User login
Foreign-Body Reaction to Orthopedic Hardware a Decade After Implantation
To the Editor:
Cutaneous reactions to implantable devices, such as dental implants, intracoronary stents, prosthetic valves, endovascular prostheses, gynecologic devices, and spinal cord stimulator devices, occur with varying frequency and include infectious, hypersensitivity, allergic, and foreign-body reactions. Manifestations have included contact dermatitis; urticarial, vasculitic, and bullous eruptions; extrusion; and granuloma formation.1,2 Immune complex reactions around implants causing pain, inflammation, and loosening of hardwarealso have been reported.3,4 Most reported cutaneous reactions typically occur within the first weeks or months after implantation; a reaction rarely presents several years after implantation. We report a cutaneous reaction to an orthopedic appliance almost 10 years after implantation.
A 67-year-old man presented with 2 painful nodules on the right clavicle that were present for several months. The patient denied fever, chills, weight loss, enlarged lymph nodes, or night sweats. Approximately 10 years prior to the appearance of the nodules, the patient fractured the right clavicle and underwent placement of a metal plate. His medical history included resection of the right tonsil and soft-palate carcinoma with radical neck dissection and postoperative radiation, which was completed approximately 4 years prior to placement of the metal plate. The patient recently completed 4 to 6 weeks of fluorouracil for shave biopsy–proven actinic keratosis overlying the entire irradiated area.
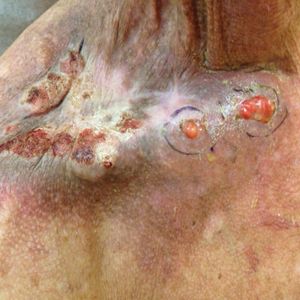
Physical examination revealed 2 pink friable nodules measuring 1.5 to 2.5 cm in diameter and leaking serous fluid within the irradiated area (Figure 1). The differential diagnosis included pyogenic granuloma, cutaneous recurrent metastasis, and atypical basal cell carcinoma. A skin biopsy specimen showed hemorrhagic ulcerated skin with acute and chronic inflammation and abscess.
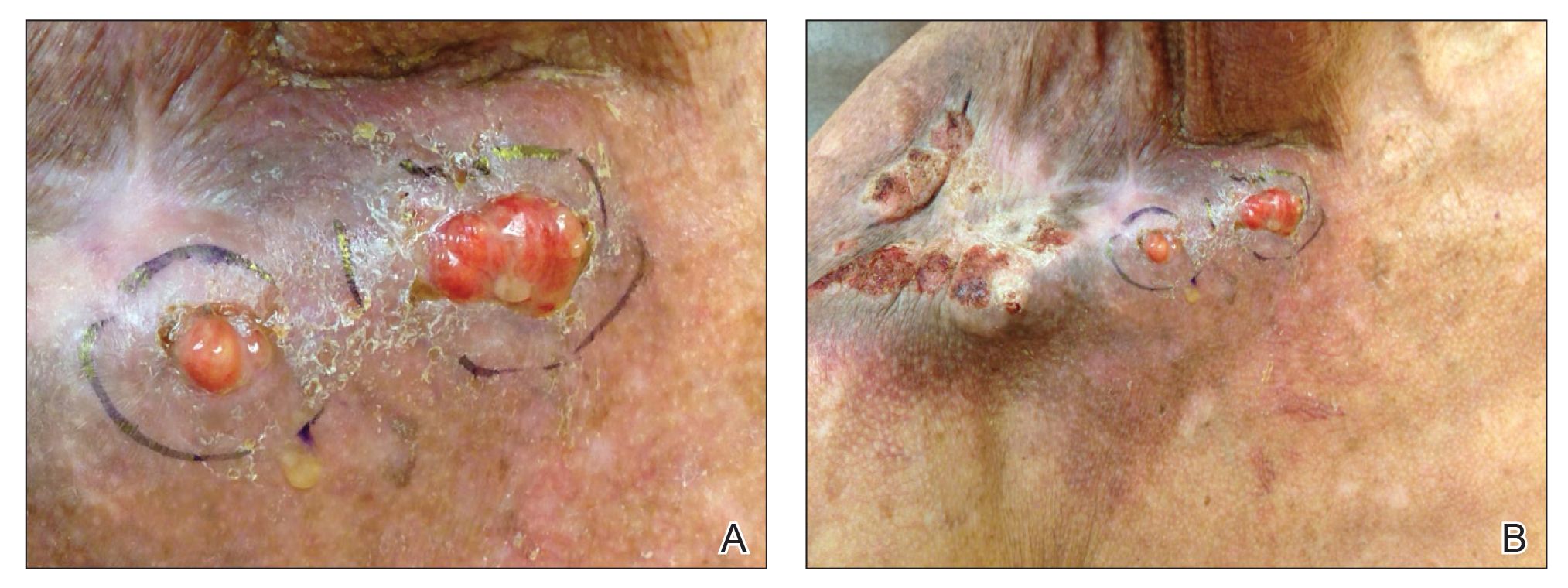
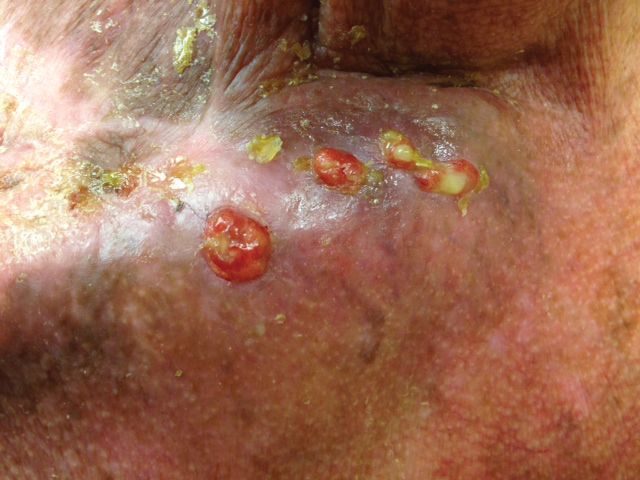
The patient presented for excisional biopsy of these areas on the right medial clavicle 1 week later. Physical examination revealed the 2 nodules had decreased in diameter; now, however, the patient had 4 discrete lesions measuring 4 to 7 mm in diameter, which were similar in appearance to the earlier nodules (Figure 2). He reported a low-grade fever, erythema, and increased tenderness of the area.
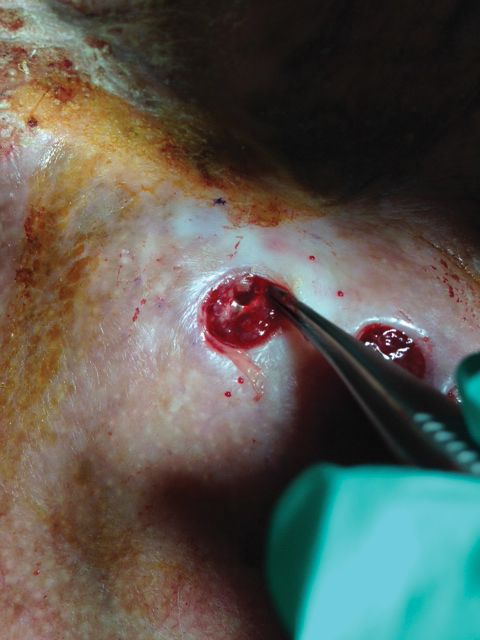
Underlying loosened orthopedic hardware screws were revealed upon punch biopsies of the involved areas (Figure 3). Wound cultures showed abundant Staphylococcus aureus and moderate group B Streptococcus; cultures for Mycobacterium were negative. The C-reactive protein level was elevated (5.47 mg/dL [reference range, ≤0.7 mg/dL]), and the erythrocyte sedimentation rate was increased (68 mm/h [reference range, 0–15 mm/h]). A complete blood cell count was within reference range, except for a mildly elevated eosinophil count (6.7% [reference range, 0%–5%]). The patient was admitted to the hospital, and antibiotics were started. Two days later, the orthopedic surgery service removed the hardware. At 3-week follow-up, physical examination revealed near closure of the wounds.
Cutaneous reactions to orthopedic implants include dermatitis, as well as urticarial, vasculitic, and bullous eruptions. Immune complex reactions can develop around implants, causing pain, inflammation, and loosening of hardware.1,3 Most inflammatory reactions take place within several months after implantation.3 Our patient’s reaction to hardware 10 years after implantation highlights the importance of taking a detailedand thorough history that includes queries about distant surgery.
- Basko-Plluska JL, Thyssen JP, Schalock PC. Cutaneous and systemic hypersensitivity reactions to metallic implants. Dermatitis. 2011;22:65-79.
- Chaudhry ZA, Najib U, Bajwa ZH, et al. Detailed analysis of allergic cutaneous reactions to spinal cord stimulator devices. J Pain Res. 2013;6:617-623.
- Huber M, Reinisch G, Trettenhahn G, et al. Presence of corrosion products and hypersensitivity-associated reactions in periprosthetic tissue after aseptic loosening of total hip replacements with metal bearing surfaces. Acta Biomater. 2009;5:172-180.
- Poncet-Wallet C, Ormezzano Y, Ernst E, et al. Study of a case of cochlear implant with recurrent cutaneous extrusion. Ann Otolaryngol Chir Cervicofac. 2009;126:264-268.
To the Editor:
Cutaneous reactions to implantable devices, such as dental implants, intracoronary stents, prosthetic valves, endovascular prostheses, gynecologic devices, and spinal cord stimulator devices, occur with varying frequency and include infectious, hypersensitivity, allergic, and foreign-body reactions. Manifestations have included contact dermatitis; urticarial, vasculitic, and bullous eruptions; extrusion; and granuloma formation.1,2 Immune complex reactions around implants causing pain, inflammation, and loosening of hardwarealso have been reported.3,4 Most reported cutaneous reactions typically occur within the first weeks or months after implantation; a reaction rarely presents several years after implantation. We report a cutaneous reaction to an orthopedic appliance almost 10 years after implantation.
A 67-year-old man presented with 2 painful nodules on the right clavicle that were present for several months. The patient denied fever, chills, weight loss, enlarged lymph nodes, or night sweats. Approximately 10 years prior to the appearance of the nodules, the patient fractured the right clavicle and underwent placement of a metal plate. His medical history included resection of the right tonsil and soft-palate carcinoma with radical neck dissection and postoperative radiation, which was completed approximately 4 years prior to placement of the metal plate. The patient recently completed 4 to 6 weeks of fluorouracil for shave biopsy–proven actinic keratosis overlying the entire irradiated area.
Physical examination revealed 2 pink friable nodules measuring 1.5 to 2.5 cm in diameter and leaking serous fluid within the irradiated area (Figure 1). The differential diagnosis included pyogenic granuloma, cutaneous recurrent metastasis, and atypical basal cell carcinoma. A skin biopsy specimen showed hemorrhagic ulcerated skin with acute and chronic inflammation and abscess.
The patient presented for excisional biopsy of these areas on the right medial clavicle 1 week later. Physical examination revealed the 2 nodules had decreased in diameter; now, however, the patient had 4 discrete lesions measuring 4 to 7 mm in diameter, which were similar in appearance to the earlier nodules (Figure 2). He reported a low-grade fever, erythema, and increased tenderness of the area.
Underlying loosened orthopedic hardware screws were revealed upon punch biopsies of the involved areas (Figure 3). Wound cultures showed abundant Staphylococcus aureus and moderate group B Streptococcus; cultures for Mycobacterium were negative. The C-reactive protein level was elevated (5.47 mg/dL [reference range, ≤0.7 mg/dL]), and the erythrocyte sedimentation rate was increased (68 mm/h [reference range, 0–15 mm/h]). A complete blood cell count was within reference range, except for a mildly elevated eosinophil count (6.7% [reference range, 0%–5%]). The patient was admitted to the hospital, and antibiotics were started. Two days later, the orthopedic surgery service removed the hardware. At 3-week follow-up, physical examination revealed near closure of the wounds.
Cutaneous reactions to orthopedic implants include dermatitis, as well as urticarial, vasculitic, and bullous eruptions. Immune complex reactions can develop around implants, causing pain, inflammation, and loosening of hardware.1,3 Most inflammatory reactions take place within several months after implantation.3 Our patient’s reaction to hardware 10 years after implantation highlights the importance of taking a detailedand thorough history that includes queries about distant surgery.
To the Editor:
Cutaneous reactions to implantable devices, such as dental implants, intracoronary stents, prosthetic valves, endovascular prostheses, gynecologic devices, and spinal cord stimulator devices, occur with varying frequency and include infectious, hypersensitivity, allergic, and foreign-body reactions. Manifestations have included contact dermatitis; urticarial, vasculitic, and bullous eruptions; extrusion; and granuloma formation.1,2 Immune complex reactions around implants causing pain, inflammation, and loosening of hardwarealso have been reported.3,4 Most reported cutaneous reactions typically occur within the first weeks or months after implantation; a reaction rarely presents several years after implantation. We report a cutaneous reaction to an orthopedic appliance almost 10 years after implantation.
A 67-year-old man presented with 2 painful nodules on the right clavicle that were present for several months. The patient denied fever, chills, weight loss, enlarged lymph nodes, or night sweats. Approximately 10 years prior to the appearance of the nodules, the patient fractured the right clavicle and underwent placement of a metal plate. His medical history included resection of the right tonsil and soft-palate carcinoma with radical neck dissection and postoperative radiation, which was completed approximately 4 years prior to placement of the metal plate. The patient recently completed 4 to 6 weeks of fluorouracil for shave biopsy–proven actinic keratosis overlying the entire irradiated area.
Physical examination revealed 2 pink friable nodules measuring 1.5 to 2.5 cm in diameter and leaking serous fluid within the irradiated area (Figure 1). The differential diagnosis included pyogenic granuloma, cutaneous recurrent metastasis, and atypical basal cell carcinoma. A skin biopsy specimen showed hemorrhagic ulcerated skin with acute and chronic inflammation and abscess.
The patient presented for excisional biopsy of these areas on the right medial clavicle 1 week later. Physical examination revealed the 2 nodules had decreased in diameter; now, however, the patient had 4 discrete lesions measuring 4 to 7 mm in diameter, which were similar in appearance to the earlier nodules (Figure 2). He reported a low-grade fever, erythema, and increased tenderness of the area.
Underlying loosened orthopedic hardware screws were revealed upon punch biopsies of the involved areas (Figure 3). Wound cultures showed abundant Staphylococcus aureus and moderate group B Streptococcus; cultures for Mycobacterium were negative. The C-reactive protein level was elevated (5.47 mg/dL [reference range, ≤0.7 mg/dL]), and the erythrocyte sedimentation rate was increased (68 mm/h [reference range, 0–15 mm/h]). A complete blood cell count was within reference range, except for a mildly elevated eosinophil count (6.7% [reference range, 0%–5%]). The patient was admitted to the hospital, and antibiotics were started. Two days later, the orthopedic surgery service removed the hardware. At 3-week follow-up, physical examination revealed near closure of the wounds.
Cutaneous reactions to orthopedic implants include dermatitis, as well as urticarial, vasculitic, and bullous eruptions. Immune complex reactions can develop around implants, causing pain, inflammation, and loosening of hardware.1,3 Most inflammatory reactions take place within several months after implantation.3 Our patient’s reaction to hardware 10 years after implantation highlights the importance of taking a detailedand thorough history that includes queries about distant surgery.
- Basko-Plluska JL, Thyssen JP, Schalock PC. Cutaneous and systemic hypersensitivity reactions to metallic implants. Dermatitis. 2011;22:65-79.
- Chaudhry ZA, Najib U, Bajwa ZH, et al. Detailed analysis of allergic cutaneous reactions to spinal cord stimulator devices. J Pain Res. 2013;6:617-623.
- Huber M, Reinisch G, Trettenhahn G, et al. Presence of corrosion products and hypersensitivity-associated reactions in periprosthetic tissue after aseptic loosening of total hip replacements with metal bearing surfaces. Acta Biomater. 2009;5:172-180.
- Poncet-Wallet C, Ormezzano Y, Ernst E, et al. Study of a case of cochlear implant with recurrent cutaneous extrusion. Ann Otolaryngol Chir Cervicofac. 2009;126:264-268.
- Basko-Plluska JL, Thyssen JP, Schalock PC. Cutaneous and systemic hypersensitivity reactions to metallic implants. Dermatitis. 2011;22:65-79.
- Chaudhry ZA, Najib U, Bajwa ZH, et al. Detailed analysis of allergic cutaneous reactions to spinal cord stimulator devices. J Pain Res. 2013;6:617-623.
- Huber M, Reinisch G, Trettenhahn G, et al. Presence of corrosion products and hypersensitivity-associated reactions in periprosthetic tissue after aseptic loosening of total hip replacements with metal bearing surfaces. Acta Biomater. 2009;5:172-180.
- Poncet-Wallet C, Ormezzano Y, Ernst E, et al. Study of a case of cochlear implant with recurrent cutaneous extrusion. Ann Otolaryngol Chir Cervicofac. 2009;126:264-268.
Practice Points
- Cutaneous reactions to implantable devices occur with varying frequency and include infectious, hypersensitivity, allergic, and foreign-body reactions.
- Most reactions typically occur within the first weeks or months after implantation; however, a reaction rarely may present several years after implantation.
Recurrent Cutaneous Exophiala Phaeohyphomycosis in an Immunosuppressed Patient
To the Editor:
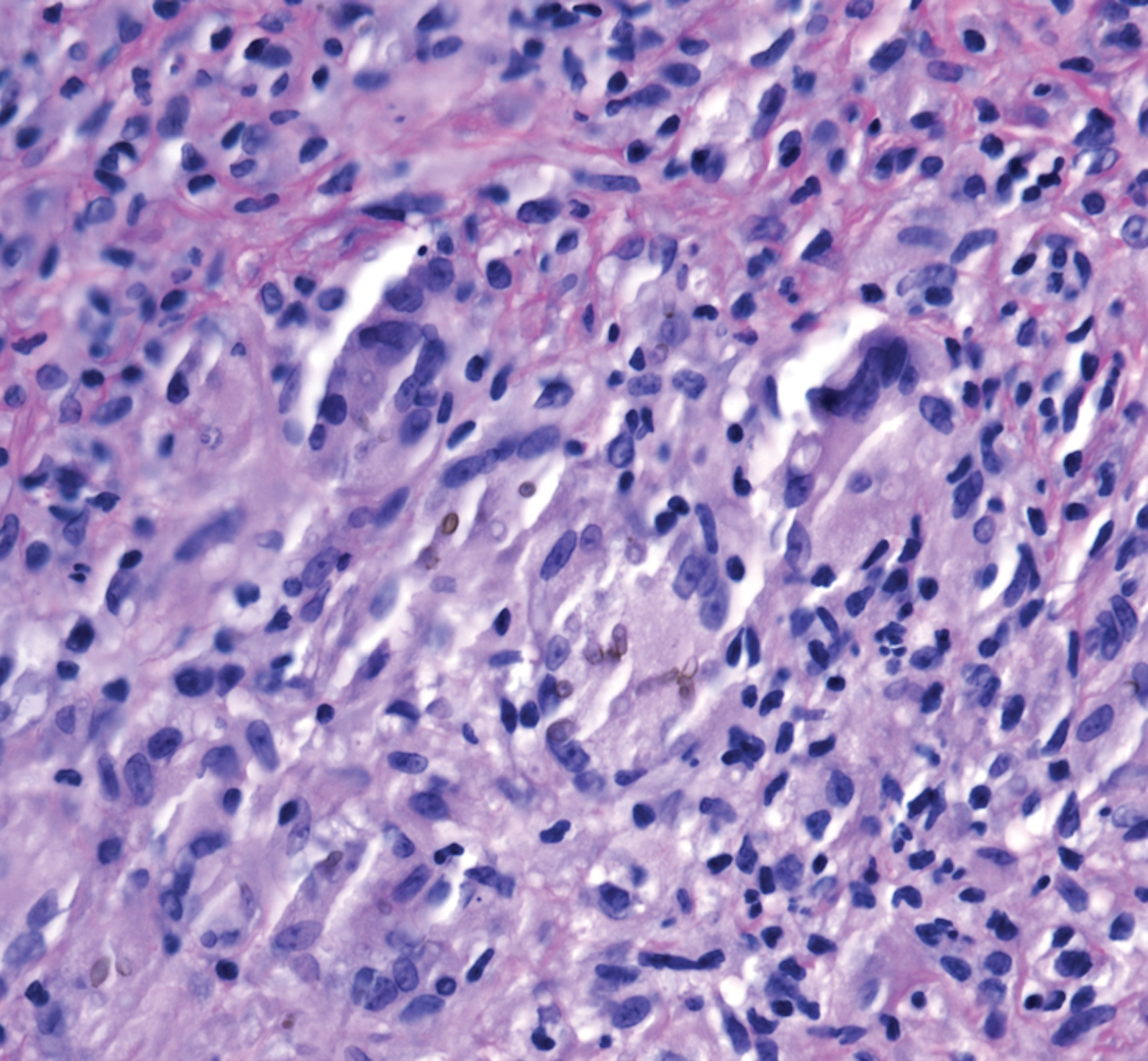
A 73-year-old man presented with a 2.5-cm, recurrent, fluctuant, multiloculated nodule on the left forearm. The lesion was nontender with occasional chalky, white to yellow discharge from multiple sinus tracts. He was otherwise well appearing without signs of systemic infection. He reported similar lesions in slightly different anatomic locations on the left forearm both 7 and 4 years prior to the current presentation. In both instances, the nodules were excised at an outside hospital without any additional treatment. Histopathology of the excised tissue from both prior occasions demonstrated brown septate hyphae surrounded by suppurative and granulomatous inflammation consistent with dematiaceous fungal infection of the dermis (Figures 1 and 2); the organisms were highlighted with periodic acid–Schiff stain.

The patient’s medical history was notable for advanced heart failure with an ejection fraction of 25% and autosomal-dominant polycystic kidney disease. He received an orthotopic kidney transplant 17 years prior to the current presentation. Medications included tacrolimus, mycophenolate mofetil, and prednisone. He denied any trauma or notable exposures to vegetation, and his travel history was unremarkable. A review of systems was negative.
At the current presentation, a sterile fungal culture was performed and found positive for Exophiala species, while bacterial and mycobacterial cultures were negative. A diagnosis of phaeohyphomycosis was made, and he was scheduled for re-excision. Out of concern for interactions with his immunosuppressive regimen, he chose to forgo any systemic antifungal therapy. He died from hospital-acquired pneumonia and volume overload unresponsive to diuretics or dialysis.
Phaeohyphomycosis is a rare fungal infection caused by several genera of dematiaceous fungi that are characterized by the presence of melaninlike cell wall pigments thought to locally hinder immune clearance by scavenging phagocyte-derived free radicals. These fungi are ubiquitous in soil and vegetation and usually penetrate the skin at sites of minor trauma.1 Phaeohyphomycosis typically affects immunosuppressed hosts, and its incidence among organ transplant recipients currently is 9%.2 The incidence in this population has been rising, however, as recent advances in immunosuppressive therapies have increased posttransplant survival.3
Subcutaneous phaeohyphomycosis can present with nodules, cysts, tumors, and/or verrucous plaques, and the diagnosis almost always requires clinicopathologic correlation.3 Rapid diagnosis can be made when septate brown hyphae and/or yeast forms are observed on hematoxylin and eosin stain. Rarely, patients present with disseminated infection, characterized by fungemia; central nervous system involvement; and/or infection of multiple deep structures including the eyes, lungs, bones, and sinuses.4 The risk for dissemination from the skin likely is related to the culprit organism’s genus; Lomentospora, Cladophialophora, and Verruconis often are associated with dissemination, while Alternaria, Exophiala, and Fonsecaea typically remain confined to the skin and subcutis.5 Due to this difference and its potential to impact management, obtaining a tissue fungal culture is advisable when phaeohyphomycosis is suspected.
There is no standard treatment of phaeohyphomycosis. Regimens typically consist of excision and prolonged courses of azole therapy, though excision alone with close follow-up may be a reasonable alternative.6 The latter is a particularly important consideration when managing phaeohyphomycosis in organ transplant recipients, as azoles are known cytochrome P450 3A4 inhibitors that can affect serum levels of common immunosuppressive medications including calcineurin inhibitors and mammalian target of rapamycin inhibitors.3 Local recurrence is common regardless of whether azole therapy is pursued,7 and dissemination of localized Exophiala infections is exceedingly rare.8 There is a strong argument to be made for our patient’s decision to forgo antifungal therapy.
This case underscores the difficulty inherent to eradicating local subcutaneous Exophiala phaeohyphomycosis while providing reassurance that with treatment, the risk of life-threatening complications is low. Obtaining tissue for both hematoxylin and eosin stain and sterile culture is crucial to ensuring prompt diagnosis and tailoring the optimal treatment and surveillance strategy to the culprit organism. To avoid delays in diagnosis and treatment, it is important for clinicians to consider phaeohyphomycosis in the differential diagnosis for recurrent nodulocystic lesions in immunosuppressed patients and to recognize that presentations may span many years.
- Bhardwaj S, Capoor MR, Kolte S, et al. Phaeohyphomycosis due to Exophiala jeanselmei: an emerging pathogen in India—case report and review. Mycopathologia. 2016;181:279-284.
- Isa-Isa R, Garcia C, Isa M, et al. Subcutaneous phaeohyphomycosis (mycotic cyst). Clin Dermatol. 2012;30:425-431.
- Tirico MCCP, Neto CF, Cruz LL, et al. Clinical spectrum of phaeohyphomycosis in solid organ transplant recipients. JAAD Case Rep. 2016;2:465-469.
- Revankar SG, Patterson JE, Sutton DA, et al. Disseminated phaeohyphomycosis: review of an emerging mycosis. Clin Infect Dis. 2002;34:467-476.
- Revankar SG, Baddley JW, Chen SC-A, et al. A mycoses study group international prospective study of phaeohyphomycosis: an analysis of 99 proven/probable cases. Open Forum Infect Dis. 2017;4:ofx200.
- Oberlin KE, Nichols AJ, Rosa R, et al. Phaeohyphomycosis due to Exophiala infections in solid organ transplant recipients: case report and literature review [published online June 26, 2017]. Transpl Infect Dis. 2017;19. doi:10.1111/tid.12723.
- Shirbur S, Telkar S, Goudar B, et al. Recurrent phaeohyphomycosis: a case report. J Clin Diagn Res. 2013;7:2015-2016.
- Li D-M, Li R-Y, de Hoog GS, et al. Fatal Exophiala infections in China, with a report of seven cases. Mycoses. 2011;54:E136-E142.
To the Editor:
A 73-year-old man presented with a 2.5-cm, recurrent, fluctuant, multiloculated nodule on the left forearm. The lesion was nontender with occasional chalky, white to yellow discharge from multiple sinus tracts. He was otherwise well appearing without signs of systemic infection. He reported similar lesions in slightly different anatomic locations on the left forearm both 7 and 4 years prior to the current presentation. In both instances, the nodules were excised at an outside hospital without any additional treatment. Histopathology of the excised tissue from both prior occasions demonstrated brown septate hyphae surrounded by suppurative and granulomatous inflammation consistent with dematiaceous fungal infection of the dermis (Figures 1 and 2); the organisms were highlighted with periodic acid–Schiff stain.
The patient’s medical history was notable for advanced heart failure with an ejection fraction of 25% and autosomal-dominant polycystic kidney disease. He received an orthotopic kidney transplant 17 years prior to the current presentation. Medications included tacrolimus, mycophenolate mofetil, and prednisone. He denied any trauma or notable exposures to vegetation, and his travel history was unremarkable. A review of systems was negative.
At the current presentation, a sterile fungal culture was performed and found positive for Exophiala species, while bacterial and mycobacterial cultures were negative. A diagnosis of phaeohyphomycosis was made, and he was scheduled for re-excision. Out of concern for interactions with his immunosuppressive regimen, he chose to forgo any systemic antifungal therapy. He died from hospital-acquired pneumonia and volume overload unresponsive to diuretics or dialysis.
Phaeohyphomycosis is a rare fungal infection caused by several genera of dematiaceous fungi that are characterized by the presence of melaninlike cell wall pigments thought to locally hinder immune clearance by scavenging phagocyte-derived free radicals. These fungi are ubiquitous in soil and vegetation and usually penetrate the skin at sites of minor trauma.1 Phaeohyphomycosis typically affects immunosuppressed hosts, and its incidence among organ transplant recipients currently is 9%.2 The incidence in this population has been rising, however, as recent advances in immunosuppressive therapies have increased posttransplant survival.3
Subcutaneous phaeohyphomycosis can present with nodules, cysts, tumors, and/or verrucous plaques, and the diagnosis almost always requires clinicopathologic correlation.3 Rapid diagnosis can be made when septate brown hyphae and/or yeast forms are observed on hematoxylin and eosin stain. Rarely, patients present with disseminated infection, characterized by fungemia; central nervous system involvement; and/or infection of multiple deep structures including the eyes, lungs, bones, and sinuses.4 The risk for dissemination from the skin likely is related to the culprit organism’s genus; Lomentospora, Cladophialophora, and Verruconis often are associated with dissemination, while Alternaria, Exophiala, and Fonsecaea typically remain confined to the skin and subcutis.5 Due to this difference and its potential to impact management, obtaining a tissue fungal culture is advisable when phaeohyphomycosis is suspected.
There is no standard treatment of phaeohyphomycosis. Regimens typically consist of excision and prolonged courses of azole therapy, though excision alone with close follow-up may be a reasonable alternative.6 The latter is a particularly important consideration when managing phaeohyphomycosis in organ transplant recipients, as azoles are known cytochrome P450 3A4 inhibitors that can affect serum levels of common immunosuppressive medications including calcineurin inhibitors and mammalian target of rapamycin inhibitors.3 Local recurrence is common regardless of whether azole therapy is pursued,7 and dissemination of localized Exophiala infections is exceedingly rare.8 There is a strong argument to be made for our patient’s decision to forgo antifungal therapy.
This case underscores the difficulty inherent to eradicating local subcutaneous Exophiala phaeohyphomycosis while providing reassurance that with treatment, the risk of life-threatening complications is low. Obtaining tissue for both hematoxylin and eosin stain and sterile culture is crucial to ensuring prompt diagnosis and tailoring the optimal treatment and surveillance strategy to the culprit organism. To avoid delays in diagnosis and treatment, it is important for clinicians to consider phaeohyphomycosis in the differential diagnosis for recurrent nodulocystic lesions in immunosuppressed patients and to recognize that presentations may span many years.
To the Editor:
A 73-year-old man presented with a 2.5-cm, recurrent, fluctuant, multiloculated nodule on the left forearm. The lesion was nontender with occasional chalky, white to yellow discharge from multiple sinus tracts. He was otherwise well appearing without signs of systemic infection. He reported similar lesions in slightly different anatomic locations on the left forearm both 7 and 4 years prior to the current presentation. In both instances, the nodules were excised at an outside hospital without any additional treatment. Histopathology of the excised tissue from both prior occasions demonstrated brown septate hyphae surrounded by suppurative and granulomatous inflammation consistent with dematiaceous fungal infection of the dermis (Figures 1 and 2); the organisms were highlighted with periodic acid–Schiff stain.
The patient’s medical history was notable for advanced heart failure with an ejection fraction of 25% and autosomal-dominant polycystic kidney disease. He received an orthotopic kidney transplant 17 years prior to the current presentation. Medications included tacrolimus, mycophenolate mofetil, and prednisone. He denied any trauma or notable exposures to vegetation, and his travel history was unremarkable. A review of systems was negative.
At the current presentation, a sterile fungal culture was performed and found positive for Exophiala species, while bacterial and mycobacterial cultures were negative. A diagnosis of phaeohyphomycosis was made, and he was scheduled for re-excision. Out of concern for interactions with his immunosuppressive regimen, he chose to forgo any systemic antifungal therapy. He died from hospital-acquired pneumonia and volume overload unresponsive to diuretics or dialysis.
Phaeohyphomycosis is a rare fungal infection caused by several genera of dematiaceous fungi that are characterized by the presence of melaninlike cell wall pigments thought to locally hinder immune clearance by scavenging phagocyte-derived free radicals. These fungi are ubiquitous in soil and vegetation and usually penetrate the skin at sites of minor trauma.1 Phaeohyphomycosis typically affects immunosuppressed hosts, and its incidence among organ transplant recipients currently is 9%.2 The incidence in this population has been rising, however, as recent advances in immunosuppressive therapies have increased posttransplant survival.3
Subcutaneous phaeohyphomycosis can present with nodules, cysts, tumors, and/or verrucous plaques, and the diagnosis almost always requires clinicopathologic correlation.3 Rapid diagnosis can be made when septate brown hyphae and/or yeast forms are observed on hematoxylin and eosin stain. Rarely, patients present with disseminated infection, characterized by fungemia; central nervous system involvement; and/or infection of multiple deep structures including the eyes, lungs, bones, and sinuses.4 The risk for dissemination from the skin likely is related to the culprit organism’s genus; Lomentospora, Cladophialophora, and Verruconis often are associated with dissemination, while Alternaria, Exophiala, and Fonsecaea typically remain confined to the skin and subcutis.5 Due to this difference and its potential to impact management, obtaining a tissue fungal culture is advisable when phaeohyphomycosis is suspected.
There is no standard treatment of phaeohyphomycosis. Regimens typically consist of excision and prolonged courses of azole therapy, though excision alone with close follow-up may be a reasonable alternative.6 The latter is a particularly important consideration when managing phaeohyphomycosis in organ transplant recipients, as azoles are known cytochrome P450 3A4 inhibitors that can affect serum levels of common immunosuppressive medications including calcineurin inhibitors and mammalian target of rapamycin inhibitors.3 Local recurrence is common regardless of whether azole therapy is pursued,7 and dissemination of localized Exophiala infections is exceedingly rare.8 There is a strong argument to be made for our patient’s decision to forgo antifungal therapy.
This case underscores the difficulty inherent to eradicating local subcutaneous Exophiala phaeohyphomycosis while providing reassurance that with treatment, the risk of life-threatening complications is low. Obtaining tissue for both hematoxylin and eosin stain and sterile culture is crucial to ensuring prompt diagnosis and tailoring the optimal treatment and surveillance strategy to the culprit organism. To avoid delays in diagnosis and treatment, it is important for clinicians to consider phaeohyphomycosis in the differential diagnosis for recurrent nodulocystic lesions in immunosuppressed patients and to recognize that presentations may span many years.
- Bhardwaj S, Capoor MR, Kolte S, et al. Phaeohyphomycosis due to Exophiala jeanselmei: an emerging pathogen in India—case report and review. Mycopathologia. 2016;181:279-284.
- Isa-Isa R, Garcia C, Isa M, et al. Subcutaneous phaeohyphomycosis (mycotic cyst). Clin Dermatol. 2012;30:425-431.
- Tirico MCCP, Neto CF, Cruz LL, et al. Clinical spectrum of phaeohyphomycosis in solid organ transplant recipients. JAAD Case Rep. 2016;2:465-469.
- Revankar SG, Patterson JE, Sutton DA, et al. Disseminated phaeohyphomycosis: review of an emerging mycosis. Clin Infect Dis. 2002;34:467-476.
- Revankar SG, Baddley JW, Chen SC-A, et al. A mycoses study group international prospective study of phaeohyphomycosis: an analysis of 99 proven/probable cases. Open Forum Infect Dis. 2017;4:ofx200.
- Oberlin KE, Nichols AJ, Rosa R, et al. Phaeohyphomycosis due to Exophiala infections in solid organ transplant recipients: case report and literature review [published online June 26, 2017]. Transpl Infect Dis. 2017;19. doi:10.1111/tid.12723.
- Shirbur S, Telkar S, Goudar B, et al. Recurrent phaeohyphomycosis: a case report. J Clin Diagn Res. 2013;7:2015-2016.
- Li D-M, Li R-Y, de Hoog GS, et al. Fatal Exophiala infections in China, with a report of seven cases. Mycoses. 2011;54:E136-E142.
- Bhardwaj S, Capoor MR, Kolte S, et al. Phaeohyphomycosis due to Exophiala jeanselmei: an emerging pathogen in India—case report and review. Mycopathologia. 2016;181:279-284.
- Isa-Isa R, Garcia C, Isa M, et al. Subcutaneous phaeohyphomycosis (mycotic cyst). Clin Dermatol. 2012;30:425-431.
- Tirico MCCP, Neto CF, Cruz LL, et al. Clinical spectrum of phaeohyphomycosis in solid organ transplant recipients. JAAD Case Rep. 2016;2:465-469.
- Revankar SG, Patterson JE, Sutton DA, et al. Disseminated phaeohyphomycosis: review of an emerging mycosis. Clin Infect Dis. 2002;34:467-476.
- Revankar SG, Baddley JW, Chen SC-A, et al. A mycoses study group international prospective study of phaeohyphomycosis: an analysis of 99 proven/probable cases. Open Forum Infect Dis. 2017;4:ofx200.
- Oberlin KE, Nichols AJ, Rosa R, et al. Phaeohyphomycosis due to Exophiala infections in solid organ transplant recipients: case report and literature review [published online June 26, 2017]. Transpl Infect Dis. 2017;19. doi:10.1111/tid.12723.
- Shirbur S, Telkar S, Goudar B, et al. Recurrent phaeohyphomycosis: a case report. J Clin Diagn Res. 2013;7:2015-2016.
- Li D-M, Li R-Y, de Hoog GS, et al. Fatal Exophiala infections in China, with a report of seven cases. Mycoses. 2011;54:E136-E142.
Practice Points
- Phaeohyphomycosis is an infection with dematiaceous fungi that most commonly affects immunosuppressed patients.
- Subcutaneous phaeohyphomycosis may present with nodulocystic lesions that recur over the course of years.
- Tissue fungal culture should be obtained when the diagnosis is suspected, as the risk for dissemination is related to the culprit organism.
- Surgical excision with close follow-up may be an appropriate management strategy for patients on immunosuppressive medications to avoid interactions with azole therapy.
Scalp Arteriovenous Fistula With Intracranial Communication
To the Editor:
A 71-year-old man presented with a nodule on the vertex of the scalp of 1 year’s duration. The lesion had become soft and tender during the week prior to presentation. He noted that he was experiencing headaches and a buzzing sound in his head. He denied all other neurologic symptoms. The patient was given amoxicillin from a primary care physician and was referred to our institution for evaluation of a presumed inflamed cyst.
The patient’s medical history included an intracranial arteriovenous fistula (AVF) treated with endovascular embolization 1 year prior to presentation, 2 substantial falls in childhood with head trauma and loss of consciousness, essential hypertension, and an aortic aneurysm. His medications included amlodipine, lisinopril, amoxicillin, a multivitamin, and grape seed extract.
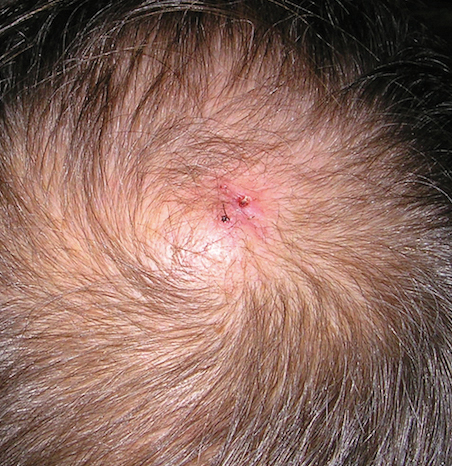
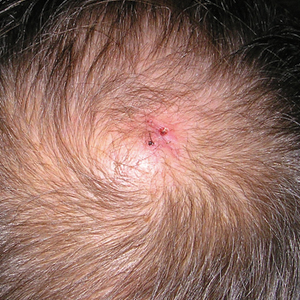
Physical examination revealed a 2-cm, pink, somewhat rubbery, subcutaneous, nonmobile nodule on the vertex of the scalp (Figure 1). The lesion was not consistent with a common pilar cyst, and an excisional biopsy was performed to exclude malignancy. Upon superficial incision, the lesion bled moderately, and the procedure was immediately discontinued. Hemostasis was obtained, and the patient was sent for ultrasonography of the lesion.
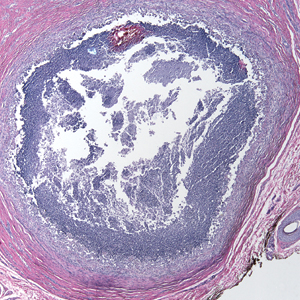
Ultrasonography demonstrated a small hypoechoic nodule measuring up to 0.5 cm containing a tangle of vessels in the subcutaneous soft tissue corresponding to the palpable abnormality. A cerebral angiogram demonstrated a dural AVF of the superior sagittal sinus with multifocal supply that connected with this scalp nodule (Figure 2). The patient was treated by interventional neuroradiology with endovascular embolization, which resulted in complete resolution of the scalp nodule.
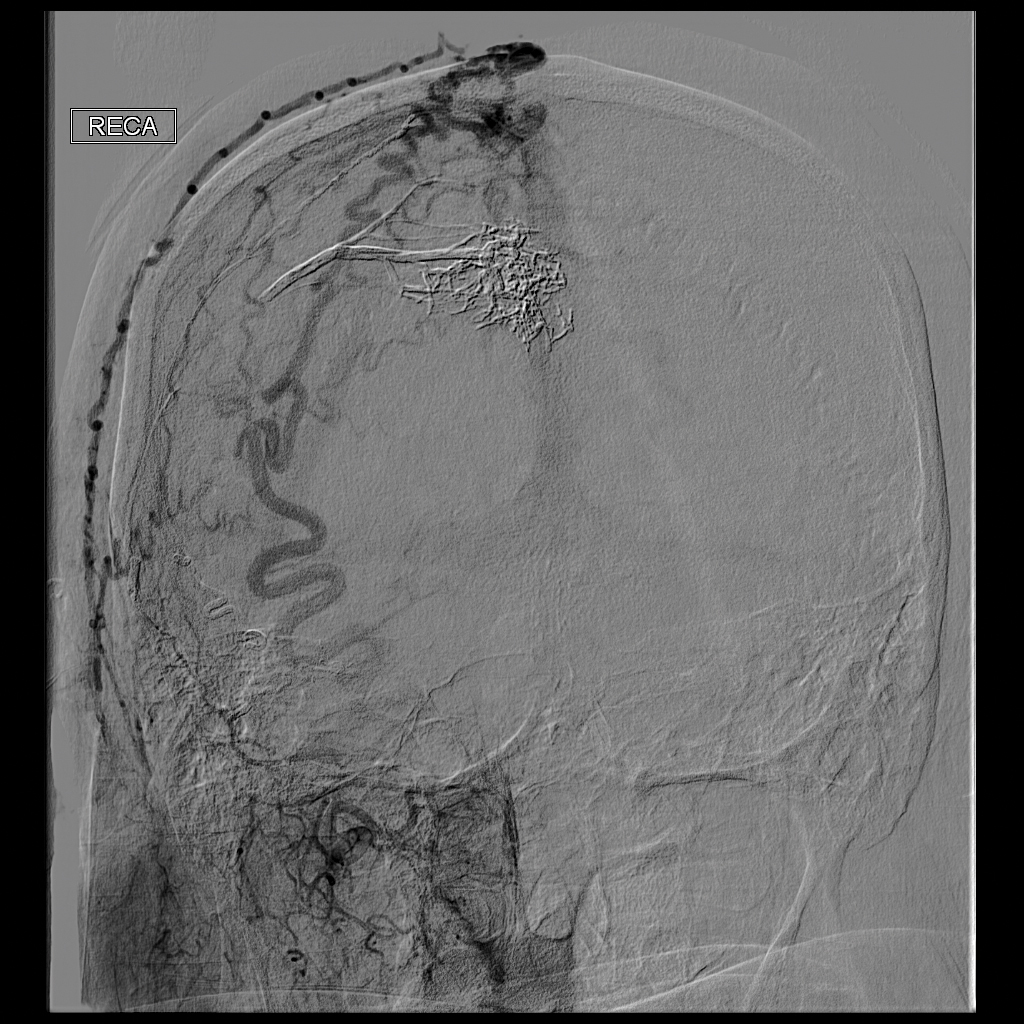
Scalp arteriovenous fistulas (S-AVFs) are characterized by abnormal connections between supplying arteries and draining veins in the subcutaneous plane of the scalp.1,2 The veins of an S-AVF undergo progressive aneurysmal dilatation from abnormal hemodynamics.1-3 Scalp arteriovenous fistulas are rare and may present as either an innocuous-looking scalp nodule or a progressively enlarging pulsatile mass on the scalp.2-4 Associated symptoms often include headache, local pain, bruits, tinnitus, and thrill.1,3,4 Recurrent hemorrhage, scalp necrosis, congestive heart failure, epilepsy, mental retardation, and intracranial ischemia also may occur.4
Scalp AVFs may occur with or without intracranial communication.4 Spontaneous S-AVFs with intracranial communication are uncommon, and their etiology is unclear. They may form as congenital malformations or may be idiopathic. Factors increasing circulation through the S-AVF such as trauma, pregnancy, hormonal changes, and inflammation prompt the development of symptoms.4 Scalp AVFs also may be caused by trauma.3 Scalp AVFs without intracranial communication have been reported following hair transplantation.1 Scalp AVFs with intracranial communication have been reported months to years after skull fracture or craniotomy.2 True spontaneous S-AVFs are difficult to differentiate from traumatic S-AVFs other than by history alone.2
Increased venous pressure has been shown to generate AVFs in rats.5 It has been suggested that S-AVFs can become enlarged by capturing subcutaneous or intracranial feeder vessels and that the consequent hemodynamic stress may induce de novo aneurysms in S-AVFs. Additionally, intracranial AVFs may alter the intracranial hemodynamics, leading to increased venous pressure in the superior sagittal sinus and the formation of communicating S-AVFs.5 Interestingly, our patient had an intracranial AVF treated with endovascular embolization 1 year prior to the formation of the S-AVF. An angiogram at the time of this embolization procedure did not demonstrate any S-AVFs. Furthermore, our patient has a history of 2 substantial falls in childhood with head trauma and loss of consciousness. Perhaps these traumas initiated a channel through the cranium where an S-AVF with intracranial communication was able to form and may have only become clinically or radiographically detectable once it enlarged due to the altered hemodynamics caused by the intracranial AVF 1 year prior.
The diagnosis of an S-AVF is confirmed with imaging studies. Doppler ultrasonography initially will help to detect that a lesion is vascular in nature. Intra-arterial digital subtraction angiography is the gold-standard imaging technique and is necessary to delineate the feeding arteries and the draining channels as well as possible communication with intracranial vasculature.1,2 There is controversy regarding the appropriate treatment of S-AVFs.2 Each S-AVF possesses unique anatomic features that dictate appropriate management. The prognosis for an S-AVF is extremely variable, and the decision to treat is based on the patient’s symptoms and risk for exsanguinating hemorrhage.2,4 Neurosurgical approaches include ligation of the feeding arteries, surgical resection, electrothrombosis, direct intralesional injection of sclerosing agents, and endovascular embolization. Endovascular intervention increasingly is utilized as a primary treatment or as a preoperative adjunct to surgery.2,4 Large S-AVFs have a high risk for recurrence after treatment with endovascular embolization alone. In cases with intracranial communication, the intracranial component is treated first.2
This case emphasizes the importance of including S-AVFs on the dermatologic differential diagnosis of a scalp nodule, especially in patients with any history of intracranial AVF. A thorough history, detailed intake of potential signs and symptoms of AVF, and palpation for bruits is recommended as part of the surgical evaluation of a scalp nodule. Imaging of scalp nodules also should be considered for patients with any history of intracranial AVF; S-AVFs should be referred to neurosurgery or interventional neuroradiology for evaluation and possible treatment.
- Bernstein J, Podnos S, Leavitt M. Arteriovenous fistula following hair transplantation. Dermatol Surg. 2011;37:873-875.
- Kumar R, Sharma G, Sharma BS. Management of scalp arterio-venous malformation: case series and review of literature. Br J Neurosurg. 2012;26:371-377.
- Gurkanlar D, Gonul M, Solmaz I, et al. Cirsoid aneurysms of the scalp. Neurosurg Rev. 2006;29:208-212.
- Senoglu M, Yasim A, Gokce M, et al. Nontraumatic scalp arteriovenous fistula in an adult: technical report on an illustrative case. Surg Neurol. 2008;70:194-197.
- Lanzino G, Passacantilli E, Lemole G, et al. Scalp arteriovenous malformation draining into the superior sagittal sinus associated with an intracranial arteriovenous malformation: just a coincidence? case report. Neurosurgery. 2003;52:440-443.
To the Editor:
A 71-year-old man presented with a nodule on the vertex of the scalp of 1 year’s duration. The lesion had become soft and tender during the week prior to presentation. He noted that he was experiencing headaches and a buzzing sound in his head. He denied all other neurologic symptoms. The patient was given amoxicillin from a primary care physician and was referred to our institution for evaluation of a presumed inflamed cyst.
The patient’s medical history included an intracranial arteriovenous fistula (AVF) treated with endovascular embolization 1 year prior to presentation, 2 substantial falls in childhood with head trauma and loss of consciousness, essential hypertension, and an aortic aneurysm. His medications included amlodipine, lisinopril, amoxicillin, a multivitamin, and grape seed extract.
Physical examination revealed a 2-cm, pink, somewhat rubbery, subcutaneous, nonmobile nodule on the vertex of the scalp (Figure 1). The lesion was not consistent with a common pilar cyst, and an excisional biopsy was performed to exclude malignancy. Upon superficial incision, the lesion bled moderately, and the procedure was immediately discontinued. Hemostasis was obtained, and the patient was sent for ultrasonography of the lesion.
Ultrasonography demonstrated a small hypoechoic nodule measuring up to 0.5 cm containing a tangle of vessels in the subcutaneous soft tissue corresponding to the palpable abnormality. A cerebral angiogram demonstrated a dural AVF of the superior sagittal sinus with multifocal supply that connected with this scalp nodule (Figure 2). The patient was treated by interventional neuroradiology with endovascular embolization, which resulted in complete resolution of the scalp nodule.
Scalp arteriovenous fistulas (S-AVFs) are characterized by abnormal connections between supplying arteries and draining veins in the subcutaneous plane of the scalp.1,2 The veins of an S-AVF undergo progressive aneurysmal dilatation from abnormal hemodynamics.1-3 Scalp arteriovenous fistulas are rare and may present as either an innocuous-looking scalp nodule or a progressively enlarging pulsatile mass on the scalp.2-4 Associated symptoms often include headache, local pain, bruits, tinnitus, and thrill.1,3,4 Recurrent hemorrhage, scalp necrosis, congestive heart failure, epilepsy, mental retardation, and intracranial ischemia also may occur.4
Scalp AVFs may occur with or without intracranial communication.4 Spontaneous S-AVFs with intracranial communication are uncommon, and their etiology is unclear. They may form as congenital malformations or may be idiopathic. Factors increasing circulation through the S-AVF such as trauma, pregnancy, hormonal changes, and inflammation prompt the development of symptoms.4 Scalp AVFs also may be caused by trauma.3 Scalp AVFs without intracranial communication have been reported following hair transplantation.1 Scalp AVFs with intracranial communication have been reported months to years after skull fracture or craniotomy.2 True spontaneous S-AVFs are difficult to differentiate from traumatic S-AVFs other than by history alone.2
Increased venous pressure has been shown to generate AVFs in rats.5 It has been suggested that S-AVFs can become enlarged by capturing subcutaneous or intracranial feeder vessels and that the consequent hemodynamic stress may induce de novo aneurysms in S-AVFs. Additionally, intracranial AVFs may alter the intracranial hemodynamics, leading to increased venous pressure in the superior sagittal sinus and the formation of communicating S-AVFs.5 Interestingly, our patient had an intracranial AVF treated with endovascular embolization 1 year prior to the formation of the S-AVF. An angiogram at the time of this embolization procedure did not demonstrate any S-AVFs. Furthermore, our patient has a history of 2 substantial falls in childhood with head trauma and loss of consciousness. Perhaps these traumas initiated a channel through the cranium where an S-AVF with intracranial communication was able to form and may have only become clinically or radiographically detectable once it enlarged due to the altered hemodynamics caused by the intracranial AVF 1 year prior.
The diagnosis of an S-AVF is confirmed with imaging studies. Doppler ultrasonography initially will help to detect that a lesion is vascular in nature. Intra-arterial digital subtraction angiography is the gold-standard imaging technique and is necessary to delineate the feeding arteries and the draining channels as well as possible communication with intracranial vasculature.1,2 There is controversy regarding the appropriate treatment of S-AVFs.2 Each S-AVF possesses unique anatomic features that dictate appropriate management. The prognosis for an S-AVF is extremely variable, and the decision to treat is based on the patient’s symptoms and risk for exsanguinating hemorrhage.2,4 Neurosurgical approaches include ligation of the feeding arteries, surgical resection, electrothrombosis, direct intralesional injection of sclerosing agents, and endovascular embolization. Endovascular intervention increasingly is utilized as a primary treatment or as a preoperative adjunct to surgery.2,4 Large S-AVFs have a high risk for recurrence after treatment with endovascular embolization alone. In cases with intracranial communication, the intracranial component is treated first.2
This case emphasizes the importance of including S-AVFs on the dermatologic differential diagnosis of a scalp nodule, especially in patients with any history of intracranial AVF. A thorough history, detailed intake of potential signs and symptoms of AVF, and palpation for bruits is recommended as part of the surgical evaluation of a scalp nodule. Imaging of scalp nodules also should be considered for patients with any history of intracranial AVF; S-AVFs should be referred to neurosurgery or interventional neuroradiology for evaluation and possible treatment.
To the Editor:
A 71-year-old man presented with a nodule on the vertex of the scalp of 1 year’s duration. The lesion had become soft and tender during the week prior to presentation. He noted that he was experiencing headaches and a buzzing sound in his head. He denied all other neurologic symptoms. The patient was given amoxicillin from a primary care physician and was referred to our institution for evaluation of a presumed inflamed cyst.
The patient’s medical history included an intracranial arteriovenous fistula (AVF) treated with endovascular embolization 1 year prior to presentation, 2 substantial falls in childhood with head trauma and loss of consciousness, essential hypertension, and an aortic aneurysm. His medications included amlodipine, lisinopril, amoxicillin, a multivitamin, and grape seed extract.
Physical examination revealed a 2-cm, pink, somewhat rubbery, subcutaneous, nonmobile nodule on the vertex of the scalp (Figure 1). The lesion was not consistent with a common pilar cyst, and an excisional biopsy was performed to exclude malignancy. Upon superficial incision, the lesion bled moderately, and the procedure was immediately discontinued. Hemostasis was obtained, and the patient was sent for ultrasonography of the lesion.
Ultrasonography demonstrated a small hypoechoic nodule measuring up to 0.5 cm containing a tangle of vessels in the subcutaneous soft tissue corresponding to the palpable abnormality. A cerebral angiogram demonstrated a dural AVF of the superior sagittal sinus with multifocal supply that connected with this scalp nodule (Figure 2). The patient was treated by interventional neuroradiology with endovascular embolization, which resulted in complete resolution of the scalp nodule.
Scalp arteriovenous fistulas (S-AVFs) are characterized by abnormal connections between supplying arteries and draining veins in the subcutaneous plane of the scalp.1,2 The veins of an S-AVF undergo progressive aneurysmal dilatation from abnormal hemodynamics.1-3 Scalp arteriovenous fistulas are rare and may present as either an innocuous-looking scalp nodule or a progressively enlarging pulsatile mass on the scalp.2-4 Associated symptoms often include headache, local pain, bruits, tinnitus, and thrill.1,3,4 Recurrent hemorrhage, scalp necrosis, congestive heart failure, epilepsy, mental retardation, and intracranial ischemia also may occur.4
Scalp AVFs may occur with or without intracranial communication.4 Spontaneous S-AVFs with intracranial communication are uncommon, and their etiology is unclear. They may form as congenital malformations or may be idiopathic. Factors increasing circulation through the S-AVF such as trauma, pregnancy, hormonal changes, and inflammation prompt the development of symptoms.4 Scalp AVFs also may be caused by trauma.3 Scalp AVFs without intracranial communication have been reported following hair transplantation.1 Scalp AVFs with intracranial communication have been reported months to years after skull fracture or craniotomy.2 True spontaneous S-AVFs are difficult to differentiate from traumatic S-AVFs other than by history alone.2
Increased venous pressure has been shown to generate AVFs in rats.5 It has been suggested that S-AVFs can become enlarged by capturing subcutaneous or intracranial feeder vessels and that the consequent hemodynamic stress may induce de novo aneurysms in S-AVFs. Additionally, intracranial AVFs may alter the intracranial hemodynamics, leading to increased venous pressure in the superior sagittal sinus and the formation of communicating S-AVFs.5 Interestingly, our patient had an intracranial AVF treated with endovascular embolization 1 year prior to the formation of the S-AVF. An angiogram at the time of this embolization procedure did not demonstrate any S-AVFs. Furthermore, our patient has a history of 2 substantial falls in childhood with head trauma and loss of consciousness. Perhaps these traumas initiated a channel through the cranium where an S-AVF with intracranial communication was able to form and may have only become clinically or radiographically detectable once it enlarged due to the altered hemodynamics caused by the intracranial AVF 1 year prior.
The diagnosis of an S-AVF is confirmed with imaging studies. Doppler ultrasonography initially will help to detect that a lesion is vascular in nature. Intra-arterial digital subtraction angiography is the gold-standard imaging technique and is necessary to delineate the feeding arteries and the draining channels as well as possible communication with intracranial vasculature.1,2 There is controversy regarding the appropriate treatment of S-AVFs.2 Each S-AVF possesses unique anatomic features that dictate appropriate management. The prognosis for an S-AVF is extremely variable, and the decision to treat is based on the patient’s symptoms and risk for exsanguinating hemorrhage.2,4 Neurosurgical approaches include ligation of the feeding arteries, surgical resection, electrothrombosis, direct intralesional injection of sclerosing agents, and endovascular embolization. Endovascular intervention increasingly is utilized as a primary treatment or as a preoperative adjunct to surgery.2,4 Large S-AVFs have a high risk for recurrence after treatment with endovascular embolization alone. In cases with intracranial communication, the intracranial component is treated first.2
This case emphasizes the importance of including S-AVFs on the dermatologic differential diagnosis of a scalp nodule, especially in patients with any history of intracranial AVF. A thorough history, detailed intake of potential signs and symptoms of AVF, and palpation for bruits is recommended as part of the surgical evaluation of a scalp nodule. Imaging of scalp nodules also should be considered for patients with any history of intracranial AVF; S-AVFs should be referred to neurosurgery or interventional neuroradiology for evaluation and possible treatment.
- Bernstein J, Podnos S, Leavitt M. Arteriovenous fistula following hair transplantation. Dermatol Surg. 2011;37:873-875.
- Kumar R, Sharma G, Sharma BS. Management of scalp arterio-venous malformation: case series and review of literature. Br J Neurosurg. 2012;26:371-377.
- Gurkanlar D, Gonul M, Solmaz I, et al. Cirsoid aneurysms of the scalp. Neurosurg Rev. 2006;29:208-212.
- Senoglu M, Yasim A, Gokce M, et al. Nontraumatic scalp arteriovenous fistula in an adult: technical report on an illustrative case. Surg Neurol. 2008;70:194-197.
- Lanzino G, Passacantilli E, Lemole G, et al. Scalp arteriovenous malformation draining into the superior sagittal sinus associated with an intracranial arteriovenous malformation: just a coincidence? case report. Neurosurgery. 2003;52:440-443.
- Bernstein J, Podnos S, Leavitt M. Arteriovenous fistula following hair transplantation. Dermatol Surg. 2011;37:873-875.
- Kumar R, Sharma G, Sharma BS. Management of scalp arterio-venous malformation: case series and review of literature. Br J Neurosurg. 2012;26:371-377.
- Gurkanlar D, Gonul M, Solmaz I, et al. Cirsoid aneurysms of the scalp. Neurosurg Rev. 2006;29:208-212.
- Senoglu M, Yasim A, Gokce M, et al. Nontraumatic scalp arteriovenous fistula in an adult: technical report on an illustrative case. Surg Neurol. 2008;70:194-197.
- Lanzino G, Passacantilli E, Lemole G, et al. Scalp arteriovenous malformation draining into the superior sagittal sinus associated with an intracranial arteriovenous malformation: just a coincidence? case report. Neurosurgery. 2003;52:440-443.
Practice Points
- Scalp arteriovenous fistulas may be traumatic or spontaneous and present as either an innocuous-looking scalp nodule or as a progressively enlarging pulsatile mass on the scalp.
- Clinical detection followed by appropriate imaging and referral to neurosurgery or interventional neuroradiology is vital to patient safety.
Oral Hairy Leukoplakia Associated With the Use of Adalimumab
To the Editor:
Oral hairy leukoplakia (OHL) is an Epstein-Barr virus (EBV)–mediated mucocutaneous disease that often involves the lingual epithelium. The lateral portions of the tongue are the most commonly affected sites. The lesions often are described as asymptomatic, white, corrugated patches or plaques that are unable to be scraped off.1 Oral hairy leukoplakia was first identified in 1984 and was considered to be associated with AIDS.2 An association between the presence of OHL and the degree of immunosuppression as well as the severity of human immunodeficiency virus (HIV) has been reported.3 Although OHL initially was considered to be pathognomonic for HIV, it has since been described in multiple other immunosuppressive conditions.4 Numerous medical conditions and combinations of immunosuppressive medications have been associated with OHL in patients who were HIV negative.5
Adalimumab is an injectable human IgG1 recombinant antibody to tumor necrosis factor α (TNF-α).6 It currently is approved by the US Food and Drug Administration for the treatment of rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, adult and pediatric Crohn disease, ulcerative colitis, noninfectious uveitis, hidradenitis suppurativa, and plaque psoriasis.7 We report a case of OHL associated with the use of adalimumab.
A 47-year-old woman initially presented with chronic plaque-type psoriasis. Her medical history was notable for bipolar disorder, migraines, hypertension, and tobacco use. The patient’s psoriasis initially was well controlled on a regimen of topical steroids and methotrexate; however, methotrexate was stopped after 2.5 years due to a mildly elevated alanine aminotransferase level, as well as an abnormal liver biopsy showing mildly active (grade 1 of 3) steatohepatitis with portal chronic inflammation, pericellular fibrosis, and portal and focal periportal fibrosis (stage 1-2 of 4). The patient and her dermatologist were uncomfortable continuing methotrexate with these findings. After baseline screening including a negative purified protein derivative skin test, adalimumab was initiated. A loading dose of 80 mg subcutaneously (SQ) was given, followed by adalimumab 40 mg SQ 1 week later and 40 mg every other week as maintenance.
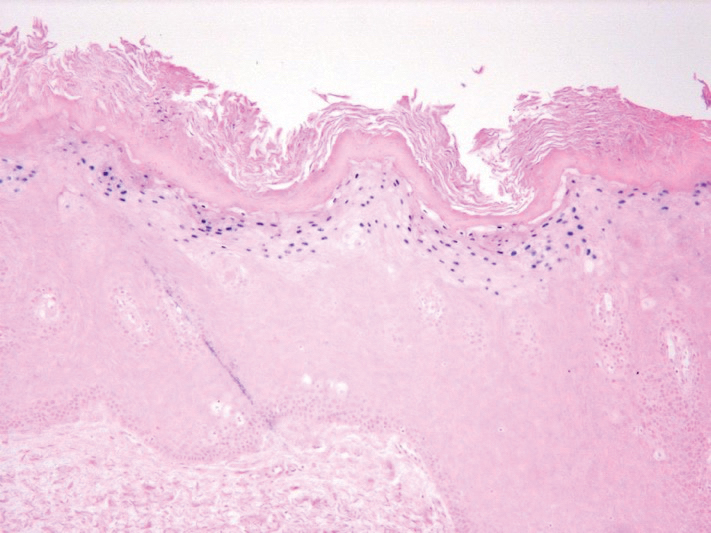
The patient’s psoriasis was well controlled with adalimumab for 22 months, but she then developed a thin white plaque on the right lateral tongue (Figure 1). An incisional biopsy of the tongue performed by an oral surgeon revealed hyperkeratosis with Candida colonization and viral cytopathic effect (Figure 2). An EBV DNA in situ hybridization stain revealed focal positivity within these cells (Figure 3), leading to a diagnosis of OHL. Laboratory evaluation demonstrated a normal complete blood cell count with differential and liver panel as well as a negative HIV test. The patient otherwise felt well and denied fevers, lymphadenopathy, and weight loss.
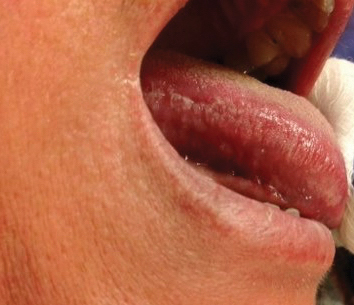
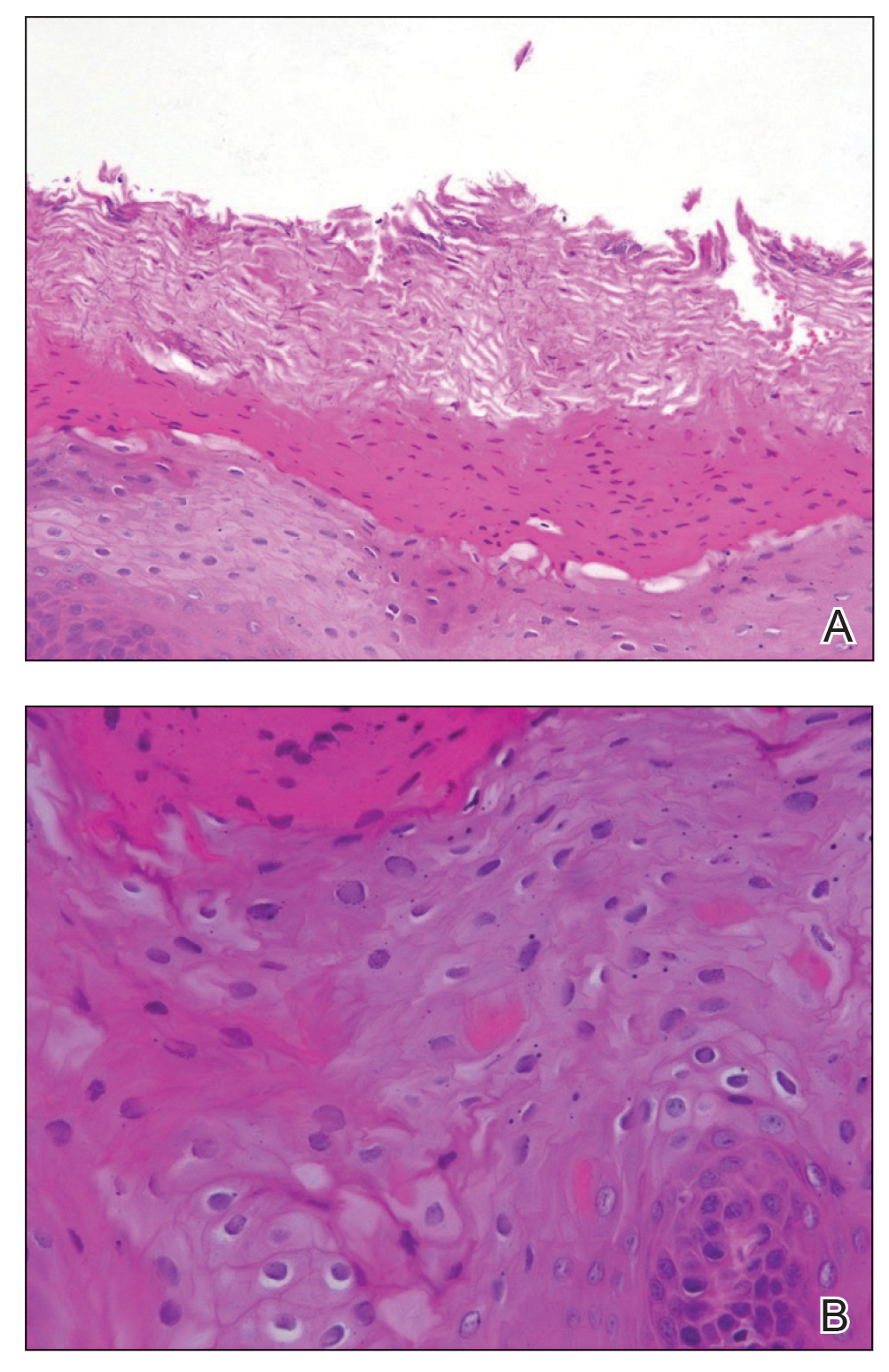
We consulted with an infectious disease and immunodeficiency specialist regarding the patient’s case. Before conducting further evaluation beyond HIV screening for immunodeficiency states, adalimumab was discontinued to see if the OHL would spontaneously resolve. Three months after discontinuation of adalimumab, the white plaque on the right lateral tongue was notably improved. The OHL continued to disappear and was completely resolved 1 year after discontinuation of adalimumab. The patient’s psoriasis had subsequently remained well controlled with diet and weight loss, smoking cessation, topical steroids, and apremilast without any recurrence of the OHL.
Oral hairy leukoplakia is associated with upregulated EBV replication and EBV-encoded proteins such as latent membrane protein 1.2 It often presents as white or gray patches on the lateral lingual margins with prominent folds and/or projections, giving a shaggy appearance. Oral hairy leukoplakia often is specific for HIV infection and rarely is associated with other immunodeficiencies.2 Prasad and Bilodeau5 performed a literature review of medical conditions and immunosuppressive medications associated with OHL in patients without HIV. Allogeneic transplant was associated with the highest incidence of OHL in HIV-negative patients (59.2% [45/76]).5 Various combinations of immunosuppressive medications (eg, prednisone, cyclosporine, azathioprine) also may be implicated in cases of HIV-negative patients with OHL. A case of OHL also has been reported with long-standing use of inhaled corticosteroids in an immunocompetent, HIV-negative patient.6 Another case was reported with long-term use of the aromatic antiepileptic lamotrigine, which resolved once stopping the medication.8 Although EBV is an oncovirus and has been associated with lymphoproliferative disorders and nasopharyngeal carcinoma, OHL is not considered to be a premalignant lesion.7 Despite the strong association between OHL and HIV, our patient was HIV negative. The only immunocompromising factor in our patient was the use of adalimumab to treat psoriasis. We did not conduct further testing for immunodeficiency states because the OHL spontaneously resolved when the adalimumab was discontinued.
PubMed and Ovid searches of articles indexed for MEDLINE using the terms adalimumab and oral hairy leukoplakia as well as TNF-alpha inhibitor and oral hairy leukoplakia with humans and English language as limitations revealed that no cases have been reported in the literature demonstrating an association between OHL and adalimumab or any other TNF-α inhibitor. However, Cetkovska et al9 reported a case of EBV hepatitis and subsequently chronic hepatitis as a complication of infliximab used for the treatment of chronic psoriasis. Because TNF-α and IFN-γ play an important role in controlling viral infections, there is an increased risk for reactivating a viral illness when depleting TNF through pharmacologic measures (ie, adalimumab, infliximab).8 Another case of EBV-associated plasmablastic lymphoma was reported after 1 year of adalimumab use in a patient with Crohn disease. The plasmablastic lymphoma resolved after 4 rounds of chemotherapy.10
The only contraindication for the use of adalimumab is a known hypersensitivity to the drug. Relative contraindications for use of adalimumab include active tuberculosis, demyelinating disease, hematologic diseases (ie, thrombocytopenia, pancytopenia), lymphoma, hepatitis C, and hepatitis B.11 The most common adverse effect of adalimumab is an injection-site reaction. Additional reported adverse effects of TNF-α inhibitors as a class are lymphoma, melanoma, nonmelanoma skin cancer, reactivation of latent tuberculosis, congestive heart failure, autoimmunity, and hematologic toxicity.11
This case demonstrates an association between adalimumab and OHL in an HIV-negative patient. Although the mechanism behind OHL and immunosuppression remains to be elucidated, this association is important to keep in mind when using adalimumab or other TNF-α inhibitors for the treatment of psoriasis or other medical conditions.
- Triantos D, Porter SR, Scully C, et al. Oral hairy leukoplakia: clinicopathologic features, pathogenesis, diagnosis, and clinical significance. Clin Infect Dis. 1997;25:1392-1396.
- Greenspan D, Greenspan JS, Conant M, et al. Oral “hairy” leucoplakia in male homosexuals: evidence of association with both papillomavirus and a herpes-group virus. Lancet. 1984;2:831-834.
- Glick M, Muzyka BC, Lurie D, et al. Oral manifestations associated with HIV-related disease as marks for immune suppression and AIDS. Oral Surg Oral Med Oral Pathol. 1994;77:344-349.
- Chambers AE, Conn B, Pemberton M, et al. Twenty-first-century oral hair leukoplakia—a non-HIV-associated entity. Oral Surg Oral Med Oral Patho Oral Radiol. 2015;119:326-332.
- Prasad JL, Bilodeau EA. Oral hairy leukoplakia in patients without HIV: presentation of 2 new cases. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:E151-E160.
- Moffat M, Jauhar S, Jones ME, et al. Oral hairy leukoplakia in an HIV-negative, immunocompetent patient. Oral Biosci Med. 2005;2:282-284.
- Greenspan JS, Greenspan D. Oral hairy leukoplakia: diagnosis and management. Oral Surg Oral Med Oral Pathol. 1989;67:396-403.
- Gordins P, Sloan P, Spickett GP, et al. Oral hairy leukoplakia in a patient on long-term anticonvulsant treatment with lamotrigine. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:E17-E23.
- Cetkovska P, Lomicova I, Mukensnabl P, et al. Anti-tumour necrosis factor treatment of severe psoriasis complicated by Epstein-Barr virus hepatitis and subsequently by chronic hepatitis. Dermatol Ther. 2015;28:369-372.
- Liu L, Charabaty A, Ozdemirli M. EBV-associated plasmablastic lymphoma in a patient with Crohn’s disease after adalimumab treatment. J Crohns Colitis. 2013;7:E118-E119.
- Humira [package insert]. North Chicago, IL: AbbVie Inc; 2018.
To the Editor:
Oral hairy leukoplakia (OHL) is an Epstein-Barr virus (EBV)–mediated mucocutaneous disease that often involves the lingual epithelium. The lateral portions of the tongue are the most commonly affected sites. The lesions often are described as asymptomatic, white, corrugated patches or plaques that are unable to be scraped off.1 Oral hairy leukoplakia was first identified in 1984 and was considered to be associated with AIDS.2 An association between the presence of OHL and the degree of immunosuppression as well as the severity of human immunodeficiency virus (HIV) has been reported.3 Although OHL initially was considered to be pathognomonic for HIV, it has since been described in multiple other immunosuppressive conditions.4 Numerous medical conditions and combinations of immunosuppressive medications have been associated with OHL in patients who were HIV negative.5
Adalimumab is an injectable human IgG1 recombinant antibody to tumor necrosis factor α (TNF-α).6 It currently is approved by the US Food and Drug Administration for the treatment of rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, adult and pediatric Crohn disease, ulcerative colitis, noninfectious uveitis, hidradenitis suppurativa, and plaque psoriasis.7 We report a case of OHL associated with the use of adalimumab.
A 47-year-old woman initially presented with chronic plaque-type psoriasis. Her medical history was notable for bipolar disorder, migraines, hypertension, and tobacco use. The patient’s psoriasis initially was well controlled on a regimen of topical steroids and methotrexate; however, methotrexate was stopped after 2.5 years due to a mildly elevated alanine aminotransferase level, as well as an abnormal liver biopsy showing mildly active (grade 1 of 3) steatohepatitis with portal chronic inflammation, pericellular fibrosis, and portal and focal periportal fibrosis (stage 1-2 of 4). The patient and her dermatologist were uncomfortable continuing methotrexate with these findings. After baseline screening including a negative purified protein derivative skin test, adalimumab was initiated. A loading dose of 80 mg subcutaneously (SQ) was given, followed by adalimumab 40 mg SQ 1 week later and 40 mg every other week as maintenance.
The patient’s psoriasis was well controlled with adalimumab for 22 months, but she then developed a thin white plaque on the right lateral tongue (Figure 1). An incisional biopsy of the tongue performed by an oral surgeon revealed hyperkeratosis with Candida colonization and viral cytopathic effect (Figure 2). An EBV DNA in situ hybridization stain revealed focal positivity within these cells (Figure 3), leading to a diagnosis of OHL. Laboratory evaluation demonstrated a normal complete blood cell count with differential and liver panel as well as a negative HIV test. The patient otherwise felt well and denied fevers, lymphadenopathy, and weight loss.
We consulted with an infectious disease and immunodeficiency specialist regarding the patient’s case. Before conducting further evaluation beyond HIV screening for immunodeficiency states, adalimumab was discontinued to see if the OHL would spontaneously resolve. Three months after discontinuation of adalimumab, the white plaque on the right lateral tongue was notably improved. The OHL continued to disappear and was completely resolved 1 year after discontinuation of adalimumab. The patient’s psoriasis had subsequently remained well controlled with diet and weight loss, smoking cessation, topical steroids, and apremilast without any recurrence of the OHL.
Oral hairy leukoplakia is associated with upregulated EBV replication and EBV-encoded proteins such as latent membrane protein 1.2 It often presents as white or gray patches on the lateral lingual margins with prominent folds and/or projections, giving a shaggy appearance. Oral hairy leukoplakia often is specific for HIV infection and rarely is associated with other immunodeficiencies.2 Prasad and Bilodeau5 performed a literature review of medical conditions and immunosuppressive medications associated with OHL in patients without HIV. Allogeneic transplant was associated with the highest incidence of OHL in HIV-negative patients (59.2% [45/76]).5 Various combinations of immunosuppressive medications (eg, prednisone, cyclosporine, azathioprine) also may be implicated in cases of HIV-negative patients with OHL. A case of OHL also has been reported with long-standing use of inhaled corticosteroids in an immunocompetent, HIV-negative patient.6 Another case was reported with long-term use of the aromatic antiepileptic lamotrigine, which resolved once stopping the medication.8 Although EBV is an oncovirus and has been associated with lymphoproliferative disorders and nasopharyngeal carcinoma, OHL is not considered to be a premalignant lesion.7 Despite the strong association between OHL and HIV, our patient was HIV negative. The only immunocompromising factor in our patient was the use of adalimumab to treat psoriasis. We did not conduct further testing for immunodeficiency states because the OHL spontaneously resolved when the adalimumab was discontinued.
PubMed and Ovid searches of articles indexed for MEDLINE using the terms adalimumab and oral hairy leukoplakia as well as TNF-alpha inhibitor and oral hairy leukoplakia with humans and English language as limitations revealed that no cases have been reported in the literature demonstrating an association between OHL and adalimumab or any other TNF-α inhibitor. However, Cetkovska et al9 reported a case of EBV hepatitis and subsequently chronic hepatitis as a complication of infliximab used for the treatment of chronic psoriasis. Because TNF-α and IFN-γ play an important role in controlling viral infections, there is an increased risk for reactivating a viral illness when depleting TNF through pharmacologic measures (ie, adalimumab, infliximab).8 Another case of EBV-associated plasmablastic lymphoma was reported after 1 year of adalimumab use in a patient with Crohn disease. The plasmablastic lymphoma resolved after 4 rounds of chemotherapy.10
The only contraindication for the use of adalimumab is a known hypersensitivity to the drug. Relative contraindications for use of adalimumab include active tuberculosis, demyelinating disease, hematologic diseases (ie, thrombocytopenia, pancytopenia), lymphoma, hepatitis C, and hepatitis B.11 The most common adverse effect of adalimumab is an injection-site reaction. Additional reported adverse effects of TNF-α inhibitors as a class are lymphoma, melanoma, nonmelanoma skin cancer, reactivation of latent tuberculosis, congestive heart failure, autoimmunity, and hematologic toxicity.11
This case demonstrates an association between adalimumab and OHL in an HIV-negative patient. Although the mechanism behind OHL and immunosuppression remains to be elucidated, this association is important to keep in mind when using adalimumab or other TNF-α inhibitors for the treatment of psoriasis or other medical conditions.
To the Editor:
Oral hairy leukoplakia (OHL) is an Epstein-Barr virus (EBV)–mediated mucocutaneous disease that often involves the lingual epithelium. The lateral portions of the tongue are the most commonly affected sites. The lesions often are described as asymptomatic, white, corrugated patches or plaques that are unable to be scraped off.1 Oral hairy leukoplakia was first identified in 1984 and was considered to be associated with AIDS.2 An association between the presence of OHL and the degree of immunosuppression as well as the severity of human immunodeficiency virus (HIV) has been reported.3 Although OHL initially was considered to be pathognomonic for HIV, it has since been described in multiple other immunosuppressive conditions.4 Numerous medical conditions and combinations of immunosuppressive medications have been associated with OHL in patients who were HIV negative.5
Adalimumab is an injectable human IgG1 recombinant antibody to tumor necrosis factor α (TNF-α).6 It currently is approved by the US Food and Drug Administration for the treatment of rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, adult and pediatric Crohn disease, ulcerative colitis, noninfectious uveitis, hidradenitis suppurativa, and plaque psoriasis.7 We report a case of OHL associated with the use of adalimumab.
A 47-year-old woman initially presented with chronic plaque-type psoriasis. Her medical history was notable for bipolar disorder, migraines, hypertension, and tobacco use. The patient’s psoriasis initially was well controlled on a regimen of topical steroids and methotrexate; however, methotrexate was stopped after 2.5 years due to a mildly elevated alanine aminotransferase level, as well as an abnormal liver biopsy showing mildly active (grade 1 of 3) steatohepatitis with portal chronic inflammation, pericellular fibrosis, and portal and focal periportal fibrosis (stage 1-2 of 4). The patient and her dermatologist were uncomfortable continuing methotrexate with these findings. After baseline screening including a negative purified protein derivative skin test, adalimumab was initiated. A loading dose of 80 mg subcutaneously (SQ) was given, followed by adalimumab 40 mg SQ 1 week later and 40 mg every other week as maintenance.
The patient’s psoriasis was well controlled with adalimumab for 22 months, but she then developed a thin white plaque on the right lateral tongue (Figure 1). An incisional biopsy of the tongue performed by an oral surgeon revealed hyperkeratosis with Candida colonization and viral cytopathic effect (Figure 2). An EBV DNA in situ hybridization stain revealed focal positivity within these cells (Figure 3), leading to a diagnosis of OHL. Laboratory evaluation demonstrated a normal complete blood cell count with differential and liver panel as well as a negative HIV test. The patient otherwise felt well and denied fevers, lymphadenopathy, and weight loss.
We consulted with an infectious disease and immunodeficiency specialist regarding the patient’s case. Before conducting further evaluation beyond HIV screening for immunodeficiency states, adalimumab was discontinued to see if the OHL would spontaneously resolve. Three months after discontinuation of adalimumab, the white plaque on the right lateral tongue was notably improved. The OHL continued to disappear and was completely resolved 1 year after discontinuation of adalimumab. The patient’s psoriasis had subsequently remained well controlled with diet and weight loss, smoking cessation, topical steroids, and apremilast without any recurrence of the OHL.
Oral hairy leukoplakia is associated with upregulated EBV replication and EBV-encoded proteins such as latent membrane protein 1.2 It often presents as white or gray patches on the lateral lingual margins with prominent folds and/or projections, giving a shaggy appearance. Oral hairy leukoplakia often is specific for HIV infection and rarely is associated with other immunodeficiencies.2 Prasad and Bilodeau5 performed a literature review of medical conditions and immunosuppressive medications associated with OHL in patients without HIV. Allogeneic transplant was associated with the highest incidence of OHL in HIV-negative patients (59.2% [45/76]).5 Various combinations of immunosuppressive medications (eg, prednisone, cyclosporine, azathioprine) also may be implicated in cases of HIV-negative patients with OHL. A case of OHL also has been reported with long-standing use of inhaled corticosteroids in an immunocompetent, HIV-negative patient.6 Another case was reported with long-term use of the aromatic antiepileptic lamotrigine, which resolved once stopping the medication.8 Although EBV is an oncovirus and has been associated with lymphoproliferative disorders and nasopharyngeal carcinoma, OHL is not considered to be a premalignant lesion.7 Despite the strong association between OHL and HIV, our patient was HIV negative. The only immunocompromising factor in our patient was the use of adalimumab to treat psoriasis. We did not conduct further testing for immunodeficiency states because the OHL spontaneously resolved when the adalimumab was discontinued.
PubMed and Ovid searches of articles indexed for MEDLINE using the terms adalimumab and oral hairy leukoplakia as well as TNF-alpha inhibitor and oral hairy leukoplakia with humans and English language as limitations revealed that no cases have been reported in the literature demonstrating an association between OHL and adalimumab or any other TNF-α inhibitor. However, Cetkovska et al9 reported a case of EBV hepatitis and subsequently chronic hepatitis as a complication of infliximab used for the treatment of chronic psoriasis. Because TNF-α and IFN-γ play an important role in controlling viral infections, there is an increased risk for reactivating a viral illness when depleting TNF through pharmacologic measures (ie, adalimumab, infliximab).8 Another case of EBV-associated plasmablastic lymphoma was reported after 1 year of adalimumab use in a patient with Crohn disease. The plasmablastic lymphoma resolved after 4 rounds of chemotherapy.10
The only contraindication for the use of adalimumab is a known hypersensitivity to the drug. Relative contraindications for use of adalimumab include active tuberculosis, demyelinating disease, hematologic diseases (ie, thrombocytopenia, pancytopenia), lymphoma, hepatitis C, and hepatitis B.11 The most common adverse effect of adalimumab is an injection-site reaction. Additional reported adverse effects of TNF-α inhibitors as a class are lymphoma, melanoma, nonmelanoma skin cancer, reactivation of latent tuberculosis, congestive heart failure, autoimmunity, and hematologic toxicity.11
This case demonstrates an association between adalimumab and OHL in an HIV-negative patient. Although the mechanism behind OHL and immunosuppression remains to be elucidated, this association is important to keep in mind when using adalimumab or other TNF-α inhibitors for the treatment of psoriasis or other medical conditions.
- Triantos D, Porter SR, Scully C, et al. Oral hairy leukoplakia: clinicopathologic features, pathogenesis, diagnosis, and clinical significance. Clin Infect Dis. 1997;25:1392-1396.
- Greenspan D, Greenspan JS, Conant M, et al. Oral “hairy” leucoplakia in male homosexuals: evidence of association with both papillomavirus and a herpes-group virus. Lancet. 1984;2:831-834.
- Glick M, Muzyka BC, Lurie D, et al. Oral manifestations associated with HIV-related disease as marks for immune suppression and AIDS. Oral Surg Oral Med Oral Pathol. 1994;77:344-349.
- Chambers AE, Conn B, Pemberton M, et al. Twenty-first-century oral hair leukoplakia—a non-HIV-associated entity. Oral Surg Oral Med Oral Patho Oral Radiol. 2015;119:326-332.
- Prasad JL, Bilodeau EA. Oral hairy leukoplakia in patients without HIV: presentation of 2 new cases. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:E151-E160.
- Moffat M, Jauhar S, Jones ME, et al. Oral hairy leukoplakia in an HIV-negative, immunocompetent patient. Oral Biosci Med. 2005;2:282-284.
- Greenspan JS, Greenspan D. Oral hairy leukoplakia: diagnosis and management. Oral Surg Oral Med Oral Pathol. 1989;67:396-403.
- Gordins P, Sloan P, Spickett GP, et al. Oral hairy leukoplakia in a patient on long-term anticonvulsant treatment with lamotrigine. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:E17-E23.
- Cetkovska P, Lomicova I, Mukensnabl P, et al. Anti-tumour necrosis factor treatment of severe psoriasis complicated by Epstein-Barr virus hepatitis and subsequently by chronic hepatitis. Dermatol Ther. 2015;28:369-372.
- Liu L, Charabaty A, Ozdemirli M. EBV-associated plasmablastic lymphoma in a patient with Crohn’s disease after adalimumab treatment. J Crohns Colitis. 2013;7:E118-E119.
- Humira [package insert]. North Chicago, IL: AbbVie Inc; 2018.
- Triantos D, Porter SR, Scully C, et al. Oral hairy leukoplakia: clinicopathologic features, pathogenesis, diagnosis, and clinical significance. Clin Infect Dis. 1997;25:1392-1396.
- Greenspan D, Greenspan JS, Conant M, et al. Oral “hairy” leucoplakia in male homosexuals: evidence of association with both papillomavirus and a herpes-group virus. Lancet. 1984;2:831-834.
- Glick M, Muzyka BC, Lurie D, et al. Oral manifestations associated with HIV-related disease as marks for immune suppression and AIDS. Oral Surg Oral Med Oral Pathol. 1994;77:344-349.
- Chambers AE, Conn B, Pemberton M, et al. Twenty-first-century oral hair leukoplakia—a non-HIV-associated entity. Oral Surg Oral Med Oral Patho Oral Radiol. 2015;119:326-332.
- Prasad JL, Bilodeau EA. Oral hairy leukoplakia in patients without HIV: presentation of 2 new cases. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:E151-E160.
- Moffat M, Jauhar S, Jones ME, et al. Oral hairy leukoplakia in an HIV-negative, immunocompetent patient. Oral Biosci Med. 2005;2:282-284.
- Greenspan JS, Greenspan D. Oral hairy leukoplakia: diagnosis and management. Oral Surg Oral Med Oral Pathol. 1989;67:396-403.
- Gordins P, Sloan P, Spickett GP, et al. Oral hairy leukoplakia in a patient on long-term anticonvulsant treatment with lamotrigine. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:E17-E23.
- Cetkovska P, Lomicova I, Mukensnabl P, et al. Anti-tumour necrosis factor treatment of severe psoriasis complicated by Epstein-Barr virus hepatitis and subsequently by chronic hepatitis. Dermatol Ther. 2015;28:369-372.
- Liu L, Charabaty A, Ozdemirli M. EBV-associated plasmablastic lymphoma in a patient with Crohn’s disease after adalimumab treatment. J Crohns Colitis. 2013;7:E118-E119.
- Humira [package insert]. North Chicago, IL: AbbVie Inc; 2018.
Practice Points
- Workup for new-onset oral hairy leukoplakia should include a comprehensive medication history.
- Oral hairy leukoplakia is an uncommon side effect of adalimumab.
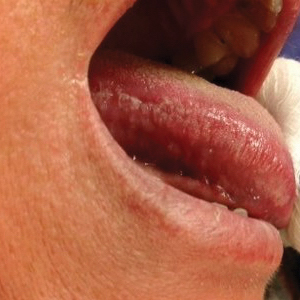
Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis Overlap Syndrome in a Patient With Relapsing Polychondritis
To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.

Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Damiani JM, Levine HL. Relapsing polychondritis—report of ten cases. Laryngoscope. 1979;89:929-46.
- Puéchal X, Terrier B, Mouthon L, et al. Relapsing polychondritis. Joint Bone Spine. 2014;81:118-24.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis. Autoimmun Rev. 2014;13:90-95.
- Harr T, French L. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;16;5:39.
To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.
Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.
To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.
Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Damiani JM, Levine HL. Relapsing polychondritis—report of ten cases. Laryngoscope. 1979;89:929-46.
- Puéchal X, Terrier B, Mouthon L, et al. Relapsing polychondritis. Joint Bone Spine. 2014;81:118-24.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis. Autoimmun Rev. 2014;13:90-95.
- Harr T, French L. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;16;5:39.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Damiani JM, Levine HL. Relapsing polychondritis—report of ten cases. Laryngoscope. 1979;89:929-46.
- Puéchal X, Terrier B, Mouthon L, et al. Relapsing polychondritis. Joint Bone Spine. 2014;81:118-24.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis. Autoimmun Rev. 2014;13:90-95.
- Harr T, French L. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;16;5:39.
Practice Points
- The clinical presentation of relapsing polychondritis (RP) may demonstrate cutaneous manifestations other than the typical inflammation of cartilage-rich structures.
- Approximately 30% of patients with RP will have another autoimmune disease.
Orbital Granuloma Formation Following Autoinjection of Paraffin Oil: Management Considerations
To the Editor:
Injectable fillers are an increasingly common means of achieving minimally invasive facial rejuvenation. In the hands of well-trained practitioners, these compounds typically are well tolerated, effective, and have a strong safety profile1; however, there have been reports of complications, including vision loss,2 orbital infarction,3 persistent inflammatory nodules,4 and infection.4,5 Paraffin, a derivative of mineral oil, currently is used in cosmetic products and medical ointments.6 In the early 1900s, it often was injected into the body for various medical procedures, such as to create prosthetic testicles, to treat bladder incontinence, and eventually to correct facial contour defects.7,8 Due to adverse effects, injection of paraffin oil was discontinued in the Western medical community around the time of World War I.7 Unfortunately, some patients continue to self-inject paraffin oil for cosmetic purposes today. We present a case of foreign-body granuloma formation mimicking periorbital cellulitis following self-injection of paraffin oil. Our patient developed serious periorbital sequelae that required surgical intervention to restore normal anatomic function.
A 60-year-old woman who was otherwise healthy presented to the emergency department with facial swelling and a rash of 2 weeks’ duration. She reported that she had purchased what she believed was a cosmetic product at a local flea market 2 weeks prior to presentation. Her purchase included needles and a syringe with verbal instructions for injection into the face. She was told the product was used to treat wrinkles and referred to the injectable material as “oil” when providing her history. She reported that she had injected the material into the bilateral lower eyelids, left lateral lip, and left lateral chin. Three days later, she developed tingling and itching with swelling and redness at the injection sites. The patient was evaluated by the emergency department team and was prescribed a 10-day course of clindamycin empirically for suspected facial cellulitis.
The patient returned to the emergency department 12 days later upon completion of the antibiotic course with worsening edema and erythema. Examination revealed indurated, erythematous, and edematous warm plaques on the face that were concentrated around the prior injection sites with substantial periorbital erythema and edema (Figure 1). A consultation with oculoplastic surgery was obtained. Mechanical ptosis of the right eyelid was noted. Visual acuity was 20/30 in both eyes with habitual correction. Intraocular pressure was soft to palpation, and the pupils were round and reactive with no evidence of a relative afferent pupillary defect. Extraocular motility was intact bilaterally. Examination of the conjunctiva and sclera revealed bilateral conjunctival injection with chemosis of the right eye. The remainder of the anterior and posterior segment examination was within normal limits bilaterally.
Computed tomography of the face showed extensive facial and periorbital swelling without abscess. A dermatology consultation was obtained. Two 4-mm punch biopsies were obtained from the left lower face and were sent for hematoxylin and eosin stain and tissue culture (bacterial, fungal, and acid-fast bacillus). Given the possibility of facial and periorbital cellulitis, empiric intravenous antibiotic therapy was initiated.
The tissue culture revealed normal skin flora. The biopsy results indicated a foreign-body reaction consistent with paraffin granuloma (Figures 2 and 3). Fite-Faraco, Grocott-Gomori methenamine-silver, and periodic acid–Schiff stains were all negative for infection. A diagnosis of foreign-body granuloma was established. Oral minocycline at a dosage of 100 mg twice daily was started, and the patient was discharged.


After 4 weeks of minocycline therapy, the patient showed no improvement and returned to the emergency department with worsening symptoms. She was readmitted and started on intravenous prednisone (1.5 mg/kg/d). Over the ensuing 5 days, the edema, erythema, conjunctival injection, and chemosis demonstrated notable improvement. She was subsequently discharged on an oral prednisone taper. Unfortunately, she did not respond to a trial of intralesional steroid injections to an area of granuloma formation on the left chin performed in the hospital before she was discharged.
In the ensuing months, she began to develop cicatricial ectropion of the right lower eyelid and mechanical ptosis of the right upper eyelid. Ten months after initial self-injection, staged surgical excision was initiated by an oculoplastic surgeon (I.V.) with the goal of debulking the periorbital region to correct the ectropion and mechanical ptosis. A transconjunctival approach was used to carefully excise the material while still maintaining the architecture of the lower eyelid. The ectropion was surgically corrected concurrently.
One month after excision, serial injections of 5-fluorouracil (5-FU) and triamcinolone acetonide 40 mg/mL were administered to the right lower eyelid and anterior orbit for 3 months. Fifteen weeks after the first surgery, a second surgery was performed to address residual medial right lower eyelid induration, right upper eyelid mechanical ptosis, and left orbital inflammation. During the postoperative period, serial monthly injections of 5-FU and triamcinolone acetonide were again performed beginning at the first postoperative month.
The surgical excisions resulted in notable improvement 3 months following excision (Figure 4). The patient noted improved ocular surface comfort with decreased foreign-body sensation and tearing. She also was pleased with the improved cosmetic outcome.
Crude substances such as paraffin, petroleum jelly, and lanolin were used for aesthetic purposes in the late 19th and early 20th centuries, initially with satisfying results; however, long-term adverse effects such as hardening of the skin, swelling, granuloma formation, ulceration, infections, and abscesses have discouraged its use by medical professionals today.5 Since paraffin is resistant to degradation and absorption, foreign-body reactions may occur upon injection. These reactions are characterized by replacement of normal subcutaneous tissue by cystic spaces of paraffin oil and/or calcification, similar to the appearance of Swiss cheese on histology and surrounded by various inflammatory cells and fibrous tissue.9,10
Clinically, there is an acute inflammatory phase followed by a latent phase of chronic granulomatous inflammation that can last for years.10 Our patient presented during the acute phase, with erythematous and edematous warm plaques around the eye mimicking an orbital infection.
The treatment of choice for paraffin granuloma is complete surgical excision to prevent recurrence.6,9 However, intralesional corticosteroids are preferred in the facial area, especially if complete removal is not possible.10 Intralesional corticosteroid injections inhibit fibroblast and macrophage activity as well as the deposition of collagen, leading to reduced pain and swelling in most cases.11 Additionally, combining antimitotic agents such as 5-FU with a corticosteroid might reduce the risk for cortisone skin atrophy.12 In our case, the patient did not respond to combined 5-FU with intralesional steroids and required oral corticosteroids while awaiting serial excisions.
Our case highlights several important points in the management of paraffin granuloma. First, the clinician must perform a thorough patient history, as surreptitious use of non–medical-grade fillers is more common than one might think.13 Second, the initial presentation of these patients can mimic an infectious process. Careful history, testing, and observation can aid in making the appropriate diagnosis. Finally, treatment of these patients is complex. The mainstays of therapy are systemic anti-inflammatory medications, time, and supportive care. In some cases, surgery may be required. When processes such as paraffin granulomas involve the periorbital region, particular care is required to avoid cicatricial lagophthalmos, ectropion, or retraction. Thoughtful surgical manipulation is required to avoid these complications, which indeed may occur even with the most appropriate interventions.
- Duker D, Erdmann R, Hartmann V, et al. The impact of adverse reactions to injectable filler substances on quality of life: results from the Berlin Injectable Filler Safety (IFS)—study. J Eur Acad Dermatol Venereol. 2016;30:1013-1020.
- Prado G, Rodriguez-Feliz J. Ocular pain and impending blindness during facial cosmetic injections: is your office prepared? [published online December 28, 2016]. Aesthetic Plast Surg. 2017;41:199-203.
- Roberts SA, Arthurs BP. Severe visual loss and orbital infarction following periorbital aesthetic poly-(L)-lactic acid (PLLA) injection. Ophthalmic Plast Reconstr Surg. 2012;28:E68-E70.
- Cassuto D, Pignatti M, Pacchioni L, et al. Management of complications caused by permanent fillers in the face: a treatment algorithm. Plast Reconstr Surg. 2016;138:215E-227E.
- Haneke E. Adverse effects of fillers and their histopathology. Facial Plast Surg. 2014;30:599-614.
- Friedrich RE, Zustin J. Paraffinoma of lips and oral mucosa: case report and brief review of literature. GMS Interdiscip Plast Reconstr Surg DGPW. 2014;3:Doc05.
- Matton G, Anseeuw A, De Keyser F. The history of injectable biomaterials and the biology of collagen. Aesthetic Plast Surg. 1985;9:133-140.
- Glicenstein J. Les premiers fillers, Vaseline et paraffine. du miracle a la catastrope. Ann Chir Plast Esthet. 2007;52:157-161.
- Cohen JL, Keoleian CM, Krull EA. Penile paraffinoma: self-injection with mineral oil. J Am Acad Dermatol 2002;47:S251-S253.
- Legaspi-Vicerra ME, Field LM. Paraffin granulomata, “witch’s chin,” and nasal deformities excision and reconstruction with reduction chinplasty and open rhinotomy resection. J Clin Aesthet Dermatol 2010;3:54-58.
- Carlos-Fabuel L, Marzal-Gamarra C, Marti-Alamo S, et al. Foreign body granulomatous reactions to cosmetic fillers. J Clin Exp Dent. 2012;4:E244-E247.
- Lemperle G, Gauthier-Hazan N. Foreign body granulomas after all injectable dermal fillers: part 2. treatment options. Plast Reconstr Surg. 2009;123:1864-1873.
- Seok J, Hong JY, Park KY, et al. Delayed immunologic complications due to injectable fillers by unlicensed practitioners: our experiences and a review of the literature. Dermatol Ther. 2016;29:41-44.
To the Editor:
Injectable fillers are an increasingly common means of achieving minimally invasive facial rejuvenation. In the hands of well-trained practitioners, these compounds typically are well tolerated, effective, and have a strong safety profile1; however, there have been reports of complications, including vision loss,2 orbital infarction,3 persistent inflammatory nodules,4 and infection.4,5 Paraffin, a derivative of mineral oil, currently is used in cosmetic products and medical ointments.6 In the early 1900s, it often was injected into the body for various medical procedures, such as to create prosthetic testicles, to treat bladder incontinence, and eventually to correct facial contour defects.7,8 Due to adverse effects, injection of paraffin oil was discontinued in the Western medical community around the time of World War I.7 Unfortunately, some patients continue to self-inject paraffin oil for cosmetic purposes today. We present a case of foreign-body granuloma formation mimicking periorbital cellulitis following self-injection of paraffin oil. Our patient developed serious periorbital sequelae that required surgical intervention to restore normal anatomic function.
A 60-year-old woman who was otherwise healthy presented to the emergency department with facial swelling and a rash of 2 weeks’ duration. She reported that she had purchased what she believed was a cosmetic product at a local flea market 2 weeks prior to presentation. Her purchase included needles and a syringe with verbal instructions for injection into the face. She was told the product was used to treat wrinkles and referred to the injectable material as “oil” when providing her history. She reported that she had injected the material into the bilateral lower eyelids, left lateral lip, and left lateral chin. Three days later, she developed tingling and itching with swelling and redness at the injection sites. The patient was evaluated by the emergency department team and was prescribed a 10-day course of clindamycin empirically for suspected facial cellulitis.
The patient returned to the emergency department 12 days later upon completion of the antibiotic course with worsening edema and erythema. Examination revealed indurated, erythematous, and edematous warm plaques on the face that were concentrated around the prior injection sites with substantial periorbital erythema and edema (Figure 1). A consultation with oculoplastic surgery was obtained. Mechanical ptosis of the right eyelid was noted. Visual acuity was 20/30 in both eyes with habitual correction. Intraocular pressure was soft to palpation, and the pupils were round and reactive with no evidence of a relative afferent pupillary defect. Extraocular motility was intact bilaterally. Examination of the conjunctiva and sclera revealed bilateral conjunctival injection with chemosis of the right eye. The remainder of the anterior and posterior segment examination was within normal limits bilaterally.
Computed tomography of the face showed extensive facial and periorbital swelling without abscess. A dermatology consultation was obtained. Two 4-mm punch biopsies were obtained from the left lower face and were sent for hematoxylin and eosin stain and tissue culture (bacterial, fungal, and acid-fast bacillus). Given the possibility of facial and periorbital cellulitis, empiric intravenous antibiotic therapy was initiated.
The tissue culture revealed normal skin flora. The biopsy results indicated a foreign-body reaction consistent with paraffin granuloma (Figures 2 and 3). Fite-Faraco, Grocott-Gomori methenamine-silver, and periodic acid–Schiff stains were all negative for infection. A diagnosis of foreign-body granuloma was established. Oral minocycline at a dosage of 100 mg twice daily was started, and the patient was discharged.
After 4 weeks of minocycline therapy, the patient showed no improvement and returned to the emergency department with worsening symptoms. She was readmitted and started on intravenous prednisone (1.5 mg/kg/d). Over the ensuing 5 days, the edema, erythema, conjunctival injection, and chemosis demonstrated notable improvement. She was subsequently discharged on an oral prednisone taper. Unfortunately, she did not respond to a trial of intralesional steroid injections to an area of granuloma formation on the left chin performed in the hospital before she was discharged.
In the ensuing months, she began to develop cicatricial ectropion of the right lower eyelid and mechanical ptosis of the right upper eyelid. Ten months after initial self-injection, staged surgical excision was initiated by an oculoplastic surgeon (I.V.) with the goal of debulking the periorbital region to correct the ectropion and mechanical ptosis. A transconjunctival approach was used to carefully excise the material while still maintaining the architecture of the lower eyelid. The ectropion was surgically corrected concurrently.
One month after excision, serial injections of 5-fluorouracil (5-FU) and triamcinolone acetonide 40 mg/mL were administered to the right lower eyelid and anterior orbit for 3 months. Fifteen weeks after the first surgery, a second surgery was performed to address residual medial right lower eyelid induration, right upper eyelid mechanical ptosis, and left orbital inflammation. During the postoperative period, serial monthly injections of 5-FU and triamcinolone acetonide were again performed beginning at the first postoperative month.
The surgical excisions resulted in notable improvement 3 months following excision (Figure 4). The patient noted improved ocular surface comfort with decreased foreign-body sensation and tearing. She also was pleased with the improved cosmetic outcome.
Crude substances such as paraffin, petroleum jelly, and lanolin were used for aesthetic purposes in the late 19th and early 20th centuries, initially with satisfying results; however, long-term adverse effects such as hardening of the skin, swelling, granuloma formation, ulceration, infections, and abscesses have discouraged its use by medical professionals today.5 Since paraffin is resistant to degradation and absorption, foreign-body reactions may occur upon injection. These reactions are characterized by replacement of normal subcutaneous tissue by cystic spaces of paraffin oil and/or calcification, similar to the appearance of Swiss cheese on histology and surrounded by various inflammatory cells and fibrous tissue.9,10
Clinically, there is an acute inflammatory phase followed by a latent phase of chronic granulomatous inflammation that can last for years.10 Our patient presented during the acute phase, with erythematous and edematous warm plaques around the eye mimicking an orbital infection.
The treatment of choice for paraffin granuloma is complete surgical excision to prevent recurrence.6,9 However, intralesional corticosteroids are preferred in the facial area, especially if complete removal is not possible.10 Intralesional corticosteroid injections inhibit fibroblast and macrophage activity as well as the deposition of collagen, leading to reduced pain and swelling in most cases.11 Additionally, combining antimitotic agents such as 5-FU with a corticosteroid might reduce the risk for cortisone skin atrophy.12 In our case, the patient did not respond to combined 5-FU with intralesional steroids and required oral corticosteroids while awaiting serial excisions.
Our case highlights several important points in the management of paraffin granuloma. First, the clinician must perform a thorough patient history, as surreptitious use of non–medical-grade fillers is more common than one might think.13 Second, the initial presentation of these patients can mimic an infectious process. Careful history, testing, and observation can aid in making the appropriate diagnosis. Finally, treatment of these patients is complex. The mainstays of therapy are systemic anti-inflammatory medications, time, and supportive care. In some cases, surgery may be required. When processes such as paraffin granulomas involve the periorbital region, particular care is required to avoid cicatricial lagophthalmos, ectropion, or retraction. Thoughtful surgical manipulation is required to avoid these complications, which indeed may occur even with the most appropriate interventions.
To the Editor:
Injectable fillers are an increasingly common means of achieving minimally invasive facial rejuvenation. In the hands of well-trained practitioners, these compounds typically are well tolerated, effective, and have a strong safety profile1; however, there have been reports of complications, including vision loss,2 orbital infarction,3 persistent inflammatory nodules,4 and infection.4,5 Paraffin, a derivative of mineral oil, currently is used in cosmetic products and medical ointments.6 In the early 1900s, it often was injected into the body for various medical procedures, such as to create prosthetic testicles, to treat bladder incontinence, and eventually to correct facial contour defects.7,8 Due to adverse effects, injection of paraffin oil was discontinued in the Western medical community around the time of World War I.7 Unfortunately, some patients continue to self-inject paraffin oil for cosmetic purposes today. We present a case of foreign-body granuloma formation mimicking periorbital cellulitis following self-injection of paraffin oil. Our patient developed serious periorbital sequelae that required surgical intervention to restore normal anatomic function.
A 60-year-old woman who was otherwise healthy presented to the emergency department with facial swelling and a rash of 2 weeks’ duration. She reported that she had purchased what she believed was a cosmetic product at a local flea market 2 weeks prior to presentation. Her purchase included needles and a syringe with verbal instructions for injection into the face. She was told the product was used to treat wrinkles and referred to the injectable material as “oil” when providing her history. She reported that she had injected the material into the bilateral lower eyelids, left lateral lip, and left lateral chin. Three days later, she developed tingling and itching with swelling and redness at the injection sites. The patient was evaluated by the emergency department team and was prescribed a 10-day course of clindamycin empirically for suspected facial cellulitis.
The patient returned to the emergency department 12 days later upon completion of the antibiotic course with worsening edema and erythema. Examination revealed indurated, erythematous, and edematous warm plaques on the face that were concentrated around the prior injection sites with substantial periorbital erythema and edema (Figure 1). A consultation with oculoplastic surgery was obtained. Mechanical ptosis of the right eyelid was noted. Visual acuity was 20/30 in both eyes with habitual correction. Intraocular pressure was soft to palpation, and the pupils were round and reactive with no evidence of a relative afferent pupillary defect. Extraocular motility was intact bilaterally. Examination of the conjunctiva and sclera revealed bilateral conjunctival injection with chemosis of the right eye. The remainder of the anterior and posterior segment examination was within normal limits bilaterally.
Computed tomography of the face showed extensive facial and periorbital swelling without abscess. A dermatology consultation was obtained. Two 4-mm punch biopsies were obtained from the left lower face and were sent for hematoxylin and eosin stain and tissue culture (bacterial, fungal, and acid-fast bacillus). Given the possibility of facial and periorbital cellulitis, empiric intravenous antibiotic therapy was initiated.
The tissue culture revealed normal skin flora. The biopsy results indicated a foreign-body reaction consistent with paraffin granuloma (Figures 2 and 3). Fite-Faraco, Grocott-Gomori methenamine-silver, and periodic acid–Schiff stains were all negative for infection. A diagnosis of foreign-body granuloma was established. Oral minocycline at a dosage of 100 mg twice daily was started, and the patient was discharged.
After 4 weeks of minocycline therapy, the patient showed no improvement and returned to the emergency department with worsening symptoms. She was readmitted and started on intravenous prednisone (1.5 mg/kg/d). Over the ensuing 5 days, the edema, erythema, conjunctival injection, and chemosis demonstrated notable improvement. She was subsequently discharged on an oral prednisone taper. Unfortunately, she did not respond to a trial of intralesional steroid injections to an area of granuloma formation on the left chin performed in the hospital before she was discharged.
In the ensuing months, she began to develop cicatricial ectropion of the right lower eyelid and mechanical ptosis of the right upper eyelid. Ten months after initial self-injection, staged surgical excision was initiated by an oculoplastic surgeon (I.V.) with the goal of debulking the periorbital region to correct the ectropion and mechanical ptosis. A transconjunctival approach was used to carefully excise the material while still maintaining the architecture of the lower eyelid. The ectropion was surgically corrected concurrently.
One month after excision, serial injections of 5-fluorouracil (5-FU) and triamcinolone acetonide 40 mg/mL were administered to the right lower eyelid and anterior orbit for 3 months. Fifteen weeks after the first surgery, a second surgery was performed to address residual medial right lower eyelid induration, right upper eyelid mechanical ptosis, and left orbital inflammation. During the postoperative period, serial monthly injections of 5-FU and triamcinolone acetonide were again performed beginning at the first postoperative month.
The surgical excisions resulted in notable improvement 3 months following excision (Figure 4). The patient noted improved ocular surface comfort with decreased foreign-body sensation and tearing. She also was pleased with the improved cosmetic outcome.
Crude substances such as paraffin, petroleum jelly, and lanolin were used for aesthetic purposes in the late 19th and early 20th centuries, initially with satisfying results; however, long-term adverse effects such as hardening of the skin, swelling, granuloma formation, ulceration, infections, and abscesses have discouraged its use by medical professionals today.5 Since paraffin is resistant to degradation and absorption, foreign-body reactions may occur upon injection. These reactions are characterized by replacement of normal subcutaneous tissue by cystic spaces of paraffin oil and/or calcification, similar to the appearance of Swiss cheese on histology and surrounded by various inflammatory cells and fibrous tissue.9,10
Clinically, there is an acute inflammatory phase followed by a latent phase of chronic granulomatous inflammation that can last for years.10 Our patient presented during the acute phase, with erythematous and edematous warm plaques around the eye mimicking an orbital infection.
The treatment of choice for paraffin granuloma is complete surgical excision to prevent recurrence.6,9 However, intralesional corticosteroids are preferred in the facial area, especially if complete removal is not possible.10 Intralesional corticosteroid injections inhibit fibroblast and macrophage activity as well as the deposition of collagen, leading to reduced pain and swelling in most cases.11 Additionally, combining antimitotic agents such as 5-FU with a corticosteroid might reduce the risk for cortisone skin atrophy.12 In our case, the patient did not respond to combined 5-FU with intralesional steroids and required oral corticosteroids while awaiting serial excisions.
Our case highlights several important points in the management of paraffin granuloma. First, the clinician must perform a thorough patient history, as surreptitious use of non–medical-grade fillers is more common than one might think.13 Second, the initial presentation of these patients can mimic an infectious process. Careful history, testing, and observation can aid in making the appropriate diagnosis. Finally, treatment of these patients is complex. The mainstays of therapy are systemic anti-inflammatory medications, time, and supportive care. In some cases, surgery may be required. When processes such as paraffin granulomas involve the periorbital region, particular care is required to avoid cicatricial lagophthalmos, ectropion, or retraction. Thoughtful surgical manipulation is required to avoid these complications, which indeed may occur even with the most appropriate interventions.
- Duker D, Erdmann R, Hartmann V, et al. The impact of adverse reactions to injectable filler substances on quality of life: results from the Berlin Injectable Filler Safety (IFS)—study. J Eur Acad Dermatol Venereol. 2016;30:1013-1020.
- Prado G, Rodriguez-Feliz J. Ocular pain and impending blindness during facial cosmetic injections: is your office prepared? [published online December 28, 2016]. Aesthetic Plast Surg. 2017;41:199-203.
- Roberts SA, Arthurs BP. Severe visual loss and orbital infarction following periorbital aesthetic poly-(L)-lactic acid (PLLA) injection. Ophthalmic Plast Reconstr Surg. 2012;28:E68-E70.
- Cassuto D, Pignatti M, Pacchioni L, et al. Management of complications caused by permanent fillers in the face: a treatment algorithm. Plast Reconstr Surg. 2016;138:215E-227E.
- Haneke E. Adverse effects of fillers and their histopathology. Facial Plast Surg. 2014;30:599-614.
- Friedrich RE, Zustin J. Paraffinoma of lips and oral mucosa: case report and brief review of literature. GMS Interdiscip Plast Reconstr Surg DGPW. 2014;3:Doc05.
- Matton G, Anseeuw A, De Keyser F. The history of injectable biomaterials and the biology of collagen. Aesthetic Plast Surg. 1985;9:133-140.
- Glicenstein J. Les premiers fillers, Vaseline et paraffine. du miracle a la catastrope. Ann Chir Plast Esthet. 2007;52:157-161.
- Cohen JL, Keoleian CM, Krull EA. Penile paraffinoma: self-injection with mineral oil. J Am Acad Dermatol 2002;47:S251-S253.
- Legaspi-Vicerra ME, Field LM. Paraffin granulomata, “witch’s chin,” and nasal deformities excision and reconstruction with reduction chinplasty and open rhinotomy resection. J Clin Aesthet Dermatol 2010;3:54-58.
- Carlos-Fabuel L, Marzal-Gamarra C, Marti-Alamo S, et al. Foreign body granulomatous reactions to cosmetic fillers. J Clin Exp Dent. 2012;4:E244-E247.
- Lemperle G, Gauthier-Hazan N. Foreign body granulomas after all injectable dermal fillers: part 2. treatment options. Plast Reconstr Surg. 2009;123:1864-1873.
- Seok J, Hong JY, Park KY, et al. Delayed immunologic complications due to injectable fillers by unlicensed practitioners: our experiences and a review of the literature. Dermatol Ther. 2016;29:41-44.
- Duker D, Erdmann R, Hartmann V, et al. The impact of adverse reactions to injectable filler substances on quality of life: results from the Berlin Injectable Filler Safety (IFS)—study. J Eur Acad Dermatol Venereol. 2016;30:1013-1020.
- Prado G, Rodriguez-Feliz J. Ocular pain and impending blindness during facial cosmetic injections: is your office prepared? [published online December 28, 2016]. Aesthetic Plast Surg. 2017;41:199-203.
- Roberts SA, Arthurs BP. Severe visual loss and orbital infarction following periorbital aesthetic poly-(L)-lactic acid (PLLA) injection. Ophthalmic Plast Reconstr Surg. 2012;28:E68-E70.
- Cassuto D, Pignatti M, Pacchioni L, et al. Management of complications caused by permanent fillers in the face: a treatment algorithm. Plast Reconstr Surg. 2016;138:215E-227E.
- Haneke E. Adverse effects of fillers and their histopathology. Facial Plast Surg. 2014;30:599-614.
- Friedrich RE, Zustin J. Paraffinoma of lips and oral mucosa: case report and brief review of literature. GMS Interdiscip Plast Reconstr Surg DGPW. 2014;3:Doc05.
- Matton G, Anseeuw A, De Keyser F. The history of injectable biomaterials and the biology of collagen. Aesthetic Plast Surg. 1985;9:133-140.
- Glicenstein J. Les premiers fillers, Vaseline et paraffine. du miracle a la catastrope. Ann Chir Plast Esthet. 2007;52:157-161.
- Cohen JL, Keoleian CM, Krull EA. Penile paraffinoma: self-injection with mineral oil. J Am Acad Dermatol 2002;47:S251-S253.
- Legaspi-Vicerra ME, Field LM. Paraffin granulomata, “witch’s chin,” and nasal deformities excision and reconstruction with reduction chinplasty and open rhinotomy resection. J Clin Aesthet Dermatol 2010;3:54-58.
- Carlos-Fabuel L, Marzal-Gamarra C, Marti-Alamo S, et al. Foreign body granulomatous reactions to cosmetic fillers. J Clin Exp Dent. 2012;4:E244-E247.
- Lemperle G, Gauthier-Hazan N. Foreign body granulomas after all injectable dermal fillers: part 2. treatment options. Plast Reconstr Surg. 2009;123:1864-1873.
- Seok J, Hong JY, Park KY, et al. Delayed immunologic complications due to injectable fillers by unlicensed practitioners: our experiences and a review of the literature. Dermatol Ther. 2016;29:41-44.
Practice Points
- The initial presentation of a foreign-body granulomatous process in a patient with surreptitious use of nonmedical filler can mimic infection; thus, careful history and diagnostic measures are paramount.
- Treatment of paraffin oil granuloma can be multifactorial and involves supportive care, systemic anti-inflammatory medications, time, and surgery.
- When a paraffin granuloma involves the orbital region, particular care is required to avoid long-term complications including cicatricial lagophthalmos, ectropion, or retractions, which can be mitigated with the help of oculoplastic surgery.

Tylosis in a Patient With Howel-Evans Syndrome: Management With Acitretin
To the Editor:
Tylosis with esophageal cancer was first described by Howel-Evans et al1 in 1958 in a family from Liverpool, England. The disease is inherited in an autosomal-dominant fashion with a mutation in the tylosis with esophageal cancer gene, TOC.2 The keratoderma associated with this syndrome has been reported to be focal in nature, painful, and primarily involving the plantar surfaces.3 Palmar involvement has been reported to manifest as calluses in patients who use their hands for manual labor.4 Oral leukoplakia also has been described in this syndrome5; however, long-term follow-up in one family demonstrated a benign course.6 Herein, we describe a case of painful tylosis in a patient with Howel-Evans syndrome who was successfully treated with acitretin.
A 50-year-old man presented to clinic for evaluation of hyperkeratosis of the palms and soles that began when he was a teenager. He reported the soles of the feet often were painful, especially without shoes (Figure, A). He used many over-the-counter emollients and tried both prescription and nonprescription keratolytics. At presentation, he was mechanically paring down some of the thickness of the calluses to decrease the pain.
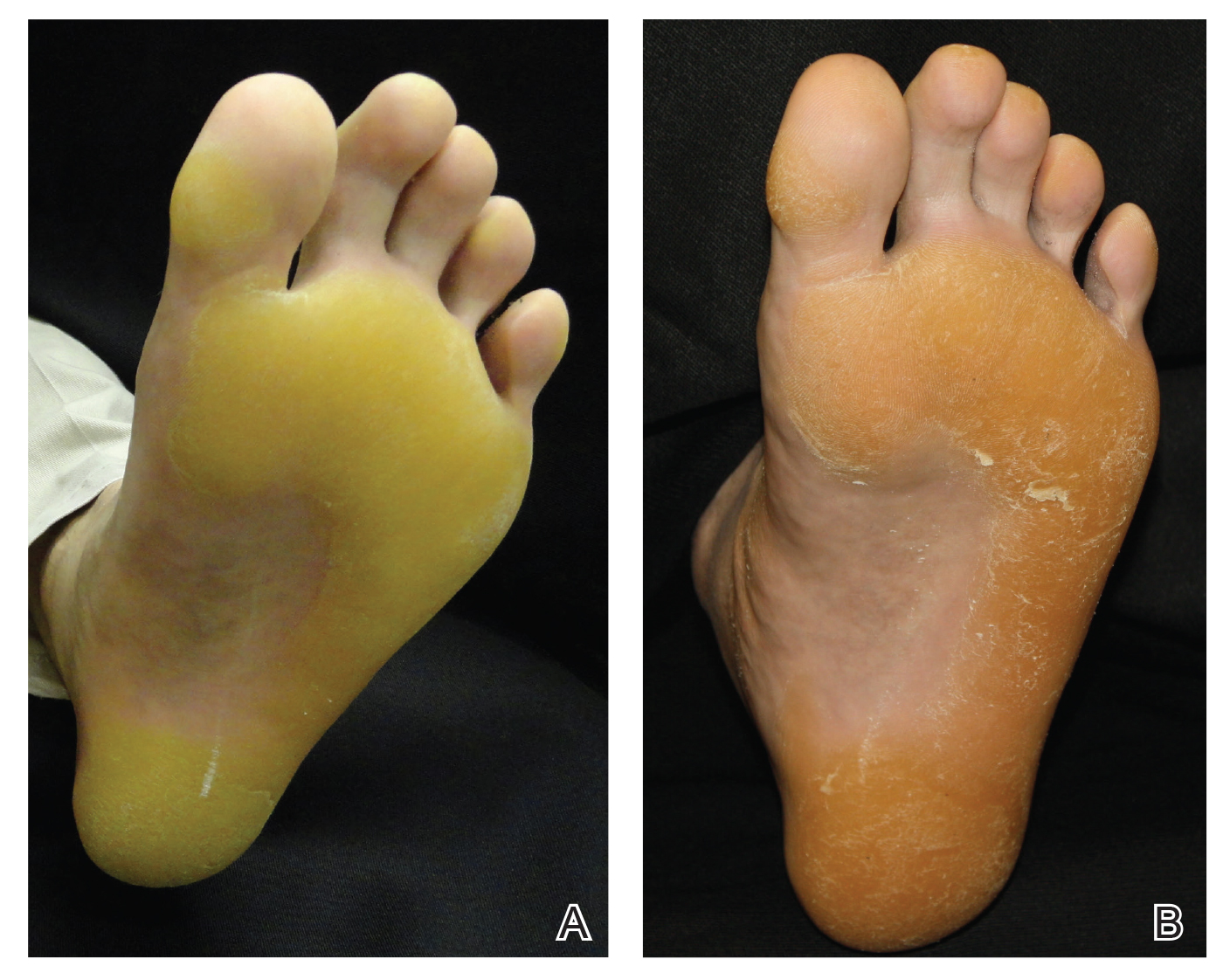
There was no relevant medical history, he had no history of smoking, he consumed more than 1 alcoholic drink per day, and he denied illicit drug use. The patient was not on any other medications. His family history revealed that his father also had the same hyperkeratosis of the palms and soles and died from esophageal carcinoma at an early age. It was determined that his father had tylosis with esophageal carcinoma (Howel-Evans syndrome). (The patient’s pedigree previously was published.3,4) Physical examination at presentation revealed plantar hyperkeratosis limited mainly to areas of pressure. His hands had mild hyperkeratosis on the distal fingers. No mucosa leukoplakia was identified.
Treatment options were discussed, and because the pain associated with the plantar keratoderma was interfering with his quality of life (QOL), acitretin was started. The initial dosage was 10 mg daily for 2 weeks and subsequently was increased to 25 mg daily. He has been maintained on this dosage for more than a year. An attempt was made to increase acitretin to 50 mg daily; however, he could not tolerate the dryness and peeling of the hands caused by the higher dosage. A fasting lipid panel and hepatic function panel performed every 3 months was within reference range. He had a remarkable decrease in the hyperkeratosis 2 months after starting therapy (Figure, B) and most importantly a decrease in pain associated with it. His QOL notably improved, enabling him to participate in sporting events with his children without severe pain. This patient was referred to gastroenterology where an esophagogastroduodenoscopy was performed and no concerning lesions were found. He was continued on this dose for 2 years. He moved to a new town, and our most recent update from him was that he was taking acitretin intermittently before big sporting events with his children.
The use of systemic retinoids has long been known to be effective in the treatment of disorders of keratinization. Recommended monitoring guidelines include a baseline complete blood cell count, renal function, hepatic function, and fasting lipid panel, which should be repeated every 3 months focusing on the hepatic function and lipid panel, as retinoids rarely cause hematologic or renal abnormalities.7 Our patient’s baseline laboratory test results were within reference range, and we repeated a fasting lipid and hepatic function panel every 3 months without any abnormalities.
Diffuse idiopathic skeletal hyperostosis (DISH), the ossification of ligaments and entheses often of the spine, is a potential complication of long-term use of oral retinoids. There are no consensus guidelines on screening for this complication, but baseline and annual radiographs seem reasonable. A 1996 study concluded that if DISH occurs, it is likely to be sporadic in a predisposed patient, as their data did not find any statistically significant relationship between the treatment or the cumulative dose and the prevalence and severity of DISH, degenerative changes, and osteoporosis.8 When annual screening is declined, imaging could be performed if a new skeletal concern were to arise in patients on long-term therapy.7 We discussed the skeletal concerns with our patient and he declined baseline or annual radiographs, but we will follow him with a rheumatologic review of systems. We feel this approach is reasonable, as our patient is a healthy adult in his 50s with no prior retinoid exposure and is on a low to moderate dose.
We report a case of Howel-Evans keratoderma successfully managed with acitretin. In patients with painful keratoderma that is interfering with QOL, low-dose acitretin can be used to diminish these symptoms.
- Howel-Evans W, McConnell RB, Clarke CA, et al. Carcinoma of the oesophagus with keratosis palmaris et plantaris (tylosis): a study of two families. Q J Med. 1958;27:413-429.
- Rogaev EI, Rogaeva EA, Ginter EK, et al. Identification of the genetic locus for keratosis palmaris et plantaris on chromosome 17 near the RARA and keratin type I genes. Nat Genet. 1993;5:158-162.
- Stevens HP, Kelsell DP, Bryant SP, et al. Linkage of an American pedigree with palmoplantar keratoderma and malignancy (palmoplantar ectodermal dysplasia type III) to 17q24. literature survey and proposed updated classification of the keratodermas. Arch Dermatol. 1996;132:640-651.
- Marger RS, Marger D. Carcinoma of the esophagus and tylosis. a lethal genetic combination. Cancer. 1993;72:17-19.
- Tyldesley WR. Oral leukoplakia associated with tylosis and esophageal carcinoma. J Oral Pathol. 1974;3:62-70.
- Ellis A, Field JK, Field EA, et al. Tylosis associated with carcinoma of the oesophagus and oral leukoplakia in a large Liverpool family—a review of six generations. Eur J Cancer B Oral Oncol. 1994;30B:102-112.
- Wu J, Wolverton S. Systemic retinoids. In: Wolverton S, ed. Comprehensive Dermatologic Drug Therapy. 4th ed. Edinburgh, Scotland: Elsevier; 2020:245-262.
- Van Dooren-Greebe RJ, Lemmens JA, De Boo T, et al. Prolonged treatment with oral retinoids in adults: no influence on the frequency and severity of spinal abnormalities. Br J Dermatol. 1996;134:71-76.
To the Editor:
Tylosis with esophageal cancer was first described by Howel-Evans et al1 in 1958 in a family from Liverpool, England. The disease is inherited in an autosomal-dominant fashion with a mutation in the tylosis with esophageal cancer gene, TOC.2 The keratoderma associated with this syndrome has been reported to be focal in nature, painful, and primarily involving the plantar surfaces.3 Palmar involvement has been reported to manifest as calluses in patients who use their hands for manual labor.4 Oral leukoplakia also has been described in this syndrome5; however, long-term follow-up in one family demonstrated a benign course.6 Herein, we describe a case of painful tylosis in a patient with Howel-Evans syndrome who was successfully treated with acitretin.
A 50-year-old man presented to clinic for evaluation of hyperkeratosis of the palms and soles that began when he was a teenager. He reported the soles of the feet often were painful, especially without shoes (Figure, A). He used many over-the-counter emollients and tried both prescription and nonprescription keratolytics. At presentation, he was mechanically paring down some of the thickness of the calluses to decrease the pain.
There was no relevant medical history, he had no history of smoking, he consumed more than 1 alcoholic drink per day, and he denied illicit drug use. The patient was not on any other medications. His family history revealed that his father also had the same hyperkeratosis of the palms and soles and died from esophageal carcinoma at an early age. It was determined that his father had tylosis with esophageal carcinoma (Howel-Evans syndrome). (The patient’s pedigree previously was published.3,4) Physical examination at presentation revealed plantar hyperkeratosis limited mainly to areas of pressure. His hands had mild hyperkeratosis on the distal fingers. No mucosa leukoplakia was identified.
Treatment options were discussed, and because the pain associated with the plantar keratoderma was interfering with his quality of life (QOL), acitretin was started. The initial dosage was 10 mg daily for 2 weeks and subsequently was increased to 25 mg daily. He has been maintained on this dosage for more than a year. An attempt was made to increase acitretin to 50 mg daily; however, he could not tolerate the dryness and peeling of the hands caused by the higher dosage. A fasting lipid panel and hepatic function panel performed every 3 months was within reference range. He had a remarkable decrease in the hyperkeratosis 2 months after starting therapy (Figure, B) and most importantly a decrease in pain associated with it. His QOL notably improved, enabling him to participate in sporting events with his children without severe pain. This patient was referred to gastroenterology where an esophagogastroduodenoscopy was performed and no concerning lesions were found. He was continued on this dose for 2 years. He moved to a new town, and our most recent update from him was that he was taking acitretin intermittently before big sporting events with his children.
The use of systemic retinoids has long been known to be effective in the treatment of disorders of keratinization. Recommended monitoring guidelines include a baseline complete blood cell count, renal function, hepatic function, and fasting lipid panel, which should be repeated every 3 months focusing on the hepatic function and lipid panel, as retinoids rarely cause hematologic or renal abnormalities.7 Our patient’s baseline laboratory test results were within reference range, and we repeated a fasting lipid and hepatic function panel every 3 months without any abnormalities.
Diffuse idiopathic skeletal hyperostosis (DISH), the ossification of ligaments and entheses often of the spine, is a potential complication of long-term use of oral retinoids. There are no consensus guidelines on screening for this complication, but baseline and annual radiographs seem reasonable. A 1996 study concluded that if DISH occurs, it is likely to be sporadic in a predisposed patient, as their data did not find any statistically significant relationship between the treatment or the cumulative dose and the prevalence and severity of DISH, degenerative changes, and osteoporosis.8 When annual screening is declined, imaging could be performed if a new skeletal concern were to arise in patients on long-term therapy.7 We discussed the skeletal concerns with our patient and he declined baseline or annual radiographs, but we will follow him with a rheumatologic review of systems. We feel this approach is reasonable, as our patient is a healthy adult in his 50s with no prior retinoid exposure and is on a low to moderate dose.
We report a case of Howel-Evans keratoderma successfully managed with acitretin. In patients with painful keratoderma that is interfering with QOL, low-dose acitretin can be used to diminish these symptoms.
To the Editor:
Tylosis with esophageal cancer was first described by Howel-Evans et al1 in 1958 in a family from Liverpool, England. The disease is inherited in an autosomal-dominant fashion with a mutation in the tylosis with esophageal cancer gene, TOC.2 The keratoderma associated with this syndrome has been reported to be focal in nature, painful, and primarily involving the plantar surfaces.3 Palmar involvement has been reported to manifest as calluses in patients who use their hands for manual labor.4 Oral leukoplakia also has been described in this syndrome5; however, long-term follow-up in one family demonstrated a benign course.6 Herein, we describe a case of painful tylosis in a patient with Howel-Evans syndrome who was successfully treated with acitretin.
A 50-year-old man presented to clinic for evaluation of hyperkeratosis of the palms and soles that began when he was a teenager. He reported the soles of the feet often were painful, especially without shoes (Figure, A). He used many over-the-counter emollients and tried both prescription and nonprescription keratolytics. At presentation, he was mechanically paring down some of the thickness of the calluses to decrease the pain.
There was no relevant medical history, he had no history of smoking, he consumed more than 1 alcoholic drink per day, and he denied illicit drug use. The patient was not on any other medications. His family history revealed that his father also had the same hyperkeratosis of the palms and soles and died from esophageal carcinoma at an early age. It was determined that his father had tylosis with esophageal carcinoma (Howel-Evans syndrome). (The patient’s pedigree previously was published.3,4) Physical examination at presentation revealed plantar hyperkeratosis limited mainly to areas of pressure. His hands had mild hyperkeratosis on the distal fingers. No mucosa leukoplakia was identified.
Treatment options were discussed, and because the pain associated with the plantar keratoderma was interfering with his quality of life (QOL), acitretin was started. The initial dosage was 10 mg daily for 2 weeks and subsequently was increased to 25 mg daily. He has been maintained on this dosage for more than a year. An attempt was made to increase acitretin to 50 mg daily; however, he could not tolerate the dryness and peeling of the hands caused by the higher dosage. A fasting lipid panel and hepatic function panel performed every 3 months was within reference range. He had a remarkable decrease in the hyperkeratosis 2 months after starting therapy (Figure, B) and most importantly a decrease in pain associated with it. His QOL notably improved, enabling him to participate in sporting events with his children without severe pain. This patient was referred to gastroenterology where an esophagogastroduodenoscopy was performed and no concerning lesions were found. He was continued on this dose for 2 years. He moved to a new town, and our most recent update from him was that he was taking acitretin intermittently before big sporting events with his children.
The use of systemic retinoids has long been known to be effective in the treatment of disorders of keratinization. Recommended monitoring guidelines include a baseline complete blood cell count, renal function, hepatic function, and fasting lipid panel, which should be repeated every 3 months focusing on the hepatic function and lipid panel, as retinoids rarely cause hematologic or renal abnormalities.7 Our patient’s baseline laboratory test results were within reference range, and we repeated a fasting lipid and hepatic function panel every 3 months without any abnormalities.
Diffuse idiopathic skeletal hyperostosis (DISH), the ossification of ligaments and entheses often of the spine, is a potential complication of long-term use of oral retinoids. There are no consensus guidelines on screening for this complication, but baseline and annual radiographs seem reasonable. A 1996 study concluded that if DISH occurs, it is likely to be sporadic in a predisposed patient, as their data did not find any statistically significant relationship between the treatment or the cumulative dose and the prevalence and severity of DISH, degenerative changes, and osteoporosis.8 When annual screening is declined, imaging could be performed if a new skeletal concern were to arise in patients on long-term therapy.7 We discussed the skeletal concerns with our patient and he declined baseline or annual radiographs, but we will follow him with a rheumatologic review of systems. We feel this approach is reasonable, as our patient is a healthy adult in his 50s with no prior retinoid exposure and is on a low to moderate dose.
We report a case of Howel-Evans keratoderma successfully managed with acitretin. In patients with painful keratoderma that is interfering with QOL, low-dose acitretin can be used to diminish these symptoms.
- Howel-Evans W, McConnell RB, Clarke CA, et al. Carcinoma of the oesophagus with keratosis palmaris et plantaris (tylosis): a study of two families. Q J Med. 1958;27:413-429.
- Rogaev EI, Rogaeva EA, Ginter EK, et al. Identification of the genetic locus for keratosis palmaris et plantaris on chromosome 17 near the RARA and keratin type I genes. Nat Genet. 1993;5:158-162.
- Stevens HP, Kelsell DP, Bryant SP, et al. Linkage of an American pedigree with palmoplantar keratoderma and malignancy (palmoplantar ectodermal dysplasia type III) to 17q24. literature survey and proposed updated classification of the keratodermas. Arch Dermatol. 1996;132:640-651.
- Marger RS, Marger D. Carcinoma of the esophagus and tylosis. a lethal genetic combination. Cancer. 1993;72:17-19.
- Tyldesley WR. Oral leukoplakia associated with tylosis and esophageal carcinoma. J Oral Pathol. 1974;3:62-70.
- Ellis A, Field JK, Field EA, et al. Tylosis associated with carcinoma of the oesophagus and oral leukoplakia in a large Liverpool family—a review of six generations. Eur J Cancer B Oral Oncol. 1994;30B:102-112.
- Wu J, Wolverton S. Systemic retinoids. In: Wolverton S, ed. Comprehensive Dermatologic Drug Therapy. 4th ed. Edinburgh, Scotland: Elsevier; 2020:245-262.
- Van Dooren-Greebe RJ, Lemmens JA, De Boo T, et al. Prolonged treatment with oral retinoids in adults: no influence on the frequency and severity of spinal abnormalities. Br J Dermatol. 1996;134:71-76.
- Howel-Evans W, McConnell RB, Clarke CA, et al. Carcinoma of the oesophagus with keratosis palmaris et plantaris (tylosis): a study of two families. Q J Med. 1958;27:413-429.
- Rogaev EI, Rogaeva EA, Ginter EK, et al. Identification of the genetic locus for keratosis palmaris et plantaris on chromosome 17 near the RARA and keratin type I genes. Nat Genet. 1993;5:158-162.
- Stevens HP, Kelsell DP, Bryant SP, et al. Linkage of an American pedigree with palmoplantar keratoderma and malignancy (palmoplantar ectodermal dysplasia type III) to 17q24. literature survey and proposed updated classification of the keratodermas. Arch Dermatol. 1996;132:640-651.
- Marger RS, Marger D. Carcinoma of the esophagus and tylosis. a lethal genetic combination. Cancer. 1993;72:17-19.
- Tyldesley WR. Oral leukoplakia associated with tylosis and esophageal carcinoma. J Oral Pathol. 1974;3:62-70.
- Ellis A, Field JK, Field EA, et al. Tylosis associated with carcinoma of the oesophagus and oral leukoplakia in a large Liverpool family—a review of six generations. Eur J Cancer B Oral Oncol. 1994;30B:102-112.
- Wu J, Wolverton S. Systemic retinoids. In: Wolverton S, ed. Comprehensive Dermatologic Drug Therapy. 4th ed. Edinburgh, Scotland: Elsevier; 2020:245-262.
- Van Dooren-Greebe RJ, Lemmens JA, De Boo T, et al. Prolonged treatment with oral retinoids in adults: no influence on the frequency and severity of spinal abnormalities. Br J Dermatol. 1996;134:71-76.
Practice Points
- Keratoderma can be especially painful for patients and can have a great impact on their quality of life. For these patients, acitretin should be considered when topical therapies have failed.
- Howel-Evans syndrome is an autosomal-dominant condition that predominantly presents with plantar keratoderma and has a high risk for esophageal cancer.
Cutaneous Filariasis in an American Traveler
To the Editor:
Cutaneous filariasis is a group of infectious diseases caused by more than 60 different nematode species and endemic to approximately 83 countries.1,2 These infections are transmitted to humans by vectors such as mosquitoes, blackflies, biting midges, or Tabanid flies.1 The blood meal taken by vectors allow microfilariae to enter the skin and develop into adult worms.1-3 It is postulated that filarial infections were first described in 2000
Filarial parasites have been categorized into groups based on the site of adult worm stage habitat. The cutaneous group includes Loa loa, Onchocerca volvulus, Mansonella perstans, and Dipetalonema streptocerca. The lymphatic group includes Wuchereria bancrofti, Brugia malayi, and Brugia timori. Lastly, the body cavity group includes Mansonella ozzardi. Because humidity is required for survival of the infective larval stage, individuals from tropical countries in Africa, Central America, and South America most commonly are affected.1 These diseases also are related to poor housing quality and inadequate sanitation.1,3,4 Travel for business, medical missions, pleasure, or emigration has caused increased filarial infections globally. In fact, dermatologic disorders with infectious etiologies cause approximately 17% of travelers to seek medical attention.3
Dermatologic manifestations indicative of a potential cutaneous filarial infection include papules, nodules, excoriations with secondary xerosis, lichenification, skin pigment changes, and/or severe pruritus. However, individuals from filarial endemic regions may not demonstrate any clinical signs or symptoms, despite having microfilariae in their blood,1 which enables the disease to propagate and poses a major public health concern. In fact, patients with chronic filarial infections are at increased risk for developing lymphedema, elephantiasis, or blindness, which are hypothesized to be the second largest cause of permanent disability worldwide.1 Although rarely seen in US citizens, cutaneous filariasis should always be a diagnostic consideration in patients who present with a pruritic eruption and have a travel history to tropical countries. We report a case of cutaneous onchocerciasis in a US citizen who developed a pruritic eczematous eruption on the right upper arm following an arthropod assault while working in an Onchocerca endemic region approximately 1.5 years prior.
A 33-year-old woman presented to our outpatient dermatology clinic with the chief concern of a pruritic rash on the right arm (Figure 1). She revealed a history of travel to Peru, associated symptoms, prior diagnoses, and attempted treatment regimens. Over a 1.5-year period, the patient traveled for work-related reasons to Madre de Dios, Peru. During that time, the patient experienced 2 bouts of severe abdominal pain, nausea, and vomiting that she attributed to poor food and water quality. She did not immediately seek medical attention. She also developed skin manifestations that began as a pruritic and irritating pinpoint-sized red papule on the right eyelid, possibly a site of an arthropod assault. She then experienced episodic eyelash loss (Figure 2) and subsequent vision changes, including blurry vision, halos around lights, and dimming around the periphery. When the patient developed circular, pink, pruritic plaques on the right upper extremity, she sought medical attention in Lima, Peru. She was diagnosed with tinea corporis and prescribed an oral antibiotic and topical antifungal.
This treatment regimen did not result in improvement. In the following months, the patient noticed worsening of the ocular symptoms, and she developed a dry cough with occasional dyspnea. She again sought medical attention in Peru and was referred to an ophthalmologist who suspected Demodex mite or fungal infection; however, workup for those pathologies was negative. The respiratory symptoms continued to worsen, particularly at night, and she began to experience palpitations.
Upon returning to the United States, the patient was evaluated by her primary care physician who ordered the following laboratory tests: a complete blood cell count with differential, comprehensive metabolic panel, vitamin D level, blood culture, lipid panel, ferritin level, and thyroid function. An electrocardiogram, throat culture, and stool culture for ova and parasites also were obtained. The electrocardiogram showed sinus tachycardia, and the stool culture revealed blastocystis, for which she was prescribed oral metronidazole and tinidazole. The other results were within reference range, and the throat culture showed normal oropharyngeal microbes.
A few days after the treatment for the blastocystis, the gastrointestinal tract symptoms improved, but dermatologic, ocular, pulmonary, and cardiac symptoms persisted. She also began experiencing night sweats and hot flashes. In between visits to the primary care physician, she sought medical attention at an urgent care clinic for worsening pulmonary and cardiac symptoms. At that time, results of a chest radiograph were normal.
The primary care physician referred her to infectious disease and dermatology for further evaluation within a few weeks of returning to the United States. Upon presentation to dermatology, the patient described a waxing and waning nature to the rash on the arm with associated intermittent pain. Physical examination revealed several erythematous patches on the triceps and a 1-cm subcutaneous nodule on the right elbow with overlying skin discoloration. The patient stated that she first noticed the inflamed painful nodule when she returned to the United States. Since its discovery, the nodule increased in size and became firm. The patient also described occasional limited range of motion of the right arm due to the discomfort of the rash and inflamed nodule. Interestingly, the patient’s partner who accompanied her to Peru also developed a similar pruritic rash on the chest. He was evaluated by dermatology and infectious disease; however, biopsies of his rash performed at a different practice revealed nonspecific results. Due to her partner’s inconclusive results, our patient initially refused a biopsy; however, she returned to the office after 1 week with worsening symptoms, and a 4-mm punch biopsy of the right arm was obtained in addition to rapid plasma reagin and QuantiFERON-TB Gold test.
Based on the patient’s travel history, clinical course, and physical examination, the clinical differential diagnosis included nummular dermatitis, panniculitis/nodular vasculitis, tinea corporis, secondary syphilis, pinta, tuberculosis, or cutaneous filariasis. Rapid plasma reagin and QuantiFERON-TB Gold test results were negative, and a periodic acid–Schiff stain of the biopsy was negative for fungal elements. Dermatopathology revealed intradermal filarial nematodes of 120 to 150 µm in diameter and 1-to 2-mm cuticles eliciting a predominantly superficial and deep lymphohistiocytic reaction (Figure 3). The histopathologic differential diagnosis based on the diameter of the nematode included M perstans, W bancrofti, and O volvulus. Clinical correlation was highly recommended to obtain a final diagnosis and management plan. A portion of the biopsy was sent to the Centers for Disease Control and Prevention, which confirmed the presence of a filarial nematode consistent with a zoonotic Brugia but also with zoonotic Onchocerca as a possible differential. Therefore, considering the history, clinical course, and histopathologic analysis, the final diagnosis of cutaneous onchocerciasis was made.
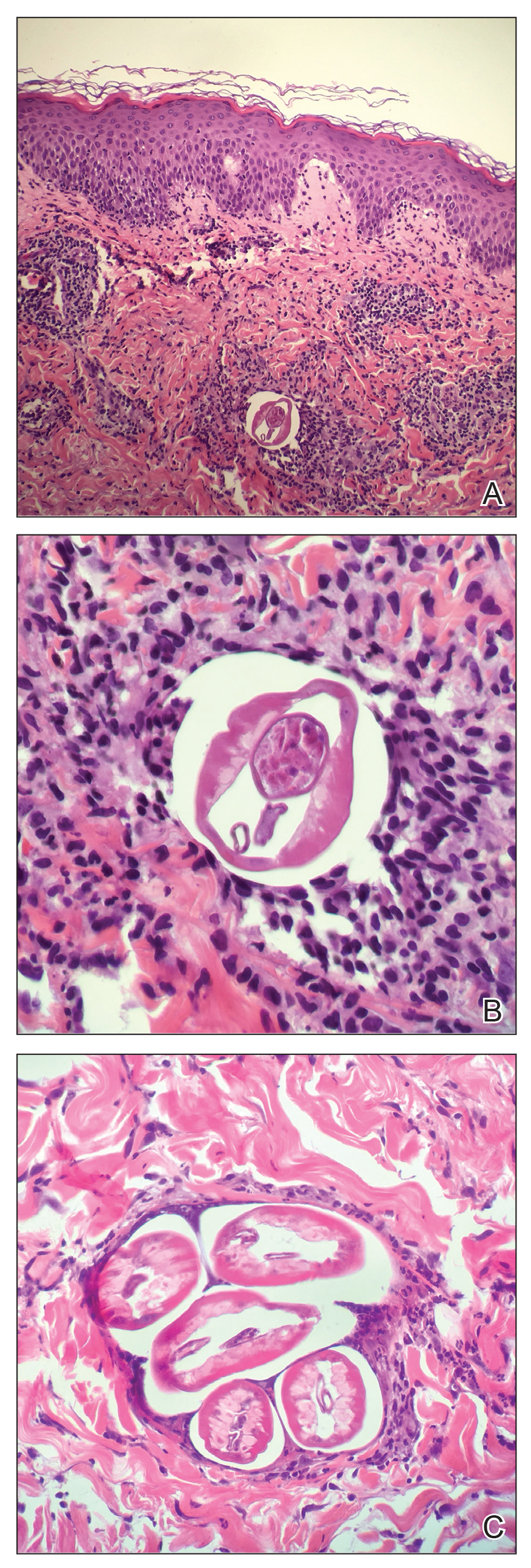
The patient was referred back to infectious disease and started on combination therapy of ivermectin (25.5 mg) plus albendazole (800 mg) taken at once followed by doxycycline (200 mg) for 6 weeks. The symptoms initially worsened, then she experienced near-complete resolution of dermatologic, pulmonary, and ocular symptoms after approximately 2 weeks of treatment. She continued to report fatigue and palpitations, for she continued to see a cardiologist. We highly encouraged her to continue following up with infectious disease to ensure complete eradication of the infection.
Onchocerciasis is common in individuals who live or work in equatorial Africa, Central America, and South America. The disease process has dermatologic, ocular, and other systemic manifestations if the infection continues long-term without treatment.2,5 Although rare, this infection has been observed in US citizens who have traveled to filarial endemic regions for a period of time ranging from 2 weeks to 39 years.2,6,7
Blackflies (genus Simulium), the intermediate host of O volvulus, breed in areas close to freely flowing water. The blackfly bites a human host, and within 7 days the microfilariae undergo 2 molts to reach the infective stage. The larvae remain in the dermis and subcutaneous tissue for 2 additional molts until they develop into adult worms. Then, adult worms may become encapsulated, eliciting and forming subcutaneous nodules, also known as onchocercomas.2,3
A fertilized female in the subcutaneous tissue releases microfilariae that may remain in the skin or travel to the eyes of the host.3 The host may demonstrate signs of infection within 3 to 15 months.2 Most commonly, patients report localized or generalized pruritus, and one extremity develops swelling, papules, lymphadenopathy, and alterations of skin pigment (hypopigmentation or hyperpigmentation).5-8 Our patient developed a pruritic eczematous eruption that only affected her right arm with an inflamed firm nodule overlying the elbow, a suspected onchocercoma. Onchocercomas are the direct result of adult worms coiled in the dermis, subcutaneous tissue, and deep fascia. These commonly are found in close proximity to bony prominences with the specific location pending the geographic area of infection.8 For example, onchocercomas more commonly are found over bony prominences in the head and upper body region in patients who acquired the disease from Central or South America, whereas individuals infected in Africa more commonly develop onchocercomas near the femoral, coccyx, or sacral regions.2
The immune response mounted by the infected host is responsible for the ocular signs and symptoms.2 Specifically, the inflammatory reaction may impact the patient’s cornea, anterior uveal tract, chorioretinal zone, or optic nerve. In fact, onchocerciasis, or river blindness, is the leading cause of blindness in the world.2 The host immune response also is responsible for the dermatologic manifestations of this disease. In addition to the dermatologic manifestations already mentioned, others include epidermal atrophy, ulcerations, femoral or inguinal lymphadenitis, lymphedema, and/or general wasting in more severe long-standing infections. Additionally, patients may experience systemic signs resulting from an underlying O volvulus infection.8 Our patient demonstrated a subjectively remarkable systemic response manifested by shortness of breath, dyspnea, cough, and palpitations, likely the result of her existing filarial infection.
Diagnosis of onchocerciasis is made by identification of microfilariae or adult worms in skin snips or punch biopsies. Histopathologic analysis of onchocerciasis has been described as several adult worms in the subcutaneous tissue surrounded by a granulomatous, fibrotic, calcified, or sometimes ossified inflammatory reaction.8 Microfilariae may migrate to the upper dermis and typically are surrounded by lymphocytes, histiocytes, plasma cells, and eosinophils with an absence of neutrophils,5,8 which directly causes the overlying epidermis to undergo reactive changes. Measurement of filarial nematodes often helps differentiate species. For example, O volvulus typically measures 230 to 500 mm in length for females and 16 to 42 mm in length for males.8 The thickness of cuticles typically measures 4 to 8 µm for females and 3 to 5 µm for males. Microfilariae of O volvulus also have been identified in patient’s sputum, urine, blood, and spinal fluid.8 An alternative diagnostic method described by Stingl et al9 is a diethylcarbamazine (DEC) patch test that was 92% accurate in onchocerciasis cases diagnosed by skin snip analysis.9 Therefore, this test may be a practical alternative when skin specimens are inconclusive.
Management of onchocerciasis should include excision of subcutaneous nodules to remove adult worms. In the past, DEC with suramin was prescribed and kills both microfilariae and adult worms.8 However, DEC may induce pruritus, chorioretinal damage, and optic neuritis and suramin is nephrotoxic.2 Therefore, oral ivermectin (0.15–0.20 mg/kg one-time dose, may be repeated in 3–12 months) is supported to be a less harmful option. Patients treated with ivermectin have a decreased risk for transmitting infection to the blackfly vector for up to 6 months after treatment, and it is a more effective microfilaricidal agent than DEC.2,3 Oral doxycycline (100 mg/d for 6 weeks) commonly is added as adjuvant therapy because it kills Wolbachia species, a bacterium that is needed for O volvulus reproduction.3
Although rare in the United States, cutaneous filariasis has become a prevalent public health concern, especially in tropical countries. Individuals with chronic cutaneous filarial infections are at increased risk for debilitating complications that negatively affect their quality of life and productivity. Although the World Health Organization has attempted to eradicate cutaneous filarial infections by mass drug administration, transmission of these diseases remains a challenge. Further research on treatment and methods to prevent transmission by controlling arthropod vectors is required to avoid short-term and long-term health consequences caused by cutaneous filariasis.
Parasitic infections should always be a diagnostic consideration in individuals who present with a pruritic eruption and a history of travel to foreign countries located in Africa, Central America, and South America. Dermatologists in the United States should increase familiarity with these infections because of increased travel, economic globalization, and the impact of global climate change on the geographic distribution of vector arthropods. To control these infections, research efforts should focus on improved sanitation; drug treatment; transmission prevention; and improved education of their clinical manifestations, especially mucocutaneous signs.
- Mendoza N, Li A, Gill A, et al. Filariasis: diagnosis and treatment. Dermatol Ther. 2009;22:475-490.
- Maso MJ, Kapila R, Schwartz RA, et al. Cutaneous onchocerciasis. Int J Dermatol. 1987;26:593-596.
- Lupi O, Downing C, Lee M, et al. Mucocutaneous manifestations of helminth infections. J Am Acad Dermatol. 2015;73:929-944.
- Bolivar-Meija A, Alarcon-Olave C, Rodriguez-Morales AJ. Skin manifestations of arthropod-borne infection in Latin America. Curr Opin. 2014;27:288-294.
- Okulicz JF, Stibich AS, Elston DM, et al. Cutaneous onchocercoma. Int J Dermatol. 2004;43:170-172.
- Nguyen JC, Murphy ME, Nutman TB, et al. Cutaneous onchocerciasis in an American traveler. Int Soc Dermatol. 2005;44:125-128.
- Toovey S, Moerman F, van Gompel A. Special infectious disease risks of expatriates and long-term travelers in tropical countries part II: infections other than malaria. J Travel Med. 2007;14:50-60.
- Meyers WM, Neafie RC, Connor DH. Bancroftian and malayan filariasis. In Binford CH, ed. Pathology of Tropical and Extraordinary Diseases. Vol 2. Fort Sam Houston, TX: Armed Forces Institute of Pathology; 1976:340-355.
- Stingl P, Ross M, Gibson DW, et al. A diagnostic “patch-test” for onchocerciasis using topical diethylcarbamazine. Trans R Soc Trop Med Hyg.
To the Editor:
Cutaneous filariasis is a group of infectious diseases caused by more than 60 different nematode species and endemic to approximately 83 countries.1,2 These infections are transmitted to humans by vectors such as mosquitoes, blackflies, biting midges, or Tabanid flies.1 The blood meal taken by vectors allow microfilariae to enter the skin and develop into adult worms.1-3 It is postulated that filarial infections were first described in 2000
Filarial parasites have been categorized into groups based on the site of adult worm stage habitat. The cutaneous group includes Loa loa, Onchocerca volvulus, Mansonella perstans, and Dipetalonema streptocerca. The lymphatic group includes Wuchereria bancrofti, Brugia malayi, and Brugia timori. Lastly, the body cavity group includes Mansonella ozzardi. Because humidity is required for survival of the infective larval stage, individuals from tropical countries in Africa, Central America, and South America most commonly are affected.1 These diseases also are related to poor housing quality and inadequate sanitation.1,3,4 Travel for business, medical missions, pleasure, or emigration has caused increased filarial infections globally. In fact, dermatologic disorders with infectious etiologies cause approximately 17% of travelers to seek medical attention.3
Dermatologic manifestations indicative of a potential cutaneous filarial infection include papules, nodules, excoriations with secondary xerosis, lichenification, skin pigment changes, and/or severe pruritus. However, individuals from filarial endemic regions may not demonstrate any clinical signs or symptoms, despite having microfilariae in their blood,1 which enables the disease to propagate and poses a major public health concern. In fact, patients with chronic filarial infections are at increased risk for developing lymphedema, elephantiasis, or blindness, which are hypothesized to be the second largest cause of permanent disability worldwide.1 Although rarely seen in US citizens, cutaneous filariasis should always be a diagnostic consideration in patients who present with a pruritic eruption and have a travel history to tropical countries. We report a case of cutaneous onchocerciasis in a US citizen who developed a pruritic eczematous eruption on the right upper arm following an arthropod assault while working in an Onchocerca endemic region approximately 1.5 years prior.
A 33-year-old woman presented to our outpatient dermatology clinic with the chief concern of a pruritic rash on the right arm (Figure 1). She revealed a history of travel to Peru, associated symptoms, prior diagnoses, and attempted treatment regimens. Over a 1.5-year period, the patient traveled for work-related reasons to Madre de Dios, Peru. During that time, the patient experienced 2 bouts of severe abdominal pain, nausea, and vomiting that she attributed to poor food and water quality. She did not immediately seek medical attention. She also developed skin manifestations that began as a pruritic and irritating pinpoint-sized red papule on the right eyelid, possibly a site of an arthropod assault. She then experienced episodic eyelash loss (Figure 2) and subsequent vision changes, including blurry vision, halos around lights, and dimming around the periphery. When the patient developed circular, pink, pruritic plaques on the right upper extremity, she sought medical attention in Lima, Peru. She was diagnosed with tinea corporis and prescribed an oral antibiotic and topical antifungal.
This treatment regimen did not result in improvement. In the following months, the patient noticed worsening of the ocular symptoms, and she developed a dry cough with occasional dyspnea. She again sought medical attention in Peru and was referred to an ophthalmologist who suspected Demodex mite or fungal infection; however, workup for those pathologies was negative. The respiratory symptoms continued to worsen, particularly at night, and she began to experience palpitations.
Upon returning to the United States, the patient was evaluated by her primary care physician who ordered the following laboratory tests: a complete blood cell count with differential, comprehensive metabolic panel, vitamin D level, blood culture, lipid panel, ferritin level, and thyroid function. An electrocardiogram, throat culture, and stool culture for ova and parasites also were obtained. The electrocardiogram showed sinus tachycardia, and the stool culture revealed blastocystis, for which she was prescribed oral metronidazole and tinidazole. The other results were within reference range, and the throat culture showed normal oropharyngeal microbes.
A few days after the treatment for the blastocystis, the gastrointestinal tract symptoms improved, but dermatologic, ocular, pulmonary, and cardiac symptoms persisted. She also began experiencing night sweats and hot flashes. In between visits to the primary care physician, she sought medical attention at an urgent care clinic for worsening pulmonary and cardiac symptoms. At that time, results of a chest radiograph were normal.
The primary care physician referred her to infectious disease and dermatology for further evaluation within a few weeks of returning to the United States. Upon presentation to dermatology, the patient described a waxing and waning nature to the rash on the arm with associated intermittent pain. Physical examination revealed several erythematous patches on the triceps and a 1-cm subcutaneous nodule on the right elbow with overlying skin discoloration. The patient stated that she first noticed the inflamed painful nodule when she returned to the United States. Since its discovery, the nodule increased in size and became firm. The patient also described occasional limited range of motion of the right arm due to the discomfort of the rash and inflamed nodule. Interestingly, the patient’s partner who accompanied her to Peru also developed a similar pruritic rash on the chest. He was evaluated by dermatology and infectious disease; however, biopsies of his rash performed at a different practice revealed nonspecific results. Due to her partner’s inconclusive results, our patient initially refused a biopsy; however, she returned to the office after 1 week with worsening symptoms, and a 4-mm punch biopsy of the right arm was obtained in addition to rapid plasma reagin and QuantiFERON-TB Gold test.
Based on the patient’s travel history, clinical course, and physical examination, the clinical differential diagnosis included nummular dermatitis, panniculitis/nodular vasculitis, tinea corporis, secondary syphilis, pinta, tuberculosis, or cutaneous filariasis. Rapid plasma reagin and QuantiFERON-TB Gold test results were negative, and a periodic acid–Schiff stain of the biopsy was negative for fungal elements. Dermatopathology revealed intradermal filarial nematodes of 120 to 150 µm in diameter and 1-to 2-mm cuticles eliciting a predominantly superficial and deep lymphohistiocytic reaction (Figure 3). The histopathologic differential diagnosis based on the diameter of the nematode included M perstans, W bancrofti, and O volvulus. Clinical correlation was highly recommended to obtain a final diagnosis and management plan. A portion of the biopsy was sent to the Centers for Disease Control and Prevention, which confirmed the presence of a filarial nematode consistent with a zoonotic Brugia but also with zoonotic Onchocerca as a possible differential. Therefore, considering the history, clinical course, and histopathologic analysis, the final diagnosis of cutaneous onchocerciasis was made.
The patient was referred back to infectious disease and started on combination therapy of ivermectin (25.5 mg) plus albendazole (800 mg) taken at once followed by doxycycline (200 mg) for 6 weeks. The symptoms initially worsened, then she experienced near-complete resolution of dermatologic, pulmonary, and ocular symptoms after approximately 2 weeks of treatment. She continued to report fatigue and palpitations, for she continued to see a cardiologist. We highly encouraged her to continue following up with infectious disease to ensure complete eradication of the infection.
Onchocerciasis is common in individuals who live or work in equatorial Africa, Central America, and South America. The disease process has dermatologic, ocular, and other systemic manifestations if the infection continues long-term without treatment.2,5 Although rare, this infection has been observed in US citizens who have traveled to filarial endemic regions for a period of time ranging from 2 weeks to 39 years.2,6,7
Blackflies (genus Simulium), the intermediate host of O volvulus, breed in areas close to freely flowing water. The blackfly bites a human host, and within 7 days the microfilariae undergo 2 molts to reach the infective stage. The larvae remain in the dermis and subcutaneous tissue for 2 additional molts until they develop into adult worms. Then, adult worms may become encapsulated, eliciting and forming subcutaneous nodules, also known as onchocercomas.2,3
A fertilized female in the subcutaneous tissue releases microfilariae that may remain in the skin or travel to the eyes of the host.3 The host may demonstrate signs of infection within 3 to 15 months.2 Most commonly, patients report localized or generalized pruritus, and one extremity develops swelling, papules, lymphadenopathy, and alterations of skin pigment (hypopigmentation or hyperpigmentation).5-8 Our patient developed a pruritic eczematous eruption that only affected her right arm with an inflamed firm nodule overlying the elbow, a suspected onchocercoma. Onchocercomas are the direct result of adult worms coiled in the dermis, subcutaneous tissue, and deep fascia. These commonly are found in close proximity to bony prominences with the specific location pending the geographic area of infection.8 For example, onchocercomas more commonly are found over bony prominences in the head and upper body region in patients who acquired the disease from Central or South America, whereas individuals infected in Africa more commonly develop onchocercomas near the femoral, coccyx, or sacral regions.2
The immune response mounted by the infected host is responsible for the ocular signs and symptoms.2 Specifically, the inflammatory reaction may impact the patient’s cornea, anterior uveal tract, chorioretinal zone, or optic nerve. In fact, onchocerciasis, or river blindness, is the leading cause of blindness in the world.2 The host immune response also is responsible for the dermatologic manifestations of this disease. In addition to the dermatologic manifestations already mentioned, others include epidermal atrophy, ulcerations, femoral or inguinal lymphadenitis, lymphedema, and/or general wasting in more severe long-standing infections. Additionally, patients may experience systemic signs resulting from an underlying O volvulus infection.8 Our patient demonstrated a subjectively remarkable systemic response manifested by shortness of breath, dyspnea, cough, and palpitations, likely the result of her existing filarial infection.
Diagnosis of onchocerciasis is made by identification of microfilariae or adult worms in skin snips or punch biopsies. Histopathologic analysis of onchocerciasis has been described as several adult worms in the subcutaneous tissue surrounded by a granulomatous, fibrotic, calcified, or sometimes ossified inflammatory reaction.8 Microfilariae may migrate to the upper dermis and typically are surrounded by lymphocytes, histiocytes, plasma cells, and eosinophils with an absence of neutrophils,5,8 which directly causes the overlying epidermis to undergo reactive changes. Measurement of filarial nematodes often helps differentiate species. For example, O volvulus typically measures 230 to 500 mm in length for females and 16 to 42 mm in length for males.8 The thickness of cuticles typically measures 4 to 8 µm for females and 3 to 5 µm for males. Microfilariae of O volvulus also have been identified in patient’s sputum, urine, blood, and spinal fluid.8 An alternative diagnostic method described by Stingl et al9 is a diethylcarbamazine (DEC) patch test that was 92% accurate in onchocerciasis cases diagnosed by skin snip analysis.9 Therefore, this test may be a practical alternative when skin specimens are inconclusive.
Management of onchocerciasis should include excision of subcutaneous nodules to remove adult worms. In the past, DEC with suramin was prescribed and kills both microfilariae and adult worms.8 However, DEC may induce pruritus, chorioretinal damage, and optic neuritis and suramin is nephrotoxic.2 Therefore, oral ivermectin (0.15–0.20 mg/kg one-time dose, may be repeated in 3–12 months) is supported to be a less harmful option. Patients treated with ivermectin have a decreased risk for transmitting infection to the blackfly vector for up to 6 months after treatment, and it is a more effective microfilaricidal agent than DEC.2,3 Oral doxycycline (100 mg/d for 6 weeks) commonly is added as adjuvant therapy because it kills Wolbachia species, a bacterium that is needed for O volvulus reproduction.3
Although rare in the United States, cutaneous filariasis has become a prevalent public health concern, especially in tropical countries. Individuals with chronic cutaneous filarial infections are at increased risk for debilitating complications that negatively affect their quality of life and productivity. Although the World Health Organization has attempted to eradicate cutaneous filarial infections by mass drug administration, transmission of these diseases remains a challenge. Further research on treatment and methods to prevent transmission by controlling arthropod vectors is required to avoid short-term and long-term health consequences caused by cutaneous filariasis.
Parasitic infections should always be a diagnostic consideration in individuals who present with a pruritic eruption and a history of travel to foreign countries located in Africa, Central America, and South America. Dermatologists in the United States should increase familiarity with these infections because of increased travel, economic globalization, and the impact of global climate change on the geographic distribution of vector arthropods. To control these infections, research efforts should focus on improved sanitation; drug treatment; transmission prevention; and improved education of their clinical manifestations, especially mucocutaneous signs.
To the Editor:
Cutaneous filariasis is a group of infectious diseases caused by more than 60 different nematode species and endemic to approximately 83 countries.1,2 These infections are transmitted to humans by vectors such as mosquitoes, blackflies, biting midges, or Tabanid flies.1 The blood meal taken by vectors allow microfilariae to enter the skin and develop into adult worms.1-3 It is postulated that filarial infections were first described in 2000
Filarial parasites have been categorized into groups based on the site of adult worm stage habitat. The cutaneous group includes Loa loa, Onchocerca volvulus, Mansonella perstans, and Dipetalonema streptocerca. The lymphatic group includes Wuchereria bancrofti, Brugia malayi, and Brugia timori. Lastly, the body cavity group includes Mansonella ozzardi. Because humidity is required for survival of the infective larval stage, individuals from tropical countries in Africa, Central America, and South America most commonly are affected.1 These diseases also are related to poor housing quality and inadequate sanitation.1,3,4 Travel for business, medical missions, pleasure, or emigration has caused increased filarial infections globally. In fact, dermatologic disorders with infectious etiologies cause approximately 17% of travelers to seek medical attention.3
Dermatologic manifestations indicative of a potential cutaneous filarial infection include papules, nodules, excoriations with secondary xerosis, lichenification, skin pigment changes, and/or severe pruritus. However, individuals from filarial endemic regions may not demonstrate any clinical signs or symptoms, despite having microfilariae in their blood,1 which enables the disease to propagate and poses a major public health concern. In fact, patients with chronic filarial infections are at increased risk for developing lymphedema, elephantiasis, or blindness, which are hypothesized to be the second largest cause of permanent disability worldwide.1 Although rarely seen in US citizens, cutaneous filariasis should always be a diagnostic consideration in patients who present with a pruritic eruption and have a travel history to tropical countries. We report a case of cutaneous onchocerciasis in a US citizen who developed a pruritic eczematous eruption on the right upper arm following an arthropod assault while working in an Onchocerca endemic region approximately 1.5 years prior.
A 33-year-old woman presented to our outpatient dermatology clinic with the chief concern of a pruritic rash on the right arm (Figure 1). She revealed a history of travel to Peru, associated symptoms, prior diagnoses, and attempted treatment regimens. Over a 1.5-year period, the patient traveled for work-related reasons to Madre de Dios, Peru. During that time, the patient experienced 2 bouts of severe abdominal pain, nausea, and vomiting that she attributed to poor food and water quality. She did not immediately seek medical attention. She also developed skin manifestations that began as a pruritic and irritating pinpoint-sized red papule on the right eyelid, possibly a site of an arthropod assault. She then experienced episodic eyelash loss (Figure 2) and subsequent vision changes, including blurry vision, halos around lights, and dimming around the periphery. When the patient developed circular, pink, pruritic plaques on the right upper extremity, she sought medical attention in Lima, Peru. She was diagnosed with tinea corporis and prescribed an oral antibiotic and topical antifungal.
This treatment regimen did not result in improvement. In the following months, the patient noticed worsening of the ocular symptoms, and she developed a dry cough with occasional dyspnea. She again sought medical attention in Peru and was referred to an ophthalmologist who suspected Demodex mite or fungal infection; however, workup for those pathologies was negative. The respiratory symptoms continued to worsen, particularly at night, and she began to experience palpitations.
Upon returning to the United States, the patient was evaluated by her primary care physician who ordered the following laboratory tests: a complete blood cell count with differential, comprehensive metabolic panel, vitamin D level, blood culture, lipid panel, ferritin level, and thyroid function. An electrocardiogram, throat culture, and stool culture for ova and parasites also were obtained. The electrocardiogram showed sinus tachycardia, and the stool culture revealed blastocystis, for which she was prescribed oral metronidazole and tinidazole. The other results were within reference range, and the throat culture showed normal oropharyngeal microbes.
A few days after the treatment for the blastocystis, the gastrointestinal tract symptoms improved, but dermatologic, ocular, pulmonary, and cardiac symptoms persisted. She also began experiencing night sweats and hot flashes. In between visits to the primary care physician, she sought medical attention at an urgent care clinic for worsening pulmonary and cardiac symptoms. At that time, results of a chest radiograph were normal.
The primary care physician referred her to infectious disease and dermatology for further evaluation within a few weeks of returning to the United States. Upon presentation to dermatology, the patient described a waxing and waning nature to the rash on the arm with associated intermittent pain. Physical examination revealed several erythematous patches on the triceps and a 1-cm subcutaneous nodule on the right elbow with overlying skin discoloration. The patient stated that she first noticed the inflamed painful nodule when she returned to the United States. Since its discovery, the nodule increased in size and became firm. The patient also described occasional limited range of motion of the right arm due to the discomfort of the rash and inflamed nodule. Interestingly, the patient’s partner who accompanied her to Peru also developed a similar pruritic rash on the chest. He was evaluated by dermatology and infectious disease; however, biopsies of his rash performed at a different practice revealed nonspecific results. Due to her partner’s inconclusive results, our patient initially refused a biopsy; however, she returned to the office after 1 week with worsening symptoms, and a 4-mm punch biopsy of the right arm was obtained in addition to rapid plasma reagin and QuantiFERON-TB Gold test.
Based on the patient’s travel history, clinical course, and physical examination, the clinical differential diagnosis included nummular dermatitis, panniculitis/nodular vasculitis, tinea corporis, secondary syphilis, pinta, tuberculosis, or cutaneous filariasis. Rapid plasma reagin and QuantiFERON-TB Gold test results were negative, and a periodic acid–Schiff stain of the biopsy was negative for fungal elements. Dermatopathology revealed intradermal filarial nematodes of 120 to 150 µm in diameter and 1-to 2-mm cuticles eliciting a predominantly superficial and deep lymphohistiocytic reaction (Figure 3). The histopathologic differential diagnosis based on the diameter of the nematode included M perstans, W bancrofti, and O volvulus. Clinical correlation was highly recommended to obtain a final diagnosis and management plan. A portion of the biopsy was sent to the Centers for Disease Control and Prevention, which confirmed the presence of a filarial nematode consistent with a zoonotic Brugia but also with zoonotic Onchocerca as a possible differential. Therefore, considering the history, clinical course, and histopathologic analysis, the final diagnosis of cutaneous onchocerciasis was made.
The patient was referred back to infectious disease and started on combination therapy of ivermectin (25.5 mg) plus albendazole (800 mg) taken at once followed by doxycycline (200 mg) for 6 weeks. The symptoms initially worsened, then she experienced near-complete resolution of dermatologic, pulmonary, and ocular symptoms after approximately 2 weeks of treatment. She continued to report fatigue and palpitations, for she continued to see a cardiologist. We highly encouraged her to continue following up with infectious disease to ensure complete eradication of the infection.
Onchocerciasis is common in individuals who live or work in equatorial Africa, Central America, and South America. The disease process has dermatologic, ocular, and other systemic manifestations if the infection continues long-term without treatment.2,5 Although rare, this infection has been observed in US citizens who have traveled to filarial endemic regions for a period of time ranging from 2 weeks to 39 years.2,6,7
Blackflies (genus Simulium), the intermediate host of O volvulus, breed in areas close to freely flowing water. The blackfly bites a human host, and within 7 days the microfilariae undergo 2 molts to reach the infective stage. The larvae remain in the dermis and subcutaneous tissue for 2 additional molts until they develop into adult worms. Then, adult worms may become encapsulated, eliciting and forming subcutaneous nodules, also known as onchocercomas.2,3
A fertilized female in the subcutaneous tissue releases microfilariae that may remain in the skin or travel to the eyes of the host.3 The host may demonstrate signs of infection within 3 to 15 months.2 Most commonly, patients report localized or generalized pruritus, and one extremity develops swelling, papules, lymphadenopathy, and alterations of skin pigment (hypopigmentation or hyperpigmentation).5-8 Our patient developed a pruritic eczematous eruption that only affected her right arm with an inflamed firm nodule overlying the elbow, a suspected onchocercoma. Onchocercomas are the direct result of adult worms coiled in the dermis, subcutaneous tissue, and deep fascia. These commonly are found in close proximity to bony prominences with the specific location pending the geographic area of infection.8 For example, onchocercomas more commonly are found over bony prominences in the head and upper body region in patients who acquired the disease from Central or South America, whereas individuals infected in Africa more commonly develop onchocercomas near the femoral, coccyx, or sacral regions.2
The immune response mounted by the infected host is responsible for the ocular signs and symptoms.2 Specifically, the inflammatory reaction may impact the patient’s cornea, anterior uveal tract, chorioretinal zone, or optic nerve. In fact, onchocerciasis, or river blindness, is the leading cause of blindness in the world.2 The host immune response also is responsible for the dermatologic manifestations of this disease. In addition to the dermatologic manifestations already mentioned, others include epidermal atrophy, ulcerations, femoral or inguinal lymphadenitis, lymphedema, and/or general wasting in more severe long-standing infections. Additionally, patients may experience systemic signs resulting from an underlying O volvulus infection.8 Our patient demonstrated a subjectively remarkable systemic response manifested by shortness of breath, dyspnea, cough, and palpitations, likely the result of her existing filarial infection.
Diagnosis of onchocerciasis is made by identification of microfilariae or adult worms in skin snips or punch biopsies. Histopathologic analysis of onchocerciasis has been described as several adult worms in the subcutaneous tissue surrounded by a granulomatous, fibrotic, calcified, or sometimes ossified inflammatory reaction.8 Microfilariae may migrate to the upper dermis and typically are surrounded by lymphocytes, histiocytes, plasma cells, and eosinophils with an absence of neutrophils,5,8 which directly causes the overlying epidermis to undergo reactive changes. Measurement of filarial nematodes often helps differentiate species. For example, O volvulus typically measures 230 to 500 mm in length for females and 16 to 42 mm in length for males.8 The thickness of cuticles typically measures 4 to 8 µm for females and 3 to 5 µm for males. Microfilariae of O volvulus also have been identified in patient’s sputum, urine, blood, and spinal fluid.8 An alternative diagnostic method described by Stingl et al9 is a diethylcarbamazine (DEC) patch test that was 92% accurate in onchocerciasis cases diagnosed by skin snip analysis.9 Therefore, this test may be a practical alternative when skin specimens are inconclusive.
Management of onchocerciasis should include excision of subcutaneous nodules to remove adult worms. In the past, DEC with suramin was prescribed and kills both microfilariae and adult worms.8 However, DEC may induce pruritus, chorioretinal damage, and optic neuritis and suramin is nephrotoxic.2 Therefore, oral ivermectin (0.15–0.20 mg/kg one-time dose, may be repeated in 3–12 months) is supported to be a less harmful option. Patients treated with ivermectin have a decreased risk for transmitting infection to the blackfly vector for up to 6 months after treatment, and it is a more effective microfilaricidal agent than DEC.2,3 Oral doxycycline (100 mg/d for 6 weeks) commonly is added as adjuvant therapy because it kills Wolbachia species, a bacterium that is needed for O volvulus reproduction.3
Although rare in the United States, cutaneous filariasis has become a prevalent public health concern, especially in tropical countries. Individuals with chronic cutaneous filarial infections are at increased risk for debilitating complications that negatively affect their quality of life and productivity. Although the World Health Organization has attempted to eradicate cutaneous filarial infections by mass drug administration, transmission of these diseases remains a challenge. Further research on treatment and methods to prevent transmission by controlling arthropod vectors is required to avoid short-term and long-term health consequences caused by cutaneous filariasis.
Parasitic infections should always be a diagnostic consideration in individuals who present with a pruritic eruption and a history of travel to foreign countries located in Africa, Central America, and South America. Dermatologists in the United States should increase familiarity with these infections because of increased travel, economic globalization, and the impact of global climate change on the geographic distribution of vector arthropods. To control these infections, research efforts should focus on improved sanitation; drug treatment; transmission prevention; and improved education of their clinical manifestations, especially mucocutaneous signs.
- Mendoza N, Li A, Gill A, et al. Filariasis: diagnosis and treatment. Dermatol Ther. 2009;22:475-490.
- Maso MJ, Kapila R, Schwartz RA, et al. Cutaneous onchocerciasis. Int J Dermatol. 1987;26:593-596.
- Lupi O, Downing C, Lee M, et al. Mucocutaneous manifestations of helminth infections. J Am Acad Dermatol. 2015;73:929-944.
- Bolivar-Meija A, Alarcon-Olave C, Rodriguez-Morales AJ. Skin manifestations of arthropod-borne infection in Latin America. Curr Opin. 2014;27:288-294.
- Okulicz JF, Stibich AS, Elston DM, et al. Cutaneous onchocercoma. Int J Dermatol. 2004;43:170-172.
- Nguyen JC, Murphy ME, Nutman TB, et al. Cutaneous onchocerciasis in an American traveler. Int Soc Dermatol. 2005;44:125-128.
- Toovey S, Moerman F, van Gompel A. Special infectious disease risks of expatriates and long-term travelers in tropical countries part II: infections other than malaria. J Travel Med. 2007;14:50-60.
- Meyers WM, Neafie RC, Connor DH. Bancroftian and malayan filariasis. In Binford CH, ed. Pathology of Tropical and Extraordinary Diseases. Vol 2. Fort Sam Houston, TX: Armed Forces Institute of Pathology; 1976:340-355.
- Stingl P, Ross M, Gibson DW, et al. A diagnostic “patch-test” for onchocerciasis using topical diethylcarbamazine. Trans R Soc Trop Med Hyg.
- Mendoza N, Li A, Gill A, et al. Filariasis: diagnosis and treatment. Dermatol Ther. 2009;22:475-490.
- Maso MJ, Kapila R, Schwartz RA, et al. Cutaneous onchocerciasis. Int J Dermatol. 1987;26:593-596.
- Lupi O, Downing C, Lee M, et al. Mucocutaneous manifestations of helminth infections. J Am Acad Dermatol. 2015;73:929-944.
- Bolivar-Meija A, Alarcon-Olave C, Rodriguez-Morales AJ. Skin manifestations of arthropod-borne infection in Latin America. Curr Opin. 2014;27:288-294.
- Okulicz JF, Stibich AS, Elston DM, et al. Cutaneous onchocercoma. Int J Dermatol. 2004;43:170-172.
- Nguyen JC, Murphy ME, Nutman TB, et al. Cutaneous onchocerciasis in an American traveler. Int Soc Dermatol. 2005;44:125-128.
- Toovey S, Moerman F, van Gompel A. Special infectious disease risks of expatriates and long-term travelers in tropical countries part II: infections other than malaria. J Travel Med. 2007;14:50-60.
- Meyers WM, Neafie RC, Connor DH. Bancroftian and malayan filariasis. In Binford CH, ed. Pathology of Tropical and Extraordinary Diseases. Vol 2. Fort Sam Houston, TX: Armed Forces Institute of Pathology; 1976:340-355.
- Stingl P, Ross M, Gibson DW, et al. A diagnostic “patch-test” for onchocerciasis using topical diethylcarbamazine. Trans R Soc Trop Med Hyg.
Practice Points
- Parasitic infections should always be a diagnostic consideration in individuals who present with a pruritic eruption and a history of travel.
- To control parasitic infections, research efforts should focus on improved sanitation, drug treatment, transmission prevention, and improved education of clinical manifestations to increase early detection.
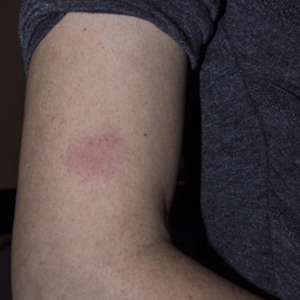
Paraneoplastic Pemphigus With Cicatricial Nail Involvement
To the Editor:
Paraneoplastic pemphigus (PNP), also known as paraneoplastic autoimmune multiorgan syndrome, is an autoimmune mucocutaneous blistering disease that typically occurs secondary to a lymphoproliferative disorder. Paraneoplastic pemphigus is characterized by severe erosive stomatitis, polymorphous skin lesions, and potential bronchiolitis obliterans that can mimic a wide array of conditions. The exact pathogenesis is unknown but is thought to be due to a combination of humoral and cell-mediated immunity. The condition usually confers a poor prognosis, with morbidity from 38% to upwards of 90%.1
A 47-year-old man developed prominent pink to dusky, ill-defined, targetoid, coalescing papules over the back; violaceous macules over the palms and soles; and numerous crusted oral erosions while hospitalized for an infection. He had a history of stage IVB follicular lymphoma (double-hit type immunoglobulin heavy chain/BCL2 fusion and rearrangement of BCL6) complicated by extensive erosive skin lesions and multiple lines of infections. The clinical differential diagnosis included Stevens-Johnson syndrome vs erythema multiforme (EM) major secondary to administration of oxacillin vs PNP. Herpes simplex virus polymerase chain reaction and Mycoplasma titers were negative. Skin biopsies from the back and right abdomen revealed severe lichenoid interface dermatitis (IFD) with numerous dyskeratotic cells mimicking EM and eosinophils; however, direct immunofluorescence of the abdomen biopsy revealed an apparent suprabasal acantholysis with intercellular C3 in the lower half of the epidermis. Histologically, PNP was favored, but indirect immunofluorescence with monkey esophagus IgG was negative.
The skin lesions progressed, and an additional skin biopsy from the left arm performed 1 month later revealed similar histologic features with intercellular IgG and C3 in the lower half of the epidermis with weak basement membrane C3 (Figure 1). Serology also confirmed elevated serum antidesmoglein 1 and 3 antibodies. Thus, in the clinical setting of an erosive mucositis with EM-like and pemphigoidlike eruptions associated with B-cell lymphoma, the patient was diagnosed with PNP.
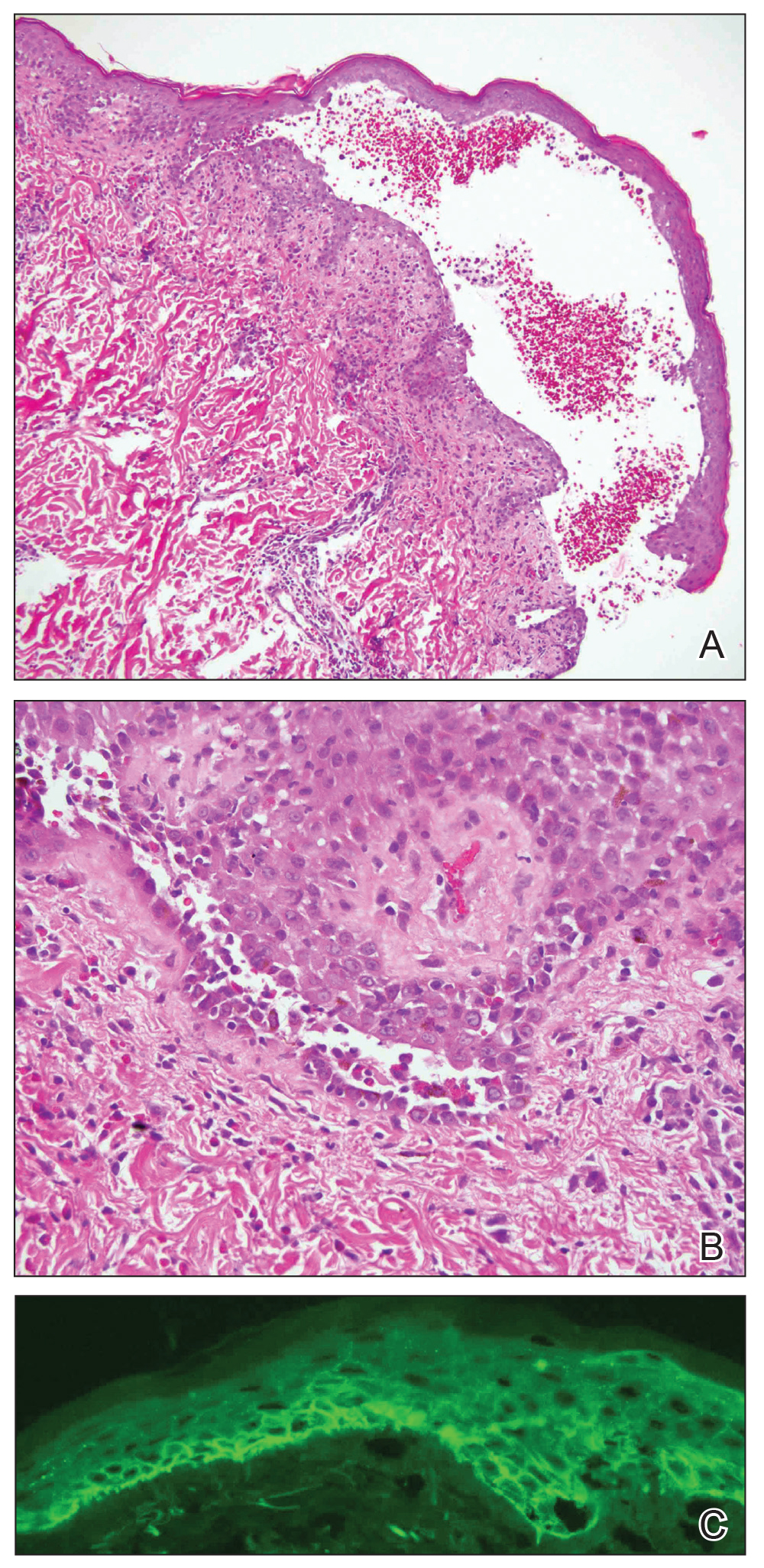
Despite multiple complications followed by intermittent treatments, the initial therapy with rituximab induction and subsequent cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone) for the B-cell lymphoma was done during his hospital stay. Toward the end of his 8-week hospitalization, the patient was noted to have new lesions involving the hands, digits, and nails. The left hand showed anonychia of several fingers with prominent scarring (Figure 2A). There were large, verrucous, crusted plaques on the distal phalanges of several fingers on the right hand (Figure 2B). At that time, he was taking 20 mg daily of prednisone (for 10 months) and had completed his 6th cycle of R-CHOP, which resulted in improvement of the skin lesions. Oral steroids were tapered, and he was maintained on rituximab infusions every 8 weeks but has since been lost to follow-up.
Paraneoplastic lymphoma is a rare condition that affects 0.012% of non-Hodgkin lymphoma and chronic lymphocytic leukemia patients.2 Reports of PNP involving the nails are even more rare, with 3 reports in the setting of underlying Castleman disease3-5 and 2 reports in patients with underlying non-Hodgkin6 and follicular1 lymphoma. These studies describe variable nail findings ranging from periungual erosions and edema, formation of dorsal pterygium, onycholysis with longitudinal furrowing, and destruction of the nail plate leading to onychomadesis and/or anonychia. These nail changes typically are seen in lichen planus or in bullous diseases affecting the basement membrane (eg, bullous pemphigoid, acquired epidermolysis bullosa) but not known in pemphigus, which is characterized by nonscarring nail changes.7
Although antidesmoglein 3 antibody was shown to be a pathologic driver in PNP, there is a weak correlation between antibody profiles and clinical presentation.8 In one case of PNP, antidesmoglein 3 antibody was negative, suggesting that lichenoid IFD may cause the phenotypic findings in PNP.9 Thus, the development of nail scarring in PNP may be explained by the presence of lichenoid IFD that is characteristic of PNP. However, the variation in antibody profile in PNP likely is a consequence of epitope spreading.
- Miest RY, Wetter DA, Drage LA, et al. A mucocutaneous eruption. Int J Dermatol. 2014;53:1425-1427.
- Anhalt GJ, Mimouni D. Paraneoplastic pemphigus. In: LA G, Katz SI, Gilchrest, eds. Fitzpatrick’s Dermatology in General Medicine 8th Edition. Vol 1. New York, NY: McGraw Hill; 2012:600.
- Chorzelski T, Hashimoto T, Maciejewska B, et al. Paraneoplastic pemphigus associated with Castleman tumor, myasthenia gravis and bronchiolitis obliterans. J Am Acad Dermatol. 1999;41:393-400.
- Lemon MA, Weston WL, Huff JC. Childhood paraneoplastic pemphigus associated with Castleman’s tumour. Br J Dermatol. 1997;136:115-117.
- Tey HL, Tang MB. A case of paraneoplastic pemphigus associated with Castleman’s disease presenting as erosive lichen planus. Clin Exp Dermatol. 2009;34:e754-e756.
- Liang JJ, Cordes SF, Witzig TE. More than skin-deep. Cleve Clin J Med. 2013;80:632-633.
- Tosti A, Andre M, Murrell DF. Nail involvement in autoimmune bullous disorders. Dermatol Clin. 2011;29:511-513, xi.
- Ohyama M, Amagai M, Hashimoto T, et al. Clinical phenotype and anti-desmoglein autoantibody profile in paraneoplastic pemphigus. J Am Acad Dermatol. 2001;44:593-598.
- Kanwar AJ, Vinay K, Varma S, et al. Anti-desmoglein antibody-negative paraneoplastic pemphigus successfully treated with rituximab. Int J Dermatol. 2015;54:576-579.
To the Editor:
Paraneoplastic pemphigus (PNP), also known as paraneoplastic autoimmune multiorgan syndrome, is an autoimmune mucocutaneous blistering disease that typically occurs secondary to a lymphoproliferative disorder. Paraneoplastic pemphigus is characterized by severe erosive stomatitis, polymorphous skin lesions, and potential bronchiolitis obliterans that can mimic a wide array of conditions. The exact pathogenesis is unknown but is thought to be due to a combination of humoral and cell-mediated immunity. The condition usually confers a poor prognosis, with morbidity from 38% to upwards of 90%.1
A 47-year-old man developed prominent pink to dusky, ill-defined, targetoid, coalescing papules over the back; violaceous macules over the palms and soles; and numerous crusted oral erosions while hospitalized for an infection. He had a history of stage IVB follicular lymphoma (double-hit type immunoglobulin heavy chain/BCL2 fusion and rearrangement of BCL6) complicated by extensive erosive skin lesions and multiple lines of infections. The clinical differential diagnosis included Stevens-Johnson syndrome vs erythema multiforme (EM) major secondary to administration of oxacillin vs PNP. Herpes simplex virus polymerase chain reaction and Mycoplasma titers were negative. Skin biopsies from the back and right abdomen revealed severe lichenoid interface dermatitis (IFD) with numerous dyskeratotic cells mimicking EM and eosinophils; however, direct immunofluorescence of the abdomen biopsy revealed an apparent suprabasal acantholysis with intercellular C3 in the lower half of the epidermis. Histologically, PNP was favored, but indirect immunofluorescence with monkey esophagus IgG was negative.
The skin lesions progressed, and an additional skin biopsy from the left arm performed 1 month later revealed similar histologic features with intercellular IgG and C3 in the lower half of the epidermis with weak basement membrane C3 (Figure 1). Serology also confirmed elevated serum antidesmoglein 1 and 3 antibodies. Thus, in the clinical setting of an erosive mucositis with EM-like and pemphigoidlike eruptions associated with B-cell lymphoma, the patient was diagnosed with PNP.
Despite multiple complications followed by intermittent treatments, the initial therapy with rituximab induction and subsequent cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone) for the B-cell lymphoma was done during his hospital stay. Toward the end of his 8-week hospitalization, the patient was noted to have new lesions involving the hands, digits, and nails. The left hand showed anonychia of several fingers with prominent scarring (Figure 2A). There were large, verrucous, crusted plaques on the distal phalanges of several fingers on the right hand (Figure 2B). At that time, he was taking 20 mg daily of prednisone (for 10 months) and had completed his 6th cycle of R-CHOP, which resulted in improvement of the skin lesions. Oral steroids were tapered, and he was maintained on rituximab infusions every 8 weeks but has since been lost to follow-up.
Paraneoplastic lymphoma is a rare condition that affects 0.012% of non-Hodgkin lymphoma and chronic lymphocytic leukemia patients.2 Reports of PNP involving the nails are even more rare, with 3 reports in the setting of underlying Castleman disease3-5 and 2 reports in patients with underlying non-Hodgkin6 and follicular1 lymphoma. These studies describe variable nail findings ranging from periungual erosions and edema, formation of dorsal pterygium, onycholysis with longitudinal furrowing, and destruction of the nail plate leading to onychomadesis and/or anonychia. These nail changes typically are seen in lichen planus or in bullous diseases affecting the basement membrane (eg, bullous pemphigoid, acquired epidermolysis bullosa) but not known in pemphigus, which is characterized by nonscarring nail changes.7
Although antidesmoglein 3 antibody was shown to be a pathologic driver in PNP, there is a weak correlation between antibody profiles and clinical presentation.8 In one case of PNP, antidesmoglein 3 antibody was negative, suggesting that lichenoid IFD may cause the phenotypic findings in PNP.9 Thus, the development of nail scarring in PNP may be explained by the presence of lichenoid IFD that is characteristic of PNP. However, the variation in antibody profile in PNP likely is a consequence of epitope spreading.
To the Editor:
Paraneoplastic pemphigus (PNP), also known as paraneoplastic autoimmune multiorgan syndrome, is an autoimmune mucocutaneous blistering disease that typically occurs secondary to a lymphoproliferative disorder. Paraneoplastic pemphigus is characterized by severe erosive stomatitis, polymorphous skin lesions, and potential bronchiolitis obliterans that can mimic a wide array of conditions. The exact pathogenesis is unknown but is thought to be due to a combination of humoral and cell-mediated immunity. The condition usually confers a poor prognosis, with morbidity from 38% to upwards of 90%.1
A 47-year-old man developed prominent pink to dusky, ill-defined, targetoid, coalescing papules over the back; violaceous macules over the palms and soles; and numerous crusted oral erosions while hospitalized for an infection. He had a history of stage IVB follicular lymphoma (double-hit type immunoglobulin heavy chain/BCL2 fusion and rearrangement of BCL6) complicated by extensive erosive skin lesions and multiple lines of infections. The clinical differential diagnosis included Stevens-Johnson syndrome vs erythema multiforme (EM) major secondary to administration of oxacillin vs PNP. Herpes simplex virus polymerase chain reaction and Mycoplasma titers were negative. Skin biopsies from the back and right abdomen revealed severe lichenoid interface dermatitis (IFD) with numerous dyskeratotic cells mimicking EM and eosinophils; however, direct immunofluorescence of the abdomen biopsy revealed an apparent suprabasal acantholysis with intercellular C3 in the lower half of the epidermis. Histologically, PNP was favored, but indirect immunofluorescence with monkey esophagus IgG was negative.
The skin lesions progressed, and an additional skin biopsy from the left arm performed 1 month later revealed similar histologic features with intercellular IgG and C3 in the lower half of the epidermis with weak basement membrane C3 (Figure 1). Serology also confirmed elevated serum antidesmoglein 1 and 3 antibodies. Thus, in the clinical setting of an erosive mucositis with EM-like and pemphigoidlike eruptions associated with B-cell lymphoma, the patient was diagnosed with PNP.
Despite multiple complications followed by intermittent treatments, the initial therapy with rituximab induction and subsequent cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone) for the B-cell lymphoma was done during his hospital stay. Toward the end of his 8-week hospitalization, the patient was noted to have new lesions involving the hands, digits, and nails. The left hand showed anonychia of several fingers with prominent scarring (Figure 2A). There were large, verrucous, crusted plaques on the distal phalanges of several fingers on the right hand (Figure 2B). At that time, he was taking 20 mg daily of prednisone (for 10 months) and had completed his 6th cycle of R-CHOP, which resulted in improvement of the skin lesions. Oral steroids were tapered, and he was maintained on rituximab infusions every 8 weeks but has since been lost to follow-up.
Paraneoplastic lymphoma is a rare condition that affects 0.012% of non-Hodgkin lymphoma and chronic lymphocytic leukemia patients.2 Reports of PNP involving the nails are even more rare, with 3 reports in the setting of underlying Castleman disease3-5 and 2 reports in patients with underlying non-Hodgkin6 and follicular1 lymphoma. These studies describe variable nail findings ranging from periungual erosions and edema, formation of dorsal pterygium, onycholysis with longitudinal furrowing, and destruction of the nail plate leading to onychomadesis and/or anonychia. These nail changes typically are seen in lichen planus or in bullous diseases affecting the basement membrane (eg, bullous pemphigoid, acquired epidermolysis bullosa) but not known in pemphigus, which is characterized by nonscarring nail changes.7
Although antidesmoglein 3 antibody was shown to be a pathologic driver in PNP, there is a weak correlation between antibody profiles and clinical presentation.8 In one case of PNP, antidesmoglein 3 antibody was negative, suggesting that lichenoid IFD may cause the phenotypic findings in PNP.9 Thus, the development of nail scarring in PNP may be explained by the presence of lichenoid IFD that is characteristic of PNP. However, the variation in antibody profile in PNP likely is a consequence of epitope spreading.
- Miest RY, Wetter DA, Drage LA, et al. A mucocutaneous eruption. Int J Dermatol. 2014;53:1425-1427.
- Anhalt GJ, Mimouni D. Paraneoplastic pemphigus. In: LA G, Katz SI, Gilchrest, eds. Fitzpatrick’s Dermatology in General Medicine 8th Edition. Vol 1. New York, NY: McGraw Hill; 2012:600.
- Chorzelski T, Hashimoto T, Maciejewska B, et al. Paraneoplastic pemphigus associated with Castleman tumor, myasthenia gravis and bronchiolitis obliterans. J Am Acad Dermatol. 1999;41:393-400.
- Lemon MA, Weston WL, Huff JC. Childhood paraneoplastic pemphigus associated with Castleman’s tumour. Br J Dermatol. 1997;136:115-117.
- Tey HL, Tang MB. A case of paraneoplastic pemphigus associated with Castleman’s disease presenting as erosive lichen planus. Clin Exp Dermatol. 2009;34:e754-e756.
- Liang JJ, Cordes SF, Witzig TE. More than skin-deep. Cleve Clin J Med. 2013;80:632-633.
- Tosti A, Andre M, Murrell DF. Nail involvement in autoimmune bullous disorders. Dermatol Clin. 2011;29:511-513, xi.
- Ohyama M, Amagai M, Hashimoto T, et al. Clinical phenotype and anti-desmoglein autoantibody profile in paraneoplastic pemphigus. J Am Acad Dermatol. 2001;44:593-598.
- Kanwar AJ, Vinay K, Varma S, et al. Anti-desmoglein antibody-negative paraneoplastic pemphigus successfully treated with rituximab. Int J Dermatol. 2015;54:576-579.
- Miest RY, Wetter DA, Drage LA, et al. A mucocutaneous eruption. Int J Dermatol. 2014;53:1425-1427.
- Anhalt GJ, Mimouni D. Paraneoplastic pemphigus. In: LA G, Katz SI, Gilchrest, eds. Fitzpatrick’s Dermatology in General Medicine 8th Edition. Vol 1. New York, NY: McGraw Hill; 2012:600.
- Chorzelski T, Hashimoto T, Maciejewska B, et al. Paraneoplastic pemphigus associated with Castleman tumor, myasthenia gravis and bronchiolitis obliterans. J Am Acad Dermatol. 1999;41:393-400.
- Lemon MA, Weston WL, Huff JC. Childhood paraneoplastic pemphigus associated with Castleman’s tumour. Br J Dermatol. 1997;136:115-117.
- Tey HL, Tang MB. A case of paraneoplastic pemphigus associated with Castleman’s disease presenting as erosive lichen planus. Clin Exp Dermatol. 2009;34:e754-e756.
- Liang JJ, Cordes SF, Witzig TE. More than skin-deep. Cleve Clin J Med. 2013;80:632-633.
- Tosti A, Andre M, Murrell DF. Nail involvement in autoimmune bullous disorders. Dermatol Clin. 2011;29:511-513, xi.
- Ohyama M, Amagai M, Hashimoto T, et al. Clinical phenotype and anti-desmoglein autoantibody profile in paraneoplastic pemphigus. J Am Acad Dermatol. 2001;44:593-598.
- Kanwar AJ, Vinay K, Varma S, et al. Anti-desmoglein antibody-negative paraneoplastic pemphigus successfully treated with rituximab. Int J Dermatol. 2015;54:576-579.
Practice Points
- Paraneoplastic pemphigus (PNP) is a rare blistering skin eruption commonly associated with an underlying malignancy.
- Paraneoplastic pemphigus generally presents with erosive stomatitis with involvement of the vermillion lip but also can involve the skin and nails.
- Nail involvement can lead to scarring of the nails and can mimic lichen planus, bullous pemphigoid, or epidermolysis bullosa of the nails. These nail changes likely are due to the pronounced lichenoid interphase dermatitis seen in PNP.
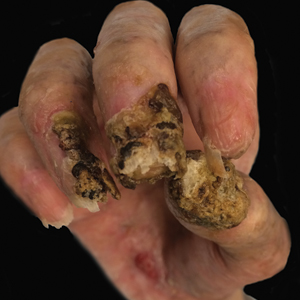
Lichen Sclerosus of the Eyelid
To the Editor:
Lichen sclerosus is a chronic inflammatory skin disease of unknown cause that predominantly affects the anogenital region, but isolated extragenital lesions occur in 6% to 15% of patients. The buttocks, thighs, neck, shoulder, upper torso, and wrists most commonly are involved; the face rarely is affected.1,2 Although the etiology of lichen sclerosus remains undetermined, there is growing evidence that autoimmunity may play a role.1 Lichen sclerosus more commonly is seen in women, and the disease can present at any age, with a bimodal onset in prepubertal children and in postmenopausal women and men in the fourth decade of life.1-3 A PubMed search of articles indexed for MEDLINE using the terms lichen and eyelid and manually screened revealed 6 cases of lichen sclerosus involving the eyelid.2-4 We describe a case of lichen sclerosus involving the eyelid and its histopathology.
A 45-year-old woman was referred to dermatology for evaluation of a right lower eyelid lesion of 3 months’ duration. She first noted a small white patch under the eyelid that had doubled in size and felt firm without bleeding or ulceration. Her medical history was unremarkable, and there was no history of ophthalmic conditions, autoimmune disease, trauma, or cancer. An ophthalmic examination was normal, except for a 20×8-mm, flat, depigmented, firm papule with scalloped borders involving the right lower eyelid margin and extending inferiorly without evidence of madarosis or ulceration (Figure 1). She underwent an incisional biopsy that revealed the diagnosis of lichen sclerosus et atrophicus (Figure 2). A full dermatologic evaluation included a genital examination and did not reveal any additional lesions. Tacrolimus ointment was started to avoid the need for long-term use of periocular steroids and their complications.
Extragenital lichen sclerosus typically is asymptomatic and only rarely presents with pruritus, in contrast to genital lichen sclerosus, which characteristically involves pruritus and dyspareunia. Although eyelid involvement is rare, ophthalmic manifestations of lichen sclerosus have included lid notching, ectropion, acquired Brown syndrome, and associated keratoconjunctivitis sicca.3-5 It characteristically appears as a well-demarcated hypopigmented papule. The differential diagnosis for a hypopigmented papule also includes amelanotic melanoma, basal cell carcinoma, vitiligo, tinea versicolor, lichen simplex chronicus, lichen planus, morphea (localized scleroderma), and systemic scleroderma with eyelid involvement.1,5
Differentiating lichen sclerosus from these conditions is of importance, as some of them can have notable morbidity and/or mortality. Of all the autoimmune connective tissue disorders, systemic sclerosus has the highest disease-specific mortality.6 Morphea, on the other hand, can have considerable morbidity. Morphea involving the head and neck notably increases the risk for neurologic complications such as seizures or central nervous system vasculitis as well as ocular complications such as anterior uveitis.6 Of note, genital lichen sclerosus carries an increased risk for squamous cell carcinoma and verrucous carcinoma; however, there have been no reported cases of malignant transformation with extragenital lesions.2
Histopathology is useful to distinguish among these entities. Although there are no specific features separating lichen sclerosus from a morphea overlap and both entities often are classified by clinical presentation, lichen sclerosus demonstrates epidermal atrophy, follicular plugging, homogenized collagen in the upper dermis with dermal edema, and lichenoid lymphocytic infiltrate (Figure 2).1 Extragenital lesions in particular also have been noted to have more epidermal atrophy and decreased rete ridges.2
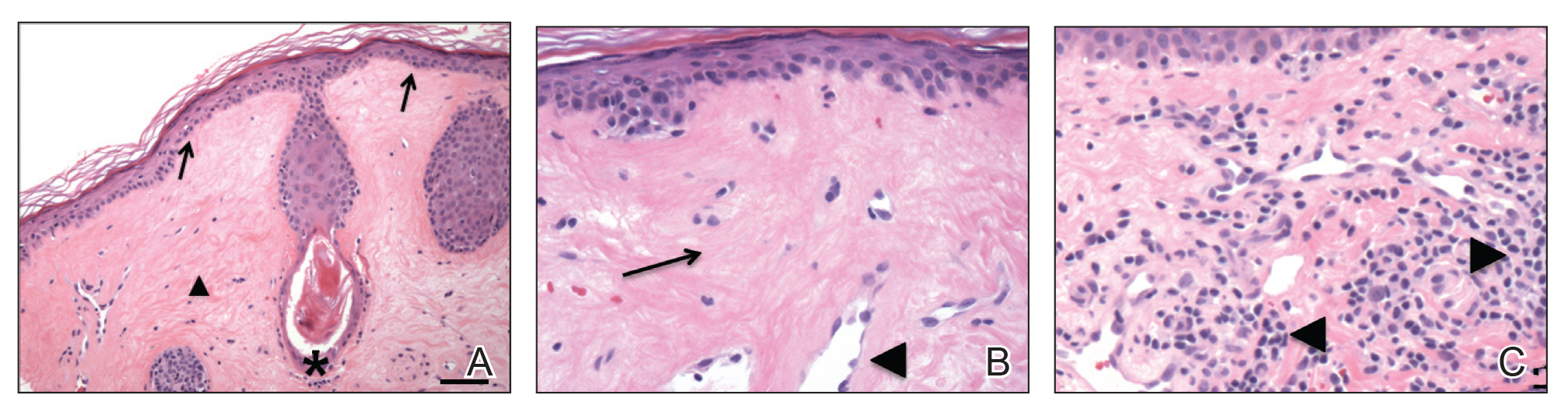
First-line treatment of lichen sclerosus includes topical corticosteroids with emollients for supportive therapy. A topical calcineurin inhibitor such as tacrolimus should be considered for patients who do not respond to corticosteroid therapy or in cases in which corticosteroid therapy is contraindicated to avoid steroid-induced glaucoma or undesirable skin atrophy and hypopigmentation.2 A collaborative approach including dermatology and internal medicine can help identify a systemic or multisystem process.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Rosenthal IM, Taube JM, Nelson DL, et al. A case of infraorbital lichen sclerosus. Dermatol Online J. 2013;19:20021.
- Rabinowitz R, Rosenthal G, Yerushalmy J, et al. Keratoconjunctivitis sicca associated with lichen sclerosus et atrophicus. Eye. 2000;14:103-104.
- Olver J, Laidler P. Acquired Brown’s syndrome in a patient with combined lichen sclerosus et atrophicus and morphea. Br J Ophthalmol. 1988;72:552-557.
- El-Baba F, Frangieh GT, Iliff WJ, et al. Morphea of the eyelids. Ophthalmology. 1982;89:125-128.
- Fett N. Scleroderma: nomenclatures, etiology, pathogenesis, prognosis, and treatment: facts and controversies. Clin Dermatol. 2013;31:432-437.
To the Editor:
Lichen sclerosus is a chronic inflammatory skin disease of unknown cause that predominantly affects the anogenital region, but isolated extragenital lesions occur in 6% to 15% of patients. The buttocks, thighs, neck, shoulder, upper torso, and wrists most commonly are involved; the face rarely is affected.1,2 Although the etiology of lichen sclerosus remains undetermined, there is growing evidence that autoimmunity may play a role.1 Lichen sclerosus more commonly is seen in women, and the disease can present at any age, with a bimodal onset in prepubertal children and in postmenopausal women and men in the fourth decade of life.1-3 A PubMed search of articles indexed for MEDLINE using the terms lichen and eyelid and manually screened revealed 6 cases of lichen sclerosus involving the eyelid.2-4 We describe a case of lichen sclerosus involving the eyelid and its histopathology.
A 45-year-old woman was referred to dermatology for evaluation of a right lower eyelid lesion of 3 months’ duration. She first noted a small white patch under the eyelid that had doubled in size and felt firm without bleeding or ulceration. Her medical history was unremarkable, and there was no history of ophthalmic conditions, autoimmune disease, trauma, or cancer. An ophthalmic examination was normal, except for a 20×8-mm, flat, depigmented, firm papule with scalloped borders involving the right lower eyelid margin and extending inferiorly without evidence of madarosis or ulceration (Figure 1). She underwent an incisional biopsy that revealed the diagnosis of lichen sclerosus et atrophicus (Figure 2). A full dermatologic evaluation included a genital examination and did not reveal any additional lesions. Tacrolimus ointment was started to avoid the need for long-term use of periocular steroids and their complications.
Extragenital lichen sclerosus typically is asymptomatic and only rarely presents with pruritus, in contrast to genital lichen sclerosus, which characteristically involves pruritus and dyspareunia. Although eyelid involvement is rare, ophthalmic manifestations of lichen sclerosus have included lid notching, ectropion, acquired Brown syndrome, and associated keratoconjunctivitis sicca.3-5 It characteristically appears as a well-demarcated hypopigmented papule. The differential diagnosis for a hypopigmented papule also includes amelanotic melanoma, basal cell carcinoma, vitiligo, tinea versicolor, lichen simplex chronicus, lichen planus, morphea (localized scleroderma), and systemic scleroderma with eyelid involvement.1,5
Differentiating lichen sclerosus from these conditions is of importance, as some of them can have notable morbidity and/or mortality. Of all the autoimmune connective tissue disorders, systemic sclerosus has the highest disease-specific mortality.6 Morphea, on the other hand, can have considerable morbidity. Morphea involving the head and neck notably increases the risk for neurologic complications such as seizures or central nervous system vasculitis as well as ocular complications such as anterior uveitis.6 Of note, genital lichen sclerosus carries an increased risk for squamous cell carcinoma and verrucous carcinoma; however, there have been no reported cases of malignant transformation with extragenital lesions.2
Histopathology is useful to distinguish among these entities. Although there are no specific features separating lichen sclerosus from a morphea overlap and both entities often are classified by clinical presentation, lichen sclerosus demonstrates epidermal atrophy, follicular plugging, homogenized collagen in the upper dermis with dermal edema, and lichenoid lymphocytic infiltrate (Figure 2).1 Extragenital lesions in particular also have been noted to have more epidermal atrophy and decreased rete ridges.2
First-line treatment of lichen sclerosus includes topical corticosteroids with emollients for supportive therapy. A topical calcineurin inhibitor such as tacrolimus should be considered for patients who do not respond to corticosteroid therapy or in cases in which corticosteroid therapy is contraindicated to avoid steroid-induced glaucoma or undesirable skin atrophy and hypopigmentation.2 A collaborative approach including dermatology and internal medicine can help identify a systemic or multisystem process.
To the Editor:
Lichen sclerosus is a chronic inflammatory skin disease of unknown cause that predominantly affects the anogenital region, but isolated extragenital lesions occur in 6% to 15% of patients. The buttocks, thighs, neck, shoulder, upper torso, and wrists most commonly are involved; the face rarely is affected.1,2 Although the etiology of lichen sclerosus remains undetermined, there is growing evidence that autoimmunity may play a role.1 Lichen sclerosus more commonly is seen in women, and the disease can present at any age, with a bimodal onset in prepubertal children and in postmenopausal women and men in the fourth decade of life.1-3 A PubMed search of articles indexed for MEDLINE using the terms lichen and eyelid and manually screened revealed 6 cases of lichen sclerosus involving the eyelid.2-4 We describe a case of lichen sclerosus involving the eyelid and its histopathology.
A 45-year-old woman was referred to dermatology for evaluation of a right lower eyelid lesion of 3 months’ duration. She first noted a small white patch under the eyelid that had doubled in size and felt firm without bleeding or ulceration. Her medical history was unremarkable, and there was no history of ophthalmic conditions, autoimmune disease, trauma, or cancer. An ophthalmic examination was normal, except for a 20×8-mm, flat, depigmented, firm papule with scalloped borders involving the right lower eyelid margin and extending inferiorly without evidence of madarosis or ulceration (Figure 1). She underwent an incisional biopsy that revealed the diagnosis of lichen sclerosus et atrophicus (Figure 2). A full dermatologic evaluation included a genital examination and did not reveal any additional lesions. Tacrolimus ointment was started to avoid the need for long-term use of periocular steroids and their complications.
Extragenital lichen sclerosus typically is asymptomatic and only rarely presents with pruritus, in contrast to genital lichen sclerosus, which characteristically involves pruritus and dyspareunia. Although eyelid involvement is rare, ophthalmic manifestations of lichen sclerosus have included lid notching, ectropion, acquired Brown syndrome, and associated keratoconjunctivitis sicca.3-5 It characteristically appears as a well-demarcated hypopigmented papule. The differential diagnosis for a hypopigmented papule also includes amelanotic melanoma, basal cell carcinoma, vitiligo, tinea versicolor, lichen simplex chronicus, lichen planus, morphea (localized scleroderma), and systemic scleroderma with eyelid involvement.1,5
Differentiating lichen sclerosus from these conditions is of importance, as some of them can have notable morbidity and/or mortality. Of all the autoimmune connective tissue disorders, systemic sclerosus has the highest disease-specific mortality.6 Morphea, on the other hand, can have considerable morbidity. Morphea involving the head and neck notably increases the risk for neurologic complications such as seizures or central nervous system vasculitis as well as ocular complications such as anterior uveitis.6 Of note, genital lichen sclerosus carries an increased risk for squamous cell carcinoma and verrucous carcinoma; however, there have been no reported cases of malignant transformation with extragenital lesions.2
Histopathology is useful to distinguish among these entities. Although there are no specific features separating lichen sclerosus from a morphea overlap and both entities often are classified by clinical presentation, lichen sclerosus demonstrates epidermal atrophy, follicular plugging, homogenized collagen in the upper dermis with dermal edema, and lichenoid lymphocytic infiltrate (Figure 2).1 Extragenital lesions in particular also have been noted to have more epidermal atrophy and decreased rete ridges.2
First-line treatment of lichen sclerosus includes topical corticosteroids with emollients for supportive therapy. A topical calcineurin inhibitor such as tacrolimus should be considered for patients who do not respond to corticosteroid therapy or in cases in which corticosteroid therapy is contraindicated to avoid steroid-induced glaucoma or undesirable skin atrophy and hypopigmentation.2 A collaborative approach including dermatology and internal medicine can help identify a systemic or multisystem process.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Rosenthal IM, Taube JM, Nelson DL, et al. A case of infraorbital lichen sclerosus. Dermatol Online J. 2013;19:20021.
- Rabinowitz R, Rosenthal G, Yerushalmy J, et al. Keratoconjunctivitis sicca associated with lichen sclerosus et atrophicus. Eye. 2000;14:103-104.
- Olver J, Laidler P. Acquired Brown’s syndrome in a patient with combined lichen sclerosus et atrophicus and morphea. Br J Ophthalmol. 1988;72:552-557.
- El-Baba F, Frangieh GT, Iliff WJ, et al. Morphea of the eyelids. Ophthalmology. 1982;89:125-128.
- Fett N. Scleroderma: nomenclatures, etiology, pathogenesis, prognosis, and treatment: facts and controversies. Clin Dermatol. 2013;31:432-437.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Rosenthal IM, Taube JM, Nelson DL, et al. A case of infraorbital lichen sclerosus. Dermatol Online J. 2013;19:20021.
- Rabinowitz R, Rosenthal G, Yerushalmy J, et al. Keratoconjunctivitis sicca associated with lichen sclerosus et atrophicus. Eye. 2000;14:103-104.
- Olver J, Laidler P. Acquired Brown’s syndrome in a patient with combined lichen sclerosus et atrophicus and morphea. Br J Ophthalmol. 1988;72:552-557.
- El-Baba F, Frangieh GT, Iliff WJ, et al. Morphea of the eyelids. Ophthalmology. 1982;89:125-128.
- Fett N. Scleroderma: nomenclatures, etiology, pathogenesis, prognosis, and treatment: facts and controversies. Clin Dermatol. 2013;31:432-437.
Practice Points
- Lichen sclerosus is not confined to only the anogenital area and can affect the face in rare cases.