User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
Powered by CHEST Physician, Clinician Reviews, MDedge Family Medicine, Internal Medicine News, and The Journal of Clinical Outcomes Management.
Pulmonary hypertension treatment gets under the skin
Pulmonary arterial hypertension (PAH) patients with moderate, stable disease can benefit from an implantable drug delivery system, based on data from a review of 60 adults with successful implantations. The findings were published in the December issue of CHEST.
“A fully implanted system offers patients the hope of returning to more normal activities such as bathing, swimming, and reduced risk of infections from externalized central venous catheter contamination or reduced subcutaneous pain from subcutaneous infusion,” wrote Aaron B. Waxman, MD, PhD, of Brigham and Women’s Hospital, Boston, and his colleagues (Chest. 2017 June 3. doi: 10.1016/j.chest.2017.04.188).
In the DelIVery Trial, clinicians at 10 locations in the United States placed a fully implantable delivery system in adults aged 18 years and older with stable PAH who were previously receiving treprostinil via an external pump at an average dose of 71 ng/kg per min.
All 60 patients were successfully implanted with a system consisting of a drug infusion pump placed in an abdominal pocket and an intravascular catheter linking the implanted pump to the superior vena cava.
“The location of the pump pocket was determined in partnership with the patient and was based on consideration of clothing styles, belt line and subcutaneous fat depth,” the researchers noted.
Procedure-related complications deemed clinically significant included one atrial fibrillation, two incidences of pneumothorax, two infections unrelated to catheter placement, and three catheter dislocations (two in the same patient). The most common patient complaints were expected implant site pain in 83% and bruising in 17%.
The findings were limited by the small number of patients, but the researchers identified several factors that contributed to the success of the procedure, including selecting patients who have shown response to treprostinil and are motivated to comply with pump refill visits, performing the procedure at centers with a high volume of PAH patients, keeping the procedure consistent for each patient, and using the same implant team in each case. “The implant procedure was successfully performed with a low complication rate by clinicians with a diverse range of specialty training,” the researchers added.
Patients reported satisfaction with the implant system at 6 weeks and 6 months, and said they spent an average of 75% less time managing their delivery system, according to previously published data on the patients’ perspective (CHEST 2016;150[1]:27-34).
Medtronic sponsored the study. The lead author, Dr. Waxman, had no financial conflicts to disclose; several coauthors reported relationships with companies including Medtronic, Actelion, Bayer, Gilead, Merck, and United Therapeutics.
The development of an implantable therapy for pulmonary hypertension could expand the use of treprostinil, a demonstrated effective treatment for PAH that has been limited in its use because of a range of side effects when given intravenously, orally, subcutaneously, or by inhalation, Joel A. Wirth, MD, FCCP, and Harold I. Palevsky, MD, FCCP, wrote in an editorial.
The use of an intravenous pump and catheter infusion system for stable PAH patients could help them return more quickly to normal activities and curb the risk of catheter-related infections, they said. “Having the potential to remove some of the burden and risk incumbent with an external delivery system may reduce several of the overall barriers to continuous intravenous prostanoid acceptance by both patients and providers,” they noted (Chest. 2017 Dec 6. doi: 10.1016/j.chest.2017.07.006).
Clinicians must be educated to perform the implant procedure itself, and care centers must be trained in identifying patient management issues and refilling the pump reservoir as needed, Dr. Wirth and Dr. Palevsky emphasized. Patients must be educated in what to expect, including how to monitor the pump and track the need for refills, they said. Although the pump is not appropriate for patients with severe PAH, “a planned staged approach of transitioning PAH patients from IV therapy to a less complex system could lend itself to employing prostanoid use earlier and for less severely affected PAH patients,” they said.
Dr. Wirth is affiliated with Tufts University, Boston. Dr. Palevsky is affiliated with the University of Pennsylvania, Philadelphia. Both Dr. Wirth and Dr. Palevsky disclosed serving as consultants and as principal investigators for United Therapeutics.
The development of an implantable therapy for pulmonary hypertension could expand the use of treprostinil, a demonstrated effective treatment for PAH that has been limited in its use because of a range of side effects when given intravenously, orally, subcutaneously, or by inhalation, Joel A. Wirth, MD, FCCP, and Harold I. Palevsky, MD, FCCP, wrote in an editorial.
The use of an intravenous pump and catheter infusion system for stable PAH patients could help them return more quickly to normal activities and curb the risk of catheter-related infections, they said. “Having the potential to remove some of the burden and risk incumbent with an external delivery system may reduce several of the overall barriers to continuous intravenous prostanoid acceptance by both patients and providers,” they noted (Chest. 2017 Dec 6. doi: 10.1016/j.chest.2017.07.006).
Clinicians must be educated to perform the implant procedure itself, and care centers must be trained in identifying patient management issues and refilling the pump reservoir as needed, Dr. Wirth and Dr. Palevsky emphasized. Patients must be educated in what to expect, including how to monitor the pump and track the need for refills, they said. Although the pump is not appropriate for patients with severe PAH, “a planned staged approach of transitioning PAH patients from IV therapy to a less complex system could lend itself to employing prostanoid use earlier and for less severely affected PAH patients,” they said.
Dr. Wirth is affiliated with Tufts University, Boston. Dr. Palevsky is affiliated with the University of Pennsylvania, Philadelphia. Both Dr. Wirth and Dr. Palevsky disclosed serving as consultants and as principal investigators for United Therapeutics.
The development of an implantable therapy for pulmonary hypertension could expand the use of treprostinil, a demonstrated effective treatment for PAH that has been limited in its use because of a range of side effects when given intravenously, orally, subcutaneously, or by inhalation, Joel A. Wirth, MD, FCCP, and Harold I. Palevsky, MD, FCCP, wrote in an editorial.
The use of an intravenous pump and catheter infusion system for stable PAH patients could help them return more quickly to normal activities and curb the risk of catheter-related infections, they said. “Having the potential to remove some of the burden and risk incumbent with an external delivery system may reduce several of the overall barriers to continuous intravenous prostanoid acceptance by both patients and providers,” they noted (Chest. 2017 Dec 6. doi: 10.1016/j.chest.2017.07.006).
Clinicians must be educated to perform the implant procedure itself, and care centers must be trained in identifying patient management issues and refilling the pump reservoir as needed, Dr. Wirth and Dr. Palevsky emphasized. Patients must be educated in what to expect, including how to monitor the pump and track the need for refills, they said. Although the pump is not appropriate for patients with severe PAH, “a planned staged approach of transitioning PAH patients from IV therapy to a less complex system could lend itself to employing prostanoid use earlier and for less severely affected PAH patients,” they said.
Dr. Wirth is affiliated with Tufts University, Boston. Dr. Palevsky is affiliated with the University of Pennsylvania, Philadelphia. Both Dr. Wirth and Dr. Palevsky disclosed serving as consultants and as principal investigators for United Therapeutics.
Pulmonary arterial hypertension (PAH) patients with moderate, stable disease can benefit from an implantable drug delivery system, based on data from a review of 60 adults with successful implantations. The findings were published in the December issue of CHEST.
“A fully implanted system offers patients the hope of returning to more normal activities such as bathing, swimming, and reduced risk of infections from externalized central venous catheter contamination or reduced subcutaneous pain from subcutaneous infusion,” wrote Aaron B. Waxman, MD, PhD, of Brigham and Women’s Hospital, Boston, and his colleagues (Chest. 2017 June 3. doi: 10.1016/j.chest.2017.04.188).
In the DelIVery Trial, clinicians at 10 locations in the United States placed a fully implantable delivery system in adults aged 18 years and older with stable PAH who were previously receiving treprostinil via an external pump at an average dose of 71 ng/kg per min.
All 60 patients were successfully implanted with a system consisting of a drug infusion pump placed in an abdominal pocket and an intravascular catheter linking the implanted pump to the superior vena cava.
“The location of the pump pocket was determined in partnership with the patient and was based on consideration of clothing styles, belt line and subcutaneous fat depth,” the researchers noted.
Procedure-related complications deemed clinically significant included one atrial fibrillation, two incidences of pneumothorax, two infections unrelated to catheter placement, and three catheter dislocations (two in the same patient). The most common patient complaints were expected implant site pain in 83% and bruising in 17%.
The findings were limited by the small number of patients, but the researchers identified several factors that contributed to the success of the procedure, including selecting patients who have shown response to treprostinil and are motivated to comply with pump refill visits, performing the procedure at centers with a high volume of PAH patients, keeping the procedure consistent for each patient, and using the same implant team in each case. “The implant procedure was successfully performed with a low complication rate by clinicians with a diverse range of specialty training,” the researchers added.
Patients reported satisfaction with the implant system at 6 weeks and 6 months, and said they spent an average of 75% less time managing their delivery system, according to previously published data on the patients’ perspective (CHEST 2016;150[1]:27-34).
Medtronic sponsored the study. The lead author, Dr. Waxman, had no financial conflicts to disclose; several coauthors reported relationships with companies including Medtronic, Actelion, Bayer, Gilead, Merck, and United Therapeutics.
Pulmonary arterial hypertension (PAH) patients with moderate, stable disease can benefit from an implantable drug delivery system, based on data from a review of 60 adults with successful implantations. The findings were published in the December issue of CHEST.
“A fully implanted system offers patients the hope of returning to more normal activities such as bathing, swimming, and reduced risk of infections from externalized central venous catheter contamination or reduced subcutaneous pain from subcutaneous infusion,” wrote Aaron B. Waxman, MD, PhD, of Brigham and Women’s Hospital, Boston, and his colleagues (Chest. 2017 June 3. doi: 10.1016/j.chest.2017.04.188).
In the DelIVery Trial, clinicians at 10 locations in the United States placed a fully implantable delivery system in adults aged 18 years and older with stable PAH who were previously receiving treprostinil via an external pump at an average dose of 71 ng/kg per min.
All 60 patients were successfully implanted with a system consisting of a drug infusion pump placed in an abdominal pocket and an intravascular catheter linking the implanted pump to the superior vena cava.
“The location of the pump pocket was determined in partnership with the patient and was based on consideration of clothing styles, belt line and subcutaneous fat depth,” the researchers noted.
Procedure-related complications deemed clinically significant included one atrial fibrillation, two incidences of pneumothorax, two infections unrelated to catheter placement, and three catheter dislocations (two in the same patient). The most common patient complaints were expected implant site pain in 83% and bruising in 17%.
The findings were limited by the small number of patients, but the researchers identified several factors that contributed to the success of the procedure, including selecting patients who have shown response to treprostinil and are motivated to comply with pump refill visits, performing the procedure at centers with a high volume of PAH patients, keeping the procedure consistent for each patient, and using the same implant team in each case. “The implant procedure was successfully performed with a low complication rate by clinicians with a diverse range of specialty training,” the researchers added.
Patients reported satisfaction with the implant system at 6 weeks and 6 months, and said they spent an average of 75% less time managing their delivery system, according to previously published data on the patients’ perspective (CHEST 2016;150[1]:27-34).
Medtronic sponsored the study. The lead author, Dr. Waxman, had no financial conflicts to disclose; several coauthors reported relationships with companies including Medtronic, Actelion, Bayer, Gilead, Merck, and United Therapeutics.
FROM CHEST
Key clinical point: An implantable drug delivery system was successfully placed in 100% of adult PAH patients with no serious complications.
Major finding: The most common complaints among patients who received an implant system to deliver treprostinil were implant site pain (83%) and bruising (17%).
Data source: A multicenter, prospective study of 60 adults with pulmonary arterial hypertension who received implantable pumps to deliver treprostinil.
Disclosures: Medtronic sponsored the study. The lead author, Dr. Waxman, had no financial conflicts to disclose; several coauthors reported relationships with companies including Medtronic, Actelion, Bayer, Gilead, Merck, and United Therapeutics.
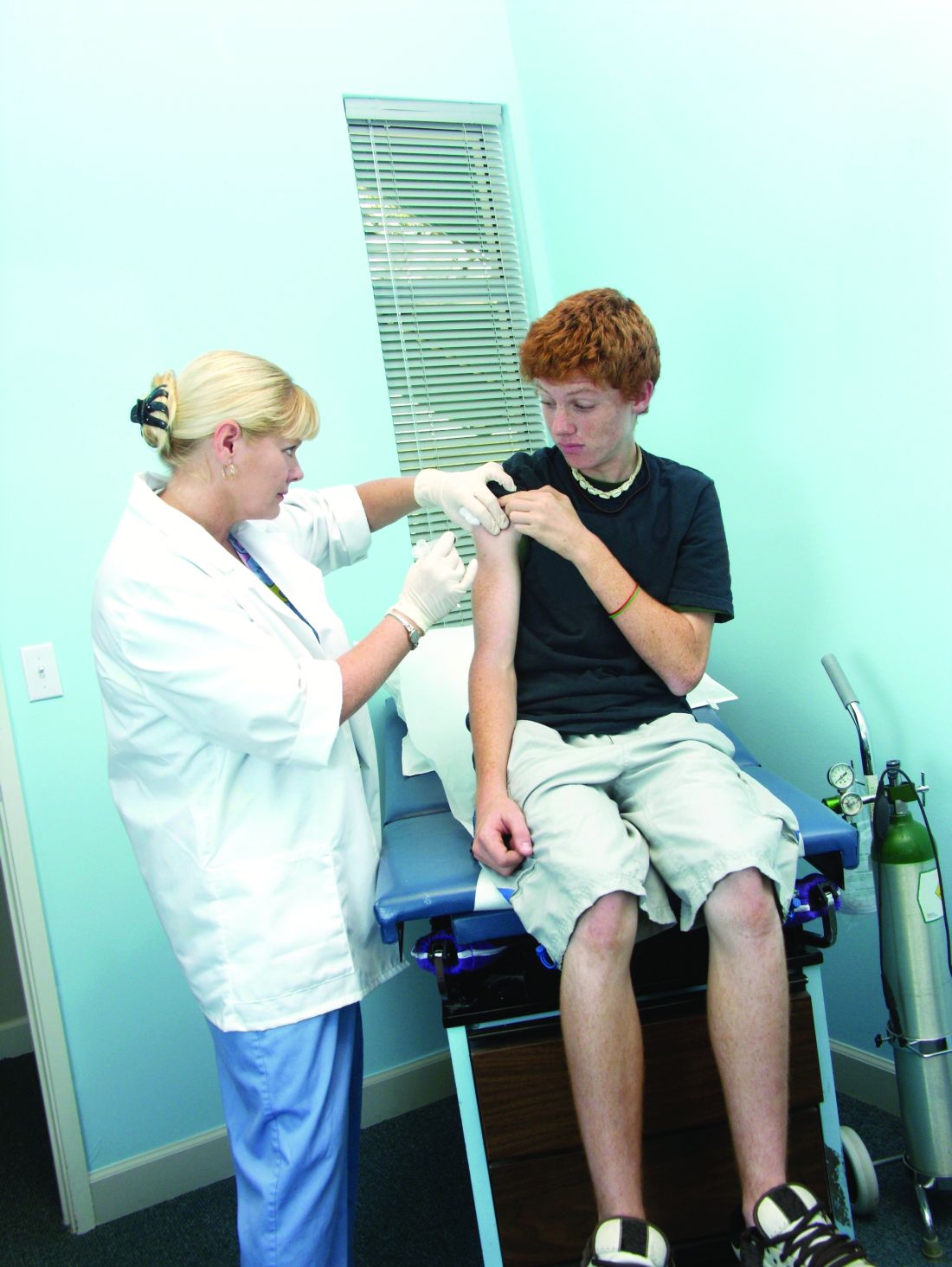
Adolescents with chronic health conditions often undervaccinated
said Annika M. Hofstetter, MD, PhD, of Columbia University, New York, and her associates.
The National Health Interview Survey on Disability in 1994-1995 estimated that chronic conditions of any type affected 15%-18% of U.S. children and adolescents. The Advisory Committee on Immunization Practices recommends that all adolescents, whether or not they have chronic medical condition, be vaccinated with human papillomavirus (HPV), Tdap, meningococcal, and flu vaccines.
Fewer adolescents with CMCs had received one more doses of HPV (81%), than did those without CMCs (85%; P less than .01). Fewer adolescents with epilepsy (63%), mental retardation (58%), cerebral palsy (54%), and autism spectrum disorder (46%) had started HPV vaccination, compared with those without each of these conditions (84%; all comparisons, P less than .001). No differences were seen for asthma or congenital heart disease, the investigators said.
More adolescents with CMCs had gotten their flu shot than did those without CMCs during the 2011-2012 season (67% vs. 50%; P less than .001) or during the 2012-2013 season (74% vs. 65%; P less than .001). More adolescents with asthma got their flu shot than did those without asthma during the 2011-2012 season (69% vs. 51%; P less than .001) or during the 2012-2013 season (74% vs. 65%; P less than .001). No differences were seen for the other common CMCs.
Nonetheless, the mean number of missed opportunities was significantly higher among unvaccinated adolescents with CMCs, compared with those without CMCs, for the first HPV vaccination, meningococcal vaccination, and influenza vaccination in both seasons measured (P less than .001 for all).
“Missed opportunities for the third HPV vaccine dose or Tdap did not differ by CMC status,” Dr. Hofstetter and her associates said.
Read more in the American Journal of Preventive Medicine (2017 Nov;53[5]:680-8).
said Annika M. Hofstetter, MD, PhD, of Columbia University, New York, and her associates.
The National Health Interview Survey on Disability in 1994-1995 estimated that chronic conditions of any type affected 15%-18% of U.S. children and adolescents. The Advisory Committee on Immunization Practices recommends that all adolescents, whether or not they have chronic medical condition, be vaccinated with human papillomavirus (HPV), Tdap, meningococcal, and flu vaccines.
Fewer adolescents with CMCs had received one more doses of HPV (81%), than did those without CMCs (85%; P less than .01). Fewer adolescents with epilepsy (63%), mental retardation (58%), cerebral palsy (54%), and autism spectrum disorder (46%) had started HPV vaccination, compared with those without each of these conditions (84%; all comparisons, P less than .001). No differences were seen for asthma or congenital heart disease, the investigators said.
More adolescents with CMCs had gotten their flu shot than did those without CMCs during the 2011-2012 season (67% vs. 50%; P less than .001) or during the 2012-2013 season (74% vs. 65%; P less than .001). More adolescents with asthma got their flu shot than did those without asthma during the 2011-2012 season (69% vs. 51%; P less than .001) or during the 2012-2013 season (74% vs. 65%; P less than .001). No differences were seen for the other common CMCs.
Nonetheless, the mean number of missed opportunities was significantly higher among unvaccinated adolescents with CMCs, compared with those without CMCs, for the first HPV vaccination, meningococcal vaccination, and influenza vaccination in both seasons measured (P less than .001 for all).
“Missed opportunities for the third HPV vaccine dose or Tdap did not differ by CMC status,” Dr. Hofstetter and her associates said.
Read more in the American Journal of Preventive Medicine (2017 Nov;53[5]:680-8).
said Annika M. Hofstetter, MD, PhD, of Columbia University, New York, and her associates.
The National Health Interview Survey on Disability in 1994-1995 estimated that chronic conditions of any type affected 15%-18% of U.S. children and adolescents. The Advisory Committee on Immunization Practices recommends that all adolescents, whether or not they have chronic medical condition, be vaccinated with human papillomavirus (HPV), Tdap, meningococcal, and flu vaccines.
Fewer adolescents with CMCs had received one more doses of HPV (81%), than did those without CMCs (85%; P less than .01). Fewer adolescents with epilepsy (63%), mental retardation (58%), cerebral palsy (54%), and autism spectrum disorder (46%) had started HPV vaccination, compared with those without each of these conditions (84%; all comparisons, P less than .001). No differences were seen for asthma or congenital heart disease, the investigators said.
More adolescents with CMCs had gotten their flu shot than did those without CMCs during the 2011-2012 season (67% vs. 50%; P less than .001) or during the 2012-2013 season (74% vs. 65%; P less than .001). More adolescents with asthma got their flu shot than did those without asthma during the 2011-2012 season (69% vs. 51%; P less than .001) or during the 2012-2013 season (74% vs. 65%; P less than .001). No differences were seen for the other common CMCs.
Nonetheless, the mean number of missed opportunities was significantly higher among unvaccinated adolescents with CMCs, compared with those without CMCs, for the first HPV vaccination, meningococcal vaccination, and influenza vaccination in both seasons measured (P less than .001 for all).
“Missed opportunities for the third HPV vaccine dose or Tdap did not differ by CMC status,” Dr. Hofstetter and her associates said.
Read more in the American Journal of Preventive Medicine (2017 Nov;53[5]:680-8).
FROM THE AMERICAN JOURNAL OF PREVENTIVE MEDICINE
Ultrathin bronchoscopy plus radial EBUS unreliable at making diagnoses
TORONTO – Ultrathin bronchoscopy plus radial endobronchial ultrasound is not a great method for determining whether a suspicious lesion is cancerous or benign, suggests new research.
In this study of patients with CT-detected solid lung lesions, the researchers were able to make a diagnosis for only 49% of those whose nodules were evaluated using ultrathin bronchoscopy plus radial endobronchial ultrasound (EBUS).

“When you do CT-guided biopsies of lung lesions, the [diagnostic] yield is about 94%. So do the math” by comparing it to the roughly 50% yield from ultrathin bronchoscopy plus radial EBUS to decide whether the latter procedure is worth doing, she noted.
The study Dr. Tanner and her associates designed compared the diagnostic yield of ultrathin bronchoscopy plus radial EBUS with standard bronchoscopy and fluoroscopy in patients with CT-detected solid lung lesions 1.5-5.0 cm in size. It ran at five U.S. centers and randomized 221 patients: 85 evaluable patients were tested using the standard methods, and 112 evaluable patients were tested using ultrathin bronchoscopy plus radial EBUS. Patients averaged 65-68 years of age and were divided evenly between women and men. Their lesions averaged slightly more than 3 cm. The ultrathin device had a 4 mm wide diameter and had a 2 mm working channel.
The diagnostic yield was 38% among patients who underwent standard bronchoscopy and fluoroscopy, and 49% among those biopsied using ultrathin bronchoscopy and radial EBUS, Dr. Tanner reported. The between-group difference in yield fell short of being statistically significant.
Forty-six of the 53 patients who were not diagnosable using standard bronchoscopy and fluoroscopy crossed over to the investigational method, which produced a diagnosis for an additional seven patients (15% of the biopsied crossover patients).
The results showed that standard bronchoscopy plus fluoroscopy is “very poor” for distinguishing cancerous and benign pulmonary lesions, Dr. Tanner concluded. The yield from ultrathin bronchoscopy plus radial EBUS in her study was similar to the diagnostic yields reported in prior studies of guided bronchoscopy, even when also using radial EBUS, she added.
Given the limitations of ultrathin bronchoscopy plus radial EBUS, Dr. Tanner suggested that the best scenario for using this diagnostic method would be in patients who need a linear EBUS procedure for mediastinal lymph node staging. Such staging often requires a biopsy of the primary tumor to make a cancer diagnosis, and in such cases, “while you’re in the neighborhood, you could do bronchoscopy with an ultrathin scope,” she suggested.
The potential also exists to augment the diagnostic yield of ultrathin bronchoscopy by applying a navigational software platform and needle biopsy, two methods not included in the study, Dr. Tanner noted. “More studies should be done using this combination,” she said.
The study was funded by Olympus. Dr. Tanner has been a consultant to and has received research funding from Olympus. She has also been a consultant to Cook Medical, Integrated Diagnostics, Oncocyte, Veracyte, and Veran Medical Technologies, and she has also received research funding from Cook, Integrated Diagnostics, Oncocyte, Oncimmune, and Veracyte.
[email protected]
On Twitter @mitchelzoler
Although bronchoscopic tools are safe and accurate to evaluate both central and peripheral lung lesions, the diagnostic yield of the different available techniques is variable. In this study, a diagnostic yield of only 49% was achieved when ultrathin bronchoscopy with radial EBUS was performed for diagnosis of solid nodules. This yield is not much better than that obtained from conventional bronchoscopy with fluoroscopic guidance and much lower than the diagnostic yield from transthoracic needle biopsy. While there is no doubt that the advances in minimally invasive technologies for diagnosing lung nodules and diagnosing and staging lung cancer have revolutionized clinical practice, pulmonologists and thoracic surgeons need to recognize not only the utility but also the limitations of the available diagnostic procedures (as well as the cost). These technologies are complimentary and multidisciplinary discussions should facilitate selection of the best procedure for each individual case.

Although bronchoscopic tools are safe and accurate to evaluate both central and peripheral lung lesions, the diagnostic yield of the different available techniques is variable. In this study, a diagnostic yield of only 49% was achieved when ultrathin bronchoscopy with radial EBUS was performed for diagnosis of solid nodules. This yield is not much better than that obtained from conventional bronchoscopy with fluoroscopic guidance and much lower than the diagnostic yield from transthoracic needle biopsy. While there is no doubt that the advances in minimally invasive technologies for diagnosing lung nodules and diagnosing and staging lung cancer have revolutionized clinical practice, pulmonologists and thoracic surgeons need to recognize not only the utility but also the limitations of the available diagnostic procedures (as well as the cost). These technologies are complimentary and multidisciplinary discussions should facilitate selection of the best procedure for each individual case.
Although bronchoscopic tools are safe and accurate to evaluate both central and peripheral lung lesions, the diagnostic yield of the different available techniques is variable. In this study, a diagnostic yield of only 49% was achieved when ultrathin bronchoscopy with radial EBUS was performed for diagnosis of solid nodules. This yield is not much better than that obtained from conventional bronchoscopy with fluoroscopic guidance and much lower than the diagnostic yield from transthoracic needle biopsy. While there is no doubt that the advances in minimally invasive technologies for diagnosing lung nodules and diagnosing and staging lung cancer have revolutionized clinical practice, pulmonologists and thoracic surgeons need to recognize not only the utility but also the limitations of the available diagnostic procedures (as well as the cost). These technologies are complimentary and multidisciplinary discussions should facilitate selection of the best procedure for each individual case.
TORONTO – Ultrathin bronchoscopy plus radial endobronchial ultrasound is not a great method for determining whether a suspicious lesion is cancerous or benign, suggests new research.
In this study of patients with CT-detected solid lung lesions, the researchers were able to make a diagnosis for only 49% of those whose nodules were evaluated using ultrathin bronchoscopy plus radial endobronchial ultrasound (EBUS).
“When you do CT-guided biopsies of lung lesions, the [diagnostic] yield is about 94%. So do the math” by comparing it to the roughly 50% yield from ultrathin bronchoscopy plus radial EBUS to decide whether the latter procedure is worth doing, she noted.
The study Dr. Tanner and her associates designed compared the diagnostic yield of ultrathin bronchoscopy plus radial EBUS with standard bronchoscopy and fluoroscopy in patients with CT-detected solid lung lesions 1.5-5.0 cm in size. It ran at five U.S. centers and randomized 221 patients: 85 evaluable patients were tested using the standard methods, and 112 evaluable patients were tested using ultrathin bronchoscopy plus radial EBUS. Patients averaged 65-68 years of age and were divided evenly between women and men. Their lesions averaged slightly more than 3 cm. The ultrathin device had a 4 mm wide diameter and had a 2 mm working channel.
The diagnostic yield was 38% among patients who underwent standard bronchoscopy and fluoroscopy, and 49% among those biopsied using ultrathin bronchoscopy and radial EBUS, Dr. Tanner reported. The between-group difference in yield fell short of being statistically significant.
Forty-six of the 53 patients who were not diagnosable using standard bronchoscopy and fluoroscopy crossed over to the investigational method, which produced a diagnosis for an additional seven patients (15% of the biopsied crossover patients).
The results showed that standard bronchoscopy plus fluoroscopy is “very poor” for distinguishing cancerous and benign pulmonary lesions, Dr. Tanner concluded. The yield from ultrathin bronchoscopy plus radial EBUS in her study was similar to the diagnostic yields reported in prior studies of guided bronchoscopy, even when also using radial EBUS, she added.
Given the limitations of ultrathin bronchoscopy plus radial EBUS, Dr. Tanner suggested that the best scenario for using this diagnostic method would be in patients who need a linear EBUS procedure for mediastinal lymph node staging. Such staging often requires a biopsy of the primary tumor to make a cancer diagnosis, and in such cases, “while you’re in the neighborhood, you could do bronchoscopy with an ultrathin scope,” she suggested.
The potential also exists to augment the diagnostic yield of ultrathin bronchoscopy by applying a navigational software platform and needle biopsy, two methods not included in the study, Dr. Tanner noted. “More studies should be done using this combination,” she said.
The study was funded by Olympus. Dr. Tanner has been a consultant to and has received research funding from Olympus. She has also been a consultant to Cook Medical, Integrated Diagnostics, Oncocyte, Veracyte, and Veran Medical Technologies, and she has also received research funding from Cook, Integrated Diagnostics, Oncocyte, Oncimmune, and Veracyte.
[email protected]
On Twitter @mitchelzoler
TORONTO – Ultrathin bronchoscopy plus radial endobronchial ultrasound is not a great method for determining whether a suspicious lesion is cancerous or benign, suggests new research.
In this study of patients with CT-detected solid lung lesions, the researchers were able to make a diagnosis for only 49% of those whose nodules were evaluated using ultrathin bronchoscopy plus radial endobronchial ultrasound (EBUS).
“When you do CT-guided biopsies of lung lesions, the [diagnostic] yield is about 94%. So do the math” by comparing it to the roughly 50% yield from ultrathin bronchoscopy plus radial EBUS to decide whether the latter procedure is worth doing, she noted.
The study Dr. Tanner and her associates designed compared the diagnostic yield of ultrathin bronchoscopy plus radial EBUS with standard bronchoscopy and fluoroscopy in patients with CT-detected solid lung lesions 1.5-5.0 cm in size. It ran at five U.S. centers and randomized 221 patients: 85 evaluable patients were tested using the standard methods, and 112 evaluable patients were tested using ultrathin bronchoscopy plus radial EBUS. Patients averaged 65-68 years of age and were divided evenly between women and men. Their lesions averaged slightly more than 3 cm. The ultrathin device had a 4 mm wide diameter and had a 2 mm working channel.
The diagnostic yield was 38% among patients who underwent standard bronchoscopy and fluoroscopy, and 49% among those biopsied using ultrathin bronchoscopy and radial EBUS, Dr. Tanner reported. The between-group difference in yield fell short of being statistically significant.
Forty-six of the 53 patients who were not diagnosable using standard bronchoscopy and fluoroscopy crossed over to the investigational method, which produced a diagnosis for an additional seven patients (15% of the biopsied crossover patients).
The results showed that standard bronchoscopy plus fluoroscopy is “very poor” for distinguishing cancerous and benign pulmonary lesions, Dr. Tanner concluded. The yield from ultrathin bronchoscopy plus radial EBUS in her study was similar to the diagnostic yields reported in prior studies of guided bronchoscopy, even when also using radial EBUS, she added.
Given the limitations of ultrathin bronchoscopy plus radial EBUS, Dr. Tanner suggested that the best scenario for using this diagnostic method would be in patients who need a linear EBUS procedure for mediastinal lymph node staging. Such staging often requires a biopsy of the primary tumor to make a cancer diagnosis, and in such cases, “while you’re in the neighborhood, you could do bronchoscopy with an ultrathin scope,” she suggested.
The potential also exists to augment the diagnostic yield of ultrathin bronchoscopy by applying a navigational software platform and needle biopsy, two methods not included in the study, Dr. Tanner noted. “More studies should be done using this combination,” she said.
The study was funded by Olympus. Dr. Tanner has been a consultant to and has received research funding from Olympus. She has also been a consultant to Cook Medical, Integrated Diagnostics, Oncocyte, Veracyte, and Veran Medical Technologies, and she has also received research funding from Cook, Integrated Diagnostics, Oncocyte, Oncimmune, and Veracyte.
[email protected]
On Twitter @mitchelzoler
AT CHEST 2017
Key clinical point:
Major finding: The diagnostic yield using ultrathin bronchoscopy with radial EBUS was 49%, while standard bronchoscopy had a 38% yield.
Data source: Multicenter, randomized study with 221 total patients and 197 evaluable patients.
Disclosures: The study was funded by Olympus. Dr. Tanner has been a consultant to and has received research funding from Olympus. She has also been a consultant to Cook Medical, Integrated Diagnostics, Oncocyte, Veracyte, and Veran Medical Technologies, and she has also received research funding from Cook, Integrated Diagnostics, Oncocyte, Oncimmune, and Veracyte.
Phrenic-nerve stimulator maintains benefits for 18 months
TORONTO – The implanted phrenic-nerve stimulation device that received Food and Drug Administration marketing approval in October 2017 for treating central sleep apnea has now shown safety and efficacy out to 18 months of continuous use in 102 patients.
After 18 months of treatment with the Remede System, patients’ outcomes remained stable and patients continued to see the improvements they had experienced after 6 and 12 months of treatment. These improvements included significant average reductions from baseline in apnea-hypopnea index and central apnea index and significant increases in oxygenation and sleep quality, Andrew C. Kao, MD, said at the CHEST annual meeting.

“We were concerned that there would be a degradation of the benefit [over time]. We are very happy that the benefit was sustained,” said Dr. Kao, a heart failure cardiologist at Saint Luke’s Health System in Kansas City, Mo.
Dr. Kao did not report an 18-month follow-up for the study’s primary endpoint, the percentage of patients after 6 months on treatment who had at least a 50% reduction from baseline in their apnea-hypopnea index. His report focused on the 6-, 12-, and 18-month changes relative to baseline for five secondary outcomes: central sleep apnea index, apnea-hypopnea index, arousal index, oxygen desaturation index, and time spent in REM sleep. For all five of these outcomes, the 102 patients showed an average, statistically significant improvement compared with baseline after 6 months on treatment that persisted virtually unchanged at 12 and 18 months.
For example, average central sleep apnea index fell from 27 events/hour at baseline to 5 per hour at 6, 12, and 18 months. Average apnea-hypopnea index fell from 46 events/hour at baseline to about 25 per hour at 6, 12, and 18 months. The average percentage of sleep spent in REM sleep improved from 12% at baseline to about 15% at 6, 12, and 18 months.
During 18 months of treatment following device implantation, four of the 102 patients had a serious adverse event. One patient required lead repositioning to relieve discomfort and three had an interaction with an implanted cardiac device. The effects resolved in all four patients without long-term impact. An additional 16 patients had discomfort that required an unscheduled medical visit, but these were not classified as serious episodes, and in 14 of these patients the discomfort resolved.
The Remede System phrenic-nerve stimulator received FDA marketing approval for moderate to severe central sleep apnea based on 6-month efficacy and 12-month safety data (Lancet. 2016 Sept 3;388[10048]:974-82). The Pivotal Trial of the Remede System enrolled 151 patients with an apnea-hypopnea index of at least 20 events/hour, about half of whom had heart failure. All patients received a device implant: In the initial intervention group of 73 patients, researchers turned on the device 1 month after implantation, and in the 78 patients randomized to the initial control arm, the device remained off for the first 7 months and then went active. The researchers followed up with 46 patients drawn from both the original treatment arm and 56 patients from the original control arm, at which point the patients had been receiving 18 months of treatment.
The Remede System pivotal trial was sponsored by Respicardia, which markets the phrenic-verse stimulator. Dr. Kao’s institution, Saint Luke’s Health System, received grant support from Respicardia.
[email protected]
On Twitter @mitchelzoler
TORONTO – The implanted phrenic-nerve stimulation device that received Food and Drug Administration marketing approval in October 2017 for treating central sleep apnea has now shown safety and efficacy out to 18 months of continuous use in 102 patients.
After 18 months of treatment with the Remede System, patients’ outcomes remained stable and patients continued to see the improvements they had experienced after 6 and 12 months of treatment. These improvements included significant average reductions from baseline in apnea-hypopnea index and central apnea index and significant increases in oxygenation and sleep quality, Andrew C. Kao, MD, said at the CHEST annual meeting.
“We were concerned that there would be a degradation of the benefit [over time]. We are very happy that the benefit was sustained,” said Dr. Kao, a heart failure cardiologist at Saint Luke’s Health System in Kansas City, Mo.
Dr. Kao did not report an 18-month follow-up for the study’s primary endpoint, the percentage of patients after 6 months on treatment who had at least a 50% reduction from baseline in their apnea-hypopnea index. His report focused on the 6-, 12-, and 18-month changes relative to baseline for five secondary outcomes: central sleep apnea index, apnea-hypopnea index, arousal index, oxygen desaturation index, and time spent in REM sleep. For all five of these outcomes, the 102 patients showed an average, statistically significant improvement compared with baseline after 6 months on treatment that persisted virtually unchanged at 12 and 18 months.
For example, average central sleep apnea index fell from 27 events/hour at baseline to 5 per hour at 6, 12, and 18 months. Average apnea-hypopnea index fell from 46 events/hour at baseline to about 25 per hour at 6, 12, and 18 months. The average percentage of sleep spent in REM sleep improved from 12% at baseline to about 15% at 6, 12, and 18 months.
During 18 months of treatment following device implantation, four of the 102 patients had a serious adverse event. One patient required lead repositioning to relieve discomfort and three had an interaction with an implanted cardiac device. The effects resolved in all four patients without long-term impact. An additional 16 patients had discomfort that required an unscheduled medical visit, but these were not classified as serious episodes, and in 14 of these patients the discomfort resolved.
The Remede System phrenic-nerve stimulator received FDA marketing approval for moderate to severe central sleep apnea based on 6-month efficacy and 12-month safety data (Lancet. 2016 Sept 3;388[10048]:974-82). The Pivotal Trial of the Remede System enrolled 151 patients with an apnea-hypopnea index of at least 20 events/hour, about half of whom had heart failure. All patients received a device implant: In the initial intervention group of 73 patients, researchers turned on the device 1 month after implantation, and in the 78 patients randomized to the initial control arm, the device remained off for the first 7 months and then went active. The researchers followed up with 46 patients drawn from both the original treatment arm and 56 patients from the original control arm, at which point the patients had been receiving 18 months of treatment.
The Remede System pivotal trial was sponsored by Respicardia, which markets the phrenic-verse stimulator. Dr. Kao’s institution, Saint Luke’s Health System, received grant support from Respicardia.
[email protected]
On Twitter @mitchelzoler
TORONTO – The implanted phrenic-nerve stimulation device that received Food and Drug Administration marketing approval in October 2017 for treating central sleep apnea has now shown safety and efficacy out to 18 months of continuous use in 102 patients.
After 18 months of treatment with the Remede System, patients’ outcomes remained stable and patients continued to see the improvements they had experienced after 6 and 12 months of treatment. These improvements included significant average reductions from baseline in apnea-hypopnea index and central apnea index and significant increases in oxygenation and sleep quality, Andrew C. Kao, MD, said at the CHEST annual meeting.
“We were concerned that there would be a degradation of the benefit [over time]. We are very happy that the benefit was sustained,” said Dr. Kao, a heart failure cardiologist at Saint Luke’s Health System in Kansas City, Mo.
Dr. Kao did not report an 18-month follow-up for the study’s primary endpoint, the percentage of patients after 6 months on treatment who had at least a 50% reduction from baseline in their apnea-hypopnea index. His report focused on the 6-, 12-, and 18-month changes relative to baseline for five secondary outcomes: central sleep apnea index, apnea-hypopnea index, arousal index, oxygen desaturation index, and time spent in REM sleep. For all five of these outcomes, the 102 patients showed an average, statistically significant improvement compared with baseline after 6 months on treatment that persisted virtually unchanged at 12 and 18 months.
For example, average central sleep apnea index fell from 27 events/hour at baseline to 5 per hour at 6, 12, and 18 months. Average apnea-hypopnea index fell from 46 events/hour at baseline to about 25 per hour at 6, 12, and 18 months. The average percentage of sleep spent in REM sleep improved from 12% at baseline to about 15% at 6, 12, and 18 months.
During 18 months of treatment following device implantation, four of the 102 patients had a serious adverse event. One patient required lead repositioning to relieve discomfort and three had an interaction with an implanted cardiac device. The effects resolved in all four patients without long-term impact. An additional 16 patients had discomfort that required an unscheduled medical visit, but these were not classified as serious episodes, and in 14 of these patients the discomfort resolved.
The Remede System phrenic-nerve stimulator received FDA marketing approval for moderate to severe central sleep apnea based on 6-month efficacy and 12-month safety data (Lancet. 2016 Sept 3;388[10048]:974-82). The Pivotal Trial of the Remede System enrolled 151 patients with an apnea-hypopnea index of at least 20 events/hour, about half of whom had heart failure. All patients received a device implant: In the initial intervention group of 73 patients, researchers turned on the device 1 month after implantation, and in the 78 patients randomized to the initial control arm, the device remained off for the first 7 months and then went active. The researchers followed up with 46 patients drawn from both the original treatment arm and 56 patients from the original control arm, at which point the patients had been receiving 18 months of treatment.
The Remede System pivotal trial was sponsored by Respicardia, which markets the phrenic-verse stimulator. Dr. Kao’s institution, Saint Luke’s Health System, received grant support from Respicardia.
[email protected]
On Twitter @mitchelzoler
AT CHEST 2017
Key clinical point:
Major finding: Average central apnea index improved from 27 events/hour at baseline to 5 events/hour after 6, 12, and 18 months of treatment.
Data source: 102 patients enrolled in the Pivotal Trial of the remede System were followed for 18 months of treatment.
Disclosures: The remede System pivotal trial was sponsored by Respicardia, which markets the phrenic-verse stimulator. Dr. Kao’s institution, Saint Luke’s Health System, received grant support from Respicardia.
First-in-class glutaminase inhibitor combats anti-PD-1/PD-L1 resistance
NATIONAL HARBOR, MD. – Combination treatment with the first-in-class glutaminase inhibitor CB-839 and nivolumab is well-tolerated and shows clinical activity in patients with advanced melanoma, renal cell carcinoma, or non-small cell lung cancer, including anti-PD-1/PD-L1 refractory patients, according to initial results from a phase 1/2 study.
Responses in melanoma patients who were progressing on nivolumab at study entry and who were refractory to multiple prior immunotherapy regimens are particularly notable, as they highlight the potential for CB-839, when added to nivolumab (Opdivo), to help overcome resistance to anti-PD-L1 therapy, Funda Meric‐Bernstam, MD, reported at the annual meeting of the Society for Immunotherapy of Cancer.
CB‐839 is highly selective and targets tumor glutamine metabolism, said Dr. Meric-Bernstam of the University of Texas MD Anderson Cancer Center, Houston.
Competition between tumor cells and immune cells for nutrients such as glutamine in the tumor microenvironment can create a metabolic checkpoint that induces local immune suppression. CB‐839 inhibits tumor glutamine consumption, thereby increasing glutamine availability to support T‐cell activity, she explained, noting that in preclinical models, CB‐839 increased intra‐tumoral glutamine and enhanced antitumor activity of PD‐1/PD‐L1 inhibitors.
In the phase 1 dose escalation study, she and her colleagues evaluated the safety and efficacy of CB-839 in combination with the PD‐1 inhibitor nivolumab in patients with melanoma, non-small cell lung cancer (NSCLC), or renal cell carcinoma (RCC). Phase 2 expansion cohorts include a melanoma rescue cohort of patients progressing on anti-PD-L1 therapy at study entry (22 patients), an NSCLC and RCC rescue cohort of patients who were progressing on anti-PD-L1 therapy at study entry or who had stable disease for 6 months or longer without a response (11 NSCLC and 11 RCC), an RCC cohort of patients with prior immunotherapy exposure and no response (10 patients), and an RCC cohort of patents who had no prior immunotherapy exposure (28 patients).
During dose escalation, patients received oral CB‐839 at 600 mg or 800 mg twice daily in combination with standard‐dose nivolumab. In the ongoing phase 2 expansion study, which continues to enroll, patients are receiving 800 mg of CB-839 twice daily with standard‐dose nivolumab, Dr. Meric-Bernstam said.
Patients in each of the cohorts were high risk and/or had intermediate or poor prognostic status at study entry. For example, 50% of patients in the melanoma rescue cohort had liver metastases, 77% had other visceral metastases, and 18% had brain metastases, and the majority of patients in the lung cancer/RCC cohort had visceral metastases. Most had progressive disease as their best response on their last line of immunotherapy.
Of 16 response-evaluable melanoma patients, 1 experienced a complete response, 2 had partial responses, and 4 had stable disease.
“So overall in this patient population that was progressing on a PD-1/PD-L1 inhibitor at enrollment, 19% had an objective response. The disease control rate in this group was 44%,” she said.
In evaluable patients in the lung cancer rescue cohort (6 patients), RCC rescue cohort (8 patients), and RCC prior exposure cohort (7 patients), disease control rates ranged from 57% to 75%, and in the immunotherapy-naive RCC cohort (19 patients), the partial response rate was 21%, and 53% had stable disease, so the overall disease control rate was 74%. Half of the patients in that group remain on study, she noted.
A closer look at the melanoma rescue cohort showed dramatic and rapid responses in two patients who each achieved a partial response in about 8 weeks with response durations of 3.7 months and 5.4 months, respectively. Additionally, pre-treatment biopsies in this cohort showed an elevated T-cell inflamed signature associated with clinical benefit from the addition of CB-839, and in one patient who had both a pretreatment and on-treatment biopsy that was evaluable, the latter showed an increase in T-cell inflamed signature and T-cell effector genes.
In all cohorts, the combination therapy was generally well tolerated. A maximum tolerated dose was not reached. Dose-limiting toxicity – a grade 3 alanine aminotransferase (ALT) increase – occurred in one patient on the 800-mg dose. The most common grade 3 or greater adverse events were fatigue, nausea, photophobia, rash, and elevated ALT, she said, noting that two patients discontinued for treatment-related adverse events (one for a grade 3 rash and one for grade 2 pneumonitis).
“Overall there appeared to be no apparent increase in immune-related adverse events, either in rate or severity, compared with [nivolumab] monotherapy,” she said.
The combination of CB-839 and nivolumab was well tolerated, and in some patients – as seen in the melanoma cohort – adding CB-839 to checkpoint blockade can overcome checkpoint blockade resistance, Dr. Meric-Bernstam concluded, noting that the disease control rates seen in the majority of lung cancer and RCC patients who were progressing on checkpoint blockade is encouraging, as is the objective response rate seen thus far in the RCC therapy-naive patients, and the stable and deep responses seen in the melanoma rescue cohort.
“Based on our encouraging signal in the melanoma rescue cohort, this [cohort] has been expanded,” she said.
Calithera Biosciences sponsored the study. Bristol-Myers Squibb provided nivolumab for the study. Dr. Meric-Bernstam has received grant or research support from Calithera Biosciences and many other companies. She also reported being a paid consultant for several companies and serving on an advisory committee or review panel, or as a board member for multiple companies.
NATIONAL HARBOR, MD. – Combination treatment with the first-in-class glutaminase inhibitor CB-839 and nivolumab is well-tolerated and shows clinical activity in patients with advanced melanoma, renal cell carcinoma, or non-small cell lung cancer, including anti-PD-1/PD-L1 refractory patients, according to initial results from a phase 1/2 study.
Responses in melanoma patients who were progressing on nivolumab at study entry and who were refractory to multiple prior immunotherapy regimens are particularly notable, as they highlight the potential for CB-839, when added to nivolumab (Opdivo), to help overcome resistance to anti-PD-L1 therapy, Funda Meric‐Bernstam, MD, reported at the annual meeting of the Society for Immunotherapy of Cancer.
CB‐839 is highly selective and targets tumor glutamine metabolism, said Dr. Meric-Bernstam of the University of Texas MD Anderson Cancer Center, Houston.
Competition between tumor cells and immune cells for nutrients such as glutamine in the tumor microenvironment can create a metabolic checkpoint that induces local immune suppression. CB‐839 inhibits tumor glutamine consumption, thereby increasing glutamine availability to support T‐cell activity, she explained, noting that in preclinical models, CB‐839 increased intra‐tumoral glutamine and enhanced antitumor activity of PD‐1/PD‐L1 inhibitors.
In the phase 1 dose escalation study, she and her colleagues evaluated the safety and efficacy of CB-839 in combination with the PD‐1 inhibitor nivolumab in patients with melanoma, non-small cell lung cancer (NSCLC), or renal cell carcinoma (RCC). Phase 2 expansion cohorts include a melanoma rescue cohort of patients progressing on anti-PD-L1 therapy at study entry (22 patients), an NSCLC and RCC rescue cohort of patients who were progressing on anti-PD-L1 therapy at study entry or who had stable disease for 6 months or longer without a response (11 NSCLC and 11 RCC), an RCC cohort of patients with prior immunotherapy exposure and no response (10 patients), and an RCC cohort of patents who had no prior immunotherapy exposure (28 patients).
During dose escalation, patients received oral CB‐839 at 600 mg or 800 mg twice daily in combination with standard‐dose nivolumab. In the ongoing phase 2 expansion study, which continues to enroll, patients are receiving 800 mg of CB-839 twice daily with standard‐dose nivolumab, Dr. Meric-Bernstam said.
Patients in each of the cohorts were high risk and/or had intermediate or poor prognostic status at study entry. For example, 50% of patients in the melanoma rescue cohort had liver metastases, 77% had other visceral metastases, and 18% had brain metastases, and the majority of patients in the lung cancer/RCC cohort had visceral metastases. Most had progressive disease as their best response on their last line of immunotherapy.
Of 16 response-evaluable melanoma patients, 1 experienced a complete response, 2 had partial responses, and 4 had stable disease.
“So overall in this patient population that was progressing on a PD-1/PD-L1 inhibitor at enrollment, 19% had an objective response. The disease control rate in this group was 44%,” she said.
In evaluable patients in the lung cancer rescue cohort (6 patients), RCC rescue cohort (8 patients), and RCC prior exposure cohort (7 patients), disease control rates ranged from 57% to 75%, and in the immunotherapy-naive RCC cohort (19 patients), the partial response rate was 21%, and 53% had stable disease, so the overall disease control rate was 74%. Half of the patients in that group remain on study, she noted.
A closer look at the melanoma rescue cohort showed dramatic and rapid responses in two patients who each achieved a partial response in about 8 weeks with response durations of 3.7 months and 5.4 months, respectively. Additionally, pre-treatment biopsies in this cohort showed an elevated T-cell inflamed signature associated with clinical benefit from the addition of CB-839, and in one patient who had both a pretreatment and on-treatment biopsy that was evaluable, the latter showed an increase in T-cell inflamed signature and T-cell effector genes.
In all cohorts, the combination therapy was generally well tolerated. A maximum tolerated dose was not reached. Dose-limiting toxicity – a grade 3 alanine aminotransferase (ALT) increase – occurred in one patient on the 800-mg dose. The most common grade 3 or greater adverse events were fatigue, nausea, photophobia, rash, and elevated ALT, she said, noting that two patients discontinued for treatment-related adverse events (one for a grade 3 rash and one for grade 2 pneumonitis).
“Overall there appeared to be no apparent increase in immune-related adverse events, either in rate or severity, compared with [nivolumab] monotherapy,” she said.
The combination of CB-839 and nivolumab was well tolerated, and in some patients – as seen in the melanoma cohort – adding CB-839 to checkpoint blockade can overcome checkpoint blockade resistance, Dr. Meric-Bernstam concluded, noting that the disease control rates seen in the majority of lung cancer and RCC patients who were progressing on checkpoint blockade is encouraging, as is the objective response rate seen thus far in the RCC therapy-naive patients, and the stable and deep responses seen in the melanoma rescue cohort.
“Based on our encouraging signal in the melanoma rescue cohort, this [cohort] has been expanded,” she said.
Calithera Biosciences sponsored the study. Bristol-Myers Squibb provided nivolumab for the study. Dr. Meric-Bernstam has received grant or research support from Calithera Biosciences and many other companies. She also reported being a paid consultant for several companies and serving on an advisory committee or review panel, or as a board member for multiple companies.
NATIONAL HARBOR, MD. – Combination treatment with the first-in-class glutaminase inhibitor CB-839 and nivolumab is well-tolerated and shows clinical activity in patients with advanced melanoma, renal cell carcinoma, or non-small cell lung cancer, including anti-PD-1/PD-L1 refractory patients, according to initial results from a phase 1/2 study.
Responses in melanoma patients who were progressing on nivolumab at study entry and who were refractory to multiple prior immunotherapy regimens are particularly notable, as they highlight the potential for CB-839, when added to nivolumab (Opdivo), to help overcome resistance to anti-PD-L1 therapy, Funda Meric‐Bernstam, MD, reported at the annual meeting of the Society for Immunotherapy of Cancer.
CB‐839 is highly selective and targets tumor glutamine metabolism, said Dr. Meric-Bernstam of the University of Texas MD Anderson Cancer Center, Houston.
Competition between tumor cells and immune cells for nutrients such as glutamine in the tumor microenvironment can create a metabolic checkpoint that induces local immune suppression. CB‐839 inhibits tumor glutamine consumption, thereby increasing glutamine availability to support T‐cell activity, she explained, noting that in preclinical models, CB‐839 increased intra‐tumoral glutamine and enhanced antitumor activity of PD‐1/PD‐L1 inhibitors.
In the phase 1 dose escalation study, she and her colleagues evaluated the safety and efficacy of CB-839 in combination with the PD‐1 inhibitor nivolumab in patients with melanoma, non-small cell lung cancer (NSCLC), or renal cell carcinoma (RCC). Phase 2 expansion cohorts include a melanoma rescue cohort of patients progressing on anti-PD-L1 therapy at study entry (22 patients), an NSCLC and RCC rescue cohort of patients who were progressing on anti-PD-L1 therapy at study entry or who had stable disease for 6 months or longer without a response (11 NSCLC and 11 RCC), an RCC cohort of patients with prior immunotherapy exposure and no response (10 patients), and an RCC cohort of patents who had no prior immunotherapy exposure (28 patients).
During dose escalation, patients received oral CB‐839 at 600 mg or 800 mg twice daily in combination with standard‐dose nivolumab. In the ongoing phase 2 expansion study, which continues to enroll, patients are receiving 800 mg of CB-839 twice daily with standard‐dose nivolumab, Dr. Meric-Bernstam said.
Patients in each of the cohorts were high risk and/or had intermediate or poor prognostic status at study entry. For example, 50% of patients in the melanoma rescue cohort had liver metastases, 77% had other visceral metastases, and 18% had brain metastases, and the majority of patients in the lung cancer/RCC cohort had visceral metastases. Most had progressive disease as their best response on their last line of immunotherapy.
Of 16 response-evaluable melanoma patients, 1 experienced a complete response, 2 had partial responses, and 4 had stable disease.
“So overall in this patient population that was progressing on a PD-1/PD-L1 inhibitor at enrollment, 19% had an objective response. The disease control rate in this group was 44%,” she said.
In evaluable patients in the lung cancer rescue cohort (6 patients), RCC rescue cohort (8 patients), and RCC prior exposure cohort (7 patients), disease control rates ranged from 57% to 75%, and in the immunotherapy-naive RCC cohort (19 patients), the partial response rate was 21%, and 53% had stable disease, so the overall disease control rate was 74%. Half of the patients in that group remain on study, she noted.
A closer look at the melanoma rescue cohort showed dramatic and rapid responses in two patients who each achieved a partial response in about 8 weeks with response durations of 3.7 months and 5.4 months, respectively. Additionally, pre-treatment biopsies in this cohort showed an elevated T-cell inflamed signature associated with clinical benefit from the addition of CB-839, and in one patient who had both a pretreatment and on-treatment biopsy that was evaluable, the latter showed an increase in T-cell inflamed signature and T-cell effector genes.
In all cohorts, the combination therapy was generally well tolerated. A maximum tolerated dose was not reached. Dose-limiting toxicity – a grade 3 alanine aminotransferase (ALT) increase – occurred in one patient on the 800-mg dose. The most common grade 3 or greater adverse events were fatigue, nausea, photophobia, rash, and elevated ALT, she said, noting that two patients discontinued for treatment-related adverse events (one for a grade 3 rash and one for grade 2 pneumonitis).
“Overall there appeared to be no apparent increase in immune-related adverse events, either in rate or severity, compared with [nivolumab] monotherapy,” she said.
The combination of CB-839 and nivolumab was well tolerated, and in some patients – as seen in the melanoma cohort – adding CB-839 to checkpoint blockade can overcome checkpoint blockade resistance, Dr. Meric-Bernstam concluded, noting that the disease control rates seen in the majority of lung cancer and RCC patients who were progressing on checkpoint blockade is encouraging, as is the objective response rate seen thus far in the RCC therapy-naive patients, and the stable and deep responses seen in the melanoma rescue cohort.
“Based on our encouraging signal in the melanoma rescue cohort, this [cohort] has been expanded,” she said.
Calithera Biosciences sponsored the study. Bristol-Myers Squibb provided nivolumab for the study. Dr. Meric-Bernstam has received grant or research support from Calithera Biosciences and many other companies. She also reported being a paid consultant for several companies and serving on an advisory committee or review panel, or as a board member for multiple companies.
AT SITC 2017
Key clinical point:
Major finding: The objective response rate in advanced melanoma patients refractory to anti-PD-1/PD-L1 therapy was 19%.
Data source: A phase 1/2 study of 82 patients.
Disclosures: Calithera Biosciences sponsored the study. Bristol-Myers Squibb provided nivolumab for the study. Dr. Meric-Bernstam has received grant or research support from Calithera Biosciences and many other companies. She also reported being a paid consultant for several companies and serving on an advisory committee or review panel or as a board member for multiple companies.
ENCORE 601 study: Entinostat shows promise in NSCLC
NATIONAL HARBOR, MD. – The oral, class I selective histone deacetylase (HDAC) inhibitor entinostat given in combination with pembrolizumab demonstrated antitumor activity and acceptable safety in patients with non–small cell lung cancer in the phase 1b/2 ENCORE 601 study.
Entinostat, which has been shown in preclinical models to enhance suppressor cells in the tumor microenvironment, was evaluated in ENCORE 601 as a treatment for non–small cell lung cancer (NSCLC), melanoma, and colorectal cancer. Previously reported phase 1 results showed that an oral dose of 5 mg weekly plus 200 mg of pembrolizumab given intravenously every 3 weeks deserved further exploration for these indications, according to Leena Gandhi, MD, who reported phase 2, stage 1 results from the lung cancer arm of the Simon two-stage study at the annual meeting of the Society for Immunotherapy of Cancer.
Treatment at that dose was studied in both anti-PD-L1–naive patients with advanced NSCLC, and in NSCLC patients who progressed on anti-PD-L1 treatment, said Dr. Gandhi of New York University Langone Medical Center.
The primary objective of stage 1 was objective response rate, and criteria for advancement were 4 or more responses out of 17 evaluable anti-PD-L1–naive patients (cohort 1), and at least 3 responses out of 31 patients who progressed on anti-PD-L1 therapy (cohort 2).
Both cohorts met the endpoint, with 4 of 17 evaluable cohort 1 patients (24%) achieving a partial response, and 3 of 31 evaluable cohort 2 patients (10%) achieving a partial response.
In cohort 1, two responses were confirmed and two were unconfirmed. One of the unconfirmed patients had malignant pericardial effusion, but remains on study with continued clinical benefit, Dr. Gandhi said, noting that three patients remain on study in all.
“The other notable thing I’d like to point out here … is that the majority of these were patients who did not have high levels of expression of PD-L1,” she said.
In cohort 2 patients, two responses were confirmed and one was unconfirmed. Three patients remain on study.
“In both of these cohorts there are a couple of patients who’ve had quite durable responses,” she said.
The best response to prior anti-PD-1therapy in the cohort 2 patients who had a response was stable disease (two patients). The response to prior therapy was unknown in one patient, she noted.
“All of them had clear regressions, after that initial PD-1 therapy, with this combination,” she said, noting that two had “essentially negative PD-L1 expression, and none had high levels of expression.”
Treatment was associated with grade 3/4 adverse events deemed drug related in 31% of patients; the most common of these events, occurring in at least 10% of patients in cohort 1, were hypophosphatemia and neutropenia, and in cohort 2 were fatigue, anemia, anorexia, and pneumonitis; 13% of patients discontinued treatment due to an adverse event, Dr. Gandhi said.
Of note, there were reductions in circulating myeloid derived suppressor cells in both cohorts following treatment.
Based on the responses seen in this first stage of the study, cohort 2 has advanced to stage 2 and has completed enrollment. Additional patients have not been enrolled in cohort 1, but that is still under consideration, she said.
Dr. Gandhi reported having no disclosures.
NATIONAL HARBOR, MD. – The oral, class I selective histone deacetylase (HDAC) inhibitor entinostat given in combination with pembrolizumab demonstrated antitumor activity and acceptable safety in patients with non–small cell lung cancer in the phase 1b/2 ENCORE 601 study.
Entinostat, which has been shown in preclinical models to enhance suppressor cells in the tumor microenvironment, was evaluated in ENCORE 601 as a treatment for non–small cell lung cancer (NSCLC), melanoma, and colorectal cancer. Previously reported phase 1 results showed that an oral dose of 5 mg weekly plus 200 mg of pembrolizumab given intravenously every 3 weeks deserved further exploration for these indications, according to Leena Gandhi, MD, who reported phase 2, stage 1 results from the lung cancer arm of the Simon two-stage study at the annual meeting of the Society for Immunotherapy of Cancer.
Treatment at that dose was studied in both anti-PD-L1–naive patients with advanced NSCLC, and in NSCLC patients who progressed on anti-PD-L1 treatment, said Dr. Gandhi of New York University Langone Medical Center.
The primary objective of stage 1 was objective response rate, and criteria for advancement were 4 or more responses out of 17 evaluable anti-PD-L1–naive patients (cohort 1), and at least 3 responses out of 31 patients who progressed on anti-PD-L1 therapy (cohort 2).
Both cohorts met the endpoint, with 4 of 17 evaluable cohort 1 patients (24%) achieving a partial response, and 3 of 31 evaluable cohort 2 patients (10%) achieving a partial response.
In cohort 1, two responses were confirmed and two were unconfirmed. One of the unconfirmed patients had malignant pericardial effusion, but remains on study with continued clinical benefit, Dr. Gandhi said, noting that three patients remain on study in all.
“The other notable thing I’d like to point out here … is that the majority of these were patients who did not have high levels of expression of PD-L1,” she said.
In cohort 2 patients, two responses were confirmed and one was unconfirmed. Three patients remain on study.
“In both of these cohorts there are a couple of patients who’ve had quite durable responses,” she said.
The best response to prior anti-PD-1therapy in the cohort 2 patients who had a response was stable disease (two patients). The response to prior therapy was unknown in one patient, she noted.
“All of them had clear regressions, after that initial PD-1 therapy, with this combination,” she said, noting that two had “essentially negative PD-L1 expression, and none had high levels of expression.”
Treatment was associated with grade 3/4 adverse events deemed drug related in 31% of patients; the most common of these events, occurring in at least 10% of patients in cohort 1, were hypophosphatemia and neutropenia, and in cohort 2 were fatigue, anemia, anorexia, and pneumonitis; 13% of patients discontinued treatment due to an adverse event, Dr. Gandhi said.
Of note, there were reductions in circulating myeloid derived suppressor cells in both cohorts following treatment.
Based on the responses seen in this first stage of the study, cohort 2 has advanced to stage 2 and has completed enrollment. Additional patients have not been enrolled in cohort 1, but that is still under consideration, she said.
Dr. Gandhi reported having no disclosures.
NATIONAL HARBOR, MD. – The oral, class I selective histone deacetylase (HDAC) inhibitor entinostat given in combination with pembrolizumab demonstrated antitumor activity and acceptable safety in patients with non–small cell lung cancer in the phase 1b/2 ENCORE 601 study.
Entinostat, which has been shown in preclinical models to enhance suppressor cells in the tumor microenvironment, was evaluated in ENCORE 601 as a treatment for non–small cell lung cancer (NSCLC), melanoma, and colorectal cancer. Previously reported phase 1 results showed that an oral dose of 5 mg weekly plus 200 mg of pembrolizumab given intravenously every 3 weeks deserved further exploration for these indications, according to Leena Gandhi, MD, who reported phase 2, stage 1 results from the lung cancer arm of the Simon two-stage study at the annual meeting of the Society for Immunotherapy of Cancer.
Treatment at that dose was studied in both anti-PD-L1–naive patients with advanced NSCLC, and in NSCLC patients who progressed on anti-PD-L1 treatment, said Dr. Gandhi of New York University Langone Medical Center.
The primary objective of stage 1 was objective response rate, and criteria for advancement were 4 or more responses out of 17 evaluable anti-PD-L1–naive patients (cohort 1), and at least 3 responses out of 31 patients who progressed on anti-PD-L1 therapy (cohort 2).
Both cohorts met the endpoint, with 4 of 17 evaluable cohort 1 patients (24%) achieving a partial response, and 3 of 31 evaluable cohort 2 patients (10%) achieving a partial response.
In cohort 1, two responses were confirmed and two were unconfirmed. One of the unconfirmed patients had malignant pericardial effusion, but remains on study with continued clinical benefit, Dr. Gandhi said, noting that three patients remain on study in all.
“The other notable thing I’d like to point out here … is that the majority of these were patients who did not have high levels of expression of PD-L1,” she said.
In cohort 2 patients, two responses were confirmed and one was unconfirmed. Three patients remain on study.
“In both of these cohorts there are a couple of patients who’ve had quite durable responses,” she said.
The best response to prior anti-PD-1therapy in the cohort 2 patients who had a response was stable disease (two patients). The response to prior therapy was unknown in one patient, she noted.
“All of them had clear regressions, after that initial PD-1 therapy, with this combination,” she said, noting that two had “essentially negative PD-L1 expression, and none had high levels of expression.”
Treatment was associated with grade 3/4 adverse events deemed drug related in 31% of patients; the most common of these events, occurring in at least 10% of patients in cohort 1, were hypophosphatemia and neutropenia, and in cohort 2 were fatigue, anemia, anorexia, and pneumonitis; 13% of patients discontinued treatment due to an adverse event, Dr. Gandhi said.
Of note, there were reductions in circulating myeloid derived suppressor cells in both cohorts following treatment.
Based on the responses seen in this first stage of the study, cohort 2 has advanced to stage 2 and has completed enrollment. Additional patients have not been enrolled in cohort 1, but that is still under consideration, she said.
Dr. Gandhi reported having no disclosures.
AT SITC 2017
Key clinical point:
Major finding: Partial responses were seen in 24% of cohort 1 patients and 10% of cohort 2 patients.
Data source: Stage 1 of a phase 2 Simon two-stage study (48 evaluable patients).
Disclosures: Dr. Gandhi reported having no disclosures.
Acute Bronchitis
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

FDA approves epinephrine autoinjector for infants, small children
The Food and Drug Administration approved an epinephrine autoinjector constructed specifically to treat life-threatening allergic reactions in infants and small children weighing 16.5-33 pounds.
The Auvi-Q 0.1 mg autoinjector by kaléo was approved after a priority review by the FDA, with features such as “a voice prompt system that guides a user with step-by-step instructions through the delivery process,” according to a written statement from the company. This auto-injector has a shorter needle length and lower dose of epinephrine than other FDA-approved 0.15-mg and 0.3-mg epinephrine autoinjectors.

In a previous study of 51 infants with a mean weight of 24 pounds who were treated with a 0.15-mg epinephrine auto-injector with a standard 12.7-mm needle length, 43% were at risk of having the needle strike the bone. Unintentional injection of epinephrine into the intraosseous space can cause systemic absorption of the epinephrine and possible cardiac complications (Ann Allergy Asthma Immunol. 2017 Jun;118[6]:719-25.e1).
This new autoinjector with a shorter needle length was designed to obviate this problem, according to kaléo’s statement.
The Auvi-Q 0.1 mg autoinjector should be available to patients in the first half of 2018, the company said.
The Food and Drug Administration approved an epinephrine autoinjector constructed specifically to treat life-threatening allergic reactions in infants and small children weighing 16.5-33 pounds.
The Auvi-Q 0.1 mg autoinjector by kaléo was approved after a priority review by the FDA, with features such as “a voice prompt system that guides a user with step-by-step instructions through the delivery process,” according to a written statement from the company. This auto-injector has a shorter needle length and lower dose of epinephrine than other FDA-approved 0.15-mg and 0.3-mg epinephrine autoinjectors.
In a previous study of 51 infants with a mean weight of 24 pounds who were treated with a 0.15-mg epinephrine auto-injector with a standard 12.7-mm needle length, 43% were at risk of having the needle strike the bone. Unintentional injection of epinephrine into the intraosseous space can cause systemic absorption of the epinephrine and possible cardiac complications (Ann Allergy Asthma Immunol. 2017 Jun;118[6]:719-25.e1).
This new autoinjector with a shorter needle length was designed to obviate this problem, according to kaléo’s statement.
The Auvi-Q 0.1 mg autoinjector should be available to patients in the first half of 2018, the company said.
The Food and Drug Administration approved an epinephrine autoinjector constructed specifically to treat life-threatening allergic reactions in infants and small children weighing 16.5-33 pounds.
The Auvi-Q 0.1 mg autoinjector by kaléo was approved after a priority review by the FDA, with features such as “a voice prompt system that guides a user with step-by-step instructions through the delivery process,” according to a written statement from the company. This auto-injector has a shorter needle length and lower dose of epinephrine than other FDA-approved 0.15-mg and 0.3-mg epinephrine autoinjectors.
In a previous study of 51 infants with a mean weight of 24 pounds who were treated with a 0.15-mg epinephrine auto-injector with a standard 12.7-mm needle length, 43% were at risk of having the needle strike the bone. Unintentional injection of epinephrine into the intraosseous space can cause systemic absorption of the epinephrine and possible cardiac complications (Ann Allergy Asthma Immunol. 2017 Jun;118[6]:719-25.e1).
This new autoinjector with a shorter needle length was designed to obviate this problem, according to kaléo’s statement.
The Auvi-Q 0.1 mg autoinjector should be available to patients in the first half of 2018, the company said.
Enteroendocrine System and Cardio-Pulmonary Health
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Chinese school-based flu vaccination program reduced outbreaks
, said Yang Pan, PhD, of the Institute for Infectious Disease and Endemic Disease Control, Beijing Center for Disease Prevention and Control, and associates.
School-based trivalent inactivated influenza vaccination programs generally occurred Oct. 15-Nov. 30 each year since 2007, with greater than 50% vaccination coverage. In an 11-year retrospective study of school outbreaks of influenza in elementary, middle, and high schools in the Beijing area during Sept. 1, 2006-March 31, 2017, there were 286 febrile outbreaks in schools, involving 6,863 children.
During the 11 years, a mismatch between circulating strains and vaccine strains was identified in two influenza seasons, such as “the A(H3N2) 3C.1 (vaccine strain)-A(H3N2) 3C.3a (circulating strains) mismatch in 2014-2015, the B(Yamagata) Clade 2 (vaccine strain)-B(Yamagata) Clade 3 (circulating strain) mismatch in the 2014-2015 influenza season, and B(Yamagata) (vaccine strain)-B(Victoria) (circulating strains) mismatch in 2015-2016,” they reported.
A combination of high flu vaccine coverage because of school-based vaccinations and a good vaccine match reduced influenza outbreaks in schools by 89% (odds ratio, 0.111), Dr. Pan and associates concluded.
“The school-based influenza vaccination program has been in operation for nearly 10 years in the Beijing area, is unique in China, and is one of the few school-based influenza programs in the world,” the researchers explained. “These data can inform and improve vaccination policy locally and nationally.”
Read more in Vaccine (2017 Nov 8. doi: 10.1016/j.vaccine.2017.10.096).
, said Yang Pan, PhD, of the Institute for Infectious Disease and Endemic Disease Control, Beijing Center for Disease Prevention and Control, and associates.
School-based trivalent inactivated influenza vaccination programs generally occurred Oct. 15-Nov. 30 each year since 2007, with greater than 50% vaccination coverage. In an 11-year retrospective study of school outbreaks of influenza in elementary, middle, and high schools in the Beijing area during Sept. 1, 2006-March 31, 2017, there were 286 febrile outbreaks in schools, involving 6,863 children.
During the 11 years, a mismatch between circulating strains and vaccine strains was identified in two influenza seasons, such as “the A(H3N2) 3C.1 (vaccine strain)-A(H3N2) 3C.3a (circulating strains) mismatch in 2014-2015, the B(Yamagata) Clade 2 (vaccine strain)-B(Yamagata) Clade 3 (circulating strain) mismatch in the 2014-2015 influenza season, and B(Yamagata) (vaccine strain)-B(Victoria) (circulating strains) mismatch in 2015-2016,” they reported.
A combination of high flu vaccine coverage because of school-based vaccinations and a good vaccine match reduced influenza outbreaks in schools by 89% (odds ratio, 0.111), Dr. Pan and associates concluded.
“The school-based influenza vaccination program has been in operation for nearly 10 years in the Beijing area, is unique in China, and is one of the few school-based influenza programs in the world,” the researchers explained. “These data can inform and improve vaccination policy locally and nationally.”
Read more in Vaccine (2017 Nov 8. doi: 10.1016/j.vaccine.2017.10.096).
, said Yang Pan, PhD, of the Institute for Infectious Disease and Endemic Disease Control, Beijing Center for Disease Prevention and Control, and associates.
School-based trivalent inactivated influenza vaccination programs generally occurred Oct. 15-Nov. 30 each year since 2007, with greater than 50% vaccination coverage. In an 11-year retrospective study of school outbreaks of influenza in elementary, middle, and high schools in the Beijing area during Sept. 1, 2006-March 31, 2017, there were 286 febrile outbreaks in schools, involving 6,863 children.
During the 11 years, a mismatch between circulating strains and vaccine strains was identified in two influenza seasons, such as “the A(H3N2) 3C.1 (vaccine strain)-A(H3N2) 3C.3a (circulating strains) mismatch in 2014-2015, the B(Yamagata) Clade 2 (vaccine strain)-B(Yamagata) Clade 3 (circulating strain) mismatch in the 2014-2015 influenza season, and B(Yamagata) (vaccine strain)-B(Victoria) (circulating strains) mismatch in 2015-2016,” they reported.
A combination of high flu vaccine coverage because of school-based vaccinations and a good vaccine match reduced influenza outbreaks in schools by 89% (odds ratio, 0.111), Dr. Pan and associates concluded.
“The school-based influenza vaccination program has been in operation for nearly 10 years in the Beijing area, is unique in China, and is one of the few school-based influenza programs in the world,” the researchers explained. “These data can inform and improve vaccination policy locally and nationally.”
Read more in Vaccine (2017 Nov 8. doi: 10.1016/j.vaccine.2017.10.096).
FROM VACCINE