User login
Improving inpatient care through antimicrobial stewardship: A case‐based approach to managing acute infections: Supplement to the Journal of Hospital Medicine
Estimated time to complete the activity: 3 hours 30 minutes
Jointly sponsored by the American Academy of CME
This activity is supported by an educational grant from Merck & Co., Inc.
There is no fee to participate in this CME‐certified activity.
Program Overview
Early and appropriate treatment of acute infections, especially in critically ill and immunocompromised patients, is paramount to successful outcomes. Appropriate empiric therapy often requires the use of multiple broad‐spectrum agents that must be used judiciously to preserve antimicrobial activity over time. Critical components of antimicrobial stewardship include the selection of appropriate antibiotics, de‐escalation of therapy after 2 or 3 days of empiric treatment, and a strategy for the duration and discontinuation of therapy. An evidence‐based approach to these essential stewardship factors will improve patient outcomes by decreasing unnecessary antimicrobial exposures and associated unwanted effects as well as reduce the risk for emergence of antimicrobial resistance.
The intent of this educational activity is to illustrate these components of antimicrobial stewardship in a practical, case‐based format. Since hospitalists and intensivists play a central role in the formation and operation of a successful antimicrobial stewardship program, special consideration will be given to strategies that they can apply in their daily practices.
Target Audience
This activity was designed to meet the needs of hospitalists and intensivists who are involved in the diagnosis, management, and treatment of infectious diseases in the hospital setting. Other healthcare professionals are also invited to participate.
Faculty and Topics
Empiric Antibiotic Selection Strategies for Healthcare‐Associated Pneumonia, Intra‐abdominal Infections, and Catheter‐Associated Bacteremia
David R. Snydman, MD, FACP, FIDSA
Chief, Division of Geographic Medicine and Infectious Diseases
Tufts Medical Center
Professor of Medicine
Tufts University School of Medicine
Boston, Massachusetts
After completing this article, learners should be better able to:
Differentiate between colonization and infection in their patients in order to devise optimal initial therapy strategies
Identify risk factors for the development of antimicrobial resistance
Select the appropriate therapeutic agent for their hospitalized patients based on the organism and site of infection
Antimicrobial De‐escalation Strategies in Hospitalized Patients with Pneumonia, Intra‐abdominal Infections, and Bacteremia
Keith S. Kaye, MD, MPH
Professor of Medicine
Wayne State University
Corporate Director, Infection Prevention, Epidemiology and Antimicrobial Stewardship
Detroit Medical Center
Detroit, Michigan
After completing this article, learners should be better able to:
Assess the rationale behind antimicrobial de‐escalation in healthcare settings and its potential healthcare benefits
Implement effective de‐escalation strategies for their patients that are pathogen‐specific and minimize the emergence of resistance
Identify common targets and opportunities for de‐escalation programs in their institution
Duration and Cessation of Antimicrobial Treatment
Thomas M. File, Jr., MD, MSc, MACP, FIDSA, FCCP
Professor, Internal Medicine
Head, Infectious Disease Section
Northeastern Ohio Universities College of Medicine and Pharmacy
Akron, Ohio
After completing this article, learners should be better able to:
Develop an evidence‐based approach to duration and cessation of antimicrobial therapy for their patients
Assess clinical data in support of a shorter course of antimicrobial therapy
Incorporate strategies for their patients to optimize antimicrobial choices, dosages, and durations of therapy in order to decrease the emergence of antimicrobial resistance
Infections, Bacterial Resistance, and Antimicrobial Stewardship: The Emerging Role of Hospitalists
David J. Rosenberg, MD, MPH, FACP, SFHM (Chairman)
Associate Chair for Hospital Operations Department of Medicine
Section Head, Hospital Medicine, Division of General Internal Medicine
North Shore University Hospital
Manhasset, New York
After completing this article, learners should be better able to:
Describe the role of the hospitalist in the successful implementation of an antimicrobial stewardship program to improve quality of care and outcomes
Identify the key elements of an antimicrobial stewardship program that promote the judicious use of antibiotics in hospital settings
Apply the critical antimicrobial stewardship elements to the care of patients in their hospital
Accreditation Statement
This activity has been planned and implemented in accordance with the Essential Areas and Policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint sponsorship of the American Academy of CME, Inc. and Global Education Exchange, Inc. American Academy of CME is accredited by the ACCME to provide continuing medical education for physicians.
Credit Designation
American Academy of CME designates this enduring material for a maximum of 3.5 AMA PRA Category 1 Credit(s). Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Disclosure of Conflict of Interest
According to the disclosure policy of the American Academy of CME, all faculty, planning committee members, editors, managers, and other individuals who are in a position to control content are required to disclose any relevant relationships with any commercial interests related to this activity. The existence of these interests or relationships is not viewed as implying bias or decreasing the value of the presentation. All educational materials were reviewed for fair balance, scientific objectivity, and levels of evidence.
Academy planner John JD Juchniewicz, MCIS, CCMEP, and GLOBEX planners and editors Meri D. Pozo, PhD and Michael L. Coco, PhD reported no financial relationships or relationships to products or devices they or their spouse/life partner have with commercial interests related to the content of this CME activity.
The faculty reported the following financial relationships or affiliations with commercial interests during the past 12 months:
David J. Rosenberg, MD, MPH, FACP, SFHM
Advisory Boardfor scientific information: Canyon Pharmaceuticals
Consultantfor marketing purposes: UCB
Grant Recipient/Research Support (PI; funds paid to Feinstein Institute): Sanofi‐Aventis
Promotional Speaker's Bureau: Sanofi‐Aventis
David R. Snydman, MD, FACP, FIDSA
Advisory Boardfor scientific information: CSL Behring, Genentech, Genzyme, Millenium, Novartis
Consultantfor clinical trial design: CSL Behring
Grant Recipient/Research Support (PI; funds paid to Tufts Medical Center): Cubist, Forest Pharmaceuticals, Johnson & Johnson, Merck & Co., Inc., Pfizer
Promotional Speaker's Bureau: CSL Behring, Merck & Co., Inc.
Keith S. Kaye, MD, MPH
Advisory Boardfor scientific information: Forest Pharmaceuticals, Merck & Co., Inc., Ortho‐McNeil, Pfizer, TheraDoc
Grant Recipient/Research Support (PI; funds paid to Wayne State University): Merck & Co., Inc., Pfizer
Promotional Speaker's Bureau: Cubist, Merck & Co., Inc., Ortho‐McNeil, Pfizer
Thomas M. File, Jr., MD, MSc, MACP, FIDSA, FCCP
Consultantfor clinical trial design: Cerexa/Forest Pharmaceuticals, Glaxo SmithKline, Merck & Co., Inc., Nabriva Therapeutics, Ortho‐McNeil, Protez/Novartis, Pfizer, Rib‐X Pharmaceuticals, Shire, Tetraphase Pharmaceuticals
Grant Recipient/Research Support (PI; funds paid to Suma Health System): Boehringer Ingelheim, Cerexa/Forest Pharmaceuticals, Gilead, Ortho‐McNeil, Pfizer, Tibotec
Independent clinical peer‐reviewer:
David Alland, MD
Professor of Medicine
Chief, Division of Infectious Disease
Interim Director, Center for Emerging and Re‐Emerging Pathogens
Assistant Dean for Clinical Research
University of Medicine and Dentistry of New JerseyThe New Jersey Medical School
Newark, New Jersey
PI for NIH STTR grant to Cepheid (to develop TB diagnostics)grant ended 9/10
Member, group of patent holders related to molecular beacon licenses
Employee (spouse): Bristol‐Myers Squibb
Shareholder/Stock options (self and spouse): Bristol‐Myers Squibb
Disclosure of Unlabeled Use
This educational activity may contain discussion of published and/or investigational uses of agents that are not indicated by the FDA. Faculty have been asked to disclose off‐label and/or investigational uses where they are mentioned. American Academy of CME (Academy), Global Education Exchange, Inc. (GLOBEX) and Merck & Co., Inc. do not recommend the use of any agent outside of the labeled indications.
The opinions expressed in the educational activity are those of the faculty and do not necessarily represent the views of the Academy, GLOBEX, Merck & Co., Inc, or any other manufacturer of pharmaceuticals or devices. Before prescribing any medication, physicians should consult primary references and full prescribing information. Please refer to the official prescribing information for each product for discussion of approved indications, contraindications, and warnings. Further, learners should appraise the information presented critically and are encouraged to consult appropriate resources for any product or device mentioned in this activity.
In addition, the American Academy of CME requires all faculty/authors to note the level of evidence for any patient care recommendation they make.
Method of Participation:
There are no fees for participating and receiving CME credit for this activity. During the period January 9, 2012 through January 9, 2013, learners must 1) review the CME information including the learning objectives and disclosure statements; 2) study the educational content of the activity; 3) go online at
Media:
Journal supplement
Disclaimer
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patient's conditions and possible contraindications on dangers in use, review of any applicable manufacturer's product information, and comparison with recommendations of other authorities.
Contact Info:
For questions or comments about this CME activity, contact:
John JD Juchniewicz, MCIS, CCMEP
American Academy of CME
2012 American Academy of CME and Global Education Exchange, Inc.
Estimated time to complete the activity: 3 hours 30 minutes
Jointly sponsored by the American Academy of CME
This activity is supported by an educational grant from Merck & Co., Inc.
There is no fee to participate in this CME‐certified activity.
Program Overview
Early and appropriate treatment of acute infections, especially in critically ill and immunocompromised patients, is paramount to successful outcomes. Appropriate empiric therapy often requires the use of multiple broad‐spectrum agents that must be used judiciously to preserve antimicrobial activity over time. Critical components of antimicrobial stewardship include the selection of appropriate antibiotics, de‐escalation of therapy after 2 or 3 days of empiric treatment, and a strategy for the duration and discontinuation of therapy. An evidence‐based approach to these essential stewardship factors will improve patient outcomes by decreasing unnecessary antimicrobial exposures and associated unwanted effects as well as reduce the risk for emergence of antimicrobial resistance.
The intent of this educational activity is to illustrate these components of antimicrobial stewardship in a practical, case‐based format. Since hospitalists and intensivists play a central role in the formation and operation of a successful antimicrobial stewardship program, special consideration will be given to strategies that they can apply in their daily practices.
Target Audience
This activity was designed to meet the needs of hospitalists and intensivists who are involved in the diagnosis, management, and treatment of infectious diseases in the hospital setting. Other healthcare professionals are also invited to participate.
Faculty and Topics
Empiric Antibiotic Selection Strategies for Healthcare‐Associated Pneumonia, Intra‐abdominal Infections, and Catheter‐Associated Bacteremia
David R. Snydman, MD, FACP, FIDSA
Chief, Division of Geographic Medicine and Infectious Diseases
Tufts Medical Center
Professor of Medicine
Tufts University School of Medicine
Boston, Massachusetts
After completing this article, learners should be better able to:
Differentiate between colonization and infection in their patients in order to devise optimal initial therapy strategies
Identify risk factors for the development of antimicrobial resistance
Select the appropriate therapeutic agent for their hospitalized patients based on the organism and site of infection
Antimicrobial De‐escalation Strategies in Hospitalized Patients with Pneumonia, Intra‐abdominal Infections, and Bacteremia
Keith S. Kaye, MD, MPH
Professor of Medicine
Wayne State University
Corporate Director, Infection Prevention, Epidemiology and Antimicrobial Stewardship
Detroit Medical Center
Detroit, Michigan
After completing this article, learners should be better able to:
Assess the rationale behind antimicrobial de‐escalation in healthcare settings and its potential healthcare benefits
Implement effective de‐escalation strategies for their patients that are pathogen‐specific and minimize the emergence of resistance
Identify common targets and opportunities for de‐escalation programs in their institution
Duration and Cessation of Antimicrobial Treatment
Thomas M. File, Jr., MD, MSc, MACP, FIDSA, FCCP
Professor, Internal Medicine
Head, Infectious Disease Section
Northeastern Ohio Universities College of Medicine and Pharmacy
Akron, Ohio
After completing this article, learners should be better able to:
Develop an evidence‐based approach to duration and cessation of antimicrobial therapy for their patients
Assess clinical data in support of a shorter course of antimicrobial therapy
Incorporate strategies for their patients to optimize antimicrobial choices, dosages, and durations of therapy in order to decrease the emergence of antimicrobial resistance
Infections, Bacterial Resistance, and Antimicrobial Stewardship: The Emerging Role of Hospitalists
David J. Rosenberg, MD, MPH, FACP, SFHM (Chairman)
Associate Chair for Hospital Operations Department of Medicine
Section Head, Hospital Medicine, Division of General Internal Medicine
North Shore University Hospital
Manhasset, New York
After completing this article, learners should be better able to:
Describe the role of the hospitalist in the successful implementation of an antimicrobial stewardship program to improve quality of care and outcomes
Identify the key elements of an antimicrobial stewardship program that promote the judicious use of antibiotics in hospital settings
Apply the critical antimicrobial stewardship elements to the care of patients in their hospital
Accreditation Statement
This activity has been planned and implemented in accordance with the Essential Areas and Policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint sponsorship of the American Academy of CME, Inc. and Global Education Exchange, Inc. American Academy of CME is accredited by the ACCME to provide continuing medical education for physicians.
Credit Designation
American Academy of CME designates this enduring material for a maximum of 3.5 AMA PRA Category 1 Credit(s). Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Disclosure of Conflict of Interest
According to the disclosure policy of the American Academy of CME, all faculty, planning committee members, editors, managers, and other individuals who are in a position to control content are required to disclose any relevant relationships with any commercial interests related to this activity. The existence of these interests or relationships is not viewed as implying bias or decreasing the value of the presentation. All educational materials were reviewed for fair balance, scientific objectivity, and levels of evidence.
Academy planner John JD Juchniewicz, MCIS, CCMEP, and GLOBEX planners and editors Meri D. Pozo, PhD and Michael L. Coco, PhD reported no financial relationships or relationships to products or devices they or their spouse/life partner have with commercial interests related to the content of this CME activity.
The faculty reported the following financial relationships or affiliations with commercial interests during the past 12 months:
David J. Rosenberg, MD, MPH, FACP, SFHM
Advisory Boardfor scientific information: Canyon Pharmaceuticals
Consultantfor marketing purposes: UCB
Grant Recipient/Research Support (PI; funds paid to Feinstein Institute): Sanofi‐Aventis
Promotional Speaker's Bureau: Sanofi‐Aventis
David R. Snydman, MD, FACP, FIDSA
Advisory Boardfor scientific information: CSL Behring, Genentech, Genzyme, Millenium, Novartis
Consultantfor clinical trial design: CSL Behring
Grant Recipient/Research Support (PI; funds paid to Tufts Medical Center): Cubist, Forest Pharmaceuticals, Johnson & Johnson, Merck & Co., Inc., Pfizer
Promotional Speaker's Bureau: CSL Behring, Merck & Co., Inc.
Keith S. Kaye, MD, MPH
Advisory Boardfor scientific information: Forest Pharmaceuticals, Merck & Co., Inc., Ortho‐McNeil, Pfizer, TheraDoc
Grant Recipient/Research Support (PI; funds paid to Wayne State University): Merck & Co., Inc., Pfizer
Promotional Speaker's Bureau: Cubist, Merck & Co., Inc., Ortho‐McNeil, Pfizer
Thomas M. File, Jr., MD, MSc, MACP, FIDSA, FCCP
Consultantfor clinical trial design: Cerexa/Forest Pharmaceuticals, Glaxo SmithKline, Merck & Co., Inc., Nabriva Therapeutics, Ortho‐McNeil, Protez/Novartis, Pfizer, Rib‐X Pharmaceuticals, Shire, Tetraphase Pharmaceuticals
Grant Recipient/Research Support (PI; funds paid to Suma Health System): Boehringer Ingelheim, Cerexa/Forest Pharmaceuticals, Gilead, Ortho‐McNeil, Pfizer, Tibotec
Independent clinical peer‐reviewer:
David Alland, MD
Professor of Medicine
Chief, Division of Infectious Disease
Interim Director, Center for Emerging and Re‐Emerging Pathogens
Assistant Dean for Clinical Research
University of Medicine and Dentistry of New JerseyThe New Jersey Medical School
Newark, New Jersey
PI for NIH STTR grant to Cepheid (to develop TB diagnostics)grant ended 9/10
Member, group of patent holders related to molecular beacon licenses
Employee (spouse): Bristol‐Myers Squibb
Shareholder/Stock options (self and spouse): Bristol‐Myers Squibb
Disclosure of Unlabeled Use
This educational activity may contain discussion of published and/or investigational uses of agents that are not indicated by the FDA. Faculty have been asked to disclose off‐label and/or investigational uses where they are mentioned. American Academy of CME (Academy), Global Education Exchange, Inc. (GLOBEX) and Merck & Co., Inc. do not recommend the use of any agent outside of the labeled indications.
The opinions expressed in the educational activity are those of the faculty and do not necessarily represent the views of the Academy, GLOBEX, Merck & Co., Inc, or any other manufacturer of pharmaceuticals or devices. Before prescribing any medication, physicians should consult primary references and full prescribing information. Please refer to the official prescribing information for each product for discussion of approved indications, contraindications, and warnings. Further, learners should appraise the information presented critically and are encouraged to consult appropriate resources for any product or device mentioned in this activity.
In addition, the American Academy of CME requires all faculty/authors to note the level of evidence for any patient care recommendation they make.
Method of Participation:
There are no fees for participating and receiving CME credit for this activity. During the period January 9, 2012 through January 9, 2013, learners must 1) review the CME information including the learning objectives and disclosure statements; 2) study the educational content of the activity; 3) go online at
Media:
Journal supplement
Disclaimer
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patient's conditions and possible contraindications on dangers in use, review of any applicable manufacturer's product information, and comparison with recommendations of other authorities.
Contact Info:
For questions or comments about this CME activity, contact:
John JD Juchniewicz, MCIS, CCMEP
American Academy of CME
2012 American Academy of CME and Global Education Exchange, Inc.
Estimated time to complete the activity: 3 hours 30 minutes
Jointly sponsored by the American Academy of CME
This activity is supported by an educational grant from Merck & Co., Inc.
There is no fee to participate in this CME‐certified activity.
Program Overview
Early and appropriate treatment of acute infections, especially in critically ill and immunocompromised patients, is paramount to successful outcomes. Appropriate empiric therapy often requires the use of multiple broad‐spectrum agents that must be used judiciously to preserve antimicrobial activity over time. Critical components of antimicrobial stewardship include the selection of appropriate antibiotics, de‐escalation of therapy after 2 or 3 days of empiric treatment, and a strategy for the duration and discontinuation of therapy. An evidence‐based approach to these essential stewardship factors will improve patient outcomes by decreasing unnecessary antimicrobial exposures and associated unwanted effects as well as reduce the risk for emergence of antimicrobial resistance.
The intent of this educational activity is to illustrate these components of antimicrobial stewardship in a practical, case‐based format. Since hospitalists and intensivists play a central role in the formation and operation of a successful antimicrobial stewardship program, special consideration will be given to strategies that they can apply in their daily practices.
Target Audience
This activity was designed to meet the needs of hospitalists and intensivists who are involved in the diagnosis, management, and treatment of infectious diseases in the hospital setting. Other healthcare professionals are also invited to participate.
Faculty and Topics
Empiric Antibiotic Selection Strategies for Healthcare‐Associated Pneumonia, Intra‐abdominal Infections, and Catheter‐Associated Bacteremia
David R. Snydman, MD, FACP, FIDSA
Chief, Division of Geographic Medicine and Infectious Diseases
Tufts Medical Center
Professor of Medicine
Tufts University School of Medicine
Boston, Massachusetts
After completing this article, learners should be better able to:
Differentiate between colonization and infection in their patients in order to devise optimal initial therapy strategies
Identify risk factors for the development of antimicrobial resistance
Select the appropriate therapeutic agent for their hospitalized patients based on the organism and site of infection
Antimicrobial De‐escalation Strategies in Hospitalized Patients with Pneumonia, Intra‐abdominal Infections, and Bacteremia
Keith S. Kaye, MD, MPH
Professor of Medicine
Wayne State University
Corporate Director, Infection Prevention, Epidemiology and Antimicrobial Stewardship
Detroit Medical Center
Detroit, Michigan
After completing this article, learners should be better able to:
Assess the rationale behind antimicrobial de‐escalation in healthcare settings and its potential healthcare benefits
Implement effective de‐escalation strategies for their patients that are pathogen‐specific and minimize the emergence of resistance
Identify common targets and opportunities for de‐escalation programs in their institution
Duration and Cessation of Antimicrobial Treatment
Thomas M. File, Jr., MD, MSc, MACP, FIDSA, FCCP
Professor, Internal Medicine
Head, Infectious Disease Section
Northeastern Ohio Universities College of Medicine and Pharmacy
Akron, Ohio
After completing this article, learners should be better able to:
Develop an evidence‐based approach to duration and cessation of antimicrobial therapy for their patients
Assess clinical data in support of a shorter course of antimicrobial therapy
Incorporate strategies for their patients to optimize antimicrobial choices, dosages, and durations of therapy in order to decrease the emergence of antimicrobial resistance
Infections, Bacterial Resistance, and Antimicrobial Stewardship: The Emerging Role of Hospitalists
David J. Rosenberg, MD, MPH, FACP, SFHM (Chairman)
Associate Chair for Hospital Operations Department of Medicine
Section Head, Hospital Medicine, Division of General Internal Medicine
North Shore University Hospital
Manhasset, New York
After completing this article, learners should be better able to:
Describe the role of the hospitalist in the successful implementation of an antimicrobial stewardship program to improve quality of care and outcomes
Identify the key elements of an antimicrobial stewardship program that promote the judicious use of antibiotics in hospital settings
Apply the critical antimicrobial stewardship elements to the care of patients in their hospital
Accreditation Statement
This activity has been planned and implemented in accordance with the Essential Areas and Policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint sponsorship of the American Academy of CME, Inc. and Global Education Exchange, Inc. American Academy of CME is accredited by the ACCME to provide continuing medical education for physicians.
Credit Designation
American Academy of CME designates this enduring material for a maximum of 3.5 AMA PRA Category 1 Credit(s). Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Disclosure of Conflict of Interest
According to the disclosure policy of the American Academy of CME, all faculty, planning committee members, editors, managers, and other individuals who are in a position to control content are required to disclose any relevant relationships with any commercial interests related to this activity. The existence of these interests or relationships is not viewed as implying bias or decreasing the value of the presentation. All educational materials were reviewed for fair balance, scientific objectivity, and levels of evidence.
Academy planner John JD Juchniewicz, MCIS, CCMEP, and GLOBEX planners and editors Meri D. Pozo, PhD and Michael L. Coco, PhD reported no financial relationships or relationships to products or devices they or their spouse/life partner have with commercial interests related to the content of this CME activity.
The faculty reported the following financial relationships or affiliations with commercial interests during the past 12 months:
David J. Rosenberg, MD, MPH, FACP, SFHM
Advisory Boardfor scientific information: Canyon Pharmaceuticals
Consultantfor marketing purposes: UCB
Grant Recipient/Research Support (PI; funds paid to Feinstein Institute): Sanofi‐Aventis
Promotional Speaker's Bureau: Sanofi‐Aventis
David R. Snydman, MD, FACP, FIDSA
Advisory Boardfor scientific information: CSL Behring, Genentech, Genzyme, Millenium, Novartis
Consultantfor clinical trial design: CSL Behring
Grant Recipient/Research Support (PI; funds paid to Tufts Medical Center): Cubist, Forest Pharmaceuticals, Johnson & Johnson, Merck & Co., Inc., Pfizer
Promotional Speaker's Bureau: CSL Behring, Merck & Co., Inc.
Keith S. Kaye, MD, MPH
Advisory Boardfor scientific information: Forest Pharmaceuticals, Merck & Co., Inc., Ortho‐McNeil, Pfizer, TheraDoc
Grant Recipient/Research Support (PI; funds paid to Wayne State University): Merck & Co., Inc., Pfizer
Promotional Speaker's Bureau: Cubist, Merck & Co., Inc., Ortho‐McNeil, Pfizer
Thomas M. File, Jr., MD, MSc, MACP, FIDSA, FCCP
Consultantfor clinical trial design: Cerexa/Forest Pharmaceuticals, Glaxo SmithKline, Merck & Co., Inc., Nabriva Therapeutics, Ortho‐McNeil, Protez/Novartis, Pfizer, Rib‐X Pharmaceuticals, Shire, Tetraphase Pharmaceuticals
Grant Recipient/Research Support (PI; funds paid to Suma Health System): Boehringer Ingelheim, Cerexa/Forest Pharmaceuticals, Gilead, Ortho‐McNeil, Pfizer, Tibotec
Independent clinical peer‐reviewer:
David Alland, MD
Professor of Medicine
Chief, Division of Infectious Disease
Interim Director, Center for Emerging and Re‐Emerging Pathogens
Assistant Dean for Clinical Research
University of Medicine and Dentistry of New JerseyThe New Jersey Medical School
Newark, New Jersey
PI for NIH STTR grant to Cepheid (to develop TB diagnostics)grant ended 9/10
Member, group of patent holders related to molecular beacon licenses
Employee (spouse): Bristol‐Myers Squibb
Shareholder/Stock options (self and spouse): Bristol‐Myers Squibb
Disclosure of Unlabeled Use
This educational activity may contain discussion of published and/or investigational uses of agents that are not indicated by the FDA. Faculty have been asked to disclose off‐label and/or investigational uses where they are mentioned. American Academy of CME (Academy), Global Education Exchange, Inc. (GLOBEX) and Merck & Co., Inc. do not recommend the use of any agent outside of the labeled indications.
The opinions expressed in the educational activity are those of the faculty and do not necessarily represent the views of the Academy, GLOBEX, Merck & Co., Inc, or any other manufacturer of pharmaceuticals or devices. Before prescribing any medication, physicians should consult primary references and full prescribing information. Please refer to the official prescribing information for each product for discussion of approved indications, contraindications, and warnings. Further, learners should appraise the information presented critically and are encouraged to consult appropriate resources for any product or device mentioned in this activity.
In addition, the American Academy of CME requires all faculty/authors to note the level of evidence for any patient care recommendation they make.
Method of Participation:
There are no fees for participating and receiving CME credit for this activity. During the period January 9, 2012 through January 9, 2013, learners must 1) review the CME information including the learning objectives and disclosure statements; 2) study the educational content of the activity; 3) go online at
Media:
Journal supplement
Disclaimer
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patient's conditions and possible contraindications on dangers in use, review of any applicable manufacturer's product information, and comparison with recommendations of other authorities.
Contact Info:
For questions or comments about this CME activity, contact:
John JD Juchniewicz, MCIS, CCMEP
American Academy of CME
2012 American Academy of CME and Global Education Exchange, Inc.
Copyright © 2012 Society of Hospital Medicine
Preface
Antibiotic resistance is a particularly troublesome problem in healthcare institutions, and is clearly linked to antibiotic usage.12 The total annual cost of antimicrobial resistance was estimated to be as high as $35 billion or more in the United States in 1989,3 and much higher in current US dollars. Approximately 5 of every 100 patients admitted to a US hospital develops a nosocomial or hospital‐associated infection (HAI),4 and many of these infections involve bacteria resistant to 1 or more antibiotics.5 This is important because hospitalized patients infected with antibiotic‐resistant bacteria spend a longer time in the hospital, at increased cost, and are at higher risk of death compared to patients with similar infections due to antibiotic‐susceptible bacteria.6 Furthermore, choice of antimicrobial agents across all hospitalized patients is being driven by the concern for these resistant organisms, potentially contributing to medication costs and hospital length of stay.3
Awareness of the problem of HAIs and their frequent association with multidrug‐resistant pathogens, led The Joint Commission of the United States to identify reduction in risk of HAIs as one of their national patient safety goals.7 While The Joint Commission's primary focus is on infection control measures (eg, hand hygiene),7 antimicrobial stewardship can and should play a key role in reducing the emergence and subsequent transmission of antimicrobial‐resistant pathogens within the hospital or other healthcare settings.
Antimicrobial stewardship has been defined as the optimal selection, dose, and duration of an antimicrobial that results in the best clinical outcome for the treatment or prevention of infection, with minimal toxicity to the patient and minimal impact on subsequent resistance. Hospitalists will instinctively find the element of an antimicrobial stewardship to be inherently valuable to their clinical practice.8 By instituting and adhering to optimal antimicrobial usage within one's own practice and across their institutions, patient care is improved through better clinical outcomes, reduced microbial resistance, and shorter hospital stays.
This supplement of the Journal of Hospital Medicine examines key aspects of antimicrobial stewardship in 4 interrelated papers with respective focuses on appropriate initiation and selection of antibiotics (Dr Snydman), antimicrobial de‐escalation strategies (Dr Kaye), duration and cessation of treatment (Dr File), and the hospitalist's role in antimicrobial stewardship (Dr Rosenberg). Three case studies, interwoven through 3 of the 4 papers, are used to highlight the application of antimicrobial stewardship principles discussed in the respective papers, in patients commonly encountered in the hospital. The clinical cases deal with healthcare‐associated pneumonia, intra‐abdominal infections (diverticulitis), and central line‐associated bacteremia. The final paper by Rosenberg reviews trends in antimicrobial resistance, costs of hospital‐acquired infections, and lays out the argument for Hospitalist participation and, at times, leadership in antimicrobial stewardship programs.
- ,,, et al.Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship.Clin Infect Dis.2007;44:159–177.
- .Antimicrobial use: risk driver of multidrug resistant microorganisms in healthcare settings.Curr Opin Infect Dis.2009;22:352–358.
- .Bug/drug resistance: sometimes less is more.Med Care.1989;27:194–203.
- .Nosocomial infection update.Emerg Infect Dis.1998;4:416–420.
- ,,, et al.NHSN annual update: antimicrobial‐resistant pathogens associated with healthcare‐associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007.Infect Control Hosp Epidemiol.2008;29:996–1011.
- .The relationship between antimicrobial resistance and patient outcomes: mortality, length of hospital stay, and health care costs.Clin Infect Dis.2006;42(suppl 2):S82–S89.
- The Joint Commission. Accreditation Program: Hospital. National Patient Safety Goals. Effective January 1, 2011. Available at http://www.jointcommission.org/assets/1/6/2011_NPSGs_HAP.pdf. Accessed January 24, 2011.
- .The search for good antimicrobial stewardship.Jt Comm J Qual Improv.2001;27:403–404.
Antibiotic resistance is a particularly troublesome problem in healthcare institutions, and is clearly linked to antibiotic usage.12 The total annual cost of antimicrobial resistance was estimated to be as high as $35 billion or more in the United States in 1989,3 and much higher in current US dollars. Approximately 5 of every 100 patients admitted to a US hospital develops a nosocomial or hospital‐associated infection (HAI),4 and many of these infections involve bacteria resistant to 1 or more antibiotics.5 This is important because hospitalized patients infected with antibiotic‐resistant bacteria spend a longer time in the hospital, at increased cost, and are at higher risk of death compared to patients with similar infections due to antibiotic‐susceptible bacteria.6 Furthermore, choice of antimicrobial agents across all hospitalized patients is being driven by the concern for these resistant organisms, potentially contributing to medication costs and hospital length of stay.3
Awareness of the problem of HAIs and their frequent association with multidrug‐resistant pathogens, led The Joint Commission of the United States to identify reduction in risk of HAIs as one of their national patient safety goals.7 While The Joint Commission's primary focus is on infection control measures (eg, hand hygiene),7 antimicrobial stewardship can and should play a key role in reducing the emergence and subsequent transmission of antimicrobial‐resistant pathogens within the hospital or other healthcare settings.
Antimicrobial stewardship has been defined as the optimal selection, dose, and duration of an antimicrobial that results in the best clinical outcome for the treatment or prevention of infection, with minimal toxicity to the patient and minimal impact on subsequent resistance. Hospitalists will instinctively find the element of an antimicrobial stewardship to be inherently valuable to their clinical practice.8 By instituting and adhering to optimal antimicrobial usage within one's own practice and across their institutions, patient care is improved through better clinical outcomes, reduced microbial resistance, and shorter hospital stays.
This supplement of the Journal of Hospital Medicine examines key aspects of antimicrobial stewardship in 4 interrelated papers with respective focuses on appropriate initiation and selection of antibiotics (Dr Snydman), antimicrobial de‐escalation strategies (Dr Kaye), duration and cessation of treatment (Dr File), and the hospitalist's role in antimicrobial stewardship (Dr Rosenberg). Three case studies, interwoven through 3 of the 4 papers, are used to highlight the application of antimicrobial stewardship principles discussed in the respective papers, in patients commonly encountered in the hospital. The clinical cases deal with healthcare‐associated pneumonia, intra‐abdominal infections (diverticulitis), and central line‐associated bacteremia. The final paper by Rosenberg reviews trends in antimicrobial resistance, costs of hospital‐acquired infections, and lays out the argument for Hospitalist participation and, at times, leadership in antimicrobial stewardship programs.
Antibiotic resistance is a particularly troublesome problem in healthcare institutions, and is clearly linked to antibiotic usage.12 The total annual cost of antimicrobial resistance was estimated to be as high as $35 billion or more in the United States in 1989,3 and much higher in current US dollars. Approximately 5 of every 100 patients admitted to a US hospital develops a nosocomial or hospital‐associated infection (HAI),4 and many of these infections involve bacteria resistant to 1 or more antibiotics.5 This is important because hospitalized patients infected with antibiotic‐resistant bacteria spend a longer time in the hospital, at increased cost, and are at higher risk of death compared to patients with similar infections due to antibiotic‐susceptible bacteria.6 Furthermore, choice of antimicrobial agents across all hospitalized patients is being driven by the concern for these resistant organisms, potentially contributing to medication costs and hospital length of stay.3
Awareness of the problem of HAIs and their frequent association with multidrug‐resistant pathogens, led The Joint Commission of the United States to identify reduction in risk of HAIs as one of their national patient safety goals.7 While The Joint Commission's primary focus is on infection control measures (eg, hand hygiene),7 antimicrobial stewardship can and should play a key role in reducing the emergence and subsequent transmission of antimicrobial‐resistant pathogens within the hospital or other healthcare settings.
Antimicrobial stewardship has been defined as the optimal selection, dose, and duration of an antimicrobial that results in the best clinical outcome for the treatment or prevention of infection, with minimal toxicity to the patient and minimal impact on subsequent resistance. Hospitalists will instinctively find the element of an antimicrobial stewardship to be inherently valuable to their clinical practice.8 By instituting and adhering to optimal antimicrobial usage within one's own practice and across their institutions, patient care is improved through better clinical outcomes, reduced microbial resistance, and shorter hospital stays.
This supplement of the Journal of Hospital Medicine examines key aspects of antimicrobial stewardship in 4 interrelated papers with respective focuses on appropriate initiation and selection of antibiotics (Dr Snydman), antimicrobial de‐escalation strategies (Dr Kaye), duration and cessation of treatment (Dr File), and the hospitalist's role in antimicrobial stewardship (Dr Rosenberg). Three case studies, interwoven through 3 of the 4 papers, are used to highlight the application of antimicrobial stewardship principles discussed in the respective papers, in patients commonly encountered in the hospital. The clinical cases deal with healthcare‐associated pneumonia, intra‐abdominal infections (diverticulitis), and central line‐associated bacteremia. The final paper by Rosenberg reviews trends in antimicrobial resistance, costs of hospital‐acquired infections, and lays out the argument for Hospitalist participation and, at times, leadership in antimicrobial stewardship programs.
- ,,, et al.Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship.Clin Infect Dis.2007;44:159–177.
- .Antimicrobial use: risk driver of multidrug resistant microorganisms in healthcare settings.Curr Opin Infect Dis.2009;22:352–358.
- .Bug/drug resistance: sometimes less is more.Med Care.1989;27:194–203.
- .Nosocomial infection update.Emerg Infect Dis.1998;4:416–420.
- ,,, et al.NHSN annual update: antimicrobial‐resistant pathogens associated with healthcare‐associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007.Infect Control Hosp Epidemiol.2008;29:996–1011.
- .The relationship between antimicrobial resistance and patient outcomes: mortality, length of hospital stay, and health care costs.Clin Infect Dis.2006;42(suppl 2):S82–S89.
- The Joint Commission. Accreditation Program: Hospital. National Patient Safety Goals. Effective January 1, 2011. Available at http://www.jointcommission.org/assets/1/6/2011_NPSGs_HAP.pdf. Accessed January 24, 2011.
- .The search for good antimicrobial stewardship.Jt Comm J Qual Improv.2001;27:403–404.
- ,,, et al.Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship.Clin Infect Dis.2007;44:159–177.
- .Antimicrobial use: risk driver of multidrug resistant microorganisms in healthcare settings.Curr Opin Infect Dis.2009;22:352–358.
- .Bug/drug resistance: sometimes less is more.Med Care.1989;27:194–203.
- .Nosocomial infection update.Emerg Infect Dis.1998;4:416–420.
- ,,, et al.NHSN annual update: antimicrobial‐resistant pathogens associated with healthcare‐associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007.Infect Control Hosp Epidemiol.2008;29:996–1011.
- .The relationship between antimicrobial resistance and patient outcomes: mortality, length of hospital stay, and health care costs.Clin Infect Dis.2006;42(suppl 2):S82–S89.
- The Joint Commission. Accreditation Program: Hospital. National Patient Safety Goals. Effective January 1, 2011. Available at http://www.jointcommission.org/assets/1/6/2011_NPSGs_HAP.pdf. Accessed January 24, 2011.
- .The search for good antimicrobial stewardship.Jt Comm J Qual Improv.2001;27:403–404.
Identifying and treating factors that put patients at risk for suicide
Managing community-acquired pneumonia during flu season
General internists need to be able to recognize community-acquired pneumonia (CAP) so that diagnostic and therapeutic interventions can be initiated promptly. It is also important to understand the most likely and possible causes of CAP so that appropriate initial antimicrobial therapy can be chosen. Especially during flu season, influenza can present as CAP and should be included in the differential diagnosis.
When managing a patient with CAP, the internist must decide which level of care, diagnostic tests, antimicrobial agents, and follow-up plans are needed. These topics will be reviewed in this article.
TWO TERMS TO REMEMBER
- CAP refers to pneumonia acquired outside a health care facility. It can be either bacterial or viral.
- CABP (community-acquired bacterial pneumonia) refers only to those cases caused by bacterial pathogens.
NUMBERS AND TRENDS
In the United States, CAP is the number-one cause of death from infection and the sixth leading cause of death overall.1 Each year, it is responsible for about 4.2 million outpatient visits, more than 60,000 deaths, and more than $17 billion in health care expenses.2
Community-acquired bacterial pneumonia: Common, serious
In a population-based US study in 1991, the incidence of CABP requiring hospitalization was 266.8 per 100,000 people.3
Estimates of overall mortality in CABP vary depending on the severity of illness and comorbid conditions. A meta-analysis published in 1996 found the overall mortality rate to be 13.7%, with a range of 5.1% to 36.5% depending on severity.4
In hospitalized patients, mortality rates and length of hospital stay appear to be declining over time. Between 1993 and 2005, the age-adjusted mortality rate decreased from 8.9% to 4.1%, and the average length of stay decreased from 7.5 to 5.7 days, with an overall reduction in hospital cost.5
CABP is more prevalent in older people than in the general population, and it increases with age from 18.2 cases per 1,000 patient-years in patients 60 to 69 years to 52.3 cases per 1,000 patient-years in those older than 85 years.6 Risk factors for pneumonia in the elderly include heart disease, chronic lung disease, immunosuppressive drugs, alcoholism, and increasing age.7 Similar to the trend in the general population, the mortality rate in elderly CABP patients appears to be decreasing over time, possibly thanks to rising rates of pneumococcal and influenza vaccination.8
Among the general population, risk factors for developing CABP also include smoking, occupational dust exposure, history of childhood pneumonia, unemployment, and single marital status.9 The incidence of CABP does not appear to be higher among pregnant women, although it is the most frequent cause of nonobstetric death in this population.10
The use of proton pump inhibitors may be an emerging risk factor for CABP.11 Also, use of nonsteroidal anti-inflammatory drugs among patients with CABP is associated with a blunted inflammatory response as well as a higher risk of pleuropulmonary complications and a delay in presentation.12
Influenza is also common, potentially severe
Influenza is also very common and potentially severe. It can cause a spectrum of disease, from mild upper respiratory tract symptoms to severe viral pneumonia that can be life-threatening and complicated by respiratory failure and the acute respiratory distress syndrome (ARDS).
Influenza infection can also be complicated by subsequent bacterial pneumonia. However, the epidemiology of influenza infection differs from that of CABP in that influenza occurs seasonally.
In the United States, seasonal influenza causes 36,000 deaths and 200,000 hospitalizations annually.13,14 As with CABP, the risk of death from influenza increases with age: it is 16 times greater in people age 85 and older than in those ages 65 to 69.13
During yearly seasonal epidemics, those at the highest risk of hospitalization and death are at the extremes of age. Risk factors for complicated influenza include heart disease, lung disease, diabetes, renal failure, rheumatologic conditions, dementia, and neurologic disease.15,16 During the 2009 H1N1 influenza pandemic, unexpected severity was seen in previously healthy young adults as well as those with obesity, neurodegenerative disease, pregnancy, and asthma.17
PATHOGENS: TYPICAL, ATYPICAL, VIRAL
Identifying the etiologic organism in CAP is confounded by limitations in the available diagnostic tests and also by poor-quality specimens that often are contaminated with bacteria that colonize the upper airways. Given these caveats, the primary pathogens responsible for CAP broadly include typical bacterial pathogens, atypical bacterial pathogens, and viruses.
Typical bacterial pathogens include Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, Moraxella catarrhalis, and, less commonly, a variety of aerobic and anaerobic gram-negative rods including Pseudomonas aeruginosa, Acinetobacter species, and Klebsiella pneumoniae.
Atypical bacterial pathogens include Mycoplasma pneumoniae, Chlamydia pneumoniae, and Legionella species.18
Viruses implicated in adult CAP include influenza A and B, parainfluenza viruses, respiratory syncytial virus, and adenovirus.19 More recently, human metapneumovirus has been described as a cause of adult CAP.20
Clues to uncommon microbes
Specific historic features or coexisting conditions that may suggest an uncommon microbiologic diagnosis include21:
- Recent travel to the southwestern United States or Southeast Asia
- Ill contacts
- Exposure to birds, bats, rabbits, or farm animals
- Alcoholism
- Chronic obstructive pulmonary disease
- Human immunodeficiency virus infection
- Structural lung disease
- Prolonged cough with whoop or posttussive vomiting
- Aspiration
- Bioterrorism.
In cases in which one or more of these conditions exist, CAP may also be caused by other agents not listed above, including Mycobacterium tuberculosis, oral anaerobes, atypical mycobacteria, Histoplasma capsulatum, Chlamydophila psittaci, Francisella tularensis, Coxiella burnettii, Pneumocystis jiroveci, Cryptococcus, Aspergillus, Coccidioides, Hantavirus, avian influenza, Burkholderia pseudomallei, severe acute respiratory syndrome virus, Bordetella pertussis, Bacillus anthracis, and Yersinia pestis.
HOW BACTERIA INVADE THE LUNGS
The pathophysiology of CABP involves both host defense and microbial virulence factors.
The airways are most commonly exposed to microbes by microaspiration of upper airway flora, although hematogenous seeding of the lungs in a bacteremic patient or contiguous spread of infection from an adjacent site can also occur.
Mucociliary clearance and the cough reflex are important initial defenses against infection and can be inhibited by neurologic diseases and conditions that impair the mucociliary mechanism. Mucosal immune cells, including macrophages and neutrophils, recognize invading pathogens and generate an antibody response.
Regulation of the host inflammatory response to infection depends on a complex interaction between immune cells, inflammatory cytokines (eg, tumor necrosis factor alpha, interleukin 1-beta, and interleukin 6), and anti-inflammatory cytokines such as interleukin 1 receptor antagonist and soluble tumor necrosis factor receptor type I.22
The interaction and timing of the inflammatory and anti-inflammatory response are essential in manifesting an appropriate host response to infection. An inadequate inflammatory response can lead to sepsis and death, but an excessive, late anti-inflammatory response can lead to a systemic inflammatory response such as ARDS. Polymorphisms within the genes coding for these factors may explain the variation in severity of illness among patients with CABP.23
HOW INFLUENZA DOES ITS DAMAGE
There are three types of influenza virus: A, B, and C. Type A causes most human infections. The influenza A virus envelope comprises a lipid bilayer that contains the projecting glycoproteins hemagglutinin and neuraminidase. Influenza viruses are named on the basis of these proteins and are designated with an H and an N, respectively, each followed by a number referring to the subtype.
Influenza infection begins when the virus makes contact with the epithelium. Hemagglutinin binds to the host cell and allows viral entry, where it begins replication. Neuraminidase prevents viral aggregation and facilitates the release of virus from infected cells.24
Mature virions are released and spread to neighboring host cells; this process is associated with desquamation and inflammation of the airways, causing cough, rhinorrhea, and sore throat. Systemic symptoms are associated with the induction of interferon, which causes fever and myalgia.25
Recovery and immunity to influenza infection occurs through both humoral and cell-mediated immunity, with antibodies directed against the specific hemagglutinin and neuraminidase antigens of the infecting virus. Immunity wanes over time and with antigenic drift of circulating viruses, making the host susceptible to recurrent influenza infection.24
Influenza is often complicated by bacterial superinfection
The influenza virus acts synergistically with certain bacteria to increase infectivity, and this may explain why influenza is often complicated by bacterial superinfection.
Mechanisms leading to bacterial superinfection include increased binding and invasion of bacteria, increased viral replication, and modification of the host inflammatory response. Some S aureus strains produce a protease that directly activates influenza virus hemagglutinin; other bacteria can activate plasminogen to promote influenza replication. The resulting increase in proteases in host tissues promotes activation of influenza through cleavage of hemagglutinin.26
The influenza virus also causes damage to the airway epithelial layer, leading to increased exposure of the binding sites necessary for adherence of S pneumoniae.27
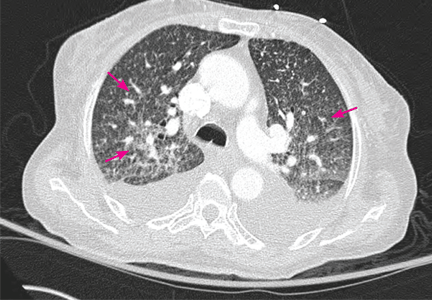
CLINICAL PRESENTATION OF COMMUNITY-ACQUIRED PNEUMONIA
Although CAP is common, agreement on its essential clinical signs and symptoms is surprisingly limited, due in part to heterogeneous patient presentations and in part to interobserver variability. The reader is referred to two excellent reviews on this topic.28,29
The diagnosis of CAP is made on clinical grounds, based on a combination of signs and symptoms. Symptoms of pneumonia can include cough, fever, chills, sputum production, dyspnea, and pleuritic pain. Physical findings can include tachypnea, tachycardia, hypoxemia, and consolidation or rales on auscultation. Laboratory data may show leukocytosis or elevated C-reactive protein, and radiographic studies may show evidence of a new infiltrate.21,30,31
Clinical presentation of influenza
Seasonal influenza as a cause of CAP is difficult to distinguish from bacterial causes. The clinical presentation of seasonal influenza most commonly includes fever or subjective feverishness, cough, myalgia, and weakness.32 In a recent multivariate analysis, five clinical features were shown to be predictive of pandemic H1N1 influenza pneumonia rather than CABP: age younger than 65 years, absence of confusion, white blood cell count less than 12 × 109/L, temperature higher than 38°C (100.4°F), and bilateral opacities on radiography.32,33
Complicated influenza infection can be either primary viral pneumonia or bacterial superinfection.
During the 1918 influenza pandemic, which predated the ability to isolate viruses, two clinical syndromes emerged: an ARDS associated with the rapid onset of cyanosis, delirium, and frothy blood-tinged sputum; and an acute bronchopneumonia characterized by necrosis, hemorrhage, edema, and vasculitis.34,35 The first syndrome has subsequently been shown to be associated with primary viral pneumonia, while the second is caused by bacterial superinfection. Modern reexamination of 1918 data has shown that bacterial superinfection was likely the reason for the distinctly fulminant presentation of that pandemic.36,37
The 2009 H1N1 influenza pandemic caused relatively mild disease in most patients. However, those with severe pneumonia more commonly developed ARDS from primary influenza pneumonia than from bacterial superinfection.17
A third influenza-associated infection is secondary bacterial pneumonia, which follows influenza infection and mimics the presentation of CABP. A typical patient presents with a recent history of influenza-like illness, followed 4 to 14 days later by a recurrence of fever, dyspnea, productive cough, and consolidation on chest radiographs.38 Leukocytosis with an increased number of immature neutrophil forms, prolonged duration of fever, and elevated erythrocyte sedimentation rate are more likely in patients with secondary bacterial pneumonia.39 Isolates from sputum samples commonly include S pneumoniae, S aureus, H influenzae, and other gram-negative rods.40
In recent flu seasons, methicillin-resistant S aureus (MRSA) has emerged as a cause of severe secondary pneumonia. Most of these isolates carry genes for the toxin Panton-Valentine leukocidin; the associated mortality rate is as high as 33%.41,42 Although community-acquired MRSA pneumonia has only been reported in case series, distinct clinical features that have been described include severe pneumonia with high fever, hypotension, shock, respiratory failure, leukopenia, and multilobar and cavitary infiltrates.43
WHEN TO SUSPECT INFLUENZA
The triad of fever, cough, and abrupt onset are the best predictors of influenza, but no single combination of signs and symptoms predict influenza infection with 100% certainty. Therefore, an understanding of local epidemiologic data regarding circulating influenza is essential to maintain a high index of suspicion.44
It is appropriate to suspect influenza in:
- Anyone who is epidemiologically linked to a known outbreak of influenza
- Children, adults, and health care workers who have fever and abrupt onset of respiratory symptoms
- Patients with fever plus exacerbation of underlying pulmonary disease
- Severely ill patients with fever or hypothermia, especially during influenza season.45
DIAGNOSTIC TESTING
Once the diagnosis of pulmonary infection is suggested by clinical features, the initial evaluation should include measurement of vital signs, physical examination, and radiographic imaging of the chest. Additional diagnostic measures to consider include viral testing, blood culture, sputum culture, urinary antigen testing for Legionella and for S pneumoniae, fungal culture, and mycobacterial smear and culture.
Chest radiography (with posterior-anterior and lateral films) is the study that usually demonstrates the presence of a pulmonary infiltrate. If initial chest radiographs do not show an infiltrate, imaging can be repeated after treatment is started if the patient’s clinical presentation still suggests pneumonia. Chest radiographs are of limited value in predicting the pathogen, but they help to determine the extent of pneumonia and to detect parapneumonic effusion.46
A caveat: anterior-posterior, posterior-anterior, and lateral views can miss more than 10% of effusions large enough to warrant thoracentesis, especially when there is lower-lobe consolidation.47
Blood cultures are recommended for patients admitted to the intensive care unit and for those with cavitary infiltrates, leukopenia, alcohol abuse, severe liver disease, asplenia, positive pneumococcal urinary antigen testing, or a pleural effusion.21 However, blood cultures are positive in only 3% to 14% of hospitalized patients with CABP, and the impact of a positive blood culture on management decisions in CABP appears to be quite small.48–50
For the highest yield, blood culture results should be obtained before antibiotics are given. Not only is this good practice, but obtaining blood culture results before starting antibiotics is one of the quality measures evaluated by the Center for Medicare and Medicaid Services.51
Sputum culture is considered optional for outpatients and patients with less-severe pneumonia.21 While it can provide a rapid diagnosis in certain cases, a good-quality sputum sample is obtained in only 39% to 54% of patients with CABP, yields a predominant morphotype in only 45% of cases, and provides a useful microbiologic diagnosis in only 14.4%.52,53 Fungal and mycobacterial cultures are only indicated in certain situations such as cavitary infiltrates or immunosuppression.
Urinary antigen testing for Legionella and S pneumoniae should be done in patients with more severe illness and in those for whom outpatient therapy has failed.21S pneumoniae testing has been shown to allow early diagnosis of pneumococcal pneumonia in 26% more patients than with Gram staining, but it fails to identify 22% of the rapid diagnoses initially identified by Gram staining.54 Thus, a sequential approach is reasonable, with urinary antigen testing for patients at high risk without useful results from sputum Gram staining. Also, recent data suggest that the pneumococcal urinary antigen test may allow optimization of antimicrobial therapy with good clinical outcomes.55
Endotracheal tests. If the patient is intubated, collection of endotracheal aspirates, bronchoscopy, or nonbronchoscopic bronchial lavage (sometimes called “mini-BAL”) should be performed.
Thoracentesis and pleural fluid cultures should be done if a pleural effusion is found. Empyema, large or loculated effusions, and parapneumonic effusions with a pH lower than 7.20, glucose levels less than 3.4 mmol/L (60 mg/dL), or positive results on microbial staining or culture should be drained by chest tube or surgically.56
Testing for influenza should be done if it will change the clinical management, such as the choice of antibiotic or infection control practices. Specimens should be obtained with either a nasopharyngeal swab or aspirate and tested with reverse transcriptase polymerase chain reaction, immunofluorescent staining, or rapid antigen detection, depending on local availability.45
Inflammatory biomarkers such as C-reactive protein and procalcitonin have been receiving interest as ways to predict the etiology and prognosis of CAP and to guide therapy. Several studies have shown that C-reactive protein can help distinguish between CAP and bronchitis, with higher values suggesting more severe pneumonia and pneumonia caused by S pneumoniae or L pneumophila.57 Procalcitonin may help discriminate between severe lower respiratory tract infections of bacterial and 2009 H1N1 origin, although less effectively than C-reactive protein. Low procalcitonin values, particularly when combined with low C-reactive protein levels, suggest that bacterial infection is unlikely.58
RISK STRATIFICATION AND SITE-OF-CARE DECISIONS
Following a presumptive diagnosis of CAP, it is important to decide not only what treatment the patient will receive but whether he or she should be hospitalized. If the patient is to be admitted to the hospital, the clinician must also decide if his or her condition warrants intensive care.
Severity-of-illness scores
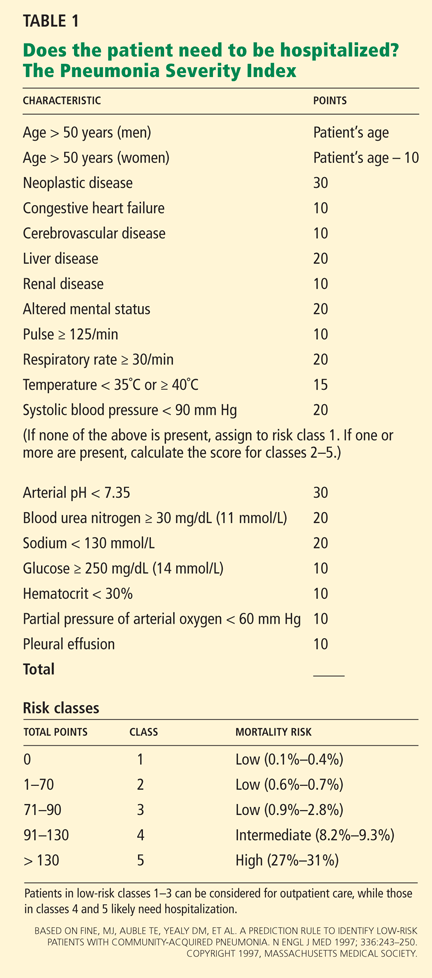
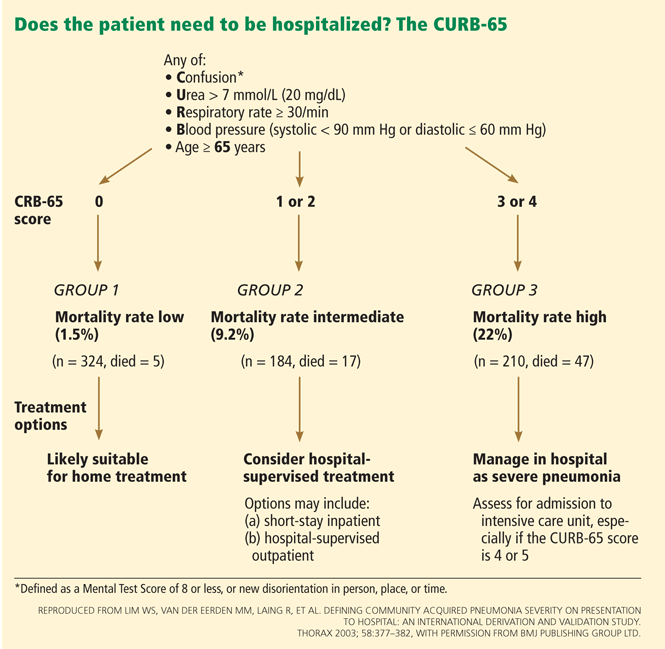
A recent meta-analysis compared the performance characteristics of the PSI and CURB-65 scores for predicting mortality in CAP and found no significant differences in overall test performance.61
Another meta-analysis found that the PSI was more sensitive than the CURB-65 and had a low false-negative rate, and so was better at showing which patients do not need to be hospitalized. Conversely, the CURB-65 was more specific and had a higher positive predictive value, and thus was more likely to correctly classify high-risk patients.62
Other scoring systems that aid in deciding about hospital admission and level of care include the CRB-6563 (which can be used instead of the CURB-65 if laboratory values are not available), SMART-COP,64 and SCAP.,65
Guidelines on when to admit to the intensive care unit
Guidelines from the Infectious Diseases Society of America (IDSA) and the American Thoracic Society (ATS) also provide guidance on when intensive care admission is advised,21 and their criteria were recently validated.66 The guidelines advocate direct admission to the intensive care unit for patients requiring vasopressors or mechanical ventilation, and intensive care unit or high-level monitoring for patients with three of the following criteria for severe CAP21:
- Respiratory rate ≥ 30
- Pao2/Fio2 ratio ≤ 250
- Multilobar infiltrates
- Confusion or disorientation
- Uremia (blood urea nitrogen ≥ 20 mg/dL)
- Leukopenia (white blood cell count < 4.0 × 109/L)
- Thrombocytopenia (platelet count < 100 × 109/L)
- Hypothermia (core temperature < 36.0°C [96.8°F])
- Hypotension requiring aggressive fluid resuscitation.
None of these scoring systems or criteria is meant to replace clinical judgment. A recent study has suggested that an oxygen saturation of less than 92% is an appropriate threshold for hospital admission, in view of higher rates of adverse events in outpatients with saturations below this value.67
TREATMENT
Multiple studies have shown that treatment of CAP in accordance with guidelines has led to improved clinical outcomes.21,68–70
How fast must antibiotics be started?
Based on studies that showed a lower mortality rate when antibiotics were started sooner, Medicare and Medicaid adopted a quality measure calling for starting antibiotics within 4 hours in patients being admitted to the hospital.50,71 However, several subsequent studies showed that the diagnosis of pneumonia is often incorrect and that rapid administration of antibiotics could lead to misdiagnosis, overuse of antibiotics, and a higher risk of Clostridium difficile infection.72,73
The current IDSA/ATS guidelines21 recommend that the first antibiotic dose be given while the patient is still in the emergency department, but do not give a specific time within which it should be given. Medicare and Medicaid later updated their quality measure to antibiotic administration within 6 hours.
Which antibiotics should be used?
The selection of antimicrobial agent depends upon the patient’s severity of illness and comorbid conditions.
Although most studies of combination antibiotic therapy have been retrospective and observational, they suggest that a macrolide (ie, one of the “mycins”) added to a beta-lactam antibiotic is beneficial, possibly by covering atypical organisms or via anti-inflammatory action.74–76 The choice of one antibiotic over another appears to be less important, and a recent Cochrane review concluded that there was no significant difference in efficacy among five antibiotic pairs studied.77
Empiric outpatient treatment of a previously healthy patient with CAP and no risk factors for drug-resistant S pneumoniae should include either a macrolide (azithromycin [Zithromax], clarithromycin [Biaxin], or erythromycin) or doxycycline. If the patient has a chronic comorbid condition such as heart, lung, liver, or renal disease, diabetes mellitus, alcoholism, malignancy, asplenia, or immunosuppression or has received antimicrobials within the preceding 3 months, then treatment should include either a respiratory fluoroquinolone (moxifloxacin [Avelox] or levofloxacin [Levaquin]) or a beta-lactam plus a macrolide.21
Overall, published data suggest that the survival rate is about the same with fluoroquinolone monotherapy as with beta-lactam plus macrolide combination therapy, and better than with beta-lactam monotherapy.78
Selection of antibiotics for inpatient treatment of CAP is influenced by severity of illness. Inpatients who do not require intensive care should be treated with either a respiratory fluoroquinolone or combination therapy with a beta-lactam (cefotaxime [Claforan], ceftriaxone [Rocephin], ampicillin, or ertapenem [Invanz]) plus a macrolide or doxycycline.21,76,79
If a specific microbiologic diagnosis is made, then treatment can be narrowed. However in certain cases, such as invasive pneumococcal infection, combination therapy may still be superior.80,81 For patients who need intensive care, treatment should always include a beta-lactam plus either azithromycin or a respiratory fluoroquinolone.21 In certain situations, additional antibiotics may be added as well, such as agents to treat Pseudomonas, community-acquired MRSA, or both.
Switching to oral therapy; short-course therapy
In the interest of avoiding unnecessary antibiotics, numerous studies have addressed the issue of an “early switch” to oral antibiotics and “short-course” therapy for CAP. In general, once clinically stable, patients with CAP, including bacteremic S pneumoniae pneumonia, can be safely switched to oral antibiotics.82
The issue of short-course therapy is more complicated, and the appropriate length of therapy for CAP is not well established. However, 5 days of levofloxacin 750 mg was shown to be as successful as 7 to 10 days of levofloxacin 500 mg.83 In another study, in patients who improved after 3 days of intravenous therapy for CAP, there was no difference in clinical outcome between those who were changed to oral therapy for 5 more days and those who received an oral placebo.84
Most patients who achieve clinical stability in the first week do not need prolonged antibiotic therapy. However, certain conditions, such as S aureus bacteremic pneumonia, complicated pneumonia, and pneumonia due to unusual organisms, may require prolonged treatment.
Other therapies
Additional therapies studied in patients with pneumonia include early mobilization, adjunctive corticosteroids, and statin drugs.
Early mobilization was shown in one study to decrease hospital length of stay without increasing adverse effects.85
Corticosteroids are not supported as a standard of care for patients with severe CAP according to current available studies.86,87 Furthermore, a randomized, controlled trial showed that prednisolone daily for a week did not improve outcomes in hospitalized patients with CAP, and it was associated with increased late failure.88
Statin trials under way. Several observational studies have suggested that statins might be beneficial in managing sepsis through their effects on endothelial cell function, antioxidant effects, anti-inflammatory effects, and immunomodulatory effects.89 However, a recent large prospective multicenter cohort study of hospitalized patients with CAP did not find evidence of a protective effect of statins on clinically meaningful outcomes in CAP or significant differences in circulating biomarkers.90 Several randomized trials of statin therapy in patients with both ventilator-associated pneumonia and CAP are under way.
INFLUENZA TREATMENT: MOST EFFECTIVE WITHIN 48 HOURS
Treatment with antiviral drugs is most effective if started within 48 hours after symptom onset, although some patients with confirmed influenza who are either not improving or who are critically ill may still benefit from treatment started later.
Treatment should be considered in patients with laboratory-confirmed or suspected influenza who are at risk of developing complicated influenza and in otherwise healthy patients who wish to reduce the duration of illness or who have close contact with patients who are at high risk of complications.
Antiviral medications are oseltamivir (Tamiflu), zanamivir (Relenza), and the adamantines amantadine (Symmetrel) and rimantadine (Flumadine).
Due to evolving viral resistance patterns, the choice of antiviral drug depends on the strain. Seasonal H1N1 is best treated with zanamivir or an adamantine, while pandemic 2009 H1N1 and H3N2 are best treated with zanamivir or oseltamivir. When strain typing is not available, empiric therapy should be with either zanamivir monotherapy or a combination of oseltamivir plus rimantadine. Influenza B viruses are resistant to adamantines and should be treated only with either zanamivir or oseltamivir.45
FOLLOW-UP AND PREVENTION
Patients with CAP can generally be expected to improve within 3 to 7 days.91 However, it may be several weeks before they return to baseline.92
Follow-up plans may be guided by the time to clinical stability. For patients who do not achieve clinical stability until more than 72 hours after admission, more aggressive follow-up on discharge is indicated, since they are more likely to experience early readmission and death.93
Pneumococcal vaccination. Because S pneumoniae remains the most common cause of CAP, efforts should be made to vaccinate patients appropriately. The Advisory Committee on Immunization Practices (ACIP) and the US Centers for Disease Control and Prevention recommend that the pneumococcal polysaccharide vaccine (Pneumovax 23; PPSV23) be given to those over age 65. Those who were vaccinated before age 65 should receive another dose at age 65 or later if at least 5 years have passed since their previous dose. Those who receive it at or after age 65 should receive only a single dose. A second dose is recommended 5 years after the first dose for people age 19 to 64 years with functional or anatomic asplenia and for those who are immunocompromised.
Influenza vaccination for all. Of note, the ACIP updated its guidelines on influenza vaccination beginning with the 2010–2011 influenza season. It no longer advocates a risk-stratified approach. Instead, it recommends universal influenza vaccination for everybody more than 6 months old.94
Smoking cessation should be addressed. Smoking cessation is a Medicare and Medicaid quality measure and should be encouraged after an episode of CAP because quitting smoking reduces the risk of pneumococcal disease by approximately 14% each year thereafter.95
- Mortensen EM, Kapoor WN, Chang CC, Fine MJ. Assessment of mortality after long-term follow-up of patients with community-acquired pneumonia. Clin Infect Dis 2003; 37:1617–1624.
- File TM, Marrie TJ. Burden of community-acquired pneumonia in North American adults. Postgrad Med 2010; 122:130–141.
- Marston BJ, Plouffe JF, File TM, et al. Incidence of community-acquired pneumonia requiring hospitalization. Results of a population-based active surveillance study in Ohio. The Community-Based Pneumonia Incidence Study Group. Arch Intern Med 1997; 157:1709–1718.
- Fine MJ, Smith MA, Carson CA, et al. Prognosis and outcomes of patients with community-acquired pneumonia. JAMA 1996; 275:134–141.
- Ruhnke GW, Coca-Perraillon M, Kitch BT, Cutler DM. Trends in mortality and medical spending in patients hospitalized for community-acquired pneumonia: 1993–2005. Med Care 2010; 48:1111–1116.
- Jackson ML, Neuzil KM, Thompson WW, et al. The burden of community-acquired pneumonia in seniors: results of a population-based study. Clin Infect Dis 2004; 39:1642–1650.
- Koivula I, Sten M, Mäkelä PH. Risk factors for pneumonia in the elderly. Am J Med 1994; 96:313–320.
- Ruhnke GW, Coca-Perraillon M, Kitch BT, Cutler DM. Marked reduction in 30-day mortality among elderly patients with community-acquired pneumonia. Am J Med 2011; 124:171–178.
- Farr BM, Bartlett CL, Wadsworth J, Miller DL. Risk factors for community-acquired pneumonia diagnosed upon hospital admission. British Thoracic Society Pneumonia Study Group. Respir Med 2000; 94:954–963.
- Graves CR. Pneumonia in pregnancy. Clin Obstet Gynecol 2010; 53:329–336.
- Eom CS, Jeon CY, Lim JW, Cho EG, Park SM, Lee KS. Use of acid-suppressive drugs and risk of pneumonia: a systematic review and meta-analysis. CMAJ 2011; 183:310–319.
- Voiriot G, Dury S, Parrot A, Mayaud C, Fartoukh M. Nonsteroidal antiinflammatory drugs may affect the presentation and course of community-acquired pneumonia. Chest 2011; 139:387–394.
- Thompson WW, Shay DK, Weintraub E, et al. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 2003; 289:179–186.
- Thompson WW, Shay DK, Weintraub E, et al. Influenza-associated hospitalizations in the United States. JAMA 2004; 292:1333–1340.
- Glezen WP, Decker M, Perrotta DM. Survey of underlying conditions of persons hospitalized with acute respiratory disease during influenza epidemics in Houston, 1978–1981. Am Rev Respir Dis 1987; 136:550–555.
- Izurieta HS, Thompson WW, Kramarz P, et al. Influenza and the rates of hospitalization for respiratory disease among infants and young children. N Engl J Med 2000; 342:232–239.
- Rothberg MB, Haessler SD. Complications of seasonal and pandemic influenza. Crit Care Med 2010; 38(suppl 4):e91–e97.
- Apisarnthanarak A, Mundy LM. Etiology of community-acquired pneumonia. Clin Chest Med 2005; 26:47–55.
- de Roux A, Marcos MA, Garcia E, et al. Viral community-acquired pneumonia in nonimmunocompromised adults. Chest 2004; 125:1343–1351.
- Hamelin ME, Côté S, Laforge J, et al. Human metapneumovirus infection in adults with community-acquired pneumonia and exacerbation of chronic obstructive pulmonary disease. Clin Infect Dis 2005; 41:498–502.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44(suppl 2):S27–S72.
- Kolling UK, Hansen F, Braun J, Rink L, Katus HA, Dalhoff K. Leucocyte response and anti-inflammatory cytokines in community acquired pneumonia. Thorax 2001; 56:121–125.
- Wunderink RG, Waterer GW. Community-acquired pneumonia: pathophysiology and host factors with focus on possible new approaches to management of lower respiratory tract infections. Infect Dis Clin North Am 2004; 18:743–759.
- Hilleman MR. Realities and enigmas of human viral influenza: pathogenesis, epidemiology and control. Vaccine 2002; 20:3068–3087.
- Bender BS, Small PA. Influenza: pathogenesis and host defense. Semin Respir Infect 1992; 7:38–45.
- Scheiblauer H, Reinacher M, Tashiro M, Rott R. Interactions between bacteria and influenza A virus in the development of influenza pneumonia. J Infect Dis 1992; 166:783–791.
- McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev 2006; 19:571–582.
- Metlay JP, Kapoor WN, Fine MJ. Does this patient have community-acquired pneumonia? Diagnosing pneumonia by history and physical examination. JAMA 1997; 278:1440–1445.
- Benbassat J, Baumal R. Narrative review: should teaching of the respiratory physical examination be restricted only to signs with proven reliability and validity? J Gen Intern Med 2010; 25:865–872.
- Kolsuz M, Erginel S, Alatas O, et al. Acute phase reactants and cytokine levels in unilateral community-acquired pneumonia. Respiration 2003; 70:615–622.
- Alves DW, Kennedy MT. Community-acquired pneumonia in casualty: etiology, clinical features, diagnosis, and management (or a look at the “new” in pneumonia since 2002). Curr Opin Pulm Med 2004; 10:166–170.
- Monto AS, Gravenstein S, Elliott M, Colopy M, Schweinle J. Clinical signs and symptoms predicting influenza infection. Arch Intern Med 2000; 160:3243–3247.
- Bewick T, Myles P, Greenwood S, et al; Influenza Clinical Information Network. Clinical and laboratory features distinguishing pandemic H1N1 influenza-related pneumonia from interpandemic community-acquired pneumonia in adults. Thorax 2011; 66:247–252.
- Morens DM, Fauci AS. The 1918 influenza pandemic: insights for the 21st century. J Infect Dis 2007; 195:1018–1028.
- Starr I. Influenza in 1918: recollections of the epidemic in Philadelphia. 1976. Ann Intern Med 2006; 145:138–140.
- Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis 2008; 198:962–970.
- Brundage JF, Shanks GD. Deaths from bacterial pneumonia during 1918–19 influenza pandemic. Emerg Infect Dis 2008; 14:1193–1199.
- Treanor J. Influenza virus. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, PA: Elsevier/Churchill Livingstone; 2005:2060–2085.
- Jarstrand C, Tunevall G. The influence of bacterial superinfection on the clinical course of influenza. Studies from the influenza epidemics in Stockholm during the winters 1969–70 and 1971–72. Scand J Infect Dis 1975; 7:243–247.
- Schwarzmann SW, Adler JL, Sullivan RJ, Marine WM. Bacterial pneumonia during the Hong Kong influenza epidemic of 1968–1969. Arch Intern Med 1971; 127:1037–1041.
- Hageman JC, Uyeki TM, Francis JS, et al. Severe community-acquired pneumonia due to Staphylococcus aureus, 2003–04 influenza season. Emerg Infect Dis 2006; 12:894–899.
- Centers for Disease Control and Prevention (CDC). Severe methicillin-resistant Staphylococcus aureus community-acquired pneumonia associated with influenza—Louisiana and Georgia, December 2006–January 2007. MMWR Morb Mortal Wkly Rep 2007; 56:325–329.
- Hidron AI, Low CE, Honig EG, Blumberg HM. Emergence of community-acquired methicillin-resistant Staphylococcus aureus strain USA300 as a cause of necrotising community-onset pneumonia. Lancet Infect Dis 2009; 9:384–392.
- Call SA, Vollenweider MA, Hornung CA, Simel DL, McKinney WP. Does this patient have influenza? JAMA 2005; 293:987–997.
- Harper SA, Bradley JS, Englund JA, et al; Expert Panel of the Infectious Diseases Society of America. Seasonal influenza in adults and children—diagnosis, treatment, chemoprophylaxis, and institutional outbreak management: clinical practice guidelines of the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:1003–1032.
- Boersma WG, Daniels JM, Löwenberg A, Boeve WJ, van de Jagt EJ. Reliability of radiographic findings and the relation to etiologic agents in community-acquired pneumonia. Respir Med 2006; 100:926–932.
- Brixey AG, Luo Y, Skouras V, Awdankiewicz A, Light RW. The efficacy of chest radiographs in detecting parapneumonic effusions. Respirology 2011; 16:1000–1004.
- Campbell SG, Marrie TJ, Anstey R, Dickinson G, Ackroyd-Stolarz S. The contribution of blood cultures to the clinical management of adult patients admitted to the hospital with community-acquired pneumonia: a prospective observational study. Chest 2003; 123:1142–1150.
- Waterer GW, Wunderink RG. The influence of the severity of community-acquired pneumonia on the usefulness of blood cultures. Respir Med 2001; 95:78–82.
- Houck PM, Bratzler DW, Nsa W, Ma A, Bartlett JG. Timing of antibiotic administration and outcomes for Medicare patients hospitalized with community-acquired pneumonia. Arch Intern Med 2004; 164:637–644.
- Information & Quality Healthcare. http://www.IQH.org/attachments/219_CoreMHelpBookletpg4_11_3.pdf. Accessed November 14, 2011.
- Rosón B, Carratalà J, Verdaguer R, Dorca J, Manresa F, Gudiol F. Prospective study of the usefulness of sputum Gram stain in the initial approach to community-acquired pneumonia requiring hospitalization. Clin Infect Dis 2000; 31:869–874.
- García-Vázquez E, Marcos MA, Mensa J, et al. Assessment of the usefulness of sputum culture for diagnosis of community-acquired pneumonia using the PORT predictive scoring system. Arch Intern Med 2004; 164:1807–1811.
- Rosón B, Fernández-Sabé N, Carratalà J, et al. Contribution of a urinary antigen assay (Binax NOW) to the early diagnosis of pneumococcal pneumonia. Clin Infect Dis 2004; 38:222–226.
- Sordé R, Falcó V, Lowak M, et al. Current and potential usefulness of pneumococcal urinary antigen detection in hospitalized patients with community-acquired pneumonia to guide antimicrobial therapy. Arch Intern Med 2011; 171:166–172.
- Koegelenberg CFN, Diacon AH, Bolliger CT. Parapneumonic pleural effusion and empyema. Respiration 2008; 75:241–250.
- Almirall J, Bolíbar I, Toran P, et al; Community-Acquired Pneumonia Maresme Study Group. Contribution of C-reactive protein to the diagnosis and assessment of severity of community-acquired pneumonia. Chest 2004; 125:1335–1342.
- Ingram PR, Inglis T, Moxon D, Speers D. Procalcitonin and C-reactive protein in severe 2009 H1N1 influenza infection. Intensive Care Med 2010; 36:528–532.
- Fine MJ, Auble TE, Yealy DM, et al. A prediction rule to identify low-risk patients with community-acquired pneumonia. N Engl J Med 1997; 336:243–250.
- Lim WS, van der Eerden MM, Laing R, et al. Defining community acquired pneumonia severity on presentation to hospital: an international derivation and validation study. Thorax 2003; 58:377–382.
- Chalmers JD, Singanayagam A, Akram AR, et al. Severity assessment tools for predicting mortality in hospitalised patients with community-acquired pneumonia. Systematic review and meta-analysis. Thorax 2010; 65:878–883.
- Loke YK, Kwok CS, Niruban A, Myint PK. Value of severity scales in predicting mortality from community-acquired pneumonia: systematic review and meta-analysis. Thorax 2010; 65:884–890.
- Capelastegui A, España PP, Quintana JM, et al. Validation of a predictive rule for the management of community-acquired pneumonia. Eur Respir J 2006; 27:151–157.
- Charles PG, Wolfe R, Whitby M, et al; Australian Community-Acquired Pneumonia Study Collaboration. SMART-COP: a tool for predicting the need for intensive respiratory or vasopressor support in community-acquired pneumonia. Clin Infect Dis 2008; 47:375–384.
- España PP, Capelastegui A, Gorordo I, et al. Development and validation of a clinical prediction rule for severe community-acquired pneumonia. Am J Respir Crit Care Med 2006; 174:1249–1256.
- Chalmers JD, Taylor JK, Mandal P, et al. Validation of the Infectious Diseases Society of America/American Thoracic Society minor criteria for intensive care unit admission in community-acquired pneumonia patients without major criteria or contraindications to intensive care unit care. Clin Infect Dis 2011; 53:503–511.
- Majumdar SR, Eurich DT, Gamble JM, Senthilselvan A, Marrie TJ. Oxygen saturations less than 92% are associated with major adverse events in outpatients with pneumonia: a population-based cohort study. Clin Infect Dis 2011; 52:325–331.
- Nathwani D, Rubinstein E, Barlow G, Davey P. Do guidelines for community-acquired pneumonia improve the cost-effectiveness of hospital care? Clin Infect Dis 2001; 32:728–741.
- Dean NC, Silver MP, Bateman KA, James B, Hadlock CJ, Hale D. Decreased mortality after implementation of a treatment guideline for community-acquired pneumonia. Am J Med 2001; 110:451–457.
- Capelastegui A, España PP, Quintana JM, et al. Improvement of process-of-care and outcomes after implementing a guideline for the management of community-acquired pneumonia: a controlled before-and-after design study. Clin Infect Dis 2004; 39:955–963.
- Silber SH, Garrett C, Singh R, et al. Early administration of antibiotics does not shorten time to clinical stability in patients with moderate-to-severe community-acquired pneumonia. Chest 2003; 124:1798–1804.
- Welker JA, Huston M, McCue JD. Antibiotic timing and errors in diagnosing pneumonia. Arch Intern Med 2008; 168:351–356.
- Polgreen PM, Chen YY, Cavanaugh JE, et al. An outbreak of severe Clostridium difficile-associated disease possibly related to inappropriate antimicrobial therapy for community-acquired pneumonia. Infect Control Hosp Epidemiol 2007; 28:212–214.
- Waterer GW, Somes GW, Wunderink RG. Monotherapy may be suboptimal for severe bacteremic pneumococcal pneumonia. Arch Intern Med 2001; 161:1837–1842.
- Lodise TP, Kwa A, Cosler L, Gupta R, Smith RP. Comparison of beta-lactam and macrolide combination therapy versus fluoroquinolone monotherapy in hospitalized Veterans Affairs patients with community-acquired pneumonia. Antimicrob Agents Chemother 2007; 51:3977–3982.
- Waterer GW, Rello J, Wunderink RG. Management of community-acquired pneumonia in adults. Am J Respir Crit Care Med 2011; 183:157–164.
- Bjerre LM, Verheij TJ, Kochen MM. Antibiotics for community acquired pneumonia in adult outpatients. Cochrane Database Syst Rev 2009; (4):CD002109.
- Frei CR, Labreche MJ, Attridge RT. Fluoroquinolones in community-acquired pneumonia: guide to selection and appropriate use. Drugs 2011; 71:757–770.
- Weiss K, Tillotson GS. The controversy of combination vs monotherapy in the treatment of hospitalized community-acquired pneumonia. Chest 2005; 128:940–946.
- Martínez JA, Horcajada JP, Almela M, et al. Addition of a macrolide to a beta-lactam-based empirical antibiotic regimen is associated with lower in-hospital mortality for patients with bacteremic pneumococcal pneumonia. Clin Infect Dis 2003; 36:389–395.
- Waterer GW, Somes GW, Wunderink RG. Monotherapy may be suboptimal for severe bacteremic pneumococcal pneumonia. Arch Intern Med 2001; 161:1837–1842.
- Ramirez JA, Bordon J. Early switch from intravenous to oral antibiotics in hospitalized patients with bacteremic community-acquired Streptococcus pneumoniae pneumonia. Arch Intern Med 2001; 161:848–850.
- Dunbar LM, Wunderink RG, Habib MP, et al. High-dose, short-course levofloxacin for community-acquired pneumonia: a new treatment paradigm. Clin Infect Dis 2003; 37:752–760.
- el Moussaoui R, de Borgie CA, van den Broek P, et al. Effectiveness of discontinuing antibiotic treatment after three days versus eight days in mild to moderate-severe community acquired pneumonia: randomised, double blind study. BMJ 2006; 332:1355.
- Mundy LM, Leet TL, Darst K, Schnitzler MA, Dunagan WC. Early mobilization of patients hospitalized with community-acquired pneumonia. Chest 2003; 124:883–889.
- Salluh JI, Póvoa P, Soares M, Castro-Faria-Neto HC, Bozza FA, Bozza PT. The role of corticosteroids in severe community-acquired pneumonia: a systematic review. Crit Care 2008; 12:R76.
- Mikami K, Suzuki M, Kitagawa H, et al. Efficacy of corticosteroids in the treatment of community-acquired pneumonia requiring hospitalization. Lung 2007; 185:249–255.
- Snijders D, Daniels JM, de Graaff CS, van der Werf TS, Boersma WG. Efficacy of corticosteroids in community-acquired pneumonia: a randomized double-blinded clinical trial. Am J Respir Crit Care Med 2010; 181:975–982.
- Chopra V, Flanders SA. Does statin use improve pneumonia outcomes? Chest 2009; 136:1381–1388.
- Yende S, Milbrandt EB, Kellum JA, et al. Understanding the potential role of statins in pneumonia and sepsis. Crit Care Med 2011; 39:1871–1878.
- Halm EA, Fine MJ, Marrie TJ, et al. Time to clinical stability in patients hospitalized with community-acquired pneumonia: implications for practice guidelines. JAMA 1998; 279:1452–1457.
- Marrie TJ, Lau CY, Wheeler SL, Wong CJ, Feagan BG. Predictors of symptom resolution in patients with community-acquired pneumonia. Clin Infect Dis 2000; 31:1362–1367.
- Aliberti S, Peyrani P, Filardo G, et al. Association between time to clinical stability and outcomes after discharge in hospitalized patients with community-acquired pneumonia. Chest 2011; 140:482–488.
- Fiore AE, Uyeki TM, Broder K, et al; Centers for Disease Control and Prevention (CDC). Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Recomm Rep 2010; 59:1–62.
- Nuorti JP, Butler JC, Farley MM, et al. Cigarette smoking and invasive pneumococcal disease. Active Bacterial Core Surveillance Team. N Engl J Med 2000; 342:681–689.
General internists need to be able to recognize community-acquired pneumonia (CAP) so that diagnostic and therapeutic interventions can be initiated promptly. It is also important to understand the most likely and possible causes of CAP so that appropriate initial antimicrobial therapy can be chosen. Especially during flu season, influenza can present as CAP and should be included in the differential diagnosis.
When managing a patient with CAP, the internist must decide which level of care, diagnostic tests, antimicrobial agents, and follow-up plans are needed. These topics will be reviewed in this article.
TWO TERMS TO REMEMBER
- CAP refers to pneumonia acquired outside a health care facility. It can be either bacterial or viral.
- CABP (community-acquired bacterial pneumonia) refers only to those cases caused by bacterial pathogens.
NUMBERS AND TRENDS
In the United States, CAP is the number-one cause of death from infection and the sixth leading cause of death overall.1 Each year, it is responsible for about 4.2 million outpatient visits, more than 60,000 deaths, and more than $17 billion in health care expenses.2
Community-acquired bacterial pneumonia: Common, serious
In a population-based US study in 1991, the incidence of CABP requiring hospitalization was 266.8 per 100,000 people.3
Estimates of overall mortality in CABP vary depending on the severity of illness and comorbid conditions. A meta-analysis published in 1996 found the overall mortality rate to be 13.7%, with a range of 5.1% to 36.5% depending on severity.4
In hospitalized patients, mortality rates and length of hospital stay appear to be declining over time. Between 1993 and 2005, the age-adjusted mortality rate decreased from 8.9% to 4.1%, and the average length of stay decreased from 7.5 to 5.7 days, with an overall reduction in hospital cost.5
CABP is more prevalent in older people than in the general population, and it increases with age from 18.2 cases per 1,000 patient-years in patients 60 to 69 years to 52.3 cases per 1,000 patient-years in those older than 85 years.6 Risk factors for pneumonia in the elderly include heart disease, chronic lung disease, immunosuppressive drugs, alcoholism, and increasing age.7 Similar to the trend in the general population, the mortality rate in elderly CABP patients appears to be decreasing over time, possibly thanks to rising rates of pneumococcal and influenza vaccination.8
Among the general population, risk factors for developing CABP also include smoking, occupational dust exposure, history of childhood pneumonia, unemployment, and single marital status.9 The incidence of CABP does not appear to be higher among pregnant women, although it is the most frequent cause of nonobstetric death in this population.10
The use of proton pump inhibitors may be an emerging risk factor for CABP.11 Also, use of nonsteroidal anti-inflammatory drugs among patients with CABP is associated with a blunted inflammatory response as well as a higher risk of pleuropulmonary complications and a delay in presentation.12
Influenza is also common, potentially severe
Influenza is also very common and potentially severe. It can cause a spectrum of disease, from mild upper respiratory tract symptoms to severe viral pneumonia that can be life-threatening and complicated by respiratory failure and the acute respiratory distress syndrome (ARDS).
Influenza infection can also be complicated by subsequent bacterial pneumonia. However, the epidemiology of influenza infection differs from that of CABP in that influenza occurs seasonally.
In the United States, seasonal influenza causes 36,000 deaths and 200,000 hospitalizations annually.13,14 As with CABP, the risk of death from influenza increases with age: it is 16 times greater in people age 85 and older than in those ages 65 to 69.13
During yearly seasonal epidemics, those at the highest risk of hospitalization and death are at the extremes of age. Risk factors for complicated influenza include heart disease, lung disease, diabetes, renal failure, rheumatologic conditions, dementia, and neurologic disease.15,16 During the 2009 H1N1 influenza pandemic, unexpected severity was seen in previously healthy young adults as well as those with obesity, neurodegenerative disease, pregnancy, and asthma.17
PATHOGENS: TYPICAL, ATYPICAL, VIRAL
Identifying the etiologic organism in CAP is confounded by limitations in the available diagnostic tests and also by poor-quality specimens that often are contaminated with bacteria that colonize the upper airways. Given these caveats, the primary pathogens responsible for CAP broadly include typical bacterial pathogens, atypical bacterial pathogens, and viruses.
Typical bacterial pathogens include Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, Moraxella catarrhalis, and, less commonly, a variety of aerobic and anaerobic gram-negative rods including Pseudomonas aeruginosa, Acinetobacter species, and Klebsiella pneumoniae.
Atypical bacterial pathogens include Mycoplasma pneumoniae, Chlamydia pneumoniae, and Legionella species.18
Viruses implicated in adult CAP include influenza A and B, parainfluenza viruses, respiratory syncytial virus, and adenovirus.19 More recently, human metapneumovirus has been described as a cause of adult CAP.20
Clues to uncommon microbes
Specific historic features or coexisting conditions that may suggest an uncommon microbiologic diagnosis include21:
- Recent travel to the southwestern United States or Southeast Asia
- Ill contacts
- Exposure to birds, bats, rabbits, or farm animals
- Alcoholism
- Chronic obstructive pulmonary disease
- Human immunodeficiency virus infection
- Structural lung disease
- Prolonged cough with whoop or posttussive vomiting
- Aspiration
- Bioterrorism.
In cases in which one or more of these conditions exist, CAP may also be caused by other agents not listed above, including Mycobacterium tuberculosis, oral anaerobes, atypical mycobacteria, Histoplasma capsulatum, Chlamydophila psittaci, Francisella tularensis, Coxiella burnettii, Pneumocystis jiroveci, Cryptococcus, Aspergillus, Coccidioides, Hantavirus, avian influenza, Burkholderia pseudomallei, severe acute respiratory syndrome virus, Bordetella pertussis, Bacillus anthracis, and Yersinia pestis.
HOW BACTERIA INVADE THE LUNGS
The pathophysiology of CABP involves both host defense and microbial virulence factors.
The airways are most commonly exposed to microbes by microaspiration of upper airway flora, although hematogenous seeding of the lungs in a bacteremic patient or contiguous spread of infection from an adjacent site can also occur.
Mucociliary clearance and the cough reflex are important initial defenses against infection and can be inhibited by neurologic diseases and conditions that impair the mucociliary mechanism. Mucosal immune cells, including macrophages and neutrophils, recognize invading pathogens and generate an antibody response.
Regulation of the host inflammatory response to infection depends on a complex interaction between immune cells, inflammatory cytokines (eg, tumor necrosis factor alpha, interleukin 1-beta, and interleukin 6), and anti-inflammatory cytokines such as interleukin 1 receptor antagonist and soluble tumor necrosis factor receptor type I.22
The interaction and timing of the inflammatory and anti-inflammatory response are essential in manifesting an appropriate host response to infection. An inadequate inflammatory response can lead to sepsis and death, but an excessive, late anti-inflammatory response can lead to a systemic inflammatory response such as ARDS. Polymorphisms within the genes coding for these factors may explain the variation in severity of illness among patients with CABP.23
HOW INFLUENZA DOES ITS DAMAGE
There are three types of influenza virus: A, B, and C. Type A causes most human infections. The influenza A virus envelope comprises a lipid bilayer that contains the projecting glycoproteins hemagglutinin and neuraminidase. Influenza viruses are named on the basis of these proteins and are designated with an H and an N, respectively, each followed by a number referring to the subtype.
Influenza infection begins when the virus makes contact with the epithelium. Hemagglutinin binds to the host cell and allows viral entry, where it begins replication. Neuraminidase prevents viral aggregation and facilitates the release of virus from infected cells.24
Mature virions are released and spread to neighboring host cells; this process is associated with desquamation and inflammation of the airways, causing cough, rhinorrhea, and sore throat. Systemic symptoms are associated with the induction of interferon, which causes fever and myalgia.25
Recovery and immunity to influenza infection occurs through both humoral and cell-mediated immunity, with antibodies directed against the specific hemagglutinin and neuraminidase antigens of the infecting virus. Immunity wanes over time and with antigenic drift of circulating viruses, making the host susceptible to recurrent influenza infection.24
Influenza is often complicated by bacterial superinfection
The influenza virus acts synergistically with certain bacteria to increase infectivity, and this may explain why influenza is often complicated by bacterial superinfection.
Mechanisms leading to bacterial superinfection include increased binding and invasion of bacteria, increased viral replication, and modification of the host inflammatory response. Some S aureus strains produce a protease that directly activates influenza virus hemagglutinin; other bacteria can activate plasminogen to promote influenza replication. The resulting increase in proteases in host tissues promotes activation of influenza through cleavage of hemagglutinin.26
The influenza virus also causes damage to the airway epithelial layer, leading to increased exposure of the binding sites necessary for adherence of S pneumoniae.27
CLINICAL PRESENTATION OF COMMUNITY-ACQUIRED PNEUMONIA
Although CAP is common, agreement on its essential clinical signs and symptoms is surprisingly limited, due in part to heterogeneous patient presentations and in part to interobserver variability. The reader is referred to two excellent reviews on this topic.28,29
The diagnosis of CAP is made on clinical grounds, based on a combination of signs and symptoms. Symptoms of pneumonia can include cough, fever, chills, sputum production, dyspnea, and pleuritic pain. Physical findings can include tachypnea, tachycardia, hypoxemia, and consolidation or rales on auscultation. Laboratory data may show leukocytosis or elevated C-reactive protein, and radiographic studies may show evidence of a new infiltrate.21,30,31
Clinical presentation of influenza
Seasonal influenza as a cause of CAP is difficult to distinguish from bacterial causes. The clinical presentation of seasonal influenza most commonly includes fever or subjective feverishness, cough, myalgia, and weakness.32 In a recent multivariate analysis, five clinical features were shown to be predictive of pandemic H1N1 influenza pneumonia rather than CABP: age younger than 65 years, absence of confusion, white blood cell count less than 12 × 109/L, temperature higher than 38°C (100.4°F), and bilateral opacities on radiography.32,33
Complicated influenza infection can be either primary viral pneumonia or bacterial superinfection.
During the 1918 influenza pandemic, which predated the ability to isolate viruses, two clinical syndromes emerged: an ARDS associated with the rapid onset of cyanosis, delirium, and frothy blood-tinged sputum; and an acute bronchopneumonia characterized by necrosis, hemorrhage, edema, and vasculitis.34,35 The first syndrome has subsequently been shown to be associated with primary viral pneumonia, while the second is caused by bacterial superinfection. Modern reexamination of 1918 data has shown that bacterial superinfection was likely the reason for the distinctly fulminant presentation of that pandemic.36,37
The 2009 H1N1 influenza pandemic caused relatively mild disease in most patients. However, those with severe pneumonia more commonly developed ARDS from primary influenza pneumonia than from bacterial superinfection.17
A third influenza-associated infection is secondary bacterial pneumonia, which follows influenza infection and mimics the presentation of CABP. A typical patient presents with a recent history of influenza-like illness, followed 4 to 14 days later by a recurrence of fever, dyspnea, productive cough, and consolidation on chest radiographs.38 Leukocytosis with an increased number of immature neutrophil forms, prolonged duration of fever, and elevated erythrocyte sedimentation rate are more likely in patients with secondary bacterial pneumonia.39 Isolates from sputum samples commonly include S pneumoniae, S aureus, H influenzae, and other gram-negative rods.40
In recent flu seasons, methicillin-resistant S aureus (MRSA) has emerged as a cause of severe secondary pneumonia. Most of these isolates carry genes for the toxin Panton-Valentine leukocidin; the associated mortality rate is as high as 33%.41,42 Although community-acquired MRSA pneumonia has only been reported in case series, distinct clinical features that have been described include severe pneumonia with high fever, hypotension, shock, respiratory failure, leukopenia, and multilobar and cavitary infiltrates.43
WHEN TO SUSPECT INFLUENZA
The triad of fever, cough, and abrupt onset are the best predictors of influenza, but no single combination of signs and symptoms predict influenza infection with 100% certainty. Therefore, an understanding of local epidemiologic data regarding circulating influenza is essential to maintain a high index of suspicion.44
It is appropriate to suspect influenza in:
- Anyone who is epidemiologically linked to a known outbreak of influenza
- Children, adults, and health care workers who have fever and abrupt onset of respiratory symptoms
- Patients with fever plus exacerbation of underlying pulmonary disease
- Severely ill patients with fever or hypothermia, especially during influenza season.45
DIAGNOSTIC TESTING
Once the diagnosis of pulmonary infection is suggested by clinical features, the initial evaluation should include measurement of vital signs, physical examination, and radiographic imaging of the chest. Additional diagnostic measures to consider include viral testing, blood culture, sputum culture, urinary antigen testing for Legionella and for S pneumoniae, fungal culture, and mycobacterial smear and culture.
Chest radiography (with posterior-anterior and lateral films) is the study that usually demonstrates the presence of a pulmonary infiltrate. If initial chest radiographs do not show an infiltrate, imaging can be repeated after treatment is started if the patient’s clinical presentation still suggests pneumonia. Chest radiographs are of limited value in predicting the pathogen, but they help to determine the extent of pneumonia and to detect parapneumonic effusion.46
A caveat: anterior-posterior, posterior-anterior, and lateral views can miss more than 10% of effusions large enough to warrant thoracentesis, especially when there is lower-lobe consolidation.47
Blood cultures are recommended for patients admitted to the intensive care unit and for those with cavitary infiltrates, leukopenia, alcohol abuse, severe liver disease, asplenia, positive pneumococcal urinary antigen testing, or a pleural effusion.21 However, blood cultures are positive in only 3% to 14% of hospitalized patients with CABP, and the impact of a positive blood culture on management decisions in CABP appears to be quite small.48–50
For the highest yield, blood culture results should be obtained before antibiotics are given. Not only is this good practice, but obtaining blood culture results before starting antibiotics is one of the quality measures evaluated by the Center for Medicare and Medicaid Services.51
Sputum culture is considered optional for outpatients and patients with less-severe pneumonia.21 While it can provide a rapid diagnosis in certain cases, a good-quality sputum sample is obtained in only 39% to 54% of patients with CABP, yields a predominant morphotype in only 45% of cases, and provides a useful microbiologic diagnosis in only 14.4%.52,53 Fungal and mycobacterial cultures are only indicated in certain situations such as cavitary infiltrates or immunosuppression.
Urinary antigen testing for Legionella and S pneumoniae should be done in patients with more severe illness and in those for whom outpatient therapy has failed.21S pneumoniae testing has been shown to allow early diagnosis of pneumococcal pneumonia in 26% more patients than with Gram staining, but it fails to identify 22% of the rapid diagnoses initially identified by Gram staining.54 Thus, a sequential approach is reasonable, with urinary antigen testing for patients at high risk without useful results from sputum Gram staining. Also, recent data suggest that the pneumococcal urinary antigen test may allow optimization of antimicrobial therapy with good clinical outcomes.55
Endotracheal tests. If the patient is intubated, collection of endotracheal aspirates, bronchoscopy, or nonbronchoscopic bronchial lavage (sometimes called “mini-BAL”) should be performed.
Thoracentesis and pleural fluid cultures should be done if a pleural effusion is found. Empyema, large or loculated effusions, and parapneumonic effusions with a pH lower than 7.20, glucose levels less than 3.4 mmol/L (60 mg/dL), or positive results on microbial staining or culture should be drained by chest tube or surgically.56
Testing for influenza should be done if it will change the clinical management, such as the choice of antibiotic or infection control practices. Specimens should be obtained with either a nasopharyngeal swab or aspirate and tested with reverse transcriptase polymerase chain reaction, immunofluorescent staining, or rapid antigen detection, depending on local availability.45
Inflammatory biomarkers such as C-reactive protein and procalcitonin have been receiving interest as ways to predict the etiology and prognosis of CAP and to guide therapy. Several studies have shown that C-reactive protein can help distinguish between CAP and bronchitis, with higher values suggesting more severe pneumonia and pneumonia caused by S pneumoniae or L pneumophila.57 Procalcitonin may help discriminate between severe lower respiratory tract infections of bacterial and 2009 H1N1 origin, although less effectively than C-reactive protein. Low procalcitonin values, particularly when combined with low C-reactive protein levels, suggest that bacterial infection is unlikely.58
RISK STRATIFICATION AND SITE-OF-CARE DECISIONS
Following a presumptive diagnosis of CAP, it is important to decide not only what treatment the patient will receive but whether he or she should be hospitalized. If the patient is to be admitted to the hospital, the clinician must also decide if his or her condition warrants intensive care.
Severity-of-illness scores
A recent meta-analysis compared the performance characteristics of the PSI and CURB-65 scores for predicting mortality in CAP and found no significant differences in overall test performance.61
Another meta-analysis found that the PSI was more sensitive than the CURB-65 and had a low false-negative rate, and so was better at showing which patients do not need to be hospitalized. Conversely, the CURB-65 was more specific and had a higher positive predictive value, and thus was more likely to correctly classify high-risk patients.62
Other scoring systems that aid in deciding about hospital admission and level of care include the CRB-6563 (which can be used instead of the CURB-65 if laboratory values are not available), SMART-COP,64 and SCAP.,65
Guidelines on when to admit to the intensive care unit
Guidelines from the Infectious Diseases Society of America (IDSA) and the American Thoracic Society (ATS) also provide guidance on when intensive care admission is advised,21 and their criteria were recently validated.66 The guidelines advocate direct admission to the intensive care unit for patients requiring vasopressors or mechanical ventilation, and intensive care unit or high-level monitoring for patients with three of the following criteria for severe CAP21:
- Respiratory rate ≥ 30
- Pao2/Fio2 ratio ≤ 250
- Multilobar infiltrates
- Confusion or disorientation
- Uremia (blood urea nitrogen ≥ 20 mg/dL)
- Leukopenia (white blood cell count < 4.0 × 109/L)
- Thrombocytopenia (platelet count < 100 × 109/L)
- Hypothermia (core temperature < 36.0°C [96.8°F])
- Hypotension requiring aggressive fluid resuscitation.
None of these scoring systems or criteria is meant to replace clinical judgment. A recent study has suggested that an oxygen saturation of less than 92% is an appropriate threshold for hospital admission, in view of higher rates of adverse events in outpatients with saturations below this value.67
TREATMENT
Multiple studies have shown that treatment of CAP in accordance with guidelines has led to improved clinical outcomes.21,68–70
How fast must antibiotics be started?
Based on studies that showed a lower mortality rate when antibiotics were started sooner, Medicare and Medicaid adopted a quality measure calling for starting antibiotics within 4 hours in patients being admitted to the hospital.50,71 However, several subsequent studies showed that the diagnosis of pneumonia is often incorrect and that rapid administration of antibiotics could lead to misdiagnosis, overuse of antibiotics, and a higher risk of Clostridium difficile infection.72,73
The current IDSA/ATS guidelines21 recommend that the first antibiotic dose be given while the patient is still in the emergency department, but do not give a specific time within which it should be given. Medicare and Medicaid later updated their quality measure to antibiotic administration within 6 hours.
Which antibiotics should be used?
The selection of antimicrobial agent depends upon the patient’s severity of illness and comorbid conditions.
Although most studies of combination antibiotic therapy have been retrospective and observational, they suggest that a macrolide (ie, one of the “mycins”) added to a beta-lactam antibiotic is beneficial, possibly by covering atypical organisms or via anti-inflammatory action.74–76 The choice of one antibiotic over another appears to be less important, and a recent Cochrane review concluded that there was no significant difference in efficacy among five antibiotic pairs studied.77
Empiric outpatient treatment of a previously healthy patient with CAP and no risk factors for drug-resistant S pneumoniae should include either a macrolide (azithromycin [Zithromax], clarithromycin [Biaxin], or erythromycin) or doxycycline. If the patient has a chronic comorbid condition such as heart, lung, liver, or renal disease, diabetes mellitus, alcoholism, malignancy, asplenia, or immunosuppression or has received antimicrobials within the preceding 3 months, then treatment should include either a respiratory fluoroquinolone (moxifloxacin [Avelox] or levofloxacin [Levaquin]) or a beta-lactam plus a macrolide.21
Overall, published data suggest that the survival rate is about the same with fluoroquinolone monotherapy as with beta-lactam plus macrolide combination therapy, and better than with beta-lactam monotherapy.78
Selection of antibiotics for inpatient treatment of CAP is influenced by severity of illness. Inpatients who do not require intensive care should be treated with either a respiratory fluoroquinolone or combination therapy with a beta-lactam (cefotaxime [Claforan], ceftriaxone [Rocephin], ampicillin, or ertapenem [Invanz]) plus a macrolide or doxycycline.21,76,79
If a specific microbiologic diagnosis is made, then treatment can be narrowed. However in certain cases, such as invasive pneumococcal infection, combination therapy may still be superior.80,81 For patients who need intensive care, treatment should always include a beta-lactam plus either azithromycin or a respiratory fluoroquinolone.21 In certain situations, additional antibiotics may be added as well, such as agents to treat Pseudomonas, community-acquired MRSA, or both.
Switching to oral therapy; short-course therapy
In the interest of avoiding unnecessary antibiotics, numerous studies have addressed the issue of an “early switch” to oral antibiotics and “short-course” therapy for CAP. In general, once clinically stable, patients with CAP, including bacteremic S pneumoniae pneumonia, can be safely switched to oral antibiotics.82
The issue of short-course therapy is more complicated, and the appropriate length of therapy for CAP is not well established. However, 5 days of levofloxacin 750 mg was shown to be as successful as 7 to 10 days of levofloxacin 500 mg.83 In another study, in patients who improved after 3 days of intravenous therapy for CAP, there was no difference in clinical outcome between those who were changed to oral therapy for 5 more days and those who received an oral placebo.84
Most patients who achieve clinical stability in the first week do not need prolonged antibiotic therapy. However, certain conditions, such as S aureus bacteremic pneumonia, complicated pneumonia, and pneumonia due to unusual organisms, may require prolonged treatment.
Other therapies
Additional therapies studied in patients with pneumonia include early mobilization, adjunctive corticosteroids, and statin drugs.
Early mobilization was shown in one study to decrease hospital length of stay without increasing adverse effects.85
Corticosteroids are not supported as a standard of care for patients with severe CAP according to current available studies.86,87 Furthermore, a randomized, controlled trial showed that prednisolone daily for a week did not improve outcomes in hospitalized patients with CAP, and it was associated with increased late failure.88
Statin trials under way. Several observational studies have suggested that statins might be beneficial in managing sepsis through their effects on endothelial cell function, antioxidant effects, anti-inflammatory effects, and immunomodulatory effects.89 However, a recent large prospective multicenter cohort study of hospitalized patients with CAP did not find evidence of a protective effect of statins on clinically meaningful outcomes in CAP or significant differences in circulating biomarkers.90 Several randomized trials of statin therapy in patients with both ventilator-associated pneumonia and CAP are under way.
INFLUENZA TREATMENT: MOST EFFECTIVE WITHIN 48 HOURS
Treatment with antiviral drugs is most effective if started within 48 hours after symptom onset, although some patients with confirmed influenza who are either not improving or who are critically ill may still benefit from treatment started later.
Treatment should be considered in patients with laboratory-confirmed or suspected influenza who are at risk of developing complicated influenza and in otherwise healthy patients who wish to reduce the duration of illness or who have close contact with patients who are at high risk of complications.
Antiviral medications are oseltamivir (Tamiflu), zanamivir (Relenza), and the adamantines amantadine (Symmetrel) and rimantadine (Flumadine).
Due to evolving viral resistance patterns, the choice of antiviral drug depends on the strain. Seasonal H1N1 is best treated with zanamivir or an adamantine, while pandemic 2009 H1N1 and H3N2 are best treated with zanamivir or oseltamivir. When strain typing is not available, empiric therapy should be with either zanamivir monotherapy or a combination of oseltamivir plus rimantadine. Influenza B viruses are resistant to adamantines and should be treated only with either zanamivir or oseltamivir.45
FOLLOW-UP AND PREVENTION
Patients with CAP can generally be expected to improve within 3 to 7 days.91 However, it may be several weeks before they return to baseline.92
Follow-up plans may be guided by the time to clinical stability. For patients who do not achieve clinical stability until more than 72 hours after admission, more aggressive follow-up on discharge is indicated, since they are more likely to experience early readmission and death.93
Pneumococcal vaccination. Because S pneumoniae remains the most common cause of CAP, efforts should be made to vaccinate patients appropriately. The Advisory Committee on Immunization Practices (ACIP) and the US Centers for Disease Control and Prevention recommend that the pneumococcal polysaccharide vaccine (Pneumovax 23; PPSV23) be given to those over age 65. Those who were vaccinated before age 65 should receive another dose at age 65 or later if at least 5 years have passed since their previous dose. Those who receive it at or after age 65 should receive only a single dose. A second dose is recommended 5 years after the first dose for people age 19 to 64 years with functional or anatomic asplenia and for those who are immunocompromised.
Influenza vaccination for all. Of note, the ACIP updated its guidelines on influenza vaccination beginning with the 2010–2011 influenza season. It no longer advocates a risk-stratified approach. Instead, it recommends universal influenza vaccination for everybody more than 6 months old.94
Smoking cessation should be addressed. Smoking cessation is a Medicare and Medicaid quality measure and should be encouraged after an episode of CAP because quitting smoking reduces the risk of pneumococcal disease by approximately 14% each year thereafter.95
General internists need to be able to recognize community-acquired pneumonia (CAP) so that diagnostic and therapeutic interventions can be initiated promptly. It is also important to understand the most likely and possible causes of CAP so that appropriate initial antimicrobial therapy can be chosen. Especially during flu season, influenza can present as CAP and should be included in the differential diagnosis.
When managing a patient with CAP, the internist must decide which level of care, diagnostic tests, antimicrobial agents, and follow-up plans are needed. These topics will be reviewed in this article.
TWO TERMS TO REMEMBER
- CAP refers to pneumonia acquired outside a health care facility. It can be either bacterial or viral.
- CABP (community-acquired bacterial pneumonia) refers only to those cases caused by bacterial pathogens.
NUMBERS AND TRENDS
In the United States, CAP is the number-one cause of death from infection and the sixth leading cause of death overall.1 Each year, it is responsible for about 4.2 million outpatient visits, more than 60,000 deaths, and more than $17 billion in health care expenses.2
Community-acquired bacterial pneumonia: Common, serious
In a population-based US study in 1991, the incidence of CABP requiring hospitalization was 266.8 per 100,000 people.3
Estimates of overall mortality in CABP vary depending on the severity of illness and comorbid conditions. A meta-analysis published in 1996 found the overall mortality rate to be 13.7%, with a range of 5.1% to 36.5% depending on severity.4
In hospitalized patients, mortality rates and length of hospital stay appear to be declining over time. Between 1993 and 2005, the age-adjusted mortality rate decreased from 8.9% to 4.1%, and the average length of stay decreased from 7.5 to 5.7 days, with an overall reduction in hospital cost.5
CABP is more prevalent in older people than in the general population, and it increases with age from 18.2 cases per 1,000 patient-years in patients 60 to 69 years to 52.3 cases per 1,000 patient-years in those older than 85 years.6 Risk factors for pneumonia in the elderly include heart disease, chronic lung disease, immunosuppressive drugs, alcoholism, and increasing age.7 Similar to the trend in the general population, the mortality rate in elderly CABP patients appears to be decreasing over time, possibly thanks to rising rates of pneumococcal and influenza vaccination.8
Among the general population, risk factors for developing CABP also include smoking, occupational dust exposure, history of childhood pneumonia, unemployment, and single marital status.9 The incidence of CABP does not appear to be higher among pregnant women, although it is the most frequent cause of nonobstetric death in this population.10
The use of proton pump inhibitors may be an emerging risk factor for CABP.11 Also, use of nonsteroidal anti-inflammatory drugs among patients with CABP is associated with a blunted inflammatory response as well as a higher risk of pleuropulmonary complications and a delay in presentation.12
Influenza is also common, potentially severe
Influenza is also very common and potentially severe. It can cause a spectrum of disease, from mild upper respiratory tract symptoms to severe viral pneumonia that can be life-threatening and complicated by respiratory failure and the acute respiratory distress syndrome (ARDS).
Influenza infection can also be complicated by subsequent bacterial pneumonia. However, the epidemiology of influenza infection differs from that of CABP in that influenza occurs seasonally.
In the United States, seasonal influenza causes 36,000 deaths and 200,000 hospitalizations annually.13,14 As with CABP, the risk of death from influenza increases with age: it is 16 times greater in people age 85 and older than in those ages 65 to 69.13
During yearly seasonal epidemics, those at the highest risk of hospitalization and death are at the extremes of age. Risk factors for complicated influenza include heart disease, lung disease, diabetes, renal failure, rheumatologic conditions, dementia, and neurologic disease.15,16 During the 2009 H1N1 influenza pandemic, unexpected severity was seen in previously healthy young adults as well as those with obesity, neurodegenerative disease, pregnancy, and asthma.17
PATHOGENS: TYPICAL, ATYPICAL, VIRAL
Identifying the etiologic organism in CAP is confounded by limitations in the available diagnostic tests and also by poor-quality specimens that often are contaminated with bacteria that colonize the upper airways. Given these caveats, the primary pathogens responsible for CAP broadly include typical bacterial pathogens, atypical bacterial pathogens, and viruses.
Typical bacterial pathogens include Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, Moraxella catarrhalis, and, less commonly, a variety of aerobic and anaerobic gram-negative rods including Pseudomonas aeruginosa, Acinetobacter species, and Klebsiella pneumoniae.
Atypical bacterial pathogens include Mycoplasma pneumoniae, Chlamydia pneumoniae, and Legionella species.18
Viruses implicated in adult CAP include influenza A and B, parainfluenza viruses, respiratory syncytial virus, and adenovirus.19 More recently, human metapneumovirus has been described as a cause of adult CAP.20
Clues to uncommon microbes
Specific historic features or coexisting conditions that may suggest an uncommon microbiologic diagnosis include21:
- Recent travel to the southwestern United States or Southeast Asia
- Ill contacts
- Exposure to birds, bats, rabbits, or farm animals
- Alcoholism
- Chronic obstructive pulmonary disease
- Human immunodeficiency virus infection
- Structural lung disease
- Prolonged cough with whoop or posttussive vomiting
- Aspiration
- Bioterrorism.
In cases in which one or more of these conditions exist, CAP may also be caused by other agents not listed above, including Mycobacterium tuberculosis, oral anaerobes, atypical mycobacteria, Histoplasma capsulatum, Chlamydophila psittaci, Francisella tularensis, Coxiella burnettii, Pneumocystis jiroveci, Cryptococcus, Aspergillus, Coccidioides, Hantavirus, avian influenza, Burkholderia pseudomallei, severe acute respiratory syndrome virus, Bordetella pertussis, Bacillus anthracis, and Yersinia pestis.
HOW BACTERIA INVADE THE LUNGS
The pathophysiology of CABP involves both host defense and microbial virulence factors.
The airways are most commonly exposed to microbes by microaspiration of upper airway flora, although hematogenous seeding of the lungs in a bacteremic patient or contiguous spread of infection from an adjacent site can also occur.
Mucociliary clearance and the cough reflex are important initial defenses against infection and can be inhibited by neurologic diseases and conditions that impair the mucociliary mechanism. Mucosal immune cells, including macrophages and neutrophils, recognize invading pathogens and generate an antibody response.
Regulation of the host inflammatory response to infection depends on a complex interaction between immune cells, inflammatory cytokines (eg, tumor necrosis factor alpha, interleukin 1-beta, and interleukin 6), and anti-inflammatory cytokines such as interleukin 1 receptor antagonist and soluble tumor necrosis factor receptor type I.22
The interaction and timing of the inflammatory and anti-inflammatory response are essential in manifesting an appropriate host response to infection. An inadequate inflammatory response can lead to sepsis and death, but an excessive, late anti-inflammatory response can lead to a systemic inflammatory response such as ARDS. Polymorphisms within the genes coding for these factors may explain the variation in severity of illness among patients with CABP.23
HOW INFLUENZA DOES ITS DAMAGE
There are three types of influenza virus: A, B, and C. Type A causes most human infections. The influenza A virus envelope comprises a lipid bilayer that contains the projecting glycoproteins hemagglutinin and neuraminidase. Influenza viruses are named on the basis of these proteins and are designated with an H and an N, respectively, each followed by a number referring to the subtype.
Influenza infection begins when the virus makes contact with the epithelium. Hemagglutinin binds to the host cell and allows viral entry, where it begins replication. Neuraminidase prevents viral aggregation and facilitates the release of virus from infected cells.24
Mature virions are released and spread to neighboring host cells; this process is associated with desquamation and inflammation of the airways, causing cough, rhinorrhea, and sore throat. Systemic symptoms are associated with the induction of interferon, which causes fever and myalgia.25
Recovery and immunity to influenza infection occurs through both humoral and cell-mediated immunity, with antibodies directed against the specific hemagglutinin and neuraminidase antigens of the infecting virus. Immunity wanes over time and with antigenic drift of circulating viruses, making the host susceptible to recurrent influenza infection.24
Influenza is often complicated by bacterial superinfection
The influenza virus acts synergistically with certain bacteria to increase infectivity, and this may explain why influenza is often complicated by bacterial superinfection.
Mechanisms leading to bacterial superinfection include increased binding and invasion of bacteria, increased viral replication, and modification of the host inflammatory response. Some S aureus strains produce a protease that directly activates influenza virus hemagglutinin; other bacteria can activate plasminogen to promote influenza replication. The resulting increase in proteases in host tissues promotes activation of influenza through cleavage of hemagglutinin.26
The influenza virus also causes damage to the airway epithelial layer, leading to increased exposure of the binding sites necessary for adherence of S pneumoniae.27
CLINICAL PRESENTATION OF COMMUNITY-ACQUIRED PNEUMONIA
Although CAP is common, agreement on its essential clinical signs and symptoms is surprisingly limited, due in part to heterogeneous patient presentations and in part to interobserver variability. The reader is referred to two excellent reviews on this topic.28,29
The diagnosis of CAP is made on clinical grounds, based on a combination of signs and symptoms. Symptoms of pneumonia can include cough, fever, chills, sputum production, dyspnea, and pleuritic pain. Physical findings can include tachypnea, tachycardia, hypoxemia, and consolidation or rales on auscultation. Laboratory data may show leukocytosis or elevated C-reactive protein, and radiographic studies may show evidence of a new infiltrate.21,30,31
Clinical presentation of influenza
Seasonal influenza as a cause of CAP is difficult to distinguish from bacterial causes. The clinical presentation of seasonal influenza most commonly includes fever or subjective feverishness, cough, myalgia, and weakness.32 In a recent multivariate analysis, five clinical features were shown to be predictive of pandemic H1N1 influenza pneumonia rather than CABP: age younger than 65 years, absence of confusion, white blood cell count less than 12 × 109/L, temperature higher than 38°C (100.4°F), and bilateral opacities on radiography.32,33
Complicated influenza infection can be either primary viral pneumonia or bacterial superinfection.
During the 1918 influenza pandemic, which predated the ability to isolate viruses, two clinical syndromes emerged: an ARDS associated with the rapid onset of cyanosis, delirium, and frothy blood-tinged sputum; and an acute bronchopneumonia characterized by necrosis, hemorrhage, edema, and vasculitis.34,35 The first syndrome has subsequently been shown to be associated with primary viral pneumonia, while the second is caused by bacterial superinfection. Modern reexamination of 1918 data has shown that bacterial superinfection was likely the reason for the distinctly fulminant presentation of that pandemic.36,37
The 2009 H1N1 influenza pandemic caused relatively mild disease in most patients. However, those with severe pneumonia more commonly developed ARDS from primary influenza pneumonia than from bacterial superinfection.17
A third influenza-associated infection is secondary bacterial pneumonia, which follows influenza infection and mimics the presentation of CABP. A typical patient presents with a recent history of influenza-like illness, followed 4 to 14 days later by a recurrence of fever, dyspnea, productive cough, and consolidation on chest radiographs.38 Leukocytosis with an increased number of immature neutrophil forms, prolonged duration of fever, and elevated erythrocyte sedimentation rate are more likely in patients with secondary bacterial pneumonia.39 Isolates from sputum samples commonly include S pneumoniae, S aureus, H influenzae, and other gram-negative rods.40
In recent flu seasons, methicillin-resistant S aureus (MRSA) has emerged as a cause of severe secondary pneumonia. Most of these isolates carry genes for the toxin Panton-Valentine leukocidin; the associated mortality rate is as high as 33%.41,42 Although community-acquired MRSA pneumonia has only been reported in case series, distinct clinical features that have been described include severe pneumonia with high fever, hypotension, shock, respiratory failure, leukopenia, and multilobar and cavitary infiltrates.43
WHEN TO SUSPECT INFLUENZA
The triad of fever, cough, and abrupt onset are the best predictors of influenza, but no single combination of signs and symptoms predict influenza infection with 100% certainty. Therefore, an understanding of local epidemiologic data regarding circulating influenza is essential to maintain a high index of suspicion.44
It is appropriate to suspect influenza in:
- Anyone who is epidemiologically linked to a known outbreak of influenza
- Children, adults, and health care workers who have fever and abrupt onset of respiratory symptoms
- Patients with fever plus exacerbation of underlying pulmonary disease
- Severely ill patients with fever or hypothermia, especially during influenza season.45
DIAGNOSTIC TESTING
Once the diagnosis of pulmonary infection is suggested by clinical features, the initial evaluation should include measurement of vital signs, physical examination, and radiographic imaging of the chest. Additional diagnostic measures to consider include viral testing, blood culture, sputum culture, urinary antigen testing for Legionella and for S pneumoniae, fungal culture, and mycobacterial smear and culture.
Chest radiography (with posterior-anterior and lateral films) is the study that usually demonstrates the presence of a pulmonary infiltrate. If initial chest radiographs do not show an infiltrate, imaging can be repeated after treatment is started if the patient’s clinical presentation still suggests pneumonia. Chest radiographs are of limited value in predicting the pathogen, but they help to determine the extent of pneumonia and to detect parapneumonic effusion.46
A caveat: anterior-posterior, posterior-anterior, and lateral views can miss more than 10% of effusions large enough to warrant thoracentesis, especially when there is lower-lobe consolidation.47
Blood cultures are recommended for patients admitted to the intensive care unit and for those with cavitary infiltrates, leukopenia, alcohol abuse, severe liver disease, asplenia, positive pneumococcal urinary antigen testing, or a pleural effusion.21 However, blood cultures are positive in only 3% to 14% of hospitalized patients with CABP, and the impact of a positive blood culture on management decisions in CABP appears to be quite small.48–50
For the highest yield, blood culture results should be obtained before antibiotics are given. Not only is this good practice, but obtaining blood culture results before starting antibiotics is one of the quality measures evaluated by the Center for Medicare and Medicaid Services.51
Sputum culture is considered optional for outpatients and patients with less-severe pneumonia.21 While it can provide a rapid diagnosis in certain cases, a good-quality sputum sample is obtained in only 39% to 54% of patients with CABP, yields a predominant morphotype in only 45% of cases, and provides a useful microbiologic diagnosis in only 14.4%.52,53 Fungal and mycobacterial cultures are only indicated in certain situations such as cavitary infiltrates or immunosuppression.
Urinary antigen testing for Legionella and S pneumoniae should be done in patients with more severe illness and in those for whom outpatient therapy has failed.21S pneumoniae testing has been shown to allow early diagnosis of pneumococcal pneumonia in 26% more patients than with Gram staining, but it fails to identify 22% of the rapid diagnoses initially identified by Gram staining.54 Thus, a sequential approach is reasonable, with urinary antigen testing for patients at high risk without useful results from sputum Gram staining. Also, recent data suggest that the pneumococcal urinary antigen test may allow optimization of antimicrobial therapy with good clinical outcomes.55
Endotracheal tests. If the patient is intubated, collection of endotracheal aspirates, bronchoscopy, or nonbronchoscopic bronchial lavage (sometimes called “mini-BAL”) should be performed.
Thoracentesis and pleural fluid cultures should be done if a pleural effusion is found. Empyema, large or loculated effusions, and parapneumonic effusions with a pH lower than 7.20, glucose levels less than 3.4 mmol/L (60 mg/dL), or positive results on microbial staining or culture should be drained by chest tube or surgically.56
Testing for influenza should be done if it will change the clinical management, such as the choice of antibiotic or infection control practices. Specimens should be obtained with either a nasopharyngeal swab or aspirate and tested with reverse transcriptase polymerase chain reaction, immunofluorescent staining, or rapid antigen detection, depending on local availability.45
Inflammatory biomarkers such as C-reactive protein and procalcitonin have been receiving interest as ways to predict the etiology and prognosis of CAP and to guide therapy. Several studies have shown that C-reactive protein can help distinguish between CAP and bronchitis, with higher values suggesting more severe pneumonia and pneumonia caused by S pneumoniae or L pneumophila.57 Procalcitonin may help discriminate between severe lower respiratory tract infections of bacterial and 2009 H1N1 origin, although less effectively than C-reactive protein. Low procalcitonin values, particularly when combined with low C-reactive protein levels, suggest that bacterial infection is unlikely.58
RISK STRATIFICATION AND SITE-OF-CARE DECISIONS
Following a presumptive diagnosis of CAP, it is important to decide not only what treatment the patient will receive but whether he or she should be hospitalized. If the patient is to be admitted to the hospital, the clinician must also decide if his or her condition warrants intensive care.
Severity-of-illness scores
A recent meta-analysis compared the performance characteristics of the PSI and CURB-65 scores for predicting mortality in CAP and found no significant differences in overall test performance.61
Another meta-analysis found that the PSI was more sensitive than the CURB-65 and had a low false-negative rate, and so was better at showing which patients do not need to be hospitalized. Conversely, the CURB-65 was more specific and had a higher positive predictive value, and thus was more likely to correctly classify high-risk patients.62
Other scoring systems that aid in deciding about hospital admission and level of care include the CRB-6563 (which can be used instead of the CURB-65 if laboratory values are not available), SMART-COP,64 and SCAP.,65
Guidelines on when to admit to the intensive care unit
Guidelines from the Infectious Diseases Society of America (IDSA) and the American Thoracic Society (ATS) also provide guidance on when intensive care admission is advised,21 and their criteria were recently validated.66 The guidelines advocate direct admission to the intensive care unit for patients requiring vasopressors or mechanical ventilation, and intensive care unit or high-level monitoring for patients with three of the following criteria for severe CAP21:
- Respiratory rate ≥ 30
- Pao2/Fio2 ratio ≤ 250
- Multilobar infiltrates
- Confusion or disorientation
- Uremia (blood urea nitrogen ≥ 20 mg/dL)
- Leukopenia (white blood cell count < 4.0 × 109/L)
- Thrombocytopenia (platelet count < 100 × 109/L)
- Hypothermia (core temperature < 36.0°C [96.8°F])
- Hypotension requiring aggressive fluid resuscitation.
None of these scoring systems or criteria is meant to replace clinical judgment. A recent study has suggested that an oxygen saturation of less than 92% is an appropriate threshold for hospital admission, in view of higher rates of adverse events in outpatients with saturations below this value.67
TREATMENT
Multiple studies have shown that treatment of CAP in accordance with guidelines has led to improved clinical outcomes.21,68–70
How fast must antibiotics be started?
Based on studies that showed a lower mortality rate when antibiotics were started sooner, Medicare and Medicaid adopted a quality measure calling for starting antibiotics within 4 hours in patients being admitted to the hospital.50,71 However, several subsequent studies showed that the diagnosis of pneumonia is often incorrect and that rapid administration of antibiotics could lead to misdiagnosis, overuse of antibiotics, and a higher risk of Clostridium difficile infection.72,73
The current IDSA/ATS guidelines21 recommend that the first antibiotic dose be given while the patient is still in the emergency department, but do not give a specific time within which it should be given. Medicare and Medicaid later updated their quality measure to antibiotic administration within 6 hours.
Which antibiotics should be used?
The selection of antimicrobial agent depends upon the patient’s severity of illness and comorbid conditions.
Although most studies of combination antibiotic therapy have been retrospective and observational, they suggest that a macrolide (ie, one of the “mycins”) added to a beta-lactam antibiotic is beneficial, possibly by covering atypical organisms or via anti-inflammatory action.74–76 The choice of one antibiotic over another appears to be less important, and a recent Cochrane review concluded that there was no significant difference in efficacy among five antibiotic pairs studied.77
Empiric outpatient treatment of a previously healthy patient with CAP and no risk factors for drug-resistant S pneumoniae should include either a macrolide (azithromycin [Zithromax], clarithromycin [Biaxin], or erythromycin) or doxycycline. If the patient has a chronic comorbid condition such as heart, lung, liver, or renal disease, diabetes mellitus, alcoholism, malignancy, asplenia, or immunosuppression or has received antimicrobials within the preceding 3 months, then treatment should include either a respiratory fluoroquinolone (moxifloxacin [Avelox] or levofloxacin [Levaquin]) or a beta-lactam plus a macrolide.21
Overall, published data suggest that the survival rate is about the same with fluoroquinolone monotherapy as with beta-lactam plus macrolide combination therapy, and better than with beta-lactam monotherapy.78
Selection of antibiotics for inpatient treatment of CAP is influenced by severity of illness. Inpatients who do not require intensive care should be treated with either a respiratory fluoroquinolone or combination therapy with a beta-lactam (cefotaxime [Claforan], ceftriaxone [Rocephin], ampicillin, or ertapenem [Invanz]) plus a macrolide or doxycycline.21,76,79
If a specific microbiologic diagnosis is made, then treatment can be narrowed. However in certain cases, such as invasive pneumococcal infection, combination therapy may still be superior.80,81 For patients who need intensive care, treatment should always include a beta-lactam plus either azithromycin or a respiratory fluoroquinolone.21 In certain situations, additional antibiotics may be added as well, such as agents to treat Pseudomonas, community-acquired MRSA, or both.
Switching to oral therapy; short-course therapy
In the interest of avoiding unnecessary antibiotics, numerous studies have addressed the issue of an “early switch” to oral antibiotics and “short-course” therapy for CAP. In general, once clinically stable, patients with CAP, including bacteremic S pneumoniae pneumonia, can be safely switched to oral antibiotics.82
The issue of short-course therapy is more complicated, and the appropriate length of therapy for CAP is not well established. However, 5 days of levofloxacin 750 mg was shown to be as successful as 7 to 10 days of levofloxacin 500 mg.83 In another study, in patients who improved after 3 days of intravenous therapy for CAP, there was no difference in clinical outcome between those who were changed to oral therapy for 5 more days and those who received an oral placebo.84
Most patients who achieve clinical stability in the first week do not need prolonged antibiotic therapy. However, certain conditions, such as S aureus bacteremic pneumonia, complicated pneumonia, and pneumonia due to unusual organisms, may require prolonged treatment.
Other therapies
Additional therapies studied in patients with pneumonia include early mobilization, adjunctive corticosteroids, and statin drugs.
Early mobilization was shown in one study to decrease hospital length of stay without increasing adverse effects.85
Corticosteroids are not supported as a standard of care for patients with severe CAP according to current available studies.86,87 Furthermore, a randomized, controlled trial showed that prednisolone daily for a week did not improve outcomes in hospitalized patients with CAP, and it was associated with increased late failure.88
Statin trials under way. Several observational studies have suggested that statins might be beneficial in managing sepsis through their effects on endothelial cell function, antioxidant effects, anti-inflammatory effects, and immunomodulatory effects.89 However, a recent large prospective multicenter cohort study of hospitalized patients with CAP did not find evidence of a protective effect of statins on clinically meaningful outcomes in CAP or significant differences in circulating biomarkers.90 Several randomized trials of statin therapy in patients with both ventilator-associated pneumonia and CAP are under way.
INFLUENZA TREATMENT: MOST EFFECTIVE WITHIN 48 HOURS
Treatment with antiviral drugs is most effective if started within 48 hours after symptom onset, although some patients with confirmed influenza who are either not improving or who are critically ill may still benefit from treatment started later.
Treatment should be considered in patients with laboratory-confirmed or suspected influenza who are at risk of developing complicated influenza and in otherwise healthy patients who wish to reduce the duration of illness or who have close contact with patients who are at high risk of complications.
Antiviral medications are oseltamivir (Tamiflu), zanamivir (Relenza), and the adamantines amantadine (Symmetrel) and rimantadine (Flumadine).
Due to evolving viral resistance patterns, the choice of antiviral drug depends on the strain. Seasonal H1N1 is best treated with zanamivir or an adamantine, while pandemic 2009 H1N1 and H3N2 are best treated with zanamivir or oseltamivir. When strain typing is not available, empiric therapy should be with either zanamivir monotherapy or a combination of oseltamivir plus rimantadine. Influenza B viruses are resistant to adamantines and should be treated only with either zanamivir or oseltamivir.45
FOLLOW-UP AND PREVENTION
Patients with CAP can generally be expected to improve within 3 to 7 days.91 However, it may be several weeks before they return to baseline.92
Follow-up plans may be guided by the time to clinical stability. For patients who do not achieve clinical stability until more than 72 hours after admission, more aggressive follow-up on discharge is indicated, since they are more likely to experience early readmission and death.93
Pneumococcal vaccination. Because S pneumoniae remains the most common cause of CAP, efforts should be made to vaccinate patients appropriately. The Advisory Committee on Immunization Practices (ACIP) and the US Centers for Disease Control and Prevention recommend that the pneumococcal polysaccharide vaccine (Pneumovax 23; PPSV23) be given to those over age 65. Those who were vaccinated before age 65 should receive another dose at age 65 or later if at least 5 years have passed since their previous dose. Those who receive it at or after age 65 should receive only a single dose. A second dose is recommended 5 years after the first dose for people age 19 to 64 years with functional or anatomic asplenia and for those who are immunocompromised.
Influenza vaccination for all. Of note, the ACIP updated its guidelines on influenza vaccination beginning with the 2010–2011 influenza season. It no longer advocates a risk-stratified approach. Instead, it recommends universal influenza vaccination for everybody more than 6 months old.94
Smoking cessation should be addressed. Smoking cessation is a Medicare and Medicaid quality measure and should be encouraged after an episode of CAP because quitting smoking reduces the risk of pneumococcal disease by approximately 14% each year thereafter.95
- Mortensen EM, Kapoor WN, Chang CC, Fine MJ. Assessment of mortality after long-term follow-up of patients with community-acquired pneumonia. Clin Infect Dis 2003; 37:1617–1624.
- File TM, Marrie TJ. Burden of community-acquired pneumonia in North American adults. Postgrad Med 2010; 122:130–141.
- Marston BJ, Plouffe JF, File TM, et al. Incidence of community-acquired pneumonia requiring hospitalization. Results of a population-based active surveillance study in Ohio. The Community-Based Pneumonia Incidence Study Group. Arch Intern Med 1997; 157:1709–1718.
- Fine MJ, Smith MA, Carson CA, et al. Prognosis and outcomes of patients with community-acquired pneumonia. JAMA 1996; 275:134–141.
- Ruhnke GW, Coca-Perraillon M, Kitch BT, Cutler DM. Trends in mortality and medical spending in patients hospitalized for community-acquired pneumonia: 1993–2005. Med Care 2010; 48:1111–1116.
- Jackson ML, Neuzil KM, Thompson WW, et al. The burden of community-acquired pneumonia in seniors: results of a population-based study. Clin Infect Dis 2004; 39:1642–1650.
- Koivula I, Sten M, Mäkelä PH. Risk factors for pneumonia in the elderly. Am J Med 1994; 96:313–320.
- Ruhnke GW, Coca-Perraillon M, Kitch BT, Cutler DM. Marked reduction in 30-day mortality among elderly patients with community-acquired pneumonia. Am J Med 2011; 124:171–178.
- Farr BM, Bartlett CL, Wadsworth J, Miller DL. Risk factors for community-acquired pneumonia diagnosed upon hospital admission. British Thoracic Society Pneumonia Study Group. Respir Med 2000; 94:954–963.
- Graves CR. Pneumonia in pregnancy. Clin Obstet Gynecol 2010; 53:329–336.
- Eom CS, Jeon CY, Lim JW, Cho EG, Park SM, Lee KS. Use of acid-suppressive drugs and risk of pneumonia: a systematic review and meta-analysis. CMAJ 2011; 183:310–319.
- Voiriot G, Dury S, Parrot A, Mayaud C, Fartoukh M. Nonsteroidal antiinflammatory drugs may affect the presentation and course of community-acquired pneumonia. Chest 2011; 139:387–394.
- Thompson WW, Shay DK, Weintraub E, et al. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 2003; 289:179–186.
- Thompson WW, Shay DK, Weintraub E, et al. Influenza-associated hospitalizations in the United States. JAMA 2004; 292:1333–1340.
- Glezen WP, Decker M, Perrotta DM. Survey of underlying conditions of persons hospitalized with acute respiratory disease during influenza epidemics in Houston, 1978–1981. Am Rev Respir Dis 1987; 136:550–555.
- Izurieta HS, Thompson WW, Kramarz P, et al. Influenza and the rates of hospitalization for respiratory disease among infants and young children. N Engl J Med 2000; 342:232–239.
- Rothberg MB, Haessler SD. Complications of seasonal and pandemic influenza. Crit Care Med 2010; 38(suppl 4):e91–e97.
- Apisarnthanarak A, Mundy LM. Etiology of community-acquired pneumonia. Clin Chest Med 2005; 26:47–55.
- de Roux A, Marcos MA, Garcia E, et al. Viral community-acquired pneumonia in nonimmunocompromised adults. Chest 2004; 125:1343–1351.
- Hamelin ME, Côté S, Laforge J, et al. Human metapneumovirus infection in adults with community-acquired pneumonia and exacerbation of chronic obstructive pulmonary disease. Clin Infect Dis 2005; 41:498–502.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44(suppl 2):S27–S72.
- Kolling UK, Hansen F, Braun J, Rink L, Katus HA, Dalhoff K. Leucocyte response and anti-inflammatory cytokines in community acquired pneumonia. Thorax 2001; 56:121–125.
- Wunderink RG, Waterer GW. Community-acquired pneumonia: pathophysiology and host factors with focus on possible new approaches to management of lower respiratory tract infections. Infect Dis Clin North Am 2004; 18:743–759.
- Hilleman MR. Realities and enigmas of human viral influenza: pathogenesis, epidemiology and control. Vaccine 2002; 20:3068–3087.
- Bender BS, Small PA. Influenza: pathogenesis and host defense. Semin Respir Infect 1992; 7:38–45.
- Scheiblauer H, Reinacher M, Tashiro M, Rott R. Interactions between bacteria and influenza A virus in the development of influenza pneumonia. J Infect Dis 1992; 166:783–791.
- McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev 2006; 19:571–582.
- Metlay JP, Kapoor WN, Fine MJ. Does this patient have community-acquired pneumonia? Diagnosing pneumonia by history and physical examination. JAMA 1997; 278:1440–1445.
- Benbassat J, Baumal R. Narrative review: should teaching of the respiratory physical examination be restricted only to signs with proven reliability and validity? J Gen Intern Med 2010; 25:865–872.
- Kolsuz M, Erginel S, Alatas O, et al. Acute phase reactants and cytokine levels in unilateral community-acquired pneumonia. Respiration 2003; 70:615–622.
- Alves DW, Kennedy MT. Community-acquired pneumonia in casualty: etiology, clinical features, diagnosis, and management (or a look at the “new” in pneumonia since 2002). Curr Opin Pulm Med 2004; 10:166–170.
- Monto AS, Gravenstein S, Elliott M, Colopy M, Schweinle J. Clinical signs and symptoms predicting influenza infection. Arch Intern Med 2000; 160:3243–3247.
- Bewick T, Myles P, Greenwood S, et al; Influenza Clinical Information Network. Clinical and laboratory features distinguishing pandemic H1N1 influenza-related pneumonia from interpandemic community-acquired pneumonia in adults. Thorax 2011; 66:247–252.
- Morens DM, Fauci AS. The 1918 influenza pandemic: insights for the 21st century. J Infect Dis 2007; 195:1018–1028.
- Starr I. Influenza in 1918: recollections of the epidemic in Philadelphia. 1976. Ann Intern Med 2006; 145:138–140.
- Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis 2008; 198:962–970.
- Brundage JF, Shanks GD. Deaths from bacterial pneumonia during 1918–19 influenza pandemic. Emerg Infect Dis 2008; 14:1193–1199.
- Treanor J. Influenza virus. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, PA: Elsevier/Churchill Livingstone; 2005:2060–2085.
- Jarstrand C, Tunevall G. The influence of bacterial superinfection on the clinical course of influenza. Studies from the influenza epidemics in Stockholm during the winters 1969–70 and 1971–72. Scand J Infect Dis 1975; 7:243–247.
- Schwarzmann SW, Adler JL, Sullivan RJ, Marine WM. Bacterial pneumonia during the Hong Kong influenza epidemic of 1968–1969. Arch Intern Med 1971; 127:1037–1041.
- Hageman JC, Uyeki TM, Francis JS, et al. Severe community-acquired pneumonia due to Staphylococcus aureus, 2003–04 influenza season. Emerg Infect Dis 2006; 12:894–899.
- Centers for Disease Control and Prevention (CDC). Severe methicillin-resistant Staphylococcus aureus community-acquired pneumonia associated with influenza—Louisiana and Georgia, December 2006–January 2007. MMWR Morb Mortal Wkly Rep 2007; 56:325–329.
- Hidron AI, Low CE, Honig EG, Blumberg HM. Emergence of community-acquired methicillin-resistant Staphylococcus aureus strain USA300 as a cause of necrotising community-onset pneumonia. Lancet Infect Dis 2009; 9:384–392.
- Call SA, Vollenweider MA, Hornung CA, Simel DL, McKinney WP. Does this patient have influenza? JAMA 2005; 293:987–997.
- Harper SA, Bradley JS, Englund JA, et al; Expert Panel of the Infectious Diseases Society of America. Seasonal influenza in adults and children—diagnosis, treatment, chemoprophylaxis, and institutional outbreak management: clinical practice guidelines of the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:1003–1032.
- Boersma WG, Daniels JM, Löwenberg A, Boeve WJ, van de Jagt EJ. Reliability of radiographic findings and the relation to etiologic agents in community-acquired pneumonia. Respir Med 2006; 100:926–932.
- Brixey AG, Luo Y, Skouras V, Awdankiewicz A, Light RW. The efficacy of chest radiographs in detecting parapneumonic effusions. Respirology 2011; 16:1000–1004.
- Campbell SG, Marrie TJ, Anstey R, Dickinson G, Ackroyd-Stolarz S. The contribution of blood cultures to the clinical management of adult patients admitted to the hospital with community-acquired pneumonia: a prospective observational study. Chest 2003; 123:1142–1150.
- Waterer GW, Wunderink RG. The influence of the severity of community-acquired pneumonia on the usefulness of blood cultures. Respir Med 2001; 95:78–82.
- Houck PM, Bratzler DW, Nsa W, Ma A, Bartlett JG. Timing of antibiotic administration and outcomes for Medicare patients hospitalized with community-acquired pneumonia. Arch Intern Med 2004; 164:637–644.
- Information & Quality Healthcare. http://www.IQH.org/attachments/219_CoreMHelpBookletpg4_11_3.pdf. Accessed November 14, 2011.
- Rosón B, Carratalà J, Verdaguer R, Dorca J, Manresa F, Gudiol F. Prospective study of the usefulness of sputum Gram stain in the initial approach to community-acquired pneumonia requiring hospitalization. Clin Infect Dis 2000; 31:869–874.
- García-Vázquez E, Marcos MA, Mensa J, et al. Assessment of the usefulness of sputum culture for diagnosis of community-acquired pneumonia using the PORT predictive scoring system. Arch Intern Med 2004; 164:1807–1811.
- Rosón B, Fernández-Sabé N, Carratalà J, et al. Contribution of a urinary antigen assay (Binax NOW) to the early diagnosis of pneumococcal pneumonia. Clin Infect Dis 2004; 38:222–226.
- Sordé R, Falcó V, Lowak M, et al. Current and potential usefulness of pneumococcal urinary antigen detection in hospitalized patients with community-acquired pneumonia to guide antimicrobial therapy. Arch Intern Med 2011; 171:166–172.
- Koegelenberg CFN, Diacon AH, Bolliger CT. Parapneumonic pleural effusion and empyema. Respiration 2008; 75:241–250.
- Almirall J, Bolíbar I, Toran P, et al; Community-Acquired Pneumonia Maresme Study Group. Contribution of C-reactive protein to the diagnosis and assessment of severity of community-acquired pneumonia. Chest 2004; 125:1335–1342.
- Ingram PR, Inglis T, Moxon D, Speers D. Procalcitonin and C-reactive protein in severe 2009 H1N1 influenza infection. Intensive Care Med 2010; 36:528–532.
- Fine MJ, Auble TE, Yealy DM, et al. A prediction rule to identify low-risk patients with community-acquired pneumonia. N Engl J Med 1997; 336:243–250.
- Lim WS, van der Eerden MM, Laing R, et al. Defining community acquired pneumonia severity on presentation to hospital: an international derivation and validation study. Thorax 2003; 58:377–382.
- Chalmers JD, Singanayagam A, Akram AR, et al. Severity assessment tools for predicting mortality in hospitalised patients with community-acquired pneumonia. Systematic review and meta-analysis. Thorax 2010; 65:878–883.
- Loke YK, Kwok CS, Niruban A, Myint PK. Value of severity scales in predicting mortality from community-acquired pneumonia: systematic review and meta-analysis. Thorax 2010; 65:884–890.
- Capelastegui A, España PP, Quintana JM, et al. Validation of a predictive rule for the management of community-acquired pneumonia. Eur Respir J 2006; 27:151–157.
- Charles PG, Wolfe R, Whitby M, et al; Australian Community-Acquired Pneumonia Study Collaboration. SMART-COP: a tool for predicting the need for intensive respiratory or vasopressor support in community-acquired pneumonia. Clin Infect Dis 2008; 47:375–384.
- España PP, Capelastegui A, Gorordo I, et al. Development and validation of a clinical prediction rule for severe community-acquired pneumonia. Am J Respir Crit Care Med 2006; 174:1249–1256.
- Chalmers JD, Taylor JK, Mandal P, et al. Validation of the Infectious Diseases Society of America/American Thoracic Society minor criteria for intensive care unit admission in community-acquired pneumonia patients without major criteria or contraindications to intensive care unit care. Clin Infect Dis 2011; 53:503–511.
- Majumdar SR, Eurich DT, Gamble JM, Senthilselvan A, Marrie TJ. Oxygen saturations less than 92% are associated with major adverse events in outpatients with pneumonia: a population-based cohort study. Clin Infect Dis 2011; 52:325–331.
- Nathwani D, Rubinstein E, Barlow G, Davey P. Do guidelines for community-acquired pneumonia improve the cost-effectiveness of hospital care? Clin Infect Dis 2001; 32:728–741.
- Dean NC, Silver MP, Bateman KA, James B, Hadlock CJ, Hale D. Decreased mortality after implementation of a treatment guideline for community-acquired pneumonia. Am J Med 2001; 110:451–457.
- Capelastegui A, España PP, Quintana JM, et al. Improvement of process-of-care and outcomes after implementing a guideline for the management of community-acquired pneumonia: a controlled before-and-after design study. Clin Infect Dis 2004; 39:955–963.
- Silber SH, Garrett C, Singh R, et al. Early administration of antibiotics does not shorten time to clinical stability in patients with moderate-to-severe community-acquired pneumonia. Chest 2003; 124:1798–1804.
- Welker JA, Huston M, McCue JD. Antibiotic timing and errors in diagnosing pneumonia. Arch Intern Med 2008; 168:351–356.
- Polgreen PM, Chen YY, Cavanaugh JE, et al. An outbreak of severe Clostridium difficile-associated disease possibly related to inappropriate antimicrobial therapy for community-acquired pneumonia. Infect Control Hosp Epidemiol 2007; 28:212–214.
- Waterer GW, Somes GW, Wunderink RG. Monotherapy may be suboptimal for severe bacteremic pneumococcal pneumonia. Arch Intern Med 2001; 161:1837–1842.
- Lodise TP, Kwa A, Cosler L, Gupta R, Smith RP. Comparison of beta-lactam and macrolide combination therapy versus fluoroquinolone monotherapy in hospitalized Veterans Affairs patients with community-acquired pneumonia. Antimicrob Agents Chemother 2007; 51:3977–3982.
- Waterer GW, Rello J, Wunderink RG. Management of community-acquired pneumonia in adults. Am J Respir Crit Care Med 2011; 183:157–164.
- Bjerre LM, Verheij TJ, Kochen MM. Antibiotics for community acquired pneumonia in adult outpatients. Cochrane Database Syst Rev 2009; (4):CD002109.
- Frei CR, Labreche MJ, Attridge RT. Fluoroquinolones in community-acquired pneumonia: guide to selection and appropriate use. Drugs 2011; 71:757–770.
- Weiss K, Tillotson GS. The controversy of combination vs monotherapy in the treatment of hospitalized community-acquired pneumonia. Chest 2005; 128:940–946.
- Martínez JA, Horcajada JP, Almela M, et al. Addition of a macrolide to a beta-lactam-based empirical antibiotic regimen is associated with lower in-hospital mortality for patients with bacteremic pneumococcal pneumonia. Clin Infect Dis 2003; 36:389–395.
- Waterer GW, Somes GW, Wunderink RG. Monotherapy may be suboptimal for severe bacteremic pneumococcal pneumonia. Arch Intern Med 2001; 161:1837–1842.
- Ramirez JA, Bordon J. Early switch from intravenous to oral antibiotics in hospitalized patients with bacteremic community-acquired Streptococcus pneumoniae pneumonia. Arch Intern Med 2001; 161:848–850.
- Dunbar LM, Wunderink RG, Habib MP, et al. High-dose, short-course levofloxacin for community-acquired pneumonia: a new treatment paradigm. Clin Infect Dis 2003; 37:752–760.
- el Moussaoui R, de Borgie CA, van den Broek P, et al. Effectiveness of discontinuing antibiotic treatment after three days versus eight days in mild to moderate-severe community acquired pneumonia: randomised, double blind study. BMJ 2006; 332:1355.
- Mundy LM, Leet TL, Darst K, Schnitzler MA, Dunagan WC. Early mobilization of patients hospitalized with community-acquired pneumonia. Chest 2003; 124:883–889.
- Salluh JI, Póvoa P, Soares M, Castro-Faria-Neto HC, Bozza FA, Bozza PT. The role of corticosteroids in severe community-acquired pneumonia: a systematic review. Crit Care 2008; 12:R76.
- Mikami K, Suzuki M, Kitagawa H, et al. Efficacy of corticosteroids in the treatment of community-acquired pneumonia requiring hospitalization. Lung 2007; 185:249–255.
- Snijders D, Daniels JM, de Graaff CS, van der Werf TS, Boersma WG. Efficacy of corticosteroids in community-acquired pneumonia: a randomized double-blinded clinical trial. Am J Respir Crit Care Med 2010; 181:975–982.
- Chopra V, Flanders SA. Does statin use improve pneumonia outcomes? Chest 2009; 136:1381–1388.
- Yende S, Milbrandt EB, Kellum JA, et al. Understanding the potential role of statins in pneumonia and sepsis. Crit Care Med 2011; 39:1871–1878.
- Halm EA, Fine MJ, Marrie TJ, et al. Time to clinical stability in patients hospitalized with community-acquired pneumonia: implications for practice guidelines. JAMA 1998; 279:1452–1457.
- Marrie TJ, Lau CY, Wheeler SL, Wong CJ, Feagan BG. Predictors of symptom resolution in patients with community-acquired pneumonia. Clin Infect Dis 2000; 31:1362–1367.
- Aliberti S, Peyrani P, Filardo G, et al. Association between time to clinical stability and outcomes after discharge in hospitalized patients with community-acquired pneumonia. Chest 2011; 140:482–488.
- Fiore AE, Uyeki TM, Broder K, et al; Centers for Disease Control and Prevention (CDC). Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Recomm Rep 2010; 59:1–62.
- Nuorti JP, Butler JC, Farley MM, et al. Cigarette smoking and invasive pneumococcal disease. Active Bacterial Core Surveillance Team. N Engl J Med 2000; 342:681–689.
- Mortensen EM, Kapoor WN, Chang CC, Fine MJ. Assessment of mortality after long-term follow-up of patients with community-acquired pneumonia. Clin Infect Dis 2003; 37:1617–1624.
- File TM, Marrie TJ. Burden of community-acquired pneumonia in North American adults. Postgrad Med 2010; 122:130–141.
- Marston BJ, Plouffe JF, File TM, et al. Incidence of community-acquired pneumonia requiring hospitalization. Results of a population-based active surveillance study in Ohio. The Community-Based Pneumonia Incidence Study Group. Arch Intern Med 1997; 157:1709–1718.
- Fine MJ, Smith MA, Carson CA, et al. Prognosis and outcomes of patients with community-acquired pneumonia. JAMA 1996; 275:134–141.
- Ruhnke GW, Coca-Perraillon M, Kitch BT, Cutler DM. Trends in mortality and medical spending in patients hospitalized for community-acquired pneumonia: 1993–2005. Med Care 2010; 48:1111–1116.
- Jackson ML, Neuzil KM, Thompson WW, et al. The burden of community-acquired pneumonia in seniors: results of a population-based study. Clin Infect Dis 2004; 39:1642–1650.
- Koivula I, Sten M, Mäkelä PH. Risk factors for pneumonia in the elderly. Am J Med 1994; 96:313–320.
- Ruhnke GW, Coca-Perraillon M, Kitch BT, Cutler DM. Marked reduction in 30-day mortality among elderly patients with community-acquired pneumonia. Am J Med 2011; 124:171–178.
- Farr BM, Bartlett CL, Wadsworth J, Miller DL. Risk factors for community-acquired pneumonia diagnosed upon hospital admission. British Thoracic Society Pneumonia Study Group. Respir Med 2000; 94:954–963.
- Graves CR. Pneumonia in pregnancy. Clin Obstet Gynecol 2010; 53:329–336.
- Eom CS, Jeon CY, Lim JW, Cho EG, Park SM, Lee KS. Use of acid-suppressive drugs and risk of pneumonia: a systematic review and meta-analysis. CMAJ 2011; 183:310–319.
- Voiriot G, Dury S, Parrot A, Mayaud C, Fartoukh M. Nonsteroidal antiinflammatory drugs may affect the presentation and course of community-acquired pneumonia. Chest 2011; 139:387–394.
- Thompson WW, Shay DK, Weintraub E, et al. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 2003; 289:179–186.
- Thompson WW, Shay DK, Weintraub E, et al. Influenza-associated hospitalizations in the United States. JAMA 2004; 292:1333–1340.
- Glezen WP, Decker M, Perrotta DM. Survey of underlying conditions of persons hospitalized with acute respiratory disease during influenza epidemics in Houston, 1978–1981. Am Rev Respir Dis 1987; 136:550–555.
- Izurieta HS, Thompson WW, Kramarz P, et al. Influenza and the rates of hospitalization for respiratory disease among infants and young children. N Engl J Med 2000; 342:232–239.
- Rothberg MB, Haessler SD. Complications of seasonal and pandemic influenza. Crit Care Med 2010; 38(suppl 4):e91–e97.
- Apisarnthanarak A, Mundy LM. Etiology of community-acquired pneumonia. Clin Chest Med 2005; 26:47–55.
- de Roux A, Marcos MA, Garcia E, et al. Viral community-acquired pneumonia in nonimmunocompromised adults. Chest 2004; 125:1343–1351.
- Hamelin ME, Côté S, Laforge J, et al. Human metapneumovirus infection in adults with community-acquired pneumonia and exacerbation of chronic obstructive pulmonary disease. Clin Infect Dis 2005; 41:498–502.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44(suppl 2):S27–S72.
- Kolling UK, Hansen F, Braun J, Rink L, Katus HA, Dalhoff K. Leucocyte response and anti-inflammatory cytokines in community acquired pneumonia. Thorax 2001; 56:121–125.
- Wunderink RG, Waterer GW. Community-acquired pneumonia: pathophysiology and host factors with focus on possible new approaches to management of lower respiratory tract infections. Infect Dis Clin North Am 2004; 18:743–759.
- Hilleman MR. Realities and enigmas of human viral influenza: pathogenesis, epidemiology and control. Vaccine 2002; 20:3068–3087.
- Bender BS, Small PA. Influenza: pathogenesis and host defense. Semin Respir Infect 1992; 7:38–45.
- Scheiblauer H, Reinacher M, Tashiro M, Rott R. Interactions between bacteria and influenza A virus in the development of influenza pneumonia. J Infect Dis 1992; 166:783–791.
- McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev 2006; 19:571–582.
- Metlay JP, Kapoor WN, Fine MJ. Does this patient have community-acquired pneumonia? Diagnosing pneumonia by history and physical examination. JAMA 1997; 278:1440–1445.
- Benbassat J, Baumal R. Narrative review: should teaching of the respiratory physical examination be restricted only to signs with proven reliability and validity? J Gen Intern Med 2010; 25:865–872.
- Kolsuz M, Erginel S, Alatas O, et al. Acute phase reactants and cytokine levels in unilateral community-acquired pneumonia. Respiration 2003; 70:615–622.
- Alves DW, Kennedy MT. Community-acquired pneumonia in casualty: etiology, clinical features, diagnosis, and management (or a look at the “new” in pneumonia since 2002). Curr Opin Pulm Med 2004; 10:166–170.
- Monto AS, Gravenstein S, Elliott M, Colopy M, Schweinle J. Clinical signs and symptoms predicting influenza infection. Arch Intern Med 2000; 160:3243–3247.
- Bewick T, Myles P, Greenwood S, et al; Influenza Clinical Information Network. Clinical and laboratory features distinguishing pandemic H1N1 influenza-related pneumonia from interpandemic community-acquired pneumonia in adults. Thorax 2011; 66:247–252.
- Morens DM, Fauci AS. The 1918 influenza pandemic: insights for the 21st century. J Infect Dis 2007; 195:1018–1028.
- Starr I. Influenza in 1918: recollections of the epidemic in Philadelphia. 1976. Ann Intern Med 2006; 145:138–140.
- Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis 2008; 198:962–970.
- Brundage JF, Shanks GD. Deaths from bacterial pneumonia during 1918–19 influenza pandemic. Emerg Infect Dis 2008; 14:1193–1199.
- Treanor J. Influenza virus. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, PA: Elsevier/Churchill Livingstone; 2005:2060–2085.
- Jarstrand C, Tunevall G. The influence of bacterial superinfection on the clinical course of influenza. Studies from the influenza epidemics in Stockholm during the winters 1969–70 and 1971–72. Scand J Infect Dis 1975; 7:243–247.
- Schwarzmann SW, Adler JL, Sullivan RJ, Marine WM. Bacterial pneumonia during the Hong Kong influenza epidemic of 1968–1969. Arch Intern Med 1971; 127:1037–1041.
- Hageman JC, Uyeki TM, Francis JS, et al. Severe community-acquired pneumonia due to Staphylococcus aureus, 2003–04 influenza season. Emerg Infect Dis 2006; 12:894–899.
- Centers for Disease Control and Prevention (CDC). Severe methicillin-resistant Staphylococcus aureus community-acquired pneumonia associated with influenza—Louisiana and Georgia, December 2006–January 2007. MMWR Morb Mortal Wkly Rep 2007; 56:325–329.
- Hidron AI, Low CE, Honig EG, Blumberg HM. Emergence of community-acquired methicillin-resistant Staphylococcus aureus strain USA300 as a cause of necrotising community-onset pneumonia. Lancet Infect Dis 2009; 9:384–392.
- Call SA, Vollenweider MA, Hornung CA, Simel DL, McKinney WP. Does this patient have influenza? JAMA 2005; 293:987–997.
- Harper SA, Bradley JS, Englund JA, et al; Expert Panel of the Infectious Diseases Society of America. Seasonal influenza in adults and children—diagnosis, treatment, chemoprophylaxis, and institutional outbreak management: clinical practice guidelines of the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:1003–1032.
- Boersma WG, Daniels JM, Löwenberg A, Boeve WJ, van de Jagt EJ. Reliability of radiographic findings and the relation to etiologic agents in community-acquired pneumonia. Respir Med 2006; 100:926–932.
- Brixey AG, Luo Y, Skouras V, Awdankiewicz A, Light RW. The efficacy of chest radiographs in detecting parapneumonic effusions. Respirology 2011; 16:1000–1004.
- Campbell SG, Marrie TJ, Anstey R, Dickinson G, Ackroyd-Stolarz S. The contribution of blood cultures to the clinical management of adult patients admitted to the hospital with community-acquired pneumonia: a prospective observational study. Chest 2003; 123:1142–1150.
- Waterer GW, Wunderink RG. The influence of the severity of community-acquired pneumonia on the usefulness of blood cultures. Respir Med 2001; 95:78–82.
- Houck PM, Bratzler DW, Nsa W, Ma A, Bartlett JG. Timing of antibiotic administration and outcomes for Medicare patients hospitalized with community-acquired pneumonia. Arch Intern Med 2004; 164:637–644.
- Information & Quality Healthcare. http://www.IQH.org/attachments/219_CoreMHelpBookletpg4_11_3.pdf. Accessed November 14, 2011.
- Rosón B, Carratalà J, Verdaguer R, Dorca J, Manresa F, Gudiol F. Prospective study of the usefulness of sputum Gram stain in the initial approach to community-acquired pneumonia requiring hospitalization. Clin Infect Dis 2000; 31:869–874.
- García-Vázquez E, Marcos MA, Mensa J, et al. Assessment of the usefulness of sputum culture for diagnosis of community-acquired pneumonia using the PORT predictive scoring system. Arch Intern Med 2004; 164:1807–1811.
- Rosón B, Fernández-Sabé N, Carratalà J, et al. Contribution of a urinary antigen assay (Binax NOW) to the early diagnosis of pneumococcal pneumonia. Clin Infect Dis 2004; 38:222–226.
- Sordé R, Falcó V, Lowak M, et al. Current and potential usefulness of pneumococcal urinary antigen detection in hospitalized patients with community-acquired pneumonia to guide antimicrobial therapy. Arch Intern Med 2011; 171:166–172.
- Koegelenberg CFN, Diacon AH, Bolliger CT. Parapneumonic pleural effusion and empyema. Respiration 2008; 75:241–250.
- Almirall J, Bolíbar I, Toran P, et al; Community-Acquired Pneumonia Maresme Study Group. Contribution of C-reactive protein to the diagnosis and assessment of severity of community-acquired pneumonia. Chest 2004; 125:1335–1342.
- Ingram PR, Inglis T, Moxon D, Speers D. Procalcitonin and C-reactive protein in severe 2009 H1N1 influenza infection. Intensive Care Med 2010; 36:528–532.
- Fine MJ, Auble TE, Yealy DM, et al. A prediction rule to identify low-risk patients with community-acquired pneumonia. N Engl J Med 1997; 336:243–250.
- Lim WS, van der Eerden MM, Laing R, et al. Defining community acquired pneumonia severity on presentation to hospital: an international derivation and validation study. Thorax 2003; 58:377–382.
- Chalmers JD, Singanayagam A, Akram AR, et al. Severity assessment tools for predicting mortality in hospitalised patients with community-acquired pneumonia. Systematic review and meta-analysis. Thorax 2010; 65:878–883.
- Loke YK, Kwok CS, Niruban A, Myint PK. Value of severity scales in predicting mortality from community-acquired pneumonia: systematic review and meta-analysis. Thorax 2010; 65:884–890.
- Capelastegui A, España PP, Quintana JM, et al. Validation of a predictive rule for the management of community-acquired pneumonia. Eur Respir J 2006; 27:151–157.
- Charles PG, Wolfe R, Whitby M, et al; Australian Community-Acquired Pneumonia Study Collaboration. SMART-COP: a tool for predicting the need for intensive respiratory or vasopressor support in community-acquired pneumonia. Clin Infect Dis 2008; 47:375–384.
- España PP, Capelastegui A, Gorordo I, et al. Development and validation of a clinical prediction rule for severe community-acquired pneumonia. Am J Respir Crit Care Med 2006; 174:1249–1256.
- Chalmers JD, Taylor JK, Mandal P, et al. Validation of the Infectious Diseases Society of America/American Thoracic Society minor criteria for intensive care unit admission in community-acquired pneumonia patients without major criteria or contraindications to intensive care unit care. Clin Infect Dis 2011; 53:503–511.
- Majumdar SR, Eurich DT, Gamble JM, Senthilselvan A, Marrie TJ. Oxygen saturations less than 92% are associated with major adverse events in outpatients with pneumonia: a population-based cohort study. Clin Infect Dis 2011; 52:325–331.
- Nathwani D, Rubinstein E, Barlow G, Davey P. Do guidelines for community-acquired pneumonia improve the cost-effectiveness of hospital care? Clin Infect Dis 2001; 32:728–741.
- Dean NC, Silver MP, Bateman KA, James B, Hadlock CJ, Hale D. Decreased mortality after implementation of a treatment guideline for community-acquired pneumonia. Am J Med 2001; 110:451–457.
- Capelastegui A, España PP, Quintana JM, et al. Improvement of process-of-care and outcomes after implementing a guideline for the management of community-acquired pneumonia: a controlled before-and-after design study. Clin Infect Dis 2004; 39:955–963.
- Silber SH, Garrett C, Singh R, et al. Early administration of antibiotics does not shorten time to clinical stability in patients with moderate-to-severe community-acquired pneumonia. Chest 2003; 124:1798–1804.
- Welker JA, Huston M, McCue JD. Antibiotic timing and errors in diagnosing pneumonia. Arch Intern Med 2008; 168:351–356.
- Polgreen PM, Chen YY, Cavanaugh JE, et al. An outbreak of severe Clostridium difficile-associated disease possibly related to inappropriate antimicrobial therapy for community-acquired pneumonia. Infect Control Hosp Epidemiol 2007; 28:212–214.
- Waterer GW, Somes GW, Wunderink RG. Monotherapy may be suboptimal for severe bacteremic pneumococcal pneumonia. Arch Intern Med 2001; 161:1837–1842.
- Lodise TP, Kwa A, Cosler L, Gupta R, Smith RP. Comparison of beta-lactam and macrolide combination therapy versus fluoroquinolone monotherapy in hospitalized Veterans Affairs patients with community-acquired pneumonia. Antimicrob Agents Chemother 2007; 51:3977–3982.
- Waterer GW, Rello J, Wunderink RG. Management of community-acquired pneumonia in adults. Am J Respir Crit Care Med 2011; 183:157–164.
- Bjerre LM, Verheij TJ, Kochen MM. Antibiotics for community acquired pneumonia in adult outpatients. Cochrane Database Syst Rev 2009; (4):CD002109.
- Frei CR, Labreche MJ, Attridge RT. Fluoroquinolones in community-acquired pneumonia: guide to selection and appropriate use. Drugs 2011; 71:757–770.
- Weiss K, Tillotson GS. The controversy of combination vs monotherapy in the treatment of hospitalized community-acquired pneumonia. Chest 2005; 128:940–946.
- Martínez JA, Horcajada JP, Almela M, et al. Addition of a macrolide to a beta-lactam-based empirical antibiotic regimen is associated with lower in-hospital mortality for patients with bacteremic pneumococcal pneumonia. Clin Infect Dis 2003; 36:389–395.
- Waterer GW, Somes GW, Wunderink RG. Monotherapy may be suboptimal for severe bacteremic pneumococcal pneumonia. Arch Intern Med 2001; 161:1837–1842.
- Ramirez JA, Bordon J. Early switch from intravenous to oral antibiotics in hospitalized patients with bacteremic community-acquired Streptococcus pneumoniae pneumonia. Arch Intern Med 2001; 161:848–850.
- Dunbar LM, Wunderink RG, Habib MP, et al. High-dose, short-course levofloxacin for community-acquired pneumonia: a new treatment paradigm. Clin Infect Dis 2003; 37:752–760.
- el Moussaoui R, de Borgie CA, van den Broek P, et al. Effectiveness of discontinuing antibiotic treatment after three days versus eight days in mild to moderate-severe community acquired pneumonia: randomised, double blind study. BMJ 2006; 332:1355.
- Mundy LM, Leet TL, Darst K, Schnitzler MA, Dunagan WC. Early mobilization of patients hospitalized with community-acquired pneumonia. Chest 2003; 124:883–889.
- Salluh JI, Póvoa P, Soares M, Castro-Faria-Neto HC, Bozza FA, Bozza PT. The role of corticosteroids in severe community-acquired pneumonia: a systematic review. Crit Care 2008; 12:R76.
- Mikami K, Suzuki M, Kitagawa H, et al. Efficacy of corticosteroids in the treatment of community-acquired pneumonia requiring hospitalization. Lung 2007; 185:249–255.
- Snijders D, Daniels JM, de Graaff CS, van der Werf TS, Boersma WG. Efficacy of corticosteroids in community-acquired pneumonia: a randomized double-blinded clinical trial. Am J Respir Crit Care Med 2010; 181:975–982.
- Chopra V, Flanders SA. Does statin use improve pneumonia outcomes? Chest 2009; 136:1381–1388.
- Yende S, Milbrandt EB, Kellum JA, et al. Understanding the potential role of statins in pneumonia and sepsis. Crit Care Med 2011; 39:1871–1878.
- Halm EA, Fine MJ, Marrie TJ, et al. Time to clinical stability in patients hospitalized with community-acquired pneumonia: implications for practice guidelines. JAMA 1998; 279:1452–1457.
- Marrie TJ, Lau CY, Wheeler SL, Wong CJ, Feagan BG. Predictors of symptom resolution in patients with community-acquired pneumonia. Clin Infect Dis 2000; 31:1362–1367.
- Aliberti S, Peyrani P, Filardo G, et al. Association between time to clinical stability and outcomes after discharge in hospitalized patients with community-acquired pneumonia. Chest 2011; 140:482–488.
- Fiore AE, Uyeki TM, Broder K, et al; Centers for Disease Control and Prevention (CDC). Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Recomm Rep 2010; 59:1–62.
- Nuorti JP, Butler JC, Farley MM, et al. Cigarette smoking and invasive pneumococcal disease. Active Bacterial Core Surveillance Team. N Engl J Med 2000; 342:681–689.
KEY POINTS
- Especially during flu season, clinicians should consider influenza in patients with respiratory symptoms.
- The diagnosis of CAP is based primarily on clinical factors: a combination of signs and symptoms such as cough, fever, chills, sputum production, dyspnea, pleuritic pain, tachypnea, tachycardia, hypoxemia, consolidation or rales on auscultation, and a new infiltrate on chest imaging.
- Empiric outpatient treatment of a previously healthy patient with CABP should include either a macrolide or doxycycline. A fluoroquinolone or beta-lactam plus a macrolide should be used for patients with comorbid conditions.
- Several indices have been validated for use in deciding on inpatient vs outpatient treatment and whether a patient with pneumonia should be admitted to an intensive care unit.
Overcoming barriers to hypertension control in African Americans
High blood pressure takes a devastating toll on African Americans. Better control can go a long way to closing the “mortality gap” between African Americans and white Americans. But which strategies are best to address this complex problem?
In this report, we review the evidence on practice-based approaches to improving blood pressure control, from new styles of patient education to home blood pressure monitoring, focusing on studies in African Americans (Table 1).1–11
BETTER CONTROL IS NEEDED


PATIENT-RELATED BARRIERS
Patient-related barriers24–40 include:
- Poor knowledge about hypertension and its consequences31,32
- Poor adherence to drug therapy (a major factor,24–26 as African Americans have poorer adherence rates than whites,27–29 which may explain some of the racial disparity in blood pressure control30)
- False health beliefs34–37
- Inability to change one’s lifestyle
- Side effects of antihypertensive drugs32
- Unrealistic expectations of treatment (eg, a cure33)
- Demographic factors (eg, socioeconomic status, educational level, age, sex).24,38–40
Perhaps the most salient and easily modifiable of these factors are patients’ reluctance to modify their lifestyle and their misconceptions about the causes, treatment, and prevention of hypertension. Patients whose beliefs are discordant with traditional biomedical concepts of hypertension have poorer blood pressure control than those whose beliefs are concordant.41 This may be more relevant to African Americans, since they are known to have cultural health beliefs that differ from those of Western culture (eg, that hypertension is a curable rather than a chronic illness, and that hypertension is a disease of nerves that often affects the blood and clogs the arteries).42
PHYSICIAN-RELATED BARRIERS
Barriers to effective blood pressure control at the physician level43–48 include:
- Nonadherence to treatment guidelines44
- Failure to intensify the regimen if goals are not met45
- Failure to emphasize therapeutic lifestyle changes.43,46–48
When primary care physicians do not follow evidence-based guidelines, the reason may be that they are not aware of them or that they do not understand them. In a national survey of 1,029 physicians that was designed to explore how well physicians know the indications for specific antihypertensive drugs and how closely their opinions and practice agreed with national guidelines, only 37.3% correctly answered all of the knowledge-related questions.49
Other reasons for nonadherence are that physicians may disagree with the guidelines, may not be able to follow the guidelines, may not believe that following them will achieve the desired effect, or may have no motivation to change their practice.50
Whatever the reason, Hyman et al51 reported that as many as 30% of physicians did not recommend treatment for patients with diastolic blood pressures of 90 to 100 mm Hg, and a higher proportion did not treat patients with systolic blood pressures of 140 to 160 mm Hg.
BARRIERS IN HEALTH CARE SYSTEMS
Although health care systems present barriers to optimal blood pressure control,20,27,31,52 there is evidence that most cases of uncontrolled hypertension occur in patients with good access to care.32,53,54 For example, an NHANES study53 suggested that most patients with uncontrolled hypertension had in fact seen a physician on average at least three times in the previous year. And this may be more pervasive in African Americans: one survey found hypertension was uncontrolled in 75% of hypertensive African American patients despite free access to care, free medications, and regular follow-up visits.41
Thus, the most significant barriers to blood pressure control appear to be patient-related and physician-related.
INTERVENTIONS AIMED AT PATIENTS
The most common approaches to improving blood pressure control at the patient level, regardless of race, are patient education,55–61 home blood pressure monitoring,62–67 and behavioral counseling to address misconceptions about hypertension,68 to improve adherence to drug therapy,69–73 and to encourage lifestyle modifications.74–78
Patient education
Patient education can improve blood pressure control.58,79–82 Its aims are to increase patients’ understanding of the disease83 and to encourage them to be more active in their own care.80,84,85
Patient education has a moderate effect on blood pressure control. The average proportion of patients whose hypertension was under control in community-based trials of various interventions ranged from 60% to 70%, compared with 38% to 46% with usual care.56,80,81
However, these strategies largely did not address misconceptions patients have about hypertension. This issue is especially critical in African Americans, who may have different perceptions of hypertension and different expectations for care41: beliefs that hypertension is “curable,” not chronic, and that medication is needed only for hypertension-related symptoms may translate to poorer rates of medication adherence.
Levine et al1 evaluated the efficacy of home visits by trained community health advisory board workers in a neighborhood in Baltimore, MD, with a high prevalence of hypertension. Participants were randomized to receive either one visit or five visits during the 40-month study period. Both groups had a statistically significant reduction in blood pressure, and in both groups the proportion of patients with adequate blood pressure control increased significantly. The results support the use of a practice- and community-based partnership to improve blood pressure control in African American patients.
Ogedegbe et al2 randomized 190 hypertensive African American patients to receive usual care or quarterly counseling sessions that used motivational interviewing focused on medication adherence. The counseled patients stayed adherent to their medications, whereas adherence declined significantly in those receiving usual care. This effect was associated with a modest, nonsignificant trend toward a net reduction in systolic blood pressure with motivational interviewing.
A novel method of health education is the use of narrative communication—ie, storytelling. It has a good amount of evidence to support it, as culturally appropriate storytelling may allow patients to identify with a story as it relates to their own lives.86–89 Examples of educational storytelling include:
- A woman with hypertension discussing what it means to have high blood pressure, and the benefits of controlling it, such as living long enough to see her grandchildren grow up
- A man discussing the importance of involving family and friends to help control blood pressure, and how dietary modifications can be made to ensure that salt alternatives are used when the family does the cooking.
Storytelling should be done in a culturally appropriate context. For example, storytellers should have the same background as the patient (ie, similar socioeconomic status and ethnic background): patients are more likely to be influenced if they identify with the storyteller and imagine themselves in a similar situation.
Houston et al3 randomized 299 hypertensive African Americans to view either three DVDs that featured patients with hypertension or three “attention-control DVDs” on topics not related to hypertension. The intervention group’s DVDs focused on storytelling and “learning more.” In the storytelling section, patients told personal stories about what it meant to have hypertension and gave advice on how to best interact with health care providers and methods to improve medication adherence. A “learning more” section focused on what high blood pressure is, addressed therapeutic lifestyle changes, and encouraged patients to communicate with their health care providers. The patients who viewed the patient narratives had significantly lower blood pressure at 3 months than those assigned to usual care. Although blood pressure subsequently increased in both groups, the benefits of the intervention still existed at the end of follow-up.
Important to note about two of the above three studies1,3 is that the interventions were done by people other than physicians, thus emphasizing the importance of a team approach to blood pressure control.
Behavioral counseling
The effectiveness of lifestyle modifications such as diet, weight loss, and physical activity in preventing and treating hypertension is well established.74–78 For example:
- In the Dietary Approaches to Stop Hypertension (DASH) trial,76 a healthy diet lowered blood pressure about as much as single drugs do, particularly in African Americans.
- The Trial of Nonpharmacologic Interventions in the Elderly (TONE)74 showed that exercise can lower blood pressure in obese hypertensive patients.
- The PREMIER trial (Lifestyle Interventions for Blood Pressure Control)75 showed that a single brief counseling session could produce substantial decreases in blood pressure in patients with stage 1 hypertension or high-normal blood pressure.
Unfortunately, these results have been hard to translate into primary care practice, especially for African American patients. Several studies have evaluated the impact of lifestyle interventions on blood pressure control in primary care practices with a large population of African American patients.
Bosworth et al,4 in a study of a practice in which almost half the patients were African American, randomized patients to receive usual care, nurse-administered tailored behavioral telephone counseling, home blood pressure monitoring, or home monitoring plus tailored behavioral telephone counseling. The combination of home monitoring and tailored behavioral telephone counseling led to a statistically significant improvement at 24 months compared with baseline.
Home blood pressure monitoring
The effectiveness of self-monitoring in improving blood pressure control is also well documented.62,63,65–67,90–95
Pickering et al62 studied patients with poorly controlled hypertension in a managed-care setting and found a reduction of 7 mm Hg systolic and 5 mm Hg diastolic pressure after 3 to 6 months of home monitoring compared with usual care.
Mengden et al,94 in a similar study, found average blood pressure reductions at 6 months of 19.3/11.9 mm Hg in the home-monitoring group vs 10.6/8.8 mm Hg in the usual-care group.
The effect of home blood pressure monitoring may be greater in African Americans.
Rogers et al93 found it to be more effective at lowering blood pressure than usual care in a group of 121 patients with poorly controlled hypertension followed in primary care practices, and these reductions were twice as large in African American patients than in white patients.93
Bondmass,92 in a study of 33 African American patients with poorly controlled hypertension, reported a 53% control rate within 4 weeks of home monitoring. All patients in the study had uncontrolled blood pressure at baseline (> 140/90 mm Hg).
Artinian et al5 evaluated the effect of nurse-managed telemonitoring on blood pressure control vs enhanced usual care. All participants were African American. The monitored group had a significantly greater reduction in systolic pressure at 12 months compared with those who received enhanced usual care.
PHYSICIAN-LEVEL INTERVENTIONS
Most interventions to improve how physicians manage patients with hypertension are designed to improve adherence to treatment guidelines. In most cases, these interventions are based on continuous quality improvement and disease management concepts such as physician education and academic detailing, reminders, feedback on performance measures, and risk-assessment tools.96,97
Physician education
Interest is increasing in physician educational interventions for blood pressure control.24,98
Inui et al,99 in an early study in a primary care practice, found that patients of physicians who received tutorials on hypertension management were more compliant with their drug regimens and had better blood pressure control than patients of physicians in the control group.
Jennett et al,100 in a similar randomized clinical trial, found that physicians who participated in an education activity were more adherent to treatment guidelines at 6 and 12 months compared with those who did not participate.
Maue et al101 showed that rates of blood pressure control improved from 41% to 52% after a 6-month educational intervention for physicians in a managed-care setting.
Tu et al102 reviewed 12 studies in which seven different physician educational interventions were used either alone or in combination and concluded that physician education improves compliance with guidelines for managing hypertension.
Unfortunately, these studies did not report outcomes separately for African American and white patients.
Hicks et al6 found that disease management approaches that target physicians whose patients with hypertension are mostly African American did not yield clinically relevant improvement in these patients, and that minority patients were significantly less likely to have their blood pressure controlled at the end of the study compared with their non-Hispanic white counterparts.
Feedback to providers
Several studies have shown that, given reminders and feedback systems, physicians will change their practice.103–106
Mashru and Lant104 combined chart audits and physician education in primary care practices and found they improved physician performance measures such as accuracy of diagnosis, number of patients who received cardiovascular risk assessment, and number of patients whose treatment was based on clinical laboratory assessments.
Feedback takes many forms but consists mostly of computerized information107 or peer-to-peer academic detailing with opinion leaders.108–110
Dickinson et al,106 for instance, showed that computer-generated listings of patients’ blood pressures combined with a physician education program on clinical management of hypertension led to increased knowledge and better follow-up on their patients.
Again, however, these studies did not distinguish between African American and white patients, which makes it difficult to judge whether or not these approaches work differently for physicians with a large proportion of African American patients.
Computerized decision-support systems
Computerized decision-support systems have proliferated in primary care practices.111
McAlister et al103 found that general practitioners randomized to manage hypertension with the assistance of a computer obtained better outcomes than with usual care.
Montgomery and Fahey,107 in a systematic review, found improved blood pressure control in two of the three trials that compared computer-generated feedback reports and reminders to usual care. Specifically, 51% of patients whose physicians received reminders either had controlled blood pressure or were at least receiving treatment vs 33% in the control group at 12 months. This difference was even higher at 24 months.
Montgomery et al7 later randomized primary care practices to use a computer-based decision-support system and a cardiovascular risk chart, the risk chart alone, or to continue as usual. Results indicated no reduction in cardiovascular risk in the computer-system or the chart-only group, whereas patients in the chart-only group had a significant reduction in systolic pressure and were prescribed more cardiovascular drugs. This study indicates that use of a computerized decision-support system is not superior to chart review and audit feedback alone.
Evidence that computerized decision systems improve blood pressure control in African Americans is scant. However, when one looks at the evidence from studies of African Americans, the outcomes do not seem to differ between African American and white patients.
Hicks et al6 examined the effectiveness of computerized decision support in improving hypertension care in a racially diverse population. Physicians were randomized to receive computerized decision support or to provide usual care without computerized support. Both groups improved significantly in prescribing appropriate drugs but not in overall blood pressure control. Furthermore, the study showed no reduction in racial disparities of care and blood pressure control.
A potential explanation for the lack of improvement in blood pressure was that the intervention dealt with making sure the appropriate drugs were prescribed rather than making sure physicians also appropriately intensified antihypertensive management when necessary.
INTERVENTIONS TARGETING PATIENTS AND PHYSICIANS
Several studies have targeted both patient and physician-level barriers to blood pressure control in practice-based settings.
Roumie et al8 randomized physicians to one of three intervention groups:
- “Provider education” consisting of an email message with a Web-based link to the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC-7)
- Provider education plus a computer alert with information about their patient’s blood pressure
- Provider education, a computer alert, and patient education (ie, patients received a letter encouraging adherence to drug therapy, changing their lifestyle, and talking with their doctor about their blood pressure).
Patients whose providers were randomized to the third group had better blood pressure control. The report did not differentiate African American vs white patients. The data, however, did show the effectiveness of adding patient education to provider education to improve blood pressure control.
Bosworth et al,112 in a study in which 40% of patients were African American, randomized patients to usual care or to bimonthly nurse-delivered behavioral telephone counseling. They also randomized providers either to receive computer-generated decision support designed to improve adherence to guidelines or to receive no support.
There were no significant differences in rates of blood pressure control in the intervention groups compared with a control group. Although differences in blood pressure control between groups were not significant, patients randomized to behavioral intervention had significantly better blood pressure control at the 24-month follow-up than at baseline.
Svetkey et al9 evaluated the effects of physician intervention, patient intervention, and physician intervention plus patient intervention compared with control on systolic blood pressure at 6 months. They found that an intensive behavioral lifestyle intervention led to a significant reduction in systolic pressure at 6 months. By itself, the physician intervention did not have a meaningful effect, but patients in the combined physician-and-patient-intervention group experienced the greatest reduction (9.7 ± 12.7 mm Hg).
It takes a team
Physicians should not be the only focus in helping patients achieve blood pressure control. Although physician and patient factors need to be addressed to improve blood pressure control in African Americans, emphasis should also be placed on interdisciplinary, team-based care utilizing health care providers such as nurses, physician assistants, and pharmacists. Team-based care has been shown to have the greatest impact of all the strategies for improving blood pressure control.113 There is a good amount of evidence involving interventions with a focus on health care providers other than physicians, although the data lack a sufficient focus on African Americans.
Carter et al,10 in a randomized controlled trial in which 26.3% of the patients were African American, found that an intervention consisting of clinical pharmacists giving physicians drug therapy recommendations based on national guidelines resulted in a significantly lower blood pressure compared with a control group: the mean reduction was 20.7/9.7 mm Hg in the intervention group vs 6.8/4.5 mm Hg in the control group.
Carter et al114 performed a meta-analysis of 37 studies and found that two strategies led to a significant reduction in blood pressure: a pharmacist-led intervention with treatment recommendations to physicians resulted in a systolic pressure reduction of 9.30 mm Hg; and nurse-led interventions resulted in a systolic pressure reduction of 4.80 mm Hg. Again, many of the studies cited in this meta-analysis lacked a focus on African Americans.
Hunt et al11 conducted a randomized controlled trial in which pharmacists actively participated in the management of blood pressure. They were involved with every aspect of care, including reviewing medications and adverse drug reactions, assessing lifestyle behaviors and barriers to adherence, making dosing adjustments, and adding medications. Patients randomized to the intervention group achieved significantly lower systolic and diastolic pressures (137/75 vs 143/78 mm Hg in the control group). However, information about race was not included.
The above studies are just a few out of a large body of evidence demonstrating the value of team-based care to improve blood pressure control. It has yet to be determined whether these models can improve blood pressure control specifically in African Americans, since so many of these trials lacked a focus on this group. Promising is an ongoing randomized prospective trial by Carter et al115 evaluating a model of collaboration between physicians and pharmacists, with a focus on patients in underrepresented minorities.
SO WHAT WORKS?
Although there is a growing body of literature on interventions to try to reduce disparities in hypertension and blood pressure control between African Americans and whites, only a few randomized controlled trials have focused on African Americans, and several have not reported their results.116 So the question remains: How should we interpret the available data, which are aggregated across racial groups, and put it into practice when caring for hypertensive African American patients?
Patient education. In trying to overcome patient-related barriers, emphasis should be on patient education, in particular addressing misconceptions about hypertension and promoting adherence to antihypertensive therapy. This is evident from the narrative storytelling intervention by Houston et al.3 Although this is the first study of its kind, this strategy may be something to consider if future studies replicate these findings. Culturally appropriate storytelling may allow patients to identify with the stories as they relate to their own personal lives. It can be an effective way to address patient education and change behaviors.
Self-monitoring with a home blood pressure monitor has also proven effective in the management of hypertension in African Americans. Indeed, the few studies that reported findings in African Americans showed impressive reductions in blood pressure. The benefits of home monitoring are well documented, and the effect on physician-related barriers such as clinical inertia are also quite impressive.117 However, most of these studies did not assess the long-term impact or cost-effectiveness of home monitoring on blood pressure control.
Behavioral counseling. Although we have good evidence of the effectiveness of behavioral counseling, whether this is sustained long-term has been less studied in African Americans. Thus, while interventions that targeted African Americans have reported impressive reductions in blood pressure, the effect tends to be greatest during the first few months of implementation, with the benefits disappearing over time.
Physician-related interventions. With regard to physician-level interventions, research has focused on physician education, utilizing alerts and computerized clinical decision-support systems. Evidence is scant on whether the use of computerized systems results in improves hypertension care in African Americans. However, a closer look at the data from studies that report outcomes in African American and white patients shows that the results do not seem to differ between these groups. Still, there is insufficient information about the impact on hypertensive African Americans.6
Strategies that address both patient- and physician-related barriers can improve overall blood pressure control; however, there is a lack of data comparing outcomes in hypertensive African Americans with those of whites, making it difficult to know if this would be an effective strategy in African American patients alone.
More studies needed that focus on African Americans
Developing interventions to improve blood pressure control in African Americans should be an ongoing priority for research if we intend to address racial disparities in cardiovascular disease. Although it is reassuring that there is a growing body of evidence and research with this focus,118–121 more research is needed to determine effective strategies that address barriers related to physician practice and to the health care system overall as they relate to blood pressure control in African Americans. More importantly, these strategies should also emphasize a team-based approach that includes nurses, pharmacists, and physician assistants. Developing targeted interventions for hypertensive African Americans will help reduce disparities in the rates of cardiovascular illness and death in this patient population.
- Levine DM, Bone LR, Hill MN, et al. The effectiveness of a community/academic health center partnership in decreasing the level of blood pressure in an urban African-American population. Ethn Dis 2003; 13:354–361.
- Ogedegbe G, Chaplin W, Schoenthaler A, et al. A practice-based trial of motivational interviewing and adherence in hypertensive African Americans. Am J Hypertens 2008; 21:1137–1143.
- Houston TK, Allison JJ, Sussman M, et al. Culturally appropriate storytelling to improve blood pressure: a randomized trial. Ann Intern Med 2011; 154:77–84.
- Bosworth HB, Olsen MK, Grubber JM, et al. Two self-management interventions to improve hypertension control: a randomized trial. Ann Intern Med 2009; 151:687–695.
- Artinian NT, Flack JM, Nordstrom CK, et al. Effects of nurse-managed telemonitoring on blood pressure at 12-month follow-up among urban African Americans. Nurs Res 2007; 56:312–322.
- Hicks LS, Sequist TD, Ayanian JZ, et al. Impact of computerized decision support on blood pressure management and control: a randomized controlled trial. J Gen Intern Med 2008; 23:429–441.
- Montgomery AA, Fahey T, Peters TJ, MacIntosh C, Sharp DJ. Evaluation of computer based clinical decision support system and risk chart for management of hypertension in primary care: randomised controlled trial. BMJ 2000; 320:686–690.
- Roumie CL, Elasy TA, Greevy R, et al. Improving blood pressure control through provider education, provider alerts, and patient education: a cluster randomized trial. Ann Intern Med 2006; 145:165–175.
- Svetkey LP, Pollak KI, Yancy WS, et al. Hypertension improvement project: randomized trial of quality improvement for physicians and lifestyle modification for patients. Hypertension 2009; 54:1226–1233.
- Carter BL, Ardery G, Dawson JD, et al. Physician and pharmacist collaboration to improve blood pressure control. Arch Intern Med 2009; 169:1996–2002.
- Hunt JS, Siemienczuk J, Pape G, et al. A randomized controlled trial of team-based care: impact of physician-pharmacist collaboration on uncontrolled hypertension. J Gen Intern Med 2008; 23:1966–1972.
- US Department of Health and Human Services: Office of Disease Prevention and Health Promotion—Healthy People 2010. Nasnewsletter 2000; 15:3.
- Egan BM, Zhao Y, Axon RN. US trends in prevalence, awareness, treatment, and control of hypertension, 1988–2008. JAMA 2010; 303:2043–2050.
- US Centers for Disease Control and Prevention. Age-specific excess deaths associated with stroke among racial/ethnic minority populations–United States, 1997. JAMA 2000; 283:2382–2383.
- Giles WH, Kittner SJ, Hebel JR, Losonczy KG, Sherwin RW. Determinants of black-white differences in the risk of cerebral infarction. The National Health and Nutrition Examination Survey Epidemiologic Follow-up Study. Arch Intern Med 1995; 155:1319–1324.
- Klag MJ, Whelton PK, Randall BL, Neaton JD, Brancati FL, Stamler J. End-stage renal disease in African-American and white men. 16-year MRFIT findings. JAMA 1997; 277:1293–1298.
- Pavlik VN, Hyman DJ, Vallbona C, Toronjo C, Louis K. Hypertension awareness and control in an inner-city African-American sample. J Hum Hypertens 1997; 11:277–283.
- Roger VL, Go AS, Lloyd-Jones DM, et al. Heart Disease and Stroke Statistics—2011 Update: A Report From the American Heart Association. Circulation Feb 1; 123( 4):e18–e209.
- Chobanian AV, Bakris GL, Black HR, et al., Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- Bone LR, Hill MN, Stallings R, et al. Community health survey in an urban African-American neighborhood: distribution and correlates of elevated blood pressure. Ethn Dis 2000; 10:87–95.
- Weber MA. Strategies for improving blood pressure control. Am J Hypertens 1998; 11:897–899.
- Hill MN, Sutton BS. Barriers to hypertension care and control. Curr Hypertens Rep 2000; 2:445–450.
- Alderman MH. Barriers to blood pressure control. Am J Hypertens 1999; 12:1268–1269.
- Chobanian AV. Control of hypertension—an important national priority. N Engl J Med 2001; 345:534–535.
- Miller NH, Hill M, Kottke T, Ockene IS. The multilevel compliance challenge: recommendations for a call to action. A statement for healthcare professionals. Circulation 1997; 95:1085–1090.
- Sackett DL, Snow JC. The magnitude of compliance and noncompliance. In:Haynes RB, Taylor DW, Sackett DL, eds. Compliance in Health Care. Baltimore, MD: John Hopkins University Press; 1979:11–22.
- Shea S, Misra D, Ehrlich MH, Field L, Francis CK. Correlates of nonadherence to hypertension treatment in an inner-city minority population. Am J Public Health 1992; 82:1607–1612.
- Kirscht JP, Rosenstock IM. Patient adherence to antihypertensive medical regimens. J Community Health. 1977; 3:115–124.
- Hershey JC, Morton BG, Davis JB, Reichgott MJ. Patient compliance with antihypertensive medication. Am J Public Health 1980; 70:1081–1089.
- Bosworth HB, Powers B, Grubber JM, et al. Racial differences in blood pressure control: potential explanatory factors. J Gen Intern Med 2008; 23:692–698.
- Douglas JG, Ferdinand KC, Bakris GL, Sowers JR. Barriers to blood pressure control in African Americans. Overcoming obstacles is challenging, but target goals can be attained. Postgrad Med 2002; 112:51–52,55,59–62passim.
- Knight EL, Bohn RL, Wang PS, Glynn RJ, Mogun H, Avorn J. Predictors of uncontrolled hypertension in ambulatory patients. Hypertension 2001; 38:809–814.
- Ogedegbe G, Mancuso CA, Allegrante JP. Expectations of blood pressure management in hypertensive African-American patients: a qualitative study. J Natl Med Assoc 2004; 96:442–449.
- Blumhagen D. Hypertension: a folk illness with a medical name. Cult Med Psychiatry 1980; 4:197–224.
- Meyer D, Leventhal H, Gutmann M. Common-sense models of illness: the example of hypertension. Health Psychol 1985; 4:115–135.
- Nelson EC, Stason WB, Neutra RR, Solomon HS, McArdle PJ. Impact of patient perceptions on compliance with treatment for hypertension. Med Care 1978; 16:893–906.
- Heurtin-Roberts S. ‘High-pertension’—the uses of a chronic folk illness for personal adaptation. Soc Sci Med 1993; 37:285–294.
- Lang T. Social and economic factors as obstacles to blood pressure control. Am J Hypertens 1998; 11:900–902.
- Lang T. Factors that appear as obstacles to the control of high blood pressure. Ethn Dis 2000; 10:125–130.
- Stamler R, Shipley M, Elliott P, Dyer A, Sans S, Stamler J. Higher blood pressure in adults with less education. Some explanations from INTERSALT. Hypertension 1992; 19:237–241.
- Heurtin-Roberts S, Reisin E. The relation of culturally influenced lay models of hypertension to compliance with treatment. Am J Hypertens 1992; 5:787–792.
- Snow LF. Folk medical beliefs and their implications for care of patients. A review bases on studies among black Americans. Ann Intern Med 1974; 81:82–96.
- Hicks LS, Shaykevich S, Bates DW, Ayanian JZ. Determinants of racial/ethnic differences in blood pressure management among hypertensive patients. BMC Cardiovasc Disord 2005; 5:16.
- Mehta SS, Wilcox CS, Schulman KA. Treatment of hypertension in patients with comorbidities: results from the study of hypertensive prescribing practices (SHyPP). Am J Hypertens 1999; 12:333–340.
- Ballard DJ, Strogatz DS, Wagner EH, et al. Hypertension control in a rural southern community: medical care process and dropping out. Am J Prev Med 1988; 4:133–139.
- Hajjar I, Miller K, Hirth V. Age-related bias in the management of hypertension: a national survey of physicians’ opinions on hypertension in elderly adults. J Gerontol A Biol Sci Med Sci 2002; 57:M487–M491.
- McAlister FA, Laupacis A, Teo KK, Hamilton PG, Montague TJ. A survey of clinician attitudes and management practices in hypertension. J Hum Hypertens 1997; 11:413–419.
- Trilling JS, Froom J. The urgent need to improve hypertension care. Arch Fam Med 2000; 9:794–801.
- Huse DM, Roht LH, Alpert JS, Hartz SC. Physicians’ knowledge, attitudes, and practice of pharmacologic treatment of hypertension. Ann Pharmacother 2001; 35:1173–1179.
- Cabana MD, Rand CS, Powe NR, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA 1999; 282:1458–1465.
- Hyman DJ, Pavlik VN, Vallbona C. Physician role in lack of awareness and control of hypertension. J Clin Hypertens (Greenwich) 2000; 2:324–330.
- Morley Kotchen J, Walker WE, Kotchen TA. Rationale for a community approach to hypertension control among inner city minority populations. Heart Dis Stroke 1994; 3:61–62.
- Hyman DJ, Pavlik VN. Characteristics of patients with uncontrolled hypertension in the United States. N Engl J Med 2001; 345:479–486.
- Lang T. Factors that appear as obstacles to the control of high blood pressure. Ethn Dis 2000; 10:125–130.
- Pierce JP, Watson DS, Knights S, Gliddon T, Williams S, Watson R. A controlled trial of health education in the physician’s office. Prev Med 1984; 13:185–194.
- Morisky DE, DeMuth NM, Field-Fass M, Green LW, Levine DM. Evaluation of family health education to build social support for long-term control of high blood pressure. Health Educ Q 1985; 12:35–50.
- Lorgelly P, Siatis I, Brooks A, et al. Is ambulatory blood pressure monitoring cost-effective in the routine surveillance of treated hypertensive patients in primary care? Br J Gen Pract 2003; 53:794–796.
- Green LW, Levine DM, Wolle J, Deeds S. Development of randomized patient education experiments with urban poor hypertensives. Patient Couns Health Educ 1979; 1:106–111.
- Gruesser M, Hartmann P, Schlottmann N, Lohmann FW, Sawicki PT, Joergens V. Structured patient education for out-patients with hypertension in general practice: a model project in Germany. J Hum Hypertens 1997; 11:501–506.
- Mühlhauser I, Sawicki PT, Didjurgeit U, Jörgens V, Trampisch HJ, Berger M. Evaluation of a structured treatment and teaching programme on hypertension in general practice. Clin Exp Hypertens 1993; 15:125–142.
- Roca B, Nadal E, Rovira RE, Valls S, Lapuebla C, Lloría N. Usefulness of a hypertension education program. South Med J 2003; 96:1133–1137.
- Pickering TG, Gerin W, Holland JK. Home blood pressure teletransmission for better diagnosis and treatment. Curr Hypertens Rep 1999; 1:489–494.
- Yarows SA, Julius S, Pickering TG. Home blood pressure monitoring. Arch Intern Med 2000; 160:1251–1257.
- Haynes RB, Sackett DL, Gibson ES, et al. Improvement of medication compliance in uncontrolled hypertension. Lancet 1976; 1:1265–1268.
- Johnson AL, Taylor DW, Sackett DL, Dunnett CW, Shimizu AG. Self-recording of blood pressure in the management of hypertension. Can Med Assoc J 1978; 119:1034–1039.
- Carnahan JE, Nugent CA. The effects of self-monitoring by patients on the control of hypertension. Am J Med Sci 1975; 269:69–73.
- Stahl SM, Kelley CR, Neill PJ, Grim CE, Mamlin J. Effects of home blood pressure measurement on long-term BP control. Am J Public Health 1984; 74:704–709.
- Boulware LE, Daumit GL, Frick KD, Minkovitz CS, Lawrence RS, Powe NR. An evidence-based review of patient-centered behavioral interventions for hypertension. Am J Prev Med 2001; 21:221–232.
- Haynes RB, Mattson ME, Engebretson TO. Patient compliance to prescribed antihypertensive medication regimens: a report to the National Heart, Lung, and Blood institute. Bethesda, MD: US Department of Health and Human Services, Public Health Service, National Institutes of Health, 1980. NIH publication 81-2102.
- Burke LE, Dunbar-Jacob JM, Hill MN. Compliance with cardiovascular disease prevention strategies: a review of the research. Ann Behav Med 1997; 19:239–263.
- Dunbar-Jacob J, Dwyer K, Dunning EJ. Compliance with antihypertensive regimen: a review of the research in the 1980s. Ann Behav Med 1991; 13:31–39.
- Haynes RB, Montague P, Oliver T, McKibbon KA, Brouwers MC, Kanani R. Interventions for helping patients to follow prescriptions for medications. Cochrane Database Syst Rev 2000; ( 2):CD000011.
- Roter DL, Hall JA, Merisca R, Nordstrom B, Cretin D, Svarstad B. Effectiveness of interventions to improve patient compliance: a meta-analysis. Med Care 1998; 36:1138–1161.
- Appel LJ, Espeland MA, Easter L, Wilson AC, Folmar S, Lacy CR. Effects of reduced sodium intake on hypertension control in older individuals: results from the Trial of Nonpharmacologic Interventions in the Elderly (TONE). Arch Intern Med 2001; 161:685–693.
- Appel LJ, Champagne CM, Harsha DW, et al; Writing Group of the PREMIER Collaborative Research Group. Effects of comprehensive lifestyle modification on blood pressure control: main results of the PREMIER clinical trial. JAMA 2003; 289:2083–2093.
- Appel LJ, Moore TJ, Obarzanek E, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med 1997; 336:1117–1124.
- Moore TJ, Conlin PR, Ard J, Svetkey LP. DASH (Dietary Approaches to Stop Hypertension) diet is effective treatment for stage 1 isolated systolic hypertension. Hypertension 2001; 38:155–158.
- Stevens VJ, Obarzanek E, Cook NR, et al; Trials for the Hypertension Prevention Research Group. Long-term weight loss and changes in blood pressure: results of the Trials of Hypertension Prevention, phase II. Ann Intern Med 2001; 134:1–11.
- Sawicki PT, Mühlhauser I, Didjurgeit U, Berger M. Improvement of hypertension care by a structured treatment and teaching programme. J Hum Hypertens 1993; 7:571–573.
- Morisky DE, Bowler MH, Finlay JS. An educational and behavioral approach toward increasing patient activation in hypertension management. J Community Health 1982; 7:171–182.
- Levine DM, Green LW, Deeds SG, Chwalow J, Russell RP, Finlay J. Health education for hypertensive patients. JAMA 1979; 241:1700–1703.
- Iso H, Shimamoto T, Yokota K, Sankai T, Jacobs DR, Komachi Y. Community-based education classes for hypertension control. A 1.5-year randomized controlled trial. Hypertension 1996; 27:968–974.
- Cuspidi C, Sampieri L, Macca G, et al. Improvement of patients’ knowledge by a single educational meeting on hypertension. J Hum Hypertens 2001; 15:57–61.
- Nessman DG, Carnahan JE, Nugent CA. Increasing compliance. Patient-operated hypertension groups. Arch Intern Med 1980; 140:1427–1430.
- Casasanta L, Patel S. Outcomes of an educational component of a disease management program for hypertension. Manag Care Interface 1999; 12:70–73.
- McAdams DP. The Stories We Live By: Personal Myths and the Making of the Self. New York NY: The Guilford Press; 1993.
- Bruner J. Acts of Meaning. Cambridge, MA: Harvard Univ Pr; 1990.
- Slater MD, Rouner D. Entertainment—education and elaboration likelihood: Understanding the processing of narrative persuasion. Commun Theory 2002; 12:173–191.
- Dal CS, Zanna MP, Fong GT. Narrative persuasion and overcoming resistance. In:Knowles ES, Linn J, eds. Resistance and Persuasion. Mahwah, NJ: Lawrence Erlbaum Assoc; 2004:175–191.
- Artinian NT, Washington OG, Templin TN. Effects of home telemonitoring and community-based monitoring on blood pressure control in urban African Americans: a pilot study. Heart Lung 2001; 30:191–199.
- Bailey B, Carney SL, Gillies AA, Smith AJ. Antihypertensive drug treatment: a comparison of usual care with self blood pressure measurement. J Hum Hypertens 1999; 13:147–150.
- Bondmass M. The effect of home monitoring and telemanagement on blood pressure control among African Americans. Telemed J 2000; 6:15–23.
- Rogers MA, Small D, Buchan DA, et al. Home monitoring service improves mean arterial pressure in patients with essential hypertension. A randomized, controlled trial. Ann Intern Med 2001; 134:1024–1032.
- Mengden T, Uen S, Baulmann J, Vetter H. Significance of blood pressure self-measurement as compared with office blood pressure measurement and ambulatory 24-hour blood pressure measurement in pharmacological studies. Blood Press Monit 2003; 8:169–172.
- Friedman RH, Kazis LE, Jette A, et al. A telecommunications system for monitoring and counseling patients with hypertension. Impact on medication adherence and blood pressure control. Am J Hypertens 1996; 9:285–292.
- Oxman AD, Thomson MA, Davis DA, Haynes RB. No magic bullets: a systematic review of 102 trials of interventions to improve professional practice. CMAJ 1995; 153:1423–1431.
- Wensing M, van der Weijden T, Grol R. Implementing guidelines and innovations in general practice: which interventions are effective? Br J Gen Pract 1998; 48:991–997.
- Davis DA, Thomson MA, Oxman AD, Haynes RB. Changing physician performance. A systematic review of the effect of continuing medical education strategies. JAMA 1995; 274:700–705.
- Inui TS, Yourtee EL, Williamson JW. Improved outcomes in hypertension after physician tutorials. A controlled trial. Ann Intern Med 1976; 84:646–651.
- Jennett PA, Wilson TW, Hayton RC, Mainprize GW, Laxdal OE. Desirable behaviours in the office management of hypertension addressed through continuing medical education. Can J Public Health 1989; 80:359–362.
- Maue SK, Rivo ML, Weiss B, Farrelly EW, Brower-Stenger S. Effect of a primary care physician-focused, population-based approach to blood pressure control. Fam Med 2002; 34:508–513.
- Tu K, Davis D. Can we alter physician behavior by educational methods? Lessons learned from studies of the management and follow-up of hypertension. J Contin Educ Health Prof 2002; 22:11–22.
- McAlister NH, Covvey HD, Tong C, Lee A, Wigle ED. Randomised controlled trial of computer assisted management of hypertension in primary care. Br Med J (Clin Res Ed) 1986; 293:670–674.
- Mashru M, Lant A. Interpractice audit of diagnosis and management of hypertension in primary care: educational intervention and review of medical records. BMJ 1997; 314:942–946.
- Degoulet P, Menard J, Berger C, Plouin PF, Devries C, Hirel JC. Hypertension management: the computer as a participant. Am J Med 1980; 68:559–567.
- Dickinson JC, Warshaw GA, Gehlbach SH, Bobula JA, Muhlbaier LH, Parkerson GR. Improving hypertension control: impact of computer feedback and physician education. Med Care 1981; 19:843–854.
- Montgomery AA, Fahey T. A systematic review of the use of computers in the management of hypertension. J Epidemiol Community Health 1998; 52:520–525.
- Coleman MT, Lott JA, Sharma S. Use of continuous quality improvement to identify barriers in the management of hypertension. Am J Med Qual 2000; 15:72–77.
- Goldberg HI, Wagner EH, Fihn SD, et al. A randomized controlled trial of CQI teams and academic detailing: can they alter compliance with guidelines? Jt Comm J Qual Improv 1998; 24:130–142.
- Horowitz CR, Goldberg HI, Martin DP, et al. Conducting a randomized controlled trial of CQI and academic detailing to implement clinical guidelines. Jt Comm J Qual Improv 1996; 22:734–750.
- Johnson B, McNair D, Kailasam K, et al. Discern—an integrated prospective decision support system. Proc Annu Symp Comput Appl Med Care 1994; 969.
- Bosworth HB, Olsen MK, Dudley T, et al. Patient education and provider decision support to control blood pressure in primary care: a cluster randomized trial. Am Heart J 2009; 157:450–456.
- Walsh JM, McDonald KM, Shojania KG, et al. Quality improvement strategies for hypertension management: a systematic review. Med Care 2006; 44:646–657.
- Carter BL, Rogers M, Daly J, Zheng S, James PA. The potency of team-based care interventions for hypertension: a meta-analysis. Arch Intern Med 2009; 169:1748–1755.
- Carter BL, Clarke W, Ardery G, et al; Collaboration Among Pharmacists Physicians To Improve Outcomes Now (CAPTION) Trial Investigators. A cluster-randomized effectiveness trial of a physician-pharmacist collaborative model to improve blood pressure control. Circ Cardiovasc Qual Outcomes 2010; 3:418–423.
- Einhorn PT. National heart, lung, and blood institute-initiated program “interventions to improve hypertension control rates in African Americans”: background and implementation. Circ Cardiovasc Qual Outcomes 2009; 2:236–240.
- Agarwal R, Bills JE, Hecht TJ, Light RP. Role of home blood pressure monitoring in overcoming therapeutic inertia and improving hypertension control: a systematic review and meta-analysis. Hypertension 2011; 57:29–38.
- Bosworth HB, Olsen MK, Neary A, et al. Take Control of Your Blood Pressure (TCYB) study: a multifactorial tailored behavioral and educational intervention for achieving blood pressure control. Patient Educ Couns 2008; 70:338–347.
- Bosworth HB, Olsen MK, Goldstein MK, et al. The veterans’ study to improve the control of hypertension (V-STITCH): design and methodology. Contemp Clin Trials 2005; 26:155–168.
- Ogedegbe G, Tobin JN, Fernandez S, et al. Counseling African Americans to Control Hypertension (CAATCH) trial: a multi-level intervention to improve blood pressure control in hypertensive blacks. Circ Cardiovasc Qual Outcomes 2009; 2:249–256.
- Bosworth HB, Almirall D, Weiner BJ, et al. The implementation of a translational study involving a primary care based behavioral program to improve blood pressure control: The HTN-IMPROVE study protocol (01295). Implement Sci 2010; 5:54.
High blood pressure takes a devastating toll on African Americans. Better control can go a long way to closing the “mortality gap” between African Americans and white Americans. But which strategies are best to address this complex problem?
In this report, we review the evidence on practice-based approaches to improving blood pressure control, from new styles of patient education to home blood pressure monitoring, focusing on studies in African Americans (Table 1).1–11
BETTER CONTROL IS NEEDED
PATIENT-RELATED BARRIERS
Patient-related barriers24–40 include:
- Poor knowledge about hypertension and its consequences31,32
- Poor adherence to drug therapy (a major factor,24–26 as African Americans have poorer adherence rates than whites,27–29 which may explain some of the racial disparity in blood pressure control30)
- False health beliefs34–37
- Inability to change one’s lifestyle
- Side effects of antihypertensive drugs32
- Unrealistic expectations of treatment (eg, a cure33)
- Demographic factors (eg, socioeconomic status, educational level, age, sex).24,38–40
Perhaps the most salient and easily modifiable of these factors are patients’ reluctance to modify their lifestyle and their misconceptions about the causes, treatment, and prevention of hypertension. Patients whose beliefs are discordant with traditional biomedical concepts of hypertension have poorer blood pressure control than those whose beliefs are concordant.41 This may be more relevant to African Americans, since they are known to have cultural health beliefs that differ from those of Western culture (eg, that hypertension is a curable rather than a chronic illness, and that hypertension is a disease of nerves that often affects the blood and clogs the arteries).42
PHYSICIAN-RELATED BARRIERS
Barriers to effective blood pressure control at the physician level43–48 include:
- Nonadherence to treatment guidelines44
- Failure to intensify the regimen if goals are not met45
- Failure to emphasize therapeutic lifestyle changes.43,46–48
When primary care physicians do not follow evidence-based guidelines, the reason may be that they are not aware of them or that they do not understand them. In a national survey of 1,029 physicians that was designed to explore how well physicians know the indications for specific antihypertensive drugs and how closely their opinions and practice agreed with national guidelines, only 37.3% correctly answered all of the knowledge-related questions.49
Other reasons for nonadherence are that physicians may disagree with the guidelines, may not be able to follow the guidelines, may not believe that following them will achieve the desired effect, or may have no motivation to change their practice.50
Whatever the reason, Hyman et al51 reported that as many as 30% of physicians did not recommend treatment for patients with diastolic blood pressures of 90 to 100 mm Hg, and a higher proportion did not treat patients with systolic blood pressures of 140 to 160 mm Hg.
BARRIERS IN HEALTH CARE SYSTEMS
Although health care systems present barriers to optimal blood pressure control,20,27,31,52 there is evidence that most cases of uncontrolled hypertension occur in patients with good access to care.32,53,54 For example, an NHANES study53 suggested that most patients with uncontrolled hypertension had in fact seen a physician on average at least three times in the previous year. And this may be more pervasive in African Americans: one survey found hypertension was uncontrolled in 75% of hypertensive African American patients despite free access to care, free medications, and regular follow-up visits.41
Thus, the most significant barriers to blood pressure control appear to be patient-related and physician-related.
INTERVENTIONS AIMED AT PATIENTS
The most common approaches to improving blood pressure control at the patient level, regardless of race, are patient education,55–61 home blood pressure monitoring,62–67 and behavioral counseling to address misconceptions about hypertension,68 to improve adherence to drug therapy,69–73 and to encourage lifestyle modifications.74–78
Patient education
Patient education can improve blood pressure control.58,79–82 Its aims are to increase patients’ understanding of the disease83 and to encourage them to be more active in their own care.80,84,85
Patient education has a moderate effect on blood pressure control. The average proportion of patients whose hypertension was under control in community-based trials of various interventions ranged from 60% to 70%, compared with 38% to 46% with usual care.56,80,81
However, these strategies largely did not address misconceptions patients have about hypertension. This issue is especially critical in African Americans, who may have different perceptions of hypertension and different expectations for care41: beliefs that hypertension is “curable,” not chronic, and that medication is needed only for hypertension-related symptoms may translate to poorer rates of medication adherence.
Levine et al1 evaluated the efficacy of home visits by trained community health advisory board workers in a neighborhood in Baltimore, MD, with a high prevalence of hypertension. Participants were randomized to receive either one visit or five visits during the 40-month study period. Both groups had a statistically significant reduction in blood pressure, and in both groups the proportion of patients with adequate blood pressure control increased significantly. The results support the use of a practice- and community-based partnership to improve blood pressure control in African American patients.
Ogedegbe et al2 randomized 190 hypertensive African American patients to receive usual care or quarterly counseling sessions that used motivational interviewing focused on medication adherence. The counseled patients stayed adherent to their medications, whereas adherence declined significantly in those receiving usual care. This effect was associated with a modest, nonsignificant trend toward a net reduction in systolic blood pressure with motivational interviewing.
A novel method of health education is the use of narrative communication—ie, storytelling. It has a good amount of evidence to support it, as culturally appropriate storytelling may allow patients to identify with a story as it relates to their own lives.86–89 Examples of educational storytelling include:
- A woman with hypertension discussing what it means to have high blood pressure, and the benefits of controlling it, such as living long enough to see her grandchildren grow up
- A man discussing the importance of involving family and friends to help control blood pressure, and how dietary modifications can be made to ensure that salt alternatives are used when the family does the cooking.
Storytelling should be done in a culturally appropriate context. For example, storytellers should have the same background as the patient (ie, similar socioeconomic status and ethnic background): patients are more likely to be influenced if they identify with the storyteller and imagine themselves in a similar situation.
Houston et al3 randomized 299 hypertensive African Americans to view either three DVDs that featured patients with hypertension or three “attention-control DVDs” on topics not related to hypertension. The intervention group’s DVDs focused on storytelling and “learning more.” In the storytelling section, patients told personal stories about what it meant to have hypertension and gave advice on how to best interact with health care providers and methods to improve medication adherence. A “learning more” section focused on what high blood pressure is, addressed therapeutic lifestyle changes, and encouraged patients to communicate with their health care providers. The patients who viewed the patient narratives had significantly lower blood pressure at 3 months than those assigned to usual care. Although blood pressure subsequently increased in both groups, the benefits of the intervention still existed at the end of follow-up.
Important to note about two of the above three studies1,3 is that the interventions were done by people other than physicians, thus emphasizing the importance of a team approach to blood pressure control.
Behavioral counseling
The effectiveness of lifestyle modifications such as diet, weight loss, and physical activity in preventing and treating hypertension is well established.74–78 For example:
- In the Dietary Approaches to Stop Hypertension (DASH) trial,76 a healthy diet lowered blood pressure about as much as single drugs do, particularly in African Americans.
- The Trial of Nonpharmacologic Interventions in the Elderly (TONE)74 showed that exercise can lower blood pressure in obese hypertensive patients.
- The PREMIER trial (Lifestyle Interventions for Blood Pressure Control)75 showed that a single brief counseling session could produce substantial decreases in blood pressure in patients with stage 1 hypertension or high-normal blood pressure.
Unfortunately, these results have been hard to translate into primary care practice, especially for African American patients. Several studies have evaluated the impact of lifestyle interventions on blood pressure control in primary care practices with a large population of African American patients.
Bosworth et al,4 in a study of a practice in which almost half the patients were African American, randomized patients to receive usual care, nurse-administered tailored behavioral telephone counseling, home blood pressure monitoring, or home monitoring plus tailored behavioral telephone counseling. The combination of home monitoring and tailored behavioral telephone counseling led to a statistically significant improvement at 24 months compared with baseline.
Home blood pressure monitoring
The effectiveness of self-monitoring in improving blood pressure control is also well documented.62,63,65–67,90–95
Pickering et al62 studied patients with poorly controlled hypertension in a managed-care setting and found a reduction of 7 mm Hg systolic and 5 mm Hg diastolic pressure after 3 to 6 months of home monitoring compared with usual care.
Mengden et al,94 in a similar study, found average blood pressure reductions at 6 months of 19.3/11.9 mm Hg in the home-monitoring group vs 10.6/8.8 mm Hg in the usual-care group.
The effect of home blood pressure monitoring may be greater in African Americans.
Rogers et al93 found it to be more effective at lowering blood pressure than usual care in a group of 121 patients with poorly controlled hypertension followed in primary care practices, and these reductions were twice as large in African American patients than in white patients.93
Bondmass,92 in a study of 33 African American patients with poorly controlled hypertension, reported a 53% control rate within 4 weeks of home monitoring. All patients in the study had uncontrolled blood pressure at baseline (> 140/90 mm Hg).
Artinian et al5 evaluated the effect of nurse-managed telemonitoring on blood pressure control vs enhanced usual care. All participants were African American. The monitored group had a significantly greater reduction in systolic pressure at 12 months compared with those who received enhanced usual care.
PHYSICIAN-LEVEL INTERVENTIONS
Most interventions to improve how physicians manage patients with hypertension are designed to improve adherence to treatment guidelines. In most cases, these interventions are based on continuous quality improvement and disease management concepts such as physician education and academic detailing, reminders, feedback on performance measures, and risk-assessment tools.96,97
Physician education
Interest is increasing in physician educational interventions for blood pressure control.24,98
Inui et al,99 in an early study in a primary care practice, found that patients of physicians who received tutorials on hypertension management were more compliant with their drug regimens and had better blood pressure control than patients of physicians in the control group.
Jennett et al,100 in a similar randomized clinical trial, found that physicians who participated in an education activity were more adherent to treatment guidelines at 6 and 12 months compared with those who did not participate.
Maue et al101 showed that rates of blood pressure control improved from 41% to 52% after a 6-month educational intervention for physicians in a managed-care setting.
Tu et al102 reviewed 12 studies in which seven different physician educational interventions were used either alone or in combination and concluded that physician education improves compliance with guidelines for managing hypertension.
Unfortunately, these studies did not report outcomes separately for African American and white patients.
Hicks et al6 found that disease management approaches that target physicians whose patients with hypertension are mostly African American did not yield clinically relevant improvement in these patients, and that minority patients were significantly less likely to have their blood pressure controlled at the end of the study compared with their non-Hispanic white counterparts.
Feedback to providers
Several studies have shown that, given reminders and feedback systems, physicians will change their practice.103–106
Mashru and Lant104 combined chart audits and physician education in primary care practices and found they improved physician performance measures such as accuracy of diagnosis, number of patients who received cardiovascular risk assessment, and number of patients whose treatment was based on clinical laboratory assessments.
Feedback takes many forms but consists mostly of computerized information107 or peer-to-peer academic detailing with opinion leaders.108–110
Dickinson et al,106 for instance, showed that computer-generated listings of patients’ blood pressures combined with a physician education program on clinical management of hypertension led to increased knowledge and better follow-up on their patients.
Again, however, these studies did not distinguish between African American and white patients, which makes it difficult to judge whether or not these approaches work differently for physicians with a large proportion of African American patients.
Computerized decision-support systems
Computerized decision-support systems have proliferated in primary care practices.111
McAlister et al103 found that general practitioners randomized to manage hypertension with the assistance of a computer obtained better outcomes than with usual care.
Montgomery and Fahey,107 in a systematic review, found improved blood pressure control in two of the three trials that compared computer-generated feedback reports and reminders to usual care. Specifically, 51% of patients whose physicians received reminders either had controlled blood pressure or were at least receiving treatment vs 33% in the control group at 12 months. This difference was even higher at 24 months.
Montgomery et al7 later randomized primary care practices to use a computer-based decision-support system and a cardiovascular risk chart, the risk chart alone, or to continue as usual. Results indicated no reduction in cardiovascular risk in the computer-system or the chart-only group, whereas patients in the chart-only group had a significant reduction in systolic pressure and were prescribed more cardiovascular drugs. This study indicates that use of a computerized decision-support system is not superior to chart review and audit feedback alone.
Evidence that computerized decision systems improve blood pressure control in African Americans is scant. However, when one looks at the evidence from studies of African Americans, the outcomes do not seem to differ between African American and white patients.
Hicks et al6 examined the effectiveness of computerized decision support in improving hypertension care in a racially diverse population. Physicians were randomized to receive computerized decision support or to provide usual care without computerized support. Both groups improved significantly in prescribing appropriate drugs but not in overall blood pressure control. Furthermore, the study showed no reduction in racial disparities of care and blood pressure control.
A potential explanation for the lack of improvement in blood pressure was that the intervention dealt with making sure the appropriate drugs were prescribed rather than making sure physicians also appropriately intensified antihypertensive management when necessary.
INTERVENTIONS TARGETING PATIENTS AND PHYSICIANS
Several studies have targeted both patient and physician-level barriers to blood pressure control in practice-based settings.
Roumie et al8 randomized physicians to one of three intervention groups:
- “Provider education” consisting of an email message with a Web-based link to the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC-7)
- Provider education plus a computer alert with information about their patient’s blood pressure
- Provider education, a computer alert, and patient education (ie, patients received a letter encouraging adherence to drug therapy, changing their lifestyle, and talking with their doctor about their blood pressure).
Patients whose providers were randomized to the third group had better blood pressure control. The report did not differentiate African American vs white patients. The data, however, did show the effectiveness of adding patient education to provider education to improve blood pressure control.
Bosworth et al,112 in a study in which 40% of patients were African American, randomized patients to usual care or to bimonthly nurse-delivered behavioral telephone counseling. They also randomized providers either to receive computer-generated decision support designed to improve adherence to guidelines or to receive no support.
There were no significant differences in rates of blood pressure control in the intervention groups compared with a control group. Although differences in blood pressure control between groups were not significant, patients randomized to behavioral intervention had significantly better blood pressure control at the 24-month follow-up than at baseline.
Svetkey et al9 evaluated the effects of physician intervention, patient intervention, and physician intervention plus patient intervention compared with control on systolic blood pressure at 6 months. They found that an intensive behavioral lifestyle intervention led to a significant reduction in systolic pressure at 6 months. By itself, the physician intervention did not have a meaningful effect, but patients in the combined physician-and-patient-intervention group experienced the greatest reduction (9.7 ± 12.7 mm Hg).
It takes a team
Physicians should not be the only focus in helping patients achieve blood pressure control. Although physician and patient factors need to be addressed to improve blood pressure control in African Americans, emphasis should also be placed on interdisciplinary, team-based care utilizing health care providers such as nurses, physician assistants, and pharmacists. Team-based care has been shown to have the greatest impact of all the strategies for improving blood pressure control.113 There is a good amount of evidence involving interventions with a focus on health care providers other than physicians, although the data lack a sufficient focus on African Americans.
Carter et al,10 in a randomized controlled trial in which 26.3% of the patients were African American, found that an intervention consisting of clinical pharmacists giving physicians drug therapy recommendations based on national guidelines resulted in a significantly lower blood pressure compared with a control group: the mean reduction was 20.7/9.7 mm Hg in the intervention group vs 6.8/4.5 mm Hg in the control group.
Carter et al114 performed a meta-analysis of 37 studies and found that two strategies led to a significant reduction in blood pressure: a pharmacist-led intervention with treatment recommendations to physicians resulted in a systolic pressure reduction of 9.30 mm Hg; and nurse-led interventions resulted in a systolic pressure reduction of 4.80 mm Hg. Again, many of the studies cited in this meta-analysis lacked a focus on African Americans.
Hunt et al11 conducted a randomized controlled trial in which pharmacists actively participated in the management of blood pressure. They were involved with every aspect of care, including reviewing medications and adverse drug reactions, assessing lifestyle behaviors and barriers to adherence, making dosing adjustments, and adding medications. Patients randomized to the intervention group achieved significantly lower systolic and diastolic pressures (137/75 vs 143/78 mm Hg in the control group). However, information about race was not included.
The above studies are just a few out of a large body of evidence demonstrating the value of team-based care to improve blood pressure control. It has yet to be determined whether these models can improve blood pressure control specifically in African Americans, since so many of these trials lacked a focus on this group. Promising is an ongoing randomized prospective trial by Carter et al115 evaluating a model of collaboration between physicians and pharmacists, with a focus on patients in underrepresented minorities.
SO WHAT WORKS?
Although there is a growing body of literature on interventions to try to reduce disparities in hypertension and blood pressure control between African Americans and whites, only a few randomized controlled trials have focused on African Americans, and several have not reported their results.116 So the question remains: How should we interpret the available data, which are aggregated across racial groups, and put it into practice when caring for hypertensive African American patients?
Patient education. In trying to overcome patient-related barriers, emphasis should be on patient education, in particular addressing misconceptions about hypertension and promoting adherence to antihypertensive therapy. This is evident from the narrative storytelling intervention by Houston et al.3 Although this is the first study of its kind, this strategy may be something to consider if future studies replicate these findings. Culturally appropriate storytelling may allow patients to identify with the stories as they relate to their own personal lives. It can be an effective way to address patient education and change behaviors.
Self-monitoring with a home blood pressure monitor has also proven effective in the management of hypertension in African Americans. Indeed, the few studies that reported findings in African Americans showed impressive reductions in blood pressure. The benefits of home monitoring are well documented, and the effect on physician-related barriers such as clinical inertia are also quite impressive.117 However, most of these studies did not assess the long-term impact or cost-effectiveness of home monitoring on blood pressure control.
Behavioral counseling. Although we have good evidence of the effectiveness of behavioral counseling, whether this is sustained long-term has been less studied in African Americans. Thus, while interventions that targeted African Americans have reported impressive reductions in blood pressure, the effect tends to be greatest during the first few months of implementation, with the benefits disappearing over time.
Physician-related interventions. With regard to physician-level interventions, research has focused on physician education, utilizing alerts and computerized clinical decision-support systems. Evidence is scant on whether the use of computerized systems results in improves hypertension care in African Americans. However, a closer look at the data from studies that report outcomes in African American and white patients shows that the results do not seem to differ between these groups. Still, there is insufficient information about the impact on hypertensive African Americans.6
Strategies that address both patient- and physician-related barriers can improve overall blood pressure control; however, there is a lack of data comparing outcomes in hypertensive African Americans with those of whites, making it difficult to know if this would be an effective strategy in African American patients alone.
More studies needed that focus on African Americans
Developing interventions to improve blood pressure control in African Americans should be an ongoing priority for research if we intend to address racial disparities in cardiovascular disease. Although it is reassuring that there is a growing body of evidence and research with this focus,118–121 more research is needed to determine effective strategies that address barriers related to physician practice and to the health care system overall as they relate to blood pressure control in African Americans. More importantly, these strategies should also emphasize a team-based approach that includes nurses, pharmacists, and physician assistants. Developing targeted interventions for hypertensive African Americans will help reduce disparities in the rates of cardiovascular illness and death in this patient population.
High blood pressure takes a devastating toll on African Americans. Better control can go a long way to closing the “mortality gap” between African Americans and white Americans. But which strategies are best to address this complex problem?
In this report, we review the evidence on practice-based approaches to improving blood pressure control, from new styles of patient education to home blood pressure monitoring, focusing on studies in African Americans (Table 1).1–11
BETTER CONTROL IS NEEDED
PATIENT-RELATED BARRIERS
Patient-related barriers24–40 include:
- Poor knowledge about hypertension and its consequences31,32
- Poor adherence to drug therapy (a major factor,24–26 as African Americans have poorer adherence rates than whites,27–29 which may explain some of the racial disparity in blood pressure control30)
- False health beliefs34–37
- Inability to change one’s lifestyle
- Side effects of antihypertensive drugs32
- Unrealistic expectations of treatment (eg, a cure33)
- Demographic factors (eg, socioeconomic status, educational level, age, sex).24,38–40
Perhaps the most salient and easily modifiable of these factors are patients’ reluctance to modify their lifestyle and their misconceptions about the causes, treatment, and prevention of hypertension. Patients whose beliefs are discordant with traditional biomedical concepts of hypertension have poorer blood pressure control than those whose beliefs are concordant.41 This may be more relevant to African Americans, since they are known to have cultural health beliefs that differ from those of Western culture (eg, that hypertension is a curable rather than a chronic illness, and that hypertension is a disease of nerves that often affects the blood and clogs the arteries).42
PHYSICIAN-RELATED BARRIERS
Barriers to effective blood pressure control at the physician level43–48 include:
- Nonadherence to treatment guidelines44
- Failure to intensify the regimen if goals are not met45
- Failure to emphasize therapeutic lifestyle changes.43,46–48
When primary care physicians do not follow evidence-based guidelines, the reason may be that they are not aware of them or that they do not understand them. In a national survey of 1,029 physicians that was designed to explore how well physicians know the indications for specific antihypertensive drugs and how closely their opinions and practice agreed with national guidelines, only 37.3% correctly answered all of the knowledge-related questions.49
Other reasons for nonadherence are that physicians may disagree with the guidelines, may not be able to follow the guidelines, may not believe that following them will achieve the desired effect, or may have no motivation to change their practice.50
Whatever the reason, Hyman et al51 reported that as many as 30% of physicians did not recommend treatment for patients with diastolic blood pressures of 90 to 100 mm Hg, and a higher proportion did not treat patients with systolic blood pressures of 140 to 160 mm Hg.
BARRIERS IN HEALTH CARE SYSTEMS
Although health care systems present barriers to optimal blood pressure control,20,27,31,52 there is evidence that most cases of uncontrolled hypertension occur in patients with good access to care.32,53,54 For example, an NHANES study53 suggested that most patients with uncontrolled hypertension had in fact seen a physician on average at least three times in the previous year. And this may be more pervasive in African Americans: one survey found hypertension was uncontrolled in 75% of hypertensive African American patients despite free access to care, free medications, and regular follow-up visits.41
Thus, the most significant barriers to blood pressure control appear to be patient-related and physician-related.
INTERVENTIONS AIMED AT PATIENTS
The most common approaches to improving blood pressure control at the patient level, regardless of race, are patient education,55–61 home blood pressure monitoring,62–67 and behavioral counseling to address misconceptions about hypertension,68 to improve adherence to drug therapy,69–73 and to encourage lifestyle modifications.74–78
Patient education
Patient education can improve blood pressure control.58,79–82 Its aims are to increase patients’ understanding of the disease83 and to encourage them to be more active in their own care.80,84,85
Patient education has a moderate effect on blood pressure control. The average proportion of patients whose hypertension was under control in community-based trials of various interventions ranged from 60% to 70%, compared with 38% to 46% with usual care.56,80,81
However, these strategies largely did not address misconceptions patients have about hypertension. This issue is especially critical in African Americans, who may have different perceptions of hypertension and different expectations for care41: beliefs that hypertension is “curable,” not chronic, and that medication is needed only for hypertension-related symptoms may translate to poorer rates of medication adherence.
Levine et al1 evaluated the efficacy of home visits by trained community health advisory board workers in a neighborhood in Baltimore, MD, with a high prevalence of hypertension. Participants were randomized to receive either one visit or five visits during the 40-month study period. Both groups had a statistically significant reduction in blood pressure, and in both groups the proportion of patients with adequate blood pressure control increased significantly. The results support the use of a practice- and community-based partnership to improve blood pressure control in African American patients.
Ogedegbe et al2 randomized 190 hypertensive African American patients to receive usual care or quarterly counseling sessions that used motivational interviewing focused on medication adherence. The counseled patients stayed adherent to their medications, whereas adherence declined significantly in those receiving usual care. This effect was associated with a modest, nonsignificant trend toward a net reduction in systolic blood pressure with motivational interviewing.
A novel method of health education is the use of narrative communication—ie, storytelling. It has a good amount of evidence to support it, as culturally appropriate storytelling may allow patients to identify with a story as it relates to their own lives.86–89 Examples of educational storytelling include:
- A woman with hypertension discussing what it means to have high blood pressure, and the benefits of controlling it, such as living long enough to see her grandchildren grow up
- A man discussing the importance of involving family and friends to help control blood pressure, and how dietary modifications can be made to ensure that salt alternatives are used when the family does the cooking.
Storytelling should be done in a culturally appropriate context. For example, storytellers should have the same background as the patient (ie, similar socioeconomic status and ethnic background): patients are more likely to be influenced if they identify with the storyteller and imagine themselves in a similar situation.
Houston et al3 randomized 299 hypertensive African Americans to view either three DVDs that featured patients with hypertension or three “attention-control DVDs” on topics not related to hypertension. The intervention group’s DVDs focused on storytelling and “learning more.” In the storytelling section, patients told personal stories about what it meant to have hypertension and gave advice on how to best interact with health care providers and methods to improve medication adherence. A “learning more” section focused on what high blood pressure is, addressed therapeutic lifestyle changes, and encouraged patients to communicate with their health care providers. The patients who viewed the patient narratives had significantly lower blood pressure at 3 months than those assigned to usual care. Although blood pressure subsequently increased in both groups, the benefits of the intervention still existed at the end of follow-up.
Important to note about two of the above three studies1,3 is that the interventions were done by people other than physicians, thus emphasizing the importance of a team approach to blood pressure control.
Behavioral counseling
The effectiveness of lifestyle modifications such as diet, weight loss, and physical activity in preventing and treating hypertension is well established.74–78 For example:
- In the Dietary Approaches to Stop Hypertension (DASH) trial,76 a healthy diet lowered blood pressure about as much as single drugs do, particularly in African Americans.
- The Trial of Nonpharmacologic Interventions in the Elderly (TONE)74 showed that exercise can lower blood pressure in obese hypertensive patients.
- The PREMIER trial (Lifestyle Interventions for Blood Pressure Control)75 showed that a single brief counseling session could produce substantial decreases in blood pressure in patients with stage 1 hypertension or high-normal blood pressure.
Unfortunately, these results have been hard to translate into primary care practice, especially for African American patients. Several studies have evaluated the impact of lifestyle interventions on blood pressure control in primary care practices with a large population of African American patients.
Bosworth et al,4 in a study of a practice in which almost half the patients were African American, randomized patients to receive usual care, nurse-administered tailored behavioral telephone counseling, home blood pressure monitoring, or home monitoring plus tailored behavioral telephone counseling. The combination of home monitoring and tailored behavioral telephone counseling led to a statistically significant improvement at 24 months compared with baseline.
Home blood pressure monitoring
The effectiveness of self-monitoring in improving blood pressure control is also well documented.62,63,65–67,90–95
Pickering et al62 studied patients with poorly controlled hypertension in a managed-care setting and found a reduction of 7 mm Hg systolic and 5 mm Hg diastolic pressure after 3 to 6 months of home monitoring compared with usual care.
Mengden et al,94 in a similar study, found average blood pressure reductions at 6 months of 19.3/11.9 mm Hg in the home-monitoring group vs 10.6/8.8 mm Hg in the usual-care group.
The effect of home blood pressure monitoring may be greater in African Americans.
Rogers et al93 found it to be more effective at lowering blood pressure than usual care in a group of 121 patients with poorly controlled hypertension followed in primary care practices, and these reductions were twice as large in African American patients than in white patients.93
Bondmass,92 in a study of 33 African American patients with poorly controlled hypertension, reported a 53% control rate within 4 weeks of home monitoring. All patients in the study had uncontrolled blood pressure at baseline (> 140/90 mm Hg).
Artinian et al5 evaluated the effect of nurse-managed telemonitoring on blood pressure control vs enhanced usual care. All participants were African American. The monitored group had a significantly greater reduction in systolic pressure at 12 months compared with those who received enhanced usual care.
PHYSICIAN-LEVEL INTERVENTIONS
Most interventions to improve how physicians manage patients with hypertension are designed to improve adherence to treatment guidelines. In most cases, these interventions are based on continuous quality improvement and disease management concepts such as physician education and academic detailing, reminders, feedback on performance measures, and risk-assessment tools.96,97
Physician education
Interest is increasing in physician educational interventions for blood pressure control.24,98
Inui et al,99 in an early study in a primary care practice, found that patients of physicians who received tutorials on hypertension management were more compliant with their drug regimens and had better blood pressure control than patients of physicians in the control group.
Jennett et al,100 in a similar randomized clinical trial, found that physicians who participated in an education activity were more adherent to treatment guidelines at 6 and 12 months compared with those who did not participate.
Maue et al101 showed that rates of blood pressure control improved from 41% to 52% after a 6-month educational intervention for physicians in a managed-care setting.
Tu et al102 reviewed 12 studies in which seven different physician educational interventions were used either alone or in combination and concluded that physician education improves compliance with guidelines for managing hypertension.
Unfortunately, these studies did not report outcomes separately for African American and white patients.
Hicks et al6 found that disease management approaches that target physicians whose patients with hypertension are mostly African American did not yield clinically relevant improvement in these patients, and that minority patients were significantly less likely to have their blood pressure controlled at the end of the study compared with their non-Hispanic white counterparts.
Feedback to providers
Several studies have shown that, given reminders and feedback systems, physicians will change their practice.103–106
Mashru and Lant104 combined chart audits and physician education in primary care practices and found they improved physician performance measures such as accuracy of diagnosis, number of patients who received cardiovascular risk assessment, and number of patients whose treatment was based on clinical laboratory assessments.
Feedback takes many forms but consists mostly of computerized information107 or peer-to-peer academic detailing with opinion leaders.108–110
Dickinson et al,106 for instance, showed that computer-generated listings of patients’ blood pressures combined with a physician education program on clinical management of hypertension led to increased knowledge and better follow-up on their patients.
Again, however, these studies did not distinguish between African American and white patients, which makes it difficult to judge whether or not these approaches work differently for physicians with a large proportion of African American patients.
Computerized decision-support systems
Computerized decision-support systems have proliferated in primary care practices.111
McAlister et al103 found that general practitioners randomized to manage hypertension with the assistance of a computer obtained better outcomes than with usual care.
Montgomery and Fahey,107 in a systematic review, found improved blood pressure control in two of the three trials that compared computer-generated feedback reports and reminders to usual care. Specifically, 51% of patients whose physicians received reminders either had controlled blood pressure or were at least receiving treatment vs 33% in the control group at 12 months. This difference was even higher at 24 months.
Montgomery et al7 later randomized primary care practices to use a computer-based decision-support system and a cardiovascular risk chart, the risk chart alone, or to continue as usual. Results indicated no reduction in cardiovascular risk in the computer-system or the chart-only group, whereas patients in the chart-only group had a significant reduction in systolic pressure and were prescribed more cardiovascular drugs. This study indicates that use of a computerized decision-support system is not superior to chart review and audit feedback alone.
Evidence that computerized decision systems improve blood pressure control in African Americans is scant. However, when one looks at the evidence from studies of African Americans, the outcomes do not seem to differ between African American and white patients.
Hicks et al6 examined the effectiveness of computerized decision support in improving hypertension care in a racially diverse population. Physicians were randomized to receive computerized decision support or to provide usual care without computerized support. Both groups improved significantly in prescribing appropriate drugs but not in overall blood pressure control. Furthermore, the study showed no reduction in racial disparities of care and blood pressure control.
A potential explanation for the lack of improvement in blood pressure was that the intervention dealt with making sure the appropriate drugs were prescribed rather than making sure physicians also appropriately intensified antihypertensive management when necessary.
INTERVENTIONS TARGETING PATIENTS AND PHYSICIANS
Several studies have targeted both patient and physician-level barriers to blood pressure control in practice-based settings.
Roumie et al8 randomized physicians to one of three intervention groups:
- “Provider education” consisting of an email message with a Web-based link to the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC-7)
- Provider education plus a computer alert with information about their patient’s blood pressure
- Provider education, a computer alert, and patient education (ie, patients received a letter encouraging adherence to drug therapy, changing their lifestyle, and talking with their doctor about their blood pressure).
Patients whose providers were randomized to the third group had better blood pressure control. The report did not differentiate African American vs white patients. The data, however, did show the effectiveness of adding patient education to provider education to improve blood pressure control.
Bosworth et al,112 in a study in which 40% of patients were African American, randomized patients to usual care or to bimonthly nurse-delivered behavioral telephone counseling. They also randomized providers either to receive computer-generated decision support designed to improve adherence to guidelines or to receive no support.
There were no significant differences in rates of blood pressure control in the intervention groups compared with a control group. Although differences in blood pressure control between groups were not significant, patients randomized to behavioral intervention had significantly better blood pressure control at the 24-month follow-up than at baseline.
Svetkey et al9 evaluated the effects of physician intervention, patient intervention, and physician intervention plus patient intervention compared with control on systolic blood pressure at 6 months. They found that an intensive behavioral lifestyle intervention led to a significant reduction in systolic pressure at 6 months. By itself, the physician intervention did not have a meaningful effect, but patients in the combined physician-and-patient-intervention group experienced the greatest reduction (9.7 ± 12.7 mm Hg).
It takes a team
Physicians should not be the only focus in helping patients achieve blood pressure control. Although physician and patient factors need to be addressed to improve blood pressure control in African Americans, emphasis should also be placed on interdisciplinary, team-based care utilizing health care providers such as nurses, physician assistants, and pharmacists. Team-based care has been shown to have the greatest impact of all the strategies for improving blood pressure control.113 There is a good amount of evidence involving interventions with a focus on health care providers other than physicians, although the data lack a sufficient focus on African Americans.
Carter et al,10 in a randomized controlled trial in which 26.3% of the patients were African American, found that an intervention consisting of clinical pharmacists giving physicians drug therapy recommendations based on national guidelines resulted in a significantly lower blood pressure compared with a control group: the mean reduction was 20.7/9.7 mm Hg in the intervention group vs 6.8/4.5 mm Hg in the control group.
Carter et al114 performed a meta-analysis of 37 studies and found that two strategies led to a significant reduction in blood pressure: a pharmacist-led intervention with treatment recommendations to physicians resulted in a systolic pressure reduction of 9.30 mm Hg; and nurse-led interventions resulted in a systolic pressure reduction of 4.80 mm Hg. Again, many of the studies cited in this meta-analysis lacked a focus on African Americans.
Hunt et al11 conducted a randomized controlled trial in which pharmacists actively participated in the management of blood pressure. They were involved with every aspect of care, including reviewing medications and adverse drug reactions, assessing lifestyle behaviors and barriers to adherence, making dosing adjustments, and adding medications. Patients randomized to the intervention group achieved significantly lower systolic and diastolic pressures (137/75 vs 143/78 mm Hg in the control group). However, information about race was not included.
The above studies are just a few out of a large body of evidence demonstrating the value of team-based care to improve blood pressure control. It has yet to be determined whether these models can improve blood pressure control specifically in African Americans, since so many of these trials lacked a focus on this group. Promising is an ongoing randomized prospective trial by Carter et al115 evaluating a model of collaboration between physicians and pharmacists, with a focus on patients in underrepresented minorities.
SO WHAT WORKS?
Although there is a growing body of literature on interventions to try to reduce disparities in hypertension and blood pressure control between African Americans and whites, only a few randomized controlled trials have focused on African Americans, and several have not reported their results.116 So the question remains: How should we interpret the available data, which are aggregated across racial groups, and put it into practice when caring for hypertensive African American patients?
Patient education. In trying to overcome patient-related barriers, emphasis should be on patient education, in particular addressing misconceptions about hypertension and promoting adherence to antihypertensive therapy. This is evident from the narrative storytelling intervention by Houston et al.3 Although this is the first study of its kind, this strategy may be something to consider if future studies replicate these findings. Culturally appropriate storytelling may allow patients to identify with the stories as they relate to their own personal lives. It can be an effective way to address patient education and change behaviors.
Self-monitoring with a home blood pressure monitor has also proven effective in the management of hypertension in African Americans. Indeed, the few studies that reported findings in African Americans showed impressive reductions in blood pressure. The benefits of home monitoring are well documented, and the effect on physician-related barriers such as clinical inertia are also quite impressive.117 However, most of these studies did not assess the long-term impact or cost-effectiveness of home monitoring on blood pressure control.
Behavioral counseling. Although we have good evidence of the effectiveness of behavioral counseling, whether this is sustained long-term has been less studied in African Americans. Thus, while interventions that targeted African Americans have reported impressive reductions in blood pressure, the effect tends to be greatest during the first few months of implementation, with the benefits disappearing over time.
Physician-related interventions. With regard to physician-level interventions, research has focused on physician education, utilizing alerts and computerized clinical decision-support systems. Evidence is scant on whether the use of computerized systems results in improves hypertension care in African Americans. However, a closer look at the data from studies that report outcomes in African American and white patients shows that the results do not seem to differ between these groups. Still, there is insufficient information about the impact on hypertensive African Americans.6
Strategies that address both patient- and physician-related barriers can improve overall blood pressure control; however, there is a lack of data comparing outcomes in hypertensive African Americans with those of whites, making it difficult to know if this would be an effective strategy in African American patients alone.
More studies needed that focus on African Americans
Developing interventions to improve blood pressure control in African Americans should be an ongoing priority for research if we intend to address racial disparities in cardiovascular disease. Although it is reassuring that there is a growing body of evidence and research with this focus,118–121 more research is needed to determine effective strategies that address barriers related to physician practice and to the health care system overall as they relate to blood pressure control in African Americans. More importantly, these strategies should also emphasize a team-based approach that includes nurses, pharmacists, and physician assistants. Developing targeted interventions for hypertensive African Americans will help reduce disparities in the rates of cardiovascular illness and death in this patient population.
- Levine DM, Bone LR, Hill MN, et al. The effectiveness of a community/academic health center partnership in decreasing the level of blood pressure in an urban African-American population. Ethn Dis 2003; 13:354–361.
- Ogedegbe G, Chaplin W, Schoenthaler A, et al. A practice-based trial of motivational interviewing and adherence in hypertensive African Americans. Am J Hypertens 2008; 21:1137–1143.
- Houston TK, Allison JJ, Sussman M, et al. Culturally appropriate storytelling to improve blood pressure: a randomized trial. Ann Intern Med 2011; 154:77–84.
- Bosworth HB, Olsen MK, Grubber JM, et al. Two self-management interventions to improve hypertension control: a randomized trial. Ann Intern Med 2009; 151:687–695.
- Artinian NT, Flack JM, Nordstrom CK, et al. Effects of nurse-managed telemonitoring on blood pressure at 12-month follow-up among urban African Americans. Nurs Res 2007; 56:312–322.
- Hicks LS, Sequist TD, Ayanian JZ, et al. Impact of computerized decision support on blood pressure management and control: a randomized controlled trial. J Gen Intern Med 2008; 23:429–441.
- Montgomery AA, Fahey T, Peters TJ, MacIntosh C, Sharp DJ. Evaluation of computer based clinical decision support system and risk chart for management of hypertension in primary care: randomised controlled trial. BMJ 2000; 320:686–690.
- Roumie CL, Elasy TA, Greevy R, et al. Improving blood pressure control through provider education, provider alerts, and patient education: a cluster randomized trial. Ann Intern Med 2006; 145:165–175.
- Svetkey LP, Pollak KI, Yancy WS, et al. Hypertension improvement project: randomized trial of quality improvement for physicians and lifestyle modification for patients. Hypertension 2009; 54:1226–1233.
- Carter BL, Ardery G, Dawson JD, et al. Physician and pharmacist collaboration to improve blood pressure control. Arch Intern Med 2009; 169:1996–2002.
- Hunt JS, Siemienczuk J, Pape G, et al. A randomized controlled trial of team-based care: impact of physician-pharmacist collaboration on uncontrolled hypertension. J Gen Intern Med 2008; 23:1966–1972.
- US Department of Health and Human Services: Office of Disease Prevention and Health Promotion—Healthy People 2010. Nasnewsletter 2000; 15:3.
- Egan BM, Zhao Y, Axon RN. US trends in prevalence, awareness, treatment, and control of hypertension, 1988–2008. JAMA 2010; 303:2043–2050.
- US Centers for Disease Control and Prevention. Age-specific excess deaths associated with stroke among racial/ethnic minority populations–United States, 1997. JAMA 2000; 283:2382–2383.
- Giles WH, Kittner SJ, Hebel JR, Losonczy KG, Sherwin RW. Determinants of black-white differences in the risk of cerebral infarction. The National Health and Nutrition Examination Survey Epidemiologic Follow-up Study. Arch Intern Med 1995; 155:1319–1324.
- Klag MJ, Whelton PK, Randall BL, Neaton JD, Brancati FL, Stamler J. End-stage renal disease in African-American and white men. 16-year MRFIT findings. JAMA 1997; 277:1293–1298.
- Pavlik VN, Hyman DJ, Vallbona C, Toronjo C, Louis K. Hypertension awareness and control in an inner-city African-American sample. J Hum Hypertens 1997; 11:277–283.
- Roger VL, Go AS, Lloyd-Jones DM, et al. Heart Disease and Stroke Statistics—2011 Update: A Report From the American Heart Association. Circulation Feb 1; 123( 4):e18–e209.
- Chobanian AV, Bakris GL, Black HR, et al., Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- Bone LR, Hill MN, Stallings R, et al. Community health survey in an urban African-American neighborhood: distribution and correlates of elevated blood pressure. Ethn Dis 2000; 10:87–95.
- Weber MA. Strategies for improving blood pressure control. Am J Hypertens 1998; 11:897–899.
- Hill MN, Sutton BS. Barriers to hypertension care and control. Curr Hypertens Rep 2000; 2:445–450.
- Alderman MH. Barriers to blood pressure control. Am J Hypertens 1999; 12:1268–1269.
- Chobanian AV. Control of hypertension—an important national priority. N Engl J Med 2001; 345:534–535.
- Miller NH, Hill M, Kottke T, Ockene IS. The multilevel compliance challenge: recommendations for a call to action. A statement for healthcare professionals. Circulation 1997; 95:1085–1090.
- Sackett DL, Snow JC. The magnitude of compliance and noncompliance. In:Haynes RB, Taylor DW, Sackett DL, eds. Compliance in Health Care. Baltimore, MD: John Hopkins University Press; 1979:11–22.
- Shea S, Misra D, Ehrlich MH, Field L, Francis CK. Correlates of nonadherence to hypertension treatment in an inner-city minority population. Am J Public Health 1992; 82:1607–1612.
- Kirscht JP, Rosenstock IM. Patient adherence to antihypertensive medical regimens. J Community Health. 1977; 3:115–124.
- Hershey JC, Morton BG, Davis JB, Reichgott MJ. Patient compliance with antihypertensive medication. Am J Public Health 1980; 70:1081–1089.
- Bosworth HB, Powers B, Grubber JM, et al. Racial differences in blood pressure control: potential explanatory factors. J Gen Intern Med 2008; 23:692–698.
- Douglas JG, Ferdinand KC, Bakris GL, Sowers JR. Barriers to blood pressure control in African Americans. Overcoming obstacles is challenging, but target goals can be attained. Postgrad Med 2002; 112:51–52,55,59–62passim.
- Knight EL, Bohn RL, Wang PS, Glynn RJ, Mogun H, Avorn J. Predictors of uncontrolled hypertension in ambulatory patients. Hypertension 2001; 38:809–814.
- Ogedegbe G, Mancuso CA, Allegrante JP. Expectations of blood pressure management in hypertensive African-American patients: a qualitative study. J Natl Med Assoc 2004; 96:442–449.
- Blumhagen D. Hypertension: a folk illness with a medical name. Cult Med Psychiatry 1980; 4:197–224.
- Meyer D, Leventhal H, Gutmann M. Common-sense models of illness: the example of hypertension. Health Psychol 1985; 4:115–135.
- Nelson EC, Stason WB, Neutra RR, Solomon HS, McArdle PJ. Impact of patient perceptions on compliance with treatment for hypertension. Med Care 1978; 16:893–906.
- Heurtin-Roberts S. ‘High-pertension’—the uses of a chronic folk illness for personal adaptation. Soc Sci Med 1993; 37:285–294.
- Lang T. Social and economic factors as obstacles to blood pressure control. Am J Hypertens 1998; 11:900–902.
- Lang T. Factors that appear as obstacles to the control of high blood pressure. Ethn Dis 2000; 10:125–130.
- Stamler R, Shipley M, Elliott P, Dyer A, Sans S, Stamler J. Higher blood pressure in adults with less education. Some explanations from INTERSALT. Hypertension 1992; 19:237–241.
- Heurtin-Roberts S, Reisin E. The relation of culturally influenced lay models of hypertension to compliance with treatment. Am J Hypertens 1992; 5:787–792.
- Snow LF. Folk medical beliefs and their implications for care of patients. A review bases on studies among black Americans. Ann Intern Med 1974; 81:82–96.
- Hicks LS, Shaykevich S, Bates DW, Ayanian JZ. Determinants of racial/ethnic differences in blood pressure management among hypertensive patients. BMC Cardiovasc Disord 2005; 5:16.
- Mehta SS, Wilcox CS, Schulman KA. Treatment of hypertension in patients with comorbidities: results from the study of hypertensive prescribing practices (SHyPP). Am J Hypertens 1999; 12:333–340.
- Ballard DJ, Strogatz DS, Wagner EH, et al. Hypertension control in a rural southern community: medical care process and dropping out. Am J Prev Med 1988; 4:133–139.
- Hajjar I, Miller K, Hirth V. Age-related bias in the management of hypertension: a national survey of physicians’ opinions on hypertension in elderly adults. J Gerontol A Biol Sci Med Sci 2002; 57:M487–M491.
- McAlister FA, Laupacis A, Teo KK, Hamilton PG, Montague TJ. A survey of clinician attitudes and management practices in hypertension. J Hum Hypertens 1997; 11:413–419.
- Trilling JS, Froom J. The urgent need to improve hypertension care. Arch Fam Med 2000; 9:794–801.
- Huse DM, Roht LH, Alpert JS, Hartz SC. Physicians’ knowledge, attitudes, and practice of pharmacologic treatment of hypertension. Ann Pharmacother 2001; 35:1173–1179.
- Cabana MD, Rand CS, Powe NR, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA 1999; 282:1458–1465.
- Hyman DJ, Pavlik VN, Vallbona C. Physician role in lack of awareness and control of hypertension. J Clin Hypertens (Greenwich) 2000; 2:324–330.
- Morley Kotchen J, Walker WE, Kotchen TA. Rationale for a community approach to hypertension control among inner city minority populations. Heart Dis Stroke 1994; 3:61–62.
- Hyman DJ, Pavlik VN. Characteristics of patients with uncontrolled hypertension in the United States. N Engl J Med 2001; 345:479–486.
- Lang T. Factors that appear as obstacles to the control of high blood pressure. Ethn Dis 2000; 10:125–130.
- Pierce JP, Watson DS, Knights S, Gliddon T, Williams S, Watson R. A controlled trial of health education in the physician’s office. Prev Med 1984; 13:185–194.
- Morisky DE, DeMuth NM, Field-Fass M, Green LW, Levine DM. Evaluation of family health education to build social support for long-term control of high blood pressure. Health Educ Q 1985; 12:35–50.
- Lorgelly P, Siatis I, Brooks A, et al. Is ambulatory blood pressure monitoring cost-effective in the routine surveillance of treated hypertensive patients in primary care? Br J Gen Pract 2003; 53:794–796.
- Green LW, Levine DM, Wolle J, Deeds S. Development of randomized patient education experiments with urban poor hypertensives. Patient Couns Health Educ 1979; 1:106–111.
- Gruesser M, Hartmann P, Schlottmann N, Lohmann FW, Sawicki PT, Joergens V. Structured patient education for out-patients with hypertension in general practice: a model project in Germany. J Hum Hypertens 1997; 11:501–506.
- Mühlhauser I, Sawicki PT, Didjurgeit U, Jörgens V, Trampisch HJ, Berger M. Evaluation of a structured treatment and teaching programme on hypertension in general practice. Clin Exp Hypertens 1993; 15:125–142.
- Roca B, Nadal E, Rovira RE, Valls S, Lapuebla C, Lloría N. Usefulness of a hypertension education program. South Med J 2003; 96:1133–1137.
- Pickering TG, Gerin W, Holland JK. Home blood pressure teletransmission for better diagnosis and treatment. Curr Hypertens Rep 1999; 1:489–494.
- Yarows SA, Julius S, Pickering TG. Home blood pressure monitoring. Arch Intern Med 2000; 160:1251–1257.
- Haynes RB, Sackett DL, Gibson ES, et al. Improvement of medication compliance in uncontrolled hypertension. Lancet 1976; 1:1265–1268.
- Johnson AL, Taylor DW, Sackett DL, Dunnett CW, Shimizu AG. Self-recording of blood pressure in the management of hypertension. Can Med Assoc J 1978; 119:1034–1039.
- Carnahan JE, Nugent CA. The effects of self-monitoring by patients on the control of hypertension. Am J Med Sci 1975; 269:69–73.
- Stahl SM, Kelley CR, Neill PJ, Grim CE, Mamlin J. Effects of home blood pressure measurement on long-term BP control. Am J Public Health 1984; 74:704–709.
- Boulware LE, Daumit GL, Frick KD, Minkovitz CS, Lawrence RS, Powe NR. An evidence-based review of patient-centered behavioral interventions for hypertension. Am J Prev Med 2001; 21:221–232.
- Haynes RB, Mattson ME, Engebretson TO. Patient compliance to prescribed antihypertensive medication regimens: a report to the National Heart, Lung, and Blood institute. Bethesda, MD: US Department of Health and Human Services, Public Health Service, National Institutes of Health, 1980. NIH publication 81-2102.
- Burke LE, Dunbar-Jacob JM, Hill MN. Compliance with cardiovascular disease prevention strategies: a review of the research. Ann Behav Med 1997; 19:239–263.
- Dunbar-Jacob J, Dwyer K, Dunning EJ. Compliance with antihypertensive regimen: a review of the research in the 1980s. Ann Behav Med 1991; 13:31–39.
- Haynes RB, Montague P, Oliver T, McKibbon KA, Brouwers MC, Kanani R. Interventions for helping patients to follow prescriptions for medications. Cochrane Database Syst Rev 2000; ( 2):CD000011.
- Roter DL, Hall JA, Merisca R, Nordstrom B, Cretin D, Svarstad B. Effectiveness of interventions to improve patient compliance: a meta-analysis. Med Care 1998; 36:1138–1161.
- Appel LJ, Espeland MA, Easter L, Wilson AC, Folmar S, Lacy CR. Effects of reduced sodium intake on hypertension control in older individuals: results from the Trial of Nonpharmacologic Interventions in the Elderly (TONE). Arch Intern Med 2001; 161:685–693.
- Appel LJ, Champagne CM, Harsha DW, et al; Writing Group of the PREMIER Collaborative Research Group. Effects of comprehensive lifestyle modification on blood pressure control: main results of the PREMIER clinical trial. JAMA 2003; 289:2083–2093.
- Appel LJ, Moore TJ, Obarzanek E, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med 1997; 336:1117–1124.
- Moore TJ, Conlin PR, Ard J, Svetkey LP. DASH (Dietary Approaches to Stop Hypertension) diet is effective treatment for stage 1 isolated systolic hypertension. Hypertension 2001; 38:155–158.
- Stevens VJ, Obarzanek E, Cook NR, et al; Trials for the Hypertension Prevention Research Group. Long-term weight loss and changes in blood pressure: results of the Trials of Hypertension Prevention, phase II. Ann Intern Med 2001; 134:1–11.
- Sawicki PT, Mühlhauser I, Didjurgeit U, Berger M. Improvement of hypertension care by a structured treatment and teaching programme. J Hum Hypertens 1993; 7:571–573.
- Morisky DE, Bowler MH, Finlay JS. An educational and behavioral approach toward increasing patient activation in hypertension management. J Community Health 1982; 7:171–182.
- Levine DM, Green LW, Deeds SG, Chwalow J, Russell RP, Finlay J. Health education for hypertensive patients. JAMA 1979; 241:1700–1703.
- Iso H, Shimamoto T, Yokota K, Sankai T, Jacobs DR, Komachi Y. Community-based education classes for hypertension control. A 1.5-year randomized controlled trial. Hypertension 1996; 27:968–974.
- Cuspidi C, Sampieri L, Macca G, et al. Improvement of patients’ knowledge by a single educational meeting on hypertension. J Hum Hypertens 2001; 15:57–61.
- Nessman DG, Carnahan JE, Nugent CA. Increasing compliance. Patient-operated hypertension groups. Arch Intern Med 1980; 140:1427–1430.
- Casasanta L, Patel S. Outcomes of an educational component of a disease management program for hypertension. Manag Care Interface 1999; 12:70–73.
- McAdams DP. The Stories We Live By: Personal Myths and the Making of the Self. New York NY: The Guilford Press; 1993.
- Bruner J. Acts of Meaning. Cambridge, MA: Harvard Univ Pr; 1990.
- Slater MD, Rouner D. Entertainment—education and elaboration likelihood: Understanding the processing of narrative persuasion. Commun Theory 2002; 12:173–191.
- Dal CS, Zanna MP, Fong GT. Narrative persuasion and overcoming resistance. In:Knowles ES, Linn J, eds. Resistance and Persuasion. Mahwah, NJ: Lawrence Erlbaum Assoc; 2004:175–191.
- Artinian NT, Washington OG, Templin TN. Effects of home telemonitoring and community-based monitoring on blood pressure control in urban African Americans: a pilot study. Heart Lung 2001; 30:191–199.
- Bailey B, Carney SL, Gillies AA, Smith AJ. Antihypertensive drug treatment: a comparison of usual care with self blood pressure measurement. J Hum Hypertens 1999; 13:147–150.
- Bondmass M. The effect of home monitoring and telemanagement on blood pressure control among African Americans. Telemed J 2000; 6:15–23.
- Rogers MA, Small D, Buchan DA, et al. Home monitoring service improves mean arterial pressure in patients with essential hypertension. A randomized, controlled trial. Ann Intern Med 2001; 134:1024–1032.
- Mengden T, Uen S, Baulmann J, Vetter H. Significance of blood pressure self-measurement as compared with office blood pressure measurement and ambulatory 24-hour blood pressure measurement in pharmacological studies. Blood Press Monit 2003; 8:169–172.
- Friedman RH, Kazis LE, Jette A, et al. A telecommunications system for monitoring and counseling patients with hypertension. Impact on medication adherence and blood pressure control. Am J Hypertens 1996; 9:285–292.
- Oxman AD, Thomson MA, Davis DA, Haynes RB. No magic bullets: a systematic review of 102 trials of interventions to improve professional practice. CMAJ 1995; 153:1423–1431.
- Wensing M, van der Weijden T, Grol R. Implementing guidelines and innovations in general practice: which interventions are effective? Br J Gen Pract 1998; 48:991–997.
- Davis DA, Thomson MA, Oxman AD, Haynes RB. Changing physician performance. A systematic review of the effect of continuing medical education strategies. JAMA 1995; 274:700–705.
- Inui TS, Yourtee EL, Williamson JW. Improved outcomes in hypertension after physician tutorials. A controlled trial. Ann Intern Med 1976; 84:646–651.
- Jennett PA, Wilson TW, Hayton RC, Mainprize GW, Laxdal OE. Desirable behaviours in the office management of hypertension addressed through continuing medical education. Can J Public Health 1989; 80:359–362.
- Maue SK, Rivo ML, Weiss B, Farrelly EW, Brower-Stenger S. Effect of a primary care physician-focused, population-based approach to blood pressure control. Fam Med 2002; 34:508–513.
- Tu K, Davis D. Can we alter physician behavior by educational methods? Lessons learned from studies of the management and follow-up of hypertension. J Contin Educ Health Prof 2002; 22:11–22.
- McAlister NH, Covvey HD, Tong C, Lee A, Wigle ED. Randomised controlled trial of computer assisted management of hypertension in primary care. Br Med J (Clin Res Ed) 1986; 293:670–674.
- Mashru M, Lant A. Interpractice audit of diagnosis and management of hypertension in primary care: educational intervention and review of medical records. BMJ 1997; 314:942–946.
- Degoulet P, Menard J, Berger C, Plouin PF, Devries C, Hirel JC. Hypertension management: the computer as a participant. Am J Med 1980; 68:559–567.
- Dickinson JC, Warshaw GA, Gehlbach SH, Bobula JA, Muhlbaier LH, Parkerson GR. Improving hypertension control: impact of computer feedback and physician education. Med Care 1981; 19:843–854.
- Montgomery AA, Fahey T. A systematic review of the use of computers in the management of hypertension. J Epidemiol Community Health 1998; 52:520–525.
- Coleman MT, Lott JA, Sharma S. Use of continuous quality improvement to identify barriers in the management of hypertension. Am J Med Qual 2000; 15:72–77.
- Goldberg HI, Wagner EH, Fihn SD, et al. A randomized controlled trial of CQI teams and academic detailing: can they alter compliance with guidelines? Jt Comm J Qual Improv 1998; 24:130–142.
- Horowitz CR, Goldberg HI, Martin DP, et al. Conducting a randomized controlled trial of CQI and academic detailing to implement clinical guidelines. Jt Comm J Qual Improv 1996; 22:734–750.
- Johnson B, McNair D, Kailasam K, et al. Discern—an integrated prospective decision support system. Proc Annu Symp Comput Appl Med Care 1994; 969.
- Bosworth HB, Olsen MK, Dudley T, et al. Patient education and provider decision support to control blood pressure in primary care: a cluster randomized trial. Am Heart J 2009; 157:450–456.
- Walsh JM, McDonald KM, Shojania KG, et al. Quality improvement strategies for hypertension management: a systematic review. Med Care 2006; 44:646–657.
- Carter BL, Rogers M, Daly J, Zheng S, James PA. The potency of team-based care interventions for hypertension: a meta-analysis. Arch Intern Med 2009; 169:1748–1755.
- Carter BL, Clarke W, Ardery G, et al; Collaboration Among Pharmacists Physicians To Improve Outcomes Now (CAPTION) Trial Investigators. A cluster-randomized effectiveness trial of a physician-pharmacist collaborative model to improve blood pressure control. Circ Cardiovasc Qual Outcomes 2010; 3:418–423.
- Einhorn PT. National heart, lung, and blood institute-initiated program “interventions to improve hypertension control rates in African Americans”: background and implementation. Circ Cardiovasc Qual Outcomes 2009; 2:236–240.
- Agarwal R, Bills JE, Hecht TJ, Light RP. Role of home blood pressure monitoring in overcoming therapeutic inertia and improving hypertension control: a systematic review and meta-analysis. Hypertension 2011; 57:29–38.
- Bosworth HB, Olsen MK, Neary A, et al. Take Control of Your Blood Pressure (TCYB) study: a multifactorial tailored behavioral and educational intervention for achieving blood pressure control. Patient Educ Couns 2008; 70:338–347.
- Bosworth HB, Olsen MK, Goldstein MK, et al. The veterans’ study to improve the control of hypertension (V-STITCH): design and methodology. Contemp Clin Trials 2005; 26:155–168.
- Ogedegbe G, Tobin JN, Fernandez S, et al. Counseling African Americans to Control Hypertension (CAATCH) trial: a multi-level intervention to improve blood pressure control in hypertensive blacks. Circ Cardiovasc Qual Outcomes 2009; 2:249–256.
- Bosworth HB, Almirall D, Weiner BJ, et al. The implementation of a translational study involving a primary care based behavioral program to improve blood pressure control: The HTN-IMPROVE study protocol (01295). Implement Sci 2010; 5:54.
- Levine DM, Bone LR, Hill MN, et al. The effectiveness of a community/academic health center partnership in decreasing the level of blood pressure in an urban African-American population. Ethn Dis 2003; 13:354–361.
- Ogedegbe G, Chaplin W, Schoenthaler A, et al. A practice-based trial of motivational interviewing and adherence in hypertensive African Americans. Am J Hypertens 2008; 21:1137–1143.
- Houston TK, Allison JJ, Sussman M, et al. Culturally appropriate storytelling to improve blood pressure: a randomized trial. Ann Intern Med 2011; 154:77–84.
- Bosworth HB, Olsen MK, Grubber JM, et al. Two self-management interventions to improve hypertension control: a randomized trial. Ann Intern Med 2009; 151:687–695.
- Artinian NT, Flack JM, Nordstrom CK, et al. Effects of nurse-managed telemonitoring on blood pressure at 12-month follow-up among urban African Americans. Nurs Res 2007; 56:312–322.
- Hicks LS, Sequist TD, Ayanian JZ, et al. Impact of computerized decision support on blood pressure management and control: a randomized controlled trial. J Gen Intern Med 2008; 23:429–441.
- Montgomery AA, Fahey T, Peters TJ, MacIntosh C, Sharp DJ. Evaluation of computer based clinical decision support system and risk chart for management of hypertension in primary care: randomised controlled trial. BMJ 2000; 320:686–690.
- Roumie CL, Elasy TA, Greevy R, et al. Improving blood pressure control through provider education, provider alerts, and patient education: a cluster randomized trial. Ann Intern Med 2006; 145:165–175.
- Svetkey LP, Pollak KI, Yancy WS, et al. Hypertension improvement project: randomized trial of quality improvement for physicians and lifestyle modification for patients. Hypertension 2009; 54:1226–1233.
- Carter BL, Ardery G, Dawson JD, et al. Physician and pharmacist collaboration to improve blood pressure control. Arch Intern Med 2009; 169:1996–2002.
- Hunt JS, Siemienczuk J, Pape G, et al. A randomized controlled trial of team-based care: impact of physician-pharmacist collaboration on uncontrolled hypertension. J Gen Intern Med 2008; 23:1966–1972.
- US Department of Health and Human Services: Office of Disease Prevention and Health Promotion—Healthy People 2010. Nasnewsletter 2000; 15:3.
- Egan BM, Zhao Y, Axon RN. US trends in prevalence, awareness, treatment, and control of hypertension, 1988–2008. JAMA 2010; 303:2043–2050.
- US Centers for Disease Control and Prevention. Age-specific excess deaths associated with stroke among racial/ethnic minority populations–United States, 1997. JAMA 2000; 283:2382–2383.
- Giles WH, Kittner SJ, Hebel JR, Losonczy KG, Sherwin RW. Determinants of black-white differences in the risk of cerebral infarction. The National Health and Nutrition Examination Survey Epidemiologic Follow-up Study. Arch Intern Med 1995; 155:1319–1324.
- Klag MJ, Whelton PK, Randall BL, Neaton JD, Brancati FL, Stamler J. End-stage renal disease in African-American and white men. 16-year MRFIT findings. JAMA 1997; 277:1293–1298.
- Pavlik VN, Hyman DJ, Vallbona C, Toronjo C, Louis K. Hypertension awareness and control in an inner-city African-American sample. J Hum Hypertens 1997; 11:277–283.
- Roger VL, Go AS, Lloyd-Jones DM, et al. Heart Disease and Stroke Statistics—2011 Update: A Report From the American Heart Association. Circulation Feb 1; 123( 4):e18–e209.
- Chobanian AV, Bakris GL, Black HR, et al., Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- Bone LR, Hill MN, Stallings R, et al. Community health survey in an urban African-American neighborhood: distribution and correlates of elevated blood pressure. Ethn Dis 2000; 10:87–95.
- Weber MA. Strategies for improving blood pressure control. Am J Hypertens 1998; 11:897–899.
- Hill MN, Sutton BS. Barriers to hypertension care and control. Curr Hypertens Rep 2000; 2:445–450.
- Alderman MH. Barriers to blood pressure control. Am J Hypertens 1999; 12:1268–1269.
- Chobanian AV. Control of hypertension—an important national priority. N Engl J Med 2001; 345:534–535.
- Miller NH, Hill M, Kottke T, Ockene IS. The multilevel compliance challenge: recommendations for a call to action. A statement for healthcare professionals. Circulation 1997; 95:1085–1090.
- Sackett DL, Snow JC. The magnitude of compliance and noncompliance. In:Haynes RB, Taylor DW, Sackett DL, eds. Compliance in Health Care. Baltimore, MD: John Hopkins University Press; 1979:11–22.
- Shea S, Misra D, Ehrlich MH, Field L, Francis CK. Correlates of nonadherence to hypertension treatment in an inner-city minority population. Am J Public Health 1992; 82:1607–1612.
- Kirscht JP, Rosenstock IM. Patient adherence to antihypertensive medical regimens. J Community Health. 1977; 3:115–124.
- Hershey JC, Morton BG, Davis JB, Reichgott MJ. Patient compliance with antihypertensive medication. Am J Public Health 1980; 70:1081–1089.
- Bosworth HB, Powers B, Grubber JM, et al. Racial differences in blood pressure control: potential explanatory factors. J Gen Intern Med 2008; 23:692–698.
- Douglas JG, Ferdinand KC, Bakris GL, Sowers JR. Barriers to blood pressure control in African Americans. Overcoming obstacles is challenging, but target goals can be attained. Postgrad Med 2002; 112:51–52,55,59–62passim.
- Knight EL, Bohn RL, Wang PS, Glynn RJ, Mogun H, Avorn J. Predictors of uncontrolled hypertension in ambulatory patients. Hypertension 2001; 38:809–814.
- Ogedegbe G, Mancuso CA, Allegrante JP. Expectations of blood pressure management in hypertensive African-American patients: a qualitative study. J Natl Med Assoc 2004; 96:442–449.
- Blumhagen D. Hypertension: a folk illness with a medical name. Cult Med Psychiatry 1980; 4:197–224.
- Meyer D, Leventhal H, Gutmann M. Common-sense models of illness: the example of hypertension. Health Psychol 1985; 4:115–135.
- Nelson EC, Stason WB, Neutra RR, Solomon HS, McArdle PJ. Impact of patient perceptions on compliance with treatment for hypertension. Med Care 1978; 16:893–906.
- Heurtin-Roberts S. ‘High-pertension’—the uses of a chronic folk illness for personal adaptation. Soc Sci Med 1993; 37:285–294.
- Lang T. Social and economic factors as obstacles to blood pressure control. Am J Hypertens 1998; 11:900–902.
- Lang T. Factors that appear as obstacles to the control of high blood pressure. Ethn Dis 2000; 10:125–130.
- Stamler R, Shipley M, Elliott P, Dyer A, Sans S, Stamler J. Higher blood pressure in adults with less education. Some explanations from INTERSALT. Hypertension 1992; 19:237–241.
- Heurtin-Roberts S, Reisin E. The relation of culturally influenced lay models of hypertension to compliance with treatment. Am J Hypertens 1992; 5:787–792.
- Snow LF. Folk medical beliefs and their implications for care of patients. A review bases on studies among black Americans. Ann Intern Med 1974; 81:82–96.
- Hicks LS, Shaykevich S, Bates DW, Ayanian JZ. Determinants of racial/ethnic differences in blood pressure management among hypertensive patients. BMC Cardiovasc Disord 2005; 5:16.
- Mehta SS, Wilcox CS, Schulman KA. Treatment of hypertension in patients with comorbidities: results from the study of hypertensive prescribing practices (SHyPP). Am J Hypertens 1999; 12:333–340.
- Ballard DJ, Strogatz DS, Wagner EH, et al. Hypertension control in a rural southern community: medical care process and dropping out. Am J Prev Med 1988; 4:133–139.
- Hajjar I, Miller K, Hirth V. Age-related bias in the management of hypertension: a national survey of physicians’ opinions on hypertension in elderly adults. J Gerontol A Biol Sci Med Sci 2002; 57:M487–M491.
- McAlister FA, Laupacis A, Teo KK, Hamilton PG, Montague TJ. A survey of clinician attitudes and management practices in hypertension. J Hum Hypertens 1997; 11:413–419.
- Trilling JS, Froom J. The urgent need to improve hypertension care. Arch Fam Med 2000; 9:794–801.
- Huse DM, Roht LH, Alpert JS, Hartz SC. Physicians’ knowledge, attitudes, and practice of pharmacologic treatment of hypertension. Ann Pharmacother 2001; 35:1173–1179.
- Cabana MD, Rand CS, Powe NR, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA 1999; 282:1458–1465.
- Hyman DJ, Pavlik VN, Vallbona C. Physician role in lack of awareness and control of hypertension. J Clin Hypertens (Greenwich) 2000; 2:324–330.
- Morley Kotchen J, Walker WE, Kotchen TA. Rationale for a community approach to hypertension control among inner city minority populations. Heart Dis Stroke 1994; 3:61–62.
- Hyman DJ, Pavlik VN. Characteristics of patients with uncontrolled hypertension in the United States. N Engl J Med 2001; 345:479–486.
- Lang T. Factors that appear as obstacles to the control of high blood pressure. Ethn Dis 2000; 10:125–130.
- Pierce JP, Watson DS, Knights S, Gliddon T, Williams S, Watson R. A controlled trial of health education in the physician’s office. Prev Med 1984; 13:185–194.
- Morisky DE, DeMuth NM, Field-Fass M, Green LW, Levine DM. Evaluation of family health education to build social support for long-term control of high blood pressure. Health Educ Q 1985; 12:35–50.
- Lorgelly P, Siatis I, Brooks A, et al. Is ambulatory blood pressure monitoring cost-effective in the routine surveillance of treated hypertensive patients in primary care? Br J Gen Pract 2003; 53:794–796.
- Green LW, Levine DM, Wolle J, Deeds S. Development of randomized patient education experiments with urban poor hypertensives. Patient Couns Health Educ 1979; 1:106–111.
- Gruesser M, Hartmann P, Schlottmann N, Lohmann FW, Sawicki PT, Joergens V. Structured patient education for out-patients with hypertension in general practice: a model project in Germany. J Hum Hypertens 1997; 11:501–506.
- Mühlhauser I, Sawicki PT, Didjurgeit U, Jörgens V, Trampisch HJ, Berger M. Evaluation of a structured treatment and teaching programme on hypertension in general practice. Clin Exp Hypertens 1993; 15:125–142.
- Roca B, Nadal E, Rovira RE, Valls S, Lapuebla C, Lloría N. Usefulness of a hypertension education program. South Med J 2003; 96:1133–1137.
- Pickering TG, Gerin W, Holland JK. Home blood pressure teletransmission for better diagnosis and treatment. Curr Hypertens Rep 1999; 1:489–494.
- Yarows SA, Julius S, Pickering TG. Home blood pressure monitoring. Arch Intern Med 2000; 160:1251–1257.
- Haynes RB, Sackett DL, Gibson ES, et al. Improvement of medication compliance in uncontrolled hypertension. Lancet 1976; 1:1265–1268.
- Johnson AL, Taylor DW, Sackett DL, Dunnett CW, Shimizu AG. Self-recording of blood pressure in the management of hypertension. Can Med Assoc J 1978; 119:1034–1039.
- Carnahan JE, Nugent CA. The effects of self-monitoring by patients on the control of hypertension. Am J Med Sci 1975; 269:69–73.
- Stahl SM, Kelley CR, Neill PJ, Grim CE, Mamlin J. Effects of home blood pressure measurement on long-term BP control. Am J Public Health 1984; 74:704–709.
- Boulware LE, Daumit GL, Frick KD, Minkovitz CS, Lawrence RS, Powe NR. An evidence-based review of patient-centered behavioral interventions for hypertension. Am J Prev Med 2001; 21:221–232.
- Haynes RB, Mattson ME, Engebretson TO. Patient compliance to prescribed antihypertensive medication regimens: a report to the National Heart, Lung, and Blood institute. Bethesda, MD: US Department of Health and Human Services, Public Health Service, National Institutes of Health, 1980. NIH publication 81-2102.
- Burke LE, Dunbar-Jacob JM, Hill MN. Compliance with cardiovascular disease prevention strategies: a review of the research. Ann Behav Med 1997; 19:239–263.
- Dunbar-Jacob J, Dwyer K, Dunning EJ. Compliance with antihypertensive regimen: a review of the research in the 1980s. Ann Behav Med 1991; 13:31–39.
- Haynes RB, Montague P, Oliver T, McKibbon KA, Brouwers MC, Kanani R. Interventions for helping patients to follow prescriptions for medications. Cochrane Database Syst Rev 2000; ( 2):CD000011.
- Roter DL, Hall JA, Merisca R, Nordstrom B, Cretin D, Svarstad B. Effectiveness of interventions to improve patient compliance: a meta-analysis. Med Care 1998; 36:1138–1161.
- Appel LJ, Espeland MA, Easter L, Wilson AC, Folmar S, Lacy CR. Effects of reduced sodium intake on hypertension control in older individuals: results from the Trial of Nonpharmacologic Interventions in the Elderly (TONE). Arch Intern Med 2001; 161:685–693.
- Appel LJ, Champagne CM, Harsha DW, et al; Writing Group of the PREMIER Collaborative Research Group. Effects of comprehensive lifestyle modification on blood pressure control: main results of the PREMIER clinical trial. JAMA 2003; 289:2083–2093.
- Appel LJ, Moore TJ, Obarzanek E, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med 1997; 336:1117–1124.
- Moore TJ, Conlin PR, Ard J, Svetkey LP. DASH (Dietary Approaches to Stop Hypertension) diet is effective treatment for stage 1 isolated systolic hypertension. Hypertension 2001; 38:155–158.
- Stevens VJ, Obarzanek E, Cook NR, et al; Trials for the Hypertension Prevention Research Group. Long-term weight loss and changes in blood pressure: results of the Trials of Hypertension Prevention, phase II. Ann Intern Med 2001; 134:1–11.
- Sawicki PT, Mühlhauser I, Didjurgeit U, Berger M. Improvement of hypertension care by a structured treatment and teaching programme. J Hum Hypertens 1993; 7:571–573.
- Morisky DE, Bowler MH, Finlay JS. An educational and behavioral approach toward increasing patient activation in hypertension management. J Community Health 1982; 7:171–182.
- Levine DM, Green LW, Deeds SG, Chwalow J, Russell RP, Finlay J. Health education for hypertensive patients. JAMA 1979; 241:1700–1703.
- Iso H, Shimamoto T, Yokota K, Sankai T, Jacobs DR, Komachi Y. Community-based education classes for hypertension control. A 1.5-year randomized controlled trial. Hypertension 1996; 27:968–974.
- Cuspidi C, Sampieri L, Macca G, et al. Improvement of patients’ knowledge by a single educational meeting on hypertension. J Hum Hypertens 2001; 15:57–61.
- Nessman DG, Carnahan JE, Nugent CA. Increasing compliance. Patient-operated hypertension groups. Arch Intern Med 1980; 140:1427–1430.
- Casasanta L, Patel S. Outcomes of an educational component of a disease management program for hypertension. Manag Care Interface 1999; 12:70–73.
- McAdams DP. The Stories We Live By: Personal Myths and the Making of the Self. New York NY: The Guilford Press; 1993.
- Bruner J. Acts of Meaning. Cambridge, MA: Harvard Univ Pr; 1990.
- Slater MD, Rouner D. Entertainment—education and elaboration likelihood: Understanding the processing of narrative persuasion. Commun Theory 2002; 12:173–191.
- Dal CS, Zanna MP, Fong GT. Narrative persuasion and overcoming resistance. In:Knowles ES, Linn J, eds. Resistance and Persuasion. Mahwah, NJ: Lawrence Erlbaum Assoc; 2004:175–191.
- Artinian NT, Washington OG, Templin TN. Effects of home telemonitoring and community-based monitoring on blood pressure control in urban African Americans: a pilot study. Heart Lung 2001; 30:191–199.
- Bailey B, Carney SL, Gillies AA, Smith AJ. Antihypertensive drug treatment: a comparison of usual care with self blood pressure measurement. J Hum Hypertens 1999; 13:147–150.
- Bondmass M. The effect of home monitoring and telemanagement on blood pressure control among African Americans. Telemed J 2000; 6:15–23.
- Rogers MA, Small D, Buchan DA, et al. Home monitoring service improves mean arterial pressure in patients with essential hypertension. A randomized, controlled trial. Ann Intern Med 2001; 134:1024–1032.
- Mengden T, Uen S, Baulmann J, Vetter H. Significance of blood pressure self-measurement as compared with office blood pressure measurement and ambulatory 24-hour blood pressure measurement in pharmacological studies. Blood Press Monit 2003; 8:169–172.
- Friedman RH, Kazis LE, Jette A, et al. A telecommunications system for monitoring and counseling patients with hypertension. Impact on medication adherence and blood pressure control. Am J Hypertens 1996; 9:285–292.
- Oxman AD, Thomson MA, Davis DA, Haynes RB. No magic bullets: a systematic review of 102 trials of interventions to improve professional practice. CMAJ 1995; 153:1423–1431.
- Wensing M, van der Weijden T, Grol R. Implementing guidelines and innovations in general practice: which interventions are effective? Br J Gen Pract 1998; 48:991–997.
- Davis DA, Thomson MA, Oxman AD, Haynes RB. Changing physician performance. A systematic review of the effect of continuing medical education strategies. JAMA 1995; 274:700–705.
- Inui TS, Yourtee EL, Williamson JW. Improved outcomes in hypertension after physician tutorials. A controlled trial. Ann Intern Med 1976; 84:646–651.
- Jennett PA, Wilson TW, Hayton RC, Mainprize GW, Laxdal OE. Desirable behaviours in the office management of hypertension addressed through continuing medical education. Can J Public Health 1989; 80:359–362.
- Maue SK, Rivo ML, Weiss B, Farrelly EW, Brower-Stenger S. Effect of a primary care physician-focused, population-based approach to blood pressure control. Fam Med 2002; 34:508–513.
- Tu K, Davis D. Can we alter physician behavior by educational methods? Lessons learned from studies of the management and follow-up of hypertension. J Contin Educ Health Prof 2002; 22:11–22.
- McAlister NH, Covvey HD, Tong C, Lee A, Wigle ED. Randomised controlled trial of computer assisted management of hypertension in primary care. Br Med J (Clin Res Ed) 1986; 293:670–674.
- Mashru M, Lant A. Interpractice audit of diagnosis and management of hypertension in primary care: educational intervention and review of medical records. BMJ 1997; 314:942–946.
- Degoulet P, Menard J, Berger C, Plouin PF, Devries C, Hirel JC. Hypertension management: the computer as a participant. Am J Med 1980; 68:559–567.
- Dickinson JC, Warshaw GA, Gehlbach SH, Bobula JA, Muhlbaier LH, Parkerson GR. Improving hypertension control: impact of computer feedback and physician education. Med Care 1981; 19:843–854.
- Montgomery AA, Fahey T. A systematic review of the use of computers in the management of hypertension. J Epidemiol Community Health 1998; 52:520–525.
- Coleman MT, Lott JA, Sharma S. Use of continuous quality improvement to identify barriers in the management of hypertension. Am J Med Qual 2000; 15:72–77.
- Goldberg HI, Wagner EH, Fihn SD, et al. A randomized controlled trial of CQI teams and academic detailing: can they alter compliance with guidelines? Jt Comm J Qual Improv 1998; 24:130–142.
- Horowitz CR, Goldberg HI, Martin DP, et al. Conducting a randomized controlled trial of CQI and academic detailing to implement clinical guidelines. Jt Comm J Qual Improv 1996; 22:734–750.
- Johnson B, McNair D, Kailasam K, et al. Discern—an integrated prospective decision support system. Proc Annu Symp Comput Appl Med Care 1994; 969.
- Bosworth HB, Olsen MK, Dudley T, et al. Patient education and provider decision support to control blood pressure in primary care: a cluster randomized trial. Am Heart J 2009; 157:450–456.
- Walsh JM, McDonald KM, Shojania KG, et al. Quality improvement strategies for hypertension management: a systematic review. Med Care 2006; 44:646–657.
- Carter BL, Rogers M, Daly J, Zheng S, James PA. The potency of team-based care interventions for hypertension: a meta-analysis. Arch Intern Med 2009; 169:1748–1755.
- Carter BL, Clarke W, Ardery G, et al; Collaboration Among Pharmacists Physicians To Improve Outcomes Now (CAPTION) Trial Investigators. A cluster-randomized effectiveness trial of a physician-pharmacist collaborative model to improve blood pressure control. Circ Cardiovasc Qual Outcomes 2010; 3:418–423.
- Einhorn PT. National heart, lung, and blood institute-initiated program “interventions to improve hypertension control rates in African Americans”: background and implementation. Circ Cardiovasc Qual Outcomes 2009; 2:236–240.
- Agarwal R, Bills JE, Hecht TJ, Light RP. Role of home blood pressure monitoring in overcoming therapeutic inertia and improving hypertension control: a systematic review and meta-analysis. Hypertension 2011; 57:29–38.
- Bosworth HB, Olsen MK, Neary A, et al. Take Control of Your Blood Pressure (TCYB) study: a multifactorial tailored behavioral and educational intervention for achieving blood pressure control. Patient Educ Couns 2008; 70:338–347.
- Bosworth HB, Olsen MK, Goldstein MK, et al. The veterans’ study to improve the control of hypertension (V-STITCH): design and methodology. Contemp Clin Trials 2005; 26:155–168.
- Ogedegbe G, Tobin JN, Fernandez S, et al. Counseling African Americans to Control Hypertension (CAATCH) trial: a multi-level intervention to improve blood pressure control in hypertensive blacks. Circ Cardiovasc Qual Outcomes 2009; 2:249–256.
- Bosworth HB, Almirall D, Weiner BJ, et al. The implementation of a translational study involving a primary care based behavioral program to improve blood pressure control: The HTN-IMPROVE study protocol (01295). Implement Sci 2010; 5:54.
KEY POINTS
- Rates of cardiovascular disease and related death are disparately high in African Americans.
- Ways to improve how physicians manage blood pressure in this patient population may include chart audit with feedback, a computerized clinical decision-support system, and keeping up-to-date with treatment guidelines. However, more data are needed to determine the effectiveness of these interventions.
- A novel method of health education is the use of narrative communication—ie, storytelling. Culturally appropriate storytelling may allow patients to identify with a story as it relates to their own lives.
- A team-based approach to blood pressure control that involves nurses, pharmacists, and physician assistants should be emphasized, even though studies that have shown positive results did not focus specifically on African Americans.
Addressing disparities in health care
In the united states, minority populations are rapidly increasing. In 1970, minorities—ie, African American, Hispanic, Asian, and Native American—accounted for 12.3% of the US population, but they now account for 25%. And this growth is expected to continue, so that by 2050 one of every two Americans will be African American, Hispanic, Asian, Pacific Islander, or Native American.1
Also, while advances in medicine over the past several decades have reduced death rates from cancer and coronary artery disease and have contributed to a longer life expectancy for Americans, minority populations have not benefited equally from these improvements.2 In fact, the growing minority populations suffer from disparities in health care compared with white patients: minority patients have a higher incidence and burden of disease, and poorer health outcomes, contributing to shorter life expectancy.
Clearly, there is an urgent need for physicians, other health care providers, health systems, and medical researchers to increase their awareness of disparities in health care and their impact on patients, as well as on the US health system and the US economy. Now more than ever, we need to equip ourselves to more effectively engage minorities and to deliver culturally competent health care that improves outcomes in our minority patients.
A MULTIFACTORIAL PROBLEM
Disparities in health care are often thought to be the result of poverty and a related lack of access to quality health care. But clinical experience and research show that this is overly simplistic. In fact, disparities result from a variety of factors. Patient-related factors can include culturally related beliefs,1 dietary preferences, and health-seeking behaviors (perhaps influenced by a distrust of doctors, researchers, and the health care system), in addition to poor health literacy. Physician-related factors include poor cultural competency, which leads to poor communication with the patient. Other factors are a continuing lack of representation of minority patients in clinical research trials, as well as biologic factors.3
TAKING ACTION
In view of the disparities in health care that affect racial and ethnic minorities, and the many factors underlying the problem, the US Department of Health and Human Services launched the initiative Healthy People 2020, a continuation of the previous 10-year Healthy People initiatives. Healthy People 2020 calls for health providers and health systems to devise effective ways to eliminate health disparities.4 It outlines high-priority health issues, sets 10-year goals for improving the health of all Americans, and suggests specific actions to take to address health disparities.4
On another front, in 2010 the National Institutes of Health formally established its National Institute of Health and Health Disparities, which funds research into the pathogenesis of health disparities in racial and ethnic minorities.5 Clearly, racial, ethnic, and cultural factors need to be considered for health care to result in better outcomes in minority populations.
OUR NEW SERIES
In this issue of the Cleveland Clinic Journal of Medicine, we launch a series we hope will provide practical tools for physicians to address the disparities in our health care system. The first installment, by Odesosu et al (page 46), addresses barriers to optimal hypertension control in African Americans by outlining potential tactics for both patients and physicians. Future articles will address the challenge of health literacy and cultural issues in medicine, slowing the progression of renal disease in African Americans (especially the complex issue of which antihypertensive agents to use), and the challenges of diabetes in Hispanics. We also plan articles on kidney transplantation in African Americans and on prostate cancer, heart failure, lupus, and diabetes.
We look forward to your comments on this series as well as suggestions for future topics. We believe that as physicians, other health providers, health systems, health insurers and policy-makers become more aware of the disparities in health care, they will embrace ways in which to deliver or promote personalized, culturally competent health care. We hope this series will provide practical tools for physicians to address these complex issues.
- Modlin CS. Culture, race, and disparities in health care. Cleve Clin J Med 2003; 70:283–288.
- Centers for Disease Control and Prevention life expectancy data. www.cdc.gov/nchs/fastats/lifexpec.htm. Accessed December 5, 2011.
- Klein JB, Nguyen CT, Saffore L, Modlin C, Modlin CS. Racial disparities in urologic health care. J Natl Med Assoc 2010: 102:108–117.
- Healthy People 2020. www.healthypeople.gov/2020/. Accessed December 5, 2011.
- National Institutes of Health. NIH announces Institute on minority health and health disparities. www.nih.gov/news/health/sep2010/nimhd-27.htm. Accessed December 5, 2011.
In the united states, minority populations are rapidly increasing. In 1970, minorities—ie, African American, Hispanic, Asian, and Native American—accounted for 12.3% of the US population, but they now account for 25%. And this growth is expected to continue, so that by 2050 one of every two Americans will be African American, Hispanic, Asian, Pacific Islander, or Native American.1
Also, while advances in medicine over the past several decades have reduced death rates from cancer and coronary artery disease and have contributed to a longer life expectancy for Americans, minority populations have not benefited equally from these improvements.2 In fact, the growing minority populations suffer from disparities in health care compared with white patients: minority patients have a higher incidence and burden of disease, and poorer health outcomes, contributing to shorter life expectancy.
Clearly, there is an urgent need for physicians, other health care providers, health systems, and medical researchers to increase their awareness of disparities in health care and their impact on patients, as well as on the US health system and the US economy. Now more than ever, we need to equip ourselves to more effectively engage minorities and to deliver culturally competent health care that improves outcomes in our minority patients.
A MULTIFACTORIAL PROBLEM
Disparities in health care are often thought to be the result of poverty and a related lack of access to quality health care. But clinical experience and research show that this is overly simplistic. In fact, disparities result from a variety of factors. Patient-related factors can include culturally related beliefs,1 dietary preferences, and health-seeking behaviors (perhaps influenced by a distrust of doctors, researchers, and the health care system), in addition to poor health literacy. Physician-related factors include poor cultural competency, which leads to poor communication with the patient. Other factors are a continuing lack of representation of minority patients in clinical research trials, as well as biologic factors.3
TAKING ACTION
In view of the disparities in health care that affect racial and ethnic minorities, and the many factors underlying the problem, the US Department of Health and Human Services launched the initiative Healthy People 2020, a continuation of the previous 10-year Healthy People initiatives. Healthy People 2020 calls for health providers and health systems to devise effective ways to eliminate health disparities.4 It outlines high-priority health issues, sets 10-year goals for improving the health of all Americans, and suggests specific actions to take to address health disparities.4
On another front, in 2010 the National Institutes of Health formally established its National Institute of Health and Health Disparities, which funds research into the pathogenesis of health disparities in racial and ethnic minorities.5 Clearly, racial, ethnic, and cultural factors need to be considered for health care to result in better outcomes in minority populations.
OUR NEW SERIES
In this issue of the Cleveland Clinic Journal of Medicine, we launch a series we hope will provide practical tools for physicians to address the disparities in our health care system. The first installment, by Odesosu et al (page 46), addresses barriers to optimal hypertension control in African Americans by outlining potential tactics for both patients and physicians. Future articles will address the challenge of health literacy and cultural issues in medicine, slowing the progression of renal disease in African Americans (especially the complex issue of which antihypertensive agents to use), and the challenges of diabetes in Hispanics. We also plan articles on kidney transplantation in African Americans and on prostate cancer, heart failure, lupus, and diabetes.
We look forward to your comments on this series as well as suggestions for future topics. We believe that as physicians, other health providers, health systems, health insurers and policy-makers become more aware of the disparities in health care, they will embrace ways in which to deliver or promote personalized, culturally competent health care. We hope this series will provide practical tools for physicians to address these complex issues.
In the united states, minority populations are rapidly increasing. In 1970, minorities—ie, African American, Hispanic, Asian, and Native American—accounted for 12.3% of the US population, but they now account for 25%. And this growth is expected to continue, so that by 2050 one of every two Americans will be African American, Hispanic, Asian, Pacific Islander, or Native American.1
Also, while advances in medicine over the past several decades have reduced death rates from cancer and coronary artery disease and have contributed to a longer life expectancy for Americans, minority populations have not benefited equally from these improvements.2 In fact, the growing minority populations suffer from disparities in health care compared with white patients: minority patients have a higher incidence and burden of disease, and poorer health outcomes, contributing to shorter life expectancy.
Clearly, there is an urgent need for physicians, other health care providers, health systems, and medical researchers to increase their awareness of disparities in health care and their impact on patients, as well as on the US health system and the US economy. Now more than ever, we need to equip ourselves to more effectively engage minorities and to deliver culturally competent health care that improves outcomes in our minority patients.
A MULTIFACTORIAL PROBLEM
Disparities in health care are often thought to be the result of poverty and a related lack of access to quality health care. But clinical experience and research show that this is overly simplistic. In fact, disparities result from a variety of factors. Patient-related factors can include culturally related beliefs,1 dietary preferences, and health-seeking behaviors (perhaps influenced by a distrust of doctors, researchers, and the health care system), in addition to poor health literacy. Physician-related factors include poor cultural competency, which leads to poor communication with the patient. Other factors are a continuing lack of representation of minority patients in clinical research trials, as well as biologic factors.3
TAKING ACTION
In view of the disparities in health care that affect racial and ethnic minorities, and the many factors underlying the problem, the US Department of Health and Human Services launched the initiative Healthy People 2020, a continuation of the previous 10-year Healthy People initiatives. Healthy People 2020 calls for health providers and health systems to devise effective ways to eliminate health disparities.4 It outlines high-priority health issues, sets 10-year goals for improving the health of all Americans, and suggests specific actions to take to address health disparities.4
On another front, in 2010 the National Institutes of Health formally established its National Institute of Health and Health Disparities, which funds research into the pathogenesis of health disparities in racial and ethnic minorities.5 Clearly, racial, ethnic, and cultural factors need to be considered for health care to result in better outcomes in minority populations.
OUR NEW SERIES
In this issue of the Cleveland Clinic Journal of Medicine, we launch a series we hope will provide practical tools for physicians to address the disparities in our health care system. The first installment, by Odesosu et al (page 46), addresses barriers to optimal hypertension control in African Americans by outlining potential tactics for both patients and physicians. Future articles will address the challenge of health literacy and cultural issues in medicine, slowing the progression of renal disease in African Americans (especially the complex issue of which antihypertensive agents to use), and the challenges of diabetes in Hispanics. We also plan articles on kidney transplantation in African Americans and on prostate cancer, heart failure, lupus, and diabetes.
We look forward to your comments on this series as well as suggestions for future topics. We believe that as physicians, other health providers, health systems, health insurers and policy-makers become more aware of the disparities in health care, they will embrace ways in which to deliver or promote personalized, culturally competent health care. We hope this series will provide practical tools for physicians to address these complex issues.
- Modlin CS. Culture, race, and disparities in health care. Cleve Clin J Med 2003; 70:283–288.
- Centers for Disease Control and Prevention life expectancy data. www.cdc.gov/nchs/fastats/lifexpec.htm. Accessed December 5, 2011.
- Klein JB, Nguyen CT, Saffore L, Modlin C, Modlin CS. Racial disparities in urologic health care. J Natl Med Assoc 2010: 102:108–117.
- Healthy People 2020. www.healthypeople.gov/2020/. Accessed December 5, 2011.
- National Institutes of Health. NIH announces Institute on minority health and health disparities. www.nih.gov/news/health/sep2010/nimhd-27.htm. Accessed December 5, 2011.
- Modlin CS. Culture, race, and disparities in health care. Cleve Clin J Med 2003; 70:283–288.
- Centers for Disease Control and Prevention life expectancy data. www.cdc.gov/nchs/fastats/lifexpec.htm. Accessed December 5, 2011.
- Klein JB, Nguyen CT, Saffore L, Modlin C, Modlin CS. Racial disparities in urologic health care. J Natl Med Assoc 2010: 102:108–117.
- Healthy People 2020. www.healthypeople.gov/2020/. Accessed December 5, 2011.
- National Institutes of Health. NIH announces Institute on minority health and health disparities. www.nih.gov/news/health/sep2010/nimhd-27.htm. Accessed December 5, 2011.
Is niacin ineffective? Or did AIM-HIGH miss its target?
The recent publication of the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides: Impact on Global Health Outcomes)1 has thrown the use of niacin as a lipid-modifying therapy into question. The trial was stopped early because an interim analysis found that the patients who took extended-release niacin had no clinical benefit. In addition, it found a trend toward more ischemic strokes, though this finding was later found not to be statistically significant.
Complicating the interpretation, while both the treatment group and the control group in the study received statin therapy, the researchers attempted to keep low-density lipoprotein cholesterol (LDL-C) levels equal, meaning that patients in the control group received more intensive statin therapy than those in the treatment group. And the placebo that the control patients received was actually a low dose of niacin, to induce flushing and thus to blind study participants and their physicians to which drug they were taking.
In the article that follows, I will explore the background, design, findings, and implications of this key trial and try to untangle the many questions about how to interpret it.
LOWERING LDL-C REDUCES RISK, BUT DOES NOT ELIMINATE IT
Large randomized controlled trials have consistently shown that lowering the level of LDL-C reduces cardiovascular event rates by 25% to 45% both in people who are known to have coronary artery disease and in those who are not.2–4 As a result, guidelines for preventing cardiovascular disease have increasingly emphasized maintaining low LDL-C levels. This has led to a proliferation in the use of inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase (statins) in patients at high cardiovascular risk.
However, these agents only reduce the risk—they do not eliminate it. Needed are additional therapies to complement existing LDL-C-lowering approaches to lower the cardiovascular risk even further.
Raising HDL-C: The next frontier
One such strategy for further lowering cardiovascular risk that has received considerable interest is to promote the biological activity of the “good” cholesterol.
Studies have consistently shown that the higher the plasma level of high-density lipoprotein cholesterol (HDL-C), the lower the risk of cardiovascular events, suggesting that raising HDL-C may be beneficial.5 Studies in animals with atherosclerosis show that raising HDL-C via genetic modification of the animal or direct infusion of the molecule has a favorable impact on both the size and the structure of experimental plaque.6,7
Accordingly, much activity has focused on developing new therapies that raise HDL-C more effectively than current ones.
Why niacin should protect the heart
For more than 50 years, niacin has been used to manage dyslipidemia.
In addition to raising HDL-C levels more effectively than any other agent available today, niacin also lowers the levels of LDL-C, triglycerides, and lipoprotein (a).8 Before statins were available, the Coronary Drug Project found that niacin reduced the rate of nonfatal myocardial infarction and the 15-year mortality rate.9 In addition, niacin has been shown to slow the progression of carotid intimal-medial thickness and coronary atherosclerosis, and even to reverse these processes in some trials.10–12
However, a number of issues remain about using niacin to prevent cardiovascular events. Nearly all patients who take it experience flushing, which limits its tolerability and, thus, our ability to titrate doses to levels needed for adequate lipid changes. While a number of modifications of niacin administration have been developed (eg, extended-release formulations and products that inhibit flushing), no large study has tested the clinical efficacy of these strategies. Furthermore, until AIM-HIGH, no large-scale trial had directly evaluated the impact of niacin therapy on a background of statin therapy.
AIM-HIGH STUDY DESIGN
The intent of the AIM-HIGH trial was to determine whether extended-release niacin (Niaspan) would reduce the risk of cardiovascular events when added to therapy with a statin—in this case, simvastatin (Zocor) supplemented with ezetimibe (Zetia).1
The trial was funded by the National Heart, Lung, and Blood Institute (NHLBI) and by Abbott Laboratories, which also supplied the extended-release niacin and the ezetimibe. Merck donated the simvastatin.
Patient characteristics
The patients were all at least 45 years of age with established, stable coronary heart disease, cerebrovascular or carotid arterial disease, or peripheral arterial disease. They also had to have low levels of HDL-C (< 40 mg/dL in men, < 50 mg/dL in women), elevated triglycerides (150–400 mg/dL), and LDL-C levels lower than 180 mg/dL if they were not taking a statin at entry.
The mean age of the patients was 64 years, 85% were men, and 92% were white. They had a high prevalence of cardiovascular risk factors: 34% had diabetes, 71% had hypertension, and 81% had metabolic syndrome. Nearly all (94%) of the patients were taking a statin at entry; 76% had been taking one for more than 1 year, and 40% had been taking one for more than 5 years.1
Simvastatin, ezetimibe, and either niacin or placebo
All lipid-modifying agents except statins and ezetimibe were stopped for least 4 weeks after enrollment.
All patients then entered a 4- to 8-week open-label period, during which they took simvastatin 40 mg daily and extended-release niacin starting at 500 mg and increased weekly up to 2,000 mg daily. Patients who could tolerate at least 1,500 mg daily were randomly assigned to treatment with either niacin 1,500 to 2,000 mg or matching placebo. Both groups continued to receive simvastatin. The placebo contained a small dose of immediate-release niacin (50 mg) in each tablet to induce flushing and to maintain blinding of treatment.
Given that niacin also lowers LDL-C, an algorithm was used to try to keep LDL-C levels roughly the same in both treatment groups. This involved adjusting the simvastatin dose and permitting the use of ezetimibe 10 mg to keep the LDL-C level between 40 and 80 mg/dL. Accordingly, participating physicians were told their patients’ LDL-C levels but were blinded to their HDL-C and triglyceride levels throughout the study.
Every 6 months, patients had a follow-up visit in the clinic, and midway through each 6-month interval they received a phone call from the investigators.1
AIM-HIGH end points
The primary end point was the composite of the first event of death due to coronary heart disease, nonfatal myocardial infarction, ischemic stroke, hospitalization for acute coronary syndrome, or symptom-driven revascularization of the coronary or cerebral arteries.
Secondary end points were:
- Death from coronary heart disease, nonfatal myocardial infarction, ischemic stroke, or hospitalization for acute coronary syndrome
- Death from coronary heart disease, nonfatal myocardial infarction, or ischemic stroke
- Death from cardiovascular causes.
Tertiary end points included:
- Death from any cause
- Individual components of the primary end point
- Prespecified subgroups according to sex, history or no history of diabetes, and presence or absence of the metabolic syndrome.1
All clinical events were adjudicated by a central committee.
STUDY HALTED EARLY
The study was planned to run for a mean of 4.6 years, during which 800 primary end point events were expected. With these numbers, the investigators calculated that the study had 85% power to detect a 25% reduction in the primary end point, at a one-sided alpha level of 0.025.
The plan called for an interim analysis when 50% of the anticipated events had occurred, with prespecified stopping boundaries based on either efficacy or futility. The boundary for lack of efficacy required an observed hazard ratio of at least 1.02 with a probability of less than .001.
In the interim analysis, after a median follow-up of only 3 years, the data and safety monitoring board recommended stopping the study early because the boundary for futility had been crossed and, unexpectedly, the rate of ischemic stroke was higher in the niacin-treated patients than in those receiving placebo.
MAJOR FINDINGS OF AIM-HIGH
Of 4,273 patients who began open-label treatment with niacin, 3,414 were randomized to treatment with niacin or placebo.1
HDL-C levels went up in both groups
At 2 years:
- HDL-C levels had increased by 25.0% (to 42 mg/dL) in the niacin group and by 9.8% (to 38 mg/dL) in the placebo group
- Triglycerides had decreased by 28.6% with niacin and by 8.1% with placebo
- LDL-C had decreased by 12.0% with niacin and by 5.5% with placebo.
Patients in the placebo group were more likely to have subsequently received the maximum dose of simvastatin, ie, 80 mg/day (24.7% vs 17.5%), and to have received ezetimibe (21.5% vs 9.5%). More patients in the niacin group required either dose reduction of the study drug (6.3% vs 3.4%) or drug discontinuation (25.4% vs 20.1%).1
No difference in the primary end point
There was no difference between the two treatment groups in the rate of the primary end point, which occurred in 282 (16.4%) of the 1,718 patients in the niacin group and 272 (16.2%) of the 1,696 patients in the placebo group (P = .79; hazard ratio 1.02, 95% confidence interval 0.87–1.21).1
However, more patients in the niacin group than in the placebo group who reached the primary end point did so by having a first ischemic stroke: 27 patients (1.6%) vs 15 patients (0.9%). Eight of these patients, all in the niacin group, had their stroke between 2 months and 4 years after they had stopped taking the study drug.
Further analysis that included all ischemic strokes revealed the same trend: 29 vs 18 patients (P = .11).1
No benefit was observed for niacin-treated patients in terms of any of the secondary or tertiary end points.
Subgroup analysis revealed no evidence of statistical heterogeneity: ie, niacin seemed to lack efficacy in all the prespecified subgroups studied (age 65 and older vs younger, men vs women, and those with or without diabetes, metabolic syndrome, prior myocardial infarction, or statin use at entry).
In general, niacin was well tolerated in the active-treatment group, with a low incidence of liver and muscle abnormalities.
PUTTING AIM-HIGH IN CONTEXT
How should practicing clinicians interpret these outcomes?
Ever since the NHLBI reported (in an urgent press release) that it was stopping the study early due to futility and a potential excess of strokes,13 there has been considerable debate as to which factors contributed to these outcomes. In the wake of the publication of more detailed information about the trial,1 this debate is likely to continue.
The AIM-HIGH results can be interpreted in several ways:
- Perhaps niacin is no good as a preventive agent
- Perhaps raising HDL-C is flawed as a preventive strategy
- Perhaps AIM-HIGH had methodologic flaws, such as looking at the wrong patient cohort or using a treatment protocol that set itself up for failure
- Perhaps statins are so good that, once you prescribe one, anything else you give provides no additional benefit.
Which of these is correct?
Is niacin no good?
In its most simple form, AIM-HIGH has always been seen as a clinical trial of niacin. While the early trials of immediate-release niacin were encouraging in terms of its effects on lipids, atherosclerotic plaque, and cardiovascular outcomes, using it in clinical practice has always been challenging, largely because many patients cannot tolerate it in doses high enough to be effective. A number of developments have improved niacin’s tolerability, but its clinical impact in the statin era has not been evaluated.
Niacin’s lack of efficacy in this trial will ultimately be viewed as a failure of the drug itself, but is this the case?
AIM-HIGH was not simply a direct comparison of niacin vs placebo on top of standard medical practice. The investigators recognized that niacin has additional effects—in particular, lowering levels of atherogenic lipids—and they attempted to control for these effects by titrating the other LDL-C-lowering therapies during the study. As a result, the trial was actually a comparison between niacin plus low-dose simvastatin on the one hand, and placebo plus high-dose simvastatin (and, more often, also ezetimibe) on the other.
Furthermore, the placebo-treated patients received small doses of immediate-release niacin to induce flushing and maintain blinding. It is therefore hard to conclude that this clinical trial was a direct evaluation of the impact of niacin.
In contrast, the Heart Protection Study 2-Treatment of HDL to Reduce the Incidence of Vascular Events (HPS2-THRIVE) study is currently evaluating extended-release niacin in combination with laropiprant, a prostaglandin receptor antagonist, vs placebo in more than 24,000 statin-treated patients.14 Without any in-trial titration of lipids, this study provides a more direct comparison of the effects of niacin in the statin era.
Niacin continues to attract interest, largely because it can raise HDL-C by 20% to 30% when given at doses of 1,500 mg or more. Also, consistent observations from population studies of an inverse relationship between HDL-C levels and cardiovascular risk5 have stimulated interest in developing novel agents that substantially raise HDL-C.
Is raising HDL-C a flawed strategy?
The failure of HDL-C-raising therapies in clinical trials15,16 has fueled concern that HDL may not be the magic elixir that many have sought. Given that niacin is the most effective HDL-C-raising agent currently available, its lack of efficacy in AIM-HIGH could be perceived as another nail in the coffin of the hypothesis that raising the HDL-C level with pharmacologic agents is beneficial.
AIM-HIGH was designed to examine the effects of raising HDL-C. To this end, it was performed exclusively in patients with low HDL-C levels, and the investigators tried to isolate the potential effects of raising HDL-C by equalizing the LDL-C levels in the treatment groups.
However, the HDL-C changes observed in AIM-HIGH are likely to have undermined the study objective. While niacin predictably increased HDL-C levels by 25%, an unexpected increase in HDL-C of 9.8% in the placebo-treated patients resulted in a difference in achieved HDL-C levels of only 4 mg/dL between the groups. This was far less than anticipated, and it likely had a major impact on an already underpowered study.
AIM-HIGH was designed to have 85% power to demonstrate a 25% reduction in clinical events, which was an optimistic estimate. On the basis of population studies, a difference of 4 mg/dL in HDL-C would be anticipated to result in no more than a 10% lower rate of clinical events, far beyond AIM-HIGH’s limit of detection.
The reasons for the increase in HDL-C in the placebo group are unknown, but they likely reflect the use of higher doses of simvastatin, some regression to the mean, and, possibly, the small doses of immediate-release niacin that the placebo contained. (Contrary to the belief of the investigators, there have been some reports of lipid changes with such doses,17 which may have contributed to the observed HDL-C-raising.)
Given that the HDL-C difference between the groups was relatively small and that niacin has additional effects beyond raising HDL-C and lowering LDL-C, it is unlikely that the futility of AIM-HIGH reflects a major indictment of HDL-C-raising. For the time being, the jury is still out on this question.
Was AIM-HIGH methodologically flawed?
A number of methodologic issues may have affected AIM-HIGH’s ability to adequately address its objectives.
The wrong cohort? In planning a study such as AIM-HIGH, the need for a relatively small sample size and the need to detect the greatest relative risk reduction with niacin would require enrollment of patients at the highest risk of cardiovascular events despite the use of statins. These needs were satisfied by only including patients who had atherosclerotic cardiovascular disease and low HDL-C levels. The inclusion of patients with low levels of HDL-C was also expected to promote greater increases in this lipid, and potentially event reduction, with niacin.
But no benefit was observed. It remains to be determined whether the inclusion of a high proportion of patients with the metabolic syndrome adversely affected the ability to detect a benefit with niacin. While post hoc analyses of studies of carotid intimal-medial thickness demonstrated no relationship between raising HDL-C with niacin and slowing of disease progression in patients with the metabolic syndrome,18 it remains to be determined whether this would translate to any effect on cardiovascular event rates.
Inadequate statistical power? An underpowered study would leave very little room for error, a pertinent point given the variability in therapeutic response in both actively treated and placebo-treated patients typically encountered in clinical trials. Giving low doses of immediate-release niacin and titrating the simvastatin dose to control LDL-C, resulting in imbalances in lipid-modifying therapies, represent additional flaws in the study design.
Stopped too soon? The early cessation of the study was somewhat questionable. The study crossed the prespecified boundary for lack of efficacy at the time of the interim analysis, and initial review by the data and safety monitoring board suggested an excess rate of ischemic stroke with niacin. The inclusion of this latter finding in the press release prompted considerable speculation regarding potential mechanisms and also concern among patients currently taking niacin. The subsequent finding that this signal was not statistically significant serves as an important warning for those conducting clinical trials not to prematurely overstate preliminary observations.
The implications for agents used in clinical practice are considerable: negative findings should not be overemphasized without robust evidence.
Do statins make everything else irrelevant?
The final factor to consider is the relative modifiability of residual clinical risk in statin-treated patients.
While residual risk is often cited as the reason to develop new antiatherosclerotic therapies, it is unknown how many of these ongoing events can be prevented. Several nonmodifiable factors such as age and concomitant disease are likely to contribute to these clinical events, which may limit our ability to further reduce event rates in patients who have already achieved low LDL-C levels with statin therapy. This may underscore the observation that no major clinical trial has demonstrated clinical benefit of an antiatherosclerotic agent on top of background medical care that included statins.
The finding that atherosclerosis continues to progress in many patients even though they take statins in high doses or achieve low LDL-C levels suggests that there is still room for improvement.
WHAT FUTURE FOR NIACIN?
So what does the future hold for niacin? The ongoing HPS2-THRIVE study provides another opportunity to evaluate the potential clinical efficacy of niacin in statin-treated patients. For now, we must wait for the results of this study.
In the meantime, it would seem reasonable to continue treatment with niacin in patients who need it for its multiple lipid-modifying effects. Whether clinicians will be less likely to initiate niacin therapy until there is clear evidence of clinical benefit remains uncertain. As for HDL-C, it remains to be determined whether any therapy targeting either quantitative or qualitative changes will be beneficial.
Over the last 3 decades, clinical trials have provided important insights into the prevention of cardiovascular events and have had a profound impact on clinical practice. Such studies simply evaluate whether one strategy is better or worse than the existing standard of care. They do not provide mechanistic insights, and when attempts have been made to address mechanisms in the study design, the trial, as in the case of AIM-HIGH, leaves more questions than answers.
Future trials will provide more clarity as to the optimal way to treat patients, but they must be based on a robust design that permits the study question to be adequately addressed.
- The AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011; 365:2255–2267.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med 1977; 62:707–714.
- Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature 1991; 353:265–267.
- Nicholls SJ, Cutri B, Worthley SG, et al. Impact of short-term administration of high-density lipoproteins and atorvastatin on atherosclerosis in rabbits. Arterioscler Thromb Vasc Biol 2005; 25:2416–2421.
- deLemos AS, Wolfe ML, Long CJ, Sivapackianathan R, Rader DJ. Identification of genetic variants in endothelial lipase in persons with elevated high-density lipoprotein cholesterol. Circulation 2002; 106:1321–1326.
- Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol 1986; 8:1245–1255.
- Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation 2004; 110:3512–3517.
- Taylor AJ, Lee HJ, Sullenberger LE. The effect of 24 months of combination statin and extended-release niacin on carotid intima-media thickness: ARBITER 3. Curr Med Res Opin 2006; 22:2243–2250.
- Brown BG, Zhao X-Q, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med 2001; 345:1583–1592.
- US Department of Health and Human Services. NIH stops clinical trial on combination cholesterol treatment. http://public.nhlbi.nih.gov/newsroom/home/GetPressRelease.aspx?id=2792. Accessed November 30, 2011.
- Brown BG, Zhao XQ. Nicotinic acid, alone and in combinations, for reduction of cardiovascular risk. Am J Cardiol 2008; 101:58B–62B.
- Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122.
- Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563–1574.
- Luria MH, Sapoznikov D. Raising HDL cholesterol with low-dose nicotinic acid and bezafibrate: preliminary experience. Postgrad Med J 1993; 69:296–299.
- Taylor AJ, Zhu D, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Relationship between glycemic status and progression of carotid intima-media thickness during treatment with combined statin and extended-release niacin in ARBITER 2. Vasc Health Risk Manag 2007; 3:159–164.
The recent publication of the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides: Impact on Global Health Outcomes)1 has thrown the use of niacin as a lipid-modifying therapy into question. The trial was stopped early because an interim analysis found that the patients who took extended-release niacin had no clinical benefit. In addition, it found a trend toward more ischemic strokes, though this finding was later found not to be statistically significant.
Complicating the interpretation, while both the treatment group and the control group in the study received statin therapy, the researchers attempted to keep low-density lipoprotein cholesterol (LDL-C) levels equal, meaning that patients in the control group received more intensive statin therapy than those in the treatment group. And the placebo that the control patients received was actually a low dose of niacin, to induce flushing and thus to blind study participants and their physicians to which drug they were taking.
In the article that follows, I will explore the background, design, findings, and implications of this key trial and try to untangle the many questions about how to interpret it.
LOWERING LDL-C REDUCES RISK, BUT DOES NOT ELIMINATE IT
Large randomized controlled trials have consistently shown that lowering the level of LDL-C reduces cardiovascular event rates by 25% to 45% both in people who are known to have coronary artery disease and in those who are not.2–4 As a result, guidelines for preventing cardiovascular disease have increasingly emphasized maintaining low LDL-C levels. This has led to a proliferation in the use of inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase (statins) in patients at high cardiovascular risk.
However, these agents only reduce the risk—they do not eliminate it. Needed are additional therapies to complement existing LDL-C-lowering approaches to lower the cardiovascular risk even further.
Raising HDL-C: The next frontier
One such strategy for further lowering cardiovascular risk that has received considerable interest is to promote the biological activity of the “good” cholesterol.
Studies have consistently shown that the higher the plasma level of high-density lipoprotein cholesterol (HDL-C), the lower the risk of cardiovascular events, suggesting that raising HDL-C may be beneficial.5 Studies in animals with atherosclerosis show that raising HDL-C via genetic modification of the animal or direct infusion of the molecule has a favorable impact on both the size and the structure of experimental plaque.6,7
Accordingly, much activity has focused on developing new therapies that raise HDL-C more effectively than current ones.
Why niacin should protect the heart
For more than 50 years, niacin has been used to manage dyslipidemia.
In addition to raising HDL-C levels more effectively than any other agent available today, niacin also lowers the levels of LDL-C, triglycerides, and lipoprotein (a).8 Before statins were available, the Coronary Drug Project found that niacin reduced the rate of nonfatal myocardial infarction and the 15-year mortality rate.9 In addition, niacin has been shown to slow the progression of carotid intimal-medial thickness and coronary atherosclerosis, and even to reverse these processes in some trials.10–12
However, a number of issues remain about using niacin to prevent cardiovascular events. Nearly all patients who take it experience flushing, which limits its tolerability and, thus, our ability to titrate doses to levels needed for adequate lipid changes. While a number of modifications of niacin administration have been developed (eg, extended-release formulations and products that inhibit flushing), no large study has tested the clinical efficacy of these strategies. Furthermore, until AIM-HIGH, no large-scale trial had directly evaluated the impact of niacin therapy on a background of statin therapy.
AIM-HIGH STUDY DESIGN
The intent of the AIM-HIGH trial was to determine whether extended-release niacin (Niaspan) would reduce the risk of cardiovascular events when added to therapy with a statin—in this case, simvastatin (Zocor) supplemented with ezetimibe (Zetia).1
The trial was funded by the National Heart, Lung, and Blood Institute (NHLBI) and by Abbott Laboratories, which also supplied the extended-release niacin and the ezetimibe. Merck donated the simvastatin.
Patient characteristics
The patients were all at least 45 years of age with established, stable coronary heart disease, cerebrovascular or carotid arterial disease, or peripheral arterial disease. They also had to have low levels of HDL-C (< 40 mg/dL in men, < 50 mg/dL in women), elevated triglycerides (150–400 mg/dL), and LDL-C levels lower than 180 mg/dL if they were not taking a statin at entry.
The mean age of the patients was 64 years, 85% were men, and 92% were white. They had a high prevalence of cardiovascular risk factors: 34% had diabetes, 71% had hypertension, and 81% had metabolic syndrome. Nearly all (94%) of the patients were taking a statin at entry; 76% had been taking one for more than 1 year, and 40% had been taking one for more than 5 years.1
Simvastatin, ezetimibe, and either niacin or placebo
All lipid-modifying agents except statins and ezetimibe were stopped for least 4 weeks after enrollment.
All patients then entered a 4- to 8-week open-label period, during which they took simvastatin 40 mg daily and extended-release niacin starting at 500 mg and increased weekly up to 2,000 mg daily. Patients who could tolerate at least 1,500 mg daily were randomly assigned to treatment with either niacin 1,500 to 2,000 mg or matching placebo. Both groups continued to receive simvastatin. The placebo contained a small dose of immediate-release niacin (50 mg) in each tablet to induce flushing and to maintain blinding of treatment.
Given that niacin also lowers LDL-C, an algorithm was used to try to keep LDL-C levels roughly the same in both treatment groups. This involved adjusting the simvastatin dose and permitting the use of ezetimibe 10 mg to keep the LDL-C level between 40 and 80 mg/dL. Accordingly, participating physicians were told their patients’ LDL-C levels but were blinded to their HDL-C and triglyceride levels throughout the study.
Every 6 months, patients had a follow-up visit in the clinic, and midway through each 6-month interval they received a phone call from the investigators.1
AIM-HIGH end points
The primary end point was the composite of the first event of death due to coronary heart disease, nonfatal myocardial infarction, ischemic stroke, hospitalization for acute coronary syndrome, or symptom-driven revascularization of the coronary or cerebral arteries.
Secondary end points were:
- Death from coronary heart disease, nonfatal myocardial infarction, ischemic stroke, or hospitalization for acute coronary syndrome
- Death from coronary heart disease, nonfatal myocardial infarction, or ischemic stroke
- Death from cardiovascular causes.
Tertiary end points included:
- Death from any cause
- Individual components of the primary end point
- Prespecified subgroups according to sex, history or no history of diabetes, and presence or absence of the metabolic syndrome.1
All clinical events were adjudicated by a central committee.
STUDY HALTED EARLY
The study was planned to run for a mean of 4.6 years, during which 800 primary end point events were expected. With these numbers, the investigators calculated that the study had 85% power to detect a 25% reduction in the primary end point, at a one-sided alpha level of 0.025.
The plan called for an interim analysis when 50% of the anticipated events had occurred, with prespecified stopping boundaries based on either efficacy or futility. The boundary for lack of efficacy required an observed hazard ratio of at least 1.02 with a probability of less than .001.
In the interim analysis, after a median follow-up of only 3 years, the data and safety monitoring board recommended stopping the study early because the boundary for futility had been crossed and, unexpectedly, the rate of ischemic stroke was higher in the niacin-treated patients than in those receiving placebo.
MAJOR FINDINGS OF AIM-HIGH
Of 4,273 patients who began open-label treatment with niacin, 3,414 were randomized to treatment with niacin or placebo.1
HDL-C levels went up in both groups
At 2 years:
- HDL-C levels had increased by 25.0% (to 42 mg/dL) in the niacin group and by 9.8% (to 38 mg/dL) in the placebo group
- Triglycerides had decreased by 28.6% with niacin and by 8.1% with placebo
- LDL-C had decreased by 12.0% with niacin and by 5.5% with placebo.
Patients in the placebo group were more likely to have subsequently received the maximum dose of simvastatin, ie, 80 mg/day (24.7% vs 17.5%), and to have received ezetimibe (21.5% vs 9.5%). More patients in the niacin group required either dose reduction of the study drug (6.3% vs 3.4%) or drug discontinuation (25.4% vs 20.1%).1
No difference in the primary end point
There was no difference between the two treatment groups in the rate of the primary end point, which occurred in 282 (16.4%) of the 1,718 patients in the niacin group and 272 (16.2%) of the 1,696 patients in the placebo group (P = .79; hazard ratio 1.02, 95% confidence interval 0.87–1.21).1
However, more patients in the niacin group than in the placebo group who reached the primary end point did so by having a first ischemic stroke: 27 patients (1.6%) vs 15 patients (0.9%). Eight of these patients, all in the niacin group, had their stroke between 2 months and 4 years after they had stopped taking the study drug.
Further analysis that included all ischemic strokes revealed the same trend: 29 vs 18 patients (P = .11).1
No benefit was observed for niacin-treated patients in terms of any of the secondary or tertiary end points.
Subgroup analysis revealed no evidence of statistical heterogeneity: ie, niacin seemed to lack efficacy in all the prespecified subgroups studied (age 65 and older vs younger, men vs women, and those with or without diabetes, metabolic syndrome, prior myocardial infarction, or statin use at entry).
In general, niacin was well tolerated in the active-treatment group, with a low incidence of liver and muscle abnormalities.
PUTTING AIM-HIGH IN CONTEXT
How should practicing clinicians interpret these outcomes?
Ever since the NHLBI reported (in an urgent press release) that it was stopping the study early due to futility and a potential excess of strokes,13 there has been considerable debate as to which factors contributed to these outcomes. In the wake of the publication of more detailed information about the trial,1 this debate is likely to continue.
The AIM-HIGH results can be interpreted in several ways:
- Perhaps niacin is no good as a preventive agent
- Perhaps raising HDL-C is flawed as a preventive strategy
- Perhaps AIM-HIGH had methodologic flaws, such as looking at the wrong patient cohort or using a treatment protocol that set itself up for failure
- Perhaps statins are so good that, once you prescribe one, anything else you give provides no additional benefit.
Which of these is correct?
Is niacin no good?
In its most simple form, AIM-HIGH has always been seen as a clinical trial of niacin. While the early trials of immediate-release niacin were encouraging in terms of its effects on lipids, atherosclerotic plaque, and cardiovascular outcomes, using it in clinical practice has always been challenging, largely because many patients cannot tolerate it in doses high enough to be effective. A number of developments have improved niacin’s tolerability, but its clinical impact in the statin era has not been evaluated.
Niacin’s lack of efficacy in this trial will ultimately be viewed as a failure of the drug itself, but is this the case?
AIM-HIGH was not simply a direct comparison of niacin vs placebo on top of standard medical practice. The investigators recognized that niacin has additional effects—in particular, lowering levels of atherogenic lipids—and they attempted to control for these effects by titrating the other LDL-C-lowering therapies during the study. As a result, the trial was actually a comparison between niacin plus low-dose simvastatin on the one hand, and placebo plus high-dose simvastatin (and, more often, also ezetimibe) on the other.
Furthermore, the placebo-treated patients received small doses of immediate-release niacin to induce flushing and maintain blinding. It is therefore hard to conclude that this clinical trial was a direct evaluation of the impact of niacin.
In contrast, the Heart Protection Study 2-Treatment of HDL to Reduce the Incidence of Vascular Events (HPS2-THRIVE) study is currently evaluating extended-release niacin in combination with laropiprant, a prostaglandin receptor antagonist, vs placebo in more than 24,000 statin-treated patients.14 Without any in-trial titration of lipids, this study provides a more direct comparison of the effects of niacin in the statin era.
Niacin continues to attract interest, largely because it can raise HDL-C by 20% to 30% when given at doses of 1,500 mg or more. Also, consistent observations from population studies of an inverse relationship between HDL-C levels and cardiovascular risk5 have stimulated interest in developing novel agents that substantially raise HDL-C.
Is raising HDL-C a flawed strategy?
The failure of HDL-C-raising therapies in clinical trials15,16 has fueled concern that HDL may not be the magic elixir that many have sought. Given that niacin is the most effective HDL-C-raising agent currently available, its lack of efficacy in AIM-HIGH could be perceived as another nail in the coffin of the hypothesis that raising the HDL-C level with pharmacologic agents is beneficial.
AIM-HIGH was designed to examine the effects of raising HDL-C. To this end, it was performed exclusively in patients with low HDL-C levels, and the investigators tried to isolate the potential effects of raising HDL-C by equalizing the LDL-C levels in the treatment groups.
However, the HDL-C changes observed in AIM-HIGH are likely to have undermined the study objective. While niacin predictably increased HDL-C levels by 25%, an unexpected increase in HDL-C of 9.8% in the placebo-treated patients resulted in a difference in achieved HDL-C levels of only 4 mg/dL between the groups. This was far less than anticipated, and it likely had a major impact on an already underpowered study.
AIM-HIGH was designed to have 85% power to demonstrate a 25% reduction in clinical events, which was an optimistic estimate. On the basis of population studies, a difference of 4 mg/dL in HDL-C would be anticipated to result in no more than a 10% lower rate of clinical events, far beyond AIM-HIGH’s limit of detection.
The reasons for the increase in HDL-C in the placebo group are unknown, but they likely reflect the use of higher doses of simvastatin, some regression to the mean, and, possibly, the small doses of immediate-release niacin that the placebo contained. (Contrary to the belief of the investigators, there have been some reports of lipid changes with such doses,17 which may have contributed to the observed HDL-C-raising.)
Given that the HDL-C difference between the groups was relatively small and that niacin has additional effects beyond raising HDL-C and lowering LDL-C, it is unlikely that the futility of AIM-HIGH reflects a major indictment of HDL-C-raising. For the time being, the jury is still out on this question.
Was AIM-HIGH methodologically flawed?
A number of methodologic issues may have affected AIM-HIGH’s ability to adequately address its objectives.
The wrong cohort? In planning a study such as AIM-HIGH, the need for a relatively small sample size and the need to detect the greatest relative risk reduction with niacin would require enrollment of patients at the highest risk of cardiovascular events despite the use of statins. These needs were satisfied by only including patients who had atherosclerotic cardiovascular disease and low HDL-C levels. The inclusion of patients with low levels of HDL-C was also expected to promote greater increases in this lipid, and potentially event reduction, with niacin.
But no benefit was observed. It remains to be determined whether the inclusion of a high proportion of patients with the metabolic syndrome adversely affected the ability to detect a benefit with niacin. While post hoc analyses of studies of carotid intimal-medial thickness demonstrated no relationship between raising HDL-C with niacin and slowing of disease progression in patients with the metabolic syndrome,18 it remains to be determined whether this would translate to any effect on cardiovascular event rates.
Inadequate statistical power? An underpowered study would leave very little room for error, a pertinent point given the variability in therapeutic response in both actively treated and placebo-treated patients typically encountered in clinical trials. Giving low doses of immediate-release niacin and titrating the simvastatin dose to control LDL-C, resulting in imbalances in lipid-modifying therapies, represent additional flaws in the study design.
Stopped too soon? The early cessation of the study was somewhat questionable. The study crossed the prespecified boundary for lack of efficacy at the time of the interim analysis, and initial review by the data and safety monitoring board suggested an excess rate of ischemic stroke with niacin. The inclusion of this latter finding in the press release prompted considerable speculation regarding potential mechanisms and also concern among patients currently taking niacin. The subsequent finding that this signal was not statistically significant serves as an important warning for those conducting clinical trials not to prematurely overstate preliminary observations.
The implications for agents used in clinical practice are considerable: negative findings should not be overemphasized without robust evidence.
Do statins make everything else irrelevant?
The final factor to consider is the relative modifiability of residual clinical risk in statin-treated patients.
While residual risk is often cited as the reason to develop new antiatherosclerotic therapies, it is unknown how many of these ongoing events can be prevented. Several nonmodifiable factors such as age and concomitant disease are likely to contribute to these clinical events, which may limit our ability to further reduce event rates in patients who have already achieved low LDL-C levels with statin therapy. This may underscore the observation that no major clinical trial has demonstrated clinical benefit of an antiatherosclerotic agent on top of background medical care that included statins.
The finding that atherosclerosis continues to progress in many patients even though they take statins in high doses or achieve low LDL-C levels suggests that there is still room for improvement.
WHAT FUTURE FOR NIACIN?
So what does the future hold for niacin? The ongoing HPS2-THRIVE study provides another opportunity to evaluate the potential clinical efficacy of niacin in statin-treated patients. For now, we must wait for the results of this study.
In the meantime, it would seem reasonable to continue treatment with niacin in patients who need it for its multiple lipid-modifying effects. Whether clinicians will be less likely to initiate niacin therapy until there is clear evidence of clinical benefit remains uncertain. As for HDL-C, it remains to be determined whether any therapy targeting either quantitative or qualitative changes will be beneficial.
Over the last 3 decades, clinical trials have provided important insights into the prevention of cardiovascular events and have had a profound impact on clinical practice. Such studies simply evaluate whether one strategy is better or worse than the existing standard of care. They do not provide mechanistic insights, and when attempts have been made to address mechanisms in the study design, the trial, as in the case of AIM-HIGH, leaves more questions than answers.
Future trials will provide more clarity as to the optimal way to treat patients, but they must be based on a robust design that permits the study question to be adequately addressed.
The recent publication of the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides: Impact on Global Health Outcomes)1 has thrown the use of niacin as a lipid-modifying therapy into question. The trial was stopped early because an interim analysis found that the patients who took extended-release niacin had no clinical benefit. In addition, it found a trend toward more ischemic strokes, though this finding was later found not to be statistically significant.
Complicating the interpretation, while both the treatment group and the control group in the study received statin therapy, the researchers attempted to keep low-density lipoprotein cholesterol (LDL-C) levels equal, meaning that patients in the control group received more intensive statin therapy than those in the treatment group. And the placebo that the control patients received was actually a low dose of niacin, to induce flushing and thus to blind study participants and their physicians to which drug they were taking.
In the article that follows, I will explore the background, design, findings, and implications of this key trial and try to untangle the many questions about how to interpret it.
LOWERING LDL-C REDUCES RISK, BUT DOES NOT ELIMINATE IT
Large randomized controlled trials have consistently shown that lowering the level of LDL-C reduces cardiovascular event rates by 25% to 45% both in people who are known to have coronary artery disease and in those who are not.2–4 As a result, guidelines for preventing cardiovascular disease have increasingly emphasized maintaining low LDL-C levels. This has led to a proliferation in the use of inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase (statins) in patients at high cardiovascular risk.
However, these agents only reduce the risk—they do not eliminate it. Needed are additional therapies to complement existing LDL-C-lowering approaches to lower the cardiovascular risk even further.
Raising HDL-C: The next frontier
One such strategy for further lowering cardiovascular risk that has received considerable interest is to promote the biological activity of the “good” cholesterol.
Studies have consistently shown that the higher the plasma level of high-density lipoprotein cholesterol (HDL-C), the lower the risk of cardiovascular events, suggesting that raising HDL-C may be beneficial.5 Studies in animals with atherosclerosis show that raising HDL-C via genetic modification of the animal or direct infusion of the molecule has a favorable impact on both the size and the structure of experimental plaque.6,7
Accordingly, much activity has focused on developing new therapies that raise HDL-C more effectively than current ones.
Why niacin should protect the heart
For more than 50 years, niacin has been used to manage dyslipidemia.
In addition to raising HDL-C levels more effectively than any other agent available today, niacin also lowers the levels of LDL-C, triglycerides, and lipoprotein (a).8 Before statins were available, the Coronary Drug Project found that niacin reduced the rate of nonfatal myocardial infarction and the 15-year mortality rate.9 In addition, niacin has been shown to slow the progression of carotid intimal-medial thickness and coronary atherosclerosis, and even to reverse these processes in some trials.10–12
However, a number of issues remain about using niacin to prevent cardiovascular events. Nearly all patients who take it experience flushing, which limits its tolerability and, thus, our ability to titrate doses to levels needed for adequate lipid changes. While a number of modifications of niacin administration have been developed (eg, extended-release formulations and products that inhibit flushing), no large study has tested the clinical efficacy of these strategies. Furthermore, until AIM-HIGH, no large-scale trial had directly evaluated the impact of niacin therapy on a background of statin therapy.
AIM-HIGH STUDY DESIGN
The intent of the AIM-HIGH trial was to determine whether extended-release niacin (Niaspan) would reduce the risk of cardiovascular events when added to therapy with a statin—in this case, simvastatin (Zocor) supplemented with ezetimibe (Zetia).1
The trial was funded by the National Heart, Lung, and Blood Institute (NHLBI) and by Abbott Laboratories, which also supplied the extended-release niacin and the ezetimibe. Merck donated the simvastatin.
Patient characteristics
The patients were all at least 45 years of age with established, stable coronary heart disease, cerebrovascular or carotid arterial disease, or peripheral arterial disease. They also had to have low levels of HDL-C (< 40 mg/dL in men, < 50 mg/dL in women), elevated triglycerides (150–400 mg/dL), and LDL-C levels lower than 180 mg/dL if they were not taking a statin at entry.
The mean age of the patients was 64 years, 85% were men, and 92% were white. They had a high prevalence of cardiovascular risk factors: 34% had diabetes, 71% had hypertension, and 81% had metabolic syndrome. Nearly all (94%) of the patients were taking a statin at entry; 76% had been taking one for more than 1 year, and 40% had been taking one for more than 5 years.1
Simvastatin, ezetimibe, and either niacin or placebo
All lipid-modifying agents except statins and ezetimibe were stopped for least 4 weeks after enrollment.
All patients then entered a 4- to 8-week open-label period, during which they took simvastatin 40 mg daily and extended-release niacin starting at 500 mg and increased weekly up to 2,000 mg daily. Patients who could tolerate at least 1,500 mg daily were randomly assigned to treatment with either niacin 1,500 to 2,000 mg or matching placebo. Both groups continued to receive simvastatin. The placebo contained a small dose of immediate-release niacin (50 mg) in each tablet to induce flushing and to maintain blinding of treatment.
Given that niacin also lowers LDL-C, an algorithm was used to try to keep LDL-C levels roughly the same in both treatment groups. This involved adjusting the simvastatin dose and permitting the use of ezetimibe 10 mg to keep the LDL-C level between 40 and 80 mg/dL. Accordingly, participating physicians were told their patients’ LDL-C levels but were blinded to their HDL-C and triglyceride levels throughout the study.
Every 6 months, patients had a follow-up visit in the clinic, and midway through each 6-month interval they received a phone call from the investigators.1
AIM-HIGH end points
The primary end point was the composite of the first event of death due to coronary heart disease, nonfatal myocardial infarction, ischemic stroke, hospitalization for acute coronary syndrome, or symptom-driven revascularization of the coronary or cerebral arteries.
Secondary end points were:
- Death from coronary heart disease, nonfatal myocardial infarction, ischemic stroke, or hospitalization for acute coronary syndrome
- Death from coronary heart disease, nonfatal myocardial infarction, or ischemic stroke
- Death from cardiovascular causes.
Tertiary end points included:
- Death from any cause
- Individual components of the primary end point
- Prespecified subgroups according to sex, history or no history of diabetes, and presence or absence of the metabolic syndrome.1
All clinical events were adjudicated by a central committee.
STUDY HALTED EARLY
The study was planned to run for a mean of 4.6 years, during which 800 primary end point events were expected. With these numbers, the investigators calculated that the study had 85% power to detect a 25% reduction in the primary end point, at a one-sided alpha level of 0.025.
The plan called for an interim analysis when 50% of the anticipated events had occurred, with prespecified stopping boundaries based on either efficacy or futility. The boundary for lack of efficacy required an observed hazard ratio of at least 1.02 with a probability of less than .001.
In the interim analysis, after a median follow-up of only 3 years, the data and safety monitoring board recommended stopping the study early because the boundary for futility had been crossed and, unexpectedly, the rate of ischemic stroke was higher in the niacin-treated patients than in those receiving placebo.
MAJOR FINDINGS OF AIM-HIGH
Of 4,273 patients who began open-label treatment with niacin, 3,414 were randomized to treatment with niacin or placebo.1
HDL-C levels went up in both groups
At 2 years:
- HDL-C levels had increased by 25.0% (to 42 mg/dL) in the niacin group and by 9.8% (to 38 mg/dL) in the placebo group
- Triglycerides had decreased by 28.6% with niacin and by 8.1% with placebo
- LDL-C had decreased by 12.0% with niacin and by 5.5% with placebo.
Patients in the placebo group were more likely to have subsequently received the maximum dose of simvastatin, ie, 80 mg/day (24.7% vs 17.5%), and to have received ezetimibe (21.5% vs 9.5%). More patients in the niacin group required either dose reduction of the study drug (6.3% vs 3.4%) or drug discontinuation (25.4% vs 20.1%).1
No difference in the primary end point
There was no difference between the two treatment groups in the rate of the primary end point, which occurred in 282 (16.4%) of the 1,718 patients in the niacin group and 272 (16.2%) of the 1,696 patients in the placebo group (P = .79; hazard ratio 1.02, 95% confidence interval 0.87–1.21).1
However, more patients in the niacin group than in the placebo group who reached the primary end point did so by having a first ischemic stroke: 27 patients (1.6%) vs 15 patients (0.9%). Eight of these patients, all in the niacin group, had their stroke between 2 months and 4 years after they had stopped taking the study drug.
Further analysis that included all ischemic strokes revealed the same trend: 29 vs 18 patients (P = .11).1
No benefit was observed for niacin-treated patients in terms of any of the secondary or tertiary end points.
Subgroup analysis revealed no evidence of statistical heterogeneity: ie, niacin seemed to lack efficacy in all the prespecified subgroups studied (age 65 and older vs younger, men vs women, and those with or without diabetes, metabolic syndrome, prior myocardial infarction, or statin use at entry).
In general, niacin was well tolerated in the active-treatment group, with a low incidence of liver and muscle abnormalities.
PUTTING AIM-HIGH IN CONTEXT
How should practicing clinicians interpret these outcomes?
Ever since the NHLBI reported (in an urgent press release) that it was stopping the study early due to futility and a potential excess of strokes,13 there has been considerable debate as to which factors contributed to these outcomes. In the wake of the publication of more detailed information about the trial,1 this debate is likely to continue.
The AIM-HIGH results can be interpreted in several ways:
- Perhaps niacin is no good as a preventive agent
- Perhaps raising HDL-C is flawed as a preventive strategy
- Perhaps AIM-HIGH had methodologic flaws, such as looking at the wrong patient cohort or using a treatment protocol that set itself up for failure
- Perhaps statins are so good that, once you prescribe one, anything else you give provides no additional benefit.
Which of these is correct?
Is niacin no good?
In its most simple form, AIM-HIGH has always been seen as a clinical trial of niacin. While the early trials of immediate-release niacin were encouraging in terms of its effects on lipids, atherosclerotic plaque, and cardiovascular outcomes, using it in clinical practice has always been challenging, largely because many patients cannot tolerate it in doses high enough to be effective. A number of developments have improved niacin’s tolerability, but its clinical impact in the statin era has not been evaluated.
Niacin’s lack of efficacy in this trial will ultimately be viewed as a failure of the drug itself, but is this the case?
AIM-HIGH was not simply a direct comparison of niacin vs placebo on top of standard medical practice. The investigators recognized that niacin has additional effects—in particular, lowering levels of atherogenic lipids—and they attempted to control for these effects by titrating the other LDL-C-lowering therapies during the study. As a result, the trial was actually a comparison between niacin plus low-dose simvastatin on the one hand, and placebo plus high-dose simvastatin (and, more often, also ezetimibe) on the other.
Furthermore, the placebo-treated patients received small doses of immediate-release niacin to induce flushing and maintain blinding. It is therefore hard to conclude that this clinical trial was a direct evaluation of the impact of niacin.
In contrast, the Heart Protection Study 2-Treatment of HDL to Reduce the Incidence of Vascular Events (HPS2-THRIVE) study is currently evaluating extended-release niacin in combination with laropiprant, a prostaglandin receptor antagonist, vs placebo in more than 24,000 statin-treated patients.14 Without any in-trial titration of lipids, this study provides a more direct comparison of the effects of niacin in the statin era.
Niacin continues to attract interest, largely because it can raise HDL-C by 20% to 30% when given at doses of 1,500 mg or more. Also, consistent observations from population studies of an inverse relationship between HDL-C levels and cardiovascular risk5 have stimulated interest in developing novel agents that substantially raise HDL-C.
Is raising HDL-C a flawed strategy?
The failure of HDL-C-raising therapies in clinical trials15,16 has fueled concern that HDL may not be the magic elixir that many have sought. Given that niacin is the most effective HDL-C-raising agent currently available, its lack of efficacy in AIM-HIGH could be perceived as another nail in the coffin of the hypothesis that raising the HDL-C level with pharmacologic agents is beneficial.
AIM-HIGH was designed to examine the effects of raising HDL-C. To this end, it was performed exclusively in patients with low HDL-C levels, and the investigators tried to isolate the potential effects of raising HDL-C by equalizing the LDL-C levels in the treatment groups.
However, the HDL-C changes observed in AIM-HIGH are likely to have undermined the study objective. While niacin predictably increased HDL-C levels by 25%, an unexpected increase in HDL-C of 9.8% in the placebo-treated patients resulted in a difference in achieved HDL-C levels of only 4 mg/dL between the groups. This was far less than anticipated, and it likely had a major impact on an already underpowered study.
AIM-HIGH was designed to have 85% power to demonstrate a 25% reduction in clinical events, which was an optimistic estimate. On the basis of population studies, a difference of 4 mg/dL in HDL-C would be anticipated to result in no more than a 10% lower rate of clinical events, far beyond AIM-HIGH’s limit of detection.
The reasons for the increase in HDL-C in the placebo group are unknown, but they likely reflect the use of higher doses of simvastatin, some regression to the mean, and, possibly, the small doses of immediate-release niacin that the placebo contained. (Contrary to the belief of the investigators, there have been some reports of lipid changes with such doses,17 which may have contributed to the observed HDL-C-raising.)
Given that the HDL-C difference between the groups was relatively small and that niacin has additional effects beyond raising HDL-C and lowering LDL-C, it is unlikely that the futility of AIM-HIGH reflects a major indictment of HDL-C-raising. For the time being, the jury is still out on this question.
Was AIM-HIGH methodologically flawed?
A number of methodologic issues may have affected AIM-HIGH’s ability to adequately address its objectives.
The wrong cohort? In planning a study such as AIM-HIGH, the need for a relatively small sample size and the need to detect the greatest relative risk reduction with niacin would require enrollment of patients at the highest risk of cardiovascular events despite the use of statins. These needs were satisfied by only including patients who had atherosclerotic cardiovascular disease and low HDL-C levels. The inclusion of patients with low levels of HDL-C was also expected to promote greater increases in this lipid, and potentially event reduction, with niacin.
But no benefit was observed. It remains to be determined whether the inclusion of a high proportion of patients with the metabolic syndrome adversely affected the ability to detect a benefit with niacin. While post hoc analyses of studies of carotid intimal-medial thickness demonstrated no relationship between raising HDL-C with niacin and slowing of disease progression in patients with the metabolic syndrome,18 it remains to be determined whether this would translate to any effect on cardiovascular event rates.
Inadequate statistical power? An underpowered study would leave very little room for error, a pertinent point given the variability in therapeutic response in both actively treated and placebo-treated patients typically encountered in clinical trials. Giving low doses of immediate-release niacin and titrating the simvastatin dose to control LDL-C, resulting in imbalances in lipid-modifying therapies, represent additional flaws in the study design.
Stopped too soon? The early cessation of the study was somewhat questionable. The study crossed the prespecified boundary for lack of efficacy at the time of the interim analysis, and initial review by the data and safety monitoring board suggested an excess rate of ischemic stroke with niacin. The inclusion of this latter finding in the press release prompted considerable speculation regarding potential mechanisms and also concern among patients currently taking niacin. The subsequent finding that this signal was not statistically significant serves as an important warning for those conducting clinical trials not to prematurely overstate preliminary observations.
The implications for agents used in clinical practice are considerable: negative findings should not be overemphasized without robust evidence.
Do statins make everything else irrelevant?
The final factor to consider is the relative modifiability of residual clinical risk in statin-treated patients.
While residual risk is often cited as the reason to develop new antiatherosclerotic therapies, it is unknown how many of these ongoing events can be prevented. Several nonmodifiable factors such as age and concomitant disease are likely to contribute to these clinical events, which may limit our ability to further reduce event rates in patients who have already achieved low LDL-C levels with statin therapy. This may underscore the observation that no major clinical trial has demonstrated clinical benefit of an antiatherosclerotic agent on top of background medical care that included statins.
The finding that atherosclerosis continues to progress in many patients even though they take statins in high doses or achieve low LDL-C levels suggests that there is still room for improvement.
WHAT FUTURE FOR NIACIN?
So what does the future hold for niacin? The ongoing HPS2-THRIVE study provides another opportunity to evaluate the potential clinical efficacy of niacin in statin-treated patients. For now, we must wait for the results of this study.
In the meantime, it would seem reasonable to continue treatment with niacin in patients who need it for its multiple lipid-modifying effects. Whether clinicians will be less likely to initiate niacin therapy until there is clear evidence of clinical benefit remains uncertain. As for HDL-C, it remains to be determined whether any therapy targeting either quantitative or qualitative changes will be beneficial.
Over the last 3 decades, clinical trials have provided important insights into the prevention of cardiovascular events and have had a profound impact on clinical practice. Such studies simply evaluate whether one strategy is better or worse than the existing standard of care. They do not provide mechanistic insights, and when attempts have been made to address mechanisms in the study design, the trial, as in the case of AIM-HIGH, leaves more questions than answers.
Future trials will provide more clarity as to the optimal way to treat patients, but they must be based on a robust design that permits the study question to be adequately addressed.
- The AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011; 365:2255–2267.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med 1977; 62:707–714.
- Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature 1991; 353:265–267.
- Nicholls SJ, Cutri B, Worthley SG, et al. Impact of short-term administration of high-density lipoproteins and atorvastatin on atherosclerosis in rabbits. Arterioscler Thromb Vasc Biol 2005; 25:2416–2421.
- deLemos AS, Wolfe ML, Long CJ, Sivapackianathan R, Rader DJ. Identification of genetic variants in endothelial lipase in persons with elevated high-density lipoprotein cholesterol. Circulation 2002; 106:1321–1326.
- Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol 1986; 8:1245–1255.
- Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation 2004; 110:3512–3517.
- Taylor AJ, Lee HJ, Sullenberger LE. The effect of 24 months of combination statin and extended-release niacin on carotid intima-media thickness: ARBITER 3. Curr Med Res Opin 2006; 22:2243–2250.
- Brown BG, Zhao X-Q, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med 2001; 345:1583–1592.
- US Department of Health and Human Services. NIH stops clinical trial on combination cholesterol treatment. http://public.nhlbi.nih.gov/newsroom/home/GetPressRelease.aspx?id=2792. Accessed November 30, 2011.
- Brown BG, Zhao XQ. Nicotinic acid, alone and in combinations, for reduction of cardiovascular risk. Am J Cardiol 2008; 101:58B–62B.
- Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122.
- Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563–1574.
- Luria MH, Sapoznikov D. Raising HDL cholesterol with low-dose nicotinic acid and bezafibrate: preliminary experience. Postgrad Med J 1993; 69:296–299.
- Taylor AJ, Zhu D, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Relationship between glycemic status and progression of carotid intima-media thickness during treatment with combined statin and extended-release niacin in ARBITER 2. Vasc Health Risk Manag 2007; 3:159–164.
- The AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011; 365:2255–2267.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med 1977; 62:707–714.
- Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature 1991; 353:265–267.
- Nicholls SJ, Cutri B, Worthley SG, et al. Impact of short-term administration of high-density lipoproteins and atorvastatin on atherosclerosis in rabbits. Arterioscler Thromb Vasc Biol 2005; 25:2416–2421.
- deLemos AS, Wolfe ML, Long CJ, Sivapackianathan R, Rader DJ. Identification of genetic variants in endothelial lipase in persons with elevated high-density lipoprotein cholesterol. Circulation 2002; 106:1321–1326.
- Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol 1986; 8:1245–1255.
- Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation 2004; 110:3512–3517.
- Taylor AJ, Lee HJ, Sullenberger LE. The effect of 24 months of combination statin and extended-release niacin on carotid intima-media thickness: ARBITER 3. Curr Med Res Opin 2006; 22:2243–2250.
- Brown BG, Zhao X-Q, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med 2001; 345:1583–1592.
- US Department of Health and Human Services. NIH stops clinical trial on combination cholesterol treatment. http://public.nhlbi.nih.gov/newsroom/home/GetPressRelease.aspx?id=2792. Accessed November 30, 2011.
- Brown BG, Zhao XQ. Nicotinic acid, alone and in combinations, for reduction of cardiovascular risk. Am J Cardiol 2008; 101:58B–62B.
- Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122.
- Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563–1574.
- Luria MH, Sapoznikov D. Raising HDL cholesterol with low-dose nicotinic acid and bezafibrate: preliminary experience. Postgrad Med J 1993; 69:296–299.
- Taylor AJ, Zhu D, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Relationship between glycemic status and progression of carotid intima-media thickness during treatment with combined statin and extended-release niacin in ARBITER 2. Vasc Health Risk Manag 2007; 3:159–164.
KEY POINTS
- The study was stopped early because of the concerns raised by the interim analysis.
- The AIM-HIGH results can be interpreted in several ways: perhaps niacin is no good as a preventive agent; perhaps raising levels of high-density lipoprotein cholesterol (HDL-C) is flawed as a preventive strategy; perhaps AIM-HIGH had methodologic flaws; or perhaps statins are so good that, once you prescribe one, anything else you do will not make much of a difference.
- It seems reasonable to continue niacin treatment in patients who need its multiple lipid-modifying effects. It is uncertain if clinicians will be less likely to prescribe niacin therapy until we have clear evidence of clinical benefit. As for HDL-C, it remains to be determined whether any therapy targeting quantitative or qualitative changes will be beneficial.
Updates in the medical management of Parkinson disease
More than a dozen drugs have been approved by the US Food and Drug Administration (FDA) for treating Parkinson disease, and more are expected in the near future. Many are currently in clinical trials, with the goals of finding ways to better control the disease with fewer adverse effects and, ultimately, to provide neuroprotection.
This article will review the features of Parkinson disease, the treatment options, and the complications in moderate to advanced disease.
PARKINSON DISEASE IS MULTIFACTORIAL
Although the cure for Parkinson disease is still elusive, much has been learned over the nearly 200 years since it was first described by James Parkinson in 1817. It is now understood to be a progressive neurodegenerative disease of multifactorial etiology: although a small proportion of patients have a direct inherited mutation that causes it, multiple genetic predisposition factors and environmental factors are more commonly involved.
The central pathology is dopaminergic loss in the basal ganglia, but other neurotransmitters are also involved and the disease extends to other areas of the brain.
CARDINAL MOTOR SYMPTOMS
In general, Parkinson disease is easy to identify. The classic patient has1:
- Tremor at rest, which can be subtle—such as only involving a thumb or a few fingers—and is absent in 20% of patients at presentation.
- Rigidity, which is felt by the examiner rather than seen by an observer.
- Bradykinesia (slow movements), which is characteristic of all Parkinson patients.
- Gait and balance problems, which usually arise after a few years, although occasionally patients present with them. Patients typically walk with small steps with occasional freezing, as if their foot were stuck. Balance problems are the most difficult to treat among the motor problems.
Asymmetry of motor problems is apparent in 75% of patients at presentation, although problems become bilateral later in the course of the disease.
NONMOTOR FEATURES CAN BE MORE DISABLING

Pain is common, but years ago it was not recognized as a specific feature of Parkinson disease. The pain from other conditions may also worsen.
Fatigue is very common and, if present, is usually one of the most disabling features.
Neuropsychiatric disturbances are among the most difficult problems, and they become increasingly common as motor symptoms are better controlled with treatment and patients live longer.
INCREASINGLY PREVALENT AS THE POPULATION AGES
Parkinson disease can present from the teenage years up to age 90, but it is most often diagnosed in patients from 60 to 70 years old (mean onset, 62.5 years). A different nomenclature is used depending on the age of onset:
- 10 to 20 years: juvenile-onset
- 21 to 40 years: young-onset.
Parkinson disease is now an epidemic, with an estimated 1 million people having it in the United States, representing 0.3% of the population and 1% of those older than 60 years.2 More people can be expected to develop it as our population ages in the next decades. It is estimated that in 2040 more people will die from Parkinson disease, Alzheimer disease, and amyotrophic lateral sclerosis (all of which are neurodegenerative diseases) than from kidney cancer, malignant melanoma, colon cancer, and lung cancer combined.
DIAGNOSIS IS STILL MAINLY CLINICAL
The diagnosis of Parkinson disease remains clinical. In addition to the motor features, the best test is a clear response to dopaminergic treatment with levodopa. If all these features are present, the diagnosis of Parkinson disease is usually correct.3
Imaging useful in select patients
The FDA recently approved a radiopharmaceutical contrast agent, DaTscan, to use with single-photon emission computed tomography (SPECT) to help diagnose Parkinson disease. DaTscan is a dopamine transporter ligand that tags presynaptic dopaminergic neurons in the basal ganglia; a patient with Parkinson disease has less signal.
The test can be used to distinguish parkinsonian syndromes from disorders that can mimic them, such as essential tremor or a psychogenic disorder. However, it cannot differentiate various Parkinson-plus syndromes (see below) such as multiple system atrophy or progressive nuclear palsy. It also cannot be used to detect drug-induced or vascular parkinsonism.
Check for Wilson disease or brain tumors in young or atypical cases
For most patients, no imaging or blood tests are needed to make the diagnosis. However, in patients younger than 50, Wilson disease, a rare inherited disorder characterized by excess copper accumulation, must be considered. Testing for Wilson disease includes serum ceruloplasmin, 24-hour urinary copper excretion, and an ophthalmologic slit-lamp examination for Kaiser-Fleischer rings.
For patients who do not quite fit the picture of Parkinson disease, such as those who have spasticity with little tremor, or who have a minimal response to levodopa, magnetic resonance imaging should be done to see if a structural lesion is present.
Consider secondary parkinsonism
Although idiopathic Parkinson disease is by far the most common form of parkinsonism in the United States and in most developing countries, secondary causes must also be considered in a patient presenting with symptoms of parkinsonism. They include:
- Dopamine-receptor blocking agents: metoclopramide (Reglan), prochlorperazine (Compazine), haloperidol (Haldol), thioridazine (Mellaril), risperidone (Risperdal), olanzapine (Zyprexa)
- Strokes in the basal ganglia
- Normal pressure hydrocephalus.
Parkinson-plus syndromes
Parkinson-plus syndromes have other features in addition to the classic features of idiopathic Parkinson disease. They occur commonly and can be difficult to distinguish from Parkinson disease and from each other.
Parkinson-plus syndromes include:
- Progressive supranuclear palsy
- Multiple system atrophy
- Corticobasal degeneration
- Lewy body dementia.
Clinical features that suggest a diagnosis other than Parkinson disease include poor response to adequate dosages of levodopa, early onset of postural instability, axial more than appendicular rigidity, early dementia, and inability to look up or down without needing to move the head (supranuclear palsy).4
MANAGING PARKINSON DISEASE
Nonpharmacologic therapy is very important. Because patients tend to live longer because of better treatment, education is particularly important. The benefits of exercise go beyond general conditioning and cardiovascular health. People who exercise vigorously at least three times a week for 30 to 45 minutes are less likely to develop Parkinson disease and, if they develop it, they tend to have slower progression.
Prevention with neuroprotective drugs is not yet an option but hopefully will be in the near future.
Drug treatment generally starts when the patient is functionally impaired. If so, either levodopa or a dopamine agonist is started, depending on the patient’s age and the severity of symptoms. With increasing severity, other drugs can be added, and when those fail to control symptoms, surgery should be considered.
Deep brain stimulation surgery can make a tremendous difference in a patient’s quality of life. Other than levodopa, it is probably the best therapy available; however, it is very expensive and is not without risks.
Levodopa: The most effective drug, until it wears off
All current drugs for Parkinson disease activate dopamine neurotransmission in the brain. The most effective—and the cheapest—is still carbidopa/levodopa (Sinemet, Parcopa, Atamet). Levodopa converts to dopamine both peripherally and after it crosses the blood-brain barrier. Carbidopa prevents the peripheral conversion of levodopa to dopamine, reducing the peripheral adverse effects of levodopa, such as nausea and vomiting. The combination drug is usually given three times a day, with different doses available (10 mg carbidopa/100 mg levodopa, 25/100, 50/200, and 25/250) and as immediate-release and controlled-release formulations as well as an orally dissolving form (Parcopa) for patients with difficulty swallowing.
The major problem with levodopa is that after 4 to 6 years of treatment, about 40% of patients develop motor fluctuations and dyskinesias.5 If treatment is started too soon or at too high a dose, these problems tend to develop even earlier, especially among younger patients.
Motor fluctuations can take many forms: slow wearing-off, abrupt loss of effectiveness, and random on-and-off effectiveness (“yo-yoing”).
Dyskinesias typically involve constant chorea (dance-like) movements and occur at peak dose. Although chorea is easily treated by lowering the dosage, patients generally prefer having these movements rather than the Parkinson symptoms that recur from underdosing.
Dopamine agonists may be best for younger patients in early stages
The next most effective class of drugs are the dopamine agonists: pramipexole (Mirapex), ropinirole (Requip), and bromocriptine (Parlodel). A fourth drug, pergolide, is no longer available because of associated valvular heart complications. Each can be used as monotherapy in mild, early Parkinson disease or as an additional drug for moderate to severe disease. They are longer-acting than levodopa and can be taken once daily. Although they are less likely than levodopa to cause wearing-off or dyskinesias, they are associated with more nonmotor side effects: nausea and vomiting, hallucinations, confusion, somnolence or sleep attacks, low blood pressure, edema, and impulse control disorders.
Multiple clinical trials have been conducted to test the efficacy of dopamine agonists vs levodopa for treating Parkinson disease.6–9 Almost always, levodopa is more effective but involves more wearing-off and dyskinesias. For this reason, for patients with milder parkinsonism who may not need the strongest drug available, trying one of the dopamine agonists first may be worthwhile.
In addition, patients younger than age 60 are more prone to develop motor fluctuations and dyskinesias, so a dopamine agonist should be tried first in patients in that age group. For patients over age 65 for whom cost may be of concern, levodopa is the preferred starting drug.
Anticholinergic drugs for tremor
Before 1969, only anticholinergic drugs were available to treat Parkinson disease. Examples include trihexyphenidyl (Artane, Trihexane) and benztropine (Cogentin). These drugs are effective for treating tremor and drooling but are much less useful against rigidity, bradykinesia, and balance problems. Side effects include confusion, dry mouth, constipation, blurred vision, urinary retention, and cognitive impairment.
Anticholinergics should only be considered for young patients in whom tremor is a large problem and who have not responded well to the traditional Parkinson drugs. Because tremor is mostly a cosmetic problem, anticholinergics can also be useful for treating actors, musicians, and other patients with a public role.
Monoamine oxidase B inhibitors are well tolerated but less effective
In the brain, dopamine is broken down by monoamine oxidase B (MAO-B); therefore, inhibiting this enzyme increases dopamine’s availability. The MAO-B inhibitors selegiline (Eldepryl, Zelapar) and rasagiline (Azilect) are effective for monotherapy for Parkinson disease but are not as effective as levodopa. Most physicians feel MAO-B inhibitors are also less effective than dopamine agonists, although double-blind, randomized clinical trials have not proven this.6,10,11
MAO-B inhibitors have a long half-life, allowing once-daily dosing, and they are very well tolerated, with a side-effect profile similar to that of placebo. As with all MAO inhibitors, caution is needed regarding drug and food interactions.
EFFECTIVE NEUROPROTECTIVE AGENTS REMAIN ELUSIVE
Although numerous drugs are now available to treat the symptoms of Parkinson disease, the ability to slow the progression of the disease remains elusive. The only factor consistently shown by epidemiologic evidence to be protective is cigarette smoking, but we don’t recommend it.
A number of agents have been tested for neuroprotective efficacy:
Coenzyme Q10 has been tested at low and high dosages but was not found to be effective.
Pramipexole, a dopamine agonist, has also been studied without success.
Creatine is currently being studied and shows promise, possibly because of its effects on complex-I, part of the electron transport chain in mitochondria, which may be disrupted in Parkinson disease.
Inosine, which elevates uric acid, is also promising. The link between high uric acid and Parkinson disease was serendipitously discovered: when evaluating numerous blood panels taken from patients with Parkinson disease who were in clinical trials (using what turned out to be ineffective agents), it was noted that patients with the slowest progression of disease tended to have the highest uric acid levels. This has led to trials evaluating the effect of elevating uric acid to a pre-gout threshold.
Calcium channel blockers may be protective, according to epidemiologic evidence. Experiments involving injecting isradipine (DynaCirc) in rat models of Parkinson disease have indicated that the drug is promising.
Rasagiline: Protective effects still unknown
A large study of the neuroprotective effects of the MAO-B inhibitor rasagiline has just been completed, but the results are uncertain.12 A unique “delayed-start” clinical trial design was used to try to evaluate whether this agent that is known to reduce symptoms may also be neuroprotective. More than 1,000 people with untreated Parkinson disease from 14 countries were randomly assigned to receive rasagiline (the early-start group) or placebo (the delayed-start group) for 36 weeks. Afterward, both groups were given rasagiline for another 36 weeks. Rasagiline was given in a daily dose of either 1 mg or 2 mg.
The investigators anticipated that if the benefits of rasagiline were purely symptomatic, the early- and delayed-start groups would have equivalent disease severity at the end of the study. If rasagiline were protective, the early-start group would be better off at the end of the study. Unfortunately, the results were ambiguous: the early- and delayed-start groups were equivalent at the end of the study if they received the 2-mg daily dose, apparently indicating no protective effect. But at the 1-mg daily dose, the delayed-start group developed more severe disease at 36 weeks and did not catch up to the early-start group after treatment with rasagiline, apparently indicating a protective benefit. As a result, no definitive conclusion can be drawn.
EXTENDING TREATMENT EFFECTS IN ADVANCED PARKINSON DISEASE
For most patients, the first 5 years after being diagnosed with Parkinson disease is the “honeymoon phase,” when almost any treatment is effective. During this time, patients tend to have enough surviving dopaminergic neurons to store levodopa, despite its very short half-life of only 60 minutes.
As the disease progresses, fewer dopaminergic neurons survive, the therapeutic window narrows, and dosing becomes a balancing act: too much dopamine causes dyskinesias, hallucinations, delusions, and impulsive behavior, and too little dopamine causes worsening of Parkinson symptoms, freezing, and wearing-off, with ensuing falls and fractures. At this stage, some patients are prescribed levodopa every 1.5 or 2 hours.
Drugs are now available that extend the half-life of levodopa by slowing the breakdown of dopamine.
Catechol-O-methyltransferase (COMT) inhibitors—including tolcapone (Tasmar) and entacapone (Comtan) (also available as combined cardidopa, entacapone, and levodopa [Stalevo])—reduce off periods by about 1 hour per day.13 Given that the price is about $2,500 per year, the cost and benefits to the patient must be considered.14–17
Rasagiline, an MAO-B inhibitor, can also be added to levodopa to extend the “on” time for about 1 hour a day and to reduce freezing of gait. Clinical trials have shown it to be well tolerated, although common side effects include worsening dyskinesias and nausea.18,19
Apomorphine (Apokyn) is a dopamine agonist given by subcutaneous injection, allowing it to avoid first-pass metabolism by the liver. The benefits start just 10 minutes after injection, but only last for about 1 hour. It is a good option for rescue therapy for patients who cannot swallow or who have severe, unpredictable, or painful off-periods. It is also useful for situations in which it is especially inconvenient to have an off-period, such as being away from home.
Many agents have been tested for improving the off-period, but most work for about 1 to 2 hours, which is not nearly as effective as deep brain stimulation.
Managing dyskinesias
Dyskinesias can be managed by giving lower doses of levodopa more often. If wearing-off is a problem, a dopamine agonist or MAO-B inhibitor can be added. For patients at this stage, a specialist should be consulted.
Amantadine (Symmetrel), an N-methyl-d-aspartate (NMDA) receptor antagonist and dopamine-releasing agent used to treat influenza, is also effective against dyskinesias. Adverse effects include anxiety, insomnia, nightmares, anticholinergic effects, and livedo reticularis.20,21
Deep brain stimulation is the best treatment for dyskinesias in a patient for whom the procedure is appropriate and who has medical insurance that covers it.
NONMOTOR FEATURES OF PARKINSON DISEASE
Dementia: One of the most limiting nonmotor features
Often the most limiting nonmotor feature of Parkinson disease is dementia, which develops at about four to six times the rate for age-matched controls. At a given time, about 40% of patients with Parkinson disease have dementia, and the risk is 80% over 15 years of the disease.
If dementia is present, many of the drugs effective against Parkinson disease cannot be used because of exacerbating side effects. Treatment is mainly restricted to levodopa.
The only FDA-approved drug to treat dementia in Parkinson disease is the same drug for Alzheimer disease, rivastigmine (Exelon). Its effects are only modest, and its cholinergic side effects may transiently worsen parkinsonian features.22
Psychosis: Also very common
About half of patients with Parkinson disease have an episode of hallucinations or delusions in their lifetime, and about 20% are actively psychotic at any time. Delusions typically have the theme of spousal infidelity. Psychosis is associated with a higher rate of death compared with patients with Parkinson disease who do not develop it. Rebound psychosis may occur on withdrawal of antipsychotic medication.23–27
Patients who develop psychosis should have a physical examination and laboratory evaluation to determine if an infection or electrolyte imbalance is the cause. Medications should be discontinued in the following order: anticholinergic drug, amantadine, MAO-B inhibitor, dopamine agonist, and COMT inhibitor. Levodopa and carbidopa should be reduced to the minimum tolerable yet effective dosages.
For a patient who still has psychosis despite a minimum Parkinson drug regimen, an atypical antipsychotic drug should be used. Although clozapine (Clozaril, FazaClo) is very effective without worsening parkinsonism, it requires weekly monitoring with a complete blood count because of the small (< 1%) risk of agranulocytosis. For that reason, the first-line drug is quetiapine (Seroquel). Most double-blind studies have not found it to be effective, yet it is the drug most often used. No other antipsychotic drugs are safe to treat Parkinson psychosis.
Many patients with Parkinson disease who are hospitalized become agitated and confused soon after they are admitted to the hospital. The best treatment is quetiapine if an oral drug can be prescribed. A benzodiazepine—eg, clonazepam (Klonopin), lorazepam (Ativan), diazepam (Valium)—at a low dose may also be effective. Haloperidol, risperidone, and olanzapine should not be given, as they block dopamine receptors and worsen rigidity.
Mood disturbances
Depression occurs in about half of patients with Parkinson disease and is a significant cause of functional impairment. About 25% of patients have anxiety, and 20% are apathetic.
Depression appears to be secondary to underlying neuroanatomic degeneration rather than a reaction to disability.28 Fortunately, most antidepressants are effective in patients with Parkinson disease.29,30 Bupropion (Wellbutrin) is a dopamine reuptake inhibitor and so increases the availability of dopamine, and it should also have antiparkinsonian effects, but unfortunately it does not. Conversely, selective serotonin reuptake inhibitors (SSRIs) theoretically can worsen or cause parkinsonism, but evidence shows that they are safe to use in patients with Parkinson disease. Some evidence indicates that tricyclic antidepressants may be superior to SSRIs for treating depression in patients with Parkinson disease, so they might be the better choice in patients who can tolerate them.
Compulsive behaviors such as punding (prolonged performance of repetitive, mechanical tasks, such as disassembling and reassembling household objects) may occur from levodopa.
In addition, impulse control disorders involving pathologic gambling, hypersexuality, compulsive shopping, or binge eating occur in about 8% of patients with Parkinson disease taking dopamine agonists. These behaviors are more likely to arise in young, single patients, who are also more likely to have a family history of impulsive control disorder.31
THE FUTURE OF DRUG THERAPY
Clinical trials are now testing new therapies that work the traditional way through dopaminergic mechanisms, as well as those that work in novel ways.
A large international trial is studying patients with newly diagnosed Parkinson disease to try to discover a biomarker. Parkinson disease is unlike many other diseases in that physicians can only use clinical features to measure improvement, which is very crude. Identifying a biomarker will make evaluating and monitoring treatment a more exact science, and will lead to faster development of effective treatments.
- Adler CH, Ahlskog JE. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for The Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Nutt JG, Wooten GF. Clinical practice. Diagnosis and initial management of Parkinson’s disease. N Engl J Med 2005; 353:1021–1027.
- Litvan I, Bhatia KP, Burn DJ, et al; Movement Disorders Society Scientific Issues Committee. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003; 18:467–486.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001; 16:448–458.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med 2000; 342:1484–1491.
- Oertel WH, Wolters E, Sampaio C, et al. Pergolide versus levodopa monotherapy in early Parkinson’s disease patients: The PELMOPET study. Mov Disord 2006; 21:343–353.
- Lees AJ, Katzenschlager R, Head J, Ben-Shlomo Y. Ten-year follow-up of three different initial treatments in de-novo PD: a randomized trial. Neurology 2001; 57:1687–1694.
- Fowler JS, Volkow ND, Logan J, et al. Slow recovery of human brain MAO B after L-deprenyl (selegeline) withdrawal. Synapse 1994; 18:86–93.
- Elmer LW, Bertoni JM. The increasing role of monoamine oxidase type B inhibitors in Parkinson’s disease therapy. Expert Opin Pharmacother 2008; 9:2759–2772.
- Olanow CW, Rascol O, Hauser R, et al; ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278. Erratum in: N Engl J Med 2011; 364:1882.
- Stocchi F, Barbato L, Nordera G, Bolner A, Caraceni T. Entacapone improves the pharmacokinetic and therapeutic response of controlled release levodopa/carbidopa in Parkinson’s patients. J Neural Transm 2004; 111:173–180.
- Brooks DJ, Sagar HUK-Irish Entacapone Study Group. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind six month study. J Neurol Neurosurg Psychiatry 2003; 74:1071–1079.
- Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M; Celomen Study Group. Efficacy and safety of entacapone in Parkinson’s disease patients with soboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol Scand 2002; 105:245–255.
- Rinne UK, Larsen JP, Siden A, Worm-Petersen J. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology 1998; 51:1309–1314.
- Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Parkinson Study Group. Ann Neurol 1997; 42:747–755.
- Parkinson Study Group. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol 2005; 62:241–248.
- Rascol O, Brooks DJ, Melamed E, et al; LARGO study group. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet 2005; 365:947–954.
- Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol 1999; 56:1383–1386.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Almaraz AC, Driver-Dunckley ED, Woodruff BK, et al. Efficacy of rivastigmine for cognitive symptoms in Parkinson disease with dementia. Neurologist 2009; 15:234–237.
- Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain 2000; 123:733–745.
- Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004; 19:831–833.
- Goetz CG, Wuu J, Curgian LM, Leurgans S. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 2005; 64:81–86.
- Tollefson GD, Dellva MA, Mattler CA, Kane JM, Wirshing DA, Kinon BJ. Controlled, double-blind investigation of the clozapine discontinuation symptoms with conversion to either olanzapine or placebo. The Collaborative Crossover Study Group. J Clin Psychopharmacol 1999; 19:435–443.
- Fernandez HH, Trieschmann ME, Okun MS. Rebound psychosis: effect of discontinuation of antipsychotics in Parkinson’s disease. Mov Disord 2005; 20:104–105.
- McDonald WM, Richard IH, DeLong MR. Prevalence, etiology, and treatment of depression in Parkinson’s disease. Biol Psychiatry 2003; 54:363–375.
- Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008; 23:850–857.
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009; 72:886–892.
- Voon V, Sohr M, Lang AE, et al. Impulse control disorders in Parkinson disease: a multicenter case-control study. Ann Neurol 2011; 69:986–996. .
More than a dozen drugs have been approved by the US Food and Drug Administration (FDA) for treating Parkinson disease, and more are expected in the near future. Many are currently in clinical trials, with the goals of finding ways to better control the disease with fewer adverse effects and, ultimately, to provide neuroprotection.
This article will review the features of Parkinson disease, the treatment options, and the complications in moderate to advanced disease.
PARKINSON DISEASE IS MULTIFACTORIAL
Although the cure for Parkinson disease is still elusive, much has been learned over the nearly 200 years since it was first described by James Parkinson in 1817. It is now understood to be a progressive neurodegenerative disease of multifactorial etiology: although a small proportion of patients have a direct inherited mutation that causes it, multiple genetic predisposition factors and environmental factors are more commonly involved.
The central pathology is dopaminergic loss in the basal ganglia, but other neurotransmitters are also involved and the disease extends to other areas of the brain.
CARDINAL MOTOR SYMPTOMS
In general, Parkinson disease is easy to identify. The classic patient has1:
- Tremor at rest, which can be subtle—such as only involving a thumb or a few fingers—and is absent in 20% of patients at presentation.
- Rigidity, which is felt by the examiner rather than seen by an observer.
- Bradykinesia (slow movements), which is characteristic of all Parkinson patients.
- Gait and balance problems, which usually arise after a few years, although occasionally patients present with them. Patients typically walk with small steps with occasional freezing, as if their foot were stuck. Balance problems are the most difficult to treat among the motor problems.
Asymmetry of motor problems is apparent in 75% of patients at presentation, although problems become bilateral later in the course of the disease.
NONMOTOR FEATURES CAN BE MORE DISABLING
Pain is common, but years ago it was not recognized as a specific feature of Parkinson disease. The pain from other conditions may also worsen.
Fatigue is very common and, if present, is usually one of the most disabling features.
Neuropsychiatric disturbances are among the most difficult problems, and they become increasingly common as motor symptoms are better controlled with treatment and patients live longer.
INCREASINGLY PREVALENT AS THE POPULATION AGES
Parkinson disease can present from the teenage years up to age 90, but it is most often diagnosed in patients from 60 to 70 years old (mean onset, 62.5 years). A different nomenclature is used depending on the age of onset:
- 10 to 20 years: juvenile-onset
- 21 to 40 years: young-onset.
Parkinson disease is now an epidemic, with an estimated 1 million people having it in the United States, representing 0.3% of the population and 1% of those older than 60 years.2 More people can be expected to develop it as our population ages in the next decades. It is estimated that in 2040 more people will die from Parkinson disease, Alzheimer disease, and amyotrophic lateral sclerosis (all of which are neurodegenerative diseases) than from kidney cancer, malignant melanoma, colon cancer, and lung cancer combined.
DIAGNOSIS IS STILL MAINLY CLINICAL
The diagnosis of Parkinson disease remains clinical. In addition to the motor features, the best test is a clear response to dopaminergic treatment with levodopa. If all these features are present, the diagnosis of Parkinson disease is usually correct.3
Imaging useful in select patients
The FDA recently approved a radiopharmaceutical contrast agent, DaTscan, to use with single-photon emission computed tomography (SPECT) to help diagnose Parkinson disease. DaTscan is a dopamine transporter ligand that tags presynaptic dopaminergic neurons in the basal ganglia; a patient with Parkinson disease has less signal.
The test can be used to distinguish parkinsonian syndromes from disorders that can mimic them, such as essential tremor or a psychogenic disorder. However, it cannot differentiate various Parkinson-plus syndromes (see below) such as multiple system atrophy or progressive nuclear palsy. It also cannot be used to detect drug-induced or vascular parkinsonism.
Check for Wilson disease or brain tumors in young or atypical cases
For most patients, no imaging or blood tests are needed to make the diagnosis. However, in patients younger than 50, Wilson disease, a rare inherited disorder characterized by excess copper accumulation, must be considered. Testing for Wilson disease includes serum ceruloplasmin, 24-hour urinary copper excretion, and an ophthalmologic slit-lamp examination for Kaiser-Fleischer rings.
For patients who do not quite fit the picture of Parkinson disease, such as those who have spasticity with little tremor, or who have a minimal response to levodopa, magnetic resonance imaging should be done to see if a structural lesion is present.
Consider secondary parkinsonism
Although idiopathic Parkinson disease is by far the most common form of parkinsonism in the United States and in most developing countries, secondary causes must also be considered in a patient presenting with symptoms of parkinsonism. They include:
- Dopamine-receptor blocking agents: metoclopramide (Reglan), prochlorperazine (Compazine), haloperidol (Haldol), thioridazine (Mellaril), risperidone (Risperdal), olanzapine (Zyprexa)
- Strokes in the basal ganglia
- Normal pressure hydrocephalus.
Parkinson-plus syndromes
Parkinson-plus syndromes have other features in addition to the classic features of idiopathic Parkinson disease. They occur commonly and can be difficult to distinguish from Parkinson disease and from each other.
Parkinson-plus syndromes include:
- Progressive supranuclear palsy
- Multiple system atrophy
- Corticobasal degeneration
- Lewy body dementia.
Clinical features that suggest a diagnosis other than Parkinson disease include poor response to adequate dosages of levodopa, early onset of postural instability, axial more than appendicular rigidity, early dementia, and inability to look up or down without needing to move the head (supranuclear palsy).4
MANAGING PARKINSON DISEASE
Nonpharmacologic therapy is very important. Because patients tend to live longer because of better treatment, education is particularly important. The benefits of exercise go beyond general conditioning and cardiovascular health. People who exercise vigorously at least three times a week for 30 to 45 minutes are less likely to develop Parkinson disease and, if they develop it, they tend to have slower progression.
Prevention with neuroprotective drugs is not yet an option but hopefully will be in the near future.
Drug treatment generally starts when the patient is functionally impaired. If so, either levodopa or a dopamine agonist is started, depending on the patient’s age and the severity of symptoms. With increasing severity, other drugs can be added, and when those fail to control symptoms, surgery should be considered.
Deep brain stimulation surgery can make a tremendous difference in a patient’s quality of life. Other than levodopa, it is probably the best therapy available; however, it is very expensive and is not without risks.
Levodopa: The most effective drug, until it wears off
All current drugs for Parkinson disease activate dopamine neurotransmission in the brain. The most effective—and the cheapest—is still carbidopa/levodopa (Sinemet, Parcopa, Atamet). Levodopa converts to dopamine both peripherally and after it crosses the blood-brain barrier. Carbidopa prevents the peripheral conversion of levodopa to dopamine, reducing the peripheral adverse effects of levodopa, such as nausea and vomiting. The combination drug is usually given three times a day, with different doses available (10 mg carbidopa/100 mg levodopa, 25/100, 50/200, and 25/250) and as immediate-release and controlled-release formulations as well as an orally dissolving form (Parcopa) for patients with difficulty swallowing.
The major problem with levodopa is that after 4 to 6 years of treatment, about 40% of patients develop motor fluctuations and dyskinesias.5 If treatment is started too soon or at too high a dose, these problems tend to develop even earlier, especially among younger patients.
Motor fluctuations can take many forms: slow wearing-off, abrupt loss of effectiveness, and random on-and-off effectiveness (“yo-yoing”).
Dyskinesias typically involve constant chorea (dance-like) movements and occur at peak dose. Although chorea is easily treated by lowering the dosage, patients generally prefer having these movements rather than the Parkinson symptoms that recur from underdosing.
Dopamine agonists may be best for younger patients in early stages
The next most effective class of drugs are the dopamine agonists: pramipexole (Mirapex), ropinirole (Requip), and bromocriptine (Parlodel). A fourth drug, pergolide, is no longer available because of associated valvular heart complications. Each can be used as monotherapy in mild, early Parkinson disease or as an additional drug for moderate to severe disease. They are longer-acting than levodopa and can be taken once daily. Although they are less likely than levodopa to cause wearing-off or dyskinesias, they are associated with more nonmotor side effects: nausea and vomiting, hallucinations, confusion, somnolence or sleep attacks, low blood pressure, edema, and impulse control disorders.
Multiple clinical trials have been conducted to test the efficacy of dopamine agonists vs levodopa for treating Parkinson disease.6–9 Almost always, levodopa is more effective but involves more wearing-off and dyskinesias. For this reason, for patients with milder parkinsonism who may not need the strongest drug available, trying one of the dopamine agonists first may be worthwhile.
In addition, patients younger than age 60 are more prone to develop motor fluctuations and dyskinesias, so a dopamine agonist should be tried first in patients in that age group. For patients over age 65 for whom cost may be of concern, levodopa is the preferred starting drug.
Anticholinergic drugs for tremor
Before 1969, only anticholinergic drugs were available to treat Parkinson disease. Examples include trihexyphenidyl (Artane, Trihexane) and benztropine (Cogentin). These drugs are effective for treating tremor and drooling but are much less useful against rigidity, bradykinesia, and balance problems. Side effects include confusion, dry mouth, constipation, blurred vision, urinary retention, and cognitive impairment.
Anticholinergics should only be considered for young patients in whom tremor is a large problem and who have not responded well to the traditional Parkinson drugs. Because tremor is mostly a cosmetic problem, anticholinergics can also be useful for treating actors, musicians, and other patients with a public role.
Monoamine oxidase B inhibitors are well tolerated but less effective
In the brain, dopamine is broken down by monoamine oxidase B (MAO-B); therefore, inhibiting this enzyme increases dopamine’s availability. The MAO-B inhibitors selegiline (Eldepryl, Zelapar) and rasagiline (Azilect) are effective for monotherapy for Parkinson disease but are not as effective as levodopa. Most physicians feel MAO-B inhibitors are also less effective than dopamine agonists, although double-blind, randomized clinical trials have not proven this.6,10,11
MAO-B inhibitors have a long half-life, allowing once-daily dosing, and they are very well tolerated, with a side-effect profile similar to that of placebo. As with all MAO inhibitors, caution is needed regarding drug and food interactions.
EFFECTIVE NEUROPROTECTIVE AGENTS REMAIN ELUSIVE
Although numerous drugs are now available to treat the symptoms of Parkinson disease, the ability to slow the progression of the disease remains elusive. The only factor consistently shown by epidemiologic evidence to be protective is cigarette smoking, but we don’t recommend it.
A number of agents have been tested for neuroprotective efficacy:
Coenzyme Q10 has been tested at low and high dosages but was not found to be effective.
Pramipexole, a dopamine agonist, has also been studied without success.
Creatine is currently being studied and shows promise, possibly because of its effects on complex-I, part of the electron transport chain in mitochondria, which may be disrupted in Parkinson disease.
Inosine, which elevates uric acid, is also promising. The link between high uric acid and Parkinson disease was serendipitously discovered: when evaluating numerous blood panels taken from patients with Parkinson disease who were in clinical trials (using what turned out to be ineffective agents), it was noted that patients with the slowest progression of disease tended to have the highest uric acid levels. This has led to trials evaluating the effect of elevating uric acid to a pre-gout threshold.
Calcium channel blockers may be protective, according to epidemiologic evidence. Experiments involving injecting isradipine (DynaCirc) in rat models of Parkinson disease have indicated that the drug is promising.
Rasagiline: Protective effects still unknown
A large study of the neuroprotective effects of the MAO-B inhibitor rasagiline has just been completed, but the results are uncertain.12 A unique “delayed-start” clinical trial design was used to try to evaluate whether this agent that is known to reduce symptoms may also be neuroprotective. More than 1,000 people with untreated Parkinson disease from 14 countries were randomly assigned to receive rasagiline (the early-start group) or placebo (the delayed-start group) for 36 weeks. Afterward, both groups were given rasagiline for another 36 weeks. Rasagiline was given in a daily dose of either 1 mg or 2 mg.
The investigators anticipated that if the benefits of rasagiline were purely symptomatic, the early- and delayed-start groups would have equivalent disease severity at the end of the study. If rasagiline were protective, the early-start group would be better off at the end of the study. Unfortunately, the results were ambiguous: the early- and delayed-start groups were equivalent at the end of the study if they received the 2-mg daily dose, apparently indicating no protective effect. But at the 1-mg daily dose, the delayed-start group developed more severe disease at 36 weeks and did not catch up to the early-start group after treatment with rasagiline, apparently indicating a protective benefit. As a result, no definitive conclusion can be drawn.
EXTENDING TREATMENT EFFECTS IN ADVANCED PARKINSON DISEASE
For most patients, the first 5 years after being diagnosed with Parkinson disease is the “honeymoon phase,” when almost any treatment is effective. During this time, patients tend to have enough surviving dopaminergic neurons to store levodopa, despite its very short half-life of only 60 minutes.
As the disease progresses, fewer dopaminergic neurons survive, the therapeutic window narrows, and dosing becomes a balancing act: too much dopamine causes dyskinesias, hallucinations, delusions, and impulsive behavior, and too little dopamine causes worsening of Parkinson symptoms, freezing, and wearing-off, with ensuing falls and fractures. At this stage, some patients are prescribed levodopa every 1.5 or 2 hours.
Drugs are now available that extend the half-life of levodopa by slowing the breakdown of dopamine.
Catechol-O-methyltransferase (COMT) inhibitors—including tolcapone (Tasmar) and entacapone (Comtan) (also available as combined cardidopa, entacapone, and levodopa [Stalevo])—reduce off periods by about 1 hour per day.13 Given that the price is about $2,500 per year, the cost and benefits to the patient must be considered.14–17
Rasagiline, an MAO-B inhibitor, can also be added to levodopa to extend the “on” time for about 1 hour a day and to reduce freezing of gait. Clinical trials have shown it to be well tolerated, although common side effects include worsening dyskinesias and nausea.18,19
Apomorphine (Apokyn) is a dopamine agonist given by subcutaneous injection, allowing it to avoid first-pass metabolism by the liver. The benefits start just 10 minutes after injection, but only last for about 1 hour. It is a good option for rescue therapy for patients who cannot swallow or who have severe, unpredictable, or painful off-periods. It is also useful for situations in which it is especially inconvenient to have an off-period, such as being away from home.
Many agents have been tested for improving the off-period, but most work for about 1 to 2 hours, which is not nearly as effective as deep brain stimulation.
Managing dyskinesias
Dyskinesias can be managed by giving lower doses of levodopa more often. If wearing-off is a problem, a dopamine agonist or MAO-B inhibitor can be added. For patients at this stage, a specialist should be consulted.
Amantadine (Symmetrel), an N-methyl-d-aspartate (NMDA) receptor antagonist and dopamine-releasing agent used to treat influenza, is also effective against dyskinesias. Adverse effects include anxiety, insomnia, nightmares, anticholinergic effects, and livedo reticularis.20,21
Deep brain stimulation is the best treatment for dyskinesias in a patient for whom the procedure is appropriate and who has medical insurance that covers it.
NONMOTOR FEATURES OF PARKINSON DISEASE
Dementia: One of the most limiting nonmotor features
Often the most limiting nonmotor feature of Parkinson disease is dementia, which develops at about four to six times the rate for age-matched controls. At a given time, about 40% of patients with Parkinson disease have dementia, and the risk is 80% over 15 years of the disease.
If dementia is present, many of the drugs effective against Parkinson disease cannot be used because of exacerbating side effects. Treatment is mainly restricted to levodopa.
The only FDA-approved drug to treat dementia in Parkinson disease is the same drug for Alzheimer disease, rivastigmine (Exelon). Its effects are only modest, and its cholinergic side effects may transiently worsen parkinsonian features.22
Psychosis: Also very common
About half of patients with Parkinson disease have an episode of hallucinations or delusions in their lifetime, and about 20% are actively psychotic at any time. Delusions typically have the theme of spousal infidelity. Psychosis is associated with a higher rate of death compared with patients with Parkinson disease who do not develop it. Rebound psychosis may occur on withdrawal of antipsychotic medication.23–27
Patients who develop psychosis should have a physical examination and laboratory evaluation to determine if an infection or electrolyte imbalance is the cause. Medications should be discontinued in the following order: anticholinergic drug, amantadine, MAO-B inhibitor, dopamine agonist, and COMT inhibitor. Levodopa and carbidopa should be reduced to the minimum tolerable yet effective dosages.
For a patient who still has psychosis despite a minimum Parkinson drug regimen, an atypical antipsychotic drug should be used. Although clozapine (Clozaril, FazaClo) is very effective without worsening parkinsonism, it requires weekly monitoring with a complete blood count because of the small (< 1%) risk of agranulocytosis. For that reason, the first-line drug is quetiapine (Seroquel). Most double-blind studies have not found it to be effective, yet it is the drug most often used. No other antipsychotic drugs are safe to treat Parkinson psychosis.
Many patients with Parkinson disease who are hospitalized become agitated and confused soon after they are admitted to the hospital. The best treatment is quetiapine if an oral drug can be prescribed. A benzodiazepine—eg, clonazepam (Klonopin), lorazepam (Ativan), diazepam (Valium)—at a low dose may also be effective. Haloperidol, risperidone, and olanzapine should not be given, as they block dopamine receptors and worsen rigidity.
Mood disturbances
Depression occurs in about half of patients with Parkinson disease and is a significant cause of functional impairment. About 25% of patients have anxiety, and 20% are apathetic.
Depression appears to be secondary to underlying neuroanatomic degeneration rather than a reaction to disability.28 Fortunately, most antidepressants are effective in patients with Parkinson disease.29,30 Bupropion (Wellbutrin) is a dopamine reuptake inhibitor and so increases the availability of dopamine, and it should also have antiparkinsonian effects, but unfortunately it does not. Conversely, selective serotonin reuptake inhibitors (SSRIs) theoretically can worsen or cause parkinsonism, but evidence shows that they are safe to use in patients with Parkinson disease. Some evidence indicates that tricyclic antidepressants may be superior to SSRIs for treating depression in patients with Parkinson disease, so they might be the better choice in patients who can tolerate them.
Compulsive behaviors such as punding (prolonged performance of repetitive, mechanical tasks, such as disassembling and reassembling household objects) may occur from levodopa.
In addition, impulse control disorders involving pathologic gambling, hypersexuality, compulsive shopping, or binge eating occur in about 8% of patients with Parkinson disease taking dopamine agonists. These behaviors are more likely to arise in young, single patients, who are also more likely to have a family history of impulsive control disorder.31
THE FUTURE OF DRUG THERAPY
Clinical trials are now testing new therapies that work the traditional way through dopaminergic mechanisms, as well as those that work in novel ways.
A large international trial is studying patients with newly diagnosed Parkinson disease to try to discover a biomarker. Parkinson disease is unlike many other diseases in that physicians can only use clinical features to measure improvement, which is very crude. Identifying a biomarker will make evaluating and monitoring treatment a more exact science, and will lead to faster development of effective treatments.
More than a dozen drugs have been approved by the US Food and Drug Administration (FDA) for treating Parkinson disease, and more are expected in the near future. Many are currently in clinical trials, with the goals of finding ways to better control the disease with fewer adverse effects and, ultimately, to provide neuroprotection.
This article will review the features of Parkinson disease, the treatment options, and the complications in moderate to advanced disease.
PARKINSON DISEASE IS MULTIFACTORIAL
Although the cure for Parkinson disease is still elusive, much has been learned over the nearly 200 years since it was first described by James Parkinson in 1817. It is now understood to be a progressive neurodegenerative disease of multifactorial etiology: although a small proportion of patients have a direct inherited mutation that causes it, multiple genetic predisposition factors and environmental factors are more commonly involved.
The central pathology is dopaminergic loss in the basal ganglia, but other neurotransmitters are also involved and the disease extends to other areas of the brain.
CARDINAL MOTOR SYMPTOMS
In general, Parkinson disease is easy to identify. The classic patient has1:
- Tremor at rest, which can be subtle—such as only involving a thumb or a few fingers—and is absent in 20% of patients at presentation.
- Rigidity, which is felt by the examiner rather than seen by an observer.
- Bradykinesia (slow movements), which is characteristic of all Parkinson patients.
- Gait and balance problems, which usually arise after a few years, although occasionally patients present with them. Patients typically walk with small steps with occasional freezing, as if their foot were stuck. Balance problems are the most difficult to treat among the motor problems.
Asymmetry of motor problems is apparent in 75% of patients at presentation, although problems become bilateral later in the course of the disease.
NONMOTOR FEATURES CAN BE MORE DISABLING
Pain is common, but years ago it was not recognized as a specific feature of Parkinson disease. The pain from other conditions may also worsen.
Fatigue is very common and, if present, is usually one of the most disabling features.
Neuropsychiatric disturbances are among the most difficult problems, and they become increasingly common as motor symptoms are better controlled with treatment and patients live longer.
INCREASINGLY PREVALENT AS THE POPULATION AGES
Parkinson disease can present from the teenage years up to age 90, but it is most often diagnosed in patients from 60 to 70 years old (mean onset, 62.5 years). A different nomenclature is used depending on the age of onset:
- 10 to 20 years: juvenile-onset
- 21 to 40 years: young-onset.
Parkinson disease is now an epidemic, with an estimated 1 million people having it in the United States, representing 0.3% of the population and 1% of those older than 60 years.2 More people can be expected to develop it as our population ages in the next decades. It is estimated that in 2040 more people will die from Parkinson disease, Alzheimer disease, and amyotrophic lateral sclerosis (all of which are neurodegenerative diseases) than from kidney cancer, malignant melanoma, colon cancer, and lung cancer combined.
DIAGNOSIS IS STILL MAINLY CLINICAL
The diagnosis of Parkinson disease remains clinical. In addition to the motor features, the best test is a clear response to dopaminergic treatment with levodopa. If all these features are present, the diagnosis of Parkinson disease is usually correct.3
Imaging useful in select patients
The FDA recently approved a radiopharmaceutical contrast agent, DaTscan, to use with single-photon emission computed tomography (SPECT) to help diagnose Parkinson disease. DaTscan is a dopamine transporter ligand that tags presynaptic dopaminergic neurons in the basal ganglia; a patient with Parkinson disease has less signal.
The test can be used to distinguish parkinsonian syndromes from disorders that can mimic them, such as essential tremor or a psychogenic disorder. However, it cannot differentiate various Parkinson-plus syndromes (see below) such as multiple system atrophy or progressive nuclear palsy. It also cannot be used to detect drug-induced or vascular parkinsonism.
Check for Wilson disease or brain tumors in young or atypical cases
For most patients, no imaging or blood tests are needed to make the diagnosis. However, in patients younger than 50, Wilson disease, a rare inherited disorder characterized by excess copper accumulation, must be considered. Testing for Wilson disease includes serum ceruloplasmin, 24-hour urinary copper excretion, and an ophthalmologic slit-lamp examination for Kaiser-Fleischer rings.
For patients who do not quite fit the picture of Parkinson disease, such as those who have spasticity with little tremor, or who have a minimal response to levodopa, magnetic resonance imaging should be done to see if a structural lesion is present.
Consider secondary parkinsonism
Although idiopathic Parkinson disease is by far the most common form of parkinsonism in the United States and in most developing countries, secondary causes must also be considered in a patient presenting with symptoms of parkinsonism. They include:
- Dopamine-receptor blocking agents: metoclopramide (Reglan), prochlorperazine (Compazine), haloperidol (Haldol), thioridazine (Mellaril), risperidone (Risperdal), olanzapine (Zyprexa)
- Strokes in the basal ganglia
- Normal pressure hydrocephalus.
Parkinson-plus syndromes
Parkinson-plus syndromes have other features in addition to the classic features of idiopathic Parkinson disease. They occur commonly and can be difficult to distinguish from Parkinson disease and from each other.
Parkinson-plus syndromes include:
- Progressive supranuclear palsy
- Multiple system atrophy
- Corticobasal degeneration
- Lewy body dementia.
Clinical features that suggest a diagnosis other than Parkinson disease include poor response to adequate dosages of levodopa, early onset of postural instability, axial more than appendicular rigidity, early dementia, and inability to look up or down without needing to move the head (supranuclear palsy).4
MANAGING PARKINSON DISEASE
Nonpharmacologic therapy is very important. Because patients tend to live longer because of better treatment, education is particularly important. The benefits of exercise go beyond general conditioning and cardiovascular health. People who exercise vigorously at least three times a week for 30 to 45 minutes are less likely to develop Parkinson disease and, if they develop it, they tend to have slower progression.
Prevention with neuroprotective drugs is not yet an option but hopefully will be in the near future.
Drug treatment generally starts when the patient is functionally impaired. If so, either levodopa or a dopamine agonist is started, depending on the patient’s age and the severity of symptoms. With increasing severity, other drugs can be added, and when those fail to control symptoms, surgery should be considered.
Deep brain stimulation surgery can make a tremendous difference in a patient’s quality of life. Other than levodopa, it is probably the best therapy available; however, it is very expensive and is not without risks.
Levodopa: The most effective drug, until it wears off
All current drugs for Parkinson disease activate dopamine neurotransmission in the brain. The most effective—and the cheapest—is still carbidopa/levodopa (Sinemet, Parcopa, Atamet). Levodopa converts to dopamine both peripherally and after it crosses the blood-brain barrier. Carbidopa prevents the peripheral conversion of levodopa to dopamine, reducing the peripheral adverse effects of levodopa, such as nausea and vomiting. The combination drug is usually given three times a day, with different doses available (10 mg carbidopa/100 mg levodopa, 25/100, 50/200, and 25/250) and as immediate-release and controlled-release formulations as well as an orally dissolving form (Parcopa) for patients with difficulty swallowing.
The major problem with levodopa is that after 4 to 6 years of treatment, about 40% of patients develop motor fluctuations and dyskinesias.5 If treatment is started too soon or at too high a dose, these problems tend to develop even earlier, especially among younger patients.
Motor fluctuations can take many forms: slow wearing-off, abrupt loss of effectiveness, and random on-and-off effectiveness (“yo-yoing”).
Dyskinesias typically involve constant chorea (dance-like) movements and occur at peak dose. Although chorea is easily treated by lowering the dosage, patients generally prefer having these movements rather than the Parkinson symptoms that recur from underdosing.
Dopamine agonists may be best for younger patients in early stages
The next most effective class of drugs are the dopamine agonists: pramipexole (Mirapex), ropinirole (Requip), and bromocriptine (Parlodel). A fourth drug, pergolide, is no longer available because of associated valvular heart complications. Each can be used as monotherapy in mild, early Parkinson disease or as an additional drug for moderate to severe disease. They are longer-acting than levodopa and can be taken once daily. Although they are less likely than levodopa to cause wearing-off or dyskinesias, they are associated with more nonmotor side effects: nausea and vomiting, hallucinations, confusion, somnolence or sleep attacks, low blood pressure, edema, and impulse control disorders.
Multiple clinical trials have been conducted to test the efficacy of dopamine agonists vs levodopa for treating Parkinson disease.6–9 Almost always, levodopa is more effective but involves more wearing-off and dyskinesias. For this reason, for patients with milder parkinsonism who may not need the strongest drug available, trying one of the dopamine agonists first may be worthwhile.
In addition, patients younger than age 60 are more prone to develop motor fluctuations and dyskinesias, so a dopamine agonist should be tried first in patients in that age group. For patients over age 65 for whom cost may be of concern, levodopa is the preferred starting drug.
Anticholinergic drugs for tremor
Before 1969, only anticholinergic drugs were available to treat Parkinson disease. Examples include trihexyphenidyl (Artane, Trihexane) and benztropine (Cogentin). These drugs are effective for treating tremor and drooling but are much less useful against rigidity, bradykinesia, and balance problems. Side effects include confusion, dry mouth, constipation, blurred vision, urinary retention, and cognitive impairment.
Anticholinergics should only be considered for young patients in whom tremor is a large problem and who have not responded well to the traditional Parkinson drugs. Because tremor is mostly a cosmetic problem, anticholinergics can also be useful for treating actors, musicians, and other patients with a public role.
Monoamine oxidase B inhibitors are well tolerated but less effective
In the brain, dopamine is broken down by monoamine oxidase B (MAO-B); therefore, inhibiting this enzyme increases dopamine’s availability. The MAO-B inhibitors selegiline (Eldepryl, Zelapar) and rasagiline (Azilect) are effective for monotherapy for Parkinson disease but are not as effective as levodopa. Most physicians feel MAO-B inhibitors are also less effective than dopamine agonists, although double-blind, randomized clinical trials have not proven this.6,10,11
MAO-B inhibitors have a long half-life, allowing once-daily dosing, and they are very well tolerated, with a side-effect profile similar to that of placebo. As with all MAO inhibitors, caution is needed regarding drug and food interactions.
EFFECTIVE NEUROPROTECTIVE AGENTS REMAIN ELUSIVE
Although numerous drugs are now available to treat the symptoms of Parkinson disease, the ability to slow the progression of the disease remains elusive. The only factor consistently shown by epidemiologic evidence to be protective is cigarette smoking, but we don’t recommend it.
A number of agents have been tested for neuroprotective efficacy:
Coenzyme Q10 has been tested at low and high dosages but was not found to be effective.
Pramipexole, a dopamine agonist, has also been studied without success.
Creatine is currently being studied and shows promise, possibly because of its effects on complex-I, part of the electron transport chain in mitochondria, which may be disrupted in Parkinson disease.
Inosine, which elevates uric acid, is also promising. The link between high uric acid and Parkinson disease was serendipitously discovered: when evaluating numerous blood panels taken from patients with Parkinson disease who were in clinical trials (using what turned out to be ineffective agents), it was noted that patients with the slowest progression of disease tended to have the highest uric acid levels. This has led to trials evaluating the effect of elevating uric acid to a pre-gout threshold.
Calcium channel blockers may be protective, according to epidemiologic evidence. Experiments involving injecting isradipine (DynaCirc) in rat models of Parkinson disease have indicated that the drug is promising.
Rasagiline: Protective effects still unknown
A large study of the neuroprotective effects of the MAO-B inhibitor rasagiline has just been completed, but the results are uncertain.12 A unique “delayed-start” clinical trial design was used to try to evaluate whether this agent that is known to reduce symptoms may also be neuroprotective. More than 1,000 people with untreated Parkinson disease from 14 countries were randomly assigned to receive rasagiline (the early-start group) or placebo (the delayed-start group) for 36 weeks. Afterward, both groups were given rasagiline for another 36 weeks. Rasagiline was given in a daily dose of either 1 mg or 2 mg.
The investigators anticipated that if the benefits of rasagiline were purely symptomatic, the early- and delayed-start groups would have equivalent disease severity at the end of the study. If rasagiline were protective, the early-start group would be better off at the end of the study. Unfortunately, the results were ambiguous: the early- and delayed-start groups were equivalent at the end of the study if they received the 2-mg daily dose, apparently indicating no protective effect. But at the 1-mg daily dose, the delayed-start group developed more severe disease at 36 weeks and did not catch up to the early-start group after treatment with rasagiline, apparently indicating a protective benefit. As a result, no definitive conclusion can be drawn.
EXTENDING TREATMENT EFFECTS IN ADVANCED PARKINSON DISEASE
For most patients, the first 5 years after being diagnosed with Parkinson disease is the “honeymoon phase,” when almost any treatment is effective. During this time, patients tend to have enough surviving dopaminergic neurons to store levodopa, despite its very short half-life of only 60 minutes.
As the disease progresses, fewer dopaminergic neurons survive, the therapeutic window narrows, and dosing becomes a balancing act: too much dopamine causes dyskinesias, hallucinations, delusions, and impulsive behavior, and too little dopamine causes worsening of Parkinson symptoms, freezing, and wearing-off, with ensuing falls and fractures. At this stage, some patients are prescribed levodopa every 1.5 or 2 hours.
Drugs are now available that extend the half-life of levodopa by slowing the breakdown of dopamine.
Catechol-O-methyltransferase (COMT) inhibitors—including tolcapone (Tasmar) and entacapone (Comtan) (also available as combined cardidopa, entacapone, and levodopa [Stalevo])—reduce off periods by about 1 hour per day.13 Given that the price is about $2,500 per year, the cost and benefits to the patient must be considered.14–17
Rasagiline, an MAO-B inhibitor, can also be added to levodopa to extend the “on” time for about 1 hour a day and to reduce freezing of gait. Clinical trials have shown it to be well tolerated, although common side effects include worsening dyskinesias and nausea.18,19
Apomorphine (Apokyn) is a dopamine agonist given by subcutaneous injection, allowing it to avoid first-pass metabolism by the liver. The benefits start just 10 minutes after injection, but only last for about 1 hour. It is a good option for rescue therapy for patients who cannot swallow or who have severe, unpredictable, or painful off-periods. It is also useful for situations in which it is especially inconvenient to have an off-period, such as being away from home.
Many agents have been tested for improving the off-period, but most work for about 1 to 2 hours, which is not nearly as effective as deep brain stimulation.
Managing dyskinesias
Dyskinesias can be managed by giving lower doses of levodopa more often. If wearing-off is a problem, a dopamine agonist or MAO-B inhibitor can be added. For patients at this stage, a specialist should be consulted.
Amantadine (Symmetrel), an N-methyl-d-aspartate (NMDA) receptor antagonist and dopamine-releasing agent used to treat influenza, is also effective against dyskinesias. Adverse effects include anxiety, insomnia, nightmares, anticholinergic effects, and livedo reticularis.20,21
Deep brain stimulation is the best treatment for dyskinesias in a patient for whom the procedure is appropriate and who has medical insurance that covers it.
NONMOTOR FEATURES OF PARKINSON DISEASE
Dementia: One of the most limiting nonmotor features
Often the most limiting nonmotor feature of Parkinson disease is dementia, which develops at about four to six times the rate for age-matched controls. At a given time, about 40% of patients with Parkinson disease have dementia, and the risk is 80% over 15 years of the disease.
If dementia is present, many of the drugs effective against Parkinson disease cannot be used because of exacerbating side effects. Treatment is mainly restricted to levodopa.
The only FDA-approved drug to treat dementia in Parkinson disease is the same drug for Alzheimer disease, rivastigmine (Exelon). Its effects are only modest, and its cholinergic side effects may transiently worsen parkinsonian features.22
Psychosis: Also very common
About half of patients with Parkinson disease have an episode of hallucinations or delusions in their lifetime, and about 20% are actively psychotic at any time. Delusions typically have the theme of spousal infidelity. Psychosis is associated with a higher rate of death compared with patients with Parkinson disease who do not develop it. Rebound psychosis may occur on withdrawal of antipsychotic medication.23–27
Patients who develop psychosis should have a physical examination and laboratory evaluation to determine if an infection or electrolyte imbalance is the cause. Medications should be discontinued in the following order: anticholinergic drug, amantadine, MAO-B inhibitor, dopamine agonist, and COMT inhibitor. Levodopa and carbidopa should be reduced to the minimum tolerable yet effective dosages.
For a patient who still has psychosis despite a minimum Parkinson drug regimen, an atypical antipsychotic drug should be used. Although clozapine (Clozaril, FazaClo) is very effective without worsening parkinsonism, it requires weekly monitoring with a complete blood count because of the small (< 1%) risk of agranulocytosis. For that reason, the first-line drug is quetiapine (Seroquel). Most double-blind studies have not found it to be effective, yet it is the drug most often used. No other antipsychotic drugs are safe to treat Parkinson psychosis.
Many patients with Parkinson disease who are hospitalized become agitated and confused soon after they are admitted to the hospital. The best treatment is quetiapine if an oral drug can be prescribed. A benzodiazepine—eg, clonazepam (Klonopin), lorazepam (Ativan), diazepam (Valium)—at a low dose may also be effective. Haloperidol, risperidone, and olanzapine should not be given, as they block dopamine receptors and worsen rigidity.
Mood disturbances
Depression occurs in about half of patients with Parkinson disease and is a significant cause of functional impairment. About 25% of patients have anxiety, and 20% are apathetic.
Depression appears to be secondary to underlying neuroanatomic degeneration rather than a reaction to disability.28 Fortunately, most antidepressants are effective in patients with Parkinson disease.29,30 Bupropion (Wellbutrin) is a dopamine reuptake inhibitor and so increases the availability of dopamine, and it should also have antiparkinsonian effects, but unfortunately it does not. Conversely, selective serotonin reuptake inhibitors (SSRIs) theoretically can worsen or cause parkinsonism, but evidence shows that they are safe to use in patients with Parkinson disease. Some evidence indicates that tricyclic antidepressants may be superior to SSRIs for treating depression in patients with Parkinson disease, so they might be the better choice in patients who can tolerate them.
Compulsive behaviors such as punding (prolonged performance of repetitive, mechanical tasks, such as disassembling and reassembling household objects) may occur from levodopa.
In addition, impulse control disorders involving pathologic gambling, hypersexuality, compulsive shopping, or binge eating occur in about 8% of patients with Parkinson disease taking dopamine agonists. These behaviors are more likely to arise in young, single patients, who are also more likely to have a family history of impulsive control disorder.31
THE FUTURE OF DRUG THERAPY
Clinical trials are now testing new therapies that work the traditional way through dopaminergic mechanisms, as well as those that work in novel ways.
A large international trial is studying patients with newly diagnosed Parkinson disease to try to discover a biomarker. Parkinson disease is unlike many other diseases in that physicians can only use clinical features to measure improvement, which is very crude. Identifying a biomarker will make evaluating and monitoring treatment a more exact science, and will lead to faster development of effective treatments.
- Adler CH, Ahlskog JE. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for The Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Nutt JG, Wooten GF. Clinical practice. Diagnosis and initial management of Parkinson’s disease. N Engl J Med 2005; 353:1021–1027.
- Litvan I, Bhatia KP, Burn DJ, et al; Movement Disorders Society Scientific Issues Committee. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003; 18:467–486.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001; 16:448–458.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med 2000; 342:1484–1491.
- Oertel WH, Wolters E, Sampaio C, et al. Pergolide versus levodopa monotherapy in early Parkinson’s disease patients: The PELMOPET study. Mov Disord 2006; 21:343–353.
- Lees AJ, Katzenschlager R, Head J, Ben-Shlomo Y. Ten-year follow-up of three different initial treatments in de-novo PD: a randomized trial. Neurology 2001; 57:1687–1694.
- Fowler JS, Volkow ND, Logan J, et al. Slow recovery of human brain MAO B after L-deprenyl (selegeline) withdrawal. Synapse 1994; 18:86–93.
- Elmer LW, Bertoni JM. The increasing role of monoamine oxidase type B inhibitors in Parkinson’s disease therapy. Expert Opin Pharmacother 2008; 9:2759–2772.
- Olanow CW, Rascol O, Hauser R, et al; ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278. Erratum in: N Engl J Med 2011; 364:1882.
- Stocchi F, Barbato L, Nordera G, Bolner A, Caraceni T. Entacapone improves the pharmacokinetic and therapeutic response of controlled release levodopa/carbidopa in Parkinson’s patients. J Neural Transm 2004; 111:173–180.
- Brooks DJ, Sagar HUK-Irish Entacapone Study Group. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind six month study. J Neurol Neurosurg Psychiatry 2003; 74:1071–1079.
- Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M; Celomen Study Group. Efficacy and safety of entacapone in Parkinson’s disease patients with soboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol Scand 2002; 105:245–255.
- Rinne UK, Larsen JP, Siden A, Worm-Petersen J. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology 1998; 51:1309–1314.
- Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Parkinson Study Group. Ann Neurol 1997; 42:747–755.
- Parkinson Study Group. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol 2005; 62:241–248.
- Rascol O, Brooks DJ, Melamed E, et al; LARGO study group. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet 2005; 365:947–954.
- Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol 1999; 56:1383–1386.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Almaraz AC, Driver-Dunckley ED, Woodruff BK, et al. Efficacy of rivastigmine for cognitive symptoms in Parkinson disease with dementia. Neurologist 2009; 15:234–237.
- Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain 2000; 123:733–745.
- Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004; 19:831–833.
- Goetz CG, Wuu J, Curgian LM, Leurgans S. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 2005; 64:81–86.
- Tollefson GD, Dellva MA, Mattler CA, Kane JM, Wirshing DA, Kinon BJ. Controlled, double-blind investigation of the clozapine discontinuation symptoms with conversion to either olanzapine or placebo. The Collaborative Crossover Study Group. J Clin Psychopharmacol 1999; 19:435–443.
- Fernandez HH, Trieschmann ME, Okun MS. Rebound psychosis: effect of discontinuation of antipsychotics in Parkinson’s disease. Mov Disord 2005; 20:104–105.
- McDonald WM, Richard IH, DeLong MR. Prevalence, etiology, and treatment of depression in Parkinson’s disease. Biol Psychiatry 2003; 54:363–375.
- Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008; 23:850–857.
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009; 72:886–892.
- Voon V, Sohr M, Lang AE, et al. Impulse control disorders in Parkinson disease: a multicenter case-control study. Ann Neurol 2011; 69:986–996. .
- Adler CH, Ahlskog JE. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for The Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Nutt JG, Wooten GF. Clinical practice. Diagnosis and initial management of Parkinson’s disease. N Engl J Med 2005; 353:1021–1027.
- Litvan I, Bhatia KP, Burn DJ, et al; Movement Disorders Society Scientific Issues Committee. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003; 18:467–486.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001; 16:448–458.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med 2000; 342:1484–1491.
- Oertel WH, Wolters E, Sampaio C, et al. Pergolide versus levodopa monotherapy in early Parkinson’s disease patients: The PELMOPET study. Mov Disord 2006; 21:343–353.
- Lees AJ, Katzenschlager R, Head J, Ben-Shlomo Y. Ten-year follow-up of three different initial treatments in de-novo PD: a randomized trial. Neurology 2001; 57:1687–1694.
- Fowler JS, Volkow ND, Logan J, et al. Slow recovery of human brain MAO B after L-deprenyl (selegeline) withdrawal. Synapse 1994; 18:86–93.
- Elmer LW, Bertoni JM. The increasing role of monoamine oxidase type B inhibitors in Parkinson’s disease therapy. Expert Opin Pharmacother 2008; 9:2759–2772.
- Olanow CW, Rascol O, Hauser R, et al; ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278. Erratum in: N Engl J Med 2011; 364:1882.
- Stocchi F, Barbato L, Nordera G, Bolner A, Caraceni T. Entacapone improves the pharmacokinetic and therapeutic response of controlled release levodopa/carbidopa in Parkinson’s patients. J Neural Transm 2004; 111:173–180.
- Brooks DJ, Sagar HUK-Irish Entacapone Study Group. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind six month study. J Neurol Neurosurg Psychiatry 2003; 74:1071–1079.
- Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M; Celomen Study Group. Efficacy and safety of entacapone in Parkinson’s disease patients with soboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol Scand 2002; 105:245–255.
- Rinne UK, Larsen JP, Siden A, Worm-Petersen J. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology 1998; 51:1309–1314.
- Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Parkinson Study Group. Ann Neurol 1997; 42:747–755.
- Parkinson Study Group. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol 2005; 62:241–248.
- Rascol O, Brooks DJ, Melamed E, et al; LARGO study group. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet 2005; 365:947–954.
- Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol 1999; 56:1383–1386.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Almaraz AC, Driver-Dunckley ED, Woodruff BK, et al. Efficacy of rivastigmine for cognitive symptoms in Parkinson disease with dementia. Neurologist 2009; 15:234–237.
- Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain 2000; 123:733–745.
- Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004; 19:831–833.
- Goetz CG, Wuu J, Curgian LM, Leurgans S. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 2005; 64:81–86.
- Tollefson GD, Dellva MA, Mattler CA, Kane JM, Wirshing DA, Kinon BJ. Controlled, double-blind investigation of the clozapine discontinuation symptoms with conversion to either olanzapine or placebo. The Collaborative Crossover Study Group. J Clin Psychopharmacol 1999; 19:435–443.
- Fernandez HH, Trieschmann ME, Okun MS. Rebound psychosis: effect of discontinuation of antipsychotics in Parkinson’s disease. Mov Disord 2005; 20:104–105.
- McDonald WM, Richard IH, DeLong MR. Prevalence, etiology, and treatment of depression in Parkinson’s disease. Biol Psychiatry 2003; 54:363–375.
- Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008; 23:850–857.
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009; 72:886–892.
- Voon V, Sohr M, Lang AE, et al. Impulse control disorders in Parkinson disease: a multicenter case-control study. Ann Neurol 2011; 69:986–996. .
KEY POINTS
- Parkinson disease can usually be diagnosed on the basis of clinical features: slow movement, resting tremor, rigidity, and asymmetrical presentation, as well as alleviation of symptoms with dopaminergic therapy.
- Early disease can be treated with levodopa, dopamine agonists, anticholinergics, and monoamine oxidase-B inhibitors.
- Advanced Parkinson disease may require a catechol-O-methyltransferase (COMT) inhibitor, apomorphine, and amantadine (Symmetrel). Side effects include motor fluctuations, dyskinesias, and cognitive problems.
Should target natriuretic peptide levels be used for outpatient management of chronic heart failure?
In the last few years, a number of randomized controlled trials have explored the value of using target levels of natriuretic peptides such as brain-type natriuretic peptide (BNP) and N-terminal BNP in the outpatient management of heart failure. Unfortunately, the results have been inconclusive.
RATIONALE FOR TARGETING NATRIURETIC PEPTIDE LEVELS
Heart failure causes devastating morbidity and death, yet its management is guided more often by subjective than by objective data.1 In other chronic conditions such as hypertension, diabetes mellitus, and hyperlipidemia, numerical targets for blood pressure, hemoglobin A1c, and low-density lipoprotein cholesterol levels are used to guide medical therapy, and lower rates of both morbidity and death have resulted.1 Extensive efforts have been undertaken to use natriuretic peptide levels to similarly guide heart failure therapy and improve outcomes.
LIMITATIONS TO TARGETING NATRIURETIC PEPTIDES
The relationship between natriuretic peptide levels and patient symptoms1 and outcomes2 is neither predictable nor linear, although the association between these levels and outcomes is stronger at the extremes, ie, at very low and very high levels.
Moreover, baseline levels vary significantly among people and within the same person, affected by factors such as genetic polymorphisms, 3 age, sex,4 body mass index,5 and other diseases, such as renal insufficiency.6
In addition, natriuretic peptide levels behave differently depending on the type of heart failure, rising much higher in systolic heart failure than in diastolic heart failure.7
ESTABLISHED USES OF MEASURING NATRIURETIC PEPTIDE LEVELS
Measuring natriuretic peptide levels has proven useful in diagnosing heart failure and in risk stratification of heart failure patients. BNP levels of less than 100 pg/mL practically exclude the diagnosis of heart failure (negative predictive value 89%),8 as do N-terminal BNP levels less than 300 pg/mL (negative predictive value 99%).9 Changes from baseline levels during acute hospitalization correlate with heart failure mortality rates, while elevated levels at discharge are associated with a higher risk of heart failure death and of readmission.10,11
NATRIURETIC PEPTIDES TO GUIDE THERAPY
Of the seven published clinical trials of therapy guided by natriuretic peptide levels, three were positive, three were negative, and one had mixed results.
Three positive trials
The Christchurch, New Zealand, trial12 (with 69 patients) found that there were fewer total cardiovascular events (death, hospital admission, or heart failure decompensation) at 9.5 months in the group randomized to receive treatment guided by the N-terminal BNP concentration than in the control group (19 vs 54, P = .02).
The STARS-BNP trial (Systolic Heart Failure Treatment Supported by BNP),13 with 220 patients, showed a significant reduction in the rate of deaths from heart failure and of readmission at 15 months in patients receiving BNP-guided treatment compared with controls (24% vs 52%, P < .001).
The PROTECT trial (Pro-B Type Natriuretic Peptide Outpatient Tailored Chronic Heart Failure Therapy),14 with 151 patients enrolled, showed a significant reduction in a composite of cardiovascular events (worsening heart failure, hospitalization for heart failure, acute coronary syndromes, ventricular arrhythmias, cerebral ischemia, and cardiovascular death) with N-terminal BNP guidance compared with standard care at a mean of 10 months of follow-up (58 events vs 100 events, P = .009). It also showed significant improvements in quality of life, left ventricular ejection fraction, and both left ventricular end-systolic and end-diastolic volume indexes with therapy guided by N-terminal BNP measurement. Moreover, therapy guided by N-terminal BNP was not associated with higher rates of renal dysfunction from more aggressive diuretic use.
Three negative trials
Conversely, three trials did not find significant reductions in rates of death or hospitalization-free survival between groups:
The STARBRITE trial (Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: Brain Natriuretic Peptide Versus the Clinical Congestion Score) (N = 130)15
The BATTLESCARRED trial (NT-proBNP-Assisted Treatment to Lessen Serial Cardiac Readmissions and Death) (N = 364)16
The PRIMA trial (Can Pro-brain-natriuretic Peptide Guided Therapy of Chronic Heart Failure Improve Heart Failure Morbidity and Mortality?) (N = 345).17
One trial with mixed results
The TIME-CHF (Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure),18 the largest of these trials to date (N = 499), did not show a survival benefit, but it did show a lower rate of hospitalization due to heart failure in the group receiving treatment guided by N-terminal BNP levels than in controls. Also, this study found that in the subset of patients younger than 75 years, therapy guided by N-terminal BNP levels reduced the risk of death and hospitalization from heart failure.
Why the different results in these studies?
Several reasons can be invoked to explain the heterogeneity of results in the studies mentioned above. Most importantly, the small sample sizes in these trials may have prevented differences from reaching statistical significance. Also, the inclusion criteria and methods varied considerably, with different natriuretic peptide targets, doses of medications, and treatment strategies.
WHAT IS THE CONCLUSION?
Although there are data to suggest that serial natriuretic peptide guidance can reduce the rates of hospitalization and death from heart failure in patients under age 75, there is not enough evidence to recommend routine measurements for the outpatient management of heart failure.
A 2009 focused update to the joint American College of Cardiology and American Heart Association 2005 guidelines19 concluded that using natriuretic peptide levels to guide heart failure therapy is not well established (class 2b, level of evidence C).
Measurement of natriuretic peptides can be useful in evaluating and risk-stratifying patients presenting in the urgent care setting in whom the clinical diagnosis of heart failure is uncertain. These measurements are to be viewed as part of the total evaluation but are not to be used in isolation to confirm or exclude the presence of heart failure or to monitor the patient for decompensation.
Natriuretic peptide measurement is not a substitute for the information derived from a good history (dyspnea, orthopnea, paroxysmal nocturnal dyspnea) and physical examination (eg, weight, jugular venous distention, crackles, a third heart sound, edema).
The consensus opinion remains that the favorable outcomes with natriuretic peptide guidance in clinical trials were due to better adherence and continuous up-titration of medications to maximally tolerated target doses of angiotensin-converting enzyme inhibitors and beta-blockers, in addition to closer follow-up of patients in those groups.20 This can be done without serial natriuretic peptide measurements.
- Bonow RO, Bennett S, Casey DE, et al; American College of Cardiology; American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures); Heart Failure Society of America. ACC/AHA clinical performance measures for adults with chronic heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures) endorsed by the Heart Failure Society of America. J Am Coll Cardiol 2005; 46:1144–1178.
- Packer M. Should B-type natriuretic peptide be measured routinely to guide the diagnosis and management of chronic heart failure? Circulation 2003; 108:2950–2953.
- Takeishi Y, Toriyama S, Takabatake N, et al. Linkage disequilibrium analyses of natriuretic peptide precursor B locus reveal risk haplotype conferring high plasma BNP levels. Biochem Biophys Res Commun 2007; 362:480–484.
- Redfield MM, Rodeheffer RJ, Jacobsen SJ, Mahoney DW, Bailey KR, Burnett JC. Plasma brain natriuretic peptide concentration: impact of age and gender. J Am Coll Cardiol 2002; 40:976–982.
- Wang TJ, Larson MG, Levy D, et al. Impact of obesity on plasma natriuretic peptide levels. Circulation 2004; 109:594–600.
- Anwaruddin S, Lloyd-Jones DM, Baggish A, et al. Renal function, congestive heart failure, and amino-terminal pro-brain natriuretic peptide measurement: results from the ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) Study. J Am Coll Cardiol 2006; 47:91–97.
- Iwanaga Y, Nishi I, Furuichi S, et al. B-type natriuretic peptide strongly reflects diastolic wall stress in patients with chronic heart failure: comparison between systolic and diastolic heart failure. J Am Coll Cardiol 2006; 47:742–748.
- Maisel AS, Krishnaswamy P, Nowak RM, et al; Breathing Not Properly Multinational Study Investigators. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002; 347:161–167.
- Januzzi JL, Camargo CA, Anwaruddin S, et al. The N-terminal Pro-BNP investigation of dyspnea in the emergency department (PRIDE) study. Am J Cardiol 2005; 95:948–954.
- Bettencourt P, Azevedo A, Pimenta J, Friões F, Ferreira S, Ferreira A. N-terminal-pro-brain natriuretic peptide predicts outcome after hospital discharge in heart failure patients. Circulation 2004; 110:2168–2174.
- Logeart D, Thabut G, Jourdain P, et al. Predischarge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. J Am Coll Cardiol 2004; 43:635–641.
- Troughton RW, Frampton CM, Yandle TG, Espiner EA, Nicholls MG, Richards AM. Treatment of heart failure guided by plasma aminoterminal brain natriuretic peptide (N-BNP) concentrations. Lancet 2000; 355:1126–1130.
- Jourdain P, Jondeau G, Funck F, et al. Plasma brain natriuretic peptide-guided therapy to improve outcome in heart failure: the STARS-BNP Multicenter Study. J Am Coll Cardiol 2007; 49:1733–1739.
- Januzzi JL, Rehman SU, Mohammed AA, et al. Use of amino-terminal pro-B-type natriuretic peptide to guide outpatient therapy of patients with chronic left ventricular systolic dysfunction. J Am Coll Cardiol 2011; 58:1881–1891.
- Shah MR, Claise KA, Bowers MT, et al. Testing new targets of therapy in advanced heart failure: the design and rationale of the Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: BRain NatrIuretic Peptide Versus the Clinical CongesTion ScorE (STARBRITE) trial. Am Heart J 2005; 150:893–898.
- Lainchbury JG, Troughton RW, Strangman KM, et al. N-terminal pro-B-type natriuretic peptide-guided treatment for chronic heart failure: results from the BATTLESCARRED (NT-proBNP-Assisted Treatment To Lessen Serial Cardiac Readmissions and Death) trial. J Am Coll Cardiol 2009; 55:53–60.
- Eurlings LW, van Pol PE, Kok WE, et al. Management of chronic heart failure guided by individual N-terminal pro-B-type natriuretic peptide targets: results of the PRIMA (Can PRo-brain-natriuretic peptide guided therapy of chronic heart failure IMprove heart fAilure morbidity and mortality?) study. J Am Coll Cardiol 2010; 56:2090–2100.
- Pfisterer M, Buser P, Rickli H, et al; TIME-CHF Investigators. BNP-guided vs symptom-guided heart failure therapy: the Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure (TIME-CHF) randomized trial. JAMA 2009; 301:383–392.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- O’Donoghue M, Braunwald E. Natriuretic peptides in heart failure: should therapy be guided by BNP levels? Nat Rev Cardiol 2010; 7:13–20.
In the last few years, a number of randomized controlled trials have explored the value of using target levels of natriuretic peptides such as brain-type natriuretic peptide (BNP) and N-terminal BNP in the outpatient management of heart failure. Unfortunately, the results have been inconclusive.
RATIONALE FOR TARGETING NATRIURETIC PEPTIDE LEVELS
Heart failure causes devastating morbidity and death, yet its management is guided more often by subjective than by objective data.1 In other chronic conditions such as hypertension, diabetes mellitus, and hyperlipidemia, numerical targets for blood pressure, hemoglobin A1c, and low-density lipoprotein cholesterol levels are used to guide medical therapy, and lower rates of both morbidity and death have resulted.1 Extensive efforts have been undertaken to use natriuretic peptide levels to similarly guide heart failure therapy and improve outcomes.
LIMITATIONS TO TARGETING NATRIURETIC PEPTIDES
The relationship between natriuretic peptide levels and patient symptoms1 and outcomes2 is neither predictable nor linear, although the association between these levels and outcomes is stronger at the extremes, ie, at very low and very high levels.
Moreover, baseline levels vary significantly among people and within the same person, affected by factors such as genetic polymorphisms, 3 age, sex,4 body mass index,5 and other diseases, such as renal insufficiency.6
In addition, natriuretic peptide levels behave differently depending on the type of heart failure, rising much higher in systolic heart failure than in diastolic heart failure.7
ESTABLISHED USES OF MEASURING NATRIURETIC PEPTIDE LEVELS
Measuring natriuretic peptide levels has proven useful in diagnosing heart failure and in risk stratification of heart failure patients. BNP levels of less than 100 pg/mL practically exclude the diagnosis of heart failure (negative predictive value 89%),8 as do N-terminal BNP levels less than 300 pg/mL (negative predictive value 99%).9 Changes from baseline levels during acute hospitalization correlate with heart failure mortality rates, while elevated levels at discharge are associated with a higher risk of heart failure death and of readmission.10,11
NATRIURETIC PEPTIDES TO GUIDE THERAPY
Of the seven published clinical trials of therapy guided by natriuretic peptide levels, three were positive, three were negative, and one had mixed results.
Three positive trials
The Christchurch, New Zealand, trial12 (with 69 patients) found that there were fewer total cardiovascular events (death, hospital admission, or heart failure decompensation) at 9.5 months in the group randomized to receive treatment guided by the N-terminal BNP concentration than in the control group (19 vs 54, P = .02).
The STARS-BNP trial (Systolic Heart Failure Treatment Supported by BNP),13 with 220 patients, showed a significant reduction in the rate of deaths from heart failure and of readmission at 15 months in patients receiving BNP-guided treatment compared with controls (24% vs 52%, P < .001).
The PROTECT trial (Pro-B Type Natriuretic Peptide Outpatient Tailored Chronic Heart Failure Therapy),14 with 151 patients enrolled, showed a significant reduction in a composite of cardiovascular events (worsening heart failure, hospitalization for heart failure, acute coronary syndromes, ventricular arrhythmias, cerebral ischemia, and cardiovascular death) with N-terminal BNP guidance compared with standard care at a mean of 10 months of follow-up (58 events vs 100 events, P = .009). It also showed significant improvements in quality of life, left ventricular ejection fraction, and both left ventricular end-systolic and end-diastolic volume indexes with therapy guided by N-terminal BNP measurement. Moreover, therapy guided by N-terminal BNP was not associated with higher rates of renal dysfunction from more aggressive diuretic use.
Three negative trials
Conversely, three trials did not find significant reductions in rates of death or hospitalization-free survival between groups:
The STARBRITE trial (Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: Brain Natriuretic Peptide Versus the Clinical Congestion Score) (N = 130)15
The BATTLESCARRED trial (NT-proBNP-Assisted Treatment to Lessen Serial Cardiac Readmissions and Death) (N = 364)16
The PRIMA trial (Can Pro-brain-natriuretic Peptide Guided Therapy of Chronic Heart Failure Improve Heart Failure Morbidity and Mortality?) (N = 345).17
One trial with mixed results
The TIME-CHF (Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure),18 the largest of these trials to date (N = 499), did not show a survival benefit, but it did show a lower rate of hospitalization due to heart failure in the group receiving treatment guided by N-terminal BNP levels than in controls. Also, this study found that in the subset of patients younger than 75 years, therapy guided by N-terminal BNP levels reduced the risk of death and hospitalization from heart failure.
Why the different results in these studies?
Several reasons can be invoked to explain the heterogeneity of results in the studies mentioned above. Most importantly, the small sample sizes in these trials may have prevented differences from reaching statistical significance. Also, the inclusion criteria and methods varied considerably, with different natriuretic peptide targets, doses of medications, and treatment strategies.
WHAT IS THE CONCLUSION?
Although there are data to suggest that serial natriuretic peptide guidance can reduce the rates of hospitalization and death from heart failure in patients under age 75, there is not enough evidence to recommend routine measurements for the outpatient management of heart failure.
A 2009 focused update to the joint American College of Cardiology and American Heart Association 2005 guidelines19 concluded that using natriuretic peptide levels to guide heart failure therapy is not well established (class 2b, level of evidence C).
Measurement of natriuretic peptides can be useful in evaluating and risk-stratifying patients presenting in the urgent care setting in whom the clinical diagnosis of heart failure is uncertain. These measurements are to be viewed as part of the total evaluation but are not to be used in isolation to confirm or exclude the presence of heart failure or to monitor the patient for decompensation.
Natriuretic peptide measurement is not a substitute for the information derived from a good history (dyspnea, orthopnea, paroxysmal nocturnal dyspnea) and physical examination (eg, weight, jugular venous distention, crackles, a third heart sound, edema).
The consensus opinion remains that the favorable outcomes with natriuretic peptide guidance in clinical trials were due to better adherence and continuous up-titration of medications to maximally tolerated target doses of angiotensin-converting enzyme inhibitors and beta-blockers, in addition to closer follow-up of patients in those groups.20 This can be done without serial natriuretic peptide measurements.
In the last few years, a number of randomized controlled trials have explored the value of using target levels of natriuretic peptides such as brain-type natriuretic peptide (BNP) and N-terminal BNP in the outpatient management of heart failure. Unfortunately, the results have been inconclusive.
RATIONALE FOR TARGETING NATRIURETIC PEPTIDE LEVELS
Heart failure causes devastating morbidity and death, yet its management is guided more often by subjective than by objective data.1 In other chronic conditions such as hypertension, diabetes mellitus, and hyperlipidemia, numerical targets for blood pressure, hemoglobin A1c, and low-density lipoprotein cholesterol levels are used to guide medical therapy, and lower rates of both morbidity and death have resulted.1 Extensive efforts have been undertaken to use natriuretic peptide levels to similarly guide heart failure therapy and improve outcomes.
LIMITATIONS TO TARGETING NATRIURETIC PEPTIDES
The relationship between natriuretic peptide levels and patient symptoms1 and outcomes2 is neither predictable nor linear, although the association between these levels and outcomes is stronger at the extremes, ie, at very low and very high levels.
Moreover, baseline levels vary significantly among people and within the same person, affected by factors such as genetic polymorphisms, 3 age, sex,4 body mass index,5 and other diseases, such as renal insufficiency.6
In addition, natriuretic peptide levels behave differently depending on the type of heart failure, rising much higher in systolic heart failure than in diastolic heart failure.7
ESTABLISHED USES OF MEASURING NATRIURETIC PEPTIDE LEVELS
Measuring natriuretic peptide levels has proven useful in diagnosing heart failure and in risk stratification of heart failure patients. BNP levels of less than 100 pg/mL practically exclude the diagnosis of heart failure (negative predictive value 89%),8 as do N-terminal BNP levels less than 300 pg/mL (negative predictive value 99%).9 Changes from baseline levels during acute hospitalization correlate with heart failure mortality rates, while elevated levels at discharge are associated with a higher risk of heart failure death and of readmission.10,11
NATRIURETIC PEPTIDES TO GUIDE THERAPY
Of the seven published clinical trials of therapy guided by natriuretic peptide levels, three were positive, three were negative, and one had mixed results.
Three positive trials
The Christchurch, New Zealand, trial12 (with 69 patients) found that there were fewer total cardiovascular events (death, hospital admission, or heart failure decompensation) at 9.5 months in the group randomized to receive treatment guided by the N-terminal BNP concentration than in the control group (19 vs 54, P = .02).
The STARS-BNP trial (Systolic Heart Failure Treatment Supported by BNP),13 with 220 patients, showed a significant reduction in the rate of deaths from heart failure and of readmission at 15 months in patients receiving BNP-guided treatment compared with controls (24% vs 52%, P < .001).
The PROTECT trial (Pro-B Type Natriuretic Peptide Outpatient Tailored Chronic Heart Failure Therapy),14 with 151 patients enrolled, showed a significant reduction in a composite of cardiovascular events (worsening heart failure, hospitalization for heart failure, acute coronary syndromes, ventricular arrhythmias, cerebral ischemia, and cardiovascular death) with N-terminal BNP guidance compared with standard care at a mean of 10 months of follow-up (58 events vs 100 events, P = .009). It also showed significant improvements in quality of life, left ventricular ejection fraction, and both left ventricular end-systolic and end-diastolic volume indexes with therapy guided by N-terminal BNP measurement. Moreover, therapy guided by N-terminal BNP was not associated with higher rates of renal dysfunction from more aggressive diuretic use.
Three negative trials
Conversely, three trials did not find significant reductions in rates of death or hospitalization-free survival between groups:
The STARBRITE trial (Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: Brain Natriuretic Peptide Versus the Clinical Congestion Score) (N = 130)15
The BATTLESCARRED trial (NT-proBNP-Assisted Treatment to Lessen Serial Cardiac Readmissions and Death) (N = 364)16
The PRIMA trial (Can Pro-brain-natriuretic Peptide Guided Therapy of Chronic Heart Failure Improve Heart Failure Morbidity and Mortality?) (N = 345).17
One trial with mixed results
The TIME-CHF (Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure),18 the largest of these trials to date (N = 499), did not show a survival benefit, but it did show a lower rate of hospitalization due to heart failure in the group receiving treatment guided by N-terminal BNP levels than in controls. Also, this study found that in the subset of patients younger than 75 years, therapy guided by N-terminal BNP levels reduced the risk of death and hospitalization from heart failure.
Why the different results in these studies?
Several reasons can be invoked to explain the heterogeneity of results in the studies mentioned above. Most importantly, the small sample sizes in these trials may have prevented differences from reaching statistical significance. Also, the inclusion criteria and methods varied considerably, with different natriuretic peptide targets, doses of medications, and treatment strategies.
WHAT IS THE CONCLUSION?
Although there are data to suggest that serial natriuretic peptide guidance can reduce the rates of hospitalization and death from heart failure in patients under age 75, there is not enough evidence to recommend routine measurements for the outpatient management of heart failure.
A 2009 focused update to the joint American College of Cardiology and American Heart Association 2005 guidelines19 concluded that using natriuretic peptide levels to guide heart failure therapy is not well established (class 2b, level of evidence C).
Measurement of natriuretic peptides can be useful in evaluating and risk-stratifying patients presenting in the urgent care setting in whom the clinical diagnosis of heart failure is uncertain. These measurements are to be viewed as part of the total evaluation but are not to be used in isolation to confirm or exclude the presence of heart failure or to monitor the patient for decompensation.
Natriuretic peptide measurement is not a substitute for the information derived from a good history (dyspnea, orthopnea, paroxysmal nocturnal dyspnea) and physical examination (eg, weight, jugular venous distention, crackles, a third heart sound, edema).
The consensus opinion remains that the favorable outcomes with natriuretic peptide guidance in clinical trials were due to better adherence and continuous up-titration of medications to maximally tolerated target doses of angiotensin-converting enzyme inhibitors and beta-blockers, in addition to closer follow-up of patients in those groups.20 This can be done without serial natriuretic peptide measurements.
- Bonow RO, Bennett S, Casey DE, et al; American College of Cardiology; American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures); Heart Failure Society of America. ACC/AHA clinical performance measures for adults with chronic heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures) endorsed by the Heart Failure Society of America. J Am Coll Cardiol 2005; 46:1144–1178.
- Packer M. Should B-type natriuretic peptide be measured routinely to guide the diagnosis and management of chronic heart failure? Circulation 2003; 108:2950–2953.
- Takeishi Y, Toriyama S, Takabatake N, et al. Linkage disequilibrium analyses of natriuretic peptide precursor B locus reveal risk haplotype conferring high plasma BNP levels. Biochem Biophys Res Commun 2007; 362:480–484.
- Redfield MM, Rodeheffer RJ, Jacobsen SJ, Mahoney DW, Bailey KR, Burnett JC. Plasma brain natriuretic peptide concentration: impact of age and gender. J Am Coll Cardiol 2002; 40:976–982.
- Wang TJ, Larson MG, Levy D, et al. Impact of obesity on plasma natriuretic peptide levels. Circulation 2004; 109:594–600.
- Anwaruddin S, Lloyd-Jones DM, Baggish A, et al. Renal function, congestive heart failure, and amino-terminal pro-brain natriuretic peptide measurement: results from the ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) Study. J Am Coll Cardiol 2006; 47:91–97.
- Iwanaga Y, Nishi I, Furuichi S, et al. B-type natriuretic peptide strongly reflects diastolic wall stress in patients with chronic heart failure: comparison between systolic and diastolic heart failure. J Am Coll Cardiol 2006; 47:742–748.
- Maisel AS, Krishnaswamy P, Nowak RM, et al; Breathing Not Properly Multinational Study Investigators. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002; 347:161–167.
- Januzzi JL, Camargo CA, Anwaruddin S, et al. The N-terminal Pro-BNP investigation of dyspnea in the emergency department (PRIDE) study. Am J Cardiol 2005; 95:948–954.
- Bettencourt P, Azevedo A, Pimenta J, Friões F, Ferreira S, Ferreira A. N-terminal-pro-brain natriuretic peptide predicts outcome after hospital discharge in heart failure patients. Circulation 2004; 110:2168–2174.
- Logeart D, Thabut G, Jourdain P, et al. Predischarge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. J Am Coll Cardiol 2004; 43:635–641.
- Troughton RW, Frampton CM, Yandle TG, Espiner EA, Nicholls MG, Richards AM. Treatment of heart failure guided by plasma aminoterminal brain natriuretic peptide (N-BNP) concentrations. Lancet 2000; 355:1126–1130.
- Jourdain P, Jondeau G, Funck F, et al. Plasma brain natriuretic peptide-guided therapy to improve outcome in heart failure: the STARS-BNP Multicenter Study. J Am Coll Cardiol 2007; 49:1733–1739.
- Januzzi JL, Rehman SU, Mohammed AA, et al. Use of amino-terminal pro-B-type natriuretic peptide to guide outpatient therapy of patients with chronic left ventricular systolic dysfunction. J Am Coll Cardiol 2011; 58:1881–1891.
- Shah MR, Claise KA, Bowers MT, et al. Testing new targets of therapy in advanced heart failure: the design and rationale of the Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: BRain NatrIuretic Peptide Versus the Clinical CongesTion ScorE (STARBRITE) trial. Am Heart J 2005; 150:893–898.
- Lainchbury JG, Troughton RW, Strangman KM, et al. N-terminal pro-B-type natriuretic peptide-guided treatment for chronic heart failure: results from the BATTLESCARRED (NT-proBNP-Assisted Treatment To Lessen Serial Cardiac Readmissions and Death) trial. J Am Coll Cardiol 2009; 55:53–60.
- Eurlings LW, van Pol PE, Kok WE, et al. Management of chronic heart failure guided by individual N-terminal pro-B-type natriuretic peptide targets: results of the PRIMA (Can PRo-brain-natriuretic peptide guided therapy of chronic heart failure IMprove heart fAilure morbidity and mortality?) study. J Am Coll Cardiol 2010; 56:2090–2100.
- Pfisterer M, Buser P, Rickli H, et al; TIME-CHF Investigators. BNP-guided vs symptom-guided heart failure therapy: the Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure (TIME-CHF) randomized trial. JAMA 2009; 301:383–392.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- O’Donoghue M, Braunwald E. Natriuretic peptides in heart failure: should therapy be guided by BNP levels? Nat Rev Cardiol 2010; 7:13–20.
- Bonow RO, Bennett S, Casey DE, et al; American College of Cardiology; American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures); Heart Failure Society of America. ACC/AHA clinical performance measures for adults with chronic heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Performance Measures (Writing Committee to Develop Heart Failure Clinical Performance Measures) endorsed by the Heart Failure Society of America. J Am Coll Cardiol 2005; 46:1144–1178.
- Packer M. Should B-type natriuretic peptide be measured routinely to guide the diagnosis and management of chronic heart failure? Circulation 2003; 108:2950–2953.
- Takeishi Y, Toriyama S, Takabatake N, et al. Linkage disequilibrium analyses of natriuretic peptide precursor B locus reveal risk haplotype conferring high plasma BNP levels. Biochem Biophys Res Commun 2007; 362:480–484.
- Redfield MM, Rodeheffer RJ, Jacobsen SJ, Mahoney DW, Bailey KR, Burnett JC. Plasma brain natriuretic peptide concentration: impact of age and gender. J Am Coll Cardiol 2002; 40:976–982.
- Wang TJ, Larson MG, Levy D, et al. Impact of obesity on plasma natriuretic peptide levels. Circulation 2004; 109:594–600.
- Anwaruddin S, Lloyd-Jones DM, Baggish A, et al. Renal function, congestive heart failure, and amino-terminal pro-brain natriuretic peptide measurement: results from the ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) Study. J Am Coll Cardiol 2006; 47:91–97.
- Iwanaga Y, Nishi I, Furuichi S, et al. B-type natriuretic peptide strongly reflects diastolic wall stress in patients with chronic heart failure: comparison between systolic and diastolic heart failure. J Am Coll Cardiol 2006; 47:742–748.
- Maisel AS, Krishnaswamy P, Nowak RM, et al; Breathing Not Properly Multinational Study Investigators. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002; 347:161–167.
- Januzzi JL, Camargo CA, Anwaruddin S, et al. The N-terminal Pro-BNP investigation of dyspnea in the emergency department (PRIDE) study. Am J Cardiol 2005; 95:948–954.
- Bettencourt P, Azevedo A, Pimenta J, Friões F, Ferreira S, Ferreira A. N-terminal-pro-brain natriuretic peptide predicts outcome after hospital discharge in heart failure patients. Circulation 2004; 110:2168–2174.
- Logeart D, Thabut G, Jourdain P, et al. Predischarge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. J Am Coll Cardiol 2004; 43:635–641.
- Troughton RW, Frampton CM, Yandle TG, Espiner EA, Nicholls MG, Richards AM. Treatment of heart failure guided by plasma aminoterminal brain natriuretic peptide (N-BNP) concentrations. Lancet 2000; 355:1126–1130.
- Jourdain P, Jondeau G, Funck F, et al. Plasma brain natriuretic peptide-guided therapy to improve outcome in heart failure: the STARS-BNP Multicenter Study. J Am Coll Cardiol 2007; 49:1733–1739.
- Januzzi JL, Rehman SU, Mohammed AA, et al. Use of amino-terminal pro-B-type natriuretic peptide to guide outpatient therapy of patients with chronic left ventricular systolic dysfunction. J Am Coll Cardiol 2011; 58:1881–1891.
- Shah MR, Claise KA, Bowers MT, et al. Testing new targets of therapy in advanced heart failure: the design and rationale of the Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: BRain NatrIuretic Peptide Versus the Clinical CongesTion ScorE (STARBRITE) trial. Am Heart J 2005; 150:893–898.
- Lainchbury JG, Troughton RW, Strangman KM, et al. N-terminal pro-B-type natriuretic peptide-guided treatment for chronic heart failure: results from the BATTLESCARRED (NT-proBNP-Assisted Treatment To Lessen Serial Cardiac Readmissions and Death) trial. J Am Coll Cardiol 2009; 55:53–60.
- Eurlings LW, van Pol PE, Kok WE, et al. Management of chronic heart failure guided by individual N-terminal pro-B-type natriuretic peptide targets: results of the PRIMA (Can PRo-brain-natriuretic peptide guided therapy of chronic heart failure IMprove heart fAilure morbidity and mortality?) study. J Am Coll Cardiol 2010; 56:2090–2100.
- Pfisterer M, Buser P, Rickli H, et al; TIME-CHF Investigators. BNP-guided vs symptom-guided heart failure therapy: the Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure (TIME-CHF) randomized trial. JAMA 2009; 301:383–392.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- O’Donoghue M, Braunwald E. Natriuretic peptides in heart failure: should therapy be guided by BNP levels? Nat Rev Cardiol 2010; 7:13–20.
Diffuse reticulonodular infiltrates
A 69-year-old woman presented to the hospital with a 3-month history of fever of unknown origin and dyspnea. Her medical history included diabetes mellitus and, many years ago, partially treated latent tuberculosis infection and pancreatic cancer, treated with Whipple surgery and chemotherapy.
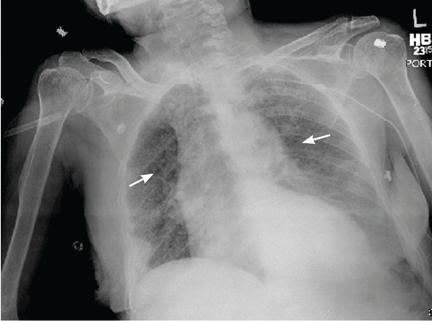
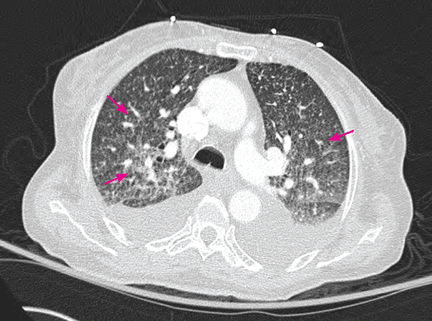
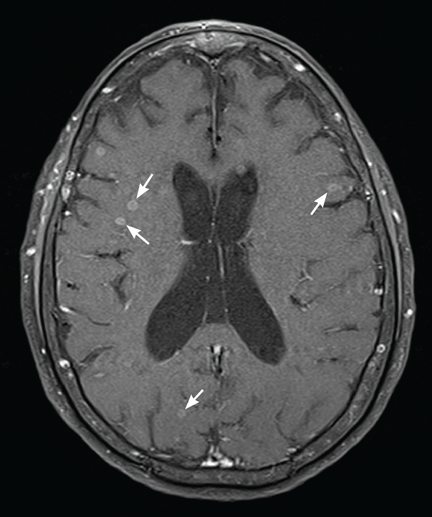
Q: What is the most likely diagnosis?
- Sarcoidosis of the lungs and central nervous system
- Hypersensitivity pneumonitis
- Miliary tuberculosis
- Cancer with pulmonary and brain metastases
- Disseminated fungal infection
A: The correct diagnosis is miliary tuberculosis, ie, progressive and widely disseminated hematogenous tuberculosis infection. Granulomas involving multiple organs suggested a broad differential diagnosis. The negative workup for infectious disease initially supported sarcoidosis by exclusion, but her condition failed to respond to steroid treatment. Multiple organ involvement is atypical for hypersensitivity pneumonitis, and antibody panels were negative. The absence of malignant cells in multiple biopsy specimens made metastasis unlikely.
Her remote history of partially treated latent tuberculosis infection raised our clinical suspicion and prompted mycobacterial antibiotic coverage. Three weeks after the initial sample collection, results from an independent laboratory revealed the presence of acid-fast bacilli in cultures of bronchoalveolar lavage fluid, cerebrospinal fluid, and blood, which were confirmed to be M tuberculosis. Despite treatment, the patient died of multiple organ failure.
Tuberculosis is rare in the United States, with 11,181 reported cases in 2010.1 Miliary tuberculosis is associated with malnutrition, HIV infection, AIDS, alcoholism, diabetes, chronic kidney failure, immunosuppressive drugs, and organ transplantation.2 Acid-fast bacilli smears and cultures confirm the diagnosis but have low sensitivity.3 Granulomas characterize miliary tuberculosis histopathologically. They may be present in sarcoidosis, hypersensitivity pneumonitis, and fungal infection but are not specific for these conditions. Tuberculin skin testing2 and interferon-gamma-release assay can be falsely negative in immunosuppressed patients,4 highlighting the emphasis on clinical suspicion. The estimated death rate is 20%,3,5 with central nervous system involvement being an independent predictor.5 Empiric therapy should not be delayed, as culture results may not be available for 6 to 8 weeks.
- US Centers for Disease Control and Prevention (CDC). Trends in tuberculosis—United States, 2010. MMWR Morb Mortal Wkly Rep 2011; 60:333–337.
- Sharma SK, Mohan A, Sharma A, Mitra DK. Miliary tuberculosis: new insights into an old disease. Lancet Infect Dis 2005; 5:415–430.
- Maartens G, Willcox PA, Benatar SR. Miliary tuberculosis: rapid diagnosis, hematologic abnormalities, and outcome in 109 treated adults. Am J Med 1990; 89:291–296.
- Mazurek GH, Jereb J, Vernon A, LoBue P, Goldberg S, Castro K; IGRA Expert Committee. Updated guidelines for using Interferon gamma release assays to detect Mycobacterium tuberculosis infection—United States, 2010. MMWR Recomm Rep 2010; 59:1–25.
- Kim JH, Langston AA, Gallis HA. Miliary tuberculosis: epidemiology, clinical manifestations, diagnosis, and outcome. Rev Infect Dis 1990; 12:583–590.
A 69-year-old woman presented to the hospital with a 3-month history of fever of unknown origin and dyspnea. Her medical history included diabetes mellitus and, many years ago, partially treated latent tuberculosis infection and pancreatic cancer, treated with Whipple surgery and chemotherapy.
Q: What is the most likely diagnosis?
- Sarcoidosis of the lungs and central nervous system
- Hypersensitivity pneumonitis
- Miliary tuberculosis
- Cancer with pulmonary and brain metastases
- Disseminated fungal infection
A: The correct diagnosis is miliary tuberculosis, ie, progressive and widely disseminated hematogenous tuberculosis infection. Granulomas involving multiple organs suggested a broad differential diagnosis. The negative workup for infectious disease initially supported sarcoidosis by exclusion, but her condition failed to respond to steroid treatment. Multiple organ involvement is atypical for hypersensitivity pneumonitis, and antibody panels were negative. The absence of malignant cells in multiple biopsy specimens made metastasis unlikely.
Her remote history of partially treated latent tuberculosis infection raised our clinical suspicion and prompted mycobacterial antibiotic coverage. Three weeks after the initial sample collection, results from an independent laboratory revealed the presence of acid-fast bacilli in cultures of bronchoalveolar lavage fluid, cerebrospinal fluid, and blood, which were confirmed to be M tuberculosis. Despite treatment, the patient died of multiple organ failure.
Tuberculosis is rare in the United States, with 11,181 reported cases in 2010.1 Miliary tuberculosis is associated with malnutrition, HIV infection, AIDS, alcoholism, diabetes, chronic kidney failure, immunosuppressive drugs, and organ transplantation.2 Acid-fast bacilli smears and cultures confirm the diagnosis but have low sensitivity.3 Granulomas characterize miliary tuberculosis histopathologically. They may be present in sarcoidosis, hypersensitivity pneumonitis, and fungal infection but are not specific for these conditions. Tuberculin skin testing2 and interferon-gamma-release assay can be falsely negative in immunosuppressed patients,4 highlighting the emphasis on clinical suspicion. The estimated death rate is 20%,3,5 with central nervous system involvement being an independent predictor.5 Empiric therapy should not be delayed, as culture results may not be available for 6 to 8 weeks.
A 69-year-old woman presented to the hospital with a 3-month history of fever of unknown origin and dyspnea. Her medical history included diabetes mellitus and, many years ago, partially treated latent tuberculosis infection and pancreatic cancer, treated with Whipple surgery and chemotherapy.
Q: What is the most likely diagnosis?
- Sarcoidosis of the lungs and central nervous system
- Hypersensitivity pneumonitis
- Miliary tuberculosis
- Cancer with pulmonary and brain metastases
- Disseminated fungal infection
A: The correct diagnosis is miliary tuberculosis, ie, progressive and widely disseminated hematogenous tuberculosis infection. Granulomas involving multiple organs suggested a broad differential diagnosis. The negative workup for infectious disease initially supported sarcoidosis by exclusion, but her condition failed to respond to steroid treatment. Multiple organ involvement is atypical for hypersensitivity pneumonitis, and antibody panels were negative. The absence of malignant cells in multiple biopsy specimens made metastasis unlikely.
Her remote history of partially treated latent tuberculosis infection raised our clinical suspicion and prompted mycobacterial antibiotic coverage. Three weeks after the initial sample collection, results from an independent laboratory revealed the presence of acid-fast bacilli in cultures of bronchoalveolar lavage fluid, cerebrospinal fluid, and blood, which were confirmed to be M tuberculosis. Despite treatment, the patient died of multiple organ failure.
Tuberculosis is rare in the United States, with 11,181 reported cases in 2010.1 Miliary tuberculosis is associated with malnutrition, HIV infection, AIDS, alcoholism, diabetes, chronic kidney failure, immunosuppressive drugs, and organ transplantation.2 Acid-fast bacilli smears and cultures confirm the diagnosis but have low sensitivity.3 Granulomas characterize miliary tuberculosis histopathologically. They may be present in sarcoidosis, hypersensitivity pneumonitis, and fungal infection but are not specific for these conditions. Tuberculin skin testing2 and interferon-gamma-release assay can be falsely negative in immunosuppressed patients,4 highlighting the emphasis on clinical suspicion. The estimated death rate is 20%,3,5 with central nervous system involvement being an independent predictor.5 Empiric therapy should not be delayed, as culture results may not be available for 6 to 8 weeks.
- US Centers for Disease Control and Prevention (CDC). Trends in tuberculosis—United States, 2010. MMWR Morb Mortal Wkly Rep 2011; 60:333–337.
- Sharma SK, Mohan A, Sharma A, Mitra DK. Miliary tuberculosis: new insights into an old disease. Lancet Infect Dis 2005; 5:415–430.
- Maartens G, Willcox PA, Benatar SR. Miliary tuberculosis: rapid diagnosis, hematologic abnormalities, and outcome in 109 treated adults. Am J Med 1990; 89:291–296.
- Mazurek GH, Jereb J, Vernon A, LoBue P, Goldberg S, Castro K; IGRA Expert Committee. Updated guidelines for using Interferon gamma release assays to detect Mycobacterium tuberculosis infection—United States, 2010. MMWR Recomm Rep 2010; 59:1–25.
- Kim JH, Langston AA, Gallis HA. Miliary tuberculosis: epidemiology, clinical manifestations, diagnosis, and outcome. Rev Infect Dis 1990; 12:583–590.
- US Centers for Disease Control and Prevention (CDC). Trends in tuberculosis—United States, 2010. MMWR Morb Mortal Wkly Rep 2011; 60:333–337.
- Sharma SK, Mohan A, Sharma A, Mitra DK. Miliary tuberculosis: new insights into an old disease. Lancet Infect Dis 2005; 5:415–430.
- Maartens G, Willcox PA, Benatar SR. Miliary tuberculosis: rapid diagnosis, hematologic abnormalities, and outcome in 109 treated adults. Am J Med 1990; 89:291–296.
- Mazurek GH, Jereb J, Vernon A, LoBue P, Goldberg S, Castro K; IGRA Expert Committee. Updated guidelines for using Interferon gamma release assays to detect Mycobacterium tuberculosis infection—United States, 2010. MMWR Recomm Rep 2010; 59:1–25.
- Kim JH, Langston AA, Gallis HA. Miliary tuberculosis: epidemiology, clinical manifestations, diagnosis, and outcome. Rev Infect Dis 1990; 12:583–590.