User login
ONLINE EXCLUSIVE: Hospitalists discuss the time-honored tradition of hospital payments to HM groups
In the Literature: HM-Related Research You Need to Know
Literature at a Glance
A guide to this month’s studies
- Risks of preoperative tobacco use
- Timing of perioperative beta-blocker use and outcomes
- Continuous vs. bolus dose diuretics in CHF
- Outcomes of carotid endearterectomy and carotid artery stenting
- Protocol for low-risk chest pain
- Effect of esomeprazole on recurrent ulcer rates in clopidogrel users
- Effect of ICU QI project on hospital mortality
- Acute kidney injury risks after coronary angiography
Smokers Have Worse Perioperative Outcomes
Clinical question: Do current smokers have worse 30-day postoperative outcomes than nonsmokers after noncardiac surgery?
Background: Approximately 20% of adults in the U.S. smoke cigarettes, and a significant fraction of surgical patients are current smokers. Despite concerns that smoking is associated with worse postoperative outcomes, these increased risks have not been quantified across multiple outcomes.
Study design: Retrospective cohort study.
Setting: Surgical patients in 200 centers throughout the United States.
Synopsis: Data from the American College of Surgeons National Surgical Quality Improvement Program from 2005 to 2008 were acquired, and 391,006 patient records were reviewed. Postoperative morbidity and mortality were significantly greater in smokers. Current smokers had a 40% increased odds of death at 30 days compared to people who had never smoked (OR 1.38, 95% CI, 1.11-1.72). Current smokers also had significantly greater odds of pulmonary complications, including pneumonia (OR 2.09, 95% CI, 1.80-2.43), unplanned intubation (OR 1.87, 95% CI, 1.58-2.21), and mechanical ventilation (OR 1.53, 95% CI, 1.31-1.79).
Furthermore, current smokers had significantly greater odds of postoperative cardiac arrest (OR 1.57, 95% CI, 1.10-2.25), myocardial infarction (OR 1.80, 95% CI, 1.11-2.25), and stroke (OR 1.73, 95% CI, 1.18-2.53). Odds of infectious complications were increased in current smokers, including deep incisional infections (OR 1.42, 95% CI, 1.21-1.68), sepsis (OR 1.30, 95% CI, 1.20-1.60), and septic shock (OR 1.55, 95% CI, 1.29-1.87).
Limitations of this study include self-reporting of smoking habits and absence of detailed smoking history just before and after surgery.
Bottom line: Current smokers have significantly increased postoperative morbidity and mortality after noncardiac surgery.
Citation: Turan A, Mascha EJ, Roberman D, et al. Smoking and perioperative outcomes. Anesthesiology. 2011;114(4):837-846.
Chronic Beta-Blockade Reduces Postoperative Myocardial Ischemia
Clinical question: Does the timing of beta-blocker exposure affect cardiovascular outcomes in patients undergoing elective, noncardiac surgery?
Background: Several studies have demonstrated that beta-blockers are associated with decreased perioperative cardiovascular morbidity and mortality. Study designs have varied greatly, and differences in dosing and timing of beta-blocker administration have caused conflicting results. The question of when to initiate beta-blockers prior to surgery remains controversial.
Study design: Prospective cohort study.
Setting: Three academic medical centers in Canada.
Synopsis: Data from 1,398 patients who had elective, noncardiac surgery with either acute (n=436) or chronic (n=962) beta-blocker exposure were analyzed. Acute exposure was defined as receiving a beta-blocker for the first time within 48 hours after surgery, whereas chronic beta-blocker exposure was defined as receiving a beta-blocker seven to 10 days prior to surgery.
Patients with chronic beta-blocker exposure were more likely to have a history of coronary disease, heart failure, or hypertension and were more likely to be receiving statins, antiplatelet agents, and angiotensin-converting enzyme inhibitors. The primary outcome was a composite of major cardiac events, including myocardial infarction, nonfatal cardiac arrest, and 30-day mortality.
Major cardiac events occurred more often in patients with acute versus chronic beta-blocker exposure in both the entire cohort (8.3% vs. 4.7%) and in the propensity-matched cohort (8.0% vs. 3.0%). Myocardial infarction accounted for the majority of cardiac events.
There are several limitations of this study: The sample size was small, the beta-blocker and dosage used varied, and the indication and exact duration of chronic beta-blocker therapy was unknown.
Bottom line: Chronic beta-blocker therapy reduces major cardiac events compared with acute beta-blocker therapy in patients undergoing elective, noncardiac surgery.
Citation: Ellenberger C, Tait G, Beattie WS. Chronic beta-blockade is associated with a better outcome after elective noncardiac surgery than acute beta-blockade: a single-center propensity-matched cohort study. Anesthesiology. 2011;114(4):817-823.
Continuous and Bolus Dosing of Furosemide Provides Similar Outcomes in Heart Failure
Clinical question: Does continuous infusion compared to bolus dosing of furosemide improve clinical outcomes in patients with acute decompensated heart failure?
Background: Diuresis with furosemide is commonly used to manage acute decompensated heart failure, but it is uncertain which dosing strategy is optimal. Continuous infusion of furosemide has been proposed as a more effective method of diuresis compared with bolus dosing, especially when higher doses are required, but data comparing the two strategies are limited.
Study design: Randomized, double-blind, controlled trial.
Setting: Twenty-six clinical sites in the U.S. and Canada.
Synopsis: Researchers randomized 308 patients with acute decompensated heart failure to either continuous or bolus intravenous dosing, which was calculated as either the equivalent of their daily oral dose (low-dose strategy) or 2.5 times their daily dose (high-dose strategy). Mean ejection fraction was 35%. Primary endpoints were patients’ assessment of symptoms based on a visual-analogue scale quantified as area under the curve, as well as change in serum creatinine level at 72 hours.
No significant differences between the continuous and bolus dosing groups were evidenced in primary endpoints at 72 hours. Patients in the bolus group had more dose increases at 48 hours (21% vs. 11%, P=0.01). Patients in the high-dose group were more likely to change from intravenous to oral doses at 48 hours (31% vs. 17%, P<0.001) and had greater net fluid loss (4.9L vs. 3.6L, P=0.01). More patients in the high-dose versus low-dose group had an increase in creatinine ≥0.3 mg/dL (23% vs. 14%, P=0.04). Hospital length of stay, readmission, and mortality rates were similar between the groups.
Bottom line: Diuretic therapy administered by continuous infusion or bolus dosing in patients with acute decompensated heart failure have equivocal effects on patients’ symptoms and kidney function.
Citation: Felker GM, Lee KL, Bull DA, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011;364(9):797-805.
Carotid Endarterectomy Is Better than Carotid Artery Stenting
Clinical question: How do the clinical outcomes of carotid artery stenting compare with those of carotid endarterectomy?
Background: Whether carotid artery stenting or carotid endarterectomy is the preferred therapy for patients with carotid artery stenosis has been highly controversial. This study was a meta-analysis of all available data from randomized trials comparing carotid endarterectomy to carotid artery stenting.
Study design: Meta-analysis.
Setting: Teaching and nonteaching hospitals.
Synopsis: Thirteen randomized trials were identified with 3,723 patients who had undergone endarterectomy and 3,754 patients who had undergone carotid artery stenting. Outcomes included stroke, myocardial infarction, cranial nerve injury, and death or stroke, and these outcomes were divided as either short-term (<30 days) or long-term (>1 year) outcomes.
Patients who had undergone carotid artery stenting had less risk of short-term myocardial infarction (OR 0.48, 95% CI, 0.29–9.78, P=0.003) and less risk of cranial nerve injury (OR 0.09, 95% CI, 0.05–0.16, P<0.001). However, carotid artery stenting had a significantly higher risk of short-term stroke and combined death or stroke, and also significantly higher long-term risk of stroke and combined death or stroke. The association between carotid artery stenting and stroke was stronger in the subgroup of patients >68 years but not in patients <68 years. There was no significant heterogeneity, and no significant modifying associations were revealed by meta-regression analysis.
Limitations include potentially unpublished small studies favoring carotid endarterectomy and a significant publication bias regarding short-term death.
Bottom line: Although carotid artery stenting has less short-term risk of myocardial infarction and cranial nerve injury, carotid endarterectomy has less short-term and long-term risks of stroke and death.
Citation: Economopoulus KP, Sergentanis TN, Tsivgoulis G, Mariolis AD, Stefanadis C. Carotid artery stenting versus carotid endarterectomy: a comprehensive meta-analysis of short-term and long-term outcomes. Stroke. 2011;42:687-692.
Chest Pain Protocol Can Identify Low-Risk Chest Pain in Emergency Departments
Clinical question: Can a two-hour accelerated diagnostic protocol (ADP) based on electrocardiogram, point-of-care biomarkers, and Thrombolysis in Myocardial Infarction (TIMI) score safely identify patients with chest pain at very low short-term risk of major cardiac events?
Background: Evaluation of patients presenting to EDs with chest pain utilize significant amounts of hospital resources. A safe, reproducible, and expeditious process to identify patients at low risk for short-term cardiac events is desired.
Study design: Prospective cohort study.
Setting: Fourteen urban EDs in nine countries across the Asia-Pacific region.
Synopsis: The study included 3,582 patients presenting to an ED with at least five minutes of chest pain suggestive of an acute coronary syndrome and for whom further evaluation with serial cardiac biomarkers was planned. A negative ADP was defined as TIMI score of 0, no new ischemic changes on initial electrocardiogram, and normal cardiac biomarkers at zero and two hours after arrival.
All components of the ADP were negative for 352 patients (9.8%). Only three low-risk patients (0.9%) by ADP had a major cardiac event during the 30-day follow-up period, yielding a negative predictive value of 99.1% (95% CI, 97.3-99.8%). Mean hospital stay for the low-risk group with a negative ADP was 43 hours with a median of 26 hours. The authors suggest that a 10% reduction in prolonged workups of patients with chest pain could be seen with implementation of this protocol.
Potential limitations include applicability only to a select cohort of patients with chest pain and the low specificity of the protocol.
Bottom line: A two-hour diagnostic protocol can help expedite discharge of patients with very-low-risk chest pain.
Citation: Than T, Cullen L, Reid CM, et al. A 2-h diagnostic protocol to assess patients with chest pain symptoms in the Asia-Pacific region (ASPECT): a prospective observational validation study. Lancet. 2011;337:1077-1084.
Esomeprazole Reduces Peptic Ulcer Recurrence in Patients on Clopidogrel
Clinical question: Does esomeprazole prevent recurrent peptic ulcers in patients with atherosclerosis on clopidogrel?
Background: Although clopidogrel is sometimes used as an alternative antiplatelet agent to aspirin, a significant rate of recurrent ulcer bleeding on clopidogrel has been described. No previous prospective trial has studied whether a proton-pump inhibitor (PPI) can reduce the risk of peptic ulcer recurrence or bleeding in atherosclerotic patients on clopidogrel.
Study design: Randomized controlled trial.
Setting: A single veterans hospital in Taiwan.
Synopsis: One hundred sixty-five patients were enrolled with a past history of peptic ulcer disease, no signs of ulcer recurrence by endoscopy, and current use of clopidogrel 37.5 mg to 75 mg per day. All patients had atherosclerosis and had been on clopidogrel for at least two weeks, without aspirin, corticosteroids, anticoagulants, or recent treatment with a PPI. Patients were randomized to clopidogrel 75 mg at night (n=82) or clopidogrel 75 mg at night plus esomeprazole 20 mg before breakfast. Follow-up endoscopy was performed at six months or as needed for symptoms.
Recurrence of ulcer was found in 1.2% of patients on clopidogrel plus esomeprazole versus 11.0% in patients on clopidogrel alone (95% CI, 2.6-17.0%; P=0.009). The pharmacodynamic study revealed no significant differences in platelet aggregation within or between treatment groups on day 1 or day 28. No significant differences were seen on the incidence of ischemic events in this setting, but the trial was underpowered to draw conclusions on this outcome.
An important limitation is that the findings of this study are applicable only to patients on clopidogrel monotherapy and not dual antiplatelet therapy.
Bottom line: A significant reduction in recurrent peptic ulcers is seen with the combination of esomeprazole plus clopidogrel, versus clopidogrel alone, in patients with atherosclerosis and a history of peptic ulcer disease.
Citation: Hsu PI, Lai KH, Liu CP. Esomeprazole with clopidogrel reduces peptic ulcer recurrence, compared with clopidogrel alone, in patients with atherosclerosis. Gastroenterology. 2011;140:791-798.
ICU Quality-Improvement Project Reduces Hospital Mortality
Clinical question: Does a quality-improvement (QI) project in the ICU reduce in-hospital mortality and length of stay among elderly adults?
Background: Previous studies have shown that ICU-acquired infections are associated with increased morbidity and mortality, and QI initiatives reduce hospital-acquired infections. However, it has not been demonstrated that QI projects in the ICU reduce in-hospital mortality or length of stay.
Study design: Retrospective cohort study.
Setting: Four hundred fifty-nine Midwestern hospitals.
Synopsis: This study included 238,937 adults age >65 who were hospitalized in an ICU from 2001 to 2006 at one of 95 hospitals invited to implement the Keystone ICU Project. The control group included 1,091,547 elderly adults at one of 364 hospitals not invited to participate in the project. The Keystone ICU Project implements evidence-based practices to reduce rates of catheter-related bloodstream infections and ventilator-associated pneumonia.
Hospital mortality was not significantly reduced during initiation or implementation of the project; however, a significant reduction in hospital mortality occurred in the study group during one to 12 months post-implementation (OR=0.83 vs. 0.88, P=0.041) and 13 to 22 months post-implementation (OR=0.76 vs. 0.84, P=0.007). In contrast, length of stay did not differ significantly between the two groups, but the study was underpowered for this outcome.
The study is limited by the complexity of the Keystone ICU Project, as well as the exclusion of smaller hospitals and nonelderly adults. The study is promising because implementing a QI project in the ICU is associated with no known harms and might confer a mortality benefit at a relatively low cost.
Bottom line: Elderly adults had lower in-hospital mortality after implementation of the Keystone QI project in ICUs.
Citation: Lipitz-Snyderman A, Steinwachs D, Needham DM, Colantuoni E, Morlock LL, Pronovost PJ. Impact of a statewide intensive care unit quality improvement initiative on hospital mortality and length of stay: retrospective comparative analysis. BMJ. 2011;342:d219.
Serious Long-Term Risks with Acute Kidney Injury after Coronary Angiography
Clinical question: Does postcoronary angiography acute kidney injury (AKI) increase the risk of poor long-term clinical outcomes?
Background: Previous studies have shown that AKI following coronary angiography increases the risk of poor short-term clinical outcomes, such as in-hospital myocardial infarction, prolonged hospital stay, and early mortality. Little is known about the long-term cardiovascular and renal outcomes following post-coronary angiography AKI.
Study design: Retrospective cohort study.
Setting: All coronary angiography centers in Alberta, Canada.
Synopsis: The study included 14,782 adults who were ≥18 years of age, underwent coronary angiography, had a baseline creatinine measurement, did not have end-stage renal disease (ESRD), and had a creatinine measurement within seven days after coronary angiography.
During a median follow-up period of 19.7 months, 1,099 (7.4%) patients developed stage 1 AKI and 321 (2.2%) developed stage 2 or 3 AKI. Mortality increased twofold with stage 1 AKI and >3-fold with stage 2 or 3 AKI. Risk of ESRD increased substantially by >11-fold in patients with stage 2 or 3 AKI. Risk of hospitalization for subsequent AKI, myocardial infarction, and heart failure also increased significantly following post-coronary angiography AKI.
Patients who experienced AKI were older, had more severe CAD, were more likely to have such comorbidities as DM, HTN, and heart failure, and had lower baseline GFRs. However, the underlying comorbidities do not completely explain the increased risk of poor long-term outcomes in the adjusted analysis.
Limitations include missing or underestimating mild cases of AKI, residual confounding from unmeasured variables, and inability of retrospective comparative studies to establish causality.
Bottom line: Adults with post-coronary angiography AKI are at increased risk of poor long-term cardiovascular and renal outcomes.
Citation: James MT, Ghali WA, Knudtson ML, et al. Associations between acute kidney injury and cardiovascular and renal outcomes after coronary angiography. Circulation. 2011;123(4):409-416. TH
Pediatric HM Literature
Proton-Pump Inhibitors Ineffective for Gastroesophageal Reflux Disease in Children

Clinical question: What is the efficacy of proton-pump inhibitors (PPIs) in children with gastroesophageal reflux disease (GERD)?
Background: Gastroesophageal reflux is both a common and normal phenomenon in infants. GERD refers to the presence of abnormal symptoms ascribed to the reflux and frequently is treated in children in a manner similar to adults with reflux esophagitis. PPIs often are prescribed as front-line treatment, and their use has increased dramatically in recent years, though their effectiveness in children remains unclear.
Study design: Systematic review of the literature.
Setting: Hawaii’s largest health insurer.
Synopsis: Medline, Embase, and the Cochrane Database of Systematic Reviews were searched for randomized controlled trials (RCTs) and crossover studies performed to evaluate the efficacy of PPIs in children 0-17 years with GERD and no complicating diseases. Ten RCTs and two crossover studies were analyzed and rated independently by two reviewers.
Due to significant heterogeneity between the studies, a meta-analysis was not possible; studies were discussed separately. PPIs offered no advantage when compared with controls (alginates, ranitidine, different dosages of PPIs), and similar rates of adverse events were reported between treatment groups.
This study is hampered by notable heterogeneity of patient type, symptoms, and study design in many of the trials. However, the results are in line with discussions at a recent FDA Gastrointestinal Drugs Advisory Committee meeting, which reviewed the lack of efficacy of PPIs in infants in four recent Phase 3 clinical trials.
Bottom line: Little evidence supports the widespread use of PPIs in children.
Citation: Van der Pol RJ, Smits MJ, van Wijk MP, Omari TI, Tabbers MM, Benninga MA. Efficacy of proton-pump inhibitors in children with gastroesophageal reflux disease: a systematic review. Pediatrics. 2011;127:925-935.
Literature at a Glance
A guide to this month’s studies
- Risks of preoperative tobacco use
- Timing of perioperative beta-blocker use and outcomes
- Continuous vs. bolus dose diuretics in CHF
- Outcomes of carotid endearterectomy and carotid artery stenting
- Protocol for low-risk chest pain
- Effect of esomeprazole on recurrent ulcer rates in clopidogrel users
- Effect of ICU QI project on hospital mortality
- Acute kidney injury risks after coronary angiography
Smokers Have Worse Perioperative Outcomes
Clinical question: Do current smokers have worse 30-day postoperative outcomes than nonsmokers after noncardiac surgery?
Background: Approximately 20% of adults in the U.S. smoke cigarettes, and a significant fraction of surgical patients are current smokers. Despite concerns that smoking is associated with worse postoperative outcomes, these increased risks have not been quantified across multiple outcomes.
Study design: Retrospective cohort study.
Setting: Surgical patients in 200 centers throughout the United States.
Synopsis: Data from the American College of Surgeons National Surgical Quality Improvement Program from 2005 to 2008 were acquired, and 391,006 patient records were reviewed. Postoperative morbidity and mortality were significantly greater in smokers. Current smokers had a 40% increased odds of death at 30 days compared to people who had never smoked (OR 1.38, 95% CI, 1.11-1.72). Current smokers also had significantly greater odds of pulmonary complications, including pneumonia (OR 2.09, 95% CI, 1.80-2.43), unplanned intubation (OR 1.87, 95% CI, 1.58-2.21), and mechanical ventilation (OR 1.53, 95% CI, 1.31-1.79).
Furthermore, current smokers had significantly greater odds of postoperative cardiac arrest (OR 1.57, 95% CI, 1.10-2.25), myocardial infarction (OR 1.80, 95% CI, 1.11-2.25), and stroke (OR 1.73, 95% CI, 1.18-2.53). Odds of infectious complications were increased in current smokers, including deep incisional infections (OR 1.42, 95% CI, 1.21-1.68), sepsis (OR 1.30, 95% CI, 1.20-1.60), and septic shock (OR 1.55, 95% CI, 1.29-1.87).
Limitations of this study include self-reporting of smoking habits and absence of detailed smoking history just before and after surgery.
Bottom line: Current smokers have significantly increased postoperative morbidity and mortality after noncardiac surgery.
Citation: Turan A, Mascha EJ, Roberman D, et al. Smoking and perioperative outcomes. Anesthesiology. 2011;114(4):837-846.
Chronic Beta-Blockade Reduces Postoperative Myocardial Ischemia
Clinical question: Does the timing of beta-blocker exposure affect cardiovascular outcomes in patients undergoing elective, noncardiac surgery?
Background: Several studies have demonstrated that beta-blockers are associated with decreased perioperative cardiovascular morbidity and mortality. Study designs have varied greatly, and differences in dosing and timing of beta-blocker administration have caused conflicting results. The question of when to initiate beta-blockers prior to surgery remains controversial.
Study design: Prospective cohort study.
Setting: Three academic medical centers in Canada.
Synopsis: Data from 1,398 patients who had elective, noncardiac surgery with either acute (n=436) or chronic (n=962) beta-blocker exposure were analyzed. Acute exposure was defined as receiving a beta-blocker for the first time within 48 hours after surgery, whereas chronic beta-blocker exposure was defined as receiving a beta-blocker seven to 10 days prior to surgery.
Patients with chronic beta-blocker exposure were more likely to have a history of coronary disease, heart failure, or hypertension and were more likely to be receiving statins, antiplatelet agents, and angiotensin-converting enzyme inhibitors. The primary outcome was a composite of major cardiac events, including myocardial infarction, nonfatal cardiac arrest, and 30-day mortality.
Major cardiac events occurred more often in patients with acute versus chronic beta-blocker exposure in both the entire cohort (8.3% vs. 4.7%) and in the propensity-matched cohort (8.0% vs. 3.0%). Myocardial infarction accounted for the majority of cardiac events.
There are several limitations of this study: The sample size was small, the beta-blocker and dosage used varied, and the indication and exact duration of chronic beta-blocker therapy was unknown.
Bottom line: Chronic beta-blocker therapy reduces major cardiac events compared with acute beta-blocker therapy in patients undergoing elective, noncardiac surgery.
Citation: Ellenberger C, Tait G, Beattie WS. Chronic beta-blockade is associated with a better outcome after elective noncardiac surgery than acute beta-blockade: a single-center propensity-matched cohort study. Anesthesiology. 2011;114(4):817-823.
Continuous and Bolus Dosing of Furosemide Provides Similar Outcomes in Heart Failure
Clinical question: Does continuous infusion compared to bolus dosing of furosemide improve clinical outcomes in patients with acute decompensated heart failure?
Background: Diuresis with furosemide is commonly used to manage acute decompensated heart failure, but it is uncertain which dosing strategy is optimal. Continuous infusion of furosemide has been proposed as a more effective method of diuresis compared with bolus dosing, especially when higher doses are required, but data comparing the two strategies are limited.
Study design: Randomized, double-blind, controlled trial.
Setting: Twenty-six clinical sites in the U.S. and Canada.
Synopsis: Researchers randomized 308 patients with acute decompensated heart failure to either continuous or bolus intravenous dosing, which was calculated as either the equivalent of their daily oral dose (low-dose strategy) or 2.5 times their daily dose (high-dose strategy). Mean ejection fraction was 35%. Primary endpoints were patients’ assessment of symptoms based on a visual-analogue scale quantified as area under the curve, as well as change in serum creatinine level at 72 hours.
No significant differences between the continuous and bolus dosing groups were evidenced in primary endpoints at 72 hours. Patients in the bolus group had more dose increases at 48 hours (21% vs. 11%, P=0.01). Patients in the high-dose group were more likely to change from intravenous to oral doses at 48 hours (31% vs. 17%, P<0.001) and had greater net fluid loss (4.9L vs. 3.6L, P=0.01). More patients in the high-dose versus low-dose group had an increase in creatinine ≥0.3 mg/dL (23% vs. 14%, P=0.04). Hospital length of stay, readmission, and mortality rates were similar between the groups.
Bottom line: Diuretic therapy administered by continuous infusion or bolus dosing in patients with acute decompensated heart failure have equivocal effects on patients’ symptoms and kidney function.
Citation: Felker GM, Lee KL, Bull DA, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011;364(9):797-805.
Carotid Endarterectomy Is Better than Carotid Artery Stenting
Clinical question: How do the clinical outcomes of carotid artery stenting compare with those of carotid endarterectomy?
Background: Whether carotid artery stenting or carotid endarterectomy is the preferred therapy for patients with carotid artery stenosis has been highly controversial. This study was a meta-analysis of all available data from randomized trials comparing carotid endarterectomy to carotid artery stenting.
Study design: Meta-analysis.
Setting: Teaching and nonteaching hospitals.
Synopsis: Thirteen randomized trials were identified with 3,723 patients who had undergone endarterectomy and 3,754 patients who had undergone carotid artery stenting. Outcomes included stroke, myocardial infarction, cranial nerve injury, and death or stroke, and these outcomes were divided as either short-term (<30 days) or long-term (>1 year) outcomes.
Patients who had undergone carotid artery stenting had less risk of short-term myocardial infarction (OR 0.48, 95% CI, 0.29–9.78, P=0.003) and less risk of cranial nerve injury (OR 0.09, 95% CI, 0.05–0.16, P<0.001). However, carotid artery stenting had a significantly higher risk of short-term stroke and combined death or stroke, and also significantly higher long-term risk of stroke and combined death or stroke. The association between carotid artery stenting and stroke was stronger in the subgroup of patients >68 years but not in patients <68 years. There was no significant heterogeneity, and no significant modifying associations were revealed by meta-regression analysis.
Limitations include potentially unpublished small studies favoring carotid endarterectomy and a significant publication bias regarding short-term death.
Bottom line: Although carotid artery stenting has less short-term risk of myocardial infarction and cranial nerve injury, carotid endarterectomy has less short-term and long-term risks of stroke and death.
Citation: Economopoulus KP, Sergentanis TN, Tsivgoulis G, Mariolis AD, Stefanadis C. Carotid artery stenting versus carotid endarterectomy: a comprehensive meta-analysis of short-term and long-term outcomes. Stroke. 2011;42:687-692.
Chest Pain Protocol Can Identify Low-Risk Chest Pain in Emergency Departments
Clinical question: Can a two-hour accelerated diagnostic protocol (ADP) based on electrocardiogram, point-of-care biomarkers, and Thrombolysis in Myocardial Infarction (TIMI) score safely identify patients with chest pain at very low short-term risk of major cardiac events?
Background: Evaluation of patients presenting to EDs with chest pain utilize significant amounts of hospital resources. A safe, reproducible, and expeditious process to identify patients at low risk for short-term cardiac events is desired.
Study design: Prospective cohort study.
Setting: Fourteen urban EDs in nine countries across the Asia-Pacific region.
Synopsis: The study included 3,582 patients presenting to an ED with at least five minutes of chest pain suggestive of an acute coronary syndrome and for whom further evaluation with serial cardiac biomarkers was planned. A negative ADP was defined as TIMI score of 0, no new ischemic changes on initial electrocardiogram, and normal cardiac biomarkers at zero and two hours after arrival.
All components of the ADP were negative for 352 patients (9.8%). Only three low-risk patients (0.9%) by ADP had a major cardiac event during the 30-day follow-up period, yielding a negative predictive value of 99.1% (95% CI, 97.3-99.8%). Mean hospital stay for the low-risk group with a negative ADP was 43 hours with a median of 26 hours. The authors suggest that a 10% reduction in prolonged workups of patients with chest pain could be seen with implementation of this protocol.
Potential limitations include applicability only to a select cohort of patients with chest pain and the low specificity of the protocol.
Bottom line: A two-hour diagnostic protocol can help expedite discharge of patients with very-low-risk chest pain.
Citation: Than T, Cullen L, Reid CM, et al. A 2-h diagnostic protocol to assess patients with chest pain symptoms in the Asia-Pacific region (ASPECT): a prospective observational validation study. Lancet. 2011;337:1077-1084.
Esomeprazole Reduces Peptic Ulcer Recurrence in Patients on Clopidogrel
Clinical question: Does esomeprazole prevent recurrent peptic ulcers in patients with atherosclerosis on clopidogrel?
Background: Although clopidogrel is sometimes used as an alternative antiplatelet agent to aspirin, a significant rate of recurrent ulcer bleeding on clopidogrel has been described. No previous prospective trial has studied whether a proton-pump inhibitor (PPI) can reduce the risk of peptic ulcer recurrence or bleeding in atherosclerotic patients on clopidogrel.
Study design: Randomized controlled trial.
Setting: A single veterans hospital in Taiwan.
Synopsis: One hundred sixty-five patients were enrolled with a past history of peptic ulcer disease, no signs of ulcer recurrence by endoscopy, and current use of clopidogrel 37.5 mg to 75 mg per day. All patients had atherosclerosis and had been on clopidogrel for at least two weeks, without aspirin, corticosteroids, anticoagulants, or recent treatment with a PPI. Patients were randomized to clopidogrel 75 mg at night (n=82) or clopidogrel 75 mg at night plus esomeprazole 20 mg before breakfast. Follow-up endoscopy was performed at six months or as needed for symptoms.
Recurrence of ulcer was found in 1.2% of patients on clopidogrel plus esomeprazole versus 11.0% in patients on clopidogrel alone (95% CI, 2.6-17.0%; P=0.009). The pharmacodynamic study revealed no significant differences in platelet aggregation within or between treatment groups on day 1 or day 28. No significant differences were seen on the incidence of ischemic events in this setting, but the trial was underpowered to draw conclusions on this outcome.
An important limitation is that the findings of this study are applicable only to patients on clopidogrel monotherapy and not dual antiplatelet therapy.
Bottom line: A significant reduction in recurrent peptic ulcers is seen with the combination of esomeprazole plus clopidogrel, versus clopidogrel alone, in patients with atherosclerosis and a history of peptic ulcer disease.
Citation: Hsu PI, Lai KH, Liu CP. Esomeprazole with clopidogrel reduces peptic ulcer recurrence, compared with clopidogrel alone, in patients with atherosclerosis. Gastroenterology. 2011;140:791-798.
ICU Quality-Improvement Project Reduces Hospital Mortality
Clinical question: Does a quality-improvement (QI) project in the ICU reduce in-hospital mortality and length of stay among elderly adults?
Background: Previous studies have shown that ICU-acquired infections are associated with increased morbidity and mortality, and QI initiatives reduce hospital-acquired infections. However, it has not been demonstrated that QI projects in the ICU reduce in-hospital mortality or length of stay.
Study design: Retrospective cohort study.
Setting: Four hundred fifty-nine Midwestern hospitals.
Synopsis: This study included 238,937 adults age >65 who were hospitalized in an ICU from 2001 to 2006 at one of 95 hospitals invited to implement the Keystone ICU Project. The control group included 1,091,547 elderly adults at one of 364 hospitals not invited to participate in the project. The Keystone ICU Project implements evidence-based practices to reduce rates of catheter-related bloodstream infections and ventilator-associated pneumonia.
Hospital mortality was not significantly reduced during initiation or implementation of the project; however, a significant reduction in hospital mortality occurred in the study group during one to 12 months post-implementation (OR=0.83 vs. 0.88, P=0.041) and 13 to 22 months post-implementation (OR=0.76 vs. 0.84, P=0.007). In contrast, length of stay did not differ significantly between the two groups, but the study was underpowered for this outcome.
The study is limited by the complexity of the Keystone ICU Project, as well as the exclusion of smaller hospitals and nonelderly adults. The study is promising because implementing a QI project in the ICU is associated with no known harms and might confer a mortality benefit at a relatively low cost.
Bottom line: Elderly adults had lower in-hospital mortality after implementation of the Keystone QI project in ICUs.
Citation: Lipitz-Snyderman A, Steinwachs D, Needham DM, Colantuoni E, Morlock LL, Pronovost PJ. Impact of a statewide intensive care unit quality improvement initiative on hospital mortality and length of stay: retrospective comparative analysis. BMJ. 2011;342:d219.
Serious Long-Term Risks with Acute Kidney Injury after Coronary Angiography
Clinical question: Does postcoronary angiography acute kidney injury (AKI) increase the risk of poor long-term clinical outcomes?
Background: Previous studies have shown that AKI following coronary angiography increases the risk of poor short-term clinical outcomes, such as in-hospital myocardial infarction, prolonged hospital stay, and early mortality. Little is known about the long-term cardiovascular and renal outcomes following post-coronary angiography AKI.
Study design: Retrospective cohort study.
Setting: All coronary angiography centers in Alberta, Canada.
Synopsis: The study included 14,782 adults who were ≥18 years of age, underwent coronary angiography, had a baseline creatinine measurement, did not have end-stage renal disease (ESRD), and had a creatinine measurement within seven days after coronary angiography.
During a median follow-up period of 19.7 months, 1,099 (7.4%) patients developed stage 1 AKI and 321 (2.2%) developed stage 2 or 3 AKI. Mortality increased twofold with stage 1 AKI and >3-fold with stage 2 or 3 AKI. Risk of ESRD increased substantially by >11-fold in patients with stage 2 or 3 AKI. Risk of hospitalization for subsequent AKI, myocardial infarction, and heart failure also increased significantly following post-coronary angiography AKI.
Patients who experienced AKI were older, had more severe CAD, were more likely to have such comorbidities as DM, HTN, and heart failure, and had lower baseline GFRs. However, the underlying comorbidities do not completely explain the increased risk of poor long-term outcomes in the adjusted analysis.
Limitations include missing or underestimating mild cases of AKI, residual confounding from unmeasured variables, and inability of retrospective comparative studies to establish causality.
Bottom line: Adults with post-coronary angiography AKI are at increased risk of poor long-term cardiovascular and renal outcomes.
Citation: James MT, Ghali WA, Knudtson ML, et al. Associations between acute kidney injury and cardiovascular and renal outcomes after coronary angiography. Circulation. 2011;123(4):409-416. TH
Pediatric HM Literature
Proton-Pump Inhibitors Ineffective for Gastroesophageal Reflux Disease in Children
Clinical question: What is the efficacy of proton-pump inhibitors (PPIs) in children with gastroesophageal reflux disease (GERD)?
Background: Gastroesophageal reflux is both a common and normal phenomenon in infants. GERD refers to the presence of abnormal symptoms ascribed to the reflux and frequently is treated in children in a manner similar to adults with reflux esophagitis. PPIs often are prescribed as front-line treatment, and their use has increased dramatically in recent years, though their effectiveness in children remains unclear.
Study design: Systematic review of the literature.
Setting: Hawaii’s largest health insurer.
Synopsis: Medline, Embase, and the Cochrane Database of Systematic Reviews were searched for randomized controlled trials (RCTs) and crossover studies performed to evaluate the efficacy of PPIs in children 0-17 years with GERD and no complicating diseases. Ten RCTs and two crossover studies were analyzed and rated independently by two reviewers.
Due to significant heterogeneity between the studies, a meta-analysis was not possible; studies were discussed separately. PPIs offered no advantage when compared with controls (alginates, ranitidine, different dosages of PPIs), and similar rates of adverse events were reported between treatment groups.
This study is hampered by notable heterogeneity of patient type, symptoms, and study design in many of the trials. However, the results are in line with discussions at a recent FDA Gastrointestinal Drugs Advisory Committee meeting, which reviewed the lack of efficacy of PPIs in infants in four recent Phase 3 clinical trials.
Bottom line: Little evidence supports the widespread use of PPIs in children.
Citation: Van der Pol RJ, Smits MJ, van Wijk MP, Omari TI, Tabbers MM, Benninga MA. Efficacy of proton-pump inhibitors in children with gastroesophageal reflux disease: a systematic review. Pediatrics. 2011;127:925-935.
Literature at a Glance
A guide to this month’s studies
- Risks of preoperative tobacco use
- Timing of perioperative beta-blocker use and outcomes
- Continuous vs. bolus dose diuretics in CHF
- Outcomes of carotid endearterectomy and carotid artery stenting
- Protocol for low-risk chest pain
- Effect of esomeprazole on recurrent ulcer rates in clopidogrel users
- Effect of ICU QI project on hospital mortality
- Acute kidney injury risks after coronary angiography
Smokers Have Worse Perioperative Outcomes
Clinical question: Do current smokers have worse 30-day postoperative outcomes than nonsmokers after noncardiac surgery?
Background: Approximately 20% of adults in the U.S. smoke cigarettes, and a significant fraction of surgical patients are current smokers. Despite concerns that smoking is associated with worse postoperative outcomes, these increased risks have not been quantified across multiple outcomes.
Study design: Retrospective cohort study.
Setting: Surgical patients in 200 centers throughout the United States.
Synopsis: Data from the American College of Surgeons National Surgical Quality Improvement Program from 2005 to 2008 were acquired, and 391,006 patient records were reviewed. Postoperative morbidity and mortality were significantly greater in smokers. Current smokers had a 40% increased odds of death at 30 days compared to people who had never smoked (OR 1.38, 95% CI, 1.11-1.72). Current smokers also had significantly greater odds of pulmonary complications, including pneumonia (OR 2.09, 95% CI, 1.80-2.43), unplanned intubation (OR 1.87, 95% CI, 1.58-2.21), and mechanical ventilation (OR 1.53, 95% CI, 1.31-1.79).
Furthermore, current smokers had significantly greater odds of postoperative cardiac arrest (OR 1.57, 95% CI, 1.10-2.25), myocardial infarction (OR 1.80, 95% CI, 1.11-2.25), and stroke (OR 1.73, 95% CI, 1.18-2.53). Odds of infectious complications were increased in current smokers, including deep incisional infections (OR 1.42, 95% CI, 1.21-1.68), sepsis (OR 1.30, 95% CI, 1.20-1.60), and septic shock (OR 1.55, 95% CI, 1.29-1.87).
Limitations of this study include self-reporting of smoking habits and absence of detailed smoking history just before and after surgery.
Bottom line: Current smokers have significantly increased postoperative morbidity and mortality after noncardiac surgery.
Citation: Turan A, Mascha EJ, Roberman D, et al. Smoking and perioperative outcomes. Anesthesiology. 2011;114(4):837-846.
Chronic Beta-Blockade Reduces Postoperative Myocardial Ischemia
Clinical question: Does the timing of beta-blocker exposure affect cardiovascular outcomes in patients undergoing elective, noncardiac surgery?
Background: Several studies have demonstrated that beta-blockers are associated with decreased perioperative cardiovascular morbidity and mortality. Study designs have varied greatly, and differences in dosing and timing of beta-blocker administration have caused conflicting results. The question of when to initiate beta-blockers prior to surgery remains controversial.
Study design: Prospective cohort study.
Setting: Three academic medical centers in Canada.
Synopsis: Data from 1,398 patients who had elective, noncardiac surgery with either acute (n=436) or chronic (n=962) beta-blocker exposure were analyzed. Acute exposure was defined as receiving a beta-blocker for the first time within 48 hours after surgery, whereas chronic beta-blocker exposure was defined as receiving a beta-blocker seven to 10 days prior to surgery.
Patients with chronic beta-blocker exposure were more likely to have a history of coronary disease, heart failure, or hypertension and were more likely to be receiving statins, antiplatelet agents, and angiotensin-converting enzyme inhibitors. The primary outcome was a composite of major cardiac events, including myocardial infarction, nonfatal cardiac arrest, and 30-day mortality.
Major cardiac events occurred more often in patients with acute versus chronic beta-blocker exposure in both the entire cohort (8.3% vs. 4.7%) and in the propensity-matched cohort (8.0% vs. 3.0%). Myocardial infarction accounted for the majority of cardiac events.
There are several limitations of this study: The sample size was small, the beta-blocker and dosage used varied, and the indication and exact duration of chronic beta-blocker therapy was unknown.
Bottom line: Chronic beta-blocker therapy reduces major cardiac events compared with acute beta-blocker therapy in patients undergoing elective, noncardiac surgery.
Citation: Ellenberger C, Tait G, Beattie WS. Chronic beta-blockade is associated with a better outcome after elective noncardiac surgery than acute beta-blockade: a single-center propensity-matched cohort study. Anesthesiology. 2011;114(4):817-823.
Continuous and Bolus Dosing of Furosemide Provides Similar Outcomes in Heart Failure
Clinical question: Does continuous infusion compared to bolus dosing of furosemide improve clinical outcomes in patients with acute decompensated heart failure?
Background: Diuresis with furosemide is commonly used to manage acute decompensated heart failure, but it is uncertain which dosing strategy is optimal. Continuous infusion of furosemide has been proposed as a more effective method of diuresis compared with bolus dosing, especially when higher doses are required, but data comparing the two strategies are limited.
Study design: Randomized, double-blind, controlled trial.
Setting: Twenty-six clinical sites in the U.S. and Canada.
Synopsis: Researchers randomized 308 patients with acute decompensated heart failure to either continuous or bolus intravenous dosing, which was calculated as either the equivalent of their daily oral dose (low-dose strategy) or 2.5 times their daily dose (high-dose strategy). Mean ejection fraction was 35%. Primary endpoints were patients’ assessment of symptoms based on a visual-analogue scale quantified as area under the curve, as well as change in serum creatinine level at 72 hours.
No significant differences between the continuous and bolus dosing groups were evidenced in primary endpoints at 72 hours. Patients in the bolus group had more dose increases at 48 hours (21% vs. 11%, P=0.01). Patients in the high-dose group were more likely to change from intravenous to oral doses at 48 hours (31% vs. 17%, P<0.001) and had greater net fluid loss (4.9L vs. 3.6L, P=0.01). More patients in the high-dose versus low-dose group had an increase in creatinine ≥0.3 mg/dL (23% vs. 14%, P=0.04). Hospital length of stay, readmission, and mortality rates were similar between the groups.
Bottom line: Diuretic therapy administered by continuous infusion or bolus dosing in patients with acute decompensated heart failure have equivocal effects on patients’ symptoms and kidney function.
Citation: Felker GM, Lee KL, Bull DA, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011;364(9):797-805.
Carotid Endarterectomy Is Better than Carotid Artery Stenting
Clinical question: How do the clinical outcomes of carotid artery stenting compare with those of carotid endarterectomy?
Background: Whether carotid artery stenting or carotid endarterectomy is the preferred therapy for patients with carotid artery stenosis has been highly controversial. This study was a meta-analysis of all available data from randomized trials comparing carotid endarterectomy to carotid artery stenting.
Study design: Meta-analysis.
Setting: Teaching and nonteaching hospitals.
Synopsis: Thirteen randomized trials were identified with 3,723 patients who had undergone endarterectomy and 3,754 patients who had undergone carotid artery stenting. Outcomes included stroke, myocardial infarction, cranial nerve injury, and death or stroke, and these outcomes were divided as either short-term (<30 days) or long-term (>1 year) outcomes.
Patients who had undergone carotid artery stenting had less risk of short-term myocardial infarction (OR 0.48, 95% CI, 0.29–9.78, P=0.003) and less risk of cranial nerve injury (OR 0.09, 95% CI, 0.05–0.16, P<0.001). However, carotid artery stenting had a significantly higher risk of short-term stroke and combined death or stroke, and also significantly higher long-term risk of stroke and combined death or stroke. The association between carotid artery stenting and stroke was stronger in the subgroup of patients >68 years but not in patients <68 years. There was no significant heterogeneity, and no significant modifying associations were revealed by meta-regression analysis.
Limitations include potentially unpublished small studies favoring carotid endarterectomy and a significant publication bias regarding short-term death.
Bottom line: Although carotid artery stenting has less short-term risk of myocardial infarction and cranial nerve injury, carotid endarterectomy has less short-term and long-term risks of stroke and death.
Citation: Economopoulus KP, Sergentanis TN, Tsivgoulis G, Mariolis AD, Stefanadis C. Carotid artery stenting versus carotid endarterectomy: a comprehensive meta-analysis of short-term and long-term outcomes. Stroke. 2011;42:687-692.
Chest Pain Protocol Can Identify Low-Risk Chest Pain in Emergency Departments
Clinical question: Can a two-hour accelerated diagnostic protocol (ADP) based on electrocardiogram, point-of-care biomarkers, and Thrombolysis in Myocardial Infarction (TIMI) score safely identify patients with chest pain at very low short-term risk of major cardiac events?
Background: Evaluation of patients presenting to EDs with chest pain utilize significant amounts of hospital resources. A safe, reproducible, and expeditious process to identify patients at low risk for short-term cardiac events is desired.
Study design: Prospective cohort study.
Setting: Fourteen urban EDs in nine countries across the Asia-Pacific region.
Synopsis: The study included 3,582 patients presenting to an ED with at least five minutes of chest pain suggestive of an acute coronary syndrome and for whom further evaluation with serial cardiac biomarkers was planned. A negative ADP was defined as TIMI score of 0, no new ischemic changes on initial electrocardiogram, and normal cardiac biomarkers at zero and two hours after arrival.
All components of the ADP were negative for 352 patients (9.8%). Only three low-risk patients (0.9%) by ADP had a major cardiac event during the 30-day follow-up period, yielding a negative predictive value of 99.1% (95% CI, 97.3-99.8%). Mean hospital stay for the low-risk group with a negative ADP was 43 hours with a median of 26 hours. The authors suggest that a 10% reduction in prolonged workups of patients with chest pain could be seen with implementation of this protocol.
Potential limitations include applicability only to a select cohort of patients with chest pain and the low specificity of the protocol.
Bottom line: A two-hour diagnostic protocol can help expedite discharge of patients with very-low-risk chest pain.
Citation: Than T, Cullen L, Reid CM, et al. A 2-h diagnostic protocol to assess patients with chest pain symptoms in the Asia-Pacific region (ASPECT): a prospective observational validation study. Lancet. 2011;337:1077-1084.
Esomeprazole Reduces Peptic Ulcer Recurrence in Patients on Clopidogrel
Clinical question: Does esomeprazole prevent recurrent peptic ulcers in patients with atherosclerosis on clopidogrel?
Background: Although clopidogrel is sometimes used as an alternative antiplatelet agent to aspirin, a significant rate of recurrent ulcer bleeding on clopidogrel has been described. No previous prospective trial has studied whether a proton-pump inhibitor (PPI) can reduce the risk of peptic ulcer recurrence or bleeding in atherosclerotic patients on clopidogrel.
Study design: Randomized controlled trial.
Setting: A single veterans hospital in Taiwan.
Synopsis: One hundred sixty-five patients were enrolled with a past history of peptic ulcer disease, no signs of ulcer recurrence by endoscopy, and current use of clopidogrel 37.5 mg to 75 mg per day. All patients had atherosclerosis and had been on clopidogrel for at least two weeks, without aspirin, corticosteroids, anticoagulants, or recent treatment with a PPI. Patients were randomized to clopidogrel 75 mg at night (n=82) or clopidogrel 75 mg at night plus esomeprazole 20 mg before breakfast. Follow-up endoscopy was performed at six months or as needed for symptoms.
Recurrence of ulcer was found in 1.2% of patients on clopidogrel plus esomeprazole versus 11.0% in patients on clopidogrel alone (95% CI, 2.6-17.0%; P=0.009). The pharmacodynamic study revealed no significant differences in platelet aggregation within or between treatment groups on day 1 or day 28. No significant differences were seen on the incidence of ischemic events in this setting, but the trial was underpowered to draw conclusions on this outcome.
An important limitation is that the findings of this study are applicable only to patients on clopidogrel monotherapy and not dual antiplatelet therapy.
Bottom line: A significant reduction in recurrent peptic ulcers is seen with the combination of esomeprazole plus clopidogrel, versus clopidogrel alone, in patients with atherosclerosis and a history of peptic ulcer disease.
Citation: Hsu PI, Lai KH, Liu CP. Esomeprazole with clopidogrel reduces peptic ulcer recurrence, compared with clopidogrel alone, in patients with atherosclerosis. Gastroenterology. 2011;140:791-798.
ICU Quality-Improvement Project Reduces Hospital Mortality
Clinical question: Does a quality-improvement (QI) project in the ICU reduce in-hospital mortality and length of stay among elderly adults?
Background: Previous studies have shown that ICU-acquired infections are associated with increased morbidity and mortality, and QI initiatives reduce hospital-acquired infections. However, it has not been demonstrated that QI projects in the ICU reduce in-hospital mortality or length of stay.
Study design: Retrospective cohort study.
Setting: Four hundred fifty-nine Midwestern hospitals.
Synopsis: This study included 238,937 adults age >65 who were hospitalized in an ICU from 2001 to 2006 at one of 95 hospitals invited to implement the Keystone ICU Project. The control group included 1,091,547 elderly adults at one of 364 hospitals not invited to participate in the project. The Keystone ICU Project implements evidence-based practices to reduce rates of catheter-related bloodstream infections and ventilator-associated pneumonia.
Hospital mortality was not significantly reduced during initiation or implementation of the project; however, a significant reduction in hospital mortality occurred in the study group during one to 12 months post-implementation (OR=0.83 vs. 0.88, P=0.041) and 13 to 22 months post-implementation (OR=0.76 vs. 0.84, P=0.007). In contrast, length of stay did not differ significantly between the two groups, but the study was underpowered for this outcome.
The study is limited by the complexity of the Keystone ICU Project, as well as the exclusion of smaller hospitals and nonelderly adults. The study is promising because implementing a QI project in the ICU is associated with no known harms and might confer a mortality benefit at a relatively low cost.
Bottom line: Elderly adults had lower in-hospital mortality after implementation of the Keystone QI project in ICUs.
Citation: Lipitz-Snyderman A, Steinwachs D, Needham DM, Colantuoni E, Morlock LL, Pronovost PJ. Impact of a statewide intensive care unit quality improvement initiative on hospital mortality and length of stay: retrospective comparative analysis. BMJ. 2011;342:d219.
Serious Long-Term Risks with Acute Kidney Injury after Coronary Angiography
Clinical question: Does postcoronary angiography acute kidney injury (AKI) increase the risk of poor long-term clinical outcomes?
Background: Previous studies have shown that AKI following coronary angiography increases the risk of poor short-term clinical outcomes, such as in-hospital myocardial infarction, prolonged hospital stay, and early mortality. Little is known about the long-term cardiovascular and renal outcomes following post-coronary angiography AKI.
Study design: Retrospective cohort study.
Setting: All coronary angiography centers in Alberta, Canada.
Synopsis: The study included 14,782 adults who were ≥18 years of age, underwent coronary angiography, had a baseline creatinine measurement, did not have end-stage renal disease (ESRD), and had a creatinine measurement within seven days after coronary angiography.
During a median follow-up period of 19.7 months, 1,099 (7.4%) patients developed stage 1 AKI and 321 (2.2%) developed stage 2 or 3 AKI. Mortality increased twofold with stage 1 AKI and >3-fold with stage 2 or 3 AKI. Risk of ESRD increased substantially by >11-fold in patients with stage 2 or 3 AKI. Risk of hospitalization for subsequent AKI, myocardial infarction, and heart failure also increased significantly following post-coronary angiography AKI.
Patients who experienced AKI were older, had more severe CAD, were more likely to have such comorbidities as DM, HTN, and heart failure, and had lower baseline GFRs. However, the underlying comorbidities do not completely explain the increased risk of poor long-term outcomes in the adjusted analysis.
Limitations include missing or underestimating mild cases of AKI, residual confounding from unmeasured variables, and inability of retrospective comparative studies to establish causality.
Bottom line: Adults with post-coronary angiography AKI are at increased risk of poor long-term cardiovascular and renal outcomes.
Citation: James MT, Ghali WA, Knudtson ML, et al. Associations between acute kidney injury and cardiovascular and renal outcomes after coronary angiography. Circulation. 2011;123(4):409-416. TH
Pediatric HM Literature
Proton-Pump Inhibitors Ineffective for Gastroesophageal Reflux Disease in Children
Clinical question: What is the efficacy of proton-pump inhibitors (PPIs) in children with gastroesophageal reflux disease (GERD)?
Background: Gastroesophageal reflux is both a common and normal phenomenon in infants. GERD refers to the presence of abnormal symptoms ascribed to the reflux and frequently is treated in children in a manner similar to adults with reflux esophagitis. PPIs often are prescribed as front-line treatment, and their use has increased dramatically in recent years, though their effectiveness in children remains unclear.
Study design: Systematic review of the literature.
Setting: Hawaii’s largest health insurer.
Synopsis: Medline, Embase, and the Cochrane Database of Systematic Reviews were searched for randomized controlled trials (RCTs) and crossover studies performed to evaluate the efficacy of PPIs in children 0-17 years with GERD and no complicating diseases. Ten RCTs and two crossover studies were analyzed and rated independently by two reviewers.
Due to significant heterogeneity between the studies, a meta-analysis was not possible; studies were discussed separately. PPIs offered no advantage when compared with controls (alginates, ranitidine, different dosages of PPIs), and similar rates of adverse events were reported between treatment groups.
This study is hampered by notable heterogeneity of patient type, symptoms, and study design in many of the trials. However, the results are in line with discussions at a recent FDA Gastrointestinal Drugs Advisory Committee meeting, which reviewed the lack of efficacy of PPIs in infants in four recent Phase 3 clinical trials.
Bottom line: Little evidence supports the widespread use of PPIs in children.
Citation: Van der Pol RJ, Smits MJ, van Wijk MP, Omari TI, Tabbers MM, Benninga MA. Efficacy of proton-pump inhibitors in children with gastroesophageal reflux disease: a systematic review. Pediatrics. 2011;127:925-935.
How Is SIADH Diagnosed and Managed?
Case
A 70-year-old woman with hypertension presents after a fall. Her medications include hydrochlorothiazide. Her blood pressure is 130/70 mm/Hg, with heart rate of 86. She has normal orthostatic vital signs. Her mucus membranes are moist and she has no jugular venous distension, edema, or ascites. Her plasma sodium (PNa) is 125 mmol/L, potassium 3.6 mmol/L, blood urea nitrogen (BUN) 30 mg/dL, and creatinine 0.8 mg/dL. Additional labs include serum thyroid stimulating hormone 1.12 mIU/L, cortisol 15 mcg/dL, serum osmolality 270 mOsm/kg, uric acid 4 mg/dL, urine osmolality 300 mOsm/kg, urine sodium (UNa) 40 mmol/L, fractional excretion of sodium 1.0%, and fractional excretion of urate (FEUrate) 13%. She receives 2 L isotonic saline intravenously over 24 hours, with resulting PNa of 127.
What is the cause of her hyponatremia, and how should her hyponatremia be managed?
Overview
Hyponatremia is one of the most common electrolyte abnormalities; it has a prevalence as high as 30% upon admission to the hospital.1 Hyponatremia is important clinically because of its high risk of mortality in the acute and symptomatic setting, and the risk of central pontine myelinolysis (CPM), or death with too rapid correction.2 Even so-called “asymptomatic” mild hyponatremia is associated with increased falls and impairments in gait and attention in the elderly.3
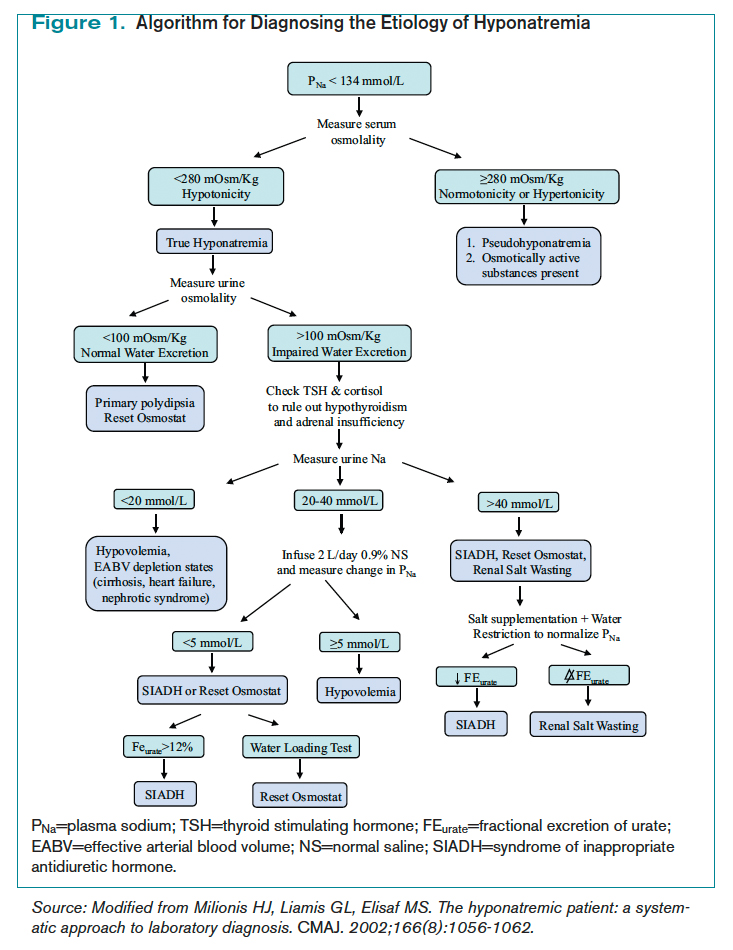
Hyponatremia is a state of excess water compared with the amount of solute in the extracellular fluid. To aid in diagnosing the etiology of hypotonic hyponatremia, the differential is traditionally divided into categories based on extracellular fluid volume (ECV) status, as shown in Table 1 (below), with syndrome of inappropriate antidiuretic hormone secretion (SIADH) being the most common cause of euvolemic hyponatremia.2 However, data show that clinical determination of volume status is often flawed,4 and an algorithmic approach to diagnosis and treatment yields improved results.5
Review of the Data
Diagnosis of SIADH. The original diagnostic criteria for SIADH, with minor modifications, are presented in Table 2, page 18).6,7,8 However, applying these criteria in clinical settings presents several difficulties, most notably a determination of ECV. The gold standard for assessing ECV status is by radioisotope, which is not practically feasible.9 Therefore, clinicians must rely on surrogate clinical markers of ECV (orthostatic hypotension, skin turgor, mucus membrane dryness, central venous pressure, BUN, BUN-creatinine ratio, and serum uric acid levels), which lack both sensitivity and specificity.4 Astoundingly, clinical assessment of ECV has been demonstrated to be accurate only 50% of the time when differentiating euvolemic patients from those with hypovolemia.4

Another challenge lies in the interpretation of UNa, which frequently is used as a surrogate for extra-arterial blood volume (EABV) status.10 Unfortunately, in the setting of diuretic use, UNa becomes inaccurate. The FEUrate, however, is unaffected by diuretic use and can be helpful in distinguishing between etiologies of hyponatremia with UNa greater than 30 mmol/L.11 The FEUrate is about 10% in normal euvolemic subjects and is reduced (usually <8%) in patients with low effective arterial blood volume.11,12 A trial of 86 patients demonstrated that a FEUrate of 12% had a specificity and positive predictive value of 100% in accurately identifying SIADH from diuretic-induced hyponatremia in patients on diuretics.11,12 Therefore, the UNa is a valid marker of EABV status when patients are not on diuretics; however, the FEUrate should be used in the setting of diuretic use.
Yet another pitfall is differentiating patients with salt depletion from those with SIADH. In these situations, measurement of the change in PNa concentration after a test infusion of isotonic saline is helpful. In salt depletion, PNa usually increases ≥5 mmol/L after 2 L saline infusion, which is not the case with SIADH.13 Incorrectly diagnosing renal salt wasting (RSW) as SIADH results in fluid restriction and, consequently, ECV depletion and increased morbidity.14 The persistence of hypouricemia and elevated FEUrate after correction of the hyponatremia in RSW differentiates it from SIADH.13, 14
Given these challenges, recommendations to use an algorithmic approach for the evaluation and diagnosis of hyponatremia have surfaced. In a study of 121 patients admitted with hyponatremia, an algorithm-based approach to the diagnosis of hyponatremia yielded an overall diagnostic accuracy of 71%, compared with an accuracy of 32% by experienced clinicians.5 This study also highlighted SIADH as the most frequent false-positive diagnosis that was expected whenever the combination of euvolemia and a UNa >30 mmol/L was present.5 Cases of diuretic-induced hyponatremia often were misclassified due to errors in the accurate assessment of ECV status, as most of these patients appeared clinically euvolemic or hypervolemic.5 Therefore, it is important to use an algorithm in identifying SIADH and to use one that does not rely solely on clinical estimation of ECV status (see Figure 1, below).
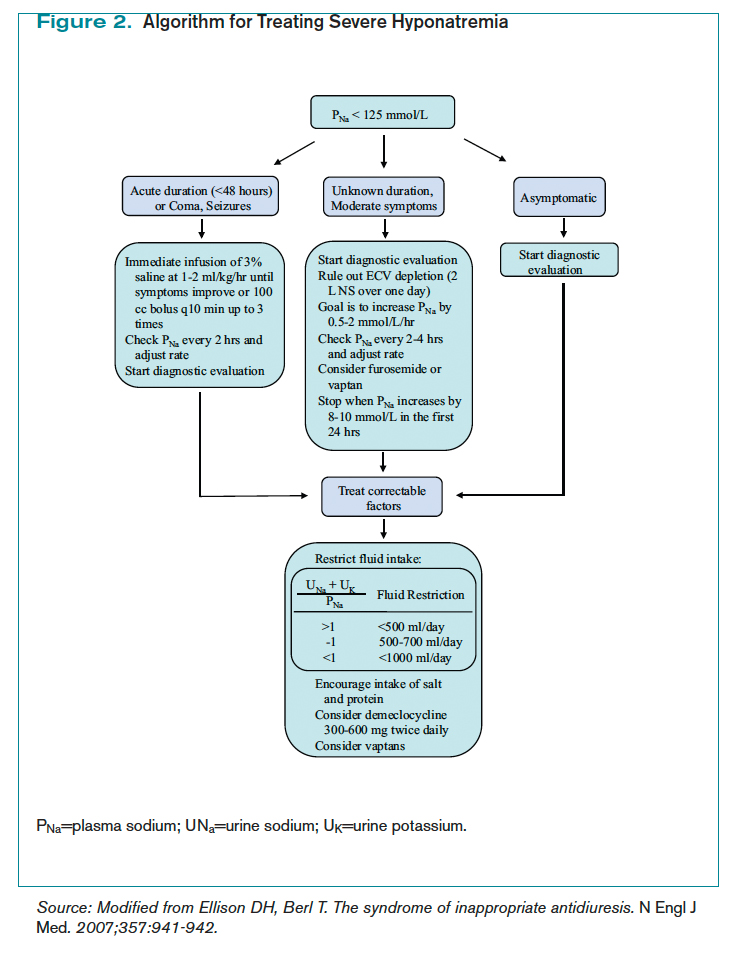
Management of acute and symptomatic hyponatremia. When hyponatremia develops acutely, urgent treatment is required (see Figure 2, below).15 Hyponatremia is considered acute when the onset is within 48 hours.15 Acute hyponatremia is most easily identified in the hospital and is commonly iatrogenic. Small case reviews in the 1980s began to associate postoperative deaths with the administration of hypotonic fluids.16 Asymptomatic patients with hyponatremia presenting from home should be considered chronic hyponatremias as the duration often is unclear.

Acute hyponatremia or neurologically symptomatic hyponatremia regardless of duration requires the use of hypertonic saline.15 Traditional sodium correction algorithms are based on early case series, which were focused on limiting neurologic complications from sodium overcorrection.17 This resulted in protocols recommending a conservative rate of correction spread over a 24- to 48-hour period.17 Infusing 3% saline at a rate of 1 ml/kg/hr to 2 ml/kg/hr results in a 1 mmol/L/hr to 2 mmol/L/hr increase in PNa.15 This simplified formula results in similar correction rates as more complex calculations.15 Correction should not exceed 8 mmol/L to 10 mmol/L within the first 24 hours, and 18 mmol/L to 25 mmol/L by 48 hours to avoid CPM.15 PNa should be checked every two hours to ensure that the correction rate is not exceeding the predicted rate, as the formulas do not take into account oral intake and ongoing losses.15
Recent observations focused on the initial four hours from onset of hyponatremia suggest a higher rate of correction can be tolerated without complications.18 Rapid sodium correction of 4 mmol/L to 6 mmol/L often is enough to stop neurologic complications.18 This can be accomplished with a bolus infusion of 100 mL of 3% saline.19 This may be repeated twice at 10-minute intervals until there is neurologic improvement.19 This might sound aggressive, but this would correspond to a rise in PNa of 5 mmol/L to 6 mmol/L in a 50 kg woman. Subsequent treatment with hypertonic fluid might not be needed if symptoms resolve.
Management of chronic hyponatremia. Hyponatremia secondary to SIADH improves with the treatment of the underlying cause, thus an active search for a causative medication or condition should be sought (see Table 1, p. 17).20
Water restriction. Restriction of fluid intake is the first-line treatment for SIADH in patients without hypovolemia. The severity of fluid restriction is guided by the concentration of the urinary solutes.15 Restriction of water intake to 500 ml/day to 1,000 ml/day is generally advised for many patients, as losses from the skin, lungs, and urine exceed this amount, leading to a gradual reduction in total body water.21 The main drawback of fluid restriction is poor compliance due to an intact thirst mechanism.
Saline infusion. The infusion of normal saline theoretically worsens hyponatremia due to SIADH because the water is retained while the salt is excreted. However, a trial of normal saline sometimes is attempted in patients in whom the differentiation between hypovolemia and euvolemia is difficult. From a study of a series of 17 patients with chronic SIADH, Musch and Decaux concluded that the infusion of intravenous normal (0.9%) saline raises PNa when the urine osmolality is less than 530 mosm/L.22
Oral solutes (urea and salt). The oral intake of salt augments water excretion23, and salt tablets are used as a second-line agent in patients with persistent hyponatremia despite fluid restriction.23 The oral administration of urea also results in increased free-water excretion via osmotic diuresis,24 but its poor palatability, lack of availability in the U.S., and limited user experience has restricted its usage.24
Demeclocycline. Demeclo-cycline is a tetracycline derivative that causes a partial nephrogenic diabetes insipidus.25 Its limitations include a slow onset of action (two to five days) and an unpredictable treatment effect with the possibility of causing profound polyuria and hypernatremia. It is also associated with reversible azotemia and sometimes nephrotoxicity, especially in patients with cirrhosis.
Lithium. Lithium also causes nephrogenic diabetes insipidus by downregulating vasopressin-stimulated aquaporin-2 expression and thus improves hyponatremia in SIADH.26 However, its use is significantly limited by its unpredictable response and the risks of interstitial nephritis and end-stage renal disease with chronic use. Therefore, it is no longer recommended for the treatment of SIADH.
Vasopressin receptor antagonists. Due to the role of excessive levels of vasopressin in the pathophysiology of most types of SIADH, antagonists of the vasopressin receptor were developed with the goal of preventing the excess water absorption that causes hyponatremia. Two vasopressin receptor antagonists, or vaptans, have been approved by the FDA for the treatment of nonemergent euvolemic and hypervolemic hyponatremia. Conivaptan is a nonselective vasopressin receptor antagonist that is for IV use only. Tolvaptan is a selective V2 receptor antagonist that is taken orally. Both conivaptan and tolvaptan successfully increase PNa levels while the drugs are being taken.27,28,29,30 Tolvaptan increases PNa levels in hyponatremia due to SIADH and CHF, and modestly so in cirrhosis.30
The most common side effects of the vaptans include dry mouth, increased thirst, and increased urination, although serious side effects (hypernatremia or too-rapid rate of increase in PNa) are possible.29 It is unclear if treating stable, asymptomatic hyponatremia with vaptans has any reduction in morbidity or mortality. One study found that tolvaptan increased the patients’ self-evaluations of mental functioning, but a study of tolvaptan used in combination with diuretics in the setting of CHF did not result in decreased mortality.29,31 Due to their expense, necessity of being started in the hospital, and unclear long-term benefit, the vaptans are only recommended when traditional measures such as fluid restriction and salt tablets have been unsuccessful.
Back to the Case
Our patient has hypotonic hyponatremia based on her low serum osmolality. The duration of her hyponatremia is unclear, but the patient is not experiencing seizures or coma. Therefore, her hyponatremia should be corrected slowly, and hypertonic saline is not indicated.
As is common in clinical practice, her true volume status is difficult to clinically ascertain. By physical exam, she appears euvolemic, but because she is on hydrochlorothiazide, she might be subtly hypovolemic. The UNa of 40 mmol/L is not consistent with hypovolemia, but its accuracy is limited in the setting of diuretics. The failure to improve her sodium by at least 5 mmol/L after a 2 L normal saline infusion argues against low effective arterial blood volume and indicates that the hydrochlorothiazide is unlikely to be the cause of her hyponatremia.
Therefore, the most likely cause of the hyponatremia is SIADH, a diagnosis further corroborated by the elevated FEUrate of 13%. Her chronic hyponatremia should be managed initially with fluid restriction while an investigation for an underlying cause of SIADH is initiated.
Bottom Line
The diagnosis of SIADH relies on the careful evaluation of laboratory values, use of an algorithm, and recognizing the limitations of clinically assessing volume status. The underlying cause of SIADH must also be sought and treated. TH
Dr. Grant is a clinical lecturer in internal medicine, Dr. Cho is a clinical instructor in internal medicine, and Dr. Nichani is an assistant professor of internal medicine at the University of Michigan Hospital and Health Systems in Ann Arbor.
References
- Upadhyay A, Jaber BL, Madias NE. Incidence and prevalence of hyponatremia. Am J Med. 2006;119(7 Suppl 1):S30-35.
- Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120(11 Suppl 1):S1-21.
- Renneboog B, Musch W, Vandemergel X, Manto MU, Decaux G. Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits. Am J Med. 2006;119(1):71.e71-78.
- Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med. 1987;83(5):905-908.
- Fenske W, Maier SK, Blechschmidt A, Allolio B, Störk S. Utility and limitations of the traditional diagnostic approach to hyponatremia: a diagnostic study. Am J Med. 2010;123(7):652-657.
- Bartter FC, Schwartz WB. The syndrome of inappropriate secretion of antidiuretic hormone. Am J Med. 1967;42(5):790-806.
- Smith DM, McKenna K, Thompson CJ. Hyponatraemia. Clin Endocrinol (Oxf). 2000;52(6):667-678.
- Verbalis JG. Hyponatraemia. Baillieres Clin Endocrinol Metab. Aug 1989;3(2):499-530.
- Maesaka JK, Imbriano LJ, Ali NM, Ilamathi E. Is it cerebral or renal salt wasting? Kidney Int. 2009;76(9):934-938.
- Verbalis JG. Disorders of body water homeostasis. Best Pract Res Clin Endocrinol Metab. 2003;17(4):471-503.
- Fenske W, Störk S, Koschker AC, et al. Value of fractional uric acid excretion in differential diagnosis of hyponatremic patients on diuretics. J Clin Endocrinol Metab. 2008;93(8):2991-2997.
- Maesaka JK, Fishbane S. Regulation of renal urate excretion: a critical review. Am J Kidney Dis. 1998;32(6):917-933.
- Milionis HJ, Liamis GL, Elisaf MS. The hyponatremic patient: a systematic approach to laboratory diagnosis. CMAJ. 2002;166(8):1056-1062.
- Bitew S, Imbriano L, Miyawaki N, Fishbane S, Maesaka JK. More on renal salt wasting without cerebral disease: response to saline infusion. Clin J Am Soc Nephrol. 2009;4(2):309-315.
- Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356(20):2064-2072.
- Arieff AI. Hyponatremia, convulsions, respiratory arrest, and permanent brain damage after elective surgery in healthy women. N Engl J Med. 1986;314(24):1529-1535.
- Ayus JC, Krothapalli RK, Arieff AI. Treatment of symptomatic hyponatremia and its relation to brain damage. A prospective study. N Engl J Med. 1987;317(19):1190-1195.
- Sterns RH, Nigwekar SU, Hix JK. The treatment of hyponatremia. Semin Nephrol. 2009;29(3):282-299.
- Hew-Butler T, Ayus JC, Kipps C, et al. Statement of the Second International Exercise-Associated Hyponatremia Consensus Development Conference, New Zealand, 2007. Clin J Sport Med. 2008;18(2):111-121.
- List AF, Hainsworth JD, Davis BW, Hande KR, Greco FA, Johnson DH. The syndrome of inappropriate secretion of antidiuretic hormone (SIADH) in small-cell lung cancer. J Clin Oncol. 1986;4(8):1191-1198.
- Verbalis JG. Managing hyponatremia in patients with syndrome of inappropriate antidiuretic hormone secretion. J Hosp Med. 2010;5 Suppl 3:S18-S26.
- Musch W, Decaux G. Treating the syndrome of inappropriate ADH secretion with isotonic saline. QJM. 1998;91(11):749-753.
- Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol. 2008;19(6):1076-1078.
- Decaux G, Brimioulle S, Genette F, Mockel J. Treatment of the syndrome of inappropriate secretion of antidiuretic hormone by urea. Am J Med. 1980;69(1):99-106.
- Forrest JN Jr., Cox M, Hong C, Morrison G, Bia M, Singer I. Superiority of demeclocycline over lithium in the treatment of chronic syndrome of inappropriate secretion of antidiuretic hormone. N Engl J Med. 1978;298(4):173-177.
- Nielsen J, Hoffert JD, Knepper MA, Agre P, Nielsen S, Fenton RA. Proteomic analysis of lithium-induced nephrogenic diabetes insipidus: mechanisms for aquaporin 2 down-regulation and cellular proliferation. Proc Natl Acad Sci U S A. 2008;105(9):3634-3639.
- Zeltser D, Rosansky S, van Rensburg H, Verbalis JG, Smith N. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. Am J Nephrol. 2007;27(5):447-457.
- Verbalis JG, Zeltser D, Smith N, Barve A, Andoh M. Assessment of the efficacy and safety of intravenous conivaptan in patients with euvolaemic hyponatraemia: subgroup analysis of a randomized, controlled study. Clin Endocrinol (Oxf). 2008;69(1):159-168.
- Schrier RW, Gross P, Gheorghiade M, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355(20):2099-2112.
- Berl T, Quittnat-Pelletier F, Verbalis JG, et al. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol. 2010;21(4):705-712.
- Konstam MA, Gheorghiade M, Burnett JC Jr., et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297(12):1319-1331.
Case
A 70-year-old woman with hypertension presents after a fall. Her medications include hydrochlorothiazide. Her blood pressure is 130/70 mm/Hg, with heart rate of 86. She has normal orthostatic vital signs. Her mucus membranes are moist and she has no jugular venous distension, edema, or ascites. Her plasma sodium (PNa) is 125 mmol/L, potassium 3.6 mmol/L, blood urea nitrogen (BUN) 30 mg/dL, and creatinine 0.8 mg/dL. Additional labs include serum thyroid stimulating hormone 1.12 mIU/L, cortisol 15 mcg/dL, serum osmolality 270 mOsm/kg, uric acid 4 mg/dL, urine osmolality 300 mOsm/kg, urine sodium (UNa) 40 mmol/L, fractional excretion of sodium 1.0%, and fractional excretion of urate (FEUrate) 13%. She receives 2 L isotonic saline intravenously over 24 hours, with resulting PNa of 127.
What is the cause of her hyponatremia, and how should her hyponatremia be managed?
Overview
Hyponatremia is one of the most common electrolyte abnormalities; it has a prevalence as high as 30% upon admission to the hospital.1 Hyponatremia is important clinically because of its high risk of mortality in the acute and symptomatic setting, and the risk of central pontine myelinolysis (CPM), or death with too rapid correction.2 Even so-called “asymptomatic” mild hyponatremia is associated with increased falls and impairments in gait and attention in the elderly.3
Hyponatremia is a state of excess water compared with the amount of solute in the extracellular fluid. To aid in diagnosing the etiology of hypotonic hyponatremia, the differential is traditionally divided into categories based on extracellular fluid volume (ECV) status, as shown in Table 1 (below), with syndrome of inappropriate antidiuretic hormone secretion (SIADH) being the most common cause of euvolemic hyponatremia.2 However, data show that clinical determination of volume status is often flawed,4 and an algorithmic approach to diagnosis and treatment yields improved results.5
Review of the Data
Diagnosis of SIADH. The original diagnostic criteria for SIADH, with minor modifications, are presented in Table 2, page 18).6,7,8 However, applying these criteria in clinical settings presents several difficulties, most notably a determination of ECV. The gold standard for assessing ECV status is by radioisotope, which is not practically feasible.9 Therefore, clinicians must rely on surrogate clinical markers of ECV (orthostatic hypotension, skin turgor, mucus membrane dryness, central venous pressure, BUN, BUN-creatinine ratio, and serum uric acid levels), which lack both sensitivity and specificity.4 Astoundingly, clinical assessment of ECV has been demonstrated to be accurate only 50% of the time when differentiating euvolemic patients from those with hypovolemia.4
Another challenge lies in the interpretation of UNa, which frequently is used as a surrogate for extra-arterial blood volume (EABV) status.10 Unfortunately, in the setting of diuretic use, UNa becomes inaccurate. The FEUrate, however, is unaffected by diuretic use and can be helpful in distinguishing between etiologies of hyponatremia with UNa greater than 30 mmol/L.11 The FEUrate is about 10% in normal euvolemic subjects and is reduced (usually <8%) in patients with low effective arterial blood volume.11,12 A trial of 86 patients demonstrated that a FEUrate of 12% had a specificity and positive predictive value of 100% in accurately identifying SIADH from diuretic-induced hyponatremia in patients on diuretics.11,12 Therefore, the UNa is a valid marker of EABV status when patients are not on diuretics; however, the FEUrate should be used in the setting of diuretic use.
Yet another pitfall is differentiating patients with salt depletion from those with SIADH. In these situations, measurement of the change in PNa concentration after a test infusion of isotonic saline is helpful. In salt depletion, PNa usually increases ≥5 mmol/L after 2 L saline infusion, which is not the case with SIADH.13 Incorrectly diagnosing renal salt wasting (RSW) as SIADH results in fluid restriction and, consequently, ECV depletion and increased morbidity.14 The persistence of hypouricemia and elevated FEUrate after correction of the hyponatremia in RSW differentiates it from SIADH.13, 14
Given these challenges, recommendations to use an algorithmic approach for the evaluation and diagnosis of hyponatremia have surfaced. In a study of 121 patients admitted with hyponatremia, an algorithm-based approach to the diagnosis of hyponatremia yielded an overall diagnostic accuracy of 71%, compared with an accuracy of 32% by experienced clinicians.5 This study also highlighted SIADH as the most frequent false-positive diagnosis that was expected whenever the combination of euvolemia and a UNa >30 mmol/L was present.5 Cases of diuretic-induced hyponatremia often were misclassified due to errors in the accurate assessment of ECV status, as most of these patients appeared clinically euvolemic or hypervolemic.5 Therefore, it is important to use an algorithm in identifying SIADH and to use one that does not rely solely on clinical estimation of ECV status (see Figure 1, below).
Management of acute and symptomatic hyponatremia. When hyponatremia develops acutely, urgent treatment is required (see Figure 2, below).15 Hyponatremia is considered acute when the onset is within 48 hours.15 Acute hyponatremia is most easily identified in the hospital and is commonly iatrogenic. Small case reviews in the 1980s began to associate postoperative deaths with the administration of hypotonic fluids.16 Asymptomatic patients with hyponatremia presenting from home should be considered chronic hyponatremias as the duration often is unclear.
Acute hyponatremia or neurologically symptomatic hyponatremia regardless of duration requires the use of hypertonic saline.15 Traditional sodium correction algorithms are based on early case series, which were focused on limiting neurologic complications from sodium overcorrection.17 This resulted in protocols recommending a conservative rate of correction spread over a 24- to 48-hour period.17 Infusing 3% saline at a rate of 1 ml/kg/hr to 2 ml/kg/hr results in a 1 mmol/L/hr to 2 mmol/L/hr increase in PNa.15 This simplified formula results in similar correction rates as more complex calculations.15 Correction should not exceed 8 mmol/L to 10 mmol/L within the first 24 hours, and 18 mmol/L to 25 mmol/L by 48 hours to avoid CPM.15 PNa should be checked every two hours to ensure that the correction rate is not exceeding the predicted rate, as the formulas do not take into account oral intake and ongoing losses.15
Recent observations focused on the initial four hours from onset of hyponatremia suggest a higher rate of correction can be tolerated without complications.18 Rapid sodium correction of 4 mmol/L to 6 mmol/L often is enough to stop neurologic complications.18 This can be accomplished with a bolus infusion of 100 mL of 3% saline.19 This may be repeated twice at 10-minute intervals until there is neurologic improvement.19 This might sound aggressive, but this would correspond to a rise in PNa of 5 mmol/L to 6 mmol/L in a 50 kg woman. Subsequent treatment with hypertonic fluid might not be needed if symptoms resolve.
Management of chronic hyponatremia. Hyponatremia secondary to SIADH improves with the treatment of the underlying cause, thus an active search for a causative medication or condition should be sought (see Table 1, p. 17).20
Water restriction. Restriction of fluid intake is the first-line treatment for SIADH in patients without hypovolemia. The severity of fluid restriction is guided by the concentration of the urinary solutes.15 Restriction of water intake to 500 ml/day to 1,000 ml/day is generally advised for many patients, as losses from the skin, lungs, and urine exceed this amount, leading to a gradual reduction in total body water.21 The main drawback of fluid restriction is poor compliance due to an intact thirst mechanism.
Saline infusion. The infusion of normal saline theoretically worsens hyponatremia due to SIADH because the water is retained while the salt is excreted. However, a trial of normal saline sometimes is attempted in patients in whom the differentiation between hypovolemia and euvolemia is difficult. From a study of a series of 17 patients with chronic SIADH, Musch and Decaux concluded that the infusion of intravenous normal (0.9%) saline raises PNa when the urine osmolality is less than 530 mosm/L.22
Oral solutes (urea and salt). The oral intake of salt augments water excretion23, and salt tablets are used as a second-line agent in patients with persistent hyponatremia despite fluid restriction.23 The oral administration of urea also results in increased free-water excretion via osmotic diuresis,24 but its poor palatability, lack of availability in the U.S., and limited user experience has restricted its usage.24
Demeclocycline. Demeclo-cycline is a tetracycline derivative that causes a partial nephrogenic diabetes insipidus.25 Its limitations include a slow onset of action (two to five days) and an unpredictable treatment effect with the possibility of causing profound polyuria and hypernatremia. It is also associated with reversible azotemia and sometimes nephrotoxicity, especially in patients with cirrhosis.
Lithium. Lithium also causes nephrogenic diabetes insipidus by downregulating vasopressin-stimulated aquaporin-2 expression and thus improves hyponatremia in SIADH.26 However, its use is significantly limited by its unpredictable response and the risks of interstitial nephritis and end-stage renal disease with chronic use. Therefore, it is no longer recommended for the treatment of SIADH.
Vasopressin receptor antagonists. Due to the role of excessive levels of vasopressin in the pathophysiology of most types of SIADH, antagonists of the vasopressin receptor were developed with the goal of preventing the excess water absorption that causes hyponatremia. Two vasopressin receptor antagonists, or vaptans, have been approved by the FDA for the treatment of nonemergent euvolemic and hypervolemic hyponatremia. Conivaptan is a nonselective vasopressin receptor antagonist that is for IV use only. Tolvaptan is a selective V2 receptor antagonist that is taken orally. Both conivaptan and tolvaptan successfully increase PNa levels while the drugs are being taken.27,28,29,30 Tolvaptan increases PNa levels in hyponatremia due to SIADH and CHF, and modestly so in cirrhosis.30
The most common side effects of the vaptans include dry mouth, increased thirst, and increased urination, although serious side effects (hypernatremia or too-rapid rate of increase in PNa) are possible.29 It is unclear if treating stable, asymptomatic hyponatremia with vaptans has any reduction in morbidity or mortality. One study found that tolvaptan increased the patients’ self-evaluations of mental functioning, but a study of tolvaptan used in combination with diuretics in the setting of CHF did not result in decreased mortality.29,31 Due to their expense, necessity of being started in the hospital, and unclear long-term benefit, the vaptans are only recommended when traditional measures such as fluid restriction and salt tablets have been unsuccessful.
Back to the Case
Our patient has hypotonic hyponatremia based on her low serum osmolality. The duration of her hyponatremia is unclear, but the patient is not experiencing seizures or coma. Therefore, her hyponatremia should be corrected slowly, and hypertonic saline is not indicated.
As is common in clinical practice, her true volume status is difficult to clinically ascertain. By physical exam, she appears euvolemic, but because she is on hydrochlorothiazide, she might be subtly hypovolemic. The UNa of 40 mmol/L is not consistent with hypovolemia, but its accuracy is limited in the setting of diuretics. The failure to improve her sodium by at least 5 mmol/L after a 2 L normal saline infusion argues against low effective arterial blood volume and indicates that the hydrochlorothiazide is unlikely to be the cause of her hyponatremia.
Therefore, the most likely cause of the hyponatremia is SIADH, a diagnosis further corroborated by the elevated FEUrate of 13%. Her chronic hyponatremia should be managed initially with fluid restriction while an investigation for an underlying cause of SIADH is initiated.
Bottom Line
The diagnosis of SIADH relies on the careful evaluation of laboratory values, use of an algorithm, and recognizing the limitations of clinically assessing volume status. The underlying cause of SIADH must also be sought and treated. TH
Dr. Grant is a clinical lecturer in internal medicine, Dr. Cho is a clinical instructor in internal medicine, and Dr. Nichani is an assistant professor of internal medicine at the University of Michigan Hospital and Health Systems in Ann Arbor.
References
- Upadhyay A, Jaber BL, Madias NE. Incidence and prevalence of hyponatremia. Am J Med. 2006;119(7 Suppl 1):S30-35.
- Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120(11 Suppl 1):S1-21.
- Renneboog B, Musch W, Vandemergel X, Manto MU, Decaux G. Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits. Am J Med. 2006;119(1):71.e71-78.
- Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med. 1987;83(5):905-908.
- Fenske W, Maier SK, Blechschmidt A, Allolio B, Störk S. Utility and limitations of the traditional diagnostic approach to hyponatremia: a diagnostic study. Am J Med. 2010;123(7):652-657.
- Bartter FC, Schwartz WB. The syndrome of inappropriate secretion of antidiuretic hormone. Am J Med. 1967;42(5):790-806.
- Smith DM, McKenna K, Thompson CJ. Hyponatraemia. Clin Endocrinol (Oxf). 2000;52(6):667-678.
- Verbalis JG. Hyponatraemia. Baillieres Clin Endocrinol Metab. Aug 1989;3(2):499-530.
- Maesaka JK, Imbriano LJ, Ali NM, Ilamathi E. Is it cerebral or renal salt wasting? Kidney Int. 2009;76(9):934-938.
- Verbalis JG. Disorders of body water homeostasis. Best Pract Res Clin Endocrinol Metab. 2003;17(4):471-503.
- Fenske W, Störk S, Koschker AC, et al. Value of fractional uric acid excretion in differential diagnosis of hyponatremic patients on diuretics. J Clin Endocrinol Metab. 2008;93(8):2991-2997.
- Maesaka JK, Fishbane S. Regulation of renal urate excretion: a critical review. Am J Kidney Dis. 1998;32(6):917-933.
- Milionis HJ, Liamis GL, Elisaf MS. The hyponatremic patient: a systematic approach to laboratory diagnosis. CMAJ. 2002;166(8):1056-1062.
- Bitew S, Imbriano L, Miyawaki N, Fishbane S, Maesaka JK. More on renal salt wasting without cerebral disease: response to saline infusion. Clin J Am Soc Nephrol. 2009;4(2):309-315.
- Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356(20):2064-2072.
- Arieff AI. Hyponatremia, convulsions, respiratory arrest, and permanent brain damage after elective surgery in healthy women. N Engl J Med. 1986;314(24):1529-1535.
- Ayus JC, Krothapalli RK, Arieff AI. Treatment of symptomatic hyponatremia and its relation to brain damage. A prospective study. N Engl J Med. 1987;317(19):1190-1195.
- Sterns RH, Nigwekar SU, Hix JK. The treatment of hyponatremia. Semin Nephrol. 2009;29(3):282-299.
- Hew-Butler T, Ayus JC, Kipps C, et al. Statement of the Second International Exercise-Associated Hyponatremia Consensus Development Conference, New Zealand, 2007. Clin J Sport Med. 2008;18(2):111-121.
- List AF, Hainsworth JD, Davis BW, Hande KR, Greco FA, Johnson DH. The syndrome of inappropriate secretion of antidiuretic hormone (SIADH) in small-cell lung cancer. J Clin Oncol. 1986;4(8):1191-1198.
- Verbalis JG. Managing hyponatremia in patients with syndrome of inappropriate antidiuretic hormone secretion. J Hosp Med. 2010;5 Suppl 3:S18-S26.
- Musch W, Decaux G. Treating the syndrome of inappropriate ADH secretion with isotonic saline. QJM. 1998;91(11):749-753.
- Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol. 2008;19(6):1076-1078.
- Decaux G, Brimioulle S, Genette F, Mockel J. Treatment of the syndrome of inappropriate secretion of antidiuretic hormone by urea. Am J Med. 1980;69(1):99-106.
- Forrest JN Jr., Cox M, Hong C, Morrison G, Bia M, Singer I. Superiority of demeclocycline over lithium in the treatment of chronic syndrome of inappropriate secretion of antidiuretic hormone. N Engl J Med. 1978;298(4):173-177.
- Nielsen J, Hoffert JD, Knepper MA, Agre P, Nielsen S, Fenton RA. Proteomic analysis of lithium-induced nephrogenic diabetes insipidus: mechanisms for aquaporin 2 down-regulation and cellular proliferation. Proc Natl Acad Sci U S A. 2008;105(9):3634-3639.
- Zeltser D, Rosansky S, van Rensburg H, Verbalis JG, Smith N. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. Am J Nephrol. 2007;27(5):447-457.
- Verbalis JG, Zeltser D, Smith N, Barve A, Andoh M. Assessment of the efficacy and safety of intravenous conivaptan in patients with euvolaemic hyponatraemia: subgroup analysis of a randomized, controlled study. Clin Endocrinol (Oxf). 2008;69(1):159-168.
- Schrier RW, Gross P, Gheorghiade M, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355(20):2099-2112.
- Berl T, Quittnat-Pelletier F, Verbalis JG, et al. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol. 2010;21(4):705-712.
- Konstam MA, Gheorghiade M, Burnett JC Jr., et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297(12):1319-1331.
Case
A 70-year-old woman with hypertension presents after a fall. Her medications include hydrochlorothiazide. Her blood pressure is 130/70 mm/Hg, with heart rate of 86. She has normal orthostatic vital signs. Her mucus membranes are moist and she has no jugular venous distension, edema, or ascites. Her plasma sodium (PNa) is 125 mmol/L, potassium 3.6 mmol/L, blood urea nitrogen (BUN) 30 mg/dL, and creatinine 0.8 mg/dL. Additional labs include serum thyroid stimulating hormone 1.12 mIU/L, cortisol 15 mcg/dL, serum osmolality 270 mOsm/kg, uric acid 4 mg/dL, urine osmolality 300 mOsm/kg, urine sodium (UNa) 40 mmol/L, fractional excretion of sodium 1.0%, and fractional excretion of urate (FEUrate) 13%. She receives 2 L isotonic saline intravenously over 24 hours, with resulting PNa of 127.
What is the cause of her hyponatremia, and how should her hyponatremia be managed?
Overview
Hyponatremia is one of the most common electrolyte abnormalities; it has a prevalence as high as 30% upon admission to the hospital.1 Hyponatremia is important clinically because of its high risk of mortality in the acute and symptomatic setting, and the risk of central pontine myelinolysis (CPM), or death with too rapid correction.2 Even so-called “asymptomatic” mild hyponatremia is associated with increased falls and impairments in gait and attention in the elderly.3
Hyponatremia is a state of excess water compared with the amount of solute in the extracellular fluid. To aid in diagnosing the etiology of hypotonic hyponatremia, the differential is traditionally divided into categories based on extracellular fluid volume (ECV) status, as shown in Table 1 (below), with syndrome of inappropriate antidiuretic hormone secretion (SIADH) being the most common cause of euvolemic hyponatremia.2 However, data show that clinical determination of volume status is often flawed,4 and an algorithmic approach to diagnosis and treatment yields improved results.5
Review of the Data
Diagnosis of SIADH. The original diagnostic criteria for SIADH, with minor modifications, are presented in Table 2, page 18).6,7,8 However, applying these criteria in clinical settings presents several difficulties, most notably a determination of ECV. The gold standard for assessing ECV status is by radioisotope, which is not practically feasible.9 Therefore, clinicians must rely on surrogate clinical markers of ECV (orthostatic hypotension, skin turgor, mucus membrane dryness, central venous pressure, BUN, BUN-creatinine ratio, and serum uric acid levels), which lack both sensitivity and specificity.4 Astoundingly, clinical assessment of ECV has been demonstrated to be accurate only 50% of the time when differentiating euvolemic patients from those with hypovolemia.4
Another challenge lies in the interpretation of UNa, which frequently is used as a surrogate for extra-arterial blood volume (EABV) status.10 Unfortunately, in the setting of diuretic use, UNa becomes inaccurate. The FEUrate, however, is unaffected by diuretic use and can be helpful in distinguishing between etiologies of hyponatremia with UNa greater than 30 mmol/L.11 The FEUrate is about 10% in normal euvolemic subjects and is reduced (usually <8%) in patients with low effective arterial blood volume.11,12 A trial of 86 patients demonstrated that a FEUrate of 12% had a specificity and positive predictive value of 100% in accurately identifying SIADH from diuretic-induced hyponatremia in patients on diuretics.11,12 Therefore, the UNa is a valid marker of EABV status when patients are not on diuretics; however, the FEUrate should be used in the setting of diuretic use.
Yet another pitfall is differentiating patients with salt depletion from those with SIADH. In these situations, measurement of the change in PNa concentration after a test infusion of isotonic saline is helpful. In salt depletion, PNa usually increases ≥5 mmol/L after 2 L saline infusion, which is not the case with SIADH.13 Incorrectly diagnosing renal salt wasting (RSW) as SIADH results in fluid restriction and, consequently, ECV depletion and increased morbidity.14 The persistence of hypouricemia and elevated FEUrate after correction of the hyponatremia in RSW differentiates it from SIADH.13, 14
Given these challenges, recommendations to use an algorithmic approach for the evaluation and diagnosis of hyponatremia have surfaced. In a study of 121 patients admitted with hyponatremia, an algorithm-based approach to the diagnosis of hyponatremia yielded an overall diagnostic accuracy of 71%, compared with an accuracy of 32% by experienced clinicians.5 This study also highlighted SIADH as the most frequent false-positive diagnosis that was expected whenever the combination of euvolemia and a UNa >30 mmol/L was present.5 Cases of diuretic-induced hyponatremia often were misclassified due to errors in the accurate assessment of ECV status, as most of these patients appeared clinically euvolemic or hypervolemic.5 Therefore, it is important to use an algorithm in identifying SIADH and to use one that does not rely solely on clinical estimation of ECV status (see Figure 1, below).
Management of acute and symptomatic hyponatremia. When hyponatremia develops acutely, urgent treatment is required (see Figure 2, below).15 Hyponatremia is considered acute when the onset is within 48 hours.15 Acute hyponatremia is most easily identified in the hospital and is commonly iatrogenic. Small case reviews in the 1980s began to associate postoperative deaths with the administration of hypotonic fluids.16 Asymptomatic patients with hyponatremia presenting from home should be considered chronic hyponatremias as the duration often is unclear.
Acute hyponatremia or neurologically symptomatic hyponatremia regardless of duration requires the use of hypertonic saline.15 Traditional sodium correction algorithms are based on early case series, which were focused on limiting neurologic complications from sodium overcorrection.17 This resulted in protocols recommending a conservative rate of correction spread over a 24- to 48-hour period.17 Infusing 3% saline at a rate of 1 ml/kg/hr to 2 ml/kg/hr results in a 1 mmol/L/hr to 2 mmol/L/hr increase in PNa.15 This simplified formula results in similar correction rates as more complex calculations.15 Correction should not exceed 8 mmol/L to 10 mmol/L within the first 24 hours, and 18 mmol/L to 25 mmol/L by 48 hours to avoid CPM.15 PNa should be checked every two hours to ensure that the correction rate is not exceeding the predicted rate, as the formulas do not take into account oral intake and ongoing losses.15
Recent observations focused on the initial four hours from onset of hyponatremia suggest a higher rate of correction can be tolerated without complications.18 Rapid sodium correction of 4 mmol/L to 6 mmol/L often is enough to stop neurologic complications.18 This can be accomplished with a bolus infusion of 100 mL of 3% saline.19 This may be repeated twice at 10-minute intervals until there is neurologic improvement.19 This might sound aggressive, but this would correspond to a rise in PNa of 5 mmol/L to 6 mmol/L in a 50 kg woman. Subsequent treatment with hypertonic fluid might not be needed if symptoms resolve.
Management of chronic hyponatremia. Hyponatremia secondary to SIADH improves with the treatment of the underlying cause, thus an active search for a causative medication or condition should be sought (see Table 1, p. 17).20
Water restriction. Restriction of fluid intake is the first-line treatment for SIADH in patients without hypovolemia. The severity of fluid restriction is guided by the concentration of the urinary solutes.15 Restriction of water intake to 500 ml/day to 1,000 ml/day is generally advised for many patients, as losses from the skin, lungs, and urine exceed this amount, leading to a gradual reduction in total body water.21 The main drawback of fluid restriction is poor compliance due to an intact thirst mechanism.
Saline infusion. The infusion of normal saline theoretically worsens hyponatremia due to SIADH because the water is retained while the salt is excreted. However, a trial of normal saline sometimes is attempted in patients in whom the differentiation between hypovolemia and euvolemia is difficult. From a study of a series of 17 patients with chronic SIADH, Musch and Decaux concluded that the infusion of intravenous normal (0.9%) saline raises PNa when the urine osmolality is less than 530 mosm/L.22
Oral solutes (urea and salt). The oral intake of salt augments water excretion23, and salt tablets are used as a second-line agent in patients with persistent hyponatremia despite fluid restriction.23 The oral administration of urea also results in increased free-water excretion via osmotic diuresis,24 but its poor palatability, lack of availability in the U.S., and limited user experience has restricted its usage.24
Demeclocycline. Demeclo-cycline is a tetracycline derivative that causes a partial nephrogenic diabetes insipidus.25 Its limitations include a slow onset of action (two to five days) and an unpredictable treatment effect with the possibility of causing profound polyuria and hypernatremia. It is also associated with reversible azotemia and sometimes nephrotoxicity, especially in patients with cirrhosis.
Lithium. Lithium also causes nephrogenic diabetes insipidus by downregulating vasopressin-stimulated aquaporin-2 expression and thus improves hyponatremia in SIADH.26 However, its use is significantly limited by its unpredictable response and the risks of interstitial nephritis and end-stage renal disease with chronic use. Therefore, it is no longer recommended for the treatment of SIADH.
Vasopressin receptor antagonists. Due to the role of excessive levels of vasopressin in the pathophysiology of most types of SIADH, antagonists of the vasopressin receptor were developed with the goal of preventing the excess water absorption that causes hyponatremia. Two vasopressin receptor antagonists, or vaptans, have been approved by the FDA for the treatment of nonemergent euvolemic and hypervolemic hyponatremia. Conivaptan is a nonselective vasopressin receptor antagonist that is for IV use only. Tolvaptan is a selective V2 receptor antagonist that is taken orally. Both conivaptan and tolvaptan successfully increase PNa levels while the drugs are being taken.27,28,29,30 Tolvaptan increases PNa levels in hyponatremia due to SIADH and CHF, and modestly so in cirrhosis.30
The most common side effects of the vaptans include dry mouth, increased thirst, and increased urination, although serious side effects (hypernatremia or too-rapid rate of increase in PNa) are possible.29 It is unclear if treating stable, asymptomatic hyponatremia with vaptans has any reduction in morbidity or mortality. One study found that tolvaptan increased the patients’ self-evaluations of mental functioning, but a study of tolvaptan used in combination with diuretics in the setting of CHF did not result in decreased mortality.29,31 Due to their expense, necessity of being started in the hospital, and unclear long-term benefit, the vaptans are only recommended when traditional measures such as fluid restriction and salt tablets have been unsuccessful.
Back to the Case
Our patient has hypotonic hyponatremia based on her low serum osmolality. The duration of her hyponatremia is unclear, but the patient is not experiencing seizures or coma. Therefore, her hyponatremia should be corrected slowly, and hypertonic saline is not indicated.
As is common in clinical practice, her true volume status is difficult to clinically ascertain. By physical exam, she appears euvolemic, but because she is on hydrochlorothiazide, she might be subtly hypovolemic. The UNa of 40 mmol/L is not consistent with hypovolemia, but its accuracy is limited in the setting of diuretics. The failure to improve her sodium by at least 5 mmol/L after a 2 L normal saline infusion argues against low effective arterial blood volume and indicates that the hydrochlorothiazide is unlikely to be the cause of her hyponatremia.
Therefore, the most likely cause of the hyponatremia is SIADH, a diagnosis further corroborated by the elevated FEUrate of 13%. Her chronic hyponatremia should be managed initially with fluid restriction while an investigation for an underlying cause of SIADH is initiated.
Bottom Line
The diagnosis of SIADH relies on the careful evaluation of laboratory values, use of an algorithm, and recognizing the limitations of clinically assessing volume status. The underlying cause of SIADH must also be sought and treated. TH
Dr. Grant is a clinical lecturer in internal medicine, Dr. Cho is a clinical instructor in internal medicine, and Dr. Nichani is an assistant professor of internal medicine at the University of Michigan Hospital and Health Systems in Ann Arbor.
References
- Upadhyay A, Jaber BL, Madias NE. Incidence and prevalence of hyponatremia. Am J Med. 2006;119(7 Suppl 1):S30-35.
- Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120(11 Suppl 1):S1-21.
- Renneboog B, Musch W, Vandemergel X, Manto MU, Decaux G. Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits. Am J Med. 2006;119(1):71.e71-78.
- Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med. 1987;83(5):905-908.
- Fenske W, Maier SK, Blechschmidt A, Allolio B, Störk S. Utility and limitations of the traditional diagnostic approach to hyponatremia: a diagnostic study. Am J Med. 2010;123(7):652-657.
- Bartter FC, Schwartz WB. The syndrome of inappropriate secretion of antidiuretic hormone. Am J Med. 1967;42(5):790-806.
- Smith DM, McKenna K, Thompson CJ. Hyponatraemia. Clin Endocrinol (Oxf). 2000;52(6):667-678.
- Verbalis JG. Hyponatraemia. Baillieres Clin Endocrinol Metab. Aug 1989;3(2):499-530.
- Maesaka JK, Imbriano LJ, Ali NM, Ilamathi E. Is it cerebral or renal salt wasting? Kidney Int. 2009;76(9):934-938.
- Verbalis JG. Disorders of body water homeostasis. Best Pract Res Clin Endocrinol Metab. 2003;17(4):471-503.
- Fenske W, Störk S, Koschker AC, et al. Value of fractional uric acid excretion in differential diagnosis of hyponatremic patients on diuretics. J Clin Endocrinol Metab. 2008;93(8):2991-2997.
- Maesaka JK, Fishbane S. Regulation of renal urate excretion: a critical review. Am J Kidney Dis. 1998;32(6):917-933.
- Milionis HJ, Liamis GL, Elisaf MS. The hyponatremic patient: a systematic approach to laboratory diagnosis. CMAJ. 2002;166(8):1056-1062.
- Bitew S, Imbriano L, Miyawaki N, Fishbane S, Maesaka JK. More on renal salt wasting without cerebral disease: response to saline infusion. Clin J Am Soc Nephrol. 2009;4(2):309-315.
- Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356(20):2064-2072.
- Arieff AI. Hyponatremia, convulsions, respiratory arrest, and permanent brain damage after elective surgery in healthy women. N Engl J Med. 1986;314(24):1529-1535.
- Ayus JC, Krothapalli RK, Arieff AI. Treatment of symptomatic hyponatremia and its relation to brain damage. A prospective study. N Engl J Med. 1987;317(19):1190-1195.
- Sterns RH, Nigwekar SU, Hix JK. The treatment of hyponatremia. Semin Nephrol. 2009;29(3):282-299.
- Hew-Butler T, Ayus JC, Kipps C, et al. Statement of the Second International Exercise-Associated Hyponatremia Consensus Development Conference, New Zealand, 2007. Clin J Sport Med. 2008;18(2):111-121.
- List AF, Hainsworth JD, Davis BW, Hande KR, Greco FA, Johnson DH. The syndrome of inappropriate secretion of antidiuretic hormone (SIADH) in small-cell lung cancer. J Clin Oncol. 1986;4(8):1191-1198.
- Verbalis JG. Managing hyponatremia in patients with syndrome of inappropriate antidiuretic hormone secretion. J Hosp Med. 2010;5 Suppl 3:S18-S26.
- Musch W, Decaux G. Treating the syndrome of inappropriate ADH secretion with isotonic saline. QJM. 1998;91(11):749-753.
- Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol. 2008;19(6):1076-1078.
- Decaux G, Brimioulle S, Genette F, Mockel J. Treatment of the syndrome of inappropriate secretion of antidiuretic hormone by urea. Am J Med. 1980;69(1):99-106.
- Forrest JN Jr., Cox M, Hong C, Morrison G, Bia M, Singer I. Superiority of demeclocycline over lithium in the treatment of chronic syndrome of inappropriate secretion of antidiuretic hormone. N Engl J Med. 1978;298(4):173-177.
- Nielsen J, Hoffert JD, Knepper MA, Agre P, Nielsen S, Fenton RA. Proteomic analysis of lithium-induced nephrogenic diabetes insipidus: mechanisms for aquaporin 2 down-regulation and cellular proliferation. Proc Natl Acad Sci U S A. 2008;105(9):3634-3639.
- Zeltser D, Rosansky S, van Rensburg H, Verbalis JG, Smith N. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. Am J Nephrol. 2007;27(5):447-457.
- Verbalis JG, Zeltser D, Smith N, Barve A, Andoh M. Assessment of the efficacy and safety of intravenous conivaptan in patients with euvolaemic hyponatraemia: subgroup analysis of a randomized, controlled study. Clin Endocrinol (Oxf). 2008;69(1):159-168.
- Schrier RW, Gross P, Gheorghiade M, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355(20):2099-2112.
- Berl T, Quittnat-Pelletier F, Verbalis JG, et al. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol. 2010;21(4):705-712.
- Konstam MA, Gheorghiade M, Burnett JC Jr., et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297(12):1319-1331.
Global Perspective
We’ve all heard the stereotypes: Other countries have socialized medicine, rationed care, endless lines, and little incentive for innovation. OK, there might be a grain of truth to the wait times. But healthcare in other developed nations is surprisingly varied in its mix of public and private providers, and it yields high-quality outcomes for a far better price than in the U.S. And yes, international innovation is alive and well.
Head-to-head comparisons can only go so far, with many countries using vastly different metrics to measure quality and efficiency. Nevertheless, the examples of bundling, reference pricing, and patient-reported outcomes offer a glimpse of how large-scale initiatives can help improve outcomes and bottom lines in the hospital and beyond.
Just the Facts
Last November, the nonprofit Commonwealth Fund in New York funded an analysis of healthcare data from the Organisation for Economic Co-operation and Development (OECD), which explains just how expensive healthcare is here.
In 2008, the U.S. spent roughly $7,500 per person on healthcare, an astonishing 50% more than the next closest country: Norway, at about $5,000 per person. And yet we lag behind Norway and almost all of our other peers in mortality rates. Cathy Schoen, senior vice president for policy, research, and evaluation at the Commonwealth Fund, says the OECD statistics also say something about how we use hospitals. Compared with its peers, the U.S. actually spends a smaller fraction of money on hospital care. We’re also on the low end of the number of acute-care hospital beds and hospital discharges per 1,000 people, and below average on the typical length of stay for acute care, at about 5.5 days. “So we’re not using the hospital more, and we’re not staying in it longer,” Schoen says. “Nor do we have way more beds, so it’s not an occupancy issue that’s driving this.”
Instead, the numbers suggest that the tests ordered, the drugs prescribed, the devices implanted, and other medical services offered are driving up costs, at least in part. So what can other countries tell us? As policymakers here debate how to bundle more healthcare payments around episodes of care (see “A Bundle of Nerves,” November 2010, p. 1), European countries including Germany and the Netherlands already are using the payment initiative on a national level to create efficiencies around hospital-based care. And they’ve done it with an American innovation: diagnostic-related groups, or DRGs. Bundling around a hip replacement, for example, includes the cost of the implant, the surgeon, and all of the hospital care. “It gives the hospital overall and all of its physicians an incentive to say, ‘If we could buy supplies cheaper, let’s do it,’ ” Schoen says.
An eye-popping 2007 McKinsey & Company study documents the relative cost of hip and knee replacement surgeries for five countries. In 2004, U.S. doctors performed just over half as many hip replacements per 100,000 people as their German counterparts. Yet the cost of each hip prosthetic averaged more than $4,800 per patient in the U.S.—four times higher than the $1,200 cost in Germany and the $1,400 cost in the United Kingdom.
Part of this difference, Schoen says, is due to supply chain management and involving doctors in the decision-making process. Many countries (and a few integrated health systems in the U.S.) are asking surgeons to help select just one or two prosthetic implants, negotiate for bulk volume pricing, and then track the clinical outcomes of those devices to flag poor performers, she says.
Setting the Bar for New Drugs
Drugs are another big-ticket item, and the U.S. pays almost twice as much per capita as the OECD average. To keep their prices lower, Schoen says, many European countries have information systems that track the relative clinical effectiveness of pharmaceuticals. “And they’re using it to inform the way they cover drugs: not to exclude them from the list of what’s covered, but to do something in Europe that’s called reference pricing,” she adds.
Let’s say a new drug costs 50% more than an older one with roughly equivalent efficacy. Under reference pricing, a doctor can still prescribe the new drug, but the patient must pay all or most of the difference. Such benchmarking has fueled an interesting dynamic. “The brand names that are coming in and want to get some market share will price themselves lower, because if they’re priced really high compared to the reference price, the chances are they just won’t ever get a market share,” Schoen says. As a result, drug prices stay lower.
The concept, although discussed in the U.S., has yet to be widely implemented here. A new study in the April issue of the Journal of Managed Care Pharmacy, however, could cause some cash-strapped governments to take a closer look.1 In the study, the Arkansas State Employee Health Plan used reference pricing for proton-pump inhibitors, using the cost of generic omeprazole as its reference point. Over the 43-month reference-pricing period, net plan costs for the drugs dropped by 49.5% per member per month.
Patient Feedback
A third lesson is that constructive feedback on quality can improve performance, even if no money is attached to outcomes. Like the U.S., Germany is placing a high priority on metrics that evaluate hospital quality. Schoen says the German performance improvement initiative is identifying outliers and providing them with feedback and technical support, but it is not built into the payment system. “They’ve had pretty rapid improvement out of that,” she says, “and I would say we’re learning the same thing in the U.S.”
Initially, Medicare data posted on its Hospital Compare website (www.hospitalcompare.hhs.gov) showed a wide hospital-to-hospital variation in mortality rates for pneumonia, heart attacks, and congestive heart failure. But since then, Schoen says, most outliers on the low end have improved dramatically, even though the only payment incentive was to encourage reporting. In fact, CMS is dropping some core measures from its hospital value-based purchasing program.
Public reporting of quality measures, especially mortality rates, is certainly not without controversy. But Schoen says that if handled properly, disseminating information that suggests a facility’s performance is subpar can tap into the professionalism of its staff and create a strong incentive among them to do better. “That’s something true both internationally and in the U.S.,” she says.
In the U.S., Schoen says, the basic questions asked of patients in the HCAHPS (Hospital Consumer Assessment of Healthcare Providers and Systems) portion of Medicare’s new VBP system represent a good start. But the experiences of other countries, she says, suggest that patient reporting should be directed more at outcomes, similar to a proposal left out of last year’s healthcare reform bill that would have created a feedback system for patients receiving implantable medical devices.
Even so, hospitals like Dartmouth-Hitchcock Medical Center in Lebanon, N.H., are instituting patient feedback systems on their own, and a National Institutes of Health (NIH) initiative called PROMIS (Patient Reported Outcomes Measurement Information System) is gaining traction. “It’s less blaming, and it’s more informing,” Schoen says. TH
Bryn Nelson is a freelance medical writer based in Seattle.
Reference
- Johnson JT, Neill KK, Davis DA. Five-year examination of utilization and drug cost outcomes associated with benefit design changes including reference pricing for proton pump inhibitors in a state employee health plan. J Manag Care Pharm. 2011;17(3):200-212.
We’ve all heard the stereotypes: Other countries have socialized medicine, rationed care, endless lines, and little incentive for innovation. OK, there might be a grain of truth to the wait times. But healthcare in other developed nations is surprisingly varied in its mix of public and private providers, and it yields high-quality outcomes for a far better price than in the U.S. And yes, international innovation is alive and well.
Head-to-head comparisons can only go so far, with many countries using vastly different metrics to measure quality and efficiency. Nevertheless, the examples of bundling, reference pricing, and patient-reported outcomes offer a glimpse of how large-scale initiatives can help improve outcomes and bottom lines in the hospital and beyond.
Just the Facts
Last November, the nonprofit Commonwealth Fund in New York funded an analysis of healthcare data from the Organisation for Economic Co-operation and Development (OECD), which explains just how expensive healthcare is here.
In 2008, the U.S. spent roughly $7,500 per person on healthcare, an astonishing 50% more than the next closest country: Norway, at about $5,000 per person. And yet we lag behind Norway and almost all of our other peers in mortality rates. Cathy Schoen, senior vice president for policy, research, and evaluation at the Commonwealth Fund, says the OECD statistics also say something about how we use hospitals. Compared with its peers, the U.S. actually spends a smaller fraction of money on hospital care. We’re also on the low end of the number of acute-care hospital beds and hospital discharges per 1,000 people, and below average on the typical length of stay for acute care, at about 5.5 days. “So we’re not using the hospital more, and we’re not staying in it longer,” Schoen says. “Nor do we have way more beds, so it’s not an occupancy issue that’s driving this.”
Instead, the numbers suggest that the tests ordered, the drugs prescribed, the devices implanted, and other medical services offered are driving up costs, at least in part. So what can other countries tell us? As policymakers here debate how to bundle more healthcare payments around episodes of care (see “A Bundle of Nerves,” November 2010, p. 1), European countries including Germany and the Netherlands already are using the payment initiative on a national level to create efficiencies around hospital-based care. And they’ve done it with an American innovation: diagnostic-related groups, or DRGs. Bundling around a hip replacement, for example, includes the cost of the implant, the surgeon, and all of the hospital care. “It gives the hospital overall and all of its physicians an incentive to say, ‘If we could buy supplies cheaper, let’s do it,’ ” Schoen says.
An eye-popping 2007 McKinsey & Company study documents the relative cost of hip and knee replacement surgeries for five countries. In 2004, U.S. doctors performed just over half as many hip replacements per 100,000 people as their German counterparts. Yet the cost of each hip prosthetic averaged more than $4,800 per patient in the U.S.—four times higher than the $1,200 cost in Germany and the $1,400 cost in the United Kingdom.
Part of this difference, Schoen says, is due to supply chain management and involving doctors in the decision-making process. Many countries (and a few integrated health systems in the U.S.) are asking surgeons to help select just one or two prosthetic implants, negotiate for bulk volume pricing, and then track the clinical outcomes of those devices to flag poor performers, she says.
Setting the Bar for New Drugs
Drugs are another big-ticket item, and the U.S. pays almost twice as much per capita as the OECD average. To keep their prices lower, Schoen says, many European countries have information systems that track the relative clinical effectiveness of pharmaceuticals. “And they’re using it to inform the way they cover drugs: not to exclude them from the list of what’s covered, but to do something in Europe that’s called reference pricing,” she adds.
Let’s say a new drug costs 50% more than an older one with roughly equivalent efficacy. Under reference pricing, a doctor can still prescribe the new drug, but the patient must pay all or most of the difference. Such benchmarking has fueled an interesting dynamic. “The brand names that are coming in and want to get some market share will price themselves lower, because if they’re priced really high compared to the reference price, the chances are they just won’t ever get a market share,” Schoen says. As a result, drug prices stay lower.
The concept, although discussed in the U.S., has yet to be widely implemented here. A new study in the April issue of the Journal of Managed Care Pharmacy, however, could cause some cash-strapped governments to take a closer look.1 In the study, the Arkansas State Employee Health Plan used reference pricing for proton-pump inhibitors, using the cost of generic omeprazole as its reference point. Over the 43-month reference-pricing period, net plan costs for the drugs dropped by 49.5% per member per month.
Patient Feedback
A third lesson is that constructive feedback on quality can improve performance, even if no money is attached to outcomes. Like the U.S., Germany is placing a high priority on metrics that evaluate hospital quality. Schoen says the German performance improvement initiative is identifying outliers and providing them with feedback and technical support, but it is not built into the payment system. “They’ve had pretty rapid improvement out of that,” she says, “and I would say we’re learning the same thing in the U.S.”
Initially, Medicare data posted on its Hospital Compare website (www.hospitalcompare.hhs.gov) showed a wide hospital-to-hospital variation in mortality rates for pneumonia, heart attacks, and congestive heart failure. But since then, Schoen says, most outliers on the low end have improved dramatically, even though the only payment incentive was to encourage reporting. In fact, CMS is dropping some core measures from its hospital value-based purchasing program.
Public reporting of quality measures, especially mortality rates, is certainly not without controversy. But Schoen says that if handled properly, disseminating information that suggests a facility’s performance is subpar can tap into the professionalism of its staff and create a strong incentive among them to do better. “That’s something true both internationally and in the U.S.,” she says.
In the U.S., Schoen says, the basic questions asked of patients in the HCAHPS (Hospital Consumer Assessment of Healthcare Providers and Systems) portion of Medicare’s new VBP system represent a good start. But the experiences of other countries, she says, suggest that patient reporting should be directed more at outcomes, similar to a proposal left out of last year’s healthcare reform bill that would have created a feedback system for patients receiving implantable medical devices.
Even so, hospitals like Dartmouth-Hitchcock Medical Center in Lebanon, N.H., are instituting patient feedback systems on their own, and a National Institutes of Health (NIH) initiative called PROMIS (Patient Reported Outcomes Measurement Information System) is gaining traction. “It’s less blaming, and it’s more informing,” Schoen says. TH
Bryn Nelson is a freelance medical writer based in Seattle.
Reference
- Johnson JT, Neill KK, Davis DA. Five-year examination of utilization and drug cost outcomes associated with benefit design changes including reference pricing for proton pump inhibitors in a state employee health plan. J Manag Care Pharm. 2011;17(3):200-212.
We’ve all heard the stereotypes: Other countries have socialized medicine, rationed care, endless lines, and little incentive for innovation. OK, there might be a grain of truth to the wait times. But healthcare in other developed nations is surprisingly varied in its mix of public and private providers, and it yields high-quality outcomes for a far better price than in the U.S. And yes, international innovation is alive and well.
Head-to-head comparisons can only go so far, with many countries using vastly different metrics to measure quality and efficiency. Nevertheless, the examples of bundling, reference pricing, and patient-reported outcomes offer a glimpse of how large-scale initiatives can help improve outcomes and bottom lines in the hospital and beyond.
Just the Facts
Last November, the nonprofit Commonwealth Fund in New York funded an analysis of healthcare data from the Organisation for Economic Co-operation and Development (OECD), which explains just how expensive healthcare is here.
In 2008, the U.S. spent roughly $7,500 per person on healthcare, an astonishing 50% more than the next closest country: Norway, at about $5,000 per person. And yet we lag behind Norway and almost all of our other peers in mortality rates. Cathy Schoen, senior vice president for policy, research, and evaluation at the Commonwealth Fund, says the OECD statistics also say something about how we use hospitals. Compared with its peers, the U.S. actually spends a smaller fraction of money on hospital care. We’re also on the low end of the number of acute-care hospital beds and hospital discharges per 1,000 people, and below average on the typical length of stay for acute care, at about 5.5 days. “So we’re not using the hospital more, and we’re not staying in it longer,” Schoen says. “Nor do we have way more beds, so it’s not an occupancy issue that’s driving this.”
Instead, the numbers suggest that the tests ordered, the drugs prescribed, the devices implanted, and other medical services offered are driving up costs, at least in part. So what can other countries tell us? As policymakers here debate how to bundle more healthcare payments around episodes of care (see “A Bundle of Nerves,” November 2010, p. 1), European countries including Germany and the Netherlands already are using the payment initiative on a national level to create efficiencies around hospital-based care. And they’ve done it with an American innovation: diagnostic-related groups, or DRGs. Bundling around a hip replacement, for example, includes the cost of the implant, the surgeon, and all of the hospital care. “It gives the hospital overall and all of its physicians an incentive to say, ‘If we could buy supplies cheaper, let’s do it,’ ” Schoen says.
An eye-popping 2007 McKinsey & Company study documents the relative cost of hip and knee replacement surgeries for five countries. In 2004, U.S. doctors performed just over half as many hip replacements per 100,000 people as their German counterparts. Yet the cost of each hip prosthetic averaged more than $4,800 per patient in the U.S.—four times higher than the $1,200 cost in Germany and the $1,400 cost in the United Kingdom.
Part of this difference, Schoen says, is due to supply chain management and involving doctors in the decision-making process. Many countries (and a few integrated health systems in the U.S.) are asking surgeons to help select just one or two prosthetic implants, negotiate for bulk volume pricing, and then track the clinical outcomes of those devices to flag poor performers, she says.
Setting the Bar for New Drugs
Drugs are another big-ticket item, and the U.S. pays almost twice as much per capita as the OECD average. To keep their prices lower, Schoen says, many European countries have information systems that track the relative clinical effectiveness of pharmaceuticals. “And they’re using it to inform the way they cover drugs: not to exclude them from the list of what’s covered, but to do something in Europe that’s called reference pricing,” she adds.
Let’s say a new drug costs 50% more than an older one with roughly equivalent efficacy. Under reference pricing, a doctor can still prescribe the new drug, but the patient must pay all or most of the difference. Such benchmarking has fueled an interesting dynamic. “The brand names that are coming in and want to get some market share will price themselves lower, because if they’re priced really high compared to the reference price, the chances are they just won’t ever get a market share,” Schoen says. As a result, drug prices stay lower.
The concept, although discussed in the U.S., has yet to be widely implemented here. A new study in the April issue of the Journal of Managed Care Pharmacy, however, could cause some cash-strapped governments to take a closer look.1 In the study, the Arkansas State Employee Health Plan used reference pricing for proton-pump inhibitors, using the cost of generic omeprazole as its reference point. Over the 43-month reference-pricing period, net plan costs for the drugs dropped by 49.5% per member per month.
Patient Feedback
A third lesson is that constructive feedback on quality can improve performance, even if no money is attached to outcomes. Like the U.S., Germany is placing a high priority on metrics that evaluate hospital quality. Schoen says the German performance improvement initiative is identifying outliers and providing them with feedback and technical support, but it is not built into the payment system. “They’ve had pretty rapid improvement out of that,” she says, “and I would say we’re learning the same thing in the U.S.”
Initially, Medicare data posted on its Hospital Compare website (www.hospitalcompare.hhs.gov) showed a wide hospital-to-hospital variation in mortality rates for pneumonia, heart attacks, and congestive heart failure. But since then, Schoen says, most outliers on the low end have improved dramatically, even though the only payment incentive was to encourage reporting. In fact, CMS is dropping some core measures from its hospital value-based purchasing program.
Public reporting of quality measures, especially mortality rates, is certainly not without controversy. But Schoen says that if handled properly, disseminating information that suggests a facility’s performance is subpar can tap into the professionalism of its staff and create a strong incentive among them to do better. “That’s something true both internationally and in the U.S.,” she says.
In the U.S., Schoen says, the basic questions asked of patients in the HCAHPS (Hospital Consumer Assessment of Healthcare Providers and Systems) portion of Medicare’s new VBP system represent a good start. But the experiences of other countries, she says, suggest that patient reporting should be directed more at outcomes, similar to a proposal left out of last year’s healthcare reform bill that would have created a feedback system for patients receiving implantable medical devices.
Even so, hospitals like Dartmouth-Hitchcock Medical Center in Lebanon, N.H., are instituting patient feedback systems on their own, and a National Institutes of Health (NIH) initiative called PROMIS (Patient Reported Outcomes Measurement Information System) is gaining traction. “It’s less blaming, and it’s more informing,” Schoen says. TH
Bryn Nelson is a freelance medical writer based in Seattle.
Reference
- Johnson JT, Neill KK, Davis DA. Five-year examination of utilization and drug cost outcomes associated with benefit design changes including reference pricing for proton pump inhibitors in a state employee health plan. J Manag Care Pharm. 2011;17(3):200-212.
ONLINE EXCLUSIVE: Quick fix eliminates indigent discharge problems
The Medical University of South Carolina (MUSC) in Charleston hasn’t solved the issue of care transitions for the indigent, “but we have thought about it a lot,” says Neal Axon, MD, MSCR, FHM, assistant professor in the Division of Hospital Medicine at MUSC. The hospital is experimenting with quality-improvement (QI) techniques learned through participation in SHM’s Project BOOST.
“The first principle I try to teach residents is that a good discharge for a patient without insurance is the same as a good discharge for a patient with insurance,” Dr. Axon says. “Many of the same principles apply.”
If the care plan fails to address basic needs of indigent patients, including access to housing, primary care, and affordable medications, that patient won’t be able to focus on their medical needs.
Affiliated primary-care clinics already see a high percentage of indigent patients, Dr. Axon says, so there might be some pushback when the hospital team attempts a new referral. “We have tried to distinguish between care that needs to be done in the first week or so after discharge versus ongoing follow-up,” he explains. “We have negotiated with the clinic so that patients can come back here for one or two visits for urgent follow-up care without being entered into the [outpatient] system permanently. We are also blessed to have federally qualified health centers in the Charleston area. We have cordial relationships with those clinics, even if it’s not as well-integrated as I might wish.”
Another service that can be helpful with care transitions for uninsured patients is a 14-bed transitional-care unit on the hospital campus. “It provides rehabilitation and long-term care for the small numbers of chronically ill patients with long-term disabilities who don’t qualify for Medicaid or Medicare and can’t be placed elsewhere,” he says. “We’re able to care for these patients in a less costly way on the unit, rather than leaving them in an acute-care bed.” The hospital, he adds, views the unit as a cost-avoidance measure.
Some have been on the unit for months; others do much better than expected and go home. “It’s always gratifying when patients come back to the hospital to visit and thank us for the care they received,” he says.
Larry Beresford is a freelance writer based in California.
The Medical University of South Carolina (MUSC) in Charleston hasn’t solved the issue of care transitions for the indigent, “but we have thought about it a lot,” says Neal Axon, MD, MSCR, FHM, assistant professor in the Division of Hospital Medicine at MUSC. The hospital is experimenting with quality-improvement (QI) techniques learned through participation in SHM’s Project BOOST.
“The first principle I try to teach residents is that a good discharge for a patient without insurance is the same as a good discharge for a patient with insurance,” Dr. Axon says. “Many of the same principles apply.”
If the care plan fails to address basic needs of indigent patients, including access to housing, primary care, and affordable medications, that patient won’t be able to focus on their medical needs.
Affiliated primary-care clinics already see a high percentage of indigent patients, Dr. Axon says, so there might be some pushback when the hospital team attempts a new referral. “We have tried to distinguish between care that needs to be done in the first week or so after discharge versus ongoing follow-up,” he explains. “We have negotiated with the clinic so that patients can come back here for one or two visits for urgent follow-up care without being entered into the [outpatient] system permanently. We are also blessed to have federally qualified health centers in the Charleston area. We have cordial relationships with those clinics, even if it’s not as well-integrated as I might wish.”
Another service that can be helpful with care transitions for uninsured patients is a 14-bed transitional-care unit on the hospital campus. “It provides rehabilitation and long-term care for the small numbers of chronically ill patients with long-term disabilities who don’t qualify for Medicaid or Medicare and can’t be placed elsewhere,” he says. “We’re able to care for these patients in a less costly way on the unit, rather than leaving them in an acute-care bed.” The hospital, he adds, views the unit as a cost-avoidance measure.
Some have been on the unit for months; others do much better than expected and go home. “It’s always gratifying when patients come back to the hospital to visit and thank us for the care they received,” he says.
Larry Beresford is a freelance writer based in California.
The Medical University of South Carolina (MUSC) in Charleston hasn’t solved the issue of care transitions for the indigent, “but we have thought about it a lot,” says Neal Axon, MD, MSCR, FHM, assistant professor in the Division of Hospital Medicine at MUSC. The hospital is experimenting with quality-improvement (QI) techniques learned through participation in SHM’s Project BOOST.
“The first principle I try to teach residents is that a good discharge for a patient without insurance is the same as a good discharge for a patient with insurance,” Dr. Axon says. “Many of the same principles apply.”
If the care plan fails to address basic needs of indigent patients, including access to housing, primary care, and affordable medications, that patient won’t be able to focus on their medical needs.
Affiliated primary-care clinics already see a high percentage of indigent patients, Dr. Axon says, so there might be some pushback when the hospital team attempts a new referral. “We have tried to distinguish between care that needs to be done in the first week or so after discharge versus ongoing follow-up,” he explains. “We have negotiated with the clinic so that patients can come back here for one or two visits for urgent follow-up care without being entered into the [outpatient] system permanently. We are also blessed to have federally qualified health centers in the Charleston area. We have cordial relationships with those clinics, even if it’s not as well-integrated as I might wish.”
Another service that can be helpful with care transitions for uninsured patients is a 14-bed transitional-care unit on the hospital campus. “It provides rehabilitation and long-term care for the small numbers of chronically ill patients with long-term disabilities who don’t qualify for Medicaid or Medicare and can’t be placed elsewhere,” he says. “We’re able to care for these patients in a less costly way on the unit, rather than leaving them in an acute-care bed.” The hospital, he adds, views the unit as a cost-avoidance measure.
Some have been on the unit for months; others do much better than expected and go home. “It’s always gratifying when patients come back to the hospital to visit and thank us for the care they received,” he says.
Larry Beresford is a freelance writer based in California.
Into the Night
The halls are quiet, the lights dimmed, the incessant ringing of telephones has fallen silent, patients slumber in their rooms, nurses sit and chart, waiting for the inevitable patient call light to glow once again. Then it happens: the overhead announcement that slices through the night like a knife.
“Code blue, code blue!”
As the code team scurries to the room, they start the protocols. However, they are waiting for someone—the conductor of the symphony, if you will. Who will answer the call? Who will whisk down the hall to take the podium? Will that patient’s primary-care physician (PCP) come? The cardiologist, maybe the pulmonologist?
No, there is one person who walks the halls at night when all others are asleep (even the ED doctors, though awake, are consumed by crowded emergency rooms and cannot help). This person is the nocturnist.
What is a nocturnist, you ask? Well, among the many titles, job descriptions, and opportunities that being a hospitalist can entail, being a nocturnist is the one that shines in the dark of night when everyone else is fast asleep. A nocturnist is a hospitalist who works the night shift. As a resident, you might have nightmares about the many nights you’ve worked, the assembly line of patients, procedures, and cross-cover calls you’ve processed.
Nocturnists are the lone wolves of the night. They wear many hats and encounter a milieu of incessant admissions, more cross-cover calls than you can swing at, more grumpy, sleepy consultant phone exchanges than you would like, and endure the chronic fatigue of a person 20 years older than their actual age. But deep down in the muck of it all, there is something about the night shift that keeps a nocturnist coming back night after night.
Nocturnist in Charge
Working as a nocturnist is the last, purest form of practicing medicine. This position affords you the perfect opportunity to get back to the patient-doctor relationship because you are not rounding on other patients, juggling staff meetings, or battling a slew of other staff pining for your patient (i.e. case workers, physical therapists, consultants, etc.). Therefore, you can spend an adequate amount of time getting to know your patient without feeling rushed.
As far as admissions are concerned, there still are those days when you feel you need more hours in a day and two extra hands to take on the flood, but as the physician in charge, you have the ability to better triage these patients and defer to a specialist if needed. It’s not like those residency days of admitting whatever they call you for.
In addition, you have the opportunity to really hone your medical skills and procedural skills, because you are the specialist at 3 a.m. There will be times when you have to make decisions without the luxury of an immediate consultation; that has its pros and cons, but it definitely makes for an exciting Friday night. Consequently, you usually are the first point of contact for the nursing staff at night, so you have the ability to formulate relationships with nurses like no other physician can, because you are there with them, side by side, handling all the emergent (and often nonemergent) cross-cover calls. The nurses learn to trust you and you them, and there is a sense of camaraderie that forms from that trust.
Night-Shift Benefits
If you are still not convinced that the nocturnist world is for you—though you will be able to spend more time and have a more meaningful relationship with patients, nursing staff, and be the hero to every consultant and PCP you allow to sleep through the night—then I must reveal that the real cherry on top is actually green. Since you are working the least desired shift in your HM group, you are somewhat of a rock star. No one wants you to be unhappy, because they really want you to keep working the night shift. It’s evident by the fact that most nocturnists are paid a 10% to 20% shift differential, according to Payscale.com. In layman’s terms, you get paid more money than everyone else.
Another benefit is that nocturnist shifts range from eight to 12 hours; some even allow you to take call from home, so you can find a position that fits your schedule. The average number of monthly shifts usually is fewer than those working the day shift (10 to 14 shifts compared with 14 to 18 shifts) on average.
Depending on what type of hospital you choose (rural or urban, community or academic), you can have a wide range of nightly responsibilities. Some nocturnists perform as many procedures as they like; others choose to perform no procedures. Patient caps might exist on the number of patients you can admit during a shift. And working as a nocturnist can afford you a terrific lifestyle, because there is an a la carte menu of hospitalist groups, shifts, and practice lifestyles to choose from. And everybody in HM knows that everyone is looking for a nocturnist, so the availability of job offers is never a problem.
Nevertheless, with more money and choices comes more responsibility. As a nocturnist, you have to be flexible and creative in order to stay informed, as you will find it challenging to make all the staff meetings. Ask your group to schedule important group meetings early, so that you can stay after your shift and attend. Sometimes you just have to dig in and stay for those later meetings, if need be. (Sleeping in the call room until your next shift makes you somewhat of a martyr.) And remind your medical director to email you any important information you might have missed.
Even though you won’t be around during the day, you must stay abreast of quality initiatives (CHF, AMI, etc.). Beware of charting requirements, which can change from day to day.
If you are looking for an exciting way of life, and the ability to practice pure medicine after residency, you might want to get “into the night” and consider an HM career as a nocturnist. TH
Dr. Cunningham has been a hospitalist since 2004 and a nocturnist the past three years at Hamilton Medical Center, a community hospital in Dalton, Ga., and locum tenens in the Tennessee area.
The halls are quiet, the lights dimmed, the incessant ringing of telephones has fallen silent, patients slumber in their rooms, nurses sit and chart, waiting for the inevitable patient call light to glow once again. Then it happens: the overhead announcement that slices through the night like a knife.
“Code blue, code blue!”
As the code team scurries to the room, they start the protocols. However, they are waiting for someone—the conductor of the symphony, if you will. Who will answer the call? Who will whisk down the hall to take the podium? Will that patient’s primary-care physician (PCP) come? The cardiologist, maybe the pulmonologist?
No, there is one person who walks the halls at night when all others are asleep (even the ED doctors, though awake, are consumed by crowded emergency rooms and cannot help). This person is the nocturnist.
What is a nocturnist, you ask? Well, among the many titles, job descriptions, and opportunities that being a hospitalist can entail, being a nocturnist is the one that shines in the dark of night when everyone else is fast asleep. A nocturnist is a hospitalist who works the night shift. As a resident, you might have nightmares about the many nights you’ve worked, the assembly line of patients, procedures, and cross-cover calls you’ve processed.
Nocturnists are the lone wolves of the night. They wear many hats and encounter a milieu of incessant admissions, more cross-cover calls than you can swing at, more grumpy, sleepy consultant phone exchanges than you would like, and endure the chronic fatigue of a person 20 years older than their actual age. But deep down in the muck of it all, there is something about the night shift that keeps a nocturnist coming back night after night.
Nocturnist in Charge
Working as a nocturnist is the last, purest form of practicing medicine. This position affords you the perfect opportunity to get back to the patient-doctor relationship because you are not rounding on other patients, juggling staff meetings, or battling a slew of other staff pining for your patient (i.e. case workers, physical therapists, consultants, etc.). Therefore, you can spend an adequate amount of time getting to know your patient without feeling rushed.
As far as admissions are concerned, there still are those days when you feel you need more hours in a day and two extra hands to take on the flood, but as the physician in charge, you have the ability to better triage these patients and defer to a specialist if needed. It’s not like those residency days of admitting whatever they call you for.
In addition, you have the opportunity to really hone your medical skills and procedural skills, because you are the specialist at 3 a.m. There will be times when you have to make decisions without the luxury of an immediate consultation; that has its pros and cons, but it definitely makes for an exciting Friday night. Consequently, you usually are the first point of contact for the nursing staff at night, so you have the ability to formulate relationships with nurses like no other physician can, because you are there with them, side by side, handling all the emergent (and often nonemergent) cross-cover calls. The nurses learn to trust you and you them, and there is a sense of camaraderie that forms from that trust.
Night-Shift Benefits
If you are still not convinced that the nocturnist world is for you—though you will be able to spend more time and have a more meaningful relationship with patients, nursing staff, and be the hero to every consultant and PCP you allow to sleep through the night—then I must reveal that the real cherry on top is actually green. Since you are working the least desired shift in your HM group, you are somewhat of a rock star. No one wants you to be unhappy, because they really want you to keep working the night shift. It’s evident by the fact that most nocturnists are paid a 10% to 20% shift differential, according to Payscale.com. In layman’s terms, you get paid more money than everyone else.
Another benefit is that nocturnist shifts range from eight to 12 hours; some even allow you to take call from home, so you can find a position that fits your schedule. The average number of monthly shifts usually is fewer than those working the day shift (10 to 14 shifts compared with 14 to 18 shifts) on average.
Depending on what type of hospital you choose (rural or urban, community or academic), you can have a wide range of nightly responsibilities. Some nocturnists perform as many procedures as they like; others choose to perform no procedures. Patient caps might exist on the number of patients you can admit during a shift. And working as a nocturnist can afford you a terrific lifestyle, because there is an a la carte menu of hospitalist groups, shifts, and practice lifestyles to choose from. And everybody in HM knows that everyone is looking for a nocturnist, so the availability of job offers is never a problem.
Nevertheless, with more money and choices comes more responsibility. As a nocturnist, you have to be flexible and creative in order to stay informed, as you will find it challenging to make all the staff meetings. Ask your group to schedule important group meetings early, so that you can stay after your shift and attend. Sometimes you just have to dig in and stay for those later meetings, if need be. (Sleeping in the call room until your next shift makes you somewhat of a martyr.) And remind your medical director to email you any important information you might have missed.
Even though you won’t be around during the day, you must stay abreast of quality initiatives (CHF, AMI, etc.). Beware of charting requirements, which can change from day to day.
If you are looking for an exciting way of life, and the ability to practice pure medicine after residency, you might want to get “into the night” and consider an HM career as a nocturnist. TH
Dr. Cunningham has been a hospitalist since 2004 and a nocturnist the past three years at Hamilton Medical Center, a community hospital in Dalton, Ga., and locum tenens in the Tennessee area.
The halls are quiet, the lights dimmed, the incessant ringing of telephones has fallen silent, patients slumber in their rooms, nurses sit and chart, waiting for the inevitable patient call light to glow once again. Then it happens: the overhead announcement that slices through the night like a knife.
“Code blue, code blue!”
As the code team scurries to the room, they start the protocols. However, they are waiting for someone—the conductor of the symphony, if you will. Who will answer the call? Who will whisk down the hall to take the podium? Will that patient’s primary-care physician (PCP) come? The cardiologist, maybe the pulmonologist?
No, there is one person who walks the halls at night when all others are asleep (even the ED doctors, though awake, are consumed by crowded emergency rooms and cannot help). This person is the nocturnist.
What is a nocturnist, you ask? Well, among the many titles, job descriptions, and opportunities that being a hospitalist can entail, being a nocturnist is the one that shines in the dark of night when everyone else is fast asleep. A nocturnist is a hospitalist who works the night shift. As a resident, you might have nightmares about the many nights you’ve worked, the assembly line of patients, procedures, and cross-cover calls you’ve processed.
Nocturnists are the lone wolves of the night. They wear many hats and encounter a milieu of incessant admissions, more cross-cover calls than you can swing at, more grumpy, sleepy consultant phone exchanges than you would like, and endure the chronic fatigue of a person 20 years older than their actual age. But deep down in the muck of it all, there is something about the night shift that keeps a nocturnist coming back night after night.
Nocturnist in Charge
Working as a nocturnist is the last, purest form of practicing medicine. This position affords you the perfect opportunity to get back to the patient-doctor relationship because you are not rounding on other patients, juggling staff meetings, or battling a slew of other staff pining for your patient (i.e. case workers, physical therapists, consultants, etc.). Therefore, you can spend an adequate amount of time getting to know your patient without feeling rushed.
As far as admissions are concerned, there still are those days when you feel you need more hours in a day and two extra hands to take on the flood, but as the physician in charge, you have the ability to better triage these patients and defer to a specialist if needed. It’s not like those residency days of admitting whatever they call you for.
In addition, you have the opportunity to really hone your medical skills and procedural skills, because you are the specialist at 3 a.m. There will be times when you have to make decisions without the luxury of an immediate consultation; that has its pros and cons, but it definitely makes for an exciting Friday night. Consequently, you usually are the first point of contact for the nursing staff at night, so you have the ability to formulate relationships with nurses like no other physician can, because you are there with them, side by side, handling all the emergent (and often nonemergent) cross-cover calls. The nurses learn to trust you and you them, and there is a sense of camaraderie that forms from that trust.
Night-Shift Benefits
If you are still not convinced that the nocturnist world is for you—though you will be able to spend more time and have a more meaningful relationship with patients, nursing staff, and be the hero to every consultant and PCP you allow to sleep through the night—then I must reveal that the real cherry on top is actually green. Since you are working the least desired shift in your HM group, you are somewhat of a rock star. No one wants you to be unhappy, because they really want you to keep working the night shift. It’s evident by the fact that most nocturnists are paid a 10% to 20% shift differential, according to Payscale.com. In layman’s terms, you get paid more money than everyone else.
Another benefit is that nocturnist shifts range from eight to 12 hours; some even allow you to take call from home, so you can find a position that fits your schedule. The average number of monthly shifts usually is fewer than those working the day shift (10 to 14 shifts compared with 14 to 18 shifts) on average.
Depending on what type of hospital you choose (rural or urban, community or academic), you can have a wide range of nightly responsibilities. Some nocturnists perform as many procedures as they like; others choose to perform no procedures. Patient caps might exist on the number of patients you can admit during a shift. And working as a nocturnist can afford you a terrific lifestyle, because there is an a la carte menu of hospitalist groups, shifts, and practice lifestyles to choose from. And everybody in HM knows that everyone is looking for a nocturnist, so the availability of job offers is never a problem.
Nevertheless, with more money and choices comes more responsibility. As a nocturnist, you have to be flexible and creative in order to stay informed, as you will find it challenging to make all the staff meetings. Ask your group to schedule important group meetings early, so that you can stay after your shift and attend. Sometimes you just have to dig in and stay for those later meetings, if need be. (Sleeping in the call room until your next shift makes you somewhat of a martyr.) And remind your medical director to email you any important information you might have missed.
Even though you won’t be around during the day, you must stay abreast of quality initiatives (CHF, AMI, etc.). Beware of charting requirements, which can change from day to day.
If you are looking for an exciting way of life, and the ability to practice pure medicine after residency, you might want to get “into the night” and consider an HM career as a nocturnist. TH
Dr. Cunningham has been a hospitalist since 2004 and a nocturnist the past three years at Hamilton Medical Center, a community hospital in Dalton, Ga., and locum tenens in the Tennessee area.
ONLINE EXCLUSIVE: Listen to experts discuss drug shortages
Click here to listen to Dr. Verma
Click here to listen to ISMP President David Cohen
Click here to listen to Dr. Verma
Click here to listen to ISMP President David Cohen
Click here to listen to Dr. Verma
Click here to listen to ISMP President David Cohen
ONLINE EXCLUSIVE: Subsidy or Investment?
Branding is defined by Merriam-Webster as the promotion of a product or service tied to a particular brand. Most hospitalists say HM has done a good job branding itself as the go-to physician specialty for patient safety and quality-improvement (QI) initiatives.
But labeling the financial support payments that help pay for that service as a subsidy?
“It’s a horrible branding exercise,” says Troy Ahlstrom, MD, SFHM, chief financial officer of Hospitalists of Northern Michigan, a hospitalist-owned and -managed group based in Traverse City.
The monies that change hands between hospitals and HM groups have long been known as subsidies, with one consulting group’s marketing materials giving advice on why subsidies are necessary. Hospitalist John Bulger, DO, FACP, FHM, of Geisinger Medical Center in Danville, Pa., says the payments must be viewed the same as financial agreements with other specialties, which rarely are viewed as subsidies.
“I would like to see us move toward more of a discussion of an investment,” Dr. Bulger says. “If you believe like I do that it’s actually a value-added tool that brings a return to the hospital, then we just have to do a better job of figuring … a methodology across the industry to showcase value.”
Todd Nelson, a technical director at the Westchester, Ill.-based Healthcare Financial Management Association, says hospitals value groups that can provide definable progress in core measures tied to patient safety and QI programs. And when it comes to funding those proactive physician groups, hospitals understand there is a cost of doing business.
“From the hospital perspective, they’re looking at it more as an investment,” says Nelson, a former chief financial officer at Iowa’s Grinnell Regional Medical Center. “They’re looking to engage the physicians. … Patient care is more than showing up and taking care of the patients.”
Richard Quinn is a freelance writer based in New Jersey.
Branding is defined by Merriam-Webster as the promotion of a product or service tied to a particular brand. Most hospitalists say HM has done a good job branding itself as the go-to physician specialty for patient safety and quality-improvement (QI) initiatives.
But labeling the financial support payments that help pay for that service as a subsidy?
“It’s a horrible branding exercise,” says Troy Ahlstrom, MD, SFHM, chief financial officer of Hospitalists of Northern Michigan, a hospitalist-owned and -managed group based in Traverse City.
The monies that change hands between hospitals and HM groups have long been known as subsidies, with one consulting group’s marketing materials giving advice on why subsidies are necessary. Hospitalist John Bulger, DO, FACP, FHM, of Geisinger Medical Center in Danville, Pa., says the payments must be viewed the same as financial agreements with other specialties, which rarely are viewed as subsidies.
“I would like to see us move toward more of a discussion of an investment,” Dr. Bulger says. “If you believe like I do that it’s actually a value-added tool that brings a return to the hospital, then we just have to do a better job of figuring … a methodology across the industry to showcase value.”
Todd Nelson, a technical director at the Westchester, Ill.-based Healthcare Financial Management Association, says hospitals value groups that can provide definable progress in core measures tied to patient safety and QI programs. And when it comes to funding those proactive physician groups, hospitals understand there is a cost of doing business.
“From the hospital perspective, they’re looking at it more as an investment,” says Nelson, a former chief financial officer at Iowa’s Grinnell Regional Medical Center. “They’re looking to engage the physicians. … Patient care is more than showing up and taking care of the patients.”
Richard Quinn is a freelance writer based in New Jersey.
Branding is defined by Merriam-Webster as the promotion of a product or service tied to a particular brand. Most hospitalists say HM has done a good job branding itself as the go-to physician specialty for patient safety and quality-improvement (QI) initiatives.
But labeling the financial support payments that help pay for that service as a subsidy?
“It’s a horrible branding exercise,” says Troy Ahlstrom, MD, SFHM, chief financial officer of Hospitalists of Northern Michigan, a hospitalist-owned and -managed group based in Traverse City.
The monies that change hands between hospitals and HM groups have long been known as subsidies, with one consulting group’s marketing materials giving advice on why subsidies are necessary. Hospitalist John Bulger, DO, FACP, FHM, of Geisinger Medical Center in Danville, Pa., says the payments must be viewed the same as financial agreements with other specialties, which rarely are viewed as subsidies.
“I would like to see us move toward more of a discussion of an investment,” Dr. Bulger says. “If you believe like I do that it’s actually a value-added tool that brings a return to the hospital, then we just have to do a better job of figuring … a methodology across the industry to showcase value.”
Todd Nelson, a technical director at the Westchester, Ill.-based Healthcare Financial Management Association, says hospitals value groups that can provide definable progress in core measures tied to patient safety and QI programs. And when it comes to funding those proactive physician groups, hospitals understand there is a cost of doing business.
“From the hospital perspective, they’re looking at it more as an investment,” says Nelson, a former chief financial officer at Iowa’s Grinnell Regional Medical Center. “They’re looking to engage the physicians. … Patient care is more than showing up and taking care of the patients.”
Richard Quinn is a freelance writer based in New Jersey.
ONLINE EXCLUSIVE: Hospitalists discuss strategies for indigent transitions
Click here to listen to Dr. Misky
Click here to listen to Jane Brock
Click here to listen to San Francisco General hospitalists Jeff Critchfield and Michelle Schneidermann
Click here to listen to Dr. Misky
Click here to listen to Jane Brock
Click here to listen to San Francisco General hospitalists Jeff Critchfield and Michelle Schneidermann
Click here to listen to Dr. Misky
Click here to listen to Jane Brock
Click here to listen to San Francisco General hospitalists Jeff Critchfield and Michelle Schneidermann
ONLINE EXCLUSIVE: TKTK
Enter text here
Enter text here
Enter text here