User login
Teaching the Teacher: Novel Faculty Development for VA Hospitalists
Educating the next generation of health professionals is 1 of 4 congressionally mandated statutory missions of the US Department of Veterans Affairs (VA).1 Even before the COVID-19 pandemic, the number of veterans accessing VA health care was increasing, and those veterans are older and more medically complex than those who seek care outside the VA.2 Almost half of medical residents reported a decline in the quality of their clinical education since the institution of the 2011 duty hours regulations, and in the past decade, more attention has been paid to the need for structured faculty development programs that focus on clinicians’ roles as medical educators.3-6 Hospitalists in particular shoulder a large portion of inpatient medicine education.7 As a result, hospitalists have adapted known frameworks for medical education to their unique clinical setting and developed novel frameworks to meet the needs of their learners.8,9
Access to technology and social media have shaped the educational experience of young learners who are accustomed to quick answers and the rapidity of change.10 The clinical teaching landscape changed again with COVID-19, requiring at least temporary abandonment of traditional in-person teaching methods, which upended well-established educational norms.11,12 In this evolving field, even seasoned preceptors may feel ill-equipped to manage the nuances of modern clinical education and may struggle to recognize which teaching skills are most critical.13,14 Baseline core teaching competencies for medical educators have been previously described and are separate from clinical competencies; however, to our knowledge, no needs assessment has previously been performed specifically for VA hospitalist clinician educators.15
Between May and June of 2020, we distributed an online needs assessment to academic VA hospitalists to identify perceived barriers to effective clinical education and preferred strategies to overcome them. We received 71 responses from 140 hospitalists (50% response rate) on the Veterans Health Administration (VHA) academic hospitalist listserv. Of respondents, 59 (83%) reported teaching health professions trainees every year. VA hospitalists reported educating a diverse group of interprofessional learners, including medical residents and students, physician assistant students, nursing students, pharmacy residents and students, and podiatry students.
Only 14 respondents (20%) were aware of faculty development training available to them through their VA facility, while 53 (75%) were aware of similar resources through academic affiliates or other outside sources. More than 95% of respondents (n = 68) reported interest in receiving VA-specific faculty development to improve skills as clinician educators. The most preferred forms of delivery were in-person or virtual real-time workshops. VA hospitalists reported the least confidence in their ability to support struggling learners, balance supervision and autonomy, and develop individualized learning plans (Table 1). 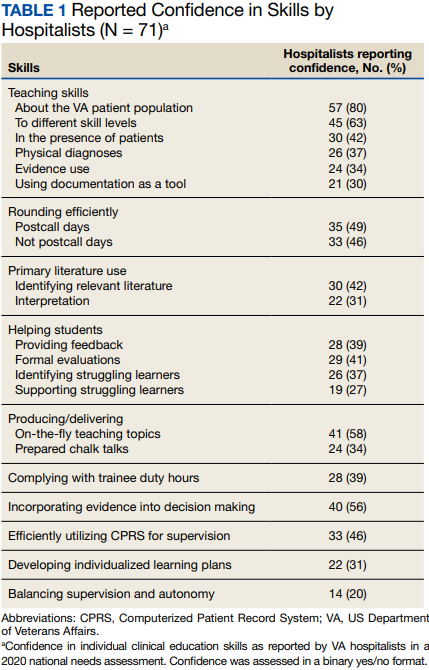
With a better understanding of the needs of academic VA hospitalists, we sought to develop, implement, and measure the impact of a faculty development program that meets the specific needs of inpatient clinicians in the VA. Here we introduce the program, its content, and the experiences of initial participants.
Teaching the Teacher
Teaching the Teacher began at a single VA institution as a series of in-person, discussion-based faculty development workshops. The series met a local need for collaborative professional development in clinical education for hospitalists and specialists who round with health professions learners on the inpatient wards. Both novice and experienced clinicians participated in the series with positive feedback. Based on the results of the national needs assessment, the program has since expanded to other sites with support from the VHA Hospital Medicine Program Office. The project’s overarching goal was to facilitate sharing of best practices across VA sites and create a network of local and national VA educators that participants could continue to access even after course completion.
Teaching the Teacher is structured into 5 facilitated hour-long sessions that can be completed either daily for 1 week or weekly for 1 month at the discretion of each institution. Each session is dedicated to a subject identified on the needs assessment as being highest yield. The hospitalist needs assessment also identified the preference for targeted faculty development that is relevant specifically to VA clinicians. To meet this need, Teaching the Teacher delivers its content through the unique lens of VA medicine. The educational mission of the VA is threaded throughout all presentations, and tips to maximize teaching in the VA’s unique clinical environments are embedded into each hour. Examples include discussions on how to incorporate veteran patients into bedside teaching, handling challenging patient-practitioner interactions as they pertain to patients, and the use of VA resources to find and teach evidence-based medicine.Each session includes a set of learning objectives; within that framework, facilitators allow participants to guide the nuances of content based on their individual and institutional priorities. The pandemic continues to shape much of the course content, as both hospitalists and their trainees grapple with mental health challenges, decreased bedside teaching, and wide variations in baseline trainee competence due to different institutional responses to teaching during a pandemic.12,16 Content is regularly updated to incorporate new literature and feedback from participants and prioritize active participation. Continuing medical education/continuing educational units credit is available through the VA for course completion.
In the first session on modern learners, participants discuss the current generation of health professions trainees, including how personality characteristics and COVID-19 have impacted their learning experiences, and strategies to improve our ability to teach them successfully (Table 2). 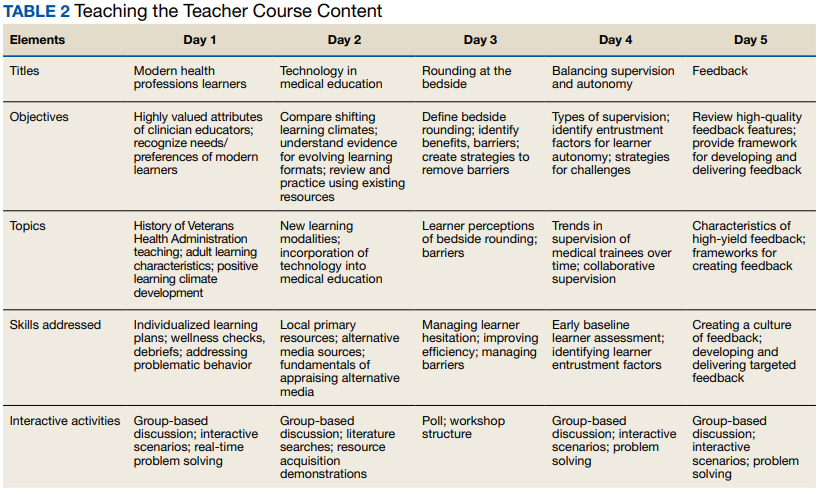
The course was originally designed to be in person, but the COVID-19 pandemic forced a shift to online format. To achieve a high-quality learning environment, the course implemented best practices in virtual synchronous instruction, including setting expectations for participation and screen use at the beginning of the series and optimizing audiovisual technology.17 During each seminar, the use of breakout rooms, polling, and the chat function fostered and sustained engagement.17 After each seminar, participants received a recording of the session, a copy of the materials reviewed, and links to referenced readings.17 The course preserved the interactive aspect of the curriculum through both these previously described techniques and our novel approaches, such as facilitated live interactions with online VA resources.
The pandemic also had an impact on curriculum content, as facilitation of online learning was a new and necessary skill set for instructors and participants. To meet this evolving need, additions in content addressed best practices in synchronous and asynchronous online learning, and augmented discussions on navigating asynchronous learning modalities such as social media. A virtual format allowed for dissemination of this course across the country and for recruitment of new course facilitators from remote sites. The team of instructors included academic hospitalist faculty from 3 VA institutions.
Program Impact
Ten academically affiliated VA hospital medicine sections across 6 states have participated in Teaching the Teacher and several more are scheduled at other sites. Of the 10, 5 completed the course in collaboration with another VA site. Ninety-seven clinicians completed < 1 session synchronously but given the asynchronous option, this number likely underestimates the total audience. Participants included physicians, nurse practitioners, and physician assistants.
Surveys were conducted before and after the program, with 58 participants completing the presurvey, 32 the postsurvey, and 27 completing both. Of the 32 postsurvey respondents, 31 (97%) would recommend the seminar to colleagues. The live, discussion-based format was the most valued aspect of the course structure, with engaging facilitators and course content also ranking highly. Just over half (n = 17) indicated specific behavioral changes they plan to enact after completing the series, such as connecting with and better understanding learners, prioritizing high-quality feedback more deliberately, and bringing medicine to the bedside. The most common critiques of the course were requests for more time for feedback skills.
Discussion
Teaching the Teacher is a VA-specific faculty development seminar for hospitalists. Participants who responded to a survey reported that it met their needs as VA clinician educators. This is the first published needs assessment of academic VA hospitalists in their roles as clinician educators and the first faculty development initiative to address those specific needs using a collaborative, multisite approach. Although this program is a pilot, the positive response it has received has set a precedent for increased development and growth.
Teaching the Teacher presents a novel approach with a condensed curriculum that is more convenient and accessible to VA clinicians than previous programs with similar goals. Hospitalists have busy and variable work schedules, and it can be difficult to find time to participate in a traditional faculty development program. While these programs are becoming more commonplace, they are often longitudinal and require a significant time and/or financial commitment from participants.18 In contrast, Teaching the Teacher is only 5 hours long, can be viewed either synchronously or asynchronously, and is no cost to participants. In the future, other specialties may similarly value an efficient faculty development curriculum, and participation from clinicians outside of hospital medicine could augment the richness of content.
Teaching the Teacher’s curriculum is not meant to be exhaustive, but rather to spark conversation among colleagues. According to survey respondents, the most lauded aspect of this program was the facilitated, discussion-based structure, wherein participants are presented with common challenges and encouraged to share their experiences and solutions with colleagues. Of particular interest to the program’s mission of greater community building are the VA facilities that chose to complete the seminar with another hospitalist section from a different institution. Within this structure lies an opportunity for seasoned educators to informally mentor junior colleagues both within and across institutions, and foster connections among educators that continue beyond the completion of the series. We envision this program growing into an enduring professional development course that begins at onboarding and is revisited at regular intervals thereafter.
Another compelling aspect of this project is the interprofessional design, bringing physicians, nurse practitioners, and physician assistants together. Health education, like clinical care, is shifting to a team approach.19 The curriculum addresses topics previously described as high priority for interprofessional faculty development, such as fostering healthy team leadership, motivating learners, and appraising evidence and online resources.20 A pilot project in VA primary care facilities found that deliberate interprofessional education improved collaboration among health care professionals.21 Prior to Teaching the Teacher, no similar faculty development program provided interprofessional learning and collaboration for VA hospitalists.
Limitations and Future Directions
There are several limitations to this preliminary study. Participation at each site was voluntary and did not always reach the full potential audience of hospitalist clinician educators. As one participant stated, future directions include doing “more to involve teachers who need to learn [these skills]. The ones who attended [from our institution] were already the best teachers.” In addition, despite the asynchronous option, lack of protected time for faculty development may be a limiting factor in participation. Support from institutional and national leadership would likely improve participation.
Measured endpoints to date consist primarily of participant satisfaction and do not yet capture objective changes in teaching. Data collection is ongoing to assess immediate and longitudinal changes in confidence and behaviors of attendees and how this might affect their health professions learners.
Last, our initial needs assessment only targeted academic hospitalists, and the needs of VA hospitalists in rural areas or at facilities without academic affiliation may be different. More research is needed to understand the diverse faculty that comprises both urban and rural VA sites, what their professional development needs are, and how those needs can be met.
Conclusions
Teaching the Teacher is a faculty development pilot, tailored to meet the needs of VA hospitalist clinician educators, that has been voluntarily adopted at multiple VA sites. The facilitated discussion format allows participants to guide the conversation and personalize content, thereby promoting a culture of discussing challenges and best practices among colleagues that we hope endures beyond the bounds of the curriculum. The program focuses on elevating the specific teaching mission of the VA and could be incorporated into onboarding and regular VA-sponsored faculty development updates. While Teaching the Teacher was originally developed for VA hospitalists, most of the content is applicable to clinicians outside hospital medicine. This project serves as a model for training clinical educators and has opportunities to expand across VA as a customizable didactic platform.
Acknowledgments
We thank Brian Schneider, MD, for his tireless support of this program, as well as all the VA clinicians who have shared their time, talents, and wisdom with us since this program’s inception.
1. US Department of Veterans Affairs, Office of Academic Affiliations. Mission of the Office of Academic Affiliations. Updated September 24, 2019. Accessed November 29, 2022. https://www.va.gov/oaa/oaa_mission.asp
2. Eibner C, Krull H, Brown KM, et al. Current and projected characteristics and unique health care needs of the patient population served by the Department of Veterans Affairs. Rand Health Q. 2016;5(4):13. Published 2016 May 9.
3. Drolet BC, Christopher DA, Fischer SA. Residents’ response to duty-hour regulations--a follow-up national survey. N Engl J Med. 2012;366(24):e35. doi:10.1056/NEJMp1202848
4. Hatem CJ, Lown BA, Newman LR. The academic health center coming of age: helping faculty become better teachers and agents of educational change. Acad Med. 2006;81(11):941-944. doi:10.1097/01.ACM.0000242490.56586.64
5. Harvey MM, Berkley HH, O’Malley PG, Durning SJ. Preparing future medical educators: development and pilot evaluation of a student-led medical education elective. Mil Med. 2020;185(1-2):e131-e137. doi:10.1093/milmed/usz175
6. Jason H. Future medical education: Preparing, priorities, possibilities. Med Teach. 2018;40(10):996-1003. doi:10.1080/0142159X.2018.1503412
7. Natarajan P, Ranji SR, Auerbach AD, Hauer KE. Effect of hospitalist attending physicians on trainee educational experiences: a systematic review. J Hosp Med. 2009;4(8):490-498. doi:10.1002/jhm.537
8. Pascoe JM, Nixon J, Lang VJ. Maximizing teaching on the wards: review and application of the One-Minute Preceptor and SNAPPS models. J Hosp Med. 2015;10(2):125-130. doi:10.1002/jhm.2302
9. Martin SK, Farnan JM, Arora VM. Future: new strategies for hospitalists to overcome challenges in teaching on today’s wards. J Hosp Med. 2013;8(7):409-413. doi:10.1002/jhm.2057
10. Waljee JF, Chopra V, Saint S. Mentoring Millennials. JAMA. 2020;323(17):1716-1717. doi:10.1001/jama.2020.3085
11. Papapanou M, Routsi E, Tsamakis K, et al. Medical education challenges and innovations during COVID-19 pandemic. Postgrad Med J. 2022;98(1159):321-327. doi:10.1136/postgradmedj-2021-140032
12. Hilburg R, Patel N, Ambruso S, Biewald MA, Farouk SS. Medical education during the Coronavirus Disease-2019 pandemic: learning from a distance. Adv Chronic Kidney Dis. 2020;27(5):412-417. doi:10.1053/j.ackd.2020.05.017
13. Simpson D, Marcdante K, Souza KH, Anderson A, Holmboe E. Job roles of the 2025 medical educator. J Grad Med Educ. 2018;10(3):243-246. doi:10.4300/JGME-D-18-00253.1
14. Armstrong EG, Mackey M, Spear SJ. Medical education as a process management problem. Acad Med. 2004;79(8):721-728. doi:10.1097/00001888-200408000-00002
15. Srinivasan M, Li ST, Meyers FJ, et al. “Teaching as a Competency”: competencies for medical educators. Acad Med. 2011;86(10):1211-1220. doi:10.1097/ACM.0b013e31822c5b9a
16. Clark E, Freytag J, Hysong SJ, Dang B, Giordano TP, Kulkarni PA. 964. Impact of the COVID-19 pandemic on bedside medical education: a mixed-methods study. Open Forum Infect Dis. 2021;8(Suppl 1):S574. Published 2021 Dec 4. doi:10.1093/ofid/ofab466.1159
17. Ohnigian S, Richards JB, Monette DL, Roberts DH. optimizing remote learning: leveraging zoom to develop and implement successful education sessions. J Med Educ Curric Dev. 2021;8:23821205211020760. Published 2021 Jun 28. doi:10.1177/23821205211020760
18. Burgess A, Matar E, Neuen B, Fox GJ. A longitudinal faculty development program: supporting a culture of teaching. BMC Med Educ. 2019;19(1):400. Published 2019 Nov 1. doi:10.1186/s12909-019-1832-3
19. Stoddard HA, Brownfield ED. Clinician-educators as dual professionals: a contemporary reappraisal. Acad Med. 2016;91(7):921-924. doi:10.1097/ACM.0000000000001210
20. Schönwetter DJ, Hamilton J, Sawatzky JA. Exploring professional development needs of educators in the health sciences professions. J Dent Educ. 2015;79(2):113-123.
21. Meyer EM, Zapatka S, Brienza RS. The development of professional identity and the formation of teams in the Veterans Affairs Connecticut Healthcare System’s Center of Excellence in Primary Care Education Program (CoEPCE). Acad Med. 2015;90(6):802-809. doi:10.1097/ACM.0000000000000594
Educating the next generation of health professionals is 1 of 4 congressionally mandated statutory missions of the US Department of Veterans Affairs (VA).1 Even before the COVID-19 pandemic, the number of veterans accessing VA health care was increasing, and those veterans are older and more medically complex than those who seek care outside the VA.2 Almost half of medical residents reported a decline in the quality of their clinical education since the institution of the 2011 duty hours regulations, and in the past decade, more attention has been paid to the need for structured faculty development programs that focus on clinicians’ roles as medical educators.3-6 Hospitalists in particular shoulder a large portion of inpatient medicine education.7 As a result, hospitalists have adapted known frameworks for medical education to their unique clinical setting and developed novel frameworks to meet the needs of their learners.8,9
Access to technology and social media have shaped the educational experience of young learners who are accustomed to quick answers and the rapidity of change.10 The clinical teaching landscape changed again with COVID-19, requiring at least temporary abandonment of traditional in-person teaching methods, which upended well-established educational norms.11,12 In this evolving field, even seasoned preceptors may feel ill-equipped to manage the nuances of modern clinical education and may struggle to recognize which teaching skills are most critical.13,14 Baseline core teaching competencies for medical educators have been previously described and are separate from clinical competencies; however, to our knowledge, no needs assessment has previously been performed specifically for VA hospitalist clinician educators.15
Between May and June of 2020, we distributed an online needs assessment to academic VA hospitalists to identify perceived barriers to effective clinical education and preferred strategies to overcome them. We received 71 responses from 140 hospitalists (50% response rate) on the Veterans Health Administration (VHA) academic hospitalist listserv. Of respondents, 59 (83%) reported teaching health professions trainees every year. VA hospitalists reported educating a diverse group of interprofessional learners, including medical residents and students, physician assistant students, nursing students, pharmacy residents and students, and podiatry students.
Only 14 respondents (20%) were aware of faculty development training available to them through their VA facility, while 53 (75%) were aware of similar resources through academic affiliates or other outside sources. More than 95% of respondents (n = 68) reported interest in receiving VA-specific faculty development to improve skills as clinician educators. The most preferred forms of delivery were in-person or virtual real-time workshops. VA hospitalists reported the least confidence in their ability to support struggling learners, balance supervision and autonomy, and develop individualized learning plans (Table 1).
With a better understanding of the needs of academic VA hospitalists, we sought to develop, implement, and measure the impact of a faculty development program that meets the specific needs of inpatient clinicians in the VA. Here we introduce the program, its content, and the experiences of initial participants.
Teaching the Teacher
Teaching the Teacher began at a single VA institution as a series of in-person, discussion-based faculty development workshops. The series met a local need for collaborative professional development in clinical education for hospitalists and specialists who round with health professions learners on the inpatient wards. Both novice and experienced clinicians participated in the series with positive feedback. Based on the results of the national needs assessment, the program has since expanded to other sites with support from the VHA Hospital Medicine Program Office. The project’s overarching goal was to facilitate sharing of best practices across VA sites and create a network of local and national VA educators that participants could continue to access even after course completion.
Teaching the Teacher is structured into 5 facilitated hour-long sessions that can be completed either daily for 1 week or weekly for 1 month at the discretion of each institution. Each session is dedicated to a subject identified on the needs assessment as being highest yield. The hospitalist needs assessment also identified the preference for targeted faculty development that is relevant specifically to VA clinicians. To meet this need, Teaching the Teacher delivers its content through the unique lens of VA medicine. The educational mission of the VA is threaded throughout all presentations, and tips to maximize teaching in the VA’s unique clinical environments are embedded into each hour. Examples include discussions on how to incorporate veteran patients into bedside teaching, handling challenging patient-practitioner interactions as they pertain to patients, and the use of VA resources to find and teach evidence-based medicine.Each session includes a set of learning objectives; within that framework, facilitators allow participants to guide the nuances of content based on their individual and institutional priorities. The pandemic continues to shape much of the course content, as both hospitalists and their trainees grapple with mental health challenges, decreased bedside teaching, and wide variations in baseline trainee competence due to different institutional responses to teaching during a pandemic.12,16 Content is regularly updated to incorporate new literature and feedback from participants and prioritize active participation. Continuing medical education/continuing educational units credit is available through the VA for course completion.
In the first session on modern learners, participants discuss the current generation of health professions trainees, including how personality characteristics and COVID-19 have impacted their learning experiences, and strategies to improve our ability to teach them successfully (Table 2).
The course was originally designed to be in person, but the COVID-19 pandemic forced a shift to online format. To achieve a high-quality learning environment, the course implemented best practices in virtual synchronous instruction, including setting expectations for participation and screen use at the beginning of the series and optimizing audiovisual technology.17 During each seminar, the use of breakout rooms, polling, and the chat function fostered and sustained engagement.17 After each seminar, participants received a recording of the session, a copy of the materials reviewed, and links to referenced readings.17 The course preserved the interactive aspect of the curriculum through both these previously described techniques and our novel approaches, such as facilitated live interactions with online VA resources.
The pandemic also had an impact on curriculum content, as facilitation of online learning was a new and necessary skill set for instructors and participants. To meet this evolving need, additions in content addressed best practices in synchronous and asynchronous online learning, and augmented discussions on navigating asynchronous learning modalities such as social media. A virtual format allowed for dissemination of this course across the country and for recruitment of new course facilitators from remote sites. The team of instructors included academic hospitalist faculty from 3 VA institutions.
Program Impact
Ten academically affiliated VA hospital medicine sections across 6 states have participated in Teaching the Teacher and several more are scheduled at other sites. Of the 10, 5 completed the course in collaboration with another VA site. Ninety-seven clinicians completed < 1 session synchronously but given the asynchronous option, this number likely underestimates the total audience. Participants included physicians, nurse practitioners, and physician assistants.
Surveys were conducted before and after the program, with 58 participants completing the presurvey, 32 the postsurvey, and 27 completing both. Of the 32 postsurvey respondents, 31 (97%) would recommend the seminar to colleagues. The live, discussion-based format was the most valued aspect of the course structure, with engaging facilitators and course content also ranking highly. Just over half (n = 17) indicated specific behavioral changes they plan to enact after completing the series, such as connecting with and better understanding learners, prioritizing high-quality feedback more deliberately, and bringing medicine to the bedside. The most common critiques of the course were requests for more time for feedback skills.
Discussion
Teaching the Teacher is a VA-specific faculty development seminar for hospitalists. Participants who responded to a survey reported that it met their needs as VA clinician educators. This is the first published needs assessment of academic VA hospitalists in their roles as clinician educators and the first faculty development initiative to address those specific needs using a collaborative, multisite approach. Although this program is a pilot, the positive response it has received has set a precedent for increased development and growth.
Teaching the Teacher presents a novel approach with a condensed curriculum that is more convenient and accessible to VA clinicians than previous programs with similar goals. Hospitalists have busy and variable work schedules, and it can be difficult to find time to participate in a traditional faculty development program. While these programs are becoming more commonplace, they are often longitudinal and require a significant time and/or financial commitment from participants.18 In contrast, Teaching the Teacher is only 5 hours long, can be viewed either synchronously or asynchronously, and is no cost to participants. In the future, other specialties may similarly value an efficient faculty development curriculum, and participation from clinicians outside of hospital medicine could augment the richness of content.
Teaching the Teacher’s curriculum is not meant to be exhaustive, but rather to spark conversation among colleagues. According to survey respondents, the most lauded aspect of this program was the facilitated, discussion-based structure, wherein participants are presented with common challenges and encouraged to share their experiences and solutions with colleagues. Of particular interest to the program’s mission of greater community building are the VA facilities that chose to complete the seminar with another hospitalist section from a different institution. Within this structure lies an opportunity for seasoned educators to informally mentor junior colleagues both within and across institutions, and foster connections among educators that continue beyond the completion of the series. We envision this program growing into an enduring professional development course that begins at onboarding and is revisited at regular intervals thereafter.
Another compelling aspect of this project is the interprofessional design, bringing physicians, nurse practitioners, and physician assistants together. Health education, like clinical care, is shifting to a team approach.19 The curriculum addresses topics previously described as high priority for interprofessional faculty development, such as fostering healthy team leadership, motivating learners, and appraising evidence and online resources.20 A pilot project in VA primary care facilities found that deliberate interprofessional education improved collaboration among health care professionals.21 Prior to Teaching the Teacher, no similar faculty development program provided interprofessional learning and collaboration for VA hospitalists.
Limitations and Future Directions
There are several limitations to this preliminary study. Participation at each site was voluntary and did not always reach the full potential audience of hospitalist clinician educators. As one participant stated, future directions include doing “more to involve teachers who need to learn [these skills]. The ones who attended [from our institution] were already the best teachers.” In addition, despite the asynchronous option, lack of protected time for faculty development may be a limiting factor in participation. Support from institutional and national leadership would likely improve participation.
Measured endpoints to date consist primarily of participant satisfaction and do not yet capture objective changes in teaching. Data collection is ongoing to assess immediate and longitudinal changes in confidence and behaviors of attendees and how this might affect their health professions learners.
Last, our initial needs assessment only targeted academic hospitalists, and the needs of VA hospitalists in rural areas or at facilities without academic affiliation may be different. More research is needed to understand the diverse faculty that comprises both urban and rural VA sites, what their professional development needs are, and how those needs can be met.
Conclusions
Teaching the Teacher is a faculty development pilot, tailored to meet the needs of VA hospitalist clinician educators, that has been voluntarily adopted at multiple VA sites. The facilitated discussion format allows participants to guide the conversation and personalize content, thereby promoting a culture of discussing challenges and best practices among colleagues that we hope endures beyond the bounds of the curriculum. The program focuses on elevating the specific teaching mission of the VA and could be incorporated into onboarding and regular VA-sponsored faculty development updates. While Teaching the Teacher was originally developed for VA hospitalists, most of the content is applicable to clinicians outside hospital medicine. This project serves as a model for training clinical educators and has opportunities to expand across VA as a customizable didactic platform.
Acknowledgments
We thank Brian Schneider, MD, for his tireless support of this program, as well as all the VA clinicians who have shared their time, talents, and wisdom with us since this program’s inception.
Educating the next generation of health professionals is 1 of 4 congressionally mandated statutory missions of the US Department of Veterans Affairs (VA).1 Even before the COVID-19 pandemic, the number of veterans accessing VA health care was increasing, and those veterans are older and more medically complex than those who seek care outside the VA.2 Almost half of medical residents reported a decline in the quality of their clinical education since the institution of the 2011 duty hours regulations, and in the past decade, more attention has been paid to the need for structured faculty development programs that focus on clinicians’ roles as medical educators.3-6 Hospitalists in particular shoulder a large portion of inpatient medicine education.7 As a result, hospitalists have adapted known frameworks for medical education to their unique clinical setting and developed novel frameworks to meet the needs of their learners.8,9
Access to technology and social media have shaped the educational experience of young learners who are accustomed to quick answers and the rapidity of change.10 The clinical teaching landscape changed again with COVID-19, requiring at least temporary abandonment of traditional in-person teaching methods, which upended well-established educational norms.11,12 In this evolving field, even seasoned preceptors may feel ill-equipped to manage the nuances of modern clinical education and may struggle to recognize which teaching skills are most critical.13,14 Baseline core teaching competencies for medical educators have been previously described and are separate from clinical competencies; however, to our knowledge, no needs assessment has previously been performed specifically for VA hospitalist clinician educators.15
Between May and June of 2020, we distributed an online needs assessment to academic VA hospitalists to identify perceived barriers to effective clinical education and preferred strategies to overcome them. We received 71 responses from 140 hospitalists (50% response rate) on the Veterans Health Administration (VHA) academic hospitalist listserv. Of respondents, 59 (83%) reported teaching health professions trainees every year. VA hospitalists reported educating a diverse group of interprofessional learners, including medical residents and students, physician assistant students, nursing students, pharmacy residents and students, and podiatry students.
Only 14 respondents (20%) were aware of faculty development training available to them through their VA facility, while 53 (75%) were aware of similar resources through academic affiliates or other outside sources. More than 95% of respondents (n = 68) reported interest in receiving VA-specific faculty development to improve skills as clinician educators. The most preferred forms of delivery were in-person or virtual real-time workshops. VA hospitalists reported the least confidence in their ability to support struggling learners, balance supervision and autonomy, and develop individualized learning plans (Table 1).
With a better understanding of the needs of academic VA hospitalists, we sought to develop, implement, and measure the impact of a faculty development program that meets the specific needs of inpatient clinicians in the VA. Here we introduce the program, its content, and the experiences of initial participants.
Teaching the Teacher
Teaching the Teacher began at a single VA institution as a series of in-person, discussion-based faculty development workshops. The series met a local need for collaborative professional development in clinical education for hospitalists and specialists who round with health professions learners on the inpatient wards. Both novice and experienced clinicians participated in the series with positive feedback. Based on the results of the national needs assessment, the program has since expanded to other sites with support from the VHA Hospital Medicine Program Office. The project’s overarching goal was to facilitate sharing of best practices across VA sites and create a network of local and national VA educators that participants could continue to access even after course completion.
Teaching the Teacher is structured into 5 facilitated hour-long sessions that can be completed either daily for 1 week or weekly for 1 month at the discretion of each institution. Each session is dedicated to a subject identified on the needs assessment as being highest yield. The hospitalist needs assessment also identified the preference for targeted faculty development that is relevant specifically to VA clinicians. To meet this need, Teaching the Teacher delivers its content through the unique lens of VA medicine. The educational mission of the VA is threaded throughout all presentations, and tips to maximize teaching in the VA’s unique clinical environments are embedded into each hour. Examples include discussions on how to incorporate veteran patients into bedside teaching, handling challenging patient-practitioner interactions as they pertain to patients, and the use of VA resources to find and teach evidence-based medicine.Each session includes a set of learning objectives; within that framework, facilitators allow participants to guide the nuances of content based on their individual and institutional priorities. The pandemic continues to shape much of the course content, as both hospitalists and their trainees grapple with mental health challenges, decreased bedside teaching, and wide variations in baseline trainee competence due to different institutional responses to teaching during a pandemic.12,16 Content is regularly updated to incorporate new literature and feedback from participants and prioritize active participation. Continuing medical education/continuing educational units credit is available through the VA for course completion.
In the first session on modern learners, participants discuss the current generation of health professions trainees, including how personality characteristics and COVID-19 have impacted their learning experiences, and strategies to improve our ability to teach them successfully (Table 2).
The course was originally designed to be in person, but the COVID-19 pandemic forced a shift to online format. To achieve a high-quality learning environment, the course implemented best practices in virtual synchronous instruction, including setting expectations for participation and screen use at the beginning of the series and optimizing audiovisual technology.17 During each seminar, the use of breakout rooms, polling, and the chat function fostered and sustained engagement.17 After each seminar, participants received a recording of the session, a copy of the materials reviewed, and links to referenced readings.17 The course preserved the interactive aspect of the curriculum through both these previously described techniques and our novel approaches, such as facilitated live interactions with online VA resources.
The pandemic also had an impact on curriculum content, as facilitation of online learning was a new and necessary skill set for instructors and participants. To meet this evolving need, additions in content addressed best practices in synchronous and asynchronous online learning, and augmented discussions on navigating asynchronous learning modalities such as social media. A virtual format allowed for dissemination of this course across the country and for recruitment of new course facilitators from remote sites. The team of instructors included academic hospitalist faculty from 3 VA institutions.
Program Impact
Ten academically affiliated VA hospital medicine sections across 6 states have participated in Teaching the Teacher and several more are scheduled at other sites. Of the 10, 5 completed the course in collaboration with another VA site. Ninety-seven clinicians completed < 1 session synchronously but given the asynchronous option, this number likely underestimates the total audience. Participants included physicians, nurse practitioners, and physician assistants.
Surveys were conducted before and after the program, with 58 participants completing the presurvey, 32 the postsurvey, and 27 completing both. Of the 32 postsurvey respondents, 31 (97%) would recommend the seminar to colleagues. The live, discussion-based format was the most valued aspect of the course structure, with engaging facilitators and course content also ranking highly. Just over half (n = 17) indicated specific behavioral changes they plan to enact after completing the series, such as connecting with and better understanding learners, prioritizing high-quality feedback more deliberately, and bringing medicine to the bedside. The most common critiques of the course were requests for more time for feedback skills.
Discussion
Teaching the Teacher is a VA-specific faculty development seminar for hospitalists. Participants who responded to a survey reported that it met their needs as VA clinician educators. This is the first published needs assessment of academic VA hospitalists in their roles as clinician educators and the first faculty development initiative to address those specific needs using a collaborative, multisite approach. Although this program is a pilot, the positive response it has received has set a precedent for increased development and growth.
Teaching the Teacher presents a novel approach with a condensed curriculum that is more convenient and accessible to VA clinicians than previous programs with similar goals. Hospitalists have busy and variable work schedules, and it can be difficult to find time to participate in a traditional faculty development program. While these programs are becoming more commonplace, they are often longitudinal and require a significant time and/or financial commitment from participants.18 In contrast, Teaching the Teacher is only 5 hours long, can be viewed either synchronously or asynchronously, and is no cost to participants. In the future, other specialties may similarly value an efficient faculty development curriculum, and participation from clinicians outside of hospital medicine could augment the richness of content.
Teaching the Teacher’s curriculum is not meant to be exhaustive, but rather to spark conversation among colleagues. According to survey respondents, the most lauded aspect of this program was the facilitated, discussion-based structure, wherein participants are presented with common challenges and encouraged to share their experiences and solutions with colleagues. Of particular interest to the program’s mission of greater community building are the VA facilities that chose to complete the seminar with another hospitalist section from a different institution. Within this structure lies an opportunity for seasoned educators to informally mentor junior colleagues both within and across institutions, and foster connections among educators that continue beyond the completion of the series. We envision this program growing into an enduring professional development course that begins at onboarding and is revisited at regular intervals thereafter.
Another compelling aspect of this project is the interprofessional design, bringing physicians, nurse practitioners, and physician assistants together. Health education, like clinical care, is shifting to a team approach.19 The curriculum addresses topics previously described as high priority for interprofessional faculty development, such as fostering healthy team leadership, motivating learners, and appraising evidence and online resources.20 A pilot project in VA primary care facilities found that deliberate interprofessional education improved collaboration among health care professionals.21 Prior to Teaching the Teacher, no similar faculty development program provided interprofessional learning and collaboration for VA hospitalists.
Limitations and Future Directions
There are several limitations to this preliminary study. Participation at each site was voluntary and did not always reach the full potential audience of hospitalist clinician educators. As one participant stated, future directions include doing “more to involve teachers who need to learn [these skills]. The ones who attended [from our institution] were already the best teachers.” In addition, despite the asynchronous option, lack of protected time for faculty development may be a limiting factor in participation. Support from institutional and national leadership would likely improve participation.
Measured endpoints to date consist primarily of participant satisfaction and do not yet capture objective changes in teaching. Data collection is ongoing to assess immediate and longitudinal changes in confidence and behaviors of attendees and how this might affect their health professions learners.
Last, our initial needs assessment only targeted academic hospitalists, and the needs of VA hospitalists in rural areas or at facilities without academic affiliation may be different. More research is needed to understand the diverse faculty that comprises both urban and rural VA sites, what their professional development needs are, and how those needs can be met.
Conclusions
Teaching the Teacher is a faculty development pilot, tailored to meet the needs of VA hospitalist clinician educators, that has been voluntarily adopted at multiple VA sites. The facilitated discussion format allows participants to guide the conversation and personalize content, thereby promoting a culture of discussing challenges and best practices among colleagues that we hope endures beyond the bounds of the curriculum. The program focuses on elevating the specific teaching mission of the VA and could be incorporated into onboarding and regular VA-sponsored faculty development updates. While Teaching the Teacher was originally developed for VA hospitalists, most of the content is applicable to clinicians outside hospital medicine. This project serves as a model for training clinical educators and has opportunities to expand across VA as a customizable didactic platform.
Acknowledgments
We thank Brian Schneider, MD, for his tireless support of this program, as well as all the VA clinicians who have shared their time, talents, and wisdom with us since this program’s inception.
1. US Department of Veterans Affairs, Office of Academic Affiliations. Mission of the Office of Academic Affiliations. Updated September 24, 2019. Accessed November 29, 2022. https://www.va.gov/oaa/oaa_mission.asp
2. Eibner C, Krull H, Brown KM, et al. Current and projected characteristics and unique health care needs of the patient population served by the Department of Veterans Affairs. Rand Health Q. 2016;5(4):13. Published 2016 May 9.
3. Drolet BC, Christopher DA, Fischer SA. Residents’ response to duty-hour regulations--a follow-up national survey. N Engl J Med. 2012;366(24):e35. doi:10.1056/NEJMp1202848
4. Hatem CJ, Lown BA, Newman LR. The academic health center coming of age: helping faculty become better teachers and agents of educational change. Acad Med. 2006;81(11):941-944. doi:10.1097/01.ACM.0000242490.56586.64
5. Harvey MM, Berkley HH, O’Malley PG, Durning SJ. Preparing future medical educators: development and pilot evaluation of a student-led medical education elective. Mil Med. 2020;185(1-2):e131-e137. doi:10.1093/milmed/usz175
6. Jason H. Future medical education: Preparing, priorities, possibilities. Med Teach. 2018;40(10):996-1003. doi:10.1080/0142159X.2018.1503412
7. Natarajan P, Ranji SR, Auerbach AD, Hauer KE. Effect of hospitalist attending physicians on trainee educational experiences: a systematic review. J Hosp Med. 2009;4(8):490-498. doi:10.1002/jhm.537
8. Pascoe JM, Nixon J, Lang VJ. Maximizing teaching on the wards: review and application of the One-Minute Preceptor and SNAPPS models. J Hosp Med. 2015;10(2):125-130. doi:10.1002/jhm.2302
9. Martin SK, Farnan JM, Arora VM. Future: new strategies for hospitalists to overcome challenges in teaching on today’s wards. J Hosp Med. 2013;8(7):409-413. doi:10.1002/jhm.2057
10. Waljee JF, Chopra V, Saint S. Mentoring Millennials. JAMA. 2020;323(17):1716-1717. doi:10.1001/jama.2020.3085
11. Papapanou M, Routsi E, Tsamakis K, et al. Medical education challenges and innovations during COVID-19 pandemic. Postgrad Med J. 2022;98(1159):321-327. doi:10.1136/postgradmedj-2021-140032
12. Hilburg R, Patel N, Ambruso S, Biewald MA, Farouk SS. Medical education during the Coronavirus Disease-2019 pandemic: learning from a distance. Adv Chronic Kidney Dis. 2020;27(5):412-417. doi:10.1053/j.ackd.2020.05.017
13. Simpson D, Marcdante K, Souza KH, Anderson A, Holmboe E. Job roles of the 2025 medical educator. J Grad Med Educ. 2018;10(3):243-246. doi:10.4300/JGME-D-18-00253.1
14. Armstrong EG, Mackey M, Spear SJ. Medical education as a process management problem. Acad Med. 2004;79(8):721-728. doi:10.1097/00001888-200408000-00002
15. Srinivasan M, Li ST, Meyers FJ, et al. “Teaching as a Competency”: competencies for medical educators. Acad Med. 2011;86(10):1211-1220. doi:10.1097/ACM.0b013e31822c5b9a
16. Clark E, Freytag J, Hysong SJ, Dang B, Giordano TP, Kulkarni PA. 964. Impact of the COVID-19 pandemic on bedside medical education: a mixed-methods study. Open Forum Infect Dis. 2021;8(Suppl 1):S574. Published 2021 Dec 4. doi:10.1093/ofid/ofab466.1159
17. Ohnigian S, Richards JB, Monette DL, Roberts DH. optimizing remote learning: leveraging zoom to develop and implement successful education sessions. J Med Educ Curric Dev. 2021;8:23821205211020760. Published 2021 Jun 28. doi:10.1177/23821205211020760
18. Burgess A, Matar E, Neuen B, Fox GJ. A longitudinal faculty development program: supporting a culture of teaching. BMC Med Educ. 2019;19(1):400. Published 2019 Nov 1. doi:10.1186/s12909-019-1832-3
19. Stoddard HA, Brownfield ED. Clinician-educators as dual professionals: a contemporary reappraisal. Acad Med. 2016;91(7):921-924. doi:10.1097/ACM.0000000000001210
20. Schönwetter DJ, Hamilton J, Sawatzky JA. Exploring professional development needs of educators in the health sciences professions. J Dent Educ. 2015;79(2):113-123.
21. Meyer EM, Zapatka S, Brienza RS. The development of professional identity and the formation of teams in the Veterans Affairs Connecticut Healthcare System’s Center of Excellence in Primary Care Education Program (CoEPCE). Acad Med. 2015;90(6):802-809. doi:10.1097/ACM.0000000000000594
1. US Department of Veterans Affairs, Office of Academic Affiliations. Mission of the Office of Academic Affiliations. Updated September 24, 2019. Accessed November 29, 2022. https://www.va.gov/oaa/oaa_mission.asp
2. Eibner C, Krull H, Brown KM, et al. Current and projected characteristics and unique health care needs of the patient population served by the Department of Veterans Affairs. Rand Health Q. 2016;5(4):13. Published 2016 May 9.
3. Drolet BC, Christopher DA, Fischer SA. Residents’ response to duty-hour regulations--a follow-up national survey. N Engl J Med. 2012;366(24):e35. doi:10.1056/NEJMp1202848
4. Hatem CJ, Lown BA, Newman LR. The academic health center coming of age: helping faculty become better teachers and agents of educational change. Acad Med. 2006;81(11):941-944. doi:10.1097/01.ACM.0000242490.56586.64
5. Harvey MM, Berkley HH, O’Malley PG, Durning SJ. Preparing future medical educators: development and pilot evaluation of a student-led medical education elective. Mil Med. 2020;185(1-2):e131-e137. doi:10.1093/milmed/usz175
6. Jason H. Future medical education: Preparing, priorities, possibilities. Med Teach. 2018;40(10):996-1003. doi:10.1080/0142159X.2018.1503412
7. Natarajan P, Ranji SR, Auerbach AD, Hauer KE. Effect of hospitalist attending physicians on trainee educational experiences: a systematic review. J Hosp Med. 2009;4(8):490-498. doi:10.1002/jhm.537
8. Pascoe JM, Nixon J, Lang VJ. Maximizing teaching on the wards: review and application of the One-Minute Preceptor and SNAPPS models. J Hosp Med. 2015;10(2):125-130. doi:10.1002/jhm.2302
9. Martin SK, Farnan JM, Arora VM. Future: new strategies for hospitalists to overcome challenges in teaching on today’s wards. J Hosp Med. 2013;8(7):409-413. doi:10.1002/jhm.2057
10. Waljee JF, Chopra V, Saint S. Mentoring Millennials. JAMA. 2020;323(17):1716-1717. doi:10.1001/jama.2020.3085
11. Papapanou M, Routsi E, Tsamakis K, et al. Medical education challenges and innovations during COVID-19 pandemic. Postgrad Med J. 2022;98(1159):321-327. doi:10.1136/postgradmedj-2021-140032
12. Hilburg R, Patel N, Ambruso S, Biewald MA, Farouk SS. Medical education during the Coronavirus Disease-2019 pandemic: learning from a distance. Adv Chronic Kidney Dis. 2020;27(5):412-417. doi:10.1053/j.ackd.2020.05.017
13. Simpson D, Marcdante K, Souza KH, Anderson A, Holmboe E. Job roles of the 2025 medical educator. J Grad Med Educ. 2018;10(3):243-246. doi:10.4300/JGME-D-18-00253.1
14. Armstrong EG, Mackey M, Spear SJ. Medical education as a process management problem. Acad Med. 2004;79(8):721-728. doi:10.1097/00001888-200408000-00002
15. Srinivasan M, Li ST, Meyers FJ, et al. “Teaching as a Competency”: competencies for medical educators. Acad Med. 2011;86(10):1211-1220. doi:10.1097/ACM.0b013e31822c5b9a
16. Clark E, Freytag J, Hysong SJ, Dang B, Giordano TP, Kulkarni PA. 964. Impact of the COVID-19 pandemic on bedside medical education: a mixed-methods study. Open Forum Infect Dis. 2021;8(Suppl 1):S574. Published 2021 Dec 4. doi:10.1093/ofid/ofab466.1159
17. Ohnigian S, Richards JB, Monette DL, Roberts DH. optimizing remote learning: leveraging zoom to develop and implement successful education sessions. J Med Educ Curric Dev. 2021;8:23821205211020760. Published 2021 Jun 28. doi:10.1177/23821205211020760
18. Burgess A, Matar E, Neuen B, Fox GJ. A longitudinal faculty development program: supporting a culture of teaching. BMC Med Educ. 2019;19(1):400. Published 2019 Nov 1. doi:10.1186/s12909-019-1832-3
19. Stoddard HA, Brownfield ED. Clinician-educators as dual professionals: a contemporary reappraisal. Acad Med. 2016;91(7):921-924. doi:10.1097/ACM.0000000000001210
20. Schönwetter DJ, Hamilton J, Sawatzky JA. Exploring professional development needs of educators in the health sciences professions. J Dent Educ. 2015;79(2):113-123.
21. Meyer EM, Zapatka S, Brienza RS. The development of professional identity and the formation of teams in the Veterans Affairs Connecticut Healthcare System’s Center of Excellence in Primary Care Education Program (CoEPCE). Acad Med. 2015;90(6):802-809. doi:10.1097/ACM.0000000000000594
Surviving CLL: Higher risk of other cancer DXs
The report, which appeared in January in Blood Cancer Journal, found that patients diagnosed with CLL between 1989 and 2019 were 63% more likely to were diagnosed with SPM than a matched population: standardized incidence ratio = 1.63, 95% confidence interval (CI), 1.59-1.68.
“Our results provide patients and their treating physicians with an overview of the risk of SPM development. This information can be used in treatment decision-making and for planning appropriate surveillance activities and interventions,” study lead author Lina van der Straten, MD, PhD, of the Albert Schweitzer Hospital and Erasmus University Medical Center in the Netherlands, said in an interview.
Ohio State University hematologist David Bond, MD, who’s familiar with the findings, said in an interview that “it’s been well-established that patients with CLL are at increased risk for second primary malignancies. This is thought to be due to impaired immune surveillance and possibly carcinogenic effects of CLL treatments.” It’s not clear, he said, “whether the rate of second cancers differs between chemoimmunotherapy-treated patients and those receiving newer oral kinase inhibitors.”
Previous research into CLL and SPM has been sparse, Dr. van der Straten said, and most studies haven’t looked at SPM over time and taken into account the widespread use of chemoimmunotherapy and agents such as ibrutinib and venetoclax.
It’s important to study this topic, she said, since “cancers diagnosed after the CLL diagnosis can outweigh the improved longevity and contribute to excess morbidity and mortality in long-term CLL survivors.”
With the help of the Netherlands Cancer Registry, researchers tracked 24,815 patients with CLL who were diagnosed over the 20-year period; 4,369 developed SPM. “We demonstrated that the risk of SPM development was higher than in the general population with an excess of 125 malignancies per 10,000 person-years in the CLL cohort,” Dr. van der Straten said. “The risk of SPM development was found to be heightened in solid and hematological cancers. Patients with CLL had an increased risk of developing cancers at the following sites or types: skin, acute myeloid leukemia, soft-tissue sarcomas, thyroid, kidney, unknown primary localization, non-Hodgkin lymphomas, lung and bronchus, and colon and rectum.”
Specifically, the study reports that “elevated risk was observed for solid (SIR = 1.67; 95% CI, 1.65-1.75) and hematological SPMs (SIR = 1.42; 95% CI, 1.24-1.62). The highest risk for SPMs was noted beyond 5 years post diagnosis (SIR = 1.70; 95% CI, 1.62-1.77), for male individuals (SIR = 1.70; 95% CI, 1.64-1.77), and patients aged 18-69 years (SR = 1.92; 95% CI, 1.79-2.05).
“Patients with CLL exposed to treatment have a higher risk of SPM development than patients who will never receive therapy,” Dr. van der Straten said. Research has shown that “treatment with fludarabine, cyclophosphamide, and rituximab has been associated with a 2.38 increased risk for SPM development, particularly acute myeloid leukemia. Indeed, we found an increased risk for hematological malignancies in patients diagnosed between 2003-2009 and 2010-2019, which might be explained by the broader administration of fludarabine-based strategies in these calendar periods.”
Multiple factors could explain the higher risk of SPM in patients with CLL, including “a dysregulated immune system, treatment-related effects, and surveillance bias,” Dr. van der Straten said. “In addition, it is proposed that the immune dysfunctional nature of CLL might enhance the effect of common carcinogens, such as UV exposure and smoking, in increasing the probability of skin and respiratory cancers.”
She added that “the risk and the spectrum of SPMs were comparable for the 2003-2009 and 2010-2019 periods, suggesting that both the introduction of chemoimmunotherapy and, in part, targeted therapies did not dramatically alter the SPM landscape. However, due to the short follow-up period for the small cohort of patients receiving targeted therapies, further research is warranted.”
Dr. Bond said the findings “are largely in line with prior studies and strengthen their conclusions. Immune surveillance appears to be critical to reducing the risk for some but not all malignancies including lung cancer and melanoma, and the treatments given for CLL can cause immune suppression and thus may increase the risk.”
Moving forward, he said, “this research highlights the importance of second cancers to patients with CLL. It also highlights the need for secondary cancer screening for CLL patients, patient education to avoid known cancer risk factors including smoking and excess UV light exposure, and the need as a field to continue to invest in research into characteristics of second cancers and mitigation strategies.”
Study funding was not reported. The authors and Dr. Bond report no disclosures.
The report, which appeared in January in Blood Cancer Journal, found that patients diagnosed with CLL between 1989 and 2019 were 63% more likely to were diagnosed with SPM than a matched population: standardized incidence ratio = 1.63, 95% confidence interval (CI), 1.59-1.68.
“Our results provide patients and their treating physicians with an overview of the risk of SPM development. This information can be used in treatment decision-making and for planning appropriate surveillance activities and interventions,” study lead author Lina van der Straten, MD, PhD, of the Albert Schweitzer Hospital and Erasmus University Medical Center in the Netherlands, said in an interview.
Ohio State University hematologist David Bond, MD, who’s familiar with the findings, said in an interview that “it’s been well-established that patients with CLL are at increased risk for second primary malignancies. This is thought to be due to impaired immune surveillance and possibly carcinogenic effects of CLL treatments.” It’s not clear, he said, “whether the rate of second cancers differs between chemoimmunotherapy-treated patients and those receiving newer oral kinase inhibitors.”
Previous research into CLL and SPM has been sparse, Dr. van der Straten said, and most studies haven’t looked at SPM over time and taken into account the widespread use of chemoimmunotherapy and agents such as ibrutinib and venetoclax.
It’s important to study this topic, she said, since “cancers diagnosed after the CLL diagnosis can outweigh the improved longevity and contribute to excess morbidity and mortality in long-term CLL survivors.”
With the help of the Netherlands Cancer Registry, researchers tracked 24,815 patients with CLL who were diagnosed over the 20-year period; 4,369 developed SPM. “We demonstrated that the risk of SPM development was higher than in the general population with an excess of 125 malignancies per 10,000 person-years in the CLL cohort,” Dr. van der Straten said. “The risk of SPM development was found to be heightened in solid and hematological cancers. Patients with CLL had an increased risk of developing cancers at the following sites or types: skin, acute myeloid leukemia, soft-tissue sarcomas, thyroid, kidney, unknown primary localization, non-Hodgkin lymphomas, lung and bronchus, and colon and rectum.”
Specifically, the study reports that “elevated risk was observed for solid (SIR = 1.67; 95% CI, 1.65-1.75) and hematological SPMs (SIR = 1.42; 95% CI, 1.24-1.62). The highest risk for SPMs was noted beyond 5 years post diagnosis (SIR = 1.70; 95% CI, 1.62-1.77), for male individuals (SIR = 1.70; 95% CI, 1.64-1.77), and patients aged 18-69 years (SR = 1.92; 95% CI, 1.79-2.05).
“Patients with CLL exposed to treatment have a higher risk of SPM development than patients who will never receive therapy,” Dr. van der Straten said. Research has shown that “treatment with fludarabine, cyclophosphamide, and rituximab has been associated with a 2.38 increased risk for SPM development, particularly acute myeloid leukemia. Indeed, we found an increased risk for hematological malignancies in patients diagnosed between 2003-2009 and 2010-2019, which might be explained by the broader administration of fludarabine-based strategies in these calendar periods.”
Multiple factors could explain the higher risk of SPM in patients with CLL, including “a dysregulated immune system, treatment-related effects, and surveillance bias,” Dr. van der Straten said. “In addition, it is proposed that the immune dysfunctional nature of CLL might enhance the effect of common carcinogens, such as UV exposure and smoking, in increasing the probability of skin and respiratory cancers.”
She added that “the risk and the spectrum of SPMs were comparable for the 2003-2009 and 2010-2019 periods, suggesting that both the introduction of chemoimmunotherapy and, in part, targeted therapies did not dramatically alter the SPM landscape. However, due to the short follow-up period for the small cohort of patients receiving targeted therapies, further research is warranted.”
Dr. Bond said the findings “are largely in line with prior studies and strengthen their conclusions. Immune surveillance appears to be critical to reducing the risk for some but not all malignancies including lung cancer and melanoma, and the treatments given for CLL can cause immune suppression and thus may increase the risk.”
Moving forward, he said, “this research highlights the importance of second cancers to patients with CLL. It also highlights the need for secondary cancer screening for CLL patients, patient education to avoid known cancer risk factors including smoking and excess UV light exposure, and the need as a field to continue to invest in research into characteristics of second cancers and mitigation strategies.”
Study funding was not reported. The authors and Dr. Bond report no disclosures.
The report, which appeared in January in Blood Cancer Journal, found that patients diagnosed with CLL between 1989 and 2019 were 63% more likely to were diagnosed with SPM than a matched population: standardized incidence ratio = 1.63, 95% confidence interval (CI), 1.59-1.68.
“Our results provide patients and their treating physicians with an overview of the risk of SPM development. This information can be used in treatment decision-making and for planning appropriate surveillance activities and interventions,” study lead author Lina van der Straten, MD, PhD, of the Albert Schweitzer Hospital and Erasmus University Medical Center in the Netherlands, said in an interview.
Ohio State University hematologist David Bond, MD, who’s familiar with the findings, said in an interview that “it’s been well-established that patients with CLL are at increased risk for second primary malignancies. This is thought to be due to impaired immune surveillance and possibly carcinogenic effects of CLL treatments.” It’s not clear, he said, “whether the rate of second cancers differs between chemoimmunotherapy-treated patients and those receiving newer oral kinase inhibitors.”
Previous research into CLL and SPM has been sparse, Dr. van der Straten said, and most studies haven’t looked at SPM over time and taken into account the widespread use of chemoimmunotherapy and agents such as ibrutinib and venetoclax.
It’s important to study this topic, she said, since “cancers diagnosed after the CLL diagnosis can outweigh the improved longevity and contribute to excess morbidity and mortality in long-term CLL survivors.”
With the help of the Netherlands Cancer Registry, researchers tracked 24,815 patients with CLL who were diagnosed over the 20-year period; 4,369 developed SPM. “We demonstrated that the risk of SPM development was higher than in the general population with an excess of 125 malignancies per 10,000 person-years in the CLL cohort,” Dr. van der Straten said. “The risk of SPM development was found to be heightened in solid and hematological cancers. Patients with CLL had an increased risk of developing cancers at the following sites or types: skin, acute myeloid leukemia, soft-tissue sarcomas, thyroid, kidney, unknown primary localization, non-Hodgkin lymphomas, lung and bronchus, and colon and rectum.”
Specifically, the study reports that “elevated risk was observed for solid (SIR = 1.67; 95% CI, 1.65-1.75) and hematological SPMs (SIR = 1.42; 95% CI, 1.24-1.62). The highest risk for SPMs was noted beyond 5 years post diagnosis (SIR = 1.70; 95% CI, 1.62-1.77), for male individuals (SIR = 1.70; 95% CI, 1.64-1.77), and patients aged 18-69 years (SR = 1.92; 95% CI, 1.79-2.05).
“Patients with CLL exposed to treatment have a higher risk of SPM development than patients who will never receive therapy,” Dr. van der Straten said. Research has shown that “treatment with fludarabine, cyclophosphamide, and rituximab has been associated with a 2.38 increased risk for SPM development, particularly acute myeloid leukemia. Indeed, we found an increased risk for hematological malignancies in patients diagnosed between 2003-2009 and 2010-2019, which might be explained by the broader administration of fludarabine-based strategies in these calendar periods.”
Multiple factors could explain the higher risk of SPM in patients with CLL, including “a dysregulated immune system, treatment-related effects, and surveillance bias,” Dr. van der Straten said. “In addition, it is proposed that the immune dysfunctional nature of CLL might enhance the effect of common carcinogens, such as UV exposure and smoking, in increasing the probability of skin and respiratory cancers.”
She added that “the risk and the spectrum of SPMs were comparable for the 2003-2009 and 2010-2019 periods, suggesting that both the introduction of chemoimmunotherapy and, in part, targeted therapies did not dramatically alter the SPM landscape. However, due to the short follow-up period for the small cohort of patients receiving targeted therapies, further research is warranted.”
Dr. Bond said the findings “are largely in line with prior studies and strengthen their conclusions. Immune surveillance appears to be critical to reducing the risk for some but not all malignancies including lung cancer and melanoma, and the treatments given for CLL can cause immune suppression and thus may increase the risk.”
Moving forward, he said, “this research highlights the importance of second cancers to patients with CLL. It also highlights the need for secondary cancer screening for CLL patients, patient education to avoid known cancer risk factors including smoking and excess UV light exposure, and the need as a field to continue to invest in research into characteristics of second cancers and mitigation strategies.”
Study funding was not reported. The authors and Dr. Bond report no disclosures.
FROM BLOOD CANCER JOURNAL
Strategy to reduce peritoneal metastases in gastric cancer
The study covered in this summary was published on researchsquare.com as a preprint and has not yet been peer reviewed.
Key takeaway
Why this matters
- Surgery and postoperative chemotherapy are standard of care for advanced gastric cancer, but up to half of patients develop peritoneal metastases with poor prognosis.
- There is no consensus on how to prevent peritoneal metastases.
- With hyperthermic intraperitoneal chemotherapy, the abdominal cavity is bathed in chemotherapy that has been heated, directly killing free cancer cells and micrometastases.
- The findings suggest that adding hyperthermic intraperitoneal chemotherapy to standard treatment greatly reduces the risk of peritoneal metastases.
Study design
- The investigators randomly assigned 134 patients with advanced gastric cancer evenly to receive either systemic chemotherapy alone or systemic chemotherapy plus hyperthermic intraperitoneal chemotherapy after radical gastrectomy.
- The hyperthermic intraperitoneal chemotherapy group had 3 L of heated saline containing 40 mg/m2 of cisplatin circulated in their peritoneal cavities for an hour. The procedure was performed twice within 72 hours of surgery.
- Systemic chemotherapy consisted of six to eight cycles of S-1 combined with oxaliplatin (SOX regimen) starting 4-6 weeks after surgery.
- Most patients (90%) had stage III disease, and the rest stage II.
- Median follow-up was 44 months.
Key results
- Overall, the 3-year DFS rate was 73.8% with hyperthermic intraperitoneal chemotherapy versus 61.2% without it (P = .031).
- In addition, 21% of patients in the hyperthermic intraperitoneal chemotherapy group developed peritoneal metastases versus 40.3% with standard care (P = .015)
- The 3-year overall survival was 73.9% in the hyperthermic intraperitoneal chemotherapy group versus 77.6% in the standard care arm, but the difference was not significant (P = .737).
- There were no serious adverse events related to hyperthermic intraperitoneal chemotherapy, and postoperative complications were similar between the groups.
- Grade 3 or 4 adverse events occurred in 14.2% of patients; there were no statistically significant between-group differences.
- Metastases to other sites, such as the liver and distant lymph nodes, were also similar between the two arms.
Limitations
- Follow-up might have been too short to detect a difference in overall survival.
- The trial was conducted at a single-center and was relatively small.
Disclosures
- The study received no external funding, and the investigators did not report any financial relationships.
This is a summary of a preprint research study, “Hyperthermic Intraperitoneal Chemotherapy (HIPEC) Plus Systemic Chemotherapy Versus Systemic Chemotherapy Alone in Locally Advanced Gastric Cancer After D2 Radical Resection: A Randomized Controlled Study,” led by Pengfei Yu of the Zhejiang Cancer Hospital, Hangzhou, China. The study has not been peer reviewed. The full text can be found at researchsquare.com.
A version of this article first appeared on Medscape.com.
The study covered in this summary was published on researchsquare.com as a preprint and has not yet been peer reviewed.
Key takeaway
Why this matters
- Surgery and postoperative chemotherapy are standard of care for advanced gastric cancer, but up to half of patients develop peritoneal metastases with poor prognosis.
- There is no consensus on how to prevent peritoneal metastases.
- With hyperthermic intraperitoneal chemotherapy, the abdominal cavity is bathed in chemotherapy that has been heated, directly killing free cancer cells and micrometastases.
- The findings suggest that adding hyperthermic intraperitoneal chemotherapy to standard treatment greatly reduces the risk of peritoneal metastases.
Study design
- The investigators randomly assigned 134 patients with advanced gastric cancer evenly to receive either systemic chemotherapy alone or systemic chemotherapy plus hyperthermic intraperitoneal chemotherapy after radical gastrectomy.
- The hyperthermic intraperitoneal chemotherapy group had 3 L of heated saline containing 40 mg/m2 of cisplatin circulated in their peritoneal cavities for an hour. The procedure was performed twice within 72 hours of surgery.
- Systemic chemotherapy consisted of six to eight cycles of S-1 combined with oxaliplatin (SOX regimen) starting 4-6 weeks after surgery.
- Most patients (90%) had stage III disease, and the rest stage II.
- Median follow-up was 44 months.
Key results
- Overall, the 3-year DFS rate was 73.8% with hyperthermic intraperitoneal chemotherapy versus 61.2% without it (P = .031).
- In addition, 21% of patients in the hyperthermic intraperitoneal chemotherapy group developed peritoneal metastases versus 40.3% with standard care (P = .015)
- The 3-year overall survival was 73.9% in the hyperthermic intraperitoneal chemotherapy group versus 77.6% in the standard care arm, but the difference was not significant (P = .737).
- There were no serious adverse events related to hyperthermic intraperitoneal chemotherapy, and postoperative complications were similar between the groups.
- Grade 3 or 4 adverse events occurred in 14.2% of patients; there were no statistically significant between-group differences.
- Metastases to other sites, such as the liver and distant lymph nodes, were also similar between the two arms.
Limitations
- Follow-up might have been too short to detect a difference in overall survival.
- The trial was conducted at a single-center and was relatively small.
Disclosures
- The study received no external funding, and the investigators did not report any financial relationships.
This is a summary of a preprint research study, “Hyperthermic Intraperitoneal Chemotherapy (HIPEC) Plus Systemic Chemotherapy Versus Systemic Chemotherapy Alone in Locally Advanced Gastric Cancer After D2 Radical Resection: A Randomized Controlled Study,” led by Pengfei Yu of the Zhejiang Cancer Hospital, Hangzhou, China. The study has not been peer reviewed. The full text can be found at researchsquare.com.
A version of this article first appeared on Medscape.com.
The study covered in this summary was published on researchsquare.com as a preprint and has not yet been peer reviewed.
Key takeaway
Why this matters
- Surgery and postoperative chemotherapy are standard of care for advanced gastric cancer, but up to half of patients develop peritoneal metastases with poor prognosis.
- There is no consensus on how to prevent peritoneal metastases.
- With hyperthermic intraperitoneal chemotherapy, the abdominal cavity is bathed in chemotherapy that has been heated, directly killing free cancer cells and micrometastases.
- The findings suggest that adding hyperthermic intraperitoneal chemotherapy to standard treatment greatly reduces the risk of peritoneal metastases.
Study design
- The investigators randomly assigned 134 patients with advanced gastric cancer evenly to receive either systemic chemotherapy alone or systemic chemotherapy plus hyperthermic intraperitoneal chemotherapy after radical gastrectomy.
- The hyperthermic intraperitoneal chemotherapy group had 3 L of heated saline containing 40 mg/m2 of cisplatin circulated in their peritoneal cavities for an hour. The procedure was performed twice within 72 hours of surgery.
- Systemic chemotherapy consisted of six to eight cycles of S-1 combined with oxaliplatin (SOX regimen) starting 4-6 weeks after surgery.
- Most patients (90%) had stage III disease, and the rest stage II.
- Median follow-up was 44 months.
Key results
- Overall, the 3-year DFS rate was 73.8% with hyperthermic intraperitoneal chemotherapy versus 61.2% without it (P = .031).
- In addition, 21% of patients in the hyperthermic intraperitoneal chemotherapy group developed peritoneal metastases versus 40.3% with standard care (P = .015)
- The 3-year overall survival was 73.9% in the hyperthermic intraperitoneal chemotherapy group versus 77.6% in the standard care arm, but the difference was not significant (P = .737).
- There were no serious adverse events related to hyperthermic intraperitoneal chemotherapy, and postoperative complications were similar between the groups.
- Grade 3 or 4 adverse events occurred in 14.2% of patients; there were no statistically significant between-group differences.
- Metastases to other sites, such as the liver and distant lymph nodes, were also similar between the two arms.
Limitations
- Follow-up might have been too short to detect a difference in overall survival.
- The trial was conducted at a single-center and was relatively small.
Disclosures
- The study received no external funding, and the investigators did not report any financial relationships.
This is a summary of a preprint research study, “Hyperthermic Intraperitoneal Chemotherapy (HIPEC) Plus Systemic Chemotherapy Versus Systemic Chemotherapy Alone in Locally Advanced Gastric Cancer After D2 Radical Resection: A Randomized Controlled Study,” led by Pengfei Yu of the Zhejiang Cancer Hospital, Hangzhou, China. The study has not been peer reviewed. The full text can be found at researchsquare.com.
A version of this article first appeared on Medscape.com.
Factors linked to higher risk for death in young cancer survivors
according to new data from the St. Jude Lifetime Cohort.
Survivors with a greater number and severity of modifiable chronic health conditions as well as those living in the most versus least resource-deprived areas had a significantly higher risk of all-cause and health-related late death.
Finding ways to mitigate these factors “will be important to improving health outcomes and developing risk-stratification strategies to optimize care delivery to survivors at varying risk of adverse health events,” the researchers wrote.
The study indicates that treating chronic health conditions alone may not be enough to increase a cancer survivor’s lifespan; improving local environments matters too.
“It is important for clinicians to ask patients about their specific situation,” first author Matthew J. Ehrhardt, MD, department of oncology, St. Jude Children’s Research Hospital, Memphis, said in a news release. “It’s easy to prescribe medications or to tell people to exercise. It takes more time and more thoughtfulness to sit and understand environments in which they are residing.”
“As clinicians, we may have limited ability to modify some of those factors. But we can work closely with the rest of the health care team, such as social workers, for example, to help survivors to identify and access local resources,” Dr. Ehrhardt added.
The study was published online in JAMA Network Open.
A growing population of childhood cancer survivors faces an increased risk for premature death in the years following their diagnosis. However, associations between social determinants of health, modifiable health conditions, and late mortality in childhood cancer survivors remain unclear.
To assess late mortality, the study team analyzed data on 9,440 participants (median age at assessment, 27.5 years; range, 5.3-71.9 years) who lived at least 5 years after being diagnosed with a childhood cancer between 1962 and 2012.
During a median follow-up of about 18 years, childhood cancer survivors had an increased rate of both all-cause and health-related late mortality (standardized mortality rate, 7.6 for both). Among specific health-related causes of death, SMRs were 16.0 for subsequent neoplasms, 9.0 for pulmonary causes, 4.2 for cardiac causes, and 4.3 for other health-related causes.
To evaluate ties between modifiable chronic health conditions, social determinants, and late mortality, the researchers restricted their analysis to 3,407 adult study participants for whom relevant data were available. Modifiable chronic health conditions included dyslipidemia, hypertension, diabetes, underweight or obesity, bone mineral deficiency, and hypothyroidism.
After adjusting for individual factors, including age at diagnosis and treatment, as well as neighborhood-level factors, the researchers observed a significantly increased risk for death among survivors with one or more modifiable chronic health conditions of grade 2 or higher (relative risk, 2.2), two chronic health conditions of grade 2 or higher (RR, 2.6) or three chronic health conditions of grade 2 or higher (RR, 3.6).
These findings suggest that “increased late mortality experienced by childhood cancer survivors in adulthood may not be predetermined by treatment-related risk factors alone,” the researchers said.
In addition, survivors living in the most disadvantaged areas, as measured by the area deprivation index (ADI), had a five- to eightfold increased risk of late death from any cause compared with those living in the least disadvantaged areas, even after adjusting for modifiable chronic health conditions, cancer treatment, demographics, and individual socioeconomic factors.
The findings have important public health implications, Dr. Ehrhardt and colleagues said. The results can, for instance, help identify and stratify cancer survivors at higher lifetime risk for specific chronic conditions and late death.
This risk-stratified approach to care, however, is “relatively static” and does not account for risk factors acquired after cancer diagnosis and treatment, such as social determinants of health.
That is why also focusing on socioeconomic factors is important, and transitional care services following cancer treatment should consider that survivors in disadvantaged neighborhoods may lack supportive resources to address health issues, potentially leading to increased risk for death, the researchers said.
The knowledge that living in a resource-poor neighborhood may raise the risk for late death in childhood cancer survivors “strengthens support for public health policies that will direct resources to such regions and facilitate a multipronged approach to risk mitigation,” the authors concluded.
This study was supported by grants from the National Institutes of Health and the American Lebanese Syrian Associated Charities. The authors reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
according to new data from the St. Jude Lifetime Cohort.
Survivors with a greater number and severity of modifiable chronic health conditions as well as those living in the most versus least resource-deprived areas had a significantly higher risk of all-cause and health-related late death.
Finding ways to mitigate these factors “will be important to improving health outcomes and developing risk-stratification strategies to optimize care delivery to survivors at varying risk of adverse health events,” the researchers wrote.
The study indicates that treating chronic health conditions alone may not be enough to increase a cancer survivor’s lifespan; improving local environments matters too.
“It is important for clinicians to ask patients about their specific situation,” first author Matthew J. Ehrhardt, MD, department of oncology, St. Jude Children’s Research Hospital, Memphis, said in a news release. “It’s easy to prescribe medications or to tell people to exercise. It takes more time and more thoughtfulness to sit and understand environments in which they are residing.”
“As clinicians, we may have limited ability to modify some of those factors. But we can work closely with the rest of the health care team, such as social workers, for example, to help survivors to identify and access local resources,” Dr. Ehrhardt added.
The study was published online in JAMA Network Open.
A growing population of childhood cancer survivors faces an increased risk for premature death in the years following their diagnosis. However, associations between social determinants of health, modifiable health conditions, and late mortality in childhood cancer survivors remain unclear.
To assess late mortality, the study team analyzed data on 9,440 participants (median age at assessment, 27.5 years; range, 5.3-71.9 years) who lived at least 5 years after being diagnosed with a childhood cancer between 1962 and 2012.
During a median follow-up of about 18 years, childhood cancer survivors had an increased rate of both all-cause and health-related late mortality (standardized mortality rate, 7.6 for both). Among specific health-related causes of death, SMRs were 16.0 for subsequent neoplasms, 9.0 for pulmonary causes, 4.2 for cardiac causes, and 4.3 for other health-related causes.
To evaluate ties between modifiable chronic health conditions, social determinants, and late mortality, the researchers restricted their analysis to 3,407 adult study participants for whom relevant data were available. Modifiable chronic health conditions included dyslipidemia, hypertension, diabetes, underweight or obesity, bone mineral deficiency, and hypothyroidism.
After adjusting for individual factors, including age at diagnosis and treatment, as well as neighborhood-level factors, the researchers observed a significantly increased risk for death among survivors with one or more modifiable chronic health conditions of grade 2 or higher (relative risk, 2.2), two chronic health conditions of grade 2 or higher (RR, 2.6) or three chronic health conditions of grade 2 or higher (RR, 3.6).
These findings suggest that “increased late mortality experienced by childhood cancer survivors in adulthood may not be predetermined by treatment-related risk factors alone,” the researchers said.
In addition, survivors living in the most disadvantaged areas, as measured by the area deprivation index (ADI), had a five- to eightfold increased risk of late death from any cause compared with those living in the least disadvantaged areas, even after adjusting for modifiable chronic health conditions, cancer treatment, demographics, and individual socioeconomic factors.
The findings have important public health implications, Dr. Ehrhardt and colleagues said. The results can, for instance, help identify and stratify cancer survivors at higher lifetime risk for specific chronic conditions and late death.
This risk-stratified approach to care, however, is “relatively static” and does not account for risk factors acquired after cancer diagnosis and treatment, such as social determinants of health.
That is why also focusing on socioeconomic factors is important, and transitional care services following cancer treatment should consider that survivors in disadvantaged neighborhoods may lack supportive resources to address health issues, potentially leading to increased risk for death, the researchers said.
The knowledge that living in a resource-poor neighborhood may raise the risk for late death in childhood cancer survivors “strengthens support for public health policies that will direct resources to such regions and facilitate a multipronged approach to risk mitigation,” the authors concluded.
This study was supported by grants from the National Institutes of Health and the American Lebanese Syrian Associated Charities. The authors reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
according to new data from the St. Jude Lifetime Cohort.
Survivors with a greater number and severity of modifiable chronic health conditions as well as those living in the most versus least resource-deprived areas had a significantly higher risk of all-cause and health-related late death.
Finding ways to mitigate these factors “will be important to improving health outcomes and developing risk-stratification strategies to optimize care delivery to survivors at varying risk of adverse health events,” the researchers wrote.
The study indicates that treating chronic health conditions alone may not be enough to increase a cancer survivor’s lifespan; improving local environments matters too.
“It is important for clinicians to ask patients about their specific situation,” first author Matthew J. Ehrhardt, MD, department of oncology, St. Jude Children’s Research Hospital, Memphis, said in a news release. “It’s easy to prescribe medications or to tell people to exercise. It takes more time and more thoughtfulness to sit and understand environments in which they are residing.”
“As clinicians, we may have limited ability to modify some of those factors. But we can work closely with the rest of the health care team, such as social workers, for example, to help survivors to identify and access local resources,” Dr. Ehrhardt added.
The study was published online in JAMA Network Open.
A growing population of childhood cancer survivors faces an increased risk for premature death in the years following their diagnosis. However, associations between social determinants of health, modifiable health conditions, and late mortality in childhood cancer survivors remain unclear.
To assess late mortality, the study team analyzed data on 9,440 participants (median age at assessment, 27.5 years; range, 5.3-71.9 years) who lived at least 5 years after being diagnosed with a childhood cancer between 1962 and 2012.
During a median follow-up of about 18 years, childhood cancer survivors had an increased rate of both all-cause and health-related late mortality (standardized mortality rate, 7.6 for both). Among specific health-related causes of death, SMRs were 16.0 for subsequent neoplasms, 9.0 for pulmonary causes, 4.2 for cardiac causes, and 4.3 for other health-related causes.
To evaluate ties between modifiable chronic health conditions, social determinants, and late mortality, the researchers restricted their analysis to 3,407 adult study participants for whom relevant data were available. Modifiable chronic health conditions included dyslipidemia, hypertension, diabetes, underweight or obesity, bone mineral deficiency, and hypothyroidism.
After adjusting for individual factors, including age at diagnosis and treatment, as well as neighborhood-level factors, the researchers observed a significantly increased risk for death among survivors with one or more modifiable chronic health conditions of grade 2 or higher (relative risk, 2.2), two chronic health conditions of grade 2 or higher (RR, 2.6) or three chronic health conditions of grade 2 or higher (RR, 3.6).
These findings suggest that “increased late mortality experienced by childhood cancer survivors in adulthood may not be predetermined by treatment-related risk factors alone,” the researchers said.
In addition, survivors living in the most disadvantaged areas, as measured by the area deprivation index (ADI), had a five- to eightfold increased risk of late death from any cause compared with those living in the least disadvantaged areas, even after adjusting for modifiable chronic health conditions, cancer treatment, demographics, and individual socioeconomic factors.
The findings have important public health implications, Dr. Ehrhardt and colleagues said. The results can, for instance, help identify and stratify cancer survivors at higher lifetime risk for specific chronic conditions and late death.
This risk-stratified approach to care, however, is “relatively static” and does not account for risk factors acquired after cancer diagnosis and treatment, such as social determinants of health.
That is why also focusing on socioeconomic factors is important, and transitional care services following cancer treatment should consider that survivors in disadvantaged neighborhoods may lack supportive resources to address health issues, potentially leading to increased risk for death, the researchers said.
The knowledge that living in a resource-poor neighborhood may raise the risk for late death in childhood cancer survivors “strengthens support for public health policies that will direct resources to such regions and facilitate a multipronged approach to risk mitigation,” the authors concluded.
This study was supported by grants from the National Institutes of Health and the American Lebanese Syrian Associated Charities. The authors reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JAMA NETWORK OPEN
Endovascular therapy benefits large infarction: ANGEL-ASPECT
The trial was stopped early because a planned interim analysis showed efficacy of endovascular therapy in this patient population.
Among patients in China with acute ischemic stroke and a large cerebral infarction, treatment with endovascular therapy within 24 hours after stroke onset “resulted in a better functional outcome at 3 months than medical management alone,” lead author Xiaochuan Huo, MD, PhD, associate chief physician, interventional neurology department, Beijing Tiantan Hospital, Capital Medical University, told this news organization.
“This trial added important evidence for the benefits of endovascular therapy,” Dr. Huo added.
The findings were presented at the International Stroke Conference and were published online in The New England Journal of Medicine. The conference was presented by the American Stroke Association, a division of the American Heart Association.
Will change practice
Commenting on the results, Tudor G. Jovin, MD, professor and chair, department of neurology, Cooper Medical School of Rowan University, Camden, N.J., said he has “little doubt” this study will change practice.
Despite previous studies showing signals of benefit from thrombectomy for patients with large-core infarcts, and some even finding a large treatment effect, “somehow the world didn’t register this,” said Dr. Jovin.
“The stroke community was perhaps reluctant to accept these signals that were there in plain sight because we have been primed for such a long time that reperfusing large infarcts was, if not detrimental, not beneficial.”
But this study, along with another study showing similar results, SELECT 2, which was also presented at this meeting and was published in the same issue of NEJM, provide “overwhelming proof” and “have finally made the community aware,” said Dr. Jovin. “This is sort of a wake-up call to say, ‘Hey, this is real; patients with large infarcts also benefit from thrombectomy.’ “
This new research suggests it’s not necessary to learn the infarct size, at least in the early time window, and doing so just wastes precious time, added Dr. Jovin.
The impact of thrombectomy on patients with “super large infarcts” is still not clear, although these are “extremely rare” in the early time window, perhaps representing only about 1% of patients, said Dr. Jovin.
The increased rate of hemorrhages in study patients receiving thrombectomy “is the price you pay” for the benefits, he said. He noted that this is not any different from the situation with tissue plasminogen activator (tPA), which is routinely used because the benefits far outweigh the risks.
ANGEL-ASPECT
As patients with large infarctions are generally excluded from studies of thrombectomy, it’s been unclear whether they benefit from this therapy, the researchers noted.
The multicenter Endovascular Therapy in Acute Anterior Circulation Large Vessel Occlusive Patients With a Large Infarct Core (ANGEL-ASPECT) trial included 455 adult patients (median age, 68 years; 38.7% women) who had a large infarct core caused by acute large-vessel occlusion in the anterior circulation (Alberta Stroke Program Early CT Score [ASPECTS] 3-5 without core volume limitations or ASPECTS 0–2 with core volume between 70 and 100 mL).
Study participants had to have a score of 6-30 on the National Institutes of Health Stroke Scale (NIHSS) and a retrospectively determined prestroke score of 0 or 1 on the Modified Rankin Scale (mRS).
The median baseline NIHSS score of study patients was 16, the median ASPECTS was 3, and the median infarct-core volume was 62 mL.
Researchers randomly assigned patients to undergo either medical management alone or medical management as well as endovascular therapy. Medical management included intravenous (IV) thrombolysis for those who were eligible.
IV thrombolysis was administered before thrombectomy for about 28% of patients in each group. Some 78.7% of all patients arrived at the hospital outside the typical 4.5-hour window and were ineligible for thrombolysis.
A greater percentage of patients in the endovascular therapy group was receiving antihypertensive medications (83.0%) than in the medical management alone group (54.0%). About 20% of patients in each group were taking an anticoagulant medication.
When the trial was halted, outcome data were available for 336 patients. An additional 120 patients had undergone randomization, and 455 had completed 90 days of follow-up.
Better functional outcome
The primary outcome was the score on the mRS at 90 days. Results showed a shift in the distribution of scores on the mRS at 90 days toward better outcomes favoring endovascular therapy over medical management alone (generalized odds ratio, 1.37; 95% confidence interval [CI], 1.11-1.69; P = .004).
The efficacy of endovascular therapy with respect to the primary outcome was similar across predefined subgroups and across all trial sites. However, the trial was not powered to allow definite conclusions based on the results of subgroup analyses.
Although patients with an ASPECT score of 0-2 (indicating very large infarct cores) are considered unlikely to benefit from endovascular treatment, the researchers did find some signals of gain for these patients.
“Although no conclusions can be drawn because the trial was not powered for this analysis and the confidence interval for the odds ratio between the trial groups included 1, there may have been a benefit with endovascular therapy in this subgroup,” the authors wrote. “More trials are warranted to determine if this benefit is valid.”
As for secondary outcomes, the percentage of patients with a score of 0-2 on the mRS at 90 days was 30.0% in the endovascular therapy group and 11.6% in the medical management group (relative risk [RR], 2.62; 95% CI, 1.69-4.06).
The percentage of patients with a score of 0-3 on the mRS at 90 days was 47.0% in the endovascular therapy group and 33.3% in the medical management group (RR, 1.50; 95% CI, 1.17-1.91).
The primary safety outcome was symptomatic intracranial hemorrhage within 48 hours, which occurred in 6.1% of the endovascular therapy group, compared to 2.7% in the medical management group (RR, 2.07; 95% CI, 0.79-5.41; P = .12)
Mortality within 90 days was 21.7% in the endovascular therapy group and 20.0% in the medical management group. Other serious adverse events occurred in 40.0% in the endovascular therapy group and 38.2% in the medical management group (P = .70).
The percentage of patients receiving IV thrombolysis was relatively low, which may have affected outcomes in the medical management group. Another potential limitation was that urokinase rather than alteplase, which is probably more effective, was used for thrombolysis in a small percentage of patients.
Further, the study did not include patients older than 80 years or those with an ASPECT value greater than 5 and infarct core volume of 70-100 mL, and it included only Chinese patients, so the results may not be generalizable, the researchers noted.
These findings will likely change clinical practice, said Dr. Huo, who noted that the current guideline doesn’t provide “a high-level recommendation” for [endovascular therapy] in patients with a low ASPECT score.
“These new results will change the guideline” to suggest endovascular therapy for large-core patients, he said.
Welcome news
An accompanying editorial by Pierre Fayad, MD, department of neurological sciences, division of vascular neurology and stroke, University of Nebraska Medical Center, Omaha, welcomed results from this and other recent related studies.
From these new results, “it is reasonable to suggest that endovascular thrombectomy be offered to patients with large strokes” if they arrive in a timely fashion at a center capable of performing the procedure and have an ASPECT value of 3-5 or an ischemic-core volume of 50 mL or greater, he wrote.
“The improved chance of independent walking and the ability to perform other daily activities in patients with the most severe strokes is welcome news for patients and for the field of stroke treatment.”
The study received funding from Covidien Healthcare International Trading (Shanghai), Johnson & Johnson MedTech, Genesis MedTech (Shanghai), and Shanghai HeartCare Medical Technology. Dr. Huo and Dr. Jovin report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The trial was stopped early because a planned interim analysis showed efficacy of endovascular therapy in this patient population.
Among patients in China with acute ischemic stroke and a large cerebral infarction, treatment with endovascular therapy within 24 hours after stroke onset “resulted in a better functional outcome at 3 months than medical management alone,” lead author Xiaochuan Huo, MD, PhD, associate chief physician, interventional neurology department, Beijing Tiantan Hospital, Capital Medical University, told this news organization.
“This trial added important evidence for the benefits of endovascular therapy,” Dr. Huo added.
The findings were presented at the International Stroke Conference and were published online in The New England Journal of Medicine. The conference was presented by the American Stroke Association, a division of the American Heart Association.
Will change practice
Commenting on the results, Tudor G. Jovin, MD, professor and chair, department of neurology, Cooper Medical School of Rowan University, Camden, N.J., said he has “little doubt” this study will change practice.
Despite previous studies showing signals of benefit from thrombectomy for patients with large-core infarcts, and some even finding a large treatment effect, “somehow the world didn’t register this,” said Dr. Jovin.
“The stroke community was perhaps reluctant to accept these signals that were there in plain sight because we have been primed for such a long time that reperfusing large infarcts was, if not detrimental, not beneficial.”
But this study, along with another study showing similar results, SELECT 2, which was also presented at this meeting and was published in the same issue of NEJM, provide “overwhelming proof” and “have finally made the community aware,” said Dr. Jovin. “This is sort of a wake-up call to say, ‘Hey, this is real; patients with large infarcts also benefit from thrombectomy.’ “
This new research suggests it’s not necessary to learn the infarct size, at least in the early time window, and doing so just wastes precious time, added Dr. Jovin.
The impact of thrombectomy on patients with “super large infarcts” is still not clear, although these are “extremely rare” in the early time window, perhaps representing only about 1% of patients, said Dr. Jovin.
The increased rate of hemorrhages in study patients receiving thrombectomy “is the price you pay” for the benefits, he said. He noted that this is not any different from the situation with tissue plasminogen activator (tPA), which is routinely used because the benefits far outweigh the risks.
ANGEL-ASPECT
As patients with large infarctions are generally excluded from studies of thrombectomy, it’s been unclear whether they benefit from this therapy, the researchers noted.
The multicenter Endovascular Therapy in Acute Anterior Circulation Large Vessel Occlusive Patients With a Large Infarct Core (ANGEL-ASPECT) trial included 455 adult patients (median age, 68 years; 38.7% women) who had a large infarct core caused by acute large-vessel occlusion in the anterior circulation (Alberta Stroke Program Early CT Score [ASPECTS] 3-5 without core volume limitations or ASPECTS 0–2 with core volume between 70 and 100 mL).
Study participants had to have a score of 6-30 on the National Institutes of Health Stroke Scale (NIHSS) and a retrospectively determined prestroke score of 0 or 1 on the Modified Rankin Scale (mRS).
The median baseline NIHSS score of study patients was 16, the median ASPECTS was 3, and the median infarct-core volume was 62 mL.
Researchers randomly assigned patients to undergo either medical management alone or medical management as well as endovascular therapy. Medical management included intravenous (IV) thrombolysis for those who were eligible.
IV thrombolysis was administered before thrombectomy for about 28% of patients in each group. Some 78.7% of all patients arrived at the hospital outside the typical 4.5-hour window and were ineligible for thrombolysis.
A greater percentage of patients in the endovascular therapy group was receiving antihypertensive medications (83.0%) than in the medical management alone group (54.0%). About 20% of patients in each group were taking an anticoagulant medication.
When the trial was halted, outcome data were available for 336 patients. An additional 120 patients had undergone randomization, and 455 had completed 90 days of follow-up.
Better functional outcome
The primary outcome was the score on the mRS at 90 days. Results showed a shift in the distribution of scores on the mRS at 90 days toward better outcomes favoring endovascular therapy over medical management alone (generalized odds ratio, 1.37; 95% confidence interval [CI], 1.11-1.69; P = .004).
The efficacy of endovascular therapy with respect to the primary outcome was similar across predefined subgroups and across all trial sites. However, the trial was not powered to allow definite conclusions based on the results of subgroup analyses.
Although patients with an ASPECT score of 0-2 (indicating very large infarct cores) are considered unlikely to benefit from endovascular treatment, the researchers did find some signals of gain for these patients.
“Although no conclusions can be drawn because the trial was not powered for this analysis and the confidence interval for the odds ratio between the trial groups included 1, there may have been a benefit with endovascular therapy in this subgroup,” the authors wrote. “More trials are warranted to determine if this benefit is valid.”
As for secondary outcomes, the percentage of patients with a score of 0-2 on the mRS at 90 days was 30.0% in the endovascular therapy group and 11.6% in the medical management group (relative risk [RR], 2.62; 95% CI, 1.69-4.06).
The percentage of patients with a score of 0-3 on the mRS at 90 days was 47.0% in the endovascular therapy group and 33.3% in the medical management group (RR, 1.50; 95% CI, 1.17-1.91).
The primary safety outcome was symptomatic intracranial hemorrhage within 48 hours, which occurred in 6.1% of the endovascular therapy group, compared to 2.7% in the medical management group (RR, 2.07; 95% CI, 0.79-5.41; P = .12)
Mortality within 90 days was 21.7% in the endovascular therapy group and 20.0% in the medical management group. Other serious adverse events occurred in 40.0% in the endovascular therapy group and 38.2% in the medical management group (P = .70).
The percentage of patients receiving IV thrombolysis was relatively low, which may have affected outcomes in the medical management group. Another potential limitation was that urokinase rather than alteplase, which is probably more effective, was used for thrombolysis in a small percentage of patients.
Further, the study did not include patients older than 80 years or those with an ASPECT value greater than 5 and infarct core volume of 70-100 mL, and it included only Chinese patients, so the results may not be generalizable, the researchers noted.
These findings will likely change clinical practice, said Dr. Huo, who noted that the current guideline doesn’t provide “a high-level recommendation” for [endovascular therapy] in patients with a low ASPECT score.
“These new results will change the guideline” to suggest endovascular therapy for large-core patients, he said.
Welcome news
An accompanying editorial by Pierre Fayad, MD, department of neurological sciences, division of vascular neurology and stroke, University of Nebraska Medical Center, Omaha, welcomed results from this and other recent related studies.
From these new results, “it is reasonable to suggest that endovascular thrombectomy be offered to patients with large strokes” if they arrive in a timely fashion at a center capable of performing the procedure and have an ASPECT value of 3-5 or an ischemic-core volume of 50 mL or greater, he wrote.
“The improved chance of independent walking and the ability to perform other daily activities in patients with the most severe strokes is welcome news for patients and for the field of stroke treatment.”
The study received funding from Covidien Healthcare International Trading (Shanghai), Johnson & Johnson MedTech, Genesis MedTech (Shanghai), and Shanghai HeartCare Medical Technology. Dr. Huo and Dr. Jovin report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The trial was stopped early because a planned interim analysis showed efficacy of endovascular therapy in this patient population.
Among patients in China with acute ischemic stroke and a large cerebral infarction, treatment with endovascular therapy within 24 hours after stroke onset “resulted in a better functional outcome at 3 months than medical management alone,” lead author Xiaochuan Huo, MD, PhD, associate chief physician, interventional neurology department, Beijing Tiantan Hospital, Capital Medical University, told this news organization.
“This trial added important evidence for the benefits of endovascular therapy,” Dr. Huo added.
The findings were presented at the International Stroke Conference and were published online in The New England Journal of Medicine. The conference was presented by the American Stroke Association, a division of the American Heart Association.
Will change practice
Commenting on the results, Tudor G. Jovin, MD, professor and chair, department of neurology, Cooper Medical School of Rowan University, Camden, N.J., said he has “little doubt” this study will change practice.
Despite previous studies showing signals of benefit from thrombectomy for patients with large-core infarcts, and some even finding a large treatment effect, “somehow the world didn’t register this,” said Dr. Jovin.
“The stroke community was perhaps reluctant to accept these signals that were there in plain sight because we have been primed for such a long time that reperfusing large infarcts was, if not detrimental, not beneficial.”
But this study, along with another study showing similar results, SELECT 2, which was also presented at this meeting and was published in the same issue of NEJM, provide “overwhelming proof” and “have finally made the community aware,” said Dr. Jovin. “This is sort of a wake-up call to say, ‘Hey, this is real; patients with large infarcts also benefit from thrombectomy.’ “
This new research suggests it’s not necessary to learn the infarct size, at least in the early time window, and doing so just wastes precious time, added Dr. Jovin.
The impact of thrombectomy on patients with “super large infarcts” is still not clear, although these are “extremely rare” in the early time window, perhaps representing only about 1% of patients, said Dr. Jovin.
The increased rate of hemorrhages in study patients receiving thrombectomy “is the price you pay” for the benefits, he said. He noted that this is not any different from the situation with tissue plasminogen activator (tPA), which is routinely used because the benefits far outweigh the risks.
ANGEL-ASPECT
As patients with large infarctions are generally excluded from studies of thrombectomy, it’s been unclear whether they benefit from this therapy, the researchers noted.
The multicenter Endovascular Therapy in Acute Anterior Circulation Large Vessel Occlusive Patients With a Large Infarct Core (ANGEL-ASPECT) trial included 455 adult patients (median age, 68 years; 38.7% women) who had a large infarct core caused by acute large-vessel occlusion in the anterior circulation (Alberta Stroke Program Early CT Score [ASPECTS] 3-5 without core volume limitations or ASPECTS 0–2 with core volume between 70 and 100 mL).
Study participants had to have a score of 6-30 on the National Institutes of Health Stroke Scale (NIHSS) and a retrospectively determined prestroke score of 0 or 1 on the Modified Rankin Scale (mRS).
The median baseline NIHSS score of study patients was 16, the median ASPECTS was 3, and the median infarct-core volume was 62 mL.
Researchers randomly assigned patients to undergo either medical management alone or medical management as well as endovascular therapy. Medical management included intravenous (IV) thrombolysis for those who were eligible.
IV thrombolysis was administered before thrombectomy for about 28% of patients in each group. Some 78.7% of all patients arrived at the hospital outside the typical 4.5-hour window and were ineligible for thrombolysis.
A greater percentage of patients in the endovascular therapy group was receiving antihypertensive medications (83.0%) than in the medical management alone group (54.0%). About 20% of patients in each group were taking an anticoagulant medication.
When the trial was halted, outcome data were available for 336 patients. An additional 120 patients had undergone randomization, and 455 had completed 90 days of follow-up.
Better functional outcome
The primary outcome was the score on the mRS at 90 days. Results showed a shift in the distribution of scores on the mRS at 90 days toward better outcomes favoring endovascular therapy over medical management alone (generalized odds ratio, 1.37; 95% confidence interval [CI], 1.11-1.69; P = .004).
The efficacy of endovascular therapy with respect to the primary outcome was similar across predefined subgroups and across all trial sites. However, the trial was not powered to allow definite conclusions based on the results of subgroup analyses.
Although patients with an ASPECT score of 0-2 (indicating very large infarct cores) are considered unlikely to benefit from endovascular treatment, the researchers did find some signals of gain for these patients.
“Although no conclusions can be drawn because the trial was not powered for this analysis and the confidence interval for the odds ratio between the trial groups included 1, there may have been a benefit with endovascular therapy in this subgroup,” the authors wrote. “More trials are warranted to determine if this benefit is valid.”
As for secondary outcomes, the percentage of patients with a score of 0-2 on the mRS at 90 days was 30.0% in the endovascular therapy group and 11.6% in the medical management group (relative risk [RR], 2.62; 95% CI, 1.69-4.06).
The percentage of patients with a score of 0-3 on the mRS at 90 days was 47.0% in the endovascular therapy group and 33.3% in the medical management group (RR, 1.50; 95% CI, 1.17-1.91).
The primary safety outcome was symptomatic intracranial hemorrhage within 48 hours, which occurred in 6.1% of the endovascular therapy group, compared to 2.7% in the medical management group (RR, 2.07; 95% CI, 0.79-5.41; P = .12)
Mortality within 90 days was 21.7% in the endovascular therapy group and 20.0% in the medical management group. Other serious adverse events occurred in 40.0% in the endovascular therapy group and 38.2% in the medical management group (P = .70).
The percentage of patients receiving IV thrombolysis was relatively low, which may have affected outcomes in the medical management group. Another potential limitation was that urokinase rather than alteplase, which is probably more effective, was used for thrombolysis in a small percentage of patients.
Further, the study did not include patients older than 80 years or those with an ASPECT value greater than 5 and infarct core volume of 70-100 mL, and it included only Chinese patients, so the results may not be generalizable, the researchers noted.
These findings will likely change clinical practice, said Dr. Huo, who noted that the current guideline doesn’t provide “a high-level recommendation” for [endovascular therapy] in patients with a low ASPECT score.
“These new results will change the guideline” to suggest endovascular therapy for large-core patients, he said.
Welcome news
An accompanying editorial by Pierre Fayad, MD, department of neurological sciences, division of vascular neurology and stroke, University of Nebraska Medical Center, Omaha, welcomed results from this and other recent related studies.
From these new results, “it is reasonable to suggest that endovascular thrombectomy be offered to patients with large strokes” if they arrive in a timely fashion at a center capable of performing the procedure and have an ASPECT value of 3-5 or an ischemic-core volume of 50 mL or greater, he wrote.
“The improved chance of independent walking and the ability to perform other daily activities in patients with the most severe strokes is welcome news for patients and for the field of stroke treatment.”
The study received funding from Covidien Healthcare International Trading (Shanghai), Johnson & Johnson MedTech, Genesis MedTech (Shanghai), and Shanghai HeartCare Medical Technology. Dr. Huo and Dr. Jovin report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ISC 2023
Similar effect of early, late BP reduction in stroke: CATIS-2
in the CATIS-2 trial.
The trial was presented by Liping Liu, MD, Beijing Tiantan Hospital, at the International Stroke Conference presented by the American Stroke Association, a division of the American Heart Association.
“Antihypertensive treatment can be delayed for at least 7 days following ischemic stroke onset, unless there are severe acute comorbidities that demand emergency blood pressure reduction to prevent serious complications,” Dr. Liu concluded.
But he acknowledged that the optimal BP management strategy in these patients remains uncertain and should be the focus of future research.
Discussing the trial at an ISC 2023 Highlights session, Lauren Sansing, MD, Yale University, New Haven, Conn., and ISC program vice chair, said: “These results seem to support waiting for a week or so before treating blood pressure in these patients.”
But Tudor Jovin, MD, Cooper Neurological Institute, Cherry Hill, N.J., and ISC program chair, countered: “To me, it’s kind of a neutral result, so what I take home from this is that you don’t necessarily have to wait.”
Dr. Jovin continued: “We used to think that it was mandatory not to treat blood pressure early because of the risk of deceasing the perfusion pressure, but this trial suggests the effects are neutral and there is probably as much benefit from lowering blood pressure for other reasons that offsets the potential harm.
“I think these are good data to rely on when we make these kinds of treatment decisions. Personally, I am a bit more aggressive with early blood pressure management and it’s good to see that you don’t get punished for that,” he added.
In his presentation, Dr. Liu explained that increased BP is common in acute stroke and is strongly associated with poor functional outcome and recurrence of ischemic stroke, but the optimal blood pressure management strategy in acute ischemic stroke remains controversial.
In the first CATIS trial (China Antihypertensive Trial in Acute Ischemic Stroke), which compared antihypertensive treatment within 48 hours of stroke onset with no antihypertensive treatment in ischemic stroke patients not receiving thrombolysis, the main results suggested that BP reduction with antihypertensive medications did not reduce the likelihood of death and major disability at 14 days or hospital discharge. But a subgroup analysis found that initiating antihypertensive treatment between 24 and 48 hours of stroke onset showed a beneficial effect on reducing death or major disability.
Current AHA/ASA guidelines suggest that, in patients with BP greater than 220/120 mm Hg who have not received thrombolysis or thrombectomy and have no comorbid conditions requiring urgent antihypertensive treatment, the benefit of initiating or reinitiating antihypertensive treatment within the first 48-72 hours is uncertain, although the guidelines say it might be reasonable to lower BP by around 15% during the first 24 hours after stroke onset, Dr. Liu noted.
The CATIS-2 trial was a multicenter, randomized, open-label, blinded-endpoints trial conducted at 106 centers in China that enrolled 4810 patients within 24-48 hours of onset of acute ischemic stroke who had elevated BP. Patients had not received thrombolytic therapy or mechanical thrombectomy.
Patients were randomly assigned to early antihypertensive therapy (initiated after randomization and aiming for a 10%-20% reduction in systolic BP) or delayed antihypertensive therapy (restarted antihypertensive therapy on day 8 of randomization, aiming for a BP of < 140/90 mm Hg).
The median age of the patients was 64 years, 65% were male, 80% had a history of hypertension, and the median National Institutes of Health Stroke Scale score was 3. Baseline BP averaged 163/92 mm Hg in both groups. The median time from stroke onset to antihypertensive treatment was 1.5 days in the early group and 8.5 days in the delayed group.
BP results showed that, at 24 hours after randomization, mean systolic pressure was reduced by 16.4 mm Hg (9.7%) in the early-treatment group and by 8.6 mm Hg (4.9%) in the delayed-treatment group (difference, –7.8 mm Hg; P < .0001).
At day 7, mean systolic pressure was 139.1 mm Hg in the early-treatment group, compared with 150.9 mm Hg in the delayed-treatment group, with a net difference in systolic BP of –11.9 mm Hg (P < .0001).
The primary outcome was the composite of death and major disability (modified Rankin Scale ≥ 3) at 3 months. This did not differ between the groups, occurring in 12.1% in the early antihypertensive treatment group versus 10.5% in the delayed antihypertensive treatment group (risk ratio, 1.15; P = .08).
There was also no difference in the major secondary outcome of shift in scores of mRS at 3 months, with a common odds ratio of 1.05 (95% confidence interval, 0.95-1.17).
There was no interaction with the composite outcome of death or major disability at 90 days in the prespecified subgroups.
Dr. Liu pointed out several limitations of the study. These included an observed primary outcome rate substantially lower than expected; the BP reduction seen within the first 7 days in the early-treatment group was moderate; and the results of the study cannot be applied to patients treated with thrombolysis or thrombectomy.
Dr. Liu has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
in the CATIS-2 trial.
The trial was presented by Liping Liu, MD, Beijing Tiantan Hospital, at the International Stroke Conference presented by the American Stroke Association, a division of the American Heart Association.
“Antihypertensive treatment can be delayed for at least 7 days following ischemic stroke onset, unless there are severe acute comorbidities that demand emergency blood pressure reduction to prevent serious complications,” Dr. Liu concluded.
But he acknowledged that the optimal BP management strategy in these patients remains uncertain and should be the focus of future research.
Discussing the trial at an ISC 2023 Highlights session, Lauren Sansing, MD, Yale University, New Haven, Conn., and ISC program vice chair, said: “These results seem to support waiting for a week or so before treating blood pressure in these patients.”
But Tudor Jovin, MD, Cooper Neurological Institute, Cherry Hill, N.J., and ISC program chair, countered: “To me, it’s kind of a neutral result, so what I take home from this is that you don’t necessarily have to wait.”
Dr. Jovin continued: “We used to think that it was mandatory not to treat blood pressure early because of the risk of deceasing the perfusion pressure, but this trial suggests the effects are neutral and there is probably as much benefit from lowering blood pressure for other reasons that offsets the potential harm.
“I think these are good data to rely on when we make these kinds of treatment decisions. Personally, I am a bit more aggressive with early blood pressure management and it’s good to see that you don’t get punished for that,” he added.
In his presentation, Dr. Liu explained that increased BP is common in acute stroke and is strongly associated with poor functional outcome and recurrence of ischemic stroke, but the optimal blood pressure management strategy in acute ischemic stroke remains controversial.
In the first CATIS trial (China Antihypertensive Trial in Acute Ischemic Stroke), which compared antihypertensive treatment within 48 hours of stroke onset with no antihypertensive treatment in ischemic stroke patients not receiving thrombolysis, the main results suggested that BP reduction with antihypertensive medications did not reduce the likelihood of death and major disability at 14 days or hospital discharge. But a subgroup analysis found that initiating antihypertensive treatment between 24 and 48 hours of stroke onset showed a beneficial effect on reducing death or major disability.
Current AHA/ASA guidelines suggest that, in patients with BP greater than 220/120 mm Hg who have not received thrombolysis or thrombectomy and have no comorbid conditions requiring urgent antihypertensive treatment, the benefit of initiating or reinitiating antihypertensive treatment within the first 48-72 hours is uncertain, although the guidelines say it might be reasonable to lower BP by around 15% during the first 24 hours after stroke onset, Dr. Liu noted.
The CATIS-2 trial was a multicenter, randomized, open-label, blinded-endpoints trial conducted at 106 centers in China that enrolled 4810 patients within 24-48 hours of onset of acute ischemic stroke who had elevated BP. Patients had not received thrombolytic therapy or mechanical thrombectomy.
Patients were randomly assigned to early antihypertensive therapy (initiated after randomization and aiming for a 10%-20% reduction in systolic BP) or delayed antihypertensive therapy (restarted antihypertensive therapy on day 8 of randomization, aiming for a BP of < 140/90 mm Hg).
The median age of the patients was 64 years, 65% were male, 80% had a history of hypertension, and the median National Institutes of Health Stroke Scale score was 3. Baseline BP averaged 163/92 mm Hg in both groups. The median time from stroke onset to antihypertensive treatment was 1.5 days in the early group and 8.5 days in the delayed group.
BP results showed that, at 24 hours after randomization, mean systolic pressure was reduced by 16.4 mm Hg (9.7%) in the early-treatment group and by 8.6 mm Hg (4.9%) in the delayed-treatment group (difference, –7.8 mm Hg; P < .0001).
At day 7, mean systolic pressure was 139.1 mm Hg in the early-treatment group, compared with 150.9 mm Hg in the delayed-treatment group, with a net difference in systolic BP of –11.9 mm Hg (P < .0001).
The primary outcome was the composite of death and major disability (modified Rankin Scale ≥ 3) at 3 months. This did not differ between the groups, occurring in 12.1% in the early antihypertensive treatment group versus 10.5% in the delayed antihypertensive treatment group (risk ratio, 1.15; P = .08).
There was also no difference in the major secondary outcome of shift in scores of mRS at 3 months, with a common odds ratio of 1.05 (95% confidence interval, 0.95-1.17).
There was no interaction with the composite outcome of death or major disability at 90 days in the prespecified subgroups.
Dr. Liu pointed out several limitations of the study. These included an observed primary outcome rate substantially lower than expected; the BP reduction seen within the first 7 days in the early-treatment group was moderate; and the results of the study cannot be applied to patients treated with thrombolysis or thrombectomy.
Dr. Liu has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
in the CATIS-2 trial.
The trial was presented by Liping Liu, MD, Beijing Tiantan Hospital, at the International Stroke Conference presented by the American Stroke Association, a division of the American Heart Association.
“Antihypertensive treatment can be delayed for at least 7 days following ischemic stroke onset, unless there are severe acute comorbidities that demand emergency blood pressure reduction to prevent serious complications,” Dr. Liu concluded.
But he acknowledged that the optimal BP management strategy in these patients remains uncertain and should be the focus of future research.
Discussing the trial at an ISC 2023 Highlights session, Lauren Sansing, MD, Yale University, New Haven, Conn., and ISC program vice chair, said: “These results seem to support waiting for a week or so before treating blood pressure in these patients.”
But Tudor Jovin, MD, Cooper Neurological Institute, Cherry Hill, N.J., and ISC program chair, countered: “To me, it’s kind of a neutral result, so what I take home from this is that you don’t necessarily have to wait.”
Dr. Jovin continued: “We used to think that it was mandatory not to treat blood pressure early because of the risk of deceasing the perfusion pressure, but this trial suggests the effects are neutral and there is probably as much benefit from lowering blood pressure for other reasons that offsets the potential harm.
“I think these are good data to rely on when we make these kinds of treatment decisions. Personally, I am a bit more aggressive with early blood pressure management and it’s good to see that you don’t get punished for that,” he added.
In his presentation, Dr. Liu explained that increased BP is common in acute stroke and is strongly associated with poor functional outcome and recurrence of ischemic stroke, but the optimal blood pressure management strategy in acute ischemic stroke remains controversial.
In the first CATIS trial (China Antihypertensive Trial in Acute Ischemic Stroke), which compared antihypertensive treatment within 48 hours of stroke onset with no antihypertensive treatment in ischemic stroke patients not receiving thrombolysis, the main results suggested that BP reduction with antihypertensive medications did not reduce the likelihood of death and major disability at 14 days or hospital discharge. But a subgroup analysis found that initiating antihypertensive treatment between 24 and 48 hours of stroke onset showed a beneficial effect on reducing death or major disability.
Current AHA/ASA guidelines suggest that, in patients with BP greater than 220/120 mm Hg who have not received thrombolysis or thrombectomy and have no comorbid conditions requiring urgent antihypertensive treatment, the benefit of initiating or reinitiating antihypertensive treatment within the first 48-72 hours is uncertain, although the guidelines say it might be reasonable to lower BP by around 15% during the first 24 hours after stroke onset, Dr. Liu noted.
The CATIS-2 trial was a multicenter, randomized, open-label, blinded-endpoints trial conducted at 106 centers in China that enrolled 4810 patients within 24-48 hours of onset of acute ischemic stroke who had elevated BP. Patients had not received thrombolytic therapy or mechanical thrombectomy.
Patients were randomly assigned to early antihypertensive therapy (initiated after randomization and aiming for a 10%-20% reduction in systolic BP) or delayed antihypertensive therapy (restarted antihypertensive therapy on day 8 of randomization, aiming for a BP of < 140/90 mm Hg).
The median age of the patients was 64 years, 65% were male, 80% had a history of hypertension, and the median National Institutes of Health Stroke Scale score was 3. Baseline BP averaged 163/92 mm Hg in both groups. The median time from stroke onset to antihypertensive treatment was 1.5 days in the early group and 8.5 days in the delayed group.
BP results showed that, at 24 hours after randomization, mean systolic pressure was reduced by 16.4 mm Hg (9.7%) in the early-treatment group and by 8.6 mm Hg (4.9%) in the delayed-treatment group (difference, –7.8 mm Hg; P < .0001).
At day 7, mean systolic pressure was 139.1 mm Hg in the early-treatment group, compared with 150.9 mm Hg in the delayed-treatment group, with a net difference in systolic BP of –11.9 mm Hg (P < .0001).
The primary outcome was the composite of death and major disability (modified Rankin Scale ≥ 3) at 3 months. This did not differ between the groups, occurring in 12.1% in the early antihypertensive treatment group versus 10.5% in the delayed antihypertensive treatment group (risk ratio, 1.15; P = .08).
There was also no difference in the major secondary outcome of shift in scores of mRS at 3 months, with a common odds ratio of 1.05 (95% confidence interval, 0.95-1.17).
There was no interaction with the composite outcome of death or major disability at 90 days in the prespecified subgroups.
Dr. Liu pointed out several limitations of the study. These included an observed primary outcome rate substantially lower than expected; the BP reduction seen within the first 7 days in the early-treatment group was moderate; and the results of the study cannot be applied to patients treated with thrombolysis or thrombectomy.
Dr. Liu has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ISC 2023
‘Infuriating’ prescription denial leaves patient without antiemetics
It was Friday, and oncologist Coral Olazagasti, MD, faced a ticking clock.
The patient – a man with HPV-related oropharyngeal cancer – was experiencing severe side effects from standard chemoradiation with weekly cisplatin. Intense nausea and grade 3 mucositis, in particular, left him struggling to swallow or take in any food or fluids.
He was on 8 mg of ondansetron (Zofran) every 8 hours, as needed, to keep the nausea at bay. The pills along with a feeding tube helped, but his symptoms were so intense, neither was quite enough.
“He still needed to be hospitalized twice for dehydration,” said Dr. Olazagasti, who specializes in head and neck medical cancer at Sylvester Comprehensive Cancer Center in Miami.
But when it came time to renew his ondansetron prescription, his insurance company denied it.
The reasoning: “The company had only approved 30 tablets a month and, for them, it was unjustifiable to approve anything above that amount,” Dr. Olazagasti explained.
After Dr. Olazagasti called the insurance company to resolve the issue, a company representative told her to fill out a prior authorization form.
But it was already after 7:30 p.m. ET on Friday.
At that point, finding the prior authorization documents, filling them out, and submitting them would take more time – and the paperwork couldn’t be filed until Monday.
“My patient was at home with zero tablets left and horrible symptoms. He couldn’t keep anything down,” Dr. Olazagasti said.
On Monday, the oncology team sent the prior authorization request, and her patient received his medication a few days later.
“My patient had to wait about 5 days to get the nausea meds he needed,” she said. In the meantime, he was in pain. “Having a refill of this simple supportive care medication rejected was infuriating.”
When Dr. Olazagasti vented her frustrations on Twitter, several people chimed in, suggesting purchasing the drug at a discount through GoodRx or Cost Plus instead of going through the insurance company.
At Cost Plus, for instance, 30 8-mg pills would cost $6.30, but ordering from the online pharmacy would mean waiting several days for delivery.
Discounts through GoodRx may provide a potentially faster solution in a pinch, but the pharmacy matters. In Miami, 30 8-mg pills would cost $19.99 at Costco with a GoodRx coupon, but $233.56 at CVS and $253.60 at Walgreens.
Although potentially useful, these options may not be the obvious choice for oncologists and patients, especially when a drug has already been approved and covered by the insurer. In this case, the denial was also a surprise, which left Dr. Olazagasti and her patient scrambling right before the weekend.
In addition, companies providing discounted generic drugs may only have a limited number of oncology-related medications. Cost Plus, for instance, now sells more than 1,000 generic prescription drugs at a fraction of what insurance companies charge, but only about 7 are cancer drugs.
On a broader level, Dr. Olazagasti noted, “insurance companies have a responsibility to cover these drugs. If we all get so fed up that we start relying on alternate routes to get patients their treatments, then insurance companies are let off the hook.”
However, using an alternative option like GoodRx or CostPlus could mean bypassing insurance company obstacles in certain cases.
“The hurdles someone may have to go through to get a generic drug approved are very frustrating,” said Stacie B. Dusetzina, PhD, professor of health policy and a professor of cancer research at Vanderbilt University in Nashville, Tenn.
In a weekend emergency situation, if the drug is discounted through GoodRx, “it can be a good backup strategy to send the prescription to the pharmacy” and more generally “worth it for patients to check if they can get a better deal on generic drugs through these companies.”
A version of this article first appeared on Medscape.com.
It was Friday, and oncologist Coral Olazagasti, MD, faced a ticking clock.
The patient – a man with HPV-related oropharyngeal cancer – was experiencing severe side effects from standard chemoradiation with weekly cisplatin. Intense nausea and grade 3 mucositis, in particular, left him struggling to swallow or take in any food or fluids.
He was on 8 mg of ondansetron (Zofran) every 8 hours, as needed, to keep the nausea at bay. The pills along with a feeding tube helped, but his symptoms were so intense, neither was quite enough.
“He still needed to be hospitalized twice for dehydration,” said Dr. Olazagasti, who specializes in head and neck medical cancer at Sylvester Comprehensive Cancer Center in Miami.
But when it came time to renew his ondansetron prescription, his insurance company denied it.
The reasoning: “The company had only approved 30 tablets a month and, for them, it was unjustifiable to approve anything above that amount,” Dr. Olazagasti explained.
After Dr. Olazagasti called the insurance company to resolve the issue, a company representative told her to fill out a prior authorization form.
But it was already after 7:30 p.m. ET on Friday.
At that point, finding the prior authorization documents, filling them out, and submitting them would take more time – and the paperwork couldn’t be filed until Monday.
“My patient was at home with zero tablets left and horrible symptoms. He couldn’t keep anything down,” Dr. Olazagasti said.
On Monday, the oncology team sent the prior authorization request, and her patient received his medication a few days later.
“My patient had to wait about 5 days to get the nausea meds he needed,” she said. In the meantime, he was in pain. “Having a refill of this simple supportive care medication rejected was infuriating.”
When Dr. Olazagasti vented her frustrations on Twitter, several people chimed in, suggesting purchasing the drug at a discount through GoodRx or Cost Plus instead of going through the insurance company.
At Cost Plus, for instance, 30 8-mg pills would cost $6.30, but ordering from the online pharmacy would mean waiting several days for delivery.
Discounts through GoodRx may provide a potentially faster solution in a pinch, but the pharmacy matters. In Miami, 30 8-mg pills would cost $19.99 at Costco with a GoodRx coupon, but $233.56 at CVS and $253.60 at Walgreens.
Although potentially useful, these options may not be the obvious choice for oncologists and patients, especially when a drug has already been approved and covered by the insurer. In this case, the denial was also a surprise, which left Dr. Olazagasti and her patient scrambling right before the weekend.
In addition, companies providing discounted generic drugs may only have a limited number of oncology-related medications. Cost Plus, for instance, now sells more than 1,000 generic prescription drugs at a fraction of what insurance companies charge, but only about 7 are cancer drugs.
On a broader level, Dr. Olazagasti noted, “insurance companies have a responsibility to cover these drugs. If we all get so fed up that we start relying on alternate routes to get patients their treatments, then insurance companies are let off the hook.”
However, using an alternative option like GoodRx or CostPlus could mean bypassing insurance company obstacles in certain cases.
“The hurdles someone may have to go through to get a generic drug approved are very frustrating,” said Stacie B. Dusetzina, PhD, professor of health policy and a professor of cancer research at Vanderbilt University in Nashville, Tenn.
In a weekend emergency situation, if the drug is discounted through GoodRx, “it can be a good backup strategy to send the prescription to the pharmacy” and more generally “worth it for patients to check if they can get a better deal on generic drugs through these companies.”
A version of this article first appeared on Medscape.com.
It was Friday, and oncologist Coral Olazagasti, MD, faced a ticking clock.
The patient – a man with HPV-related oropharyngeal cancer – was experiencing severe side effects from standard chemoradiation with weekly cisplatin. Intense nausea and grade 3 mucositis, in particular, left him struggling to swallow or take in any food or fluids.
He was on 8 mg of ondansetron (Zofran) every 8 hours, as needed, to keep the nausea at bay. The pills along with a feeding tube helped, but his symptoms were so intense, neither was quite enough.
“He still needed to be hospitalized twice for dehydration,” said Dr. Olazagasti, who specializes in head and neck medical cancer at Sylvester Comprehensive Cancer Center in Miami.
But when it came time to renew his ondansetron prescription, his insurance company denied it.
The reasoning: “The company had only approved 30 tablets a month and, for them, it was unjustifiable to approve anything above that amount,” Dr. Olazagasti explained.
After Dr. Olazagasti called the insurance company to resolve the issue, a company representative told her to fill out a prior authorization form.
But it was already after 7:30 p.m. ET on Friday.
At that point, finding the prior authorization documents, filling them out, and submitting them would take more time – and the paperwork couldn’t be filed until Monday.
“My patient was at home with zero tablets left and horrible symptoms. He couldn’t keep anything down,” Dr. Olazagasti said.
On Monday, the oncology team sent the prior authorization request, and her patient received his medication a few days later.
“My patient had to wait about 5 days to get the nausea meds he needed,” she said. In the meantime, he was in pain. “Having a refill of this simple supportive care medication rejected was infuriating.”
When Dr. Olazagasti vented her frustrations on Twitter, several people chimed in, suggesting purchasing the drug at a discount through GoodRx or Cost Plus instead of going through the insurance company.
At Cost Plus, for instance, 30 8-mg pills would cost $6.30, but ordering from the online pharmacy would mean waiting several days for delivery.
Discounts through GoodRx may provide a potentially faster solution in a pinch, but the pharmacy matters. In Miami, 30 8-mg pills would cost $19.99 at Costco with a GoodRx coupon, but $233.56 at CVS and $253.60 at Walgreens.
Although potentially useful, these options may not be the obvious choice for oncologists and patients, especially when a drug has already been approved and covered by the insurer. In this case, the denial was also a surprise, which left Dr. Olazagasti and her patient scrambling right before the weekend.
In addition, companies providing discounted generic drugs may only have a limited number of oncology-related medications. Cost Plus, for instance, now sells more than 1,000 generic prescription drugs at a fraction of what insurance companies charge, but only about 7 are cancer drugs.
On a broader level, Dr. Olazagasti noted, “insurance companies have a responsibility to cover these drugs. If we all get so fed up that we start relying on alternate routes to get patients their treatments, then insurance companies are let off the hook.”
However, using an alternative option like GoodRx or CostPlus could mean bypassing insurance company obstacles in certain cases.
“The hurdles someone may have to go through to get a generic drug approved are very frustrating,” said Stacie B. Dusetzina, PhD, professor of health policy and a professor of cancer research at Vanderbilt University in Nashville, Tenn.
In a weekend emergency situation, if the drug is discounted through GoodRx, “it can be a good backup strategy to send the prescription to the pharmacy” and more generally “worth it for patients to check if they can get a better deal on generic drugs through these companies.”
A version of this article first appeared on Medscape.com.
Omit radiation in older women with low-risk, ER+ breast cancer
say researchers reporting 10-year outcomes from the large phase 3 trial known as PRIME II.
“Our trial provides robust evidence indicating that irradiation can be safely omitted in women 65 years of age or older who have grade 1 or 2 ER-high cancers treated by breast-conserving therapy, provided that they receive 5 years of adjuvant endocrine therapy,” concluded investigators led by Ian Kunkler, MB, a clinical oncology professor at the University of Edinburgh.
The trial randomly assigned 1,326 women who had undergone a lumpectomy to either whole-breast irradiation or no radiation on a background of tamoxifen.
The incidence of local recurrence was lower with radiation (0.9% vs. 9.5%), but there was no significant difference in distant metastases or breast cancer–specific or overall survival.
The findings will “help clinicians guide older patients on whether this particular aspect of early breast cancer treatment can be omitted,” Dr. Kunkler said in a press release. Radiation carries risks of heart and lung damage, and these results show that skipping it does not increase the odds of dying from breast cancer.
The new study was published in the New England Journal of Medicine.
“Any doubt that radiotherapy cannot be omitted in women” who meet the criteria “can be put to rest,” commented breast radiation oncologists Alice Ho, MD, of Duke University in Durham, N.C., and Jennifer Bellon, MD, of Harvard Medical School, Boston, in an accompanying editorial.
Clinical guidelines already support omitting radiation therapy in older women with low-risk tumors treated with lumpectomy and endocrine therapy, but the move has been controversial owing to a lack of long-term data, and use of radiation for such women remains common in the United States, the investigators explain.
The “highly anticipated” results for 10-year outcomes from this trial should help address that issue, as well as “the long-standing problem of overtreatment in older women with low-risk breast cancer,” the editorialists comment.
Study details
PRIME II was conducted from 2003 to 2009 mainly in the United Kingdom. Participants were aged 65 years or older and had T1 or T2 ER-positive tumors no larger than 3 cm and were without nodal involvement.
Following lumpectomies with clear margins, the women underwent endocrine therapy; the investigators recommended tamoxifen at 20 mg/day for 5 years.
Women who were randomly assigned to radiation also received 40-50 Gy of whole-breast irradiation in 20-25 fractions over 3-5 weeks.
At 10 years, 1.6% of women in the no-radiation arm had distant metastases as their first recurrence vs. 3% of women who underwent radiation.
Ten-year breast cancer–specific survival was 97.9% with radiation and 97.4% with no radiation. Ten-year overall survival was 80.7% in the radiotherapy arm vs. 80.8% in the no-radiotherapy group.
In addition, the recurrence rate was lower after radiation. The investigators suggest that lower adherence to endocrine therapy and lower levels of ER positivity increased the risk of local recurrence among women who didn’t receive radiation.
Almost 10% of the women who did not receive radiation had local recurrences by 10 years, but the investigators note that if tumors do recur locally, women still have the option of a second lumpectomy, and if they so choose, they can then receive radiation, so local recurrence “does not necessarily mean loss of the breast.”
PRIME II was funded by the Scottish Government’s chief scientist office and the Breast Cancer Institute at Western General Hospital, Edinburgh. Dr. Kunkler reported no conflicts of interest. A coauthor has acted as a speaker, adviser, and/or researcher for many companies, including Hoffmann-La Roche, Exact Sciences, and Eli Lilly. Dr. Ho reported grants from and/or being a consultant for GlaxoSmithKline, Roche, Merck, and others. Dr. Bellon reported ties to Varian Medical Systems and Veracyte.
A version of this article originally appeared on Medscape.com.
say researchers reporting 10-year outcomes from the large phase 3 trial known as PRIME II.
“Our trial provides robust evidence indicating that irradiation can be safely omitted in women 65 years of age or older who have grade 1 or 2 ER-high cancers treated by breast-conserving therapy, provided that they receive 5 years of adjuvant endocrine therapy,” concluded investigators led by Ian Kunkler, MB, a clinical oncology professor at the University of Edinburgh.
The trial randomly assigned 1,326 women who had undergone a lumpectomy to either whole-breast irradiation or no radiation on a background of tamoxifen.
The incidence of local recurrence was lower with radiation (0.9% vs. 9.5%), but there was no significant difference in distant metastases or breast cancer–specific or overall survival.
The findings will “help clinicians guide older patients on whether this particular aspect of early breast cancer treatment can be omitted,” Dr. Kunkler said in a press release. Radiation carries risks of heart and lung damage, and these results show that skipping it does not increase the odds of dying from breast cancer.
The new study was published in the New England Journal of Medicine.
“Any doubt that radiotherapy cannot be omitted in women” who meet the criteria “can be put to rest,” commented breast radiation oncologists Alice Ho, MD, of Duke University in Durham, N.C., and Jennifer Bellon, MD, of Harvard Medical School, Boston, in an accompanying editorial.
Clinical guidelines already support omitting radiation therapy in older women with low-risk tumors treated with lumpectomy and endocrine therapy, but the move has been controversial owing to a lack of long-term data, and use of radiation for such women remains common in the United States, the investigators explain.
The “highly anticipated” results for 10-year outcomes from this trial should help address that issue, as well as “the long-standing problem of overtreatment in older women with low-risk breast cancer,” the editorialists comment.
Study details
PRIME II was conducted from 2003 to 2009 mainly in the United Kingdom. Participants were aged 65 years or older and had T1 or T2 ER-positive tumors no larger than 3 cm and were without nodal involvement.
Following lumpectomies with clear margins, the women underwent endocrine therapy; the investigators recommended tamoxifen at 20 mg/day for 5 years.
Women who were randomly assigned to radiation also received 40-50 Gy of whole-breast irradiation in 20-25 fractions over 3-5 weeks.
At 10 years, 1.6% of women in the no-radiation arm had distant metastases as their first recurrence vs. 3% of women who underwent radiation.
Ten-year breast cancer–specific survival was 97.9% with radiation and 97.4% with no radiation. Ten-year overall survival was 80.7% in the radiotherapy arm vs. 80.8% in the no-radiotherapy group.
In addition, the recurrence rate was lower after radiation. The investigators suggest that lower adherence to endocrine therapy and lower levels of ER positivity increased the risk of local recurrence among women who didn’t receive radiation.
Almost 10% of the women who did not receive radiation had local recurrences by 10 years, but the investigators note that if tumors do recur locally, women still have the option of a second lumpectomy, and if they so choose, they can then receive radiation, so local recurrence “does not necessarily mean loss of the breast.”
PRIME II was funded by the Scottish Government’s chief scientist office and the Breast Cancer Institute at Western General Hospital, Edinburgh. Dr. Kunkler reported no conflicts of interest. A coauthor has acted as a speaker, adviser, and/or researcher for many companies, including Hoffmann-La Roche, Exact Sciences, and Eli Lilly. Dr. Ho reported grants from and/or being a consultant for GlaxoSmithKline, Roche, Merck, and others. Dr. Bellon reported ties to Varian Medical Systems and Veracyte.
A version of this article originally appeared on Medscape.com.
say researchers reporting 10-year outcomes from the large phase 3 trial known as PRIME II.
“Our trial provides robust evidence indicating that irradiation can be safely omitted in women 65 years of age or older who have grade 1 or 2 ER-high cancers treated by breast-conserving therapy, provided that they receive 5 years of adjuvant endocrine therapy,” concluded investigators led by Ian Kunkler, MB, a clinical oncology professor at the University of Edinburgh.
The trial randomly assigned 1,326 women who had undergone a lumpectomy to either whole-breast irradiation or no radiation on a background of tamoxifen.
The incidence of local recurrence was lower with radiation (0.9% vs. 9.5%), but there was no significant difference in distant metastases or breast cancer–specific or overall survival.
The findings will “help clinicians guide older patients on whether this particular aspect of early breast cancer treatment can be omitted,” Dr. Kunkler said in a press release. Radiation carries risks of heart and lung damage, and these results show that skipping it does not increase the odds of dying from breast cancer.
The new study was published in the New England Journal of Medicine.
“Any doubt that radiotherapy cannot be omitted in women” who meet the criteria “can be put to rest,” commented breast radiation oncologists Alice Ho, MD, of Duke University in Durham, N.C., and Jennifer Bellon, MD, of Harvard Medical School, Boston, in an accompanying editorial.
Clinical guidelines already support omitting radiation therapy in older women with low-risk tumors treated with lumpectomy and endocrine therapy, but the move has been controversial owing to a lack of long-term data, and use of radiation for such women remains common in the United States, the investigators explain.
The “highly anticipated” results for 10-year outcomes from this trial should help address that issue, as well as “the long-standing problem of overtreatment in older women with low-risk breast cancer,” the editorialists comment.
Study details
PRIME II was conducted from 2003 to 2009 mainly in the United Kingdom. Participants were aged 65 years or older and had T1 or T2 ER-positive tumors no larger than 3 cm and were without nodal involvement.
Following lumpectomies with clear margins, the women underwent endocrine therapy; the investigators recommended tamoxifen at 20 mg/day for 5 years.
Women who were randomly assigned to radiation also received 40-50 Gy of whole-breast irradiation in 20-25 fractions over 3-5 weeks.
At 10 years, 1.6% of women in the no-radiation arm had distant metastases as their first recurrence vs. 3% of women who underwent radiation.
Ten-year breast cancer–specific survival was 97.9% with radiation and 97.4% with no radiation. Ten-year overall survival was 80.7% in the radiotherapy arm vs. 80.8% in the no-radiotherapy group.
In addition, the recurrence rate was lower after radiation. The investigators suggest that lower adherence to endocrine therapy and lower levels of ER positivity increased the risk of local recurrence among women who didn’t receive radiation.
Almost 10% of the women who did not receive radiation had local recurrences by 10 years, but the investigators note that if tumors do recur locally, women still have the option of a second lumpectomy, and if they so choose, they can then receive radiation, so local recurrence “does not necessarily mean loss of the breast.”
PRIME II was funded by the Scottish Government’s chief scientist office and the Breast Cancer Institute at Western General Hospital, Edinburgh. Dr. Kunkler reported no conflicts of interest. A coauthor has acted as a speaker, adviser, and/or researcher for many companies, including Hoffmann-La Roche, Exact Sciences, and Eli Lilly. Dr. Ho reported grants from and/or being a consultant for GlaxoSmithKline, Roche, Merck, and others. Dr. Bellon reported ties to Varian Medical Systems and Veracyte.
A version of this article originally appeared on Medscape.com.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Thrombolysis not necessary in mild nondisabling stroke: ARAMIS
in the ARAMIS trial.
The trial was presented by Thanh Nguyen, MD, Boston Medical Center, at the International Stroke Conference presented by the American Stroke Association, a division of the American Heart Association.
“Given the ease of administration, less intensive monitoring, low cost, and safety profile of dual antiplatelet therapy, the current findings support the use of dual antiplatelet in this population,” Dr. Nguyen concluded.
In a comment on the trial, Pooja Khatri, MD, professor of neurology at the University of Cincinnati, and lead investigator of the previous PRISMS study of tissue plasminogen activator (tPA) or alteplase in mild stroke, said the results reinforced the current recommendations of giving dual antiplatelet therapy but not alteplase to these patients.
Noting that the standard of care is now to give dual antiplatelet therapy to these patients, Dr. Khatri said: “These data reassure that this remains the right way to go.”
She added that her take-home message from the study would be: “Keep giving dual antiplatelet therapy, and we may be doing more harm than good with alteplase in this patient population.”
Introducing her presentation, Dr. Nguyen explained that mild ischemic stroke, defined as having a National Institutes of Health Stroke Scale (NIHSS) score of 5 or less, comprises half of ischemic stroke patients in the United States. But the benefit of thrombolysis in patients with minor ischemic stroke that is not disabling is unknown.
A subgroup analysis of one of the major thrombolysis trials (IST-3) found that a higher proportion of patients with mild ischemic stroke who were treated within 3 hours of symptom onset were alive and independent at 6 months if they had been given thrombolysis (84%), compared to 65% in the control group who received standard medical treatment.
This led to the first randomized trial (PRISMS) dedicated to patients with mild nondisabling stroke, which found that alteplase given within 3 hours of symptom onset did not increase the likelihood of a good functional outcome at 90 days in comparison with single-agent aspirin. The study was unfortunately terminated early for administrative reasons, and no definitive conclusions could be drawn on the basis of these results, Dr. Nguyen reported.
In 2018, the American Heart Association/American Stroke Association guidelines indicated that for patients who present within 3 hours of symptom onset with mild ischemic stroke that was judged to be nondisabling, thrombolysis with intravenous alteplase could be considered, she noted.
In the meantime, dual antiplatelet therapy was shown to be safe and effective in the POINT and CHANCE trials in patients presenting with minor stroke within 12 or 24 hours, and the CHANCE trial also found a benefit in reducing recurrent stroke that was most effective in the first 2 weeks.
The current ARAMIS trial was therefore conducted to evaluate dual antiplatelet therapy in comparison with thrombolysis for patients with acute minor stroke (NIHSS 5 or less) who presented within 4.5 hours of symptom onset and were without clearly disabling deficit.
The trial was conducted in 38 hospitals in China and included 760 patients (median NIHSS score of 2) who were randomly assigned to receive intravenous alteplase at the standard dose of 0.9 mg/kg, followed by guideline-based antiplatelet treatment, or dual antiplatelet therapy (clopidogrel 300 mg plus 100 mg aspirin loading dose followed by 10 to 14 days of aspirin 100 mg and clopidogrel 75 mg).
The trial was designed to assess noninferiority of dual antiplatelet therapy to alteplase with noninferiority margin of –4.5%.
In the modified intention-to-treat analysis, which included 722 patients, the primary outcome (excellent functional outcome, defined as a Modified Rankin Scale score of 0 or 1 at 90 days) occurred in 93.8% of patients in the dual antiplatelet therapy group and in 91.4% of the alteplase group. This gave a difference of 2.4%, which fell within the limits for noninferiority (P = .0002 for noninferiority test).
“Therefore, this was a positive trial,” Dr. Nguyen stated.
About 20% of patients crossed over from the dual antiplatelet group to the thrombolysis group, and about 16% of patients crossed over from the thrombolysis group to the dual antiplatelet group. But a per-protocol and an “as treated” analysis showed results similar to those of the main intention-to-treat analysis.
Secondary outcomes were largely similar between the two groups other than early neurologic deterioration, which was less common in the dual antiplatelet therapy group.
In terms of safety, symptomatic intracranial hemorrhage occurred in 0.3% (1/369) in the dual antiplatelet group and in 0.9% (3/350) in the alteplase group, a nonsignificant difference.
Events of “any bleeding” occurred in more patients in the thrombolysis group (5.4%) than in the dual antiplatelet therapy group (1.6%), and this difference was significant (P = .01).
Subgroup analysis showed a trend toward benefit of alteplase for patients with higher NIHSS score at baseline (NIHSS > 3). Otherwise, the other subgroups looked similar to the main results.
Dr. Nguyen pointed out one limitation of the study – that dual antiplatelet therapy was updated to standard treatment in this target population in the 2019 AHA/ASA guidelines.
In her discussion of the study, Dr. Khatri suggested that the ARAMIS results were what might have been expected.
“Dual antiplatelet therapy is designed to prevent stroke. Even in the POINT trial, dual antiplatelet therapy showed no effect on 90-day functional outcome. It was really about prevention. The PRISMS trial suggested that alteplase was also unlikely to improve 90-day functional outcome in this population of patients with mild and not clearly disabling stroke. So, it is not surprising that dual antiplatelet therapy was noninferior to alteplase for 90-day functional outcome for both those reasons,” she explained.
“That being said, while designed as a noninferiority study, it is interesting to note that alteplase again showed no evidence of treatment effect compared to antiplatelet therapy, affirming what was observed in the prematurely terminated PRISMS trial,” Dr. Khatri added.
In a discussion of the study at an ISC 2023 highlights session, ISC program chair Tudor Jovin, MD, Cooper Neurological Institute, Cherry Hill, N.J., said: “This is very important data and it’s actually the first completed trial that examines this question.”
But, he added, “I think we need to refine our knowledge about what a nondisabling stroke actually is. You could argue that every stroke is disabling. I think we need more clarity on this definition, as in practice, many clinicians still give tPA on account of these mild strokes still being disabling.”
The ARAMIS trial was funded by the National Key R&D Program of China and the Science and Technology Project Plan of Liaoning Province. Dr. Nguyen reports research support from Medtronic that was not related to the current study.
A version of this article first appeared on Medscape.com.
in the ARAMIS trial.
The trial was presented by Thanh Nguyen, MD, Boston Medical Center, at the International Stroke Conference presented by the American Stroke Association, a division of the American Heart Association.
“Given the ease of administration, less intensive monitoring, low cost, and safety profile of dual antiplatelet therapy, the current findings support the use of dual antiplatelet in this population,” Dr. Nguyen concluded.
In a comment on the trial, Pooja Khatri, MD, professor of neurology at the University of Cincinnati, and lead investigator of the previous PRISMS study of tissue plasminogen activator (tPA) or alteplase in mild stroke, said the results reinforced the current recommendations of giving dual antiplatelet therapy but not alteplase to these patients.
Noting that the standard of care is now to give dual antiplatelet therapy to these patients, Dr. Khatri said: “These data reassure that this remains the right way to go.”
She added that her take-home message from the study would be: “Keep giving dual antiplatelet therapy, and we may be doing more harm than good with alteplase in this patient population.”
Introducing her presentation, Dr. Nguyen explained that mild ischemic stroke, defined as having a National Institutes of Health Stroke Scale (NIHSS) score of 5 or less, comprises half of ischemic stroke patients in the United States. But the benefit of thrombolysis in patients with minor ischemic stroke that is not disabling is unknown.
A subgroup analysis of one of the major thrombolysis trials (IST-3) found that a higher proportion of patients with mild ischemic stroke who were treated within 3 hours of symptom onset were alive and independent at 6 months if they had been given thrombolysis (84%), compared to 65% in the control group who received standard medical treatment.
This led to the first randomized trial (PRISMS) dedicated to patients with mild nondisabling stroke, which found that alteplase given within 3 hours of symptom onset did not increase the likelihood of a good functional outcome at 90 days in comparison with single-agent aspirin. The study was unfortunately terminated early for administrative reasons, and no definitive conclusions could be drawn on the basis of these results, Dr. Nguyen reported.
In 2018, the American Heart Association/American Stroke Association guidelines indicated that for patients who present within 3 hours of symptom onset with mild ischemic stroke that was judged to be nondisabling, thrombolysis with intravenous alteplase could be considered, she noted.
In the meantime, dual antiplatelet therapy was shown to be safe and effective in the POINT and CHANCE trials in patients presenting with minor stroke within 12 or 24 hours, and the CHANCE trial also found a benefit in reducing recurrent stroke that was most effective in the first 2 weeks.
The current ARAMIS trial was therefore conducted to evaluate dual antiplatelet therapy in comparison with thrombolysis for patients with acute minor stroke (NIHSS 5 or less) who presented within 4.5 hours of symptom onset and were without clearly disabling deficit.
The trial was conducted in 38 hospitals in China and included 760 patients (median NIHSS score of 2) who were randomly assigned to receive intravenous alteplase at the standard dose of 0.9 mg/kg, followed by guideline-based antiplatelet treatment, or dual antiplatelet therapy (clopidogrel 300 mg plus 100 mg aspirin loading dose followed by 10 to 14 days of aspirin 100 mg and clopidogrel 75 mg).
The trial was designed to assess noninferiority of dual antiplatelet therapy to alteplase with noninferiority margin of –4.5%.
In the modified intention-to-treat analysis, which included 722 patients, the primary outcome (excellent functional outcome, defined as a Modified Rankin Scale score of 0 or 1 at 90 days) occurred in 93.8% of patients in the dual antiplatelet therapy group and in 91.4% of the alteplase group. This gave a difference of 2.4%, which fell within the limits for noninferiority (P = .0002 for noninferiority test).
“Therefore, this was a positive trial,” Dr. Nguyen stated.
About 20% of patients crossed over from the dual antiplatelet group to the thrombolysis group, and about 16% of patients crossed over from the thrombolysis group to the dual antiplatelet group. But a per-protocol and an “as treated” analysis showed results similar to those of the main intention-to-treat analysis.
Secondary outcomes were largely similar between the two groups other than early neurologic deterioration, which was less common in the dual antiplatelet therapy group.
In terms of safety, symptomatic intracranial hemorrhage occurred in 0.3% (1/369) in the dual antiplatelet group and in 0.9% (3/350) in the alteplase group, a nonsignificant difference.
Events of “any bleeding” occurred in more patients in the thrombolysis group (5.4%) than in the dual antiplatelet therapy group (1.6%), and this difference was significant (P = .01).
Subgroup analysis showed a trend toward benefit of alteplase for patients with higher NIHSS score at baseline (NIHSS > 3). Otherwise, the other subgroups looked similar to the main results.
Dr. Nguyen pointed out one limitation of the study – that dual antiplatelet therapy was updated to standard treatment in this target population in the 2019 AHA/ASA guidelines.
In her discussion of the study, Dr. Khatri suggested that the ARAMIS results were what might have been expected.
“Dual antiplatelet therapy is designed to prevent stroke. Even in the POINT trial, dual antiplatelet therapy showed no effect on 90-day functional outcome. It was really about prevention. The PRISMS trial suggested that alteplase was also unlikely to improve 90-day functional outcome in this population of patients with mild and not clearly disabling stroke. So, it is not surprising that dual antiplatelet therapy was noninferior to alteplase for 90-day functional outcome for both those reasons,” she explained.
“That being said, while designed as a noninferiority study, it is interesting to note that alteplase again showed no evidence of treatment effect compared to antiplatelet therapy, affirming what was observed in the prematurely terminated PRISMS trial,” Dr. Khatri added.
In a discussion of the study at an ISC 2023 highlights session, ISC program chair Tudor Jovin, MD, Cooper Neurological Institute, Cherry Hill, N.J., said: “This is very important data and it’s actually the first completed trial that examines this question.”
But, he added, “I think we need to refine our knowledge about what a nondisabling stroke actually is. You could argue that every stroke is disabling. I think we need more clarity on this definition, as in practice, many clinicians still give tPA on account of these mild strokes still being disabling.”
The ARAMIS trial was funded by the National Key R&D Program of China and the Science and Technology Project Plan of Liaoning Province. Dr. Nguyen reports research support from Medtronic that was not related to the current study.
A version of this article first appeared on Medscape.com.
in the ARAMIS trial.
The trial was presented by Thanh Nguyen, MD, Boston Medical Center, at the International Stroke Conference presented by the American Stroke Association, a division of the American Heart Association.
“Given the ease of administration, less intensive monitoring, low cost, and safety profile of dual antiplatelet therapy, the current findings support the use of dual antiplatelet in this population,” Dr. Nguyen concluded.
In a comment on the trial, Pooja Khatri, MD, professor of neurology at the University of Cincinnati, and lead investigator of the previous PRISMS study of tissue plasminogen activator (tPA) or alteplase in mild stroke, said the results reinforced the current recommendations of giving dual antiplatelet therapy but not alteplase to these patients.
Noting that the standard of care is now to give dual antiplatelet therapy to these patients, Dr. Khatri said: “These data reassure that this remains the right way to go.”
She added that her take-home message from the study would be: “Keep giving dual antiplatelet therapy, and we may be doing more harm than good with alteplase in this patient population.”
Introducing her presentation, Dr. Nguyen explained that mild ischemic stroke, defined as having a National Institutes of Health Stroke Scale (NIHSS) score of 5 or less, comprises half of ischemic stroke patients in the United States. But the benefit of thrombolysis in patients with minor ischemic stroke that is not disabling is unknown.
A subgroup analysis of one of the major thrombolysis trials (IST-3) found that a higher proportion of patients with mild ischemic stroke who were treated within 3 hours of symptom onset were alive and independent at 6 months if they had been given thrombolysis (84%), compared to 65% in the control group who received standard medical treatment.
This led to the first randomized trial (PRISMS) dedicated to patients with mild nondisabling stroke, which found that alteplase given within 3 hours of symptom onset did not increase the likelihood of a good functional outcome at 90 days in comparison with single-agent aspirin. The study was unfortunately terminated early for administrative reasons, and no definitive conclusions could be drawn on the basis of these results, Dr. Nguyen reported.
In 2018, the American Heart Association/American Stroke Association guidelines indicated that for patients who present within 3 hours of symptom onset with mild ischemic stroke that was judged to be nondisabling, thrombolysis with intravenous alteplase could be considered, she noted.
In the meantime, dual antiplatelet therapy was shown to be safe and effective in the POINT and CHANCE trials in patients presenting with minor stroke within 12 or 24 hours, and the CHANCE trial also found a benefit in reducing recurrent stroke that was most effective in the first 2 weeks.
The current ARAMIS trial was therefore conducted to evaluate dual antiplatelet therapy in comparison with thrombolysis for patients with acute minor stroke (NIHSS 5 or less) who presented within 4.5 hours of symptom onset and were without clearly disabling deficit.
The trial was conducted in 38 hospitals in China and included 760 patients (median NIHSS score of 2) who were randomly assigned to receive intravenous alteplase at the standard dose of 0.9 mg/kg, followed by guideline-based antiplatelet treatment, or dual antiplatelet therapy (clopidogrel 300 mg plus 100 mg aspirin loading dose followed by 10 to 14 days of aspirin 100 mg and clopidogrel 75 mg).
The trial was designed to assess noninferiority of dual antiplatelet therapy to alteplase with noninferiority margin of –4.5%.
In the modified intention-to-treat analysis, which included 722 patients, the primary outcome (excellent functional outcome, defined as a Modified Rankin Scale score of 0 or 1 at 90 days) occurred in 93.8% of patients in the dual antiplatelet therapy group and in 91.4% of the alteplase group. This gave a difference of 2.4%, which fell within the limits for noninferiority (P = .0002 for noninferiority test).
“Therefore, this was a positive trial,” Dr. Nguyen stated.
About 20% of patients crossed over from the dual antiplatelet group to the thrombolysis group, and about 16% of patients crossed over from the thrombolysis group to the dual antiplatelet group. But a per-protocol and an “as treated” analysis showed results similar to those of the main intention-to-treat analysis.
Secondary outcomes were largely similar between the two groups other than early neurologic deterioration, which was less common in the dual antiplatelet therapy group.
In terms of safety, symptomatic intracranial hemorrhage occurred in 0.3% (1/369) in the dual antiplatelet group and in 0.9% (3/350) in the alteplase group, a nonsignificant difference.
Events of “any bleeding” occurred in more patients in the thrombolysis group (5.4%) than in the dual antiplatelet therapy group (1.6%), and this difference was significant (P = .01).
Subgroup analysis showed a trend toward benefit of alteplase for patients with higher NIHSS score at baseline (NIHSS > 3). Otherwise, the other subgroups looked similar to the main results.
Dr. Nguyen pointed out one limitation of the study – that dual antiplatelet therapy was updated to standard treatment in this target population in the 2019 AHA/ASA guidelines.
In her discussion of the study, Dr. Khatri suggested that the ARAMIS results were what might have been expected.
“Dual antiplatelet therapy is designed to prevent stroke. Even in the POINT trial, dual antiplatelet therapy showed no effect on 90-day functional outcome. It was really about prevention. The PRISMS trial suggested that alteplase was also unlikely to improve 90-day functional outcome in this population of patients with mild and not clearly disabling stroke. So, it is not surprising that dual antiplatelet therapy was noninferior to alteplase for 90-day functional outcome for both those reasons,” she explained.
“That being said, while designed as a noninferiority study, it is interesting to note that alteplase again showed no evidence of treatment effect compared to antiplatelet therapy, affirming what was observed in the prematurely terminated PRISMS trial,” Dr. Khatri added.
In a discussion of the study at an ISC 2023 highlights session, ISC program chair Tudor Jovin, MD, Cooper Neurological Institute, Cherry Hill, N.J., said: “This is very important data and it’s actually the first completed trial that examines this question.”
But, he added, “I think we need to refine our knowledge about what a nondisabling stroke actually is. You could argue that every stroke is disabling. I think we need more clarity on this definition, as in practice, many clinicians still give tPA on account of these mild strokes still being disabling.”
The ARAMIS trial was funded by the National Key R&D Program of China and the Science and Technology Project Plan of Liaoning Province. Dr. Nguyen reports research support from Medtronic that was not related to the current study.
A version of this article first appeared on Medscape.com.
FROM ISC 2023
Diabetes drug tied to lower dementia risk
new research suggests.
Overall, in a large cohort study from South Korea, patients who took pioglitazone were 16% less likely to develop dementia over an average of 10 years than peers who did not take the drug.
However, the dementia risk reduction was 54% among those with ischemic heart disease and 43% among those with a history of stroke.
“Our study was to see the association between pioglitazone use and incidence of dementia, not how (with what mechanisms) this drug can suppress dementia pathology,” coinvestigator Eosu Kim, MD, PhD, Yonsei University, Seoul, South Korea, said in an interview.
However, “as we found this drug is more effective in diabetic patients who have blood circulation problems in the heart or brain than in those without such problems, we speculate that pioglitazone’s antidementia action may be related to improving blood vessel’s health,” Dr. Kim said.
This finding suggests that pioglitazone could be used as a personalized treatment approach for dementia prevention in this subgroup of patients with diabetes, the researchers noted.
The results were published online in Neurology.
Dose-response relationship
Risk for dementia is doubled in adults with T2DM, the investigators wrote. Prior studies have suggested that pioglitazone may protect against dementia, as well as a first or recurrent stroke, in patients with T2DM.
This led Dr. Kim and colleagues to examine the effects of pioglitazone on dementia risk overall and in relation to stroke and ischemic heart disease.
Using the national Korean health database, the researchers identified 91,218 adults aged 50 and older with new-onset T2DM who did not have dementia. A total of 3,467 were treated with pioglitazone.
Pioglitazone exposure was defined as a total cumulative daily dose of 90 or more calculated from all dispensations during 4 years after T2DM diagnosis, with outcomes assessed after this period.
Over an average of 10 years, 8.3% of pioglitazone users developed dementia, compared with 10.0% of nonusers.
There was a statistically significant 16% lower risk for developing all-cause dementia among pioglitazone users than among nonusers (adjusted hazard ratio, 0.84; 95% confidence interval, 0.75-0.95).
A dose-response relationship was evident; pioglitazone users who received the highest cumulative daily dose were at lower risk for dementia (aHR, 0.72; 95% CI, 0.55-0.94).
Several limitations
The reduced risk for dementia was more pronounced among patients who used pioglitazone for 4 years in comparison with patients who did not use the drug (aHR, 0.63; 95% CI, 0.44-0.90).
The apparent protective effect of pioglitazone with regard to dementia was greater among those with a history of ischemic heart disease (aHR, 0.46; 95% CI, 0.24-0.90) or stroke (aHR, 0.57; 95% CI, 0.38-0.86) before diabetes diagnosis.
The incidence of stroke was also reduced with pioglitazone use (aHR, 0.81; 95% CI, 0.66-1.0).
“These results provide valuable information on who could potentially benefit from pioglitazone use for prevention of dementia,” Dr. Kim said in a news release.
However, “the risk and benefit balance of long-term use of this drug to prevent dementia should be prospectively assessed,” he said in an interview.
The researchers cautioned that the study was observational; hence, the reported associations cannot address causal relationships. Also, because of the use of claims data, drug compliance could not be guaranteed, and exposure may have been overestimated.
There is also the potential for selection bias, and no information on apolipoprotein E was available, they noted.
More data needed
In an accompanying editorial, Colleen J. Maxwell, PhD, University of Waterloo (Ont.), and colleagues wrote that the results “not only support previous studies showing the potential cognitive benefit of pioglitazone but also extend our understanding of this benefit through the mediating effect of reducing ischemic stroke.”
However, because of their associated risks, which include fractures, weight gain, heart failure, and bladder cancer, thiazolidinediones are not currently favored in diabetes management guidelines – and their use has significantly declined since the mid to late 2000s, the editorialists noted.
They agreed that it will be important to reassess the risk-benefit profile of pioglitazone in T2DM as additional findings emerge.
They also noted that sodium-glucose cotransporter-2 inhibitors, which have significant cardiovascular and renal benefits and minimal side effects, may also lower the risk for dementia.
“As both pioglitazone and SGLT-2 inhibitors are second-line options for physicians, the current decision would easily be in favor of SGLT-2 inhibitors given their safety profile,” Dr. Maxwell and colleagues wrote.
For now, pioglitazone “should not be used to prevent dementia in patients with T2DM,” they concluded.
The study was supported by grants from the National Research Foundation of Korea funded by the Korean government and the Ministry of Health and Welfare. The investigators and editorialists report no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
new research suggests.
Overall, in a large cohort study from South Korea, patients who took pioglitazone were 16% less likely to develop dementia over an average of 10 years than peers who did not take the drug.
However, the dementia risk reduction was 54% among those with ischemic heart disease and 43% among those with a history of stroke.
“Our study was to see the association between pioglitazone use and incidence of dementia, not how (with what mechanisms) this drug can suppress dementia pathology,” coinvestigator Eosu Kim, MD, PhD, Yonsei University, Seoul, South Korea, said in an interview.
However, “as we found this drug is more effective in diabetic patients who have blood circulation problems in the heart or brain than in those without such problems, we speculate that pioglitazone’s antidementia action may be related to improving blood vessel’s health,” Dr. Kim said.
This finding suggests that pioglitazone could be used as a personalized treatment approach for dementia prevention in this subgroup of patients with diabetes, the researchers noted.
The results were published online in Neurology.
Dose-response relationship
Risk for dementia is doubled in adults with T2DM, the investigators wrote. Prior studies have suggested that pioglitazone may protect against dementia, as well as a first or recurrent stroke, in patients with T2DM.
This led Dr. Kim and colleagues to examine the effects of pioglitazone on dementia risk overall and in relation to stroke and ischemic heart disease.
Using the national Korean health database, the researchers identified 91,218 adults aged 50 and older with new-onset T2DM who did not have dementia. A total of 3,467 were treated with pioglitazone.
Pioglitazone exposure was defined as a total cumulative daily dose of 90 or more calculated from all dispensations during 4 years after T2DM diagnosis, with outcomes assessed after this period.
Over an average of 10 years, 8.3% of pioglitazone users developed dementia, compared with 10.0% of nonusers.
There was a statistically significant 16% lower risk for developing all-cause dementia among pioglitazone users than among nonusers (adjusted hazard ratio, 0.84; 95% confidence interval, 0.75-0.95).
A dose-response relationship was evident; pioglitazone users who received the highest cumulative daily dose were at lower risk for dementia (aHR, 0.72; 95% CI, 0.55-0.94).
Several limitations
The reduced risk for dementia was more pronounced among patients who used pioglitazone for 4 years in comparison with patients who did not use the drug (aHR, 0.63; 95% CI, 0.44-0.90).
The apparent protective effect of pioglitazone with regard to dementia was greater among those with a history of ischemic heart disease (aHR, 0.46; 95% CI, 0.24-0.90) or stroke (aHR, 0.57; 95% CI, 0.38-0.86) before diabetes diagnosis.
The incidence of stroke was also reduced with pioglitazone use (aHR, 0.81; 95% CI, 0.66-1.0).
“These results provide valuable information on who could potentially benefit from pioglitazone use for prevention of dementia,” Dr. Kim said in a news release.
However, “the risk and benefit balance of long-term use of this drug to prevent dementia should be prospectively assessed,” he said in an interview.
The researchers cautioned that the study was observational; hence, the reported associations cannot address causal relationships. Also, because of the use of claims data, drug compliance could not be guaranteed, and exposure may have been overestimated.
There is also the potential for selection bias, and no information on apolipoprotein E was available, they noted.
More data needed
In an accompanying editorial, Colleen J. Maxwell, PhD, University of Waterloo (Ont.), and colleagues wrote that the results “not only support previous studies showing the potential cognitive benefit of pioglitazone but also extend our understanding of this benefit through the mediating effect of reducing ischemic stroke.”
However, because of their associated risks, which include fractures, weight gain, heart failure, and bladder cancer, thiazolidinediones are not currently favored in diabetes management guidelines – and their use has significantly declined since the mid to late 2000s, the editorialists noted.
They agreed that it will be important to reassess the risk-benefit profile of pioglitazone in T2DM as additional findings emerge.
They also noted that sodium-glucose cotransporter-2 inhibitors, which have significant cardiovascular and renal benefits and minimal side effects, may also lower the risk for dementia.
“As both pioglitazone and SGLT-2 inhibitors are second-line options for physicians, the current decision would easily be in favor of SGLT-2 inhibitors given their safety profile,” Dr. Maxwell and colleagues wrote.
For now, pioglitazone “should not be used to prevent dementia in patients with T2DM,” they concluded.
The study was supported by grants from the National Research Foundation of Korea funded by the Korean government and the Ministry of Health and Welfare. The investigators and editorialists report no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
new research suggests.
Overall, in a large cohort study from South Korea, patients who took pioglitazone were 16% less likely to develop dementia over an average of 10 years than peers who did not take the drug.
However, the dementia risk reduction was 54% among those with ischemic heart disease and 43% among those with a history of stroke.
“Our study was to see the association between pioglitazone use and incidence of dementia, not how (with what mechanisms) this drug can suppress dementia pathology,” coinvestigator Eosu Kim, MD, PhD, Yonsei University, Seoul, South Korea, said in an interview.
However, “as we found this drug is more effective in diabetic patients who have blood circulation problems in the heart or brain than in those without such problems, we speculate that pioglitazone’s antidementia action may be related to improving blood vessel’s health,” Dr. Kim said.
This finding suggests that pioglitazone could be used as a personalized treatment approach for dementia prevention in this subgroup of patients with diabetes, the researchers noted.
The results were published online in Neurology.
Dose-response relationship
Risk for dementia is doubled in adults with T2DM, the investigators wrote. Prior studies have suggested that pioglitazone may protect against dementia, as well as a first or recurrent stroke, in patients with T2DM.
This led Dr. Kim and colleagues to examine the effects of pioglitazone on dementia risk overall and in relation to stroke and ischemic heart disease.
Using the national Korean health database, the researchers identified 91,218 adults aged 50 and older with new-onset T2DM who did not have dementia. A total of 3,467 were treated with pioglitazone.
Pioglitazone exposure was defined as a total cumulative daily dose of 90 or more calculated from all dispensations during 4 years after T2DM diagnosis, with outcomes assessed after this period.
Over an average of 10 years, 8.3% of pioglitazone users developed dementia, compared with 10.0% of nonusers.
There was a statistically significant 16% lower risk for developing all-cause dementia among pioglitazone users than among nonusers (adjusted hazard ratio, 0.84; 95% confidence interval, 0.75-0.95).
A dose-response relationship was evident; pioglitazone users who received the highest cumulative daily dose were at lower risk for dementia (aHR, 0.72; 95% CI, 0.55-0.94).
Several limitations
The reduced risk for dementia was more pronounced among patients who used pioglitazone for 4 years in comparison with patients who did not use the drug (aHR, 0.63; 95% CI, 0.44-0.90).
The apparent protective effect of pioglitazone with regard to dementia was greater among those with a history of ischemic heart disease (aHR, 0.46; 95% CI, 0.24-0.90) or stroke (aHR, 0.57; 95% CI, 0.38-0.86) before diabetes diagnosis.
The incidence of stroke was also reduced with pioglitazone use (aHR, 0.81; 95% CI, 0.66-1.0).
“These results provide valuable information on who could potentially benefit from pioglitazone use for prevention of dementia,” Dr. Kim said in a news release.
However, “the risk and benefit balance of long-term use of this drug to prevent dementia should be prospectively assessed,” he said in an interview.
The researchers cautioned that the study was observational; hence, the reported associations cannot address causal relationships. Also, because of the use of claims data, drug compliance could not be guaranteed, and exposure may have been overestimated.
There is also the potential for selection bias, and no information on apolipoprotein E was available, they noted.
More data needed
In an accompanying editorial, Colleen J. Maxwell, PhD, University of Waterloo (Ont.), and colleagues wrote that the results “not only support previous studies showing the potential cognitive benefit of pioglitazone but also extend our understanding of this benefit through the mediating effect of reducing ischemic stroke.”
However, because of their associated risks, which include fractures, weight gain, heart failure, and bladder cancer, thiazolidinediones are not currently favored in diabetes management guidelines – and their use has significantly declined since the mid to late 2000s, the editorialists noted.
They agreed that it will be important to reassess the risk-benefit profile of pioglitazone in T2DM as additional findings emerge.
They also noted that sodium-glucose cotransporter-2 inhibitors, which have significant cardiovascular and renal benefits and minimal side effects, may also lower the risk for dementia.
“As both pioglitazone and SGLT-2 inhibitors are second-line options for physicians, the current decision would easily be in favor of SGLT-2 inhibitors given their safety profile,” Dr. Maxwell and colleagues wrote.
For now, pioglitazone “should not be used to prevent dementia in patients with T2DM,” they concluded.
The study was supported by grants from the National Research Foundation of Korea funded by the Korean government and the Ministry of Health and Welfare. The investigators and editorialists report no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
FROM NEUROLOGY