User login
Aspergillus nidulans Causing Primary Cutaneous Aspergillosis in an Immunocompetent Patient
To the Editor:
Cutaneous aspergillosis mostly has been reported in immunosuppressed hosts and usually is caused by Aspergillus flavus or Aspergillus fumigatus. We report the occurrence of primary cutaneous aspergillosis (PCA) caused by a relatively rare species, Aspergillus nidulans, in a middle-aged patient without overt immunosuppression or history of trauma.
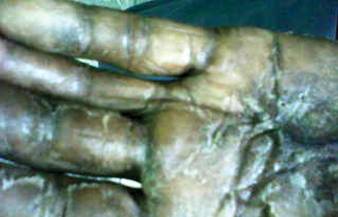
A 57-year-old woman was referred to the dermatology outpatient department for evaluation of a lesion on the right hand of 1 month's duration. On examination the lesion measured approximately 4×3 cm with central necrosis (Figure 1). Her medical history was unremarkable and routine laboratory test results were within reference range.

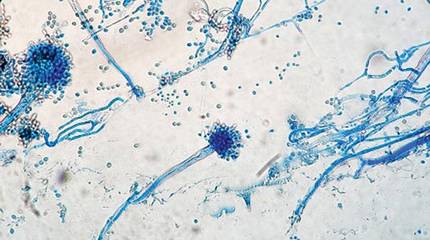
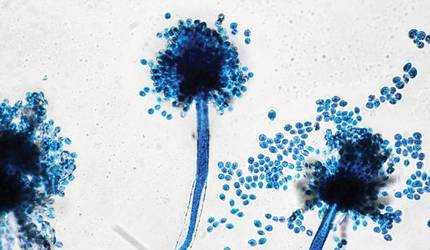
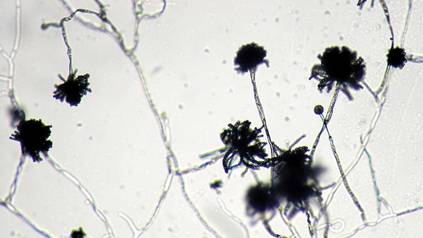
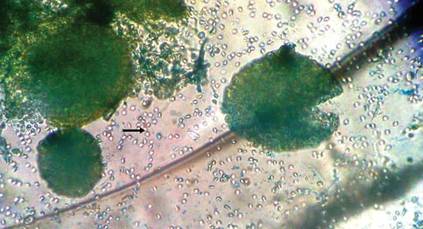
The patient was an agricultural worker with no history of trauma. Her history was unremarkable. A 20% potassium hydroxide mount of the tissue revealed septate, branched, hyaline hyphae. A soft, wooly, greenish brown growth was observed after 3 days of incubation on Sabouraud dextrose agar (Figure 2). No growth was observed on dermatophyte test medium. A lactophenol cotton blue mount revealed columnar conidial heads with brown, short, smooth-walled conidiophores (Figures 3–6). Vesicles were hemispheric and small (8–12 µm in diameter), with metulae and phialides occurring in the upper portion. Conidia were globose (3–4 µm) and rough. Based on these findings the fungus was identified as A nidulans. The patient did not respond to daily oral ketoconazole, and after 1 month of therapy the lesion did not regress. She was eventually treated with oral itraconazole and the lesion completely healed within 15 weeks.
An overwhelming majority of the cases of cutaneous aspergillosis have been reported either in immunocompromised hosts (ie, leukemia, cutaneous T-cell lymphoma, Hodgkin disease, human immunodeficiency virus/AIDS, solid-organ or hematopoietic stem cell transplant recipients) or in patients with contributing risk factors (ie, severe burns, diabetes mellitus, preterm or underweight neonates, elderly patients). Two outbreaks of this condition have been reported in neonatal intensive care units, with the source of contamination being linked to nonsterile disposable gloves, incubators, and humidity chambers.1,2 However, PCA is a relatively rare condition and often is associated with disruption of dermal integrity by trauma or maceration, followed by colonization of the wound by Aspergillus spores that are ubiquitously present in soil and decomposed vegetation.3-5 Our case was remarkable, as the patient was not immunosuppressed and did not have a history of trauma. However, we surmise that fungal inoculation might have inadvertently occurred through some trivial trauma sustained through her professional work.
The 2 species that have most commonly been associated with PCA are A flavus and A fumigatus.6,7 There have been isolated reports of PCA caused by other organisms such as Aspergillus niger,8,9 Aspergillus terreus,10Aspergillus ustus,11 or Aspergillus calidoustus.12 In a report of a neutropenic 56-year-old patient suffering from acute myeloblastic leukemia, PCA developed in association with a double-lumen Hickman catheter after a period of prolonged hospitalization.13 A study by the National Institutes of Health (1976-1997) revealed 6 life-threatening cases of A nidulans infection in patients with chronic granulomatous disease.14
We did not perform antifungal susceptibility testing on the isolate in our patient. However, we observed disease that was refractory to ketoconazole therapy but successfully resolved with oral itraconazole. Antifungal susceptibility was noted in a large number of reported cases of Aspergillus infections that were resistant to conventional treatment, such as voriconazole, itraconazole, and amphotericin B.15 Thus antifungal susceptibility testing is necessary before starting treatment. There also have been reports of recurrence of cutaneous aspergillosis following incomplete and irregular treatment.16 Our case of PCA also failed to respond to ketoconazole therapy, thus stressing the need for thorough mycological characterization, including the determination of an antifungal susceptibility profile, for successful and complete management of this condition.
Acknowledgment
The authors would like to thank Arunaloke Chakraborti, MD, Chandigarh, India, for the help extended for identification of the fungus.
- Stock C, Veyrier M, Raberin H, et al. Severe cutaneous aspergillosis in a premature neonate linked to nonsterile disposable glove contamination [published online ahead of print August 31, 2011]? Am J Infect Control. 2012;40:465-467.
- Etienne KA, Subudhi CP, Chadwick PR, et al. Investigation of a cluster of cutaneous aspergillosis in a neonatal intensive care unit [published online ahead of print August 12, 2011]. J Hosp Infect. 2011;79:344-348.
- Isaac M. Cutaneous aspergillosis. Dermatol Clin. 1996;14:137-140.
- Cahill KM, Mofty AM, Kawaguchi TP. Primary cutaneous aspergillosis. Arch Dermatol. 1967;96:545-547.
- Carlile JR, Millet RE, Cho CT, et al. Primary cutaneous aspergillosis in a leukemic child. Arch Dermatol. 1978;114:78-80.
- John PU, Shadomy HJ. Deep fungal infections. In: Fitzpatrick TB, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. New York, NY: McGraw Hill; 1987:2266-2268.
- Chakrabarti A, Gupta V, Biswas G, et al. Primary cutaneous aspergillosis: our experience in 10 years. J Infect. 1998;37:24-27.
- Robinson A, Fien S, Grassi MA. Nonhealing scalp wound infected with Aspergillus niger in an elderly patient. Cutis. 2011;87:197-200.
- Thomas LM, Rand HK, Miller JL, et al. Primary cutaneous aspergillosis in a patient with a solid organ transplant: case report and review of the literature. Cutis. 2008;81:127-130.
- Yuanjie Z, Jingxia D, Hai W, et al. Primary cutaneous aspergillosis in a patient with cutaneous T-cell lymphoma [published online ahead of print October 22, 2008]. Mycoses. 2009;52:462-464.
- Krishnan-Natesan S, Chandrasekar PH, Manavathu EK, et al. Successful treatment of primary cutaneous Aspergillus ustus infection with surgical debridement and a combination of voriconazole and terbinafine [published online ahead of print October 7, 2008]. Diagn Microbiol Infect Dis. 2008;62:443-446.
- Sato Y, Suzino K, Suzuki A, et al. Case of primary cutaneous Aspergillus calidoustus infection caused by nerve block therapy [in Japanese]. Med Mycol J. 2011;52:239-244.
- Lucas GM, Tucker P, Merz WG. Primary cutaneous Aspergillus nidulans infection associated with a Hickman catheter in a patient with neutropenia. Clin Infect Dis. 1999;29:1594-1596.
- Segal BH, DeCarlo ES, Kwon-Chung KJ, et al. Aspergillus nidulans infection in chronic granulomatous disease. Medicine (Baltimore). 1998;77:345-354.
- Woodruff CA, Hebert AA. Neonatal primary cutaneous aspergillosis: case report and review of the literature. Pediatr Dermatol. 2002;19:439-444.
- Mohapatra S, Xess I, Swetha JV, et al. Primary cutaneous aspergillosis due to Aspergillus niger in an immunocompetent patient. Indian J Med Microbiol. 2009;27:367-370.
To the Editor:
Cutaneous aspergillosis mostly has been reported in immunosuppressed hosts and usually is caused by Aspergillus flavus or Aspergillus fumigatus. We report the occurrence of primary cutaneous aspergillosis (PCA) caused by a relatively rare species, Aspergillus nidulans, in a middle-aged patient without overt immunosuppression or history of trauma.
A 57-year-old woman was referred to the dermatology outpatient department for evaluation of a lesion on the right hand of 1 month's duration. On examination the lesion measured approximately 4×3 cm with central necrosis (Figure 1). Her medical history was unremarkable and routine laboratory test results were within reference range.
The patient was an agricultural worker with no history of trauma. Her history was unremarkable. A 20% potassium hydroxide mount of the tissue revealed septate, branched, hyaline hyphae. A soft, wooly, greenish brown growth was observed after 3 days of incubation on Sabouraud dextrose agar (Figure 2). No growth was observed on dermatophyte test medium. A lactophenol cotton blue mount revealed columnar conidial heads with brown, short, smooth-walled conidiophores (Figures 3–6). Vesicles were hemispheric and small (8–12 µm in diameter), with metulae and phialides occurring in the upper portion. Conidia were globose (3–4 µm) and rough. Based on these findings the fungus was identified as A nidulans. The patient did not respond to daily oral ketoconazole, and after 1 month of therapy the lesion did not regress. She was eventually treated with oral itraconazole and the lesion completely healed within 15 weeks.
An overwhelming majority of the cases of cutaneous aspergillosis have been reported either in immunocompromised hosts (ie, leukemia, cutaneous T-cell lymphoma, Hodgkin disease, human immunodeficiency virus/AIDS, solid-organ or hematopoietic stem cell transplant recipients) or in patients with contributing risk factors (ie, severe burns, diabetes mellitus, preterm or underweight neonates, elderly patients). Two outbreaks of this condition have been reported in neonatal intensive care units, with the source of contamination being linked to nonsterile disposable gloves, incubators, and humidity chambers.1,2 However, PCA is a relatively rare condition and often is associated with disruption of dermal integrity by trauma or maceration, followed by colonization of the wound by Aspergillus spores that are ubiquitously present in soil and decomposed vegetation.3-5 Our case was remarkable, as the patient was not immunosuppressed and did not have a history of trauma. However, we surmise that fungal inoculation might have inadvertently occurred through some trivial trauma sustained through her professional work.
The 2 species that have most commonly been associated with PCA are A flavus and A fumigatus.6,7 There have been isolated reports of PCA caused by other organisms such as Aspergillus niger,8,9 Aspergillus terreus,10Aspergillus ustus,11 or Aspergillus calidoustus.12 In a report of a neutropenic 56-year-old patient suffering from acute myeloblastic leukemia, PCA developed in association with a double-lumen Hickman catheter after a period of prolonged hospitalization.13 A study by the National Institutes of Health (1976-1997) revealed 6 life-threatening cases of A nidulans infection in patients with chronic granulomatous disease.14
We did not perform antifungal susceptibility testing on the isolate in our patient. However, we observed disease that was refractory to ketoconazole therapy but successfully resolved with oral itraconazole. Antifungal susceptibility was noted in a large number of reported cases of Aspergillus infections that were resistant to conventional treatment, such as voriconazole, itraconazole, and amphotericin B.15 Thus antifungal susceptibility testing is necessary before starting treatment. There also have been reports of recurrence of cutaneous aspergillosis following incomplete and irregular treatment.16 Our case of PCA also failed to respond to ketoconazole therapy, thus stressing the need for thorough mycological characterization, including the determination of an antifungal susceptibility profile, for successful and complete management of this condition.
Acknowledgment
The authors would like to thank Arunaloke Chakraborti, MD, Chandigarh, India, for the help extended for identification of the fungus.
To the Editor:
Cutaneous aspergillosis mostly has been reported in immunosuppressed hosts and usually is caused by Aspergillus flavus or Aspergillus fumigatus. We report the occurrence of primary cutaneous aspergillosis (PCA) caused by a relatively rare species, Aspergillus nidulans, in a middle-aged patient without overt immunosuppression or history of trauma.
A 57-year-old woman was referred to the dermatology outpatient department for evaluation of a lesion on the right hand of 1 month's duration. On examination the lesion measured approximately 4×3 cm with central necrosis (Figure 1). Her medical history was unremarkable and routine laboratory test results were within reference range.
The patient was an agricultural worker with no history of trauma. Her history was unremarkable. A 20% potassium hydroxide mount of the tissue revealed septate, branched, hyaline hyphae. A soft, wooly, greenish brown growth was observed after 3 days of incubation on Sabouraud dextrose agar (Figure 2). No growth was observed on dermatophyte test medium. A lactophenol cotton blue mount revealed columnar conidial heads with brown, short, smooth-walled conidiophores (Figures 3–6). Vesicles were hemispheric and small (8–12 µm in diameter), with metulae and phialides occurring in the upper portion. Conidia were globose (3–4 µm) and rough. Based on these findings the fungus was identified as A nidulans. The patient did not respond to daily oral ketoconazole, and after 1 month of therapy the lesion did not regress. She was eventually treated with oral itraconazole and the lesion completely healed within 15 weeks.
An overwhelming majority of the cases of cutaneous aspergillosis have been reported either in immunocompromised hosts (ie, leukemia, cutaneous T-cell lymphoma, Hodgkin disease, human immunodeficiency virus/AIDS, solid-organ or hematopoietic stem cell transplant recipients) or in patients with contributing risk factors (ie, severe burns, diabetes mellitus, preterm or underweight neonates, elderly patients). Two outbreaks of this condition have been reported in neonatal intensive care units, with the source of contamination being linked to nonsterile disposable gloves, incubators, and humidity chambers.1,2 However, PCA is a relatively rare condition and often is associated with disruption of dermal integrity by trauma or maceration, followed by colonization of the wound by Aspergillus spores that are ubiquitously present in soil and decomposed vegetation.3-5 Our case was remarkable, as the patient was not immunosuppressed and did not have a history of trauma. However, we surmise that fungal inoculation might have inadvertently occurred through some trivial trauma sustained through her professional work.
The 2 species that have most commonly been associated with PCA are A flavus and A fumigatus.6,7 There have been isolated reports of PCA caused by other organisms such as Aspergillus niger,8,9 Aspergillus terreus,10Aspergillus ustus,11 or Aspergillus calidoustus.12 In a report of a neutropenic 56-year-old patient suffering from acute myeloblastic leukemia, PCA developed in association with a double-lumen Hickman catheter after a period of prolonged hospitalization.13 A study by the National Institutes of Health (1976-1997) revealed 6 life-threatening cases of A nidulans infection in patients with chronic granulomatous disease.14
We did not perform antifungal susceptibility testing on the isolate in our patient. However, we observed disease that was refractory to ketoconazole therapy but successfully resolved with oral itraconazole. Antifungal susceptibility was noted in a large number of reported cases of Aspergillus infections that were resistant to conventional treatment, such as voriconazole, itraconazole, and amphotericin B.15 Thus antifungal susceptibility testing is necessary before starting treatment. There also have been reports of recurrence of cutaneous aspergillosis following incomplete and irregular treatment.16 Our case of PCA also failed to respond to ketoconazole therapy, thus stressing the need for thorough mycological characterization, including the determination of an antifungal susceptibility profile, for successful and complete management of this condition.
Acknowledgment
The authors would like to thank Arunaloke Chakraborti, MD, Chandigarh, India, for the help extended for identification of the fungus.
- Stock C, Veyrier M, Raberin H, et al. Severe cutaneous aspergillosis in a premature neonate linked to nonsterile disposable glove contamination [published online ahead of print August 31, 2011]? Am J Infect Control. 2012;40:465-467.
- Etienne KA, Subudhi CP, Chadwick PR, et al. Investigation of a cluster of cutaneous aspergillosis in a neonatal intensive care unit [published online ahead of print August 12, 2011]. J Hosp Infect. 2011;79:344-348.
- Isaac M. Cutaneous aspergillosis. Dermatol Clin. 1996;14:137-140.
- Cahill KM, Mofty AM, Kawaguchi TP. Primary cutaneous aspergillosis. Arch Dermatol. 1967;96:545-547.
- Carlile JR, Millet RE, Cho CT, et al. Primary cutaneous aspergillosis in a leukemic child. Arch Dermatol. 1978;114:78-80.
- John PU, Shadomy HJ. Deep fungal infections. In: Fitzpatrick TB, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. New York, NY: McGraw Hill; 1987:2266-2268.
- Chakrabarti A, Gupta V, Biswas G, et al. Primary cutaneous aspergillosis: our experience in 10 years. J Infect. 1998;37:24-27.
- Robinson A, Fien S, Grassi MA. Nonhealing scalp wound infected with Aspergillus niger in an elderly patient. Cutis. 2011;87:197-200.
- Thomas LM, Rand HK, Miller JL, et al. Primary cutaneous aspergillosis in a patient with a solid organ transplant: case report and review of the literature. Cutis. 2008;81:127-130.
- Yuanjie Z, Jingxia D, Hai W, et al. Primary cutaneous aspergillosis in a patient with cutaneous T-cell lymphoma [published online ahead of print October 22, 2008]. Mycoses. 2009;52:462-464.
- Krishnan-Natesan S, Chandrasekar PH, Manavathu EK, et al. Successful treatment of primary cutaneous Aspergillus ustus infection with surgical debridement and a combination of voriconazole and terbinafine [published online ahead of print October 7, 2008]. Diagn Microbiol Infect Dis. 2008;62:443-446.
- Sato Y, Suzino K, Suzuki A, et al. Case of primary cutaneous Aspergillus calidoustus infection caused by nerve block therapy [in Japanese]. Med Mycol J. 2011;52:239-244.
- Lucas GM, Tucker P, Merz WG. Primary cutaneous Aspergillus nidulans infection associated with a Hickman catheter in a patient with neutropenia. Clin Infect Dis. 1999;29:1594-1596.
- Segal BH, DeCarlo ES, Kwon-Chung KJ, et al. Aspergillus nidulans infection in chronic granulomatous disease. Medicine (Baltimore). 1998;77:345-354.
- Woodruff CA, Hebert AA. Neonatal primary cutaneous aspergillosis: case report and review of the literature. Pediatr Dermatol. 2002;19:439-444.
- Mohapatra S, Xess I, Swetha JV, et al. Primary cutaneous aspergillosis due to Aspergillus niger in an immunocompetent patient. Indian J Med Microbiol. 2009;27:367-370.
- Stock C, Veyrier M, Raberin H, et al. Severe cutaneous aspergillosis in a premature neonate linked to nonsterile disposable glove contamination [published online ahead of print August 31, 2011]? Am J Infect Control. 2012;40:465-467.
- Etienne KA, Subudhi CP, Chadwick PR, et al. Investigation of a cluster of cutaneous aspergillosis in a neonatal intensive care unit [published online ahead of print August 12, 2011]. J Hosp Infect. 2011;79:344-348.
- Isaac M. Cutaneous aspergillosis. Dermatol Clin. 1996;14:137-140.
- Cahill KM, Mofty AM, Kawaguchi TP. Primary cutaneous aspergillosis. Arch Dermatol. 1967;96:545-547.
- Carlile JR, Millet RE, Cho CT, et al. Primary cutaneous aspergillosis in a leukemic child. Arch Dermatol. 1978;114:78-80.
- John PU, Shadomy HJ. Deep fungal infections. In: Fitzpatrick TB, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. New York, NY: McGraw Hill; 1987:2266-2268.
- Chakrabarti A, Gupta V, Biswas G, et al. Primary cutaneous aspergillosis: our experience in 10 years. J Infect. 1998;37:24-27.
- Robinson A, Fien S, Grassi MA. Nonhealing scalp wound infected with Aspergillus niger in an elderly patient. Cutis. 2011;87:197-200.
- Thomas LM, Rand HK, Miller JL, et al. Primary cutaneous aspergillosis in a patient with a solid organ transplant: case report and review of the literature. Cutis. 2008;81:127-130.
- Yuanjie Z, Jingxia D, Hai W, et al. Primary cutaneous aspergillosis in a patient with cutaneous T-cell lymphoma [published online ahead of print October 22, 2008]. Mycoses. 2009;52:462-464.
- Krishnan-Natesan S, Chandrasekar PH, Manavathu EK, et al. Successful treatment of primary cutaneous Aspergillus ustus infection with surgical debridement and a combination of voriconazole and terbinafine [published online ahead of print October 7, 2008]. Diagn Microbiol Infect Dis. 2008;62:443-446.
- Sato Y, Suzino K, Suzuki A, et al. Case of primary cutaneous Aspergillus calidoustus infection caused by nerve block therapy [in Japanese]. Med Mycol J. 2011;52:239-244.
- Lucas GM, Tucker P, Merz WG. Primary cutaneous Aspergillus nidulans infection associated with a Hickman catheter in a patient with neutropenia. Clin Infect Dis. 1999;29:1594-1596.
- Segal BH, DeCarlo ES, Kwon-Chung KJ, et al. Aspergillus nidulans infection in chronic granulomatous disease. Medicine (Baltimore). 1998;77:345-354.
- Woodruff CA, Hebert AA. Neonatal primary cutaneous aspergillosis: case report and review of the literature. Pediatr Dermatol. 2002;19:439-444.
- Mohapatra S, Xess I, Swetha JV, et al. Primary cutaneous aspergillosis due to Aspergillus niger in an immunocompetent patient. Indian J Med Microbiol. 2009;27:367-370.
Onychomycosis: Current and Investigational Therapies
To the Editor:
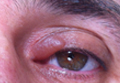
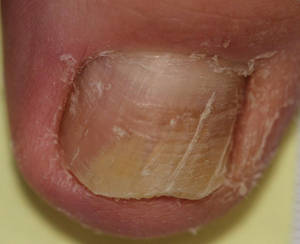
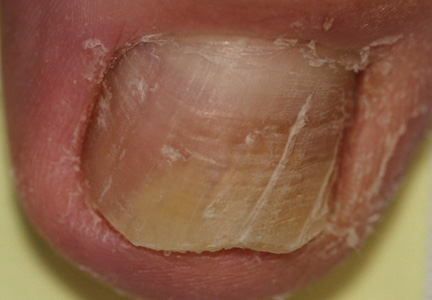
Onychomycosis is a fungal infection of the nail plate by dermatophytes, yeasts, and nondermatophyte molds. It is a common problem with a prevalence of 10% to 12% in the United States.1,2 The clinical presentation of onychomycosis is shown in the Figure. Although some patients may have mild asymptomatic cases of onychomycosis and do not inquire about treatment, many will have more advanced cases, presenting with pain and discomfort, secondary infection, unattractive appearance, or problems performing everyday functions. The goal of onychomycosis treatment is to eliminate the fungus, if possible, which usually restores the nail to its normal state when it fully grows out. Patients should be counseled that it is a long process that may take 6 months or more for fingernails and 12 to 18 months for toenails. These estimates are based on a growth rate of 2 to 3 mm per month for fingernails and 1 to 2 mm per month for toenails.3 Nails grow fastest during the teenaged years and slow down with advancing age.4 It should be noted that advanced cases of onychomycosis affecting the nail matrix may cause permanent scarring; therefore, the nail unit may still appear dystrophic after the causative organism is eliminated. The US Food and Drug Administration (FDA) defines a complete cure as negative potassium hydroxide preparation and negative fungal culture plus a completely normal appearance of the nail.
Treatment of onychomycosis poses a number of challenges. First, hyperkeratosis and the fungal mass may limit the delivery of topical and systemic drugs to the source of the infection. In addition, high rates of relapse and reinfection after treatment may be due to residual hyphae or spores.5 Furthermore, the extended length of treatment limits patient adherence and many patients are unwilling to forego wearing nail cosmetics during the course of some of the treatments.
There are 4 approved classes of antifungal drugs for the treatment of onychomycosis: allylamines, azoles, morpholines, and hydroxypyridinones.6 The allylamines (eg, terbinafine) inhibit squalene epoxidase.7 Oral terbinafine (250 mg daily) taken for 6 weeks for fingernails and 12 weeks for toenails is considered the current systemic treatment preference in onychomycosis therapy8 with complete cure rates in 12-week studies of approximately 38%9 and 49%.10
The second class of drugs is the azoles, which inhibit lanosterol 14a-demethylase, a step in the ergosterol biosynthesis pathway.6 Two members of this class that are widely used in treating onychomycosis are oral itraconazole11 and off-label oral fluconazole.12 The approved dose for oral itraconazole is 200 mg daily for 3 months (or an alternative pulse regimen) with a reported complete cure rate of 14%.11 Although fluconazole is not FDA approved for the treatment of onychomycosis in the United States, it is used extensively in other countries and to some extent off label in the United States. In a study of 362 patients with onychomycosis treated with oral fluconazole, complete cure rates were 48% in patients who received 450 mg weekly, 46% in those who received 300 mg weekly, and 37% in those who received 150 mg weekly for up to 9 months.12 It should be noted that several oral triazole antifungals, namely albaconazole,13 posaconazole,14 and ravuconazole,15 have undergone phase 1 and 2 studies for the treatment of onychomycosis and have shown some efficacy.
Another class of antifungals are the morpholines including topical amorolfine, which is approved for use in Europe but not in North America.16 Amorolfine inhibits D14 reductase and D7-D8 isomerase, thus depleting ergosterol.17 In one randomized controlled study, the combination of amorolfine nail lacquer and oral terbinafine compared to oral terbinafine alone resulted in a higher clinical cure rate with the combination (59.2% vs 46%); complete cure rate was not reported.16
Finally, the hydroxypyridinone class includes topical ciclopirox, which has a poorly understood mechanism of action but may involve iron chelation or oxidative damage.18,19 Ciclopirox nail lacquer 8% was approved by the FDA in 1999 and has reported complete cure rates of 5.5% to 8.5% with monthly nail debridement.20
Based on the poor efficacy of many of the currently available treatments and time-consuming treatment courses, it is clear that there is a need for alternative and novel therapies. There has been a greater emphasis on topical agents due to their more favorable side-effect profile and lower risk for drug-drug interactions. Although there are many agents for the treatment of onychomycosis currently in development, many are in vitro studies or phase 1 and 2 studies. However, we will focus on drugs that are further along in phase 3 studies and those that were recently FDA approved.
Efinaconazole is a member of the azole class of drugs and has completed 2 phase 3 clinical trials (study 1, N=870; study 2, N=785).21 Patients in these 2 studies were randomized to receive either efinaconazole nail solution 10% or vehicle for 48 weeks followed by a 4-week washout period. Complete cure rates in the 2 studies were 17.8% and 15.2% in the treated group and 3.3% and 5.5% in the control group. The mycological cure rates were 55.2% and 53.4% in the treated group and 16.8% and 16.9% in the control group. The side-effect profile was minimal, with the most common adverse events being application-site dermatitis and vesiculation, which were not significantly higher in the treated group versus the control group.21 Efinaconazole received FDA approval for the treatment of toenail onychomycosis in June 2014.
There are some notable differences between ciclopirox and efinaconazole that may improve patient compliance with the latter. First, treatment with ciclopirox includes monthly nail debridement, which is not required with efinaconazole. Secondly, although ciclopirox lacquer must be removed weekly, efinaconazole is a solution, so no removal is necessary.
Terbinafine nail solution (TNS) is a member of the allylamine class and has completed phase 3 clinical trials.22 Three studies—2 vehicle controlled and 1 active comparator—were performed. The first compared TNS and vehicle, both applied daily for 24 weeks; the second study repeated the same for 48 weeks; and the third study compared TNS to amorolfine nail lacquer 5% daily for 48 weeks. The best results for complete cure were achieved with TNS for 48 weeks in the vehicle-controlled study with a rate of 2.2% versus 0%. The authors also concluded TNS was not more effective than amorolfine, as complete cure rates were 1.2% for TNS and 0.96% for amorolfine. The most common side effects were headache, nasopharyngitis, and influenza.22
Tavaborole is a member of the new benzoxaborole class, which inhibits protein synthesis by forming an adduct with the aminoacyl–transfer RNA synthetase.23 The topical solution was engineered to have improved penetration through the nail plate. In vitro studies showed better penetration than both ciclopirox and amorolfine.24 Two identical phase 3 randomized, double-blind, vehicle-controlled studies were completed involving 1197 patients who were treated with tavaborole topical solution 5% daily compared to vehicle for 48 weeks followed by a 4-week washout period with promising results.25 The incidence of treatment-related side effects was comparable to the vehicle. The most common adverse events were exfoliation, erythema, and dermatitis, all occurring at the application site.25 Tavaborole was approved by the FDA for the treatment of toenail onychomycosis in July 2014.
Luliconazole is a member of the azole class and a phase 2b/3 clinical trial with a 10% solution involving 334 patients was completed in June 2013.26 Results from this trial are expected in early 2015.
Lasers are a developing area for onychomycosis therapy and the appeal stems from their ability to selectively deliver energy to the target tissue, thus avoiding systemic side effects. Since 2010, the FDA has approved numerous laser devices for the temporary cosmetic improvement of onychomycosis, all of which are Nd:YAG 1064-nm lasers.27,28 It was previously thought that the mechanism of action for the fungicidal effect was achieved with heat,29 but newer in vitro studies have shown that the amount of time and level of heat required to kill Trichophyton rubrum would not be tolerable to patients.30 Although the mechanism of action is poorly understood, some clinical trials have shown success using the Nd:YAG 1064-nm laser for treatment of onychomycosis. However, in a study of 8 patients treated with the Nd:YAG 1064-nm laser for 5 treatment sessions, none had a mycological or clinical cure and there was only mild clinical improvement. In addition, most patients had pain and burning during the treatments requiring many short breaks.30 Although not yet FDA approved for the treatment of onychomycosis, other types of lasers are currently being studied, including CO2, near-infrared diode, and femtosecond-infrared laser systems.3
Plasma therapy is a developing area for the treatment of onychomycosis. Plasma was shown to be fungicidal to T rubrum in an in vitro model (MOE Medical Devices, written communication, July 2012), and a clinical trial to evaluate the safety, tolerability, and efficacy of plasma in human subjects is ongoing (registered on March 22, 2013, at www.clinicaltrials.gov with the identifier NCT01819051).
Onychomycosis is a common problem that increases in prevalence with advancing age. Oral terbinafine is considered the first-line treatment at this point in time.31 Two new topical agents, efinaconazole and tavaborole, were recently FDA approved and may be used for the treatment of toenail onychomycosis without the need for nail debridement. The Nd:YAG laser has shown some promise in earlier clinical studies but was ineffective in a more recent study.
1. Ghannoum MA, Hajjeh RA, Scher R, et al. A large-scale North American study of fungal isolates from nails: the frequency of onychomycosis, fungal distribution, and antifungal susceptibility patterns. J Am Acad Dermatol. 2000;43:641-648.
2. Heikkila H, Stubb S. The prevalence of onychomycosis in Finland. Br J Dermatol. 1995;133:699-703.
3. Scher RK, Rich P, Pariser D, et al. The epidemiology, etiology, and pathophysiology of onychomycosis. Semin Cutan Med Surg. 2013;32(2, suppl 1):S2-S4.
4. Abdullah L, Abbas O. Common nail changes and disorders in older people: diagnosis and management. Can Fam Physician. 2011;57:173-181.
5. Scher RK, Baran R. Onychomycosis in clinical practice: factors contributing to recurrence. Br J Dermatol. 2003;149(suppl 65):5-9.
6. Welsh O, Vera-Cabrera L, Welsh E. Onychomycosis. Clin Dermatol. 2010;28:151-159.
7. Gupta AK, Sauder DN, Shear NH. Antifungal agents: an overview. part II. J Am Acad Dermatol. 1994;30:911-933.
8. Gupta AK, Paquet M, Simpson F, et al. Terbinafine in the treatment of dermatophyte toenail onychomycosis: a meta-analysis of efficacy for continuous and intermittent regimens. J Eur Acad Dermatol Venereol. 2013;27:267-272.
9. Drake LA, Shear NH, Arlette JP, et al. Oral terbinafine in the treatment of toenail onychomycosis: North American multicenter trial. J Am Acad Dermatol. 1997;37:740-745.
10. Evans EG, Sigurgeirsson B. Double blind, randomised study of continuous terbinafine compared with intermittent itraconazole in treatment of toenail onychomycosis. the LION Study Group. BMJ. 1999;318:1031-1035.
11. Sporanox [package insert]. Macquarie Park, Australia: Janssen-Cilag Pty Ltd; 2014.
12. Scher RK, Breneman D, Rich P, et al. Once-weekly fluconazole (150, 300, or 450 mg) in the treatment of distal subungual onychomycosis of the toenail. J Am Acad Dermatol. 1998;38(6, pt 2):S77-S86.
13. Sigurgeirsson B, van Rossem K, Malahias S, et al. A phase II, randomized, double-blind, placebo-controlled, parallel group, dose-ranging study to investigate the efficacy and safety of 4 dose regimens of oral albaconazole in patients with distal subungual onychomycosis. J Am Acad Dermatol. 2013;69:416-425.
14. Elewski B, Pollak R, Ashton S, et al. A randomized, placebo- and active-controlled, parallel-group, multicentre, investigator-blinded study of four treatment regimens of posaconazole in adults with toenail onychomycosis. Br J Dermatol. 2012;166:389-398.
15. Gupta AK, Leonardi C, Stoltz RR, et al. A phase I/II randomized, double-blind, placebo-controlled, dose-ranging study evaluating the efficacy, safety and pharmacokinetics of ravuconazole in the treatment of onychomycosis. J Eur Acad Dermatol Venereol. 2005;19:437-443.
16. Baran R, Sigurgeirsson B, de Berker D, et al. A multicentre, randomized, controlled study of the efficacy, safety and cost-effectiveness of a combination therapy with amorolfine nail lacquer and oral terbinafine compared with oral terbinafine alone for the treatment of onychomycosis with matrix involvement. Br J Dermatol. 2007;157:149-157.
17. Polak A. Preclinical data and mode of action of amorolfine. Dermatology. 1992;184(suppl 1):3-7.
18. Belenky P, Camacho D, Collins JJ. Fungicidal drugs induce a common oxidative-damage cellular death pathway. Cell Rep. 2013;3:350-358.
19. Lee RE, Liu TT, Barker KS, et al. Genome-wide expression profiling of the response to ciclopirox olamine in Candida albicans. J Antimicrob Chemother. 2005;55:655-662.
20. Penlac [package insert]. Bridgewater, NJ: sanofi-aventis; 2006.
21. Elewski BE, Rich P, Pollak R, et al. Efinaconazole 10% solution in the treatment of toenail onychomycosis: two phase III multicenter, randomized, double-blind studies. J Am Acad Dermatol. 2013;68:600-608.
22. Elewski BE, Ghannoum MA, Mayser P, et al. Efficacy, safety and tolerability of topical terbinafine nail solution in patients with mild-to-moderate toenail onychomycosis: results from three randomized studies using double-blind vehicle-controlled and open-label active-controlled designs. J Eur Acad Dermatol Venereol. 2013;27:287-294.
23. Rock FL, Mao W, Yaremchuk A, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759-1761.
24. Hui X, Baker SJ, Wester RC, et al. In vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. J Pharm Sci. 2007;96:2622-2631.
25. Elewski BE, Rich P, Wiltz H, et al. Effectiveness and safety of tavaborole, a novel born-based molecule for the treatment of onychomycosis: results from two phase 3 studies. Poster presented at: Women’s & Pediatric Dermatology Seminar; October 4-6, 2013; Newport Beach, CA.
26. The solution study: Topica’s phase 2b/3 clinical trial. Topica Pharmaceuticals Inc Web site. http://www.
topicapharma.com/phase-2b3. Accessed December 2, 2014.
27. Gupta AK, Simpson FC. Medical devices for the treatment of onychomycosis. Dermatol Ther. 2012;25:574-581.
28. Ortiz AE, Avram MM, Wanner MA. A review of lasers and light for the treatment of onychomycosis. Lasers Surg Med. 2014;46:117-124.
29. Vural E, Winfield HL, Shingleton AW, et al. The effects of laser irradiation on Trichophyton rubrum growth. Lasers Med Sci. 2008;23:349-353.
30. Carney C, Cantrell W, Warner J, et al. Treatment of onychomycosis using a submillisecond 1064-nm neodymium:yttrium-aluminum-garnet laser. J Am Acad Dermatol. 2013;69:578-582.
31. Gupta AK, Daigle D, Paquet M. Therapies for onychomycosis: a systematic review and network meta-analysis of mycological cure [published online ahead of print July 17, 2014]. J Am Podiatr Med Assoc. doi:10.7547/13-110.1.
To the Editor:
Onychomycosis is a fungal infection of the nail plate by dermatophytes, yeasts, and nondermatophyte molds. It is a common problem with a prevalence of 10% to 12% in the United States.1,2 The clinical presentation of onychomycosis is shown in the Figure. Although some patients may have mild asymptomatic cases of onychomycosis and do not inquire about treatment, many will have more advanced cases, presenting with pain and discomfort, secondary infection, unattractive appearance, or problems performing everyday functions. The goal of onychomycosis treatment is to eliminate the fungus, if possible, which usually restores the nail to its normal state when it fully grows out. Patients should be counseled that it is a long process that may take 6 months or more for fingernails and 12 to 18 months for toenails. These estimates are based on a growth rate of 2 to 3 mm per month for fingernails and 1 to 2 mm per month for toenails.3 Nails grow fastest during the teenaged years and slow down with advancing age.4 It should be noted that advanced cases of onychomycosis affecting the nail matrix may cause permanent scarring; therefore, the nail unit may still appear dystrophic after the causative organism is eliminated. The US Food and Drug Administration (FDA) defines a complete cure as negative potassium hydroxide preparation and negative fungal culture plus a completely normal appearance of the nail.
Treatment of onychomycosis poses a number of challenges. First, hyperkeratosis and the fungal mass may limit the delivery of topical and systemic drugs to the source of the infection. In addition, high rates of relapse and reinfection after treatment may be due to residual hyphae or spores.5 Furthermore, the extended length of treatment limits patient adherence and many patients are unwilling to forego wearing nail cosmetics during the course of some of the treatments.
There are 4 approved classes of antifungal drugs for the treatment of onychomycosis: allylamines, azoles, morpholines, and hydroxypyridinones.6 The allylamines (eg, terbinafine) inhibit squalene epoxidase.7 Oral terbinafine (250 mg daily) taken for 6 weeks for fingernails and 12 weeks for toenails is considered the current systemic treatment preference in onychomycosis therapy8 with complete cure rates in 12-week studies of approximately 38%9 and 49%.10
The second class of drugs is the azoles, which inhibit lanosterol 14a-demethylase, a step in the ergosterol biosynthesis pathway.6 Two members of this class that are widely used in treating onychomycosis are oral itraconazole11 and off-label oral fluconazole.12 The approved dose for oral itraconazole is 200 mg daily for 3 months (or an alternative pulse regimen) with a reported complete cure rate of 14%.11 Although fluconazole is not FDA approved for the treatment of onychomycosis in the United States, it is used extensively in other countries and to some extent off label in the United States. In a study of 362 patients with onychomycosis treated with oral fluconazole, complete cure rates were 48% in patients who received 450 mg weekly, 46% in those who received 300 mg weekly, and 37% in those who received 150 mg weekly for up to 9 months.12 It should be noted that several oral triazole antifungals, namely albaconazole,13 posaconazole,14 and ravuconazole,15 have undergone phase 1 and 2 studies for the treatment of onychomycosis and have shown some efficacy.
Another class of antifungals are the morpholines including topical amorolfine, which is approved for use in Europe but not in North America.16 Amorolfine inhibits D14 reductase and D7-D8 isomerase, thus depleting ergosterol.17 In one randomized controlled study, the combination of amorolfine nail lacquer and oral terbinafine compared to oral terbinafine alone resulted in a higher clinical cure rate with the combination (59.2% vs 46%); complete cure rate was not reported.16
Finally, the hydroxypyridinone class includes topical ciclopirox, which has a poorly understood mechanism of action but may involve iron chelation or oxidative damage.18,19 Ciclopirox nail lacquer 8% was approved by the FDA in 1999 and has reported complete cure rates of 5.5% to 8.5% with monthly nail debridement.20
Based on the poor efficacy of many of the currently available treatments and time-consuming treatment courses, it is clear that there is a need for alternative and novel therapies. There has been a greater emphasis on topical agents due to their more favorable side-effect profile and lower risk for drug-drug interactions. Although there are many agents for the treatment of onychomycosis currently in development, many are in vitro studies or phase 1 and 2 studies. However, we will focus on drugs that are further along in phase 3 studies and those that were recently FDA approved.
Efinaconazole is a member of the azole class of drugs and has completed 2 phase 3 clinical trials (study 1, N=870; study 2, N=785).21 Patients in these 2 studies were randomized to receive either efinaconazole nail solution 10% or vehicle for 48 weeks followed by a 4-week washout period. Complete cure rates in the 2 studies were 17.8% and 15.2% in the treated group and 3.3% and 5.5% in the control group. The mycological cure rates were 55.2% and 53.4% in the treated group and 16.8% and 16.9% in the control group. The side-effect profile was minimal, with the most common adverse events being application-site dermatitis and vesiculation, which were not significantly higher in the treated group versus the control group.21 Efinaconazole received FDA approval for the treatment of toenail onychomycosis in June 2014.
There are some notable differences between ciclopirox and efinaconazole that may improve patient compliance with the latter. First, treatment with ciclopirox includes monthly nail debridement, which is not required with efinaconazole. Secondly, although ciclopirox lacquer must be removed weekly, efinaconazole is a solution, so no removal is necessary.
Terbinafine nail solution (TNS) is a member of the allylamine class and has completed phase 3 clinical trials.22 Three studies—2 vehicle controlled and 1 active comparator—were performed. The first compared TNS and vehicle, both applied daily for 24 weeks; the second study repeated the same for 48 weeks; and the third study compared TNS to amorolfine nail lacquer 5% daily for 48 weeks. The best results for complete cure were achieved with TNS for 48 weeks in the vehicle-controlled study with a rate of 2.2% versus 0%. The authors also concluded TNS was not more effective than amorolfine, as complete cure rates were 1.2% for TNS and 0.96% for amorolfine. The most common side effects were headache, nasopharyngitis, and influenza.22
Tavaborole is a member of the new benzoxaborole class, which inhibits protein synthesis by forming an adduct with the aminoacyl–transfer RNA synthetase.23 The topical solution was engineered to have improved penetration through the nail plate. In vitro studies showed better penetration than both ciclopirox and amorolfine.24 Two identical phase 3 randomized, double-blind, vehicle-controlled studies were completed involving 1197 patients who were treated with tavaborole topical solution 5% daily compared to vehicle for 48 weeks followed by a 4-week washout period with promising results.25 The incidence of treatment-related side effects was comparable to the vehicle. The most common adverse events were exfoliation, erythema, and dermatitis, all occurring at the application site.25 Tavaborole was approved by the FDA for the treatment of toenail onychomycosis in July 2014.
Luliconazole is a member of the azole class and a phase 2b/3 clinical trial with a 10% solution involving 334 patients was completed in June 2013.26 Results from this trial are expected in early 2015.
Lasers are a developing area for onychomycosis therapy and the appeal stems from their ability to selectively deliver energy to the target tissue, thus avoiding systemic side effects. Since 2010, the FDA has approved numerous laser devices for the temporary cosmetic improvement of onychomycosis, all of which are Nd:YAG 1064-nm lasers.27,28 It was previously thought that the mechanism of action for the fungicidal effect was achieved with heat,29 but newer in vitro studies have shown that the amount of time and level of heat required to kill Trichophyton rubrum would not be tolerable to patients.30 Although the mechanism of action is poorly understood, some clinical trials have shown success using the Nd:YAG 1064-nm laser for treatment of onychomycosis. However, in a study of 8 patients treated with the Nd:YAG 1064-nm laser for 5 treatment sessions, none had a mycological or clinical cure and there was only mild clinical improvement. In addition, most patients had pain and burning during the treatments requiring many short breaks.30 Although not yet FDA approved for the treatment of onychomycosis, other types of lasers are currently being studied, including CO2, near-infrared diode, and femtosecond-infrared laser systems.3
Plasma therapy is a developing area for the treatment of onychomycosis. Plasma was shown to be fungicidal to T rubrum in an in vitro model (MOE Medical Devices, written communication, July 2012), and a clinical trial to evaluate the safety, tolerability, and efficacy of plasma in human subjects is ongoing (registered on March 22, 2013, at www.clinicaltrials.gov with the identifier NCT01819051).
Onychomycosis is a common problem that increases in prevalence with advancing age. Oral terbinafine is considered the first-line treatment at this point in time.31 Two new topical agents, efinaconazole and tavaborole, were recently FDA approved and may be used for the treatment of toenail onychomycosis without the need for nail debridement. The Nd:YAG laser has shown some promise in earlier clinical studies but was ineffective in a more recent study.
To the Editor:
Onychomycosis is a fungal infection of the nail plate by dermatophytes, yeasts, and nondermatophyte molds. It is a common problem with a prevalence of 10% to 12% in the United States.1,2 The clinical presentation of onychomycosis is shown in the Figure. Although some patients may have mild asymptomatic cases of onychomycosis and do not inquire about treatment, many will have more advanced cases, presenting with pain and discomfort, secondary infection, unattractive appearance, or problems performing everyday functions. The goal of onychomycosis treatment is to eliminate the fungus, if possible, which usually restores the nail to its normal state when it fully grows out. Patients should be counseled that it is a long process that may take 6 months or more for fingernails and 12 to 18 months for toenails. These estimates are based on a growth rate of 2 to 3 mm per month for fingernails and 1 to 2 mm per month for toenails.3 Nails grow fastest during the teenaged years and slow down with advancing age.4 It should be noted that advanced cases of onychomycosis affecting the nail matrix may cause permanent scarring; therefore, the nail unit may still appear dystrophic after the causative organism is eliminated. The US Food and Drug Administration (FDA) defines a complete cure as negative potassium hydroxide preparation and negative fungal culture plus a completely normal appearance of the nail.
Treatment of onychomycosis poses a number of challenges. First, hyperkeratosis and the fungal mass may limit the delivery of topical and systemic drugs to the source of the infection. In addition, high rates of relapse and reinfection after treatment may be due to residual hyphae or spores.5 Furthermore, the extended length of treatment limits patient adherence and many patients are unwilling to forego wearing nail cosmetics during the course of some of the treatments.
There are 4 approved classes of antifungal drugs for the treatment of onychomycosis: allylamines, azoles, morpholines, and hydroxypyridinones.6 The allylamines (eg, terbinafine) inhibit squalene epoxidase.7 Oral terbinafine (250 mg daily) taken for 6 weeks for fingernails and 12 weeks for toenails is considered the current systemic treatment preference in onychomycosis therapy8 with complete cure rates in 12-week studies of approximately 38%9 and 49%.10
The second class of drugs is the azoles, which inhibit lanosterol 14a-demethylase, a step in the ergosterol biosynthesis pathway.6 Two members of this class that are widely used in treating onychomycosis are oral itraconazole11 and off-label oral fluconazole.12 The approved dose for oral itraconazole is 200 mg daily for 3 months (or an alternative pulse regimen) with a reported complete cure rate of 14%.11 Although fluconazole is not FDA approved for the treatment of onychomycosis in the United States, it is used extensively in other countries and to some extent off label in the United States. In a study of 362 patients with onychomycosis treated with oral fluconazole, complete cure rates were 48% in patients who received 450 mg weekly, 46% in those who received 300 mg weekly, and 37% in those who received 150 mg weekly for up to 9 months.12 It should be noted that several oral triazole antifungals, namely albaconazole,13 posaconazole,14 and ravuconazole,15 have undergone phase 1 and 2 studies for the treatment of onychomycosis and have shown some efficacy.
Another class of antifungals are the morpholines including topical amorolfine, which is approved for use in Europe but not in North America.16 Amorolfine inhibits D14 reductase and D7-D8 isomerase, thus depleting ergosterol.17 In one randomized controlled study, the combination of amorolfine nail lacquer and oral terbinafine compared to oral terbinafine alone resulted in a higher clinical cure rate with the combination (59.2% vs 46%); complete cure rate was not reported.16
Finally, the hydroxypyridinone class includes topical ciclopirox, which has a poorly understood mechanism of action but may involve iron chelation or oxidative damage.18,19 Ciclopirox nail lacquer 8% was approved by the FDA in 1999 and has reported complete cure rates of 5.5% to 8.5% with monthly nail debridement.20
Based on the poor efficacy of many of the currently available treatments and time-consuming treatment courses, it is clear that there is a need for alternative and novel therapies. There has been a greater emphasis on topical agents due to their more favorable side-effect profile and lower risk for drug-drug interactions. Although there are many agents for the treatment of onychomycosis currently in development, many are in vitro studies or phase 1 and 2 studies. However, we will focus on drugs that are further along in phase 3 studies and those that were recently FDA approved.
Efinaconazole is a member of the azole class of drugs and has completed 2 phase 3 clinical trials (study 1, N=870; study 2, N=785).21 Patients in these 2 studies were randomized to receive either efinaconazole nail solution 10% or vehicle for 48 weeks followed by a 4-week washout period. Complete cure rates in the 2 studies were 17.8% and 15.2% in the treated group and 3.3% and 5.5% in the control group. The mycological cure rates were 55.2% and 53.4% in the treated group and 16.8% and 16.9% in the control group. The side-effect profile was minimal, with the most common adverse events being application-site dermatitis and vesiculation, which were not significantly higher in the treated group versus the control group.21 Efinaconazole received FDA approval for the treatment of toenail onychomycosis in June 2014.
There are some notable differences between ciclopirox and efinaconazole that may improve patient compliance with the latter. First, treatment with ciclopirox includes monthly nail debridement, which is not required with efinaconazole. Secondly, although ciclopirox lacquer must be removed weekly, efinaconazole is a solution, so no removal is necessary.
Terbinafine nail solution (TNS) is a member of the allylamine class and has completed phase 3 clinical trials.22 Three studies—2 vehicle controlled and 1 active comparator—were performed. The first compared TNS and vehicle, both applied daily for 24 weeks; the second study repeated the same for 48 weeks; and the third study compared TNS to amorolfine nail lacquer 5% daily for 48 weeks. The best results for complete cure were achieved with TNS for 48 weeks in the vehicle-controlled study with a rate of 2.2% versus 0%. The authors also concluded TNS was not more effective than amorolfine, as complete cure rates were 1.2% for TNS and 0.96% for amorolfine. The most common side effects were headache, nasopharyngitis, and influenza.22
Tavaborole is a member of the new benzoxaborole class, which inhibits protein synthesis by forming an adduct with the aminoacyl–transfer RNA synthetase.23 The topical solution was engineered to have improved penetration through the nail plate. In vitro studies showed better penetration than both ciclopirox and amorolfine.24 Two identical phase 3 randomized, double-blind, vehicle-controlled studies were completed involving 1197 patients who were treated with tavaborole topical solution 5% daily compared to vehicle for 48 weeks followed by a 4-week washout period with promising results.25 The incidence of treatment-related side effects was comparable to the vehicle. The most common adverse events were exfoliation, erythema, and dermatitis, all occurring at the application site.25 Tavaborole was approved by the FDA for the treatment of toenail onychomycosis in July 2014.
Luliconazole is a member of the azole class and a phase 2b/3 clinical trial with a 10% solution involving 334 patients was completed in June 2013.26 Results from this trial are expected in early 2015.
Lasers are a developing area for onychomycosis therapy and the appeal stems from their ability to selectively deliver energy to the target tissue, thus avoiding systemic side effects. Since 2010, the FDA has approved numerous laser devices for the temporary cosmetic improvement of onychomycosis, all of which are Nd:YAG 1064-nm lasers.27,28 It was previously thought that the mechanism of action for the fungicidal effect was achieved with heat,29 but newer in vitro studies have shown that the amount of time and level of heat required to kill Trichophyton rubrum would not be tolerable to patients.30 Although the mechanism of action is poorly understood, some clinical trials have shown success using the Nd:YAG 1064-nm laser for treatment of onychomycosis. However, in a study of 8 patients treated with the Nd:YAG 1064-nm laser for 5 treatment sessions, none had a mycological or clinical cure and there was only mild clinical improvement. In addition, most patients had pain and burning during the treatments requiring many short breaks.30 Although not yet FDA approved for the treatment of onychomycosis, other types of lasers are currently being studied, including CO2, near-infrared diode, and femtosecond-infrared laser systems.3
Plasma therapy is a developing area for the treatment of onychomycosis. Plasma was shown to be fungicidal to T rubrum in an in vitro model (MOE Medical Devices, written communication, July 2012), and a clinical trial to evaluate the safety, tolerability, and efficacy of plasma in human subjects is ongoing (registered on March 22, 2013, at www.clinicaltrials.gov with the identifier NCT01819051).
Onychomycosis is a common problem that increases in prevalence with advancing age. Oral terbinafine is considered the first-line treatment at this point in time.31 Two new topical agents, efinaconazole and tavaborole, were recently FDA approved and may be used for the treatment of toenail onychomycosis without the need for nail debridement. The Nd:YAG laser has shown some promise in earlier clinical studies but was ineffective in a more recent study.
1. Ghannoum MA, Hajjeh RA, Scher R, et al. A large-scale North American study of fungal isolates from nails: the frequency of onychomycosis, fungal distribution, and antifungal susceptibility patterns. J Am Acad Dermatol. 2000;43:641-648.
2. Heikkila H, Stubb S. The prevalence of onychomycosis in Finland. Br J Dermatol. 1995;133:699-703.
3. Scher RK, Rich P, Pariser D, et al. The epidemiology, etiology, and pathophysiology of onychomycosis. Semin Cutan Med Surg. 2013;32(2, suppl 1):S2-S4.
4. Abdullah L, Abbas O. Common nail changes and disorders in older people: diagnosis and management. Can Fam Physician. 2011;57:173-181.
5. Scher RK, Baran R. Onychomycosis in clinical practice: factors contributing to recurrence. Br J Dermatol. 2003;149(suppl 65):5-9.
6. Welsh O, Vera-Cabrera L, Welsh E. Onychomycosis. Clin Dermatol. 2010;28:151-159.
7. Gupta AK, Sauder DN, Shear NH. Antifungal agents: an overview. part II. J Am Acad Dermatol. 1994;30:911-933.
8. Gupta AK, Paquet M, Simpson F, et al. Terbinafine in the treatment of dermatophyte toenail onychomycosis: a meta-analysis of efficacy for continuous and intermittent regimens. J Eur Acad Dermatol Venereol. 2013;27:267-272.
9. Drake LA, Shear NH, Arlette JP, et al. Oral terbinafine in the treatment of toenail onychomycosis: North American multicenter trial. J Am Acad Dermatol. 1997;37:740-745.
10. Evans EG, Sigurgeirsson B. Double blind, randomised study of continuous terbinafine compared with intermittent itraconazole in treatment of toenail onychomycosis. the LION Study Group. BMJ. 1999;318:1031-1035.
11. Sporanox [package insert]. Macquarie Park, Australia: Janssen-Cilag Pty Ltd; 2014.
12. Scher RK, Breneman D, Rich P, et al. Once-weekly fluconazole (150, 300, or 450 mg) in the treatment of distal subungual onychomycosis of the toenail. J Am Acad Dermatol. 1998;38(6, pt 2):S77-S86.
13. Sigurgeirsson B, van Rossem K, Malahias S, et al. A phase II, randomized, double-blind, placebo-controlled, parallel group, dose-ranging study to investigate the efficacy and safety of 4 dose regimens of oral albaconazole in patients with distal subungual onychomycosis. J Am Acad Dermatol. 2013;69:416-425.
14. Elewski B, Pollak R, Ashton S, et al. A randomized, placebo- and active-controlled, parallel-group, multicentre, investigator-blinded study of four treatment regimens of posaconazole in adults with toenail onychomycosis. Br J Dermatol. 2012;166:389-398.
15. Gupta AK, Leonardi C, Stoltz RR, et al. A phase I/II randomized, double-blind, placebo-controlled, dose-ranging study evaluating the efficacy, safety and pharmacokinetics of ravuconazole in the treatment of onychomycosis. J Eur Acad Dermatol Venereol. 2005;19:437-443.
16. Baran R, Sigurgeirsson B, de Berker D, et al. A multicentre, randomized, controlled study of the efficacy, safety and cost-effectiveness of a combination therapy with amorolfine nail lacquer and oral terbinafine compared with oral terbinafine alone for the treatment of onychomycosis with matrix involvement. Br J Dermatol. 2007;157:149-157.
17. Polak A. Preclinical data and mode of action of amorolfine. Dermatology. 1992;184(suppl 1):3-7.
18. Belenky P, Camacho D, Collins JJ. Fungicidal drugs induce a common oxidative-damage cellular death pathway. Cell Rep. 2013;3:350-358.
19. Lee RE, Liu TT, Barker KS, et al. Genome-wide expression profiling of the response to ciclopirox olamine in Candida albicans. J Antimicrob Chemother. 2005;55:655-662.
20. Penlac [package insert]. Bridgewater, NJ: sanofi-aventis; 2006.
21. Elewski BE, Rich P, Pollak R, et al. Efinaconazole 10% solution in the treatment of toenail onychomycosis: two phase III multicenter, randomized, double-blind studies. J Am Acad Dermatol. 2013;68:600-608.
22. Elewski BE, Ghannoum MA, Mayser P, et al. Efficacy, safety and tolerability of topical terbinafine nail solution in patients with mild-to-moderate toenail onychomycosis: results from three randomized studies using double-blind vehicle-controlled and open-label active-controlled designs. J Eur Acad Dermatol Venereol. 2013;27:287-294.
23. Rock FL, Mao W, Yaremchuk A, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759-1761.
24. Hui X, Baker SJ, Wester RC, et al. In vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. J Pharm Sci. 2007;96:2622-2631.
25. Elewski BE, Rich P, Wiltz H, et al. Effectiveness and safety of tavaborole, a novel born-based molecule for the treatment of onychomycosis: results from two phase 3 studies. Poster presented at: Women’s & Pediatric Dermatology Seminar; October 4-6, 2013; Newport Beach, CA.
26. The solution study: Topica’s phase 2b/3 clinical trial. Topica Pharmaceuticals Inc Web site. http://www.
topicapharma.com/phase-2b3. Accessed December 2, 2014.
27. Gupta AK, Simpson FC. Medical devices for the treatment of onychomycosis. Dermatol Ther. 2012;25:574-581.
28. Ortiz AE, Avram MM, Wanner MA. A review of lasers and light for the treatment of onychomycosis. Lasers Surg Med. 2014;46:117-124.
29. Vural E, Winfield HL, Shingleton AW, et al. The effects of laser irradiation on Trichophyton rubrum growth. Lasers Med Sci. 2008;23:349-353.
30. Carney C, Cantrell W, Warner J, et al. Treatment of onychomycosis using a submillisecond 1064-nm neodymium:yttrium-aluminum-garnet laser. J Am Acad Dermatol. 2013;69:578-582.
31. Gupta AK, Daigle D, Paquet M. Therapies for onychomycosis: a systematic review and network meta-analysis of mycological cure [published online ahead of print July 17, 2014]. J Am Podiatr Med Assoc. doi:10.7547/13-110.1.
1. Ghannoum MA, Hajjeh RA, Scher R, et al. A large-scale North American study of fungal isolates from nails: the frequency of onychomycosis, fungal distribution, and antifungal susceptibility patterns. J Am Acad Dermatol. 2000;43:641-648.
2. Heikkila H, Stubb S. The prevalence of onychomycosis in Finland. Br J Dermatol. 1995;133:699-703.
3. Scher RK, Rich P, Pariser D, et al. The epidemiology, etiology, and pathophysiology of onychomycosis. Semin Cutan Med Surg. 2013;32(2, suppl 1):S2-S4.
4. Abdullah L, Abbas O. Common nail changes and disorders in older people: diagnosis and management. Can Fam Physician. 2011;57:173-181.
5. Scher RK, Baran R. Onychomycosis in clinical practice: factors contributing to recurrence. Br J Dermatol. 2003;149(suppl 65):5-9.
6. Welsh O, Vera-Cabrera L, Welsh E. Onychomycosis. Clin Dermatol. 2010;28:151-159.
7. Gupta AK, Sauder DN, Shear NH. Antifungal agents: an overview. part II. J Am Acad Dermatol. 1994;30:911-933.
8. Gupta AK, Paquet M, Simpson F, et al. Terbinafine in the treatment of dermatophyte toenail onychomycosis: a meta-analysis of efficacy for continuous and intermittent regimens. J Eur Acad Dermatol Venereol. 2013;27:267-272.
9. Drake LA, Shear NH, Arlette JP, et al. Oral terbinafine in the treatment of toenail onychomycosis: North American multicenter trial. J Am Acad Dermatol. 1997;37:740-745.
10. Evans EG, Sigurgeirsson B. Double blind, randomised study of continuous terbinafine compared with intermittent itraconazole in treatment of toenail onychomycosis. the LION Study Group. BMJ. 1999;318:1031-1035.
11. Sporanox [package insert]. Macquarie Park, Australia: Janssen-Cilag Pty Ltd; 2014.
12. Scher RK, Breneman D, Rich P, et al. Once-weekly fluconazole (150, 300, or 450 mg) in the treatment of distal subungual onychomycosis of the toenail. J Am Acad Dermatol. 1998;38(6, pt 2):S77-S86.
13. Sigurgeirsson B, van Rossem K, Malahias S, et al. A phase II, randomized, double-blind, placebo-controlled, parallel group, dose-ranging study to investigate the efficacy and safety of 4 dose regimens of oral albaconazole in patients with distal subungual onychomycosis. J Am Acad Dermatol. 2013;69:416-425.
14. Elewski B, Pollak R, Ashton S, et al. A randomized, placebo- and active-controlled, parallel-group, multicentre, investigator-blinded study of four treatment regimens of posaconazole in adults with toenail onychomycosis. Br J Dermatol. 2012;166:389-398.
15. Gupta AK, Leonardi C, Stoltz RR, et al. A phase I/II randomized, double-blind, placebo-controlled, dose-ranging study evaluating the efficacy, safety and pharmacokinetics of ravuconazole in the treatment of onychomycosis. J Eur Acad Dermatol Venereol. 2005;19:437-443.
16. Baran R, Sigurgeirsson B, de Berker D, et al. A multicentre, randomized, controlled study of the efficacy, safety and cost-effectiveness of a combination therapy with amorolfine nail lacquer and oral terbinafine compared with oral terbinafine alone for the treatment of onychomycosis with matrix involvement. Br J Dermatol. 2007;157:149-157.
17. Polak A. Preclinical data and mode of action of amorolfine. Dermatology. 1992;184(suppl 1):3-7.
18. Belenky P, Camacho D, Collins JJ. Fungicidal drugs induce a common oxidative-damage cellular death pathway. Cell Rep. 2013;3:350-358.
19. Lee RE, Liu TT, Barker KS, et al. Genome-wide expression profiling of the response to ciclopirox olamine in Candida albicans. J Antimicrob Chemother. 2005;55:655-662.
20. Penlac [package insert]. Bridgewater, NJ: sanofi-aventis; 2006.
21. Elewski BE, Rich P, Pollak R, et al. Efinaconazole 10% solution in the treatment of toenail onychomycosis: two phase III multicenter, randomized, double-blind studies. J Am Acad Dermatol. 2013;68:600-608.
22. Elewski BE, Ghannoum MA, Mayser P, et al. Efficacy, safety and tolerability of topical terbinafine nail solution in patients with mild-to-moderate toenail onychomycosis: results from three randomized studies using double-blind vehicle-controlled and open-label active-controlled designs. J Eur Acad Dermatol Venereol. 2013;27:287-294.
23. Rock FL, Mao W, Yaremchuk A, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759-1761.
24. Hui X, Baker SJ, Wester RC, et al. In vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. J Pharm Sci. 2007;96:2622-2631.
25. Elewski BE, Rich P, Wiltz H, et al. Effectiveness and safety of tavaborole, a novel born-based molecule for the treatment of onychomycosis: results from two phase 3 studies. Poster presented at: Women’s & Pediatric Dermatology Seminar; October 4-6, 2013; Newport Beach, CA.
26. The solution study: Topica’s phase 2b/3 clinical trial. Topica Pharmaceuticals Inc Web site. http://www.
topicapharma.com/phase-2b3. Accessed December 2, 2014.
27. Gupta AK, Simpson FC. Medical devices for the treatment of onychomycosis. Dermatol Ther. 2012;25:574-581.
28. Ortiz AE, Avram MM, Wanner MA. A review of lasers and light for the treatment of onychomycosis. Lasers Surg Med. 2014;46:117-124.
29. Vural E, Winfield HL, Shingleton AW, et al. The effects of laser irradiation on Trichophyton rubrum growth. Lasers Med Sci. 2008;23:349-353.
30. Carney C, Cantrell W, Warner J, et al. Treatment of onychomycosis using a submillisecond 1064-nm neodymium:yttrium-aluminum-garnet laser. J Am Acad Dermatol. 2013;69:578-582.
31. Gupta AK, Daigle D, Paquet M. Therapies for onychomycosis: a systematic review and network meta-analysis of mycological cure [published online ahead of print July 17, 2014]. J Am Podiatr Med Assoc. doi:10.7547/13-110.1.
Hemorrhagic Bullous Lesions Due to Bacillus cereus in a Cirrhotic Patient
To the Editor:
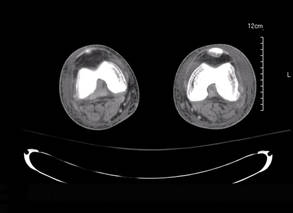
A 42-year-old man with hypertension, hypothyroidism, and alcohol-related cirrhosis was admitted for evaluation of rapidly deteriorating mental status. He was referred from a rehabilitation facility where he had been admitted 4 days earlier after a hospitalization for hepatorenal syndrome and pneumonia. He was alert and ambulating until the day of the current admission. On arrival he was hypotensive(54/42 mm Hg); hypothermic (35°C, rectally); and unresponsive, except to painful stimuli. Jaundice, hepatosplenomegaly, ascites, and bilateral lower extremity edema were noted. There were multiple tense and flaccid bullous lesions containing serosanguineous fluid over both tibias and calves, without crepitus (Figure 1).

|
|
|
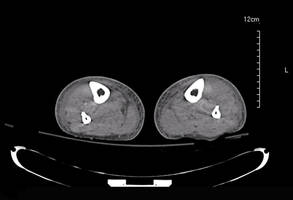
Laboratory test results revealed leukocytosis (total leukocytes, 10,900/mm3 [reference range, 4500–10,800/mm3), hypoglycemia (glucose, <20 mg/dL [reference range, 74–106 mg/dL]), renal insufficiency (serum creatinine, 2.5 mg/dL [reference range, 0.66–1.25 mg/dL]), metabolic acidosis (pH, 7.1 [reference range, 7.35–7.45]; bicarbonate, 13 mmol/L [reference range, 22–30 mmol/L]; lactic acid, 11.9 mmol/L [reference range, 0.7–2.1 mmol/L]), liver dysfunction (aspartate aminotransferase, 576 IU/L [reference range, 15–46 IU/L]), and coagulopathy with evidence of diffuse intravascular coagulation (total platelets, 75,000/mm3 [reference range, 150,000–450,000/mm3]; international normalized ratio, 9.5 [reference range, 0.8–1.2]; partial thromboplastin time, 108 seconds [reference range 23.0–35.0 seconds]; fibrinogen, 145 mg/dL [reference range, 228–501 mg/dL]; D-dimer, >20 µg/mL [reference range, 0.01–0.58 μg/mL]). Computed tomography of the pelvis and legs showed ascites, extensive subcutaneous edema, and cutaneous blisterlike lesions superior to the level of the ankles bilaterally. No gas, foreign bodies, collections, asymmetric facial thickening, or evidence of infection across tissue planes was present (Figures 2 and 3).
Specimens of blood and aspirates from the bullae at multiple lower leg sites were sent for microbiologic evaluation. The blood specimens were inoculated at bedside into aerobic and anaerobic blood culture bottles and incubated in an automated blood culture system. The aspirate samples were inoculated to trypticase soy agar with 5% sheep blood, Columbia-nalidixic acid agar, chocolate agar, MacConkey agar, and thioglycollate broth, which were incubated at 37ºC in air supplemented with 5% CO2, and to CDC anaerobic blood agar, which was incubated under anaerobic conditions. Gram-stained smears of the aspirates from the bullae demonstrated few granulocytes and numerous large gram-positive bacilli (Figure 4). By the next day, growth of large gram-positive bacilli was detected in both aerobic and anaerobic blood culture bottles and in pure culture from all the bullae samples. The bacterial colonies on sheep blood agar were opaque and white-gray in color, with a rough surface, undulate margins, and surrounding β hemolysis. The isolate was a motile, catalase-positive, arginine-positive, salicin-positive, lecithinase-positive, and penicillin-resistant organism that was identified as Bacillus cereus.
Antimicrobial susceptibility testing for B cereus has not been standardized, but evaluation by broth microdilution suggested decreased susceptibility to penicillin (minimum inhibitory concentration [MIC], 2 µg/mL) and clindamycin (MIC, 2 µg/mL), but retained susceptibility to ciprofloxacin (MIC, ≤0.25 µg/mL), tetracycline (MIC, ≤1 µg/mL), rifampin (MIC, ≤1 µg/mL), and vancomycin (MIC, ≤2 µg/mL).
The patient was admitted to the intensive care unit and was treated initially with fluid resuscitation; transfusions; ventilatory support; and intravenous vancomycin, clindamycin, and imipenem. This regimen was changed to vancomycin and ciprofloxacin when culture and susceptibility results became available to complete a 14-day course. Signs of sepsis resolved and the mental status and skin lesions improved. Ultimately, the patient died due to complications of hepatic failure.
Bacillus cereus is a rod-shaped, gram-positive, facultative, aerobic organism that is widely distributed in the environment.1 Spore formation makes B cereus resistant to most physical and chemical disinfection methods; as a consequence, it is a frequent contaminant in materials (eg, plants, dust, soil, sediment), foodstuffs, and clinical specimens.1
Traditionally considered in the context of foodborne illness, B cereus is recognized increasingly as a cause of systemic and local infections in both immunosuppressed and immunocompetent patients. Nongastrointestinal infections reported include fulminant bacteremia, pneumonia, meningitis, brain abscesses, endophthalmitis, necrotizing fasciitis, and central line catheter–related and cutaneous infections.1,2
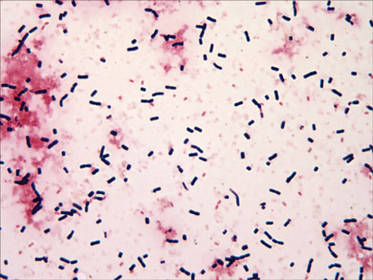
Cutaneous lesions may have a variety of forms and appearance at initial presentation, including small papules or vesicles that progress into a rapidly spreading cellulitis1,2 with a characteristic serosanguineous draining fluid,2 single necrotic bullae,3 and gas-gangrenelike infections with extensive soft tissue involvement resembling clostridial myonecrosis.1,4 Single or multiple papulovesicular lesions can even mimic cutaneous anthrax.1-4 Necrotic or hemorrhagic bullous lesions,3 such as those observed in our patient, are rare.
Exposed areas such as extremities and digits are most often affected, presumably due to entrance of spores from soil, water, decaying organic material, or fomites through skin microabrasions or trauma-induced wounds.1 Once in the tissue, the crystalline surface protein layer (S-layer) of the bacilli promotes adhesion to human epithelial cells and neutrophils,5 followed by release of virulence factors including proteases, collagenases, lecithinaselike enzymes, necrotizing exotoxinlike hemolysins, phospholipases, and most importantly a dermonecrotic vascular permeability factor.1,5 Toxins produced by B cereus are similar to those closely related to Bacillus anthracis, the agent of anthrax.1,2
When large gram-positive bacilli are observed in tissue or wound specimens, initial therapy should address both aerobic (Bacillus species) and anaerobic (Clostridium species) organisms.1,4,6 Once B cereus is recovered, treatment should rely on susceptibility testing of the isolate. Bacillus cereus produces ß-lactamase, thus penicillin and cephalosporin should be avoided.1 Vancomycin, clindamycin, aminoglycosides, and fluoroquinolones are the drugs of choice.1,3,4,6 Daptomycin and linezolid also are active in vitro,1 but clinical experience with these agents is limited. Necrotic infection or deep tissue involvement requires surgical intervention.
Numerous other organisms can cause cellulitis and soft tissue infections with hemorrhagic bullae.1,3,6 Streptococci, particularly Streptococcus pyogenes, and occasionally staphylococci are the primary consideration in normal hosts without trauma.3,6 In immunocompromised patients, including those with cirrhosis, diabetes mellitus, and malignancy, Clostridium perfringens and gram-negative organisms such as Escherichia coli, other enteric bacteria including Pseudomonas aeruginosa, Aeromonas, and halophilic Vibrio species are more frequent.3,6
We describe a patient with underlying cirrhosis who developed bilateral lower extremity hemorrhagic bullous lesions and sepsis due to infection with B cereus, an emerging cause of serious infections in patients with underlying immunocompromising conditions such as cirrhosis, diabetes mellitus, and malignancy. Hemorrhagic bullae in immunocompromised patients are associated with sepsis and rapidly progressive illness, and rapid treatment is essential. Bacillus cereus should be included as a consideration in the differential diagnosis and management of patients presenting with bullous cellulitis and sepsis.
1. Bottone EJ. Bacillus cereus, a volatile human pathogen. Clin Microbiol Rev. 2010;23:382-398.
2. Henrickson KJ. A second species of bacillus causing primary cutaneous disease. Int J Dermatol. 1990;29:19-20.
3. Liu BM, Hsiao CT, Chung KJ, et al. Hemorrhagic bullae represent an ominous sign for cirrhotic patients [published online ahead of print November 5, 2007]. J Emer Med. 2008;34:277-281.
4. Meredith FT, Fowler VG, Gautier M, et al. Bacillus cereus necrotizing cellulitis mimicking clostridial myonecrosis: case report and review of the literature. Scand J Infect Dis. 1997;29:528-529.
5. Kotiranta A, Lounatmaa K, Haapasalo M. Epidemiology and pathogenesis of Bacillus cereus infections. Microbes Infect. 2000;2:189-198.
6. Lee CC, Chi CH, Lee NY, et al. Necrotizing fasciitis in patients with liver cirrhosis: predominance of monomicrobial gram-negative bacillary infections [published online ahead of print July 23, 2008]. Diagn Microbiol Infect Dis. 2008;62:219-225.
To the Editor:
A 42-year-old man with hypertension, hypothyroidism, and alcohol-related cirrhosis was admitted for evaluation of rapidly deteriorating mental status. He was referred from a rehabilitation facility where he had been admitted 4 days earlier after a hospitalization for hepatorenal syndrome and pneumonia. He was alert and ambulating until the day of the current admission. On arrival he was hypotensive(54/42 mm Hg); hypothermic (35°C, rectally); and unresponsive, except to painful stimuli. Jaundice, hepatosplenomegaly, ascites, and bilateral lower extremity edema were noted. There were multiple tense and flaccid bullous lesions containing serosanguineous fluid over both tibias and calves, without crepitus (Figure 1).
|
|
|
|
|
|
Laboratory test results revealed leukocytosis (total leukocytes, 10,900/mm3 [reference range, 4500–10,800/mm3), hypoglycemia (glucose, <20 mg/dL [reference range, 74–106 mg/dL]), renal insufficiency (serum creatinine, 2.5 mg/dL [reference range, 0.66–1.25 mg/dL]), metabolic acidosis (pH, 7.1 [reference range, 7.35–7.45]; bicarbonate, 13 mmol/L [reference range, 22–30 mmol/L]; lactic acid, 11.9 mmol/L [reference range, 0.7–2.1 mmol/L]), liver dysfunction (aspartate aminotransferase, 576 IU/L [reference range, 15–46 IU/L]), and coagulopathy with evidence of diffuse intravascular coagulation (total platelets, 75,000/mm3 [reference range, 150,000–450,000/mm3]; international normalized ratio, 9.5 [reference range, 0.8–1.2]; partial thromboplastin time, 108 seconds [reference range 23.0–35.0 seconds]; fibrinogen, 145 mg/dL [reference range, 228–501 mg/dL]; D-dimer, >20 µg/mL [reference range, 0.01–0.58 μg/mL]). Computed tomography of the pelvis and legs showed ascites, extensive subcutaneous edema, and cutaneous blisterlike lesions superior to the level of the ankles bilaterally. No gas, foreign bodies, collections, asymmetric facial thickening, or evidence of infection across tissue planes was present (Figures 2 and 3).
Specimens of blood and aspirates from the bullae at multiple lower leg sites were sent for microbiologic evaluation. The blood specimens were inoculated at bedside into aerobic and anaerobic blood culture bottles and incubated in an automated blood culture system. The aspirate samples were inoculated to trypticase soy agar with 5% sheep blood, Columbia-nalidixic acid agar, chocolate agar, MacConkey agar, and thioglycollate broth, which were incubated at 37ºC in air supplemented with 5% CO2, and to CDC anaerobic blood agar, which was incubated under anaerobic conditions. Gram-stained smears of the aspirates from the bullae demonstrated few granulocytes and numerous large gram-positive bacilli (Figure 4). By the next day, growth of large gram-positive bacilli was detected in both aerobic and anaerobic blood culture bottles and in pure culture from all the bullae samples. The bacterial colonies on sheep blood agar were opaque and white-gray in color, with a rough surface, undulate margins, and surrounding β hemolysis. The isolate was a motile, catalase-positive, arginine-positive, salicin-positive, lecithinase-positive, and penicillin-resistant organism that was identified as Bacillus cereus.
Antimicrobial susceptibility testing for B cereus has not been standardized, but evaluation by broth microdilution suggested decreased susceptibility to penicillin (minimum inhibitory concentration [MIC], 2 µg/mL) and clindamycin (MIC, 2 µg/mL), but retained susceptibility to ciprofloxacin (MIC, ≤0.25 µg/mL), tetracycline (MIC, ≤1 µg/mL), rifampin (MIC, ≤1 µg/mL), and vancomycin (MIC, ≤2 µg/mL).
The patient was admitted to the intensive care unit and was treated initially with fluid resuscitation; transfusions; ventilatory support; and intravenous vancomycin, clindamycin, and imipenem. This regimen was changed to vancomycin and ciprofloxacin when culture and susceptibility results became available to complete a 14-day course. Signs of sepsis resolved and the mental status and skin lesions improved. Ultimately, the patient died due to complications of hepatic failure.
Bacillus cereus is a rod-shaped, gram-positive, facultative, aerobic organism that is widely distributed in the environment.1 Spore formation makes B cereus resistant to most physical and chemical disinfection methods; as a consequence, it is a frequent contaminant in materials (eg, plants, dust, soil, sediment), foodstuffs, and clinical specimens.1
Traditionally considered in the context of foodborne illness, B cereus is recognized increasingly as a cause of systemic and local infections in both immunosuppressed and immunocompetent patients. Nongastrointestinal infections reported include fulminant bacteremia, pneumonia, meningitis, brain abscesses, endophthalmitis, necrotizing fasciitis, and central line catheter–related and cutaneous infections.1,2
Cutaneous lesions may have a variety of forms and appearance at initial presentation, including small papules or vesicles that progress into a rapidly spreading cellulitis1,2 with a characteristic serosanguineous draining fluid,2 single necrotic bullae,3 and gas-gangrenelike infections with extensive soft tissue involvement resembling clostridial myonecrosis.1,4 Single or multiple papulovesicular lesions can even mimic cutaneous anthrax.1-4 Necrotic or hemorrhagic bullous lesions,3 such as those observed in our patient, are rare.
Exposed areas such as extremities and digits are most often affected, presumably due to entrance of spores from soil, water, decaying organic material, or fomites through skin microabrasions or trauma-induced wounds.1 Once in the tissue, the crystalline surface protein layer (S-layer) of the bacilli promotes adhesion to human epithelial cells and neutrophils,5 followed by release of virulence factors including proteases, collagenases, lecithinaselike enzymes, necrotizing exotoxinlike hemolysins, phospholipases, and most importantly a dermonecrotic vascular permeability factor.1,5 Toxins produced by B cereus are similar to those closely related to Bacillus anthracis, the agent of anthrax.1,2
When large gram-positive bacilli are observed in tissue or wound specimens, initial therapy should address both aerobic (Bacillus species) and anaerobic (Clostridium species) organisms.1,4,6 Once B cereus is recovered, treatment should rely on susceptibility testing of the isolate. Bacillus cereus produces ß-lactamase, thus penicillin and cephalosporin should be avoided.1 Vancomycin, clindamycin, aminoglycosides, and fluoroquinolones are the drugs of choice.1,3,4,6 Daptomycin and linezolid also are active in vitro,1 but clinical experience with these agents is limited. Necrotic infection or deep tissue involvement requires surgical intervention.
Numerous other organisms can cause cellulitis and soft tissue infections with hemorrhagic bullae.1,3,6 Streptococci, particularly Streptococcus pyogenes, and occasionally staphylococci are the primary consideration in normal hosts without trauma.3,6 In immunocompromised patients, including those with cirrhosis, diabetes mellitus, and malignancy, Clostridium perfringens and gram-negative organisms such as Escherichia coli, other enteric bacteria including Pseudomonas aeruginosa, Aeromonas, and halophilic Vibrio species are more frequent.3,6
We describe a patient with underlying cirrhosis who developed bilateral lower extremity hemorrhagic bullous lesions and sepsis due to infection with B cereus, an emerging cause of serious infections in patients with underlying immunocompromising conditions such as cirrhosis, diabetes mellitus, and malignancy. Hemorrhagic bullae in immunocompromised patients are associated with sepsis and rapidly progressive illness, and rapid treatment is essential. Bacillus cereus should be included as a consideration in the differential diagnosis and management of patients presenting with bullous cellulitis and sepsis.
To the Editor:
A 42-year-old man with hypertension, hypothyroidism, and alcohol-related cirrhosis was admitted for evaluation of rapidly deteriorating mental status. He was referred from a rehabilitation facility where he had been admitted 4 days earlier after a hospitalization for hepatorenal syndrome and pneumonia. He was alert and ambulating until the day of the current admission. On arrival he was hypotensive(54/42 mm Hg); hypothermic (35°C, rectally); and unresponsive, except to painful stimuli. Jaundice, hepatosplenomegaly, ascites, and bilateral lower extremity edema were noted. There were multiple tense and flaccid bullous lesions containing serosanguineous fluid over both tibias and calves, without crepitus (Figure 1).
|
|
|
|
|
|
Laboratory test results revealed leukocytosis (total leukocytes, 10,900/mm3 [reference range, 4500–10,800/mm3), hypoglycemia (glucose, <20 mg/dL [reference range, 74–106 mg/dL]), renal insufficiency (serum creatinine, 2.5 mg/dL [reference range, 0.66–1.25 mg/dL]), metabolic acidosis (pH, 7.1 [reference range, 7.35–7.45]; bicarbonate, 13 mmol/L [reference range, 22–30 mmol/L]; lactic acid, 11.9 mmol/L [reference range, 0.7–2.1 mmol/L]), liver dysfunction (aspartate aminotransferase, 576 IU/L [reference range, 15–46 IU/L]), and coagulopathy with evidence of diffuse intravascular coagulation (total platelets, 75,000/mm3 [reference range, 150,000–450,000/mm3]; international normalized ratio, 9.5 [reference range, 0.8–1.2]; partial thromboplastin time, 108 seconds [reference range 23.0–35.0 seconds]; fibrinogen, 145 mg/dL [reference range, 228–501 mg/dL]; D-dimer, >20 µg/mL [reference range, 0.01–0.58 μg/mL]). Computed tomography of the pelvis and legs showed ascites, extensive subcutaneous edema, and cutaneous blisterlike lesions superior to the level of the ankles bilaterally. No gas, foreign bodies, collections, asymmetric facial thickening, or evidence of infection across tissue planes was present (Figures 2 and 3).
Specimens of blood and aspirates from the bullae at multiple lower leg sites were sent for microbiologic evaluation. The blood specimens were inoculated at bedside into aerobic and anaerobic blood culture bottles and incubated in an automated blood culture system. The aspirate samples were inoculated to trypticase soy agar with 5% sheep blood, Columbia-nalidixic acid agar, chocolate agar, MacConkey agar, and thioglycollate broth, which were incubated at 37ºC in air supplemented with 5% CO2, and to CDC anaerobic blood agar, which was incubated under anaerobic conditions. Gram-stained smears of the aspirates from the bullae demonstrated few granulocytes and numerous large gram-positive bacilli (Figure 4). By the next day, growth of large gram-positive bacilli was detected in both aerobic and anaerobic blood culture bottles and in pure culture from all the bullae samples. The bacterial colonies on sheep blood agar were opaque and white-gray in color, with a rough surface, undulate margins, and surrounding β hemolysis. The isolate was a motile, catalase-positive, arginine-positive, salicin-positive, lecithinase-positive, and penicillin-resistant organism that was identified as Bacillus cereus.
Antimicrobial susceptibility testing for B cereus has not been standardized, but evaluation by broth microdilution suggested decreased susceptibility to penicillin (minimum inhibitory concentration [MIC], 2 µg/mL) and clindamycin (MIC, 2 µg/mL), but retained susceptibility to ciprofloxacin (MIC, ≤0.25 µg/mL), tetracycline (MIC, ≤1 µg/mL), rifampin (MIC, ≤1 µg/mL), and vancomycin (MIC, ≤2 µg/mL).
The patient was admitted to the intensive care unit and was treated initially with fluid resuscitation; transfusions; ventilatory support; and intravenous vancomycin, clindamycin, and imipenem. This regimen was changed to vancomycin and ciprofloxacin when culture and susceptibility results became available to complete a 14-day course. Signs of sepsis resolved and the mental status and skin lesions improved. Ultimately, the patient died due to complications of hepatic failure.
Bacillus cereus is a rod-shaped, gram-positive, facultative, aerobic organism that is widely distributed in the environment.1 Spore formation makes B cereus resistant to most physical and chemical disinfection methods; as a consequence, it is a frequent contaminant in materials (eg, plants, dust, soil, sediment), foodstuffs, and clinical specimens.1
Traditionally considered in the context of foodborne illness, B cereus is recognized increasingly as a cause of systemic and local infections in both immunosuppressed and immunocompetent patients. Nongastrointestinal infections reported include fulminant bacteremia, pneumonia, meningitis, brain abscesses, endophthalmitis, necrotizing fasciitis, and central line catheter–related and cutaneous infections.1,2
Cutaneous lesions may have a variety of forms and appearance at initial presentation, including small papules or vesicles that progress into a rapidly spreading cellulitis1,2 with a characteristic serosanguineous draining fluid,2 single necrotic bullae,3 and gas-gangrenelike infections with extensive soft tissue involvement resembling clostridial myonecrosis.1,4 Single or multiple papulovesicular lesions can even mimic cutaneous anthrax.1-4 Necrotic or hemorrhagic bullous lesions,3 such as those observed in our patient, are rare.
Exposed areas such as extremities and digits are most often affected, presumably due to entrance of spores from soil, water, decaying organic material, or fomites through skin microabrasions or trauma-induced wounds.1 Once in the tissue, the crystalline surface protein layer (S-layer) of the bacilli promotes adhesion to human epithelial cells and neutrophils,5 followed by release of virulence factors including proteases, collagenases, lecithinaselike enzymes, necrotizing exotoxinlike hemolysins, phospholipases, and most importantly a dermonecrotic vascular permeability factor.1,5 Toxins produced by B cereus are similar to those closely related to Bacillus anthracis, the agent of anthrax.1,2
When large gram-positive bacilli are observed in tissue or wound specimens, initial therapy should address both aerobic (Bacillus species) and anaerobic (Clostridium species) organisms.1,4,6 Once B cereus is recovered, treatment should rely on susceptibility testing of the isolate. Bacillus cereus produces ß-lactamase, thus penicillin and cephalosporin should be avoided.1 Vancomycin, clindamycin, aminoglycosides, and fluoroquinolones are the drugs of choice.1,3,4,6 Daptomycin and linezolid also are active in vitro,1 but clinical experience with these agents is limited. Necrotic infection or deep tissue involvement requires surgical intervention.
Numerous other organisms can cause cellulitis and soft tissue infections with hemorrhagic bullae.1,3,6 Streptococci, particularly Streptococcus pyogenes, and occasionally staphylococci are the primary consideration in normal hosts without trauma.3,6 In immunocompromised patients, including those with cirrhosis, diabetes mellitus, and malignancy, Clostridium perfringens and gram-negative organisms such as Escherichia coli, other enteric bacteria including Pseudomonas aeruginosa, Aeromonas, and halophilic Vibrio species are more frequent.3,6
We describe a patient with underlying cirrhosis who developed bilateral lower extremity hemorrhagic bullous lesions and sepsis due to infection with B cereus, an emerging cause of serious infections in patients with underlying immunocompromising conditions such as cirrhosis, diabetes mellitus, and malignancy. Hemorrhagic bullae in immunocompromised patients are associated with sepsis and rapidly progressive illness, and rapid treatment is essential. Bacillus cereus should be included as a consideration in the differential diagnosis and management of patients presenting with bullous cellulitis and sepsis.
1. Bottone EJ. Bacillus cereus, a volatile human pathogen. Clin Microbiol Rev. 2010;23:382-398.
2. Henrickson KJ. A second species of bacillus causing primary cutaneous disease. Int J Dermatol. 1990;29:19-20.
3. Liu BM, Hsiao CT, Chung KJ, et al. Hemorrhagic bullae represent an ominous sign for cirrhotic patients [published online ahead of print November 5, 2007]. J Emer Med. 2008;34:277-281.
4. Meredith FT, Fowler VG, Gautier M, et al. Bacillus cereus necrotizing cellulitis mimicking clostridial myonecrosis: case report and review of the literature. Scand J Infect Dis. 1997;29:528-529.
5. Kotiranta A, Lounatmaa K, Haapasalo M. Epidemiology and pathogenesis of Bacillus cereus infections. Microbes Infect. 2000;2:189-198.
6. Lee CC, Chi CH, Lee NY, et al. Necrotizing fasciitis in patients with liver cirrhosis: predominance of monomicrobial gram-negative bacillary infections [published online ahead of print July 23, 2008]. Diagn Microbiol Infect Dis. 2008;62:219-225.
1. Bottone EJ. Bacillus cereus, a volatile human pathogen. Clin Microbiol Rev. 2010;23:382-398.
2. Henrickson KJ. A second species of bacillus causing primary cutaneous disease. Int J Dermatol. 1990;29:19-20.
3. Liu BM, Hsiao CT, Chung KJ, et al. Hemorrhagic bullae represent an ominous sign for cirrhotic patients [published online ahead of print November 5, 2007]. J Emer Med. 2008;34:277-281.
4. Meredith FT, Fowler VG, Gautier M, et al. Bacillus cereus necrotizing cellulitis mimicking clostridial myonecrosis: case report and review of the literature. Scand J Infect Dis. 1997;29:528-529.
5. Kotiranta A, Lounatmaa K, Haapasalo M. Epidemiology and pathogenesis of Bacillus cereus infections. Microbes Infect. 2000;2:189-198.
6. Lee CC, Chi CH, Lee NY, et al. Necrotizing fasciitis in patients with liver cirrhosis: predominance of monomicrobial gram-negative bacillary infections [published online ahead of print July 23, 2008]. Diagn Microbiol Infect Dis. 2008;62:219-225.
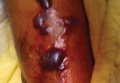
Late-Onset Nevus Comedonicus on Both Eyelids With Hypothyroidism
To the Editor:
A 62-year-old woman was referred to the dermatology clinic for papules on both eyelids of 6 months’ duration. She underwent surgery for a thyroid gland adenoma 3 years prior and subsequently experienced hypothyroidism. Levothyroxine sodium was administered daily (100 µg initially; 50 µg over the last 1.5 years). Papules occurred on both eyelids 6 months prior to presentation and gradually increased in number. The center of each papule was filled with a black keratinous plug. The skin lesions became raised after the patient ate fatty foods. The lesions remained entirely asymptomatic and there was no family history of a similar disorder.
Physical examinations showed no systemic abnormalities. Dermatologic examination showed clustered 3- to 4-mm flesh-colored papules on both upper eyelids; the centers of the papules were filled with 1- to 2-mm black keratinous plugs (Figure 1A). Several similar skin lesions existed on the lower eyelids, nasal root, and right side of the nasal dorsum. On laboratory examination, the results of routine blood, urine, and stool tests, as well as renal and hepatic functions, electrolytes, and blood sugar levels, were within reference range. Indicators including triglyceride of 2.50 mmol/L (reference range, 0.40–1.90 mmol/L), total cholesterol of 6.31 mmol/L (reference range, 3.00–5.70 mmol/L), serum total thyroxine (T4) of 5.32 µg/dL (reference range, 6.09–12.23 µg/dL), total triiodothyronine (T3) of 64 ng/dL (reference range, 87–178 ng/dL), serum free thyroxine (FT4) of 0.41 ng/dL (reference range, 0.61–1.12 ng/dL), serum free triiodothyronine (FT3) of 182 pg/dL (reference range, 250–390 pg/dL), and thyrotropin of 33.75 µIU/mL (reference range, 0.34–5.60 µIU/mL) were not within reference range; however, thyroperoxidase antibodies, thyrotropin receptor antibodies, thyroglobulin antibodies, thyroglobulin, and calcitonin were within reference range. Color ultrasonography indicated post–subtotal resection of the bilateral thyroid glands.
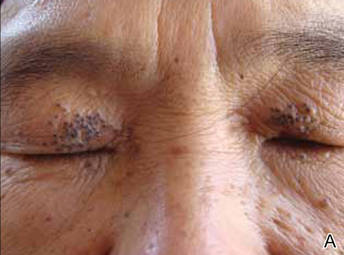
|
Histopathologic analysis of the skin lesions showed that the epidermis became atrophic and thinner, and several atrophic and cyst-dilated follicular structures existed inside the dermis. Some structures opened through the epidermis; the walls were squamous epithelium and keratin filled the structures (Figure 2). The condition was diagnosed as nevus comedonicus (NC).
The patient was referred to the endocrinology department and treated with levothyroxine sodium (100 µg daily). At 5-month follow-up, the T4, T3, FT4, FT3, and thyrotropin levels were within reference range and most of the skin lesions had resolved (Figure 1B).
Nevus comedonicus is an unusual skin lesion with a predilection for the face, neck, shoulders, upper arms, and trunk. The clinical manifestations include comedonelike papules with centers that are characterized by large, black, solid keratinous plugs. When the plugs are peeled off, volcanic craterlike pits will be left. The skin lesions usually are ribbonlike and clustered on 1 side of the body. Pathologic examination often shows that the epidermis is pitted downward, and the dilated follicular ostia are plugged with keratin.1,2 Paige and Mendelson3 divided NC into 2 types: inflammatory and noninflammatory. Approximately half of NC patients experience cysts, abscesses, fistulae, and scars.4
The exact etiology of NC is unclear. Some researchers believe that it is a congenital hair follicle deformity; more specifically, that it is caused by a developmental defect in the hair follicles in the embryonic stage (ie, abnormal differentiation of epithelial stem cells that differentiate into follicles). Most incidences of NC occur at birth or before growth and development. However, few studies have reported late-onset NC.5
|
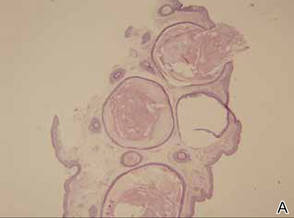
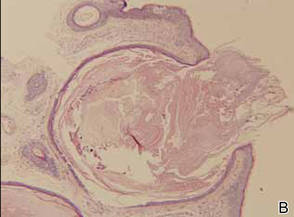
The relationship between NC and thyroid disease is unique. Clinical research has shown that hypothyroidism can result in hair loss and cracks.6 In animal experiments, hypothyroidism model mice often experienced degenerative changes of their hair follicles and hair papillae as well as changes in the telogen phase, such as thinning of the outer and inner root sheaths.7 Meanwhile, decreased cell proliferation activity in the hair follicles was observed. Therefore, it is reasonable to conclude that thyroid hormones have regulatory effects on the growth and development of hair follicles.7
A study on human hair follicles found that thyroid hormone receptor β1 is expressed in human hair follicles.8 Research on in vitro–cultured human hair follicles showed that thyroid hormones T3 and T4 upregulated the proliferation of hair matrix cells and downregulated their apoptosis. Thyroid hormones also prolonged the duration of the hair growth phase (anagen).9 Furthermore, expression of thyrotropin receptor was detected in human hair follicles. Because increased serum thyrotropin levels can lead to clinical hair loss, thyrotropin may inhibit the growth of hair follicles via thyrotropin receptor.10 In our patient, NC occurred on both eyelids when the patient experienced hypothyroidism following thyroid gland adenoma surgery. Following treatment with levothyroxine sodium, the T4, T3, FT4, FT3, and thyrotropin levels were within reference range and most of the skin lesions resolved. Therefore, the occurrence of NC may be related to hypothyroidism in this patient. The low thyroid hormone levels and elevated thyrotropin level possibly induced degenerative changes and injuries to the hair matrix cells, resulting in hair follicle obstruction and accumulation of keratin, which ultimately led to NC. However, the exact relationship between NC and thyroid diseases requires elucidation in future studies.
1. Engber PB. The nevus comedonicus syndrome: a case report with emphasis on associated internal manifestations. Int J Dermatol. 1978;17:745-749.
2. Kirtak N, Inaloz HS, Karakok M, et al. Extensive inflammatory nevus comedonicus involving half of the body. Int J Dermatol. 2004;43:434-436.
3. Paige TN, Mendelson CG. Bilateral nevus comedonicus. Arch Dermatol. 1967;96:172-175.
4. James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: WB Saunders; 2006.
5. Ahn SY, Oh Y, Bak H, et al. Co-occurrence of nevus comedonicus with accessory breast tissue. Int J Dermatol. 2008;47:530-531.
6. Freinkel RK, Freinkel N. Hair growth and alopecia in hypothyroidism. Arch Dermatol. 1972;106:349-352.
7. Tsujio M, Yoshioka K, Satoh M, et al. Skin morphology of thyroidectomized rats. Vet Pathol. 2008;45:505-511.
8. Billoni N, Buan B, Gautier B, et al. Thyroid hormone receptor β1 is expressed in the human hair follicle. Br J Dermatol. 2000;142:645-652.
9. van Beek N, Bodó E, Kromminga A, et al. Thyroid hormones directly alter human hair follicle functions: anagen prolongation and stimulation of both hair matrix keratinocyte proliferation and hair pigmentation. J Clin Endocrinol Metab. 2008;93:4381-4388.
10. Bodó E, Kromminga A, Bíró T, et al. Human female hair follicles are a direct, nonclassical target for thyroid-stimulating hormone. J Invest Dermatol. 2009;129:1126-1139.
To the Editor:
A 62-year-old woman was referred to the dermatology clinic for papules on both eyelids of 6 months’ duration. She underwent surgery for a thyroid gland adenoma 3 years prior and subsequently experienced hypothyroidism. Levothyroxine sodium was administered daily (100 µg initially; 50 µg over the last 1.5 years). Papules occurred on both eyelids 6 months prior to presentation and gradually increased in number. The center of each papule was filled with a black keratinous plug. The skin lesions became raised after the patient ate fatty foods. The lesions remained entirely asymptomatic and there was no family history of a similar disorder.
Physical examinations showed no systemic abnormalities. Dermatologic examination showed clustered 3- to 4-mm flesh-colored papules on both upper eyelids; the centers of the papules were filled with 1- to 2-mm black keratinous plugs (Figure 1A). Several similar skin lesions existed on the lower eyelids, nasal root, and right side of the nasal dorsum. On laboratory examination, the results of routine blood, urine, and stool tests, as well as renal and hepatic functions, electrolytes, and blood sugar levels, were within reference range. Indicators including triglyceride of 2.50 mmol/L (reference range, 0.40–1.90 mmol/L), total cholesterol of 6.31 mmol/L (reference range, 3.00–5.70 mmol/L), serum total thyroxine (T4) of 5.32 µg/dL (reference range, 6.09–12.23 µg/dL), total triiodothyronine (T3) of 64 ng/dL (reference range, 87–178 ng/dL), serum free thyroxine (FT4) of 0.41 ng/dL (reference range, 0.61–1.12 ng/dL), serum free triiodothyronine (FT3) of 182 pg/dL (reference range, 250–390 pg/dL), and thyrotropin of 33.75 µIU/mL (reference range, 0.34–5.60 µIU/mL) were not within reference range; however, thyroperoxidase antibodies, thyrotropin receptor antibodies, thyroglobulin antibodies, thyroglobulin, and calcitonin were within reference range. Color ultrasonography indicated post–subtotal resection of the bilateral thyroid glands.
|
|
Histopathologic analysis of the skin lesions showed that the epidermis became atrophic and thinner, and several atrophic and cyst-dilated follicular structures existed inside the dermis. Some structures opened through the epidermis; the walls were squamous epithelium and keratin filled the structures (Figure 2). The condition was diagnosed as nevus comedonicus (NC).
The patient was referred to the endocrinology department and treated with levothyroxine sodium (100 µg daily). At 5-month follow-up, the T4, T3, FT4, FT3, and thyrotropin levels were within reference range and most of the skin lesions had resolved (Figure 1B).
Nevus comedonicus is an unusual skin lesion with a predilection for the face, neck, shoulders, upper arms, and trunk. The clinical manifestations include comedonelike papules with centers that are characterized by large, black, solid keratinous plugs. When the plugs are peeled off, volcanic craterlike pits will be left. The skin lesions usually are ribbonlike and clustered on 1 side of the body. Pathologic examination often shows that the epidermis is pitted downward, and the dilated follicular ostia are plugged with keratin.1,2 Paige and Mendelson3 divided NC into 2 types: inflammatory and noninflammatory. Approximately half of NC patients experience cysts, abscesses, fistulae, and scars.4
The exact etiology of NC is unclear. Some researchers believe that it is a congenital hair follicle deformity; more specifically, that it is caused by a developmental defect in the hair follicles in the embryonic stage (ie, abnormal differentiation of epithelial stem cells that differentiate into follicles). Most incidences of NC occur at birth or before growth and development. However, few studies have reported late-onset NC.5
|
The relationship between NC and thyroid disease is unique. Clinical research has shown that hypothyroidism can result in hair loss and cracks.6 In animal experiments, hypothyroidism model mice often experienced degenerative changes of their hair follicles and hair papillae as well as changes in the telogen phase, such as thinning of the outer and inner root sheaths.7 Meanwhile, decreased cell proliferation activity in the hair follicles was observed. Therefore, it is reasonable to conclude that thyroid hormones have regulatory effects on the growth and development of hair follicles.7
A study on human hair follicles found that thyroid hormone receptor β1 is expressed in human hair follicles.8 Research on in vitro–cultured human hair follicles showed that thyroid hormones T3 and T4 upregulated the proliferation of hair matrix cells and downregulated their apoptosis. Thyroid hormones also prolonged the duration of the hair growth phase (anagen).9 Furthermore, expression of thyrotropin receptor was detected in human hair follicles. Because increased serum thyrotropin levels can lead to clinical hair loss, thyrotropin may inhibit the growth of hair follicles via thyrotropin receptor.10 In our patient, NC occurred on both eyelids when the patient experienced hypothyroidism following thyroid gland adenoma surgery. Following treatment with levothyroxine sodium, the T4, T3, FT4, FT3, and thyrotropin levels were within reference range and most of the skin lesions resolved. Therefore, the occurrence of NC may be related to hypothyroidism in this patient. The low thyroid hormone levels and elevated thyrotropin level possibly induced degenerative changes and injuries to the hair matrix cells, resulting in hair follicle obstruction and accumulation of keratin, which ultimately led to NC. However, the exact relationship between NC and thyroid diseases requires elucidation in future studies.
To the Editor:
A 62-year-old woman was referred to the dermatology clinic for papules on both eyelids of 6 months’ duration. She underwent surgery for a thyroid gland adenoma 3 years prior and subsequently experienced hypothyroidism. Levothyroxine sodium was administered daily (100 µg initially; 50 µg over the last 1.5 years). Papules occurred on both eyelids 6 months prior to presentation and gradually increased in number. The center of each papule was filled with a black keratinous plug. The skin lesions became raised after the patient ate fatty foods. The lesions remained entirely asymptomatic and there was no family history of a similar disorder.
Physical examinations showed no systemic abnormalities. Dermatologic examination showed clustered 3- to 4-mm flesh-colored papules on both upper eyelids; the centers of the papules were filled with 1- to 2-mm black keratinous plugs (Figure 1A). Several similar skin lesions existed on the lower eyelids, nasal root, and right side of the nasal dorsum. On laboratory examination, the results of routine blood, urine, and stool tests, as well as renal and hepatic functions, electrolytes, and blood sugar levels, were within reference range. Indicators including triglyceride of 2.50 mmol/L (reference range, 0.40–1.90 mmol/L), total cholesterol of 6.31 mmol/L (reference range, 3.00–5.70 mmol/L), serum total thyroxine (T4) of 5.32 µg/dL (reference range, 6.09–12.23 µg/dL), total triiodothyronine (T3) of 64 ng/dL (reference range, 87–178 ng/dL), serum free thyroxine (FT4) of 0.41 ng/dL (reference range, 0.61–1.12 ng/dL), serum free triiodothyronine (FT3) of 182 pg/dL (reference range, 250–390 pg/dL), and thyrotropin of 33.75 µIU/mL (reference range, 0.34–5.60 µIU/mL) were not within reference range; however, thyroperoxidase antibodies, thyrotropin receptor antibodies, thyroglobulin antibodies, thyroglobulin, and calcitonin were within reference range. Color ultrasonography indicated post–subtotal resection of the bilateral thyroid glands.
|
|
Histopathologic analysis of the skin lesions showed that the epidermis became atrophic and thinner, and several atrophic and cyst-dilated follicular structures existed inside the dermis. Some structures opened through the epidermis; the walls were squamous epithelium and keratin filled the structures (Figure 2). The condition was diagnosed as nevus comedonicus (NC).
The patient was referred to the endocrinology department and treated with levothyroxine sodium (100 µg daily). At 5-month follow-up, the T4, T3, FT4, FT3, and thyrotropin levels were within reference range and most of the skin lesions had resolved (Figure 1B).
Nevus comedonicus is an unusual skin lesion with a predilection for the face, neck, shoulders, upper arms, and trunk. The clinical manifestations include comedonelike papules with centers that are characterized by large, black, solid keratinous plugs. When the plugs are peeled off, volcanic craterlike pits will be left. The skin lesions usually are ribbonlike and clustered on 1 side of the body. Pathologic examination often shows that the epidermis is pitted downward, and the dilated follicular ostia are plugged with keratin.1,2 Paige and Mendelson3 divided NC into 2 types: inflammatory and noninflammatory. Approximately half of NC patients experience cysts, abscesses, fistulae, and scars.4
The exact etiology of NC is unclear. Some researchers believe that it is a congenital hair follicle deformity; more specifically, that it is caused by a developmental defect in the hair follicles in the embryonic stage (ie, abnormal differentiation of epithelial stem cells that differentiate into follicles). Most incidences of NC occur at birth or before growth and development. However, few studies have reported late-onset NC.5
|
The relationship between NC and thyroid disease is unique. Clinical research has shown that hypothyroidism can result in hair loss and cracks.6 In animal experiments, hypothyroidism model mice often experienced degenerative changes of their hair follicles and hair papillae as well as changes in the telogen phase, such as thinning of the outer and inner root sheaths.7 Meanwhile, decreased cell proliferation activity in the hair follicles was observed. Therefore, it is reasonable to conclude that thyroid hormones have regulatory effects on the growth and development of hair follicles.7
A study on human hair follicles found that thyroid hormone receptor β1 is expressed in human hair follicles.8 Research on in vitro–cultured human hair follicles showed that thyroid hormones T3 and T4 upregulated the proliferation of hair matrix cells and downregulated their apoptosis. Thyroid hormones also prolonged the duration of the hair growth phase (anagen).9 Furthermore, expression of thyrotropin receptor was detected in human hair follicles. Because increased serum thyrotropin levels can lead to clinical hair loss, thyrotropin may inhibit the growth of hair follicles via thyrotropin receptor.10 In our patient, NC occurred on both eyelids when the patient experienced hypothyroidism following thyroid gland adenoma surgery. Following treatment with levothyroxine sodium, the T4, T3, FT4, FT3, and thyrotropin levels were within reference range and most of the skin lesions resolved. Therefore, the occurrence of NC may be related to hypothyroidism in this patient. The low thyroid hormone levels and elevated thyrotropin level possibly induced degenerative changes and injuries to the hair matrix cells, resulting in hair follicle obstruction and accumulation of keratin, which ultimately led to NC. However, the exact relationship between NC and thyroid diseases requires elucidation in future studies.
1. Engber PB. The nevus comedonicus syndrome: a case report with emphasis on associated internal manifestations. Int J Dermatol. 1978;17:745-749.
2. Kirtak N, Inaloz HS, Karakok M, et al. Extensive inflammatory nevus comedonicus involving half of the body. Int J Dermatol. 2004;43:434-436.
3. Paige TN, Mendelson CG. Bilateral nevus comedonicus. Arch Dermatol. 1967;96:172-175.
4. James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: WB Saunders; 2006.
5. Ahn SY, Oh Y, Bak H, et al. Co-occurrence of nevus comedonicus with accessory breast tissue. Int J Dermatol. 2008;47:530-531.
6. Freinkel RK, Freinkel N. Hair growth and alopecia in hypothyroidism. Arch Dermatol. 1972;106:349-352.
7. Tsujio M, Yoshioka K, Satoh M, et al. Skin morphology of thyroidectomized rats. Vet Pathol. 2008;45:505-511.
8. Billoni N, Buan B, Gautier B, et al. Thyroid hormone receptor β1 is expressed in the human hair follicle. Br J Dermatol. 2000;142:645-652.
9. van Beek N, Bodó E, Kromminga A, et al. Thyroid hormones directly alter human hair follicle functions: anagen prolongation and stimulation of both hair matrix keratinocyte proliferation and hair pigmentation. J Clin Endocrinol Metab. 2008;93:4381-4388.
10. Bodó E, Kromminga A, Bíró T, et al. Human female hair follicles are a direct, nonclassical target for thyroid-stimulating hormone. J Invest Dermatol. 2009;129:1126-1139.
1. Engber PB. The nevus comedonicus syndrome: a case report with emphasis on associated internal manifestations. Int J Dermatol. 1978;17:745-749.
2. Kirtak N, Inaloz HS, Karakok M, et al. Extensive inflammatory nevus comedonicus involving half of the body. Int J Dermatol. 2004;43:434-436.
3. Paige TN, Mendelson CG. Bilateral nevus comedonicus. Arch Dermatol. 1967;96:172-175.
4. James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: WB Saunders; 2006.
5. Ahn SY, Oh Y, Bak H, et al. Co-occurrence of nevus comedonicus with accessory breast tissue. Int J Dermatol. 2008;47:530-531.
6. Freinkel RK, Freinkel N. Hair growth and alopecia in hypothyroidism. Arch Dermatol. 1972;106:349-352.
7. Tsujio M, Yoshioka K, Satoh M, et al. Skin morphology of thyroidectomized rats. Vet Pathol. 2008;45:505-511.
8. Billoni N, Buan B, Gautier B, et al. Thyroid hormone receptor β1 is expressed in the human hair follicle. Br J Dermatol. 2000;142:645-652.
9. van Beek N, Bodó E, Kromminga A, et al. Thyroid hormones directly alter human hair follicle functions: anagen prolongation and stimulation of both hair matrix keratinocyte proliferation and hair pigmentation. J Clin Endocrinol Metab. 2008;93:4381-4388.
10. Bodó E, Kromminga A, Bíró T, et al. Human female hair follicles are a direct, nonclassical target for thyroid-stimulating hormone. J Invest Dermatol. 2009;129:1126-1139.
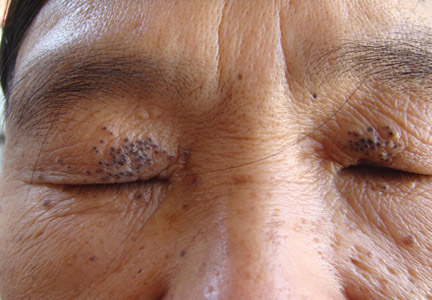
Leser-Trélat Sign: A Paraneoplastic Process?
To the Editor:
Leser-Trélat sign is a rare skin condition characterized by the sudden appearance of seborrheic keratoses that rapidly increase in number and size within weeks to months. Co-occurrence has been reported with a large number of malignancies, particularly adenocarcinoma and lymphoma. We present a case of Leser-Trélat sign that was not associated with an underlying malignancy.
A 44-year-old man was admitted to our dermatology outpatient department with a serpigo on the neck that had grown rapidly in the last month. His medical history and family history were unremarkable. Dermatologic examination revealed numerous 3- to 4-mm brown and slightly verrucous papules on the neck (Figure 1). A punch biopsy of the lesion showed acanthosis of predominantly basaloid cells, papillomatosis, and hyperkeratosis, as well as the presence of characteristic horn cysts (Figure 2). He was tested for possible underlying internal malignancy. Liver and kidney function tests, electrolyte count, protein electrophoresis, and whole blood and urine tests were within reference range. Chest radiography and abdominal ultrasonography revealed no signs of pathology. The erythrocyte sedimentation rate was 20 mm/h (reference range, 0–20 mm/h) and tests for hepatitis, human immunodeficiency virus, and syphilis were negative. Abdominal, cranial, and thorax computed tomography revealed no abnormalities. Otolaryngologic examinations also were negative. Additional endoscopic analyses, esophagogastroduodenoscopy, and colonoscopy revealed no abnormalities. At 1-year follow-up, the seborrheic keratoses remained unchanged. He has remained in good health without specific signs or symptoms suggestive of an underlying malignancy.
Paraneoplastic syndromes are associated with malignancy but progress without connection to a primary tumor or metastasis and form a group of clinical manifestations. The characteristic progress of paraneoplastic syndromes shows parallelism with the progression of the tumor. The mechanism underlying the development is not known, though the actions of bioactive substances that cause responses in the tumor, such as polypeptide hormones, hormonelike peptides, antibodies or immune complexes, and cytokines or growth factors, have been implicated.1
Although the term paraneoplastic syndrome commonly is used for Leser-Trélat sign, we do not believe it is accurate. As Fink et al2 and Schwengle et al3 indicated, the possibility of the co-occurrence being fortuitous is high. Showing a parallel progress of malignancy with paraneoplastic dermatosis requires that the paraneoplastic syndrome also diminish when the tumor is cured.4 It should then reappear with cancer recurrence or metastasis, which has not been exhibited in many case presentations in the literature.3 Disease regression was observed in only 1 of 3 seborrheic keratosis cases after primary cancer treatment.5
In patients with a malignancy, the sudden increase in seborrheic keratosis is based exclusively on the subjective evaluation of the patient, which may not be reliable. Schwengle et al3 stated that this sudden increase can be related to the awareness level of the patient who had a cancer diagnosis. Bräuer et al6 stated that no plausible definition distinguishes eruptive versus common seborrheic keratoses.
As a result, the results regarding the relationship between malignancy and Leser-Trélat sign are conflicting, and no strong evidence supports the presence of the sign. Only case reports have suggested that Leser-Trélat sign accompanies malignancy. Studies investigating its etiopathogenesis have not revealed a substance that has been released from or as a response to a tumor.
We believe that the presence of eruptive seborrheic keratosis does not necessitate screening for underlying internal malignancies.
- Cohen PR. Paraneoplastic dermatopathology: cutaneous paraneoplastic syndromes. Adv Dermatol. 1996;11:215-252.
- Fink AM, Filz D, Krajnik G, et al. Seborrhoeic keratoses in patients with internal malignancies: a case-control study with a prospective accrual of patients. J Eur Acad Dermatol Venereol. 2009;23:1316-1319.
- Schwengle LE, Rampen FH, Wobbes T. Seborrhoeic keratoses and internal malignancies. a case control study. Clin Exp Dermatol. 1988;13:177-179.
- Curth HO. Skin lesions and internal carcinoma. In: Andrade S, Gumport S, Popkin GL, et al, eds. Cancer of the Skin: Biology, Diagnosis, and Management. Vol 2. Philadelphia, PA: WB Saunders; 1976:1308-1341.
- Heaphy MR Jr, Millns JL, Schroeter AL. The sign of Leser-Trélat in a case of adenocarcinoma of the lung. J Am Acad Dermatol. 2000;43(2, pt 2):386-390.
- Bräuer J, Happle R, Gieler U. The sign of Leser-Trélat: fact or myth? J Eur Acad Dermatol Venereol. 1992;1:77-80.
To the Editor:
Leser-Trélat sign is a rare skin condition characterized by the sudden appearance of seborrheic keratoses that rapidly increase in number and size within weeks to months. Co-occurrence has been reported with a large number of malignancies, particularly adenocarcinoma and lymphoma. We present a case of Leser-Trélat sign that was not associated with an underlying malignancy.
A 44-year-old man was admitted to our dermatology outpatient department with a serpigo on the neck that had grown rapidly in the last month. His medical history and family history were unremarkable. Dermatologic examination revealed numerous 3- to 4-mm brown and slightly verrucous papules on the neck (Figure 1). A punch biopsy of the lesion showed acanthosis of predominantly basaloid cells, papillomatosis, and hyperkeratosis, as well as the presence of characteristic horn cysts (Figure 2). He was tested for possible underlying internal malignancy. Liver and kidney function tests, electrolyte count, protein electrophoresis, and whole blood and urine tests were within reference range. Chest radiography and abdominal ultrasonography revealed no signs of pathology. The erythrocyte sedimentation rate was 20 mm/h (reference range, 0–20 mm/h) and tests for hepatitis, human immunodeficiency virus, and syphilis were negative. Abdominal, cranial, and thorax computed tomography revealed no abnormalities. Otolaryngologic examinations also were negative. Additional endoscopic analyses, esophagogastroduodenoscopy, and colonoscopy revealed no abnormalities. At 1-year follow-up, the seborrheic keratoses remained unchanged. He has remained in good health without specific signs or symptoms suggestive of an underlying malignancy.
Paraneoplastic syndromes are associated with malignancy but progress without connection to a primary tumor or metastasis and form a group of clinical manifestations. The characteristic progress of paraneoplastic syndromes shows parallelism with the progression of the tumor. The mechanism underlying the development is not known, though the actions of bioactive substances that cause responses in the tumor, such as polypeptide hormones, hormonelike peptides, antibodies or immune complexes, and cytokines or growth factors, have been implicated.1
Although the term paraneoplastic syndrome commonly is used for Leser-Trélat sign, we do not believe it is accurate. As Fink et al2 and Schwengle et al3 indicated, the possibility of the co-occurrence being fortuitous is high. Showing a parallel progress of malignancy with paraneoplastic dermatosis requires that the paraneoplastic syndrome also diminish when the tumor is cured.4 It should then reappear with cancer recurrence or metastasis, which has not been exhibited in many case presentations in the literature.3 Disease regression was observed in only 1 of 3 seborrheic keratosis cases after primary cancer treatment.5
In patients with a malignancy, the sudden increase in seborrheic keratosis is based exclusively on the subjective evaluation of the patient, which may not be reliable. Schwengle et al3 stated that this sudden increase can be related to the awareness level of the patient who had a cancer diagnosis. Bräuer et al6 stated that no plausible definition distinguishes eruptive versus common seborrheic keratoses.
As a result, the results regarding the relationship between malignancy and Leser-Trélat sign are conflicting, and no strong evidence supports the presence of the sign. Only case reports have suggested that Leser-Trélat sign accompanies malignancy. Studies investigating its etiopathogenesis have not revealed a substance that has been released from or as a response to a tumor.
We believe that the presence of eruptive seborrheic keratosis does not necessitate screening for underlying internal malignancies.
To the Editor:
Leser-Trélat sign is a rare skin condition characterized by the sudden appearance of seborrheic keratoses that rapidly increase in number and size within weeks to months. Co-occurrence has been reported with a large number of malignancies, particularly adenocarcinoma and lymphoma. We present a case of Leser-Trélat sign that was not associated with an underlying malignancy.
A 44-year-old man was admitted to our dermatology outpatient department with a serpigo on the neck that had grown rapidly in the last month. His medical history and family history were unremarkable. Dermatologic examination revealed numerous 3- to 4-mm brown and slightly verrucous papules on the neck (Figure 1). A punch biopsy of the lesion showed acanthosis of predominantly basaloid cells, papillomatosis, and hyperkeratosis, as well as the presence of characteristic horn cysts (Figure 2). He was tested for possible underlying internal malignancy. Liver and kidney function tests, electrolyte count, protein electrophoresis, and whole blood and urine tests were within reference range. Chest radiography and abdominal ultrasonography revealed no signs of pathology. The erythrocyte sedimentation rate was 20 mm/h (reference range, 0–20 mm/h) and tests for hepatitis, human immunodeficiency virus, and syphilis were negative. Abdominal, cranial, and thorax computed tomography revealed no abnormalities. Otolaryngologic examinations also were negative. Additional endoscopic analyses, esophagogastroduodenoscopy, and colonoscopy revealed no abnormalities. At 1-year follow-up, the seborrheic keratoses remained unchanged. He has remained in good health without specific signs or symptoms suggestive of an underlying malignancy.
Paraneoplastic syndromes are associated with malignancy but progress without connection to a primary tumor or metastasis and form a group of clinical manifestations. The characteristic progress of paraneoplastic syndromes shows parallelism with the progression of the tumor. The mechanism underlying the development is not known, though the actions of bioactive substances that cause responses in the tumor, such as polypeptide hormones, hormonelike peptides, antibodies or immune complexes, and cytokines or growth factors, have been implicated.1
Although the term paraneoplastic syndrome commonly is used for Leser-Trélat sign, we do not believe it is accurate. As Fink et al2 and Schwengle et al3 indicated, the possibility of the co-occurrence being fortuitous is high. Showing a parallel progress of malignancy with paraneoplastic dermatosis requires that the paraneoplastic syndrome also diminish when the tumor is cured.4 It should then reappear with cancer recurrence or metastasis, which has not been exhibited in many case presentations in the literature.3 Disease regression was observed in only 1 of 3 seborrheic keratosis cases after primary cancer treatment.5
In patients with a malignancy, the sudden increase in seborrheic keratosis is based exclusively on the subjective evaluation of the patient, which may not be reliable. Schwengle et al3 stated that this sudden increase can be related to the awareness level of the patient who had a cancer diagnosis. Bräuer et al6 stated that no plausible definition distinguishes eruptive versus common seborrheic keratoses.
As a result, the results regarding the relationship between malignancy and Leser-Trélat sign are conflicting, and no strong evidence supports the presence of the sign. Only case reports have suggested that Leser-Trélat sign accompanies malignancy. Studies investigating its etiopathogenesis have not revealed a substance that has been released from or as a response to a tumor.
We believe that the presence of eruptive seborrheic keratosis does not necessitate screening for underlying internal malignancies.
- Cohen PR. Paraneoplastic dermatopathology: cutaneous paraneoplastic syndromes. Adv Dermatol. 1996;11:215-252.
- Fink AM, Filz D, Krajnik G, et al. Seborrhoeic keratoses in patients with internal malignancies: a case-control study with a prospective accrual of patients. J Eur Acad Dermatol Venereol. 2009;23:1316-1319.
- Schwengle LE, Rampen FH, Wobbes T. Seborrhoeic keratoses and internal malignancies. a case control study. Clin Exp Dermatol. 1988;13:177-179.
- Curth HO. Skin lesions and internal carcinoma. In: Andrade S, Gumport S, Popkin GL, et al, eds. Cancer of the Skin: Biology, Diagnosis, and Management. Vol 2. Philadelphia, PA: WB Saunders; 1976:1308-1341.
- Heaphy MR Jr, Millns JL, Schroeter AL. The sign of Leser-Trélat in a case of adenocarcinoma of the lung. J Am Acad Dermatol. 2000;43(2, pt 2):386-390.
- Bräuer J, Happle R, Gieler U. The sign of Leser-Trélat: fact or myth? J Eur Acad Dermatol Venereol. 1992;1:77-80.
- Cohen PR. Paraneoplastic dermatopathology: cutaneous paraneoplastic syndromes. Adv Dermatol. 1996;11:215-252.
- Fink AM, Filz D, Krajnik G, et al. Seborrhoeic keratoses in patients with internal malignancies: a case-control study with a prospective accrual of patients. J Eur Acad Dermatol Venereol. 2009;23:1316-1319.
- Schwengle LE, Rampen FH, Wobbes T. Seborrhoeic keratoses and internal malignancies. a case control study. Clin Exp Dermatol. 1988;13:177-179.
- Curth HO. Skin lesions and internal carcinoma. In: Andrade S, Gumport S, Popkin GL, et al, eds. Cancer of the Skin: Biology, Diagnosis, and Management. Vol 2. Philadelphia, PA: WB Saunders; 1976:1308-1341.
- Heaphy MR Jr, Millns JL, Schroeter AL. The sign of Leser-Trélat in a case of adenocarcinoma of the lung. J Am Acad Dermatol. 2000;43(2, pt 2):386-390.
- Bräuer J, Happle R, Gieler U. The sign of Leser-Trélat: fact or myth? J Eur Acad Dermatol Venereol. 1992;1:77-80.
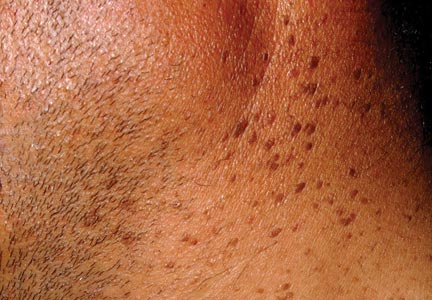
Hypertrophic Scar Treatment With Intralesional Triamcinolone Acetonide and Pulsed Dye Laser Results in Necrosis
To the Editor:
Intralesional corticosteroids and pulsed dye laser (PDL)(585–595 nm), either as monotherapy or combination therapy, are commonly used to treat hypertrophic scars.1 We describe an unusual adverse effect of protracted necrosis following combination therapy with intralesional triamcinolone acetonide and PDL for hypertrophic scar revision.
A 29-year-old healthy man presented after sustaining a contaminated crush trauma to the medial aspect of the left foot (Figure, A), requiring staged reconstruction with split-thickness skin grafting. The site healed with a hypertrophic scar (Figure, B), and treatment was pursued 18 months later with a community dermatologist with laser experience. Intralesional triamcinolone acetonide from a new vial diluted with 2% lidocaine was injected into the hypertrophic portions of the scar (20 mg/mL; 1.5 cc injected). The scar was then treated with a PDL at settings of 595 nm, 7-mm spot size, 10 J/cm2, pulse duration of 1.5 milliseconds, 30-millisecond spray with a 20-millisecond delay of cryogen spray, and double-stacked pulses with a 2- to 3-second delay between pulses. The patient reported that purpura was present in treated areas immediately after treatment.
At 6 days posttreatment, the first evidence of possible necrosis with tissue depression in mid posterior portions of the scar appeared (Figure, C). At day 14, deep ulcerations became evident (Figure, D), and by day 21, an exudative, yellow, fibrinopurulent membrane appeared (Figure, E). By 7 weeks posttreatment, ulceration size was only slightly reduced (Figure, F).
The necrosis was treated with debridement and local wound care using collagen matrix dressings (protease modulating matrix). Seven months after attempted scar revision, a deep 4-mm ulcer with a narrow tunnel-like connection to the skin surface remained anteriorly, disconnected from earlier mid posterior ulcerations, which was indicative of delayed onset of necrosis (Figure, G).
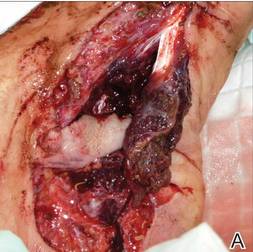
| 
| 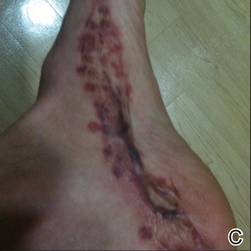
| ||
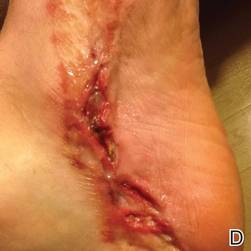
| 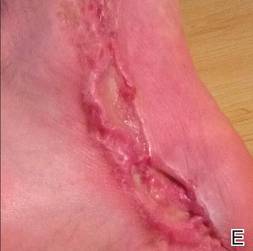
| The medial aspect of the left foot sustained a contaminated crush trauma (A). Following reconstruction, the site healed with a hypertrophic scar (B). Six days after treatment with intralesional triamcinolone acetonide and pulsed dye laser, purpura and tissue depression were observed in mid posterior portions of the scar (C). At 14 days posttreatment, deep ulcerations were evident (D). At 21 days posttreatment, exudative fibrinopurulent membrane appeared (E). At 7 weeks posttreatment, ulceration size was slightly reduced (F). At 7 months posttreatment, a delayed-onset, deep ulceration remained anteriorly (G). | ||
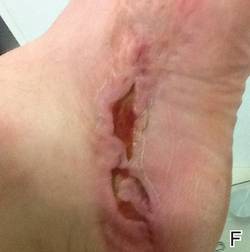
| 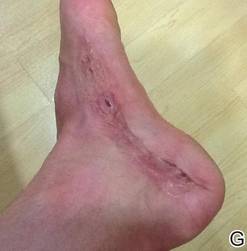
|
Intralesional corticosteroids have been associated with several side effects including pain, dermal atrophy, infection, pigmentation changes, and telangiectases.2 Pulsed dye laser therapy is generally described as safe with mild and transient side effects including purpura (resolves in 7–10 days), edema (diminishes in 48 hours), hyperpigmentation, and pain.3 More serious complications such as ulceration and scarring were initially described with PDL for purely vascular cutaneous lesions,4 and reports of necrosis and scarring have followed with PDL treatment utilizing higher fluences, shorter pulse durations, and stacking of pulses.5
Combination therapy for scar revision with intralesional triamcinolone acetonide and PDL reduces the corticosteroid dose and has been shown to be more efficacious than either modality alone with minimal to no complications.6 In our case, we believe the therapeutic combination resulted in several effects including bulk heating, vascular compromise, and inhibition of wound healing that together contributed to protracted necrosis. Triamcinolone acetonide was injected first; the corticosteroid fluid bolus likely conducted heat energy from the PDL spot leading to tissue damage, which corresponded clinically to the deep, delayed-onset necrosis. The PDL’s destructive effect on the vasculature after stacked pulses likely led to the initial necrosis, with wound healing time potentially extended by the anti-inflammatory effect of the corticosteroids. The location on the lower extremity, an area with limited vasculature and subcutaneous tissue, may have further contributed to impaired wound healing. Finally, the underlying scar tissue was by nature devoid of adnexal structures to expedite wound healing. Although the combination of intralesional corticosteroids and PDL is widely used and considered safe, we suggest that dermatologists carefully choose PDL parameters and pulse delivery methods when utilizing these modalities for scar revision.
1. Tziotzios C, Profyris C, Sterling J. Cutaneous scarring: pathophysiology, molecular mechanisms, and scar reduction therapeutics part II. strategies to reduce scar formation after dermatologic procedures. J Am Acad Dermatol.2012;66:13-24.
2. Chowdri NA, Masarat M, Mattoo A, et al. Keloids and hypertrophic scars: results with intraoperative and serial postoperative corticosteroid injection therapy. Aust N Z J Surg. 1999;69:655-659.
3. Liu A, Moy RL, Ross EV, et al. Pulsed dye laser and pulsed dye laser-mediated photodynamic therapy in the treatment of dermatologic disorders. Dermatol Surg. 2012;38:351-366.
4. Lamb SR, Sheehan-Dare RA. Leg ulceration after pulsed dye laser treatment of a vascular malformation. Lasers Surg Med. 2003;32:396-398.
5. Witman PM, Wagner AM, Scherer K, et al. Complications following pulsed dye laser treatment of superficial hemangiomas. Lasers Surg Med. 2006;38:116-123.
6. Asilian A, Darougheh A, Shariati F. New combination of triamcinolone, 5-fluorouracil, and pulsed-dye laser for treatment of keloid and hypertrophic scars. Dermatol Surg. 2006;32:907-915.
To the Editor:
Intralesional corticosteroids and pulsed dye laser (PDL)(585–595 nm), either as monotherapy or combination therapy, are commonly used to treat hypertrophic scars.1 We describe an unusual adverse effect of protracted necrosis following combination therapy with intralesional triamcinolone acetonide and PDL for hypertrophic scar revision.
A 29-year-old healthy man presented after sustaining a contaminated crush trauma to the medial aspect of the left foot (Figure, A), requiring staged reconstruction with split-thickness skin grafting. The site healed with a hypertrophic scar (Figure, B), and treatment was pursued 18 months later with a community dermatologist with laser experience. Intralesional triamcinolone acetonide from a new vial diluted with 2% lidocaine was injected into the hypertrophic portions of the scar (20 mg/mL; 1.5 cc injected). The scar was then treated with a PDL at settings of 595 nm, 7-mm spot size, 10 J/cm2, pulse duration of 1.5 milliseconds, 30-millisecond spray with a 20-millisecond delay of cryogen spray, and double-stacked pulses with a 2- to 3-second delay between pulses. The patient reported that purpura was present in treated areas immediately after treatment.
At 6 days posttreatment, the first evidence of possible necrosis with tissue depression in mid posterior portions of the scar appeared (Figure, C). At day 14, deep ulcerations became evident (Figure, D), and by day 21, an exudative, yellow, fibrinopurulent membrane appeared (Figure, E). By 7 weeks posttreatment, ulceration size was only slightly reduced (Figure, F).
The necrosis was treated with debridement and local wound care using collagen matrix dressings (protease modulating matrix). Seven months after attempted scar revision, a deep 4-mm ulcer with a narrow tunnel-like connection to the skin surface remained anteriorly, disconnected from earlier mid posterior ulcerations, which was indicative of delayed onset of necrosis (Figure, G).
|
|
|
| ||
|
|
| The medial aspect of the left foot sustained a contaminated crush trauma (A). Following reconstruction, the site healed with a hypertrophic scar (B). Six days after treatment with intralesional triamcinolone acetonide and pulsed dye laser, purpura and tissue depression were observed in mid posterior portions of the scar (C). At 14 days posttreatment, deep ulcerations were evident (D). At 21 days posttreatment, exudative fibrinopurulent membrane appeared (E). At 7 weeks posttreatment, ulceration size was slightly reduced (F). At 7 months posttreatment, a delayed-onset, deep ulceration remained anteriorly (G). | ||
|
|
|
Intralesional corticosteroids have been associated with several side effects including pain, dermal atrophy, infection, pigmentation changes, and telangiectases.2 Pulsed dye laser therapy is generally described as safe with mild and transient side effects including purpura (resolves in 7–10 days), edema (diminishes in 48 hours), hyperpigmentation, and pain.3 More serious complications such as ulceration and scarring were initially described with PDL for purely vascular cutaneous lesions,4 and reports of necrosis and scarring have followed with PDL treatment utilizing higher fluences, shorter pulse durations, and stacking of pulses.5
Combination therapy for scar revision with intralesional triamcinolone acetonide and PDL reduces the corticosteroid dose and has been shown to be more efficacious than either modality alone with minimal to no complications.6 In our case, we believe the therapeutic combination resulted in several effects including bulk heating, vascular compromise, and inhibition of wound healing that together contributed to protracted necrosis. Triamcinolone acetonide was injected first; the corticosteroid fluid bolus likely conducted heat energy from the PDL spot leading to tissue damage, which corresponded clinically to the deep, delayed-onset necrosis. The PDL’s destructive effect on the vasculature after stacked pulses likely led to the initial necrosis, with wound healing time potentially extended by the anti-inflammatory effect of the corticosteroids. The location on the lower extremity, an area with limited vasculature and subcutaneous tissue, may have further contributed to impaired wound healing. Finally, the underlying scar tissue was by nature devoid of adnexal structures to expedite wound healing. Although the combination of intralesional corticosteroids and PDL is widely used and considered safe, we suggest that dermatologists carefully choose PDL parameters and pulse delivery methods when utilizing these modalities for scar revision.
To the Editor:
Intralesional corticosteroids and pulsed dye laser (PDL)(585–595 nm), either as monotherapy or combination therapy, are commonly used to treat hypertrophic scars.1 We describe an unusual adverse effect of protracted necrosis following combination therapy with intralesional triamcinolone acetonide and PDL for hypertrophic scar revision.
A 29-year-old healthy man presented after sustaining a contaminated crush trauma to the medial aspect of the left foot (Figure, A), requiring staged reconstruction with split-thickness skin grafting. The site healed with a hypertrophic scar (Figure, B), and treatment was pursued 18 months later with a community dermatologist with laser experience. Intralesional triamcinolone acetonide from a new vial diluted with 2% lidocaine was injected into the hypertrophic portions of the scar (20 mg/mL; 1.5 cc injected). The scar was then treated with a PDL at settings of 595 nm, 7-mm spot size, 10 J/cm2, pulse duration of 1.5 milliseconds, 30-millisecond spray with a 20-millisecond delay of cryogen spray, and double-stacked pulses with a 2- to 3-second delay between pulses. The patient reported that purpura was present in treated areas immediately after treatment.
At 6 days posttreatment, the first evidence of possible necrosis with tissue depression in mid posterior portions of the scar appeared (Figure, C). At day 14, deep ulcerations became evident (Figure, D), and by day 21, an exudative, yellow, fibrinopurulent membrane appeared (Figure, E). By 7 weeks posttreatment, ulceration size was only slightly reduced (Figure, F).
The necrosis was treated with debridement and local wound care using collagen matrix dressings (protease modulating matrix). Seven months after attempted scar revision, a deep 4-mm ulcer with a narrow tunnel-like connection to the skin surface remained anteriorly, disconnected from earlier mid posterior ulcerations, which was indicative of delayed onset of necrosis (Figure, G).
|
|
|
| ||
|
|
| The medial aspect of the left foot sustained a contaminated crush trauma (A). Following reconstruction, the site healed with a hypertrophic scar (B). Six days after treatment with intralesional triamcinolone acetonide and pulsed dye laser, purpura and tissue depression were observed in mid posterior portions of the scar (C). At 14 days posttreatment, deep ulcerations were evident (D). At 21 days posttreatment, exudative fibrinopurulent membrane appeared (E). At 7 weeks posttreatment, ulceration size was slightly reduced (F). At 7 months posttreatment, a delayed-onset, deep ulceration remained anteriorly (G). | ||
|
|
|
Intralesional corticosteroids have been associated with several side effects including pain, dermal atrophy, infection, pigmentation changes, and telangiectases.2 Pulsed dye laser therapy is generally described as safe with mild and transient side effects including purpura (resolves in 7–10 days), edema (diminishes in 48 hours), hyperpigmentation, and pain.3 More serious complications such as ulceration and scarring were initially described with PDL for purely vascular cutaneous lesions,4 and reports of necrosis and scarring have followed with PDL treatment utilizing higher fluences, shorter pulse durations, and stacking of pulses.5
Combination therapy for scar revision with intralesional triamcinolone acetonide and PDL reduces the corticosteroid dose and has been shown to be more efficacious than either modality alone with minimal to no complications.6 In our case, we believe the therapeutic combination resulted in several effects including bulk heating, vascular compromise, and inhibition of wound healing that together contributed to protracted necrosis. Triamcinolone acetonide was injected first; the corticosteroid fluid bolus likely conducted heat energy from the PDL spot leading to tissue damage, which corresponded clinically to the deep, delayed-onset necrosis. The PDL’s destructive effect on the vasculature after stacked pulses likely led to the initial necrosis, with wound healing time potentially extended by the anti-inflammatory effect of the corticosteroids. The location on the lower extremity, an area with limited vasculature and subcutaneous tissue, may have further contributed to impaired wound healing. Finally, the underlying scar tissue was by nature devoid of adnexal structures to expedite wound healing. Although the combination of intralesional corticosteroids and PDL is widely used and considered safe, we suggest that dermatologists carefully choose PDL parameters and pulse delivery methods when utilizing these modalities for scar revision.
1. Tziotzios C, Profyris C, Sterling J. Cutaneous scarring: pathophysiology, molecular mechanisms, and scar reduction therapeutics part II. strategies to reduce scar formation after dermatologic procedures. J Am Acad Dermatol.2012;66:13-24.
2. Chowdri NA, Masarat M, Mattoo A, et al. Keloids and hypertrophic scars: results with intraoperative and serial postoperative corticosteroid injection therapy. Aust N Z J Surg. 1999;69:655-659.
3. Liu A, Moy RL, Ross EV, et al. Pulsed dye laser and pulsed dye laser-mediated photodynamic therapy in the treatment of dermatologic disorders. Dermatol Surg. 2012;38:351-366.
4. Lamb SR, Sheehan-Dare RA. Leg ulceration after pulsed dye laser treatment of a vascular malformation. Lasers Surg Med. 2003;32:396-398.
5. Witman PM, Wagner AM, Scherer K, et al. Complications following pulsed dye laser treatment of superficial hemangiomas. Lasers Surg Med. 2006;38:116-123.
6. Asilian A, Darougheh A, Shariati F. New combination of triamcinolone, 5-fluorouracil, and pulsed-dye laser for treatment of keloid and hypertrophic scars. Dermatol Surg. 2006;32:907-915.
1. Tziotzios C, Profyris C, Sterling J. Cutaneous scarring: pathophysiology, molecular mechanisms, and scar reduction therapeutics part II. strategies to reduce scar formation after dermatologic procedures. J Am Acad Dermatol.2012;66:13-24.
2. Chowdri NA, Masarat M, Mattoo A, et al. Keloids and hypertrophic scars: results with intraoperative and serial postoperative corticosteroid injection therapy. Aust N Z J Surg. 1999;69:655-659.
3. Liu A, Moy RL, Ross EV, et al. Pulsed dye laser and pulsed dye laser-mediated photodynamic therapy in the treatment of dermatologic disorders. Dermatol Surg. 2012;38:351-366.
4. Lamb SR, Sheehan-Dare RA. Leg ulceration after pulsed dye laser treatment of a vascular malformation. Lasers Surg Med. 2003;32:396-398.
5. Witman PM, Wagner AM, Scherer K, et al. Complications following pulsed dye laser treatment of superficial hemangiomas. Lasers Surg Med. 2006;38:116-123.
6. Asilian A, Darougheh A, Shariati F. New combination of triamcinolone, 5-fluorouracil, and pulsed-dye laser for treatment of keloid and hypertrophic scars. Dermatol Surg. 2006;32:907-915.
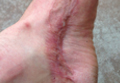
Photosensitivity Reaction From Dronedarone for Atrial Fibrillation
To the Editor:
A 61-year-old woman with a history of atrial fibrillation, type 2 diabetes mellitus, and hyperlipidemia presented with an erythematous, edematous, pruritic eruption on the chest, neck, and arms of 2 weeks’ duration. The patient had no history of considerable sun exposure or reports of photosensitivity. One month prior to presentation she had started taking dronedarone for improved control of atrial fibrillation. She had no known history of drug allergies. Other medications included valsartan, digoxin, pioglitazone, simvastatin, aspirin, hydrocodone, and zolpidem, all of which were unchanged for years. There were no changes in topical products used.
Physical examination revealed confluent, well-demarcated, erythematous and edematous papules and plaques over the anterior aspect of the neck, bilateral forearms, and dorsal aspect of the hands, with a v-shaped distribution on the chest (Figure). There was notable sparing of the submental region, upper arms, abdomen, back, and legs. Dronedarone was discontinued and she was started on fluocinonide ointment 0.05% and oral hydroxyzine for pruritus. Her rash resolved within the following few weeks.
|
|
| Confluent, well-demarcated, erythematous and edematous papules and plaques in a v-shaped distribution on the chest (A) and dorsal aspect of the hands (B). |
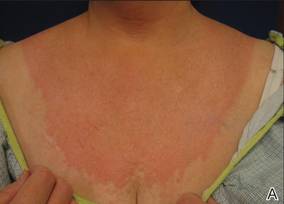
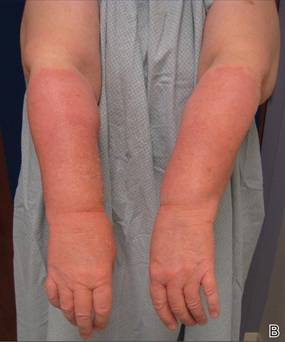
Dronedarone is a noniodinated benzofuran derivative. It is structurally similar to and shares the antiarrhythmic properties of amiodarone,1 and thus it is used in the treatment of atrial fibrillation and atrial flutter. However, the pulmonary and thyroid toxicities sometimes associated with amiodarone have not been observed with dronedarone. The primary side effect of dronedarone is gastrointestinal distress, specifically nausea, vomiting, and diarrhea. Dronedarone has been associated with severe liver injury and hepatic failure.2 Cutaneous reactions appear to be an uncommon side effect of dronedarone therapy. Across 5 clinical studies (N=6285), adverse events involving skin and subcutaneous tissue including eczema, allergic dermatitis, pruritus, and nonspecific rash occurred in 5% of dronedarone and 3% of placebo patients. Photosensitivity reactions occurred in less than 1% of dronedarone recipients.3
Although the lack of a biopsy leaves the possibility of a contact or photocontact dermatitis, our patient demonstrated the potential for dronedarone to cause a photodistributed drug eruption that resolved after cessation of the medication.
1. Hoy SM, Keam SJ. Dronedarone. Drugs. 2009;69:1647-1663.
2. In brief: FDA warning on dronedarone (Multaq). Med Lett Drugs Ther. 2011;53:17.
3. Multaq [package insert]. Bridgewater, NJ: sanofi-aventis; 2009.
To the Editor:
A 61-year-old woman with a history of atrial fibrillation, type 2 diabetes mellitus, and hyperlipidemia presented with an erythematous, edematous, pruritic eruption on the chest, neck, and arms of 2 weeks’ duration. The patient had no history of considerable sun exposure or reports of photosensitivity. One month prior to presentation she had started taking dronedarone for improved control of atrial fibrillation. She had no known history of drug allergies. Other medications included valsartan, digoxin, pioglitazone, simvastatin, aspirin, hydrocodone, and zolpidem, all of which were unchanged for years. There were no changes in topical products used.
Physical examination revealed confluent, well-demarcated, erythematous and edematous papules and plaques over the anterior aspect of the neck, bilateral forearms, and dorsal aspect of the hands, with a v-shaped distribution on the chest (Figure). There was notable sparing of the submental region, upper arms, abdomen, back, and legs. Dronedarone was discontinued and she was started on fluocinonide ointment 0.05% and oral hydroxyzine for pruritus. Her rash resolved within the following few weeks.
|
|
| Confluent, well-demarcated, erythematous and edematous papules and plaques in a v-shaped distribution on the chest (A) and dorsal aspect of the hands (B). |
Dronedarone is a noniodinated benzofuran derivative. It is structurally similar to and shares the antiarrhythmic properties of amiodarone,1 and thus it is used in the treatment of atrial fibrillation and atrial flutter. However, the pulmonary and thyroid toxicities sometimes associated with amiodarone have not been observed with dronedarone. The primary side effect of dronedarone is gastrointestinal distress, specifically nausea, vomiting, and diarrhea. Dronedarone has been associated with severe liver injury and hepatic failure.2 Cutaneous reactions appear to be an uncommon side effect of dronedarone therapy. Across 5 clinical studies (N=6285), adverse events involving skin and subcutaneous tissue including eczema, allergic dermatitis, pruritus, and nonspecific rash occurred in 5% of dronedarone and 3% of placebo patients. Photosensitivity reactions occurred in less than 1% of dronedarone recipients.3
Although the lack of a biopsy leaves the possibility of a contact or photocontact dermatitis, our patient demonstrated the potential for dronedarone to cause a photodistributed drug eruption that resolved after cessation of the medication.
To the Editor:
A 61-year-old woman with a history of atrial fibrillation, type 2 diabetes mellitus, and hyperlipidemia presented with an erythematous, edematous, pruritic eruption on the chest, neck, and arms of 2 weeks’ duration. The patient had no history of considerable sun exposure or reports of photosensitivity. One month prior to presentation she had started taking dronedarone for improved control of atrial fibrillation. She had no known history of drug allergies. Other medications included valsartan, digoxin, pioglitazone, simvastatin, aspirin, hydrocodone, and zolpidem, all of which were unchanged for years. There were no changes in topical products used.
Physical examination revealed confluent, well-demarcated, erythematous and edematous papules and plaques over the anterior aspect of the neck, bilateral forearms, and dorsal aspect of the hands, with a v-shaped distribution on the chest (Figure). There was notable sparing of the submental region, upper arms, abdomen, back, and legs. Dronedarone was discontinued and she was started on fluocinonide ointment 0.05% and oral hydroxyzine for pruritus. Her rash resolved within the following few weeks.
|
|
| Confluent, well-demarcated, erythematous and edematous papules and plaques in a v-shaped distribution on the chest (A) and dorsal aspect of the hands (B). |
Dronedarone is a noniodinated benzofuran derivative. It is structurally similar to and shares the antiarrhythmic properties of amiodarone,1 and thus it is used in the treatment of atrial fibrillation and atrial flutter. However, the pulmonary and thyroid toxicities sometimes associated with amiodarone have not been observed with dronedarone. The primary side effect of dronedarone is gastrointestinal distress, specifically nausea, vomiting, and diarrhea. Dronedarone has been associated with severe liver injury and hepatic failure.2 Cutaneous reactions appear to be an uncommon side effect of dronedarone therapy. Across 5 clinical studies (N=6285), adverse events involving skin and subcutaneous tissue including eczema, allergic dermatitis, pruritus, and nonspecific rash occurred in 5% of dronedarone and 3% of placebo patients. Photosensitivity reactions occurred in less than 1% of dronedarone recipients.3
Although the lack of a biopsy leaves the possibility of a contact or photocontact dermatitis, our patient demonstrated the potential for dronedarone to cause a photodistributed drug eruption that resolved after cessation of the medication.
1. Hoy SM, Keam SJ. Dronedarone. Drugs. 2009;69:1647-1663.
2. In brief: FDA warning on dronedarone (Multaq). Med Lett Drugs Ther. 2011;53:17.
3. Multaq [package insert]. Bridgewater, NJ: sanofi-aventis; 2009.
1. Hoy SM, Keam SJ. Dronedarone. Drugs. 2009;69:1647-1663.
2. In brief: FDA warning on dronedarone (Multaq). Med Lett Drugs Ther. 2011;53:17.
3. Multaq [package insert]. Bridgewater, NJ: sanofi-aventis; 2009.
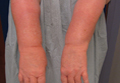
An Unusual Presentation of Congenital Dermal Melanocytosis Fitting the Rare Diagnosis of Dermal Melanocyte Hamartoma
To the Editor:
Dermal melanocytosis is thought to be the result of a defect in melanoblast migration during embryogenesis and is characterized by the presence of functional fusiform and dendritic melanocytes in the dermis. Congenital dermal melanocytosis is classified into various subtypes based on the distribution, morphology, natural history of lesions, and distinctive histologic findings. We present an unusual case of congenital dermal melanocytosis that might fit the rare entity of dermal melanocyte hamartoma (DMH).
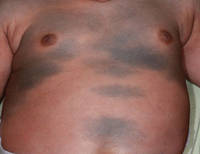
|
| Figure 1. Uniform grayish blue patches over the torso with sharp demarcation lines that followed a dermatomal distribution. |
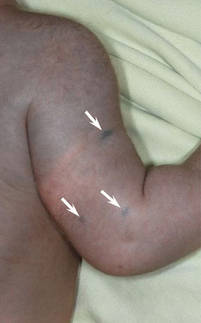
|
| Figure 2. Three conspicuous darker blue macules (arrows) within a bluish patch. |
|
| Figure 3. Intradermal dendritic pigmented melanocytes localized in the upper dermis and arranged parallel to the skin surface (H&E, original magnification ×100 [inset, original magnification ×400]). |
A 4-month-old girl presented with bilateral bluish patches over the trunk and upper extremities. The lesions were present since birth, and remained entirely unchanged during a follow-up period of 18 months. Mental and physical development was normal. There was no family history of pigmentary disorders. On physical examination uniform grayish blue patches were seen over the torso and upper extremities. The patches seemed to follow a dermatomal distribution along the trunk and extremities (Figure 1). Several well-circumscribed, much darker blue macules were scattered within the bluish patches (Figure 2). The rest of the physical examination was unremarkable. Complete blood cell count and blood chemistry values were within reference range. Skin biopsy specimens from both the grayish blue patches and the conspicuous darker blue macules showed a fair amount of dermal bipolar dendritic pigmented melanocytes arranged parallel to the skin surface with no apparent disturbance of collagen bundles (Figure 3). No melanophages were seen. The clinical, histologic, and laboratory findings were consistent with a diagnosis of congenital dermal melanocytosis. An underlying lysosomal storage disease was ruled out through metabolic screening that included liver function tests, abdominal sonography, and neurologic and ophthalmologic examinations.
The clinical spectrum of congenital dermal melanocytosis includes several clinical entities such as mongolian spots, Ota nevus, Ito nevus, blue nevi, and DMH.1 The differentiation between these different types of dermal melanocytosis can be challenging, especially when the process of melanocytosis is extensive. Among the several types of congenital dermal melanocytosis, Ota or Ito nevi can be ruled out in our patient based on the clinical presentation.
The term dermal melanocyte hamartoma was introduced by Burkhart and Gohara.2 It is characterized by congenital dermal melanocytosis that follows a dermatomal pattern. The original case report included speckled, darker blue macules with a background of grayish blue patches similar to our patient.2 Melanocytes in DMH typically are located in the upper half of the reticular dermis, as opposed to extensive mongolian spots in which ectopic melanocytes usually are found in the lower half of the dermis. The pigmentation in our patient did not change during 22 months of follow-up, which is more consistent with the diagnosis of DMH versus extensive mongolian spots. Our patient did not present with any systemic anomalies. However, a case of congenital melanocytosis and neuroectodermal malformation has been described in the literature.3
Little is known about the etiology of dermal melanocytosis. Mutations in the guanine nucleotide binding protein q polypeptide gene, GNAQ, or guanine nucleotide binding protein alpha 11 gene, GNA11, were found to cause dermal melanocytosis in mice.4 Also, GNAQ mutations have been demonstrated in humans with dermal melanocytosis as well as uveal melanoma.5 Progress in our understanding of the pathogenesis of dermal melanocytosis is expected to lead to a more accurate classification of dermal pigmentation disorders.
1. Stanford DG, Georgouras KE. Dermal melanocytosis: a clinical spectrum. Australas J Dermatol. 1996;37:19-25.
2. Burkhart CG, Gohara A. Dermal melanocyte hamartoma: a distinctive new form of dermal melanocytosis.
Arch Dermatol. 1981;117:102-104.
3. Schwartz RA, Cohen-Addad N, Lambert MW, et al. Congenital melanocytosis with myelomeningocele and hydrocephalus. Cutis. 1986;37:37-39.
4. Van Raamsdonk CD, Fitch KR, Fuchs H, et al. Effects of G-protein mutations on skin color. Nat Genet. 2004;36:961-968.
5. Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue nevi. Nature. 2009;457:599-602.
To the Editor:
Dermal melanocytosis is thought to be the result of a defect in melanoblast migration during embryogenesis and is characterized by the presence of functional fusiform and dendritic melanocytes in the dermis. Congenital dermal melanocytosis is classified into various subtypes based on the distribution, morphology, natural history of lesions, and distinctive histologic findings. We present an unusual case of congenital dermal melanocytosis that might fit the rare entity of dermal melanocyte hamartoma (DMH).
|
|
| Figure 1. Uniform grayish blue patches over the torso with sharp demarcation lines that followed a dermatomal distribution. |
|
|
| Figure 2. Three conspicuous darker blue macules (arrows) within a bluish patch. |
|
|
| Figure 3. Intradermal dendritic pigmented melanocytes localized in the upper dermis and arranged parallel to the skin surface (H&E, original magnification ×100 [inset, original magnification ×400]). |
A 4-month-old girl presented with bilateral bluish patches over the trunk and upper extremities. The lesions were present since birth, and remained entirely unchanged during a follow-up period of 18 months. Mental and physical development was normal. There was no family history of pigmentary disorders. On physical examination uniform grayish blue patches were seen over the torso and upper extremities. The patches seemed to follow a dermatomal distribution along the trunk and extremities (Figure 1). Several well-circumscribed, much darker blue macules were scattered within the bluish patches (Figure 2). The rest of the physical examination was unremarkable. Complete blood cell count and blood chemistry values were within reference range. Skin biopsy specimens from both the grayish blue patches and the conspicuous darker blue macules showed a fair amount of dermal bipolar dendritic pigmented melanocytes arranged parallel to the skin surface with no apparent disturbance of collagen bundles (Figure 3). No melanophages were seen. The clinical, histologic, and laboratory findings were consistent with a diagnosis of congenital dermal melanocytosis. An underlying lysosomal storage disease was ruled out through metabolic screening that included liver function tests, abdominal sonography, and neurologic and ophthalmologic examinations.
The clinical spectrum of congenital dermal melanocytosis includes several clinical entities such as mongolian spots, Ota nevus, Ito nevus, blue nevi, and DMH.1 The differentiation between these different types of dermal melanocytosis can be challenging, especially when the process of melanocytosis is extensive. Among the several types of congenital dermal melanocytosis, Ota or Ito nevi can be ruled out in our patient based on the clinical presentation.
The term dermal melanocyte hamartoma was introduced by Burkhart and Gohara.2 It is characterized by congenital dermal melanocytosis that follows a dermatomal pattern. The original case report included speckled, darker blue macules with a background of grayish blue patches similar to our patient.2 Melanocytes in DMH typically are located in the upper half of the reticular dermis, as opposed to extensive mongolian spots in which ectopic melanocytes usually are found in the lower half of the dermis. The pigmentation in our patient did not change during 22 months of follow-up, which is more consistent with the diagnosis of DMH versus extensive mongolian spots. Our patient did not present with any systemic anomalies. However, a case of congenital melanocytosis and neuroectodermal malformation has been described in the literature.3
Little is known about the etiology of dermal melanocytosis. Mutations in the guanine nucleotide binding protein q polypeptide gene, GNAQ, or guanine nucleotide binding protein alpha 11 gene, GNA11, were found to cause dermal melanocytosis in mice.4 Also, GNAQ mutations have been demonstrated in humans with dermal melanocytosis as well as uveal melanoma.5 Progress in our understanding of the pathogenesis of dermal melanocytosis is expected to lead to a more accurate classification of dermal pigmentation disorders.
To the Editor:
Dermal melanocytosis is thought to be the result of a defect in melanoblast migration during embryogenesis and is characterized by the presence of functional fusiform and dendritic melanocytes in the dermis. Congenital dermal melanocytosis is classified into various subtypes based on the distribution, morphology, natural history of lesions, and distinctive histologic findings. We present an unusual case of congenital dermal melanocytosis that might fit the rare entity of dermal melanocyte hamartoma (DMH).
|
|
| Figure 1. Uniform grayish blue patches over the torso with sharp demarcation lines that followed a dermatomal distribution. |
|
|
| Figure 2. Three conspicuous darker blue macules (arrows) within a bluish patch. |
|
|
| Figure 3. Intradermal dendritic pigmented melanocytes localized in the upper dermis and arranged parallel to the skin surface (H&E, original magnification ×100 [inset, original magnification ×400]). |
A 4-month-old girl presented with bilateral bluish patches over the trunk and upper extremities. The lesions were present since birth, and remained entirely unchanged during a follow-up period of 18 months. Mental and physical development was normal. There was no family history of pigmentary disorders. On physical examination uniform grayish blue patches were seen over the torso and upper extremities. The patches seemed to follow a dermatomal distribution along the trunk and extremities (Figure 1). Several well-circumscribed, much darker blue macules were scattered within the bluish patches (Figure 2). The rest of the physical examination was unremarkable. Complete blood cell count and blood chemistry values were within reference range. Skin biopsy specimens from both the grayish blue patches and the conspicuous darker blue macules showed a fair amount of dermal bipolar dendritic pigmented melanocytes arranged parallel to the skin surface with no apparent disturbance of collagen bundles (Figure 3). No melanophages were seen. The clinical, histologic, and laboratory findings were consistent with a diagnosis of congenital dermal melanocytosis. An underlying lysosomal storage disease was ruled out through metabolic screening that included liver function tests, abdominal sonography, and neurologic and ophthalmologic examinations.
The clinical spectrum of congenital dermal melanocytosis includes several clinical entities such as mongolian spots, Ota nevus, Ito nevus, blue nevi, and DMH.1 The differentiation between these different types of dermal melanocytosis can be challenging, especially when the process of melanocytosis is extensive. Among the several types of congenital dermal melanocytosis, Ota or Ito nevi can be ruled out in our patient based on the clinical presentation.
The term dermal melanocyte hamartoma was introduced by Burkhart and Gohara.2 It is characterized by congenital dermal melanocytosis that follows a dermatomal pattern. The original case report included speckled, darker blue macules with a background of grayish blue patches similar to our patient.2 Melanocytes in DMH typically are located in the upper half of the reticular dermis, as opposed to extensive mongolian spots in which ectopic melanocytes usually are found in the lower half of the dermis. The pigmentation in our patient did not change during 22 months of follow-up, which is more consistent with the diagnosis of DMH versus extensive mongolian spots. Our patient did not present with any systemic anomalies. However, a case of congenital melanocytosis and neuroectodermal malformation has been described in the literature.3
Little is known about the etiology of dermal melanocytosis. Mutations in the guanine nucleotide binding protein q polypeptide gene, GNAQ, or guanine nucleotide binding protein alpha 11 gene, GNA11, were found to cause dermal melanocytosis in mice.4 Also, GNAQ mutations have been demonstrated in humans with dermal melanocytosis as well as uveal melanoma.5 Progress in our understanding of the pathogenesis of dermal melanocytosis is expected to lead to a more accurate classification of dermal pigmentation disorders.
1. Stanford DG, Georgouras KE. Dermal melanocytosis: a clinical spectrum. Australas J Dermatol. 1996;37:19-25.
2. Burkhart CG, Gohara A. Dermal melanocyte hamartoma: a distinctive new form of dermal melanocytosis.
Arch Dermatol. 1981;117:102-104.
3. Schwartz RA, Cohen-Addad N, Lambert MW, et al. Congenital melanocytosis with myelomeningocele and hydrocephalus. Cutis. 1986;37:37-39.
4. Van Raamsdonk CD, Fitch KR, Fuchs H, et al. Effects of G-protein mutations on skin color. Nat Genet. 2004;36:961-968.
5. Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue nevi. Nature. 2009;457:599-602.
1. Stanford DG, Georgouras KE. Dermal melanocytosis: a clinical spectrum. Australas J Dermatol. 1996;37:19-25.
2. Burkhart CG, Gohara A. Dermal melanocyte hamartoma: a distinctive new form of dermal melanocytosis.
Arch Dermatol. 1981;117:102-104.
3. Schwartz RA, Cohen-Addad N, Lambert MW, et al. Congenital melanocytosis with myelomeningocele and hydrocephalus. Cutis. 1986;37:37-39.
4. Van Raamsdonk CD, Fitch KR, Fuchs H, et al. Effects of G-protein mutations on skin color. Nat Genet. 2004;36:961-968.
5. Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue nevi. Nature. 2009;457:599-602.
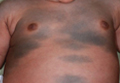
Shave Removal Plus Electrodesiccation for the Treatment of Cutaneous Neurofibromas
To the Editor:
Cutaneous neurofibromas are a clinical feature of both neurofibromatosis type I (NF-1) and neurofibromatosis type II (NF-2). Neurofibromatosis type I occurs in 1 in 3000 live births and NF-2 occurs in 1 in 40,000 live births.1 Neurofibromas are discrete masses that arise from peripheral nerves and are composed of Schwann cells, mast cells, fibroblasts, and perineural cells. They commonly appear during or after puberty in the majority of patients and increase in number as patients age.1,2 Cutaneous neurofibromas can be painful and pruritic as well as cosmetically unappealing. Current treatment of neurofibromas consists primarily of standard surgical excision or laser therapy. Reports of loop electrocoagulation in the operating room, combination erbium:YAG and CO2 laser, and Nd:YAG laser treatments of neurofibromas have been published in the literature.3-5 These treatments can be effective, but the bothersome lesions in patients with neurofibromatosis often are multiple, making these procedures time consuming and costly. A removal technique that is time efficient, cost effective, and poses little discomfort or functional impairment for the patient is desirable. We describe the technique of shave removal with electrodesiccation for multiple neurofibromas.
A 24-year-old woman with NF-1 presented to the dermatology clinic for treatment of several pruritic neurofibromas of the trunk, including the bilateral breasts (Figure, A). We performed shave removal of each lesion with electrodesiccation of the lesion base with successful treatment of symptoms and acceptable cosmetic outcome (Figure, B). The patient identified symptomatic and cosmetically bothersome neurofibromas. The lesions were disinfected with an alcohol swab and anesthetized with 1% lidocaine and a 1:200,000 dilution of epinephrine. A shave biopsy blade was used to remove each neurofibroma at the level of the surrounding skin. Toothed forceps were utilized to lift the lesion and provide traction. Electrodesiccation was then used for destruction of the base or any residual gelatinous component of the lesion to the level of the surrounding skin, thus achieving hemostasis. Lastly, petrolatum was applied to the wound and covered with an adhesive dressing. Daily dressing change was performed for 2 weeks or until healed.
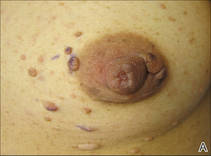
|
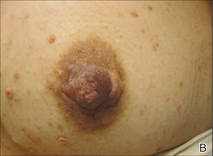
|
| A patient with neurofibromatosis type I with neurofibromas on the breast before (A) and approximately 6 months after treatment with shave removal plus electrodesiccation of the lesions (B). |
Patients with neurofibromatoses often present to dermatologists for management of symptomatic or cosmetically bothersome neurofibromas, which are often numerous and diffusely distributed. There are few published practical descriptions of removal techniques to manage these multiple and recurrent manifestations of the disease. The shave removal plus electrodesiccation technique we described is straightforward for practitioners and provides a well-tolerated mechanism for treating multiple lesions at once, while also offering patients acceptable functional and cosmetic results.
1. Listernick R, Charrow J. The neurofibromatoses. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill Companies; 2007:1331-1339.
2. Pinson S, Wolkenstein P. Neurofibromatosis type 1 or Von Recklinghausen’s disease [in French]. Rev Med Interne. 2005;26:196-215.
3. Elwakil TF, Samy NA, Elbasiouny MS. Non-excision treatment of multiple cutaneous neurofibromas by laser photocoagulation [published online ahead of print August 15, 2007]. Lasers Med Sci. 2008;23:301-306.
4. Meissner M, Ochsendorf F, Kaufmann R. Quick and effective treatment of small neurofibromas [published online ahead of print November 23, 2010]. J Dtsch Dermatol Ges. 2011;9:167-168.
5. Roberts AH, Crockett DJ. An operation for the treatment of cutaneous neurofibromatosis. Br J Plast Surg. 1985;38:292-293.
To the Editor:
Cutaneous neurofibromas are a clinical feature of both neurofibromatosis type I (NF-1) and neurofibromatosis type II (NF-2). Neurofibromatosis type I occurs in 1 in 3000 live births and NF-2 occurs in 1 in 40,000 live births.1 Neurofibromas are discrete masses that arise from peripheral nerves and are composed of Schwann cells, mast cells, fibroblasts, and perineural cells. They commonly appear during or after puberty in the majority of patients and increase in number as patients age.1,2 Cutaneous neurofibromas can be painful and pruritic as well as cosmetically unappealing. Current treatment of neurofibromas consists primarily of standard surgical excision or laser therapy. Reports of loop electrocoagulation in the operating room, combination erbium:YAG and CO2 laser, and Nd:YAG laser treatments of neurofibromas have been published in the literature.3-5 These treatments can be effective, but the bothersome lesions in patients with neurofibromatosis often are multiple, making these procedures time consuming and costly. A removal technique that is time efficient, cost effective, and poses little discomfort or functional impairment for the patient is desirable. We describe the technique of shave removal with electrodesiccation for multiple neurofibromas.
A 24-year-old woman with NF-1 presented to the dermatology clinic for treatment of several pruritic neurofibromas of the trunk, including the bilateral breasts (Figure, A). We performed shave removal of each lesion with electrodesiccation of the lesion base with successful treatment of symptoms and acceptable cosmetic outcome (Figure, B). The patient identified symptomatic and cosmetically bothersome neurofibromas. The lesions were disinfected with an alcohol swab and anesthetized with 1% lidocaine and a 1:200,000 dilution of epinephrine. A shave biopsy blade was used to remove each neurofibroma at the level of the surrounding skin. Toothed forceps were utilized to lift the lesion and provide traction. Electrodesiccation was then used for destruction of the base or any residual gelatinous component of the lesion to the level of the surrounding skin, thus achieving hemostasis. Lastly, petrolatum was applied to the wound and covered with an adhesive dressing. Daily dressing change was performed for 2 weeks or until healed.
|
|
|
|
| A patient with neurofibromatosis type I with neurofibromas on the breast before (A) and approximately 6 months after treatment with shave removal plus electrodesiccation of the lesions (B). |
Patients with neurofibromatoses often present to dermatologists for management of symptomatic or cosmetically bothersome neurofibromas, which are often numerous and diffusely distributed. There are few published practical descriptions of removal techniques to manage these multiple and recurrent manifestations of the disease. The shave removal plus electrodesiccation technique we described is straightforward for practitioners and provides a well-tolerated mechanism for treating multiple lesions at once, while also offering patients acceptable functional and cosmetic results.
To the Editor:
Cutaneous neurofibromas are a clinical feature of both neurofibromatosis type I (NF-1) and neurofibromatosis type II (NF-2). Neurofibromatosis type I occurs in 1 in 3000 live births and NF-2 occurs in 1 in 40,000 live births.1 Neurofibromas are discrete masses that arise from peripheral nerves and are composed of Schwann cells, mast cells, fibroblasts, and perineural cells. They commonly appear during or after puberty in the majority of patients and increase in number as patients age.1,2 Cutaneous neurofibromas can be painful and pruritic as well as cosmetically unappealing. Current treatment of neurofibromas consists primarily of standard surgical excision or laser therapy. Reports of loop electrocoagulation in the operating room, combination erbium:YAG and CO2 laser, and Nd:YAG laser treatments of neurofibromas have been published in the literature.3-5 These treatments can be effective, but the bothersome lesions in patients with neurofibromatosis often are multiple, making these procedures time consuming and costly. A removal technique that is time efficient, cost effective, and poses little discomfort or functional impairment for the patient is desirable. We describe the technique of shave removal with electrodesiccation for multiple neurofibromas.
A 24-year-old woman with NF-1 presented to the dermatology clinic for treatment of several pruritic neurofibromas of the trunk, including the bilateral breasts (Figure, A). We performed shave removal of each lesion with electrodesiccation of the lesion base with successful treatment of symptoms and acceptable cosmetic outcome (Figure, B). The patient identified symptomatic and cosmetically bothersome neurofibromas. The lesions were disinfected with an alcohol swab and anesthetized with 1% lidocaine and a 1:200,000 dilution of epinephrine. A shave biopsy blade was used to remove each neurofibroma at the level of the surrounding skin. Toothed forceps were utilized to lift the lesion and provide traction. Electrodesiccation was then used for destruction of the base or any residual gelatinous component of the lesion to the level of the surrounding skin, thus achieving hemostasis. Lastly, petrolatum was applied to the wound and covered with an adhesive dressing. Daily dressing change was performed for 2 weeks or until healed.
|
|
|
|
| A patient with neurofibromatosis type I with neurofibromas on the breast before (A) and approximately 6 months after treatment with shave removal plus electrodesiccation of the lesions (B). |
Patients with neurofibromatoses often present to dermatologists for management of symptomatic or cosmetically bothersome neurofibromas, which are often numerous and diffusely distributed. There are few published practical descriptions of removal techniques to manage these multiple and recurrent manifestations of the disease. The shave removal plus electrodesiccation technique we described is straightforward for practitioners and provides a well-tolerated mechanism for treating multiple lesions at once, while also offering patients acceptable functional and cosmetic results.
1. Listernick R, Charrow J. The neurofibromatoses. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill Companies; 2007:1331-1339.
2. Pinson S, Wolkenstein P. Neurofibromatosis type 1 or Von Recklinghausen’s disease [in French]. Rev Med Interne. 2005;26:196-215.
3. Elwakil TF, Samy NA, Elbasiouny MS. Non-excision treatment of multiple cutaneous neurofibromas by laser photocoagulation [published online ahead of print August 15, 2007]. Lasers Med Sci. 2008;23:301-306.
4. Meissner M, Ochsendorf F, Kaufmann R. Quick and effective treatment of small neurofibromas [published online ahead of print November 23, 2010]. J Dtsch Dermatol Ges. 2011;9:167-168.
5. Roberts AH, Crockett DJ. An operation for the treatment of cutaneous neurofibromatosis. Br J Plast Surg. 1985;38:292-293.
1. Listernick R, Charrow J. The neurofibromatoses. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill Companies; 2007:1331-1339.
2. Pinson S, Wolkenstein P. Neurofibromatosis type 1 or Von Recklinghausen’s disease [in French]. Rev Med Interne. 2005;26:196-215.
3. Elwakil TF, Samy NA, Elbasiouny MS. Non-excision treatment of multiple cutaneous neurofibromas by laser photocoagulation [published online ahead of print August 15, 2007]. Lasers Med Sci. 2008;23:301-306.
4. Meissner M, Ochsendorf F, Kaufmann R. Quick and effective treatment of small neurofibromas [published online ahead of print November 23, 2010]. J Dtsch Dermatol Ges. 2011;9:167-168.
5. Roberts AH, Crockett DJ. An operation for the treatment of cutaneous neurofibromatosis. Br J Plast Surg. 1985;38:292-293.
Flurbiprofen-Induced Unilateral Eyelid Angioedema
To the Editor:
Flurbiprofen, a member of the phenylalkanoic acid derivative group of nonsteroidal anti-inflammatory drugs (NSAIDs), are commonly used to treat fever, inflammation, and pain of arthritis.1 The exact prevalence of allergic reactions to NSAIDs in the general population is not known. Rhinoconjunctivitis, bronchospasm, urticaria, angioedema, and anaphylaxis can occur as an allergic reaction to NSAIDs. Isolated angioedema following NSAID ingestion typically involves the face, particularly the periorbital skin, lips, and mouth.2 These patients may develop urticaria and/or angioedema only after NSAID ingestion, but they do not have underlying chronic urticaria. We report a rare case of isolated unilateral eyelid angioedema with flurbiprofen.
A 39-year-old man presented with the onset of unilateral angioedema of the left upper eyelid that had developed approximately 30 minutes after taking flurbiprofen (100 mg). He reported frequent use of flurbiprofen for headaches. The patient also had a history of taking aspirin, ibuprofen, diclofenac, etodolac, and naproxen sodium as needed for migraines with no prior angioedema. He had no history of chronic urticaria or allergic disease. The patient was treated with oral pheniramine hydrogen maleate and angioedema resolved after 12 hours. Three days later, the patient used flurbiprofen again for a headache. He was readmitted to our clinic with unilateral angioedema of the left upper eyelid (Figure). The symptoms started approximately 30 minutes after taking flurbiprofen. Angioedema resolved within 1 day with oral pheniramine.
Nonsteroidal anti-inflammatory drugs are the most commonly prescribed class of drugs in the world and are the most common cause of all adverse drug reactions.3 Urticaria, angioedema, and anaphylaxis are common adverse reactions to NSAIDs. The prevalence of urticaria and angioedema to NSAIDs has been reported to be 0.1% to 3% worldwide.4
Angioedema is an abrupt localized swelling of the skin and mucous membranes of the face, lips, mouth, throat, larynx, extremities, and genitalia. Angioedema generally develops over minutes to hours and resolves in 24 to 48 hours.5 Angioedema without urticaria is the clinical syndrome that can be caused by an adverse drug reaction. In an Italian review of 2137 reactions, NSAIDs were causative agents in 33.6% of patients with drug-induced angioedema.6 In another study, Leeyaphan et al5 reported that 50% of patients with drug-induced angioedema resulted from NSAIDs, commonly with ibuprofen and diclofenac. Although angioedema is due to inhibition of cyclooxygenase 1, overproduction of leukotrienes, and possibly IgE-mediated reactions to single drugs,7 localized unilateral eyelid angioedema with NSAIDs is rare. The exact mechanism of localized eyelid edema is not known.8 We believe that the unilateral eyelid angioedema in our patient was caused by flurbiprofen use because the reaction recurred when the drug was used again.
1. Roszkowski MT, Swift JQ, Hargreaves KM. Effect of NSAID administration on tissue levels of immunoreactive prostaglandin E2, leukotriene B4, and (S)-flurbiprofen following extraction of impacted third molars. Pain. 1997;73:339-345.
2. Asero R. Multiple sensitivity to NSAID. Allergy. 2000;55:893-894.
3. Nettis E, Colanardi MC, Ferrannini A, et al. Update on sensitivity to nonsteroidal anti-inflammatory drugs. Curr Drug Targets Immune Endocr Metabol Disord. 2001;1:233-240.
4. Kulthanan K, Jiamton S, Boochangkool K, et al. Angioedema: clinical and etiological aspects. Clin Dev Immunol. 2007;2007:26438.
5. Leeyaphan C, Kulthanan K, Jongiarearnprasert K, et al. Drug-induced angioedema without urticaria: prevalence and clinical features [published online ahead of print November 17, 2009]. J Eur Acad Dermatol Venereol. 2010;24:685-691.
6. Cutaneous reactions to analgesic-antipyretics and nonsteroidal anti-inflammatory drugs. analysis of reports to the spontaneous reporting system of the Gruppo Italiano Studi Epidemiologici in Dermatologia. Dermatology. 1993;186:164-169.
7. Stevenson OE, Finch TM. Allergic contact dermatitis from rectified camphor oil in Earex ear drops. Contact Dermatitis. 2003;49:51.
8. Tsuruta D, Oshimo T, Sowa J, et al. Unilateral eyelid angioedema with congestion of the right bulbar conjunctiva due to loxoprofen sodium. Cutis. 2011;87:41-43.
To the Editor:
Flurbiprofen, a member of the phenylalkanoic acid derivative group of nonsteroidal anti-inflammatory drugs (NSAIDs), are commonly used to treat fever, inflammation, and pain of arthritis.1 The exact prevalence of allergic reactions to NSAIDs in the general population is not known. Rhinoconjunctivitis, bronchospasm, urticaria, angioedema, and anaphylaxis can occur as an allergic reaction to NSAIDs. Isolated angioedema following NSAID ingestion typically involves the face, particularly the periorbital skin, lips, and mouth.2 These patients may develop urticaria and/or angioedema only after NSAID ingestion, but they do not have underlying chronic urticaria. We report a rare case of isolated unilateral eyelid angioedema with flurbiprofen.
A 39-year-old man presented with the onset of unilateral angioedema of the left upper eyelid that had developed approximately 30 minutes after taking flurbiprofen (100 mg). He reported frequent use of flurbiprofen for headaches. The patient also had a history of taking aspirin, ibuprofen, diclofenac, etodolac, and naproxen sodium as needed for migraines with no prior angioedema. He had no history of chronic urticaria or allergic disease. The patient was treated with oral pheniramine hydrogen maleate and angioedema resolved after 12 hours. Three days later, the patient used flurbiprofen again for a headache. He was readmitted to our clinic with unilateral angioedema of the left upper eyelid (Figure). The symptoms started approximately 30 minutes after taking flurbiprofen. Angioedema resolved within 1 day with oral pheniramine.
Nonsteroidal anti-inflammatory drugs are the most commonly prescribed class of drugs in the world and are the most common cause of all adverse drug reactions.3 Urticaria, angioedema, and anaphylaxis are common adverse reactions to NSAIDs. The prevalence of urticaria and angioedema to NSAIDs has been reported to be 0.1% to 3% worldwide.4
Angioedema is an abrupt localized swelling of the skin and mucous membranes of the face, lips, mouth, throat, larynx, extremities, and genitalia. Angioedema generally develops over minutes to hours and resolves in 24 to 48 hours.5 Angioedema without urticaria is the clinical syndrome that can be caused by an adverse drug reaction. In an Italian review of 2137 reactions, NSAIDs were causative agents in 33.6% of patients with drug-induced angioedema.6 In another study, Leeyaphan et al5 reported that 50% of patients with drug-induced angioedema resulted from NSAIDs, commonly with ibuprofen and diclofenac. Although angioedema is due to inhibition of cyclooxygenase 1, overproduction of leukotrienes, and possibly IgE-mediated reactions to single drugs,7 localized unilateral eyelid angioedema with NSAIDs is rare. The exact mechanism of localized eyelid edema is not known.8 We believe that the unilateral eyelid angioedema in our patient was caused by flurbiprofen use because the reaction recurred when the drug was used again.
To the Editor:
Flurbiprofen, a member of the phenylalkanoic acid derivative group of nonsteroidal anti-inflammatory drugs (NSAIDs), are commonly used to treat fever, inflammation, and pain of arthritis.1 The exact prevalence of allergic reactions to NSAIDs in the general population is not known. Rhinoconjunctivitis, bronchospasm, urticaria, angioedema, and anaphylaxis can occur as an allergic reaction to NSAIDs. Isolated angioedema following NSAID ingestion typically involves the face, particularly the periorbital skin, lips, and mouth.2 These patients may develop urticaria and/or angioedema only after NSAID ingestion, but they do not have underlying chronic urticaria. We report a rare case of isolated unilateral eyelid angioedema with flurbiprofen.
A 39-year-old man presented with the onset of unilateral angioedema of the left upper eyelid that had developed approximately 30 minutes after taking flurbiprofen (100 mg). He reported frequent use of flurbiprofen for headaches. The patient also had a history of taking aspirin, ibuprofen, diclofenac, etodolac, and naproxen sodium as needed for migraines with no prior angioedema. He had no history of chronic urticaria or allergic disease. The patient was treated with oral pheniramine hydrogen maleate and angioedema resolved after 12 hours. Three days later, the patient used flurbiprofen again for a headache. He was readmitted to our clinic with unilateral angioedema of the left upper eyelid (Figure). The symptoms started approximately 30 minutes after taking flurbiprofen. Angioedema resolved within 1 day with oral pheniramine.
Nonsteroidal anti-inflammatory drugs are the most commonly prescribed class of drugs in the world and are the most common cause of all adverse drug reactions.3 Urticaria, angioedema, and anaphylaxis are common adverse reactions to NSAIDs. The prevalence of urticaria and angioedema to NSAIDs has been reported to be 0.1% to 3% worldwide.4
Angioedema is an abrupt localized swelling of the skin and mucous membranes of the face, lips, mouth, throat, larynx, extremities, and genitalia. Angioedema generally develops over minutes to hours and resolves in 24 to 48 hours.5 Angioedema without urticaria is the clinical syndrome that can be caused by an adverse drug reaction. In an Italian review of 2137 reactions, NSAIDs were causative agents in 33.6% of patients with drug-induced angioedema.6 In another study, Leeyaphan et al5 reported that 50% of patients with drug-induced angioedema resulted from NSAIDs, commonly with ibuprofen and diclofenac. Although angioedema is due to inhibition of cyclooxygenase 1, overproduction of leukotrienes, and possibly IgE-mediated reactions to single drugs,7 localized unilateral eyelid angioedema with NSAIDs is rare. The exact mechanism of localized eyelid edema is not known.8 We believe that the unilateral eyelid angioedema in our patient was caused by flurbiprofen use because the reaction recurred when the drug was used again.
1. Roszkowski MT, Swift JQ, Hargreaves KM. Effect of NSAID administration on tissue levels of immunoreactive prostaglandin E2, leukotriene B4, and (S)-flurbiprofen following extraction of impacted third molars. Pain. 1997;73:339-345.
2. Asero R. Multiple sensitivity to NSAID. Allergy. 2000;55:893-894.
3. Nettis E, Colanardi MC, Ferrannini A, et al. Update on sensitivity to nonsteroidal anti-inflammatory drugs. Curr Drug Targets Immune Endocr Metabol Disord. 2001;1:233-240.
4. Kulthanan K, Jiamton S, Boochangkool K, et al. Angioedema: clinical and etiological aspects. Clin Dev Immunol. 2007;2007:26438.
5. Leeyaphan C, Kulthanan K, Jongiarearnprasert K, et al. Drug-induced angioedema without urticaria: prevalence and clinical features [published online ahead of print November 17, 2009]. J Eur Acad Dermatol Venereol. 2010;24:685-691.
6. Cutaneous reactions to analgesic-antipyretics and nonsteroidal anti-inflammatory drugs. analysis of reports to the spontaneous reporting system of the Gruppo Italiano Studi Epidemiologici in Dermatologia. Dermatology. 1993;186:164-169.
7. Stevenson OE, Finch TM. Allergic contact dermatitis from rectified camphor oil in Earex ear drops. Contact Dermatitis. 2003;49:51.
8. Tsuruta D, Oshimo T, Sowa J, et al. Unilateral eyelid angioedema with congestion of the right bulbar conjunctiva due to loxoprofen sodium. Cutis. 2011;87:41-43.
1. Roszkowski MT, Swift JQ, Hargreaves KM. Effect of NSAID administration on tissue levels of immunoreactive prostaglandin E2, leukotriene B4, and (S)-flurbiprofen following extraction of impacted third molars. Pain. 1997;73:339-345.
2. Asero R. Multiple sensitivity to NSAID. Allergy. 2000;55:893-894.
3. Nettis E, Colanardi MC, Ferrannini A, et al. Update on sensitivity to nonsteroidal anti-inflammatory drugs. Curr Drug Targets Immune Endocr Metabol Disord. 2001;1:233-240.
4. Kulthanan K, Jiamton S, Boochangkool K, et al. Angioedema: clinical and etiological aspects. Clin Dev Immunol. 2007;2007:26438.
5. Leeyaphan C, Kulthanan K, Jongiarearnprasert K, et al. Drug-induced angioedema without urticaria: prevalence and clinical features [published online ahead of print November 17, 2009]. J Eur Acad Dermatol Venereol. 2010;24:685-691.
6. Cutaneous reactions to analgesic-antipyretics and nonsteroidal anti-inflammatory drugs. analysis of reports to the spontaneous reporting system of the Gruppo Italiano Studi Epidemiologici in Dermatologia. Dermatology. 1993;186:164-169.
7. Stevenson OE, Finch TM. Allergic contact dermatitis from rectified camphor oil in Earex ear drops. Contact Dermatitis. 2003;49:51.
8. Tsuruta D, Oshimo T, Sowa J, et al. Unilateral eyelid angioedema with congestion of the right bulbar conjunctiva due to loxoprofen sodium. Cutis. 2011;87:41-43.