User login
Academic Institutions
Hospitalists work in many types of facilities, including academic centers that utilize residents (including interns) in healthcare delivery. Medical and surgical services furnished by a resident within the scope of the training program are covered as provider services and paid by Medicare through direct Graduate Medical Education (GME) and Indirect Medical Education (IME) payments; the services of the resident may not be billed or paid for using the Medicare Physician Fee Schedule.
Similarly, the teaching physician is not paid for the resident’s work. The teaching physician is paid for their participation in patient care. In other words, payment is provided to the teaching physician for services that are:
- Furnished by a physician who is not a resident; or
- Furnished by a resident with a teaching physician physically present during the critical or key portion(s) of the service.
Teaching physicians participate in evaluation and management (E/M) services with residents in several ways. Consider the following teaching physician scenarios:
Scenario 1: “Stand-Alone” Service
The resident sees a patient in the morning. The teaching physician independently sees the patient later that same day, performing all required elements to support their own bill (e.g. 99233: subsequent hospital care, per day, which requires at least two of these three key components: a detailed interval history, a detailed examination, or high-complexity medical decision-making). When documenting, the teaching physician can write their own note with or without any of the residents’ information. The attending note “stands alone” in support of the reported visit level. Alternatively, the teaching physician might “link to” the resident note, instead of personally redocumenting the entire service.
Appropriate documentation includes teaching physician notation of the provided critical or key portion(s) of the service and the involvement in patient management. The visit level is based upon the combined documentation, both teaching physician and resident.
Using Medicare-approved linkage statements will ensure compliance with teaching physician rules. Examples:
- “I performed a history and physical examination of the patient and discussed his management with the resident. I reviewed the resident’s note and agree with the documented findings and plan of care.”
- “I saw and evaluated the patient. I agree with the findings and the plan of care as documented in the resident’s note.”
- “I saw and examined the patient. I agree with the resident’s note, except the heart murmur is louder, so I will obtain an echo to evaluate.”
Each of the above linkage statements is acceptable, and “more is always better.” The last example best identifies the teaching physician’s involvement in patient management and best supports other regulatory goals and quality initiatives of the current healthcare environment.
Scenario 2: “Supervised” Service
The resident and the teaching physician see the patient at the same time. The teaching physician supervises the resident’s performance of the required service elements or personally performs elements separate from those completed by the resident. Despite personal supervision, the attending still must document their presence during the encounter, performance of the critical or key portion(s) of the service, and involvement in patient management. The visit level is based upon the combined documentation.
Medicare-accepted teaching physician statements associated with this scenario include:
- “I was present with the resident during the history and exam. I discussed the case with the resident and agree with the findings and plan as documented in the resident’s note.”
- “I saw the patient with the resident and agree with the resident’s findings and plan.”
These generalized statements will be accepted for billing under teaching physician rules. However, documenting patient-specific elements of the assessment and plan unequivocally demonstrates teaching- physician involvement in patient care and the quality of care provided.
Scenario 3: The “Shared” Service
The resident performs a portion or all of the required service elements without teaching-physician presence and documents this service. The teaching physician then independently performs only the critical or key portion(s) of the service and, as appropriate, discusses the case with the resident. As in the other scenarios, the attending documents the presence and performance of the critical or key portion(s) of the service, as well as involvement in patient management. The teaching physician selects the visit level based upon the combined documentation of the teaching physician and resident.
Such Medicare-approved statements for use by teaching physicians under this scenario include:
- “I saw and evaluated the patient. I reviewed the resident’s note and agree, except that picture is more consistent with pericarditis than myocardial ischemia. Will begin NSAIDs.”
- “I saw and evaluated the patient. Discussed with resident and agree with resident’s findings and plan as documented in the resident’s note.”
- “See resident’s note for details. I saw and evaluated the patient and agree with the resident’s finding and plans as written.”
- “I saw and evaluated the patient. Agree with resident’s note, but lower extremities are weaker, now 3/5; MRI of L/S spine today.”
Regardless of the timing between the attending and the resident encounter represented in each scenario, the teaching physician cannot “link to” a resident note that has not been written. More specifically, if the resident’s note has not been documented at the time the teaching physician writes their note, the teaching physician can’t link to the resident’s note or consider it for billing purposes.
Time-Based Exception
Time-based E/M services (e.g. critical-care services, discharge-day management, prolonged care, etc.) do not follow the same guideline as the standard E/M services, which are selected upon the level of history, exam, and decision-making. Only the billing provider’s time counts toward the reported visit level. This means that the teaching physician must be present for the entire period of time for which the claim is made. Documentation should identify the teaching physician’s total visit time (spent on the unit/floor for inpatient services), including face-to-face time with the patient. Time spent by the resident without the presence of the teaching physician does not count toward the teaching physician’s reported time. Additionally, time spent “teaching” the resident cannot be attributed to the teaching physician’s visit time.
Student Notes
Per Medicare guidelines, students (medical, nurse practitioner, etc.) can document services in the medical record. However, the teaching physician can only refer to medical student documentation associated with the review of systems and/or past/family/social history. The teaching physician cannot refer to a student’s documentation of physical exam findings or medical decision-making.
If the medical student documents E/M services, the teaching physician must verify and redocument the history of present illness, as well as perform and redocument the physical exam and medical decision-making activities of the service. The teaching physician then selects the visit level and documents service. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
References
- Guidelines for Teaching Physicians, Interns, Residents. Centers for Medicare & Medicaid Services website. Available at: http://www.cms.gov/MLNProducts/downloads/gdelinesteachgresfctsht.pdf. Accessed May 6, 2011.
- Medicare Claims Processing Manual: Chapter 12, Section 100. Centers for Medicare & Medicaid Services website. Available at: http://www.cms.hhs.gov/manuals/downloads/clm104c12.pdf. Accessed May 6, 2011.
- Medicare Benefit Policy Manual: Chapter 15, Section 30.2. Centers for Medicare & Medicaid Services website. Available at: http://www.cms.hhs.gov/manuals/Downloads/bp102c15.pdf. Accessed May 6, 2011.
- Manaker, S. Teaching Physician Regulations. In: Coding for Chest Medicine 2008. Northbrook, IL: American College of Chest Physicians, 2008; 279-285.
- Pohlig, C. Evaluation & Management Services: An Overview. In: Coding for Chest Medicine 2011. Northbrook, IL: American College of Chest Physicians, 2010; 323-330.
- Abraham M, Ahlman J, Boudreau A, Connelly J, Evans D. Current Procedural Terminology Professional Edition. Chicago: American Medical Association Press; 2011.
Hospitalists work in many types of facilities, including academic centers that utilize residents (including interns) in healthcare delivery. Medical and surgical services furnished by a resident within the scope of the training program are covered as provider services and paid by Medicare through direct Graduate Medical Education (GME) and Indirect Medical Education (IME) payments; the services of the resident may not be billed or paid for using the Medicare Physician Fee Schedule.
Similarly, the teaching physician is not paid for the resident’s work. The teaching physician is paid for their participation in patient care. In other words, payment is provided to the teaching physician for services that are:
- Furnished by a physician who is not a resident; or
- Furnished by a resident with a teaching physician physically present during the critical or key portion(s) of the service.
Teaching physicians participate in evaluation and management (E/M) services with residents in several ways. Consider the following teaching physician scenarios:
Scenario 1: “Stand-Alone” Service
The resident sees a patient in the morning. The teaching physician independently sees the patient later that same day, performing all required elements to support their own bill (e.g. 99233: subsequent hospital care, per day, which requires at least two of these three key components: a detailed interval history, a detailed examination, or high-complexity medical decision-making). When documenting, the teaching physician can write their own note with or without any of the residents’ information. The attending note “stands alone” in support of the reported visit level. Alternatively, the teaching physician might “link to” the resident note, instead of personally redocumenting the entire service.
Appropriate documentation includes teaching physician notation of the provided critical or key portion(s) of the service and the involvement in patient management. The visit level is based upon the combined documentation, both teaching physician and resident.
Using Medicare-approved linkage statements will ensure compliance with teaching physician rules. Examples:
- “I performed a history and physical examination of the patient and discussed his management with the resident. I reviewed the resident’s note and agree with the documented findings and plan of care.”
- “I saw and evaluated the patient. I agree with the findings and the plan of care as documented in the resident’s note.”
- “I saw and examined the patient. I agree with the resident’s note, except the heart murmur is louder, so I will obtain an echo to evaluate.”
Each of the above linkage statements is acceptable, and “more is always better.” The last example best identifies the teaching physician’s involvement in patient management and best supports other regulatory goals and quality initiatives of the current healthcare environment.
Scenario 2: “Supervised” Service
The resident and the teaching physician see the patient at the same time. The teaching physician supervises the resident’s performance of the required service elements or personally performs elements separate from those completed by the resident. Despite personal supervision, the attending still must document their presence during the encounter, performance of the critical or key portion(s) of the service, and involvement in patient management. The visit level is based upon the combined documentation.
Medicare-accepted teaching physician statements associated with this scenario include:
- “I was present with the resident during the history and exam. I discussed the case with the resident and agree with the findings and plan as documented in the resident’s note.”
- “I saw the patient with the resident and agree with the resident’s findings and plan.”
These generalized statements will be accepted for billing under teaching physician rules. However, documenting patient-specific elements of the assessment and plan unequivocally demonstrates teaching- physician involvement in patient care and the quality of care provided.
Scenario 3: The “Shared” Service
The resident performs a portion or all of the required service elements without teaching-physician presence and documents this service. The teaching physician then independently performs only the critical or key portion(s) of the service and, as appropriate, discusses the case with the resident. As in the other scenarios, the attending documents the presence and performance of the critical or key portion(s) of the service, as well as involvement in patient management. The teaching physician selects the visit level based upon the combined documentation of the teaching physician and resident.
Such Medicare-approved statements for use by teaching physicians under this scenario include:
- “I saw and evaluated the patient. I reviewed the resident’s note and agree, except that picture is more consistent with pericarditis than myocardial ischemia. Will begin NSAIDs.”
- “I saw and evaluated the patient. Discussed with resident and agree with resident’s findings and plan as documented in the resident’s note.”
- “See resident’s note for details. I saw and evaluated the patient and agree with the resident’s finding and plans as written.”
- “I saw and evaluated the patient. Agree with resident’s note, but lower extremities are weaker, now 3/5; MRI of L/S spine today.”
Regardless of the timing between the attending and the resident encounter represented in each scenario, the teaching physician cannot “link to” a resident note that has not been written. More specifically, if the resident’s note has not been documented at the time the teaching physician writes their note, the teaching physician can’t link to the resident’s note or consider it for billing purposes.
Time-Based Exception
Time-based E/M services (e.g. critical-care services, discharge-day management, prolonged care, etc.) do not follow the same guideline as the standard E/M services, which are selected upon the level of history, exam, and decision-making. Only the billing provider’s time counts toward the reported visit level. This means that the teaching physician must be present for the entire period of time for which the claim is made. Documentation should identify the teaching physician’s total visit time (spent on the unit/floor for inpatient services), including face-to-face time with the patient. Time spent by the resident without the presence of the teaching physician does not count toward the teaching physician’s reported time. Additionally, time spent “teaching” the resident cannot be attributed to the teaching physician’s visit time.
Student Notes
Per Medicare guidelines, students (medical, nurse practitioner, etc.) can document services in the medical record. However, the teaching physician can only refer to medical student documentation associated with the review of systems and/or past/family/social history. The teaching physician cannot refer to a student’s documentation of physical exam findings or medical decision-making.
If the medical student documents E/M services, the teaching physician must verify and redocument the history of present illness, as well as perform and redocument the physical exam and medical decision-making activities of the service. The teaching physician then selects the visit level and documents service. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
References
- Guidelines for Teaching Physicians, Interns, Residents. Centers for Medicare & Medicaid Services website. Available at: http://www.cms.gov/MLNProducts/downloads/gdelinesteachgresfctsht.pdf. Accessed May 6, 2011.
- Medicare Claims Processing Manual: Chapter 12, Section 100. Centers for Medicare & Medicaid Services website. Available at: http://www.cms.hhs.gov/manuals/downloads/clm104c12.pdf. Accessed May 6, 2011.
- Medicare Benefit Policy Manual: Chapter 15, Section 30.2. Centers for Medicare & Medicaid Services website. Available at: http://www.cms.hhs.gov/manuals/Downloads/bp102c15.pdf. Accessed May 6, 2011.
- Manaker, S. Teaching Physician Regulations. In: Coding for Chest Medicine 2008. Northbrook, IL: American College of Chest Physicians, 2008; 279-285.
- Pohlig, C. Evaluation & Management Services: An Overview. In: Coding for Chest Medicine 2011. Northbrook, IL: American College of Chest Physicians, 2010; 323-330.
- Abraham M, Ahlman J, Boudreau A, Connelly J, Evans D. Current Procedural Terminology Professional Edition. Chicago: American Medical Association Press; 2011.
Hospitalists work in many types of facilities, including academic centers that utilize residents (including interns) in healthcare delivery. Medical and surgical services furnished by a resident within the scope of the training program are covered as provider services and paid by Medicare through direct Graduate Medical Education (GME) and Indirect Medical Education (IME) payments; the services of the resident may not be billed or paid for using the Medicare Physician Fee Schedule.
Similarly, the teaching physician is not paid for the resident’s work. The teaching physician is paid for their participation in patient care. In other words, payment is provided to the teaching physician for services that are:
- Furnished by a physician who is not a resident; or
- Furnished by a resident with a teaching physician physically present during the critical or key portion(s) of the service.
Teaching physicians participate in evaluation and management (E/M) services with residents in several ways. Consider the following teaching physician scenarios:
Scenario 1: “Stand-Alone” Service
The resident sees a patient in the morning. The teaching physician independently sees the patient later that same day, performing all required elements to support their own bill (e.g. 99233: subsequent hospital care, per day, which requires at least two of these three key components: a detailed interval history, a detailed examination, or high-complexity medical decision-making). When documenting, the teaching physician can write their own note with or without any of the residents’ information. The attending note “stands alone” in support of the reported visit level. Alternatively, the teaching physician might “link to” the resident note, instead of personally redocumenting the entire service.
Appropriate documentation includes teaching physician notation of the provided critical or key portion(s) of the service and the involvement in patient management. The visit level is based upon the combined documentation, both teaching physician and resident.
Using Medicare-approved linkage statements will ensure compliance with teaching physician rules. Examples:
- “I performed a history and physical examination of the patient and discussed his management with the resident. I reviewed the resident’s note and agree with the documented findings and plan of care.”
- “I saw and evaluated the patient. I agree with the findings and the plan of care as documented in the resident’s note.”
- “I saw and examined the patient. I agree with the resident’s note, except the heart murmur is louder, so I will obtain an echo to evaluate.”
Each of the above linkage statements is acceptable, and “more is always better.” The last example best identifies the teaching physician’s involvement in patient management and best supports other regulatory goals and quality initiatives of the current healthcare environment.
Scenario 2: “Supervised” Service
The resident and the teaching physician see the patient at the same time. The teaching physician supervises the resident’s performance of the required service elements or personally performs elements separate from those completed by the resident. Despite personal supervision, the attending still must document their presence during the encounter, performance of the critical or key portion(s) of the service, and involvement in patient management. The visit level is based upon the combined documentation.
Medicare-accepted teaching physician statements associated with this scenario include:
- “I was present with the resident during the history and exam. I discussed the case with the resident and agree with the findings and plan as documented in the resident’s note.”
- “I saw the patient with the resident and agree with the resident’s findings and plan.”
These generalized statements will be accepted for billing under teaching physician rules. However, documenting patient-specific elements of the assessment and plan unequivocally demonstrates teaching- physician involvement in patient care and the quality of care provided.
Scenario 3: The “Shared” Service
The resident performs a portion or all of the required service elements without teaching-physician presence and documents this service. The teaching physician then independently performs only the critical or key portion(s) of the service and, as appropriate, discusses the case with the resident. As in the other scenarios, the attending documents the presence and performance of the critical or key portion(s) of the service, as well as involvement in patient management. The teaching physician selects the visit level based upon the combined documentation of the teaching physician and resident.
Such Medicare-approved statements for use by teaching physicians under this scenario include:
- “I saw and evaluated the patient. I reviewed the resident’s note and agree, except that picture is more consistent with pericarditis than myocardial ischemia. Will begin NSAIDs.”
- “I saw and evaluated the patient. Discussed with resident and agree with resident’s findings and plan as documented in the resident’s note.”
- “See resident’s note for details. I saw and evaluated the patient and agree with the resident’s finding and plans as written.”
- “I saw and evaluated the patient. Agree with resident’s note, but lower extremities are weaker, now 3/5; MRI of L/S spine today.”
Regardless of the timing between the attending and the resident encounter represented in each scenario, the teaching physician cannot “link to” a resident note that has not been written. More specifically, if the resident’s note has not been documented at the time the teaching physician writes their note, the teaching physician can’t link to the resident’s note or consider it for billing purposes.
Time-Based Exception
Time-based E/M services (e.g. critical-care services, discharge-day management, prolonged care, etc.) do not follow the same guideline as the standard E/M services, which are selected upon the level of history, exam, and decision-making. Only the billing provider’s time counts toward the reported visit level. This means that the teaching physician must be present for the entire period of time for which the claim is made. Documentation should identify the teaching physician’s total visit time (spent on the unit/floor for inpatient services), including face-to-face time with the patient. Time spent by the resident without the presence of the teaching physician does not count toward the teaching physician’s reported time. Additionally, time spent “teaching” the resident cannot be attributed to the teaching physician’s visit time.
Student Notes
Per Medicare guidelines, students (medical, nurse practitioner, etc.) can document services in the medical record. However, the teaching physician can only refer to medical student documentation associated with the review of systems and/or past/family/social history. The teaching physician cannot refer to a student’s documentation of physical exam findings or medical decision-making.
If the medical student documents E/M services, the teaching physician must verify and redocument the history of present illness, as well as perform and redocument the physical exam and medical decision-making activities of the service. The teaching physician then selects the visit level and documents service. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
References
- Guidelines for Teaching Physicians, Interns, Residents. Centers for Medicare & Medicaid Services website. Available at: http://www.cms.gov/MLNProducts/downloads/gdelinesteachgresfctsht.pdf. Accessed May 6, 2011.
- Medicare Claims Processing Manual: Chapter 12, Section 100. Centers for Medicare & Medicaid Services website. Available at: http://www.cms.hhs.gov/manuals/downloads/clm104c12.pdf. Accessed May 6, 2011.
- Medicare Benefit Policy Manual: Chapter 15, Section 30.2. Centers for Medicare & Medicaid Services website. Available at: http://www.cms.hhs.gov/manuals/Downloads/bp102c15.pdf. Accessed May 6, 2011.
- Manaker, S. Teaching Physician Regulations. In: Coding for Chest Medicine 2008. Northbrook, IL: American College of Chest Physicians, 2008; 279-285.
- Pohlig, C. Evaluation & Management Services: An Overview. In: Coding for Chest Medicine 2011. Northbrook, IL: American College of Chest Physicians, 2010; 323-330.
- Abraham M, Ahlman J, Boudreau A, Connelly J, Evans D. Current Procedural Terminology Professional Edition. Chicago: American Medical Association Press; 2011.
Ultrasound More Common at the Bedside
A recent “Current Concepts” article in the New England Journal of Medicine (2011;364:749) by a pair of Yale University physicians asserts that the day is close at hand when ultrasound interpretations by clinicians at the patient’s bedside will become as routine in hospital care as the trusty stethoscope. Ultrasound, a noninvasive form of imaging related to oceanographic sonar, has moved beyond its traditional home in radiology to myriad other medical specialties and practice areas. The technology has become smaller, less expensive, and higher in resolution in recent years, the authors note, adding that it has been used on Mount Everest and the international space station, as well as in battlefield situations.
“It’s becoming more accessible, and more training is available to physicians who aren’t radiologists,” says Diane Sliwka, MD, a hospitalist at the University of California at San Francisco (UCSF).
Dr. Sliwka says the NEJM article represents a milestone in the dissemination of bedside ultrasound. She conducts monthly faculty development training in procedural ultrasound at UCSF, workshops at HM and internal-medicine conferences, and training sessions for other hospitals.
The most common uses for bedside, “point of care” ultrasound include guiding procedures, such as thoracentesis and paracentesis, with improved safety over doing such insertions “blind.” Emerging procedural uses include lumbar puncture and arthrocentesis. Diagnostically, bedside ultrasound can provide quick screening and assessment, for example, of fluid buildup around the heart; previously, it could take hours to get the results from a formal heart study.
As with the stethoscope, Dr. Sliwka says, training in its correct use and scope of appropriate bedside practice is essential: “My advice is to learn from the experts at your facility, including the radiologists, critical care, or emergency physicians.” Ultrasound courses are increasing at hospitalist conferences, but space often is limited, and further supervised practice back home is needed.
The next step for hospitalists could be the definition of appropriate scope of practice, training, and competencies for its use. “Creating a niche in this area can be a nice change of pace from our traditional work as hospitalists,” Dr. Sliwka says. —LB
Technology
Video Chat Takes Off for Physicians

A recent study of digital adoption trends found that 7% of U.S. physicians now use video consultations to communicate with patients.
Manhattan Research’s 2011 “Taking the Pulse” survey of 2,000 physicians’ use of technology found that video chat is emerging as a way to consult with patients about nonurgent issues and follow-up questions or with geographically dispersed patients. Psychiatrists and oncologists are more likely to use the new technology. Doctors’ concerns regarding reimbursement, liability, and HIPAA privacy rules remain barriers to adoption.
For more information, visit ManhattanResearch.com/News-and-Events/Press-Releases/physician-patient-online-video-conferencing.—LB
Legal
Positive Outcomes from Full Disclosure of Medical Errors
The University of Michigan Health System’s (UMHS) risk-management model of full disclosure with offer of compensation for medical errors sparked hospitalist Allen Kachalia, MD, JD, of Brigham & Women’s Hospital in Boston to retrospectively study the outcomes of malpractice-claims-related performance before and after UMHS implemented the system in 2001.
Among the results Dr. Kachalia reported in his research abstract plenary at HM10, and subsequently published in Annals of Internal Medicine (2010;153(4):213-221), the mean monthly rate of new claims per 100,000 patient contacts decreased 36% after the full-disclosure model was adopted, while the rate of claims resulting in lawsuits declined by 65%. Claims also were resolved more quickly with the full-disclosure model.
Disclosure of medical error, Dr. Kachalia says, means “if someone is injured by medical care caused by medical error, the physician tells the patient they made the error, how it happened, and, often, what they’ll do to fix it.” An apology is somewhat different, he adds, and there’s no generic script for an apology. “What patients want is sincerity,” he says.
How can hospitalists work with full disclosure? “The general advice most institutions give is that when you want to disclose a medical error, first get your risk-management and patient-safety officers involved. They can help during every step of the process of investigating the event and disclosing,” Dr. Kachalia explains. “Assure patients that you are going to look into their concerns. Then make sure that a thorough investigation is done.”—LB
Practice Management
AMA-MGMA Toolkit Sorts Transitional-Care Software Options
HM practices with physicians in outpatient settings—be they discharge clinics or transitional-care centers—don’t always know how to determine the most useful practice-management software for their needs. So for those not helped by informatics staff, consider the new “Practice Management System Software Directory” from AMA and the Medical Group Management Association (MGMA).
The online repository, which launched in May, is a companion to the “Selecting a Practice Management System” toolkit the joint venture unveiled last fall. While the system is geared toward ambulatory-care settings, Robert Tennant, a senior policy advisor with MGMA, says any HM group with practitioners working on transitional care would find it useful.
Overall, the directory’s goal is to guide providers on how to navigate the increasingly complex world of practice-management options as new guidelines for “meaningful use” are defined, as well as new rules governing electronic claims processing. A new claims standard, known as HIPAA version 5010, is going live Jan. 1, 2012, so Tennant believes the directory is timely.
“It’s very difficult, whether in a practice or a hospital, to know the best software to pick,” he says. “There are plenty of vendors out there telling you they’re the best. There’s no easy way to comparison-shop.”
Now physicians can use the toolkit to measure basic functions. The directory, which will be updated on a rolling basis, will catalogue price range (excluding implementation costs), the number of installed customers, the target market for the product, what year the software was first offered, and whether the vendor also offers an electronic health record (EHR) system. That last point is of particular note to hospitalists as a link between practice management and medical records can help make a practice more efficient, Tennant says.
“What we’ve seen,” he adds, “is those that have that seamless integration between practice-management systems and EHR have higher productivity and higher levels of satisfaction.” —RQ
By The Numbers
$131,564
The average amount of money HM groups received in support per full-time equivalent (FTE) in fiscal year 2010, according to new SHM-MGMA survey data. The data point—the so-called “subsidy”—was first revealed at HM11 in Dallas.
After several years of leveling off at roughly $100,000, some hospitalists say they were surprised to see the figure rise so quickly. The report also shows that 19% of hospitalist practices receive no support, a finding that prompted new SHM President Joseph Li, MD, SFHM, to ask: “Are we looking at two business models or two care models?”—RQ
A recent “Current Concepts” article in the New England Journal of Medicine (2011;364:749) by a pair of Yale University physicians asserts that the day is close at hand when ultrasound interpretations by clinicians at the patient’s bedside will become as routine in hospital care as the trusty stethoscope. Ultrasound, a noninvasive form of imaging related to oceanographic sonar, has moved beyond its traditional home in radiology to myriad other medical specialties and practice areas. The technology has become smaller, less expensive, and higher in resolution in recent years, the authors note, adding that it has been used on Mount Everest and the international space station, as well as in battlefield situations.
“It’s becoming more accessible, and more training is available to physicians who aren’t radiologists,” says Diane Sliwka, MD, a hospitalist at the University of California at San Francisco (UCSF).
Dr. Sliwka says the NEJM article represents a milestone in the dissemination of bedside ultrasound. She conducts monthly faculty development training in procedural ultrasound at UCSF, workshops at HM and internal-medicine conferences, and training sessions for other hospitals.
The most common uses for bedside, “point of care” ultrasound include guiding procedures, such as thoracentesis and paracentesis, with improved safety over doing such insertions “blind.” Emerging procedural uses include lumbar puncture and arthrocentesis. Diagnostically, bedside ultrasound can provide quick screening and assessment, for example, of fluid buildup around the heart; previously, it could take hours to get the results from a formal heart study.
As with the stethoscope, Dr. Sliwka says, training in its correct use and scope of appropriate bedside practice is essential: “My advice is to learn from the experts at your facility, including the radiologists, critical care, or emergency physicians.” Ultrasound courses are increasing at hospitalist conferences, but space often is limited, and further supervised practice back home is needed.
The next step for hospitalists could be the definition of appropriate scope of practice, training, and competencies for its use. “Creating a niche in this area can be a nice change of pace from our traditional work as hospitalists,” Dr. Sliwka says. —LB
Technology
Video Chat Takes Off for Physicians
A recent study of digital adoption trends found that 7% of U.S. physicians now use video consultations to communicate with patients.
Manhattan Research’s 2011 “Taking the Pulse” survey of 2,000 physicians’ use of technology found that video chat is emerging as a way to consult with patients about nonurgent issues and follow-up questions or with geographically dispersed patients. Psychiatrists and oncologists are more likely to use the new technology. Doctors’ concerns regarding reimbursement, liability, and HIPAA privacy rules remain barriers to adoption.
For more information, visit ManhattanResearch.com/News-and-Events/Press-Releases/physician-patient-online-video-conferencing.—LB
Legal
Positive Outcomes from Full Disclosure of Medical Errors
The University of Michigan Health System’s (UMHS) risk-management model of full disclosure with offer of compensation for medical errors sparked hospitalist Allen Kachalia, MD, JD, of Brigham & Women’s Hospital in Boston to retrospectively study the outcomes of malpractice-claims-related performance before and after UMHS implemented the system in 2001.
Among the results Dr. Kachalia reported in his research abstract plenary at HM10, and subsequently published in Annals of Internal Medicine (2010;153(4):213-221), the mean monthly rate of new claims per 100,000 patient contacts decreased 36% after the full-disclosure model was adopted, while the rate of claims resulting in lawsuits declined by 65%. Claims also were resolved more quickly with the full-disclosure model.
Disclosure of medical error, Dr. Kachalia says, means “if someone is injured by medical care caused by medical error, the physician tells the patient they made the error, how it happened, and, often, what they’ll do to fix it.” An apology is somewhat different, he adds, and there’s no generic script for an apology. “What patients want is sincerity,” he says.
How can hospitalists work with full disclosure? “The general advice most institutions give is that when you want to disclose a medical error, first get your risk-management and patient-safety officers involved. They can help during every step of the process of investigating the event and disclosing,” Dr. Kachalia explains. “Assure patients that you are going to look into their concerns. Then make sure that a thorough investigation is done.”—LB
Practice Management
AMA-MGMA Toolkit Sorts Transitional-Care Software Options
HM practices with physicians in outpatient settings—be they discharge clinics or transitional-care centers—don’t always know how to determine the most useful practice-management software for their needs. So for those not helped by informatics staff, consider the new “Practice Management System Software Directory” from AMA and the Medical Group Management Association (MGMA).
The online repository, which launched in May, is a companion to the “Selecting a Practice Management System” toolkit the joint venture unveiled last fall. While the system is geared toward ambulatory-care settings, Robert Tennant, a senior policy advisor with MGMA, says any HM group with practitioners working on transitional care would find it useful.
Overall, the directory’s goal is to guide providers on how to navigate the increasingly complex world of practice-management options as new guidelines for “meaningful use” are defined, as well as new rules governing electronic claims processing. A new claims standard, known as HIPAA version 5010, is going live Jan. 1, 2012, so Tennant believes the directory is timely.
“It’s very difficult, whether in a practice or a hospital, to know the best software to pick,” he says. “There are plenty of vendors out there telling you they’re the best. There’s no easy way to comparison-shop.”
Now physicians can use the toolkit to measure basic functions. The directory, which will be updated on a rolling basis, will catalogue price range (excluding implementation costs), the number of installed customers, the target market for the product, what year the software was first offered, and whether the vendor also offers an electronic health record (EHR) system. That last point is of particular note to hospitalists as a link between practice management and medical records can help make a practice more efficient, Tennant says.
“What we’ve seen,” he adds, “is those that have that seamless integration between practice-management systems and EHR have higher productivity and higher levels of satisfaction.” —RQ
By The Numbers
$131,564
The average amount of money HM groups received in support per full-time equivalent (FTE) in fiscal year 2010, according to new SHM-MGMA survey data. The data point—the so-called “subsidy”—was first revealed at HM11 in Dallas.
After several years of leveling off at roughly $100,000, some hospitalists say they were surprised to see the figure rise so quickly. The report also shows that 19% of hospitalist practices receive no support, a finding that prompted new SHM President Joseph Li, MD, SFHM, to ask: “Are we looking at two business models or two care models?”—RQ
A recent “Current Concepts” article in the New England Journal of Medicine (2011;364:749) by a pair of Yale University physicians asserts that the day is close at hand when ultrasound interpretations by clinicians at the patient’s bedside will become as routine in hospital care as the trusty stethoscope. Ultrasound, a noninvasive form of imaging related to oceanographic sonar, has moved beyond its traditional home in radiology to myriad other medical specialties and practice areas. The technology has become smaller, less expensive, and higher in resolution in recent years, the authors note, adding that it has been used on Mount Everest and the international space station, as well as in battlefield situations.
“It’s becoming more accessible, and more training is available to physicians who aren’t radiologists,” says Diane Sliwka, MD, a hospitalist at the University of California at San Francisco (UCSF).
Dr. Sliwka says the NEJM article represents a milestone in the dissemination of bedside ultrasound. She conducts monthly faculty development training in procedural ultrasound at UCSF, workshops at HM and internal-medicine conferences, and training sessions for other hospitals.
The most common uses for bedside, “point of care” ultrasound include guiding procedures, such as thoracentesis and paracentesis, with improved safety over doing such insertions “blind.” Emerging procedural uses include lumbar puncture and arthrocentesis. Diagnostically, bedside ultrasound can provide quick screening and assessment, for example, of fluid buildup around the heart; previously, it could take hours to get the results from a formal heart study.
As with the stethoscope, Dr. Sliwka says, training in its correct use and scope of appropriate bedside practice is essential: “My advice is to learn from the experts at your facility, including the radiologists, critical care, or emergency physicians.” Ultrasound courses are increasing at hospitalist conferences, but space often is limited, and further supervised practice back home is needed.
The next step for hospitalists could be the definition of appropriate scope of practice, training, and competencies for its use. “Creating a niche in this area can be a nice change of pace from our traditional work as hospitalists,” Dr. Sliwka says. —LB
Technology
Video Chat Takes Off for Physicians
A recent study of digital adoption trends found that 7% of U.S. physicians now use video consultations to communicate with patients.
Manhattan Research’s 2011 “Taking the Pulse” survey of 2,000 physicians’ use of technology found that video chat is emerging as a way to consult with patients about nonurgent issues and follow-up questions or with geographically dispersed patients. Psychiatrists and oncologists are more likely to use the new technology. Doctors’ concerns regarding reimbursement, liability, and HIPAA privacy rules remain barriers to adoption.
For more information, visit ManhattanResearch.com/News-and-Events/Press-Releases/physician-patient-online-video-conferencing.—LB
Legal
Positive Outcomes from Full Disclosure of Medical Errors
The University of Michigan Health System’s (UMHS) risk-management model of full disclosure with offer of compensation for medical errors sparked hospitalist Allen Kachalia, MD, JD, of Brigham & Women’s Hospital in Boston to retrospectively study the outcomes of malpractice-claims-related performance before and after UMHS implemented the system in 2001.
Among the results Dr. Kachalia reported in his research abstract plenary at HM10, and subsequently published in Annals of Internal Medicine (2010;153(4):213-221), the mean monthly rate of new claims per 100,000 patient contacts decreased 36% after the full-disclosure model was adopted, while the rate of claims resulting in lawsuits declined by 65%. Claims also were resolved more quickly with the full-disclosure model.
Disclosure of medical error, Dr. Kachalia says, means “if someone is injured by medical care caused by medical error, the physician tells the patient they made the error, how it happened, and, often, what they’ll do to fix it.” An apology is somewhat different, he adds, and there’s no generic script for an apology. “What patients want is sincerity,” he says.
How can hospitalists work with full disclosure? “The general advice most institutions give is that when you want to disclose a medical error, first get your risk-management and patient-safety officers involved. They can help during every step of the process of investigating the event and disclosing,” Dr. Kachalia explains. “Assure patients that you are going to look into their concerns. Then make sure that a thorough investigation is done.”—LB
Practice Management
AMA-MGMA Toolkit Sorts Transitional-Care Software Options
HM practices with physicians in outpatient settings—be they discharge clinics or transitional-care centers—don’t always know how to determine the most useful practice-management software for their needs. So for those not helped by informatics staff, consider the new “Practice Management System Software Directory” from AMA and the Medical Group Management Association (MGMA).
The online repository, which launched in May, is a companion to the “Selecting a Practice Management System” toolkit the joint venture unveiled last fall. While the system is geared toward ambulatory-care settings, Robert Tennant, a senior policy advisor with MGMA, says any HM group with practitioners working on transitional care would find it useful.
Overall, the directory’s goal is to guide providers on how to navigate the increasingly complex world of practice-management options as new guidelines for “meaningful use” are defined, as well as new rules governing electronic claims processing. A new claims standard, known as HIPAA version 5010, is going live Jan. 1, 2012, so Tennant believes the directory is timely.
“It’s very difficult, whether in a practice or a hospital, to know the best software to pick,” he says. “There are plenty of vendors out there telling you they’re the best. There’s no easy way to comparison-shop.”
Now physicians can use the toolkit to measure basic functions. The directory, which will be updated on a rolling basis, will catalogue price range (excluding implementation costs), the number of installed customers, the target market for the product, what year the software was first offered, and whether the vendor also offers an electronic health record (EHR) system. That last point is of particular note to hospitalists as a link between practice management and medical records can help make a practice more efficient, Tennant says.
“What we’ve seen,” he adds, “is those that have that seamless integration between practice-management systems and EHR have higher productivity and higher levels of satisfaction.” —RQ
By The Numbers
$131,564
The average amount of money HM groups received in support per full-time equivalent (FTE) in fiscal year 2010, according to new SHM-MGMA survey data. The data point—the so-called “subsidy”—was first revealed at HM11 in Dallas.
After several years of leveling off at roughly $100,000, some hospitalists say they were surprised to see the figure rise so quickly. The report also shows that 19% of hospitalist practices receive no support, a finding that prompted new SHM President Joseph Li, MD, SFHM, to ask: “Are we looking at two business models or two care models?”—RQ
Fast and Furious
Every May, Mayo Clinic hospitalist Jason Persoff, MD, SFHM, sheds his doctor’s gear, grabs his camera and camcorder, and heads to the Midwest in search of ferocious weather for two weeks. “My wife jokingly calls it my ‘midlife crisis prevention program,’ ” says Dr. Persoff, who works in Jacksonville, Fla.
This year, he put his doctor’s gear back on sooner than he expected.
After 20 years of chasing storms, Dr. Persoff found himself in what might have been considered an inevitable situation: helping people injured in a tornado. When a monstrous twister with winds of more than 200 mph barreled through Joplin, Mo., on May 22, Dr. Persoff was less than a mile from its path. He and a “chase partner,” Robert Balogh, MD, an Oklahoma-based internist and former hospitalist, were able to rush to the scene and assist in the aftermath.
In the moments after the fast-forming storm, Dr. Persoff hoped that the damage wouldn’t be so devastating, despite the first ominous signs he saw along the highway.
“We were dealing with a raining sky of debris,” he says. “There was Styrofoam insulation falling from the sky, papers, there was a Barbie doll in the middle of the road, but I have no idea where that came from. There were trees and twigs and leaves, so I knew that the destruction to Joplin had been significant. But I hoped that it would be very limited.”
As he traveled along another road, he saw two dozen flipped-over semi-trucks.
“There was no decision,” Dr. Balogh says. “We knew right then that the chase was over for us.”
One hospital serving the area, St. John’s Regional Medical Center, was destroyed, its roof ripped off, he learned. At press time, the tornado had killed more than 150 and caused an estimated $3 billion in damage.
Dr. Persoff checked in at the ED of another hospital, Freeman Health System, and offered his help. He spent 10 hours there, first treating trauma patients.
“We were immediately put to work because there were just so many people coming in,” he says. “The initial trauma that came in was pretty fast and furious. If somebody could be saved, and it wasn’t going to require an effort that would jeopardize resources, they did everything they could to save people. They put in chest tubes, ventilated them, [performed] other procedures.
"If somebody was dying and that was pretty obvious, it required us to rethink how we were going to approach things. And I made a diligent effort to help the dying with low doses of pain medication to help them through.”
There were amputations, impalements, eviscerations.
“We had patients who were covered in glass, and by covered I don’t mean they just had glass in their skin—they were covered with it,” he says. “When you’d examine them, there was a risk of your glove getting torn doing an exam.”
Dr. Balogh describes the patient influx as an “absolutely overwhelming” onslaught, with ambulances, cars, and pickup trucks that had rescued strangers on the roadside arriving seemingly nonstop.
It was so frantic, he says, that he was worried “if I even take time to talk to one patient .. I’ve missed the next 15.”
When the patients from St. John’s began to arrive at Freeman, Dr. Persoff treated them, too. He wrote admission orders on 24 patients.
“The patients weren’t able to provide history,” he says. “Some of the medical records fell as far as, I think, Kansas City (160 miles to the north), from the air,” he explains. “So we had no medical records. We had patients who were demented or delirious. We had patients who’d undergone routine procedures, several patients who were postoperative.”
Leaving the hospital, he said, was gut-wrenching.
“I felt like a loser. I felt like I was handing patient-care responsibilities to a completely overtaxed system because I was tired,” he says. “When I started not making good decisions, I knew that I wasn’t helping anybody and it was time for me to step aside. But that was a very hard decision to make.”
Dr. Persoff says he’ll never forget the triage nurse on duty. She was there when he arrived, about 6:30 p.m., and was perfectly orchestrating the trauma care, even though there was no way for any of the hospital staff to know what had become of their own families and homes. And she was still there when he left at 4 a.m., so efficient and fresh it was as if she’d “just come in from having showered.”

“I don’t know what she knew or where her house was or where her family was,” he says. “I just knew that she was there working like there was no tomorrow and doing it in a way that I couldn’t. That was one of the times where I was like, ‘Wow, this is really humbling.’ ”
Dr. Persoff, who writes about his hobby at Stormdoctor.blogspot.com, continued his storm chasing; he even helped provide assistance two days later, after storms near Oklahoma City exacted a human toll that was not nearly as severe. But first, he says, he had to do some soul-searching. After all, he had hoped for a tornado to form in the Joplin area.
“My chase partners and I were talking about how can the rational person want to continue storm-chasing after having seen what we’d seen. And it took me a while to sort of figure out where my own conscience was on this,” he says. “I felt very guilty for having even wanted [a tornado] earlier in the day. Then I also felt like, had the storm not formed where it did, I wouldn’t have been there, my partner Dr. Balogh wouldn’t have been there, and we would not have been able to assist in that disaster.
“So in many ways it was karma. It happened. We were there at a time when Joplin needed some help.”
After the storm, Dr. Persoff received words of thanks from the town.
Jane Culver, a floor nurse with whom he worked, told him via email: “People often say to me, ‘Doctors are just in it for the money, they really don’t really care about me.’ Well, I say they don’t know the Dr. Jason Persoffs of the world. You are a true humanitarian, and the people of Joplin are lucky you were in our midst at our hour of need.”

Stephanie Conrad, whose grandmother Clara had her broken hip cared for by Dr. Persoff, called him “the angel doctor.”
“Thank you so much for using your knowledge, skills, and expertise during this crisis,” Conrad wrote in an email. “It is physicians like you that make a difference in the lives of others. You were truly a blessing that night.”
Tom Collins is a freelance medical writer based in Florida.
Every May, Mayo Clinic hospitalist Jason Persoff, MD, SFHM, sheds his doctor’s gear, grabs his camera and camcorder, and heads to the Midwest in search of ferocious weather for two weeks. “My wife jokingly calls it my ‘midlife crisis prevention program,’ ” says Dr. Persoff, who works in Jacksonville, Fla.
This year, he put his doctor’s gear back on sooner than he expected.
After 20 years of chasing storms, Dr. Persoff found himself in what might have been considered an inevitable situation: helping people injured in a tornado. When a monstrous twister with winds of more than 200 mph barreled through Joplin, Mo., on May 22, Dr. Persoff was less than a mile from its path. He and a “chase partner,” Robert Balogh, MD, an Oklahoma-based internist and former hospitalist, were able to rush to the scene and assist in the aftermath.
In the moments after the fast-forming storm, Dr. Persoff hoped that the damage wouldn’t be so devastating, despite the first ominous signs he saw along the highway.
“We were dealing with a raining sky of debris,” he says. “There was Styrofoam insulation falling from the sky, papers, there was a Barbie doll in the middle of the road, but I have no idea where that came from. There were trees and twigs and leaves, so I knew that the destruction to Joplin had been significant. But I hoped that it would be very limited.”
As he traveled along another road, he saw two dozen flipped-over semi-trucks.
“There was no decision,” Dr. Balogh says. “We knew right then that the chase was over for us.”
One hospital serving the area, St. John’s Regional Medical Center, was destroyed, its roof ripped off, he learned. At press time, the tornado had killed more than 150 and caused an estimated $3 billion in damage.
Dr. Persoff checked in at the ED of another hospital, Freeman Health System, and offered his help. He spent 10 hours there, first treating trauma patients.
“We were immediately put to work because there were just so many people coming in,” he says. “The initial trauma that came in was pretty fast and furious. If somebody could be saved, and it wasn’t going to require an effort that would jeopardize resources, they did everything they could to save people. They put in chest tubes, ventilated them, [performed] other procedures.
"If somebody was dying and that was pretty obvious, it required us to rethink how we were going to approach things. And I made a diligent effort to help the dying with low doses of pain medication to help them through.”
There were amputations, impalements, eviscerations.
“We had patients who were covered in glass, and by covered I don’t mean they just had glass in their skin—they were covered with it,” he says. “When you’d examine them, there was a risk of your glove getting torn doing an exam.”
Dr. Balogh describes the patient influx as an “absolutely overwhelming” onslaught, with ambulances, cars, and pickup trucks that had rescued strangers on the roadside arriving seemingly nonstop.
It was so frantic, he says, that he was worried “if I even take time to talk to one patient .. I’ve missed the next 15.”
When the patients from St. John’s began to arrive at Freeman, Dr. Persoff treated them, too. He wrote admission orders on 24 patients.
“The patients weren’t able to provide history,” he says. “Some of the medical records fell as far as, I think, Kansas City (160 miles to the north), from the air,” he explains. “So we had no medical records. We had patients who were demented or delirious. We had patients who’d undergone routine procedures, several patients who were postoperative.”
Leaving the hospital, he said, was gut-wrenching.
“I felt like a loser. I felt like I was handing patient-care responsibilities to a completely overtaxed system because I was tired,” he says. “When I started not making good decisions, I knew that I wasn’t helping anybody and it was time for me to step aside. But that was a very hard decision to make.”
Dr. Persoff says he’ll never forget the triage nurse on duty. She was there when he arrived, about 6:30 p.m., and was perfectly orchestrating the trauma care, even though there was no way for any of the hospital staff to know what had become of their own families and homes. And she was still there when he left at 4 a.m., so efficient and fresh it was as if she’d “just come in from having showered.”
“I don’t know what she knew or where her house was or where her family was,” he says. “I just knew that she was there working like there was no tomorrow and doing it in a way that I couldn’t. That was one of the times where I was like, ‘Wow, this is really humbling.’ ”
Dr. Persoff, who writes about his hobby at Stormdoctor.blogspot.com, continued his storm chasing; he even helped provide assistance two days later, after storms near Oklahoma City exacted a human toll that was not nearly as severe. But first, he says, he had to do some soul-searching. After all, he had hoped for a tornado to form in the Joplin area.
“My chase partners and I were talking about how can the rational person want to continue storm-chasing after having seen what we’d seen. And it took me a while to sort of figure out where my own conscience was on this,” he says. “I felt very guilty for having even wanted [a tornado] earlier in the day. Then I also felt like, had the storm not formed where it did, I wouldn’t have been there, my partner Dr. Balogh wouldn’t have been there, and we would not have been able to assist in that disaster.
“So in many ways it was karma. It happened. We were there at a time when Joplin needed some help.”
After the storm, Dr. Persoff received words of thanks from the town.
Jane Culver, a floor nurse with whom he worked, told him via email: “People often say to me, ‘Doctors are just in it for the money, they really don’t really care about me.’ Well, I say they don’t know the Dr. Jason Persoffs of the world. You are a true humanitarian, and the people of Joplin are lucky you were in our midst at our hour of need.”
Stephanie Conrad, whose grandmother Clara had her broken hip cared for by Dr. Persoff, called him “the angel doctor.”
“Thank you so much for using your knowledge, skills, and expertise during this crisis,” Conrad wrote in an email. “It is physicians like you that make a difference in the lives of others. You were truly a blessing that night.”
Tom Collins is a freelance medical writer based in Florida.
Every May, Mayo Clinic hospitalist Jason Persoff, MD, SFHM, sheds his doctor’s gear, grabs his camera and camcorder, and heads to the Midwest in search of ferocious weather for two weeks. “My wife jokingly calls it my ‘midlife crisis prevention program,’ ” says Dr. Persoff, who works in Jacksonville, Fla.
This year, he put his doctor’s gear back on sooner than he expected.
After 20 years of chasing storms, Dr. Persoff found himself in what might have been considered an inevitable situation: helping people injured in a tornado. When a monstrous twister with winds of more than 200 mph barreled through Joplin, Mo., on May 22, Dr. Persoff was less than a mile from its path. He and a “chase partner,” Robert Balogh, MD, an Oklahoma-based internist and former hospitalist, were able to rush to the scene and assist in the aftermath.
In the moments after the fast-forming storm, Dr. Persoff hoped that the damage wouldn’t be so devastating, despite the first ominous signs he saw along the highway.
“We were dealing with a raining sky of debris,” he says. “There was Styrofoam insulation falling from the sky, papers, there was a Barbie doll in the middle of the road, but I have no idea where that came from. There were trees and twigs and leaves, so I knew that the destruction to Joplin had been significant. But I hoped that it would be very limited.”
As he traveled along another road, he saw two dozen flipped-over semi-trucks.
“There was no decision,” Dr. Balogh says. “We knew right then that the chase was over for us.”
One hospital serving the area, St. John’s Regional Medical Center, was destroyed, its roof ripped off, he learned. At press time, the tornado had killed more than 150 and caused an estimated $3 billion in damage.
Dr. Persoff checked in at the ED of another hospital, Freeman Health System, and offered his help. He spent 10 hours there, first treating trauma patients.
“We were immediately put to work because there were just so many people coming in,” he says. “The initial trauma that came in was pretty fast and furious. If somebody could be saved, and it wasn’t going to require an effort that would jeopardize resources, they did everything they could to save people. They put in chest tubes, ventilated them, [performed] other procedures.
"If somebody was dying and that was pretty obvious, it required us to rethink how we were going to approach things. And I made a diligent effort to help the dying with low doses of pain medication to help them through.”
There were amputations, impalements, eviscerations.
“We had patients who were covered in glass, and by covered I don’t mean they just had glass in their skin—they were covered with it,” he says. “When you’d examine them, there was a risk of your glove getting torn doing an exam.”
Dr. Balogh describes the patient influx as an “absolutely overwhelming” onslaught, with ambulances, cars, and pickup trucks that had rescued strangers on the roadside arriving seemingly nonstop.
It was so frantic, he says, that he was worried “if I even take time to talk to one patient .. I’ve missed the next 15.”
When the patients from St. John’s began to arrive at Freeman, Dr. Persoff treated them, too. He wrote admission orders on 24 patients.
“The patients weren’t able to provide history,” he says. “Some of the medical records fell as far as, I think, Kansas City (160 miles to the north), from the air,” he explains. “So we had no medical records. We had patients who were demented or delirious. We had patients who’d undergone routine procedures, several patients who were postoperative.”
Leaving the hospital, he said, was gut-wrenching.
“I felt like a loser. I felt like I was handing patient-care responsibilities to a completely overtaxed system because I was tired,” he says. “When I started not making good decisions, I knew that I wasn’t helping anybody and it was time for me to step aside. But that was a very hard decision to make.”
Dr. Persoff says he’ll never forget the triage nurse on duty. She was there when he arrived, about 6:30 p.m., and was perfectly orchestrating the trauma care, even though there was no way for any of the hospital staff to know what had become of their own families and homes. And she was still there when he left at 4 a.m., so efficient and fresh it was as if she’d “just come in from having showered.”
“I don’t know what she knew or where her house was or where her family was,” he says. “I just knew that she was there working like there was no tomorrow and doing it in a way that I couldn’t. That was one of the times where I was like, ‘Wow, this is really humbling.’ ”
Dr. Persoff, who writes about his hobby at Stormdoctor.blogspot.com, continued his storm chasing; he even helped provide assistance two days later, after storms near Oklahoma City exacted a human toll that was not nearly as severe. But first, he says, he had to do some soul-searching. After all, he had hoped for a tornado to form in the Joplin area.
“My chase partners and I were talking about how can the rational person want to continue storm-chasing after having seen what we’d seen. And it took me a while to sort of figure out where my own conscience was on this,” he says. “I felt very guilty for having even wanted [a tornado] earlier in the day. Then I also felt like, had the storm not formed where it did, I wouldn’t have been there, my partner Dr. Balogh wouldn’t have been there, and we would not have been able to assist in that disaster.
“So in many ways it was karma. It happened. We were there at a time when Joplin needed some help.”
After the storm, Dr. Persoff received words of thanks from the town.
Jane Culver, a floor nurse with whom he worked, told him via email: “People often say to me, ‘Doctors are just in it for the money, they really don’t really care about me.’ Well, I say they don’t know the Dr. Jason Persoffs of the world. You are a true humanitarian, and the people of Joplin are lucky you were in our midst at our hour of need.”
Stephanie Conrad, whose grandmother Clara had her broken hip cared for by Dr. Persoff, called him “the angel doctor.”
“Thank you so much for using your knowledge, skills, and expertise during this crisis,” Conrad wrote in an email. “It is physicians like you that make a difference in the lives of others. You were truly a blessing that night.”
Tom Collins is a freelance medical writer based in Florida.
Cause For Concern
When a drug is in short supply at Beth Israel Deaconess Medical Center in Boston, a message goes out to the physicians on the hospital’s intranet system. When the shortage gets close to being critically short in supply, a message will be embedded into the physician order-entry system recommending that the physicians use an alternate drug—if there is an alternate.
It’s an alert system that has been put to frequent use lately, says Joseph Li, MD, SFHM, director of the hospital medicine program at Beth Israel Deaconess, associate professor of medicine at Harvard Medical School, and president of SHM.
The rate of drug shortages has been rising steadily in recent years due to quality questions at manufacturers, consolidation in the drug-manufacturing industry, and other factors, according to data from the U.S. Food and Drug Administration and other sources.
“It does seem like there’s more today than previous years,” says Dr. Li, who was a pharmacist before he trained in internal medicine.
Some of the recent shortages at Beth Israel Deaconess have involved the diuretic furosemide, the antiemetic Compazine, and the anticoagulant heparin. “More often than not, there’s a reasonable alternative that can be chosen,” he says. “Not necessarily exactly the same drug, but usually in the same therapeutic class.”
While actual cases of patient harm due to drug shortages appear to be relatively uncommon, having drugs in short supply can lead to a safety problem hovering over a medical center and its hospitalists. In addition to the potential of simply not having an alternate to give to a patient, hospitalists and their pharmacists sometimes have to adjust to a new dosage that comes with a replacement medication.
Plus, having to manage the problem when a drug shortage hits can be a headache, with time and resources spent trying to obtain updates from drug manufacturers and find other drugs that can be used in the meantime, experts say.
With hospitalists now treating so many patients, many of them complex and on multiple medications, it is an important issue for hospitalists to stay aware of and to be prepared for, Dr. Li says. More than 90% of all medical patients at Beth Israel Deaconess are now cared for by hospitalists, he says, and it’s a similar situation for many acute-care hospitals around the country.
If a drug is in short supply, balancing availability with patient needs can be especially tricky for a hospitalist caring for patients with a multitude of demands, Dr. Li says. “There is an effort to make sure that our most vulnerable population of patients receive these treatments before the general population of patients have access to it,” he adds.
However, the very existence of hospitalists makes it easier to navigate a shortage compared to the days when hundreds of providers would be caring for a pool of patients.
“If you’re trying to notify a group of providers about shortages and have an impact on their prescribing habits, I think it’s easier today,” he says.
Troubled Waters
The FDA says it confirmed a record 178 cases of drug shortages in 2010 (www.fda.gov/drugs/drugsafety/drugshortages/default.htm). That was up from 55 shortages five years ago. And according to the University of Utah Drug Information Service, the problem is actually more pervasive than that, reporting 120 shortages in the U.S. in 2001, with a reported 211 in 2010. And through March of this year, there were 80 reported cases of shortages, on pace for another record year.
“In the past couple of years, it’s just been exponential,” says Diane Ginsburg, president of the American Society of Health-System Pharmacists and clinical professor and assistant dean for student affairs at the University of Texas’ College of Pharmacy in Austin.
According to the FDA, 77% of the shortages in 2010 involved sterile injectable drugs.
“There are fewer and fewer firms making these older sterile injectables, and they are often discontinued for newer, more profitable agents,” FDA spokeswoman Yolanda Fultz-Morris said in an email. “When one firm has a delay or a manufacturing problem, it is extremely difficult for the remaining firms to quickly increase production.”
The biggest cause for the shortages in those drugs has been product quality issues, namely microbial contamination and newly identified impurities, according to the FDA. From January to October of 2010, 42% of drug shortages were due to quality problems.
Eighteen percent were due to product discontinuation by the manufacturer and another 18% were due to delays and capacity problems. Nine percent were due to difficulties getting raw materials, and 4% of the sterile injectable shortages were due to increased demand because there was a shortage of another injectable medication. In other words, one shortage led directly to another.
Kevin Schweers, a spokesman for the National Community Pharmacists Association, says generic drugs, especially Schedule II substances, have been in short supply. But there can be problems even when one generic is available to replace another generic.
An example, he says, is when a “new generic substituted in place of the old one is made by a different manufacturer and may come in a different color or shape. That can leave patients”—including those just released from hospitals—“wondering and asking the pharmacist why their medication is different or if a mistake was made.”
Patient Safety and Communication Errors
Lalit Verma, MD, director of the hospital medicine program at Durham Regional Medical Center in North Carolina and assistant professor of medicine at the Duke University School of Medicine, is unaware of any situations in which a shortage put patients in jeopardy at his hospital. He says the pharmacy at Durham Regional, which has seen recent shortages in morphine and heparin, among other drugs, keeps doctors up to date and has adjusted doses appropriately when replacements are used.
“It’s probably been more than I’ve experienced in my 10 years as a hospitalist,” Dr. Verma says. “We have a very good pharmacy program that updates us regularly on drug shortages and offers alternatives.”
Dr. Li also says no patient’s safety has been jeopardized by a shortage.
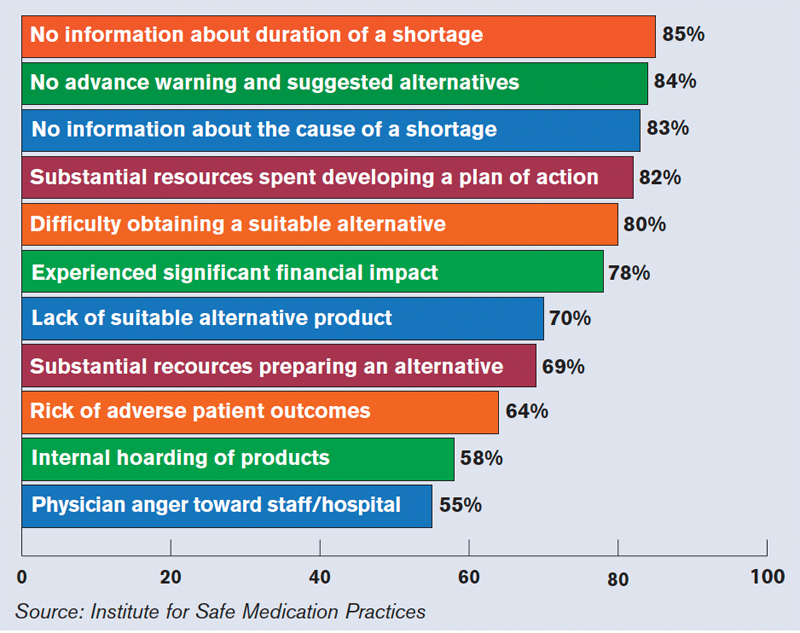
Others say patient safety has been affected, according to 1,800 healthcare practitioners who participated in a survey last year conducted by the Institute for Safe Medication Practices (ISMP), a nonprofit group. Twenty percent of the respondents said drug-shortage-related errors were made, while 32% said they had “near misses” related to drug shortages. Nineteen percent said there had been adverse patient outcomes as a result of drug shortages.
The study noted two instances in which patients died when they were switched to dilaudid because morphine was in short supply; both patients were given morphine doses instead of adjusted doses for dilaudid.
“It’s about six- or sevenfold more potent than morphine,” says Michael Cohen, ISMP president. “And so when that drug is prescribed in a morphine dose, that would be a massive overdose for some patients.”
He adds that hospitals have tried to stay on top of the drug shortage problem, but that “it’s very difficult.”
“A lot of this happens last-minute,” Cohen says. “Physicians aren’t given a chance to even realize that a certain drug isn’t available, so it causes an interruption in the whole flow of things in the hospital.” Some hospitals have had to hire staffers who handle just the inevitable daily drug shortages, he adds.
A law has been proposed in the U.S. Senate that would require drug manufacturers to notify the FDA when circumstances arise that might reasonably lead to a drug shortage (see “Senate Bill Would Require Advance Notice of Potential Shortages,” p. 41).
Cohen says another concern is that some hospitals, faced with shortages in electrolytes, such as potassium phosphate and sodium acetate, have been turning to less-regulated sterile compounding pharmacies for the products.
Dr. Verma, of Durham Regional, says perhaps the biggest challenge is staying on top of changing doses. “I think there was a learning curve for physicians in using dilaudid [rather than morphine] because the dosing is quite different, so that can cause challenges for patient care when you’re switching in and out of drug classes,” he says. “It’s not a perfect science. It doesn’t cripple us, but it does make it more challenging to fine-tune patient care.”
Ginsburg, of the ASHP, urges hospitalists to stay in close contact with the pharmacists at their hospitals and to be diligent about reporting shortages to the ASHP.
“Please work closely with the pharmacists, because we’re the ones that can really help,” she says. “We’re in it together with them, in terms of trying to provide care for their patients.” TH
Thomas R. Collins a freelance medical writer based in Florida.
When a drug is in short supply at Beth Israel Deaconess Medical Center in Boston, a message goes out to the physicians on the hospital’s intranet system. When the shortage gets close to being critically short in supply, a message will be embedded into the physician order-entry system recommending that the physicians use an alternate drug—if there is an alternate.
It’s an alert system that has been put to frequent use lately, says Joseph Li, MD, SFHM, director of the hospital medicine program at Beth Israel Deaconess, associate professor of medicine at Harvard Medical School, and president of SHM.
The rate of drug shortages has been rising steadily in recent years due to quality questions at manufacturers, consolidation in the drug-manufacturing industry, and other factors, according to data from the U.S. Food and Drug Administration and other sources.
“It does seem like there’s more today than previous years,” says Dr. Li, who was a pharmacist before he trained in internal medicine.
Some of the recent shortages at Beth Israel Deaconess have involved the diuretic furosemide, the antiemetic Compazine, and the anticoagulant heparin. “More often than not, there’s a reasonable alternative that can be chosen,” he says. “Not necessarily exactly the same drug, but usually in the same therapeutic class.”
While actual cases of patient harm due to drug shortages appear to be relatively uncommon, having drugs in short supply can lead to a safety problem hovering over a medical center and its hospitalists. In addition to the potential of simply not having an alternate to give to a patient, hospitalists and their pharmacists sometimes have to adjust to a new dosage that comes with a replacement medication.
Plus, having to manage the problem when a drug shortage hits can be a headache, with time and resources spent trying to obtain updates from drug manufacturers and find other drugs that can be used in the meantime, experts say.
With hospitalists now treating so many patients, many of them complex and on multiple medications, it is an important issue for hospitalists to stay aware of and to be prepared for, Dr. Li says. More than 90% of all medical patients at Beth Israel Deaconess are now cared for by hospitalists, he says, and it’s a similar situation for many acute-care hospitals around the country.
If a drug is in short supply, balancing availability with patient needs can be especially tricky for a hospitalist caring for patients with a multitude of demands, Dr. Li says. “There is an effort to make sure that our most vulnerable population of patients receive these treatments before the general population of patients have access to it,” he adds.
However, the very existence of hospitalists makes it easier to navigate a shortage compared to the days when hundreds of providers would be caring for a pool of patients.
“If you’re trying to notify a group of providers about shortages and have an impact on their prescribing habits, I think it’s easier today,” he says.
Troubled Waters
The FDA says it confirmed a record 178 cases of drug shortages in 2010 (www.fda.gov/drugs/drugsafety/drugshortages/default.htm). That was up from 55 shortages five years ago. And according to the University of Utah Drug Information Service, the problem is actually more pervasive than that, reporting 120 shortages in the U.S. in 2001, with a reported 211 in 2010. And through March of this year, there were 80 reported cases of shortages, on pace for another record year.
“In the past couple of years, it’s just been exponential,” says Diane Ginsburg, president of the American Society of Health-System Pharmacists and clinical professor and assistant dean for student affairs at the University of Texas’ College of Pharmacy in Austin.
According to the FDA, 77% of the shortages in 2010 involved sterile injectable drugs.
“There are fewer and fewer firms making these older sterile injectables, and they are often discontinued for newer, more profitable agents,” FDA spokeswoman Yolanda Fultz-Morris said in an email. “When one firm has a delay or a manufacturing problem, it is extremely difficult for the remaining firms to quickly increase production.”
The biggest cause for the shortages in those drugs has been product quality issues, namely microbial contamination and newly identified impurities, according to the FDA. From January to October of 2010, 42% of drug shortages were due to quality problems.
Eighteen percent were due to product discontinuation by the manufacturer and another 18% were due to delays and capacity problems. Nine percent were due to difficulties getting raw materials, and 4% of the sterile injectable shortages were due to increased demand because there was a shortage of another injectable medication. In other words, one shortage led directly to another.
Kevin Schweers, a spokesman for the National Community Pharmacists Association, says generic drugs, especially Schedule II substances, have been in short supply. But there can be problems even when one generic is available to replace another generic.
An example, he says, is when a “new generic substituted in place of the old one is made by a different manufacturer and may come in a different color or shape. That can leave patients”—including those just released from hospitals—“wondering and asking the pharmacist why their medication is different or if a mistake was made.”
Patient Safety and Communication Errors
Lalit Verma, MD, director of the hospital medicine program at Durham Regional Medical Center in North Carolina and assistant professor of medicine at the Duke University School of Medicine, is unaware of any situations in which a shortage put patients in jeopardy at his hospital. He says the pharmacy at Durham Regional, which has seen recent shortages in morphine and heparin, among other drugs, keeps doctors up to date and has adjusted doses appropriately when replacements are used.
“It’s probably been more than I’ve experienced in my 10 years as a hospitalist,” Dr. Verma says. “We have a very good pharmacy program that updates us regularly on drug shortages and offers alternatives.”
Dr. Li also says no patient’s safety has been jeopardized by a shortage.
Others say patient safety has been affected, according to 1,800 healthcare practitioners who participated in a survey last year conducted by the Institute for Safe Medication Practices (ISMP), a nonprofit group. Twenty percent of the respondents said drug-shortage-related errors were made, while 32% said they had “near misses” related to drug shortages. Nineteen percent said there had been adverse patient outcomes as a result of drug shortages.
The study noted two instances in which patients died when they were switched to dilaudid because morphine was in short supply; both patients were given morphine doses instead of adjusted doses for dilaudid.
“It’s about six- or sevenfold more potent than morphine,” says Michael Cohen, ISMP president. “And so when that drug is prescribed in a morphine dose, that would be a massive overdose for some patients.”
He adds that hospitals have tried to stay on top of the drug shortage problem, but that “it’s very difficult.”
“A lot of this happens last-minute,” Cohen says. “Physicians aren’t given a chance to even realize that a certain drug isn’t available, so it causes an interruption in the whole flow of things in the hospital.” Some hospitals have had to hire staffers who handle just the inevitable daily drug shortages, he adds.
A law has been proposed in the U.S. Senate that would require drug manufacturers to notify the FDA when circumstances arise that might reasonably lead to a drug shortage (see “Senate Bill Would Require Advance Notice of Potential Shortages,” p. 41).
Cohen says another concern is that some hospitals, faced with shortages in electrolytes, such as potassium phosphate and sodium acetate, have been turning to less-regulated sterile compounding pharmacies for the products.
Dr. Verma, of Durham Regional, says perhaps the biggest challenge is staying on top of changing doses. “I think there was a learning curve for physicians in using dilaudid [rather than morphine] because the dosing is quite different, so that can cause challenges for patient care when you’re switching in and out of drug classes,” he says. “It’s not a perfect science. It doesn’t cripple us, but it does make it more challenging to fine-tune patient care.”
Ginsburg, of the ASHP, urges hospitalists to stay in close contact with the pharmacists at their hospitals and to be diligent about reporting shortages to the ASHP.
“Please work closely with the pharmacists, because we’re the ones that can really help,” she says. “We’re in it together with them, in terms of trying to provide care for their patients.” TH
Thomas R. Collins a freelance medical writer based in Florida.
When a drug is in short supply at Beth Israel Deaconess Medical Center in Boston, a message goes out to the physicians on the hospital’s intranet system. When the shortage gets close to being critically short in supply, a message will be embedded into the physician order-entry system recommending that the physicians use an alternate drug—if there is an alternate.
It’s an alert system that has been put to frequent use lately, says Joseph Li, MD, SFHM, director of the hospital medicine program at Beth Israel Deaconess, associate professor of medicine at Harvard Medical School, and president of SHM.
The rate of drug shortages has been rising steadily in recent years due to quality questions at manufacturers, consolidation in the drug-manufacturing industry, and other factors, according to data from the U.S. Food and Drug Administration and other sources.
“It does seem like there’s more today than previous years,” says Dr. Li, who was a pharmacist before he trained in internal medicine.
Some of the recent shortages at Beth Israel Deaconess have involved the diuretic furosemide, the antiemetic Compazine, and the anticoagulant heparin. “More often than not, there’s a reasonable alternative that can be chosen,” he says. “Not necessarily exactly the same drug, but usually in the same therapeutic class.”
While actual cases of patient harm due to drug shortages appear to be relatively uncommon, having drugs in short supply can lead to a safety problem hovering over a medical center and its hospitalists. In addition to the potential of simply not having an alternate to give to a patient, hospitalists and their pharmacists sometimes have to adjust to a new dosage that comes with a replacement medication.
Plus, having to manage the problem when a drug shortage hits can be a headache, with time and resources spent trying to obtain updates from drug manufacturers and find other drugs that can be used in the meantime, experts say.
With hospitalists now treating so many patients, many of them complex and on multiple medications, it is an important issue for hospitalists to stay aware of and to be prepared for, Dr. Li says. More than 90% of all medical patients at Beth Israel Deaconess are now cared for by hospitalists, he says, and it’s a similar situation for many acute-care hospitals around the country.
If a drug is in short supply, balancing availability with patient needs can be especially tricky for a hospitalist caring for patients with a multitude of demands, Dr. Li says. “There is an effort to make sure that our most vulnerable population of patients receive these treatments before the general population of patients have access to it,” he adds.
However, the very existence of hospitalists makes it easier to navigate a shortage compared to the days when hundreds of providers would be caring for a pool of patients.
“If you’re trying to notify a group of providers about shortages and have an impact on their prescribing habits, I think it’s easier today,” he says.
Troubled Waters
The FDA says it confirmed a record 178 cases of drug shortages in 2010 (www.fda.gov/drugs/drugsafety/drugshortages/default.htm). That was up from 55 shortages five years ago. And according to the University of Utah Drug Information Service, the problem is actually more pervasive than that, reporting 120 shortages in the U.S. in 2001, with a reported 211 in 2010. And through March of this year, there were 80 reported cases of shortages, on pace for another record year.
“In the past couple of years, it’s just been exponential,” says Diane Ginsburg, president of the American Society of Health-System Pharmacists and clinical professor and assistant dean for student affairs at the University of Texas’ College of Pharmacy in Austin.
According to the FDA, 77% of the shortages in 2010 involved sterile injectable drugs.
“There are fewer and fewer firms making these older sterile injectables, and they are often discontinued for newer, more profitable agents,” FDA spokeswoman Yolanda Fultz-Morris said in an email. “When one firm has a delay or a manufacturing problem, it is extremely difficult for the remaining firms to quickly increase production.”
The biggest cause for the shortages in those drugs has been product quality issues, namely microbial contamination and newly identified impurities, according to the FDA. From January to October of 2010, 42% of drug shortages were due to quality problems.
Eighteen percent were due to product discontinuation by the manufacturer and another 18% were due to delays and capacity problems. Nine percent were due to difficulties getting raw materials, and 4% of the sterile injectable shortages were due to increased demand because there was a shortage of another injectable medication. In other words, one shortage led directly to another.
Kevin Schweers, a spokesman for the National Community Pharmacists Association, says generic drugs, especially Schedule II substances, have been in short supply. But there can be problems even when one generic is available to replace another generic.
An example, he says, is when a “new generic substituted in place of the old one is made by a different manufacturer and may come in a different color or shape. That can leave patients”—including those just released from hospitals—“wondering and asking the pharmacist why their medication is different or if a mistake was made.”
Patient Safety and Communication Errors
Lalit Verma, MD, director of the hospital medicine program at Durham Regional Medical Center in North Carolina and assistant professor of medicine at the Duke University School of Medicine, is unaware of any situations in which a shortage put patients in jeopardy at his hospital. He says the pharmacy at Durham Regional, which has seen recent shortages in morphine and heparin, among other drugs, keeps doctors up to date and has adjusted doses appropriately when replacements are used.
“It’s probably been more than I’ve experienced in my 10 years as a hospitalist,” Dr. Verma says. “We have a very good pharmacy program that updates us regularly on drug shortages and offers alternatives.”
Dr. Li also says no patient’s safety has been jeopardized by a shortage.
Others say patient safety has been affected, according to 1,800 healthcare practitioners who participated in a survey last year conducted by the Institute for Safe Medication Practices (ISMP), a nonprofit group. Twenty percent of the respondents said drug-shortage-related errors were made, while 32% said they had “near misses” related to drug shortages. Nineteen percent said there had been adverse patient outcomes as a result of drug shortages.
The study noted two instances in which patients died when they were switched to dilaudid because morphine was in short supply; both patients were given morphine doses instead of adjusted doses for dilaudid.
“It’s about six- or sevenfold more potent than morphine,” says Michael Cohen, ISMP president. “And so when that drug is prescribed in a morphine dose, that would be a massive overdose for some patients.”
He adds that hospitals have tried to stay on top of the drug shortage problem, but that “it’s very difficult.”
“A lot of this happens last-minute,” Cohen says. “Physicians aren’t given a chance to even realize that a certain drug isn’t available, so it causes an interruption in the whole flow of things in the hospital.” Some hospitals have had to hire staffers who handle just the inevitable daily drug shortages, he adds.
A law has been proposed in the U.S. Senate that would require drug manufacturers to notify the FDA when circumstances arise that might reasonably lead to a drug shortage (see “Senate Bill Would Require Advance Notice of Potential Shortages,” p. 41).
Cohen says another concern is that some hospitals, faced with shortages in electrolytes, such as potassium phosphate and sodium acetate, have been turning to less-regulated sterile compounding pharmacies for the products.
Dr. Verma, of Durham Regional, says perhaps the biggest challenge is staying on top of changing doses. “I think there was a learning curve for physicians in using dilaudid [rather than morphine] because the dosing is quite different, so that can cause challenges for patient care when you’re switching in and out of drug classes,” he says. “It’s not a perfect science. It doesn’t cripple us, but it does make it more challenging to fine-tune patient care.”
Ginsburg, of the ASHP, urges hospitalists to stay in close contact with the pharmacists at their hospitals and to be diligent about reporting shortages to the ASHP.
“Please work closely with the pharmacists, because we’re the ones that can really help,” she says. “We’re in it together with them, in terms of trying to provide care for their patients.” TH
Thomas R. Collins a freelance medical writer based in Florida.
What Is Your Value?

For those of you who attended Bob Wachter’s talk at HM11 in Dallas, you learned that Bob drives a particular model of a popular SUV made by a well-known Japanese manufacturer. When he was in the market for a vehicle, he decided he wanted to buy an SUV. He acknowledged there were certainly less expensive SUVs on the market, along with more expensive alternatives.
So why did he choose to purchase that particular model? Was it the color, the seat warmers, or the keyless entry system? The answer is simple: He decided to purchase the popular SUV because he thought it was the best value for his dollar.
I have this vision of Bob, head cocked to one side, with his index finger resting against his chin and a text bubble above his head reading, “What is the quality of this vehicle and what is the price tag?”
These are decisions all of us make in our everyday lives. I make the same value judgment when I pull into the gasoline station to purchase gas (regular or premium?) or when I go to the grocery store (brand-name or generic orange juice?). But we know that higher cost doesn’t always mean higher quality. Think American-made automobiles versus Japanese-made vehicles in the 1970s and ’80s.
Along those same lines, let’s think about the U.S. healthcare system in 2011. America is trying to move its healthcare toward a value-based system. How do we receive the best healthcare for the—many times taxpayer—dollar? I am a taxpayer and I am all for higher-quality healthcare for my dollars.
At HM11, I heard from many supporters of healthcare reform, but I also heard many people vilify the government’s efforts at reforming our healthcare system. Just about everyone agreed that the future is uncertain. The current healthcare system certainly values hospitalists. It is hard to argue with the facts. In less than 15 years, our healthcare system has created jobs for more than 30,000 hospitalists, the majority of whom require nonclinical revenue from hospitals to meet expenses. The latest SHM-MGMA data show that the average hospitalist full-time equivalent (FTE) receives more than $131,500 of nonclinical revenue (primarily from hospitals) annually.
Payors of healthcare are no different than Bob when it comes to purchasing a car, or me when it comes to purchasing orange juice. Payors will pay for hospitalists as long as they perceive value in their investment.
But what is the basis of this notion that hospitalists are high-value healthcare providers, and is it justified? At HM11, I heard about the continued rise in hospitalist salaries. Higher costs mean we will have to increase quality if we hope to achieve the same value (value=quality/cost).
Don’t Worry, Share Your Data
I have listened to many presentations about healthcare value, quality, and cost. My perception is that it makes the most sense if it is personal. I live in Massachusetts, and my state government has been aggressive at helping everyone understand the quality and the cost of care being delivered at our hospitals. For example, our state government generates a massive annual report that describes the quality and cost of healthcare being delivered at individual hospitals; a PDF of the report is available at www.mass.gov. (For full disclosure, I work at Beth Israel Deaconess Medical Center [BIDMC] in Boston and I serve on a Massachusetts Department of Public Health Stroke Advisory Committee.)
The annual report shows there is not as much of a direct relationship between quality and cost as one would like to see. But I applaud Massachusetts for producing this report. Recognizing and understanding a problem is the first step in creating a solution to the problem. One cannot create a value-based system without understanding the existing quality and cost.
This is one of the reasons why, several years ago, the BIDMC leadership posted my hospital’s quality data online for public consumption (www.bidmc.org/QualityandSafety.aspx). The BIDMC website even features a short video of hospitalist Ken Sands, MD, who also happens to be the vice president of quality at BIDMC, telling you about the hospital quality data. Before the hospital posted this data online, most of our hospital staff and providers, let alone our patients and their families, were unaware of the data. BIDMC is not the only hospital who does this. I understand Cedars-Sinai Medical Center in Los Angeles and Dartmouth-Hitchcock Medical Center in New Hampshire have long shared their quality data publicly.
But the truth is, if you look hard enough, you can find these data for just about all acute-care hospitals in the country. Start with Medicare’s Hospital Compare website (www.hospitalcompare.hhs.gov). However, BIDMC and others have simply made it easier to find the data by putting it directly on their websites.
Policy of Transparency
An interesting thing happened over the past decade at BIDMC. In 1997, there were no hospitalists who cared for BIDMC patients. Today, hospitalists manage nearly 100% of the patients hospitalized on our large medical service.
When you look at the data being reported by BIDMC and the state of Massachusetts about nonsurgical conditions, doesn’t that reflect the care being provided by the hospitalists who work at BIDMC? I imagine that is what will run through my CEO and CFO’s minds when we discuss the hospitalist budget this summer. They will ask themselves, “What is the value of our hospitalists? What is the quality of their care? How much do they cost us?”
Some of you might be in a similar position. Do your hospitalists now provide the bulk of the care at your hospital? Are your hospital’s data being publicly reported? I think the answer is a resounding “yes” for many of you.
Allow me to ask this question: What are you doing to collect data to understand the quality and cost of your hospitalist program? Wouldn’t you rather know this information before your hospital or state government tells you?
As the director of my hospitalist group, I spearhead our group efforts to better understand the quality of care we provide. This proactive, introspective approach is essential, especially if hospitalist groups around the country hope to continue being perceived as “high value” providers.
I am interested in hearing from you about your efforts to understand the care being provided by your hospitalists. Feel free to email me at [email protected]. TH
Dr. Li is president of SHM.
For those of you who attended Bob Wachter’s talk at HM11 in Dallas, you learned that Bob drives a particular model of a popular SUV made by a well-known Japanese manufacturer. When he was in the market for a vehicle, he decided he wanted to buy an SUV. He acknowledged there were certainly less expensive SUVs on the market, along with more expensive alternatives.
So why did he choose to purchase that particular model? Was it the color, the seat warmers, or the keyless entry system? The answer is simple: He decided to purchase the popular SUV because he thought it was the best value for his dollar.
I have this vision of Bob, head cocked to one side, with his index finger resting against his chin and a text bubble above his head reading, “What is the quality of this vehicle and what is the price tag?”
These are decisions all of us make in our everyday lives. I make the same value judgment when I pull into the gasoline station to purchase gas (regular or premium?) or when I go to the grocery store (brand-name or generic orange juice?). But we know that higher cost doesn’t always mean higher quality. Think American-made automobiles versus Japanese-made vehicles in the 1970s and ’80s.
Along those same lines, let’s think about the U.S. healthcare system in 2011. America is trying to move its healthcare toward a value-based system. How do we receive the best healthcare for the—many times taxpayer—dollar? I am a taxpayer and I am all for higher-quality healthcare for my dollars.
At HM11, I heard from many supporters of healthcare reform, but I also heard many people vilify the government’s efforts at reforming our healthcare system. Just about everyone agreed that the future is uncertain. The current healthcare system certainly values hospitalists. It is hard to argue with the facts. In less than 15 years, our healthcare system has created jobs for more than 30,000 hospitalists, the majority of whom require nonclinical revenue from hospitals to meet expenses. The latest SHM-MGMA data show that the average hospitalist full-time equivalent (FTE) receives more than $131,500 of nonclinical revenue (primarily from hospitals) annually.
Payors of healthcare are no different than Bob when it comes to purchasing a car, or me when it comes to purchasing orange juice. Payors will pay for hospitalists as long as they perceive value in their investment.
But what is the basis of this notion that hospitalists are high-value healthcare providers, and is it justified? At HM11, I heard about the continued rise in hospitalist salaries. Higher costs mean we will have to increase quality if we hope to achieve the same value (value=quality/cost).
Don’t Worry, Share Your Data
I have listened to many presentations about healthcare value, quality, and cost. My perception is that it makes the most sense if it is personal. I live in Massachusetts, and my state government has been aggressive at helping everyone understand the quality and the cost of care being delivered at our hospitals. For example, our state government generates a massive annual report that describes the quality and cost of healthcare being delivered at individual hospitals; a PDF of the report is available at www.mass.gov. (For full disclosure, I work at Beth Israel Deaconess Medical Center [BIDMC] in Boston and I serve on a Massachusetts Department of Public Health Stroke Advisory Committee.)
The annual report shows there is not as much of a direct relationship between quality and cost as one would like to see. But I applaud Massachusetts for producing this report. Recognizing and understanding a problem is the first step in creating a solution to the problem. One cannot create a value-based system without understanding the existing quality and cost.
This is one of the reasons why, several years ago, the BIDMC leadership posted my hospital’s quality data online for public consumption (www.bidmc.org/QualityandSafety.aspx). The BIDMC website even features a short video of hospitalist Ken Sands, MD, who also happens to be the vice president of quality at BIDMC, telling you about the hospital quality data. Before the hospital posted this data online, most of our hospital staff and providers, let alone our patients and their families, were unaware of the data. BIDMC is not the only hospital who does this. I understand Cedars-Sinai Medical Center in Los Angeles and Dartmouth-Hitchcock Medical Center in New Hampshire have long shared their quality data publicly.
But the truth is, if you look hard enough, you can find these data for just about all acute-care hospitals in the country. Start with Medicare’s Hospital Compare website (www.hospitalcompare.hhs.gov). However, BIDMC and others have simply made it easier to find the data by putting it directly on their websites.
Policy of Transparency
An interesting thing happened over the past decade at BIDMC. In 1997, there were no hospitalists who cared for BIDMC patients. Today, hospitalists manage nearly 100% of the patients hospitalized on our large medical service.
When you look at the data being reported by BIDMC and the state of Massachusetts about nonsurgical conditions, doesn’t that reflect the care being provided by the hospitalists who work at BIDMC? I imagine that is what will run through my CEO and CFO’s minds when we discuss the hospitalist budget this summer. They will ask themselves, “What is the value of our hospitalists? What is the quality of their care? How much do they cost us?”
Some of you might be in a similar position. Do your hospitalists now provide the bulk of the care at your hospital? Are your hospital’s data being publicly reported? I think the answer is a resounding “yes” for many of you.
Allow me to ask this question: What are you doing to collect data to understand the quality and cost of your hospitalist program? Wouldn’t you rather know this information before your hospital or state government tells you?
As the director of my hospitalist group, I spearhead our group efforts to better understand the quality of care we provide. This proactive, introspective approach is essential, especially if hospitalist groups around the country hope to continue being perceived as “high value” providers.
I am interested in hearing from you about your efforts to understand the care being provided by your hospitalists. Feel free to email me at [email protected]. TH
Dr. Li is president of SHM.
For those of you who attended Bob Wachter’s talk at HM11 in Dallas, you learned that Bob drives a particular model of a popular SUV made by a well-known Japanese manufacturer. When he was in the market for a vehicle, he decided he wanted to buy an SUV. He acknowledged there were certainly less expensive SUVs on the market, along with more expensive alternatives.
So why did he choose to purchase that particular model? Was it the color, the seat warmers, or the keyless entry system? The answer is simple: He decided to purchase the popular SUV because he thought it was the best value for his dollar.
I have this vision of Bob, head cocked to one side, with his index finger resting against his chin and a text bubble above his head reading, “What is the quality of this vehicle and what is the price tag?”
These are decisions all of us make in our everyday lives. I make the same value judgment when I pull into the gasoline station to purchase gas (regular or premium?) or when I go to the grocery store (brand-name or generic orange juice?). But we know that higher cost doesn’t always mean higher quality. Think American-made automobiles versus Japanese-made vehicles in the 1970s and ’80s.
Along those same lines, let’s think about the U.S. healthcare system in 2011. America is trying to move its healthcare toward a value-based system. How do we receive the best healthcare for the—many times taxpayer—dollar? I am a taxpayer and I am all for higher-quality healthcare for my dollars.
At HM11, I heard from many supporters of healthcare reform, but I also heard many people vilify the government’s efforts at reforming our healthcare system. Just about everyone agreed that the future is uncertain. The current healthcare system certainly values hospitalists. It is hard to argue with the facts. In less than 15 years, our healthcare system has created jobs for more than 30,000 hospitalists, the majority of whom require nonclinical revenue from hospitals to meet expenses. The latest SHM-MGMA data show that the average hospitalist full-time equivalent (FTE) receives more than $131,500 of nonclinical revenue (primarily from hospitals) annually.
Payors of healthcare are no different than Bob when it comes to purchasing a car, or me when it comes to purchasing orange juice. Payors will pay for hospitalists as long as they perceive value in their investment.
But what is the basis of this notion that hospitalists are high-value healthcare providers, and is it justified? At HM11, I heard about the continued rise in hospitalist salaries. Higher costs mean we will have to increase quality if we hope to achieve the same value (value=quality/cost).
Don’t Worry, Share Your Data
I have listened to many presentations about healthcare value, quality, and cost. My perception is that it makes the most sense if it is personal. I live in Massachusetts, and my state government has been aggressive at helping everyone understand the quality and the cost of care being delivered at our hospitals. For example, our state government generates a massive annual report that describes the quality and cost of healthcare being delivered at individual hospitals; a PDF of the report is available at www.mass.gov. (For full disclosure, I work at Beth Israel Deaconess Medical Center [BIDMC] in Boston and I serve on a Massachusetts Department of Public Health Stroke Advisory Committee.)
The annual report shows there is not as much of a direct relationship between quality and cost as one would like to see. But I applaud Massachusetts for producing this report. Recognizing and understanding a problem is the first step in creating a solution to the problem. One cannot create a value-based system without understanding the existing quality and cost.
This is one of the reasons why, several years ago, the BIDMC leadership posted my hospital’s quality data online for public consumption (www.bidmc.org/QualityandSafety.aspx). The BIDMC website even features a short video of hospitalist Ken Sands, MD, who also happens to be the vice president of quality at BIDMC, telling you about the hospital quality data. Before the hospital posted this data online, most of our hospital staff and providers, let alone our patients and their families, were unaware of the data. BIDMC is not the only hospital who does this. I understand Cedars-Sinai Medical Center in Los Angeles and Dartmouth-Hitchcock Medical Center in New Hampshire have long shared their quality data publicly.
But the truth is, if you look hard enough, you can find these data for just about all acute-care hospitals in the country. Start with Medicare’s Hospital Compare website (www.hospitalcompare.hhs.gov). However, BIDMC and others have simply made it easier to find the data by putting it directly on their websites.
Policy of Transparency
An interesting thing happened over the past decade at BIDMC. In 1997, there were no hospitalists who cared for BIDMC patients. Today, hospitalists manage nearly 100% of the patients hospitalized on our large medical service.
When you look at the data being reported by BIDMC and the state of Massachusetts about nonsurgical conditions, doesn’t that reflect the care being provided by the hospitalists who work at BIDMC? I imagine that is what will run through my CEO and CFO’s minds when we discuss the hospitalist budget this summer. They will ask themselves, “What is the value of our hospitalists? What is the quality of their care? How much do they cost us?”
Some of you might be in a similar position. Do your hospitalists now provide the bulk of the care at your hospital? Are your hospital’s data being publicly reported? I think the answer is a resounding “yes” for many of you.
Allow me to ask this question: What are you doing to collect data to understand the quality and cost of your hospitalist program? Wouldn’t you rather know this information before your hospital or state government tells you?
As the director of my hospitalist group, I spearhead our group efforts to better understand the quality of care we provide. This proactive, introspective approach is essential, especially if hospitalist groups around the country hope to continue being perceived as “high value” providers.
I am interested in hearing from you about your efforts to understand the care being provided by your hospitalists. Feel free to email me at [email protected]. TH
Dr. Li is president of SHM.
Subsidy or Payment?

Question: Before hospitalists, who cared for hospitalized patients?
Answer: Generalists—in other words, internists, family physicians, pediatricians.
Q: How much did that system cost hospitals?
A: Nothing, or very little. In some cases, support dollars were available for weekend, night, or uninsured patient coverage, but by and large this system cost hospitals little. Physicians admitted their patients to the hospital because the alternatives (sending a hypoxic pneumonia patient home from clinic, turning out the office lights and hoping the patient survived the night, or bringing the patient home with them) offered uncomfortable ethical, malpractice, or alimony consequences. So doctors admitted these patients to the hospital and visited them daily.
Q: The average amount of support per hospitalist is $131,564, or about $1.7 million per HM group seeing adult patients. The bulk of those dollars come from the hospital. If we assume that the people running hospitals are smart, then why would those smart businesspeople pay $1.7 million for something they used to get for free?
A: Because there is something they get in return for that money. Or, perhaps, something they think they are getting in return for those dollars.
Q: What?
A: I often go through this exercise with the residents in our hospitalist training program when we discuss the drivers of the HM movement. I usually discuss the reasons why a hospital should fund these groups; it always seems like such a no-brainer to me.
Enter a recent news item from Montana. The story from the Helena Independent Record (see “Unsustainable Growth?” p. 1) noted that a multispecialty group practice in Helena announced they were no longer admitting their patients to a local hospital in protest over a new hospital policy to charge the clinic practice. The fee was to defray some of the costs of the HM program. A hospital representative was quoted as saying “physicians are responsible for obtaining hospital coverage for their own patients, not the hospital.”
I can’t really argue with the logic of that statement. Surely a clinic has responsibility to ensure that their patients get cared for while they are inpatients. If an internist is going to see a patient in the clinic and admit them to the hospital, shouldn’t an internist then see the patient in the hospital?
If I’m a hospital CEO, the answer is no.
To retrench a bit, yes, I’d want a board-certified internal-medicine (or pediatric or family medicine) physician to see the hospitalized patient. But in the process, I wouldn’t want them to only practice internal medicine. That was the model hospitals had 25 years ago—a model that cost them very little, a model that they played a large part in exterminating. The fact that most hospitals are willing to pay millions or more per year to not have that system tells me that they don’t want that system.
Q: So, what do hospitals want?
A: Hospitalists, not internists in the hospital.
What’s the difference? Well, it’s a perception issue. Many, if not most, believe that all it takes to be a great hospitalist is to show up for your shift, provide great care to your 15 patients, and go home. That is, the job is defined by the clinical effort—the internist part. Although there is tremendous benefit to this and I recognize its importance (and let’s not forget the weekend, night, and holiday coverage), this sells us short and puts our financial stability in peril.
To be great, to best help our patients, to give our hospitals what they want and need, we have to evolve from “internists in the hospital” to hospitalists. Hospitalists are defined not by our clinical effort but rather by our nonclinical effort. This is what hospitals are paying $1.7 million per year for. They had the internist in the hospital model and chose to pay more—they chose the hospitalist model.
To be a great hospitalist group means embracing the nonclinical work that envelops the clinical practice—the process and quality improvement (QI). That is, fundamentally changing the unsafe systems that surround our patients. Making them safer, more efficient and of higher quality.
This takes time.
Time = Money
It takes time to implement a QI project to reduce central line infections in the ICU. Or to develop and implement a VTE prophylaxis order set or an insulin or heparin drip protocol. Or to work closely with nursing to reduce falls on a medical unit. It takes time to be at the pneumonia core measures meeting every Monday at 7 a.m. and the hospital credentialing committee meeting every other Friday at 3 p.m. It also takes time to implement a new electronic health record or roll out the new LEAN project to reduce ED wait times.
This takes time, effort, and bandwidth—the kind that can’t be shoehorned into the average clinical day. This is work that needs to be done primarily during nonclinical hours. It’s the kind of work that defines HM as a field; the kind of work that increasingly determines your hospital’s bottom line; the kind of work that has tremendous value; the kind of work that requires remuneration.
In paying for the hospitalist model, your hospital is paying for the clinical (internist) and nonclinical (hospitalist) work you do. The $1.7 million per year is not a subsidy they pay to keep you in business. It’s the price they must pay to compensate your group for all the nonclinical work you do around quality, safety, efficiency, and leadership.
Q: But what if my group isn’t doing these kinds of things?
A: Then your hospital funding is at risk. The Montana story addresses just such a scenario. Clearly the hospital C-suite in this instance only valued (or was presented with) clinical work. Therefore, they felt that others should subsidize the hospitalist salaries—in this case, the clinic. I don’t know the particulars of this case but deduce this because it would be ludicrous to expect the clinic to pay for the part of the hospitalists’ time spent improving the hospital’s systems of care.
Writing the Final Chapter
At the core of the HM funding model is the concept of subsidy versus compensation. If we are only providing clinical care, then the offset dollars from the hospital to support our salaries is functionally a subsidy—a dollar amount to make up for our collections shortfall. However, if it is support for the nonclinical work we are doing, then it is compensation.
As the story of hospitalist funding is written, the report from Montana should serve as a cautionary tale. Hospital financial pressures likely will focus more scrutiny on the hospitalist financial support model. And as this story plays out, HM groups will be expected to bring more to the table than patient care.
Those that do will live happily ever after.
Those that don’t will be forced to answer the tough question: What’s the difference between an internist in the hospital and a hospitalist? If the answer is nothing, that story will have a decidedly and predictably less happy ending. TH
Dr. Glasheen is associate professor of medicine at the University of Colorado Denver, where he serves as director of the Hospital Medicine Program and the Hospitalist Training Program, and as associate program director of the Internal Medicine Residency Program.
Question: Before hospitalists, who cared for hospitalized patients?
Answer: Generalists—in other words, internists, family physicians, pediatricians.
Q: How much did that system cost hospitals?
A: Nothing, or very little. In some cases, support dollars were available for weekend, night, or uninsured patient coverage, but by and large this system cost hospitals little. Physicians admitted their patients to the hospital because the alternatives (sending a hypoxic pneumonia patient home from clinic, turning out the office lights and hoping the patient survived the night, or bringing the patient home with them) offered uncomfortable ethical, malpractice, or alimony consequences. So doctors admitted these patients to the hospital and visited them daily.
Q: The average amount of support per hospitalist is $131,564, or about $1.7 million per HM group seeing adult patients. The bulk of those dollars come from the hospital. If we assume that the people running hospitals are smart, then why would those smart businesspeople pay $1.7 million for something they used to get for free?
A: Because there is something they get in return for that money. Or, perhaps, something they think they are getting in return for those dollars.
Q: What?
A: I often go through this exercise with the residents in our hospitalist training program when we discuss the drivers of the HM movement. I usually discuss the reasons why a hospital should fund these groups; it always seems like such a no-brainer to me.
Enter a recent news item from Montana. The story from the Helena Independent Record (see “Unsustainable Growth?” p. 1) noted that a multispecialty group practice in Helena announced they were no longer admitting their patients to a local hospital in protest over a new hospital policy to charge the clinic practice. The fee was to defray some of the costs of the HM program. A hospital representative was quoted as saying “physicians are responsible for obtaining hospital coverage for their own patients, not the hospital.”
I can’t really argue with the logic of that statement. Surely a clinic has responsibility to ensure that their patients get cared for while they are inpatients. If an internist is going to see a patient in the clinic and admit them to the hospital, shouldn’t an internist then see the patient in the hospital?
If I’m a hospital CEO, the answer is no.
To retrench a bit, yes, I’d want a board-certified internal-medicine (or pediatric or family medicine) physician to see the hospitalized patient. But in the process, I wouldn’t want them to only practice internal medicine. That was the model hospitals had 25 years ago—a model that cost them very little, a model that they played a large part in exterminating. The fact that most hospitals are willing to pay millions or more per year to not have that system tells me that they don’t want that system.
Q: So, what do hospitals want?
A: Hospitalists, not internists in the hospital.
What’s the difference? Well, it’s a perception issue. Many, if not most, believe that all it takes to be a great hospitalist is to show up for your shift, provide great care to your 15 patients, and go home. That is, the job is defined by the clinical effort—the internist part. Although there is tremendous benefit to this and I recognize its importance (and let’s not forget the weekend, night, and holiday coverage), this sells us short and puts our financial stability in peril.
To be great, to best help our patients, to give our hospitals what they want and need, we have to evolve from “internists in the hospital” to hospitalists. Hospitalists are defined not by our clinical effort but rather by our nonclinical effort. This is what hospitals are paying $1.7 million per year for. They had the internist in the hospital model and chose to pay more—they chose the hospitalist model.
To be a great hospitalist group means embracing the nonclinical work that envelops the clinical practice—the process and quality improvement (QI). That is, fundamentally changing the unsafe systems that surround our patients. Making them safer, more efficient and of higher quality.
This takes time.
Time = Money
It takes time to implement a QI project to reduce central line infections in the ICU. Or to develop and implement a VTE prophylaxis order set or an insulin or heparin drip protocol. Or to work closely with nursing to reduce falls on a medical unit. It takes time to be at the pneumonia core measures meeting every Monday at 7 a.m. and the hospital credentialing committee meeting every other Friday at 3 p.m. It also takes time to implement a new electronic health record or roll out the new LEAN project to reduce ED wait times.
This takes time, effort, and bandwidth—the kind that can’t be shoehorned into the average clinical day. This is work that needs to be done primarily during nonclinical hours. It’s the kind of work that defines HM as a field; the kind of work that increasingly determines your hospital’s bottom line; the kind of work that has tremendous value; the kind of work that requires remuneration.
In paying for the hospitalist model, your hospital is paying for the clinical (internist) and nonclinical (hospitalist) work you do. The $1.7 million per year is not a subsidy they pay to keep you in business. It’s the price they must pay to compensate your group for all the nonclinical work you do around quality, safety, efficiency, and leadership.
Q: But what if my group isn’t doing these kinds of things?
A: Then your hospital funding is at risk. The Montana story addresses just such a scenario. Clearly the hospital C-suite in this instance only valued (or was presented with) clinical work. Therefore, they felt that others should subsidize the hospitalist salaries—in this case, the clinic. I don’t know the particulars of this case but deduce this because it would be ludicrous to expect the clinic to pay for the part of the hospitalists’ time spent improving the hospital’s systems of care.
Writing the Final Chapter
At the core of the HM funding model is the concept of subsidy versus compensation. If we are only providing clinical care, then the offset dollars from the hospital to support our salaries is functionally a subsidy—a dollar amount to make up for our collections shortfall. However, if it is support for the nonclinical work we are doing, then it is compensation.
As the story of hospitalist funding is written, the report from Montana should serve as a cautionary tale. Hospital financial pressures likely will focus more scrutiny on the hospitalist financial support model. And as this story plays out, HM groups will be expected to bring more to the table than patient care.
Those that do will live happily ever after.
Those that don’t will be forced to answer the tough question: What’s the difference between an internist in the hospital and a hospitalist? If the answer is nothing, that story will have a decidedly and predictably less happy ending. TH
Dr. Glasheen is associate professor of medicine at the University of Colorado Denver, where he serves as director of the Hospital Medicine Program and the Hospitalist Training Program, and as associate program director of the Internal Medicine Residency Program.
Question: Before hospitalists, who cared for hospitalized patients?
Answer: Generalists—in other words, internists, family physicians, pediatricians.
Q: How much did that system cost hospitals?
A: Nothing, or very little. In some cases, support dollars were available for weekend, night, or uninsured patient coverage, but by and large this system cost hospitals little. Physicians admitted their patients to the hospital because the alternatives (sending a hypoxic pneumonia patient home from clinic, turning out the office lights and hoping the patient survived the night, or bringing the patient home with them) offered uncomfortable ethical, malpractice, or alimony consequences. So doctors admitted these patients to the hospital and visited them daily.
Q: The average amount of support per hospitalist is $131,564, or about $1.7 million per HM group seeing adult patients. The bulk of those dollars come from the hospital. If we assume that the people running hospitals are smart, then why would those smart businesspeople pay $1.7 million for something they used to get for free?
A: Because there is something they get in return for that money. Or, perhaps, something they think they are getting in return for those dollars.
Q: What?
A: I often go through this exercise with the residents in our hospitalist training program when we discuss the drivers of the HM movement. I usually discuss the reasons why a hospital should fund these groups; it always seems like such a no-brainer to me.
Enter a recent news item from Montana. The story from the Helena Independent Record (see “Unsustainable Growth?” p. 1) noted that a multispecialty group practice in Helena announced they were no longer admitting their patients to a local hospital in protest over a new hospital policy to charge the clinic practice. The fee was to defray some of the costs of the HM program. A hospital representative was quoted as saying “physicians are responsible for obtaining hospital coverage for their own patients, not the hospital.”
I can’t really argue with the logic of that statement. Surely a clinic has responsibility to ensure that their patients get cared for while they are inpatients. If an internist is going to see a patient in the clinic and admit them to the hospital, shouldn’t an internist then see the patient in the hospital?
If I’m a hospital CEO, the answer is no.
To retrench a bit, yes, I’d want a board-certified internal-medicine (or pediatric or family medicine) physician to see the hospitalized patient. But in the process, I wouldn’t want them to only practice internal medicine. That was the model hospitals had 25 years ago—a model that cost them very little, a model that they played a large part in exterminating. The fact that most hospitals are willing to pay millions or more per year to not have that system tells me that they don’t want that system.
Q: So, what do hospitals want?
A: Hospitalists, not internists in the hospital.
What’s the difference? Well, it’s a perception issue. Many, if not most, believe that all it takes to be a great hospitalist is to show up for your shift, provide great care to your 15 patients, and go home. That is, the job is defined by the clinical effort—the internist part. Although there is tremendous benefit to this and I recognize its importance (and let’s not forget the weekend, night, and holiday coverage), this sells us short and puts our financial stability in peril.
To be great, to best help our patients, to give our hospitals what they want and need, we have to evolve from “internists in the hospital” to hospitalists. Hospitalists are defined not by our clinical effort but rather by our nonclinical effort. This is what hospitals are paying $1.7 million per year for. They had the internist in the hospital model and chose to pay more—they chose the hospitalist model.
To be a great hospitalist group means embracing the nonclinical work that envelops the clinical practice—the process and quality improvement (QI). That is, fundamentally changing the unsafe systems that surround our patients. Making them safer, more efficient and of higher quality.
This takes time.
Time = Money
It takes time to implement a QI project to reduce central line infections in the ICU. Or to develop and implement a VTE prophylaxis order set or an insulin or heparin drip protocol. Or to work closely with nursing to reduce falls on a medical unit. It takes time to be at the pneumonia core measures meeting every Monday at 7 a.m. and the hospital credentialing committee meeting every other Friday at 3 p.m. It also takes time to implement a new electronic health record or roll out the new LEAN project to reduce ED wait times.
This takes time, effort, and bandwidth—the kind that can’t be shoehorned into the average clinical day. This is work that needs to be done primarily during nonclinical hours. It’s the kind of work that defines HM as a field; the kind of work that increasingly determines your hospital’s bottom line; the kind of work that has tremendous value; the kind of work that requires remuneration.
In paying for the hospitalist model, your hospital is paying for the clinical (internist) and nonclinical (hospitalist) work you do. The $1.7 million per year is not a subsidy they pay to keep you in business. It’s the price they must pay to compensate your group for all the nonclinical work you do around quality, safety, efficiency, and leadership.
Q: But what if my group isn’t doing these kinds of things?
A: Then your hospital funding is at risk. The Montana story addresses just such a scenario. Clearly the hospital C-suite in this instance only valued (or was presented with) clinical work. Therefore, they felt that others should subsidize the hospitalist salaries—in this case, the clinic. I don’t know the particulars of this case but deduce this because it would be ludicrous to expect the clinic to pay for the part of the hospitalists’ time spent improving the hospital’s systems of care.
Writing the Final Chapter
At the core of the HM funding model is the concept of subsidy versus compensation. If we are only providing clinical care, then the offset dollars from the hospital to support our salaries is functionally a subsidy—a dollar amount to make up for our collections shortfall. However, if it is support for the nonclinical work we are doing, then it is compensation.
As the story of hospitalist funding is written, the report from Montana should serve as a cautionary tale. Hospital financial pressures likely will focus more scrutiny on the hospitalist financial support model. And as this story plays out, HM groups will be expected to bring more to the table than patient care.
Those that do will live happily ever after.
Those that don’t will be forced to answer the tough question: What’s the difference between an internist in the hospital and a hospitalist? If the answer is nothing, that story will have a decidedly and predictably less happy ending. TH
Dr. Glasheen is associate professor of medicine at the University of Colorado Denver, where he serves as director of the Hospital Medicine Program and the Hospitalist Training Program, and as associate program director of the Internal Medicine Residency Program.
New Developments

HM11 and the publication of the SHM-MGMA survey on hospitalist productivity and compensation occur every summer, and they always provide lots of new information to get me thinking. Two things stand out this year: Hospitalist demand remains high, and hospitals are paying a lot to have hospitalist services.
Supply and Demand
Along with SHM President Joe Li and Rob Bessler, who is CEO of Sound Physicians, I had the pleasure of presenting a preview of some data from the latest SHM-MGMA survey at the annual meeting May 11 in Dallas. During the session, I asked the large crowd of hospitalists how many were from practices that are actively recruiting additional hospitalists. About 40% of the hands went up.
If 40% of HM groups are actively recruiting, some for more than one open position, that’s a lot of recruiting. But it is dramatically less than the response I got when I asked the same question just three years ago at HM08 in San Diego. At that meeting, nearly every hand in the room went up, indicating everybody was recruiting (see “We’re Hiring,” July 2008, p. 62).
Of course, my show-of-hands survey of attendees at SHM meetings is not a perfect method to assess hospitalist supply and demand. But I think the dramatic change in responses from 2008 to 2011 is meaningful; it also matches what I’m seeing in the marketplace. I hear repeatedly that the years of rapid growth in hospitalist staffing have ended in many or most major metropolitan areas. For example, in places like Seattle (where I practice), Minneapolis, and Boston, there are far fewer open positions now than just two years ago, and most are to replace a departing doctor rather than to increase the overall staffing level.
But the far more numerous smaller markets are still recruiting aggressively in an effort to increase the overall staffing of the practice (and not just replace departing doctors). And changes in resident work-hour limitations are requiring teaching hospitals to increase hospitalist staffing to offset the reduction in resident availability. But it’s possible that if the larger markets are indeed becoming somewhat saturated with hospitalists, then there will be a trickledown effect, which should make more candidates available everywhere.
What will be the side effects if indeed the supply of hospitalists catches up to the demand, or even exceeds demand, in some places? It is easy to imagine that greater competition among candidates might mean that practices are increasingly able to hire the more talented and committed doctors, which should improve the overall performance of hospitalist practices.
Although I don’t have proof, I think this phenomenon has been in play in the field of emergency medicine for many years. When I was a resident in the 1980s, ED doctors typically were not the best and brightest at their hospitals. But the way I see it, the field began to attract better candidates, and as ED residencies and practices began to “fill up,” they could be more selective in new hires. Therefore, the average talent of the average ED doctor went up.
I think the average hospitalist today is pretty talented, but I also think it could get even better if the supply of hospitalists exceeds demand. I just hope I continue to make the cut!
If typical market forces are operative for hospitalists (far from a guarantee in any healthcare enterprise), then an oversupply of hospitalists could mean a flattening of the historical trend in hospitalist incomes. To this point, in our relatively young field, incomes have risen faster than can be explained solely by inflation or increases in hospitalist productivity. A relative shortage of hospitalists might be one of the main forces pushing incomes up, and it might go away.
We’ll see.
Hospital Support Trends Up
The most remarkable number in the 2011 SHM-MGMA survey is the financial support provided to practices per FTE hospitalist annually. This support nearly always comes from a hospital, and is often colloquially, and misleadingly, referred to as the “subsidy.”
In 2001, hospital support was about $65,000 per FTE. In the 2008 and 2010 surveys, the median financial support per FTE was $97,000 and $98,000, respectively. But it jumped to $136,403 this year. That is a really huge jump in one year. (Note: The surveys changed from biannual to annual in 2010, and the new SHM-MGMA survey uses a different financial support question/methodology and has a different respondent pool than the previous SHM surveys.)
Some of the increased dollars probably went to pay rising hospitalist compensation, which rose about 3% over the prior year without any significant increase in productivity. But that 3% salary increase translates to only about $5,000 (median compensation rose from roughly $215,000 to $220,000), and could be explained in part by such factors as removing academicians from this data set. (Starting in 2010, academic hospitalists are surveyed and reported separately, so aren’t included here.) So I don’t think the change in hospitalist incomes seen in this survey has much to do with the dramatic, near-40% increase in financial support.
The survey showed that hospitalist productivity hasn’t declined, so the other most likely culprit is declining professional fee collections, which might be due to an increasing portion of hospitalized patients who are uninsured or underinsured. Many hospitals report that their “payor mix” has worsened since the economic crisis of the last few years. And because hospitals typically hold the risk for the financial performance of their hospitalists, then if the latter see more uninsured patients and collect less in professional fees, the hospital will make up the difference. This phenomenon might explain much of the increased financial support.
But I’m not satisfied that a worsening payor mix explains everything. For example, if this were the most significant reason for increasing financial support, I think we would have seen this effect in the prior survey. Why did it “hit” so suddenly in this year alone?
We will get more information about collection rates when the second part of the survey is published in September. For example, we’ll be able to compare the dollars collected per encounter or per wRVU in the current survey to the prior one. If there was a significant drop, then it will require only a little math to see how much overall collections dropped per FTE and see if it is similar to the rise in financial support provided.
Of course, it will be very informative to see what the financial support turns out to be in the next survey (check back in late spring 2012). Will it stay around $136,000 per FTE or be something very different? TH
Dr. Nelson has been a practicing hospitalist since 1988 and is co-founder and past president of SHM. He is a principal in Nelson Flores Hospital Medicine Consultants, a national hospitalist practice management consulting firm (www.nelsonflores.com). He is course co-director and faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official position of SHM.
HM11 and the publication of the SHM-MGMA survey on hospitalist productivity and compensation occur every summer, and they always provide lots of new information to get me thinking. Two things stand out this year: Hospitalist demand remains high, and hospitals are paying a lot to have hospitalist services.
Supply and Demand
Along with SHM President Joe Li and Rob Bessler, who is CEO of Sound Physicians, I had the pleasure of presenting a preview of some data from the latest SHM-MGMA survey at the annual meeting May 11 in Dallas. During the session, I asked the large crowd of hospitalists how many were from practices that are actively recruiting additional hospitalists. About 40% of the hands went up.
If 40% of HM groups are actively recruiting, some for more than one open position, that’s a lot of recruiting. But it is dramatically less than the response I got when I asked the same question just three years ago at HM08 in San Diego. At that meeting, nearly every hand in the room went up, indicating everybody was recruiting (see “We’re Hiring,” July 2008, p. 62).
Of course, my show-of-hands survey of attendees at SHM meetings is not a perfect method to assess hospitalist supply and demand. But I think the dramatic change in responses from 2008 to 2011 is meaningful; it also matches what I’m seeing in the marketplace. I hear repeatedly that the years of rapid growth in hospitalist staffing have ended in many or most major metropolitan areas. For example, in places like Seattle (where I practice), Minneapolis, and Boston, there are far fewer open positions now than just two years ago, and most are to replace a departing doctor rather than to increase the overall staffing level.
But the far more numerous smaller markets are still recruiting aggressively in an effort to increase the overall staffing of the practice (and not just replace departing doctors). And changes in resident work-hour limitations are requiring teaching hospitals to increase hospitalist staffing to offset the reduction in resident availability. But it’s possible that if the larger markets are indeed becoming somewhat saturated with hospitalists, then there will be a trickledown effect, which should make more candidates available everywhere.
What will be the side effects if indeed the supply of hospitalists catches up to the demand, or even exceeds demand, in some places? It is easy to imagine that greater competition among candidates might mean that practices are increasingly able to hire the more talented and committed doctors, which should improve the overall performance of hospitalist practices.
Although I don’t have proof, I think this phenomenon has been in play in the field of emergency medicine for many years. When I was a resident in the 1980s, ED doctors typically were not the best and brightest at their hospitals. But the way I see it, the field began to attract better candidates, and as ED residencies and practices began to “fill up,” they could be more selective in new hires. Therefore, the average talent of the average ED doctor went up.
I think the average hospitalist today is pretty talented, but I also think it could get even better if the supply of hospitalists exceeds demand. I just hope I continue to make the cut!
If typical market forces are operative for hospitalists (far from a guarantee in any healthcare enterprise), then an oversupply of hospitalists could mean a flattening of the historical trend in hospitalist incomes. To this point, in our relatively young field, incomes have risen faster than can be explained solely by inflation or increases in hospitalist productivity. A relative shortage of hospitalists might be one of the main forces pushing incomes up, and it might go away.
We’ll see.
Hospital Support Trends Up
The most remarkable number in the 2011 SHM-MGMA survey is the financial support provided to practices per FTE hospitalist annually. This support nearly always comes from a hospital, and is often colloquially, and misleadingly, referred to as the “subsidy.”
In 2001, hospital support was about $65,000 per FTE. In the 2008 and 2010 surveys, the median financial support per FTE was $97,000 and $98,000, respectively. But it jumped to $136,403 this year. That is a really huge jump in one year. (Note: The surveys changed from biannual to annual in 2010, and the new SHM-MGMA survey uses a different financial support question/methodology and has a different respondent pool than the previous SHM surveys.)
Some of the increased dollars probably went to pay rising hospitalist compensation, which rose about 3% over the prior year without any significant increase in productivity. But that 3% salary increase translates to only about $5,000 (median compensation rose from roughly $215,000 to $220,000), and could be explained in part by such factors as removing academicians from this data set. (Starting in 2010, academic hospitalists are surveyed and reported separately, so aren’t included here.) So I don’t think the change in hospitalist incomes seen in this survey has much to do with the dramatic, near-40% increase in financial support.
The survey showed that hospitalist productivity hasn’t declined, so the other most likely culprit is declining professional fee collections, which might be due to an increasing portion of hospitalized patients who are uninsured or underinsured. Many hospitals report that their “payor mix” has worsened since the economic crisis of the last few years. And because hospitals typically hold the risk for the financial performance of their hospitalists, then if the latter see more uninsured patients and collect less in professional fees, the hospital will make up the difference. This phenomenon might explain much of the increased financial support.
But I’m not satisfied that a worsening payor mix explains everything. For example, if this were the most significant reason for increasing financial support, I think we would have seen this effect in the prior survey. Why did it “hit” so suddenly in this year alone?
We will get more information about collection rates when the second part of the survey is published in September. For example, we’ll be able to compare the dollars collected per encounter or per wRVU in the current survey to the prior one. If there was a significant drop, then it will require only a little math to see how much overall collections dropped per FTE and see if it is similar to the rise in financial support provided.
Of course, it will be very informative to see what the financial support turns out to be in the next survey (check back in late spring 2012). Will it stay around $136,000 per FTE or be something very different? TH
Dr. Nelson has been a practicing hospitalist since 1988 and is co-founder and past president of SHM. He is a principal in Nelson Flores Hospital Medicine Consultants, a national hospitalist practice management consulting firm (www.nelsonflores.com). He is course co-director and faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official position of SHM.
HM11 and the publication of the SHM-MGMA survey on hospitalist productivity and compensation occur every summer, and they always provide lots of new information to get me thinking. Two things stand out this year: Hospitalist demand remains high, and hospitals are paying a lot to have hospitalist services.
Supply and Demand
Along with SHM President Joe Li and Rob Bessler, who is CEO of Sound Physicians, I had the pleasure of presenting a preview of some data from the latest SHM-MGMA survey at the annual meeting May 11 in Dallas. During the session, I asked the large crowd of hospitalists how many were from practices that are actively recruiting additional hospitalists. About 40% of the hands went up.
If 40% of HM groups are actively recruiting, some for more than one open position, that’s a lot of recruiting. But it is dramatically less than the response I got when I asked the same question just three years ago at HM08 in San Diego. At that meeting, nearly every hand in the room went up, indicating everybody was recruiting (see “We’re Hiring,” July 2008, p. 62).
Of course, my show-of-hands survey of attendees at SHM meetings is not a perfect method to assess hospitalist supply and demand. But I think the dramatic change in responses from 2008 to 2011 is meaningful; it also matches what I’m seeing in the marketplace. I hear repeatedly that the years of rapid growth in hospitalist staffing have ended in many or most major metropolitan areas. For example, in places like Seattle (where I practice), Minneapolis, and Boston, there are far fewer open positions now than just two years ago, and most are to replace a departing doctor rather than to increase the overall staffing level.
But the far more numerous smaller markets are still recruiting aggressively in an effort to increase the overall staffing of the practice (and not just replace departing doctors). And changes in resident work-hour limitations are requiring teaching hospitals to increase hospitalist staffing to offset the reduction in resident availability. But it’s possible that if the larger markets are indeed becoming somewhat saturated with hospitalists, then there will be a trickledown effect, which should make more candidates available everywhere.
What will be the side effects if indeed the supply of hospitalists catches up to the demand, or even exceeds demand, in some places? It is easy to imagine that greater competition among candidates might mean that practices are increasingly able to hire the more talented and committed doctors, which should improve the overall performance of hospitalist practices.
Although I don’t have proof, I think this phenomenon has been in play in the field of emergency medicine for many years. When I was a resident in the 1980s, ED doctors typically were not the best and brightest at their hospitals. But the way I see it, the field began to attract better candidates, and as ED residencies and practices began to “fill up,” they could be more selective in new hires. Therefore, the average talent of the average ED doctor went up.
I think the average hospitalist today is pretty talented, but I also think it could get even better if the supply of hospitalists exceeds demand. I just hope I continue to make the cut!
If typical market forces are operative for hospitalists (far from a guarantee in any healthcare enterprise), then an oversupply of hospitalists could mean a flattening of the historical trend in hospitalist incomes. To this point, in our relatively young field, incomes have risen faster than can be explained solely by inflation or increases in hospitalist productivity. A relative shortage of hospitalists might be one of the main forces pushing incomes up, and it might go away.
We’ll see.
Hospital Support Trends Up
The most remarkable number in the 2011 SHM-MGMA survey is the financial support provided to practices per FTE hospitalist annually. This support nearly always comes from a hospital, and is often colloquially, and misleadingly, referred to as the “subsidy.”
In 2001, hospital support was about $65,000 per FTE. In the 2008 and 2010 surveys, the median financial support per FTE was $97,000 and $98,000, respectively. But it jumped to $136,403 this year. That is a really huge jump in one year. (Note: The surveys changed from biannual to annual in 2010, and the new SHM-MGMA survey uses a different financial support question/methodology and has a different respondent pool than the previous SHM surveys.)
Some of the increased dollars probably went to pay rising hospitalist compensation, which rose about 3% over the prior year without any significant increase in productivity. But that 3% salary increase translates to only about $5,000 (median compensation rose from roughly $215,000 to $220,000), and could be explained in part by such factors as removing academicians from this data set. (Starting in 2010, academic hospitalists are surveyed and reported separately, so aren’t included here.) So I don’t think the change in hospitalist incomes seen in this survey has much to do with the dramatic, near-40% increase in financial support.
The survey showed that hospitalist productivity hasn’t declined, so the other most likely culprit is declining professional fee collections, which might be due to an increasing portion of hospitalized patients who are uninsured or underinsured. Many hospitals report that their “payor mix” has worsened since the economic crisis of the last few years. And because hospitals typically hold the risk for the financial performance of their hospitalists, then if the latter see more uninsured patients and collect less in professional fees, the hospital will make up the difference. This phenomenon might explain much of the increased financial support.
But I’m not satisfied that a worsening payor mix explains everything. For example, if this were the most significant reason for increasing financial support, I think we would have seen this effect in the prior survey. Why did it “hit” so suddenly in this year alone?
We will get more information about collection rates when the second part of the survey is published in September. For example, we’ll be able to compare the dollars collected per encounter or per wRVU in the current survey to the prior one. If there was a significant drop, then it will require only a little math to see how much overall collections dropped per FTE and see if it is similar to the rise in financial support provided.
Of course, it will be very informative to see what the financial support turns out to be in the next survey (check back in late spring 2012). Will it stay around $136,000 per FTE or be something very different? TH
Dr. Nelson has been a practicing hospitalist since 1988 and is co-founder and past president of SHM. He is a principal in Nelson Flores Hospital Medicine Consultants, a national hospitalist practice management consulting firm (www.nelsonflores.com). He is course co-director and faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official position of SHM.
How to assess for possible drug-drug interactions
Bronchial thermoplasty: A promising therapy, still in its infancy
Treating severe, refractory asthma is an ever-evolving challenge and a major source of frustration for patients and clinicians. Failure of inhaler treatment often results in debilitation of the patient and leads to long-term use of corticosteroids, with their insidious side effects.1–3
Most asthma research continues to focus on inhibiting the cytokine cascade to reduce inflammation. However, inflammation is not the only pathophysiologic process underlying asthma.
Bronchial thermoplasty takes a novel approach and offers reason for some optimism.4–6 The aim of this minimally invasive bronchoscopic procedure is to attenuate bronchoconstriction by reducing airway smooth muscle mass.
In this issue of the Cleveland Clinic Journal of Medicine, Dr. Thomas Gildea and colleagues7 review the pathophysiology of asthma and the utility of decreasing airway smooth muscle via bronchial thermoplasty, its logistics, and the clinical trials that led to its approval by the US Food and Drug Administration (FDA) for the treatment of severe refractory asthma.
EVIDENCE FROM CLINICAL TRIALS
After studies in animals showed that bronchial thermoplasty was feasible, several randomized trials in humans—the Asthma Intervention Research (AIR) trial,6 the Research in Severe Asthma (RISA) trial,8 and the Asthma Intervention Research 2 (AIR2) trial9—found that the complication rates were acceptable, quality of life was improved, and health care utilization was reduced after the procedure during a 12- to 36-month period. These study results were essential in paving the way for FDA approval.
AIR2: A randomized controlled trial
The latest study to evaluate bronchial thermoplasty, the AIR2 trial,9 was designed with a feature that is used relatively infrequently in trials of invasive procedures: a sham control. A sham procedure can be defined as one performed on control-group participants to ensure that they experience the same incidental effects of the procedure as do participants who actually undergo the procedure.10
Thus, the patients in the control group received the same medications before and after the procedure, they were taken to the procedure room, and the bronchoscope was actually inserted into their lungs—but thermoplasty was not performed. All of this was done in a double-blind manner: neither the patients nor the physicians caring for them before and after the procedure knew which group they were in.
The aim of this exercise was to reduce bias, namely, the placebo effect, and to reinforce results that depend on subjective symptoms, such as the Asthma Quality of Life Questionnaire (AQLQ) score. Clinical trials in severe asthma are notoriously marred by the placebo effect, resulting in spurious improvements in lung function and symptoms.
The AIR2 trial found a significant reduction in severe exacerbations and emergency department visits, and a clinically meaningful improvement in AQLQ score from baseline at 6, 9, and 12 months in the bronchial thermoplasty group. However, 16 patients needed to be hospitalized after the procedure in the bronchial thermoplasty group, compared with two patients in the sham-procedure group.
The AIR2 trial, through the use of a sham-procedure control group, was able to minimize multiple forms of bias and thus provides the most reliable data for clinicians to extrapolate the good and the bad effects of bronchial thermoplasty.
THE PROCEDURE IS STILL IN ITS INFANCY
With any new therapy, we need to look at the benefits and complications not only in the short term but also the long term, ie, to determine whether the benefit is sustainable.
Long-term data on the benefits and side effects of bronchial thermoplasty have yet to be reported. However, radiofrequency ablation has been used in lung cancer therapy during the past decade, with favorable periprocedure complication profiles. Additionally, 5-year follow-up data have shown superior outcomes in stage I non-small-cell lung cancer survival rates with radiofrequency ablation compared with external-beam radiation.11
Ongoing studies will eventually provide insight on long-term outcomes of bronchial thermoplasty in asthma patients. Until such time, patients who have reached the limits of step-up therapy for severe refractory asthma should be informed that clinicians do not yet have a complete understanding of clinical benefits or sustainability of thermoplasty. Still, confidence in bronchial thermoplasty should be grounded in the simplicity of the procedure, the low short-term complication rates, and the long-term success of comparable medical procedures such as radiofrequency ablation in lung cancer, which utilizes similar technology.
Although this procedure is still in its infancy, the potential for long-term effectiveness in improving pulmonary function and quality of life in patients with severe asthma are undeniable. The body of data supporting its use will continue to evolve and hopefully point the way to better control of severe refractory asthma.
- Bollet AJ, Black R, Bunim JJ. Major undesirable side-effects resulting from prednisolone and prednisone. J Am Med Assoc 1955; 158:459–463.
- Olgaard K, Storm T, van Wowern N, et al. Glucocorticoid-induced osteoporosis in the lumbar spine, forearm, and mandible of nephrotic patients: a double-blind study on the high-dose, long-term effects of prednisone versus deflazacort. Calcif Tissue Int 1992; 50:490–497.
- Krasner AS. Glucocorticoid-induced adrenal insufficiency. JAMA 1999; 282:671–676.
- Cox G, Miller JD, McWilliams A, Fitzgerald JM, Lam S. Bronchial thermoplasty for asthma. Am J Respir Crit Care Med 2006; 173:965–969.
- Miller JD, Cox G, Vincic L, Lombard CM, Loomas BE, Danek CJ. A prospective feasibility study of bronchial thermoplasty in the human airway. Chest 2005; 127:1999–2006.
- Cox G, Thomson NC, Rubin AS, et al; AIR Trial Study Group. Asthma control during the year after bronchial thermoplasty. N Engl J Med 2007; 356:1327–1337.
- Gildea TR, Khatri SB, Castro M. Bronchial thermoplasty: a new treatment for severe refractory asthma. Cleve Clin J Med 2011; 78:477–485.
- Pavord ID, Cox G, Thomson NC, et al; RISA Trial Study Group. Safety and efficacy of bronchial thermoplasty in symptomatic, severe asthma. Am J Respir Crit Care Med 2007; 176:1185–1191.
- Castro M, Rubin AS, Laviolette M, et al; AIR2 Trial Study Group. Effectiveness and safety of bronchial thermoplasty in the treatment of severe asthma: a multicenter, randomized, double-blind, sham-controlled clinical trial. Am J Respir Crit Care Med 2010; 181:116–124.
- Simpson JA, Weiner ESC, editors. Oxford English Dictionary. 2nd ed. New York, NY: Oxford University Press; 1989.
- Sibley GS, Jamieson TA, Marks LB, Anscher MS, Prosnitz LR. Radiotherapy alone for medically inoperable stage I non-small-cell lung cancer: the Duke experience. Int J Radiat Oncol Biol Phys 1998; 40:149–154.
Treating severe, refractory asthma is an ever-evolving challenge and a major source of frustration for patients and clinicians. Failure of inhaler treatment often results in debilitation of the patient and leads to long-term use of corticosteroids, with their insidious side effects.1–3
Most asthma research continues to focus on inhibiting the cytokine cascade to reduce inflammation. However, inflammation is not the only pathophysiologic process underlying asthma.
Bronchial thermoplasty takes a novel approach and offers reason for some optimism.4–6 The aim of this minimally invasive bronchoscopic procedure is to attenuate bronchoconstriction by reducing airway smooth muscle mass.
In this issue of the Cleveland Clinic Journal of Medicine, Dr. Thomas Gildea and colleagues7 review the pathophysiology of asthma and the utility of decreasing airway smooth muscle via bronchial thermoplasty, its logistics, and the clinical trials that led to its approval by the US Food and Drug Administration (FDA) for the treatment of severe refractory asthma.
EVIDENCE FROM CLINICAL TRIALS
After studies in animals showed that bronchial thermoplasty was feasible, several randomized trials in humans—the Asthma Intervention Research (AIR) trial,6 the Research in Severe Asthma (RISA) trial,8 and the Asthma Intervention Research 2 (AIR2) trial9—found that the complication rates were acceptable, quality of life was improved, and health care utilization was reduced after the procedure during a 12- to 36-month period. These study results were essential in paving the way for FDA approval.
AIR2: A randomized controlled trial
The latest study to evaluate bronchial thermoplasty, the AIR2 trial,9 was designed with a feature that is used relatively infrequently in trials of invasive procedures: a sham control. A sham procedure can be defined as one performed on control-group participants to ensure that they experience the same incidental effects of the procedure as do participants who actually undergo the procedure.10
Thus, the patients in the control group received the same medications before and after the procedure, they were taken to the procedure room, and the bronchoscope was actually inserted into their lungs—but thermoplasty was not performed. All of this was done in a double-blind manner: neither the patients nor the physicians caring for them before and after the procedure knew which group they were in.
The aim of this exercise was to reduce bias, namely, the placebo effect, and to reinforce results that depend on subjective symptoms, such as the Asthma Quality of Life Questionnaire (AQLQ) score. Clinical trials in severe asthma are notoriously marred by the placebo effect, resulting in spurious improvements in lung function and symptoms.
The AIR2 trial found a significant reduction in severe exacerbations and emergency department visits, and a clinically meaningful improvement in AQLQ score from baseline at 6, 9, and 12 months in the bronchial thermoplasty group. However, 16 patients needed to be hospitalized after the procedure in the bronchial thermoplasty group, compared with two patients in the sham-procedure group.
The AIR2 trial, through the use of a sham-procedure control group, was able to minimize multiple forms of bias and thus provides the most reliable data for clinicians to extrapolate the good and the bad effects of bronchial thermoplasty.
THE PROCEDURE IS STILL IN ITS INFANCY
With any new therapy, we need to look at the benefits and complications not only in the short term but also the long term, ie, to determine whether the benefit is sustainable.
Long-term data on the benefits and side effects of bronchial thermoplasty have yet to be reported. However, radiofrequency ablation has been used in lung cancer therapy during the past decade, with favorable periprocedure complication profiles. Additionally, 5-year follow-up data have shown superior outcomes in stage I non-small-cell lung cancer survival rates with radiofrequency ablation compared with external-beam radiation.11
Ongoing studies will eventually provide insight on long-term outcomes of bronchial thermoplasty in asthma patients. Until such time, patients who have reached the limits of step-up therapy for severe refractory asthma should be informed that clinicians do not yet have a complete understanding of clinical benefits or sustainability of thermoplasty. Still, confidence in bronchial thermoplasty should be grounded in the simplicity of the procedure, the low short-term complication rates, and the long-term success of comparable medical procedures such as radiofrequency ablation in lung cancer, which utilizes similar technology.
Although this procedure is still in its infancy, the potential for long-term effectiveness in improving pulmonary function and quality of life in patients with severe asthma are undeniable. The body of data supporting its use will continue to evolve and hopefully point the way to better control of severe refractory asthma.
Treating severe, refractory asthma is an ever-evolving challenge and a major source of frustration for patients and clinicians. Failure of inhaler treatment often results in debilitation of the patient and leads to long-term use of corticosteroids, with their insidious side effects.1–3
Most asthma research continues to focus on inhibiting the cytokine cascade to reduce inflammation. However, inflammation is not the only pathophysiologic process underlying asthma.
Bronchial thermoplasty takes a novel approach and offers reason for some optimism.4–6 The aim of this minimally invasive bronchoscopic procedure is to attenuate bronchoconstriction by reducing airway smooth muscle mass.
In this issue of the Cleveland Clinic Journal of Medicine, Dr. Thomas Gildea and colleagues7 review the pathophysiology of asthma and the utility of decreasing airway smooth muscle via bronchial thermoplasty, its logistics, and the clinical trials that led to its approval by the US Food and Drug Administration (FDA) for the treatment of severe refractory asthma.
EVIDENCE FROM CLINICAL TRIALS
After studies in animals showed that bronchial thermoplasty was feasible, several randomized trials in humans—the Asthma Intervention Research (AIR) trial,6 the Research in Severe Asthma (RISA) trial,8 and the Asthma Intervention Research 2 (AIR2) trial9—found that the complication rates were acceptable, quality of life was improved, and health care utilization was reduced after the procedure during a 12- to 36-month period. These study results were essential in paving the way for FDA approval.
AIR2: A randomized controlled trial
The latest study to evaluate bronchial thermoplasty, the AIR2 trial,9 was designed with a feature that is used relatively infrequently in trials of invasive procedures: a sham control. A sham procedure can be defined as one performed on control-group participants to ensure that they experience the same incidental effects of the procedure as do participants who actually undergo the procedure.10
Thus, the patients in the control group received the same medications before and after the procedure, they were taken to the procedure room, and the bronchoscope was actually inserted into their lungs—but thermoplasty was not performed. All of this was done in a double-blind manner: neither the patients nor the physicians caring for them before and after the procedure knew which group they were in.
The aim of this exercise was to reduce bias, namely, the placebo effect, and to reinforce results that depend on subjective symptoms, such as the Asthma Quality of Life Questionnaire (AQLQ) score. Clinical trials in severe asthma are notoriously marred by the placebo effect, resulting in spurious improvements in lung function and symptoms.
The AIR2 trial found a significant reduction in severe exacerbations and emergency department visits, and a clinically meaningful improvement in AQLQ score from baseline at 6, 9, and 12 months in the bronchial thermoplasty group. However, 16 patients needed to be hospitalized after the procedure in the bronchial thermoplasty group, compared with two patients in the sham-procedure group.
The AIR2 trial, through the use of a sham-procedure control group, was able to minimize multiple forms of bias and thus provides the most reliable data for clinicians to extrapolate the good and the bad effects of bronchial thermoplasty.
THE PROCEDURE IS STILL IN ITS INFANCY
With any new therapy, we need to look at the benefits and complications not only in the short term but also the long term, ie, to determine whether the benefit is sustainable.
Long-term data on the benefits and side effects of bronchial thermoplasty have yet to be reported. However, radiofrequency ablation has been used in lung cancer therapy during the past decade, with favorable periprocedure complication profiles. Additionally, 5-year follow-up data have shown superior outcomes in stage I non-small-cell lung cancer survival rates with radiofrequency ablation compared with external-beam radiation.11
Ongoing studies will eventually provide insight on long-term outcomes of bronchial thermoplasty in asthma patients. Until such time, patients who have reached the limits of step-up therapy for severe refractory asthma should be informed that clinicians do not yet have a complete understanding of clinical benefits or sustainability of thermoplasty. Still, confidence in bronchial thermoplasty should be grounded in the simplicity of the procedure, the low short-term complication rates, and the long-term success of comparable medical procedures such as radiofrequency ablation in lung cancer, which utilizes similar technology.
Although this procedure is still in its infancy, the potential for long-term effectiveness in improving pulmonary function and quality of life in patients with severe asthma are undeniable. The body of data supporting its use will continue to evolve and hopefully point the way to better control of severe refractory asthma.
- Bollet AJ, Black R, Bunim JJ. Major undesirable side-effects resulting from prednisolone and prednisone. J Am Med Assoc 1955; 158:459–463.
- Olgaard K, Storm T, van Wowern N, et al. Glucocorticoid-induced osteoporosis in the lumbar spine, forearm, and mandible of nephrotic patients: a double-blind study on the high-dose, long-term effects of prednisone versus deflazacort. Calcif Tissue Int 1992; 50:490–497.
- Krasner AS. Glucocorticoid-induced adrenal insufficiency. JAMA 1999; 282:671–676.
- Cox G, Miller JD, McWilliams A, Fitzgerald JM, Lam S. Bronchial thermoplasty for asthma. Am J Respir Crit Care Med 2006; 173:965–969.
- Miller JD, Cox G, Vincic L, Lombard CM, Loomas BE, Danek CJ. A prospective feasibility study of bronchial thermoplasty in the human airway. Chest 2005; 127:1999–2006.
- Cox G, Thomson NC, Rubin AS, et al; AIR Trial Study Group. Asthma control during the year after bronchial thermoplasty. N Engl J Med 2007; 356:1327–1337.
- Gildea TR, Khatri SB, Castro M. Bronchial thermoplasty: a new treatment for severe refractory asthma. Cleve Clin J Med 2011; 78:477–485.
- Pavord ID, Cox G, Thomson NC, et al; RISA Trial Study Group. Safety and efficacy of bronchial thermoplasty in symptomatic, severe asthma. Am J Respir Crit Care Med 2007; 176:1185–1191.
- Castro M, Rubin AS, Laviolette M, et al; AIR2 Trial Study Group. Effectiveness and safety of bronchial thermoplasty in the treatment of severe asthma: a multicenter, randomized, double-blind, sham-controlled clinical trial. Am J Respir Crit Care Med 2010; 181:116–124.
- Simpson JA, Weiner ESC, editors. Oxford English Dictionary. 2nd ed. New York, NY: Oxford University Press; 1989.
- Sibley GS, Jamieson TA, Marks LB, Anscher MS, Prosnitz LR. Radiotherapy alone for medically inoperable stage I non-small-cell lung cancer: the Duke experience. Int J Radiat Oncol Biol Phys 1998; 40:149–154.
- Bollet AJ, Black R, Bunim JJ. Major undesirable side-effects resulting from prednisolone and prednisone. J Am Med Assoc 1955; 158:459–463.
- Olgaard K, Storm T, van Wowern N, et al. Glucocorticoid-induced osteoporosis in the lumbar spine, forearm, and mandible of nephrotic patients: a double-blind study on the high-dose, long-term effects of prednisone versus deflazacort. Calcif Tissue Int 1992; 50:490–497.
- Krasner AS. Glucocorticoid-induced adrenal insufficiency. JAMA 1999; 282:671–676.
- Cox G, Miller JD, McWilliams A, Fitzgerald JM, Lam S. Bronchial thermoplasty for asthma. Am J Respir Crit Care Med 2006; 173:965–969.
- Miller JD, Cox G, Vincic L, Lombard CM, Loomas BE, Danek CJ. A prospective feasibility study of bronchial thermoplasty in the human airway. Chest 2005; 127:1999–2006.
- Cox G, Thomson NC, Rubin AS, et al; AIR Trial Study Group. Asthma control during the year after bronchial thermoplasty. N Engl J Med 2007; 356:1327–1337.
- Gildea TR, Khatri SB, Castro M. Bronchial thermoplasty: a new treatment for severe refractory asthma. Cleve Clin J Med 2011; 78:477–485.
- Pavord ID, Cox G, Thomson NC, et al; RISA Trial Study Group. Safety and efficacy of bronchial thermoplasty in symptomatic, severe asthma. Am J Respir Crit Care Med 2007; 176:1185–1191.
- Castro M, Rubin AS, Laviolette M, et al; AIR2 Trial Study Group. Effectiveness and safety of bronchial thermoplasty in the treatment of severe asthma: a multicenter, randomized, double-blind, sham-controlled clinical trial. Am J Respir Crit Care Med 2010; 181:116–124.
- Simpson JA, Weiner ESC, editors. Oxford English Dictionary. 2nd ed. New York, NY: Oxford University Press; 1989.
- Sibley GS, Jamieson TA, Marks LB, Anscher MS, Prosnitz LR. Radiotherapy alone for medically inoperable stage I non-small-cell lung cancer: the Duke experience. Int J Radiat Oncol Biol Phys 1998; 40:149–154.
Bronchial thermoplasty: A new treatment for severe refractory asthma
Asthma now has a new treatment, but it isn’t for everybody. Called bronchial thermoplasty, it is reserved for patients whose asthma is severe and refractory, as it involves three sessions of bronchoscopy, each lasting up to 1 hour, during which the smooth muscle layer is methodically ablated from the airway using radiofrequency energy.1,2
The US Food and Drug Administration (FDA) has approved bronchial thermoplasty,3 and although it does not cure asthma or completely eliminate its symptoms, patients with severe asthma that was not well controlled with medical therapy who underwent this procedure in clinical trials subsequently had fewer symptoms, enjoyed better quality of life, and needed less intensive health care (such as emergency room visits) than patients who did not undergo the procedure.4–6
Here, we present an overview of the pathophysiology of severe refractory asthma and the clinical trials of bronchial thermoplasty, its current protocols, and the status of this new treatment.
WHAT IS SEVERE REFRACTORY ASTHMA?
Asthma is a chronic inflammatory condition of the airways characterized by episodic symptoms of breathlessness, cough, and wheezing, which can wax and wane over time. Approximately 8.2% of the general population is affected.7
Our understanding of the pathophysiology of asthma has improved over the past 20 years, and with the publication of clinical guidelines from the National Asthma Education and Prevention Program in 1991,8 1997,9 and 2002,10 outcomes have improved. Most people with asthma can control their symptoms if they adhere to anti-inflammatory therapies and avoid triggers. Yet 5% to 10% of asthma patients have severe refractory disease, and asthma accounts for nearly half a million hospitalizations every year.11
The latest guidelines, published in 2007, emphasize the importance of assessing the severity of asthma, including the patient’s impairment (symptoms and limitations) and risk (likelihood of exacerbations).12
Workshop consensus definition of severe refractory asthma
A consensus group convened by the American Thoracic Society13 defined asthma as severe and refractory if the patient meets at least one of the following major criteria:
- Takes oral corticosteroids continuously or nearly continuously (> 50% of year)
- Takes high-dose inhaled corticosteroids.
In addition, the patient must meet at least two minor criteria, ie:
- Takes a controller medication such as a long-acting beta-agonist, theophylline, or a leukotriene antagonist every day
- Takes a short-acting beta agonist every day or nearly every day
- Has persistent airway obstruction, ie, a forced expiratory volume in 1 second (FEV1) less than 80% of predicted, or a peak expiratory flow that has a diurnal variability greater than 20%
- Has one or more urgent care visits for asthma per year
- Needs three or more oral corticosteroid “bursts” per year
- Has prompt deterioration when the dose of oral or inhaled corticosteroid is reduced by 25% or less
- Has had a near-fatal asthma event in the past.
Compared with people with mild asthma, people who have severe refractory asthma tend to be older, have fewer allergies, and make more use of intensive and urgent health care.14
Asthma is due to both inflammation and bronchoconstriction
The pathophysiology of asthma involves both chronic airway inflammation and bronchoconstriction, the latter characterized by a greater response to methacholine. Histologic findings include excessive mucus secretion, epithelial cell injury, and smooth muscle hypertrophy. These changes can lead to persistent airflow obstruction that can be difficult to control with medical therapies.12
Bronchoconstriction can be reversed temporarily with bronchodilators, but no longlasting therapy to reduce it has been available until now. Bronchial thermoplasty targets this gap in asthma management.
STUDIES OF BRONCHIAL THERMOPLASTY
Radiofrequency ablation has been used to treat other medical conditions such as lung cancer and cardiac arrhythmias.15,16 Its use to treat asthma by eradicating smooth muscle cells from the airway wall began with studies in animals.1 Later, studies were done in people without asthma,2 then in patients with mild to moderate asthma,17 and finally in patients with moderate to severe refractory asthma.4–6
These studies helped clarify which type of patients would be appropriate candidates and the outcomes to be anticipated, including adverse events.
Early studies
Danek et al,1 in a study in nonasthmatic dogs, found that thermoplasty at 65°C or 75°C (149°F or 167°F) attenuated the airway’s response to methacholine up to 3 years after treatment. As early as 1 week after treatment, airway smooth muscle was seen to be degenerating or absent, and the effect was inversely proportional to airway responsiveness.
Adverse effects of the procedure were cough, inflammatory edema of the airway wall, retained mucus, and blanching of the airway wall at the site of catheter contact. Three years later, there was no evidence of smooth muscle regeneration.
Miller et al2 next performed a feasibility study in eight patients, mean age 58 ± 8.3 years, who were scheduled to undergo lung resection for lung cancer. Five to 20 days before surgery, the investigators performed thermoplasty at 55°C or 65°C (131°F or 149°F) in three to nine sites per patient, 1 cm from known tumors but within areas to be resected. There were no significant adverse events such as hemoptysis, respiratory infections, or excess bronchial irritation.
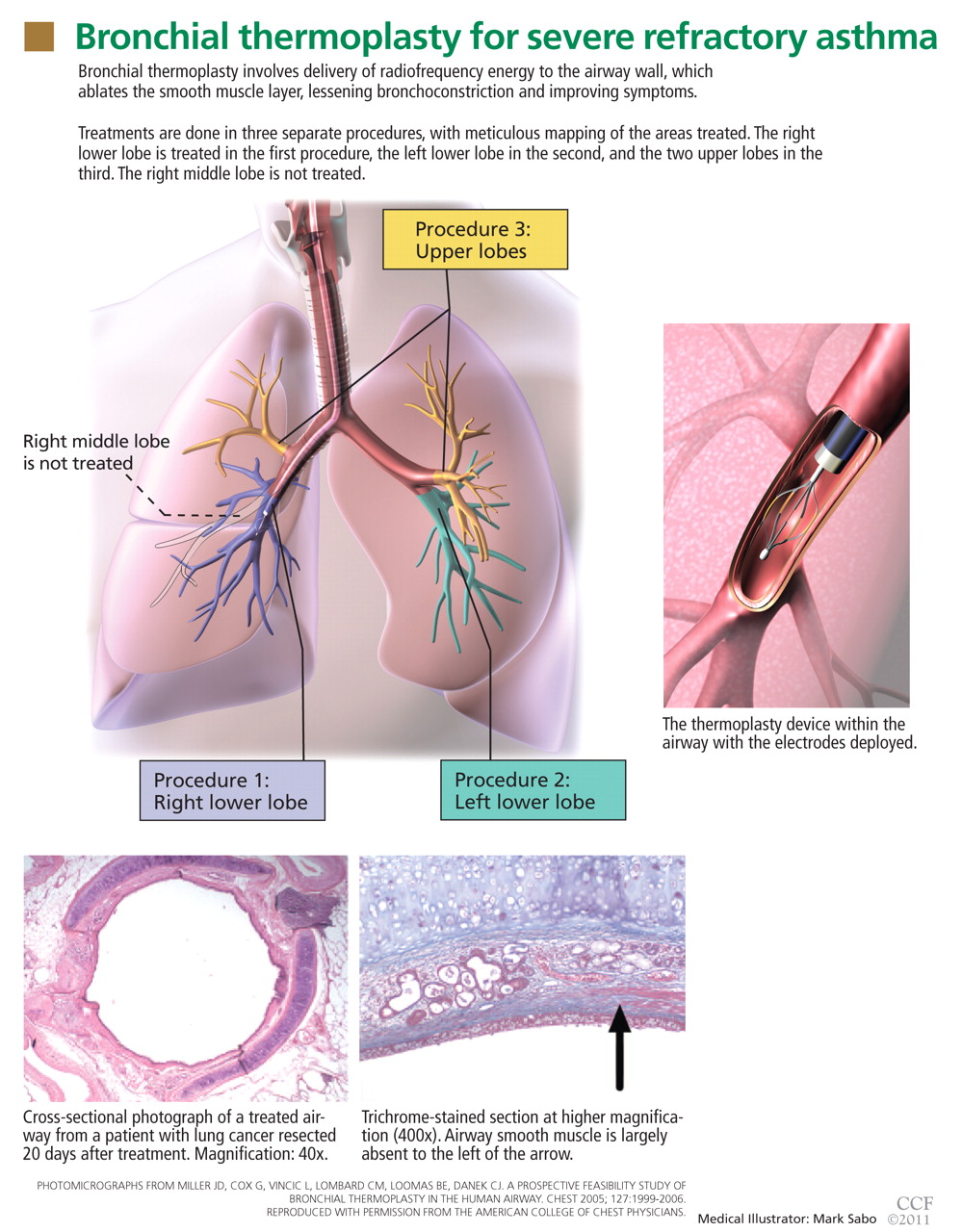
A pilot study in mild to moderate asthma
Cox et al17 performed the first study of bronchial thermoplasty in patients with mild to moderate asthma. This was a prospective observational study in 16 patients who were younger than the patients in the previous study, with an average age of 30 years (range 24–58). They were given prednisone 30 or 50 mg the day before the procedure and on the day of the procedure. Three treatments were done, 3 weeks apart. The right middle lobe was not treated because the bronchus leading to it is relatively long and narrow, raising concern about damaging it.18
Results. The most frequent side effects were symptoms of airway irritation such as cough, dyspnea, wheezing, and bronchospasm. The mean time to onset was less than 1.7 days, and the mean time to resolution was 4.6 days after the most recent procedure. None of the patients needed to be hospitalized in the immediate postprocedure period.
In the 2 years after the procedure, there were 312 adverse events, mainly mild. Three (1%) of the adverse events were reported as severe, but they were deemed not related to the procedure. Yearly computed tomographic scans of the chest showed no structural changes such as bronchiectasis in the parenchyma or bronchial wall.
The FEV1 was higher at 12 weeks and at 1 year after thermoplasty than at baseline but was not significantly different from baseline at 2 years.
At baseline, the patients reported that 50% of their days were symptom-free; this increased to 73% at 12 weeks (P = .015).
In addition, airway hyperresponsiveness decreased significantly, and the effect persisted over 2 years. The provocative concentration of methacholine that caused a 20% reduction in FEV1 (the PC20) was:
- 0.92 mg/mL at baseline (95% confidence interval 0.42–1.99)
- 4.75 mg/mL at 12 weeks (2.51–8.85)
- 5.45 mg/mL at 1 year (1.54–19.32)
- 3.40 mg/mL at 2 years (1.35–8.52).
Limitations of this study include the relatively small number of patients enrolled and their relatively stable asthma.
The AIR trial: A randomized trial in moderate or persistent asthma
The first large multicenter trial of bronchial thermoplasty, the Asthma Intervention Research (AIR) trial,4 was prospective and randomized but not blinded. The aim was to determine whether bronchial thermoplasty would improve asthma control after long-acting beta agonists were discontinued.
Patients could be enrolled if they were 18 to 65 years old, had moderate or persistent asthma, and needed to take an inhaled corticosteroid (beclomethasone [Qvar] 200 μg or more or an equivalent drug) and a long-acting beta agonist (salmeterol [Serevent] 100 μg or more or an equivalent drug) every day. They also needed to have FEV1 values of 60% to 85% of predicted and airway reactivity (PC20 < 8 mg/mL), and their asthma had to have been stable for 6 weeks.
At baseline, the long-acting beta agonist was withdrawn temporarily; the final criterion for entry was that their asthma had to become worse when this was done.
Then, 112 patients were randomized to receive either bronchial thermoplasty with medical care (inhaled corticosteroids and long-acting beta agonists) or usual care, ie, medical therapy alone. Treatments were done in three sessions over 9 weeks, followed by attempts to discontinue their long-acting beta agonists at 3, 6, and 9 months after the procedure without exacerbations.
An exacerbation was defined as at least one of the following for 2 consecutive days: a reduction of peak flow by 20% of baseline average, the need for more than three additional puffs of rescue inhaler, or nocturnal awakenings caused by asthma symptoms. The patients kept a daily diary of their symptoms and rescue inhaler use, and they completed the Asthma Quality of Life Questionnaire (AQLQ) and the Asthma Control Questionnaire (ACQ).
Results. The number of mild (but not severe) exacerbations per week was significantly lower at 3 and 12 months than at baseline in the thermoplasty group, with 10 fewer mild exacerbations per patient per year, but was unchanged in the control group. There were significantly greater improvements in morning peak flow at 3, 6, and 12 months from baseline in the treatment group than in the usual-care group. Rescue medication use was also significantly less at 3 and 12 months. Symptom scores, AQLQ scores, and ACQ scores were all significantly better than at baseline as well.
Not surprisingly, in this cohort with unstable asthma, there were 407 adverse events in the treatment group and 106 adverse events in the control group. Most of these occurred within 1 day and resolved within 7 days after the procedure. There were more hospitalizations in the treatment group as well, for reasons that included exacerbations of asthma, collapse of the left lower lobe, and pleurisy.4
Therefore, this trial found that thermoplasty improved asthma symptoms within 3 months and that the effect lasted 1 year, with an encouraging reduction in the number of mild exacerbations. However, it was not blinded, and there is a strong placebo effect in asthma. Needed was a randomized trial in which the control group would undergo a sham treatment.
The RISA trial: A randomized trial in severe asthma
The Research in Severe Asthma (RISA) trial5 included patients with more severe asthma than those in the AIR trial. Entry criteria were:
- Taking high doses of an inhaled corticosteroid (> 750 μg of fluticasone or its equivalent per day)
- Taking prednisone (≤ 30 mg/day)
- An FEV1 of at least 50% of predicted without a bronchodilator
- A positive methacholine test.
Seventeen patients were randomized to undergo bronchial thermoplasty, and another 17 were randomized to receive medical treatment.
After a 2-week run-in period, the thermoplasty patients underwent three treatments, performed 3 weeks apart. For the next 16 weeks, the corticosteroid doses were kept stable in both groups, followed by a 14-week corticosteroid-weaning phase and then a 16-week reduced-corticosteroid phase. During this time, attempts were made to decrease the oral or inhaled corticosteroid doses according to a protocol (eg, a 20%–25% reduction every 2–4 weeks) unless there were mild exacerbations lasting more than 7 days.
Results. There were more adverse events in the thermoplasty group than in the medical management group in the treatment period, including seven hospitalizations for exacerbations of asthma and a partial collapse of the left lower lobe. There were no significant differences in adverse events between groups in the posttreatment period (up to 6 weeks after the last treatment). Forty-nine percent of the events were mild in each group; 10% of the events were severe in the thermoplasty group vs 4% in the control group.
During the steroid-stable phase, patients in the thermoplasty group used rescue inhalers significantly less than those in the control group, and their prebronchodilator FEV1 and AQLQ and ACQ scores were better. The differences in rescue inhaler use and questionnaire scores remained significant at 1 year.
Comment. As expected, serious adverse events occurred more often in patients with severe asthma in the treatment group than in the control group. However, 1 year after the procedure, the adverse-event rates were similar in the treatment and control groups, suggesting that this procedure can be safely performed in similar patient populations. Although there was significant potential for a placebo effect, these patients with severe persistent asthma showed significant improvement in clinical measures of asthma compared with the control group.
AIR2: A randomized, double-blind trial
The latest trial of this new therapy in severe asthma was the AIR2 trial.6 A major difference in its design compared with the earlier ones was that the control group underwent sham thermoplasty, allowing the trial to be truly double-blinded. (The bronchoscopy team knew which patients got which treatment, but the patients and the study physicians following them did not).
The primary outcome was the change in AQLQ score from baseline at 6, 9, and 12 months. Secondary outcomes included absolute changes in the asthma control scores, symptom scores, peak flows, rescue medication use, and FEV1.
The randomized groups (196 patients in the thermoplasty group and 101 in the sham treatment group) were well matched, and more than 80% in each group met the American Thoracic Society criteria for severe refractory asthma.
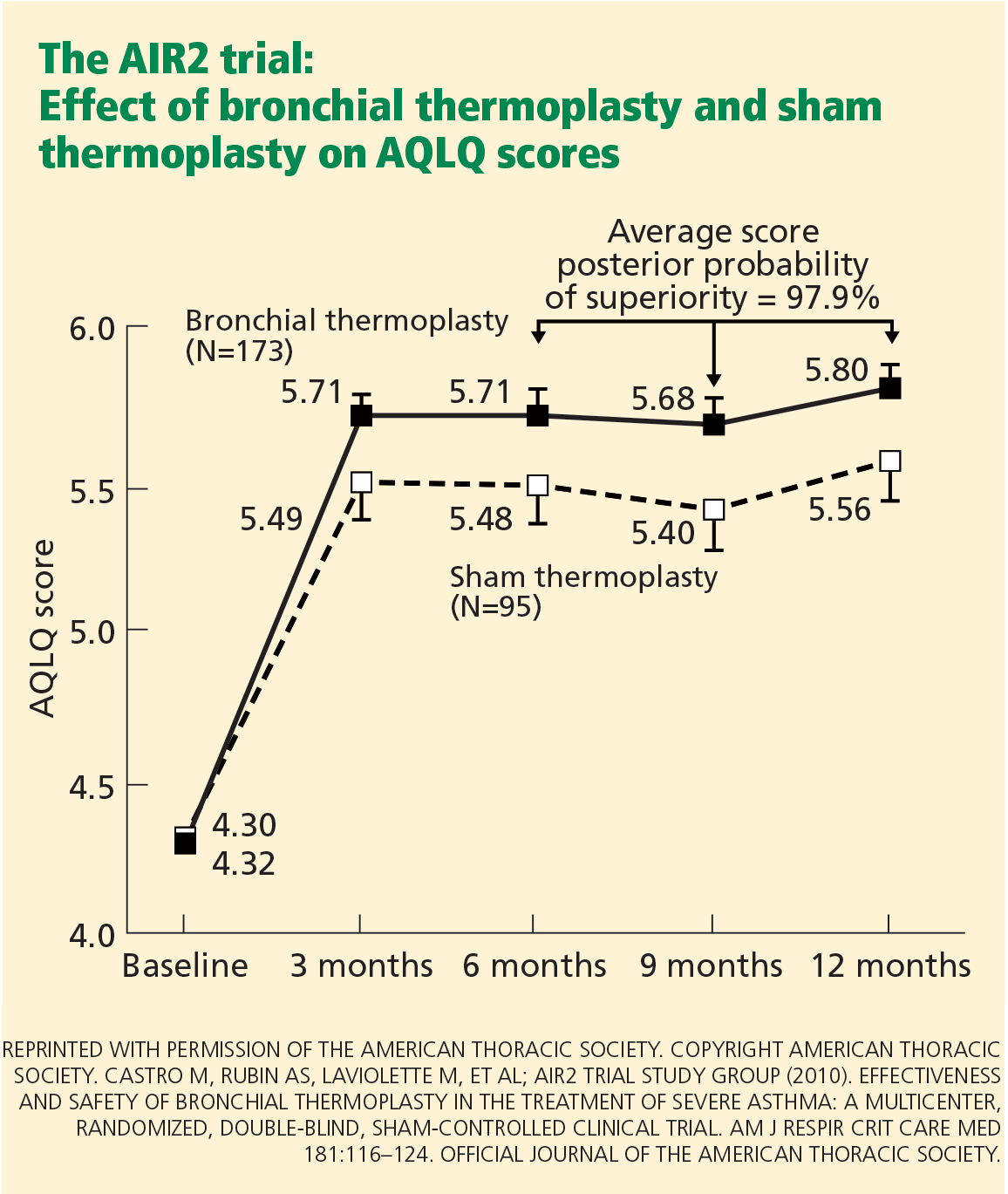
On the AQLQ, a change of more than 0.5 is considered clinically meaningful. Interestingly, there was a significant and clinically meaningful improvement in AQLQ in 64% of the sham treatment group, highlighting the placebo effect in asthma treatment.19 However, a larger proportion (79%) of the treated group had a clinically meaningful improvement on the AQLQ than in the sham treatment group.
Adverse events occurred in both groups; however, during the treatment phase, 16 patients in the bronchial thermoplasty group needed to be hospitalized for respiratory symptoms including worsening asthma, atelectasis, lower respiratory tract infections, decreased FEV1, and an aspirated tooth. One episode of hemoptysis required bronchial artery embolization. In contrast, only two patients in the sham treatment group needed hospitalization.
Therefore, this trial showed that patients with severe asthma treated with bronchial thermoplasty had a long-term improvement in quality of life and needed less health care.6
Translating these trials into practice
To summarize, these clinical trials showed that bronchial thermoplasty was feasible, was relatively safe, and produced better clinical outcomes in patients with severe asthma when medical therapies did not control their symptoms.
In practice, patient selection is likely to be important. A key question will be, Does the patient truly have severe refractory asthma, or is the patient not taking his or her medication? Adherence to therapy should be evaluated.
In addition, patients need to be observed and monitored closely during and after the treatment period, as airway complications and asthma exacerbations can occur up to 6 weeks after the last procedure. About 80% of all study patients had multiple symptoms of asthma and other symptoms in the treatment period. Rarely did these symptoms result in hospitalization, but they were more common in the treatment group in the AIR2 trial.
Long-term studies have evaluated the duration of effect and the safety of bronchial thermoplasty, and outcomes appear favorable.20,21
WHY DOES IT WORK?
The role of airway smooth muscle in asthma is yet to be fully elucidated. The trials outlined here showed that although asthma is a disease of the airways, including the small airways, treatment of airways 3 mm or larger improves asthma symptoms, quality of life, and health care utilization.6 Thus, the role of airway smooth muscle in asthma and as a target of therapy has not previously been fully realized.21
Early investigations into the mechanisms of airflow obstruction and airway resistance found that 75% of postnasal resistance occurs in the first six to eight generations (ie, branchings) of the airways, indicating that larger airways are involved.22 (The number of generations varies depending on the size of the person but it typically is 10 to 12.) Findings from the study in dogs introduced the idea that smooth muscle alterations contributed to the changes in airway resistance, and that subtle changes in airway smooth muscle could clinically benefit asthma patients.1
The speculated purpose of the airway smooth muscle layer is to support the airway, allow gas exchange, propel mucus for clearance, defend the airway, enhance cough, and promote lymphatic flow. However, the airway smooth muscle layer may also be vestigial. In asthma, airway smooth muscle adds to bronchoconstriction and hyperresponsiveness, and has a role in mediating inflammation and airway remodeling.21 No definitive studies have shown that eliminating airway smooth muscle greatly inhibits normal airway function.18
What exactly does thermoplasty do to the smooth muscle? Studies in smooth muscle from cows showed that high temperatures directly disrupt the actin-myosin interaction, likely through denaturation of motor proteins.23 This immediate loss of muscle cell function is not likely to be the result of apoptosis, autophagy, or necrosis, or mediated by heat-shock proteins, in view of the relatively quick muscle response and lack of progressive changes. Tissue responsiveness is substantially reduced a few seconds after application of 60°C of heat and is subsequently abolished within 5 minutes after treatment.23
The intervention appears to be dose-dependent. Responsiveness to cholinergic stimulation is lessened by treatment, and the desired effect is seen within seconds and does not progress.
Therefore, we can surmise that disruption of myosin function is likely the mechanism of the therapeutic effect, breaking the cascade of airway smooth muscle spasm. Now that we know about the airway smooth muscle as a possible target of therapy, and that it may play only a vestigial role, we can think about other therapies that focus on it.18,23
BRONCHIAL THERMOPLASTY PROTOCOLS
Patients are assessed before and on the day of the procedure to make sure their disease is stable (ie, their postbronchodilator FEV1 is within 15% of baseline values, and they have no evidence of asthma exacerbation or active infection), similar to the protocol used in the AIR2 trial,6 before proceeding with the treatment.
Patients are given 50 mg of prednisone 3 days before and again on the day of the procedure. Nebulized albuterol (2.5–5.0 mg) is given before the patients undergo screening spirometry and again before the procedure. If the preprocedure FEV1 is lower than 15% below baseline, we postpone the procedure to another day.
The procedure is performed with the patient under moderate conscious sedation, typically using fentanyl (Sublimaze), midazolam (Versed), and topical lidocaine in a monitored environment. The bronchoscope is inserted via either the mouth or nose, and supplemental oxygen is provided.
Thermoplasty is performed with the Alair system (Asthmatx, Inc., Sunnyvale, CA), which delivers a specific amount of radiofrequency (thermal) energy through a dedicated catheter. The catheter is deployed through a 2.0-mm channel of a flexible bronchoscope, starting in distal airways as small as 3 mm in diameter and working proximally to sequentially treat all airways to the mainstem lobar bronchi. The sites treated are meticulously recorded on a bronchial airway map to ensure that treatment sites are not skipped or overlapped (FIGURE 1).
An array of four electrodes is manually expanded to make contact with the airway walls; each electrode has 5 mm of exposed wire. As the energy is delivered, the control unit measures electrical resistance converted to thermal energy and turns off the current when an appropriate dosage is given. This thermal energy is what is responsible for altering the airway smooth muscle.
A full course of treatment requires three separate bronchoscopy sessions, each separated by 2 to 3 weeks. The left lower lobe and the right lower lobe are treated in separate procedures, and then both upper lobes are treated in a third procedure to minimize any respiratory symptoms. Each procedure usually requires 50 to 75 activations of the device and takes up to 60 minutes.
After each procedure the patient should be observed for 3 to 4 hours, and spirometry should be repeated to make sure the FEV1 (percent predicted) is within 20% of the baseline value. An additional 50-mg dose of prednisone is prescribed for the day after the procedure.24
FDA CLEARANCE AND LONG-TERM FOLLOW-UP
The FDA approved the Alair device for treating severe refractory asthma in early 2010.3 The indications for it are based on the study populations in the published trials. Patients can be evaluated for this treatment if they have well-documented severe persistent asthma not well controlled on inhaled corticosteroids and long-acting beta agonists and have no significant contraindications to bronchoscopy.
As part of the conditions of approval, the FDA required a postapproval study based on the long-term follow-up of the AIR2 trial. They specifically wanted to compare patients who have desirable long-term outcomes and those in whom any treatment effect wanes with time. Since we have only a few years of follow-up data, we still do not know all the possible late effects of the treatment; we have an opportunity to learn more.
Another question that needs to be studied is whether thermoplasty will help other forms of bronchospastic lung disease, such as chronic obstructive pulmonary disease.
A second postapproval study will be a prospective, open-label, single-arm, multicenter study conducted in the United States to assess the treatment effect and short-term and long-term safety profile of thermoplasty in asthma.
As experience with the procedure increases, we will be better able to characterize which patients may benefit from it. In addition, the knowledge gained by the longer-term study of airway smooth muscle function alterations will potentially drive the discovery of other innovative therapies for severe asthma.
- Danek CJ, Lombard CM, Dungworth DL, et al. Reduction in airway hyperresponsiveness to methacholine by the application of RF energy in dogs. J Appl Physiol 2004; 97:1946–1953.
- Miller JD, Cox G, Vincic L, Lombard CM, Loomas BE, Danek CJ. A prospective feasibility study of bronchial thermoplasty in the human airway. Chest 2005; 127:1999–2006.
- US Food and Drug Administration (FDA). Approval of Alair Bronchial Thermoplasty System: Alair Catheter and Alair RF Controller. 2010. www.accessdata.fda.gov/cdrh_docs/pdf8/P080032a.pdf. Accessed June 1, 2011.
- Cox G, Thomson NC, Rubin AS, et al; AIR Trial Study Group. Asthma control during the year after bronchial thermoplasty. N Engl J Med 2007; 356:1327–1337.
- Pavord ID, Cox G, Thomson NC, et al; RISA Trial Study Group. Safety and efficacy of bronchial thermoplasty in symptomatic, severe asthma. Am J Respir Crit Care Med 2007; 176:1185–1191.
- Castro M, Rubin AS, Laviolette M, et al; AIR2 Trial Study Group. Effectiveness and safety of bronchial thermoplasty in the treatment of severe asthma: a multicenter, randomized, double-blind, sham-controlled clinical trial. Am J Respir Crit Care Med 2010; 181:116–124.
- Centers for Disease Control and Prevention. Vital signs: asthma prevalence, disease characteristics, and self-management education—United States, 2001–2009. MMWR Morb Mortal Wkly Rep 2011; 60( 17):547–552.
- Guidelines for the diagnosis and management of asthma. National Heart, Lung, and Blood Institute. National Asthma Education Program. Expert Panel Report. J Allergy Clin Immunol 1991; 88:425–534.
- US Department of Health and Human Services. Expert panel report 2 (EPR-2): Guidelines for the diagnosis and management of asthma, 1997. www.nhlbi.nih.gov/guidelines/archives/epr-2/index.htm. Accessed June 1, 2011.
- US Department of Health and Human Services. Expert panel report: Guidelines for the diagnosis and management of asthma—Update on selected topics 2002. www.nhlbi.nih.gov/guidelines/archives/epr-2_upd/index.htm. Accessed June 1, 2011.
- Akinbami L. Asthma prevalence, health care use and mortality: United States 2003–05, CDC National Center for Health Statistics, 2006. www.cdc.gov/nchs/data/hestat/asthma03-05/asthma03-05.htm. Accessed June 1, 2011.
- US Department of Health and Human Services. Expert panel report 3 (EPR-3): Guidelines for the diagnosis and management of asthma full report, 2007. www.nhlbi.nih.gov/guidelines/asthma/asthgdln.htm. Accessed June 1, 2011.
- Proceedings of the ATS workshop on refractory asthma: current understanding, recommendations, and unanswered questions. American Thoracic Society Am J Respir Crit Care Med 2000; 162:2341–2351.
- Moore WC, Bleecker ER, Curran-Everett D, et al; National Heart, Lung, and Blood Institute’s Severe Asthma Research Program. Characterization of the severe asthma phenotype by the National Heart, Lung, and Blood Institute’s Severe Asthma Research Program. J Allergy Clin Immunol 2007; 119:405–413.
- Ambrogi MC, Fanucchi O, Lencioni R, Cioni R, Mussi A. Pulmonary radiofrequency ablation in a single lung patient. Thorax 2006; 61:828–829.
- Benussi S, Cini R, Gaynor SL, Alfieri O, Calafiore AM. Bipolar radiofrequency maze procedure through a transseptal approach. Ann Thorac Surg 2010; 90:1025–1027.
- Cox G, Miller JD, McWilliams A, Fitzgerald JM, Lam S. Bronchial thermoplasty for asthma. Am J Respir Crit Care Med 2006; 173:965–969.
- Cox PG, Miller J, Mitzner W, Leff AR. Radiofrequency ablation of airway smooth muscle for sustained treatment of asthma: preliminary investigations. Eur Respir J 2004; 24:659–663.
- Wise RA, Bartlett SJ, Brown ED, et al; American Lung Association Asthma Clinical Research Centers. Randomized trial of the effect of drug presentation on asthma outcomes: the American Lung Association Asthma Clinical Research Centers. J Allergy Clin Immunol 2009; 124:436–444.
- Castro M, Rubin A, Laviolette M, Hanania NA, Armstrong B, Cox G; AIR2 Trial Study Group. Persistence of effectiveness of bronchial thermoplasty in patients with severe asthma. Ann Allergy Asthma Immunol 2011. doi: 10.1016/j.anai.2011.03.005.
- Thomson NC, Rubin AS, Niven RM, et al; AIR Trial Study Group. Long-term (5 year) safety of bronchial thermoplasty: Asthma Intervention Research (AIR) trial. BMC Pulm Med 2011; 11:8.
- Solway J, Irvin CG. Airway smooth muscle as a target for asthma therapy. N Engl J Med 2007; 356:1367–1369.
- Ingram RH, McFadden ER. Localization and mechanisms of airway responses. N Engl J Med 1977; 297:596–600.
- Dyrda P, Tazzeo T, DoHarris L, et al. Acute response of airway muscle to extreme temperature includes disruption of actin-myosin interaction. Am J Respir Cell Mol Biol 2011; 44:213–221.
- Mayse ML, Laviolette M, Rubin AS, et al. Clinical pearls for bronchial thermoplasty. J Bronchol 2007; 14:115–123.
Asthma now has a new treatment, but it isn’t for everybody. Called bronchial thermoplasty, it is reserved for patients whose asthma is severe and refractory, as it involves three sessions of bronchoscopy, each lasting up to 1 hour, during which the smooth muscle layer is methodically ablated from the airway using radiofrequency energy.1,2
The US Food and Drug Administration (FDA) has approved bronchial thermoplasty,3 and although it does not cure asthma or completely eliminate its symptoms, patients with severe asthma that was not well controlled with medical therapy who underwent this procedure in clinical trials subsequently had fewer symptoms, enjoyed better quality of life, and needed less intensive health care (such as emergency room visits) than patients who did not undergo the procedure.4–6
Here, we present an overview of the pathophysiology of severe refractory asthma and the clinical trials of bronchial thermoplasty, its current protocols, and the status of this new treatment.
WHAT IS SEVERE REFRACTORY ASTHMA?
Asthma is a chronic inflammatory condition of the airways characterized by episodic symptoms of breathlessness, cough, and wheezing, which can wax and wane over time. Approximately 8.2% of the general population is affected.7
Our understanding of the pathophysiology of asthma has improved over the past 20 years, and with the publication of clinical guidelines from the National Asthma Education and Prevention Program in 1991,8 1997,9 and 2002,10 outcomes have improved. Most people with asthma can control their symptoms if they adhere to anti-inflammatory therapies and avoid triggers. Yet 5% to 10% of asthma patients have severe refractory disease, and asthma accounts for nearly half a million hospitalizations every year.11
The latest guidelines, published in 2007, emphasize the importance of assessing the severity of asthma, including the patient’s impairment (symptoms and limitations) and risk (likelihood of exacerbations).12
Workshop consensus definition of severe refractory asthma
A consensus group convened by the American Thoracic Society13 defined asthma as severe and refractory if the patient meets at least one of the following major criteria:
- Takes oral corticosteroids continuously or nearly continuously (> 50% of year)
- Takes high-dose inhaled corticosteroids.
In addition, the patient must meet at least two minor criteria, ie:
- Takes a controller medication such as a long-acting beta-agonist, theophylline, or a leukotriene antagonist every day
- Takes a short-acting beta agonist every day or nearly every day
- Has persistent airway obstruction, ie, a forced expiratory volume in 1 second (FEV1) less than 80% of predicted, or a peak expiratory flow that has a diurnal variability greater than 20%
- Has one or more urgent care visits for asthma per year
- Needs three or more oral corticosteroid “bursts” per year
- Has prompt deterioration when the dose of oral or inhaled corticosteroid is reduced by 25% or less
- Has had a near-fatal asthma event in the past.
Compared with people with mild asthma, people who have severe refractory asthma tend to be older, have fewer allergies, and make more use of intensive and urgent health care.14
Asthma is due to both inflammation and bronchoconstriction
The pathophysiology of asthma involves both chronic airway inflammation and bronchoconstriction, the latter characterized by a greater response to methacholine. Histologic findings include excessive mucus secretion, epithelial cell injury, and smooth muscle hypertrophy. These changes can lead to persistent airflow obstruction that can be difficult to control with medical therapies.12
Bronchoconstriction can be reversed temporarily with bronchodilators, but no longlasting therapy to reduce it has been available until now. Bronchial thermoplasty targets this gap in asthma management.
STUDIES OF BRONCHIAL THERMOPLASTY
Radiofrequency ablation has been used to treat other medical conditions such as lung cancer and cardiac arrhythmias.15,16 Its use to treat asthma by eradicating smooth muscle cells from the airway wall began with studies in animals.1 Later, studies were done in people without asthma,2 then in patients with mild to moderate asthma,17 and finally in patients with moderate to severe refractory asthma.4–6
These studies helped clarify which type of patients would be appropriate candidates and the outcomes to be anticipated, including adverse events.
Early studies
Danek et al,1 in a study in nonasthmatic dogs, found that thermoplasty at 65°C or 75°C (149°F or 167°F) attenuated the airway’s response to methacholine up to 3 years after treatment. As early as 1 week after treatment, airway smooth muscle was seen to be degenerating or absent, and the effect was inversely proportional to airway responsiveness.
Adverse effects of the procedure were cough, inflammatory edema of the airway wall, retained mucus, and blanching of the airway wall at the site of catheter contact. Three years later, there was no evidence of smooth muscle regeneration.
Miller et al2 next performed a feasibility study in eight patients, mean age 58 ± 8.3 years, who were scheduled to undergo lung resection for lung cancer. Five to 20 days before surgery, the investigators performed thermoplasty at 55°C or 65°C (131°F or 149°F) in three to nine sites per patient, 1 cm from known tumors but within areas to be resected. There were no significant adverse events such as hemoptysis, respiratory infections, or excess bronchial irritation.
A pilot study in mild to moderate asthma
Cox et al17 performed the first study of bronchial thermoplasty in patients with mild to moderate asthma. This was a prospective observational study in 16 patients who were younger than the patients in the previous study, with an average age of 30 years (range 24–58). They were given prednisone 30 or 50 mg the day before the procedure and on the day of the procedure. Three treatments were done, 3 weeks apart. The right middle lobe was not treated because the bronchus leading to it is relatively long and narrow, raising concern about damaging it.18
Results. The most frequent side effects were symptoms of airway irritation such as cough, dyspnea, wheezing, and bronchospasm. The mean time to onset was less than 1.7 days, and the mean time to resolution was 4.6 days after the most recent procedure. None of the patients needed to be hospitalized in the immediate postprocedure period.
In the 2 years after the procedure, there were 312 adverse events, mainly mild. Three (1%) of the adverse events were reported as severe, but they were deemed not related to the procedure. Yearly computed tomographic scans of the chest showed no structural changes such as bronchiectasis in the parenchyma or bronchial wall.
The FEV1 was higher at 12 weeks and at 1 year after thermoplasty than at baseline but was not significantly different from baseline at 2 years.
At baseline, the patients reported that 50% of their days were symptom-free; this increased to 73% at 12 weeks (P = .015).
In addition, airway hyperresponsiveness decreased significantly, and the effect persisted over 2 years. The provocative concentration of methacholine that caused a 20% reduction in FEV1 (the PC20) was:
- 0.92 mg/mL at baseline (95% confidence interval 0.42–1.99)
- 4.75 mg/mL at 12 weeks (2.51–8.85)
- 5.45 mg/mL at 1 year (1.54–19.32)
- 3.40 mg/mL at 2 years (1.35–8.52).
Limitations of this study include the relatively small number of patients enrolled and their relatively stable asthma.
The AIR trial: A randomized trial in moderate or persistent asthma
The first large multicenter trial of bronchial thermoplasty, the Asthma Intervention Research (AIR) trial,4 was prospective and randomized but not blinded. The aim was to determine whether bronchial thermoplasty would improve asthma control after long-acting beta agonists were discontinued.
Patients could be enrolled if they were 18 to 65 years old, had moderate or persistent asthma, and needed to take an inhaled corticosteroid (beclomethasone [Qvar] 200 μg or more or an equivalent drug) and a long-acting beta agonist (salmeterol [Serevent] 100 μg or more or an equivalent drug) every day. They also needed to have FEV1 values of 60% to 85% of predicted and airway reactivity (PC20 < 8 mg/mL), and their asthma had to have been stable for 6 weeks.
At baseline, the long-acting beta agonist was withdrawn temporarily; the final criterion for entry was that their asthma had to become worse when this was done.
Then, 112 patients were randomized to receive either bronchial thermoplasty with medical care (inhaled corticosteroids and long-acting beta agonists) or usual care, ie, medical therapy alone. Treatments were done in three sessions over 9 weeks, followed by attempts to discontinue their long-acting beta agonists at 3, 6, and 9 months after the procedure without exacerbations.
An exacerbation was defined as at least one of the following for 2 consecutive days: a reduction of peak flow by 20% of baseline average, the need for more than three additional puffs of rescue inhaler, or nocturnal awakenings caused by asthma symptoms. The patients kept a daily diary of their symptoms and rescue inhaler use, and they completed the Asthma Quality of Life Questionnaire (AQLQ) and the Asthma Control Questionnaire (ACQ).
Results. The number of mild (but not severe) exacerbations per week was significantly lower at 3 and 12 months than at baseline in the thermoplasty group, with 10 fewer mild exacerbations per patient per year, but was unchanged in the control group. There were significantly greater improvements in morning peak flow at 3, 6, and 12 months from baseline in the treatment group than in the usual-care group. Rescue medication use was also significantly less at 3 and 12 months. Symptom scores, AQLQ scores, and ACQ scores were all significantly better than at baseline as well.
Not surprisingly, in this cohort with unstable asthma, there were 407 adverse events in the treatment group and 106 adverse events in the control group. Most of these occurred within 1 day and resolved within 7 days after the procedure. There were more hospitalizations in the treatment group as well, for reasons that included exacerbations of asthma, collapse of the left lower lobe, and pleurisy.4
Therefore, this trial found that thermoplasty improved asthma symptoms within 3 months and that the effect lasted 1 year, with an encouraging reduction in the number of mild exacerbations. However, it was not blinded, and there is a strong placebo effect in asthma. Needed was a randomized trial in which the control group would undergo a sham treatment.
The RISA trial: A randomized trial in severe asthma
The Research in Severe Asthma (RISA) trial5 included patients with more severe asthma than those in the AIR trial. Entry criteria were:
- Taking high doses of an inhaled corticosteroid (> 750 μg of fluticasone or its equivalent per day)
- Taking prednisone (≤ 30 mg/day)
- An FEV1 of at least 50% of predicted without a bronchodilator
- A positive methacholine test.
Seventeen patients were randomized to undergo bronchial thermoplasty, and another 17 were randomized to receive medical treatment.
After a 2-week run-in period, the thermoplasty patients underwent three treatments, performed 3 weeks apart. For the next 16 weeks, the corticosteroid doses were kept stable in both groups, followed by a 14-week corticosteroid-weaning phase and then a 16-week reduced-corticosteroid phase. During this time, attempts were made to decrease the oral or inhaled corticosteroid doses according to a protocol (eg, a 20%–25% reduction every 2–4 weeks) unless there were mild exacerbations lasting more than 7 days.
Results. There were more adverse events in the thermoplasty group than in the medical management group in the treatment period, including seven hospitalizations for exacerbations of asthma and a partial collapse of the left lower lobe. There were no significant differences in adverse events between groups in the posttreatment period (up to 6 weeks after the last treatment). Forty-nine percent of the events were mild in each group; 10% of the events were severe in the thermoplasty group vs 4% in the control group.
During the steroid-stable phase, patients in the thermoplasty group used rescue inhalers significantly less than those in the control group, and their prebronchodilator FEV1 and AQLQ and ACQ scores were better. The differences in rescue inhaler use and questionnaire scores remained significant at 1 year.
Comment. As expected, serious adverse events occurred more often in patients with severe asthma in the treatment group than in the control group. However, 1 year after the procedure, the adverse-event rates were similar in the treatment and control groups, suggesting that this procedure can be safely performed in similar patient populations. Although there was significant potential for a placebo effect, these patients with severe persistent asthma showed significant improvement in clinical measures of asthma compared with the control group.
AIR2: A randomized, double-blind trial
The latest trial of this new therapy in severe asthma was the AIR2 trial.6 A major difference in its design compared with the earlier ones was that the control group underwent sham thermoplasty, allowing the trial to be truly double-blinded. (The bronchoscopy team knew which patients got which treatment, but the patients and the study physicians following them did not).
The primary outcome was the change in AQLQ score from baseline at 6, 9, and 12 months. Secondary outcomes included absolute changes in the asthma control scores, symptom scores, peak flows, rescue medication use, and FEV1.
The randomized groups (196 patients in the thermoplasty group and 101 in the sham treatment group) were well matched, and more than 80% in each group met the American Thoracic Society criteria for severe refractory asthma.
On the AQLQ, a change of more than 0.5 is considered clinically meaningful. Interestingly, there was a significant and clinically meaningful improvement in AQLQ in 64% of the sham treatment group, highlighting the placebo effect in asthma treatment.19 However, a larger proportion (79%) of the treated group had a clinically meaningful improvement on the AQLQ than in the sham treatment group.
Adverse events occurred in both groups; however, during the treatment phase, 16 patients in the bronchial thermoplasty group needed to be hospitalized for respiratory symptoms including worsening asthma, atelectasis, lower respiratory tract infections, decreased FEV1, and an aspirated tooth. One episode of hemoptysis required bronchial artery embolization. In contrast, only two patients in the sham treatment group needed hospitalization.
Therefore, this trial showed that patients with severe asthma treated with bronchial thermoplasty had a long-term improvement in quality of life and needed less health care.6
Translating these trials into practice
To summarize, these clinical trials showed that bronchial thermoplasty was feasible, was relatively safe, and produced better clinical outcomes in patients with severe asthma when medical therapies did not control their symptoms.
In practice, patient selection is likely to be important. A key question will be, Does the patient truly have severe refractory asthma, or is the patient not taking his or her medication? Adherence to therapy should be evaluated.
In addition, patients need to be observed and monitored closely during and after the treatment period, as airway complications and asthma exacerbations can occur up to 6 weeks after the last procedure. About 80% of all study patients had multiple symptoms of asthma and other symptoms in the treatment period. Rarely did these symptoms result in hospitalization, but they were more common in the treatment group in the AIR2 trial.
Long-term studies have evaluated the duration of effect and the safety of bronchial thermoplasty, and outcomes appear favorable.20,21
WHY DOES IT WORK?
The role of airway smooth muscle in asthma is yet to be fully elucidated. The trials outlined here showed that although asthma is a disease of the airways, including the small airways, treatment of airways 3 mm or larger improves asthma symptoms, quality of life, and health care utilization.6 Thus, the role of airway smooth muscle in asthma and as a target of therapy has not previously been fully realized.21
Early investigations into the mechanisms of airflow obstruction and airway resistance found that 75% of postnasal resistance occurs in the first six to eight generations (ie, branchings) of the airways, indicating that larger airways are involved.22 (The number of generations varies depending on the size of the person but it typically is 10 to 12.) Findings from the study in dogs introduced the idea that smooth muscle alterations contributed to the changes in airway resistance, and that subtle changes in airway smooth muscle could clinically benefit asthma patients.1
The speculated purpose of the airway smooth muscle layer is to support the airway, allow gas exchange, propel mucus for clearance, defend the airway, enhance cough, and promote lymphatic flow. However, the airway smooth muscle layer may also be vestigial. In asthma, airway smooth muscle adds to bronchoconstriction and hyperresponsiveness, and has a role in mediating inflammation and airway remodeling.21 No definitive studies have shown that eliminating airway smooth muscle greatly inhibits normal airway function.18
What exactly does thermoplasty do to the smooth muscle? Studies in smooth muscle from cows showed that high temperatures directly disrupt the actin-myosin interaction, likely through denaturation of motor proteins.23 This immediate loss of muscle cell function is not likely to be the result of apoptosis, autophagy, or necrosis, or mediated by heat-shock proteins, in view of the relatively quick muscle response and lack of progressive changes. Tissue responsiveness is substantially reduced a few seconds after application of 60°C of heat and is subsequently abolished within 5 minutes after treatment.23
The intervention appears to be dose-dependent. Responsiveness to cholinergic stimulation is lessened by treatment, and the desired effect is seen within seconds and does not progress.
Therefore, we can surmise that disruption of myosin function is likely the mechanism of the therapeutic effect, breaking the cascade of airway smooth muscle spasm. Now that we know about the airway smooth muscle as a possible target of therapy, and that it may play only a vestigial role, we can think about other therapies that focus on it.18,23
BRONCHIAL THERMOPLASTY PROTOCOLS
Patients are assessed before and on the day of the procedure to make sure their disease is stable (ie, their postbronchodilator FEV1 is within 15% of baseline values, and they have no evidence of asthma exacerbation or active infection), similar to the protocol used in the AIR2 trial,6 before proceeding with the treatment.
Patients are given 50 mg of prednisone 3 days before and again on the day of the procedure. Nebulized albuterol (2.5–5.0 mg) is given before the patients undergo screening spirometry and again before the procedure. If the preprocedure FEV1 is lower than 15% below baseline, we postpone the procedure to another day.
The procedure is performed with the patient under moderate conscious sedation, typically using fentanyl (Sublimaze), midazolam (Versed), and topical lidocaine in a monitored environment. The bronchoscope is inserted via either the mouth or nose, and supplemental oxygen is provided.
Thermoplasty is performed with the Alair system (Asthmatx, Inc., Sunnyvale, CA), which delivers a specific amount of radiofrequency (thermal) energy through a dedicated catheter. The catheter is deployed through a 2.0-mm channel of a flexible bronchoscope, starting in distal airways as small as 3 mm in diameter and working proximally to sequentially treat all airways to the mainstem lobar bronchi. The sites treated are meticulously recorded on a bronchial airway map to ensure that treatment sites are not skipped or overlapped (FIGURE 1).
An array of four electrodes is manually expanded to make contact with the airway walls; each electrode has 5 mm of exposed wire. As the energy is delivered, the control unit measures electrical resistance converted to thermal energy and turns off the current when an appropriate dosage is given. This thermal energy is what is responsible for altering the airway smooth muscle.
A full course of treatment requires three separate bronchoscopy sessions, each separated by 2 to 3 weeks. The left lower lobe and the right lower lobe are treated in separate procedures, and then both upper lobes are treated in a third procedure to minimize any respiratory symptoms. Each procedure usually requires 50 to 75 activations of the device and takes up to 60 minutes.
After each procedure the patient should be observed for 3 to 4 hours, and spirometry should be repeated to make sure the FEV1 (percent predicted) is within 20% of the baseline value. An additional 50-mg dose of prednisone is prescribed for the day after the procedure.24
FDA CLEARANCE AND LONG-TERM FOLLOW-UP
The FDA approved the Alair device for treating severe refractory asthma in early 2010.3 The indications for it are based on the study populations in the published trials. Patients can be evaluated for this treatment if they have well-documented severe persistent asthma not well controlled on inhaled corticosteroids and long-acting beta agonists and have no significant contraindications to bronchoscopy.
As part of the conditions of approval, the FDA required a postapproval study based on the long-term follow-up of the AIR2 trial. They specifically wanted to compare patients who have desirable long-term outcomes and those in whom any treatment effect wanes with time. Since we have only a few years of follow-up data, we still do not know all the possible late effects of the treatment; we have an opportunity to learn more.
Another question that needs to be studied is whether thermoplasty will help other forms of bronchospastic lung disease, such as chronic obstructive pulmonary disease.
A second postapproval study will be a prospective, open-label, single-arm, multicenter study conducted in the United States to assess the treatment effect and short-term and long-term safety profile of thermoplasty in asthma.
As experience with the procedure increases, we will be better able to characterize which patients may benefit from it. In addition, the knowledge gained by the longer-term study of airway smooth muscle function alterations will potentially drive the discovery of other innovative therapies for severe asthma.
Asthma now has a new treatment, but it isn’t for everybody. Called bronchial thermoplasty, it is reserved for patients whose asthma is severe and refractory, as it involves three sessions of bronchoscopy, each lasting up to 1 hour, during which the smooth muscle layer is methodically ablated from the airway using radiofrequency energy.1,2
The US Food and Drug Administration (FDA) has approved bronchial thermoplasty,3 and although it does not cure asthma or completely eliminate its symptoms, patients with severe asthma that was not well controlled with medical therapy who underwent this procedure in clinical trials subsequently had fewer symptoms, enjoyed better quality of life, and needed less intensive health care (such as emergency room visits) than patients who did not undergo the procedure.4–6
Here, we present an overview of the pathophysiology of severe refractory asthma and the clinical trials of bronchial thermoplasty, its current protocols, and the status of this new treatment.
WHAT IS SEVERE REFRACTORY ASTHMA?
Asthma is a chronic inflammatory condition of the airways characterized by episodic symptoms of breathlessness, cough, and wheezing, which can wax and wane over time. Approximately 8.2% of the general population is affected.7
Our understanding of the pathophysiology of asthma has improved over the past 20 years, and with the publication of clinical guidelines from the National Asthma Education and Prevention Program in 1991,8 1997,9 and 2002,10 outcomes have improved. Most people with asthma can control their symptoms if they adhere to anti-inflammatory therapies and avoid triggers. Yet 5% to 10% of asthma patients have severe refractory disease, and asthma accounts for nearly half a million hospitalizations every year.11
The latest guidelines, published in 2007, emphasize the importance of assessing the severity of asthma, including the patient’s impairment (symptoms and limitations) and risk (likelihood of exacerbations).12
Workshop consensus definition of severe refractory asthma
A consensus group convened by the American Thoracic Society13 defined asthma as severe and refractory if the patient meets at least one of the following major criteria:
- Takes oral corticosteroids continuously or nearly continuously (> 50% of year)
- Takes high-dose inhaled corticosteroids.
In addition, the patient must meet at least two minor criteria, ie:
- Takes a controller medication such as a long-acting beta-agonist, theophylline, or a leukotriene antagonist every day
- Takes a short-acting beta agonist every day or nearly every day
- Has persistent airway obstruction, ie, a forced expiratory volume in 1 second (FEV1) less than 80% of predicted, or a peak expiratory flow that has a diurnal variability greater than 20%
- Has one or more urgent care visits for asthma per year
- Needs three or more oral corticosteroid “bursts” per year
- Has prompt deterioration when the dose of oral or inhaled corticosteroid is reduced by 25% or less
- Has had a near-fatal asthma event in the past.
Compared with people with mild asthma, people who have severe refractory asthma tend to be older, have fewer allergies, and make more use of intensive and urgent health care.14
Asthma is due to both inflammation and bronchoconstriction
The pathophysiology of asthma involves both chronic airway inflammation and bronchoconstriction, the latter characterized by a greater response to methacholine. Histologic findings include excessive mucus secretion, epithelial cell injury, and smooth muscle hypertrophy. These changes can lead to persistent airflow obstruction that can be difficult to control with medical therapies.12
Bronchoconstriction can be reversed temporarily with bronchodilators, but no longlasting therapy to reduce it has been available until now. Bronchial thermoplasty targets this gap in asthma management.
STUDIES OF BRONCHIAL THERMOPLASTY
Radiofrequency ablation has been used to treat other medical conditions such as lung cancer and cardiac arrhythmias.15,16 Its use to treat asthma by eradicating smooth muscle cells from the airway wall began with studies in animals.1 Later, studies were done in people without asthma,2 then in patients with mild to moderate asthma,17 and finally in patients with moderate to severe refractory asthma.4–6
These studies helped clarify which type of patients would be appropriate candidates and the outcomes to be anticipated, including adverse events.
Early studies
Danek et al,1 in a study in nonasthmatic dogs, found that thermoplasty at 65°C or 75°C (149°F or 167°F) attenuated the airway’s response to methacholine up to 3 years after treatment. As early as 1 week after treatment, airway smooth muscle was seen to be degenerating or absent, and the effect was inversely proportional to airway responsiveness.
Adverse effects of the procedure were cough, inflammatory edema of the airway wall, retained mucus, and blanching of the airway wall at the site of catheter contact. Three years later, there was no evidence of smooth muscle regeneration.
Miller et al2 next performed a feasibility study in eight patients, mean age 58 ± 8.3 years, who were scheduled to undergo lung resection for lung cancer. Five to 20 days before surgery, the investigators performed thermoplasty at 55°C or 65°C (131°F or 149°F) in three to nine sites per patient, 1 cm from known tumors but within areas to be resected. There were no significant adverse events such as hemoptysis, respiratory infections, or excess bronchial irritation.
A pilot study in mild to moderate asthma
Cox et al17 performed the first study of bronchial thermoplasty in patients with mild to moderate asthma. This was a prospective observational study in 16 patients who were younger than the patients in the previous study, with an average age of 30 years (range 24–58). They were given prednisone 30 or 50 mg the day before the procedure and on the day of the procedure. Three treatments were done, 3 weeks apart. The right middle lobe was not treated because the bronchus leading to it is relatively long and narrow, raising concern about damaging it.18
Results. The most frequent side effects were symptoms of airway irritation such as cough, dyspnea, wheezing, and bronchospasm. The mean time to onset was less than 1.7 days, and the mean time to resolution was 4.6 days after the most recent procedure. None of the patients needed to be hospitalized in the immediate postprocedure period.
In the 2 years after the procedure, there were 312 adverse events, mainly mild. Three (1%) of the adverse events were reported as severe, but they were deemed not related to the procedure. Yearly computed tomographic scans of the chest showed no structural changes such as bronchiectasis in the parenchyma or bronchial wall.
The FEV1 was higher at 12 weeks and at 1 year after thermoplasty than at baseline but was not significantly different from baseline at 2 years.
At baseline, the patients reported that 50% of their days were symptom-free; this increased to 73% at 12 weeks (P = .015).
In addition, airway hyperresponsiveness decreased significantly, and the effect persisted over 2 years. The provocative concentration of methacholine that caused a 20% reduction in FEV1 (the PC20) was:
- 0.92 mg/mL at baseline (95% confidence interval 0.42–1.99)
- 4.75 mg/mL at 12 weeks (2.51–8.85)
- 5.45 mg/mL at 1 year (1.54–19.32)
- 3.40 mg/mL at 2 years (1.35–8.52).
Limitations of this study include the relatively small number of patients enrolled and their relatively stable asthma.
The AIR trial: A randomized trial in moderate or persistent asthma
The first large multicenter trial of bronchial thermoplasty, the Asthma Intervention Research (AIR) trial,4 was prospective and randomized but not blinded. The aim was to determine whether bronchial thermoplasty would improve asthma control after long-acting beta agonists were discontinued.
Patients could be enrolled if they were 18 to 65 years old, had moderate or persistent asthma, and needed to take an inhaled corticosteroid (beclomethasone [Qvar] 200 μg or more or an equivalent drug) and a long-acting beta agonist (salmeterol [Serevent] 100 μg or more or an equivalent drug) every day. They also needed to have FEV1 values of 60% to 85% of predicted and airway reactivity (PC20 < 8 mg/mL), and their asthma had to have been stable for 6 weeks.
At baseline, the long-acting beta agonist was withdrawn temporarily; the final criterion for entry was that their asthma had to become worse when this was done.
Then, 112 patients were randomized to receive either bronchial thermoplasty with medical care (inhaled corticosteroids and long-acting beta agonists) or usual care, ie, medical therapy alone. Treatments were done in three sessions over 9 weeks, followed by attempts to discontinue their long-acting beta agonists at 3, 6, and 9 months after the procedure without exacerbations.
An exacerbation was defined as at least one of the following for 2 consecutive days: a reduction of peak flow by 20% of baseline average, the need for more than three additional puffs of rescue inhaler, or nocturnal awakenings caused by asthma symptoms. The patients kept a daily diary of their symptoms and rescue inhaler use, and they completed the Asthma Quality of Life Questionnaire (AQLQ) and the Asthma Control Questionnaire (ACQ).
Results. The number of mild (but not severe) exacerbations per week was significantly lower at 3 and 12 months than at baseline in the thermoplasty group, with 10 fewer mild exacerbations per patient per year, but was unchanged in the control group. There were significantly greater improvements in morning peak flow at 3, 6, and 12 months from baseline in the treatment group than in the usual-care group. Rescue medication use was also significantly less at 3 and 12 months. Symptom scores, AQLQ scores, and ACQ scores were all significantly better than at baseline as well.
Not surprisingly, in this cohort with unstable asthma, there were 407 adverse events in the treatment group and 106 adverse events in the control group. Most of these occurred within 1 day and resolved within 7 days after the procedure. There were more hospitalizations in the treatment group as well, for reasons that included exacerbations of asthma, collapse of the left lower lobe, and pleurisy.4
Therefore, this trial found that thermoplasty improved asthma symptoms within 3 months and that the effect lasted 1 year, with an encouraging reduction in the number of mild exacerbations. However, it was not blinded, and there is a strong placebo effect in asthma. Needed was a randomized trial in which the control group would undergo a sham treatment.
The RISA trial: A randomized trial in severe asthma
The Research in Severe Asthma (RISA) trial5 included patients with more severe asthma than those in the AIR trial. Entry criteria were:
- Taking high doses of an inhaled corticosteroid (> 750 μg of fluticasone or its equivalent per day)
- Taking prednisone (≤ 30 mg/day)
- An FEV1 of at least 50% of predicted without a bronchodilator
- A positive methacholine test.
Seventeen patients were randomized to undergo bronchial thermoplasty, and another 17 were randomized to receive medical treatment.
After a 2-week run-in period, the thermoplasty patients underwent three treatments, performed 3 weeks apart. For the next 16 weeks, the corticosteroid doses were kept stable in both groups, followed by a 14-week corticosteroid-weaning phase and then a 16-week reduced-corticosteroid phase. During this time, attempts were made to decrease the oral or inhaled corticosteroid doses according to a protocol (eg, a 20%–25% reduction every 2–4 weeks) unless there were mild exacerbations lasting more than 7 days.
Results. There were more adverse events in the thermoplasty group than in the medical management group in the treatment period, including seven hospitalizations for exacerbations of asthma and a partial collapse of the left lower lobe. There were no significant differences in adverse events between groups in the posttreatment period (up to 6 weeks after the last treatment). Forty-nine percent of the events were mild in each group; 10% of the events were severe in the thermoplasty group vs 4% in the control group.
During the steroid-stable phase, patients in the thermoplasty group used rescue inhalers significantly less than those in the control group, and their prebronchodilator FEV1 and AQLQ and ACQ scores were better. The differences in rescue inhaler use and questionnaire scores remained significant at 1 year.
Comment. As expected, serious adverse events occurred more often in patients with severe asthma in the treatment group than in the control group. However, 1 year after the procedure, the adverse-event rates were similar in the treatment and control groups, suggesting that this procedure can be safely performed in similar patient populations. Although there was significant potential for a placebo effect, these patients with severe persistent asthma showed significant improvement in clinical measures of asthma compared with the control group.
AIR2: A randomized, double-blind trial
The latest trial of this new therapy in severe asthma was the AIR2 trial.6 A major difference in its design compared with the earlier ones was that the control group underwent sham thermoplasty, allowing the trial to be truly double-blinded. (The bronchoscopy team knew which patients got which treatment, but the patients and the study physicians following them did not).
The primary outcome was the change in AQLQ score from baseline at 6, 9, and 12 months. Secondary outcomes included absolute changes in the asthma control scores, symptom scores, peak flows, rescue medication use, and FEV1.
The randomized groups (196 patients in the thermoplasty group and 101 in the sham treatment group) were well matched, and more than 80% in each group met the American Thoracic Society criteria for severe refractory asthma.
On the AQLQ, a change of more than 0.5 is considered clinically meaningful. Interestingly, there was a significant and clinically meaningful improvement in AQLQ in 64% of the sham treatment group, highlighting the placebo effect in asthma treatment.19 However, a larger proportion (79%) of the treated group had a clinically meaningful improvement on the AQLQ than in the sham treatment group.
Adverse events occurred in both groups; however, during the treatment phase, 16 patients in the bronchial thermoplasty group needed to be hospitalized for respiratory symptoms including worsening asthma, atelectasis, lower respiratory tract infections, decreased FEV1, and an aspirated tooth. One episode of hemoptysis required bronchial artery embolization. In contrast, only two patients in the sham treatment group needed hospitalization.
Therefore, this trial showed that patients with severe asthma treated with bronchial thermoplasty had a long-term improvement in quality of life and needed less health care.6
Translating these trials into practice
To summarize, these clinical trials showed that bronchial thermoplasty was feasible, was relatively safe, and produced better clinical outcomes in patients with severe asthma when medical therapies did not control their symptoms.
In practice, patient selection is likely to be important. A key question will be, Does the patient truly have severe refractory asthma, or is the patient not taking his or her medication? Adherence to therapy should be evaluated.
In addition, patients need to be observed and monitored closely during and after the treatment period, as airway complications and asthma exacerbations can occur up to 6 weeks after the last procedure. About 80% of all study patients had multiple symptoms of asthma and other symptoms in the treatment period. Rarely did these symptoms result in hospitalization, but they were more common in the treatment group in the AIR2 trial.
Long-term studies have evaluated the duration of effect and the safety of bronchial thermoplasty, and outcomes appear favorable.20,21
WHY DOES IT WORK?
The role of airway smooth muscle in asthma is yet to be fully elucidated. The trials outlined here showed that although asthma is a disease of the airways, including the small airways, treatment of airways 3 mm or larger improves asthma symptoms, quality of life, and health care utilization.6 Thus, the role of airway smooth muscle in asthma and as a target of therapy has not previously been fully realized.21
Early investigations into the mechanisms of airflow obstruction and airway resistance found that 75% of postnasal resistance occurs in the first six to eight generations (ie, branchings) of the airways, indicating that larger airways are involved.22 (The number of generations varies depending on the size of the person but it typically is 10 to 12.) Findings from the study in dogs introduced the idea that smooth muscle alterations contributed to the changes in airway resistance, and that subtle changes in airway smooth muscle could clinically benefit asthma patients.1
The speculated purpose of the airway smooth muscle layer is to support the airway, allow gas exchange, propel mucus for clearance, defend the airway, enhance cough, and promote lymphatic flow. However, the airway smooth muscle layer may also be vestigial. In asthma, airway smooth muscle adds to bronchoconstriction and hyperresponsiveness, and has a role in mediating inflammation and airway remodeling.21 No definitive studies have shown that eliminating airway smooth muscle greatly inhibits normal airway function.18
What exactly does thermoplasty do to the smooth muscle? Studies in smooth muscle from cows showed that high temperatures directly disrupt the actin-myosin interaction, likely through denaturation of motor proteins.23 This immediate loss of muscle cell function is not likely to be the result of apoptosis, autophagy, or necrosis, or mediated by heat-shock proteins, in view of the relatively quick muscle response and lack of progressive changes. Tissue responsiveness is substantially reduced a few seconds after application of 60°C of heat and is subsequently abolished within 5 minutes after treatment.23
The intervention appears to be dose-dependent. Responsiveness to cholinergic stimulation is lessened by treatment, and the desired effect is seen within seconds and does not progress.
Therefore, we can surmise that disruption of myosin function is likely the mechanism of the therapeutic effect, breaking the cascade of airway smooth muscle spasm. Now that we know about the airway smooth muscle as a possible target of therapy, and that it may play only a vestigial role, we can think about other therapies that focus on it.18,23
BRONCHIAL THERMOPLASTY PROTOCOLS
Patients are assessed before and on the day of the procedure to make sure their disease is stable (ie, their postbronchodilator FEV1 is within 15% of baseline values, and they have no evidence of asthma exacerbation or active infection), similar to the protocol used in the AIR2 trial,6 before proceeding with the treatment.
Patients are given 50 mg of prednisone 3 days before and again on the day of the procedure. Nebulized albuterol (2.5–5.0 mg) is given before the patients undergo screening spirometry and again before the procedure. If the preprocedure FEV1 is lower than 15% below baseline, we postpone the procedure to another day.
The procedure is performed with the patient under moderate conscious sedation, typically using fentanyl (Sublimaze), midazolam (Versed), and topical lidocaine in a monitored environment. The bronchoscope is inserted via either the mouth or nose, and supplemental oxygen is provided.
Thermoplasty is performed with the Alair system (Asthmatx, Inc., Sunnyvale, CA), which delivers a specific amount of radiofrequency (thermal) energy through a dedicated catheter. The catheter is deployed through a 2.0-mm channel of a flexible bronchoscope, starting in distal airways as small as 3 mm in diameter and working proximally to sequentially treat all airways to the mainstem lobar bronchi. The sites treated are meticulously recorded on a bronchial airway map to ensure that treatment sites are not skipped or overlapped (FIGURE 1).
An array of four electrodes is manually expanded to make contact with the airway walls; each electrode has 5 mm of exposed wire. As the energy is delivered, the control unit measures electrical resistance converted to thermal energy and turns off the current when an appropriate dosage is given. This thermal energy is what is responsible for altering the airway smooth muscle.
A full course of treatment requires three separate bronchoscopy sessions, each separated by 2 to 3 weeks. The left lower lobe and the right lower lobe are treated in separate procedures, and then both upper lobes are treated in a third procedure to minimize any respiratory symptoms. Each procedure usually requires 50 to 75 activations of the device and takes up to 60 minutes.
After each procedure the patient should be observed for 3 to 4 hours, and spirometry should be repeated to make sure the FEV1 (percent predicted) is within 20% of the baseline value. An additional 50-mg dose of prednisone is prescribed for the day after the procedure.24
FDA CLEARANCE AND LONG-TERM FOLLOW-UP
The FDA approved the Alair device for treating severe refractory asthma in early 2010.3 The indications for it are based on the study populations in the published trials. Patients can be evaluated for this treatment if they have well-documented severe persistent asthma not well controlled on inhaled corticosteroids and long-acting beta agonists and have no significant contraindications to bronchoscopy.
As part of the conditions of approval, the FDA required a postapproval study based on the long-term follow-up of the AIR2 trial. They specifically wanted to compare patients who have desirable long-term outcomes and those in whom any treatment effect wanes with time. Since we have only a few years of follow-up data, we still do not know all the possible late effects of the treatment; we have an opportunity to learn more.
Another question that needs to be studied is whether thermoplasty will help other forms of bronchospastic lung disease, such as chronic obstructive pulmonary disease.
A second postapproval study will be a prospective, open-label, single-arm, multicenter study conducted in the United States to assess the treatment effect and short-term and long-term safety profile of thermoplasty in asthma.
As experience with the procedure increases, we will be better able to characterize which patients may benefit from it. In addition, the knowledge gained by the longer-term study of airway smooth muscle function alterations will potentially drive the discovery of other innovative therapies for severe asthma.
- Danek CJ, Lombard CM, Dungworth DL, et al. Reduction in airway hyperresponsiveness to methacholine by the application of RF energy in dogs. J Appl Physiol 2004; 97:1946–1953.
- Miller JD, Cox G, Vincic L, Lombard CM, Loomas BE, Danek CJ. A prospective feasibility study of bronchial thermoplasty in the human airway. Chest 2005; 127:1999–2006.
- US Food and Drug Administration (FDA). Approval of Alair Bronchial Thermoplasty System: Alair Catheter and Alair RF Controller. 2010. www.accessdata.fda.gov/cdrh_docs/pdf8/P080032a.pdf. Accessed June 1, 2011.
- Cox G, Thomson NC, Rubin AS, et al; AIR Trial Study Group. Asthma control during the year after bronchial thermoplasty. N Engl J Med 2007; 356:1327–1337.
- Pavord ID, Cox G, Thomson NC, et al; RISA Trial Study Group. Safety and efficacy of bronchial thermoplasty in symptomatic, severe asthma. Am J Respir Crit Care Med 2007; 176:1185–1191.
- Castro M, Rubin AS, Laviolette M, et al; AIR2 Trial Study Group. Effectiveness and safety of bronchial thermoplasty in the treatment of severe asthma: a multicenter, randomized, double-blind, sham-controlled clinical trial. Am J Respir Crit Care Med 2010; 181:116–124.
- Centers for Disease Control and Prevention. Vital signs: asthma prevalence, disease characteristics, and self-management education—United States, 2001–2009. MMWR Morb Mortal Wkly Rep 2011; 60( 17):547–552.
- Guidelines for the diagnosis and management of asthma. National Heart, Lung, and Blood Institute. National Asthma Education Program. Expert Panel Report. J Allergy Clin Immunol 1991; 88:425–534.
- US Department of Health and Human Services. Expert panel report 2 (EPR-2): Guidelines for the diagnosis and management of asthma, 1997. www.nhlbi.nih.gov/guidelines/archives/epr-2/index.htm. Accessed June 1, 2011.
- US Department of Health and Human Services. Expert panel report: Guidelines for the diagnosis and management of asthma—Update on selected topics 2002. www.nhlbi.nih.gov/guidelines/archives/epr-2_upd/index.htm. Accessed June 1, 2011.
- Akinbami L. Asthma prevalence, health care use and mortality: United States 2003–05, CDC National Center for Health Statistics, 2006. www.cdc.gov/nchs/data/hestat/asthma03-05/asthma03-05.htm. Accessed June 1, 2011.
- US Department of Health and Human Services. Expert panel report 3 (EPR-3): Guidelines for the diagnosis and management of asthma full report, 2007. www.nhlbi.nih.gov/guidelines/asthma/asthgdln.htm. Accessed June 1, 2011.
- Proceedings of the ATS workshop on refractory asthma: current understanding, recommendations, and unanswered questions. American Thoracic Society Am J Respir Crit Care Med 2000; 162:2341–2351.
- Moore WC, Bleecker ER, Curran-Everett D, et al; National Heart, Lung, and Blood Institute’s Severe Asthma Research Program. Characterization of the severe asthma phenotype by the National Heart, Lung, and Blood Institute’s Severe Asthma Research Program. J Allergy Clin Immunol 2007; 119:405–413.
- Ambrogi MC, Fanucchi O, Lencioni R, Cioni R, Mussi A. Pulmonary radiofrequency ablation in a single lung patient. Thorax 2006; 61:828–829.
- Benussi S, Cini R, Gaynor SL, Alfieri O, Calafiore AM. Bipolar radiofrequency maze procedure through a transseptal approach. Ann Thorac Surg 2010; 90:1025–1027.
- Cox G, Miller JD, McWilliams A, Fitzgerald JM, Lam S. Bronchial thermoplasty for asthma. Am J Respir Crit Care Med 2006; 173:965–969.
- Cox PG, Miller J, Mitzner W, Leff AR. Radiofrequency ablation of airway smooth muscle for sustained treatment of asthma: preliminary investigations. Eur Respir J 2004; 24:659–663.
- Wise RA, Bartlett SJ, Brown ED, et al; American Lung Association Asthma Clinical Research Centers. Randomized trial of the effect of drug presentation on asthma outcomes: the American Lung Association Asthma Clinical Research Centers. J Allergy Clin Immunol 2009; 124:436–444.
- Castro M, Rubin A, Laviolette M, Hanania NA, Armstrong B, Cox G; AIR2 Trial Study Group. Persistence of effectiveness of bronchial thermoplasty in patients with severe asthma. Ann Allergy Asthma Immunol 2011. doi: 10.1016/j.anai.2011.03.005.
- Thomson NC, Rubin AS, Niven RM, et al; AIR Trial Study Group. Long-term (5 year) safety of bronchial thermoplasty: Asthma Intervention Research (AIR) trial. BMC Pulm Med 2011; 11:8.
- Solway J, Irvin CG. Airway smooth muscle as a target for asthma therapy. N Engl J Med 2007; 356:1367–1369.
- Ingram RH, McFadden ER. Localization and mechanisms of airway responses. N Engl J Med 1977; 297:596–600.
- Dyrda P, Tazzeo T, DoHarris L, et al. Acute response of airway muscle to extreme temperature includes disruption of actin-myosin interaction. Am J Respir Cell Mol Biol 2011; 44:213–221.
- Mayse ML, Laviolette M, Rubin AS, et al. Clinical pearls for bronchial thermoplasty. J Bronchol 2007; 14:115–123.
- Danek CJ, Lombard CM, Dungworth DL, et al. Reduction in airway hyperresponsiveness to methacholine by the application of RF energy in dogs. J Appl Physiol 2004; 97:1946–1953.
- Miller JD, Cox G, Vincic L, Lombard CM, Loomas BE, Danek CJ. A prospective feasibility study of bronchial thermoplasty in the human airway. Chest 2005; 127:1999–2006.
- US Food and Drug Administration (FDA). Approval of Alair Bronchial Thermoplasty System: Alair Catheter and Alair RF Controller. 2010. www.accessdata.fda.gov/cdrh_docs/pdf8/P080032a.pdf. Accessed June 1, 2011.
- Cox G, Thomson NC, Rubin AS, et al; AIR Trial Study Group. Asthma control during the year after bronchial thermoplasty. N Engl J Med 2007; 356:1327–1337.
- Pavord ID, Cox G, Thomson NC, et al; RISA Trial Study Group. Safety and efficacy of bronchial thermoplasty in symptomatic, severe asthma. Am J Respir Crit Care Med 2007; 176:1185–1191.
- Castro M, Rubin AS, Laviolette M, et al; AIR2 Trial Study Group. Effectiveness and safety of bronchial thermoplasty in the treatment of severe asthma: a multicenter, randomized, double-blind, sham-controlled clinical trial. Am J Respir Crit Care Med 2010; 181:116–124.
- Centers for Disease Control and Prevention. Vital signs: asthma prevalence, disease characteristics, and self-management education—United States, 2001–2009. MMWR Morb Mortal Wkly Rep 2011; 60( 17):547–552.
- Guidelines for the diagnosis and management of asthma. National Heart, Lung, and Blood Institute. National Asthma Education Program. Expert Panel Report. J Allergy Clin Immunol 1991; 88:425–534.
- US Department of Health and Human Services. Expert panel report 2 (EPR-2): Guidelines for the diagnosis and management of asthma, 1997. www.nhlbi.nih.gov/guidelines/archives/epr-2/index.htm. Accessed June 1, 2011.
- US Department of Health and Human Services. Expert panel report: Guidelines for the diagnosis and management of asthma—Update on selected topics 2002. www.nhlbi.nih.gov/guidelines/archives/epr-2_upd/index.htm. Accessed June 1, 2011.
- Akinbami L. Asthma prevalence, health care use and mortality: United States 2003–05, CDC National Center for Health Statistics, 2006. www.cdc.gov/nchs/data/hestat/asthma03-05/asthma03-05.htm. Accessed June 1, 2011.
- US Department of Health and Human Services. Expert panel report 3 (EPR-3): Guidelines for the diagnosis and management of asthma full report, 2007. www.nhlbi.nih.gov/guidelines/asthma/asthgdln.htm. Accessed June 1, 2011.
- Proceedings of the ATS workshop on refractory asthma: current understanding, recommendations, and unanswered questions. American Thoracic Society Am J Respir Crit Care Med 2000; 162:2341–2351.
- Moore WC, Bleecker ER, Curran-Everett D, et al; National Heart, Lung, and Blood Institute’s Severe Asthma Research Program. Characterization of the severe asthma phenotype by the National Heart, Lung, and Blood Institute’s Severe Asthma Research Program. J Allergy Clin Immunol 2007; 119:405–413.
- Ambrogi MC, Fanucchi O, Lencioni R, Cioni R, Mussi A. Pulmonary radiofrequency ablation in a single lung patient. Thorax 2006; 61:828–829.
- Benussi S, Cini R, Gaynor SL, Alfieri O, Calafiore AM. Bipolar radiofrequency maze procedure through a transseptal approach. Ann Thorac Surg 2010; 90:1025–1027.
- Cox G, Miller JD, McWilliams A, Fitzgerald JM, Lam S. Bronchial thermoplasty for asthma. Am J Respir Crit Care Med 2006; 173:965–969.
- Cox PG, Miller J, Mitzner W, Leff AR. Radiofrequency ablation of airway smooth muscle for sustained treatment of asthma: preliminary investigations. Eur Respir J 2004; 24:659–663.
- Wise RA, Bartlett SJ, Brown ED, et al; American Lung Association Asthma Clinical Research Centers. Randomized trial of the effect of drug presentation on asthma outcomes: the American Lung Association Asthma Clinical Research Centers. J Allergy Clin Immunol 2009; 124:436–444.
- Castro M, Rubin A, Laviolette M, Hanania NA, Armstrong B, Cox G; AIR2 Trial Study Group. Persistence of effectiveness of bronchial thermoplasty in patients with severe asthma. Ann Allergy Asthma Immunol 2011. doi: 10.1016/j.anai.2011.03.005.
- Thomson NC, Rubin AS, Niven RM, et al; AIR Trial Study Group. Long-term (5 year) safety of bronchial thermoplasty: Asthma Intervention Research (AIR) trial. BMC Pulm Med 2011; 11:8.
- Solway J, Irvin CG. Airway smooth muscle as a target for asthma therapy. N Engl J Med 2007; 356:1367–1369.
- Ingram RH, McFadden ER. Localization and mechanisms of airway responses. N Engl J Med 1977; 297:596–600.
- Dyrda P, Tazzeo T, DoHarris L, et al. Acute response of airway muscle to extreme temperature includes disruption of actin-myosin interaction. Am J Respir Cell Mol Biol 2011; 44:213–221.
- Mayse ML, Laviolette M, Rubin AS, et al. Clinical pearls for bronchial thermoplasty. J Bronchol 2007; 14:115–123.
KEY POINTS
- Bronchial thermoplasty involves the application of radiofrequency energy to the airways distal to the mainstem bronchi down to airways as small as 3 mm in diameter.
- Treatments are done in three separate sessions, with careful monitoring before and after for respiratory complications that can occur in severe asthma. Airway complications and asthma exacerbations can occur up to 6 weeks after the last procedure, thus requiring close patient follow-up.
- In clinical trials, including a randomized trial in which the control group underwent sham thermoplasty, bronchial thermoplasty had an acceptable safety profile while improving asthma quality-of-life scores, symptoms, and health care utilization.