User login
BRCA1/2 testing and cancer risk management in underserved women at a public hospital
Background and objective Genetic test uptake and cancer risk management have been understudied in medically underserved populations. Study aims were to quantify rates of BRCA1/2 genetic testing and evidence-based cancer risk management (ie, prophylactic surgeries and surveillance practices) in women who were seen for breast and ovarian cancer genetic counseling in a public, safety net health system.
Methods We conducted a retrospective medical record abstraction of 195 women who presented for breast or ovarian genetic counseling within a 2-year period (2008-2009) at Parkland Health & Hospital System in Dallas, Texas.
Results The identified women represented a racially and ethnically diverse population: 48% Hispanic, 37% non-Hispanic black, 12% non-Hispanic white, and 3% Asian. Among the 158 women who were medically eligible for genetic testing, 134 (84.8%) received BRCA1/2 results, with most tests funded through a financial assistance program. In all, 29 women (22%) tested positive for BRCA1/2 mutations. Financial and funding barriers were identified for 20 of the untested women. Among the identified high-risk women (mutation carriers, selected variants, and noncarriers with pretest BRCAPRO scores 30 or more), 26% had prophylactic breast surgeries and 33% had prophylactic ovarian surgeries within the follow-up period averaging 35 months. Of those who opted for surveillance, 71% had at least 1 mammogram or MRI and 38% had CA-125 tests. Trends indicated lower rates of all risk management behaviors, except for mammogram or MRI, among non-Hispanic black women.
Conclusions Within this racially and ethnically diverse sample, BRCA1/2 test uptake was high, but financial barriers were identified for nontested women. The rates of breast cancer risk management were generally comparable with other studies, but risk management for ovarian cancer was limited, especially among non-Hispanic black women. The reasons for these apparen disparities should be further explored.
Click on the PDF icon at the top of this introduction to read the full article.
Background and objective Genetic test uptake and cancer risk management have been understudied in medically underserved populations. Study aims were to quantify rates of BRCA1/2 genetic testing and evidence-based cancer risk management (ie, prophylactic surgeries and surveillance practices) in women who were seen for breast and ovarian cancer genetic counseling in a public, safety net health system.
Methods We conducted a retrospective medical record abstraction of 195 women who presented for breast or ovarian genetic counseling within a 2-year period (2008-2009) at Parkland Health & Hospital System in Dallas, Texas.
Results The identified women represented a racially and ethnically diverse population: 48% Hispanic, 37% non-Hispanic black, 12% non-Hispanic white, and 3% Asian. Among the 158 women who were medically eligible for genetic testing, 134 (84.8%) received BRCA1/2 results, with most tests funded through a financial assistance program. In all, 29 women (22%) tested positive for BRCA1/2 mutations. Financial and funding barriers were identified for 20 of the untested women. Among the identified high-risk women (mutation carriers, selected variants, and noncarriers with pretest BRCAPRO scores 30 or more), 26% had prophylactic breast surgeries and 33% had prophylactic ovarian surgeries within the follow-up period averaging 35 months. Of those who opted for surveillance, 71% had at least 1 mammogram or MRI and 38% had CA-125 tests. Trends indicated lower rates of all risk management behaviors, except for mammogram or MRI, among non-Hispanic black women.
Conclusions Within this racially and ethnically diverse sample, BRCA1/2 test uptake was high, but financial barriers were identified for nontested women. The rates of breast cancer risk management were generally comparable with other studies, but risk management for ovarian cancer was limited, especially among non-Hispanic black women. The reasons for these apparen disparities should be further explored.
Click on the PDF icon at the top of this introduction to read the full article.
Background and objective Genetic test uptake and cancer risk management have been understudied in medically underserved populations. Study aims were to quantify rates of BRCA1/2 genetic testing and evidence-based cancer risk management (ie, prophylactic surgeries and surveillance practices) in women who were seen for breast and ovarian cancer genetic counseling in a public, safety net health system.
Methods We conducted a retrospective medical record abstraction of 195 women who presented for breast or ovarian genetic counseling within a 2-year period (2008-2009) at Parkland Health & Hospital System in Dallas, Texas.
Results The identified women represented a racially and ethnically diverse population: 48% Hispanic, 37% non-Hispanic black, 12% non-Hispanic white, and 3% Asian. Among the 158 women who were medically eligible for genetic testing, 134 (84.8%) received BRCA1/2 results, with most tests funded through a financial assistance program. In all, 29 women (22%) tested positive for BRCA1/2 mutations. Financial and funding barriers were identified for 20 of the untested women. Among the identified high-risk women (mutation carriers, selected variants, and noncarriers with pretest BRCAPRO scores 30 or more), 26% had prophylactic breast surgeries and 33% had prophylactic ovarian surgeries within the follow-up period averaging 35 months. Of those who opted for surveillance, 71% had at least 1 mammogram or MRI and 38% had CA-125 tests. Trends indicated lower rates of all risk management behaviors, except for mammogram or MRI, among non-Hispanic black women.
Conclusions Within this racially and ethnically diverse sample, BRCA1/2 test uptake was high, but financial barriers were identified for nontested women. The rates of breast cancer risk management were generally comparable with other studies, but risk management for ovarian cancer was limited, especially among non-Hispanic black women. The reasons for these apparen disparities should be further explored.
Click on the PDF icon at the top of this introduction to read the full article.
Community Oncology Podcast - Pazopanib in soft tissue sarcoma
Dr. David Henry's podcast covers highlights of the November issue including pazopanib in soft tissue sarcoma and dasatinib in first-line treatment of chronic myeloid leukemia.
Dr. David Henry's podcast covers highlights of the November issue including pazopanib in soft tissue sarcoma and dasatinib in first-line treatment of chronic myeloid leukemia.
Dr. David Henry's podcast covers highlights of the November issue including pazopanib in soft tissue sarcoma and dasatinib in first-line treatment of chronic myeloid leukemia.
Federal Grant Supports "eHospitalist" Pilot Program in Wisconsin
John Almquist, MD, FHM, director of hospitalist services for Ministry Health Care, a 15-hospital system serving rural Wisconsin, believes that an "e-hospitalist" pilot project now being tested at Ministry St. Mary's Hospital in Rhinelander, Wis., could be a boon for rural communities that have difficulty recruiting primary-care physicians (PCPs).
When the hospitals in those communities are unable to offer hospitalist coverage, it makes the setting less attractive to PCPs because they might have to follow their patients in the hospital day and night, he explains.
Ministry recruited and trained two nurse practitioners who will soon be deployed at a critical-access hospital in Eagle River, population 1,443, supported remotely by the eight-member HM group in Rhinelander for consultations, supervision, and multidisciplinary rounds. The training is bolstered by written order sets focused on 30 common medical conditions that lead to admissions to rural hospitals.
"The hospitalist in Rhinelander is also able to talk directly to the patient at the remote site," Dr. Almquist says.
The e-hospitalist program uses a telehealth network developed by Marshfield Clinic, a multispecialty physician group practice based in Marshfield, Wis. The clinic recently received a $1 million grant from the federal government to expand its 15-year-old telemedicine program. Part of the grant money is being used to expand the ehospitalist approach to new sites.
Visit our website for more information about hospitalists and telemedicine.
John Almquist, MD, FHM, director of hospitalist services for Ministry Health Care, a 15-hospital system serving rural Wisconsin, believes that an "e-hospitalist" pilot project now being tested at Ministry St. Mary's Hospital in Rhinelander, Wis., could be a boon for rural communities that have difficulty recruiting primary-care physicians (PCPs).
When the hospitals in those communities are unable to offer hospitalist coverage, it makes the setting less attractive to PCPs because they might have to follow their patients in the hospital day and night, he explains.
Ministry recruited and trained two nurse practitioners who will soon be deployed at a critical-access hospital in Eagle River, population 1,443, supported remotely by the eight-member HM group in Rhinelander for consultations, supervision, and multidisciplinary rounds. The training is bolstered by written order sets focused on 30 common medical conditions that lead to admissions to rural hospitals.
"The hospitalist in Rhinelander is also able to talk directly to the patient at the remote site," Dr. Almquist says.
The e-hospitalist program uses a telehealth network developed by Marshfield Clinic, a multispecialty physician group practice based in Marshfield, Wis. The clinic recently received a $1 million grant from the federal government to expand its 15-year-old telemedicine program. Part of the grant money is being used to expand the ehospitalist approach to new sites.
Visit our website for more information about hospitalists and telemedicine.
John Almquist, MD, FHM, director of hospitalist services for Ministry Health Care, a 15-hospital system serving rural Wisconsin, believes that an "e-hospitalist" pilot project now being tested at Ministry St. Mary's Hospital in Rhinelander, Wis., could be a boon for rural communities that have difficulty recruiting primary-care physicians (PCPs).
When the hospitals in those communities are unable to offer hospitalist coverage, it makes the setting less attractive to PCPs because they might have to follow their patients in the hospital day and night, he explains.
Ministry recruited and trained two nurse practitioners who will soon be deployed at a critical-access hospital in Eagle River, population 1,443, supported remotely by the eight-member HM group in Rhinelander for consultations, supervision, and multidisciplinary rounds. The training is bolstered by written order sets focused on 30 common medical conditions that lead to admissions to rural hospitals.
"The hospitalist in Rhinelander is also able to talk directly to the patient at the remote site," Dr. Almquist says.
The e-hospitalist program uses a telehealth network developed by Marshfield Clinic, a multispecialty physician group practice based in Marshfield, Wis. The clinic recently received a $1 million grant from the federal government to expand its 15-year-old telemedicine program. Part of the grant money is being used to expand the ehospitalist approach to new sites.
Visit our website for more information about hospitalists and telemedicine.
ITL: Physician Reviews of HM-Relevant Research
Clinical question: Does the addition of clopidogrel to aspirin reduce the risk of any type of recurrent stroke, or affect the risk of bleeding or death, in patients who recently suffered a lacunar stroke?
Background: There are no prior randomized, multicenter trials on secondary prevention of lacunar stroke; aspirin is the standard antiplatelet therapy in this setting.
Study design: Double-blind, randomized, multicenter trial.
Setting: Eighty-two clinical centers in North America, Latin America, and Spain.
Synopsis: Researchers enrolled 3,020 patients from 2003 to 2011; criteria included age >30 years old and symptomatic lacunar stroke (proven by MRI) in the preceding 180 days.
Results showed no significant difference between recurrent strokes (any type) in the aspirin-only group (2.7% per year) versus the aspirin-plus-clopidogrel group (2.5% per year). Major hemorrhage risk was much higher in the aspirin-plus-clopidogrel group (2.1% per year) versus aspirin-only group (1.1% per year). All-cause mortality also was much higher in the aspirin-plus-clopidogrel group (N=113) versus the aspirin-only group (N=77).
Bottom line: The addition of clopidogrel to aspirin for secondary prevention does not significantly reduce the risk of recurrent stroke, but it does significantly increase the risk of bleeding and death.
Citation: Benavente OR, Hart RG, McClure LA, et al. Effects of clopidogrel added to aspirin in patients with recent lacunar stroke. N Engl J Med. 2012;367:817-825.
For more physician reviews of recent HM-relevant literature, visit our website.
Clinical question: Does the addition of clopidogrel to aspirin reduce the risk of any type of recurrent stroke, or affect the risk of bleeding or death, in patients who recently suffered a lacunar stroke?
Background: There are no prior randomized, multicenter trials on secondary prevention of lacunar stroke; aspirin is the standard antiplatelet therapy in this setting.
Study design: Double-blind, randomized, multicenter trial.
Setting: Eighty-two clinical centers in North America, Latin America, and Spain.
Synopsis: Researchers enrolled 3,020 patients from 2003 to 2011; criteria included age >30 years old and symptomatic lacunar stroke (proven by MRI) in the preceding 180 days.
Results showed no significant difference between recurrent strokes (any type) in the aspirin-only group (2.7% per year) versus the aspirin-plus-clopidogrel group (2.5% per year). Major hemorrhage risk was much higher in the aspirin-plus-clopidogrel group (2.1% per year) versus aspirin-only group (1.1% per year). All-cause mortality also was much higher in the aspirin-plus-clopidogrel group (N=113) versus the aspirin-only group (N=77).
Bottom line: The addition of clopidogrel to aspirin for secondary prevention does not significantly reduce the risk of recurrent stroke, but it does significantly increase the risk of bleeding and death.
Citation: Benavente OR, Hart RG, McClure LA, et al. Effects of clopidogrel added to aspirin in patients with recent lacunar stroke. N Engl J Med. 2012;367:817-825.
For more physician reviews of recent HM-relevant literature, visit our website.
Clinical question: Does the addition of clopidogrel to aspirin reduce the risk of any type of recurrent stroke, or affect the risk of bleeding or death, in patients who recently suffered a lacunar stroke?
Background: There are no prior randomized, multicenter trials on secondary prevention of lacunar stroke; aspirin is the standard antiplatelet therapy in this setting.
Study design: Double-blind, randomized, multicenter trial.
Setting: Eighty-two clinical centers in North America, Latin America, and Spain.
Synopsis: Researchers enrolled 3,020 patients from 2003 to 2011; criteria included age >30 years old and symptomatic lacunar stroke (proven by MRI) in the preceding 180 days.
Results showed no significant difference between recurrent strokes (any type) in the aspirin-only group (2.7% per year) versus the aspirin-plus-clopidogrel group (2.5% per year). Major hemorrhage risk was much higher in the aspirin-plus-clopidogrel group (2.1% per year) versus aspirin-only group (1.1% per year). All-cause mortality also was much higher in the aspirin-plus-clopidogrel group (N=113) versus the aspirin-only group (N=77).
Bottom line: The addition of clopidogrel to aspirin for secondary prevention does not significantly reduce the risk of recurrent stroke, but it does significantly increase the risk of bleeding and death.
Citation: Benavente OR, Hart RG, McClure LA, et al. Effects of clopidogrel added to aspirin in patients with recent lacunar stroke. N Engl J Med. 2012;367:817-825.
For more physician reviews of recent HM-relevant literature, visit our website.
Woman with “Dull, Achy” Back Pain and Shortness of Breath
ANSWER
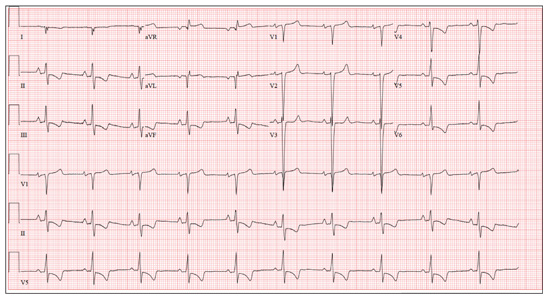
This ECG demonstrates normal sinus rhythm, right-axis deviation, evidence of a lateral MI, and inferolateral ST- and T-wave abnormalities.
Right-axis deviation is indicated by an R-wave axis between 90° and 180° and QS or QR complexes in lead I and/or aVL. While the most common cause of a right-axis deviation is right ventricular hypertrophy, it is also evident in a lateral MI. Evidence for the latter includes the presence of significant Q waves in leads I and aVL. Finally, inferolateral ST- and T-wave changes are evidenced by inverted T waves in leads II, III, aVF, and precordial leads V4 to V6.
ECG evidence of a lateral MI not present on a previous scan (eight months ago), in the presence of a normal troponin level, suggests a recent MI.
ANSWER
This ECG demonstrates normal sinus rhythm, right-axis deviation, evidence of a lateral MI, and inferolateral ST- and T-wave abnormalities.
Right-axis deviation is indicated by an R-wave axis between 90° and 180° and QS or QR complexes in lead I and/or aVL. While the most common cause of a right-axis deviation is right ventricular hypertrophy, it is also evident in a lateral MI. Evidence for the latter includes the presence of significant Q waves in leads I and aVL. Finally, inferolateral ST- and T-wave changes are evidenced by inverted T waves in leads II, III, aVF, and precordial leads V4 to V6.
ECG evidence of a lateral MI not present on a previous scan (eight months ago), in the presence of a normal troponin level, suggests a recent MI.
ANSWER
This ECG demonstrates normal sinus rhythm, right-axis deviation, evidence of a lateral MI, and inferolateral ST- and T-wave abnormalities.
Right-axis deviation is indicated by an R-wave axis between 90° and 180° and QS or QR complexes in lead I and/or aVL. While the most common cause of a right-axis deviation is right ventricular hypertrophy, it is also evident in a lateral MI. Evidence for the latter includes the presence of significant Q waves in leads I and aVL. Finally, inferolateral ST- and T-wave changes are evidenced by inverted T waves in leads II, III, aVF, and precordial leads V4 to V6.
ECG evidence of a lateral MI not present on a previous scan (eight months ago), in the presence of a normal troponin level, suggests a recent MI.
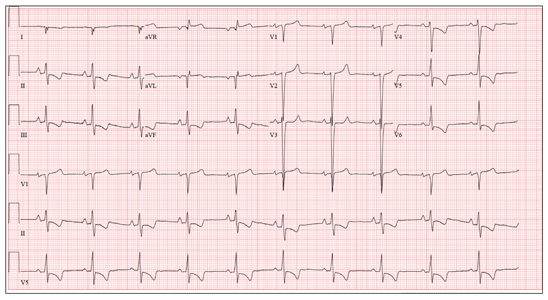
A 70-year-old woman has a 10-year history of a dilated nonischemic cardiomyopathy and New York Heart Association Class II heart failure. She presents with a one-week history of back pain and shortness of breath. She describes the pain as a “dull, achy” pressure, exacerbated by exertion and relieved with rest. She says the pain is localized in the back between her scapulas and does not radiate. She denies substernal chest pain, nausea, vomiting, or diaphoresis; the only associated symptom is dyspnea. Her most recent echocardiogram showed a dilated left ventricle, with a left ventricular ejection fraction of 29%, and a normal right ventricle, with mild hypertrophy and mildly reduced systolic function. She was also noted to have atherosclerotic changes in her ascending and descending thoracic aorta. Medical history is remarkable for diabetes, hypertension, chronic renal insufficiency, hyperlipidemia, and cataracts. Her current medications include aspirin, fer-rous sulfate, furosemide, hydralazine, glargine insulin, isosorbide dinitrate, lisinopril, metoprolol, and raloxifene. She is allergic to codeine, amiodarone, and radi-ographic contrast. Family history is positive for coronary artery disease, diabetes, and stroke. The patient is widowed, does not smoke, and does not consume alcohol. She is very active in her local quilting club. The review of systems is positive for increased weakness and diarrhea. She states that approximately two weeks ago, she experienced vague epigastric pain and diaphoresis; she did not seek medical attention, as it resolved. The physical exam reveals a thin, elderly woman in mild distress. Blood pressure is 139/82 mm Hg; pulse, 66 beats/min; respiratory rate, 21 breaths/¬min-1; and temperature, 35.9°C. Her weight is 108 lb. Pertinent physical findings include a grade II/VI diastolic murmur at the left lower sternal border, 2+ peripheral pulses with a bruit present in the right femoral artery, occasional late expiratory wheezes in both lung bases, vertebral tenderness at the T6-T7 level with no evidence of scoliosis or kyphosis, and no evidence of peripheral edema. She is intact from a neurologic standpoint. Significant laboratory data include a serum glucose level of 294 mg/dL; blood urea nitrogen (BUN), 68 mg/dL; creatinine, 1.75 mg/dL; glomerular filtration rate, 30 mL/min; B-type natriuretic peptide, 984 pg/mL; and serum troponin, 0.11 ng/mL. An ECG is obtained that reveals the following: a ventricular rate of 62 beats/min; PR interval, 160 ms; QRS duration, 94 ms; QT/QTc interval, 404/410 ms; P ax-is, 84°; R axis, 151°; and T axis, 253°. What is your interpretation of this ECG?
Topical Steroids: the Solution or the Cause?
ANSWER
The correct answer is all of the above (choice “d”). Prolonged injudicious use of topical steroids can cause a number of problems, including these; they are collectively termed iatrogenic since they are ultimately caused by prescribed medication. One of the more difficult aspects of this problem to deal with is the “addictive” state, in which withdrawal symptoms compel the patient to continue applying the offending steroid cream.
DISCUSSION
This is a relatively common scenario in dermatology offices. The misuse of topical steroids is well known, and something we strive to prevent—but with mixed results. It’s one of the reasons we’re stingy with refills of such medications, requiring the patient to be seen at least once a year. Unfortunately, this patient had been getting “refills” from friends in Mexico; patients often “borrow” steroid creams from household members or friends, or use products prescribed for one condition to treat others for which they were not intended.
The primary mode of action of topical steroids is vasoconstriction, a positive thing in terms of reduction of inflammation. The bad news is that continuous use of class 1 (the most powerful) steroids, such as clobetasol, can cause such profound and prolonged vasoconstriction that the skin effectively loses its blood supply and withers, sometimes down to adipose tissue. As one might suspect, this is more likely in already thin-skinned areas, including the antecubital area, face, neck, eyelids, and genitals, where the creation of striae is especially common.
Fairly early on in this process, before frank atrophy occurs, the condition being treated usually resolves. However, when the steroid is stopped, stinging and itching immediately return—which, of course, causes the patient to reapply the medication, perpetuating the vicious cycle.
The cycle is ultimately broken by gradual reduction in the frequency of application of successively weaker steroids. Usually, the skin gradually regenerates and returns to normal. In this case, the process will be lengthy and will almost certainly result in significant scarring.
Even injudicious application of weaker classes of steroids (eg, hydrocortisone 2.5% cream) to areas such as the face can result in a range of deleterious effects, including localized rosacea-like eruption or erythema. It has been reported that approximately 75% of cases of perioral dermatitis are either caused by or exacerbated by the application of topical steroids.
Topical application of even mid-strength steroids can also have systemic effects (eg, adrenal suppression, hyperglycemia) if applied over large areas. This is especially true when pediatric patients are involved.
Prevention of these iatrogenic effects lies in selecting the lowest strength steroid for the condition and area in question, then using them sparingly: no more than twice a day, and for no more than five days in a row, stopping for two consecutive days to allow the skin to regenerate. Even more caution should be exercised in treating children and when applying the product to intertriginous areas (skin-on-skin areas, such as the groin, in axillae, or under the breasts). Covering steroid-treated areas with anything—bandages, socks, even skin—effectively potentiates the positive and negative effects of steroids.
ANSWER
The correct answer is all of the above (choice “d”). Prolonged injudicious use of topical steroids can cause a number of problems, including these; they are collectively termed iatrogenic since they are ultimately caused by prescribed medication. One of the more difficult aspects of this problem to deal with is the “addictive” state, in which withdrawal symptoms compel the patient to continue applying the offending steroid cream.
DISCUSSION
This is a relatively common scenario in dermatology offices. The misuse of topical steroids is well known, and something we strive to prevent—but with mixed results. It’s one of the reasons we’re stingy with refills of such medications, requiring the patient to be seen at least once a year. Unfortunately, this patient had been getting “refills” from friends in Mexico; patients often “borrow” steroid creams from household members or friends, or use products prescribed for one condition to treat others for which they were not intended.
The primary mode of action of topical steroids is vasoconstriction, a positive thing in terms of reduction of inflammation. The bad news is that continuous use of class 1 (the most powerful) steroids, such as clobetasol, can cause such profound and prolonged vasoconstriction that the skin effectively loses its blood supply and withers, sometimes down to adipose tissue. As one might suspect, this is more likely in already thin-skinned areas, including the antecubital area, face, neck, eyelids, and genitals, where the creation of striae is especially common.
Fairly early on in this process, before frank atrophy occurs, the condition being treated usually resolves. However, when the steroid is stopped, stinging and itching immediately return—which, of course, causes the patient to reapply the medication, perpetuating the vicious cycle.
The cycle is ultimately broken by gradual reduction in the frequency of application of successively weaker steroids. Usually, the skin gradually regenerates and returns to normal. In this case, the process will be lengthy and will almost certainly result in significant scarring.
Even injudicious application of weaker classes of steroids (eg, hydrocortisone 2.5% cream) to areas such as the face can result in a range of deleterious effects, including localized rosacea-like eruption or erythema. It has been reported that approximately 75% of cases of perioral dermatitis are either caused by or exacerbated by the application of topical steroids.
Topical application of even mid-strength steroids can also have systemic effects (eg, adrenal suppression, hyperglycemia) if applied over large areas. This is especially true when pediatric patients are involved.
Prevention of these iatrogenic effects lies in selecting the lowest strength steroid for the condition and area in question, then using them sparingly: no more than twice a day, and for no more than five days in a row, stopping for two consecutive days to allow the skin to regenerate. Even more caution should be exercised in treating children and when applying the product to intertriginous areas (skin-on-skin areas, such as the groin, in axillae, or under the breasts). Covering steroid-treated areas with anything—bandages, socks, even skin—effectively potentiates the positive and negative effects of steroids.
ANSWER
The correct answer is all of the above (choice “d”). Prolonged injudicious use of topical steroids can cause a number of problems, including these; they are collectively termed iatrogenic since they are ultimately caused by prescribed medication. One of the more difficult aspects of this problem to deal with is the “addictive” state, in which withdrawal symptoms compel the patient to continue applying the offending steroid cream.
DISCUSSION
This is a relatively common scenario in dermatology offices. The misuse of topical steroids is well known, and something we strive to prevent—but with mixed results. It’s one of the reasons we’re stingy with refills of such medications, requiring the patient to be seen at least once a year. Unfortunately, this patient had been getting “refills” from friends in Mexico; patients often “borrow” steroid creams from household members or friends, or use products prescribed for one condition to treat others for which they were not intended.
The primary mode of action of topical steroids is vasoconstriction, a positive thing in terms of reduction of inflammation. The bad news is that continuous use of class 1 (the most powerful) steroids, such as clobetasol, can cause such profound and prolonged vasoconstriction that the skin effectively loses its blood supply and withers, sometimes down to adipose tissue. As one might suspect, this is more likely in already thin-skinned areas, including the antecubital area, face, neck, eyelids, and genitals, where the creation of striae is especially common.
Fairly early on in this process, before frank atrophy occurs, the condition being treated usually resolves. However, when the steroid is stopped, stinging and itching immediately return—which, of course, causes the patient to reapply the medication, perpetuating the vicious cycle.
The cycle is ultimately broken by gradual reduction in the frequency of application of successively weaker steroids. Usually, the skin gradually regenerates and returns to normal. In this case, the process will be lengthy and will almost certainly result in significant scarring.
Even injudicious application of weaker classes of steroids (eg, hydrocortisone 2.5% cream) to areas such as the face can result in a range of deleterious effects, including localized rosacea-like eruption or erythema. It has been reported that approximately 75% of cases of perioral dermatitis are either caused by or exacerbated by the application of topical steroids.
Topical application of even mid-strength steroids can also have systemic effects (eg, adrenal suppression, hyperglycemia) if applied over large areas. This is especially true when pediatric patients are involved.
Prevention of these iatrogenic effects lies in selecting the lowest strength steroid for the condition and area in question, then using them sparingly: no more than twice a day, and for no more than five days in a row, stopping for two consecutive days to allow the skin to regenerate. Even more caution should be exercised in treating children and when applying the product to intertriginous areas (skin-on-skin areas, such as the groin, in axillae, or under the breasts). Covering steroid-treated areas with anything—bandages, socks, even skin—effectively potentiates the positive and negative effects of steroids.
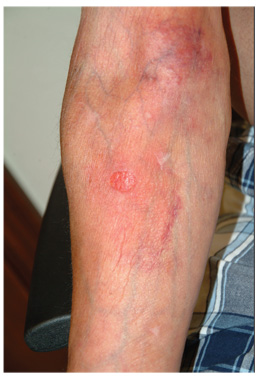
A 59-year-old man presents with skin changes on both antecubital areas. For more than a year, he has applied clobetasol 0.05% cream at least twice daily to the area ostensibly for treatment of long-standing eczema, which has affected not only the antecubital areas but also the patient’s legs. In addition to the eczema, he has a history of atopy, marked by seasonal allergies and asthma. He notes that his stress level has increased in the past several months, which he suspects has contributed to his itching. On examination, marked epidermal atrophy is seen in both antecubital areas, along with extensive purpura. Surface adnexal structures, such as hair, follicles, and skin lines, are sparse at best, but dermal and subdermal vasculature are readily visible. In the midst of the affected area on the right arm, a nickel-sized, full-thickness defect is noted. Beneath it, adipose tissue can be seen. Clearly, these changes are attributable to the effects of the clobetasol, which the patient is advised to stop. But he replies that when he does, the treated areas burn and itch even more, until he obtains relief by applying more clobetasol.

Cold weather and diarrhea: Don't forget yersiniosis
The genus Yersinia includes 11 species. Three species are generally associated with human disease; Y. enterocolitica, Y. pestis, and Y. pseudotuberculosis. Yersinia pestis is the causative agent of plague. Yersinia pseudotuberculosis can manifest with fever, abdominal pain, and scarlatiniform rash. Additional symptoms include diarrhea, sterile joint effusions, erythema nodosum, and septicemia; these symptoms can be indistinguishable from Kawasaki Disease. By report, almost 10 % of cases of Kawasaki Disease in Japan have serologic or bacteriologic evidence of Y. pseudotuberculosis infection [Redbook: 2012 Report of the Committee on Infectious Diseases, 795-7]. Y. enterocolitica is most often associated with yersiniosis.
Although Y. enterocolitica is not the most common cause of diarrheal illness in the United States, it is one of the nine pathogens that have been monitored by the Foodborne Diseases Active Surveillance Network (FoodNet) since 1996. In the United States, it is estimated that Y. enterocolitica causes slightly over 115,000 infections annually (Emerg. Infect. Dis. 2011;17:7-15). The disease is more common in cooler months. It is transmitted by consumption of contaminated food, especially raw or undercooked pork products.

Only a few outbreaks have been reported in the United States, and these were usually associated with consumption of pork, specifically chitterlings (pig intestines), a winter holiday dish prepared most frequently in black households in the South (MMWR 1990;39:819-20). Transmission to infants and young children is thought to occur from caretakers preparing chitterlings who have not adequately cleaned their hands prior to touching objects subsequently handled by the child.
The incubation period is usually 4-6 days (range, 1-14 days). The duration of diarrhea is variable and can persist up to 3 weeks. Organisms can be excreted an average of 6 weeks. Clinical manifestations vary by age. Younger children usually present with fever and diarrhea. Stools frequently contain blood and leucocytes. Vomiting is also reported in most series. In contrast, older children and adults often present with a pseudoappendicitis syndrome with right-sided abdominal pain and fever. Leukocytosis is often present. At surgery, mesenteric adenitis is observed, and the appendix generally is normal.
Bacteremia can occur and is usually associated with infection in children less than 1 year of age and in those with iron-overloaded states, including persons with sickle cell disease, beta-thalassemia, and those receiving deferoxamine therapy. While uncommon, focal manifestations including pharyngitis, osteomyelitis, pyomyositis, pneumonia, empyema, and meningitis may occur.

Diagnosis is confirmed by isolation of the organism from stool, blood, peritoneal fluid, lymph nodes, and throat cultures. Most laboratories do not routinely test for Yersinia in stool cultures. If Y. enterocolitica is suspected, you should notify the laboratory so the stool can be plated on appropriate media (CIN agar). Serologic tests to detect a rise in serum antibody titers to confirm infection are available in reference and research laboratories, but are not generally used for diagnosis. Cross reactivity with Brucella, Salmonella, Vibrio, and Rickettsia may lead to false positive titer results. Y. enterocolitica antibodies also have antigenic similarity with thyroid tissue. You may see persistent elevation of titers in patients with thyroid disease.
Benefit of antimicrobial therapy for isolated Y. enterocolitica gastrointestinal disease and Y. pseudotuberculosis has not been established. Therapy may decrease the duration of fecal shedding. Treatment is indicated for immunocompromised hosts and persons with septicemia and focal infections. Y. enterocolitica and Y. pseudotuberculosis are usually sensitive to trimethoprim-sulfamethoxazole, aminoglycosides, cefotaxime, fluoroquinolones (persons greater than 18 years of age or older), and tetracycline or doxycycline (for children at least 8 years of age and older).
So what is the actual incidence and when should the practitioner be concerned? Initial population based surveillance data for Y. enterocolitica infections in FoodNet sites between 1996 and 1999 reported an overall incidence of 0.9 cases per 100,000 population. The highest incidence was among black and Asian individuals and was 3.2 cases and 1.5 cases per 100,000 population, respectively. The incidence in Hispanics and whites was 0.6 and 0.4 cases per 100,000 respectively. Incidence increased with decreasing age in all racial/ethnic groups. Blacks infants had the highest incidence, 141.9 cases/100,000 population, and the highest incidence in infants was reported from Georgia (207 cases/100,000). Seasonal variation in incidence was noted only in black individuals with peak activity occurring in December (Clin. Infect. Dis. 2004;38[Suppl 3]:S181-9).

The most recent data from FoodNet (1996-2009) reveals an overall incidence of 0.5/100,000. There was a decline in incidence in all racial and ethnic groups. The highest incidence is still observed in black and Asians (0.9 and 0.7 per 100,000). The most dramatic decline occurred in black individuals (3.2 vs. 0.9 per 100,000). In 1998, an educational campaign was initiated in Georgia that targeted high-risk individuals and provided information on the safe handling and preparation of chitterlings. The state of Georgia reported the greatest decline to 0.4/100,000, which has almost eliminated the racial disparity reported in 2009. It is unclear if this campaign was the only reason for the decline in Georgia. The incidence in whites is 0.2/100,000. Since 2007, the incidence in Asian children less than 5 years of ages has been the highest amongst all racial and ethnic groups. Pork consumption is still assumed to be the major source. Seasonal variability persists amongst Black children less 5 years of age, implying that chitterlings may still be the source of infection for individuals in this group (Clin. Infect. Dis. 2012:54 [Suppl 5]:S385-S90).
In general, yersiniosis should be included in the differential of a febrile diarrheal illness, particularly during the cooler months and holiday season. It is prudent to determine if consumption and/or preparation of chitterlings or other pork products by the patient or caretakers has occurred. This will enable you to alert the laboratory so stool specimens can be cultured on the appropriate medium (CIN agar). Consumption of chitterlings is not limited to any specific racial or ethnic group. Individuals from rural and farming areas may also consume this product.
Dr. Word is a pediatric infectious disease specialist and director of the Houston Travel Medicine Clinic. She said she had no relevant financial disclosures. Write to Dr. Word at [email protected].
The genus Yersinia includes 11 species. Three species are generally associated with human disease; Y. enterocolitica, Y. pestis, and Y. pseudotuberculosis. Yersinia pestis is the causative agent of plague. Yersinia pseudotuberculosis can manifest with fever, abdominal pain, and scarlatiniform rash. Additional symptoms include diarrhea, sterile joint effusions, erythema nodosum, and septicemia; these symptoms can be indistinguishable from Kawasaki Disease. By report, almost 10 % of cases of Kawasaki Disease in Japan have serologic or bacteriologic evidence of Y. pseudotuberculosis infection [Redbook: 2012 Report of the Committee on Infectious Diseases, 795-7]. Y. enterocolitica is most often associated with yersiniosis.
Although Y. enterocolitica is not the most common cause of diarrheal illness in the United States, it is one of the nine pathogens that have been monitored by the Foodborne Diseases Active Surveillance Network (FoodNet) since 1996. In the United States, it is estimated that Y. enterocolitica causes slightly over 115,000 infections annually (Emerg. Infect. Dis. 2011;17:7-15). The disease is more common in cooler months. It is transmitted by consumption of contaminated food, especially raw or undercooked pork products.
Only a few outbreaks have been reported in the United States, and these were usually associated with consumption of pork, specifically chitterlings (pig intestines), a winter holiday dish prepared most frequently in black households in the South (MMWR 1990;39:819-20). Transmission to infants and young children is thought to occur from caretakers preparing chitterlings who have not adequately cleaned their hands prior to touching objects subsequently handled by the child.
The incubation period is usually 4-6 days (range, 1-14 days). The duration of diarrhea is variable and can persist up to 3 weeks. Organisms can be excreted an average of 6 weeks. Clinical manifestations vary by age. Younger children usually present with fever and diarrhea. Stools frequently contain blood and leucocytes. Vomiting is also reported in most series. In contrast, older children and adults often present with a pseudoappendicitis syndrome with right-sided abdominal pain and fever. Leukocytosis is often present. At surgery, mesenteric adenitis is observed, and the appendix generally is normal.
Bacteremia can occur and is usually associated with infection in children less than 1 year of age and in those with iron-overloaded states, including persons with sickle cell disease, beta-thalassemia, and those receiving deferoxamine therapy. While uncommon, focal manifestations including pharyngitis, osteomyelitis, pyomyositis, pneumonia, empyema, and meningitis may occur.
Diagnosis is confirmed by isolation of the organism from stool, blood, peritoneal fluid, lymph nodes, and throat cultures. Most laboratories do not routinely test for Yersinia in stool cultures. If Y. enterocolitica is suspected, you should notify the laboratory so the stool can be plated on appropriate media (CIN agar). Serologic tests to detect a rise in serum antibody titers to confirm infection are available in reference and research laboratories, but are not generally used for diagnosis. Cross reactivity with Brucella, Salmonella, Vibrio, and Rickettsia may lead to false positive titer results. Y. enterocolitica antibodies also have antigenic similarity with thyroid tissue. You may see persistent elevation of titers in patients with thyroid disease.
Benefit of antimicrobial therapy for isolated Y. enterocolitica gastrointestinal disease and Y. pseudotuberculosis has not been established. Therapy may decrease the duration of fecal shedding. Treatment is indicated for immunocompromised hosts and persons with septicemia and focal infections. Y. enterocolitica and Y. pseudotuberculosis are usually sensitive to trimethoprim-sulfamethoxazole, aminoglycosides, cefotaxime, fluoroquinolones (persons greater than 18 years of age or older), and tetracycline or doxycycline (for children at least 8 years of age and older).
So what is the actual incidence and when should the practitioner be concerned? Initial population based surveillance data for Y. enterocolitica infections in FoodNet sites between 1996 and 1999 reported an overall incidence of 0.9 cases per 100,000 population. The highest incidence was among black and Asian individuals and was 3.2 cases and 1.5 cases per 100,000 population, respectively. The incidence in Hispanics and whites was 0.6 and 0.4 cases per 100,000 respectively. Incidence increased with decreasing age in all racial/ethnic groups. Blacks infants had the highest incidence, 141.9 cases/100,000 population, and the highest incidence in infants was reported from Georgia (207 cases/100,000). Seasonal variation in incidence was noted only in black individuals with peak activity occurring in December (Clin. Infect. Dis. 2004;38[Suppl 3]:S181-9).
The most recent data from FoodNet (1996-2009) reveals an overall incidence of 0.5/100,000. There was a decline in incidence in all racial and ethnic groups. The highest incidence is still observed in black and Asians (0.9 and 0.7 per 100,000). The most dramatic decline occurred in black individuals (3.2 vs. 0.9 per 100,000). In 1998, an educational campaign was initiated in Georgia that targeted high-risk individuals and provided information on the safe handling and preparation of chitterlings. The state of Georgia reported the greatest decline to 0.4/100,000, which has almost eliminated the racial disparity reported in 2009. It is unclear if this campaign was the only reason for the decline in Georgia. The incidence in whites is 0.2/100,000. Since 2007, the incidence in Asian children less than 5 years of ages has been the highest amongst all racial and ethnic groups. Pork consumption is still assumed to be the major source. Seasonal variability persists amongst Black children less 5 years of age, implying that chitterlings may still be the source of infection for individuals in this group (Clin. Infect. Dis. 2012:54 [Suppl 5]:S385-S90).
In general, yersiniosis should be included in the differential of a febrile diarrheal illness, particularly during the cooler months and holiday season. It is prudent to determine if consumption and/or preparation of chitterlings or other pork products by the patient or caretakers has occurred. This will enable you to alert the laboratory so stool specimens can be cultured on the appropriate medium (CIN agar). Consumption of chitterlings is not limited to any specific racial or ethnic group. Individuals from rural and farming areas may also consume this product.
Dr. Word is a pediatric infectious disease specialist and director of the Houston Travel Medicine Clinic. She said she had no relevant financial disclosures. Write to Dr. Word at [email protected].
The genus Yersinia includes 11 species. Three species are generally associated with human disease; Y. enterocolitica, Y. pestis, and Y. pseudotuberculosis. Yersinia pestis is the causative agent of plague. Yersinia pseudotuberculosis can manifest with fever, abdominal pain, and scarlatiniform rash. Additional symptoms include diarrhea, sterile joint effusions, erythema nodosum, and septicemia; these symptoms can be indistinguishable from Kawasaki Disease. By report, almost 10 % of cases of Kawasaki Disease in Japan have serologic or bacteriologic evidence of Y. pseudotuberculosis infection [Redbook: 2012 Report of the Committee on Infectious Diseases, 795-7]. Y. enterocolitica is most often associated with yersiniosis.
Although Y. enterocolitica is not the most common cause of diarrheal illness in the United States, it is one of the nine pathogens that have been monitored by the Foodborne Diseases Active Surveillance Network (FoodNet) since 1996. In the United States, it is estimated that Y. enterocolitica causes slightly over 115,000 infections annually (Emerg. Infect. Dis. 2011;17:7-15). The disease is more common in cooler months. It is transmitted by consumption of contaminated food, especially raw or undercooked pork products.
Only a few outbreaks have been reported in the United States, and these were usually associated with consumption of pork, specifically chitterlings (pig intestines), a winter holiday dish prepared most frequently in black households in the South (MMWR 1990;39:819-20). Transmission to infants and young children is thought to occur from caretakers preparing chitterlings who have not adequately cleaned their hands prior to touching objects subsequently handled by the child.
The incubation period is usually 4-6 days (range, 1-14 days). The duration of diarrhea is variable and can persist up to 3 weeks. Organisms can be excreted an average of 6 weeks. Clinical manifestations vary by age. Younger children usually present with fever and diarrhea. Stools frequently contain blood and leucocytes. Vomiting is also reported in most series. In contrast, older children and adults often present with a pseudoappendicitis syndrome with right-sided abdominal pain and fever. Leukocytosis is often present. At surgery, mesenteric adenitis is observed, and the appendix generally is normal.
Bacteremia can occur and is usually associated with infection in children less than 1 year of age and in those with iron-overloaded states, including persons with sickle cell disease, beta-thalassemia, and those receiving deferoxamine therapy. While uncommon, focal manifestations including pharyngitis, osteomyelitis, pyomyositis, pneumonia, empyema, and meningitis may occur.
Diagnosis is confirmed by isolation of the organism from stool, blood, peritoneal fluid, lymph nodes, and throat cultures. Most laboratories do not routinely test for Yersinia in stool cultures. If Y. enterocolitica is suspected, you should notify the laboratory so the stool can be plated on appropriate media (CIN agar). Serologic tests to detect a rise in serum antibody titers to confirm infection are available in reference and research laboratories, but are not generally used for diagnosis. Cross reactivity with Brucella, Salmonella, Vibrio, and Rickettsia may lead to false positive titer results. Y. enterocolitica antibodies also have antigenic similarity with thyroid tissue. You may see persistent elevation of titers in patients with thyroid disease.
Benefit of antimicrobial therapy for isolated Y. enterocolitica gastrointestinal disease and Y. pseudotuberculosis has not been established. Therapy may decrease the duration of fecal shedding. Treatment is indicated for immunocompromised hosts and persons with septicemia and focal infections. Y. enterocolitica and Y. pseudotuberculosis are usually sensitive to trimethoprim-sulfamethoxazole, aminoglycosides, cefotaxime, fluoroquinolones (persons greater than 18 years of age or older), and tetracycline or doxycycline (for children at least 8 years of age and older).
So what is the actual incidence and when should the practitioner be concerned? Initial population based surveillance data for Y. enterocolitica infections in FoodNet sites between 1996 and 1999 reported an overall incidence of 0.9 cases per 100,000 population. The highest incidence was among black and Asian individuals and was 3.2 cases and 1.5 cases per 100,000 population, respectively. The incidence in Hispanics and whites was 0.6 and 0.4 cases per 100,000 respectively. Incidence increased with decreasing age in all racial/ethnic groups. Blacks infants had the highest incidence, 141.9 cases/100,000 population, and the highest incidence in infants was reported from Georgia (207 cases/100,000). Seasonal variation in incidence was noted only in black individuals with peak activity occurring in December (Clin. Infect. Dis. 2004;38[Suppl 3]:S181-9).
The most recent data from FoodNet (1996-2009) reveals an overall incidence of 0.5/100,000. There was a decline in incidence in all racial and ethnic groups. The highest incidence is still observed in black and Asians (0.9 and 0.7 per 100,000). The most dramatic decline occurred in black individuals (3.2 vs. 0.9 per 100,000). In 1998, an educational campaign was initiated in Georgia that targeted high-risk individuals and provided information on the safe handling and preparation of chitterlings. The state of Georgia reported the greatest decline to 0.4/100,000, which has almost eliminated the racial disparity reported in 2009. It is unclear if this campaign was the only reason for the decline in Georgia. The incidence in whites is 0.2/100,000. Since 2007, the incidence in Asian children less than 5 years of ages has been the highest amongst all racial and ethnic groups. Pork consumption is still assumed to be the major source. Seasonal variability persists amongst Black children less 5 years of age, implying that chitterlings may still be the source of infection for individuals in this group (Clin. Infect. Dis. 2012:54 [Suppl 5]:S385-S90).
In general, yersiniosis should be included in the differential of a febrile diarrheal illness, particularly during the cooler months and holiday season. It is prudent to determine if consumption and/or preparation of chitterlings or other pork products by the patient or caretakers has occurred. This will enable you to alert the laboratory so stool specimens can be cultured on the appropriate medium (CIN agar). Consumption of chitterlings is not limited to any specific racial or ethnic group. Individuals from rural and farming areas may also consume this product.
Dr. Word is a pediatric infectious disease specialist and director of the Houston Travel Medicine Clinic. She said she had no relevant financial disclosures. Write to Dr. Word at [email protected].

Is the Relational Approach to Diagnosis Possible or Desirable?
The American Family Therapy Academy recently issued a policy statement protesting the DSM-5, and asks the American Psychiatric Association to consider the importance of relational and family context to psychiatric diagnoses.
AFTA, a multidisciplinary group, does not support the current revision of the DSM, stating that it "continues a long history of ignoring research and excluding vital contributions of nonpsychiatric mental health disciplines." This statement refers to the substantial body of research concerning the role of relational factors in mental health and mental illness, and also refers to the large number of effective family treatments, including, but not limited to, family therapy.
The academy criticizes the DSM’s use of the biomedical model and its omission of the role of family and sociocultural contexts on well-being. AFTA states that the DSM "delegitimizes the focus on relationship, life stage, community, and access to power and resources." AFTA points out that the DSM fails to take into account culture, class and ‘destructive unjust social factors,’ such as poverty, hunger, homelessness, violence, racism, and other forms of oppression. AFTA considers these factors to be important in reaching a diagnosis that accurately describes patients.
Many psychiatrists, especially family, social, and cultural psychiatrists, agree with AFTA’s position. Several family researchers and family psychiatrists have been pushing for many years to get relational diagnoses included in the DSM-IV and the DSM-5 (J. Fam. Psychol. 2006;20:359-68), citing decades of excellent research into relational diagnoses. Their attempts are supported by nonmedical health care professionals who complain that they cannot get paid by insurance companies for treating families. However, putting any diagnosis in the DSM so the insurance companies get paid is a backward way of thinking. Any diagnostic system of American psychiatry should not be framed or influenced by financial organizations that want to ration health care.
Some psychiatrists who contributed to the DSM offer the disclaimer that "they do not mean this to be a bible." However, the DSM is frequently used "as a bible," for example, in the courts. More importantly, reductionist diagnostic descriptions in the DSM narrow the public’s and the professionals’ thinking about psychological difficulties, and, by extrapolation, limit the conceptualization of what types of interventions might be helpful.
For example, describing psychiatric illnesses as biological leads to the assumption that biological interventions are needed. If an illness is defined using a biopsychosocial explanation, however, this broader understanding leads to a wider array of possible treatments. A psychiatric diagnostic system should recognize all the factors that are known to contribute to psychological health and illness to be of most use in patient care.
There is also a strong argument for not including relational diagnoses in the DSM. The argument is this: Relational factors are process factors, rather than static factors. For example, expressed emotion (EE) is not a characteristic of a family but rather a description of family distress that arises as a result of living with a disease. It is a description of a family process. Providing psychoeducation to a distressed family substantially reduces the level of EE and the subsequent risk of patient relapse. EE is a measure of relational process. If EE is entered into the DSM, there is a danger of its being seen as a static entity.
A delicate balance exists between the utilitarian need for a system of diagnoses and the risk of overdefining people and their relationships as "pathological." It was not that long ago that we pathologized homosexuality and described the entity of the "schizophrenogenic mother."
Dr. Larry Freeman, a member of the Association of Family Psychiatrists, adds: "Be wary of a pressure beyond medical circles to utilize psychiatry as a force for social control. I do a great deal of workers’ [compensation], and so-called ‘preexisting conditions’ are commonly framed as the "cause" of a worker’s emotional response to injury, and therefore, [the worker’s] current psychiatric conditions are not accepted as a consequence of the original injury event.
"Be careful that we do not enable this distortion further in our efforts to include context and history."
How should we include patient contexts such as violence, abuse, trauma, poverty, injustice, or relational dysfunction? How do we acknowledge that these factors play a significant role in the lives of our patients? For children, this is especially important as treatment often focuses on changing or stabilizing their environment, and ensuring that there is adequate attachment and nurturance.
How do we ensure that these relationships and contexts are adequately defined so we can monitor the effectiveness (or not) of interventions? AFTA supports the creation of a work group that will focus on developing an alternative to the DSM for the conceptualization of emotional distress. David Elkins, Ph.D., is planning an international summit in 2013 with representatives from all therapist groups to discuss the feasibility of such a system.
Another way forward is to develop a diagnostic system that focuses on health. The Global Assessment of Functioning (GAF), describes with reasonable accuracy a person’s individual level of functioning on a scale of 1 to 100. The Global Assessment of Relational Functioning (GARF) describes the health of a relationship on a scale of 1-100. Using these scales, pathology and health coexist on a continuum, with anchors throughout the scale. These systems are currently crude instruments, but imagine how much better they could become if they were the focus of research, clinical trials, etc.
There will always be the need for individual diagnoses, where the melancholic continues to suffer despite having an excellent social and family context, and there will always be cases where we cannot decide if the patient is ill unto himself or if his illness is informed by the context of his life.
But consider the inverse, the person who is optimistic and functional in spite of the dire context of his life, people who hold beliefs, convictions, and so on that raise them above their life circumstances. (Think of visionaries like Gandhi or Mandela). In the same way, there are relationships that function well, despite the presence of adversity. How do we develop a system that aspires to "health" instead of pathology? The American health care system (or rather its illness care system) needs to morph into true health care with a focus on prevention on both an individual and relational front.
For additional information, see Relational Processes and DSM-V: Neuroscience, Assessment, Prevention, and Treatment (Washington: American Psychiatric Association Publishing, 2006).
Dr. Alison Heru is with the department of psychiatry at the University of Colorado at Denver, Aurora. She has been a member of the Association of Family Psychiatrists since 2002 and currently serves as the organization’s treasurer. In addition, she is the coauthor of two books on working with families and is the author of numerous articles on this topic.
The American Family Therapy Academy recently issued a policy statement protesting the DSM-5, and asks the American Psychiatric Association to consider the importance of relational and family context to psychiatric diagnoses.
AFTA, a multidisciplinary group, does not support the current revision of the DSM, stating that it "continues a long history of ignoring research and excluding vital contributions of nonpsychiatric mental health disciplines." This statement refers to the substantial body of research concerning the role of relational factors in mental health and mental illness, and also refers to the large number of effective family treatments, including, but not limited to, family therapy.
The academy criticizes the DSM’s use of the biomedical model and its omission of the role of family and sociocultural contexts on well-being. AFTA states that the DSM "delegitimizes the focus on relationship, life stage, community, and access to power and resources." AFTA points out that the DSM fails to take into account culture, class and ‘destructive unjust social factors,’ such as poverty, hunger, homelessness, violence, racism, and other forms of oppression. AFTA considers these factors to be important in reaching a diagnosis that accurately describes patients.
Many psychiatrists, especially family, social, and cultural psychiatrists, agree with AFTA’s position. Several family researchers and family psychiatrists have been pushing for many years to get relational diagnoses included in the DSM-IV and the DSM-5 (J. Fam. Psychol. 2006;20:359-68), citing decades of excellent research into relational diagnoses. Their attempts are supported by nonmedical health care professionals who complain that they cannot get paid by insurance companies for treating families. However, putting any diagnosis in the DSM so the insurance companies get paid is a backward way of thinking. Any diagnostic system of American psychiatry should not be framed or influenced by financial organizations that want to ration health care.
Some psychiatrists who contributed to the DSM offer the disclaimer that "they do not mean this to be a bible." However, the DSM is frequently used "as a bible," for example, in the courts. More importantly, reductionist diagnostic descriptions in the DSM narrow the public’s and the professionals’ thinking about psychological difficulties, and, by extrapolation, limit the conceptualization of what types of interventions might be helpful.
For example, describing psychiatric illnesses as biological leads to the assumption that biological interventions are needed. If an illness is defined using a biopsychosocial explanation, however, this broader understanding leads to a wider array of possible treatments. A psychiatric diagnostic system should recognize all the factors that are known to contribute to psychological health and illness to be of most use in patient care.
There is also a strong argument for not including relational diagnoses in the DSM. The argument is this: Relational factors are process factors, rather than static factors. For example, expressed emotion (EE) is not a characteristic of a family but rather a description of family distress that arises as a result of living with a disease. It is a description of a family process. Providing psychoeducation to a distressed family substantially reduces the level of EE and the subsequent risk of patient relapse. EE is a measure of relational process. If EE is entered into the DSM, there is a danger of its being seen as a static entity.
A delicate balance exists between the utilitarian need for a system of diagnoses and the risk of overdefining people and their relationships as "pathological." It was not that long ago that we pathologized homosexuality and described the entity of the "schizophrenogenic mother."
Dr. Larry Freeman, a member of the Association of Family Psychiatrists, adds: "Be wary of a pressure beyond medical circles to utilize psychiatry as a force for social control. I do a great deal of workers’ [compensation], and so-called ‘preexisting conditions’ are commonly framed as the "cause" of a worker’s emotional response to injury, and therefore, [the worker’s] current psychiatric conditions are not accepted as a consequence of the original injury event.
"Be careful that we do not enable this distortion further in our efforts to include context and history."
How should we include patient contexts such as violence, abuse, trauma, poverty, injustice, or relational dysfunction? How do we acknowledge that these factors play a significant role in the lives of our patients? For children, this is especially important as treatment often focuses on changing or stabilizing their environment, and ensuring that there is adequate attachment and nurturance.
How do we ensure that these relationships and contexts are adequately defined so we can monitor the effectiveness (or not) of interventions? AFTA supports the creation of a work group that will focus on developing an alternative to the DSM for the conceptualization of emotional distress. David Elkins, Ph.D., is planning an international summit in 2013 with representatives from all therapist groups to discuss the feasibility of such a system.
Another way forward is to develop a diagnostic system that focuses on health. The Global Assessment of Functioning (GAF), describes with reasonable accuracy a person’s individual level of functioning on a scale of 1 to 100. The Global Assessment of Relational Functioning (GARF) describes the health of a relationship on a scale of 1-100. Using these scales, pathology and health coexist on a continuum, with anchors throughout the scale. These systems are currently crude instruments, but imagine how much better they could become if they were the focus of research, clinical trials, etc.
There will always be the need for individual diagnoses, where the melancholic continues to suffer despite having an excellent social and family context, and there will always be cases where we cannot decide if the patient is ill unto himself or if his illness is informed by the context of his life.
But consider the inverse, the person who is optimistic and functional in spite of the dire context of his life, people who hold beliefs, convictions, and so on that raise them above their life circumstances. (Think of visionaries like Gandhi or Mandela). In the same way, there are relationships that function well, despite the presence of adversity. How do we develop a system that aspires to "health" instead of pathology? The American health care system (or rather its illness care system) needs to morph into true health care with a focus on prevention on both an individual and relational front.
For additional information, see Relational Processes and DSM-V: Neuroscience, Assessment, Prevention, and Treatment (Washington: American Psychiatric Association Publishing, 2006).
Dr. Alison Heru is with the department of psychiatry at the University of Colorado at Denver, Aurora. She has been a member of the Association of Family Psychiatrists since 2002 and currently serves as the organization’s treasurer. In addition, she is the coauthor of two books on working with families and is the author of numerous articles on this topic.
The American Family Therapy Academy recently issued a policy statement protesting the DSM-5, and asks the American Psychiatric Association to consider the importance of relational and family context to psychiatric diagnoses.
AFTA, a multidisciplinary group, does not support the current revision of the DSM, stating that it "continues a long history of ignoring research and excluding vital contributions of nonpsychiatric mental health disciplines." This statement refers to the substantial body of research concerning the role of relational factors in mental health and mental illness, and also refers to the large number of effective family treatments, including, but not limited to, family therapy.
The academy criticizes the DSM’s use of the biomedical model and its omission of the role of family and sociocultural contexts on well-being. AFTA states that the DSM "delegitimizes the focus on relationship, life stage, community, and access to power and resources." AFTA points out that the DSM fails to take into account culture, class and ‘destructive unjust social factors,’ such as poverty, hunger, homelessness, violence, racism, and other forms of oppression. AFTA considers these factors to be important in reaching a diagnosis that accurately describes patients.
Many psychiatrists, especially family, social, and cultural psychiatrists, agree with AFTA’s position. Several family researchers and family psychiatrists have been pushing for many years to get relational diagnoses included in the DSM-IV and the DSM-5 (J. Fam. Psychol. 2006;20:359-68), citing decades of excellent research into relational diagnoses. Their attempts are supported by nonmedical health care professionals who complain that they cannot get paid by insurance companies for treating families. However, putting any diagnosis in the DSM so the insurance companies get paid is a backward way of thinking. Any diagnostic system of American psychiatry should not be framed or influenced by financial organizations that want to ration health care.
Some psychiatrists who contributed to the DSM offer the disclaimer that "they do not mean this to be a bible." However, the DSM is frequently used "as a bible," for example, in the courts. More importantly, reductionist diagnostic descriptions in the DSM narrow the public’s and the professionals’ thinking about psychological difficulties, and, by extrapolation, limit the conceptualization of what types of interventions might be helpful.
For example, describing psychiatric illnesses as biological leads to the assumption that biological interventions are needed. If an illness is defined using a biopsychosocial explanation, however, this broader understanding leads to a wider array of possible treatments. A psychiatric diagnostic system should recognize all the factors that are known to contribute to psychological health and illness to be of most use in patient care.
There is also a strong argument for not including relational diagnoses in the DSM. The argument is this: Relational factors are process factors, rather than static factors. For example, expressed emotion (EE) is not a characteristic of a family but rather a description of family distress that arises as a result of living with a disease. It is a description of a family process. Providing psychoeducation to a distressed family substantially reduces the level of EE and the subsequent risk of patient relapse. EE is a measure of relational process. If EE is entered into the DSM, there is a danger of its being seen as a static entity.
A delicate balance exists between the utilitarian need for a system of diagnoses and the risk of overdefining people and their relationships as "pathological." It was not that long ago that we pathologized homosexuality and described the entity of the "schizophrenogenic mother."
Dr. Larry Freeman, a member of the Association of Family Psychiatrists, adds: "Be wary of a pressure beyond medical circles to utilize psychiatry as a force for social control. I do a great deal of workers’ [compensation], and so-called ‘preexisting conditions’ are commonly framed as the "cause" of a worker’s emotional response to injury, and therefore, [the worker’s] current psychiatric conditions are not accepted as a consequence of the original injury event.
"Be careful that we do not enable this distortion further in our efforts to include context and history."
How should we include patient contexts such as violence, abuse, trauma, poverty, injustice, or relational dysfunction? How do we acknowledge that these factors play a significant role in the lives of our patients? For children, this is especially important as treatment often focuses on changing or stabilizing their environment, and ensuring that there is adequate attachment and nurturance.
How do we ensure that these relationships and contexts are adequately defined so we can monitor the effectiveness (or not) of interventions? AFTA supports the creation of a work group that will focus on developing an alternative to the DSM for the conceptualization of emotional distress. David Elkins, Ph.D., is planning an international summit in 2013 with representatives from all therapist groups to discuss the feasibility of such a system.
Another way forward is to develop a diagnostic system that focuses on health. The Global Assessment of Functioning (GAF), describes with reasonable accuracy a person’s individual level of functioning on a scale of 1 to 100. The Global Assessment of Relational Functioning (GARF) describes the health of a relationship on a scale of 1-100. Using these scales, pathology and health coexist on a continuum, with anchors throughout the scale. These systems are currently crude instruments, but imagine how much better they could become if they were the focus of research, clinical trials, etc.
There will always be the need for individual diagnoses, where the melancholic continues to suffer despite having an excellent social and family context, and there will always be cases where we cannot decide if the patient is ill unto himself or if his illness is informed by the context of his life.
But consider the inverse, the person who is optimistic and functional in spite of the dire context of his life, people who hold beliefs, convictions, and so on that raise them above their life circumstances. (Think of visionaries like Gandhi or Mandela). In the same way, there are relationships that function well, despite the presence of adversity. How do we develop a system that aspires to "health" instead of pathology? The American health care system (or rather its illness care system) needs to morph into true health care with a focus on prevention on both an individual and relational front.
For additional information, see Relational Processes and DSM-V: Neuroscience, Assessment, Prevention, and Treatment (Washington: American Psychiatric Association Publishing, 2006).
Dr. Alison Heru is with the department of psychiatry at the University of Colorado at Denver, Aurora. She has been a member of the Association of Family Psychiatrists since 2002 and currently serves as the organization’s treasurer. In addition, she is the coauthor of two books on working with families and is the author of numerous articles on this topic.

Preventing VTE with Decision Support
Over 900,000 incident and recurrent venous thromboembolism (VTE) events occur in the United States each year, resulting in nearly 300,000 fatalities.[1] VTE, including deep vein thrombosis (DVT) and pulmonary embolism (PE), is among the most common causes of death in the United States, with more people dying annually from VTE than motor vehicle accidents and breast cancer.[2]
Accordingly, healthcare policy makers and regulators have placed greater emphasis on VTE prevention, including use of VTE prophylaxis measures in the Centers for Medicare and Medicaid Services (CMS) value‐based purchasing (pay for performance) program and the Joint Commission's adoption of a national hospital patient safety goal related to anticoagulation therapy.[3, 4] Beginning in 2008, VTE events following hip and knee procedures were included as 1 of 10 hospital‐acquired conditions for which CMS would not pay for associated additional costs of care.[5]
A typical 300‐bed hospital can expect roughly 150 cases of hospital‐acquired VTE annually.[6] Up to 75% of these cases will occur on the medicine service, where nearly every patient has 1 or more VTE risk factor.[7] Although effective preventive modalities exist, prophylaxis rates among medical patients have been noted to be <50%.[8, 9] While quality improvement interventions have been shown to be effective in improving compliance with VTE prophylaxis, there are few studies describing effectiveness of these interventions in electronic health record (EHR) environments.[10] As EHR implementation accelerates, it will be essential to define the strengths and limitations of various decision support approaches to optimally improve patient safety.
We sought to evaluate the effectiveness and safety of a computerized decision support application, which was designed as part of a quality improvement initiative to improve rates of VTE prophylaxis rates on the medicine services at 2 hospital sites.
METHODS
Setting
The initiative was conducted at Montefiore Medical Center, an academic medical center in the Bronx, New York. This article describes results from an effort to improve inpatient VTE prophylaxis rates as part of an overall medical center initiative to improve anticoagulation management beginning in 2007. The initiative was led by an interdisciplinary committee consisting of administrators, medical and surgical physicians, nursing staff, and information technology and performance improvement personnel.
Intervention
As part of the initial quality improvement project, the group analyzed factors associated with and rates of hospital‐acquired VTE. Among the findings was a predominance of hospital‐acquired VTE cases and suboptimal rates of VTE prophylaxis on medicine services. Accordingly, the medicine service, whose discharge volume was 36,500 in 2010, was the population of focus for the improvement effort. The analysis also demonstrated a 99% agreement rate between administratively coded VTE events and VTE diagnoses verified from chart review, validating the utility of institutional administrative data for ongoing study of VTE events. As the hospital sites had computerized physician order entry, the group sought to develop an electronic clinical decision support module. The primary objective of the quality improvement effort was to increase VTE prophylaxis rates and decrease VTE incidence among medicine patients.
A range of clinical decision support approaches was explored. Based on team review, key decision support design objectives were to:
- Minimize alert fatigue
Utilize existing clinical information system variables to:
- Avoid de novo physician data entry solely to support the application
- Automatically identify and exclude patients in whom pharmacologic VTE prophylaxis was contraindicated
- Utilize the 8th edition of the American College of Chest Physicians VTE guidelines[8] as a basis for recommendations (as the study was conducted prior to the 9th edition release)
The VTE decision support module was comprised of order sets with the following features:
- Patients were identified as on the medicine service based on admitting service designation.
- An order set was populated from this triggering mechanism offering pharmacologic VTE prophylaxis options, or alternately, options to document lack of a clinical indication for pharmacologic VTE prophylaxis, planned therapeutic anticoagulation, or contraindication to VTE prophylaxis.
- Alternate order sets were offered with mechanical VTE prophylaxis options if the physician indicated pharmacologic VTE prophylaxis was contraindicated or if the information system identified a clinical contraindication.
- If pharmacologic VTE prophylaxis was not prescribed, the rules logic was repeated every 5 days.
Analyses
The evaluation sought to assess the effectiveness and safety of the decision support module. VTE processes and outcomes for the 6‐month periods immediately before and after full scale decision support go‐live on September 9, 2009, were evaluated. This time window was chosen in relation to CMS' requirement that hospitals use present on admission codes for discharge diagnoses (including VTE) on October 1, 2007, and first implementation of a hospital‐acquired condition policy on October 1, 2008.[5] The 6‐month period prior to September 2009 was within the first calendar year where both CMS policies were in effect.
Effectiveness of the decision support module was measured by evaluating the proportion of medicine service discharges before and after module deployment who:
- Received any VTE prophylaxis modality
- Received a pharmacologic VTE prophylaxis modality
- Developed a hospital‐acquired VTE
Successful receipt of any VTE prophylaxis modality was defined as use of compression stockings, pneumatic compression devices, or pharmacologic VTE prophylaxis modalities, including therapeutic anticoagulation (eg, mechanical heart valve). Medications counting toward the definition of pharmacologic agents included unfractionated heparin, dalteparin, warfarin, fondaparinux, lepirudin, argatroban, or bivalirudin, which are all on formulary at the medical center. Heparin used as an intravenous flush or associated with dialysis was excluded. Hospital‐acquired VTE was defined by the numerator International Classification of Diseases, 9th Revision (ICD‐9) discharge diagnosis codes for DVT or PE events as specified in the Agency for Healthcare Research and Quality (AHRQ) postoperative PE or DVT Patient Safety Indicator 12, and where the codes were not present on admission.[11]
Patient discharges excluded from analyses were those with patient age <18 years, length of stay 1 day or less, VTE diagnosis present on admission, or patient with an inferior vena cava filter during the stay. For evaluation of pharmacologic VTE prophylaxis, patients were additionally excluded if they had a platelet count <50,000/L during their stay, were a neurosurgical patient, or had a discharge diagnosis that included gastrointestinal bleeding or coagulopathy.
The safety of the decision support application was measured by assessing the proportion of medicine service discharges before and after decision support deployment who developed bleeding or thrombocytopenia. Bleeding was defined as receipt of 1 or more packed red blood cell units following administration of an anticoagulant medication at a VTE prophylaxis dosage range. Exclusion criteria for bleeding evaluation were patients aged <18 years, with length of stay 1 day or less, VTE diagnosis present on admission, platelet count <50,000/L during the stay, were a neurosurgical patient, or had diagnoses of anemia, hematologic malignancy, or inferior vena cava filter during the stay, or diagnoses of gastrointestinal bleeding, hemorrhage, or hematoma on admission.
Thrombocytopenia was defined as a >50% decrease from the initial platelet count during the hospital stay, or a decrease from an admission platelet count of >100,000/L to <100,000/L during the hospital stay. Criteria for exclusion from these analyses were age <18 years, length of stay 1 day or less, diagnosis of VTE present on admission, and vena cava filter during the stay.
A medical center comparison group was defined to contrast the magnitude of change in study end points on medicine services where the intervention was deployed with the change on other services where decision support was not used, and to distinguish potential changes observed on medicine services from secular trends. The comparison group consisted of discharges from cardiology, cardiothoracic surgery, family medicine, general surgery, surgical subspecialty, oncology, psychiatry, and rehabilitation medicine services. Newborn, neurosurgery, obstetrics, and pediatrics service discharges were excluded from the comparison group because of their being at low risk for VTE or in a high‐risk group in whom pharmacologic VTE prophylaxis was frequently contraindicated. All parameters described above were evaluated in the comparison group using inclusion and exclusion criteria similar to the intervention group. Outcomes (hospital‐acquired VTE, bleeding, thrombocytopenia) were assessed similarly across index admissions and readmissions.
The significance of change in rates of prescribing, VTE incidence, and adverse event occurrence, were tested by comparing event proportions before and after decision support module implementation in both groups. As all variables were categorical, significance was assessed using 2‐sided Pearson 2 tests at an level of 0.05. Statistical analyses were performed using SPSS software (IBM, Armonk, NY). This project was reviewed by the Albert Einstein College of Medicine/Montefiore Medical Center institutional review board (protocol number 12‐02‐058X) and deemed exempt. Design of the decision support module and definition of the implementation and evaluation plan required approximately 1 year of monthly interdisciplinary team meetings and 200 hours of programmer development time.
RESULTS
Table 1 compares the effectiveness of the decision support module intervention in medicine intervention and in nonmedicine (nonintervention) services. Among medicine service patients, any VTE prophylaxis ordering increased from 61.9% to 82.1% (P < 0.001), and pharmacologic VTE prophylaxis increased from 59.0% to 74.5% (P < 0.001). Smaller but significant increases were observed on nonmedicine services. Hospital‐acquired VTE incidence on medicine services decreased significantly, from 0.65% to 0.42% (P = 0.008) and nonsignificantly on nonmedicine services.
| Medicine Service | Nonmedicine Services | |||||||
|---|---|---|---|---|---|---|---|---|
| Pre | Post | Pre | Post | |||||
| % (n) | % (n) | Relative Change | Significance | % (n) | % (n) | Relative Change | Significance | |
| ||||||||
| Any VTE prophylaxis | ||||||||
| Eligible | N = 15,254 | N = 15,065 | N/A | N/A | N = 8566 | N = 8162 | N/A | N/A |
| Received | 61.9 (9443) | 82.1 (12,372) | +32.7% | P < 0.001 | 70.5 (6040) | 73.6 (6010) | +4.4% | P < 0.001 |
| Pharmacologic VTE prophylaxis | ||||||||
| Eligible | N = 14,768 | N = 14,588 | N/A | N/A | N = 7883 | N = 7567 | N/A | N/A |
| Received | 59.0 (8712) | 74.5 (10,869) | +26.3% | P < 0.001 | 59.3 (4677) | 63.3 (4791) | +6.7% | P < 0.001 |
| Hospital‐acquired VTE incidence | ||||||||
| Susceptible | N = 15,254 | N = 15,065 | N/A | N/A | N = 8566 | N = 8162 | N/A | N/A |
| Developed | 0.65 (99) | 0.42 (64) | 34.5% | P = 0.008 | 0.82 (70) | 0.72 (59) | 11.5% | P = 0.486 |
Table 2 shows ordering patterns for major VTE prophylaxis modalities. Among eligible medicine service patients, rates of low molecular weight heparin prophylaxis increased from 13.0% to 23.7% (P < 0.001), and of unfractionated heparin prophylaxis from 35.1% to 40.7% (P < 0.001). On nonmedicine services, there was no significant change in low molecular weight heparin use, and unfractionated heparin use increased significantly from 37.2% to 40.9% (P < 0.001). Proportions of patients receiving mechanical prophylaxis or not receiving prophylaxis decreased significantly by 37.8% on medicine services and by 9.8% on nonmedicine services Table 3 shows the safety of the decision support module. Bleeding rates increased on medicine services from 2.9% to 4.0% (P < 0.001) and on nonmedicine services from 7.7% to 8.6% (P = 0.043). Nonsignificant changes in thrombocytopenia rates were observed on both services.
| Medicine Service | Nonmedicine Services | |||||||
|---|---|---|---|---|---|---|---|---|
| Pre % (n) | Post % (n) | Relative Change | Significance | Pre % (n) | Post % (n) | Relative Change | Significance | |
| ||||||||
| Eligible for pharmacologic VTE prophylaxis | N = 14,768 | N = 14,588 | N/A | N = 7883 | N = 7567 | N/A | ||
| Low molecular weight heparin | 13.0 (1922) | 23.7 (3463) | +82.4% | P < 0.001 | 15.3 (1206) | 15.9 (1204) | +4.0% | P = 0.294 |
| Unfractionated heparin | 35.1 (5181) | 40.7 (5936) | +16.0% | P < 0.001 | 37.2 (2932) | 40.9 (3093) | +9.9% | P < 0.001 |
| Warfarin | 10.8 (1594) | 10.0 (1461) | 7.2% | P = 0.029 | 6.8 (532) | 6.4 (483) | 5.4% | P = 0.359 |
| Other agent | 0.1 (15) | 0.1 (9) | 39.3% | P = 0.232 | 0.1 (7) | 0.2 (11) | +63.7 | P = 0.303 |
| Mechanical prophylaxis or did not receive | 41.0 (6056) | 25.5 (3719) | 37.8% | P < 0.001 | 40.7 (3206) | 36.7 (2776) | 9.8% | P < 0.001 |
| Medicine Service | Nonmedicine Services | |||||||
|---|---|---|---|---|---|---|---|---|
| Pre % (n) | Post % (n) | Relative Change | Significance | Pre % (n) | Post % (n) | Relative Change | Significance | |
| ||||||||
| Bleeding | ||||||||
| Susceptible | N = 13,614 | N = 13,445 | N/A | N = 7372 | N = 7061 | N/A | ||
| Developed | 2.9 (401) | 4.0 (534) | +34.8% | P < 0.001 | 7.7 (565) | 8.6 (606) | +12.0% | P = 0.043 |
| Thrombocytopenia | ||||||||
| Susceptible | N = 15,254 | N = 15,065 | N/A | N = 8566 | N = 8162 | N/A | ||
| Developed | 7.4 (1123) | 6.9 (1047) | 5.6% | P = 0.164 | 8.7 (749) | 8.8 (716) | +0.3% | P = 0.948 |
DISCUSSION
Following implementation of a computerized decision support application to improve VTE prophylaxis on 2 hospital medicine services, we observed a significant increase in the rate of overall and pharmacologic VTE prophylaxis use and a significant decrease in the incidence of hospital‐acquired VTE. Changes were of greater magnitude and significance on medicine services where the intervention was deployed.
Rates of any VTE prophylaxis and pharmacologic VTE prophylaxis ordering on medicine services increased significantly by 32.7% and 26.3%, respectively. These rates increased on nonmedicine comparison services by a more modest 4.4% for any VTE prophylaxis and 6.7% for pharmacologic VTE prophylaxis. Although the medicine service intervention was designed to be agnostic to the type of prophylactic heparin preparation, the intervention resulted in a significant 82.4% increase in low molecular weight heparin use and a significant 16.0% increase in unfractionated heparin use. With respect to outcomes, we observed a 34.5% decrease (P < 0.001) in hospital‐acquired VTE incidence on medicine services and a nonsignificant decrease on nonmedicine services.
In assessing intervention safety, increased usage of VTE prophylaxis was not accompanied by an increase in thrombocytopenia, but was associated with an increase in bleeding from 2.9% to 4.0% (P < 0.001) on medicine services and from 7.7% to 8.6% (P = 0.043) on non‐medicine services. As our intervention was a quality improvement project, we conducted a brief post hoc analysis to evaluate the increased bleeding rate on the medicine service following intervention. A random sample of 50 records of medicine patients who had received VTE prophylaxis and had a subsequent bleeding event was reviewed. Findings are summarized in Table 4. Prophylaxis was used appropriately in 100% of cases. Bleeding episodes were minor in that no case required more than 2 U of packed red blood cells. The most common clinical scenario was a patient with baseline anemia, typically with chronic kidney disease, who had a slight decrease in hematocrit of unclear etiology requiring 1 U of blood.
| Characteristic | % (N = 50) |
|---|---|
| |
| Prophylaxis indication | |
| Pharmacologic VTE prophylaxis indicateda | 100.0 |
| Clinical characteristic | |
| Anemia upon admission | 92.0 |
| Chronic kidney disease | 66.0 |
| Suspected bleeding source | |
| Unclear | 62.0 |
| Gastrointestinal | 18.0 |
| Catheter/external device site | 8.0 |
| Operative | 6.0 |
| Epistaxis | 4.0 |
| Gynecologic | 2.0 |
| Medication use | |
| Prophylactic agent associated with bleeding | |
| Unfractionated heparin | 66.0 |
| Dalteparin | 34.0 |
| On antiplatelet agent at time of bleedb | 52.0 |
| Transfusion outcome | |
| Required >2 packed red blood cell units | 0.0 |
Although the intervention occurred on medicine services, favorable albeit smaller changes were observed on nonmedicine services. We expected this favorable secular trend because of VTE prophylaxis awareness efforts across the organization as a whole. There was also ongoing focus on VTE prevention and outcomes by policymakers, regulatory agencies, and professional societies during the time period of study.[3, 4, 5, 12] Public reporting of CMS inpatient surgical VTE prophylaxis measures was required throughout the study period.[13] Changes observed on medicine services occurred during a period where there were no publicly reported measures of VTE prophylaxis for inpatient medicine services.
Our study had several limitations. We derived our eligibility criteria for VTE prophylaxis based on administrative data. To address this, we incorporated accepted standardized definitions,[11] used clinical data elements in our queries beyond ICD‐9 codes (eg, platelet count), and applied pertinent exclusion criteria (eg, length of stay 1 day or less). VTE events that were present on admission were excluded from analyses. However, as these community‐acquired VTE events may be caused by inadequate VTE prophylaxis during a prior hospitalization, the overall true incidence of hospital‐acquired VTE was likely underestimated.
With respect to the hospital‐acquired VTE outcome, we did not distinguish superficial from deep VTE. A consistent AHRQ definition of 13 ICD‐9 VTE codes was used to identify clinically significant VTE events for the periods before and after the intervention. Although the present on admission code identified VTE events that were hospital acquired, 1 new acute VTE ICD‐9 code was added in October 2009, allowing for more specific coding of acute, isolated, upper extremity VTE. Accordingly, our postintervention hospital‐acquired VTE rate may have slightly underestimated the true hospital‐acquired VTE incidence by omitting some coded acute, isolated, upper extremity VTE cases (if not coded using the prior Other VTE codes). In a study in a teaching hospital setting, isolated upper extremity VTE accounted for up to 21% of all symptomatic VTE events among adults.[14]
With respect to VTE prophylaxis, the study evaluated use in a dichotomous fashion but did not assess appropriateness, or adequacy of dosing of pharmacologic agents. We did not employ the intervention in a randomized fashion on the medicine service. As our project was a quality improvement intervention, we used a concurrent control group of nonmedicine service patients to assess potential secular trend bias.
With respect to the safety of the intervention, the record review we performed supported the appropriateness of prophylaxis use following the intervention, but was not designed to establish whether the increase in prophylaxis use was the proximate cause of bleeding events observed. Similarly, as specific testing for heparin‐induced thrombocytopenia was not used, the lack of significant change in thrombocytopenia rates before and after the intervention cannot directly establish the intervention's safety. Finally, our study also included only in‐hospital end points.
The rate of VTE prophylaxis use in hospitals has been noted to be disappointing.[15] Two large multinational studies found that VTE prophylaxis rates in at‐risk hospitalized medical patients in the United States were 48% and 52%.[9, 16] Amin and colleagues found the overall rate of VTE prophylaxis among 227 US hospitals to be 62%.[17] Accordingly, our intervention, which resulted in an 82% compliance rate on a large medical service and was associated with a significantly reduced VTE incidence, appears to be highly effective. Our results are likely more favorable in that beyond length of stay criteria, we did not exclude less acutely ill medical patients from analyses.
Michota summarized quality improvement studies for VTE prevention.[10] Among 9 studies attempting to improve VTE prophylaxis, 2 used electronic decision support as a primary strategy, and only 1, by Kucher et al., used a computerized approach on a medical service.[18] This study showed significant improvement in VTE prophylaxis and incidence in patients randomized to a provider computer program. The intervention was complex, requiring specification of 8 patient‐level risk factors via a customized database, and the physician to recommend specific prophylactic regimens accordingly. Our findings, using a more basic approach, similarly support the effectiveness of using automated decision support, which can be readily modified as evidence‐based guidelines evolve.
Overall adoption of information technology systems in US hospitals is low: only 7.6% of hospitals have a basic system, and 17% have computerized physician order entry.[19] As hospitals have been financially incentivized to adopt such systems, our relatively simple intervention may prove to be readily generalizable across varied vendor systems.[20] The intervention involved order sets triggered by automated logic, corollary information, and a hard stop to prompt VTE prophylaxis. Within the context of intensified emphasis on reducing harm in the inpatient setting and various pay for performance programs, our intervention is also of importance to payers.[3, 5] Using national data in year 2000, Zhan and Miller calculated the excess charges per case associated with VTE to be $21,709.[21]
In conclusion, a relatively simple automated clinical decision support application significantly improved rates of VTE prophylaxis and was associated with significantly lower hospital‐acquired VTE incidence in hospitalized medicine patients, with a reasonable safety profile.
Acknowledgments
The authors acknowledge the roles of Gillian Wendt and Maggie Feng in data acquisition.
Disclosure: The authors declare no conflict of interest related to the research, analyses, or preparation of this manuscript. M. J. Sinnett reports receiving payment for speaking on behalf of Amgen, which was not a funder of this study. All coauthors have seen and agree with the contents of the manuscript, and all coauthors fulfill the authorship criteria specified by the Journal of Hospital Medicine. Rohit Bhalla, MD, MPH, takes responsibility for the entire manuscript. This submission is not under review by any other publication. Development of the electronic decision support application was supported in part by funding under the 2008 Cardinal Health Foundation Patient Safety Grant Program.
- . The epidemiology of venous thromboembolism: implications for prevention and management. Paper presented at: Surgeon General's Workshop on Deep Vein Thrombosis; May 8, 2006; Bethesda, MD. Available at: http://www.surgeongeneral.gov/topics/deepvein/workshop/agenda.html. Accessed February 17,2012.
- American Public Health Association White Paper. Deep‐vein thrombosis: advancing awareness to protect patient lives. Public Health Leadership Conference on Deep‐Vein Thrombosis. Washington, D.C.; February 26,2003. Available at: http://www.apha.org/NR/rdonlyres/A209F84A‐7C0E‐4761–9ECF‐61D22E1E11F7/0/DVT_ White_Paper.pdf. Accessed February 27, 2012.
- Department of Health and Human Services. Centers for Medicare and Medicaid Services. Medicare program: hospital inpatient value based purchasing program. Fed Regist.2011;76(88):26490–26547.
- The Joint Commission. 2011 hospital national patient safety goals. Available at: http://www.jointcommission.org/assets/1/6/HAP_NPSG_6–10‐11.pdf. Accessed October 23,2011.
- US Department of Health and Human Services. Centers for Medicare and Medicaid Services. Medicare Learning Network. Hospital acquired conditions in acute inpatient prospective payment system (IPPS) hospitals. Available at: https://www.cms.gov/HospitalAcqCond/downloads/HACFactsheet.pdf. Accessed February 17,2012.
- , .Preventing Hospital‐Acquired Venous Thromboembolism: A Guide For Effective Quality Improvement. Society of Hospital Medicine. AHRQ Publication No. 08–0075.Rockville, MD:Agency for Healthcare Research and Quality;2008.
- . Prophylaxis for thromboembolism in hospitalized medical patients. N Engl J Med.2007;356:1438–1444.
- , , , et al. Prevention of venous thromboembolism: American College of Chest Physicians Evidence‐Based Clinical Practice Guidelines (8th Edition). Chest.2008;133;381S–453S.
- , , , et al. Venous thromboembolism risk and prophylaxis in the acute hospital care setting (ENDORSE study): a multinational cross‐sectional study. Lancet.2008;371(9610):387–394.
- . Bridging the gap between evidence and practice in venous thromboembolism prophylaxis: the quality improvement process. J Gen Intern Med.2007;22(12):1762–1770.
- Agency for Healthcare Research and Quality. PSI #12: Postoperative pulmonary embolism or deep vein thrombosis. Version 4.1.; 2009. Available at: http://www.qualityindicators.ahrq.gov/Downloads/Modules/PSI/V41/TechSpecs/PSI%2012%20Postoperative%20Pulmonary %20Embolism%20or%20Deep%20Vein%20Thrombosis.pdf. Accessed February 19,2012.
- , . The CMS ruling on venous thromboembolism after total knee or hip arthroplasty: weighing risks and benefits. JAMA.2009;301(10):1063–1065.
- US Department of Health and Human Services. Centers for Medicare and Medicaid Services. Hospital compare. Available at: http://www.hospitalcompare.hhs.gov. Accessed October 23,2011.
- , , , , , . Upper extremity deep venous thrombosis. Chest.2003;123;1953–1956.
- , . Thromboprophylaxis for adults in hospital: an intervention that would save many lives is still not being implemented. BMJ.2007;334:1017–1018.
- , , , et al. Venous thromboembolism prophylaxis in acutely ill hospitalized medical patients: findings from the International Medical Prevention Registry on Venous Thromboembolism. Chest.2007:132:936–945.
- , , , . Thromboprophylaxis rates in US medical centers: success or failure?J Thromb Haemost.2007;5:1610–1616.
- , , , et al. Electronic alerts to prevent venous thromboembolism among hospitalized patients. N Engl J Med.2005;352:969–977.
- , , , et al. Use of electronic health records in U.S. hospitals. N Engl J Med.2009;360:1628–1638.
- , . The “meaningful use” regulation for electronic health records. N Engl J Med.2010;363(6):501–504.
- , . Excess length of stay, charges, and mortality attributable to medical injuries during hospitalization. JAMA.2003;290(14):1868–1874.
Over 900,000 incident and recurrent venous thromboembolism (VTE) events occur in the United States each year, resulting in nearly 300,000 fatalities.[1] VTE, including deep vein thrombosis (DVT) and pulmonary embolism (PE), is among the most common causes of death in the United States, with more people dying annually from VTE than motor vehicle accidents and breast cancer.[2]
Accordingly, healthcare policy makers and regulators have placed greater emphasis on VTE prevention, including use of VTE prophylaxis measures in the Centers for Medicare and Medicaid Services (CMS) value‐based purchasing (pay for performance) program and the Joint Commission's adoption of a national hospital patient safety goal related to anticoagulation therapy.[3, 4] Beginning in 2008, VTE events following hip and knee procedures were included as 1 of 10 hospital‐acquired conditions for which CMS would not pay for associated additional costs of care.[5]
A typical 300‐bed hospital can expect roughly 150 cases of hospital‐acquired VTE annually.[6] Up to 75% of these cases will occur on the medicine service, where nearly every patient has 1 or more VTE risk factor.[7] Although effective preventive modalities exist, prophylaxis rates among medical patients have been noted to be <50%.[8, 9] While quality improvement interventions have been shown to be effective in improving compliance with VTE prophylaxis, there are few studies describing effectiveness of these interventions in electronic health record (EHR) environments.[10] As EHR implementation accelerates, it will be essential to define the strengths and limitations of various decision support approaches to optimally improve patient safety.
We sought to evaluate the effectiveness and safety of a computerized decision support application, which was designed as part of a quality improvement initiative to improve rates of VTE prophylaxis rates on the medicine services at 2 hospital sites.
METHODS
Setting
The initiative was conducted at Montefiore Medical Center, an academic medical center in the Bronx, New York. This article describes results from an effort to improve inpatient VTE prophylaxis rates as part of an overall medical center initiative to improve anticoagulation management beginning in 2007. The initiative was led by an interdisciplinary committee consisting of administrators, medical and surgical physicians, nursing staff, and information technology and performance improvement personnel.
Intervention
As part of the initial quality improvement project, the group analyzed factors associated with and rates of hospital‐acquired VTE. Among the findings was a predominance of hospital‐acquired VTE cases and suboptimal rates of VTE prophylaxis on medicine services. Accordingly, the medicine service, whose discharge volume was 36,500 in 2010, was the population of focus for the improvement effort. The analysis also demonstrated a 99% agreement rate between administratively coded VTE events and VTE diagnoses verified from chart review, validating the utility of institutional administrative data for ongoing study of VTE events. As the hospital sites had computerized physician order entry, the group sought to develop an electronic clinical decision support module. The primary objective of the quality improvement effort was to increase VTE prophylaxis rates and decrease VTE incidence among medicine patients.
A range of clinical decision support approaches was explored. Based on team review, key decision support design objectives were to:
- Minimize alert fatigue
Utilize existing clinical information system variables to:
- Avoid de novo physician data entry solely to support the application
- Automatically identify and exclude patients in whom pharmacologic VTE prophylaxis was contraindicated
- Utilize the 8th edition of the American College of Chest Physicians VTE guidelines[8] as a basis for recommendations (as the study was conducted prior to the 9th edition release)
The VTE decision support module was comprised of order sets with the following features:
- Patients were identified as on the medicine service based on admitting service designation.
- An order set was populated from this triggering mechanism offering pharmacologic VTE prophylaxis options, or alternately, options to document lack of a clinical indication for pharmacologic VTE prophylaxis, planned therapeutic anticoagulation, or contraindication to VTE prophylaxis.
- Alternate order sets were offered with mechanical VTE prophylaxis options if the physician indicated pharmacologic VTE prophylaxis was contraindicated or if the information system identified a clinical contraindication.
- If pharmacologic VTE prophylaxis was not prescribed, the rules logic was repeated every 5 days.
Analyses
The evaluation sought to assess the effectiveness and safety of the decision support module. VTE processes and outcomes for the 6‐month periods immediately before and after full scale decision support go‐live on September 9, 2009, were evaluated. This time window was chosen in relation to CMS' requirement that hospitals use present on admission codes for discharge diagnoses (including VTE) on October 1, 2007, and first implementation of a hospital‐acquired condition policy on October 1, 2008.[5] The 6‐month period prior to September 2009 was within the first calendar year where both CMS policies were in effect.
Effectiveness of the decision support module was measured by evaluating the proportion of medicine service discharges before and after module deployment who:
- Received any VTE prophylaxis modality
- Received a pharmacologic VTE prophylaxis modality
- Developed a hospital‐acquired VTE
Successful receipt of any VTE prophylaxis modality was defined as use of compression stockings, pneumatic compression devices, or pharmacologic VTE prophylaxis modalities, including therapeutic anticoagulation (eg, mechanical heart valve). Medications counting toward the definition of pharmacologic agents included unfractionated heparin, dalteparin, warfarin, fondaparinux, lepirudin, argatroban, or bivalirudin, which are all on formulary at the medical center. Heparin used as an intravenous flush or associated with dialysis was excluded. Hospital‐acquired VTE was defined by the numerator International Classification of Diseases, 9th Revision (ICD‐9) discharge diagnosis codes for DVT or PE events as specified in the Agency for Healthcare Research and Quality (AHRQ) postoperative PE or DVT Patient Safety Indicator 12, and where the codes were not present on admission.[11]
Patient discharges excluded from analyses were those with patient age <18 years, length of stay 1 day or less, VTE diagnosis present on admission, or patient with an inferior vena cava filter during the stay. For evaluation of pharmacologic VTE prophylaxis, patients were additionally excluded if they had a platelet count <50,000/L during their stay, were a neurosurgical patient, or had a discharge diagnosis that included gastrointestinal bleeding or coagulopathy.
The safety of the decision support application was measured by assessing the proportion of medicine service discharges before and after decision support deployment who developed bleeding or thrombocytopenia. Bleeding was defined as receipt of 1 or more packed red blood cell units following administration of an anticoagulant medication at a VTE prophylaxis dosage range. Exclusion criteria for bleeding evaluation were patients aged <18 years, with length of stay 1 day or less, VTE diagnosis present on admission, platelet count <50,000/L during the stay, were a neurosurgical patient, or had diagnoses of anemia, hematologic malignancy, or inferior vena cava filter during the stay, or diagnoses of gastrointestinal bleeding, hemorrhage, or hematoma on admission.
Thrombocytopenia was defined as a >50% decrease from the initial platelet count during the hospital stay, or a decrease from an admission platelet count of >100,000/L to <100,000/L during the hospital stay. Criteria for exclusion from these analyses were age <18 years, length of stay 1 day or less, diagnosis of VTE present on admission, and vena cava filter during the stay.
A medical center comparison group was defined to contrast the magnitude of change in study end points on medicine services where the intervention was deployed with the change on other services where decision support was not used, and to distinguish potential changes observed on medicine services from secular trends. The comparison group consisted of discharges from cardiology, cardiothoracic surgery, family medicine, general surgery, surgical subspecialty, oncology, psychiatry, and rehabilitation medicine services. Newborn, neurosurgery, obstetrics, and pediatrics service discharges were excluded from the comparison group because of their being at low risk for VTE or in a high‐risk group in whom pharmacologic VTE prophylaxis was frequently contraindicated. All parameters described above were evaluated in the comparison group using inclusion and exclusion criteria similar to the intervention group. Outcomes (hospital‐acquired VTE, bleeding, thrombocytopenia) were assessed similarly across index admissions and readmissions.
The significance of change in rates of prescribing, VTE incidence, and adverse event occurrence, were tested by comparing event proportions before and after decision support module implementation in both groups. As all variables were categorical, significance was assessed using 2‐sided Pearson 2 tests at an level of 0.05. Statistical analyses were performed using SPSS software (IBM, Armonk, NY). This project was reviewed by the Albert Einstein College of Medicine/Montefiore Medical Center institutional review board (protocol number 12‐02‐058X) and deemed exempt. Design of the decision support module and definition of the implementation and evaluation plan required approximately 1 year of monthly interdisciplinary team meetings and 200 hours of programmer development time.
RESULTS
Table 1 compares the effectiveness of the decision support module intervention in medicine intervention and in nonmedicine (nonintervention) services. Among medicine service patients, any VTE prophylaxis ordering increased from 61.9% to 82.1% (P < 0.001), and pharmacologic VTE prophylaxis increased from 59.0% to 74.5% (P < 0.001). Smaller but significant increases were observed on nonmedicine services. Hospital‐acquired VTE incidence on medicine services decreased significantly, from 0.65% to 0.42% (P = 0.008) and nonsignificantly on nonmedicine services.
| Medicine Service | Nonmedicine Services | |||||||
|---|---|---|---|---|---|---|---|---|
| Pre | Post | Pre | Post | |||||
| % (n) | % (n) | Relative Change | Significance | % (n) | % (n) | Relative Change | Significance | |
| ||||||||
| Any VTE prophylaxis | ||||||||
| Eligible | N = 15,254 | N = 15,065 | N/A | N/A | N = 8566 | N = 8162 | N/A | N/A |
| Received | 61.9 (9443) | 82.1 (12,372) | +32.7% | P < 0.001 | 70.5 (6040) | 73.6 (6010) | +4.4% | P < 0.001 |
| Pharmacologic VTE prophylaxis | ||||||||
| Eligible | N = 14,768 | N = 14,588 | N/A | N/A | N = 7883 | N = 7567 | N/A | N/A |
| Received | 59.0 (8712) | 74.5 (10,869) | +26.3% | P < 0.001 | 59.3 (4677) | 63.3 (4791) | +6.7% | P < 0.001 |
| Hospital‐acquired VTE incidence | ||||||||
| Susceptible | N = 15,254 | N = 15,065 | N/A | N/A | N = 8566 | N = 8162 | N/A | N/A |
| Developed | 0.65 (99) | 0.42 (64) | 34.5% | P = 0.008 | 0.82 (70) | 0.72 (59) | 11.5% | P = 0.486 |
Table 2 shows ordering patterns for major VTE prophylaxis modalities. Among eligible medicine service patients, rates of low molecular weight heparin prophylaxis increased from 13.0% to 23.7% (P < 0.001), and of unfractionated heparin prophylaxis from 35.1% to 40.7% (P < 0.001). On nonmedicine services, there was no significant change in low molecular weight heparin use, and unfractionated heparin use increased significantly from 37.2% to 40.9% (P < 0.001). Proportions of patients receiving mechanical prophylaxis or not receiving prophylaxis decreased significantly by 37.8% on medicine services and by 9.8% on nonmedicine services Table 3 shows the safety of the decision support module. Bleeding rates increased on medicine services from 2.9% to 4.0% (P < 0.001) and on nonmedicine services from 7.7% to 8.6% (P = 0.043). Nonsignificant changes in thrombocytopenia rates were observed on both services.
| Medicine Service | Nonmedicine Services | |||||||
|---|---|---|---|---|---|---|---|---|
| Pre % (n) | Post % (n) | Relative Change | Significance | Pre % (n) | Post % (n) | Relative Change | Significance | |
| ||||||||
| Eligible for pharmacologic VTE prophylaxis | N = 14,768 | N = 14,588 | N/A | N = 7883 | N = 7567 | N/A | ||
| Low molecular weight heparin | 13.0 (1922) | 23.7 (3463) | +82.4% | P < 0.001 | 15.3 (1206) | 15.9 (1204) | +4.0% | P = 0.294 |
| Unfractionated heparin | 35.1 (5181) | 40.7 (5936) | +16.0% | P < 0.001 | 37.2 (2932) | 40.9 (3093) | +9.9% | P < 0.001 |
| Warfarin | 10.8 (1594) | 10.0 (1461) | 7.2% | P = 0.029 | 6.8 (532) | 6.4 (483) | 5.4% | P = 0.359 |
| Other agent | 0.1 (15) | 0.1 (9) | 39.3% | P = 0.232 | 0.1 (7) | 0.2 (11) | +63.7 | P = 0.303 |
| Mechanical prophylaxis or did not receive | 41.0 (6056) | 25.5 (3719) | 37.8% | P < 0.001 | 40.7 (3206) | 36.7 (2776) | 9.8% | P < 0.001 |
| Medicine Service | Nonmedicine Services | |||||||
|---|---|---|---|---|---|---|---|---|
| Pre % (n) | Post % (n) | Relative Change | Significance | Pre % (n) | Post % (n) | Relative Change | Significance | |
| ||||||||
| Bleeding | ||||||||
| Susceptible | N = 13,614 | N = 13,445 | N/A | N = 7372 | N = 7061 | N/A | ||
| Developed | 2.9 (401) | 4.0 (534) | +34.8% | P < 0.001 | 7.7 (565) | 8.6 (606) | +12.0% | P = 0.043 |
| Thrombocytopenia | ||||||||
| Susceptible | N = 15,254 | N = 15,065 | N/A | N = 8566 | N = 8162 | N/A | ||
| Developed | 7.4 (1123) | 6.9 (1047) | 5.6% | P = 0.164 | 8.7 (749) | 8.8 (716) | +0.3% | P = 0.948 |
DISCUSSION
Following implementation of a computerized decision support application to improve VTE prophylaxis on 2 hospital medicine services, we observed a significant increase in the rate of overall and pharmacologic VTE prophylaxis use and a significant decrease in the incidence of hospital‐acquired VTE. Changes were of greater magnitude and significance on medicine services where the intervention was deployed.
Rates of any VTE prophylaxis and pharmacologic VTE prophylaxis ordering on medicine services increased significantly by 32.7% and 26.3%, respectively. These rates increased on nonmedicine comparison services by a more modest 4.4% for any VTE prophylaxis and 6.7% for pharmacologic VTE prophylaxis. Although the medicine service intervention was designed to be agnostic to the type of prophylactic heparin preparation, the intervention resulted in a significant 82.4% increase in low molecular weight heparin use and a significant 16.0% increase in unfractionated heparin use. With respect to outcomes, we observed a 34.5% decrease (P < 0.001) in hospital‐acquired VTE incidence on medicine services and a nonsignificant decrease on nonmedicine services.
In assessing intervention safety, increased usage of VTE prophylaxis was not accompanied by an increase in thrombocytopenia, but was associated with an increase in bleeding from 2.9% to 4.0% (P < 0.001) on medicine services and from 7.7% to 8.6% (P = 0.043) on non‐medicine services. As our intervention was a quality improvement project, we conducted a brief post hoc analysis to evaluate the increased bleeding rate on the medicine service following intervention. A random sample of 50 records of medicine patients who had received VTE prophylaxis and had a subsequent bleeding event was reviewed. Findings are summarized in Table 4. Prophylaxis was used appropriately in 100% of cases. Bleeding episodes were minor in that no case required more than 2 U of packed red blood cells. The most common clinical scenario was a patient with baseline anemia, typically with chronic kidney disease, who had a slight decrease in hematocrit of unclear etiology requiring 1 U of blood.
| Characteristic | % (N = 50) |
|---|---|
| |
| Prophylaxis indication | |
| Pharmacologic VTE prophylaxis indicateda | 100.0 |
| Clinical characteristic | |
| Anemia upon admission | 92.0 |
| Chronic kidney disease | 66.0 |
| Suspected bleeding source | |
| Unclear | 62.0 |
| Gastrointestinal | 18.0 |
| Catheter/external device site | 8.0 |
| Operative | 6.0 |
| Epistaxis | 4.0 |
| Gynecologic | 2.0 |
| Medication use | |
| Prophylactic agent associated with bleeding | |
| Unfractionated heparin | 66.0 |
| Dalteparin | 34.0 |
| On antiplatelet agent at time of bleedb | 52.0 |
| Transfusion outcome | |
| Required >2 packed red blood cell units | 0.0 |
Although the intervention occurred on medicine services, favorable albeit smaller changes were observed on nonmedicine services. We expected this favorable secular trend because of VTE prophylaxis awareness efforts across the organization as a whole. There was also ongoing focus on VTE prevention and outcomes by policymakers, regulatory agencies, and professional societies during the time period of study.[3, 4, 5, 12] Public reporting of CMS inpatient surgical VTE prophylaxis measures was required throughout the study period.[13] Changes observed on medicine services occurred during a period where there were no publicly reported measures of VTE prophylaxis for inpatient medicine services.
Our study had several limitations. We derived our eligibility criteria for VTE prophylaxis based on administrative data. To address this, we incorporated accepted standardized definitions,[11] used clinical data elements in our queries beyond ICD‐9 codes (eg, platelet count), and applied pertinent exclusion criteria (eg, length of stay 1 day or less). VTE events that were present on admission were excluded from analyses. However, as these community‐acquired VTE events may be caused by inadequate VTE prophylaxis during a prior hospitalization, the overall true incidence of hospital‐acquired VTE was likely underestimated.
With respect to the hospital‐acquired VTE outcome, we did not distinguish superficial from deep VTE. A consistent AHRQ definition of 13 ICD‐9 VTE codes was used to identify clinically significant VTE events for the periods before and after the intervention. Although the present on admission code identified VTE events that were hospital acquired, 1 new acute VTE ICD‐9 code was added in October 2009, allowing for more specific coding of acute, isolated, upper extremity VTE. Accordingly, our postintervention hospital‐acquired VTE rate may have slightly underestimated the true hospital‐acquired VTE incidence by omitting some coded acute, isolated, upper extremity VTE cases (if not coded using the prior Other VTE codes). In a study in a teaching hospital setting, isolated upper extremity VTE accounted for up to 21% of all symptomatic VTE events among adults.[14]
With respect to VTE prophylaxis, the study evaluated use in a dichotomous fashion but did not assess appropriateness, or adequacy of dosing of pharmacologic agents. We did not employ the intervention in a randomized fashion on the medicine service. As our project was a quality improvement intervention, we used a concurrent control group of nonmedicine service patients to assess potential secular trend bias.
With respect to the safety of the intervention, the record review we performed supported the appropriateness of prophylaxis use following the intervention, but was not designed to establish whether the increase in prophylaxis use was the proximate cause of bleeding events observed. Similarly, as specific testing for heparin‐induced thrombocytopenia was not used, the lack of significant change in thrombocytopenia rates before and after the intervention cannot directly establish the intervention's safety. Finally, our study also included only in‐hospital end points.
The rate of VTE prophylaxis use in hospitals has been noted to be disappointing.[15] Two large multinational studies found that VTE prophylaxis rates in at‐risk hospitalized medical patients in the United States were 48% and 52%.[9, 16] Amin and colleagues found the overall rate of VTE prophylaxis among 227 US hospitals to be 62%.[17] Accordingly, our intervention, which resulted in an 82% compliance rate on a large medical service and was associated with a significantly reduced VTE incidence, appears to be highly effective. Our results are likely more favorable in that beyond length of stay criteria, we did not exclude less acutely ill medical patients from analyses.
Michota summarized quality improvement studies for VTE prevention.[10] Among 9 studies attempting to improve VTE prophylaxis, 2 used electronic decision support as a primary strategy, and only 1, by Kucher et al., used a computerized approach on a medical service.[18] This study showed significant improvement in VTE prophylaxis and incidence in patients randomized to a provider computer program. The intervention was complex, requiring specification of 8 patient‐level risk factors via a customized database, and the physician to recommend specific prophylactic regimens accordingly. Our findings, using a more basic approach, similarly support the effectiveness of using automated decision support, which can be readily modified as evidence‐based guidelines evolve.
Overall adoption of information technology systems in US hospitals is low: only 7.6% of hospitals have a basic system, and 17% have computerized physician order entry.[19] As hospitals have been financially incentivized to adopt such systems, our relatively simple intervention may prove to be readily generalizable across varied vendor systems.[20] The intervention involved order sets triggered by automated logic, corollary information, and a hard stop to prompt VTE prophylaxis. Within the context of intensified emphasis on reducing harm in the inpatient setting and various pay for performance programs, our intervention is also of importance to payers.[3, 5] Using national data in year 2000, Zhan and Miller calculated the excess charges per case associated with VTE to be $21,709.[21]
In conclusion, a relatively simple automated clinical decision support application significantly improved rates of VTE prophylaxis and was associated with significantly lower hospital‐acquired VTE incidence in hospitalized medicine patients, with a reasonable safety profile.
Acknowledgments
The authors acknowledge the roles of Gillian Wendt and Maggie Feng in data acquisition.
Disclosure: The authors declare no conflict of interest related to the research, analyses, or preparation of this manuscript. M. J. Sinnett reports receiving payment for speaking on behalf of Amgen, which was not a funder of this study. All coauthors have seen and agree with the contents of the manuscript, and all coauthors fulfill the authorship criteria specified by the Journal of Hospital Medicine. Rohit Bhalla, MD, MPH, takes responsibility for the entire manuscript. This submission is not under review by any other publication. Development of the electronic decision support application was supported in part by funding under the 2008 Cardinal Health Foundation Patient Safety Grant Program.
Over 900,000 incident and recurrent venous thromboembolism (VTE) events occur in the United States each year, resulting in nearly 300,000 fatalities.[1] VTE, including deep vein thrombosis (DVT) and pulmonary embolism (PE), is among the most common causes of death in the United States, with more people dying annually from VTE than motor vehicle accidents and breast cancer.[2]
Accordingly, healthcare policy makers and regulators have placed greater emphasis on VTE prevention, including use of VTE prophylaxis measures in the Centers for Medicare and Medicaid Services (CMS) value‐based purchasing (pay for performance) program and the Joint Commission's adoption of a national hospital patient safety goal related to anticoagulation therapy.[3, 4] Beginning in 2008, VTE events following hip and knee procedures were included as 1 of 10 hospital‐acquired conditions for which CMS would not pay for associated additional costs of care.[5]
A typical 300‐bed hospital can expect roughly 150 cases of hospital‐acquired VTE annually.[6] Up to 75% of these cases will occur on the medicine service, where nearly every patient has 1 or more VTE risk factor.[7] Although effective preventive modalities exist, prophylaxis rates among medical patients have been noted to be <50%.[8, 9] While quality improvement interventions have been shown to be effective in improving compliance with VTE prophylaxis, there are few studies describing effectiveness of these interventions in electronic health record (EHR) environments.[10] As EHR implementation accelerates, it will be essential to define the strengths and limitations of various decision support approaches to optimally improve patient safety.
We sought to evaluate the effectiveness and safety of a computerized decision support application, which was designed as part of a quality improvement initiative to improve rates of VTE prophylaxis rates on the medicine services at 2 hospital sites.
METHODS
Setting
The initiative was conducted at Montefiore Medical Center, an academic medical center in the Bronx, New York. This article describes results from an effort to improve inpatient VTE prophylaxis rates as part of an overall medical center initiative to improve anticoagulation management beginning in 2007. The initiative was led by an interdisciplinary committee consisting of administrators, medical and surgical physicians, nursing staff, and information technology and performance improvement personnel.
Intervention
As part of the initial quality improvement project, the group analyzed factors associated with and rates of hospital‐acquired VTE. Among the findings was a predominance of hospital‐acquired VTE cases and suboptimal rates of VTE prophylaxis on medicine services. Accordingly, the medicine service, whose discharge volume was 36,500 in 2010, was the population of focus for the improvement effort. The analysis also demonstrated a 99% agreement rate between administratively coded VTE events and VTE diagnoses verified from chart review, validating the utility of institutional administrative data for ongoing study of VTE events. As the hospital sites had computerized physician order entry, the group sought to develop an electronic clinical decision support module. The primary objective of the quality improvement effort was to increase VTE prophylaxis rates and decrease VTE incidence among medicine patients.
A range of clinical decision support approaches was explored. Based on team review, key decision support design objectives were to:
- Minimize alert fatigue
Utilize existing clinical information system variables to:
- Avoid de novo physician data entry solely to support the application
- Automatically identify and exclude patients in whom pharmacologic VTE prophylaxis was contraindicated
- Utilize the 8th edition of the American College of Chest Physicians VTE guidelines[8] as a basis for recommendations (as the study was conducted prior to the 9th edition release)
The VTE decision support module was comprised of order sets with the following features:
- Patients were identified as on the medicine service based on admitting service designation.
- An order set was populated from this triggering mechanism offering pharmacologic VTE prophylaxis options, or alternately, options to document lack of a clinical indication for pharmacologic VTE prophylaxis, planned therapeutic anticoagulation, or contraindication to VTE prophylaxis.
- Alternate order sets were offered with mechanical VTE prophylaxis options if the physician indicated pharmacologic VTE prophylaxis was contraindicated or if the information system identified a clinical contraindication.
- If pharmacologic VTE prophylaxis was not prescribed, the rules logic was repeated every 5 days.
Analyses
The evaluation sought to assess the effectiveness and safety of the decision support module. VTE processes and outcomes for the 6‐month periods immediately before and after full scale decision support go‐live on September 9, 2009, were evaluated. This time window was chosen in relation to CMS' requirement that hospitals use present on admission codes for discharge diagnoses (including VTE) on October 1, 2007, and first implementation of a hospital‐acquired condition policy on October 1, 2008.[5] The 6‐month period prior to September 2009 was within the first calendar year where both CMS policies were in effect.
Effectiveness of the decision support module was measured by evaluating the proportion of medicine service discharges before and after module deployment who:
- Received any VTE prophylaxis modality
- Received a pharmacologic VTE prophylaxis modality
- Developed a hospital‐acquired VTE
Successful receipt of any VTE prophylaxis modality was defined as use of compression stockings, pneumatic compression devices, or pharmacologic VTE prophylaxis modalities, including therapeutic anticoagulation (eg, mechanical heart valve). Medications counting toward the definition of pharmacologic agents included unfractionated heparin, dalteparin, warfarin, fondaparinux, lepirudin, argatroban, or bivalirudin, which are all on formulary at the medical center. Heparin used as an intravenous flush or associated with dialysis was excluded. Hospital‐acquired VTE was defined by the numerator International Classification of Diseases, 9th Revision (ICD‐9) discharge diagnosis codes for DVT or PE events as specified in the Agency for Healthcare Research and Quality (AHRQ) postoperative PE or DVT Patient Safety Indicator 12, and where the codes were not present on admission.[11]
Patient discharges excluded from analyses were those with patient age <18 years, length of stay 1 day or less, VTE diagnosis present on admission, or patient with an inferior vena cava filter during the stay. For evaluation of pharmacologic VTE prophylaxis, patients were additionally excluded if they had a platelet count <50,000/L during their stay, were a neurosurgical patient, or had a discharge diagnosis that included gastrointestinal bleeding or coagulopathy.
The safety of the decision support application was measured by assessing the proportion of medicine service discharges before and after decision support deployment who developed bleeding or thrombocytopenia. Bleeding was defined as receipt of 1 or more packed red blood cell units following administration of an anticoagulant medication at a VTE prophylaxis dosage range. Exclusion criteria for bleeding evaluation were patients aged <18 years, with length of stay 1 day or less, VTE diagnosis present on admission, platelet count <50,000/L during the stay, were a neurosurgical patient, or had diagnoses of anemia, hematologic malignancy, or inferior vena cava filter during the stay, or diagnoses of gastrointestinal bleeding, hemorrhage, or hematoma on admission.
Thrombocytopenia was defined as a >50% decrease from the initial platelet count during the hospital stay, or a decrease from an admission platelet count of >100,000/L to <100,000/L during the hospital stay. Criteria for exclusion from these analyses were age <18 years, length of stay 1 day or less, diagnosis of VTE present on admission, and vena cava filter during the stay.
A medical center comparison group was defined to contrast the magnitude of change in study end points on medicine services where the intervention was deployed with the change on other services where decision support was not used, and to distinguish potential changes observed on medicine services from secular trends. The comparison group consisted of discharges from cardiology, cardiothoracic surgery, family medicine, general surgery, surgical subspecialty, oncology, psychiatry, and rehabilitation medicine services. Newborn, neurosurgery, obstetrics, and pediatrics service discharges were excluded from the comparison group because of their being at low risk for VTE or in a high‐risk group in whom pharmacologic VTE prophylaxis was frequently contraindicated. All parameters described above were evaluated in the comparison group using inclusion and exclusion criteria similar to the intervention group. Outcomes (hospital‐acquired VTE, bleeding, thrombocytopenia) were assessed similarly across index admissions and readmissions.
The significance of change in rates of prescribing, VTE incidence, and adverse event occurrence, were tested by comparing event proportions before and after decision support module implementation in both groups. As all variables were categorical, significance was assessed using 2‐sided Pearson 2 tests at an level of 0.05. Statistical analyses were performed using SPSS software (IBM, Armonk, NY). This project was reviewed by the Albert Einstein College of Medicine/Montefiore Medical Center institutional review board (protocol number 12‐02‐058X) and deemed exempt. Design of the decision support module and definition of the implementation and evaluation plan required approximately 1 year of monthly interdisciplinary team meetings and 200 hours of programmer development time.
RESULTS
Table 1 compares the effectiveness of the decision support module intervention in medicine intervention and in nonmedicine (nonintervention) services. Among medicine service patients, any VTE prophylaxis ordering increased from 61.9% to 82.1% (P < 0.001), and pharmacologic VTE prophylaxis increased from 59.0% to 74.5% (P < 0.001). Smaller but significant increases were observed on nonmedicine services. Hospital‐acquired VTE incidence on medicine services decreased significantly, from 0.65% to 0.42% (P = 0.008) and nonsignificantly on nonmedicine services.
| Medicine Service | Nonmedicine Services | |||||||
|---|---|---|---|---|---|---|---|---|
| Pre | Post | Pre | Post | |||||
| % (n) | % (n) | Relative Change | Significance | % (n) | % (n) | Relative Change | Significance | |
| ||||||||
| Any VTE prophylaxis | ||||||||
| Eligible | N = 15,254 | N = 15,065 | N/A | N/A | N = 8566 | N = 8162 | N/A | N/A |
| Received | 61.9 (9443) | 82.1 (12,372) | +32.7% | P < 0.001 | 70.5 (6040) | 73.6 (6010) | +4.4% | P < 0.001 |
| Pharmacologic VTE prophylaxis | ||||||||
| Eligible | N = 14,768 | N = 14,588 | N/A | N/A | N = 7883 | N = 7567 | N/A | N/A |
| Received | 59.0 (8712) | 74.5 (10,869) | +26.3% | P < 0.001 | 59.3 (4677) | 63.3 (4791) | +6.7% | P < 0.001 |
| Hospital‐acquired VTE incidence | ||||||||
| Susceptible | N = 15,254 | N = 15,065 | N/A | N/A | N = 8566 | N = 8162 | N/A | N/A |
| Developed | 0.65 (99) | 0.42 (64) | 34.5% | P = 0.008 | 0.82 (70) | 0.72 (59) | 11.5% | P = 0.486 |
Table 2 shows ordering patterns for major VTE prophylaxis modalities. Among eligible medicine service patients, rates of low molecular weight heparin prophylaxis increased from 13.0% to 23.7% (P < 0.001), and of unfractionated heparin prophylaxis from 35.1% to 40.7% (P < 0.001). On nonmedicine services, there was no significant change in low molecular weight heparin use, and unfractionated heparin use increased significantly from 37.2% to 40.9% (P < 0.001). Proportions of patients receiving mechanical prophylaxis or not receiving prophylaxis decreased significantly by 37.8% on medicine services and by 9.8% on nonmedicine services Table 3 shows the safety of the decision support module. Bleeding rates increased on medicine services from 2.9% to 4.0% (P < 0.001) and on nonmedicine services from 7.7% to 8.6% (P = 0.043). Nonsignificant changes in thrombocytopenia rates were observed on both services.
| Medicine Service | Nonmedicine Services | |||||||
|---|---|---|---|---|---|---|---|---|
| Pre % (n) | Post % (n) | Relative Change | Significance | Pre % (n) | Post % (n) | Relative Change | Significance | |
| ||||||||
| Eligible for pharmacologic VTE prophylaxis | N = 14,768 | N = 14,588 | N/A | N = 7883 | N = 7567 | N/A | ||
| Low molecular weight heparin | 13.0 (1922) | 23.7 (3463) | +82.4% | P < 0.001 | 15.3 (1206) | 15.9 (1204) | +4.0% | P = 0.294 |
| Unfractionated heparin | 35.1 (5181) | 40.7 (5936) | +16.0% | P < 0.001 | 37.2 (2932) | 40.9 (3093) | +9.9% | P < 0.001 |
| Warfarin | 10.8 (1594) | 10.0 (1461) | 7.2% | P = 0.029 | 6.8 (532) | 6.4 (483) | 5.4% | P = 0.359 |
| Other agent | 0.1 (15) | 0.1 (9) | 39.3% | P = 0.232 | 0.1 (7) | 0.2 (11) | +63.7 | P = 0.303 |
| Mechanical prophylaxis or did not receive | 41.0 (6056) | 25.5 (3719) | 37.8% | P < 0.001 | 40.7 (3206) | 36.7 (2776) | 9.8% | P < 0.001 |
| Medicine Service | Nonmedicine Services | |||||||
|---|---|---|---|---|---|---|---|---|
| Pre % (n) | Post % (n) | Relative Change | Significance | Pre % (n) | Post % (n) | Relative Change | Significance | |
| ||||||||
| Bleeding | ||||||||
| Susceptible | N = 13,614 | N = 13,445 | N/A | N = 7372 | N = 7061 | N/A | ||
| Developed | 2.9 (401) | 4.0 (534) | +34.8% | P < 0.001 | 7.7 (565) | 8.6 (606) | +12.0% | P = 0.043 |
| Thrombocytopenia | ||||||||
| Susceptible | N = 15,254 | N = 15,065 | N/A | N = 8566 | N = 8162 | N/A | ||
| Developed | 7.4 (1123) | 6.9 (1047) | 5.6% | P = 0.164 | 8.7 (749) | 8.8 (716) | +0.3% | P = 0.948 |
DISCUSSION
Following implementation of a computerized decision support application to improve VTE prophylaxis on 2 hospital medicine services, we observed a significant increase in the rate of overall and pharmacologic VTE prophylaxis use and a significant decrease in the incidence of hospital‐acquired VTE. Changes were of greater magnitude and significance on medicine services where the intervention was deployed.
Rates of any VTE prophylaxis and pharmacologic VTE prophylaxis ordering on medicine services increased significantly by 32.7% and 26.3%, respectively. These rates increased on nonmedicine comparison services by a more modest 4.4% for any VTE prophylaxis and 6.7% for pharmacologic VTE prophylaxis. Although the medicine service intervention was designed to be agnostic to the type of prophylactic heparin preparation, the intervention resulted in a significant 82.4% increase in low molecular weight heparin use and a significant 16.0% increase in unfractionated heparin use. With respect to outcomes, we observed a 34.5% decrease (P < 0.001) in hospital‐acquired VTE incidence on medicine services and a nonsignificant decrease on nonmedicine services.
In assessing intervention safety, increased usage of VTE prophylaxis was not accompanied by an increase in thrombocytopenia, but was associated with an increase in bleeding from 2.9% to 4.0% (P < 0.001) on medicine services and from 7.7% to 8.6% (P = 0.043) on non‐medicine services. As our intervention was a quality improvement project, we conducted a brief post hoc analysis to evaluate the increased bleeding rate on the medicine service following intervention. A random sample of 50 records of medicine patients who had received VTE prophylaxis and had a subsequent bleeding event was reviewed. Findings are summarized in Table 4. Prophylaxis was used appropriately in 100% of cases. Bleeding episodes were minor in that no case required more than 2 U of packed red blood cells. The most common clinical scenario was a patient with baseline anemia, typically with chronic kidney disease, who had a slight decrease in hematocrit of unclear etiology requiring 1 U of blood.
| Characteristic | % (N = 50) |
|---|---|
| |
| Prophylaxis indication | |
| Pharmacologic VTE prophylaxis indicateda | 100.0 |
| Clinical characteristic | |
| Anemia upon admission | 92.0 |
| Chronic kidney disease | 66.0 |
| Suspected bleeding source | |
| Unclear | 62.0 |
| Gastrointestinal | 18.0 |
| Catheter/external device site | 8.0 |
| Operative | 6.0 |
| Epistaxis | 4.0 |
| Gynecologic | 2.0 |
| Medication use | |
| Prophylactic agent associated with bleeding | |
| Unfractionated heparin | 66.0 |
| Dalteparin | 34.0 |
| On antiplatelet agent at time of bleedb | 52.0 |
| Transfusion outcome | |
| Required >2 packed red blood cell units | 0.0 |
Although the intervention occurred on medicine services, favorable albeit smaller changes were observed on nonmedicine services. We expected this favorable secular trend because of VTE prophylaxis awareness efforts across the organization as a whole. There was also ongoing focus on VTE prevention and outcomes by policymakers, regulatory agencies, and professional societies during the time period of study.[3, 4, 5, 12] Public reporting of CMS inpatient surgical VTE prophylaxis measures was required throughout the study period.[13] Changes observed on medicine services occurred during a period where there were no publicly reported measures of VTE prophylaxis for inpatient medicine services.
Our study had several limitations. We derived our eligibility criteria for VTE prophylaxis based on administrative data. To address this, we incorporated accepted standardized definitions,[11] used clinical data elements in our queries beyond ICD‐9 codes (eg, platelet count), and applied pertinent exclusion criteria (eg, length of stay 1 day or less). VTE events that were present on admission were excluded from analyses. However, as these community‐acquired VTE events may be caused by inadequate VTE prophylaxis during a prior hospitalization, the overall true incidence of hospital‐acquired VTE was likely underestimated.
With respect to the hospital‐acquired VTE outcome, we did not distinguish superficial from deep VTE. A consistent AHRQ definition of 13 ICD‐9 VTE codes was used to identify clinically significant VTE events for the periods before and after the intervention. Although the present on admission code identified VTE events that were hospital acquired, 1 new acute VTE ICD‐9 code was added in October 2009, allowing for more specific coding of acute, isolated, upper extremity VTE. Accordingly, our postintervention hospital‐acquired VTE rate may have slightly underestimated the true hospital‐acquired VTE incidence by omitting some coded acute, isolated, upper extremity VTE cases (if not coded using the prior Other VTE codes). In a study in a teaching hospital setting, isolated upper extremity VTE accounted for up to 21% of all symptomatic VTE events among adults.[14]
With respect to VTE prophylaxis, the study evaluated use in a dichotomous fashion but did not assess appropriateness, or adequacy of dosing of pharmacologic agents. We did not employ the intervention in a randomized fashion on the medicine service. As our project was a quality improvement intervention, we used a concurrent control group of nonmedicine service patients to assess potential secular trend bias.
With respect to the safety of the intervention, the record review we performed supported the appropriateness of prophylaxis use following the intervention, but was not designed to establish whether the increase in prophylaxis use was the proximate cause of bleeding events observed. Similarly, as specific testing for heparin‐induced thrombocytopenia was not used, the lack of significant change in thrombocytopenia rates before and after the intervention cannot directly establish the intervention's safety. Finally, our study also included only in‐hospital end points.
The rate of VTE prophylaxis use in hospitals has been noted to be disappointing.[15] Two large multinational studies found that VTE prophylaxis rates in at‐risk hospitalized medical patients in the United States were 48% and 52%.[9, 16] Amin and colleagues found the overall rate of VTE prophylaxis among 227 US hospitals to be 62%.[17] Accordingly, our intervention, which resulted in an 82% compliance rate on a large medical service and was associated with a significantly reduced VTE incidence, appears to be highly effective. Our results are likely more favorable in that beyond length of stay criteria, we did not exclude less acutely ill medical patients from analyses.
Michota summarized quality improvement studies for VTE prevention.[10] Among 9 studies attempting to improve VTE prophylaxis, 2 used electronic decision support as a primary strategy, and only 1, by Kucher et al., used a computerized approach on a medical service.[18] This study showed significant improvement in VTE prophylaxis and incidence in patients randomized to a provider computer program. The intervention was complex, requiring specification of 8 patient‐level risk factors via a customized database, and the physician to recommend specific prophylactic regimens accordingly. Our findings, using a more basic approach, similarly support the effectiveness of using automated decision support, which can be readily modified as evidence‐based guidelines evolve.
Overall adoption of information technology systems in US hospitals is low: only 7.6% of hospitals have a basic system, and 17% have computerized physician order entry.[19] As hospitals have been financially incentivized to adopt such systems, our relatively simple intervention may prove to be readily generalizable across varied vendor systems.[20] The intervention involved order sets triggered by automated logic, corollary information, and a hard stop to prompt VTE prophylaxis. Within the context of intensified emphasis on reducing harm in the inpatient setting and various pay for performance programs, our intervention is also of importance to payers.[3, 5] Using national data in year 2000, Zhan and Miller calculated the excess charges per case associated with VTE to be $21,709.[21]
In conclusion, a relatively simple automated clinical decision support application significantly improved rates of VTE prophylaxis and was associated with significantly lower hospital‐acquired VTE incidence in hospitalized medicine patients, with a reasonable safety profile.
Acknowledgments
The authors acknowledge the roles of Gillian Wendt and Maggie Feng in data acquisition.
Disclosure: The authors declare no conflict of interest related to the research, analyses, or preparation of this manuscript. M. J. Sinnett reports receiving payment for speaking on behalf of Amgen, which was not a funder of this study. All coauthors have seen and agree with the contents of the manuscript, and all coauthors fulfill the authorship criteria specified by the Journal of Hospital Medicine. Rohit Bhalla, MD, MPH, takes responsibility for the entire manuscript. This submission is not under review by any other publication. Development of the electronic decision support application was supported in part by funding under the 2008 Cardinal Health Foundation Patient Safety Grant Program.
- . The epidemiology of venous thromboembolism: implications for prevention and management. Paper presented at: Surgeon General's Workshop on Deep Vein Thrombosis; May 8, 2006; Bethesda, MD. Available at: http://www.surgeongeneral.gov/topics/deepvein/workshop/agenda.html. Accessed February 17,2012.
- American Public Health Association White Paper. Deep‐vein thrombosis: advancing awareness to protect patient lives. Public Health Leadership Conference on Deep‐Vein Thrombosis. Washington, D.C.; February 26,2003. Available at: http://www.apha.org/NR/rdonlyres/A209F84A‐7C0E‐4761–9ECF‐61D22E1E11F7/0/DVT_ White_Paper.pdf. Accessed February 27, 2012.
- Department of Health and Human Services. Centers for Medicare and Medicaid Services. Medicare program: hospital inpatient value based purchasing program. Fed Regist.2011;76(88):26490–26547.
- The Joint Commission. 2011 hospital national patient safety goals. Available at: http://www.jointcommission.org/assets/1/6/HAP_NPSG_6–10‐11.pdf. Accessed October 23,2011.
- US Department of Health and Human Services. Centers for Medicare and Medicaid Services. Medicare Learning Network. Hospital acquired conditions in acute inpatient prospective payment system (IPPS) hospitals. Available at: https://www.cms.gov/HospitalAcqCond/downloads/HACFactsheet.pdf. Accessed February 17,2012.
- , .Preventing Hospital‐Acquired Venous Thromboembolism: A Guide For Effective Quality Improvement. Society of Hospital Medicine. AHRQ Publication No. 08–0075.Rockville, MD:Agency for Healthcare Research and Quality;2008.
- . Prophylaxis for thromboembolism in hospitalized medical patients. N Engl J Med.2007;356:1438–1444.
- , , , et al. Prevention of venous thromboembolism: American College of Chest Physicians Evidence‐Based Clinical Practice Guidelines (8th Edition). Chest.2008;133;381S–453S.
- , , , et al. Venous thromboembolism risk and prophylaxis in the acute hospital care setting (ENDORSE study): a multinational cross‐sectional study. Lancet.2008;371(9610):387–394.
- . Bridging the gap between evidence and practice in venous thromboembolism prophylaxis: the quality improvement process. J Gen Intern Med.2007;22(12):1762–1770.
- Agency for Healthcare Research and Quality. PSI #12: Postoperative pulmonary embolism or deep vein thrombosis. Version 4.1.; 2009. Available at: http://www.qualityindicators.ahrq.gov/Downloads/Modules/PSI/V41/TechSpecs/PSI%2012%20Postoperative%20Pulmonary %20Embolism%20or%20Deep%20Vein%20Thrombosis.pdf. Accessed February 19,2012.
- , . The CMS ruling on venous thromboembolism after total knee or hip arthroplasty: weighing risks and benefits. JAMA.2009;301(10):1063–1065.
- US Department of Health and Human Services. Centers for Medicare and Medicaid Services. Hospital compare. Available at: http://www.hospitalcompare.hhs.gov. Accessed October 23,2011.
- , , , , , . Upper extremity deep venous thrombosis. Chest.2003;123;1953–1956.
- , . Thromboprophylaxis for adults in hospital: an intervention that would save many lives is still not being implemented. BMJ.2007;334:1017–1018.
- , , , et al. Venous thromboembolism prophylaxis in acutely ill hospitalized medical patients: findings from the International Medical Prevention Registry on Venous Thromboembolism. Chest.2007:132:936–945.
- , , , . Thromboprophylaxis rates in US medical centers: success or failure?J Thromb Haemost.2007;5:1610–1616.
- , , , et al. Electronic alerts to prevent venous thromboembolism among hospitalized patients. N Engl J Med.2005;352:969–977.
- , , , et al. Use of electronic health records in U.S. hospitals. N Engl J Med.2009;360:1628–1638.
- , . The “meaningful use” regulation for electronic health records. N Engl J Med.2010;363(6):501–504.
- , . Excess length of stay, charges, and mortality attributable to medical injuries during hospitalization. JAMA.2003;290(14):1868–1874.
- . The epidemiology of venous thromboembolism: implications for prevention and management. Paper presented at: Surgeon General's Workshop on Deep Vein Thrombosis; May 8, 2006; Bethesda, MD. Available at: http://www.surgeongeneral.gov/topics/deepvein/workshop/agenda.html. Accessed February 17,2012.
- American Public Health Association White Paper. Deep‐vein thrombosis: advancing awareness to protect patient lives. Public Health Leadership Conference on Deep‐Vein Thrombosis. Washington, D.C.; February 26,2003. Available at: http://www.apha.org/NR/rdonlyres/A209F84A‐7C0E‐4761–9ECF‐61D22E1E11F7/0/DVT_ White_Paper.pdf. Accessed February 27, 2012.
- Department of Health and Human Services. Centers for Medicare and Medicaid Services. Medicare program: hospital inpatient value based purchasing program. Fed Regist.2011;76(88):26490–26547.
- The Joint Commission. 2011 hospital national patient safety goals. Available at: http://www.jointcommission.org/assets/1/6/HAP_NPSG_6–10‐11.pdf. Accessed October 23,2011.
- US Department of Health and Human Services. Centers for Medicare and Medicaid Services. Medicare Learning Network. Hospital acquired conditions in acute inpatient prospective payment system (IPPS) hospitals. Available at: https://www.cms.gov/HospitalAcqCond/downloads/HACFactsheet.pdf. Accessed February 17,2012.
- , .Preventing Hospital‐Acquired Venous Thromboembolism: A Guide For Effective Quality Improvement. Society of Hospital Medicine. AHRQ Publication No. 08–0075.Rockville, MD:Agency for Healthcare Research and Quality;2008.
- . Prophylaxis for thromboembolism in hospitalized medical patients. N Engl J Med.2007;356:1438–1444.
- , , , et al. Prevention of venous thromboembolism: American College of Chest Physicians Evidence‐Based Clinical Practice Guidelines (8th Edition). Chest.2008;133;381S–453S.
- , , , et al. Venous thromboembolism risk and prophylaxis in the acute hospital care setting (ENDORSE study): a multinational cross‐sectional study. Lancet.2008;371(9610):387–394.
- . Bridging the gap between evidence and practice in venous thromboembolism prophylaxis: the quality improvement process. J Gen Intern Med.2007;22(12):1762–1770.
- Agency for Healthcare Research and Quality. PSI #12: Postoperative pulmonary embolism or deep vein thrombosis. Version 4.1.; 2009. Available at: http://www.qualityindicators.ahrq.gov/Downloads/Modules/PSI/V41/TechSpecs/PSI%2012%20Postoperative%20Pulmonary %20Embolism%20or%20Deep%20Vein%20Thrombosis.pdf. Accessed February 19,2012.
- , . The CMS ruling on venous thromboembolism after total knee or hip arthroplasty: weighing risks and benefits. JAMA.2009;301(10):1063–1065.
- US Department of Health and Human Services. Centers for Medicare and Medicaid Services. Hospital compare. Available at: http://www.hospitalcompare.hhs.gov. Accessed October 23,2011.
- , , , , , . Upper extremity deep venous thrombosis. Chest.2003;123;1953–1956.
- , . Thromboprophylaxis for adults in hospital: an intervention that would save many lives is still not being implemented. BMJ.2007;334:1017–1018.
- , , , et al. Venous thromboembolism prophylaxis in acutely ill hospitalized medical patients: findings from the International Medical Prevention Registry on Venous Thromboembolism. Chest.2007:132:936–945.
- , , , . Thromboprophylaxis rates in US medical centers: success or failure?J Thromb Haemost.2007;5:1610–1616.
- , , , et al. Electronic alerts to prevent venous thromboembolism among hospitalized patients. N Engl J Med.2005;352:969–977.
- , , , et al. Use of electronic health records in U.S. hospitals. N Engl J Med.2009;360:1628–1638.
- , . The “meaningful use” regulation for electronic health records. N Engl J Med.2010;363(6):501–504.
- , . Excess length of stay, charges, and mortality attributable to medical injuries during hospitalization. JAMA.2003;290(14):1868–1874.
Copyright © 2012 Society of Hospital Medicine
Steroids in Pneumonia
Community‐acquired pneumonia (CAP) is the most common lower respiratory tract infection in adults and a leading cause of infection‐related deaths in the United States.[1] According to a survey, pneumonia was the most common reason for hospital admissions through the emergency department in 2003.[2] CAP is associated with significant morbidity and mortality among those sick enough to require hospitalization. In a prospective study, hospital mortality rates ranged from 5% to 18% and length of stay from 9 to 23 days depending on patient location (intensive care unit [ICU] vs elsewhere) and severity of illness.[3]
Empirical evidence suggests that host inflammatory response contributes significantly to lung injury in pneumonia.[4] Studies have demonstrated reduction in the host inflammatory response as well as in mortality among animals with bacterial pneumonia when exposed to glucocorticoids.[5, 6] Furthermore, the efficacy of adjunctive steroid therapy in severe pneumonia caused by Pneumocystis jirovecii[7] and in pneumococcal meningitis[8, 9] is already established. However, due to equivocal, and at times conflicting, human clinical trial data on the impact of steroid therapy in CAP, the 2007 consensus guidelines (jointly published by the Infectious Diseases Society of America and American Thoracic Society) do not provide recommendations for or against use of steroids in CAP, except in the setting of hypotension secondary to adrenal insufficiency.[10]
In their meta‐analysis, Chen et al. analyzed data from 6 randomized clinical trials (RCTs) published between 1972 and 2007 (including 2 on pediatric patients) and concluded that adding steroids to current standard of care was not beneficial.[11] Earlier, Lamontagne et al.'s meta‐analysis included RCTs on hospitalized CAP patients as well as those on patients with acute lung injury (ALI) or acute respiratory distress syndrome (ARDS) from any cause.[12] They concluded that low‐dose corticosteroid therapy reduced all‐cause in‐hospital mortality in this mixed patient population (relative risk [RR]: 0.68 [95% confidence interval (CI): 0.49 to 0.96]). Recently, data from a number of additional RCTs have become available.[13, 14, 15, 16, 17] Therefore, an updated review of RCTs evaluating the role of adjunctive steroid therapy among adults hospitalized with CAP was warranted.
MATERIALS AND METHODS
We conducted this systematic review and meta‐analysis in accordance with the recommendations published in the Cochrane Handbook for Systematic Reviews of Interventions[18] and reported our findings according to the Preferred Reporting Items for Systematic Reviews and Meta‐analyses guidelines.[19] The overall quality of evidence was judged using the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) framework.[20]
Data Sources and Search Strategies
A comprehensive search of several databases including PubMed, Ovid MEDLINE In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE, Ovid EMBASE, Ovid Cochrane Database of Systematic Reviews, Ovid Cochrane Central Register of Controlled Trials, and Scopus was conducted. The time range for search started from each database's earliest inclusive dates up to July 2011. An experienced institutional librarian assisted with the design and conduct of our literature search. Controlled vocabulary, supplemented with keywords, was used to search for the topic: steroid therapy for community‐acquired pneumonia. We consulted expert colleagues to ensure the inclusion of all eligible reports and also checked the bibliographies of previously published systematic reviews.[12, 21]
Eligibility Criteria
Studies deemed eligible for inclusion were RCTs that met the following patients, intervention, control, outcomes (PICO) criteria: P, adults hospitalized with CAP; I, administration of systemic corticosteroids plus standard treatment; C, standard treatment without corticosteroids; O, primary outcome: hospital mortality; secondary outcomes, length of hospital stay, length of ICU stay and duration of mechanical ventilation. Under the P criterion we included RCTs that defined CAP as a lung infection (based on a reasonable combination of history, physical examination, imaging, and/or other investigative data, such as per the American Thoracic Society definition)[22] of presumed or proven bacterial etiology, in a patient who was not immunocompromised and had no exposure to a healthcare facility in the past 90 days.
Study Selection and Quality Assessment
Two reviewers independently performed study selection, data extraction, and quality assessment. Data were abstracted using standardized data collection instruments. Kappa statistic was calculated to assess the reviewers' level of agreement.
We perused full texts of all articles whose abstracts met selection criteria, performing an appraisal of their quality using the Cochrane risk‐of‐bias tool.[23] We also reviewed the baseline characteristics of patients in each study cohort.
Analysis
We estimated RR and weighted mean differences along with the respective 95% confidence intervals by pooling data using a random effects model.[24] Study heterogeneity was assessed using the I2 statistic, which estimates the percentage of variation that is not attributable to chance.[25] We performed a priori subgroup analyses based on the location (ICU vs non‐ICU) and mean age group of study participants (based on a cutoff of 50 years). A significant (P < 0.05) test of interaction would provide an explanation for any heterogeneity.[26] We also performed an a priori sensitivity analysis excluding any studies published before the year 2000 to exclude the impact of changing standards of care for inpatient management of CAP over time.
The original investigators were not contacted for purposes of obtaining raw data.
RESULTS
Eight RCTs, comprising 1119 subjects, were eventually chosen.[14]. Seven shortlisted studies were excluded due to methodological limitations, failure to fully meet PICO criteria, or gross insufficiency of descriptive data on subjects or methodology.[18, 31, 32, 33, 34, 35, 36] Figure 1 illustrates the study selection process.
Table 1 summarizes the baseline characteristics of patient populations from each study. Mean ages in 7 RCTs were between 60 years and 80 years. In Marik et al., the mean age of the intervention group was 31.7 years, whereas that of the control group was 40.6 years (P value not reported).[30] Three RCTs included ICU patients only,[17, 28, 30] whereas 4 only included general medical ward patients.[14, 15, 29, 31] Disease severity scores at admission were similar between the 2 groups in all RCTs except Sabry and Omar,[17] which was the only clinical trial to use a chest radiograph score. Only Sabry and Omar,[17] and Mikami et al.[29] excluded chronic obstructive pulmonary disease patients. Where possible, the serum C‐reactive protein (CRP) value on day one was subtracted from that on day eight to generate a one week delta CRP.
| Author, Year | Number of Patients | Gender: Males (% Age) | Age (y) | Steroids Used (Daily Dose and Duration) | COPD (% of Total) | Diabetes (% of Total) | Mean PaO2/FiO2 Ratio | Severity Score (Score: Mean) | Patients Already in ICU (% of Total) | One‐week Delta CRP (mg/dL) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | ||
| ||||||||||||||||||||
| McHardy 1972[31] | 126 | 40 | 86 | 45 | 50 | 62 | 59 | Prednisolone 20 mg, 7 d | 40 | 35 | Not reported | Not reported | Not reported | 0 | 0 | N/A | N/A | |||
| Marik 1993[30] | 30 | 14 | 16 | Not reported | 32 | 41 | Hydrocortisone 10 mg/kg 1 | Not reported | Not reported | 213 | 214 | APACHE II | 100 | 100 | N/A | N/A | ||||
| 11 | 14 | |||||||||||||||||||
| Confalonieri 2005[28] | 46 | 23 | 23 | 74 | 65 | 60 | 67 | Hydrocortisone 200 mg bolus, then 10 mg/h, 7 d | Not reported | Not reported | 141a | 178a | APACHE II | 100 | 100 | 37a | +5a | |||
| 17 | 18 | |||||||||||||||||||
| Mikami 2007[29] | 31 | 15 | 16 | 73.3 | 75 | 76 | 68 | Prednisolone 40 mg IV, 3 d | 0 | 0 | Not reported | PaO2 (FiO2 not reported) | PSI | 0 | 0 | N/A | N/A | |||
| 61 | 64 | 95 | 86 | |||||||||||||||||
| Snijders 2010[16] | 213 | 104 | 109 | 52.9 | 63.3 | 63 | 64 | Prednisolone 40 mg IV/PO, 7 d | 18 | 22 | 10 | 11 | Not reported | PSI class V (% of total) | 14.4 | 6.4 | N/A | N/A | ||
| 13 | 16 | |||||||||||||||||||
| Fernandez‐Serrano 2011[15] | 45 | 23 | 22 | 69.6 | 63.6 | 66 | 61 | Methylprednisone 200 mg IV; then 20 mg/6 h, 3 d; then 20 mg/12 h, 3 d; then 20 mg/d, 3 d | 17 | 9 | 9 | 18 | 200 | 257 | SAPS classes IV + V (% of total) | 0 | 0 | N/A | N/A | |
| 65 | 54 | |||||||||||||||||||
| Meijvis 2011[14] | 304 | 151 | 153 | 57 | 56 | 65 | 63 | Dexamethasone 5 mg/d, 4 d | 13 | 9 | 15 | 14 | Not reported | PSI class V (% of total) | 0 | 0 | N/A | N/A | ||
| 17 | 14 | |||||||||||||||||||
| Sabry 2011[17] | 80 | 40 | 40 | 30 | 28 | 62 | 63 | Hydrocortisone 200 mg IV, then 12.5 mg/h, 7 d | 0 | 0 | Not reported | 338a | 243a | Chest radiograph score | 100 | 100 | 38a | 23a | ||
| 1a | 3a | |||||||||||||||||||
The mean ICU length of stay was 12.7 days for the steroid group and 12.3 days for the control group. The mean hospital lengths of stay were 10.2 days and 13.6 days, respectively. Quality of the studies was moderate (see Supporting Information, Appendix I, in the online version of this article). Kappa score was >0.90.
Meta‐analysis
Figure 2 illustrates the results of our meta‐analyses. Although adjunctive steroid therapy had no effect on hospital mortality or ICU length of stay, it was associated with reduced hospital length of stay (RR: 1.21 days [95% CI: 2.12 to 0.29]). Of note, Mikami et al.[29] did not report mortality in their article, whereas in McHardy and Schonell,[31] using the factorial design, each of the 2 treatment groups were further subdivided into those patients who received 1 g of ampicillin and those who received 2 g of ampicillin (Figure 2A).
Analysis of other outcomes was limited by the fact that data were pooled from only a few studies. These included the need for and duration of mechanical ventilation, development of new ARDS and ICU admission rate, neither of which was associated with steroid therapy. However, steroid use was associated with lower incidence of delayed shock (ie, shock occurring after enrollment (RR: 0.12 [95% CI: 0.03 to 0.41]) and lower incidence of persistent chest x‐ray abnormalities at 1 week (RR: 0.13 [95% CI: 0.06 to 0.27]).
Subgroup and Sensitivity Analyses
Heterogeneity (I2 statistic) was <50% for all outcomes except ICU length of stay (74%). There were no significant interactions to suggest a subgroup effect based on older vs younger or ICU vs non‐ICU based patients (Table 2). In a priori sensitivity analysis that excluded McHardy and Schonell (published in 1972) and Marik et al. (published in 1993),[30] the results were not different from the main analysis.
| No. of Studies | Effect Size | LL | UL | P for Interaction (Difference Between Subgroups) | |
|---|---|---|---|---|---|
| |||||
| Mortality | |||||
| ICU | 3 | 0.27 | 0.08 | 0.83 | 0.06 |
| Non‐ICU | 4 | 0.96 | 0.45 | 2.05 | |
| Older | 7 | 0.75 | 0.40 | 1.38 | 0.56 |
| Young | 1 | 0.38 | 0.04 | 3.26 | |
| Need for mechanical ventilation | |||||
| ICU | 1 | 0.57 | 0.12 | 2.66 | 0.39 |
| Non‐ICU | 1 | 0.14 | 0.01 | 2.51 | |
| Older | 1 | 0.14 | 0.01 | 2.51 | 0.39 |
| Younger | 1 | 0.57 | 0.12 | 2.66 | |
| LOS | |||||
| ICU | 1 | 8.00 | 16.41 | 0.41 | 0.11 |
| Non‐ICU | 3 | 1.14 | 1.97 | 0.31 | |
| ICU LOS | |||||
| ICU | 2 | 3.91 | 11.44 | 3.62 | 0.45 |
| Non‐ICU | 2 | 0.81 | 8.98 | 10.60 | |
| Older | 3 | 2.26 | 9.95 | 5.43 | 0.65 |
| Younger | 1 | 0.30 | 3.91 | 3.31 | |
Quality of Evidence
Using the GRADE framework, the overall quality of evidence (confidence in the estimates) was judged to be moderate with the following main limitations: 1) methodological limitations among included studies (prognostic imbalance), 2) imprecision (small number of events and wide confidence intervals), and 3) inconsistency in the outcome ICU length of stay (as reflected by the I2 statistic).
Other Reported Outcomes
Four studies[15, 16, 29, 31] provided descriptive details of microbiologic data, whereas 1 study[16] provided analytical data on microbiology. In the latter, patients with Streptococcus pneumoniae infection (identified variably by sputum, pleural fluid, urine, or blood samples), had lower clinical cure rates in the steroid group at day 30 (P = 0.01) and higher numbers of late failures (defined as recurrence of signs and symptoms of pneumonia, P = 0.02).
Three[28, 30, 31] studies did not provide data on glycemic trends, whereas Fernandez‐Serrano et al., Mikami et al., and Snijders et al. reported that rates of hyperglycemia were not different across the 2 groups.[15, 16, 29] Meijvis et al.[14] reported more frequent hyperglycemia in the steroid group (44% vs 23%, P < 0.001) but no difference in the need for glucose‐lowering treatment (5% vs 3%, P = 0.57). Sabry and Omar[17] reported a higher incidence of hyperglycemia in the steroid group (no numerical data reported). Snijders et al.,[16] Meijvis et al.,[14] and Sabry and Omar[17] reported that the rates of super‐infection were not different between the 2 groups. No other adverse effects were consistently reported.
DISCUSSION
In this meta‐analysis of 8 RCTs, we found no significant association between steroid therapy and our primary outcome of interest (hospital mortality). However, length of hospital stay was shorter in the steroid group. These findings were not altered in various sensitivity and subgroup analyses. Although adverse effects of steroid therapy were not consistently reported, most of the RCTs reported that hyperglycemia was either no more common in the steroid group or did not require additional treatment.
Previous meta‐analyses have also concluded that adding corticosteroids to conventional therapy does not impact mortality among adults hospitalized with CAP.[12, 22] This may or may not be a consequence of inadequate statistical power. Although Lamontagne et al.[13] reported that low‐dose corticosteroid therapy (2 mg/kg/day or less of methylprednisolone or equivalent) was associated with reduced hospital mortality (RR: 0.68 [95% CI: 0.49 to 0.96]), this result was obtained by pooling data from 5 RCTs on adults hospitalized with CAP and 4 on adults with ALI/ARDS from any cause. In a subgroup analysis of RCTs conducted only on CAP patients, no impact on mortality was found. Of note, all RCTs involving CAP patients had used low‐dose steroids; the 3 RCTs using high‐dose steroids were carried out on ALI/ARDS patients.[36, 37, 38] Similarly, all RCTs in our meta‐analysis were also characterized by steroid doses under 2 mg/kg/day of methylprednisolone or equivalent.
Our study is the first to demonstrate decreased length of hospital stay in this patient population. Importantly, each of the 5 studies that reported this outcome (including 3 relatively recent RCTs) showed the same trend. However, it is not inconceivable that steroid use led to a quicker decline in cytokine levels resulting in an earlier resolution of fever and hence earlier discharge without a faster cure per se. The two studies whose data permitted calculation of delta CRP also demonstrated a faster CRP decline in the steroid group (Table 1).
Our analysis also suggested reduced incidence of delayed shock. However, these data were pooled from only 2 RCTs,[17, 28] and each of them used hydrocortisone, whose direct mineralocorticoid effect is an obvious confounder. Similarly, according to data pooled from 2 RCTs, steroid use was associated with fewer cases of persistent chest x‐ray abnormalities by day 8. Of note, although calculation of the I2 statistic was not possible because of too few studies, visual inspection of the forest plots suggested low levels of heterogeneity.
It is plausible that the impact of adjunctive steroids in CAP may vary based on the causative pathogen. This pathogen‐specific association has been observed in patients with bacterial meningitis, where most of the benefit is seemingly limited to pneumococcal meningitis.[9, 10] Unfortunately, as demonstrated by Snijders et al.,[16] establishing microbiologic etiology in CAP can be difficult, and most patients are treated empirically.
Our analysis showed no difference in duration of ventilation among patients who required ventilatory support on admission. However, only 2 studies reported this outcome.[17, 28] Second, in Confalonieri et al.,[28] the steroid group had a more severe baseline inflammatory response as illustrated by higher serum CRP levels (P = 0.04). Moreover, while mechanical ventilation was defined as either invasive or noninvasive ventilation, the steroid group had a higher number of patients who required noninvasive ventilation (P = 0.03), thus introducing selection bias. This study had additional areas of concern too, including a mortality of 0 among its 46 ICU patients, in contrast to established mortality rates of up to around 20%.[4] Unlike this study, Sabry and Omar[17] reported that none of their patients was on noninvasive ventilation. It may be pertinent to compare our findings with those of Steinberg et al.,[40] who studied patients with ARDS (pneumonia being the most common cause) who received methylprednisolone. This group had an early increase in ventilator‐free days, but that effect became less pronounced (though still significant) when the study end point was prolonged from 30 to 90 days.[41]
The 2 studies that were published before 2000 (McHardy and Schonell,[31] and Marik et al.[30]) were excluded in our a priori sensitivity analysis. A number of considerations led to this decision. First, standards of care for inpatient management of pneumoniaincluding pharmacologic therapies and ventilation strategieshave changed considerably over time. For instance, newer generation macrolides became available for clinical use in the early 1990s and meropenem in 1996.[41] Therefore, it would be hard to assume constancy of effect from that time period. Furthermore, the study by McHardy and Schonell[31] suffered from significant differences in the baseline characteristics of its 2 arms. There was incomplete randomization; patients with diabetes were excluded from only the steroid arm. Another issue with Marik et al.[30] was the considerably younger age of participants compared to other studies (Table 1).
Limitations
In spite of our relatively stringent selection criteria and a number of subgroup and sensitivity analyses, the overall quality of evidence was only moderate (Table 2). Key issues with the findings reported by Confalonieri et al.,[28] McHardy and Schonell,[31] and Marik et al.[30] were discussed earlier. Baseline severity of illness, patient comorbidities, and length of follow‐up were variable both within and across various studies. Another major limitation was that the intervention of interest (ie, steroid therapy) was not uniformly applied as the regimens varied considerably even though all regimens fit the designation of low‐dose steroids as previously noted (Table 1).
In conclusion, although evidence suggests that adjunctive steroid therapy is associated with reduced hospital length of stay, the data are not strong enough to recommend routine use of steroids among all adults hospitalized with CAP. However, considering that there was no increase in mortality or hospital length of stay with steroid use, it is reasonable to continue steroids if warranted for treatment of underlying comorbid conditions.
Due to the aforementioned limitations in RCTs published to date, we believe that additional studies that are more robustly designed and sufficiently powered to detect differences in key outcomes (including mortality) are warranted. Investigators should ensure appropriate randomization of groups, taking into account severity of illness, comorbid conditions and prior use of steroid therapy. Standardizing the intervention (including dose and duration of steroid therapy and time to first antibiotic dose) would be essential. Concurrent measurement of inflammatory markers such as delta CRP would be useful too. Finally, accurate measurement of all secondary outcomes of interest, including adverse effects and duration of both invasive and noninvasive mechanical ventilation, would be important to accurately study the benefit of steroids among the most likely beneficiaries: those patients who are the sickest.
Acknowledgments
The authors gratefully acknowledge the assistance of Dr. Jon Ebbert (Department of Medicine, Mayo Clinic, Rochester, MN) with proofreading the manuscript and providing thoughtful editorial suggestions.
Disclosures
The authors report no conflicts of interest.
- Centers for Disease Control and Prevention 2008. CDC/NCHS, National Vital Statistics System. Leading causes of Death. Available at: http://www.cdc.gov/nchs/nvss/mortality_tables.htm. Accessed August 14,2011.
- , . Reasons for being admitted to the hospital through the emergency department, 2003. HCUP Statistical Brief #2. February 2006. Agency for Healthcare Research and Quality, Rockville, MD. Available at: http://www.hcup‐us.ahrq.gov/reports/statbriefs/sb2.pdf. Accessed August 14,2011.
- , , , et al. Processes of care and outcomes for community‐acquired pneumonia. Am J Med. 2011;124(12):1175.e9–17.
- , , , , , . Cytokine kinetics and other host factors in response to pneumococcal pulmonary infection in mice. Infect Immun. 1998;66(3):912–922.
- , , , et al. Effects of glucocorticoids in ventilated piglets with severe pneumonia. Eur Respir J. 2008;32(4):1037–1046.
- , , , et al. Risk of death does not alter the efficacy of hydrocortisone therapy in a mouse E. coli pneumonia model: risk and corticosteroids in sepsis. Intensive Care Med. 2008;34(3):568–577.
- , , , . Adjunctive corticosteroids for Pneumocystis jiroveci pneumonia in patients with HIV‐infection. Cochrane Database Syst Rev. 2006;(3):CD006150.
- , . Dexamethasone in adults with bacterial meningitis. N Engl J Med. 2002;347(20):1549–1556.
- , , , . Steroids in adults with acute bacterial meningitis: a systematic review. Lancet Infect Dis. 2004;4(3):139–143.
- , , , et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community‐acquired pneumonia in adults. Clin Infect Dis. 2007;44(suppl 2):S27–S72.
- , , , . Corticosteroids for pneumonia. Cochrane Database Syst Rev. 2011;(3):CD007720.
- , , , , , . Corticosteroid therapy for acute lung injury, acute respiratory distress syndrome, and severe pneumonia: a meta‐analysis of randomized controlled trials. J Crit Care. 2010;25(3):420–435.
- , , , et al. Dexamethasone and length of hospital stay in patients with community‐acquired pneumonia: a randomised, double‐blind, placebo‐controlled trial. Lancet. 2011;377(9782):2023–2030.
- , , , et al. Effect of corticosteroids on the clinical course of community‐acquired pneumonia: a randomized controlled trial. Crit Care. 2011;15(2):R96.
- , , , et al. Efficacy of corticosteroids in community‐acquired pneumonia: a randomized double‐blinded clinical trial. Am J Respir Crit Care Med. 2010;181:975–978.
- , . Corticosteroids and ICU course of community acquired pneumonia in Egyptian settings. Pharmacol Pharm. 2011;2(2):73–81.
- , , , , . Corticosteroid treatment in severe community‐acquired pneumonia: duration of treatment affects control of systemic inflammation and clinical improvement. Intensive Care Med. 2011;37(9):1553–1554.
- Higgins JPT, Green S, eds. Cochrane Handbook for Systematic Reviews of Interventions. West Sussex, UK:Wiley‐Blackwell;2008.
- , , , ;PRISMA Group. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. J Clin Epidemiol. 2009;62:1006–1012.
- , , , et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401–406.
- , , , , , . The role of corticosteroids in severe community‐acquired pneumonia: a systematic review. Crit Care. 2008;12(3):R76.
- , , , et al. Severe community‐acquired pneumonia: assessment of severity criteria. Am J Respir Crit Care Med. 1998;158(4):1102–1108.
- , .Assessing risk of bias in included studies. In: Higgins JPT, Green S eds. Cochrane Handbook for Systematic Reviews of Interventions. Chichester, UK:John Wiley 2009.
- , . Meta‐analysis in clinical trials. Control Clin Trials. 1986;7(3):177–188.
- , , , . Measuring inconsistency in meta‐analyses. BMJ. 2003;327(7414):557–560.
- , . Interaction revisited: the difference between two estimates. BMJ. 2003;326(7382):219.
- , , , et al. Hydrocortisone infusion for severe community‐acquired pneumonia: a preliminary randomized study. Am J Respir Crit Care Med. 2005;171(3):242–248.
- , , , , , . Efficacy of corticosteroids in the treatment of community‐acquired pneumonia requiring hospitalisation. Lung. 2007;185(5):249–255.
- , , , , , . Hydrocortisone and tumour necrosis factor in severe community acquired pneumonia. A randomised controlled study. Chest. 1993;104(2):389–392.
- , . Ampicillin dosage and use of prednisolone in treatment of pneumonia: co‐operative controlled trial. Br Med J. 1972;4:569–573.
- , , , et al. Impact of systemic corticosteroids on the clinical course and outcomes of patients with severe community‐acquired pneumonia: a cohort study. J Crit Care. 2011;26(2):193–200.
- , , , . Factors of importance for the long term prognosis after hospital treated pneumonia. Thorax. 1993;48(8):785–789.
- , , , . Assessment of mortality after long‐term follow‐up of patients with community‐acquired pneumonia. Clin Infect Dis. 2003;37(12):1617–1624.
- , , , , . Community‐acquired pneumonia on the intensive care unit: secondary analysis of 17,869 cases in the ICNARC Case Mix Programme Database. Crit Care. 2006;10(suppl 2):S1.
- , , , , , . Effects of systemic steroids in patients with severe community‐acquired pneumonia. Eur Respir J. 2007;30(5):951–956.
- , , , . Analysis of systemic corticosteroid usage and survival in patients requiring mechanical ventilation for severe community‐acquired pneumonia. J Infect Chemother. 2011;17(4):449–455.
- , , , et al. Effect of high‐dose prednisolone on lung fluid in patients with non‐cardiogenic lung edema [in German]. Wien Klin Wochenschr. 1987;99:245–249.
- , , , et al. Early steroid therapy for respiratory failure. Arch Surg. 1985;120:536–540.
- , , , et al. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med. 2006;354(16):1671–1684.
- , , , et al. High‐dose corticosteroids in patients with the adult respiratory distress syndrome. N Engl J Med. 1987;317:1565–1570.
- . Antibiotics—past, present, and future. Med Clin North Am. 2006;90(6):1049–1076.
Community‐acquired pneumonia (CAP) is the most common lower respiratory tract infection in adults and a leading cause of infection‐related deaths in the United States.[1] According to a survey, pneumonia was the most common reason for hospital admissions through the emergency department in 2003.[2] CAP is associated with significant morbidity and mortality among those sick enough to require hospitalization. In a prospective study, hospital mortality rates ranged from 5% to 18% and length of stay from 9 to 23 days depending on patient location (intensive care unit [ICU] vs elsewhere) and severity of illness.[3]
Empirical evidence suggests that host inflammatory response contributes significantly to lung injury in pneumonia.[4] Studies have demonstrated reduction in the host inflammatory response as well as in mortality among animals with bacterial pneumonia when exposed to glucocorticoids.[5, 6] Furthermore, the efficacy of adjunctive steroid therapy in severe pneumonia caused by Pneumocystis jirovecii[7] and in pneumococcal meningitis[8, 9] is already established. However, due to equivocal, and at times conflicting, human clinical trial data on the impact of steroid therapy in CAP, the 2007 consensus guidelines (jointly published by the Infectious Diseases Society of America and American Thoracic Society) do not provide recommendations for or against use of steroids in CAP, except in the setting of hypotension secondary to adrenal insufficiency.[10]
In their meta‐analysis, Chen et al. analyzed data from 6 randomized clinical trials (RCTs) published between 1972 and 2007 (including 2 on pediatric patients) and concluded that adding steroids to current standard of care was not beneficial.[11] Earlier, Lamontagne et al.'s meta‐analysis included RCTs on hospitalized CAP patients as well as those on patients with acute lung injury (ALI) or acute respiratory distress syndrome (ARDS) from any cause.[12] They concluded that low‐dose corticosteroid therapy reduced all‐cause in‐hospital mortality in this mixed patient population (relative risk [RR]: 0.68 [95% confidence interval (CI): 0.49 to 0.96]). Recently, data from a number of additional RCTs have become available.[13, 14, 15, 16, 17] Therefore, an updated review of RCTs evaluating the role of adjunctive steroid therapy among adults hospitalized with CAP was warranted.
MATERIALS AND METHODS
We conducted this systematic review and meta‐analysis in accordance with the recommendations published in the Cochrane Handbook for Systematic Reviews of Interventions[18] and reported our findings according to the Preferred Reporting Items for Systematic Reviews and Meta‐analyses guidelines.[19] The overall quality of evidence was judged using the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) framework.[20]
Data Sources and Search Strategies
A comprehensive search of several databases including PubMed, Ovid MEDLINE In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE, Ovid EMBASE, Ovid Cochrane Database of Systematic Reviews, Ovid Cochrane Central Register of Controlled Trials, and Scopus was conducted. The time range for search started from each database's earliest inclusive dates up to July 2011. An experienced institutional librarian assisted with the design and conduct of our literature search. Controlled vocabulary, supplemented with keywords, was used to search for the topic: steroid therapy for community‐acquired pneumonia. We consulted expert colleagues to ensure the inclusion of all eligible reports and also checked the bibliographies of previously published systematic reviews.[12, 21]
Eligibility Criteria
Studies deemed eligible for inclusion were RCTs that met the following patients, intervention, control, outcomes (PICO) criteria: P, adults hospitalized with CAP; I, administration of systemic corticosteroids plus standard treatment; C, standard treatment without corticosteroids; O, primary outcome: hospital mortality; secondary outcomes, length of hospital stay, length of ICU stay and duration of mechanical ventilation. Under the P criterion we included RCTs that defined CAP as a lung infection (based on a reasonable combination of history, physical examination, imaging, and/or other investigative data, such as per the American Thoracic Society definition)[22] of presumed or proven bacterial etiology, in a patient who was not immunocompromised and had no exposure to a healthcare facility in the past 90 days.
Study Selection and Quality Assessment
Two reviewers independently performed study selection, data extraction, and quality assessment. Data were abstracted using standardized data collection instruments. Kappa statistic was calculated to assess the reviewers' level of agreement.
We perused full texts of all articles whose abstracts met selection criteria, performing an appraisal of their quality using the Cochrane risk‐of‐bias tool.[23] We also reviewed the baseline characteristics of patients in each study cohort.
Analysis
We estimated RR and weighted mean differences along with the respective 95% confidence intervals by pooling data using a random effects model.[24] Study heterogeneity was assessed using the I2 statistic, which estimates the percentage of variation that is not attributable to chance.[25] We performed a priori subgroup analyses based on the location (ICU vs non‐ICU) and mean age group of study participants (based on a cutoff of 50 years). A significant (P < 0.05) test of interaction would provide an explanation for any heterogeneity.[26] We also performed an a priori sensitivity analysis excluding any studies published before the year 2000 to exclude the impact of changing standards of care for inpatient management of CAP over time.
The original investigators were not contacted for purposes of obtaining raw data.
RESULTS
Eight RCTs, comprising 1119 subjects, were eventually chosen.[14]. Seven shortlisted studies were excluded due to methodological limitations, failure to fully meet PICO criteria, or gross insufficiency of descriptive data on subjects or methodology.[18, 31, 32, 33, 34, 35, 36] Figure 1 illustrates the study selection process.
Table 1 summarizes the baseline characteristics of patient populations from each study. Mean ages in 7 RCTs were between 60 years and 80 years. In Marik et al., the mean age of the intervention group was 31.7 years, whereas that of the control group was 40.6 years (P value not reported).[30] Three RCTs included ICU patients only,[17, 28, 30] whereas 4 only included general medical ward patients.[14, 15, 29, 31] Disease severity scores at admission were similar between the 2 groups in all RCTs except Sabry and Omar,[17] which was the only clinical trial to use a chest radiograph score. Only Sabry and Omar,[17] and Mikami et al.[29] excluded chronic obstructive pulmonary disease patients. Where possible, the serum C‐reactive protein (CRP) value on day one was subtracted from that on day eight to generate a one week delta CRP.
| Author, Year | Number of Patients | Gender: Males (% Age) | Age (y) | Steroids Used (Daily Dose and Duration) | COPD (% of Total) | Diabetes (% of Total) | Mean PaO2/FiO2 Ratio | Severity Score (Score: Mean) | Patients Already in ICU (% of Total) | One‐week Delta CRP (mg/dL) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | ||
| ||||||||||||||||||||
| McHardy 1972[31] | 126 | 40 | 86 | 45 | 50 | 62 | 59 | Prednisolone 20 mg, 7 d | 40 | 35 | Not reported | Not reported | Not reported | 0 | 0 | N/A | N/A | |||
| Marik 1993[30] | 30 | 14 | 16 | Not reported | 32 | 41 | Hydrocortisone 10 mg/kg 1 | Not reported | Not reported | 213 | 214 | APACHE II | 100 | 100 | N/A | N/A | ||||
| 11 | 14 | |||||||||||||||||||
| Confalonieri 2005[28] | 46 | 23 | 23 | 74 | 65 | 60 | 67 | Hydrocortisone 200 mg bolus, then 10 mg/h, 7 d | Not reported | Not reported | 141a | 178a | APACHE II | 100 | 100 | 37a | +5a | |||
| 17 | 18 | |||||||||||||||||||
| Mikami 2007[29] | 31 | 15 | 16 | 73.3 | 75 | 76 | 68 | Prednisolone 40 mg IV, 3 d | 0 | 0 | Not reported | PaO2 (FiO2 not reported) | PSI | 0 | 0 | N/A | N/A | |||
| 61 | 64 | 95 | 86 | |||||||||||||||||
| Snijders 2010[16] | 213 | 104 | 109 | 52.9 | 63.3 | 63 | 64 | Prednisolone 40 mg IV/PO, 7 d | 18 | 22 | 10 | 11 | Not reported | PSI class V (% of total) | 14.4 | 6.4 | N/A | N/A | ||
| 13 | 16 | |||||||||||||||||||
| Fernandez‐Serrano 2011[15] | 45 | 23 | 22 | 69.6 | 63.6 | 66 | 61 | Methylprednisone 200 mg IV; then 20 mg/6 h, 3 d; then 20 mg/12 h, 3 d; then 20 mg/d, 3 d | 17 | 9 | 9 | 18 | 200 | 257 | SAPS classes IV + V (% of total) | 0 | 0 | N/A | N/A | |
| 65 | 54 | |||||||||||||||||||
| Meijvis 2011[14] | 304 | 151 | 153 | 57 | 56 | 65 | 63 | Dexamethasone 5 mg/d, 4 d | 13 | 9 | 15 | 14 | Not reported | PSI class V (% of total) | 0 | 0 | N/A | N/A | ||
| 17 | 14 | |||||||||||||||||||
| Sabry 2011[17] | 80 | 40 | 40 | 30 | 28 | 62 | 63 | Hydrocortisone 200 mg IV, then 12.5 mg/h, 7 d | 0 | 0 | Not reported | 338a | 243a | Chest radiograph score | 100 | 100 | 38a | 23a | ||
| 1a | 3a | |||||||||||||||||||
The mean ICU length of stay was 12.7 days for the steroid group and 12.3 days for the control group. The mean hospital lengths of stay were 10.2 days and 13.6 days, respectively. Quality of the studies was moderate (see Supporting Information, Appendix I, in the online version of this article). Kappa score was >0.90.
Meta‐analysis
Figure 2 illustrates the results of our meta‐analyses. Although adjunctive steroid therapy had no effect on hospital mortality or ICU length of stay, it was associated with reduced hospital length of stay (RR: 1.21 days [95% CI: 2.12 to 0.29]). Of note, Mikami et al.[29] did not report mortality in their article, whereas in McHardy and Schonell,[31] using the factorial design, each of the 2 treatment groups were further subdivided into those patients who received 1 g of ampicillin and those who received 2 g of ampicillin (Figure 2A).
Analysis of other outcomes was limited by the fact that data were pooled from only a few studies. These included the need for and duration of mechanical ventilation, development of new ARDS and ICU admission rate, neither of which was associated with steroid therapy. However, steroid use was associated with lower incidence of delayed shock (ie, shock occurring after enrollment (RR: 0.12 [95% CI: 0.03 to 0.41]) and lower incidence of persistent chest x‐ray abnormalities at 1 week (RR: 0.13 [95% CI: 0.06 to 0.27]).
Subgroup and Sensitivity Analyses
Heterogeneity (I2 statistic) was <50% for all outcomes except ICU length of stay (74%). There were no significant interactions to suggest a subgroup effect based on older vs younger or ICU vs non‐ICU based patients (Table 2). In a priori sensitivity analysis that excluded McHardy and Schonell (published in 1972) and Marik et al. (published in 1993),[30] the results were not different from the main analysis.
| No. of Studies | Effect Size | LL | UL | P for Interaction (Difference Between Subgroups) | |
|---|---|---|---|---|---|
| |||||
| Mortality | |||||
| ICU | 3 | 0.27 | 0.08 | 0.83 | 0.06 |
| Non‐ICU | 4 | 0.96 | 0.45 | 2.05 | |
| Older | 7 | 0.75 | 0.40 | 1.38 | 0.56 |
| Young | 1 | 0.38 | 0.04 | 3.26 | |
| Need for mechanical ventilation | |||||
| ICU | 1 | 0.57 | 0.12 | 2.66 | 0.39 |
| Non‐ICU | 1 | 0.14 | 0.01 | 2.51 | |
| Older | 1 | 0.14 | 0.01 | 2.51 | 0.39 |
| Younger | 1 | 0.57 | 0.12 | 2.66 | |
| LOS | |||||
| ICU | 1 | 8.00 | 16.41 | 0.41 | 0.11 |
| Non‐ICU | 3 | 1.14 | 1.97 | 0.31 | |
| ICU LOS | |||||
| ICU | 2 | 3.91 | 11.44 | 3.62 | 0.45 |
| Non‐ICU | 2 | 0.81 | 8.98 | 10.60 | |
| Older | 3 | 2.26 | 9.95 | 5.43 | 0.65 |
| Younger | 1 | 0.30 | 3.91 | 3.31 | |
Quality of Evidence
Using the GRADE framework, the overall quality of evidence (confidence in the estimates) was judged to be moderate with the following main limitations: 1) methodological limitations among included studies (prognostic imbalance), 2) imprecision (small number of events and wide confidence intervals), and 3) inconsistency in the outcome ICU length of stay (as reflected by the I2 statistic).
Other Reported Outcomes
Four studies[15, 16, 29, 31] provided descriptive details of microbiologic data, whereas 1 study[16] provided analytical data on microbiology. In the latter, patients with Streptococcus pneumoniae infection (identified variably by sputum, pleural fluid, urine, or blood samples), had lower clinical cure rates in the steroid group at day 30 (P = 0.01) and higher numbers of late failures (defined as recurrence of signs and symptoms of pneumonia, P = 0.02).
Three[28, 30, 31] studies did not provide data on glycemic trends, whereas Fernandez‐Serrano et al., Mikami et al., and Snijders et al. reported that rates of hyperglycemia were not different across the 2 groups.[15, 16, 29] Meijvis et al.[14] reported more frequent hyperglycemia in the steroid group (44% vs 23%, P < 0.001) but no difference in the need for glucose‐lowering treatment (5% vs 3%, P = 0.57). Sabry and Omar[17] reported a higher incidence of hyperglycemia in the steroid group (no numerical data reported). Snijders et al.,[16] Meijvis et al.,[14] and Sabry and Omar[17] reported that the rates of super‐infection were not different between the 2 groups. No other adverse effects were consistently reported.
DISCUSSION
In this meta‐analysis of 8 RCTs, we found no significant association between steroid therapy and our primary outcome of interest (hospital mortality). However, length of hospital stay was shorter in the steroid group. These findings were not altered in various sensitivity and subgroup analyses. Although adverse effects of steroid therapy were not consistently reported, most of the RCTs reported that hyperglycemia was either no more common in the steroid group or did not require additional treatment.
Previous meta‐analyses have also concluded that adding corticosteroids to conventional therapy does not impact mortality among adults hospitalized with CAP.[12, 22] This may or may not be a consequence of inadequate statistical power. Although Lamontagne et al.[13] reported that low‐dose corticosteroid therapy (2 mg/kg/day or less of methylprednisolone or equivalent) was associated with reduced hospital mortality (RR: 0.68 [95% CI: 0.49 to 0.96]), this result was obtained by pooling data from 5 RCTs on adults hospitalized with CAP and 4 on adults with ALI/ARDS from any cause. In a subgroup analysis of RCTs conducted only on CAP patients, no impact on mortality was found. Of note, all RCTs involving CAP patients had used low‐dose steroids; the 3 RCTs using high‐dose steroids were carried out on ALI/ARDS patients.[36, 37, 38] Similarly, all RCTs in our meta‐analysis were also characterized by steroid doses under 2 mg/kg/day of methylprednisolone or equivalent.
Our study is the first to demonstrate decreased length of hospital stay in this patient population. Importantly, each of the 5 studies that reported this outcome (including 3 relatively recent RCTs) showed the same trend. However, it is not inconceivable that steroid use led to a quicker decline in cytokine levels resulting in an earlier resolution of fever and hence earlier discharge without a faster cure per se. The two studies whose data permitted calculation of delta CRP also demonstrated a faster CRP decline in the steroid group (Table 1).
Our analysis also suggested reduced incidence of delayed shock. However, these data were pooled from only 2 RCTs,[17, 28] and each of them used hydrocortisone, whose direct mineralocorticoid effect is an obvious confounder. Similarly, according to data pooled from 2 RCTs, steroid use was associated with fewer cases of persistent chest x‐ray abnormalities by day 8. Of note, although calculation of the I2 statistic was not possible because of too few studies, visual inspection of the forest plots suggested low levels of heterogeneity.
It is plausible that the impact of adjunctive steroids in CAP may vary based on the causative pathogen. This pathogen‐specific association has been observed in patients with bacterial meningitis, where most of the benefit is seemingly limited to pneumococcal meningitis.[9, 10] Unfortunately, as demonstrated by Snijders et al.,[16] establishing microbiologic etiology in CAP can be difficult, and most patients are treated empirically.
Our analysis showed no difference in duration of ventilation among patients who required ventilatory support on admission. However, only 2 studies reported this outcome.[17, 28] Second, in Confalonieri et al.,[28] the steroid group had a more severe baseline inflammatory response as illustrated by higher serum CRP levels (P = 0.04). Moreover, while mechanical ventilation was defined as either invasive or noninvasive ventilation, the steroid group had a higher number of patients who required noninvasive ventilation (P = 0.03), thus introducing selection bias. This study had additional areas of concern too, including a mortality of 0 among its 46 ICU patients, in contrast to established mortality rates of up to around 20%.[4] Unlike this study, Sabry and Omar[17] reported that none of their patients was on noninvasive ventilation. It may be pertinent to compare our findings with those of Steinberg et al.,[40] who studied patients with ARDS (pneumonia being the most common cause) who received methylprednisolone. This group had an early increase in ventilator‐free days, but that effect became less pronounced (though still significant) when the study end point was prolonged from 30 to 90 days.[41]
The 2 studies that were published before 2000 (McHardy and Schonell,[31] and Marik et al.[30]) were excluded in our a priori sensitivity analysis. A number of considerations led to this decision. First, standards of care for inpatient management of pneumoniaincluding pharmacologic therapies and ventilation strategieshave changed considerably over time. For instance, newer generation macrolides became available for clinical use in the early 1990s and meropenem in 1996.[41] Therefore, it would be hard to assume constancy of effect from that time period. Furthermore, the study by McHardy and Schonell[31] suffered from significant differences in the baseline characteristics of its 2 arms. There was incomplete randomization; patients with diabetes were excluded from only the steroid arm. Another issue with Marik et al.[30] was the considerably younger age of participants compared to other studies (Table 1).
Limitations
In spite of our relatively stringent selection criteria and a number of subgroup and sensitivity analyses, the overall quality of evidence was only moderate (Table 2). Key issues with the findings reported by Confalonieri et al.,[28] McHardy and Schonell,[31] and Marik et al.[30] were discussed earlier. Baseline severity of illness, patient comorbidities, and length of follow‐up were variable both within and across various studies. Another major limitation was that the intervention of interest (ie, steroid therapy) was not uniformly applied as the regimens varied considerably even though all regimens fit the designation of low‐dose steroids as previously noted (Table 1).
In conclusion, although evidence suggests that adjunctive steroid therapy is associated with reduced hospital length of stay, the data are not strong enough to recommend routine use of steroids among all adults hospitalized with CAP. However, considering that there was no increase in mortality or hospital length of stay with steroid use, it is reasonable to continue steroids if warranted for treatment of underlying comorbid conditions.
Due to the aforementioned limitations in RCTs published to date, we believe that additional studies that are more robustly designed and sufficiently powered to detect differences in key outcomes (including mortality) are warranted. Investigators should ensure appropriate randomization of groups, taking into account severity of illness, comorbid conditions and prior use of steroid therapy. Standardizing the intervention (including dose and duration of steroid therapy and time to first antibiotic dose) would be essential. Concurrent measurement of inflammatory markers such as delta CRP would be useful too. Finally, accurate measurement of all secondary outcomes of interest, including adverse effects and duration of both invasive and noninvasive mechanical ventilation, would be important to accurately study the benefit of steroids among the most likely beneficiaries: those patients who are the sickest.
Acknowledgments
The authors gratefully acknowledge the assistance of Dr. Jon Ebbert (Department of Medicine, Mayo Clinic, Rochester, MN) with proofreading the manuscript and providing thoughtful editorial suggestions.
Disclosures
The authors report no conflicts of interest.
Community‐acquired pneumonia (CAP) is the most common lower respiratory tract infection in adults and a leading cause of infection‐related deaths in the United States.[1] According to a survey, pneumonia was the most common reason for hospital admissions through the emergency department in 2003.[2] CAP is associated with significant morbidity and mortality among those sick enough to require hospitalization. In a prospective study, hospital mortality rates ranged from 5% to 18% and length of stay from 9 to 23 days depending on patient location (intensive care unit [ICU] vs elsewhere) and severity of illness.[3]
Empirical evidence suggests that host inflammatory response contributes significantly to lung injury in pneumonia.[4] Studies have demonstrated reduction in the host inflammatory response as well as in mortality among animals with bacterial pneumonia when exposed to glucocorticoids.[5, 6] Furthermore, the efficacy of adjunctive steroid therapy in severe pneumonia caused by Pneumocystis jirovecii[7] and in pneumococcal meningitis[8, 9] is already established. However, due to equivocal, and at times conflicting, human clinical trial data on the impact of steroid therapy in CAP, the 2007 consensus guidelines (jointly published by the Infectious Diseases Society of America and American Thoracic Society) do not provide recommendations for or against use of steroids in CAP, except in the setting of hypotension secondary to adrenal insufficiency.[10]
In their meta‐analysis, Chen et al. analyzed data from 6 randomized clinical trials (RCTs) published between 1972 and 2007 (including 2 on pediatric patients) and concluded that adding steroids to current standard of care was not beneficial.[11] Earlier, Lamontagne et al.'s meta‐analysis included RCTs on hospitalized CAP patients as well as those on patients with acute lung injury (ALI) or acute respiratory distress syndrome (ARDS) from any cause.[12] They concluded that low‐dose corticosteroid therapy reduced all‐cause in‐hospital mortality in this mixed patient population (relative risk [RR]: 0.68 [95% confidence interval (CI): 0.49 to 0.96]). Recently, data from a number of additional RCTs have become available.[13, 14, 15, 16, 17] Therefore, an updated review of RCTs evaluating the role of adjunctive steroid therapy among adults hospitalized with CAP was warranted.
MATERIALS AND METHODS
We conducted this systematic review and meta‐analysis in accordance with the recommendations published in the Cochrane Handbook for Systematic Reviews of Interventions[18] and reported our findings according to the Preferred Reporting Items for Systematic Reviews and Meta‐analyses guidelines.[19] The overall quality of evidence was judged using the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) framework.[20]
Data Sources and Search Strategies
A comprehensive search of several databases including PubMed, Ovid MEDLINE In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE, Ovid EMBASE, Ovid Cochrane Database of Systematic Reviews, Ovid Cochrane Central Register of Controlled Trials, and Scopus was conducted. The time range for search started from each database's earliest inclusive dates up to July 2011. An experienced institutional librarian assisted with the design and conduct of our literature search. Controlled vocabulary, supplemented with keywords, was used to search for the topic: steroid therapy for community‐acquired pneumonia. We consulted expert colleagues to ensure the inclusion of all eligible reports and also checked the bibliographies of previously published systematic reviews.[12, 21]
Eligibility Criteria
Studies deemed eligible for inclusion were RCTs that met the following patients, intervention, control, outcomes (PICO) criteria: P, adults hospitalized with CAP; I, administration of systemic corticosteroids plus standard treatment; C, standard treatment without corticosteroids; O, primary outcome: hospital mortality; secondary outcomes, length of hospital stay, length of ICU stay and duration of mechanical ventilation. Under the P criterion we included RCTs that defined CAP as a lung infection (based on a reasonable combination of history, physical examination, imaging, and/or other investigative data, such as per the American Thoracic Society definition)[22] of presumed or proven bacterial etiology, in a patient who was not immunocompromised and had no exposure to a healthcare facility in the past 90 days.
Study Selection and Quality Assessment
Two reviewers independently performed study selection, data extraction, and quality assessment. Data were abstracted using standardized data collection instruments. Kappa statistic was calculated to assess the reviewers' level of agreement.
We perused full texts of all articles whose abstracts met selection criteria, performing an appraisal of their quality using the Cochrane risk‐of‐bias tool.[23] We also reviewed the baseline characteristics of patients in each study cohort.
Analysis
We estimated RR and weighted mean differences along with the respective 95% confidence intervals by pooling data using a random effects model.[24] Study heterogeneity was assessed using the I2 statistic, which estimates the percentage of variation that is not attributable to chance.[25] We performed a priori subgroup analyses based on the location (ICU vs non‐ICU) and mean age group of study participants (based on a cutoff of 50 years). A significant (P < 0.05) test of interaction would provide an explanation for any heterogeneity.[26] We also performed an a priori sensitivity analysis excluding any studies published before the year 2000 to exclude the impact of changing standards of care for inpatient management of CAP over time.
The original investigators were not contacted for purposes of obtaining raw data.
RESULTS
Eight RCTs, comprising 1119 subjects, were eventually chosen.[14]. Seven shortlisted studies were excluded due to methodological limitations, failure to fully meet PICO criteria, or gross insufficiency of descriptive data on subjects or methodology.[18, 31, 32, 33, 34, 35, 36] Figure 1 illustrates the study selection process.
Table 1 summarizes the baseline characteristics of patient populations from each study. Mean ages in 7 RCTs were between 60 years and 80 years. In Marik et al., the mean age of the intervention group was 31.7 years, whereas that of the control group was 40.6 years (P value not reported).[30] Three RCTs included ICU patients only,[17, 28, 30] whereas 4 only included general medical ward patients.[14, 15, 29, 31] Disease severity scores at admission were similar between the 2 groups in all RCTs except Sabry and Omar,[17] which was the only clinical trial to use a chest radiograph score. Only Sabry and Omar,[17] and Mikami et al.[29] excluded chronic obstructive pulmonary disease patients. Where possible, the serum C‐reactive protein (CRP) value on day one was subtracted from that on day eight to generate a one week delta CRP.
| Author, Year | Number of Patients | Gender: Males (% Age) | Age (y) | Steroids Used (Daily Dose and Duration) | COPD (% of Total) | Diabetes (% of Total) | Mean PaO2/FiO2 Ratio | Severity Score (Score: Mean) | Patients Already in ICU (% of Total) | One‐week Delta CRP (mg/dL) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | Steroid | Control | ||
| ||||||||||||||||||||
| McHardy 1972[31] | 126 | 40 | 86 | 45 | 50 | 62 | 59 | Prednisolone 20 mg, 7 d | 40 | 35 | Not reported | Not reported | Not reported | 0 | 0 | N/A | N/A | |||
| Marik 1993[30] | 30 | 14 | 16 | Not reported | 32 | 41 | Hydrocortisone 10 mg/kg 1 | Not reported | Not reported | 213 | 214 | APACHE II | 100 | 100 | N/A | N/A | ||||
| 11 | 14 | |||||||||||||||||||
| Confalonieri 2005[28] | 46 | 23 | 23 | 74 | 65 | 60 | 67 | Hydrocortisone 200 mg bolus, then 10 mg/h, 7 d | Not reported | Not reported | 141a | 178a | APACHE II | 100 | 100 | 37a | +5a | |||
| 17 | 18 | |||||||||||||||||||
| Mikami 2007[29] | 31 | 15 | 16 | 73.3 | 75 | 76 | 68 | Prednisolone 40 mg IV, 3 d | 0 | 0 | Not reported | PaO2 (FiO2 not reported) | PSI | 0 | 0 | N/A | N/A | |||
| 61 | 64 | 95 | 86 | |||||||||||||||||
| Snijders 2010[16] | 213 | 104 | 109 | 52.9 | 63.3 | 63 | 64 | Prednisolone 40 mg IV/PO, 7 d | 18 | 22 | 10 | 11 | Not reported | PSI class V (% of total) | 14.4 | 6.4 | N/A | N/A | ||
| 13 | 16 | |||||||||||||||||||
| Fernandez‐Serrano 2011[15] | 45 | 23 | 22 | 69.6 | 63.6 | 66 | 61 | Methylprednisone 200 mg IV; then 20 mg/6 h, 3 d; then 20 mg/12 h, 3 d; then 20 mg/d, 3 d | 17 | 9 | 9 | 18 | 200 | 257 | SAPS classes IV + V (% of total) | 0 | 0 | N/A | N/A | |
| 65 | 54 | |||||||||||||||||||
| Meijvis 2011[14] | 304 | 151 | 153 | 57 | 56 | 65 | 63 | Dexamethasone 5 mg/d, 4 d | 13 | 9 | 15 | 14 | Not reported | PSI class V (% of total) | 0 | 0 | N/A | N/A | ||
| 17 | 14 | |||||||||||||||||||
| Sabry 2011[17] | 80 | 40 | 40 | 30 | 28 | 62 | 63 | Hydrocortisone 200 mg IV, then 12.5 mg/h, 7 d | 0 | 0 | Not reported | 338a | 243a | Chest radiograph score | 100 | 100 | 38a | 23a | ||
| 1a | 3a | |||||||||||||||||||
The mean ICU length of stay was 12.7 days for the steroid group and 12.3 days for the control group. The mean hospital lengths of stay were 10.2 days and 13.6 days, respectively. Quality of the studies was moderate (see Supporting Information, Appendix I, in the online version of this article). Kappa score was >0.90.
Meta‐analysis
Figure 2 illustrates the results of our meta‐analyses. Although adjunctive steroid therapy had no effect on hospital mortality or ICU length of stay, it was associated with reduced hospital length of stay (RR: 1.21 days [95% CI: 2.12 to 0.29]). Of note, Mikami et al.[29] did not report mortality in their article, whereas in McHardy and Schonell,[31] using the factorial design, each of the 2 treatment groups were further subdivided into those patients who received 1 g of ampicillin and those who received 2 g of ampicillin (Figure 2A).
Analysis of other outcomes was limited by the fact that data were pooled from only a few studies. These included the need for and duration of mechanical ventilation, development of new ARDS and ICU admission rate, neither of which was associated with steroid therapy. However, steroid use was associated with lower incidence of delayed shock (ie, shock occurring after enrollment (RR: 0.12 [95% CI: 0.03 to 0.41]) and lower incidence of persistent chest x‐ray abnormalities at 1 week (RR: 0.13 [95% CI: 0.06 to 0.27]).
Subgroup and Sensitivity Analyses
Heterogeneity (I2 statistic) was <50% for all outcomes except ICU length of stay (74%). There were no significant interactions to suggest a subgroup effect based on older vs younger or ICU vs non‐ICU based patients (Table 2). In a priori sensitivity analysis that excluded McHardy and Schonell (published in 1972) and Marik et al. (published in 1993),[30] the results were not different from the main analysis.
| No. of Studies | Effect Size | LL | UL | P for Interaction (Difference Between Subgroups) | |
|---|---|---|---|---|---|
| |||||
| Mortality | |||||
| ICU | 3 | 0.27 | 0.08 | 0.83 | 0.06 |
| Non‐ICU | 4 | 0.96 | 0.45 | 2.05 | |
| Older | 7 | 0.75 | 0.40 | 1.38 | 0.56 |
| Young | 1 | 0.38 | 0.04 | 3.26 | |
| Need for mechanical ventilation | |||||
| ICU | 1 | 0.57 | 0.12 | 2.66 | 0.39 |
| Non‐ICU | 1 | 0.14 | 0.01 | 2.51 | |
| Older | 1 | 0.14 | 0.01 | 2.51 | 0.39 |
| Younger | 1 | 0.57 | 0.12 | 2.66 | |
| LOS | |||||
| ICU | 1 | 8.00 | 16.41 | 0.41 | 0.11 |
| Non‐ICU | 3 | 1.14 | 1.97 | 0.31 | |
| ICU LOS | |||||
| ICU | 2 | 3.91 | 11.44 | 3.62 | 0.45 |
| Non‐ICU | 2 | 0.81 | 8.98 | 10.60 | |
| Older | 3 | 2.26 | 9.95 | 5.43 | 0.65 |
| Younger | 1 | 0.30 | 3.91 | 3.31 | |
Quality of Evidence
Using the GRADE framework, the overall quality of evidence (confidence in the estimates) was judged to be moderate with the following main limitations: 1) methodological limitations among included studies (prognostic imbalance), 2) imprecision (small number of events and wide confidence intervals), and 3) inconsistency in the outcome ICU length of stay (as reflected by the I2 statistic).
Other Reported Outcomes
Four studies[15, 16, 29, 31] provided descriptive details of microbiologic data, whereas 1 study[16] provided analytical data on microbiology. In the latter, patients with Streptococcus pneumoniae infection (identified variably by sputum, pleural fluid, urine, or blood samples), had lower clinical cure rates in the steroid group at day 30 (P = 0.01) and higher numbers of late failures (defined as recurrence of signs and symptoms of pneumonia, P = 0.02).
Three[28, 30, 31] studies did not provide data on glycemic trends, whereas Fernandez‐Serrano et al., Mikami et al., and Snijders et al. reported that rates of hyperglycemia were not different across the 2 groups.[15, 16, 29] Meijvis et al.[14] reported more frequent hyperglycemia in the steroid group (44% vs 23%, P < 0.001) but no difference in the need for glucose‐lowering treatment (5% vs 3%, P = 0.57). Sabry and Omar[17] reported a higher incidence of hyperglycemia in the steroid group (no numerical data reported). Snijders et al.,[16] Meijvis et al.,[14] and Sabry and Omar[17] reported that the rates of super‐infection were not different between the 2 groups. No other adverse effects were consistently reported.
DISCUSSION
In this meta‐analysis of 8 RCTs, we found no significant association between steroid therapy and our primary outcome of interest (hospital mortality). However, length of hospital stay was shorter in the steroid group. These findings were not altered in various sensitivity and subgroup analyses. Although adverse effects of steroid therapy were not consistently reported, most of the RCTs reported that hyperglycemia was either no more common in the steroid group or did not require additional treatment.
Previous meta‐analyses have also concluded that adding corticosteroids to conventional therapy does not impact mortality among adults hospitalized with CAP.[12, 22] This may or may not be a consequence of inadequate statistical power. Although Lamontagne et al.[13] reported that low‐dose corticosteroid therapy (2 mg/kg/day or less of methylprednisolone or equivalent) was associated with reduced hospital mortality (RR: 0.68 [95% CI: 0.49 to 0.96]), this result was obtained by pooling data from 5 RCTs on adults hospitalized with CAP and 4 on adults with ALI/ARDS from any cause. In a subgroup analysis of RCTs conducted only on CAP patients, no impact on mortality was found. Of note, all RCTs involving CAP patients had used low‐dose steroids; the 3 RCTs using high‐dose steroids were carried out on ALI/ARDS patients.[36, 37, 38] Similarly, all RCTs in our meta‐analysis were also characterized by steroid doses under 2 mg/kg/day of methylprednisolone or equivalent.
Our study is the first to demonstrate decreased length of hospital stay in this patient population. Importantly, each of the 5 studies that reported this outcome (including 3 relatively recent RCTs) showed the same trend. However, it is not inconceivable that steroid use led to a quicker decline in cytokine levels resulting in an earlier resolution of fever and hence earlier discharge without a faster cure per se. The two studies whose data permitted calculation of delta CRP also demonstrated a faster CRP decline in the steroid group (Table 1).
Our analysis also suggested reduced incidence of delayed shock. However, these data were pooled from only 2 RCTs,[17, 28] and each of them used hydrocortisone, whose direct mineralocorticoid effect is an obvious confounder. Similarly, according to data pooled from 2 RCTs, steroid use was associated with fewer cases of persistent chest x‐ray abnormalities by day 8. Of note, although calculation of the I2 statistic was not possible because of too few studies, visual inspection of the forest plots suggested low levels of heterogeneity.
It is plausible that the impact of adjunctive steroids in CAP may vary based on the causative pathogen. This pathogen‐specific association has been observed in patients with bacterial meningitis, where most of the benefit is seemingly limited to pneumococcal meningitis.[9, 10] Unfortunately, as demonstrated by Snijders et al.,[16] establishing microbiologic etiology in CAP can be difficult, and most patients are treated empirically.
Our analysis showed no difference in duration of ventilation among patients who required ventilatory support on admission. However, only 2 studies reported this outcome.[17, 28] Second, in Confalonieri et al.,[28] the steroid group had a more severe baseline inflammatory response as illustrated by higher serum CRP levels (P = 0.04). Moreover, while mechanical ventilation was defined as either invasive or noninvasive ventilation, the steroid group had a higher number of patients who required noninvasive ventilation (P = 0.03), thus introducing selection bias. This study had additional areas of concern too, including a mortality of 0 among its 46 ICU patients, in contrast to established mortality rates of up to around 20%.[4] Unlike this study, Sabry and Omar[17] reported that none of their patients was on noninvasive ventilation. It may be pertinent to compare our findings with those of Steinberg et al.,[40] who studied patients with ARDS (pneumonia being the most common cause) who received methylprednisolone. This group had an early increase in ventilator‐free days, but that effect became less pronounced (though still significant) when the study end point was prolonged from 30 to 90 days.[41]
The 2 studies that were published before 2000 (McHardy and Schonell,[31] and Marik et al.[30]) were excluded in our a priori sensitivity analysis. A number of considerations led to this decision. First, standards of care for inpatient management of pneumoniaincluding pharmacologic therapies and ventilation strategieshave changed considerably over time. For instance, newer generation macrolides became available for clinical use in the early 1990s and meropenem in 1996.[41] Therefore, it would be hard to assume constancy of effect from that time period. Furthermore, the study by McHardy and Schonell[31] suffered from significant differences in the baseline characteristics of its 2 arms. There was incomplete randomization; patients with diabetes were excluded from only the steroid arm. Another issue with Marik et al.[30] was the considerably younger age of participants compared to other studies (Table 1).
Limitations
In spite of our relatively stringent selection criteria and a number of subgroup and sensitivity analyses, the overall quality of evidence was only moderate (Table 2). Key issues with the findings reported by Confalonieri et al.,[28] McHardy and Schonell,[31] and Marik et al.[30] were discussed earlier. Baseline severity of illness, patient comorbidities, and length of follow‐up were variable both within and across various studies. Another major limitation was that the intervention of interest (ie, steroid therapy) was not uniformly applied as the regimens varied considerably even though all regimens fit the designation of low‐dose steroids as previously noted (Table 1).
In conclusion, although evidence suggests that adjunctive steroid therapy is associated with reduced hospital length of stay, the data are not strong enough to recommend routine use of steroids among all adults hospitalized with CAP. However, considering that there was no increase in mortality or hospital length of stay with steroid use, it is reasonable to continue steroids if warranted for treatment of underlying comorbid conditions.
Due to the aforementioned limitations in RCTs published to date, we believe that additional studies that are more robustly designed and sufficiently powered to detect differences in key outcomes (including mortality) are warranted. Investigators should ensure appropriate randomization of groups, taking into account severity of illness, comorbid conditions and prior use of steroid therapy. Standardizing the intervention (including dose and duration of steroid therapy and time to first antibiotic dose) would be essential. Concurrent measurement of inflammatory markers such as delta CRP would be useful too. Finally, accurate measurement of all secondary outcomes of interest, including adverse effects and duration of both invasive and noninvasive mechanical ventilation, would be important to accurately study the benefit of steroids among the most likely beneficiaries: those patients who are the sickest.
Acknowledgments
The authors gratefully acknowledge the assistance of Dr. Jon Ebbert (Department of Medicine, Mayo Clinic, Rochester, MN) with proofreading the manuscript and providing thoughtful editorial suggestions.
Disclosures
The authors report no conflicts of interest.
- Centers for Disease Control and Prevention 2008. CDC/NCHS, National Vital Statistics System. Leading causes of Death. Available at: http://www.cdc.gov/nchs/nvss/mortality_tables.htm. Accessed August 14,2011.
- , . Reasons for being admitted to the hospital through the emergency department, 2003. HCUP Statistical Brief #2. February 2006. Agency for Healthcare Research and Quality, Rockville, MD. Available at: http://www.hcup‐us.ahrq.gov/reports/statbriefs/sb2.pdf. Accessed August 14,2011.
- , , , et al. Processes of care and outcomes for community‐acquired pneumonia. Am J Med. 2011;124(12):1175.e9–17.
- , , , , , . Cytokine kinetics and other host factors in response to pneumococcal pulmonary infection in mice. Infect Immun. 1998;66(3):912–922.
- , , , et al. Effects of glucocorticoids in ventilated piglets with severe pneumonia. Eur Respir J. 2008;32(4):1037–1046.
- , , , et al. Risk of death does not alter the efficacy of hydrocortisone therapy in a mouse E. coli pneumonia model: risk and corticosteroids in sepsis. Intensive Care Med. 2008;34(3):568–577.
- , , , . Adjunctive corticosteroids for Pneumocystis jiroveci pneumonia in patients with HIV‐infection. Cochrane Database Syst Rev. 2006;(3):CD006150.
- , . Dexamethasone in adults with bacterial meningitis. N Engl J Med. 2002;347(20):1549–1556.
- , , , . Steroids in adults with acute bacterial meningitis: a systematic review. Lancet Infect Dis. 2004;4(3):139–143.
- , , , et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community‐acquired pneumonia in adults. Clin Infect Dis. 2007;44(suppl 2):S27–S72.
- , , , . Corticosteroids for pneumonia. Cochrane Database Syst Rev. 2011;(3):CD007720.
- , , , , , . Corticosteroid therapy for acute lung injury, acute respiratory distress syndrome, and severe pneumonia: a meta‐analysis of randomized controlled trials. J Crit Care. 2010;25(3):420–435.
- , , , et al. Dexamethasone and length of hospital stay in patients with community‐acquired pneumonia: a randomised, double‐blind, placebo‐controlled trial. Lancet. 2011;377(9782):2023–2030.
- , , , et al. Effect of corticosteroids on the clinical course of community‐acquired pneumonia: a randomized controlled trial. Crit Care. 2011;15(2):R96.
- , , , et al. Efficacy of corticosteroids in community‐acquired pneumonia: a randomized double‐blinded clinical trial. Am J Respir Crit Care Med. 2010;181:975–978.
- , . Corticosteroids and ICU course of community acquired pneumonia in Egyptian settings. Pharmacol Pharm. 2011;2(2):73–81.
- , , , , . Corticosteroid treatment in severe community‐acquired pneumonia: duration of treatment affects control of systemic inflammation and clinical improvement. Intensive Care Med. 2011;37(9):1553–1554.
- Higgins JPT, Green S, eds. Cochrane Handbook for Systematic Reviews of Interventions. West Sussex, UK:Wiley‐Blackwell;2008.
- , , , ;PRISMA Group. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. J Clin Epidemiol. 2009;62:1006–1012.
- , , , et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401–406.
- , , , , , . The role of corticosteroids in severe community‐acquired pneumonia: a systematic review. Crit Care. 2008;12(3):R76.
- , , , et al. Severe community‐acquired pneumonia: assessment of severity criteria. Am J Respir Crit Care Med. 1998;158(4):1102–1108.
- , .Assessing risk of bias in included studies. In: Higgins JPT, Green S eds. Cochrane Handbook for Systematic Reviews of Interventions. Chichester, UK:John Wiley 2009.
- , . Meta‐analysis in clinical trials. Control Clin Trials. 1986;7(3):177–188.
- , , , . Measuring inconsistency in meta‐analyses. BMJ. 2003;327(7414):557–560.
- , . Interaction revisited: the difference between two estimates. BMJ. 2003;326(7382):219.
- , , , et al. Hydrocortisone infusion for severe community‐acquired pneumonia: a preliminary randomized study. Am J Respir Crit Care Med. 2005;171(3):242–248.
- , , , , , . Efficacy of corticosteroids in the treatment of community‐acquired pneumonia requiring hospitalisation. Lung. 2007;185(5):249–255.
- , , , , , . Hydrocortisone and tumour necrosis factor in severe community acquired pneumonia. A randomised controlled study. Chest. 1993;104(2):389–392.
- , . Ampicillin dosage and use of prednisolone in treatment of pneumonia: co‐operative controlled trial. Br Med J. 1972;4:569–573.
- , , , et al. Impact of systemic corticosteroids on the clinical course and outcomes of patients with severe community‐acquired pneumonia: a cohort study. J Crit Care. 2011;26(2):193–200.
- , , , . Factors of importance for the long term prognosis after hospital treated pneumonia. Thorax. 1993;48(8):785–789.
- , , , . Assessment of mortality after long‐term follow‐up of patients with community‐acquired pneumonia. Clin Infect Dis. 2003;37(12):1617–1624.
- , , , , . Community‐acquired pneumonia on the intensive care unit: secondary analysis of 17,869 cases in the ICNARC Case Mix Programme Database. Crit Care. 2006;10(suppl 2):S1.
- , , , , , . Effects of systemic steroids in patients with severe community‐acquired pneumonia. Eur Respir J. 2007;30(5):951–956.
- , , , . Analysis of systemic corticosteroid usage and survival in patients requiring mechanical ventilation for severe community‐acquired pneumonia. J Infect Chemother. 2011;17(4):449–455.
- , , , et al. Effect of high‐dose prednisolone on lung fluid in patients with non‐cardiogenic lung edema [in German]. Wien Klin Wochenschr. 1987;99:245–249.
- , , , et al. Early steroid therapy for respiratory failure. Arch Surg. 1985;120:536–540.
- , , , et al. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med. 2006;354(16):1671–1684.
- , , , et al. High‐dose corticosteroids in patients with the adult respiratory distress syndrome. N Engl J Med. 1987;317:1565–1570.
- . Antibiotics—past, present, and future. Med Clin North Am. 2006;90(6):1049–1076.
- Centers for Disease Control and Prevention 2008. CDC/NCHS, National Vital Statistics System. Leading causes of Death. Available at: http://www.cdc.gov/nchs/nvss/mortality_tables.htm. Accessed August 14,2011.
- , . Reasons for being admitted to the hospital through the emergency department, 2003. HCUP Statistical Brief #2. February 2006. Agency for Healthcare Research and Quality, Rockville, MD. Available at: http://www.hcup‐us.ahrq.gov/reports/statbriefs/sb2.pdf. Accessed August 14,2011.
- , , , et al. Processes of care and outcomes for community‐acquired pneumonia. Am J Med. 2011;124(12):1175.e9–17.
- , , , , , . Cytokine kinetics and other host factors in response to pneumococcal pulmonary infection in mice. Infect Immun. 1998;66(3):912–922.
- , , , et al. Effects of glucocorticoids in ventilated piglets with severe pneumonia. Eur Respir J. 2008;32(4):1037–1046.
- , , , et al. Risk of death does not alter the efficacy of hydrocortisone therapy in a mouse E. coli pneumonia model: risk and corticosteroids in sepsis. Intensive Care Med. 2008;34(3):568–577.
- , , , . Adjunctive corticosteroids for Pneumocystis jiroveci pneumonia in patients with HIV‐infection. Cochrane Database Syst Rev. 2006;(3):CD006150.
- , . Dexamethasone in adults with bacterial meningitis. N Engl J Med. 2002;347(20):1549–1556.
- , , , . Steroids in adults with acute bacterial meningitis: a systematic review. Lancet Infect Dis. 2004;4(3):139–143.
- , , , et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community‐acquired pneumonia in adults. Clin Infect Dis. 2007;44(suppl 2):S27–S72.
- , , , . Corticosteroids for pneumonia. Cochrane Database Syst Rev. 2011;(3):CD007720.
- , , , , , . Corticosteroid therapy for acute lung injury, acute respiratory distress syndrome, and severe pneumonia: a meta‐analysis of randomized controlled trials. J Crit Care. 2010;25(3):420–435.
- , , , et al. Dexamethasone and length of hospital stay in patients with community‐acquired pneumonia: a randomised, double‐blind, placebo‐controlled trial. Lancet. 2011;377(9782):2023–2030.
- , , , et al. Effect of corticosteroids on the clinical course of community‐acquired pneumonia: a randomized controlled trial. Crit Care. 2011;15(2):R96.
- , , , et al. Efficacy of corticosteroids in community‐acquired pneumonia: a randomized double‐blinded clinical trial. Am J Respir Crit Care Med. 2010;181:975–978.
- , . Corticosteroids and ICU course of community acquired pneumonia in Egyptian settings. Pharmacol Pharm. 2011;2(2):73–81.
- , , , , . Corticosteroid treatment in severe community‐acquired pneumonia: duration of treatment affects control of systemic inflammation and clinical improvement. Intensive Care Med. 2011;37(9):1553–1554.
- Higgins JPT, Green S, eds. Cochrane Handbook for Systematic Reviews of Interventions. West Sussex, UK:Wiley‐Blackwell;2008.
- , , , ;PRISMA Group. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. J Clin Epidemiol. 2009;62:1006–1012.
- , , , et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401–406.
- , , , , , . The role of corticosteroids in severe community‐acquired pneumonia: a systematic review. Crit Care. 2008;12(3):R76.
- , , , et al. Severe community‐acquired pneumonia: assessment of severity criteria. Am J Respir Crit Care Med. 1998;158(4):1102–1108.
- , .Assessing risk of bias in included studies. In: Higgins JPT, Green S eds. Cochrane Handbook for Systematic Reviews of Interventions. Chichester, UK:John Wiley 2009.
- , . Meta‐analysis in clinical trials. Control Clin Trials. 1986;7(3):177–188.
- , , , . Measuring inconsistency in meta‐analyses. BMJ. 2003;327(7414):557–560.
- , . Interaction revisited: the difference between two estimates. BMJ. 2003;326(7382):219.
- , , , et al. Hydrocortisone infusion for severe community‐acquired pneumonia: a preliminary randomized study. Am J Respir Crit Care Med. 2005;171(3):242–248.
- , , , , , . Efficacy of corticosteroids in the treatment of community‐acquired pneumonia requiring hospitalisation. Lung. 2007;185(5):249–255.
- , , , , , . Hydrocortisone and tumour necrosis factor in severe community acquired pneumonia. A randomised controlled study. Chest. 1993;104(2):389–392.
- , . Ampicillin dosage and use of prednisolone in treatment of pneumonia: co‐operative controlled trial. Br Med J. 1972;4:569–573.
- , , , et al. Impact of systemic corticosteroids on the clinical course and outcomes of patients with severe community‐acquired pneumonia: a cohort study. J Crit Care. 2011;26(2):193–200.
- , , , . Factors of importance for the long term prognosis after hospital treated pneumonia. Thorax. 1993;48(8):785–789.
- , , , . Assessment of mortality after long‐term follow‐up of patients with community‐acquired pneumonia. Clin Infect Dis. 2003;37(12):1617–1624.
- , , , , . Community‐acquired pneumonia on the intensive care unit: secondary analysis of 17,869 cases in the ICNARC Case Mix Programme Database. Crit Care. 2006;10(suppl 2):S1.
- , , , , , . Effects of systemic steroids in patients with severe community‐acquired pneumonia. Eur Respir J. 2007;30(5):951–956.
- , , , . Analysis of systemic corticosteroid usage and survival in patients requiring mechanical ventilation for severe community‐acquired pneumonia. J Infect Chemother. 2011;17(4):449–455.
- , , , et al. Effect of high‐dose prednisolone on lung fluid in patients with non‐cardiogenic lung edema [in German]. Wien Klin Wochenschr. 1987;99:245–249.
- , , , et al. Early steroid therapy for respiratory failure. Arch Surg. 1985;120:536–540.
- , , , et al. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med. 2006;354(16):1671–1684.
- , , , et al. High‐dose corticosteroids in patients with the adult respiratory distress syndrome. N Engl J Med. 1987;317:1565–1570.
- . Antibiotics—past, present, and future. Med Clin North Am. 2006;90(6):1049–1076.
Copyright © 2012 Society of Hospital Medicine