User login
Oral Fixed Drug Eruption Due to Tinidazole
To the Editor:
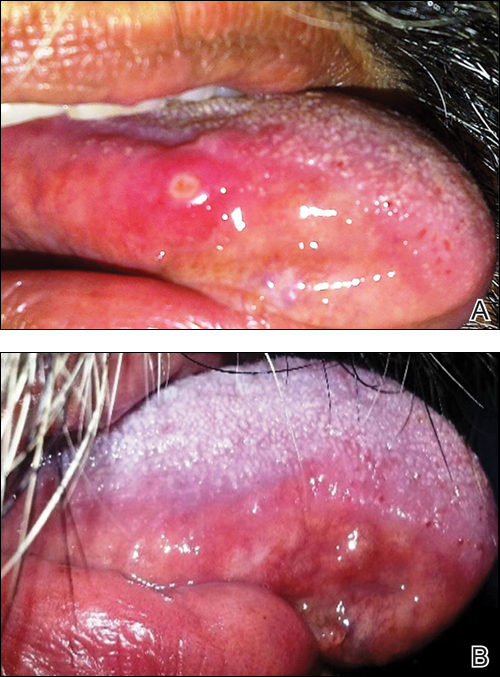
A 50-year-old man presented with a painful ulcer and a burning sensation on the tongue of 2 days’ duration (Figure, A). The ulcer had a yellowish white appearance with erythematous borders. The patient started taking tinidazole 500 mg twice daily 2 days prior, which was prescribed by his primary care physician for an episode of gastroenteritis. He was not taking any other medications and did not smoke or drink. Routine laboratory test results did not reveal any abnormalities. Based on the physical examination as well as the patient’s medical and medication history, a provisional diagnosis of fixed drug eruption (FDE) due to tinidazole was made. Tinidazole was immediately withdrawn and the patient was prescribed beclomethasone dipropionate ointment twice daily to relieve the burning sensation. A punch biopsy of the lesion was recommended; however, the patient opted to wait a week after discontinuing the drug. At follow-up 1 week later, complete healing of the ulcer was observed with no scarring and the burning sensation had resolved (Figure, B). After obtaining informed consent from the patient, an oral challenge test was conducted in the office with 50 mg of tinidazole. Four hours after taking the drug orally, the patient felt a burning sensation and a small ulcerative lesion was observed on the tongue at the same site the next day. The patient was informed of the fixed drug reaction to tinidazole, a drug belonging to the nitroimidazole group, and this information also was conveyed to the patient’s primary care physician.
Tinidazole is a synthetic antiprotozoal and antibacterial agent used primarily in infections such as amebiasis, giardiasis, and trichomoniasis.1 Tinidazole may be a therapeutic alternative to metronidazole. Fixed drug eruption is a distinctive variant of drug eruption with characteristic recurrence at the same site of skin or mucous membranes. It is characterized by onset of round/oval, erythematous, well-defined macules on the skin and/or mucosa associated with itching and burning.1 Fixed drug eruption generally is restricted to the mucous membrane and skin, with the lips, palms, soles, glans penis, and groin area being the most common sites. Intraoral involvement, excluding the lips, of FDE is rare. The tongue is a rare site of an FDE.2 Fixed drug eruption on the tongue has been reported with clarithromycin.3 Dental clinicians have to be aware of the possibility of FDE due to commonly used drugs such tinidazole, which would help in prompt diagnosis of these lesions.
- Prieto A, De Barrio M, Infante S, et al. Recurrent fixed drug eruption due to metronidazole elicited by patch test with tinidazole. Contact Dermatitis. 2005;53:169-170.
- Dhar S, Kanwar AJ. Fixed drug eruption on the tongue of a 4-year-old boy. Pediatr Dermatol. 1995;12:51-52.
- Alonso JC, Melgosa AC, Gonzalo MJ, et al. Fixed drug eruption on the tongue due to clarithromycin. Contact Dermatitis. 2005;53:121-122.
To the Editor:
A 50-year-old man presented with a painful ulcer and a burning sensation on the tongue of 2 days’ duration (Figure, A). The ulcer had a yellowish white appearance with erythematous borders. The patient started taking tinidazole 500 mg twice daily 2 days prior, which was prescribed by his primary care physician for an episode of gastroenteritis. He was not taking any other medications and did not smoke or drink. Routine laboratory test results did not reveal any abnormalities. Based on the physical examination as well as the patient’s medical and medication history, a provisional diagnosis of fixed drug eruption (FDE) due to tinidazole was made. Tinidazole was immediately withdrawn and the patient was prescribed beclomethasone dipropionate ointment twice daily to relieve the burning sensation. A punch biopsy of the lesion was recommended; however, the patient opted to wait a week after discontinuing the drug. At follow-up 1 week later, complete healing of the ulcer was observed with no scarring and the burning sensation had resolved (Figure, B). After obtaining informed consent from the patient, an oral challenge test was conducted in the office with 50 mg of tinidazole. Four hours after taking the drug orally, the patient felt a burning sensation and a small ulcerative lesion was observed on the tongue at the same site the next day. The patient was informed of the fixed drug reaction to tinidazole, a drug belonging to the nitroimidazole group, and this information also was conveyed to the patient’s primary care physician.
Tinidazole is a synthetic antiprotozoal and antibacterial agent used primarily in infections such as amebiasis, giardiasis, and trichomoniasis.1 Tinidazole may be a therapeutic alternative to metronidazole. Fixed drug eruption is a distinctive variant of drug eruption with characteristic recurrence at the same site of skin or mucous membranes. It is characterized by onset of round/oval, erythematous, well-defined macules on the skin and/or mucosa associated with itching and burning.1 Fixed drug eruption generally is restricted to the mucous membrane and skin, with the lips, palms, soles, glans penis, and groin area being the most common sites. Intraoral involvement, excluding the lips, of FDE is rare. The tongue is a rare site of an FDE.2 Fixed drug eruption on the tongue has been reported with clarithromycin.3 Dental clinicians have to be aware of the possibility of FDE due to commonly used drugs such tinidazole, which would help in prompt diagnosis of these lesions.
To the Editor:
A 50-year-old man presented with a painful ulcer and a burning sensation on the tongue of 2 days’ duration (Figure, A). The ulcer had a yellowish white appearance with erythematous borders. The patient started taking tinidazole 500 mg twice daily 2 days prior, which was prescribed by his primary care physician for an episode of gastroenteritis. He was not taking any other medications and did not smoke or drink. Routine laboratory test results did not reveal any abnormalities. Based on the physical examination as well as the patient’s medical and medication history, a provisional diagnosis of fixed drug eruption (FDE) due to tinidazole was made. Tinidazole was immediately withdrawn and the patient was prescribed beclomethasone dipropionate ointment twice daily to relieve the burning sensation. A punch biopsy of the lesion was recommended; however, the patient opted to wait a week after discontinuing the drug. At follow-up 1 week later, complete healing of the ulcer was observed with no scarring and the burning sensation had resolved (Figure, B). After obtaining informed consent from the patient, an oral challenge test was conducted in the office with 50 mg of tinidazole. Four hours after taking the drug orally, the patient felt a burning sensation and a small ulcerative lesion was observed on the tongue at the same site the next day. The patient was informed of the fixed drug reaction to tinidazole, a drug belonging to the nitroimidazole group, and this information also was conveyed to the patient’s primary care physician.
Tinidazole is a synthetic antiprotozoal and antibacterial agent used primarily in infections such as amebiasis, giardiasis, and trichomoniasis.1 Tinidazole may be a therapeutic alternative to metronidazole. Fixed drug eruption is a distinctive variant of drug eruption with characteristic recurrence at the same site of skin or mucous membranes. It is characterized by onset of round/oval, erythematous, well-defined macules on the skin and/or mucosa associated with itching and burning.1 Fixed drug eruption generally is restricted to the mucous membrane and skin, with the lips, palms, soles, glans penis, and groin area being the most common sites. Intraoral involvement, excluding the lips, of FDE is rare. The tongue is a rare site of an FDE.2 Fixed drug eruption on the tongue has been reported with clarithromycin.3 Dental clinicians have to be aware of the possibility of FDE due to commonly used drugs such tinidazole, which would help in prompt diagnosis of these lesions.
- Prieto A, De Barrio M, Infante S, et al. Recurrent fixed drug eruption due to metronidazole elicited by patch test with tinidazole. Contact Dermatitis. 2005;53:169-170.
- Dhar S, Kanwar AJ. Fixed drug eruption on the tongue of a 4-year-old boy. Pediatr Dermatol. 1995;12:51-52.
- Alonso JC, Melgosa AC, Gonzalo MJ, et al. Fixed drug eruption on the tongue due to clarithromycin. Contact Dermatitis. 2005;53:121-122.
- Prieto A, De Barrio M, Infante S, et al. Recurrent fixed drug eruption due to metronidazole elicited by patch test with tinidazole. Contact Dermatitis. 2005;53:169-170.
- Dhar S, Kanwar AJ. Fixed drug eruption on the tongue of a 4-year-old boy. Pediatr Dermatol. 1995;12:51-52.
- Alonso JC, Melgosa AC, Gonzalo MJ, et al. Fixed drug eruption on the tongue due to clarithromycin. Contact Dermatitis. 2005;53:121-122.
Practice Points
- Fixed drug eruption (FDE) is characterized by onset of round/oval, erythematous, well-defined macules on the skin and/or mucosa associated with itching and burning.
- Intraoral involvement of FDE is rare.
- Tinidazole may cause FDE and should be suspected in patients with a spontaneous eruption of macules on mucous membranes.
Autoimmune Progesterone Dermatitis Presenting With Purpura
To the Editor:
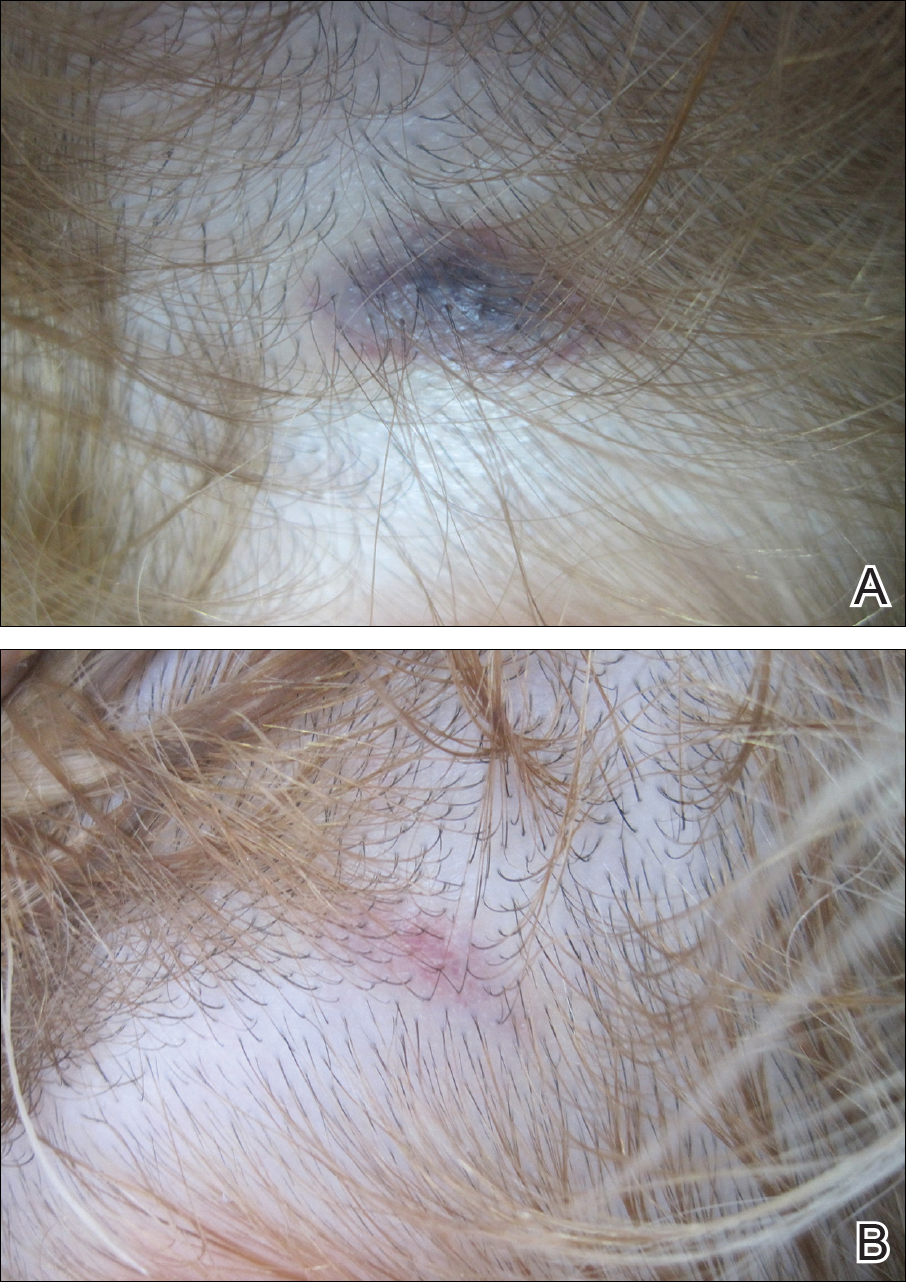
A 32-year-old woman presented with a recurrent painful eruption on the scalp of 1 year's duration. The lesion occurred on the left temporal region 1 week prior to menstruation and spontaneously resolved following menses; it recurred every month for 1 year. She had no notable medical history. She had taken oral contraceptive pills for 4 years and stopped 2 years prior to the development of the lesions. Dermatologic examination revealed a purple-colored, violaceous, centrally elevated, painful plaque that measured 2 cm in diameter in the left temporal region of the scalp (Figure, A). Laboratory test results were within reference range. The lesion spontaneously resolved with mild residual erythema at a follow-up visit after menstruation (Figure, B).
Because the eruption occurred and relapsed with the patient's menstrual cycle, we suspected progesterone hypersensitivity. An intradermal skin test was performed on the forearm with 0.05 mL of medroxyprogesterone acetate, and saline was used as a negative control. An indurated erythematous nodule occurred on the progesterone-treated side within 6 hours. Based on these findings and the patient's history, she was diagnosed with autoimmune progesterone dermatitis (APD). We recommended her to use gonadotropin-releasing hormone agonists as treatment, but the patient refused. At 6-month follow-up she had recurrent lesions but did not report any concerns.
Autoimmune progesterone dermatitis is a rare condition that is characterized by cyclical skin eruptions, typically occurring in the luteal phase of the menstrual cycle with spontaneous resolution after menses.1,2 It was first described by Geber3 in a patient with cyclical urticarial lesions. In 1964, Shelley et al4 characterized APD in a 27-year-old woman with a pruritic vesicular eruption with cyclical premenstrual exacerbations. Although it is believed there is no genetic predisposition to APD, a case series involving 3 sisters demonstrated that genetic susceptibility might play a role in the etiology.5 The etiology of APD is still unknown. It is thought to represent an autoimmune reaction to endogenous or exogenous progesterone.1 Our patient also had used oral contraceptives for 4 years and this exogenous progesterone might have played a role in the sensitization of the patient and the development of this autoimmune reaction.
The clinical features of APD usually begin 3 to 10 days prior to menstruation and end 1 to 2 days after menses. Autoimmune progesterone dermatitis can present in a variety of forms including eczema, erythema multiforme, erythema annulare centrifugum, fixed drug eruption, stomatitis, folliculitis, urticaria, and angioedema.6 A case of APD presenting with petechiae and purpura has been reported.7 There are no specific histologic findings for APD.8 Demonstration of progesterone sensitivity with a progesterone challenge test is the mainstay of diagnosis. Immediate urticaria may occur in some patients, with others experiencing a delayed reaction peaking at 24 to 96 hours.9 The main criteria of APD include the following: recurrent cyclic lesions related to the menstrual cycle; positive intradermal progesterone skin test; and prevention of lesions by inhibiting ovulation.1 Two of these criteria were positive in our patient, but we did not use any medications to prevent ovulation at the patient's request.
Current treatment modalities often attempt to inhibit the secretion of endogenous progesterone by suppressing ovulation. Oral contraceptives and conjugated estrogens have limited efficacy rates.8 Gonadotropin-releasing hormone agonists (ie, buserelin, triptorelin) have been used with success.1,6 Tamoxifen and danazol are other treatment options. For cases refractory to medical treatments, bilateral oophorectomy can be considered a definitive treatment.6
Autoimmune progesterone dermatitis may present in many different clinical forms. It should be considered in the differential diagnosis in patients with recurrent skin lesions related to menstrual cycle both in women of childbearing age and in men taking synthetic progesterone.
- Lee MK, Lee WY, Yong SJ, et al. A case of autoimmune progesterone dermatitis misdiagnosed as allergic contact dermatitis. Allergy Asthma Immunol Res. 2011;3:141-144.
- García-Ortega P, Scorza E. Progesterone autoimmune dermatitis with positive autologous serum skin test result. Obstet Gynecol. 2011;117:495-498.
- Geber J. Desensitization in the treatment of menstrual intoxication and other allergic symptoms. Br J Dermatol. 1930;51:265-268.
- Shelley WB, Preucel RW, Spoont SS. Autoimmune progesterone dermatitis: cure by oophorectomy. JAMA. 1964;190:35-38.
- Chawla SV, Quirk C, Sondheimer SJ, et al. Autoimmune progesterone dermatitis. Arch Dermatol. 2009;145:341-342.
- Medeiros S, Rodrigues-Alves R, Costa M, et al. Autoimmune progesterone dermatitis: treatment with oophorectomy. Clin Exp Dermatol. 2010;35:e12-e13.
- Wintzen M, Goor-van Egmond MB, Noz KC. Autoimmune progesterone dermatitis presenting with purpura and petechiae. Clin Exp Dermatol. 2004;29:316.
- Baptist AP, Baldwin JL. Autoimmune progesterone dermatitis in a patient with endometriosis: case report and review of the literature. Clin Mol Allergy. 2004;2:10.
- Le K, Wood G. A case of autoimmune progesterone dermatitis diagnosed by progesterone pessary. Australas J Dermatol. 2011;52:139-141.
To the Editor:
A 32-year-old woman presented with a recurrent painful eruption on the scalp of 1 year's duration. The lesion occurred on the left temporal region 1 week prior to menstruation and spontaneously resolved following menses; it recurred every month for 1 year. She had no notable medical history. She had taken oral contraceptive pills for 4 years and stopped 2 years prior to the development of the lesions. Dermatologic examination revealed a purple-colored, violaceous, centrally elevated, painful plaque that measured 2 cm in diameter in the left temporal region of the scalp (Figure, A). Laboratory test results were within reference range. The lesion spontaneously resolved with mild residual erythema at a follow-up visit after menstruation (Figure, B).
Because the eruption occurred and relapsed with the patient's menstrual cycle, we suspected progesterone hypersensitivity. An intradermal skin test was performed on the forearm with 0.05 mL of medroxyprogesterone acetate, and saline was used as a negative control. An indurated erythematous nodule occurred on the progesterone-treated side within 6 hours. Based on these findings and the patient's history, she was diagnosed with autoimmune progesterone dermatitis (APD). We recommended her to use gonadotropin-releasing hormone agonists as treatment, but the patient refused. At 6-month follow-up she had recurrent lesions but did not report any concerns.
Autoimmune progesterone dermatitis is a rare condition that is characterized by cyclical skin eruptions, typically occurring in the luteal phase of the menstrual cycle with spontaneous resolution after menses.1,2 It was first described by Geber3 in a patient with cyclical urticarial lesions. In 1964, Shelley et al4 characterized APD in a 27-year-old woman with a pruritic vesicular eruption with cyclical premenstrual exacerbations. Although it is believed there is no genetic predisposition to APD, a case series involving 3 sisters demonstrated that genetic susceptibility might play a role in the etiology.5 The etiology of APD is still unknown. It is thought to represent an autoimmune reaction to endogenous or exogenous progesterone.1 Our patient also had used oral contraceptives for 4 years and this exogenous progesterone might have played a role in the sensitization of the patient and the development of this autoimmune reaction.
The clinical features of APD usually begin 3 to 10 days prior to menstruation and end 1 to 2 days after menses. Autoimmune progesterone dermatitis can present in a variety of forms including eczema, erythema multiforme, erythema annulare centrifugum, fixed drug eruption, stomatitis, folliculitis, urticaria, and angioedema.6 A case of APD presenting with petechiae and purpura has been reported.7 There are no specific histologic findings for APD.8 Demonstration of progesterone sensitivity with a progesterone challenge test is the mainstay of diagnosis. Immediate urticaria may occur in some patients, with others experiencing a delayed reaction peaking at 24 to 96 hours.9 The main criteria of APD include the following: recurrent cyclic lesions related to the menstrual cycle; positive intradermal progesterone skin test; and prevention of lesions by inhibiting ovulation.1 Two of these criteria were positive in our patient, but we did not use any medications to prevent ovulation at the patient's request.
Current treatment modalities often attempt to inhibit the secretion of endogenous progesterone by suppressing ovulation. Oral contraceptives and conjugated estrogens have limited efficacy rates.8 Gonadotropin-releasing hormone agonists (ie, buserelin, triptorelin) have been used with success.1,6 Tamoxifen and danazol are other treatment options. For cases refractory to medical treatments, bilateral oophorectomy can be considered a definitive treatment.6
Autoimmune progesterone dermatitis may present in many different clinical forms. It should be considered in the differential diagnosis in patients with recurrent skin lesions related to menstrual cycle both in women of childbearing age and in men taking synthetic progesterone.
To the Editor:
A 32-year-old woman presented with a recurrent painful eruption on the scalp of 1 year's duration. The lesion occurred on the left temporal region 1 week prior to menstruation and spontaneously resolved following menses; it recurred every month for 1 year. She had no notable medical history. She had taken oral contraceptive pills for 4 years and stopped 2 years prior to the development of the lesions. Dermatologic examination revealed a purple-colored, violaceous, centrally elevated, painful plaque that measured 2 cm in diameter in the left temporal region of the scalp (Figure, A). Laboratory test results were within reference range. The lesion spontaneously resolved with mild residual erythema at a follow-up visit after menstruation (Figure, B).
Because the eruption occurred and relapsed with the patient's menstrual cycle, we suspected progesterone hypersensitivity. An intradermal skin test was performed on the forearm with 0.05 mL of medroxyprogesterone acetate, and saline was used as a negative control. An indurated erythematous nodule occurred on the progesterone-treated side within 6 hours. Based on these findings and the patient's history, she was diagnosed with autoimmune progesterone dermatitis (APD). We recommended her to use gonadotropin-releasing hormone agonists as treatment, but the patient refused. At 6-month follow-up she had recurrent lesions but did not report any concerns.
Autoimmune progesterone dermatitis is a rare condition that is characterized by cyclical skin eruptions, typically occurring in the luteal phase of the menstrual cycle with spontaneous resolution after menses.1,2 It was first described by Geber3 in a patient with cyclical urticarial lesions. In 1964, Shelley et al4 characterized APD in a 27-year-old woman with a pruritic vesicular eruption with cyclical premenstrual exacerbations. Although it is believed there is no genetic predisposition to APD, a case series involving 3 sisters demonstrated that genetic susceptibility might play a role in the etiology.5 The etiology of APD is still unknown. It is thought to represent an autoimmune reaction to endogenous or exogenous progesterone.1 Our patient also had used oral contraceptives for 4 years and this exogenous progesterone might have played a role in the sensitization of the patient and the development of this autoimmune reaction.
The clinical features of APD usually begin 3 to 10 days prior to menstruation and end 1 to 2 days after menses. Autoimmune progesterone dermatitis can present in a variety of forms including eczema, erythema multiforme, erythema annulare centrifugum, fixed drug eruption, stomatitis, folliculitis, urticaria, and angioedema.6 A case of APD presenting with petechiae and purpura has been reported.7 There are no specific histologic findings for APD.8 Demonstration of progesterone sensitivity with a progesterone challenge test is the mainstay of diagnosis. Immediate urticaria may occur in some patients, with others experiencing a delayed reaction peaking at 24 to 96 hours.9 The main criteria of APD include the following: recurrent cyclic lesions related to the menstrual cycle; positive intradermal progesterone skin test; and prevention of lesions by inhibiting ovulation.1 Two of these criteria were positive in our patient, but we did not use any medications to prevent ovulation at the patient's request.
Current treatment modalities often attempt to inhibit the secretion of endogenous progesterone by suppressing ovulation. Oral contraceptives and conjugated estrogens have limited efficacy rates.8 Gonadotropin-releasing hormone agonists (ie, buserelin, triptorelin) have been used with success.1,6 Tamoxifen and danazol are other treatment options. For cases refractory to medical treatments, bilateral oophorectomy can be considered a definitive treatment.6
Autoimmune progesterone dermatitis may present in many different clinical forms. It should be considered in the differential diagnosis in patients with recurrent skin lesions related to menstrual cycle both in women of childbearing age and in men taking synthetic progesterone.
- Lee MK, Lee WY, Yong SJ, et al. A case of autoimmune progesterone dermatitis misdiagnosed as allergic contact dermatitis. Allergy Asthma Immunol Res. 2011;3:141-144.
- García-Ortega P, Scorza E. Progesterone autoimmune dermatitis with positive autologous serum skin test result. Obstet Gynecol. 2011;117:495-498.
- Geber J. Desensitization in the treatment of menstrual intoxication and other allergic symptoms. Br J Dermatol. 1930;51:265-268.
- Shelley WB, Preucel RW, Spoont SS. Autoimmune progesterone dermatitis: cure by oophorectomy. JAMA. 1964;190:35-38.
- Chawla SV, Quirk C, Sondheimer SJ, et al. Autoimmune progesterone dermatitis. Arch Dermatol. 2009;145:341-342.
- Medeiros S, Rodrigues-Alves R, Costa M, et al. Autoimmune progesterone dermatitis: treatment with oophorectomy. Clin Exp Dermatol. 2010;35:e12-e13.
- Wintzen M, Goor-van Egmond MB, Noz KC. Autoimmune progesterone dermatitis presenting with purpura and petechiae. Clin Exp Dermatol. 2004;29:316.
- Baptist AP, Baldwin JL. Autoimmune progesterone dermatitis in a patient with endometriosis: case report and review of the literature. Clin Mol Allergy. 2004;2:10.
- Le K, Wood G. A case of autoimmune progesterone dermatitis diagnosed by progesterone pessary. Australas J Dermatol. 2011;52:139-141.
- Lee MK, Lee WY, Yong SJ, et al. A case of autoimmune progesterone dermatitis misdiagnosed as allergic contact dermatitis. Allergy Asthma Immunol Res. 2011;3:141-144.
- García-Ortega P, Scorza E. Progesterone autoimmune dermatitis with positive autologous serum skin test result. Obstet Gynecol. 2011;117:495-498.
- Geber J. Desensitization in the treatment of menstrual intoxication and other allergic symptoms. Br J Dermatol. 1930;51:265-268.
- Shelley WB, Preucel RW, Spoont SS. Autoimmune progesterone dermatitis: cure by oophorectomy. JAMA. 1964;190:35-38.
- Chawla SV, Quirk C, Sondheimer SJ, et al. Autoimmune progesterone dermatitis. Arch Dermatol. 2009;145:341-342.
- Medeiros S, Rodrigues-Alves R, Costa M, et al. Autoimmune progesterone dermatitis: treatment with oophorectomy. Clin Exp Dermatol. 2010;35:e12-e13.
- Wintzen M, Goor-van Egmond MB, Noz KC. Autoimmune progesterone dermatitis presenting with purpura and petechiae. Clin Exp Dermatol. 2004;29:316.
- Baptist AP, Baldwin JL. Autoimmune progesterone dermatitis in a patient with endometriosis: case report and review of the literature. Clin Mol Allergy. 2004;2:10.
- Le K, Wood G. A case of autoimmune progesterone dermatitis diagnosed by progesterone pessary. Australas J Dermatol. 2011;52:139-141.
Practice Points
- Autoimmune progesterone dermatitis is characterized by cyclical skin eruptions, typically occurring in the second half of the menstrual cycle.
- Autoimmune progesterone dermatitis is thought to be an autoimmune reaction to endogenous or exogenous progesterone.
- This condition should be considered in female patients with recurrent skin lesions related to their menstrual cycle.
Acute Localized Exanthematous Pustulosis Caused by Flurbiprofen
To the Editor:
Acute generalized exanthematous pustulosis (AGEP) is an acute skin reaction that is characterized by generalized, nonfollicular, pinhead-sized, sterile pustules on an erythematous and edematous background. The eruption can be accompanied by fever and neutrophilic leukocytosis. Skin symptoms arise quickly (within a few hours), most commonly following drug administration. The medications most frequently responsible are beta-lactam antibiotics, macrolides, calcium channel blockers, and antimalarials. Pustules spontaneously resolve in 15 days and generalized desquamation occurs approximately 2 weeks later. The estimated incidence rate of AGEP is approximately 1 to 5 cases per million per year. Acute localized exanthematous pustulosis (ALEP) is a less common form of AGEP. We report a case of ALEP localized on the face that was caused by flurbiprofen, a propionic acid derivative from the family of nonsteroidal anti-inflammatory drugs (NSAIDs).
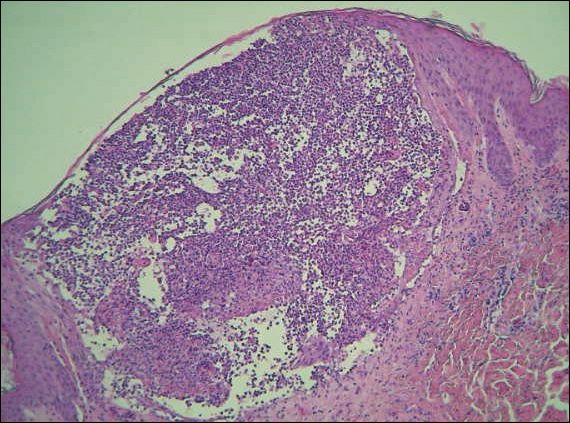
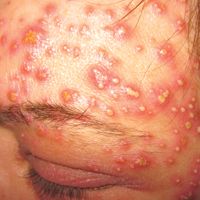
A 40-year-old woman was referred to the dermatology department due to the sudden onset of multiple pustules on the face. One week earlier she started oral flurbiprofen (8.75 mg daily) for a sore throat. After 3 days of therapy, multiple pruritic, erythematous and edematous lesions appeared abruptly on the face with associated multiple small nonfollicular pustules. At presentation the patient was febrile (temperature, 38.2°C) and presented with bilateral ocular edema and superficial small nonfollicular pustules on an erythematous background over the face, scalp, and oral mucosa (Figure 1). The rest of the body was not involved. The patient denied prior adverse reactions to other drugs. The white blood cell count was 15,000/μL (reference range, 4500–11,000/μL), with an increased neutrophil count (12,000/μL [reference range, 1800–7800/μL]). The erythrocyte sedimentation rate and C-reactive protein level was elevated (erythrocyte sedimentation rate, 53 mm/h [reference range, 0–20 mm/h]; C-reactive protein, 98 mg/dL [reference range, 0–5 mg/dL]). Bacterial and fungal cultures of skin lesions were negative. The results of a viral polymerase chain reaction analysis proved the absence of varicella-zoster virus or herpes simplex virus. Histopathology of a skin biopsy specimen showed subcorneal pustules composed of neutrophils and eosinophils, epidermal spongiosis, some necrotic keratinocytes, vacuolization of the basal layer, papillary edema, and a perivascular neutrophil and lymphocyte infiltrate (Figure 2). A leukocytoclastic infiltrate within and around the walls of blood vessels at the superficial level of the dermis and red cell extravasation in the epidermis was present. She discontinued use of flurbiprofen and was treated with a systemic corticosteroid (methylprednisolone 0.5 mg/kg daily). The pustules rapidly resolved within 7 days after discontinuation of flurbiprofen and were followed by transient scaling and discrete residual hyperpigmentation.
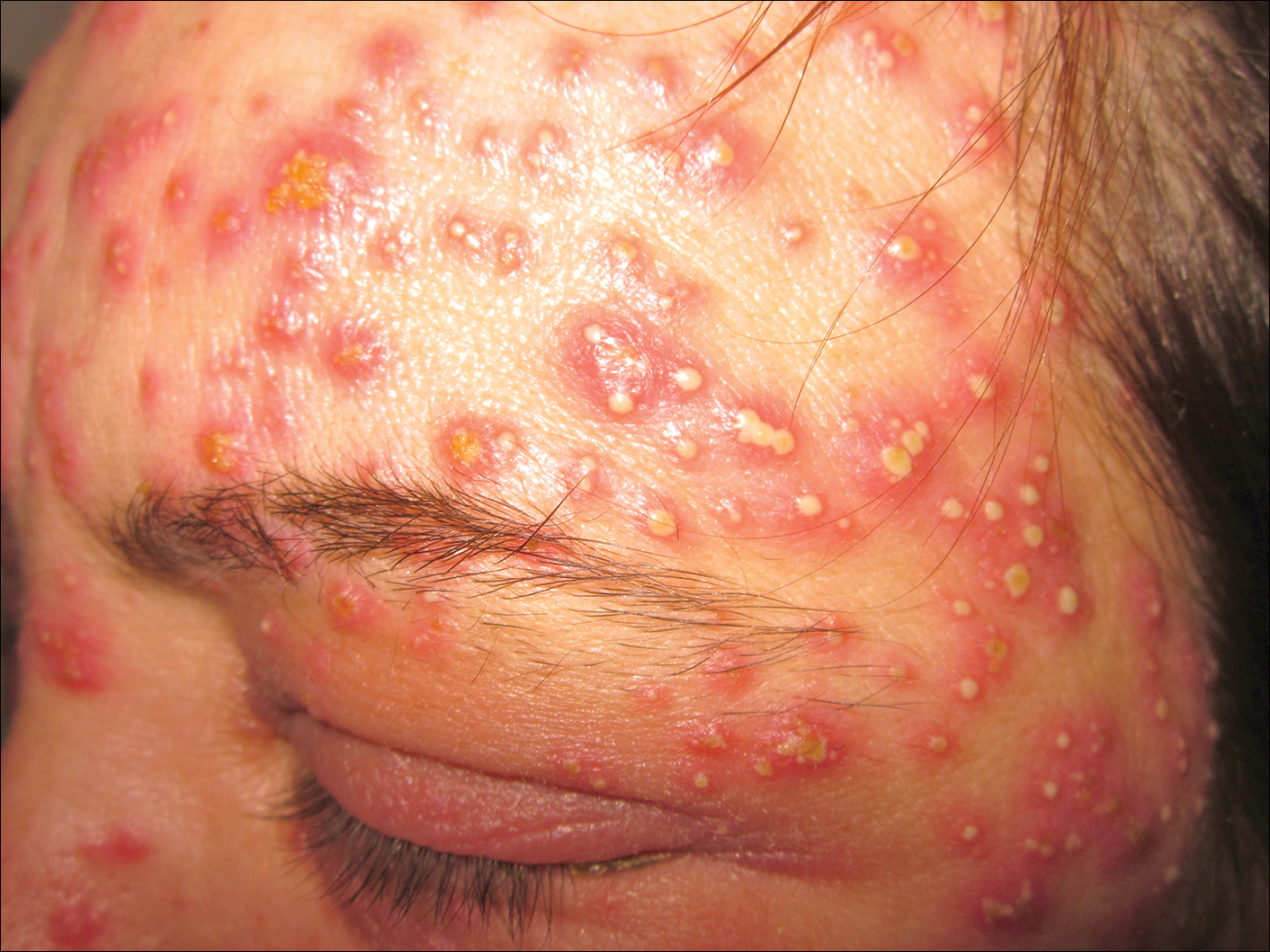
Acute localized exanthematous pustulosis is a less common form of a pustular drug eruption in which lesions are consistent with AGEP but typically are localized to the face, neck, or chest. The definition of ALEP was introduced by Prange et al1 to describe a woman who was diagnosed with a localized pustular eruption on the face without a generalized distribution as in AGEP. In the past, this localized eruption was described under different names (eg, localized pustular eruption, localized toxin follicular pustuloderma, nongeneralized acute exanthematic pustulosis).2-5 According to a PubMed search of articles indexed for MEDLINE using the terms localized pustulosis, localized pustular eruption, and localized pustuloderma, only 16 separate cases of ALEP have been documented since the report by Prange et al.1 The medications most frequently responsible are antibiotics. Three cases developed following administration of amoxicillin2,5,6; 2 cases of amoxicillin–clavulanic acid7,8; 1 of penicillin1; 1 of azithromycin9; 1 of levofloxacin10; and 1 of combination of cephalosporin, sulfamethoxazole-trimethoprim, and vancomycin.11 Other nonantibiotic causative drugs include sulfamethoxazole-trimethoprim,12 infliximab,13 sorafenib,14 docetaxel,15 finasteride,16 ibuprofen,17 and paracetamol.18 In reported cases, the lesions are consistent with the characteristics of AGEP both clinically and histopathologically but are localized typically to the face, neck, or chest. In the majority of patients with ALEP, the absence of fever has been observed, but it does not appear distinctive for diagnosis. Our patient represents another case of ALEP with flurbiprofen as the causative drug. The close relationship between the administration of the drug and the development of the pustules, the rapid acute resolution as soon as treatment was interrupted, and the histologic findings all supported the diagnosis of ALEP following administration of flurbiprofen. This NSAID—2-fluoro-α-methyl-(1,1'-biphenyl)-4-acetic acid—is a prostaglandin synthetase inhibitor with anti-inflammatory activity. It is a propionic acid derivative that is similar to ibuprofen, which was once involved in the occurrence of ALEP.17 In 2009, Rastogi et al17 reported a case of a 64-year-old woman with an acute outbreak of multiple pustular lesions and underlying erythema affecting the cheeks and chin without fever who had been taking ibuprofen for a toothache. The case is similar to ours and confirms that NSAIDs can induce ALEP. Compared with other NSAIDs, propionic acid derivatives are usually well tolerated and serious adverse reactions rarely have been documented.19
The physiopathologic mechanisms of ALEP are unknown but likely are similar to AGEP. The demonstration of drug-specific positive patch test responses and in vitro lymphocyte proliferative responses in patients with a history of AGEP strongly suggests that this adverse cutaneous reaction occurs via a drug-specific T cell–mediated process.20
Further study is needed to understand the etiopathogenesis of the localized form of the disease and to facilitate a correct diagnosis of this rare disorder.
- Prange B, Marini A, Kalke A, et al. Acute localized exanthematous pustulosis (ALEP). J Dtsch Dermatol Ges. 2005;3:210-212.
- Shuttleworth D. A localized, recurrent pustular eruption following amoxycillin administration. Clin Exp Dermatol. 1989;14:367-368.
- De Argila D, Ortiz-Frutos J, Rodriguez-Peralto JL, et al. An atypical case of non-generalized acute exanthematic pustulosis. Actas Dermosifiliogr. 1996;87:475-478.
- Corbalan-Velez R, Peon G, Ara M, et al. Localized toxic follicular pustuloderma. Int J Dermatol. 2000;39:209-211.
- Prieto A, de Barrio M, López-Sáez P, et al. Recurrent localized pustular eruption induced by amoxicillin. Allergy. 1997;52:777-778.
- Vickers JL, Matherne RJ, Mainous EG, et al. Acute localized exanthematous pustulosis: a cutaneous drug reaction in a dental setting. J Am Dent Assoc. 2008;139:1200-1203.
- Betto P, Germi L, Bonoldi E, et al. Acute localized exanthematous pustulosis (ALEP) caused by amoxicillin-clavulanic acid. Int J Dermatol. 2008;47:295-296.
- Ozkaya-Parlakay A, Azkur D, Kara A, et al. Localized acute generalized exanthematous pustulosis with amoxicillin and clavulanic acid. Turk J Pediatr. 2011;53:229-232.
- Zweegers J, Bovenschen HJ. A woman with skin abnormalities around the mouth [in Dutch]. Ned Tijdschr Geneeskd. 2012;156:A4613.
- Corral de la Calle M, Martín Díaz MA, Flores CR, et al. Acute localized exanthematous pustulosis secondary to levofloxacin. Br J Dermatol. 2005;152:1076-1077.
- Sim HS, Seol JE, Chun JS, et al. Acute localized exanthematous pustulosis on the face. Ann Dermatol. 2011;23(suppl 3):S3368-S3370.
- Lee I, Turner M, Lee CC. Acute patchy exanthematous pustulosis caused by sulfamethoxazole-trimethoprim. J Am Acad Dermatol. 2010;63:e41-e43.
- Lee HY, Pelivani N, Beltraminelli H, et al. Amicrobial pustulosis-like rash in a patient with Crohn’s disease under anti-TNF-alpha blocker. Dermatology. 2011;222:304-310.
- Liang CP, Yang CS, Shen JL, et al. Sorafenib-induced acute localized exanthematous pustulosis in a patient with hepatocellular carcinoma. Br J Dermatol. 2011;165:443-445.
- Kim SW, Lee UH, Jang SJ, et al. Acute localized exanthematous pustulosis induced by docetaxel. J Am Acad Dermatol. 2010;63:e44-e46.
- Tresch S, Cozzio A, Kamarashev J, et al. T cell-mediated acute localized exanthematous pustulosis caused by finasteride. J Allergy Clin Immunol. 2012;129:589-594.
- Rastogi S, Modi M, Dhawan V. Acute localized exanthematous pustulosis (ALEP) caused by Ibuprofen. a case report. Br J Oral Maxillofac Surg. 2009;47:132-134.
- Wohl Y, Goldberg I, Sharazi I, et al. A case of paracetamol-induced acute generalized exanthematous pustulosis in a pregnant woman localized in the neck region. Skinmed. 2004;3:47-49.
- Mehra KK, Rupawala AH, Gogtay NJ. Immediate hypersensitivity reaction to a single oral dose of flurbiprofen. J Postgrad Med. 2010;56:36-37.
- Girardi M, Duncan KO, Tigelaar RE, et al. Cross comparison of patch-test and lymphocyte proliferation responses in patients with a history of acute generalized exanthematous pustulosis. Am J Dermatopathol. 2005;27:343-346.
To the Editor:
Acute generalized exanthematous pustulosis (AGEP) is an acute skin reaction that is characterized by generalized, nonfollicular, pinhead-sized, sterile pustules on an erythematous and edematous background. The eruption can be accompanied by fever and neutrophilic leukocytosis. Skin symptoms arise quickly (within a few hours), most commonly following drug administration. The medications most frequently responsible are beta-lactam antibiotics, macrolides, calcium channel blockers, and antimalarials. Pustules spontaneously resolve in 15 days and generalized desquamation occurs approximately 2 weeks later. The estimated incidence rate of AGEP is approximately 1 to 5 cases per million per year. Acute localized exanthematous pustulosis (ALEP) is a less common form of AGEP. We report a case of ALEP localized on the face that was caused by flurbiprofen, a propionic acid derivative from the family of nonsteroidal anti-inflammatory drugs (NSAIDs).
A 40-year-old woman was referred to the dermatology department due to the sudden onset of multiple pustules on the face. One week earlier she started oral flurbiprofen (8.75 mg daily) for a sore throat. After 3 days of therapy, multiple pruritic, erythematous and edematous lesions appeared abruptly on the face with associated multiple small nonfollicular pustules. At presentation the patient was febrile (temperature, 38.2°C) and presented with bilateral ocular edema and superficial small nonfollicular pustules on an erythematous background over the face, scalp, and oral mucosa (Figure 1). The rest of the body was not involved. The patient denied prior adverse reactions to other drugs. The white blood cell count was 15,000/μL (reference range, 4500–11,000/μL), with an increased neutrophil count (12,000/μL [reference range, 1800–7800/μL]). The erythrocyte sedimentation rate and C-reactive protein level was elevated (erythrocyte sedimentation rate, 53 mm/h [reference range, 0–20 mm/h]; C-reactive protein, 98 mg/dL [reference range, 0–5 mg/dL]). Bacterial and fungal cultures of skin lesions were negative. The results of a viral polymerase chain reaction analysis proved the absence of varicella-zoster virus or herpes simplex virus. Histopathology of a skin biopsy specimen showed subcorneal pustules composed of neutrophils and eosinophils, epidermal spongiosis, some necrotic keratinocytes, vacuolization of the basal layer, papillary edema, and a perivascular neutrophil and lymphocyte infiltrate (Figure 2). A leukocytoclastic infiltrate within and around the walls of blood vessels at the superficial level of the dermis and red cell extravasation in the epidermis was present. She discontinued use of flurbiprofen and was treated with a systemic corticosteroid (methylprednisolone 0.5 mg/kg daily). The pustules rapidly resolved within 7 days after discontinuation of flurbiprofen and were followed by transient scaling and discrete residual hyperpigmentation.
Acute localized exanthematous pustulosis is a less common form of a pustular drug eruption in which lesions are consistent with AGEP but typically are localized to the face, neck, or chest. The definition of ALEP was introduced by Prange et al1 to describe a woman who was diagnosed with a localized pustular eruption on the face without a generalized distribution as in AGEP. In the past, this localized eruption was described under different names (eg, localized pustular eruption, localized toxin follicular pustuloderma, nongeneralized acute exanthematic pustulosis).2-5 According to a PubMed search of articles indexed for MEDLINE using the terms localized pustulosis, localized pustular eruption, and localized pustuloderma, only 16 separate cases of ALEP have been documented since the report by Prange et al.1 The medications most frequently responsible are antibiotics. Three cases developed following administration of amoxicillin2,5,6; 2 cases of amoxicillin–clavulanic acid7,8; 1 of penicillin1; 1 of azithromycin9; 1 of levofloxacin10; and 1 of combination of cephalosporin, sulfamethoxazole-trimethoprim, and vancomycin.11 Other nonantibiotic causative drugs include sulfamethoxazole-trimethoprim,12 infliximab,13 sorafenib,14 docetaxel,15 finasteride,16 ibuprofen,17 and paracetamol.18 In reported cases, the lesions are consistent with the characteristics of AGEP both clinically and histopathologically but are localized typically to the face, neck, or chest. In the majority of patients with ALEP, the absence of fever has been observed, but it does not appear distinctive for diagnosis. Our patient represents another case of ALEP with flurbiprofen as the causative drug. The close relationship between the administration of the drug and the development of the pustules, the rapid acute resolution as soon as treatment was interrupted, and the histologic findings all supported the diagnosis of ALEP following administration of flurbiprofen. This NSAID—2-fluoro-α-methyl-(1,1'-biphenyl)-4-acetic acid—is a prostaglandin synthetase inhibitor with anti-inflammatory activity. It is a propionic acid derivative that is similar to ibuprofen, which was once involved in the occurrence of ALEP.17 In 2009, Rastogi et al17 reported a case of a 64-year-old woman with an acute outbreak of multiple pustular lesions and underlying erythema affecting the cheeks and chin without fever who had been taking ibuprofen for a toothache. The case is similar to ours and confirms that NSAIDs can induce ALEP. Compared with other NSAIDs, propionic acid derivatives are usually well tolerated and serious adverse reactions rarely have been documented.19
The physiopathologic mechanisms of ALEP are unknown but likely are similar to AGEP. The demonstration of drug-specific positive patch test responses and in vitro lymphocyte proliferative responses in patients with a history of AGEP strongly suggests that this adverse cutaneous reaction occurs via a drug-specific T cell–mediated process.20
Further study is needed to understand the etiopathogenesis of the localized form of the disease and to facilitate a correct diagnosis of this rare disorder.
To the Editor:
Acute generalized exanthematous pustulosis (AGEP) is an acute skin reaction that is characterized by generalized, nonfollicular, pinhead-sized, sterile pustules on an erythematous and edematous background. The eruption can be accompanied by fever and neutrophilic leukocytosis. Skin symptoms arise quickly (within a few hours), most commonly following drug administration. The medications most frequently responsible are beta-lactam antibiotics, macrolides, calcium channel blockers, and antimalarials. Pustules spontaneously resolve in 15 days and generalized desquamation occurs approximately 2 weeks later. The estimated incidence rate of AGEP is approximately 1 to 5 cases per million per year. Acute localized exanthematous pustulosis (ALEP) is a less common form of AGEP. We report a case of ALEP localized on the face that was caused by flurbiprofen, a propionic acid derivative from the family of nonsteroidal anti-inflammatory drugs (NSAIDs).
A 40-year-old woman was referred to the dermatology department due to the sudden onset of multiple pustules on the face. One week earlier she started oral flurbiprofen (8.75 mg daily) for a sore throat. After 3 days of therapy, multiple pruritic, erythematous and edematous lesions appeared abruptly on the face with associated multiple small nonfollicular pustules. At presentation the patient was febrile (temperature, 38.2°C) and presented with bilateral ocular edema and superficial small nonfollicular pustules on an erythematous background over the face, scalp, and oral mucosa (Figure 1). The rest of the body was not involved. The patient denied prior adverse reactions to other drugs. The white blood cell count was 15,000/μL (reference range, 4500–11,000/μL), with an increased neutrophil count (12,000/μL [reference range, 1800–7800/μL]). The erythrocyte sedimentation rate and C-reactive protein level was elevated (erythrocyte sedimentation rate, 53 mm/h [reference range, 0–20 mm/h]; C-reactive protein, 98 mg/dL [reference range, 0–5 mg/dL]). Bacterial and fungal cultures of skin lesions were negative. The results of a viral polymerase chain reaction analysis proved the absence of varicella-zoster virus or herpes simplex virus. Histopathology of a skin biopsy specimen showed subcorneal pustules composed of neutrophils and eosinophils, epidermal spongiosis, some necrotic keratinocytes, vacuolization of the basal layer, papillary edema, and a perivascular neutrophil and lymphocyte infiltrate (Figure 2). A leukocytoclastic infiltrate within and around the walls of blood vessels at the superficial level of the dermis and red cell extravasation in the epidermis was present. She discontinued use of flurbiprofen and was treated with a systemic corticosteroid (methylprednisolone 0.5 mg/kg daily). The pustules rapidly resolved within 7 days after discontinuation of flurbiprofen and were followed by transient scaling and discrete residual hyperpigmentation.
Acute localized exanthematous pustulosis is a less common form of a pustular drug eruption in which lesions are consistent with AGEP but typically are localized to the face, neck, or chest. The definition of ALEP was introduced by Prange et al1 to describe a woman who was diagnosed with a localized pustular eruption on the face without a generalized distribution as in AGEP. In the past, this localized eruption was described under different names (eg, localized pustular eruption, localized toxin follicular pustuloderma, nongeneralized acute exanthematic pustulosis).2-5 According to a PubMed search of articles indexed for MEDLINE using the terms localized pustulosis, localized pustular eruption, and localized pustuloderma, only 16 separate cases of ALEP have been documented since the report by Prange et al.1 The medications most frequently responsible are antibiotics. Three cases developed following administration of amoxicillin2,5,6; 2 cases of amoxicillin–clavulanic acid7,8; 1 of penicillin1; 1 of azithromycin9; 1 of levofloxacin10; and 1 of combination of cephalosporin, sulfamethoxazole-trimethoprim, and vancomycin.11 Other nonantibiotic causative drugs include sulfamethoxazole-trimethoprim,12 infliximab,13 sorafenib,14 docetaxel,15 finasteride,16 ibuprofen,17 and paracetamol.18 In reported cases, the lesions are consistent with the characteristics of AGEP both clinically and histopathologically but are localized typically to the face, neck, or chest. In the majority of patients with ALEP, the absence of fever has been observed, but it does not appear distinctive for diagnosis. Our patient represents another case of ALEP with flurbiprofen as the causative drug. The close relationship between the administration of the drug and the development of the pustules, the rapid acute resolution as soon as treatment was interrupted, and the histologic findings all supported the diagnosis of ALEP following administration of flurbiprofen. This NSAID—2-fluoro-α-methyl-(1,1'-biphenyl)-4-acetic acid—is a prostaglandin synthetase inhibitor with anti-inflammatory activity. It is a propionic acid derivative that is similar to ibuprofen, which was once involved in the occurrence of ALEP.17 In 2009, Rastogi et al17 reported a case of a 64-year-old woman with an acute outbreak of multiple pustular lesions and underlying erythema affecting the cheeks and chin without fever who had been taking ibuprofen for a toothache. The case is similar to ours and confirms that NSAIDs can induce ALEP. Compared with other NSAIDs, propionic acid derivatives are usually well tolerated and serious adverse reactions rarely have been documented.19
The physiopathologic mechanisms of ALEP are unknown but likely are similar to AGEP. The demonstration of drug-specific positive patch test responses and in vitro lymphocyte proliferative responses in patients with a history of AGEP strongly suggests that this adverse cutaneous reaction occurs via a drug-specific T cell–mediated process.20
Further study is needed to understand the etiopathogenesis of the localized form of the disease and to facilitate a correct diagnosis of this rare disorder.
- Prange B, Marini A, Kalke A, et al. Acute localized exanthematous pustulosis (ALEP). J Dtsch Dermatol Ges. 2005;3:210-212.
- Shuttleworth D. A localized, recurrent pustular eruption following amoxycillin administration. Clin Exp Dermatol. 1989;14:367-368.
- De Argila D, Ortiz-Frutos J, Rodriguez-Peralto JL, et al. An atypical case of non-generalized acute exanthematic pustulosis. Actas Dermosifiliogr. 1996;87:475-478.
- Corbalan-Velez R, Peon G, Ara M, et al. Localized toxic follicular pustuloderma. Int J Dermatol. 2000;39:209-211.
- Prieto A, de Barrio M, López-Sáez P, et al. Recurrent localized pustular eruption induced by amoxicillin. Allergy. 1997;52:777-778.
- Vickers JL, Matherne RJ, Mainous EG, et al. Acute localized exanthematous pustulosis: a cutaneous drug reaction in a dental setting. J Am Dent Assoc. 2008;139:1200-1203.
- Betto P, Germi L, Bonoldi E, et al. Acute localized exanthematous pustulosis (ALEP) caused by amoxicillin-clavulanic acid. Int J Dermatol. 2008;47:295-296.
- Ozkaya-Parlakay A, Azkur D, Kara A, et al. Localized acute generalized exanthematous pustulosis with amoxicillin and clavulanic acid. Turk J Pediatr. 2011;53:229-232.
- Zweegers J, Bovenschen HJ. A woman with skin abnormalities around the mouth [in Dutch]. Ned Tijdschr Geneeskd. 2012;156:A4613.
- Corral de la Calle M, Martín Díaz MA, Flores CR, et al. Acute localized exanthematous pustulosis secondary to levofloxacin. Br J Dermatol. 2005;152:1076-1077.
- Sim HS, Seol JE, Chun JS, et al. Acute localized exanthematous pustulosis on the face. Ann Dermatol. 2011;23(suppl 3):S3368-S3370.
- Lee I, Turner M, Lee CC. Acute patchy exanthematous pustulosis caused by sulfamethoxazole-trimethoprim. J Am Acad Dermatol. 2010;63:e41-e43.
- Lee HY, Pelivani N, Beltraminelli H, et al. Amicrobial pustulosis-like rash in a patient with Crohn’s disease under anti-TNF-alpha blocker. Dermatology. 2011;222:304-310.
- Liang CP, Yang CS, Shen JL, et al. Sorafenib-induced acute localized exanthematous pustulosis in a patient with hepatocellular carcinoma. Br J Dermatol. 2011;165:443-445.
- Kim SW, Lee UH, Jang SJ, et al. Acute localized exanthematous pustulosis induced by docetaxel. J Am Acad Dermatol. 2010;63:e44-e46.
- Tresch S, Cozzio A, Kamarashev J, et al. T cell-mediated acute localized exanthematous pustulosis caused by finasteride. J Allergy Clin Immunol. 2012;129:589-594.
- Rastogi S, Modi M, Dhawan V. Acute localized exanthematous pustulosis (ALEP) caused by Ibuprofen. a case report. Br J Oral Maxillofac Surg. 2009;47:132-134.
- Wohl Y, Goldberg I, Sharazi I, et al. A case of paracetamol-induced acute generalized exanthematous pustulosis in a pregnant woman localized in the neck region. Skinmed. 2004;3:47-49.
- Mehra KK, Rupawala AH, Gogtay NJ. Immediate hypersensitivity reaction to a single oral dose of flurbiprofen. J Postgrad Med. 2010;56:36-37.
- Girardi M, Duncan KO, Tigelaar RE, et al. Cross comparison of patch-test and lymphocyte proliferation responses in patients with a history of acute generalized exanthematous pustulosis. Am J Dermatopathol. 2005;27:343-346.
- Prange B, Marini A, Kalke A, et al. Acute localized exanthematous pustulosis (ALEP). J Dtsch Dermatol Ges. 2005;3:210-212.
- Shuttleworth D. A localized, recurrent pustular eruption following amoxycillin administration. Clin Exp Dermatol. 1989;14:367-368.
- De Argila D, Ortiz-Frutos J, Rodriguez-Peralto JL, et al. An atypical case of non-generalized acute exanthematic pustulosis. Actas Dermosifiliogr. 1996;87:475-478.
- Corbalan-Velez R, Peon G, Ara M, et al. Localized toxic follicular pustuloderma. Int J Dermatol. 2000;39:209-211.
- Prieto A, de Barrio M, López-Sáez P, et al. Recurrent localized pustular eruption induced by amoxicillin. Allergy. 1997;52:777-778.
- Vickers JL, Matherne RJ, Mainous EG, et al. Acute localized exanthematous pustulosis: a cutaneous drug reaction in a dental setting. J Am Dent Assoc. 2008;139:1200-1203.
- Betto P, Germi L, Bonoldi E, et al. Acute localized exanthematous pustulosis (ALEP) caused by amoxicillin-clavulanic acid. Int J Dermatol. 2008;47:295-296.
- Ozkaya-Parlakay A, Azkur D, Kara A, et al. Localized acute generalized exanthematous pustulosis with amoxicillin and clavulanic acid. Turk J Pediatr. 2011;53:229-232.
- Zweegers J, Bovenschen HJ. A woman with skin abnormalities around the mouth [in Dutch]. Ned Tijdschr Geneeskd. 2012;156:A4613.
- Corral de la Calle M, Martín Díaz MA, Flores CR, et al. Acute localized exanthematous pustulosis secondary to levofloxacin. Br J Dermatol. 2005;152:1076-1077.
- Sim HS, Seol JE, Chun JS, et al. Acute localized exanthematous pustulosis on the face. Ann Dermatol. 2011;23(suppl 3):S3368-S3370.
- Lee I, Turner M, Lee CC. Acute patchy exanthematous pustulosis caused by sulfamethoxazole-trimethoprim. J Am Acad Dermatol. 2010;63:e41-e43.
- Lee HY, Pelivani N, Beltraminelli H, et al. Amicrobial pustulosis-like rash in a patient with Crohn’s disease under anti-TNF-alpha blocker. Dermatology. 2011;222:304-310.
- Liang CP, Yang CS, Shen JL, et al. Sorafenib-induced acute localized exanthematous pustulosis in a patient with hepatocellular carcinoma. Br J Dermatol. 2011;165:443-445.
- Kim SW, Lee UH, Jang SJ, et al. Acute localized exanthematous pustulosis induced by docetaxel. J Am Acad Dermatol. 2010;63:e44-e46.
- Tresch S, Cozzio A, Kamarashev J, et al. T cell-mediated acute localized exanthematous pustulosis caused by finasteride. J Allergy Clin Immunol. 2012;129:589-594.
- Rastogi S, Modi M, Dhawan V. Acute localized exanthematous pustulosis (ALEP) caused by Ibuprofen. a case report. Br J Oral Maxillofac Surg. 2009;47:132-134.
- Wohl Y, Goldberg I, Sharazi I, et al. A case of paracetamol-induced acute generalized exanthematous pustulosis in a pregnant woman localized in the neck region. Skinmed. 2004;3:47-49.
- Mehra KK, Rupawala AH, Gogtay NJ. Immediate hypersensitivity reaction to a single oral dose of flurbiprofen. J Postgrad Med. 2010;56:36-37.
- Girardi M, Duncan KO, Tigelaar RE, et al. Cross comparison of patch-test and lymphocyte proliferation responses in patients with a history of acute generalized exanthematous pustulosis. Am J Dermatopathol. 2005;27:343-346.
Practice Points
- Acute localized exanthematous pustulosis is a form of a pustular drug eruption in which lesions are consistent with acute generalized exanthematous pustulosis but typically localized in a single area.
- The medications most frequently responsible are antibiotics. Flurbiprofen, a propionic acid derivative, could be a rare causative agent of this disease.
Contact Allergy to Poliglecaprone 25 Sutures
To the Editor:
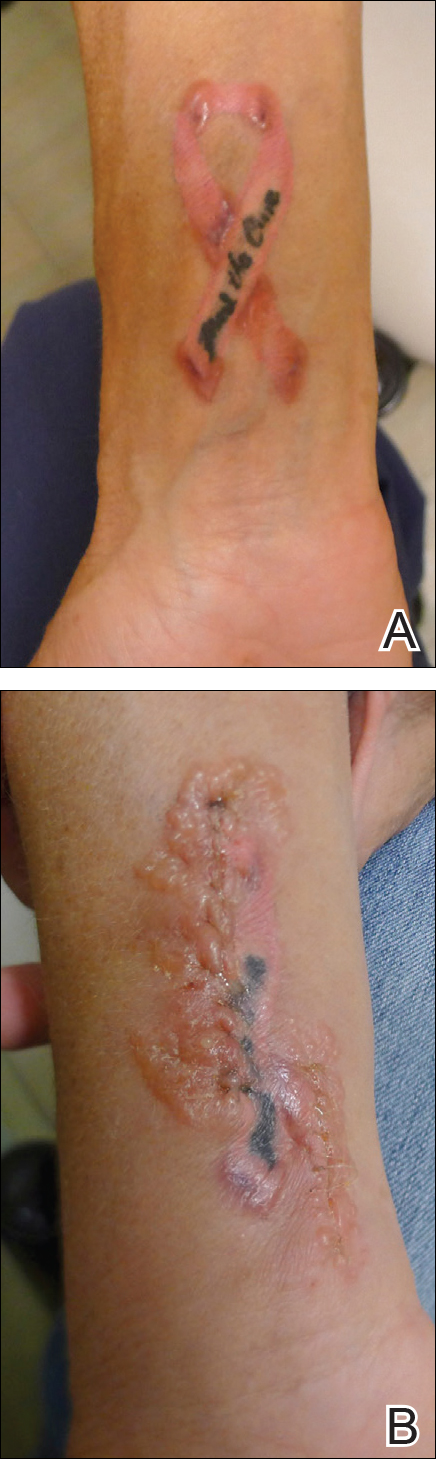
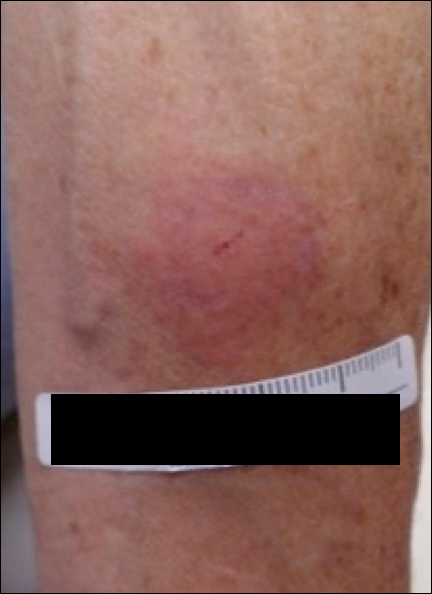
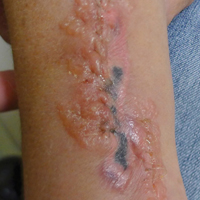
A 42-year-old woman who had a tattoo on the right wrist surgically removed 2 days prior developed severe erythema and swelling at the incision site (Figure 1). Exposure at the incision site was limited to bacitracin, poliglecaprone 25 suture, and plain cotton gauze. Patch testing of bacitracin was performed, which was ++ (moderately positive reaction) at the 96-hour reading, indicating that part of the reaction was due to the topical antibiotic. Testing of the suture was performed by tying the suture to the skin of the forearm and removing it at 48 hours. There was a ++ reaction to the suture prior to removal at 48 hours, which increased to +++ (severely positive reaction) after suture removal at 96 hours (Figure 2). Therefore, it appears that allergy to the suture also was partially responsible for the postsurgical reaction.
Poliglecaprone 25 suture is a monofilament synthetic absorbable material that is a copolymer of glycolide and ε-caprolactone. One case report of oral contact allergy to this suture material resulted in failure of an oral graft; however, no testing was performed to verify the contact allergy.1 Caprolactam ([CH2]5C[O]NH) is a related chemical that can be synthesized by treating caprolactone ([CH2]5CO2) with ammonia at elevated temperatures.2 Contact allergy has been reported to polyamide 6 suture, which is obtained by polymerizing ε-caprolactam. This report stated that contact allergy to ε-caprolactam also has been reported occupationally during manufacture and from its use in fishing nets, socks, gloves, and stockings.3
The package insert for the poliglecaprone 25 suture states that the material is “nonantigenic, nonpyrogenic and elicits only a slight tissue reaction during absorption.”4 We present a case of contact allergy to poliglecaprone 25 suture that was confirmed by allergy testing.
- Mawardi H. Oral contact allergy to suture material results in connective tissue graft failure: a case report. J Periodontol Online. 2014;4:155-160.
- Buntara T, Noel S, Phua PH, et al. Caprolactam from renewable resources: catalytic conversion of 5-hydroxymethylfurfural into caprolactone. Angew Chem Int Ed Engl. 2011;50:7083-7087.
- Hausen BM. Allergic contact dermatitis from colored surgical suture material: contact allergy to epsilon-caprolactam and acid blue 158. Am J Contact Dermat. 2003;14:174-175.
- Monocryl [package insert]. Somerville, NJ: Ethicon, Inc; 1996.
To the Editor:
A 42-year-old woman who had a tattoo on the right wrist surgically removed 2 days prior developed severe erythema and swelling at the incision site (Figure 1). Exposure at the incision site was limited to bacitracin, poliglecaprone 25 suture, and plain cotton gauze. Patch testing of bacitracin was performed, which was ++ (moderately positive reaction) at the 96-hour reading, indicating that part of the reaction was due to the topical antibiotic. Testing of the suture was performed by tying the suture to the skin of the forearm and removing it at 48 hours. There was a ++ reaction to the suture prior to removal at 48 hours, which increased to +++ (severely positive reaction) after suture removal at 96 hours (Figure 2). Therefore, it appears that allergy to the suture also was partially responsible for the postsurgical reaction.
Poliglecaprone 25 suture is a monofilament synthetic absorbable material that is a copolymer of glycolide and ε-caprolactone. One case report of oral contact allergy to this suture material resulted in failure of an oral graft; however, no testing was performed to verify the contact allergy.1 Caprolactam ([CH2]5C[O]NH) is a related chemical that can be synthesized by treating caprolactone ([CH2]5CO2) with ammonia at elevated temperatures.2 Contact allergy has been reported to polyamide 6 suture, which is obtained by polymerizing ε-caprolactam. This report stated that contact allergy to ε-caprolactam also has been reported occupationally during manufacture and from its use in fishing nets, socks, gloves, and stockings.3
The package insert for the poliglecaprone 25 suture states that the material is “nonantigenic, nonpyrogenic and elicits only a slight tissue reaction during absorption.”4 We present a case of contact allergy to poliglecaprone 25 suture that was confirmed by allergy testing.
To the Editor:
A 42-year-old woman who had a tattoo on the right wrist surgically removed 2 days prior developed severe erythema and swelling at the incision site (Figure 1). Exposure at the incision site was limited to bacitracin, poliglecaprone 25 suture, and plain cotton gauze. Patch testing of bacitracin was performed, which was ++ (moderately positive reaction) at the 96-hour reading, indicating that part of the reaction was due to the topical antibiotic. Testing of the suture was performed by tying the suture to the skin of the forearm and removing it at 48 hours. There was a ++ reaction to the suture prior to removal at 48 hours, which increased to +++ (severely positive reaction) after suture removal at 96 hours (Figure 2). Therefore, it appears that allergy to the suture also was partially responsible for the postsurgical reaction.
Poliglecaprone 25 suture is a monofilament synthetic absorbable material that is a copolymer of glycolide and ε-caprolactone. One case report of oral contact allergy to this suture material resulted in failure of an oral graft; however, no testing was performed to verify the contact allergy.1 Caprolactam ([CH2]5C[O]NH) is a related chemical that can be synthesized by treating caprolactone ([CH2]5CO2) with ammonia at elevated temperatures.2 Contact allergy has been reported to polyamide 6 suture, which is obtained by polymerizing ε-caprolactam. This report stated that contact allergy to ε-caprolactam also has been reported occupationally during manufacture and from its use in fishing nets, socks, gloves, and stockings.3
The package insert for the poliglecaprone 25 suture states that the material is “nonantigenic, nonpyrogenic and elicits only a slight tissue reaction during absorption.”4 We present a case of contact allergy to poliglecaprone 25 suture that was confirmed by allergy testing.
- Mawardi H. Oral contact allergy to suture material results in connective tissue graft failure: a case report. J Periodontol Online. 2014;4:155-160.
- Buntara T, Noel S, Phua PH, et al. Caprolactam from renewable resources: catalytic conversion of 5-hydroxymethylfurfural into caprolactone. Angew Chem Int Ed Engl. 2011;50:7083-7087.
- Hausen BM. Allergic contact dermatitis from colored surgical suture material: contact allergy to epsilon-caprolactam and acid blue 158. Am J Contact Dermat. 2003;14:174-175.
- Monocryl [package insert]. Somerville, NJ: Ethicon, Inc; 1996.
- Mawardi H. Oral contact allergy to suture material results in connective tissue graft failure: a case report. J Periodontol Online. 2014;4:155-160.
- Buntara T, Noel S, Phua PH, et al. Caprolactam from renewable resources: catalytic conversion of 5-hydroxymethylfurfural into caprolactone. Angew Chem Int Ed Engl. 2011;50:7083-7087.
- Hausen BM. Allergic contact dermatitis from colored surgical suture material: contact allergy to epsilon-caprolactam and acid blue 158. Am J Contact Dermat. 2003;14:174-175.
- Monocryl [package insert]. Somerville, NJ: Ethicon, Inc; 1996.
Practice Point
- Physicians should be aware that rare contact reactions can occur with certain types of sutures.
Recurrent Cerebriform Connective Tissue Nevus on the Foot of a Patient With Proteus Syndrome
To the Editor:
A 12-year-old girl presented with discomfort and walking limitation caused by cutaneous masses on the plantar aspects of the feet with associated bone abnormalities that had started as several flesh-colored papules on the plantar surface of both feet at the age of 1 year. Over time the lesions gradually enlarged and formed irregular masses, more prominently on the right foot. At the age of 6 years, surgical correction was performed due to increased walking impairment and a skin examination that suggested connective tissue nevus. The results were good. However, the local tissue overgrowth recurred after 1 year. Slowly growing lesions were found at the surgical site, which necessitated hospitalization. Her medical history was negative for other disease. There was no family history of similar skin conditions and her parents were nonconsanguineous.
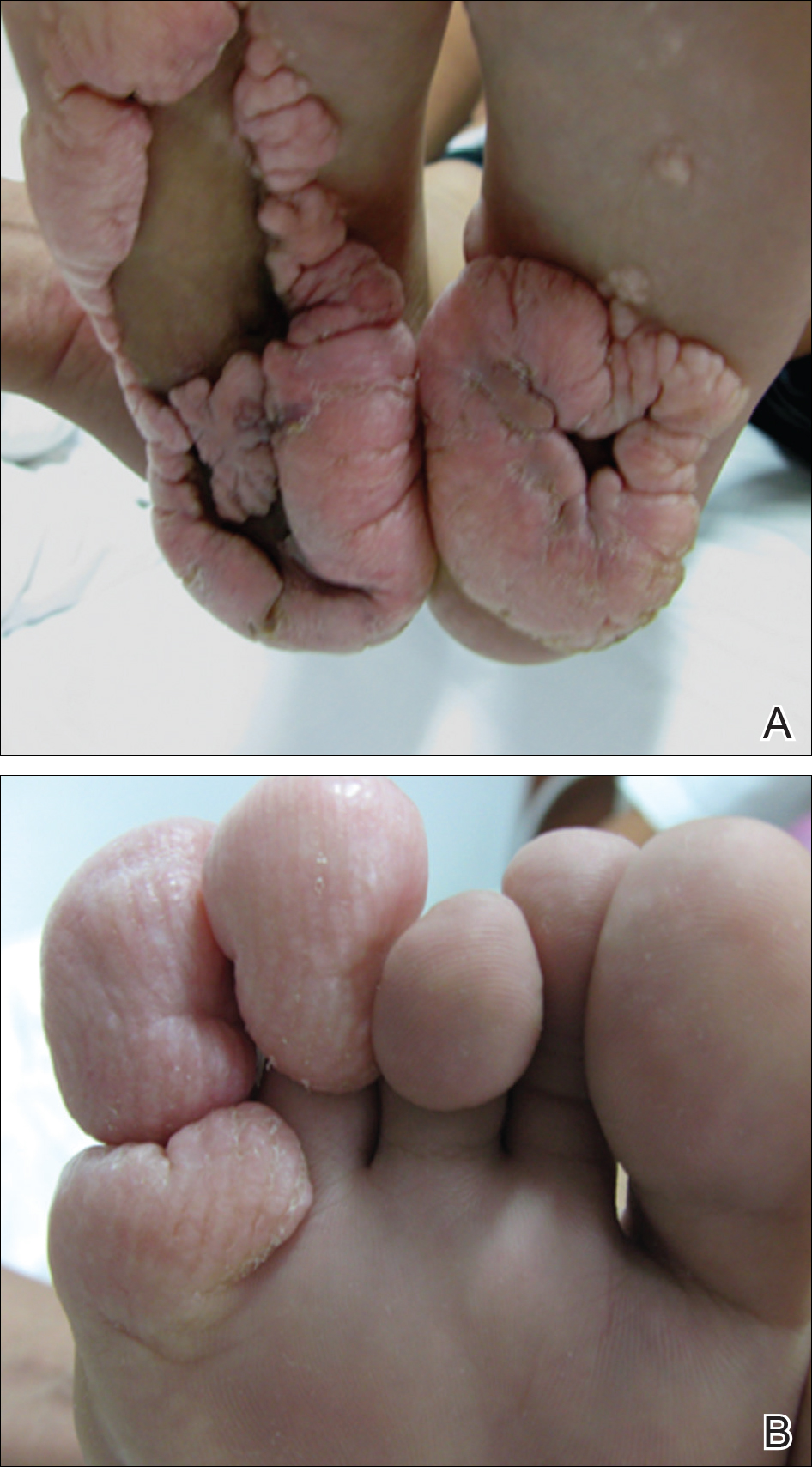
Physical examination revealed malnutrition and poor development in height as well as difficulty walking. She also had moderate scoliosis with a curve to the left. Dermatological examination showed multiple reddish cerebriform hyperplasia in both plantar areas; the right side was more severely involved (Figure 1A). There was macrodactyly of 2 toes on the right foot (Figure 1B). All results of routine blood examinations were within reference range. There were no abnormalities noted in the abdominal ultrasound and cardiac examinations. Plain radiographs of the spine and feet demonstrated scoliosis and exostosis on the calcaneus and bottom of the scaphoid. Histopathologic examination of tissue from the plantar cerebriform hyperplasia revealed hyperkeratosis, slight acanthosis and papillomatosis in the epidermis, and dense collagen bands and sparse elastic fibers in the dermis (Figure 2).

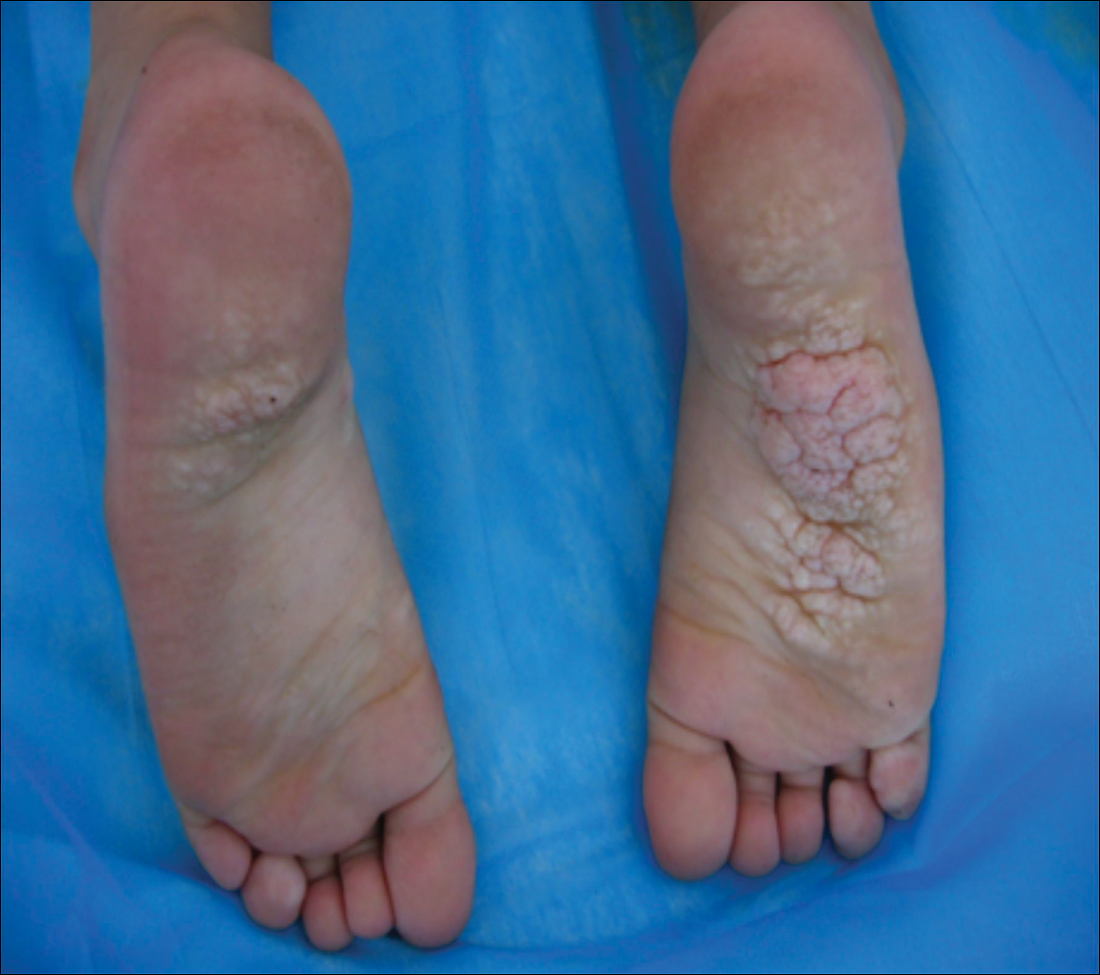
Given the clinical and radiologic manifestation, the diagnosis of Proteus syndrome (PS) was established. After taking into account the severe discomfort and the success of the first surgery, we performed a resection and full-thickness skin graft surgery once again. The feet recovered without any discomfort in daily life. The appearance of the skin graft area was normal 1 year following surgery (Figure 3). She was treated with spinal plate fixation at another institution, progressed well for 2 years, and was subsequently lost to follow-up.
Proteus syndrome is a multisystem disorder with a difficult diagnosis due to the variability of its manifestations. The worldwide incidence of this rare disorder is less than 1 per 1 million individuals, and it is thought to be caused by a somatic genetic alteration.1 Clinical characteristics include bone abnormalities, vascular malformations, dysregulation of fatty tissue, linear verrucous epidermal nevus, and cerebriform connective tissue nevus (CCTN). Although CCTN is not a common finding in patients with PS, it is considered a fairly specific sign with the greatest impact for the diagnosis of PS.2
The general feature of PS--asymmetric disproportionate overgrowth of tissues--appears at 6 to 18 months of age, which makes it challenging to diagnose disease earlier. The CCTN in our patient was present since 1 year of age.
To make a diagnosis of PS, one must have all the general criteria and various specific criteria. The revised diagnostic criteria for PS are given in the Table.3 According to the diagnostic criteria, our patient fulfilled the mandatory general criteria and had plantar CCTN, epidermal nevus, and dysregulated adipose tissue. The CCTN has notable diagnostic value in mildly affected patients, as it is absent in diseases included in the differential diagnosis such as neurofibromatosis, Klippel-Trenaunay-Weber syndrome, Maffucci syndrome, and Bannayan-Riley-Ruvalcaba syndrome. Hemihyperplasia-multiple lipomatosis syndrome and CLOVES (congenital, lipomatous overgrowth, vascular malformations, epidermal nevi, and scoliosis/spinal/skeletal anomalies) syndrome also can present on the plantar surfaces, and lesions may be overgrown at birth but are softer and compressible, have wrinkles instead of deep folds, and tend to grow with the child rather than disproportionately as in PS.4
The epidermal nevi and vascular malformations generally do not spread or increase in number. In contrast, CCTN in PS grows throughout childhood but tends to remain stable in adulthood.4 Postponing surgical treatment until skin lesions stabilize appears to be the best option. However, for practical purposes, surgical intervention may be required at an earlier phase to address the severe functional and cosmetic consequences. Some patients require multiple orthopedic procedures over the ensuing years or decades to control the hyperplasia.3 New CCTN that developed from the prior surgical incision, macrodactyly of the fourth and fifth right toes, and scoliosis appeared when the patient came to our clinic for retreatment 1 year after the initial presentation. The asymmetrical and disproportionate overgrowth of tissues had moderately accelerated in that period. Considering the increasingly impaired walking, we performed a second surgery. On follow-up visits, the patient expressed improvement in daily life.

Studies had been performed to clarify the genetic bases of PS, and the somatic activating mutation in AKT1 (AKT serine/threonine kinase 1) was reported to be the cause of the disease.5,6 Germline PTEN (phosphatase and tensin homolog) mutations have been identified in some patients with overgrowth abnormalities of PS. However, given the misdiagnosis of PS with PTEN mutations and the notion that a gene alone cannot result in PS, the loss-of-function mutations of LEMD3 that have been reported in familial cutaneous collagenomas also may be related to the abnormal growth of connective and bone tissues that are typical of PS.7,8 Lindhurst et al5 concluded that PS is caused by a somatic activating mutation in AKT1, which proved the hypothesis of somatic mosaicism and implicated activation of the PI3K-AKT pathway in the characteristic clinical findings of overgrowth and tumor susceptibility in this disorder. AKT1 is activated by loss-of-function mutations in PTEN, which explains why patients with these mutations (eg, those with the segmental overgrowth, lipomatosis, arteriovenous malformation, epidermal nevus, SOLAMEN [segmental overgrowth, lipomatosis, arteriovenous malformation, and epidermal nevus] syndrome) and patients with activating mutations in AKT1 (eg, those with PS) have overlapping but distinct clinical manifestations. Molecular genetic testing may be useful to confirm the diagnosis in individuals who meet clinical criteria and to establish the diagnosis in individuals with clinical findings that are ambiguous or mild. Further studies are necessary to progress the understanding and management of PS, which will require cooperation of geneticists, surgeons, and other specialists.
- Popescu MD, Burnei G, Draghici L, et al. Proteus syndrome: a difficult diagnosis and management plan. J Med Life. 2014;7:563-566.
- Schepis C, Greco D, Siragusa M, et al. Cerebriform plantar hyperplasia: the major cutaneous feature of Proteus syndrome. Int J Dermatol. 2008;47:374-376.
- Biesecker L. The challenges of Proteus syndrome: diagnosis and management. Eur J Hum Genet. 2006;14:1151-1157.
- Beachkofsky TM, Sapp JC, Biesecker LG, et al. Progressive overgrowth of the cerebriform connective tissue nevus in patients with Proteus syndrome. J Am Acad Dermatol. 2010;63:799-804.
- Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365:611-619.
- Wieland I, Tinschert S, Zenker M. High-level somatic mosaicism of AKT1 c.49G>A mutation in skin scrapings from epidermal nevi enables non-invasive molecular diagnosis in patients with Proteus syndrome. Am J Med Genet A. 2013;161A:889-891.
- Cohen MJ, Turner JT, Biesecker LG. Proteus syndrome: misdiagnosis with PTEN mutations. Am J Med Genet A. 2003;122A:323-324.
- Di Stefani A, Gabellini M, Ferlosio A, et al. Cerebriform plantar hyperplasia: the clinico-pathological hallmark of Proteus syndrome. Acta Derm Venereol. 2011;91:580-581.
To the Editor:
A 12-year-old girl presented with discomfort and walking limitation caused by cutaneous masses on the plantar aspects of the feet with associated bone abnormalities that had started as several flesh-colored papules on the plantar surface of both feet at the age of 1 year. Over time the lesions gradually enlarged and formed irregular masses, more prominently on the right foot. At the age of 6 years, surgical correction was performed due to increased walking impairment and a skin examination that suggested connective tissue nevus. The results were good. However, the local tissue overgrowth recurred after 1 year. Slowly growing lesions were found at the surgical site, which necessitated hospitalization. Her medical history was negative for other disease. There was no family history of similar skin conditions and her parents were nonconsanguineous.
Physical examination revealed malnutrition and poor development in height as well as difficulty walking. She also had moderate scoliosis with a curve to the left. Dermatological examination showed multiple reddish cerebriform hyperplasia in both plantar areas; the right side was more severely involved (Figure 1A). There was macrodactyly of 2 toes on the right foot (Figure 1B). All results of routine blood examinations were within reference range. There were no abnormalities noted in the abdominal ultrasound and cardiac examinations. Plain radiographs of the spine and feet demonstrated scoliosis and exostosis on the calcaneus and bottom of the scaphoid. Histopathologic examination of tissue from the plantar cerebriform hyperplasia revealed hyperkeratosis, slight acanthosis and papillomatosis in the epidermis, and dense collagen bands and sparse elastic fibers in the dermis (Figure 2).
Given the clinical and radiologic manifestation, the diagnosis of Proteus syndrome (PS) was established. After taking into account the severe discomfort and the success of the first surgery, we performed a resection and full-thickness skin graft surgery once again. The feet recovered without any discomfort in daily life. The appearance of the skin graft area was normal 1 year following surgery (Figure 3). She was treated with spinal plate fixation at another institution, progressed well for 2 years, and was subsequently lost to follow-up.
Proteus syndrome is a multisystem disorder with a difficult diagnosis due to the variability of its manifestations. The worldwide incidence of this rare disorder is less than 1 per 1 million individuals, and it is thought to be caused by a somatic genetic alteration.1 Clinical characteristics include bone abnormalities, vascular malformations, dysregulation of fatty tissue, linear verrucous epidermal nevus, and cerebriform connective tissue nevus (CCTN). Although CCTN is not a common finding in patients with PS, it is considered a fairly specific sign with the greatest impact for the diagnosis of PS.2
The general feature of PS--asymmetric disproportionate overgrowth of tissues--appears at 6 to 18 months of age, which makes it challenging to diagnose disease earlier. The CCTN in our patient was present since 1 year of age.
To make a diagnosis of PS, one must have all the general criteria and various specific criteria. The revised diagnostic criteria for PS are given in the Table.3 According to the diagnostic criteria, our patient fulfilled the mandatory general criteria and had plantar CCTN, epidermal nevus, and dysregulated adipose tissue. The CCTN has notable diagnostic value in mildly affected patients, as it is absent in diseases included in the differential diagnosis such as neurofibromatosis, Klippel-Trenaunay-Weber syndrome, Maffucci syndrome, and Bannayan-Riley-Ruvalcaba syndrome. Hemihyperplasia-multiple lipomatosis syndrome and CLOVES (congenital, lipomatous overgrowth, vascular malformations, epidermal nevi, and scoliosis/spinal/skeletal anomalies) syndrome also can present on the plantar surfaces, and lesions may be overgrown at birth but are softer and compressible, have wrinkles instead of deep folds, and tend to grow with the child rather than disproportionately as in PS.4
The epidermal nevi and vascular malformations generally do not spread or increase in number. In contrast, CCTN in PS grows throughout childhood but tends to remain stable in adulthood.4 Postponing surgical treatment until skin lesions stabilize appears to be the best option. However, for practical purposes, surgical intervention may be required at an earlier phase to address the severe functional and cosmetic consequences. Some patients require multiple orthopedic procedures over the ensuing years or decades to control the hyperplasia.3 New CCTN that developed from the prior surgical incision, macrodactyly of the fourth and fifth right toes, and scoliosis appeared when the patient came to our clinic for retreatment 1 year after the initial presentation. The asymmetrical and disproportionate overgrowth of tissues had moderately accelerated in that period. Considering the increasingly impaired walking, we performed a second surgery. On follow-up visits, the patient expressed improvement in daily life.
Studies had been performed to clarify the genetic bases of PS, and the somatic activating mutation in AKT1 (AKT serine/threonine kinase 1) was reported to be the cause of the disease.5,6 Germline PTEN (phosphatase and tensin homolog) mutations have been identified in some patients with overgrowth abnormalities of PS. However, given the misdiagnosis of PS with PTEN mutations and the notion that a gene alone cannot result in PS, the loss-of-function mutations of LEMD3 that have been reported in familial cutaneous collagenomas also may be related to the abnormal growth of connective and bone tissues that are typical of PS.7,8 Lindhurst et al5 concluded that PS is caused by a somatic activating mutation in AKT1, which proved the hypothesis of somatic mosaicism and implicated activation of the PI3K-AKT pathway in the characteristic clinical findings of overgrowth and tumor susceptibility in this disorder. AKT1 is activated by loss-of-function mutations in PTEN, which explains why patients with these mutations (eg, those with the segmental overgrowth, lipomatosis, arteriovenous malformation, epidermal nevus, SOLAMEN [segmental overgrowth, lipomatosis, arteriovenous malformation, and epidermal nevus] syndrome) and patients with activating mutations in AKT1 (eg, those with PS) have overlapping but distinct clinical manifestations. Molecular genetic testing may be useful to confirm the diagnosis in individuals who meet clinical criteria and to establish the diagnosis in individuals with clinical findings that are ambiguous or mild. Further studies are necessary to progress the understanding and management of PS, which will require cooperation of geneticists, surgeons, and other specialists.
To the Editor:
A 12-year-old girl presented with discomfort and walking limitation caused by cutaneous masses on the plantar aspects of the feet with associated bone abnormalities that had started as several flesh-colored papules on the plantar surface of both feet at the age of 1 year. Over time the lesions gradually enlarged and formed irregular masses, more prominently on the right foot. At the age of 6 years, surgical correction was performed due to increased walking impairment and a skin examination that suggested connective tissue nevus. The results were good. However, the local tissue overgrowth recurred after 1 year. Slowly growing lesions were found at the surgical site, which necessitated hospitalization. Her medical history was negative for other disease. There was no family history of similar skin conditions and her parents were nonconsanguineous.
Physical examination revealed malnutrition and poor development in height as well as difficulty walking. She also had moderate scoliosis with a curve to the left. Dermatological examination showed multiple reddish cerebriform hyperplasia in both plantar areas; the right side was more severely involved (Figure 1A). There was macrodactyly of 2 toes on the right foot (Figure 1B). All results of routine blood examinations were within reference range. There were no abnormalities noted in the abdominal ultrasound and cardiac examinations. Plain radiographs of the spine and feet demonstrated scoliosis and exostosis on the calcaneus and bottom of the scaphoid. Histopathologic examination of tissue from the plantar cerebriform hyperplasia revealed hyperkeratosis, slight acanthosis and papillomatosis in the epidermis, and dense collagen bands and sparse elastic fibers in the dermis (Figure 2).
Given the clinical and radiologic manifestation, the diagnosis of Proteus syndrome (PS) was established. After taking into account the severe discomfort and the success of the first surgery, we performed a resection and full-thickness skin graft surgery once again. The feet recovered without any discomfort in daily life. The appearance of the skin graft area was normal 1 year following surgery (Figure 3). She was treated with spinal plate fixation at another institution, progressed well for 2 years, and was subsequently lost to follow-up.
Proteus syndrome is a multisystem disorder with a difficult diagnosis due to the variability of its manifestations. The worldwide incidence of this rare disorder is less than 1 per 1 million individuals, and it is thought to be caused by a somatic genetic alteration.1 Clinical characteristics include bone abnormalities, vascular malformations, dysregulation of fatty tissue, linear verrucous epidermal nevus, and cerebriform connective tissue nevus (CCTN). Although CCTN is not a common finding in patients with PS, it is considered a fairly specific sign with the greatest impact for the diagnosis of PS.2
The general feature of PS--asymmetric disproportionate overgrowth of tissues--appears at 6 to 18 months of age, which makes it challenging to diagnose disease earlier. The CCTN in our patient was present since 1 year of age.
To make a diagnosis of PS, one must have all the general criteria and various specific criteria. The revised diagnostic criteria for PS are given in the Table.3 According to the diagnostic criteria, our patient fulfilled the mandatory general criteria and had plantar CCTN, epidermal nevus, and dysregulated adipose tissue. The CCTN has notable diagnostic value in mildly affected patients, as it is absent in diseases included in the differential diagnosis such as neurofibromatosis, Klippel-Trenaunay-Weber syndrome, Maffucci syndrome, and Bannayan-Riley-Ruvalcaba syndrome. Hemihyperplasia-multiple lipomatosis syndrome and CLOVES (congenital, lipomatous overgrowth, vascular malformations, epidermal nevi, and scoliosis/spinal/skeletal anomalies) syndrome also can present on the plantar surfaces, and lesions may be overgrown at birth but are softer and compressible, have wrinkles instead of deep folds, and tend to grow with the child rather than disproportionately as in PS.4
The epidermal nevi and vascular malformations generally do not spread or increase in number. In contrast, CCTN in PS grows throughout childhood but tends to remain stable in adulthood.4 Postponing surgical treatment until skin lesions stabilize appears to be the best option. However, for practical purposes, surgical intervention may be required at an earlier phase to address the severe functional and cosmetic consequences. Some patients require multiple orthopedic procedures over the ensuing years or decades to control the hyperplasia.3 New CCTN that developed from the prior surgical incision, macrodactyly of the fourth and fifth right toes, and scoliosis appeared when the patient came to our clinic for retreatment 1 year after the initial presentation. The asymmetrical and disproportionate overgrowth of tissues had moderately accelerated in that period. Considering the increasingly impaired walking, we performed a second surgery. On follow-up visits, the patient expressed improvement in daily life.
Studies had been performed to clarify the genetic bases of PS, and the somatic activating mutation in AKT1 (AKT serine/threonine kinase 1) was reported to be the cause of the disease.5,6 Germline PTEN (phosphatase and tensin homolog) mutations have been identified in some patients with overgrowth abnormalities of PS. However, given the misdiagnosis of PS with PTEN mutations and the notion that a gene alone cannot result in PS, the loss-of-function mutations of LEMD3 that have been reported in familial cutaneous collagenomas also may be related to the abnormal growth of connective and bone tissues that are typical of PS.7,8 Lindhurst et al5 concluded that PS is caused by a somatic activating mutation in AKT1, which proved the hypothesis of somatic mosaicism and implicated activation of the PI3K-AKT pathway in the characteristic clinical findings of overgrowth and tumor susceptibility in this disorder. AKT1 is activated by loss-of-function mutations in PTEN, which explains why patients with these mutations (eg, those with the segmental overgrowth, lipomatosis, arteriovenous malformation, epidermal nevus, SOLAMEN [segmental overgrowth, lipomatosis, arteriovenous malformation, and epidermal nevus] syndrome) and patients with activating mutations in AKT1 (eg, those with PS) have overlapping but distinct clinical manifestations. Molecular genetic testing may be useful to confirm the diagnosis in individuals who meet clinical criteria and to establish the diagnosis in individuals with clinical findings that are ambiguous or mild. Further studies are necessary to progress the understanding and management of PS, which will require cooperation of geneticists, surgeons, and other specialists.
- Popescu MD, Burnei G, Draghici L, et al. Proteus syndrome: a difficult diagnosis and management plan. J Med Life. 2014;7:563-566.
- Schepis C, Greco D, Siragusa M, et al. Cerebriform plantar hyperplasia: the major cutaneous feature of Proteus syndrome. Int J Dermatol. 2008;47:374-376.
- Biesecker L. The challenges of Proteus syndrome: diagnosis and management. Eur J Hum Genet. 2006;14:1151-1157.
- Beachkofsky TM, Sapp JC, Biesecker LG, et al. Progressive overgrowth of the cerebriform connective tissue nevus in patients with Proteus syndrome. J Am Acad Dermatol. 2010;63:799-804.
- Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365:611-619.
- Wieland I, Tinschert S, Zenker M. High-level somatic mosaicism of AKT1 c.49G>A mutation in skin scrapings from epidermal nevi enables non-invasive molecular diagnosis in patients with Proteus syndrome. Am J Med Genet A. 2013;161A:889-891.
- Cohen MJ, Turner JT, Biesecker LG. Proteus syndrome: misdiagnosis with PTEN mutations. Am J Med Genet A. 2003;122A:323-324.
- Di Stefani A, Gabellini M, Ferlosio A, et al. Cerebriform plantar hyperplasia: the clinico-pathological hallmark of Proteus syndrome. Acta Derm Venereol. 2011;91:580-581.
- Popescu MD, Burnei G, Draghici L, et al. Proteus syndrome: a difficult diagnosis and management plan. J Med Life. 2014;7:563-566.
- Schepis C, Greco D, Siragusa M, et al. Cerebriform plantar hyperplasia: the major cutaneous feature of Proteus syndrome. Int J Dermatol. 2008;47:374-376.
- Biesecker L. The challenges of Proteus syndrome: diagnosis and management. Eur J Hum Genet. 2006;14:1151-1157.
- Beachkofsky TM, Sapp JC, Biesecker LG, et al. Progressive overgrowth of the cerebriform connective tissue nevus in patients with Proteus syndrome. J Am Acad Dermatol. 2010;63:799-804.
- Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365:611-619.
- Wieland I, Tinschert S, Zenker M. High-level somatic mosaicism of AKT1 c.49G>A mutation in skin scrapings from epidermal nevi enables non-invasive molecular diagnosis in patients with Proteus syndrome. Am J Med Genet A. 2013;161A:889-891.
- Cohen MJ, Turner JT, Biesecker LG. Proteus syndrome: misdiagnosis with PTEN mutations. Am J Med Genet A. 2003;122A:323-324.
- Di Stefani A, Gabellini M, Ferlosio A, et al. Cerebriform plantar hyperplasia: the clinico-pathological hallmark of Proteus syndrome. Acta Derm Venereol. 2011;91:580-581.
Practice Points
- Proteus syndrome (PS) is a rare mosaic condition characterized by progressive overgrowth of skin, connective tissue, brain tissue, and other tissues.
- A somatic activating mutation of the AKT1 gene has been identified as a cause for developing PS.
- Distinct cutaneous features, including cerebriform connective tissue nevi (CCTN), epidermal nevi, vascular malformations, and adipose abnormalities, can alert the dermatologist to the underlying condition before the onset of asymmetric skeletal overgrowth.
- The CCTN in PS grows throughout childhood but tends to remain stable in adulthood. Postponing surgical treatment until skin lesions stabilize appears to be the best option. However, for practical purposes, surgical intervention may be required at an earlier phase to address the severe functional and cosmetic consequences.
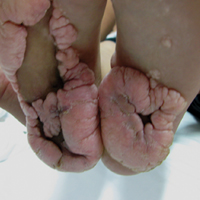
Acute Inflammatory Skin Reaction During Neutrophil Recovery After Antileukemic Therapy
To the Editor:
A 34-year-old man presented with fever, easy bruising, and pancytopenia with increased peripheral blasts of 77%. Bone marrow biopsy showed hypercellular marrow with 80% to 90% involvement by acute promyelocytic leukemia (APL) with complex cytogenetics: 47,XY,t(4;17;18)(p16;q21,q25;q21.1),+8, ins(15;17)(q22;q21q25). He underwent induction chemotherapy with all-trans retinoic acid (ATRA) and idarubicin, which was complicated by differentiation syndrome that presented with fever and fluid retention. Discontinuation of ATRA and initiation of dexamethasone led to resolution of the symptoms. Complete hematologic and molecular remission was achieved after the induction chemotherapy.
Following a risk-adapted treatment protocol for consolidation therapy,1 he underwent an uneventful first cycle of consolidation therapy. On day 15 of the second cycle of consolidation therapy with ATRA and mitoxantrone he was hospitalized with a fever (temperature, 38°C) in a setting of neutropenia (absolute neutrophil count [ANC], 0/µL [reference range, 1500–7200/µL]). He was empirically treated with ceftazidime and vancomycin and maintained on prophylactic acyclovir and fluconazole. Routine workup was negative for infection. He became afebrile within 24 hours. With negative infectious workup, vancomycin was discontinued on day 17. On day 33 he again developed a fever (temperature, 38.8°C) when the ANC started to recover (570/µL). A new skin rash was noted at this time. Physical examination revealed generalized, nonpruritic, tender, pink papules and plaques with dusky centers and central pustules on the trunk as well as the upper and lower extremities. The palms and soles were spared. The rash was somewhat reminiscent of Sweet syndrome (SS). No vesicles, bullae, or erosions were seen (Figure 1). Repeat blood and urine cultures and chest radiograph were unremarkable. Ceftazidime was discontinued due to concern of drug-associated rash. Within the next 48 hours, the patient developed rigors and a worsening rash that led to reinitiation of broad-spectrum antibiotic coverage with meropenem and vancomycin. Computed tomography of the chest, abdomen, and pelvis did not show any evidence of infection or other abnormalities. Skin biopsy showed an acute folliculitis and multiple foci of mixed granulomatous inflammation consisting of histiocytes, lymphocytes, and neutrophils with focal necrosis present in the dermis, dermis-subcutis junction, and subcutis (Figure 2). Diagnostic features of vasculitis were not seen. Viral cytopathic features were not identified. Tissue culture and special stains including Gram, acid-fast bacteria, and Grocott methenamine silver stains were negative for infectious organisms in the biopsy. Both direct fluorescent antibody study and cell cultures for varicella-zoster virus, cytomegalovirus, and herpes simplex virus also were negative.
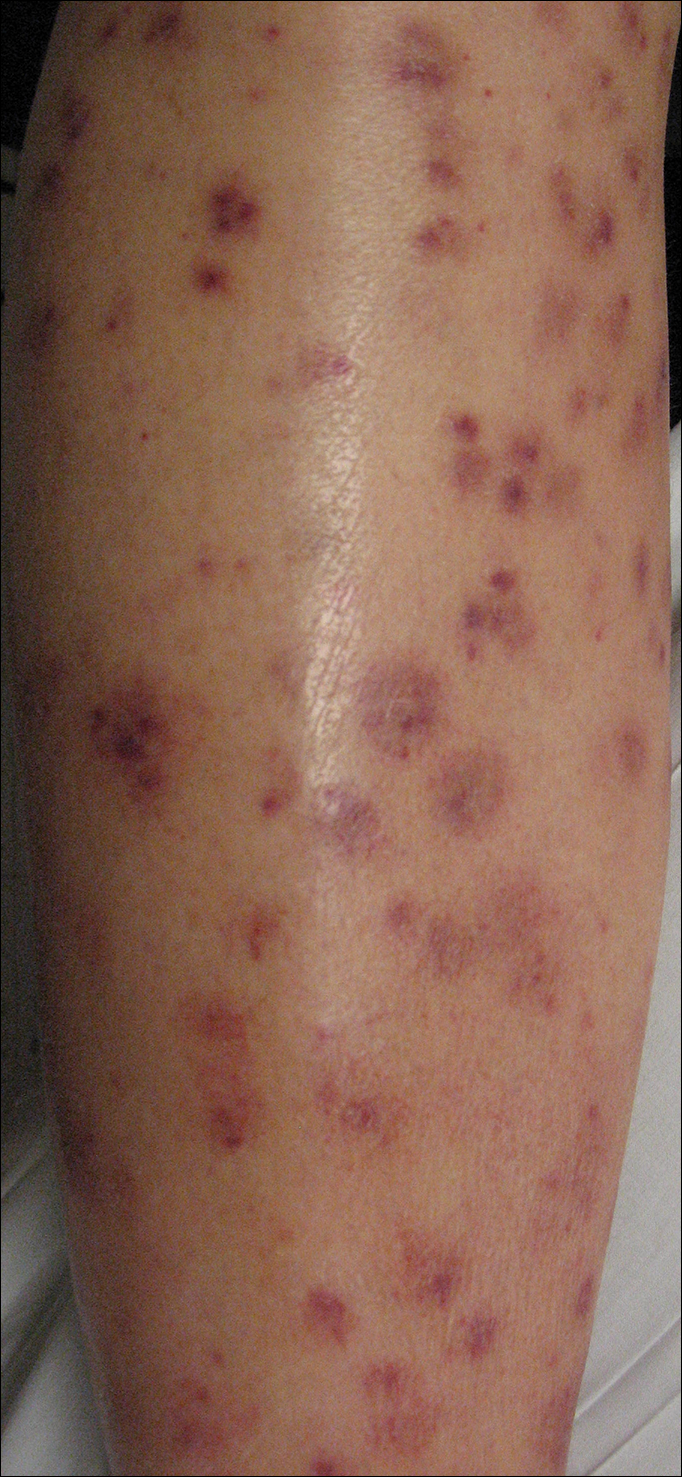
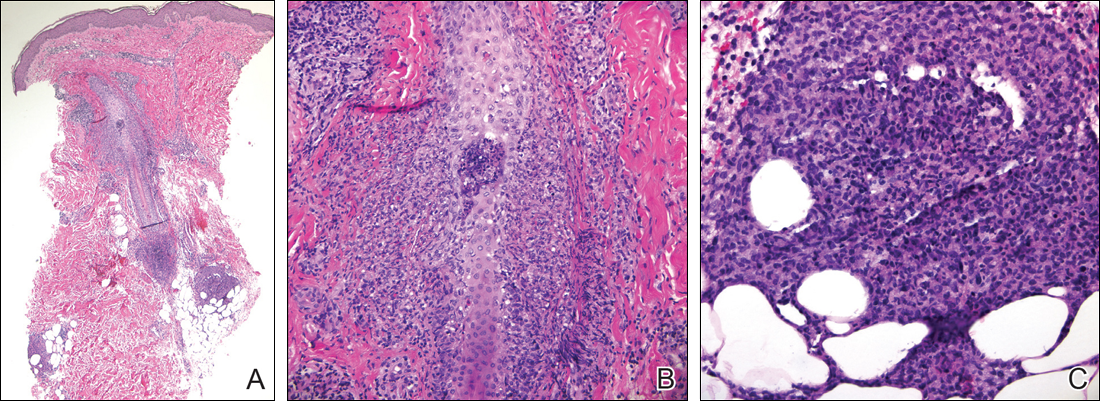
In the absence of microorganisms on skin biopsy and low clinical suspicion of infection, vancomycin and meropenem were discontinued on day 35 and empiric treatment with oral prednisone 40 mg daily was initiated on day 38, which resulted in a rapid improvement of the patient’s rash within 24 hours with complete resolution after a 7-day course of prednisone. Notably, the patient manifested concomitant recovery of the ANC. The patient completed his last cycle of consolidation therapy with ATRA and idarubicin without further complications and remains in molecular remission.
Neutrophilic dermatoses (NDs) are a group of disorders characterized by neutrophilic cutaneous infiltration without evidence of infection. These entities include SS, pyoderma gangrenosum, subcorneal pustular dermatosis, erythema elevatum diutinum, and neutrophilic eccrine hidradenitis.2 Neutrophilic dermatoses commonly present with acute onset of skin lesions and fever. Underlying systemic disease such as malignancy, inflammatory disease, autoimmune disease, pregnancy, and medications are known to be associated with ND. Although the rash clinically was reminiscent of SS, the histopathologic features were inconsistent with SS. Sweet syndrome typically presents with extensive monotonous neutrophilic infiltrates in the dermis. In this case, the neutrophilic infiltrates were localized and associated with the hair follicle, in the dermis and subcutis, and were accompanied by a granulomatous inflammation. Neutrophilic eccrine hidradenitis clinically is similar to SS and the distinction usually is made on the basis of histopathologic examination. Lack of the neutrophilic infiltrates within the eccrine secretary coils in our case did not support the diagnosis of neutrophilic eccrine hidradenitis.
Although the histopathologic features of the presented case were inconsistent with a particular subtype of ND, the clinical presentation and response to corticosteroids suggested that this unusual mixed inflammatory skin reaction might share a similar pathophysiologic mechanism.
A review of 20 patients with sterile neutrophilic folliculitis demonstrated an association with systemic diseases including cutaneous T-cell lymphoma, monoclonal gammopathy, Crohn disease, and autoimmune disorders.3 In acute myeloid leukemia, sterile neutrophilic folliculitis may be part of the initial presentation and responds to induction chemotherapy.4 An extensive search of PubMed articles indexed for MEDLINE using the search terms folliculitis, APL, and neutrophilic dermatoses did not reveal any prior reports of isolated neutrophilic folliculitis or mixed granulomatous reaction in patients with APL in molecular remission.
Although rare, cases of ATRA-induced SS have been reported. Some authors believe that SS in APL may represent a partial form of differentiation syndrome.5 Those cases usually occur during first induction. However, a recurrent episode of differentiation syndrome cannot be excluded in this patient.
A cutaneous reaction to chemotherapy with mitoxantrone as a cause also should be considered, given that the rash occurred only during the second cycle of consolidation therapy when mitoxantrone was used. However, this rash is rare in patients receiving mitoxantrone. The late onset of the rash from the time of last mitoxantrone administration argues against this diagnosis.
In summary, we describe an unusual presentation of a sterile mixed inflammatory skin reaction that occurred in a setting of neutrophil recovery following a second cycle of induction chemotherapy with ATRA and mitoxantrone for APL.
- Sanz MA, Montesinos P, Rayón C, et al; PETHEMA and HOVON Groups. Risk-adapted treatment of acute promyelocytic leukemia based on all-trans retinoic acid and anthracycline with addition of cytarabine in consolidation therapy for high-risk patients: further improvements in treatment outcome [published online April 14, 2010]. Blood. 2010;115:5137-5146.
- Hensley CD, Caughman SW. Neutrophilic dermatoses associated with hematologic disorders. Clin Dermatol. 2000;18:355-367.
- Margro CM, Crowson AN. Sterile neutrophilic folliculitis with perifollicular vasculopathy: a distinctive cutaneous reaction pattern reflecting systemic disease. J Cutan Pathol. 1998;25:215-221.
- Inuzuka M, Tokura Y. Sterile suppurative folliculitis associated with acute myeloblastic leukaemia. Br J Dermatol. 2002;146:904-907.
- Astudillo L, Loche F, Reynish W, et al. Sweet’s syndrome associated with retinoic acid syndrome in a patient with promyelocytic leukemia [published online January 10, 2002]. Ann Hematol. 2002;81:111-114.
To the Editor:
A 34-year-old man presented with fever, easy bruising, and pancytopenia with increased peripheral blasts of 77%. Bone marrow biopsy showed hypercellular marrow with 80% to 90% involvement by acute promyelocytic leukemia (APL) with complex cytogenetics: 47,XY,t(4;17;18)(p16;q21,q25;q21.1),+8, ins(15;17)(q22;q21q25). He underwent induction chemotherapy with all-trans retinoic acid (ATRA) and idarubicin, which was complicated by differentiation syndrome that presented with fever and fluid retention. Discontinuation of ATRA and initiation of dexamethasone led to resolution of the symptoms. Complete hematologic and molecular remission was achieved after the induction chemotherapy.
Following a risk-adapted treatment protocol for consolidation therapy,1 he underwent an uneventful first cycle of consolidation therapy. On day 15 of the second cycle of consolidation therapy with ATRA and mitoxantrone he was hospitalized with a fever (temperature, 38°C) in a setting of neutropenia (absolute neutrophil count [ANC], 0/µL [reference range, 1500–7200/µL]). He was empirically treated with ceftazidime and vancomycin and maintained on prophylactic acyclovir and fluconazole. Routine workup was negative for infection. He became afebrile within 24 hours. With negative infectious workup, vancomycin was discontinued on day 17. On day 33 he again developed a fever (temperature, 38.8°C) when the ANC started to recover (570/µL). A new skin rash was noted at this time. Physical examination revealed generalized, nonpruritic, tender, pink papules and plaques with dusky centers and central pustules on the trunk as well as the upper and lower extremities. The palms and soles were spared. The rash was somewhat reminiscent of Sweet syndrome (SS). No vesicles, bullae, or erosions were seen (Figure 1). Repeat blood and urine cultures and chest radiograph were unremarkable. Ceftazidime was discontinued due to concern of drug-associated rash. Within the next 48 hours, the patient developed rigors and a worsening rash that led to reinitiation of broad-spectrum antibiotic coverage with meropenem and vancomycin. Computed tomography of the chest, abdomen, and pelvis did not show any evidence of infection or other abnormalities. Skin biopsy showed an acute folliculitis and multiple foci of mixed granulomatous inflammation consisting of histiocytes, lymphocytes, and neutrophils with focal necrosis present in the dermis, dermis-subcutis junction, and subcutis (Figure 2). Diagnostic features of vasculitis were not seen. Viral cytopathic features were not identified. Tissue culture and special stains including Gram, acid-fast bacteria, and Grocott methenamine silver stains were negative for infectious organisms in the biopsy. Both direct fluorescent antibody study and cell cultures for varicella-zoster virus, cytomegalovirus, and herpes simplex virus also were negative.
In the absence of microorganisms on skin biopsy and low clinical suspicion of infection, vancomycin and meropenem were discontinued on day 35 and empiric treatment with oral prednisone 40 mg daily was initiated on day 38, which resulted in a rapid improvement of the patient’s rash within 24 hours with complete resolution after a 7-day course of prednisone. Notably, the patient manifested concomitant recovery of the ANC. The patient completed his last cycle of consolidation therapy with ATRA and idarubicin without further complications and remains in molecular remission.
Neutrophilic dermatoses (NDs) are a group of disorders characterized by neutrophilic cutaneous infiltration without evidence of infection. These entities include SS, pyoderma gangrenosum, subcorneal pustular dermatosis, erythema elevatum diutinum, and neutrophilic eccrine hidradenitis.2 Neutrophilic dermatoses commonly present with acute onset of skin lesions and fever. Underlying systemic disease such as malignancy, inflammatory disease, autoimmune disease, pregnancy, and medications are known to be associated with ND. Although the rash clinically was reminiscent of SS, the histopathologic features were inconsistent with SS. Sweet syndrome typically presents with extensive monotonous neutrophilic infiltrates in the dermis. In this case, the neutrophilic infiltrates were localized and associated with the hair follicle, in the dermis and subcutis, and were accompanied by a granulomatous inflammation. Neutrophilic eccrine hidradenitis clinically is similar to SS and the distinction usually is made on the basis of histopathologic examination. Lack of the neutrophilic infiltrates within the eccrine secretary coils in our case did not support the diagnosis of neutrophilic eccrine hidradenitis.
Although the histopathologic features of the presented case were inconsistent with a particular subtype of ND, the clinical presentation and response to corticosteroids suggested that this unusual mixed inflammatory skin reaction might share a similar pathophysiologic mechanism.
A review of 20 patients with sterile neutrophilic folliculitis demonstrated an association with systemic diseases including cutaneous T-cell lymphoma, monoclonal gammopathy, Crohn disease, and autoimmune disorders.3 In acute myeloid leukemia, sterile neutrophilic folliculitis may be part of the initial presentation and responds to induction chemotherapy.4 An extensive search of PubMed articles indexed for MEDLINE using the search terms folliculitis, APL, and neutrophilic dermatoses did not reveal any prior reports of isolated neutrophilic folliculitis or mixed granulomatous reaction in patients with APL in molecular remission.
Although rare, cases of ATRA-induced SS have been reported. Some authors believe that SS in APL may represent a partial form of differentiation syndrome.5 Those cases usually occur during first induction. However, a recurrent episode of differentiation syndrome cannot be excluded in this patient.
A cutaneous reaction to chemotherapy with mitoxantrone as a cause also should be considered, given that the rash occurred only during the second cycle of consolidation therapy when mitoxantrone was used. However, this rash is rare in patients receiving mitoxantrone. The late onset of the rash from the time of last mitoxantrone administration argues against this diagnosis.
In summary, we describe an unusual presentation of a sterile mixed inflammatory skin reaction that occurred in a setting of neutrophil recovery following a second cycle of induction chemotherapy with ATRA and mitoxantrone for APL.
To the Editor:
A 34-year-old man presented with fever, easy bruising, and pancytopenia with increased peripheral blasts of 77%. Bone marrow biopsy showed hypercellular marrow with 80% to 90% involvement by acute promyelocytic leukemia (APL) with complex cytogenetics: 47,XY,t(4;17;18)(p16;q21,q25;q21.1),+8, ins(15;17)(q22;q21q25). He underwent induction chemotherapy with all-trans retinoic acid (ATRA) and idarubicin, which was complicated by differentiation syndrome that presented with fever and fluid retention. Discontinuation of ATRA and initiation of dexamethasone led to resolution of the symptoms. Complete hematologic and molecular remission was achieved after the induction chemotherapy.
Following a risk-adapted treatment protocol for consolidation therapy,1 he underwent an uneventful first cycle of consolidation therapy. On day 15 of the second cycle of consolidation therapy with ATRA and mitoxantrone he was hospitalized with a fever (temperature, 38°C) in a setting of neutropenia (absolute neutrophil count [ANC], 0/µL [reference range, 1500–7200/µL]). He was empirically treated with ceftazidime and vancomycin and maintained on prophylactic acyclovir and fluconazole. Routine workup was negative for infection. He became afebrile within 24 hours. With negative infectious workup, vancomycin was discontinued on day 17. On day 33 he again developed a fever (temperature, 38.8°C) when the ANC started to recover (570/µL). A new skin rash was noted at this time. Physical examination revealed generalized, nonpruritic, tender, pink papules and plaques with dusky centers and central pustules on the trunk as well as the upper and lower extremities. The palms and soles were spared. The rash was somewhat reminiscent of Sweet syndrome (SS). No vesicles, bullae, or erosions were seen (Figure 1). Repeat blood and urine cultures and chest radiograph were unremarkable. Ceftazidime was discontinued due to concern of drug-associated rash. Within the next 48 hours, the patient developed rigors and a worsening rash that led to reinitiation of broad-spectrum antibiotic coverage with meropenem and vancomycin. Computed tomography of the chest, abdomen, and pelvis did not show any evidence of infection or other abnormalities. Skin biopsy showed an acute folliculitis and multiple foci of mixed granulomatous inflammation consisting of histiocytes, lymphocytes, and neutrophils with focal necrosis present in the dermis, dermis-subcutis junction, and subcutis (Figure 2). Diagnostic features of vasculitis were not seen. Viral cytopathic features were not identified. Tissue culture and special stains including Gram, acid-fast bacteria, and Grocott methenamine silver stains were negative for infectious organisms in the biopsy. Both direct fluorescent antibody study and cell cultures for varicella-zoster virus, cytomegalovirus, and herpes simplex virus also were negative.
In the absence of microorganisms on skin biopsy and low clinical suspicion of infection, vancomycin and meropenem were discontinued on day 35 and empiric treatment with oral prednisone 40 mg daily was initiated on day 38, which resulted in a rapid improvement of the patient’s rash within 24 hours with complete resolution after a 7-day course of prednisone. Notably, the patient manifested concomitant recovery of the ANC. The patient completed his last cycle of consolidation therapy with ATRA and idarubicin without further complications and remains in molecular remission.
Neutrophilic dermatoses (NDs) are a group of disorders characterized by neutrophilic cutaneous infiltration without evidence of infection. These entities include SS, pyoderma gangrenosum, subcorneal pustular dermatosis, erythema elevatum diutinum, and neutrophilic eccrine hidradenitis.2 Neutrophilic dermatoses commonly present with acute onset of skin lesions and fever. Underlying systemic disease such as malignancy, inflammatory disease, autoimmune disease, pregnancy, and medications are known to be associated with ND. Although the rash clinically was reminiscent of SS, the histopathologic features were inconsistent with SS. Sweet syndrome typically presents with extensive monotonous neutrophilic infiltrates in the dermis. In this case, the neutrophilic infiltrates were localized and associated with the hair follicle, in the dermis and subcutis, and were accompanied by a granulomatous inflammation. Neutrophilic eccrine hidradenitis clinically is similar to SS and the distinction usually is made on the basis of histopathologic examination. Lack of the neutrophilic infiltrates within the eccrine secretary coils in our case did not support the diagnosis of neutrophilic eccrine hidradenitis.
Although the histopathologic features of the presented case were inconsistent with a particular subtype of ND, the clinical presentation and response to corticosteroids suggested that this unusual mixed inflammatory skin reaction might share a similar pathophysiologic mechanism.
A review of 20 patients with sterile neutrophilic folliculitis demonstrated an association with systemic diseases including cutaneous T-cell lymphoma, monoclonal gammopathy, Crohn disease, and autoimmune disorders.3 In acute myeloid leukemia, sterile neutrophilic folliculitis may be part of the initial presentation and responds to induction chemotherapy.4 An extensive search of PubMed articles indexed for MEDLINE using the search terms folliculitis, APL, and neutrophilic dermatoses did not reveal any prior reports of isolated neutrophilic folliculitis or mixed granulomatous reaction in patients with APL in molecular remission.
Although rare, cases of ATRA-induced SS have been reported. Some authors believe that SS in APL may represent a partial form of differentiation syndrome.5 Those cases usually occur during first induction. However, a recurrent episode of differentiation syndrome cannot be excluded in this patient.
A cutaneous reaction to chemotherapy with mitoxantrone as a cause also should be considered, given that the rash occurred only during the second cycle of consolidation therapy when mitoxantrone was used. However, this rash is rare in patients receiving mitoxantrone. The late onset of the rash from the time of last mitoxantrone administration argues against this diagnosis.
In summary, we describe an unusual presentation of a sterile mixed inflammatory skin reaction that occurred in a setting of neutrophil recovery following a second cycle of induction chemotherapy with ATRA and mitoxantrone for APL.
- Sanz MA, Montesinos P, Rayón C, et al; PETHEMA and HOVON Groups. Risk-adapted treatment of acute promyelocytic leukemia based on all-trans retinoic acid and anthracycline with addition of cytarabine in consolidation therapy for high-risk patients: further improvements in treatment outcome [published online April 14, 2010]. Blood. 2010;115:5137-5146.
- Hensley CD, Caughman SW. Neutrophilic dermatoses associated with hematologic disorders. Clin Dermatol. 2000;18:355-367.
- Margro CM, Crowson AN. Sterile neutrophilic folliculitis with perifollicular vasculopathy: a distinctive cutaneous reaction pattern reflecting systemic disease. J Cutan Pathol. 1998;25:215-221.
- Inuzuka M, Tokura Y. Sterile suppurative folliculitis associated with acute myeloblastic leukaemia. Br J Dermatol. 2002;146:904-907.
- Astudillo L, Loche F, Reynish W, et al. Sweet’s syndrome associated with retinoic acid syndrome in a patient with promyelocytic leukemia [published online January 10, 2002]. Ann Hematol. 2002;81:111-114.
- Sanz MA, Montesinos P, Rayón C, et al; PETHEMA and HOVON Groups. Risk-adapted treatment of acute promyelocytic leukemia based on all-trans retinoic acid and anthracycline with addition of cytarabine in consolidation therapy for high-risk patients: further improvements in treatment outcome [published online April 14, 2010]. Blood. 2010;115:5137-5146.
- Hensley CD, Caughman SW. Neutrophilic dermatoses associated with hematologic disorders. Clin Dermatol. 2000;18:355-367.
- Margro CM, Crowson AN. Sterile neutrophilic folliculitis with perifollicular vasculopathy: a distinctive cutaneous reaction pattern reflecting systemic disease. J Cutan Pathol. 1998;25:215-221.
- Inuzuka M, Tokura Y. Sterile suppurative folliculitis associated with acute myeloblastic leukaemia. Br J Dermatol. 2002;146:904-907.
- Astudillo L, Loche F, Reynish W, et al. Sweet’s syndrome associated with retinoic acid syndrome in a patient with promyelocytic leukemia [published online January 10, 2002]. Ann Hematol. 2002;81:111-114.
Practice Point
- Sterile mixed inflammatory skin reactions reminiscent of neutrophilic dermatoses may occur during neutrophil recovery in patients undergoing therapy for leukemias and need to be considered as part of the differential diagnosis.
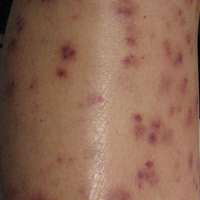
Epidermodysplasia Verruciformis and the Risk for Malignancy
To the Editor:
Epidermodysplasia verruciformis (EV) is a rare autosomal-recessive genodermatosis characterized by widespread infection with specific strains of human papillomavirus (HPV). Patients with EV have a unique susceptibility to acquire HPV due to defects in cellular immunity to the presenting antigens.1 These defects may be related to mutations of the EVER genes or due to acquisition of an immunosuppressive condition.2,3 Infections with HPV-3 and HPV-10 do not lead to the development of malignancies. However, infection with HPV-5, HPV-8, and HPV-14 can lead to the development of nonmelanoma skin cancers, usually squamous cell carcinomas (SCCs), in approximately 60% of patients.3,4 This viral condition lasts throughout the patient’s lifetime and presents as tinea versicolor–like macules and patches. These lesions may be confused with seborrheic keratosis or verruca plana.5 Lesions typically are hypopigmented but occasionally may be hyperpigmented or erythematous. They often are found on the trunk, but lesions on the face, arms, palms, legs, and soles have been reported.5 Mucous membranes are always spared. Epidermodysplasia verruciformis often presents in childhood, except in cases related to acquired immunosuppression. The condition has no sex or racial predilection and no geographical preference.5
A 7-year-old boy (Fitzpatrick skin type V) presented with an asymptomatic rash on the trunk (Figure 1), dorsal aspect of the hands, and forehead. The lesions first appeared 5 years prior on the upper back and upper chest and had recently spread to the forehead and frontal aspect of the scalp. The patient had a history of myelomeningocele, which was corrected at birth with surgical placement of a ventriculoperitoneal shunt. The patient was otherwise healthy and met all appropriate developmental milestones for his age group. Family history revealed consanguinity of the patient’s paternal grandparents who were first cousins. The patient’s mother denied any other family member having similar rashes or lesions.
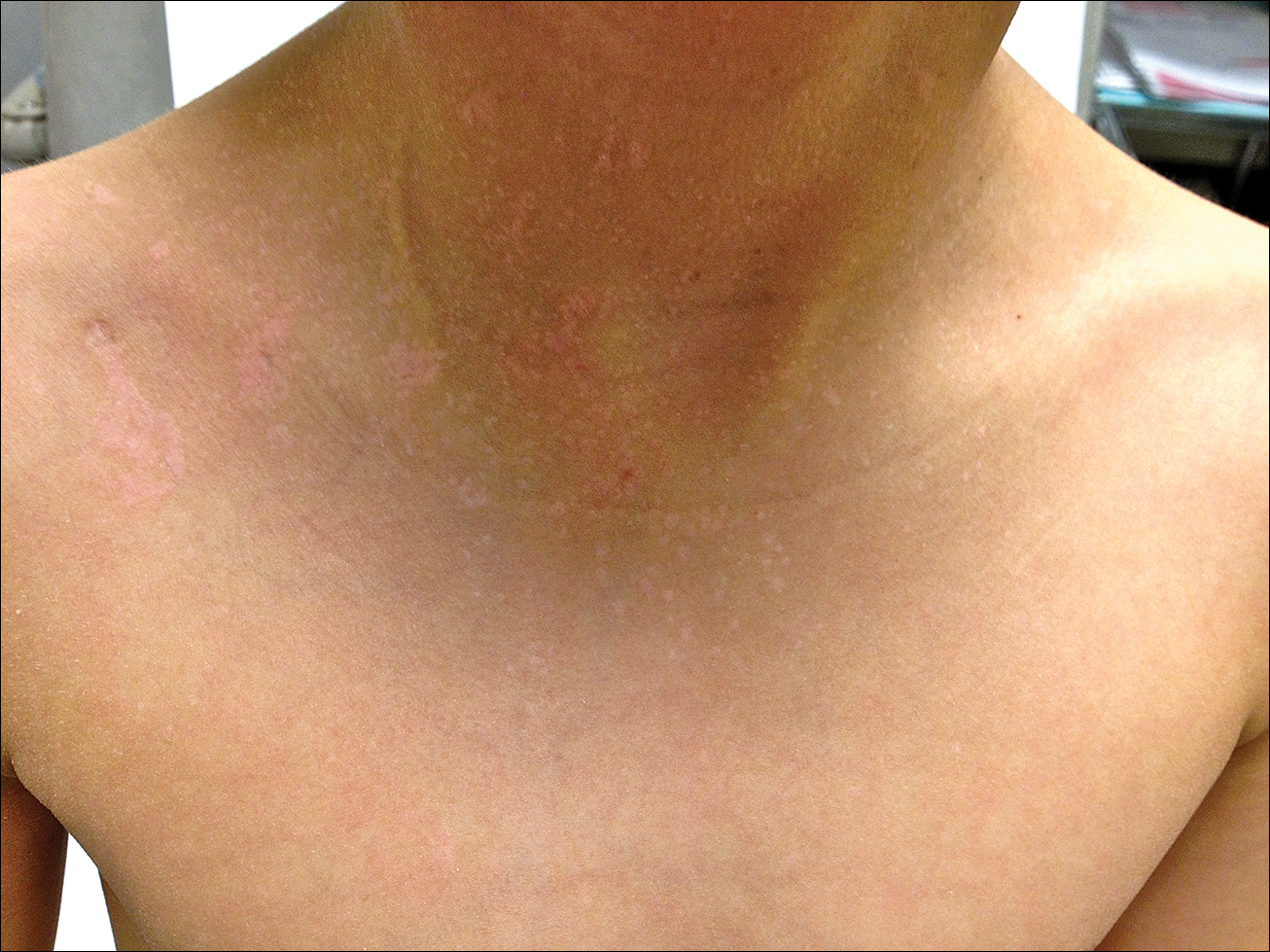
The patient had been treated for pityriasis versicolor on and off for 2 years by another dermatologist. His mother reported faithfully applying ketoconazole cream twice daily for several months with no improvement. She also reported using topical steroids, which did not provide any benefit. The patient and mother denied any associated pruritus, bleeding, burning, or physical discomfort.
Skin examination revealed diffuse, flat, polymorphous, hypopigmented and salmon-colored hyperkeratotic macules and patches with mild scaling on the upper region of the anterior aspect of the chest and upper back (Figure 2A). Additionally, the patient had an extensive number of lesions on the forehead and frontal aspect of the scalp (Figure 2B).

A shave biopsy demonstrated a thick basket weave stratum corneum, koilocytes, and large pale keratinocytes with characteristic blue cytoplasm. These findings were characteristic for EV.
At the patient’s 3-month follow-up visit, he again denied any symptoms associated with the lesions and reported that the appearance was diminishing in severity. On examination there was no evidence of SCC. The mother was advised to discontinue all topical treatments for the patient and return to the office every 3 to 6 months for regular skin surveillance. The mother was further advised to protect the patient from UV radiation with sunscreen and sun-protective clothing.
Epidermodysplasia verruciformis was first reported by Lewandowsky and Lutz6 in 1922. This rare condition often presents in childhood and is characterized by a persistent HPV infection and an autosomal-recessive inheritance pattern. Reports in the literature frequently involve kindreds. Often, patients with EV have a family history of first-degree or second-degree consanguinity.7
The clinical presentation of EV often resembles a pityriasis versicolor–like eruption. However, pityriasis versicolor is less commonly seen in childhood and is more prevalent in patients aged 21 to 30 years, likely due to increased sebum production and changing hormone levels. Furthermore, it is unusual to see pityriasis versicolor affect the face and scalp.8 Lesions of EV vary from hypopigmented and pinkish red macules to confluent patches and hyperkeratotic verrucalike lesions.3 Clinical characteristics also may include dyschromic patches; lesions that resemble flat warts on the trunk, face, and distal arms; and/or lesions that appear similar to seborrheic keratoses on the dorsal aspect of the hands.9,10
Mutations of the EVER gene downregulate a cell’s ability to adequately attack the HPV antigens.11 Although some patients with EV are found to have mutations of the EVER1 and EVER2 genes, a notable portion of patients with EV lack these mutations. Three other causes of EV include acquisition of immunosuppressive conditions including lymphoma, solid organ transplant, and human immunodeficiency virus. If one suspects autosomal-recessive inheritance of EV, genetic testing such as polymerase chain reaction DNA fragment analysis can be performed to determine if there are mutations on the EVER1 or EVER2 genes.12
The inability of patients with EV to mount an immune response to multiple types of HPV increases the risk for developing cutaneous malignancies.7 Additionally, it is known that UV radiation diminishes skin cell immunity, and the combination of EV and UV radiation further increases the risk for developing SCCs.11 The development of nonmelanoma skin cancers usually occurs on sun-exposed skin 20 to 30 years after the onset of lesions, with the highest occurrence of SCCs presenting in the fourth decade of life.1
Protection from UV light exposure is critical to reduce the risk for malignancy. Treatment options for EV lesions have included topical imiquimod 5%, 5-fluorouracil, oral isotretinoin, and intralesional interferon alfa, but patients are often refractory to these interventions. Curettage, surgical excision, electrosurgery, and laser ablation can be effective for individual lesions but carry a greater risk for scarring.1 Photodynamic therapy with aminolevulinic acid and blue light represents a promising option that deserves further study.
Epidermodysplasia verruciformis should be considered as a differential diagnosis in all patients presenting with disseminated lesions resembling pityriasis versicolor that are unresponsive to treatment. A biopsy will help to establish the diagnosis. Patients should minimize sun exposure and report any skin lesions that are changing in appearance.
- Hoffner MV, Camacho FM. Surgical treatment of epidermodysplasia verruciformis. Dermatol Surg. 2010;36:363-367.
- McDermott D, Gammon B, Snijders P. Autosomal dominant epidermodysplasia verruciformis lacking a known EVER1 or EVER2 mutation. Pediatr Dermatol. 2009;26:306-310.
- Patel T, Morrison K, Rady P, et al. Epidermodysplasia verruciformis and susceptibility to HPV. Dis Markers. 2010;29:199-206.
- Hultgren TL, Srinivasan SK, DiMaio DJ. Epidermodysplasia verruciformis occurring in a patient with human immunodeficiency virus: a case report. Cutis. 2007;79:308-311.
- Oliveira W, Netu C, Rady P, et al. Clinical aspects of epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2003;17:394-398.
- Lewandowsky F, Lutz W. Ein Fall einer bisher nicht beschriebenen Hauterkrankung (epidermodysplasia verruciformis). Arch Dermatol Syphilol. 1922;141:193-203.
- Prystowsky S, Herndon J, Freeman R, et al. Epidermodysplasia verruciformis. Am J Dis Child. 1976;130:437-440.
- Kyriakis KP, Terzoudi S, Palamaras I, et al. Pityriasis versicolor prevalence by age and gender. Mycoses. 2006;49:517-518.
- Nuovo G, Ishag M. The histologic spectrum of epidermodysplasia verruciformis. Am J Surg Pathol. 2000;24:1400-1406.
- Jacobelli S, Laude H, Carlotti A, et al. Epidermodysplasia verruciformis in human immunodeficiency virus-infected patients: a marker of human papillomavirus-related disorders not affected by antiretroviral therapy. Arch Dermatol. 2011;147:590-596.
- Rogers HD, MacGregor JL, Nord KM, et al. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:316-320.
- Gober MD, Rady PL, He Q, et al. Novel homozygous frameshift mutation of EVER1 gene in an epidermodysplasia verruciformis patient. J Invest Dermatol. 2007;127:817-820.
To the Editor:
Epidermodysplasia verruciformis (EV) is a rare autosomal-recessive genodermatosis characterized by widespread infection with specific strains of human papillomavirus (HPV). Patients with EV have a unique susceptibility to acquire HPV due to defects in cellular immunity to the presenting antigens.1 These defects may be related to mutations of the EVER genes or due to acquisition of an immunosuppressive condition.2,3 Infections with HPV-3 and HPV-10 do not lead to the development of malignancies. However, infection with HPV-5, HPV-8, and HPV-14 can lead to the development of nonmelanoma skin cancers, usually squamous cell carcinomas (SCCs), in approximately 60% of patients.3,4 This viral condition lasts throughout the patient’s lifetime and presents as tinea versicolor–like macules and patches. These lesions may be confused with seborrheic keratosis or verruca plana.5 Lesions typically are hypopigmented but occasionally may be hyperpigmented or erythematous. They often are found on the trunk, but lesions on the face, arms, palms, legs, and soles have been reported.5 Mucous membranes are always spared. Epidermodysplasia verruciformis often presents in childhood, except in cases related to acquired immunosuppression. The condition has no sex or racial predilection and no geographical preference.5
A 7-year-old boy (Fitzpatrick skin type V) presented with an asymptomatic rash on the trunk (Figure 1), dorsal aspect of the hands, and forehead. The lesions first appeared 5 years prior on the upper back and upper chest and had recently spread to the forehead and frontal aspect of the scalp. The patient had a history of myelomeningocele, which was corrected at birth with surgical placement of a ventriculoperitoneal shunt. The patient was otherwise healthy and met all appropriate developmental milestones for his age group. Family history revealed consanguinity of the patient’s paternal grandparents who were first cousins. The patient’s mother denied any other family member having similar rashes or lesions.
The patient had been treated for pityriasis versicolor on and off for 2 years by another dermatologist. His mother reported faithfully applying ketoconazole cream twice daily for several months with no improvement. She also reported using topical steroids, which did not provide any benefit. The patient and mother denied any associated pruritus, bleeding, burning, or physical discomfort.
Skin examination revealed diffuse, flat, polymorphous, hypopigmented and salmon-colored hyperkeratotic macules and patches with mild scaling on the upper region of the anterior aspect of the chest and upper back (Figure 2A). Additionally, the patient had an extensive number of lesions on the forehead and frontal aspect of the scalp (Figure 2B).
A shave biopsy demonstrated a thick basket weave stratum corneum, koilocytes, and large pale keratinocytes with characteristic blue cytoplasm. These findings were characteristic for EV.
At the patient’s 3-month follow-up visit, he again denied any symptoms associated with the lesions and reported that the appearance was diminishing in severity. On examination there was no evidence of SCC. The mother was advised to discontinue all topical treatments for the patient and return to the office every 3 to 6 months for regular skin surveillance. The mother was further advised to protect the patient from UV radiation with sunscreen and sun-protective clothing.
Epidermodysplasia verruciformis was first reported by Lewandowsky and Lutz6 in 1922. This rare condition often presents in childhood and is characterized by a persistent HPV infection and an autosomal-recessive inheritance pattern. Reports in the literature frequently involve kindreds. Often, patients with EV have a family history of first-degree or second-degree consanguinity.7
The clinical presentation of EV often resembles a pityriasis versicolor–like eruption. However, pityriasis versicolor is less commonly seen in childhood and is more prevalent in patients aged 21 to 30 years, likely due to increased sebum production and changing hormone levels. Furthermore, it is unusual to see pityriasis versicolor affect the face and scalp.8 Lesions of EV vary from hypopigmented and pinkish red macules to confluent patches and hyperkeratotic verrucalike lesions.3 Clinical characteristics also may include dyschromic patches; lesions that resemble flat warts on the trunk, face, and distal arms; and/or lesions that appear similar to seborrheic keratoses on the dorsal aspect of the hands.9,10
Mutations of the EVER gene downregulate a cell’s ability to adequately attack the HPV antigens.11 Although some patients with EV are found to have mutations of the EVER1 and EVER2 genes, a notable portion of patients with EV lack these mutations. Three other causes of EV include acquisition of immunosuppressive conditions including lymphoma, solid organ transplant, and human immunodeficiency virus. If one suspects autosomal-recessive inheritance of EV, genetic testing such as polymerase chain reaction DNA fragment analysis can be performed to determine if there are mutations on the EVER1 or EVER2 genes.12
The inability of patients with EV to mount an immune response to multiple types of HPV increases the risk for developing cutaneous malignancies.7 Additionally, it is known that UV radiation diminishes skin cell immunity, and the combination of EV and UV radiation further increases the risk for developing SCCs.11 The development of nonmelanoma skin cancers usually occurs on sun-exposed skin 20 to 30 years after the onset of lesions, with the highest occurrence of SCCs presenting in the fourth decade of life.1
Protection from UV light exposure is critical to reduce the risk for malignancy. Treatment options for EV lesions have included topical imiquimod 5%, 5-fluorouracil, oral isotretinoin, and intralesional interferon alfa, but patients are often refractory to these interventions. Curettage, surgical excision, electrosurgery, and laser ablation can be effective for individual lesions but carry a greater risk for scarring.1 Photodynamic therapy with aminolevulinic acid and blue light represents a promising option that deserves further study.
Epidermodysplasia verruciformis should be considered as a differential diagnosis in all patients presenting with disseminated lesions resembling pityriasis versicolor that are unresponsive to treatment. A biopsy will help to establish the diagnosis. Patients should minimize sun exposure and report any skin lesions that are changing in appearance.
To the Editor:
Epidermodysplasia verruciformis (EV) is a rare autosomal-recessive genodermatosis characterized by widespread infection with specific strains of human papillomavirus (HPV). Patients with EV have a unique susceptibility to acquire HPV due to defects in cellular immunity to the presenting antigens.1 These defects may be related to mutations of the EVER genes or due to acquisition of an immunosuppressive condition.2,3 Infections with HPV-3 and HPV-10 do not lead to the development of malignancies. However, infection with HPV-5, HPV-8, and HPV-14 can lead to the development of nonmelanoma skin cancers, usually squamous cell carcinomas (SCCs), in approximately 60% of patients.3,4 This viral condition lasts throughout the patient’s lifetime and presents as tinea versicolor–like macules and patches. These lesions may be confused with seborrheic keratosis or verruca plana.5 Lesions typically are hypopigmented but occasionally may be hyperpigmented or erythematous. They often are found on the trunk, but lesions on the face, arms, palms, legs, and soles have been reported.5 Mucous membranes are always spared. Epidermodysplasia verruciformis often presents in childhood, except in cases related to acquired immunosuppression. The condition has no sex or racial predilection and no geographical preference.5
A 7-year-old boy (Fitzpatrick skin type V) presented with an asymptomatic rash on the trunk (Figure 1), dorsal aspect of the hands, and forehead. The lesions first appeared 5 years prior on the upper back and upper chest and had recently spread to the forehead and frontal aspect of the scalp. The patient had a history of myelomeningocele, which was corrected at birth with surgical placement of a ventriculoperitoneal shunt. The patient was otherwise healthy and met all appropriate developmental milestones for his age group. Family history revealed consanguinity of the patient’s paternal grandparents who were first cousins. The patient’s mother denied any other family member having similar rashes or lesions.
The patient had been treated for pityriasis versicolor on and off for 2 years by another dermatologist. His mother reported faithfully applying ketoconazole cream twice daily for several months with no improvement. She also reported using topical steroids, which did not provide any benefit. The patient and mother denied any associated pruritus, bleeding, burning, or physical discomfort.
Skin examination revealed diffuse, flat, polymorphous, hypopigmented and salmon-colored hyperkeratotic macules and patches with mild scaling on the upper region of the anterior aspect of the chest and upper back (Figure 2A). Additionally, the patient had an extensive number of lesions on the forehead and frontal aspect of the scalp (Figure 2B).
A shave biopsy demonstrated a thick basket weave stratum corneum, koilocytes, and large pale keratinocytes with characteristic blue cytoplasm. These findings were characteristic for EV.
At the patient’s 3-month follow-up visit, he again denied any symptoms associated with the lesions and reported that the appearance was diminishing in severity. On examination there was no evidence of SCC. The mother was advised to discontinue all topical treatments for the patient and return to the office every 3 to 6 months for regular skin surveillance. The mother was further advised to protect the patient from UV radiation with sunscreen and sun-protective clothing.
Epidermodysplasia verruciformis was first reported by Lewandowsky and Lutz6 in 1922. This rare condition often presents in childhood and is characterized by a persistent HPV infection and an autosomal-recessive inheritance pattern. Reports in the literature frequently involve kindreds. Often, patients with EV have a family history of first-degree or second-degree consanguinity.7
The clinical presentation of EV often resembles a pityriasis versicolor–like eruption. However, pityriasis versicolor is less commonly seen in childhood and is more prevalent in patients aged 21 to 30 years, likely due to increased sebum production and changing hormone levels. Furthermore, it is unusual to see pityriasis versicolor affect the face and scalp.8 Lesions of EV vary from hypopigmented and pinkish red macules to confluent patches and hyperkeratotic verrucalike lesions.3 Clinical characteristics also may include dyschromic patches; lesions that resemble flat warts on the trunk, face, and distal arms; and/or lesions that appear similar to seborrheic keratoses on the dorsal aspect of the hands.9,10
Mutations of the EVER gene downregulate a cell’s ability to adequately attack the HPV antigens.11 Although some patients with EV are found to have mutations of the EVER1 and EVER2 genes, a notable portion of patients with EV lack these mutations. Three other causes of EV include acquisition of immunosuppressive conditions including lymphoma, solid organ transplant, and human immunodeficiency virus. If one suspects autosomal-recessive inheritance of EV, genetic testing such as polymerase chain reaction DNA fragment analysis can be performed to determine if there are mutations on the EVER1 or EVER2 genes.12
The inability of patients with EV to mount an immune response to multiple types of HPV increases the risk for developing cutaneous malignancies.7 Additionally, it is known that UV radiation diminishes skin cell immunity, and the combination of EV and UV radiation further increases the risk for developing SCCs.11 The development of nonmelanoma skin cancers usually occurs on sun-exposed skin 20 to 30 years after the onset of lesions, with the highest occurrence of SCCs presenting in the fourth decade of life.1
Protection from UV light exposure is critical to reduce the risk for malignancy. Treatment options for EV lesions have included topical imiquimod 5%, 5-fluorouracil, oral isotretinoin, and intralesional interferon alfa, but patients are often refractory to these interventions. Curettage, surgical excision, electrosurgery, and laser ablation can be effective for individual lesions but carry a greater risk for scarring.1 Photodynamic therapy with aminolevulinic acid and blue light represents a promising option that deserves further study.
Epidermodysplasia verruciformis should be considered as a differential diagnosis in all patients presenting with disseminated lesions resembling pityriasis versicolor that are unresponsive to treatment. A biopsy will help to establish the diagnosis. Patients should minimize sun exposure and report any skin lesions that are changing in appearance.
- Hoffner MV, Camacho FM. Surgical treatment of epidermodysplasia verruciformis. Dermatol Surg. 2010;36:363-367.
- McDermott D, Gammon B, Snijders P. Autosomal dominant epidermodysplasia verruciformis lacking a known EVER1 or EVER2 mutation. Pediatr Dermatol. 2009;26:306-310.
- Patel T, Morrison K, Rady P, et al. Epidermodysplasia verruciformis and susceptibility to HPV. Dis Markers. 2010;29:199-206.
- Hultgren TL, Srinivasan SK, DiMaio DJ. Epidermodysplasia verruciformis occurring in a patient with human immunodeficiency virus: a case report. Cutis. 2007;79:308-311.
- Oliveira W, Netu C, Rady P, et al. Clinical aspects of epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2003;17:394-398.
- Lewandowsky F, Lutz W. Ein Fall einer bisher nicht beschriebenen Hauterkrankung (epidermodysplasia verruciformis). Arch Dermatol Syphilol. 1922;141:193-203.
- Prystowsky S, Herndon J, Freeman R, et al. Epidermodysplasia verruciformis. Am J Dis Child. 1976;130:437-440.
- Kyriakis KP, Terzoudi S, Palamaras I, et al. Pityriasis versicolor prevalence by age and gender. Mycoses. 2006;49:517-518.
- Nuovo G, Ishag M. The histologic spectrum of epidermodysplasia verruciformis. Am J Surg Pathol. 2000;24:1400-1406.
- Jacobelli S, Laude H, Carlotti A, et al. Epidermodysplasia verruciformis in human immunodeficiency virus-infected patients: a marker of human papillomavirus-related disorders not affected by antiretroviral therapy. Arch Dermatol. 2011;147:590-596.
- Rogers HD, MacGregor JL, Nord KM, et al. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:316-320.
- Gober MD, Rady PL, He Q, et al. Novel homozygous frameshift mutation of EVER1 gene in an epidermodysplasia verruciformis patient. J Invest Dermatol. 2007;127:817-820.
- Hoffner MV, Camacho FM. Surgical treatment of epidermodysplasia verruciformis. Dermatol Surg. 2010;36:363-367.
- McDermott D, Gammon B, Snijders P. Autosomal dominant epidermodysplasia verruciformis lacking a known EVER1 or EVER2 mutation. Pediatr Dermatol. 2009;26:306-310.
- Patel T, Morrison K, Rady P, et al. Epidermodysplasia verruciformis and susceptibility to HPV. Dis Markers. 2010;29:199-206.
- Hultgren TL, Srinivasan SK, DiMaio DJ. Epidermodysplasia verruciformis occurring in a patient with human immunodeficiency virus: a case report. Cutis. 2007;79:308-311.
- Oliveira W, Netu C, Rady P, et al. Clinical aspects of epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2003;17:394-398.
- Lewandowsky F, Lutz W. Ein Fall einer bisher nicht beschriebenen Hauterkrankung (epidermodysplasia verruciformis). Arch Dermatol Syphilol. 1922;141:193-203.
- Prystowsky S, Herndon J, Freeman R, et al. Epidermodysplasia verruciformis. Am J Dis Child. 1976;130:437-440.
- Kyriakis KP, Terzoudi S, Palamaras I, et al. Pityriasis versicolor prevalence by age and gender. Mycoses. 2006;49:517-518.
- Nuovo G, Ishag M. The histologic spectrum of epidermodysplasia verruciformis. Am J Surg Pathol. 2000;24:1400-1406.
- Jacobelli S, Laude H, Carlotti A, et al. Epidermodysplasia verruciformis in human immunodeficiency virus-infected patients: a marker of human papillomavirus-related disorders not affected by antiretroviral therapy. Arch Dermatol. 2011;147:590-596.
- Rogers HD, MacGregor JL, Nord KM, et al. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:316-320.
- Gober MD, Rady PL, He Q, et al. Novel homozygous frameshift mutation of EVER1 gene in an epidermodysplasia verruciformis patient. J Invest Dermatol. 2007;127:817-820.
Practice Points
- Epidermodysplasia verruciformis (EV) is a rare genodermatosis that usually presents in early childhood and presents as verrucous papules and plaques most commonly on the skin of the head, neck, and upper extremities. It often is misdiagnosed at pityriasis versicolor.
- Mutations of the EVER1 and EVER2 genes have been identified as a source for developing EV.
- Epidermodysplasia verruciformis produces wartlike lesions in individuals who have a unique susceptibility to acquiring the human papillomavirus and early onset of nonmelanoma skin cancers, most commonly squamous cell carcinomas related to viral oncogenesis.
- Avoidance and protection from UV exposure is a critical component of treatment plans for patients with EV.
Diagnosis of a Rapidly Growing Preauricular Nodule: Chondroid Syringoma or Pleomorphic Adenoma?
To the Editor:
Chondroid syringoma is a rare benign mixed tumor that originates from the sweat glands, typically presenting with both epithelial and mesenchymal components.1 It differs from pleomorphic adenoma, which arises from salivary glands. The surgical approach for complete excision is different for the 2 tumors; therefore, definitive diagnosis is important. For chondroid syringoma, total excision is recommended,2 while for pleomorphic adenoma, extracapsular dissection or superficial parotidectomy is commonly indicated. We report a case of a preauricular nodule at presentation and highlight the importance of differentiating a chondroid syringoma from a pleomorphic adenoma. This case is unique because of the anatomic location of the nodule, making diagnosis difficult because the tumor was abutting the parotid gland and a biopsy included normal salivary gland cells.
A 19-year-old man with a history of moderate acne on the shoulders, back, and face presented with a rapidly growing, painless nodule on right preauricular region of 6 months’ duration. The nodule was originally thought to be acne related and monitored, as the patient was asymptomatic. On examination the patient was found to have a firm, fixed, nontender, subcutaneous nodule overlying the right temporomandibular joint just anterior to the right tragus (Figure 1). Laboratory results were unremarkable. Computed tomography showed a subcutaneous nonaggressive-appearing soft-tissue mass measuring 16×17×12 mm just anterior and inferior to the external auditory canal cartilaginous segment with no bony abnormalities. The patient was initially treated with incomplete excision of the area for a presumed sebaceous cyst; 2 months later, an abnormal biopsy prompted a complete excisional biopsy.
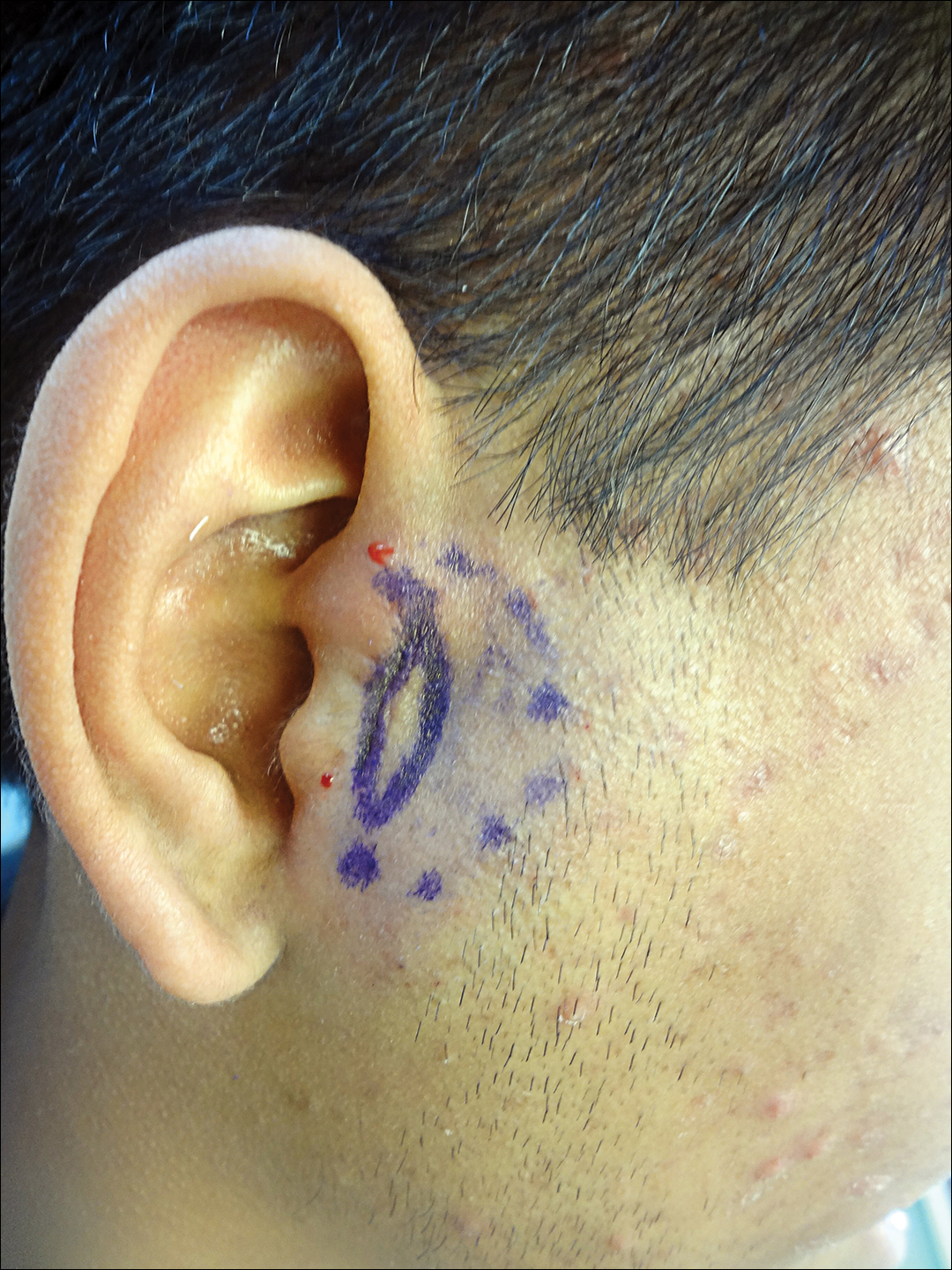
Histologically, the initial incomplete excision biopsy revealed myxoid and chondroid tissue with glandular elements and adjacent lymph node with strong positivity for S-100 protein and moderate positivity for glial fibrillary acid protein, consistent with chondroid syringoma (Figure 2). Histological findings on second excision biopsy performed 2 months later showed tumor cells surrounded by normal salivary gland cells; therefore, it was difficult to define the origin of this tumor. Subsequent magnetic resonance imaging showed no evidence of the tumor and normal parotid gland borders.
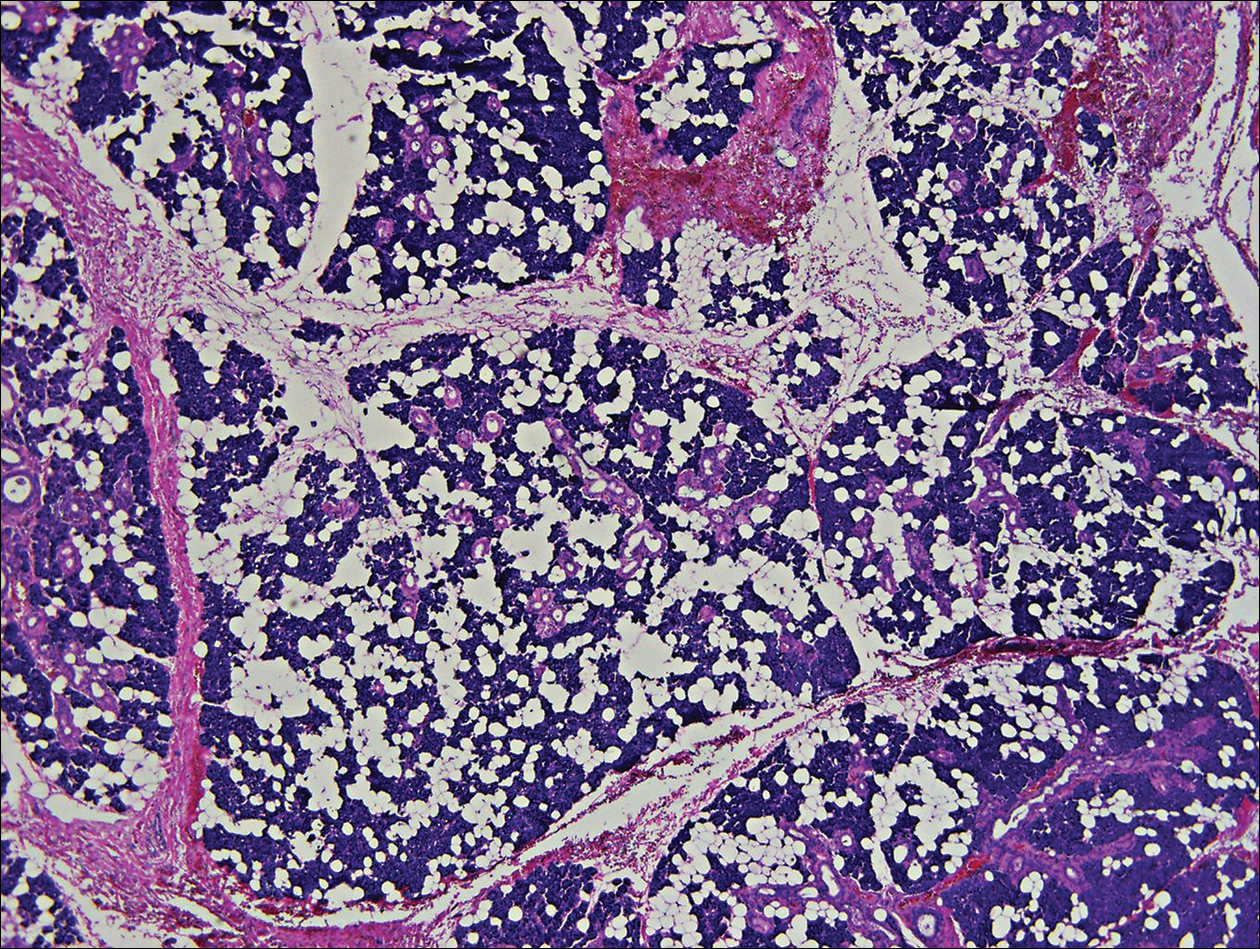
Chondroid syringoma is a rare nonmelanoma skin tumor of the head and neck, mostly benign in nature but with malignant potential. Predominantly, it presents in males as an asymptomatic, slow-growing, nontender nodule.2 Malignant chondroid syringomas are more rare, typically appear on the trunk and legs of females, and present as rapidly growing hard nodules. They can arise de novo or from a preexisting chondroid syringoma and can metastasize.3,4
Clinically and histologically, chondroid syringoma resembles a pleomorphic adenoma. Its diagnosis is dependent on the clinical location to exclude origin in a salivary gland.5 Folliculosebaceous and myoepithelial differentiation within the tumor has been reported.6 Immunocytochemistry is the same in both types and is used to identify 2 prominent components—epithelial and mesenchymal—found in both chondroid syringoma and pleomorphic adenoma. Immunocytochemistry differentiates the epithelial component, which expresses cytokeratin, epithelial membrane antigen, and carcinoembryonic antigen. In contrast, the mesenchymal component expresses S-100, vimentin, and neuron‐specific enolase, and less often glial fibrillary acidic protein, smooth muscle actin, calponin, or p63.5,7,8 Identification of both layers is a distinctive trait of both tumors, rendering it apart from other conditions in the differential diagnosis.5
Typical treatment options include excision, electrodesiccation, dermabrasion, and argon or CO2 laser. Total excision is recommended if there is a benign tumor and complete excision is a cure.2 One case of recurrent benign chondroid syringoma was treated by Mohs micrographic surgery on the eyebrow9; however, Mohs surgery was not recommended in our case due to concerns of spread if malignant as well as an unknown tumor depth, as these tumors have a tendency for deep infiltration.
Due to its anatomical location and presentation as an anterior preauricular mass, it was difficult to differentiate between chondroid syringoma from sweat gland origin and pleomorphic adenoma from the salivary gland. As seen in our case, it is important for physicians to be aware of the differential diagnosis for mixed tumors because it can have a notable effect on the type of surgical therapy and follow-up management.
- Hirsch P, Helwig EB. Chondroid syringoma. Arch Dermatol. 1961;84:835-847.
- Turhan-Haktanir N, Sahin O, Bukulmez A, et al. Chondroid syringoma in a child. Pediatr Dermatol. 2007;24:505-507.
- Mathiasen RA, Rasgon BM, Rumore G. Malignant chondroid syringoma of the face: a first reported case. Otolaryngol Head Neck Surg. 2005;133:305-307.
- Barnett MD, Wallack MK, Zuretti A, et al. Recurrent malignant chondroid syringoma of the foot: a case report and review of the literature. Am J Clin Oncol. 2000;23:227-232.
- Dubb M, Michelow P. Cytologic features of chondroid syringoma in fine needle aspiration biopsies a report of 3 cases. Acta Cytol. 2010;54:183-186.
- Rauso R, Santagata M, Tartaro G, et al. Chondroid syringoma: rare tumor of orofacial region. Minerva Stomatol. 2009;58:383-388.
- Metzler G, Schaumburg-Lever G, Hornstein O, et al. Malignant chondroid syringoma: immunohistopathology. Am J Dermatopathol. 1996;18:83-89.
- Argenyi ZB, Balogh K, Goeken JA. Immunohistochemical characterization of chondroid syringomas. Am J Clin Pathol. 1988;90:662-669.
- Walls AC, Deng A, Geist DE. Mohs micrographic surgery for recurrent chondroid syringoma of the eyebrow. Dermatol Surg. 2012;38:800-802.
To the Editor:
Chondroid syringoma is a rare benign mixed tumor that originates from the sweat glands, typically presenting with both epithelial and mesenchymal components.1 It differs from pleomorphic adenoma, which arises from salivary glands. The surgical approach for complete excision is different for the 2 tumors; therefore, definitive diagnosis is important. For chondroid syringoma, total excision is recommended,2 while for pleomorphic adenoma, extracapsular dissection or superficial parotidectomy is commonly indicated. We report a case of a preauricular nodule at presentation and highlight the importance of differentiating a chondroid syringoma from a pleomorphic adenoma. This case is unique because of the anatomic location of the nodule, making diagnosis difficult because the tumor was abutting the parotid gland and a biopsy included normal salivary gland cells.
A 19-year-old man with a history of moderate acne on the shoulders, back, and face presented with a rapidly growing, painless nodule on right preauricular region of 6 months’ duration. The nodule was originally thought to be acne related and monitored, as the patient was asymptomatic. On examination the patient was found to have a firm, fixed, nontender, subcutaneous nodule overlying the right temporomandibular joint just anterior to the right tragus (Figure 1). Laboratory results were unremarkable. Computed tomography showed a subcutaneous nonaggressive-appearing soft-tissue mass measuring 16×17×12 mm just anterior and inferior to the external auditory canal cartilaginous segment with no bony abnormalities. The patient was initially treated with incomplete excision of the area for a presumed sebaceous cyst; 2 months later, an abnormal biopsy prompted a complete excisional biopsy.
Histologically, the initial incomplete excision biopsy revealed myxoid and chondroid tissue with glandular elements and adjacent lymph node with strong positivity for S-100 protein and moderate positivity for glial fibrillary acid protein, consistent with chondroid syringoma (Figure 2). Histological findings on second excision biopsy performed 2 months later showed tumor cells surrounded by normal salivary gland cells; therefore, it was difficult to define the origin of this tumor. Subsequent magnetic resonance imaging showed no evidence of the tumor and normal parotid gland borders.
Chondroid syringoma is a rare nonmelanoma skin tumor of the head and neck, mostly benign in nature but with malignant potential. Predominantly, it presents in males as an asymptomatic, slow-growing, nontender nodule.2 Malignant chondroid syringomas are more rare, typically appear on the trunk and legs of females, and present as rapidly growing hard nodules. They can arise de novo or from a preexisting chondroid syringoma and can metastasize.3,4
Clinically and histologically, chondroid syringoma resembles a pleomorphic adenoma. Its diagnosis is dependent on the clinical location to exclude origin in a salivary gland.5 Folliculosebaceous and myoepithelial differentiation within the tumor has been reported.6 Immunocytochemistry is the same in both types and is used to identify 2 prominent components—epithelial and mesenchymal—found in both chondroid syringoma and pleomorphic adenoma. Immunocytochemistry differentiates the epithelial component, which expresses cytokeratin, epithelial membrane antigen, and carcinoembryonic antigen. In contrast, the mesenchymal component expresses S-100, vimentin, and neuron‐specific enolase, and less often glial fibrillary acidic protein, smooth muscle actin, calponin, or p63.5,7,8 Identification of both layers is a distinctive trait of both tumors, rendering it apart from other conditions in the differential diagnosis.5
Typical treatment options include excision, electrodesiccation, dermabrasion, and argon or CO2 laser. Total excision is recommended if there is a benign tumor and complete excision is a cure.2 One case of recurrent benign chondroid syringoma was treated by Mohs micrographic surgery on the eyebrow9; however, Mohs surgery was not recommended in our case due to concerns of spread if malignant as well as an unknown tumor depth, as these tumors have a tendency for deep infiltration.
Due to its anatomical location and presentation as an anterior preauricular mass, it was difficult to differentiate between chondroid syringoma from sweat gland origin and pleomorphic adenoma from the salivary gland. As seen in our case, it is important for physicians to be aware of the differential diagnosis for mixed tumors because it can have a notable effect on the type of surgical therapy and follow-up management.
To the Editor:
Chondroid syringoma is a rare benign mixed tumor that originates from the sweat glands, typically presenting with both epithelial and mesenchymal components.1 It differs from pleomorphic adenoma, which arises from salivary glands. The surgical approach for complete excision is different for the 2 tumors; therefore, definitive diagnosis is important. For chondroid syringoma, total excision is recommended,2 while for pleomorphic adenoma, extracapsular dissection or superficial parotidectomy is commonly indicated. We report a case of a preauricular nodule at presentation and highlight the importance of differentiating a chondroid syringoma from a pleomorphic adenoma. This case is unique because of the anatomic location of the nodule, making diagnosis difficult because the tumor was abutting the parotid gland and a biopsy included normal salivary gland cells.
A 19-year-old man with a history of moderate acne on the shoulders, back, and face presented with a rapidly growing, painless nodule on right preauricular region of 6 months’ duration. The nodule was originally thought to be acne related and monitored, as the patient was asymptomatic. On examination the patient was found to have a firm, fixed, nontender, subcutaneous nodule overlying the right temporomandibular joint just anterior to the right tragus (Figure 1). Laboratory results were unremarkable. Computed tomography showed a subcutaneous nonaggressive-appearing soft-tissue mass measuring 16×17×12 mm just anterior and inferior to the external auditory canal cartilaginous segment with no bony abnormalities. The patient was initially treated with incomplete excision of the area for a presumed sebaceous cyst; 2 months later, an abnormal biopsy prompted a complete excisional biopsy.
Histologically, the initial incomplete excision biopsy revealed myxoid and chondroid tissue with glandular elements and adjacent lymph node with strong positivity for S-100 protein and moderate positivity for glial fibrillary acid protein, consistent with chondroid syringoma (Figure 2). Histological findings on second excision biopsy performed 2 months later showed tumor cells surrounded by normal salivary gland cells; therefore, it was difficult to define the origin of this tumor. Subsequent magnetic resonance imaging showed no evidence of the tumor and normal parotid gland borders.
Chondroid syringoma is a rare nonmelanoma skin tumor of the head and neck, mostly benign in nature but with malignant potential. Predominantly, it presents in males as an asymptomatic, slow-growing, nontender nodule.2 Malignant chondroid syringomas are more rare, typically appear on the trunk and legs of females, and present as rapidly growing hard nodules. They can arise de novo or from a preexisting chondroid syringoma and can metastasize.3,4
Clinically and histologically, chondroid syringoma resembles a pleomorphic adenoma. Its diagnosis is dependent on the clinical location to exclude origin in a salivary gland.5 Folliculosebaceous and myoepithelial differentiation within the tumor has been reported.6 Immunocytochemistry is the same in both types and is used to identify 2 prominent components—epithelial and mesenchymal—found in both chondroid syringoma and pleomorphic adenoma. Immunocytochemistry differentiates the epithelial component, which expresses cytokeratin, epithelial membrane antigen, and carcinoembryonic antigen. In contrast, the mesenchymal component expresses S-100, vimentin, and neuron‐specific enolase, and less often glial fibrillary acidic protein, smooth muscle actin, calponin, or p63.5,7,8 Identification of both layers is a distinctive trait of both tumors, rendering it apart from other conditions in the differential diagnosis.5
Typical treatment options include excision, electrodesiccation, dermabrasion, and argon or CO2 laser. Total excision is recommended if there is a benign tumor and complete excision is a cure.2 One case of recurrent benign chondroid syringoma was treated by Mohs micrographic surgery on the eyebrow9; however, Mohs surgery was not recommended in our case due to concerns of spread if malignant as well as an unknown tumor depth, as these tumors have a tendency for deep infiltration.
Due to its anatomical location and presentation as an anterior preauricular mass, it was difficult to differentiate between chondroid syringoma from sweat gland origin and pleomorphic adenoma from the salivary gland. As seen in our case, it is important for physicians to be aware of the differential diagnosis for mixed tumors because it can have a notable effect on the type of surgical therapy and follow-up management.
- Hirsch P, Helwig EB. Chondroid syringoma. Arch Dermatol. 1961;84:835-847.
- Turhan-Haktanir N, Sahin O, Bukulmez A, et al. Chondroid syringoma in a child. Pediatr Dermatol. 2007;24:505-507.
- Mathiasen RA, Rasgon BM, Rumore G. Malignant chondroid syringoma of the face: a first reported case. Otolaryngol Head Neck Surg. 2005;133:305-307.
- Barnett MD, Wallack MK, Zuretti A, et al. Recurrent malignant chondroid syringoma of the foot: a case report and review of the literature. Am J Clin Oncol. 2000;23:227-232.
- Dubb M, Michelow P. Cytologic features of chondroid syringoma in fine needle aspiration biopsies a report of 3 cases. Acta Cytol. 2010;54:183-186.
- Rauso R, Santagata M, Tartaro G, et al. Chondroid syringoma: rare tumor of orofacial region. Minerva Stomatol. 2009;58:383-388.
- Metzler G, Schaumburg-Lever G, Hornstein O, et al. Malignant chondroid syringoma: immunohistopathology. Am J Dermatopathol. 1996;18:83-89.
- Argenyi ZB, Balogh K, Goeken JA. Immunohistochemical characterization of chondroid syringomas. Am J Clin Pathol. 1988;90:662-669.
- Walls AC, Deng A, Geist DE. Mohs micrographic surgery for recurrent chondroid syringoma of the eyebrow. Dermatol Surg. 2012;38:800-802.
- Hirsch P, Helwig EB. Chondroid syringoma. Arch Dermatol. 1961;84:835-847.
- Turhan-Haktanir N, Sahin O, Bukulmez A, et al. Chondroid syringoma in a child. Pediatr Dermatol. 2007;24:505-507.
- Mathiasen RA, Rasgon BM, Rumore G. Malignant chondroid syringoma of the face: a first reported case. Otolaryngol Head Neck Surg. 2005;133:305-307.
- Barnett MD, Wallack MK, Zuretti A, et al. Recurrent malignant chondroid syringoma of the foot: a case report and review of the literature. Am J Clin Oncol. 2000;23:227-232.
- Dubb M, Michelow P. Cytologic features of chondroid syringoma in fine needle aspiration biopsies a report of 3 cases. Acta Cytol. 2010;54:183-186.
- Rauso R, Santagata M, Tartaro G, et al. Chondroid syringoma: rare tumor of orofacial region. Minerva Stomatol. 2009;58:383-388.
- Metzler G, Schaumburg-Lever G, Hornstein O, et al. Malignant chondroid syringoma: immunohistopathology. Am J Dermatopathol. 1996;18:83-89.
- Argenyi ZB, Balogh K, Goeken JA. Immunohistochemical characterization of chondroid syringomas. Am J Clin Pathol. 1988;90:662-669.
- Walls AC, Deng A, Geist DE. Mohs micrographic surgery for recurrent chondroid syringoma of the eyebrow. Dermatol Surg. 2012;38:800-802.
Practice Points
- Clinically and histologically, pleomorphic adenomas and chondroid syringoma both have identical presentations. Differentiation can be determined by knowing where the mixed tumor originated.
- Both lesions warrant different surgical management techniques. Pleomorphic adenoma requires extracapsular dissection or superficial parotidectomy, while complete excision is recommended for chondroid syringoma.
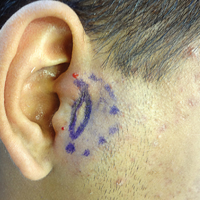
Mucous Membrane Pemphigoid Involving the Trachea and Bronchi: An Extremely Rare and Life-Threatening Presentation
To the Editor:
Mucous membrane pemphigoid (MMP) is an autoimmune blistering disorder that causes subepithelial damage and scarring of mucosal surfaces with or without skin involvement.1 The clinical presentation is highly variable. The oropharynx is the most common site of initial presentation, followed by ocular, nasopharyngeal, anogenital, skin, laryngeal, and esophageal involvement.2 Patients often present to a variety of specialists depending on initial symptoms, and due to the diverse clinical manifestations, MMP often is misdiagnosed. Our patient presented an even greater challenge because the disease progressed to tracheal and bronchial involvement.
A 37-year-old man presented to his primary care physician with a chief concern of a sore throat and oral ulcers. The patient was treated with a course of antibiotics followed by a nystatin oral solution. He continued to develop ulcerative lesions on the soft palate, posterior pharynx, and nasal mucosae. He sought treatment from 2 otolaryngologists (ENTs) and a gastroenterologist, and continued to be treated with multiple oral antibiotics, fluconazole, and topical nystatin. Despite treatment, the patient developed pansinusitis and laryngitis and presented to the ENT department at our institution with severe hoarseness and dyspnea on exertion. Examination by the ENT department revealed ulcerative lesions of the nares with stenosis and ulcers along the soft palate. Videolaryngostroboscopy showed remarkable supraglottic edema with thick endolaryngeal mucus. The patient worked as a funeral director and had notable formaldehyde exposure. He also hunted wild game and performed taxidermy regularly.
The patient was admitted and treated with intravenous dexamethasone for a compromised airway. Subsequently, he was taken to the operating room and had biopsies performed of the posterior pharynx. Given his exposure history, the infectious disease department was consulted and he was evaluated for multiple viral, bacterial, and fungal suspects including leishmania and tularemia. Age-appropriate screening, physical examination, and review of systems were negative for an underlying neoplasm. Histopathologic examination revealed a subepithelial vesicular mucositis with a mixed infiltrate of lymphocytes and histiocytes. Direct immunofluorescence microscopy demonstrated strong linear fluorescence along the epithelial-subepithelial junction with IgG and C3. Based on these findings, the diagnosis of MMP was made.
Further testing for bullous pemphigoid antigen 1 (BP230) and bullous pemphigoid antigen 2 (BP180) were negative. On one occasion the patient tested positive for anti-BP230 IgG, but it was at a level judged to be insignificant (7.5 [reference range, <9]). The patient also was negative for autoantibodies against desmoglein 1 and 3. Indirect immunofluorescence using rat bladder epithelium was not performed.
The patient was started on methotrexate and oral prednisone by the rheumatology department, but after 1 week, he presented in respiratory distress and was taken for an emergency tracheostomy. The patient eventually was referred to the dermatology department where methotrexate was discontinued and the patient was started on titrating doses of prednisone and mycophenolate mofetil. Eight weeks later, the patient became completely aphonic and was taken by ENT for dilation of the supraglottic, glottic, and subglottic stenosis with mucosal triamcinolone injections. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily was initiated in addition to mycophenolate mofetil 3 g and prednisone 80 mg, but again the patient developed near-complete tracheal stenosis just proximal to the tracheostomy entry site. At 16 weeks, balloon dilation was repeated with dexamethasone injections and topical mitomycin C. Subsequently, the patient regained some use of his voice. Although the next several laryngoscopes showed improvement in the patient’s epiglottis and glottis, the trachea continued to require debridement and dilation.
Despite maximal medical therapy and surgical interventions, the patient had little improvement in his voice and large clots of blood obstructed his tracheostomy daily. He was unable to sleep in his preferred position on the stomach (prone) due to dyspnea but had less distress sleeping on his back (supine). The patient was referred to the pulmonology department for an endotracheobronchoscopy to further evaluate the airway. It was discovered that the mucosa of the trachea from the level of the tracheostomy to the carina was friable with active erosions and thick bloody secretions (Figure 1). Lesions extended as far as the scope was able to visualize to the left upper lobe takeoff and the right mainstem bronchus (Figure 2). Biopsies of the carinal mucosa showed 3+/3+ linear fluorescence with IgG along the dermoepidermal junction. Salt-split studies were performed, but because the specimen was fragmented, it was not possible to assess if the fluorescence was present at the floor or at the roof of the split.

Given the severity of disease and failure to respond to other aggressive immunosuppressive therapies as well as having been with a tracheostomy for 22 months, the patient was started on 2 doses of intravenous rituximab 1 g 2 weeks apart along with trimethoprim-sulfamethoxazole (3 times weekly) for pneumocystis pneumonia prophylaxis. No complications were observed during infusions. After 2 rituximab infusions, he was weaned off of prednisone and a repeat bronchoscopy showed no airway ulcers beyond the distal trachea or endobronchial obstruction. However, the subglottic space and area above the tracheostomy showed remarkable stenosis with a cobblestone pattern and granulation tissue with continued narrowing of the subglottic area. The ENT performed further dilation and after 34 months, the tracheostomy was removed and a T-tube was placed. The patient required cleaning out of the T-tube approximately every 3 months, and after 2 years the original T-tube was replaced with a new one. At the time of this report, the ENT recommended removing the T-tube, but the patient was reluctant to do so; therefore, a second T-tube replacement is planned. He continues to do well without relapse and has been off all medical therapy for nearly 4 years.
Mucous membrane pemphigoid is an acquired autoimmune subepithelial blistering disease that predominantly affects mucous membranes with or without skin involvement. This condition has been referred to as cicatricial pemphigoid, oral pemphigoid, and ocular cicatricial pemphigoid, among other names. It is characterized by linear deposition of IgG, IgA, or C3 along the epithelial basement membrane zone. According to the international consensus on MMP, the target antigens identified in the epithelial basement membrane zone include bullous pemphigoid antigen 1 (BP230), bullous pemphigoid antigen 2 (BP180), laminin 5 (α3, β3, γ2 chains), laminin 6 (α3 chain), type VII collagen, and integrin β4 subunit.3 Not all patients with MMP will have circulating autoantibodies to the above components, and although our patient did have detectable anti-BP230 IgG, it was not considered clinically significant. Furthermore, the type of autoantibody does not impact decisions regarding therapy selection.3
Although rare, MMP is well-known to dermatologists and ophthalmologists who manage a large majority of MMP patients depending on which mucosa is involved. Mucous membrane pemphigoid is extremely rare in the lower respiratory tract, and when these lesions are discovered, it often is in the face of life-threatening respiratory distress. Mucous membrane pemphigoid is a challenging disease to treat, even more so when the primary specialty physician is unable to visualize the affected areas. Our patient’s disease was limited primarily to the pharynx, larynx, trachea, and bronchi with few oral lesions. According to a PubMed search of articles indexed for MEDLINE using the terms mucous membrane pemphigus, cicatricial pemphigoid, trachea, bronchus, and fatal, 8 reports (7 case reports and 1 prospective study) of MMP involving the lower respiratory tract have been published.4-11 Of the case reports, each patient also presented with involvement of the eyes or skin.4,5,7-11 Four of these cases were fatal secondary to cardiopulmonary arrest.5,7,9,10 In the prospective study, 110 consecutive patients with clinical, histologic, and immunologic criteria of MMP were examined with a flexible nasopharyngolaryngoscope.6 Thirty-eight patients had nose or throat symptoms but only 10 had laryngeal involvement and 5 had acute dyspnea. The nasal valves, choanae, pharynx, and/or larynx were severely scarred in 7 patients, which was fatal in 3.6
Medical treatment should be based on the following factors of the patient’s disease: site, severity, and rapidity of progression.3 High-risk patients can be defined as those who have lesions at any of the following sites: ocular, genital, nasopharyngeal, esophageal, and laryngeal mucosae. As our patient had involvement at several high-risk sites, in particular sites only visualized by various scoping procedures, a team of physicians including dermatologists, ENT physicians, pulmonologists, and oncologists was necessary to facilitate his care. Scarring is the hallmark of MMP and prevention of scarring is the most important aspect of treatment of MMP. Surgical repair of the previously involved mucosa is difficult, as the tissue is prone to re-scarring and difficult to heal. Over the last several years, there has been increasing evidence for the use of rituximab in autoimmune bullous skin diseases including pemphigus vulgaris, epidermolysis bullosa acquisita, and MMP.12-14 After 2 infusions of rituximab, our patient had clearance of his disease and currently is doing well with a T-tube.
Acknowledgments
We thank Kim Yancey, MD (Dallas, Texas), for providing access to the patient’s diagnostic laboratory immunology and reviewing biopsy specimens; Luis Angel, MD (San Antonio, Texas), for providing bronchoscopy photographs; and C. Blake Simpson, MD (San Antonio, Texas), for co-managing this challenging case.
- James WD, Berger TG, Elston D. Chronic blistering diseases. In: James WD, Berger TG, Elston D. Andrews’ Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Sanders Elsevier; 2010:448-467.
- Neff AG, Turner M, Mutasim DF. Treatment strategies in mucous membrane pemphigoid. Ther Clin Risk Manag. 2008;4:617-626.
- Chan LS, Ahmed AR, Anhalt GJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-379.
- Kato K, Moriyama Y, Saito H, et al. A case of mucous membrane pemphigoid involving the trachea and bronchus with autoantibodies to β3 subunit of laminin-332. Acta Derm Venereol. 2014;94:237-238.
- Gamm DM, Harris A, Mehran RJ, et al. Mucous membrane pemphigoid with fatal bronchial involvement in a seventeen-year-old girl. Cornea. 2006;25:474-478.
- Alexandre M, Brette MD, Pascal F, et al. A prospective study of upper aerodigestive tract manifestations of mucous membrane pemphigoid. Medicine (Baltimore). 2006;85:239-252.
- de Carvalho CR, Amato MB, Da Silva LM, et al. Obstructive respiratory failure in cicatricial pemphigoid. Thorax. 1989;44:601-602.
- Müller LC, Salzer GM. Stenosis of left mainstem bronchus in a case of cicatricial pemphigoid. Eur J Cardiothorac Surg. 1988;2:284-286.
- Camisa C, Allen CM. Death from CP in a young woman with oral, laryngeal, and bronchial involvement. Cutis. 1987;40:426-429.
- Derbes VJ, Pitot HC, Chernosky ME. Fatal cicatricial mucous membrane pemphigoid of the trachea. Dermatol Trop Ecol Geogr. 1962;1:114-117.
- Wieme N, Lambert J, Moerman M, et al. Epidermolysis bullosa acquisita with combined features of bullous pemphigoid and cicatricial pemphigoid. Dermatology. 1999;198:310-313.
- Taylor J, McMillan R, Shephard M, et al. World Workshop on Oral Medicine VI: a systematic review of the treatment of mucous membrane pemphigoid [published online March 11, 2015]. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;120:161.e20-171.e20.
- Sobolewska B, Deuter C, Zierhut M. Current medical treatment of ocular mucous membrane pemphigoid [published online July 9, 2013]. Ocul Surf. 2013;11:259-266.
- Maley A, Warren M, Haberman I, et al. Rituximab combined with conventional therapy versus conventional therapy alone for the treatment of mucous membrane pemphigoid (MMP) [published online February 28, 2016]. J Am Acad Dermatol. 2016;74:835-840.
To the Editor:
Mucous membrane pemphigoid (MMP) is an autoimmune blistering disorder that causes subepithelial damage and scarring of mucosal surfaces with or without skin involvement.1 The clinical presentation is highly variable. The oropharynx is the most common site of initial presentation, followed by ocular, nasopharyngeal, anogenital, skin, laryngeal, and esophageal involvement.2 Patients often present to a variety of specialists depending on initial symptoms, and due to the diverse clinical manifestations, MMP often is misdiagnosed. Our patient presented an even greater challenge because the disease progressed to tracheal and bronchial involvement.
A 37-year-old man presented to his primary care physician with a chief concern of a sore throat and oral ulcers. The patient was treated with a course of antibiotics followed by a nystatin oral solution. He continued to develop ulcerative lesions on the soft palate, posterior pharynx, and nasal mucosae. He sought treatment from 2 otolaryngologists (ENTs) and a gastroenterologist, and continued to be treated with multiple oral antibiotics, fluconazole, and topical nystatin. Despite treatment, the patient developed pansinusitis and laryngitis and presented to the ENT department at our institution with severe hoarseness and dyspnea on exertion. Examination by the ENT department revealed ulcerative lesions of the nares with stenosis and ulcers along the soft palate. Videolaryngostroboscopy showed remarkable supraglottic edema with thick endolaryngeal mucus. The patient worked as a funeral director and had notable formaldehyde exposure. He also hunted wild game and performed taxidermy regularly.
The patient was admitted and treated with intravenous dexamethasone for a compromised airway. Subsequently, he was taken to the operating room and had biopsies performed of the posterior pharynx. Given his exposure history, the infectious disease department was consulted and he was evaluated for multiple viral, bacterial, and fungal suspects including leishmania and tularemia. Age-appropriate screening, physical examination, and review of systems were negative for an underlying neoplasm. Histopathologic examination revealed a subepithelial vesicular mucositis with a mixed infiltrate of lymphocytes and histiocytes. Direct immunofluorescence microscopy demonstrated strong linear fluorescence along the epithelial-subepithelial junction with IgG and C3. Based on these findings, the diagnosis of MMP was made.
Further testing for bullous pemphigoid antigen 1 (BP230) and bullous pemphigoid antigen 2 (BP180) were negative. On one occasion the patient tested positive for anti-BP230 IgG, but it was at a level judged to be insignificant (7.5 [reference range, <9]). The patient also was negative for autoantibodies against desmoglein 1 and 3. Indirect immunofluorescence using rat bladder epithelium was not performed.
The patient was started on methotrexate and oral prednisone by the rheumatology department, but after 1 week, he presented in respiratory distress and was taken for an emergency tracheostomy. The patient eventually was referred to the dermatology department where methotrexate was discontinued and the patient was started on titrating doses of prednisone and mycophenolate mofetil. Eight weeks later, the patient became completely aphonic and was taken by ENT for dilation of the supraglottic, glottic, and subglottic stenosis with mucosal triamcinolone injections. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily was initiated in addition to mycophenolate mofetil 3 g and prednisone 80 mg, but again the patient developed near-complete tracheal stenosis just proximal to the tracheostomy entry site. At 16 weeks, balloon dilation was repeated with dexamethasone injections and topical mitomycin C. Subsequently, the patient regained some use of his voice. Although the next several laryngoscopes showed improvement in the patient’s epiglottis and glottis, the trachea continued to require debridement and dilation.
Despite maximal medical therapy and surgical interventions, the patient had little improvement in his voice and large clots of blood obstructed his tracheostomy daily. He was unable to sleep in his preferred position on the stomach (prone) due to dyspnea but had less distress sleeping on his back (supine). The patient was referred to the pulmonology department for an endotracheobronchoscopy to further evaluate the airway. It was discovered that the mucosa of the trachea from the level of the tracheostomy to the carina was friable with active erosions and thick bloody secretions (Figure 1). Lesions extended as far as the scope was able to visualize to the left upper lobe takeoff and the right mainstem bronchus (Figure 2). Biopsies of the carinal mucosa showed 3+/3+ linear fluorescence with IgG along the dermoepidermal junction. Salt-split studies were performed, but because the specimen was fragmented, it was not possible to assess if the fluorescence was present at the floor or at the roof of the split.
Given the severity of disease and failure to respond to other aggressive immunosuppressive therapies as well as having been with a tracheostomy for 22 months, the patient was started on 2 doses of intravenous rituximab 1 g 2 weeks apart along with trimethoprim-sulfamethoxazole (3 times weekly) for pneumocystis pneumonia prophylaxis. No complications were observed during infusions. After 2 rituximab infusions, he was weaned off of prednisone and a repeat bronchoscopy showed no airway ulcers beyond the distal trachea or endobronchial obstruction. However, the subglottic space and area above the tracheostomy showed remarkable stenosis with a cobblestone pattern and granulation tissue with continued narrowing of the subglottic area. The ENT performed further dilation and after 34 months, the tracheostomy was removed and a T-tube was placed. The patient required cleaning out of the T-tube approximately every 3 months, and after 2 years the original T-tube was replaced with a new one. At the time of this report, the ENT recommended removing the T-tube, but the patient was reluctant to do so; therefore, a second T-tube replacement is planned. He continues to do well without relapse and has been off all medical therapy for nearly 4 years.
Mucous membrane pemphigoid is an acquired autoimmune subepithelial blistering disease that predominantly affects mucous membranes with or without skin involvement. This condition has been referred to as cicatricial pemphigoid, oral pemphigoid, and ocular cicatricial pemphigoid, among other names. It is characterized by linear deposition of IgG, IgA, or C3 along the epithelial basement membrane zone. According to the international consensus on MMP, the target antigens identified in the epithelial basement membrane zone include bullous pemphigoid antigen 1 (BP230), bullous pemphigoid antigen 2 (BP180), laminin 5 (α3, β3, γ2 chains), laminin 6 (α3 chain), type VII collagen, and integrin β4 subunit.3 Not all patients with MMP will have circulating autoantibodies to the above components, and although our patient did have detectable anti-BP230 IgG, it was not considered clinically significant. Furthermore, the type of autoantibody does not impact decisions regarding therapy selection.3
Although rare, MMP is well-known to dermatologists and ophthalmologists who manage a large majority of MMP patients depending on which mucosa is involved. Mucous membrane pemphigoid is extremely rare in the lower respiratory tract, and when these lesions are discovered, it often is in the face of life-threatening respiratory distress. Mucous membrane pemphigoid is a challenging disease to treat, even more so when the primary specialty physician is unable to visualize the affected areas. Our patient’s disease was limited primarily to the pharynx, larynx, trachea, and bronchi with few oral lesions. According to a PubMed search of articles indexed for MEDLINE using the terms mucous membrane pemphigus, cicatricial pemphigoid, trachea, bronchus, and fatal, 8 reports (7 case reports and 1 prospective study) of MMP involving the lower respiratory tract have been published.4-11 Of the case reports, each patient also presented with involvement of the eyes or skin.4,5,7-11 Four of these cases were fatal secondary to cardiopulmonary arrest.5,7,9,10 In the prospective study, 110 consecutive patients with clinical, histologic, and immunologic criteria of MMP were examined with a flexible nasopharyngolaryngoscope.6 Thirty-eight patients had nose or throat symptoms but only 10 had laryngeal involvement and 5 had acute dyspnea. The nasal valves, choanae, pharynx, and/or larynx were severely scarred in 7 patients, which was fatal in 3.6
Medical treatment should be based on the following factors of the patient’s disease: site, severity, and rapidity of progression.3 High-risk patients can be defined as those who have lesions at any of the following sites: ocular, genital, nasopharyngeal, esophageal, and laryngeal mucosae. As our patient had involvement at several high-risk sites, in particular sites only visualized by various scoping procedures, a team of physicians including dermatologists, ENT physicians, pulmonologists, and oncologists was necessary to facilitate his care. Scarring is the hallmark of MMP and prevention of scarring is the most important aspect of treatment of MMP. Surgical repair of the previously involved mucosa is difficult, as the tissue is prone to re-scarring and difficult to heal. Over the last several years, there has been increasing evidence for the use of rituximab in autoimmune bullous skin diseases including pemphigus vulgaris, epidermolysis bullosa acquisita, and MMP.12-14 After 2 infusions of rituximab, our patient had clearance of his disease and currently is doing well with a T-tube.
Acknowledgments
We thank Kim Yancey, MD (Dallas, Texas), for providing access to the patient’s diagnostic laboratory immunology and reviewing biopsy specimens; Luis Angel, MD (San Antonio, Texas), for providing bronchoscopy photographs; and C. Blake Simpson, MD (San Antonio, Texas), for co-managing this challenging case.
To the Editor:
Mucous membrane pemphigoid (MMP) is an autoimmune blistering disorder that causes subepithelial damage and scarring of mucosal surfaces with or without skin involvement.1 The clinical presentation is highly variable. The oropharynx is the most common site of initial presentation, followed by ocular, nasopharyngeal, anogenital, skin, laryngeal, and esophageal involvement.2 Patients often present to a variety of specialists depending on initial symptoms, and due to the diverse clinical manifestations, MMP often is misdiagnosed. Our patient presented an even greater challenge because the disease progressed to tracheal and bronchial involvement.
A 37-year-old man presented to his primary care physician with a chief concern of a sore throat and oral ulcers. The patient was treated with a course of antibiotics followed by a nystatin oral solution. He continued to develop ulcerative lesions on the soft palate, posterior pharynx, and nasal mucosae. He sought treatment from 2 otolaryngologists (ENTs) and a gastroenterologist, and continued to be treated with multiple oral antibiotics, fluconazole, and topical nystatin. Despite treatment, the patient developed pansinusitis and laryngitis and presented to the ENT department at our institution with severe hoarseness and dyspnea on exertion. Examination by the ENT department revealed ulcerative lesions of the nares with stenosis and ulcers along the soft palate. Videolaryngostroboscopy showed remarkable supraglottic edema with thick endolaryngeal mucus. The patient worked as a funeral director and had notable formaldehyde exposure. He also hunted wild game and performed taxidermy regularly.
The patient was admitted and treated with intravenous dexamethasone for a compromised airway. Subsequently, he was taken to the operating room and had biopsies performed of the posterior pharynx. Given his exposure history, the infectious disease department was consulted and he was evaluated for multiple viral, bacterial, and fungal suspects including leishmania and tularemia. Age-appropriate screening, physical examination, and review of systems were negative for an underlying neoplasm. Histopathologic examination revealed a subepithelial vesicular mucositis with a mixed infiltrate of lymphocytes and histiocytes. Direct immunofluorescence microscopy demonstrated strong linear fluorescence along the epithelial-subepithelial junction with IgG and C3. Based on these findings, the diagnosis of MMP was made.
Further testing for bullous pemphigoid antigen 1 (BP230) and bullous pemphigoid antigen 2 (BP180) were negative. On one occasion the patient tested positive for anti-BP230 IgG, but it was at a level judged to be insignificant (7.5 [reference range, <9]). The patient also was negative for autoantibodies against desmoglein 1 and 3. Indirect immunofluorescence using rat bladder epithelium was not performed.
The patient was started on methotrexate and oral prednisone by the rheumatology department, but after 1 week, he presented in respiratory distress and was taken for an emergency tracheostomy. The patient eventually was referred to the dermatology department where methotrexate was discontinued and the patient was started on titrating doses of prednisone and mycophenolate mofetil. Eight weeks later, the patient became completely aphonic and was taken by ENT for dilation of the supraglottic, glottic, and subglottic stenosis with mucosal triamcinolone injections. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily was initiated in addition to mycophenolate mofetil 3 g and prednisone 80 mg, but again the patient developed near-complete tracheal stenosis just proximal to the tracheostomy entry site. At 16 weeks, balloon dilation was repeated with dexamethasone injections and topical mitomycin C. Subsequently, the patient regained some use of his voice. Although the next several laryngoscopes showed improvement in the patient’s epiglottis and glottis, the trachea continued to require debridement and dilation.
Despite maximal medical therapy and surgical interventions, the patient had little improvement in his voice and large clots of blood obstructed his tracheostomy daily. He was unable to sleep in his preferred position on the stomach (prone) due to dyspnea but had less distress sleeping on his back (supine). The patient was referred to the pulmonology department for an endotracheobronchoscopy to further evaluate the airway. It was discovered that the mucosa of the trachea from the level of the tracheostomy to the carina was friable with active erosions and thick bloody secretions (Figure 1). Lesions extended as far as the scope was able to visualize to the left upper lobe takeoff and the right mainstem bronchus (Figure 2). Biopsies of the carinal mucosa showed 3+/3+ linear fluorescence with IgG along the dermoepidermal junction. Salt-split studies were performed, but because the specimen was fragmented, it was not possible to assess if the fluorescence was present at the floor or at the roof of the split.
Given the severity of disease and failure to respond to other aggressive immunosuppressive therapies as well as having been with a tracheostomy for 22 months, the patient was started on 2 doses of intravenous rituximab 1 g 2 weeks apart along with trimethoprim-sulfamethoxazole (3 times weekly) for pneumocystis pneumonia prophylaxis. No complications were observed during infusions. After 2 rituximab infusions, he was weaned off of prednisone and a repeat bronchoscopy showed no airway ulcers beyond the distal trachea or endobronchial obstruction. However, the subglottic space and area above the tracheostomy showed remarkable stenosis with a cobblestone pattern and granulation tissue with continued narrowing of the subglottic area. The ENT performed further dilation and after 34 months, the tracheostomy was removed and a T-tube was placed. The patient required cleaning out of the T-tube approximately every 3 months, and after 2 years the original T-tube was replaced with a new one. At the time of this report, the ENT recommended removing the T-tube, but the patient was reluctant to do so; therefore, a second T-tube replacement is planned. He continues to do well without relapse and has been off all medical therapy for nearly 4 years.
Mucous membrane pemphigoid is an acquired autoimmune subepithelial blistering disease that predominantly affects mucous membranes with or without skin involvement. This condition has been referred to as cicatricial pemphigoid, oral pemphigoid, and ocular cicatricial pemphigoid, among other names. It is characterized by linear deposition of IgG, IgA, or C3 along the epithelial basement membrane zone. According to the international consensus on MMP, the target antigens identified in the epithelial basement membrane zone include bullous pemphigoid antigen 1 (BP230), bullous pemphigoid antigen 2 (BP180), laminin 5 (α3, β3, γ2 chains), laminin 6 (α3 chain), type VII collagen, and integrin β4 subunit.3 Not all patients with MMP will have circulating autoantibodies to the above components, and although our patient did have detectable anti-BP230 IgG, it was not considered clinically significant. Furthermore, the type of autoantibody does not impact decisions regarding therapy selection.3
Although rare, MMP is well-known to dermatologists and ophthalmologists who manage a large majority of MMP patients depending on which mucosa is involved. Mucous membrane pemphigoid is extremely rare in the lower respiratory tract, and when these lesions are discovered, it often is in the face of life-threatening respiratory distress. Mucous membrane pemphigoid is a challenging disease to treat, even more so when the primary specialty physician is unable to visualize the affected areas. Our patient’s disease was limited primarily to the pharynx, larynx, trachea, and bronchi with few oral lesions. According to a PubMed search of articles indexed for MEDLINE using the terms mucous membrane pemphigus, cicatricial pemphigoid, trachea, bronchus, and fatal, 8 reports (7 case reports and 1 prospective study) of MMP involving the lower respiratory tract have been published.4-11 Of the case reports, each patient also presented with involvement of the eyes or skin.4,5,7-11 Four of these cases were fatal secondary to cardiopulmonary arrest.5,7,9,10 In the prospective study, 110 consecutive patients with clinical, histologic, and immunologic criteria of MMP were examined with a flexible nasopharyngolaryngoscope.6 Thirty-eight patients had nose or throat symptoms but only 10 had laryngeal involvement and 5 had acute dyspnea. The nasal valves, choanae, pharynx, and/or larynx were severely scarred in 7 patients, which was fatal in 3.6
Medical treatment should be based on the following factors of the patient’s disease: site, severity, and rapidity of progression.3 High-risk patients can be defined as those who have lesions at any of the following sites: ocular, genital, nasopharyngeal, esophageal, and laryngeal mucosae. As our patient had involvement at several high-risk sites, in particular sites only visualized by various scoping procedures, a team of physicians including dermatologists, ENT physicians, pulmonologists, and oncologists was necessary to facilitate his care. Scarring is the hallmark of MMP and prevention of scarring is the most important aspect of treatment of MMP. Surgical repair of the previously involved mucosa is difficult, as the tissue is prone to re-scarring and difficult to heal. Over the last several years, there has been increasing evidence for the use of rituximab in autoimmune bullous skin diseases including pemphigus vulgaris, epidermolysis bullosa acquisita, and MMP.12-14 After 2 infusions of rituximab, our patient had clearance of his disease and currently is doing well with a T-tube.
Acknowledgments
We thank Kim Yancey, MD (Dallas, Texas), for providing access to the patient’s diagnostic laboratory immunology and reviewing biopsy specimens; Luis Angel, MD (San Antonio, Texas), for providing bronchoscopy photographs; and C. Blake Simpson, MD (San Antonio, Texas), for co-managing this challenging case.
- James WD, Berger TG, Elston D. Chronic blistering diseases. In: James WD, Berger TG, Elston D. Andrews’ Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Sanders Elsevier; 2010:448-467.
- Neff AG, Turner M, Mutasim DF. Treatment strategies in mucous membrane pemphigoid. Ther Clin Risk Manag. 2008;4:617-626.
- Chan LS, Ahmed AR, Anhalt GJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-379.
- Kato K, Moriyama Y, Saito H, et al. A case of mucous membrane pemphigoid involving the trachea and bronchus with autoantibodies to β3 subunit of laminin-332. Acta Derm Venereol. 2014;94:237-238.
- Gamm DM, Harris A, Mehran RJ, et al. Mucous membrane pemphigoid with fatal bronchial involvement in a seventeen-year-old girl. Cornea. 2006;25:474-478.
- Alexandre M, Brette MD, Pascal F, et al. A prospective study of upper aerodigestive tract manifestations of mucous membrane pemphigoid. Medicine (Baltimore). 2006;85:239-252.
- de Carvalho CR, Amato MB, Da Silva LM, et al. Obstructive respiratory failure in cicatricial pemphigoid. Thorax. 1989;44:601-602.
- Müller LC, Salzer GM. Stenosis of left mainstem bronchus in a case of cicatricial pemphigoid. Eur J Cardiothorac Surg. 1988;2:284-286.
- Camisa C, Allen CM. Death from CP in a young woman with oral, laryngeal, and bronchial involvement. Cutis. 1987;40:426-429.
- Derbes VJ, Pitot HC, Chernosky ME. Fatal cicatricial mucous membrane pemphigoid of the trachea. Dermatol Trop Ecol Geogr. 1962;1:114-117.
- Wieme N, Lambert J, Moerman M, et al. Epidermolysis bullosa acquisita with combined features of bullous pemphigoid and cicatricial pemphigoid. Dermatology. 1999;198:310-313.
- Taylor J, McMillan R, Shephard M, et al. World Workshop on Oral Medicine VI: a systematic review of the treatment of mucous membrane pemphigoid [published online March 11, 2015]. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;120:161.e20-171.e20.
- Sobolewska B, Deuter C, Zierhut M. Current medical treatment of ocular mucous membrane pemphigoid [published online July 9, 2013]. Ocul Surf. 2013;11:259-266.
- Maley A, Warren M, Haberman I, et al. Rituximab combined with conventional therapy versus conventional therapy alone for the treatment of mucous membrane pemphigoid (MMP) [published online February 28, 2016]. J Am Acad Dermatol. 2016;74:835-840.
- James WD, Berger TG, Elston D. Chronic blistering diseases. In: James WD, Berger TG, Elston D. Andrews’ Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Sanders Elsevier; 2010:448-467.
- Neff AG, Turner M, Mutasim DF. Treatment strategies in mucous membrane pemphigoid. Ther Clin Risk Manag. 2008;4:617-626.
- Chan LS, Ahmed AR, Anhalt GJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-379.
- Kato K, Moriyama Y, Saito H, et al. A case of mucous membrane pemphigoid involving the trachea and bronchus with autoantibodies to β3 subunit of laminin-332. Acta Derm Venereol. 2014;94:237-238.
- Gamm DM, Harris A, Mehran RJ, et al. Mucous membrane pemphigoid with fatal bronchial involvement in a seventeen-year-old girl. Cornea. 2006;25:474-478.
- Alexandre M, Brette MD, Pascal F, et al. A prospective study of upper aerodigestive tract manifestations of mucous membrane pemphigoid. Medicine (Baltimore). 2006;85:239-252.
- de Carvalho CR, Amato MB, Da Silva LM, et al. Obstructive respiratory failure in cicatricial pemphigoid. Thorax. 1989;44:601-602.
- Müller LC, Salzer GM. Stenosis of left mainstem bronchus in a case of cicatricial pemphigoid. Eur J Cardiothorac Surg. 1988;2:284-286.
- Camisa C, Allen CM. Death from CP in a young woman with oral, laryngeal, and bronchial involvement. Cutis. 1987;40:426-429.
- Derbes VJ, Pitot HC, Chernosky ME. Fatal cicatricial mucous membrane pemphigoid of the trachea. Dermatol Trop Ecol Geogr. 1962;1:114-117.
- Wieme N, Lambert J, Moerman M, et al. Epidermolysis bullosa acquisita with combined features of bullous pemphigoid and cicatricial pemphigoid. Dermatology. 1999;198:310-313.
- Taylor J, McMillan R, Shephard M, et al. World Workshop on Oral Medicine VI: a systematic review of the treatment of mucous membrane pemphigoid [published online March 11, 2015]. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;120:161.e20-171.e20.
- Sobolewska B, Deuter C, Zierhut M. Current medical treatment of ocular mucous membrane pemphigoid [published online July 9, 2013]. Ocul Surf. 2013;11:259-266.
- Maley A, Warren M, Haberman I, et al. Rituximab combined with conventional therapy versus conventional therapy alone for the treatment of mucous membrane pemphigoid (MMP) [published online February 28, 2016]. J Am Acad Dermatol. 2016;74:835-840.
Practice Points
- Mucous membrane pemphigoid (MMP) can present with diverse clinical manifestations, making the diagnosis challenging for many clinicians, including experienced dermatologists.
- If not treated early and aggressively, MMP can lead to scarring and is a potentially life-threatening disease.

Novel De Novo Heterozygous Frameshift Mutation of the ADAR1 Gene in Heavy Dyschromatosis Symmetrica Hereditaria
To the Editor:
Dyschromatosis symmetrica hereditaria (DSH)(Online Mendelian Inheritance in Man 127400), also called reticulate acropigmentation of Dohi, is a pigmentary genodermatosis characterized by a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Linkage analysis has revealed that the DSH gene locus resides on chromosome 1q11-q21,1 and the adenosine deaminase RNA specific gene, ADAR1 (also called DSRAD), in this region has been identified as being responsible for the development of DSH.2 We report a sporadic case of severe DSH with the ADAR1 gene detected in a mutation analysis.
A 6-year-old girl presented with a mixture of hyperpigmented and hypopigmented macules on the dorsal aspects of the hands and feet and the curved side of the wrists, heels, and knees, as well as scattered frecklelike and depigmented spots on the face, ears, neck, arms, and upper back (Figure 1). Her parents noted that hyperpigmented and hypopigmented macules on the dorsal aspects of the hands developed at 5 months of age. Exacerbation after exposure to sunlight resulted in the eruption becoming remarkable in summer and fainter in winter. The skin lesions gradually became more progressive. Physical examination revealed that the patient generally was healthy.
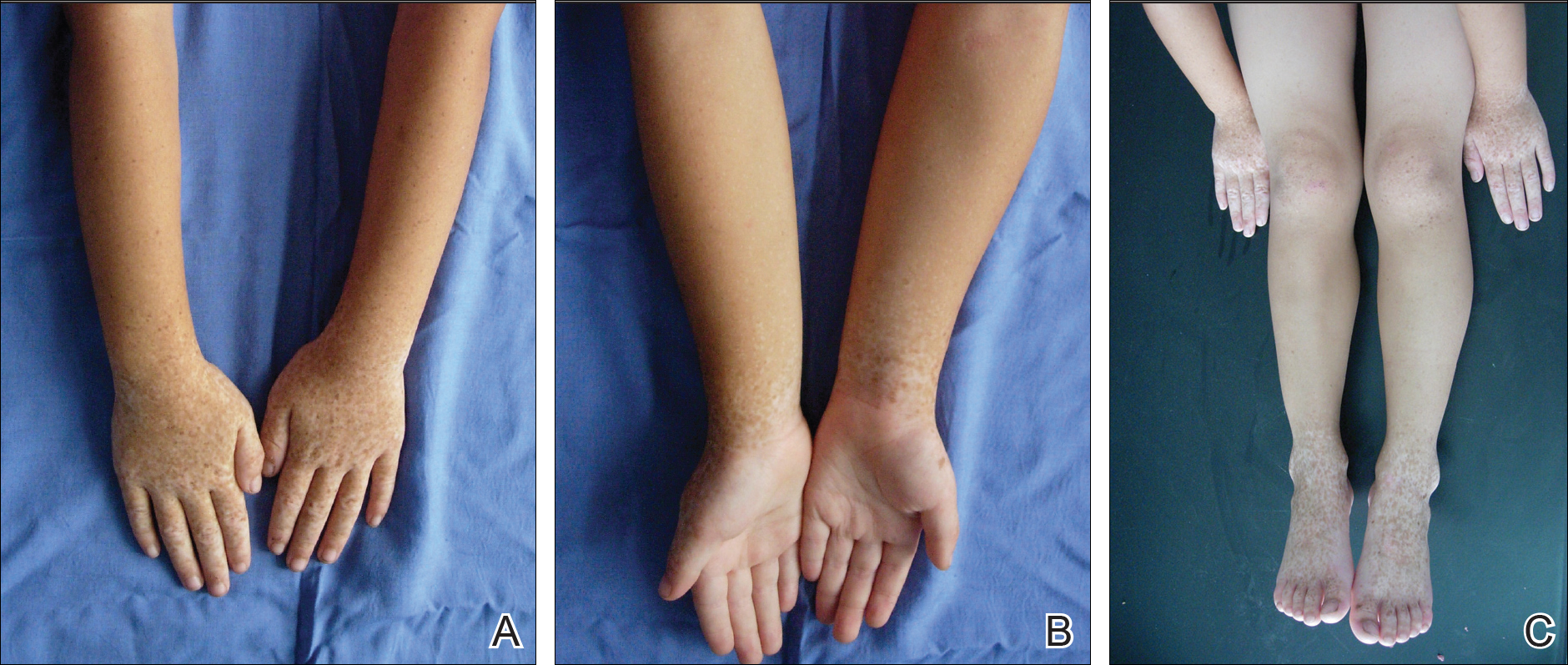
After obtaining informed consent, we performed a mutation analysis of the ADAR1 gene in our patient and her parents. We used a kit to extract genomic DNA from peripheral blood, which was then used to amplify the exons of the ADAR1 gene with intronic flanking sequences by polymerase chain reaction with the primer.3 After amplification, polymerase chain reaction products were purified. We sequenced the ADAR1 gene. Sequence comparisons and analysis found that the patient (proband) carried a heterozygous insertional mutation c.2253insG in exon 6 of the ADAR1 gene. This mutation was not detected in the proband’s healthy parents and 100 normal individuals (Figure 2).
Dyschromatosis symmetrica hereditaria is acquired by autosomal-dominant inheritance and is mainly reported in Asians, especially in Japan and China. Oyama et al4 reviewed 185 cases of DSH in Japan and found the onset of this disease usually was during infancy or childhood; 73% of patients developed the skin lesions before 6 years of age. Suzuki et al5 reported 10 unrelated Japanese patients and found the onset of disease ranged from 1 year of age to childhood. Zhang et al1,6 investigated 78 Chinese patients with DSH including 8 multigenerational families and 2 sporadic patients and found the age of disease onset ranged from 6 months to 15 years of age. The age of onset in our patient (5 months) was younger than these prior reports.
Patients with DSH have a characteristic appearance including a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Few patients have similar lesions on the knees and elbows. Many patients have frecklelike macules on the face and arms.1-6 One patient has been described with scattered depigmented spots on the face and chest.1 Our patient had a characteristic appearance as well as some special manifestations including skin lesions on the curved side of the wrist, ears, neck, and upper back.
The human ADAR1 gene spans 30 kilobase and contains 15 exons. It encodes RNA-specific adenosine deaminase composed of 1226 amino acid residues. This enzyme is important for various functions such as site-specific RNA editing and nuclear translation. This enzyme has 2 Z-alpha domains, 3 double-stranded RNA–binding domains, and the putative deaminase domain corresponding to exon 2, exons 2 to 7, and exons 9 to 14 of ADAR1, respectively.6
Mutation analysis of the ADAR1 gene in this case showed heterozygous insertion mutation c.2253insG in exon 6 of the ADAR1 gene, which changed the reading frame, and 475 amino acid residues in C-terminus are replaced by 90 amino acid residues (TSSRAQVRLPSKSWGSLVPSRLRTQQEA RQAGSSRCGSPCLDWGEREGRTHGFHRG NPSDRGQSQKNYAPPLKVPRSTAKT DTPSHWQHLP). This mutation was not detected in the proband’s healthy parents and the 100 control individuals, which indicated that it was a de novo mutation and the pathogenic mutation of DSH rather than a common polymorphism.
In conclusion, we report a novel mutation of the ADAR1 gene with a heavy clinical phenotype in DSH. This study expands the spectrum of clinical manifestations and demonstrates the ADAR1 mutation in DSH.
Acknowledgments
We are most grateful to the patient and her family for taking part in our study.
- Zhang XJ, Gao M, Li M, et al. Identification of a locus for dyschromatosis symmetrica hereditaria at chromosome 1q11-1q21. J Invest Dermatol. 2003;120:776-780.
- Miyamura Y, Suzuki T, Kono M, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria [published online August 11, 2003]. Am J Hum Genet. 2003;73:693-699.
- Li M, Li C, Hua H, et al. Identification of two novel mutations in Chinese patients with dyschromatosis symmetrica hereditaria [published online October 8, 2005]. Arch Dermatol Res. 2005;297:196-200.
- Oyama M, Shimizu H, Ohata Y, et al. Dyschromatosis symmetrica hereditaria (reticulate acropigmentation of Dohi): report of a Japanese family with the condition and a literature review of 185 cases. Br J Dermatol. 1999;140:491-496.
- Suzuki N, Suzuki T, Inagaki K, et al. Ten novel mutations of the ADAR1 gene in Japanese patients with dyschromatosis symmetrica hereditaria [published online August 17, 2006]. J Invest Dermatol. 2007;127:309-311.
- Zhang XJ, He PP, Li M, et al. Seven novel mutations of the ADAR gene in Chinese families and sporadic patients with dyschromatosis symmetrica hereditaria (DSH). Hum Mutat. 2004;23:629-630.
To the Editor:
Dyschromatosis symmetrica hereditaria (DSH)(Online Mendelian Inheritance in Man 127400), also called reticulate acropigmentation of Dohi, is a pigmentary genodermatosis characterized by a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Linkage analysis has revealed that the DSH gene locus resides on chromosome 1q11-q21,1 and the adenosine deaminase RNA specific gene, ADAR1 (also called DSRAD), in this region has been identified as being responsible for the development of DSH.2 We report a sporadic case of severe DSH with the ADAR1 gene detected in a mutation analysis.
A 6-year-old girl presented with a mixture of hyperpigmented and hypopigmented macules on the dorsal aspects of the hands and feet and the curved side of the wrists, heels, and knees, as well as scattered frecklelike and depigmented spots on the face, ears, neck, arms, and upper back (Figure 1). Her parents noted that hyperpigmented and hypopigmented macules on the dorsal aspects of the hands developed at 5 months of age. Exacerbation after exposure to sunlight resulted in the eruption becoming remarkable in summer and fainter in winter. The skin lesions gradually became more progressive. Physical examination revealed that the patient generally was healthy.
After obtaining informed consent, we performed a mutation analysis of the ADAR1 gene in our patient and her parents. We used a kit to extract genomic DNA from peripheral blood, which was then used to amplify the exons of the ADAR1 gene with intronic flanking sequences by polymerase chain reaction with the primer.3 After amplification, polymerase chain reaction products were purified. We sequenced the ADAR1 gene. Sequence comparisons and analysis found that the patient (proband) carried a heterozygous insertional mutation c.2253insG in exon 6 of the ADAR1 gene. This mutation was not detected in the proband’s healthy parents and 100 normal individuals (Figure 2).
Dyschromatosis symmetrica hereditaria is acquired by autosomal-dominant inheritance and is mainly reported in Asians, especially in Japan and China. Oyama et al4 reviewed 185 cases of DSH in Japan and found the onset of this disease usually was during infancy or childhood; 73% of patients developed the skin lesions before 6 years of age. Suzuki et al5 reported 10 unrelated Japanese patients and found the onset of disease ranged from 1 year of age to childhood. Zhang et al1,6 investigated 78 Chinese patients with DSH including 8 multigenerational families and 2 sporadic patients and found the age of disease onset ranged from 6 months to 15 years of age. The age of onset in our patient (5 months) was younger than these prior reports.
Patients with DSH have a characteristic appearance including a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Few patients have similar lesions on the knees and elbows. Many patients have frecklelike macules on the face and arms.1-6 One patient has been described with scattered depigmented spots on the face and chest.1 Our patient had a characteristic appearance as well as some special manifestations including skin lesions on the curved side of the wrist, ears, neck, and upper back.
The human ADAR1 gene spans 30 kilobase and contains 15 exons. It encodes RNA-specific adenosine deaminase composed of 1226 amino acid residues. This enzyme is important for various functions such as site-specific RNA editing and nuclear translation. This enzyme has 2 Z-alpha domains, 3 double-stranded RNA–binding domains, and the putative deaminase domain corresponding to exon 2, exons 2 to 7, and exons 9 to 14 of ADAR1, respectively.6
Mutation analysis of the ADAR1 gene in this case showed heterozygous insertion mutation c.2253insG in exon 6 of the ADAR1 gene, which changed the reading frame, and 475 amino acid residues in C-terminus are replaced by 90 amino acid residues (TSSRAQVRLPSKSWGSLVPSRLRTQQEA RQAGSSRCGSPCLDWGEREGRTHGFHRG NPSDRGQSQKNYAPPLKVPRSTAKT DTPSHWQHLP). This mutation was not detected in the proband’s healthy parents and the 100 control individuals, which indicated that it was a de novo mutation and the pathogenic mutation of DSH rather than a common polymorphism.
In conclusion, we report a novel mutation of the ADAR1 gene with a heavy clinical phenotype in DSH. This study expands the spectrum of clinical manifestations and demonstrates the ADAR1 mutation in DSH.
Acknowledgments
We are most grateful to the patient and her family for taking part in our study.
To the Editor:
Dyschromatosis symmetrica hereditaria (DSH)(Online Mendelian Inheritance in Man 127400), also called reticulate acropigmentation of Dohi, is a pigmentary genodermatosis characterized by a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Linkage analysis has revealed that the DSH gene locus resides on chromosome 1q11-q21,1 and the adenosine deaminase RNA specific gene, ADAR1 (also called DSRAD), in this region has been identified as being responsible for the development of DSH.2 We report a sporadic case of severe DSH with the ADAR1 gene detected in a mutation analysis.
A 6-year-old girl presented with a mixture of hyperpigmented and hypopigmented macules on the dorsal aspects of the hands and feet and the curved side of the wrists, heels, and knees, as well as scattered frecklelike and depigmented spots on the face, ears, neck, arms, and upper back (Figure 1). Her parents noted that hyperpigmented and hypopigmented macules on the dorsal aspects of the hands developed at 5 months of age. Exacerbation after exposure to sunlight resulted in the eruption becoming remarkable in summer and fainter in winter. The skin lesions gradually became more progressive. Physical examination revealed that the patient generally was healthy.
After obtaining informed consent, we performed a mutation analysis of the ADAR1 gene in our patient and her parents. We used a kit to extract genomic DNA from peripheral blood, which was then used to amplify the exons of the ADAR1 gene with intronic flanking sequences by polymerase chain reaction with the primer.3 After amplification, polymerase chain reaction products were purified. We sequenced the ADAR1 gene. Sequence comparisons and analysis found that the patient (proband) carried a heterozygous insertional mutation c.2253insG in exon 6 of the ADAR1 gene. This mutation was not detected in the proband’s healthy parents and 100 normal individuals (Figure 2).
Dyschromatosis symmetrica hereditaria is acquired by autosomal-dominant inheritance and is mainly reported in Asians, especially in Japan and China. Oyama et al4 reviewed 185 cases of DSH in Japan and found the onset of this disease usually was during infancy or childhood; 73% of patients developed the skin lesions before 6 years of age. Suzuki et al5 reported 10 unrelated Japanese patients and found the onset of disease ranged from 1 year of age to childhood. Zhang et al1,6 investigated 78 Chinese patients with DSH including 8 multigenerational families and 2 sporadic patients and found the age of disease onset ranged from 6 months to 15 years of age. The age of onset in our patient (5 months) was younger than these prior reports.
Patients with DSH have a characteristic appearance including a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Few patients have similar lesions on the knees and elbows. Many patients have frecklelike macules on the face and arms.1-6 One patient has been described with scattered depigmented spots on the face and chest.1 Our patient had a characteristic appearance as well as some special manifestations including skin lesions on the curved side of the wrist, ears, neck, and upper back.
The human ADAR1 gene spans 30 kilobase and contains 15 exons. It encodes RNA-specific adenosine deaminase composed of 1226 amino acid residues. This enzyme is important for various functions such as site-specific RNA editing and nuclear translation. This enzyme has 2 Z-alpha domains, 3 double-stranded RNA–binding domains, and the putative deaminase domain corresponding to exon 2, exons 2 to 7, and exons 9 to 14 of ADAR1, respectively.6
Mutation analysis of the ADAR1 gene in this case showed heterozygous insertion mutation c.2253insG in exon 6 of the ADAR1 gene, which changed the reading frame, and 475 amino acid residues in C-terminus are replaced by 90 amino acid residues (TSSRAQVRLPSKSWGSLVPSRLRTQQEA RQAGSSRCGSPCLDWGEREGRTHGFHRG NPSDRGQSQKNYAPPLKVPRSTAKT DTPSHWQHLP). This mutation was not detected in the proband’s healthy parents and the 100 control individuals, which indicated that it was a de novo mutation and the pathogenic mutation of DSH rather than a common polymorphism.
In conclusion, we report a novel mutation of the ADAR1 gene with a heavy clinical phenotype in DSH. This study expands the spectrum of clinical manifestations and demonstrates the ADAR1 mutation in DSH.
Acknowledgments
We are most grateful to the patient and her family for taking part in our study.
- Zhang XJ, Gao M, Li M, et al. Identification of a locus for dyschromatosis symmetrica hereditaria at chromosome 1q11-1q21. J Invest Dermatol. 2003;120:776-780.
- Miyamura Y, Suzuki T, Kono M, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria [published online August 11, 2003]. Am J Hum Genet. 2003;73:693-699.
- Li M, Li C, Hua H, et al. Identification of two novel mutations in Chinese patients with dyschromatosis symmetrica hereditaria [published online October 8, 2005]. Arch Dermatol Res. 2005;297:196-200.
- Oyama M, Shimizu H, Ohata Y, et al. Dyschromatosis symmetrica hereditaria (reticulate acropigmentation of Dohi): report of a Japanese family with the condition and a literature review of 185 cases. Br J Dermatol. 1999;140:491-496.
- Suzuki N, Suzuki T, Inagaki K, et al. Ten novel mutations of the ADAR1 gene in Japanese patients with dyschromatosis symmetrica hereditaria [published online August 17, 2006]. J Invest Dermatol. 2007;127:309-311.
- Zhang XJ, He PP, Li M, et al. Seven novel mutations of the ADAR gene in Chinese families and sporadic patients with dyschromatosis symmetrica hereditaria (DSH). Hum Mutat. 2004;23:629-630.
- Zhang XJ, Gao M, Li M, et al. Identification of a locus for dyschromatosis symmetrica hereditaria at chromosome 1q11-1q21. J Invest Dermatol. 2003;120:776-780.
- Miyamura Y, Suzuki T, Kono M, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria [published online August 11, 2003]. Am J Hum Genet. 2003;73:693-699.
- Li M, Li C, Hua H, et al. Identification of two novel mutations in Chinese patients with dyschromatosis symmetrica hereditaria [published online October 8, 2005]. Arch Dermatol Res. 2005;297:196-200.
- Oyama M, Shimizu H, Ohata Y, et al. Dyschromatosis symmetrica hereditaria (reticulate acropigmentation of Dohi): report of a Japanese family with the condition and a literature review of 185 cases. Br J Dermatol. 1999;140:491-496.
- Suzuki N, Suzuki T, Inagaki K, et al. Ten novel mutations of the ADAR1 gene in Japanese patients with dyschromatosis symmetrica hereditaria [published online August 17, 2006]. J Invest Dermatol. 2007;127:309-311.
- Zhang XJ, He PP, Li M, et al. Seven novel mutations of the ADAR gene in Chinese families and sporadic patients with dyschromatosis symmetrica hereditaria (DSH). Hum Mutat. 2004;23:629-630.
Practice Points
- The adenosine deaminase RNA specific gene, ADAR1, has been identified as being responsible for the development of dyschromatosis symmetrica hereditaria (DSH).
- The characteristic appearance of DSH is a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet.