User login
Reasons for Discontinuation Vary by Psoriasis Treatment
Primary Systemic Amyloidosis
Varicella-Zoster Virus in Children Immunized With the Varicella Vaccine
Faulty equipment blamed for improper diagnosis: $78M verdict … and more
AT 36 WEEKS’ GESTATION, a woman went to the emergency department (ED) with abdominal pain. After ultrasonography (US), a nurse told her the fetus had died in utero, but the mother continued to feel fetal movement. The ED physician requested a second US, but it took 75 minutes for a radiology technician to arrive. This US showed a beating fetal heart with placental abruption. After cesarean delivery, the child was found to have cerebral palsy.
PATIENT’S CLAIM The first US was performed by an inexperienced technician using outdated equipment and the wrong transducer. An experienced technician with newer equipment should have been immediately available. The ED physician did not react when fetal distress was first identified.
DEFENDANTS’ DEFENSE The ED physician was told that the baby had died. Perhaps the child’s heart had started again by the time the second US was performed and a heartbeat found. The hospital denied negligence.
VERDICT A Pennsylvania jury found the ED physician not negligent; the hospital was 100% at fault. A $78.5 million verdict included $1.5 million in emotional distress to the mother, $10 million in pain and suffering for the child, $2million in lost future earnings, and the rest in future medical expenses.
Ligated ureter found after hysterectomy
A 50-YEAR-OLD WOMAN underwent laparoscopically assisted vaginal hysterectomy. She went to the ED with pain 6 days later. Imaging studies indicated a ligated ureter; a nephrostomy tube was placed. She required a nephrostomy bag for 4 months and underwent two repair operations.
PATIENT’S CLAIM The patient’s ureter was ligated and/or constricted during surgery. The gynecologist was negligent in failing to recognize and repair the injury during surgery.
PHYSICIAN’S DEFENSE The ureter was not ligated during surgery; therefore it could not have been discovered. In addition, injury to a ureter is a known risk of the procedure.
VERDICT An Arizona defense verdict was returned.
Myomectomy after cesarean; mother dies
IMMEDIATELY AFTER A WOMAN with preeclampsia had a cesarean delivery, she underwent a myomectomy. The day before discharge, her abdominal incision opened and a clear liquid drained. The day after discharge, she went to the ED with intense abdominal pain. Necrotizing fasciitis was found and debridement surgery performed. She was transferred to another hospital but died of sepsis several days later.
ESTATE’S CLAIM The infection occurred because the myomectomy was performed immediately following cesarean delivery. Prophylactic antibiotics were not prescribed before surgery. The mother was not fully informed as to the risks of concurrent operations. The ObGyns failed to recognize the infection before discharging the patient.
PHYSICIANS’ DEFENSE The patient was fully informed of the risks of surgery; it was reasonable to perform myomectomy immediately following cesarean delivery. There were no signs or symptoms of infection before discharge. Cesarean incisions open about 30% of the time—not a cause for concern. Prophylactic antibiotics for cesarean procedures are not standard of care. The patient’s infection was caused by a rapidly spreading, rare bacterium.
VERDICT A Michigan defense verdict was returned.
TOLAC to cesarean: baby has cerebral palsy
A WOMAN WANTED A TRIAL OF LABOR after a previous cesarean delivery (TOLAC). During labor, fetal distress was noted, and a cesarean delivery was performed. Uterine rupture had occurred. The baby has spastic cerebral palsy with significantly impaired neuromotor and cognitive abilities.
PARENTS’ CLAIM The hospital staff and physicians overlooked earlier fetal distress. A timelier delivery would have prevented the child’s injuries.
DEFENDANTs’ DEFENSE The hospital reached a confidential settlement. The ObGyns claimed fetal tracings were not suggestive of uterine rupture; they met the standard of care.
VERDICT A Texas defense verdict was returned.
WHEN GESTATIONAL DIABETES WAS DIAGNOSED at 33 weeks’ gestation, a family practitioner (FP) referred the mother to an ObGyn practice. Two ObGyns performed amniocentesis to check fetal lung maturity. After the procedure, fetal distress was noted, and the ObGyns instructed the FP to induce labor.
The baby suffered brain damage, had seizures, and has cerebral palsy. She was born without kidney function. By age 10, she had 2 kidney transplant operations and functions at a pre-kindergarten level.
PATIENT’S CLAIM The mother was not fully informed of the risks of and alternatives to amniocentesis. Although complications arose before amniocentesis, the test proceeded. The ObGyns were negligent in not performing cesarean delivery when fetal distress was detected.
DEFENDANTS’ DEFENSE The FP and hospital settled prior to trial. The ObGyns claimed that their care was an appropriate alternative to the actions the patient claimed should have been taken.
VERDICT Costs for the child’s care had reached $1.4 million before trial. A $9 million Virginia verdict was returned that included $7 million for the child and $2 million for the mother, but the settlement was reduced by the state cap.
Did HT cause breast cancer?
A 52-YEAR-OLD WOMAN was prescribed conjugated estrogens/medroxyprogesterone acetate (Prempro, Wyeth, Inc.) for hormone therapy by her gynecologist.
After taking the drug for 5 years, the patient developed invasive breast cancer. She underwent a lumpectomy, chemotherapy, and three reconstructive surgeries.
PATIENT’S CLAIM The manufacturer failed to warn of a woman’s risk of developing breast cancer while taking the product.
DEFENDANT’S DEFENSE Prempro alone does not cause cancer. The drug is just one of many contributing factors that may or may not increase the risk of developing breast cancer.
VERDICT A $3.75 million Connecticut verdict was returned for the patient plus $250,000 to her husband for loss of consortium.
Failure to diagnose preeclampsia—twice
AT 28 WEEKS’ GESTATION, a woman with a history of hypertension went to an ED with headache, nausea, vomiting, cramping, and ringing in her ears. After waiting 4 hours before being seen, her BP was 150/108 mm Hg and normal fetal heart tones were heard. The ED physician diagnosed otitis media and discharged her.
Later that evening, the patient returned to the ED with similar symptoms. A urine specimen showed significant proteinuria and the fetal heart rate was 158 bpm. A second ED physician diagnosed a urinary tract infection, prescribed antibiotics and pain medication, and sent her home.
A few hours later, she returned to the ED by ambulance suffering from eclamptic seizures. Her BP was 174/121 mm Hg, and no fetal heart tones were heard. She delivered a stillborn child by cesarean delivery.
PATIENT’S CLAIM The ED physicians were negligent in failing to diagnose preeclampsia at the first two visits.
PHYSICIANS’ DEFENSE The first ED physician settled for $45,000 while serving a prison sentence after conviction on two sex-abuse felonies related to his treatment of female patients at the same ED. The second ED physician denied negligence.
VERDICT A $50,000 Alabama verdict was returned for compensatory damages for the mother and $600,000 punitive damages for the stillborn child.
Inflated Foley catheter injures mother
DURING A LONG AND DIFFICULT LABOR, an ObGyn used forceps to complete delivery of a 31-year-old woman’s first child. The baby was healthy, but the mother has suffered urinary incontinence since delivery. Despite several repair operations, the condition remains.
PATIENT’S CLAIM The ObGyn was negligent in failing to remove a fully inflated Foley catheter before beginning delivery, leading to a urethral sphincter injury.
PHYSICIAN’S DEFENSE The decision regarding removal of the catheter was a matter of hospital policy. The ObGyn blamed improper catheter placement on the nurses.
VERDICT A Kentucky defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
AT 36 WEEKS’ GESTATION, a woman went to the emergency department (ED) with abdominal pain. After ultrasonography (US), a nurse told her the fetus had died in utero, but the mother continued to feel fetal movement. The ED physician requested a second US, but it took 75 minutes for a radiology technician to arrive. This US showed a beating fetal heart with placental abruption. After cesarean delivery, the child was found to have cerebral palsy.
PATIENT’S CLAIM The first US was performed by an inexperienced technician using outdated equipment and the wrong transducer. An experienced technician with newer equipment should have been immediately available. The ED physician did not react when fetal distress was first identified.
DEFENDANTS’ DEFENSE The ED physician was told that the baby had died. Perhaps the child’s heart had started again by the time the second US was performed and a heartbeat found. The hospital denied negligence.
VERDICT A Pennsylvania jury found the ED physician not negligent; the hospital was 100% at fault. A $78.5 million verdict included $1.5 million in emotional distress to the mother, $10 million in pain and suffering for the child, $2million in lost future earnings, and the rest in future medical expenses.
Ligated ureter found after hysterectomy
A 50-YEAR-OLD WOMAN underwent laparoscopically assisted vaginal hysterectomy. She went to the ED with pain 6 days later. Imaging studies indicated a ligated ureter; a nephrostomy tube was placed. She required a nephrostomy bag for 4 months and underwent two repair operations.
PATIENT’S CLAIM The patient’s ureter was ligated and/or constricted during surgery. The gynecologist was negligent in failing to recognize and repair the injury during surgery.
PHYSICIAN’S DEFENSE The ureter was not ligated during surgery; therefore it could not have been discovered. In addition, injury to a ureter is a known risk of the procedure.
VERDICT An Arizona defense verdict was returned.
Myomectomy after cesarean; mother dies
IMMEDIATELY AFTER A WOMAN with preeclampsia had a cesarean delivery, she underwent a myomectomy. The day before discharge, her abdominal incision opened and a clear liquid drained. The day after discharge, she went to the ED with intense abdominal pain. Necrotizing fasciitis was found and debridement surgery performed. She was transferred to another hospital but died of sepsis several days later.
ESTATE’S CLAIM The infection occurred because the myomectomy was performed immediately following cesarean delivery. Prophylactic antibiotics were not prescribed before surgery. The mother was not fully informed as to the risks of concurrent operations. The ObGyns failed to recognize the infection before discharging the patient.
PHYSICIANS’ DEFENSE The patient was fully informed of the risks of surgery; it was reasonable to perform myomectomy immediately following cesarean delivery. There were no signs or symptoms of infection before discharge. Cesarean incisions open about 30% of the time—not a cause for concern. Prophylactic antibiotics for cesarean procedures are not standard of care. The patient’s infection was caused by a rapidly spreading, rare bacterium.
VERDICT A Michigan defense verdict was returned.
TOLAC to cesarean: baby has cerebral palsy
A WOMAN WANTED A TRIAL OF LABOR after a previous cesarean delivery (TOLAC). During labor, fetal distress was noted, and a cesarean delivery was performed. Uterine rupture had occurred. The baby has spastic cerebral palsy with significantly impaired neuromotor and cognitive abilities.
PARENTS’ CLAIM The hospital staff and physicians overlooked earlier fetal distress. A timelier delivery would have prevented the child’s injuries.
DEFENDANTs’ DEFENSE The hospital reached a confidential settlement. The ObGyns claimed fetal tracings were not suggestive of uterine rupture; they met the standard of care.
VERDICT A Texas defense verdict was returned.
WHEN GESTATIONAL DIABETES WAS DIAGNOSED at 33 weeks’ gestation, a family practitioner (FP) referred the mother to an ObGyn practice. Two ObGyns performed amniocentesis to check fetal lung maturity. After the procedure, fetal distress was noted, and the ObGyns instructed the FP to induce labor.
The baby suffered brain damage, had seizures, and has cerebral palsy. She was born without kidney function. By age 10, she had 2 kidney transplant operations and functions at a pre-kindergarten level.
PATIENT’S CLAIM The mother was not fully informed of the risks of and alternatives to amniocentesis. Although complications arose before amniocentesis, the test proceeded. The ObGyns were negligent in not performing cesarean delivery when fetal distress was detected.
DEFENDANTS’ DEFENSE The FP and hospital settled prior to trial. The ObGyns claimed that their care was an appropriate alternative to the actions the patient claimed should have been taken.
VERDICT Costs for the child’s care had reached $1.4 million before trial. A $9 million Virginia verdict was returned that included $7 million for the child and $2 million for the mother, but the settlement was reduced by the state cap.
Did HT cause breast cancer?
A 52-YEAR-OLD WOMAN was prescribed conjugated estrogens/medroxyprogesterone acetate (Prempro, Wyeth, Inc.) for hormone therapy by her gynecologist.
After taking the drug for 5 years, the patient developed invasive breast cancer. She underwent a lumpectomy, chemotherapy, and three reconstructive surgeries.
PATIENT’S CLAIM The manufacturer failed to warn of a woman’s risk of developing breast cancer while taking the product.
DEFENDANT’S DEFENSE Prempro alone does not cause cancer. The drug is just one of many contributing factors that may or may not increase the risk of developing breast cancer.
VERDICT A $3.75 million Connecticut verdict was returned for the patient plus $250,000 to her husband for loss of consortium.
Failure to diagnose preeclampsia—twice
AT 28 WEEKS’ GESTATION, a woman with a history of hypertension went to an ED with headache, nausea, vomiting, cramping, and ringing in her ears. After waiting 4 hours before being seen, her BP was 150/108 mm Hg and normal fetal heart tones were heard. The ED physician diagnosed otitis media and discharged her.
Later that evening, the patient returned to the ED with similar symptoms. A urine specimen showed significant proteinuria and the fetal heart rate was 158 bpm. A second ED physician diagnosed a urinary tract infection, prescribed antibiotics and pain medication, and sent her home.
A few hours later, she returned to the ED by ambulance suffering from eclamptic seizures. Her BP was 174/121 mm Hg, and no fetal heart tones were heard. She delivered a stillborn child by cesarean delivery.
PATIENT’S CLAIM The ED physicians were negligent in failing to diagnose preeclampsia at the first two visits.
PHYSICIANS’ DEFENSE The first ED physician settled for $45,000 while serving a prison sentence after conviction on two sex-abuse felonies related to his treatment of female patients at the same ED. The second ED physician denied negligence.
VERDICT A $50,000 Alabama verdict was returned for compensatory damages for the mother and $600,000 punitive damages for the stillborn child.
Inflated Foley catheter injures mother
DURING A LONG AND DIFFICULT LABOR, an ObGyn used forceps to complete delivery of a 31-year-old woman’s first child. The baby was healthy, but the mother has suffered urinary incontinence since delivery. Despite several repair operations, the condition remains.
PATIENT’S CLAIM The ObGyn was negligent in failing to remove a fully inflated Foley catheter before beginning delivery, leading to a urethral sphincter injury.
PHYSICIAN’S DEFENSE The decision regarding removal of the catheter was a matter of hospital policy. The ObGyn blamed improper catheter placement on the nurses.
VERDICT A Kentucky defense verdict was returned.
AT 36 WEEKS’ GESTATION, a woman went to the emergency department (ED) with abdominal pain. After ultrasonography (US), a nurse told her the fetus had died in utero, but the mother continued to feel fetal movement. The ED physician requested a second US, but it took 75 minutes for a radiology technician to arrive. This US showed a beating fetal heart with placental abruption. After cesarean delivery, the child was found to have cerebral palsy.
PATIENT’S CLAIM The first US was performed by an inexperienced technician using outdated equipment and the wrong transducer. An experienced technician with newer equipment should have been immediately available. The ED physician did not react when fetal distress was first identified.
DEFENDANTS’ DEFENSE The ED physician was told that the baby had died. Perhaps the child’s heart had started again by the time the second US was performed and a heartbeat found. The hospital denied negligence.
VERDICT A Pennsylvania jury found the ED physician not negligent; the hospital was 100% at fault. A $78.5 million verdict included $1.5 million in emotional distress to the mother, $10 million in pain and suffering for the child, $2million in lost future earnings, and the rest in future medical expenses.
Ligated ureter found after hysterectomy
A 50-YEAR-OLD WOMAN underwent laparoscopically assisted vaginal hysterectomy. She went to the ED with pain 6 days later. Imaging studies indicated a ligated ureter; a nephrostomy tube was placed. She required a nephrostomy bag for 4 months and underwent two repair operations.
PATIENT’S CLAIM The patient’s ureter was ligated and/or constricted during surgery. The gynecologist was negligent in failing to recognize and repair the injury during surgery.
PHYSICIAN’S DEFENSE The ureter was not ligated during surgery; therefore it could not have been discovered. In addition, injury to a ureter is a known risk of the procedure.
VERDICT An Arizona defense verdict was returned.
Myomectomy after cesarean; mother dies
IMMEDIATELY AFTER A WOMAN with preeclampsia had a cesarean delivery, she underwent a myomectomy. The day before discharge, her abdominal incision opened and a clear liquid drained. The day after discharge, she went to the ED with intense abdominal pain. Necrotizing fasciitis was found and debridement surgery performed. She was transferred to another hospital but died of sepsis several days later.
ESTATE’S CLAIM The infection occurred because the myomectomy was performed immediately following cesarean delivery. Prophylactic antibiotics were not prescribed before surgery. The mother was not fully informed as to the risks of concurrent operations. The ObGyns failed to recognize the infection before discharging the patient.
PHYSICIANS’ DEFENSE The patient was fully informed of the risks of surgery; it was reasonable to perform myomectomy immediately following cesarean delivery. There were no signs or symptoms of infection before discharge. Cesarean incisions open about 30% of the time—not a cause for concern. Prophylactic antibiotics for cesarean procedures are not standard of care. The patient’s infection was caused by a rapidly spreading, rare bacterium.
VERDICT A Michigan defense verdict was returned.
TOLAC to cesarean: baby has cerebral palsy
A WOMAN WANTED A TRIAL OF LABOR after a previous cesarean delivery (TOLAC). During labor, fetal distress was noted, and a cesarean delivery was performed. Uterine rupture had occurred. The baby has spastic cerebral palsy with significantly impaired neuromotor and cognitive abilities.
PARENTS’ CLAIM The hospital staff and physicians overlooked earlier fetal distress. A timelier delivery would have prevented the child’s injuries.
DEFENDANTs’ DEFENSE The hospital reached a confidential settlement. The ObGyns claimed fetal tracings were not suggestive of uterine rupture; they met the standard of care.
VERDICT A Texas defense verdict was returned.
WHEN GESTATIONAL DIABETES WAS DIAGNOSED at 33 weeks’ gestation, a family practitioner (FP) referred the mother to an ObGyn practice. Two ObGyns performed amniocentesis to check fetal lung maturity. After the procedure, fetal distress was noted, and the ObGyns instructed the FP to induce labor.
The baby suffered brain damage, had seizures, and has cerebral palsy. She was born without kidney function. By age 10, she had 2 kidney transplant operations and functions at a pre-kindergarten level.
PATIENT’S CLAIM The mother was not fully informed of the risks of and alternatives to amniocentesis. Although complications arose before amniocentesis, the test proceeded. The ObGyns were negligent in not performing cesarean delivery when fetal distress was detected.
DEFENDANTS’ DEFENSE The FP and hospital settled prior to trial. The ObGyns claimed that their care was an appropriate alternative to the actions the patient claimed should have been taken.
VERDICT Costs for the child’s care had reached $1.4 million before trial. A $9 million Virginia verdict was returned that included $7 million for the child and $2 million for the mother, but the settlement was reduced by the state cap.
Did HT cause breast cancer?
A 52-YEAR-OLD WOMAN was prescribed conjugated estrogens/medroxyprogesterone acetate (Prempro, Wyeth, Inc.) for hormone therapy by her gynecologist.
After taking the drug for 5 years, the patient developed invasive breast cancer. She underwent a lumpectomy, chemotherapy, and three reconstructive surgeries.
PATIENT’S CLAIM The manufacturer failed to warn of a woman’s risk of developing breast cancer while taking the product.
DEFENDANT’S DEFENSE Prempro alone does not cause cancer. The drug is just one of many contributing factors that may or may not increase the risk of developing breast cancer.
VERDICT A $3.75 million Connecticut verdict was returned for the patient plus $250,000 to her husband for loss of consortium.
Failure to diagnose preeclampsia—twice
AT 28 WEEKS’ GESTATION, a woman with a history of hypertension went to an ED with headache, nausea, vomiting, cramping, and ringing in her ears. After waiting 4 hours before being seen, her BP was 150/108 mm Hg and normal fetal heart tones were heard. The ED physician diagnosed otitis media and discharged her.
Later that evening, the patient returned to the ED with similar symptoms. A urine specimen showed significant proteinuria and the fetal heart rate was 158 bpm. A second ED physician diagnosed a urinary tract infection, prescribed antibiotics and pain medication, and sent her home.
A few hours later, she returned to the ED by ambulance suffering from eclamptic seizures. Her BP was 174/121 mm Hg, and no fetal heart tones were heard. She delivered a stillborn child by cesarean delivery.
PATIENT’S CLAIM The ED physicians were negligent in failing to diagnose preeclampsia at the first two visits.
PHYSICIANS’ DEFENSE The first ED physician settled for $45,000 while serving a prison sentence after conviction on two sex-abuse felonies related to his treatment of female patients at the same ED. The second ED physician denied negligence.
VERDICT A $50,000 Alabama verdict was returned for compensatory damages for the mother and $600,000 punitive damages for the stillborn child.
Inflated Foley catheter injures mother
DURING A LONG AND DIFFICULT LABOR, an ObGyn used forceps to complete delivery of a 31-year-old woman’s first child. The baby was healthy, but the mother has suffered urinary incontinence since delivery. Despite several repair operations, the condition remains.
PATIENT’S CLAIM The ObGyn was negligent in failing to remove a fully inflated Foley catheter before beginning delivery, leading to a urethral sphincter injury.
PHYSICIAN’S DEFENSE The decision regarding removal of the catheter was a matter of hospital policy. The ObGyn blamed improper catheter placement on the nurses.
VERDICT A Kentucky defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
Ins and outs of straight-stick laparoscopic myomectomy
Watch 3 videos illustrating laparoscopic myomectomy
These videos were provided by Gaby Moawad, MD, and James Robinson, MD, MS.
By age 50, almost 70% of white women and more than 80% of black women will have a uterine leiomyoma.1 These benign, hormone-sensitive neoplasms1 are asymptomatic in the majority of women, but they can cause infertility, abnormal uterine bleeding, and bulk symptoms.2
When symptomatic, fibroids are amenable to multiple management options, ranging from expectant management to medical therapy to uterine artery embolization to myomectomy to hysterectomy. Myomectomy remains the surgical option of choice for women with symptomatic fibroids who wish to retain their fertility. It is also an option for some women who may not desire fertility retention but who do wish to maintain their uterus.
Compared with traditional myomectomy by laparotomy, laparoscopic myomectomy offers the advantages of:
- less blood loss
- less postoperative pain
- less postoperative adhesions formation
- faster recovery
- better cosmesis.3,4
Current technology makes performing laparoscopic myomectomy by either “straight-stick” or robotic assistance a viable option for most women.
In this article, we describe our technique in performing straight-stick laparoscopic myomectomy.
Preoperative evaluation: The first key to success
Laparoscopic myomectomy is an advanced, delicate, and challenging surgery. Preoperative evaluation is integral to its planning and a successful outcome.
Imaging
We recommend magnetic resonance imaging (MRI) as a standard order whenever a laparoscopic approach to myomectomy is being considered, for several reasons. First, MRI of the abdomen and pelvis with contrast allows for a precise map of the location of fibroids in relation to the myometrium and the uterine cavity. Reviewing the MRI results with the patient preoperatively gives both you and the patient a clearer picture of the challenges ahead. Patients tell us they appreciate these easier-to-understand images of their anatomy and, in cases when the decision is made to proceed abdominally, it is more clear to the patient why the decision is being made.
Surgically, the MRI helps compensate for the lack of tactile feedback when faced with deep intramural fibroids laparoscopically. The MRI also helps avoid operative surprises. Experience teaches us that, when relying on transvaginal ultrasound alone, adenomyotic regions can be identified mistakenly as fibroids. Preoperative MRI can help you avoid this discovery at the time of surgery.
A flexible office hysteroscopy serves as an adjunct to MRI for precise preoperative cavitary evaluation, especially when fibroids are present in close proximity to the endometrial cavity or the patient reports menorrhagia as a component of her symptomatology. When small submucosal fibroids exist in addition to larger fibroids, we frequently perform a combined hysteroscopic and laparoscopic approach to myomectomy.
Although bowel preparation does not diminish complications from bowel surgery or improve outcome,5 we generally use laxative suppositories the night prior to surgery to improve access to the posterior cul-de-sac and reduce bulk resulting from a sigmoid full of feces.
Preoperative laboratory evaluation should always include complete blood count, beta hCG, blood type testing, and antibody screen. In patients with known anemia or large intramural fibroids, we typically match the patient for 2 units of packed red blood cells. Additionally, if significant blood loss is anticipated, cell saver technology can be modified to accommodate a laparoscopic suction tip, allowing the patient’s own blood to be collected and readministered.
Aside from the standard risks of surgery, including bleeding, transfusion, infection, and injury to adjacent organs, myomectomy has its own unique risks that need to be made clear to patients preoperatively.
Surgery timing. Initially, women with symptomatic fibroids are at significant risk for developing more fibroids in the future. In fact, 25% of women who undergo myomectomy will require a second surgery at some point in their lives to address recurrent symptoms.1 If women are young, not yet ready to conceive, and are still relatively asymptomatic, waiting to perform myomectomy may be the most prudent course.
Future uterine rupture. There are no good myomectomy data to guide us with respect to the risk of uterine rupture at future pregnancy. When we perform deep intramural myomectomy (regardless of endometrial disruption), we extrapolate from classical cesarean section data and counsel our patients to have planned cesarean sections for all future pregnancies. Patients are also counseled that uterine rupture has been well described after laparoscopic myomectomy prior to the onset of labor so any sudden onset of pain or bleeding during the late second or third trimester of pregnancy has to be regarded as a medical emergency.
Pregnancy. Again, no good data exist to guide us with respect to postoperative timing of future pregnancy. We typically suggest patients refrain from conceiving following myomectomy for at least 6 months. We are aware that other well-respected surgeons have different thresholds.
Reference
1. Andiani GB, Fedele L, Parazzini F, Villa L. Risk of recurrence after myomectomy. Br J Obstet Gynaecol. 1991;98(4):385-389.
How to minimize blood loss
Blood loss during laparoscopic myomectomy generally is less than during laparotomy due to venous compression from pneumoperitoneum. However, blood loss remains a chief concern when performing laparoscopic myomectomy. A variety of techniques have been described to minimize blood loss, including injection of dilute vasopressin6 and tourniquet placement around uterine vessels.7
Injection. To temporarily minimize bleeding in the surgical field, we routinely utilize subserosal injection of dilute vasopressin (20 IU in 100 mL of normal saline) until visible vessels blanch. This practice is more effective than deep myoma or myometrial injection.
Tourniquet. We selectively use a laparoscopically placed tourniquet to compress uterine arteries at the mid-cervix during surgery. One approach is as follows (see VIDEO 1):
- Open windows in bilateral broad ligaments lateral to the uterine pedicles and medial to the ureters.
- Pass the end of a 14-16 French red rubber catheter through one of the low lateral port sites with the port removed.
- Tag the trailing end of the tourniquet outside the abdomen and replace the port alongside the catheter.
- Pass the end of the catheter down through the ipsilateral broad ligament window and under the posterior cervix.
- Pass the end of the catheter up through the contralateral broad ligament window and over the anterior cervix.
- Pass the end of the catheter through each of the broad ligament windows a second time and then out through the contralateral port site.
- Pull the tourniquet tight from both port sites (which will occlude the uterine arteries). Place Kelly clamps on the catheter ends where they exit the port sites to maintain tension until the end of the uterine repair.
Lateral ports can still be utilized with the tourniquet in place.
Permanent occlusion. In women undergoing laparoscopic myomectomy who have completed child bearing, we advocate permanent uterine artery occlusion at the origin of the uterine arteries retroperitoneally. This can be performed in a number of ways— utilizing clips, suture, or transection. Uterine artery occlusion not only leads to less operative blood loss but preliminary studies also suggest it decreases the risk of fibroid recurrence.8
After the patient is prepped and draped in low lithotomy position with her arms tucked at her sides, drain the bladder with an indwelling catheter. Insert a uterine manipulator, such as the VCare (Conmed Corporation), into the uterus.
Obtain umbilical entry for a 30° optic scope, and place the patient in steep Trendelenburg position. We use two 5-mm lateral ports and one 12-mm suprapubic port. The level of placement of the lateral ports is tailored to the size of the fibroids; it can be anywhere from the level of the anterior iliac spine to the level of the umbilicus for fibroids contained in the pelvis or in the abdomen, respectively.
Uterine incision
After vasopressin injection, tourniquet placement, or permanent uterine artery occlusion is performed as described above, we advocate a transverse uterine incision. We do so mainly because:
- The transverse incision runs parallel to the arcuate vessels of the myometrium, leading to less bleeding.
- We suture from the lateral ports so the transverse incision facilitates a more ergonomic repair.
Perform the uterine incision (we use the Harmonic Ace [Ethicon Endo-Surgery, Inc]) through the uterine serosa deep toward the myoma. Incision size should be appropriate to the diameter of the fibroid; smaller incisions result in unnecessary struggling during enucleation of the fibroid. Incision depth should reach the fibroid capsule, and this incision should be developed over the entire fibroid. Tunneling in the myometrium is undesirable and should be avoided because it increases myometrial injury as well as the risk of hematoma.
Fibroid enucleation
Once the initial uterine incision is complete, enucleate the fibroid using a combination of traction, countertraction, sharp, and blunt dissection (see VIDEO 2). Pearls to successful enucleation include:
- Maintain traction and countertraction when cutting tissue. This helps to identify appropriate planes and allows tissue to separate quickly, minimizing thermal energy spread.
- Replace the tenaculum or myoma screw regularly at the border of the myoma and myometrium. The ultrasonic scalpel blade can be drilled into the myoma in order to create traction on the myoma.
- Bluntly peel tissue from the myoma outward. Ideally all myometrium and vessels should stay with the uterus. A properly enucleated fibroid will be pearly in appearance and avascular.
- Be particularly careful when in contact with the endometrium. Even submucous fibroids can be enucleated regularly without entering the endometrial cavity.
Uterine repair
If the endometrial cavity is inadvertently entered, close the defect (we use a 2-0 monocryl or Vicryl suture). Next, imbricate the endometrium over with successive layers, taking special care not to pass a needle into the endometrial cavity. In cases in which significant endometrial disruption cannot be avoided, use a postoperative intrauterine balloon stent. This is placed postoperatively and left in place for 2 weeks. To stimulate endometrial proliferation, we prescribe oral estradiol 1 mg twice per day for 4 weeks. Following endometrial stimulation, we prescribe a 10-day progestin withdrawal and ask the patient to return to the office for a flexible diagnostic hysteroscopy following her first menses to ensure cavitary integrity.
Once fibroid enucleation is complete, perform a multilayer closure of the defect using an absorbable, unidirectional barbed suture (V-Loc, Covidien). Eliminate all dead space in the closure. The last two throws of each barbed suture should be in the direction of the prior two throws to secure the suture. Finally, cut the suture at the tissue edge without leaving any trailing tail or knot.
Why use a barbed suture? The advantages of using absorbable barbed suture include:
- elimination of knot tying
- shorter closure time
- better tension distribution throughout the wound.
Close the seromuscularis layer in a hemostatic baseball stitch fashion to minimize suture exposure and subsequent adhesions (FIGURE). Use of the suprapubic port for the needle retrieval device facilitates placement of the alternating “inside-out” baseball stitch (see VIDEO 3).

FIGURE Use of a hemostatic baseball stitch (with absorbable, unidirectional barbed suture) to close the seromuscularis layer of the uterus, thereby minimizing suture exposure and subsequent adhesions.
Morcellation
Mechanically morcellate the fibroids through the suprapubic port site. We use an electrical morcellator (Karl Storz Endoscopy). In cases of massive fibroids (>15 cm) we utilize cold- knife morcellation with a # 10 scalpel through an extended 3-cm suprapubic incision with a vertical fascial incision protected by a self-retaining wound retractor. It is important to be vigilant to remove all fibroid pieces as postoperative disseminated leiomyomatosis is well described.
Adhesion prevention
Myomectomy is notorious for creating dense and challenging postoperative adhesions. Given the high rate of repeat surgery for patients undergoing the procedure, anything you can do to limit the adhesion load will be appreciated by both your patient and her next surgeon. Without exception, the most important adhesion-prevention strategy is meticulous attention to tissue handling and hemostasis. In general, laparoscopy leads to fewer adhesions than laparotomy, but a bloody field and raw uterine serosa will create an environment ripe for adhesions regardless of surgical approach. If the operative field is dry, use commercial adhesion prevention aids according to manufacturers’ recommendations.
- Use preoperative MRI to tailor your surgical approach
- When menorrhagia is a presenting symptom, assess the endometrial cavity preoperatively and consider combining the laparoscopic myomectomy with hysteroscopic myomectomy
- Minimize blood loss with vasopressin or a laparoscopic tourniquet
- Utilize a transverse uterine incision and a lateral suturing technique
- Use delayed absorbable barbed suture to close the myometrial defect
- Bury the seromuscular closure suture by utilizing an “inside-out” baseball stitch
- If the risk for postoperative cavitary adhesions is high, consider postoperative balloon placement with close postoperative follow-up
- Advise patients to wait 6 months prior to attempting to conceive and have a low threshold for scheduled cesarean delivery to minimize the risk of uterine rupture
Concluding thoughts, from experience
Laparoscopic myomectomy is a challenging yet rewarding procedure. For essential points to our approach, see “Laparoscopic myomectomy: Key takeaways” on this page.
Other important things to keep in mind:
- Fibroid presentation is as varied as the women who have them—meticulous preoperative preparation is an absolute must.
- Utilize well-established approaches to preventing blood loss, removing fibroids, and repairing the uterine defects. The accomplished gynecologic laparoscopist will be successful in the majority of cases.
- Practice suturing in a box-trainer setting before taking on initial cases. Early cases should focus on straightforward subserosal fibroids and, as skills progress, more and more difficult cases will become reasonable.
- Do not place any hard and fast limit on either the number or size of fibroids you are willing to remove laparoscopically. Rather, rely on sound surgical judgment, an honest assessment of your limitations, and a healthy dose of caution as you approach every new patient. Never sacrifice the quality of your repair for a less invasive approach to surgery.
Laparoscopic myomectomy: 8 pearls
Jon I. Einarsson, MD, MPH (March 2010)
Give vasopressin to reduce bleeding in gynecologic surgery
Robert L. Barbieri, MD (Editorial, March 2010)
Barbed suture, now in the toolbox of minimally invasive gyn surgery
Jon I. Einarsson, MD, MPH; James A. Greenberg, MD (September 2009)
When necessity calls for treating uterine fibroids
William H. Parker, MD (Surgical Techniques, June 2008)
Advising your patients–Uterine fibroids: Childbearing, cancer, and hormone effects
William H. Parker, MD (May 2008)
We want to hear from you! Tell us what you think.
1. Baird DD, Dunson DB, Hill MC, Cousins D, Schectman JM. High cumulative incidence of uterine leiomyoma in black and white women: ultrasound evidence. Am J Obstet Gynecol. 2003;188(1):100-107.
2. Wallach EE, Vlahos NF. Uterine myomas: an overview of development clinical features, and management. Obstet Gynecol. 2004;104(2):393-406.
3. Stringer NH, Walker JC, Meyer PM. Comparison of 49 laparoscopic myomec- tomies and 49 open myomectomies. J Am Assoc Gynecol Laparosc. 1997;4(4):457-464.
4. Bulletti C, Polli V, Negrini V, Giacomucci E, Flamigni C. Adhesion formation after laparoscopic myomectomy. J Am Assoc Gynecol Laparosc. 1996;3(4):533-536.
5. Ram E, Sherman Y, Weil R, Vishne T, Kravarusic D, Dreznik Z. Is mechanical bowel preparation mandatory for elective colon surgery? A prospective randomized study. Arch Surg. 2005;140(3):285-288.
6. Frederick J, Fletcher H, Simeon D, Mullings A, Hardie M. Intramyometrial vasopressin as a haemostatic agent during myomectomy. Br J Obstet Gynaecol. 1994;101(5):435-437.
7. Taylor A, Sharma M, Tsirkas P, Di Spiezio Sardo A, Setchell M, Magos A. Reducing blood loss at open myomectomy using triple tourniquets: a randomised controlled trial. BJOG. 2005;112(3):340-345.
8. Burbank F, Hutchins FL, Jr. Uterine artery occlusion by embolization or surgery for the treatment of fibroids: A unifying hypothesis-transient uterine ischemia. J Am Assoc Gynecol Laparosc. 2000;7(suppl 4):S1-S49.
Watch 3 videos illustrating laparoscopic myomectomy
These videos were provided by Gaby Moawad, MD, and James Robinson, MD, MS.
By age 50, almost 70% of white women and more than 80% of black women will have a uterine leiomyoma.1 These benign, hormone-sensitive neoplasms1 are asymptomatic in the majority of women, but they can cause infertility, abnormal uterine bleeding, and bulk symptoms.2
When symptomatic, fibroids are amenable to multiple management options, ranging from expectant management to medical therapy to uterine artery embolization to myomectomy to hysterectomy. Myomectomy remains the surgical option of choice for women with symptomatic fibroids who wish to retain their fertility. It is also an option for some women who may not desire fertility retention but who do wish to maintain their uterus.
Compared with traditional myomectomy by laparotomy, laparoscopic myomectomy offers the advantages of:
- less blood loss
- less postoperative pain
- less postoperative adhesions formation
- faster recovery
- better cosmesis.3,4
Current technology makes performing laparoscopic myomectomy by either “straight-stick” or robotic assistance a viable option for most women.
In this article, we describe our technique in performing straight-stick laparoscopic myomectomy.
Preoperative evaluation: The first key to success
Laparoscopic myomectomy is an advanced, delicate, and challenging surgery. Preoperative evaluation is integral to its planning and a successful outcome.
Imaging
We recommend magnetic resonance imaging (MRI) as a standard order whenever a laparoscopic approach to myomectomy is being considered, for several reasons. First, MRI of the abdomen and pelvis with contrast allows for a precise map of the location of fibroids in relation to the myometrium and the uterine cavity. Reviewing the MRI results with the patient preoperatively gives both you and the patient a clearer picture of the challenges ahead. Patients tell us they appreciate these easier-to-understand images of their anatomy and, in cases when the decision is made to proceed abdominally, it is more clear to the patient why the decision is being made.
Surgically, the MRI helps compensate for the lack of tactile feedback when faced with deep intramural fibroids laparoscopically. The MRI also helps avoid operative surprises. Experience teaches us that, when relying on transvaginal ultrasound alone, adenomyotic regions can be identified mistakenly as fibroids. Preoperative MRI can help you avoid this discovery at the time of surgery.
A flexible office hysteroscopy serves as an adjunct to MRI for precise preoperative cavitary evaluation, especially when fibroids are present in close proximity to the endometrial cavity or the patient reports menorrhagia as a component of her symptomatology. When small submucosal fibroids exist in addition to larger fibroids, we frequently perform a combined hysteroscopic and laparoscopic approach to myomectomy.
Although bowel preparation does not diminish complications from bowel surgery or improve outcome,5 we generally use laxative suppositories the night prior to surgery to improve access to the posterior cul-de-sac and reduce bulk resulting from a sigmoid full of feces.
Preoperative laboratory evaluation should always include complete blood count, beta hCG, blood type testing, and antibody screen. In patients with known anemia or large intramural fibroids, we typically match the patient for 2 units of packed red blood cells. Additionally, if significant blood loss is anticipated, cell saver technology can be modified to accommodate a laparoscopic suction tip, allowing the patient’s own blood to be collected and readministered.
Aside from the standard risks of surgery, including bleeding, transfusion, infection, and injury to adjacent organs, myomectomy has its own unique risks that need to be made clear to patients preoperatively.
Surgery timing. Initially, women with symptomatic fibroids are at significant risk for developing more fibroids in the future. In fact, 25% of women who undergo myomectomy will require a second surgery at some point in their lives to address recurrent symptoms.1 If women are young, not yet ready to conceive, and are still relatively asymptomatic, waiting to perform myomectomy may be the most prudent course.
Future uterine rupture. There are no good myomectomy data to guide us with respect to the risk of uterine rupture at future pregnancy. When we perform deep intramural myomectomy (regardless of endometrial disruption), we extrapolate from classical cesarean section data and counsel our patients to have planned cesarean sections for all future pregnancies. Patients are also counseled that uterine rupture has been well described after laparoscopic myomectomy prior to the onset of labor so any sudden onset of pain or bleeding during the late second or third trimester of pregnancy has to be regarded as a medical emergency.
Pregnancy. Again, no good data exist to guide us with respect to postoperative timing of future pregnancy. We typically suggest patients refrain from conceiving following myomectomy for at least 6 months. We are aware that other well-respected surgeons have different thresholds.
Reference
1. Andiani GB, Fedele L, Parazzini F, Villa L. Risk of recurrence after myomectomy. Br J Obstet Gynaecol. 1991;98(4):385-389.
How to minimize blood loss
Blood loss during laparoscopic myomectomy generally is less than during laparotomy due to venous compression from pneumoperitoneum. However, blood loss remains a chief concern when performing laparoscopic myomectomy. A variety of techniques have been described to minimize blood loss, including injection of dilute vasopressin6 and tourniquet placement around uterine vessels.7
Injection. To temporarily minimize bleeding in the surgical field, we routinely utilize subserosal injection of dilute vasopressin (20 IU in 100 mL of normal saline) until visible vessels blanch. This practice is more effective than deep myoma or myometrial injection.
Tourniquet. We selectively use a laparoscopically placed tourniquet to compress uterine arteries at the mid-cervix during surgery. One approach is as follows (see VIDEO 1):
- Open windows in bilateral broad ligaments lateral to the uterine pedicles and medial to the ureters.
- Pass the end of a 14-16 French red rubber catheter through one of the low lateral port sites with the port removed.
- Tag the trailing end of the tourniquet outside the abdomen and replace the port alongside the catheter.
- Pass the end of the catheter down through the ipsilateral broad ligament window and under the posterior cervix.
- Pass the end of the catheter up through the contralateral broad ligament window and over the anterior cervix.
- Pass the end of the catheter through each of the broad ligament windows a second time and then out through the contralateral port site.
- Pull the tourniquet tight from both port sites (which will occlude the uterine arteries). Place Kelly clamps on the catheter ends where they exit the port sites to maintain tension until the end of the uterine repair.
Lateral ports can still be utilized with the tourniquet in place.
Permanent occlusion. In women undergoing laparoscopic myomectomy who have completed child bearing, we advocate permanent uterine artery occlusion at the origin of the uterine arteries retroperitoneally. This can be performed in a number of ways— utilizing clips, suture, or transection. Uterine artery occlusion not only leads to less operative blood loss but preliminary studies also suggest it decreases the risk of fibroid recurrence.8
After the patient is prepped and draped in low lithotomy position with her arms tucked at her sides, drain the bladder with an indwelling catheter. Insert a uterine manipulator, such as the VCare (Conmed Corporation), into the uterus.
Obtain umbilical entry for a 30° optic scope, and place the patient in steep Trendelenburg position. We use two 5-mm lateral ports and one 12-mm suprapubic port. The level of placement of the lateral ports is tailored to the size of the fibroids; it can be anywhere from the level of the anterior iliac spine to the level of the umbilicus for fibroids contained in the pelvis or in the abdomen, respectively.
Uterine incision
After vasopressin injection, tourniquet placement, or permanent uterine artery occlusion is performed as described above, we advocate a transverse uterine incision. We do so mainly because:
- The transverse incision runs parallel to the arcuate vessels of the myometrium, leading to less bleeding.
- We suture from the lateral ports so the transverse incision facilitates a more ergonomic repair.
Perform the uterine incision (we use the Harmonic Ace [Ethicon Endo-Surgery, Inc]) through the uterine serosa deep toward the myoma. Incision size should be appropriate to the diameter of the fibroid; smaller incisions result in unnecessary struggling during enucleation of the fibroid. Incision depth should reach the fibroid capsule, and this incision should be developed over the entire fibroid. Tunneling in the myometrium is undesirable and should be avoided because it increases myometrial injury as well as the risk of hematoma.
Fibroid enucleation
Once the initial uterine incision is complete, enucleate the fibroid using a combination of traction, countertraction, sharp, and blunt dissection (see VIDEO 2). Pearls to successful enucleation include:
- Maintain traction and countertraction when cutting tissue. This helps to identify appropriate planes and allows tissue to separate quickly, minimizing thermal energy spread.
- Replace the tenaculum or myoma screw regularly at the border of the myoma and myometrium. The ultrasonic scalpel blade can be drilled into the myoma in order to create traction on the myoma.
- Bluntly peel tissue from the myoma outward. Ideally all myometrium and vessels should stay with the uterus. A properly enucleated fibroid will be pearly in appearance and avascular.
- Be particularly careful when in contact with the endometrium. Even submucous fibroids can be enucleated regularly without entering the endometrial cavity.
Uterine repair
If the endometrial cavity is inadvertently entered, close the defect (we use a 2-0 monocryl or Vicryl suture). Next, imbricate the endometrium over with successive layers, taking special care not to pass a needle into the endometrial cavity. In cases in which significant endometrial disruption cannot be avoided, use a postoperative intrauterine balloon stent. This is placed postoperatively and left in place for 2 weeks. To stimulate endometrial proliferation, we prescribe oral estradiol 1 mg twice per day for 4 weeks. Following endometrial stimulation, we prescribe a 10-day progestin withdrawal and ask the patient to return to the office for a flexible diagnostic hysteroscopy following her first menses to ensure cavitary integrity.
Once fibroid enucleation is complete, perform a multilayer closure of the defect using an absorbable, unidirectional barbed suture (V-Loc, Covidien). Eliminate all dead space in the closure. The last two throws of each barbed suture should be in the direction of the prior two throws to secure the suture. Finally, cut the suture at the tissue edge without leaving any trailing tail or knot.
Why use a barbed suture? The advantages of using absorbable barbed suture include:
- elimination of knot tying
- shorter closure time
- better tension distribution throughout the wound.
Close the seromuscularis layer in a hemostatic baseball stitch fashion to minimize suture exposure and subsequent adhesions (FIGURE). Use of the suprapubic port for the needle retrieval device facilitates placement of the alternating “inside-out” baseball stitch (see VIDEO 3).
FIGURE Use of a hemostatic baseball stitch (with absorbable, unidirectional barbed suture) to close the seromuscularis layer of the uterus, thereby minimizing suture exposure and subsequent adhesions.
Morcellation
Mechanically morcellate the fibroids through the suprapubic port site. We use an electrical morcellator (Karl Storz Endoscopy). In cases of massive fibroids (>15 cm) we utilize cold- knife morcellation with a # 10 scalpel through an extended 3-cm suprapubic incision with a vertical fascial incision protected by a self-retaining wound retractor. It is important to be vigilant to remove all fibroid pieces as postoperative disseminated leiomyomatosis is well described.
Adhesion prevention
Myomectomy is notorious for creating dense and challenging postoperative adhesions. Given the high rate of repeat surgery for patients undergoing the procedure, anything you can do to limit the adhesion load will be appreciated by both your patient and her next surgeon. Without exception, the most important adhesion-prevention strategy is meticulous attention to tissue handling and hemostasis. In general, laparoscopy leads to fewer adhesions than laparotomy, but a bloody field and raw uterine serosa will create an environment ripe for adhesions regardless of surgical approach. If the operative field is dry, use commercial adhesion prevention aids according to manufacturers’ recommendations.
- Use preoperative MRI to tailor your surgical approach
- When menorrhagia is a presenting symptom, assess the endometrial cavity preoperatively and consider combining the laparoscopic myomectomy with hysteroscopic myomectomy
- Minimize blood loss with vasopressin or a laparoscopic tourniquet
- Utilize a transverse uterine incision and a lateral suturing technique
- Use delayed absorbable barbed suture to close the myometrial defect
- Bury the seromuscular closure suture by utilizing an “inside-out” baseball stitch
- If the risk for postoperative cavitary adhesions is high, consider postoperative balloon placement with close postoperative follow-up
- Advise patients to wait 6 months prior to attempting to conceive and have a low threshold for scheduled cesarean delivery to minimize the risk of uterine rupture
Concluding thoughts, from experience
Laparoscopic myomectomy is a challenging yet rewarding procedure. For essential points to our approach, see “Laparoscopic myomectomy: Key takeaways” on this page.
Other important things to keep in mind:
- Fibroid presentation is as varied as the women who have them—meticulous preoperative preparation is an absolute must.
- Utilize well-established approaches to preventing blood loss, removing fibroids, and repairing the uterine defects. The accomplished gynecologic laparoscopist will be successful in the majority of cases.
- Practice suturing in a box-trainer setting before taking on initial cases. Early cases should focus on straightforward subserosal fibroids and, as skills progress, more and more difficult cases will become reasonable.
- Do not place any hard and fast limit on either the number or size of fibroids you are willing to remove laparoscopically. Rather, rely on sound surgical judgment, an honest assessment of your limitations, and a healthy dose of caution as you approach every new patient. Never sacrifice the quality of your repair for a less invasive approach to surgery.
Laparoscopic myomectomy: 8 pearls
Jon I. Einarsson, MD, MPH (March 2010)
Give vasopressin to reduce bleeding in gynecologic surgery
Robert L. Barbieri, MD (Editorial, March 2010)
Barbed suture, now in the toolbox of minimally invasive gyn surgery
Jon I. Einarsson, MD, MPH; James A. Greenberg, MD (September 2009)
When necessity calls for treating uterine fibroids
William H. Parker, MD (Surgical Techniques, June 2008)
Advising your patients–Uterine fibroids: Childbearing, cancer, and hormone effects
William H. Parker, MD (May 2008)
We want to hear from you! Tell us what you think.
Watch 3 videos illustrating laparoscopic myomectomy
These videos were provided by Gaby Moawad, MD, and James Robinson, MD, MS.
By age 50, almost 70% of white women and more than 80% of black women will have a uterine leiomyoma.1 These benign, hormone-sensitive neoplasms1 are asymptomatic in the majority of women, but they can cause infertility, abnormal uterine bleeding, and bulk symptoms.2
When symptomatic, fibroids are amenable to multiple management options, ranging from expectant management to medical therapy to uterine artery embolization to myomectomy to hysterectomy. Myomectomy remains the surgical option of choice for women with symptomatic fibroids who wish to retain their fertility. It is also an option for some women who may not desire fertility retention but who do wish to maintain their uterus.
Compared with traditional myomectomy by laparotomy, laparoscopic myomectomy offers the advantages of:
- less blood loss
- less postoperative pain
- less postoperative adhesions formation
- faster recovery
- better cosmesis.3,4
Current technology makes performing laparoscopic myomectomy by either “straight-stick” or robotic assistance a viable option for most women.
In this article, we describe our technique in performing straight-stick laparoscopic myomectomy.
Preoperative evaluation: The first key to success
Laparoscopic myomectomy is an advanced, delicate, and challenging surgery. Preoperative evaluation is integral to its planning and a successful outcome.
Imaging
We recommend magnetic resonance imaging (MRI) as a standard order whenever a laparoscopic approach to myomectomy is being considered, for several reasons. First, MRI of the abdomen and pelvis with contrast allows for a precise map of the location of fibroids in relation to the myometrium and the uterine cavity. Reviewing the MRI results with the patient preoperatively gives both you and the patient a clearer picture of the challenges ahead. Patients tell us they appreciate these easier-to-understand images of their anatomy and, in cases when the decision is made to proceed abdominally, it is more clear to the patient why the decision is being made.
Surgically, the MRI helps compensate for the lack of tactile feedback when faced with deep intramural fibroids laparoscopically. The MRI also helps avoid operative surprises. Experience teaches us that, when relying on transvaginal ultrasound alone, adenomyotic regions can be identified mistakenly as fibroids. Preoperative MRI can help you avoid this discovery at the time of surgery.
A flexible office hysteroscopy serves as an adjunct to MRI for precise preoperative cavitary evaluation, especially when fibroids are present in close proximity to the endometrial cavity or the patient reports menorrhagia as a component of her symptomatology. When small submucosal fibroids exist in addition to larger fibroids, we frequently perform a combined hysteroscopic and laparoscopic approach to myomectomy.
Although bowel preparation does not diminish complications from bowel surgery or improve outcome,5 we generally use laxative suppositories the night prior to surgery to improve access to the posterior cul-de-sac and reduce bulk resulting from a sigmoid full of feces.
Preoperative laboratory evaluation should always include complete blood count, beta hCG, blood type testing, and antibody screen. In patients with known anemia or large intramural fibroids, we typically match the patient for 2 units of packed red blood cells. Additionally, if significant blood loss is anticipated, cell saver technology can be modified to accommodate a laparoscopic suction tip, allowing the patient’s own blood to be collected and readministered.
Aside from the standard risks of surgery, including bleeding, transfusion, infection, and injury to adjacent organs, myomectomy has its own unique risks that need to be made clear to patients preoperatively.
Surgery timing. Initially, women with symptomatic fibroids are at significant risk for developing more fibroids in the future. In fact, 25% of women who undergo myomectomy will require a second surgery at some point in their lives to address recurrent symptoms.1 If women are young, not yet ready to conceive, and are still relatively asymptomatic, waiting to perform myomectomy may be the most prudent course.
Future uterine rupture. There are no good myomectomy data to guide us with respect to the risk of uterine rupture at future pregnancy. When we perform deep intramural myomectomy (regardless of endometrial disruption), we extrapolate from classical cesarean section data and counsel our patients to have planned cesarean sections for all future pregnancies. Patients are also counseled that uterine rupture has been well described after laparoscopic myomectomy prior to the onset of labor so any sudden onset of pain or bleeding during the late second or third trimester of pregnancy has to be regarded as a medical emergency.
Pregnancy. Again, no good data exist to guide us with respect to postoperative timing of future pregnancy. We typically suggest patients refrain from conceiving following myomectomy for at least 6 months. We are aware that other well-respected surgeons have different thresholds.
Reference
1. Andiani GB, Fedele L, Parazzini F, Villa L. Risk of recurrence after myomectomy. Br J Obstet Gynaecol. 1991;98(4):385-389.
How to minimize blood loss
Blood loss during laparoscopic myomectomy generally is less than during laparotomy due to venous compression from pneumoperitoneum. However, blood loss remains a chief concern when performing laparoscopic myomectomy. A variety of techniques have been described to minimize blood loss, including injection of dilute vasopressin6 and tourniquet placement around uterine vessels.7
Injection. To temporarily minimize bleeding in the surgical field, we routinely utilize subserosal injection of dilute vasopressin (20 IU in 100 mL of normal saline) until visible vessels blanch. This practice is more effective than deep myoma or myometrial injection.
Tourniquet. We selectively use a laparoscopically placed tourniquet to compress uterine arteries at the mid-cervix during surgery. One approach is as follows (see VIDEO 1):
- Open windows in bilateral broad ligaments lateral to the uterine pedicles and medial to the ureters.
- Pass the end of a 14-16 French red rubber catheter through one of the low lateral port sites with the port removed.
- Tag the trailing end of the tourniquet outside the abdomen and replace the port alongside the catheter.
- Pass the end of the catheter down through the ipsilateral broad ligament window and under the posterior cervix.
- Pass the end of the catheter up through the contralateral broad ligament window and over the anterior cervix.
- Pass the end of the catheter through each of the broad ligament windows a second time and then out through the contralateral port site.
- Pull the tourniquet tight from both port sites (which will occlude the uterine arteries). Place Kelly clamps on the catheter ends where they exit the port sites to maintain tension until the end of the uterine repair.
Lateral ports can still be utilized with the tourniquet in place.
Permanent occlusion. In women undergoing laparoscopic myomectomy who have completed child bearing, we advocate permanent uterine artery occlusion at the origin of the uterine arteries retroperitoneally. This can be performed in a number of ways— utilizing clips, suture, or transection. Uterine artery occlusion not only leads to less operative blood loss but preliminary studies also suggest it decreases the risk of fibroid recurrence.8
After the patient is prepped and draped in low lithotomy position with her arms tucked at her sides, drain the bladder with an indwelling catheter. Insert a uterine manipulator, such as the VCare (Conmed Corporation), into the uterus.
Obtain umbilical entry for a 30° optic scope, and place the patient in steep Trendelenburg position. We use two 5-mm lateral ports and one 12-mm suprapubic port. The level of placement of the lateral ports is tailored to the size of the fibroids; it can be anywhere from the level of the anterior iliac spine to the level of the umbilicus for fibroids contained in the pelvis or in the abdomen, respectively.
Uterine incision
After vasopressin injection, tourniquet placement, or permanent uterine artery occlusion is performed as described above, we advocate a transverse uterine incision. We do so mainly because:
- The transverse incision runs parallel to the arcuate vessels of the myometrium, leading to less bleeding.
- We suture from the lateral ports so the transverse incision facilitates a more ergonomic repair.
Perform the uterine incision (we use the Harmonic Ace [Ethicon Endo-Surgery, Inc]) through the uterine serosa deep toward the myoma. Incision size should be appropriate to the diameter of the fibroid; smaller incisions result in unnecessary struggling during enucleation of the fibroid. Incision depth should reach the fibroid capsule, and this incision should be developed over the entire fibroid. Tunneling in the myometrium is undesirable and should be avoided because it increases myometrial injury as well as the risk of hematoma.
Fibroid enucleation
Once the initial uterine incision is complete, enucleate the fibroid using a combination of traction, countertraction, sharp, and blunt dissection (see VIDEO 2). Pearls to successful enucleation include:
- Maintain traction and countertraction when cutting tissue. This helps to identify appropriate planes and allows tissue to separate quickly, minimizing thermal energy spread.
- Replace the tenaculum or myoma screw regularly at the border of the myoma and myometrium. The ultrasonic scalpel blade can be drilled into the myoma in order to create traction on the myoma.
- Bluntly peel tissue from the myoma outward. Ideally all myometrium and vessels should stay with the uterus. A properly enucleated fibroid will be pearly in appearance and avascular.
- Be particularly careful when in contact with the endometrium. Even submucous fibroids can be enucleated regularly without entering the endometrial cavity.
Uterine repair
If the endometrial cavity is inadvertently entered, close the defect (we use a 2-0 monocryl or Vicryl suture). Next, imbricate the endometrium over with successive layers, taking special care not to pass a needle into the endometrial cavity. In cases in which significant endometrial disruption cannot be avoided, use a postoperative intrauterine balloon stent. This is placed postoperatively and left in place for 2 weeks. To stimulate endometrial proliferation, we prescribe oral estradiol 1 mg twice per day for 4 weeks. Following endometrial stimulation, we prescribe a 10-day progestin withdrawal and ask the patient to return to the office for a flexible diagnostic hysteroscopy following her first menses to ensure cavitary integrity.
Once fibroid enucleation is complete, perform a multilayer closure of the defect using an absorbable, unidirectional barbed suture (V-Loc, Covidien). Eliminate all dead space in the closure. The last two throws of each barbed suture should be in the direction of the prior two throws to secure the suture. Finally, cut the suture at the tissue edge without leaving any trailing tail or knot.
Why use a barbed suture? The advantages of using absorbable barbed suture include:
- elimination of knot tying
- shorter closure time
- better tension distribution throughout the wound.
Close the seromuscularis layer in a hemostatic baseball stitch fashion to minimize suture exposure and subsequent adhesions (FIGURE). Use of the suprapubic port for the needle retrieval device facilitates placement of the alternating “inside-out” baseball stitch (see VIDEO 3).
FIGURE Use of a hemostatic baseball stitch (with absorbable, unidirectional barbed suture) to close the seromuscularis layer of the uterus, thereby minimizing suture exposure and subsequent adhesions.
Morcellation
Mechanically morcellate the fibroids through the suprapubic port site. We use an electrical morcellator (Karl Storz Endoscopy). In cases of massive fibroids (>15 cm) we utilize cold- knife morcellation with a # 10 scalpel through an extended 3-cm suprapubic incision with a vertical fascial incision protected by a self-retaining wound retractor. It is important to be vigilant to remove all fibroid pieces as postoperative disseminated leiomyomatosis is well described.
Adhesion prevention
Myomectomy is notorious for creating dense and challenging postoperative adhesions. Given the high rate of repeat surgery for patients undergoing the procedure, anything you can do to limit the adhesion load will be appreciated by both your patient and her next surgeon. Without exception, the most important adhesion-prevention strategy is meticulous attention to tissue handling and hemostasis. In general, laparoscopy leads to fewer adhesions than laparotomy, but a bloody field and raw uterine serosa will create an environment ripe for adhesions regardless of surgical approach. If the operative field is dry, use commercial adhesion prevention aids according to manufacturers’ recommendations.
- Use preoperative MRI to tailor your surgical approach
- When menorrhagia is a presenting symptom, assess the endometrial cavity preoperatively and consider combining the laparoscopic myomectomy with hysteroscopic myomectomy
- Minimize blood loss with vasopressin or a laparoscopic tourniquet
- Utilize a transverse uterine incision and a lateral suturing technique
- Use delayed absorbable barbed suture to close the myometrial defect
- Bury the seromuscular closure suture by utilizing an “inside-out” baseball stitch
- If the risk for postoperative cavitary adhesions is high, consider postoperative balloon placement with close postoperative follow-up
- Advise patients to wait 6 months prior to attempting to conceive and have a low threshold for scheduled cesarean delivery to minimize the risk of uterine rupture
Concluding thoughts, from experience
Laparoscopic myomectomy is a challenging yet rewarding procedure. For essential points to our approach, see “Laparoscopic myomectomy: Key takeaways” on this page.
Other important things to keep in mind:
- Fibroid presentation is as varied as the women who have them—meticulous preoperative preparation is an absolute must.
- Utilize well-established approaches to preventing blood loss, removing fibroids, and repairing the uterine defects. The accomplished gynecologic laparoscopist will be successful in the majority of cases.
- Practice suturing in a box-trainer setting before taking on initial cases. Early cases should focus on straightforward subserosal fibroids and, as skills progress, more and more difficult cases will become reasonable.
- Do not place any hard and fast limit on either the number or size of fibroids you are willing to remove laparoscopically. Rather, rely on sound surgical judgment, an honest assessment of your limitations, and a healthy dose of caution as you approach every new patient. Never sacrifice the quality of your repair for a less invasive approach to surgery.
Laparoscopic myomectomy: 8 pearls
Jon I. Einarsson, MD, MPH (March 2010)
Give vasopressin to reduce bleeding in gynecologic surgery
Robert L. Barbieri, MD (Editorial, March 2010)
Barbed suture, now in the toolbox of minimally invasive gyn surgery
Jon I. Einarsson, MD, MPH; James A. Greenberg, MD (September 2009)
When necessity calls for treating uterine fibroids
William H. Parker, MD (Surgical Techniques, June 2008)
Advising your patients–Uterine fibroids: Childbearing, cancer, and hormone effects
William H. Parker, MD (May 2008)
We want to hear from you! Tell us what you think.
1. Baird DD, Dunson DB, Hill MC, Cousins D, Schectman JM. High cumulative incidence of uterine leiomyoma in black and white women: ultrasound evidence. Am J Obstet Gynecol. 2003;188(1):100-107.
2. Wallach EE, Vlahos NF. Uterine myomas: an overview of development clinical features, and management. Obstet Gynecol. 2004;104(2):393-406.
3. Stringer NH, Walker JC, Meyer PM. Comparison of 49 laparoscopic myomec- tomies and 49 open myomectomies. J Am Assoc Gynecol Laparosc. 1997;4(4):457-464.
4. Bulletti C, Polli V, Negrini V, Giacomucci E, Flamigni C. Adhesion formation after laparoscopic myomectomy. J Am Assoc Gynecol Laparosc. 1996;3(4):533-536.
5. Ram E, Sherman Y, Weil R, Vishne T, Kravarusic D, Dreznik Z. Is mechanical bowel preparation mandatory for elective colon surgery? A prospective randomized study. Arch Surg. 2005;140(3):285-288.
6. Frederick J, Fletcher H, Simeon D, Mullings A, Hardie M. Intramyometrial vasopressin as a haemostatic agent during myomectomy. Br J Obstet Gynaecol. 1994;101(5):435-437.
7. Taylor A, Sharma M, Tsirkas P, Di Spiezio Sardo A, Setchell M, Magos A. Reducing blood loss at open myomectomy using triple tourniquets: a randomised controlled trial. BJOG. 2005;112(3):340-345.
8. Burbank F, Hutchins FL, Jr. Uterine artery occlusion by embolization or surgery for the treatment of fibroids: A unifying hypothesis-transient uterine ischemia. J Am Assoc Gynecol Laparosc. 2000;7(suppl 4):S1-S49.
1. Baird DD, Dunson DB, Hill MC, Cousins D, Schectman JM. High cumulative incidence of uterine leiomyoma in black and white women: ultrasound evidence. Am J Obstet Gynecol. 2003;188(1):100-107.
2. Wallach EE, Vlahos NF. Uterine myomas: an overview of development clinical features, and management. Obstet Gynecol. 2004;104(2):393-406.
3. Stringer NH, Walker JC, Meyer PM. Comparison of 49 laparoscopic myomec- tomies and 49 open myomectomies. J Am Assoc Gynecol Laparosc. 1997;4(4):457-464.
4. Bulletti C, Polli V, Negrini V, Giacomucci E, Flamigni C. Adhesion formation after laparoscopic myomectomy. J Am Assoc Gynecol Laparosc. 1996;3(4):533-536.
5. Ram E, Sherman Y, Weil R, Vishne T, Kravarusic D, Dreznik Z. Is mechanical bowel preparation mandatory for elective colon surgery? A prospective randomized study. Arch Surg. 2005;140(3):285-288.
6. Frederick J, Fletcher H, Simeon D, Mullings A, Hardie M. Intramyometrial vasopressin as a haemostatic agent during myomectomy. Br J Obstet Gynaecol. 1994;101(5):435-437.
7. Taylor A, Sharma M, Tsirkas P, Di Spiezio Sardo A, Setchell M, Magos A. Reducing blood loss at open myomectomy using triple tourniquets: a randomised controlled trial. BJOG. 2005;112(3):340-345.
8. Burbank F, Hutchins FL, Jr. Uterine artery occlusion by embolization or surgery for the treatment of fibroids: A unifying hypothesis-transient uterine ischemia. J Am Assoc Gynecol Laparosc. 2000;7(suppl 4):S1-S49.
DEXA screening—are we doing too much?
Reconsider the intervals at which you recommend rescreening for osteoporosis; for post-menopausal women with a baseline of normal bone mineral density (BMD) or mild osteopenia, a 15-year interval is probably sufficient.1
STRENGTH OF RECOMMENDATION
B: Based on a single cohort study
Gourlay ML, Fine JP, Preisser JS, et al. Bone density testing interval and transition to osteoporosis in older women. N Engl J Med. 2012;366: 225-233
ILLUSTRATIVE CASE
A 67-year-old woman whose recent dual-energy x-ray absorptiometry (DEXA) scan showed mild osteopenia asks when she should have her next bone scan. What should you tell her?
One in 5 people who sustain a hip fracture die within a year,2 and as many as 36% die prematurely.3 Osteoporosis is the primary predictor of fracture risk and, in older white women in particular, low bone mineral density (BMD) increases the likelihood of fracture by 70% to 80%.4
Optimal screening frequency not known
The US Preventive Services Task Force (USPSTF) guideline for osteoporosis screening concludes that there is a lack of evidence about optimal rescreening intervals and states that intervals >2 years may be necessary to better predict fracture risk.5 In addition, the USPSTF cites a prospective study showing that repeat measurement of BMD after 8 years added little predictive value compared with baseline DEXA scan results.6
The prospective cohort study detailed below was undertaken to help guide decisions about how frequently to screen
STUDY SUMMARY: Longer intervals are reasonable for those at low risk
Gourlay et al followed 4957 women age ≥67 years with normal BMD or osteopenia and no history of hip or clinical vertebral fracture or osteoporosis treatment. The primary outcome was the estimated time it would take for 10% of the women to develop osteoporosis. The time until 2% of the women developed such a fracture was the secondary outcome
Participants had baseline DEXA scans, which were repeated at years 2, 6, 8, 10, and 16. The researchers followed the women until they were diagnosed with osteoporosis, started on medication for osteoporosis, or developed a hip or clinical vertebral fracture
After adjusting for multiple covariates (age, body mass index, smoking status, use of glucocorticoids, fracture after age 50, estrogen use, and rheumatoid arthritis), the intervals between baseline testing and the development of osteoporosis were:
- 16.8 years (95% confidence interval [CI], 11.5-24.6) for women with normal BMD
- 17.3 years (95% CI, 13.9-21.5) for women with mild osteopenia
- 4.7 years (95% CI, 4.2-5.2) for women with moderate osteopenia
- 1.1 year (95% CI, 1.0-1.3) for women with advanced osteopenia
Intervals until 2% of the cohort developed fractures were similar
Overall, the authors used a sensible approach to estimate reasonable intervals between DEXA screenings (TABLE)
TABLE
Suggested rescreening intervals based on DEXA scan results1
| DEXA result (T-score) | Rescreening interval* |
|---|---|
| Normal/mild osteopenia (> -1.50) | 15 years |
| Moderate osteopenia (-1.50 to -1.99) | 5 years |
| Advanced osteopenia (-2.0 to -2.49) | 1 year |
| *Consider reducing these intervals by one-third for women older than 80 years. | |
WHAT’S NEW: Many DEXA scans can be eliminated
Rescreening all postmenopausal women every 2 years is unlikely to reduce osteoporotic fractures. This cohort study provides evidence that rescreening can often be delayed for many years, depending on the patient’s baseline risk. Changing practice based on these findings can reduce resource utilization without adversely affecting women’s health
CAVEATS: Questions about applicability may remain
This analysis was limited to women ≥67 years, so different results might be obtained from analyses that included younger postmenopausal women. In addition, 99% of the participants were white. Because the prevalence of osteoporosis of the hip among white women is equal to or slightly higher than it is among nonwhite women, it is likely that the suggested intervals are reasonable estimates for women of all races
In women >80 years, the interval between baseline testing and the development of osteoporosis was shorter than that of their younger counterparts. Thus, it might be reasonable to reduce rescreening intervals by a third for women in their 80s
CHALLENGES TO IMPLEMENTATION: Education needed for patients and docs
This study is the best so far to address the frequency of rescreening. In order to implement it, patients as well as clinicians will need to be educated. Effective long-term (>10 y) reminder systems would improve implementation
The recommendations of professional associations may also be a factor. The National Osteoporosis Foundation recommends assessing BMD every 2 years, but notes that more frequent testing may sometimes be warranted.7 The American College of Preventive Medicine recommends that screening for osteoporosis not occur more often than every 2 years.8
Acknowledgement
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of health.
1. Gourlay ML, Fine JP, Preisser JS, et al. Bone-density testing interval and transition to osteoporosis in older women. N Engl J Med. 2012;366:225-233.
2. Leibson CL, Tosteson AN, Gabriel SE, et al. Mortality, disability, and nursing home use for persons with and without hip fracture. J Am Geriatr Soc. 2002;50:1644-1650.
3. Abrahamsen B, van Staa T, Ariely R, et al. Excess mortality following hip fracture: a systematic epidemiological review. Osteoporosis Int. 2009;20:1633-1650.
4. Smith J, Shoukri K. Diagnosis of osteoporosis. Clin Cornerstone. 2000;2:22-33.
5. US Preventive Services Task Force. Screening for osteoporosis: U.S. Preventive Services Task Force recommendation statement. Available at: http://www.uspreventiveservicestaskforce.org/uspstf10/osteoporosis/osteors.htm. Accessed June 15, 2012.
6. Hillier TA, Stone KL, Bauer DC, et al. Evaluating the value of repeat bone mineral density measurement and prediction of fractures in older women. Arch Intern Med. 2007;167:155-160.
7. National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. 2010. Available at: http://www.nof.org/sites/default/files/pdfs/NOF_ClinicianGuide2009_v7.pdf. Accessed June 30, 2012.
8. Lim LS, Hoeksema LJ, Sherin K. ACPM Prevention Practice Committee. Screening for osteoporosis in the adult US population: ACPM position statement on preventive practice. Am J Prev Med. 2009;36:366-375.
Reconsider the intervals at which you recommend rescreening for osteoporosis; for post-menopausal women with a baseline of normal bone mineral density (BMD) or mild osteopenia, a 15-year interval is probably sufficient.1
STRENGTH OF RECOMMENDATION
B: Based on a single cohort study
Gourlay ML, Fine JP, Preisser JS, et al. Bone density testing interval and transition to osteoporosis in older women. N Engl J Med. 2012;366: 225-233
ILLUSTRATIVE CASE
A 67-year-old woman whose recent dual-energy x-ray absorptiometry (DEXA) scan showed mild osteopenia asks when she should have her next bone scan. What should you tell her?
One in 5 people who sustain a hip fracture die within a year,2 and as many as 36% die prematurely.3 Osteoporosis is the primary predictor of fracture risk and, in older white women in particular, low bone mineral density (BMD) increases the likelihood of fracture by 70% to 80%.4
Optimal screening frequency not known
The US Preventive Services Task Force (USPSTF) guideline for osteoporosis screening concludes that there is a lack of evidence about optimal rescreening intervals and states that intervals >2 years may be necessary to better predict fracture risk.5 In addition, the USPSTF cites a prospective study showing that repeat measurement of BMD after 8 years added little predictive value compared with baseline DEXA scan results.6
The prospective cohort study detailed below was undertaken to help guide decisions about how frequently to screen
STUDY SUMMARY: Longer intervals are reasonable for those at low risk
Gourlay et al followed 4957 women age ≥67 years with normal BMD or osteopenia and no history of hip or clinical vertebral fracture or osteoporosis treatment. The primary outcome was the estimated time it would take for 10% of the women to develop osteoporosis. The time until 2% of the women developed such a fracture was the secondary outcome
Participants had baseline DEXA scans, which were repeated at years 2, 6, 8, 10, and 16. The researchers followed the women until they were diagnosed with osteoporosis, started on medication for osteoporosis, or developed a hip or clinical vertebral fracture
After adjusting for multiple covariates (age, body mass index, smoking status, use of glucocorticoids, fracture after age 50, estrogen use, and rheumatoid arthritis), the intervals between baseline testing and the development of osteoporosis were:
- 16.8 years (95% confidence interval [CI], 11.5-24.6) for women with normal BMD
- 17.3 years (95% CI, 13.9-21.5) for women with mild osteopenia
- 4.7 years (95% CI, 4.2-5.2) for women with moderate osteopenia
- 1.1 year (95% CI, 1.0-1.3) for women with advanced osteopenia
Intervals until 2% of the cohort developed fractures were similar
Overall, the authors used a sensible approach to estimate reasonable intervals between DEXA screenings (TABLE)
TABLE
Suggested rescreening intervals based on DEXA scan results1
| DEXA result (T-score) | Rescreening interval* |
|---|---|
| Normal/mild osteopenia (> -1.50) | 15 years |
| Moderate osteopenia (-1.50 to -1.99) | 5 years |
| Advanced osteopenia (-2.0 to -2.49) | 1 year |
| *Consider reducing these intervals by one-third for women older than 80 years. | |
WHAT’S NEW: Many DEXA scans can be eliminated
Rescreening all postmenopausal women every 2 years is unlikely to reduce osteoporotic fractures. This cohort study provides evidence that rescreening can often be delayed for many years, depending on the patient’s baseline risk. Changing practice based on these findings can reduce resource utilization without adversely affecting women’s health
CAVEATS: Questions about applicability may remain
This analysis was limited to women ≥67 years, so different results might be obtained from analyses that included younger postmenopausal women. In addition, 99% of the participants were white. Because the prevalence of osteoporosis of the hip among white women is equal to or slightly higher than it is among nonwhite women, it is likely that the suggested intervals are reasonable estimates for women of all races
In women >80 years, the interval between baseline testing and the development of osteoporosis was shorter than that of their younger counterparts. Thus, it might be reasonable to reduce rescreening intervals by a third for women in their 80s
CHALLENGES TO IMPLEMENTATION: Education needed for patients and docs
This study is the best so far to address the frequency of rescreening. In order to implement it, patients as well as clinicians will need to be educated. Effective long-term (>10 y) reminder systems would improve implementation
The recommendations of professional associations may also be a factor. The National Osteoporosis Foundation recommends assessing BMD every 2 years, but notes that more frequent testing may sometimes be warranted.7 The American College of Preventive Medicine recommends that screening for osteoporosis not occur more often than every 2 years.8
Acknowledgement
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of health.
Reconsider the intervals at which you recommend rescreening for osteoporosis; for post-menopausal women with a baseline of normal bone mineral density (BMD) or mild osteopenia, a 15-year interval is probably sufficient.1
STRENGTH OF RECOMMENDATION
B: Based on a single cohort study
Gourlay ML, Fine JP, Preisser JS, et al. Bone density testing interval and transition to osteoporosis in older women. N Engl J Med. 2012;366: 225-233
ILLUSTRATIVE CASE
A 67-year-old woman whose recent dual-energy x-ray absorptiometry (DEXA) scan showed mild osteopenia asks when she should have her next bone scan. What should you tell her?
One in 5 people who sustain a hip fracture die within a year,2 and as many as 36% die prematurely.3 Osteoporosis is the primary predictor of fracture risk and, in older white women in particular, low bone mineral density (BMD) increases the likelihood of fracture by 70% to 80%.4
Optimal screening frequency not known
The US Preventive Services Task Force (USPSTF) guideline for osteoporosis screening concludes that there is a lack of evidence about optimal rescreening intervals and states that intervals >2 years may be necessary to better predict fracture risk.5 In addition, the USPSTF cites a prospective study showing that repeat measurement of BMD after 8 years added little predictive value compared with baseline DEXA scan results.6
The prospective cohort study detailed below was undertaken to help guide decisions about how frequently to screen
STUDY SUMMARY: Longer intervals are reasonable for those at low risk
Gourlay et al followed 4957 women age ≥67 years with normal BMD or osteopenia and no history of hip or clinical vertebral fracture or osteoporosis treatment. The primary outcome was the estimated time it would take for 10% of the women to develop osteoporosis. The time until 2% of the women developed such a fracture was the secondary outcome
Participants had baseline DEXA scans, which were repeated at years 2, 6, 8, 10, and 16. The researchers followed the women until they were diagnosed with osteoporosis, started on medication for osteoporosis, or developed a hip or clinical vertebral fracture
After adjusting for multiple covariates (age, body mass index, smoking status, use of glucocorticoids, fracture after age 50, estrogen use, and rheumatoid arthritis), the intervals between baseline testing and the development of osteoporosis were:
- 16.8 years (95% confidence interval [CI], 11.5-24.6) for women with normal BMD
- 17.3 years (95% CI, 13.9-21.5) for women with mild osteopenia
- 4.7 years (95% CI, 4.2-5.2) for women with moderate osteopenia
- 1.1 year (95% CI, 1.0-1.3) for women with advanced osteopenia
Intervals until 2% of the cohort developed fractures were similar
Overall, the authors used a sensible approach to estimate reasonable intervals between DEXA screenings (TABLE)
TABLE
Suggested rescreening intervals based on DEXA scan results1
| DEXA result (T-score) | Rescreening interval* |
|---|---|
| Normal/mild osteopenia (> -1.50) | 15 years |
| Moderate osteopenia (-1.50 to -1.99) | 5 years |
| Advanced osteopenia (-2.0 to -2.49) | 1 year |
| *Consider reducing these intervals by one-third for women older than 80 years. | |
WHAT’S NEW: Many DEXA scans can be eliminated
Rescreening all postmenopausal women every 2 years is unlikely to reduce osteoporotic fractures. This cohort study provides evidence that rescreening can often be delayed for many years, depending on the patient’s baseline risk. Changing practice based on these findings can reduce resource utilization without adversely affecting women’s health
CAVEATS: Questions about applicability may remain
This analysis was limited to women ≥67 years, so different results might be obtained from analyses that included younger postmenopausal women. In addition, 99% of the participants were white. Because the prevalence of osteoporosis of the hip among white women is equal to or slightly higher than it is among nonwhite women, it is likely that the suggested intervals are reasonable estimates for women of all races
In women >80 years, the interval between baseline testing and the development of osteoporosis was shorter than that of their younger counterparts. Thus, it might be reasonable to reduce rescreening intervals by a third for women in their 80s
CHALLENGES TO IMPLEMENTATION: Education needed for patients and docs
This study is the best so far to address the frequency of rescreening. In order to implement it, patients as well as clinicians will need to be educated. Effective long-term (>10 y) reminder systems would improve implementation
The recommendations of professional associations may also be a factor. The National Osteoporosis Foundation recommends assessing BMD every 2 years, but notes that more frequent testing may sometimes be warranted.7 The American College of Preventive Medicine recommends that screening for osteoporosis not occur more often than every 2 years.8
Acknowledgement
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of health.
1. Gourlay ML, Fine JP, Preisser JS, et al. Bone-density testing interval and transition to osteoporosis in older women. N Engl J Med. 2012;366:225-233.
2. Leibson CL, Tosteson AN, Gabriel SE, et al. Mortality, disability, and nursing home use for persons with and without hip fracture. J Am Geriatr Soc. 2002;50:1644-1650.
3. Abrahamsen B, van Staa T, Ariely R, et al. Excess mortality following hip fracture: a systematic epidemiological review. Osteoporosis Int. 2009;20:1633-1650.
4. Smith J, Shoukri K. Diagnosis of osteoporosis. Clin Cornerstone. 2000;2:22-33.
5. US Preventive Services Task Force. Screening for osteoporosis: U.S. Preventive Services Task Force recommendation statement. Available at: http://www.uspreventiveservicestaskforce.org/uspstf10/osteoporosis/osteors.htm. Accessed June 15, 2012.
6. Hillier TA, Stone KL, Bauer DC, et al. Evaluating the value of repeat bone mineral density measurement and prediction of fractures in older women. Arch Intern Med. 2007;167:155-160.
7. National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. 2010. Available at: http://www.nof.org/sites/default/files/pdfs/NOF_ClinicianGuide2009_v7.pdf. Accessed June 30, 2012.
8. Lim LS, Hoeksema LJ, Sherin K. ACPM Prevention Practice Committee. Screening for osteoporosis in the adult US population: ACPM position statement on preventive practice. Am J Prev Med. 2009;36:366-375.
1. Gourlay ML, Fine JP, Preisser JS, et al. Bone-density testing interval and transition to osteoporosis in older women. N Engl J Med. 2012;366:225-233.
2. Leibson CL, Tosteson AN, Gabriel SE, et al. Mortality, disability, and nursing home use for persons with and without hip fracture. J Am Geriatr Soc. 2002;50:1644-1650.
3. Abrahamsen B, van Staa T, Ariely R, et al. Excess mortality following hip fracture: a systematic epidemiological review. Osteoporosis Int. 2009;20:1633-1650.
4. Smith J, Shoukri K. Diagnosis of osteoporosis. Clin Cornerstone. 2000;2:22-33.
5. US Preventive Services Task Force. Screening for osteoporosis: U.S. Preventive Services Task Force recommendation statement. Available at: http://www.uspreventiveservicestaskforce.org/uspstf10/osteoporosis/osteors.htm. Accessed June 15, 2012.
6. Hillier TA, Stone KL, Bauer DC, et al. Evaluating the value of repeat bone mineral density measurement and prediction of fractures in older women. Arch Intern Med. 2007;167:155-160.
7. National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. 2010. Available at: http://www.nof.org/sites/default/files/pdfs/NOF_ClinicianGuide2009_v7.pdf. Accessed June 30, 2012.
8. Lim LS, Hoeksema LJ, Sherin K. ACPM Prevention Practice Committee. Screening for osteoporosis in the adult US population: ACPM position statement on preventive practice. Am J Prev Med. 2009;36:366-375.
Copyright © 2012 The Family Physicians Inquiries Network. All rights reserved.
An evidence-based approach to treating pediatric anxiety disorders
Anxiety disorders are remarkably common among pediatric patients1,2 and are associated with significant morbidity3 and increased risk of suicidality in adolescents.4,5 Effective diagnosis and treatment of pediatric anxiety disorders are critical for reducing psychosocial morbidity,3,6 suicidality, and the risk of secondary mood disorders.7
This article summarizes open-label studies and randomized controlled trials (RCTs) of selective serotonin reuptake inhibitors (SSRIs), selective serotonin-norepinephrine reuptake inhibitors, atypical anxiolytics, and benzodiazepines in children and adolescents with generalized anxiety disorder (GAD), social phobia, separation anxiety disorder, and panic disorder. Although we focus on psychopharmacologic treatments, the best outcomes generally are observed with multimodal treatments that combine psychotherapy and pharmacotherapy.
Generalized anxiety disorder
Researchers have evaluated SSRIs, benzodiazepines, and buspirone in pediatric patients with GAD. In a double-blind, placebo-controlled trial of 22 patients age 5 to 17, sertraline, 50 mg/d, was associated with improvement in Hamilton Anxiety Rating Scale (HAM-A), Clinical Global Impression-Severity (CGI-S), and Clinical Global Impression-Improvement (CGI-I) scores over 9 weeks.8 The Child-Adolescent Anxiety Multimodal Study compared cognitive-behavioral therapy (CBT) to sertraline or sertraline plus CBT in 488 patients age 7 to 17, 78% of whom had GAD.9 Sertraline monotherapy was superior to placebo and not statistically different from CBT, while combination treatment was superior to both monotherapy conditions in improving CGI score. In both trials, sertraline was well tolerated.
One study evaluated fluoxetine, 5 to 40 mg/d, or CBT in 14 youths with GAD; both treatments improved symptoms.10 In a study of 320 GAD patients age 6 to 17, venlafaxine extended-release (XR) initiated at 37.5 mg/d was associated with improved HAM-A scores.11 In general, venlafaxine was well tolerated; adverse effects included increased blood pressure, asthenia, pain, anorexia, somnolence, weight loss, and possibly treatment-emergent suicidal ideation.
Two RCTs of buspirone, 15 to 60 mg/d, that evaluated 559 children and adolescents age 6 to 17 with GAD did not observe significant differences between buspirone and placebo.12 By contrast, 2 open-label studies of youths with anxiety suggested improvement associated with buspirone.12 Treatment-emergent adverse events included nausea, stomachache, and headache.
Clinical trials of benzodiazepines in anxious children and adolescents have yielded mixed results. A 4-week, open-label trial of alprazolam, 0.5 mg to 1.5 mg/d, in 12 adolescents with overanxious disorder—the DSM-III forerunner of GAD—found improvements in anxiety, depression, psychomotor excitation, and hyperactivity, but patients experienced sedation, activation, headache, and nausea.13 However, a double-blind RCT in 30 youths age 8 to 16 found no statistically significant difference between alprazolam and placebo.14 Alprazolam generally was well tolerated; fatigue and dry mouth were reported, but no withdrawal symptoms. Additionally, benzodiazepine use may be associated with tolerance and—in young children—disinhibition.
Social phobia
Researchers have evaluated paroxetine, citalopram, fluoxetine, and venlafaxine for treating social phobia in pediatric patients. In an RCT, 78% of paroxetine-treated patients with social phobia responded compared with 38% for placebo over 16 weeks. Adverse events—including withdrawal symptoms—were twice as likely in patients who received paroxetine. Additionally, 4 paroxetine patients exhibited suicidal ideation vs 0 patients who received placebo.15
In an RCT of 293 children and adolescents age 8 to 17 with social phobia, venlafaxine XR was initiated at 37.5 mg/d and titrated to 112.5 mg/d, 150 mg/d, or 225 mg/d, depending on body weight.16 The venlafaxine group experienced significantly improved anxiety symptoms and the medication generally was well tolerated, although 3 venlafaxine-treated patients developed suicidal ideation compared with 0 in the placebo group.
An RCT compared Social Effectiveness Therapy for Children (SET-C) and fluoxetine, 10 to 40 mg/d, for 139 patients age 7 to 17 with social phobia.17 SET-C is a CBT for children and adolescents that focuses on increasing interpersonal skills and becoming more comfortable in social situations; it involves psychoeducation, social skills training, and exposure exercises. At endpoint, 53% of patients in the SET-C group no longer met diagnostic criteria for social phobia. Fluoxetine was well tolerated; no severe adverse events were reported.
In an open-label study of sertraline (mean dose = 123 mg/d) for 14 young persons with social phobia, 36% of patients responded and 29% partially responded at 8 weeks.18 Adverse events generally were mild and included nausea, diarrhea, and headache. In a 12-week study, 12 pediatric patients with social phobia received citalopram, 10 to 40 mg/d, and eight 15-minute counseling sessions. At endpoint, clinicians rated 83% of patients as much improved or very much improved. The medication generally was well tolerated.19
Separation anxiety disorder
In a 4-week, double-blind crossover pilot study, researchers randomly assigned 15 children age 7 to 13 with separation anxiety disorder to clonazepam, up to 2 mg/d, or placebo.20 There was no significant difference in CGI-I score between clonazepam and placebo. Side effects—including drowsiness, irritability and “oppositional behavior”—were more frequent in patients treated with clonazepam.
Panic disorder
Only 2 open-label studies of SSRIs have been conducted in pediatric patients with panic disorder. The first evaluated the effectiveness and tolerability of fluoxetine, sertraline, or paroxetine over 6 months in 12 patients; 67% no longer met criteria for panic disorder at endpoint.21 In this study, benzodiazepines—including clonazepam and lorazepam—were used in 67% of patients at the start of SSRI treatment. The authors suggested this strategy may be clinically useful for patients with panic disorder.
In the second study, Fairbanks et al22 examined the use of fluoxetine for 6 to 9 weeks in 16 outpatients with mixed anxiety disorders who did not respond to psychotherapy. Patients age ≤12 were given 5 to 40 mg/d and those age ≥13 received 5 to 80 mg/d. Fluoxetine was associated with clinically significant improvement in 3 of the 5 patients who had panic disorder. Although overall fluoxetine was well tolerated, drowsiness, dyssomnia, decreased appetite, nausea, and abdominal pain were the most common side effects. Fluoxetine was not associated with suicidal ideation.
Mixed anxiety disorders
Most trials of pediatric anxiety have evaluated patients with “mixed anxiety disorders” because GAD, social phobia, and separation anxiety disorder are highly comorbid and share diagnostic features (Figure 1).9 An RCT of fluvoxamine, up to 300 mg/d, in 128 pediatric patients with ≥1 anxiety disorders found significant differences in CGI-I and endpoint Pediatric Anxiety Rating Scale (PARS) scores.23 Fluvoxamine was well tolerated but associated with increased motor activity and abdominal discomfort compared with placebo.
Two open-label trials of pediatric patients with mixed anxiety disorders suggested fluoxetine may be beneficial. Fairbanks et al22 documented clinical improvement in 10 of 10 patients with separation anxiety disorder, 8 of 10 with social phobia, 4 of 6 with specific phobia, 3 of 5 with panic disorder, and 1 of 7 with GAD. Birmaher et al24 evaluated 21 pediatric patients with overanxious disorder, social phobia, or separation anxiety who had not responded to psychotherapy and were not depressed; all patients received flexibly-dosed fluoxetine for up to 10 months. Fluoxetine was well tolerated and 81% of patients improved.
Finally, in a 12-week RCT of 74 patients age 7 to 17 with GAD, separation anxiety disorder, and/or social phobia, fluoxetine, 10 to 20 mg/d, was associated with improved scores on the Screen for Anxiety Related Emotional Disorders, PARS, CGI-I, CGI-S, and Children’s Global Assessment Scale.25 A follow-up open-label trial suggested that maintenance treatment is associated with sustained improvement.26
Figure 1: The pediatric anxiety disorders triad: Comorbidity is common
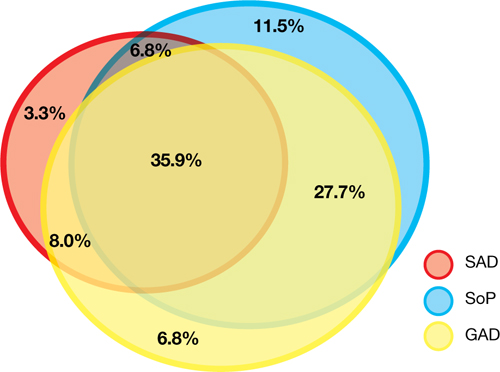
In the Child-Adolescent Multimodal Treatment Study, GAD was the most common disorder; however, GAD, SAD, and SoP were highly comorbid
GAD: generalized anxiety disorder; SAD: separation anxiety disorder; SoP: social phobia
Source: Reference 9
Anxiety disorders with ADHD
Anxiety disorders often are comorbid with attention-deficit/hyperactivity disorder (ADHD). An RCT of patients age 8 to 17 with ADHD and comorbid anxiety found that atomoxetine was associated with improved PARS scores and ADHD symptoms.27 The target dose was 1.2 mg/kg/d. Atomoxetine was well-tolerated; decreased appetite was the only significant adverse event in the treatment group vs placebo.
Multimodal treatment
Although this article reviews evidence for psychopharmacologic treatments, psychotherapeutic treatment of young patients with anxiety disorders has seen significant advances.28 Most psychotherapy studies have evaluated the efficacy of CBT,29-31 although there is evidence for psychodynamic therapy and interpersonal therapy.32 The American Academy of Child & Adolescent Psychiatry recommends a multimodal treatment approach because combination treatment appears to be more effective than monotherapy.8,28,33 Also, clinicians who treat pediatric patients who have an anxiety disorder should evaluate the family’s role on anxiety symptoms and may consider family therapy.
Treatment considerations
Evidence supports the efficacy of sertraline, citalopram, paroxetine, fluvoxamine, fluoxetine, and venlafaxine for treating children and adolescents with anxiety disorders (Figure 2).8,9,11,15,16,23,25 Some practitioners suggest using differing dosing strategies for pediatric anxiety disorders compared with those used to treat adults (Table).34 When considering SSRIs for children and adolescents, keep in mind the “black-box” warning regarding suicidality in these patients. Carefully monitor patients for treatment-emergent suicidality and routinely reassess for the presence and severity of suicidal ideation and suicide risk.
Figure 2: Number needed to treat for SSRIs and SNRIs in pediatric anxiety disorders

GAD: generalized anxiety disorder; RUPP: Research Unit on Pediatric Psychopharmacology; SAD: separation anxiety disorder; SNRI: serotonin-norepinephrine reuptake inhibitor; SoP: social phobia; SSRI: selective serotonin reuptake inhibitorTable
Practical dosing of SSRIs and SNRIs in pediatric patients with anxietya
| Medication | Initial child dose (age <12; mg/d) | Initial adolescent dose (age 12 to 17; mg/d) | Target dose (mg/d) |
|---|---|---|---|
| Citalopram | 5 to 10 | 10 | 20 to 40 |
| Escitalopram | 2.5 to 5 | 5 to 10 | 10 to 20 |
| Fluoxetineb | 10 | 20 | 20 to 40 (children), 40 to 60 (adolescents) |
| Paroxetineb | 5 to 10 | 10 | 20 |
| Sertralinec | 10 to 12.5 | 25 | 150 |
| Venlafaxine | 37.5 | 37.5 | 150 |
| aGeneralized anxiety disorder, social phobia, and separation anxiety disorder bMay consider cytochrome P450 genotyping for 2D6, which may suggest an alternate dosing strategy cSertraline is available in a liquid formulation (20 mg/mL) SNRI: serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor Source: Adapted from reference 34 | |||
Related Resources
- Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
- Anxiety and Depression Association of America. www.adaa.org.
- American Academy of Child & Adolescent Psychiatry. www.aacap.org.
Drug Brand Names
- Alprazolam • Xanax
- Atomoxetine • Strattera
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Fluoxetine • Prozac
- Fluvoxamine • Luvox, Luvox CR
- Lorazepam • Ativan
- Paroxetine • Paxil, Paxil CR
- Sertraline • Zoloft
- Venlafaxine • Effexor, Effexor XR
Disclosures
Dr. Strawn has received research support from the American Academy of Child & Adolescent Psychiatry, Eli Lilly and Company, and Shire, and is an employee of the University of Cincinnati, Cincinnati, OH.
Dr. McReynolds was employed by Eli Lilly and Company from 1997 to 2005.
1. Beesdo K, Knappe S, Pine DS. Anxiety and anxiety disorders in children and adolescents: developmental issues and implications for DSM-V. Psychiatr Clin North Am. 2009;32(3):483-524.
2. Beesdo K, Pine DS, Lieb R, et al. Incidence and risk patterns of anxiety and depressive disorders and categorization of generalized anxiety disorder. Arch Gen Psychiatry. 2010;67(1):47-57.
3. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first grade children: prediction to anxious symptoms and adaptive functioning in fifth grade. J Child Psychol Psychiatry. 1995;36(3):427-437.
4. Foley DL, Goldston DB, Costello EJ, et al. Proximal psychiatric risk factors for suicidality in youth: the Great Smoky Mountains Study. Arch Gen Psychiatry. 2006;63(9):1017-1024.
5. Jacobson CM, Muehlenkamp JJ, Miller AL, et al. Psychiatric impairment among adolescents engaging in different types of deliberate self-harm. J Clin Child Adolesc Psychol. 2008;37(2):363-375.
6. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first-grade children. J Abnorm Child Psychol. 1994;22(4):441-455.
7. Pine DS, Cohen P, Gurley D, et al. The risk for early-adulthood anxiety and depressive disorders in adolescents with anxiety and depressive disorders. Arch Gen Psychiatry. 1998;55(1):56-64.
8. Rynn MA, Siqueland L, Rickels K. Placebo-controlled trial of sertraline in the treatment of children with generalized anxiety disorders. Am J Psychiatry. 2001;158(12):2008-2014.
9. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
10. Maslowsky J, Mogg K, Bradley BP, et al. A preliminary investigation of neural correlates of treatment in adolescents with generalized anxiety disorder. J Child Adolesc Psychopharmacol. 2010;20(2):105-111.
11. Rynn MA, Riddle MA, Yeung PP, et al. Efficacy and safety of extended-release venlafaxine in the treatment of generalized anxiety disorder in children and adolescents: two placebo-controlled trials. Am J Psychiatry. 2007;164(2):290-300.
12. BuSpar [package insert] Princeton NJ: Bristol-Myers Squibb; 2010.
13. Simeon JG, Ferguson HB. Alprazolam effects in children with anxiety disorders. Can J Psychiatry. 1987;32(7):570-574.
14. Simeon JG, Ferguson HB, Knott V, et al. Clinical, cognitive, and neurophysiological effects of alprazolam in children and adolescents with overanxious and avoidant disorders. J Am Acad Child Adolesc Psychiatry. 1992;31(1):29-33.
15. Wagner KD, Berard R, Stein MB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of paroxetine in children and adolescents with social anxiety disorder. Arch Gen Psychiatry. 2004;61(11):1153-1162.
16. March JS, Entusah AR, Rynn M, et al. A randomized controlled trial of venlafaxine ER versus placebo in pediatric social anxiety disorder. Biol Psychiatry. 2007;62(10):1149-1154.
17. Beidel DC, Turner SM, Sallee FR, et al. SET-C versus fluoxetine in the treatment of childhood social phobia. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1622-1632.
18. Compton SN, Grant PJ, Chrisman AK, et al. Sertraline in children and adolescents with social anxiety disorder: an open trial. J Am Acad Child Adolesc Psychiatry. 2001;40(5):564-571.
19. Chavira DA, Stein MB. Combined psychoeducation and treatment with selective serotonin reuptake inhibitors for youth with generalized social anxiety disorder. J Child Adolesc Psychopharmacol. 2002;12(1):47-54.
20. Graae F, Milner J, Rizzotto L, et al. Clonazepam in childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(3):372-376.
21. Renaud J, Birmaher B, Wassick SC, et al. Use of selective serotonin reuptake inhibitors for the treatment of childhood panic disorder: a pilot study. J Child Adolesc Psychopharmacol. 1999;9(2):73-83.
22. Fairbanks JM, Pine DS, Tancer NK, et al. Open fluoxetine treatment of mixed anxiety disorders in children and adolescents. J Child Adolesc Psychopharmacol. 1997;7(1):17-29.
23. The Research Unit on Pediatric Psychopharmacology Anxiety Study Group. Fluvoxamine for the treatment of anxiety disorders in children and adolescents. N Engl J Med. 2001;344(17):1279-1285.
24. Birmaher B, Waterman GS, Ryan N, et al. Fluoxetine for childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(7):993-999.
25. Birmaher B, Axelson DA, Monk K, et al. Fluoxetine for the treatment of childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2003;42(4):415-423.
26. Clark DB, Birmaher B, Axelson D, et al. Fluoxetine for the treatment of childhood anxiety disorders: open-label, long-term extension to a controlled trial. J Am Acad Child Adolesc Psychiatry. 2005;44(12):1263-1270.
27. Geller D, Donnelly C, Lopez F, et al. Atomoxetine treatment for pediatric patients with attention-deficit/hyperactivity disorder with comorbid anxiety disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(9):1119-1127.
28. Connolly SD, Bernstein GA. Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
29. Kendall PC. Treating anxiety disorders in children: results of a randomized clinical trial. J Consult Clin Psychol. 1994;62(1):100-110.
30. Kendall PC, Flannery-Schroeder E, Panichelli-Mindel SM, et al. Therapy for youths with anxiety disorders: a second randomized clinical trial. J Consult Clin Psychol. 1997;65(3):366-380.
31. Reynolds S, Wilson C, Austin J, et al. Effects of psychotherapy for anxiety in children and adolescents: a meta-analytic review. Clin Psychol Rev. 2012;32(4):251-262.
32. Strawn JR, Wehry AM, DelBello MP, et al. Establishing the neurobiologic basis of treatment in children and adolescents with generalized anxiety disorder. Depress Anxiety. 2012;29(4):328-339.
33. Ginsburg GS, Kendall PC, Sakolsky D, et al. Remission after acute treatment in children and adolescents with anxiety disorders: findings from the CAMS. J Consult Clin Psychol. 2011;79(6):806-813.
34. Findling RL, Kowatch RA. How (not) to dose antidepressants and antipsychotics for children. Current Psychiatry. 2007;6(6):79-83.
Anxiety disorders are remarkably common among pediatric patients1,2 and are associated with significant morbidity3 and increased risk of suicidality in adolescents.4,5 Effective diagnosis and treatment of pediatric anxiety disorders are critical for reducing psychosocial morbidity,3,6 suicidality, and the risk of secondary mood disorders.7
This article summarizes open-label studies and randomized controlled trials (RCTs) of selective serotonin reuptake inhibitors (SSRIs), selective serotonin-norepinephrine reuptake inhibitors, atypical anxiolytics, and benzodiazepines in children and adolescents with generalized anxiety disorder (GAD), social phobia, separation anxiety disorder, and panic disorder. Although we focus on psychopharmacologic treatments, the best outcomes generally are observed with multimodal treatments that combine psychotherapy and pharmacotherapy.
Generalized anxiety disorder
Researchers have evaluated SSRIs, benzodiazepines, and buspirone in pediatric patients with GAD. In a double-blind, placebo-controlled trial of 22 patients age 5 to 17, sertraline, 50 mg/d, was associated with improvement in Hamilton Anxiety Rating Scale (HAM-A), Clinical Global Impression-Severity (CGI-S), and Clinical Global Impression-Improvement (CGI-I) scores over 9 weeks.8 The Child-Adolescent Anxiety Multimodal Study compared cognitive-behavioral therapy (CBT) to sertraline or sertraline plus CBT in 488 patients age 7 to 17, 78% of whom had GAD.9 Sertraline monotherapy was superior to placebo and not statistically different from CBT, while combination treatment was superior to both monotherapy conditions in improving CGI score. In both trials, sertraline was well tolerated.
One study evaluated fluoxetine, 5 to 40 mg/d, or CBT in 14 youths with GAD; both treatments improved symptoms.10 In a study of 320 GAD patients age 6 to 17, venlafaxine extended-release (XR) initiated at 37.5 mg/d was associated with improved HAM-A scores.11 In general, venlafaxine was well tolerated; adverse effects included increased blood pressure, asthenia, pain, anorexia, somnolence, weight loss, and possibly treatment-emergent suicidal ideation.
Two RCTs of buspirone, 15 to 60 mg/d, that evaluated 559 children and adolescents age 6 to 17 with GAD did not observe significant differences between buspirone and placebo.12 By contrast, 2 open-label studies of youths with anxiety suggested improvement associated with buspirone.12 Treatment-emergent adverse events included nausea, stomachache, and headache.
Clinical trials of benzodiazepines in anxious children and adolescents have yielded mixed results. A 4-week, open-label trial of alprazolam, 0.5 mg to 1.5 mg/d, in 12 adolescents with overanxious disorder—the DSM-III forerunner of GAD—found improvements in anxiety, depression, psychomotor excitation, and hyperactivity, but patients experienced sedation, activation, headache, and nausea.13 However, a double-blind RCT in 30 youths age 8 to 16 found no statistically significant difference between alprazolam and placebo.14 Alprazolam generally was well tolerated; fatigue and dry mouth were reported, but no withdrawal symptoms. Additionally, benzodiazepine use may be associated with tolerance and—in young children—disinhibition.
Social phobia
Researchers have evaluated paroxetine, citalopram, fluoxetine, and venlafaxine for treating social phobia in pediatric patients. In an RCT, 78% of paroxetine-treated patients with social phobia responded compared with 38% for placebo over 16 weeks. Adverse events—including withdrawal symptoms—were twice as likely in patients who received paroxetine. Additionally, 4 paroxetine patients exhibited suicidal ideation vs 0 patients who received placebo.15
In an RCT of 293 children and adolescents age 8 to 17 with social phobia, venlafaxine XR was initiated at 37.5 mg/d and titrated to 112.5 mg/d, 150 mg/d, or 225 mg/d, depending on body weight.16 The venlafaxine group experienced significantly improved anxiety symptoms and the medication generally was well tolerated, although 3 venlafaxine-treated patients developed suicidal ideation compared with 0 in the placebo group.
An RCT compared Social Effectiveness Therapy for Children (SET-C) and fluoxetine, 10 to 40 mg/d, for 139 patients age 7 to 17 with social phobia.17 SET-C is a CBT for children and adolescents that focuses on increasing interpersonal skills and becoming more comfortable in social situations; it involves psychoeducation, social skills training, and exposure exercises. At endpoint, 53% of patients in the SET-C group no longer met diagnostic criteria for social phobia. Fluoxetine was well tolerated; no severe adverse events were reported.
In an open-label study of sertraline (mean dose = 123 mg/d) for 14 young persons with social phobia, 36% of patients responded and 29% partially responded at 8 weeks.18 Adverse events generally were mild and included nausea, diarrhea, and headache. In a 12-week study, 12 pediatric patients with social phobia received citalopram, 10 to 40 mg/d, and eight 15-minute counseling sessions. At endpoint, clinicians rated 83% of patients as much improved or very much improved. The medication generally was well tolerated.19
Separation anxiety disorder
In a 4-week, double-blind crossover pilot study, researchers randomly assigned 15 children age 7 to 13 with separation anxiety disorder to clonazepam, up to 2 mg/d, or placebo.20 There was no significant difference in CGI-I score between clonazepam and placebo. Side effects—including drowsiness, irritability and “oppositional behavior”—were more frequent in patients treated with clonazepam.
Panic disorder
Only 2 open-label studies of SSRIs have been conducted in pediatric patients with panic disorder. The first evaluated the effectiveness and tolerability of fluoxetine, sertraline, or paroxetine over 6 months in 12 patients; 67% no longer met criteria for panic disorder at endpoint.21 In this study, benzodiazepines—including clonazepam and lorazepam—were used in 67% of patients at the start of SSRI treatment. The authors suggested this strategy may be clinically useful for patients with panic disorder.
In the second study, Fairbanks et al22 examined the use of fluoxetine for 6 to 9 weeks in 16 outpatients with mixed anxiety disorders who did not respond to psychotherapy. Patients age ≤12 were given 5 to 40 mg/d and those age ≥13 received 5 to 80 mg/d. Fluoxetine was associated with clinically significant improvement in 3 of the 5 patients who had panic disorder. Although overall fluoxetine was well tolerated, drowsiness, dyssomnia, decreased appetite, nausea, and abdominal pain were the most common side effects. Fluoxetine was not associated with suicidal ideation.
Mixed anxiety disorders
Most trials of pediatric anxiety have evaluated patients with “mixed anxiety disorders” because GAD, social phobia, and separation anxiety disorder are highly comorbid and share diagnostic features (Figure 1).9 An RCT of fluvoxamine, up to 300 mg/d, in 128 pediatric patients with ≥1 anxiety disorders found significant differences in CGI-I and endpoint Pediatric Anxiety Rating Scale (PARS) scores.23 Fluvoxamine was well tolerated but associated with increased motor activity and abdominal discomfort compared with placebo.
Two open-label trials of pediatric patients with mixed anxiety disorders suggested fluoxetine may be beneficial. Fairbanks et al22 documented clinical improvement in 10 of 10 patients with separation anxiety disorder, 8 of 10 with social phobia, 4 of 6 with specific phobia, 3 of 5 with panic disorder, and 1 of 7 with GAD. Birmaher et al24 evaluated 21 pediatric patients with overanxious disorder, social phobia, or separation anxiety who had not responded to psychotherapy and were not depressed; all patients received flexibly-dosed fluoxetine for up to 10 months. Fluoxetine was well tolerated and 81% of patients improved.
Finally, in a 12-week RCT of 74 patients age 7 to 17 with GAD, separation anxiety disorder, and/or social phobia, fluoxetine, 10 to 20 mg/d, was associated with improved scores on the Screen for Anxiety Related Emotional Disorders, PARS, CGI-I, CGI-S, and Children’s Global Assessment Scale.25 A follow-up open-label trial suggested that maintenance treatment is associated with sustained improvement.26
Figure 1: The pediatric anxiety disorders triad: Comorbidity is common
In the Child-Adolescent Multimodal Treatment Study, GAD was the most common disorder; however, GAD, SAD, and SoP were highly comorbid
GAD: generalized anxiety disorder; SAD: separation anxiety disorder; SoP: social phobia
Source: Reference 9
Anxiety disorders with ADHD
Anxiety disorders often are comorbid with attention-deficit/hyperactivity disorder (ADHD). An RCT of patients age 8 to 17 with ADHD and comorbid anxiety found that atomoxetine was associated with improved PARS scores and ADHD symptoms.27 The target dose was 1.2 mg/kg/d. Atomoxetine was well-tolerated; decreased appetite was the only significant adverse event in the treatment group vs placebo.
Multimodal treatment
Although this article reviews evidence for psychopharmacologic treatments, psychotherapeutic treatment of young patients with anxiety disorders has seen significant advances.28 Most psychotherapy studies have evaluated the efficacy of CBT,29-31 although there is evidence for psychodynamic therapy and interpersonal therapy.32 The American Academy of Child & Adolescent Psychiatry recommends a multimodal treatment approach because combination treatment appears to be more effective than monotherapy.8,28,33 Also, clinicians who treat pediatric patients who have an anxiety disorder should evaluate the family’s role on anxiety symptoms and may consider family therapy.
Treatment considerations
Evidence supports the efficacy of sertraline, citalopram, paroxetine, fluvoxamine, fluoxetine, and venlafaxine for treating children and adolescents with anxiety disorders (Figure 2).8,9,11,15,16,23,25 Some practitioners suggest using differing dosing strategies for pediatric anxiety disorders compared with those used to treat adults (Table).34 When considering SSRIs for children and adolescents, keep in mind the “black-box” warning regarding suicidality in these patients. Carefully monitor patients for treatment-emergent suicidality and routinely reassess for the presence and severity of suicidal ideation and suicide risk.
Figure 2: Number needed to treat for SSRIs and SNRIs in pediatric anxiety disorders
GAD: generalized anxiety disorder; RUPP: Research Unit on Pediatric Psychopharmacology; SAD: separation anxiety disorder; SNRI: serotonin-norepinephrine reuptake inhibitor; SoP: social phobia; SSRI: selective serotonin reuptake inhibitorTable
Practical dosing of SSRIs and SNRIs in pediatric patients with anxietya
| Medication | Initial child dose (age <12; mg/d) | Initial adolescent dose (age 12 to 17; mg/d) | Target dose (mg/d) |
|---|---|---|---|
| Citalopram | 5 to 10 | 10 | 20 to 40 |
| Escitalopram | 2.5 to 5 | 5 to 10 | 10 to 20 |
| Fluoxetineb | 10 | 20 | 20 to 40 (children), 40 to 60 (adolescents) |
| Paroxetineb | 5 to 10 | 10 | 20 |
| Sertralinec | 10 to 12.5 | 25 | 150 |
| Venlafaxine | 37.5 | 37.5 | 150 |
| aGeneralized anxiety disorder, social phobia, and separation anxiety disorder bMay consider cytochrome P450 genotyping for 2D6, which may suggest an alternate dosing strategy cSertraline is available in a liquid formulation (20 mg/mL) SNRI: serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor Source: Adapted from reference 34 | |||
Related Resources
- Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
- Anxiety and Depression Association of America. www.adaa.org.
- American Academy of Child & Adolescent Psychiatry. www.aacap.org.
Drug Brand Names
- Alprazolam • Xanax
- Atomoxetine • Strattera
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Fluoxetine • Prozac
- Fluvoxamine • Luvox, Luvox CR
- Lorazepam • Ativan
- Paroxetine • Paxil, Paxil CR
- Sertraline • Zoloft
- Venlafaxine • Effexor, Effexor XR
Disclosures
Dr. Strawn has received research support from the American Academy of Child & Adolescent Psychiatry, Eli Lilly and Company, and Shire, and is an employee of the University of Cincinnati, Cincinnati, OH.
Dr. McReynolds was employed by Eli Lilly and Company from 1997 to 2005.
Anxiety disorders are remarkably common among pediatric patients1,2 and are associated with significant morbidity3 and increased risk of suicidality in adolescents.4,5 Effective diagnosis and treatment of pediatric anxiety disorders are critical for reducing psychosocial morbidity,3,6 suicidality, and the risk of secondary mood disorders.7
This article summarizes open-label studies and randomized controlled trials (RCTs) of selective serotonin reuptake inhibitors (SSRIs), selective serotonin-norepinephrine reuptake inhibitors, atypical anxiolytics, and benzodiazepines in children and adolescents with generalized anxiety disorder (GAD), social phobia, separation anxiety disorder, and panic disorder. Although we focus on psychopharmacologic treatments, the best outcomes generally are observed with multimodal treatments that combine psychotherapy and pharmacotherapy.
Generalized anxiety disorder
Researchers have evaluated SSRIs, benzodiazepines, and buspirone in pediatric patients with GAD. In a double-blind, placebo-controlled trial of 22 patients age 5 to 17, sertraline, 50 mg/d, was associated with improvement in Hamilton Anxiety Rating Scale (HAM-A), Clinical Global Impression-Severity (CGI-S), and Clinical Global Impression-Improvement (CGI-I) scores over 9 weeks.8 The Child-Adolescent Anxiety Multimodal Study compared cognitive-behavioral therapy (CBT) to sertraline or sertraline plus CBT in 488 patients age 7 to 17, 78% of whom had GAD.9 Sertraline monotherapy was superior to placebo and not statistically different from CBT, while combination treatment was superior to both monotherapy conditions in improving CGI score. In both trials, sertraline was well tolerated.
One study evaluated fluoxetine, 5 to 40 mg/d, or CBT in 14 youths with GAD; both treatments improved symptoms.10 In a study of 320 GAD patients age 6 to 17, venlafaxine extended-release (XR) initiated at 37.5 mg/d was associated with improved HAM-A scores.11 In general, venlafaxine was well tolerated; adverse effects included increased blood pressure, asthenia, pain, anorexia, somnolence, weight loss, and possibly treatment-emergent suicidal ideation.
Two RCTs of buspirone, 15 to 60 mg/d, that evaluated 559 children and adolescents age 6 to 17 with GAD did not observe significant differences between buspirone and placebo.12 By contrast, 2 open-label studies of youths with anxiety suggested improvement associated with buspirone.12 Treatment-emergent adverse events included nausea, stomachache, and headache.
Clinical trials of benzodiazepines in anxious children and adolescents have yielded mixed results. A 4-week, open-label trial of alprazolam, 0.5 mg to 1.5 mg/d, in 12 adolescents with overanxious disorder—the DSM-III forerunner of GAD—found improvements in anxiety, depression, psychomotor excitation, and hyperactivity, but patients experienced sedation, activation, headache, and nausea.13 However, a double-blind RCT in 30 youths age 8 to 16 found no statistically significant difference between alprazolam and placebo.14 Alprazolam generally was well tolerated; fatigue and dry mouth were reported, but no withdrawal symptoms. Additionally, benzodiazepine use may be associated with tolerance and—in young children—disinhibition.
Social phobia
Researchers have evaluated paroxetine, citalopram, fluoxetine, and venlafaxine for treating social phobia in pediatric patients. In an RCT, 78% of paroxetine-treated patients with social phobia responded compared with 38% for placebo over 16 weeks. Adverse events—including withdrawal symptoms—were twice as likely in patients who received paroxetine. Additionally, 4 paroxetine patients exhibited suicidal ideation vs 0 patients who received placebo.15
In an RCT of 293 children and adolescents age 8 to 17 with social phobia, venlafaxine XR was initiated at 37.5 mg/d and titrated to 112.5 mg/d, 150 mg/d, or 225 mg/d, depending on body weight.16 The venlafaxine group experienced significantly improved anxiety symptoms and the medication generally was well tolerated, although 3 venlafaxine-treated patients developed suicidal ideation compared with 0 in the placebo group.
An RCT compared Social Effectiveness Therapy for Children (SET-C) and fluoxetine, 10 to 40 mg/d, for 139 patients age 7 to 17 with social phobia.17 SET-C is a CBT for children and adolescents that focuses on increasing interpersonal skills and becoming more comfortable in social situations; it involves psychoeducation, social skills training, and exposure exercises. At endpoint, 53% of patients in the SET-C group no longer met diagnostic criteria for social phobia. Fluoxetine was well tolerated; no severe adverse events were reported.
In an open-label study of sertraline (mean dose = 123 mg/d) for 14 young persons with social phobia, 36% of patients responded and 29% partially responded at 8 weeks.18 Adverse events generally were mild and included nausea, diarrhea, and headache. In a 12-week study, 12 pediatric patients with social phobia received citalopram, 10 to 40 mg/d, and eight 15-minute counseling sessions. At endpoint, clinicians rated 83% of patients as much improved or very much improved. The medication generally was well tolerated.19
Separation anxiety disorder
In a 4-week, double-blind crossover pilot study, researchers randomly assigned 15 children age 7 to 13 with separation anxiety disorder to clonazepam, up to 2 mg/d, or placebo.20 There was no significant difference in CGI-I score between clonazepam and placebo. Side effects—including drowsiness, irritability and “oppositional behavior”—were more frequent in patients treated with clonazepam.
Panic disorder
Only 2 open-label studies of SSRIs have been conducted in pediatric patients with panic disorder. The first evaluated the effectiveness and tolerability of fluoxetine, sertraline, or paroxetine over 6 months in 12 patients; 67% no longer met criteria for panic disorder at endpoint.21 In this study, benzodiazepines—including clonazepam and lorazepam—were used in 67% of patients at the start of SSRI treatment. The authors suggested this strategy may be clinically useful for patients with panic disorder.
In the second study, Fairbanks et al22 examined the use of fluoxetine for 6 to 9 weeks in 16 outpatients with mixed anxiety disorders who did not respond to psychotherapy. Patients age ≤12 were given 5 to 40 mg/d and those age ≥13 received 5 to 80 mg/d. Fluoxetine was associated with clinically significant improvement in 3 of the 5 patients who had panic disorder. Although overall fluoxetine was well tolerated, drowsiness, dyssomnia, decreased appetite, nausea, and abdominal pain were the most common side effects. Fluoxetine was not associated with suicidal ideation.
Mixed anxiety disorders
Most trials of pediatric anxiety have evaluated patients with “mixed anxiety disorders” because GAD, social phobia, and separation anxiety disorder are highly comorbid and share diagnostic features (Figure 1).9 An RCT of fluvoxamine, up to 300 mg/d, in 128 pediatric patients with ≥1 anxiety disorders found significant differences in CGI-I and endpoint Pediatric Anxiety Rating Scale (PARS) scores.23 Fluvoxamine was well tolerated but associated with increased motor activity and abdominal discomfort compared with placebo.
Two open-label trials of pediatric patients with mixed anxiety disorders suggested fluoxetine may be beneficial. Fairbanks et al22 documented clinical improvement in 10 of 10 patients with separation anxiety disorder, 8 of 10 with social phobia, 4 of 6 with specific phobia, 3 of 5 with panic disorder, and 1 of 7 with GAD. Birmaher et al24 evaluated 21 pediatric patients with overanxious disorder, social phobia, or separation anxiety who had not responded to psychotherapy and were not depressed; all patients received flexibly-dosed fluoxetine for up to 10 months. Fluoxetine was well tolerated and 81% of patients improved.
Finally, in a 12-week RCT of 74 patients age 7 to 17 with GAD, separation anxiety disorder, and/or social phobia, fluoxetine, 10 to 20 mg/d, was associated with improved scores on the Screen for Anxiety Related Emotional Disorders, PARS, CGI-I, CGI-S, and Children’s Global Assessment Scale.25 A follow-up open-label trial suggested that maintenance treatment is associated with sustained improvement.26
Figure 1: The pediatric anxiety disorders triad: Comorbidity is common
In the Child-Adolescent Multimodal Treatment Study, GAD was the most common disorder; however, GAD, SAD, and SoP were highly comorbid
GAD: generalized anxiety disorder; SAD: separation anxiety disorder; SoP: social phobia
Source: Reference 9
Anxiety disorders with ADHD
Anxiety disorders often are comorbid with attention-deficit/hyperactivity disorder (ADHD). An RCT of patients age 8 to 17 with ADHD and comorbid anxiety found that atomoxetine was associated with improved PARS scores and ADHD symptoms.27 The target dose was 1.2 mg/kg/d. Atomoxetine was well-tolerated; decreased appetite was the only significant adverse event in the treatment group vs placebo.
Multimodal treatment
Although this article reviews evidence for psychopharmacologic treatments, psychotherapeutic treatment of young patients with anxiety disorders has seen significant advances.28 Most psychotherapy studies have evaluated the efficacy of CBT,29-31 although there is evidence for psychodynamic therapy and interpersonal therapy.32 The American Academy of Child & Adolescent Psychiatry recommends a multimodal treatment approach because combination treatment appears to be more effective than monotherapy.8,28,33 Also, clinicians who treat pediatric patients who have an anxiety disorder should evaluate the family’s role on anxiety symptoms and may consider family therapy.
Treatment considerations
Evidence supports the efficacy of sertraline, citalopram, paroxetine, fluvoxamine, fluoxetine, and venlafaxine for treating children and adolescents with anxiety disorders (Figure 2).8,9,11,15,16,23,25 Some practitioners suggest using differing dosing strategies for pediatric anxiety disorders compared with those used to treat adults (Table).34 When considering SSRIs for children and adolescents, keep in mind the “black-box” warning regarding suicidality in these patients. Carefully monitor patients for treatment-emergent suicidality and routinely reassess for the presence and severity of suicidal ideation and suicide risk.
Figure 2: Number needed to treat for SSRIs and SNRIs in pediatric anxiety disorders
GAD: generalized anxiety disorder; RUPP: Research Unit on Pediatric Psychopharmacology; SAD: separation anxiety disorder; SNRI: serotonin-norepinephrine reuptake inhibitor; SoP: social phobia; SSRI: selective serotonin reuptake inhibitorTable
Practical dosing of SSRIs and SNRIs in pediatric patients with anxietya
| Medication | Initial child dose (age <12; mg/d) | Initial adolescent dose (age 12 to 17; mg/d) | Target dose (mg/d) |
|---|---|---|---|
| Citalopram | 5 to 10 | 10 | 20 to 40 |
| Escitalopram | 2.5 to 5 | 5 to 10 | 10 to 20 |
| Fluoxetineb | 10 | 20 | 20 to 40 (children), 40 to 60 (adolescents) |
| Paroxetineb | 5 to 10 | 10 | 20 |
| Sertralinec | 10 to 12.5 | 25 | 150 |
| Venlafaxine | 37.5 | 37.5 | 150 |
| aGeneralized anxiety disorder, social phobia, and separation anxiety disorder bMay consider cytochrome P450 genotyping for 2D6, which may suggest an alternate dosing strategy cSertraline is available in a liquid formulation (20 mg/mL) SNRI: serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor Source: Adapted from reference 34 | |||
Related Resources
- Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
- Anxiety and Depression Association of America. www.adaa.org.
- American Academy of Child & Adolescent Psychiatry. www.aacap.org.
Drug Brand Names
- Alprazolam • Xanax
- Atomoxetine • Strattera
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Fluoxetine • Prozac
- Fluvoxamine • Luvox, Luvox CR
- Lorazepam • Ativan
- Paroxetine • Paxil, Paxil CR
- Sertraline • Zoloft
- Venlafaxine • Effexor, Effexor XR
Disclosures
Dr. Strawn has received research support from the American Academy of Child & Adolescent Psychiatry, Eli Lilly and Company, and Shire, and is an employee of the University of Cincinnati, Cincinnati, OH.
Dr. McReynolds was employed by Eli Lilly and Company from 1997 to 2005.
1. Beesdo K, Knappe S, Pine DS. Anxiety and anxiety disorders in children and adolescents: developmental issues and implications for DSM-V. Psychiatr Clin North Am. 2009;32(3):483-524.
2. Beesdo K, Pine DS, Lieb R, et al. Incidence and risk patterns of anxiety and depressive disorders and categorization of generalized anxiety disorder. Arch Gen Psychiatry. 2010;67(1):47-57.
3. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first grade children: prediction to anxious symptoms and adaptive functioning in fifth grade. J Child Psychol Psychiatry. 1995;36(3):427-437.
4. Foley DL, Goldston DB, Costello EJ, et al. Proximal psychiatric risk factors for suicidality in youth: the Great Smoky Mountains Study. Arch Gen Psychiatry. 2006;63(9):1017-1024.
5. Jacobson CM, Muehlenkamp JJ, Miller AL, et al. Psychiatric impairment among adolescents engaging in different types of deliberate self-harm. J Clin Child Adolesc Psychol. 2008;37(2):363-375.
6. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first-grade children. J Abnorm Child Psychol. 1994;22(4):441-455.
7. Pine DS, Cohen P, Gurley D, et al. The risk for early-adulthood anxiety and depressive disorders in adolescents with anxiety and depressive disorders. Arch Gen Psychiatry. 1998;55(1):56-64.
8. Rynn MA, Siqueland L, Rickels K. Placebo-controlled trial of sertraline in the treatment of children with generalized anxiety disorders. Am J Psychiatry. 2001;158(12):2008-2014.
9. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
10. Maslowsky J, Mogg K, Bradley BP, et al. A preliminary investigation of neural correlates of treatment in adolescents with generalized anxiety disorder. J Child Adolesc Psychopharmacol. 2010;20(2):105-111.
11. Rynn MA, Riddle MA, Yeung PP, et al. Efficacy and safety of extended-release venlafaxine in the treatment of generalized anxiety disorder in children and adolescents: two placebo-controlled trials. Am J Psychiatry. 2007;164(2):290-300.
12. BuSpar [package insert] Princeton NJ: Bristol-Myers Squibb; 2010.
13. Simeon JG, Ferguson HB. Alprazolam effects in children with anxiety disorders. Can J Psychiatry. 1987;32(7):570-574.
14. Simeon JG, Ferguson HB, Knott V, et al. Clinical, cognitive, and neurophysiological effects of alprazolam in children and adolescents with overanxious and avoidant disorders. J Am Acad Child Adolesc Psychiatry. 1992;31(1):29-33.
15. Wagner KD, Berard R, Stein MB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of paroxetine in children and adolescents with social anxiety disorder. Arch Gen Psychiatry. 2004;61(11):1153-1162.
16. March JS, Entusah AR, Rynn M, et al. A randomized controlled trial of venlafaxine ER versus placebo in pediatric social anxiety disorder. Biol Psychiatry. 2007;62(10):1149-1154.
17. Beidel DC, Turner SM, Sallee FR, et al. SET-C versus fluoxetine in the treatment of childhood social phobia. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1622-1632.
18. Compton SN, Grant PJ, Chrisman AK, et al. Sertraline in children and adolescents with social anxiety disorder: an open trial. J Am Acad Child Adolesc Psychiatry. 2001;40(5):564-571.
19. Chavira DA, Stein MB. Combined psychoeducation and treatment with selective serotonin reuptake inhibitors for youth with generalized social anxiety disorder. J Child Adolesc Psychopharmacol. 2002;12(1):47-54.
20. Graae F, Milner J, Rizzotto L, et al. Clonazepam in childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(3):372-376.
21. Renaud J, Birmaher B, Wassick SC, et al. Use of selective serotonin reuptake inhibitors for the treatment of childhood panic disorder: a pilot study. J Child Adolesc Psychopharmacol. 1999;9(2):73-83.
22. Fairbanks JM, Pine DS, Tancer NK, et al. Open fluoxetine treatment of mixed anxiety disorders in children and adolescents. J Child Adolesc Psychopharmacol. 1997;7(1):17-29.
23. The Research Unit on Pediatric Psychopharmacology Anxiety Study Group. Fluvoxamine for the treatment of anxiety disorders in children and adolescents. N Engl J Med. 2001;344(17):1279-1285.
24. Birmaher B, Waterman GS, Ryan N, et al. Fluoxetine for childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(7):993-999.
25. Birmaher B, Axelson DA, Monk K, et al. Fluoxetine for the treatment of childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2003;42(4):415-423.
26. Clark DB, Birmaher B, Axelson D, et al. Fluoxetine for the treatment of childhood anxiety disorders: open-label, long-term extension to a controlled trial. J Am Acad Child Adolesc Psychiatry. 2005;44(12):1263-1270.
27. Geller D, Donnelly C, Lopez F, et al. Atomoxetine treatment for pediatric patients with attention-deficit/hyperactivity disorder with comorbid anxiety disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(9):1119-1127.
28. Connolly SD, Bernstein GA. Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
29. Kendall PC. Treating anxiety disorders in children: results of a randomized clinical trial. J Consult Clin Psychol. 1994;62(1):100-110.
30. Kendall PC, Flannery-Schroeder E, Panichelli-Mindel SM, et al. Therapy for youths with anxiety disorders: a second randomized clinical trial. J Consult Clin Psychol. 1997;65(3):366-380.
31. Reynolds S, Wilson C, Austin J, et al. Effects of psychotherapy for anxiety in children and adolescents: a meta-analytic review. Clin Psychol Rev. 2012;32(4):251-262.
32. Strawn JR, Wehry AM, DelBello MP, et al. Establishing the neurobiologic basis of treatment in children and adolescents with generalized anxiety disorder. Depress Anxiety. 2012;29(4):328-339.
33. Ginsburg GS, Kendall PC, Sakolsky D, et al. Remission after acute treatment in children and adolescents with anxiety disorders: findings from the CAMS. J Consult Clin Psychol. 2011;79(6):806-813.
34. Findling RL, Kowatch RA. How (not) to dose antidepressants and antipsychotics for children. Current Psychiatry. 2007;6(6):79-83.
1. Beesdo K, Knappe S, Pine DS. Anxiety and anxiety disorders in children and adolescents: developmental issues and implications for DSM-V. Psychiatr Clin North Am. 2009;32(3):483-524.
2. Beesdo K, Pine DS, Lieb R, et al. Incidence and risk patterns of anxiety and depressive disorders and categorization of generalized anxiety disorder. Arch Gen Psychiatry. 2010;67(1):47-57.
3. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first grade children: prediction to anxious symptoms and adaptive functioning in fifth grade. J Child Psychol Psychiatry. 1995;36(3):427-437.
4. Foley DL, Goldston DB, Costello EJ, et al. Proximal psychiatric risk factors for suicidality in youth: the Great Smoky Mountains Study. Arch Gen Psychiatry. 2006;63(9):1017-1024.
5. Jacobson CM, Muehlenkamp JJ, Miller AL, et al. Psychiatric impairment among adolescents engaging in different types of deliberate self-harm. J Clin Child Adolesc Psychol. 2008;37(2):363-375.
6. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first-grade children. J Abnorm Child Psychol. 1994;22(4):441-455.
7. Pine DS, Cohen P, Gurley D, et al. The risk for early-adulthood anxiety and depressive disorders in adolescents with anxiety and depressive disorders. Arch Gen Psychiatry. 1998;55(1):56-64.
8. Rynn MA, Siqueland L, Rickels K. Placebo-controlled trial of sertraline in the treatment of children with generalized anxiety disorders. Am J Psychiatry. 2001;158(12):2008-2014.
9. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
10. Maslowsky J, Mogg K, Bradley BP, et al. A preliminary investigation of neural correlates of treatment in adolescents with generalized anxiety disorder. J Child Adolesc Psychopharmacol. 2010;20(2):105-111.
11. Rynn MA, Riddle MA, Yeung PP, et al. Efficacy and safety of extended-release venlafaxine in the treatment of generalized anxiety disorder in children and adolescents: two placebo-controlled trials. Am J Psychiatry. 2007;164(2):290-300.
12. BuSpar [package insert] Princeton NJ: Bristol-Myers Squibb; 2010.
13. Simeon JG, Ferguson HB. Alprazolam effects in children with anxiety disorders. Can J Psychiatry. 1987;32(7):570-574.
14. Simeon JG, Ferguson HB, Knott V, et al. Clinical, cognitive, and neurophysiological effects of alprazolam in children and adolescents with overanxious and avoidant disorders. J Am Acad Child Adolesc Psychiatry. 1992;31(1):29-33.
15. Wagner KD, Berard R, Stein MB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of paroxetine in children and adolescents with social anxiety disorder. Arch Gen Psychiatry. 2004;61(11):1153-1162.
16. March JS, Entusah AR, Rynn M, et al. A randomized controlled trial of venlafaxine ER versus placebo in pediatric social anxiety disorder. Biol Psychiatry. 2007;62(10):1149-1154.
17. Beidel DC, Turner SM, Sallee FR, et al. SET-C versus fluoxetine in the treatment of childhood social phobia. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1622-1632.
18. Compton SN, Grant PJ, Chrisman AK, et al. Sertraline in children and adolescents with social anxiety disorder: an open trial. J Am Acad Child Adolesc Psychiatry. 2001;40(5):564-571.
19. Chavira DA, Stein MB. Combined psychoeducation and treatment with selective serotonin reuptake inhibitors for youth with generalized social anxiety disorder. J Child Adolesc Psychopharmacol. 2002;12(1):47-54.
20. Graae F, Milner J, Rizzotto L, et al. Clonazepam in childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(3):372-376.
21. Renaud J, Birmaher B, Wassick SC, et al. Use of selective serotonin reuptake inhibitors for the treatment of childhood panic disorder: a pilot study. J Child Adolesc Psychopharmacol. 1999;9(2):73-83.
22. Fairbanks JM, Pine DS, Tancer NK, et al. Open fluoxetine treatment of mixed anxiety disorders in children and adolescents. J Child Adolesc Psychopharmacol. 1997;7(1):17-29.
23. The Research Unit on Pediatric Psychopharmacology Anxiety Study Group. Fluvoxamine for the treatment of anxiety disorders in children and adolescents. N Engl J Med. 2001;344(17):1279-1285.
24. Birmaher B, Waterman GS, Ryan N, et al. Fluoxetine for childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(7):993-999.
25. Birmaher B, Axelson DA, Monk K, et al. Fluoxetine for the treatment of childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2003;42(4):415-423.
26. Clark DB, Birmaher B, Axelson D, et al. Fluoxetine for the treatment of childhood anxiety disorders: open-label, long-term extension to a controlled trial. J Am Acad Child Adolesc Psychiatry. 2005;44(12):1263-1270.
27. Geller D, Donnelly C, Lopez F, et al. Atomoxetine treatment for pediatric patients with attention-deficit/hyperactivity disorder with comorbid anxiety disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(9):1119-1127.
28. Connolly SD, Bernstein GA. Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
29. Kendall PC. Treating anxiety disorders in children: results of a randomized clinical trial. J Consult Clin Psychol. 1994;62(1):100-110.
30. Kendall PC, Flannery-Schroeder E, Panichelli-Mindel SM, et al. Therapy for youths with anxiety disorders: a second randomized clinical trial. J Consult Clin Psychol. 1997;65(3):366-380.
31. Reynolds S, Wilson C, Austin J, et al. Effects of psychotherapy for anxiety in children and adolescents: a meta-analytic review. Clin Psychol Rev. 2012;32(4):251-262.
32. Strawn JR, Wehry AM, DelBello MP, et al. Establishing the neurobiologic basis of treatment in children and adolescents with generalized anxiety disorder. Depress Anxiety. 2012;29(4):328-339.
33. Ginsburg GS, Kendall PC, Sakolsky D, et al. Remission after acute treatment in children and adolescents with anxiety disorders: findings from the CAMS. J Consult Clin Psychol. 2011;79(6):806-813.
34. Findling RL, Kowatch RA. How (not) to dose antidepressants and antipsychotics for children. Current Psychiatry. 2007;6(6):79-83.
Sleep terrors in adults: How to help control this potentially dangerous condition
Sleep terrors (STs)—also known as night terrors—are characterized by sudden arousal accompanied by a piercing scream or cry in the first few hours after falling asleep. These parasomnias arise out of slow-wave sleep (stages 3 and 4 of nonrapid eye movement [non-REM] sleep) and affect approximately 5% of adults.1 The condition is twice as common in men than women, and usually affects children but may not develop until adulthood.1
During STs, a patient may act scared, afraid, agitated, anxious, or panicky without being fully aware of his or her surroundings. The episode may last 30 seconds to 5 minutes; most patients don’t remember the event the next morning. STs may leave individuals feeling exhausted and perplexed the next day. Verbalization during the episode is incoherent and a patient’s perception of the environment seems altered. Tachycardia, tachypnea, sweating, flushed skin, or mydriasis are prominent. When ST patients walk, they may do so violently and can cause harm to themselves or others.
The differential diagnosis of STs includes posttraumatic stress disorder; nocturnal seizures characterized by excessive motor activity and organic CNS lesions; REM sleep behavior disorder; sleep choking syndrome; and nocturnal panic attacks. Patients with STs report high rates of stressful events—eg, divorce or bereavement—in the previous year. They are more likely to have a history of mood and anxiety disorders and high levels of depression, anxiety, and obsessive-compulsive and phobic traits. One study found patients with STs were 4.3 times more likely to have had a car accident in the past year.2
Evaluating and treating STs
Rule out comorbid conditions such as obstructive sleep apnea and periodic limb movement disorder. Encourage your patient to improve his or her sleep hygiene by maintaining a regular sleep/wake cycle, exercising, and limiting caffeine and alcohol and exposure to bright light before bedtime.
Self-help techniques. To avoid injury, encourage your patient to remove dangerous objects from their sleeping area. Suggest locking the doors to the room or home, and putting medications in a secure place. Patients also may consider keeping their mattress close to the floor to limit the risk of injury.
Pharmacotherapy and psychotherapy. Along with counseling and support, your patient may benefit from cognitive-behavioral therapy, relaxation therapy, or hypnosis.3 Anticipatory arousal therapy may help by interrupting the altered underlying electrophysiology of partial arousal.
If your patient is concerned about physical injury during STs, consider prescribing clonazepam, temazepam, or diazepam.4 Trazodone and selective serotonin reuptake inhibitors such as paroxetine5 also have been used to treat STs.
Disclosure
Dr. Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Crisp AH. The sleepwalking/night terrors syndrome in adults. Postgrad Med J. 1996;72(852):599-604.
2. Oudiette D, Leu S, Pottier M, et al. Dreamlike mentations during sleepwalking and sleep terrors in adults. Sleep. 2009;32(12):1621-1627.
3. Lowe P, Humphreys C, Williams SJ. Night terrors: women’s experiences of (not) sleeping where there is domestic violence. Violence Against Women. 2007;13(6):549-561.
4. Schenck CH, Mahowald MW. Long-term nightly benzodiazepine treatment of injurious parasomnias and other disorders of disrupted nocturnal sleep in 170 adults. Am J Med. 1996;100(3):333-337.
5. Lillywhite AR, Wilson SJ, Nutt DJ. Successful treatment of night terrors and somnambulism with paroxetine. Br J Psychiatry. 1994;164(4):551-554.
Sleep terrors (STs)—also known as night terrors—are characterized by sudden arousal accompanied by a piercing scream or cry in the first few hours after falling asleep. These parasomnias arise out of slow-wave sleep (stages 3 and 4 of nonrapid eye movement [non-REM] sleep) and affect approximately 5% of adults.1 The condition is twice as common in men than women, and usually affects children but may not develop until adulthood.1
During STs, a patient may act scared, afraid, agitated, anxious, or panicky without being fully aware of his or her surroundings. The episode may last 30 seconds to 5 minutes; most patients don’t remember the event the next morning. STs may leave individuals feeling exhausted and perplexed the next day. Verbalization during the episode is incoherent and a patient’s perception of the environment seems altered. Tachycardia, tachypnea, sweating, flushed skin, or mydriasis are prominent. When ST patients walk, they may do so violently and can cause harm to themselves or others.
The differential diagnosis of STs includes posttraumatic stress disorder; nocturnal seizures characterized by excessive motor activity and organic CNS lesions; REM sleep behavior disorder; sleep choking syndrome; and nocturnal panic attacks. Patients with STs report high rates of stressful events—eg, divorce or bereavement—in the previous year. They are more likely to have a history of mood and anxiety disorders and high levels of depression, anxiety, and obsessive-compulsive and phobic traits. One study found patients with STs were 4.3 times more likely to have had a car accident in the past year.2
Evaluating and treating STs
Rule out comorbid conditions such as obstructive sleep apnea and periodic limb movement disorder. Encourage your patient to improve his or her sleep hygiene by maintaining a regular sleep/wake cycle, exercising, and limiting caffeine and alcohol and exposure to bright light before bedtime.
Self-help techniques. To avoid injury, encourage your patient to remove dangerous objects from their sleeping area. Suggest locking the doors to the room or home, and putting medications in a secure place. Patients also may consider keeping their mattress close to the floor to limit the risk of injury.
Pharmacotherapy and psychotherapy. Along with counseling and support, your patient may benefit from cognitive-behavioral therapy, relaxation therapy, or hypnosis.3 Anticipatory arousal therapy may help by interrupting the altered underlying electrophysiology of partial arousal.
If your patient is concerned about physical injury during STs, consider prescribing clonazepam, temazepam, or diazepam.4 Trazodone and selective serotonin reuptake inhibitors such as paroxetine5 also have been used to treat STs.
Disclosure
Dr. Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Sleep terrors (STs)—also known as night terrors—are characterized by sudden arousal accompanied by a piercing scream or cry in the first few hours after falling asleep. These parasomnias arise out of slow-wave sleep (stages 3 and 4 of nonrapid eye movement [non-REM] sleep) and affect approximately 5% of adults.1 The condition is twice as common in men than women, and usually affects children but may not develop until adulthood.1
During STs, a patient may act scared, afraid, agitated, anxious, or panicky without being fully aware of his or her surroundings. The episode may last 30 seconds to 5 minutes; most patients don’t remember the event the next morning. STs may leave individuals feeling exhausted and perplexed the next day. Verbalization during the episode is incoherent and a patient’s perception of the environment seems altered. Tachycardia, tachypnea, sweating, flushed skin, or mydriasis are prominent. When ST patients walk, they may do so violently and can cause harm to themselves or others.
The differential diagnosis of STs includes posttraumatic stress disorder; nocturnal seizures characterized by excessive motor activity and organic CNS lesions; REM sleep behavior disorder; sleep choking syndrome; and nocturnal panic attacks. Patients with STs report high rates of stressful events—eg, divorce or bereavement—in the previous year. They are more likely to have a history of mood and anxiety disorders and high levels of depression, anxiety, and obsessive-compulsive and phobic traits. One study found patients with STs were 4.3 times more likely to have had a car accident in the past year.2
Evaluating and treating STs
Rule out comorbid conditions such as obstructive sleep apnea and periodic limb movement disorder. Encourage your patient to improve his or her sleep hygiene by maintaining a regular sleep/wake cycle, exercising, and limiting caffeine and alcohol and exposure to bright light before bedtime.
Self-help techniques. To avoid injury, encourage your patient to remove dangerous objects from their sleeping area. Suggest locking the doors to the room or home, and putting medications in a secure place. Patients also may consider keeping their mattress close to the floor to limit the risk of injury.
Pharmacotherapy and psychotherapy. Along with counseling and support, your patient may benefit from cognitive-behavioral therapy, relaxation therapy, or hypnosis.3 Anticipatory arousal therapy may help by interrupting the altered underlying electrophysiology of partial arousal.
If your patient is concerned about physical injury during STs, consider prescribing clonazepam, temazepam, or diazepam.4 Trazodone and selective serotonin reuptake inhibitors such as paroxetine5 also have been used to treat STs.
Disclosure
Dr. Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Crisp AH. The sleepwalking/night terrors syndrome in adults. Postgrad Med J. 1996;72(852):599-604.
2. Oudiette D, Leu S, Pottier M, et al. Dreamlike mentations during sleepwalking and sleep terrors in adults. Sleep. 2009;32(12):1621-1627.
3. Lowe P, Humphreys C, Williams SJ. Night terrors: women’s experiences of (not) sleeping where there is domestic violence. Violence Against Women. 2007;13(6):549-561.
4. Schenck CH, Mahowald MW. Long-term nightly benzodiazepine treatment of injurious parasomnias and other disorders of disrupted nocturnal sleep in 170 adults. Am J Med. 1996;100(3):333-337.
5. Lillywhite AR, Wilson SJ, Nutt DJ. Successful treatment of night terrors and somnambulism with paroxetine. Br J Psychiatry. 1994;164(4):551-554.
1. Crisp AH. The sleepwalking/night terrors syndrome in adults. Postgrad Med J. 1996;72(852):599-604.
2. Oudiette D, Leu S, Pottier M, et al. Dreamlike mentations during sleepwalking and sleep terrors in adults. Sleep. 2009;32(12):1621-1627.
3. Lowe P, Humphreys C, Williams SJ. Night terrors: women’s experiences of (not) sleeping where there is domestic violence. Violence Against Women. 2007;13(6):549-561.
4. Schenck CH, Mahowald MW. Long-term nightly benzodiazepine treatment of injurious parasomnias and other disorders of disrupted nocturnal sleep in 170 adults. Am J Med. 1996;100(3):333-337.
5. Lillywhite AR, Wilson SJ, Nutt DJ. Successful treatment of night terrors and somnambulism with paroxetine. Br J Psychiatry. 1994;164(4):551-554.
Omega-3 fatty acids for psychiatric illness
Discuss this article at www.facebook.com/CurrentPsychiatry
Epidemiologic data suggest that people who consume diets rich in omega-3 fatty acids (FAs)—long-chain polyunsaturated FAs such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)—have a decreased risk of major depressive disorder (MDD), postpartum depression, and bipolar disorder (BD).1-5 Omega-3 FA concentration may impact serotonin and dopamine transmission via effects on cell membrane fluidity.6 Therefore, decreased intake may increase the risk of several psychiatric disorders. As the average Western diet has changed over the last 2 centuries, omega-3 FA consumption has decreased.7 Omega-3 FAs cannot be synthesized by the body and must come from exogenous sources, such as fish and nuts. For a discussion of different types of dietary fats, see Box 1.8
Should we advise our patients to increase their omega-3 FA consumption? The American Psychiatric Association (APA) and the American Heart Association (AHA) recommend omega-3 FA consumption for the general population and in some cases, supplementation for specific disorders (Box 2).9-12 New data has been published since Current Psychiatry last reviewed the evidence for using omega-3 FAs for psychiatric conditions in 2004.8 This article looks at the latest evidence on the use of omega-3 FAs to treat mood disorders, schizophrenia, dementia, and other psychiatric conditions.
Dietary fat is saturated or unsaturated. Unsaturated fats are further categorized as monounsaturated or polyunsaturated (PUFA). PUFAs contain a hydrocarbon chain with ≥2 double bonds.8 The position of this double bond relative to the methyl end carbon—or “omega” carbon—groups the PUFAs into 2 categories:8
- omega-6 fatty acids, including arachidonic acid (AA) and linoleic acid (LA)
- omega-3 fatty acids, including eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and alpha-linolenic acid (ALA). ALA is a metabolic precursor to EPA and DHA.
PUFAs—in particular AA and DHA—are thought to contribute to cell membrane fluidity, modulation of neurotransmitters, and signal transduction pathways. As precursors to eicosanoids and cytokines, PUFAs may affect anti-inflammatory response systems.
Consumption of omega-3 fatty acids (FAs) reduces risk for arrhythmia, thrombosis, and atherosclerotic plaque, according to American Heart Association (AHA) guidelines. Omega-3 FA intake also may improve endothelial function, slightly lower blood pressure, and reduce inflammatory response. Replacing dietary saturated fat with polyunsaturated fat reduces coronary heart disease risk by 19%.9 The AHA recommends that all adults eat fish, particularly oily fish such as salmon or tuna, ≥2 times per week. Patients with documented coronary heart disease should consume 1 g/d eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) combined10 either via oily fish or omega-3 FA capsules. Side effects of omega-3 FA supplements are minor and include mild gastrointestinal discomfort, mostly burping or an unpleasant aftertaste; no cases of bleeding have been reported.11
For patients with hypertriglyceridemia, 2 to 4 g/d may be useful. Because of a theoretical risk of bleeding, doses >3 g/d should be supervised by a physician.
Because psychiatric illnesses and cardiovascular disease may be comorbid, the Omega-3 FA Subcommittee of the American Psychiatric Association supports the AHA’s guidelines regarding fish consumption, and further recommends that patients with mood, impulse control, or psychotic disorders consume ≥1 g/d of combined EPA and DHA.12
Limitations of the data
Reviewing the literature on omega-3 FAs to treat psychiatric disorders is hampered by several difficulties:13
- studies may evaluate the use of EPA alone, EPA combined with DHA, or DHA alone
- the doses of EPA and DHA and ratio of EPA to DHA of the supplements used in clinical trials varies greatly
- patients’ dietary consumption of omega-3 FAs is difficult to control
- DSM diagnostic criteria, as well as severity of illness, differ within studies.
In addition, studies may use omega-3 FAs as monotherapy or as adjuncts. All of these factors lead to difficulty interpreting the literature, as well as trouble in extracting data for meta-analysis.
Omega-3 FAs for mood disorders
MDD and other depressive diagnoses. Several meta-analyses examining the use of omega-3 FAs for treating depressive disorders have had equivocal findings. Variability in results might be partially explained by differences in the severity of baseline depression among diverse study populations, diagnostic variation, differing omega-3 supplementation protocols, or other issues.13 In addition, publication bias also may affect results.
In a 2011 literature review and meta-analysis of omega-3 FAs as monotherapy or an adjunct to antidepressants to treat MDD, Bloch and Hannestad6 concluded that omega-3 FAs offer a small but nonsignificant benefit in treating MDD. This review suggested that omega-3 FAs may be more effective in patients with more severe depression. The effects of varying levels of EPA vs DHA were not examined.
In a systematic review and meta-analysis, Appleton et al14 concluded that omega-3 FA supplements have little beneficial effect on depressed mood in individuals who do not have a depressive illness diagnosis (eg, MDD). However, this study did not consider the differential effects of EPA vs DHA on treatment response. Patients diagnosed with a depressive illness received greater benefits from omega-3 FA supplementation, although the patients in this study were heterogeneous. Similar to Bloch and Hannestad, Appleton et al14 found that omega-3 FA supplementation may be most beneficial for depressed patients with more severe symptoms, but is unlikely to help those with mild-to-moderate symptoms or individuals without symptoms who aim to prevent depression.
A meta-analysis by Martins15 looked at EPA vs DHA to treat depressive illness and found that only supplements that were mostly or completely EPA effectively treated depressive symptoms. Martins also found that severity of illness is key for positive treatment outcomes; there was a significant relationship between higher baseline depression levels and efficacy.15 Martins noted that omega-3 FA therapy was more effective as a treatment than a preventive strategy, and that adding omega-3 FAs to antidepressants was more efficacious than omega-3 FAs alone.15
A meta-analysis of clinical trials of omega-3 FAs for depressive illness suggested EPA should be ≥60% of total EPA + DHA.16
BD. A recent meta-analysis of 6 randomized controlled trials (RCTs) found that adding omega-3 supplements to mood stabilizers in patients with BD was associated with a statistically significant reduction of depressive symptoms, but was not effective for treating mania.17 The authors suggested patients with BD—especially those with comorbid cardiovascular or metabolic conditions— increase their dietary consumption of foods containing omega-3 FAs (Table)18 and, if necessary, take a supplement of 1 to 1.5 g/d of mixed EPA and DHA, with a higher ratio of EPA.19 See Box 3 for a box on how to read omega-3 supplement labels.
In a small RCT of 51 children and adolescents (age 6 to 17) with symptomatic bipolar I or bipolar II disorder, supplementation with flax oil (alpha-linolenic acid, a polyunsaturated omega-3 FA that is a precursor to EPA and DHA) did not affect symptoms as measured by several rating scales.20
Perinatal and postpartum depression. Omega-3 FAs are considered a safe treatment for depressive disorders during pregnancy because they provide neurodevelopmental benefits for neonates and have few contraindications during pregnancy.21 RCTs of omega-3 FA monotherapy for perinatal depression have been small (≤51 patients) and produced mixed findings.21 A pilot study (N = 16) of patients with postpartum depression found a significant decrease in depressive symptoms with EPA treatment.22 More research is needed before omega-3 FA supplementation can be recommended during pregnancy.
Table
Foods with healthy fats: From best to worst
| Polyunsaturated fats | Omega-3 | Fish-based: oily fish, including salmon, tuna, mackerel, lake trout, herring, and sardines Plant-based: tofu and other forms of soybeans; walnuts and flaxseed and their oils, and canola oil |
| Omega-6 | Only available in plant-based form: corn, soy, and safflower oil | |
| Monosaturated fats | Olive and peanut oil | |
| Saturated fats | Red meats, high-fat dairy, and partially hydrogenated oils | |
| Source: Reference 18 | ||
Because nutritional supplements vary, advise patients to look at the supplement facts on the back of a bottle of omega-3 fatty acids. The American Psychiatric Association recommends patients take a total eicosapentaenoic acid (EPA) + docosahexaenoic acid (DHA) of 1 g/d; EPA should be ≥60% of total EPA + DHA.
This image is an example of a label that would meet the appropriate criteria. Total EPA + DHA = 1,490 mg and EPA is 60% of this combined total.

Source: Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584
Schizophrenia
In a Cochrane review of 8 studies of patients with schizophrenia, adjunctive treatment with omega-3 FAs led to >25% reduction in the Positive and Negative Syndrome Scale, but this improvement was not statistically significant.23 Omega-3 FAs did not decrease tardive dyskinesia symptoms as measured by the Abnormal Involuntary Movement Scale. The authors stated that results were inconclusive, and use of omega-3 FAs in patients with schizophrenia remains experimental. In a separate meta-analysis that included 335 patients with schizophrenia, EPA augmentation had no beneficial effect on psychotic symptoms.24
In a double-blind RCT of 81 adolescents and young adults (age 13 to 25) at ultra-high risk of psychotic illness, 5% of patients who received 1.2 g/d of omega-3 FAs developed a psychotic disorder compared with 28% of patients receiving placebo.25 The authors concluded that supplementation with omega-3 FAs may be a safe and effective strategy for young patients with subthreshold psychotic symptoms.
Dementia
Studies evaluating the relationship between omega-3 FAs and dementia risk have revealed mixed findings.26,27 In a pilot study of 10 geriatric patients with moderately severe dementia related to thrombotic cerebrovascular disorder, DHA supplementation led to improved Hamilton Depression Rating Scale and Mini-Mental State Examination (MMSE) scores compared with controls.28 In another study, administering EPA to 64 patients with Alzheimer’s disease significantly improved MMSE scores, with maximum improvement at 3 months, but this benefit dissipated after 6 months of treatment.29 In a study of 22 patients with various types of dementia, Suzuki et al30 found that DHA supplementation improved scores on a Japanese dementia scale. These studies show promise, but more evidence is necessary before recommendations can be made.
Other psychiatric disorders
Omega-3 FAs as monotherapy or an adjunct to psychostimulants does not seem to improve symptoms in children who meet DSM-IV-TR criteria for attention-deficit/hyperactivity disorder (ADHD).31-33 Studies of omega-3 FAs as treatment for anxiety and personality disorders are limited. To date, omega-3 FAs as adjunctive treatment in obsessive-compulsive disorder (OCD) and monotherapy in borderline personality disorder have not shown efficacy.34,35
Using omega-3 FAs in practice
Based on new data and several recent meta-analyses, clinical recommendations have emerged. Sarris et al17 suggested patients with BD increase dietary intake of omega-3 FAs or take a supplement with 1 to 1.5 g/d of mixed EPA and DHA (with a higher ratio of EPA). In MDD, the type of omega-3 FA supplementation seems to be important; EPA seems to be the primary component for efficacy.15,19 Additionally, the more severe the depression, the more likely symptoms will respond to omega-3 FAs.6,14,15 Omega-3 FAs are not effective at preventing depression14,15 and evidence is equivocal for treating perinatal depression.21 Omega-3 FA supplementation has not shown efficacy for patients with schizophrenia,23,24 although it may prevent transition to psychosis in adolescents and young adults at ultra-high risk for a psychotic disorder.25 Data examining omega-3 FA supplementation in postpartum depression22 and dementia28,29 are limited but show promise. Omega-3 FAs appear to lack efficacy in ADHD,31-33 OCD,34 and borderline personality disorder.35
Related Resources
- National Center for Complementary and Alternative Medicine. Omega-3 fatty acids. http://nccam.nih.gov/health/omega3.
- National Institutes of Health. Office of Dietary Supplements. Working group report: Omega-3 fatty acids and cardiovascular disease. http://ods.od.nih.gov/Health_Information/omega_3_fatty_acids.aspx.
Disclosure
Dr. Morreale reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Hibbeln JR. Fish consumption and major depression. Lancet. 1998;351(9110):1213.-
2. Tanskanen A, Hibbeln JR, Tuomilehto J, et al. Fish consumption and depressive symptoms in the general population in Finland. Psychiatr Serv. 2001;52(4):529-531.
3. Silvers KM, Scott KM. Fish consumption and self-reported physical and mental health status. Public Health Nutr. 2002;5(3):427-431.
4. Timonen M, Horrobin DF, Jokelaienen J, et al. Fish consumption and depression: the northern Finland 1966 birth cohort study. J Affect Disord. 2004;82(3):447-452.
5. Freeman MP, Rapaport MH. Omega-3 fatty acids and depression: from cellular mechanisms to clinical care. J Clin Psychiatry. 2011;72(2):258-259.
6. Bloch MH, Hannestad J. Omega-3 fatty acids for the treatment of depression: systematic review and meta-analysis [published online ahead of print September 20 2011]. Mol Psychiatry. doi: 10.1038/mp.2011.100.
7. Parker G, Gibson NA, Brotchie H, et al. Omega-3 fatty acids and mood disorders. Am J Psychiatry. 2006;163(6):969-978.
8. Martinez JM, Marangell LB. Omega-3 fatty acids: do ‘fish oils’ have a therapeutic role in psychiatry? Current Psychiatry. 2004;3(1):25-52.
9. Mozaffarian D, Micha R, Wallace S. Effects of coronary heart disease of increasing polyunsaturated fat in place of saturated fat: a systematic review and meta-analysis of randomized controlled trials. PLoS Med. 2010;7(3):e1000252.-
10. Kris-Etherton PM, Harris WS, Appel LJ. AHA Nutrition Committee. American Heart Association. Omega-3 fatty acids and cardiovascular disease: new recommendations from the American Heart Association. Arterioscler Thromb Vasc Biol. 2003;23(2):151-152.
11. Freeman MP, Fava M, Lake J, et al. Complementary and alternative medicine in major depressive disorder: the American Psychiatric Association Task Force report. J Clin Psychiatry. 2010;71(6):669-681.
12. Freeman MP, Hibbeln J, Wisner KL, et al. Omega-3 fatty acids: evidence basis for treatment and future research in psychiatry. J Clin Psychiatry. 2006;67(12):1954-1967.
13. Mischoulon D. The impact of omega-3 fatty acids on depressive disorders and suicidality: can we reconcile 2 studies with seemingly contradictory results? J Clin Psychiatry. 2011;72(12):1574-1576.
14. Appleton KM, Rogers PJ, Andrew RN. Updated systematic review and meta-analysis of the effects of n-3 long-chain polyunsaturated fatty acids on depressed mood. Am J Clin Nutr. 2010;91(31):757-770.
15. Martins JG. EPA but not DHA appears to be responsible for the efficacy of omega-3 long chain polyunsaturated fatty acid supplementation in depression: evidence from a meta-analysis of randomized controlled trials. J Am Coll Nutr. 2009;28(5):525-542.
16. Young G, Conquer J. Omega-3 fatty acids and neuropsychiatric disorders. Reprod Nutr Dev. 2005;45(1):1-28.
17. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
18. Sacks F. Ask the expert: omega-3 fatty acids. The Nutrition Source.http://www.hsph.harvard.edu/nutritionsource/questions/omega-3/index.html. Accessed July 23 2012.
19. Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584.
20. Gracious BL, Chirieac MC, Costescu S, et al. Randomized, placebo-controlled trial of flax oil in pediatric bipolar disorder. Bipolar Disord. 2010;12(2):142-154.
21. Freeman MP. Omega-3 fatty acids in major depressive disorder. J Clin Psychiatry. 2009;70(suppl 5):7-11.
22. Freeman MP, Hibbeln JR, Wisner KL, et al. Randomized dose-ranging pilot trial of omega-3 fatty acids for postpartum depression. Acta Psychiatr Scand. 2006;113(1):31-35.
23. Joy CB, Mumby-Croft R, Joy LA. Polyunsaturated fatty acid supplementation for schizophrenia. Cochrane Database Syst Rev. 2006;(3):CD001257.-
24. Fusar-Poli P, Berger G. Eicosapentaenoic acid interventions in schizophrenia: meta-analysis of randomized placebo-controlled studies. J Clin Psychopharmacol. 2012;32(2):179-185.
25. Amminger GP, Schäfer MR, Papageorgiou K, et al. Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2010;67(2):146-154.
26. Morris MC, Evans DA, Bienias JL, et al. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60(7):940-946.
27. Engelhart MJ, Geerlings MI, Ruitenberg A, et al. Diet and risk of dementia: does fat matter? The Rotterdam Study. Neurology. 2002;59(12):1915-1921.
28. Terano T, Fujishiro S, Ban T, et al. Docosahexaenoic acid supplementation improves the moderately severe dementia from thrombotic cerebrovascular diseases. Lipids. 1999;34 suppl:S345-S346.
29. Otsuka M. Analysis of dietary factors in Alzheimer’s disease: clinical use of nutritional intervention for prevention and treatment of dementia [in Japanese]. Nihon Ronen Igakkai Zasshi. 2000;37(12):970-973.
30. Suzuki H, Morikawa Y, Takahashi H. Effect of DHA oil supplementation in intelligence and visual acuity in the elderly. World Rev Nutr Diet. 2001;88:68-71.
31. Joshi K, Lad S, Kale M, et al. Supplementation with flax oil and vitamin C improves the outcome of attention deficit hyperactivity disorder (ADHD). Prostaglandins Leukot Essent Fatty Acids. 2006;74(1):17-21.
32. Voigt RG, Llorente AM, Jensen CL, et al. A randomized, double-blind, placebo-controlled trial of docosahexaenoic acid supplementation in children with attention-deficit/hyperactivity disorder. J Pediatr. 2001;139(2):189-196.
33. Hirayama S, Hamazaki T, Terasawa K. Effect of docosahexaenoic acid-containing food administration on symptoms of attention-deficit/hyperactivity disorder - a placebo-controlled double-blind study. Eur J Clin Nutr. 2004;58(3):467-473.
34. Fux M, Benjamin J, Nemets B. A placebo-controlled cross-over trial of adjunctive EPA in OCD. J Psychiatr Res. 2004;38(3):323-325.
35. Zanarini MC, Frankenburg FR. Omega-3 Fatty acid treatment of women with borderline personality disorder: a double-blind placebo-controlled pilot study. Am J Psychiatry. 2003;160(1):167-169.
Discuss this article at www.facebook.com/CurrentPsychiatry
Epidemiologic data suggest that people who consume diets rich in omega-3 fatty acids (FAs)—long-chain polyunsaturated FAs such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)—have a decreased risk of major depressive disorder (MDD), postpartum depression, and bipolar disorder (BD).1-5 Omega-3 FA concentration may impact serotonin and dopamine transmission via effects on cell membrane fluidity.6 Therefore, decreased intake may increase the risk of several psychiatric disorders. As the average Western diet has changed over the last 2 centuries, omega-3 FA consumption has decreased.7 Omega-3 FAs cannot be synthesized by the body and must come from exogenous sources, such as fish and nuts. For a discussion of different types of dietary fats, see Box 1.8
Should we advise our patients to increase their omega-3 FA consumption? The American Psychiatric Association (APA) and the American Heart Association (AHA) recommend omega-3 FA consumption for the general population and in some cases, supplementation for specific disorders (Box 2).9-12 New data has been published since Current Psychiatry last reviewed the evidence for using omega-3 FAs for psychiatric conditions in 2004.8 This article looks at the latest evidence on the use of omega-3 FAs to treat mood disorders, schizophrenia, dementia, and other psychiatric conditions.
Dietary fat is saturated or unsaturated. Unsaturated fats are further categorized as monounsaturated or polyunsaturated (PUFA). PUFAs contain a hydrocarbon chain with ≥2 double bonds.8 The position of this double bond relative to the methyl end carbon—or “omega” carbon—groups the PUFAs into 2 categories:8
- omega-6 fatty acids, including arachidonic acid (AA) and linoleic acid (LA)
- omega-3 fatty acids, including eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and alpha-linolenic acid (ALA). ALA is a metabolic precursor to EPA and DHA.
PUFAs—in particular AA and DHA—are thought to contribute to cell membrane fluidity, modulation of neurotransmitters, and signal transduction pathways. As precursors to eicosanoids and cytokines, PUFAs may affect anti-inflammatory response systems.
Consumption of omega-3 fatty acids (FAs) reduces risk for arrhythmia, thrombosis, and atherosclerotic plaque, according to American Heart Association (AHA) guidelines. Omega-3 FA intake also may improve endothelial function, slightly lower blood pressure, and reduce inflammatory response. Replacing dietary saturated fat with polyunsaturated fat reduces coronary heart disease risk by 19%.9 The AHA recommends that all adults eat fish, particularly oily fish such as salmon or tuna, ≥2 times per week. Patients with documented coronary heart disease should consume 1 g/d eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) combined10 either via oily fish or omega-3 FA capsules. Side effects of omega-3 FA supplements are minor and include mild gastrointestinal discomfort, mostly burping or an unpleasant aftertaste; no cases of bleeding have been reported.11
For patients with hypertriglyceridemia, 2 to 4 g/d may be useful. Because of a theoretical risk of bleeding, doses >3 g/d should be supervised by a physician.
Because psychiatric illnesses and cardiovascular disease may be comorbid, the Omega-3 FA Subcommittee of the American Psychiatric Association supports the AHA’s guidelines regarding fish consumption, and further recommends that patients with mood, impulse control, or psychotic disorders consume ≥1 g/d of combined EPA and DHA.12
Limitations of the data
Reviewing the literature on omega-3 FAs to treat psychiatric disorders is hampered by several difficulties:13
- studies may evaluate the use of EPA alone, EPA combined with DHA, or DHA alone
- the doses of EPA and DHA and ratio of EPA to DHA of the supplements used in clinical trials varies greatly
- patients’ dietary consumption of omega-3 FAs is difficult to control
- DSM diagnostic criteria, as well as severity of illness, differ within studies.
In addition, studies may use omega-3 FAs as monotherapy or as adjuncts. All of these factors lead to difficulty interpreting the literature, as well as trouble in extracting data for meta-analysis.
Omega-3 FAs for mood disorders
MDD and other depressive diagnoses. Several meta-analyses examining the use of omega-3 FAs for treating depressive disorders have had equivocal findings. Variability in results might be partially explained by differences in the severity of baseline depression among diverse study populations, diagnostic variation, differing omega-3 supplementation protocols, or other issues.13 In addition, publication bias also may affect results.
In a 2011 literature review and meta-analysis of omega-3 FAs as monotherapy or an adjunct to antidepressants to treat MDD, Bloch and Hannestad6 concluded that omega-3 FAs offer a small but nonsignificant benefit in treating MDD. This review suggested that omega-3 FAs may be more effective in patients with more severe depression. The effects of varying levels of EPA vs DHA were not examined.
In a systematic review and meta-analysis, Appleton et al14 concluded that omega-3 FA supplements have little beneficial effect on depressed mood in individuals who do not have a depressive illness diagnosis (eg, MDD). However, this study did not consider the differential effects of EPA vs DHA on treatment response. Patients diagnosed with a depressive illness received greater benefits from omega-3 FA supplementation, although the patients in this study were heterogeneous. Similar to Bloch and Hannestad, Appleton et al14 found that omega-3 FA supplementation may be most beneficial for depressed patients with more severe symptoms, but is unlikely to help those with mild-to-moderate symptoms or individuals without symptoms who aim to prevent depression.
A meta-analysis by Martins15 looked at EPA vs DHA to treat depressive illness and found that only supplements that were mostly or completely EPA effectively treated depressive symptoms. Martins also found that severity of illness is key for positive treatment outcomes; there was a significant relationship between higher baseline depression levels and efficacy.15 Martins noted that omega-3 FA therapy was more effective as a treatment than a preventive strategy, and that adding omega-3 FAs to antidepressants was more efficacious than omega-3 FAs alone.15
A meta-analysis of clinical trials of omega-3 FAs for depressive illness suggested EPA should be ≥60% of total EPA + DHA.16
BD. A recent meta-analysis of 6 randomized controlled trials (RCTs) found that adding omega-3 supplements to mood stabilizers in patients with BD was associated with a statistically significant reduction of depressive symptoms, but was not effective for treating mania.17 The authors suggested patients with BD—especially those with comorbid cardiovascular or metabolic conditions— increase their dietary consumption of foods containing omega-3 FAs (Table)18 and, if necessary, take a supplement of 1 to 1.5 g/d of mixed EPA and DHA, with a higher ratio of EPA.19 See Box 3 for a box on how to read omega-3 supplement labels.
In a small RCT of 51 children and adolescents (age 6 to 17) with symptomatic bipolar I or bipolar II disorder, supplementation with flax oil (alpha-linolenic acid, a polyunsaturated omega-3 FA that is a precursor to EPA and DHA) did not affect symptoms as measured by several rating scales.20
Perinatal and postpartum depression. Omega-3 FAs are considered a safe treatment for depressive disorders during pregnancy because they provide neurodevelopmental benefits for neonates and have few contraindications during pregnancy.21 RCTs of omega-3 FA monotherapy for perinatal depression have been small (≤51 patients) and produced mixed findings.21 A pilot study (N = 16) of patients with postpartum depression found a significant decrease in depressive symptoms with EPA treatment.22 More research is needed before omega-3 FA supplementation can be recommended during pregnancy.
Table
Foods with healthy fats: From best to worst
| Polyunsaturated fats | Omega-3 | Fish-based: oily fish, including salmon, tuna, mackerel, lake trout, herring, and sardines Plant-based: tofu and other forms of soybeans; walnuts and flaxseed and their oils, and canola oil |
| Omega-6 | Only available in plant-based form: corn, soy, and safflower oil | |
| Monosaturated fats | Olive and peanut oil | |
| Saturated fats | Red meats, high-fat dairy, and partially hydrogenated oils | |
| Source: Reference 18 | ||
Because nutritional supplements vary, advise patients to look at the supplement facts on the back of a bottle of omega-3 fatty acids. The American Psychiatric Association recommends patients take a total eicosapentaenoic acid (EPA) + docosahexaenoic acid (DHA) of 1 g/d; EPA should be ≥60% of total EPA + DHA.
This image is an example of a label that would meet the appropriate criteria. Total EPA + DHA = 1,490 mg and EPA is 60% of this combined total.
Source: Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584
Schizophrenia
In a Cochrane review of 8 studies of patients with schizophrenia, adjunctive treatment with omega-3 FAs led to >25% reduction in the Positive and Negative Syndrome Scale, but this improvement was not statistically significant.23 Omega-3 FAs did not decrease tardive dyskinesia symptoms as measured by the Abnormal Involuntary Movement Scale. The authors stated that results were inconclusive, and use of omega-3 FAs in patients with schizophrenia remains experimental. In a separate meta-analysis that included 335 patients with schizophrenia, EPA augmentation had no beneficial effect on psychotic symptoms.24
In a double-blind RCT of 81 adolescents and young adults (age 13 to 25) at ultra-high risk of psychotic illness, 5% of patients who received 1.2 g/d of omega-3 FAs developed a psychotic disorder compared with 28% of patients receiving placebo.25 The authors concluded that supplementation with omega-3 FAs may be a safe and effective strategy for young patients with subthreshold psychotic symptoms.
Dementia
Studies evaluating the relationship between omega-3 FAs and dementia risk have revealed mixed findings.26,27 In a pilot study of 10 geriatric patients with moderately severe dementia related to thrombotic cerebrovascular disorder, DHA supplementation led to improved Hamilton Depression Rating Scale and Mini-Mental State Examination (MMSE) scores compared with controls.28 In another study, administering EPA to 64 patients with Alzheimer’s disease significantly improved MMSE scores, with maximum improvement at 3 months, but this benefit dissipated after 6 months of treatment.29 In a study of 22 patients with various types of dementia, Suzuki et al30 found that DHA supplementation improved scores on a Japanese dementia scale. These studies show promise, but more evidence is necessary before recommendations can be made.
Other psychiatric disorders
Omega-3 FAs as monotherapy or an adjunct to psychostimulants does not seem to improve symptoms in children who meet DSM-IV-TR criteria for attention-deficit/hyperactivity disorder (ADHD).31-33 Studies of omega-3 FAs as treatment for anxiety and personality disorders are limited. To date, omega-3 FAs as adjunctive treatment in obsessive-compulsive disorder (OCD) and monotherapy in borderline personality disorder have not shown efficacy.34,35
Using omega-3 FAs in practice
Based on new data and several recent meta-analyses, clinical recommendations have emerged. Sarris et al17 suggested patients with BD increase dietary intake of omega-3 FAs or take a supplement with 1 to 1.5 g/d of mixed EPA and DHA (with a higher ratio of EPA). In MDD, the type of omega-3 FA supplementation seems to be important; EPA seems to be the primary component for efficacy.15,19 Additionally, the more severe the depression, the more likely symptoms will respond to omega-3 FAs.6,14,15 Omega-3 FAs are not effective at preventing depression14,15 and evidence is equivocal for treating perinatal depression.21 Omega-3 FA supplementation has not shown efficacy for patients with schizophrenia,23,24 although it may prevent transition to psychosis in adolescents and young adults at ultra-high risk for a psychotic disorder.25 Data examining omega-3 FA supplementation in postpartum depression22 and dementia28,29 are limited but show promise. Omega-3 FAs appear to lack efficacy in ADHD,31-33 OCD,34 and borderline personality disorder.35
Related Resources
- National Center for Complementary and Alternative Medicine. Omega-3 fatty acids. http://nccam.nih.gov/health/omega3.
- National Institutes of Health. Office of Dietary Supplements. Working group report: Omega-3 fatty acids and cardiovascular disease. http://ods.od.nih.gov/Health_Information/omega_3_fatty_acids.aspx.
Disclosure
Dr. Morreale reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Epidemiologic data suggest that people who consume diets rich in omega-3 fatty acids (FAs)—long-chain polyunsaturated FAs such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)—have a decreased risk of major depressive disorder (MDD), postpartum depression, and bipolar disorder (BD).1-5 Omega-3 FA concentration may impact serotonin and dopamine transmission via effects on cell membrane fluidity.6 Therefore, decreased intake may increase the risk of several psychiatric disorders. As the average Western diet has changed over the last 2 centuries, omega-3 FA consumption has decreased.7 Omega-3 FAs cannot be synthesized by the body and must come from exogenous sources, such as fish and nuts. For a discussion of different types of dietary fats, see Box 1.8
Should we advise our patients to increase their omega-3 FA consumption? The American Psychiatric Association (APA) and the American Heart Association (AHA) recommend omega-3 FA consumption for the general population and in some cases, supplementation for specific disorders (Box 2).9-12 New data has been published since Current Psychiatry last reviewed the evidence for using omega-3 FAs for psychiatric conditions in 2004.8 This article looks at the latest evidence on the use of omega-3 FAs to treat mood disorders, schizophrenia, dementia, and other psychiatric conditions.
Dietary fat is saturated or unsaturated. Unsaturated fats are further categorized as monounsaturated or polyunsaturated (PUFA). PUFAs contain a hydrocarbon chain with ≥2 double bonds.8 The position of this double bond relative to the methyl end carbon—or “omega” carbon—groups the PUFAs into 2 categories:8
- omega-6 fatty acids, including arachidonic acid (AA) and linoleic acid (LA)
- omega-3 fatty acids, including eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and alpha-linolenic acid (ALA). ALA is a metabolic precursor to EPA and DHA.
PUFAs—in particular AA and DHA—are thought to contribute to cell membrane fluidity, modulation of neurotransmitters, and signal transduction pathways. As precursors to eicosanoids and cytokines, PUFAs may affect anti-inflammatory response systems.
Consumption of omega-3 fatty acids (FAs) reduces risk for arrhythmia, thrombosis, and atherosclerotic plaque, according to American Heart Association (AHA) guidelines. Omega-3 FA intake also may improve endothelial function, slightly lower blood pressure, and reduce inflammatory response. Replacing dietary saturated fat with polyunsaturated fat reduces coronary heart disease risk by 19%.9 The AHA recommends that all adults eat fish, particularly oily fish such as salmon or tuna, ≥2 times per week. Patients with documented coronary heart disease should consume 1 g/d eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) combined10 either via oily fish or omega-3 FA capsules. Side effects of omega-3 FA supplements are minor and include mild gastrointestinal discomfort, mostly burping or an unpleasant aftertaste; no cases of bleeding have been reported.11
For patients with hypertriglyceridemia, 2 to 4 g/d may be useful. Because of a theoretical risk of bleeding, doses >3 g/d should be supervised by a physician.
Because psychiatric illnesses and cardiovascular disease may be comorbid, the Omega-3 FA Subcommittee of the American Psychiatric Association supports the AHA’s guidelines regarding fish consumption, and further recommends that patients with mood, impulse control, or psychotic disorders consume ≥1 g/d of combined EPA and DHA.12
Limitations of the data
Reviewing the literature on omega-3 FAs to treat psychiatric disorders is hampered by several difficulties:13
- studies may evaluate the use of EPA alone, EPA combined with DHA, or DHA alone
- the doses of EPA and DHA and ratio of EPA to DHA of the supplements used in clinical trials varies greatly
- patients’ dietary consumption of omega-3 FAs is difficult to control
- DSM diagnostic criteria, as well as severity of illness, differ within studies.
In addition, studies may use omega-3 FAs as monotherapy or as adjuncts. All of these factors lead to difficulty interpreting the literature, as well as trouble in extracting data for meta-analysis.
Omega-3 FAs for mood disorders
MDD and other depressive diagnoses. Several meta-analyses examining the use of omega-3 FAs for treating depressive disorders have had equivocal findings. Variability in results might be partially explained by differences in the severity of baseline depression among diverse study populations, diagnostic variation, differing omega-3 supplementation protocols, or other issues.13 In addition, publication bias also may affect results.
In a 2011 literature review and meta-analysis of omega-3 FAs as monotherapy or an adjunct to antidepressants to treat MDD, Bloch and Hannestad6 concluded that omega-3 FAs offer a small but nonsignificant benefit in treating MDD. This review suggested that omega-3 FAs may be more effective in patients with more severe depression. The effects of varying levels of EPA vs DHA were not examined.
In a systematic review and meta-analysis, Appleton et al14 concluded that omega-3 FA supplements have little beneficial effect on depressed mood in individuals who do not have a depressive illness diagnosis (eg, MDD). However, this study did not consider the differential effects of EPA vs DHA on treatment response. Patients diagnosed with a depressive illness received greater benefits from omega-3 FA supplementation, although the patients in this study were heterogeneous. Similar to Bloch and Hannestad, Appleton et al14 found that omega-3 FA supplementation may be most beneficial for depressed patients with more severe symptoms, but is unlikely to help those with mild-to-moderate symptoms or individuals without symptoms who aim to prevent depression.
A meta-analysis by Martins15 looked at EPA vs DHA to treat depressive illness and found that only supplements that were mostly or completely EPA effectively treated depressive symptoms. Martins also found that severity of illness is key for positive treatment outcomes; there was a significant relationship between higher baseline depression levels and efficacy.15 Martins noted that omega-3 FA therapy was more effective as a treatment than a preventive strategy, and that adding omega-3 FAs to antidepressants was more efficacious than omega-3 FAs alone.15
A meta-analysis of clinical trials of omega-3 FAs for depressive illness suggested EPA should be ≥60% of total EPA + DHA.16
BD. A recent meta-analysis of 6 randomized controlled trials (RCTs) found that adding omega-3 supplements to mood stabilizers in patients with BD was associated with a statistically significant reduction of depressive symptoms, but was not effective for treating mania.17 The authors suggested patients with BD—especially those with comorbid cardiovascular or metabolic conditions— increase their dietary consumption of foods containing omega-3 FAs (Table)18 and, if necessary, take a supplement of 1 to 1.5 g/d of mixed EPA and DHA, with a higher ratio of EPA.19 See Box 3 for a box on how to read omega-3 supplement labels.
In a small RCT of 51 children and adolescents (age 6 to 17) with symptomatic bipolar I or bipolar II disorder, supplementation with flax oil (alpha-linolenic acid, a polyunsaturated omega-3 FA that is a precursor to EPA and DHA) did not affect symptoms as measured by several rating scales.20
Perinatal and postpartum depression. Omega-3 FAs are considered a safe treatment for depressive disorders during pregnancy because they provide neurodevelopmental benefits for neonates and have few contraindications during pregnancy.21 RCTs of omega-3 FA monotherapy for perinatal depression have been small (≤51 patients) and produced mixed findings.21 A pilot study (N = 16) of patients with postpartum depression found a significant decrease in depressive symptoms with EPA treatment.22 More research is needed before omega-3 FA supplementation can be recommended during pregnancy.
Table
Foods with healthy fats: From best to worst
| Polyunsaturated fats | Omega-3 | Fish-based: oily fish, including salmon, tuna, mackerel, lake trout, herring, and sardines Plant-based: tofu and other forms of soybeans; walnuts and flaxseed and their oils, and canola oil |
| Omega-6 | Only available in plant-based form: corn, soy, and safflower oil | |
| Monosaturated fats | Olive and peanut oil | |
| Saturated fats | Red meats, high-fat dairy, and partially hydrogenated oils | |
| Source: Reference 18 | ||
Because nutritional supplements vary, advise patients to look at the supplement facts on the back of a bottle of omega-3 fatty acids. The American Psychiatric Association recommends patients take a total eicosapentaenoic acid (EPA) + docosahexaenoic acid (DHA) of 1 g/d; EPA should be ≥60% of total EPA + DHA.
This image is an example of a label that would meet the appropriate criteria. Total EPA + DHA = 1,490 mg and EPA is 60% of this combined total.
Source: Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584
Schizophrenia
In a Cochrane review of 8 studies of patients with schizophrenia, adjunctive treatment with omega-3 FAs led to >25% reduction in the Positive and Negative Syndrome Scale, but this improvement was not statistically significant.23 Omega-3 FAs did not decrease tardive dyskinesia symptoms as measured by the Abnormal Involuntary Movement Scale. The authors stated that results were inconclusive, and use of omega-3 FAs in patients with schizophrenia remains experimental. In a separate meta-analysis that included 335 patients with schizophrenia, EPA augmentation had no beneficial effect on psychotic symptoms.24
In a double-blind RCT of 81 adolescents and young adults (age 13 to 25) at ultra-high risk of psychotic illness, 5% of patients who received 1.2 g/d of omega-3 FAs developed a psychotic disorder compared with 28% of patients receiving placebo.25 The authors concluded that supplementation with omega-3 FAs may be a safe and effective strategy for young patients with subthreshold psychotic symptoms.
Dementia
Studies evaluating the relationship between omega-3 FAs and dementia risk have revealed mixed findings.26,27 In a pilot study of 10 geriatric patients with moderately severe dementia related to thrombotic cerebrovascular disorder, DHA supplementation led to improved Hamilton Depression Rating Scale and Mini-Mental State Examination (MMSE) scores compared with controls.28 In another study, administering EPA to 64 patients with Alzheimer’s disease significantly improved MMSE scores, with maximum improvement at 3 months, but this benefit dissipated after 6 months of treatment.29 In a study of 22 patients with various types of dementia, Suzuki et al30 found that DHA supplementation improved scores on a Japanese dementia scale. These studies show promise, but more evidence is necessary before recommendations can be made.
Other psychiatric disorders
Omega-3 FAs as monotherapy or an adjunct to psychostimulants does not seem to improve symptoms in children who meet DSM-IV-TR criteria for attention-deficit/hyperactivity disorder (ADHD).31-33 Studies of omega-3 FAs as treatment for anxiety and personality disorders are limited. To date, omega-3 FAs as adjunctive treatment in obsessive-compulsive disorder (OCD) and monotherapy in borderline personality disorder have not shown efficacy.34,35
Using omega-3 FAs in practice
Based on new data and several recent meta-analyses, clinical recommendations have emerged. Sarris et al17 suggested patients with BD increase dietary intake of omega-3 FAs or take a supplement with 1 to 1.5 g/d of mixed EPA and DHA (with a higher ratio of EPA). In MDD, the type of omega-3 FA supplementation seems to be important; EPA seems to be the primary component for efficacy.15,19 Additionally, the more severe the depression, the more likely symptoms will respond to omega-3 FAs.6,14,15 Omega-3 FAs are not effective at preventing depression14,15 and evidence is equivocal for treating perinatal depression.21 Omega-3 FA supplementation has not shown efficacy for patients with schizophrenia,23,24 although it may prevent transition to psychosis in adolescents and young adults at ultra-high risk for a psychotic disorder.25 Data examining omega-3 FA supplementation in postpartum depression22 and dementia28,29 are limited but show promise. Omega-3 FAs appear to lack efficacy in ADHD,31-33 OCD,34 and borderline personality disorder.35
Related Resources
- National Center for Complementary and Alternative Medicine. Omega-3 fatty acids. http://nccam.nih.gov/health/omega3.
- National Institutes of Health. Office of Dietary Supplements. Working group report: Omega-3 fatty acids and cardiovascular disease. http://ods.od.nih.gov/Health_Information/omega_3_fatty_acids.aspx.
Disclosure
Dr. Morreale reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Hibbeln JR. Fish consumption and major depression. Lancet. 1998;351(9110):1213.-
2. Tanskanen A, Hibbeln JR, Tuomilehto J, et al. Fish consumption and depressive symptoms in the general population in Finland. Psychiatr Serv. 2001;52(4):529-531.
3. Silvers KM, Scott KM. Fish consumption and self-reported physical and mental health status. Public Health Nutr. 2002;5(3):427-431.
4. Timonen M, Horrobin DF, Jokelaienen J, et al. Fish consumption and depression: the northern Finland 1966 birth cohort study. J Affect Disord. 2004;82(3):447-452.
5. Freeman MP, Rapaport MH. Omega-3 fatty acids and depression: from cellular mechanisms to clinical care. J Clin Psychiatry. 2011;72(2):258-259.
6. Bloch MH, Hannestad J. Omega-3 fatty acids for the treatment of depression: systematic review and meta-analysis [published online ahead of print September 20 2011]. Mol Psychiatry. doi: 10.1038/mp.2011.100.
7. Parker G, Gibson NA, Brotchie H, et al. Omega-3 fatty acids and mood disorders. Am J Psychiatry. 2006;163(6):969-978.
8. Martinez JM, Marangell LB. Omega-3 fatty acids: do ‘fish oils’ have a therapeutic role in psychiatry? Current Psychiatry. 2004;3(1):25-52.
9. Mozaffarian D, Micha R, Wallace S. Effects of coronary heart disease of increasing polyunsaturated fat in place of saturated fat: a systematic review and meta-analysis of randomized controlled trials. PLoS Med. 2010;7(3):e1000252.-
10. Kris-Etherton PM, Harris WS, Appel LJ. AHA Nutrition Committee. American Heart Association. Omega-3 fatty acids and cardiovascular disease: new recommendations from the American Heart Association. Arterioscler Thromb Vasc Biol. 2003;23(2):151-152.
11. Freeman MP, Fava M, Lake J, et al. Complementary and alternative medicine in major depressive disorder: the American Psychiatric Association Task Force report. J Clin Psychiatry. 2010;71(6):669-681.
12. Freeman MP, Hibbeln J, Wisner KL, et al. Omega-3 fatty acids: evidence basis for treatment and future research in psychiatry. J Clin Psychiatry. 2006;67(12):1954-1967.
13. Mischoulon D. The impact of omega-3 fatty acids on depressive disorders and suicidality: can we reconcile 2 studies with seemingly contradictory results? J Clin Psychiatry. 2011;72(12):1574-1576.
14. Appleton KM, Rogers PJ, Andrew RN. Updated systematic review and meta-analysis of the effects of n-3 long-chain polyunsaturated fatty acids on depressed mood. Am J Clin Nutr. 2010;91(31):757-770.
15. Martins JG. EPA but not DHA appears to be responsible for the efficacy of omega-3 long chain polyunsaturated fatty acid supplementation in depression: evidence from a meta-analysis of randomized controlled trials. J Am Coll Nutr. 2009;28(5):525-542.
16. Young G, Conquer J. Omega-3 fatty acids and neuropsychiatric disorders. Reprod Nutr Dev. 2005;45(1):1-28.
17. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
18. Sacks F. Ask the expert: omega-3 fatty acids. The Nutrition Source.http://www.hsph.harvard.edu/nutritionsource/questions/omega-3/index.html. Accessed July 23 2012.
19. Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584.
20. Gracious BL, Chirieac MC, Costescu S, et al. Randomized, placebo-controlled trial of flax oil in pediatric bipolar disorder. Bipolar Disord. 2010;12(2):142-154.
21. Freeman MP. Omega-3 fatty acids in major depressive disorder. J Clin Psychiatry. 2009;70(suppl 5):7-11.
22. Freeman MP, Hibbeln JR, Wisner KL, et al. Randomized dose-ranging pilot trial of omega-3 fatty acids for postpartum depression. Acta Psychiatr Scand. 2006;113(1):31-35.
23. Joy CB, Mumby-Croft R, Joy LA. Polyunsaturated fatty acid supplementation for schizophrenia. Cochrane Database Syst Rev. 2006;(3):CD001257.-
24. Fusar-Poli P, Berger G. Eicosapentaenoic acid interventions in schizophrenia: meta-analysis of randomized placebo-controlled studies. J Clin Psychopharmacol. 2012;32(2):179-185.
25. Amminger GP, Schäfer MR, Papageorgiou K, et al. Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2010;67(2):146-154.
26. Morris MC, Evans DA, Bienias JL, et al. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60(7):940-946.
27. Engelhart MJ, Geerlings MI, Ruitenberg A, et al. Diet and risk of dementia: does fat matter? The Rotterdam Study. Neurology. 2002;59(12):1915-1921.
28. Terano T, Fujishiro S, Ban T, et al. Docosahexaenoic acid supplementation improves the moderately severe dementia from thrombotic cerebrovascular diseases. Lipids. 1999;34 suppl:S345-S346.
29. Otsuka M. Analysis of dietary factors in Alzheimer’s disease: clinical use of nutritional intervention for prevention and treatment of dementia [in Japanese]. Nihon Ronen Igakkai Zasshi. 2000;37(12):970-973.
30. Suzuki H, Morikawa Y, Takahashi H. Effect of DHA oil supplementation in intelligence and visual acuity in the elderly. World Rev Nutr Diet. 2001;88:68-71.
31. Joshi K, Lad S, Kale M, et al. Supplementation with flax oil and vitamin C improves the outcome of attention deficit hyperactivity disorder (ADHD). Prostaglandins Leukot Essent Fatty Acids. 2006;74(1):17-21.
32. Voigt RG, Llorente AM, Jensen CL, et al. A randomized, double-blind, placebo-controlled trial of docosahexaenoic acid supplementation in children with attention-deficit/hyperactivity disorder. J Pediatr. 2001;139(2):189-196.
33. Hirayama S, Hamazaki T, Terasawa K. Effect of docosahexaenoic acid-containing food administration on symptoms of attention-deficit/hyperactivity disorder - a placebo-controlled double-blind study. Eur J Clin Nutr. 2004;58(3):467-473.
34. Fux M, Benjamin J, Nemets B. A placebo-controlled cross-over trial of adjunctive EPA in OCD. J Psychiatr Res. 2004;38(3):323-325.
35. Zanarini MC, Frankenburg FR. Omega-3 Fatty acid treatment of women with borderline personality disorder: a double-blind placebo-controlled pilot study. Am J Psychiatry. 2003;160(1):167-169.
1. Hibbeln JR. Fish consumption and major depression. Lancet. 1998;351(9110):1213.-
2. Tanskanen A, Hibbeln JR, Tuomilehto J, et al. Fish consumption and depressive symptoms in the general population in Finland. Psychiatr Serv. 2001;52(4):529-531.
3. Silvers KM, Scott KM. Fish consumption and self-reported physical and mental health status. Public Health Nutr. 2002;5(3):427-431.
4. Timonen M, Horrobin DF, Jokelaienen J, et al. Fish consumption and depression: the northern Finland 1966 birth cohort study. J Affect Disord. 2004;82(3):447-452.
5. Freeman MP, Rapaport MH. Omega-3 fatty acids and depression: from cellular mechanisms to clinical care. J Clin Psychiatry. 2011;72(2):258-259.
6. Bloch MH, Hannestad J. Omega-3 fatty acids for the treatment of depression: systematic review and meta-analysis [published online ahead of print September 20 2011]. Mol Psychiatry. doi: 10.1038/mp.2011.100.
7. Parker G, Gibson NA, Brotchie H, et al. Omega-3 fatty acids and mood disorders. Am J Psychiatry. 2006;163(6):969-978.
8. Martinez JM, Marangell LB. Omega-3 fatty acids: do ‘fish oils’ have a therapeutic role in psychiatry? Current Psychiatry. 2004;3(1):25-52.
9. Mozaffarian D, Micha R, Wallace S. Effects of coronary heart disease of increasing polyunsaturated fat in place of saturated fat: a systematic review and meta-analysis of randomized controlled trials. PLoS Med. 2010;7(3):e1000252.-
10. Kris-Etherton PM, Harris WS, Appel LJ. AHA Nutrition Committee. American Heart Association. Omega-3 fatty acids and cardiovascular disease: new recommendations from the American Heart Association. Arterioscler Thromb Vasc Biol. 2003;23(2):151-152.
11. Freeman MP, Fava M, Lake J, et al. Complementary and alternative medicine in major depressive disorder: the American Psychiatric Association Task Force report. J Clin Psychiatry. 2010;71(6):669-681.
12. Freeman MP, Hibbeln J, Wisner KL, et al. Omega-3 fatty acids: evidence basis for treatment and future research in psychiatry. J Clin Psychiatry. 2006;67(12):1954-1967.
13. Mischoulon D. The impact of omega-3 fatty acids on depressive disorders and suicidality: can we reconcile 2 studies with seemingly contradictory results? J Clin Psychiatry. 2011;72(12):1574-1576.
14. Appleton KM, Rogers PJ, Andrew RN. Updated systematic review and meta-analysis of the effects of n-3 long-chain polyunsaturated fatty acids on depressed mood. Am J Clin Nutr. 2010;91(31):757-770.
15. Martins JG. EPA but not DHA appears to be responsible for the efficacy of omega-3 long chain polyunsaturated fatty acid supplementation in depression: evidence from a meta-analysis of randomized controlled trials. J Am Coll Nutr. 2009;28(5):525-542.
16. Young G, Conquer J. Omega-3 fatty acids and neuropsychiatric disorders. Reprod Nutr Dev. 2005;45(1):1-28.
17. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
18. Sacks F. Ask the expert: omega-3 fatty acids. The Nutrition Source.http://www.hsph.harvard.edu/nutritionsource/questions/omega-3/index.html. Accessed July 23 2012.
19. Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584.
20. Gracious BL, Chirieac MC, Costescu S, et al. Randomized, placebo-controlled trial of flax oil in pediatric bipolar disorder. Bipolar Disord. 2010;12(2):142-154.
21. Freeman MP. Omega-3 fatty acids in major depressive disorder. J Clin Psychiatry. 2009;70(suppl 5):7-11.
22. Freeman MP, Hibbeln JR, Wisner KL, et al. Randomized dose-ranging pilot trial of omega-3 fatty acids for postpartum depression. Acta Psychiatr Scand. 2006;113(1):31-35.
23. Joy CB, Mumby-Croft R, Joy LA. Polyunsaturated fatty acid supplementation for schizophrenia. Cochrane Database Syst Rev. 2006;(3):CD001257.-
24. Fusar-Poli P, Berger G. Eicosapentaenoic acid interventions in schizophrenia: meta-analysis of randomized placebo-controlled studies. J Clin Psychopharmacol. 2012;32(2):179-185.
25. Amminger GP, Schäfer MR, Papageorgiou K, et al. Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2010;67(2):146-154.
26. Morris MC, Evans DA, Bienias JL, et al. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60(7):940-946.
27. Engelhart MJ, Geerlings MI, Ruitenberg A, et al. Diet and risk of dementia: does fat matter? The Rotterdam Study. Neurology. 2002;59(12):1915-1921.
28. Terano T, Fujishiro S, Ban T, et al. Docosahexaenoic acid supplementation improves the moderately severe dementia from thrombotic cerebrovascular diseases. Lipids. 1999;34 suppl:S345-S346.
29. Otsuka M. Analysis of dietary factors in Alzheimer’s disease: clinical use of nutritional intervention for prevention and treatment of dementia [in Japanese]. Nihon Ronen Igakkai Zasshi. 2000;37(12):970-973.
30. Suzuki H, Morikawa Y, Takahashi H. Effect of DHA oil supplementation in intelligence and visual acuity in the elderly. World Rev Nutr Diet. 2001;88:68-71.
31. Joshi K, Lad S, Kale M, et al. Supplementation with flax oil and vitamin C improves the outcome of attention deficit hyperactivity disorder (ADHD). Prostaglandins Leukot Essent Fatty Acids. 2006;74(1):17-21.
32. Voigt RG, Llorente AM, Jensen CL, et al. A randomized, double-blind, placebo-controlled trial of docosahexaenoic acid supplementation in children with attention-deficit/hyperactivity disorder. J Pediatr. 2001;139(2):189-196.
33. Hirayama S, Hamazaki T, Terasawa K. Effect of docosahexaenoic acid-containing food administration on symptoms of attention-deficit/hyperactivity disorder - a placebo-controlled double-blind study. Eur J Clin Nutr. 2004;58(3):467-473.
34. Fux M, Benjamin J, Nemets B. A placebo-controlled cross-over trial of adjunctive EPA in OCD. J Psychiatr Res. 2004;38(3):323-325.
35. Zanarini MC, Frankenburg FR. Omega-3 Fatty acid treatment of women with borderline personality disorder: a double-blind placebo-controlled pilot study. Am J Psychiatry. 2003;160(1):167-169.
How to target psychiatric symptoms of Huntington’s disease
Discuss this article at www.facebook.com/CurrentPsychiatry
Psychiatric symptoms are a common and debilitating manifestation of Huntington’s disease (HD), a progressive, inherited neurodegenerative disorder also characterized by chorea (involuntary, nonrepetitive movements) and cognitive decline. The prevalence of HD is 4 to 8 patients per 100,000 persons in most populations of European descent, with lower prevalence among non-Europeans.1 HD is caused by an abnormal expansion of a trinucleotide (CAG) repeat sequence on chromosome 4, and is inherited in an autosomal dominant fashion, meaning a HD patient’s child has a 50% chance of inheriting the mutation. The expansion is located in the gene that encodes the “huntingtin” protein, the normal function of which is not well understood.
There’s no cure for HD, and treatments primarily are directed at symptom control. Psychiatric symptoms include depression, apathy, anxiety, and psychosis (Table).2-4 Treating patients with HD can be challenging because most psychiatrists will see only a handful of patients with this multifaceted illness during their careers. See Box 1 for a case study of a patient with HD.
Table
Psychiatric symptoms of HD
| Anxiety |
| Apathy |
| Delusions |
| Disinhibitions, impulsivity, aggressive behavior |
| Dysphoria |
| Euphoria |
| Hallucinations |
| Irritability |
| Obsessions and compulsions |
| HD: Huntington’s disease Source: References 2-4 |
Mr. M, age 50, was diagnosed with Huntington’s disease (HD) 1 year ago. He returns to our psychiatric clinic for treatment of depressive symptoms and temper. Previously, he was prescribed citalopram, 40 mg/d; eventually low-dose olanzapine, 2.5 mg at night, was added. Mr. M reported better temper control, but his low mood, irritability, hopelessness, and amotivation were not significantly improved.
Mr. M left his job at a software company because he had difficulty completing tasks as the result of mood and cognitive changes. He wants to return to work, but feels that he would be unable to complete his job duties.
He begins a trial of bupropion, 150 mg/d, to improve the vegetative component of his mood symptoms to help him return to work. Mr. M now complains of worsening chorea, irritability, and insomnia, with continued difficulty completing tasks. He is intermittently tearful throughout the interview.
Mr. M continues to struggle with mood symptoms that likely are related to the stressful experience of declining function and the intrinsic evolution of HD. His chorea worsens on bupropion; this agent is discontinued and replaced with mirtazapine, 15 mg at night, for his depressive symptoms and insomnia. Citalopram and olanzapine are unchanged. Mr. M is advised to follow up with our HD psychiatry team in 1 month, and is referred for brief psychotherapy. We remind him—as we do for all of our HD patients—to call the HD clinic or 911 if he becomes suicidal. Ongoing treatment efforts likely will be complex, given the multifaceted and progressive nature of his disease.
Psychiatric sequelae
In general, psychiatric symptoms of HD become increasingly prevalent over time (Box 2).3,5 In a 2001 study of 52 HD patients by Paulsen et al,2 51 patients had ≥1 psychiatric symptom, such as dysphoria (69.2%), agitation (67.3%), irritability (65.4%), apathy (55.8%), and anxiety (51.9%); delusions (11.5%) and hallucinations (1.9%) were less prevalent.2 Similarly, Thompson et al3 followed 111 HD patients for ≥3 years and all experienced psychiatric symptoms.
According to Thompson et al,3 the presence and severity of apathy, irritability, and depression trend differently across the course of Huntington’s disease (HD). Apathy worsens with disease progression, closely following cognitive and motor symptoms. Irritability increases significantly, but this effect seems confined to early stages of HD. Depressive symptoms appear to decline slightly as HD advances, although it is unclear if this is because of antidepressants’ effects, increasing emotional blunting, and waning insight in later stages of HD, or another unknown factor.3 This study did not examine psychotic symptoms over time because few patients were experiencing delusions or hallucinations.
Similar to Thompson et al, Naarding et al5 found that apathy and depression in HD follow distinct time courses. Depression is a feature of early HD and apathy worsens with overall disease progression.
Depressed mood and functional ability—not cognitive or motor symptoms6—are the 2 most critical factors linked to health-related quality of life in HD. Hamilton et al7 found that apathy or executive dysfunction in HD patients is strongly related to decline in ability to complete activities of daily living, and may be severely debilitating.
Apathy. Often mistaken for a symptom of depression, apathy’s presentation may resemble anhedonia or fatigue; however, research suggests that depression and apathy are distinct conditions. Naarding et al
5 noted that apathy is more common than depressive symptoms in HD patients and may be a hallmark symptom of HD.
Depression affects most HD patients, and often is most severe early in the disease course. Hubers et al8 found that 20% of 100 HD patients had suicidal ideation. The strongest predictor was depressed mood.
Sleep disturbances and daytime somnolence are common among HD patients, and patients with comorbid depression report more disturbed sleep. Managing disturbed sleep and daytime somnolence in HD, with emphasis on comorbid depression, may improve the quality of life of patients and their caregivers.9
Anxiety was present in >50% of HD patients in a study by Paulsen et al2 and 37% evaluated by Craufurd et al.10 Craufurd et al10 also reported that 61% of patients were “physically tense and unable to relax.”
Among HD patients, 5% report obsessions and 10% report compulsive behaviors; these symptoms appear to become increasingly common as HD progresses.4,10
Impulsivity and disinhibition. Craufurd et al10 found that 71% of HD patients experienced poor judgment and self-monitoring, 40% had poor temper control and verbal outbursts, 22% exhibited threatening behavior or violence, and 6% had disinhibited or inappropriate sexual behavior.10
Recent studies have shown higher rates of disinhibition in “presymptomatic” gene-positive subjects vs gene-negative controls, suggesting that these symptoms may arise early in HD.11 Further, researchers demonstrated that patients lack symptom awareness and rate themselves as less impaired than their caregivers do.11
In our clinical experience, impulsivity frequently is encountered and creates significant conflict between patients and their caregivers. We speculate that when coupled with depressive symptoms of HD, impulsivity and disinhibition may play an important role in the high rates of suicidality seen in these patients.
Psychosis. Delusions and hallucinations are less common in HD than other psychiatric symptoms. Craufurd et al10 reported 3% of HD patients had delusions, 3% had auditory hallucinations, 2% had tactile hallucinations, and no patients had visual hallucinations.
A few case reports and a small study by Tsuang et al12 suggested that psychotic features in HD may be similar to those seen in paranoid schizophrenia. Tsuang et al12 also noted that more severe HD-related psychosis tends to cluster in families, which suggests that susceptibility to HD psychosis may be heritable.
Treating psychiatric symptoms
High-quality randomized controlled trials of pharmacotherapies for psychiatric symptoms in HD patients are lacking. Decisions regarding which agents to use often are based on case reports or clinical experience. The suggestions below are based on available evidence and our clinical experience.
Depression. Depressive symptoms in HD seem to respond to conventional pharmacologic treatments for major depressive disorder (MDD). A small trial of venlafaxine extended-release (XR) in 26 HD patients with MDD showed statistically significant improvements in depressive symptoms; however, this trial was not blinded and did not have a placebo group.13 In addition, 1 in 5 patients developed significant side effects—nausea, irritability, or worsening chorea.13
Evidence for selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors, and tricyclic antidepressants (TCAs) is lacking. Antidepressant choice should be based on patient response, side effect profile, and the need for secondary therapeutic effects.14
We often prescribe sertraline, citalopram, or escitalopram for our HD patients because of the relative absence of drug-drug interactions and favorable safety profile in medically and surgically ill patients. However, it’s important to tailor the treatment approach to your patient’s needs—eg, patients prone to forgetting their medicine may benefit from a drug with a longer half-life, such as fluoxetine. We avoid TCAs because of their anticholinergic effects, which may worsen dementia symptoms. Because HD patients have high rates of suicidality, agents that are highly toxic when taken in overdose should be used with caution.
One small study of HD patients with MDD or bipolar disorder showed clinical improvement in depressive symptoms after electroconvulsive therapy (ECT).15 Patients who suffered from comorbid delusions had the best improvements in mood.15 ECT likely is a good choice for HD patients who have failed several antidepressants, are suicidal, or who have depression with psychotic features.16
Apathy. A 2011 review concluded that no evidence-based recommendations regarding pharmacologic treatment for apathy in HD can be made because of lack of research.7 The Huntington’s Disease Society of America’s (HDSA) A Physician’s Guide to Managing Huntington’s Disease includes recommendations for treating apathy based on clinical experience.16 It suggests a nonsedating SSRI, followed by a trial of methylphenidate, pemoline, or dextroamphetamine if SSRIs were unsuccessful.
16 The HDSA guide notes psychostimulants may worsen irritability in HD and have a high potential for abuse. ECT appears to have little effect on apathy.15
Anxiety. A small, open-label study of 11 patients found that olanzapine, 5 mg/d, significantly improved depression, anxiety, irritability, and obsessive behavior in HD patients.17
The HDSA guide suggests treating anxiety and obsessive-compulsive symptoms as you would in patients without HD. For anxiety, SSRIs and possibly a short-term trial of a low-dose benzodiazepine (ie, lorazepam, clonazepam) are suggested.16 Benzodiazepines may increase the risk of falls and delirium in this population. Anecdotally, buspirone is helpful in some patients, with a starting dose of 5 mg 2 to 3 times per day and increased to 20 to 30 mg/d in divided doses.16 For obsessive-compulsive symptoms, SSRIs are recommended; atypical antipsychotics are reserved for severe or refractory symptoms.16
Disinhibition and impulsivity. There’s no research on treating disinhibition and impulsivity in HD. In our clinical experience, atypical antipsychotics are the most helpful. Factors regarding choosing an agent and dosing levels are similar to those for psychotic symptoms.
Psychotic symptoms. Most studies of typical and atypical antipsychotics for HD psychosis have shown beneficial effects.14,16-21 Neurologists frequently use these agents for managing chorea. Both neurologic and psychiatric features of the patient’s presentation must be considered when selecting a drug because treatment directed at 1 component of the disease may inadvertently exacerbate another. Specifically, higher potency antipsychotics (eg, haloperidol) are effective for chorea but can dramatically worsen bradykinesia; lower potency agents (eg, quetiapine) are less helpful for chorea but do not significantly worsen rigidity symptoms.
Olanzapine has been shown to improve chorea, anxiety, irritability, depression, sleep dysfunction, and weight loss in addition to psychotic symptoms.14,17 We find that olanzapine treats a constellation of symptoms common among HD patients, and we prescribe it frequently. Because olanzapine is considered a mid-potency agent, we find it’s best suited for concurrent control of psychotic symptoms and mild to moderate chorea in patients with minimal bradykinesia. Start olanzapine at 2.5 mg/d and gradually increase to 5 to 10 mg/d as tolerated.14
Risperidone is effective for treating psychosis and chorea. It can be started at 0.5 to 1 mg/d, and gradually increased to 6 to 8 mg/d.14 The depot formulation of risperidone has been shown to be effective in HD, which may help patients adhere to their medication.18 Risperidone is a mid-high potency antipsychotic, and in our experience is best used to control psychotic symptoms in patients with moderate chorea and few or no symptoms of bradykinesia or rigidity.
Quetiapine reduces psychotic symptoms, agitation, irritability, and insomnia without worsening bradykinesia or rigidity,19 but it is not beneficial for chorea. It can be started at 12.5 mg/d and gradually increased for effect as tolerated, up to 600 mg/d (depending on indication), in 2 or 3 divided doses.14
Haloperidol is a high-potency typical antipsychotic and may help psychotic patients with severe chorea; it should not be used in patients with bradykinesia. Start haloperidol at 0.5 to 1 mg/d and gradually increase to 6 to 8 mg/d as tolerated.14 Because of higher likelihood of side effects with typical antipsychotics, we often reserve its use for patients whose psychosis does not respond to atypical agents.
Other antipsychotics. Aripiprazole in HD has been examined in only 2 single- patient case reports20,21; the drug appeared to reduce psychosis and possibly chorea. Clozapine’s effectiveness for HD psychosis is not well known. It does not appear to be helpful for chorea and can cause agranulocytosis.22
Because one of the hallmarks of HD is dementia, it is worth noting that the FDA has issued a “black-box” warning on the use of antipsychotic drugs in patients with dementia because of concerns regarding increased mortality. However, drawing specific conclusions is difficult because the FDA warning is based on studies that looked primarily at Alzheimer’s disease and vascular dementia, not HD.
Other pharmacotherapies
Tetrabenazine is the only FDA-approved drug for treating HD. However, it carries a “black-box” warning for increased risk of depression and suicidal ideation and is contraindicated in suicidal patients and those with untreated or inadequately treated depression.
Although several small trials have had conflicting results regarding its benefit, amantadine sometimes is used to treat chorea.23-25 For more information about tetrabenazine and amantadine, see Box 3.
Tetrabenazine, the only FDA-approved drug for treating Huntington’s disease (HD), is a dopamine-depleting agent given to control chorea. In a 12-week, randomized, double-blind, placebo-controlled clinical trial, tetrabenazine was shown to be effective in HD patients.a Treatment with tetrabenazine results in symptomatic improvement of chorea, but does not slow or alter the course of the disease. Tetrabenazine can provide relief from choreiform movements, but these benefits should be balanced with the risks of depression and suicidality.a Tetrabenazine is known to prolong QTc interval, and should be used with caution in combination with other drugs that have the potential to do the same (eg, antipsychotics).a
Several case reports have found an association between tetrabenazine and development of neuroleptic malignant syndrome (NMS).b-d Be aware of the clinical characteristics of NMS—mental status change, rigidity, fever, and dysautonomia—and use caution when starting patients taking tetrabenazine on antipsychotics or other agents known to cause NMS.
Amantadine also has been used to treat chorea in HD patients who are unable to tolerate tetrabenazine or antipsychotics. Our neurologists sometimes have found it to be beneficial in patients with juvenile-onset HD because these patients often have debilitating dystonia. Be aware that amantadine is known to precipitate or worsen psychosis.e
References
- Food and Drug Administration. NDA 21-894 Xenazine® (tetrabenazine). Risk evaluation and mitigation strategy (REMS). Click here. Published August 15, 2008. Updated April 2011. Accessed June 20, 2012.
- Stevens E, Roman A, Houa M, et al. Severe hyperthermia during tetrabenazine therapy for tardive dyskinesia. Intensive Care Med. 1998;24(4):369-371.
- Petzinger GM, Bressman SB. A case of tetrabenazine-induced neuroleptic malignant syndrome after prolonged treatment. Mov Disord. 1997;12(2):246-248.
- Ossemann M, Sindic CJ, Laterre C. Tetrabenazine as a cause of neuroleptic malignant syndrome. Mov Disord. 1996;11(1):95.
- Wolters EC. Dopaminomimetic psychosis in Parkinson’s disease patients: diagnosis and treatment. Neurology. 1999;52 (7 suppl 3):S10-S13.
Related Resources
- Huntington’s Disease Society of America. www.hdsa.org.
- Family Caregiver Alliance. Huntington’s disease. www.caregiver.org/caregiver/jsp/content_node.jsp?nodeid=574.
- Huntington Study Group. www.huntington-study-group.org.
- Huntington’s Disease Advocacy Center. www.hdac.org.
Drug Brand Names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Bupropion • Wellbutrin, Wellbutrin XL, others
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Dextroamphetamine • Dexedrine
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lorazepam • Ativan
- Methylphenidate • Concerta, Ritalin, others
- Mirtazapine • Remeron
- Olanzapine • Zyprexa
- Pemoline • Cylert
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tetrabenazine • Xenazine
- Venlafaxine XR • Effexor XR
Disclosures
Dr. Scher is a consultant to the advisory board for Lundbeck.
Ms. Kocsis reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Harper PS. The epidemiology of Huntington’s disease. Hum Genet. 1992;89(4):365-376.
2. Paulsen JS, Ready RE, Hamilton JM, et al. Neuropsychiatric aspects of Huntington’s disease. J Neurol Neurosurg Psychiatry. 2001;71(3):310-314.
3. Thompson JC, Harris J, Sollom AC, et al. Longitudinal evaluation of neuropsychiatric symptoms in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2012;24(1):53-60.
4. Beglinger LJ, Langbehn DR, Duff K, et al. Probability of obsessive and compulsive symptoms in Huntington’s disease. Biol Psychiatry. 2007;61(3):415-418.
5. Naarding P, Janzing JG, Eling P, et al. Apathy is not depression in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2009;21(3):266-270.
6. Ho AK, Gilbert AS, Mason SL, et al. Health-related quality of life in Huntington’s disease: which factors matter most? Mov Disord. 2009;24(4):574-578.
7. Hamilton JM, Salmon DP, Corey-Bloom J, et al. Behavioural abnormalities contribute to functional decline in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2003;74(1):120-122.
8. Hubers AA, Reedeker N, Giltay EJ, et al. Suicidality in Huntington’s disease. J Affect Disord. 2012;136(3):550-557.
9. Videnovic A, Leurgans S, Fan W, et al. Daytime somnolence and nocturnal sleep disturbances in Huntington disease. Parkinsonism Relat Disord. 2009;15(6):471-474.
10. Craufurd D, Thompson JC, Snowden JS. Behavioral changes in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14(4):219-226.
11. Duff K, Paulsen JS, Beglinger LJ, et al. “Frontal” behaviors before the diagnosis of Huntington’s disease and their relationship to markers of disease progression: evidence of early lack of awareness. J Neuropsychiatry Clin Neurosci. 2010;22(2):196-207.
12. Tsuang D, Almqvist EW, Lipe H, et al. Familial aggregation of psychotic symptoms in Huntington’s disease. Am J Psychiatry. 2000;157(12):1955-1959.
13. Holl AK, Wilkinson L, Painold A, et al. Combating depression in Huntington’s disease: effective antidepressive treatment with venlafaxine XR. Int Clin Psychopharmacol. 2010;25(1):46-50.
14. Killoran A, Biglan KM. Therapeutics in Huntington’s disease. Curr Treat Options Neurol. 2012;14(2):137-149.
15. Ranen NG, Peyser CE, Folstein SE. ECT as a treatment for depression in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 1994;6(2):154-159.
16. Rosenblatt A, Ranen NG, Nance MA, et al. A physician’s guide to the management of Huntington’s disease. 2nd edition. http://www.hdsa.org/images/content/1/1/11289.pdf. Published 1999. Accessed July 27, 2012.
17. Squitieri F, Cannella M, Piorcellini A, et al. Short-term effects of olanzapine in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14(1):69-72.
18. Johnston TG. Risperidone long-acting injection and Huntington’s disease: case series with significant psychiatric and behavioural symptoms. Int Clin Psychopharmacol. 2011;26(2):114-119.
19. Alpay M, Koroshetz WJ. Quetiapine in the treatment of behavioral disturbances in patients with Huntington’s disease. Psychosomatics. 2006;47(1):70-72.
20. Lin WC, Chou YH. Aripiprazole effects on psychosis and chorea in a patient with Huntington’s disease. Am J Psychiatry. 2008;165(9):1207-1208.
21. Oulis P, Mourikis I, Konstantakopoulos G, et al. Aripiprazole in the treatment of olanzapine-resistant psychotic and motor symptoms of Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2010;22(3):352c.e4-352c.e5.
22. van Vugt JP, Siesling S, Vergeer M, et al. Clozapine versus placebo in Huntington’s disease: a double blind randomised comparative study. J Neurol Neurosurg Psychiatry. 1997;63(1):35-39.
23. Verhagen Metman L, Morris MJ, Farmer C, et al. Huntington’s disease: a randomized, controlled trial using the NMDA-antagonist amantadine. Neurology. 2002;59(5):694-699.
24. Lucetti C, Del Dotto P, Gambaccini G, et al. IV amantadine improves chorea in Huntington’s disease: an acute randomized, controlled study. Neurology. 2003;60(12):1995-1997.
25. O’Suilleabhain P, Dewey RB, Jr. A randomized trial of amantadine in Huntington disease. Arch Neurol. 2003;60(7):996-998.
Discuss this article at www.facebook.com/CurrentPsychiatry
Psychiatric symptoms are a common and debilitating manifestation of Huntington’s disease (HD), a progressive, inherited neurodegenerative disorder also characterized by chorea (involuntary, nonrepetitive movements) and cognitive decline. The prevalence of HD is 4 to 8 patients per 100,000 persons in most populations of European descent, with lower prevalence among non-Europeans.1 HD is caused by an abnormal expansion of a trinucleotide (CAG) repeat sequence on chromosome 4, and is inherited in an autosomal dominant fashion, meaning a HD patient’s child has a 50% chance of inheriting the mutation. The expansion is located in the gene that encodes the “huntingtin” protein, the normal function of which is not well understood.
There’s no cure for HD, and treatments primarily are directed at symptom control. Psychiatric symptoms include depression, apathy, anxiety, and psychosis (Table).2-4 Treating patients with HD can be challenging because most psychiatrists will see only a handful of patients with this multifaceted illness during their careers. See Box 1 for a case study of a patient with HD.
Table
Psychiatric symptoms of HD
| Anxiety |
| Apathy |
| Delusions |
| Disinhibitions, impulsivity, aggressive behavior |
| Dysphoria |
| Euphoria |
| Hallucinations |
| Irritability |
| Obsessions and compulsions |
| HD: Huntington’s disease Source: References 2-4 |
Mr. M, age 50, was diagnosed with Huntington’s disease (HD) 1 year ago. He returns to our psychiatric clinic for treatment of depressive symptoms and temper. Previously, he was prescribed citalopram, 40 mg/d; eventually low-dose olanzapine, 2.5 mg at night, was added. Mr. M reported better temper control, but his low mood, irritability, hopelessness, and amotivation were not significantly improved.
Mr. M left his job at a software company because he had difficulty completing tasks as the result of mood and cognitive changes. He wants to return to work, but feels that he would be unable to complete his job duties.
He begins a trial of bupropion, 150 mg/d, to improve the vegetative component of his mood symptoms to help him return to work. Mr. M now complains of worsening chorea, irritability, and insomnia, with continued difficulty completing tasks. He is intermittently tearful throughout the interview.
Mr. M continues to struggle with mood symptoms that likely are related to the stressful experience of declining function and the intrinsic evolution of HD. His chorea worsens on bupropion; this agent is discontinued and replaced with mirtazapine, 15 mg at night, for his depressive symptoms and insomnia. Citalopram and olanzapine are unchanged. Mr. M is advised to follow up with our HD psychiatry team in 1 month, and is referred for brief psychotherapy. We remind him—as we do for all of our HD patients—to call the HD clinic or 911 if he becomes suicidal. Ongoing treatment efforts likely will be complex, given the multifaceted and progressive nature of his disease.
Psychiatric sequelae
In general, psychiatric symptoms of HD become increasingly prevalent over time (Box 2).3,5 In a 2001 study of 52 HD patients by Paulsen et al,2 51 patients had ≥1 psychiatric symptom, such as dysphoria (69.2%), agitation (67.3%), irritability (65.4%), apathy (55.8%), and anxiety (51.9%); delusions (11.5%) and hallucinations (1.9%) were less prevalent.2 Similarly, Thompson et al3 followed 111 HD patients for ≥3 years and all experienced psychiatric symptoms.
According to Thompson et al,3 the presence and severity of apathy, irritability, and depression trend differently across the course of Huntington’s disease (HD). Apathy worsens with disease progression, closely following cognitive and motor symptoms. Irritability increases significantly, but this effect seems confined to early stages of HD. Depressive symptoms appear to decline slightly as HD advances, although it is unclear if this is because of antidepressants’ effects, increasing emotional blunting, and waning insight in later stages of HD, or another unknown factor.3 This study did not examine psychotic symptoms over time because few patients were experiencing delusions or hallucinations.
Similar to Thompson et al, Naarding et al5 found that apathy and depression in HD follow distinct time courses. Depression is a feature of early HD and apathy worsens with overall disease progression.
Depressed mood and functional ability—not cognitive or motor symptoms6—are the 2 most critical factors linked to health-related quality of life in HD. Hamilton et al7 found that apathy or executive dysfunction in HD patients is strongly related to decline in ability to complete activities of daily living, and may be severely debilitating.
Apathy. Often mistaken for a symptom of depression, apathy’s presentation may resemble anhedonia or fatigue; however, research suggests that depression and apathy are distinct conditions. Naarding et al
5 noted that apathy is more common than depressive symptoms in HD patients and may be a hallmark symptom of HD.
Depression affects most HD patients, and often is most severe early in the disease course. Hubers et al8 found that 20% of 100 HD patients had suicidal ideation. The strongest predictor was depressed mood.
Sleep disturbances and daytime somnolence are common among HD patients, and patients with comorbid depression report more disturbed sleep. Managing disturbed sleep and daytime somnolence in HD, with emphasis on comorbid depression, may improve the quality of life of patients and their caregivers.9
Anxiety was present in >50% of HD patients in a study by Paulsen et al2 and 37% evaluated by Craufurd et al.10 Craufurd et al10 also reported that 61% of patients were “physically tense and unable to relax.”
Among HD patients, 5% report obsessions and 10% report compulsive behaviors; these symptoms appear to become increasingly common as HD progresses.4,10
Impulsivity and disinhibition. Craufurd et al10 found that 71% of HD patients experienced poor judgment and self-monitoring, 40% had poor temper control and verbal outbursts, 22% exhibited threatening behavior or violence, and 6% had disinhibited or inappropriate sexual behavior.10
Recent studies have shown higher rates of disinhibition in “presymptomatic” gene-positive subjects vs gene-negative controls, suggesting that these symptoms may arise early in HD.11 Further, researchers demonstrated that patients lack symptom awareness and rate themselves as less impaired than their caregivers do.11
In our clinical experience, impulsivity frequently is encountered and creates significant conflict between patients and their caregivers. We speculate that when coupled with depressive symptoms of HD, impulsivity and disinhibition may play an important role in the high rates of suicidality seen in these patients.
Psychosis. Delusions and hallucinations are less common in HD than other psychiatric symptoms. Craufurd et al10 reported 3% of HD patients had delusions, 3% had auditory hallucinations, 2% had tactile hallucinations, and no patients had visual hallucinations.
A few case reports and a small study by Tsuang et al12 suggested that psychotic features in HD may be similar to those seen in paranoid schizophrenia. Tsuang et al12 also noted that more severe HD-related psychosis tends to cluster in families, which suggests that susceptibility to HD psychosis may be heritable.
Treating psychiatric symptoms
High-quality randomized controlled trials of pharmacotherapies for psychiatric symptoms in HD patients are lacking. Decisions regarding which agents to use often are based on case reports or clinical experience. The suggestions below are based on available evidence and our clinical experience.
Depression. Depressive symptoms in HD seem to respond to conventional pharmacologic treatments for major depressive disorder (MDD). A small trial of venlafaxine extended-release (XR) in 26 HD patients with MDD showed statistically significant improvements in depressive symptoms; however, this trial was not blinded and did not have a placebo group.13 In addition, 1 in 5 patients developed significant side effects—nausea, irritability, or worsening chorea.13
Evidence for selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors, and tricyclic antidepressants (TCAs) is lacking. Antidepressant choice should be based on patient response, side effect profile, and the need for secondary therapeutic effects.14
We often prescribe sertraline, citalopram, or escitalopram for our HD patients because of the relative absence of drug-drug interactions and favorable safety profile in medically and surgically ill patients. However, it’s important to tailor the treatment approach to your patient’s needs—eg, patients prone to forgetting their medicine may benefit from a drug with a longer half-life, such as fluoxetine. We avoid TCAs because of their anticholinergic effects, which may worsen dementia symptoms. Because HD patients have high rates of suicidality, agents that are highly toxic when taken in overdose should be used with caution.
One small study of HD patients with MDD or bipolar disorder showed clinical improvement in depressive symptoms after electroconvulsive therapy (ECT).15 Patients who suffered from comorbid delusions had the best improvements in mood.15 ECT likely is a good choice for HD patients who have failed several antidepressants, are suicidal, or who have depression with psychotic features.16
Apathy. A 2011 review concluded that no evidence-based recommendations regarding pharmacologic treatment for apathy in HD can be made because of lack of research.7 The Huntington’s Disease Society of America’s (HDSA) A Physician’s Guide to Managing Huntington’s Disease includes recommendations for treating apathy based on clinical experience.16 It suggests a nonsedating SSRI, followed by a trial of methylphenidate, pemoline, or dextroamphetamine if SSRIs were unsuccessful.
16 The HDSA guide notes psychostimulants may worsen irritability in HD and have a high potential for abuse. ECT appears to have little effect on apathy.15
Anxiety. A small, open-label study of 11 patients found that olanzapine, 5 mg/d, significantly improved depression, anxiety, irritability, and obsessive behavior in HD patients.17
The HDSA guide suggests treating anxiety and obsessive-compulsive symptoms as you would in patients without HD. For anxiety, SSRIs and possibly a short-term trial of a low-dose benzodiazepine (ie, lorazepam, clonazepam) are suggested.16 Benzodiazepines may increase the risk of falls and delirium in this population. Anecdotally, buspirone is helpful in some patients, with a starting dose of 5 mg 2 to 3 times per day and increased to 20 to 30 mg/d in divided doses.16 For obsessive-compulsive symptoms, SSRIs are recommended; atypical antipsychotics are reserved for severe or refractory symptoms.16
Disinhibition and impulsivity. There’s no research on treating disinhibition and impulsivity in HD. In our clinical experience, atypical antipsychotics are the most helpful. Factors regarding choosing an agent and dosing levels are similar to those for psychotic symptoms.
Psychotic symptoms. Most studies of typical and atypical antipsychotics for HD psychosis have shown beneficial effects.14,16-21 Neurologists frequently use these agents for managing chorea. Both neurologic and psychiatric features of the patient’s presentation must be considered when selecting a drug because treatment directed at 1 component of the disease may inadvertently exacerbate another. Specifically, higher potency antipsychotics (eg, haloperidol) are effective for chorea but can dramatically worsen bradykinesia; lower potency agents (eg, quetiapine) are less helpful for chorea but do not significantly worsen rigidity symptoms.
Olanzapine has been shown to improve chorea, anxiety, irritability, depression, sleep dysfunction, and weight loss in addition to psychotic symptoms.14,17 We find that olanzapine treats a constellation of symptoms common among HD patients, and we prescribe it frequently. Because olanzapine is considered a mid-potency agent, we find it’s best suited for concurrent control of psychotic symptoms and mild to moderate chorea in patients with minimal bradykinesia. Start olanzapine at 2.5 mg/d and gradually increase to 5 to 10 mg/d as tolerated.14
Risperidone is effective for treating psychosis and chorea. It can be started at 0.5 to 1 mg/d, and gradually increased to 6 to 8 mg/d.14 The depot formulation of risperidone has been shown to be effective in HD, which may help patients adhere to their medication.18 Risperidone is a mid-high potency antipsychotic, and in our experience is best used to control psychotic symptoms in patients with moderate chorea and few or no symptoms of bradykinesia or rigidity.
Quetiapine reduces psychotic symptoms, agitation, irritability, and insomnia without worsening bradykinesia or rigidity,19 but it is not beneficial for chorea. It can be started at 12.5 mg/d and gradually increased for effect as tolerated, up to 600 mg/d (depending on indication), in 2 or 3 divided doses.14
Haloperidol is a high-potency typical antipsychotic and may help psychotic patients with severe chorea; it should not be used in patients with bradykinesia. Start haloperidol at 0.5 to 1 mg/d and gradually increase to 6 to 8 mg/d as tolerated.14 Because of higher likelihood of side effects with typical antipsychotics, we often reserve its use for patients whose psychosis does not respond to atypical agents.
Other antipsychotics. Aripiprazole in HD has been examined in only 2 single- patient case reports20,21; the drug appeared to reduce psychosis and possibly chorea. Clozapine’s effectiveness for HD psychosis is not well known. It does not appear to be helpful for chorea and can cause agranulocytosis.22
Because one of the hallmarks of HD is dementia, it is worth noting that the FDA has issued a “black-box” warning on the use of antipsychotic drugs in patients with dementia because of concerns regarding increased mortality. However, drawing specific conclusions is difficult because the FDA warning is based on studies that looked primarily at Alzheimer’s disease and vascular dementia, not HD.
Other pharmacotherapies
Tetrabenazine is the only FDA-approved drug for treating HD. However, it carries a “black-box” warning for increased risk of depression and suicidal ideation and is contraindicated in suicidal patients and those with untreated or inadequately treated depression.
Although several small trials have had conflicting results regarding its benefit, amantadine sometimes is used to treat chorea.23-25 For more information about tetrabenazine and amantadine, see Box 3.
Tetrabenazine, the only FDA-approved drug for treating Huntington’s disease (HD), is a dopamine-depleting agent given to control chorea. In a 12-week, randomized, double-blind, placebo-controlled clinical trial, tetrabenazine was shown to be effective in HD patients.a Treatment with tetrabenazine results in symptomatic improvement of chorea, but does not slow or alter the course of the disease. Tetrabenazine can provide relief from choreiform movements, but these benefits should be balanced with the risks of depression and suicidality.a Tetrabenazine is known to prolong QTc interval, and should be used with caution in combination with other drugs that have the potential to do the same (eg, antipsychotics).a
Several case reports have found an association between tetrabenazine and development of neuroleptic malignant syndrome (NMS).b-d Be aware of the clinical characteristics of NMS—mental status change, rigidity, fever, and dysautonomia—and use caution when starting patients taking tetrabenazine on antipsychotics or other agents known to cause NMS.
Amantadine also has been used to treat chorea in HD patients who are unable to tolerate tetrabenazine or antipsychotics. Our neurologists sometimes have found it to be beneficial in patients with juvenile-onset HD because these patients often have debilitating dystonia. Be aware that amantadine is known to precipitate or worsen psychosis.e
References
- Food and Drug Administration. NDA 21-894 Xenazine® (tetrabenazine). Risk evaluation and mitigation strategy (REMS). Click here. Published August 15, 2008. Updated April 2011. Accessed June 20, 2012.
- Stevens E, Roman A, Houa M, et al. Severe hyperthermia during tetrabenazine therapy for tardive dyskinesia. Intensive Care Med. 1998;24(4):369-371.
- Petzinger GM, Bressman SB. A case of tetrabenazine-induced neuroleptic malignant syndrome after prolonged treatment. Mov Disord. 1997;12(2):246-248.
- Ossemann M, Sindic CJ, Laterre C. Tetrabenazine as a cause of neuroleptic malignant syndrome. Mov Disord. 1996;11(1):95.
- Wolters EC. Dopaminomimetic psychosis in Parkinson’s disease patients: diagnosis and treatment. Neurology. 1999;52 (7 suppl 3):S10-S13.
Related Resources
- Huntington’s Disease Society of America. www.hdsa.org.
- Family Caregiver Alliance. Huntington’s disease. www.caregiver.org/caregiver/jsp/content_node.jsp?nodeid=574.
- Huntington Study Group. www.huntington-study-group.org.
- Huntington’s Disease Advocacy Center. www.hdac.org.
Drug Brand Names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Bupropion • Wellbutrin, Wellbutrin XL, others
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Dextroamphetamine • Dexedrine
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lorazepam • Ativan
- Methylphenidate • Concerta, Ritalin, others
- Mirtazapine • Remeron
- Olanzapine • Zyprexa
- Pemoline • Cylert
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tetrabenazine • Xenazine
- Venlafaxine XR • Effexor XR
Disclosures
Dr. Scher is a consultant to the advisory board for Lundbeck.
Ms. Kocsis reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Psychiatric symptoms are a common and debilitating manifestation of Huntington’s disease (HD), a progressive, inherited neurodegenerative disorder also characterized by chorea (involuntary, nonrepetitive movements) and cognitive decline. The prevalence of HD is 4 to 8 patients per 100,000 persons in most populations of European descent, with lower prevalence among non-Europeans.1 HD is caused by an abnormal expansion of a trinucleotide (CAG) repeat sequence on chromosome 4, and is inherited in an autosomal dominant fashion, meaning a HD patient’s child has a 50% chance of inheriting the mutation. The expansion is located in the gene that encodes the “huntingtin” protein, the normal function of which is not well understood.
There’s no cure for HD, and treatments primarily are directed at symptom control. Psychiatric symptoms include depression, apathy, anxiety, and psychosis (Table).2-4 Treating patients with HD can be challenging because most psychiatrists will see only a handful of patients with this multifaceted illness during their careers. See Box 1 for a case study of a patient with HD.
Table
Psychiatric symptoms of HD
| Anxiety |
| Apathy |
| Delusions |
| Disinhibitions, impulsivity, aggressive behavior |
| Dysphoria |
| Euphoria |
| Hallucinations |
| Irritability |
| Obsessions and compulsions |
| HD: Huntington’s disease Source: References 2-4 |
Mr. M, age 50, was diagnosed with Huntington’s disease (HD) 1 year ago. He returns to our psychiatric clinic for treatment of depressive symptoms and temper. Previously, he was prescribed citalopram, 40 mg/d; eventually low-dose olanzapine, 2.5 mg at night, was added. Mr. M reported better temper control, but his low mood, irritability, hopelessness, and amotivation were not significantly improved.
Mr. M left his job at a software company because he had difficulty completing tasks as the result of mood and cognitive changes. He wants to return to work, but feels that he would be unable to complete his job duties.
He begins a trial of bupropion, 150 mg/d, to improve the vegetative component of his mood symptoms to help him return to work. Mr. M now complains of worsening chorea, irritability, and insomnia, with continued difficulty completing tasks. He is intermittently tearful throughout the interview.
Mr. M continues to struggle with mood symptoms that likely are related to the stressful experience of declining function and the intrinsic evolution of HD. His chorea worsens on bupropion; this agent is discontinued and replaced with mirtazapine, 15 mg at night, for his depressive symptoms and insomnia. Citalopram and olanzapine are unchanged. Mr. M is advised to follow up with our HD psychiatry team in 1 month, and is referred for brief psychotherapy. We remind him—as we do for all of our HD patients—to call the HD clinic or 911 if he becomes suicidal. Ongoing treatment efforts likely will be complex, given the multifaceted and progressive nature of his disease.
Psychiatric sequelae
In general, psychiatric symptoms of HD become increasingly prevalent over time (Box 2).3,5 In a 2001 study of 52 HD patients by Paulsen et al,2 51 patients had ≥1 psychiatric symptom, such as dysphoria (69.2%), agitation (67.3%), irritability (65.4%), apathy (55.8%), and anxiety (51.9%); delusions (11.5%) and hallucinations (1.9%) were less prevalent.2 Similarly, Thompson et al3 followed 111 HD patients for ≥3 years and all experienced psychiatric symptoms.
According to Thompson et al,3 the presence and severity of apathy, irritability, and depression trend differently across the course of Huntington’s disease (HD). Apathy worsens with disease progression, closely following cognitive and motor symptoms. Irritability increases significantly, but this effect seems confined to early stages of HD. Depressive symptoms appear to decline slightly as HD advances, although it is unclear if this is because of antidepressants’ effects, increasing emotional blunting, and waning insight in later stages of HD, or another unknown factor.3 This study did not examine psychotic symptoms over time because few patients were experiencing delusions or hallucinations.
Similar to Thompson et al, Naarding et al5 found that apathy and depression in HD follow distinct time courses. Depression is a feature of early HD and apathy worsens with overall disease progression.
Depressed mood and functional ability—not cognitive or motor symptoms6—are the 2 most critical factors linked to health-related quality of life in HD. Hamilton et al7 found that apathy or executive dysfunction in HD patients is strongly related to decline in ability to complete activities of daily living, and may be severely debilitating.
Apathy. Often mistaken for a symptom of depression, apathy’s presentation may resemble anhedonia or fatigue; however, research suggests that depression and apathy are distinct conditions. Naarding et al
5 noted that apathy is more common than depressive symptoms in HD patients and may be a hallmark symptom of HD.
Depression affects most HD patients, and often is most severe early in the disease course. Hubers et al8 found that 20% of 100 HD patients had suicidal ideation. The strongest predictor was depressed mood.
Sleep disturbances and daytime somnolence are common among HD patients, and patients with comorbid depression report more disturbed sleep. Managing disturbed sleep and daytime somnolence in HD, with emphasis on comorbid depression, may improve the quality of life of patients and their caregivers.9
Anxiety was present in >50% of HD patients in a study by Paulsen et al2 and 37% evaluated by Craufurd et al.10 Craufurd et al10 also reported that 61% of patients were “physically tense and unable to relax.”
Among HD patients, 5% report obsessions and 10% report compulsive behaviors; these symptoms appear to become increasingly common as HD progresses.4,10
Impulsivity and disinhibition. Craufurd et al10 found that 71% of HD patients experienced poor judgment and self-monitoring, 40% had poor temper control and verbal outbursts, 22% exhibited threatening behavior or violence, and 6% had disinhibited or inappropriate sexual behavior.10
Recent studies have shown higher rates of disinhibition in “presymptomatic” gene-positive subjects vs gene-negative controls, suggesting that these symptoms may arise early in HD.11 Further, researchers demonstrated that patients lack symptom awareness and rate themselves as less impaired than their caregivers do.11
In our clinical experience, impulsivity frequently is encountered and creates significant conflict between patients and their caregivers. We speculate that when coupled with depressive symptoms of HD, impulsivity and disinhibition may play an important role in the high rates of suicidality seen in these patients.
Psychosis. Delusions and hallucinations are less common in HD than other psychiatric symptoms. Craufurd et al10 reported 3% of HD patients had delusions, 3% had auditory hallucinations, 2% had tactile hallucinations, and no patients had visual hallucinations.
A few case reports and a small study by Tsuang et al12 suggested that psychotic features in HD may be similar to those seen in paranoid schizophrenia. Tsuang et al12 also noted that more severe HD-related psychosis tends to cluster in families, which suggests that susceptibility to HD psychosis may be heritable.
Treating psychiatric symptoms
High-quality randomized controlled trials of pharmacotherapies for psychiatric symptoms in HD patients are lacking. Decisions regarding which agents to use often are based on case reports or clinical experience. The suggestions below are based on available evidence and our clinical experience.
Depression. Depressive symptoms in HD seem to respond to conventional pharmacologic treatments for major depressive disorder (MDD). A small trial of venlafaxine extended-release (XR) in 26 HD patients with MDD showed statistically significant improvements in depressive symptoms; however, this trial was not blinded and did not have a placebo group.13 In addition, 1 in 5 patients developed significant side effects—nausea, irritability, or worsening chorea.13
Evidence for selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors, and tricyclic antidepressants (TCAs) is lacking. Antidepressant choice should be based on patient response, side effect profile, and the need for secondary therapeutic effects.14
We often prescribe sertraline, citalopram, or escitalopram for our HD patients because of the relative absence of drug-drug interactions and favorable safety profile in medically and surgically ill patients. However, it’s important to tailor the treatment approach to your patient’s needs—eg, patients prone to forgetting their medicine may benefit from a drug with a longer half-life, such as fluoxetine. We avoid TCAs because of their anticholinergic effects, which may worsen dementia symptoms. Because HD patients have high rates of suicidality, agents that are highly toxic when taken in overdose should be used with caution.
One small study of HD patients with MDD or bipolar disorder showed clinical improvement in depressive symptoms after electroconvulsive therapy (ECT).15 Patients who suffered from comorbid delusions had the best improvements in mood.15 ECT likely is a good choice for HD patients who have failed several antidepressants, are suicidal, or who have depression with psychotic features.16
Apathy. A 2011 review concluded that no evidence-based recommendations regarding pharmacologic treatment for apathy in HD can be made because of lack of research.7 The Huntington’s Disease Society of America’s (HDSA) A Physician’s Guide to Managing Huntington’s Disease includes recommendations for treating apathy based on clinical experience.16 It suggests a nonsedating SSRI, followed by a trial of methylphenidate, pemoline, or dextroamphetamine if SSRIs were unsuccessful.
16 The HDSA guide notes psychostimulants may worsen irritability in HD and have a high potential for abuse. ECT appears to have little effect on apathy.15
Anxiety. A small, open-label study of 11 patients found that olanzapine, 5 mg/d, significantly improved depression, anxiety, irritability, and obsessive behavior in HD patients.17
The HDSA guide suggests treating anxiety and obsessive-compulsive symptoms as you would in patients without HD. For anxiety, SSRIs and possibly a short-term trial of a low-dose benzodiazepine (ie, lorazepam, clonazepam) are suggested.16 Benzodiazepines may increase the risk of falls and delirium in this population. Anecdotally, buspirone is helpful in some patients, with a starting dose of 5 mg 2 to 3 times per day and increased to 20 to 30 mg/d in divided doses.16 For obsessive-compulsive symptoms, SSRIs are recommended; atypical antipsychotics are reserved for severe or refractory symptoms.16
Disinhibition and impulsivity. There’s no research on treating disinhibition and impulsivity in HD. In our clinical experience, atypical antipsychotics are the most helpful. Factors regarding choosing an agent and dosing levels are similar to those for psychotic symptoms.
Psychotic symptoms. Most studies of typical and atypical antipsychotics for HD psychosis have shown beneficial effects.14,16-21 Neurologists frequently use these agents for managing chorea. Both neurologic and psychiatric features of the patient’s presentation must be considered when selecting a drug because treatment directed at 1 component of the disease may inadvertently exacerbate another. Specifically, higher potency antipsychotics (eg, haloperidol) are effective for chorea but can dramatically worsen bradykinesia; lower potency agents (eg, quetiapine) are less helpful for chorea but do not significantly worsen rigidity symptoms.
Olanzapine has been shown to improve chorea, anxiety, irritability, depression, sleep dysfunction, and weight loss in addition to psychotic symptoms.14,17 We find that olanzapine treats a constellation of symptoms common among HD patients, and we prescribe it frequently. Because olanzapine is considered a mid-potency agent, we find it’s best suited for concurrent control of psychotic symptoms and mild to moderate chorea in patients with minimal bradykinesia. Start olanzapine at 2.5 mg/d and gradually increase to 5 to 10 mg/d as tolerated.14
Risperidone is effective for treating psychosis and chorea. It can be started at 0.5 to 1 mg/d, and gradually increased to 6 to 8 mg/d.14 The depot formulation of risperidone has been shown to be effective in HD, which may help patients adhere to their medication.18 Risperidone is a mid-high potency antipsychotic, and in our experience is best used to control psychotic symptoms in patients with moderate chorea and few or no symptoms of bradykinesia or rigidity.
Quetiapine reduces psychotic symptoms, agitation, irritability, and insomnia without worsening bradykinesia or rigidity,19 but it is not beneficial for chorea. It can be started at 12.5 mg/d and gradually increased for effect as tolerated, up to 600 mg/d (depending on indication), in 2 or 3 divided doses.14
Haloperidol is a high-potency typical antipsychotic and may help psychotic patients with severe chorea; it should not be used in patients with bradykinesia. Start haloperidol at 0.5 to 1 mg/d and gradually increase to 6 to 8 mg/d as tolerated.14 Because of higher likelihood of side effects with typical antipsychotics, we often reserve its use for patients whose psychosis does not respond to atypical agents.
Other antipsychotics. Aripiprazole in HD has been examined in only 2 single- patient case reports20,21; the drug appeared to reduce psychosis and possibly chorea. Clozapine’s effectiveness for HD psychosis is not well known. It does not appear to be helpful for chorea and can cause agranulocytosis.22
Because one of the hallmarks of HD is dementia, it is worth noting that the FDA has issued a “black-box” warning on the use of antipsychotic drugs in patients with dementia because of concerns regarding increased mortality. However, drawing specific conclusions is difficult because the FDA warning is based on studies that looked primarily at Alzheimer’s disease and vascular dementia, not HD.
Other pharmacotherapies
Tetrabenazine is the only FDA-approved drug for treating HD. However, it carries a “black-box” warning for increased risk of depression and suicidal ideation and is contraindicated in suicidal patients and those with untreated or inadequately treated depression.
Although several small trials have had conflicting results regarding its benefit, amantadine sometimes is used to treat chorea.23-25 For more information about tetrabenazine and amantadine, see Box 3.
Tetrabenazine, the only FDA-approved drug for treating Huntington’s disease (HD), is a dopamine-depleting agent given to control chorea. In a 12-week, randomized, double-blind, placebo-controlled clinical trial, tetrabenazine was shown to be effective in HD patients.a Treatment with tetrabenazine results in symptomatic improvement of chorea, but does not slow or alter the course of the disease. Tetrabenazine can provide relief from choreiform movements, but these benefits should be balanced with the risks of depression and suicidality.a Tetrabenazine is known to prolong QTc interval, and should be used with caution in combination with other drugs that have the potential to do the same (eg, antipsychotics).a
Several case reports have found an association between tetrabenazine and development of neuroleptic malignant syndrome (NMS).b-d Be aware of the clinical characteristics of NMS—mental status change, rigidity, fever, and dysautonomia—and use caution when starting patients taking tetrabenazine on antipsychotics or other agents known to cause NMS.
Amantadine also has been used to treat chorea in HD patients who are unable to tolerate tetrabenazine or antipsychotics. Our neurologists sometimes have found it to be beneficial in patients with juvenile-onset HD because these patients often have debilitating dystonia. Be aware that amantadine is known to precipitate or worsen psychosis.e
References
- Food and Drug Administration. NDA 21-894 Xenazine® (tetrabenazine). Risk evaluation and mitigation strategy (REMS). Click here. Published August 15, 2008. Updated April 2011. Accessed June 20, 2012.
- Stevens E, Roman A, Houa M, et al. Severe hyperthermia during tetrabenazine therapy for tardive dyskinesia. Intensive Care Med. 1998;24(4):369-371.
- Petzinger GM, Bressman SB. A case of tetrabenazine-induced neuroleptic malignant syndrome after prolonged treatment. Mov Disord. 1997;12(2):246-248.
- Ossemann M, Sindic CJ, Laterre C. Tetrabenazine as a cause of neuroleptic malignant syndrome. Mov Disord. 1996;11(1):95.
- Wolters EC. Dopaminomimetic psychosis in Parkinson’s disease patients: diagnosis and treatment. Neurology. 1999;52 (7 suppl 3):S10-S13.
Related Resources
- Huntington’s Disease Society of America. www.hdsa.org.
- Family Caregiver Alliance. Huntington’s disease. www.caregiver.org/caregiver/jsp/content_node.jsp?nodeid=574.
- Huntington Study Group. www.huntington-study-group.org.
- Huntington’s Disease Advocacy Center. www.hdac.org.
Drug Brand Names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Bupropion • Wellbutrin, Wellbutrin XL, others
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Dextroamphetamine • Dexedrine
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lorazepam • Ativan
- Methylphenidate • Concerta, Ritalin, others
- Mirtazapine • Remeron
- Olanzapine • Zyprexa
- Pemoline • Cylert
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tetrabenazine • Xenazine
- Venlafaxine XR • Effexor XR
Disclosures
Dr. Scher is a consultant to the advisory board for Lundbeck.
Ms. Kocsis reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Harper PS. The epidemiology of Huntington’s disease. Hum Genet. 1992;89(4):365-376.
2. Paulsen JS, Ready RE, Hamilton JM, et al. Neuropsychiatric aspects of Huntington’s disease. J Neurol Neurosurg Psychiatry. 2001;71(3):310-314.
3. Thompson JC, Harris J, Sollom AC, et al. Longitudinal evaluation of neuropsychiatric symptoms in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2012;24(1):53-60.
4. Beglinger LJ, Langbehn DR, Duff K, et al. Probability of obsessive and compulsive symptoms in Huntington’s disease. Biol Psychiatry. 2007;61(3):415-418.
5. Naarding P, Janzing JG, Eling P, et al. Apathy is not depression in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2009;21(3):266-270.
6. Ho AK, Gilbert AS, Mason SL, et al. Health-related quality of life in Huntington’s disease: which factors matter most? Mov Disord. 2009;24(4):574-578.
7. Hamilton JM, Salmon DP, Corey-Bloom J, et al. Behavioural abnormalities contribute to functional decline in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2003;74(1):120-122.
8. Hubers AA, Reedeker N, Giltay EJ, et al. Suicidality in Huntington’s disease. J Affect Disord. 2012;136(3):550-557.
9. Videnovic A, Leurgans S, Fan W, et al. Daytime somnolence and nocturnal sleep disturbances in Huntington disease. Parkinsonism Relat Disord. 2009;15(6):471-474.
10. Craufurd D, Thompson JC, Snowden JS. Behavioral changes in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14(4):219-226.
11. Duff K, Paulsen JS, Beglinger LJ, et al. “Frontal” behaviors before the diagnosis of Huntington’s disease and their relationship to markers of disease progression: evidence of early lack of awareness. J Neuropsychiatry Clin Neurosci. 2010;22(2):196-207.
12. Tsuang D, Almqvist EW, Lipe H, et al. Familial aggregation of psychotic symptoms in Huntington’s disease. Am J Psychiatry. 2000;157(12):1955-1959.
13. Holl AK, Wilkinson L, Painold A, et al. Combating depression in Huntington’s disease: effective antidepressive treatment with venlafaxine XR. Int Clin Psychopharmacol. 2010;25(1):46-50.
14. Killoran A, Biglan KM. Therapeutics in Huntington’s disease. Curr Treat Options Neurol. 2012;14(2):137-149.
15. Ranen NG, Peyser CE, Folstein SE. ECT as a treatment for depression in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 1994;6(2):154-159.
16. Rosenblatt A, Ranen NG, Nance MA, et al. A physician’s guide to the management of Huntington’s disease. 2nd edition. http://www.hdsa.org/images/content/1/1/11289.pdf. Published 1999. Accessed July 27, 2012.
17. Squitieri F, Cannella M, Piorcellini A, et al. Short-term effects of olanzapine in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14(1):69-72.
18. Johnston TG. Risperidone long-acting injection and Huntington’s disease: case series with significant psychiatric and behavioural symptoms. Int Clin Psychopharmacol. 2011;26(2):114-119.
19. Alpay M, Koroshetz WJ. Quetiapine in the treatment of behavioral disturbances in patients with Huntington’s disease. Psychosomatics. 2006;47(1):70-72.
20. Lin WC, Chou YH. Aripiprazole effects on psychosis and chorea in a patient with Huntington’s disease. Am J Psychiatry. 2008;165(9):1207-1208.
21. Oulis P, Mourikis I, Konstantakopoulos G, et al. Aripiprazole in the treatment of olanzapine-resistant psychotic and motor symptoms of Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2010;22(3):352c.e4-352c.e5.
22. van Vugt JP, Siesling S, Vergeer M, et al. Clozapine versus placebo in Huntington’s disease: a double blind randomised comparative study. J Neurol Neurosurg Psychiatry. 1997;63(1):35-39.
23. Verhagen Metman L, Morris MJ, Farmer C, et al. Huntington’s disease: a randomized, controlled trial using the NMDA-antagonist amantadine. Neurology. 2002;59(5):694-699.
24. Lucetti C, Del Dotto P, Gambaccini G, et al. IV amantadine improves chorea in Huntington’s disease: an acute randomized, controlled study. Neurology. 2003;60(12):1995-1997.
25. O’Suilleabhain P, Dewey RB, Jr. A randomized trial of amantadine in Huntington disease. Arch Neurol. 2003;60(7):996-998.
1. Harper PS. The epidemiology of Huntington’s disease. Hum Genet. 1992;89(4):365-376.
2. Paulsen JS, Ready RE, Hamilton JM, et al. Neuropsychiatric aspects of Huntington’s disease. J Neurol Neurosurg Psychiatry. 2001;71(3):310-314.
3. Thompson JC, Harris J, Sollom AC, et al. Longitudinal evaluation of neuropsychiatric symptoms in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2012;24(1):53-60.
4. Beglinger LJ, Langbehn DR, Duff K, et al. Probability of obsessive and compulsive symptoms in Huntington’s disease. Biol Psychiatry. 2007;61(3):415-418.
5. Naarding P, Janzing JG, Eling P, et al. Apathy is not depression in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2009;21(3):266-270.
6. Ho AK, Gilbert AS, Mason SL, et al. Health-related quality of life in Huntington’s disease: which factors matter most? Mov Disord. 2009;24(4):574-578.
7. Hamilton JM, Salmon DP, Corey-Bloom J, et al. Behavioural abnormalities contribute to functional decline in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2003;74(1):120-122.
8. Hubers AA, Reedeker N, Giltay EJ, et al. Suicidality in Huntington’s disease. J Affect Disord. 2012;136(3):550-557.
9. Videnovic A, Leurgans S, Fan W, et al. Daytime somnolence and nocturnal sleep disturbances in Huntington disease. Parkinsonism Relat Disord. 2009;15(6):471-474.
10. Craufurd D, Thompson JC, Snowden JS. Behavioral changes in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14(4):219-226.
11. Duff K, Paulsen JS, Beglinger LJ, et al. “Frontal” behaviors before the diagnosis of Huntington’s disease and their relationship to markers of disease progression: evidence of early lack of awareness. J Neuropsychiatry Clin Neurosci. 2010;22(2):196-207.
12. Tsuang D, Almqvist EW, Lipe H, et al. Familial aggregation of psychotic symptoms in Huntington’s disease. Am J Psychiatry. 2000;157(12):1955-1959.
13. Holl AK, Wilkinson L, Painold A, et al. Combating depression in Huntington’s disease: effective antidepressive treatment with venlafaxine XR. Int Clin Psychopharmacol. 2010;25(1):46-50.
14. Killoran A, Biglan KM. Therapeutics in Huntington’s disease. Curr Treat Options Neurol. 2012;14(2):137-149.
15. Ranen NG, Peyser CE, Folstein SE. ECT as a treatment for depression in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 1994;6(2):154-159.
16. Rosenblatt A, Ranen NG, Nance MA, et al. A physician’s guide to the management of Huntington’s disease. 2nd edition. http://www.hdsa.org/images/content/1/1/11289.pdf. Published 1999. Accessed July 27, 2012.
17. Squitieri F, Cannella M, Piorcellini A, et al. Short-term effects of olanzapine in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14(1):69-72.
18. Johnston TG. Risperidone long-acting injection and Huntington’s disease: case series with significant psychiatric and behavioural symptoms. Int Clin Psychopharmacol. 2011;26(2):114-119.
19. Alpay M, Koroshetz WJ. Quetiapine in the treatment of behavioral disturbances in patients with Huntington’s disease. Psychosomatics. 2006;47(1):70-72.
20. Lin WC, Chou YH. Aripiprazole effects on psychosis and chorea in a patient with Huntington’s disease. Am J Psychiatry. 2008;165(9):1207-1208.
21. Oulis P, Mourikis I, Konstantakopoulos G, et al. Aripiprazole in the treatment of olanzapine-resistant psychotic and motor symptoms of Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2010;22(3):352c.e4-352c.e5.
22. van Vugt JP, Siesling S, Vergeer M, et al. Clozapine versus placebo in Huntington’s disease: a double blind randomised comparative study. J Neurol Neurosurg Psychiatry. 1997;63(1):35-39.
23. Verhagen Metman L, Morris MJ, Farmer C, et al. Huntington’s disease: a randomized, controlled trial using the NMDA-antagonist amantadine. Neurology. 2002;59(5):694-699.
24. Lucetti C, Del Dotto P, Gambaccini G, et al. IV amantadine improves chorea in Huntington’s disease: an acute randomized, controlled study. Neurology. 2003;60(12):1995-1997.
25. O’Suilleabhain P, Dewey RB, Jr. A randomized trial of amantadine in Huntington disease. Arch Neurol. 2003;60(7):996-998.