with sebaceous, pigmented, rippled patterns and clear cell differentiation on histology.3,5
The genetic basis for trichoblastoma remains unclear. Although these primitive hair follicle tumors share histologic features with BCC and trichoepithelioma, which have both demonstrated PTCH (patched) mutations, no classic mutations in the PTCH gene were seen in 11 cases of trichoblastoma.6 In contrast, there have been reports of trichoblastoma occurring in Brooke-Spiegler syndrome and a case of a somatic CYLD (cylindromatosis) frameshift mutation in a trichoblastoma in a patient with Brooke-Spiegler syndrome7; however, the significance of CYLD mutation in thepathogenesis of trichoblastoma has not been fully elucidated.8
Trichoblastomas generally behave in an indolent asymptomatic manner, with malignant transformation being a rare occurrence.9 However, complete excision of these tumors is recommended given the potential for continued slow growth if incompletely removed. In the case of our patient, the lesion was completely excised at the outset. We plan to clinically monitor for evidence of disease recurrence as part of an annual full-body examination and have noted no evidence of recurrence in more than 2 years.
1. Brenn T, McKee PH. Tumors of the hair follicle. In: McKee PH, Calonje E, Granter SR, eds. Pathology of the Skin With Clinical Correlations. 3rd ed. London, England: Elsevier Mosby; 2005:1552-1557.
2. Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceous of Jadassohn: a clinicopathologic study of series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
3. Kim DW, Lee JH, Kim I. Giant melanotrichoblastoma. Am J Dermatopathol. 2011;33:e37-e40.
4. Schulz T, Hartschuh W. Merkel cells are absent in basal cell carcinomas but frequently found in trichoblastomas. an immunohistochemical study. J Cutan Pathol. 1997;24:14-24.
5. Swick BL, Baum CL, Walling HW. Rippled-patterntrichoblastoma with apocrine differentiation arising in a nevus sebaceus: report of a case and review of the literature. J Cutan Pathol. 2009;36:1200-1205.
6. Hafner C, Schmiemann V, Ruetten A, et al. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas [published online ahead of print June 26, 2007]. Hum Pathol. 2007;38:1496-1500.
7. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27-33.
8. Kazakov DV, Schaller J, Vanecek T, et al. Brooke-Spiegler syndrome: report of a case with a novel mutation in the CYLD gene and different types of somatic mutations in benign and malignant tumors [published online ahead of print February 4, 2010]. J Cutan Pathol. 2010;37:886-890.
9. Schulz T, Proske S, Hartschuh W, et al. High-grade trichoblastic carcinoma arising in trichoblastoma: a rare adnexal neoplasm often showing metastatic spread. Am J Dermatopathol. 2005;27:9-16
1. Brenn T, McKee PH. Tumors of the hair follicle. In: McKee PH, Calonje E, Granter SR, eds. Pathology of the Skin With Clinical Correlations. 3rd ed. London, England: Elsevier Mosby; 2005:1552-1557.
2. Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceous of Jadassohn: a clinicopathologic study of series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
3. Kim DW, Lee JH, Kim I. Giant melanotrichoblastoma. Am J Dermatopathol. 2011;33:e37-e40.
4. Schulz T, Hartschuh W. Merkel cells are absent in basal cell carcinomas but frequently found in trichoblastomas. an immunohistochemical study. J Cutan Pathol. 1997;24:14-24.
5. Swick BL, Baum CL, Walling HW. Rippled-patterntrichoblastoma with apocrine differentiation arising in a nevus sebaceus: report of a case and review of the literature. J Cutan Pathol. 2009;36:1200-1205.
6. Hafner C, Schmiemann V, Ruetten A, et al. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas [published online ahead of print June 26, 2007]. Hum Pathol. 2007;38:1496-1500.
7. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27-33.
8. Kazakov DV, Schaller J, Vanecek T, et al. Brooke-Spiegler syndrome: report of a case with a novel mutation in the CYLD gene and different types of somatic mutations in benign and malignant tumors [published online ahead of print February 4, 2010]. J Cutan Pathol. 2010;37:886-890.
9. Schulz T, Proske S, Hartschuh W, et al. High-grade trichoblastic carcinoma arising in trichoblastoma: a rare adnexal neoplasm often showing metastatic spread. Am J Dermatopathol. 2005;27:9-16
Lepromatous Leprosy Associated With Erythema Nodosum Leprosum
To the Editor:
A 41-year-old man presented to our clinic with concerns of ulceration, skin thickening, and loss of eyebrows. The ulceration developed on the knees approximately 1 year prior to presentation; however, he reported loss of sensation in the knees after an accident involving his back 7 years prior. Loss of the eyebrows and skin thickening on the abdomen occurred within the same time frame of the knee ulceration. He also experienced generalized loss of skin sensation. The patient had a history of hunting and fishing but denied any contact with an armadillo.
On clinical examination thin yellow plaques were present on both cheeks, as well as typical leonine facies, a saddle nose deformity, loss of bilateral eyebrows, slate gray color on the face, and infiltrated plaques with telangiectases on the nose. Subcutaneous nodules developed on the arms, chest, and abdomen. He also had angulated ulcers with granulation tissue on the knees.
Laboratory data collected included a complete metabolic panel and complete blood cell count with differential. The complete metabolic panel was within reference range excluding a moderately high glucose level of 114 mg/dL (reference range, 70–100 mg/dL) and a low creatinine level of 0.7 mg/dL (reference range, 0.5–1.2 mg/dL). The complete blood cell count showed mild anemia with a hemoglobin to hematocrit ratio of 11.0 g/dL (reference range, 12.0–16.0 g/dL) to 32.6% (reference range, 37.0%–48.5%) and a nonspecific elevated mean corpuscular volume of 94.4 fL (reference range, 82–95 fL). The differential revealed a low percentage of lymphocytes (11.4% [reference range, 21%–44%]) and a high percentage of granulocytes (85.6% [reference range, 38%–73%]).
A total of 3 biopsies were taken from the abdomen, upper chest, and right wrist. Histology showed a diffuse infiltrate of epithelioid histiocytes forming ill-defined nodules in the dermis, extending into the represented subcutis (Figure 1). Many of the histiocytes had prominent foamy cytoplasms (Figure 2). Scattered lymphocytes and plasma cells also were present. Fite-modified acid-fast staining was performed on all of the biopsies and yielded large numbers of acid-fast bacilli dispersed throughout the specimens (Figure 3), which confirmed the diagnosis of lepromatous leprosy. The patient was referred to the National Hansen’s Disease (Leprosy) Program in Baton Rouge, Louisiana. The physician team started him on high-dose clofazimine, dapsone, and rifampin. He also was diagnosed with erythema nodosum leprosum for which he was given thalidomide.
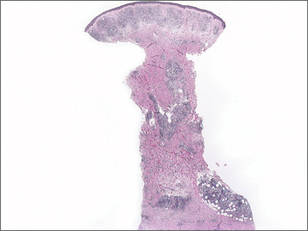
Hansen disease, also known as leprosy, is a chronic inflammatory disease caused by Mycobacterium leprae and Mycobacterium lepromatosis. The mode of transmission is postulated to be through respiratory droplets and nasal secretions. Armadillo exposure, poor sanitary conditions, endemic location, and infected family members are considered risk factors for the development of leprosy. The incubation period is approximately 5 years, with symptoms arising up to 20 years after acquisition of the bacillus. Replication tends to begin in cooler regions of the body. The disease spectrum ranges from lepromatous to tuberculoid.1-3 Lepromatous leprosy consists of an uncontrolled high-titer replication of the mycobacteria, resulting mostly in cutaneous changes and late nerve damage. Histologically, foamy or undifferentiated macrophages predominate the field; they often include many bacilli that can be demonstrated by the modified Fite-Faraco method, a clinically relevant carbol-fuchsin stain.3-5 Acid-fast organisms stain red. A Ziehl-Neelsen stain and Harada modified Allochrome method also may be used.
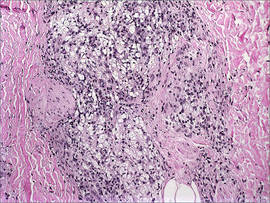
The damage to peripheral nerves and the cutis can cause substantial loss of function and lead to painless ulceration of the skin. Changes in the skin may include nodules, hypopigmented macules, sores, and skin thickening. Leonine facies and disfigurement also may be observed. A type III hypersensitivity reaction may occur, resulting in erythema nodosum leprosum, a type II lepra reaction characterized by elevated tumor necrosis factor α.6
Treatment requires multidrug therapy involving dapsone, rifampin, and clofazimine.4 The World Health Organization is targeted at eliminating the disease and providing free multidrug therapy to patients.7

- Leprosy (Hansen’s disease): Technical Information. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/nczved/divisions/dfbmd/diseases/hansens_disease/technical.html/. Updated May 17, 2010. Accessed July 2, 2014.
- Esfandbod M. Images in clinical medicine. tuberculoid leprosy. N Engl J Med. 2011;364:1657.
- Robati RM, Rahimi H, Asadi-Kani Z, et al. Photoclinic. lepromatous leprosy. Arch Iran Med. 2010;13:443-444.
- Moschella SL. An update on the diagnosis and treatment of leprosy. J Am Acad Dermatol. 2004;51:417-426.
- Wolff K, Johnson RA, Saavedra AP, eds. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013.
- Walker SL, Lockwood DN. Leprosy. Clin Dermatol. 2007;25:165-172.
- World Health Organization Committee on Leprosy. WHO Expert Committee on Leprosy: Seventh Report. Geneva, Switzerland: World Health Organization Committee on Leprosy; 1997.
To the Editor:
A 41-year-old man presented to our clinic with concerns of ulceration, skin thickening, and loss of eyebrows. The ulceration developed on the knees approximately 1 year prior to presentation; however, he reported loss of sensation in the knees after an accident involving his back 7 years prior. Loss of the eyebrows and skin thickening on the abdomen occurred within the same time frame of the knee ulceration. He also experienced generalized loss of skin sensation. The patient had a history of hunting and fishing but denied any contact with an armadillo.
On clinical examination thin yellow plaques were present on both cheeks, as well as typical leonine facies, a saddle nose deformity, loss of bilateral eyebrows, slate gray color on the face, and infiltrated plaques with telangiectases on the nose. Subcutaneous nodules developed on the arms, chest, and abdomen. He also had angulated ulcers with granulation tissue on the knees.
Laboratory data collected included a complete metabolic panel and complete blood cell count with differential. The complete metabolic panel was within reference range excluding a moderately high glucose level of 114 mg/dL (reference range, 70–100 mg/dL) and a low creatinine level of 0.7 mg/dL (reference range, 0.5–1.2 mg/dL). The complete blood cell count showed mild anemia with a hemoglobin to hematocrit ratio of 11.0 g/dL (reference range, 12.0–16.0 g/dL) to 32.6% (reference range, 37.0%–48.5%) and a nonspecific elevated mean corpuscular volume of 94.4 fL (reference range, 82–95 fL). The differential revealed a low percentage of lymphocytes (11.4% [reference range, 21%–44%]) and a high percentage of granulocytes (85.6% [reference range, 38%–73%]).
A total of 3 biopsies were taken from the abdomen, upper chest, and right wrist. Histology showed a diffuse infiltrate of epithelioid histiocytes forming ill-defined nodules in the dermis, extending into the represented subcutis (Figure 1). Many of the histiocytes had prominent foamy cytoplasms (Figure 2). Scattered lymphocytes and plasma cells also were present. Fite-modified acid-fast staining was performed on all of the biopsies and yielded large numbers of acid-fast bacilli dispersed throughout the specimens (Figure 3), which confirmed the diagnosis of lepromatous leprosy. The patient was referred to the National Hansen’s Disease (Leprosy) Program in Baton Rouge, Louisiana. The physician team started him on high-dose clofazimine, dapsone, and rifampin. He also was diagnosed with erythema nodosum leprosum for which he was given thalidomide.
Hansen disease, also known as leprosy, is a chronic inflammatory disease caused by Mycobacterium leprae and Mycobacterium lepromatosis. The mode of transmission is postulated to be through respiratory droplets and nasal secretions. Armadillo exposure, poor sanitary conditions, endemic location, and infected family members are considered risk factors for the development of leprosy. The incubation period is approximately 5 years, with symptoms arising up to 20 years after acquisition of the bacillus. Replication tends to begin in cooler regions of the body. The disease spectrum ranges from lepromatous to tuberculoid.1-3 Lepromatous leprosy consists of an uncontrolled high-titer replication of the mycobacteria, resulting mostly in cutaneous changes and late nerve damage. Histologically, foamy or undifferentiated macrophages predominate the field; they often include many bacilli that can be demonstrated by the modified Fite-Faraco method, a clinically relevant carbol-fuchsin stain.3-5 Acid-fast organisms stain red. A Ziehl-Neelsen stain and Harada modified Allochrome method also may be used.
The damage to peripheral nerves and the cutis can cause substantial loss of function and lead to painless ulceration of the skin. Changes in the skin may include nodules, hypopigmented macules, sores, and skin thickening. Leonine facies and disfigurement also may be observed. A type III hypersensitivity reaction may occur, resulting in erythema nodosum leprosum, a type II lepra reaction characterized by elevated tumor necrosis factor α.6
Treatment requires multidrug therapy involving dapsone, rifampin, and clofazimine.4 The World Health Organization is targeted at eliminating the disease and providing free multidrug therapy to patients.7
To the Editor:
A 41-year-old man presented to our clinic with concerns of ulceration, skin thickening, and loss of eyebrows. The ulceration developed on the knees approximately 1 year prior to presentation; however, he reported loss of sensation in the knees after an accident involving his back 7 years prior. Loss of the eyebrows and skin thickening on the abdomen occurred within the same time frame of the knee ulceration. He also experienced generalized loss of skin sensation. The patient had a history of hunting and fishing but denied any contact with an armadillo.
On clinical examination thin yellow plaques were present on both cheeks, as well as typical leonine facies, a saddle nose deformity, loss of bilateral eyebrows, slate gray color on the face, and infiltrated plaques with telangiectases on the nose. Subcutaneous nodules developed on the arms, chest, and abdomen. He also had angulated ulcers with granulation tissue on the knees.
Laboratory data collected included a complete metabolic panel and complete blood cell count with differential. The complete metabolic panel was within reference range excluding a moderately high glucose level of 114 mg/dL (reference range, 70–100 mg/dL) and a low creatinine level of 0.7 mg/dL (reference range, 0.5–1.2 mg/dL). The complete blood cell count showed mild anemia with a hemoglobin to hematocrit ratio of 11.0 g/dL (reference range, 12.0–16.0 g/dL) to 32.6% (reference range, 37.0%–48.5%) and a nonspecific elevated mean corpuscular volume of 94.4 fL (reference range, 82–95 fL). The differential revealed a low percentage of lymphocytes (11.4% [reference range, 21%–44%]) and a high percentage of granulocytes (85.6% [reference range, 38%–73%]).
A total of 3 biopsies were taken from the abdomen, upper chest, and right wrist. Histology showed a diffuse infiltrate of epithelioid histiocytes forming ill-defined nodules in the dermis, extending into the represented subcutis (Figure 1). Many of the histiocytes had prominent foamy cytoplasms (Figure 2). Scattered lymphocytes and plasma cells also were present. Fite-modified acid-fast staining was performed on all of the biopsies and yielded large numbers of acid-fast bacilli dispersed throughout the specimens (Figure 3), which confirmed the diagnosis of lepromatous leprosy. The patient was referred to the National Hansen’s Disease (Leprosy) Program in Baton Rouge, Louisiana. The physician team started him on high-dose clofazimine, dapsone, and rifampin. He also was diagnosed with erythema nodosum leprosum for which he was given thalidomide.
Hansen disease, also known as leprosy, is a chronic inflammatory disease caused by Mycobacterium leprae and Mycobacterium lepromatosis. The mode of transmission is postulated to be through respiratory droplets and nasal secretions. Armadillo exposure, poor sanitary conditions, endemic location, and infected family members are considered risk factors for the development of leprosy. The incubation period is approximately 5 years, with symptoms arising up to 20 years after acquisition of the bacillus. Replication tends to begin in cooler regions of the body. The disease spectrum ranges from lepromatous to tuberculoid.1-3 Lepromatous leprosy consists of an uncontrolled high-titer replication of the mycobacteria, resulting mostly in cutaneous changes and late nerve damage. Histologically, foamy or undifferentiated macrophages predominate the field; they often include many bacilli that can be demonstrated by the modified Fite-Faraco method, a clinically relevant carbol-fuchsin stain.3-5 Acid-fast organisms stain red. A Ziehl-Neelsen stain and Harada modified Allochrome method also may be used.
The damage to peripheral nerves and the cutis can cause substantial loss of function and lead to painless ulceration of the skin. Changes in the skin may include nodules, hypopigmented macules, sores, and skin thickening. Leonine facies and disfigurement also may be observed. A type III hypersensitivity reaction may occur, resulting in erythema nodosum leprosum, a type II lepra reaction characterized by elevated tumor necrosis factor α.6
Treatment requires multidrug therapy involving dapsone, rifampin, and clofazimine.4 The World Health Organization is targeted at eliminating the disease and providing free multidrug therapy to patients.7
- Leprosy (Hansen’s disease): Technical Information. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/nczved/divisions/dfbmd/diseases/hansens_disease/technical.html/. Updated May 17, 2010. Accessed July 2, 2014.
- Esfandbod M. Images in clinical medicine. tuberculoid leprosy. N Engl J Med. 2011;364:1657.
- Robati RM, Rahimi H, Asadi-Kani Z, et al. Photoclinic. lepromatous leprosy. Arch Iran Med. 2010;13:443-444.
- Moschella SL. An update on the diagnosis and treatment of leprosy. J Am Acad Dermatol. 2004;51:417-426.
- Wolff K, Johnson RA, Saavedra AP, eds. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013.
- Walker SL, Lockwood DN. Leprosy. Clin Dermatol. 2007;25:165-172.
- World Health Organization Committee on Leprosy. WHO Expert Committee on Leprosy: Seventh Report. Geneva, Switzerland: World Health Organization Committee on Leprosy; 1997.
- Leprosy (Hansen’s disease): Technical Information. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/nczved/divisions/dfbmd/diseases/hansens_disease/technical.html/. Updated May 17, 2010. Accessed July 2, 2014.
- Esfandbod M. Images in clinical medicine. tuberculoid leprosy. N Engl J Med. 2011;364:1657.
- Robati RM, Rahimi H, Asadi-Kani Z, et al. Photoclinic. lepromatous leprosy. Arch Iran Med. 2010;13:443-444.
- Moschella SL. An update on the diagnosis and treatment of leprosy. J Am Acad Dermatol. 2004;51:417-426.
- Wolff K, Johnson RA, Saavedra AP, eds. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013.
- Walker SL, Lockwood DN. Leprosy. Clin Dermatol. 2007;25:165-172.
- World Health Organization Committee on Leprosy. WHO Expert Committee on Leprosy: Seventh Report. Geneva, Switzerland: World Health Organization Committee on Leprosy; 1997.