User login
Collagenous and Elastotic Marginal Plaques of the Hands
To the Editor:
Collagenous and elastotic marginal plaques of the hands (CEMPHs) has several names including degenerative collagenous plaques of the hands, keratoelastoidosis marginalis, and digital papular calcific elastosis. This rare disorder is an acquired, slowly progressive, asymptomatic, dermal connective tissue abnormality that is underrecognized and underdiagnosed. Clinical presentation includes hyperkeratotic translucent papules arranged linearly on the radial aspect of the hands.
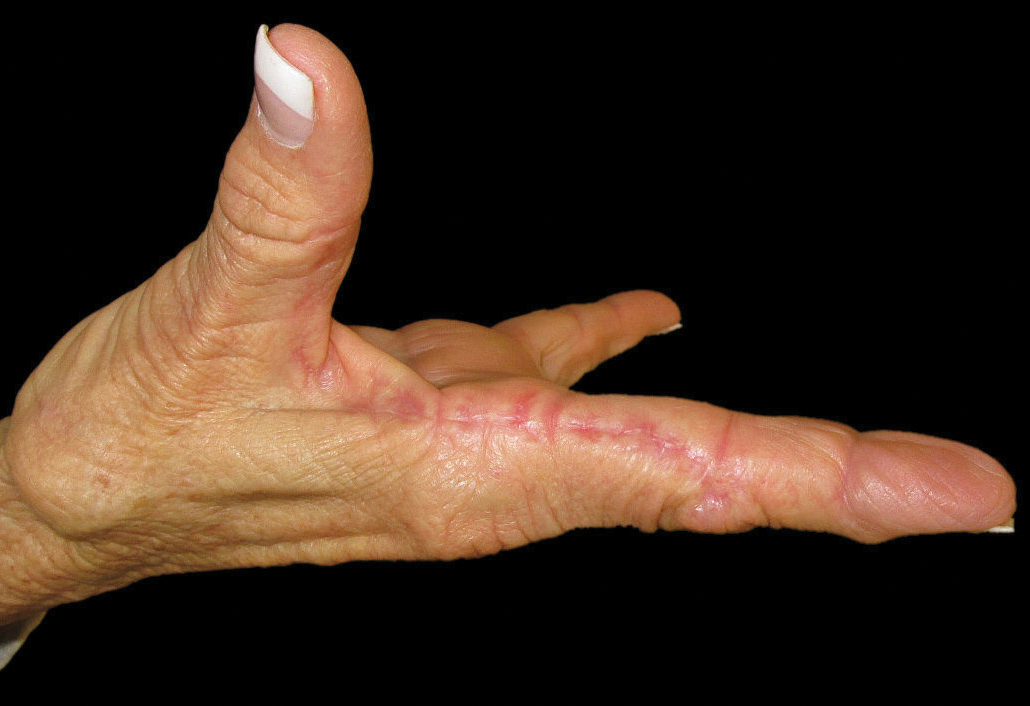
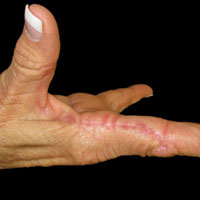
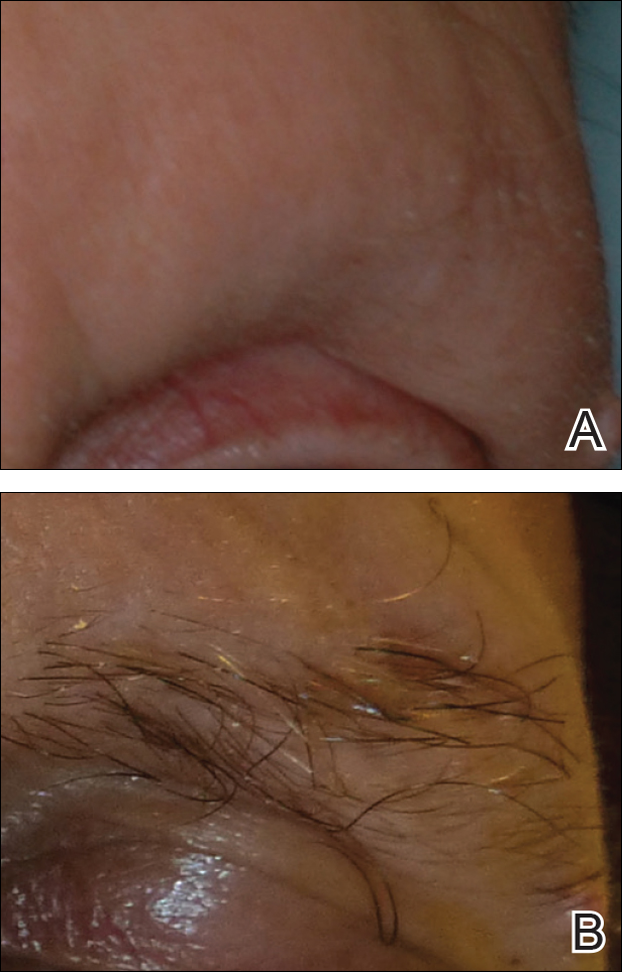
A 74-year-old woman described having "rough hands" of more than 20 years' duration. She presented with 4-cm wide longitudinal, erythematous, firm, depressed plaques along the lateral edge of the second finger and extending to the medial thumb in both hands (Figure 1). She had attempted multiple treatments by her primary care physician, including topical and oral medications unknown to the patient and light therapy, all without benefit over a period of several years. We have attempted salicylic acid 40%, clobetasol cream 0.05%, and emollient creams containing α-hydroxy acid. At best the condition fluctuated between a subtle raised scale at the edge to smooth and occasionally more red-pink, seemingly unrelated to any treatments.
The patient did not have plaques elsewhere on the body, and notably, the feet were clear. She did not have a history of repeated trauma to the hands and did not engage in manual labor. She denied excessive sun exposure, though she had Fitzpatrick skin type III and a history of multiple precancers and nonmelanoma skin cancers 7 years prior to presentation.
Histology of CEMPH reveals a hyperkeratotic epidermis with an avascular and acellular replacement of the superficial reticular dermis by haphazardly arranged, thickened collagen fibers (Figure 2A-2C). Collagen fibers were oriented perpendicularly to the epidermal surface. Intervening amorphous basophilic elastotic masses were present in the upper dermis with occasional calcification and degenerative elastic fibers (Figure 2D).

Collagenous and elastotic marginal plaques of the hands is a chronic, asymptomatic, sclerotic skin disorder described in a 1960 case series of 5 patients reported by Burks et al.1 Although it has many names, the most common is CEMPH. Collagenous and elastotic marginal plaques of the hands most often presents in white men aged 50 to 60 years.2 Patients typically are asymptomatic with plaques limited to the junction of the palmar and dorsal surfaces of the hands with only minimal intermittent stiffness around the flexor creases. Lesions begin as discrete yellow papules that coalesce to form hyperkeratotic linear plaques with occasional telangiectasia.3
The etiology of CEMPH is attributed to collagen and elastin degeneration by chronic actinic damage, pressure, or trauma.4,5 The 3 stages of degeneration include an initial linear padded stage, an intermediate padded plaque stage, and an advanced padded hyperkeratotic plaque stage.4 Vascular compromise is seen from the enlarged and fused thickened collagen and elastic fibers that in turn lead to ischemic changes, hyperkeratosis with epidermal atrophy, and papillary dermis telangiectasia. Absence or weak expression of keratins 14 and 10 and strong expression of keratin 16 have been reported in the epidermis of CEMPH patients.4
Collagenous and elastotic marginal plaques of the hands do not have a specific treatment, as it is a benign, slowly progressive condition. Several treatments such as laser therapy, high-potency topical corticosteroids, topical tazarotene and tretinoin, oral isotretinoin, and cryotherapy have been tried with little long-term success.4 Moisturizing may help reduce fissuring, and patients are advised to avoid the sun and repeated trauma to the hands.
The differential diagnosis of CEMPH is summarized in the Table. Two genodermatoses—acrokeratoelastoidosis of Costa and focal acral hyperkeratosis—clinically resemble CEMPH. Acrokeratoelastoidosis of Costa is an autosomal-dominant condition that occurs without trauma in children and young adults. Histopathology shows orthokeratotic hyperkeratosis due to an overproduction of filaggrin in the granular layer of the epidermis. The reticular dermis shows basophilic, thick, curled and fragmented elastic fibers with dilated capillaries that can be seen with Weigert elastic, Verhoeff-van Gieson, or orcein stains. Focal acral hyperkeratosis occurs on the hands and feet, predominantly in black patients. On histology, the epidermis shows a characteristic orthohyperkeratosis, moderate acanthosis, and slight hypergranulosis with no dermal involvment.6
Chronic hyperkeratotic eczematous dermatitis is another common entity in the differential characterized by hyperkeratotic plaques that scale and fissure. Biopsy demonstrates a spongiotic acanthotic epidermis.7,8
Psoriasis of the hands, specifically hyperkeratotic palmoplantar psoriasis, is associated with manual labor, similar to CEMPH. Histology shows epidermal hyperplasia; regular acanthosis; loss of the granular skin layer with prominent dermal capillaries; and a mixed dermal infiltrate of lymphocytes, macrophages, and neutrophils.9 Hyperkeratotic palmoplantar lichen planus presents with pruritic papules in the third and fifth decades of life. Histologically, hyperkeratosis, acanthosis, and wedge-shaped hypergranulosis with a lichenoid lymphocytic infiltration at the dermoepidermal junction is seen.10
Palmoplantar keratodermas due to inflammatory reactive dermatoses include callosities that develop in response to repeated trauma or friction on the skin. On histology, there is prominent hyperkeratosis and acanthosis with moderate papillomatosis.11 Drug-related palmoplantar keratodermas such as those from arsenic exposure can lead to multiple, irregular, verrucous, keratotic, and pigmented lesions on the palms and soles. Histologically, atypical keratinocytes are seen in the epidermis with thick hyperkeratosis and vacuolated cells without solar elastosis.12
In conclusion, CEMPH is an underdiagnosed and underrecognized condition characterized by asymptomatic hyperkeratotic linear plaques along the medial aspect of the thumb and radial aspect of the index finger. It is important to keep CEMPH in mind when dealing with occupational cases of repeated long-term trauma or pressure to the hands as well as excessive sun exposure. It also is imperative to separate it from other diseases and avoid misdiagnosing this degenerative collagenous and elastotic disease as a malignant lesion.
- Burks JW, Wise LJ, Clark WH. Degenerative collagenous plaques of the hands. Arch Dermatol. 1960;82:362-366.
- Jordaan HF, Rossouw DJ. Digital papular calcific elastosis: a histopathological, histochemical and ultrastructural study of 20 patients. J Cutan Pathol. 1990;17:358-370.
- Mortimore RJ, Conrad RJ. Collagenous and elastotic marginal plaques of the hands. Australas J Dermatol. 2001;42:211-213.
- Tieu KD, Satter EK. Thickened plaques on the hands. Collagenous and elastotic marginal plaques of the hands (CEMPH). Arch Dermatol. 2011;147:499-504.
- Todd D, Al-Aboosi M, Hameed O, et al. The role of UV light in the pathogenesis of digital papular calcific elastosis. Arch Dermatol. 2001;137:379-381.
- Mengesha YM, Kayal JD, Swerlick RA. Keratoelastoidosis marginalis. J Cutan Med Surg. 2002;6:23-25.
- MacKee MG, Lewis MG. Keratolysis exfoliativa and the mosaic fungus. Arch Dermatol. 1931;23:445-447.
- Walling HW, Swick BL, Storrs FJ, et al. Frictional hyperkeratotic hand dermatitis responding to Grenz ray therapy. Contact Dermatitis. 2008;58:49-51.
- Farley E, Masrour S, McKey J, et al. Palmoplantar psoriasis: a phenotypical and clinical review with introduction of a new quality-of-life assessment tool. J Am Acad Dermatol. 2009;60:1024-1031.
- Rotunda AM, Craft N, Haley JC. Hyperkeratotic plaques on the palms and soles. palmoplantar lichen planus, hyperkeratotic variant. Arch Dermatol. 2004;140:1275-1280.
- Unal VS, Sevin A, Dayican A. Palmar callus formation as a result of mechanical trauma during sailing. Plast Reconstr Surg. 2005;115:2161-2162.
- Cöl M, Cöl C, Soran A, et al. Arsenic-related Bowen's disease, palmar keratosis, and skin cancer. Environ Health Perspect. 1999;107:687-689.
To the Editor:
Collagenous and elastotic marginal plaques of the hands (CEMPHs) has several names including degenerative collagenous plaques of the hands, keratoelastoidosis marginalis, and digital papular calcific elastosis. This rare disorder is an acquired, slowly progressive, asymptomatic, dermal connective tissue abnormality that is underrecognized and underdiagnosed. Clinical presentation includes hyperkeratotic translucent papules arranged linearly on the radial aspect of the hands.
A 74-year-old woman described having "rough hands" of more than 20 years' duration. She presented with 4-cm wide longitudinal, erythematous, firm, depressed plaques along the lateral edge of the second finger and extending to the medial thumb in both hands (Figure 1). She had attempted multiple treatments by her primary care physician, including topical and oral medications unknown to the patient and light therapy, all without benefit over a period of several years. We have attempted salicylic acid 40%, clobetasol cream 0.05%, and emollient creams containing α-hydroxy acid. At best the condition fluctuated between a subtle raised scale at the edge to smooth and occasionally more red-pink, seemingly unrelated to any treatments.
The patient did not have plaques elsewhere on the body, and notably, the feet were clear. She did not have a history of repeated trauma to the hands and did not engage in manual labor. She denied excessive sun exposure, though she had Fitzpatrick skin type III and a history of multiple precancers and nonmelanoma skin cancers 7 years prior to presentation.
Histology of CEMPH reveals a hyperkeratotic epidermis with an avascular and acellular replacement of the superficial reticular dermis by haphazardly arranged, thickened collagen fibers (Figure 2A-2C). Collagen fibers were oriented perpendicularly to the epidermal surface. Intervening amorphous basophilic elastotic masses were present in the upper dermis with occasional calcification and degenerative elastic fibers (Figure 2D).
Collagenous and elastotic marginal plaques of the hands is a chronic, asymptomatic, sclerotic skin disorder described in a 1960 case series of 5 patients reported by Burks et al.1 Although it has many names, the most common is CEMPH. Collagenous and elastotic marginal plaques of the hands most often presents in white men aged 50 to 60 years.2 Patients typically are asymptomatic with plaques limited to the junction of the palmar and dorsal surfaces of the hands with only minimal intermittent stiffness around the flexor creases. Lesions begin as discrete yellow papules that coalesce to form hyperkeratotic linear plaques with occasional telangiectasia.3
The etiology of CEMPH is attributed to collagen and elastin degeneration by chronic actinic damage, pressure, or trauma.4,5 The 3 stages of degeneration include an initial linear padded stage, an intermediate padded plaque stage, and an advanced padded hyperkeratotic plaque stage.4 Vascular compromise is seen from the enlarged and fused thickened collagen and elastic fibers that in turn lead to ischemic changes, hyperkeratosis with epidermal atrophy, and papillary dermis telangiectasia. Absence or weak expression of keratins 14 and 10 and strong expression of keratin 16 have been reported in the epidermis of CEMPH patients.4
Collagenous and elastotic marginal plaques of the hands do not have a specific treatment, as it is a benign, slowly progressive condition. Several treatments such as laser therapy, high-potency topical corticosteroids, topical tazarotene and tretinoin, oral isotretinoin, and cryotherapy have been tried with little long-term success.4 Moisturizing may help reduce fissuring, and patients are advised to avoid the sun and repeated trauma to the hands.
The differential diagnosis of CEMPH is summarized in the Table. Two genodermatoses—acrokeratoelastoidosis of Costa and focal acral hyperkeratosis—clinically resemble CEMPH. Acrokeratoelastoidosis of Costa is an autosomal-dominant condition that occurs without trauma in children and young adults. Histopathology shows orthokeratotic hyperkeratosis due to an overproduction of filaggrin in the granular layer of the epidermis. The reticular dermis shows basophilic, thick, curled and fragmented elastic fibers with dilated capillaries that can be seen with Weigert elastic, Verhoeff-van Gieson, or orcein stains. Focal acral hyperkeratosis occurs on the hands and feet, predominantly in black patients. On histology, the epidermis shows a characteristic orthohyperkeratosis, moderate acanthosis, and slight hypergranulosis with no dermal involvment.6
Chronic hyperkeratotic eczematous dermatitis is another common entity in the differential characterized by hyperkeratotic plaques that scale and fissure. Biopsy demonstrates a spongiotic acanthotic epidermis.7,8
Psoriasis of the hands, specifically hyperkeratotic palmoplantar psoriasis, is associated with manual labor, similar to CEMPH. Histology shows epidermal hyperplasia; regular acanthosis; loss of the granular skin layer with prominent dermal capillaries; and a mixed dermal infiltrate of lymphocytes, macrophages, and neutrophils.9 Hyperkeratotic palmoplantar lichen planus presents with pruritic papules in the third and fifth decades of life. Histologically, hyperkeratosis, acanthosis, and wedge-shaped hypergranulosis with a lichenoid lymphocytic infiltration at the dermoepidermal junction is seen.10
Palmoplantar keratodermas due to inflammatory reactive dermatoses include callosities that develop in response to repeated trauma or friction on the skin. On histology, there is prominent hyperkeratosis and acanthosis with moderate papillomatosis.11 Drug-related palmoplantar keratodermas such as those from arsenic exposure can lead to multiple, irregular, verrucous, keratotic, and pigmented lesions on the palms and soles. Histologically, atypical keratinocytes are seen in the epidermis with thick hyperkeratosis and vacuolated cells without solar elastosis.12
In conclusion, CEMPH is an underdiagnosed and underrecognized condition characterized by asymptomatic hyperkeratotic linear plaques along the medial aspect of the thumb and radial aspect of the index finger. It is important to keep CEMPH in mind when dealing with occupational cases of repeated long-term trauma or pressure to the hands as well as excessive sun exposure. It also is imperative to separate it from other diseases and avoid misdiagnosing this degenerative collagenous and elastotic disease as a malignant lesion.
To the Editor:
Collagenous and elastotic marginal plaques of the hands (CEMPHs) has several names including degenerative collagenous plaques of the hands, keratoelastoidosis marginalis, and digital papular calcific elastosis. This rare disorder is an acquired, slowly progressive, asymptomatic, dermal connective tissue abnormality that is underrecognized and underdiagnosed. Clinical presentation includes hyperkeratotic translucent papules arranged linearly on the radial aspect of the hands.
A 74-year-old woman described having "rough hands" of more than 20 years' duration. She presented with 4-cm wide longitudinal, erythematous, firm, depressed plaques along the lateral edge of the second finger and extending to the medial thumb in both hands (Figure 1). She had attempted multiple treatments by her primary care physician, including topical and oral medications unknown to the patient and light therapy, all without benefit over a period of several years. We have attempted salicylic acid 40%, clobetasol cream 0.05%, and emollient creams containing α-hydroxy acid. At best the condition fluctuated between a subtle raised scale at the edge to smooth and occasionally more red-pink, seemingly unrelated to any treatments.
The patient did not have plaques elsewhere on the body, and notably, the feet were clear. She did not have a history of repeated trauma to the hands and did not engage in manual labor. She denied excessive sun exposure, though she had Fitzpatrick skin type III and a history of multiple precancers and nonmelanoma skin cancers 7 years prior to presentation.
Histology of CEMPH reveals a hyperkeratotic epidermis with an avascular and acellular replacement of the superficial reticular dermis by haphazardly arranged, thickened collagen fibers (Figure 2A-2C). Collagen fibers were oriented perpendicularly to the epidermal surface. Intervening amorphous basophilic elastotic masses were present in the upper dermis with occasional calcification and degenerative elastic fibers (Figure 2D).
Collagenous and elastotic marginal plaques of the hands is a chronic, asymptomatic, sclerotic skin disorder described in a 1960 case series of 5 patients reported by Burks et al.1 Although it has many names, the most common is CEMPH. Collagenous and elastotic marginal plaques of the hands most often presents in white men aged 50 to 60 years.2 Patients typically are asymptomatic with plaques limited to the junction of the palmar and dorsal surfaces of the hands with only minimal intermittent stiffness around the flexor creases. Lesions begin as discrete yellow papules that coalesce to form hyperkeratotic linear plaques with occasional telangiectasia.3
The etiology of CEMPH is attributed to collagen and elastin degeneration by chronic actinic damage, pressure, or trauma.4,5 The 3 stages of degeneration include an initial linear padded stage, an intermediate padded plaque stage, and an advanced padded hyperkeratotic plaque stage.4 Vascular compromise is seen from the enlarged and fused thickened collagen and elastic fibers that in turn lead to ischemic changes, hyperkeratosis with epidermal atrophy, and papillary dermis telangiectasia. Absence or weak expression of keratins 14 and 10 and strong expression of keratin 16 have been reported in the epidermis of CEMPH patients.4
Collagenous and elastotic marginal plaques of the hands do not have a specific treatment, as it is a benign, slowly progressive condition. Several treatments such as laser therapy, high-potency topical corticosteroids, topical tazarotene and tretinoin, oral isotretinoin, and cryotherapy have been tried with little long-term success.4 Moisturizing may help reduce fissuring, and patients are advised to avoid the sun and repeated trauma to the hands.
The differential diagnosis of CEMPH is summarized in the Table. Two genodermatoses—acrokeratoelastoidosis of Costa and focal acral hyperkeratosis—clinically resemble CEMPH. Acrokeratoelastoidosis of Costa is an autosomal-dominant condition that occurs without trauma in children and young adults. Histopathology shows orthokeratotic hyperkeratosis due to an overproduction of filaggrin in the granular layer of the epidermis. The reticular dermis shows basophilic, thick, curled and fragmented elastic fibers with dilated capillaries that can be seen with Weigert elastic, Verhoeff-van Gieson, or orcein stains. Focal acral hyperkeratosis occurs on the hands and feet, predominantly in black patients. On histology, the epidermis shows a characteristic orthohyperkeratosis, moderate acanthosis, and slight hypergranulosis with no dermal involvment.6
Chronic hyperkeratotic eczematous dermatitis is another common entity in the differential characterized by hyperkeratotic plaques that scale and fissure. Biopsy demonstrates a spongiotic acanthotic epidermis.7,8
Psoriasis of the hands, specifically hyperkeratotic palmoplantar psoriasis, is associated with manual labor, similar to CEMPH. Histology shows epidermal hyperplasia; regular acanthosis; loss of the granular skin layer with prominent dermal capillaries; and a mixed dermal infiltrate of lymphocytes, macrophages, and neutrophils.9 Hyperkeratotic palmoplantar lichen planus presents with pruritic papules in the third and fifth decades of life. Histologically, hyperkeratosis, acanthosis, and wedge-shaped hypergranulosis with a lichenoid lymphocytic infiltration at the dermoepidermal junction is seen.10
Palmoplantar keratodermas due to inflammatory reactive dermatoses include callosities that develop in response to repeated trauma or friction on the skin. On histology, there is prominent hyperkeratosis and acanthosis with moderate papillomatosis.11 Drug-related palmoplantar keratodermas such as those from arsenic exposure can lead to multiple, irregular, verrucous, keratotic, and pigmented lesions on the palms and soles. Histologically, atypical keratinocytes are seen in the epidermis with thick hyperkeratosis and vacuolated cells without solar elastosis.12
In conclusion, CEMPH is an underdiagnosed and underrecognized condition characterized by asymptomatic hyperkeratotic linear plaques along the medial aspect of the thumb and radial aspect of the index finger. It is important to keep CEMPH in mind when dealing with occupational cases of repeated long-term trauma or pressure to the hands as well as excessive sun exposure. It also is imperative to separate it from other diseases and avoid misdiagnosing this degenerative collagenous and elastotic disease as a malignant lesion.
- Burks JW, Wise LJ, Clark WH. Degenerative collagenous plaques of the hands. Arch Dermatol. 1960;82:362-366.
- Jordaan HF, Rossouw DJ. Digital papular calcific elastosis: a histopathological, histochemical and ultrastructural study of 20 patients. J Cutan Pathol. 1990;17:358-370.
- Mortimore RJ, Conrad RJ. Collagenous and elastotic marginal plaques of the hands. Australas J Dermatol. 2001;42:211-213.
- Tieu KD, Satter EK. Thickened plaques on the hands. Collagenous and elastotic marginal plaques of the hands (CEMPH). Arch Dermatol. 2011;147:499-504.
- Todd D, Al-Aboosi M, Hameed O, et al. The role of UV light in the pathogenesis of digital papular calcific elastosis. Arch Dermatol. 2001;137:379-381.
- Mengesha YM, Kayal JD, Swerlick RA. Keratoelastoidosis marginalis. J Cutan Med Surg. 2002;6:23-25.
- MacKee MG, Lewis MG. Keratolysis exfoliativa and the mosaic fungus. Arch Dermatol. 1931;23:445-447.
- Walling HW, Swick BL, Storrs FJ, et al. Frictional hyperkeratotic hand dermatitis responding to Grenz ray therapy. Contact Dermatitis. 2008;58:49-51.
- Farley E, Masrour S, McKey J, et al. Palmoplantar psoriasis: a phenotypical and clinical review with introduction of a new quality-of-life assessment tool. J Am Acad Dermatol. 2009;60:1024-1031.
- Rotunda AM, Craft N, Haley JC. Hyperkeratotic plaques on the palms and soles. palmoplantar lichen planus, hyperkeratotic variant. Arch Dermatol. 2004;140:1275-1280.
- Unal VS, Sevin A, Dayican A. Palmar callus formation as a result of mechanical trauma during sailing. Plast Reconstr Surg. 2005;115:2161-2162.
- Cöl M, Cöl C, Soran A, et al. Arsenic-related Bowen's disease, palmar keratosis, and skin cancer. Environ Health Perspect. 1999;107:687-689.
- Burks JW, Wise LJ, Clark WH. Degenerative collagenous plaques of the hands. Arch Dermatol. 1960;82:362-366.
- Jordaan HF, Rossouw DJ. Digital papular calcific elastosis: a histopathological, histochemical and ultrastructural study of 20 patients. J Cutan Pathol. 1990;17:358-370.
- Mortimore RJ, Conrad RJ. Collagenous and elastotic marginal plaques of the hands. Australas J Dermatol. 2001;42:211-213.
- Tieu KD, Satter EK. Thickened plaques on the hands. Collagenous and elastotic marginal plaques of the hands (CEMPH). Arch Dermatol. 2011;147:499-504.
- Todd D, Al-Aboosi M, Hameed O, et al. The role of UV light in the pathogenesis of digital papular calcific elastosis. Arch Dermatol. 2001;137:379-381.
- Mengesha YM, Kayal JD, Swerlick RA. Keratoelastoidosis marginalis. J Cutan Med Surg. 2002;6:23-25.
- MacKee MG, Lewis MG. Keratolysis exfoliativa and the mosaic fungus. Arch Dermatol. 1931;23:445-447.
- Walling HW, Swick BL, Storrs FJ, et al. Frictional hyperkeratotic hand dermatitis responding to Grenz ray therapy. Contact Dermatitis. 2008;58:49-51.
- Farley E, Masrour S, McKey J, et al. Palmoplantar psoriasis: a phenotypical and clinical review with introduction of a new quality-of-life assessment tool. J Am Acad Dermatol. 2009;60:1024-1031.
- Rotunda AM, Craft N, Haley JC. Hyperkeratotic plaques on the palms and soles. palmoplantar lichen planus, hyperkeratotic variant. Arch Dermatol. 2004;140:1275-1280.
- Unal VS, Sevin A, Dayican A. Palmar callus formation as a result of mechanical trauma during sailing. Plast Reconstr Surg. 2005;115:2161-2162.
- Cöl M, Cöl C, Soran A, et al. Arsenic-related Bowen's disease, palmar keratosis, and skin cancer. Environ Health Perspect. 1999;107:687-689.
Practice Points
- The etiology of collagenous and elastotic marginal plaques of the hands (CEMPHs) is attributed to collagen and elastin degeneration by chronic actinic damage, pressure, or trauma.
- It is important to keep CEMPH in mind when dealing with occupational cases of repeated long-term trauma or pressure to the hands as well as excessive sun exposure. It should be separated from other diseases and avoid being misdiagnosed as a malignant lesion.
Reversible Cutaneous Side Effects of Vismodegib Treatment
To the Editor:
Vismodegib, a first-in-class inhibitor of the hedgehog signaling pathway, is useful in the treatment of advanced basal cell carcinomas (BCCs).1 Common side effects of vismodegib include alopecia (58%), muscle spasms (71%), and dysgeusia (71%).2 Some of these side effects have been hypothesized to be mechanism related.3,4 Keratoacanthomas have been reported to occur after vismodegib treatment of BCC.5 We report 3 cases illustrating reversible cutaneous side effects of vismodegib: alopecia, follicular dermatitis, and drug hypersensitivity reaction.
A 53-year-old man with a locally advanced BCC of the right medial canthus began experiencing progressive and diffuse hair loss on the beard area, parietal scalp, eyelashes, and eyebrows after 2 months of vismodegib treatment. At 12 months of treatment, he had complete loss of eyelashes and eyebrows (Figure, A). After vismodegib was discontinued due to disease progression, all of his hair began regrowing within several months, with complete hair regrowth observed at 20 months after the last dose (Figure, B).
A 55-year-old man with several locally advanced BCCs developed new-onset mildly pruritic, acneform lesions on the chest and back after 4 months of vismodegib treatment. Biopsy of the lesions showed a folliculocentric mixed dermal infiltrate. The patient did not have a history of follicular dermatitis. The dermatitis resolved several months after onset without treatment, despite continued vismodegib.
A 55-year-old man with locally advanced BCCs developed erythematous dermal plaques on the arms and chest after 2 months of vismodegib treatment. Lesions were asymptomatic. He was not using any other medications and did not have any contact allergen exposures. Punch biopsy showed superficial and deep perivascular dermatitis with occasional eosinophils, consistent with drug hypersensitivity. Although lesions spontaneously resolved without treatment after 1 month, he experienced a couple more bouts of these lesions over the next year. He continued vismodegib for 2 years without return of this eruption.
The average time frame for hair regrowth after vismodegib cessation has not been characterized and awaits future larger studies. The frequency of follicular dermatitis and drug eruption also has not been determined and may require careful observation by dermatologists in larger numbers of treated patients.
Because the hedgehog pathway is critical for normal hair follicle function, follicle-based toxicities of vismodegib including alopecia and folliculitis could be hypothesized to reflect effective blockade of the pathway.6 Currently, there are no data that these changes correlate with tumor response.
Although alopecia is a recognized side effect of vismodegib, regrowth has not been previously reported.1,2 Knowledge of the reversibility of alopecia as well as other toxicities has the potential to influence patient decision-making on drug initiation and adherence.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Chang AL, Solomon JA, Hainsworth JD, et al. Expanded access study of patients with advanced basal cell carcinoma treated with the Hedgehog pathway inhibitor, vismodegib. J Am Acad Dermatol. 2014;70:60-69.
- St-Jacques B, Dassule HR, Karavanova I, et al. Sonic hedgehog signaling is essential for hair development. Curr Biol. 1998;8:1058-1068.
- Hall JM, Bell ML, Finger TE. Disruption of sonic hedgehog signaling alters growth and patterning of lingual taste papillae. Dev Biol. 2003;255:263-277.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Rittie L, Stoll SW, Kang S, et al. Hedgehog signaling maintains hair follicle stem cell phenotype in young and aged human skin. Aging Cell. 2009;8:738-751.
To the Editor:
Vismodegib, a first-in-class inhibitor of the hedgehog signaling pathway, is useful in the treatment of advanced basal cell carcinomas (BCCs).1 Common side effects of vismodegib include alopecia (58%), muscle spasms (71%), and dysgeusia (71%).2 Some of these side effects have been hypothesized to be mechanism related.3,4 Keratoacanthomas have been reported to occur after vismodegib treatment of BCC.5 We report 3 cases illustrating reversible cutaneous side effects of vismodegib: alopecia, follicular dermatitis, and drug hypersensitivity reaction.
A 53-year-old man with a locally advanced BCC of the right medial canthus began experiencing progressive and diffuse hair loss on the beard area, parietal scalp, eyelashes, and eyebrows after 2 months of vismodegib treatment. At 12 months of treatment, he had complete loss of eyelashes and eyebrows (Figure, A). After vismodegib was discontinued due to disease progression, all of his hair began regrowing within several months, with complete hair regrowth observed at 20 months after the last dose (Figure, B).
A 55-year-old man with several locally advanced BCCs developed new-onset mildly pruritic, acneform lesions on the chest and back after 4 months of vismodegib treatment. Biopsy of the lesions showed a folliculocentric mixed dermal infiltrate. The patient did not have a history of follicular dermatitis. The dermatitis resolved several months after onset without treatment, despite continued vismodegib.
A 55-year-old man with locally advanced BCCs developed erythematous dermal plaques on the arms and chest after 2 months of vismodegib treatment. Lesions were asymptomatic. He was not using any other medications and did not have any contact allergen exposures. Punch biopsy showed superficial and deep perivascular dermatitis with occasional eosinophils, consistent with drug hypersensitivity. Although lesions spontaneously resolved without treatment after 1 month, he experienced a couple more bouts of these lesions over the next year. He continued vismodegib for 2 years without return of this eruption.
The average time frame for hair regrowth after vismodegib cessation has not been characterized and awaits future larger studies. The frequency of follicular dermatitis and drug eruption also has not been determined and may require careful observation by dermatologists in larger numbers of treated patients.
Because the hedgehog pathway is critical for normal hair follicle function, follicle-based toxicities of vismodegib including alopecia and folliculitis could be hypothesized to reflect effective blockade of the pathway.6 Currently, there are no data that these changes correlate with tumor response.
Although alopecia is a recognized side effect of vismodegib, regrowth has not been previously reported.1,2 Knowledge of the reversibility of alopecia as well as other toxicities has the potential to influence patient decision-making on drug initiation and adherence.
To the Editor:
Vismodegib, a first-in-class inhibitor of the hedgehog signaling pathway, is useful in the treatment of advanced basal cell carcinomas (BCCs).1 Common side effects of vismodegib include alopecia (58%), muscle spasms (71%), and dysgeusia (71%).2 Some of these side effects have been hypothesized to be mechanism related.3,4 Keratoacanthomas have been reported to occur after vismodegib treatment of BCC.5 We report 3 cases illustrating reversible cutaneous side effects of vismodegib: alopecia, follicular dermatitis, and drug hypersensitivity reaction.
A 53-year-old man with a locally advanced BCC of the right medial canthus began experiencing progressive and diffuse hair loss on the beard area, parietal scalp, eyelashes, and eyebrows after 2 months of vismodegib treatment. At 12 months of treatment, he had complete loss of eyelashes and eyebrows (Figure, A). After vismodegib was discontinued due to disease progression, all of his hair began regrowing within several months, with complete hair regrowth observed at 20 months after the last dose (Figure, B).
A 55-year-old man with several locally advanced BCCs developed new-onset mildly pruritic, acneform lesions on the chest and back after 4 months of vismodegib treatment. Biopsy of the lesions showed a folliculocentric mixed dermal infiltrate. The patient did not have a history of follicular dermatitis. The dermatitis resolved several months after onset without treatment, despite continued vismodegib.
A 55-year-old man with locally advanced BCCs developed erythematous dermal plaques on the arms and chest after 2 months of vismodegib treatment. Lesions were asymptomatic. He was not using any other medications and did not have any contact allergen exposures. Punch biopsy showed superficial and deep perivascular dermatitis with occasional eosinophils, consistent with drug hypersensitivity. Although lesions spontaneously resolved without treatment after 1 month, he experienced a couple more bouts of these lesions over the next year. He continued vismodegib for 2 years without return of this eruption.
The average time frame for hair regrowth after vismodegib cessation has not been characterized and awaits future larger studies. The frequency of follicular dermatitis and drug eruption also has not been determined and may require careful observation by dermatologists in larger numbers of treated patients.
Because the hedgehog pathway is critical for normal hair follicle function, follicle-based toxicities of vismodegib including alopecia and folliculitis could be hypothesized to reflect effective blockade of the pathway.6 Currently, there are no data that these changes correlate with tumor response.
Although alopecia is a recognized side effect of vismodegib, regrowth has not been previously reported.1,2 Knowledge of the reversibility of alopecia as well as other toxicities has the potential to influence patient decision-making on drug initiation and adherence.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Chang AL, Solomon JA, Hainsworth JD, et al. Expanded access study of patients with advanced basal cell carcinoma treated with the Hedgehog pathway inhibitor, vismodegib. J Am Acad Dermatol. 2014;70:60-69.
- St-Jacques B, Dassule HR, Karavanova I, et al. Sonic hedgehog signaling is essential for hair development. Curr Biol. 1998;8:1058-1068.
- Hall JM, Bell ML, Finger TE. Disruption of sonic hedgehog signaling alters growth and patterning of lingual taste papillae. Dev Biol. 2003;255:263-277.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Rittie L, Stoll SW, Kang S, et al. Hedgehog signaling maintains hair follicle stem cell phenotype in young and aged human skin. Aging Cell. 2009;8:738-751.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Chang AL, Solomon JA, Hainsworth JD, et al. Expanded access study of patients with advanced basal cell carcinoma treated with the Hedgehog pathway inhibitor, vismodegib. J Am Acad Dermatol. 2014;70:60-69.
- St-Jacques B, Dassule HR, Karavanova I, et al. Sonic hedgehog signaling is essential for hair development. Curr Biol. 1998;8:1058-1068.
- Hall JM, Bell ML, Finger TE. Disruption of sonic hedgehog signaling alters growth and patterning of lingual taste papillae. Dev Biol. 2003;255:263-277.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Rittie L, Stoll SW, Kang S, et al. Hedgehog signaling maintains hair follicle stem cell phenotype in young and aged human skin. Aging Cell. 2009;8:738-751.
Practice Points
- Hair loss is a common late side effect of vismodegib usage and is reversible, but regrowth takes many months.
- Mild folliculitis that resolves spontaneously has been observed in patients using vismodegib.
- Dermal hypersensitivity has been observed in patients on vismodegib, though the exact frequency of this type of dermatitis is not known.
Primary Cutaneous Cryptococcosis Presenting as an Extensive Eroded Plaque
To the Editor:
Primary cutaneous cryptococcal infection is rare. Cryptococcal skin infections, either primary or disseminated, can be highly pleomorphic and mimic entities such as basal cell carcinoma or even severe dermatitis, as in our case.
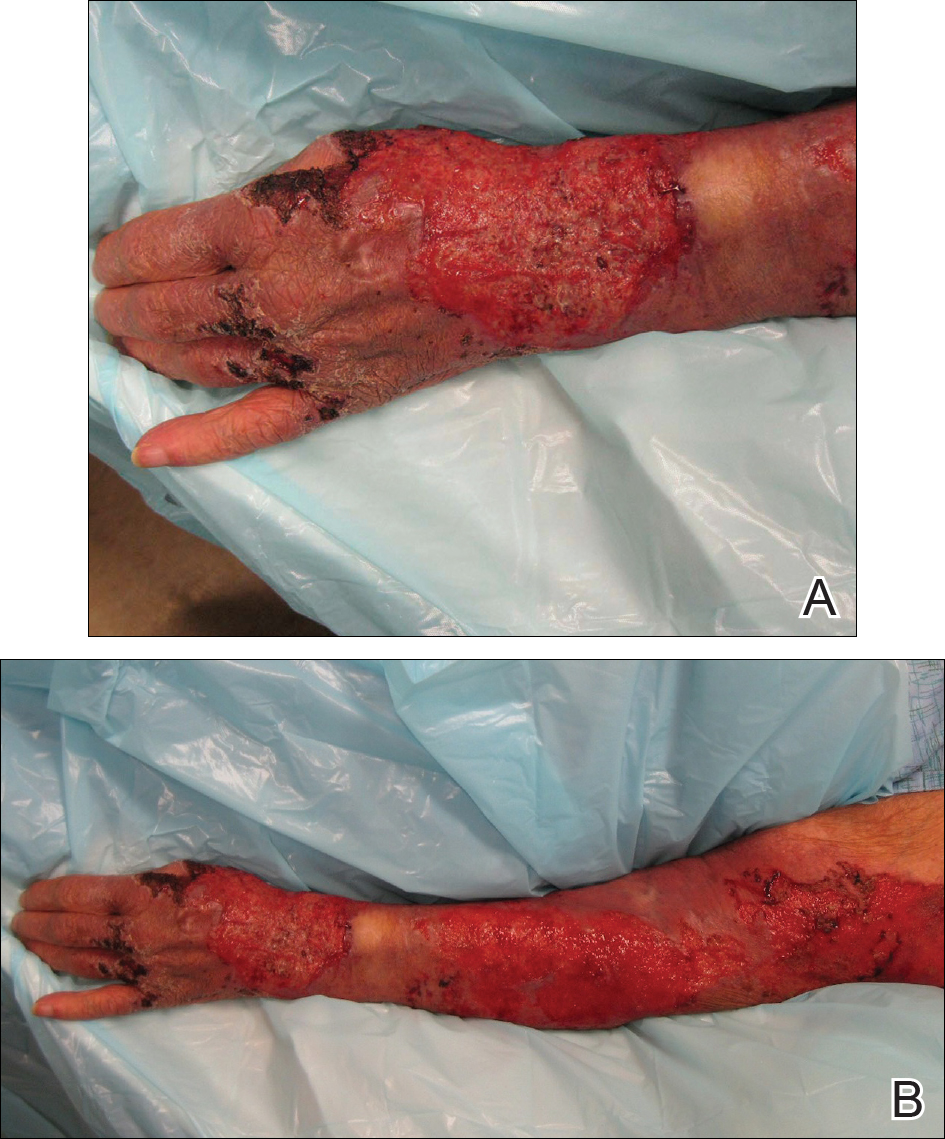
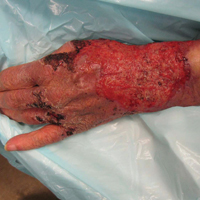
An 80-year-old woman who was residing in a nursing facility presented to the emergency department with an itchy nontender rash on the left arm of 2 to 3 weeks' duration that gradually spread. The patient had not started any new topical or oral medications and was otherwise healthy. A review of symptoms was negative for fever, weight loss, or new cough. Her medical history was notable for congestive heart failure, chronic obstructive pulmonary disease requiring chronic low-dose prednisone, hypothyroidism, atrial fibrillation, hypertension, and dementia. On physical examination the patient had a large, well-demarcated, pink, scaly plaque with areas of ulceration extending from the dorsal aspect of the hand and fingers to the mid upper arm. There was minimal overlying yellow-brown crust (Figure 1). A potassium hydroxide preparation from a superficial scraping was negative. A punch biopsy specimen was obtained from the lesion and microscopic examination revealed histiocytes with innumerable intracytoplasmic yeast forms demonstrating small buds (Figure 2). The organisms were highlighted by periodic acid-Schiff and Grocott-Gomori methenamine-silver stains (Figure 3), while acid-fast bacillus and Fite stains were negative. The presumptive diagnosis of cutaneous cryptococcosis was made, and subsequent culture and latex agglutination test was positive for Cryptococcus neoformans. A chest radiograph showed no evidence of active disease. Infectious disease specialists were consulted and ordered additional laboratory studies, which were negative for human immunodeficiency virus, hepatitis, and fungemia. The patient had a low CD4 count of 119 cells/μL (reference range, 496-2186 cells/μL). Workup for systemic Cryptococcus, including head computed tomography, cerebral spinal fluid analysis, and bone marrow biopsy were all negative. Epstein-Barr virus and human T lymphotropic virus tests were both negative. The source of the patient's low CD4 count was never discovered. She gradually began to improve with diligent wound care and continued fluconazole 400 mg daily. The patient's history did reveal working on a chicken farm as an adult many years ago.
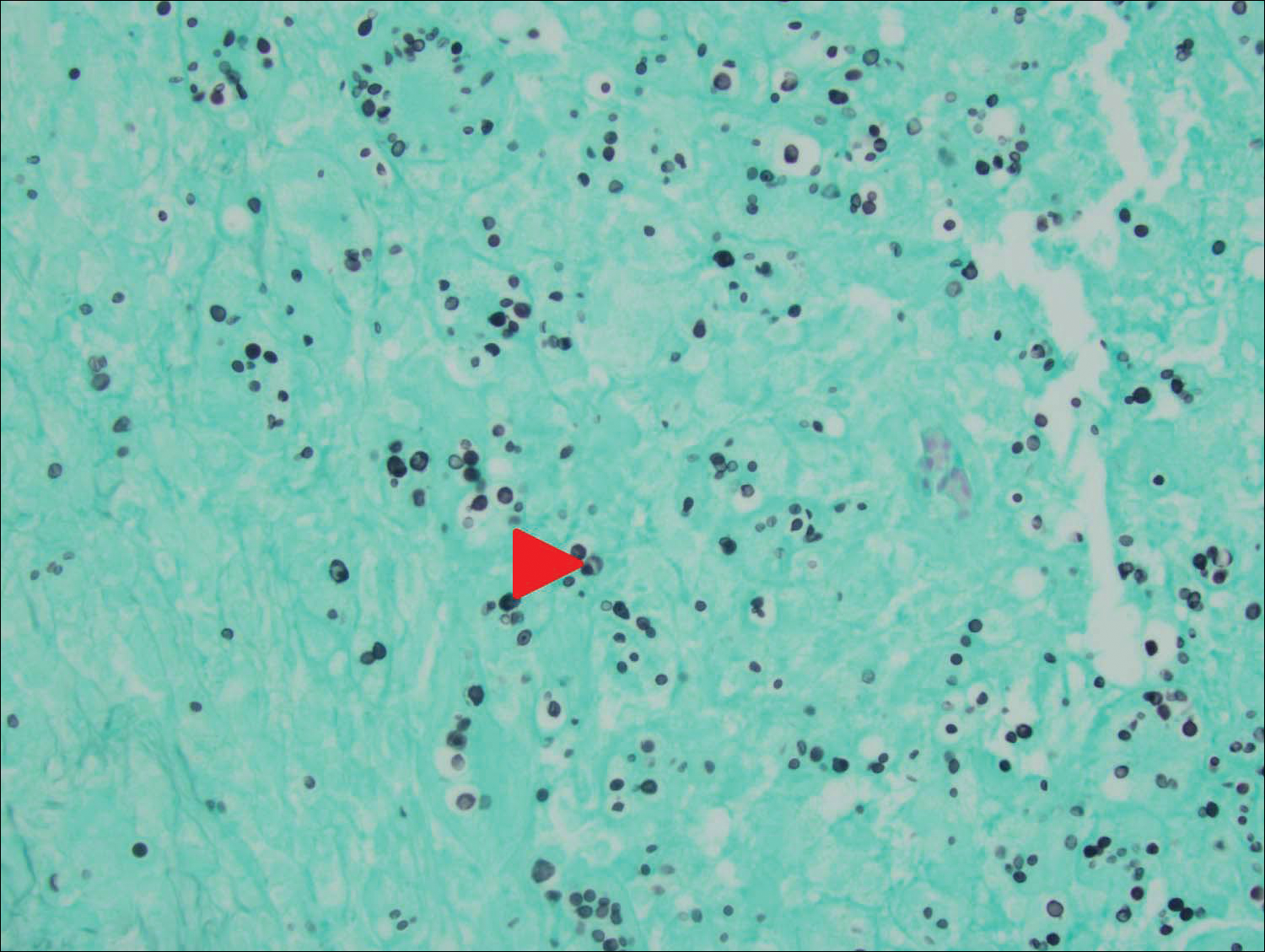
Cryptococcus is a yeast that causes infection primarily through airborne spores that lead to pulmonary infection. Cryptococcus neoformans is the most common pathogenic strain, though infection with other strains such as Cryptococcus albidus1 and Cryptococcus laurentii2 have been reported. Primary cutaneous cryptococcosis is an exceedingly rare entity, with the majority of cases of cutaneous cryptococcosis originating from primary pulmonary infection with hematogenous dissemination to the skin. Primary cutaneous cryptococcosis rarely can be caused by inoculation in nonimmunosuppressed hosts and infection of nonimmunosuppressed hosts is more common in men than in women.3 Manifestations of cutaneous cryptococcosis can be incredibly varied and diagnosis requires a high index of suspicion along with appropriate histological and serological confirmation. Cutaneous cryptococcosis can present in various clinical ways, including molluscumlike lesions, which are more common in patients with AIDS; acneform lesions; vesicles; dermal plaques or nodules; and rarely cellulitis with ulcerations, as in our patient. Cryptococcosis also can imitate basal cell carcinoma, nummular and follicular eczema, and Kaposi sarcoma.4
Histologic examination reveals either a gelatinous or granulomatous pattern based on the number of organisms present. The gelatinous pattern is characterized by little inflammation and a large number of phagocytosed organisms floating in mucin. The granulomatous pattern shows prominent inflammation with lymphocytes, histiocytes, and giant cells, as well as associated necrosis.
Treatment depends on the type of infection and host immunological status. Immunocompetent hosts with cutaneous infection may spontaneously heal. Treatment consists of surgical excision, if possible, followed by fluconazole or itraconazole. For disseminated cryptococcal infections in immunosuppressed hosts, the standard of care is amphotericin B with or without flucytosine.3
- Hoang JK, Burruss J. Localized cutaneous Cryptococcus albidus infection in a 14-year-old boy on etanercept therapy [published online June 5, 2007]. Pediatr Dermatol. 2007;24:285-288. doi:10.1111/j.1525-1470.2007.00404.x.
- Vlchkova-Lashkoska M, Kamberova S, Starova A, et al. Cutaneous Cryptococcus laurentii infection in a human immunodeficiency virus-negative subject. J Eur Acad Dermatol Venereol. 2004;18:99-100.
- Antony SA, Antony SJ. Primary cutaneous Cryptococcus in nonimmunocompromised patients. Cutis. 1995;56:96-98.
- Murakawa GJ, Kerschmann R, Berger T. Cutaneous Cryptococcus infection and AIDS. report of 12 cases and review of the literature. Arch Dermatol. 1996;132:545-548.
To the Editor:
Primary cutaneous cryptococcal infection is rare. Cryptococcal skin infections, either primary or disseminated, can be highly pleomorphic and mimic entities such as basal cell carcinoma or even severe dermatitis, as in our case.
An 80-year-old woman who was residing in a nursing facility presented to the emergency department with an itchy nontender rash on the left arm of 2 to 3 weeks' duration that gradually spread. The patient had not started any new topical or oral medications and was otherwise healthy. A review of symptoms was negative for fever, weight loss, or new cough. Her medical history was notable for congestive heart failure, chronic obstructive pulmonary disease requiring chronic low-dose prednisone, hypothyroidism, atrial fibrillation, hypertension, and dementia. On physical examination the patient had a large, well-demarcated, pink, scaly plaque with areas of ulceration extending from the dorsal aspect of the hand and fingers to the mid upper arm. There was minimal overlying yellow-brown crust (Figure 1). A potassium hydroxide preparation from a superficial scraping was negative. A punch biopsy specimen was obtained from the lesion and microscopic examination revealed histiocytes with innumerable intracytoplasmic yeast forms demonstrating small buds (Figure 2). The organisms were highlighted by periodic acid-Schiff and Grocott-Gomori methenamine-silver stains (Figure 3), while acid-fast bacillus and Fite stains were negative. The presumptive diagnosis of cutaneous cryptococcosis was made, and subsequent culture and latex agglutination test was positive for Cryptococcus neoformans. A chest radiograph showed no evidence of active disease. Infectious disease specialists were consulted and ordered additional laboratory studies, which were negative for human immunodeficiency virus, hepatitis, and fungemia. The patient had a low CD4 count of 119 cells/μL (reference range, 496-2186 cells/μL). Workup for systemic Cryptococcus, including head computed tomography, cerebral spinal fluid analysis, and bone marrow biopsy were all negative. Epstein-Barr virus and human T lymphotropic virus tests were both negative. The source of the patient's low CD4 count was never discovered. She gradually began to improve with diligent wound care and continued fluconazole 400 mg daily. The patient's history did reveal working on a chicken farm as an adult many years ago.
Cryptococcus is a yeast that causes infection primarily through airborne spores that lead to pulmonary infection. Cryptococcus neoformans is the most common pathogenic strain, though infection with other strains such as Cryptococcus albidus1 and Cryptococcus laurentii2 have been reported. Primary cutaneous cryptococcosis is an exceedingly rare entity, with the majority of cases of cutaneous cryptococcosis originating from primary pulmonary infection with hematogenous dissemination to the skin. Primary cutaneous cryptococcosis rarely can be caused by inoculation in nonimmunosuppressed hosts and infection of nonimmunosuppressed hosts is more common in men than in women.3 Manifestations of cutaneous cryptococcosis can be incredibly varied and diagnosis requires a high index of suspicion along with appropriate histological and serological confirmation. Cutaneous cryptococcosis can present in various clinical ways, including molluscumlike lesions, which are more common in patients with AIDS; acneform lesions; vesicles; dermal plaques or nodules; and rarely cellulitis with ulcerations, as in our patient. Cryptococcosis also can imitate basal cell carcinoma, nummular and follicular eczema, and Kaposi sarcoma.4
Histologic examination reveals either a gelatinous or granulomatous pattern based on the number of organisms present. The gelatinous pattern is characterized by little inflammation and a large number of phagocytosed organisms floating in mucin. The granulomatous pattern shows prominent inflammation with lymphocytes, histiocytes, and giant cells, as well as associated necrosis.
Treatment depends on the type of infection and host immunological status. Immunocompetent hosts with cutaneous infection may spontaneously heal. Treatment consists of surgical excision, if possible, followed by fluconazole or itraconazole. For disseminated cryptococcal infections in immunosuppressed hosts, the standard of care is amphotericin B with or without flucytosine.3
To the Editor:
Primary cutaneous cryptococcal infection is rare. Cryptococcal skin infections, either primary or disseminated, can be highly pleomorphic and mimic entities such as basal cell carcinoma or even severe dermatitis, as in our case.
An 80-year-old woman who was residing in a nursing facility presented to the emergency department with an itchy nontender rash on the left arm of 2 to 3 weeks' duration that gradually spread. The patient had not started any new topical or oral medications and was otherwise healthy. A review of symptoms was negative for fever, weight loss, or new cough. Her medical history was notable for congestive heart failure, chronic obstructive pulmonary disease requiring chronic low-dose prednisone, hypothyroidism, atrial fibrillation, hypertension, and dementia. On physical examination the patient had a large, well-demarcated, pink, scaly plaque with areas of ulceration extending from the dorsal aspect of the hand and fingers to the mid upper arm. There was minimal overlying yellow-brown crust (Figure 1). A potassium hydroxide preparation from a superficial scraping was negative. A punch biopsy specimen was obtained from the lesion and microscopic examination revealed histiocytes with innumerable intracytoplasmic yeast forms demonstrating small buds (Figure 2). The organisms were highlighted by periodic acid-Schiff and Grocott-Gomori methenamine-silver stains (Figure 3), while acid-fast bacillus and Fite stains were negative. The presumptive diagnosis of cutaneous cryptococcosis was made, and subsequent culture and latex agglutination test was positive for Cryptococcus neoformans. A chest radiograph showed no evidence of active disease. Infectious disease specialists were consulted and ordered additional laboratory studies, which were negative for human immunodeficiency virus, hepatitis, and fungemia. The patient had a low CD4 count of 119 cells/μL (reference range, 496-2186 cells/μL). Workup for systemic Cryptococcus, including head computed tomography, cerebral spinal fluid analysis, and bone marrow biopsy were all negative. Epstein-Barr virus and human T lymphotropic virus tests were both negative. The source of the patient's low CD4 count was never discovered. She gradually began to improve with diligent wound care and continued fluconazole 400 mg daily. The patient's history did reveal working on a chicken farm as an adult many years ago.
Cryptococcus is a yeast that causes infection primarily through airborne spores that lead to pulmonary infection. Cryptococcus neoformans is the most common pathogenic strain, though infection with other strains such as Cryptococcus albidus1 and Cryptococcus laurentii2 have been reported. Primary cutaneous cryptococcosis is an exceedingly rare entity, with the majority of cases of cutaneous cryptococcosis originating from primary pulmonary infection with hematogenous dissemination to the skin. Primary cutaneous cryptococcosis rarely can be caused by inoculation in nonimmunosuppressed hosts and infection of nonimmunosuppressed hosts is more common in men than in women.3 Manifestations of cutaneous cryptococcosis can be incredibly varied and diagnosis requires a high index of suspicion along with appropriate histological and serological confirmation. Cutaneous cryptococcosis can present in various clinical ways, including molluscumlike lesions, which are more common in patients with AIDS; acneform lesions; vesicles; dermal plaques or nodules; and rarely cellulitis with ulcerations, as in our patient. Cryptococcosis also can imitate basal cell carcinoma, nummular and follicular eczema, and Kaposi sarcoma.4
Histologic examination reveals either a gelatinous or granulomatous pattern based on the number of organisms present. The gelatinous pattern is characterized by little inflammation and a large number of phagocytosed organisms floating in mucin. The granulomatous pattern shows prominent inflammation with lymphocytes, histiocytes, and giant cells, as well as associated necrosis.
Treatment depends on the type of infection and host immunological status. Immunocompetent hosts with cutaneous infection may spontaneously heal. Treatment consists of surgical excision, if possible, followed by fluconazole or itraconazole. For disseminated cryptococcal infections in immunosuppressed hosts, the standard of care is amphotericin B with or without flucytosine.3
- Hoang JK, Burruss J. Localized cutaneous Cryptococcus albidus infection in a 14-year-old boy on etanercept therapy [published online June 5, 2007]. Pediatr Dermatol. 2007;24:285-288. doi:10.1111/j.1525-1470.2007.00404.x.
- Vlchkova-Lashkoska M, Kamberova S, Starova A, et al. Cutaneous Cryptococcus laurentii infection in a human immunodeficiency virus-negative subject. J Eur Acad Dermatol Venereol. 2004;18:99-100.
- Antony SA, Antony SJ. Primary cutaneous Cryptococcus in nonimmunocompromised patients. Cutis. 1995;56:96-98.
- Murakawa GJ, Kerschmann R, Berger T. Cutaneous Cryptococcus infection and AIDS. report of 12 cases and review of the literature. Arch Dermatol. 1996;132:545-548.
- Hoang JK, Burruss J. Localized cutaneous Cryptococcus albidus infection in a 14-year-old boy on etanercept therapy [published online June 5, 2007]. Pediatr Dermatol. 2007;24:285-288. doi:10.1111/j.1525-1470.2007.00404.x.
- Vlchkova-Lashkoska M, Kamberova S, Starova A, et al. Cutaneous Cryptococcus laurentii infection in a human immunodeficiency virus-negative subject. J Eur Acad Dermatol Venereol. 2004;18:99-100.
- Antony SA, Antony SJ. Primary cutaneous Cryptococcus in nonimmunocompromised patients. Cutis. 1995;56:96-98.
- Murakawa GJ, Kerschmann R, Berger T. Cutaneous Cryptococcus infection and AIDS. report of 12 cases and review of the literature. Arch Dermatol. 1996;132:545-548.
Practice Points
- Primary cutaneous cryptococcosis is rare in nonimmunosuppressed patients.
- Primary cutaneous cryptococcosis secondary to inoculation can have a clinical presentation similar to more common conditions, such as molluscum, acne, and dermatitis.
Postoperative Henoch-Schönlein Purpura
To the Editor:
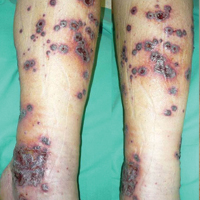
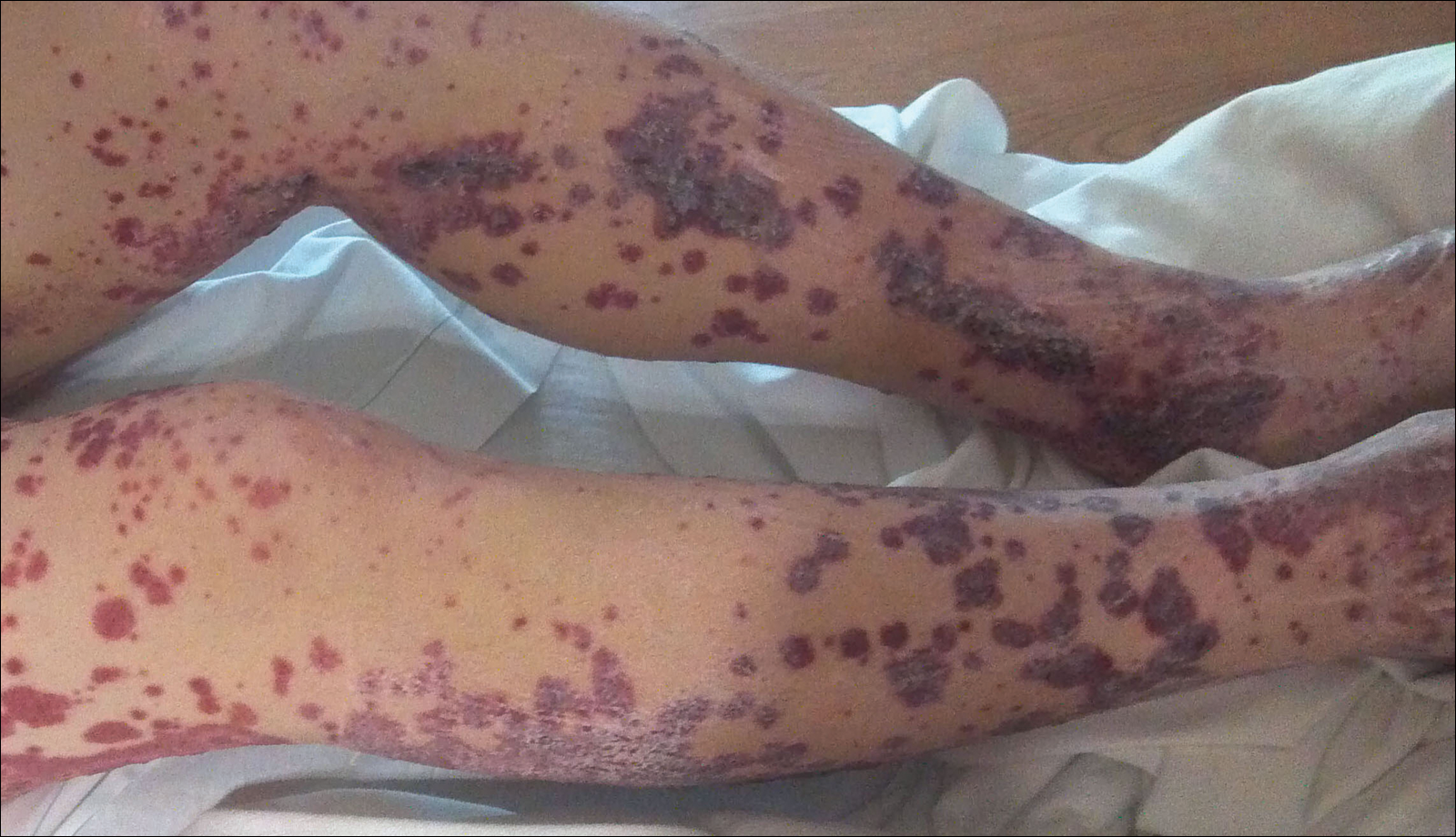
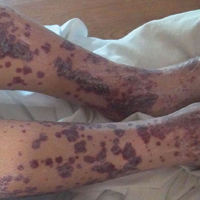
A 57-year-old man with a history of type 2 diabetes mellitus and hypertension was hospitalized for heart disease resulting in an aortic valve replacement and multiple-vessel bypass grafting. He experienced a stormy septic postoperative course during which he developed numerous palpable purplish plaques (Figure 1). The lesions were bilateral and more heavily involved the lower legs and buttocks. The head and neck remained free of skin lesions. Additionally, the patient reported a bilateral burning sensation from the knees to the feet.
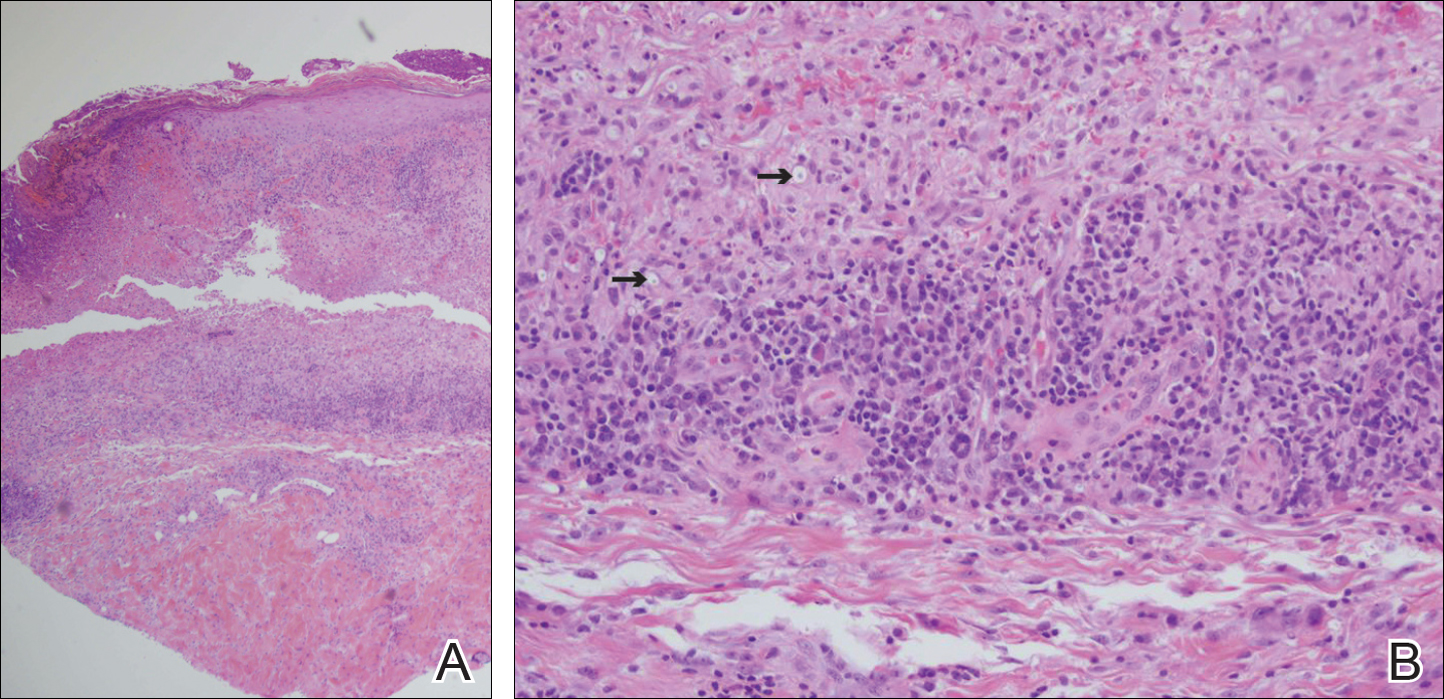
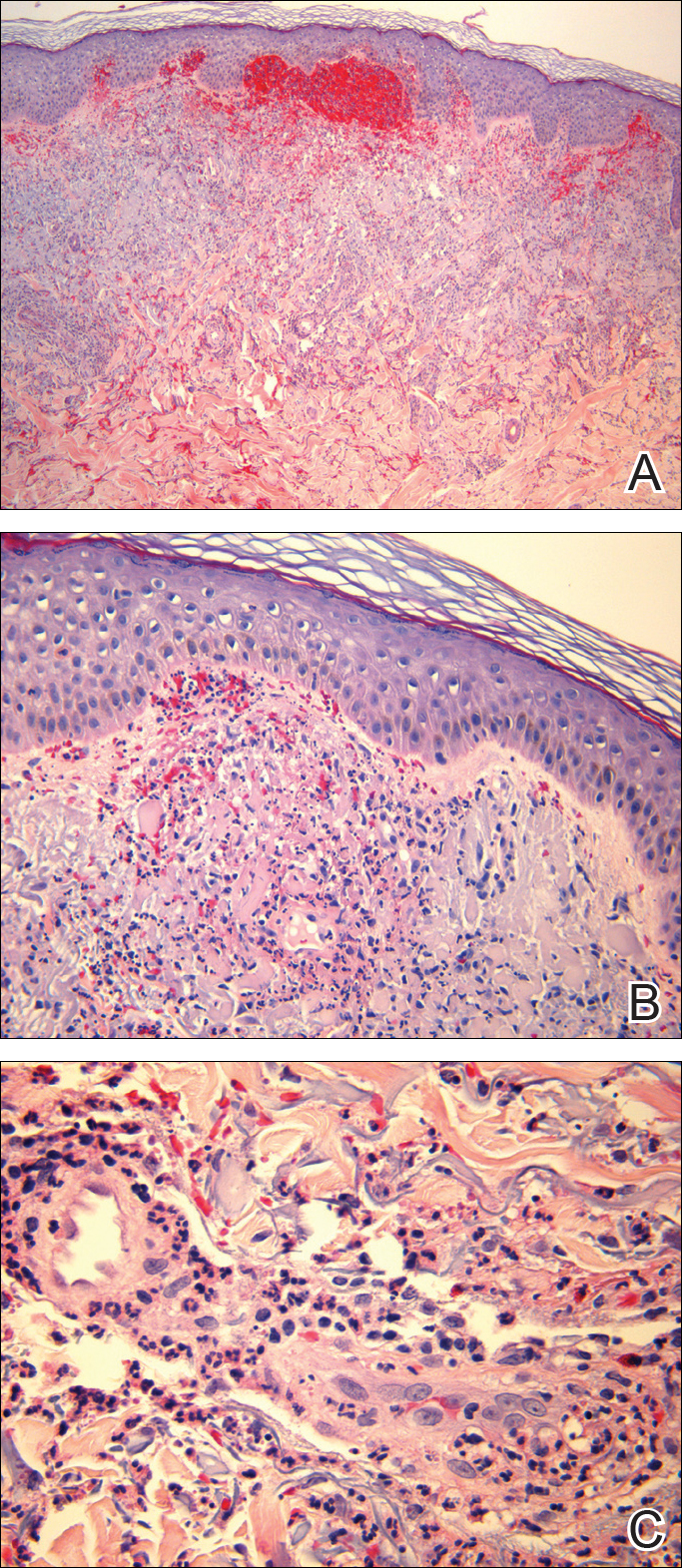
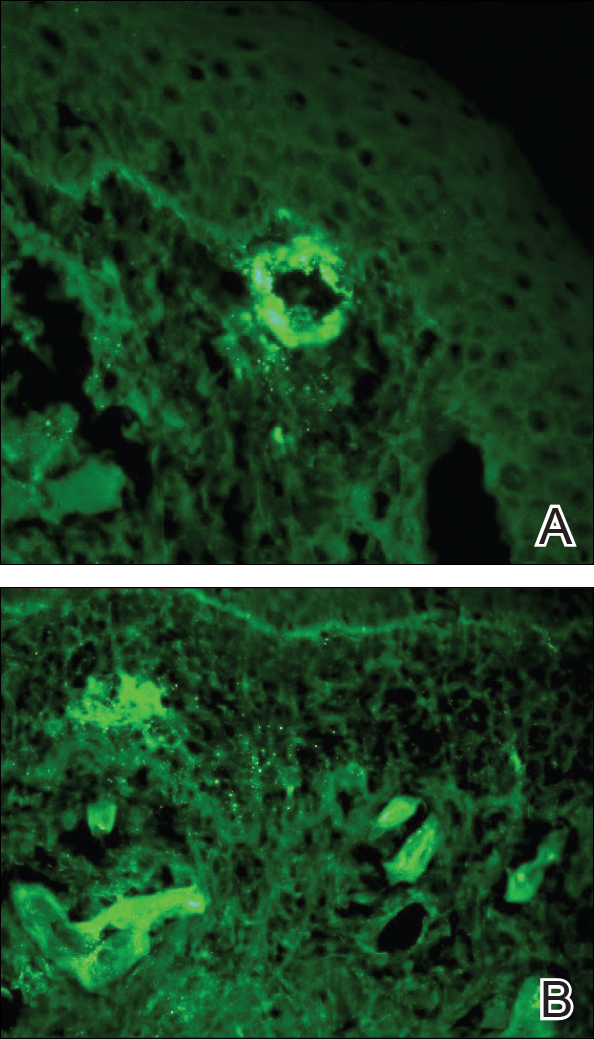
Punch biopsies of lesions from the right upper arm were obtained. Hematoxylin and eosin staining revealed neutrophilic-predominant small vessel vasculitis (Figure 2A) with the upper dermal location more heavily involved, as demonstrated by involvement of a superficial vascular plexus (Figures 2B and 2C) that was consistent with Henoch-Schönlein purpura (HSP). The diagnosis later was confirmed with immunofluorescence. Direct immunofluorescence revealed granular IgA deposition around the superficial vascular plexus (Figure 3). No IgG, IgM, C3, C5b-9 complement complex, or fibrinogen deposition was seen. Additionally, periodic acid-Schiff staining failed to show microorganisms, thrombi, or intravascular hyaline material.
At our initial consultation, we observed an ill-appearing afebrile man with purplish plaques. Our impression was that he had vasculitis and not warfarin necrosis, which had been suspected by the cardiovascular team. The burning sensation noted by the patient lent credence to our vasculitic diagnosis. Proteinuria and hematuria were present; however, the values for blood urea nitrogen, creatinine, and glomerular filtration rate all remained within reference range. His signs and symptoms responded dramatically to prednisone. He remains on 1 mg of prednisone daily and a nephrologist continues to monitor renal function as an outpatient.
Henoch-Schönlein purpura is a systemic leukocytoclastic vasculitis involving small vessels. The small vessel vasculitis is associated with IgA antigen-antibody complex deposition in areas throughout the body. Palpable purpura typically is seen on the skin, which characteristically involves dependent areas such as the legs and the buttocks. Lesions normally are present bilaterally in a symmetric distribution. Initially, the lesions develop as erythematous macules that progress to purple, nonblanching, palpable, and purpuric plaques.1 Henoch-Schönlein purpura most commonly involves the skin; however, other locations for the immune complexes include the gastrointestinal tract, joints, and kidneys.2 The cause for the body's immunogenic deposition response is unknown in a majority of cases.
Henoch-Schönlein purpura most commonly is seen in the pediatric population with a predilection for males.3 The incidence in the pediatric population is 13.5 to 20 per 100,000 children per year; HSP is more rare in adults.4-6 Henoch-Schönlein purpura most often is a self-limiting disease that requires only supportive treatment. The signs and symptoms last 4 to 6 weeks in most patients and resolve completely in 94% of children and 89% of adults.7 Renal involvement carries a worse prognosis. Adult patients have a higher incidence of renal involvement, renal insufficiency, and subsequent progression to end-stage renal disease.3,8-10 In a study by Hung et al8 of 65 children and 22 adult HSP patients, 12 adults presented with renal involvement in which hematuria or proteinuria were present. Of them, 6 progressed to renal insufficiency (defined as having a plasma creatinine concentration>1.2 mg/dL).8 Fogazzi et al11 reported similar findings; 8 of 16 patients affected with HSP progressed to renal insufficiency with creatinine clearances ranging from 31 to 60 mL/min, and 3 patients required chronic dialysis. Pillebout et al9 evaluated 250 adults with HSP and 32% reached renal insufficiency with creatinine clearances of less than 50 mL/min, with 11% of patients developing end-stage renal disease. The degree of hematuria and/or proteinuria has been shown to be an effective prognostic indicator.9,10 Coppo et al10 found a similar prognosis among children and adults with HSP-related nephritis.
Our patient described the burning sensation as occurring bilaterally from the knees down to the feet, which provided an additional clue that small vessel vasculitis was involved, as occluded blood vessels can cause ischemia to nerves and perivascular involvement can affect nearby neural structures. Sais et al12 demonstrated that paresthesia in the setting of HSP was a risk factor for systemic involvement. Of note, our patient's paresthesia lasted only several days.
The cause of HSP is not always as evident in the adult population as in the pediatric population. Early diagnosis of HSP in adults may allow for the proper instatement of treatment to deter long-term renal complications. Follow-up with urinalysis is recommended because a small percentage of patients have a late progression to renal failure.13,14
Because the dermatologists involved in this case knew where and what types of biopsies to perform, a correct diagnosis was obtained quickly, allowing for the correct therapeutic intervention. After the diagnosis of HSP is made in an adult, nephrology should be consulted early in the treatment course.
- Rai A, Nast C, Adler S. Henoch-Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637-2644.
- Helander SD, De Castro FR, Gibson LE. Henoch-Schönlein purpura: clinicopathologic correlation of cutaneous vascular IgA deposits and the relationship to leukocytoclastic vasculitis. Acta Derm Venereol. 1995;75:125-129.
- Garcia-Porrua C, Calvino MC, Llorca J, et al. Henoch-Schönlein purpura in children and adults: clinical differences in a defined population. Semin Arthritis Rheum. 2002;32:149-156.
- Stewart M, Savage JM, Bell B, et al. Long term renal prognosis of Henoch-Schönlein purpura in an unselected childhood population. Eur J Pediatr. 1988;147:113-115.
- Watts RA, Scott DG. Epidemiology of the vasculitides. Semin Respir Crit Care. 2004;25:455-464.
- Gardner-Medwin JM, Dolezalova P, Cummins C, et al. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet. 2002;360:1197-1202.
- Blanco R, Martínez-Taboada VM, Rodríguez-Valverde V, et al. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum. 1997;40:859-864.
- Hung SP, Yang YH, Lin YT, et al. Clinical manifestations and outcomes of Henoch-Schönlein purpura: comparison between adults and children. Pediatr Neonatol. 2009;50:162-168.
- Pillebout E, Thervet E, Hill G, et al. Henoch-Schönlein purpura in adults: outcomes and prognostic factors. J Am Soc Nephrol. 2002;13:1271-1278.
- Coppo R, Mazzucco G, Cagnoli L, et al. Long-term prognosis of Henoch-Schönlein nephritis in adults and children. Italian Group of Renal Immunopathology collaborative study on Henoch-Schönlein purpura. Nephrol Dial Transplant. 1997;12:2277-2283.
- Fogazzi GB, Pasquali S, Moriggi M, et al. Long-term outcome of Schönlein-Henoch nephritis in the adult. Clin Nephrol. 1989;31:60-66.
- Sais G, Vidaller A, Jucgla A. Prognostic factors in leukocytoclastic vasculitis. a clinicopathologic study of 160 patients. Arch Dermatol. 1998;134:309-315.
- Kraft DM, McKee D, Scott C. Henoch-Schönlein purpura: a review. Am Fam Physician. 1998;58:405-408.
- Narchi H. Risk of long-term renal impairment and duration of follow up recommended for Henoch-Schönlein purpura with normal or minimal urinary findings: a systematic review. Arch Dis Child. 2005;90:916-920.
To the Editor:
A 57-year-old man with a history of type 2 diabetes mellitus and hypertension was hospitalized for heart disease resulting in an aortic valve replacement and multiple-vessel bypass grafting. He experienced a stormy septic postoperative course during which he developed numerous palpable purplish plaques (Figure 1). The lesions were bilateral and more heavily involved the lower legs and buttocks. The head and neck remained free of skin lesions. Additionally, the patient reported a bilateral burning sensation from the knees to the feet.
Punch biopsies of lesions from the right upper arm were obtained. Hematoxylin and eosin staining revealed neutrophilic-predominant small vessel vasculitis (Figure 2A) with the upper dermal location more heavily involved, as demonstrated by involvement of a superficial vascular plexus (Figures 2B and 2C) that was consistent with Henoch-Schönlein purpura (HSP). The diagnosis later was confirmed with immunofluorescence. Direct immunofluorescence revealed granular IgA deposition around the superficial vascular plexus (Figure 3). No IgG, IgM, C3, C5b-9 complement complex, or fibrinogen deposition was seen. Additionally, periodic acid-Schiff staining failed to show microorganisms, thrombi, or intravascular hyaline material.
At our initial consultation, we observed an ill-appearing afebrile man with purplish plaques. Our impression was that he had vasculitis and not warfarin necrosis, which had been suspected by the cardiovascular team. The burning sensation noted by the patient lent credence to our vasculitic diagnosis. Proteinuria and hematuria were present; however, the values for blood urea nitrogen, creatinine, and glomerular filtration rate all remained within reference range. His signs and symptoms responded dramatically to prednisone. He remains on 1 mg of prednisone daily and a nephrologist continues to monitor renal function as an outpatient.
Henoch-Schönlein purpura is a systemic leukocytoclastic vasculitis involving small vessels. The small vessel vasculitis is associated with IgA antigen-antibody complex deposition in areas throughout the body. Palpable purpura typically is seen on the skin, which characteristically involves dependent areas such as the legs and the buttocks. Lesions normally are present bilaterally in a symmetric distribution. Initially, the lesions develop as erythematous macules that progress to purple, nonblanching, palpable, and purpuric plaques.1 Henoch-Schönlein purpura most commonly involves the skin; however, other locations for the immune complexes include the gastrointestinal tract, joints, and kidneys.2 The cause for the body's immunogenic deposition response is unknown in a majority of cases.
Henoch-Schönlein purpura most commonly is seen in the pediatric population with a predilection for males.3 The incidence in the pediatric population is 13.5 to 20 per 100,000 children per year; HSP is more rare in adults.4-6 Henoch-Schönlein purpura most often is a self-limiting disease that requires only supportive treatment. The signs and symptoms last 4 to 6 weeks in most patients and resolve completely in 94% of children and 89% of adults.7 Renal involvement carries a worse prognosis. Adult patients have a higher incidence of renal involvement, renal insufficiency, and subsequent progression to end-stage renal disease.3,8-10 In a study by Hung et al8 of 65 children and 22 adult HSP patients, 12 adults presented with renal involvement in which hematuria or proteinuria were present. Of them, 6 progressed to renal insufficiency (defined as having a plasma creatinine concentration>1.2 mg/dL).8 Fogazzi et al11 reported similar findings; 8 of 16 patients affected with HSP progressed to renal insufficiency with creatinine clearances ranging from 31 to 60 mL/min, and 3 patients required chronic dialysis. Pillebout et al9 evaluated 250 adults with HSP and 32% reached renal insufficiency with creatinine clearances of less than 50 mL/min, with 11% of patients developing end-stage renal disease. The degree of hematuria and/or proteinuria has been shown to be an effective prognostic indicator.9,10 Coppo et al10 found a similar prognosis among children and adults with HSP-related nephritis.
Our patient described the burning sensation as occurring bilaterally from the knees down to the feet, which provided an additional clue that small vessel vasculitis was involved, as occluded blood vessels can cause ischemia to nerves and perivascular involvement can affect nearby neural structures. Sais et al12 demonstrated that paresthesia in the setting of HSP was a risk factor for systemic involvement. Of note, our patient's paresthesia lasted only several days.
The cause of HSP is not always as evident in the adult population as in the pediatric population. Early diagnosis of HSP in adults may allow for the proper instatement of treatment to deter long-term renal complications. Follow-up with urinalysis is recommended because a small percentage of patients have a late progression to renal failure.13,14
Because the dermatologists involved in this case knew where and what types of biopsies to perform, a correct diagnosis was obtained quickly, allowing for the correct therapeutic intervention. After the diagnosis of HSP is made in an adult, nephrology should be consulted early in the treatment course.
To the Editor:
A 57-year-old man with a history of type 2 diabetes mellitus and hypertension was hospitalized for heart disease resulting in an aortic valve replacement and multiple-vessel bypass grafting. He experienced a stormy septic postoperative course during which he developed numerous palpable purplish plaques (Figure 1). The lesions were bilateral and more heavily involved the lower legs and buttocks. The head and neck remained free of skin lesions. Additionally, the patient reported a bilateral burning sensation from the knees to the feet.
Punch biopsies of lesions from the right upper arm were obtained. Hematoxylin and eosin staining revealed neutrophilic-predominant small vessel vasculitis (Figure 2A) with the upper dermal location more heavily involved, as demonstrated by involvement of a superficial vascular plexus (Figures 2B and 2C) that was consistent with Henoch-Schönlein purpura (HSP). The diagnosis later was confirmed with immunofluorescence. Direct immunofluorescence revealed granular IgA deposition around the superficial vascular plexus (Figure 3). No IgG, IgM, C3, C5b-9 complement complex, or fibrinogen deposition was seen. Additionally, periodic acid-Schiff staining failed to show microorganisms, thrombi, or intravascular hyaline material.
At our initial consultation, we observed an ill-appearing afebrile man with purplish plaques. Our impression was that he had vasculitis and not warfarin necrosis, which had been suspected by the cardiovascular team. The burning sensation noted by the patient lent credence to our vasculitic diagnosis. Proteinuria and hematuria were present; however, the values for blood urea nitrogen, creatinine, and glomerular filtration rate all remained within reference range. His signs and symptoms responded dramatically to prednisone. He remains on 1 mg of prednisone daily and a nephrologist continues to monitor renal function as an outpatient.
Henoch-Schönlein purpura is a systemic leukocytoclastic vasculitis involving small vessels. The small vessel vasculitis is associated with IgA antigen-antibody complex deposition in areas throughout the body. Palpable purpura typically is seen on the skin, which characteristically involves dependent areas such as the legs and the buttocks. Lesions normally are present bilaterally in a symmetric distribution. Initially, the lesions develop as erythematous macules that progress to purple, nonblanching, palpable, and purpuric plaques.1 Henoch-Schönlein purpura most commonly involves the skin; however, other locations for the immune complexes include the gastrointestinal tract, joints, and kidneys.2 The cause for the body's immunogenic deposition response is unknown in a majority of cases.
Henoch-Schönlein purpura most commonly is seen in the pediatric population with a predilection for males.3 The incidence in the pediatric population is 13.5 to 20 per 100,000 children per year; HSP is more rare in adults.4-6 Henoch-Schönlein purpura most often is a self-limiting disease that requires only supportive treatment. The signs and symptoms last 4 to 6 weeks in most patients and resolve completely in 94% of children and 89% of adults.7 Renal involvement carries a worse prognosis. Adult patients have a higher incidence of renal involvement, renal insufficiency, and subsequent progression to end-stage renal disease.3,8-10 In a study by Hung et al8 of 65 children and 22 adult HSP patients, 12 adults presented with renal involvement in which hematuria or proteinuria were present. Of them, 6 progressed to renal insufficiency (defined as having a plasma creatinine concentration>1.2 mg/dL).8 Fogazzi et al11 reported similar findings; 8 of 16 patients affected with HSP progressed to renal insufficiency with creatinine clearances ranging from 31 to 60 mL/min, and 3 patients required chronic dialysis. Pillebout et al9 evaluated 250 adults with HSP and 32% reached renal insufficiency with creatinine clearances of less than 50 mL/min, with 11% of patients developing end-stage renal disease. The degree of hematuria and/or proteinuria has been shown to be an effective prognostic indicator.9,10 Coppo et al10 found a similar prognosis among children and adults with HSP-related nephritis.
Our patient described the burning sensation as occurring bilaterally from the knees down to the feet, which provided an additional clue that small vessel vasculitis was involved, as occluded blood vessels can cause ischemia to nerves and perivascular involvement can affect nearby neural structures. Sais et al12 demonstrated that paresthesia in the setting of HSP was a risk factor for systemic involvement. Of note, our patient's paresthesia lasted only several days.
The cause of HSP is not always as evident in the adult population as in the pediatric population. Early diagnosis of HSP in adults may allow for the proper instatement of treatment to deter long-term renal complications. Follow-up with urinalysis is recommended because a small percentage of patients have a late progression to renal failure.13,14
Because the dermatologists involved in this case knew where and what types of biopsies to perform, a correct diagnosis was obtained quickly, allowing for the correct therapeutic intervention. After the diagnosis of HSP is made in an adult, nephrology should be consulted early in the treatment course.
- Rai A, Nast C, Adler S. Henoch-Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637-2644.
- Helander SD, De Castro FR, Gibson LE. Henoch-Schönlein purpura: clinicopathologic correlation of cutaneous vascular IgA deposits and the relationship to leukocytoclastic vasculitis. Acta Derm Venereol. 1995;75:125-129.
- Garcia-Porrua C, Calvino MC, Llorca J, et al. Henoch-Schönlein purpura in children and adults: clinical differences in a defined population. Semin Arthritis Rheum. 2002;32:149-156.
- Stewart M, Savage JM, Bell B, et al. Long term renal prognosis of Henoch-Schönlein purpura in an unselected childhood population. Eur J Pediatr. 1988;147:113-115.
- Watts RA, Scott DG. Epidemiology of the vasculitides. Semin Respir Crit Care. 2004;25:455-464.
- Gardner-Medwin JM, Dolezalova P, Cummins C, et al. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet. 2002;360:1197-1202.
- Blanco R, Martínez-Taboada VM, Rodríguez-Valverde V, et al. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum. 1997;40:859-864.
- Hung SP, Yang YH, Lin YT, et al. Clinical manifestations and outcomes of Henoch-Schönlein purpura: comparison between adults and children. Pediatr Neonatol. 2009;50:162-168.
- Pillebout E, Thervet E, Hill G, et al. Henoch-Schönlein purpura in adults: outcomes and prognostic factors. J Am Soc Nephrol. 2002;13:1271-1278.
- Coppo R, Mazzucco G, Cagnoli L, et al. Long-term prognosis of Henoch-Schönlein nephritis in adults and children. Italian Group of Renal Immunopathology collaborative study on Henoch-Schönlein purpura. Nephrol Dial Transplant. 1997;12:2277-2283.
- Fogazzi GB, Pasquali S, Moriggi M, et al. Long-term outcome of Schönlein-Henoch nephritis in the adult. Clin Nephrol. 1989;31:60-66.
- Sais G, Vidaller A, Jucgla A. Prognostic factors in leukocytoclastic vasculitis. a clinicopathologic study of 160 patients. Arch Dermatol. 1998;134:309-315.
- Kraft DM, McKee D, Scott C. Henoch-Schönlein purpura: a review. Am Fam Physician. 1998;58:405-408.
- Narchi H. Risk of long-term renal impairment and duration of follow up recommended for Henoch-Schönlein purpura with normal or minimal urinary findings: a systematic review. Arch Dis Child. 2005;90:916-920.
- Rai A, Nast C, Adler S. Henoch-Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637-2644.
- Helander SD, De Castro FR, Gibson LE. Henoch-Schönlein purpura: clinicopathologic correlation of cutaneous vascular IgA deposits and the relationship to leukocytoclastic vasculitis. Acta Derm Venereol. 1995;75:125-129.
- Garcia-Porrua C, Calvino MC, Llorca J, et al. Henoch-Schönlein purpura in children and adults: clinical differences in a defined population. Semin Arthritis Rheum. 2002;32:149-156.
- Stewart M, Savage JM, Bell B, et al. Long term renal prognosis of Henoch-Schönlein purpura in an unselected childhood population. Eur J Pediatr. 1988;147:113-115.
- Watts RA, Scott DG. Epidemiology of the vasculitides. Semin Respir Crit Care. 2004;25:455-464.
- Gardner-Medwin JM, Dolezalova P, Cummins C, et al. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet. 2002;360:1197-1202.
- Blanco R, Martínez-Taboada VM, Rodríguez-Valverde V, et al. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum. 1997;40:859-864.
- Hung SP, Yang YH, Lin YT, et al. Clinical manifestations and outcomes of Henoch-Schönlein purpura: comparison between adults and children. Pediatr Neonatol. 2009;50:162-168.
- Pillebout E, Thervet E, Hill G, et al. Henoch-Schönlein purpura in adults: outcomes and prognostic factors. J Am Soc Nephrol. 2002;13:1271-1278.
- Coppo R, Mazzucco G, Cagnoli L, et al. Long-term prognosis of Henoch-Schönlein nephritis in adults and children. Italian Group of Renal Immunopathology collaborative study on Henoch-Schönlein purpura. Nephrol Dial Transplant. 1997;12:2277-2283.
- Fogazzi GB, Pasquali S, Moriggi M, et al. Long-term outcome of Schönlein-Henoch nephritis in the adult. Clin Nephrol. 1989;31:60-66.
- Sais G, Vidaller A, Jucgla A. Prognostic factors in leukocytoclastic vasculitis. a clinicopathologic study of 160 patients. Arch Dermatol. 1998;134:309-315.
- Kraft DM, McKee D, Scott C. Henoch-Schönlein purpura: a review. Am Fam Physician. 1998;58:405-408.
- Narchi H. Risk of long-term renal impairment and duration of follow up recommended for Henoch-Schönlein purpura with normal or minimal urinary findings: a systematic review. Arch Dis Child. 2005;90:916-920.
Practice Points
- Henoch-Schönlein purpura is a multidisciplinary problem.
- Henoch-Schönlein purpura is an IgA-mediated disorder that is more common in children and has a more severe course in adults.
Phacomatosis Cesioflammea in Association With von Recklinghausen Disease (Neurofibromatosis Type I)
To the Editor:
Vascular lesions associated with melanocytic nevi were first described by Ota et al1 in 1947 and given the name phacomatosis pigmentovascularis. In 2005, Happle2 reclassified phacomatosis pigmentovascularis into 3 well-defined types: (1) phacomatosis cesioflammea: blue spots (caesius means bluish gray in Latin) and nevus flammeus; (2) phacomatosis spilorosea: nevus spilus coexisting with a pale pink telangiectatic nevus; and (3) phacomatosis cesiomarmorata: blue spots and cutis marmorata telangiectatica congenita. In 2011 Joshi et al3 described a case of a 31-year-old woman who had a port-wine stain in association with neurofibromatosis type I (NF-1). We present a case of phacomatosis cesioflammea in association with NF-1.
A 20-year-old woman presented to our outpatient section with a bluish black birthmark on the left side of the face since birth with the onset of multiple painless flesh-colored nodules on the trunk and arms of 1 year’s duration. She reported having occasional pruritus over the nodular lesions. Cutaneous examination showed multiple well-defined café au lait macules (0.5–3.0 cm) with regular margins. Multiple flesh-colored nodules were evident on the upper arms (Figure 1) and trunk. The nodules were firm in consistency and showed buttonholing phenomenon with some of the lesions demonstrating bag-of-worms consistency on palpation. Both palms showed multiple brownish frecklelike macules (Figure 2). A single bluish patch extended from the left ala of the nose to the sideburns. Adjoining the bluish patch was a subtle, ill-defined, nonblanchable red patch extending from the lower margin of the bluish patch to the mandibular ridge (Figure 3). Ocular examination showed melanosis bulbi of the left sclera and a few iris hamartomas (Lisch nodules) in both eyes. A biopsy of the skin nodule was obtained under local anesthesia after obtaining the patient’s informed consent; the specimen was fixed in 10% buffered formalin. A hematoxylin and eosin–stained section showed a well-circumscribed nonencapsulated tumor in the dermis composed of loosely spaced spindle cells and wavy collagenous strands (Figure 4). Routine hemogram and blood biochemistry including urinalysis were within reference range. Radiologic examination of the long bones was unremarkable. Our patient had 3 of 6 criteria defined by the National Institutes of Health for diagnosis of NF-1.4 On clinicopathological correlation we made a diagnosis of phacomatosis cesioflammea in association with NF-1. We have reassured the patient about the benign nature of vascular nevus. She was informed that the skin nodules could increase in size during pregnancy and to regularly follow-up with an eye specialist if any visual abnormalities occur.
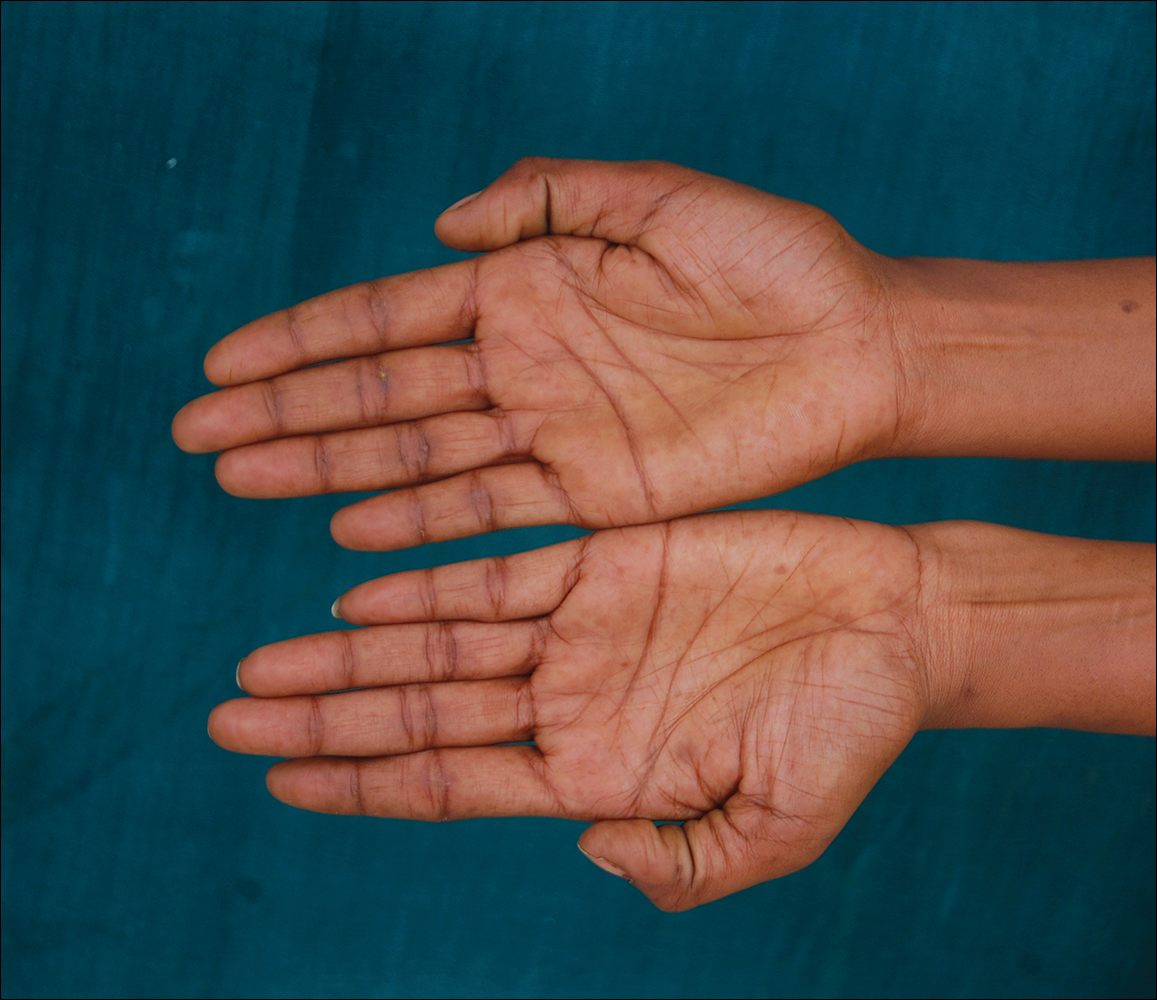
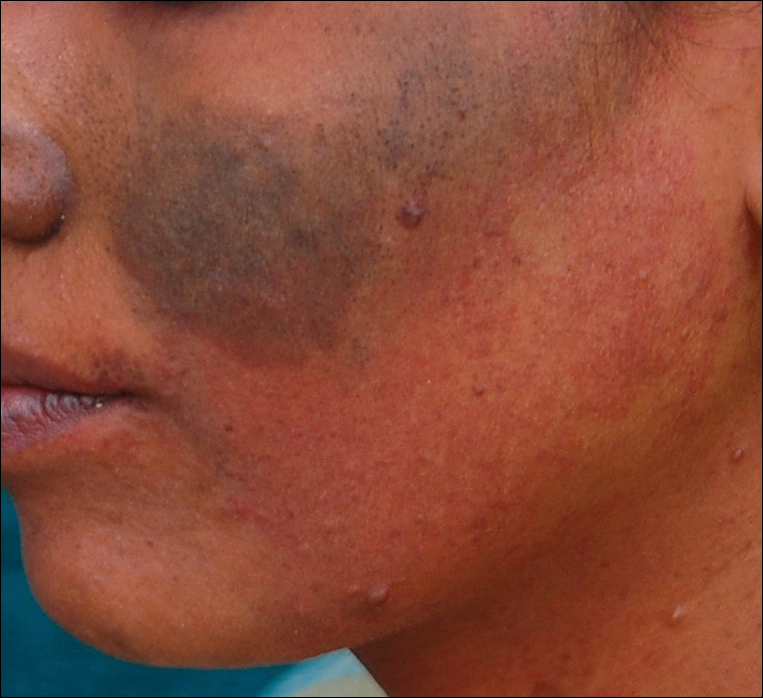
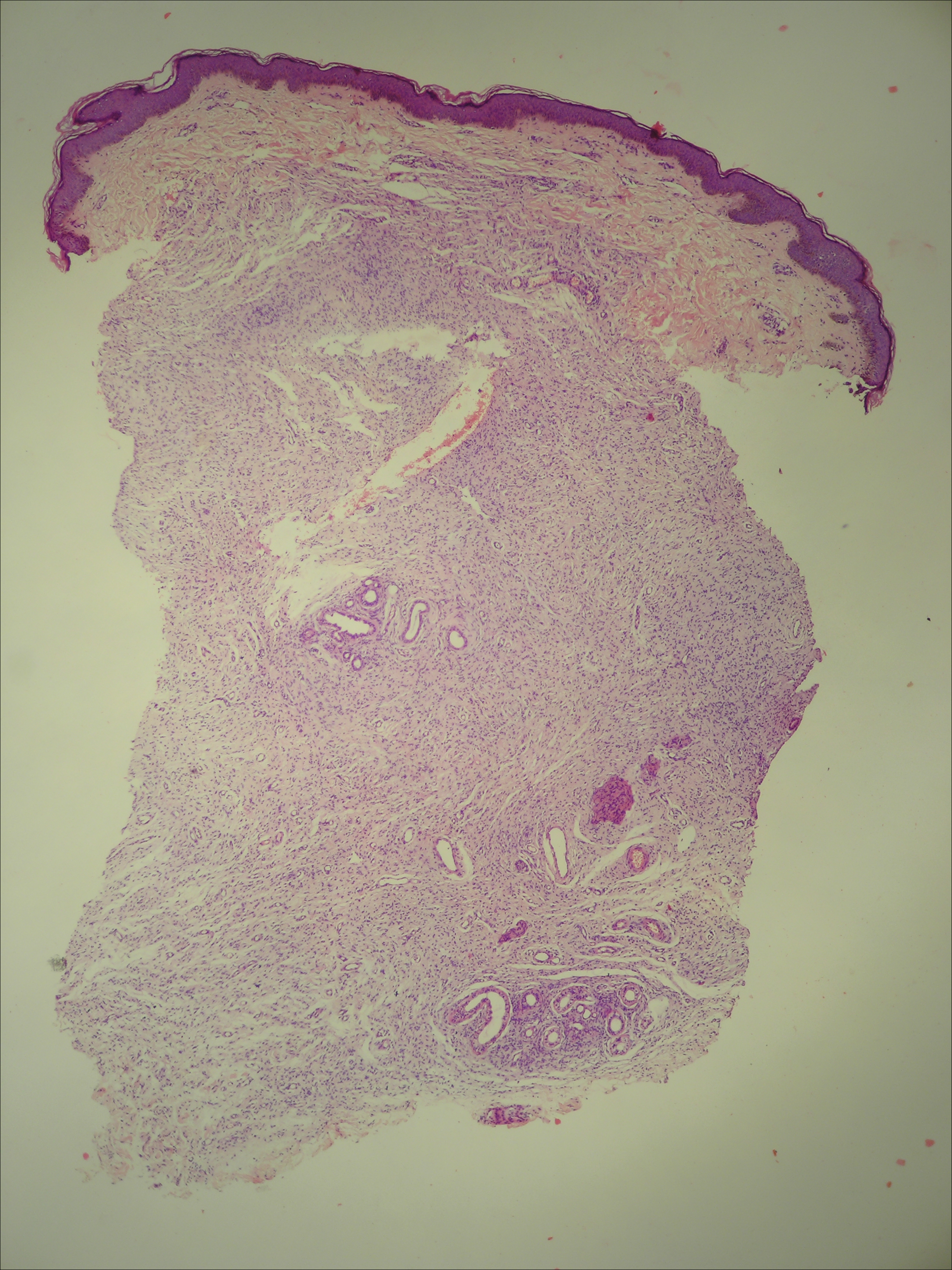
The term phacomatosis is applied to genetically determined disorders of tissue derived from ectodermal origin (eg, skin, central nervous system, eyes) and commonly includes NF-1, tuberous sclerosis, and von Hippel-Lindau syndrome. Neurofibromatosis type I was first described by German pathologist Friedrich Daniel von Recklinghausen.5 Phacomatosis pigmentovascularis has been defined as the association of vascular nevus with a pigmentary nevus. Its pathogenesis can be explained by the twin spotting phenomenon.6 Twin spots are paired patches of mutant tissue that differ from each other and from the surrounding normal background skin. They can occur as 2 clinical types: allelic and nonallelic twin spotting. Our patient had nonallelic twin spots for 2 nevoid conditions: vascular (nevus flammeus) and pigmentary (nevus of Ota). Nevus of Ota was distributed in the V2 segment (maxillary nerve) of the fifth cranial nerve along with classical melanosis bulbi, which is considered a characteristic clinical feature of nevus of Ota (nevus cesius).7 Nevus flammeus (port-wine stain) is a vascular malformation presenting with flat lesions that persists throughout a patient’s life. The phenomenon of twin spotting, or didymosis (didymos means twin in Greek), has been proposed for co-occurrence of vascular and pigmented nevi.8 The association of NF-1 along with phacomatosis cesioflammea (a twin spot) could be explained from mosaicism of tissues derived from neuroectodermal and mesenchymal elements. Neurofibromatosis type I can occur as a mosaic disorder due to either postzygotic germ line or somatic mutations in the NF1 gene located on the proximal long arm of chromosome 17.9 Irrespective of the mutational event, a mosaic patient has a mixture of cells, some have normal copies of a particular gene and others have an abnormal copy of the same gene. Somatic mutation can lead to segmental (localized), generalized, or gonadal mosaicism. Somatic mutations occurring early during embryonic development produce generalized mosaicism, and generalized mosaics clinically appear similar to nonmosaic NF-1 cases.10,11 However, due to a lack of adequate facilities for mutation analysis and financial constraints, we were unable to confirm our case as generalized somatic mosaic for NF1 gene.
Several morphologic abnormalities have been reported with phacomatosis cesioflammea. Wu et al12 reported a single case of phacomatosis cesioflammea associated with pectus excavatum in a 9-month-old infant. Shields et al13 suggested that a thorough ocular examination on a periodic basis is essential to rule out melanoma of ocular tissues in patients with nevus flammeus and ocular melanosis.
Phacomatosis cesioflammea can occur in association with NF-1. The exact incidence of association is not known. The nevoid condition can be treated with appropriate lasers.
- Ota M, Kawamura T, Ito N. Phacomatosis pigmentovascularis (Ota). Jpn J Dermatol. 1947;52:1-3.
- Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol. 2005;141:385-388.
- Joshi A, Manchanda Y, Rijhwani M. Port-wine-stain with rare associations in two cases from Kuwait: phakomatosis pigmentovascularis redefined. Gulf J Dermatol Venereol. 2011;18:59-64.
- Neurofibromatosis. Conference Statement. National Institutes of Health Consensus. Arch Neurol. 1988;45:575-578.
- Gerber PA, Antal AS, Neumann NJ, et al. Neurofibromatosis. Eur J Med Res. 2009;14:102-105.
- Goyal T, Varshney A. Phacomatosis cesioflammea: first case report from India. Indian J Dermatol Venereol Leprol. 2010;76:307.
- Happle R. Didymosis cesioanemica: an unusual counterpart of phakomatosis cesioflammea. Eur J Dermatol. 2011;21:471.
- Happle R, Steijlen PM. Phacomatosis pigmentovascularis interpreted as a phenomenon of twin spots [in German]. Hautarzt. 1989;40:721-724.
- Adigun CG, Stein J. Segmental neurofibromatosis. Dermatol Online J. 2011;17:25.
- Ruggieri M, Huson SM. The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology. 2001;56:1433-1443.
- Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61:1-14.
- Wu CY, Chen PH, Chen GS. Phacomatosis cesioflammea associated with pectus excavatum. Acta Derm Venereol. 2009;89:309-310.
- Shields CL, Kligman BE, Suriano M, et al. Phacomatosis pigmentovascularis of cesioflammea type in 7 patients: combination of ocular pigmentation (melanocytosis or melanosis) and nevus flammeus with risk for melanoma. Arch Ophthalmol. 2011;129:746-750.
To the Editor:
Vascular lesions associated with melanocytic nevi were first described by Ota et al1 in 1947 and given the name phacomatosis pigmentovascularis. In 2005, Happle2 reclassified phacomatosis pigmentovascularis into 3 well-defined types: (1) phacomatosis cesioflammea: blue spots (caesius means bluish gray in Latin) and nevus flammeus; (2) phacomatosis spilorosea: nevus spilus coexisting with a pale pink telangiectatic nevus; and (3) phacomatosis cesiomarmorata: blue spots and cutis marmorata telangiectatica congenita. In 2011 Joshi et al3 described a case of a 31-year-old woman who had a port-wine stain in association with neurofibromatosis type I (NF-1). We present a case of phacomatosis cesioflammea in association with NF-1.
A 20-year-old woman presented to our outpatient section with a bluish black birthmark on the left side of the face since birth with the onset of multiple painless flesh-colored nodules on the trunk and arms of 1 year’s duration. She reported having occasional pruritus over the nodular lesions. Cutaneous examination showed multiple well-defined café au lait macules (0.5–3.0 cm) with regular margins. Multiple flesh-colored nodules were evident on the upper arms (Figure 1) and trunk. The nodules were firm in consistency and showed buttonholing phenomenon with some of the lesions demonstrating bag-of-worms consistency on palpation. Both palms showed multiple brownish frecklelike macules (Figure 2). A single bluish patch extended from the left ala of the nose to the sideburns. Adjoining the bluish patch was a subtle, ill-defined, nonblanchable red patch extending from the lower margin of the bluish patch to the mandibular ridge (Figure 3). Ocular examination showed melanosis bulbi of the left sclera and a few iris hamartomas (Lisch nodules) in both eyes. A biopsy of the skin nodule was obtained under local anesthesia after obtaining the patient’s informed consent; the specimen was fixed in 10% buffered formalin. A hematoxylin and eosin–stained section showed a well-circumscribed nonencapsulated tumor in the dermis composed of loosely spaced spindle cells and wavy collagenous strands (Figure 4). Routine hemogram and blood biochemistry including urinalysis were within reference range. Radiologic examination of the long bones was unremarkable. Our patient had 3 of 6 criteria defined by the National Institutes of Health for diagnosis of NF-1.4 On clinicopathological correlation we made a diagnosis of phacomatosis cesioflammea in association with NF-1. We have reassured the patient about the benign nature of vascular nevus. She was informed that the skin nodules could increase in size during pregnancy and to regularly follow-up with an eye specialist if any visual abnormalities occur.
The term phacomatosis is applied to genetically determined disorders of tissue derived from ectodermal origin (eg, skin, central nervous system, eyes) and commonly includes NF-1, tuberous sclerosis, and von Hippel-Lindau syndrome. Neurofibromatosis type I was first described by German pathologist Friedrich Daniel von Recklinghausen.5 Phacomatosis pigmentovascularis has been defined as the association of vascular nevus with a pigmentary nevus. Its pathogenesis can be explained by the twin spotting phenomenon.6 Twin spots are paired patches of mutant tissue that differ from each other and from the surrounding normal background skin. They can occur as 2 clinical types: allelic and nonallelic twin spotting. Our patient had nonallelic twin spots for 2 nevoid conditions: vascular (nevus flammeus) and pigmentary (nevus of Ota). Nevus of Ota was distributed in the V2 segment (maxillary nerve) of the fifth cranial nerve along with classical melanosis bulbi, which is considered a characteristic clinical feature of nevus of Ota (nevus cesius).7 Nevus flammeus (port-wine stain) is a vascular malformation presenting with flat lesions that persists throughout a patient’s life. The phenomenon of twin spotting, or didymosis (didymos means twin in Greek), has been proposed for co-occurrence of vascular and pigmented nevi.8 The association of NF-1 along with phacomatosis cesioflammea (a twin spot) could be explained from mosaicism of tissues derived from neuroectodermal and mesenchymal elements. Neurofibromatosis type I can occur as a mosaic disorder due to either postzygotic germ line or somatic mutations in the NF1 gene located on the proximal long arm of chromosome 17.9 Irrespective of the mutational event, a mosaic patient has a mixture of cells, some have normal copies of a particular gene and others have an abnormal copy of the same gene. Somatic mutation can lead to segmental (localized), generalized, or gonadal mosaicism. Somatic mutations occurring early during embryonic development produce generalized mosaicism, and generalized mosaics clinically appear similar to nonmosaic NF-1 cases.10,11 However, due to a lack of adequate facilities for mutation analysis and financial constraints, we were unable to confirm our case as generalized somatic mosaic for NF1 gene.
Several morphologic abnormalities have been reported with phacomatosis cesioflammea. Wu et al12 reported a single case of phacomatosis cesioflammea associated with pectus excavatum in a 9-month-old infant. Shields et al13 suggested that a thorough ocular examination on a periodic basis is essential to rule out melanoma of ocular tissues in patients with nevus flammeus and ocular melanosis.
Phacomatosis cesioflammea can occur in association with NF-1. The exact incidence of association is not known. The nevoid condition can be treated with appropriate lasers.
To the Editor:
Vascular lesions associated with melanocytic nevi were first described by Ota et al1 in 1947 and given the name phacomatosis pigmentovascularis. In 2005, Happle2 reclassified phacomatosis pigmentovascularis into 3 well-defined types: (1) phacomatosis cesioflammea: blue spots (caesius means bluish gray in Latin) and nevus flammeus; (2) phacomatosis spilorosea: nevus spilus coexisting with a pale pink telangiectatic nevus; and (3) phacomatosis cesiomarmorata: blue spots and cutis marmorata telangiectatica congenita. In 2011 Joshi et al3 described a case of a 31-year-old woman who had a port-wine stain in association with neurofibromatosis type I (NF-1). We present a case of phacomatosis cesioflammea in association with NF-1.
A 20-year-old woman presented to our outpatient section with a bluish black birthmark on the left side of the face since birth with the onset of multiple painless flesh-colored nodules on the trunk and arms of 1 year’s duration. She reported having occasional pruritus over the nodular lesions. Cutaneous examination showed multiple well-defined café au lait macules (0.5–3.0 cm) with regular margins. Multiple flesh-colored nodules were evident on the upper arms (Figure 1) and trunk. The nodules were firm in consistency and showed buttonholing phenomenon with some of the lesions demonstrating bag-of-worms consistency on palpation. Both palms showed multiple brownish frecklelike macules (Figure 2). A single bluish patch extended from the left ala of the nose to the sideburns. Adjoining the bluish patch was a subtle, ill-defined, nonblanchable red patch extending from the lower margin of the bluish patch to the mandibular ridge (Figure 3). Ocular examination showed melanosis bulbi of the left sclera and a few iris hamartomas (Lisch nodules) in both eyes. A biopsy of the skin nodule was obtained under local anesthesia after obtaining the patient’s informed consent; the specimen was fixed in 10% buffered formalin. A hematoxylin and eosin–stained section showed a well-circumscribed nonencapsulated tumor in the dermis composed of loosely spaced spindle cells and wavy collagenous strands (Figure 4). Routine hemogram and blood biochemistry including urinalysis were within reference range. Radiologic examination of the long bones was unremarkable. Our patient had 3 of 6 criteria defined by the National Institutes of Health for diagnosis of NF-1.4 On clinicopathological correlation we made a diagnosis of phacomatosis cesioflammea in association with NF-1. We have reassured the patient about the benign nature of vascular nevus. She was informed that the skin nodules could increase in size during pregnancy and to regularly follow-up with an eye specialist if any visual abnormalities occur.
The term phacomatosis is applied to genetically determined disorders of tissue derived from ectodermal origin (eg, skin, central nervous system, eyes) and commonly includes NF-1, tuberous sclerosis, and von Hippel-Lindau syndrome. Neurofibromatosis type I was first described by German pathologist Friedrich Daniel von Recklinghausen.5 Phacomatosis pigmentovascularis has been defined as the association of vascular nevus with a pigmentary nevus. Its pathogenesis can be explained by the twin spotting phenomenon.6 Twin spots are paired patches of mutant tissue that differ from each other and from the surrounding normal background skin. They can occur as 2 clinical types: allelic and nonallelic twin spotting. Our patient had nonallelic twin spots for 2 nevoid conditions: vascular (nevus flammeus) and pigmentary (nevus of Ota). Nevus of Ota was distributed in the V2 segment (maxillary nerve) of the fifth cranial nerve along with classical melanosis bulbi, which is considered a characteristic clinical feature of nevus of Ota (nevus cesius).7 Nevus flammeus (port-wine stain) is a vascular malformation presenting with flat lesions that persists throughout a patient’s life. The phenomenon of twin spotting, or didymosis (didymos means twin in Greek), has been proposed for co-occurrence of vascular and pigmented nevi.8 The association of NF-1 along with phacomatosis cesioflammea (a twin spot) could be explained from mosaicism of tissues derived from neuroectodermal and mesenchymal elements. Neurofibromatosis type I can occur as a mosaic disorder due to either postzygotic germ line or somatic mutations in the NF1 gene located on the proximal long arm of chromosome 17.9 Irrespective of the mutational event, a mosaic patient has a mixture of cells, some have normal copies of a particular gene and others have an abnormal copy of the same gene. Somatic mutation can lead to segmental (localized), generalized, or gonadal mosaicism. Somatic mutations occurring early during embryonic development produce generalized mosaicism, and generalized mosaics clinically appear similar to nonmosaic NF-1 cases.10,11 However, due to a lack of adequate facilities for mutation analysis and financial constraints, we were unable to confirm our case as generalized somatic mosaic for NF1 gene.
Several morphologic abnormalities have been reported with phacomatosis cesioflammea. Wu et al12 reported a single case of phacomatosis cesioflammea associated with pectus excavatum in a 9-month-old infant. Shields et al13 suggested that a thorough ocular examination on a periodic basis is essential to rule out melanoma of ocular tissues in patients with nevus flammeus and ocular melanosis.
Phacomatosis cesioflammea can occur in association with NF-1. The exact incidence of association is not known. The nevoid condition can be treated with appropriate lasers.
- Ota M, Kawamura T, Ito N. Phacomatosis pigmentovascularis (Ota). Jpn J Dermatol. 1947;52:1-3.
- Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol. 2005;141:385-388.
- Joshi A, Manchanda Y, Rijhwani M. Port-wine-stain with rare associations in two cases from Kuwait: phakomatosis pigmentovascularis redefined. Gulf J Dermatol Venereol. 2011;18:59-64.
- Neurofibromatosis. Conference Statement. National Institutes of Health Consensus. Arch Neurol. 1988;45:575-578.
- Gerber PA, Antal AS, Neumann NJ, et al. Neurofibromatosis. Eur J Med Res. 2009;14:102-105.
- Goyal T, Varshney A. Phacomatosis cesioflammea: first case report from India. Indian J Dermatol Venereol Leprol. 2010;76:307.
- Happle R. Didymosis cesioanemica: an unusual counterpart of phakomatosis cesioflammea. Eur J Dermatol. 2011;21:471.
- Happle R, Steijlen PM. Phacomatosis pigmentovascularis interpreted as a phenomenon of twin spots [in German]. Hautarzt. 1989;40:721-724.
- Adigun CG, Stein J. Segmental neurofibromatosis. Dermatol Online J. 2011;17:25.
- Ruggieri M, Huson SM. The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology. 2001;56:1433-1443.
- Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61:1-14.
- Wu CY, Chen PH, Chen GS. Phacomatosis cesioflammea associated with pectus excavatum. Acta Derm Venereol. 2009;89:309-310.
- Shields CL, Kligman BE, Suriano M, et al. Phacomatosis pigmentovascularis of cesioflammea type in 7 patients: combination of ocular pigmentation (melanocytosis or melanosis) and nevus flammeus with risk for melanoma. Arch Ophthalmol. 2011;129:746-750.
- Ota M, Kawamura T, Ito N. Phacomatosis pigmentovascularis (Ota). Jpn J Dermatol. 1947;52:1-3.
- Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol. 2005;141:385-388.
- Joshi A, Manchanda Y, Rijhwani M. Port-wine-stain with rare associations in two cases from Kuwait: phakomatosis pigmentovascularis redefined. Gulf J Dermatol Venereol. 2011;18:59-64.
- Neurofibromatosis. Conference Statement. National Institutes of Health Consensus. Arch Neurol. 1988;45:575-578.
- Gerber PA, Antal AS, Neumann NJ, et al. Neurofibromatosis. Eur J Med Res. 2009;14:102-105.
- Goyal T, Varshney A. Phacomatosis cesioflammea: first case report from India. Indian J Dermatol Venereol Leprol. 2010;76:307.
- Happle R. Didymosis cesioanemica: an unusual counterpart of phakomatosis cesioflammea. Eur J Dermatol. 2011;21:471.
- Happle R, Steijlen PM. Phacomatosis pigmentovascularis interpreted as a phenomenon of twin spots [in German]. Hautarzt. 1989;40:721-724.
- Adigun CG, Stein J. Segmental neurofibromatosis. Dermatol Online J. 2011;17:25.
- Ruggieri M, Huson SM. The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology. 2001;56:1433-1443.
- Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61:1-14.
- Wu CY, Chen PH, Chen GS. Phacomatosis cesioflammea associated with pectus excavatum. Acta Derm Venereol. 2009;89:309-310.
- Shields CL, Kligman BE, Suriano M, et al. Phacomatosis pigmentovascularis of cesioflammea type in 7 patients: combination of ocular pigmentation (melanocytosis or melanosis) and nevus flammeus with risk for melanoma. Arch Ophthalmol. 2011;129:746-750.
Practice Points
- Phacomatosis cesioflammea can be associated with neurofibromatosis type I.
- The port-wine stain component of phacomatosis cesioflammea may develop nodularity in long-standing cases.
- The Nd:YAG laser is beneficial for treating blue spots of phacomatosis cesioflammea.
Verrucous Carcinoma of the Buccal Mucosa With Extension to the Cheek
To the Editor:
Verrucous carcinoma is an uncommon type of squamous cell carcinoma (SCC) and was first described by Ackerman1 in 1948. Rock and Fisher2 called this condition oral florid papillomatosis. The distinctive features of this tumor are low-grade malignancy, slow growth, local invasiveness, and rarely intraoral and extraoral metastasis. Extraorally, it can occur in any part of the body,3 a common site being the anogenital region. Depending on the area of occurrence, the condition also is known as Buschke-Lowenstein tumor4 or giant condyloma acuminatum (anogenital region) and carcinoma cuniculatum5 (plantar region). The exact etiology of the condition is unknown, though it is associated with human papillomavirus infection, traumatic scars, chronic infection, tobacco, and chemical carcinogens.3 We report a rare case of verrucous carcinoma originating from the buccal mucosa that subsequently spread to involve the lip and cheek as a large cauliflowerlike growth, which is an unusual presentation.
A 65-year-old man presented to the dermatology department with a painless growth inside the left side of the oral cavity that had developed 5 years prior as a growth on the left buccal mucosa. The lesion gradually increased in size to involve the left oral commissure including the upper and lower lips and the skin of the left cheek; it extended beyond the nasolabial fold in a cauliflowerlike pattern. The lesion was insidious in onset and was not associated with pain, itching, or bleeding. The patient chewed tobacco for the last 40 years, with no similar lesions on any part of the body. On physical examination a warty papilliform lesion was seen on the left buccal mucosa with extension to 2 cm of the upper and lower lip on the left side including the left oral commissure and the skin of the left cheek beyond the nasolabial fold where it appeared as a cauliflowerlike growth measuring 4×5 cm in size (Figure 1). No notable lymphadenopathy was present.
Digital radiographs of the skull (posteroanterior oblique view)(Figure 2) and mandible (left oblique view) showed a lobulated soft-tissue density lesion overlying the left half of the mandible (near the mandibular angle) with involvement of both the upper and lower lips on the left side. However, no obvious underlying bony erosion was noted.
Computed tomography revealed a large soft-tissue mass (41.3×35.3 mm)(Figure 3A) involving the left buccal mucosa with extension into overlying muscle, subcutaneous tissue, and skin. Externally, the lesion was exophytic, irregular, and polypoidal with surface ulceration. Medially, the lesion involved the left oral commissure and parts of the adjoining upper and lower lips. No underlying bony erosion was seen. An enlarged lymph node measuring 20×15 mm was noted in the left upper deep cervical group in the submandibular region (Figure 3B).
Our clinical differential diagnosis included verrucous carcinoma and hypertrophic variety of lupus vulgaris. A 1×2-cm diagnostic incisional biopsy was performed from the cauliflowerlike growth and ultrasound-guided fine-needle aspiration was done from the lymph node. Histopathology revealed a hyperplastic stratified squamous epithelium with upward extension of verrucous projections, which was largely superficial to the adjacent epithelium (Figure 4A). In addition to the surface verrucous projections, there was lesion extension into the subepithelial zone in the form of round club-shaped protrusions (Figure 4B). There was no loss of polarity in these downward proliferations. No horn pearl formation was present. Fine-needle aspiration revealed reactive lymphadenitis.

The final diagnosis of verrucous carcinoma was made and the patient was referred to the oncosurgery department for further management.
Verrucous carcinoma is a rare, low-grade, well-differentiated SCC of the skin or mucosa presenting with a verrucoid or cauliflowerlike appearance. It shows locally aggressive behavior and has low metastatic potential,6 a low degree of dysplasia, and a good prognosis. Because it is a tumor with predominantly horizontal growth, it tends to erode more than infiltrate. It does not present with remote metastasis.7 It has been known by several different names, usually related to anatomic sites (eg, Ackerman tumor, oral florid papillomatosis, carcinoma cuniculatum).
In the oral cavity, verrucous carcinoma constitutes 2% to 4.5% of all forms of SCC seen mainly in men older than 50 years and also is associated with a high incidence (37.7%) of a second primary tumor mainly in the oral mucosa (eg, tongue, lips, palate, salivary gland).8 Indudharan et al9 reported a case of verrucous carcinoma of the maxillary antrum in a young male patient, which also was a rare entity. Verrucous carcinoma is thought to predominantly affect elderly men. Walvekar et al10 reported a male to female ratio of 3.6 to 1 in patients with verrucous carcinoma, with a mean age of 53.9 years. According to Varshney et al,11 patients may range in age from the fourth to eighth decades of life, with a mean age of 60 years; 80% are male. The etiopathogenesis of verrucous carcinoma is related to the following carcinogens: biologic (eg, human papillomavirus), chemical (eg, smoking), and physical (eg, constant trauma).
Verrucous carcinoma should be considered in the differential diagnosis of slow-growing, locally spreading tumors. Oral tumors, especially in tobacco chewers, should raise suspicion of verrucous carcinoma, which will enable prompt management of the tumor.
- Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
- Rock JA, Fisher ER. Florid papillomatosis of the oral cavity and larynx. Arch Otolaryngol. 1960;72:593-598.
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2007;56:506-507.
- Buschke A, Lowenstein L. Uber carcinomahnliche condylomata acuminata despenis. Klin Wochenschr. 1925;4:1726-1728.
- Aird I, Johnson HD, Lennox B, et al. Epithelioma cuniculatum: a variety of squamous carcinoma peculiar to the foot. Br J Surg. 1954;42:245-250.
- Schwartz RA. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-21.
- Zanini M, Wulkan C, Paschoal FM, et al. Verrucous carcinoma: a clinical histopathologic variant of squamous cell carcinoma. An Bras Dermatol. 2004;79:619-621.
- Kalsotra P, Manhas M, Sood R. Verrucous carcinoma of hard palate. JK Science. 2000;2:52-54.
- Indudharan R, Das PK, Thida T. Verrucous carcinoma of maxillary antrum. Singapore Med J. 1996;37:559-561.
- Walvekar RR, Chaukar DA, Deshpande MS, et al. Verrucous carcinoma of the oral cavity: a clinical and pathological study of 101 cases. Oral Oncol. 2009;45:47-51.
- Varshney S, Singh J, Saxena RK, et al. Verrucous carcinoma of larynx. Indian J Otolaryngol Head Neck Surg. 2004;56:54-56.
To the Editor:
Verrucous carcinoma is an uncommon type of squamous cell carcinoma (SCC) and was first described by Ackerman1 in 1948. Rock and Fisher2 called this condition oral florid papillomatosis. The distinctive features of this tumor are low-grade malignancy, slow growth, local invasiveness, and rarely intraoral and extraoral metastasis. Extraorally, it can occur in any part of the body,3 a common site being the anogenital region. Depending on the area of occurrence, the condition also is known as Buschke-Lowenstein tumor4 or giant condyloma acuminatum (anogenital region) and carcinoma cuniculatum5 (plantar region). The exact etiology of the condition is unknown, though it is associated with human papillomavirus infection, traumatic scars, chronic infection, tobacco, and chemical carcinogens.3 We report a rare case of verrucous carcinoma originating from the buccal mucosa that subsequently spread to involve the lip and cheek as a large cauliflowerlike growth, which is an unusual presentation.
A 65-year-old man presented to the dermatology department with a painless growth inside the left side of the oral cavity that had developed 5 years prior as a growth on the left buccal mucosa. The lesion gradually increased in size to involve the left oral commissure including the upper and lower lips and the skin of the left cheek; it extended beyond the nasolabial fold in a cauliflowerlike pattern. The lesion was insidious in onset and was not associated with pain, itching, or bleeding. The patient chewed tobacco for the last 40 years, with no similar lesions on any part of the body. On physical examination a warty papilliform lesion was seen on the left buccal mucosa with extension to 2 cm of the upper and lower lip on the left side including the left oral commissure and the skin of the left cheek beyond the nasolabial fold where it appeared as a cauliflowerlike growth measuring 4×5 cm in size (Figure 1). No notable lymphadenopathy was present.
Digital radiographs of the skull (posteroanterior oblique view)(Figure 2) and mandible (left oblique view) showed a lobulated soft-tissue density lesion overlying the left half of the mandible (near the mandibular angle) with involvement of both the upper and lower lips on the left side. However, no obvious underlying bony erosion was noted.
Computed tomography revealed a large soft-tissue mass (41.3×35.3 mm)(Figure 3A) involving the left buccal mucosa with extension into overlying muscle, subcutaneous tissue, and skin. Externally, the lesion was exophytic, irregular, and polypoidal with surface ulceration. Medially, the lesion involved the left oral commissure and parts of the adjoining upper and lower lips. No underlying bony erosion was seen. An enlarged lymph node measuring 20×15 mm was noted in the left upper deep cervical group in the submandibular region (Figure 3B).
Our clinical differential diagnosis included verrucous carcinoma and hypertrophic variety of lupus vulgaris. A 1×2-cm diagnostic incisional biopsy was performed from the cauliflowerlike growth and ultrasound-guided fine-needle aspiration was done from the lymph node. Histopathology revealed a hyperplastic stratified squamous epithelium with upward extension of verrucous projections, which was largely superficial to the adjacent epithelium (Figure 4A). In addition to the surface verrucous projections, there was lesion extension into the subepithelial zone in the form of round club-shaped protrusions (Figure 4B). There was no loss of polarity in these downward proliferations. No horn pearl formation was present. Fine-needle aspiration revealed reactive lymphadenitis.
The final diagnosis of verrucous carcinoma was made and the patient was referred to the oncosurgery department for further management.
Verrucous carcinoma is a rare, low-grade, well-differentiated SCC of the skin or mucosa presenting with a verrucoid or cauliflowerlike appearance. It shows locally aggressive behavior and has low metastatic potential,6 a low degree of dysplasia, and a good prognosis. Because it is a tumor with predominantly horizontal growth, it tends to erode more than infiltrate. It does not present with remote metastasis.7 It has been known by several different names, usually related to anatomic sites (eg, Ackerman tumor, oral florid papillomatosis, carcinoma cuniculatum).
In the oral cavity, verrucous carcinoma constitutes 2% to 4.5% of all forms of SCC seen mainly in men older than 50 years and also is associated with a high incidence (37.7%) of a second primary tumor mainly in the oral mucosa (eg, tongue, lips, palate, salivary gland).8 Indudharan et al9 reported a case of verrucous carcinoma of the maxillary antrum in a young male patient, which also was a rare entity. Verrucous carcinoma is thought to predominantly affect elderly men. Walvekar et al10 reported a male to female ratio of 3.6 to 1 in patients with verrucous carcinoma, with a mean age of 53.9 years. According to Varshney et al,11 patients may range in age from the fourth to eighth decades of life, with a mean age of 60 years; 80% are male. The etiopathogenesis of verrucous carcinoma is related to the following carcinogens: biologic (eg, human papillomavirus), chemical (eg, smoking), and physical (eg, constant trauma).
Verrucous carcinoma should be considered in the differential diagnosis of slow-growing, locally spreading tumors. Oral tumors, especially in tobacco chewers, should raise suspicion of verrucous carcinoma, which will enable prompt management of the tumor.
To the Editor:
Verrucous carcinoma is an uncommon type of squamous cell carcinoma (SCC) and was first described by Ackerman1 in 1948. Rock and Fisher2 called this condition oral florid papillomatosis. The distinctive features of this tumor are low-grade malignancy, slow growth, local invasiveness, and rarely intraoral and extraoral metastasis. Extraorally, it can occur in any part of the body,3 a common site being the anogenital region. Depending on the area of occurrence, the condition also is known as Buschke-Lowenstein tumor4 or giant condyloma acuminatum (anogenital region) and carcinoma cuniculatum5 (plantar region). The exact etiology of the condition is unknown, though it is associated with human papillomavirus infection, traumatic scars, chronic infection, tobacco, and chemical carcinogens.3 We report a rare case of verrucous carcinoma originating from the buccal mucosa that subsequently spread to involve the lip and cheek as a large cauliflowerlike growth, which is an unusual presentation.
A 65-year-old man presented to the dermatology department with a painless growth inside the left side of the oral cavity that had developed 5 years prior as a growth on the left buccal mucosa. The lesion gradually increased in size to involve the left oral commissure including the upper and lower lips and the skin of the left cheek; it extended beyond the nasolabial fold in a cauliflowerlike pattern. The lesion was insidious in onset and was not associated with pain, itching, or bleeding. The patient chewed tobacco for the last 40 years, with no similar lesions on any part of the body. On physical examination a warty papilliform lesion was seen on the left buccal mucosa with extension to 2 cm of the upper and lower lip on the left side including the left oral commissure and the skin of the left cheek beyond the nasolabial fold where it appeared as a cauliflowerlike growth measuring 4×5 cm in size (Figure 1). No notable lymphadenopathy was present.
Digital radiographs of the skull (posteroanterior oblique view)(Figure 2) and mandible (left oblique view) showed a lobulated soft-tissue density lesion overlying the left half of the mandible (near the mandibular angle) with involvement of both the upper and lower lips on the left side. However, no obvious underlying bony erosion was noted.
Computed tomography revealed a large soft-tissue mass (41.3×35.3 mm)(Figure 3A) involving the left buccal mucosa with extension into overlying muscle, subcutaneous tissue, and skin. Externally, the lesion was exophytic, irregular, and polypoidal with surface ulceration. Medially, the lesion involved the left oral commissure and parts of the adjoining upper and lower lips. No underlying bony erosion was seen. An enlarged lymph node measuring 20×15 mm was noted in the left upper deep cervical group in the submandibular region (Figure 3B).
Our clinical differential diagnosis included verrucous carcinoma and hypertrophic variety of lupus vulgaris. A 1×2-cm diagnostic incisional biopsy was performed from the cauliflowerlike growth and ultrasound-guided fine-needle aspiration was done from the lymph node. Histopathology revealed a hyperplastic stratified squamous epithelium with upward extension of verrucous projections, which was largely superficial to the adjacent epithelium (Figure 4A). In addition to the surface verrucous projections, there was lesion extension into the subepithelial zone in the form of round club-shaped protrusions (Figure 4B). There was no loss of polarity in these downward proliferations. No horn pearl formation was present. Fine-needle aspiration revealed reactive lymphadenitis.
The final diagnosis of verrucous carcinoma was made and the patient was referred to the oncosurgery department for further management.
Verrucous carcinoma is a rare, low-grade, well-differentiated SCC of the skin or mucosa presenting with a verrucoid or cauliflowerlike appearance. It shows locally aggressive behavior and has low metastatic potential,6 a low degree of dysplasia, and a good prognosis. Because it is a tumor with predominantly horizontal growth, it tends to erode more than infiltrate. It does not present with remote metastasis.7 It has been known by several different names, usually related to anatomic sites (eg, Ackerman tumor, oral florid papillomatosis, carcinoma cuniculatum).
In the oral cavity, verrucous carcinoma constitutes 2% to 4.5% of all forms of SCC seen mainly in men older than 50 years and also is associated with a high incidence (37.7%) of a second primary tumor mainly in the oral mucosa (eg, tongue, lips, palate, salivary gland).8 Indudharan et al9 reported a case of verrucous carcinoma of the maxillary antrum in a young male patient, which also was a rare entity. Verrucous carcinoma is thought to predominantly affect elderly men. Walvekar et al10 reported a male to female ratio of 3.6 to 1 in patients with verrucous carcinoma, with a mean age of 53.9 years. According to Varshney et al,11 patients may range in age from the fourth to eighth decades of life, with a mean age of 60 years; 80% are male. The etiopathogenesis of verrucous carcinoma is related to the following carcinogens: biologic (eg, human papillomavirus), chemical (eg, smoking), and physical (eg, constant trauma).
Verrucous carcinoma should be considered in the differential diagnosis of slow-growing, locally spreading tumors. Oral tumors, especially in tobacco chewers, should raise suspicion of verrucous carcinoma, which will enable prompt management of the tumor.
- Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
- Rock JA, Fisher ER. Florid papillomatosis of the oral cavity and larynx. Arch Otolaryngol. 1960;72:593-598.
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2007;56:506-507.
- Buschke A, Lowenstein L. Uber carcinomahnliche condylomata acuminata despenis. Klin Wochenschr. 1925;4:1726-1728.
- Aird I, Johnson HD, Lennox B, et al. Epithelioma cuniculatum: a variety of squamous carcinoma peculiar to the foot. Br J Surg. 1954;42:245-250.
- Schwartz RA. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-21.
- Zanini M, Wulkan C, Paschoal FM, et al. Verrucous carcinoma: a clinical histopathologic variant of squamous cell carcinoma. An Bras Dermatol. 2004;79:619-621.
- Kalsotra P, Manhas M, Sood R. Verrucous carcinoma of hard palate. JK Science. 2000;2:52-54.
- Indudharan R, Das PK, Thida T. Verrucous carcinoma of maxillary antrum. Singapore Med J. 1996;37:559-561.
- Walvekar RR, Chaukar DA, Deshpande MS, et al. Verrucous carcinoma of the oral cavity: a clinical and pathological study of 101 cases. Oral Oncol. 2009;45:47-51.
- Varshney S, Singh J, Saxena RK, et al. Verrucous carcinoma of larynx. Indian J Otolaryngol Head Neck Surg. 2004;56:54-56.
- Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
- Rock JA, Fisher ER. Florid papillomatosis of the oral cavity and larynx. Arch Otolaryngol. 1960;72:593-598.
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2007;56:506-507.
- Buschke A, Lowenstein L. Uber carcinomahnliche condylomata acuminata despenis. Klin Wochenschr. 1925;4:1726-1728.
- Aird I, Johnson HD, Lennox B, et al. Epithelioma cuniculatum: a variety of squamous carcinoma peculiar to the foot. Br J Surg. 1954;42:245-250.
- Schwartz RA. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-21.
- Zanini M, Wulkan C, Paschoal FM, et al. Verrucous carcinoma: a clinical histopathologic variant of squamous cell carcinoma. An Bras Dermatol. 2004;79:619-621.
- Kalsotra P, Manhas M, Sood R. Verrucous carcinoma of hard palate. JK Science. 2000;2:52-54.
- Indudharan R, Das PK, Thida T. Verrucous carcinoma of maxillary antrum. Singapore Med J. 1996;37:559-561.
- Walvekar RR, Chaukar DA, Deshpande MS, et al. Verrucous carcinoma of the oral cavity: a clinical and pathological study of 101 cases. Oral Oncol. 2009;45:47-51.
- Varshney S, Singh J, Saxena RK, et al. Verrucous carcinoma of larynx. Indian J Otolaryngol Head Neck Surg. 2004;56:54-56.
Practice Points
- Verrucous carcinoma is a slow-growing tumor that often presents in advanced clinical stages because it is poorly understood and underrecognized, especially in developing countries.
- Good clinicopathological correlation is required in cases of verrucous carcinoma to avoid misdiagnosis and provide appropriate treatment.
- Case-specific management should be considered, as presentation of verrucous carcinoma varies.
- Radiography should be considered to assess for lymph node involvement.
Widespread Poikilodermatous Dermatomyositis Associated With Chronic Lymphocytic Leukemia
To the Editor:
Dermatomyositis represents a rare idiopathic inflammatory process presenting with cutaneous lesions and muscular weakness. It often represents a paraneoplastic syndrome. We report the case of a 62-year-old man with a history of total-body poikiloderma and a recent diagnosis of chronic lymphocytic leukemia (CLL). Despite lacking typical features of the disease, a diagnosis of dermatomyositis was made. Our patient may represent a distinct poikilodermatous variant of dermatomyositis, sharing the generalized distribution of the erythrodermic subtype.
A 62-year-old man presented with pruritic poikiloderma involving the neck, arms, legs, abdomen, chest, and back of 2 years’ duration (Figure). He also experienced dysphagia and weakness of the legs. The rash was previously treated by other dermatologists with a combination of high-potency topical steroids and topical tacrolimus 0.1% without success. His history was notable for CLL, which had been diagnosed by a dermatologist 6 months prior to the current presentation. Prior to his visit to the dermatologist, the patient had received 6 chemotherapeutic sessions with a combination of rituximab and cyclophosphamide for the treatment of CLL. The rash did not improve with chemotherapy.

Repeat biopsies of affected regions only demonstrated features of mild interface dermatitis. Direct immunofluorescence studies showed scattered colloid body fluorescence for IgM. Because of bilateral weakness of the legs, a muscle biopsy was taken, which demonstrated severe atrophy and interstitial fibrosis, with neurogenic abnormalities detected in areas of lesser atrophy via abnormal muscle fiber–type grouping. Metabolic panel showed elevated muscle enzymes in the blood: creatine kinase, 243 U/L (reference range, 10–225 U/L); serum aldolase, 16 U/L (reference range, ≤8.1 U/L); lactate dehydrogenase, 314 U/L (reference range, 60–200 U/L). An autoimmune panel was negative for Jo-1, Scl-70, U1 ribonucleoprotein, DNA, desmoglein 1 and 3, and antiacetylcholine receptor antibodies. An elevated erythrocyte sedimentation rate was measured at 16 mm/h (reference range, 0–10 mm/h). Given these findings, the lesions were confirmed as a widespread poikilodermatous variant of dermatomyositis.
The patient was placed on a daily 50-mg dose of prednisone, which produced rapid improvement in scaling and erythema. Creatine kinase and serum aldolase levels normalized and motor strength increased. After 1 week the prednisone dosage was reduced to a daily 30-mg dose, and then 20 mg a week later. The skin lesions completely resolved within 4 to 5 months and the patient is currently on a prednisone dose of 5 mg, alternating with 2.5 mg of prednisone and rituximab infusion every 2 months.
Dermatomyositis is a rare entity with an incidence of approximately 0.5 to 1 per 100,000 individuals.1 It presents with a characteristic rash composed of Gottron papules; pathognomonic flat violaceous papules on the dorsal interphalangeal joints, elbows, or knees; and a heliotrope rash, a violaceous erythema involving the eyelids. Poikiloderma frequently is reported to present in a shawl-like distribution, encompassing the shoulders, arms, and upper back.1,2 Dermatomyositis of the poikilodermatous type can present in nonphotoexposed areas and photoexposed areas. The unusual feature is the total-body involvement, which is analogous to erythroderma.3
Our case may represent a distinct poikilodermatous manifestation sharing the distribution of the erythrodermic subtype. We believe that the skin lesions may have represented a paraneoplastic event presenting prior to diagnosis with CLL. Dermatomyositis has a strong association with cancer, with patients 3 times more likely to develop internal malignancy.4 Association is strongest for non-Hodgkin lymphoma, as well as ovarian, lung, colorectal, pancreatic, and gastric cancer. When associated with malignancy, symptoms of dermatomyositis or myositis typically precede the discovery of malignancy by an average of 1.9 years.5 Dermatomyositis has been previously reported to present as a paraneoplastic manifestation of CLL.6 One case has been reported of a patient with CLL who developed leukemia cutis presenting with poikiloderma in the characteristic dermatomyositis shawl-like distribution.7 The lack of dermal infiltration with leukemic cells in our patient, however, makes a paraneoplastic etiology much more likely.
Our patient’s rash did not initially improve with treatment of CLL, but dermatomyositis associated with hematological malignancy may precede, occur simultaneously, or follow the diagnosis of malignancy.8 Additionally, symptoms of dermatomyositis do not always parallel the course of hematological malignancy outcome. However, rituximab has been used as a treatment of dermatomyositis and may have contributed some synergistic effect in combination with prednisone in our patient.9
- Dourmishev LA, Dourmishev AL, Schwartz RA. Dermatomyositis: cutaneous manifestations of its variants. Int J Dermatol. 2002;41:625-630.
- Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-920; quiz 921-992.
- Liu ZH, Wang XD. Acute-onset adult dermatomyositis presenting with erythroderma and diplopia. Clin Exp Dermatol. 2007;32:751-752.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Bohan A, Peter JB, Bowman RL, et al. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255-286.
- Ishida T, Aikawa K, Tamura T, et al. Chronic lymphocytic leukemia associated with nephrotic syndrome and dermatomyositis. Intern Med. 1995;34:15-17.
- Nousari HC, Kimyai-Asadi A, Huang CH, et al. T-cell chronic lymphocytic leukemia mimicking dermatomyositis. Int J Dermatol. 2000;39:144-146.
- Marie I, Guillevin L, Menard JF, et al. Hematological malignancy associated with polymyositis and dermatomyositis. Autoimmun Rev. 2012;11:615-620.
- Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601-607.
To the Editor:
Dermatomyositis represents a rare idiopathic inflammatory process presenting with cutaneous lesions and muscular weakness. It often represents a paraneoplastic syndrome. We report the case of a 62-year-old man with a history of total-body poikiloderma and a recent diagnosis of chronic lymphocytic leukemia (CLL). Despite lacking typical features of the disease, a diagnosis of dermatomyositis was made. Our patient may represent a distinct poikilodermatous variant of dermatomyositis, sharing the generalized distribution of the erythrodermic subtype.
A 62-year-old man presented with pruritic poikiloderma involving the neck, arms, legs, abdomen, chest, and back of 2 years’ duration (Figure). He also experienced dysphagia and weakness of the legs. The rash was previously treated by other dermatologists with a combination of high-potency topical steroids and topical tacrolimus 0.1% without success. His history was notable for CLL, which had been diagnosed by a dermatologist 6 months prior to the current presentation. Prior to his visit to the dermatologist, the patient had received 6 chemotherapeutic sessions with a combination of rituximab and cyclophosphamide for the treatment of CLL. The rash did not improve with chemotherapy.
Repeat biopsies of affected regions only demonstrated features of mild interface dermatitis. Direct immunofluorescence studies showed scattered colloid body fluorescence for IgM. Because of bilateral weakness of the legs, a muscle biopsy was taken, which demonstrated severe atrophy and interstitial fibrosis, with neurogenic abnormalities detected in areas of lesser atrophy via abnormal muscle fiber–type grouping. Metabolic panel showed elevated muscle enzymes in the blood: creatine kinase, 243 U/L (reference range, 10–225 U/L); serum aldolase, 16 U/L (reference range, ≤8.1 U/L); lactate dehydrogenase, 314 U/L (reference range, 60–200 U/L). An autoimmune panel was negative for Jo-1, Scl-70, U1 ribonucleoprotein, DNA, desmoglein 1 and 3, and antiacetylcholine receptor antibodies. An elevated erythrocyte sedimentation rate was measured at 16 mm/h (reference range, 0–10 mm/h). Given these findings, the lesions were confirmed as a widespread poikilodermatous variant of dermatomyositis.
The patient was placed on a daily 50-mg dose of prednisone, which produced rapid improvement in scaling and erythema. Creatine kinase and serum aldolase levels normalized and motor strength increased. After 1 week the prednisone dosage was reduced to a daily 30-mg dose, and then 20 mg a week later. The skin lesions completely resolved within 4 to 5 months and the patient is currently on a prednisone dose of 5 mg, alternating with 2.5 mg of prednisone and rituximab infusion every 2 months.
Dermatomyositis is a rare entity with an incidence of approximately 0.5 to 1 per 100,000 individuals.1 It presents with a characteristic rash composed of Gottron papules; pathognomonic flat violaceous papules on the dorsal interphalangeal joints, elbows, or knees; and a heliotrope rash, a violaceous erythema involving the eyelids. Poikiloderma frequently is reported to present in a shawl-like distribution, encompassing the shoulders, arms, and upper back.1,2 Dermatomyositis of the poikilodermatous type can present in nonphotoexposed areas and photoexposed areas. The unusual feature is the total-body involvement, which is analogous to erythroderma.3
Our case may represent a distinct poikilodermatous manifestation sharing the distribution of the erythrodermic subtype. We believe that the skin lesions may have represented a paraneoplastic event presenting prior to diagnosis with CLL. Dermatomyositis has a strong association with cancer, with patients 3 times more likely to develop internal malignancy.4 Association is strongest for non-Hodgkin lymphoma, as well as ovarian, lung, colorectal, pancreatic, and gastric cancer. When associated with malignancy, symptoms of dermatomyositis or myositis typically precede the discovery of malignancy by an average of 1.9 years.5 Dermatomyositis has been previously reported to present as a paraneoplastic manifestation of CLL.6 One case has been reported of a patient with CLL who developed leukemia cutis presenting with poikiloderma in the characteristic dermatomyositis shawl-like distribution.7 The lack of dermal infiltration with leukemic cells in our patient, however, makes a paraneoplastic etiology much more likely.
Our patient’s rash did not initially improve with treatment of CLL, but dermatomyositis associated with hematological malignancy may precede, occur simultaneously, or follow the diagnosis of malignancy.8 Additionally, symptoms of dermatomyositis do not always parallel the course of hematological malignancy outcome. However, rituximab has been used as a treatment of dermatomyositis and may have contributed some synergistic effect in combination with prednisone in our patient.9
To the Editor:
Dermatomyositis represents a rare idiopathic inflammatory process presenting with cutaneous lesions and muscular weakness. It often represents a paraneoplastic syndrome. We report the case of a 62-year-old man with a history of total-body poikiloderma and a recent diagnosis of chronic lymphocytic leukemia (CLL). Despite lacking typical features of the disease, a diagnosis of dermatomyositis was made. Our patient may represent a distinct poikilodermatous variant of dermatomyositis, sharing the generalized distribution of the erythrodermic subtype.
A 62-year-old man presented with pruritic poikiloderma involving the neck, arms, legs, abdomen, chest, and back of 2 years’ duration (Figure). He also experienced dysphagia and weakness of the legs. The rash was previously treated by other dermatologists with a combination of high-potency topical steroids and topical tacrolimus 0.1% without success. His history was notable for CLL, which had been diagnosed by a dermatologist 6 months prior to the current presentation. Prior to his visit to the dermatologist, the patient had received 6 chemotherapeutic sessions with a combination of rituximab and cyclophosphamide for the treatment of CLL. The rash did not improve with chemotherapy.
Repeat biopsies of affected regions only demonstrated features of mild interface dermatitis. Direct immunofluorescence studies showed scattered colloid body fluorescence for IgM. Because of bilateral weakness of the legs, a muscle biopsy was taken, which demonstrated severe atrophy and interstitial fibrosis, with neurogenic abnormalities detected in areas of lesser atrophy via abnormal muscle fiber–type grouping. Metabolic panel showed elevated muscle enzymes in the blood: creatine kinase, 243 U/L (reference range, 10–225 U/L); serum aldolase, 16 U/L (reference range, ≤8.1 U/L); lactate dehydrogenase, 314 U/L (reference range, 60–200 U/L). An autoimmune panel was negative for Jo-1, Scl-70, U1 ribonucleoprotein, DNA, desmoglein 1 and 3, and antiacetylcholine receptor antibodies. An elevated erythrocyte sedimentation rate was measured at 16 mm/h (reference range, 0–10 mm/h). Given these findings, the lesions were confirmed as a widespread poikilodermatous variant of dermatomyositis.
The patient was placed on a daily 50-mg dose of prednisone, which produced rapid improvement in scaling and erythema. Creatine kinase and serum aldolase levels normalized and motor strength increased. After 1 week the prednisone dosage was reduced to a daily 30-mg dose, and then 20 mg a week later. The skin lesions completely resolved within 4 to 5 months and the patient is currently on a prednisone dose of 5 mg, alternating with 2.5 mg of prednisone and rituximab infusion every 2 months.
Dermatomyositis is a rare entity with an incidence of approximately 0.5 to 1 per 100,000 individuals.1 It presents with a characteristic rash composed of Gottron papules; pathognomonic flat violaceous papules on the dorsal interphalangeal joints, elbows, or knees; and a heliotrope rash, a violaceous erythema involving the eyelids. Poikiloderma frequently is reported to present in a shawl-like distribution, encompassing the shoulders, arms, and upper back.1,2 Dermatomyositis of the poikilodermatous type can present in nonphotoexposed areas and photoexposed areas. The unusual feature is the total-body involvement, which is analogous to erythroderma.3
Our case may represent a distinct poikilodermatous manifestation sharing the distribution of the erythrodermic subtype. We believe that the skin lesions may have represented a paraneoplastic event presenting prior to diagnosis with CLL. Dermatomyositis has a strong association with cancer, with patients 3 times more likely to develop internal malignancy.4 Association is strongest for non-Hodgkin lymphoma, as well as ovarian, lung, colorectal, pancreatic, and gastric cancer. When associated with malignancy, symptoms of dermatomyositis or myositis typically precede the discovery of malignancy by an average of 1.9 years.5 Dermatomyositis has been previously reported to present as a paraneoplastic manifestation of CLL.6 One case has been reported of a patient with CLL who developed leukemia cutis presenting with poikiloderma in the characteristic dermatomyositis shawl-like distribution.7 The lack of dermal infiltration with leukemic cells in our patient, however, makes a paraneoplastic etiology much more likely.
Our patient’s rash did not initially improve with treatment of CLL, but dermatomyositis associated with hematological malignancy may precede, occur simultaneously, or follow the diagnosis of malignancy.8 Additionally, symptoms of dermatomyositis do not always parallel the course of hematological malignancy outcome. However, rituximab has been used as a treatment of dermatomyositis and may have contributed some synergistic effect in combination with prednisone in our patient.9
- Dourmishev LA, Dourmishev AL, Schwartz RA. Dermatomyositis: cutaneous manifestations of its variants. Int J Dermatol. 2002;41:625-630.
- Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-920; quiz 921-992.
- Liu ZH, Wang XD. Acute-onset adult dermatomyositis presenting with erythroderma and diplopia. Clin Exp Dermatol. 2007;32:751-752.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Bohan A, Peter JB, Bowman RL, et al. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255-286.
- Ishida T, Aikawa K, Tamura T, et al. Chronic lymphocytic leukemia associated with nephrotic syndrome and dermatomyositis. Intern Med. 1995;34:15-17.
- Nousari HC, Kimyai-Asadi A, Huang CH, et al. T-cell chronic lymphocytic leukemia mimicking dermatomyositis. Int J Dermatol. 2000;39:144-146.
- Marie I, Guillevin L, Menard JF, et al. Hematological malignancy associated with polymyositis and dermatomyositis. Autoimmun Rev. 2012;11:615-620.
- Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601-607.
- Dourmishev LA, Dourmishev AL, Schwartz RA. Dermatomyositis: cutaneous manifestations of its variants. Int J Dermatol. 2002;41:625-630.
- Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-920; quiz 921-992.
- Liu ZH, Wang XD. Acute-onset adult dermatomyositis presenting with erythroderma and diplopia. Clin Exp Dermatol. 2007;32:751-752.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Bohan A, Peter JB, Bowman RL, et al. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255-286.
- Ishida T, Aikawa K, Tamura T, et al. Chronic lymphocytic leukemia associated with nephrotic syndrome and dermatomyositis. Intern Med. 1995;34:15-17.
- Nousari HC, Kimyai-Asadi A, Huang CH, et al. T-cell chronic lymphocytic leukemia mimicking dermatomyositis. Int J Dermatol. 2000;39:144-146.
- Marie I, Guillevin L, Menard JF, et al. Hematological malignancy associated with polymyositis and dermatomyositis. Autoimmun Rev. 2012;11:615-620.
- Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601-607.
Practice Points
- Poikiloderma, even with an unusual clinical presentation, can be a useful clinical clue for the diagnosis of dermatomyositis or other collagen vascular disease.
- Dermatomyositis can be paraneoplastic and though often associated with epithelial malignancies and solid tumors can also be associated with leukemias.
Hyperkeratotic Papules on the Medial Aspects of the Feet
To the Editor:
A 43-year-old woman with recently diagnosed diabetes mellitus and a history of thrombotic thrombocytopenic purpura on chronic oral steroids presented with a several-year history of small bumps and bilateral hyperpigmentation on the feet. On physical examination 2- to 3-mm dark brown, hyperkeratotic, firm papules were present on the medial aspects of the feet as well as the dorsal and medial aspects of the thumbs (Figure 1). There also were brown thickened firm plaques on the heels and soles of the feet.
A punch biopsy of the medial aspect of the right foot was performed (Figure 2). Microscopic examination revealed acral skin with hyperkeratosis, parakeratosis, mild hypergranulosis, mild basilar pigmentation, and mild dermal fibrosis (Figure 2A). A periodic acid–Schiff stain for fungus was negative. An elastic van Gieson stain showed fragmentation of the dermal elastic fibers (Figure 2B). The patient was diagnosed with acrokeratoelastoidosis (AKE).
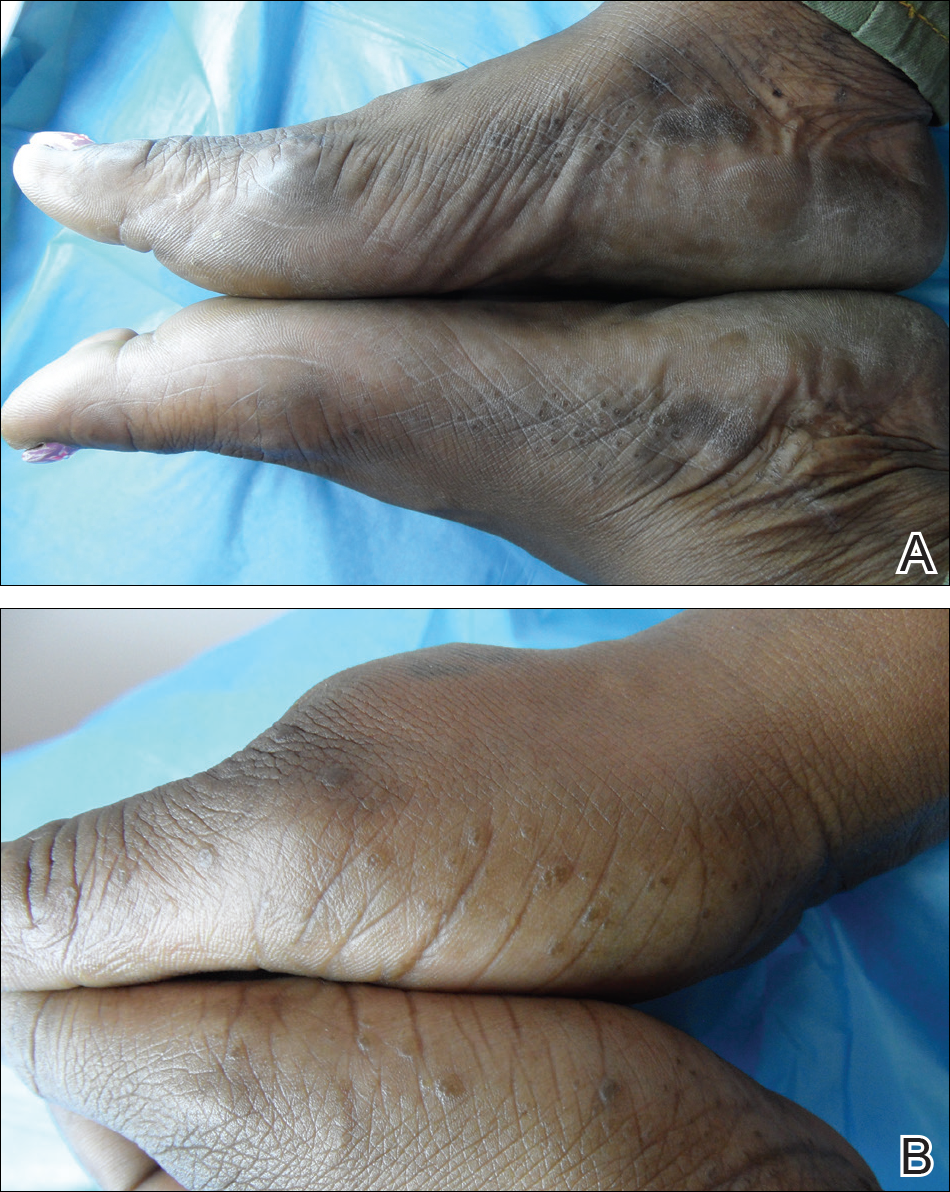
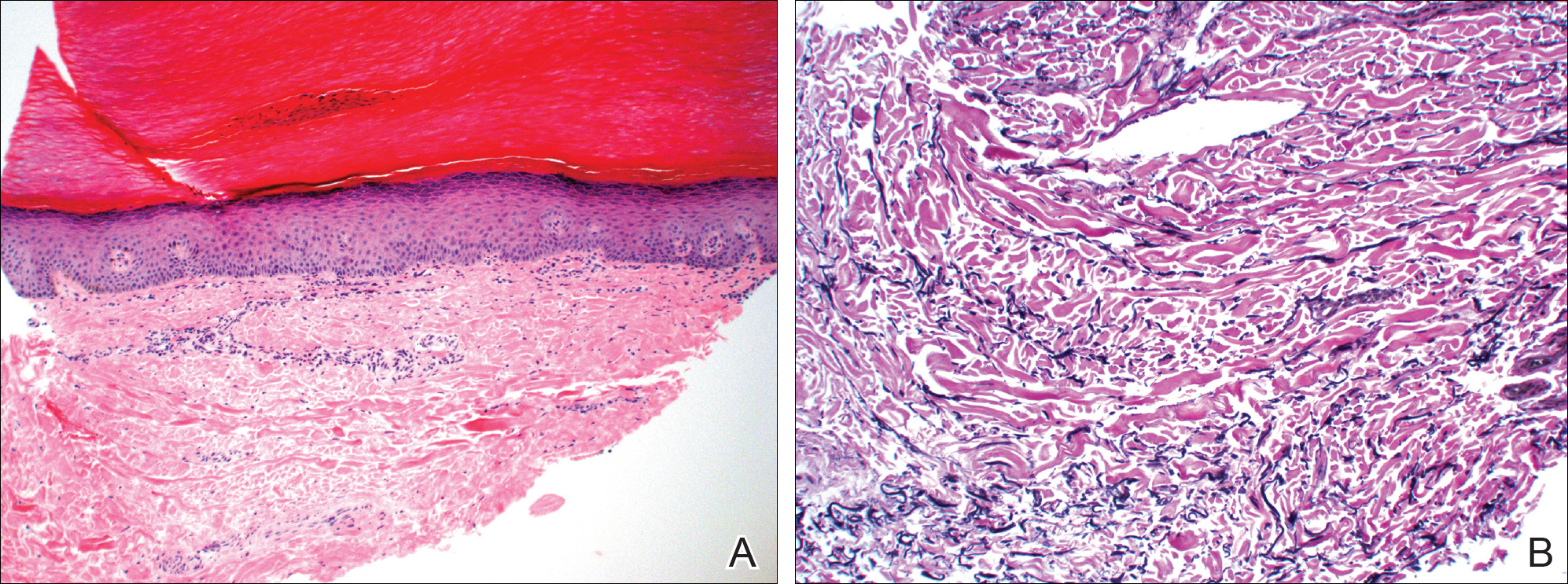
Acrokeratoelastoidosis is a rare autosomal-dominant genodermatosis characterized by firm yellow papules and plaques that appear along the margins of the hands and feet and increase in number over time.1 Histopathologically, hyperkeratosis with hypergranulosis and acanthosis can be seen. Elastorrhexis, resulting in fragmentation of elastic fibers within the dermis, typically is present, a feature that distinguishes AKE from focal acral hyperkeratosis.2 Also, the dermis may be normal with hematoxylin and eosin stain or slightly thickened with mild depression and thin elastic fibers. There is no reported racial or sex predilection, but rapid progression of the disease during pregnancy has been observed.3
The pathogenesis of AKE is not completely understood. However, it has been implicated that abnormalities in the secretion of elastic fibers from fibroblasts may be involved in disease pathogenesis.4,5 Electron microscopy has demonstrated fibroblasts with dense granules at the periphery of their cytoplasm and an absence of surrounding elastic fibers. Genetic studies have linked underlying mutations in chromosome 2 to the disease.6 Defects in keratinization and overproduction of filaggrin also may be involved in the disease process.7
Most therapies generally are ineffective but have included urea, salicylic acid, prednisone, and tretinoin.8 Six-month treatment with etretinate 25 to 50 mg has shown promising results, though recurrences occurred with dosage reduction or discontinuation.9 Our patient demonstrated mild improvement with urea cream 30%.
- Meziane M, Senouci K, Ouidane Y, et al. Acrokeratoelastoidosis. Dermatol Online J. 2008;14:11.
- Lewis KG, Bercovitch L, Dill SW, et al. Acquired disorders of elastic tissue: part II. decreased elastic tissue. J Am Acad Dermatol. 2004;51:165-185; quiz 186-188.
- Tsai S, Kageyama N, Warthan M, et al. Acrokeratoelastoidosis. Int J Dermatol. 2005;44:406-407.
- Johansson EA, Kariniemi AL, Niemi KM. Palmoplantar keratoderma of punctate type: acrokeratoelastoidosis Costa. Acta Derm Venereol. 1980;60:149-153.
- Fiallo P, Pesce C, Brusasco A, et al. Acrokeratoelastoidosis of Costa: a primary disease of the elastic tissue? J Cutan Pathol. 1998;25:580-582.
- Shbaklo Z, Jamaleddine NF, Kibbi AG, et al. Acrokeratoelastoidosis. Int J Dermatol. 1990;29:333-336.
- Abulafia J, Vignale RA. Degenerative collagenous plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
- Hu W, Cook TF, Vicki GJ, et al. Acrokeratoelastoidosis. Pediatr Dermatol. 2002;19:320-322.
- Handfield-Jones S, Kennedy CT. Acrokeratoelastoidosis treated with etretinate. J Am Acad Dermatol. 1987;17(5, pt 2):881-882.
To the Editor:
A 43-year-old woman with recently diagnosed diabetes mellitus and a history of thrombotic thrombocytopenic purpura on chronic oral steroids presented with a several-year history of small bumps and bilateral hyperpigmentation on the feet. On physical examination 2- to 3-mm dark brown, hyperkeratotic, firm papules were present on the medial aspects of the feet as well as the dorsal and medial aspects of the thumbs (Figure 1). There also were brown thickened firm plaques on the heels and soles of the feet.
A punch biopsy of the medial aspect of the right foot was performed (Figure 2). Microscopic examination revealed acral skin with hyperkeratosis, parakeratosis, mild hypergranulosis, mild basilar pigmentation, and mild dermal fibrosis (Figure 2A). A periodic acid–Schiff stain for fungus was negative. An elastic van Gieson stain showed fragmentation of the dermal elastic fibers (Figure 2B). The patient was diagnosed with acrokeratoelastoidosis (AKE).
Acrokeratoelastoidosis is a rare autosomal-dominant genodermatosis characterized by firm yellow papules and plaques that appear along the margins of the hands and feet and increase in number over time.1 Histopathologically, hyperkeratosis with hypergranulosis and acanthosis can be seen. Elastorrhexis, resulting in fragmentation of elastic fibers within the dermis, typically is present, a feature that distinguishes AKE from focal acral hyperkeratosis.2 Also, the dermis may be normal with hematoxylin and eosin stain or slightly thickened with mild depression and thin elastic fibers. There is no reported racial or sex predilection, but rapid progression of the disease during pregnancy has been observed.3
The pathogenesis of AKE is not completely understood. However, it has been implicated that abnormalities in the secretion of elastic fibers from fibroblasts may be involved in disease pathogenesis.4,5 Electron microscopy has demonstrated fibroblasts with dense granules at the periphery of their cytoplasm and an absence of surrounding elastic fibers. Genetic studies have linked underlying mutations in chromosome 2 to the disease.6 Defects in keratinization and overproduction of filaggrin also may be involved in the disease process.7
Most therapies generally are ineffective but have included urea, salicylic acid, prednisone, and tretinoin.8 Six-month treatment with etretinate 25 to 50 mg has shown promising results, though recurrences occurred with dosage reduction or discontinuation.9 Our patient demonstrated mild improvement with urea cream 30%.
To the Editor:
A 43-year-old woman with recently diagnosed diabetes mellitus and a history of thrombotic thrombocytopenic purpura on chronic oral steroids presented with a several-year history of small bumps and bilateral hyperpigmentation on the feet. On physical examination 2- to 3-mm dark brown, hyperkeratotic, firm papules were present on the medial aspects of the feet as well as the dorsal and medial aspects of the thumbs (Figure 1). There also were brown thickened firm plaques on the heels and soles of the feet.
A punch biopsy of the medial aspect of the right foot was performed (Figure 2). Microscopic examination revealed acral skin with hyperkeratosis, parakeratosis, mild hypergranulosis, mild basilar pigmentation, and mild dermal fibrosis (Figure 2A). A periodic acid–Schiff stain for fungus was negative. An elastic van Gieson stain showed fragmentation of the dermal elastic fibers (Figure 2B). The patient was diagnosed with acrokeratoelastoidosis (AKE).
Acrokeratoelastoidosis is a rare autosomal-dominant genodermatosis characterized by firm yellow papules and plaques that appear along the margins of the hands and feet and increase in number over time.1 Histopathologically, hyperkeratosis with hypergranulosis and acanthosis can be seen. Elastorrhexis, resulting in fragmentation of elastic fibers within the dermis, typically is present, a feature that distinguishes AKE from focal acral hyperkeratosis.2 Also, the dermis may be normal with hematoxylin and eosin stain or slightly thickened with mild depression and thin elastic fibers. There is no reported racial or sex predilection, but rapid progression of the disease during pregnancy has been observed.3
The pathogenesis of AKE is not completely understood. However, it has been implicated that abnormalities in the secretion of elastic fibers from fibroblasts may be involved in disease pathogenesis.4,5 Electron microscopy has demonstrated fibroblasts with dense granules at the periphery of their cytoplasm and an absence of surrounding elastic fibers. Genetic studies have linked underlying mutations in chromosome 2 to the disease.6 Defects in keratinization and overproduction of filaggrin also may be involved in the disease process.7
Most therapies generally are ineffective but have included urea, salicylic acid, prednisone, and tretinoin.8 Six-month treatment with etretinate 25 to 50 mg has shown promising results, though recurrences occurred with dosage reduction or discontinuation.9 Our patient demonstrated mild improvement with urea cream 30%.
- Meziane M, Senouci K, Ouidane Y, et al. Acrokeratoelastoidosis. Dermatol Online J. 2008;14:11.
- Lewis KG, Bercovitch L, Dill SW, et al. Acquired disorders of elastic tissue: part II. decreased elastic tissue. J Am Acad Dermatol. 2004;51:165-185; quiz 186-188.
- Tsai S, Kageyama N, Warthan M, et al. Acrokeratoelastoidosis. Int J Dermatol. 2005;44:406-407.
- Johansson EA, Kariniemi AL, Niemi KM. Palmoplantar keratoderma of punctate type: acrokeratoelastoidosis Costa. Acta Derm Venereol. 1980;60:149-153.
- Fiallo P, Pesce C, Brusasco A, et al. Acrokeratoelastoidosis of Costa: a primary disease of the elastic tissue? J Cutan Pathol. 1998;25:580-582.
- Shbaklo Z, Jamaleddine NF, Kibbi AG, et al. Acrokeratoelastoidosis. Int J Dermatol. 1990;29:333-336.
- Abulafia J, Vignale RA. Degenerative collagenous plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
- Hu W, Cook TF, Vicki GJ, et al. Acrokeratoelastoidosis. Pediatr Dermatol. 2002;19:320-322.
- Handfield-Jones S, Kennedy CT. Acrokeratoelastoidosis treated with etretinate. J Am Acad Dermatol. 1987;17(5, pt 2):881-882.
- Meziane M, Senouci K, Ouidane Y, et al. Acrokeratoelastoidosis. Dermatol Online J. 2008;14:11.
- Lewis KG, Bercovitch L, Dill SW, et al. Acquired disorders of elastic tissue: part II. decreased elastic tissue. J Am Acad Dermatol. 2004;51:165-185; quiz 186-188.
- Tsai S, Kageyama N, Warthan M, et al. Acrokeratoelastoidosis. Int J Dermatol. 2005;44:406-407.
- Johansson EA, Kariniemi AL, Niemi KM. Palmoplantar keratoderma of punctate type: acrokeratoelastoidosis Costa. Acta Derm Venereol. 1980;60:149-153.
- Fiallo P, Pesce C, Brusasco A, et al. Acrokeratoelastoidosis of Costa: a primary disease of the elastic tissue? J Cutan Pathol. 1998;25:580-582.
- Shbaklo Z, Jamaleddine NF, Kibbi AG, et al. Acrokeratoelastoidosis. Int J Dermatol. 1990;29:333-336.
- Abulafia J, Vignale RA. Degenerative collagenous plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
- Hu W, Cook TF, Vicki GJ, et al. Acrokeratoelastoidosis. Pediatr Dermatol. 2002;19:320-322.
- Handfield-Jones S, Kennedy CT. Acrokeratoelastoidosis treated with etretinate. J Am Acad Dermatol. 1987;17(5, pt 2):881-882.
Practice Points
- Acrokeratoelastoidosis is a rare autosomal-dominant genodermatosis characterized by firm yellow papules along the margins of the hands and feet.
- Most therapies generally are ineffective but have included urea, salicylic acid, and tretinoin.
Localized Pemphigus Foliaceus
To the Editor:
Pemphigus foliaceus is a rare autoimmune blistering disorder that typically presents with crusted scaly erosions in a seborrheic distribution. We describe a case of pemphigus foliaceus localized to the right cheek of 10 years’ duration that spread to other areas. With a PubMed search of articles indexed for MEDLINE yielding only 14 cases of localized pemphigus foliaceus (Table), it represents an extremely rare entity that often is a diagnostic challenge and may be a harbinger for disseminated disease months to years after the inciting lesion appears.
A 51-year-old woman presented with an asymptomatic cutaneous eruption that had remained localized to the right cheek for 10 years before it increased in size and new lesions developed on the left cheek, chest, and upper back. No inciting factors, such as contactants, insect bites, infections, medications, or recent travel were identified. On physical examination a well-demarcated, hypertrophic, verrucouslike plaque with central pink atrophy and exfoliative scale involved the right malar and submalar regions but spared the mucocutaneous junctions of the face (Figure 1). Subtle dark brown papules, some with overlying scale, speckled the left cheek, right jawline, chest, and upper back. The oral cavity was clear.
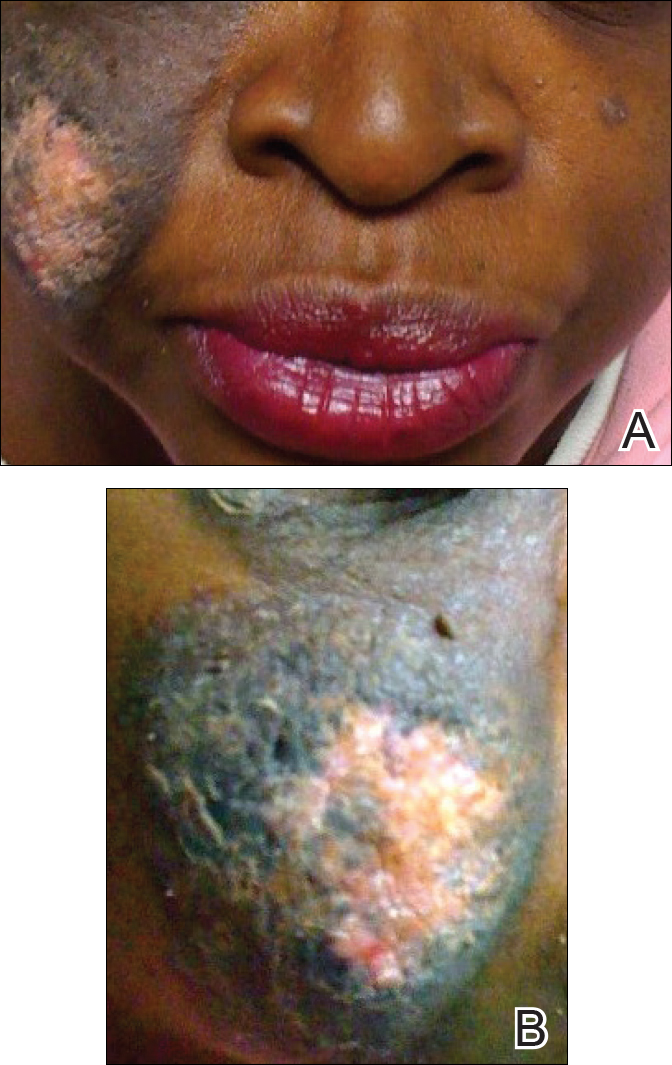
Leading differentials included hypertrophic discoid lupus erythematosus and pemphigus vegetans. Other considerations included sarcoidosis, granuloma faciale, lupus vulgaris, disseminated coccidioidomycosis or blastomycosis, and squamous cell carcinoma.
An initial biopsy revealed a lymphocytic lichenoid dermatitis with epidermal hyperplasia and scattered eosinophils for which the following differentials were provided: insect bite, hypertrophic lichen planus, prurigo nodularis superimposed on rosacea, and allergic contact dermatitis. Under these histologic diagnoses, tacrolimus ointment 0.03%, topical mid-potency corticosteroid, and a combination of oral doxycycline and metronidazole gel 1% were prescribed but failed to ameliorate her condition.
Because the clinical differentials were vast and noncorrelative with the original pathology, additional biopsies were performed: one from the edge of the large malar plaque, which was transected for hematoxylin and eosin (H&E) and tissue cultures; one perilesional to the large malar plaque for direct immunofluorescence (DIF); and one from the papule on the right jawline for H&E. Tissue cultures were negative for fungal and mycobacterial organisms. Both specimens submitted for H&E showed the prominent epidermal hyperplasia and lymphocytic dermal infiltrate noted on the original H&E but also demonstrated intragranular acantholysis (Figure 2). The DIF revealed intercellular IgG and C3 deposition throughout the epidermis (Figure 3). Indirect immunofluorescence was negative, but enzyme-linked immunosorbent assay detected circulating antidesmoglein-1 but not antidesmoglein-3 autoantibodies. Other serologies including antinuclear antibody, anti–double-stranded DNA, antihistone, anti–Sjögren syndrome A, and anti–Sjögren syndrome B antibodies were negative.
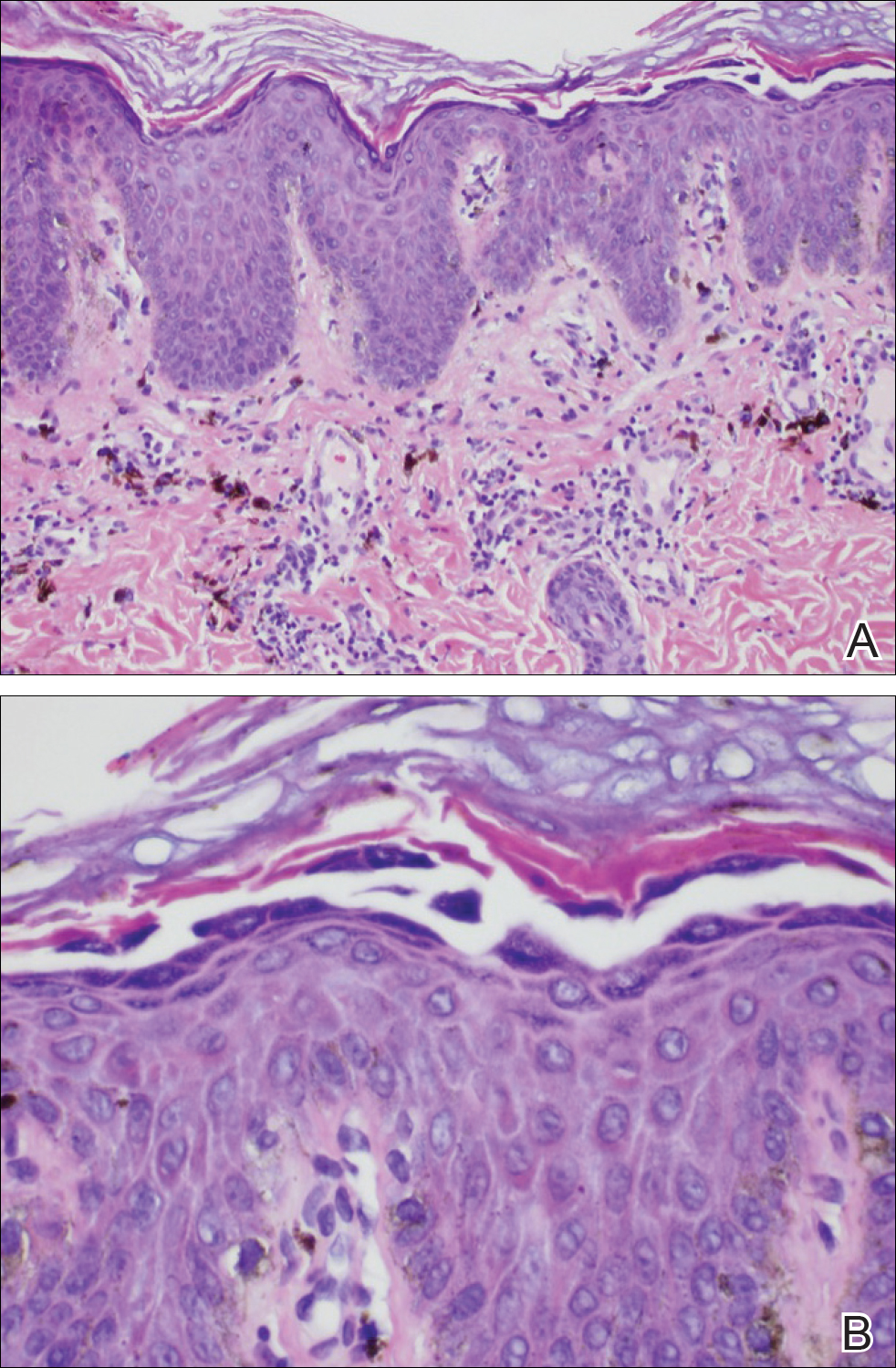

The diagnosis of localized pemphigus foliaceus was made and management with oral prednisone and mycophenolate mofetil resulted in improvement within weeks.
Localized pemphigus foliaceus is extremely rare with only 14 cases reported in the literature (Table).1-10 Its diagnosis is challenging, as the clinical presentation simulates various entities and the histological features and serological markers are difficult to capture.
Localized pemphigus foliaceus typically presents as an isolated, erythematous, scaly, crusted plaque involving the nose, cheek, or scalp and may mimic several conditions including contact dermatitis, seborrheic dermatitis, rosacea, cutaneous sarcoidosis, discoid lupus erythematosus, lupus vulgaris, impetigo contagiosa, solar keratosis, and nonmelanoma skin cancer.1-10
The predilection for sun-exposed areas suggests UV radiation may induce binding of antidesmoglein-1 autoantibodies with subsequent cytokine-mediated inflammation and acantholysis at these sites.11-13 Similarly, the immunomodulatory agent imiquimod has been reported to induce pemphigus foliaceus at its application sites.6
When pemphigus foliaceus is clinically discernible, the histology and DIF are in accordance with the clinical diagnosis 53.8% of the time.13 In cases of localized pemphigus foliaceus in which the diagnosis is more elusive, many biopsies often are needed to capture the characteristic intragranular acantholysis; this feature often is so subtle that unless the diagnosis is suspected, it is underappreciated or undetectable. In chronic lesions, it may be masked by secondary changes such as acanthosis, hyperkeratosis, and parakeratosis.14
In pemphigus foliaceus, detection of circulating antidesmoglein-1 autoantibodies by enzyme-linked immunosorbent assay is slightly more sensitive and specific compared to indirect immunofluorescence, but both correlate with disease activity.15,16 The low or absent autoantibody titers in localized pemphigus foliaceus may reflect its limited involvement, but dissemination of the disease with subsequent elevation of autoantibody titers may occur months to years after initial presentation,1,2,9 as was the case with our patient.
The majority of localized pemphigus foliaceus cases require systemic prednisone, sometimes in conjunction with nonsteroidal immunosuppressants or topical high-potency corticosteroids.1-3,5,6,8-10 One case was efficaciously managed with tacrolimus ointment 0.1%.7
Localized pemphigus foliaceus is a rare and challenging entity that must be a diagnostic consideration for any chronic focal plaque on the face or scalp, as it may herald disseminated disease.
- Paramsothy Y, Lawrence CM. “Tin-tack” sign in localized pemphigus foliaceus. Br J Dermatol. 1987;116:127-129.
- Newton JA, McGibbon DH, Monk B, et al. Pemphigus foliaceus localized to the nose. Br J Dermatol. 1988;118:303-312.
- Koide M, Kokura N, Takano N. Pemphigus foliaceus localized on the face [in Japanese]. Jpn J Dermatol. 1989;97:1262.
- Yamamoto S, Kanekura T, Gushi A, et al. A case of localized pemphigus foliaceus. J Dermatol. 1996;23:893-895.
- Kishibe M, Kinouchi M, Ishida-Yamamoto A, et al. Pemphigus foliaceus localized to the nose. Clin Exp Dermatol. 2003;28:560-562.
- Lin R, Ladd DJ, Powell DJ, et al. Localized pemphigus foliaceus induced by topical imiquimod treatment. Arch Dermatol. 2004;140:889-890.
- Termeer CC, Technau K, Augustin M, et al. Topical tacrolimus (Protopic) for the treatment of a localized pemphigus foliaceus. J Eur Acad Dermatol Venereol. 2004;18:636-637.
- Zaraa I, El Euch D, Kort R, et al. Localized pemphigus: a report of three cases. Int J Dermatol 2010;49:715-716.
- Ohata C, Akamatsu K, Imai N, et al. Localized pemphigus foliaceus exclusively involving the follicular infundibulum: a novel peau d’orange appearance. Eur J Dermatol. 2011;21:392-395.
- Maderal AD, Miner A, Nousari C, et al. Localized pemphigus foliaceus with unilateral facial involvement. Actas Dermosifiliogr. 2014;105:413-417.
- Cram DL, Winkelmann RK. Ultraviolet-induced acantholysis in pemphigus. Arch Dermatol. 1965;92:7-13.
- Kano Y, Shimosegawa M, Mizukawa Y, et al. Pemphigus foliaceus induced by exposure to sunlight. Dermatology. 2000;201:132-138.
- Lebe B, Gül Nıflıoğlu G, Seyrek S, et al. Evaluation of clinical and histopathologic/direct immunofluorescence diagnosis in autoimmune vesiculobullous dermatitis: utility of direct immunofluorescence. Turk Patoloji Derg. 2012;28:11-16.
- Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol. 2011;29:432-436.
- Ishii K, Amagai M, Hall RP, et al. Characterization of autoantibodies in pemphigus using antigen specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. 1997;159:2010-2017.
- Ng PP, Thng ST, Mohamed K, et al. Comparison of desmoglein ELISA and indirect immunofluorescence using two substrates (monkey esophagus and normal human skin) in the diagnosis of pemphigus. Australas J Dermatol. 2005;46:239-241.
To the Editor:
Pemphigus foliaceus is a rare autoimmune blistering disorder that typically presents with crusted scaly erosions in a seborrheic distribution. We describe a case of pemphigus foliaceus localized to the right cheek of 10 years’ duration that spread to other areas. With a PubMed search of articles indexed for MEDLINE yielding only 14 cases of localized pemphigus foliaceus (Table), it represents an extremely rare entity that often is a diagnostic challenge and may be a harbinger for disseminated disease months to years after the inciting lesion appears.
A 51-year-old woman presented with an asymptomatic cutaneous eruption that had remained localized to the right cheek for 10 years before it increased in size and new lesions developed on the left cheek, chest, and upper back. No inciting factors, such as contactants, insect bites, infections, medications, or recent travel were identified. On physical examination a well-demarcated, hypertrophic, verrucouslike plaque with central pink atrophy and exfoliative scale involved the right malar and submalar regions but spared the mucocutaneous junctions of the face (Figure 1). Subtle dark brown papules, some with overlying scale, speckled the left cheek, right jawline, chest, and upper back. The oral cavity was clear.
Leading differentials included hypertrophic discoid lupus erythematosus and pemphigus vegetans. Other considerations included sarcoidosis, granuloma faciale, lupus vulgaris, disseminated coccidioidomycosis or blastomycosis, and squamous cell carcinoma.
An initial biopsy revealed a lymphocytic lichenoid dermatitis with epidermal hyperplasia and scattered eosinophils for which the following differentials were provided: insect bite, hypertrophic lichen planus, prurigo nodularis superimposed on rosacea, and allergic contact dermatitis. Under these histologic diagnoses, tacrolimus ointment 0.03%, topical mid-potency corticosteroid, and a combination of oral doxycycline and metronidazole gel 1% were prescribed but failed to ameliorate her condition.
Because the clinical differentials were vast and noncorrelative with the original pathology, additional biopsies were performed: one from the edge of the large malar plaque, which was transected for hematoxylin and eosin (H&E) and tissue cultures; one perilesional to the large malar plaque for direct immunofluorescence (DIF); and one from the papule on the right jawline for H&E. Tissue cultures were negative for fungal and mycobacterial organisms. Both specimens submitted for H&E showed the prominent epidermal hyperplasia and lymphocytic dermal infiltrate noted on the original H&E but also demonstrated intragranular acantholysis (Figure 2). The DIF revealed intercellular IgG and C3 deposition throughout the epidermis (Figure 3). Indirect immunofluorescence was negative, but enzyme-linked immunosorbent assay detected circulating antidesmoglein-1 but not antidesmoglein-3 autoantibodies. Other serologies including antinuclear antibody, anti–double-stranded DNA, antihistone, anti–Sjögren syndrome A, and anti–Sjögren syndrome B antibodies were negative.
The diagnosis of localized pemphigus foliaceus was made and management with oral prednisone and mycophenolate mofetil resulted in improvement within weeks.
Localized pemphigus foliaceus is extremely rare with only 14 cases reported in the literature (Table).1-10 Its diagnosis is challenging, as the clinical presentation simulates various entities and the histological features and serological markers are difficult to capture.
Localized pemphigus foliaceus typically presents as an isolated, erythematous, scaly, crusted plaque involving the nose, cheek, or scalp and may mimic several conditions including contact dermatitis, seborrheic dermatitis, rosacea, cutaneous sarcoidosis, discoid lupus erythematosus, lupus vulgaris, impetigo contagiosa, solar keratosis, and nonmelanoma skin cancer.1-10
The predilection for sun-exposed areas suggests UV radiation may induce binding of antidesmoglein-1 autoantibodies with subsequent cytokine-mediated inflammation and acantholysis at these sites.11-13 Similarly, the immunomodulatory agent imiquimod has been reported to induce pemphigus foliaceus at its application sites.6
When pemphigus foliaceus is clinically discernible, the histology and DIF are in accordance with the clinical diagnosis 53.8% of the time.13 In cases of localized pemphigus foliaceus in which the diagnosis is more elusive, many biopsies often are needed to capture the characteristic intragranular acantholysis; this feature often is so subtle that unless the diagnosis is suspected, it is underappreciated or undetectable. In chronic lesions, it may be masked by secondary changes such as acanthosis, hyperkeratosis, and parakeratosis.14
In pemphigus foliaceus, detection of circulating antidesmoglein-1 autoantibodies by enzyme-linked immunosorbent assay is slightly more sensitive and specific compared to indirect immunofluorescence, but both correlate with disease activity.15,16 The low or absent autoantibody titers in localized pemphigus foliaceus may reflect its limited involvement, but dissemination of the disease with subsequent elevation of autoantibody titers may occur months to years after initial presentation,1,2,9 as was the case with our patient.
The majority of localized pemphigus foliaceus cases require systemic prednisone, sometimes in conjunction with nonsteroidal immunosuppressants or topical high-potency corticosteroids.1-3,5,6,8-10 One case was efficaciously managed with tacrolimus ointment 0.1%.7
Localized pemphigus foliaceus is a rare and challenging entity that must be a diagnostic consideration for any chronic focal plaque on the face or scalp, as it may herald disseminated disease.
To the Editor:
Pemphigus foliaceus is a rare autoimmune blistering disorder that typically presents with crusted scaly erosions in a seborrheic distribution. We describe a case of pemphigus foliaceus localized to the right cheek of 10 years’ duration that spread to other areas. With a PubMed search of articles indexed for MEDLINE yielding only 14 cases of localized pemphigus foliaceus (Table), it represents an extremely rare entity that often is a diagnostic challenge and may be a harbinger for disseminated disease months to years after the inciting lesion appears.
A 51-year-old woman presented with an asymptomatic cutaneous eruption that had remained localized to the right cheek for 10 years before it increased in size and new lesions developed on the left cheek, chest, and upper back. No inciting factors, such as contactants, insect bites, infections, medications, or recent travel were identified. On physical examination a well-demarcated, hypertrophic, verrucouslike plaque with central pink atrophy and exfoliative scale involved the right malar and submalar regions but spared the mucocutaneous junctions of the face (Figure 1). Subtle dark brown papules, some with overlying scale, speckled the left cheek, right jawline, chest, and upper back. The oral cavity was clear.
Leading differentials included hypertrophic discoid lupus erythematosus and pemphigus vegetans. Other considerations included sarcoidosis, granuloma faciale, lupus vulgaris, disseminated coccidioidomycosis or blastomycosis, and squamous cell carcinoma.
An initial biopsy revealed a lymphocytic lichenoid dermatitis with epidermal hyperplasia and scattered eosinophils for which the following differentials were provided: insect bite, hypertrophic lichen planus, prurigo nodularis superimposed on rosacea, and allergic contact dermatitis. Under these histologic diagnoses, tacrolimus ointment 0.03%, topical mid-potency corticosteroid, and a combination of oral doxycycline and metronidazole gel 1% were prescribed but failed to ameliorate her condition.
Because the clinical differentials were vast and noncorrelative with the original pathology, additional biopsies were performed: one from the edge of the large malar plaque, which was transected for hematoxylin and eosin (H&E) and tissue cultures; one perilesional to the large malar plaque for direct immunofluorescence (DIF); and one from the papule on the right jawline for H&E. Tissue cultures were negative for fungal and mycobacterial organisms. Both specimens submitted for H&E showed the prominent epidermal hyperplasia and lymphocytic dermal infiltrate noted on the original H&E but also demonstrated intragranular acantholysis (Figure 2). The DIF revealed intercellular IgG and C3 deposition throughout the epidermis (Figure 3). Indirect immunofluorescence was negative, but enzyme-linked immunosorbent assay detected circulating antidesmoglein-1 but not antidesmoglein-3 autoantibodies. Other serologies including antinuclear antibody, anti–double-stranded DNA, antihistone, anti–Sjögren syndrome A, and anti–Sjögren syndrome B antibodies were negative.
The diagnosis of localized pemphigus foliaceus was made and management with oral prednisone and mycophenolate mofetil resulted in improvement within weeks.
Localized pemphigus foliaceus is extremely rare with only 14 cases reported in the literature (Table).1-10 Its diagnosis is challenging, as the clinical presentation simulates various entities and the histological features and serological markers are difficult to capture.
Localized pemphigus foliaceus typically presents as an isolated, erythematous, scaly, crusted plaque involving the nose, cheek, or scalp and may mimic several conditions including contact dermatitis, seborrheic dermatitis, rosacea, cutaneous sarcoidosis, discoid lupus erythematosus, lupus vulgaris, impetigo contagiosa, solar keratosis, and nonmelanoma skin cancer.1-10
The predilection for sun-exposed areas suggests UV radiation may induce binding of antidesmoglein-1 autoantibodies with subsequent cytokine-mediated inflammation and acantholysis at these sites.11-13 Similarly, the immunomodulatory agent imiquimod has been reported to induce pemphigus foliaceus at its application sites.6
When pemphigus foliaceus is clinically discernible, the histology and DIF are in accordance with the clinical diagnosis 53.8% of the time.13 In cases of localized pemphigus foliaceus in which the diagnosis is more elusive, many biopsies often are needed to capture the characteristic intragranular acantholysis; this feature often is so subtle that unless the diagnosis is suspected, it is underappreciated or undetectable. In chronic lesions, it may be masked by secondary changes such as acanthosis, hyperkeratosis, and parakeratosis.14
In pemphigus foliaceus, detection of circulating antidesmoglein-1 autoantibodies by enzyme-linked immunosorbent assay is slightly more sensitive and specific compared to indirect immunofluorescence, but both correlate with disease activity.15,16 The low or absent autoantibody titers in localized pemphigus foliaceus may reflect its limited involvement, but dissemination of the disease with subsequent elevation of autoantibody titers may occur months to years after initial presentation,1,2,9 as was the case with our patient.
The majority of localized pemphigus foliaceus cases require systemic prednisone, sometimes in conjunction with nonsteroidal immunosuppressants or topical high-potency corticosteroids.1-3,5,6,8-10 One case was efficaciously managed with tacrolimus ointment 0.1%.7
Localized pemphigus foliaceus is a rare and challenging entity that must be a diagnostic consideration for any chronic focal plaque on the face or scalp, as it may herald disseminated disease.
- Paramsothy Y, Lawrence CM. “Tin-tack” sign in localized pemphigus foliaceus. Br J Dermatol. 1987;116:127-129.
- Newton JA, McGibbon DH, Monk B, et al. Pemphigus foliaceus localized to the nose. Br J Dermatol. 1988;118:303-312.
- Koide M, Kokura N, Takano N. Pemphigus foliaceus localized on the face [in Japanese]. Jpn J Dermatol. 1989;97:1262.
- Yamamoto S, Kanekura T, Gushi A, et al. A case of localized pemphigus foliaceus. J Dermatol. 1996;23:893-895.
- Kishibe M, Kinouchi M, Ishida-Yamamoto A, et al. Pemphigus foliaceus localized to the nose. Clin Exp Dermatol. 2003;28:560-562.
- Lin R, Ladd DJ, Powell DJ, et al. Localized pemphigus foliaceus induced by topical imiquimod treatment. Arch Dermatol. 2004;140:889-890.
- Termeer CC, Technau K, Augustin M, et al. Topical tacrolimus (Protopic) for the treatment of a localized pemphigus foliaceus. J Eur Acad Dermatol Venereol. 2004;18:636-637.
- Zaraa I, El Euch D, Kort R, et al. Localized pemphigus: a report of three cases. Int J Dermatol 2010;49:715-716.
- Ohata C, Akamatsu K, Imai N, et al. Localized pemphigus foliaceus exclusively involving the follicular infundibulum: a novel peau d’orange appearance. Eur J Dermatol. 2011;21:392-395.
- Maderal AD, Miner A, Nousari C, et al. Localized pemphigus foliaceus with unilateral facial involvement. Actas Dermosifiliogr. 2014;105:413-417.
- Cram DL, Winkelmann RK. Ultraviolet-induced acantholysis in pemphigus. Arch Dermatol. 1965;92:7-13.
- Kano Y, Shimosegawa M, Mizukawa Y, et al. Pemphigus foliaceus induced by exposure to sunlight. Dermatology. 2000;201:132-138.
- Lebe B, Gül Nıflıoğlu G, Seyrek S, et al. Evaluation of clinical and histopathologic/direct immunofluorescence diagnosis in autoimmune vesiculobullous dermatitis: utility of direct immunofluorescence. Turk Patoloji Derg. 2012;28:11-16.
- Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol. 2011;29:432-436.
- Ishii K, Amagai M, Hall RP, et al. Characterization of autoantibodies in pemphigus using antigen specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. 1997;159:2010-2017.
- Ng PP, Thng ST, Mohamed K, et al. Comparison of desmoglein ELISA and indirect immunofluorescence using two substrates (monkey esophagus and normal human skin) in the diagnosis of pemphigus. Australas J Dermatol. 2005;46:239-241.
- Paramsothy Y, Lawrence CM. “Tin-tack” sign in localized pemphigus foliaceus. Br J Dermatol. 1987;116:127-129.
- Newton JA, McGibbon DH, Monk B, et al. Pemphigus foliaceus localized to the nose. Br J Dermatol. 1988;118:303-312.
- Koide M, Kokura N, Takano N. Pemphigus foliaceus localized on the face [in Japanese]. Jpn J Dermatol. 1989;97:1262.
- Yamamoto S, Kanekura T, Gushi A, et al. A case of localized pemphigus foliaceus. J Dermatol. 1996;23:893-895.
- Kishibe M, Kinouchi M, Ishida-Yamamoto A, et al. Pemphigus foliaceus localized to the nose. Clin Exp Dermatol. 2003;28:560-562.
- Lin R, Ladd DJ, Powell DJ, et al. Localized pemphigus foliaceus induced by topical imiquimod treatment. Arch Dermatol. 2004;140:889-890.
- Termeer CC, Technau K, Augustin M, et al. Topical tacrolimus (Protopic) for the treatment of a localized pemphigus foliaceus. J Eur Acad Dermatol Venereol. 2004;18:636-637.
- Zaraa I, El Euch D, Kort R, et al. Localized pemphigus: a report of three cases. Int J Dermatol 2010;49:715-716.
- Ohata C, Akamatsu K, Imai N, et al. Localized pemphigus foliaceus exclusively involving the follicular infundibulum: a novel peau d’orange appearance. Eur J Dermatol. 2011;21:392-395.
- Maderal AD, Miner A, Nousari C, et al. Localized pemphigus foliaceus with unilateral facial involvement. Actas Dermosifiliogr. 2014;105:413-417.
- Cram DL, Winkelmann RK. Ultraviolet-induced acantholysis in pemphigus. Arch Dermatol. 1965;92:7-13.
- Kano Y, Shimosegawa M, Mizukawa Y, et al. Pemphigus foliaceus induced by exposure to sunlight. Dermatology. 2000;201:132-138.
- Lebe B, Gül Nıflıoğlu G, Seyrek S, et al. Evaluation of clinical and histopathologic/direct immunofluorescence diagnosis in autoimmune vesiculobullous dermatitis: utility of direct immunofluorescence. Turk Patoloji Derg. 2012;28:11-16.
- Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol. 2011;29:432-436.
- Ishii K, Amagai M, Hall RP, et al. Characterization of autoantibodies in pemphigus using antigen specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. 1997;159:2010-2017.
- Ng PP, Thng ST, Mohamed K, et al. Comparison of desmoglein ELISA and indirect immunofluorescence using two substrates (monkey esophagus and normal human skin) in the diagnosis of pemphigus. Australas J Dermatol. 2005;46:239-241.
Practice Points
- The diagnosis of pemphigus foliceus is challenging, as the clinical presentation simulates various entities.
- Clinicopathological correlation is important. If pathology and other diagnostics do not support clinical findings, trust your clinical assessment and consider repeating or adjusting the workup.
Severe Henoch-Schönlein Purpura Complicating Infliximab Therapy for Ulcerative Colitis
To the Editor:
Anti–tumor necrosis factor (TNF) α treatments have radically improved the management of chronic inflammatory conditions, including rheumatoid arthritis, ankylosing spondylitis, psoriasis and psoriatic arthritis, and bowel diseases (eg, Crohn disease, ulcerative colitis [UC]). Because the number of patients treated with these agents has increased, uncommon adverse reactions have increasingly occurred. Cutaneous adverse reactions that have been reported with anti-TNF agents include immediate injection-site reaction, systemic infusion reactions, and delayed reactions.1 Among the delayed adverse reactions, psoriatic and eczematous eruptions as well as cutaneous infections are the most common, while cutaneous adverse effects related to an immune imbalance syndrome including vasculitis; lupuslike, lichenlike, and granulomatous eruptions; and skin cancer rarely are observed.1 Although most of the cutaneous adverse effects do not require anti-TNF treatment discontinuation and are resolved with symptomatic treatment, anti-TNF therapy must be stopped in more severe cases. We report the case of severe Henoch-Schönlein purpura (HSP) following treatment with infliximab.
A 46-year-old man who was a nonsmoker with quiescent UC on infliximab for 30 months presented with palpable necrotic purpura on both legs (Figure) and arms as well as the abdomen of 10 days’ duration, along with diffuse joint pain and swelling. He had no history of infectious or gastrointestinal symptoms. The last infliximab infusion was performed 6 weeks prior to developing the purpura. His UC was diagnosed 10 years prior to the current presentation and was not associated with any extragastrointestinal manifestations. Since diagnosis, UC had failed to respond to therapies such as azathioprine, cyclosporine, and purinethol. The complete blood cell count was normal. The C-reactive protein level was 18.7 mg/L (reference range, <5 mg/L) and the erythrocyte sedimentation rate was 30 mm/h (reference range, 0–20 mm/h). Electrolytes, urea, creatinine clearance, and liver function were normal, and a chest radiograph and radiographs of the swollen joints were unremarkable. The total IgA level was elevated at 4 g/L (reference range, 0.7–4 g/L), with IgG and IgM levels within reference range. There was no hematuria or proteinuria on urinalysis. Tests for antinuclear antibodies, rheumatoid factor, circulating immune complexes, and antineutrophil cytoplasmic antibody were negative. Total complement, C3, and C4 levels also were normal. A skin biopsy confirmed a leukocytoclastic vasculitis of small vessels with C3 deposition. Serologic tests for hepatitis B virus, hepatitis C virus, and human immunodeficiency virus were negative. Based on these findings, the diagnosis of HSP was made. Systemic corticosteroids—120 mg daily of intravenous methylprednisolone for 3 days, followed by 1 mg/kg daily of oral prednisone for 2 weeks—were then introduced with rapid clinical improvement. Henoch-Schönlein purpura and joint symptoms completely resolved, but UC relapsed with bloody diarrhea and severe abdominal pain. Oral prednisone was maintained (1 mg/kg daily). Because of the severity of cutaneous vasculitis (HSP), a multidisciplinary decision was taken to definitively stop the anti-TNF agents and to first add azathioprine (2 mg/kg daily for 2 months), then subcutaneous methotrexate (25 mg weekly). Colonoscopy did not show any dysplasia or adenocarcinoma and confirmed the diagnosis of UC. After 6 months of combined therapy, UC was still active and we decided to perform a total colectomy with ileostomy formation. Complete remission of UC was obtained and maintained after 28 months of follow-up.
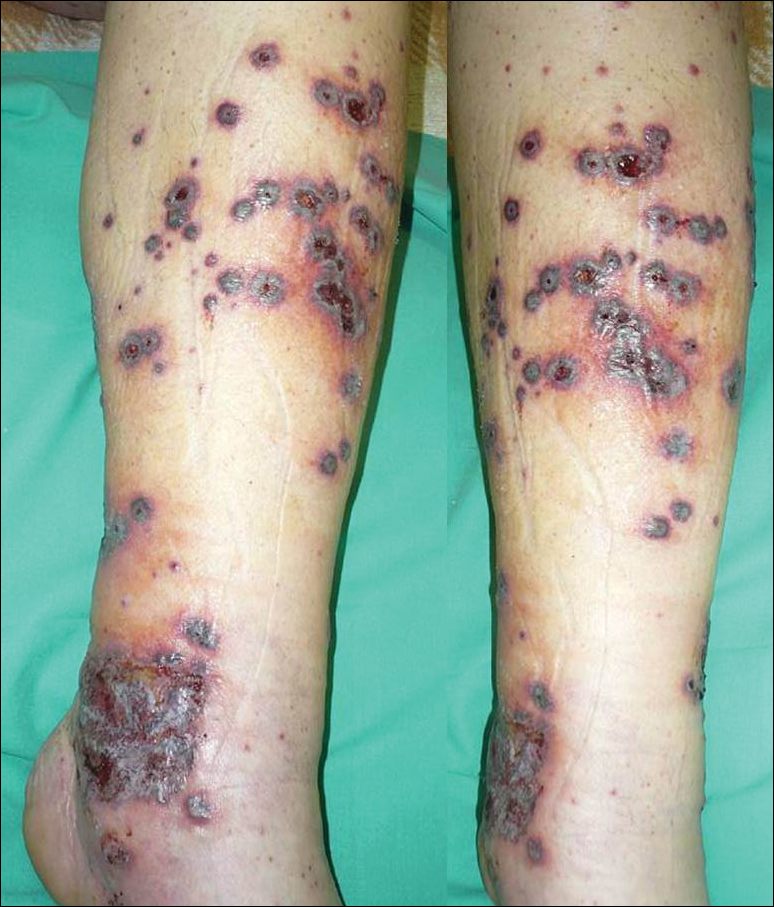
Henoch-Schönlein purpura is a multisystem small vessel leukocytoclastic vasculitis with the deposition of immune complexes containing IgA. Clinical manifestations may include palpable purpura, arthritis, enteritis, and nephritis. Henoch-Schönlein purpura usually affects children. Adult onset is rare but associated with more severe symptoms and a poor prognosis.2 The criteria for HSP, as defined by the American College of Rheumatology,3 include palpable purpura, 20 years or younger at disease onset, bowel angina, and presence of vascular wall granulocytes on biopsy. At least 2 of these criteria are required for HSP diagnosis. Various viral or bacterial infections and drugs can trigger HSP, which also can be associated with autoinflammatory or autoimmune diseases. The association of HSP and UC is a rare event, as demonstrated by de Oliveira et al.4 Although only 2 cases of cutaneous vasculitis mimicking HSP have been described in UC,4 we cannot exclude a possible association between HSP and UC. However, our patient had UC for 10 years and never had clinical manifestations of vasculitis.
There are 5 reports of HSP following etanercept5,6 or adalimumab7-9 therapy and 1 following infliximab therapy.10 In all cases, HSP occurred after several months of anti-TNF therapy. However, there also are reports of cutaneous vasculitis associated with arthralgia and glomerulonephritis that resolved after withdrawal of anti-TNF agents.11,12 It is possible that some of these reactions may have been manifestations of undiagnosed HSP. In a series of 113 patients who developed cutaneous vasculitis after anti-TNF agents, visceral vasculitis was observed in 24% of patients. Treatment of vasculitis involved withdrawal of the anti-TNF therapy in 101 cases (89%).13 In these UC patients with few therapeutic alternatives, the continuation of anti-TNF agents should be discussed. In the previous series,13 of 16 patients who were rechallenged with the same or a different TNF antagonist, 12 (75%) experienced vasculitis relapse, suggesting a class effect of TNF inhibition. Because of the severity of cutaneous vasculitis and as previously suggested in a recent analytical and comprehensive overview on paradoxical reactions under TNF blockers,1 we decided not to re-expose our patient to infliximab or to other anti-TNF agents.
In conclusion, HSP may occur during anti-TNF therapy and physicians need to be aware of this potentially serious complication.
- Toussirot É, Aubin F. Paradoxical reactions under TNF-α blocking agents and other biological agents given for chronic immune-mediated diseases: an analytical and comprehensive overview. RMD Open. 2016;2:e000239.
- Saulsbury FT. Henoch-Schönlein purpura. Curr Opin Rheumatol. 2001;13:35-40.
- Ortiz-Sanjuán F, Blanco R, Hernández JL, et al. Applicability of the 2006 European League Against Rheumatism (EULAR) criteria for the classification ofHenoch-Schönlein purpura. an analysis based on 766 patients with cutaneous vasculitis. Clin Exp Rheumatol. 2015;33(2, suppl 89):S44-S47.
- de Oliveira GT, Martins SS, Deboni M, et al. Cutaneous vasculitis in ulcerative colitis mimicking Henoch-Schönlein purpura [published online May 22, 2012]. J Crohns Colitis. 2013;7:e69-e73.
- Marques I, Lagos A, Reis J, et al. Reversible Henoch-Schönlein purpura complicating adalimumab therapy. J Crohns Colitis. 2012;6:796-799.
- Rahman FZ, Takhar GK, Roy O, et al. Henoch-Schönlein purpura complicating adalimumab therapy for Crohn’s disease. World J Gastrointest Pharmacol Ther. 2010;1:119-122.
- Lee A, Kasama R, Evangelisto A, et al. Henoch-Schönlein purpura after etanercept therapy for psoriasis. J Clin Rheumatol. 2006;12:249-251.
- Duffy TN, Genta M, Moll S, et al. Henoch Schönlein purpura following etanercept treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2006;24(2, suppl 41):S106.
- LaConti JJ, Donet JA, Cho-Vega JH, et al. Henoch-Schönlein purpura with adalimumab therapy for ulcerative colitis: a case report and review of the literature. Case Rep Rheumatol. 2016:2812980.
- Nobile S, Catassi C, Felici L. Herpes zoster infection followed by Henoch-Schönlein purpura in a girl receiving infliximab for ulcerative colitis. J Clin Rheumatol. 2009;15:101.
- Mohan N, Edwards ET, Cupps TR, et al. Leukocytoclastic vasculitis associated with tumor necrosis factor-alpha blocking agents. J Rheumatol. 2004;31:1955-1958.
- Simms R, Kipgen D, Dahill S, et al. ANCA-associated renal vasculitis following anti-tumor necrosis factor alpha therapy. Am J Kidney Dis. 2008;51:e11-e14.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore). 2007;86:242-251.
To the Editor:
Anti–tumor necrosis factor (TNF) α treatments have radically improved the management of chronic inflammatory conditions, including rheumatoid arthritis, ankylosing spondylitis, psoriasis and psoriatic arthritis, and bowel diseases (eg, Crohn disease, ulcerative colitis [UC]). Because the number of patients treated with these agents has increased, uncommon adverse reactions have increasingly occurred. Cutaneous adverse reactions that have been reported with anti-TNF agents include immediate injection-site reaction, systemic infusion reactions, and delayed reactions.1 Among the delayed adverse reactions, psoriatic and eczematous eruptions as well as cutaneous infections are the most common, while cutaneous adverse effects related to an immune imbalance syndrome including vasculitis; lupuslike, lichenlike, and granulomatous eruptions; and skin cancer rarely are observed.1 Although most of the cutaneous adverse effects do not require anti-TNF treatment discontinuation and are resolved with symptomatic treatment, anti-TNF therapy must be stopped in more severe cases. We report the case of severe Henoch-Schönlein purpura (HSP) following treatment with infliximab.
A 46-year-old man who was a nonsmoker with quiescent UC on infliximab for 30 months presented with palpable necrotic purpura on both legs (Figure) and arms as well as the abdomen of 10 days’ duration, along with diffuse joint pain and swelling. He had no history of infectious or gastrointestinal symptoms. The last infliximab infusion was performed 6 weeks prior to developing the purpura. His UC was diagnosed 10 years prior to the current presentation and was not associated with any extragastrointestinal manifestations. Since diagnosis, UC had failed to respond to therapies such as azathioprine, cyclosporine, and purinethol. The complete blood cell count was normal. The C-reactive protein level was 18.7 mg/L (reference range, <5 mg/L) and the erythrocyte sedimentation rate was 30 mm/h (reference range, 0–20 mm/h). Electrolytes, urea, creatinine clearance, and liver function were normal, and a chest radiograph and radiographs of the swollen joints were unremarkable. The total IgA level was elevated at 4 g/L (reference range, 0.7–4 g/L), with IgG and IgM levels within reference range. There was no hematuria or proteinuria on urinalysis. Tests for antinuclear antibodies, rheumatoid factor, circulating immune complexes, and antineutrophil cytoplasmic antibody were negative. Total complement, C3, and C4 levels also were normal. A skin biopsy confirmed a leukocytoclastic vasculitis of small vessels with C3 deposition. Serologic tests for hepatitis B virus, hepatitis C virus, and human immunodeficiency virus were negative. Based on these findings, the diagnosis of HSP was made. Systemic corticosteroids—120 mg daily of intravenous methylprednisolone for 3 days, followed by 1 mg/kg daily of oral prednisone for 2 weeks—were then introduced with rapid clinical improvement. Henoch-Schönlein purpura and joint symptoms completely resolved, but UC relapsed with bloody diarrhea and severe abdominal pain. Oral prednisone was maintained (1 mg/kg daily). Because of the severity of cutaneous vasculitis (HSP), a multidisciplinary decision was taken to definitively stop the anti-TNF agents and to first add azathioprine (2 mg/kg daily for 2 months), then subcutaneous methotrexate (25 mg weekly). Colonoscopy did not show any dysplasia or adenocarcinoma and confirmed the diagnosis of UC. After 6 months of combined therapy, UC was still active and we decided to perform a total colectomy with ileostomy formation. Complete remission of UC was obtained and maintained after 28 months of follow-up.
Henoch-Schönlein purpura is a multisystem small vessel leukocytoclastic vasculitis with the deposition of immune complexes containing IgA. Clinical manifestations may include palpable purpura, arthritis, enteritis, and nephritis. Henoch-Schönlein purpura usually affects children. Adult onset is rare but associated with more severe symptoms and a poor prognosis.2 The criteria for HSP, as defined by the American College of Rheumatology,3 include palpable purpura, 20 years or younger at disease onset, bowel angina, and presence of vascular wall granulocytes on biopsy. At least 2 of these criteria are required for HSP diagnosis. Various viral or bacterial infections and drugs can trigger HSP, which also can be associated with autoinflammatory or autoimmune diseases. The association of HSP and UC is a rare event, as demonstrated by de Oliveira et al.4 Although only 2 cases of cutaneous vasculitis mimicking HSP have been described in UC,4 we cannot exclude a possible association between HSP and UC. However, our patient had UC for 10 years and never had clinical manifestations of vasculitis.
There are 5 reports of HSP following etanercept5,6 or adalimumab7-9 therapy and 1 following infliximab therapy.10 In all cases, HSP occurred after several months of anti-TNF therapy. However, there also are reports of cutaneous vasculitis associated with arthralgia and glomerulonephritis that resolved after withdrawal of anti-TNF agents.11,12 It is possible that some of these reactions may have been manifestations of undiagnosed HSP. In a series of 113 patients who developed cutaneous vasculitis after anti-TNF agents, visceral vasculitis was observed in 24% of patients. Treatment of vasculitis involved withdrawal of the anti-TNF therapy in 101 cases (89%).13 In these UC patients with few therapeutic alternatives, the continuation of anti-TNF agents should be discussed. In the previous series,13 of 16 patients who were rechallenged with the same or a different TNF antagonist, 12 (75%) experienced vasculitis relapse, suggesting a class effect of TNF inhibition. Because of the severity of cutaneous vasculitis and as previously suggested in a recent analytical and comprehensive overview on paradoxical reactions under TNF blockers,1 we decided not to re-expose our patient to infliximab or to other anti-TNF agents.
In conclusion, HSP may occur during anti-TNF therapy and physicians need to be aware of this potentially serious complication.
To the Editor:
Anti–tumor necrosis factor (TNF) α treatments have radically improved the management of chronic inflammatory conditions, including rheumatoid arthritis, ankylosing spondylitis, psoriasis and psoriatic arthritis, and bowel diseases (eg, Crohn disease, ulcerative colitis [UC]). Because the number of patients treated with these agents has increased, uncommon adverse reactions have increasingly occurred. Cutaneous adverse reactions that have been reported with anti-TNF agents include immediate injection-site reaction, systemic infusion reactions, and delayed reactions.1 Among the delayed adverse reactions, psoriatic and eczematous eruptions as well as cutaneous infections are the most common, while cutaneous adverse effects related to an immune imbalance syndrome including vasculitis; lupuslike, lichenlike, and granulomatous eruptions; and skin cancer rarely are observed.1 Although most of the cutaneous adverse effects do not require anti-TNF treatment discontinuation and are resolved with symptomatic treatment, anti-TNF therapy must be stopped in more severe cases. We report the case of severe Henoch-Schönlein purpura (HSP) following treatment with infliximab.
A 46-year-old man who was a nonsmoker with quiescent UC on infliximab for 30 months presented with palpable necrotic purpura on both legs (Figure) and arms as well as the abdomen of 10 days’ duration, along with diffuse joint pain and swelling. He had no history of infectious or gastrointestinal symptoms. The last infliximab infusion was performed 6 weeks prior to developing the purpura. His UC was diagnosed 10 years prior to the current presentation and was not associated with any extragastrointestinal manifestations. Since diagnosis, UC had failed to respond to therapies such as azathioprine, cyclosporine, and purinethol. The complete blood cell count was normal. The C-reactive protein level was 18.7 mg/L (reference range, <5 mg/L) and the erythrocyte sedimentation rate was 30 mm/h (reference range, 0–20 mm/h). Electrolytes, urea, creatinine clearance, and liver function were normal, and a chest radiograph and radiographs of the swollen joints were unremarkable. The total IgA level was elevated at 4 g/L (reference range, 0.7–4 g/L), with IgG and IgM levels within reference range. There was no hematuria or proteinuria on urinalysis. Tests for antinuclear antibodies, rheumatoid factor, circulating immune complexes, and antineutrophil cytoplasmic antibody were negative. Total complement, C3, and C4 levels also were normal. A skin biopsy confirmed a leukocytoclastic vasculitis of small vessels with C3 deposition. Serologic tests for hepatitis B virus, hepatitis C virus, and human immunodeficiency virus were negative. Based on these findings, the diagnosis of HSP was made. Systemic corticosteroids—120 mg daily of intravenous methylprednisolone for 3 days, followed by 1 mg/kg daily of oral prednisone for 2 weeks—were then introduced with rapid clinical improvement. Henoch-Schönlein purpura and joint symptoms completely resolved, but UC relapsed with bloody diarrhea and severe abdominal pain. Oral prednisone was maintained (1 mg/kg daily). Because of the severity of cutaneous vasculitis (HSP), a multidisciplinary decision was taken to definitively stop the anti-TNF agents and to first add azathioprine (2 mg/kg daily for 2 months), then subcutaneous methotrexate (25 mg weekly). Colonoscopy did not show any dysplasia or adenocarcinoma and confirmed the diagnosis of UC. After 6 months of combined therapy, UC was still active and we decided to perform a total colectomy with ileostomy formation. Complete remission of UC was obtained and maintained after 28 months of follow-up.
Henoch-Schönlein purpura is a multisystem small vessel leukocytoclastic vasculitis with the deposition of immune complexes containing IgA. Clinical manifestations may include palpable purpura, arthritis, enteritis, and nephritis. Henoch-Schönlein purpura usually affects children. Adult onset is rare but associated with more severe symptoms and a poor prognosis.2 The criteria for HSP, as defined by the American College of Rheumatology,3 include palpable purpura, 20 years or younger at disease onset, bowel angina, and presence of vascular wall granulocytes on biopsy. At least 2 of these criteria are required for HSP diagnosis. Various viral or bacterial infections and drugs can trigger HSP, which also can be associated with autoinflammatory or autoimmune diseases. The association of HSP and UC is a rare event, as demonstrated by de Oliveira et al.4 Although only 2 cases of cutaneous vasculitis mimicking HSP have been described in UC,4 we cannot exclude a possible association between HSP and UC. However, our patient had UC for 10 years and never had clinical manifestations of vasculitis.
There are 5 reports of HSP following etanercept5,6 or adalimumab7-9 therapy and 1 following infliximab therapy.10 In all cases, HSP occurred after several months of anti-TNF therapy. However, there also are reports of cutaneous vasculitis associated with arthralgia and glomerulonephritis that resolved after withdrawal of anti-TNF agents.11,12 It is possible that some of these reactions may have been manifestations of undiagnosed HSP. In a series of 113 patients who developed cutaneous vasculitis after anti-TNF agents, visceral vasculitis was observed in 24% of patients. Treatment of vasculitis involved withdrawal of the anti-TNF therapy in 101 cases (89%).13 In these UC patients with few therapeutic alternatives, the continuation of anti-TNF agents should be discussed. In the previous series,13 of 16 patients who were rechallenged with the same or a different TNF antagonist, 12 (75%) experienced vasculitis relapse, suggesting a class effect of TNF inhibition. Because of the severity of cutaneous vasculitis and as previously suggested in a recent analytical and comprehensive overview on paradoxical reactions under TNF blockers,1 we decided not to re-expose our patient to infliximab or to other anti-TNF agents.
In conclusion, HSP may occur during anti-TNF therapy and physicians need to be aware of this potentially serious complication.
- Toussirot É, Aubin F. Paradoxical reactions under TNF-α blocking agents and other biological agents given for chronic immune-mediated diseases: an analytical and comprehensive overview. RMD Open. 2016;2:e000239.
- Saulsbury FT. Henoch-Schönlein purpura. Curr Opin Rheumatol. 2001;13:35-40.
- Ortiz-Sanjuán F, Blanco R, Hernández JL, et al. Applicability of the 2006 European League Against Rheumatism (EULAR) criteria for the classification ofHenoch-Schönlein purpura. an analysis based on 766 patients with cutaneous vasculitis. Clin Exp Rheumatol. 2015;33(2, suppl 89):S44-S47.
- de Oliveira GT, Martins SS, Deboni M, et al. Cutaneous vasculitis in ulcerative colitis mimicking Henoch-Schönlein purpura [published online May 22, 2012]. J Crohns Colitis. 2013;7:e69-e73.
- Marques I, Lagos A, Reis J, et al. Reversible Henoch-Schönlein purpura complicating adalimumab therapy. J Crohns Colitis. 2012;6:796-799.
- Rahman FZ, Takhar GK, Roy O, et al. Henoch-Schönlein purpura complicating adalimumab therapy for Crohn’s disease. World J Gastrointest Pharmacol Ther. 2010;1:119-122.
- Lee A, Kasama R, Evangelisto A, et al. Henoch-Schönlein purpura after etanercept therapy for psoriasis. J Clin Rheumatol. 2006;12:249-251.
- Duffy TN, Genta M, Moll S, et al. Henoch Schönlein purpura following etanercept treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2006;24(2, suppl 41):S106.
- LaConti JJ, Donet JA, Cho-Vega JH, et al. Henoch-Schönlein purpura with adalimumab therapy for ulcerative colitis: a case report and review of the literature. Case Rep Rheumatol. 2016:2812980.
- Nobile S, Catassi C, Felici L. Herpes zoster infection followed by Henoch-Schönlein purpura in a girl receiving infliximab for ulcerative colitis. J Clin Rheumatol. 2009;15:101.
- Mohan N, Edwards ET, Cupps TR, et al. Leukocytoclastic vasculitis associated with tumor necrosis factor-alpha blocking agents. J Rheumatol. 2004;31:1955-1958.
- Simms R, Kipgen D, Dahill S, et al. ANCA-associated renal vasculitis following anti-tumor necrosis factor alpha therapy. Am J Kidney Dis. 2008;51:e11-e14.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore). 2007;86:242-251.
- Toussirot É, Aubin F. Paradoxical reactions under TNF-α blocking agents and other biological agents given for chronic immune-mediated diseases: an analytical and comprehensive overview. RMD Open. 2016;2:e000239.
- Saulsbury FT. Henoch-Schönlein purpura. Curr Opin Rheumatol. 2001;13:35-40.
- Ortiz-Sanjuán F, Blanco R, Hernández JL, et al. Applicability of the 2006 European League Against Rheumatism (EULAR) criteria for the classification ofHenoch-Schönlein purpura. an analysis based on 766 patients with cutaneous vasculitis. Clin Exp Rheumatol. 2015;33(2, suppl 89):S44-S47.
- de Oliveira GT, Martins SS, Deboni M, et al. Cutaneous vasculitis in ulcerative colitis mimicking Henoch-Schönlein purpura [published online May 22, 2012]. J Crohns Colitis. 2013;7:e69-e73.
- Marques I, Lagos A, Reis J, et al. Reversible Henoch-Schönlein purpura complicating adalimumab therapy. J Crohns Colitis. 2012;6:796-799.
- Rahman FZ, Takhar GK, Roy O, et al. Henoch-Schönlein purpura complicating adalimumab therapy for Crohn’s disease. World J Gastrointest Pharmacol Ther. 2010;1:119-122.
- Lee A, Kasama R, Evangelisto A, et al. Henoch-Schönlein purpura after etanercept therapy for psoriasis. J Clin Rheumatol. 2006;12:249-251.
- Duffy TN, Genta M, Moll S, et al. Henoch Schönlein purpura following etanercept treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2006;24(2, suppl 41):S106.
- LaConti JJ, Donet JA, Cho-Vega JH, et al. Henoch-Schönlein purpura with adalimumab therapy for ulcerative colitis: a case report and review of the literature. Case Rep Rheumatol. 2016:2812980.
- Nobile S, Catassi C, Felici L. Herpes zoster infection followed by Henoch-Schönlein purpura in a girl receiving infliximab for ulcerative colitis. J Clin Rheumatol. 2009;15:101.
- Mohan N, Edwards ET, Cupps TR, et al. Leukocytoclastic vasculitis associated with tumor necrosis factor-alpha blocking agents. J Rheumatol. 2004;31:1955-1958.
- Simms R, Kipgen D, Dahill S, et al. ANCA-associated renal vasculitis following anti-tumor necrosis factor alpha therapy. Am J Kidney Dis. 2008;51:e11-e14.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore). 2007;86:242-251.
Practice Points
- Cutaneous adverse effects may occur in approximately 20% of patients treated with anti–tumor necrosis factor (TNF) drugs.
- Henoch-Schönlein purpura (HSP), a small-vessel vasculitis, is an extremely rare complication of anti-TNF treatment.
- Although most cutaneous adverse effects do not require anti-TNF treatment discontinuation and are resolved with symptomatic treatment, anti-TNF therapy must be stopped in more severe cases.