User login
National Groups Promote Efficiency Agenda
A number of national organizations are helping hospitals and hospitalists get a better handle on their efficiency. One such group, a Charlotte, N.C.-based performance improvement alliance of 2,600 hospitals called Premier, recently completed the third year of its ongoing collaborative, QUEST (QUality, Efficiency, Safety, and Transparency). Three-year results found 157 charter-member hospitals saving an estimated 25,000 patient lives (based on 29% lower mortality rates than risk-adjusted national averages) and $4.5 billion in costs, compared with hospitals not participating in the initiative.
The high-performing hospitals in the collaborative use an efficiency dashboard to pinpoint and quantify saving opportunities, says Richard Bankowitz, MD, MBA, FACP, an internist and medical information specialist who serves as Premier’s enterprisewide chief medical officer. Collaborators participate in education and training, consultation, conference calls, a national meeting, and an online performance-improvement portal, with a commitment to transparently share their data and a focus on quality in areas of mortality rates, harm avoidance, readmissions, costs, and patient-reported experience.
“We’ve shown quite a lot of improvement,” Dr. Bankowitz says. “We’ve been able to look at hospitals that appear to have excellent readmissions rates or nursing strategies, and then try to figure out their secrets.”
Even the best-performing hospitals have opportunities to pinpoint and eliminate inefficiency. “But we need to be more than efficient,” he adds. “We also need to be effective. Having perfect efficiency in providing unnecessary procedures doesn’t do anybody any good.”
Numerous tools and methods are freely available, he says, but he also encourages hospitalist groups to stay focused on what provides value and will impact efficiency in hospitals.
“Look for processes of care that bring value, versus waste,” he says. “Have we ever stepped back and thought about the way we provide care as a whole—from end to end? Really look at the utilization—of tests, of consultations, of pharmaceuticals—and consider all of the inputs. Are they really adding value? Do you know which patients account for the most costs?”
He also encourages hospitalists to pull together interdisciplinary quality teams and focus on the patients who are more frequently admitted or problematic and costly, such as heart-failure patients. “Get the team to design a process of care that includes inpatient, outpatient, and the skilled nursing facility,” he says, adding there is potential for waste in transitions of care.
Hospitals are in an increasingly tough position, Dr. Bankowitz admits. “They’re no longer able to just cut their way out of financial problems. Hospitalists have an important role,” he notes. “They can take more of a systems view, seeing care processes from end to end.”
Larry Beresford is a freelance author in Oakland, Calif.
A number of national organizations are helping hospitals and hospitalists get a better handle on their efficiency. One such group, a Charlotte, N.C.-based performance improvement alliance of 2,600 hospitals called Premier, recently completed the third year of its ongoing collaborative, QUEST (QUality, Efficiency, Safety, and Transparency). Three-year results found 157 charter-member hospitals saving an estimated 25,000 patient lives (based on 29% lower mortality rates than risk-adjusted national averages) and $4.5 billion in costs, compared with hospitals not participating in the initiative.
The high-performing hospitals in the collaborative use an efficiency dashboard to pinpoint and quantify saving opportunities, says Richard Bankowitz, MD, MBA, FACP, an internist and medical information specialist who serves as Premier’s enterprisewide chief medical officer. Collaborators participate in education and training, consultation, conference calls, a national meeting, and an online performance-improvement portal, with a commitment to transparently share their data and a focus on quality in areas of mortality rates, harm avoidance, readmissions, costs, and patient-reported experience.
“We’ve shown quite a lot of improvement,” Dr. Bankowitz says. “We’ve been able to look at hospitals that appear to have excellent readmissions rates or nursing strategies, and then try to figure out their secrets.”
Even the best-performing hospitals have opportunities to pinpoint and eliminate inefficiency. “But we need to be more than efficient,” he adds. “We also need to be effective. Having perfect efficiency in providing unnecessary procedures doesn’t do anybody any good.”
Numerous tools and methods are freely available, he says, but he also encourages hospitalist groups to stay focused on what provides value and will impact efficiency in hospitals.
“Look for processes of care that bring value, versus waste,” he says. “Have we ever stepped back and thought about the way we provide care as a whole—from end to end? Really look at the utilization—of tests, of consultations, of pharmaceuticals—and consider all of the inputs. Are they really adding value? Do you know which patients account for the most costs?”
He also encourages hospitalists to pull together interdisciplinary quality teams and focus on the patients who are more frequently admitted or problematic and costly, such as heart-failure patients. “Get the team to design a process of care that includes inpatient, outpatient, and the skilled nursing facility,” he says, adding there is potential for waste in transitions of care.
Hospitals are in an increasingly tough position, Dr. Bankowitz admits. “They’re no longer able to just cut their way out of financial problems. Hospitalists have an important role,” he notes. “They can take more of a systems view, seeing care processes from end to end.”
Larry Beresford is a freelance author in Oakland, Calif.
A number of national organizations are helping hospitals and hospitalists get a better handle on their efficiency. One such group, a Charlotte, N.C.-based performance improvement alliance of 2,600 hospitals called Premier, recently completed the third year of its ongoing collaborative, QUEST (QUality, Efficiency, Safety, and Transparency). Three-year results found 157 charter-member hospitals saving an estimated 25,000 patient lives (based on 29% lower mortality rates than risk-adjusted national averages) and $4.5 billion in costs, compared with hospitals not participating in the initiative.
The high-performing hospitals in the collaborative use an efficiency dashboard to pinpoint and quantify saving opportunities, says Richard Bankowitz, MD, MBA, FACP, an internist and medical information specialist who serves as Premier’s enterprisewide chief medical officer. Collaborators participate in education and training, consultation, conference calls, a national meeting, and an online performance-improvement portal, with a commitment to transparently share their data and a focus on quality in areas of mortality rates, harm avoidance, readmissions, costs, and patient-reported experience.
“We’ve shown quite a lot of improvement,” Dr. Bankowitz says. “We’ve been able to look at hospitals that appear to have excellent readmissions rates or nursing strategies, and then try to figure out their secrets.”
Even the best-performing hospitals have opportunities to pinpoint and eliminate inefficiency. “But we need to be more than efficient,” he adds. “We also need to be effective. Having perfect efficiency in providing unnecessary procedures doesn’t do anybody any good.”
Numerous tools and methods are freely available, he says, but he also encourages hospitalist groups to stay focused on what provides value and will impact efficiency in hospitals.
“Look for processes of care that bring value, versus waste,” he says. “Have we ever stepped back and thought about the way we provide care as a whole—from end to end? Really look at the utilization—of tests, of consultations, of pharmaceuticals—and consider all of the inputs. Are they really adding value? Do you know which patients account for the most costs?”
He also encourages hospitalists to pull together interdisciplinary quality teams and focus on the patients who are more frequently admitted or problematic and costly, such as heart-failure patients. “Get the team to design a process of care that includes inpatient, outpatient, and the skilled nursing facility,” he says, adding there is potential for waste in transitions of care.
Hospitals are in an increasingly tough position, Dr. Bankowitz admits. “They’re no longer able to just cut their way out of financial problems. Hospitalists have an important role,” he notes. “They can take more of a systems view, seeing care processes from end to end.”
Larry Beresford is a freelance author in Oakland, Calif.
Remediation, Attrition Rates High in Surgery Residents
Almost one-third of general surgery residents required remediation over an 11-year period, most often because of a deficiency in medical knowledge, judging from findings in a retrospective study of remediation and attrition rates among general surgery residents at six academic surgical residency programs in California.
The high remediation rate identified in this study "begs the question of whether we are falling short in the education of surgical residents," said Dr. Arezou Yaghoubian of the department of surgery, Harbor-UCLA Medical Center, Los Angeles, and associates (Arch. Surg. 2012;147:829-33).
They conducted the study to determine which of the six Accreditation Council for Graduate Medical Education (ACGME) competencies (patient care, medical knowledge, practice-based learning, interpersonal and communication skills, professionalism, and system-based practice), most often require remediation and to identify predictors of remediation. There is a scarcity of data on how well surgical residency programs have been achieving these competencies, and this information may provide insight into how to modify the surgical curriculum more effectively in this new era of limited hours, they said.
In the study of 348 general surgery residents at the six training programs between 1999 and 2010, the most common reason for remediation was medical knowledge in 74%, followed by interpersonal and communication skills in 24%, patient care in 22%, professionalism in 18%, system-based practice in 14%, and practice-based learning in 8%.
Of the 107 residents who required remediation, 27 required remediation more than once. Almost 16% of the residents left their programs, but most (53 of 55 residents) left voluntarily. The other two failed remediation and had to leave the program.
Monthly meetings with faculty was the most common form of remediation, in 79%, followed by specific reading assignments (72%), required attendance at review courses and/or conferences (27%), evaluation by a therapist, psychologist, or psychiatrist (12%), and having to repeat a clinical year (6.5%).
More than half of the remediations were initiated during the first 2 postgraduate years (25% in the first and 35% in the second year), followed by 21% during the third year, 16% during the fourth year, and 4% during the fifth year.
A predictor of remediation was having received honors during the third-year surgery clerkship (58% of those who were subject to remediation vs. 45% of those who were not remediated, a statistically significant difference), which, the authors noted, was counterintuitive.
United States Medical Licensing Examination (USMLE) step 1 and/or step 2 scores and American Board of Surgery In-Training Examination (ABSITE) scores at postgraduate years 1 through 4 were also predictive of remediation. The median USMLE step 1 and step 2 scores were 225 and 223 among the residents subject to remediation, vs. 232 for step 1 and step 2 scores among those who were not remediated, statistically significant differences.
The ABSITE scores during postgraduate years 1 through 4 were significantly lower among those who were subject to remediation, but the differences in median scores in years 5 through 7 were not significantly different.
But remediation was not a predictor of attrition. The only predictor of attrition was the ABSITE score at the third postgraduate year, which was a median of 34 among those who left the program and 62 among those who stayed.
Possible explanations for the high remediation rate is that residents are not well prepared for the demands of a surgical residency, they need to be more efficient with their time because of the 80-hour work week, and they may not necessarily be spending their increased time outside of the hospital studying at home, the authors said. Possible reasons for the attrition rate among the residents, an "ongoing concern" in general surgery, may be marital, family, and personal issues and a need for a less stressful environment, they added.
Acknowledging the study's limitations, including the retrospective design and lack of information on how many residents passed the American Board of Surgery boards, the authors concluded that the high remediation rate "should give surgical educators pause as we should closely examine the potential sources of these deficiencies."
They called on surgical societies to "take the initiative to encourage the restructuring of medical school education, such that future surgeons are better prepared to enter surgical residencies," and for residency programs to "determine whether current educational methods are adequate to prepare future surgeons."
The authors reported they had no financial disclosures.
The surgical residents in this study were intelligent high achievers and the mean USMLE step 1 score of the residents who were remediated was a "quite respectable 225," Dr. Karen Deveney said in an editorial. "A substantial portion of our very bright residents who have a history of great success in everything they do may have difficulty keeping up with the fast pace and high workload demands," while some may opt for a less stressful career path, and others may need more help from faculty to meet expectations and will "persevere," she said.
| Dr. Deveney |
"It is incumbent on those of us in more senior positions to create educational systems that eliminate nonessential tasks so that residents can devote more attention during the compressed work hours to learning what they need to become competent surgeons," she wrote. "Only then can we have a better chance of training and retaining the best and the brightest" (Arch. Surg. 2012;147; 833).
Dr. Karen Deveney is professor of surgery and vice chair of education and program director, department of surgery, Oregon Health and Science University, Portland. She had no financial disclosures to report.
The surgical residents in this study were intelligent high achievers and the mean USMLE step 1 score of the residents who were remediated was a "quite respectable 225," Dr. Karen Deveney said in an editorial. "A substantial portion of our very bright residents who have a history of great success in everything they do may have difficulty keeping up with the fast pace and high workload demands," while some may opt for a less stressful career path, and others may need more help from faculty to meet expectations and will "persevere," she said.
| Dr. Deveney |
"It is incumbent on those of us in more senior positions to create educational systems that eliminate nonessential tasks so that residents can devote more attention during the compressed work hours to learning what they need to become competent surgeons," she wrote. "Only then can we have a better chance of training and retaining the best and the brightest" (Arch. Surg. 2012;147; 833).
Dr. Karen Deveney is professor of surgery and vice chair of education and program director, department of surgery, Oregon Health and Science University, Portland. She had no financial disclosures to report.
The surgical residents in this study were intelligent high achievers and the mean USMLE step 1 score of the residents who were remediated was a "quite respectable 225," Dr. Karen Deveney said in an editorial. "A substantial portion of our very bright residents who have a history of great success in everything they do may have difficulty keeping up with the fast pace and high workload demands," while some may opt for a less stressful career path, and others may need more help from faculty to meet expectations and will "persevere," she said.
| Dr. Deveney |
"It is incumbent on those of us in more senior positions to create educational systems that eliminate nonessential tasks so that residents can devote more attention during the compressed work hours to learning what they need to become competent surgeons," she wrote. "Only then can we have a better chance of training and retaining the best and the brightest" (Arch. Surg. 2012;147; 833).
Dr. Karen Deveney is professor of surgery and vice chair of education and program director, department of surgery, Oregon Health and Science University, Portland. She had no financial disclosures to report.
Almost one-third of general surgery residents required remediation over an 11-year period, most often because of a deficiency in medical knowledge, judging from findings in a retrospective study of remediation and attrition rates among general surgery residents at six academic surgical residency programs in California.
The high remediation rate identified in this study "begs the question of whether we are falling short in the education of surgical residents," said Dr. Arezou Yaghoubian of the department of surgery, Harbor-UCLA Medical Center, Los Angeles, and associates (Arch. Surg. 2012;147:829-33).
They conducted the study to determine which of the six Accreditation Council for Graduate Medical Education (ACGME) competencies (patient care, medical knowledge, practice-based learning, interpersonal and communication skills, professionalism, and system-based practice), most often require remediation and to identify predictors of remediation. There is a scarcity of data on how well surgical residency programs have been achieving these competencies, and this information may provide insight into how to modify the surgical curriculum more effectively in this new era of limited hours, they said.
In the study of 348 general surgery residents at the six training programs between 1999 and 2010, the most common reason for remediation was medical knowledge in 74%, followed by interpersonal and communication skills in 24%, patient care in 22%, professionalism in 18%, system-based practice in 14%, and practice-based learning in 8%.
Of the 107 residents who required remediation, 27 required remediation more than once. Almost 16% of the residents left their programs, but most (53 of 55 residents) left voluntarily. The other two failed remediation and had to leave the program.
Monthly meetings with faculty was the most common form of remediation, in 79%, followed by specific reading assignments (72%), required attendance at review courses and/or conferences (27%), evaluation by a therapist, psychologist, or psychiatrist (12%), and having to repeat a clinical year (6.5%).
More than half of the remediations were initiated during the first 2 postgraduate years (25% in the first and 35% in the second year), followed by 21% during the third year, 16% during the fourth year, and 4% during the fifth year.
A predictor of remediation was having received honors during the third-year surgery clerkship (58% of those who were subject to remediation vs. 45% of those who were not remediated, a statistically significant difference), which, the authors noted, was counterintuitive.
United States Medical Licensing Examination (USMLE) step 1 and/or step 2 scores and American Board of Surgery In-Training Examination (ABSITE) scores at postgraduate years 1 through 4 were also predictive of remediation. The median USMLE step 1 and step 2 scores were 225 and 223 among the residents subject to remediation, vs. 232 for step 1 and step 2 scores among those who were not remediated, statistically significant differences.
The ABSITE scores during postgraduate years 1 through 4 were significantly lower among those who were subject to remediation, but the differences in median scores in years 5 through 7 were not significantly different.
But remediation was not a predictor of attrition. The only predictor of attrition was the ABSITE score at the third postgraduate year, which was a median of 34 among those who left the program and 62 among those who stayed.
Possible explanations for the high remediation rate is that residents are not well prepared for the demands of a surgical residency, they need to be more efficient with their time because of the 80-hour work week, and they may not necessarily be spending their increased time outside of the hospital studying at home, the authors said. Possible reasons for the attrition rate among the residents, an "ongoing concern" in general surgery, may be marital, family, and personal issues and a need for a less stressful environment, they added.
Acknowledging the study's limitations, including the retrospective design and lack of information on how many residents passed the American Board of Surgery boards, the authors concluded that the high remediation rate "should give surgical educators pause as we should closely examine the potential sources of these deficiencies."
They called on surgical societies to "take the initiative to encourage the restructuring of medical school education, such that future surgeons are better prepared to enter surgical residencies," and for residency programs to "determine whether current educational methods are adequate to prepare future surgeons."
The authors reported they had no financial disclosures.
Almost one-third of general surgery residents required remediation over an 11-year period, most often because of a deficiency in medical knowledge, judging from findings in a retrospective study of remediation and attrition rates among general surgery residents at six academic surgical residency programs in California.
The high remediation rate identified in this study "begs the question of whether we are falling short in the education of surgical residents," said Dr. Arezou Yaghoubian of the department of surgery, Harbor-UCLA Medical Center, Los Angeles, and associates (Arch. Surg. 2012;147:829-33).
They conducted the study to determine which of the six Accreditation Council for Graduate Medical Education (ACGME) competencies (patient care, medical knowledge, practice-based learning, interpersonal and communication skills, professionalism, and system-based practice), most often require remediation and to identify predictors of remediation. There is a scarcity of data on how well surgical residency programs have been achieving these competencies, and this information may provide insight into how to modify the surgical curriculum more effectively in this new era of limited hours, they said.
In the study of 348 general surgery residents at the six training programs between 1999 and 2010, the most common reason for remediation was medical knowledge in 74%, followed by interpersonal and communication skills in 24%, patient care in 22%, professionalism in 18%, system-based practice in 14%, and practice-based learning in 8%.
Of the 107 residents who required remediation, 27 required remediation more than once. Almost 16% of the residents left their programs, but most (53 of 55 residents) left voluntarily. The other two failed remediation and had to leave the program.
Monthly meetings with faculty was the most common form of remediation, in 79%, followed by specific reading assignments (72%), required attendance at review courses and/or conferences (27%), evaluation by a therapist, psychologist, or psychiatrist (12%), and having to repeat a clinical year (6.5%).
More than half of the remediations were initiated during the first 2 postgraduate years (25% in the first and 35% in the second year), followed by 21% during the third year, 16% during the fourth year, and 4% during the fifth year.
A predictor of remediation was having received honors during the third-year surgery clerkship (58% of those who were subject to remediation vs. 45% of those who were not remediated, a statistically significant difference), which, the authors noted, was counterintuitive.
United States Medical Licensing Examination (USMLE) step 1 and/or step 2 scores and American Board of Surgery In-Training Examination (ABSITE) scores at postgraduate years 1 through 4 were also predictive of remediation. The median USMLE step 1 and step 2 scores were 225 and 223 among the residents subject to remediation, vs. 232 for step 1 and step 2 scores among those who were not remediated, statistically significant differences.
The ABSITE scores during postgraduate years 1 through 4 were significantly lower among those who were subject to remediation, but the differences in median scores in years 5 through 7 were not significantly different.
But remediation was not a predictor of attrition. The only predictor of attrition was the ABSITE score at the third postgraduate year, which was a median of 34 among those who left the program and 62 among those who stayed.
Possible explanations for the high remediation rate is that residents are not well prepared for the demands of a surgical residency, they need to be more efficient with their time because of the 80-hour work week, and they may not necessarily be spending their increased time outside of the hospital studying at home, the authors said. Possible reasons for the attrition rate among the residents, an "ongoing concern" in general surgery, may be marital, family, and personal issues and a need for a less stressful environment, they added.
Acknowledging the study's limitations, including the retrospective design and lack of information on how many residents passed the American Board of Surgery boards, the authors concluded that the high remediation rate "should give surgical educators pause as we should closely examine the potential sources of these deficiencies."
They called on surgical societies to "take the initiative to encourage the restructuring of medical school education, such that future surgeons are better prepared to enter surgical residencies," and for residency programs to "determine whether current educational methods are adequate to prepare future surgeons."
The authors reported they had no financial disclosures.
Major Finding: Remediation was required for 31% of the general surgery residents in the study, most often initiated because of a deficiency in medical knowledge (74%). All but 2 of the 55 residents who left the program left voluntarily, not because of failed remediation.
Data Source: A retrospective study of 348 general surgery residents at six academic surgical training programs in California between 1999 and 2010, which evaluated the rates and predictors of remediation and attrition.
Disclosures: The authors of the study had no disclosures.

ONLINE EXCLUSIVE: Anticoagulant's Receives FDA Approval to Treat Deep Vein Thrombosis, Pulmonary Embolism
Rivaroxaban (Xarelto) has won another approval from the U.S. Food and Drug Administration (FDA). Already green-lighted for use to reduce the risk of DVT and pulmonary embolism (PE) after knee or hip replacement surgery—and reduce the risk of stroke in non-valvular atrial fibrillation patients—the anticoagulant therapy has been approved for use in the treatment of acute DVT and PE, and to reduce the risk of recurrent DVT and PE after initial treatment. It’s a landmark step that will likely have big implications for hospitalists.
“Xarelto is the first oral anti-clotting drug approved to treat and reduce the recurrence of blood clots since the approval of warfarin nearly 60 years ago,” Richard Pazdur, MD, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a news release.
—Hiren Shah, MD, assistant professor of medicine, Northwestern University Feinberg School of Medicine, medical director, hospital medicine, Northwestern Memorial Hospital, Chicago
“Single-drug therapy without the need for parental bridging treatment, or drug-level monitoring, is a breakthrough in the treatment of VTE, and represents a paradigm shift that we have not seen in a long time for a very common emergency room and hospital-based medical condition,” says Hiren Shah, MD, assistant professor of medicine at Northwestern University’s Feinberg School of Medicine and medical director of hospital medicine at Northwestern Memorial Hospital in Chicago.
Ian Jenkins, assistant professor in the Division of Hospital Medicine at the University of California at San Diego, says factors that will help determine whether a patient is a candidate for rivaroxaban include the ability to pay for it; compliance, because the duration of effect is shorter than it is for warfarin; and good and stable renal function.
“We now have the first approved oral warfarin alternative for VTE, and for appropriate candidates, it's a more convenient if not better treatment,” Dr. Jenkins says. “The main downside is that warfarin remains reversible, and the new drugs are minimally so.”
Dr. Shah predicts a more efficient discharge process, which, for rivaroxaban patients, will no longer include arranging for international normalized ratio (INR) monitoring or time-consuming counseling on taking injections and drug interactions with vitamin-K antagonists.
“That’s a very complex, 30-minute process,” says Dr. Shah, who also who runs Northwestern’s VTE-prevention program. “With a single agent, I think the value here is you don’t need that complex care coordination anymore, and that’s time-saving for a hospitalist.”
Dr. Shah notes coordination of care will still be very important with this indication, especially because the dose for rivaroxaban in the treatment of acute DVT changes from twice a day to once a day starting at Day 21. “Whatever education initiatives we undertake, they have to extend that entire spectrum,” he adds.
Visit our website for more information about treating acute DVT.
Rivaroxaban (Xarelto) has won another approval from the U.S. Food and Drug Administration (FDA). Already green-lighted for use to reduce the risk of DVT and pulmonary embolism (PE) after knee or hip replacement surgery—and reduce the risk of stroke in non-valvular atrial fibrillation patients—the anticoagulant therapy has been approved for use in the treatment of acute DVT and PE, and to reduce the risk of recurrent DVT and PE after initial treatment. It’s a landmark step that will likely have big implications for hospitalists.
“Xarelto is the first oral anti-clotting drug approved to treat and reduce the recurrence of blood clots since the approval of warfarin nearly 60 years ago,” Richard Pazdur, MD, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a news release.
—Hiren Shah, MD, assistant professor of medicine, Northwestern University Feinberg School of Medicine, medical director, hospital medicine, Northwestern Memorial Hospital, Chicago
“Single-drug therapy without the need for parental bridging treatment, or drug-level monitoring, is a breakthrough in the treatment of VTE, and represents a paradigm shift that we have not seen in a long time for a very common emergency room and hospital-based medical condition,” says Hiren Shah, MD, assistant professor of medicine at Northwestern University’s Feinberg School of Medicine and medical director of hospital medicine at Northwestern Memorial Hospital in Chicago.
Ian Jenkins, assistant professor in the Division of Hospital Medicine at the University of California at San Diego, says factors that will help determine whether a patient is a candidate for rivaroxaban include the ability to pay for it; compliance, because the duration of effect is shorter than it is for warfarin; and good and stable renal function.
“We now have the first approved oral warfarin alternative for VTE, and for appropriate candidates, it's a more convenient if not better treatment,” Dr. Jenkins says. “The main downside is that warfarin remains reversible, and the new drugs are minimally so.”
Dr. Shah predicts a more efficient discharge process, which, for rivaroxaban patients, will no longer include arranging for international normalized ratio (INR) monitoring or time-consuming counseling on taking injections and drug interactions with vitamin-K antagonists.
“That’s a very complex, 30-minute process,” says Dr. Shah, who also who runs Northwestern’s VTE-prevention program. “With a single agent, I think the value here is you don’t need that complex care coordination anymore, and that’s time-saving for a hospitalist.”
Dr. Shah notes coordination of care will still be very important with this indication, especially because the dose for rivaroxaban in the treatment of acute DVT changes from twice a day to once a day starting at Day 21. “Whatever education initiatives we undertake, they have to extend that entire spectrum,” he adds.
Visit our website for more information about treating acute DVT.
Rivaroxaban (Xarelto) has won another approval from the U.S. Food and Drug Administration (FDA). Already green-lighted for use to reduce the risk of DVT and pulmonary embolism (PE) after knee or hip replacement surgery—and reduce the risk of stroke in non-valvular atrial fibrillation patients—the anticoagulant therapy has been approved for use in the treatment of acute DVT and PE, and to reduce the risk of recurrent DVT and PE after initial treatment. It’s a landmark step that will likely have big implications for hospitalists.
“Xarelto is the first oral anti-clotting drug approved to treat and reduce the recurrence of blood clots since the approval of warfarin nearly 60 years ago,” Richard Pazdur, MD, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a news release.
—Hiren Shah, MD, assistant professor of medicine, Northwestern University Feinberg School of Medicine, medical director, hospital medicine, Northwestern Memorial Hospital, Chicago
“Single-drug therapy without the need for parental bridging treatment, or drug-level monitoring, is a breakthrough in the treatment of VTE, and represents a paradigm shift that we have not seen in a long time for a very common emergency room and hospital-based medical condition,” says Hiren Shah, MD, assistant professor of medicine at Northwestern University’s Feinberg School of Medicine and medical director of hospital medicine at Northwestern Memorial Hospital in Chicago.
Ian Jenkins, assistant professor in the Division of Hospital Medicine at the University of California at San Diego, says factors that will help determine whether a patient is a candidate for rivaroxaban include the ability to pay for it; compliance, because the duration of effect is shorter than it is for warfarin; and good and stable renal function.
“We now have the first approved oral warfarin alternative for VTE, and for appropriate candidates, it's a more convenient if not better treatment,” Dr. Jenkins says. “The main downside is that warfarin remains reversible, and the new drugs are minimally so.”
Dr. Shah predicts a more efficient discharge process, which, for rivaroxaban patients, will no longer include arranging for international normalized ratio (INR) monitoring or time-consuming counseling on taking injections and drug interactions with vitamin-K antagonists.
“That’s a very complex, 30-minute process,” says Dr. Shah, who also who runs Northwestern’s VTE-prevention program. “With a single agent, I think the value here is you don’t need that complex care coordination anymore, and that’s time-saving for a hospitalist.”
Dr. Shah notes coordination of care will still be very important with this indication, especially because the dose for rivaroxaban in the treatment of acute DVT changes from twice a day to once a day starting at Day 21. “Whatever education initiatives we undertake, they have to extend that entire spectrum,” he adds.
Visit our website for more information about treating acute DVT.
Melissa Officinalis
Used in foods, some traditional medicines, herbal tea, herbal toothpastes, and aromatherapy, Melissa officinalis (lemon balm) is a perennial herb in the Lamiaceae (mint) family found in southern Europe and the Mediterranean area. The medicinal use of lemon balm dates back at least 2,000 years (Ann. N. Y. Acad. Sci. 1965;130:474-82). Lower abdominal distress and nervous conditions are some of the ailments treated with lemon balm in folk medicine; herpes lesions are a modern indication (Nat. Prod. Res. 2008;22:1433-40). The essential oil and phenylpropanoid derivatives are thought to be the two primary groups of active constituents in lemon balm (Phytochemistry. 2011;72:572-8).

The main individual components of M. officinalis essential oil have been identified as the monoterpenaldehydes citral a, citral b, and citronellal (Phytomedicine. 2008;15:734-40). The chief phenolic compounds are rosmarinic acid, which is an ester of caffeic acid and 3,4-dihydroxyphenyllactic acid, as well as caffeic acid, which is isolated from the fresh leaves and stems (J. Nat. Prod. 2009;72:1512-5Phytochemistry. 2011;72:572-8). Six flavonoids, including luteolin and apigenin, have also been isolated from the leaves of lemon balm (Acta. Pol. Pharm. 2002;59:139-43; J. Nat. Prod. 2007;70:1889-94). Given the presence of such ingredients known to exhibit antioxidant properties, it is not surprising that such a capacity is considered one of the main medicinal benefits of M. officinalis. Indeed, lemon balm is reputed to display significant antioxidant, anxiolytic (Med. J. Nutrition. Metab. 2011;4:211-8; Phytomedicine. 2010;17:397-403; Psychosom. Med. 2004;66:607-13), and antiviral (particularly antiherpetic) activity (Proc. Soc. Exp. Biol. Med. 1964;117:431-4; Virol. J. 2011;8:188). M. officinalis is also a component, with two other herbs, in a mixture (Ob-X) recently shown to lower body weight gain and adipose tissue mass in genetically obese mice (Pharm. Biol. 2011;49:614-9).
Antioxidant Activity
In a 2009 study, investigators examined the antioxidant potential of three plants (M. officinalis, Matricaria recutita (German chamomile), and Cymbopogon citrus [lemon grass]) used in Brazil to treat neurologic conditions. M. officinalis was found to deliver the greatest reduction in thiobarbituric acid reactive species (TBARS) and the most salient antioxidant effect as evaluated by the 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay. The investigators concluded that M. officinalis warrants consideration as a treatment for oxidative stress–associated neurologic diseases (Neurochem. Res. 2009;34:973-83).
Additional evidence of its antioxidant activity is emerging. In early 2012, Martins et al. reported on their study in which an aqueous extract of M. officinalis significantly mitigated manganese-induced brain oxidative stress in mice. They found that the extract attenuated oxidative damage (TBARS) and reduced total thiol levels, and concluded that their findings show the potent antioxidant activity of M. officinalis (Brain. Res. Bull. 2012;87;74-9). In addition, a recent study found that lemon balm infusion in a tea, after 30 days of daily consumption, significantly lowered oxidative stress and DNA damage in radiology staff exposed to low doses of radiation at work (Toxicol. Ind. Health. 2011;27:205-12).
Antiviral Activity
In 2006, Gaby reported on various natural substances, used in the diet or topically, that exert activity against herpes simplex lesions and prevent recurrences, serving as effective alternatives to acyclovir and its attendant side effects. He cited lemon balm as having exhibited antiviral properties in two studies in the 1990s (Altern. Med. Rev. 2006;11:93-101).
In 1994, 116 patients with acute herpes simplex applied a standardized lemon balm cream (containing 1% Lo-701) or a placebo cream two to four times daily in a randomized, double-blind trial over a 5- to 10-day period within 72 hours of symptom onset. While only 19% of the placebo group reported satisfactory healing, 41% of the active treatment group was satisfied (Phytomedicine. 1994;1:25-31). In 1999, a double-blind, placebo-controlled trial randomized 66 patients with a minimum of four herpes simplex episodes per year to treatment (four times daily for 5 days) with the same standardized lemon balm cream or placebo. Symptom scores were significantly lower in the treatment group than the control group by the second day of the protocol, though the trend supporting active treatment over 5 days was not significant (Phytomedicine 1999;6:225-30).
In 2008, Mazzanti et al. evaluated the antiviral activity against herpes simplex virus type 2 (HSV-2) of a hydroalcoholic extract of lemon balm leaves using a cytopathic effect inhibition assay on Vero cells. They found that lemon balm diminished the cytopathic effect of HSV-2 on Vero cells, with a maximum suppression effect with 0.5 mg/mL. The extract, shown through NMR (nuclear magnetic resonance) and HPLC (high-performance liquid chromatography) analysis to contain rosmarinic acid (4.1% w/w), did not prevent the entry of HSV-2 into cells, indicating postpenetration activity by the botanical agent. The investigators concluded that their work supports the use of lemon balm for treating herpes lesions, and justifies its further study in clinical trials (Nat. Prod. Res. 2008;22:1433-40).
Also that year, Schnitzler et al. evaluated the antiviral effect of lemon balm oil on HSV-1 and HSV-2 in vitro on monkey kidney cells. They found that plaque formation was significantly lowered (by 98.8% for HSV-1 and 97.2% for HSV-2) by noncytotoxic lemon balm oil concentrations, with higher concentrations nearly eradicating infections. Using time-on-addition assays, the investigators determined that pretreatment with lemon balm oil significantly suppressed both viruses before infection of cells, suggesting that the oil impacted the virus prior to adsorption, but not after reaching the host cell. They concluded that this implies the capacity for direct antiviral activity. The authors added that the lipophilic nature of lemon balm oil allows for its penetration into the skin, further supporting its suitability as a topical treatment of herpes (Phytomedicine. 2008;15:734-40).
In a more recent in vitro experiment evaluating antiviral activity against HSV-1, Astani et al. compared an aqueous extract of M. officinalis and phenolic extract compounds (caffeic acid, p-coumaric acid, and rosmarinic acid). The lemon balm extract exhibited high virucidal activity against HSV-1, even at concentrations of 1.5 mcg/mL; phenolic compounds showed similar results only at concentrations 100 times greater. Further, lemon balm extract and rosmarinic acid dose-dependently suppressed HSV-1 attachment to host cells. The researchers concluded that rosmarinic acid was the primary constituent responsible for the antiviral activity displayed by lemon balm, but noted that M. officinalis extract, which imparted virucidal activity against HSV-1 in vitro with low toxicity, has a greater selectivity index against HSV than that of its constituents alone (Chemotherapy. 2012;58:70-7).
In 2008, Geuenich et al. investigated several species of the Lamiaceae family (including lemon balm) for their potency in suppressing HIV-1 infection. The aqueous extracts from the leaves of lemon balm (as well as peppermint and sage) dose-dependently displayed substantial activity against HIV-1 infection in T-cell lines, primary macrophages, and in ex vivo tonsil histocultures. The investigators also found that exposure of extracts to free virions strongly and quickly suppressed infections, though no antiviral effect was seen in exposure to surface-bound virions or target cells alone. Noting the antiherpetic activity of these Lamiaceae family extracts, the investigators suggested that the development of virucidal topical microbicides using such ingredients is warranted (Retrovirology. 2008;5:27).
Hypopigmentary Potential
A potential hypopigmentary application of lemon balm also may be emerging. In 2011, Fujita et al. isolated 16-hydroxy-9-oxo-10E,12E,14E-octadecatrienoic acid (also called Corchorifatty acid B [CFAB]) from the ethanol extracts of the aerial parts of M. officinalis, and found that it suppresses pigmentation in human melanocytes and murine melanoma B16 cells, probably by promoting accelerated degradation of tyrosinase in B16 cells. Further, they noted that the mechanism of action of CFAB is markedly different from those of many other hypopigmentary agents, which facilitate tyrosinase degradation in proteasomes or lysosomes. That is, the reductions in tyrosinase caused by CFAB are thought to take place in post–Golgi complex areas, not in proteasomal or lysosomal ones (Exp. Dermatol. 2011;20(5):420-4).
Conclusions
Like many botanical ingredients studied and harnessed in our modern pharmacopeia, lemon balm has a history of use in traditional medicine. Recent studies suggest antioxidant, anxiolytic, and, especially, antiviral properties, notably in the treatment of herpes viruses. More research is necessary, however, to establish a broader role for M. officinalis in the dermatologic armamentarium.
Dr. Baumann is in private practice in Miami Beach. She did not disclose any conflicts of interest. To respond to this column, or to suggest topics for future columns, write to Dr. Baumann at [email protected].
Used in foods, some traditional medicines, herbal tea, herbal toothpastes, and aromatherapy, Melissa officinalis (lemon balm) is a perennial herb in the Lamiaceae (mint) family found in southern Europe and the Mediterranean area. The medicinal use of lemon balm dates back at least 2,000 years (Ann. N. Y. Acad. Sci. 1965;130:474-82). Lower abdominal distress and nervous conditions are some of the ailments treated with lemon balm in folk medicine; herpes lesions are a modern indication (Nat. Prod. Res. 2008;22:1433-40). The essential oil and phenylpropanoid derivatives are thought to be the two primary groups of active constituents in lemon balm (Phytochemistry. 2011;72:572-8).
The main individual components of M. officinalis essential oil have been identified as the monoterpenaldehydes citral a, citral b, and citronellal (Phytomedicine. 2008;15:734-40). The chief phenolic compounds are rosmarinic acid, which is an ester of caffeic acid and 3,4-dihydroxyphenyllactic acid, as well as caffeic acid, which is isolated from the fresh leaves and stems (J. Nat. Prod. 2009;72:1512-5Phytochemistry. 2011;72:572-8). Six flavonoids, including luteolin and apigenin, have also been isolated from the leaves of lemon balm (Acta. Pol. Pharm. 2002;59:139-43; J. Nat. Prod. 2007;70:1889-94). Given the presence of such ingredients known to exhibit antioxidant properties, it is not surprising that such a capacity is considered one of the main medicinal benefits of M. officinalis. Indeed, lemon balm is reputed to display significant antioxidant, anxiolytic (Med. J. Nutrition. Metab. 2011;4:211-8; Phytomedicine. 2010;17:397-403; Psychosom. Med. 2004;66:607-13), and antiviral (particularly antiherpetic) activity (Proc. Soc. Exp. Biol. Med. 1964;117:431-4; Virol. J. 2011;8:188). M. officinalis is also a component, with two other herbs, in a mixture (Ob-X) recently shown to lower body weight gain and adipose tissue mass in genetically obese mice (Pharm. Biol. 2011;49:614-9).
Antioxidant Activity
In a 2009 study, investigators examined the antioxidant potential of three plants (M. officinalis, Matricaria recutita (German chamomile), and Cymbopogon citrus [lemon grass]) used in Brazil to treat neurologic conditions. M. officinalis was found to deliver the greatest reduction in thiobarbituric acid reactive species (TBARS) and the most salient antioxidant effect as evaluated by the 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay. The investigators concluded that M. officinalis warrants consideration as a treatment for oxidative stress–associated neurologic diseases (Neurochem. Res. 2009;34:973-83).
Additional evidence of its antioxidant activity is emerging. In early 2012, Martins et al. reported on their study in which an aqueous extract of M. officinalis significantly mitigated manganese-induced brain oxidative stress in mice. They found that the extract attenuated oxidative damage (TBARS) and reduced total thiol levels, and concluded that their findings show the potent antioxidant activity of M. officinalis (Brain. Res. Bull. 2012;87;74-9). In addition, a recent study found that lemon balm infusion in a tea, after 30 days of daily consumption, significantly lowered oxidative stress and DNA damage in radiology staff exposed to low doses of radiation at work (Toxicol. Ind. Health. 2011;27:205-12).
Antiviral Activity
In 2006, Gaby reported on various natural substances, used in the diet or topically, that exert activity against herpes simplex lesions and prevent recurrences, serving as effective alternatives to acyclovir and its attendant side effects. He cited lemon balm as having exhibited antiviral properties in two studies in the 1990s (Altern. Med. Rev. 2006;11:93-101).
In 1994, 116 patients with acute herpes simplex applied a standardized lemon balm cream (containing 1% Lo-701) or a placebo cream two to four times daily in a randomized, double-blind trial over a 5- to 10-day period within 72 hours of symptom onset. While only 19% of the placebo group reported satisfactory healing, 41% of the active treatment group was satisfied (Phytomedicine. 1994;1:25-31). In 1999, a double-blind, placebo-controlled trial randomized 66 patients with a minimum of four herpes simplex episodes per year to treatment (four times daily for 5 days) with the same standardized lemon balm cream or placebo. Symptom scores were significantly lower in the treatment group than the control group by the second day of the protocol, though the trend supporting active treatment over 5 days was not significant (Phytomedicine 1999;6:225-30).
In 2008, Mazzanti et al. evaluated the antiviral activity against herpes simplex virus type 2 (HSV-2) of a hydroalcoholic extract of lemon balm leaves using a cytopathic effect inhibition assay on Vero cells. They found that lemon balm diminished the cytopathic effect of HSV-2 on Vero cells, with a maximum suppression effect with 0.5 mg/mL. The extract, shown through NMR (nuclear magnetic resonance) and HPLC (high-performance liquid chromatography) analysis to contain rosmarinic acid (4.1% w/w), did not prevent the entry of HSV-2 into cells, indicating postpenetration activity by the botanical agent. The investigators concluded that their work supports the use of lemon balm for treating herpes lesions, and justifies its further study in clinical trials (Nat. Prod. Res. 2008;22:1433-40).
Also that year, Schnitzler et al. evaluated the antiviral effect of lemon balm oil on HSV-1 and HSV-2 in vitro on monkey kidney cells. They found that plaque formation was significantly lowered (by 98.8% for HSV-1 and 97.2% for HSV-2) by noncytotoxic lemon balm oil concentrations, with higher concentrations nearly eradicating infections. Using time-on-addition assays, the investigators determined that pretreatment with lemon balm oil significantly suppressed both viruses before infection of cells, suggesting that the oil impacted the virus prior to adsorption, but not after reaching the host cell. They concluded that this implies the capacity for direct antiviral activity. The authors added that the lipophilic nature of lemon balm oil allows for its penetration into the skin, further supporting its suitability as a topical treatment of herpes (Phytomedicine. 2008;15:734-40).
In a more recent in vitro experiment evaluating antiviral activity against HSV-1, Astani et al. compared an aqueous extract of M. officinalis and phenolic extract compounds (caffeic acid, p-coumaric acid, and rosmarinic acid). The lemon balm extract exhibited high virucidal activity against HSV-1, even at concentrations of 1.5 mcg/mL; phenolic compounds showed similar results only at concentrations 100 times greater. Further, lemon balm extract and rosmarinic acid dose-dependently suppressed HSV-1 attachment to host cells. The researchers concluded that rosmarinic acid was the primary constituent responsible for the antiviral activity displayed by lemon balm, but noted that M. officinalis extract, which imparted virucidal activity against HSV-1 in vitro with low toxicity, has a greater selectivity index against HSV than that of its constituents alone (Chemotherapy. 2012;58:70-7).
In 2008, Geuenich et al. investigated several species of the Lamiaceae family (including lemon balm) for their potency in suppressing HIV-1 infection. The aqueous extracts from the leaves of lemon balm (as well as peppermint and sage) dose-dependently displayed substantial activity against HIV-1 infection in T-cell lines, primary macrophages, and in ex vivo tonsil histocultures. The investigators also found that exposure of extracts to free virions strongly and quickly suppressed infections, though no antiviral effect was seen in exposure to surface-bound virions or target cells alone. Noting the antiherpetic activity of these Lamiaceae family extracts, the investigators suggested that the development of virucidal topical microbicides using such ingredients is warranted (Retrovirology. 2008;5:27).
Hypopigmentary Potential
A potential hypopigmentary application of lemon balm also may be emerging. In 2011, Fujita et al. isolated 16-hydroxy-9-oxo-10E,12E,14E-octadecatrienoic acid (also called Corchorifatty acid B [CFAB]) from the ethanol extracts of the aerial parts of M. officinalis, and found that it suppresses pigmentation in human melanocytes and murine melanoma B16 cells, probably by promoting accelerated degradation of tyrosinase in B16 cells. Further, they noted that the mechanism of action of CFAB is markedly different from those of many other hypopigmentary agents, which facilitate tyrosinase degradation in proteasomes or lysosomes. That is, the reductions in tyrosinase caused by CFAB are thought to take place in post–Golgi complex areas, not in proteasomal or lysosomal ones (Exp. Dermatol. 2011;20(5):420-4).
Conclusions
Like many botanical ingredients studied and harnessed in our modern pharmacopeia, lemon balm has a history of use in traditional medicine. Recent studies suggest antioxidant, anxiolytic, and, especially, antiviral properties, notably in the treatment of herpes viruses. More research is necessary, however, to establish a broader role for M. officinalis in the dermatologic armamentarium.
Dr. Baumann is in private practice in Miami Beach. She did not disclose any conflicts of interest. To respond to this column, or to suggest topics for future columns, write to Dr. Baumann at [email protected].
Used in foods, some traditional medicines, herbal tea, herbal toothpastes, and aromatherapy, Melissa officinalis (lemon balm) is a perennial herb in the Lamiaceae (mint) family found in southern Europe and the Mediterranean area. The medicinal use of lemon balm dates back at least 2,000 years (Ann. N. Y. Acad. Sci. 1965;130:474-82). Lower abdominal distress and nervous conditions are some of the ailments treated with lemon balm in folk medicine; herpes lesions are a modern indication (Nat. Prod. Res. 2008;22:1433-40). The essential oil and phenylpropanoid derivatives are thought to be the two primary groups of active constituents in lemon balm (Phytochemistry. 2011;72:572-8).
The main individual components of M. officinalis essential oil have been identified as the monoterpenaldehydes citral a, citral b, and citronellal (Phytomedicine. 2008;15:734-40). The chief phenolic compounds are rosmarinic acid, which is an ester of caffeic acid and 3,4-dihydroxyphenyllactic acid, as well as caffeic acid, which is isolated from the fresh leaves and stems (J. Nat. Prod. 2009;72:1512-5Phytochemistry. 2011;72:572-8). Six flavonoids, including luteolin and apigenin, have also been isolated from the leaves of lemon balm (Acta. Pol. Pharm. 2002;59:139-43; J. Nat. Prod. 2007;70:1889-94). Given the presence of such ingredients known to exhibit antioxidant properties, it is not surprising that such a capacity is considered one of the main medicinal benefits of M. officinalis. Indeed, lemon balm is reputed to display significant antioxidant, anxiolytic (Med. J. Nutrition. Metab. 2011;4:211-8; Phytomedicine. 2010;17:397-403; Psychosom. Med. 2004;66:607-13), and antiviral (particularly antiherpetic) activity (Proc. Soc. Exp. Biol. Med. 1964;117:431-4; Virol. J. 2011;8:188). M. officinalis is also a component, with two other herbs, in a mixture (Ob-X) recently shown to lower body weight gain and adipose tissue mass in genetically obese mice (Pharm. Biol. 2011;49:614-9).
Antioxidant Activity
In a 2009 study, investigators examined the antioxidant potential of three plants (M. officinalis, Matricaria recutita (German chamomile), and Cymbopogon citrus [lemon grass]) used in Brazil to treat neurologic conditions. M. officinalis was found to deliver the greatest reduction in thiobarbituric acid reactive species (TBARS) and the most salient antioxidant effect as evaluated by the 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay. The investigators concluded that M. officinalis warrants consideration as a treatment for oxidative stress–associated neurologic diseases (Neurochem. Res. 2009;34:973-83).
Additional evidence of its antioxidant activity is emerging. In early 2012, Martins et al. reported on their study in which an aqueous extract of M. officinalis significantly mitigated manganese-induced brain oxidative stress in mice. They found that the extract attenuated oxidative damage (TBARS) and reduced total thiol levels, and concluded that their findings show the potent antioxidant activity of M. officinalis (Brain. Res. Bull. 2012;87;74-9). In addition, a recent study found that lemon balm infusion in a tea, after 30 days of daily consumption, significantly lowered oxidative stress and DNA damage in radiology staff exposed to low doses of radiation at work (Toxicol. Ind. Health. 2011;27:205-12).
Antiviral Activity
In 2006, Gaby reported on various natural substances, used in the diet or topically, that exert activity against herpes simplex lesions and prevent recurrences, serving as effective alternatives to acyclovir and its attendant side effects. He cited lemon balm as having exhibited antiviral properties in two studies in the 1990s (Altern. Med. Rev. 2006;11:93-101).
In 1994, 116 patients with acute herpes simplex applied a standardized lemon balm cream (containing 1% Lo-701) or a placebo cream two to four times daily in a randomized, double-blind trial over a 5- to 10-day period within 72 hours of symptom onset. While only 19% of the placebo group reported satisfactory healing, 41% of the active treatment group was satisfied (Phytomedicine. 1994;1:25-31). In 1999, a double-blind, placebo-controlled trial randomized 66 patients with a minimum of four herpes simplex episodes per year to treatment (four times daily for 5 days) with the same standardized lemon balm cream or placebo. Symptom scores were significantly lower in the treatment group than the control group by the second day of the protocol, though the trend supporting active treatment over 5 days was not significant (Phytomedicine 1999;6:225-30).
In 2008, Mazzanti et al. evaluated the antiviral activity against herpes simplex virus type 2 (HSV-2) of a hydroalcoholic extract of lemon balm leaves using a cytopathic effect inhibition assay on Vero cells. They found that lemon balm diminished the cytopathic effect of HSV-2 on Vero cells, with a maximum suppression effect with 0.5 mg/mL. The extract, shown through NMR (nuclear magnetic resonance) and HPLC (high-performance liquid chromatography) analysis to contain rosmarinic acid (4.1% w/w), did not prevent the entry of HSV-2 into cells, indicating postpenetration activity by the botanical agent. The investigators concluded that their work supports the use of lemon balm for treating herpes lesions, and justifies its further study in clinical trials (Nat. Prod. Res. 2008;22:1433-40).
Also that year, Schnitzler et al. evaluated the antiviral effect of lemon balm oil on HSV-1 and HSV-2 in vitro on monkey kidney cells. They found that plaque formation was significantly lowered (by 98.8% for HSV-1 and 97.2% for HSV-2) by noncytotoxic lemon balm oil concentrations, with higher concentrations nearly eradicating infections. Using time-on-addition assays, the investigators determined that pretreatment with lemon balm oil significantly suppressed both viruses before infection of cells, suggesting that the oil impacted the virus prior to adsorption, but not after reaching the host cell. They concluded that this implies the capacity for direct antiviral activity. The authors added that the lipophilic nature of lemon balm oil allows for its penetration into the skin, further supporting its suitability as a topical treatment of herpes (Phytomedicine. 2008;15:734-40).
In a more recent in vitro experiment evaluating antiviral activity against HSV-1, Astani et al. compared an aqueous extract of M. officinalis and phenolic extract compounds (caffeic acid, p-coumaric acid, and rosmarinic acid). The lemon balm extract exhibited high virucidal activity against HSV-1, even at concentrations of 1.5 mcg/mL; phenolic compounds showed similar results only at concentrations 100 times greater. Further, lemon balm extract and rosmarinic acid dose-dependently suppressed HSV-1 attachment to host cells. The researchers concluded that rosmarinic acid was the primary constituent responsible for the antiviral activity displayed by lemon balm, but noted that M. officinalis extract, which imparted virucidal activity against HSV-1 in vitro with low toxicity, has a greater selectivity index against HSV than that of its constituents alone (Chemotherapy. 2012;58:70-7).
In 2008, Geuenich et al. investigated several species of the Lamiaceae family (including lemon balm) for their potency in suppressing HIV-1 infection. The aqueous extracts from the leaves of lemon balm (as well as peppermint and sage) dose-dependently displayed substantial activity against HIV-1 infection in T-cell lines, primary macrophages, and in ex vivo tonsil histocultures. The investigators also found that exposure of extracts to free virions strongly and quickly suppressed infections, though no antiviral effect was seen in exposure to surface-bound virions or target cells alone. Noting the antiherpetic activity of these Lamiaceae family extracts, the investigators suggested that the development of virucidal topical microbicides using such ingredients is warranted (Retrovirology. 2008;5:27).
Hypopigmentary Potential
A potential hypopigmentary application of lemon balm also may be emerging. In 2011, Fujita et al. isolated 16-hydroxy-9-oxo-10E,12E,14E-octadecatrienoic acid (also called Corchorifatty acid B [CFAB]) from the ethanol extracts of the aerial parts of M. officinalis, and found that it suppresses pigmentation in human melanocytes and murine melanoma B16 cells, probably by promoting accelerated degradation of tyrosinase in B16 cells. Further, they noted that the mechanism of action of CFAB is markedly different from those of many other hypopigmentary agents, which facilitate tyrosinase degradation in proteasomes or lysosomes. That is, the reductions in tyrosinase caused by CFAB are thought to take place in post–Golgi complex areas, not in proteasomal or lysosomal ones (Exp. Dermatol. 2011;20(5):420-4).
Conclusions
Like many botanical ingredients studied and harnessed in our modern pharmacopeia, lemon balm has a history of use in traditional medicine. Recent studies suggest antioxidant, anxiolytic, and, especially, antiviral properties, notably in the treatment of herpes viruses. More research is necessary, however, to establish a broader role for M. officinalis in the dermatologic armamentarium.
Dr. Baumann is in private practice in Miami Beach. She did not disclose any conflicts of interest. To respond to this column, or to suggest topics for future columns, write to Dr. Baumann at [email protected].

Botulinum Toxin Threading Yields More Uniform Result
ATLANTA – The use of a threading technique, rather than the standard depot injection technique, when using botulinum toxin A to treat perioral and glabellar rhytides provides a more uniform and natural cosmetic result, according to Dr. H. William Higgins II.
Threading involves injecting the muscle along its normal anatomic course to paralyze the related muscle more evenly, he explained at the annual meeting of the American Society for Dermatologic Surgery.
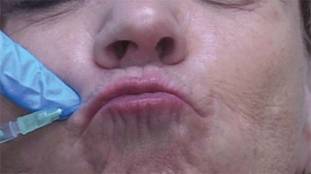
For upper and lower lip treatment, for example, injections are made at a 20- to 30-degree angle, entering the skin at a location just lateral to the targeted rhytid. The toxin is dispensed while withdrawing, thereby threading the injection along the length of the orbicularis oris. This differs from the typical approach, which often involves a depot injection at an angle more perpendicular to the skin, said Dr. Higgins of Brown University in Providence, R.I.
For the glabellar lines, the threading technique involves four symmetrical injection points, with two points targeting each corrugator. Injections at the more medial points are made directly above the inner canthus, with intramuscular injections made perpendicularly to the skin in the traditional depot manner.
At the two lateral injection points, however, the needle is inserted in most cases just medial to the mid-pupillary lines, thereby targeting the "tail" of the corrugators, he explained.
"Similar to our approach at the orbicularis oris, rather than injecting at an angle more perpendicular to the skin, we inject at an angle of roughly 20-30 degrees, entering the skin at a location just medial to the glabellar rhytid we intend to treat. The needle is then directed laterally and slightly superiorly in order to follow the anatomy of the corrugator supercilii, and the injection is threaded along the muscle’s length while withdrawing," he explained.
This approach corrects for the inadequate responses sometimes seen when using the typical method of placing subepidermal blebs to produce localized microparesis of the targeted muscle, and could reduce the need for touch-up injections.
Cosmetic outcomes have been excellent and patient satisfaction high with the use of this technique, he said. In his experience, the technique has dramatically reduced the incidence of adverse effects.
"It has been documented that, even with conservative dosing, neuromodulator treatment of perioral rhytides can affect mouth function by weakening the lip sphincter, but this has not been the case in our patient population when using this technique," he said.
Similarly, when treating glabellar rhytides, the injection of the toxin at a more precise depth – and more evenly along the tail of the corrugators, has resulted in a reduced incidence of brow ptosis as well as more natural smoothing.
"This approach helps prevent the undesirable appearance of a 'forehead freeze,' " he said.
The threading technique also results in fewer needle sticks, which means less pain and bruising for the patients.
The use of a longer 1- or 1.5-inch needle could potentially allow for even fewer injections without compromising the result, Dr. Higgins noted.
"Furthermore, this technique could conceivably be applied on other areas of the face. Crow's feet, for example, could be treated with fewer threading injections rather than with multiple depot injections," he said.
Dr. Higgins reported having no relevant financial disclosures.
ATLANTA – The use of a threading technique, rather than the standard depot injection technique, when using botulinum toxin A to treat perioral and glabellar rhytides provides a more uniform and natural cosmetic result, according to Dr. H. William Higgins II.
Threading involves injecting the muscle along its normal anatomic course to paralyze the related muscle more evenly, he explained at the annual meeting of the American Society for Dermatologic Surgery.
For upper and lower lip treatment, for example, injections are made at a 20- to 30-degree angle, entering the skin at a location just lateral to the targeted rhytid. The toxin is dispensed while withdrawing, thereby threading the injection along the length of the orbicularis oris. This differs from the typical approach, which often involves a depot injection at an angle more perpendicular to the skin, said Dr. Higgins of Brown University in Providence, R.I.
For the glabellar lines, the threading technique involves four symmetrical injection points, with two points targeting each corrugator. Injections at the more medial points are made directly above the inner canthus, with intramuscular injections made perpendicularly to the skin in the traditional depot manner.
At the two lateral injection points, however, the needle is inserted in most cases just medial to the mid-pupillary lines, thereby targeting the "tail" of the corrugators, he explained.
"Similar to our approach at the orbicularis oris, rather than injecting at an angle more perpendicular to the skin, we inject at an angle of roughly 20-30 degrees, entering the skin at a location just medial to the glabellar rhytid we intend to treat. The needle is then directed laterally and slightly superiorly in order to follow the anatomy of the corrugator supercilii, and the injection is threaded along the muscle’s length while withdrawing," he explained.
This approach corrects for the inadequate responses sometimes seen when using the typical method of placing subepidermal blebs to produce localized microparesis of the targeted muscle, and could reduce the need for touch-up injections.
Cosmetic outcomes have been excellent and patient satisfaction high with the use of this technique, he said. In his experience, the technique has dramatically reduced the incidence of adverse effects.
"It has been documented that, even with conservative dosing, neuromodulator treatment of perioral rhytides can affect mouth function by weakening the lip sphincter, but this has not been the case in our patient population when using this technique," he said.
Similarly, when treating glabellar rhytides, the injection of the toxin at a more precise depth – and more evenly along the tail of the corrugators, has resulted in a reduced incidence of brow ptosis as well as more natural smoothing.
"This approach helps prevent the undesirable appearance of a 'forehead freeze,' " he said.
The threading technique also results in fewer needle sticks, which means less pain and bruising for the patients.
The use of a longer 1- or 1.5-inch needle could potentially allow for even fewer injections without compromising the result, Dr. Higgins noted.
"Furthermore, this technique could conceivably be applied on other areas of the face. Crow's feet, for example, could be treated with fewer threading injections rather than with multiple depot injections," he said.
Dr. Higgins reported having no relevant financial disclosures.
ATLANTA – The use of a threading technique, rather than the standard depot injection technique, when using botulinum toxin A to treat perioral and glabellar rhytides provides a more uniform and natural cosmetic result, according to Dr. H. William Higgins II.
Threading involves injecting the muscle along its normal anatomic course to paralyze the related muscle more evenly, he explained at the annual meeting of the American Society for Dermatologic Surgery.
For upper and lower lip treatment, for example, injections are made at a 20- to 30-degree angle, entering the skin at a location just lateral to the targeted rhytid. The toxin is dispensed while withdrawing, thereby threading the injection along the length of the orbicularis oris. This differs from the typical approach, which often involves a depot injection at an angle more perpendicular to the skin, said Dr. Higgins of Brown University in Providence, R.I.
For the glabellar lines, the threading technique involves four symmetrical injection points, with two points targeting each corrugator. Injections at the more medial points are made directly above the inner canthus, with intramuscular injections made perpendicularly to the skin in the traditional depot manner.
At the two lateral injection points, however, the needle is inserted in most cases just medial to the mid-pupillary lines, thereby targeting the "tail" of the corrugators, he explained.
"Similar to our approach at the orbicularis oris, rather than injecting at an angle more perpendicular to the skin, we inject at an angle of roughly 20-30 degrees, entering the skin at a location just medial to the glabellar rhytid we intend to treat. The needle is then directed laterally and slightly superiorly in order to follow the anatomy of the corrugator supercilii, and the injection is threaded along the muscle’s length while withdrawing," he explained.
This approach corrects for the inadequate responses sometimes seen when using the typical method of placing subepidermal blebs to produce localized microparesis of the targeted muscle, and could reduce the need for touch-up injections.
Cosmetic outcomes have been excellent and patient satisfaction high with the use of this technique, he said. In his experience, the technique has dramatically reduced the incidence of adverse effects.
"It has been documented that, even with conservative dosing, neuromodulator treatment of perioral rhytides can affect mouth function by weakening the lip sphincter, but this has not been the case in our patient population when using this technique," he said.
Similarly, when treating glabellar rhytides, the injection of the toxin at a more precise depth – and more evenly along the tail of the corrugators, has resulted in a reduced incidence of brow ptosis as well as more natural smoothing.
"This approach helps prevent the undesirable appearance of a 'forehead freeze,' " he said.
The threading technique also results in fewer needle sticks, which means less pain and bruising for the patients.
The use of a longer 1- or 1.5-inch needle could potentially allow for even fewer injections without compromising the result, Dr. Higgins noted.
"Furthermore, this technique could conceivably be applied on other areas of the face. Crow's feet, for example, could be treated with fewer threading injections rather than with multiple depot injections," he said.
Dr. Higgins reported having no relevant financial disclosures.
EXPERT ANALYSIS FROM THE ANNUAL MEETING OF THE AMERICAN SOCIETY FOR DERMATOLOGIC SURGERY
Blogging Best Practices
Most physicians have a practice website, a static "digital storefront" that provides patients with basic information such as location, hours, and staff bios. That was sufficient once; it’s not any longer.
A blog, short for web-log, gives you the opportunity to create a dynamic site, with fresh, accurate, current information. If you’re on Twitter and Facebook, you might think a blog is unnecessary. You’d be wrong. In fact, it’s the ideal source for material to share on social sites like Facebook.
Starting a blog is easy and inexpensive. It can even be free with sites like WordPress or Tumblr (both of which I use). I’m unable to go into details here, but tutorials can easily be found online. Blogging sites worth reading include Copyblogger, The Minimalists, and Chris Brogan.
So why should you blog? The top reasons include:
• Patient education. Eighty percent of people who are online have searched for health information. I’m sure you have had patients share erroneous medical information with you that they’ve found online. Instead of just complaining about it, we can do something to change it: Create and share good content for both your current and prospective patients.
• Become a trusted spokesperson or expert. Blogging regularly and sharing content on social sites like Twitter and Facebook, provides writers, editors, and producers the opportunity to contact you. Being quoted in a national magazine or appearing on a local television show is also a great way to reach new patients. Remember, too, that regularly updated blogs are frequently crawled by search engines, which means over time, more traffic will come to your blog.
• Become a valued member of the community. Whether it’s to help promote a local race for psoriasis or to educate the community about a measles outbreak, you can use your blog to reach out in a positive way. This is also a great way for doctors new to a neighborhood to find patients.
• Show your personable side. More than ever before, patients are searching online to find the right physician. When patients read your blog and watch you in a video, they begin to establish trust.
• Reduce workload. No, I’m not being sarcastic. We all have instructions and advice that we repeat verbatim to our patients over and over. Instead of having to do this all the time, write a blog post or do a short video that will live forever. This is especially important for postoperative instructions that patients may like to watch at home. It’s also helpful for caregivers who weren’t at the visit.
As for blogging best practices, if you can’t do it alone (and most of us can’t because of time constraints), enlist the help of trusted office staff. Assign a blog manager who is responsible for an editorial calendar, updates, and responding to comments in a timely manner.
Use your blog for patient education and outreach, not marketing. Readers want value. Bombard them with product and procedure pushing, and they’ll run away.
Be authentic, honest, and transparent.
Be conversational and engaging. Patients don’t want to read doctor speak. However, that doesn’t mean you can’t include studies, statistics, and the like.
Never write about a specific patient by name or in a way that he or she could be re-identified or that violates HIPAA.
Tell stories. Readers remember them.
Write clearly and concisely, keeping blog posts under 400 words.
Post a minimum of once a week; however, two to three times a week is best for search engines.
Respond to both positive and negative comments in a professional, nonconfrontational manner.
Offer an RSS feed so people can easily follow along.
Be patient. It may take time for people to find your blog, but once they do, you’ll feel both personally and professionally rewarded.
Dr. Benabio is in private practice in San Diego. Visit his consumer health blog at http://thedermblog.com; connect with him on Twitter @Dermdoc and on Facebook (DermDoc).
Most physicians have a practice website, a static "digital storefront" that provides patients with basic information such as location, hours, and staff bios. That was sufficient once; it’s not any longer.
A blog, short for web-log, gives you the opportunity to create a dynamic site, with fresh, accurate, current information. If you’re on Twitter and Facebook, you might think a blog is unnecessary. You’d be wrong. In fact, it’s the ideal source for material to share on social sites like Facebook.
Starting a blog is easy and inexpensive. It can even be free with sites like WordPress or Tumblr (both of which I use). I’m unable to go into details here, but tutorials can easily be found online. Blogging sites worth reading include Copyblogger, The Minimalists, and Chris Brogan.
So why should you blog? The top reasons include:
• Patient education. Eighty percent of people who are online have searched for health information. I’m sure you have had patients share erroneous medical information with you that they’ve found online. Instead of just complaining about it, we can do something to change it: Create and share good content for both your current and prospective patients.
• Become a trusted spokesperson or expert. Blogging regularly and sharing content on social sites like Twitter and Facebook, provides writers, editors, and producers the opportunity to contact you. Being quoted in a national magazine or appearing on a local television show is also a great way to reach new patients. Remember, too, that regularly updated blogs are frequently crawled by search engines, which means over time, more traffic will come to your blog.
• Become a valued member of the community. Whether it’s to help promote a local race for psoriasis or to educate the community about a measles outbreak, you can use your blog to reach out in a positive way. This is also a great way for doctors new to a neighborhood to find patients.
• Show your personable side. More than ever before, patients are searching online to find the right physician. When patients read your blog and watch you in a video, they begin to establish trust.
• Reduce workload. No, I’m not being sarcastic. We all have instructions and advice that we repeat verbatim to our patients over and over. Instead of having to do this all the time, write a blog post or do a short video that will live forever. This is especially important for postoperative instructions that patients may like to watch at home. It’s also helpful for caregivers who weren’t at the visit.
As for blogging best practices, if you can’t do it alone (and most of us can’t because of time constraints), enlist the help of trusted office staff. Assign a blog manager who is responsible for an editorial calendar, updates, and responding to comments in a timely manner.
Use your blog for patient education and outreach, not marketing. Readers want value. Bombard them with product and procedure pushing, and they’ll run away.
Be authentic, honest, and transparent.
Be conversational and engaging. Patients don’t want to read doctor speak. However, that doesn’t mean you can’t include studies, statistics, and the like.
Never write about a specific patient by name or in a way that he or she could be re-identified or that violates HIPAA.
Tell stories. Readers remember them.
Write clearly and concisely, keeping blog posts under 400 words.
Post a minimum of once a week; however, two to three times a week is best for search engines.
Respond to both positive and negative comments in a professional, nonconfrontational manner.
Offer an RSS feed so people can easily follow along.
Be patient. It may take time for people to find your blog, but once they do, you’ll feel both personally and professionally rewarded.
Dr. Benabio is in private practice in San Diego. Visit his consumer health blog at http://thedermblog.com; connect with him on Twitter @Dermdoc and on Facebook (DermDoc).
Most physicians have a practice website, a static "digital storefront" that provides patients with basic information such as location, hours, and staff bios. That was sufficient once; it’s not any longer.
A blog, short for web-log, gives you the opportunity to create a dynamic site, with fresh, accurate, current information. If you’re on Twitter and Facebook, you might think a blog is unnecessary. You’d be wrong. In fact, it’s the ideal source for material to share on social sites like Facebook.
Starting a blog is easy and inexpensive. It can even be free with sites like WordPress or Tumblr (both of which I use). I’m unable to go into details here, but tutorials can easily be found online. Blogging sites worth reading include Copyblogger, The Minimalists, and Chris Brogan.
So why should you blog? The top reasons include:
• Patient education. Eighty percent of people who are online have searched for health information. I’m sure you have had patients share erroneous medical information with you that they’ve found online. Instead of just complaining about it, we can do something to change it: Create and share good content for both your current and prospective patients.
• Become a trusted spokesperson or expert. Blogging regularly and sharing content on social sites like Twitter and Facebook, provides writers, editors, and producers the opportunity to contact you. Being quoted in a national magazine or appearing on a local television show is also a great way to reach new patients. Remember, too, that regularly updated blogs are frequently crawled by search engines, which means over time, more traffic will come to your blog.
• Become a valued member of the community. Whether it’s to help promote a local race for psoriasis or to educate the community about a measles outbreak, you can use your blog to reach out in a positive way. This is also a great way for doctors new to a neighborhood to find patients.
• Show your personable side. More than ever before, patients are searching online to find the right physician. When patients read your blog and watch you in a video, they begin to establish trust.
• Reduce workload. No, I’m not being sarcastic. We all have instructions and advice that we repeat verbatim to our patients over and over. Instead of having to do this all the time, write a blog post or do a short video that will live forever. This is especially important for postoperative instructions that patients may like to watch at home. It’s also helpful for caregivers who weren’t at the visit.
As for blogging best practices, if you can’t do it alone (and most of us can’t because of time constraints), enlist the help of trusted office staff. Assign a blog manager who is responsible for an editorial calendar, updates, and responding to comments in a timely manner.
Use your blog for patient education and outreach, not marketing. Readers want value. Bombard them with product and procedure pushing, and they’ll run away.
Be authentic, honest, and transparent.
Be conversational and engaging. Patients don’t want to read doctor speak. However, that doesn’t mean you can’t include studies, statistics, and the like.
Never write about a specific patient by name or in a way that he or she could be re-identified or that violates HIPAA.
Tell stories. Readers remember them.
Write clearly and concisely, keeping blog posts under 400 words.
Post a minimum of once a week; however, two to three times a week is best for search engines.
Respond to both positive and negative comments in a professional, nonconfrontational manner.
Offer an RSS feed so people can easily follow along.
Be patient. It may take time for people to find your blog, but once they do, you’ll feel both personally and professionally rewarded.
Dr. Benabio is in private practice in San Diego. Visit his consumer health blog at http://thedermblog.com; connect with him on Twitter @Dermdoc and on Facebook (DermDoc).

Pediatric Psychiatry Services Infiltrate Primary Care
SAN FRANCISCO – After more than 2 decades as a primary care pediatrician, Dr. Teresa M. Hargrave was so frustrated by the lack of psychiatric services for her patients that she retrained as a child and adolescent psychiatrist. Now, she’s part of a New York state program that spreads her psychiatric skills to more patients than she imagined could be possible.
"If this program had been in place when I was a pediatrician, I would never have had to switch," said Dr. Hargrave of the State University of New York (SUNY) in Syracuse.

Today, New York primary care physicians can call 855-227-7272 toll free on weekdays for an immediate consultation with a master’s level therapist in the Child and Adolescent Psychiatry for Primary Care program (CAP PC). If a patient seems to need psychotropic medication, the therapist connects the pediatrician with a psychiatrist on the program’s team, such as Dr. Hargrave, who helps the primary care physician manage treatment through phone consultations and, if needed, in-person assessments.
Dozens of similar efforts – in a variety of formats – have sprung up across the country. They’re all trying to address a fundamental mismatch: There are only 7,400 practicing child and adolescent psychiatrists in the United States but more than 15 million young people in those age groups who need psychiatric care, according to data analyses from the American Academy of Child and Adolescent Psychiatry.
The National Network of Child Psychiatry Access Programs acts as a hub for these programs in 24 states, with programs in 4 more states set to take their first calls soon.
These model programs are making great inroads in getting care to the estimated 15%-25% of children seen in primary care offices who have behavioral health disorders, but reimbursement problems create a roadblock that must be overcome in the years ahead for the programs to be fully effective, several experts said in interviews at the annual meeting of the American Academy of Child and Adolescent Psychiatry.
New York Program
New York’s CAP PC program modeled itself after one of the first state-wide programs, the Massachusetts Child Psychiatry Access Project, with some key changes. The CAP PC program covers 95% of the New York state population but uses the same toll-free number everywhere, compared with multiple different phone numbers being used in different regions in Massachusetts. New York’s program also added an educational component for primary care physicians – a free 15-hour "Mini-Fellowship" weekend program followed by a dozen 1-hour biweekly case-based conference calls.

Primary care physicians seem to love the help, Dr. David Kaye said at a poster presentation at the meeting. In its 2 years of operation, the CAP PC program has registered 829 primary care physicians (80% pediatricians, 20% family physicians), 292 of whom took the training sessions. The program handled 1,016 intake and follow-up calls, provided 993 consultations with a psychiatrist, conducted 94 face-to-face evaluations, and referred 305 patients to other services, reported Dr. Kaye, professor of psychiatry and director of child and adolescent psychiatry training at SUNY in Buffalo, N.Y.
Among 325 primary care physicians surveyed 2 weeks after contact with the CAP PC program, 94% said the consultations were very or extremely helpful, and 99% said they would recommend the program to other primary care physicians.
The program has greatly increased the number of children accessing psychiatric services compared with a previous pilot program in central New York that provided immediate telephone referrals and psychiatric consultation within 24 hours of a request, Dr. Hargrave said in a separate poster presentation at the meeting.
The CAP PC program improved upon the pilot by offering psychiatric consultation within 2 hours of a request, occasional in-person consultations, the education program, and a centralized computer database that allows the therapists and psychiatrists on different shifts to access patient records quickly, she said.
Compared with data from 2 years of the pilot program, data from the CAP PC program in the central New York area showed an increase in the number of children served from 6 to 14 per month (a 133% gain), an increase in the number of clinicians involved from 77 to 116 per month (a 51% gain), and an increase in the proportion of patients managed within the primary care office because of a decrease in the rate of referrals to more expensive specialists from 39% to 22%, Dr. Hargrave reported.
"The amount of morbidity that primary care physicians are coping with is amazing," especially in rural areas, she said.
Texas Model
A different model in Texas significantly decreased psychiatric symptoms and improved quality of life in children and adolescents participating in the program, Dr. Steven R. Pliszka reported in another poster presentation.

The Services Uniting Pediatrics and Psychiatry Outreaching to Texas (SUPPORT) program, funded by the Department of State Health Services, placed master’s level licensed therapists into primary care pediatric practices in six regions across the state. These therapists tried to see patients the same day that pediatricians referred them, and typically saw each patient for one to six sessions of practical, problem-focused therapies. A consulting child and adolescent psychiatrist helped determine which patients might need psychotropic medication and advised pediatricians on drug choice, dosing, and monitoring.
The SUPPORT program enrolled 145 pediatricians and 14,582 children covered by Medicaid. The outcomes evaluation involved a subset of 4,047 patients who were assessed at baseline, 3 month, and 6 months using the Child Behavior Checklist (CBCL) and the Pediatric Quality of Life Inventory (PedsQL).
In both younger (1.5-5 years of age) and older children (5-18 years), scores significantly decreased on the internalizing, externalizing, and total scales of the CBCL as well as on the individual symptom scales. Scores on the PedsQL improved significantly in each of four age groups (2-4 years, 5-7 years, 8-12 years, and 13-18 years), said Dr. Pliszka, professor and chair of child and adolescent psychiatry at the University of Texas at San Antonio.
Mean total scores on the CBCL, for example, decreased from approximately 63 to about 53 at 6 months. Mean PedsQL scores at baseline ranged approximately from 68 to 71 at baseline (depending on the age group) and increased to a range of about 77-81.
Data on diagnoses and prescriptions tracked by the program suggest that the pediatricians prescribed appropriate medications to the 2,207 patients who received at least one psychotropic medication (15% of all patients), Dr. Pliszka said.
"So, kids with ADHD got treated with a stimulant, kids with depression got an antidepressant, [and] kids with bipolar disorder got combinations of different medications. We also did not have any really bad outcomes. There were no suicides, no serious adverse drug effects. It shows that the model is a way to treat even fairly serious mental illnesses in the primary care setting," he said.
Dr. Pliszka and his associates next plan to compare outcomes for patients managed through SUPPORT and usual care (referral by primary care physicians to mental health clinics in the community).
Reimbursement Issues
Government and academic funds support these programs for now, but better funding mechanisms for collaborative care are needed for long-term sustainability, each of the physicians interviewed said.
New York’s CAP PC is a collaboration among five academic centers that is funded by a grant from the State Office of Mental Health. The SUPPORT program received Medicaid support in Texas.
While there probably are enough master’s level therapists to expand SUPPORT beyond the Medicaid population, "what’s lacking is that it’s difficult for both the pediatrician and the master’s level person to get reimbursed for that type of activity because they use completely different codes," Dr. Pliszka said. "Projects of this type would make the argument for modifying the reimbursement system to allow more integrated care."
Part of CAP PC’s education program helps New York primary care physicians get comfortable with coding for their mental health work, but there are gaps in that approach, Dr. Kaye said. "In some of our regions, docs can be paid reasonably for what they’re doing, but in lots of places, they can’t put in a code for ADHD or depression and get reimbursed" because insurers say they’re not credentialed mental health providers.
"There’s got to be a way on the payment side that Medicaid and/or the insurers figure out how to pay primary care docs to do this work, and to pay them fairly," he said. "I think this is going to be a huge part of the future of primary care. The numbers are that mental health problems are the most common chronic condition that kids get."
Even for the psychiatrists involved, the current model is not sustainable, he added. The New York grant pays each of the five academic centers for a 10-hour day of consultation each week, which is far less than the actual hours contributed.
"We’re all university based. We believe in the project, so we’ve been able to sustain that. Can we do that for 20 years? I don’t know," Dr. Kaye said.
"The major drawback is that it takes time, and insurance does not reimburse for that time. To really get such a system as this off the ground or well integrated" will require reimbursement for the time spent by all the health care providers involved, Dr. Hargrave said.
She said she hopes that in the future, all children and primary care clinicians will have access to mental health care, advice and support, "and that the clinicians – whether primary care or psychiatric – could be paid adequately for the work that we do."
Dr. Pliszka reported financial associations with Shire Pharmaceuticals and Ortho-McNeil-Janssen Pharmaceuticals. Dr. Kaye and Dr. Hargrave received research support from the New York State Office of Mental Health. Some of their coinvestigators reported financial associations with the Resource for Advancing Children’s Health Institute, American Psychiatric Publishing, Marriott Foundation, Shire Pharmaceuticals, and Ortho-McNeil-Janssen.
SAN FRANCISCO – After more than 2 decades as a primary care pediatrician, Dr. Teresa M. Hargrave was so frustrated by the lack of psychiatric services for her patients that she retrained as a child and adolescent psychiatrist. Now, she’s part of a New York state program that spreads her psychiatric skills to more patients than she imagined could be possible.
"If this program had been in place when I was a pediatrician, I would never have had to switch," said Dr. Hargrave of the State University of New York (SUNY) in Syracuse.
Today, New York primary care physicians can call 855-227-7272 toll free on weekdays for an immediate consultation with a master’s level therapist in the Child and Adolescent Psychiatry for Primary Care program (CAP PC). If a patient seems to need psychotropic medication, the therapist connects the pediatrician with a psychiatrist on the program’s team, such as Dr. Hargrave, who helps the primary care physician manage treatment through phone consultations and, if needed, in-person assessments.
Dozens of similar efforts – in a variety of formats – have sprung up across the country. They’re all trying to address a fundamental mismatch: There are only 7,400 practicing child and adolescent psychiatrists in the United States but more than 15 million young people in those age groups who need psychiatric care, according to data analyses from the American Academy of Child and Adolescent Psychiatry.
The National Network of Child Psychiatry Access Programs acts as a hub for these programs in 24 states, with programs in 4 more states set to take their first calls soon.
These model programs are making great inroads in getting care to the estimated 15%-25% of children seen in primary care offices who have behavioral health disorders, but reimbursement problems create a roadblock that must be overcome in the years ahead for the programs to be fully effective, several experts said in interviews at the annual meeting of the American Academy of Child and Adolescent Psychiatry.
New York Program
New York’s CAP PC program modeled itself after one of the first state-wide programs, the Massachusetts Child Psychiatry Access Project, with some key changes. The CAP PC program covers 95% of the New York state population but uses the same toll-free number everywhere, compared with multiple different phone numbers being used in different regions in Massachusetts. New York’s program also added an educational component for primary care physicians – a free 15-hour "Mini-Fellowship" weekend program followed by a dozen 1-hour biweekly case-based conference calls.
Primary care physicians seem to love the help, Dr. David Kaye said at a poster presentation at the meeting. In its 2 years of operation, the CAP PC program has registered 829 primary care physicians (80% pediatricians, 20% family physicians), 292 of whom took the training sessions. The program handled 1,016 intake and follow-up calls, provided 993 consultations with a psychiatrist, conducted 94 face-to-face evaluations, and referred 305 patients to other services, reported Dr. Kaye, professor of psychiatry and director of child and adolescent psychiatry training at SUNY in Buffalo, N.Y.
Among 325 primary care physicians surveyed 2 weeks after contact with the CAP PC program, 94% said the consultations were very or extremely helpful, and 99% said they would recommend the program to other primary care physicians.
The program has greatly increased the number of children accessing psychiatric services compared with a previous pilot program in central New York that provided immediate telephone referrals and psychiatric consultation within 24 hours of a request, Dr. Hargrave said in a separate poster presentation at the meeting.
The CAP PC program improved upon the pilot by offering psychiatric consultation within 2 hours of a request, occasional in-person consultations, the education program, and a centralized computer database that allows the therapists and psychiatrists on different shifts to access patient records quickly, she said.
Compared with data from 2 years of the pilot program, data from the CAP PC program in the central New York area showed an increase in the number of children served from 6 to 14 per month (a 133% gain), an increase in the number of clinicians involved from 77 to 116 per month (a 51% gain), and an increase in the proportion of patients managed within the primary care office because of a decrease in the rate of referrals to more expensive specialists from 39% to 22%, Dr. Hargrave reported.
"The amount of morbidity that primary care physicians are coping with is amazing," especially in rural areas, she said.
Texas Model
A different model in Texas significantly decreased psychiatric symptoms and improved quality of life in children and adolescents participating in the program, Dr. Steven R. Pliszka reported in another poster presentation.
The Services Uniting Pediatrics and Psychiatry Outreaching to Texas (SUPPORT) program, funded by the Department of State Health Services, placed master’s level licensed therapists into primary care pediatric practices in six regions across the state. These therapists tried to see patients the same day that pediatricians referred them, and typically saw each patient for one to six sessions of practical, problem-focused therapies. A consulting child and adolescent psychiatrist helped determine which patients might need psychotropic medication and advised pediatricians on drug choice, dosing, and monitoring.
The SUPPORT program enrolled 145 pediatricians and 14,582 children covered by Medicaid. The outcomes evaluation involved a subset of 4,047 patients who were assessed at baseline, 3 month, and 6 months using the Child Behavior Checklist (CBCL) and the Pediatric Quality of Life Inventory (PedsQL).
In both younger (1.5-5 years of age) and older children (5-18 years), scores significantly decreased on the internalizing, externalizing, and total scales of the CBCL as well as on the individual symptom scales. Scores on the PedsQL improved significantly in each of four age groups (2-4 years, 5-7 years, 8-12 years, and 13-18 years), said Dr. Pliszka, professor and chair of child and adolescent psychiatry at the University of Texas at San Antonio.
Mean total scores on the CBCL, for example, decreased from approximately 63 to about 53 at 6 months. Mean PedsQL scores at baseline ranged approximately from 68 to 71 at baseline (depending on the age group) and increased to a range of about 77-81.
Data on diagnoses and prescriptions tracked by the program suggest that the pediatricians prescribed appropriate medications to the 2,207 patients who received at least one psychotropic medication (15% of all patients), Dr. Pliszka said.
"So, kids with ADHD got treated with a stimulant, kids with depression got an antidepressant, [and] kids with bipolar disorder got combinations of different medications. We also did not have any really bad outcomes. There were no suicides, no serious adverse drug effects. It shows that the model is a way to treat even fairly serious mental illnesses in the primary care setting," he said.
Dr. Pliszka and his associates next plan to compare outcomes for patients managed through SUPPORT and usual care (referral by primary care physicians to mental health clinics in the community).
Reimbursement Issues
Government and academic funds support these programs for now, but better funding mechanisms for collaborative care are needed for long-term sustainability, each of the physicians interviewed said.
New York’s CAP PC is a collaboration among five academic centers that is funded by a grant from the State Office of Mental Health. The SUPPORT program received Medicaid support in Texas.
While there probably are enough master’s level therapists to expand SUPPORT beyond the Medicaid population, "what’s lacking is that it’s difficult for both the pediatrician and the master’s level person to get reimbursed for that type of activity because they use completely different codes," Dr. Pliszka said. "Projects of this type would make the argument for modifying the reimbursement system to allow more integrated care."
Part of CAP PC’s education program helps New York primary care physicians get comfortable with coding for their mental health work, but there are gaps in that approach, Dr. Kaye said. "In some of our regions, docs can be paid reasonably for what they’re doing, but in lots of places, they can’t put in a code for ADHD or depression and get reimbursed" because insurers say they’re not credentialed mental health providers.
"There’s got to be a way on the payment side that Medicaid and/or the insurers figure out how to pay primary care docs to do this work, and to pay them fairly," he said. "I think this is going to be a huge part of the future of primary care. The numbers are that mental health problems are the most common chronic condition that kids get."
Even for the psychiatrists involved, the current model is not sustainable, he added. The New York grant pays each of the five academic centers for a 10-hour day of consultation each week, which is far less than the actual hours contributed.
"We’re all university based. We believe in the project, so we’ve been able to sustain that. Can we do that for 20 years? I don’t know," Dr. Kaye said.
"The major drawback is that it takes time, and insurance does not reimburse for that time. To really get such a system as this off the ground or well integrated" will require reimbursement for the time spent by all the health care providers involved, Dr. Hargrave said.
She said she hopes that in the future, all children and primary care clinicians will have access to mental health care, advice and support, "and that the clinicians – whether primary care or psychiatric – could be paid adequately for the work that we do."
Dr. Pliszka reported financial associations with Shire Pharmaceuticals and Ortho-McNeil-Janssen Pharmaceuticals. Dr. Kaye and Dr. Hargrave received research support from the New York State Office of Mental Health. Some of their coinvestigators reported financial associations with the Resource for Advancing Children’s Health Institute, American Psychiatric Publishing, Marriott Foundation, Shire Pharmaceuticals, and Ortho-McNeil-Janssen.
SAN FRANCISCO – After more than 2 decades as a primary care pediatrician, Dr. Teresa M. Hargrave was so frustrated by the lack of psychiatric services for her patients that she retrained as a child and adolescent psychiatrist. Now, she’s part of a New York state program that spreads her psychiatric skills to more patients than she imagined could be possible.
"If this program had been in place when I was a pediatrician, I would never have had to switch," said Dr. Hargrave of the State University of New York (SUNY) in Syracuse.
Today, New York primary care physicians can call 855-227-7272 toll free on weekdays for an immediate consultation with a master’s level therapist in the Child and Adolescent Psychiatry for Primary Care program (CAP PC). If a patient seems to need psychotropic medication, the therapist connects the pediatrician with a psychiatrist on the program’s team, such as Dr. Hargrave, who helps the primary care physician manage treatment through phone consultations and, if needed, in-person assessments.
Dozens of similar efforts – in a variety of formats – have sprung up across the country. They’re all trying to address a fundamental mismatch: There are only 7,400 practicing child and adolescent psychiatrists in the United States but more than 15 million young people in those age groups who need psychiatric care, according to data analyses from the American Academy of Child and Adolescent Psychiatry.
The National Network of Child Psychiatry Access Programs acts as a hub for these programs in 24 states, with programs in 4 more states set to take their first calls soon.
These model programs are making great inroads in getting care to the estimated 15%-25% of children seen in primary care offices who have behavioral health disorders, but reimbursement problems create a roadblock that must be overcome in the years ahead for the programs to be fully effective, several experts said in interviews at the annual meeting of the American Academy of Child and Adolescent Psychiatry.
New York Program
New York’s CAP PC program modeled itself after one of the first state-wide programs, the Massachusetts Child Psychiatry Access Project, with some key changes. The CAP PC program covers 95% of the New York state population but uses the same toll-free number everywhere, compared with multiple different phone numbers being used in different regions in Massachusetts. New York’s program also added an educational component for primary care physicians – a free 15-hour "Mini-Fellowship" weekend program followed by a dozen 1-hour biweekly case-based conference calls.
Primary care physicians seem to love the help, Dr. David Kaye said at a poster presentation at the meeting. In its 2 years of operation, the CAP PC program has registered 829 primary care physicians (80% pediatricians, 20% family physicians), 292 of whom took the training sessions. The program handled 1,016 intake and follow-up calls, provided 993 consultations with a psychiatrist, conducted 94 face-to-face evaluations, and referred 305 patients to other services, reported Dr. Kaye, professor of psychiatry and director of child and adolescent psychiatry training at SUNY in Buffalo, N.Y.
Among 325 primary care physicians surveyed 2 weeks after contact with the CAP PC program, 94% said the consultations were very or extremely helpful, and 99% said they would recommend the program to other primary care physicians.
The program has greatly increased the number of children accessing psychiatric services compared with a previous pilot program in central New York that provided immediate telephone referrals and psychiatric consultation within 24 hours of a request, Dr. Hargrave said in a separate poster presentation at the meeting.
The CAP PC program improved upon the pilot by offering psychiatric consultation within 2 hours of a request, occasional in-person consultations, the education program, and a centralized computer database that allows the therapists and psychiatrists on different shifts to access patient records quickly, she said.
Compared with data from 2 years of the pilot program, data from the CAP PC program in the central New York area showed an increase in the number of children served from 6 to 14 per month (a 133% gain), an increase in the number of clinicians involved from 77 to 116 per month (a 51% gain), and an increase in the proportion of patients managed within the primary care office because of a decrease in the rate of referrals to more expensive specialists from 39% to 22%, Dr. Hargrave reported.
"The amount of morbidity that primary care physicians are coping with is amazing," especially in rural areas, she said.
Texas Model
A different model in Texas significantly decreased psychiatric symptoms and improved quality of life in children and adolescents participating in the program, Dr. Steven R. Pliszka reported in another poster presentation.
The Services Uniting Pediatrics and Psychiatry Outreaching to Texas (SUPPORT) program, funded by the Department of State Health Services, placed master’s level licensed therapists into primary care pediatric practices in six regions across the state. These therapists tried to see patients the same day that pediatricians referred them, and typically saw each patient for one to six sessions of practical, problem-focused therapies. A consulting child and adolescent psychiatrist helped determine which patients might need psychotropic medication and advised pediatricians on drug choice, dosing, and monitoring.
The SUPPORT program enrolled 145 pediatricians and 14,582 children covered by Medicaid. The outcomes evaluation involved a subset of 4,047 patients who were assessed at baseline, 3 month, and 6 months using the Child Behavior Checklist (CBCL) and the Pediatric Quality of Life Inventory (PedsQL).
In both younger (1.5-5 years of age) and older children (5-18 years), scores significantly decreased on the internalizing, externalizing, and total scales of the CBCL as well as on the individual symptom scales. Scores on the PedsQL improved significantly in each of four age groups (2-4 years, 5-7 years, 8-12 years, and 13-18 years), said Dr. Pliszka, professor and chair of child and adolescent psychiatry at the University of Texas at San Antonio.
Mean total scores on the CBCL, for example, decreased from approximately 63 to about 53 at 6 months. Mean PedsQL scores at baseline ranged approximately from 68 to 71 at baseline (depending on the age group) and increased to a range of about 77-81.
Data on diagnoses and prescriptions tracked by the program suggest that the pediatricians prescribed appropriate medications to the 2,207 patients who received at least one psychotropic medication (15% of all patients), Dr. Pliszka said.
"So, kids with ADHD got treated with a stimulant, kids with depression got an antidepressant, [and] kids with bipolar disorder got combinations of different medications. We also did not have any really bad outcomes. There were no suicides, no serious adverse drug effects. It shows that the model is a way to treat even fairly serious mental illnesses in the primary care setting," he said.
Dr. Pliszka and his associates next plan to compare outcomes for patients managed through SUPPORT and usual care (referral by primary care physicians to mental health clinics in the community).
Reimbursement Issues
Government and academic funds support these programs for now, but better funding mechanisms for collaborative care are needed for long-term sustainability, each of the physicians interviewed said.
New York’s CAP PC is a collaboration among five academic centers that is funded by a grant from the State Office of Mental Health. The SUPPORT program received Medicaid support in Texas.
While there probably are enough master’s level therapists to expand SUPPORT beyond the Medicaid population, "what’s lacking is that it’s difficult for both the pediatrician and the master’s level person to get reimbursed for that type of activity because they use completely different codes," Dr. Pliszka said. "Projects of this type would make the argument for modifying the reimbursement system to allow more integrated care."
Part of CAP PC’s education program helps New York primary care physicians get comfortable with coding for their mental health work, but there are gaps in that approach, Dr. Kaye said. "In some of our regions, docs can be paid reasonably for what they’re doing, but in lots of places, they can’t put in a code for ADHD or depression and get reimbursed" because insurers say they’re not credentialed mental health providers.
"There’s got to be a way on the payment side that Medicaid and/or the insurers figure out how to pay primary care docs to do this work, and to pay them fairly," he said. "I think this is going to be a huge part of the future of primary care. The numbers are that mental health problems are the most common chronic condition that kids get."
Even for the psychiatrists involved, the current model is not sustainable, he added. The New York grant pays each of the five academic centers for a 10-hour day of consultation each week, which is far less than the actual hours contributed.
"We’re all university based. We believe in the project, so we’ve been able to sustain that. Can we do that for 20 years? I don’t know," Dr. Kaye said.
"The major drawback is that it takes time, and insurance does not reimburse for that time. To really get such a system as this off the ground or well integrated" will require reimbursement for the time spent by all the health care providers involved, Dr. Hargrave said.
She said she hopes that in the future, all children and primary care clinicians will have access to mental health care, advice and support, "and that the clinicians – whether primary care or psychiatric – could be paid adequately for the work that we do."
Dr. Pliszka reported financial associations with Shire Pharmaceuticals and Ortho-McNeil-Janssen Pharmaceuticals. Dr. Kaye and Dr. Hargrave received research support from the New York State Office of Mental Health. Some of their coinvestigators reported financial associations with the Resource for Advancing Children’s Health Institute, American Psychiatric Publishing, Marriott Foundation, Shire Pharmaceuticals, and Ortho-McNeil-Janssen.
AT THE ANNUAL MEETING OF THE AMERICAN ACADEMY OF CHILD AND ADOLESCENT PSYCHIATRY
Major Finding: Ninety-four percent of 325 New York primary care physicians who were surveyed rated the CAP PC program as very or extremely helpful. The program increased the number of children served by 133% in one region. The SUPPORT program significantly decreased psychiatric symptoms and improved quality of life scores in Texas children and adolescents.
Data Source: Data are from New York and Texas programs aimed at giving primary care physicians greater access to pediatric and adolescent psychiatric services.
Disclosures: Dr. Pliszka reported financial associations with Shire Pharmaceuticals and Ortho-McNeil-Janssen Pharmaceuticals. Dr. Kaye and Dr. Hargrave received research support from the New York State Office of Mental Health. Some of their coinvestigators reported financial associations with the Resource for Advancing Children’s Health Institute, American Psychiatric Publishing, Marriott Foundation, Shire Pharmaceuticals, and Ortho-McNeil-Janssen.

Treating herpes zoster and postherpetic neuralgia: An evidence-based approach
Postherpetic neuralgia (PHN) is a management challenge—because of its severity, long duration, and potential for debilitation, often in the highly vulnerable elderly population. And, as the most common complication of an acute episode of herpes zoster (shingles) in an immunocompetent person, PHN is likely no stranger to your practice.
Herpes zoster is one of the most common neurological problems, with an incidence of up to 1 million new cases per year in the United States.1 Although the precise number for the prevalence of PHN in the United States is unknown, investigators estimate it at 500,000 to 1 million.2
Major risk factors for development of PHN after an episode of herpes zoster include:
older age
greater acute pain during herpes zoster
greater severity of rash.3,4
PHN is commonly defined as “dermatomal pain that persists 120 days or more after the onset of rash.”5 The pain of PHN has been characterized as a stimulus-dependent continuous burning, throbbing, or episodic sharp electric shock-like sensation6 and as a stimulus-dependent tactile allodynia (ie, pain after normally nonpainful stimulus) and hyperalgesia (exaggerated response to a painful stimulus). In addition, some patients experience myofascial pain secondary to excessive muscle guarding. Chronic pruritus can be present.
More than 90% of patients who have PHN have allodynia,7 which tends to occur in areas where sensation is relatively preserved. Patients also feel spontaneous pain in areas where sensation is lost or impaired.
In this article, we review the evidence for the range of treatments for acute herpes zoster and PHN, as well offer preventive strategies for herpes zoster.
Acute herpes zoster: Start antivirals early
Evidence-based treatment of acute herpes zoster includes antiviral drugs and analgesics.
Antiviral agents suppress viral replication and have a beneficial effect on acute and chronic pain. Acyclovir (800 mg, 5 times a day), valacyclovir (1000 mg, every 8 hours), and famciclovir (500 mg, every 8 hours) are antivirals commonly used to treat herpes zoster. All 3 drugs have comparable efficacy and safety profiles.
In a meta-analysis of patients older than 50 years who were treated with acyclovir or placebo, pain persisted in 15% of the acyclovir-treated group, compared with 35% of the placebo group.8 In terms of duration, a study comparing famciclovir treatment with placebo showed that subjects in the placebo group had persistent pain for 163 days, whereas famciclovir-treated patients had pain for 63 days.9
Based on this evidence, antiviral medications are strongly recommended for treating herpes zoster, especially for patients at increased risk of developing PHN. Antiviral treatment should be started within 72 hours of the onset of the rash.
No good evidence supports the efficacy of antiviral treatment administered 72 hours after the onset of rash. One uncontrolled trial, however, examined the effectiveness of acyclovir started before vs after 72 hours; the difference in pain persistence was not significant between the groups, suggesting acyclovir has benefit even when given after 72 hours.10
In clinical practice, the diagnosis of herpes zoster is often not made within 72 hours of symptom onset; nevertheless, it is important to identify patients who could still benefit from antiviral medication even when treatment is started relatively late in the disease course. This is especially true in ocular zoster, because viral shedding may continue beyond 72 hours.11
Analgesics are part of a practical approach for managing herpes zoster–associated pain that begins with a short-acting opioid in combination with acetaminophen or a nonsteroidal anti-inflammatory (NSAID) agent. Gabapentin or pregabalin, followed by a tricyclic antidepressant, can be added if conventional analgesics are not entirely effective. The analgesic regimen should be tailored to the patient’s needs and tolerance of adverse effects. If pain control is inadequate or adverse effects are intolerable, consider referring the patient to a pain management center for possible interventional modalities.
Key Point Gabapentin or pregabalin, followed by a tricyclic antidepressant, can be added if conventional analgesics are not effective for herpes zoster pain. |
Corticosteroids are not recommended routinely for treatment of herpes zoster; you can try them in otherwise healthy older adults, however, if antiviral therapy and analgesics do not relieve pain. In 2 double-blind controlled trials, a combination of acyclovir and corticosteroids for 21 days did not decrease the incidence of PHN—although some benefit was seen in terms of patients’ return to normal activities, cessation of analgesic therapy, and improved sleep.12,13
Evidence-based treatment options for PHN
Pharmacotherapy for PHN includes anticonvulsants, tricyclic antidepressants, opioids, and topical agents. Invasive interventions have a limited but important role in the management of PHN pain in clinical practice.
Calcium channel-blocking anticonvulsants gabapentin and pregabalin are safe and relatively well tolerated. They can be used as first-line agents for PHN, starting with a low dosage and titrating up, based on effectiveness and tolerability.
Gabapentin is FDA approved for the treatment of PHN. The starting dosage is 100 to 300 mg taken at night, titrated as needed by 100 to 300 mg every 3 to 5 days, to as high a dosage as 1800 to 3600 mg/d in 3 or 4 divided doses. In 2 large, randomized controlled trials, gabapentin produced a statistically significant reduction in pain ratings and improved sleep and quality of life.14,15 Adverse effects include somnolence, dizziness, peripheral edema, visual adverse effects, and gait and balance problems.
Because gabapentin is excreted by the kidneys, take care when using it in patients with renal insufficiency. Gabapentin clearance is linearly related to creatinine clearance and is decreased in the elderly and in individuals with impaired renal function. Hence, the gabapentin dose and the frequency of dosing must be adjusted in these patients.
In patients on hemodialysis, plasma gabapentin levels can be maintained by giving a dose of 200 to 300 mg 4 hours after hemodialysis.16
Extended-release gabapentin. The FDA recently approved an extended-release gabapentin formulation for PHN. Approval was based on a 12-week pivotal study and 2 adjunct studies. In a multicenter, randomized, double-blind, parallel-group, placebo-controlled, 12-week study evaluating the efficacy, safety, and dose response of 3 doses, extended-release gabapentin was effective at 1200 mg/d dosing. The initial recommended dose is 600 mg, once daily for 3 days, followed by 600 mg, twice daily, beginning on Day 4.17 The premise is that the extended-release preparation improves bioavailability of the active drug and, therefore, reduces the incidence of adverse effects, compared with regular gabapentin.
Overall, evidence is mixed. Two randomized controlled trials of extended-release gabapentin showed benefit (ie, reduced pain score on a numerical rating scale) with twice-a-day dosing (600 mg in the morning and 1200 mg at night), compared with a once-daily 1800-mg dose as well as placebo, for reduction in intensity of pain18 and specific pain quality.19 In another trial, however, extended-release gabapentin, 1800 mg once daily, did not show any benefit compared with placebo.20
Pregabalin is also FDA approved for PHN. The effective dosage range is 150 to 600 mg/d. Pregabalin provided significantly superior pain relief and improved sleep scores compared with placebo in 776 patients with PHN.21 Adverse effects include weight gain, dizziness, and somnolence. Titrate the dosage slowly in the elderly.
Sodium channel-blocking anticonvulsants topiramate, lamotrigine, carbamazepine, oxcarbazepine, levetriacetam, and valproic acid are not FDA approved for PHN. These agents may be a treatment option, however, for patients with PHN who do not respond to conventional therapy. In an 8-week randomized controlled trial, patients treated with divalproex sodium (valproic acid and sodium valproate), 1000 mg/d, experienced significant pain relief compared with placebo-treated patients.22 Adverse effects included vertigo, hair loss, headache, nausea, and diarrhea.
Tricyclic antidepressants, including amitriptyline, desipramine, and nortriptyline, might work by (1) inhibiting norepinephrine and serotonin uptake, (2) sodium-channel blockade, or (3) another mechanism that is unclear. Although amitriptyline is the most studied tricyclic antidepressant for PHN, available evidence and clinical experience suggest that nortriptyline and desipramine have comparable efficacy and are better tolerated.23,24
Key Point Available evidence and clinical experience suggest that nortriptyline and desipramine have comparable efficacy and are better tolerated than amitriptyline for PHN. |
Nortriptyline and desipramine are preferred in frail and elderly patients. Start therapy with 10 to 25 mg nightly, titrating as tolerated every 2 weeks to 75 to 150 mg as a single daily dose. Adverse effects include dry mouth, fatigue, dizziness, sedation, urinary retention, orthostatic hypotension, weight gain, blurred vision, QT interval prolongation, constipation, and sexual dysfunction.
Serotonin-norepinephrine reuptake inhibitor (SNRI) antidepressants. Use of such agents as duloxetine and venlafaxine in PHN patients is extrapolated from their proven efficacy in treating diabetic neuropathy and other neuropathic pain conditions. Try duloxetine if your patient does not respond to or tolerate a tricyclic. The recommended dosage is 60 to 120 mg/d in 2 divided doses.24
Two randomized, 12-week, double-blind, placebo-controlled trials using duloxetine 60 mg once a day and 60 mg twice a day for diabetic peripheral neuropathy concluded that 120 mg was safe and effective in treating diabetic peripheral neuropathy, but 120 mg was not as well tolerated as 60 mg once a day.25
Monitor liver function periodically in patients taking duloxetine. Alternatively, you can give venlafaxine; the recommended dosage is 75 to 225 mg/d.26
Opioid analgesics are recommended as second- and third-line agents for PHN. Adverse effects include nausea, pruritus, sedation, confusion, constipation, hypogonadism, and risk of developing tolerance and abuse.
A double-blind crossover trial evaluated the analgesic efficacy of oral oxycodone; treatment resulted in significant reduction of allodynia, steady pain, and spontaneous paroxysmal pain. Oxycodone treatment resulted in superior scores of global effectiveness, disability reduction, and patient preference, compared with placebo.27
In a randomized crossover trial, the combination of gabapentin and morphine was superior to either of these medications alone in relieving pain in PHN.28
Tramadol, an atypical opioid, has a weak μ-opioid receptor agonist effect and inhibits reuptake of serotonin and norepinephrine. Avoid using it in patients with a history of seizures. The maximum recommended dosage is 400 mg/d. An extended-release formulation of tramadol is also available.
Tramadol provided superior pain relief and improved quality of life in PHN patients in a randomized placebo-controlled trial.29
Tapentadol has weak μ-opioid receptor agonist activity; norepinephrine reuptake inhibition is more predominant than serotonin reuptake inhibition. This drug is also available as an extended-release formulation. The maximum recommended dosage is 600 mg/d.
Avoid using tapentadol in patients with a history of seizures. Note: Although there is no scientific evidence regarding the use of tapentadol in neuropathic pain, we use it often in our practice.
Topical therapies
Treating PHN with a topical agent is associated with relatively fewer adverse effects than what has been seen with oral therapy because systemic absorption is minimal.
Lidocaine is available as a transdermal patch and as a topical gel ointment. The 5% lidocaine patch is FDA approved for treating PHN. Lidocaine, a sodium-channel blocker, is useful for treating patients with clinical evidence of allodynia. You can cut a patch to fit the affected area; a maximum of 3 patches can be used simultaneously for 12 hours on, 12 hours off. If helpful, the patch can be left in place for 18 hours.30
In 2 open-labeled, nonrandomized prospective studies, patients treated with the lidocaine patch had reduced intensity of pain and improved quality of life.31,32
If lidocaine patches are not available, or affordable, or if a patient has difficulty applying them, use 5% lidocaine gel instead.
Capsaicin topical cream is sold in 2 concentrations: 0.025% and 0.075%. An extract of hot chili peppers, capsaicin acts as an agonist at the vanilloid receptors. The recommended dosage is 3 or 4 times a day. Initial application causes burning to become worse, but repeated use results in diminished pain and hyperalgesia.
A 6-week, blinded parallel study, followed by a 2-year open label follow-up, showed that the 0.075% dose of topical capsaicin cream relieved pain in 64% of patients; pain was relieved in 25% of placebo-treated patients.33
An 8% capsaicin patch is FDA approved for treating PHN. The patch must be applied by a health care professional in a monitored setting. Prepare the affected area by pretreating it with a local anesthetic cream; then apply the patch and leave it in place for 1 hour. As many as 4 patches can be used at once. A single application can provide pain relief for as long as 12 weeks. Adverse effects are mostly mild and transient.
In a double-blind, randomized, placebo-controlled trial with an open-label extension, the score on a numeric pain-rating scale declined from baseline in both the high-concentration capsaicin group and the placebo group during Week 1; however, the capsaicin-treated group experienced long-term improvement through Week 12.34
(See TABLE 114-21, 23, 24, 27-34 for a summary of pharmacotherapeutic options.)
TABLE 1
Pharmacotherapeutic options for managing postherpetic neuralgia14-21, 23, 24, 27-34
| *Obtain baseline EKG in patients with history of cardiac disease. †May need to start a patient on short-acting opioid medications before changing over to a fentanyl patch. ‡Has a long and unpredictable half-life, hence the need for extra caution in elderly patients. §Has not been studied in neuropathic pain; found to be effective in PHN and other chronic pain conditions. IISingle application has been found to be effective for about 3 months. MAOI, monoamine oxidase inhibitor; PHN, postherpetic neuralgia; SNRI, serotonin-norepinephrine reuptake inhibitor; SSRI, selective serotonin reuptake inhibitor; TCA, tricyclic antidepressant. | ||||
| Medication | Starting dose | Dose titration | Common adverse effects | Cautions and comments |
| Anticonvulsants | ||||
| Gabapentin | 100-300 mg | Start at bedtime and increase to tid dosing; increase by 100-300 mg every 3-5 days to total dose of 1800-3600 mg/d in 3 or 4 divided doses | Somnolence, dizziness, fatigue, ataxia, peripheral edema, weight gain, visual adverse effects | Decrease dose in patients with renal impairment. Dialysis patients: Every-other-day dosing; dosed on the day of dialysis. Avoid sudden discontinuation |
| Extended-release gabapentin | 600 mg daily for 3 days, then 600 mg bid beginning Day 4 | 600 mg bid | Somnolence, dizziness | Recently approved by FDA for PHN; not much clinical experience as yet |
| Pregabalin | 50 mg tid or 75 mg bid | 300-600 mg/d in 2 divided doses for 7-10 days | Somnolence, fatigue, dizziness, peripheral edema and weight gain, blurred vision, and euphoria | Decrease dose in patients with renal impairment. Titrate dosage slowly in elderly patients |
| Tricyclic antidepressants* | ||||
| Amitriptyline Desipramine Nortriptyline | 10-25 mg at bedtime. Start at a lower dose in elderly | Increase as tolerated every 2 weeks, with a target dose of 75-150 mg as a single daily dose | Sedation, dry mouth, blurred vision, weight gain, urinary retention, constipation, sexual dysfunction | Cardiac arrhythmic disease, glaucoma, suicide risk, seizure disorder. Risk of serotonin syndrome with concomitant use of tramadol, SSRIs, or SNRIs. Amitriptyline has the most anticholinergic effects |
| Opioids | ||||
| Fentanyl patch† Methadone‡ Morphine Oxycodone | 12 μg/hour 2.5 mg tid 15 mg q 6 hours prn 5 mg q 6 hours prn | Titrate at weekly intervals balancing analgesia and adverse effects. If patient tolerates the medications, can titrate faster | Nausea and vomiting, constipation, sedation, itching, risk of tolerance and abuse | Driving impairment and cognitive dysfunction during treatment initiation. Be careful in patients with sleep apnea. Additive effects of sedation with neuromodulating medications |
| Atypical opioids | ||||
| Tapentadol§ | 50 mg every 4-6 hours prn | Can titrate up to 100 mg q 4 hours. Maximum daily dose is 600 mg | Nausea and vomiting, constipation, drowsiness, and dizziness | Be careful in patients taking SSRIs, SNRIs, MAOIs, and TCAs. Decrease dose in patients with moderate hepatic and renal impairment. Avoid use in patients with a history of seizures |
| Tramadol | 50 mg every 6 hours prn | Can titrate up to 100 mg q 6 hours. Maximum daily dose: 400 mg. Extended-release dosing once a day | Nausea and vomiting, constipation, drowsiness, dizziness | Be careful in patients with seizure disorder and concomitant use of SSRIs, SNRIs, and TCAs. Decrease dose in patients with hepatic or renal disease |
| Topical agents | ||||
| Lidocaine patch | 5% lidocaine patch | Can use up to 3 patches 12 hours/d | Local erythema, rash, blisters | Contraindicated in patients with known hypersensitivity to amide local anesthetics (eg, bupivacaine, mepivacaine). Do not use on skin with open lesions |
| Topical capsaicin | 0.025% and 0.075% cream | Apply 3-4 times a day over affected region | No systemic adverse effects. Burning and stinging sensation at the application site | Avoid contact with eyes, nose, and mouth. Application of lidocaine gel locally may be helpful prior to capsaicin cream application |
| Capsaicin patchII | 8% single application patch | Need topical local anesthetic application prior to patch application. Patch applied for 1 hour | Local site irritation, burning, temporary increase in pain | Done in a physician’s office under monitored circumstances. Patient may need oral analgesics for a short period following application of the patch |
Alternative modalities to reduce pain
Acupuncture and transcutaneous electrical nerve stimulation (TENS) have been tried for the relief of PHN without consistent evidence of efficacy. There are no significant adverse effects associated with these therapies; however, the cost of treatment may be an issue. Acupuncture is not covered by many insurance carriers. Mental-health interventions, including cognitive and behavioral therapy, might help with overall physical and emotional functioning and quality of life.
Key Point Acupuncture and transcutaneous electrical nerve stimulation do not appear to be effective for PHN relief. |
Invasive interventions
Researchers have examined several interventional modalities for treating PHN that is refractory to medication.
Sympathetic nerve blocks. Retrospective studies have shown that sympathetic nerve block provides short-term improvement in pain in 40% to 50% of patients with PHN.35
Intercostal nerve block has been reported to provide long-lasting pain relief in patients with thoracic PHN.36
Neuraxial use of intrathecal methylprednisone is supported by moderately good evidence of benefit in patients with intractable PHN.37 Because this intervention poses significant risk of neurologic sequelae, we do not recommend that it be used in clinical practice.
Spinal cord stimulation was studied prospectively in a case series of 28 patients.38 Long-term pain relief was obtained in 82%. Patients serve as their own controls by switching off the spinal cord stimulator and monitoring pain. Consider spinal cord stimulation for patients with well-established PHN that is refractory to conventional management.
Cryotherapy was used for facial neuralgia pain, without significant benefit.39 Another trial showed short-term benefit in 11 of 14 patients who underwent cryotherapy of the intercostal nerves for thoracic PHN.40
Botulinium toxin A injection. An abstract presented at the February 2010 meeting of the American Academy of Pain Medicine described how subcutaneous injection of botulinium toxin A reduced pain in patients with PHN, compared with lidocaine and placebo injections. The pain relief was noted in 1 week and persisted for 90 days.41
Surgery. Many surgical interventions have been described and used to treat PHN, but none has a role in clinical practice.
Key Point Many surgical interventions have been used to treat PHN, but none has a role in clinical practice. |
When should you refer to a pain management center?
Dermatomal pain that lasts for longer than 180 days after a herpes zoster rash can be considered “well-established PHN” to denote its refractory nature. As a primary care clinician, you can refer a patient with PHN to a pain management center at any stage of disease but especially when the:
patient has a significant medical comorbidity and you think that he or she requires the services of a specialist to manage multimodal pharmacotherapy
PHN pain is refractory to conventional treatment modalities
- patient needs an invasive intervention
- patient needs treatment with a high-dose capsaicin patch and you have not been trained to apply it.
Preventing herpes zoster and PHN
Obviously, preventing PHN is closely tied to preventing herpes zoster. To help prevent herpes zoster:
vaccinate children with varicella vaccine to prevent primary varicella infection42
use varicella-zoster immunoglobulin, as recommended by the CDC’s Advisory Committee on Immunization Practices (ACIP), in immunocompromised, seronegative patients who were exposed recently to a person with chickenpox or herpes zoster42
administer the herpes zoster vaccine to patients 60 years and older, as recommended by ACIP.43 The FDA recently approved use of this vaccine for people 50 through 59 years, but ACIP has not changed its recommendations.44
As we’ve discussed, herpes zoster vaccination, antiviral therapy, and aggressive pain control can reduce the incidence, severity, and duration of acute herpes zoster and PHN.
A large multicenter, randomized, placebo-controlled trial demonstrated that herpes zoster vaccine decreases the likelihood of developing herpes zoster in immunocompetent individuals 60 years and older.45 The vaccine reduced the incidence of herpes zoster by 51.3%; reduced the burden of illness by 61.1%; and reduced the incidence of PHN by 66.5%.45 The live, attenuated vaccine is contraindicated in children, pregnant women, and immunocompromised individuals.
The number needed to treat for herpes zoster vaccine is 175; that is, 1 case of herpes zoster is avoided for every 175 people vaccinated.1
Key Point One case of herpes zoster is avoided for every 175 people vaccinated. |
Newer tools mean a better outcome
We have improved our ability to diminish the incidence of herpes zoster and PHN and to manage postherpetic pain more effectively. These advances include the development of a herpes zoster vaccine; consensus that antiviral therapy and aggressive pain management can reduce the burden of PHN; identification of efficacious treatments for PHN; and recognition of PHN as a study model for neuropathic pain research.
Disclosure
The authors reported no potential conflict of interest relevant to this article.
References
- Oxman MN, Levin MJ, Johnson GR, et al; Shingles Prevention Study Group. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271–2284.
- Bennett GJ. Neuropathic pain: An overview. In: Borsook D, ed. Molecular Neurobiology of Pain. Seattle, WA: IASP Press; 1997:109-113.
- Jung BF, Johnson RW, Griffin DR, Dworkin RH. Risk factors for postherpetic neuralgia in patients with herpes zoster. Neurology. 2004;62:1545–1551.
- Dworkin RH, Boon RJ, Griffin DR, Phung D. Postherpetic neuralgia: impact of famciclovir, age, rash severity, and acute pain in herpes zoster patients. J Infect Dis. 1998;178(suppl 1):S76–S80.
- Volpi A, Gross G, Hercogova J, Johnson RW. Current management of herpes zoster: the European view. Am J Clin Dermatol. 2005;6:317–325.
- Dworkin RH, Portenoy RK. Pain and its persistence in herpes zoster. Pain. 1996;67:241–251.
- Bowsher D. Pathophysiology of postherpetic neuralgia: towards a rational treatment. Neurology. 1995;45(12 suppl 8):S56–S57.
- Dworkin RH, Schmader KE. Epidemiology and natural history of herpes zoster and postherpetic neuralgia. In Watson CPN, Gershon AA, eds. Herpes Zoster and Postherpetic Neuralgia. 2nd ed. New York, NY: Elsevier Press; 2001:39-64.
- Tyring S, Barbarash RA, Nahlik JE, et al. Famciclovir for the treatment of acute herpes zoster: effects on acute disease and postherpetic neuralgia. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:89–96.
- Kurokawa I, Kumano K, Murkawa K. Clinical correlates of prolonged pain in Japanese patients with acute herpes zoster. J Int Med Res. 2002;30:56–65.
- Zaal MJ, Volker-Dieben HJ, Wienesen M, et al. Longitudinal analysis of varicella-zoster virus DNA on the ocular surface associated with herpes zoster ophthalmicus. Am J Ophthalmol. 2001;131:25–29.
- Wood MJ, Johnson RW, McKendrick MW, et al. A randomized trial of acyclovir for 7 days or 21 days with and without prednisolone for treatment of acute herpes zoster. N Engl J Med. 1994;330:896–900.
- Whitley RJ, Weiss H, Gnann JW Jr, et al. Acyclovir with and without prednisone for the treatment of herpes zoster: a randomized, placebo-controlled trial. Ann Intern Med. 1996;125:376–383.
- Rice AS, Maton S. Gabapentin in postherpetic neuralgia: a randomised, double blind, placebo controlled study. Pain. 2001;94:215–224.
- Collins SL, Moore RA, McQuay HJ, Wiffen P. Antidepressants and anticonvulsants for diabetic neuropathy and postherpetic neuralgia: a quantitative systematic review. J Pain Symptom Manage. 2000;20:449–458.
- Wong MO, Eldon MA, Keane WF, et al. Disposition of gabapentin in anuric subjects on hemodialysis. J Clin Pharmacol. 1995;35:622–626.
- Horizant (gabapentin encarbil) extended-release tablets [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2012.
- Irving G, Jensen M, Cramer M, et al. Efficacy and tolerability of gastric-retentive gabapentin for the treatment of postherpetic neuralgia: results of a double-blind, randomized, placebo-controlled clinical trial. Clin J Pain. 2009;25:185–192.
- Jensen MP, Chiang YK, Wu J. Assessment of pain quality in a clinical trial of gabapentin extended release for postherpetic neuralgia. Clin J Pain. 2009;25:286–292.
- Wallace MS, Irving G, Cowles VE. Gabapentin extended-release tablets for the treatment of patients with postherpetic neuralgia: a randomized, double-blind, placebo-controlled, multicenter study. Clin Drug Investig. 2010;30:765–776.
- Frampton JE, Foster RH. Pregabalin in the treatment of postherpetic neuralgia. Drugs. 2005;65:111–118.
- Kochar D, Garg P, Bumb RA, et al. Divalproex sodium in the management of postherpetic neuralgia: a randomized double-blind placebo-controlled study. QJM. 2005;98:29–34.
- Watson CP, Vernich L, Chipman M, Reed K. Nortriptyline vs amitriptyline in postherpetic neuralgia: a randomized trial. Neurology. 1998;51:1166–1171.
- Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132:237–251.
- Cymbalta (duloxetine hydrochloride) delayed-release capsules [package insert]. Indianapolis, IN: Lilly USA; 2011.
- Rowbotham MC, Goli V, Kunz NR, Lei D. Venlafaxine extended release in the treatment of painful diabetic neuropathy: a double-blind, placebo-controlled study. Pain. 2004;110:697–706.
- Watson CP, Babul N. Efficacy of oxycodone in neuropathic pain: a randomized trial in postherpetic neuralgia. Neurology. 1998;50:1837–1841.
- Gilron I, Bailey JM, Tu D, et al. Morphine, gabapentin, or their combination for neuropathic pain. N Engl J Med. 2005;352:1324–1334.
- Boureau F, Legallicier P, Kabir-Ahmadi M. Tramadol in post-herpetic neuralgia: a randomized, double-blind, placebo-controlled trial. Pain. 2003;104:323–331.
- Hermann DN, Barbano RL, Hart-Gouleau S, et al. An open-label study of the lidocaine patch 5% in painful idiopathic sensory polyneuropathy. Pain Med. 2005;379–384.
- Davies PS, Galer BS. Review of lidocaine patch 5% studies in the treatment of postherpetic neuralgia. Drugs. 2004;64:937–947.
- Gammaitoni AR, Alvarez NA, Galer BS. Safety and tolerability of the lidocaine patch 5%, a targeted peripheral analgesic: a review of literature. J Clin Pharmacol. 2003;43:111–117.
- Watson CP, Tyler KL, Bickers DR, et al. A randomized vehicle-controlled trial of topical capsaicin in the treatment of postherpetic neuralgia. Clin Ther. 1993;15:510–526.
- Backonja MM, Malan TP, Vanhove GF, Tobias JK. C102/106 Study Group. NGX-4010, a high concentration capsaicin patch, for the treatment of postherpetic neuralgia: a randomized, double-blind, controlled study with an open-label extension. Pain Med. 2010;11:600–608.
- Kumar V, Krone K, Mathieu A. Neuraxial and sympathetic blocks in herpes zoster and postherpetic neuralgia: an appraisal of current evidence. Reg Anesth Pain Med. 2004;29:454–461.
- Doi K, Nikai T, Sakura S, Saito Y. Intercostal nerve block with 5% tetracaine for chronic pain syndromes. J Clin Anesth. 2002;14:39–41.
- Kotani N, Kushikata T, Hashimoto H, et al. Intrathecal methylprednisolone for intractable postherpetic neuralgia. N Engl J Med. 2000;343:1514–1519.
- Harke H, Gretenkort P, Ladleif HU, et al. Spinal cord stimulation in postherpetic neuralgia and in acute herpes zoster pain. Anesth Anal. 2002;94:694–700.
- Barnard D, Lloyd J, Evans J. Cryoanalgesia in the management of chronic facial pain. J Maxillofac Surg. 1981;9:101–102.
- Jones MJ, Murrin KR. Intercostal block with cryotherapy. Ann R Coll Surg Engl. 1987;69:261–262.
- Xiao L, Hui H. Therapeutic effect of botulinium toxin A in the treatment of postherpetic neuralgia by subcutaneous injection. Presented at: 26th Annual Meeting of the American Academy of Pain Medicine; February 3-6, 2010; San Antonio, TX.
- Marin M, Güris D, Chaves SS, et al. Prevention of varicella: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2007;56(RR-4):1–40.
- Harpaz R, Ortega-Sanchez IR, Seward JF. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1–30.
- Centers for Disease Control and Prevention (CDC). Update on herpes zoster vaccine: licensure for persons aged 50 through 59 years. MMWR Morb Mortal Wkly Rep. 2011;60(44):1528.
- Gnann JW Jr. Vaccination to prevent herpes zoster in older adults. J Pain. 2008;9(1 suppl 1):S31–S36.
Postherpetic neuralgia (PHN) is a management challenge—because of its severity, long duration, and potential for debilitation, often in the highly vulnerable elderly population. And, as the most common complication of an acute episode of herpes zoster (shingles) in an immunocompetent person, PHN is likely no stranger to your practice.
Herpes zoster is one of the most common neurological problems, with an incidence of up to 1 million new cases per year in the United States.1 Although the precise number for the prevalence of PHN in the United States is unknown, investigators estimate it at 500,000 to 1 million.2
Major risk factors for development of PHN after an episode of herpes zoster include:
older age
greater acute pain during herpes zoster
greater severity of rash.3,4
PHN is commonly defined as “dermatomal pain that persists 120 days or more after the onset of rash.”5 The pain of PHN has been characterized as a stimulus-dependent continuous burning, throbbing, or episodic sharp electric shock-like sensation6 and as a stimulus-dependent tactile allodynia (ie, pain after normally nonpainful stimulus) and hyperalgesia (exaggerated response to a painful stimulus). In addition, some patients experience myofascial pain secondary to excessive muscle guarding. Chronic pruritus can be present.
More than 90% of patients who have PHN have allodynia,7 which tends to occur in areas where sensation is relatively preserved. Patients also feel spontaneous pain in areas where sensation is lost or impaired.
In this article, we review the evidence for the range of treatments for acute herpes zoster and PHN, as well offer preventive strategies for herpes zoster.
Acute herpes zoster: Start antivirals early
Evidence-based treatment of acute herpes zoster includes antiviral drugs and analgesics.
Antiviral agents suppress viral replication and have a beneficial effect on acute and chronic pain. Acyclovir (800 mg, 5 times a day), valacyclovir (1000 mg, every 8 hours), and famciclovir (500 mg, every 8 hours) are antivirals commonly used to treat herpes zoster. All 3 drugs have comparable efficacy and safety profiles.
In a meta-analysis of patients older than 50 years who were treated with acyclovir or placebo, pain persisted in 15% of the acyclovir-treated group, compared with 35% of the placebo group.8 In terms of duration, a study comparing famciclovir treatment with placebo showed that subjects in the placebo group had persistent pain for 163 days, whereas famciclovir-treated patients had pain for 63 days.9
Based on this evidence, antiviral medications are strongly recommended for treating herpes zoster, especially for patients at increased risk of developing PHN. Antiviral treatment should be started within 72 hours of the onset of the rash.
No good evidence supports the efficacy of antiviral treatment administered 72 hours after the onset of rash. One uncontrolled trial, however, examined the effectiveness of acyclovir started before vs after 72 hours; the difference in pain persistence was not significant between the groups, suggesting acyclovir has benefit even when given after 72 hours.10
In clinical practice, the diagnosis of herpes zoster is often not made within 72 hours of symptom onset; nevertheless, it is important to identify patients who could still benefit from antiviral medication even when treatment is started relatively late in the disease course. This is especially true in ocular zoster, because viral shedding may continue beyond 72 hours.11
Analgesics are part of a practical approach for managing herpes zoster–associated pain that begins with a short-acting opioid in combination with acetaminophen or a nonsteroidal anti-inflammatory (NSAID) agent. Gabapentin or pregabalin, followed by a tricyclic antidepressant, can be added if conventional analgesics are not entirely effective. The analgesic regimen should be tailored to the patient’s needs and tolerance of adverse effects. If pain control is inadequate or adverse effects are intolerable, consider referring the patient to a pain management center for possible interventional modalities.
Key Point Gabapentin or pregabalin, followed by a tricyclic antidepressant, can be added if conventional analgesics are not effective for herpes zoster pain. |
Corticosteroids are not recommended routinely for treatment of herpes zoster; you can try them in otherwise healthy older adults, however, if antiviral therapy and analgesics do not relieve pain. In 2 double-blind controlled trials, a combination of acyclovir and corticosteroids for 21 days did not decrease the incidence of PHN—although some benefit was seen in terms of patients’ return to normal activities, cessation of analgesic therapy, and improved sleep.12,13
Evidence-based treatment options for PHN
Pharmacotherapy for PHN includes anticonvulsants, tricyclic antidepressants, opioids, and topical agents. Invasive interventions have a limited but important role in the management of PHN pain in clinical practice.
Calcium channel-blocking anticonvulsants gabapentin and pregabalin are safe and relatively well tolerated. They can be used as first-line agents for PHN, starting with a low dosage and titrating up, based on effectiveness and tolerability.
Gabapentin is FDA approved for the treatment of PHN. The starting dosage is 100 to 300 mg taken at night, titrated as needed by 100 to 300 mg every 3 to 5 days, to as high a dosage as 1800 to 3600 mg/d in 3 or 4 divided doses. In 2 large, randomized controlled trials, gabapentin produced a statistically significant reduction in pain ratings and improved sleep and quality of life.14,15 Adverse effects include somnolence, dizziness, peripheral edema, visual adverse effects, and gait and balance problems.
Because gabapentin is excreted by the kidneys, take care when using it in patients with renal insufficiency. Gabapentin clearance is linearly related to creatinine clearance and is decreased in the elderly and in individuals with impaired renal function. Hence, the gabapentin dose and the frequency of dosing must be adjusted in these patients.
In patients on hemodialysis, plasma gabapentin levels can be maintained by giving a dose of 200 to 300 mg 4 hours after hemodialysis.16
Extended-release gabapentin. The FDA recently approved an extended-release gabapentin formulation for PHN. Approval was based on a 12-week pivotal study and 2 adjunct studies. In a multicenter, randomized, double-blind, parallel-group, placebo-controlled, 12-week study evaluating the efficacy, safety, and dose response of 3 doses, extended-release gabapentin was effective at 1200 mg/d dosing. The initial recommended dose is 600 mg, once daily for 3 days, followed by 600 mg, twice daily, beginning on Day 4.17 The premise is that the extended-release preparation improves bioavailability of the active drug and, therefore, reduces the incidence of adverse effects, compared with regular gabapentin.
Overall, evidence is mixed. Two randomized controlled trials of extended-release gabapentin showed benefit (ie, reduced pain score on a numerical rating scale) with twice-a-day dosing (600 mg in the morning and 1200 mg at night), compared with a once-daily 1800-mg dose as well as placebo, for reduction in intensity of pain18 and specific pain quality.19 In another trial, however, extended-release gabapentin, 1800 mg once daily, did not show any benefit compared with placebo.20
Pregabalin is also FDA approved for PHN. The effective dosage range is 150 to 600 mg/d. Pregabalin provided significantly superior pain relief and improved sleep scores compared with placebo in 776 patients with PHN.21 Adverse effects include weight gain, dizziness, and somnolence. Titrate the dosage slowly in the elderly.
Sodium channel-blocking anticonvulsants topiramate, lamotrigine, carbamazepine, oxcarbazepine, levetriacetam, and valproic acid are not FDA approved for PHN. These agents may be a treatment option, however, for patients with PHN who do not respond to conventional therapy. In an 8-week randomized controlled trial, patients treated with divalproex sodium (valproic acid and sodium valproate), 1000 mg/d, experienced significant pain relief compared with placebo-treated patients.22 Adverse effects included vertigo, hair loss, headache, nausea, and diarrhea.
Tricyclic antidepressants, including amitriptyline, desipramine, and nortriptyline, might work by (1) inhibiting norepinephrine and serotonin uptake, (2) sodium-channel blockade, or (3) another mechanism that is unclear. Although amitriptyline is the most studied tricyclic antidepressant for PHN, available evidence and clinical experience suggest that nortriptyline and desipramine have comparable efficacy and are better tolerated.23,24
Key Point Available evidence and clinical experience suggest that nortriptyline and desipramine have comparable efficacy and are better tolerated than amitriptyline for PHN. |
Nortriptyline and desipramine are preferred in frail and elderly patients. Start therapy with 10 to 25 mg nightly, titrating as tolerated every 2 weeks to 75 to 150 mg as a single daily dose. Adverse effects include dry mouth, fatigue, dizziness, sedation, urinary retention, orthostatic hypotension, weight gain, blurred vision, QT interval prolongation, constipation, and sexual dysfunction.
Serotonin-norepinephrine reuptake inhibitor (SNRI) antidepressants. Use of such agents as duloxetine and venlafaxine in PHN patients is extrapolated from their proven efficacy in treating diabetic neuropathy and other neuropathic pain conditions. Try duloxetine if your patient does not respond to or tolerate a tricyclic. The recommended dosage is 60 to 120 mg/d in 2 divided doses.24
Two randomized, 12-week, double-blind, placebo-controlled trials using duloxetine 60 mg once a day and 60 mg twice a day for diabetic peripheral neuropathy concluded that 120 mg was safe and effective in treating diabetic peripheral neuropathy, but 120 mg was not as well tolerated as 60 mg once a day.25
Monitor liver function periodically in patients taking duloxetine. Alternatively, you can give venlafaxine; the recommended dosage is 75 to 225 mg/d.26
Opioid analgesics are recommended as second- and third-line agents for PHN. Adverse effects include nausea, pruritus, sedation, confusion, constipation, hypogonadism, and risk of developing tolerance and abuse.
A double-blind crossover trial evaluated the analgesic efficacy of oral oxycodone; treatment resulted in significant reduction of allodynia, steady pain, and spontaneous paroxysmal pain. Oxycodone treatment resulted in superior scores of global effectiveness, disability reduction, and patient preference, compared with placebo.27
In a randomized crossover trial, the combination of gabapentin and morphine was superior to either of these medications alone in relieving pain in PHN.28
Tramadol, an atypical opioid, has a weak μ-opioid receptor agonist effect and inhibits reuptake of serotonin and norepinephrine. Avoid using it in patients with a history of seizures. The maximum recommended dosage is 400 mg/d. An extended-release formulation of tramadol is also available.
Tramadol provided superior pain relief and improved quality of life in PHN patients in a randomized placebo-controlled trial.29
Tapentadol has weak μ-opioid receptor agonist activity; norepinephrine reuptake inhibition is more predominant than serotonin reuptake inhibition. This drug is also available as an extended-release formulation. The maximum recommended dosage is 600 mg/d.
Avoid using tapentadol in patients with a history of seizures. Note: Although there is no scientific evidence regarding the use of tapentadol in neuropathic pain, we use it often in our practice.
Topical therapies
Treating PHN with a topical agent is associated with relatively fewer adverse effects than what has been seen with oral therapy because systemic absorption is minimal.
Lidocaine is available as a transdermal patch and as a topical gel ointment. The 5% lidocaine patch is FDA approved for treating PHN. Lidocaine, a sodium-channel blocker, is useful for treating patients with clinical evidence of allodynia. You can cut a patch to fit the affected area; a maximum of 3 patches can be used simultaneously for 12 hours on, 12 hours off. If helpful, the patch can be left in place for 18 hours.30
In 2 open-labeled, nonrandomized prospective studies, patients treated with the lidocaine patch had reduced intensity of pain and improved quality of life.31,32
If lidocaine patches are not available, or affordable, or if a patient has difficulty applying them, use 5% lidocaine gel instead.
Capsaicin topical cream is sold in 2 concentrations: 0.025% and 0.075%. An extract of hot chili peppers, capsaicin acts as an agonist at the vanilloid receptors. The recommended dosage is 3 or 4 times a day. Initial application causes burning to become worse, but repeated use results in diminished pain and hyperalgesia.
A 6-week, blinded parallel study, followed by a 2-year open label follow-up, showed that the 0.075% dose of topical capsaicin cream relieved pain in 64% of patients; pain was relieved in 25% of placebo-treated patients.33
An 8% capsaicin patch is FDA approved for treating PHN. The patch must be applied by a health care professional in a monitored setting. Prepare the affected area by pretreating it with a local anesthetic cream; then apply the patch and leave it in place for 1 hour. As many as 4 patches can be used at once. A single application can provide pain relief for as long as 12 weeks. Adverse effects are mostly mild and transient.
In a double-blind, randomized, placebo-controlled trial with an open-label extension, the score on a numeric pain-rating scale declined from baseline in both the high-concentration capsaicin group and the placebo group during Week 1; however, the capsaicin-treated group experienced long-term improvement through Week 12.34
(See TABLE 114-21, 23, 24, 27-34 for a summary of pharmacotherapeutic options.)
TABLE 1
Pharmacotherapeutic options for managing postherpetic neuralgia14-21, 23, 24, 27-34
| *Obtain baseline EKG in patients with history of cardiac disease. †May need to start a patient on short-acting opioid medications before changing over to a fentanyl patch. ‡Has a long and unpredictable half-life, hence the need for extra caution in elderly patients. §Has not been studied in neuropathic pain; found to be effective in PHN and other chronic pain conditions. IISingle application has been found to be effective for about 3 months. MAOI, monoamine oxidase inhibitor; PHN, postherpetic neuralgia; SNRI, serotonin-norepinephrine reuptake inhibitor; SSRI, selective serotonin reuptake inhibitor; TCA, tricyclic antidepressant. | ||||
| Medication | Starting dose | Dose titration | Common adverse effects | Cautions and comments |
| Anticonvulsants | ||||
| Gabapentin | 100-300 mg | Start at bedtime and increase to tid dosing; increase by 100-300 mg every 3-5 days to total dose of 1800-3600 mg/d in 3 or 4 divided doses | Somnolence, dizziness, fatigue, ataxia, peripheral edema, weight gain, visual adverse effects | Decrease dose in patients with renal impairment. Dialysis patients: Every-other-day dosing; dosed on the day of dialysis. Avoid sudden discontinuation |
| Extended-release gabapentin | 600 mg daily for 3 days, then 600 mg bid beginning Day 4 | 600 mg bid | Somnolence, dizziness | Recently approved by FDA for PHN; not much clinical experience as yet |
| Pregabalin | 50 mg tid or 75 mg bid | 300-600 mg/d in 2 divided doses for 7-10 days | Somnolence, fatigue, dizziness, peripheral edema and weight gain, blurred vision, and euphoria | Decrease dose in patients with renal impairment. Titrate dosage slowly in elderly patients |
| Tricyclic antidepressants* | ||||
| Amitriptyline Desipramine Nortriptyline | 10-25 mg at bedtime. Start at a lower dose in elderly | Increase as tolerated every 2 weeks, with a target dose of 75-150 mg as a single daily dose | Sedation, dry mouth, blurred vision, weight gain, urinary retention, constipation, sexual dysfunction | Cardiac arrhythmic disease, glaucoma, suicide risk, seizure disorder. Risk of serotonin syndrome with concomitant use of tramadol, SSRIs, or SNRIs. Amitriptyline has the most anticholinergic effects |
| Opioids | ||||
| Fentanyl patch† Methadone‡ Morphine Oxycodone | 12 μg/hour 2.5 mg tid 15 mg q 6 hours prn 5 mg q 6 hours prn | Titrate at weekly intervals balancing analgesia and adverse effects. If patient tolerates the medications, can titrate faster | Nausea and vomiting, constipation, sedation, itching, risk of tolerance and abuse | Driving impairment and cognitive dysfunction during treatment initiation. Be careful in patients with sleep apnea. Additive effects of sedation with neuromodulating medications |
| Atypical opioids | ||||
| Tapentadol§ | 50 mg every 4-6 hours prn | Can titrate up to 100 mg q 4 hours. Maximum daily dose is 600 mg | Nausea and vomiting, constipation, drowsiness, and dizziness | Be careful in patients taking SSRIs, SNRIs, MAOIs, and TCAs. Decrease dose in patients with moderate hepatic and renal impairment. Avoid use in patients with a history of seizures |
| Tramadol | 50 mg every 6 hours prn | Can titrate up to 100 mg q 6 hours. Maximum daily dose: 400 mg. Extended-release dosing once a day | Nausea and vomiting, constipation, drowsiness, dizziness | Be careful in patients with seizure disorder and concomitant use of SSRIs, SNRIs, and TCAs. Decrease dose in patients with hepatic or renal disease |
| Topical agents | ||||
| Lidocaine patch | 5% lidocaine patch | Can use up to 3 patches 12 hours/d | Local erythema, rash, blisters | Contraindicated in patients with known hypersensitivity to amide local anesthetics (eg, bupivacaine, mepivacaine). Do not use on skin with open lesions |
| Topical capsaicin | 0.025% and 0.075% cream | Apply 3-4 times a day over affected region | No systemic adverse effects. Burning and stinging sensation at the application site | Avoid contact with eyes, nose, and mouth. Application of lidocaine gel locally may be helpful prior to capsaicin cream application |
| Capsaicin patchII | 8% single application patch | Need topical local anesthetic application prior to patch application. Patch applied for 1 hour | Local site irritation, burning, temporary increase in pain | Done in a physician’s office under monitored circumstances. Patient may need oral analgesics for a short period following application of the patch |
Alternative modalities to reduce pain
Acupuncture and transcutaneous electrical nerve stimulation (TENS) have been tried for the relief of PHN without consistent evidence of efficacy. There are no significant adverse effects associated with these therapies; however, the cost of treatment may be an issue. Acupuncture is not covered by many insurance carriers. Mental-health interventions, including cognitive and behavioral therapy, might help with overall physical and emotional functioning and quality of life.
Key Point Acupuncture and transcutaneous electrical nerve stimulation do not appear to be effective for PHN relief. |
Invasive interventions
Researchers have examined several interventional modalities for treating PHN that is refractory to medication.
Sympathetic nerve blocks. Retrospective studies have shown that sympathetic nerve block provides short-term improvement in pain in 40% to 50% of patients with PHN.35
Intercostal nerve block has been reported to provide long-lasting pain relief in patients with thoracic PHN.36
Neuraxial use of intrathecal methylprednisone is supported by moderately good evidence of benefit in patients with intractable PHN.37 Because this intervention poses significant risk of neurologic sequelae, we do not recommend that it be used in clinical practice.
Spinal cord stimulation was studied prospectively in a case series of 28 patients.38 Long-term pain relief was obtained in 82%. Patients serve as their own controls by switching off the spinal cord stimulator and monitoring pain. Consider spinal cord stimulation for patients with well-established PHN that is refractory to conventional management.
Cryotherapy was used for facial neuralgia pain, without significant benefit.39 Another trial showed short-term benefit in 11 of 14 patients who underwent cryotherapy of the intercostal nerves for thoracic PHN.40
Botulinium toxin A injection. An abstract presented at the February 2010 meeting of the American Academy of Pain Medicine described how subcutaneous injection of botulinium toxin A reduced pain in patients with PHN, compared with lidocaine and placebo injections. The pain relief was noted in 1 week and persisted for 90 days.41
Surgery. Many surgical interventions have been described and used to treat PHN, but none has a role in clinical practice.
Key Point Many surgical interventions have been used to treat PHN, but none has a role in clinical practice. |
When should you refer to a pain management center?
Dermatomal pain that lasts for longer than 180 days after a herpes zoster rash can be considered “well-established PHN” to denote its refractory nature. As a primary care clinician, you can refer a patient with PHN to a pain management center at any stage of disease but especially when the:
patient has a significant medical comorbidity and you think that he or she requires the services of a specialist to manage multimodal pharmacotherapy
PHN pain is refractory to conventional treatment modalities
- patient needs an invasive intervention
- patient needs treatment with a high-dose capsaicin patch and you have not been trained to apply it.
Preventing herpes zoster and PHN
Obviously, preventing PHN is closely tied to preventing herpes zoster. To help prevent herpes zoster:
vaccinate children with varicella vaccine to prevent primary varicella infection42
use varicella-zoster immunoglobulin, as recommended by the CDC’s Advisory Committee on Immunization Practices (ACIP), in immunocompromised, seronegative patients who were exposed recently to a person with chickenpox or herpes zoster42
administer the herpes zoster vaccine to patients 60 years and older, as recommended by ACIP.43 The FDA recently approved use of this vaccine for people 50 through 59 years, but ACIP has not changed its recommendations.44
As we’ve discussed, herpes zoster vaccination, antiviral therapy, and aggressive pain control can reduce the incidence, severity, and duration of acute herpes zoster and PHN.
A large multicenter, randomized, placebo-controlled trial demonstrated that herpes zoster vaccine decreases the likelihood of developing herpes zoster in immunocompetent individuals 60 years and older.45 The vaccine reduced the incidence of herpes zoster by 51.3%; reduced the burden of illness by 61.1%; and reduced the incidence of PHN by 66.5%.45 The live, attenuated vaccine is contraindicated in children, pregnant women, and immunocompromised individuals.
The number needed to treat for herpes zoster vaccine is 175; that is, 1 case of herpes zoster is avoided for every 175 people vaccinated.1
Key Point One case of herpes zoster is avoided for every 175 people vaccinated. |
Newer tools mean a better outcome
We have improved our ability to diminish the incidence of herpes zoster and PHN and to manage postherpetic pain more effectively. These advances include the development of a herpes zoster vaccine; consensus that antiviral therapy and aggressive pain management can reduce the burden of PHN; identification of efficacious treatments for PHN; and recognition of PHN as a study model for neuropathic pain research.
Disclosure
The authors reported no potential conflict of interest relevant to this article.
References
- Oxman MN, Levin MJ, Johnson GR, et al; Shingles Prevention Study Group. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271–2284.
- Bennett GJ. Neuropathic pain: An overview. In: Borsook D, ed. Molecular Neurobiology of Pain. Seattle, WA: IASP Press; 1997:109-113.
- Jung BF, Johnson RW, Griffin DR, Dworkin RH. Risk factors for postherpetic neuralgia in patients with herpes zoster. Neurology. 2004;62:1545–1551.
- Dworkin RH, Boon RJ, Griffin DR, Phung D. Postherpetic neuralgia: impact of famciclovir, age, rash severity, and acute pain in herpes zoster patients. J Infect Dis. 1998;178(suppl 1):S76–S80.
- Volpi A, Gross G, Hercogova J, Johnson RW. Current management of herpes zoster: the European view. Am J Clin Dermatol. 2005;6:317–325.
- Dworkin RH, Portenoy RK. Pain and its persistence in herpes zoster. Pain. 1996;67:241–251.
- Bowsher D. Pathophysiology of postherpetic neuralgia: towards a rational treatment. Neurology. 1995;45(12 suppl 8):S56–S57.
- Dworkin RH, Schmader KE. Epidemiology and natural history of herpes zoster and postherpetic neuralgia. In Watson CPN, Gershon AA, eds. Herpes Zoster and Postherpetic Neuralgia. 2nd ed. New York, NY: Elsevier Press; 2001:39-64.
- Tyring S, Barbarash RA, Nahlik JE, et al. Famciclovir for the treatment of acute herpes zoster: effects on acute disease and postherpetic neuralgia. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:89–96.
- Kurokawa I, Kumano K, Murkawa K. Clinical correlates of prolonged pain in Japanese patients with acute herpes zoster. J Int Med Res. 2002;30:56–65.
- Zaal MJ, Volker-Dieben HJ, Wienesen M, et al. Longitudinal analysis of varicella-zoster virus DNA on the ocular surface associated with herpes zoster ophthalmicus. Am J Ophthalmol. 2001;131:25–29.
- Wood MJ, Johnson RW, McKendrick MW, et al. A randomized trial of acyclovir for 7 days or 21 days with and without prednisolone for treatment of acute herpes zoster. N Engl J Med. 1994;330:896–900.
- Whitley RJ, Weiss H, Gnann JW Jr, et al. Acyclovir with and without prednisone for the treatment of herpes zoster: a randomized, placebo-controlled trial. Ann Intern Med. 1996;125:376–383.
- Rice AS, Maton S. Gabapentin in postherpetic neuralgia: a randomised, double blind, placebo controlled study. Pain. 2001;94:215–224.
- Collins SL, Moore RA, McQuay HJ, Wiffen P. Antidepressants and anticonvulsants for diabetic neuropathy and postherpetic neuralgia: a quantitative systematic review. J Pain Symptom Manage. 2000;20:449–458.
- Wong MO, Eldon MA, Keane WF, et al. Disposition of gabapentin in anuric subjects on hemodialysis. J Clin Pharmacol. 1995;35:622–626.
- Horizant (gabapentin encarbil) extended-release tablets [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2012.
- Irving G, Jensen M, Cramer M, et al. Efficacy and tolerability of gastric-retentive gabapentin for the treatment of postherpetic neuralgia: results of a double-blind, randomized, placebo-controlled clinical trial. Clin J Pain. 2009;25:185–192.
- Jensen MP, Chiang YK, Wu J. Assessment of pain quality in a clinical trial of gabapentin extended release for postherpetic neuralgia. Clin J Pain. 2009;25:286–292.
- Wallace MS, Irving G, Cowles VE. Gabapentin extended-release tablets for the treatment of patients with postherpetic neuralgia: a randomized, double-blind, placebo-controlled, multicenter study. Clin Drug Investig. 2010;30:765–776.
- Frampton JE, Foster RH. Pregabalin in the treatment of postherpetic neuralgia. Drugs. 2005;65:111–118.
- Kochar D, Garg P, Bumb RA, et al. Divalproex sodium in the management of postherpetic neuralgia: a randomized double-blind placebo-controlled study. QJM. 2005;98:29–34.
- Watson CP, Vernich L, Chipman M, Reed K. Nortriptyline vs amitriptyline in postherpetic neuralgia: a randomized trial. Neurology. 1998;51:1166–1171.
- Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132:237–251.
- Cymbalta (duloxetine hydrochloride) delayed-release capsules [package insert]. Indianapolis, IN: Lilly USA; 2011.
- Rowbotham MC, Goli V, Kunz NR, Lei D. Venlafaxine extended release in the treatment of painful diabetic neuropathy: a double-blind, placebo-controlled study. Pain. 2004;110:697–706.
- Watson CP, Babul N. Efficacy of oxycodone in neuropathic pain: a randomized trial in postherpetic neuralgia. Neurology. 1998;50:1837–1841.
- Gilron I, Bailey JM, Tu D, et al. Morphine, gabapentin, or their combination for neuropathic pain. N Engl J Med. 2005;352:1324–1334.
- Boureau F, Legallicier P, Kabir-Ahmadi M. Tramadol in post-herpetic neuralgia: a randomized, double-blind, placebo-controlled trial. Pain. 2003;104:323–331.
- Hermann DN, Barbano RL, Hart-Gouleau S, et al. An open-label study of the lidocaine patch 5% in painful idiopathic sensory polyneuropathy. Pain Med. 2005;379–384.
- Davies PS, Galer BS. Review of lidocaine patch 5% studies in the treatment of postherpetic neuralgia. Drugs. 2004;64:937–947.
- Gammaitoni AR, Alvarez NA, Galer BS. Safety and tolerability of the lidocaine patch 5%, a targeted peripheral analgesic: a review of literature. J Clin Pharmacol. 2003;43:111–117.
- Watson CP, Tyler KL, Bickers DR, et al. A randomized vehicle-controlled trial of topical capsaicin in the treatment of postherpetic neuralgia. Clin Ther. 1993;15:510–526.
- Backonja MM, Malan TP, Vanhove GF, Tobias JK. C102/106 Study Group. NGX-4010, a high concentration capsaicin patch, for the treatment of postherpetic neuralgia: a randomized, double-blind, controlled study with an open-label extension. Pain Med. 2010;11:600–608.
- Kumar V, Krone K, Mathieu A. Neuraxial and sympathetic blocks in herpes zoster and postherpetic neuralgia: an appraisal of current evidence. Reg Anesth Pain Med. 2004;29:454–461.
- Doi K, Nikai T, Sakura S, Saito Y. Intercostal nerve block with 5% tetracaine for chronic pain syndromes. J Clin Anesth. 2002;14:39–41.
- Kotani N, Kushikata T, Hashimoto H, et al. Intrathecal methylprednisolone for intractable postherpetic neuralgia. N Engl J Med. 2000;343:1514–1519.
- Harke H, Gretenkort P, Ladleif HU, et al. Spinal cord stimulation in postherpetic neuralgia and in acute herpes zoster pain. Anesth Anal. 2002;94:694–700.
- Barnard D, Lloyd J, Evans J. Cryoanalgesia in the management of chronic facial pain. J Maxillofac Surg. 1981;9:101–102.
- Jones MJ, Murrin KR. Intercostal block with cryotherapy. Ann R Coll Surg Engl. 1987;69:261–262.
- Xiao L, Hui H. Therapeutic effect of botulinium toxin A in the treatment of postherpetic neuralgia by subcutaneous injection. Presented at: 26th Annual Meeting of the American Academy of Pain Medicine; February 3-6, 2010; San Antonio, TX.
- Marin M, Güris D, Chaves SS, et al. Prevention of varicella: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2007;56(RR-4):1–40.
- Harpaz R, Ortega-Sanchez IR, Seward JF. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1–30.
- Centers for Disease Control and Prevention (CDC). Update on herpes zoster vaccine: licensure for persons aged 50 through 59 years. MMWR Morb Mortal Wkly Rep. 2011;60(44):1528.
- Gnann JW Jr. Vaccination to prevent herpes zoster in older adults. J Pain. 2008;9(1 suppl 1):S31–S36.
Postherpetic neuralgia (PHN) is a management challenge—because of its severity, long duration, and potential for debilitation, often in the highly vulnerable elderly population. And, as the most common complication of an acute episode of herpes zoster (shingles) in an immunocompetent person, PHN is likely no stranger to your practice.
Herpes zoster is one of the most common neurological problems, with an incidence of up to 1 million new cases per year in the United States.1 Although the precise number for the prevalence of PHN in the United States is unknown, investigators estimate it at 500,000 to 1 million.2
Major risk factors for development of PHN after an episode of herpes zoster include:
older age
greater acute pain during herpes zoster
greater severity of rash.3,4
PHN is commonly defined as “dermatomal pain that persists 120 days or more after the onset of rash.”5 The pain of PHN has been characterized as a stimulus-dependent continuous burning, throbbing, or episodic sharp electric shock-like sensation6 and as a stimulus-dependent tactile allodynia (ie, pain after normally nonpainful stimulus) and hyperalgesia (exaggerated response to a painful stimulus). In addition, some patients experience myofascial pain secondary to excessive muscle guarding. Chronic pruritus can be present.
More than 90% of patients who have PHN have allodynia,7 which tends to occur in areas where sensation is relatively preserved. Patients also feel spontaneous pain in areas where sensation is lost or impaired.
In this article, we review the evidence for the range of treatments for acute herpes zoster and PHN, as well offer preventive strategies for herpes zoster.
Acute herpes zoster: Start antivirals early
Evidence-based treatment of acute herpes zoster includes antiviral drugs and analgesics.
Antiviral agents suppress viral replication and have a beneficial effect on acute and chronic pain. Acyclovir (800 mg, 5 times a day), valacyclovir (1000 mg, every 8 hours), and famciclovir (500 mg, every 8 hours) are antivirals commonly used to treat herpes zoster. All 3 drugs have comparable efficacy and safety profiles.
In a meta-analysis of patients older than 50 years who were treated with acyclovir or placebo, pain persisted in 15% of the acyclovir-treated group, compared with 35% of the placebo group.8 In terms of duration, a study comparing famciclovir treatment with placebo showed that subjects in the placebo group had persistent pain for 163 days, whereas famciclovir-treated patients had pain for 63 days.9
Based on this evidence, antiviral medications are strongly recommended for treating herpes zoster, especially for patients at increased risk of developing PHN. Antiviral treatment should be started within 72 hours of the onset of the rash.
No good evidence supports the efficacy of antiviral treatment administered 72 hours after the onset of rash. One uncontrolled trial, however, examined the effectiveness of acyclovir started before vs after 72 hours; the difference in pain persistence was not significant between the groups, suggesting acyclovir has benefit even when given after 72 hours.10
In clinical practice, the diagnosis of herpes zoster is often not made within 72 hours of symptom onset; nevertheless, it is important to identify patients who could still benefit from antiviral medication even when treatment is started relatively late in the disease course. This is especially true in ocular zoster, because viral shedding may continue beyond 72 hours.11
Analgesics are part of a practical approach for managing herpes zoster–associated pain that begins with a short-acting opioid in combination with acetaminophen or a nonsteroidal anti-inflammatory (NSAID) agent. Gabapentin or pregabalin, followed by a tricyclic antidepressant, can be added if conventional analgesics are not entirely effective. The analgesic regimen should be tailored to the patient’s needs and tolerance of adverse effects. If pain control is inadequate or adverse effects are intolerable, consider referring the patient to a pain management center for possible interventional modalities.
Key Point Gabapentin or pregabalin, followed by a tricyclic antidepressant, can be added if conventional analgesics are not effective for herpes zoster pain. |
Corticosteroids are not recommended routinely for treatment of herpes zoster; you can try them in otherwise healthy older adults, however, if antiviral therapy and analgesics do not relieve pain. In 2 double-blind controlled trials, a combination of acyclovir and corticosteroids for 21 days did not decrease the incidence of PHN—although some benefit was seen in terms of patients’ return to normal activities, cessation of analgesic therapy, and improved sleep.12,13
Evidence-based treatment options for PHN
Pharmacotherapy for PHN includes anticonvulsants, tricyclic antidepressants, opioids, and topical agents. Invasive interventions have a limited but important role in the management of PHN pain in clinical practice.
Calcium channel-blocking anticonvulsants gabapentin and pregabalin are safe and relatively well tolerated. They can be used as first-line agents for PHN, starting with a low dosage and titrating up, based on effectiveness and tolerability.
Gabapentin is FDA approved for the treatment of PHN. The starting dosage is 100 to 300 mg taken at night, titrated as needed by 100 to 300 mg every 3 to 5 days, to as high a dosage as 1800 to 3600 mg/d in 3 or 4 divided doses. In 2 large, randomized controlled trials, gabapentin produced a statistically significant reduction in pain ratings and improved sleep and quality of life.14,15 Adverse effects include somnolence, dizziness, peripheral edema, visual adverse effects, and gait and balance problems.
Because gabapentin is excreted by the kidneys, take care when using it in patients with renal insufficiency. Gabapentin clearance is linearly related to creatinine clearance and is decreased in the elderly and in individuals with impaired renal function. Hence, the gabapentin dose and the frequency of dosing must be adjusted in these patients.
In patients on hemodialysis, plasma gabapentin levels can be maintained by giving a dose of 200 to 300 mg 4 hours after hemodialysis.16
Extended-release gabapentin. The FDA recently approved an extended-release gabapentin formulation for PHN. Approval was based on a 12-week pivotal study and 2 adjunct studies. In a multicenter, randomized, double-blind, parallel-group, placebo-controlled, 12-week study evaluating the efficacy, safety, and dose response of 3 doses, extended-release gabapentin was effective at 1200 mg/d dosing. The initial recommended dose is 600 mg, once daily for 3 days, followed by 600 mg, twice daily, beginning on Day 4.17 The premise is that the extended-release preparation improves bioavailability of the active drug and, therefore, reduces the incidence of adverse effects, compared with regular gabapentin.
Overall, evidence is mixed. Two randomized controlled trials of extended-release gabapentin showed benefit (ie, reduced pain score on a numerical rating scale) with twice-a-day dosing (600 mg in the morning and 1200 mg at night), compared with a once-daily 1800-mg dose as well as placebo, for reduction in intensity of pain18 and specific pain quality.19 In another trial, however, extended-release gabapentin, 1800 mg once daily, did not show any benefit compared with placebo.20
Pregabalin is also FDA approved for PHN. The effective dosage range is 150 to 600 mg/d. Pregabalin provided significantly superior pain relief and improved sleep scores compared with placebo in 776 patients with PHN.21 Adverse effects include weight gain, dizziness, and somnolence. Titrate the dosage slowly in the elderly.
Sodium channel-blocking anticonvulsants topiramate, lamotrigine, carbamazepine, oxcarbazepine, levetriacetam, and valproic acid are not FDA approved for PHN. These agents may be a treatment option, however, for patients with PHN who do not respond to conventional therapy. In an 8-week randomized controlled trial, patients treated with divalproex sodium (valproic acid and sodium valproate), 1000 mg/d, experienced significant pain relief compared with placebo-treated patients.22 Adverse effects included vertigo, hair loss, headache, nausea, and diarrhea.
Tricyclic antidepressants, including amitriptyline, desipramine, and nortriptyline, might work by (1) inhibiting norepinephrine and serotonin uptake, (2) sodium-channel blockade, or (3) another mechanism that is unclear. Although amitriptyline is the most studied tricyclic antidepressant for PHN, available evidence and clinical experience suggest that nortriptyline and desipramine have comparable efficacy and are better tolerated.23,24
Key Point Available evidence and clinical experience suggest that nortriptyline and desipramine have comparable efficacy and are better tolerated than amitriptyline for PHN. |
Nortriptyline and desipramine are preferred in frail and elderly patients. Start therapy with 10 to 25 mg nightly, titrating as tolerated every 2 weeks to 75 to 150 mg as a single daily dose. Adverse effects include dry mouth, fatigue, dizziness, sedation, urinary retention, orthostatic hypotension, weight gain, blurred vision, QT interval prolongation, constipation, and sexual dysfunction.
Serotonin-norepinephrine reuptake inhibitor (SNRI) antidepressants. Use of such agents as duloxetine and venlafaxine in PHN patients is extrapolated from their proven efficacy in treating diabetic neuropathy and other neuropathic pain conditions. Try duloxetine if your patient does not respond to or tolerate a tricyclic. The recommended dosage is 60 to 120 mg/d in 2 divided doses.24
Two randomized, 12-week, double-blind, placebo-controlled trials using duloxetine 60 mg once a day and 60 mg twice a day for diabetic peripheral neuropathy concluded that 120 mg was safe and effective in treating diabetic peripheral neuropathy, but 120 mg was not as well tolerated as 60 mg once a day.25
Monitor liver function periodically in patients taking duloxetine. Alternatively, you can give venlafaxine; the recommended dosage is 75 to 225 mg/d.26
Opioid analgesics are recommended as second- and third-line agents for PHN. Adverse effects include nausea, pruritus, sedation, confusion, constipation, hypogonadism, and risk of developing tolerance and abuse.
A double-blind crossover trial evaluated the analgesic efficacy of oral oxycodone; treatment resulted in significant reduction of allodynia, steady pain, and spontaneous paroxysmal pain. Oxycodone treatment resulted in superior scores of global effectiveness, disability reduction, and patient preference, compared with placebo.27
In a randomized crossover trial, the combination of gabapentin and morphine was superior to either of these medications alone in relieving pain in PHN.28
Tramadol, an atypical opioid, has a weak μ-opioid receptor agonist effect and inhibits reuptake of serotonin and norepinephrine. Avoid using it in patients with a history of seizures. The maximum recommended dosage is 400 mg/d. An extended-release formulation of tramadol is also available.
Tramadol provided superior pain relief and improved quality of life in PHN patients in a randomized placebo-controlled trial.29
Tapentadol has weak μ-opioid receptor agonist activity; norepinephrine reuptake inhibition is more predominant than serotonin reuptake inhibition. This drug is also available as an extended-release formulation. The maximum recommended dosage is 600 mg/d.
Avoid using tapentadol in patients with a history of seizures. Note: Although there is no scientific evidence regarding the use of tapentadol in neuropathic pain, we use it often in our practice.
Topical therapies
Treating PHN with a topical agent is associated with relatively fewer adverse effects than what has been seen with oral therapy because systemic absorption is minimal.
Lidocaine is available as a transdermal patch and as a topical gel ointment. The 5% lidocaine patch is FDA approved for treating PHN. Lidocaine, a sodium-channel blocker, is useful for treating patients with clinical evidence of allodynia. You can cut a patch to fit the affected area; a maximum of 3 patches can be used simultaneously for 12 hours on, 12 hours off. If helpful, the patch can be left in place for 18 hours.30
In 2 open-labeled, nonrandomized prospective studies, patients treated with the lidocaine patch had reduced intensity of pain and improved quality of life.31,32
If lidocaine patches are not available, or affordable, or if a patient has difficulty applying them, use 5% lidocaine gel instead.
Capsaicin topical cream is sold in 2 concentrations: 0.025% and 0.075%. An extract of hot chili peppers, capsaicin acts as an agonist at the vanilloid receptors. The recommended dosage is 3 or 4 times a day. Initial application causes burning to become worse, but repeated use results in diminished pain and hyperalgesia.
A 6-week, blinded parallel study, followed by a 2-year open label follow-up, showed that the 0.075% dose of topical capsaicin cream relieved pain in 64% of patients; pain was relieved in 25% of placebo-treated patients.33
An 8% capsaicin patch is FDA approved for treating PHN. The patch must be applied by a health care professional in a monitored setting. Prepare the affected area by pretreating it with a local anesthetic cream; then apply the patch and leave it in place for 1 hour. As many as 4 patches can be used at once. A single application can provide pain relief for as long as 12 weeks. Adverse effects are mostly mild and transient.
In a double-blind, randomized, placebo-controlled trial with an open-label extension, the score on a numeric pain-rating scale declined from baseline in both the high-concentration capsaicin group and the placebo group during Week 1; however, the capsaicin-treated group experienced long-term improvement through Week 12.34
(See TABLE 114-21, 23, 24, 27-34 for a summary of pharmacotherapeutic options.)
TABLE 1
Pharmacotherapeutic options for managing postherpetic neuralgia14-21, 23, 24, 27-34
| *Obtain baseline EKG in patients with history of cardiac disease. †May need to start a patient on short-acting opioid medications before changing over to a fentanyl patch. ‡Has a long and unpredictable half-life, hence the need for extra caution in elderly patients. §Has not been studied in neuropathic pain; found to be effective in PHN and other chronic pain conditions. IISingle application has been found to be effective for about 3 months. MAOI, monoamine oxidase inhibitor; PHN, postherpetic neuralgia; SNRI, serotonin-norepinephrine reuptake inhibitor; SSRI, selective serotonin reuptake inhibitor; TCA, tricyclic antidepressant. | ||||
| Medication | Starting dose | Dose titration | Common adverse effects | Cautions and comments |
| Anticonvulsants | ||||
| Gabapentin | 100-300 mg | Start at bedtime and increase to tid dosing; increase by 100-300 mg every 3-5 days to total dose of 1800-3600 mg/d in 3 or 4 divided doses | Somnolence, dizziness, fatigue, ataxia, peripheral edema, weight gain, visual adverse effects | Decrease dose in patients with renal impairment. Dialysis patients: Every-other-day dosing; dosed on the day of dialysis. Avoid sudden discontinuation |
| Extended-release gabapentin | 600 mg daily for 3 days, then 600 mg bid beginning Day 4 | 600 mg bid | Somnolence, dizziness | Recently approved by FDA for PHN; not much clinical experience as yet |
| Pregabalin | 50 mg tid or 75 mg bid | 300-600 mg/d in 2 divided doses for 7-10 days | Somnolence, fatigue, dizziness, peripheral edema and weight gain, blurred vision, and euphoria | Decrease dose in patients with renal impairment. Titrate dosage slowly in elderly patients |
| Tricyclic antidepressants* | ||||
| Amitriptyline Desipramine Nortriptyline | 10-25 mg at bedtime. Start at a lower dose in elderly | Increase as tolerated every 2 weeks, with a target dose of 75-150 mg as a single daily dose | Sedation, dry mouth, blurred vision, weight gain, urinary retention, constipation, sexual dysfunction | Cardiac arrhythmic disease, glaucoma, suicide risk, seizure disorder. Risk of serotonin syndrome with concomitant use of tramadol, SSRIs, or SNRIs. Amitriptyline has the most anticholinergic effects |
| Opioids | ||||
| Fentanyl patch† Methadone‡ Morphine Oxycodone | 12 μg/hour 2.5 mg tid 15 mg q 6 hours prn 5 mg q 6 hours prn | Titrate at weekly intervals balancing analgesia and adverse effects. If patient tolerates the medications, can titrate faster | Nausea and vomiting, constipation, sedation, itching, risk of tolerance and abuse | Driving impairment and cognitive dysfunction during treatment initiation. Be careful in patients with sleep apnea. Additive effects of sedation with neuromodulating medications |
| Atypical opioids | ||||
| Tapentadol§ | 50 mg every 4-6 hours prn | Can titrate up to 100 mg q 4 hours. Maximum daily dose is 600 mg | Nausea and vomiting, constipation, drowsiness, and dizziness | Be careful in patients taking SSRIs, SNRIs, MAOIs, and TCAs. Decrease dose in patients with moderate hepatic and renal impairment. Avoid use in patients with a history of seizures |
| Tramadol | 50 mg every 6 hours prn | Can titrate up to 100 mg q 6 hours. Maximum daily dose: 400 mg. Extended-release dosing once a day | Nausea and vomiting, constipation, drowsiness, dizziness | Be careful in patients with seizure disorder and concomitant use of SSRIs, SNRIs, and TCAs. Decrease dose in patients with hepatic or renal disease |
| Topical agents | ||||
| Lidocaine patch | 5% lidocaine patch | Can use up to 3 patches 12 hours/d | Local erythema, rash, blisters | Contraindicated in patients with known hypersensitivity to amide local anesthetics (eg, bupivacaine, mepivacaine). Do not use on skin with open lesions |
| Topical capsaicin | 0.025% and 0.075% cream | Apply 3-4 times a day over affected region | No systemic adverse effects. Burning and stinging sensation at the application site | Avoid contact with eyes, nose, and mouth. Application of lidocaine gel locally may be helpful prior to capsaicin cream application |
| Capsaicin patchII | 8% single application patch | Need topical local anesthetic application prior to patch application. Patch applied for 1 hour | Local site irritation, burning, temporary increase in pain | Done in a physician’s office under monitored circumstances. Patient may need oral analgesics for a short period following application of the patch |
Alternative modalities to reduce pain
Acupuncture and transcutaneous electrical nerve stimulation (TENS) have been tried for the relief of PHN without consistent evidence of efficacy. There are no significant adverse effects associated with these therapies; however, the cost of treatment may be an issue. Acupuncture is not covered by many insurance carriers. Mental-health interventions, including cognitive and behavioral therapy, might help with overall physical and emotional functioning and quality of life.
Key Point Acupuncture and transcutaneous electrical nerve stimulation do not appear to be effective for PHN relief. |
Invasive interventions
Researchers have examined several interventional modalities for treating PHN that is refractory to medication.
Sympathetic nerve blocks. Retrospective studies have shown that sympathetic nerve block provides short-term improvement in pain in 40% to 50% of patients with PHN.35
Intercostal nerve block has been reported to provide long-lasting pain relief in patients with thoracic PHN.36
Neuraxial use of intrathecal methylprednisone is supported by moderately good evidence of benefit in patients with intractable PHN.37 Because this intervention poses significant risk of neurologic sequelae, we do not recommend that it be used in clinical practice.
Spinal cord stimulation was studied prospectively in a case series of 28 patients.38 Long-term pain relief was obtained in 82%. Patients serve as their own controls by switching off the spinal cord stimulator and monitoring pain. Consider spinal cord stimulation for patients with well-established PHN that is refractory to conventional management.
Cryotherapy was used for facial neuralgia pain, without significant benefit.39 Another trial showed short-term benefit in 11 of 14 patients who underwent cryotherapy of the intercostal nerves for thoracic PHN.40
Botulinium toxin A injection. An abstract presented at the February 2010 meeting of the American Academy of Pain Medicine described how subcutaneous injection of botulinium toxin A reduced pain in patients with PHN, compared with lidocaine and placebo injections. The pain relief was noted in 1 week and persisted for 90 days.41
Surgery. Many surgical interventions have been described and used to treat PHN, but none has a role in clinical practice.
Key Point Many surgical interventions have been used to treat PHN, but none has a role in clinical practice. |
When should you refer to a pain management center?
Dermatomal pain that lasts for longer than 180 days after a herpes zoster rash can be considered “well-established PHN” to denote its refractory nature. As a primary care clinician, you can refer a patient with PHN to a pain management center at any stage of disease but especially when the:
patient has a significant medical comorbidity and you think that he or she requires the services of a specialist to manage multimodal pharmacotherapy
PHN pain is refractory to conventional treatment modalities
- patient needs an invasive intervention
- patient needs treatment with a high-dose capsaicin patch and you have not been trained to apply it.
Preventing herpes zoster and PHN
Obviously, preventing PHN is closely tied to preventing herpes zoster. To help prevent herpes zoster:
vaccinate children with varicella vaccine to prevent primary varicella infection42
use varicella-zoster immunoglobulin, as recommended by the CDC’s Advisory Committee on Immunization Practices (ACIP), in immunocompromised, seronegative patients who were exposed recently to a person with chickenpox or herpes zoster42
administer the herpes zoster vaccine to patients 60 years and older, as recommended by ACIP.43 The FDA recently approved use of this vaccine for people 50 through 59 years, but ACIP has not changed its recommendations.44
As we’ve discussed, herpes zoster vaccination, antiviral therapy, and aggressive pain control can reduce the incidence, severity, and duration of acute herpes zoster and PHN.
A large multicenter, randomized, placebo-controlled trial demonstrated that herpes zoster vaccine decreases the likelihood of developing herpes zoster in immunocompetent individuals 60 years and older.45 The vaccine reduced the incidence of herpes zoster by 51.3%; reduced the burden of illness by 61.1%; and reduced the incidence of PHN by 66.5%.45 The live, attenuated vaccine is contraindicated in children, pregnant women, and immunocompromised individuals.
The number needed to treat for herpes zoster vaccine is 175; that is, 1 case of herpes zoster is avoided for every 175 people vaccinated.1
Key Point One case of herpes zoster is avoided for every 175 people vaccinated. |
Newer tools mean a better outcome
We have improved our ability to diminish the incidence of herpes zoster and PHN and to manage postherpetic pain more effectively. These advances include the development of a herpes zoster vaccine; consensus that antiviral therapy and aggressive pain management can reduce the burden of PHN; identification of efficacious treatments for PHN; and recognition of PHN as a study model for neuropathic pain research.
Disclosure
The authors reported no potential conflict of interest relevant to this article.
References
- Oxman MN, Levin MJ, Johnson GR, et al; Shingles Prevention Study Group. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271–2284.
- Bennett GJ. Neuropathic pain: An overview. In: Borsook D, ed. Molecular Neurobiology of Pain. Seattle, WA: IASP Press; 1997:109-113.
- Jung BF, Johnson RW, Griffin DR, Dworkin RH. Risk factors for postherpetic neuralgia in patients with herpes zoster. Neurology. 2004;62:1545–1551.
- Dworkin RH, Boon RJ, Griffin DR, Phung D. Postherpetic neuralgia: impact of famciclovir, age, rash severity, and acute pain in herpes zoster patients. J Infect Dis. 1998;178(suppl 1):S76–S80.
- Volpi A, Gross G, Hercogova J, Johnson RW. Current management of herpes zoster: the European view. Am J Clin Dermatol. 2005;6:317–325.
- Dworkin RH, Portenoy RK. Pain and its persistence in herpes zoster. Pain. 1996;67:241–251.
- Bowsher D. Pathophysiology of postherpetic neuralgia: towards a rational treatment. Neurology. 1995;45(12 suppl 8):S56–S57.
- Dworkin RH, Schmader KE. Epidemiology and natural history of herpes zoster and postherpetic neuralgia. In Watson CPN, Gershon AA, eds. Herpes Zoster and Postherpetic Neuralgia. 2nd ed. New York, NY: Elsevier Press; 2001:39-64.
- Tyring S, Barbarash RA, Nahlik JE, et al. Famciclovir for the treatment of acute herpes zoster: effects on acute disease and postherpetic neuralgia. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:89–96.
- Kurokawa I, Kumano K, Murkawa K. Clinical correlates of prolonged pain in Japanese patients with acute herpes zoster. J Int Med Res. 2002;30:56–65.
- Zaal MJ, Volker-Dieben HJ, Wienesen M, et al. Longitudinal analysis of varicella-zoster virus DNA on the ocular surface associated with herpes zoster ophthalmicus. Am J Ophthalmol. 2001;131:25–29.
- Wood MJ, Johnson RW, McKendrick MW, et al. A randomized trial of acyclovir for 7 days or 21 days with and without prednisolone for treatment of acute herpes zoster. N Engl J Med. 1994;330:896–900.
- Whitley RJ, Weiss H, Gnann JW Jr, et al. Acyclovir with and without prednisone for the treatment of herpes zoster: a randomized, placebo-controlled trial. Ann Intern Med. 1996;125:376–383.
- Rice AS, Maton S. Gabapentin in postherpetic neuralgia: a randomised, double blind, placebo controlled study. Pain. 2001;94:215–224.
- Collins SL, Moore RA, McQuay HJ, Wiffen P. Antidepressants and anticonvulsants for diabetic neuropathy and postherpetic neuralgia: a quantitative systematic review. J Pain Symptom Manage. 2000;20:449–458.
- Wong MO, Eldon MA, Keane WF, et al. Disposition of gabapentin in anuric subjects on hemodialysis. J Clin Pharmacol. 1995;35:622–626.
- Horizant (gabapentin encarbil) extended-release tablets [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2012.
- Irving G, Jensen M, Cramer M, et al. Efficacy and tolerability of gastric-retentive gabapentin for the treatment of postherpetic neuralgia: results of a double-blind, randomized, placebo-controlled clinical trial. Clin J Pain. 2009;25:185–192.
- Jensen MP, Chiang YK, Wu J. Assessment of pain quality in a clinical trial of gabapentin extended release for postherpetic neuralgia. Clin J Pain. 2009;25:286–292.
- Wallace MS, Irving G, Cowles VE. Gabapentin extended-release tablets for the treatment of patients with postherpetic neuralgia: a randomized, double-blind, placebo-controlled, multicenter study. Clin Drug Investig. 2010;30:765–776.
- Frampton JE, Foster RH. Pregabalin in the treatment of postherpetic neuralgia. Drugs. 2005;65:111–118.
- Kochar D, Garg P, Bumb RA, et al. Divalproex sodium in the management of postherpetic neuralgia: a randomized double-blind placebo-controlled study. QJM. 2005;98:29–34.
- Watson CP, Vernich L, Chipman M, Reed K. Nortriptyline vs amitriptyline in postherpetic neuralgia: a randomized trial. Neurology. 1998;51:1166–1171.
- Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132:237–251.
- Cymbalta (duloxetine hydrochloride) delayed-release capsules [package insert]. Indianapolis, IN: Lilly USA; 2011.
- Rowbotham MC, Goli V, Kunz NR, Lei D. Venlafaxine extended release in the treatment of painful diabetic neuropathy: a double-blind, placebo-controlled study. Pain. 2004;110:697–706.
- Watson CP, Babul N. Efficacy of oxycodone in neuropathic pain: a randomized trial in postherpetic neuralgia. Neurology. 1998;50:1837–1841.
- Gilron I, Bailey JM, Tu D, et al. Morphine, gabapentin, or their combination for neuropathic pain. N Engl J Med. 2005;352:1324–1334.
- Boureau F, Legallicier P, Kabir-Ahmadi M. Tramadol in post-herpetic neuralgia: a randomized, double-blind, placebo-controlled trial. Pain. 2003;104:323–331.
- Hermann DN, Barbano RL, Hart-Gouleau S, et al. An open-label study of the lidocaine patch 5% in painful idiopathic sensory polyneuropathy. Pain Med. 2005;379–384.
- Davies PS, Galer BS. Review of lidocaine patch 5% studies in the treatment of postherpetic neuralgia. Drugs. 2004;64:937–947.
- Gammaitoni AR, Alvarez NA, Galer BS. Safety and tolerability of the lidocaine patch 5%, a targeted peripheral analgesic: a review of literature. J Clin Pharmacol. 2003;43:111–117.
- Watson CP, Tyler KL, Bickers DR, et al. A randomized vehicle-controlled trial of topical capsaicin in the treatment of postherpetic neuralgia. Clin Ther. 1993;15:510–526.
- Backonja MM, Malan TP, Vanhove GF, Tobias JK. C102/106 Study Group. NGX-4010, a high concentration capsaicin patch, for the treatment of postherpetic neuralgia: a randomized, double-blind, controlled study with an open-label extension. Pain Med. 2010;11:600–608.
- Kumar V, Krone K, Mathieu A. Neuraxial and sympathetic blocks in herpes zoster and postherpetic neuralgia: an appraisal of current evidence. Reg Anesth Pain Med. 2004;29:454–461.
- Doi K, Nikai T, Sakura S, Saito Y. Intercostal nerve block with 5% tetracaine for chronic pain syndromes. J Clin Anesth. 2002;14:39–41.
- Kotani N, Kushikata T, Hashimoto H, et al. Intrathecal methylprednisolone for intractable postherpetic neuralgia. N Engl J Med. 2000;343:1514–1519.
- Harke H, Gretenkort P, Ladleif HU, et al. Spinal cord stimulation in postherpetic neuralgia and in acute herpes zoster pain. Anesth Anal. 2002;94:694–700.
- Barnard D, Lloyd J, Evans J. Cryoanalgesia in the management of chronic facial pain. J Maxillofac Surg. 1981;9:101–102.
- Jones MJ, Murrin KR. Intercostal block with cryotherapy. Ann R Coll Surg Engl. 1987;69:261–262.
- Xiao L, Hui H. Therapeutic effect of botulinium toxin A in the treatment of postherpetic neuralgia by subcutaneous injection. Presented at: 26th Annual Meeting of the American Academy of Pain Medicine; February 3-6, 2010; San Antonio, TX.
- Marin M, Güris D, Chaves SS, et al. Prevention of varicella: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2007;56(RR-4):1–40.
- Harpaz R, Ortega-Sanchez IR, Seward JF. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1–30.
- Centers for Disease Control and Prevention (CDC). Update on herpes zoster vaccine: licensure for persons aged 50 through 59 years. MMWR Morb Mortal Wkly Rep. 2011;60(44):1528.
- Gnann JW Jr. Vaccination to prevent herpes zoster in older adults. J Pain. 2008;9(1 suppl 1):S31–S36.
Holiday Bonuses Revisited
As I’ve been writing for several years, holiday bonuses have increasingly become a thing of the past in the business world. Most companies have replaced them with various types of structured, incentive-based reward systems.
Yet an informal poll of dermatologists around the country on the RxDerm-L listserv reveals that a substantial percentage of private practitioners continue to offer their employees no-strings-attached holiday bonuses. This is somewhat understandable in the sense that, once employees come to expect a particular benefit, it is difficult to take it away. Given the uncertainties of the fluctuating economy, though, and the impending changes in the status of health care in this country, many practices may have to follow the national trend and begin tying their awards programs to performance.
Holiday bonuses have been falling out of favor in the business world because companies face increased pressure to reduce costs and so have become more focused on growth and performance. Since this is also increasingly true in the world of private practice, it makes sense for medical practices to heed this trend.
Instead of giving arbitrary, across-the-board bonuses, you may want to find ways to appropriately reward your highest-performing employees. This can be done by reserving more bonus budgets (called "variable compensation" in business lingo, as opposed to guaranteed, salaried compensation) for bonuses that are based on performance and must be re-earned each year.
First, however, you must decide what targets and goals you wish your employees to achieve during the year. Specific performance goals will vary by the type of practice: Cosmetic practices might set financial goals, such as a profit and revenue targets, while offices primarily practicing medical dermatology might measure performance according to output and quality of work.
Whatever changes you decide to make, be sure to share them with your staff from the outset. Employees must be aware of what they need to accomplish to earn a performance-based bonus.
Commonly cited guidelines for effectively rewarding employees include the following:
• Reward your employees in ways that they find rewarding. (This does not necessarily mean cash.) The reward should be matched to the achievement, so bigger achievements earn bigger rewards.
• To be effective, rewards must be given as soon as possible after a specific laudable behavior or achievement, and the employee should always be told why he or she is receiving it. A reward coming weeks or months later – say, during the holiday season – has little or no effect as a performance incentive.
So how do you know what rewards your employees will find rewarding? Ask them! In my office, their ideas have been surprisingly creative – and cheap. For example, my employees are required to park their cars each day on the other side of the hospital campus from my office building. One of them suggested that a closer parking space would be a good reward, so I obtained an extra access card for the doctors’ lot right next to my building and each month one "Employee of the Month" gets to park there. This reward – which costs me nothing – has become the most hotly contested in the office.
One of the strongest motivators is the confidence that you, the boss, have taken the time to notice a job well done and praise it publicly, in a timely manner.
Time off is another powerful motivator: Who (including you) doesn’t appreciate a bit more free time?
This is not to say, of course, that you cannot also give your employees a gift at holiday time – as long as you (and they) understand that it is a one-time gift, with no guarantees or expectations of annual repetition. Such gifts usually consist of either cash (or gift certificates/cards) or a non-cash gift such as a fruit basket or baked goods.
Keep in mind that bonuses and gifts nearly always qualify as a tax write-off for employers, but they may or may not count as taxable income for employees. As a general rule, cash bonuses tend to be taxable while non-cash awards are not, but check your state’s applicable laws to be sure.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. To respond to this column, e-mail him at our editorial offices at [email protected].
As I’ve been writing for several years, holiday bonuses have increasingly become a thing of the past in the business world. Most companies have replaced them with various types of structured, incentive-based reward systems.
Yet an informal poll of dermatologists around the country on the RxDerm-L listserv reveals that a substantial percentage of private practitioners continue to offer their employees no-strings-attached holiday bonuses. This is somewhat understandable in the sense that, once employees come to expect a particular benefit, it is difficult to take it away. Given the uncertainties of the fluctuating economy, though, and the impending changes in the status of health care in this country, many practices may have to follow the national trend and begin tying their awards programs to performance.
Holiday bonuses have been falling out of favor in the business world because companies face increased pressure to reduce costs and so have become more focused on growth and performance. Since this is also increasingly true in the world of private practice, it makes sense for medical practices to heed this trend.
Instead of giving arbitrary, across-the-board bonuses, you may want to find ways to appropriately reward your highest-performing employees. This can be done by reserving more bonus budgets (called "variable compensation" in business lingo, as opposed to guaranteed, salaried compensation) for bonuses that are based on performance and must be re-earned each year.
First, however, you must decide what targets and goals you wish your employees to achieve during the year. Specific performance goals will vary by the type of practice: Cosmetic practices might set financial goals, such as a profit and revenue targets, while offices primarily practicing medical dermatology might measure performance according to output and quality of work.
Whatever changes you decide to make, be sure to share them with your staff from the outset. Employees must be aware of what they need to accomplish to earn a performance-based bonus.
Commonly cited guidelines for effectively rewarding employees include the following:
• Reward your employees in ways that they find rewarding. (This does not necessarily mean cash.) The reward should be matched to the achievement, so bigger achievements earn bigger rewards.
• To be effective, rewards must be given as soon as possible after a specific laudable behavior or achievement, and the employee should always be told why he or she is receiving it. A reward coming weeks or months later – say, during the holiday season – has little or no effect as a performance incentive.
So how do you know what rewards your employees will find rewarding? Ask them! In my office, their ideas have been surprisingly creative – and cheap. For example, my employees are required to park their cars each day on the other side of the hospital campus from my office building. One of them suggested that a closer parking space would be a good reward, so I obtained an extra access card for the doctors’ lot right next to my building and each month one "Employee of the Month" gets to park there. This reward – which costs me nothing – has become the most hotly contested in the office.
One of the strongest motivators is the confidence that you, the boss, have taken the time to notice a job well done and praise it publicly, in a timely manner.
Time off is another powerful motivator: Who (including you) doesn’t appreciate a bit more free time?
This is not to say, of course, that you cannot also give your employees a gift at holiday time – as long as you (and they) understand that it is a one-time gift, with no guarantees or expectations of annual repetition. Such gifts usually consist of either cash (or gift certificates/cards) or a non-cash gift such as a fruit basket or baked goods.
Keep in mind that bonuses and gifts nearly always qualify as a tax write-off for employers, but they may or may not count as taxable income for employees. As a general rule, cash bonuses tend to be taxable while non-cash awards are not, but check your state’s applicable laws to be sure.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. To respond to this column, e-mail him at our editorial offices at [email protected].
As I’ve been writing for several years, holiday bonuses have increasingly become a thing of the past in the business world. Most companies have replaced them with various types of structured, incentive-based reward systems.
Yet an informal poll of dermatologists around the country on the RxDerm-L listserv reveals that a substantial percentage of private practitioners continue to offer their employees no-strings-attached holiday bonuses. This is somewhat understandable in the sense that, once employees come to expect a particular benefit, it is difficult to take it away. Given the uncertainties of the fluctuating economy, though, and the impending changes in the status of health care in this country, many practices may have to follow the national trend and begin tying their awards programs to performance.
Holiday bonuses have been falling out of favor in the business world because companies face increased pressure to reduce costs and so have become more focused on growth and performance. Since this is also increasingly true in the world of private practice, it makes sense for medical practices to heed this trend.
Instead of giving arbitrary, across-the-board bonuses, you may want to find ways to appropriately reward your highest-performing employees. This can be done by reserving more bonus budgets (called "variable compensation" in business lingo, as opposed to guaranteed, salaried compensation) for bonuses that are based on performance and must be re-earned each year.
First, however, you must decide what targets and goals you wish your employees to achieve during the year. Specific performance goals will vary by the type of practice: Cosmetic practices might set financial goals, such as a profit and revenue targets, while offices primarily practicing medical dermatology might measure performance according to output and quality of work.
Whatever changes you decide to make, be sure to share them with your staff from the outset. Employees must be aware of what they need to accomplish to earn a performance-based bonus.
Commonly cited guidelines for effectively rewarding employees include the following:
• Reward your employees in ways that they find rewarding. (This does not necessarily mean cash.) The reward should be matched to the achievement, so bigger achievements earn bigger rewards.
• To be effective, rewards must be given as soon as possible after a specific laudable behavior or achievement, and the employee should always be told why he or she is receiving it. A reward coming weeks or months later – say, during the holiday season – has little or no effect as a performance incentive.
So how do you know what rewards your employees will find rewarding? Ask them! In my office, their ideas have been surprisingly creative – and cheap. For example, my employees are required to park their cars each day on the other side of the hospital campus from my office building. One of them suggested that a closer parking space would be a good reward, so I obtained an extra access card for the doctors’ lot right next to my building and each month one "Employee of the Month" gets to park there. This reward – which costs me nothing – has become the most hotly contested in the office.
One of the strongest motivators is the confidence that you, the boss, have taken the time to notice a job well done and praise it publicly, in a timely manner.
Time off is another powerful motivator: Who (including you) doesn’t appreciate a bit more free time?
This is not to say, of course, that you cannot also give your employees a gift at holiday time – as long as you (and they) understand that it is a one-time gift, with no guarantees or expectations of annual repetition. Such gifts usually consist of either cash (or gift certificates/cards) or a non-cash gift such as a fruit basket or baked goods.
Keep in mind that bonuses and gifts nearly always qualify as a tax write-off for employers, but they may or may not count as taxable income for employees. As a general rule, cash bonuses tend to be taxable while non-cash awards are not, but check your state’s applicable laws to be sure.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. To respond to this column, e-mail him at our editorial offices at [email protected].

Comparison Shopping
My friend had a cyst removed from the end of her right fourth finger. She told me that it’s healing well and she’s happy with the doctor’s work.
"Who removed it?" I asked her.
She gave me the name of a hand surgeon at a local teaching hospital.
"Did you compare alternatives for cost?" I asked her.
She had gotten another surgical opinion, but didn’t really understand my question about cost comparison. My friend is a retired attorney who knows me well enough to realize I’m not always totally serious.
I told her about the current thrust to make consumers (i.e., patients) more cost conscious by giving them "more skin in the game" (making it worth their while to get the best deal they can, as they would when, say, buying a flat-screen TV).
When I explained to my friend what I meant, she told me she wasn’t sure how well that would work. I told her that although I have skin in the skin game, I’m not so sure either.
Changes are in the wind. I got an e-mail the other day from a patient whose leg I had recently biopsied. I had told him that the result showed a fairly large basal cell and recommended excision, suggesting either of two surgeons.
His answer was: "Thanks. I will compare their costs and let you know which one I pick."
I responded that doing that would be fine, but might be complicated by the fact that one of the surgeons does Mohs, so to compare prices he would need to consult that doctor first and find out which technique he would use.
Perhaps I shouldn’t have been put off by his e-mail, but I was. It seemed to imply that a professional recommendation is on the same plane as advice about picking a lawn mower. Is it? I was saying (or thought I was saying): "Here are two doctors whose work I know and trust." Reducing that to dollars and cents makes it something else, something less.
What it actually does is to reduce my professional opinion to shopping advice, which is in fact exactly what market-based incentives are supposed to do.
I thought of this push for cost consciousness a few months ago, when my wife had back surgery. We got several opinions: Laminectomy? Laminectomy with fusion? Even when surgeons at different hospitals recommended the same procedure, we did not ask what reimbursement rate the respective institution had negotiated with our insurer. (There are, of course, big differences, based not on quality – whatever that is – but on each hospital network’s market clout.)
The surgeon we picked arranged for several preoperative visits. At one of them the nurse said my wife would need a CT scan. We went right up one floor, and she had it done.
But should we have? There had already been an MRI. Was a CT scan really needed? And if it was, would we get the best price upstairs, or maybe across town?
Did my wife know? As a dermatologist, did I? Of course not. What would it have meant for us to say, "Hold on now, we’ve got skin in this game. We’d like to know why you need a CT scan. Is it really necessary? Is this the most cost-effective place to do it?"
Would that have been a shopping question, or a professional challenge? What would my friend’s hand surgeon have said if she asked him for a quote, or brought in one from another surgeon? Is buying surgery similar to bringing in a competitor’s coupon to Wal-Mart, or scanning a bookstore’s barcode on the Amazon app to see if they can match or beat it?
The powers shaping health care are not likely to be moved by such questions. They will point out – correctly – that medical costs are out of control, and therefore something must be done. They will therefore do something, as they are doing with electronic health records and will shortly do with ICD-10. The consequences of all these actions, intended and otherwise, remain to be seen both to doctors as providers and to all of us as consumers.
In the meantime, I have downloaded a discount coupon from the web: 10% off on any cystoscopy, but only if I act now and bring along 10 friends.
Let’s go, guys!
Dr. Rockoff practices dermatology in Brookline, Mass. To respond to this column, e-mail Dr. Rockoff at at [email protected].
My friend had a cyst removed from the end of her right fourth finger. She told me that it’s healing well and she’s happy with the doctor’s work.
"Who removed it?" I asked her.
She gave me the name of a hand surgeon at a local teaching hospital.
"Did you compare alternatives for cost?" I asked her.
She had gotten another surgical opinion, but didn’t really understand my question about cost comparison. My friend is a retired attorney who knows me well enough to realize I’m not always totally serious.
I told her about the current thrust to make consumers (i.e., patients) more cost conscious by giving them "more skin in the game" (making it worth their while to get the best deal they can, as they would when, say, buying a flat-screen TV).
When I explained to my friend what I meant, she told me she wasn’t sure how well that would work. I told her that although I have skin in the skin game, I’m not so sure either.
Changes are in the wind. I got an e-mail the other day from a patient whose leg I had recently biopsied. I had told him that the result showed a fairly large basal cell and recommended excision, suggesting either of two surgeons.
His answer was: "Thanks. I will compare their costs and let you know which one I pick."
I responded that doing that would be fine, but might be complicated by the fact that one of the surgeons does Mohs, so to compare prices he would need to consult that doctor first and find out which technique he would use.
Perhaps I shouldn’t have been put off by his e-mail, but I was. It seemed to imply that a professional recommendation is on the same plane as advice about picking a lawn mower. Is it? I was saying (or thought I was saying): "Here are two doctors whose work I know and trust." Reducing that to dollars and cents makes it something else, something less.
What it actually does is to reduce my professional opinion to shopping advice, which is in fact exactly what market-based incentives are supposed to do.
I thought of this push for cost consciousness a few months ago, when my wife had back surgery. We got several opinions: Laminectomy? Laminectomy with fusion? Even when surgeons at different hospitals recommended the same procedure, we did not ask what reimbursement rate the respective institution had negotiated with our insurer. (There are, of course, big differences, based not on quality – whatever that is – but on each hospital network’s market clout.)
The surgeon we picked arranged for several preoperative visits. At one of them the nurse said my wife would need a CT scan. We went right up one floor, and she had it done.
But should we have? There had already been an MRI. Was a CT scan really needed? And if it was, would we get the best price upstairs, or maybe across town?
Did my wife know? As a dermatologist, did I? Of course not. What would it have meant for us to say, "Hold on now, we’ve got skin in this game. We’d like to know why you need a CT scan. Is it really necessary? Is this the most cost-effective place to do it?"
Would that have been a shopping question, or a professional challenge? What would my friend’s hand surgeon have said if she asked him for a quote, or brought in one from another surgeon? Is buying surgery similar to bringing in a competitor’s coupon to Wal-Mart, or scanning a bookstore’s barcode on the Amazon app to see if they can match or beat it?
The powers shaping health care are not likely to be moved by such questions. They will point out – correctly – that medical costs are out of control, and therefore something must be done. They will therefore do something, as they are doing with electronic health records and will shortly do with ICD-10. The consequences of all these actions, intended and otherwise, remain to be seen both to doctors as providers and to all of us as consumers.
In the meantime, I have downloaded a discount coupon from the web: 10% off on any cystoscopy, but only if I act now and bring along 10 friends.
Let’s go, guys!
Dr. Rockoff practices dermatology in Brookline, Mass. To respond to this column, e-mail Dr. Rockoff at at [email protected].
My friend had a cyst removed from the end of her right fourth finger. She told me that it’s healing well and she’s happy with the doctor’s work.
"Who removed it?" I asked her.
She gave me the name of a hand surgeon at a local teaching hospital.
"Did you compare alternatives for cost?" I asked her.
She had gotten another surgical opinion, but didn’t really understand my question about cost comparison. My friend is a retired attorney who knows me well enough to realize I’m not always totally serious.
I told her about the current thrust to make consumers (i.e., patients) more cost conscious by giving them "more skin in the game" (making it worth their while to get the best deal they can, as they would when, say, buying a flat-screen TV).
When I explained to my friend what I meant, she told me she wasn’t sure how well that would work. I told her that although I have skin in the skin game, I’m not so sure either.
Changes are in the wind. I got an e-mail the other day from a patient whose leg I had recently biopsied. I had told him that the result showed a fairly large basal cell and recommended excision, suggesting either of two surgeons.
His answer was: "Thanks. I will compare their costs and let you know which one I pick."
I responded that doing that would be fine, but might be complicated by the fact that one of the surgeons does Mohs, so to compare prices he would need to consult that doctor first and find out which technique he would use.
Perhaps I shouldn’t have been put off by his e-mail, but I was. It seemed to imply that a professional recommendation is on the same plane as advice about picking a lawn mower. Is it? I was saying (or thought I was saying): "Here are two doctors whose work I know and trust." Reducing that to dollars and cents makes it something else, something less.
What it actually does is to reduce my professional opinion to shopping advice, which is in fact exactly what market-based incentives are supposed to do.
I thought of this push for cost consciousness a few months ago, when my wife had back surgery. We got several opinions: Laminectomy? Laminectomy with fusion? Even when surgeons at different hospitals recommended the same procedure, we did not ask what reimbursement rate the respective institution had negotiated with our insurer. (There are, of course, big differences, based not on quality – whatever that is – but on each hospital network’s market clout.)
The surgeon we picked arranged for several preoperative visits. At one of them the nurse said my wife would need a CT scan. We went right up one floor, and she had it done.
But should we have? There had already been an MRI. Was a CT scan really needed? And if it was, would we get the best price upstairs, or maybe across town?
Did my wife know? As a dermatologist, did I? Of course not. What would it have meant for us to say, "Hold on now, we’ve got skin in this game. We’d like to know why you need a CT scan. Is it really necessary? Is this the most cost-effective place to do it?"
Would that have been a shopping question, or a professional challenge? What would my friend’s hand surgeon have said if she asked him for a quote, or brought in one from another surgeon? Is buying surgery similar to bringing in a competitor’s coupon to Wal-Mart, or scanning a bookstore’s barcode on the Amazon app to see if they can match or beat it?
The powers shaping health care are not likely to be moved by such questions. They will point out – correctly – that medical costs are out of control, and therefore something must be done. They will therefore do something, as they are doing with electronic health records and will shortly do with ICD-10. The consequences of all these actions, intended and otherwise, remain to be seen both to doctors as providers and to all of us as consumers.
In the meantime, I have downloaded a discount coupon from the web: 10% off on any cystoscopy, but only if I act now and bring along 10 friends.
Let’s go, guys!
Dr. Rockoff practices dermatology in Brookline, Mass. To respond to this column, e-mail Dr. Rockoff at at [email protected].
