User login
Isolated Scrotal Granular Parakeratosis: An Atypical Clinical Presentation
To the Editor:
Granular parakeratosis is a rare condition with an unclear etiology that results from a myriad of factors, including exposure to irritants, friction, moisture, and heat. The diagnosis is made based on a distinct histologic reaction pattern that may be protective against the triggers. We present a case of isolated scrotal granular parakeratosis in a patient with compensatory hyperhidrosis after endoscopic thoracic sympathectomy.
A 52-year-old man presented with a 5-year history of a recurrent rash affecting the scrotum. He experienced monthly flares that were exacerbated by inguinal hyperhidrosis. His symptoms included a burning sensation and pruritus followed by superficial desquamation, with gradual yet temporary improvement. His medical history was remarkable for primary axillary and palmoplantar hyperhidrosis, with compensatory inguinal hyperhidrosis after endoscopic thoracic sympathectomy 8 years prior to presentation.
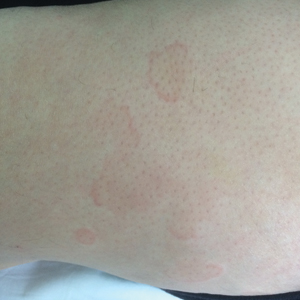
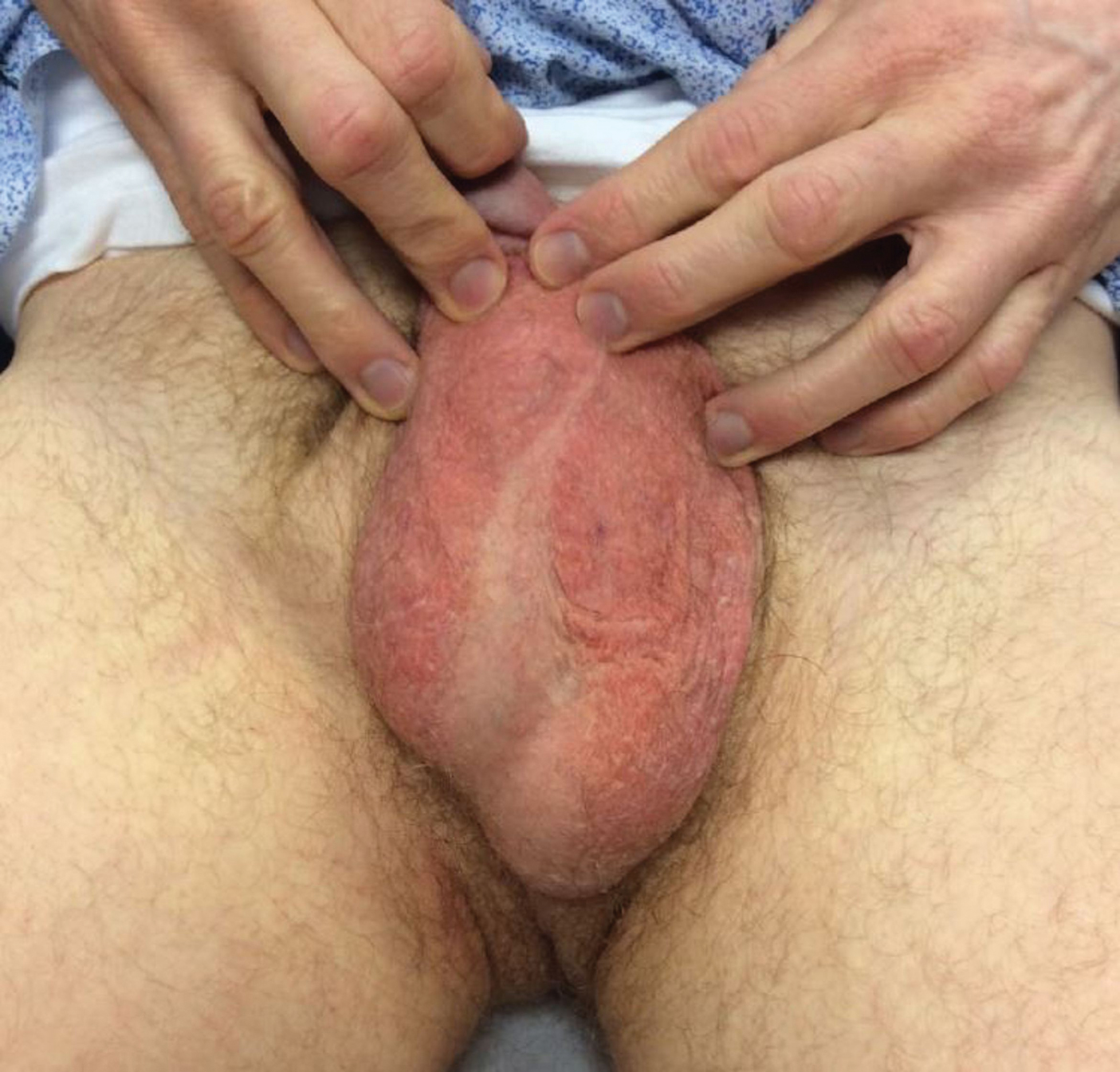
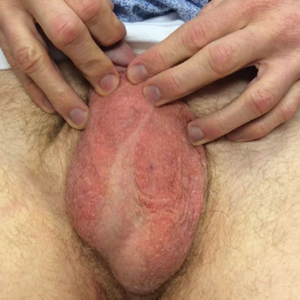
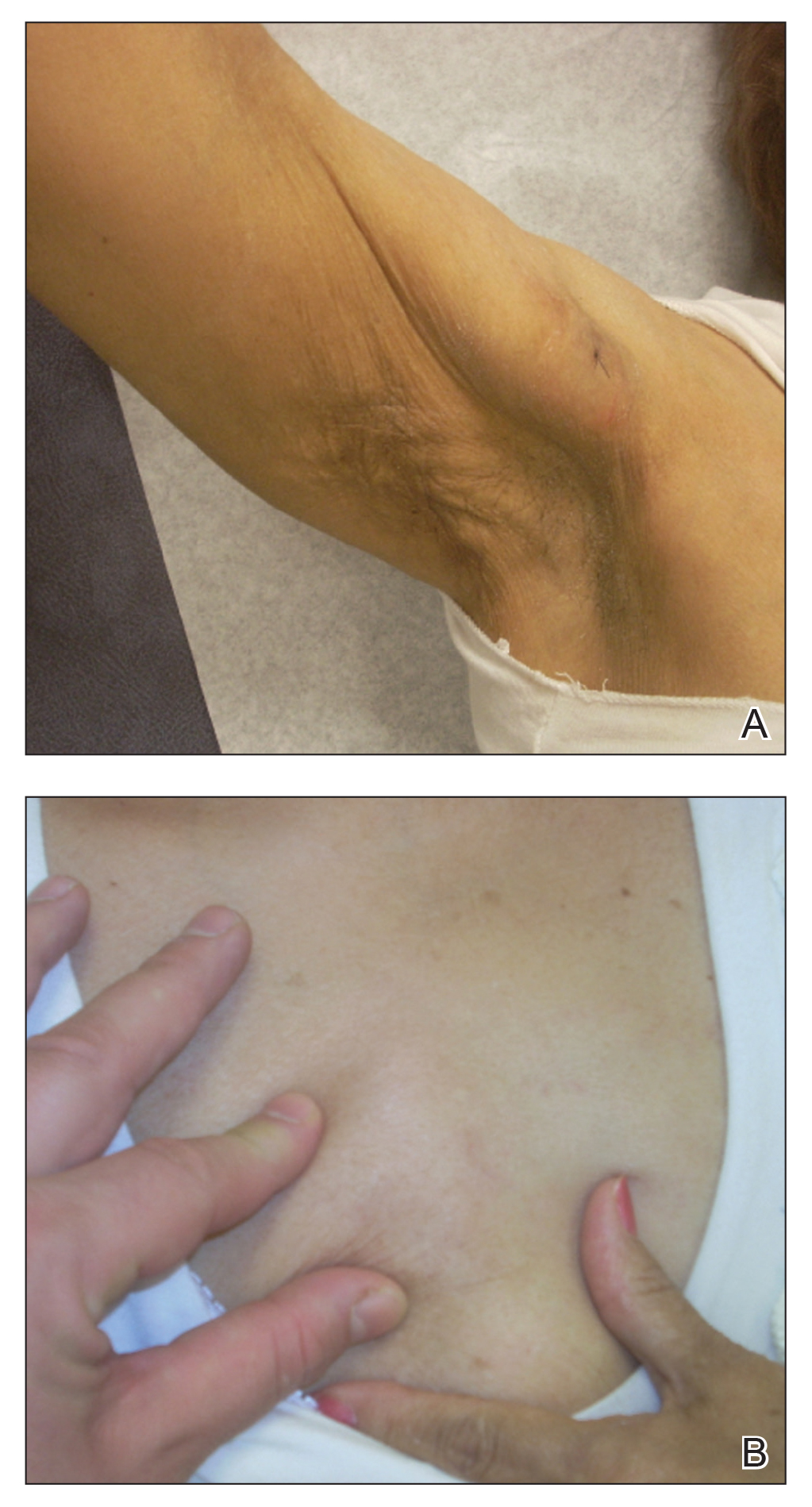
Physical examination revealed a well-demarcated, scaly, erythematous plaque affecting the scrotal skin with sparing of the median raphe, penis, and inguinal folds (Figure 1). There were no other lesions noted in the axillary region or other skin folds.
Prior treatments prescribed by other providers included topical pimecrolimus, antifungal creams, topical corticosteroids, zinc oxide ointment, and daily application of an over-the-counter medicated powder with no resolution.
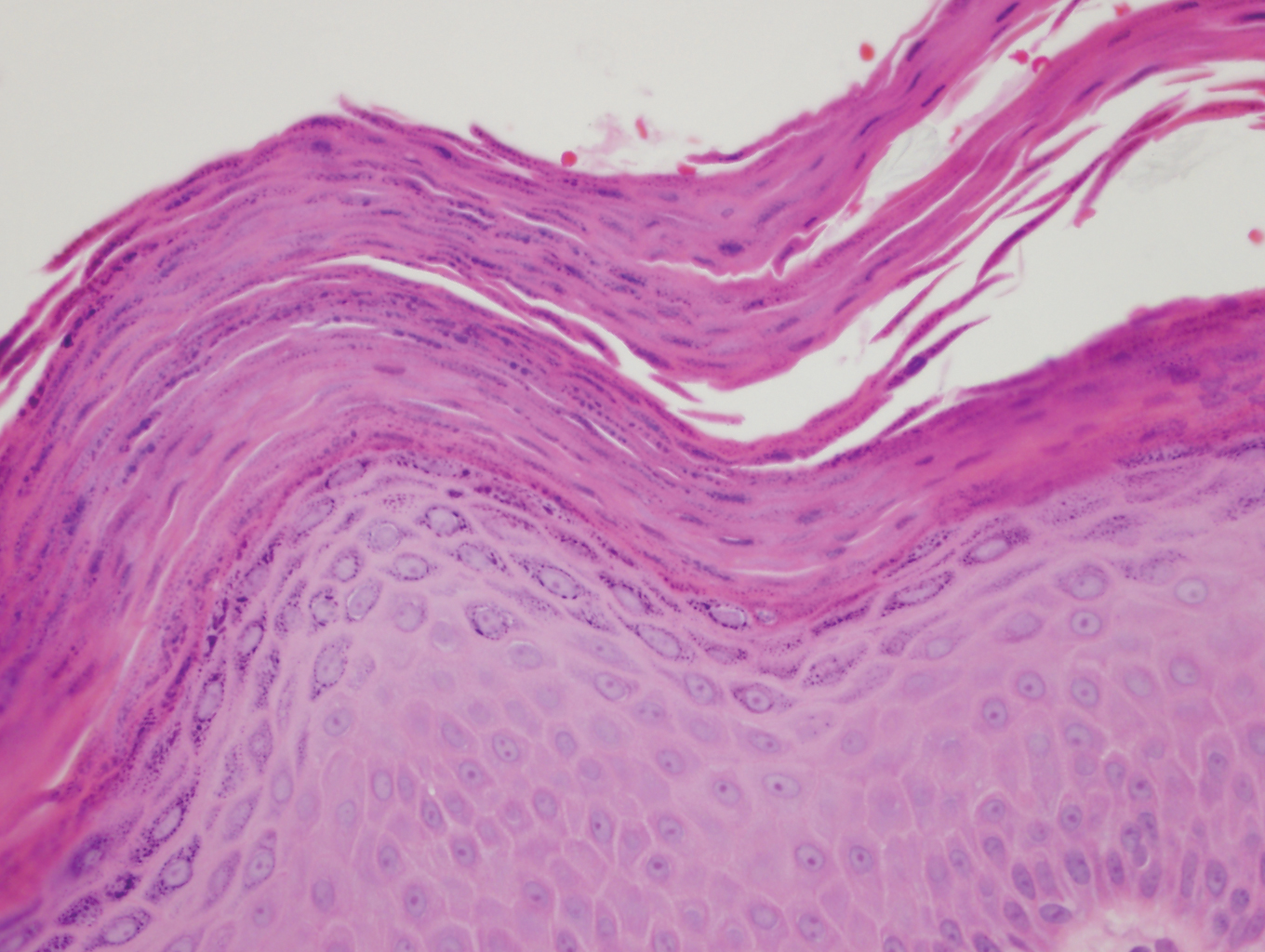
A punch biopsy performed at the current presentation showed psoriasiform hyperplasia of the epidermis with only a focally diminished granular layer. There was overlying thick parakeratosis and retention of keratohyalin granules (Figure 2). Grocott-Gomori methenamine- silver staining was negative for fungal elements in the sections examined. Clinical history, morphology of the eruption, and histologic features were consistent with granular parakeratosis.
Since the first reported incident of granular parakeratosis of the axilla in 1991,1 granular parakeratosis has been reported in other intertriginous areas, including the inframammary folds, inguinal folds, genitalia, perianal skin, and beneath the abdominal pannus.2 One case study in 1998 reported a patient with isolated involvement of the inguinal region3; however, this presentation is rare.4 This condition has been reported in both sexes and all age groups, including children.5
Granular parakeratosis classically presents as erythematous to brown hyperkeratotic papules that coalesce into plaques.6 It is thought to be a reactive inflammatory condition secondary to aggravating factors such as exposure to heat,7 moisture, and friction; skin occlusion; repeated washing; irritation from external agents; antiperspirants; and use of depilatory creams.8 Histopathology is characteristic and consists of retained nuclei and keratohyalin granules within the stratum corneum, beneath which there is a retained stratum granulosum. Epidermal changes may be varied and include atrophy or hyperplasia.
Murine models have postulated that granular parakeratosis may result from a deficiency in caspase 14, a protease vital to the formation of a well-functioning skin barrier.9 A cornified envelope often is noted in granular parakeratotic cells with no defects in desmosomes and cell membranes, suggesting that the pathogenesis lies within processing of profilaggrin to filaggrin, resulting in a failure to degrade keratohyalin granules and aggregation of keratin filaments.10 Granular parakeratosis is not known to be associated with other medical conditions, but it has been observed in patients receiving chemotherapy for breast11 and ovarian12 carcinomas. In infants with atopic dermatitis, granular parakeratosis was reported in 5 out of 7 cases.6 In our patient with secondary inguinal hyperhidrosis after thoracic sympathectomy, granular parakeratosis may be reactive to excess sweating and friction in the scrotal area.
Granular parakeratosis follows a waxing and waning pattern that may spontaneously resolve without any treatment; it also can follow a protracted course, as in a case with associated facial papules that persisted for 20 years.13 Topical corticosteroids alone or in combination with topical antifungal agents have been used for the treatment of granular parakeratosis with the goal of accelerating resolution.2,14 However, the efficacy of these therapeutic interventions is limited, and no controlled trials are underway. Topical vitamin D analogues15,16 and topical retinoids17 also have been reported with successful outcomes. Spontaneous resolution also has been observed in 2 different cases after previously being unresponsive to topical treatment.18,19 Treatment with Clostridium botulinum toxin A resulted in complete remission of the disease observed at 6-month follow-up. The pharmacologic action of the neurotoxin disrupts the stimulation of eccrine sweat glands, resulting in decreased sweating, a known exacerbating factor of granular parakeratosis.20
In summary, our case represents a unique clinical presentation of granular parakeratosis with classic histopathologic features. A high index of suspicion and a biopsy are vital to arriving at the correct diagnosis.
- Northcutt AD, Nelson DM, Tschen JA. Axillary granular parakeratosis. J Am Acad Dermatol. 1991;24:541-544.
- Burford C. Granular parakeratosis of multiple intertriginous areas. Australas J Dermatol. 2008;49:35-38.
- Mehregan DA, Thomas JE, Mehregan DR. Intertriginous granular parakeratosis. J Am Acad Dermatol. 1998;39:495-496.
- Leclerc-Mercier S, Prost-Squarcioni C, Hamel-Teillac D, et al. A case of congenital granular parakeratosis. Am J Dermatopathol. 2011;33:531-533.
- Scheinfeld NS, Mones J. Granular parakeratosis: pathologic and clinical correlation of 18 cases of granular parakeratosis. J Am Acad Dermatol. 2005;52:863-867.
- Akkaya AD, Oram Y, Aydin O. Infantile granular parakeratosis: cytologic examination of superficial scrapings as an aid to diagnosis. Pediatr Dermatol. 2015;32:392-396.
- Rodríguez G. Axillary granular parakeratosis [in Spanish]. Biomedica. 2002;22:519-523.
- Samrao A, Reis M, Niedt G, et al. Granular parakeratosis: response to calcipotriene and brief review of current therapeutic options. Skinmed. 2010;8:357-359.
- Hoste E, Denecker G, Gilbert B, et al. Caspase-14-deficient mice are more prone to the development of parakeratosis. J Invest Dermatol. 2013;133:742-750.
- Metze D, Rutten A. Granular parakeratosis—a unique acquired disorder of keratinization. J Cutan Pathol. 1999;26:339-352.
- Wallace CA, Pichardo RO, Yosipovitch G, et al. Granular parakeratosis: a case report and literature review. J Cutan Pathol. 2003;30:332-335.
- Jaconelli L, Doebelin B, Kanitakis J, et al. Granular parakeratosis in a patient treated with liposomal doxorubicin for ovarian carcinoma. J Am Acad Dermatol. 2008;58(5 suppl 1):S84-S87.
- Reddy IS, Swarnalata G, Mody T. Intertriginous granular parakeratosis persisting for 20 years. Indian J Dermatol Venereol Leprol. 2008;74:405-407.
- Dearden C, al-Nakib W, Andries K, et al. Drug resistant rhinoviruses from the nose of experimentally treated volunteers. Arch Virol. 1989;109:71-81.
- Patel U, Patel T, Skinner RB Jr. Resolution of granular parakeratosis with topical calcitriol. Arch Dermatol. 2011;147:997-998.
- Contreras ME, Gottfried LC, Bang RH, et al. Axillary intertriginous granular parakeratosis responsive to topical calcipotriene and ammonium lactate. Int J Dermatol. 2003;42:382-383.
- Brown SK, Heilman ER. Granular parakeratosis: resolution with topical tretinoin. J Am Acad Dermatol. 2002;47(5 suppl):S279-S280.
- Compton AK, Jackson JM. Isotretinoin as a treatment for axillary granular parakeratosis. Cutis. 2007;80:55-56.
- Webster CG, Resnik KS, Webster GF. Axillary granular parakeratosis: response to isotretinoin. J Am Acad Dermatol. 1997; 37:789-790.
- Ravitskiy L, Heymann WR. Botulinum toxin-induced resolution of axillary granular parakeratosis. Skinmed. 2005;4:118-120.
To the Editor:
Granular parakeratosis is a rare condition with an unclear etiology that results from a myriad of factors, including exposure to irritants, friction, moisture, and heat. The diagnosis is made based on a distinct histologic reaction pattern that may be protective against the triggers. We present a case of isolated scrotal granular parakeratosis in a patient with compensatory hyperhidrosis after endoscopic thoracic sympathectomy.
A 52-year-old man presented with a 5-year history of a recurrent rash affecting the scrotum. He experienced monthly flares that were exacerbated by inguinal hyperhidrosis. His symptoms included a burning sensation and pruritus followed by superficial desquamation, with gradual yet temporary improvement. His medical history was remarkable for primary axillary and palmoplantar hyperhidrosis, with compensatory inguinal hyperhidrosis after endoscopic thoracic sympathectomy 8 years prior to presentation.
Physical examination revealed a well-demarcated, scaly, erythematous plaque affecting the scrotal skin with sparing of the median raphe, penis, and inguinal folds (Figure 1). There were no other lesions noted in the axillary region or other skin folds.
Prior treatments prescribed by other providers included topical pimecrolimus, antifungal creams, topical corticosteroids, zinc oxide ointment, and daily application of an over-the-counter medicated powder with no resolution.
A punch biopsy performed at the current presentation showed psoriasiform hyperplasia of the epidermis with only a focally diminished granular layer. There was overlying thick parakeratosis and retention of keratohyalin granules (Figure 2). Grocott-Gomori methenamine- silver staining was negative for fungal elements in the sections examined. Clinical history, morphology of the eruption, and histologic features were consistent with granular parakeratosis.
Since the first reported incident of granular parakeratosis of the axilla in 1991,1 granular parakeratosis has been reported in other intertriginous areas, including the inframammary folds, inguinal folds, genitalia, perianal skin, and beneath the abdominal pannus.2 One case study in 1998 reported a patient with isolated involvement of the inguinal region3; however, this presentation is rare.4 This condition has been reported in both sexes and all age groups, including children.5
Granular parakeratosis classically presents as erythematous to brown hyperkeratotic papules that coalesce into plaques.6 It is thought to be a reactive inflammatory condition secondary to aggravating factors such as exposure to heat,7 moisture, and friction; skin occlusion; repeated washing; irritation from external agents; antiperspirants; and use of depilatory creams.8 Histopathology is characteristic and consists of retained nuclei and keratohyalin granules within the stratum corneum, beneath which there is a retained stratum granulosum. Epidermal changes may be varied and include atrophy or hyperplasia.
Murine models have postulated that granular parakeratosis may result from a deficiency in caspase 14, a protease vital to the formation of a well-functioning skin barrier.9 A cornified envelope often is noted in granular parakeratotic cells with no defects in desmosomes and cell membranes, suggesting that the pathogenesis lies within processing of profilaggrin to filaggrin, resulting in a failure to degrade keratohyalin granules and aggregation of keratin filaments.10 Granular parakeratosis is not known to be associated with other medical conditions, but it has been observed in patients receiving chemotherapy for breast11 and ovarian12 carcinomas. In infants with atopic dermatitis, granular parakeratosis was reported in 5 out of 7 cases.6 In our patient with secondary inguinal hyperhidrosis after thoracic sympathectomy, granular parakeratosis may be reactive to excess sweating and friction in the scrotal area.
Granular parakeratosis follows a waxing and waning pattern that may spontaneously resolve without any treatment; it also can follow a protracted course, as in a case with associated facial papules that persisted for 20 years.13 Topical corticosteroids alone or in combination with topical antifungal agents have been used for the treatment of granular parakeratosis with the goal of accelerating resolution.2,14 However, the efficacy of these therapeutic interventions is limited, and no controlled trials are underway. Topical vitamin D analogues15,16 and topical retinoids17 also have been reported with successful outcomes. Spontaneous resolution also has been observed in 2 different cases after previously being unresponsive to topical treatment.18,19 Treatment with Clostridium botulinum toxin A resulted in complete remission of the disease observed at 6-month follow-up. The pharmacologic action of the neurotoxin disrupts the stimulation of eccrine sweat glands, resulting in decreased sweating, a known exacerbating factor of granular parakeratosis.20
In summary, our case represents a unique clinical presentation of granular parakeratosis with classic histopathologic features. A high index of suspicion and a biopsy are vital to arriving at the correct diagnosis.
To the Editor:
Granular parakeratosis is a rare condition with an unclear etiology that results from a myriad of factors, including exposure to irritants, friction, moisture, and heat. The diagnosis is made based on a distinct histologic reaction pattern that may be protective against the triggers. We present a case of isolated scrotal granular parakeratosis in a patient with compensatory hyperhidrosis after endoscopic thoracic sympathectomy.
A 52-year-old man presented with a 5-year history of a recurrent rash affecting the scrotum. He experienced monthly flares that were exacerbated by inguinal hyperhidrosis. His symptoms included a burning sensation and pruritus followed by superficial desquamation, with gradual yet temporary improvement. His medical history was remarkable for primary axillary and palmoplantar hyperhidrosis, with compensatory inguinal hyperhidrosis after endoscopic thoracic sympathectomy 8 years prior to presentation.
Physical examination revealed a well-demarcated, scaly, erythematous plaque affecting the scrotal skin with sparing of the median raphe, penis, and inguinal folds (Figure 1). There were no other lesions noted in the axillary region or other skin folds.
Prior treatments prescribed by other providers included topical pimecrolimus, antifungal creams, topical corticosteroids, zinc oxide ointment, and daily application of an over-the-counter medicated powder with no resolution.
A punch biopsy performed at the current presentation showed psoriasiform hyperplasia of the epidermis with only a focally diminished granular layer. There was overlying thick parakeratosis and retention of keratohyalin granules (Figure 2). Grocott-Gomori methenamine- silver staining was negative for fungal elements in the sections examined. Clinical history, morphology of the eruption, and histologic features were consistent with granular parakeratosis.
Since the first reported incident of granular parakeratosis of the axilla in 1991,1 granular parakeratosis has been reported in other intertriginous areas, including the inframammary folds, inguinal folds, genitalia, perianal skin, and beneath the abdominal pannus.2 One case study in 1998 reported a patient with isolated involvement of the inguinal region3; however, this presentation is rare.4 This condition has been reported in both sexes and all age groups, including children.5
Granular parakeratosis classically presents as erythematous to brown hyperkeratotic papules that coalesce into plaques.6 It is thought to be a reactive inflammatory condition secondary to aggravating factors such as exposure to heat,7 moisture, and friction; skin occlusion; repeated washing; irritation from external agents; antiperspirants; and use of depilatory creams.8 Histopathology is characteristic and consists of retained nuclei and keratohyalin granules within the stratum corneum, beneath which there is a retained stratum granulosum. Epidermal changes may be varied and include atrophy or hyperplasia.
Murine models have postulated that granular parakeratosis may result from a deficiency in caspase 14, a protease vital to the formation of a well-functioning skin barrier.9 A cornified envelope often is noted in granular parakeratotic cells with no defects in desmosomes and cell membranes, suggesting that the pathogenesis lies within processing of profilaggrin to filaggrin, resulting in a failure to degrade keratohyalin granules and aggregation of keratin filaments.10 Granular parakeratosis is not known to be associated with other medical conditions, but it has been observed in patients receiving chemotherapy for breast11 and ovarian12 carcinomas. In infants with atopic dermatitis, granular parakeratosis was reported in 5 out of 7 cases.6 In our patient with secondary inguinal hyperhidrosis after thoracic sympathectomy, granular parakeratosis may be reactive to excess sweating and friction in the scrotal area.
Granular parakeratosis follows a waxing and waning pattern that may spontaneously resolve without any treatment; it also can follow a protracted course, as in a case with associated facial papules that persisted for 20 years.13 Topical corticosteroids alone or in combination with topical antifungal agents have been used for the treatment of granular parakeratosis with the goal of accelerating resolution.2,14 However, the efficacy of these therapeutic interventions is limited, and no controlled trials are underway. Topical vitamin D analogues15,16 and topical retinoids17 also have been reported with successful outcomes. Spontaneous resolution also has been observed in 2 different cases after previously being unresponsive to topical treatment.18,19 Treatment with Clostridium botulinum toxin A resulted in complete remission of the disease observed at 6-month follow-up. The pharmacologic action of the neurotoxin disrupts the stimulation of eccrine sweat glands, resulting in decreased sweating, a known exacerbating factor of granular parakeratosis.20
In summary, our case represents a unique clinical presentation of granular parakeratosis with classic histopathologic features. A high index of suspicion and a biopsy are vital to arriving at the correct diagnosis.
- Northcutt AD, Nelson DM, Tschen JA. Axillary granular parakeratosis. J Am Acad Dermatol. 1991;24:541-544.
- Burford C. Granular parakeratosis of multiple intertriginous areas. Australas J Dermatol. 2008;49:35-38.
- Mehregan DA, Thomas JE, Mehregan DR. Intertriginous granular parakeratosis. J Am Acad Dermatol. 1998;39:495-496.
- Leclerc-Mercier S, Prost-Squarcioni C, Hamel-Teillac D, et al. A case of congenital granular parakeratosis. Am J Dermatopathol. 2011;33:531-533.
- Scheinfeld NS, Mones J. Granular parakeratosis: pathologic and clinical correlation of 18 cases of granular parakeratosis. J Am Acad Dermatol. 2005;52:863-867.
- Akkaya AD, Oram Y, Aydin O. Infantile granular parakeratosis: cytologic examination of superficial scrapings as an aid to diagnosis. Pediatr Dermatol. 2015;32:392-396.
- Rodríguez G. Axillary granular parakeratosis [in Spanish]. Biomedica. 2002;22:519-523.
- Samrao A, Reis M, Niedt G, et al. Granular parakeratosis: response to calcipotriene and brief review of current therapeutic options. Skinmed. 2010;8:357-359.
- Hoste E, Denecker G, Gilbert B, et al. Caspase-14-deficient mice are more prone to the development of parakeratosis. J Invest Dermatol. 2013;133:742-750.
- Metze D, Rutten A. Granular parakeratosis—a unique acquired disorder of keratinization. J Cutan Pathol. 1999;26:339-352.
- Wallace CA, Pichardo RO, Yosipovitch G, et al. Granular parakeratosis: a case report and literature review. J Cutan Pathol. 2003;30:332-335.
- Jaconelli L, Doebelin B, Kanitakis J, et al. Granular parakeratosis in a patient treated with liposomal doxorubicin for ovarian carcinoma. J Am Acad Dermatol. 2008;58(5 suppl 1):S84-S87.
- Reddy IS, Swarnalata G, Mody T. Intertriginous granular parakeratosis persisting for 20 years. Indian J Dermatol Venereol Leprol. 2008;74:405-407.
- Dearden C, al-Nakib W, Andries K, et al. Drug resistant rhinoviruses from the nose of experimentally treated volunteers. Arch Virol. 1989;109:71-81.
- Patel U, Patel T, Skinner RB Jr. Resolution of granular parakeratosis with topical calcitriol. Arch Dermatol. 2011;147:997-998.
- Contreras ME, Gottfried LC, Bang RH, et al. Axillary intertriginous granular parakeratosis responsive to topical calcipotriene and ammonium lactate. Int J Dermatol. 2003;42:382-383.
- Brown SK, Heilman ER. Granular parakeratosis: resolution with topical tretinoin. J Am Acad Dermatol. 2002;47(5 suppl):S279-S280.
- Compton AK, Jackson JM. Isotretinoin as a treatment for axillary granular parakeratosis. Cutis. 2007;80:55-56.
- Webster CG, Resnik KS, Webster GF. Axillary granular parakeratosis: response to isotretinoin. J Am Acad Dermatol. 1997; 37:789-790.
- Ravitskiy L, Heymann WR. Botulinum toxin-induced resolution of axillary granular parakeratosis. Skinmed. 2005;4:118-120.
- Northcutt AD, Nelson DM, Tschen JA. Axillary granular parakeratosis. J Am Acad Dermatol. 1991;24:541-544.
- Burford C. Granular parakeratosis of multiple intertriginous areas. Australas J Dermatol. 2008;49:35-38.
- Mehregan DA, Thomas JE, Mehregan DR. Intertriginous granular parakeratosis. J Am Acad Dermatol. 1998;39:495-496.
- Leclerc-Mercier S, Prost-Squarcioni C, Hamel-Teillac D, et al. A case of congenital granular parakeratosis. Am J Dermatopathol. 2011;33:531-533.
- Scheinfeld NS, Mones J. Granular parakeratosis: pathologic and clinical correlation of 18 cases of granular parakeratosis. J Am Acad Dermatol. 2005;52:863-867.
- Akkaya AD, Oram Y, Aydin O. Infantile granular parakeratosis: cytologic examination of superficial scrapings as an aid to diagnosis. Pediatr Dermatol. 2015;32:392-396.
- Rodríguez G. Axillary granular parakeratosis [in Spanish]. Biomedica. 2002;22:519-523.
- Samrao A, Reis M, Niedt G, et al. Granular parakeratosis: response to calcipotriene and brief review of current therapeutic options. Skinmed. 2010;8:357-359.
- Hoste E, Denecker G, Gilbert B, et al. Caspase-14-deficient mice are more prone to the development of parakeratosis. J Invest Dermatol. 2013;133:742-750.
- Metze D, Rutten A. Granular parakeratosis—a unique acquired disorder of keratinization. J Cutan Pathol. 1999;26:339-352.
- Wallace CA, Pichardo RO, Yosipovitch G, et al. Granular parakeratosis: a case report and literature review. J Cutan Pathol. 2003;30:332-335.
- Jaconelli L, Doebelin B, Kanitakis J, et al. Granular parakeratosis in a patient treated with liposomal doxorubicin for ovarian carcinoma. J Am Acad Dermatol. 2008;58(5 suppl 1):S84-S87.
- Reddy IS, Swarnalata G, Mody T. Intertriginous granular parakeratosis persisting for 20 years. Indian J Dermatol Venereol Leprol. 2008;74:405-407.
- Dearden C, al-Nakib W, Andries K, et al. Drug resistant rhinoviruses from the nose of experimentally treated volunteers. Arch Virol. 1989;109:71-81.
- Patel U, Patel T, Skinner RB Jr. Resolution of granular parakeratosis with topical calcitriol. Arch Dermatol. 2011;147:997-998.
- Contreras ME, Gottfried LC, Bang RH, et al. Axillary intertriginous granular parakeratosis responsive to topical calcipotriene and ammonium lactate. Int J Dermatol. 2003;42:382-383.
- Brown SK, Heilman ER. Granular parakeratosis: resolution with topical tretinoin. J Am Acad Dermatol. 2002;47(5 suppl):S279-S280.
- Compton AK, Jackson JM. Isotretinoin as a treatment for axillary granular parakeratosis. Cutis. 2007;80:55-56.
- Webster CG, Resnik KS, Webster GF. Axillary granular parakeratosis: response to isotretinoin. J Am Acad Dermatol. 1997; 37:789-790.
- Ravitskiy L, Heymann WR. Botulinum toxin-induced resolution of axillary granular parakeratosis. Skinmed. 2005;4:118-120.
Practice Points
- Granular parakeratosis can occur in response to triggers such as irritants, friction, hyperhidrosis, and heat.
- Granular parakeratosis can have an atypical presentation; therefore, a high index of suspicion and punch biopsy are vital to arrive at the correct diagnosis.
- Classic histopathology demonstrates retained nuclei and keratohyalin granules within the stratum corneum beneath which there is a retained stratum granulosum.
Fulminant Hemorrhagic Bullae of the Upper Extremities Arising in the Setting of IV Placement During Severe COVID-19 Infection: Observations From a Major Consultative Practice
To the Editor:
A range of dermatologic manifestations of COVID-19 have been reported, including nonspecific maculopapular exanthems, urticaria, and varicellalike eruptions.1 Additionally, there have been sporadic accounts of cutaneous vasculopathic signs such as perniolike lesions, acro-ischemia, livedo reticularis, and retiform purpura.2 We describe exuberant hemorrhagic bullae occurring on the extremities of 2 critically ill patients with COVID-19. We hypothesized that the bullae were vasculopathic in nature and possibly exacerbated by peripheral intravenous (IV)–related injury.
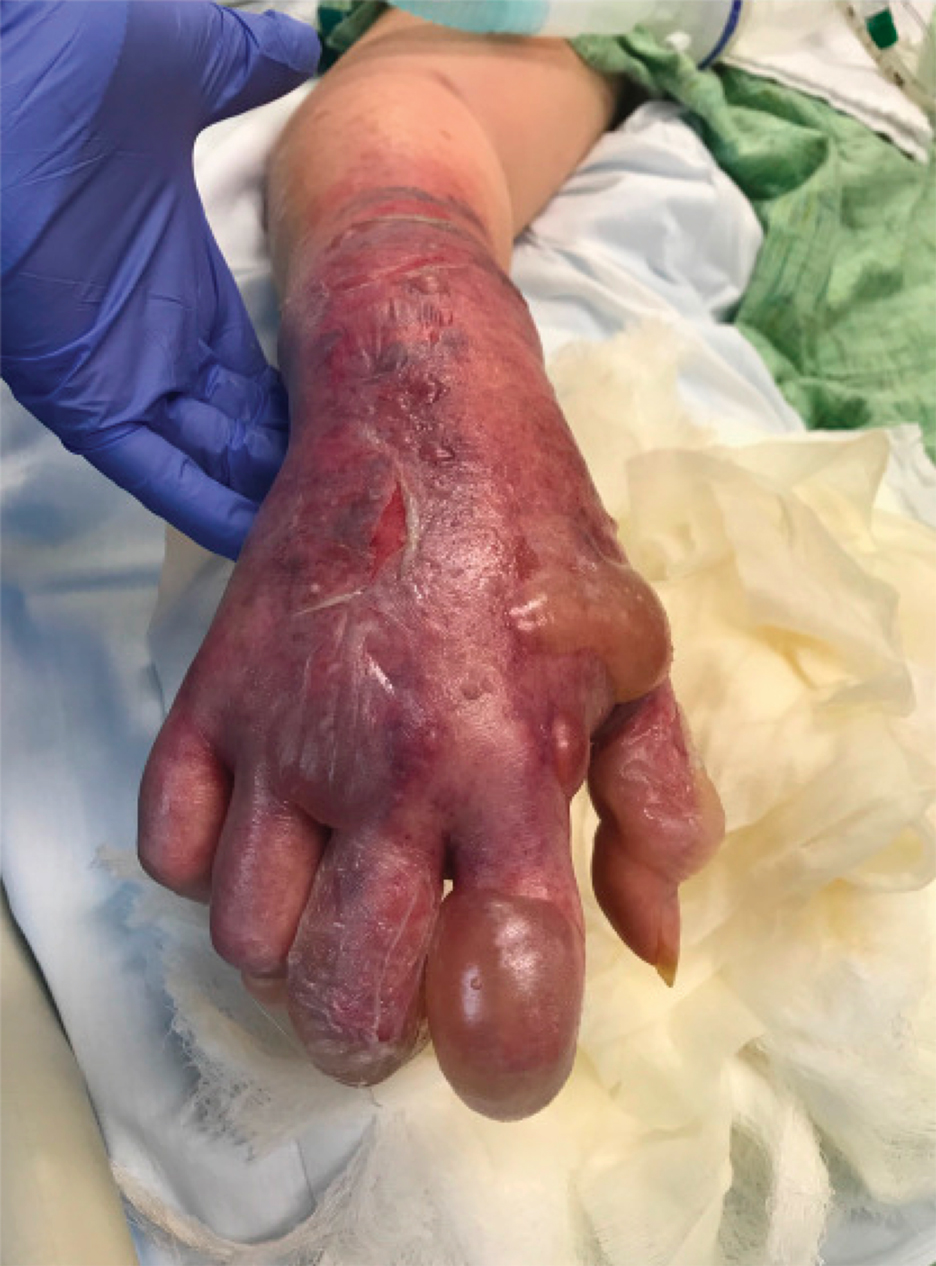
A 62-year-old woman with a history of diabetes mellitus and chronic obstructive pulmonary disease was admitted to the intensive care unit for acute hypoxemic respiratory failure secondary to COVID-19 infection. Dermatology was consulted for evaluation of blisters on the right arm. A new peripheral IV line was inserted into the patient’s right forearm for treatment of secondary methicillin-resistant Staphylococcus aureus pneumonia. The peripheral IV was inserted into the right proximal forearm for 2 days prior to development of ecchymosis and blisters. Intravenous medications included vancomycin, cefepime, methylprednisolone, and famotidine, as well as maintenance fluids (normal saline). Physical examination revealed extensive confluent ecchymoses with overlying tense bullae (Figure 1). Notable laboratory findings included an elevated D-dimer (peak of 8.67 μg/mL fibrinogen-equivalent units [FEUs], reference range <0.5 μg/mL FEU) and fibrinogen (789 mg/dL, reference range 200–400 mg/dL) levels. Three days later she developed worsening edema of the right arm, accompanied by more extensive bullae formation (Figure 2). Computed tomography of the right arm showed extensive subcutaneous stranding and subcutaneous edema. An orthopedic consultation determined that there was no compartment syndrome, and surgical intervention was not recommended. The patient’s course was complicated by multiorgan failure, and she died 18 days after admission.
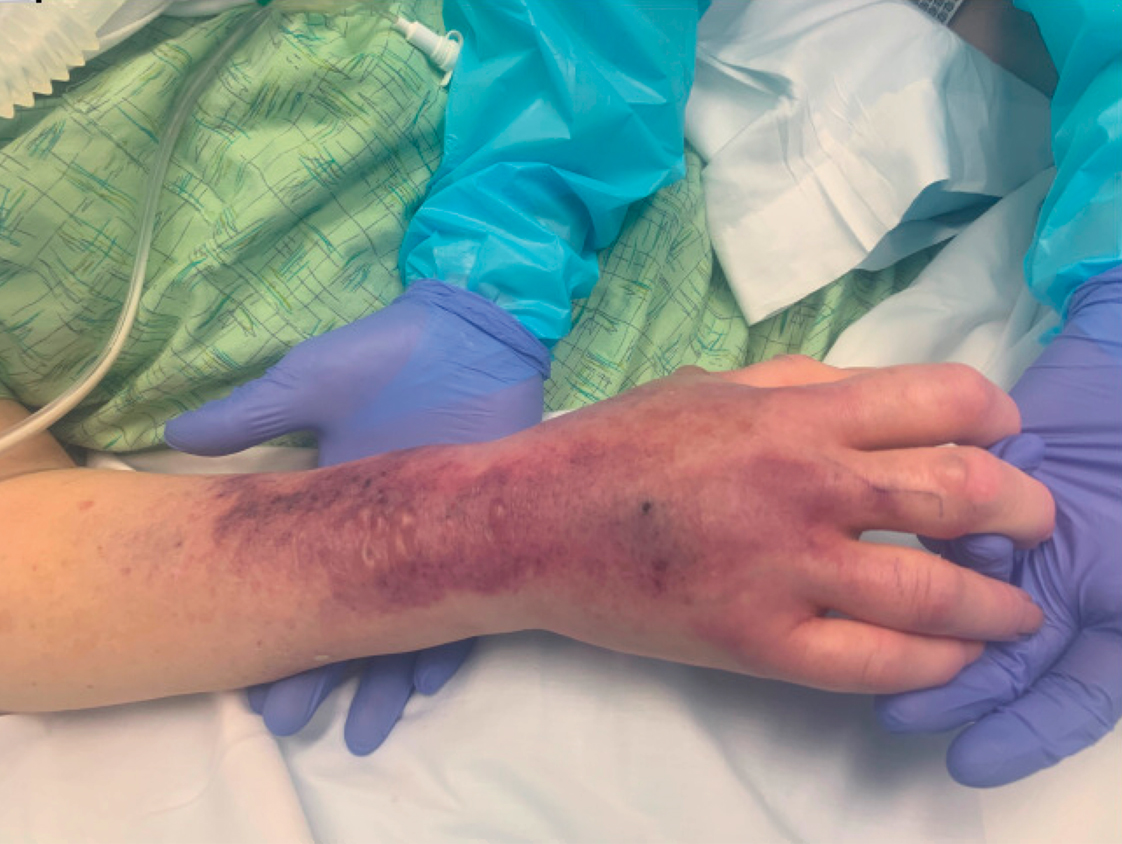
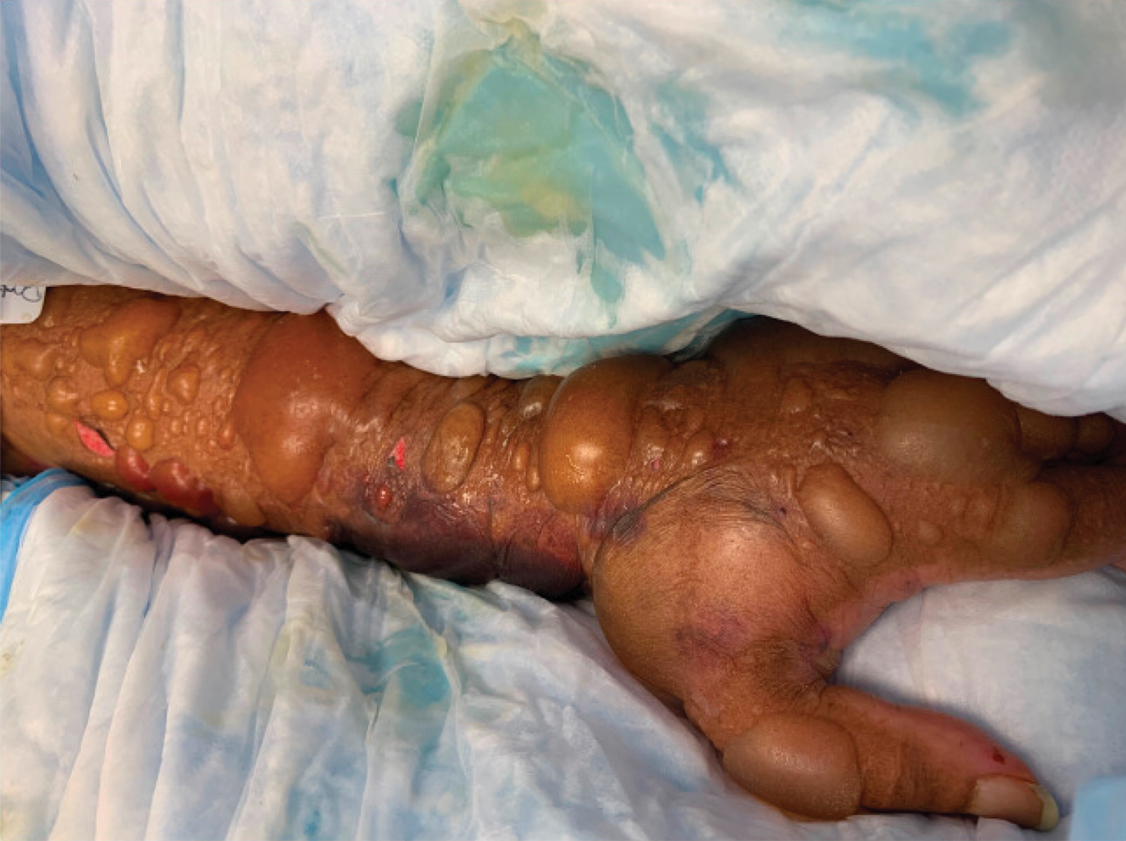
A 67-year-old man with coronary artery disease, diabetes mellitus, and hemiparesis secondary to stroke was admitted to the intensive care unit due to hypoxemia secondary to COVID-19 pneumonia. Dermatology was consulted for the evaluation of blisters on both arms. The right forearm peripheral IV line was used for 4 days prior to the development of cutaneous symptoms. Intravenous medications included cefepime, famotidine, and methylprednisolone. The left forearm peripheral IV line was in place for 1 day prior to the development of blisters and was used for the infusion of maintenance fluids (lactated Ringer’s solution). On the first day of the eruption, small bullae were noted at sites of prior peripheral IV lines (Figure 3). On day 3 of admission, the eruption progressed to larger and more confluent tense bullae with ecchymosis (Figure 4). Additionally, laboratory test results were notable for an elevated D-dimer (peak of >20.00 ug/mL FEU) and fibrinogen (748 mg/dL) levels. Computed tomography of the arms showed extensive subcutaneous stranding and fluid along the fascial planes of the arms, with no gas or abscess formation. Surgical intervention was not recommended following an orthopedic consultation. The patient’s course was complicated by acute kidney injury and rhabdomyolysis; he was later discharged to a skilled nursing facility in stable condition.

Reports from China indicate that approximately 50% of COVID-19 patients have elevated D-dimer levels and are at risk for thrombosis.3 We hypothesize that the exuberant hemorrhagic bullous eruptions in our 2 cases may be mediated in part by a hypercoagulable state secondary to COVID-19 infection combined with IV-related trauma or extravasation injury. However, a direct cytotoxic effect of the virus cannot be entirely excluded as a potential inciting factor. Other entities considered in the differential for localized bullae included trauma-induced bullous pemphigoid as well as bullous cellulitis. Both patients were treated with high-dose steroids as well as broad-spectrum antibiotics, which were expected to lead to improvement in symptoms of bullous pemphigoid and cellulitis, respectively; however, they did not lead to symptom improvement.
Extravasation injury results from unintentional administration of potentially vesicant substances into tissues surrounding the intended vascular channel.4 The mechanism of action of these injuries is postulated to arise from direct tissue injury from cytotoxic substances, elevated osmotic pressure, and reduced blood supply if vasoconstrictive substances are infused.5 In our patients, these injuries also may have promoted vascular occlusion leading to the brisk reaction observed. Although ecchymoses typically are associated with hypocoagulable states, both of our patients were noted to have normal platelet levels throughout hospitalization. Additionally, findings of elevated D-dimer and fibrinogen levels point to a hypercoagulable state. However, there is a possibility of platelet dysfunction leading to the observed cutaneous findings of ecchymoses. Thrombocytopenia is a common finding in patients with COVID-19 and is found to be associated with increased in-hospital mortality.6 Additional study of these reactions is needed given the propensity for multiorgan failure and death in patients with COVID-19 from suspected diffuse microvascular damage.3
- Recalcati S. Cutaneous manifestations in COVID-19: a first perspective [published online March 26, 2020]. J Eur Acad Dermatol Venereol. doi:10.1111/jdv.16387
- Zhang Y, Cao W, Xiao M, et al. Clinical and coagulation characteristics of 7 patients with critical COVID-19 pneumonia and acro-ischemia [in Chinese][published online March 28, 2020]. Zhonghua Xue Ye Xue Za Zhi. 2020;41:E006.
- Mei H, Hu Y. Characteristics, causes, diagnosis and treatment of coagulation dysfunction in patients with COVID-19 [in Chinese][published online March 14, 2020]. Zhonghua Xue Ye Xue Za Zhi. 2020;41:E002.
- Sauerland C, Engelking C, Wickham R, et al. Vesicant extravasation part I: mechanisms, pathogenesis, and nursing care to reduce risk. Oncol Nurs Forum. 2006;33:1134-1141.
- Reynolds PM, MacLaren R, Mueller SW, et al. Management of extravasation injuries: a focused evaluation of noncytotoxic medications. Pharmacotherapy. 2014;34:617-632.
- Yang X, Yang Q, Wang Y, et al. Thrombocytopenia and its association with mortality in patients with COVID-19. J Thromb Haemost. 2020;18:1469‐1472.
To the Editor:
A range of dermatologic manifestations of COVID-19 have been reported, including nonspecific maculopapular exanthems, urticaria, and varicellalike eruptions.1 Additionally, there have been sporadic accounts of cutaneous vasculopathic signs such as perniolike lesions, acro-ischemia, livedo reticularis, and retiform purpura.2 We describe exuberant hemorrhagic bullae occurring on the extremities of 2 critically ill patients with COVID-19. We hypothesized that the bullae were vasculopathic in nature and possibly exacerbated by peripheral intravenous (IV)–related injury.
A 62-year-old woman with a history of diabetes mellitus and chronic obstructive pulmonary disease was admitted to the intensive care unit for acute hypoxemic respiratory failure secondary to COVID-19 infection. Dermatology was consulted for evaluation of blisters on the right arm. A new peripheral IV line was inserted into the patient’s right forearm for treatment of secondary methicillin-resistant Staphylococcus aureus pneumonia. The peripheral IV was inserted into the right proximal forearm for 2 days prior to development of ecchymosis and blisters. Intravenous medications included vancomycin, cefepime, methylprednisolone, and famotidine, as well as maintenance fluids (normal saline). Physical examination revealed extensive confluent ecchymoses with overlying tense bullae (Figure 1). Notable laboratory findings included an elevated D-dimer (peak of 8.67 μg/mL fibrinogen-equivalent units [FEUs], reference range <0.5 μg/mL FEU) and fibrinogen (789 mg/dL, reference range 200–400 mg/dL) levels. Three days later she developed worsening edema of the right arm, accompanied by more extensive bullae formation (Figure 2). Computed tomography of the right arm showed extensive subcutaneous stranding and subcutaneous edema. An orthopedic consultation determined that there was no compartment syndrome, and surgical intervention was not recommended. The patient’s course was complicated by multiorgan failure, and she died 18 days after admission.
A 67-year-old man with coronary artery disease, diabetes mellitus, and hemiparesis secondary to stroke was admitted to the intensive care unit due to hypoxemia secondary to COVID-19 pneumonia. Dermatology was consulted for the evaluation of blisters on both arms. The right forearm peripheral IV line was used for 4 days prior to the development of cutaneous symptoms. Intravenous medications included cefepime, famotidine, and methylprednisolone. The left forearm peripheral IV line was in place for 1 day prior to the development of blisters and was used for the infusion of maintenance fluids (lactated Ringer’s solution). On the first day of the eruption, small bullae were noted at sites of prior peripheral IV lines (Figure 3). On day 3 of admission, the eruption progressed to larger and more confluent tense bullae with ecchymosis (Figure 4). Additionally, laboratory test results were notable for an elevated D-dimer (peak of >20.00 ug/mL FEU) and fibrinogen (748 mg/dL) levels. Computed tomography of the arms showed extensive subcutaneous stranding and fluid along the fascial planes of the arms, with no gas or abscess formation. Surgical intervention was not recommended following an orthopedic consultation. The patient’s course was complicated by acute kidney injury and rhabdomyolysis; he was later discharged to a skilled nursing facility in stable condition.
Reports from China indicate that approximately 50% of COVID-19 patients have elevated D-dimer levels and are at risk for thrombosis.3 We hypothesize that the exuberant hemorrhagic bullous eruptions in our 2 cases may be mediated in part by a hypercoagulable state secondary to COVID-19 infection combined with IV-related trauma or extravasation injury. However, a direct cytotoxic effect of the virus cannot be entirely excluded as a potential inciting factor. Other entities considered in the differential for localized bullae included trauma-induced bullous pemphigoid as well as bullous cellulitis. Both patients were treated with high-dose steroids as well as broad-spectrum antibiotics, which were expected to lead to improvement in symptoms of bullous pemphigoid and cellulitis, respectively; however, they did not lead to symptom improvement.
Extravasation injury results from unintentional administration of potentially vesicant substances into tissues surrounding the intended vascular channel.4 The mechanism of action of these injuries is postulated to arise from direct tissue injury from cytotoxic substances, elevated osmotic pressure, and reduced blood supply if vasoconstrictive substances are infused.5 In our patients, these injuries also may have promoted vascular occlusion leading to the brisk reaction observed. Although ecchymoses typically are associated with hypocoagulable states, both of our patients were noted to have normal platelet levels throughout hospitalization. Additionally, findings of elevated D-dimer and fibrinogen levels point to a hypercoagulable state. However, there is a possibility of platelet dysfunction leading to the observed cutaneous findings of ecchymoses. Thrombocytopenia is a common finding in patients with COVID-19 and is found to be associated with increased in-hospital mortality.6 Additional study of these reactions is needed given the propensity for multiorgan failure and death in patients with COVID-19 from suspected diffuse microvascular damage.3
To the Editor:
A range of dermatologic manifestations of COVID-19 have been reported, including nonspecific maculopapular exanthems, urticaria, and varicellalike eruptions.1 Additionally, there have been sporadic accounts of cutaneous vasculopathic signs such as perniolike lesions, acro-ischemia, livedo reticularis, and retiform purpura.2 We describe exuberant hemorrhagic bullae occurring on the extremities of 2 critically ill patients with COVID-19. We hypothesized that the bullae were vasculopathic in nature and possibly exacerbated by peripheral intravenous (IV)–related injury.
A 62-year-old woman with a history of diabetes mellitus and chronic obstructive pulmonary disease was admitted to the intensive care unit for acute hypoxemic respiratory failure secondary to COVID-19 infection. Dermatology was consulted for evaluation of blisters on the right arm. A new peripheral IV line was inserted into the patient’s right forearm for treatment of secondary methicillin-resistant Staphylococcus aureus pneumonia. The peripheral IV was inserted into the right proximal forearm for 2 days prior to development of ecchymosis and blisters. Intravenous medications included vancomycin, cefepime, methylprednisolone, and famotidine, as well as maintenance fluids (normal saline). Physical examination revealed extensive confluent ecchymoses with overlying tense bullae (Figure 1). Notable laboratory findings included an elevated D-dimer (peak of 8.67 μg/mL fibrinogen-equivalent units [FEUs], reference range <0.5 μg/mL FEU) and fibrinogen (789 mg/dL, reference range 200–400 mg/dL) levels. Three days later she developed worsening edema of the right arm, accompanied by more extensive bullae formation (Figure 2). Computed tomography of the right arm showed extensive subcutaneous stranding and subcutaneous edema. An orthopedic consultation determined that there was no compartment syndrome, and surgical intervention was not recommended. The patient’s course was complicated by multiorgan failure, and she died 18 days after admission.
A 67-year-old man with coronary artery disease, diabetes mellitus, and hemiparesis secondary to stroke was admitted to the intensive care unit due to hypoxemia secondary to COVID-19 pneumonia. Dermatology was consulted for the evaluation of blisters on both arms. The right forearm peripheral IV line was used for 4 days prior to the development of cutaneous symptoms. Intravenous medications included cefepime, famotidine, and methylprednisolone. The left forearm peripheral IV line was in place for 1 day prior to the development of blisters and was used for the infusion of maintenance fluids (lactated Ringer’s solution). On the first day of the eruption, small bullae were noted at sites of prior peripheral IV lines (Figure 3). On day 3 of admission, the eruption progressed to larger and more confluent tense bullae with ecchymosis (Figure 4). Additionally, laboratory test results were notable for an elevated D-dimer (peak of >20.00 ug/mL FEU) and fibrinogen (748 mg/dL) levels. Computed tomography of the arms showed extensive subcutaneous stranding and fluid along the fascial planes of the arms, with no gas or abscess formation. Surgical intervention was not recommended following an orthopedic consultation. The patient’s course was complicated by acute kidney injury and rhabdomyolysis; he was later discharged to a skilled nursing facility in stable condition.
Reports from China indicate that approximately 50% of COVID-19 patients have elevated D-dimer levels and are at risk for thrombosis.3 We hypothesize that the exuberant hemorrhagic bullous eruptions in our 2 cases may be mediated in part by a hypercoagulable state secondary to COVID-19 infection combined with IV-related trauma or extravasation injury. However, a direct cytotoxic effect of the virus cannot be entirely excluded as a potential inciting factor. Other entities considered in the differential for localized bullae included trauma-induced bullous pemphigoid as well as bullous cellulitis. Both patients were treated with high-dose steroids as well as broad-spectrum antibiotics, which were expected to lead to improvement in symptoms of bullous pemphigoid and cellulitis, respectively; however, they did not lead to symptom improvement.
Extravasation injury results from unintentional administration of potentially vesicant substances into tissues surrounding the intended vascular channel.4 The mechanism of action of these injuries is postulated to arise from direct tissue injury from cytotoxic substances, elevated osmotic pressure, and reduced blood supply if vasoconstrictive substances are infused.5 In our patients, these injuries also may have promoted vascular occlusion leading to the brisk reaction observed. Although ecchymoses typically are associated with hypocoagulable states, both of our patients were noted to have normal platelet levels throughout hospitalization. Additionally, findings of elevated D-dimer and fibrinogen levels point to a hypercoagulable state. However, there is a possibility of platelet dysfunction leading to the observed cutaneous findings of ecchymoses. Thrombocytopenia is a common finding in patients with COVID-19 and is found to be associated with increased in-hospital mortality.6 Additional study of these reactions is needed given the propensity for multiorgan failure and death in patients with COVID-19 from suspected diffuse microvascular damage.3
- Recalcati S. Cutaneous manifestations in COVID-19: a first perspective [published online March 26, 2020]. J Eur Acad Dermatol Venereol. doi:10.1111/jdv.16387
- Zhang Y, Cao W, Xiao M, et al. Clinical and coagulation characteristics of 7 patients with critical COVID-19 pneumonia and acro-ischemia [in Chinese][published online March 28, 2020]. Zhonghua Xue Ye Xue Za Zhi. 2020;41:E006.
- Mei H, Hu Y. Characteristics, causes, diagnosis and treatment of coagulation dysfunction in patients with COVID-19 [in Chinese][published online March 14, 2020]. Zhonghua Xue Ye Xue Za Zhi. 2020;41:E002.
- Sauerland C, Engelking C, Wickham R, et al. Vesicant extravasation part I: mechanisms, pathogenesis, and nursing care to reduce risk. Oncol Nurs Forum. 2006;33:1134-1141.
- Reynolds PM, MacLaren R, Mueller SW, et al. Management of extravasation injuries: a focused evaluation of noncytotoxic medications. Pharmacotherapy. 2014;34:617-632.
- Yang X, Yang Q, Wang Y, et al. Thrombocytopenia and its association with mortality in patients with COVID-19. J Thromb Haemost. 2020;18:1469‐1472.
- Recalcati S. Cutaneous manifestations in COVID-19: a first perspective [published online March 26, 2020]. J Eur Acad Dermatol Venereol. doi:10.1111/jdv.16387
- Zhang Y, Cao W, Xiao M, et al. Clinical and coagulation characteristics of 7 patients with critical COVID-19 pneumonia and acro-ischemia [in Chinese][published online March 28, 2020]. Zhonghua Xue Ye Xue Za Zhi. 2020;41:E006.
- Mei H, Hu Y. Characteristics, causes, diagnosis and treatment of coagulation dysfunction in patients with COVID-19 [in Chinese][published online March 14, 2020]. Zhonghua Xue Ye Xue Za Zhi. 2020;41:E002.
- Sauerland C, Engelking C, Wickham R, et al. Vesicant extravasation part I: mechanisms, pathogenesis, and nursing care to reduce risk. Oncol Nurs Forum. 2006;33:1134-1141.
- Reynolds PM, MacLaren R, Mueller SW, et al. Management of extravasation injuries: a focused evaluation of noncytotoxic medications. Pharmacotherapy. 2014;34:617-632.
- Yang X, Yang Q, Wang Y, et al. Thrombocytopenia and its association with mortality in patients with COVID-19. J Thromb Haemost. 2020;18:1469‐1472.
Practice Points
- Hemorrhagic bullae are an uncommon cutaneous manifestation of COVID-19 infection in hospitalized individuals.
- Although there is no reported treatment for COVID-19–associated hemorrhagic bullae, we recommend supportive care and management of underlying etiology.
Persistent Panniculitis in Dermatomyositis
To the Editor:
A 62-year-old woman with a history of dermatomyositis (DM) presented to dermatology clinic for evaluation of multiple subcutaneous nodules. Two years prior to the current presentation, the patient was diagnosed by her primary care physician with DM based on clinical presentation. She initially developed body aches, muscle pain, and weakness of the upper extremities, specifically around the shoulders, and later the lower extremities, specifically around the thighs. The initial physical examination revealed pain with movement, tenderness to palpation, and proximal extremity weakness. The patient also noted a 50-lb weight loss. Over the next year, she noted dysphagia and developed multiple subcutaneous nodules on the right arm, chest, and left axilla. Subsequently, she developed a violaceous, hyperpigmented, periorbital rash and erythema of the anterior chest. She did not experience hair loss, oral ulcers, photosensitivity, or joint pain.
Laboratory testing in the months following the initial presentation revealed a creatine phosphokinase level of 436 U/L (reference range, 20–200 U/L), an erythrocyte sedimentation rate of 60 mm/h (reference range, <31 mm/h), and an aldolase level of 10.4 U/L (reference range, 1.0–8.0 U/L). Lactate dehydrogenase and thyroid function tests were within normal limits. Antinuclear antibodies, anti–double-stranded DNA, anti-Smith antibodies, anti-ribonucleoprotein, anti–Jo-1 antibodies, and anti–smooth muscle antibodies all were negative. Total blood complement levels were elevated, but complement C3 and C4 were within normal limits. Imaging demonstrated normal chest radiographs, and a modified barium swallow confirmed swallowing dysfunction. A right quadricep muscle biopsy confirmed the diagnosis of DM. A malignancy work-up including mammography, colonoscopy, and computed tomography of the chest, abdomen, and pelvis was negative aside from nodular opacities in the chest. She was treated with prednisone (60 mg, 0.9 mg/kg) daily and methotrexate (15–20 mg) weekly for several months. While the treatment attenuated the rash and improved weakness, the nodules persisted, prompting a referral to dermatology.
Physical examination at the dermatology clinic demonstrated the persistent subcutaneous nodules were indurated and bilaterally located on the arms, axillae, chest, abdomen, buttocks, and thighs with no pain or erythema (Figure). Laboratory tests demonstrated a normal creatine phosphokinase level, elevated erythrocyte sedimentation rate (70 mm/h), and elevated aldolase level (9.3 U/L). Complement levels were elevated, though complement C3 and C4 remained within normal limits. Histopathology of nodules from the medial right upper arm and left thigh showed lobular panniculitis with fat necrosis, calcification, and interface changes. The patient was treated for several months with daily mycophenolate mofetil (1 g increased to 3 g) and daily hydroxychloroquine (200 mg) without any effect on the nodules.
The histologic features of panniculitis in lupus and DM are similar and include multifocal hyalinization of the subcuticular fat and diffuse lobular infiltrates of mature lymphocytes without nuclear atypia.1 Though clinical panniculitis is a rare finding in DM, histologic panniculitis is a relatively common finding.2 Despite the similar histopathology of lupus and DM, the presence of typical DM clinical and laboratory features in our patient (body aches, muscle pain, proximal weakness, cutaneous manifestations, elevated creatine phosphokinase, normal complement C3 and C4) made a diagnosis of DM more likely.
Clinical panniculitis is a rare subcutaneous manifestation of DM with around 50 cases reported in the literature (Table). A PubMed search of articles indexed for MEDLINE was conducted using the terms dermatomyositis and panniculitis through July 2019. Additionally, a full-text review and search of references within these articles was used to identify all cases of patients presenting with panniculitis in the setting of DM. Exclusion criteria were cases in which another etiology was considered likely (infectious panniculitis and lupus panniculitis) as well as those without an English translation. We identified 43 cases; the average age of the patients was 39.6 years, and 36 (83.7%) of the cases were women. Patients typically presented with persistent, indurated, painful, erythematous, nodular lesions localized to the arms, abdomen, buttocks, and thighs.
While panniculitis has been reported preceding and concurrent with a diagnosis of DM, a number of cases described presentation as late as 5 years following onset of classic DM symptoms.12,13,31 In some cases (3/43 [7.0%]), panniculitis was the only cutaneous manifestation of DM.15,33,36 However, it occurred more commonly with other characteristic skin findings, such as heliotrope rash or Gottron sign.Some investigators have recommended that panniculitis be included as a diagnostic feature of DM and that DM be considered in the differential diagnosis in isolated cases of panniculitis.25,33
Though it seems panniculitis in DM may correlate with a better prognosis, we identified underlying malignancies in 3 cases. Malignancies associated with panniculitis in DM included ovarian adenocarcinoma, nasopharyngeal carcinoma, and parotid carcinoma, indicating that appropriate cancer screening still is critical in the diagnostic workup.2,11,22
A majority of the reported panniculitis cases in DM have responded to treatment with prednisone; however, treatment with prednisone has been more recalcitrant in other cases. Reports of successful additional therapies include methotrexate, cyclosporine, azathioprine, hydroxychloroquine, intravenous immunoglobulin, mepacrine, or a combination of these entities.19,22 In most cases, improvement of the panniculitis and other DM symptoms occurred simultaneously.25 It is noteworthy that the muscular symptoms often resolved more rapidly than cutaneous manifestations.33 Few reported cases (6 including the current case) found a persistent panniculitis despite improvement and remission of the myositis.3,5,10,11,30
Our patient was treated with both prednisone and methotrexate for several months, leading to remission of muscular symptoms (along with return to baseline of creatine phosphokinase), yet the panniculitis did not improve. The subcutaneous nodules also did not respond to treatment with mycophenolate mofetil and hydroxychloroquine.
Recent immunohistochemical studies have suggested that panniculitic lesions show better outcomes with immunosuppressive therapy when compared with other DM-related skin lesions.40 However, this was not the case for our patient, who after months of immunosuppressive therapy showed complete resolution of the periorbital and chest rashes with persistence of multiple indurated subcutaneous nodules.
Our case adds to a number of reports of DM presenting with panniculitis. Our patient fit the classic demographic of previously reported cases, as she was an adult woman without evidence of underlying malignancy; however, our case remains an example of the therapeutic challenge that exists when encountering a persistent, treatment-resistant panniculitis despite resolution of all other features of DM.
- Wick MR. Panniculitis: a summary. Semin Diagn Pathol. 2017;34:261-272.
- Girouard SD, Velez NF, Penson RT, et al. Panniculitis associated with dermatomyositis and recurrent ovarian cancer. Arch Dermatol. 2012;148:740-744.
- van Dongen HM, van Vugt RM, Stoof TJ. Extensive persistent panniculitis in the context of dermatomyositis. J Clin Rheumatol. 2020;26:E187-E188.
- Choi YJ, Yoo WH. Panniculitis, a rare presentation of onset and exacerbation of juvenile dermatomyositis: a case report and literature review. Arch Rheumatol. 2018;33:367-371.
- Azevedo PO, Castellen NR, Salai AF, et al. Panniculitis associated with amyopathic dermatomyositis. An Bras Dermatol. 2018;93:119-121.
- Agulló A, Hinds B, Larrea M, et al. Livedo racemosa, reticulated ulcerations, panniculitis and violaceous plaques in a 46-year-old woman. Indian Dermatol Online J. 2018;9:47-49.
- Hattori Y, Matsuyama K, Takahashi T, et al. Anti-MDA5 antibody-positive dermatomyositis presenting with cellulitis-like erythema on the mandible as an initial symptom. Case Rep Dermatol. 2018;10:110-114.
- Hasegawa A, Shimomura Y, Kibune N, et al. Panniculitis as the initial manifestation of dermatomyositis with anti-MDA5 antibody. Clin Exp Dermatol. 2017;42:551-553.
- Salman A, Kasapcopur O, Ergun T, et al. Panniculitis in juvenile dermatomyositis: report of a case and review of the published work. J Dermatol. 2016;43:951-953.
- Carroll M, Mellick N, Wagner G. Dermatomyositis panniculitis: a case report. Australas J Dermatol. 2015;56:224‐226.
- Chairatchaneeboon M, Kulthanan K, Manapajon A. Calcific panniculitis and nasopharyngeal cancer-associated adult-onset dermatomyositis: a case report and literature review. Springerplus. 2015;4:201.
- Otero Rivas MM, Vicente Villa A, González Lara L, et al. Panniculitis in juvenile dermatomyositis. Clin Exp Dermatol. 2015;40:574-575.
- Yanaba K, Tanito K, Hamaguchi Y, et al. Anti‐transcription intermediary factor‐1γ/α/β antibody‐positive dermatomyositis associated with multiple panniculitis lesions. Int J Rheum Dis. 2015;20:1831-1834.
- Pau-Charles I, Moreno PJ, Ortiz-Ibanez K, et al. Anti-MDA5 positive clinically amyopathic dermatomyositis presenting with severe cardiomyopathy. J Eur Acad Dermatol Venereol. 2014;28:1097-1102.
- Lamb R, Digby S, Stewart W, et al. Cutaneous ulceration: more than skin deep? Clin Exp Dermatol. 2013;38:443-445.
- Arias M, Hernández MI, Cunha LG, et al. Panniculitis in a patient with dermatomyositis. An Bras Dermatol. 2011;86:146-148.
- Hemmi S, Kushida R, Nishimura H, et al. Magnetic resonance imaging diagnosis of panniculitis in dermatomyositis. Muscle Nerve. 2010;41:151-153.
- Geddes MR, Sinnreich M, Chalk C. Minocycline-induced dermatomyositis. Muscle Nerve. 2010;41:547-549.
- Abdul‐Wahab A, Holden CA, Harland C, et al Calcific panniculitis in adult‐onset dermatomyositis. Clin Exp Dermatol. 2009;34:E854-E856.
- Carneiro S, Alvim G, Resende P, et al. Dermatomyositis with panniculitis. Skinmed. 2007;6:46-47.
- Carrera E, Lobrinus JA, Spertini O, et al. Dermatomyositis, lobarpanniculitis and inflammatory myopathy with abundant macrophages. Neuromuscul Disord. 2006;16:468-471.
- Lin JH, Chu CY, Lin RY. Panniculitis in adult onset dermatomyositis: report of two cases and review of the literature. Dermatol Sinica. 2006;24:194-200.
- Chen GY, Liu MF, Lee JY, et al. Combination of massive mucinosis, dermatomyositis, pyoderma gangrenosum-like ulcer, bullae and fatal intestinal vasculopathy in a young female. Eur J Dermatol. 2005;15:396-400.
- Nakamori A, Yamaguchi Y, Kurimoto I, et al. Vesiculobullous dermatomyositis with panniculitis without muscle disease. J Am Acad Dermatol. 2003;49:1136-1139.
- Solans R, Cortés J, Selva A, et al. Panniculitis: a cutaneous manifestation of dermatomyositis. J Am Acad Dermatol. 2002;46:S148-S150.
- Chao YY, Yang LJ. Dermatomyositis presenting as panniculitis. Int J Dermatol. 2000;39:141-144.
- Lee MW, Lim YS, Choi JH, et al. Panniculitis showing membranocystic changes in the dermatomyositis. J Dermatol. 1999;26:608‐610.
- Ghali FE, Reed AM, Groben PA, et al. Panniculitis in juvenile dermatomyositis. Pediatr Dermatol. 1999;16:270-272.
- Molnar K, Kemeny L, Korom I, et al. Panniculitis in dermatomyositis: report of two cases. Br J Dermatol. 1998;139:161‐163.
- Ishikawa O, Tamura A, Ryuzaki K, et al. Membranocystic changes in the panniculitis of dermatomyositis. Br J Dermatol. 1996;134:773-776.
- Sabroe RA, Wallington TB, Kennedy CT. Dermatomyositis treated with high-dose intravenous immunoglobulins and associated with panniculitis. Clin Exp Dermatol. 1995;20:164-167.
- Neidenbach PJ, Sahn EE, Helton J. Panniculitis in juvenile dermatomyositis. J Am Acad Dermatol. 1995;33:305-307.
- Fusade T, Belanyi P, Joly P, et al. Subcutaneous changes in dermatomyositis. Br J Dermatol. 1993;128:451-453.
- Winkelmann WJ, Billick RC, Srolovitz H. Dermatomyositis presenting as panniculitis. J Am Acad Dermatol. 1990;23:127-128.
- Commens C, O’Neill P, Walker G. Dermatomyositis associated with multifocal lipoatrophy. J Am Acad Dermatol. 1990;22:966-969.
- Raimer SS, Solomon AR, Daniels JC. Polymyositis presenting with panniculitis. J Am Acad Dermatol. 1985;13(2 pt 2):366‐369.
- Feldman D, Hochberg MC, Zizic TM, et al. Cutaneous vasculitis in adult polymyositis/dermatomyositis. J Rheumatol. 1983;10:85-89.
- Kimura S, Fukuyama Y. Tubular cytoplasmic inclusions in a case of childhood dermatomyositis with migratory subcutaneous nodules. Eur J Pediatr. 1977;125:275-283.
- Weber FP, Gray AMH. Chronic relapsing polydermatomyositis with predominant involvement of the subcutaneous fat. Br J Dermatol. 1924;36:544-560.
- Santos‐Briz A, Calle A, Linos K, et al. Dermatomyositis panniculitis: a clinicopathological and immunohistochemical study of 18 cases. J Eur Acad Dermatol Venereol. 2018;32:1352-1359.
To the Editor:
A 62-year-old woman with a history of dermatomyositis (DM) presented to dermatology clinic for evaluation of multiple subcutaneous nodules. Two years prior to the current presentation, the patient was diagnosed by her primary care physician with DM based on clinical presentation. She initially developed body aches, muscle pain, and weakness of the upper extremities, specifically around the shoulders, and later the lower extremities, specifically around the thighs. The initial physical examination revealed pain with movement, tenderness to palpation, and proximal extremity weakness. The patient also noted a 50-lb weight loss. Over the next year, she noted dysphagia and developed multiple subcutaneous nodules on the right arm, chest, and left axilla. Subsequently, she developed a violaceous, hyperpigmented, periorbital rash and erythema of the anterior chest. She did not experience hair loss, oral ulcers, photosensitivity, or joint pain.
Laboratory testing in the months following the initial presentation revealed a creatine phosphokinase level of 436 U/L (reference range, 20–200 U/L), an erythrocyte sedimentation rate of 60 mm/h (reference range, <31 mm/h), and an aldolase level of 10.4 U/L (reference range, 1.0–8.0 U/L). Lactate dehydrogenase and thyroid function tests were within normal limits. Antinuclear antibodies, anti–double-stranded DNA, anti-Smith antibodies, anti-ribonucleoprotein, anti–Jo-1 antibodies, and anti–smooth muscle antibodies all were negative. Total blood complement levels were elevated, but complement C3 and C4 were within normal limits. Imaging demonstrated normal chest radiographs, and a modified barium swallow confirmed swallowing dysfunction. A right quadricep muscle biopsy confirmed the diagnosis of DM. A malignancy work-up including mammography, colonoscopy, and computed tomography of the chest, abdomen, and pelvis was negative aside from nodular opacities in the chest. She was treated with prednisone (60 mg, 0.9 mg/kg) daily and methotrexate (15–20 mg) weekly for several months. While the treatment attenuated the rash and improved weakness, the nodules persisted, prompting a referral to dermatology.
Physical examination at the dermatology clinic demonstrated the persistent subcutaneous nodules were indurated and bilaterally located on the arms, axillae, chest, abdomen, buttocks, and thighs with no pain or erythema (Figure). Laboratory tests demonstrated a normal creatine phosphokinase level, elevated erythrocyte sedimentation rate (70 mm/h), and elevated aldolase level (9.3 U/L). Complement levels were elevated, though complement C3 and C4 remained within normal limits. Histopathology of nodules from the medial right upper arm and left thigh showed lobular panniculitis with fat necrosis, calcification, and interface changes. The patient was treated for several months with daily mycophenolate mofetil (1 g increased to 3 g) and daily hydroxychloroquine (200 mg) without any effect on the nodules.
The histologic features of panniculitis in lupus and DM are similar and include multifocal hyalinization of the subcuticular fat and diffuse lobular infiltrates of mature lymphocytes without nuclear atypia.1 Though clinical panniculitis is a rare finding in DM, histologic panniculitis is a relatively common finding.2 Despite the similar histopathology of lupus and DM, the presence of typical DM clinical and laboratory features in our patient (body aches, muscle pain, proximal weakness, cutaneous manifestations, elevated creatine phosphokinase, normal complement C3 and C4) made a diagnosis of DM more likely.
Clinical panniculitis is a rare subcutaneous manifestation of DM with around 50 cases reported in the literature (Table). A PubMed search of articles indexed for MEDLINE was conducted using the terms dermatomyositis and panniculitis through July 2019. Additionally, a full-text review and search of references within these articles was used to identify all cases of patients presenting with panniculitis in the setting of DM. Exclusion criteria were cases in which another etiology was considered likely (infectious panniculitis and lupus panniculitis) as well as those without an English translation. We identified 43 cases; the average age of the patients was 39.6 years, and 36 (83.7%) of the cases were women. Patients typically presented with persistent, indurated, painful, erythematous, nodular lesions localized to the arms, abdomen, buttocks, and thighs.
While panniculitis has been reported preceding and concurrent with a diagnosis of DM, a number of cases described presentation as late as 5 years following onset of classic DM symptoms.12,13,31 In some cases (3/43 [7.0%]), panniculitis was the only cutaneous manifestation of DM.15,33,36 However, it occurred more commonly with other characteristic skin findings, such as heliotrope rash or Gottron sign.Some investigators have recommended that panniculitis be included as a diagnostic feature of DM and that DM be considered in the differential diagnosis in isolated cases of panniculitis.25,33
Though it seems panniculitis in DM may correlate with a better prognosis, we identified underlying malignancies in 3 cases. Malignancies associated with panniculitis in DM included ovarian adenocarcinoma, nasopharyngeal carcinoma, and parotid carcinoma, indicating that appropriate cancer screening still is critical in the diagnostic workup.2,11,22
A majority of the reported panniculitis cases in DM have responded to treatment with prednisone; however, treatment with prednisone has been more recalcitrant in other cases. Reports of successful additional therapies include methotrexate, cyclosporine, azathioprine, hydroxychloroquine, intravenous immunoglobulin, mepacrine, or a combination of these entities.19,22 In most cases, improvement of the panniculitis and other DM symptoms occurred simultaneously.25 It is noteworthy that the muscular symptoms often resolved more rapidly than cutaneous manifestations.33 Few reported cases (6 including the current case) found a persistent panniculitis despite improvement and remission of the myositis.3,5,10,11,30
Our patient was treated with both prednisone and methotrexate for several months, leading to remission of muscular symptoms (along with return to baseline of creatine phosphokinase), yet the panniculitis did not improve. The subcutaneous nodules also did not respond to treatment with mycophenolate mofetil and hydroxychloroquine.
Recent immunohistochemical studies have suggested that panniculitic lesions show better outcomes with immunosuppressive therapy when compared with other DM-related skin lesions.40 However, this was not the case for our patient, who after months of immunosuppressive therapy showed complete resolution of the periorbital and chest rashes with persistence of multiple indurated subcutaneous nodules.
Our case adds to a number of reports of DM presenting with panniculitis. Our patient fit the classic demographic of previously reported cases, as she was an adult woman without evidence of underlying malignancy; however, our case remains an example of the therapeutic challenge that exists when encountering a persistent, treatment-resistant panniculitis despite resolution of all other features of DM.
To the Editor:
A 62-year-old woman with a history of dermatomyositis (DM) presented to dermatology clinic for evaluation of multiple subcutaneous nodules. Two years prior to the current presentation, the patient was diagnosed by her primary care physician with DM based on clinical presentation. She initially developed body aches, muscle pain, and weakness of the upper extremities, specifically around the shoulders, and later the lower extremities, specifically around the thighs. The initial physical examination revealed pain with movement, tenderness to palpation, and proximal extremity weakness. The patient also noted a 50-lb weight loss. Over the next year, she noted dysphagia and developed multiple subcutaneous nodules on the right arm, chest, and left axilla. Subsequently, she developed a violaceous, hyperpigmented, periorbital rash and erythema of the anterior chest. She did not experience hair loss, oral ulcers, photosensitivity, or joint pain.
Laboratory testing in the months following the initial presentation revealed a creatine phosphokinase level of 436 U/L (reference range, 20–200 U/L), an erythrocyte sedimentation rate of 60 mm/h (reference range, <31 mm/h), and an aldolase level of 10.4 U/L (reference range, 1.0–8.0 U/L). Lactate dehydrogenase and thyroid function tests were within normal limits. Antinuclear antibodies, anti–double-stranded DNA, anti-Smith antibodies, anti-ribonucleoprotein, anti–Jo-1 antibodies, and anti–smooth muscle antibodies all were negative. Total blood complement levels were elevated, but complement C3 and C4 were within normal limits. Imaging demonstrated normal chest radiographs, and a modified barium swallow confirmed swallowing dysfunction. A right quadricep muscle biopsy confirmed the diagnosis of DM. A malignancy work-up including mammography, colonoscopy, and computed tomography of the chest, abdomen, and pelvis was negative aside from nodular opacities in the chest. She was treated with prednisone (60 mg, 0.9 mg/kg) daily and methotrexate (15–20 mg) weekly for several months. While the treatment attenuated the rash and improved weakness, the nodules persisted, prompting a referral to dermatology.
Physical examination at the dermatology clinic demonstrated the persistent subcutaneous nodules were indurated and bilaterally located on the arms, axillae, chest, abdomen, buttocks, and thighs with no pain or erythema (Figure). Laboratory tests demonstrated a normal creatine phosphokinase level, elevated erythrocyte sedimentation rate (70 mm/h), and elevated aldolase level (9.3 U/L). Complement levels were elevated, though complement C3 and C4 remained within normal limits. Histopathology of nodules from the medial right upper arm and left thigh showed lobular panniculitis with fat necrosis, calcification, and interface changes. The patient was treated for several months with daily mycophenolate mofetil (1 g increased to 3 g) and daily hydroxychloroquine (200 mg) without any effect on the nodules.
The histologic features of panniculitis in lupus and DM are similar and include multifocal hyalinization of the subcuticular fat and diffuse lobular infiltrates of mature lymphocytes without nuclear atypia.1 Though clinical panniculitis is a rare finding in DM, histologic panniculitis is a relatively common finding.2 Despite the similar histopathology of lupus and DM, the presence of typical DM clinical and laboratory features in our patient (body aches, muscle pain, proximal weakness, cutaneous manifestations, elevated creatine phosphokinase, normal complement C3 and C4) made a diagnosis of DM more likely.
Clinical panniculitis is a rare subcutaneous manifestation of DM with around 50 cases reported in the literature (Table). A PubMed search of articles indexed for MEDLINE was conducted using the terms dermatomyositis and panniculitis through July 2019. Additionally, a full-text review and search of references within these articles was used to identify all cases of patients presenting with panniculitis in the setting of DM. Exclusion criteria were cases in which another etiology was considered likely (infectious panniculitis and lupus panniculitis) as well as those without an English translation. We identified 43 cases; the average age of the patients was 39.6 years, and 36 (83.7%) of the cases were women. Patients typically presented with persistent, indurated, painful, erythematous, nodular lesions localized to the arms, abdomen, buttocks, and thighs.
While panniculitis has been reported preceding and concurrent with a diagnosis of DM, a number of cases described presentation as late as 5 years following onset of classic DM symptoms.12,13,31 In some cases (3/43 [7.0%]), panniculitis was the only cutaneous manifestation of DM.15,33,36 However, it occurred more commonly with other characteristic skin findings, such as heliotrope rash or Gottron sign.Some investigators have recommended that panniculitis be included as a diagnostic feature of DM and that DM be considered in the differential diagnosis in isolated cases of panniculitis.25,33
Though it seems panniculitis in DM may correlate with a better prognosis, we identified underlying malignancies in 3 cases. Malignancies associated with panniculitis in DM included ovarian adenocarcinoma, nasopharyngeal carcinoma, and parotid carcinoma, indicating that appropriate cancer screening still is critical in the diagnostic workup.2,11,22
A majority of the reported panniculitis cases in DM have responded to treatment with prednisone; however, treatment with prednisone has been more recalcitrant in other cases. Reports of successful additional therapies include methotrexate, cyclosporine, azathioprine, hydroxychloroquine, intravenous immunoglobulin, mepacrine, or a combination of these entities.19,22 In most cases, improvement of the panniculitis and other DM symptoms occurred simultaneously.25 It is noteworthy that the muscular symptoms often resolved more rapidly than cutaneous manifestations.33 Few reported cases (6 including the current case) found a persistent panniculitis despite improvement and remission of the myositis.3,5,10,11,30
Our patient was treated with both prednisone and methotrexate for several months, leading to remission of muscular symptoms (along with return to baseline of creatine phosphokinase), yet the panniculitis did not improve. The subcutaneous nodules also did not respond to treatment with mycophenolate mofetil and hydroxychloroquine.
Recent immunohistochemical studies have suggested that panniculitic lesions show better outcomes with immunosuppressive therapy when compared with other DM-related skin lesions.40 However, this was not the case for our patient, who after months of immunosuppressive therapy showed complete resolution of the periorbital and chest rashes with persistence of multiple indurated subcutaneous nodules.
Our case adds to a number of reports of DM presenting with panniculitis. Our patient fit the classic demographic of previously reported cases, as she was an adult woman without evidence of underlying malignancy; however, our case remains an example of the therapeutic challenge that exists when encountering a persistent, treatment-resistant panniculitis despite resolution of all other features of DM.
- Wick MR. Panniculitis: a summary. Semin Diagn Pathol. 2017;34:261-272.
- Girouard SD, Velez NF, Penson RT, et al. Panniculitis associated with dermatomyositis and recurrent ovarian cancer. Arch Dermatol. 2012;148:740-744.
- van Dongen HM, van Vugt RM, Stoof TJ. Extensive persistent panniculitis in the context of dermatomyositis. J Clin Rheumatol. 2020;26:E187-E188.
- Choi YJ, Yoo WH. Panniculitis, a rare presentation of onset and exacerbation of juvenile dermatomyositis: a case report and literature review. Arch Rheumatol. 2018;33:367-371.
- Azevedo PO, Castellen NR, Salai AF, et al. Panniculitis associated with amyopathic dermatomyositis. An Bras Dermatol. 2018;93:119-121.
- Agulló A, Hinds B, Larrea M, et al. Livedo racemosa, reticulated ulcerations, panniculitis and violaceous plaques in a 46-year-old woman. Indian Dermatol Online J. 2018;9:47-49.
- Hattori Y, Matsuyama K, Takahashi T, et al. Anti-MDA5 antibody-positive dermatomyositis presenting with cellulitis-like erythema on the mandible as an initial symptom. Case Rep Dermatol. 2018;10:110-114.
- Hasegawa A, Shimomura Y, Kibune N, et al. Panniculitis as the initial manifestation of dermatomyositis with anti-MDA5 antibody. Clin Exp Dermatol. 2017;42:551-553.
- Salman A, Kasapcopur O, Ergun T, et al. Panniculitis in juvenile dermatomyositis: report of a case and review of the published work. J Dermatol. 2016;43:951-953.
- Carroll M, Mellick N, Wagner G. Dermatomyositis panniculitis: a case report. Australas J Dermatol. 2015;56:224‐226.
- Chairatchaneeboon M, Kulthanan K, Manapajon A. Calcific panniculitis and nasopharyngeal cancer-associated adult-onset dermatomyositis: a case report and literature review. Springerplus. 2015;4:201.
- Otero Rivas MM, Vicente Villa A, González Lara L, et al. Panniculitis in juvenile dermatomyositis. Clin Exp Dermatol. 2015;40:574-575.
- Yanaba K, Tanito K, Hamaguchi Y, et al. Anti‐transcription intermediary factor‐1γ/α/β antibody‐positive dermatomyositis associated with multiple panniculitis lesions. Int J Rheum Dis. 2015;20:1831-1834.
- Pau-Charles I, Moreno PJ, Ortiz-Ibanez K, et al. Anti-MDA5 positive clinically amyopathic dermatomyositis presenting with severe cardiomyopathy. J Eur Acad Dermatol Venereol. 2014;28:1097-1102.
- Lamb R, Digby S, Stewart W, et al. Cutaneous ulceration: more than skin deep? Clin Exp Dermatol. 2013;38:443-445.
- Arias M, Hernández MI, Cunha LG, et al. Panniculitis in a patient with dermatomyositis. An Bras Dermatol. 2011;86:146-148.
- Hemmi S, Kushida R, Nishimura H, et al. Magnetic resonance imaging diagnosis of panniculitis in dermatomyositis. Muscle Nerve. 2010;41:151-153.
- Geddes MR, Sinnreich M, Chalk C. Minocycline-induced dermatomyositis. Muscle Nerve. 2010;41:547-549.
- Abdul‐Wahab A, Holden CA, Harland C, et al Calcific panniculitis in adult‐onset dermatomyositis. Clin Exp Dermatol. 2009;34:E854-E856.
- Carneiro S, Alvim G, Resende P, et al. Dermatomyositis with panniculitis. Skinmed. 2007;6:46-47.
- Carrera E, Lobrinus JA, Spertini O, et al. Dermatomyositis, lobarpanniculitis and inflammatory myopathy with abundant macrophages. Neuromuscul Disord. 2006;16:468-471.
- Lin JH, Chu CY, Lin RY. Panniculitis in adult onset dermatomyositis: report of two cases and review of the literature. Dermatol Sinica. 2006;24:194-200.
- Chen GY, Liu MF, Lee JY, et al. Combination of massive mucinosis, dermatomyositis, pyoderma gangrenosum-like ulcer, bullae and fatal intestinal vasculopathy in a young female. Eur J Dermatol. 2005;15:396-400.
- Nakamori A, Yamaguchi Y, Kurimoto I, et al. Vesiculobullous dermatomyositis with panniculitis without muscle disease. J Am Acad Dermatol. 2003;49:1136-1139.
- Solans R, Cortés J, Selva A, et al. Panniculitis: a cutaneous manifestation of dermatomyositis. J Am Acad Dermatol. 2002;46:S148-S150.
- Chao YY, Yang LJ. Dermatomyositis presenting as panniculitis. Int J Dermatol. 2000;39:141-144.
- Lee MW, Lim YS, Choi JH, et al. Panniculitis showing membranocystic changes in the dermatomyositis. J Dermatol. 1999;26:608‐610.
- Ghali FE, Reed AM, Groben PA, et al. Panniculitis in juvenile dermatomyositis. Pediatr Dermatol. 1999;16:270-272.
- Molnar K, Kemeny L, Korom I, et al. Panniculitis in dermatomyositis: report of two cases. Br J Dermatol. 1998;139:161‐163.
- Ishikawa O, Tamura A, Ryuzaki K, et al. Membranocystic changes in the panniculitis of dermatomyositis. Br J Dermatol. 1996;134:773-776.
- Sabroe RA, Wallington TB, Kennedy CT. Dermatomyositis treated with high-dose intravenous immunoglobulins and associated with panniculitis. Clin Exp Dermatol. 1995;20:164-167.
- Neidenbach PJ, Sahn EE, Helton J. Panniculitis in juvenile dermatomyositis. J Am Acad Dermatol. 1995;33:305-307.
- Fusade T, Belanyi P, Joly P, et al. Subcutaneous changes in dermatomyositis. Br J Dermatol. 1993;128:451-453.
- Winkelmann WJ, Billick RC, Srolovitz H. Dermatomyositis presenting as panniculitis. J Am Acad Dermatol. 1990;23:127-128.
- Commens C, O’Neill P, Walker G. Dermatomyositis associated with multifocal lipoatrophy. J Am Acad Dermatol. 1990;22:966-969.
- Raimer SS, Solomon AR, Daniels JC. Polymyositis presenting with panniculitis. J Am Acad Dermatol. 1985;13(2 pt 2):366‐369.
- Feldman D, Hochberg MC, Zizic TM, et al. Cutaneous vasculitis in adult polymyositis/dermatomyositis. J Rheumatol. 1983;10:85-89.
- Kimura S, Fukuyama Y. Tubular cytoplasmic inclusions in a case of childhood dermatomyositis with migratory subcutaneous nodules. Eur J Pediatr. 1977;125:275-283.
- Weber FP, Gray AMH. Chronic relapsing polydermatomyositis with predominant involvement of the subcutaneous fat. Br J Dermatol. 1924;36:544-560.
- Santos‐Briz A, Calle A, Linos K, et al. Dermatomyositis panniculitis: a clinicopathological and immunohistochemical study of 18 cases. J Eur Acad Dermatol Venereol. 2018;32:1352-1359.
- Wick MR. Panniculitis: a summary. Semin Diagn Pathol. 2017;34:261-272.
- Girouard SD, Velez NF, Penson RT, et al. Panniculitis associated with dermatomyositis and recurrent ovarian cancer. Arch Dermatol. 2012;148:740-744.
- van Dongen HM, van Vugt RM, Stoof TJ. Extensive persistent panniculitis in the context of dermatomyositis. J Clin Rheumatol. 2020;26:E187-E188.
- Choi YJ, Yoo WH. Panniculitis, a rare presentation of onset and exacerbation of juvenile dermatomyositis: a case report and literature review. Arch Rheumatol. 2018;33:367-371.
- Azevedo PO, Castellen NR, Salai AF, et al. Panniculitis associated with amyopathic dermatomyositis. An Bras Dermatol. 2018;93:119-121.
- Agulló A, Hinds B, Larrea M, et al. Livedo racemosa, reticulated ulcerations, panniculitis and violaceous plaques in a 46-year-old woman. Indian Dermatol Online J. 2018;9:47-49.
- Hattori Y, Matsuyama K, Takahashi T, et al. Anti-MDA5 antibody-positive dermatomyositis presenting with cellulitis-like erythema on the mandible as an initial symptom. Case Rep Dermatol. 2018;10:110-114.
- Hasegawa A, Shimomura Y, Kibune N, et al. Panniculitis as the initial manifestation of dermatomyositis with anti-MDA5 antibody. Clin Exp Dermatol. 2017;42:551-553.
- Salman A, Kasapcopur O, Ergun T, et al. Panniculitis in juvenile dermatomyositis: report of a case and review of the published work. J Dermatol. 2016;43:951-953.
- Carroll M, Mellick N, Wagner G. Dermatomyositis panniculitis: a case report. Australas J Dermatol. 2015;56:224‐226.
- Chairatchaneeboon M, Kulthanan K, Manapajon A. Calcific panniculitis and nasopharyngeal cancer-associated adult-onset dermatomyositis: a case report and literature review. Springerplus. 2015;4:201.
- Otero Rivas MM, Vicente Villa A, González Lara L, et al. Panniculitis in juvenile dermatomyositis. Clin Exp Dermatol. 2015;40:574-575.
- Yanaba K, Tanito K, Hamaguchi Y, et al. Anti‐transcription intermediary factor‐1γ/α/β antibody‐positive dermatomyositis associated with multiple panniculitis lesions. Int J Rheum Dis. 2015;20:1831-1834.
- Pau-Charles I, Moreno PJ, Ortiz-Ibanez K, et al. Anti-MDA5 positive clinically amyopathic dermatomyositis presenting with severe cardiomyopathy. J Eur Acad Dermatol Venereol. 2014;28:1097-1102.
- Lamb R, Digby S, Stewart W, et al. Cutaneous ulceration: more than skin deep? Clin Exp Dermatol. 2013;38:443-445.
- Arias M, Hernández MI, Cunha LG, et al. Panniculitis in a patient with dermatomyositis. An Bras Dermatol. 2011;86:146-148.
- Hemmi S, Kushida R, Nishimura H, et al. Magnetic resonance imaging diagnosis of panniculitis in dermatomyositis. Muscle Nerve. 2010;41:151-153.
- Geddes MR, Sinnreich M, Chalk C. Minocycline-induced dermatomyositis. Muscle Nerve. 2010;41:547-549.
- Abdul‐Wahab A, Holden CA, Harland C, et al Calcific panniculitis in adult‐onset dermatomyositis. Clin Exp Dermatol. 2009;34:E854-E856.
- Carneiro S, Alvim G, Resende P, et al. Dermatomyositis with panniculitis. Skinmed. 2007;6:46-47.
- Carrera E, Lobrinus JA, Spertini O, et al. Dermatomyositis, lobarpanniculitis and inflammatory myopathy with abundant macrophages. Neuromuscul Disord. 2006;16:468-471.
- Lin JH, Chu CY, Lin RY. Panniculitis in adult onset dermatomyositis: report of two cases and review of the literature. Dermatol Sinica. 2006;24:194-200.
- Chen GY, Liu MF, Lee JY, et al. Combination of massive mucinosis, dermatomyositis, pyoderma gangrenosum-like ulcer, bullae and fatal intestinal vasculopathy in a young female. Eur J Dermatol. 2005;15:396-400.
- Nakamori A, Yamaguchi Y, Kurimoto I, et al. Vesiculobullous dermatomyositis with panniculitis without muscle disease. J Am Acad Dermatol. 2003;49:1136-1139.
- Solans R, Cortés J, Selva A, et al. Panniculitis: a cutaneous manifestation of dermatomyositis. J Am Acad Dermatol. 2002;46:S148-S150.
- Chao YY, Yang LJ. Dermatomyositis presenting as panniculitis. Int J Dermatol. 2000;39:141-144.
- Lee MW, Lim YS, Choi JH, et al. Panniculitis showing membranocystic changes in the dermatomyositis. J Dermatol. 1999;26:608‐610.
- Ghali FE, Reed AM, Groben PA, et al. Panniculitis in juvenile dermatomyositis. Pediatr Dermatol. 1999;16:270-272.
- Molnar K, Kemeny L, Korom I, et al. Panniculitis in dermatomyositis: report of two cases. Br J Dermatol. 1998;139:161‐163.
- Ishikawa O, Tamura A, Ryuzaki K, et al. Membranocystic changes in the panniculitis of dermatomyositis. Br J Dermatol. 1996;134:773-776.
- Sabroe RA, Wallington TB, Kennedy CT. Dermatomyositis treated with high-dose intravenous immunoglobulins and associated with panniculitis. Clin Exp Dermatol. 1995;20:164-167.
- Neidenbach PJ, Sahn EE, Helton J. Panniculitis in juvenile dermatomyositis. J Am Acad Dermatol. 1995;33:305-307.
- Fusade T, Belanyi P, Joly P, et al. Subcutaneous changes in dermatomyositis. Br J Dermatol. 1993;128:451-453.
- Winkelmann WJ, Billick RC, Srolovitz H. Dermatomyositis presenting as panniculitis. J Am Acad Dermatol. 1990;23:127-128.
- Commens C, O’Neill P, Walker G. Dermatomyositis associated with multifocal lipoatrophy. J Am Acad Dermatol. 1990;22:966-969.
- Raimer SS, Solomon AR, Daniels JC. Polymyositis presenting with panniculitis. J Am Acad Dermatol. 1985;13(2 pt 2):366‐369.
- Feldman D, Hochberg MC, Zizic TM, et al. Cutaneous vasculitis in adult polymyositis/dermatomyositis. J Rheumatol. 1983;10:85-89.
- Kimura S, Fukuyama Y. Tubular cytoplasmic inclusions in a case of childhood dermatomyositis with migratory subcutaneous nodules. Eur J Pediatr. 1977;125:275-283.
- Weber FP, Gray AMH. Chronic relapsing polydermatomyositis with predominant involvement of the subcutaneous fat. Br J Dermatol. 1924;36:544-560.
- Santos‐Briz A, Calle A, Linos K, et al. Dermatomyositis panniculitis: a clinicopathological and immunohistochemical study of 18 cases. J Eur Acad Dermatol Venereol. 2018;32:1352-1359.
Practice Points
- Clinical panniculitis is a rare subcutaneous manifestation of dermatomyositis (DM) that dermatologists must consider when evaluating patients with this condition.
- Panniculitis can precede, occur simultaneously with, or develop up to 5 years after onset of DM.
- Many patients suffer from treatment-resistant panniculitis in DM, suggesting that therapeutic management of this condition may require long-term and more aggressive treatment modalities.
Etanercept-Induced Squamous Proliferations in a Patient With Porokeratosis
To the Editor:
Etanercept is an immune-modulating drug used for the treatment of a variety of diseases including psoriasis, rheumatoid arthritis, and ankylosing spondylitis. It is an anti–tumor necrosis factor (TNF) fusion protein consisting of an extracellular domain of the p75 TNF receptor and the Fc portion of human IgG.1 Etanercept is well known for its immunosuppressive side effects. A handful of case reports have provided evidence of squamous cell cancers in the setting of etanercept therapy. The most comprehensive description was a case series by Brewer et al2 describing 4 patients with squamous cell carcinoma (SCC) that developed 1 to 17 months after the initiation of etanercept therapy. We present a case of a patient diagnosed with psoriasis and concomitant porokeratosis who developed multiple SCCs and squamous proliferations after initiation of etanercept therapy.
A 66-year-old man was referred to our clinic for treatment of psoriasis, as noted on a biopsy of the right ankle diagnosed several years prior. He was being treated with etanercept 50 mg twice weekly. Other treatments included calcipotriene–betamethasone dipropionate, salicylic acid gel, intralesional triamcinolone, clobetasol, and urea 40%. Physical examination revealed multiple erythematous tender nodules with hyperkeratotic scale distributed on the right arm and leg (Figure 1) that were concerning for SCC. Biopsies from 6 lesions revealed multiple SCC/keratoacanthomas (KAs) with verrucous features (Figure 2). Primers for human papillomavirus (HPV) 6, 11, 16, 18, 31, 33, and 51 were all negative. At that time, etanercept was discontinued. The patient was referred for Mohs micrographic surgery and underwent excision of several SCC lesions including an approximately 7-cm SCC on the right ankle (Figure 1B). Positron emission tomography/computed tomography found hypermetabolic lymphadenopathy. A follow-up biopsy of the inguinal nodes identified no malignant cells. Given their multiplicity, the patient was initiated on a prolonged course of a retinoid with acitretin 35 mg daily. The clearance of the large 7-cm lesion with a single stage of Mohs micrographic surgery directed suspicion to a pseudoepitheliomatous or HPV-induced cause for the lesions. Rereview of the original 6 biopsies indicated 1 definitive SCC on the right wrist, 2 KAs, and 3 that were most consistent with verruca vulgaris. At 1-year follow-up, most of the hyperkeratotic lesions had resolved with continued acitretin. Baseline porokeratosis lesions that were abundantly present on the arms and legs resolved by 1-year follow-up (Figure 3A).
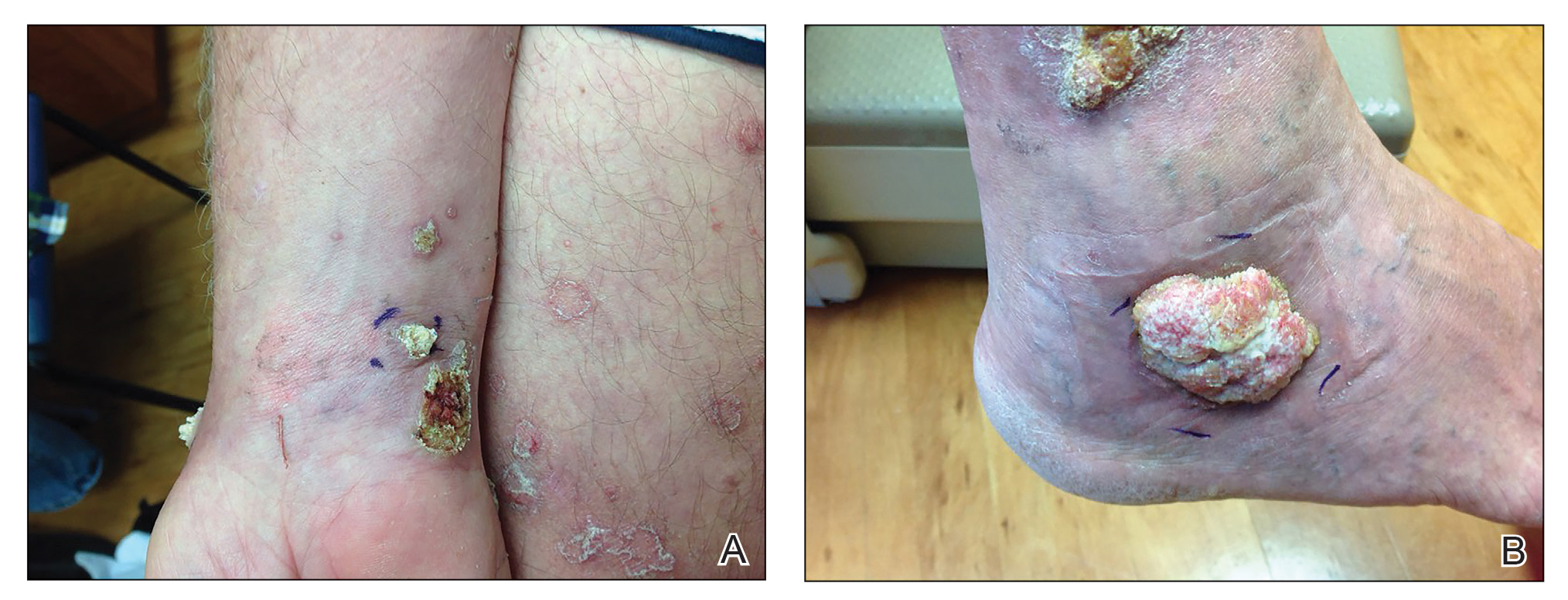
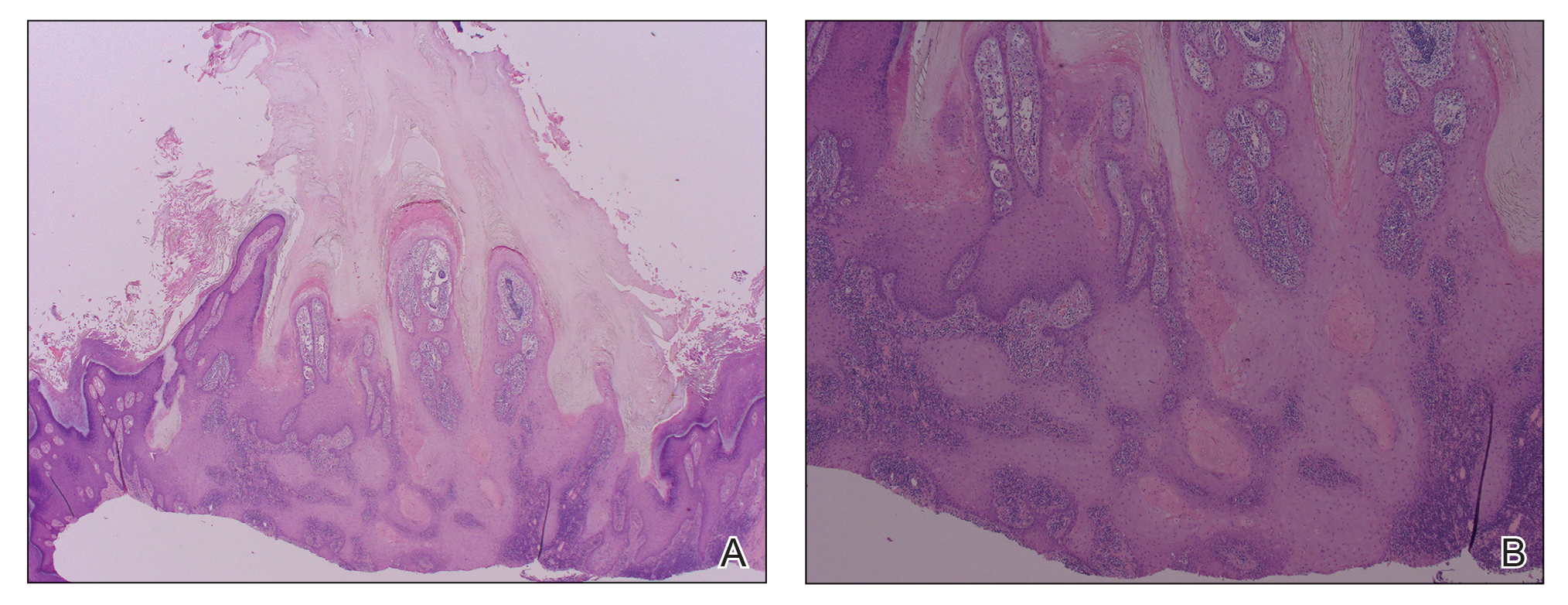
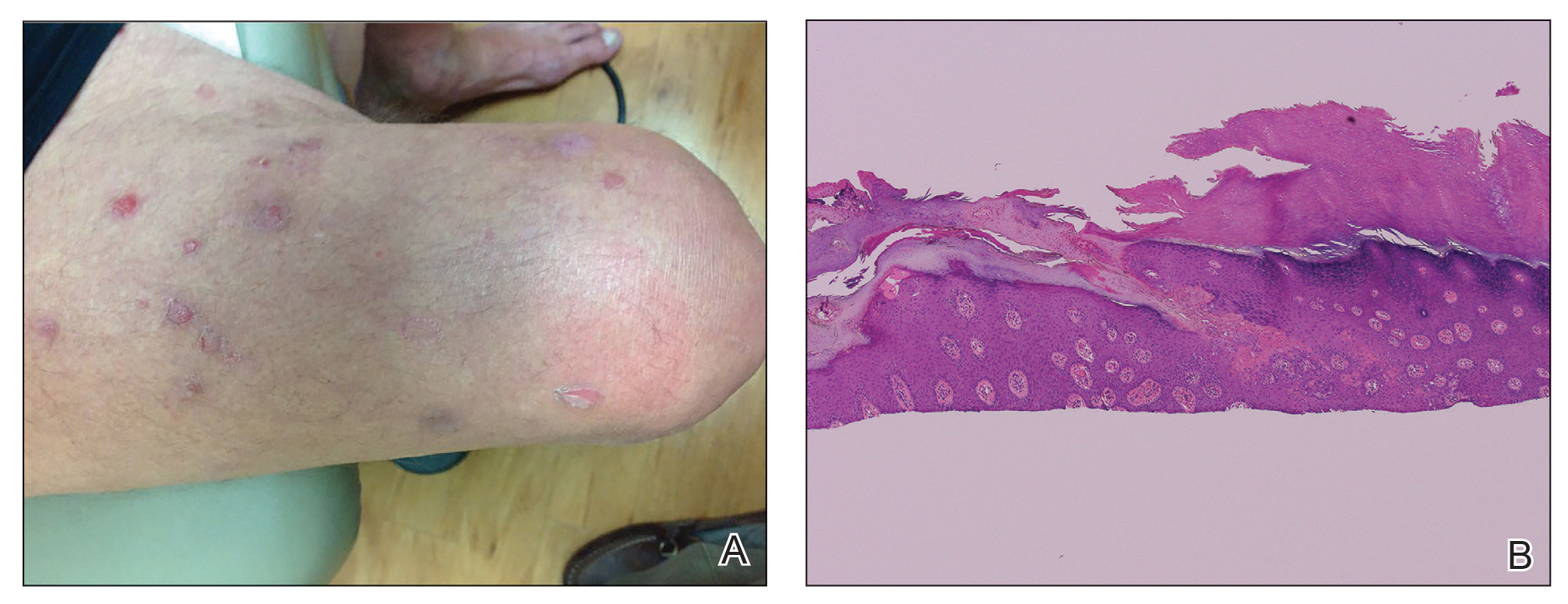
The link between classic porokeratosis and the development of squamous cell proliferations is well established. Ninomiya et al3 noted a possible mechanism of p53 overexpression in the epidermis of porokeratotic lesions that may make the lesions particularly susceptible to the development of immunosuppression-induced SCC. Etanercept is an immune-modulating drug with well-known immunosuppressive side effects including reactivation of HPV as well as the development of SCCs.
Our patient initially was diagnosed with psoriasis and etanercept was initiated. The presence of coexistent porokeratosis likely predisposed him to etanercept-induced squamous proliferations including 2 SCCs and verrucous lesions, with histologic features suggesting SCC/KA. Histopathology revealed a cornoid lamella in SCC (Figure 3B), suggesting development of malignancy within epithelial clones, as noted by Lee et al.4
Targeted systemic therapies may lead to the formation of SCCs. The association between epidermal growth factor receptor (EGFR) kinase inhibitors and SCC formation is well known. For instance, sorafenib—a multikinase inhibitor that is downstream in the EGFR pathway—has been noted to induce epidermal growths including KAs and SCCs.5 There has been no definitive causal relationship identified between the development of SCC and TNF-α inhibitors. It has been suggested that perhaps there is an unmasking effect, as subclinical SCC manifests after TNF-α inhibition that leads to SCC development. Discontinuation of etanercept and resolution of lesions highlights a potential role of TNF-α inhibition and tumorigenesis of SCCs, especially in the background of porokeratosis. Vigilance for development of immunosuppression-induced malignancy, especially squamous cell proliferations, has become exceedingly important with exponentially increasing use of biologic therapies in medicine.
- Feldmann M, Charles P, Taylor P, et al. Biological insights from clinical trials with anti-TNF therapy. Springer Semin Immunopathol Springer Sem Immunopathol. 1998;20:211-228.
- Brewer JD, Schott ARH, Roenigk RK. Multiple squamous cell carcinomas in the setting of psoriasis treated with etanercept: a report of four cases and review of the literature. Int J Dermatol. 2011;50:1555-1559.
- Ninomiya Y, Urano Y, Yoshimoto K, et al. p53 gene mutation analysis in porokeratosis and porokeratosis-associated squamous cell carcinoma. J Dermatol Sci. 1997;14:173-178.
- Lee HR, Han TY, Son S-J, et al. Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis. Ann Dermatol. 2011;23:536.
- Kwon EJ, Kish LS, Jaworsky C. The histologic spectrum of epithelial neoplasms induced by sorafenib. J Am Acad Dermatol. 2009;61:522-527.
To the Editor:
Etanercept is an immune-modulating drug used for the treatment of a variety of diseases including psoriasis, rheumatoid arthritis, and ankylosing spondylitis. It is an anti–tumor necrosis factor (TNF) fusion protein consisting of an extracellular domain of the p75 TNF receptor and the Fc portion of human IgG.1 Etanercept is well known for its immunosuppressive side effects. A handful of case reports have provided evidence of squamous cell cancers in the setting of etanercept therapy. The most comprehensive description was a case series by Brewer et al2 describing 4 patients with squamous cell carcinoma (SCC) that developed 1 to 17 months after the initiation of etanercept therapy. We present a case of a patient diagnosed with psoriasis and concomitant porokeratosis who developed multiple SCCs and squamous proliferations after initiation of etanercept therapy.
A 66-year-old man was referred to our clinic for treatment of psoriasis, as noted on a biopsy of the right ankle diagnosed several years prior. He was being treated with etanercept 50 mg twice weekly. Other treatments included calcipotriene–betamethasone dipropionate, salicylic acid gel, intralesional triamcinolone, clobetasol, and urea 40%. Physical examination revealed multiple erythematous tender nodules with hyperkeratotic scale distributed on the right arm and leg (Figure 1) that were concerning for SCC. Biopsies from 6 lesions revealed multiple SCC/keratoacanthomas (KAs) with verrucous features (Figure 2). Primers for human papillomavirus (HPV) 6, 11, 16, 18, 31, 33, and 51 were all negative. At that time, etanercept was discontinued. The patient was referred for Mohs micrographic surgery and underwent excision of several SCC lesions including an approximately 7-cm SCC on the right ankle (Figure 1B). Positron emission tomography/computed tomography found hypermetabolic lymphadenopathy. A follow-up biopsy of the inguinal nodes identified no malignant cells. Given their multiplicity, the patient was initiated on a prolonged course of a retinoid with acitretin 35 mg daily. The clearance of the large 7-cm lesion with a single stage of Mohs micrographic surgery directed suspicion to a pseudoepitheliomatous or HPV-induced cause for the lesions. Rereview of the original 6 biopsies indicated 1 definitive SCC on the right wrist, 2 KAs, and 3 that were most consistent with verruca vulgaris. At 1-year follow-up, most of the hyperkeratotic lesions had resolved with continued acitretin. Baseline porokeratosis lesions that were abundantly present on the arms and legs resolved by 1-year follow-up (Figure 3A).
The link between classic porokeratosis and the development of squamous cell proliferations is well established. Ninomiya et al3 noted a possible mechanism of p53 overexpression in the epidermis of porokeratotic lesions that may make the lesions particularly susceptible to the development of immunosuppression-induced SCC. Etanercept is an immune-modulating drug with well-known immunosuppressive side effects including reactivation of HPV as well as the development of SCCs.
Our patient initially was diagnosed with psoriasis and etanercept was initiated. The presence of coexistent porokeratosis likely predisposed him to etanercept-induced squamous proliferations including 2 SCCs and verrucous lesions, with histologic features suggesting SCC/KA. Histopathology revealed a cornoid lamella in SCC (Figure 3B), suggesting development of malignancy within epithelial clones, as noted by Lee et al.4
Targeted systemic therapies may lead to the formation of SCCs. The association between epidermal growth factor receptor (EGFR) kinase inhibitors and SCC formation is well known. For instance, sorafenib—a multikinase inhibitor that is downstream in the EGFR pathway—has been noted to induce epidermal growths including KAs and SCCs.5 There has been no definitive causal relationship identified between the development of SCC and TNF-α inhibitors. It has been suggested that perhaps there is an unmasking effect, as subclinical SCC manifests after TNF-α inhibition that leads to SCC development. Discontinuation of etanercept and resolution of lesions highlights a potential role of TNF-α inhibition and tumorigenesis of SCCs, especially in the background of porokeratosis. Vigilance for development of immunosuppression-induced malignancy, especially squamous cell proliferations, has become exceedingly important with exponentially increasing use of biologic therapies in medicine.
To the Editor:
Etanercept is an immune-modulating drug used for the treatment of a variety of diseases including psoriasis, rheumatoid arthritis, and ankylosing spondylitis. It is an anti–tumor necrosis factor (TNF) fusion protein consisting of an extracellular domain of the p75 TNF receptor and the Fc portion of human IgG.1 Etanercept is well known for its immunosuppressive side effects. A handful of case reports have provided evidence of squamous cell cancers in the setting of etanercept therapy. The most comprehensive description was a case series by Brewer et al2 describing 4 patients with squamous cell carcinoma (SCC) that developed 1 to 17 months after the initiation of etanercept therapy. We present a case of a patient diagnosed with psoriasis and concomitant porokeratosis who developed multiple SCCs and squamous proliferations after initiation of etanercept therapy.
A 66-year-old man was referred to our clinic for treatment of psoriasis, as noted on a biopsy of the right ankle diagnosed several years prior. He was being treated with etanercept 50 mg twice weekly. Other treatments included calcipotriene–betamethasone dipropionate, salicylic acid gel, intralesional triamcinolone, clobetasol, and urea 40%. Physical examination revealed multiple erythematous tender nodules with hyperkeratotic scale distributed on the right arm and leg (Figure 1) that were concerning for SCC. Biopsies from 6 lesions revealed multiple SCC/keratoacanthomas (KAs) with verrucous features (Figure 2). Primers for human papillomavirus (HPV) 6, 11, 16, 18, 31, 33, and 51 were all negative. At that time, etanercept was discontinued. The patient was referred for Mohs micrographic surgery and underwent excision of several SCC lesions including an approximately 7-cm SCC on the right ankle (Figure 1B). Positron emission tomography/computed tomography found hypermetabolic lymphadenopathy. A follow-up biopsy of the inguinal nodes identified no malignant cells. Given their multiplicity, the patient was initiated on a prolonged course of a retinoid with acitretin 35 mg daily. The clearance of the large 7-cm lesion with a single stage of Mohs micrographic surgery directed suspicion to a pseudoepitheliomatous or HPV-induced cause for the lesions. Rereview of the original 6 biopsies indicated 1 definitive SCC on the right wrist, 2 KAs, and 3 that were most consistent with verruca vulgaris. At 1-year follow-up, most of the hyperkeratotic lesions had resolved with continued acitretin. Baseline porokeratosis lesions that were abundantly present on the arms and legs resolved by 1-year follow-up (Figure 3A).
The link between classic porokeratosis and the development of squamous cell proliferations is well established. Ninomiya et al3 noted a possible mechanism of p53 overexpression in the epidermis of porokeratotic lesions that may make the lesions particularly susceptible to the development of immunosuppression-induced SCC. Etanercept is an immune-modulating drug with well-known immunosuppressive side effects including reactivation of HPV as well as the development of SCCs.
Our patient initially was diagnosed with psoriasis and etanercept was initiated. The presence of coexistent porokeratosis likely predisposed him to etanercept-induced squamous proliferations including 2 SCCs and verrucous lesions, with histologic features suggesting SCC/KA. Histopathology revealed a cornoid lamella in SCC (Figure 3B), suggesting development of malignancy within epithelial clones, as noted by Lee et al.4
Targeted systemic therapies may lead to the formation of SCCs. The association between epidermal growth factor receptor (EGFR) kinase inhibitors and SCC formation is well known. For instance, sorafenib—a multikinase inhibitor that is downstream in the EGFR pathway—has been noted to induce epidermal growths including KAs and SCCs.5 There has been no definitive causal relationship identified between the development of SCC and TNF-α inhibitors. It has been suggested that perhaps there is an unmasking effect, as subclinical SCC manifests after TNF-α inhibition that leads to SCC development. Discontinuation of etanercept and resolution of lesions highlights a potential role of TNF-α inhibition and tumorigenesis of SCCs, especially in the background of porokeratosis. Vigilance for development of immunosuppression-induced malignancy, especially squamous cell proliferations, has become exceedingly important with exponentially increasing use of biologic therapies in medicine.
- Feldmann M, Charles P, Taylor P, et al. Biological insights from clinical trials with anti-TNF therapy. Springer Semin Immunopathol Springer Sem Immunopathol. 1998;20:211-228.
- Brewer JD, Schott ARH, Roenigk RK. Multiple squamous cell carcinomas in the setting of psoriasis treated with etanercept: a report of four cases and review of the literature. Int J Dermatol. 2011;50:1555-1559.
- Ninomiya Y, Urano Y, Yoshimoto K, et al. p53 gene mutation analysis in porokeratosis and porokeratosis-associated squamous cell carcinoma. J Dermatol Sci. 1997;14:173-178.
- Lee HR, Han TY, Son S-J, et al. Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis. Ann Dermatol. 2011;23:536.
- Kwon EJ, Kish LS, Jaworsky C. The histologic spectrum of epithelial neoplasms induced by sorafenib. J Am Acad Dermatol. 2009;61:522-527.
- Feldmann M, Charles P, Taylor P, et al. Biological insights from clinical trials with anti-TNF therapy. Springer Semin Immunopathol Springer Sem Immunopathol. 1998;20:211-228.
- Brewer JD, Schott ARH, Roenigk RK. Multiple squamous cell carcinomas in the setting of psoriasis treated with etanercept: a report of four cases and review of the literature. Int J Dermatol. 2011;50:1555-1559.
- Ninomiya Y, Urano Y, Yoshimoto K, et al. p53 gene mutation analysis in porokeratosis and porokeratosis-associated squamous cell carcinoma. J Dermatol Sci. 1997;14:173-178.
- Lee HR, Han TY, Son S-J, et al. Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis. Ann Dermatol. 2011;23:536.
- Kwon EJ, Kish LS, Jaworsky C. The histologic spectrum of epithelial neoplasms induced by sorafenib. J Am Acad Dermatol. 2009;61:522-527.
Practice Points
- The use of biologics, particularly tumor necrosis factor α blockers, rarely are reported to induce skin cancer.
- Squamous cell carcinoma in the setting of biologic treatment would warrant a change of systemic medication.
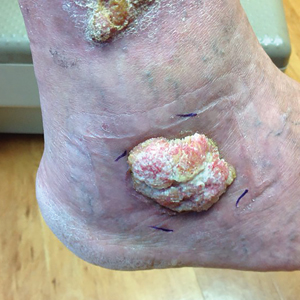
Cutaneous Carcinomatous Arteriopathy and Retiform Purpura Secondary to Metastatic Penile Carcinoma
To the Editor:
A 56-year-old man with a history of stage IV metastatic penile squamous cell carcinoma treated with penectomy and chemotherapy with 5-fluorouracil and cisplatin presented with several painful ulcerations in the groin, abdomen, and thighs. The lesions initially appeared in the groin and were treated as bacterial abscesses with antibiotics. Over the next few weeks, new lesions appeared on the abdomen and thighs. An additional cycle of chemotherapy led to a reduction in number; however, they again increased within a few weeks. Medications included enoxaparin followed by 3 weeks of warfarin use due to a right leg deep vein thrombosis.
Physical examination revealed multiple 1- to 4-cm, firm, ulcerated nodules on the bilateral inguinal folds, abdomen, and upper thighs, as well as areas of livedo racemosa and noninflammatory retiform purpura with central ulceration (Figures 1 and 2). This retiform purpura was both perilesional and in areas without ulcerations. Laboratory values included the following: sodium, 127 mmol/L (reference range, 136–145 mmol/L); prothrombin time, 16.1 seconds (reference range, 11–15 seconds); white blood cell count, 20.69×109/L (reference range, 4.5–11.0×109/L) with 87% neutrophils (reference range, 54%–62%); hemoglobin, 6.1 g/dL (reference range, 13.5–17.5 g/dL); hematocrit, 18.8% (reference range, 41%–53%); platelets, 474×109/L (reference range, 150–400×109/L); D-dimer, 0.77 mg/L (reference range, ≤0.50 mg/L); fibrinogen, 489 mg/dL (reference range, 150–400 mg/dL); prior urine culture positive for Pseudomonas aeruginosa. He was negative for hepatitis B and hepatitis C viruses as well as HIV, and the lesions were not clinically consistent with herpes simplex virus, as they were not scalloped or circinate. Punch biopsies were obtained from a nodule on the left leg and a purpuric patch on the right leg.
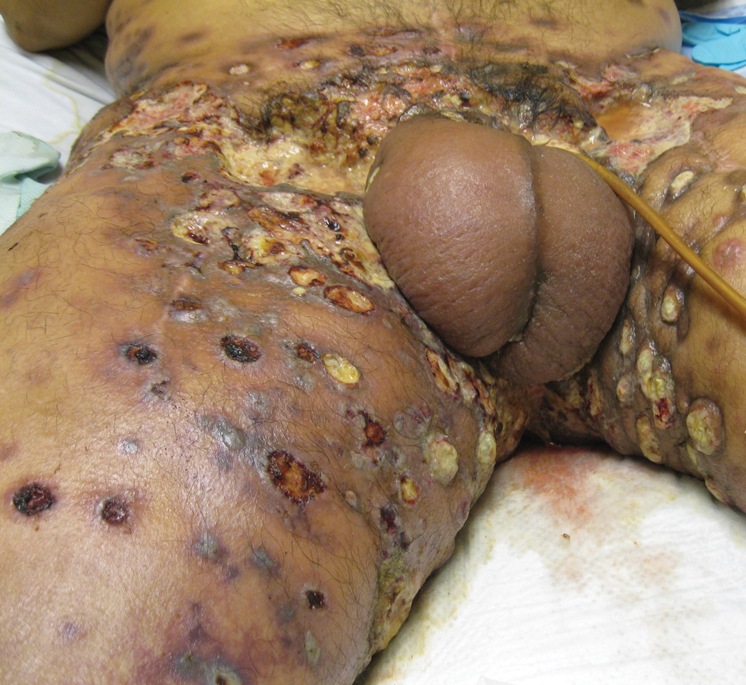
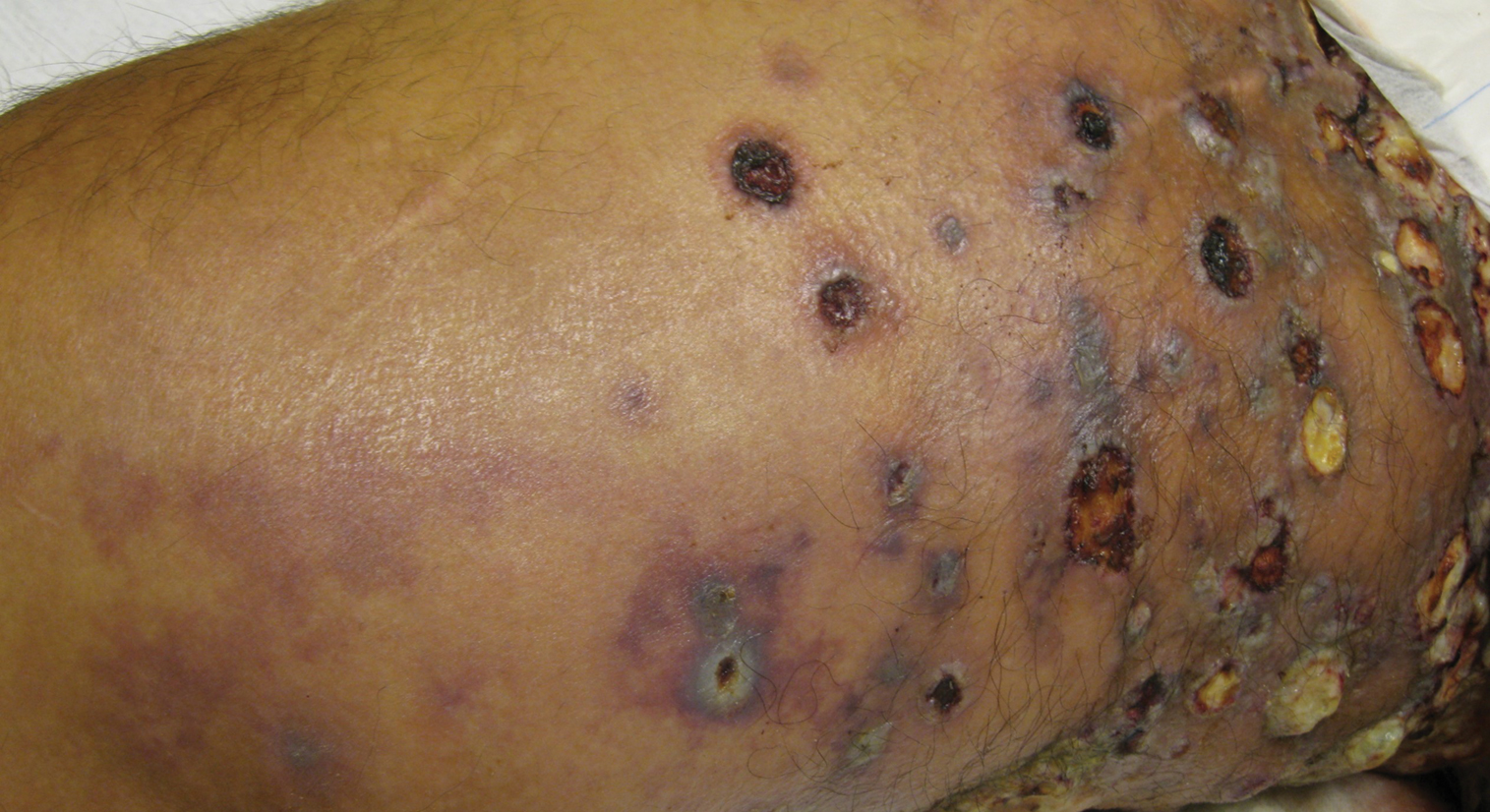
Histopathology of the ulcerated nodule revealed a proliferation of atypical keratinocytes with hyperchromatic and pleomorphic nuclei in the dermis without involvement of the overlying epidermis, consistent with metastatic squamous cell carcinoma (Figure 3). Histopathology of the purpuric patch demonstrated a thrombotic vasculopathy with numerous fibrin thrombi in the lumina of superficial dermal capillaries (Figure 4). No atypical cells, calcifications, or organisms were seen in the vessels. Periodic acid–Schiff, Fite, and Gram stains also were negative. The extent of the disease portended a poor prognosis, and additional vasculopathic workup was not pursued. Following antibiotic treatment and palliative care consultation, he died from subsequent infectious complications 1 month after presentation.
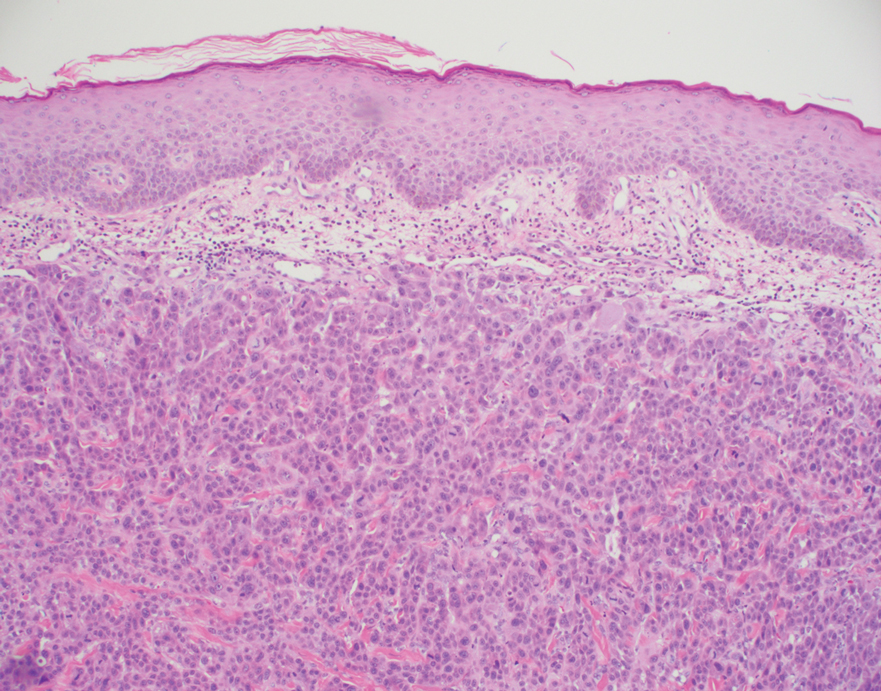
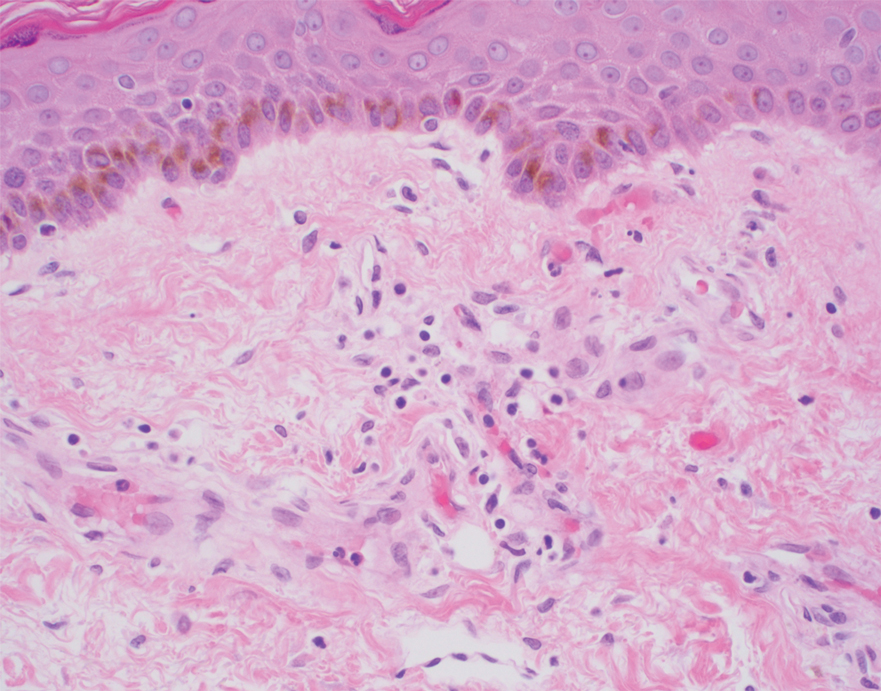
Cutaneous metastases may occur in the setting of multiple malignancies including breast, lung, melanoma, and various gastrointestinal cancers.1 These may present in multiple ways, including firm nontender nodules or as plaques with one of the following morphologies: carcinoma erysipeloides: erythematous, occasionally tender areas resembling cellulitis due to lymphatic obstruction by tumor cells2; carcinoma en cuirasse: indurated sclerotic scarlike plaques due to collagen infiltration3; or carcinoma telangiectoides: telangiectatic, thin erythematous plaques due to dermal capillary infiltration by malignant cells.3
Ischemic cutaneous lesions less commonly occur in the setting of malignancy and can be the result of both direct and indirect systemic effects from the cancer. Malignancies are known to directly trigger vasculopathies in other organs, most commonly the lungs, through 2 primary mechanisms. First, in carcinomatous arteriopathy, metastatic cells promote fibrocellular intimal proliferation of small pulmonary arteries and arterioles leading to stenosis, thrombosis, and obliteration. This mechanism has been described in pulmonary thrombotic microangiopathy secondary to lung carcinoma.4 This pathophysiology likely is also what underlies paraneoplastic acral vascular syndromes, which culminate in digital ischemia. Hypothesized mechanisms for this ischemia also range from vasospasm to thromboembolism.5 Secondly, in vasculitis carcinomatosa, metastatic tumor cells damage or block vessel walls, resulting in end-organ ischemia. Vasculitis carcinomatosa is a well-known phenomenon in angiocentric and intravascular lymphoid malignancies (typically of B-T or natural killer/T-cell origin) but also has been reported in a case of gastric adenocarcinoma with arterial invasion.6 This process is different than carcinoma telangiectoides where malignant cells may be present in the vasculature on histopathology but not trigger thrombosis and ischemic skin necrosis.
Systemic coagulopathies such as disseminated intravascular coagulation (DIC), thrombotic thrombocytopenia purpura, and catastrophic antiphospholipid antibody syndrome can occur in the setting of malignancies.7 Clinically, all may present with livedo racemosa, noninflammatory retiform purpura, and widespread skin necrosis. In adult patients, purpura fulminans most often is seen in the setting of sepsis and DIC, with accompanying evidence of microangiopathy.8 Catastrophic antiphospholipid antibody syndrome can be triggered by malignancy and is characterized by central nervous system, renal, pulmonary, and gastrointestinal complications. Skin involvement such as ulcers, livedo reticularis, and gangrene have been reported.9 Other causes of thrombotic vasculopathy include warfarin necrosis, heparin-induced thrombotic thrombocytopenia, calciphylaxis, and angioinvasive infections.8 Warfarin necrosis and heparin-induced thrombotic thrombocytopenia typically present days after initiating therapy with the respective medication. Calciphylaxis typically occurs in patients on dialysis, though it may occur in nonuremic patients including those with malignancy.8 Patients with malignancies on chemotherapy can become neutropenic and are at risk for ecthyma gangrenosum due to P aeruginosa and other gram-negative rods, Staphylococcus aureus, and angioinvasive fungi.10
Based on clinical, histopathological, and laboratory data, we favored a diagnosis of cutaneous carcinomatous arteriopathy. Vasculitis carcinomatosa was a possibility despite the lack of vasculotropism on histopathology, which may have been due to biopsy site selection. Systemic coagulopathies such as DIC, thrombotic thrombocytopenia purpura, and catastrophic antiphospholipid antibody syndrome were unlikely, as the ischemic skin lesions and livedo racemosa were limited to areas adjacent to cutaneous metastases, and the patient lacked other common multiorgan manifestations or laboratory findings. Although our patient was on warfarin, he was on a stable dose for weeks and histopathologic features of subcutaneous thrombosis were not seen. The biopsy also was not consistent with calciphylaxis. Ecthyma gangrenosum was unlikely given the lack of organisms on histopathology and negative skin and blood cultures. Although additional laboratory testing in this patient may have included cryoglobulins and cryofibrinogens, both entities were unlikely due to a lack of ischemic acral lesions.
In conclusion, we present a case of localized thrombotic vasculopathy that likely was secondary to cutaneous carcinomatous arteriopathy in the setting of cutaneous metastatic penile squamous cell carcinoma. The differential diagnosis of retiform purpura, livedo racemosa, and other signs of cutaneous ischemia in patients with metastatic cancer is broad and can be the result of both direct and indirect systemic effects from the cancer. Appropriate workup in these cases should include skin biopsies for histopathology and culture, medication review, and laboratory evaluation for systemic coagulopathies.
- Alcaraz I, Cerroni L, Ruetten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Prat L, Chouaid C, Kettaneh A, et al. Cutaneous lymphangitis carcinomatosa in a patient with lung adenocarcinoma: case report and literature review. Lung Cancer. 2013;79:91-93.
- Marneros AG, Blanco F, Husain S, et al. Classification of cutaneous intravascular breast cancer metastases based on immunolabeling for blood and lymph vessels. J Am Acad Dermatol. 2009;60:633-638.
- von Herbay A, Illes A, Waldherr R, et al. Pulmonary tumor thrombotic microangiopathy with pulmonary hypertension. Cancer. 1990;66:587-592.
- Besnerais ML, Miranda S, Cailleux N, et al. Digital ischemia associated with cancer. Medicine. 2014;93:E47.
- Sweeney S, Utzschneider R, Fraire AE. Vasculitis carcinomatosa occurring in association with adenocarcinoma of the stomach. Ann Diagn Pathol. 1998;2:247-249.
- Zwicker JI, Furie BC, Furie B. Cancer-associated thrombosis. Crit Rev Oncol Hematol. 2007;62:126-136.
- Thornsberry LA, LoSicco KI, English JC. The skin and hypercoagulable states. J Am Acad Dermatol. 2013;69:450-462.
- Miesbach W, Asherson RA, Cervera R, et al; CAPS Registry Group. The role of malignancies in patients with catastrophic anti-phospholipid (Asherson’s) syndrome. Clin Rheumatol. 2007;26:2109-2114.
- Pozo D. Ecthyma gangrenosum‐like eruption associated with Morganella morganii infection. Br J Dermatol. 1998;139:520-521.
To the Editor:
A 56-year-old man with a history of stage IV metastatic penile squamous cell carcinoma treated with penectomy and chemotherapy with 5-fluorouracil and cisplatin presented with several painful ulcerations in the groin, abdomen, and thighs. The lesions initially appeared in the groin and were treated as bacterial abscesses with antibiotics. Over the next few weeks, new lesions appeared on the abdomen and thighs. An additional cycle of chemotherapy led to a reduction in number; however, they again increased within a few weeks. Medications included enoxaparin followed by 3 weeks of warfarin use due to a right leg deep vein thrombosis.
Physical examination revealed multiple 1- to 4-cm, firm, ulcerated nodules on the bilateral inguinal folds, abdomen, and upper thighs, as well as areas of livedo racemosa and noninflammatory retiform purpura with central ulceration (Figures 1 and 2). This retiform purpura was both perilesional and in areas without ulcerations. Laboratory values included the following: sodium, 127 mmol/L (reference range, 136–145 mmol/L); prothrombin time, 16.1 seconds (reference range, 11–15 seconds); white blood cell count, 20.69×109/L (reference range, 4.5–11.0×109/L) with 87% neutrophils (reference range, 54%–62%); hemoglobin, 6.1 g/dL (reference range, 13.5–17.5 g/dL); hematocrit, 18.8% (reference range, 41%–53%); platelets, 474×109/L (reference range, 150–400×109/L); D-dimer, 0.77 mg/L (reference range, ≤0.50 mg/L); fibrinogen, 489 mg/dL (reference range, 150–400 mg/dL); prior urine culture positive for Pseudomonas aeruginosa. He was negative for hepatitis B and hepatitis C viruses as well as HIV, and the lesions were not clinically consistent with herpes simplex virus, as they were not scalloped or circinate. Punch biopsies were obtained from a nodule on the left leg and a purpuric patch on the right leg.
Histopathology of the ulcerated nodule revealed a proliferation of atypical keratinocytes with hyperchromatic and pleomorphic nuclei in the dermis without involvement of the overlying epidermis, consistent with metastatic squamous cell carcinoma (Figure 3). Histopathology of the purpuric patch demonstrated a thrombotic vasculopathy with numerous fibrin thrombi in the lumina of superficial dermal capillaries (Figure 4). No atypical cells, calcifications, or organisms were seen in the vessels. Periodic acid–Schiff, Fite, and Gram stains also were negative. The extent of the disease portended a poor prognosis, and additional vasculopathic workup was not pursued. Following antibiotic treatment and palliative care consultation, he died from subsequent infectious complications 1 month after presentation.
Cutaneous metastases may occur in the setting of multiple malignancies including breast, lung, melanoma, and various gastrointestinal cancers.1 These may present in multiple ways, including firm nontender nodules or as plaques with one of the following morphologies: carcinoma erysipeloides: erythematous, occasionally tender areas resembling cellulitis due to lymphatic obstruction by tumor cells2; carcinoma en cuirasse: indurated sclerotic scarlike plaques due to collagen infiltration3; or carcinoma telangiectoides: telangiectatic, thin erythematous plaques due to dermal capillary infiltration by malignant cells.3
Ischemic cutaneous lesions less commonly occur in the setting of malignancy and can be the result of both direct and indirect systemic effects from the cancer. Malignancies are known to directly trigger vasculopathies in other organs, most commonly the lungs, through 2 primary mechanisms. First, in carcinomatous arteriopathy, metastatic cells promote fibrocellular intimal proliferation of small pulmonary arteries and arterioles leading to stenosis, thrombosis, and obliteration. This mechanism has been described in pulmonary thrombotic microangiopathy secondary to lung carcinoma.4 This pathophysiology likely is also what underlies paraneoplastic acral vascular syndromes, which culminate in digital ischemia. Hypothesized mechanisms for this ischemia also range from vasospasm to thromboembolism.5 Secondly, in vasculitis carcinomatosa, metastatic tumor cells damage or block vessel walls, resulting in end-organ ischemia. Vasculitis carcinomatosa is a well-known phenomenon in angiocentric and intravascular lymphoid malignancies (typically of B-T or natural killer/T-cell origin) but also has been reported in a case of gastric adenocarcinoma with arterial invasion.6 This process is different than carcinoma telangiectoides where malignant cells may be present in the vasculature on histopathology but not trigger thrombosis and ischemic skin necrosis.
Systemic coagulopathies such as disseminated intravascular coagulation (DIC), thrombotic thrombocytopenia purpura, and catastrophic antiphospholipid antibody syndrome can occur in the setting of malignancies.7 Clinically, all may present with livedo racemosa, noninflammatory retiform purpura, and widespread skin necrosis. In adult patients, purpura fulminans most often is seen in the setting of sepsis and DIC, with accompanying evidence of microangiopathy.8 Catastrophic antiphospholipid antibody syndrome can be triggered by malignancy and is characterized by central nervous system, renal, pulmonary, and gastrointestinal complications. Skin involvement such as ulcers, livedo reticularis, and gangrene have been reported.9 Other causes of thrombotic vasculopathy include warfarin necrosis, heparin-induced thrombotic thrombocytopenia, calciphylaxis, and angioinvasive infections.8 Warfarin necrosis and heparin-induced thrombotic thrombocytopenia typically present days after initiating therapy with the respective medication. Calciphylaxis typically occurs in patients on dialysis, though it may occur in nonuremic patients including those with malignancy.8 Patients with malignancies on chemotherapy can become neutropenic and are at risk for ecthyma gangrenosum due to P aeruginosa and other gram-negative rods, Staphylococcus aureus, and angioinvasive fungi.10
Based on clinical, histopathological, and laboratory data, we favored a diagnosis of cutaneous carcinomatous arteriopathy. Vasculitis carcinomatosa was a possibility despite the lack of vasculotropism on histopathology, which may have been due to biopsy site selection. Systemic coagulopathies such as DIC, thrombotic thrombocytopenia purpura, and catastrophic antiphospholipid antibody syndrome were unlikely, as the ischemic skin lesions and livedo racemosa were limited to areas adjacent to cutaneous metastases, and the patient lacked other common multiorgan manifestations or laboratory findings. Although our patient was on warfarin, he was on a stable dose for weeks and histopathologic features of subcutaneous thrombosis were not seen. The biopsy also was not consistent with calciphylaxis. Ecthyma gangrenosum was unlikely given the lack of organisms on histopathology and negative skin and blood cultures. Although additional laboratory testing in this patient may have included cryoglobulins and cryofibrinogens, both entities were unlikely due to a lack of ischemic acral lesions.
In conclusion, we present a case of localized thrombotic vasculopathy that likely was secondary to cutaneous carcinomatous arteriopathy in the setting of cutaneous metastatic penile squamous cell carcinoma. The differential diagnosis of retiform purpura, livedo racemosa, and other signs of cutaneous ischemia in patients with metastatic cancer is broad and can be the result of both direct and indirect systemic effects from the cancer. Appropriate workup in these cases should include skin biopsies for histopathology and culture, medication review, and laboratory evaluation for systemic coagulopathies.
To the Editor:
A 56-year-old man with a history of stage IV metastatic penile squamous cell carcinoma treated with penectomy and chemotherapy with 5-fluorouracil and cisplatin presented with several painful ulcerations in the groin, abdomen, and thighs. The lesions initially appeared in the groin and were treated as bacterial abscesses with antibiotics. Over the next few weeks, new lesions appeared on the abdomen and thighs. An additional cycle of chemotherapy led to a reduction in number; however, they again increased within a few weeks. Medications included enoxaparin followed by 3 weeks of warfarin use due to a right leg deep vein thrombosis.
Physical examination revealed multiple 1- to 4-cm, firm, ulcerated nodules on the bilateral inguinal folds, abdomen, and upper thighs, as well as areas of livedo racemosa and noninflammatory retiform purpura with central ulceration (Figures 1 and 2). This retiform purpura was both perilesional and in areas without ulcerations. Laboratory values included the following: sodium, 127 mmol/L (reference range, 136–145 mmol/L); prothrombin time, 16.1 seconds (reference range, 11–15 seconds); white blood cell count, 20.69×109/L (reference range, 4.5–11.0×109/L) with 87% neutrophils (reference range, 54%–62%); hemoglobin, 6.1 g/dL (reference range, 13.5–17.5 g/dL); hematocrit, 18.8% (reference range, 41%–53%); platelets, 474×109/L (reference range, 150–400×109/L); D-dimer, 0.77 mg/L (reference range, ≤0.50 mg/L); fibrinogen, 489 mg/dL (reference range, 150–400 mg/dL); prior urine culture positive for Pseudomonas aeruginosa. He was negative for hepatitis B and hepatitis C viruses as well as HIV, and the lesions were not clinically consistent with herpes simplex virus, as they were not scalloped or circinate. Punch biopsies were obtained from a nodule on the left leg and a purpuric patch on the right leg.
Histopathology of the ulcerated nodule revealed a proliferation of atypical keratinocytes with hyperchromatic and pleomorphic nuclei in the dermis without involvement of the overlying epidermis, consistent with metastatic squamous cell carcinoma (Figure 3). Histopathology of the purpuric patch demonstrated a thrombotic vasculopathy with numerous fibrin thrombi in the lumina of superficial dermal capillaries (Figure 4). No atypical cells, calcifications, or organisms were seen in the vessels. Periodic acid–Schiff, Fite, and Gram stains also were negative. The extent of the disease portended a poor prognosis, and additional vasculopathic workup was not pursued. Following antibiotic treatment and palliative care consultation, he died from subsequent infectious complications 1 month after presentation.
Cutaneous metastases may occur in the setting of multiple malignancies including breast, lung, melanoma, and various gastrointestinal cancers.1 These may present in multiple ways, including firm nontender nodules or as plaques with one of the following morphologies: carcinoma erysipeloides: erythematous, occasionally tender areas resembling cellulitis due to lymphatic obstruction by tumor cells2; carcinoma en cuirasse: indurated sclerotic scarlike plaques due to collagen infiltration3; or carcinoma telangiectoides: telangiectatic, thin erythematous plaques due to dermal capillary infiltration by malignant cells.3
Ischemic cutaneous lesions less commonly occur in the setting of malignancy and can be the result of both direct and indirect systemic effects from the cancer. Malignancies are known to directly trigger vasculopathies in other organs, most commonly the lungs, through 2 primary mechanisms. First, in carcinomatous arteriopathy, metastatic cells promote fibrocellular intimal proliferation of small pulmonary arteries and arterioles leading to stenosis, thrombosis, and obliteration. This mechanism has been described in pulmonary thrombotic microangiopathy secondary to lung carcinoma.4 This pathophysiology likely is also what underlies paraneoplastic acral vascular syndromes, which culminate in digital ischemia. Hypothesized mechanisms for this ischemia also range from vasospasm to thromboembolism.5 Secondly, in vasculitis carcinomatosa, metastatic tumor cells damage or block vessel walls, resulting in end-organ ischemia. Vasculitis carcinomatosa is a well-known phenomenon in angiocentric and intravascular lymphoid malignancies (typically of B-T or natural killer/T-cell origin) but also has been reported in a case of gastric adenocarcinoma with arterial invasion.6 This process is different than carcinoma telangiectoides where malignant cells may be present in the vasculature on histopathology but not trigger thrombosis and ischemic skin necrosis.
Systemic coagulopathies such as disseminated intravascular coagulation (DIC), thrombotic thrombocytopenia purpura, and catastrophic antiphospholipid antibody syndrome can occur in the setting of malignancies.7 Clinically, all may present with livedo racemosa, noninflammatory retiform purpura, and widespread skin necrosis. In adult patients, purpura fulminans most often is seen in the setting of sepsis and DIC, with accompanying evidence of microangiopathy.8 Catastrophic antiphospholipid antibody syndrome can be triggered by malignancy and is characterized by central nervous system, renal, pulmonary, and gastrointestinal complications. Skin involvement such as ulcers, livedo reticularis, and gangrene have been reported.9 Other causes of thrombotic vasculopathy include warfarin necrosis, heparin-induced thrombotic thrombocytopenia, calciphylaxis, and angioinvasive infections.8 Warfarin necrosis and heparin-induced thrombotic thrombocytopenia typically present days after initiating therapy with the respective medication. Calciphylaxis typically occurs in patients on dialysis, though it may occur in nonuremic patients including those with malignancy.8 Patients with malignancies on chemotherapy can become neutropenic and are at risk for ecthyma gangrenosum due to P aeruginosa and other gram-negative rods, Staphylococcus aureus, and angioinvasive fungi.10
Based on clinical, histopathological, and laboratory data, we favored a diagnosis of cutaneous carcinomatous arteriopathy. Vasculitis carcinomatosa was a possibility despite the lack of vasculotropism on histopathology, which may have been due to biopsy site selection. Systemic coagulopathies such as DIC, thrombotic thrombocytopenia purpura, and catastrophic antiphospholipid antibody syndrome were unlikely, as the ischemic skin lesions and livedo racemosa were limited to areas adjacent to cutaneous metastases, and the patient lacked other common multiorgan manifestations or laboratory findings. Although our patient was on warfarin, he was on a stable dose for weeks and histopathologic features of subcutaneous thrombosis were not seen. The biopsy also was not consistent with calciphylaxis. Ecthyma gangrenosum was unlikely given the lack of organisms on histopathology and negative skin and blood cultures. Although additional laboratory testing in this patient may have included cryoglobulins and cryofibrinogens, both entities were unlikely due to a lack of ischemic acral lesions.
In conclusion, we present a case of localized thrombotic vasculopathy that likely was secondary to cutaneous carcinomatous arteriopathy in the setting of cutaneous metastatic penile squamous cell carcinoma. The differential diagnosis of retiform purpura, livedo racemosa, and other signs of cutaneous ischemia in patients with metastatic cancer is broad and can be the result of both direct and indirect systemic effects from the cancer. Appropriate workup in these cases should include skin biopsies for histopathology and culture, medication review, and laboratory evaluation for systemic coagulopathies.
- Alcaraz I, Cerroni L, Ruetten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Prat L, Chouaid C, Kettaneh A, et al. Cutaneous lymphangitis carcinomatosa in a patient with lung adenocarcinoma: case report and literature review. Lung Cancer. 2013;79:91-93.
- Marneros AG, Blanco F, Husain S, et al. Classification of cutaneous intravascular breast cancer metastases based on immunolabeling for blood and lymph vessels. J Am Acad Dermatol. 2009;60:633-638.
- von Herbay A, Illes A, Waldherr R, et al. Pulmonary tumor thrombotic microangiopathy with pulmonary hypertension. Cancer. 1990;66:587-592.
- Besnerais ML, Miranda S, Cailleux N, et al. Digital ischemia associated with cancer. Medicine. 2014;93:E47.
- Sweeney S, Utzschneider R, Fraire AE. Vasculitis carcinomatosa occurring in association with adenocarcinoma of the stomach. Ann Diagn Pathol. 1998;2:247-249.
- Zwicker JI, Furie BC, Furie B. Cancer-associated thrombosis. Crit Rev Oncol Hematol. 2007;62:126-136.
- Thornsberry LA, LoSicco KI, English JC. The skin and hypercoagulable states. J Am Acad Dermatol. 2013;69:450-462.
- Miesbach W, Asherson RA, Cervera R, et al; CAPS Registry Group. The role of malignancies in patients with catastrophic anti-phospholipid (Asherson’s) syndrome. Clin Rheumatol. 2007;26:2109-2114.
- Pozo D. Ecthyma gangrenosum‐like eruption associated with Morganella morganii infection. Br J Dermatol. 1998;139:520-521.
- Alcaraz I, Cerroni L, Ruetten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Prat L, Chouaid C, Kettaneh A, et al. Cutaneous lymphangitis carcinomatosa in a patient with lung adenocarcinoma: case report and literature review. Lung Cancer. 2013;79:91-93.
- Marneros AG, Blanco F, Husain S, et al. Classification of cutaneous intravascular breast cancer metastases based on immunolabeling for blood and lymph vessels. J Am Acad Dermatol. 2009;60:633-638.
- von Herbay A, Illes A, Waldherr R, et al. Pulmonary tumor thrombotic microangiopathy with pulmonary hypertension. Cancer. 1990;66:587-592.
- Besnerais ML, Miranda S, Cailleux N, et al. Digital ischemia associated with cancer. Medicine. 2014;93:E47.
- Sweeney S, Utzschneider R, Fraire AE. Vasculitis carcinomatosa occurring in association with adenocarcinoma of the stomach. Ann Diagn Pathol. 1998;2:247-249.
- Zwicker JI, Furie BC, Furie B. Cancer-associated thrombosis. Crit Rev Oncol Hematol. 2007;62:126-136.
- Thornsberry LA, LoSicco KI, English JC. The skin and hypercoagulable states. J Am Acad Dermatol. 2013;69:450-462.
- Miesbach W, Asherson RA, Cervera R, et al; CAPS Registry Group. The role of malignancies in patients with catastrophic anti-phospholipid (Asherson’s) syndrome. Clin Rheumatol. 2007;26:2109-2114.
- Pozo D. Ecthyma gangrenosum‐like eruption associated with Morganella morganii infection. Br J Dermatol. 1998;139:520-521.
Practice Points
- Cutaneous metastases may present in multiple ways, including carcinoma erysipeloides, carcinoma en cuirasse, or carcinoma telangiectoides.
- Ischemic cutaneous lesions, characterized by livedoid skin changes and retiform purpura, occur less commonly in the setting of malignancy.
- Direct mechanisms include carcinomatous arteriopathy and vasculitis carcinomatosa. Indirect systemic processes include coagulopathies such as disseminated intravascular coagulation, thrombotic thrombocytopenia purpura, catastrophic antiphospholipid antibody syndrome, calciphylaxis, and cryoglobulinemia.
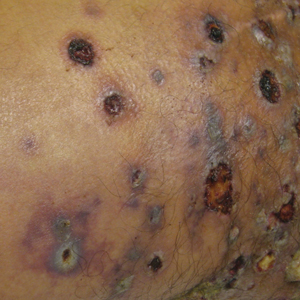
Wiping Away Cellulitis: A Case of Factitious Disorder
To the Editor:
Patients with psychocutaneous disorders present unique challenges to physicians. We illustrate the critical role that dermoscopy may play to illuminate exogenous skin pathology.
A 50-year-old woman with a reported medical history of systemic lupus erythematosus, chronic pain, and nonhealing leg ulcers presented to the emergency department with severe pain of the left lower leg and redness that was concerning for cellulitis. She sought treatment at an outside hospital for cellulitis 2 weeks prior but left against medical advice. Symptomatic review revealed chest pain, shortness of breath, nausea, vomiting, and diarrhea. The primary team started her on intravenous clindamycin and vancomycin for the presumed infection and scheduled narcotic medications due to concerns of intractable pain in the left leg. The dermatology department was consulted after failure to improve with 1 week of systemic antibiotics.
Physical examination revealed a geometric, atrophic, purple plaque on the left anterior shin from a prior leg ulcer as well as a diffuse red-pink patch extending from the knee to the ankle. Notably, the cellulitis spared the left posterior calf resting against the sheet and had a sharp line of demarcation at the distal shin. The leg was cool to the touch while the patient was distractible. She later reported that the leg was extremely tender to palpation. Dermoscopy revealed linear red pigments within skin furrows that accentuated skin lines (Figure). These findings raised suspicions of an external manipulation. The skin was wiped with an alcohol pad that removed a shimmering pink substance consistent in appearance to a cosmetic product. The skin beneath the cellulitis appeared normal.
On further review of the patient’s medical record, it was noted that she was admitted several months ago for ulcers of the left leg. She had been to multiple hospitals and had numerous rounds of antibiotics. Biopsy of an ulcer revealed dermal fibrosis consistent with scarring. Aerobic bacteria, atypical mycobacteria, and fungal cultures were all negative. The physicians suspected a self-induced etiology consistent with dermatitis artefacta. The patient emphasized multiple psychosocial stressors as well as having frequent lupus flares despite repeated negative workup. Given the exaggerated symptoms and unnecessary hospital visits, she was given the diagnosis of factitious disorder (malingering or Munchausen syndrome). After extensive discussion, the patient was amenable to outpatient mental health counseling.
Dermoscopy is not a standard method to diagnose cellulitis of the skin; however, when patients present with an atypical response to appropriate care, the presumed diagnosis must be challenged. This patient had dramatized symptoms, false medical history, and numerous hospitalizations that were suspicious for factitious disorder.1 Furthermore, the physical examination was inconsistent with the classic course of cellulitis. In this case, dermoscopy had advantages over biopsies because it was noninvasive, gave immediate feedback, and provided a macroscopic view of the morphology. Via dermoscopy, we had an objective lens to distinguish cellulitis from cosmetic product and to obtain the correct diagnosis.
- Harth W, Taube KM, Gieler U. Facticious disorders in dermatology. J Dtsch Dermatol Ges. 2010;8:361-372.
To the Editor:
Patients with psychocutaneous disorders present unique challenges to physicians. We illustrate the critical role that dermoscopy may play to illuminate exogenous skin pathology.
A 50-year-old woman with a reported medical history of systemic lupus erythematosus, chronic pain, and nonhealing leg ulcers presented to the emergency department with severe pain of the left lower leg and redness that was concerning for cellulitis. She sought treatment at an outside hospital for cellulitis 2 weeks prior but left against medical advice. Symptomatic review revealed chest pain, shortness of breath, nausea, vomiting, and diarrhea. The primary team started her on intravenous clindamycin and vancomycin for the presumed infection and scheduled narcotic medications due to concerns of intractable pain in the left leg. The dermatology department was consulted after failure to improve with 1 week of systemic antibiotics.
Physical examination revealed a geometric, atrophic, purple plaque on the left anterior shin from a prior leg ulcer as well as a diffuse red-pink patch extending from the knee to the ankle. Notably, the cellulitis spared the left posterior calf resting against the sheet and had a sharp line of demarcation at the distal shin. The leg was cool to the touch while the patient was distractible. She later reported that the leg was extremely tender to palpation. Dermoscopy revealed linear red pigments within skin furrows that accentuated skin lines (Figure). These findings raised suspicions of an external manipulation. The skin was wiped with an alcohol pad that removed a shimmering pink substance consistent in appearance to a cosmetic product. The skin beneath the cellulitis appeared normal.
On further review of the patient’s medical record, it was noted that she was admitted several months ago for ulcers of the left leg. She had been to multiple hospitals and had numerous rounds of antibiotics. Biopsy of an ulcer revealed dermal fibrosis consistent with scarring. Aerobic bacteria, atypical mycobacteria, and fungal cultures were all negative. The physicians suspected a self-induced etiology consistent with dermatitis artefacta. The patient emphasized multiple psychosocial stressors as well as having frequent lupus flares despite repeated negative workup. Given the exaggerated symptoms and unnecessary hospital visits, she was given the diagnosis of factitious disorder (malingering or Munchausen syndrome). After extensive discussion, the patient was amenable to outpatient mental health counseling.
Dermoscopy is not a standard method to diagnose cellulitis of the skin; however, when patients present with an atypical response to appropriate care, the presumed diagnosis must be challenged. This patient had dramatized symptoms, false medical history, and numerous hospitalizations that were suspicious for factitious disorder.1 Furthermore, the physical examination was inconsistent with the classic course of cellulitis. In this case, dermoscopy had advantages over biopsies because it was noninvasive, gave immediate feedback, and provided a macroscopic view of the morphology. Via dermoscopy, we had an objective lens to distinguish cellulitis from cosmetic product and to obtain the correct diagnosis.
To the Editor:
Patients with psychocutaneous disorders present unique challenges to physicians. We illustrate the critical role that dermoscopy may play to illuminate exogenous skin pathology.
A 50-year-old woman with a reported medical history of systemic lupus erythematosus, chronic pain, and nonhealing leg ulcers presented to the emergency department with severe pain of the left lower leg and redness that was concerning for cellulitis. She sought treatment at an outside hospital for cellulitis 2 weeks prior but left against medical advice. Symptomatic review revealed chest pain, shortness of breath, nausea, vomiting, and diarrhea. The primary team started her on intravenous clindamycin and vancomycin for the presumed infection and scheduled narcotic medications due to concerns of intractable pain in the left leg. The dermatology department was consulted after failure to improve with 1 week of systemic antibiotics.
Physical examination revealed a geometric, atrophic, purple plaque on the left anterior shin from a prior leg ulcer as well as a diffuse red-pink patch extending from the knee to the ankle. Notably, the cellulitis spared the left posterior calf resting against the sheet and had a sharp line of demarcation at the distal shin. The leg was cool to the touch while the patient was distractible. She later reported that the leg was extremely tender to palpation. Dermoscopy revealed linear red pigments within skin furrows that accentuated skin lines (Figure). These findings raised suspicions of an external manipulation. The skin was wiped with an alcohol pad that removed a shimmering pink substance consistent in appearance to a cosmetic product. The skin beneath the cellulitis appeared normal.
On further review of the patient’s medical record, it was noted that she was admitted several months ago for ulcers of the left leg. She had been to multiple hospitals and had numerous rounds of antibiotics. Biopsy of an ulcer revealed dermal fibrosis consistent with scarring. Aerobic bacteria, atypical mycobacteria, and fungal cultures were all negative. The physicians suspected a self-induced etiology consistent with dermatitis artefacta. The patient emphasized multiple psychosocial stressors as well as having frequent lupus flares despite repeated negative workup. Given the exaggerated symptoms and unnecessary hospital visits, she was given the diagnosis of factitious disorder (malingering or Munchausen syndrome). After extensive discussion, the patient was amenable to outpatient mental health counseling.
Dermoscopy is not a standard method to diagnose cellulitis of the skin; however, when patients present with an atypical response to appropriate care, the presumed diagnosis must be challenged. This patient had dramatized symptoms, false medical history, and numerous hospitalizations that were suspicious for factitious disorder.1 Furthermore, the physical examination was inconsistent with the classic course of cellulitis. In this case, dermoscopy had advantages over biopsies because it was noninvasive, gave immediate feedback, and provided a macroscopic view of the morphology. Via dermoscopy, we had an objective lens to distinguish cellulitis from cosmetic product and to obtain the correct diagnosis.
- Harth W, Taube KM, Gieler U. Facticious disorders in dermatology. J Dtsch Dermatol Ges. 2010;8:361-372.
- Harth W, Taube KM, Gieler U. Facticious disorders in dermatology. J Dtsch Dermatol Ges. 2010;8:361-372.
Practice Points
- Consider exogenous factors or alternative diagnoses when a patient does not respond to appropriate care.
- Although dermoscopy is not used to diagnose cellulitis, it could be helpful in distinguishing cosmetic products used in dermatitis artefacta.
Pigmented Basal Cell Carcinoma With Annular Leukoderma
To the Editor:
Annular leukoderma, or the halo phenomenon, is a circular reaction of hypopigmentation that most commonly is observed alongside congenital nevi, acquired melanocytic nevi, blue nevi, Spitz nevi, vitiligo, and rarely melanoma.1 There is limited literature on the mechanism of the halo phenomenon. Most of the literature proposes a T cell–mediated immune response to antigens, which causes not only surrounding pigment loss but also heralds the regression of central lesions.2 Others have suggested a vascular mechanism, with blood shunted away from the lesions.3 Because guidelines discourage biopsy of typical halo nevi, it becomes important to evaluate lesions for worrisome features such as ulceration or asymmetry, especially in older patients. We present a case of a pigmented basal cell carcinoma (BCC) that exhibited the halo phenomenon. Four other cases have been described in the literature.3-6
A 53-year-old man presented for evaluation of an asymptomatic lesion on the left side of the abdomen of approximately 8 months’ duration. He had no personal or family history of skin cancer. Physical examination revealed a central 1-cm, pink, verrucous papule surrounded by a 2×1.2-cm, depigmented, circular patch on the left side of the inferior abdomen (Figure 1). Upon questioning, the patient produced cell phone photographs of the trunk from 3 years prior, which did not show any lesions present. Full-body skin examination did not reveal any other concerning pigmented lesions. Excisional biopsy was performed due to concern for amelanotic melanoma, and histopathology revealed a superficial and pigmented BCC (Figure 2). Immunohistochemistry with Melan-A was negative for atypical melanocytes, with no uptake in the leukoderma areas.
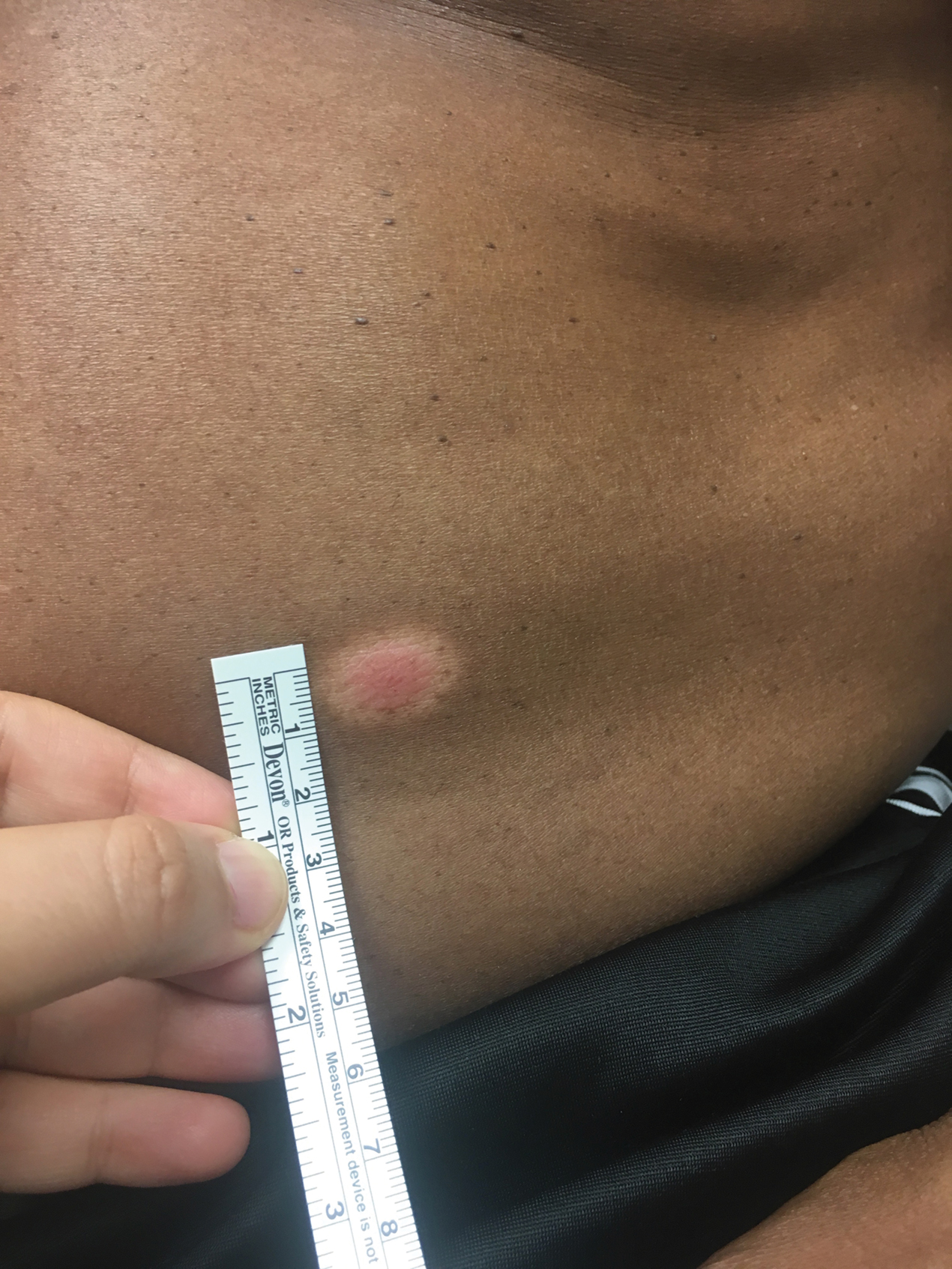
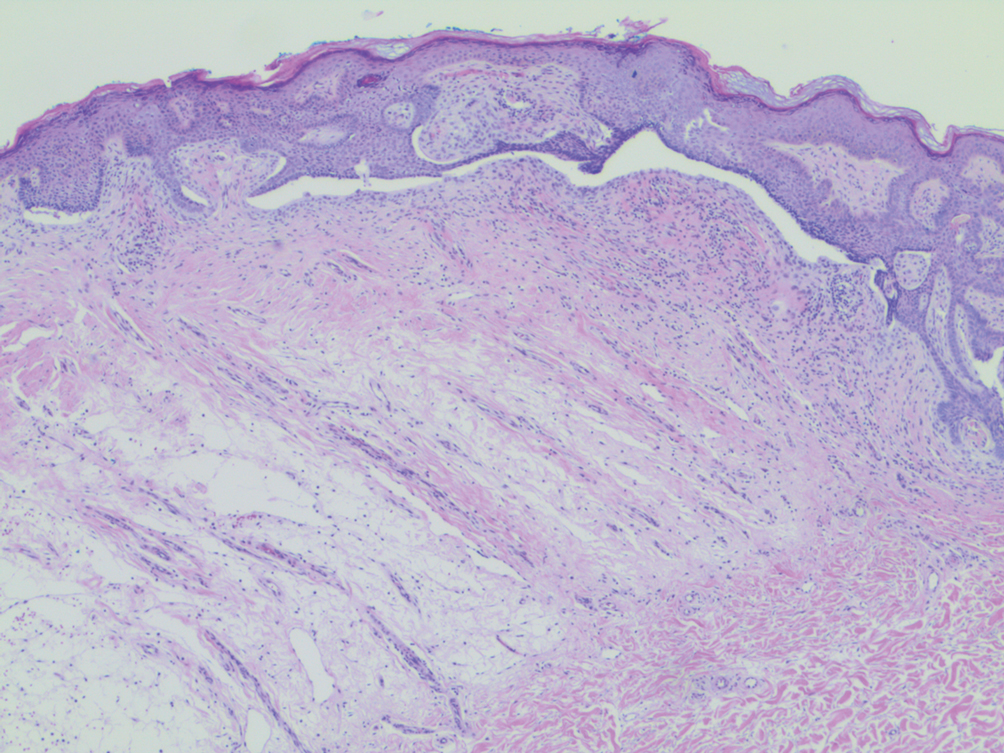
The clinical presentation initially was concerning for amelanotic melanoma. All melanoma subtypes may appear as hypomelanotic lesions, though these most commonly are observed in the desmoplastic or nodular subtypes. Amelanotic melanomas may present as well-defined red or pink macules, plaques, or nodules, with some tumors presenting with light brown pigmentation.7
The differential diagnosis for lesions with the halo phenomenon is large. In adults, the halo phenomenon may be concerning for malignant or regressing melanoma. As an immunogenic tumor, melanoma’s immunogenic melanocytes may incite a cell-mediated immune response to antigens common to neoplastic and normal melanocytes, which can clinically manifest not only as local annular leukoderma but also as distant vitiligo or halo nevi.7 The halo phenomenon more commonly is associated with benign processes such as vitiligo and halo nevi in children. In most children, halo nevi occur as an isolated phenomenon but still warrant a complete skin examination for melanoma and vitiligo. The presence of halo nevi has been associated with distant vitiligo—possibly through shared immunologic mechanisms—especially if patients present with the Koebner phenomenon, multiple halo nevi, or a family history of vitiligo.8 A prospective study also found that the presence of halo nevi was an independent risk factor for the progression of segmental vitiligo to mixed vitiligo.9 Hormones also may play a role in the leukoderma acquisitum centrifugum, or halo, nevi. Halo nevi most commonly affect adolescents and pregnant women. It has been postulated that congenital nevi may be unique in their response to altered estrogen levels, increasing the rate not only of halo nevi but also of melanoma in pregnant women.10
Our patient’s final histologic diagnosis was pigmented BCC, which comprises only 6% of all BCCs.3 The proposed mechanism is that melanocytes colonize the tumor in the surrounding stroma and produce excess melanin. Basal cell carcinoma with halo phenomenon is a rare presentation. As in our case, 2 prior BCC reports also involved patients older than 50 years,3,5 with the 2 other cases describing women in their late twenties and early thirties.4,6 Additionally, 2 of 4 reports described patients with a history of multiple BCCs.3,5
In summary, the seemingly benign halo phenomenon may accompany malignant processes such as nonmelanoma skin cancer. Careful consideration of lesion time course and atypia is imperative for proper clinical suspicion in such cases.
- Mooney MA, Barr RJ, Buxton MG. Halo nevus or halo phenomenon? a study of 142 cases. J Cutan Pathol. 1995;22:342-348.
- Zeff RA, Freitag A, Grin CM, et al. The immune response in halo nevi. J Am Acad Dermatol. 1997;37:620-624.
- Johnson DB, Ceilley RI. Basal cell carcinoma with annular leukoderma mimicking leukoderma acquisitum centrifugum. Arch Dermatol. 1980;116:352-353.
- Basak PY, Meric G, Ciris M. Basal cell carcinoma with halo phenomenon in a young female: significance of dermatoscopy in early diagnosis. Indian J Dermatol. 2015;60:214.
- Pembroke AC, Liddell K. Basal cell epithelioma with a hypopigmented halo. Arch Dermatol. 1981;117:317.
- Rustemeyer J, Günther L, Deichert L. A rare association: basal cell carcinoma in a vitiliginous macula. Oral Maxillofac Surg. 2011;15:175-177.
- Naveh HP, Rao UN, Butterfield LH. Melanoma‐associated leukoderma—immunology in black and white? Pigment Cell Melanoma Res. 2013;26:796-804.
- Zhou H, Wu L-C, Chen M-K, et al. Factors associated with development of vitiligo in patients with halo nevus. Chinese Med J. 2017;130:2703.
- Ezzedine K, Diallo A, Léauté‐Labrèze C, et al. Halo naevi and leukotrichia are strong predictors of the passage to mixed vitiligo in a subgroup of segmental vitiligo. Br J Dermatol. 2012;166:539-544.
- Nading MA, Nanney LB, Ellis DL. Pregnancy and estrogen receptor β expression in a large congenital nevus. Arch Dermatol. 2009;145:691-694.
To the Editor:
Annular leukoderma, or the halo phenomenon, is a circular reaction of hypopigmentation that most commonly is observed alongside congenital nevi, acquired melanocytic nevi, blue nevi, Spitz nevi, vitiligo, and rarely melanoma.1 There is limited literature on the mechanism of the halo phenomenon. Most of the literature proposes a T cell–mediated immune response to antigens, which causes not only surrounding pigment loss but also heralds the regression of central lesions.2 Others have suggested a vascular mechanism, with blood shunted away from the lesions.3 Because guidelines discourage biopsy of typical halo nevi, it becomes important to evaluate lesions for worrisome features such as ulceration or asymmetry, especially in older patients. We present a case of a pigmented basal cell carcinoma (BCC) that exhibited the halo phenomenon. Four other cases have been described in the literature.3-6
A 53-year-old man presented for evaluation of an asymptomatic lesion on the left side of the abdomen of approximately 8 months’ duration. He had no personal or family history of skin cancer. Physical examination revealed a central 1-cm, pink, verrucous papule surrounded by a 2×1.2-cm, depigmented, circular patch on the left side of the inferior abdomen (Figure 1). Upon questioning, the patient produced cell phone photographs of the trunk from 3 years prior, which did not show any lesions present. Full-body skin examination did not reveal any other concerning pigmented lesions. Excisional biopsy was performed due to concern for amelanotic melanoma, and histopathology revealed a superficial and pigmented BCC (Figure 2). Immunohistochemistry with Melan-A was negative for atypical melanocytes, with no uptake in the leukoderma areas.
The clinical presentation initially was concerning for amelanotic melanoma. All melanoma subtypes may appear as hypomelanotic lesions, though these most commonly are observed in the desmoplastic or nodular subtypes. Amelanotic melanomas may present as well-defined red or pink macules, plaques, or nodules, with some tumors presenting with light brown pigmentation.7
The differential diagnosis for lesions with the halo phenomenon is large. In adults, the halo phenomenon may be concerning for malignant or regressing melanoma. As an immunogenic tumor, melanoma’s immunogenic melanocytes may incite a cell-mediated immune response to antigens common to neoplastic and normal melanocytes, which can clinically manifest not only as local annular leukoderma but also as distant vitiligo or halo nevi.7 The halo phenomenon more commonly is associated with benign processes such as vitiligo and halo nevi in children. In most children, halo nevi occur as an isolated phenomenon but still warrant a complete skin examination for melanoma and vitiligo. The presence of halo nevi has been associated with distant vitiligo—possibly through shared immunologic mechanisms—especially if patients present with the Koebner phenomenon, multiple halo nevi, or a family history of vitiligo.8 A prospective study also found that the presence of halo nevi was an independent risk factor for the progression of segmental vitiligo to mixed vitiligo.9 Hormones also may play a role in the leukoderma acquisitum centrifugum, or halo, nevi. Halo nevi most commonly affect adolescents and pregnant women. It has been postulated that congenital nevi may be unique in their response to altered estrogen levels, increasing the rate not only of halo nevi but also of melanoma in pregnant women.10
Our patient’s final histologic diagnosis was pigmented BCC, which comprises only 6% of all BCCs.3 The proposed mechanism is that melanocytes colonize the tumor in the surrounding stroma and produce excess melanin. Basal cell carcinoma with halo phenomenon is a rare presentation. As in our case, 2 prior BCC reports also involved patients older than 50 years,3,5 with the 2 other cases describing women in their late twenties and early thirties.4,6 Additionally, 2 of 4 reports described patients with a history of multiple BCCs.3,5
In summary, the seemingly benign halo phenomenon may accompany malignant processes such as nonmelanoma skin cancer. Careful consideration of lesion time course and atypia is imperative for proper clinical suspicion in such cases.
To the Editor:
Annular leukoderma, or the halo phenomenon, is a circular reaction of hypopigmentation that most commonly is observed alongside congenital nevi, acquired melanocytic nevi, blue nevi, Spitz nevi, vitiligo, and rarely melanoma.1 There is limited literature on the mechanism of the halo phenomenon. Most of the literature proposes a T cell–mediated immune response to antigens, which causes not only surrounding pigment loss but also heralds the regression of central lesions.2 Others have suggested a vascular mechanism, with blood shunted away from the lesions.3 Because guidelines discourage biopsy of typical halo nevi, it becomes important to evaluate lesions for worrisome features such as ulceration or asymmetry, especially in older patients. We present a case of a pigmented basal cell carcinoma (BCC) that exhibited the halo phenomenon. Four other cases have been described in the literature.3-6
A 53-year-old man presented for evaluation of an asymptomatic lesion on the left side of the abdomen of approximately 8 months’ duration. He had no personal or family history of skin cancer. Physical examination revealed a central 1-cm, pink, verrucous papule surrounded by a 2×1.2-cm, depigmented, circular patch on the left side of the inferior abdomen (Figure 1). Upon questioning, the patient produced cell phone photographs of the trunk from 3 years prior, which did not show any lesions present. Full-body skin examination did not reveal any other concerning pigmented lesions. Excisional biopsy was performed due to concern for amelanotic melanoma, and histopathology revealed a superficial and pigmented BCC (Figure 2). Immunohistochemistry with Melan-A was negative for atypical melanocytes, with no uptake in the leukoderma areas.
The clinical presentation initially was concerning for amelanotic melanoma. All melanoma subtypes may appear as hypomelanotic lesions, though these most commonly are observed in the desmoplastic or nodular subtypes. Amelanotic melanomas may present as well-defined red or pink macules, plaques, or nodules, with some tumors presenting with light brown pigmentation.7
The differential diagnosis for lesions with the halo phenomenon is large. In adults, the halo phenomenon may be concerning for malignant or regressing melanoma. As an immunogenic tumor, melanoma’s immunogenic melanocytes may incite a cell-mediated immune response to antigens common to neoplastic and normal melanocytes, which can clinically manifest not only as local annular leukoderma but also as distant vitiligo or halo nevi.7 The halo phenomenon more commonly is associated with benign processes such as vitiligo and halo nevi in children. In most children, halo nevi occur as an isolated phenomenon but still warrant a complete skin examination for melanoma and vitiligo. The presence of halo nevi has been associated with distant vitiligo—possibly through shared immunologic mechanisms—especially if patients present with the Koebner phenomenon, multiple halo nevi, or a family history of vitiligo.8 A prospective study also found that the presence of halo nevi was an independent risk factor for the progression of segmental vitiligo to mixed vitiligo.9 Hormones also may play a role in the leukoderma acquisitum centrifugum, or halo, nevi. Halo nevi most commonly affect adolescents and pregnant women. It has been postulated that congenital nevi may be unique in their response to altered estrogen levels, increasing the rate not only of halo nevi but also of melanoma in pregnant women.10
Our patient’s final histologic diagnosis was pigmented BCC, which comprises only 6% of all BCCs.3 The proposed mechanism is that melanocytes colonize the tumor in the surrounding stroma and produce excess melanin. Basal cell carcinoma with halo phenomenon is a rare presentation. As in our case, 2 prior BCC reports also involved patients older than 50 years,3,5 with the 2 other cases describing women in their late twenties and early thirties.4,6 Additionally, 2 of 4 reports described patients with a history of multiple BCCs.3,5
In summary, the seemingly benign halo phenomenon may accompany malignant processes such as nonmelanoma skin cancer. Careful consideration of lesion time course and atypia is imperative for proper clinical suspicion in such cases.
- Mooney MA, Barr RJ, Buxton MG. Halo nevus or halo phenomenon? a study of 142 cases. J Cutan Pathol. 1995;22:342-348.
- Zeff RA, Freitag A, Grin CM, et al. The immune response in halo nevi. J Am Acad Dermatol. 1997;37:620-624.
- Johnson DB, Ceilley RI. Basal cell carcinoma with annular leukoderma mimicking leukoderma acquisitum centrifugum. Arch Dermatol. 1980;116:352-353.
- Basak PY, Meric G, Ciris M. Basal cell carcinoma with halo phenomenon in a young female: significance of dermatoscopy in early diagnosis. Indian J Dermatol. 2015;60:214.
- Pembroke AC, Liddell K. Basal cell epithelioma with a hypopigmented halo. Arch Dermatol. 1981;117:317.
- Rustemeyer J, Günther L, Deichert L. A rare association: basal cell carcinoma in a vitiliginous macula. Oral Maxillofac Surg. 2011;15:175-177.
- Naveh HP, Rao UN, Butterfield LH. Melanoma‐associated leukoderma—immunology in black and white? Pigment Cell Melanoma Res. 2013;26:796-804.
- Zhou H, Wu L-C, Chen M-K, et al. Factors associated with development of vitiligo in patients with halo nevus. Chinese Med J. 2017;130:2703.
- Ezzedine K, Diallo A, Léauté‐Labrèze C, et al. Halo naevi and leukotrichia are strong predictors of the passage to mixed vitiligo in a subgroup of segmental vitiligo. Br J Dermatol. 2012;166:539-544.
- Nading MA, Nanney LB, Ellis DL. Pregnancy and estrogen receptor β expression in a large congenital nevus. Arch Dermatol. 2009;145:691-694.
- Mooney MA, Barr RJ, Buxton MG. Halo nevus or halo phenomenon? a study of 142 cases. J Cutan Pathol. 1995;22:342-348.
- Zeff RA, Freitag A, Grin CM, et al. The immune response in halo nevi. J Am Acad Dermatol. 1997;37:620-624.
- Johnson DB, Ceilley RI. Basal cell carcinoma with annular leukoderma mimicking leukoderma acquisitum centrifugum. Arch Dermatol. 1980;116:352-353.
- Basak PY, Meric G, Ciris M. Basal cell carcinoma with halo phenomenon in a young female: significance of dermatoscopy in early diagnosis. Indian J Dermatol. 2015;60:214.
- Pembroke AC, Liddell K. Basal cell epithelioma with a hypopigmented halo. Arch Dermatol. 1981;117:317.
- Rustemeyer J, Günther L, Deichert L. A rare association: basal cell carcinoma in a vitiliginous macula. Oral Maxillofac Surg. 2011;15:175-177.
- Naveh HP, Rao UN, Butterfield LH. Melanoma‐associated leukoderma—immunology in black and white? Pigment Cell Melanoma Res. 2013;26:796-804.
- Zhou H, Wu L-C, Chen M-K, et al. Factors associated with development of vitiligo in patients with halo nevus. Chinese Med J. 2017;130:2703.
- Ezzedine K, Diallo A, Léauté‐Labrèze C, et al. Halo naevi and leukotrichia are strong predictors of the passage to mixed vitiligo in a subgroup of segmental vitiligo. Br J Dermatol. 2012;166:539-544.
- Nading MA, Nanney LB, Ellis DL. Pregnancy and estrogen receptor β expression in a large congenital nevus. Arch Dermatol. 2009;145:691-694.
Practice Points
- Annular leukoderma, or the halo phenomenon, is a circular reaction of hypopigmentation that more commonly is associated with benign processes such as halo nevi.
- The halo phenomenon may accompany malignant processes, such as nonmelanoma skin cancer. Careful consideration of lesion time course and atypia is imperative for proper clinical suspicion in such cases.
Argyria From a Topical Home Remedy
To the Editor:
Argyria is a rare disease caused by chronic exposure to products with high silver content (eg, oral ingestion, inhalation, percutaneous absorption). With time, the blood levels of silver surpass the body’s renal and hepatic excretory capacities that lead to silver granules being deposited in the skin and internal organs, including the liver, spleen, adrenal glands, and bone marrow.1 The cutaneous deposition results in a blue or blue-gray pigmentation of the skin, mucous membranes, and nails. Intervals of exposure that span from 8 months to 5 years prior to symptom onset have been described in the literature.2 The discoloration that results often is permanent, with no established way of effectively removing silver deposits from the tissue.3
A 22-year-old autistic man, who was completely dependent on his mother’s care, presented to the emergency department with a primary concern of abdominal pain. The mother reported that he was indicating abdominal pain by motioning to his stomach for the last 5 days. The mother also reported he did not have a bowel movement during this time, and she noticed his hands were shaking. Prior to presentation, the mother had given him 2 enemas and had him on a 3-day strict liquid fast consisting of water, lemon juice, cayenne pepper, honey, and orange juice. Notably, the mother had a strong history of using naturopathic remedies for treatment of her son’s ailments.
On admission, the patient was stable. There was a 2-point decrease in the patient’s body mass index over the last month. Initial serum electrolytes were highly abnormal with a serum sodium level of 124 mEq/L (reference range, 135–145 mEq/L), blood urea nitrogen of 3 mg/dL (reference range, 7–20 mg/dL), creatinine of 0.77 mg/dL (reference range, 0.74–1.35 mg/dL), and lactic acid of 2.1 mEq/L (reference range, 0.5–1 mEq/L). Serum osmolality was 272 mOsm/kg (reference range, 275–295 mOsm/kg). Urine osmolality was 114 mOsm/kg (reference range, 500–850 mOsm/kg) with a low-normal urine sodium level of 41 mmol/24 hr (reference range, 40–220 mmol/24 hr). Abnormalities were felt to be secondary to malnutrition from the strict liquid diet (blood urea nitrogen and creatinine ratio of 3:1 suggestive of notable protein calorie malnutrition). The patient was given 1 L of normal saline in the emergency department, with further fluids held so as not to increase serum sodium level too rapidly. A regular diet was started.
Physical examination revealed dry mucosal membranes but otherwise was unremarkable. Active bowel sounds were noted, as well as a soft, nontender, and nondistended abdomen; however, when examining the patient’s hands for reported shaking, a distinct abnormality of the nails was noticed. The patient had slate blue discoloration of the lunula, along with hyperpigmented violaceous discoloration of the proximal nail bed on all 10 fingernails (Figure 1). No abnormalities were seen on the toenails. The mother had a distinct bluish gray discoloration of the face as well as similar nail findings (Figure 2), strongly suggestive of colloidal silver use. An urgent serum silver level was ordered on the patient as well as a heavy metal panel. The mother was found applying numerous “natural remedies” to the patient’s skin while in the hospital, including a liquid spray and lotion, both in unmarked bottles. At that time, the mother was informed that no external supplements should be applied to her son. The serum silver level was elevated substantially at 94.3 ng/mL (reference range, <1.0 ng/mL). When the mother was confronted, she initially denied use of silver but later admitted to notable silver content in the cream she was applying to her son’s skin. The mother reported that she read online that colloidal silver had been historically used to cure numerous ailments and she was ordering products from an online company. She was counseled on the dangers of both topical application and ingestion of silver, and all supplements were removed from the home.
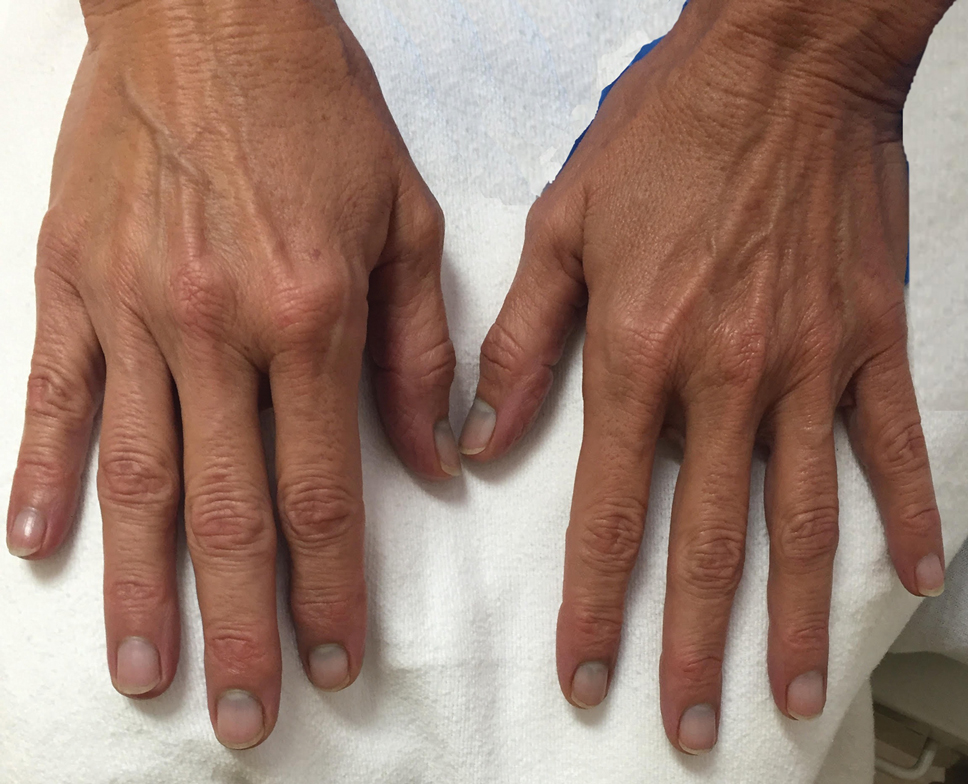
Argyria is a rare condition caused by chronic exposure to silver and is characterized by a blue-gray pigmentation in the skin and appendages, mucous membranes, and internal organs.4 Clinically, argyria is classified as generalized or localized. Generalized argyria results from ingestion or inhalation of silver compounds, where granules deposit preferentially in sun-exposed areas of skin as well as internal organs, with the highest concentration in the liver, spleen, and adrenal glands; discoloration often is permanent.5 On the contrary, localized argyria results from direct external contact with silver and granules deposited in the hands, eyes, and mucosa.5 Although the exact mechanism of penetration from topical silver remains unknown, it is thought to enter via the eccrine sweat ducts, as histopathology reveals silver granules found in highest concentration surrounding sweat glands in the dermis.6
Initial differential diagnoses for altered nail pigmentation include drug-induced causes, systemic diseases, cyanosis, and exposure to metals.7 The most commonly indicated medications resulting in blue nail pigment changes include antimalarials, minocycline, zidovudine, and phenothiazine. Systemic diseases that may cause blue nail color change include Wilson disease, hemochromatosis, Addison disease, methemoglobinemia, and alkaptonuria.7 Metals include gold, mercury, arsenic, bismuth, lead, and silver.4 After a thorough review of the patient’s medications and lack of support for any underlying disease process, contact with metals, particularly silver, was ranked highly on our differential list. In support of this theory, the mother’s bluish gray facial skin led to high clinical suspicion that she was ingesting colloidal silver and also was exposing her son to silver.
Treatment of argyria is challenging but first and foremost involves discontinuation of the source of chronic silver exposure. Unfortunately, the discoloration of generalized argyria often is permanent. Sunscreen can be used to help prevent any further darkening of pigment. The pigment in localized argyria has been reported to slowly fade with time, and there also have been reports of successful treatment using a low-fluence Q-switched 1064-nm Nd:YAG laser.8
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Lencastre A, Lobo M, João A. Argyria—case report. An Bras Dermatol. 2013;88:413-416.
- Park S-W, Kim J-H, Shin H-T, et al. An effective modality for argyria treatment: Q-switched 1,064-nm Nd:YAG laser. Ann Dermatol. 2013;25:511-512.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Garcias-Ladaria J, Hernandez-Bel P, Torregrosa-Calatayud JL, et al. Localized cutaneous argyria: a report of 2 cases. Actas Dermosifiliogr. 2013;104:253-254.
- Kapur N, Landon G, Yu RC. Localized argyria in an antique restorer. Br J Dermatol. 2001;144:191-192.
- Kubba A, Kubba R, Batrani M, Pal T. Argyria an unrecognized cause of cutaneous pigmentation in Indian patients: a case series and review of the literature. Indian J Dermatol Venereol Leprol. 2013;79:805-811.
- Han TY, Chang HS, Lee HK, et al. Successful treatment of argyria using a low-fluence Q-switched 1064-nm Nd:YAG laser. Int J Dermatol. 2011;50:751-753.
To the Editor:
Argyria is a rare disease caused by chronic exposure to products with high silver content (eg, oral ingestion, inhalation, percutaneous absorption). With time, the blood levels of silver surpass the body’s renal and hepatic excretory capacities that lead to silver granules being deposited in the skin and internal organs, including the liver, spleen, adrenal glands, and bone marrow.1 The cutaneous deposition results in a blue or blue-gray pigmentation of the skin, mucous membranes, and nails. Intervals of exposure that span from 8 months to 5 years prior to symptom onset have been described in the literature.2 The discoloration that results often is permanent, with no established way of effectively removing silver deposits from the tissue.3
A 22-year-old autistic man, who was completely dependent on his mother’s care, presented to the emergency department with a primary concern of abdominal pain. The mother reported that he was indicating abdominal pain by motioning to his stomach for the last 5 days. The mother also reported he did not have a bowel movement during this time, and she noticed his hands were shaking. Prior to presentation, the mother had given him 2 enemas and had him on a 3-day strict liquid fast consisting of water, lemon juice, cayenne pepper, honey, and orange juice. Notably, the mother had a strong history of using naturopathic remedies for treatment of her son’s ailments.
On admission, the patient was stable. There was a 2-point decrease in the patient’s body mass index over the last month. Initial serum electrolytes were highly abnormal with a serum sodium level of 124 mEq/L (reference range, 135–145 mEq/L), blood urea nitrogen of 3 mg/dL (reference range, 7–20 mg/dL), creatinine of 0.77 mg/dL (reference range, 0.74–1.35 mg/dL), and lactic acid of 2.1 mEq/L (reference range, 0.5–1 mEq/L). Serum osmolality was 272 mOsm/kg (reference range, 275–295 mOsm/kg). Urine osmolality was 114 mOsm/kg (reference range, 500–850 mOsm/kg) with a low-normal urine sodium level of 41 mmol/24 hr (reference range, 40–220 mmol/24 hr). Abnormalities were felt to be secondary to malnutrition from the strict liquid diet (blood urea nitrogen and creatinine ratio of 3:1 suggestive of notable protein calorie malnutrition). The patient was given 1 L of normal saline in the emergency department, with further fluids held so as not to increase serum sodium level too rapidly. A regular diet was started.
Physical examination revealed dry mucosal membranes but otherwise was unremarkable. Active bowel sounds were noted, as well as a soft, nontender, and nondistended abdomen; however, when examining the patient’s hands for reported shaking, a distinct abnormality of the nails was noticed. The patient had slate blue discoloration of the lunula, along with hyperpigmented violaceous discoloration of the proximal nail bed on all 10 fingernails (Figure 1). No abnormalities were seen on the toenails. The mother had a distinct bluish gray discoloration of the face as well as similar nail findings (Figure 2), strongly suggestive of colloidal silver use. An urgent serum silver level was ordered on the patient as well as a heavy metal panel. The mother was found applying numerous “natural remedies” to the patient’s skin while in the hospital, including a liquid spray and lotion, both in unmarked bottles. At that time, the mother was informed that no external supplements should be applied to her son. The serum silver level was elevated substantially at 94.3 ng/mL (reference range, <1.0 ng/mL). When the mother was confronted, she initially denied use of silver but later admitted to notable silver content in the cream she was applying to her son’s skin. The mother reported that she read online that colloidal silver had been historically used to cure numerous ailments and she was ordering products from an online company. She was counseled on the dangers of both topical application and ingestion of silver, and all supplements were removed from the home.
Argyria is a rare condition caused by chronic exposure to silver and is characterized by a blue-gray pigmentation in the skin and appendages, mucous membranes, and internal organs.4 Clinically, argyria is classified as generalized or localized. Generalized argyria results from ingestion or inhalation of silver compounds, where granules deposit preferentially in sun-exposed areas of skin as well as internal organs, with the highest concentration in the liver, spleen, and adrenal glands; discoloration often is permanent.5 On the contrary, localized argyria results from direct external contact with silver and granules deposited in the hands, eyes, and mucosa.5 Although the exact mechanism of penetration from topical silver remains unknown, it is thought to enter via the eccrine sweat ducts, as histopathology reveals silver granules found in highest concentration surrounding sweat glands in the dermis.6
Initial differential diagnoses for altered nail pigmentation include drug-induced causes, systemic diseases, cyanosis, and exposure to metals.7 The most commonly indicated medications resulting in blue nail pigment changes include antimalarials, minocycline, zidovudine, and phenothiazine. Systemic diseases that may cause blue nail color change include Wilson disease, hemochromatosis, Addison disease, methemoglobinemia, and alkaptonuria.7 Metals include gold, mercury, arsenic, bismuth, lead, and silver.4 After a thorough review of the patient’s medications and lack of support for any underlying disease process, contact with metals, particularly silver, was ranked highly on our differential list. In support of this theory, the mother’s bluish gray facial skin led to high clinical suspicion that she was ingesting colloidal silver and also was exposing her son to silver.
Treatment of argyria is challenging but first and foremost involves discontinuation of the source of chronic silver exposure. Unfortunately, the discoloration of generalized argyria often is permanent. Sunscreen can be used to help prevent any further darkening of pigment. The pigment in localized argyria has been reported to slowly fade with time, and there also have been reports of successful treatment using a low-fluence Q-switched 1064-nm Nd:YAG laser.8
To the Editor:
Argyria is a rare disease caused by chronic exposure to products with high silver content (eg, oral ingestion, inhalation, percutaneous absorption). With time, the blood levels of silver surpass the body’s renal and hepatic excretory capacities that lead to silver granules being deposited in the skin and internal organs, including the liver, spleen, adrenal glands, and bone marrow.1 The cutaneous deposition results in a blue or blue-gray pigmentation of the skin, mucous membranes, and nails. Intervals of exposure that span from 8 months to 5 years prior to symptom onset have been described in the literature.2 The discoloration that results often is permanent, with no established way of effectively removing silver deposits from the tissue.3
A 22-year-old autistic man, who was completely dependent on his mother’s care, presented to the emergency department with a primary concern of abdominal pain. The mother reported that he was indicating abdominal pain by motioning to his stomach for the last 5 days. The mother also reported he did not have a bowel movement during this time, and she noticed his hands were shaking. Prior to presentation, the mother had given him 2 enemas and had him on a 3-day strict liquid fast consisting of water, lemon juice, cayenne pepper, honey, and orange juice. Notably, the mother had a strong history of using naturopathic remedies for treatment of her son’s ailments.
On admission, the patient was stable. There was a 2-point decrease in the patient’s body mass index over the last month. Initial serum electrolytes were highly abnormal with a serum sodium level of 124 mEq/L (reference range, 135–145 mEq/L), blood urea nitrogen of 3 mg/dL (reference range, 7–20 mg/dL), creatinine of 0.77 mg/dL (reference range, 0.74–1.35 mg/dL), and lactic acid of 2.1 mEq/L (reference range, 0.5–1 mEq/L). Serum osmolality was 272 mOsm/kg (reference range, 275–295 mOsm/kg). Urine osmolality was 114 mOsm/kg (reference range, 500–850 mOsm/kg) with a low-normal urine sodium level of 41 mmol/24 hr (reference range, 40–220 mmol/24 hr). Abnormalities were felt to be secondary to malnutrition from the strict liquid diet (blood urea nitrogen and creatinine ratio of 3:1 suggestive of notable protein calorie malnutrition). The patient was given 1 L of normal saline in the emergency department, with further fluids held so as not to increase serum sodium level too rapidly. A regular diet was started.
Physical examination revealed dry mucosal membranes but otherwise was unremarkable. Active bowel sounds were noted, as well as a soft, nontender, and nondistended abdomen; however, when examining the patient’s hands for reported shaking, a distinct abnormality of the nails was noticed. The patient had slate blue discoloration of the lunula, along with hyperpigmented violaceous discoloration of the proximal nail bed on all 10 fingernails (Figure 1). No abnormalities were seen on the toenails. The mother had a distinct bluish gray discoloration of the face as well as similar nail findings (Figure 2), strongly suggestive of colloidal silver use. An urgent serum silver level was ordered on the patient as well as a heavy metal panel. The mother was found applying numerous “natural remedies” to the patient’s skin while in the hospital, including a liquid spray and lotion, both in unmarked bottles. At that time, the mother was informed that no external supplements should be applied to her son. The serum silver level was elevated substantially at 94.3 ng/mL (reference range, <1.0 ng/mL). When the mother was confronted, she initially denied use of silver but later admitted to notable silver content in the cream she was applying to her son’s skin. The mother reported that she read online that colloidal silver had been historically used to cure numerous ailments and she was ordering products from an online company. She was counseled on the dangers of both topical application and ingestion of silver, and all supplements were removed from the home.
Argyria is a rare condition caused by chronic exposure to silver and is characterized by a blue-gray pigmentation in the skin and appendages, mucous membranes, and internal organs.4 Clinically, argyria is classified as generalized or localized. Generalized argyria results from ingestion or inhalation of silver compounds, where granules deposit preferentially in sun-exposed areas of skin as well as internal organs, with the highest concentration in the liver, spleen, and adrenal glands; discoloration often is permanent.5 On the contrary, localized argyria results from direct external contact with silver and granules deposited in the hands, eyes, and mucosa.5 Although the exact mechanism of penetration from topical silver remains unknown, it is thought to enter via the eccrine sweat ducts, as histopathology reveals silver granules found in highest concentration surrounding sweat glands in the dermis.6
Initial differential diagnoses for altered nail pigmentation include drug-induced causes, systemic diseases, cyanosis, and exposure to metals.7 The most commonly indicated medications resulting in blue nail pigment changes include antimalarials, minocycline, zidovudine, and phenothiazine. Systemic diseases that may cause blue nail color change include Wilson disease, hemochromatosis, Addison disease, methemoglobinemia, and alkaptonuria.7 Metals include gold, mercury, arsenic, bismuth, lead, and silver.4 After a thorough review of the patient’s medications and lack of support for any underlying disease process, contact with metals, particularly silver, was ranked highly on our differential list. In support of this theory, the mother’s bluish gray facial skin led to high clinical suspicion that she was ingesting colloidal silver and also was exposing her son to silver.
Treatment of argyria is challenging but first and foremost involves discontinuation of the source of chronic silver exposure. Unfortunately, the discoloration of generalized argyria often is permanent. Sunscreen can be used to help prevent any further darkening of pigment. The pigment in localized argyria has been reported to slowly fade with time, and there also have been reports of successful treatment using a low-fluence Q-switched 1064-nm Nd:YAG laser.8
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Lencastre A, Lobo M, João A. Argyria—case report. An Bras Dermatol. 2013;88:413-416.
- Park S-W, Kim J-H, Shin H-T, et al. An effective modality for argyria treatment: Q-switched 1,064-nm Nd:YAG laser. Ann Dermatol. 2013;25:511-512.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Garcias-Ladaria J, Hernandez-Bel P, Torregrosa-Calatayud JL, et al. Localized cutaneous argyria: a report of 2 cases. Actas Dermosifiliogr. 2013;104:253-254.
- Kapur N, Landon G, Yu RC. Localized argyria in an antique restorer. Br J Dermatol. 2001;144:191-192.
- Kubba A, Kubba R, Batrani M, Pal T. Argyria an unrecognized cause of cutaneous pigmentation in Indian patients: a case series and review of the literature. Indian J Dermatol Venereol Leprol. 2013;79:805-811.
- Han TY, Chang HS, Lee HK, et al. Successful treatment of argyria using a low-fluence Q-switched 1064-nm Nd:YAG laser. Int J Dermatol. 2011;50:751-753.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Lencastre A, Lobo M, João A. Argyria—case report. An Bras Dermatol. 2013;88:413-416.
- Park S-W, Kim J-H, Shin H-T, et al. An effective modality for argyria treatment: Q-switched 1,064-nm Nd:YAG laser. Ann Dermatol. 2013;25:511-512.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Garcias-Ladaria J, Hernandez-Bel P, Torregrosa-Calatayud JL, et al. Localized cutaneous argyria: a report of 2 cases. Actas Dermosifiliogr. 2013;104:253-254.
- Kapur N, Landon G, Yu RC. Localized argyria in an antique restorer. Br J Dermatol. 2001;144:191-192.
- Kubba A, Kubba R, Batrani M, Pal T. Argyria an unrecognized cause of cutaneous pigmentation in Indian patients: a case series and review of the literature. Indian J Dermatol Venereol Leprol. 2013;79:805-811.
- Han TY, Chang HS, Lee HK, et al. Successful treatment of argyria using a low-fluence Q-switched 1064-nm Nd:YAG laser. Int J Dermatol. 2011;50:751-753.
Practice Points
- Argyria results from chronic exposure to products with a high silver content and may result in abnormalities of the skin and internal organs.
- Examination of the fingernails can provide important clues to underlying systemic conditions or external exposures.
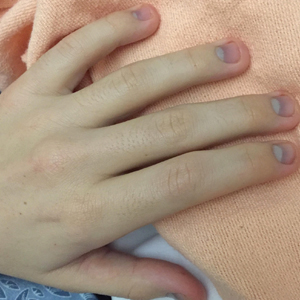
Phacomatosis Pigmentokeratotica Associated With Raynaud Phenomenon, Segmental Nevi, Hyperhidrosis, and Scoliosis
To the Editor:
Phacomatosis pigmentokeratotica (PPK) is a rare epidermal nevus syndrome complicated by multiple extracutaneous anomalies, including skeletal defects and neurologic anomalies. Less common associations include lateral curvature of the spine and hyperhidrosis. We present a patient with PPK and unilateral Raynaud phenomenon in addition to a segmental distribution of melanocytic nevi, hyperhidrosis, and scoliosis.
A 9-year-old girl was born with a yellow-orange alopecic plaque on the right side of the scalp (Figure 1). There also were 2 large, irregularly pigmented patches localized on the right side of the upper back and buttock. Over 3 years, numerous papular nevi developed within these pigmented patches and were diagnosed as speckled lentiginous nevi (Figure 2). In addition, numerous nevi of various sizes affected the right face, right shoulder, right arm (Figure 3), and right neck and were clearly demarcated along the midline. Several nevi also were noted within the nevus sebaceous on the right scalp. These skin lesions expanded progressively with age. At 6 years of age, she was diagnosed with hyperhidrosis of the right half of the body, which was most pronounced on the face. Raynaud phenomenon restricted to the right hand also was noted (Figure 4). Upon cold exposure, the digits become pale white, cold, and numb; then blue; and finally red. She lacked other features of connective tissue disease, and autoantibody testing was negative. She also was noted to have an abnormal lateral curvature of the spine (scoliosis). Auditory, ocular, and neurologic examinations were normal. Cranial and cerebral magnetic resonance imaging showed no central nervous system abnormalities. Her family history was negative for nevus spilus, nevus sebaceous, and neurofibromatosis. The clinical findings in our patient led to the diagnosis of PPK.
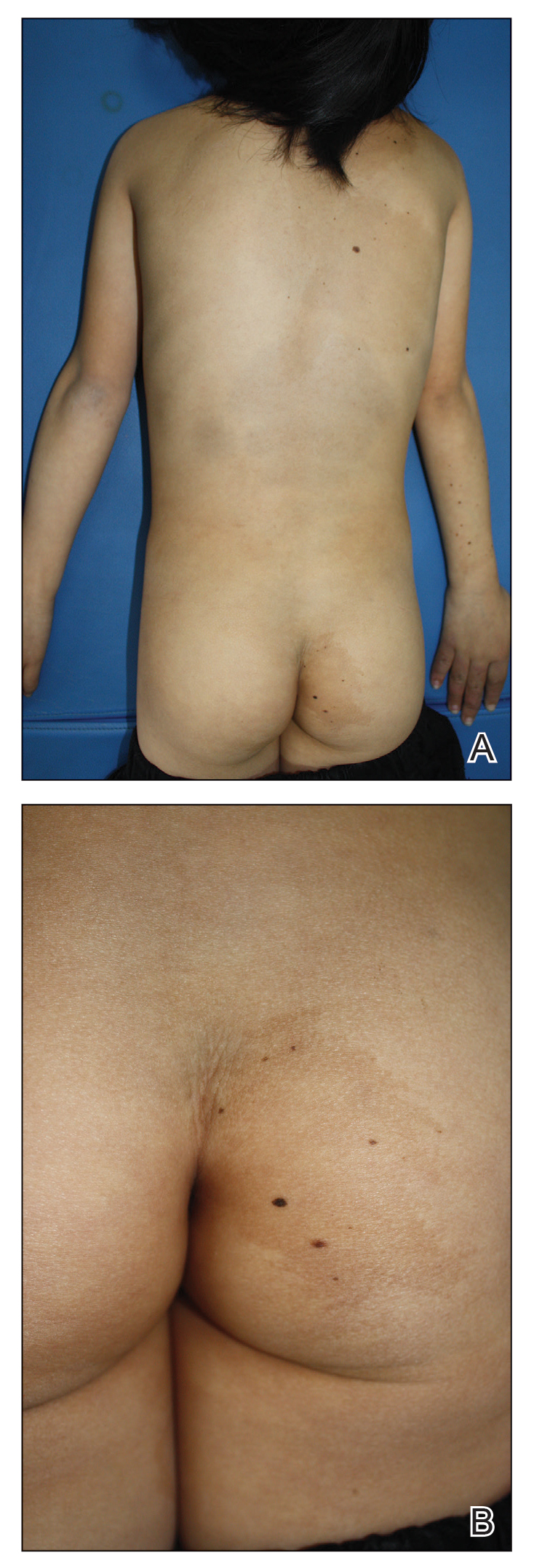
Phacomatosis pigmentokeratotica is a distinctive epidermal nevus syndrome characterized by the coexistence of a speckled lentiginous nevus, also known as a nevus spilus, and a nevus sebaceous1; PPK frequently is complicated by skeletal, ophthalmic, or neurologic abnormalities.2 Most cases reported are sporadic, and a postzygotic mosaic HRas proto-oncogene, GTPase, HRAS, mutation has been demonstrated in some patients and may contribute to the phenotype of PPK.3,4
Other anomalies have included ichthyosislike diffuse hyperkeratosis, laxity of the hands, pelvic hypoplasia, glaucoma, psychomotor retardation, and hypophosphatemic rickets. These patients also should be monitored for the development of malignant neoplasms within the nevus sebaceous.5 Segmental hyperhidrosis may be seen in association with the nevus spilus component.2
Raynaud phenomenon involving only the right hand was a unique finding in our patient. In 3 years of follow-up, our patient developed no evidence of connective tissue disease or other systemic illness. We speculate that Raynaud phenomenon of the right hand along with hyperhidrosis of the right side of the body could be a result of dysfunction of the autonomic nervous system. We propose that Raynaud phenomenon represents an unusual manifestation of PPK and may broaden the spectrum of extracutaneous anomalies associated with the disease. The finding of segmental nevi outside of the confines of the nevus spilus was another unusual manifestation of mosaicism.
- Happle R, Hoffmann R, Restano L, et al. Phacomatosis pigmentokeratotica: a melanocytic-epidermal twin nevus syndrome. Am J Med Genet. 1996;65:363-365.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22, 23-24.
- Groesser L, Herschberger E, Sagrera A, et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Invest Dermatol. 2013;133:1998-2003.
- Martin RJ, Arefi M, Splitt M, et al. Phacomatosis pigmentokeratotica and precocious puberty associated with HRAS mutation. Br J Dermatol. 2018;178:289-291.
- Chu GY, Wu CY. Phacomatosis pigmentokeratotica: a follow-up report with fatal outcome. Acta Derm Venereol. 2014;94:467-468.
To the Editor:
Phacomatosis pigmentokeratotica (PPK) is a rare epidermal nevus syndrome complicated by multiple extracutaneous anomalies, including skeletal defects and neurologic anomalies. Less common associations include lateral curvature of the spine and hyperhidrosis. We present a patient with PPK and unilateral Raynaud phenomenon in addition to a segmental distribution of melanocytic nevi, hyperhidrosis, and scoliosis.
A 9-year-old girl was born with a yellow-orange alopecic plaque on the right side of the scalp (Figure 1). There also were 2 large, irregularly pigmented patches localized on the right side of the upper back and buttock. Over 3 years, numerous papular nevi developed within these pigmented patches and were diagnosed as speckled lentiginous nevi (Figure 2). In addition, numerous nevi of various sizes affected the right face, right shoulder, right arm (Figure 3), and right neck and were clearly demarcated along the midline. Several nevi also were noted within the nevus sebaceous on the right scalp. These skin lesions expanded progressively with age. At 6 years of age, she was diagnosed with hyperhidrosis of the right half of the body, which was most pronounced on the face. Raynaud phenomenon restricted to the right hand also was noted (Figure 4). Upon cold exposure, the digits become pale white, cold, and numb; then blue; and finally red. She lacked other features of connective tissue disease, and autoantibody testing was negative. She also was noted to have an abnormal lateral curvature of the spine (scoliosis). Auditory, ocular, and neurologic examinations were normal. Cranial and cerebral magnetic resonance imaging showed no central nervous system abnormalities. Her family history was negative for nevus spilus, nevus sebaceous, and neurofibromatosis. The clinical findings in our patient led to the diagnosis of PPK.
Phacomatosis pigmentokeratotica is a distinctive epidermal nevus syndrome characterized by the coexistence of a speckled lentiginous nevus, also known as a nevus spilus, and a nevus sebaceous1; PPK frequently is complicated by skeletal, ophthalmic, or neurologic abnormalities.2 Most cases reported are sporadic, and a postzygotic mosaic HRas proto-oncogene, GTPase, HRAS, mutation has been demonstrated in some patients and may contribute to the phenotype of PPK.3,4
Other anomalies have included ichthyosislike diffuse hyperkeratosis, laxity of the hands, pelvic hypoplasia, glaucoma, psychomotor retardation, and hypophosphatemic rickets. These patients also should be monitored for the development of malignant neoplasms within the nevus sebaceous.5 Segmental hyperhidrosis may be seen in association with the nevus spilus component.2
Raynaud phenomenon involving only the right hand was a unique finding in our patient. In 3 years of follow-up, our patient developed no evidence of connective tissue disease or other systemic illness. We speculate that Raynaud phenomenon of the right hand along with hyperhidrosis of the right side of the body could be a result of dysfunction of the autonomic nervous system. We propose that Raynaud phenomenon represents an unusual manifestation of PPK and may broaden the spectrum of extracutaneous anomalies associated with the disease. The finding of segmental nevi outside of the confines of the nevus spilus was another unusual manifestation of mosaicism.
To the Editor:
Phacomatosis pigmentokeratotica (PPK) is a rare epidermal nevus syndrome complicated by multiple extracutaneous anomalies, including skeletal defects and neurologic anomalies. Less common associations include lateral curvature of the spine and hyperhidrosis. We present a patient with PPK and unilateral Raynaud phenomenon in addition to a segmental distribution of melanocytic nevi, hyperhidrosis, and scoliosis.
A 9-year-old girl was born with a yellow-orange alopecic plaque on the right side of the scalp (Figure 1). There also were 2 large, irregularly pigmented patches localized on the right side of the upper back and buttock. Over 3 years, numerous papular nevi developed within these pigmented patches and were diagnosed as speckled lentiginous nevi (Figure 2). In addition, numerous nevi of various sizes affected the right face, right shoulder, right arm (Figure 3), and right neck and were clearly demarcated along the midline. Several nevi also were noted within the nevus sebaceous on the right scalp. These skin lesions expanded progressively with age. At 6 years of age, she was diagnosed with hyperhidrosis of the right half of the body, which was most pronounced on the face. Raynaud phenomenon restricted to the right hand also was noted (Figure 4). Upon cold exposure, the digits become pale white, cold, and numb; then blue; and finally red. She lacked other features of connective tissue disease, and autoantibody testing was negative. She also was noted to have an abnormal lateral curvature of the spine (scoliosis). Auditory, ocular, and neurologic examinations were normal. Cranial and cerebral magnetic resonance imaging showed no central nervous system abnormalities. Her family history was negative for nevus spilus, nevus sebaceous, and neurofibromatosis. The clinical findings in our patient led to the diagnosis of PPK.
Phacomatosis pigmentokeratotica is a distinctive epidermal nevus syndrome characterized by the coexistence of a speckled lentiginous nevus, also known as a nevus spilus, and a nevus sebaceous1; PPK frequently is complicated by skeletal, ophthalmic, or neurologic abnormalities.2 Most cases reported are sporadic, and a postzygotic mosaic HRas proto-oncogene, GTPase, HRAS, mutation has been demonstrated in some patients and may contribute to the phenotype of PPK.3,4
Other anomalies have included ichthyosislike diffuse hyperkeratosis, laxity of the hands, pelvic hypoplasia, glaucoma, psychomotor retardation, and hypophosphatemic rickets. These patients also should be monitored for the development of malignant neoplasms within the nevus sebaceous.5 Segmental hyperhidrosis may be seen in association with the nevus spilus component.2
Raynaud phenomenon involving only the right hand was a unique finding in our patient. In 3 years of follow-up, our patient developed no evidence of connective tissue disease or other systemic illness. We speculate that Raynaud phenomenon of the right hand along with hyperhidrosis of the right side of the body could be a result of dysfunction of the autonomic nervous system. We propose that Raynaud phenomenon represents an unusual manifestation of PPK and may broaden the spectrum of extracutaneous anomalies associated with the disease. The finding of segmental nevi outside of the confines of the nevus spilus was another unusual manifestation of mosaicism.
- Happle R, Hoffmann R, Restano L, et al. Phacomatosis pigmentokeratotica: a melanocytic-epidermal twin nevus syndrome. Am J Med Genet. 1996;65:363-365.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22, 23-24.
- Groesser L, Herschberger E, Sagrera A, et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Invest Dermatol. 2013;133:1998-2003.
- Martin RJ, Arefi M, Splitt M, et al. Phacomatosis pigmentokeratotica and precocious puberty associated with HRAS mutation. Br J Dermatol. 2018;178:289-291.
- Chu GY, Wu CY. Phacomatosis pigmentokeratotica: a follow-up report with fatal outcome. Acta Derm Venereol. 2014;94:467-468.
- Happle R, Hoffmann R, Restano L, et al. Phacomatosis pigmentokeratotica: a melanocytic-epidermal twin nevus syndrome. Am J Med Genet. 1996;65:363-365.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22, 23-24.
- Groesser L, Herschberger E, Sagrera A, et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Invest Dermatol. 2013;133:1998-2003.
- Martin RJ, Arefi M, Splitt M, et al. Phacomatosis pigmentokeratotica and precocious puberty associated with HRAS mutation. Br J Dermatol. 2018;178:289-291.
- Chu GY, Wu CY. Phacomatosis pigmentokeratotica: a follow-up report with fatal outcome. Acta Derm Venereol. 2014;94:467-468.
Practice Points
- Phacomatosis pigmentokeratotica (PPK) is characterized by the coexistence of speckled lentiginous nevus and nevus sebaceous.
- Raynaud phenomenon may be an unreported association with PPK.
Urticarial Vasculitis Successfully Treated With Omalizumab
To the Editor:
Urticarial vasculitis (UV) is a clinicopathologic entity. It manifests as an eruption of erythematous wheals that clinically resemble urticaria, but the lesions of UV last longer, may leave residual hyperpigmentation, and may or may not be pruritic.1 Therapies most often employed include oral antihistamines and systemic immunosuppressant drugs such as corticosteroids, dapsone, colchicine, or hydroxychloroquine.2 We present a woman with UV who successfully was treated with omalizumab.
A 49-year-old woman presented to our outpatient clinic with generalized pruritic skin rashes of 2 years’ duration. She also described swelling on the upper eyelids 2 times monthly. She used several antihistamines (up to 4 times daily) and was taking systemic corticosteroids and antidepressants. Physical examination revealed generalized erythematous and edematous papules and plaques on the trunk and extremities (Figure 1). At follow-up a few days later, we observed that the lesions were lasting for more than 24 hours, but there was no residual pigmentation. According to clinical concerns and the association with angioedema, we initially thought the diagnosis was chronic urticaria and angioedema. The patient had no extracutaneous manifestations such as fever, arthralgia, or lymphadenopathy. Routine laboratory examinations including antinuclear antibodies were within reference range. She had normal C3 and C4 levels and an elevated total IgE level (344 IU/mL [reference range, 0–170 IU/mL]). Because the IgE level was elevated and she had no response to the highest dosages of antihistamines, we decided to start omalizumab therapy. Prior to starting omalizumab, we performed a skin biopsy for histopathologic and direct immunofluorescence examinations for UV, as the duration of the lesions was more than 24 hours. Histopathologic examination revealed lymphocytes within the vessel wall and perivascular lymphocytic infiltration with eosinophils (Figure 2). On direct immunofluorescence, perivascular IgA deposition was observed (Figure 3). Histopathologic findings were associated with lymphocytic vasculitis. Systemic involvement was not detected on detailed laboratory and radiologic examinations.
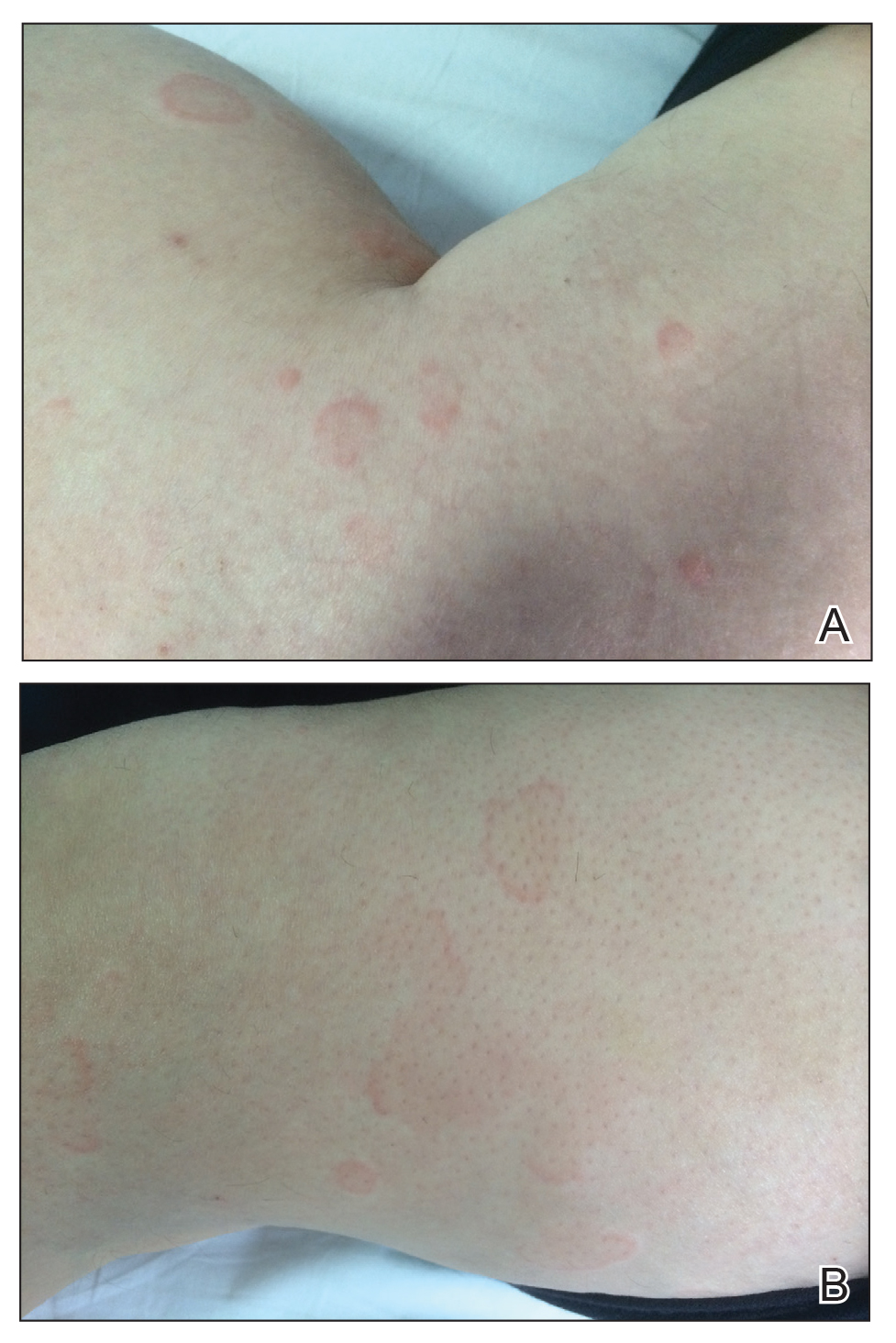


After the first application of omalizumab, the lesions disappeared within a few days. She was treated with subcutaneous omalizumab 300 mg every 4 weeks for 6 months, and we did not observe any adverse effects related to the drug. There was no relapse after therapy cessation.
Omalizumab is a recombinant humanized anti-IgE monoclonal antibody that is approved by the US Food and Drug Administration for treatment of chronic idiopathic urticaria.3-5 Studies have suggested that omalizumab might play an important role in the treatment of other potentially IgE-mediated disease processes including allergic asthma, atopic dermatitis, allergic rhinitis, nasal polyposis, and severe ocular allergies.6 The proposed mechanism of action of omalizumab includes reduction of free IgE through the reversible formation of tiny, biologically inert complexes; targeting IgE-expressing B cells; and inhibiting production of IgE. Because it reduces free IgE, omalizumab has been used in normal IgE or hyper-IgE situations. Omalizumab also induces eosinophil apoptosis; increases IL-2, IL-3, tumor necrosis factor α, and IFN-γ; and reduces IL-4.7 A number of off-label uses have been described such as atopic dermatitis, bullous pemphigoid, hyper-IgE syndrome, cutaneous mastocytosis, toxic epidermal necrolysis, and eosinophilic granulomatosis with polyangitis.8 There are no clinical studies of omalizumab for UV, and only a few case reports have shown that omalizumab also might be beneficial for this condition.2-4 Diez et al4 reported 3 cases of women aged 28, 51, and 54 years with spontaneous chronic urticaria with autoimmune and pressure components as well as vasculitis whose symptoms completely improved after starting omalizumab. Kai et al3 successfully treated a patient with normocomplementemic UV with omalizumab and suggested that omalizumab markedly improved the patient’s quality of life with chronic urticaria and UV. Ghazanfar and Thomsen2 reported the case of a 68-year-old man diagnosed with histopathologically confirmed leukocytoclastic vasculitis. He had used systemic corticosteroid therapy and dapsone without notable improvement. The patient was switched to subcutaneous omalizumab 300 mg once every 4 weeks; after 1 month, he observed complete remission of the UV and symptoms.2
Our case suggests that omalizumab has a beneficial effect on patients with UV. Omalizumab may be effective in UV through its reduction of IgE, as in chronic urticaria, and through downstream effects on cellular activation mechanisms (possibly a reduction in chemotaxis or immune complex formation). However, the mechanism of action of omalizumab for UV remains, in part, unresolved. It is not known whether omalizumab is efficacious against both normocomplementemic and hypocomplementemic UV. Further studies with a greater number of patients are needed to confirm the effects of omalizumab for vasculitic patients.
- Chang S, Carr W. Urticarial vasculitis. Allergy Asthma Proc. 2007;28:97-100.
- Ghazanfar MN, Thomsen SF. Omalizumab for urticarial vasculitis: case report and review of the literature. Case Rep Dermatol Med. 2015:576893.
- Kai AC, Flohr C, Grattan CE. Improvement in quality of life impairment followed by relapse with 6-monthly periodic administration of omalizumab for severe treatment-refractory chronic urticaria and urticarial vasculitis. Clin Exp Dermatol. 2014;39:651-652.
- Diez LS, Tamayo LM, Cardona R. Omalizumab: therapeutic option in chronic spontaneous urticaria difficult to control with associated vasculitis, report of three cases. Biomedica. 2013;33:503-512.
- Maurer M, Rosen K, Hsieh HJ. Omalizumab for chronic urticaria. N Engl J Med. 2013;368:2530.
- Ben Shoshan M. Omalizumab: not only for asthma. Recent Pat Inflamm Allergy Drug Discov. 2008;2:191-201.
- Fueyo-Casado A, Campos-Munoz L, Gonzalez-Guerra E, et al. Effectiveness of omalizumab in a case of urticarial vasculitis. Clin Exp Dermatol. Published March 1, 2017. doi:10.1111/ced.13076
- Chia JC, Mydlarski PR. Dermatologic uses of omalizumab. J Dermatol Treat. Published November 7, 2016. doi:10.1080/09546634.2016.1249819
To the Editor:
Urticarial vasculitis (UV) is a clinicopathologic entity. It manifests as an eruption of erythematous wheals that clinically resemble urticaria, but the lesions of UV last longer, may leave residual hyperpigmentation, and may or may not be pruritic.1 Therapies most often employed include oral antihistamines and systemic immunosuppressant drugs such as corticosteroids, dapsone, colchicine, or hydroxychloroquine.2 We present a woman with UV who successfully was treated with omalizumab.
A 49-year-old woman presented to our outpatient clinic with generalized pruritic skin rashes of 2 years’ duration. She also described swelling on the upper eyelids 2 times monthly. She used several antihistamines (up to 4 times daily) and was taking systemic corticosteroids and antidepressants. Physical examination revealed generalized erythematous and edematous papules and plaques on the trunk and extremities (Figure 1). At follow-up a few days later, we observed that the lesions were lasting for more than 24 hours, but there was no residual pigmentation. According to clinical concerns and the association with angioedema, we initially thought the diagnosis was chronic urticaria and angioedema. The patient had no extracutaneous manifestations such as fever, arthralgia, or lymphadenopathy. Routine laboratory examinations including antinuclear antibodies were within reference range. She had normal C3 and C4 levels and an elevated total IgE level (344 IU/mL [reference range, 0–170 IU/mL]). Because the IgE level was elevated and she had no response to the highest dosages of antihistamines, we decided to start omalizumab therapy. Prior to starting omalizumab, we performed a skin biopsy for histopathologic and direct immunofluorescence examinations for UV, as the duration of the lesions was more than 24 hours. Histopathologic examination revealed lymphocytes within the vessel wall and perivascular lymphocytic infiltration with eosinophils (Figure 2). On direct immunofluorescence, perivascular IgA deposition was observed (Figure 3). Histopathologic findings were associated with lymphocytic vasculitis. Systemic involvement was not detected on detailed laboratory and radiologic examinations.
After the first application of omalizumab, the lesions disappeared within a few days. She was treated with subcutaneous omalizumab 300 mg every 4 weeks for 6 months, and we did not observe any adverse effects related to the drug. There was no relapse after therapy cessation.
Omalizumab is a recombinant humanized anti-IgE monoclonal antibody that is approved by the US Food and Drug Administration for treatment of chronic idiopathic urticaria.3-5 Studies have suggested that omalizumab might play an important role in the treatment of other potentially IgE-mediated disease processes including allergic asthma, atopic dermatitis, allergic rhinitis, nasal polyposis, and severe ocular allergies.6 The proposed mechanism of action of omalizumab includes reduction of free IgE through the reversible formation of tiny, biologically inert complexes; targeting IgE-expressing B cells; and inhibiting production of IgE. Because it reduces free IgE, omalizumab has been used in normal IgE or hyper-IgE situations. Omalizumab also induces eosinophil apoptosis; increases IL-2, IL-3, tumor necrosis factor α, and IFN-γ; and reduces IL-4.7 A number of off-label uses have been described such as atopic dermatitis, bullous pemphigoid, hyper-IgE syndrome, cutaneous mastocytosis, toxic epidermal necrolysis, and eosinophilic granulomatosis with polyangitis.8 There are no clinical studies of omalizumab for UV, and only a few case reports have shown that omalizumab also might be beneficial for this condition.2-4 Diez et al4 reported 3 cases of women aged 28, 51, and 54 years with spontaneous chronic urticaria with autoimmune and pressure components as well as vasculitis whose symptoms completely improved after starting omalizumab. Kai et al3 successfully treated a patient with normocomplementemic UV with omalizumab and suggested that omalizumab markedly improved the patient’s quality of life with chronic urticaria and UV. Ghazanfar and Thomsen2 reported the case of a 68-year-old man diagnosed with histopathologically confirmed leukocytoclastic vasculitis. He had used systemic corticosteroid therapy and dapsone without notable improvement. The patient was switched to subcutaneous omalizumab 300 mg once every 4 weeks; after 1 month, he observed complete remission of the UV and symptoms.2
Our case suggests that omalizumab has a beneficial effect on patients with UV. Omalizumab may be effective in UV through its reduction of IgE, as in chronic urticaria, and through downstream effects on cellular activation mechanisms (possibly a reduction in chemotaxis or immune complex formation). However, the mechanism of action of omalizumab for UV remains, in part, unresolved. It is not known whether omalizumab is efficacious against both normocomplementemic and hypocomplementemic UV. Further studies with a greater number of patients are needed to confirm the effects of omalizumab for vasculitic patients.
To the Editor:
Urticarial vasculitis (UV) is a clinicopathologic entity. It manifests as an eruption of erythematous wheals that clinically resemble urticaria, but the lesions of UV last longer, may leave residual hyperpigmentation, and may or may not be pruritic.1 Therapies most often employed include oral antihistamines and systemic immunosuppressant drugs such as corticosteroids, dapsone, colchicine, or hydroxychloroquine.2 We present a woman with UV who successfully was treated with omalizumab.
A 49-year-old woman presented to our outpatient clinic with generalized pruritic skin rashes of 2 years’ duration. She also described swelling on the upper eyelids 2 times monthly. She used several antihistamines (up to 4 times daily) and was taking systemic corticosteroids and antidepressants. Physical examination revealed generalized erythematous and edematous papules and plaques on the trunk and extremities (Figure 1). At follow-up a few days later, we observed that the lesions were lasting for more than 24 hours, but there was no residual pigmentation. According to clinical concerns and the association with angioedema, we initially thought the diagnosis was chronic urticaria and angioedema. The patient had no extracutaneous manifestations such as fever, arthralgia, or lymphadenopathy. Routine laboratory examinations including antinuclear antibodies were within reference range. She had normal C3 and C4 levels and an elevated total IgE level (344 IU/mL [reference range, 0–170 IU/mL]). Because the IgE level was elevated and she had no response to the highest dosages of antihistamines, we decided to start omalizumab therapy. Prior to starting omalizumab, we performed a skin biopsy for histopathologic and direct immunofluorescence examinations for UV, as the duration of the lesions was more than 24 hours. Histopathologic examination revealed lymphocytes within the vessel wall and perivascular lymphocytic infiltration with eosinophils (Figure 2). On direct immunofluorescence, perivascular IgA deposition was observed (Figure 3). Histopathologic findings were associated with lymphocytic vasculitis. Systemic involvement was not detected on detailed laboratory and radiologic examinations.
After the first application of omalizumab, the lesions disappeared within a few days. She was treated with subcutaneous omalizumab 300 mg every 4 weeks for 6 months, and we did not observe any adverse effects related to the drug. There was no relapse after therapy cessation.
Omalizumab is a recombinant humanized anti-IgE monoclonal antibody that is approved by the US Food and Drug Administration for treatment of chronic idiopathic urticaria.3-5 Studies have suggested that omalizumab might play an important role in the treatment of other potentially IgE-mediated disease processes including allergic asthma, atopic dermatitis, allergic rhinitis, nasal polyposis, and severe ocular allergies.6 The proposed mechanism of action of omalizumab includes reduction of free IgE through the reversible formation of tiny, biologically inert complexes; targeting IgE-expressing B cells; and inhibiting production of IgE. Because it reduces free IgE, omalizumab has been used in normal IgE or hyper-IgE situations. Omalizumab also induces eosinophil apoptosis; increases IL-2, IL-3, tumor necrosis factor α, and IFN-γ; and reduces IL-4.7 A number of off-label uses have been described such as atopic dermatitis, bullous pemphigoid, hyper-IgE syndrome, cutaneous mastocytosis, toxic epidermal necrolysis, and eosinophilic granulomatosis with polyangitis.8 There are no clinical studies of omalizumab for UV, and only a few case reports have shown that omalizumab also might be beneficial for this condition.2-4 Diez et al4 reported 3 cases of women aged 28, 51, and 54 years with spontaneous chronic urticaria with autoimmune and pressure components as well as vasculitis whose symptoms completely improved after starting omalizumab. Kai et al3 successfully treated a patient with normocomplementemic UV with omalizumab and suggested that omalizumab markedly improved the patient’s quality of life with chronic urticaria and UV. Ghazanfar and Thomsen2 reported the case of a 68-year-old man diagnosed with histopathologically confirmed leukocytoclastic vasculitis. He had used systemic corticosteroid therapy and dapsone without notable improvement. The patient was switched to subcutaneous omalizumab 300 mg once every 4 weeks; after 1 month, he observed complete remission of the UV and symptoms.2
Our case suggests that omalizumab has a beneficial effect on patients with UV. Omalizumab may be effective in UV through its reduction of IgE, as in chronic urticaria, and through downstream effects on cellular activation mechanisms (possibly a reduction in chemotaxis or immune complex formation). However, the mechanism of action of omalizumab for UV remains, in part, unresolved. It is not known whether omalizumab is efficacious against both normocomplementemic and hypocomplementemic UV. Further studies with a greater number of patients are needed to confirm the effects of omalizumab for vasculitic patients.
- Chang S, Carr W. Urticarial vasculitis. Allergy Asthma Proc. 2007;28:97-100.
- Ghazanfar MN, Thomsen SF. Omalizumab for urticarial vasculitis: case report and review of the literature. Case Rep Dermatol Med. 2015:576893.
- Kai AC, Flohr C, Grattan CE. Improvement in quality of life impairment followed by relapse with 6-monthly periodic administration of omalizumab for severe treatment-refractory chronic urticaria and urticarial vasculitis. Clin Exp Dermatol. 2014;39:651-652.
- Diez LS, Tamayo LM, Cardona R. Omalizumab: therapeutic option in chronic spontaneous urticaria difficult to control with associated vasculitis, report of three cases. Biomedica. 2013;33:503-512.
- Maurer M, Rosen K, Hsieh HJ. Omalizumab for chronic urticaria. N Engl J Med. 2013;368:2530.
- Ben Shoshan M. Omalizumab: not only for asthma. Recent Pat Inflamm Allergy Drug Discov. 2008;2:191-201.
- Fueyo-Casado A, Campos-Munoz L, Gonzalez-Guerra E, et al. Effectiveness of omalizumab in a case of urticarial vasculitis. Clin Exp Dermatol. Published March 1, 2017. doi:10.1111/ced.13076
- Chia JC, Mydlarski PR. Dermatologic uses of omalizumab. J Dermatol Treat. Published November 7, 2016. doi:10.1080/09546634.2016.1249819
- Chang S, Carr W. Urticarial vasculitis. Allergy Asthma Proc. 2007;28:97-100.
- Ghazanfar MN, Thomsen SF. Omalizumab for urticarial vasculitis: case report and review of the literature. Case Rep Dermatol Med. 2015:576893.
- Kai AC, Flohr C, Grattan CE. Improvement in quality of life impairment followed by relapse with 6-monthly periodic administration of omalizumab for severe treatment-refractory chronic urticaria and urticarial vasculitis. Clin Exp Dermatol. 2014;39:651-652.
- Diez LS, Tamayo LM, Cardona R. Omalizumab: therapeutic option in chronic spontaneous urticaria difficult to control with associated vasculitis, report of three cases. Biomedica. 2013;33:503-512.
- Maurer M, Rosen K, Hsieh HJ. Omalizumab for chronic urticaria. N Engl J Med. 2013;368:2530.
- Ben Shoshan M. Omalizumab: not only for asthma. Recent Pat Inflamm Allergy Drug Discov. 2008;2:191-201.
- Fueyo-Casado A, Campos-Munoz L, Gonzalez-Guerra E, et al. Effectiveness of omalizumab in a case of urticarial vasculitis. Clin Exp Dermatol. Published March 1, 2017. doi:10.1111/ced.13076
- Chia JC, Mydlarski PR. Dermatologic uses of omalizumab. J Dermatol Treat. Published November 7, 2016. doi:10.1080/09546634.2016.1249819
Practice Points
- The differential diagnosis of urticaria and urticarial vasculitis may be complicated.
- Omalizumab is an effective urticaria treatment and also can be an alternative treatment choice in resistant urticarial vasculitis.