User login
No Easy Task
In college, while most of her fellow students were staying up late and sleeping in, Alice Marshbanks, MD, FHM, was an early riser. Now she regularly works from 4 p.m. to 2 a.m., and she sleeps in most mornings. "I’m sleeping later and living more of a teenage lifestyle," she jokes. "I’m actually getting younger."
Dr. Marshbanks might be an anomaly among established hospitalists. A physician since 1989 and a hospitalist since 1995, she actually prefers working the swing shift, and she says she’s the only one in her group at WakeMed Hospital in Raleigh, N.C., who does. Although Dr. Marshbanks is not a true nocturnist—she doesn’t work the typical 7 p.m. to 7 a.m. graveyard shift—her contracted position provides valuable transition coverage for night admissions, which have increased as the HM program at WakeMed has grown.
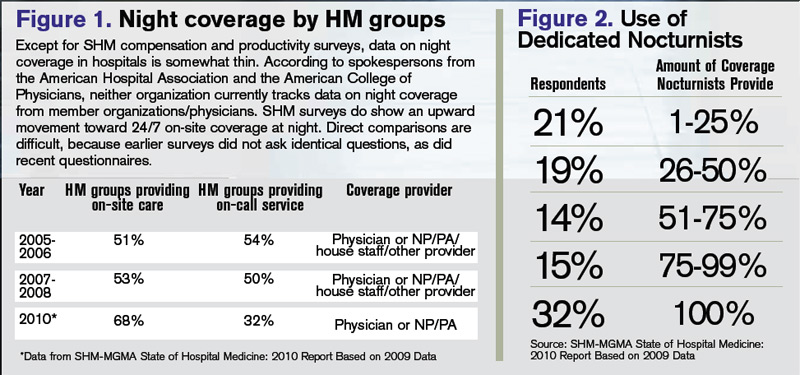
Surveys indicate that HM groups continue to move toward in-house coverage models to provide 24/7 hospitalist responsiveness. In the 2011 SHM-MGMA State of Hospital Medicine report, which will be released next month, 81% of responding nonteaching hospitalist practices reported providing on-site care at night. That’s up from 68% of responding HM practices that reported furnishing that service in the 2010 report. Only 53% of HM groups reported providing on-site night hospitalists in the 2007-2008 State of Hospital Medicine survey, which was produced solely by SHM.
Kenneth R. Epstein, MD, MBA, FACP, FHM, chief medical officer for Hospitalist Consultants Inc., headquartered in Traverse City, Mich., has observed this trend first-hand. In academic hospitals, due to new Accreditation Council for Graduate Medical Education (ACGME) and Resident Review Committee (RRC) regulations, "the only safety valve to handle admissions after the house staff numbers are capped is the hospitalist."
The need for such a safety valve will increase again this summer, as new ACGME duty-hour regulations on resident hours and supervision kick in.
Nonteaching hospitals are not exempt from these pressures. To deal with increasing demands for night coverage, HM groups across the country are using a variety of practice models, such as hiring dedicated nocturnists or moonlighters to cover nights, rotating shifts among team members, or using midlevel providers (physician assistants or nurse practitioners) as night staffers. On-call or in-house coverage models are determined by a variety of factors, including the size of the HM group, patient volume and acuity, and staff availability. Sustainability continues to be a challenge for most groups; however, the in-house coverage model seems to increase nursing and ED satisfaction, most experts say, and is an added value for hospital administration, although financial returns vary.
Continuity of care is at the heart of the night-coverage issue. Some experts worry that patient outcomes will suffer if there isn’t an in-house presence, but studies looking at this issue have been inconclusive, asserts Patti VanDort, RN, MSN, NEA-BC, vice president of nursing and chief nursing officer at Holland Hospital in southwestern Michigan.
"You’ve got to have the same level and quality of care during nights and weekends that you have during the weekdays," she says. "It’s got to be the same for all."
That said, some hospitals don’t have the volume to justify in-house night staffing. Hospitalists and program directors have described the ways in which they handle night staffing, balancing demand, program size, and physician satisfaction.
Tailored to Fit
"Hospitalist programs have different scale and scope depending on the needs of the institution," says Michael R. Humphrey, MD, vice president and chief clinical officer for Emergency and Ambulatory Services at St. Rita’s Medical Center in Lima, Ohio. A 365-bed community hospital, St. Rita’s employs nocturnists as part of its 24-hour hospitalist program. Dr. Humphrey still works as an ED physician and reports that the hospitalists are invaluable for admitting, providing cross-cover, covering the ICU, and handling code blue and rapid responses. "As a Level II trauma center, we can’t have ED physicians leave the department to run upstairs and do codes," he says. "They typically don’t get back within five minutes."
Holland Hospital, a 213-bed facility, provides around-the-clock hospitalist coverage in its eight-bed ICU, according to VanDort. That change was precipitated by the nursing staff’s decision to pursue Magnet Status, which was awarded in 2007 by the American Nurses Credentialing Center (ANCC). For inpatient coverage, the hospital-owned HM group Lakeshore Health Partners, headed by Bart D. Sak, MD, MA, FHM, maintains six FTE hospitalists on a rotating block schedule. Each night, one physician works from 4 p.m. until midnight, overlapping with a nonphysician provider (NPP), a member of the hospitalist group, who works a 7 p.m. to 7 a.m. shift.
"We have two providers in-house when admissions from the ED are heating up, and then we have an NPP in-house to cover the one to three additional admissions that may come in after midnight and to field floor calls," Dr. Sak says.
The physician who worked until midnight is on call for backup support and might come back to the hospital if things get too intense in the pre-dawn hours. "This arrangement works quite well for a program of our size," Dr. Sak says. "It takes a team-oriented approach and experienced NPPs who can work independently."
The Holland approach simply wouldn’t work at Kaiser Permanente’s East Bay site in Oakland, Calif., where Tom Baudendistel, MD, FACP, is part of a 50-member hospitalist group and director of the internal-medicine residency program. "Between codes, cross-cover, ICU, and floor admissions, there is simply too much acuity and volume," he says.
The peak hours for East Bay admissions are mid-afternoon to midnight. Two overnight hospitalist shifts (one from 8 a.m. to 8 p.m., another from 7 a.m. to 7 p.m.) are supplemented with two swing shifts (one from 2 to 10 p.m., another from 4 p.m. to midnight). Four full-time nocturnists cover 10 of the 14 overnight shifts per week, which allows for vacation and some protected administrative time. The balance of the overnight shifts are covered by the rest of the hospitalist group, which has 50 members.
The contracted nocturnists are incentivized with additional compensation at the end of the year, when the chief of hospitalists allocates bonuses. They also work fewer shifts a month than the other members of the group. "One thing our group agrees on is that the night docs should get a little more," Dr. Baudendistel says. "It’s a very fair tradeoff for everyone."
A Mile in Their Shoes
Medical directors must balance a variety of factors when scheduling around-the-clock coverage. From day one, the hospitalist program at Albany Memorial Hospital in New York, where John Krisa, MD, is medical director, has been an in-house 24/7 program. Dr. Krisa’s group uses per diem physicians or fellows on their days off to cover most of the nights. The other hospitalists on the team do not escape occasional night duty, and they cover what is left after plugging in the moonlighters. This leaves from zero to five nights per month for each full-time hospitalist. Even the medical director covers night shifts, something Dr. Krisa thinks is valuable to his leadership.
"You, as the leader, still have to walk a mile in that other person’s shoes," he says. "There are different challenges associated with both day and night shifts, so you have to appreciate what your colleagues are going through on the other shifts."
Hospitalist Consultants’ Dr. Epstein agrees with that concept.
"Whenever medical directors have personal experience of how the system is working, they are better able to recommend and make changes," he says.
It’s also valuable, Dr. Krisa explains, for the group leader to interact with ED staff and hear their concerns. Working night shifts helps avoid the night team versus day team schisms, which can lead to group disunity, he says.
Different Skill Set, Different Mindset?
The fact of the matter, though, is that pulling night shifts does not appeal to most established hospitalists. Sleep researchers have found that humans’ body clocks prefer office hours. Even if night-shift hours are consistent, those who work nights never really catch up on the sleep they need during the daytime.
Even so, some physicians embrace the graveyard shift. Working the night swing shift agrees with Dr. Marshbanks’ schedule. The hours are consistent, she works fewer shifts to qualify for FTE pay, and her shift is time-limited, as opposed to work-limited. She’s also filling a niche that others in her group eschew. "It’s a shift that most people with children don’t like because the hours are very disruptive to family life," she says.
The workload at night is different. Instead of the routine rounding typical in day shifts, her work is more urgent. She does more admissions because she works the busiest ED hours, covers acute-stroke codes, and provides cross-cover. And, she says, night staff tends to be "a solid group, so we interact more on a regular basis, since there are fewer of us."
The nocturnists at St. Rita’s Hospital are not held to the same meeting schedule as their daytime hospitalist colleagues, but they’re expected to read meeting minutes and to be responsible for any changes in guidelines or operational information, Dr. Humphrey says. Also stipulated in their hospitalist contracts is the requirement that they maintain competency in procedures, such as central-line placement and airway management.
What’s Better for Patients?
Experts have raised concerns that patient care can be compromised during off-hours, when staffing levels are reduced.1 The Leapfrog Group’s ICU Physician Safety (IPS) Standard argues for high-intensity ICU staffing to reduce patient mortality.2 A number of investigators have tried to determine whether patients admitted off-hours (weekends, nights, holidays) fare worse than those admitted during weekdays. Peter Cram, MD, MBA, acting director of the division of general internal medicine and associate professor of medicine at the Carver College of Medicine at the University of Iowa in Iowa City, found in a 2004 study that patients admitted to hospitals on weekends experienced slightly higher risk-adjusted mortality than did patients admitted on weekdays.3
But here’s the problem with studies such as this, says Dr. Cram: "Patients admitted on evenings and weekends are not the same as those admitted 9 to 5 on weekdays."
During weekdays, admissions combine patients with emergent issues and those scheduled for elective procedures. On weekends, "you get only emergencies—you don’t have low-risk patients," he points out. "So, even with optimal 24/7 staffing, you would still expect those patients coming in at night, and on holidays, to have worse outcomes because they are coming in with more acute problems. It remains an open question whether 24/7 staffing will improve off-hours outcomes." More research, Dr. Cram adds, is needed to establish whether full in-house staffing is the best solution.
Dr. Epstein has compared on-call versus in-house night staffing. In a 2007 study, he found no difference when using indicators such as length of stay, readmission rates, and patient satisfaction.4 However, he noticed positives from in-house coverage. "Although there are no data supporting the value of hospitalists on these parameters, having a nocturnist in-house increases nursing satisfaction, because they are responsive to pages when there is a question about a patient," he says. "It’s also a service to hospital medical staff, because they can handle rapid responses and codes."
There is some evidence that working nights can be deleterious to physicians’ and nurses’ health. One study found that interns were more likely to be involved in collisions after leaving extended night shifts; another found an increased risk of needle-stick injury at the end of a long night shift; and data from the long-running Nurses’ Health Study indicate that long-term night work can result in increased risk of colorectal and breast cancers.5,6,7,8 The increased risks of cancer could be related to lack of exposure to light at night and the body’s decreased production of melatonin, although this remains a topic of ongoing research.
"No Easy Answers"
VanDort, the nursing director, is "passionate" about having 24/7 coverage and reports that her nursing staff is happy with the hybrid model currently used at Holland Hospital. "I do envision a day when we’ll have physicians here around the clock," she says. "Patients are sick during the middle of the night, so you can’t staff your system one way during the daytime hours and your nighttime differently. It’s not fair to those patients."
Dr. Cram, who is a hospitalist, outcomes researcher, and division director, says that in an ideal world, it would make more business sense to have the hospital operating at full capacity around the clock, seven days a week. "But we don’t live in that world," he admits. "It is hard to find ways to achieve ’round-the-clock staffing at the levels we’d like."
He also concludes that there are "no easy answers" to the night-coverage conundrum. "But it might be prudent to think about incentives," he says. "Perhaps we should pay more for staffing weekends, evenings, and holidays, or we could reduce the annual number of shifts we expect our nocturnists to do, relative to those physicians who staff days."
Dr. Krisa says he, too, is biased toward an in-house coverage model, especially when programs reach a critical volume. "There is no substitute for the immediate ability to evaluate a sick patient," he explains. "My feeling is that an in-house, 24/7 presence will become the standard." TH
Gretchen Henkel is a freelance writer based in California.
References
- Wong HJ, Morra D. Excellent hospital care for all: open and operating 24/7. J Gen Intern Med. 2011.
- Pronovost PJ, Angus DC, Dorman T, Robinson KA, Dremsizov TT, Young TL. Physician staffing patterns and clinical outcomes in critically ill patients: a systemic review. JAMA. 2002;288(17):2151-2162.
- Cram P, Hillis SL, Barnett M, Rosenthal GE. Effects of weekend admission and hospital teaching status on in-hospital mortality. Am J Med. 2004;117(3):151-157.
- Epstein KR, Juarez E, Loya K, Gorman MJ, Singer A. The effect of 24-7 hospitalist coverage on clinical metrics. Presented May 2007, annual meeting, Society of Hospital Medicine, Dallas.
- Barger LK, Cade BE, Ayas NT, et al. Extended work shifts and the risk of motor vehicle crashes among interns. N Engl J Med. 2005;352:125-134.
- Ayas NT, Barger LK, Cade BE, et al. Extended work duration and the risk of self-reported percutaneous injuries in interns. JAMA. 2006;296(9):1055-1062.
- Schernhammer ES, Laden F, Speizer FE, et al. Night-shift work and risk of colorectal cancer in the nurses’ health study. J Natl Cancer Inst. 2003;95(11):825-828.
- Schernhammer ES, Laden F, Speizer FE, et al. Rotating night shifts and risk of breast cancer in women partici-pating in the nurses’ health study. J Natl Cancer Inst. 2001;93(20):1563-1568.
In college, while most of her fellow students were staying up late and sleeping in, Alice Marshbanks, MD, FHM, was an early riser. Now she regularly works from 4 p.m. to 2 a.m., and she sleeps in most mornings. "I’m sleeping later and living more of a teenage lifestyle," she jokes. "I’m actually getting younger."
Dr. Marshbanks might be an anomaly among established hospitalists. A physician since 1989 and a hospitalist since 1995, she actually prefers working the swing shift, and she says she’s the only one in her group at WakeMed Hospital in Raleigh, N.C., who does. Although Dr. Marshbanks is not a true nocturnist—she doesn’t work the typical 7 p.m. to 7 a.m. graveyard shift—her contracted position provides valuable transition coverage for night admissions, which have increased as the HM program at WakeMed has grown.
Surveys indicate that HM groups continue to move toward in-house coverage models to provide 24/7 hospitalist responsiveness. In the 2011 SHM-MGMA State of Hospital Medicine report, which will be released next month, 81% of responding nonteaching hospitalist practices reported providing on-site care at night. That’s up from 68% of responding HM practices that reported furnishing that service in the 2010 report. Only 53% of HM groups reported providing on-site night hospitalists in the 2007-2008 State of Hospital Medicine survey, which was produced solely by SHM.
Kenneth R. Epstein, MD, MBA, FACP, FHM, chief medical officer for Hospitalist Consultants Inc., headquartered in Traverse City, Mich., has observed this trend first-hand. In academic hospitals, due to new Accreditation Council for Graduate Medical Education (ACGME) and Resident Review Committee (RRC) regulations, "the only safety valve to handle admissions after the house staff numbers are capped is the hospitalist."
The need for such a safety valve will increase again this summer, as new ACGME duty-hour regulations on resident hours and supervision kick in.
Nonteaching hospitals are not exempt from these pressures. To deal with increasing demands for night coverage, HM groups across the country are using a variety of practice models, such as hiring dedicated nocturnists or moonlighters to cover nights, rotating shifts among team members, or using midlevel providers (physician assistants or nurse practitioners) as night staffers. On-call or in-house coverage models are determined by a variety of factors, including the size of the HM group, patient volume and acuity, and staff availability. Sustainability continues to be a challenge for most groups; however, the in-house coverage model seems to increase nursing and ED satisfaction, most experts say, and is an added value for hospital administration, although financial returns vary.
Continuity of care is at the heart of the night-coverage issue. Some experts worry that patient outcomes will suffer if there isn’t an in-house presence, but studies looking at this issue have been inconclusive, asserts Patti VanDort, RN, MSN, NEA-BC, vice president of nursing and chief nursing officer at Holland Hospital in southwestern Michigan.
"You’ve got to have the same level and quality of care during nights and weekends that you have during the weekdays," she says. "It’s got to be the same for all."
That said, some hospitals don’t have the volume to justify in-house night staffing. Hospitalists and program directors have described the ways in which they handle night staffing, balancing demand, program size, and physician satisfaction.
Tailored to Fit
"Hospitalist programs have different scale and scope depending on the needs of the institution," says Michael R. Humphrey, MD, vice president and chief clinical officer for Emergency and Ambulatory Services at St. Rita’s Medical Center in Lima, Ohio. A 365-bed community hospital, St. Rita’s employs nocturnists as part of its 24-hour hospitalist program. Dr. Humphrey still works as an ED physician and reports that the hospitalists are invaluable for admitting, providing cross-cover, covering the ICU, and handling code blue and rapid responses. "As a Level II trauma center, we can’t have ED physicians leave the department to run upstairs and do codes," he says. "They typically don’t get back within five minutes."
Holland Hospital, a 213-bed facility, provides around-the-clock hospitalist coverage in its eight-bed ICU, according to VanDort. That change was precipitated by the nursing staff’s decision to pursue Magnet Status, which was awarded in 2007 by the American Nurses Credentialing Center (ANCC). For inpatient coverage, the hospital-owned HM group Lakeshore Health Partners, headed by Bart D. Sak, MD, MA, FHM, maintains six FTE hospitalists on a rotating block schedule. Each night, one physician works from 4 p.m. until midnight, overlapping with a nonphysician provider (NPP), a member of the hospitalist group, who works a 7 p.m. to 7 a.m. shift.
"We have two providers in-house when admissions from the ED are heating up, and then we have an NPP in-house to cover the one to three additional admissions that may come in after midnight and to field floor calls," Dr. Sak says.
The physician who worked until midnight is on call for backup support and might come back to the hospital if things get too intense in the pre-dawn hours. "This arrangement works quite well for a program of our size," Dr. Sak says. "It takes a team-oriented approach and experienced NPPs who can work independently."
The Holland approach simply wouldn’t work at Kaiser Permanente’s East Bay site in Oakland, Calif., where Tom Baudendistel, MD, FACP, is part of a 50-member hospitalist group and director of the internal-medicine residency program. "Between codes, cross-cover, ICU, and floor admissions, there is simply too much acuity and volume," he says.
The peak hours for East Bay admissions are mid-afternoon to midnight. Two overnight hospitalist shifts (one from 8 a.m. to 8 p.m., another from 7 a.m. to 7 p.m.) are supplemented with two swing shifts (one from 2 to 10 p.m., another from 4 p.m. to midnight). Four full-time nocturnists cover 10 of the 14 overnight shifts per week, which allows for vacation and some protected administrative time. The balance of the overnight shifts are covered by the rest of the hospitalist group, which has 50 members.
The contracted nocturnists are incentivized with additional compensation at the end of the year, when the chief of hospitalists allocates bonuses. They also work fewer shifts a month than the other members of the group. "One thing our group agrees on is that the night docs should get a little more," Dr. Baudendistel says. "It’s a very fair tradeoff for everyone."
A Mile in Their Shoes
Medical directors must balance a variety of factors when scheduling around-the-clock coverage. From day one, the hospitalist program at Albany Memorial Hospital in New York, where John Krisa, MD, is medical director, has been an in-house 24/7 program. Dr. Krisa’s group uses per diem physicians or fellows on their days off to cover most of the nights. The other hospitalists on the team do not escape occasional night duty, and they cover what is left after plugging in the moonlighters. This leaves from zero to five nights per month for each full-time hospitalist. Even the medical director covers night shifts, something Dr. Krisa thinks is valuable to his leadership.
"You, as the leader, still have to walk a mile in that other person’s shoes," he says. "There are different challenges associated with both day and night shifts, so you have to appreciate what your colleagues are going through on the other shifts."
Hospitalist Consultants’ Dr. Epstein agrees with that concept.
"Whenever medical directors have personal experience of how the system is working, they are better able to recommend and make changes," he says.
It’s also valuable, Dr. Krisa explains, for the group leader to interact with ED staff and hear their concerns. Working night shifts helps avoid the night team versus day team schisms, which can lead to group disunity, he says.
Different Skill Set, Different Mindset?
The fact of the matter, though, is that pulling night shifts does not appeal to most established hospitalists. Sleep researchers have found that humans’ body clocks prefer office hours. Even if night-shift hours are consistent, those who work nights never really catch up on the sleep they need during the daytime.
Even so, some physicians embrace the graveyard shift. Working the night swing shift agrees with Dr. Marshbanks’ schedule. The hours are consistent, she works fewer shifts to qualify for FTE pay, and her shift is time-limited, as opposed to work-limited. She’s also filling a niche that others in her group eschew. "It’s a shift that most people with children don’t like because the hours are very disruptive to family life," she says.
The workload at night is different. Instead of the routine rounding typical in day shifts, her work is more urgent. She does more admissions because she works the busiest ED hours, covers acute-stroke codes, and provides cross-cover. And, she says, night staff tends to be "a solid group, so we interact more on a regular basis, since there are fewer of us."
The nocturnists at St. Rita’s Hospital are not held to the same meeting schedule as their daytime hospitalist colleagues, but they’re expected to read meeting minutes and to be responsible for any changes in guidelines or operational information, Dr. Humphrey says. Also stipulated in their hospitalist contracts is the requirement that they maintain competency in procedures, such as central-line placement and airway management.
What’s Better for Patients?
Experts have raised concerns that patient care can be compromised during off-hours, when staffing levels are reduced.1 The Leapfrog Group’s ICU Physician Safety (IPS) Standard argues for high-intensity ICU staffing to reduce patient mortality.2 A number of investigators have tried to determine whether patients admitted off-hours (weekends, nights, holidays) fare worse than those admitted during weekdays. Peter Cram, MD, MBA, acting director of the division of general internal medicine and associate professor of medicine at the Carver College of Medicine at the University of Iowa in Iowa City, found in a 2004 study that patients admitted to hospitals on weekends experienced slightly higher risk-adjusted mortality than did patients admitted on weekdays.3
But here’s the problem with studies such as this, says Dr. Cram: "Patients admitted on evenings and weekends are not the same as those admitted 9 to 5 on weekdays."
During weekdays, admissions combine patients with emergent issues and those scheduled for elective procedures. On weekends, "you get only emergencies—you don’t have low-risk patients," he points out. "So, even with optimal 24/7 staffing, you would still expect those patients coming in at night, and on holidays, to have worse outcomes because they are coming in with more acute problems. It remains an open question whether 24/7 staffing will improve off-hours outcomes." More research, Dr. Cram adds, is needed to establish whether full in-house staffing is the best solution.
Dr. Epstein has compared on-call versus in-house night staffing. In a 2007 study, he found no difference when using indicators such as length of stay, readmission rates, and patient satisfaction.4 However, he noticed positives from in-house coverage. "Although there are no data supporting the value of hospitalists on these parameters, having a nocturnist in-house increases nursing satisfaction, because they are responsive to pages when there is a question about a patient," he says. "It’s also a service to hospital medical staff, because they can handle rapid responses and codes."
There is some evidence that working nights can be deleterious to physicians’ and nurses’ health. One study found that interns were more likely to be involved in collisions after leaving extended night shifts; another found an increased risk of needle-stick injury at the end of a long night shift; and data from the long-running Nurses’ Health Study indicate that long-term night work can result in increased risk of colorectal and breast cancers.5,6,7,8 The increased risks of cancer could be related to lack of exposure to light at night and the body’s decreased production of melatonin, although this remains a topic of ongoing research.
"No Easy Answers"
VanDort, the nursing director, is "passionate" about having 24/7 coverage and reports that her nursing staff is happy with the hybrid model currently used at Holland Hospital. "I do envision a day when we’ll have physicians here around the clock," she says. "Patients are sick during the middle of the night, so you can’t staff your system one way during the daytime hours and your nighttime differently. It’s not fair to those patients."
Dr. Cram, who is a hospitalist, outcomes researcher, and division director, says that in an ideal world, it would make more business sense to have the hospital operating at full capacity around the clock, seven days a week. "But we don’t live in that world," he admits. "It is hard to find ways to achieve ’round-the-clock staffing at the levels we’d like."
He also concludes that there are "no easy answers" to the night-coverage conundrum. "But it might be prudent to think about incentives," he says. "Perhaps we should pay more for staffing weekends, evenings, and holidays, or we could reduce the annual number of shifts we expect our nocturnists to do, relative to those physicians who staff days."
Dr. Krisa says he, too, is biased toward an in-house coverage model, especially when programs reach a critical volume. "There is no substitute for the immediate ability to evaluate a sick patient," he explains. "My feeling is that an in-house, 24/7 presence will become the standard." TH
Gretchen Henkel is a freelance writer based in California.
References
- Wong HJ, Morra D. Excellent hospital care for all: open and operating 24/7. J Gen Intern Med. 2011.
- Pronovost PJ, Angus DC, Dorman T, Robinson KA, Dremsizov TT, Young TL. Physician staffing patterns and clinical outcomes in critically ill patients: a systemic review. JAMA. 2002;288(17):2151-2162.
- Cram P, Hillis SL, Barnett M, Rosenthal GE. Effects of weekend admission and hospital teaching status on in-hospital mortality. Am J Med. 2004;117(3):151-157.
- Epstein KR, Juarez E, Loya K, Gorman MJ, Singer A. The effect of 24-7 hospitalist coverage on clinical metrics. Presented May 2007, annual meeting, Society of Hospital Medicine, Dallas.
- Barger LK, Cade BE, Ayas NT, et al. Extended work shifts and the risk of motor vehicle crashes among interns. N Engl J Med. 2005;352:125-134.
- Ayas NT, Barger LK, Cade BE, et al. Extended work duration and the risk of self-reported percutaneous injuries in interns. JAMA. 2006;296(9):1055-1062.
- Schernhammer ES, Laden F, Speizer FE, et al. Night-shift work and risk of colorectal cancer in the nurses’ health study. J Natl Cancer Inst. 2003;95(11):825-828.
- Schernhammer ES, Laden F, Speizer FE, et al. Rotating night shifts and risk of breast cancer in women partici-pating in the nurses’ health study. J Natl Cancer Inst. 2001;93(20):1563-1568.
In college, while most of her fellow students were staying up late and sleeping in, Alice Marshbanks, MD, FHM, was an early riser. Now she regularly works from 4 p.m. to 2 a.m., and she sleeps in most mornings. "I’m sleeping later and living more of a teenage lifestyle," she jokes. "I’m actually getting younger."
Dr. Marshbanks might be an anomaly among established hospitalists. A physician since 1989 and a hospitalist since 1995, she actually prefers working the swing shift, and she says she’s the only one in her group at WakeMed Hospital in Raleigh, N.C., who does. Although Dr. Marshbanks is not a true nocturnist—she doesn’t work the typical 7 p.m. to 7 a.m. graveyard shift—her contracted position provides valuable transition coverage for night admissions, which have increased as the HM program at WakeMed has grown.
Surveys indicate that HM groups continue to move toward in-house coverage models to provide 24/7 hospitalist responsiveness. In the 2011 SHM-MGMA State of Hospital Medicine report, which will be released next month, 81% of responding nonteaching hospitalist practices reported providing on-site care at night. That’s up from 68% of responding HM practices that reported furnishing that service in the 2010 report. Only 53% of HM groups reported providing on-site night hospitalists in the 2007-2008 State of Hospital Medicine survey, which was produced solely by SHM.
Kenneth R. Epstein, MD, MBA, FACP, FHM, chief medical officer for Hospitalist Consultants Inc., headquartered in Traverse City, Mich., has observed this trend first-hand. In academic hospitals, due to new Accreditation Council for Graduate Medical Education (ACGME) and Resident Review Committee (RRC) regulations, "the only safety valve to handle admissions after the house staff numbers are capped is the hospitalist."
The need for such a safety valve will increase again this summer, as new ACGME duty-hour regulations on resident hours and supervision kick in.
Nonteaching hospitals are not exempt from these pressures. To deal with increasing demands for night coverage, HM groups across the country are using a variety of practice models, such as hiring dedicated nocturnists or moonlighters to cover nights, rotating shifts among team members, or using midlevel providers (physician assistants or nurse practitioners) as night staffers. On-call or in-house coverage models are determined by a variety of factors, including the size of the HM group, patient volume and acuity, and staff availability. Sustainability continues to be a challenge for most groups; however, the in-house coverage model seems to increase nursing and ED satisfaction, most experts say, and is an added value for hospital administration, although financial returns vary.
Continuity of care is at the heart of the night-coverage issue. Some experts worry that patient outcomes will suffer if there isn’t an in-house presence, but studies looking at this issue have been inconclusive, asserts Patti VanDort, RN, MSN, NEA-BC, vice president of nursing and chief nursing officer at Holland Hospital in southwestern Michigan.
"You’ve got to have the same level and quality of care during nights and weekends that you have during the weekdays," she says. "It’s got to be the same for all."
That said, some hospitals don’t have the volume to justify in-house night staffing. Hospitalists and program directors have described the ways in which they handle night staffing, balancing demand, program size, and physician satisfaction.
Tailored to Fit
"Hospitalist programs have different scale and scope depending on the needs of the institution," says Michael R. Humphrey, MD, vice president and chief clinical officer for Emergency and Ambulatory Services at St. Rita’s Medical Center in Lima, Ohio. A 365-bed community hospital, St. Rita’s employs nocturnists as part of its 24-hour hospitalist program. Dr. Humphrey still works as an ED physician and reports that the hospitalists are invaluable for admitting, providing cross-cover, covering the ICU, and handling code blue and rapid responses. "As a Level II trauma center, we can’t have ED physicians leave the department to run upstairs and do codes," he says. "They typically don’t get back within five minutes."
Holland Hospital, a 213-bed facility, provides around-the-clock hospitalist coverage in its eight-bed ICU, according to VanDort. That change was precipitated by the nursing staff’s decision to pursue Magnet Status, which was awarded in 2007 by the American Nurses Credentialing Center (ANCC). For inpatient coverage, the hospital-owned HM group Lakeshore Health Partners, headed by Bart D. Sak, MD, MA, FHM, maintains six FTE hospitalists on a rotating block schedule. Each night, one physician works from 4 p.m. until midnight, overlapping with a nonphysician provider (NPP), a member of the hospitalist group, who works a 7 p.m. to 7 a.m. shift.
"We have two providers in-house when admissions from the ED are heating up, and then we have an NPP in-house to cover the one to three additional admissions that may come in after midnight and to field floor calls," Dr. Sak says.
The physician who worked until midnight is on call for backup support and might come back to the hospital if things get too intense in the pre-dawn hours. "This arrangement works quite well for a program of our size," Dr. Sak says. "It takes a team-oriented approach and experienced NPPs who can work independently."
The Holland approach simply wouldn’t work at Kaiser Permanente’s East Bay site in Oakland, Calif., where Tom Baudendistel, MD, FACP, is part of a 50-member hospitalist group and director of the internal-medicine residency program. "Between codes, cross-cover, ICU, and floor admissions, there is simply too much acuity and volume," he says.
The peak hours for East Bay admissions are mid-afternoon to midnight. Two overnight hospitalist shifts (one from 8 a.m. to 8 p.m., another from 7 a.m. to 7 p.m.) are supplemented with two swing shifts (one from 2 to 10 p.m., another from 4 p.m. to midnight). Four full-time nocturnists cover 10 of the 14 overnight shifts per week, which allows for vacation and some protected administrative time. The balance of the overnight shifts are covered by the rest of the hospitalist group, which has 50 members.
The contracted nocturnists are incentivized with additional compensation at the end of the year, when the chief of hospitalists allocates bonuses. They also work fewer shifts a month than the other members of the group. "One thing our group agrees on is that the night docs should get a little more," Dr. Baudendistel says. "It’s a very fair tradeoff for everyone."
A Mile in Their Shoes
Medical directors must balance a variety of factors when scheduling around-the-clock coverage. From day one, the hospitalist program at Albany Memorial Hospital in New York, where John Krisa, MD, is medical director, has been an in-house 24/7 program. Dr. Krisa’s group uses per diem physicians or fellows on their days off to cover most of the nights. The other hospitalists on the team do not escape occasional night duty, and they cover what is left after plugging in the moonlighters. This leaves from zero to five nights per month for each full-time hospitalist. Even the medical director covers night shifts, something Dr. Krisa thinks is valuable to his leadership.
"You, as the leader, still have to walk a mile in that other person’s shoes," he says. "There are different challenges associated with both day and night shifts, so you have to appreciate what your colleagues are going through on the other shifts."
Hospitalist Consultants’ Dr. Epstein agrees with that concept.
"Whenever medical directors have personal experience of how the system is working, they are better able to recommend and make changes," he says.
It’s also valuable, Dr. Krisa explains, for the group leader to interact with ED staff and hear their concerns. Working night shifts helps avoid the night team versus day team schisms, which can lead to group disunity, he says.
Different Skill Set, Different Mindset?
The fact of the matter, though, is that pulling night shifts does not appeal to most established hospitalists. Sleep researchers have found that humans’ body clocks prefer office hours. Even if night-shift hours are consistent, those who work nights never really catch up on the sleep they need during the daytime.
Even so, some physicians embrace the graveyard shift. Working the night swing shift agrees with Dr. Marshbanks’ schedule. The hours are consistent, she works fewer shifts to qualify for FTE pay, and her shift is time-limited, as opposed to work-limited. She’s also filling a niche that others in her group eschew. "It’s a shift that most people with children don’t like because the hours are very disruptive to family life," she says.
The workload at night is different. Instead of the routine rounding typical in day shifts, her work is more urgent. She does more admissions because she works the busiest ED hours, covers acute-stroke codes, and provides cross-cover. And, she says, night staff tends to be "a solid group, so we interact more on a regular basis, since there are fewer of us."
The nocturnists at St. Rita’s Hospital are not held to the same meeting schedule as their daytime hospitalist colleagues, but they’re expected to read meeting minutes and to be responsible for any changes in guidelines or operational information, Dr. Humphrey says. Also stipulated in their hospitalist contracts is the requirement that they maintain competency in procedures, such as central-line placement and airway management.
What’s Better for Patients?
Experts have raised concerns that patient care can be compromised during off-hours, when staffing levels are reduced.1 The Leapfrog Group’s ICU Physician Safety (IPS) Standard argues for high-intensity ICU staffing to reduce patient mortality.2 A number of investigators have tried to determine whether patients admitted off-hours (weekends, nights, holidays) fare worse than those admitted during weekdays. Peter Cram, MD, MBA, acting director of the division of general internal medicine and associate professor of medicine at the Carver College of Medicine at the University of Iowa in Iowa City, found in a 2004 study that patients admitted to hospitals on weekends experienced slightly higher risk-adjusted mortality than did patients admitted on weekdays.3
But here’s the problem with studies such as this, says Dr. Cram: "Patients admitted on evenings and weekends are not the same as those admitted 9 to 5 on weekdays."
During weekdays, admissions combine patients with emergent issues and those scheduled for elective procedures. On weekends, "you get only emergencies—you don’t have low-risk patients," he points out. "So, even with optimal 24/7 staffing, you would still expect those patients coming in at night, and on holidays, to have worse outcomes because they are coming in with more acute problems. It remains an open question whether 24/7 staffing will improve off-hours outcomes." More research, Dr. Cram adds, is needed to establish whether full in-house staffing is the best solution.
Dr. Epstein has compared on-call versus in-house night staffing. In a 2007 study, he found no difference when using indicators such as length of stay, readmission rates, and patient satisfaction.4 However, he noticed positives from in-house coverage. "Although there are no data supporting the value of hospitalists on these parameters, having a nocturnist in-house increases nursing satisfaction, because they are responsive to pages when there is a question about a patient," he says. "It’s also a service to hospital medical staff, because they can handle rapid responses and codes."
There is some evidence that working nights can be deleterious to physicians’ and nurses’ health. One study found that interns were more likely to be involved in collisions after leaving extended night shifts; another found an increased risk of needle-stick injury at the end of a long night shift; and data from the long-running Nurses’ Health Study indicate that long-term night work can result in increased risk of colorectal and breast cancers.5,6,7,8 The increased risks of cancer could be related to lack of exposure to light at night and the body’s decreased production of melatonin, although this remains a topic of ongoing research.
"No Easy Answers"
VanDort, the nursing director, is "passionate" about having 24/7 coverage and reports that her nursing staff is happy with the hybrid model currently used at Holland Hospital. "I do envision a day when we’ll have physicians here around the clock," she says. "Patients are sick during the middle of the night, so you can’t staff your system one way during the daytime hours and your nighttime differently. It’s not fair to those patients."
Dr. Cram, who is a hospitalist, outcomes researcher, and division director, says that in an ideal world, it would make more business sense to have the hospital operating at full capacity around the clock, seven days a week. "But we don’t live in that world," he admits. "It is hard to find ways to achieve ’round-the-clock staffing at the levels we’d like."
He also concludes that there are "no easy answers" to the night-coverage conundrum. "But it might be prudent to think about incentives," he says. "Perhaps we should pay more for staffing weekends, evenings, and holidays, or we could reduce the annual number of shifts we expect our nocturnists to do, relative to those physicians who staff days."
Dr. Krisa says he, too, is biased toward an in-house coverage model, especially when programs reach a critical volume. "There is no substitute for the immediate ability to evaluate a sick patient," he explains. "My feeling is that an in-house, 24/7 presence will become the standard." TH
Gretchen Henkel is a freelance writer based in California.
References
- Wong HJ, Morra D. Excellent hospital care for all: open and operating 24/7. J Gen Intern Med. 2011.
- Pronovost PJ, Angus DC, Dorman T, Robinson KA, Dremsizov TT, Young TL. Physician staffing patterns and clinical outcomes in critically ill patients: a systemic review. JAMA. 2002;288(17):2151-2162.
- Cram P, Hillis SL, Barnett M, Rosenthal GE. Effects of weekend admission and hospital teaching status on in-hospital mortality. Am J Med. 2004;117(3):151-157.
- Epstein KR, Juarez E, Loya K, Gorman MJ, Singer A. The effect of 24-7 hospitalist coverage on clinical metrics. Presented May 2007, annual meeting, Society of Hospital Medicine, Dallas.
- Barger LK, Cade BE, Ayas NT, et al. Extended work shifts and the risk of motor vehicle crashes among interns. N Engl J Med. 2005;352:125-134.
- Ayas NT, Barger LK, Cade BE, et al. Extended work duration and the risk of self-reported percutaneous injuries in interns. JAMA. 2006;296(9):1055-1062.
- Schernhammer ES, Laden F, Speizer FE, et al. Night-shift work and risk of colorectal cancer in the nurses’ health study. J Natl Cancer Inst. 2003;95(11):825-828.
- Schernhammer ES, Laden F, Speizer FE, et al. Rotating night shifts and risk of breast cancer in women partici-pating in the nurses’ health study. J Natl Cancer Inst. 2001;93(20):1563-1568.
What’s Next for Hospital Medicine?
At the Medical University of South Carolina (MUSC) in Charleston, a familiar scene plays out in the hospitalist program. New hospitalists express an interest in a certain area and the university tries to accommodate them, making time for them to pursue additional training as they juggle the daily demands of treating patients, says Patrick Cawley, MD, MBA, SFHM, associate professor at the university and a former SHM president.
"We try to have a personal growth plan for each hospitalist that aligns with their interest," Dr. Cawley says. "So if we have a hospitalist that’s very, very interested in quality improvement, we’ll seek out opportunities to get that hospitalist experience, and start with smaller projects and then bigger projects."
As the field of HM hits a notable mark in its history—it’s been 15 years since the term "hospitalist" was coined—more advanced training will continue to emerge as a key issue and obstacle in the field, say experts who were asked to take a look into HM’s crystal ball.
They also predict continued growth of the field, with tens of thousands of new hospitalists emerging in the next decade or so. They also say that hospitalists will emerge as leaders in the application and use of new technology, and that there will be more demands placed on hospitalists to show their worth in hard data.
There also promises to be a growing presence of private management firms providing hospitalists to hospitals, which doctors both inside and outside of those firms say could have a beneficial effect on the overall quality of patient care.

-Patrick Cawley, MD, MBA, SFHM, associate professor, Medical University of South Carolina, Charleston, former SHM president
Father Time
For now, Dr. Cawley says, at MUSC and elsewhere, hospitalist programs are scrambling for time to enhance the skills needed to tend to increased demands.
"You have to carve out time. That’s literally what you have to do," he explains. "That’s expensive to take a doctor away from clinical service for a week, or an even an hour or two a week. I mean, somebody’s got to pay for that."
Training on hospitalist-specific management topics, he says, needs to evolve further. "I think there’s a recognition that this stuff is important and that hospitals and hospitalists need to get better aligned," he says. "This is something that will continue to mature over the next 10 years."
The range of tasks is growing ever broader for the hospitalist, and so the need for enhanced training is greater, says Larry Wellikson, MD, SFHM, CEO of SHM.
"They’re being asked to do bedside patient care, but they’re being asked to do more. They’re asked to be systems engineers, they’re asked to be safety experts, they’re asked to be the information manager, if you will, the IT guys," he says. "These skills they have not been trained to do and they need … either to say, ‘No, I can’t do that because I haven’t been trained,’ or they need to go and look where they can get that expertise.
"That’s what we try to do at SHM, with our Leadership Academy and our Practice Management Academy."
Frank Michota, MD, FHM, director of academic affairs in the Department of Hospital Medicine at The Cleveland Clinic, says that one of the biggest challenges the field needs to tackle over the next several years is to better standardize the education of hospitalists, saying there is "incredible inconsistency from hospitalist to hospitalist in terms of knowledge base, experience and … understanding the scope of practice."
"We continue to have significant variation in hospital practice models and the types of measurements that are available to those hospitalists for practice improvement," he says. "We continue to see significant turnover in the field with kind of a lack of maturity"—and not the kind of experience base that "you would like to see 15 years in."
"There is really no confidence that everyone at the base of that iceberg will ever make it to the tip because it’s still not viewed by many who entered the field as being a long-term career choice," Dr. Michota says. For many, he said, it is "a look-and-see proposition."
All of this, he says, points to the need for a full certification process by an HM board.
"I don’t want to make it sound like it has not been an impressive evolution to this point, but I think if we are going to meet the expectations, we do have to do more than we’re doing now," Dr. Michota says.
Some of the gaps in training might be able to be filled by private hospital management groups, which have training programs for their doctors that are made possible by their scale and whose presence is predicted to grow over the next 15 years.
Robert Bessler, MD, who in 2001 founded Tacoma, Wash.-based Sound Physicians, which has become one of the largest private hospitalist organizations in the country, says private companies are able to conduct training that is impossible for many hospitals to conduct themselves.
"You’re going to get good people who are all of good training and good knowledge, but they’re not all going to have experience," he says, "and so what are the hospitals that are employing 50% of the hospitalists in this country going to do about that? It’s pretty much nothing. They’re going to occasionally send some people to conferences and hope—because they don’t have that infrastructure."
At teaching institutes like those at private firms, the process is sped up, Dr. Bessler adds.
"That’s why we built our hospitalists’ institute at Sound—to turn really good, quality doctors into effective hospitalists in a much more rapid fashion," he says. "Because before we built this, it was just get them involved and hope after a couple of years they’ve really become efficient. Our hospital partners and the patients can’t wait that long."
Robert Reynolds, MD, founder of PrimeDoc, an Asheville, N.C.-based company that provides doctors for 12 hospitalist programs and employs about 100 doctors, says there needs to be more focus on teaching the "realistic side of the business of medicine," as well as on quality outcomes and patient satisfaction. But he also doubts there will be much change in training.
"[From] my cynical side and the voice of experience, I don’t see any change in the near future," he says. "What we’re seeing now is physicians come out of residency with a good clinical base, but really having no idea of how the healthcare system works in a bigger picture, how it works as an industry. So we’re having to spend a lot of time and effort training physicians to start thinking like practicing physicians."
The experts all agree that there will be an increase in hospitalists being provided by private corporations. Dr. Reynolds says that trend will continue in part due to healthcare reform’s emphasis on outcomes for reimbursement and a corporation’s ability to assist with physician training, as well as data and reporting needs.
"More and more hospital compensation and physician compensation is going to be based on actual data, performance data," he says. "And in order to really do a good job of capturing and reporting that kind of data, you need enough size to support an IT system and training systems that will produce and capture the kind of data that will be necessary."
Erin Fisher, MD, MHM, a pediatric hospitalist at Rady Children’s Hospital in San Diego, says a major goal of the future should be to change the reimbursement structure "so that you have something that is reasonable and encourages appropriate testing, treatments, and coordination of our healthcare system in a systematic way, rather than pieces." In such a system, hospitalists might see something to prompt them to intervene in a preventive way.
"The bigger question is, can our healthcare system, in five to 10 years, change itself enough that it uses every episode of care as an opportunity to do preventive care and coordinate care in the best way?" says Dr. Fisher, an SHM board member.
Continued Growth?
There is agreement that the field will continue to expand, with SHM predicting that the number of hospitalists in the U.S. will reach 40,000 in the next several years, up from today’s 30,000 figure.
Dr. Wellikson says that the figure could rise to as many as 70,000 or more if specialty hospitalists—such as surgical hospitalists, neuro-hospitalists, and laborists—are included. Those hospital-based specialties are now only in their infancy.
"Everything you can see shows that people are still flocking into hospital medicine," Dr. Wellikson adds.
Hospitalists numbered in the hundreds just 15 years ago, so growth has been explosive the past decade. Dr. Cawley, however, says the pace of growth might be starting to slow already, shifting to undeveloped or underserved areas. "Hospitalist programs are at almost all the large [hospitals] and really the growth has been at the smaller hospitals in the last several years," he says.
With the projected rise of Medicare beneficiaries due to the aging of the baby-boom generation, use of hospitals is expected to skyrocket, meaning more hospitalists will be needed, Dr. Bessler says. He also cites data from the National Rural Health Association noting that 25% of the U.S. population lives in areas considered rural, but that only 10% of the physicians live in those areas, indicating a potential growth area for hospitalists.
"That would tell me that demand will continue to outpace supply," he says.
Mike Tarwater, a member of the board of the American Hospital Association and CEO of Carolinas Medical Center in Charlotte, N.C., agrees with Dr. Bessler. Even with the move toward more outpatient care, Tarwater says, the aging of the population will mean a higher demand for hospitalists.

-Mike Tarwater, board member, American Hospital Association, CEO, Carolinas Medical Center, Charlotte, N.C.
"I think that the primary-care physicians—either because of their love for it or their belief that it’s the better way to go with the treatment of their patients—are going to be really stretched to keep that ambulatory practice going and to get to round on patients in the hospitals," he says. "I think there’s going to be a continued growth of the trend that we’ve seen over the last 15 years."
That growth also will mean a greater emphasis on technology use, whether it’s technology used for quick diagnostics like portable ultrasound or more widely used and refined electronic health records (EHR)—or, as Tarwater describes, "probably things we don’t imagine today."
"Our doctors, more than any other doctors, are tech-savvy; they’re early adopters," Dr. Wellikson says.
Hospitalists likely will emerge as leaders in the adoption of new technology, several experts predict.
Without a doubt, I think that hospitalists are going to be a driving force in the adaptation of the electronic [health] record to the clinical care within their hospitals," Dr. Michota says.
As the needs of HM grow, and the field grows more complex, there will inevitably be more divisions and departments of hospital medicine in places where it is now only a section, Dr. Cawley says.
"When you’re a division or a department, you have more autonomy over your own future, so I see this happening," he says. "I think more and more will carve themselves out of general internal medicine, and a lot of that will come because of a demand for more independence and greater autonomy." TH
Thomas R. Collins is a medical writer based in Florida.
More Value, More PATIENTs, More Technology
HM Pioneer Looks Into Crystal Ball

"We’re that camel’s nose."
With that in mind, what does Dr. Wachter predict for the next 15 years of HM? Here are his top three prognostications:
- A shift from the pressure to improve quality and safety to pressure to improve value, with more emphasis on cost and waste reduction.
"Hospitalist groups that are effective at [quality and safety] will continue to be popular in their organization, while hospitalist groups that aren’t will find that their standing is compromised," he says.
How that is done will vary from institution to institution. It will require a complex process of literature and creation of algorithms, he adds, "but also rolling up your sleeves and meeting with the right people, and working through the politics and diplomacy in order to get this work done."
- At teaching hospitals, a greater role for hospitalists to take care of patients who have traditionally been cared for by residents.
This is borne of the new Accreditation Council for Graduate Medical Education (ACGME) rules for residents’ work hours and supervision, which will require more hospitalists to fill voids in patient care and supervisory roles.
"If you like growth, that’s a great trend for hospitalists," Dr. Wachter says. "But if you’re in the business of trying to hire enough hospitalists to fill all your needs, it’s not that great because the demand curve is just tremendous; there’s a national shortage of hospitalists."
The trend also will affect what "a faculty job looks like," because there will be more clinical needs that will take time away from more traditionally academic work, he explains.
"Academic hospitals are not very good at creating satisfying, sustainable jobs that are largely clinical," Dr. Wachter says. "So how do you make sure that those people have fulfilling jobs, that they don’t feel like second-class citizens, that they can get promoted if they do what you’ve asked them to do well? I think that’s a huge challenge for the field, but it is a challenge borne of a new imperative."
- A revolutionary move from a pen-and-paper hospital environment to a technology-driven workplace.
"It will change the way we do our work," Dr. Wachter says, adding it will also mean other, more subtle changes.
"It takes away the importance of geography to some extent," he adds. "I can be off in the doctor’s lounge or in my house and still do my work.
"And so how do you retain or enhance the relationships that are so fundamental to providing good care? That’s not only between doctors and patients and families, but also between doctors and nurses, and each other."-TC
At the Medical University of South Carolina (MUSC) in Charleston, a familiar scene plays out in the hospitalist program. New hospitalists express an interest in a certain area and the university tries to accommodate them, making time for them to pursue additional training as they juggle the daily demands of treating patients, says Patrick Cawley, MD, MBA, SFHM, associate professor at the university and a former SHM president.
"We try to have a personal growth plan for each hospitalist that aligns with their interest," Dr. Cawley says. "So if we have a hospitalist that’s very, very interested in quality improvement, we’ll seek out opportunities to get that hospitalist experience, and start with smaller projects and then bigger projects."
As the field of HM hits a notable mark in its history—it’s been 15 years since the term "hospitalist" was coined—more advanced training will continue to emerge as a key issue and obstacle in the field, say experts who were asked to take a look into HM’s crystal ball.
They also predict continued growth of the field, with tens of thousands of new hospitalists emerging in the next decade or so. They also say that hospitalists will emerge as leaders in the application and use of new technology, and that there will be more demands placed on hospitalists to show their worth in hard data.
There also promises to be a growing presence of private management firms providing hospitalists to hospitals, which doctors both inside and outside of those firms say could have a beneficial effect on the overall quality of patient care.
-Patrick Cawley, MD, MBA, SFHM, associate professor, Medical University of South Carolina, Charleston, former SHM president
Father Time
For now, Dr. Cawley says, at MUSC and elsewhere, hospitalist programs are scrambling for time to enhance the skills needed to tend to increased demands.
"You have to carve out time. That’s literally what you have to do," he explains. "That’s expensive to take a doctor away from clinical service for a week, or an even an hour or two a week. I mean, somebody’s got to pay for that."
Training on hospitalist-specific management topics, he says, needs to evolve further. "I think there’s a recognition that this stuff is important and that hospitals and hospitalists need to get better aligned," he says. "This is something that will continue to mature over the next 10 years."
The range of tasks is growing ever broader for the hospitalist, and so the need for enhanced training is greater, says Larry Wellikson, MD, SFHM, CEO of SHM.
"They’re being asked to do bedside patient care, but they’re being asked to do more. They’re asked to be systems engineers, they’re asked to be safety experts, they’re asked to be the information manager, if you will, the IT guys," he says. "These skills they have not been trained to do and they need … either to say, ‘No, I can’t do that because I haven’t been trained,’ or they need to go and look where they can get that expertise.
"That’s what we try to do at SHM, with our Leadership Academy and our Practice Management Academy."
Frank Michota, MD, FHM, director of academic affairs in the Department of Hospital Medicine at The Cleveland Clinic, says that one of the biggest challenges the field needs to tackle over the next several years is to better standardize the education of hospitalists, saying there is "incredible inconsistency from hospitalist to hospitalist in terms of knowledge base, experience and … understanding the scope of practice."
"We continue to have significant variation in hospital practice models and the types of measurements that are available to those hospitalists for practice improvement," he says. "We continue to see significant turnover in the field with kind of a lack of maturity"—and not the kind of experience base that "you would like to see 15 years in."
"There is really no confidence that everyone at the base of that iceberg will ever make it to the tip because it’s still not viewed by many who entered the field as being a long-term career choice," Dr. Michota says. For many, he said, it is "a look-and-see proposition."
All of this, he says, points to the need for a full certification process by an HM board.
"I don’t want to make it sound like it has not been an impressive evolution to this point, but I think if we are going to meet the expectations, we do have to do more than we’re doing now," Dr. Michota says.
Some of the gaps in training might be able to be filled by private hospital management groups, which have training programs for their doctors that are made possible by their scale and whose presence is predicted to grow over the next 15 years.
Robert Bessler, MD, who in 2001 founded Tacoma, Wash.-based Sound Physicians, which has become one of the largest private hospitalist organizations in the country, says private companies are able to conduct training that is impossible for many hospitals to conduct themselves.
"You’re going to get good people who are all of good training and good knowledge, but they’re not all going to have experience," he says, "and so what are the hospitals that are employing 50% of the hospitalists in this country going to do about that? It’s pretty much nothing. They’re going to occasionally send some people to conferences and hope—because they don’t have that infrastructure."
At teaching institutes like those at private firms, the process is sped up, Dr. Bessler adds.
"That’s why we built our hospitalists’ institute at Sound—to turn really good, quality doctors into effective hospitalists in a much more rapid fashion," he says. "Because before we built this, it was just get them involved and hope after a couple of years they’ve really become efficient. Our hospital partners and the patients can’t wait that long."
Robert Reynolds, MD, founder of PrimeDoc, an Asheville, N.C.-based company that provides doctors for 12 hospitalist programs and employs about 100 doctors, says there needs to be more focus on teaching the "realistic side of the business of medicine," as well as on quality outcomes and patient satisfaction. But he also doubts there will be much change in training.
"[From] my cynical side and the voice of experience, I don’t see any change in the near future," he says. "What we’re seeing now is physicians come out of residency with a good clinical base, but really having no idea of how the healthcare system works in a bigger picture, how it works as an industry. So we’re having to spend a lot of time and effort training physicians to start thinking like practicing physicians."
The experts all agree that there will be an increase in hospitalists being provided by private corporations. Dr. Reynolds says that trend will continue in part due to healthcare reform’s emphasis on outcomes for reimbursement and a corporation’s ability to assist with physician training, as well as data and reporting needs.
"More and more hospital compensation and physician compensation is going to be based on actual data, performance data," he says. "And in order to really do a good job of capturing and reporting that kind of data, you need enough size to support an IT system and training systems that will produce and capture the kind of data that will be necessary."
Erin Fisher, MD, MHM, a pediatric hospitalist at Rady Children’s Hospital in San Diego, says a major goal of the future should be to change the reimbursement structure "so that you have something that is reasonable and encourages appropriate testing, treatments, and coordination of our healthcare system in a systematic way, rather than pieces." In such a system, hospitalists might see something to prompt them to intervene in a preventive way.
"The bigger question is, can our healthcare system, in five to 10 years, change itself enough that it uses every episode of care as an opportunity to do preventive care and coordinate care in the best way?" says Dr. Fisher, an SHM board member.
Continued Growth?
There is agreement that the field will continue to expand, with SHM predicting that the number of hospitalists in the U.S. will reach 40,000 in the next several years, up from today’s 30,000 figure.
Dr. Wellikson says that the figure could rise to as many as 70,000 or more if specialty hospitalists—such as surgical hospitalists, neuro-hospitalists, and laborists—are included. Those hospital-based specialties are now only in their infancy.
"Everything you can see shows that people are still flocking into hospital medicine," Dr. Wellikson adds.
Hospitalists numbered in the hundreds just 15 years ago, so growth has been explosive the past decade. Dr. Cawley, however, says the pace of growth might be starting to slow already, shifting to undeveloped or underserved areas. "Hospitalist programs are at almost all the large [hospitals] and really the growth has been at the smaller hospitals in the last several years," he says.
With the projected rise of Medicare beneficiaries due to the aging of the baby-boom generation, use of hospitals is expected to skyrocket, meaning more hospitalists will be needed, Dr. Bessler says. He also cites data from the National Rural Health Association noting that 25% of the U.S. population lives in areas considered rural, but that only 10% of the physicians live in those areas, indicating a potential growth area for hospitalists.
"That would tell me that demand will continue to outpace supply," he says.
Mike Tarwater, a member of the board of the American Hospital Association and CEO of Carolinas Medical Center in Charlotte, N.C., agrees with Dr. Bessler. Even with the move toward more outpatient care, Tarwater says, the aging of the population will mean a higher demand for hospitalists.
-Mike Tarwater, board member, American Hospital Association, CEO, Carolinas Medical Center, Charlotte, N.C.
"I think that the primary-care physicians—either because of their love for it or their belief that it’s the better way to go with the treatment of their patients—are going to be really stretched to keep that ambulatory practice going and to get to round on patients in the hospitals," he says. "I think there’s going to be a continued growth of the trend that we’ve seen over the last 15 years."
That growth also will mean a greater emphasis on technology use, whether it’s technology used for quick diagnostics like portable ultrasound or more widely used and refined electronic health records (EHR)—or, as Tarwater describes, "probably things we don’t imagine today."
"Our doctors, more than any other doctors, are tech-savvy; they’re early adopters," Dr. Wellikson says.
Hospitalists likely will emerge as leaders in the adoption of new technology, several experts predict.
Without a doubt, I think that hospitalists are going to be a driving force in the adaptation of the electronic [health] record to the clinical care within their hospitals," Dr. Michota says.
As the needs of HM grow, and the field grows more complex, there will inevitably be more divisions and departments of hospital medicine in places where it is now only a section, Dr. Cawley says.
"When you’re a division or a department, you have more autonomy over your own future, so I see this happening," he says. "I think more and more will carve themselves out of general internal medicine, and a lot of that will come because of a demand for more independence and greater autonomy." TH
Thomas R. Collins is a medical writer based in Florida.
More Value, More PATIENTs, More Technology
HM Pioneer Looks Into Crystal Ball
"We’re that camel’s nose."
With that in mind, what does Dr. Wachter predict for the next 15 years of HM? Here are his top three prognostications:
- A shift from the pressure to improve quality and safety to pressure to improve value, with more emphasis on cost and waste reduction.
"Hospitalist groups that are effective at [quality and safety] will continue to be popular in their organization, while hospitalist groups that aren’t will find that their standing is compromised," he says.
How that is done will vary from institution to institution. It will require a complex process of literature and creation of algorithms, he adds, "but also rolling up your sleeves and meeting with the right people, and working through the politics and diplomacy in order to get this work done."
- At teaching hospitals, a greater role for hospitalists to take care of patients who have traditionally been cared for by residents.
This is borne of the new Accreditation Council for Graduate Medical Education (ACGME) rules for residents’ work hours and supervision, which will require more hospitalists to fill voids in patient care and supervisory roles.
"If you like growth, that’s a great trend for hospitalists," Dr. Wachter says. "But if you’re in the business of trying to hire enough hospitalists to fill all your needs, it’s not that great because the demand curve is just tremendous; there’s a national shortage of hospitalists."
The trend also will affect what "a faculty job looks like," because there will be more clinical needs that will take time away from more traditionally academic work, he explains.
"Academic hospitals are not very good at creating satisfying, sustainable jobs that are largely clinical," Dr. Wachter says. "So how do you make sure that those people have fulfilling jobs, that they don’t feel like second-class citizens, that they can get promoted if they do what you’ve asked them to do well? I think that’s a huge challenge for the field, but it is a challenge borne of a new imperative."
- A revolutionary move from a pen-and-paper hospital environment to a technology-driven workplace.
"It will change the way we do our work," Dr. Wachter says, adding it will also mean other, more subtle changes.
"It takes away the importance of geography to some extent," he adds. "I can be off in the doctor’s lounge or in my house and still do my work.
"And so how do you retain or enhance the relationships that are so fundamental to providing good care? That’s not only between doctors and patients and families, but also between doctors and nurses, and each other."-TC
At the Medical University of South Carolina (MUSC) in Charleston, a familiar scene plays out in the hospitalist program. New hospitalists express an interest in a certain area and the university tries to accommodate them, making time for them to pursue additional training as they juggle the daily demands of treating patients, says Patrick Cawley, MD, MBA, SFHM, associate professor at the university and a former SHM president.
"We try to have a personal growth plan for each hospitalist that aligns with their interest," Dr. Cawley says. "So if we have a hospitalist that’s very, very interested in quality improvement, we’ll seek out opportunities to get that hospitalist experience, and start with smaller projects and then bigger projects."
As the field of HM hits a notable mark in its history—it’s been 15 years since the term "hospitalist" was coined—more advanced training will continue to emerge as a key issue and obstacle in the field, say experts who were asked to take a look into HM’s crystal ball.
They also predict continued growth of the field, with tens of thousands of new hospitalists emerging in the next decade or so. They also say that hospitalists will emerge as leaders in the application and use of new technology, and that there will be more demands placed on hospitalists to show their worth in hard data.
There also promises to be a growing presence of private management firms providing hospitalists to hospitals, which doctors both inside and outside of those firms say could have a beneficial effect on the overall quality of patient care.
-Patrick Cawley, MD, MBA, SFHM, associate professor, Medical University of South Carolina, Charleston, former SHM president
Father Time
For now, Dr. Cawley says, at MUSC and elsewhere, hospitalist programs are scrambling for time to enhance the skills needed to tend to increased demands.
"You have to carve out time. That’s literally what you have to do," he explains. "That’s expensive to take a doctor away from clinical service for a week, or an even an hour or two a week. I mean, somebody’s got to pay for that."
Training on hospitalist-specific management topics, he says, needs to evolve further. "I think there’s a recognition that this stuff is important and that hospitals and hospitalists need to get better aligned," he says. "This is something that will continue to mature over the next 10 years."
The range of tasks is growing ever broader for the hospitalist, and so the need for enhanced training is greater, says Larry Wellikson, MD, SFHM, CEO of SHM.
"They’re being asked to do bedside patient care, but they’re being asked to do more. They’re asked to be systems engineers, they’re asked to be safety experts, they’re asked to be the information manager, if you will, the IT guys," he says. "These skills they have not been trained to do and they need … either to say, ‘No, I can’t do that because I haven’t been trained,’ or they need to go and look where they can get that expertise.
"That’s what we try to do at SHM, with our Leadership Academy and our Practice Management Academy."
Frank Michota, MD, FHM, director of academic affairs in the Department of Hospital Medicine at The Cleveland Clinic, says that one of the biggest challenges the field needs to tackle over the next several years is to better standardize the education of hospitalists, saying there is "incredible inconsistency from hospitalist to hospitalist in terms of knowledge base, experience and … understanding the scope of practice."
"We continue to have significant variation in hospital practice models and the types of measurements that are available to those hospitalists for practice improvement," he says. "We continue to see significant turnover in the field with kind of a lack of maturity"—and not the kind of experience base that "you would like to see 15 years in."
"There is really no confidence that everyone at the base of that iceberg will ever make it to the tip because it’s still not viewed by many who entered the field as being a long-term career choice," Dr. Michota says. For many, he said, it is "a look-and-see proposition."
All of this, he says, points to the need for a full certification process by an HM board.
"I don’t want to make it sound like it has not been an impressive evolution to this point, but I think if we are going to meet the expectations, we do have to do more than we’re doing now," Dr. Michota says.
Some of the gaps in training might be able to be filled by private hospital management groups, which have training programs for their doctors that are made possible by their scale and whose presence is predicted to grow over the next 15 years.
Robert Bessler, MD, who in 2001 founded Tacoma, Wash.-based Sound Physicians, which has become one of the largest private hospitalist organizations in the country, says private companies are able to conduct training that is impossible for many hospitals to conduct themselves.
"You’re going to get good people who are all of good training and good knowledge, but they’re not all going to have experience," he says, "and so what are the hospitals that are employing 50% of the hospitalists in this country going to do about that? It’s pretty much nothing. They’re going to occasionally send some people to conferences and hope—because they don’t have that infrastructure."
At teaching institutes like those at private firms, the process is sped up, Dr. Bessler adds.
"That’s why we built our hospitalists’ institute at Sound—to turn really good, quality doctors into effective hospitalists in a much more rapid fashion," he says. "Because before we built this, it was just get them involved and hope after a couple of years they’ve really become efficient. Our hospital partners and the patients can’t wait that long."
Robert Reynolds, MD, founder of PrimeDoc, an Asheville, N.C.-based company that provides doctors for 12 hospitalist programs and employs about 100 doctors, says there needs to be more focus on teaching the "realistic side of the business of medicine," as well as on quality outcomes and patient satisfaction. But he also doubts there will be much change in training.
"[From] my cynical side and the voice of experience, I don’t see any change in the near future," he says. "What we’re seeing now is physicians come out of residency with a good clinical base, but really having no idea of how the healthcare system works in a bigger picture, how it works as an industry. So we’re having to spend a lot of time and effort training physicians to start thinking like practicing physicians."
The experts all agree that there will be an increase in hospitalists being provided by private corporations. Dr. Reynolds says that trend will continue in part due to healthcare reform’s emphasis on outcomes for reimbursement and a corporation’s ability to assist with physician training, as well as data and reporting needs.
"More and more hospital compensation and physician compensation is going to be based on actual data, performance data," he says. "And in order to really do a good job of capturing and reporting that kind of data, you need enough size to support an IT system and training systems that will produce and capture the kind of data that will be necessary."
Erin Fisher, MD, MHM, a pediatric hospitalist at Rady Children’s Hospital in San Diego, says a major goal of the future should be to change the reimbursement structure "so that you have something that is reasonable and encourages appropriate testing, treatments, and coordination of our healthcare system in a systematic way, rather than pieces." In such a system, hospitalists might see something to prompt them to intervene in a preventive way.
"The bigger question is, can our healthcare system, in five to 10 years, change itself enough that it uses every episode of care as an opportunity to do preventive care and coordinate care in the best way?" says Dr. Fisher, an SHM board member.
Continued Growth?
There is agreement that the field will continue to expand, with SHM predicting that the number of hospitalists in the U.S. will reach 40,000 in the next several years, up from today’s 30,000 figure.
Dr. Wellikson says that the figure could rise to as many as 70,000 or more if specialty hospitalists—such as surgical hospitalists, neuro-hospitalists, and laborists—are included. Those hospital-based specialties are now only in their infancy.
"Everything you can see shows that people are still flocking into hospital medicine," Dr. Wellikson adds.
Hospitalists numbered in the hundreds just 15 years ago, so growth has been explosive the past decade. Dr. Cawley, however, says the pace of growth might be starting to slow already, shifting to undeveloped or underserved areas. "Hospitalist programs are at almost all the large [hospitals] and really the growth has been at the smaller hospitals in the last several years," he says.
With the projected rise of Medicare beneficiaries due to the aging of the baby-boom generation, use of hospitals is expected to skyrocket, meaning more hospitalists will be needed, Dr. Bessler says. He also cites data from the National Rural Health Association noting that 25% of the U.S. population lives in areas considered rural, but that only 10% of the physicians live in those areas, indicating a potential growth area for hospitalists.
"That would tell me that demand will continue to outpace supply," he says.
Mike Tarwater, a member of the board of the American Hospital Association and CEO of Carolinas Medical Center in Charlotte, N.C., agrees with Dr. Bessler. Even with the move toward more outpatient care, Tarwater says, the aging of the population will mean a higher demand for hospitalists.
-Mike Tarwater, board member, American Hospital Association, CEO, Carolinas Medical Center, Charlotte, N.C.
"I think that the primary-care physicians—either because of their love for it or their belief that it’s the better way to go with the treatment of their patients—are going to be really stretched to keep that ambulatory practice going and to get to round on patients in the hospitals," he says. "I think there’s going to be a continued growth of the trend that we’ve seen over the last 15 years."
That growth also will mean a greater emphasis on technology use, whether it’s technology used for quick diagnostics like portable ultrasound or more widely used and refined electronic health records (EHR)—or, as Tarwater describes, "probably things we don’t imagine today."
"Our doctors, more than any other doctors, are tech-savvy; they’re early adopters," Dr. Wellikson says.
Hospitalists likely will emerge as leaders in the adoption of new technology, several experts predict.
Without a doubt, I think that hospitalists are going to be a driving force in the adaptation of the electronic [health] record to the clinical care within their hospitals," Dr. Michota says.
As the needs of HM grow, and the field grows more complex, there will inevitably be more divisions and departments of hospital medicine in places where it is now only a section, Dr. Cawley says.
"When you’re a division or a department, you have more autonomy over your own future, so I see this happening," he says. "I think more and more will carve themselves out of general internal medicine, and a lot of that will come because of a demand for more independence and greater autonomy." TH
Thomas R. Collins is a medical writer based in Florida.
More Value, More PATIENTs, More Technology
HM Pioneer Looks Into Crystal Ball
"We’re that camel’s nose."
With that in mind, what does Dr. Wachter predict for the next 15 years of HM? Here are his top three prognostications:
- A shift from the pressure to improve quality and safety to pressure to improve value, with more emphasis on cost and waste reduction.
"Hospitalist groups that are effective at [quality and safety] will continue to be popular in their organization, while hospitalist groups that aren’t will find that their standing is compromised," he says.
How that is done will vary from institution to institution. It will require a complex process of literature and creation of algorithms, he adds, "but also rolling up your sleeves and meeting with the right people, and working through the politics and diplomacy in order to get this work done."
- At teaching hospitals, a greater role for hospitalists to take care of patients who have traditionally been cared for by residents.
This is borne of the new Accreditation Council for Graduate Medical Education (ACGME) rules for residents’ work hours and supervision, which will require more hospitalists to fill voids in patient care and supervisory roles.
"If you like growth, that’s a great trend for hospitalists," Dr. Wachter says. "But if you’re in the business of trying to hire enough hospitalists to fill all your needs, it’s not that great because the demand curve is just tremendous; there’s a national shortage of hospitalists."
The trend also will affect what "a faculty job looks like," because there will be more clinical needs that will take time away from more traditionally academic work, he explains.
"Academic hospitals are not very good at creating satisfying, sustainable jobs that are largely clinical," Dr. Wachter says. "So how do you make sure that those people have fulfilling jobs, that they don’t feel like second-class citizens, that they can get promoted if they do what you’ve asked them to do well? I think that’s a huge challenge for the field, but it is a challenge borne of a new imperative."
- A revolutionary move from a pen-and-paper hospital environment to a technology-driven workplace.
"It will change the way we do our work," Dr. Wachter says, adding it will also mean other, more subtle changes.
"It takes away the importance of geography to some extent," he adds. "I can be off in the doctor’s lounge or in my house and still do my work.
"And so how do you retain or enhance the relationships that are so fundamental to providing good care? That’s not only between doctors and patients and families, but also between doctors and nurses, and each other."-TC
Maternity Management
Editor's Note: Second in a two-part series
Lest anyone forget, it is essential to support workers having children for one reason—the continuation of the human species, says Rachel Lovins, MD, SFHM, who directs the hospitalist program at Waterbury Hospital in Waterbury, Conn. For HM program directors, that means following pregnancy labor laws. But it also should involve reasonably accommodating hospitalists who are balancing their new baby’s needs with the demands of their profession, says Dr. Lovins and other HM leaders.
"As there are more women in medicine, everybody needs to be more aware of this issue. We don’t want to make good talent feel uncomfortable with the process of taking maternity leave and reducing time," says Michelle Marks, DO, FAAP, SFHM, director of the Center for Pediatric Hospital Medicine at the Cleveland Clinic.
All HM program directors need to be aware of such federal laws as the Pregnancy Discrimination Act and the Family and Medical Leave Act (www.eeoc.gov/laws/types/pregnancy.cfm), as well as the corresponding laws of the state in which they work. Directors can contact their human resources (HR) department for assistance.
"Calling them upfront will save a lot of headaches later on," says Jasen Gundersen, MD, MBA, CPE, SFHM, chief medical officer of the hospital medicine division in Fort Lauderdale, Fla., for Knoxville, Tenn.-based TeamHealth.
Here are some other recommendations on how HM directors can best manage pregnancy issues affecting their team:
The "R" in Relationship
There are many reasons why the director of a hospitalist group should develop a good relationship with the providers in their group, but one of them is that a hospitalist is more likely to tell her director sooner rather than later that she is pregnant, Dr. Marks says.
"Knowing your staff well and knowing them personally helps a lot, too, because you can gauge where they are going personally, as far as marriage, children, that type of thing," she adds.
The earlier a group leader knows a staff member is pregnant, the more time they have to plan for maternity leave. And the better the plan, the easier the leave is on the entire group, says Dr. Gundersen.
Generally, finding out that a physician is pregnant within three to five months of conception provides enough time to make adequate arrangements for coverage, Drs. Marks and Lovins note.

The Conversation
Before scheduling a meeting to discuss maternity leave and plans for returning to work with the hospitalist, the group leader should call HR to see if such a conversation is permissible, says Dr. Marks. A better approach might be to wait until the hospitalist broaches the subject.
"So many times the hospitalist will ask for counseling as far as what are her options of coming back," Dr. Marks says. "That opens the door for an open discussion."
Once the conversation starts, the group leader should gauge the length of maternity leave, her plans for coming back full time or part time, and the anticipated scheduling limitations or childcare considerations, Dr. Gundersen says.
"That’s not to say the pregnant woman can really predict all the time what’s going to happen," says Kerry Weiner, MD, MPH, chief clinical officer for North Hollywood, Calif.-based IPC: The Hospitalist Company, Inc. "Obviously, it’s a medical condition that can change and everyone understands that. It’s getting a feel of what you can actually know at the time."
If it’s the HM director’s intent to call the physician while she is on leave to see how she and the baby are doing and how the maternity leave is going, that should be discussed during the conversation, Dr. Gundersen says.
"If you establish upfront that you are going to make that phone call, I think that’s fine to do," he explains. "If you’re calling constantly and pressuring the person, I don’t think that that’s kosher at all."

—Rachel Lovins, MD, SFHM, director, hospitalist program, Waterbury (Conn.) Hospital
The Coverage Plan
Most maternity leaves are from eight to 12 weeks, although the length varies by HM program and individual. It is essential to have your group’s coverage plan outlined well in advance of the maternity leave.
In a private-practice model in which hospitalists work weekdays and have a call-coverage schedule for nights and weekends, a group leader can spread the extra work among the other hospitalists in the group because there are more hospitalists working during the day when patient census is higher, Dr. Weiner says.
Shifting the workload in other schedule models isn’t always as easy. "In the seven-day-on, seven-day-off model, because of that maximum patient-to-doctor ratio, I don’t think there’s any way to do it without hiring help," Dr. Lovins says. "It’s important to recruit per diems all the time. When you’re in a bind is the worst time to do it."
To limit the disruption to patient care and operations quality, the goal when using outside hospitalists is to contract with physicians who have worked with the group before and who know the community, hospital, systems, and patients, Dr. Weiner says.
For HM groups that use a flexible schedule, maternity coverage plans aren’t really needed, says Reuben Tovar, MD, chairman of Hospital Internists of Austin, a physician-owned and -managed hospitalist practice in Texas.
"We’re not salary, so that changes the dynamic completely. People who work more make more, and people who work less make less," he explains. "We are much more liberal about time off, because if a person is taking off to do what is important to them, like taking care of a child, then the rest of us feel better about doing extra work."
—Michelle Marks, DO, FAAP, SFHM, director, Center for Pediatric Hospital Medicine, Cleveland Clinic
Things Change
Plans discussed at the outset with a pregnant hospitalist can change after the child is born, HM group directors caution.
"Particularly for the first child, people say, ‘I’ll come back full blast. Don’t worry about it.’ And they figure out how hard all that is in the first couple of weeks, and then I get a different answer," Dr. Tovar says. "I think the whole mom/wife/doctor thing is tough. I recognize how hard that is. Even though I am not in that role, I can see it."
Dr. Gundersen suggests group directors have a backup plan, in case the maternity leave lasts longer than expected or the transition back to work is delayed. "It really prevents you from putting pressure on the physician," he says.
If a hospitalist who had planned to come back full time decides that she wants to work less, a director should check with HR to see what the process would entail.
"Generally, we have to negotiate a time frame for when they can drop down" to part-time hours, Dr. Marks says. "It usually takes three to four months for me to be able to adjust staffing to make it work."
Back to Work
Physicians can return from maternity leave in a reduced role, but they very rarely drop out of medicine entirely, Dr. Marks says.
"[They] have put in a lot of time to get where they are," she says. "Plus, women in medicine are usually high achievers and very interested in their careers."
Yet hospitalist leaders should recognize that returning to work after having a baby is stressful. It will take some time for the returning hospitalist to develop a rhythm between her duties as a mother and a doctor.
Directors can review the hospitalist’s nonclinical roles, help with priorities, and perhaps reassign some of the responsibilities to colleagues, Dr. Marks says. With more women breastfeeding, it is important to provide a convenient space with a door that locks for women to breast-pump at work, she and the other directors say.
"The best thing in the world is to have colleagues that you trust and can rely on," Dr. Lovins says. "That way, people can help each other out in emergencies, like if someone has to take their kid to the doctor. That’s the kind of program I want to have and would want to be part of."
Lisa Ryan is a freelance writer based in New Jersey.
Editor's Note: Second in a two-part series
Lest anyone forget, it is essential to support workers having children for one reason—the continuation of the human species, says Rachel Lovins, MD, SFHM, who directs the hospitalist program at Waterbury Hospital in Waterbury, Conn. For HM program directors, that means following pregnancy labor laws. But it also should involve reasonably accommodating hospitalists who are balancing their new baby’s needs with the demands of their profession, says Dr. Lovins and other HM leaders.
"As there are more women in medicine, everybody needs to be more aware of this issue. We don’t want to make good talent feel uncomfortable with the process of taking maternity leave and reducing time," says Michelle Marks, DO, FAAP, SFHM, director of the Center for Pediatric Hospital Medicine at the Cleveland Clinic.
All HM program directors need to be aware of such federal laws as the Pregnancy Discrimination Act and the Family and Medical Leave Act (www.eeoc.gov/laws/types/pregnancy.cfm), as well as the corresponding laws of the state in which they work. Directors can contact their human resources (HR) department for assistance.
"Calling them upfront will save a lot of headaches later on," says Jasen Gundersen, MD, MBA, CPE, SFHM, chief medical officer of the hospital medicine division in Fort Lauderdale, Fla., for Knoxville, Tenn.-based TeamHealth.
Here are some other recommendations on how HM directors can best manage pregnancy issues affecting their team:
The "R" in Relationship
There are many reasons why the director of a hospitalist group should develop a good relationship with the providers in their group, but one of them is that a hospitalist is more likely to tell her director sooner rather than later that she is pregnant, Dr. Marks says.
"Knowing your staff well and knowing them personally helps a lot, too, because you can gauge where they are going personally, as far as marriage, children, that type of thing," she adds.
The earlier a group leader knows a staff member is pregnant, the more time they have to plan for maternity leave. And the better the plan, the easier the leave is on the entire group, says Dr. Gundersen.
Generally, finding out that a physician is pregnant within three to five months of conception provides enough time to make adequate arrangements for coverage, Drs. Marks and Lovins note.
The Conversation
Before scheduling a meeting to discuss maternity leave and plans for returning to work with the hospitalist, the group leader should call HR to see if such a conversation is permissible, says Dr. Marks. A better approach might be to wait until the hospitalist broaches the subject.
"So many times the hospitalist will ask for counseling as far as what are her options of coming back," Dr. Marks says. "That opens the door for an open discussion."
Once the conversation starts, the group leader should gauge the length of maternity leave, her plans for coming back full time or part time, and the anticipated scheduling limitations or childcare considerations, Dr. Gundersen says.
"That’s not to say the pregnant woman can really predict all the time what’s going to happen," says Kerry Weiner, MD, MPH, chief clinical officer for North Hollywood, Calif.-based IPC: The Hospitalist Company, Inc. "Obviously, it’s a medical condition that can change and everyone understands that. It’s getting a feel of what you can actually know at the time."
If it’s the HM director’s intent to call the physician while she is on leave to see how she and the baby are doing and how the maternity leave is going, that should be discussed during the conversation, Dr. Gundersen says.
"If you establish upfront that you are going to make that phone call, I think that’s fine to do," he explains. "If you’re calling constantly and pressuring the person, I don’t think that that’s kosher at all."
—Rachel Lovins, MD, SFHM, director, hospitalist program, Waterbury (Conn.) Hospital
The Coverage Plan
Most maternity leaves are from eight to 12 weeks, although the length varies by HM program and individual. It is essential to have your group’s coverage plan outlined well in advance of the maternity leave.
In a private-practice model in which hospitalists work weekdays and have a call-coverage schedule for nights and weekends, a group leader can spread the extra work among the other hospitalists in the group because there are more hospitalists working during the day when patient census is higher, Dr. Weiner says.
Shifting the workload in other schedule models isn’t always as easy. "In the seven-day-on, seven-day-off model, because of that maximum patient-to-doctor ratio, I don’t think there’s any way to do it without hiring help," Dr. Lovins says. "It’s important to recruit per diems all the time. When you’re in a bind is the worst time to do it."
To limit the disruption to patient care and operations quality, the goal when using outside hospitalists is to contract with physicians who have worked with the group before and who know the community, hospital, systems, and patients, Dr. Weiner says.
For HM groups that use a flexible schedule, maternity coverage plans aren’t really needed, says Reuben Tovar, MD, chairman of Hospital Internists of Austin, a physician-owned and -managed hospitalist practice in Texas.
"We’re not salary, so that changes the dynamic completely. People who work more make more, and people who work less make less," he explains. "We are much more liberal about time off, because if a person is taking off to do what is important to them, like taking care of a child, then the rest of us feel better about doing extra work."
—Michelle Marks, DO, FAAP, SFHM, director, Center for Pediatric Hospital Medicine, Cleveland Clinic
Things Change
Plans discussed at the outset with a pregnant hospitalist can change after the child is born, HM group directors caution.
"Particularly for the first child, people say, ‘I’ll come back full blast. Don’t worry about it.’ And they figure out how hard all that is in the first couple of weeks, and then I get a different answer," Dr. Tovar says. "I think the whole mom/wife/doctor thing is tough. I recognize how hard that is. Even though I am not in that role, I can see it."
Dr. Gundersen suggests group directors have a backup plan, in case the maternity leave lasts longer than expected or the transition back to work is delayed. "It really prevents you from putting pressure on the physician," he says.
If a hospitalist who had planned to come back full time decides that she wants to work less, a director should check with HR to see what the process would entail.
"Generally, we have to negotiate a time frame for when they can drop down" to part-time hours, Dr. Marks says. "It usually takes three to four months for me to be able to adjust staffing to make it work."
Back to Work
Physicians can return from maternity leave in a reduced role, but they very rarely drop out of medicine entirely, Dr. Marks says.
"[They] have put in a lot of time to get where they are," she says. "Plus, women in medicine are usually high achievers and very interested in their careers."
Yet hospitalist leaders should recognize that returning to work after having a baby is stressful. It will take some time for the returning hospitalist to develop a rhythm between her duties as a mother and a doctor.
Directors can review the hospitalist’s nonclinical roles, help with priorities, and perhaps reassign some of the responsibilities to colleagues, Dr. Marks says. With more women breastfeeding, it is important to provide a convenient space with a door that locks for women to breast-pump at work, she and the other directors say.
"The best thing in the world is to have colleagues that you trust and can rely on," Dr. Lovins says. "That way, people can help each other out in emergencies, like if someone has to take their kid to the doctor. That’s the kind of program I want to have and would want to be part of."
Lisa Ryan is a freelance writer based in New Jersey.
Editor's Note: Second in a two-part series
Lest anyone forget, it is essential to support workers having children for one reason—the continuation of the human species, says Rachel Lovins, MD, SFHM, who directs the hospitalist program at Waterbury Hospital in Waterbury, Conn. For HM program directors, that means following pregnancy labor laws. But it also should involve reasonably accommodating hospitalists who are balancing their new baby’s needs with the demands of their profession, says Dr. Lovins and other HM leaders.
"As there are more women in medicine, everybody needs to be more aware of this issue. We don’t want to make good talent feel uncomfortable with the process of taking maternity leave and reducing time," says Michelle Marks, DO, FAAP, SFHM, director of the Center for Pediatric Hospital Medicine at the Cleveland Clinic.
All HM program directors need to be aware of such federal laws as the Pregnancy Discrimination Act and the Family and Medical Leave Act (www.eeoc.gov/laws/types/pregnancy.cfm), as well as the corresponding laws of the state in which they work. Directors can contact their human resources (HR) department for assistance.
"Calling them upfront will save a lot of headaches later on," says Jasen Gundersen, MD, MBA, CPE, SFHM, chief medical officer of the hospital medicine division in Fort Lauderdale, Fla., for Knoxville, Tenn.-based TeamHealth.
Here are some other recommendations on how HM directors can best manage pregnancy issues affecting their team:
The "R" in Relationship
There are many reasons why the director of a hospitalist group should develop a good relationship with the providers in their group, but one of them is that a hospitalist is more likely to tell her director sooner rather than later that she is pregnant, Dr. Marks says.
"Knowing your staff well and knowing them personally helps a lot, too, because you can gauge where they are going personally, as far as marriage, children, that type of thing," she adds.
The earlier a group leader knows a staff member is pregnant, the more time they have to plan for maternity leave. And the better the plan, the easier the leave is on the entire group, says Dr. Gundersen.
Generally, finding out that a physician is pregnant within three to five months of conception provides enough time to make adequate arrangements for coverage, Drs. Marks and Lovins note.
The Conversation
Before scheduling a meeting to discuss maternity leave and plans for returning to work with the hospitalist, the group leader should call HR to see if such a conversation is permissible, says Dr. Marks. A better approach might be to wait until the hospitalist broaches the subject.
"So many times the hospitalist will ask for counseling as far as what are her options of coming back," Dr. Marks says. "That opens the door for an open discussion."
Once the conversation starts, the group leader should gauge the length of maternity leave, her plans for coming back full time or part time, and the anticipated scheduling limitations or childcare considerations, Dr. Gundersen says.
"That’s not to say the pregnant woman can really predict all the time what’s going to happen," says Kerry Weiner, MD, MPH, chief clinical officer for North Hollywood, Calif.-based IPC: The Hospitalist Company, Inc. "Obviously, it’s a medical condition that can change and everyone understands that. It’s getting a feel of what you can actually know at the time."
If it’s the HM director’s intent to call the physician while she is on leave to see how she and the baby are doing and how the maternity leave is going, that should be discussed during the conversation, Dr. Gundersen says.
"If you establish upfront that you are going to make that phone call, I think that’s fine to do," he explains. "If you’re calling constantly and pressuring the person, I don’t think that that’s kosher at all."
—Rachel Lovins, MD, SFHM, director, hospitalist program, Waterbury (Conn.) Hospital
The Coverage Plan
Most maternity leaves are from eight to 12 weeks, although the length varies by HM program and individual. It is essential to have your group’s coverage plan outlined well in advance of the maternity leave.
In a private-practice model in which hospitalists work weekdays and have a call-coverage schedule for nights and weekends, a group leader can spread the extra work among the other hospitalists in the group because there are more hospitalists working during the day when patient census is higher, Dr. Weiner says.
Shifting the workload in other schedule models isn’t always as easy. "In the seven-day-on, seven-day-off model, because of that maximum patient-to-doctor ratio, I don’t think there’s any way to do it without hiring help," Dr. Lovins says. "It’s important to recruit per diems all the time. When you’re in a bind is the worst time to do it."
To limit the disruption to patient care and operations quality, the goal when using outside hospitalists is to contract with physicians who have worked with the group before and who know the community, hospital, systems, and patients, Dr. Weiner says.
For HM groups that use a flexible schedule, maternity coverage plans aren’t really needed, says Reuben Tovar, MD, chairman of Hospital Internists of Austin, a physician-owned and -managed hospitalist practice in Texas.
"We’re not salary, so that changes the dynamic completely. People who work more make more, and people who work less make less," he explains. "We are much more liberal about time off, because if a person is taking off to do what is important to them, like taking care of a child, then the rest of us feel better about doing extra work."
—Michelle Marks, DO, FAAP, SFHM, director, Center for Pediatric Hospital Medicine, Cleveland Clinic
Things Change
Plans discussed at the outset with a pregnant hospitalist can change after the child is born, HM group directors caution.
"Particularly for the first child, people say, ‘I’ll come back full blast. Don’t worry about it.’ And they figure out how hard all that is in the first couple of weeks, and then I get a different answer," Dr. Tovar says. "I think the whole mom/wife/doctor thing is tough. I recognize how hard that is. Even though I am not in that role, I can see it."
Dr. Gundersen suggests group directors have a backup plan, in case the maternity leave lasts longer than expected or the transition back to work is delayed. "It really prevents you from putting pressure on the physician," he says.
If a hospitalist who had planned to come back full time decides that she wants to work less, a director should check with HR to see what the process would entail.
"Generally, we have to negotiate a time frame for when they can drop down" to part-time hours, Dr. Marks says. "It usually takes three to four months for me to be able to adjust staffing to make it work."
Back to Work
Physicians can return from maternity leave in a reduced role, but they very rarely drop out of medicine entirely, Dr. Marks says.
"[They] have put in a lot of time to get where they are," she says. "Plus, women in medicine are usually high achievers and very interested in their careers."
Yet hospitalist leaders should recognize that returning to work after having a baby is stressful. It will take some time for the returning hospitalist to develop a rhythm between her duties as a mother and a doctor.
Directors can review the hospitalist’s nonclinical roles, help with priorities, and perhaps reassign some of the responsibilities to colleagues, Dr. Marks says. With more women breastfeeding, it is important to provide a convenient space with a door that locks for women to breast-pump at work, she and the other directors say.
"The best thing in the world is to have colleagues that you trust and can rely on," Dr. Lovins says. "That way, people can help each other out in emergencies, like if someone has to take their kid to the doctor. That’s the kind of program I want to have and would want to be part of."
Lisa Ryan is a freelance writer based in New Jersey.
Pediatric HM Literature

Background: The management of young febrile infants with UTIs is marked by uncertainty and variation. Although recent studies have demonstrated the safety and efficacy of oral antibiotics as primary treatment in infants younger than six months of age, younger infants tend to receive a longer duration of intravenous antibiotics. This might reflect a lack of clear delineation of the risk of adverse events and bacteremia in this population.
Study design: Retrospective chart review.
Setting: Twenty primarily tertiary-care EDs.
Synopsis: Infants aged 29 to 60 days with febrile UTIs were identified through laboratory and chart review at the participating centers. Bacteremia and adverse events (death, shock, bacterial meningitis, intensive care, surgical intervention, or other substantial clinical complications) were identified, as well as patients with a high-risk past medical history (PMH) or who were clinically ill on examination, based on a priori definitions of chart wording.
Adverse events occurred in 2.8% of the 1,895 patients; bacteremia occurred in 6.5%. Recursive partitioning analysis was used to identify a very-low-risk population for adverse events—those who were not clinically ill and without high-risk PMH (prediction model sensitivity 98% and negative predictive value 99.9%)—but it was not as successful in accurately identifying infants at very low risk for bacteremia.
Limitations of this study include the lack of a clear description of the adverse events identified (and their presumed relationship to the UTIs), the reliance on ED documentation, and conservative definitions of bacterial meningitis. Nonetheless, this is a study of significant magnitude in a population marked by uncertainty. Results of this study further strengthen data that support the feasibility of outpatient antibiotic therapy in well-appearing infants.
Bottom line: Well-appearing infants aged 29-60 days and without significant past medical history are at very low risk for adverse events.
Citation: Schnadower D, Kupperman N, Macias CG, et al. Febrile infants with urinary tract infections at very low risk for adverse events and bacteremia. Pediatrics. 2010;126:1074-1083.
Background: The management of young febrile infants with UTIs is marked by uncertainty and variation. Although recent studies have demonstrated the safety and efficacy of oral antibiotics as primary treatment in infants younger than six months of age, younger infants tend to receive a longer duration of intravenous antibiotics. This might reflect a lack of clear delineation of the risk of adverse events and bacteremia in this population.
Study design: Retrospective chart review.
Setting: Twenty primarily tertiary-care EDs.
Synopsis: Infants aged 29 to 60 days with febrile UTIs were identified through laboratory and chart review at the participating centers. Bacteremia and adverse events (death, shock, bacterial meningitis, intensive care, surgical intervention, or other substantial clinical complications) were identified, as well as patients with a high-risk past medical history (PMH) or who were clinically ill on examination, based on a priori definitions of chart wording.
Adverse events occurred in 2.8% of the 1,895 patients; bacteremia occurred in 6.5%. Recursive partitioning analysis was used to identify a very-low-risk population for adverse events—those who were not clinically ill and without high-risk PMH (prediction model sensitivity 98% and negative predictive value 99.9%)—but it was not as successful in accurately identifying infants at very low risk for bacteremia.
Limitations of this study include the lack of a clear description of the adverse events identified (and their presumed relationship to the UTIs), the reliance on ED documentation, and conservative definitions of bacterial meningitis. Nonetheless, this is a study of significant magnitude in a population marked by uncertainty. Results of this study further strengthen data that support the feasibility of outpatient antibiotic therapy in well-appearing infants.
Bottom line: Well-appearing infants aged 29-60 days and without significant past medical history are at very low risk for adverse events.
Citation: Schnadower D, Kupperman N, Macias CG, et al. Febrile infants with urinary tract infections at very low risk for adverse events and bacteremia. Pediatrics. 2010;126:1074-1083.
Background: The management of young febrile infants with UTIs is marked by uncertainty and variation. Although recent studies have demonstrated the safety and efficacy of oral antibiotics as primary treatment in infants younger than six months of age, younger infants tend to receive a longer duration of intravenous antibiotics. This might reflect a lack of clear delineation of the risk of adverse events and bacteremia in this population.
Study design: Retrospective chart review.
Setting: Twenty primarily tertiary-care EDs.
Synopsis: Infants aged 29 to 60 days with febrile UTIs were identified through laboratory and chart review at the participating centers. Bacteremia and adverse events (death, shock, bacterial meningitis, intensive care, surgical intervention, or other substantial clinical complications) were identified, as well as patients with a high-risk past medical history (PMH) or who were clinically ill on examination, based on a priori definitions of chart wording.
Adverse events occurred in 2.8% of the 1,895 patients; bacteremia occurred in 6.5%. Recursive partitioning analysis was used to identify a very-low-risk population for adverse events—those who were not clinically ill and without high-risk PMH (prediction model sensitivity 98% and negative predictive value 99.9%)—but it was not as successful in accurately identifying infants at very low risk for bacteremia.
Limitations of this study include the lack of a clear description of the adverse events identified (and their presumed relationship to the UTIs), the reliance on ED documentation, and conservative definitions of bacterial meningitis. Nonetheless, this is a study of significant magnitude in a population marked by uncertainty. Results of this study further strengthen data that support the feasibility of outpatient antibiotic therapy in well-appearing infants.
Bottom line: Well-appearing infants aged 29-60 days and without significant past medical history are at very low risk for adverse events.
Citation: Schnadower D, Kupperman N, Macias CG, et al. Febrile infants with urinary tract infections at very low risk for adverse events and bacteremia. Pediatrics. 2010;126:1074-1083.
ONLINE EXCLUSIVE: Listen to Pat Cawley and Frank Michota discuss what's next for HM
Click here to listen to Dr. Cawley
Click here to listen to Dr. Michota
Click here to listen to Dr. Cawley
Click here to listen to Dr. Michota
Click here to listen to Dr. Cawley
Click here to listen to Dr. Michota
ONLINE EXCLUSIVE: Listen to program directors discuss managing a group when a hospitalist is out on maternity leave
Click here to listen to Dr. Weiner
Click here to listen to Dr. Marks
Click here to listen to Dr. Weiner
Click here to listen to Dr. Marks
Click here to listen to Dr. Weiner
Click here to listen to Dr. Marks
ONLINE EXCLUSIVE: Listen to HM pioneers John Nelson, Bob Wachter, and Win Whitcomb ruminate on HM's 15-year anniversary
Click here to listen to Dr. Nelson
Click here to listen to Dr. Wachter
Click here to listen to Dr. Whitcomb
Click here to listen to Dr. Nelson
Click here to listen to Dr. Wachter
Click here to listen to Dr. Whitcomb
Click here to listen to Dr. Nelson
Click here to listen to Dr. Wachter
Click here to listen to Dr. Whitcomb
ONLINE EXCLUSIVE: How to minimize the adverse affects of working night shifts
Click here to listen to Dr. Landrigan
Click here to listen to Dr. Landrigan
Click here to listen to Dr. Landrigan
Managing seizures: Achieving control while minimizing risk
• Prescribe an antiepileptic drug (AED) after a first unprovoked seizure only if the seizure was prolonged or there is a risk of recurrence. C
• Use monotherapy whenever possible; if seizures continue and potential adverse effects prevent an increase in dosage, switch to a different AED and taper off the first agent. A
• Consider gradual withdrawal of AEDs from patients who have been seizure-free for 2 to 5 years. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Joe G, a 44-year-old man who has been your patient for years, comes to your office 48 hours after having a seizure. He has no history of seizures, had no warning signs or symptoms, and felt fine all day, but simply collapsed when the seizure occurred. He was transported to the emergency department (ED), and found to be postictal, with no further seizure activity. The ED work-up included a hemogram, comprehensive metabolic panel, and computed tomography brain scan, all of which were normal. An hour later, Joe had a normal neurological exam, then underwent electroencephalography (EEG) and magnetic resonance imaging (MRI) and was discharged home without medication.
How would you treat this patient?
About 10% of Americans will experience a seizure at some point in their lives,1,2 and more than 3 million have epilepsy.3 The incidence ranges from 1% among 20-year-olds to more than 3% by the age of 75.1,2
To adequately care for such patients—whether they have had multiple seizures or only one—you need to know whether they’re at risk for recurrences, when (or if) to prescribe an AED, and which agents provide optimal seizure control with the fewest adverse effects. You also need to know when a referral to an epilepsy specialist is indicated, when or whether it’s safe for patients to stop taking antiseizure medication, and how to address lifestyle issues that patients with epilepsy often need help with.
This review addresses these and other questions.
Is it epilepsy? How to respond to a single seizure
A seizure—a transient occurrence of signs or symptoms due to abnormal excessive or synchronous neural activity in the brain—can be either focal (partial) or generalized. In addition, seizures can be broadly divided into 2 categories, based on etiology:
Provoked seizures are caused by an acute structural, toxic, or metabolic insult to the brain, and, presumably, would not have occurred if the underlying medical condition did not exist. Treating the cause—eg, alcohol withdrawal, hyponatremia, or hypoglycemia—should prevent a recurrence.
Unprovoked seizures have no apparent underlying cause. Epilepsy is defined as a chronic condition characterized by ≥2 unprovoked seizures at least 24 hours apart, and epilepsy syndromes are classified as localization-related or generalized (TABLE 1).1,4,5
Generally, epileptologists do not recommend symptomatic treatment of a first unprovoked seizure6—a consensus based on several randomized controlled trials that found immediate treatment with an AED reduced the risk of a subsequent seizure in the short term, but did not affect long-term outcomes or the development of epilepsy.7
Treatment should begin after a single seizure, however, if the seizure was prolonged or there is an increased risk of recurrence.6 Factors that increase this risk include an abnormal EEG, particularly if the abnormality is epileptiform; the presence of a brain lesion; a localized (focal) seizure; and an abnormal neurologic exam.8 A history of status epilepticus—a single, unremitting seizure lasting ≥5 to 10 minutes or frequent seizures without a return to neurologic baseline in between—or complex febrile seizures, and a family history of epilepsy are risk factors for recurrence, as well.7
When the patient is a child. Prescribing an AED for a child after a first unprovoked seizure is not indicated to prevent the development of epilepsy, but may be considered, as for adults, in circumstances where the benefit of reducing the risk of a second seizure outweighs the risk of pharmacologic and psychosocial adverse effects.9
CASE Joe’s ED records show that his MRI was normal, but his EEG revealed an epileptogenic focus on the right temporal region—a finding that indicates that he has an elevated risk of recurrence and is a candidate for an AED. Before selecting a particular agent, you review his chart.
Joe is taking a thiazide diuretic and a calcium channel blocker for hypertension. He was a heavy drinker until he had an episode of pancreatitis 10 years ago, and has been abstinent ever since. About 5 years ago, he suffered from depression and was treated with sertraline, but the depression resolved and the drug was discontinued 3 years ago. The patient’s mother and brother have type 2 diabetes and his father had a myocardial infarction before the age of 60. Joe was laid off from his sales job 18 months ago and is actively seeking employment. At this point, you consider a broad-spectrum AED that would not interact with his current medications or adversely affect his medical conditions, and would be relatively inexpensive.
TABLE 1
Identifying seizures and types of epilepsy:1,4,5 International League Against Epilepsy classification
| Type of seizure |
Focal
Generalized
|
| Type of epilepsy syndrome* |
Localization related (partial or focal)
Generalized
|
| *This is a partial listing, with selected examples of epilepsy syndromes. |
What to consider in a first-line drug
The number of AEDs on the market has increased sharply in the past few years, giving physicians many medications to choose from. Selecting the optimal drug is particularly important for the initial treatment, as many patients remain on the first AED for years. Second-generation AEDs have been found to be as effective as, and better tolerated than, first-generation antiseizure drugs. But all AEDs carry a warning of a potential increase in suicide risk and the need to monitor patients for behavior changes.10
Before selecting an AED for a particular patient, consider the following questions:
What type of seizure? AEDs are generally classified by spectrum of activity into “narrow-spectrum” and “broad-spectrum.” Narrow-spectrum drugs are more effective for controlling partial seizures, but have the potential to exacerbate generalized seizures; broad-spectrum AEDs can be used for both. (TABLE 211-18 lists indications for first- and second-generation AEDs based on type of epilepsy.) If there’s no definitive diagnosis of the type of epilepsy a patient has, use a broad-spectrum drug.
What other drugs is the patient taking? If the AED will be added to the patient’s current medication regimen, look closely at potential pharmacodynamic drug-drug interactions, and consider whether a dosage adjustment is needed. Determine, too, whether the patient has any comorbidities that could affect his or her response to the AED.
Side effects, such as weight gain or loss, urolithiasis, and hepatic enzyme induction, are key considerations. (TABLE W1,19-24 which details dose, side effects, and costs of first- and second-generation AEDs, can be found at jfponline.com.)
Is the patient elderly? AED clearance is reduced in the elderly, so lower doses are needed. Reduction in serum albumin increases the free or active component of highly protein-bound drugs, increasing the likelihood of adverse effects.
Is the patient female? Some AEDs may have effects on women’s hormonal function, sexuality, bone health, and pregnancy.25 Hepatic enzyme inducers increase the clearance of oral contraceptives, reducing their efficacy. Vitamin D and calcium metabolism can also be affected, which can lead to osteomalacia. Valproate treatment in women is associated with higher levels of insulin, testosterone, and triglycerides.26 Cytochrome P-450-activating AEDs in general are associated with higher testosterone levels and reduced libido.27
Potential pregnancy is another consideration. Women with epilepsy are able to bear healthy children. What’s more, patients whose seizures are controlled with AEDs should be maintained on medication throughout pregnancy, as the risk of fetal harm from seizures generally outweighs the teratogenicity of the drug.28
Although large studies are limited, a study of 1532 infants exposed to AEDs in the first trimester did not find an increase in major birth defects compared with infants without such exposure.29 More recently, a large observational cohort study conducted in more than 40 countries found that the possibility of harm to a developing fetus is not only drug-specific but also dose-related.30 (To learn more, see “Pregnancy and epilepsy—when you’re managing both,” in the December 2010 issue of The Journal of Family Practice.)
Is cost a factor? Finally, consider the cost of the AED you would like to prescribe, and whether the patient has a prescription drug plan or the means to pay for his prescription.
CASE After a discussion of potential side effects, including the potential for suicidal ideation associated with AEDs, you prescribe carbamazepine for Joe as seizure prophylaxis, because it is the least expensive of the broad-spectrum AEDs and is unlikely to exacerbate his previous pancreatitis or interact with his current medications.
TABLE 2
Choosing an AED: What to consider11-18
| Epilepsy type | |||||
| Localization-related (focal/partial) | Idiopathic (generalized) | Nonidiopathic (generalized) | |||
| Anticonvulsant* | Tonic-clonic | Absence | Myoclonic | ||
| First generation | |||||
| Carbamazepine† | √ | √ | |||
| Ethosuximide† | √ | ||||
| Phenobarbital† | √ | √ | √ | ||
| Phenytoin† | √ | √ | √ | ||
| Primidone | √ | √ | √ | ||
| Valproate† | √ | √ | √ | √ | √ |
| Second generation | |||||
| Felbamate | √ | √ | |||
| Gabapentin† | √ | ||||
| Lacosamide | √ | ||||
| Lamotrigine | √† | √ | √‡ | √ | |
| Levetiracetam | √ | √ | √ | ||
| Oxcarbazepine† | √ | ||||
| Pregabalin | √ | ||||
| Rufinamide | √ | √ | |||
| Tiagabine | √ | ||||
| Topiramate | √‡ | √ | √ | ||
| Vigabatrin | √ | √ | |||
| Zonisamide | √ | √ | |||
| *Bold type indicates broad-spectrum antiepileptic drugs. †Supported by American Academy of Neurology (AAN) evidence-based guideline level A or B recommendation for monotherapy in newly diagnosed epilepsy patients. ‡Supported by AAN evidence-based guideline level B recommendation for monotherapy in newly diagnosed absence epilepsy. | |||||
TABLE W1
A closer look at antiepileptic drugs19-24
| Drug name | Maintenance dosage | Adverse effects | Cost (30-day supply)* | |
| Common | Rare/idiosyncratic | |||
| First generation | ||||
| Carbamazepine | 800-1200 mg/d | Dizziness, drowsiness, diplopia, nausea, vomiting, diarrhea, rash, pruritus, SIADH | Aplastic anemia, agranulocytosis, hyponatremia, SJS, hepatic failure, pancreatitis, suicidal ideation | $4-$50 (XR: $200) |
| Ethosuximide | 20 mg/kg per day | Sleep disturbance, drowsiness, hyperactivity, behavior changes, headache, nausea, vomiting, hiccups | Agranulocytosis, aplastic anemia, SJS, hepatic failure, serum sickness, suicidal ideation | $40-150 |
| Phenobarbital | 1-4 mg/kg per day; 120-400 mg/d | Altered sleep cycles, sedation, ataxia, lethargy, behavior changes, hyperactivity, nausea, rash | Agranulocytosis, dermatitis, SJS, hepatic failure, serum sickness, connective tissue disorders, metabolic bone disease, intellect blunting, suicidal ideation | $4-$10 |
| Phenytoin | 300-600 mg/d | Confusion, slurred speech, double vision, ataxia, nystagmus, neuropathy, hirsutism, acne, gingival hyperplasia | Neuropathy, agranulocytosis, SJS, immune reactions/serum sickness, hepatic failure, skin thickening, metabolic bone disease, suicidal ideation | $35 |
| Valproic acid | 60-350 mg/kg per day | Tremor, weight gain, PCOS, nausea, vomiting, alopecia, easy bruising | Hepatic failure, pancreatitis, hearing loss, blood dyscrasias/thrombocytopenia, hyperammonemia, encephalopathy, osteoporosis, suicidal ideation | $40 (ER: $150) |
| Second generation | ||||
| Felbamate | 2400-3600 mg/d | Somnolence, nausea, vomiting, weight loss, anorexia | Aplastic anemia (>13 years), hepatic failure, suicidal ideation | $300-$500† |
| Gabapentin | 900-1800 mg/d | Somnolence, fatigue, weight gain, nystagmus | Pedal edema, suicidal ideation | $4-$100 |
| Lacosamide | 200-400 mg/d | Headache, dizziness, ataxia, nausea, diplopia | Euphoria, prolongation of PR interval, heart block, suicidal ideation | $420† |
| Lamotrigine | 300-500 mg/d | Dizziness, ataxia, nausea, somnolence, rash | SJS, hypersensitivity reactions (renal/hepatic failure), DIC, suicidal ideation | $30-$100 |
| Levetiracetam | 3000 mg/d | Somnolence, dizziness, aggression, agitation, anxiety, weight loss | Infection, pancytopenia, liver failure, suicidal ideation | $30-$100 (XR: $245†) |
| Oxcarbazepine | 1200 mg/d | Somnolence, fatigue, headache, ataxia, nausea, rash | Hyponatremia, SJS, TEN, angioedema | $250-$1000 |
| Pregabalin | 150-600 mg/d | Peripheral edema, dry mouth, dizziness, ataxia, diplopia, weight gain | Angioedema, CK elevation, mild PR interval prolongation, suicidal ideation | $100-$350† |
| Rufinamide | 3200 mg/d | Headache, dizziness, fatigue, nausea | Shortened QT interval, hypersensitivity rash, suicidal ideation | $400-$750† |
| Tiagabine | 32-56 mg/d | Difficulty concentrating, dizziness, headache, somnolence, nervousness | Spike-wave stupor, sudden death, suicidal ideation | $140-$650† |
| Topiramate | 200-400 mg/d | Somnolence, dizziness, fatigue, weight loss, difficulty concentrating, speech problems, paresthesias, diarrhea, nausea | Acute myopia and glaucoma, hyperthermia (children); metabolic acidosis, hyperammonemia, liver failure, oligohydrosis, SJS/TEN, kidney stones, suicidal ideation | $40 - $100 |
| Vigabatrin | 1500 mg/d | Fatigue, somnolence, nystagmus, tremor, weight gain | Vision loss (30% of patients) blurred vision, arthralgia, suicidal ideation | :$50 -$100† |
| Zonisamide | 400- 600 mg/d | Somnolence, difficulty concentrating, anorexia, nausea | SJS, TEN, aplastic anemia, agranulocytosis, nephrolithiasis/, oligohydrosis, acidosis, suicidal ideation | $50-$200 |
| CK, creatine kinase; DIC, disseminated intravascular coagulation; ER, extended release; IV, intravenous; PCOS, polycystic ovarian syndrome; SIADH, syndrome of inappropriate antidiuretic hormone hypersecretion; SJS, Stevens-Johnson syndrome; TEN, toxic epidermal necrolysis, XR, extended release. *Costs from www.drugstore.com, www.savewithgenericdrugs.com, and www.pharmacychecker.com. †No generic available. | ||||
When to add a second AED
Monotherapy is the preferred method of epilepsy treatment, and controls seizures for 70% to 90% of patients.31,32 If seizures continue and potential adverse effects prevent you from increasing the dosage, switching to a different AED, then tapering off the first agent, is recommended.33,34
If the new AED fails to provide adequate seizure control, consider combination therapy. An additional 10% to 15% of patients with epilepsy achieve control with dual therapy.33,34
Many second-generation agents are approved for adjunctive therapy. However, the use of 2 AEDs increases the risk of toxicities and drug interactions, and requires complex dosage adjustments, which should be done slowly and cautiously. Combination therapy also increases costs and may cause a decrease in compliance.33,34
Noncompliance is the single most common reason for treatment failure in patients with epilepsy, occurring at an estimated rate of up to 60%.35,36 The complexity of the drug regimen is the major cause, regardless of patient age, sex, psychomotor development, seizure type, or seizure frequency.35,36
Because of the lack of good clinical trials of combination antiepilepsy therapy, no evidence is available to indicate which AEDs are safe and effective when taken together. There is, however, evidence that certain combinations should be avoided due to the risk of increased adverse effects. These include phenobarbital/valproate, phenytoin/carbamazepine, and carbamazepine/lamotrigine.25
Managing the patient who is seizure-free
After a patient has been seizure-free for 2 to 5 years, consider a reduction in, or a discontinuation of, his or her AED. The relapse rate varies from 10% to 70%, with meta-analyses showing a rate of 25% in the first year and 29% in the second year.19,37 The American Academy of Neurology (AAN) has published an evidence-based guideline for discontinuing AEDs in seizure-free patients, available at www.aan.com/professionals/practice/pdfs/gl0007.pdf.
Withdrawal should be gradual and, for patients on combination therapy, carried out one drug at a time to prevent a recurrence of seizures or status epilepticus. The AAN recommends a 2- to- 3-month withdrawal period for AEDs (and longer for benzodiazepines), although relapse rates have been found to be lower when the medication is withdrawn more slowly, over about 6 months.19,34 If seizures recur after withdrawal, restart the AEDs at previous dosages.19,34,38
Should the patient drive?
For patients with epilepsy, loss of independence related to driving restrictions is a major source of stress. A 10-year follow-up study of Danish patients with epilepsy found a 7-fold increase in motor vehicle accidents (MVAs) in patients with seizure disorders.39 Other studies have shown that the seizure-free interval is the best predictor of involvement in an MVA.40
The risk of driving accidents decreases as the seizure-free interval increases. Unfortunately, however, a decline in patient compliance is also associated with longer seizure-free intervals—creating the potential for recurrence and driving risk. Because of this discrepancy, a consensus statement from the AAN, American Epilepsy Society, and Epilepsy Foundation of America recommends a minimum 3-month seizure-free interval before patients are allowed to drive.41
Use clinical judgment in deciding whether to extend the seizure-free period. State laws vary widely regarding the need to report patients with seizure disorders, limitations on professional drivers, and seizure-free intervals required, so it is important to be familiar with the laws in your state. The Epilepsy Foundation has a helpful online resource with a database detailing individual state statutes (http://www.epilepsyfoundation.org/living/wellness/transportation/driverlicensing.cfm).
The danger of uncontrolled seizures
Overall, AEDs effectively control 70% of 80% of cases; the remaining 20% to 30% are considered medically refractory.38 What’s more, after 2 AED failures, a patient’s chances of achieving full seizure control with additional drugs are no better than 10% to 20%.42 And, as more drugs are tried, the likelihood of full control declines even further.43
Patients with uncontrolled seizures have a cumulative risk of sudden unexpected death in epilepsy (SUDEP) of 0.5% per year.44 Cognitive decline is associated with uncontrolled epilepsy, as well. In children, frequent seizures may significantly alter neuronal networks, affecting cognitive and motor development.
Is your patient a candidate for surgery?
Patients with disabling complex partial seizures that remain uncontrolled after 2 or more AED trials (either as monotherapy or in combination) should be referred to an epilepsy specialty center for evaluation for surgery.45 This should be considered as early as possible to afford the patient the best chance of achieving seizure control.
Successful epilepsy surgery—in which the portion of the brain causing the misfiring that causes the seizures is removed—often results in a better quality of life; it is also cost effective.46 Not everyone with refractory epilepsy is a candidate for surgery, of course. Among those who are, however, 50% to 70% of patients can expect to have improved seizure control.47
Status epilepticus is a medical emergency
A patient who develops status epilepticus is at high risk and requires immediate, and simultaneous, evaluation and treatment. Status epilepticus carries nearly a 20% mortality from the first episode,48 and the 10-year mortality rate after an episode of status epilepticus is as high as 40%.49
Although most of the deaths associated with status epilepticus are due to the underlying pathology, early treatment can prevent or ameliorate complications from rhabdomyolysis and irreversible anoxic neuronal damage.50
A benzodiazepine (typically, a 10-mg IV bolus of diazepam) is the initial treatment for status epilepticus, followed by or concurrent with fosphenytoin (15-18 mg/kg). If status epilepticus remains refractory to first-line drugs (lasting >30 minutes), intubation and transfer to an intensive care setting may be required, and a neurological consult should be obtained.
Pharmacologic treatment of status epilepticus falls into 3 main classes: benzodiazepines, standard AEDs, and general anesthetics such as propofol. Benzodiazepines act very rapidly to control most prolonged seizures, and are the first-line treatment choice. Diazepam has long been the mainstay of treatment, and is usually readily available. However, in both a large systematic review and a head-to-head trial, lorazepam was found to be superior to diazepam in ending seizure activity and maintaining seizure control without the use of other medications51,52—and is now the drug of choice for initial treatment of status epilepticus.
CASE You continue to see Joe every 3 to 4 months to monitor his basic blood work and mood. A year after his seizure, he remains seizure-free and is tolerating the AED without any adverse effects.
CORRESPONDENCE
William J. Geiger, MD, FAAFP, Medical College of Wisconsin, Columbia St. Mary’s Family Medicine Residency, 1121 East North Avenue, Milwaukee, WI 53212; [email protected]
1. Epilepsy Foundation of America. Epilepsy and seizure statistics. Available at: http://www.epilepsyfoundation.org/about/statistics.cfm. Accessed June 15, 2009.
2. Centers for Disease Control and Prevention (CDC). Prevalence and most common causes of disability among adults—United States, 2005. MMWR Morb Mortal Wkly Rep. 2009;58:421-426. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm5816a2.htm. Accessed June 15, 2009.
3. Hirtz D, Thurman DJ, Gwinn-Hardy K, et al. How common are the “common” neurologic disorders? Neurology. 2007;68:326-337.
4. Engel J Jr. ILAE classification of epilepsy syndromes. Epilepsy Res. 2006;70(suppl 1):S5-S10.
5. Rudzinski LA, Shih JJ. Continuum: lifelong learning in neurology. Epilepsia. 2010;16:15-35.
6. Chaves J, Sander JW. Seizure aggravation in idiopathic generalized epilepsies. Epilepsia. 2005;46(suppl 9):S133-S139.
7. Beghi E. Management of first seizure. General conclusions and recommendations. Epilepsia. 2008;49(suppl 1):S58-S61.
8. Berg A. Risk of recurrence after a first unprovoked seizure. Epilepsia. 2008;49(suppl 1):S13-S18.
9. Hirtz D, Ashwal S, Berg A, et al. Practice parameter: evaluating a first non-febrile seizure in children: report of the Quality Standards Subcommittee of the American Academy of Neurology, the Child Neurology Society, and the American Epilepsy Society. Neurology. 2000;55:616-623.
10. US Food and Drug Administration. Suicidal behavior and ideation and antiepileptic drugs. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm100190.htm. Updated May 5, 2009. Accessed June 28, 2009.
11. French JA, Kanner AM, Bautista J, et al. Efficacy and tolerability of the new antiepileptic drugs I: treatment of new epilepsy, report of the therapeutic and technology assessment subcommittee and quality standards subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2004;62:1252-1260.
12. French J, Smith M, Faught E, et al. Practice advisory: the use of felbamate in the treatment of patients with intractable epilepsy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 1999;52:1540-1545.
13. Glauser T, Kluger G, Sachdeo R, et al. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology. 2008;70:1950-1958.
14. Suzuki Y, Nagai T, Ono J, et al. Zonisamide monotherapy in newly-diagnosed infantile spasms. Epilepsia. 1997;38:1035-1038.
15. Kochak GM, Page JG, Buchanan RA, et al. Steady-state pharmacokinetics of zonisamide, an antiepileptic agent for treatment of refractory complex partial seizures. J Clin Pharmacol. 1998;38:166-171.
16. Arroyo S, Anhut H, Kugler AR, et al. Pregabalin 1008-011 International Study Group. Pregabalin add-on treatment: a randomized, double-blind, placebo-controlled, dose-response study in adults with partial seizures. Epilepsia. 2004;45:20-27.
17. Brodie MJ, Rosenfeld WE, Vazquez B, et al. Rufinamide for the adjunctive treatment of partial seizures in adults and adolescents: a randomized placebo-controlled trial. Epilepsia. 2009;50:1899-1909.
18. Ben-Menachem E, Biton V, Jatuzis D, et al. Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia. 2007;48:1308-1317.
19. Gidal B, Garnett W. Epilepsy. In: Dipiro J, et al, eds. Pharmacotherapy: A Pathophysiologic Approach. 6th ed. New York: McGraw-Hill; 2005:1023-1048.
20. Pellock JM, Treatment of epilepsy in the new millennium. Pharmacotherapy. 2000;20:129S-138S.
21. Schachter S. Pharmacology of antiepileptic drugs. Available at: http://www.utdonline.com/online/content/topic.do?topicKey=epil_eeg/5220. Accessed July 15, 2009.
22. Woelfel J. Comparison of antiepileptic drugs. Pharmacist’s Letter/Prescriber's Letter. July 2009;25:1-24.
23. Wolters Kluwer Health Inc. Anticonvulsants. Drug facts and comparisons online. Available at: http://www.efactsonline.com. Accessed July 10, 2009.
24. US Food and Drug Administration. Information for healthcare professionals. Suicidality and antiepileptic drugs [FDA alert]. Available at: http://www.fda.gov/Drugs/DrugSafety/PublicHealthAdvisories/ucm054709.htm. January 31, 2008. Accessed June 30, 2009.
25. French J. Treatment with antiepileptic drugs, new and old. Continuum. 2007;13:71-90.
26. Sheehan M. Polycystic ovarian syndrome: diagnosis and management. Clin Med Res. 2004;2:13-27.
27. Harden CL. Sexual dysfunction in women with epilepsy. Seizure. 2008;17:131-135.
28. Harden CL, Hopp J, Ting TY, et al. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): obstetrical complications and change in seizure frequency. Neurology. 2009;73:126-132.
29. Molgaard-Nielsen D, Hviid A. Newer-generation antiepileptic drugs and the risk of major birth defects. JAMA. 2011;305:1996-2002.
30. Tomson T, Battino D, Bonizonni E, et al. Dose-dependent risk of malformations with antiepileptic drugs: an analysis of data from the EURAP epilepsy and pregnancy registry. Lancet Neurol. 2011;10:609-617.
31. Callaghan BC, Anand K, Hesdorffer D, et al. Likelihood of seizure remission in an adult population with refractory epilepsy. Ann Neurol. 2007;62:382-389.
32. Luciano AL, Shorvon SD. Results of treatment changes in patients with apparently drug-resistant chronic epilepsy. Ann Neurol. 2007;62:375-381.
33. Abramowicz M, ed. Drugs for epilepsy [treatment guidelines]. The Medical Letter. 2008;70:1-12.
34.Stokes T, Shaw EJ, Juarez-Garcia A, et al. Clinical guidelines and evidence review for the epilepsies: diagnosis and management in adults and children in primary and secondary care. London: Royal College of General Practitioners. Available at: www.nice.org.uk/CG020fullguideline. Published October 2004. Accessed July 10, 2009.
35. Garnett WR. Antiepileptic drug treatment: outcomes and adherence. Pharmacotherapy. 2000;20:191S-199S.
36. Briesacher BA, Andrade SE, Fouayzi H, et al. Comparison of drug adherence rates among patients with seven different medical conditions. Pharmacotherapy. 2008;28:437-443.
37. Shinnar S, Gross-Tsur V. Discontinuing antiepileptic drug therapy. In: Wyllie E, ed. The Treatment of Epilepsy. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2001:811-819.
38. Kwan P, Brodie J. Refractory epilepsy: a progressive, intractable but preventable condition? Seizures. 2002;11:77-84.
39. Lings S. Increased driving accident frequency in Danish patients with epilepsy. Neurology. 2001;57:435-439.
40. Krauss GL, Krumholz A, Carter RC, et al. Risk factors for seizure-related motor vehicle crashes in patients with epilepsy. Neurology. 1999;52:1324-1329.
41. American Academy of Neurology, American Epilepsy Society, and Epilepsy Foundation of America. Consensus statements, sample statutory provisions, and model regulations regarding driver licensing and epilepsy. Epilepsia. 1994;35:696-705.
42. Thadani VM, Taylor J. Surgical treatments for epilepsy. Continuum. 2007;13:152-176.
43. Brodie MJ, Kwan P. Staged approach to epilepsy management. Neurology. 2002;58(8 suppl 5):S2-S8.
44. Sillanpaa M, Jalava M, Kaleva O, et al. Long-term prognosis of seizures with onset in childhood. N Engl J Med. 1998;338:1715-1722.
45. Engel J Jr, Wiebe S, French J, et al. Practice parameter: temporal lobe and localized neocortical resections for epilepsy. Neurology. 2003;60:538-547.
46. Boon P, D'Have M, Van Walleghen P, et al. Direct medical costs of refractory epilepsy incurred by three different treatment modalities: a prospective assessment. Epilepsia. 2002;43:96-102.
47. Passaro EA. Outcome of epilepsy surgery. Available at: http://emedicine.medscape.com/article/1185416-overview. Updated May 16, 2011. Accessed June 28, 2011.
48. DeLorenzo RJ, Pellock JM, Towne AR, et al. Epidemiology of status epilepticus. J Clin Neurophysiol. 1995;12:316-325.
49. Logroscino G, Hesdorffer DC, Cascino GD, et al. Long-term mortality after a first episode of status epilepticus. Neurology. 2002; 58:537-541.
50. Kalviaine R. Treatment of status epilepticus. Essential Evidence Plus. Wiley-Blackwell. Available at: http://www.essentialevidenceplus.com/content/ebmg_ebm/766. Accessed July 15, 2009.
51. Prasad K, Al-Roomi K, Krishnan PR, et al. Anticonvulsant therapy for status epilepticus. Cochrane Database Syst Rev. 2005;(4):CD003723.
52. Treiman DM, Meyers PD, Walton NY, et al. A comparison of four treatments for generalized convulsive status epilepticus. N Engl J Med. 1998;339:792-798.
• Prescribe an antiepileptic drug (AED) after a first unprovoked seizure only if the seizure was prolonged or there is a risk of recurrence. C
• Use monotherapy whenever possible; if seizures continue and potential adverse effects prevent an increase in dosage, switch to a different AED and taper off the first agent. A
• Consider gradual withdrawal of AEDs from patients who have been seizure-free for 2 to 5 years. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Joe G, a 44-year-old man who has been your patient for years, comes to your office 48 hours after having a seizure. He has no history of seizures, had no warning signs or symptoms, and felt fine all day, but simply collapsed when the seizure occurred. He was transported to the emergency department (ED), and found to be postictal, with no further seizure activity. The ED work-up included a hemogram, comprehensive metabolic panel, and computed tomography brain scan, all of which were normal. An hour later, Joe had a normal neurological exam, then underwent electroencephalography (EEG) and magnetic resonance imaging (MRI) and was discharged home without medication.
How would you treat this patient?
About 10% of Americans will experience a seizure at some point in their lives,1,2 and more than 3 million have epilepsy.3 The incidence ranges from 1% among 20-year-olds to more than 3% by the age of 75.1,2
To adequately care for such patients—whether they have had multiple seizures or only one—you need to know whether they’re at risk for recurrences, when (or if) to prescribe an AED, and which agents provide optimal seizure control with the fewest adverse effects. You also need to know when a referral to an epilepsy specialist is indicated, when or whether it’s safe for patients to stop taking antiseizure medication, and how to address lifestyle issues that patients with epilepsy often need help with.
This review addresses these and other questions.
Is it epilepsy? How to respond to a single seizure
A seizure—a transient occurrence of signs or symptoms due to abnormal excessive or synchronous neural activity in the brain—can be either focal (partial) or generalized. In addition, seizures can be broadly divided into 2 categories, based on etiology:
Provoked seizures are caused by an acute structural, toxic, or metabolic insult to the brain, and, presumably, would not have occurred if the underlying medical condition did not exist. Treating the cause—eg, alcohol withdrawal, hyponatremia, or hypoglycemia—should prevent a recurrence.
Unprovoked seizures have no apparent underlying cause. Epilepsy is defined as a chronic condition characterized by ≥2 unprovoked seizures at least 24 hours apart, and epilepsy syndromes are classified as localization-related or generalized (TABLE 1).1,4,5
Generally, epileptologists do not recommend symptomatic treatment of a first unprovoked seizure6—a consensus based on several randomized controlled trials that found immediate treatment with an AED reduced the risk of a subsequent seizure in the short term, but did not affect long-term outcomes or the development of epilepsy.7
Treatment should begin after a single seizure, however, if the seizure was prolonged or there is an increased risk of recurrence.6 Factors that increase this risk include an abnormal EEG, particularly if the abnormality is epileptiform; the presence of a brain lesion; a localized (focal) seizure; and an abnormal neurologic exam.8 A history of status epilepticus—a single, unremitting seizure lasting ≥5 to 10 minutes or frequent seizures without a return to neurologic baseline in between—or complex febrile seizures, and a family history of epilepsy are risk factors for recurrence, as well.7
When the patient is a child. Prescribing an AED for a child after a first unprovoked seizure is not indicated to prevent the development of epilepsy, but may be considered, as for adults, in circumstances where the benefit of reducing the risk of a second seizure outweighs the risk of pharmacologic and psychosocial adverse effects.9
CASE Joe’s ED records show that his MRI was normal, but his EEG revealed an epileptogenic focus on the right temporal region—a finding that indicates that he has an elevated risk of recurrence and is a candidate for an AED. Before selecting a particular agent, you review his chart.
Joe is taking a thiazide diuretic and a calcium channel blocker for hypertension. He was a heavy drinker until he had an episode of pancreatitis 10 years ago, and has been abstinent ever since. About 5 years ago, he suffered from depression and was treated with sertraline, but the depression resolved and the drug was discontinued 3 years ago. The patient’s mother and brother have type 2 diabetes and his father had a myocardial infarction before the age of 60. Joe was laid off from his sales job 18 months ago and is actively seeking employment. At this point, you consider a broad-spectrum AED that would not interact with his current medications or adversely affect his medical conditions, and would be relatively inexpensive.
TABLE 1
Identifying seizures and types of epilepsy:1,4,5 International League Against Epilepsy classification
| Type of seizure |
Focal
Generalized
|
| Type of epilepsy syndrome* |
Localization related (partial or focal)
Generalized
|
| *This is a partial listing, with selected examples of epilepsy syndromes. |
What to consider in a first-line drug
The number of AEDs on the market has increased sharply in the past few years, giving physicians many medications to choose from. Selecting the optimal drug is particularly important for the initial treatment, as many patients remain on the first AED for years. Second-generation AEDs have been found to be as effective as, and better tolerated than, first-generation antiseizure drugs. But all AEDs carry a warning of a potential increase in suicide risk and the need to monitor patients for behavior changes.10
Before selecting an AED for a particular patient, consider the following questions:
What type of seizure? AEDs are generally classified by spectrum of activity into “narrow-spectrum” and “broad-spectrum.” Narrow-spectrum drugs are more effective for controlling partial seizures, but have the potential to exacerbate generalized seizures; broad-spectrum AEDs can be used for both. (TABLE 211-18 lists indications for first- and second-generation AEDs based on type of epilepsy.) If there’s no definitive diagnosis of the type of epilepsy a patient has, use a broad-spectrum drug.
What other drugs is the patient taking? If the AED will be added to the patient’s current medication regimen, look closely at potential pharmacodynamic drug-drug interactions, and consider whether a dosage adjustment is needed. Determine, too, whether the patient has any comorbidities that could affect his or her response to the AED.
Side effects, such as weight gain or loss, urolithiasis, and hepatic enzyme induction, are key considerations. (TABLE W1,19-24 which details dose, side effects, and costs of first- and second-generation AEDs, can be found at jfponline.com.)
Is the patient elderly? AED clearance is reduced in the elderly, so lower doses are needed. Reduction in serum albumin increases the free or active component of highly protein-bound drugs, increasing the likelihood of adverse effects.
Is the patient female? Some AEDs may have effects on women’s hormonal function, sexuality, bone health, and pregnancy.25 Hepatic enzyme inducers increase the clearance of oral contraceptives, reducing their efficacy. Vitamin D and calcium metabolism can also be affected, which can lead to osteomalacia. Valproate treatment in women is associated with higher levels of insulin, testosterone, and triglycerides.26 Cytochrome P-450-activating AEDs in general are associated with higher testosterone levels and reduced libido.27
Potential pregnancy is another consideration. Women with epilepsy are able to bear healthy children. What’s more, patients whose seizures are controlled with AEDs should be maintained on medication throughout pregnancy, as the risk of fetal harm from seizures generally outweighs the teratogenicity of the drug.28
Although large studies are limited, a study of 1532 infants exposed to AEDs in the first trimester did not find an increase in major birth defects compared with infants without such exposure.29 More recently, a large observational cohort study conducted in more than 40 countries found that the possibility of harm to a developing fetus is not only drug-specific but also dose-related.30 (To learn more, see “Pregnancy and epilepsy—when you’re managing both,” in the December 2010 issue of The Journal of Family Practice.)
Is cost a factor? Finally, consider the cost of the AED you would like to prescribe, and whether the patient has a prescription drug plan or the means to pay for his prescription.
CASE After a discussion of potential side effects, including the potential for suicidal ideation associated with AEDs, you prescribe carbamazepine for Joe as seizure prophylaxis, because it is the least expensive of the broad-spectrum AEDs and is unlikely to exacerbate his previous pancreatitis or interact with his current medications.
TABLE 2
Choosing an AED: What to consider11-18
| Epilepsy type | |||||
| Localization-related (focal/partial) | Idiopathic (generalized) | Nonidiopathic (generalized) | |||
| Anticonvulsant* | Tonic-clonic | Absence | Myoclonic | ||
| First generation | |||||
| Carbamazepine† | √ | √ | |||
| Ethosuximide† | √ | ||||
| Phenobarbital† | √ | √ | √ | ||
| Phenytoin† | √ | √ | √ | ||
| Primidone | √ | √ | √ | ||
| Valproate† | √ | √ | √ | √ | √ |
| Second generation | |||||
| Felbamate | √ | √ | |||
| Gabapentin† | √ | ||||
| Lacosamide | √ | ||||
| Lamotrigine | √† | √ | √‡ | √ | |
| Levetiracetam | √ | √ | √ | ||
| Oxcarbazepine† | √ | ||||
| Pregabalin | √ | ||||
| Rufinamide | √ | √ | |||
| Tiagabine | √ | ||||
| Topiramate | √‡ | √ | √ | ||
| Vigabatrin | √ | √ | |||
| Zonisamide | √ | √ | |||
| *Bold type indicates broad-spectrum antiepileptic drugs. †Supported by American Academy of Neurology (AAN) evidence-based guideline level A or B recommendation for monotherapy in newly diagnosed epilepsy patients. ‡Supported by AAN evidence-based guideline level B recommendation for monotherapy in newly diagnosed absence epilepsy. | |||||
TABLE W1
A closer look at antiepileptic drugs19-24
| Drug name | Maintenance dosage | Adverse effects | Cost (30-day supply)* | |
| Common | Rare/idiosyncratic | |||
| First generation | ||||
| Carbamazepine | 800-1200 mg/d | Dizziness, drowsiness, diplopia, nausea, vomiting, diarrhea, rash, pruritus, SIADH | Aplastic anemia, agranulocytosis, hyponatremia, SJS, hepatic failure, pancreatitis, suicidal ideation | $4-$50 (XR: $200) |
| Ethosuximide | 20 mg/kg per day | Sleep disturbance, drowsiness, hyperactivity, behavior changes, headache, nausea, vomiting, hiccups | Agranulocytosis, aplastic anemia, SJS, hepatic failure, serum sickness, suicidal ideation | $40-150 |
| Phenobarbital | 1-4 mg/kg per day; 120-400 mg/d | Altered sleep cycles, sedation, ataxia, lethargy, behavior changes, hyperactivity, nausea, rash | Agranulocytosis, dermatitis, SJS, hepatic failure, serum sickness, connective tissue disorders, metabolic bone disease, intellect blunting, suicidal ideation | $4-$10 |
| Phenytoin | 300-600 mg/d | Confusion, slurred speech, double vision, ataxia, nystagmus, neuropathy, hirsutism, acne, gingival hyperplasia | Neuropathy, agranulocytosis, SJS, immune reactions/serum sickness, hepatic failure, skin thickening, metabolic bone disease, suicidal ideation | $35 |
| Valproic acid | 60-350 mg/kg per day | Tremor, weight gain, PCOS, nausea, vomiting, alopecia, easy bruising | Hepatic failure, pancreatitis, hearing loss, blood dyscrasias/thrombocytopenia, hyperammonemia, encephalopathy, osteoporosis, suicidal ideation | $40 (ER: $150) |
| Second generation | ||||
| Felbamate | 2400-3600 mg/d | Somnolence, nausea, vomiting, weight loss, anorexia | Aplastic anemia (>13 years), hepatic failure, suicidal ideation | $300-$500† |
| Gabapentin | 900-1800 mg/d | Somnolence, fatigue, weight gain, nystagmus | Pedal edema, suicidal ideation | $4-$100 |
| Lacosamide | 200-400 mg/d | Headache, dizziness, ataxia, nausea, diplopia | Euphoria, prolongation of PR interval, heart block, suicidal ideation | $420† |
| Lamotrigine | 300-500 mg/d | Dizziness, ataxia, nausea, somnolence, rash | SJS, hypersensitivity reactions (renal/hepatic failure), DIC, suicidal ideation | $30-$100 |
| Levetiracetam | 3000 mg/d | Somnolence, dizziness, aggression, agitation, anxiety, weight loss | Infection, pancytopenia, liver failure, suicidal ideation | $30-$100 (XR: $245†) |
| Oxcarbazepine | 1200 mg/d | Somnolence, fatigue, headache, ataxia, nausea, rash | Hyponatremia, SJS, TEN, angioedema | $250-$1000 |
| Pregabalin | 150-600 mg/d | Peripheral edema, dry mouth, dizziness, ataxia, diplopia, weight gain | Angioedema, CK elevation, mild PR interval prolongation, suicidal ideation | $100-$350† |
| Rufinamide | 3200 mg/d | Headache, dizziness, fatigue, nausea | Shortened QT interval, hypersensitivity rash, suicidal ideation | $400-$750† |
| Tiagabine | 32-56 mg/d | Difficulty concentrating, dizziness, headache, somnolence, nervousness | Spike-wave stupor, sudden death, suicidal ideation | $140-$650† |
| Topiramate | 200-400 mg/d | Somnolence, dizziness, fatigue, weight loss, difficulty concentrating, speech problems, paresthesias, diarrhea, nausea | Acute myopia and glaucoma, hyperthermia (children); metabolic acidosis, hyperammonemia, liver failure, oligohydrosis, SJS/TEN, kidney stones, suicidal ideation | $40 - $100 |
| Vigabatrin | 1500 mg/d | Fatigue, somnolence, nystagmus, tremor, weight gain | Vision loss (30% of patients) blurred vision, arthralgia, suicidal ideation | :$50 -$100† |
| Zonisamide | 400- 600 mg/d | Somnolence, difficulty concentrating, anorexia, nausea | SJS, TEN, aplastic anemia, agranulocytosis, nephrolithiasis/, oligohydrosis, acidosis, suicidal ideation | $50-$200 |
| CK, creatine kinase; DIC, disseminated intravascular coagulation; ER, extended release; IV, intravenous; PCOS, polycystic ovarian syndrome; SIADH, syndrome of inappropriate antidiuretic hormone hypersecretion; SJS, Stevens-Johnson syndrome; TEN, toxic epidermal necrolysis, XR, extended release. *Costs from www.drugstore.com, www.savewithgenericdrugs.com, and www.pharmacychecker.com. †No generic available. | ||||
When to add a second AED
Monotherapy is the preferred method of epilepsy treatment, and controls seizures for 70% to 90% of patients.31,32 If seizures continue and potential adverse effects prevent you from increasing the dosage, switching to a different AED, then tapering off the first agent, is recommended.33,34
If the new AED fails to provide adequate seizure control, consider combination therapy. An additional 10% to 15% of patients with epilepsy achieve control with dual therapy.33,34
Many second-generation agents are approved for adjunctive therapy. However, the use of 2 AEDs increases the risk of toxicities and drug interactions, and requires complex dosage adjustments, which should be done slowly and cautiously. Combination therapy also increases costs and may cause a decrease in compliance.33,34
Noncompliance is the single most common reason for treatment failure in patients with epilepsy, occurring at an estimated rate of up to 60%.35,36 The complexity of the drug regimen is the major cause, regardless of patient age, sex, psychomotor development, seizure type, or seizure frequency.35,36
Because of the lack of good clinical trials of combination antiepilepsy therapy, no evidence is available to indicate which AEDs are safe and effective when taken together. There is, however, evidence that certain combinations should be avoided due to the risk of increased adverse effects. These include phenobarbital/valproate, phenytoin/carbamazepine, and carbamazepine/lamotrigine.25
Managing the patient who is seizure-free
After a patient has been seizure-free for 2 to 5 years, consider a reduction in, or a discontinuation of, his or her AED. The relapse rate varies from 10% to 70%, with meta-analyses showing a rate of 25% in the first year and 29% in the second year.19,37 The American Academy of Neurology (AAN) has published an evidence-based guideline for discontinuing AEDs in seizure-free patients, available at www.aan.com/professionals/practice/pdfs/gl0007.pdf.
Withdrawal should be gradual and, for patients on combination therapy, carried out one drug at a time to prevent a recurrence of seizures or status epilepticus. The AAN recommends a 2- to- 3-month withdrawal period for AEDs (and longer for benzodiazepines), although relapse rates have been found to be lower when the medication is withdrawn more slowly, over about 6 months.19,34 If seizures recur after withdrawal, restart the AEDs at previous dosages.19,34,38
Should the patient drive?
For patients with epilepsy, loss of independence related to driving restrictions is a major source of stress. A 10-year follow-up study of Danish patients with epilepsy found a 7-fold increase in motor vehicle accidents (MVAs) in patients with seizure disorders.39 Other studies have shown that the seizure-free interval is the best predictor of involvement in an MVA.40
The risk of driving accidents decreases as the seizure-free interval increases. Unfortunately, however, a decline in patient compliance is also associated with longer seizure-free intervals—creating the potential for recurrence and driving risk. Because of this discrepancy, a consensus statement from the AAN, American Epilepsy Society, and Epilepsy Foundation of America recommends a minimum 3-month seizure-free interval before patients are allowed to drive.41
Use clinical judgment in deciding whether to extend the seizure-free period. State laws vary widely regarding the need to report patients with seizure disorders, limitations on professional drivers, and seizure-free intervals required, so it is important to be familiar with the laws in your state. The Epilepsy Foundation has a helpful online resource with a database detailing individual state statutes (http://www.epilepsyfoundation.org/living/wellness/transportation/driverlicensing.cfm).
The danger of uncontrolled seizures
Overall, AEDs effectively control 70% of 80% of cases; the remaining 20% to 30% are considered medically refractory.38 What’s more, after 2 AED failures, a patient’s chances of achieving full seizure control with additional drugs are no better than 10% to 20%.42 And, as more drugs are tried, the likelihood of full control declines even further.43
Patients with uncontrolled seizures have a cumulative risk of sudden unexpected death in epilepsy (SUDEP) of 0.5% per year.44 Cognitive decline is associated with uncontrolled epilepsy, as well. In children, frequent seizures may significantly alter neuronal networks, affecting cognitive and motor development.
Is your patient a candidate for surgery?
Patients with disabling complex partial seizures that remain uncontrolled after 2 or more AED trials (either as monotherapy or in combination) should be referred to an epilepsy specialty center for evaluation for surgery.45 This should be considered as early as possible to afford the patient the best chance of achieving seizure control.
Successful epilepsy surgery—in which the portion of the brain causing the misfiring that causes the seizures is removed—often results in a better quality of life; it is also cost effective.46 Not everyone with refractory epilepsy is a candidate for surgery, of course. Among those who are, however, 50% to 70% of patients can expect to have improved seizure control.47
Status epilepticus is a medical emergency
A patient who develops status epilepticus is at high risk and requires immediate, and simultaneous, evaluation and treatment. Status epilepticus carries nearly a 20% mortality from the first episode,48 and the 10-year mortality rate after an episode of status epilepticus is as high as 40%.49
Although most of the deaths associated with status epilepticus are due to the underlying pathology, early treatment can prevent or ameliorate complications from rhabdomyolysis and irreversible anoxic neuronal damage.50
A benzodiazepine (typically, a 10-mg IV bolus of diazepam) is the initial treatment for status epilepticus, followed by or concurrent with fosphenytoin (15-18 mg/kg). If status epilepticus remains refractory to first-line drugs (lasting >30 minutes), intubation and transfer to an intensive care setting may be required, and a neurological consult should be obtained.
Pharmacologic treatment of status epilepticus falls into 3 main classes: benzodiazepines, standard AEDs, and general anesthetics such as propofol. Benzodiazepines act very rapidly to control most prolonged seizures, and are the first-line treatment choice. Diazepam has long been the mainstay of treatment, and is usually readily available. However, in both a large systematic review and a head-to-head trial, lorazepam was found to be superior to diazepam in ending seizure activity and maintaining seizure control without the use of other medications51,52—and is now the drug of choice for initial treatment of status epilepticus.
CASE You continue to see Joe every 3 to 4 months to monitor his basic blood work and mood. A year after his seizure, he remains seizure-free and is tolerating the AED without any adverse effects.
CORRESPONDENCE
William J. Geiger, MD, FAAFP, Medical College of Wisconsin, Columbia St. Mary’s Family Medicine Residency, 1121 East North Avenue, Milwaukee, WI 53212; [email protected]
• Prescribe an antiepileptic drug (AED) after a first unprovoked seizure only if the seizure was prolonged or there is a risk of recurrence. C
• Use monotherapy whenever possible; if seizures continue and potential adverse effects prevent an increase in dosage, switch to a different AED and taper off the first agent. A
• Consider gradual withdrawal of AEDs from patients who have been seizure-free for 2 to 5 years. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Joe G, a 44-year-old man who has been your patient for years, comes to your office 48 hours after having a seizure. He has no history of seizures, had no warning signs or symptoms, and felt fine all day, but simply collapsed when the seizure occurred. He was transported to the emergency department (ED), and found to be postictal, with no further seizure activity. The ED work-up included a hemogram, comprehensive metabolic panel, and computed tomography brain scan, all of which were normal. An hour later, Joe had a normal neurological exam, then underwent electroencephalography (EEG) and magnetic resonance imaging (MRI) and was discharged home without medication.
How would you treat this patient?
About 10% of Americans will experience a seizure at some point in their lives,1,2 and more than 3 million have epilepsy.3 The incidence ranges from 1% among 20-year-olds to more than 3% by the age of 75.1,2
To adequately care for such patients—whether they have had multiple seizures or only one—you need to know whether they’re at risk for recurrences, when (or if) to prescribe an AED, and which agents provide optimal seizure control with the fewest adverse effects. You also need to know when a referral to an epilepsy specialist is indicated, when or whether it’s safe for patients to stop taking antiseizure medication, and how to address lifestyle issues that patients with epilepsy often need help with.
This review addresses these and other questions.
Is it epilepsy? How to respond to a single seizure
A seizure—a transient occurrence of signs or symptoms due to abnormal excessive or synchronous neural activity in the brain—can be either focal (partial) or generalized. In addition, seizures can be broadly divided into 2 categories, based on etiology:
Provoked seizures are caused by an acute structural, toxic, or metabolic insult to the brain, and, presumably, would not have occurred if the underlying medical condition did not exist. Treating the cause—eg, alcohol withdrawal, hyponatremia, or hypoglycemia—should prevent a recurrence.
Unprovoked seizures have no apparent underlying cause. Epilepsy is defined as a chronic condition characterized by ≥2 unprovoked seizures at least 24 hours apart, and epilepsy syndromes are classified as localization-related or generalized (TABLE 1).1,4,5
Generally, epileptologists do not recommend symptomatic treatment of a first unprovoked seizure6—a consensus based on several randomized controlled trials that found immediate treatment with an AED reduced the risk of a subsequent seizure in the short term, but did not affect long-term outcomes or the development of epilepsy.7
Treatment should begin after a single seizure, however, if the seizure was prolonged or there is an increased risk of recurrence.6 Factors that increase this risk include an abnormal EEG, particularly if the abnormality is epileptiform; the presence of a brain lesion; a localized (focal) seizure; and an abnormal neurologic exam.8 A history of status epilepticus—a single, unremitting seizure lasting ≥5 to 10 minutes or frequent seizures without a return to neurologic baseline in between—or complex febrile seizures, and a family history of epilepsy are risk factors for recurrence, as well.7
When the patient is a child. Prescribing an AED for a child after a first unprovoked seizure is not indicated to prevent the development of epilepsy, but may be considered, as for adults, in circumstances where the benefit of reducing the risk of a second seizure outweighs the risk of pharmacologic and psychosocial adverse effects.9
CASE Joe’s ED records show that his MRI was normal, but his EEG revealed an epileptogenic focus on the right temporal region—a finding that indicates that he has an elevated risk of recurrence and is a candidate for an AED. Before selecting a particular agent, you review his chart.
Joe is taking a thiazide diuretic and a calcium channel blocker for hypertension. He was a heavy drinker until he had an episode of pancreatitis 10 years ago, and has been abstinent ever since. About 5 years ago, he suffered from depression and was treated with sertraline, but the depression resolved and the drug was discontinued 3 years ago. The patient’s mother and brother have type 2 diabetes and his father had a myocardial infarction before the age of 60. Joe was laid off from his sales job 18 months ago and is actively seeking employment. At this point, you consider a broad-spectrum AED that would not interact with his current medications or adversely affect his medical conditions, and would be relatively inexpensive.
TABLE 1
Identifying seizures and types of epilepsy:1,4,5 International League Against Epilepsy classification
| Type of seizure |
Focal
Generalized
|
| Type of epilepsy syndrome* |
Localization related (partial or focal)
Generalized
|
| *This is a partial listing, with selected examples of epilepsy syndromes. |
What to consider in a first-line drug
The number of AEDs on the market has increased sharply in the past few years, giving physicians many medications to choose from. Selecting the optimal drug is particularly important for the initial treatment, as many patients remain on the first AED for years. Second-generation AEDs have been found to be as effective as, and better tolerated than, first-generation antiseizure drugs. But all AEDs carry a warning of a potential increase in suicide risk and the need to monitor patients for behavior changes.10
Before selecting an AED for a particular patient, consider the following questions:
What type of seizure? AEDs are generally classified by spectrum of activity into “narrow-spectrum” and “broad-spectrum.” Narrow-spectrum drugs are more effective for controlling partial seizures, but have the potential to exacerbate generalized seizures; broad-spectrum AEDs can be used for both. (TABLE 211-18 lists indications for first- and second-generation AEDs based on type of epilepsy.) If there’s no definitive diagnosis of the type of epilepsy a patient has, use a broad-spectrum drug.
What other drugs is the patient taking? If the AED will be added to the patient’s current medication regimen, look closely at potential pharmacodynamic drug-drug interactions, and consider whether a dosage adjustment is needed. Determine, too, whether the patient has any comorbidities that could affect his or her response to the AED.
Side effects, such as weight gain or loss, urolithiasis, and hepatic enzyme induction, are key considerations. (TABLE W1,19-24 which details dose, side effects, and costs of first- and second-generation AEDs, can be found at jfponline.com.)
Is the patient elderly? AED clearance is reduced in the elderly, so lower doses are needed. Reduction in serum albumin increases the free or active component of highly protein-bound drugs, increasing the likelihood of adverse effects.
Is the patient female? Some AEDs may have effects on women’s hormonal function, sexuality, bone health, and pregnancy.25 Hepatic enzyme inducers increase the clearance of oral contraceptives, reducing their efficacy. Vitamin D and calcium metabolism can also be affected, which can lead to osteomalacia. Valproate treatment in women is associated with higher levels of insulin, testosterone, and triglycerides.26 Cytochrome P-450-activating AEDs in general are associated with higher testosterone levels and reduced libido.27
Potential pregnancy is another consideration. Women with epilepsy are able to bear healthy children. What’s more, patients whose seizures are controlled with AEDs should be maintained on medication throughout pregnancy, as the risk of fetal harm from seizures generally outweighs the teratogenicity of the drug.28
Although large studies are limited, a study of 1532 infants exposed to AEDs in the first trimester did not find an increase in major birth defects compared with infants without such exposure.29 More recently, a large observational cohort study conducted in more than 40 countries found that the possibility of harm to a developing fetus is not only drug-specific but also dose-related.30 (To learn more, see “Pregnancy and epilepsy—when you’re managing both,” in the December 2010 issue of The Journal of Family Practice.)
Is cost a factor? Finally, consider the cost of the AED you would like to prescribe, and whether the patient has a prescription drug plan or the means to pay for his prescription.
CASE After a discussion of potential side effects, including the potential for suicidal ideation associated with AEDs, you prescribe carbamazepine for Joe as seizure prophylaxis, because it is the least expensive of the broad-spectrum AEDs and is unlikely to exacerbate his previous pancreatitis or interact with his current medications.
TABLE 2
Choosing an AED: What to consider11-18
| Epilepsy type | |||||
| Localization-related (focal/partial) | Idiopathic (generalized) | Nonidiopathic (generalized) | |||
| Anticonvulsant* | Tonic-clonic | Absence | Myoclonic | ||
| First generation | |||||
| Carbamazepine† | √ | √ | |||
| Ethosuximide† | √ | ||||
| Phenobarbital† | √ | √ | √ | ||
| Phenytoin† | √ | √ | √ | ||
| Primidone | √ | √ | √ | ||
| Valproate† | √ | √ | √ | √ | √ |
| Second generation | |||||
| Felbamate | √ | √ | |||
| Gabapentin† | √ | ||||
| Lacosamide | √ | ||||
| Lamotrigine | √† | √ | √‡ | √ | |
| Levetiracetam | √ | √ | √ | ||
| Oxcarbazepine† | √ | ||||
| Pregabalin | √ | ||||
| Rufinamide | √ | √ | |||
| Tiagabine | √ | ||||
| Topiramate | √‡ | √ | √ | ||
| Vigabatrin | √ | √ | |||
| Zonisamide | √ | √ | |||
| *Bold type indicates broad-spectrum antiepileptic drugs. †Supported by American Academy of Neurology (AAN) evidence-based guideline level A or B recommendation for monotherapy in newly diagnosed epilepsy patients. ‡Supported by AAN evidence-based guideline level B recommendation for monotherapy in newly diagnosed absence epilepsy. | |||||
TABLE W1
A closer look at antiepileptic drugs19-24
| Drug name | Maintenance dosage | Adverse effects | Cost (30-day supply)* | |
| Common | Rare/idiosyncratic | |||
| First generation | ||||
| Carbamazepine | 800-1200 mg/d | Dizziness, drowsiness, diplopia, nausea, vomiting, diarrhea, rash, pruritus, SIADH | Aplastic anemia, agranulocytosis, hyponatremia, SJS, hepatic failure, pancreatitis, suicidal ideation | $4-$50 (XR: $200) |
| Ethosuximide | 20 mg/kg per day | Sleep disturbance, drowsiness, hyperactivity, behavior changes, headache, nausea, vomiting, hiccups | Agranulocytosis, aplastic anemia, SJS, hepatic failure, serum sickness, suicidal ideation | $40-150 |
| Phenobarbital | 1-4 mg/kg per day; 120-400 mg/d | Altered sleep cycles, sedation, ataxia, lethargy, behavior changes, hyperactivity, nausea, rash | Agranulocytosis, dermatitis, SJS, hepatic failure, serum sickness, connective tissue disorders, metabolic bone disease, intellect blunting, suicidal ideation | $4-$10 |
| Phenytoin | 300-600 mg/d | Confusion, slurred speech, double vision, ataxia, nystagmus, neuropathy, hirsutism, acne, gingival hyperplasia | Neuropathy, agranulocytosis, SJS, immune reactions/serum sickness, hepatic failure, skin thickening, metabolic bone disease, suicidal ideation | $35 |
| Valproic acid | 60-350 mg/kg per day | Tremor, weight gain, PCOS, nausea, vomiting, alopecia, easy bruising | Hepatic failure, pancreatitis, hearing loss, blood dyscrasias/thrombocytopenia, hyperammonemia, encephalopathy, osteoporosis, suicidal ideation | $40 (ER: $150) |
| Second generation | ||||
| Felbamate | 2400-3600 mg/d | Somnolence, nausea, vomiting, weight loss, anorexia | Aplastic anemia (>13 years), hepatic failure, suicidal ideation | $300-$500† |
| Gabapentin | 900-1800 mg/d | Somnolence, fatigue, weight gain, nystagmus | Pedal edema, suicidal ideation | $4-$100 |
| Lacosamide | 200-400 mg/d | Headache, dizziness, ataxia, nausea, diplopia | Euphoria, prolongation of PR interval, heart block, suicidal ideation | $420† |
| Lamotrigine | 300-500 mg/d | Dizziness, ataxia, nausea, somnolence, rash | SJS, hypersensitivity reactions (renal/hepatic failure), DIC, suicidal ideation | $30-$100 |
| Levetiracetam | 3000 mg/d | Somnolence, dizziness, aggression, agitation, anxiety, weight loss | Infection, pancytopenia, liver failure, suicidal ideation | $30-$100 (XR: $245†) |
| Oxcarbazepine | 1200 mg/d | Somnolence, fatigue, headache, ataxia, nausea, rash | Hyponatremia, SJS, TEN, angioedema | $250-$1000 |
| Pregabalin | 150-600 mg/d | Peripheral edema, dry mouth, dizziness, ataxia, diplopia, weight gain | Angioedema, CK elevation, mild PR interval prolongation, suicidal ideation | $100-$350† |
| Rufinamide | 3200 mg/d | Headache, dizziness, fatigue, nausea | Shortened QT interval, hypersensitivity rash, suicidal ideation | $400-$750† |
| Tiagabine | 32-56 mg/d | Difficulty concentrating, dizziness, headache, somnolence, nervousness | Spike-wave stupor, sudden death, suicidal ideation | $140-$650† |
| Topiramate | 200-400 mg/d | Somnolence, dizziness, fatigue, weight loss, difficulty concentrating, speech problems, paresthesias, diarrhea, nausea | Acute myopia and glaucoma, hyperthermia (children); metabolic acidosis, hyperammonemia, liver failure, oligohydrosis, SJS/TEN, kidney stones, suicidal ideation | $40 - $100 |
| Vigabatrin | 1500 mg/d | Fatigue, somnolence, nystagmus, tremor, weight gain | Vision loss (30% of patients) blurred vision, arthralgia, suicidal ideation | :$50 -$100† |
| Zonisamide | 400- 600 mg/d | Somnolence, difficulty concentrating, anorexia, nausea | SJS, TEN, aplastic anemia, agranulocytosis, nephrolithiasis/, oligohydrosis, acidosis, suicidal ideation | $50-$200 |
| CK, creatine kinase; DIC, disseminated intravascular coagulation; ER, extended release; IV, intravenous; PCOS, polycystic ovarian syndrome; SIADH, syndrome of inappropriate antidiuretic hormone hypersecretion; SJS, Stevens-Johnson syndrome; TEN, toxic epidermal necrolysis, XR, extended release. *Costs from www.drugstore.com, www.savewithgenericdrugs.com, and www.pharmacychecker.com. †No generic available. | ||||
When to add a second AED
Monotherapy is the preferred method of epilepsy treatment, and controls seizures for 70% to 90% of patients.31,32 If seizures continue and potential adverse effects prevent you from increasing the dosage, switching to a different AED, then tapering off the first agent, is recommended.33,34
If the new AED fails to provide adequate seizure control, consider combination therapy. An additional 10% to 15% of patients with epilepsy achieve control with dual therapy.33,34
Many second-generation agents are approved for adjunctive therapy. However, the use of 2 AEDs increases the risk of toxicities and drug interactions, and requires complex dosage adjustments, which should be done slowly and cautiously. Combination therapy also increases costs and may cause a decrease in compliance.33,34
Noncompliance is the single most common reason for treatment failure in patients with epilepsy, occurring at an estimated rate of up to 60%.35,36 The complexity of the drug regimen is the major cause, regardless of patient age, sex, psychomotor development, seizure type, or seizure frequency.35,36
Because of the lack of good clinical trials of combination antiepilepsy therapy, no evidence is available to indicate which AEDs are safe and effective when taken together. There is, however, evidence that certain combinations should be avoided due to the risk of increased adverse effects. These include phenobarbital/valproate, phenytoin/carbamazepine, and carbamazepine/lamotrigine.25
Managing the patient who is seizure-free
After a patient has been seizure-free for 2 to 5 years, consider a reduction in, or a discontinuation of, his or her AED. The relapse rate varies from 10% to 70%, with meta-analyses showing a rate of 25% in the first year and 29% in the second year.19,37 The American Academy of Neurology (AAN) has published an evidence-based guideline for discontinuing AEDs in seizure-free patients, available at www.aan.com/professionals/practice/pdfs/gl0007.pdf.
Withdrawal should be gradual and, for patients on combination therapy, carried out one drug at a time to prevent a recurrence of seizures or status epilepticus. The AAN recommends a 2- to- 3-month withdrawal period for AEDs (and longer for benzodiazepines), although relapse rates have been found to be lower when the medication is withdrawn more slowly, over about 6 months.19,34 If seizures recur after withdrawal, restart the AEDs at previous dosages.19,34,38
Should the patient drive?
For patients with epilepsy, loss of independence related to driving restrictions is a major source of stress. A 10-year follow-up study of Danish patients with epilepsy found a 7-fold increase in motor vehicle accidents (MVAs) in patients with seizure disorders.39 Other studies have shown that the seizure-free interval is the best predictor of involvement in an MVA.40
The risk of driving accidents decreases as the seizure-free interval increases. Unfortunately, however, a decline in patient compliance is also associated with longer seizure-free intervals—creating the potential for recurrence and driving risk. Because of this discrepancy, a consensus statement from the AAN, American Epilepsy Society, and Epilepsy Foundation of America recommends a minimum 3-month seizure-free interval before patients are allowed to drive.41
Use clinical judgment in deciding whether to extend the seizure-free period. State laws vary widely regarding the need to report patients with seizure disorders, limitations on professional drivers, and seizure-free intervals required, so it is important to be familiar with the laws in your state. The Epilepsy Foundation has a helpful online resource with a database detailing individual state statutes (http://www.epilepsyfoundation.org/living/wellness/transportation/driverlicensing.cfm).
The danger of uncontrolled seizures
Overall, AEDs effectively control 70% of 80% of cases; the remaining 20% to 30% are considered medically refractory.38 What’s more, after 2 AED failures, a patient’s chances of achieving full seizure control with additional drugs are no better than 10% to 20%.42 And, as more drugs are tried, the likelihood of full control declines even further.43
Patients with uncontrolled seizures have a cumulative risk of sudden unexpected death in epilepsy (SUDEP) of 0.5% per year.44 Cognitive decline is associated with uncontrolled epilepsy, as well. In children, frequent seizures may significantly alter neuronal networks, affecting cognitive and motor development.
Is your patient a candidate for surgery?
Patients with disabling complex partial seizures that remain uncontrolled after 2 or more AED trials (either as monotherapy or in combination) should be referred to an epilepsy specialty center for evaluation for surgery.45 This should be considered as early as possible to afford the patient the best chance of achieving seizure control.
Successful epilepsy surgery—in which the portion of the brain causing the misfiring that causes the seizures is removed—often results in a better quality of life; it is also cost effective.46 Not everyone with refractory epilepsy is a candidate for surgery, of course. Among those who are, however, 50% to 70% of patients can expect to have improved seizure control.47
Status epilepticus is a medical emergency
A patient who develops status epilepticus is at high risk and requires immediate, and simultaneous, evaluation and treatment. Status epilepticus carries nearly a 20% mortality from the first episode,48 and the 10-year mortality rate after an episode of status epilepticus is as high as 40%.49
Although most of the deaths associated with status epilepticus are due to the underlying pathology, early treatment can prevent or ameliorate complications from rhabdomyolysis and irreversible anoxic neuronal damage.50
A benzodiazepine (typically, a 10-mg IV bolus of diazepam) is the initial treatment for status epilepticus, followed by or concurrent with fosphenytoin (15-18 mg/kg). If status epilepticus remains refractory to first-line drugs (lasting >30 minutes), intubation and transfer to an intensive care setting may be required, and a neurological consult should be obtained.
Pharmacologic treatment of status epilepticus falls into 3 main classes: benzodiazepines, standard AEDs, and general anesthetics such as propofol. Benzodiazepines act very rapidly to control most prolonged seizures, and are the first-line treatment choice. Diazepam has long been the mainstay of treatment, and is usually readily available. However, in both a large systematic review and a head-to-head trial, lorazepam was found to be superior to diazepam in ending seizure activity and maintaining seizure control without the use of other medications51,52—and is now the drug of choice for initial treatment of status epilepticus.
CASE You continue to see Joe every 3 to 4 months to monitor his basic blood work and mood. A year after his seizure, he remains seizure-free and is tolerating the AED without any adverse effects.
CORRESPONDENCE
William J. Geiger, MD, FAAFP, Medical College of Wisconsin, Columbia St. Mary’s Family Medicine Residency, 1121 East North Avenue, Milwaukee, WI 53212; [email protected]
1. Epilepsy Foundation of America. Epilepsy and seizure statistics. Available at: http://www.epilepsyfoundation.org/about/statistics.cfm. Accessed June 15, 2009.
2. Centers for Disease Control and Prevention (CDC). Prevalence and most common causes of disability among adults—United States, 2005. MMWR Morb Mortal Wkly Rep. 2009;58:421-426. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm5816a2.htm. Accessed June 15, 2009.
3. Hirtz D, Thurman DJ, Gwinn-Hardy K, et al. How common are the “common” neurologic disorders? Neurology. 2007;68:326-337.
4. Engel J Jr. ILAE classification of epilepsy syndromes. Epilepsy Res. 2006;70(suppl 1):S5-S10.
5. Rudzinski LA, Shih JJ. Continuum: lifelong learning in neurology. Epilepsia. 2010;16:15-35.
6. Chaves J, Sander JW. Seizure aggravation in idiopathic generalized epilepsies. Epilepsia. 2005;46(suppl 9):S133-S139.
7. Beghi E. Management of first seizure. General conclusions and recommendations. Epilepsia. 2008;49(suppl 1):S58-S61.
8. Berg A. Risk of recurrence after a first unprovoked seizure. Epilepsia. 2008;49(suppl 1):S13-S18.
9. Hirtz D, Ashwal S, Berg A, et al. Practice parameter: evaluating a first non-febrile seizure in children: report of the Quality Standards Subcommittee of the American Academy of Neurology, the Child Neurology Society, and the American Epilepsy Society. Neurology. 2000;55:616-623.
10. US Food and Drug Administration. Suicidal behavior and ideation and antiepileptic drugs. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm100190.htm. Updated May 5, 2009. Accessed June 28, 2009.
11. French JA, Kanner AM, Bautista J, et al. Efficacy and tolerability of the new antiepileptic drugs I: treatment of new epilepsy, report of the therapeutic and technology assessment subcommittee and quality standards subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2004;62:1252-1260.
12. French J, Smith M, Faught E, et al. Practice advisory: the use of felbamate in the treatment of patients with intractable epilepsy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 1999;52:1540-1545.
13. Glauser T, Kluger G, Sachdeo R, et al. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology. 2008;70:1950-1958.
14. Suzuki Y, Nagai T, Ono J, et al. Zonisamide monotherapy in newly-diagnosed infantile spasms. Epilepsia. 1997;38:1035-1038.
15. Kochak GM, Page JG, Buchanan RA, et al. Steady-state pharmacokinetics of zonisamide, an antiepileptic agent for treatment of refractory complex partial seizures. J Clin Pharmacol. 1998;38:166-171.
16. Arroyo S, Anhut H, Kugler AR, et al. Pregabalin 1008-011 International Study Group. Pregabalin add-on treatment: a randomized, double-blind, placebo-controlled, dose-response study in adults with partial seizures. Epilepsia. 2004;45:20-27.
17. Brodie MJ, Rosenfeld WE, Vazquez B, et al. Rufinamide for the adjunctive treatment of partial seizures in adults and adolescents: a randomized placebo-controlled trial. Epilepsia. 2009;50:1899-1909.
18. Ben-Menachem E, Biton V, Jatuzis D, et al. Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia. 2007;48:1308-1317.
19. Gidal B, Garnett W. Epilepsy. In: Dipiro J, et al, eds. Pharmacotherapy: A Pathophysiologic Approach. 6th ed. New York: McGraw-Hill; 2005:1023-1048.
20. Pellock JM, Treatment of epilepsy in the new millennium. Pharmacotherapy. 2000;20:129S-138S.
21. Schachter S. Pharmacology of antiepileptic drugs. Available at: http://www.utdonline.com/online/content/topic.do?topicKey=epil_eeg/5220. Accessed July 15, 2009.
22. Woelfel J. Comparison of antiepileptic drugs. Pharmacist’s Letter/Prescriber's Letter. July 2009;25:1-24.
23. Wolters Kluwer Health Inc. Anticonvulsants. Drug facts and comparisons online. Available at: http://www.efactsonline.com. Accessed July 10, 2009.
24. US Food and Drug Administration. Information for healthcare professionals. Suicidality and antiepileptic drugs [FDA alert]. Available at: http://www.fda.gov/Drugs/DrugSafety/PublicHealthAdvisories/ucm054709.htm. January 31, 2008. Accessed June 30, 2009.
25. French J. Treatment with antiepileptic drugs, new and old. Continuum. 2007;13:71-90.
26. Sheehan M. Polycystic ovarian syndrome: diagnosis and management. Clin Med Res. 2004;2:13-27.
27. Harden CL. Sexual dysfunction in women with epilepsy. Seizure. 2008;17:131-135.
28. Harden CL, Hopp J, Ting TY, et al. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): obstetrical complications and change in seizure frequency. Neurology. 2009;73:126-132.
29. Molgaard-Nielsen D, Hviid A. Newer-generation antiepileptic drugs and the risk of major birth defects. JAMA. 2011;305:1996-2002.
30. Tomson T, Battino D, Bonizonni E, et al. Dose-dependent risk of malformations with antiepileptic drugs: an analysis of data from the EURAP epilepsy and pregnancy registry. Lancet Neurol. 2011;10:609-617.
31. Callaghan BC, Anand K, Hesdorffer D, et al. Likelihood of seizure remission in an adult population with refractory epilepsy. Ann Neurol. 2007;62:382-389.
32. Luciano AL, Shorvon SD. Results of treatment changes in patients with apparently drug-resistant chronic epilepsy. Ann Neurol. 2007;62:375-381.
33. Abramowicz M, ed. Drugs for epilepsy [treatment guidelines]. The Medical Letter. 2008;70:1-12.
34.Stokes T, Shaw EJ, Juarez-Garcia A, et al. Clinical guidelines and evidence review for the epilepsies: diagnosis and management in adults and children in primary and secondary care. London: Royal College of General Practitioners. Available at: www.nice.org.uk/CG020fullguideline. Published October 2004. Accessed July 10, 2009.
35. Garnett WR. Antiepileptic drug treatment: outcomes and adherence. Pharmacotherapy. 2000;20:191S-199S.
36. Briesacher BA, Andrade SE, Fouayzi H, et al. Comparison of drug adherence rates among patients with seven different medical conditions. Pharmacotherapy. 2008;28:437-443.
37. Shinnar S, Gross-Tsur V. Discontinuing antiepileptic drug therapy. In: Wyllie E, ed. The Treatment of Epilepsy. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2001:811-819.
38. Kwan P, Brodie J. Refractory epilepsy: a progressive, intractable but preventable condition? Seizures. 2002;11:77-84.
39. Lings S. Increased driving accident frequency in Danish patients with epilepsy. Neurology. 2001;57:435-439.
40. Krauss GL, Krumholz A, Carter RC, et al. Risk factors for seizure-related motor vehicle crashes in patients with epilepsy. Neurology. 1999;52:1324-1329.
41. American Academy of Neurology, American Epilepsy Society, and Epilepsy Foundation of America. Consensus statements, sample statutory provisions, and model regulations regarding driver licensing and epilepsy. Epilepsia. 1994;35:696-705.
42. Thadani VM, Taylor J. Surgical treatments for epilepsy. Continuum. 2007;13:152-176.
43. Brodie MJ, Kwan P. Staged approach to epilepsy management. Neurology. 2002;58(8 suppl 5):S2-S8.
44. Sillanpaa M, Jalava M, Kaleva O, et al. Long-term prognosis of seizures with onset in childhood. N Engl J Med. 1998;338:1715-1722.
45. Engel J Jr, Wiebe S, French J, et al. Practice parameter: temporal lobe and localized neocortical resections for epilepsy. Neurology. 2003;60:538-547.
46. Boon P, D'Have M, Van Walleghen P, et al. Direct medical costs of refractory epilepsy incurred by three different treatment modalities: a prospective assessment. Epilepsia. 2002;43:96-102.
47. Passaro EA. Outcome of epilepsy surgery. Available at: http://emedicine.medscape.com/article/1185416-overview. Updated May 16, 2011. Accessed June 28, 2011.
48. DeLorenzo RJ, Pellock JM, Towne AR, et al. Epidemiology of status epilepticus. J Clin Neurophysiol. 1995;12:316-325.
49. Logroscino G, Hesdorffer DC, Cascino GD, et al. Long-term mortality after a first episode of status epilepticus. Neurology. 2002; 58:537-541.
50. Kalviaine R. Treatment of status epilepticus. Essential Evidence Plus. Wiley-Blackwell. Available at: http://www.essentialevidenceplus.com/content/ebmg_ebm/766. Accessed July 15, 2009.
51. Prasad K, Al-Roomi K, Krishnan PR, et al. Anticonvulsant therapy for status epilepticus. Cochrane Database Syst Rev. 2005;(4):CD003723.
52. Treiman DM, Meyers PD, Walton NY, et al. A comparison of four treatments for generalized convulsive status epilepticus. N Engl J Med. 1998;339:792-798.
1. Epilepsy Foundation of America. Epilepsy and seizure statistics. Available at: http://www.epilepsyfoundation.org/about/statistics.cfm. Accessed June 15, 2009.
2. Centers for Disease Control and Prevention (CDC). Prevalence and most common causes of disability among adults—United States, 2005. MMWR Morb Mortal Wkly Rep. 2009;58:421-426. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm5816a2.htm. Accessed June 15, 2009.
3. Hirtz D, Thurman DJ, Gwinn-Hardy K, et al. How common are the “common” neurologic disorders? Neurology. 2007;68:326-337.
4. Engel J Jr. ILAE classification of epilepsy syndromes. Epilepsy Res. 2006;70(suppl 1):S5-S10.
5. Rudzinski LA, Shih JJ. Continuum: lifelong learning in neurology. Epilepsia. 2010;16:15-35.
6. Chaves J, Sander JW. Seizure aggravation in idiopathic generalized epilepsies. Epilepsia. 2005;46(suppl 9):S133-S139.
7. Beghi E. Management of first seizure. General conclusions and recommendations. Epilepsia. 2008;49(suppl 1):S58-S61.
8. Berg A. Risk of recurrence after a first unprovoked seizure. Epilepsia. 2008;49(suppl 1):S13-S18.
9. Hirtz D, Ashwal S, Berg A, et al. Practice parameter: evaluating a first non-febrile seizure in children: report of the Quality Standards Subcommittee of the American Academy of Neurology, the Child Neurology Society, and the American Epilepsy Society. Neurology. 2000;55:616-623.
10. US Food and Drug Administration. Suicidal behavior and ideation and antiepileptic drugs. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm100190.htm. Updated May 5, 2009. Accessed June 28, 2009.
11. French JA, Kanner AM, Bautista J, et al. Efficacy and tolerability of the new antiepileptic drugs I: treatment of new epilepsy, report of the therapeutic and technology assessment subcommittee and quality standards subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2004;62:1252-1260.
12. French J, Smith M, Faught E, et al. Practice advisory: the use of felbamate in the treatment of patients with intractable epilepsy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 1999;52:1540-1545.
13. Glauser T, Kluger G, Sachdeo R, et al. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology. 2008;70:1950-1958.
14. Suzuki Y, Nagai T, Ono J, et al. Zonisamide monotherapy in newly-diagnosed infantile spasms. Epilepsia. 1997;38:1035-1038.
15. Kochak GM, Page JG, Buchanan RA, et al. Steady-state pharmacokinetics of zonisamide, an antiepileptic agent for treatment of refractory complex partial seizures. J Clin Pharmacol. 1998;38:166-171.
16. Arroyo S, Anhut H, Kugler AR, et al. Pregabalin 1008-011 International Study Group. Pregabalin add-on treatment: a randomized, double-blind, placebo-controlled, dose-response study in adults with partial seizures. Epilepsia. 2004;45:20-27.
17. Brodie MJ, Rosenfeld WE, Vazquez B, et al. Rufinamide for the adjunctive treatment of partial seizures in adults and adolescents: a randomized placebo-controlled trial. Epilepsia. 2009;50:1899-1909.
18. Ben-Menachem E, Biton V, Jatuzis D, et al. Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia. 2007;48:1308-1317.
19. Gidal B, Garnett W. Epilepsy. In: Dipiro J, et al, eds. Pharmacotherapy: A Pathophysiologic Approach. 6th ed. New York: McGraw-Hill; 2005:1023-1048.
20. Pellock JM, Treatment of epilepsy in the new millennium. Pharmacotherapy. 2000;20:129S-138S.
21. Schachter S. Pharmacology of antiepileptic drugs. Available at: http://www.utdonline.com/online/content/topic.do?topicKey=epil_eeg/5220. Accessed July 15, 2009.
22. Woelfel J. Comparison of antiepileptic drugs. Pharmacist’s Letter/Prescriber's Letter. July 2009;25:1-24.
23. Wolters Kluwer Health Inc. Anticonvulsants. Drug facts and comparisons online. Available at: http://www.efactsonline.com. Accessed July 10, 2009.
24. US Food and Drug Administration. Information for healthcare professionals. Suicidality and antiepileptic drugs [FDA alert]. Available at: http://www.fda.gov/Drugs/DrugSafety/PublicHealthAdvisories/ucm054709.htm. January 31, 2008. Accessed June 30, 2009.
25. French J. Treatment with antiepileptic drugs, new and old. Continuum. 2007;13:71-90.
26. Sheehan M. Polycystic ovarian syndrome: diagnosis and management. Clin Med Res. 2004;2:13-27.
27. Harden CL. Sexual dysfunction in women with epilepsy. Seizure. 2008;17:131-135.
28. Harden CL, Hopp J, Ting TY, et al. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): obstetrical complications and change in seizure frequency. Neurology. 2009;73:126-132.
29. Molgaard-Nielsen D, Hviid A. Newer-generation antiepileptic drugs and the risk of major birth defects. JAMA. 2011;305:1996-2002.
30. Tomson T, Battino D, Bonizonni E, et al. Dose-dependent risk of malformations with antiepileptic drugs: an analysis of data from the EURAP epilepsy and pregnancy registry. Lancet Neurol. 2011;10:609-617.
31. Callaghan BC, Anand K, Hesdorffer D, et al. Likelihood of seizure remission in an adult population with refractory epilepsy. Ann Neurol. 2007;62:382-389.
32. Luciano AL, Shorvon SD. Results of treatment changes in patients with apparently drug-resistant chronic epilepsy. Ann Neurol. 2007;62:375-381.
33. Abramowicz M, ed. Drugs for epilepsy [treatment guidelines]. The Medical Letter. 2008;70:1-12.
34.Stokes T, Shaw EJ, Juarez-Garcia A, et al. Clinical guidelines and evidence review for the epilepsies: diagnosis and management in adults and children in primary and secondary care. London: Royal College of General Practitioners. Available at: www.nice.org.uk/CG020fullguideline. Published October 2004. Accessed July 10, 2009.
35. Garnett WR. Antiepileptic drug treatment: outcomes and adherence. Pharmacotherapy. 2000;20:191S-199S.
36. Briesacher BA, Andrade SE, Fouayzi H, et al. Comparison of drug adherence rates among patients with seven different medical conditions. Pharmacotherapy. 2008;28:437-443.
37. Shinnar S, Gross-Tsur V. Discontinuing antiepileptic drug therapy. In: Wyllie E, ed. The Treatment of Epilepsy. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2001:811-819.
38. Kwan P, Brodie J. Refractory epilepsy: a progressive, intractable but preventable condition? Seizures. 2002;11:77-84.
39. Lings S. Increased driving accident frequency in Danish patients with epilepsy. Neurology. 2001;57:435-439.
40. Krauss GL, Krumholz A, Carter RC, et al. Risk factors for seizure-related motor vehicle crashes in patients with epilepsy. Neurology. 1999;52:1324-1329.
41. American Academy of Neurology, American Epilepsy Society, and Epilepsy Foundation of America. Consensus statements, sample statutory provisions, and model regulations regarding driver licensing and epilepsy. Epilepsia. 1994;35:696-705.
42. Thadani VM, Taylor J. Surgical treatments for epilepsy. Continuum. 2007;13:152-176.
43. Brodie MJ, Kwan P. Staged approach to epilepsy management. Neurology. 2002;58(8 suppl 5):S2-S8.
44. Sillanpaa M, Jalava M, Kaleva O, et al. Long-term prognosis of seizures with onset in childhood. N Engl J Med. 1998;338:1715-1722.
45. Engel J Jr, Wiebe S, French J, et al. Practice parameter: temporal lobe and localized neocortical resections for epilepsy. Neurology. 2003;60:538-547.
46. Boon P, D'Have M, Van Walleghen P, et al. Direct medical costs of refractory epilepsy incurred by three different treatment modalities: a prospective assessment. Epilepsia. 2002;43:96-102.
47. Passaro EA. Outcome of epilepsy surgery. Available at: http://emedicine.medscape.com/article/1185416-overview. Updated May 16, 2011. Accessed June 28, 2011.
48. DeLorenzo RJ, Pellock JM, Towne AR, et al. Epidemiology of status epilepticus. J Clin Neurophysiol. 1995;12:316-325.
49. Logroscino G, Hesdorffer DC, Cascino GD, et al. Long-term mortality after a first episode of status epilepticus. Neurology. 2002; 58:537-541.
50. Kalviaine R. Treatment of status epilepticus. Essential Evidence Plus. Wiley-Blackwell. Available at: http://www.essentialevidenceplus.com/content/ebmg_ebm/766. Accessed July 15, 2009.
51. Prasad K, Al-Roomi K, Krishnan PR, et al. Anticonvulsant therapy for status epilepticus. Cochrane Database Syst Rev. 2005;(4):CD003723.
52. Treiman DM, Meyers PD, Walton NY, et al. A comparison of four treatments for generalized convulsive status epilepticus. N Engl J Med. 1998;339:792-798.
The old and the new
I recently attended the 14th World Conference on Lung Cancer (WCLC), a biennial multidisciplinary meeting for medical oncologists, surgeons, pulmonologists, radiation oncologists, and pathologists. The medical oncology portion of this conference was abuzz with excitement about the prospects of molecularly targeted therapies.Five years ago, few would have predicted that lung cancer would be the disease leading the way into the personalized medicine era in oncology. The recent discovery of a small number of critical genes that act as driving mutations for non-small cell lung cancer (NSCLC) has set the stage for the development of targeted agents against these mutations.
Gene mutations and molecular targeting
The Lung Cancer Mutation Consortium, comprised of 14 US-based cancer centers and sponsored by the National Cancer Institute, reported at the conference that mutations could be identified in 54% of adenocarcinomas, including genes such as KRAS, EGFR, BRAF, HER2, PI3KCA, ALK,MET, and others. Each of these genes has drugs either in clinical development or already marketed for other diseases with the same genetic alterations.Of note is that 97% of these mutations were mutually exclusive, suggesting that only one drug will be necessary to treat each of the subgroups. Proof of this concept is the development of crizotinib, a small molecule that inhibits the EML4-ALK fusion gene/protein with remarkable activity—over 80% of patients respond to this drug. Its approval is eagerly awaited.
Another exciting report presented at the WCLC investigated genetic abnormalities in the second most common subtype of NSCLC—squamous cell. Investigators used a combination of methods to identify genetic mutations, amplifications, or deletions in almost two-thirds of patients with this disease, setting the stage for molecularly targeted treatment in this group as well.
We already have adopted pathway inhibition as a standard in lung cancer patients who harbor an epidermal growth factor receptor (EGFR) mutation, with increasing evidence suggesting that tyrosine kinase inhibitors such as erlotinib (Tarceva) are superior for first-line treatment of EGFR-mutated adenocarcinoma. Molecular diagnostics to guide treatment in the community setting is now firmly established in the most common diseases we see—breast, colon, and lung cancers.
And yet amid all of this excitement regarding novel pathways,validated targets, next-generation massively parallel sequencing, and so on, we must not forget that the majority of cancers are treated in both the adjuvant and metastatic setting with tried-and-true chemotherapeutic or endocrine agents. I even make a point of telling the fellows training with me that I am fairly confident that they will be giving chemotherapy throughout their careers, although it will certainly not dominate as it does today.
Revisiting mechanisms of action
All oncologists need to refamiliarize themselves with the mechanisms of action for the drugs that we use daily. In truth, each of the traditional chemotherapy agents are in fact targeting a cellular molecular pathway. It’s just that we previously lacked the technology and knowledge to identify the specific target. For that reason, I am excited about two comprehensive reviews in this issue of Community Oncology.
The first is a discussion of the estrogen receptor signaling pathway by Adam Brufsky (page 343).Much exciting knowledge has been gained over the past decade in understanding mechanisms of resistance to this oldest of validated targets. Now, trying to block alternative pathways of estrogen receptor activation in conjunction with aromatase inhibitors or other endocrine agents is the focus of much active research.
Also in this issue is a comprehensive review by Michael Trigg and Anne Flanagan-Minick of the mechanisms of action of commonly used anticancer agents (page 357). This is essential reading, as it discusses both classic cytotoxic agents and newer signal transduction modifiers. But perhaps most importantly, this review emphasizes the current thinking that most advanced epithelial tumors will not be brought under control with a single therapeutic agent, a lesson we learned in the era of cytoxic drugs only. In fact, it is likely that the landscape will be dramatically more complex as agents from different classes are necessarily combined to achieve maximum effect.
More and more it appears that integrating personalized medicine into a system of practice-based guidelines will be a formidable challenge. Still, there is a great opportunity for community oncologists to prove value to their third-party payers and directly to patients for the high-level decision making required to provide optimal care. Such decision making must be part of the value equation as reimbursement moves away from margins on drug acquisition and to oncologists providing the best care based on their knowledge and informatics resources.
I recently attended the 14th World Conference on Lung Cancer (WCLC), a biennial multidisciplinary meeting for medical oncologists, surgeons, pulmonologists, radiation oncologists, and pathologists. The medical oncology portion of this conference was abuzz with excitement about the prospects of molecularly targeted therapies.Five years ago, few would have predicted that lung cancer would be the disease leading the way into the personalized medicine era in oncology. The recent discovery of a small number of critical genes that act as driving mutations for non-small cell lung cancer (NSCLC) has set the stage for the development of targeted agents against these mutations.
Gene mutations and molecular targeting
The Lung Cancer Mutation Consortium, comprised of 14 US-based cancer centers and sponsored by the National Cancer Institute, reported at the conference that mutations could be identified in 54% of adenocarcinomas, including genes such as KRAS, EGFR, BRAF, HER2, PI3KCA, ALK,MET, and others. Each of these genes has drugs either in clinical development or already marketed for other diseases with the same genetic alterations.Of note is that 97% of these mutations were mutually exclusive, suggesting that only one drug will be necessary to treat each of the subgroups. Proof of this concept is the development of crizotinib, a small molecule that inhibits the EML4-ALK fusion gene/protein with remarkable activity—over 80% of patients respond to this drug. Its approval is eagerly awaited.
Another exciting report presented at the WCLC investigated genetic abnormalities in the second most common subtype of NSCLC—squamous cell. Investigators used a combination of methods to identify genetic mutations, amplifications, or deletions in almost two-thirds of patients with this disease, setting the stage for molecularly targeted treatment in this group as well.
We already have adopted pathway inhibition as a standard in lung cancer patients who harbor an epidermal growth factor receptor (EGFR) mutation, with increasing evidence suggesting that tyrosine kinase inhibitors such as erlotinib (Tarceva) are superior for first-line treatment of EGFR-mutated adenocarcinoma. Molecular diagnostics to guide treatment in the community setting is now firmly established in the most common diseases we see—breast, colon, and lung cancers.
And yet amid all of this excitement regarding novel pathways,validated targets, next-generation massively parallel sequencing, and so on, we must not forget that the majority of cancers are treated in both the adjuvant and metastatic setting with tried-and-true chemotherapeutic or endocrine agents. I even make a point of telling the fellows training with me that I am fairly confident that they will be giving chemotherapy throughout their careers, although it will certainly not dominate as it does today.
Revisiting mechanisms of action
All oncologists need to refamiliarize themselves with the mechanisms of action for the drugs that we use daily. In truth, each of the traditional chemotherapy agents are in fact targeting a cellular molecular pathway. It’s just that we previously lacked the technology and knowledge to identify the specific target. For that reason, I am excited about two comprehensive reviews in this issue of Community Oncology.
The first is a discussion of the estrogen receptor signaling pathway by Adam Brufsky (page 343).Much exciting knowledge has been gained over the past decade in understanding mechanisms of resistance to this oldest of validated targets. Now, trying to block alternative pathways of estrogen receptor activation in conjunction with aromatase inhibitors or other endocrine agents is the focus of much active research.
Also in this issue is a comprehensive review by Michael Trigg and Anne Flanagan-Minick of the mechanisms of action of commonly used anticancer agents (page 357). This is essential reading, as it discusses both classic cytotoxic agents and newer signal transduction modifiers. But perhaps most importantly, this review emphasizes the current thinking that most advanced epithelial tumors will not be brought under control with a single therapeutic agent, a lesson we learned in the era of cytoxic drugs only. In fact, it is likely that the landscape will be dramatically more complex as agents from different classes are necessarily combined to achieve maximum effect.
More and more it appears that integrating personalized medicine into a system of practice-based guidelines will be a formidable challenge. Still, there is a great opportunity for community oncologists to prove value to their third-party payers and directly to patients for the high-level decision making required to provide optimal care. Such decision making must be part of the value equation as reimbursement moves away from margins on drug acquisition and to oncologists providing the best care based on their knowledge and informatics resources.
I recently attended the 14th World Conference on Lung Cancer (WCLC), a biennial multidisciplinary meeting for medical oncologists, surgeons, pulmonologists, radiation oncologists, and pathologists. The medical oncology portion of this conference was abuzz with excitement about the prospects of molecularly targeted therapies.Five years ago, few would have predicted that lung cancer would be the disease leading the way into the personalized medicine era in oncology. The recent discovery of a small number of critical genes that act as driving mutations for non-small cell lung cancer (NSCLC) has set the stage for the development of targeted agents against these mutations.
Gene mutations and molecular targeting
The Lung Cancer Mutation Consortium, comprised of 14 US-based cancer centers and sponsored by the National Cancer Institute, reported at the conference that mutations could be identified in 54% of adenocarcinomas, including genes such as KRAS, EGFR, BRAF, HER2, PI3KCA, ALK,MET, and others. Each of these genes has drugs either in clinical development or already marketed for other diseases with the same genetic alterations.Of note is that 97% of these mutations were mutually exclusive, suggesting that only one drug will be necessary to treat each of the subgroups. Proof of this concept is the development of crizotinib, a small molecule that inhibits the EML4-ALK fusion gene/protein with remarkable activity—over 80% of patients respond to this drug. Its approval is eagerly awaited.
Another exciting report presented at the WCLC investigated genetic abnormalities in the second most common subtype of NSCLC—squamous cell. Investigators used a combination of methods to identify genetic mutations, amplifications, or deletions in almost two-thirds of patients with this disease, setting the stage for molecularly targeted treatment in this group as well.
We already have adopted pathway inhibition as a standard in lung cancer patients who harbor an epidermal growth factor receptor (EGFR) mutation, with increasing evidence suggesting that tyrosine kinase inhibitors such as erlotinib (Tarceva) are superior for first-line treatment of EGFR-mutated adenocarcinoma. Molecular diagnostics to guide treatment in the community setting is now firmly established in the most common diseases we see—breast, colon, and lung cancers.
And yet amid all of this excitement regarding novel pathways,validated targets, next-generation massively parallel sequencing, and so on, we must not forget that the majority of cancers are treated in both the adjuvant and metastatic setting with tried-and-true chemotherapeutic or endocrine agents. I even make a point of telling the fellows training with me that I am fairly confident that they will be giving chemotherapy throughout their careers, although it will certainly not dominate as it does today.
Revisiting mechanisms of action
All oncologists need to refamiliarize themselves with the mechanisms of action for the drugs that we use daily. In truth, each of the traditional chemotherapy agents are in fact targeting a cellular molecular pathway. It’s just that we previously lacked the technology and knowledge to identify the specific target. For that reason, I am excited about two comprehensive reviews in this issue of Community Oncology.
The first is a discussion of the estrogen receptor signaling pathway by Adam Brufsky (page 343).Much exciting knowledge has been gained over the past decade in understanding mechanisms of resistance to this oldest of validated targets. Now, trying to block alternative pathways of estrogen receptor activation in conjunction with aromatase inhibitors or other endocrine agents is the focus of much active research.
Also in this issue is a comprehensive review by Michael Trigg and Anne Flanagan-Minick of the mechanisms of action of commonly used anticancer agents (page 357). This is essential reading, as it discusses both classic cytotoxic agents and newer signal transduction modifiers. But perhaps most importantly, this review emphasizes the current thinking that most advanced epithelial tumors will not be brought under control with a single therapeutic agent, a lesson we learned in the era of cytoxic drugs only. In fact, it is likely that the landscape will be dramatically more complex as agents from different classes are necessarily combined to achieve maximum effect.
More and more it appears that integrating personalized medicine into a system of practice-based guidelines will be a formidable challenge. Still, there is a great opportunity for community oncologists to prove value to their third-party payers and directly to patients for the high-level decision making required to provide optimal care. Such decision making must be part of the value equation as reimbursement moves away from margins on drug acquisition and to oncologists providing the best care based on their knowledge and informatics resources.