User login
Reports of dysuria and nocturia
The history and findings in this case are suggestive of small cell carcinoma of the prostate (SCCP).
SCCP is a rare and aggressive cancer that comprises 1%–5% of all prostate cancers (if mixed cases with adenocarcinoma are included). Similar to small cell carcinoma of the lung or other small cell primaries, SCCP is characterized by a primary tumor of the prostate gland that expresses small cell morphology and high-grade features, including minimal cytoplasm, nuclear molding, fine chromatin pattern, extensive tumor necrosis and apoptosis, variable tumor giant cells, and a high mitotic rate. Patients often have disproportionally low PSA levels despite having large metastatic burden and visceral disease. Pathologic diagnosis is made on the basis of prostate biopsy using characteristics of small cell tumors and immunohistochemical staining for neuroendocrine markers, such as CD56, chromogranin A, synaptophysin, and neuron-specific enolase.
SCCP arises de novo in approximately 50% of cases; it also occurs in patients with previous or concomitant prostate adenocarcinoma. Patients are often symptomatic at diagnosis because of the extent of the tumor. The aggressive nature and high proliferation rate associated with SCCP result in an increased risk for lytic or blastic bone, visceral, and brain metastases. In addition, paraneoplastic syndromes (eg, the syndrome of inappropriate antidiuretic hormone secretion, Cushing syndrome, and hypercalcemia) frequently occur as a result of the release of peptides.
SCCP metastasizes early in its course and is associated with a poor prognosis. It has a median survival of < 1 year. Fluorodeoxyglucose PET-CT are useful for staging and monitoring treatment response; in addition, given the disease's predilection for brain metastases, MRI of the brain should be considered.
The optimal treatment for patients with metastatic SCCP has not yet been determined. Localized SCCP is treated aggressively, typically with a multimodality approach involving chemotherapy with concurrent or consolidative radiotherapy.
According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), platinum-based combination chemotherapy (cisplatin-etoposide, carboplatin-etoposide, docetaxel-carboplatin, cabazitaxel-carboplatin) is the first-line approach for patients with metastatic disease.
Physicians are also advised to consult the NCCN guidelines for small cell lung cancer because the behavior of SCCP is similar to that of small cell carcinoma of the lung. Immunotherapy with pembrolizumab may be used for platinum-resistant extrapulmonary small cell carcinoma. However, sipuleucel-T is not recommended for patients with SCCP.
Chad R. Tracy, MD, Professor; Director, Minimally Invasive Surgery, Department of Urology, University of Iowa Hospitals and Clinics, Iowa City, Iowa
Chad R. Tracy, MD, has disclosed the following relevant financial relationships:
Serve(d) as a consultant for: CVICO Medical Solutions.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are suggestive of small cell carcinoma of the prostate (SCCP).
SCCP is a rare and aggressive cancer that comprises 1%–5% of all prostate cancers (if mixed cases with adenocarcinoma are included). Similar to small cell carcinoma of the lung or other small cell primaries, SCCP is characterized by a primary tumor of the prostate gland that expresses small cell morphology and high-grade features, including minimal cytoplasm, nuclear molding, fine chromatin pattern, extensive tumor necrosis and apoptosis, variable tumor giant cells, and a high mitotic rate. Patients often have disproportionally low PSA levels despite having large metastatic burden and visceral disease. Pathologic diagnosis is made on the basis of prostate biopsy using characteristics of small cell tumors and immunohistochemical staining for neuroendocrine markers, such as CD56, chromogranin A, synaptophysin, and neuron-specific enolase.
SCCP arises de novo in approximately 50% of cases; it also occurs in patients with previous or concomitant prostate adenocarcinoma. Patients are often symptomatic at diagnosis because of the extent of the tumor. The aggressive nature and high proliferation rate associated with SCCP result in an increased risk for lytic or blastic bone, visceral, and brain metastases. In addition, paraneoplastic syndromes (eg, the syndrome of inappropriate antidiuretic hormone secretion, Cushing syndrome, and hypercalcemia) frequently occur as a result of the release of peptides.
SCCP metastasizes early in its course and is associated with a poor prognosis. It has a median survival of < 1 year. Fluorodeoxyglucose PET-CT are useful for staging and monitoring treatment response; in addition, given the disease's predilection for brain metastases, MRI of the brain should be considered.
The optimal treatment for patients with metastatic SCCP has not yet been determined. Localized SCCP is treated aggressively, typically with a multimodality approach involving chemotherapy with concurrent or consolidative radiotherapy.
According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), platinum-based combination chemotherapy (cisplatin-etoposide, carboplatin-etoposide, docetaxel-carboplatin, cabazitaxel-carboplatin) is the first-line approach for patients with metastatic disease.
Physicians are also advised to consult the NCCN guidelines for small cell lung cancer because the behavior of SCCP is similar to that of small cell carcinoma of the lung. Immunotherapy with pembrolizumab may be used for platinum-resistant extrapulmonary small cell carcinoma. However, sipuleucel-T is not recommended for patients with SCCP.
Chad R. Tracy, MD, Professor; Director, Minimally Invasive Surgery, Department of Urology, University of Iowa Hospitals and Clinics, Iowa City, Iowa
Chad R. Tracy, MD, has disclosed the following relevant financial relationships:
Serve(d) as a consultant for: CVICO Medical Solutions.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are suggestive of small cell carcinoma of the prostate (SCCP).
SCCP is a rare and aggressive cancer that comprises 1%–5% of all prostate cancers (if mixed cases with adenocarcinoma are included). Similar to small cell carcinoma of the lung or other small cell primaries, SCCP is characterized by a primary tumor of the prostate gland that expresses small cell morphology and high-grade features, including minimal cytoplasm, nuclear molding, fine chromatin pattern, extensive tumor necrosis and apoptosis, variable tumor giant cells, and a high mitotic rate. Patients often have disproportionally low PSA levels despite having large metastatic burden and visceral disease. Pathologic diagnosis is made on the basis of prostate biopsy using characteristics of small cell tumors and immunohistochemical staining for neuroendocrine markers, such as CD56, chromogranin A, synaptophysin, and neuron-specific enolase.
SCCP arises de novo in approximately 50% of cases; it also occurs in patients with previous or concomitant prostate adenocarcinoma. Patients are often symptomatic at diagnosis because of the extent of the tumor. The aggressive nature and high proliferation rate associated with SCCP result in an increased risk for lytic or blastic bone, visceral, and brain metastases. In addition, paraneoplastic syndromes (eg, the syndrome of inappropriate antidiuretic hormone secretion, Cushing syndrome, and hypercalcemia) frequently occur as a result of the release of peptides.
SCCP metastasizes early in its course and is associated with a poor prognosis. It has a median survival of < 1 year. Fluorodeoxyglucose PET-CT are useful for staging and monitoring treatment response; in addition, given the disease's predilection for brain metastases, MRI of the brain should be considered.
The optimal treatment for patients with metastatic SCCP has not yet been determined. Localized SCCP is treated aggressively, typically with a multimodality approach involving chemotherapy with concurrent or consolidative radiotherapy.
According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), platinum-based combination chemotherapy (cisplatin-etoposide, carboplatin-etoposide, docetaxel-carboplatin, cabazitaxel-carboplatin) is the first-line approach for patients with metastatic disease.
Physicians are also advised to consult the NCCN guidelines for small cell lung cancer because the behavior of SCCP is similar to that of small cell carcinoma of the lung. Immunotherapy with pembrolizumab may be used for platinum-resistant extrapulmonary small cell carcinoma. However, sipuleucel-T is not recommended for patients with SCCP.
Chad R. Tracy, MD, Professor; Director, Minimally Invasive Surgery, Department of Urology, University of Iowa Hospitals and Clinics, Iowa City, Iowa
Chad R. Tracy, MD, has disclosed the following relevant financial relationships:
Serve(d) as a consultant for: CVICO Medical Solutions.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
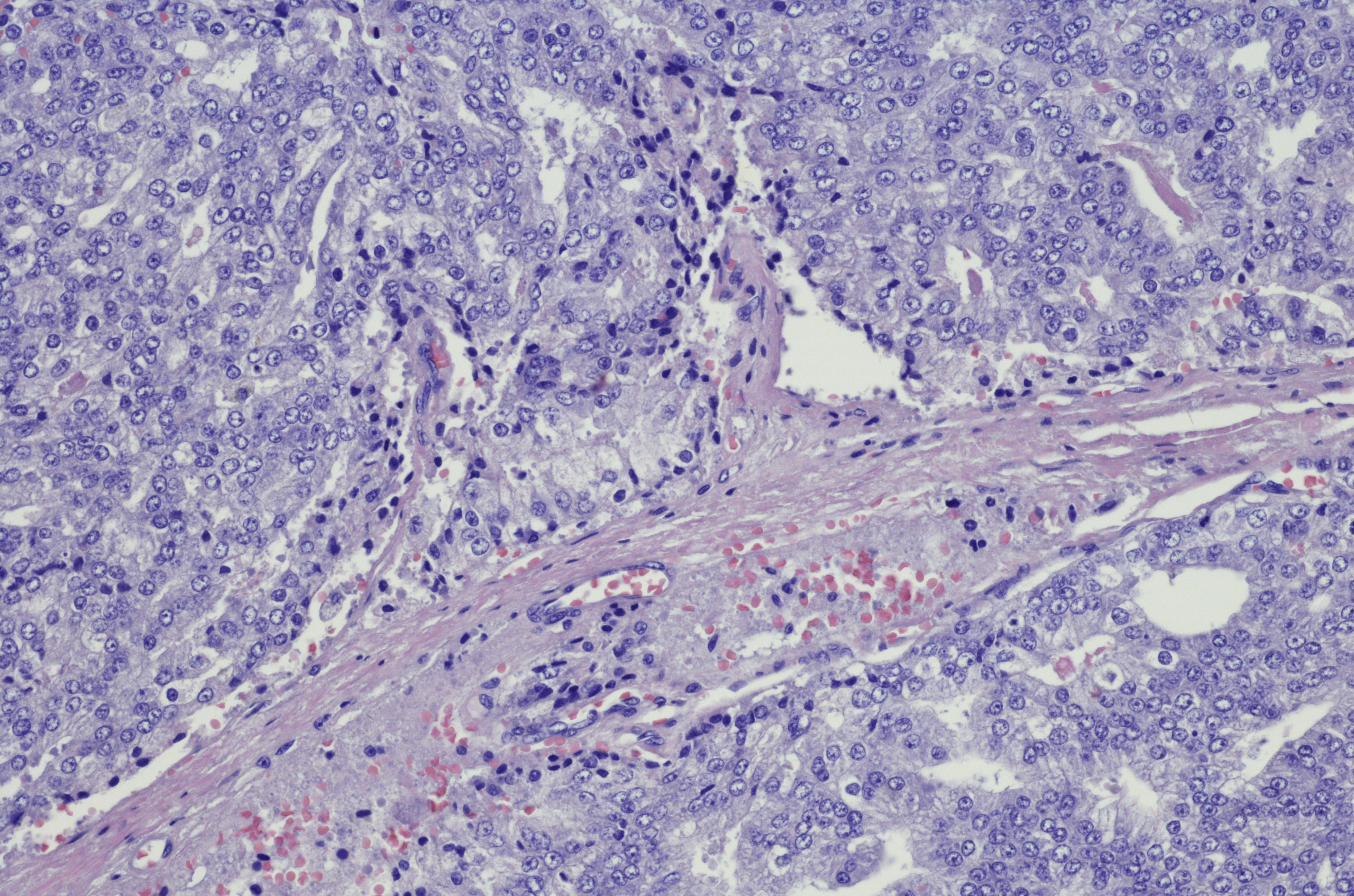
A 69-year-old nonsmoking African American man presents with reports of dysuria, nocturia, and unintentional weight loss. He reveals no other lower urinary tract symptoms, pelvic pain, night sweats, back pain, or excessive fatigue. Digital rectal exam reveals an enlarged prostate with a firm, irregular nodule at the right side of the gland. Laboratory tests reveal a prostate-specific antigen (PSA) level of 2.22 ng/mL; a comprehensive metabolic panel and CBC are within normal limits. The patient is 6 ft 1 in and weighs 187 lb.
A transrectal ultrasound-guided prostate biopsy is performed. Histologic examination reveals immunoreactivity for the neuroendocrine markers synaptophysin, chromogranin A, and expression of transcription factor 1. A proliferation of small cells (> 4 lymphocytes in diameter) is noted, with scant cytoplasm, poorly defined borders, finely granular salt-and-pepper chromatin, inconspicuous nucleoli, and a high mitotic count. Evidence of perineural invasion is noted.
A little education goes a long way for advocacy
If you are reading this, you probably know what a PBM is or at least know what the acronym stands for (pharmacy benefit manager). But don’t be surprised if many people, even physicians, still have never heard the term or don’t know (or really care) what it stands for. This past weekend, I saw how important even a little bit of education on this seemingly boring topic can create passionate advocates in less than an hour.

On March 10, the Coalition of State Rheumatology Organizations had its Fellows Conference on real-life topics such as evaluating a contract, malpractice troubleshooting, getting out of debt and creating wealth, and learning about the latest coding issues, among others. We had a record-breaking number of fellows in attendance this year. I gave a presentation on formulary construction (list of drugs that insurance will cover), what tools are used to keep the formulary profitable, and what are the potential consequences for patients with the use of these tools, such as step therapy and nonmedical switching. Remember that if you have a condition requiring an expensive drug that is not covered on the formulary, you will not have access to it unless it is given to you for free by some type of assistance program, or you happen to be very wealthy.
It was the first time I gave this talk at our Fellows Conference, and I realized fairly quickly that a decent proportion of the audience did not know what PBM stood for, much less the power that PBMs have in setting up the list of expensive drugs that they will pay for. I wasn’t so surprised by how little they knew about the particulars of this topic – for example, that lower-priced medications are often shunned by PBMs because they are not as profitable for the PBM as higher-priced drugs. However, I was very pleasantly surprised at the number of fellows who came to me after my talk with almost as much passion as I have for this topic. Many asked how they could get involved and what they could do right now to support advocacy for their patients. It all seemed to fall in place for them as they began telling me stories of the problems they had in getting medications for their patients – adults and kids alike.
The “meme” on the street is that drug pricing, patient access, and the drug supply channel is “much too complex” for the non-economist to understand. That was not the case at the Fellows Conference. It started off with me moving back and forth across the stage explaining how the system is run by entities whose fiduciary responsibility is to their shareholders, not our patients. I explained the fierce competition, the bidding process, the “rebate equation,” and many stories of egregious policies and behaviors by an oligopoly of health insurers and their powerful PBMs. I repeated over and over that “If you make an expensive drug that is not on the formulary, no one will take to your drug, unless you give it away for free.”
It became clear to the room that the competition among expensive drug makers to get preferred status on the formulary is fierce. I explained how to win that coveted spot on the formulary by legally kicking back the most money, in the form of rebates and fees, to the PBM. Unfortunately, these rebates and fees are generally a percentage of the list price, so often it is the highest-priced drug that wins the coveted spot. I explained that patients get no benefit from the money kicked back to the PBM, and in fact, because their coinsurance is often based on the list price of the drug, patients’ cost share will go up when PBMs pick the drug with the highest price. I gave the example of a major PBM placing a $10,000 brand-name drug on the formulary and excluding the $400 generic version of the same drug. I told them that PBMs call these the “lowest cost” drugs – for them. This made them angry. I also explained to the fellows that these kickbacks are legal because PBMs have “safe harbor” from the antikickback statute. And yes, that made them even angrier. The more I spoke about the harm done to patients both physically and monetarily by utilization management tools such as step therapy and nonmedical switching, the angrier and more passionate they became.
What started as a room full of fellows wondering whether they really were interested in a talk about PBMs and formulary construction turned, in less than an hour, into a room filled with passion and fury: Rheumatology fellows ready to go and fight for their patients. It’s not as complicated as everyone wants you to believe. In that short time, fellows who had walked into that conference hall, not knowing what to expect from me, walked out with a new attitude and passion, hungry for the next step they could take to advocate for their patients. My slogan on Twitter has always been that I will continue to educate and advocate as long as my passion stays ahead of my cynicism. My passion certainly got a boost as I watched the fellows in the conference hall turn into “Rheums for Action” before my eyes.
Dr. Feldman is a rheumatologist in private practice with The Rheumatology Group in New Orleans. She is the CSRO’s Vice President of Advocacy and Government Affairs and its immediate Past President, as well as past chair of the Alliance for Safe Biologic Medicines and a past member of the American College of Rheumatology insurance subcommittee. You can reach her at [email protected].
If you are reading this, you probably know what a PBM is or at least know what the acronym stands for (pharmacy benefit manager). But don’t be surprised if many people, even physicians, still have never heard the term or don’t know (or really care) what it stands for. This past weekend, I saw how important even a little bit of education on this seemingly boring topic can create passionate advocates in less than an hour.
On March 10, the Coalition of State Rheumatology Organizations had its Fellows Conference on real-life topics such as evaluating a contract, malpractice troubleshooting, getting out of debt and creating wealth, and learning about the latest coding issues, among others. We had a record-breaking number of fellows in attendance this year. I gave a presentation on formulary construction (list of drugs that insurance will cover), what tools are used to keep the formulary profitable, and what are the potential consequences for patients with the use of these tools, such as step therapy and nonmedical switching. Remember that if you have a condition requiring an expensive drug that is not covered on the formulary, you will not have access to it unless it is given to you for free by some type of assistance program, or you happen to be very wealthy.
It was the first time I gave this talk at our Fellows Conference, and I realized fairly quickly that a decent proportion of the audience did not know what PBM stood for, much less the power that PBMs have in setting up the list of expensive drugs that they will pay for. I wasn’t so surprised by how little they knew about the particulars of this topic – for example, that lower-priced medications are often shunned by PBMs because they are not as profitable for the PBM as higher-priced drugs. However, I was very pleasantly surprised at the number of fellows who came to me after my talk with almost as much passion as I have for this topic. Many asked how they could get involved and what they could do right now to support advocacy for their patients. It all seemed to fall in place for them as they began telling me stories of the problems they had in getting medications for their patients – adults and kids alike.
The “meme” on the street is that drug pricing, patient access, and the drug supply channel is “much too complex” for the non-economist to understand. That was not the case at the Fellows Conference. It started off with me moving back and forth across the stage explaining how the system is run by entities whose fiduciary responsibility is to their shareholders, not our patients. I explained the fierce competition, the bidding process, the “rebate equation,” and many stories of egregious policies and behaviors by an oligopoly of health insurers and their powerful PBMs. I repeated over and over that “If you make an expensive drug that is not on the formulary, no one will take to your drug, unless you give it away for free.”
It became clear to the room that the competition among expensive drug makers to get preferred status on the formulary is fierce. I explained how to win that coveted spot on the formulary by legally kicking back the most money, in the form of rebates and fees, to the PBM. Unfortunately, these rebates and fees are generally a percentage of the list price, so often it is the highest-priced drug that wins the coveted spot. I explained that patients get no benefit from the money kicked back to the PBM, and in fact, because their coinsurance is often based on the list price of the drug, patients’ cost share will go up when PBMs pick the drug with the highest price. I gave the example of a major PBM placing a $10,000 brand-name drug on the formulary and excluding the $400 generic version of the same drug. I told them that PBMs call these the “lowest cost” drugs – for them. This made them angry. I also explained to the fellows that these kickbacks are legal because PBMs have “safe harbor” from the antikickback statute. And yes, that made them even angrier. The more I spoke about the harm done to patients both physically and monetarily by utilization management tools such as step therapy and nonmedical switching, the angrier and more passionate they became.
What started as a room full of fellows wondering whether they really were interested in a talk about PBMs and formulary construction turned, in less than an hour, into a room filled with passion and fury: Rheumatology fellows ready to go and fight for their patients. It’s not as complicated as everyone wants you to believe. In that short time, fellows who had walked into that conference hall, not knowing what to expect from me, walked out with a new attitude and passion, hungry for the next step they could take to advocate for their patients. My slogan on Twitter has always been that I will continue to educate and advocate as long as my passion stays ahead of my cynicism. My passion certainly got a boost as I watched the fellows in the conference hall turn into “Rheums for Action” before my eyes.
Dr. Feldman is a rheumatologist in private practice with The Rheumatology Group in New Orleans. She is the CSRO’s Vice President of Advocacy and Government Affairs and its immediate Past President, as well as past chair of the Alliance for Safe Biologic Medicines and a past member of the American College of Rheumatology insurance subcommittee. You can reach her at [email protected].
If you are reading this, you probably know what a PBM is or at least know what the acronym stands for (pharmacy benefit manager). But don’t be surprised if many people, even physicians, still have never heard the term or don’t know (or really care) what it stands for. This past weekend, I saw how important even a little bit of education on this seemingly boring topic can create passionate advocates in less than an hour.
On March 10, the Coalition of State Rheumatology Organizations had its Fellows Conference on real-life topics such as evaluating a contract, malpractice troubleshooting, getting out of debt and creating wealth, and learning about the latest coding issues, among others. We had a record-breaking number of fellows in attendance this year. I gave a presentation on formulary construction (list of drugs that insurance will cover), what tools are used to keep the formulary profitable, and what are the potential consequences for patients with the use of these tools, such as step therapy and nonmedical switching. Remember that if you have a condition requiring an expensive drug that is not covered on the formulary, you will not have access to it unless it is given to you for free by some type of assistance program, or you happen to be very wealthy.
It was the first time I gave this talk at our Fellows Conference, and I realized fairly quickly that a decent proportion of the audience did not know what PBM stood for, much less the power that PBMs have in setting up the list of expensive drugs that they will pay for. I wasn’t so surprised by how little they knew about the particulars of this topic – for example, that lower-priced medications are often shunned by PBMs because they are not as profitable for the PBM as higher-priced drugs. However, I was very pleasantly surprised at the number of fellows who came to me after my talk with almost as much passion as I have for this topic. Many asked how they could get involved and what they could do right now to support advocacy for their patients. It all seemed to fall in place for them as they began telling me stories of the problems they had in getting medications for their patients – adults and kids alike.
The “meme” on the street is that drug pricing, patient access, and the drug supply channel is “much too complex” for the non-economist to understand. That was not the case at the Fellows Conference. It started off with me moving back and forth across the stage explaining how the system is run by entities whose fiduciary responsibility is to their shareholders, not our patients. I explained the fierce competition, the bidding process, the “rebate equation,” and many stories of egregious policies and behaviors by an oligopoly of health insurers and their powerful PBMs. I repeated over and over that “If you make an expensive drug that is not on the formulary, no one will take to your drug, unless you give it away for free.”
It became clear to the room that the competition among expensive drug makers to get preferred status on the formulary is fierce. I explained how to win that coveted spot on the formulary by legally kicking back the most money, in the form of rebates and fees, to the PBM. Unfortunately, these rebates and fees are generally a percentage of the list price, so often it is the highest-priced drug that wins the coveted spot. I explained that patients get no benefit from the money kicked back to the PBM, and in fact, because their coinsurance is often based on the list price of the drug, patients’ cost share will go up when PBMs pick the drug with the highest price. I gave the example of a major PBM placing a $10,000 brand-name drug on the formulary and excluding the $400 generic version of the same drug. I told them that PBMs call these the “lowest cost” drugs – for them. This made them angry. I also explained to the fellows that these kickbacks are legal because PBMs have “safe harbor” from the antikickback statute. And yes, that made them even angrier. The more I spoke about the harm done to patients both physically and monetarily by utilization management tools such as step therapy and nonmedical switching, the angrier and more passionate they became.
What started as a room full of fellows wondering whether they really were interested in a talk about PBMs and formulary construction turned, in less than an hour, into a room filled with passion and fury: Rheumatology fellows ready to go and fight for their patients. It’s not as complicated as everyone wants you to believe. In that short time, fellows who had walked into that conference hall, not knowing what to expect from me, walked out with a new attitude and passion, hungry for the next step they could take to advocate for their patients. My slogan on Twitter has always been that I will continue to educate and advocate as long as my passion stays ahead of my cynicism. My passion certainly got a boost as I watched the fellows in the conference hall turn into “Rheums for Action” before my eyes.
Dr. Feldman is a rheumatologist in private practice with The Rheumatology Group in New Orleans. She is the CSRO’s Vice President of Advocacy and Government Affairs and its immediate Past President, as well as past chair of the Alliance for Safe Biologic Medicines and a past member of the American College of Rheumatology insurance subcommittee. You can reach her at [email protected].
Upadacitinib shows positive endoscopic outcomes in Crohn’s disease at 1 year
The findings of this subanalysis come from two phase 3 induction trials (U-EXCEL and U-EXCEED) and one maintenance study (U-ENDURE) of upadacitinib in this patient population.
“Upadacitinib shows large differences relative to placebo in endoscopic response and remission ... in a difficult-to-treat population of patients, the majority of whom had failed an advanced therapy,” lead investigator Brian Feagan, MD, senior scientific director of the GI contract research firm Alimentiv in London, Ontario, said in an interview.
“The absolute magnitude of the finding was unanticipated – a greater treatment effect than might be anticipated for these outcomes compared with other advanced treatments for Crohn’s disease in these higher-risk patients,” he said.
Dr. Feagan presented the research at the annual congress of the European Crohn’s and Colitis Organisation.
Research methodology
At baseline, participants had an average daily stool frequency of 4 or more and/or an abdominal pain score of 2 or greater. They also had a Simple Endoscopic Score for Crohn’s disease of 6 or more, excluding a narrowing component, or a score of 4 or more for isolated ileal Crohn’s disease.
In the treatment induction phase, patients were randomly assigned 2:1, with 674 people receiving 45 mg upadacitinib and 347 taking a placebo once daily for 12 weeks.
Participants who experienced at least a 30% decrease in stool frequency and/or daily abdominal pain scores were enrolled in the maintenance phase of the study. For this phase, patients were randomly assigned again, with 168 receiving 30 mg upadacitinib, 169 receiving 15 mg upadacitinib, and 165 taking a placebo once daily for 52 weeks.
In each induction and maintenance cohort, more than 70% of patients had failed one prior biologic therapy, with failure defined as inadequate response or intolerance. Among those who failed a previous biologic in induction, 96% had also failed prior treatment with an anti–tumor necrosis factor (anti-TNF) inhibitor.
Participants’ mean age was 38-40 years, and 52%-55% were men. Patients who had not failed previous therapy had Crohn’s disease for a median of 6-7 years. In contrast, the prior-failure group had Crohn’s disease for a median of 9-10 years.
Key outcomes
At 12 weeks, endoscopic response among patients who had not failed a prior biologic was 52% in the treatment group versus 16% of the placebo group. In the prior-failure group, endoscopic response was observed in 36% and 5%, respectively.
Endoscopic remission at 12 weeks among patients who had not failed a prior biologic was 36% in the treatment group versus 10% in the placebo group. In the prior-failure group, endoscopic remission was 20% in the treatment group versus 3% in those who took placebo.
Participants in the treatment groups of the 52-week maintenance phase of the study experienced higher endoscopic response and endoscopic remission rates compared with those who received placebo.
Endoscopic response in the group without prior biologic failure was 44% in the 30-mg upadacitinib group, 40% in the 15-mg group, and 18% in the placebo group. Among those with prior biologic failure, endoscopic response was seen in 39% of the 30-mg upadacitinib group, 23% of the 15-mg group, and 4% of the placebo group.
There is a “very striking difference in endoscopic response rates between the high dose and placebo,” Dr. Feagan said. “That difference here is in the response rate. You see dose separation.”
Endoscopic remission among those without prior biologic failure was observed in 34% of the 30-mg upadacitinib group, 27% of the 15-mg group, and 16% of the placebo group. Among those with prior biologic failure, endoscopic remission was seen in 27% of the 30-mg upadacitinib group, 16% of the 15-mg group, and 2% of the placebo group.
The results show “a clear advantage for the 30-mg dose versus the 15-mg in the maintenance component, especially in patients who had failed an advanced therapy,” Dr. Feagan said.
Safety signals
Upadacitinib was well tolerated in the induction and maintenance phases, and no new safety risks were observed compared with the known safety profile of the drug, the researchers noted.
For example, during the induction studies, the rate of any adverse event among patients without prior biologic failure was 60% in the 45-mg upadacitinib group and 53% in the placebo group. Among those who failed a prior biologic, the rates were 67% in the 45-mg upadacitinib group and 66% in the placebo group.
The adverse events were “issues that have already been identified with JAK inhibitors, the biochemical abnormalities with CPK [creatine phosphokinase] elevations and transaminase elevations,” Dr. Feagan said.
There were no cases of herpes zoster among patients who received placebo compared with five cases in the 45-mg upadacitinib group without prior biologic failure and 10 cases in the prior biologic failure group.
“The zoster signal is there even at induction with the 45-mg dose versus placebo,” Dr. Feagan said.
‘Encouraging’ results
The study indicates that upadacitinib is effective in improving endoscopic outcomes for patients with Crohn’s disease, regardless of their prior biologic treatments, Robin L. Dalal, MD, assistant professor of medicine at Vanderbilt University in Nashville, Tenn., said when asked to comment on the study.
“This is important because, as the treatment landscape for Crohn’s disease has expanded, sequencing of therapies has become more complex,” added Dr. Dalal, who was not involved in the research. “For upadacitinib in Crohn’s disease, prior biologic use may not be a factor in endoscopic response rates.”
The findings are “very encouraging for physicians and practitioners who treat IBD [inflammatory bowel disease] patients,” Maithili Chitnavis, MD, of the inflammatory bowel disease section at Atrium Health Gastroenterology in Charlotte, N.C., said when asked for comment.
“We clearly care about how patients feel overall, but endoscopic and histologic outcomes are important to investigate because we want to ensure there is internal healing to prevent a lot of the longstanding complications of Crohn’s disease, such as malignancy, strictures, fistulizing/penetrating disease, and need for surgery,” said Dr. Chitnavis, who was not involved with the study.
Upadacitinib is an oral agent, which distinguishes it from the injectable or infusion-based biologic therapies for Crohn’s disease, Dr. Chitnavis noted.
The finding that the medication works in patients with or without prior biologic failure is important, she said.
“With its anticipated ... approval for Crohn’s disease [by the Food and Drug Administration], it is expected that patients will have had to have demonstrated a lack of or loss of response to another biologic, specifically in the anti-TNF category (for example, infliximab, adalimumab, certolizumab) prior to starting upadacitinib due to concerns of potential side effects associated with the class of medications to which it belongs,” Dr. Chitnavis said. “Therefore, it makes it even more relevant to know how patients who have failed a prior biologic respond to this therapy.”
Dr. Feagan has reported serving as a consultant and speaker for AbbVie. Dr. Dalal has reported being a consultant for AbbVie in 2021. Dr. Chitnavis has reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
The findings of this subanalysis come from two phase 3 induction trials (U-EXCEL and U-EXCEED) and one maintenance study (U-ENDURE) of upadacitinib in this patient population.
“Upadacitinib shows large differences relative to placebo in endoscopic response and remission ... in a difficult-to-treat population of patients, the majority of whom had failed an advanced therapy,” lead investigator Brian Feagan, MD, senior scientific director of the GI contract research firm Alimentiv in London, Ontario, said in an interview.
“The absolute magnitude of the finding was unanticipated – a greater treatment effect than might be anticipated for these outcomes compared with other advanced treatments for Crohn’s disease in these higher-risk patients,” he said.
Dr. Feagan presented the research at the annual congress of the European Crohn’s and Colitis Organisation.
Research methodology
At baseline, participants had an average daily stool frequency of 4 or more and/or an abdominal pain score of 2 or greater. They also had a Simple Endoscopic Score for Crohn’s disease of 6 or more, excluding a narrowing component, or a score of 4 or more for isolated ileal Crohn’s disease.
In the treatment induction phase, patients were randomly assigned 2:1, with 674 people receiving 45 mg upadacitinib and 347 taking a placebo once daily for 12 weeks.
Participants who experienced at least a 30% decrease in stool frequency and/or daily abdominal pain scores were enrolled in the maintenance phase of the study. For this phase, patients were randomly assigned again, with 168 receiving 30 mg upadacitinib, 169 receiving 15 mg upadacitinib, and 165 taking a placebo once daily for 52 weeks.
In each induction and maintenance cohort, more than 70% of patients had failed one prior biologic therapy, with failure defined as inadequate response or intolerance. Among those who failed a previous biologic in induction, 96% had also failed prior treatment with an anti–tumor necrosis factor (anti-TNF) inhibitor.
Participants’ mean age was 38-40 years, and 52%-55% were men. Patients who had not failed previous therapy had Crohn’s disease for a median of 6-7 years. In contrast, the prior-failure group had Crohn’s disease for a median of 9-10 years.
Key outcomes
At 12 weeks, endoscopic response among patients who had not failed a prior biologic was 52% in the treatment group versus 16% of the placebo group. In the prior-failure group, endoscopic response was observed in 36% and 5%, respectively.
Endoscopic remission at 12 weeks among patients who had not failed a prior biologic was 36% in the treatment group versus 10% in the placebo group. In the prior-failure group, endoscopic remission was 20% in the treatment group versus 3% in those who took placebo.
Participants in the treatment groups of the 52-week maintenance phase of the study experienced higher endoscopic response and endoscopic remission rates compared with those who received placebo.
Endoscopic response in the group without prior biologic failure was 44% in the 30-mg upadacitinib group, 40% in the 15-mg group, and 18% in the placebo group. Among those with prior biologic failure, endoscopic response was seen in 39% of the 30-mg upadacitinib group, 23% of the 15-mg group, and 4% of the placebo group.
There is a “very striking difference in endoscopic response rates between the high dose and placebo,” Dr. Feagan said. “That difference here is in the response rate. You see dose separation.”
Endoscopic remission among those without prior biologic failure was observed in 34% of the 30-mg upadacitinib group, 27% of the 15-mg group, and 16% of the placebo group. Among those with prior biologic failure, endoscopic remission was seen in 27% of the 30-mg upadacitinib group, 16% of the 15-mg group, and 2% of the placebo group.
The results show “a clear advantage for the 30-mg dose versus the 15-mg in the maintenance component, especially in patients who had failed an advanced therapy,” Dr. Feagan said.
Safety signals
Upadacitinib was well tolerated in the induction and maintenance phases, and no new safety risks were observed compared with the known safety profile of the drug, the researchers noted.
For example, during the induction studies, the rate of any adverse event among patients without prior biologic failure was 60% in the 45-mg upadacitinib group and 53% in the placebo group. Among those who failed a prior biologic, the rates were 67% in the 45-mg upadacitinib group and 66% in the placebo group.
The adverse events were “issues that have already been identified with JAK inhibitors, the biochemical abnormalities with CPK [creatine phosphokinase] elevations and transaminase elevations,” Dr. Feagan said.
There were no cases of herpes zoster among patients who received placebo compared with five cases in the 45-mg upadacitinib group without prior biologic failure and 10 cases in the prior biologic failure group.
“The zoster signal is there even at induction with the 45-mg dose versus placebo,” Dr. Feagan said.
‘Encouraging’ results
The study indicates that upadacitinib is effective in improving endoscopic outcomes for patients with Crohn’s disease, regardless of their prior biologic treatments, Robin L. Dalal, MD, assistant professor of medicine at Vanderbilt University in Nashville, Tenn., said when asked to comment on the study.
“This is important because, as the treatment landscape for Crohn’s disease has expanded, sequencing of therapies has become more complex,” added Dr. Dalal, who was not involved in the research. “For upadacitinib in Crohn’s disease, prior biologic use may not be a factor in endoscopic response rates.”
The findings are “very encouraging for physicians and practitioners who treat IBD [inflammatory bowel disease] patients,” Maithili Chitnavis, MD, of the inflammatory bowel disease section at Atrium Health Gastroenterology in Charlotte, N.C., said when asked for comment.
“We clearly care about how patients feel overall, but endoscopic and histologic outcomes are important to investigate because we want to ensure there is internal healing to prevent a lot of the longstanding complications of Crohn’s disease, such as malignancy, strictures, fistulizing/penetrating disease, and need for surgery,” said Dr. Chitnavis, who was not involved with the study.
Upadacitinib is an oral agent, which distinguishes it from the injectable or infusion-based biologic therapies for Crohn’s disease, Dr. Chitnavis noted.
The finding that the medication works in patients with or without prior biologic failure is important, she said.
“With its anticipated ... approval for Crohn’s disease [by the Food and Drug Administration], it is expected that patients will have had to have demonstrated a lack of or loss of response to another biologic, specifically in the anti-TNF category (for example, infliximab, adalimumab, certolizumab) prior to starting upadacitinib due to concerns of potential side effects associated with the class of medications to which it belongs,” Dr. Chitnavis said. “Therefore, it makes it even more relevant to know how patients who have failed a prior biologic respond to this therapy.”
Dr. Feagan has reported serving as a consultant and speaker for AbbVie. Dr. Dalal has reported being a consultant for AbbVie in 2021. Dr. Chitnavis has reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
The findings of this subanalysis come from two phase 3 induction trials (U-EXCEL and U-EXCEED) and one maintenance study (U-ENDURE) of upadacitinib in this patient population.
“Upadacitinib shows large differences relative to placebo in endoscopic response and remission ... in a difficult-to-treat population of patients, the majority of whom had failed an advanced therapy,” lead investigator Brian Feagan, MD, senior scientific director of the GI contract research firm Alimentiv in London, Ontario, said in an interview.
“The absolute magnitude of the finding was unanticipated – a greater treatment effect than might be anticipated for these outcomes compared with other advanced treatments for Crohn’s disease in these higher-risk patients,” he said.
Dr. Feagan presented the research at the annual congress of the European Crohn’s and Colitis Organisation.
Research methodology
At baseline, participants had an average daily stool frequency of 4 or more and/or an abdominal pain score of 2 or greater. They also had a Simple Endoscopic Score for Crohn’s disease of 6 or more, excluding a narrowing component, or a score of 4 or more for isolated ileal Crohn’s disease.
In the treatment induction phase, patients were randomly assigned 2:1, with 674 people receiving 45 mg upadacitinib and 347 taking a placebo once daily for 12 weeks.
Participants who experienced at least a 30% decrease in stool frequency and/or daily abdominal pain scores were enrolled in the maintenance phase of the study. For this phase, patients were randomly assigned again, with 168 receiving 30 mg upadacitinib, 169 receiving 15 mg upadacitinib, and 165 taking a placebo once daily for 52 weeks.
In each induction and maintenance cohort, more than 70% of patients had failed one prior biologic therapy, with failure defined as inadequate response or intolerance. Among those who failed a previous biologic in induction, 96% had also failed prior treatment with an anti–tumor necrosis factor (anti-TNF) inhibitor.
Participants’ mean age was 38-40 years, and 52%-55% were men. Patients who had not failed previous therapy had Crohn’s disease for a median of 6-7 years. In contrast, the prior-failure group had Crohn’s disease for a median of 9-10 years.
Key outcomes
At 12 weeks, endoscopic response among patients who had not failed a prior biologic was 52% in the treatment group versus 16% of the placebo group. In the prior-failure group, endoscopic response was observed in 36% and 5%, respectively.
Endoscopic remission at 12 weeks among patients who had not failed a prior biologic was 36% in the treatment group versus 10% in the placebo group. In the prior-failure group, endoscopic remission was 20% in the treatment group versus 3% in those who took placebo.
Participants in the treatment groups of the 52-week maintenance phase of the study experienced higher endoscopic response and endoscopic remission rates compared with those who received placebo.
Endoscopic response in the group without prior biologic failure was 44% in the 30-mg upadacitinib group, 40% in the 15-mg group, and 18% in the placebo group. Among those with prior biologic failure, endoscopic response was seen in 39% of the 30-mg upadacitinib group, 23% of the 15-mg group, and 4% of the placebo group.
There is a “very striking difference in endoscopic response rates between the high dose and placebo,” Dr. Feagan said. “That difference here is in the response rate. You see dose separation.”
Endoscopic remission among those without prior biologic failure was observed in 34% of the 30-mg upadacitinib group, 27% of the 15-mg group, and 16% of the placebo group. Among those with prior biologic failure, endoscopic remission was seen in 27% of the 30-mg upadacitinib group, 16% of the 15-mg group, and 2% of the placebo group.
The results show “a clear advantage for the 30-mg dose versus the 15-mg in the maintenance component, especially in patients who had failed an advanced therapy,” Dr. Feagan said.
Safety signals
Upadacitinib was well tolerated in the induction and maintenance phases, and no new safety risks were observed compared with the known safety profile of the drug, the researchers noted.
For example, during the induction studies, the rate of any adverse event among patients without prior biologic failure was 60% in the 45-mg upadacitinib group and 53% in the placebo group. Among those who failed a prior biologic, the rates were 67% in the 45-mg upadacitinib group and 66% in the placebo group.
The adverse events were “issues that have already been identified with JAK inhibitors, the biochemical abnormalities with CPK [creatine phosphokinase] elevations and transaminase elevations,” Dr. Feagan said.
There were no cases of herpes zoster among patients who received placebo compared with five cases in the 45-mg upadacitinib group without prior biologic failure and 10 cases in the prior biologic failure group.
“The zoster signal is there even at induction with the 45-mg dose versus placebo,” Dr. Feagan said.
‘Encouraging’ results
The study indicates that upadacitinib is effective in improving endoscopic outcomes for patients with Crohn’s disease, regardless of their prior biologic treatments, Robin L. Dalal, MD, assistant professor of medicine at Vanderbilt University in Nashville, Tenn., said when asked to comment on the study.
“This is important because, as the treatment landscape for Crohn’s disease has expanded, sequencing of therapies has become more complex,” added Dr. Dalal, who was not involved in the research. “For upadacitinib in Crohn’s disease, prior biologic use may not be a factor in endoscopic response rates.”
The findings are “very encouraging for physicians and practitioners who treat IBD [inflammatory bowel disease] patients,” Maithili Chitnavis, MD, of the inflammatory bowel disease section at Atrium Health Gastroenterology in Charlotte, N.C., said when asked for comment.
“We clearly care about how patients feel overall, but endoscopic and histologic outcomes are important to investigate because we want to ensure there is internal healing to prevent a lot of the longstanding complications of Crohn’s disease, such as malignancy, strictures, fistulizing/penetrating disease, and need for surgery,” said Dr. Chitnavis, who was not involved with the study.
Upadacitinib is an oral agent, which distinguishes it from the injectable or infusion-based biologic therapies for Crohn’s disease, Dr. Chitnavis noted.
The finding that the medication works in patients with or without prior biologic failure is important, she said.
“With its anticipated ... approval for Crohn’s disease [by the Food and Drug Administration], it is expected that patients will have had to have demonstrated a lack of or loss of response to another biologic, specifically in the anti-TNF category (for example, infliximab, adalimumab, certolizumab) prior to starting upadacitinib due to concerns of potential side effects associated with the class of medications to which it belongs,” Dr. Chitnavis said. “Therefore, it makes it even more relevant to know how patients who have failed a prior biologic respond to this therapy.”
Dr. Feagan has reported serving as a consultant and speaker for AbbVie. Dr. Dalal has reported being a consultant for AbbVie in 2021. Dr. Chitnavis has reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
FROM ECCO 2023
From PERT to AI, high-risk PE care evolves
In 2012, a small group of specialists, consisting of a critical care pulmonologist, cardiologist, cardiac surgeon, and vascular specialist, at Massachusetts General Hospital, Boston, met to Monday morning quarterback an acute pulmonary embolism case that didn’t go as well as they’d hoped. They came up with a concept known as the pulmonary embolism response team – PERT for short – an idea that soon took hold in other centers and served as the vanguard to other innovative approaches to managing critical care patients with PE, which is the third-leading cause of cardiovascular death in the United States (Intern Emerg Med. 2023. doi: 10.1007/s11739-022-03180-w).
Three years later the PERT Consortium came together, which today has 102 members, according to the organization’s website (www.pertconsortium.org), and members in South America, Europe, Asia, and Australia. Since then, and apps to expedite diagnosis and treatment. The PERT Consortium, meanwhile, is in the process of creating the PE Centers of Excellence program to certify centers that meet certain requirements.

“Part of the reason we recognized that a discussion across specialties was important was because there weren’t the large clinical trials that could tell us exactly what to do for any given case,” said Christopher Kabrhel, MD, MPH, director of the Center for Vascular Emergencies at Mass General and a professor at Harvard Medical School in Boston, who assembled that formative meeting. “Without a clear basis in data, it was really important to have all the different specialists weigh in and give their perspective and talk about what was the best approach for the patient’s care.”
Filling data gaps
Some of those data gaps persist today, Dr. Kabrhel said. “It’s precisely that lack of head-to-head data that existed in 2012, and to a great extent still exists today, that led us to create this system.” The American Heart Association just this January issued a scientific statement on surgical management and mechanical circulatory support in high-risk PE (Circulation. 2023;147:e628-47).
But the intervening research has been uneven. The Pulmonary Embolism Thrombolysis (PEITHO) trial in 2014 evaluated systemic thrombolysis and anticoagulation alone (N Engl J Med. 2014;370:1402-11), but head-to-head studies of catheter-directed thrombolysis (CDT), which was just emerging in 2012, and either systemic thrombolysis or anticoagulation have been lacking, Dr. Kabrhel said. The Hi-PEITHO trial in high-risk PE patients is evaluating ultrasound-guided CDT plus anticoagulation vs. anticoagulation alone (Am Heart J. 2022:251:43-54), but it isn’t complete.
“The therapeutic landscape for PE is evolving incredibly rapidly,” he said. “When we first started PERT we were just starting to see CDT. Since then, we’ve seen several new thrombolytic catheters come onto the market, but there’s also been a proliferation of suction embolectomy catheters and we’ve seen a potentially larger role for surgery and the use of ECMO [extracorporeal membrane oxygenation] or cardiac bypass to bridge patients to definitive therapy. With the rapid evolution and the seemingly daily addition of new therapeutic options, I think the need for PERT is only increasing.”
A recent study out of the University of Michigan reported that the PERT there led to a decrease in the use of advanced therapies given to acute PE patients without reducing mortality or extending hospital stays (Thromb Res. 2023;221:73-8). A study in Spain reported that patients with high-risk and intermediate high-risk PE who had PERT-coordinated care had half the 12-month mortality rate of non-PERT counterparts, 9% vs. 22.2% (P = .02) (Med Clin [Barc]. 2023;S0025-7753(23)00017-9). And a 2021 study at University Hospitals in Cleveland reported that PERT-managed PE patients had a 60% lower rate of adverse outcomes at 90 days than non–PERT-managed patients (J Invasive Cardiol. 2021;33:E173-E180).

Nelish Ardeshna, MD, MA, the lead author of the Michigan study, said the PERT there was formed in 2017. Besides the multispecialty team that can be summoned to a teleconference on short notice, the protocol includes having at least one noninvasive specialist, such as a cardiologist or hospitalist, and one interventionalist, such as a radiologist, always on call. The PERT gets activated through the paging system after a hospital or emergency department physician identifies a suspected or established high-risk PE.
“High-risk PE patients can present in all settings, including the emergency department, ICU, surgical floor, or medical floor,” said Dr. Ardeshna, an internal medicine resident. “Management for these patients is equally varied from anticoagulation to systemic thrombolytics. Not all providers may be familiar with current guidelines to select the optimal therapy for high-risk pulmonary embolism patients. PERT aims to bridge that gap by providing a multidisciplinary discussion with PE specialists that can help identify the correct therapeutic options for optimal outcomes.”

At Cleveland Clinic, where the PERT has been in place since 2012, the PERT can consist of six to eight different specialties and involve up to 15 providers on a conference call, said Leben Tefera, MD, a vascular specialist and head of the PERT team there.
“Each patient will come in and have certain comorbidities,” Dr. Tefera said. “The unfortunate thing about a majority of the PEs that we see, in particular ones [in patients] that are very sick and require inpatient treatment, is that they don’t really fit into a box; you can’t come up with one kind of generic care routine or care path that treats the majority of patients with PE.”
Evolving to follow-up care
As the PERT protocol led to better inpatient outcomes, the teams became more aware that discharged PE patients were struggling with mental health and other quality-of-life issues – symptoms that have been understudied, according to a protocol Dr. Tefera coauthored for a prospective observational study of psychological distress symptoms in PE survivors. By contrast, the protocol noted, these symptoms have been studied extensively in myocardial infarction and stroke patients (Res Pract Thromb Hemost. 2023. doi: 10.1016/j.rpth.2023.10045). Other studies have found that 35%-50% of patients reported mental health symptoms 3 months after PE (Chest. 2021;159:2428-38; Qual Life Res. 2019;28:2111-24).
“A lot of physicians have known it for quite some time, but it wasn’t really until the last couple of years that physicians started saying psychological stress is something that we need to quantify and that we need to actually treat, that we actually need to address,” Dr. Tefera said. That led Dr. Tefera and his Cleveland Clinic PERT colleagues to set up a follow-up clinic for PE patients.
At their follow-up visits, patients complete validated questionnaires about anxiety, depression, fear of recurrence, PE-specific quality of life, and posttraumatic stress disorder. “If they flag as positive, we give them a referral to an in-house psychologist,” he said. “One thing I can report is that patients absolutely, positively love this, because it’s something that they are all experiencing that a lot of physicians just aren’t addressing.”
Artificial intelligence emerges
At the University of Pittsburgh Medical Center, the PERT has started evaluating artificial intelligence to aid in PE diagnosis. Belinda Rivera-Lebron, MD, director of the acute and chronic embolism program at Pitt, explained that the AI protocol hasn’t been adopted yet, but the concept is to have a platform that’s compatible with the hospital system’s electronic medical record.

She described how AI would work once the PERT is activated. “Once the patient goes through the CT scanner, within 60 seconds of that scan being completed, the scan gets uploaded into the cloud and the app or the platform is able to tell you whether there is PE present or absent, and whether there is right ventricle dilation on that scan. This is even before you probably even think about opening up the computer to look at the scan, and even before radiology opens up the scan to read,” she said. “It’s so fast.”
The idea is to send the scans rapidly to the PERT. “It will send you a text, a notification on your phone that will tell you Mr. Smith is PE positive,” Dr. Rivera-Lebron said. “Then you open it and you are able to scroll through the CT scan in your phone. So, it’s really remarkable.”
Clinical trials worth watching
Meanwhile, a number of clinical trials have started to enroll patients, or will soon, that Dr. Rivera-Lebron said are worth paying attention to.
PEITHO-3 is a randomized, placebo-controlled trial with long-term follow-up comparing the efficacy of a reduced-dose alteplase regimen or standard heparin anticoagulation in patients with intermediate to high-risk PE (Thromb Haemost. 2022;122:867-66).
PEERLESS is a prospective randomized trial comparing mechanical thrombectomy and CDT (ClinicalTrials.gov identifier NCT05111613).
PE-Thrombus Removal with Catheter-directed Therapy (PE-TRACT) is an open-label Phase 3 trial comparing anticoagulation and CDT that’s not yet recruiting (ClinicalTrials.gov identifier NCT05591118).
FlowTriever for Acute Massive Pulmonary Embolism (FLAME) is a prospective cohort study evaluating a clot-retrieving device in high-risk PE patients (ClinicalTrials.gov identifier NCT04795167).
When completed and published, these trials could provide PERTs more evidence for their decision-making.
Dr. Ardeshna and Dr. Tefera have no relevant relationships to disclose. Dr. Rivera-Lebron disclosed relationships with INARI Catheter and Johnson & Johnson. Dr. Kabrhel disclosed relationships with Bristol Myers Squibb and Pfizer.
In 2012, a small group of specialists, consisting of a critical care pulmonologist, cardiologist, cardiac surgeon, and vascular specialist, at Massachusetts General Hospital, Boston, met to Monday morning quarterback an acute pulmonary embolism case that didn’t go as well as they’d hoped. They came up with a concept known as the pulmonary embolism response team – PERT for short – an idea that soon took hold in other centers and served as the vanguard to other innovative approaches to managing critical care patients with PE, which is the third-leading cause of cardiovascular death in the United States (Intern Emerg Med. 2023. doi: 10.1007/s11739-022-03180-w).
Three years later the PERT Consortium came together, which today has 102 members, according to the organization’s website (www.pertconsortium.org), and members in South America, Europe, Asia, and Australia. Since then, and apps to expedite diagnosis and treatment. The PERT Consortium, meanwhile, is in the process of creating the PE Centers of Excellence program to certify centers that meet certain requirements.
“Part of the reason we recognized that a discussion across specialties was important was because there weren’t the large clinical trials that could tell us exactly what to do for any given case,” said Christopher Kabrhel, MD, MPH, director of the Center for Vascular Emergencies at Mass General and a professor at Harvard Medical School in Boston, who assembled that formative meeting. “Without a clear basis in data, it was really important to have all the different specialists weigh in and give their perspective and talk about what was the best approach for the patient’s care.”
Filling data gaps
Some of those data gaps persist today, Dr. Kabrhel said. “It’s precisely that lack of head-to-head data that existed in 2012, and to a great extent still exists today, that led us to create this system.” The American Heart Association just this January issued a scientific statement on surgical management and mechanical circulatory support in high-risk PE (Circulation. 2023;147:e628-47).
But the intervening research has been uneven. The Pulmonary Embolism Thrombolysis (PEITHO) trial in 2014 evaluated systemic thrombolysis and anticoagulation alone (N Engl J Med. 2014;370:1402-11), but head-to-head studies of catheter-directed thrombolysis (CDT), which was just emerging in 2012, and either systemic thrombolysis or anticoagulation have been lacking, Dr. Kabrhel said. The Hi-PEITHO trial in high-risk PE patients is evaluating ultrasound-guided CDT plus anticoagulation vs. anticoagulation alone (Am Heart J. 2022:251:43-54), but it isn’t complete.
“The therapeutic landscape for PE is evolving incredibly rapidly,” he said. “When we first started PERT we were just starting to see CDT. Since then, we’ve seen several new thrombolytic catheters come onto the market, but there’s also been a proliferation of suction embolectomy catheters and we’ve seen a potentially larger role for surgery and the use of ECMO [extracorporeal membrane oxygenation] or cardiac bypass to bridge patients to definitive therapy. With the rapid evolution and the seemingly daily addition of new therapeutic options, I think the need for PERT is only increasing.”
A recent study out of the University of Michigan reported that the PERT there led to a decrease in the use of advanced therapies given to acute PE patients without reducing mortality or extending hospital stays (Thromb Res. 2023;221:73-8). A study in Spain reported that patients with high-risk and intermediate high-risk PE who had PERT-coordinated care had half the 12-month mortality rate of non-PERT counterparts, 9% vs. 22.2% (P = .02) (Med Clin [Barc]. 2023;S0025-7753(23)00017-9). And a 2021 study at University Hospitals in Cleveland reported that PERT-managed PE patients had a 60% lower rate of adverse outcomes at 90 days than non–PERT-managed patients (J Invasive Cardiol. 2021;33:E173-E180).
Nelish Ardeshna, MD, MA, the lead author of the Michigan study, said the PERT there was formed in 2017. Besides the multispecialty team that can be summoned to a teleconference on short notice, the protocol includes having at least one noninvasive specialist, such as a cardiologist or hospitalist, and one interventionalist, such as a radiologist, always on call. The PERT gets activated through the paging system after a hospital or emergency department physician identifies a suspected or established high-risk PE.
“High-risk PE patients can present in all settings, including the emergency department, ICU, surgical floor, or medical floor,” said Dr. Ardeshna, an internal medicine resident. “Management for these patients is equally varied from anticoagulation to systemic thrombolytics. Not all providers may be familiar with current guidelines to select the optimal therapy for high-risk pulmonary embolism patients. PERT aims to bridge that gap by providing a multidisciplinary discussion with PE specialists that can help identify the correct therapeutic options for optimal outcomes.”
At Cleveland Clinic, where the PERT has been in place since 2012, the PERT can consist of six to eight different specialties and involve up to 15 providers on a conference call, said Leben Tefera, MD, a vascular specialist and head of the PERT team there.
“Each patient will come in and have certain comorbidities,” Dr. Tefera said. “The unfortunate thing about a majority of the PEs that we see, in particular ones [in patients] that are very sick and require inpatient treatment, is that they don’t really fit into a box; you can’t come up with one kind of generic care routine or care path that treats the majority of patients with PE.”
Evolving to follow-up care
As the PERT protocol led to better inpatient outcomes, the teams became more aware that discharged PE patients were struggling with mental health and other quality-of-life issues – symptoms that have been understudied, according to a protocol Dr. Tefera coauthored for a prospective observational study of psychological distress symptoms in PE survivors. By contrast, the protocol noted, these symptoms have been studied extensively in myocardial infarction and stroke patients (Res Pract Thromb Hemost. 2023. doi: 10.1016/j.rpth.2023.10045). Other studies have found that 35%-50% of patients reported mental health symptoms 3 months after PE (Chest. 2021;159:2428-38; Qual Life Res. 2019;28:2111-24).
“A lot of physicians have known it for quite some time, but it wasn’t really until the last couple of years that physicians started saying psychological stress is something that we need to quantify and that we need to actually treat, that we actually need to address,” Dr. Tefera said. That led Dr. Tefera and his Cleveland Clinic PERT colleagues to set up a follow-up clinic for PE patients.
At their follow-up visits, patients complete validated questionnaires about anxiety, depression, fear of recurrence, PE-specific quality of life, and posttraumatic stress disorder. “If they flag as positive, we give them a referral to an in-house psychologist,” he said. “One thing I can report is that patients absolutely, positively love this, because it’s something that they are all experiencing that a lot of physicians just aren’t addressing.”
Artificial intelligence emerges
At the University of Pittsburgh Medical Center, the PERT has started evaluating artificial intelligence to aid in PE diagnosis. Belinda Rivera-Lebron, MD, director of the acute and chronic embolism program at Pitt, explained that the AI protocol hasn’t been adopted yet, but the concept is to have a platform that’s compatible with the hospital system’s electronic medical record.
She described how AI would work once the PERT is activated. “Once the patient goes through the CT scanner, within 60 seconds of that scan being completed, the scan gets uploaded into the cloud and the app or the platform is able to tell you whether there is PE present or absent, and whether there is right ventricle dilation on that scan. This is even before you probably even think about opening up the computer to look at the scan, and even before radiology opens up the scan to read,” she said. “It’s so fast.”
The idea is to send the scans rapidly to the PERT. “It will send you a text, a notification on your phone that will tell you Mr. Smith is PE positive,” Dr. Rivera-Lebron said. “Then you open it and you are able to scroll through the CT scan in your phone. So, it’s really remarkable.”
Clinical trials worth watching
Meanwhile, a number of clinical trials have started to enroll patients, or will soon, that Dr. Rivera-Lebron said are worth paying attention to.
PEITHO-3 is a randomized, placebo-controlled trial with long-term follow-up comparing the efficacy of a reduced-dose alteplase regimen or standard heparin anticoagulation in patients with intermediate to high-risk PE (Thromb Haemost. 2022;122:867-66).
PEERLESS is a prospective randomized trial comparing mechanical thrombectomy and CDT (ClinicalTrials.gov identifier NCT05111613).
PE-Thrombus Removal with Catheter-directed Therapy (PE-TRACT) is an open-label Phase 3 trial comparing anticoagulation and CDT that’s not yet recruiting (ClinicalTrials.gov identifier NCT05591118).
FlowTriever for Acute Massive Pulmonary Embolism (FLAME) is a prospective cohort study evaluating a clot-retrieving device in high-risk PE patients (ClinicalTrials.gov identifier NCT04795167).
When completed and published, these trials could provide PERTs more evidence for their decision-making.
Dr. Ardeshna and Dr. Tefera have no relevant relationships to disclose. Dr. Rivera-Lebron disclosed relationships with INARI Catheter and Johnson & Johnson. Dr. Kabrhel disclosed relationships with Bristol Myers Squibb and Pfizer.
In 2012, a small group of specialists, consisting of a critical care pulmonologist, cardiologist, cardiac surgeon, and vascular specialist, at Massachusetts General Hospital, Boston, met to Monday morning quarterback an acute pulmonary embolism case that didn’t go as well as they’d hoped. They came up with a concept known as the pulmonary embolism response team – PERT for short – an idea that soon took hold in other centers and served as the vanguard to other innovative approaches to managing critical care patients with PE, which is the third-leading cause of cardiovascular death in the United States (Intern Emerg Med. 2023. doi: 10.1007/s11739-022-03180-w).
Three years later the PERT Consortium came together, which today has 102 members, according to the organization’s website (www.pertconsortium.org), and members in South America, Europe, Asia, and Australia. Since then, and apps to expedite diagnosis and treatment. The PERT Consortium, meanwhile, is in the process of creating the PE Centers of Excellence program to certify centers that meet certain requirements.
“Part of the reason we recognized that a discussion across specialties was important was because there weren’t the large clinical trials that could tell us exactly what to do for any given case,” said Christopher Kabrhel, MD, MPH, director of the Center for Vascular Emergencies at Mass General and a professor at Harvard Medical School in Boston, who assembled that formative meeting. “Without a clear basis in data, it was really important to have all the different specialists weigh in and give their perspective and talk about what was the best approach for the patient’s care.”
Filling data gaps
Some of those data gaps persist today, Dr. Kabrhel said. “It’s precisely that lack of head-to-head data that existed in 2012, and to a great extent still exists today, that led us to create this system.” The American Heart Association just this January issued a scientific statement on surgical management and mechanical circulatory support in high-risk PE (Circulation. 2023;147:e628-47).
But the intervening research has been uneven. The Pulmonary Embolism Thrombolysis (PEITHO) trial in 2014 evaluated systemic thrombolysis and anticoagulation alone (N Engl J Med. 2014;370:1402-11), but head-to-head studies of catheter-directed thrombolysis (CDT), which was just emerging in 2012, and either systemic thrombolysis or anticoagulation have been lacking, Dr. Kabrhel said. The Hi-PEITHO trial in high-risk PE patients is evaluating ultrasound-guided CDT plus anticoagulation vs. anticoagulation alone (Am Heart J. 2022:251:43-54), but it isn’t complete.
“The therapeutic landscape for PE is evolving incredibly rapidly,” he said. “When we first started PERT we were just starting to see CDT. Since then, we’ve seen several new thrombolytic catheters come onto the market, but there’s also been a proliferation of suction embolectomy catheters and we’ve seen a potentially larger role for surgery and the use of ECMO [extracorporeal membrane oxygenation] or cardiac bypass to bridge patients to definitive therapy. With the rapid evolution and the seemingly daily addition of new therapeutic options, I think the need for PERT is only increasing.”
A recent study out of the University of Michigan reported that the PERT there led to a decrease in the use of advanced therapies given to acute PE patients without reducing mortality or extending hospital stays (Thromb Res. 2023;221:73-8). A study in Spain reported that patients with high-risk and intermediate high-risk PE who had PERT-coordinated care had half the 12-month mortality rate of non-PERT counterparts, 9% vs. 22.2% (P = .02) (Med Clin [Barc]. 2023;S0025-7753(23)00017-9). And a 2021 study at University Hospitals in Cleveland reported that PERT-managed PE patients had a 60% lower rate of adverse outcomes at 90 days than non–PERT-managed patients (J Invasive Cardiol. 2021;33:E173-E180).
Nelish Ardeshna, MD, MA, the lead author of the Michigan study, said the PERT there was formed in 2017. Besides the multispecialty team that can be summoned to a teleconference on short notice, the protocol includes having at least one noninvasive specialist, such as a cardiologist or hospitalist, and one interventionalist, such as a radiologist, always on call. The PERT gets activated through the paging system after a hospital or emergency department physician identifies a suspected or established high-risk PE.
“High-risk PE patients can present in all settings, including the emergency department, ICU, surgical floor, or medical floor,” said Dr. Ardeshna, an internal medicine resident. “Management for these patients is equally varied from anticoagulation to systemic thrombolytics. Not all providers may be familiar with current guidelines to select the optimal therapy for high-risk pulmonary embolism patients. PERT aims to bridge that gap by providing a multidisciplinary discussion with PE specialists that can help identify the correct therapeutic options for optimal outcomes.”
At Cleveland Clinic, where the PERT has been in place since 2012, the PERT can consist of six to eight different specialties and involve up to 15 providers on a conference call, said Leben Tefera, MD, a vascular specialist and head of the PERT team there.
“Each patient will come in and have certain comorbidities,” Dr. Tefera said. “The unfortunate thing about a majority of the PEs that we see, in particular ones [in patients] that are very sick and require inpatient treatment, is that they don’t really fit into a box; you can’t come up with one kind of generic care routine or care path that treats the majority of patients with PE.”
Evolving to follow-up care
As the PERT protocol led to better inpatient outcomes, the teams became more aware that discharged PE patients were struggling with mental health and other quality-of-life issues – symptoms that have been understudied, according to a protocol Dr. Tefera coauthored for a prospective observational study of psychological distress symptoms in PE survivors. By contrast, the protocol noted, these symptoms have been studied extensively in myocardial infarction and stroke patients (Res Pract Thromb Hemost. 2023. doi: 10.1016/j.rpth.2023.10045). Other studies have found that 35%-50% of patients reported mental health symptoms 3 months after PE (Chest. 2021;159:2428-38; Qual Life Res. 2019;28:2111-24).
“A lot of physicians have known it for quite some time, but it wasn’t really until the last couple of years that physicians started saying psychological stress is something that we need to quantify and that we need to actually treat, that we actually need to address,” Dr. Tefera said. That led Dr. Tefera and his Cleveland Clinic PERT colleagues to set up a follow-up clinic for PE patients.
At their follow-up visits, patients complete validated questionnaires about anxiety, depression, fear of recurrence, PE-specific quality of life, and posttraumatic stress disorder. “If they flag as positive, we give them a referral to an in-house psychologist,” he said. “One thing I can report is that patients absolutely, positively love this, because it’s something that they are all experiencing that a lot of physicians just aren’t addressing.”
Artificial intelligence emerges
At the University of Pittsburgh Medical Center, the PERT has started evaluating artificial intelligence to aid in PE diagnosis. Belinda Rivera-Lebron, MD, director of the acute and chronic embolism program at Pitt, explained that the AI protocol hasn’t been adopted yet, but the concept is to have a platform that’s compatible with the hospital system’s electronic medical record.
She described how AI would work once the PERT is activated. “Once the patient goes through the CT scanner, within 60 seconds of that scan being completed, the scan gets uploaded into the cloud and the app or the platform is able to tell you whether there is PE present or absent, and whether there is right ventricle dilation on that scan. This is even before you probably even think about opening up the computer to look at the scan, and even before radiology opens up the scan to read,” she said. “It’s so fast.”
The idea is to send the scans rapidly to the PERT. “It will send you a text, a notification on your phone that will tell you Mr. Smith is PE positive,” Dr. Rivera-Lebron said. “Then you open it and you are able to scroll through the CT scan in your phone. So, it’s really remarkable.”
Clinical trials worth watching
Meanwhile, a number of clinical trials have started to enroll patients, or will soon, that Dr. Rivera-Lebron said are worth paying attention to.
PEITHO-3 is a randomized, placebo-controlled trial with long-term follow-up comparing the efficacy of a reduced-dose alteplase regimen or standard heparin anticoagulation in patients with intermediate to high-risk PE (Thromb Haemost. 2022;122:867-66).
PEERLESS is a prospective randomized trial comparing mechanical thrombectomy and CDT (ClinicalTrials.gov identifier NCT05111613).
PE-Thrombus Removal with Catheter-directed Therapy (PE-TRACT) is an open-label Phase 3 trial comparing anticoagulation and CDT that’s not yet recruiting (ClinicalTrials.gov identifier NCT05591118).
FlowTriever for Acute Massive Pulmonary Embolism (FLAME) is a prospective cohort study evaluating a clot-retrieving device in high-risk PE patients (ClinicalTrials.gov identifier NCT04795167).
When completed and published, these trials could provide PERTs more evidence for their decision-making.
Dr. Ardeshna and Dr. Tefera have no relevant relationships to disclose. Dr. Rivera-Lebron disclosed relationships with INARI Catheter and Johnson & Johnson. Dr. Kabrhel disclosed relationships with Bristol Myers Squibb and Pfizer.
Artificial pancreas ‘superior’ in young kids with type 1 diabetes
A hybrid closed-loop automated insulin delivery system improved time-in-range for blood glucose, compared with standard care, for children with type 1 diabetes in a 13-week trial.
The hybrid closed-loop system, also called automated insulin delivery or artificial pancreas, was composed of a t:slim X2 insulin pump, a Dexcom G6 continuous glucose monitor (CGM), and Control-IQ technology system algorithm software (Tandem Diabetes Care). The system was approved in the United States in 2018 for adults and children as young as 6 years.
Type 1 diabetes treatment is particularly challenging in children younger than 6 because of their small insulin dosing requirements and unpredictable eating and activity habits, lead author R. Paul Wadwa, MD, of the Barbara Davis Center for Diabetes, University of Colorado at Denver, Aurora, and colleagues wrote.
Thus far in the United States, only the Medtronic MiniMed 770G and the Omnipod 5 automated insulin delivery systems are approved for children as young as 2 years, they noted.
In the current study of 102 children with type 1 diabetes aged at least 2 years but younger than 6 years, time-in-range over 13 weeks was higher for those randomized to the hybrid closed-loop system, compared with standard of care; the latter included either an insulin pump or multiple daily injections plus a separate Dexcom G6 CGM.
The hybrid closed-loop system added an average of about 3 hours in ideal blood glucose range over the 13 weeks, compared with no change with standard care.
Moreover, the trial was conducted during the COVID-19 pandemic, necessitating virtual care for most of the study participants. As a result, more than 80% of the training on use of the system and over 90% of all the visits were conducted virtually.
“Successful use of the closed-loop system under these conditions is an important finding that could affect the approach to initiating and monitoring the use of the closed-loop system and expand the use of such systems, particularly in patients living in areas without an endocrinologist but with reliable internet access,” the investigators wrote.
Their findings were published online in the New England Journal of Medicine.
“These results suggest that, in very young children, closed-loop systems are superior to standard care with respect to glucose control,” Daniela Bruttomesso, MD, PhD, of the University of Padua (Italy) wrote in an accompanying editorial.
“Moreover, they show that the closed-loop system can be started remotely in children in this age range, with results that are similar to those obtained when parents or guardians receive face-to-face education about the use of these systems. The closed-loop system used in this trial appeared to be safe and effective.”
Dr. Bruttomesso added: “Although the results were solid, the trial period was only 13 weeks, and there were more unscheduled contacts in the closed-loop group than in the standard care group. In addition, the authors compared a closed-loop system with standard care, rather than in-person initiation of a closed-loop system with remote initiation.”
More time-in-range, no hypoglycemia with automated system
The 102 children were enrolled in the trial between April 28, 2021, and Jan. 13, 2022, at three different U.S. study sites; 68 children were randomized to the closed-loop system and 34 children to standard care. All but one participant completed the 13-week study.
Both groups had virtual or in-person trial visits at 2, 6, and 13 weeks after randomization, and telephone contact at 1 and 10 weeks. Training was virtual for 55 of the 68 children in the closed-loop group (81%). A total of 91% of 407 study visits in the closed-loop and 96% of 204 study visits in the standard-care group were also virtual.
The mean percentage of time spent in target glucose range (70-180 mg/dL) increased from 56.9% at baseline to 69.3% at 13 weeks for the closed-loop group, compared with virtually no change, from 54.9% to 55.9%, in the standard-care group. The mean adjusted difference between the two groups was significant (P < .001).
The closed-loop group also spent significantly less time than the standard-care group with glucose levels above 250 mg/dL during the study period (8.4% vs. 15.0%; P < .001), had lower mean glucose levels (155 vs. 174 mg/dL; P < .001), and lower hemoglobin A1c (7.0% vs. 7.5%; P < .001).
However, time spent with glucose levels below 70 mg/dL (3.0% vs. 3.0%; P = .57) and below 54 mg/dL (0.6% vs. 0.5%) didn’t differ between the groups.
There were two cases of severe hypoglycemia in the closed-loop group and one in the standard-care group. One case of diabetic ketoacidosis related to infusion set failure occurred in the closed-loop group versus none in the standard-care group.
Dr. Bruttomesso commented that a virtual approach has several advantages over in-person visits, including “a more relaxed environment, lower travel costs, and greater ease of contact with clinicians.”
At the same time, though, “patient preferences, possible legal issues, and accessibility to technology ... are all important considerations in choosing the most appropriate way to communicate with patients at the initiation of a closed-loop system or during routine follow-up.” The families of the patients in this trial had above-average incomes, she pointed out.
Ultimately, she said, “A mix of face-to-face visits and virtual clinic meetings may become routine in the management of diabetes in young children.”
The study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases. Dr. Wadwa reported receiving grants/contracts from Beta Bionics, Dexcom, Eli Lilly, and MannKind, travel fees from Eli Lilly, and lecture fees from Tandem Diabetes Care, and serves as a consultant for Dexcom. Dr. Bruttomesso reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A hybrid closed-loop automated insulin delivery system improved time-in-range for blood glucose, compared with standard care, for children with type 1 diabetes in a 13-week trial.
The hybrid closed-loop system, also called automated insulin delivery or artificial pancreas, was composed of a t:slim X2 insulin pump, a Dexcom G6 continuous glucose monitor (CGM), and Control-IQ technology system algorithm software (Tandem Diabetes Care). The system was approved in the United States in 2018 for adults and children as young as 6 years.
Type 1 diabetes treatment is particularly challenging in children younger than 6 because of their small insulin dosing requirements and unpredictable eating and activity habits, lead author R. Paul Wadwa, MD, of the Barbara Davis Center for Diabetes, University of Colorado at Denver, Aurora, and colleagues wrote.
Thus far in the United States, only the Medtronic MiniMed 770G and the Omnipod 5 automated insulin delivery systems are approved for children as young as 2 years, they noted.
In the current study of 102 children with type 1 diabetes aged at least 2 years but younger than 6 years, time-in-range over 13 weeks was higher for those randomized to the hybrid closed-loop system, compared with standard of care; the latter included either an insulin pump or multiple daily injections plus a separate Dexcom G6 CGM.
The hybrid closed-loop system added an average of about 3 hours in ideal blood glucose range over the 13 weeks, compared with no change with standard care.
Moreover, the trial was conducted during the COVID-19 pandemic, necessitating virtual care for most of the study participants. As a result, more than 80% of the training on use of the system and over 90% of all the visits were conducted virtually.
“Successful use of the closed-loop system under these conditions is an important finding that could affect the approach to initiating and monitoring the use of the closed-loop system and expand the use of such systems, particularly in patients living in areas without an endocrinologist but with reliable internet access,” the investigators wrote.
Their findings were published online in the New England Journal of Medicine.
“These results suggest that, in very young children, closed-loop systems are superior to standard care with respect to glucose control,” Daniela Bruttomesso, MD, PhD, of the University of Padua (Italy) wrote in an accompanying editorial.
“Moreover, they show that the closed-loop system can be started remotely in children in this age range, with results that are similar to those obtained when parents or guardians receive face-to-face education about the use of these systems. The closed-loop system used in this trial appeared to be safe and effective.”
Dr. Bruttomesso added: “Although the results were solid, the trial period was only 13 weeks, and there were more unscheduled contacts in the closed-loop group than in the standard care group. In addition, the authors compared a closed-loop system with standard care, rather than in-person initiation of a closed-loop system with remote initiation.”
More time-in-range, no hypoglycemia with automated system
The 102 children were enrolled in the trial between April 28, 2021, and Jan. 13, 2022, at three different U.S. study sites; 68 children were randomized to the closed-loop system and 34 children to standard care. All but one participant completed the 13-week study.
Both groups had virtual or in-person trial visits at 2, 6, and 13 weeks after randomization, and telephone contact at 1 and 10 weeks. Training was virtual for 55 of the 68 children in the closed-loop group (81%). A total of 91% of 407 study visits in the closed-loop and 96% of 204 study visits in the standard-care group were also virtual.
The mean percentage of time spent in target glucose range (70-180 mg/dL) increased from 56.9% at baseline to 69.3% at 13 weeks for the closed-loop group, compared with virtually no change, from 54.9% to 55.9%, in the standard-care group. The mean adjusted difference between the two groups was significant (P < .001).
The closed-loop group also spent significantly less time than the standard-care group with glucose levels above 250 mg/dL during the study period (8.4% vs. 15.0%; P < .001), had lower mean glucose levels (155 vs. 174 mg/dL; P < .001), and lower hemoglobin A1c (7.0% vs. 7.5%; P < .001).
However, time spent with glucose levels below 70 mg/dL (3.0% vs. 3.0%; P = .57) and below 54 mg/dL (0.6% vs. 0.5%) didn’t differ between the groups.
There were two cases of severe hypoglycemia in the closed-loop group and one in the standard-care group. One case of diabetic ketoacidosis related to infusion set failure occurred in the closed-loop group versus none in the standard-care group.
Dr. Bruttomesso commented that a virtual approach has several advantages over in-person visits, including “a more relaxed environment, lower travel costs, and greater ease of contact with clinicians.”
At the same time, though, “patient preferences, possible legal issues, and accessibility to technology ... are all important considerations in choosing the most appropriate way to communicate with patients at the initiation of a closed-loop system or during routine follow-up.” The families of the patients in this trial had above-average incomes, she pointed out.
Ultimately, she said, “A mix of face-to-face visits and virtual clinic meetings may become routine in the management of diabetes in young children.”
The study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases. Dr. Wadwa reported receiving grants/contracts from Beta Bionics, Dexcom, Eli Lilly, and MannKind, travel fees from Eli Lilly, and lecture fees from Tandem Diabetes Care, and serves as a consultant for Dexcom. Dr. Bruttomesso reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A hybrid closed-loop automated insulin delivery system improved time-in-range for blood glucose, compared with standard care, for children with type 1 diabetes in a 13-week trial.
The hybrid closed-loop system, also called automated insulin delivery or artificial pancreas, was composed of a t:slim X2 insulin pump, a Dexcom G6 continuous glucose monitor (CGM), and Control-IQ technology system algorithm software (Tandem Diabetes Care). The system was approved in the United States in 2018 for adults and children as young as 6 years.
Type 1 diabetes treatment is particularly challenging in children younger than 6 because of their small insulin dosing requirements and unpredictable eating and activity habits, lead author R. Paul Wadwa, MD, of the Barbara Davis Center for Diabetes, University of Colorado at Denver, Aurora, and colleagues wrote.
Thus far in the United States, only the Medtronic MiniMed 770G and the Omnipod 5 automated insulin delivery systems are approved for children as young as 2 years, they noted.
In the current study of 102 children with type 1 diabetes aged at least 2 years but younger than 6 years, time-in-range over 13 weeks was higher for those randomized to the hybrid closed-loop system, compared with standard of care; the latter included either an insulin pump or multiple daily injections plus a separate Dexcom G6 CGM.
The hybrid closed-loop system added an average of about 3 hours in ideal blood glucose range over the 13 weeks, compared with no change with standard care.
Moreover, the trial was conducted during the COVID-19 pandemic, necessitating virtual care for most of the study participants. As a result, more than 80% of the training on use of the system and over 90% of all the visits were conducted virtually.
“Successful use of the closed-loop system under these conditions is an important finding that could affect the approach to initiating and monitoring the use of the closed-loop system and expand the use of such systems, particularly in patients living in areas without an endocrinologist but with reliable internet access,” the investigators wrote.
Their findings were published online in the New England Journal of Medicine.
“These results suggest that, in very young children, closed-loop systems are superior to standard care with respect to glucose control,” Daniela Bruttomesso, MD, PhD, of the University of Padua (Italy) wrote in an accompanying editorial.
“Moreover, they show that the closed-loop system can be started remotely in children in this age range, with results that are similar to those obtained when parents or guardians receive face-to-face education about the use of these systems. The closed-loop system used in this trial appeared to be safe and effective.”
Dr. Bruttomesso added: “Although the results were solid, the trial period was only 13 weeks, and there were more unscheduled contacts in the closed-loop group than in the standard care group. In addition, the authors compared a closed-loop system with standard care, rather than in-person initiation of a closed-loop system with remote initiation.”
More time-in-range, no hypoglycemia with automated system
The 102 children were enrolled in the trial between April 28, 2021, and Jan. 13, 2022, at three different U.S. study sites; 68 children were randomized to the closed-loop system and 34 children to standard care. All but one participant completed the 13-week study.
Both groups had virtual or in-person trial visits at 2, 6, and 13 weeks after randomization, and telephone contact at 1 and 10 weeks. Training was virtual for 55 of the 68 children in the closed-loop group (81%). A total of 91% of 407 study visits in the closed-loop and 96% of 204 study visits in the standard-care group were also virtual.
The mean percentage of time spent in target glucose range (70-180 mg/dL) increased from 56.9% at baseline to 69.3% at 13 weeks for the closed-loop group, compared with virtually no change, from 54.9% to 55.9%, in the standard-care group. The mean adjusted difference between the two groups was significant (P < .001).
The closed-loop group also spent significantly less time than the standard-care group with glucose levels above 250 mg/dL during the study period (8.4% vs. 15.0%; P < .001), had lower mean glucose levels (155 vs. 174 mg/dL; P < .001), and lower hemoglobin A1c (7.0% vs. 7.5%; P < .001).
However, time spent with glucose levels below 70 mg/dL (3.0% vs. 3.0%; P = .57) and below 54 mg/dL (0.6% vs. 0.5%) didn’t differ between the groups.
There were two cases of severe hypoglycemia in the closed-loop group and one in the standard-care group. One case of diabetic ketoacidosis related to infusion set failure occurred in the closed-loop group versus none in the standard-care group.
Dr. Bruttomesso commented that a virtual approach has several advantages over in-person visits, including “a more relaxed environment, lower travel costs, and greater ease of contact with clinicians.”
At the same time, though, “patient preferences, possible legal issues, and accessibility to technology ... are all important considerations in choosing the most appropriate way to communicate with patients at the initiation of a closed-loop system or during routine follow-up.” The families of the patients in this trial had above-average incomes, she pointed out.
Ultimately, she said, “A mix of face-to-face visits and virtual clinic meetings may become routine in the management of diabetes in young children.”
The study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases. Dr. Wadwa reported receiving grants/contracts from Beta Bionics, Dexcom, Eli Lilly, and MannKind, travel fees from Eli Lilly, and lecture fees from Tandem Diabetes Care, and serves as a consultant for Dexcom. Dr. Bruttomesso reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Physician suicide: Investigating its prevalence and cause
Physicians are admired for their sacrifice and dedication. Yet beneath the surface lies a painful, quiet reality:
The Physicians Foundation says that 55% of physicians know a doctor who considered, attempted, or died by suicide. Doctor’s Burden: Medscape Physician Suicide Report 2023 asked more than 9,000 doctors if they had suicidal thoughts. Nine percent of male physicians and 11% of female physicians said yes.
Why do so many doctors take their own lives?
“It’s not a new phenomenon,” says Rajnish Jaiswal, MD, associate chief of emergency medicine at NYC H+H Metropolitan Hospital and assistant professor of emergency medicine at New York Medical College. “There was a paper 150 years ago, published in England, which commented on the high rates of physician suicides compared to other professionals, and that trend has continued.”
Dr. Jaiswal says that the feeling in the physician community is that the numbers are even higher than what’s reported, unfortunately, which is an opinion echoed by other doctors this news organization spoke with for this story.
A perfect storm
Jodie Eckleberry-Hunt, PhD, a board-certified health psychologist, executive coach, and author, says the most significant culprit historically may be a rigid mindset that many physicians have. “There’s black and white, there’s a right answer and a wrong answer, there’s good and bad, and some physicians have a really hard time flexing,” she says.
Psychological flexibility underlies resilience. Dr. Eckleberry-Hunt says, “Think about your bounce factor and how that resilience is protective. Life isn’t always going to go well. You have to be able to flex and bounce, and some physicians (not all of them, of course) tend to be lower on cognitive flexibility.”
Brad Fern, coach and psychotherapist at Fern Executive and Physician Consulting, Minneapolis, says he uses two analogies that help when he works with physicians. One is the evil twins, and the other is the pressure cooker.
Mr. Fern says that the evil twins are silence and isolation and that several professions, including physicians, fall prey to these. To put any dent in suicidal ideations and suicide, Mr. Fern says, these must be addressed.
“Physicians tend not to talk about what’s bothering them, and that’s for many different reasons. They disproportionally tend to be great at helping other people but not great at receiving help themselves.”
On top of that, there’s a pressure cooker where they work. Mr. Fern doesn’t think anyone would argue that the health care system in the United States is not dysfunctional, at least to some degree. He says that this dysfunction acts like the physicians’ pressure cooker.
Add in circumstances, cultures, and day-to-day issues everyone has, like relational issues, parenting issues, and mental health problems. Then, toss in an individual’s lower resiliency, the inability to receive help, and a predicament for good measure – a loss, a divorce, or financial woes, for instance, which can overwhelm. Mr. Fern says it can be a mathematical equation for suicidal ideation.
Is there a why?
“Some people think there’s a reason for suicide, but often, there’s a spectrum of reasons,” says Mr. Fern. He says that some physicians are trying to escape emotional pain. For others, it can be fear or a revenge thing, like, “the hell with you, I’m going to kill myself.” It can be getting attention the way teens do, as professionals have seen. Then there’s the organic component, like brain trauma, brain imbalance, depression, anxiety, or bipolar disorder. And finally, a drug or alcohol issue.
“But the reason why physician suicide is elevated, I think, is because there’s this ethos around being silent and, ‘I’m going to listen to and solve everyone else’s problems, but I’m not going to reach out and get help for my own,’ ” says Mr. Fern. “If you take advantage of mental health services, you’re implying that you’re mentally ill. And most physicians aren’t going to do that.”
On the positive side, Dr. Eckleberry-Hunt says that she sees many younger physicians discussing trauma. As a result, they’re more open to receiving help than previous generations. She speculates whether physicians have always had trauma from their past and whether current-day issues are now triggering it or whether they have more trauma these days. “Are they talking about it more, or is it experienced more?”
The failure of the system
The building blocks for physician suicide may have been there from the beginning. “From your first day of medical school and throughout your career, there was a very rigid system in place that is quite unforgiving, is quite stressful, and demands a lot,” says Dr. Jaiswal. And it’s within this system that physicians must operate.
“You have all the corporations, entities, organizations, [and] medical societies talking about physician wellness, burnout, and suicide, but the reality is it’s not making that much of a difference,” he says.
In her report, “What I’ve Learned From 1,710 Doctor Suicides,” Pamelia Wible, MD, who runs a physician suicide helpline that physicians can email and get an immediate callback, likens the current system to assembly line medicine.
Dr. Eckleberry-Hunt thinks the message has been bungled in health care. Everyone discusses burnout, meditation, self-care, and other essential constructs. “But we don’t deal with the root cause [of suicide]. Instead, we teach you soothing strategies.”
Further, Dr. Jaiswal says that not all physicians who commit suicide experience burnout or are experiencing burnout and that the vast majority of physicians who experience burnout don’t have suicidal ideation. “In the sense, that ‘let’s address physician burnout and that will hopefully translate to a reduced number of physician suicides’ – there is a very tenuous argument to be made for that because that is just one aspect in this complex system,” he says.
We need more than just lip service on suicide
Overall, the experts interviewed for this article acknowledged that the system is at least talking about physician suicide, which is a big first step. However, most agree that where big health entities go wrong is that they set up wellness or mental health programs, they implement a wellness officer, they write up talking points for physicians who need mental health care to get that care, and they think they’ve done their job, that they’ve done what’s required to address the problem.
But Dr. Jaiswal thinks these are often mostly public-relations rebuttals. Mr. Fern suggests, “It’s a show that’s not effective.” And Dr. Eckleberry-Hunt says that “even if you had a legit, well-funded well-being program for health care providers, you would still have a baseline rate of physician suicide, and that gets down to having drug and alcohol education and talking about having a system for physicians to access that doesn’t come along with insurance billing” – one that doesn’t create a paper trail and follow physician licensure and job applications for the rest of their career; one that doesn’t associate their mental health care with their work institution; one that offers confidentiality.
“For most folks, there is still a big distrust in the system. As physicians, very few of them feel that the system that they’re operating in has their best interest at heart. And that is why very few physicians will self-report any mental health issues, depression, or even ideation to colleagues, superiors, or managers,” says Dr. Jaiswal. Many more feel skeptical about the confidentiality of the programs in place.
The experts acknowledge that many people are trying to work on this and bring about change on multiple levels – grassroots, department levels, state, and federal. “But I think the biggest thing that the system has to do is earn back the trust of the physician,” Dr. Jaiswal adds.
“Physician suicide is a very visible problem in a very broken system. So, it’ll be very difficult in isolation to treat it without making any systemic changes, because that’s happening right now, and it’s not working,” says Dr. Jaiswal.
“The thing that I am most hopeful about is that I am seeing an influx of younger physicians who seek me out, and granted, their training programs tell them to come and see me, but they are ready and willing to talk about their mental health separate from work. They’re not coming in saying, ‘Here are all the people who I blame.’ They’re saying, ‘These are my struggles, and I want to be a better, happier physician,’ ” says Dr. Eckleberry-Hunt.
A version of this article originally appeared on Medscape.com.
Physicians are admired for their sacrifice and dedication. Yet beneath the surface lies a painful, quiet reality:
The Physicians Foundation says that 55% of physicians know a doctor who considered, attempted, or died by suicide. Doctor’s Burden: Medscape Physician Suicide Report 2023 asked more than 9,000 doctors if they had suicidal thoughts. Nine percent of male physicians and 11% of female physicians said yes.
Why do so many doctors take their own lives?
“It’s not a new phenomenon,” says Rajnish Jaiswal, MD, associate chief of emergency medicine at NYC H+H Metropolitan Hospital and assistant professor of emergency medicine at New York Medical College. “There was a paper 150 years ago, published in England, which commented on the high rates of physician suicides compared to other professionals, and that trend has continued.”
Dr. Jaiswal says that the feeling in the physician community is that the numbers are even higher than what’s reported, unfortunately, which is an opinion echoed by other doctors this news organization spoke with for this story.
A perfect storm
Jodie Eckleberry-Hunt, PhD, a board-certified health psychologist, executive coach, and author, says the most significant culprit historically may be a rigid mindset that many physicians have. “There’s black and white, there’s a right answer and a wrong answer, there’s good and bad, and some physicians have a really hard time flexing,” she says.
Psychological flexibility underlies resilience. Dr. Eckleberry-Hunt says, “Think about your bounce factor and how that resilience is protective. Life isn’t always going to go well. You have to be able to flex and bounce, and some physicians (not all of them, of course) tend to be lower on cognitive flexibility.”
Brad Fern, coach and psychotherapist at Fern Executive and Physician Consulting, Minneapolis, says he uses two analogies that help when he works with physicians. One is the evil twins, and the other is the pressure cooker.
Mr. Fern says that the evil twins are silence and isolation and that several professions, including physicians, fall prey to these. To put any dent in suicidal ideations and suicide, Mr. Fern says, these must be addressed.
“Physicians tend not to talk about what’s bothering them, and that’s for many different reasons. They disproportionally tend to be great at helping other people but not great at receiving help themselves.”
On top of that, there’s a pressure cooker where they work. Mr. Fern doesn’t think anyone would argue that the health care system in the United States is not dysfunctional, at least to some degree. He says that this dysfunction acts like the physicians’ pressure cooker.
Add in circumstances, cultures, and day-to-day issues everyone has, like relational issues, parenting issues, and mental health problems. Then, toss in an individual’s lower resiliency, the inability to receive help, and a predicament for good measure – a loss, a divorce, or financial woes, for instance, which can overwhelm. Mr. Fern says it can be a mathematical equation for suicidal ideation.
Is there a why?
“Some people think there’s a reason for suicide, but often, there’s a spectrum of reasons,” says Mr. Fern. He says that some physicians are trying to escape emotional pain. For others, it can be fear or a revenge thing, like, “the hell with you, I’m going to kill myself.” It can be getting attention the way teens do, as professionals have seen. Then there’s the organic component, like brain trauma, brain imbalance, depression, anxiety, or bipolar disorder. And finally, a drug or alcohol issue.
“But the reason why physician suicide is elevated, I think, is because there’s this ethos around being silent and, ‘I’m going to listen to and solve everyone else’s problems, but I’m not going to reach out and get help for my own,’ ” says Mr. Fern. “If you take advantage of mental health services, you’re implying that you’re mentally ill. And most physicians aren’t going to do that.”
On the positive side, Dr. Eckleberry-Hunt says that she sees many younger physicians discussing trauma. As a result, they’re more open to receiving help than previous generations. She speculates whether physicians have always had trauma from their past and whether current-day issues are now triggering it or whether they have more trauma these days. “Are they talking about it more, or is it experienced more?”
The failure of the system
The building blocks for physician suicide may have been there from the beginning. “From your first day of medical school and throughout your career, there was a very rigid system in place that is quite unforgiving, is quite stressful, and demands a lot,” says Dr. Jaiswal. And it’s within this system that physicians must operate.
“You have all the corporations, entities, organizations, [and] medical societies talking about physician wellness, burnout, and suicide, but the reality is it’s not making that much of a difference,” he says.
In her report, “What I’ve Learned From 1,710 Doctor Suicides,” Pamelia Wible, MD, who runs a physician suicide helpline that physicians can email and get an immediate callback, likens the current system to assembly line medicine.
Dr. Eckleberry-Hunt thinks the message has been bungled in health care. Everyone discusses burnout, meditation, self-care, and other essential constructs. “But we don’t deal with the root cause [of suicide]. Instead, we teach you soothing strategies.”
Further, Dr. Jaiswal says that not all physicians who commit suicide experience burnout or are experiencing burnout and that the vast majority of physicians who experience burnout don’t have suicidal ideation. “In the sense, that ‘let’s address physician burnout and that will hopefully translate to a reduced number of physician suicides’ – there is a very tenuous argument to be made for that because that is just one aspect in this complex system,” he says.
We need more than just lip service on suicide
Overall, the experts interviewed for this article acknowledged that the system is at least talking about physician suicide, which is a big first step. However, most agree that where big health entities go wrong is that they set up wellness or mental health programs, they implement a wellness officer, they write up talking points for physicians who need mental health care to get that care, and they think they’ve done their job, that they’ve done what’s required to address the problem.
But Dr. Jaiswal thinks these are often mostly public-relations rebuttals. Mr. Fern suggests, “It’s a show that’s not effective.” And Dr. Eckleberry-Hunt says that “even if you had a legit, well-funded well-being program for health care providers, you would still have a baseline rate of physician suicide, and that gets down to having drug and alcohol education and talking about having a system for physicians to access that doesn’t come along with insurance billing” – one that doesn’t create a paper trail and follow physician licensure and job applications for the rest of their career; one that doesn’t associate their mental health care with their work institution; one that offers confidentiality.
“For most folks, there is still a big distrust in the system. As physicians, very few of them feel that the system that they’re operating in has their best interest at heart. And that is why very few physicians will self-report any mental health issues, depression, or even ideation to colleagues, superiors, or managers,” says Dr. Jaiswal. Many more feel skeptical about the confidentiality of the programs in place.
The experts acknowledge that many people are trying to work on this and bring about change on multiple levels – grassroots, department levels, state, and federal. “But I think the biggest thing that the system has to do is earn back the trust of the physician,” Dr. Jaiswal adds.
“Physician suicide is a very visible problem in a very broken system. So, it’ll be very difficult in isolation to treat it without making any systemic changes, because that’s happening right now, and it’s not working,” says Dr. Jaiswal.
“The thing that I am most hopeful about is that I am seeing an influx of younger physicians who seek me out, and granted, their training programs tell them to come and see me, but they are ready and willing to talk about their mental health separate from work. They’re not coming in saying, ‘Here are all the people who I blame.’ They’re saying, ‘These are my struggles, and I want to be a better, happier physician,’ ” says Dr. Eckleberry-Hunt.
A version of this article originally appeared on Medscape.com.
Physicians are admired for their sacrifice and dedication. Yet beneath the surface lies a painful, quiet reality:
The Physicians Foundation says that 55% of physicians know a doctor who considered, attempted, or died by suicide. Doctor’s Burden: Medscape Physician Suicide Report 2023 asked more than 9,000 doctors if they had suicidal thoughts. Nine percent of male physicians and 11% of female physicians said yes.
Why do so many doctors take their own lives?
“It’s not a new phenomenon,” says Rajnish Jaiswal, MD, associate chief of emergency medicine at NYC H+H Metropolitan Hospital and assistant professor of emergency medicine at New York Medical College. “There was a paper 150 years ago, published in England, which commented on the high rates of physician suicides compared to other professionals, and that trend has continued.”
Dr. Jaiswal says that the feeling in the physician community is that the numbers are even higher than what’s reported, unfortunately, which is an opinion echoed by other doctors this news organization spoke with for this story.
A perfect storm
Jodie Eckleberry-Hunt, PhD, a board-certified health psychologist, executive coach, and author, says the most significant culprit historically may be a rigid mindset that many physicians have. “There’s black and white, there’s a right answer and a wrong answer, there’s good and bad, and some physicians have a really hard time flexing,” she says.
Psychological flexibility underlies resilience. Dr. Eckleberry-Hunt says, “Think about your bounce factor and how that resilience is protective. Life isn’t always going to go well. You have to be able to flex and bounce, and some physicians (not all of them, of course) tend to be lower on cognitive flexibility.”
Brad Fern, coach and psychotherapist at Fern Executive and Physician Consulting, Minneapolis, says he uses two analogies that help when he works with physicians. One is the evil twins, and the other is the pressure cooker.
Mr. Fern says that the evil twins are silence and isolation and that several professions, including physicians, fall prey to these. To put any dent in suicidal ideations and suicide, Mr. Fern says, these must be addressed.
“Physicians tend not to talk about what’s bothering them, and that’s for many different reasons. They disproportionally tend to be great at helping other people but not great at receiving help themselves.”
On top of that, there’s a pressure cooker where they work. Mr. Fern doesn’t think anyone would argue that the health care system in the United States is not dysfunctional, at least to some degree. He says that this dysfunction acts like the physicians’ pressure cooker.
Add in circumstances, cultures, and day-to-day issues everyone has, like relational issues, parenting issues, and mental health problems. Then, toss in an individual’s lower resiliency, the inability to receive help, and a predicament for good measure – a loss, a divorce, or financial woes, for instance, which can overwhelm. Mr. Fern says it can be a mathematical equation for suicidal ideation.
Is there a why?
“Some people think there’s a reason for suicide, but often, there’s a spectrum of reasons,” says Mr. Fern. He says that some physicians are trying to escape emotional pain. For others, it can be fear or a revenge thing, like, “the hell with you, I’m going to kill myself.” It can be getting attention the way teens do, as professionals have seen. Then there’s the organic component, like brain trauma, brain imbalance, depression, anxiety, or bipolar disorder. And finally, a drug or alcohol issue.
“But the reason why physician suicide is elevated, I think, is because there’s this ethos around being silent and, ‘I’m going to listen to and solve everyone else’s problems, but I’m not going to reach out and get help for my own,’ ” says Mr. Fern. “If you take advantage of mental health services, you’re implying that you’re mentally ill. And most physicians aren’t going to do that.”
On the positive side, Dr. Eckleberry-Hunt says that she sees many younger physicians discussing trauma. As a result, they’re more open to receiving help than previous generations. She speculates whether physicians have always had trauma from their past and whether current-day issues are now triggering it or whether they have more trauma these days. “Are they talking about it more, or is it experienced more?”
The failure of the system
The building blocks for physician suicide may have been there from the beginning. “From your first day of medical school and throughout your career, there was a very rigid system in place that is quite unforgiving, is quite stressful, and demands a lot,” says Dr. Jaiswal. And it’s within this system that physicians must operate.
“You have all the corporations, entities, organizations, [and] medical societies talking about physician wellness, burnout, and suicide, but the reality is it’s not making that much of a difference,” he says.
In her report, “What I’ve Learned From 1,710 Doctor Suicides,” Pamelia Wible, MD, who runs a physician suicide helpline that physicians can email and get an immediate callback, likens the current system to assembly line medicine.
Dr. Eckleberry-Hunt thinks the message has been bungled in health care. Everyone discusses burnout, meditation, self-care, and other essential constructs. “But we don’t deal with the root cause [of suicide]. Instead, we teach you soothing strategies.”
Further, Dr. Jaiswal says that not all physicians who commit suicide experience burnout or are experiencing burnout and that the vast majority of physicians who experience burnout don’t have suicidal ideation. “In the sense, that ‘let’s address physician burnout and that will hopefully translate to a reduced number of physician suicides’ – there is a very tenuous argument to be made for that because that is just one aspect in this complex system,” he says.
We need more than just lip service on suicide
Overall, the experts interviewed for this article acknowledged that the system is at least talking about physician suicide, which is a big first step. However, most agree that where big health entities go wrong is that they set up wellness or mental health programs, they implement a wellness officer, they write up talking points for physicians who need mental health care to get that care, and they think they’ve done their job, that they’ve done what’s required to address the problem.
But Dr. Jaiswal thinks these are often mostly public-relations rebuttals. Mr. Fern suggests, “It’s a show that’s not effective.” And Dr. Eckleberry-Hunt says that “even if you had a legit, well-funded well-being program for health care providers, you would still have a baseline rate of physician suicide, and that gets down to having drug and alcohol education and talking about having a system for physicians to access that doesn’t come along with insurance billing” – one that doesn’t create a paper trail and follow physician licensure and job applications for the rest of their career; one that doesn’t associate their mental health care with their work institution; one that offers confidentiality.
“For most folks, there is still a big distrust in the system. As physicians, very few of them feel that the system that they’re operating in has their best interest at heart. And that is why very few physicians will self-report any mental health issues, depression, or even ideation to colleagues, superiors, or managers,” says Dr. Jaiswal. Many more feel skeptical about the confidentiality of the programs in place.
The experts acknowledge that many people are trying to work on this and bring about change on multiple levels – grassroots, department levels, state, and federal. “But I think the biggest thing that the system has to do is earn back the trust of the physician,” Dr. Jaiswal adds.
“Physician suicide is a very visible problem in a very broken system. So, it’ll be very difficult in isolation to treat it without making any systemic changes, because that’s happening right now, and it’s not working,” says Dr. Jaiswal.
“The thing that I am most hopeful about is that I am seeing an influx of younger physicians who seek me out, and granted, their training programs tell them to come and see me, but they are ready and willing to talk about their mental health separate from work. They’re not coming in saying, ‘Here are all the people who I blame.’ They’re saying, ‘These are my struggles, and I want to be a better, happier physician,’ ” says Dr. Eckleberry-Hunt.
A version of this article originally appeared on Medscape.com.
Ulcerative colitis cases projected to top 2 million in eight countries by 2031
The data and analytics company’s report offers projections for diagnosed incident and prevalent cases of UC in the United States, United Kingdom, Germany, Spain, Japan, Italy, France, and Canada.
In 2031, the United States will have the highest number of diagnosed prevalent cases of UC, with 655,317 cases, whereas Canada will have the fewest, with 91,186 cases, the company projects.
“UC can occur at any age, although most people are diagnosed in their mid-thirties. Men and women are equally likely to be affected, but older men are more likely to be diagnosed than older women,” Bharti Prabhakar, MPH, associate project manager at GlobalData, said in a statement.
In all eight countries, adults aged 30-69 years accounted for more than 65% of the diagnosed prevalent cases of UC, whereas those younger than 20 years made up less than 3% of the cases, GlobalData noted.
Incidence also rising
Diagnosed incident cases of UC in the eight countries are expected to increase from 160,122 cases in 2021 to 168,467 cases in 2031, at an annual growth rate of 0.52%, the company said.
In 2031, the United States will have the highest number of diagnosed incident cases of UC, with 104,795 cases, and France will have the fewest, with 2972 cases, the company predicted.
GlobalData epidemiologists attribute the predicted increases in UC prevalence and incidence to changes in population dynamics in each country.
The forecast is supported by historical data obtained from peer-reviewed articles and population-based studies, the firm noted.
The methodology was kept consistent across the eight countries to allow for a meaningful comparison of the forecast incident and prevalent cases of UC across these markets, GlobalData added.
“UC can affect people of any racial or ethnic group,” Ms. Prabhakar stated. “Genes, abnormal immune reactions, the microbiome, diet, stress, and the environment have all been suggested as triggers, but there is no definite evidence that any one of these factors is the cause of UC.”
Western countries have reported high incidence and prevalence of UC, Ms. Prabhaker noted. “Therefore, environmental factors may either suppress or reinforce inherent predispositions for UC and might also be crucial in triggering disease onset.”
A version of this article originally appeared on Medscape.com.
The data and analytics company’s report offers projections for diagnosed incident and prevalent cases of UC in the United States, United Kingdom, Germany, Spain, Japan, Italy, France, and Canada.
In 2031, the United States will have the highest number of diagnosed prevalent cases of UC, with 655,317 cases, whereas Canada will have the fewest, with 91,186 cases, the company projects.
“UC can occur at any age, although most people are diagnosed in their mid-thirties. Men and women are equally likely to be affected, but older men are more likely to be diagnosed than older women,” Bharti Prabhakar, MPH, associate project manager at GlobalData, said in a statement.
In all eight countries, adults aged 30-69 years accounted for more than 65% of the diagnosed prevalent cases of UC, whereas those younger than 20 years made up less than 3% of the cases, GlobalData noted.
Incidence also rising
Diagnosed incident cases of UC in the eight countries are expected to increase from 160,122 cases in 2021 to 168,467 cases in 2031, at an annual growth rate of 0.52%, the company said.
In 2031, the United States will have the highest number of diagnosed incident cases of UC, with 104,795 cases, and France will have the fewest, with 2972 cases, the company predicted.
GlobalData epidemiologists attribute the predicted increases in UC prevalence and incidence to changes in population dynamics in each country.
The forecast is supported by historical data obtained from peer-reviewed articles and population-based studies, the firm noted.
The methodology was kept consistent across the eight countries to allow for a meaningful comparison of the forecast incident and prevalent cases of UC across these markets, GlobalData added.
“UC can affect people of any racial or ethnic group,” Ms. Prabhakar stated. “Genes, abnormal immune reactions, the microbiome, diet, stress, and the environment have all been suggested as triggers, but there is no definite evidence that any one of these factors is the cause of UC.”
Western countries have reported high incidence and prevalence of UC, Ms. Prabhaker noted. “Therefore, environmental factors may either suppress or reinforce inherent predispositions for UC and might also be crucial in triggering disease onset.”
A version of this article originally appeared on Medscape.com.
The data and analytics company’s report offers projections for diagnosed incident and prevalent cases of UC in the United States, United Kingdom, Germany, Spain, Japan, Italy, France, and Canada.
In 2031, the United States will have the highest number of diagnosed prevalent cases of UC, with 655,317 cases, whereas Canada will have the fewest, with 91,186 cases, the company projects.
“UC can occur at any age, although most people are diagnosed in their mid-thirties. Men and women are equally likely to be affected, but older men are more likely to be diagnosed than older women,” Bharti Prabhakar, MPH, associate project manager at GlobalData, said in a statement.
In all eight countries, adults aged 30-69 years accounted for more than 65% of the diagnosed prevalent cases of UC, whereas those younger than 20 years made up less than 3% of the cases, GlobalData noted.
Incidence also rising
Diagnosed incident cases of UC in the eight countries are expected to increase from 160,122 cases in 2021 to 168,467 cases in 2031, at an annual growth rate of 0.52%, the company said.
In 2031, the United States will have the highest number of diagnosed incident cases of UC, with 104,795 cases, and France will have the fewest, with 2972 cases, the company predicted.
GlobalData epidemiologists attribute the predicted increases in UC prevalence and incidence to changes in population dynamics in each country.
The forecast is supported by historical data obtained from peer-reviewed articles and population-based studies, the firm noted.
The methodology was kept consistent across the eight countries to allow for a meaningful comparison of the forecast incident and prevalent cases of UC across these markets, GlobalData added.
“UC can affect people of any racial or ethnic group,” Ms. Prabhakar stated. “Genes, abnormal immune reactions, the microbiome, diet, stress, and the environment have all been suggested as triggers, but there is no definite evidence that any one of these factors is the cause of UC.”
Western countries have reported high incidence and prevalence of UC, Ms. Prabhaker noted. “Therefore, environmental factors may either suppress or reinforce inherent predispositions for UC and might also be crucial in triggering disease onset.”
A version of this article originally appeared on Medscape.com.
Oral PCSK9 inhibitor shows encouraging LDL lowering
A new oral formulation of a PCSK9-inhibiting, cholesterol-lowering drug in development by Merck has shown encouraging results in a phase 2 study.
The study was presented by Christie Ballantyne, MD, Baylor College of Medicine, Houston, at the joint scientific sessions of the American College of Cardiology and the World Heart Federation.

“In this diverse population of hypercholesterolemic patients, all doses of MK-0616 showed superior reduction of LDL vs. placebo up to a 60.9% placebo-adjusted reduction from baseline to week 8, which was consistent across subgroups,” Dr. Ballantyne reported.
“Reduction in ApoB and non-HDL cholesterol were consistent with that of LDL cholesterol, with up to a 51.8% reduction in ApoB and a 55.8% reduction in non-HDL,” he noted.
He added that the drug was well tolerated with no difference in adverse events across the treatment groups, compared with placebo.
“These data support the further development of MK-0616, an oral PCSK9 inhibitor that may improve access to effective LDL-cholesterol lowering therapies and improve attainment of guideline-recommended LDL goals aimed at reducing cardiovascular risk,” Dr. Ballantyne concluded. “The results are encouraging for a phase 3 program that is now being designed.”
He explained that elevated LDL is a primary causative factor for atherosclerotic cardiovascular disease (ASCVD), and despite effective treatments (statins), a large proportion of patients fail to achieve guideline-recommended LDL levels. Injectable treatments targeting PCSK9 have demonstrated large reductions in LDL and decreased risk of ASCVD events, but access barriers and need for repeat injections have led to poor adoption. An oral PCSK9 inhibitor may widen access and improve attainment of guideline-recommended treatment goals.
Dr. Ballantyne described the new drug, MK-0616, as a “macrocyclic peptide that can bind PCSK9 with monoclonal antibody-like affinity at 1/100th of the molecular weight.”
The current phase 2 study included 381 adult patients (49% female; median age 62 years) with a wide range of ASCVD risk. Average LDL-C level was 119.5 mg/dL at baseline. Around 40% of patients were not taking statins, 35% were on low- to moderate-intensity statin therapy, and 26% were on high-intensity statin therapy.
They were randomly assigned to four different doses of MK-0616 (6, 12, 18, or 30 mg once daily) or matching placebo.
Results showed that all doses of MK-0616 demonstrated statistically significant differences in percentage change in LDL-C from baseline to week 8 vs. placebo: –41.2% (6 mg), –55.7% (12 mg), –59.1% (18 mg), and –60.9% (30 mg).
The mean percentage changes in ApoB from baseline vs. placebo were –32.8%, –45.8%, –48.7%, and 51.8% for the four escalating doses of the drug. And non-HDL cholesterol changes were –35.9%, –50.5%, –53.2%, and –55.8% respectively.
The proportion of participants at protocol-defined goals for LDL reduction was 80.5%, 85.5%, 90.8%, and 90.8% with MK-0616 at the 6-mg, 12-mg, 18-mg, and 30-mg doses, compared with 9.3% with placebo.
Dr. Ballantyne reported that the efficacy looked similar in all subgroups, and regardless of baseline therapy.
“This was a dose-finding study, which will help select a dose to be taken forward in larger studies, and it looks from these results as though you get most of the efficacy by 12 mg,” he added.
Adverse events occurred in a proportion of participants in the MK-0616 groups (39.5% to 43.4%) similar to that of placebo (44.0%), and discontinuations as a result of adverse events occurred in two or fewer participants in any treatment group.
‘Super exciting’
Putting the results of his study into perspective at an ACC press conference, Rhonda Cooper-DeHoff, PharmD, associate professor in the department of pharmacotherapy and translational research at the University of Florida in Gainesville, commented.

“For the last quarter of a century we have had statins available to treat elevated LDL and atherosclerosis and despite that we have many patients who refuse to take statins or are afraid to take statins,” she said. “This is not about cost as the statins are all available generically now. But many patients claim to be intolerant or unresponsive.”
She noted that in 2015/2016 the first injectable PCSK9 inhibitors became available “which really were very exciting molecules, but they have a high cost and access issues, and patients often do not like injections so there are still a lot of issues.”
Dr. Cooper-DeHoff pointed out that this oral PCSK9 inhibitor seems to be as effective at lowering LDL as the injectable products regardless of whether statins are on board or not, which she said was “super exciting.”
She added: “We are all going to be waiting excitedly for the outcome data with this oral PCSK9 inhibitor.”
She also noted that another study (CLEAR Outcomes) presented at the ACC meeting showed good lipid-lowering results and a reduction in cardiovascular outcomes in statin-intolerant patients with another oral lipid lowering drug, bempedoic acid (Nexletol).
She said the two oral drugs promised a “very bright for the future for LDL lowering and the treatment of atherosclerosis in our patients,” adding that “we are now really chipping away at the barriers to achieving the holy grail of guideline-directed LDL lowering to prevent hard outcomes.”
The results were published online in the Journal of the American College of Cardiology at the time of presentation.
This study was funded by Merck. Dr. Ballantyne has received grant/research support through his institution from Abbott Diagnostic, Akcea, Amgen, Arrowhead, Esperion, Ionis, Merck, New Amsterdam, Novartis, Novo Nordisk, Regeneron, and Roche Diagnostics and has been a consultant for 89Bio, Abbott Diagnostics, Alnylam Pharmaceuticals, Althera, Amarin, Amgen, Arrowhead, AstraZeneca, Denka Seiken, Esperion, Genentech, Gilead, Illumina, Ionis, Matinas BioPharma, Merck, New Amsterdam, Novartis, Novo Nordisk, Pfizer, Regeneron, and Roche Diagnostics.
A version of this article first appeared on Medscape.com.
A new oral formulation of a PCSK9-inhibiting, cholesterol-lowering drug in development by Merck has shown encouraging results in a phase 2 study.
The study was presented by Christie Ballantyne, MD, Baylor College of Medicine, Houston, at the joint scientific sessions of the American College of Cardiology and the World Heart Federation.
“In this diverse population of hypercholesterolemic patients, all doses of MK-0616 showed superior reduction of LDL vs. placebo up to a 60.9% placebo-adjusted reduction from baseline to week 8, which was consistent across subgroups,” Dr. Ballantyne reported.
“Reduction in ApoB and non-HDL cholesterol were consistent with that of LDL cholesterol, with up to a 51.8% reduction in ApoB and a 55.8% reduction in non-HDL,” he noted.
He added that the drug was well tolerated with no difference in adverse events across the treatment groups, compared with placebo.
“These data support the further development of MK-0616, an oral PCSK9 inhibitor that may improve access to effective LDL-cholesterol lowering therapies and improve attainment of guideline-recommended LDL goals aimed at reducing cardiovascular risk,” Dr. Ballantyne concluded. “The results are encouraging for a phase 3 program that is now being designed.”
He explained that elevated LDL is a primary causative factor for atherosclerotic cardiovascular disease (ASCVD), and despite effective treatments (statins), a large proportion of patients fail to achieve guideline-recommended LDL levels. Injectable treatments targeting PCSK9 have demonstrated large reductions in LDL and decreased risk of ASCVD events, but access barriers and need for repeat injections have led to poor adoption. An oral PCSK9 inhibitor may widen access and improve attainment of guideline-recommended treatment goals.
Dr. Ballantyne described the new drug, MK-0616, as a “macrocyclic peptide that can bind PCSK9 with monoclonal antibody-like affinity at 1/100th of the molecular weight.”
The current phase 2 study included 381 adult patients (49% female; median age 62 years) with a wide range of ASCVD risk. Average LDL-C level was 119.5 mg/dL at baseline. Around 40% of patients were not taking statins, 35% were on low- to moderate-intensity statin therapy, and 26% were on high-intensity statin therapy.
They were randomly assigned to four different doses of MK-0616 (6, 12, 18, or 30 mg once daily) or matching placebo.
Results showed that all doses of MK-0616 demonstrated statistically significant differences in percentage change in LDL-C from baseline to week 8 vs. placebo: –41.2% (6 mg), –55.7% (12 mg), –59.1% (18 mg), and –60.9% (30 mg).
The mean percentage changes in ApoB from baseline vs. placebo were –32.8%, –45.8%, –48.7%, and 51.8% for the four escalating doses of the drug. And non-HDL cholesterol changes were –35.9%, –50.5%, –53.2%, and –55.8% respectively.
The proportion of participants at protocol-defined goals for LDL reduction was 80.5%, 85.5%, 90.8%, and 90.8% with MK-0616 at the 6-mg, 12-mg, 18-mg, and 30-mg doses, compared with 9.3% with placebo.
Dr. Ballantyne reported that the efficacy looked similar in all subgroups, and regardless of baseline therapy.
“This was a dose-finding study, which will help select a dose to be taken forward in larger studies, and it looks from these results as though you get most of the efficacy by 12 mg,” he added.
Adverse events occurred in a proportion of participants in the MK-0616 groups (39.5% to 43.4%) similar to that of placebo (44.0%), and discontinuations as a result of adverse events occurred in two or fewer participants in any treatment group.
‘Super exciting’
Putting the results of his study into perspective at an ACC press conference, Rhonda Cooper-DeHoff, PharmD, associate professor in the department of pharmacotherapy and translational research at the University of Florida in Gainesville, commented.
“For the last quarter of a century we have had statins available to treat elevated LDL and atherosclerosis and despite that we have many patients who refuse to take statins or are afraid to take statins,” she said. “This is not about cost as the statins are all available generically now. But many patients claim to be intolerant or unresponsive.”
She noted that in 2015/2016 the first injectable PCSK9 inhibitors became available “which really were very exciting molecules, but they have a high cost and access issues, and patients often do not like injections so there are still a lot of issues.”
Dr. Cooper-DeHoff pointed out that this oral PCSK9 inhibitor seems to be as effective at lowering LDL as the injectable products regardless of whether statins are on board or not, which she said was “super exciting.”
She added: “We are all going to be waiting excitedly for the outcome data with this oral PCSK9 inhibitor.”
She also noted that another study (CLEAR Outcomes) presented at the ACC meeting showed good lipid-lowering results and a reduction in cardiovascular outcomes in statin-intolerant patients with another oral lipid lowering drug, bempedoic acid (Nexletol).
She said the two oral drugs promised a “very bright for the future for LDL lowering and the treatment of atherosclerosis in our patients,” adding that “we are now really chipping away at the barriers to achieving the holy grail of guideline-directed LDL lowering to prevent hard outcomes.”
The results were published online in the Journal of the American College of Cardiology at the time of presentation.
This study was funded by Merck. Dr. Ballantyne has received grant/research support through his institution from Abbott Diagnostic, Akcea, Amgen, Arrowhead, Esperion, Ionis, Merck, New Amsterdam, Novartis, Novo Nordisk, Regeneron, and Roche Diagnostics and has been a consultant for 89Bio, Abbott Diagnostics, Alnylam Pharmaceuticals, Althera, Amarin, Amgen, Arrowhead, AstraZeneca, Denka Seiken, Esperion, Genentech, Gilead, Illumina, Ionis, Matinas BioPharma, Merck, New Amsterdam, Novartis, Novo Nordisk, Pfizer, Regeneron, and Roche Diagnostics.
A version of this article first appeared on Medscape.com.
A new oral formulation of a PCSK9-inhibiting, cholesterol-lowering drug in development by Merck has shown encouraging results in a phase 2 study.
The study was presented by Christie Ballantyne, MD, Baylor College of Medicine, Houston, at the joint scientific sessions of the American College of Cardiology and the World Heart Federation.
“In this diverse population of hypercholesterolemic patients, all doses of MK-0616 showed superior reduction of LDL vs. placebo up to a 60.9% placebo-adjusted reduction from baseline to week 8, which was consistent across subgroups,” Dr. Ballantyne reported.
“Reduction in ApoB and non-HDL cholesterol were consistent with that of LDL cholesterol, with up to a 51.8% reduction in ApoB and a 55.8% reduction in non-HDL,” he noted.
He added that the drug was well tolerated with no difference in adverse events across the treatment groups, compared with placebo.
“These data support the further development of MK-0616, an oral PCSK9 inhibitor that may improve access to effective LDL-cholesterol lowering therapies and improve attainment of guideline-recommended LDL goals aimed at reducing cardiovascular risk,” Dr. Ballantyne concluded. “The results are encouraging for a phase 3 program that is now being designed.”
He explained that elevated LDL is a primary causative factor for atherosclerotic cardiovascular disease (ASCVD), and despite effective treatments (statins), a large proportion of patients fail to achieve guideline-recommended LDL levels. Injectable treatments targeting PCSK9 have demonstrated large reductions in LDL and decreased risk of ASCVD events, but access barriers and need for repeat injections have led to poor adoption. An oral PCSK9 inhibitor may widen access and improve attainment of guideline-recommended treatment goals.
Dr. Ballantyne described the new drug, MK-0616, as a “macrocyclic peptide that can bind PCSK9 with monoclonal antibody-like affinity at 1/100th of the molecular weight.”
The current phase 2 study included 381 adult patients (49% female; median age 62 years) with a wide range of ASCVD risk. Average LDL-C level was 119.5 mg/dL at baseline. Around 40% of patients were not taking statins, 35% were on low- to moderate-intensity statin therapy, and 26% were on high-intensity statin therapy.
They were randomly assigned to four different doses of MK-0616 (6, 12, 18, or 30 mg once daily) or matching placebo.
Results showed that all doses of MK-0616 demonstrated statistically significant differences in percentage change in LDL-C from baseline to week 8 vs. placebo: –41.2% (6 mg), –55.7% (12 mg), –59.1% (18 mg), and –60.9% (30 mg).
The mean percentage changes in ApoB from baseline vs. placebo were –32.8%, –45.8%, –48.7%, and 51.8% for the four escalating doses of the drug. And non-HDL cholesterol changes were –35.9%, –50.5%, –53.2%, and –55.8% respectively.
The proportion of participants at protocol-defined goals for LDL reduction was 80.5%, 85.5%, 90.8%, and 90.8% with MK-0616 at the 6-mg, 12-mg, 18-mg, and 30-mg doses, compared with 9.3% with placebo.
Dr. Ballantyne reported that the efficacy looked similar in all subgroups, and regardless of baseline therapy.
“This was a dose-finding study, which will help select a dose to be taken forward in larger studies, and it looks from these results as though you get most of the efficacy by 12 mg,” he added.
Adverse events occurred in a proportion of participants in the MK-0616 groups (39.5% to 43.4%) similar to that of placebo (44.0%), and discontinuations as a result of adverse events occurred in two or fewer participants in any treatment group.
‘Super exciting’
Putting the results of his study into perspective at an ACC press conference, Rhonda Cooper-DeHoff, PharmD, associate professor in the department of pharmacotherapy and translational research at the University of Florida in Gainesville, commented.
“For the last quarter of a century we have had statins available to treat elevated LDL and atherosclerosis and despite that we have many patients who refuse to take statins or are afraid to take statins,” she said. “This is not about cost as the statins are all available generically now. But many patients claim to be intolerant or unresponsive.”
She noted that in 2015/2016 the first injectable PCSK9 inhibitors became available “which really were very exciting molecules, but they have a high cost and access issues, and patients often do not like injections so there are still a lot of issues.”
Dr. Cooper-DeHoff pointed out that this oral PCSK9 inhibitor seems to be as effective at lowering LDL as the injectable products regardless of whether statins are on board or not, which she said was “super exciting.”
She added: “We are all going to be waiting excitedly for the outcome data with this oral PCSK9 inhibitor.”
She also noted that another study (CLEAR Outcomes) presented at the ACC meeting showed good lipid-lowering results and a reduction in cardiovascular outcomes in statin-intolerant patients with another oral lipid lowering drug, bempedoic acid (Nexletol).
She said the two oral drugs promised a “very bright for the future for LDL lowering and the treatment of atherosclerosis in our patients,” adding that “we are now really chipping away at the barriers to achieving the holy grail of guideline-directed LDL lowering to prevent hard outcomes.”
The results were published online in the Journal of the American College of Cardiology at the time of presentation.
This study was funded by Merck. Dr. Ballantyne has received grant/research support through his institution from Abbott Diagnostic, Akcea, Amgen, Arrowhead, Esperion, Ionis, Merck, New Amsterdam, Novartis, Novo Nordisk, Regeneron, and Roche Diagnostics and has been a consultant for 89Bio, Abbott Diagnostics, Alnylam Pharmaceuticals, Althera, Amarin, Amgen, Arrowhead, AstraZeneca, Denka Seiken, Esperion, Genentech, Gilead, Illumina, Ionis, Matinas BioPharma, Merck, New Amsterdam, Novartis, Novo Nordisk, Pfizer, Regeneron, and Roche Diagnostics.
A version of this article first appeared on Medscape.com.
FROM ACC 2023
Some, not all, ultraprocessed foods linked to type 2 diabetes
High total intake of ultraprocessed food (UPF) is associated with an increased risk of developing type 2 diabetes, suggests a large-scale analysis that nevertheless revealed that the risk applies only to certain such foods.

The research was recently published in Diabetes Care by Zhangling Chen, PhD, Erasmus MC Rotterdam, Netherlands, and colleagues.
Examining almost 200,000 participants in three U.S. studies, yielding more than 5 million person-years of follow-up, the scientists found that high intake of UPF was associated with a 28% increased risk of type 2 diabetes, after statistical adjustments.
However, the increased risk was restricted to certain UPFs, including ready meals, refined breads, sweetened beverages, and sauces and condiments, with other foods considered UPFs, such as cereals, dark- and whole grain breads, and packaged sweet and savory snacks, among others, associated with a reduced risk of diabetes.
Senior author Jean-Philippe Drouin-Chartier, PhD, Nutrition Center, Laval University, Quebec City, told this news organization: “While whole grain breads can be considered as ultraprocessed foods, their consumption should not be discouraged. In our study, we observed that whole grain breads consumption is inversely associated with type 2 diabetes risk. This is supported by many studies linking dietary fiber consumption to better cardiometabolic health.”
Ultraprocessed food intake higher in the U.S. than in Europe
The researchers note that a handful of European studies have also reported an association between UPF consumption and increased type 2 diabetes risk, with the effect ranging from 15% to 53%, depending on the level of intake and the cohort of patients studied.
They note, however, that total UPF intake in the U.S. is “much higher than in Europe,” particularly in the case of ultraprocessed breads and cereals and artificially or sugar-sweetened beverages.
In the current study, they examined data on 71,781 women from the Nurses’ Health Study, 87,918 women from the NHS II, and 38,847 men from the Health Professional Follow-up Study, none of whom had cardiovascular disease, cancer, or diabetes at baseline.
In all three studies, questionnaires were administered every 2 years to collect demographic, lifestyle, and medical information, and a validated food frequency questionnaire was used every 2-4 years to assess participants’ diets over 30 years of follow-up.
Using the NOVA Food Classification system, the items on the food frequency questionnaire were categorized into one of four groups: unprocessed or minimally processed foods; processed culinary ingredients; processed foods; or UPFs, which were subdivided into nine mutually exclusive subgroups.
Servings per day were then used to determine individual UPF intake.
Higher total UPF intake was associated with a greater total energy intake, body mass index, and prevalence of hypercholesterolemia and/or hypertension, as well as lower healthy eating scores and physical activity.
The researchers calculated that, over 5,187,678 person-years of follow-up, there were 19,503 cases of type 2 diabetes across the three study cohorts.
Multivariate analysis taking into consideration a range of potential risk factors, including BMI, revealed that, across the three study cohorts, the highest quintile of UPF intake was associated with a significantly increased risk of type 2 diabetes.
Compared with the lowest quintile of UPF intake, the hazard ratio for incident type 2 diabetes was 1.28 (P < .0001), with an increase in risk per additional serving per day of 3%.
The UPFs associated with a higher type 2 diabetes risk were as previously described and also included animal-based products and ready-to-eat mixed dishes.
In contrast, intake of UPFs including cereals, dark and whole grain breads, packaged sweet and savory snacks, fruit-based products, and yogurt and dairy-based desserts were linked to a reduced risk of type 2 diabetes.
Then to further validate their findings, the researchers conducted a meta-analysis of their own and four additional studies, comprising 415,554 participants and 21,932 events, with a follow-up of 3.4-32.0 years.
They determined that the pooled relative risk of type 2 diabetes with the highest versus lowest levels of UPF consumption was 1.40, with each 10% increase in total UPF intake associated with a 12% increase in diabetes risk.
Ideal is to have access to minimally processed foods
The NOVA food classification system states that UPFs are industrial formulations “made mostly or entirely with substances extracted from foods, often chemically modified, with additives and with little, if any, whole foods added.”
A recent study questioned the value of the NOVA classification after finding that it had “low consistency” when assigning foods.
Previous studies have nevertheless revealed that UPFs and their constituents negatively affect the gut microbiota and can cause systemic inflammation, insulin resistance, and increased body weight.
Dr. Drouin-Chartier concluded: “There is a need to facilitate ... access to minimally processed foods. This encompasses [appropriate] pricing and physical access [to such foods], that is, addressing the issue of food deserts.”
The NHS I and II and HPFS studies are supported by National Institutes of Health. Dr. Drouin-Chartier has reported a relationship with the Dairy Farmers of Canada. No other financial relationships were declared.
A version of this article first appeared on Medscape.com.
High total intake of ultraprocessed food (UPF) is associated with an increased risk of developing type 2 diabetes, suggests a large-scale analysis that nevertheless revealed that the risk applies only to certain such foods.
The research was recently published in Diabetes Care by Zhangling Chen, PhD, Erasmus MC Rotterdam, Netherlands, and colleagues.
Examining almost 200,000 participants in three U.S. studies, yielding more than 5 million person-years of follow-up, the scientists found that high intake of UPF was associated with a 28% increased risk of type 2 diabetes, after statistical adjustments.
However, the increased risk was restricted to certain UPFs, including ready meals, refined breads, sweetened beverages, and sauces and condiments, with other foods considered UPFs, such as cereals, dark- and whole grain breads, and packaged sweet and savory snacks, among others, associated with a reduced risk of diabetes.
Senior author Jean-Philippe Drouin-Chartier, PhD, Nutrition Center, Laval University, Quebec City, told this news organization: “While whole grain breads can be considered as ultraprocessed foods, their consumption should not be discouraged. In our study, we observed that whole grain breads consumption is inversely associated with type 2 diabetes risk. This is supported by many studies linking dietary fiber consumption to better cardiometabolic health.”
Ultraprocessed food intake higher in the U.S. than in Europe
The researchers note that a handful of European studies have also reported an association between UPF consumption and increased type 2 diabetes risk, with the effect ranging from 15% to 53%, depending on the level of intake and the cohort of patients studied.
They note, however, that total UPF intake in the U.S. is “much higher than in Europe,” particularly in the case of ultraprocessed breads and cereals and artificially or sugar-sweetened beverages.
In the current study, they examined data on 71,781 women from the Nurses’ Health Study, 87,918 women from the NHS II, and 38,847 men from the Health Professional Follow-up Study, none of whom had cardiovascular disease, cancer, or diabetes at baseline.
In all three studies, questionnaires were administered every 2 years to collect demographic, lifestyle, and medical information, and a validated food frequency questionnaire was used every 2-4 years to assess participants’ diets over 30 years of follow-up.
Using the NOVA Food Classification system, the items on the food frequency questionnaire were categorized into one of four groups: unprocessed or minimally processed foods; processed culinary ingredients; processed foods; or UPFs, which were subdivided into nine mutually exclusive subgroups.
Servings per day were then used to determine individual UPF intake.
Higher total UPF intake was associated with a greater total energy intake, body mass index, and prevalence of hypercholesterolemia and/or hypertension, as well as lower healthy eating scores and physical activity.
The researchers calculated that, over 5,187,678 person-years of follow-up, there were 19,503 cases of type 2 diabetes across the three study cohorts.
Multivariate analysis taking into consideration a range of potential risk factors, including BMI, revealed that, across the three study cohorts, the highest quintile of UPF intake was associated with a significantly increased risk of type 2 diabetes.
Compared with the lowest quintile of UPF intake, the hazard ratio for incident type 2 diabetes was 1.28 (P < .0001), with an increase in risk per additional serving per day of 3%.
The UPFs associated with a higher type 2 diabetes risk were as previously described and also included animal-based products and ready-to-eat mixed dishes.
In contrast, intake of UPFs including cereals, dark and whole grain breads, packaged sweet and savory snacks, fruit-based products, and yogurt and dairy-based desserts were linked to a reduced risk of type 2 diabetes.
Then to further validate their findings, the researchers conducted a meta-analysis of their own and four additional studies, comprising 415,554 participants and 21,932 events, with a follow-up of 3.4-32.0 years.
They determined that the pooled relative risk of type 2 diabetes with the highest versus lowest levels of UPF consumption was 1.40, with each 10% increase in total UPF intake associated with a 12% increase in diabetes risk.
Ideal is to have access to minimally processed foods
The NOVA food classification system states that UPFs are industrial formulations “made mostly or entirely with substances extracted from foods, often chemically modified, with additives and with little, if any, whole foods added.”
A recent study questioned the value of the NOVA classification after finding that it had “low consistency” when assigning foods.
Previous studies have nevertheless revealed that UPFs and their constituents negatively affect the gut microbiota and can cause systemic inflammation, insulin resistance, and increased body weight.
Dr. Drouin-Chartier concluded: “There is a need to facilitate ... access to minimally processed foods. This encompasses [appropriate] pricing and physical access [to such foods], that is, addressing the issue of food deserts.”
The NHS I and II and HPFS studies are supported by National Institutes of Health. Dr. Drouin-Chartier has reported a relationship with the Dairy Farmers of Canada. No other financial relationships were declared.
A version of this article first appeared on Medscape.com.
High total intake of ultraprocessed food (UPF) is associated with an increased risk of developing type 2 diabetes, suggests a large-scale analysis that nevertheless revealed that the risk applies only to certain such foods.
The research was recently published in Diabetes Care by Zhangling Chen, PhD, Erasmus MC Rotterdam, Netherlands, and colleagues.
Examining almost 200,000 participants in three U.S. studies, yielding more than 5 million person-years of follow-up, the scientists found that high intake of UPF was associated with a 28% increased risk of type 2 diabetes, after statistical adjustments.
However, the increased risk was restricted to certain UPFs, including ready meals, refined breads, sweetened beverages, and sauces and condiments, with other foods considered UPFs, such as cereals, dark- and whole grain breads, and packaged sweet and savory snacks, among others, associated with a reduced risk of diabetes.
Senior author Jean-Philippe Drouin-Chartier, PhD, Nutrition Center, Laval University, Quebec City, told this news organization: “While whole grain breads can be considered as ultraprocessed foods, their consumption should not be discouraged. In our study, we observed that whole grain breads consumption is inversely associated with type 2 diabetes risk. This is supported by many studies linking dietary fiber consumption to better cardiometabolic health.”
Ultraprocessed food intake higher in the U.S. than in Europe
The researchers note that a handful of European studies have also reported an association between UPF consumption and increased type 2 diabetes risk, with the effect ranging from 15% to 53%, depending on the level of intake and the cohort of patients studied.
They note, however, that total UPF intake in the U.S. is “much higher than in Europe,” particularly in the case of ultraprocessed breads and cereals and artificially or sugar-sweetened beverages.
In the current study, they examined data on 71,781 women from the Nurses’ Health Study, 87,918 women from the NHS II, and 38,847 men from the Health Professional Follow-up Study, none of whom had cardiovascular disease, cancer, or diabetes at baseline.
In all three studies, questionnaires were administered every 2 years to collect demographic, lifestyle, and medical information, and a validated food frequency questionnaire was used every 2-4 years to assess participants’ diets over 30 years of follow-up.
Using the NOVA Food Classification system, the items on the food frequency questionnaire were categorized into one of four groups: unprocessed or minimally processed foods; processed culinary ingredients; processed foods; or UPFs, which were subdivided into nine mutually exclusive subgroups.
Servings per day were then used to determine individual UPF intake.
Higher total UPF intake was associated with a greater total energy intake, body mass index, and prevalence of hypercholesterolemia and/or hypertension, as well as lower healthy eating scores and physical activity.
The researchers calculated that, over 5,187,678 person-years of follow-up, there were 19,503 cases of type 2 diabetes across the three study cohorts.
Multivariate analysis taking into consideration a range of potential risk factors, including BMI, revealed that, across the three study cohorts, the highest quintile of UPF intake was associated with a significantly increased risk of type 2 diabetes.
Compared with the lowest quintile of UPF intake, the hazard ratio for incident type 2 diabetes was 1.28 (P < .0001), with an increase in risk per additional serving per day of 3%.
The UPFs associated with a higher type 2 diabetes risk were as previously described and also included animal-based products and ready-to-eat mixed dishes.
In contrast, intake of UPFs including cereals, dark and whole grain breads, packaged sweet and savory snacks, fruit-based products, and yogurt and dairy-based desserts were linked to a reduced risk of type 2 diabetes.
Then to further validate their findings, the researchers conducted a meta-analysis of their own and four additional studies, comprising 415,554 participants and 21,932 events, with a follow-up of 3.4-32.0 years.
They determined that the pooled relative risk of type 2 diabetes with the highest versus lowest levels of UPF consumption was 1.40, with each 10% increase in total UPF intake associated with a 12% increase in diabetes risk.
Ideal is to have access to minimally processed foods
The NOVA food classification system states that UPFs are industrial formulations “made mostly or entirely with substances extracted from foods, often chemically modified, with additives and with little, if any, whole foods added.”
A recent study questioned the value of the NOVA classification after finding that it had “low consistency” when assigning foods.
Previous studies have nevertheless revealed that UPFs and their constituents negatively affect the gut microbiota and can cause systemic inflammation, insulin resistance, and increased body weight.
Dr. Drouin-Chartier concluded: “There is a need to facilitate ... access to minimally processed foods. This encompasses [appropriate] pricing and physical access [to such foods], that is, addressing the issue of food deserts.”
The NHS I and II and HPFS studies are supported by National Institutes of Health. Dr. Drouin-Chartier has reported a relationship with the Dairy Farmers of Canada. No other financial relationships were declared.
A version of this article first appeared on Medscape.com.
FROM DIABETES CARE