User login
Society of Hospital Medicine’s 2015 Fellows Class Applications Welcome
Don’t wait until the last minute to apply for the Fellow and Senior Fellow in Hospital Medicine designation. Start your application today at www.hospitalmedicine.org/fellows.
The FHM and SFHM designation are open to all hospitalists, not just physicians. Physician assistants, nurse practitioners, and practice administrators are all eligible candidates for Fellow status. All inductees will be recognized at a plenary session at HM15 in National Harbor, Md.
The deadline for the 2015 class of Fellows is Jan. 9, 2015.
Don’t wait until the last minute to apply for the Fellow and Senior Fellow in Hospital Medicine designation. Start your application today at www.hospitalmedicine.org/fellows.
The FHM and SFHM designation are open to all hospitalists, not just physicians. Physician assistants, nurse practitioners, and practice administrators are all eligible candidates for Fellow status. All inductees will be recognized at a plenary session at HM15 in National Harbor, Md.
The deadline for the 2015 class of Fellows is Jan. 9, 2015.
Don’t wait until the last minute to apply for the Fellow and Senior Fellow in Hospital Medicine designation. Start your application today at www.hospitalmedicine.org/fellows.
The FHM and SFHM designation are open to all hospitalists, not just physicians. Physician assistants, nurse practitioners, and practice administrators are all eligible candidates for Fellow status. All inductees will be recognized at a plenary session at HM15 in National Harbor, Md.
The deadline for the 2015 class of Fellows is Jan. 9, 2015.
Common Coding Mistakes Hospitalists Should Avoid

Medical decision-making (MDM) mistakes are common. Here are the coding and documentation mistakes hospitalists make most often, along with some tips on how to avoid them.
Listing the problem without a plan. Healthcare professionals are able to infer the acuity and severity of a case without superfluous or redundant documentation, but auditors may not have this ability. Adequate documentation for every service date helps to convey patient complexity during a medical record review. Although the problem list may not change dramatically from day to day during a hospitalization, the auditor only reviews the service date in question, not the entire medical record.
Hospitalists should be sure to formulate a complete and accurate description of the patient’s condition with an analogous plan of care for each encounter. Listing problems without a corresponding plan of care does not corroborate physician management of that problem and could cause a downgrade of complexity. Listing problems with a brief, generalized comment (e.g. “DM, CKD, CHF: Continue current treatment plan”) equally diminishes the complexity and effort put forth by the physician.
Clearly document the plan. The care plan represents problems the physician personally manages, along with those that must also be considered when he or she formulates the management options, even if another physician is primarily managing the problem. For example, the hospitalist can monitor the patient’s diabetic management while the nephrologist oversees the chronic kidney disease (CKD). Since the CKD impacts the hospitalist’s diabetic care plan, the hospitalist may also receive credit for any CKD consideration if the documentation supports a hospitalist-related care plan, or comment about CKD that does not overlap or replicate the nephrologist’s plan. In other words, there must be some “value-added” input by the hospitalist.
Credit is given for the quantity of problems addressed as well as the quality. For inpatient care, an established problem is defined as one in which a care plan has been generated by the physician (or same specialty group practice member) during the current hospitalization. Established problems are less complex than new problems, for which a diagnosis, prognosis, or care plan has not been developed. Severity of the problem also influences complexity. A “worsening” problem is considered more complex than an “improving” problem, since the worsening problem likely requires revisions to the current care plan and, thus, more physician effort. Physician documentation should always:
- Identify all problems managed or addressed during each encounter;
- Identify problems as stable or progressing, when appropriate;
- Indicate differential diagnoses when the problem remains undefined;
- Indicate the management/treatment option(s) for each problem; and
- Note management options to be continued somewhere in the progress note for that encounter (e.g. medication list) when documentation indicates a continuation of current management options (e.g. “continue meds”).
Considering relevant data. “Data” is organized as pathology/laboratory testing, radiology, and medicine-based diagnostic testing that contributes to diagnosing or managing patient problems. Pertinent orders or results may appear in the medical record, but most of the background interactions and communications involving testing are undetected when reviewing the progress note. To receive credit:
- Specify tests ordered and rationale in the physician’s progress note, or make an entry that refers to another auditor-accessible location for ordered tests and studies; however, this latter option jeopardizes a medical record review due to potential lack of awareness of the need to submit this extraneous information during a payer record request or appeal.
- Document test review by including a brief entry in the progress note (e.g. “elevated glucose levels” or “CXR shows RLL infiltrates”); credit is not given for entries lacking a comment on the findings (e.g. “CXR reviewed”).
- Summarize key points when reviewing old records or obtaining history from someone other than the patient, as necessary; be sure to identify the increased efforts of reviewing the considerable number of old records by stating, “OSH (outside hospital) records reviewed and shows…” or “Records from previous hospitalization(s) reveal….”
- Indicate when images, tracings, or specimens are “personally reviewed,” or the auditor will assume the physician merely reviewed the written report; be sure to include a comment on the findings.
- Summarize any discussions of unexpected or contradictory test results with the physician performing the procedure or diagnostic study.
Data credit may be more substantial during the initial investigative phase of the hospitalization, before diagnoses or treatment options have been confirmed. Routine monitoring of the stabilized patient may not yield as many “points.”
Undervaluing the patient’s complexity. A general lack of understanding of the MDM component of the documentation guidelines often results in physicians undervaluing their services. Some physicians may consider a case “low complexity” simply because of the frequency with which they encounter the case type. The speed with which the care plan is developed should have no bearing on how complex the patient’s condition really is. Hospitalists need to better identify the risk involved for the patient.
Patient risk is categorized as minimal, low, moderate, or high based on pre-assigned items pertaining to the presenting problem, diagnostic procedures ordered, and management options selected. The single highest-rated item detected on the Table of Risk determines the overall patient risk for an encounter.1 Chronic conditions with exacerbations and invasive procedures offer more patient risk than acute, uncomplicated illnesses or noninvasive procedures. Stable or improving problems are considered “less risky” than progressing problems; conditions that pose a threat to life/bodily function outweigh undiagnosed problems where it is difficult to determine the patient’s prognosis; and medication risk varies with the administration (e.g. oral vs. parenteral), type, and potential for adverse effects. Medication risk for a particular drug is invariable whether the dosage is increased, decreased, or continued without change. Physicians should:
- Provide status for all problems in the plan of care and identify them as stable, worsening, or progressing (mild or severe), when applicable; don’t assume that the auditor can infer this from the documentation details.
- Document all diagnostic or therapeutic procedures considered.
- Identify surgical risk factors involving co-morbid conditions that place the patient at greater risk than the average patient, when appropriate.
- Associate the labs ordered to monitor for medication toxicity with the corresponding medication; don’t assume that the auditor knows which labs are used to check for toxicity.
Varying levels of complexity. Remember that decision-making is just one of three components in evaluation and management (E&M) services, along with history and exam. MDM is identical for both the 1995 and 1997 guidelines, rooted in the complexity of the patient’s problem(s) addressed during a given encounter.1,2 Complexity is categorized as straightforward, low, moderate, or high, and directly correlates to the content of physician documentation.
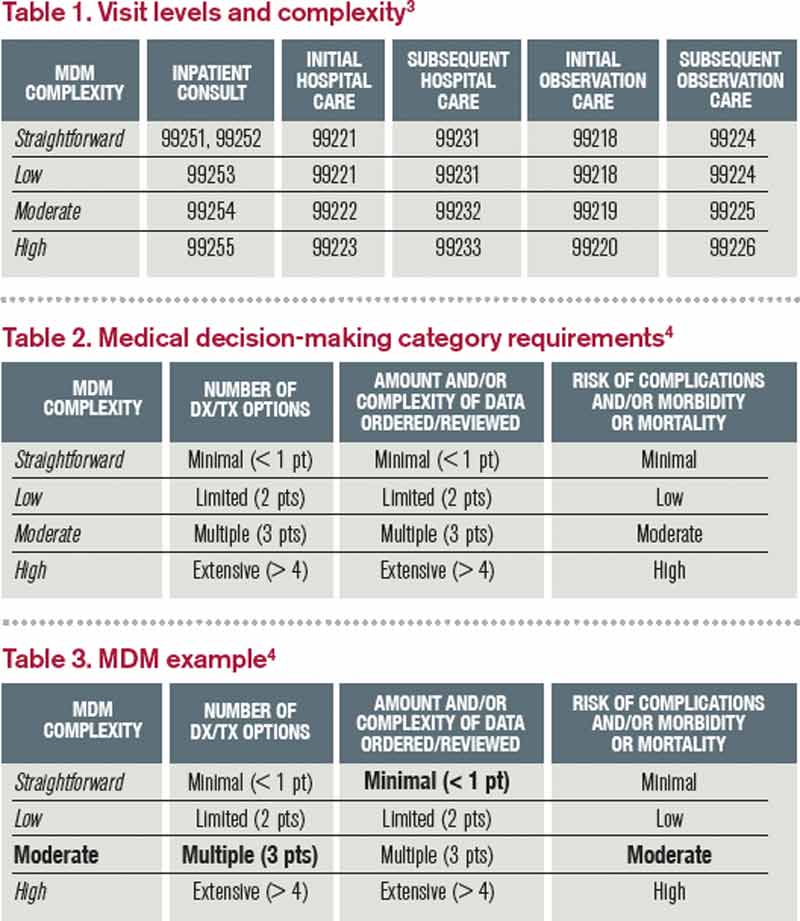
Each visit level represents a particular level of complexity (see Table 1). Auditors only consider the care plan for a given service date when reviewing MDM. More specifically, the auditor reviews three areas of MDM for each encounter (see Table 2), and the physician receives credit for: a) the number of diagnoses and/or treatment options; b) the amount and/or complexity of data ordered/reviewed; c) the risk of complications/morbidity/mortality.
To determine MDM complexity, each MDM category is assigned a point level. Complexity correlates to the second-highest MDM category. For example, if the auditor assigns “multiple” diagnoses/treatment options, “minimal” data, and “high” risk, the physician attains moderate complexity decision-making (see Table 3).
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
References
- Centers for Medicare and Medicaid Services. 1995 Documentation Guidelines for Evaluation and Management Services. Available at: www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNEdWebGuide/Downloads/95Docguidelines.pdf. Accessed July 7, 2014.
- Centers for Medicare and Medicaid Services. 1997 Documentation Guidelines for Evaluation and Management Services. Available at: http://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNEdWebGuide/Downloads/97Docguidelines.pdf. Accessed July 7, 2014.
- American Medical Association. Current Procedural Terminology: 2014 Professional Edition. Chicago: American Medical Association; 2013:14-21.
- Novitas Solutions. Novitas Solutions documentation worksheet. Available at: www.novitas-solutions.com/webcenter/content/conn/UCM_Repository/uuid/dDocName:00004966. Accessed July 7, 2014.
Medical decision-making (MDM) mistakes are common. Here are the coding and documentation mistakes hospitalists make most often, along with some tips on how to avoid them.
Listing the problem without a plan. Healthcare professionals are able to infer the acuity and severity of a case without superfluous or redundant documentation, but auditors may not have this ability. Adequate documentation for every service date helps to convey patient complexity during a medical record review. Although the problem list may not change dramatically from day to day during a hospitalization, the auditor only reviews the service date in question, not the entire medical record.
Hospitalists should be sure to formulate a complete and accurate description of the patient’s condition with an analogous plan of care for each encounter. Listing problems without a corresponding plan of care does not corroborate physician management of that problem and could cause a downgrade of complexity. Listing problems with a brief, generalized comment (e.g. “DM, CKD, CHF: Continue current treatment plan”) equally diminishes the complexity and effort put forth by the physician.
Clearly document the plan. The care plan represents problems the physician personally manages, along with those that must also be considered when he or she formulates the management options, even if another physician is primarily managing the problem. For example, the hospitalist can monitor the patient’s diabetic management while the nephrologist oversees the chronic kidney disease (CKD). Since the CKD impacts the hospitalist’s diabetic care plan, the hospitalist may also receive credit for any CKD consideration if the documentation supports a hospitalist-related care plan, or comment about CKD that does not overlap or replicate the nephrologist’s plan. In other words, there must be some “value-added” input by the hospitalist.
Credit is given for the quantity of problems addressed as well as the quality. For inpatient care, an established problem is defined as one in which a care plan has been generated by the physician (or same specialty group practice member) during the current hospitalization. Established problems are less complex than new problems, for which a diagnosis, prognosis, or care plan has not been developed. Severity of the problem also influences complexity. A “worsening” problem is considered more complex than an “improving” problem, since the worsening problem likely requires revisions to the current care plan and, thus, more physician effort. Physician documentation should always:
- Identify all problems managed or addressed during each encounter;
- Identify problems as stable or progressing, when appropriate;
- Indicate differential diagnoses when the problem remains undefined;
- Indicate the management/treatment option(s) for each problem; and
- Note management options to be continued somewhere in the progress note for that encounter (e.g. medication list) when documentation indicates a continuation of current management options (e.g. “continue meds”).
Considering relevant data. “Data” is organized as pathology/laboratory testing, radiology, and medicine-based diagnostic testing that contributes to diagnosing or managing patient problems. Pertinent orders or results may appear in the medical record, but most of the background interactions and communications involving testing are undetected when reviewing the progress note. To receive credit:
- Specify tests ordered and rationale in the physician’s progress note, or make an entry that refers to another auditor-accessible location for ordered tests and studies; however, this latter option jeopardizes a medical record review due to potential lack of awareness of the need to submit this extraneous information during a payer record request or appeal.
- Document test review by including a brief entry in the progress note (e.g. “elevated glucose levels” or “CXR shows RLL infiltrates”); credit is not given for entries lacking a comment on the findings (e.g. “CXR reviewed”).
- Summarize key points when reviewing old records or obtaining history from someone other than the patient, as necessary; be sure to identify the increased efforts of reviewing the considerable number of old records by stating, “OSH (outside hospital) records reviewed and shows…” or “Records from previous hospitalization(s) reveal….”
- Indicate when images, tracings, or specimens are “personally reviewed,” or the auditor will assume the physician merely reviewed the written report; be sure to include a comment on the findings.
- Summarize any discussions of unexpected or contradictory test results with the physician performing the procedure or diagnostic study.
Data credit may be more substantial during the initial investigative phase of the hospitalization, before diagnoses or treatment options have been confirmed. Routine monitoring of the stabilized patient may not yield as many “points.”
Undervaluing the patient’s complexity. A general lack of understanding of the MDM component of the documentation guidelines often results in physicians undervaluing their services. Some physicians may consider a case “low complexity” simply because of the frequency with which they encounter the case type. The speed with which the care plan is developed should have no bearing on how complex the patient’s condition really is. Hospitalists need to better identify the risk involved for the patient.
Patient risk is categorized as minimal, low, moderate, or high based on pre-assigned items pertaining to the presenting problem, diagnostic procedures ordered, and management options selected. The single highest-rated item detected on the Table of Risk determines the overall patient risk for an encounter.1 Chronic conditions with exacerbations and invasive procedures offer more patient risk than acute, uncomplicated illnesses or noninvasive procedures. Stable or improving problems are considered “less risky” than progressing problems; conditions that pose a threat to life/bodily function outweigh undiagnosed problems where it is difficult to determine the patient’s prognosis; and medication risk varies with the administration (e.g. oral vs. parenteral), type, and potential for adverse effects. Medication risk for a particular drug is invariable whether the dosage is increased, decreased, or continued without change. Physicians should:
- Provide status for all problems in the plan of care and identify them as stable, worsening, or progressing (mild or severe), when applicable; don’t assume that the auditor can infer this from the documentation details.
- Document all diagnostic or therapeutic procedures considered.
- Identify surgical risk factors involving co-morbid conditions that place the patient at greater risk than the average patient, when appropriate.
- Associate the labs ordered to monitor for medication toxicity with the corresponding medication; don’t assume that the auditor knows which labs are used to check for toxicity.
Varying levels of complexity. Remember that decision-making is just one of three components in evaluation and management (E&M) services, along with history and exam. MDM is identical for both the 1995 and 1997 guidelines, rooted in the complexity of the patient’s problem(s) addressed during a given encounter.1,2 Complexity is categorized as straightforward, low, moderate, or high, and directly correlates to the content of physician documentation.
Each visit level represents a particular level of complexity (see Table 1). Auditors only consider the care plan for a given service date when reviewing MDM. More specifically, the auditor reviews three areas of MDM for each encounter (see Table 2), and the physician receives credit for: a) the number of diagnoses and/or treatment options; b) the amount and/or complexity of data ordered/reviewed; c) the risk of complications/morbidity/mortality.
To determine MDM complexity, each MDM category is assigned a point level. Complexity correlates to the second-highest MDM category. For example, if the auditor assigns “multiple” diagnoses/treatment options, “minimal” data, and “high” risk, the physician attains moderate complexity decision-making (see Table 3).
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
References
- Centers for Medicare and Medicaid Services. 1995 Documentation Guidelines for Evaluation and Management Services. Available at: www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNEdWebGuide/Downloads/95Docguidelines.pdf. Accessed July 7, 2014.
- Centers for Medicare and Medicaid Services. 1997 Documentation Guidelines for Evaluation and Management Services. Available at: http://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNEdWebGuide/Downloads/97Docguidelines.pdf. Accessed July 7, 2014.
- American Medical Association. Current Procedural Terminology: 2014 Professional Edition. Chicago: American Medical Association; 2013:14-21.
- Novitas Solutions. Novitas Solutions documentation worksheet. Available at: www.novitas-solutions.com/webcenter/content/conn/UCM_Repository/uuid/dDocName:00004966. Accessed July 7, 2014.
Medical decision-making (MDM) mistakes are common. Here are the coding and documentation mistakes hospitalists make most often, along with some tips on how to avoid them.
Listing the problem without a plan. Healthcare professionals are able to infer the acuity and severity of a case without superfluous or redundant documentation, but auditors may not have this ability. Adequate documentation for every service date helps to convey patient complexity during a medical record review. Although the problem list may not change dramatically from day to day during a hospitalization, the auditor only reviews the service date in question, not the entire medical record.
Hospitalists should be sure to formulate a complete and accurate description of the patient’s condition with an analogous plan of care for each encounter. Listing problems without a corresponding plan of care does not corroborate physician management of that problem and could cause a downgrade of complexity. Listing problems with a brief, generalized comment (e.g. “DM, CKD, CHF: Continue current treatment plan”) equally diminishes the complexity and effort put forth by the physician.
Clearly document the plan. The care plan represents problems the physician personally manages, along with those that must also be considered when he or she formulates the management options, even if another physician is primarily managing the problem. For example, the hospitalist can monitor the patient’s diabetic management while the nephrologist oversees the chronic kidney disease (CKD). Since the CKD impacts the hospitalist’s diabetic care plan, the hospitalist may also receive credit for any CKD consideration if the documentation supports a hospitalist-related care plan, or comment about CKD that does not overlap or replicate the nephrologist’s plan. In other words, there must be some “value-added” input by the hospitalist.
Credit is given for the quantity of problems addressed as well as the quality. For inpatient care, an established problem is defined as one in which a care plan has been generated by the physician (or same specialty group practice member) during the current hospitalization. Established problems are less complex than new problems, for which a diagnosis, prognosis, or care plan has not been developed. Severity of the problem also influences complexity. A “worsening” problem is considered more complex than an “improving” problem, since the worsening problem likely requires revisions to the current care plan and, thus, more physician effort. Physician documentation should always:
- Identify all problems managed or addressed during each encounter;
- Identify problems as stable or progressing, when appropriate;
- Indicate differential diagnoses when the problem remains undefined;
- Indicate the management/treatment option(s) for each problem; and
- Note management options to be continued somewhere in the progress note for that encounter (e.g. medication list) when documentation indicates a continuation of current management options (e.g. “continue meds”).
Considering relevant data. “Data” is organized as pathology/laboratory testing, radiology, and medicine-based diagnostic testing that contributes to diagnosing or managing patient problems. Pertinent orders or results may appear in the medical record, but most of the background interactions and communications involving testing are undetected when reviewing the progress note. To receive credit:
- Specify tests ordered and rationale in the physician’s progress note, or make an entry that refers to another auditor-accessible location for ordered tests and studies; however, this latter option jeopardizes a medical record review due to potential lack of awareness of the need to submit this extraneous information during a payer record request or appeal.
- Document test review by including a brief entry in the progress note (e.g. “elevated glucose levels” or “CXR shows RLL infiltrates”); credit is not given for entries lacking a comment on the findings (e.g. “CXR reviewed”).
- Summarize key points when reviewing old records or obtaining history from someone other than the patient, as necessary; be sure to identify the increased efforts of reviewing the considerable number of old records by stating, “OSH (outside hospital) records reviewed and shows…” or “Records from previous hospitalization(s) reveal….”
- Indicate when images, tracings, or specimens are “personally reviewed,” or the auditor will assume the physician merely reviewed the written report; be sure to include a comment on the findings.
- Summarize any discussions of unexpected or contradictory test results with the physician performing the procedure or diagnostic study.
Data credit may be more substantial during the initial investigative phase of the hospitalization, before diagnoses or treatment options have been confirmed. Routine monitoring of the stabilized patient may not yield as many “points.”
Undervaluing the patient’s complexity. A general lack of understanding of the MDM component of the documentation guidelines often results in physicians undervaluing their services. Some physicians may consider a case “low complexity” simply because of the frequency with which they encounter the case type. The speed with which the care plan is developed should have no bearing on how complex the patient’s condition really is. Hospitalists need to better identify the risk involved for the patient.
Patient risk is categorized as minimal, low, moderate, or high based on pre-assigned items pertaining to the presenting problem, diagnostic procedures ordered, and management options selected. The single highest-rated item detected on the Table of Risk determines the overall patient risk for an encounter.1 Chronic conditions with exacerbations and invasive procedures offer more patient risk than acute, uncomplicated illnesses or noninvasive procedures. Stable or improving problems are considered “less risky” than progressing problems; conditions that pose a threat to life/bodily function outweigh undiagnosed problems where it is difficult to determine the patient’s prognosis; and medication risk varies with the administration (e.g. oral vs. parenteral), type, and potential for adverse effects. Medication risk for a particular drug is invariable whether the dosage is increased, decreased, or continued without change. Physicians should:
- Provide status for all problems in the plan of care and identify them as stable, worsening, or progressing (mild or severe), when applicable; don’t assume that the auditor can infer this from the documentation details.
- Document all diagnostic or therapeutic procedures considered.
- Identify surgical risk factors involving co-morbid conditions that place the patient at greater risk than the average patient, when appropriate.
- Associate the labs ordered to monitor for medication toxicity with the corresponding medication; don’t assume that the auditor knows which labs are used to check for toxicity.
Varying levels of complexity. Remember that decision-making is just one of three components in evaluation and management (E&M) services, along with history and exam. MDM is identical for both the 1995 and 1997 guidelines, rooted in the complexity of the patient’s problem(s) addressed during a given encounter.1,2 Complexity is categorized as straightforward, low, moderate, or high, and directly correlates to the content of physician documentation.
Each visit level represents a particular level of complexity (see Table 1). Auditors only consider the care plan for a given service date when reviewing MDM. More specifically, the auditor reviews three areas of MDM for each encounter (see Table 2), and the physician receives credit for: a) the number of diagnoses and/or treatment options; b) the amount and/or complexity of data ordered/reviewed; c) the risk of complications/morbidity/mortality.
To determine MDM complexity, each MDM category is assigned a point level. Complexity correlates to the second-highest MDM category. For example, if the auditor assigns “multiple” diagnoses/treatment options, “minimal” data, and “high” risk, the physician attains moderate complexity decision-making (see Table 3).
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
References
- Centers for Medicare and Medicaid Services. 1995 Documentation Guidelines for Evaluation and Management Services. Available at: www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNEdWebGuide/Downloads/95Docguidelines.pdf. Accessed July 7, 2014.
- Centers for Medicare and Medicaid Services. 1997 Documentation Guidelines for Evaluation and Management Services. Available at: http://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNEdWebGuide/Downloads/97Docguidelines.pdf. Accessed July 7, 2014.
- American Medical Association. Current Procedural Terminology: 2014 Professional Edition. Chicago: American Medical Association; 2013:14-21.
- Novitas Solutions. Novitas Solutions documentation worksheet. Available at: www.novitas-solutions.com/webcenter/content/conn/UCM_Repository/uuid/dDocName:00004966. Accessed July 7, 2014.
Society of Hospital Medicine Accepting Pre-Orders for 2014 HM Report
How does your HM group’s productivity stack up against others on productivity? How about compensation? These are the questions that can guide major business decisions for a hospital medicine group, and SHM’s State of Hospital Medicine Report, published every two years, can answer them. The 2014 issue is now available for pre-order and delivery in September.
To order, visit www.hospitalmedicine.org/survey.
How does your HM group’s productivity stack up against others on productivity? How about compensation? These are the questions that can guide major business decisions for a hospital medicine group, and SHM’s State of Hospital Medicine Report, published every two years, can answer them. The 2014 issue is now available for pre-order and delivery in September.
To order, visit www.hospitalmedicine.org/survey.
How does your HM group’s productivity stack up against others on productivity? How about compensation? These are the questions that can guide major business decisions for a hospital medicine group, and SHM’s State of Hospital Medicine Report, published every two years, can answer them. The 2014 issue is now available for pre-order and delivery in September.
To order, visit www.hospitalmedicine.org/survey.
Society of Hospital Medicine’s New Membership Ambassador Program Perks
Do you know someone who should be a part of the HM movement but hasn’t joined SHM? Now you can both win: Your colleague can enjoy all the benefits of SHM membership, and you can receive credits against your future dues. Plus, you’ll get the chance to win a free registration to HM15.
Now through December 31, all active SHM members can earn dues credits and special recognition for recruiting new physician, allied health, or affiliate members. Active members will be eligible for:
- A $35 credit toward 2015-2016 dues when recruiting one new member;
- A $50 credit toward 2015-2016 dues when recruiting two to four new members;
- A $75 credit toward 2015-2016 dues when recruiting five to nine new members; or
- A $125 credit toward 2015-2016 dues when recruiting 10+ new members.
For EVERY member recruited, individuals will receive one entry into a grand prize drawing to receive complimentary registration to Hospital Medicine 2015 in National Harbor, Md.
For details, visit www.hospitalmedicine.org/membership.
Do you know someone who should be a part of the HM movement but hasn’t joined SHM? Now you can both win: Your colleague can enjoy all the benefits of SHM membership, and you can receive credits against your future dues. Plus, you’ll get the chance to win a free registration to HM15.
Now through December 31, all active SHM members can earn dues credits and special recognition for recruiting new physician, allied health, or affiliate members. Active members will be eligible for:
- A $35 credit toward 2015-2016 dues when recruiting one new member;
- A $50 credit toward 2015-2016 dues when recruiting two to four new members;
- A $75 credit toward 2015-2016 dues when recruiting five to nine new members; or
- A $125 credit toward 2015-2016 dues when recruiting 10+ new members.
For EVERY member recruited, individuals will receive one entry into a grand prize drawing to receive complimentary registration to Hospital Medicine 2015 in National Harbor, Md.
For details, visit www.hospitalmedicine.org/membership.
Do you know someone who should be a part of the HM movement but hasn’t joined SHM? Now you can both win: Your colleague can enjoy all the benefits of SHM membership, and you can receive credits against your future dues. Plus, you’ll get the chance to win a free registration to HM15.
Now through December 31, all active SHM members can earn dues credits and special recognition for recruiting new physician, allied health, or affiliate members. Active members will be eligible for:
- A $35 credit toward 2015-2016 dues when recruiting one new member;
- A $50 credit toward 2015-2016 dues when recruiting two to four new members;
- A $75 credit toward 2015-2016 dues when recruiting five to nine new members; or
- A $125 credit toward 2015-2016 dues when recruiting 10+ new members.
For EVERY member recruited, individuals will receive one entry into a grand prize drawing to receive complimentary registration to Hospital Medicine 2015 in National Harbor, Md.
For details, visit www.hospitalmedicine.org/membership.
CODE-H Medical Coding Education Program Becomes Interactive
SHM’s coding education program, CODE-H, now has an interactive component through the SHM Learning Portal. CODE-H originally was developed as a series of live and on-demand webinars complemented by online forums; today, CODE-H Interactive brings the same expertise to an interactive platform ideal for new hospitalists learning the nuances of coding, hospital medicine groups assessing the coding skills of their caregivers, or even coders using it as a training tool for conducting audits of hospital medicine groups.
To learn more about CODE-H and CODE-H Interactive, visit www.hospitalmedicine.org/codeh.
SHM’s coding education program, CODE-H, now has an interactive component through the SHM Learning Portal. CODE-H originally was developed as a series of live and on-demand webinars complemented by online forums; today, CODE-H Interactive brings the same expertise to an interactive platform ideal for new hospitalists learning the nuances of coding, hospital medicine groups assessing the coding skills of their caregivers, or even coders using it as a training tool for conducting audits of hospital medicine groups.
To learn more about CODE-H and CODE-H Interactive, visit www.hospitalmedicine.org/codeh.
SHM’s coding education program, CODE-H, now has an interactive component through the SHM Learning Portal. CODE-H originally was developed as a series of live and on-demand webinars complemented by online forums; today, CODE-H Interactive brings the same expertise to an interactive platform ideal for new hospitalists learning the nuances of coding, hospital medicine groups assessing the coding skills of their caregivers, or even coders using it as a training tool for conducting audits of hospital medicine groups.
To learn more about CODE-H and CODE-H Interactive, visit www.hospitalmedicine.org/codeh.
Adult Hospital Medicine Boot Camp for Physician Assistants, Nurse Practitioners
Nurse practitioners and physician assistants are a critical part of the hospitalist care team. Together with the American Academy of Physician Assistants, SHM is hosting the annual Adult Hospital Medicine Boot Camp (www.aapa.org/bootcamp) specifically for nurse practitioners (NPs) and physician assistants (PAs).
The four-day program helps PAs and NPs stay up to date on the most common diagnoses, diseases, and treatments for hospitalized patients (27.75 hours Category 1 CME). A pre-course for PAs and NPs new to hospital medicine introduces them to the unique demands of inpatient care (eight hours Category 1 CME).
Adult Hospital Medicine Boot Camp October 2-5, 2014
The Westin Peachtree Plaza, Atlanta
Hospital Medicine 101
October 1, 2014
The Westin Peachtree Plaza, Atlanta
Nurse practitioners and physician assistants are a critical part of the hospitalist care team. Together with the American Academy of Physician Assistants, SHM is hosting the annual Adult Hospital Medicine Boot Camp (www.aapa.org/bootcamp) specifically for nurse practitioners (NPs) and physician assistants (PAs).
The four-day program helps PAs and NPs stay up to date on the most common diagnoses, diseases, and treatments for hospitalized patients (27.75 hours Category 1 CME). A pre-course for PAs and NPs new to hospital medicine introduces them to the unique demands of inpatient care (eight hours Category 1 CME).
Adult Hospital Medicine Boot Camp October 2-5, 2014
The Westin Peachtree Plaza, Atlanta
Hospital Medicine 101
October 1, 2014
The Westin Peachtree Plaza, Atlanta
Nurse practitioners and physician assistants are a critical part of the hospitalist care team. Together with the American Academy of Physician Assistants, SHM is hosting the annual Adult Hospital Medicine Boot Camp (www.aapa.org/bootcamp) specifically for nurse practitioners (NPs) and physician assistants (PAs).
The four-day program helps PAs and NPs stay up to date on the most common diagnoses, diseases, and treatments for hospitalized patients (27.75 hours Category 1 CME). A pre-course for PAs and NPs new to hospital medicine introduces them to the unique demands of inpatient care (eight hours Category 1 CME).
Adult Hospital Medicine Boot Camp October 2-5, 2014
The Westin Peachtree Plaza, Atlanta
Hospital Medicine 101
October 1, 2014
The Westin Peachtree Plaza, Atlanta
TeamHealth Hospital Medicine Shares Performance Stats
In February, SHM published the first performance assessment tool for HM groups. Now, HMGs across the country are using the “Key Principles and Characteristics of an Effective Hospital Medicine Group” to better understand their organizations’ strengths and areas needing improvement. Knoxville-based TeamHealth is the first to share its findings with SHM and The Hospitalist.
Before SHM published the assessment tool, there were very few objective attempts to provide guidelines that define an effective HMG. At TeamHealth, we viewed this tool as a way to proactively analyze our HMGs—a starting point if you will, to measure our performance against the principles identified in this assessment.
To this end, we allocated an internal analyst to work with our regional leadership teams. We felt it was important to have one person coordinating the analysis in order to ensure consistency with regard to how performance was defined. The analyst, along with the regional medical director and vice president of client services, went through each of the 47 key characteristics and identified the program’s status by evaluating the following statements:
- This characteristic does not apply to our HMG;
- Yes, we fully address the characteristic;
- Yes, we partially address the characteristic; or
- No, we do not materially address the characteristic.
For purposes of scoring, we then assigned a weight to each of the characteristics: three points if “fully addressed”; two points if “partially addressed”; one point if not addressed. We did not find that any of the characteristics fell under the “does not apply to our HMG” category.
A “100% effective” HMG was defined as scoring the highest possible score of 141 (i.e., three points for “fully addressing” each of the 47 characteristics).
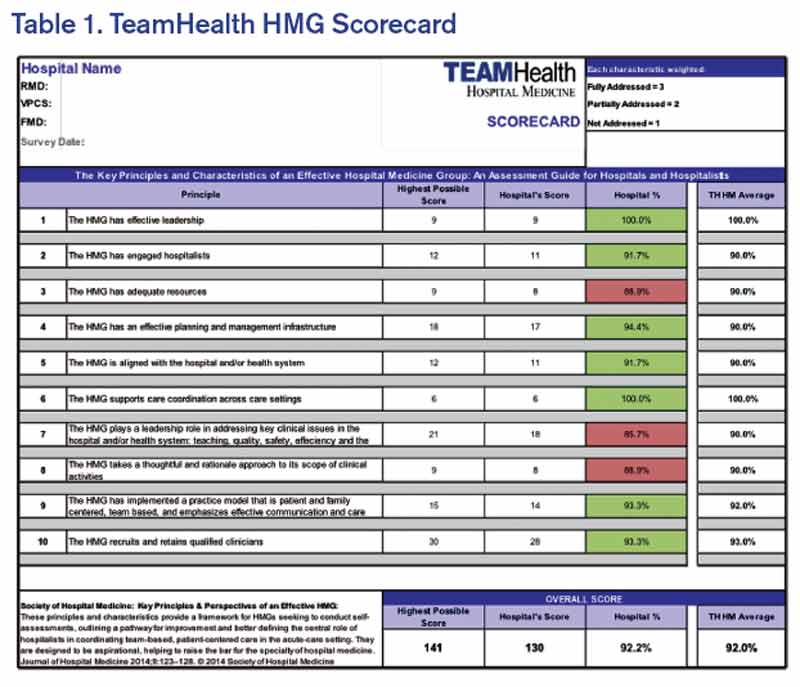
We are currently at the next step in our assessment process. This step involves completion of a scorecard for each individual HMG (see Table 1). Additionally, the individual HMG score will be benchmarked against TeamHealth Hospital Medicine performance overall.
Finally, our regional teams will take the scorecard and meet with their hospital administrators to review the assessment tool, our methodology for completion, and the hospital’s performance.
We fully recognize that some of our hospital partners have measurement standards that differ from those presented by SHM in this assessment; nonetheless, TeamHealth feels the tool in its present state is a significant first step toward quantifying a high-functioning HMG—and will ultimately help improve both hospitalists and hospital performance.
Roberta P. Himebaugh is executive vice president of TeamHealth Hospital Medicine.
In February, SHM published the first performance assessment tool for HM groups. Now, HMGs across the country are using the “Key Principles and Characteristics of an Effective Hospital Medicine Group” to better understand their organizations’ strengths and areas needing improvement. Knoxville-based TeamHealth is the first to share its findings with SHM and The Hospitalist.
Before SHM published the assessment tool, there were very few objective attempts to provide guidelines that define an effective HMG. At TeamHealth, we viewed this tool as a way to proactively analyze our HMGs—a starting point if you will, to measure our performance against the principles identified in this assessment.
To this end, we allocated an internal analyst to work with our regional leadership teams. We felt it was important to have one person coordinating the analysis in order to ensure consistency with regard to how performance was defined. The analyst, along with the regional medical director and vice president of client services, went through each of the 47 key characteristics and identified the program’s status by evaluating the following statements:
- This characteristic does not apply to our HMG;
- Yes, we fully address the characteristic;
- Yes, we partially address the characteristic; or
- No, we do not materially address the characteristic.
For purposes of scoring, we then assigned a weight to each of the characteristics: three points if “fully addressed”; two points if “partially addressed”; one point if not addressed. We did not find that any of the characteristics fell under the “does not apply to our HMG” category.
A “100% effective” HMG was defined as scoring the highest possible score of 141 (i.e., three points for “fully addressing” each of the 47 characteristics).
We are currently at the next step in our assessment process. This step involves completion of a scorecard for each individual HMG (see Table 1). Additionally, the individual HMG score will be benchmarked against TeamHealth Hospital Medicine performance overall.
Finally, our regional teams will take the scorecard and meet with their hospital administrators to review the assessment tool, our methodology for completion, and the hospital’s performance.
We fully recognize that some of our hospital partners have measurement standards that differ from those presented by SHM in this assessment; nonetheless, TeamHealth feels the tool in its present state is a significant first step toward quantifying a high-functioning HMG—and will ultimately help improve both hospitalists and hospital performance.
Roberta P. Himebaugh is executive vice president of TeamHealth Hospital Medicine.
In February, SHM published the first performance assessment tool for HM groups. Now, HMGs across the country are using the “Key Principles and Characteristics of an Effective Hospital Medicine Group” to better understand their organizations’ strengths and areas needing improvement. Knoxville-based TeamHealth is the first to share its findings with SHM and The Hospitalist.
Before SHM published the assessment tool, there were very few objective attempts to provide guidelines that define an effective HMG. At TeamHealth, we viewed this tool as a way to proactively analyze our HMGs—a starting point if you will, to measure our performance against the principles identified in this assessment.
To this end, we allocated an internal analyst to work with our regional leadership teams. We felt it was important to have one person coordinating the analysis in order to ensure consistency with regard to how performance was defined. The analyst, along with the regional medical director and vice president of client services, went through each of the 47 key characteristics and identified the program’s status by evaluating the following statements:
- This characteristic does not apply to our HMG;
- Yes, we fully address the characteristic;
- Yes, we partially address the characteristic; or
- No, we do not materially address the characteristic.
For purposes of scoring, we then assigned a weight to each of the characteristics: three points if “fully addressed”; two points if “partially addressed”; one point if not addressed. We did not find that any of the characteristics fell under the “does not apply to our HMG” category.
A “100% effective” HMG was defined as scoring the highest possible score of 141 (i.e., three points for “fully addressing” each of the 47 characteristics).
We are currently at the next step in our assessment process. This step involves completion of a scorecard for each individual HMG (see Table 1). Additionally, the individual HMG score will be benchmarked against TeamHealth Hospital Medicine performance overall.
Finally, our regional teams will take the scorecard and meet with their hospital administrators to review the assessment tool, our methodology for completion, and the hospital’s performance.
We fully recognize that some of our hospital partners have measurement standards that differ from those presented by SHM in this assessment; nonetheless, TeamHealth feels the tool in its present state is a significant first step toward quantifying a high-functioning HMG—and will ultimately help improve both hospitalists and hospital performance.
Roberta P. Himebaugh is executive vice president of TeamHealth Hospital Medicine.
Are topical nitrates safe and effective for upper extremity tendinopathies?
Topical nitrates provide short-term relief with some side effects, especially headache. Topical nitroglycerin (NTG) patches improve subjective pain scores by about 30% and range of motion over 3 days in patients with acute shoulder tendinopathy (strength of recommendation [SOR]: C, small randomized controlled trial [RCT] with no methodologic flaws).
NTG patches, when combined with tendon rehabilitation, improve subjective pain ratings by about 30% and shoulder strength by about 10% in patients with chronic shoulder tendinopathy over 3 to 6 months, but not in the long term (SOR: C, RCTs with methodologic flaws). They improve pain and strength 15% to 50% for chronic extensor tendinosis of the elbow over a 6-month period (SOR: C, small RCT with methodologic flaws).
NTG patches used without tendon rehabilitation don’t improve pain or strength in chronic lateral epicondylitis over 8 weeks (SOR: C, RCT).
Topical NTG patches commonly produce headaches and rashes (SOR: B, multiple RCTs).
EVIDENCE SUMMARY
A small RCT found that NTG therapy improved short-term pain and joint mobility in patients with acute supraspinatus tendinitis.1 Investigators randomized 10 men and 10 women with acute shoulder tendonitis (fewer than 7 days’ duration) to use either 5-mg NTG patches or placebo patches daily for 3 days. Patients rated pain on a 10-point scale, and investigators measured joint mobility on a 4-point scale.
After 48 hours of treatment, NTG patches significantly reduced pain ratings from baseline (from 7 to 2 points; P<.001), whereas placebo didn’t (6 vs 6 points; P not significant). NTG patches also improved joint mobility from baseline (from 2 points “moderately restricted” to .1 points “not restricted”; P<.001), but placebo didn’t (1.2 points “mildly restricted” vs 1.2 points; P not significant). The placebo group had less pain and joint restriction than the NTG group at the start of the study. Two patients reported headache 24 hours after starting treatment.
NTG plus rehabilitation improves chronic shoulder pain, range of motion
A double-blind RCT evaluating NTG patches for 53 patients (57 shoulders) with chronic supraspinatus tendinopathy (shoulder pain lasting longer than 3 months) found that they improved pain, strength, and range of motion at 3 to 6 months.2 Investigators randomized patients to receive one-quarter of a 5-mg 24-hour NTG patch or placebo patch daily and enrolled all patients in a rehabilitation program. They assessed subjective pain (at night and with activity), strength, and external rotation at baseline and at 2, 6, 12, and 24 weeks.
NTG patches improved nighttime pain about 30% (at 12 and 24 weeks), pain with activity about 60% (at 24 weeks), strength about 10% (at 12 and 24 weeks), and range of motion about 20% (at 24 weeks; P<.05 for all comparisons). The placebo group initially had more pain, less strength, and less mobility than the NTG group. Investigators reported no adverse effects.
NTG and rehab improve elbow pain, but with side effects
Another RCT comparing topical NTG patches in patients with chronic extensor tendinosis of the elbow found that they improved most parameters.3 Investigators randomized 86 patients with elbow tendonitis (longer than 3 months) to NTG patches (one-quarter of a 5-mg 24-hour patch) or placebo patches and enrolled all patients in a tendon rehabilitation program. They assessed subjective pain, extensor tendon tenderness, and muscle strength at baseline and at 2, 6, 12, and 24 weeks.
NTG patches improved subjective pain, tendon tenderness, and strength significantly more than placebo at all follow-up points, by 15% to 50% (P<.05 for all comparisons). The study was flawed because the control group started with more pain, tenderness, and weakness than the NTG group. Five patients discontinued NTG because of adverse effects (headache, dermatitis, and facial flushing).
A follow-up study done 5 years after discontinuation of therapy found equal outcomes with NTG and placebo.4 Investigators evaluated, by phone or in person, 58 of the 86 patients in the original study. NTG and placebo therapy produced equivalent reductions in subjective 0 to 4 elbow pain scores over baseline (average pain 2.5 initially, 1.5 at 12 weeks, and 1.0 at 5 years; P<.01 for all comparisons with baseline, no significant difference between nitrates and placebo).
NTG without rehab works no better than placebo
Another RCT that evaluated 3 different doses of NTG patches for 8 weeks in 154 patients with chronic lateral epicondylosis found NTG treatment was no better than placebo for pain or strength.5 Investigators randomized patients with more than 3 months of symptoms to 3 NTG patch doses (.72 mg/24 h, 1.44 mg/ 24 h, or 3.6 mg/24 h) compared with placebo and evaluated subjective pain (at rest, with activity, and at night), grip strength, and force, at baseline and 8 weeks.
The study lacked a formal wrist strengthening rehabilitation program. Patients in the placebo group had lower baseline pain scores than the NTG groups. Seven patients dropped out of the study because of headaches.
RECOMMENDATIONS
We found no authoritative recommendations regarding the use of topical nitrates for upper extremity tendinopathies.
An online reference text doesn’t make a recommendation, but references the studies described previously.6 The authors state that headache is the most common adverse effect of topical nitrates, but it becomes less severe over the course of treatment. They recommend caution in patients with hypotension, pregnancy, or migraines, and those who take diuretics. The authors also note that nitrates are relatively contraindicated in patients with ischemic heart disease, anemia, phosphodiesterase inhibitor therapy (such as sildenafil), angle-closure glaucoma, and allergy to nitrates.
1. Berrazueta JR, Losada A, Poveda J, et al. Successful treatment of shoulder pain syndrome due to supraspinatus tendinitis with transdermal nitroglycerin. A double blind study. Pain. 1996;66:63-67.
2. Paoloni JA, Appleyard RC, Nelson J, et al. Topical glyceryl trinitrate application in the treatment of chronic supraspinatus tendinopathy: a randomized, double-blinded, placebo-controlled clinical trial. Am J Sports Med. 2005;33:806-813.
3. Paoloni JA, Appleyard RC, Nelson J, et al. Topical nitric oxide application in the treatment of chronic extensor tendinosis at the elbow: a randomized, double-blinded, placebo-controlled clinical trial. Am J Sports Med. 2003;31:915-920.
4. McCallum SD, Paoloni JA, Murrell GA, et al. Five-year prospective comparison study of topical glyceryl trinitrate treatment of chronic lateral epicondylosis at the elbow. Br J Sports Med. 2011;45:416-420.
5. Paolini JA, Murrell GA, Burch RM, et al. Randomised, double-blind, placebo-controlled clinical trial of a new topical glyceryl trinitrate patch for chronic lateral epicondylosis. Br J Sports Med. 2009;43:299-302.
6. Simons SM, Kruse D. Rotator cuff tendinopathy. UpToDate Web site. Available at: www.uptodate.com/contents/rotator-cuff-tendinopathy. Accessed February 19, 2014.
Topical nitrates provide short-term relief with some side effects, especially headache. Topical nitroglycerin (NTG) patches improve subjective pain scores by about 30% and range of motion over 3 days in patients with acute shoulder tendinopathy (strength of recommendation [SOR]: C, small randomized controlled trial [RCT] with no methodologic flaws).
NTG patches, when combined with tendon rehabilitation, improve subjective pain ratings by about 30% and shoulder strength by about 10% in patients with chronic shoulder tendinopathy over 3 to 6 months, but not in the long term (SOR: C, RCTs with methodologic flaws). They improve pain and strength 15% to 50% for chronic extensor tendinosis of the elbow over a 6-month period (SOR: C, small RCT with methodologic flaws).
NTG patches used without tendon rehabilitation don’t improve pain or strength in chronic lateral epicondylitis over 8 weeks (SOR: C, RCT).
Topical NTG patches commonly produce headaches and rashes (SOR: B, multiple RCTs).
EVIDENCE SUMMARY
A small RCT found that NTG therapy improved short-term pain and joint mobility in patients with acute supraspinatus tendinitis.1 Investigators randomized 10 men and 10 women with acute shoulder tendonitis (fewer than 7 days’ duration) to use either 5-mg NTG patches or placebo patches daily for 3 days. Patients rated pain on a 10-point scale, and investigators measured joint mobility on a 4-point scale.
After 48 hours of treatment, NTG patches significantly reduced pain ratings from baseline (from 7 to 2 points; P<.001), whereas placebo didn’t (6 vs 6 points; P not significant). NTG patches also improved joint mobility from baseline (from 2 points “moderately restricted” to .1 points “not restricted”; P<.001), but placebo didn’t (1.2 points “mildly restricted” vs 1.2 points; P not significant). The placebo group had less pain and joint restriction than the NTG group at the start of the study. Two patients reported headache 24 hours after starting treatment.
NTG plus rehabilitation improves chronic shoulder pain, range of motion
A double-blind RCT evaluating NTG patches for 53 patients (57 shoulders) with chronic supraspinatus tendinopathy (shoulder pain lasting longer than 3 months) found that they improved pain, strength, and range of motion at 3 to 6 months.2 Investigators randomized patients to receive one-quarter of a 5-mg 24-hour NTG patch or placebo patch daily and enrolled all patients in a rehabilitation program. They assessed subjective pain (at night and with activity), strength, and external rotation at baseline and at 2, 6, 12, and 24 weeks.
NTG patches improved nighttime pain about 30% (at 12 and 24 weeks), pain with activity about 60% (at 24 weeks), strength about 10% (at 12 and 24 weeks), and range of motion about 20% (at 24 weeks; P<.05 for all comparisons). The placebo group initially had more pain, less strength, and less mobility than the NTG group. Investigators reported no adverse effects.
NTG and rehab improve elbow pain, but with side effects
Another RCT comparing topical NTG patches in patients with chronic extensor tendinosis of the elbow found that they improved most parameters.3 Investigators randomized 86 patients with elbow tendonitis (longer than 3 months) to NTG patches (one-quarter of a 5-mg 24-hour patch) or placebo patches and enrolled all patients in a tendon rehabilitation program. They assessed subjective pain, extensor tendon tenderness, and muscle strength at baseline and at 2, 6, 12, and 24 weeks.
NTG patches improved subjective pain, tendon tenderness, and strength significantly more than placebo at all follow-up points, by 15% to 50% (P<.05 for all comparisons). The study was flawed because the control group started with more pain, tenderness, and weakness than the NTG group. Five patients discontinued NTG because of adverse effects (headache, dermatitis, and facial flushing).
A follow-up study done 5 years after discontinuation of therapy found equal outcomes with NTG and placebo.4 Investigators evaluated, by phone or in person, 58 of the 86 patients in the original study. NTG and placebo therapy produced equivalent reductions in subjective 0 to 4 elbow pain scores over baseline (average pain 2.5 initially, 1.5 at 12 weeks, and 1.0 at 5 years; P<.01 for all comparisons with baseline, no significant difference between nitrates and placebo).
NTG without rehab works no better than placebo
Another RCT that evaluated 3 different doses of NTG patches for 8 weeks in 154 patients with chronic lateral epicondylosis found NTG treatment was no better than placebo for pain or strength.5 Investigators randomized patients with more than 3 months of symptoms to 3 NTG patch doses (.72 mg/24 h, 1.44 mg/ 24 h, or 3.6 mg/24 h) compared with placebo and evaluated subjective pain (at rest, with activity, and at night), grip strength, and force, at baseline and 8 weeks.
The study lacked a formal wrist strengthening rehabilitation program. Patients in the placebo group had lower baseline pain scores than the NTG groups. Seven patients dropped out of the study because of headaches.
RECOMMENDATIONS
We found no authoritative recommendations regarding the use of topical nitrates for upper extremity tendinopathies.
An online reference text doesn’t make a recommendation, but references the studies described previously.6 The authors state that headache is the most common adverse effect of topical nitrates, but it becomes less severe over the course of treatment. They recommend caution in patients with hypotension, pregnancy, or migraines, and those who take diuretics. The authors also note that nitrates are relatively contraindicated in patients with ischemic heart disease, anemia, phosphodiesterase inhibitor therapy (such as sildenafil), angle-closure glaucoma, and allergy to nitrates.
Topical nitrates provide short-term relief with some side effects, especially headache. Topical nitroglycerin (NTG) patches improve subjective pain scores by about 30% and range of motion over 3 days in patients with acute shoulder tendinopathy (strength of recommendation [SOR]: C, small randomized controlled trial [RCT] with no methodologic flaws).
NTG patches, when combined with tendon rehabilitation, improve subjective pain ratings by about 30% and shoulder strength by about 10% in patients with chronic shoulder tendinopathy over 3 to 6 months, but not in the long term (SOR: C, RCTs with methodologic flaws). They improve pain and strength 15% to 50% for chronic extensor tendinosis of the elbow over a 6-month period (SOR: C, small RCT with methodologic flaws).
NTG patches used without tendon rehabilitation don’t improve pain or strength in chronic lateral epicondylitis over 8 weeks (SOR: C, RCT).
Topical NTG patches commonly produce headaches and rashes (SOR: B, multiple RCTs).
EVIDENCE SUMMARY
A small RCT found that NTG therapy improved short-term pain and joint mobility in patients with acute supraspinatus tendinitis.1 Investigators randomized 10 men and 10 women with acute shoulder tendonitis (fewer than 7 days’ duration) to use either 5-mg NTG patches or placebo patches daily for 3 days. Patients rated pain on a 10-point scale, and investigators measured joint mobility on a 4-point scale.
After 48 hours of treatment, NTG patches significantly reduced pain ratings from baseline (from 7 to 2 points; P<.001), whereas placebo didn’t (6 vs 6 points; P not significant). NTG patches also improved joint mobility from baseline (from 2 points “moderately restricted” to .1 points “not restricted”; P<.001), but placebo didn’t (1.2 points “mildly restricted” vs 1.2 points; P not significant). The placebo group had less pain and joint restriction than the NTG group at the start of the study. Two patients reported headache 24 hours after starting treatment.
NTG plus rehabilitation improves chronic shoulder pain, range of motion
A double-blind RCT evaluating NTG patches for 53 patients (57 shoulders) with chronic supraspinatus tendinopathy (shoulder pain lasting longer than 3 months) found that they improved pain, strength, and range of motion at 3 to 6 months.2 Investigators randomized patients to receive one-quarter of a 5-mg 24-hour NTG patch or placebo patch daily and enrolled all patients in a rehabilitation program. They assessed subjective pain (at night and with activity), strength, and external rotation at baseline and at 2, 6, 12, and 24 weeks.
NTG patches improved nighttime pain about 30% (at 12 and 24 weeks), pain with activity about 60% (at 24 weeks), strength about 10% (at 12 and 24 weeks), and range of motion about 20% (at 24 weeks; P<.05 for all comparisons). The placebo group initially had more pain, less strength, and less mobility than the NTG group. Investigators reported no adverse effects.
NTG and rehab improve elbow pain, but with side effects
Another RCT comparing topical NTG patches in patients with chronic extensor tendinosis of the elbow found that they improved most parameters.3 Investigators randomized 86 patients with elbow tendonitis (longer than 3 months) to NTG patches (one-quarter of a 5-mg 24-hour patch) or placebo patches and enrolled all patients in a tendon rehabilitation program. They assessed subjective pain, extensor tendon tenderness, and muscle strength at baseline and at 2, 6, 12, and 24 weeks.
NTG patches improved subjective pain, tendon tenderness, and strength significantly more than placebo at all follow-up points, by 15% to 50% (P<.05 for all comparisons). The study was flawed because the control group started with more pain, tenderness, and weakness than the NTG group. Five patients discontinued NTG because of adverse effects (headache, dermatitis, and facial flushing).
A follow-up study done 5 years after discontinuation of therapy found equal outcomes with NTG and placebo.4 Investigators evaluated, by phone or in person, 58 of the 86 patients in the original study. NTG and placebo therapy produced equivalent reductions in subjective 0 to 4 elbow pain scores over baseline (average pain 2.5 initially, 1.5 at 12 weeks, and 1.0 at 5 years; P<.01 for all comparisons with baseline, no significant difference between nitrates and placebo).
NTG without rehab works no better than placebo
Another RCT that evaluated 3 different doses of NTG patches for 8 weeks in 154 patients with chronic lateral epicondylosis found NTG treatment was no better than placebo for pain or strength.5 Investigators randomized patients with more than 3 months of symptoms to 3 NTG patch doses (.72 mg/24 h, 1.44 mg/ 24 h, or 3.6 mg/24 h) compared with placebo and evaluated subjective pain (at rest, with activity, and at night), grip strength, and force, at baseline and 8 weeks.
The study lacked a formal wrist strengthening rehabilitation program. Patients in the placebo group had lower baseline pain scores than the NTG groups. Seven patients dropped out of the study because of headaches.
RECOMMENDATIONS
We found no authoritative recommendations regarding the use of topical nitrates for upper extremity tendinopathies.
An online reference text doesn’t make a recommendation, but references the studies described previously.6 The authors state that headache is the most common adverse effect of topical nitrates, but it becomes less severe over the course of treatment. They recommend caution in patients with hypotension, pregnancy, or migraines, and those who take diuretics. The authors also note that nitrates are relatively contraindicated in patients with ischemic heart disease, anemia, phosphodiesterase inhibitor therapy (such as sildenafil), angle-closure glaucoma, and allergy to nitrates.
1. Berrazueta JR, Losada A, Poveda J, et al. Successful treatment of shoulder pain syndrome due to supraspinatus tendinitis with transdermal nitroglycerin. A double blind study. Pain. 1996;66:63-67.
2. Paoloni JA, Appleyard RC, Nelson J, et al. Topical glyceryl trinitrate application in the treatment of chronic supraspinatus tendinopathy: a randomized, double-blinded, placebo-controlled clinical trial. Am J Sports Med. 2005;33:806-813.
3. Paoloni JA, Appleyard RC, Nelson J, et al. Topical nitric oxide application in the treatment of chronic extensor tendinosis at the elbow: a randomized, double-blinded, placebo-controlled clinical trial. Am J Sports Med. 2003;31:915-920.
4. McCallum SD, Paoloni JA, Murrell GA, et al. Five-year prospective comparison study of topical glyceryl trinitrate treatment of chronic lateral epicondylosis at the elbow. Br J Sports Med. 2011;45:416-420.
5. Paolini JA, Murrell GA, Burch RM, et al. Randomised, double-blind, placebo-controlled clinical trial of a new topical glyceryl trinitrate patch for chronic lateral epicondylosis. Br J Sports Med. 2009;43:299-302.
6. Simons SM, Kruse D. Rotator cuff tendinopathy. UpToDate Web site. Available at: www.uptodate.com/contents/rotator-cuff-tendinopathy. Accessed February 19, 2014.
1. Berrazueta JR, Losada A, Poveda J, et al. Successful treatment of shoulder pain syndrome due to supraspinatus tendinitis with transdermal nitroglycerin. A double blind study. Pain. 1996;66:63-67.
2. Paoloni JA, Appleyard RC, Nelson J, et al. Topical glyceryl trinitrate application in the treatment of chronic supraspinatus tendinopathy: a randomized, double-blinded, placebo-controlled clinical trial. Am J Sports Med. 2005;33:806-813.
3. Paoloni JA, Appleyard RC, Nelson J, et al. Topical nitric oxide application in the treatment of chronic extensor tendinosis at the elbow: a randomized, double-blinded, placebo-controlled clinical trial. Am J Sports Med. 2003;31:915-920.
4. McCallum SD, Paoloni JA, Murrell GA, et al. Five-year prospective comparison study of topical glyceryl trinitrate treatment of chronic lateral epicondylosis at the elbow. Br J Sports Med. 2011;45:416-420.
5. Paolini JA, Murrell GA, Burch RM, et al. Randomised, double-blind, placebo-controlled clinical trial of a new topical glyceryl trinitrate patch for chronic lateral epicondylosis. Br J Sports Med. 2009;43:299-302.
6. Simons SM, Kruse D. Rotator cuff tendinopathy. UpToDate Web site. Available at: www.uptodate.com/contents/rotator-cuff-tendinopathy. Accessed February 19, 2014.
Evidence-based answers from the Family Physicians Inquiries Network
Medical Decision-Making: Avoid These Common Coding & Documentation Mistakes
Medical decision-making (MDM) mistakes are common. Here are the coding and documentation mistakes hospitalists make most often, along with some tips on how to avoid them.
Listing the problem without a plan. Healthcare professionals are able to infer the acuity and severity of a case without superfluous or redundant documentation, but auditors may not have this ability. Adequate documentation for every service date helps to convey patient complexity during a medical record review. Although the problem list may not change dramatically from day to day during a hospitalization, the auditor only reviews the service date in question, not the entire medical record.
Hospitalists should be sure to formulate a complete and accurate description of the patient’s condition with an analogous plan of care for each encounter. Listing problems without a corresponding plan of care does not corroborate physician management of that problem and could cause a downgrade of complexity. Listing problems with a brief, generalized comment (e.g. “DM, CKD, CHF: Continue current treatment plan”) equally diminishes the complexity and effort put forth by the physician.
Clearly document the plan. The care plan represents problems the physician personally manages, along with those that must also be considered when he or she formulates the management options, even if another physician is primarily managing the problem. For example, the hospitalist can monitor the patient’s diabetic management while the nephrologist oversees the chronic kidney disease (CKD). Since the CKD impacts the hospitalist’s diabetic care plan, the hospitalist may also receive credit for any CKD consideration if the documentation supports a hospitalist-related care plan, or comment about CKD that does not overlap or replicate the nephrologist’s plan. In other words, there must be some “value-added” input by the hospitalist.
Credit is given for the quantity of problems addressed as well as the quality. For inpatient care, an established problem is defined as one in which a care plan has been generated by the physician (or same specialty group practice member) during the current hospitalization. Established problems are less complex than new problems, for which a diagnosis, prognosis, or care plan has not been developed. Severity of the problem also influences complexity. A “worsening” problem is considered more complex than an “improving” problem, since the worsening problem likely requires revisions to the current care plan and, thus, more physician effort. Physician documentation should always:
- Identify all problems managed or addressed during each encounter;
- Identify problems as stable or progressing, when appropriate;
- Indicate differential diagnoses when the problem remains undefined;
- Indicate the management/treatment option(s) for each problem; and
- Note management options to be continued somewhere in the progress note for that encounter (e.g. medication list) when documentation indicates a continuation of current management options (e.g. “continue meds”).
Considering relevant data. “Data” is organized as pathology/laboratory testing, radiology, and medicine-based diagnostic testing that contributes to diagnosing or managing patient problems. Pertinent orders or results may appear in the medical record, but most of the background interactions and communications involving testing are undetected when reviewing the progress note. To receive credit:
- Specify tests ordered and rationale in the physician’s progress note, or make an entry that refers to another auditor-accessible location for ordered tests and studies; however, this latter option jeopardizes a medical record review due to potential lack of awareness of the need to submit this extraneous information during a payer record request or appeal.
- Document test review by including a brief entry in the progress note (e.g. “elevated glucose levels” or “CXR shows RLL infiltrates”); credit is not given for entries lacking a comment on the findings (e.g. “CXR reviewed”).
- Summarize key points when reviewing old records or obtaining history from someone other than the patient, as necessary; be sure to identify the increased efforts of reviewing the considerable number of old records by stating, “OSH (outside hospital) records reviewed and shows…” or “Records from previous hospitalization(s) reveal….”
- Indicate when images, tracings, or specimens are “personally reviewed,” or the auditor will assume the physician merely reviewed the written report; be sure to include a comment on the findings.
- Summarize any discussions of unexpected or contradictory test results with the physician performing the procedure or diagnostic study.
Data credit may be more substantial during the initial investigative phase of the hospitalization, before diagnoses or treatment options have been confirmed. Routine monitoring of the stabilized patient may not yield as many “points.”
Undervaluing the patient’s complexity. A general lack of understanding of the MDM component of the documentation guidelines often results in physicians undervaluing their services. Some physicians may consider a case “low complexity” simply because of the frequency with which they encounter the case type. The speed with which the care plan is developed should have no bearing on how complex the patient’s condition really is. Hospitalists need to better identify the risk involved for the patient.
Patient risk is categorized as minimal, low, moderate, or high based on pre-assigned items pertaining to the presenting problem, diagnostic procedures ordered, and management options selected. The single highest-rated item detected on the Table of Risk determines the overall patient risk for an encounter.1 Chronic conditions with exacerbations and invasive procedures offer more patient risk than acute, uncomplicated illnesses or noninvasive procedures. Stable or improving problems are considered “less risky” than progressing problems; conditions that pose a threat to life/bodily function outweigh undiagnosed problems where it is difficult to determine the patient’s prognosis; and medication risk varies with the administration (e.g. oral vs. parenteral), type, and potential for adverse effects. Medication risk for a particular drug is invariable whether the dosage is increased, decreased, or continued without change. Physicians should:
- Provide status for all problems in the plan of care and identify them as stable, worsening, or progressing (mild or severe), when applicable; don’t assume that the auditor can infer this from the documentation details.
- Document all diagnostic or therapeutic procedures considered.
- Identify surgical risk factors involving co-morbid conditions that place the patient at greater risk than the average patient, when appropriate.
- Associate the labs ordered to monitor for medication toxicity with the corresponding medication; don’t assume that the auditor knows which labs are used to check for toxicity.
Varying levels of complexity. Remember that decision-making is just one of three components in evaluation and management (E&M) services, along with history and exam. MDM is identical for both the 1995 and 1997 guidelines, rooted in the complexity of the patient’s problem(s) addressed during a given encounter.1,2 Complexity is categorized as straightforward, low, moderate, or high, and directly correlates to the content of physician documentation.
Each visit level represents a particular level of complexity (see Table 1). Auditors only consider the care plan for a given service date when reviewing MDM. More specifically, the auditor reviews three areas of MDM for each encounter (see Table 2), and the physician receives credit for: a) the number of diagnoses and/or treatment options; b) the amount and/or complexity of data ordered/reviewed; c) the risk of complications/morbidity/mortality.
To determine MDM complexity, each MDM category is assigned a point level. Complexity correlates to the second-highest MDM category. For example, if the auditor assigns “multiple” diagnoses/treatment options, “minimal” data, and “high” risk, the physician attains moderate complexity decision-making (see Table 3).
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
References
- Centers for Medicare and Medicaid Services. 1995 Documentation Guidelines for Evaluation and Management Services. Available at: www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNEdWebGuide/Downloads/95Docguidelines.pdf. Accessed July 7, 2014.
- Centers for Medicare and Medicaid Services. 1997 Documentation Guidelines for Evaluation and Management Services. Available at: http://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNEdWebGuide/Downloads/97Docguidelines.pdf. Accessed July 7, 2014.
- American Medical Association. Current Procedural Terminology: 2014 Professional Edition. Chicago: American Medical Association; 2013:14-21.
- Novitas Solutions. Novitas Solutions documentation worksheet. Available at: www.novitas-solutions.com/webcenter/content/conn/UCM_Repository/uuid/dDocName:00004966. Accessed July 7, 2014.
Medical decision-making (MDM) mistakes are common. Here are the coding and documentation mistakes hospitalists make most often, along with some tips on how to avoid them.
Listing the problem without a plan. Healthcare professionals are able to infer the acuity and severity of a case without superfluous or redundant documentation, but auditors may not have this ability. Adequate documentation for every service date helps to convey patient complexity during a medical record review. Although the problem list may not change dramatically from day to day during a hospitalization, the auditor only reviews the service date in question, not the entire medical record.
Hospitalists should be sure to formulate a complete and accurate description of the patient’s condition with an analogous plan of care for each encounter. Listing problems without a corresponding plan of care does not corroborate physician management of that problem and could cause a downgrade of complexity. Listing problems with a brief, generalized comment (e.g. “DM, CKD, CHF: Continue current treatment plan”) equally diminishes the complexity and effort put forth by the physician.
Clearly document the plan. The care plan represents problems the physician personally manages, along with those that must also be considered when he or she formulates the management options, even if another physician is primarily managing the problem. For example, the hospitalist can monitor the patient’s diabetic management while the nephrologist oversees the chronic kidney disease (CKD). Since the CKD impacts the hospitalist’s diabetic care plan, the hospitalist may also receive credit for any CKD consideration if the documentation supports a hospitalist-related care plan, or comment about CKD that does not overlap or replicate the nephrologist’s plan. In other words, there must be some “value-added” input by the hospitalist.
Credit is given for the quantity of problems addressed as well as the quality. For inpatient care, an established problem is defined as one in which a care plan has been generated by the physician (or same specialty group practice member) during the current hospitalization. Established problems are less complex than new problems, for which a diagnosis, prognosis, or care plan has not been developed. Severity of the problem also influences complexity. A “worsening” problem is considered more complex than an “improving” problem, since the worsening problem likely requires revisions to the current care plan and, thus, more physician effort. Physician documentation should always:
- Identify all problems managed or addressed during each encounter;
- Identify problems as stable or progressing, when appropriate;
- Indicate differential diagnoses when the problem remains undefined;
- Indicate the management/treatment option(s) for each problem; and
- Note management options to be continued somewhere in the progress note for that encounter (e.g. medication list) when documentation indicates a continuation of current management options (e.g. “continue meds”).
Considering relevant data. “Data” is organized as pathology/laboratory testing, radiology, and medicine-based diagnostic testing that contributes to diagnosing or managing patient problems. Pertinent orders or results may appear in the medical record, but most of the background interactions and communications involving testing are undetected when reviewing the progress note. To receive credit:
- Specify tests ordered and rationale in the physician’s progress note, or make an entry that refers to another auditor-accessible location for ordered tests and studies; however, this latter option jeopardizes a medical record review due to potential lack of awareness of the need to submit this extraneous information during a payer record request or appeal.
- Document test review by including a brief entry in the progress note (e.g. “elevated glucose levels” or “CXR shows RLL infiltrates”); credit is not given for entries lacking a comment on the findings (e.g. “CXR reviewed”).
- Summarize key points when reviewing old records or obtaining history from someone other than the patient, as necessary; be sure to identify the increased efforts of reviewing the considerable number of old records by stating, “OSH (outside hospital) records reviewed and shows…” or “Records from previous hospitalization(s) reveal….”
- Indicate when images, tracings, or specimens are “personally reviewed,” or the auditor will assume the physician merely reviewed the written report; be sure to include a comment on the findings.
- Summarize any discussions of unexpected or contradictory test results with the physician performing the procedure or diagnostic study.
Data credit may be more substantial during the initial investigative phase of the hospitalization, before diagnoses or treatment options have been confirmed. Routine monitoring of the stabilized patient may not yield as many “points.”
Undervaluing the patient’s complexity. A general lack of understanding of the MDM component of the documentation guidelines often results in physicians undervaluing their services. Some physicians may consider a case “low complexity” simply because of the frequency with which they encounter the case type. The speed with which the care plan is developed should have no bearing on how complex the patient’s condition really is. Hospitalists need to better identify the risk involved for the patient.
Patient risk is categorized as minimal, low, moderate, or high based on pre-assigned items pertaining to the presenting problem, diagnostic procedures ordered, and management options selected. The single highest-rated item detected on the Table of Risk determines the overall patient risk for an encounter.1 Chronic conditions with exacerbations and invasive procedures offer more patient risk than acute, uncomplicated illnesses or noninvasive procedures. Stable or improving problems are considered “less risky” than progressing problems; conditions that pose a threat to life/bodily function outweigh undiagnosed problems where it is difficult to determine the patient’s prognosis; and medication risk varies with the administration (e.g. oral vs. parenteral), type, and potential for adverse effects. Medication risk for a particular drug is invariable whether the dosage is increased, decreased, or continued without change. Physicians should:
- Provide status for all problems in the plan of care and identify them as stable, worsening, or progressing (mild or severe), when applicable; don’t assume that the auditor can infer this from the documentation details.
- Document all diagnostic or therapeutic procedures considered.
- Identify surgical risk factors involving co-morbid conditions that place the patient at greater risk than the average patient, when appropriate.
- Associate the labs ordered to monitor for medication toxicity with the corresponding medication; don’t assume that the auditor knows which labs are used to check for toxicity.
Varying levels of complexity. Remember that decision-making is just one of three components in evaluation and management (E&M) services, along with history and exam. MDM is identical for both the 1995 and 1997 guidelines, rooted in the complexity of the patient’s problem(s) addressed during a given encounter.1,2 Complexity is categorized as straightforward, low, moderate, or high, and directly correlates to the content of physician documentation.
Each visit level represents a particular level of complexity (see Table 1). Auditors only consider the care plan for a given service date when reviewing MDM. More specifically, the auditor reviews three areas of MDM for each encounter (see Table 2), and the physician receives credit for: a) the number of diagnoses and/or treatment options; b) the amount and/or complexity of data ordered/reviewed; c) the risk of complications/morbidity/mortality.
To determine MDM complexity, each MDM category is assigned a point level. Complexity correlates to the second-highest MDM category. For example, if the auditor assigns “multiple” diagnoses/treatment options, “minimal” data, and “high” risk, the physician attains moderate complexity decision-making (see Table 3).
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
References
- Centers for Medicare and Medicaid Services. 1995 Documentation Guidelines for Evaluation and Management Services. Available at: www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNEdWebGuide/Downloads/95Docguidelines.pdf. Accessed July 7, 2014.
- Centers for Medicare and Medicaid Services. 1997 Documentation Guidelines for Evaluation and Management Services. Available at: http://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNEdWebGuide/Downloads/97Docguidelines.pdf. Accessed July 7, 2014.
- American Medical Association. Current Procedural Terminology: 2014 Professional Edition. Chicago: American Medical Association; 2013:14-21.
- Novitas Solutions. Novitas Solutions documentation worksheet. Available at: www.novitas-solutions.com/webcenter/content/conn/UCM_Repository/uuid/dDocName:00004966. Accessed July 7, 2014.
Medical decision-making (MDM) mistakes are common. Here are the coding and documentation mistakes hospitalists make most often, along with some tips on how to avoid them.
Listing the problem without a plan. Healthcare professionals are able to infer the acuity and severity of a case without superfluous or redundant documentation, but auditors may not have this ability. Adequate documentation for every service date helps to convey patient complexity during a medical record review. Although the problem list may not change dramatically from day to day during a hospitalization, the auditor only reviews the service date in question, not the entire medical record.
Hospitalists should be sure to formulate a complete and accurate description of the patient’s condition with an analogous plan of care for each encounter. Listing problems without a corresponding plan of care does not corroborate physician management of that problem and could cause a downgrade of complexity. Listing problems with a brief, generalized comment (e.g. “DM, CKD, CHF: Continue current treatment plan”) equally diminishes the complexity and effort put forth by the physician.
Clearly document the plan. The care plan represents problems the physician personally manages, along with those that must also be considered when he or she formulates the management options, even if another physician is primarily managing the problem. For example, the hospitalist can monitor the patient’s diabetic management while the nephrologist oversees the chronic kidney disease (CKD). Since the CKD impacts the hospitalist’s diabetic care plan, the hospitalist may also receive credit for any CKD consideration if the documentation supports a hospitalist-related care plan, or comment about CKD that does not overlap or replicate the nephrologist’s plan. In other words, there must be some “value-added” input by the hospitalist.
Credit is given for the quantity of problems addressed as well as the quality. For inpatient care, an established problem is defined as one in which a care plan has been generated by the physician (or same specialty group practice member) during the current hospitalization. Established problems are less complex than new problems, for which a diagnosis, prognosis, or care plan has not been developed. Severity of the problem also influences complexity. A “worsening” problem is considered more complex than an “improving” problem, since the worsening problem likely requires revisions to the current care plan and, thus, more physician effort. Physician documentation should always:
- Identify all problems managed or addressed during each encounter;
- Identify problems as stable or progressing, when appropriate;
- Indicate differential diagnoses when the problem remains undefined;
- Indicate the management/treatment option(s) for each problem; and
- Note management options to be continued somewhere in the progress note for that encounter (e.g. medication list) when documentation indicates a continuation of current management options (e.g. “continue meds”).
Considering relevant data. “Data” is organized as pathology/laboratory testing, radiology, and medicine-based diagnostic testing that contributes to diagnosing or managing patient problems. Pertinent orders or results may appear in the medical record, but most of the background interactions and communications involving testing are undetected when reviewing the progress note. To receive credit:
- Specify tests ordered and rationale in the physician’s progress note, or make an entry that refers to another auditor-accessible location for ordered tests and studies; however, this latter option jeopardizes a medical record review due to potential lack of awareness of the need to submit this extraneous information during a payer record request or appeal.
- Document test review by including a brief entry in the progress note (e.g. “elevated glucose levels” or “CXR shows RLL infiltrates”); credit is not given for entries lacking a comment on the findings (e.g. “CXR reviewed”).
- Summarize key points when reviewing old records or obtaining history from someone other than the patient, as necessary; be sure to identify the increased efforts of reviewing the considerable number of old records by stating, “OSH (outside hospital) records reviewed and shows…” or “Records from previous hospitalization(s) reveal….”
- Indicate when images, tracings, or specimens are “personally reviewed,” or the auditor will assume the physician merely reviewed the written report; be sure to include a comment on the findings.
- Summarize any discussions of unexpected or contradictory test results with the physician performing the procedure or diagnostic study.
Data credit may be more substantial during the initial investigative phase of the hospitalization, before diagnoses or treatment options have been confirmed. Routine monitoring of the stabilized patient may not yield as many “points.”
Undervaluing the patient’s complexity. A general lack of understanding of the MDM component of the documentation guidelines often results in physicians undervaluing their services. Some physicians may consider a case “low complexity” simply because of the frequency with which they encounter the case type. The speed with which the care plan is developed should have no bearing on how complex the patient’s condition really is. Hospitalists need to better identify the risk involved for the patient.
Patient risk is categorized as minimal, low, moderate, or high based on pre-assigned items pertaining to the presenting problem, diagnostic procedures ordered, and management options selected. The single highest-rated item detected on the Table of Risk determines the overall patient risk for an encounter.1 Chronic conditions with exacerbations and invasive procedures offer more patient risk than acute, uncomplicated illnesses or noninvasive procedures. Stable or improving problems are considered “less risky” than progressing problems; conditions that pose a threat to life/bodily function outweigh undiagnosed problems where it is difficult to determine the patient’s prognosis; and medication risk varies with the administration (e.g. oral vs. parenteral), type, and potential for adverse effects. Medication risk for a particular drug is invariable whether the dosage is increased, decreased, or continued without change. Physicians should:
- Provide status for all problems in the plan of care and identify them as stable, worsening, or progressing (mild or severe), when applicable; don’t assume that the auditor can infer this from the documentation details.
- Document all diagnostic or therapeutic procedures considered.
- Identify surgical risk factors involving co-morbid conditions that place the patient at greater risk than the average patient, when appropriate.
- Associate the labs ordered to monitor for medication toxicity with the corresponding medication; don’t assume that the auditor knows which labs are used to check for toxicity.
Varying levels of complexity. Remember that decision-making is just one of three components in evaluation and management (E&M) services, along with history and exam. MDM is identical for both the 1995 and 1997 guidelines, rooted in the complexity of the patient’s problem(s) addressed during a given encounter.1,2 Complexity is categorized as straightforward, low, moderate, or high, and directly correlates to the content of physician documentation.
Each visit level represents a particular level of complexity (see Table 1). Auditors only consider the care plan for a given service date when reviewing MDM. More specifically, the auditor reviews three areas of MDM for each encounter (see Table 2), and the physician receives credit for: a) the number of diagnoses and/or treatment options; b) the amount and/or complexity of data ordered/reviewed; c) the risk of complications/morbidity/mortality.
To determine MDM complexity, each MDM category is assigned a point level. Complexity correlates to the second-highest MDM category. For example, if the auditor assigns “multiple” diagnoses/treatment options, “minimal” data, and “high” risk, the physician attains moderate complexity decision-making (see Table 3).
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
References
- Centers for Medicare and Medicaid Services. 1995 Documentation Guidelines for Evaluation and Management Services. Available at: www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNEdWebGuide/Downloads/95Docguidelines.pdf. Accessed July 7, 2014.
- Centers for Medicare and Medicaid Services. 1997 Documentation Guidelines for Evaluation and Management Services. Available at: http://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNEdWebGuide/Downloads/97Docguidelines.pdf. Accessed July 7, 2014.
- American Medical Association. Current Procedural Terminology: 2014 Professional Edition. Chicago: American Medical Association; 2013:14-21.
- Novitas Solutions. Novitas Solutions documentation worksheet. Available at: www.novitas-solutions.com/webcenter/content/conn/UCM_Repository/uuid/dDocName:00004966. Accessed July 7, 2014.
How to avoid 3 common errors in dementia screening
› Use age- and education-corrected normative data when using dementia screening tools. C
› Use verbatim instructions and the same size stimuli and response pages provided in a test’s manual. C
› Ensure that norms used for comparisons are current. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Treatment options for dementia are expanding and improving, giving extra impetus to detecting this progressive disease as early as possible. For example, research on the cholinesterase inhibitor donepezil has shown it can delay cognitive decline by 6 months or more compared with controls1,2 and possibly postpone institutionalization. With the number of elderly individuals and cases of dementia projected to grow significantly over the next 20 years,3 primary care physicians will increasingly be screening for cognitive impairment. Given the time constraints and patient loads in today’s practices, it’s not surprising that physicians tend to use evaluation tools that are brief and simple to administer. However, there are also serious pitfalls in the use of these tools.
When to screen. Many health-related organizations address screening for dementia4,5 and offer screening criteria (eg, the Alzheimer’s Association,6 the US Preventive Services Task Force7). Our experience suggests that specific behavioral changes are reasonable indicators of suspected dementia that should prompt cognitive screening. Using the Kingston Standardized Behavioural Assessment,8 we demonstrated a consistent pattern of earliest behavior change in a community-dwelling group with dementia.9 Meaningful clues are a decreased ability to engage in specific functional activities (including participation in favorite pastimes, ability to eat properly if left to prepare one’s own food, handling of personal finances, word finding, and reading) and unsteadiness. These specific behavioral changes reported by family or a caregiver suggest the need for cognitive screening.
Pitfalls associated with common screening tools, if not taken into account, can seriously limit the usefulness of information gained during assessment and potentially lead to a wrong conclusion. Screening tools are just that: a means of detecting the possible existence of a condition. Results are based on probability and subject to error. Therefore, a single test score is insufficient to render a diagnosis of dementia, and is one variable in a set of diagnostic criteria.
The purpose of this article is to review some of the most commonly used tools and procedures for dementia screening, identify procedural or interpretive errors made in everyday clinical practice, and suggest practical yet simple strategies to address these problems and improve the accuracy of assessments. We illustrate key points with clinical examples and vignettes using the Mini-Mental State Examination (MMSE),10 an Animal Naming Task, and the Trail Making Test.11
Common error #1: Reliance on simple, single cutoff scores
There are a number of important considerations to keep in mind when trying to make sense of scores from the many available cognitive tests.
The range of normal test results is wide. The normal range for most physiologic measures, such as glucose levels or hemoglobin counts, is relatively narrow. However, human cognitive functions can naturally differ from person to person, and the range of normal can be extremely large.
A single, all-purpose cutoff score ignores critical factors. Very often, clinicians have dealt with the issue of wide variance in cognition scores by establishing a general cutoff point to serve as a pass-fail mark. But this practice can result in both under- and overidentification of dementia, and it ignores the 2 components that chiefly determine how individuals differ cognitively: age and intelligence.
Practical fix: Use age-, intelligence-corrected normative data
Level of cognitive performance can be revealing when adjustments are made for age and intelligence. Not taking these factors into account can lead to many errors in clinical decision making.
Age matters. Many cognitive capacities decline as part of normal aging even in otherwise healthy individuals (eg, reaction time, spatial abilities, flexibility in novel problem solving).12 With this in mind, psychologists often have made the distinction between “hold” tests (remaining stable or even improving with age) and “no-hold” tests (declining with age).13 Therefore it is critical to ask, “What is normal, given a particular patient’s age?” If normative data corrected for age are available for a given test, use them.
Intelligence is a factor, too. Intelligence, like most human qualities, is distributed along a bell-shaped curve of normal distribution, wherein most people fall somewhere in the middle and a smaller number will be at the lower and higher tails of the curve. Not all of us fall into the average range of intelligence; indeed, psychometrically, only half of us do. The other half are found somewhere in the more extreme ends. In evaluating a person for dementia, it is critical to compare test results with those found in the appropriate intellectual group. But how does the physician looking for a brief assessment strategy determine a patient’s premorbid level of intellectual functioning?
A widely used and accepted heuristic for gauging intelligence is “years of education.” Of course, education is not perfectly correlated with intelligence, particularly as those who are now elderly may have been denied the opportunity to attend school due to the Great Depression, war, or other life events. Nevertheless, with these limitations in mind, level of education is a reasonable approximation of intelligence. In practical application, premorbid intellectual level is determined by using education-corrected normative data.
Typically with cognitive tests, cutoff scores and score ranges are defined for general levels of education (eg, less than grade 12 or more than grade 12; elementary school, high school, post-secondary, etc). Adjusted norms for age and education are usually determined by taking large samples of subjects and stratifying the distribution by subgroups—eg, 5-year age groups; levels of education such as elementary school or high school—and then statistically analyzing each group and noting the relative differences between them.
Illustration: MMSE. Although not designed for the overall measurement of cognitive impairment in dementia, the MMSE10 has become widely used for that purpose. It is fairly insensitive to cognitive changes associated with earlier stages of dementia,14 and is intended only as a means of identifying patients in need of more comprehensive assessment. However, the MMSE is increasingly used to make a diagnosis of dementia.15 In some areas (eg, Ontario, Canada), it is used to justify paying for treatment with cognitive enhancers.

The universal cutoff score proves inadequate. Although several dementia cutoff scores for the MMSE have been proposed, it is common practice to use an MMSE score ≥24 to rule out dementia.16 In our clinical practice, however, many patients who ultimately are diagnosed with early dementia often perform well on the MMSE, although rather poorly on other dementia screens, such as the Kingston Standardized Cognitive Assessment-Revised (KSCAr)17 or the mini-KSCAr.18
Recently, we reviewed cases of >70 individuals from our outpatient clinic who were given the MMSE and were also diagnosed as having dementia by both DSM-IV (Diagnostic and Statistical Manual of Mental Disorders)19 and the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association20 criteria. Over three-quarters (78%) of these cases had an MMSE score of ≥24. Based on MMSE scores alone, these individuals would have been declared “not demented.”17
Correcting for age and intelligence increases accuracy. Published age and education norms are available for the MMSE.21 In applying these norms to our sample described above, the number of misidentified patients drops to approximately one-third (35.7%). This means that instead of misidentifying 2 out of 3 cases, the age and education corrections reduced this to about one out of 3, thereby increasing sensitivity and specificity. While this is still an unacceptably high rate of false negatives, it shows the considerable value of using age and education corrections.
The challenge of optimizing sensitivity and specificity of dementia screening tools is ongoing. As a matter of interest, we include TABLE 1,4,18,22-24 which shows calculated sensitivities and specificities of some commonly used screening tests.
Another practical fix: Use distributions and percentile-based normative data
Instead of simple cutoff scores, test scores can be, and often are, translated into percentiles to provide a meaningful context for evaluation and to make it easier to compare scores between patients. Someone with a score at the 70th percentile has performed as well as or better than 70% of others in the group who have taken the test. Usually, the average range of a normal population is defined as being between the 25th to 75th percentiles, encompassing 50% of that population. In general, percentiles make interpreting performance easier. Percentile-based test norms can also help determine with increased accuracy if there has been a decline over time.
Illustration: Animal naming task. In a common version of this task, patients are asked to name as many animals as they can in 60 seconds. This task has its roots in neuro- psychological tests of verbal fluency, such as the Controlled Oral Word Association Task.25 Verbal fluency tasks such as naming animals tap verbal generativity/problem-solving and self-monitoring, but are also highly dependent on vocabulary (word knowledge), a cognitive ability that is quite stable and even improves as one ages until individuals are well into their 80s.26
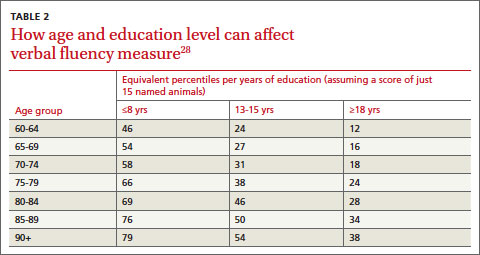
It is common practice with this procedure to consider a cutoff score of 15 as a minimally acceptable level of performance.27 Here again, there are potentially great differences in expected performance based on age and intelligence. TABLE 2 shows the effect of age and education on verbal fluency, expressed as percentiles, using a raw score of 15.28 For an individual in their early 60s who has a university degree, naming just 15 animals puts their performance at the 12th percentile (below average). The same performance for someone in their 90s who has only 8 years of education puts them in the 79th percentile (above the average range of 25th-75th percentiles). This performance would indicate impairment for the 60-year-old university-educated individual, but strong cognitive function for the 90-year-old.
Common error #2: Deviating from standardized procedures
While clinicians specifically trained in cognitive measurement are familiar with the rigor by which tests are constructed, those with less training are often unaware that even seemingly minor deviations in procedure can contaminate results as surely as using nonsterile containers in biologic testing, leading to inaccurate interpretations of cognition.
Practical fix: Administer tests using verbatim instructions
Failing to follow instructions can significantly bias acquired data, particularly when using performance tests that are timed.
Illustration: Trail Making Test. Trail Making is an old 2-part test developed for the United States Army in the 1940s,11 and used in the Halstead-Reitan neuropsychological battery. Part A is a timed measure of an individual’s ability to join up a series of numbered circles in ascending order. Part B measures the ability to alternately switch between 2 related tasks: namely, alternately joining numbered and lettered circles, in ascending order. This is considered a measure of complex attention, which is often disrupted in early dementia.29
The test uses a specific standardized set of instructions, and Part B’s interpretation depends on having first administered Part A. Anecdotally, we have increasingly seen clinician reports using only Part B. Eliminating Part A removes a significant opportunity for patients to become familiar with the task’s demands, placing them at a considerable disadvantage on Part B and thereby invalidating the normative data.
In addition, follow the exact phrasing of the instructions and use stimuli and response pages that are the same size as those provided in the manual. If a patient errs at any point, it’s important that the test administrator reads, verbatim, the provided correction statements because these statements influence the amount of time spent correcting an error and therefore the final score.
Common error #3: Using outdated normative data
Neglecting to use updated norms that reflect current cohort differences can compromise screening accuracy.
Practical fix: Ensure current norms are used for comparisons
Societal influences—computers and other technologies, nutrition, etc—have led to steady improvements in cognitive and physical abilities. In basic psychology, this pattern of improving cognition, documented as an approximate increase of 3 IQ points per decade, is referred to as the Flynn effect.30 Therefore, not only do age and education need to be controlled for, but normative data must be current.
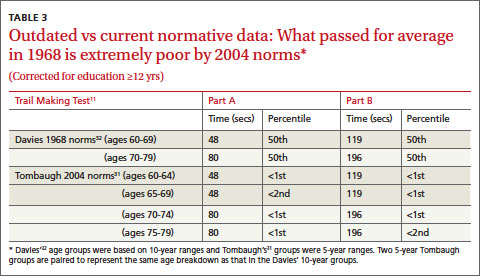
Cognitive screening tools are usually published with norms compiled at the time of the test’s development. However, scores are periodically “re-normed” to reflect current levels of ability. These updated norms are readily available in published journal articles or online. (Current norms for each of the tests used as examples in this article are provided in the references).21,28,31
Illustration: Trail Making Test. The normative data for this test are not only age- and education-sensitive, but are also highly sensitive to cohort effects. Early norms such as those of Davies,32 while often still quoted in literature and even in some training initiatives, are now seriously outdated and should not be used for interpretation. TABLE 3 shows how an average individual (ie, 50th percentile) in the 1960s, in one of 2 age groups, would compare in speed to an individual of similar age today.31 A time score that was at the 50th percentile in 1968 is now at or below the 1st percentile. More recent norms are also usually corrected for education, as are those provided by Tombaugh.31
In “A 'case' for using optimal procedures” (below), TABLE 4 shows the results of using outdated Trail Making norms vs current Trail Making norms.
George is a 77-year-old retired school teacher with >15 years of education who was referred to us for complaints of memory loss and suspicion of progressive cognitive deficits. On cognitive screening he scored 26/30 on the Mini-Mental State Examination, generated 16 animal names in 60 seconds, and completed Parts A and B of the Trail Making test in 80 seconds and 196 seconds, respectively. TABLE 4 summarizes test scores and interpretation with and without appropriate corrections.
George’s case dramatically illustrates the clinical impact of using (or not using) optimal interpretive procedures—ie, age and education corrections and current (not outdated) norms. Using the basic cutoff scores without corrections, George’s performance is within acceptable limits on all 3 screening tests, and he is sent home with the comforting news that his performance was within normal limits. However, by using appropriate comparative data, the same scores on all 3 screens indicate impairment. A likely next step would be referral for specialized testing. Monitoring for progressive deterioration is advisable, and perhaps initiation of medication.
TABLE 4
Trail Making: Outdated norms vs current norms
| Version 1 – No corrections for age or education for MMSE or COWAT; outdated Trail Making norms | |||
| Test | Score | Results | Suggests dementia |
| MMSE | 26 | ≥24 within normal limits10 | No |
| COWAT | 16 | >15 within normal limits25 | No |
| Trail Making A | 80 secs | 50th percentile32 | No |
| Trail Making B | 196 secs | 50th percentile32 | No |
| Decision: Negative for dementia | |||
|
|
|
|
|
| Version 2 – Applied age and education corrections for MMSE and COWAT; current Trail Making norms | |||
| Test | Score | Results | Suggests dementia |
| MMSE | 26 | Expected = 2822 | Yes |
| COWAT | 16 | 38th percentile28 | Yes |
| Trail Making A | 80 secs | <1st percentile31 | Yes |
| Trail Making B | 196 secs | <2nd percentile31 | Yes |
| Decision: Positive for dementia | |||
COWAT, Controlled Oral Word Association Task; MMSE, Mini-Mental State Examination.
Patients deserve an accurate assessment
A diagnosis of dementia profoundly affects patients and families. Progressive dementia such as Alzheimer’s disease means an individual will spend the rest of his or her life (usually 8-10 years) with decreasing cognitive capacity and quality of life.33-35 It also means families will spend years providing or arranging for care, and watching their family member deteriorate. Early detection can afford affected individuals and families the opportunity to make plans for fulfilling wishes and dreams before increased impairment makes such plans unattainable. The importance of rigor in assessment is therefore essential.
Optimizing accuracy in screening for dementia also can enable physicians to reasonably reassure patients that they likely do not suffer from a dementia at the present time, or to at least recommend that they be further assessed by a specialist. Without rigor, time and resources are wasted and the important question that triggered the referral is neither satisfactorily—nor accurately—addressed. Thus, there is a need to use not just simple cutoff scores but to apply the most current age and education normative data, and adhere to administrative instructions verbatim.
CORRESPONDENCE
Lindy A. Kilik, PhD, Geriatric Psychiatry Program, Providence Care Mental Health Services, PO Bag 603, Kingston, Ontario, Canada K7L 4X3; [email protected]
1. Loveman E, Green C, Kirby J, et al. The clinical and cost-effectiveness of donepezil, rivastigmine, galantamine and memantine for Alzheimer’s disease. Health Technol Assess. 2006;10:iii-iv,ix- xi,1-160.
2. Medical Care Corporation. Delaying the onset and progression of Alzheimer’s disease. Prevent AD Web site. Available at: http://www.preventad.com/pdf/support/article/DelayingADProgression.pdf. Accessed June 18, 2014.
3. Hopkins RW. Dementia projections for the counties, regional municipalities and districts of Ontario. Geriatric Psychiatry Unit Clinical/Research Bulletin, No. 16. Providence Care Web site. Available at: http://www.providencecare.ca/clinical-tools/Documents/Ontario-Dementia-Projections-2010.pdf. Accessed June 18, 2014.
4. Simmons BB, Hartmann B, Dejoseph D. Evaluation of suspected dementia. Am Fam Physician. 2011;84:895-902.
5. McCarten JR, Borson S. Should family physicians routinely screen patients for cognitive impairment? Yes: screening is the first step toward improving care. Am Fam Physician. 2014;89: 861-862.
6. Alzheimer’s Association. Health Care Professionals and Alzheimer’s. Alzheimer’s Association Web site. Available at: http://www.alz.org/health-care-professionals/cognitive-tests-patient-assessment.asp. Accessed June 18, 2014.
7. US Preventive Services Task Force. Screening for cognitive impairment in older adults. US Preventive Services Task Force Web site. Available at: http://www.uspreventiveservicestaskforce.org/uspstf/uspsdeme.htm. Accessed June 18, 2014.
8. Hopkins RW, Kilik LA, Day D, et al. Kingston Standardized Behavioural Assessment. Am J Alzheimers Dis Other Demen. 2006;21:339-346.
9. Kilik LA, Hopkins RW, Day D, et al. The progression of behaviour in dementia: an in-office guide for clinicians. Am J Alzheimers Dis Other Demen. 2008;23:242-249.
10. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189-198.
11. Army Individual Test Battery. Manual of Directions and Scoring. Washington, DC: War Department, Adjutant General’s Office; 1944.
12. Wechsler D. The Measurement and Appraisal of Adult Intelligence. 4th ed. The Williams & Wilkins Company: Baltimore, MD; 1958.
13. Larrabee GJ, Largen JW, Levin HS. Sensitivity of age-decline resistant (“hold”) WAIS subtests to Alzheimer’s disease. J Clin Exp Neuropsychol. 1985;7:497-504.
14. Herndon RM. Assessment of the elderly with dementia. In: Handbook of Neurologic Rating Scales. 2nd ed. Demos Medical Publishing LLC: New York, NY; 2006:199.
15. Brugnolo A, Nobili F, Barbieri MP, et al. The factorial structure of the mini mental state examination (MMSE) in Alzheimer’s disease. Arch Gerontol Geriatr. 2009;49:180-185.
16. Folstein M, Anthony JC, Parhad I, et al. The meaning of cognitive impairment in the elderly. J Am Geriatr Soc. 1985;33:228-235.
17. Hopkins RW, Kilik LA, Day DJ, et al. The Revised Kingston Standardized Cognitive Assessment. Int J Geriatr Psychiatry. 2004;19:320-326.
18. Hopkins R, Kilik L. The mini-Kingston Standardized Cognitive Assessment. Kingston Scales Web site. Available at: http://www.kingstonscales.org. Accessed June 18, 2014.
19. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: American Psychiatric Association; 1994.
20. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34: 939-944.
21. Crum RM, Anthony JC, Bassett SS, et al. Population-based norms for the Mini-Mental State Examination by age and educational level. JAMA. 1993;269:2386-2391.
22. O’Bryant SE, Humphreys JD, Smith GE, et al. Detecting dementia with the mini-mental state examination in highly educated individuals. Arch Neurol. 2008;65:963-967.
23. O’Sullivan M, Morris RG, Markus HS. Brief cognitive assessment for patients with cerebral small vessel disease. J Neurol Neurosurg Psychiatry. 2005;76:1140-1145.
24. Nasreddine ZS, Phillips NA, Bédirian V, et al. The Montreal Cognitive Assessment (MoCA): a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53:695-699.
25. Benton AL, Hamsher K. Multilingual Aphasia Examination. 2nd ed. Iowa City, OA: AJA Associates, Inc; 1976.
26. Wechsler D. WAIS-III Administration and Scoring Manual. San Antonio, TX: The Psychological Corporation; 1997.
27. Morris JC, Heyman A, Mohs RC, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part I. Clinical and neuropsychological assesment of Alzheimer’s disease. Neurology. 1989;39:1159-1165.
28. Gladsjo JA, Miller SW, Heaton RK. Norms for Letter and Category Fluency: Demographic Corrections for Age, Education and Ethnicity. Odessa, FL: Psychological Assessment Resources; 1999.
29. Perry R, Hodges J. Attention and executive deficits in Alzheimer’s disease. A critical review. Brain. 1999;122(pt 3):383-404.
30. Flynn JR. The mean IQ of Americans: Massive gains 1932 to 1978. Psychol Bull. 1984;95:29-51.
31. Tombaugh TN. Trail Making Test A and B: normative data stratified by age and education. Arch Clin Neuropsychol. 2004;19: 203-214.
32. Davies A. The influence of age on trail making test performance. J Clin Psychol. 1968;24:96-98.
33. Bianchetti A, Trabucch M. Clinical aspects of Alzheimer’s disease. Aging (Milano). 2001;13:221-230.
34. Kay D, Forster DP, Newens AJ. Long-term survival, place of death, and death certification in clinically diagnosed pre-senile dementia in northern England. Follow-up after 8-12 years. Br J Psychiatry. 2000;177:156-162.
35. Chaussalet T, Thompson WA. Data requirements in a model of the natural history of Alzheimer’s disease. Health Care Manag Sci. 2001;4:13-19.
› Use age- and education-corrected normative data when using dementia screening tools. C
› Use verbatim instructions and the same size stimuli and response pages provided in a test’s manual. C
› Ensure that norms used for comparisons are current. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Treatment options for dementia are expanding and improving, giving extra impetus to detecting this progressive disease as early as possible. For example, research on the cholinesterase inhibitor donepezil has shown it can delay cognitive decline by 6 months or more compared with controls1,2 and possibly postpone institutionalization. With the number of elderly individuals and cases of dementia projected to grow significantly over the next 20 years,3 primary care physicians will increasingly be screening for cognitive impairment. Given the time constraints and patient loads in today’s practices, it’s not surprising that physicians tend to use evaluation tools that are brief and simple to administer. However, there are also serious pitfalls in the use of these tools.
When to screen. Many health-related organizations address screening for dementia4,5 and offer screening criteria (eg, the Alzheimer’s Association,6 the US Preventive Services Task Force7). Our experience suggests that specific behavioral changes are reasonable indicators of suspected dementia that should prompt cognitive screening. Using the Kingston Standardized Behavioural Assessment,8 we demonstrated a consistent pattern of earliest behavior change in a community-dwelling group with dementia.9 Meaningful clues are a decreased ability to engage in specific functional activities (including participation in favorite pastimes, ability to eat properly if left to prepare one’s own food, handling of personal finances, word finding, and reading) and unsteadiness. These specific behavioral changes reported by family or a caregiver suggest the need for cognitive screening.
Pitfalls associated with common screening tools, if not taken into account, can seriously limit the usefulness of information gained during assessment and potentially lead to a wrong conclusion. Screening tools are just that: a means of detecting the possible existence of a condition. Results are based on probability and subject to error. Therefore, a single test score is insufficient to render a diagnosis of dementia, and is one variable in a set of diagnostic criteria.
The purpose of this article is to review some of the most commonly used tools and procedures for dementia screening, identify procedural or interpretive errors made in everyday clinical practice, and suggest practical yet simple strategies to address these problems and improve the accuracy of assessments. We illustrate key points with clinical examples and vignettes using the Mini-Mental State Examination (MMSE),10 an Animal Naming Task, and the Trail Making Test.11
Common error #1: Reliance on simple, single cutoff scores
There are a number of important considerations to keep in mind when trying to make sense of scores from the many available cognitive tests.
The range of normal test results is wide. The normal range for most physiologic measures, such as glucose levels or hemoglobin counts, is relatively narrow. However, human cognitive functions can naturally differ from person to person, and the range of normal can be extremely large.
A single, all-purpose cutoff score ignores critical factors. Very often, clinicians have dealt with the issue of wide variance in cognition scores by establishing a general cutoff point to serve as a pass-fail mark. But this practice can result in both under- and overidentification of dementia, and it ignores the 2 components that chiefly determine how individuals differ cognitively: age and intelligence.
Practical fix: Use age-, intelligence-corrected normative data
Level of cognitive performance can be revealing when adjustments are made for age and intelligence. Not taking these factors into account can lead to many errors in clinical decision making.
Age matters. Many cognitive capacities decline as part of normal aging even in otherwise healthy individuals (eg, reaction time, spatial abilities, flexibility in novel problem solving).12 With this in mind, psychologists often have made the distinction between “hold” tests (remaining stable or even improving with age) and “no-hold” tests (declining with age).13 Therefore it is critical to ask, “What is normal, given a particular patient’s age?” If normative data corrected for age are available for a given test, use them.
Intelligence is a factor, too. Intelligence, like most human qualities, is distributed along a bell-shaped curve of normal distribution, wherein most people fall somewhere in the middle and a smaller number will be at the lower and higher tails of the curve. Not all of us fall into the average range of intelligence; indeed, psychometrically, only half of us do. The other half are found somewhere in the more extreme ends. In evaluating a person for dementia, it is critical to compare test results with those found in the appropriate intellectual group. But how does the physician looking for a brief assessment strategy determine a patient’s premorbid level of intellectual functioning?
A widely used and accepted heuristic for gauging intelligence is “years of education.” Of course, education is not perfectly correlated with intelligence, particularly as those who are now elderly may have been denied the opportunity to attend school due to the Great Depression, war, or other life events. Nevertheless, with these limitations in mind, level of education is a reasonable approximation of intelligence. In practical application, premorbid intellectual level is determined by using education-corrected normative data.
Typically with cognitive tests, cutoff scores and score ranges are defined for general levels of education (eg, less than grade 12 or more than grade 12; elementary school, high school, post-secondary, etc). Adjusted norms for age and education are usually determined by taking large samples of subjects and stratifying the distribution by subgroups—eg, 5-year age groups; levels of education such as elementary school or high school—and then statistically analyzing each group and noting the relative differences between them.
Illustration: MMSE. Although not designed for the overall measurement of cognitive impairment in dementia, the MMSE10 has become widely used for that purpose. It is fairly insensitive to cognitive changes associated with earlier stages of dementia,14 and is intended only as a means of identifying patients in need of more comprehensive assessment. However, the MMSE is increasingly used to make a diagnosis of dementia.15 In some areas (eg, Ontario, Canada), it is used to justify paying for treatment with cognitive enhancers.
The universal cutoff score proves inadequate. Although several dementia cutoff scores for the MMSE have been proposed, it is common practice to use an MMSE score ≥24 to rule out dementia.16 In our clinical practice, however, many patients who ultimately are diagnosed with early dementia often perform well on the MMSE, although rather poorly on other dementia screens, such as the Kingston Standardized Cognitive Assessment-Revised (KSCAr)17 or the mini-KSCAr.18
Recently, we reviewed cases of >70 individuals from our outpatient clinic who were given the MMSE and were also diagnosed as having dementia by both DSM-IV (Diagnostic and Statistical Manual of Mental Disorders)19 and the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association20 criteria. Over three-quarters (78%) of these cases had an MMSE score of ≥24. Based on MMSE scores alone, these individuals would have been declared “not demented.”17
Correcting for age and intelligence increases accuracy. Published age and education norms are available for the MMSE.21 In applying these norms to our sample described above, the number of misidentified patients drops to approximately one-third (35.7%). This means that instead of misidentifying 2 out of 3 cases, the age and education corrections reduced this to about one out of 3, thereby increasing sensitivity and specificity. While this is still an unacceptably high rate of false negatives, it shows the considerable value of using age and education corrections.
The challenge of optimizing sensitivity and specificity of dementia screening tools is ongoing. As a matter of interest, we include TABLE 1,4,18,22-24 which shows calculated sensitivities and specificities of some commonly used screening tests.
Another practical fix: Use distributions and percentile-based normative data
Instead of simple cutoff scores, test scores can be, and often are, translated into percentiles to provide a meaningful context for evaluation and to make it easier to compare scores between patients. Someone with a score at the 70th percentile has performed as well as or better than 70% of others in the group who have taken the test. Usually, the average range of a normal population is defined as being between the 25th to 75th percentiles, encompassing 50% of that population. In general, percentiles make interpreting performance easier. Percentile-based test norms can also help determine with increased accuracy if there has been a decline over time.
Illustration: Animal naming task. In a common version of this task, patients are asked to name as many animals as they can in 60 seconds. This task has its roots in neuro- psychological tests of verbal fluency, such as the Controlled Oral Word Association Task.25 Verbal fluency tasks such as naming animals tap verbal generativity/problem-solving and self-monitoring, but are also highly dependent on vocabulary (word knowledge), a cognitive ability that is quite stable and even improves as one ages until individuals are well into their 80s.26
It is common practice with this procedure to consider a cutoff score of 15 as a minimally acceptable level of performance.27 Here again, there are potentially great differences in expected performance based on age and intelligence. TABLE 2 shows the effect of age and education on verbal fluency, expressed as percentiles, using a raw score of 15.28 For an individual in their early 60s who has a university degree, naming just 15 animals puts their performance at the 12th percentile (below average). The same performance for someone in their 90s who has only 8 years of education puts them in the 79th percentile (above the average range of 25th-75th percentiles). This performance would indicate impairment for the 60-year-old university-educated individual, but strong cognitive function for the 90-year-old.
Common error #2: Deviating from standardized procedures
While clinicians specifically trained in cognitive measurement are familiar with the rigor by which tests are constructed, those with less training are often unaware that even seemingly minor deviations in procedure can contaminate results as surely as using nonsterile containers in biologic testing, leading to inaccurate interpretations of cognition.
Practical fix: Administer tests using verbatim instructions
Failing to follow instructions can significantly bias acquired data, particularly when using performance tests that are timed.
Illustration: Trail Making Test. Trail Making is an old 2-part test developed for the United States Army in the 1940s,11 and used in the Halstead-Reitan neuropsychological battery. Part A is a timed measure of an individual’s ability to join up a series of numbered circles in ascending order. Part B measures the ability to alternately switch between 2 related tasks: namely, alternately joining numbered and lettered circles, in ascending order. This is considered a measure of complex attention, which is often disrupted in early dementia.29
The test uses a specific standardized set of instructions, and Part B’s interpretation depends on having first administered Part A. Anecdotally, we have increasingly seen clinician reports using only Part B. Eliminating Part A removes a significant opportunity for patients to become familiar with the task’s demands, placing them at a considerable disadvantage on Part B and thereby invalidating the normative data.
In addition, follow the exact phrasing of the instructions and use stimuli and response pages that are the same size as those provided in the manual. If a patient errs at any point, it’s important that the test administrator reads, verbatim, the provided correction statements because these statements influence the amount of time spent correcting an error and therefore the final score.
Common error #3: Using outdated normative data
Neglecting to use updated norms that reflect current cohort differences can compromise screening accuracy.
Practical fix: Ensure current norms are used for comparisons
Societal influences—computers and other technologies, nutrition, etc—have led to steady improvements in cognitive and physical abilities. In basic psychology, this pattern of improving cognition, documented as an approximate increase of 3 IQ points per decade, is referred to as the Flynn effect.30 Therefore, not only do age and education need to be controlled for, but normative data must be current.
Cognitive screening tools are usually published with norms compiled at the time of the test’s development. However, scores are periodically “re-normed” to reflect current levels of ability. These updated norms are readily available in published journal articles or online. (Current norms for each of the tests used as examples in this article are provided in the references).21,28,31
Illustration: Trail Making Test. The normative data for this test are not only age- and education-sensitive, but are also highly sensitive to cohort effects. Early norms such as those of Davies,32 while often still quoted in literature and even in some training initiatives, are now seriously outdated and should not be used for interpretation. TABLE 3 shows how an average individual (ie, 50th percentile) in the 1960s, in one of 2 age groups, would compare in speed to an individual of similar age today.31 A time score that was at the 50th percentile in 1968 is now at or below the 1st percentile. More recent norms are also usually corrected for education, as are those provided by Tombaugh.31
In “A 'case' for using optimal procedures” (below), TABLE 4 shows the results of using outdated Trail Making norms vs current Trail Making norms.
George is a 77-year-old retired school teacher with >15 years of education who was referred to us for complaints of memory loss and suspicion of progressive cognitive deficits. On cognitive screening he scored 26/30 on the Mini-Mental State Examination, generated 16 animal names in 60 seconds, and completed Parts A and B of the Trail Making test in 80 seconds and 196 seconds, respectively. TABLE 4 summarizes test scores and interpretation with and without appropriate corrections.
George’s case dramatically illustrates the clinical impact of using (or not using) optimal interpretive procedures—ie, age and education corrections and current (not outdated) norms. Using the basic cutoff scores without corrections, George’s performance is within acceptable limits on all 3 screening tests, and he is sent home with the comforting news that his performance was within normal limits. However, by using appropriate comparative data, the same scores on all 3 screens indicate impairment. A likely next step would be referral for specialized testing. Monitoring for progressive deterioration is advisable, and perhaps initiation of medication.
TABLE 4
Trail Making: Outdated norms vs current norms
| Version 1 – No corrections for age or education for MMSE or COWAT; outdated Trail Making norms | |||
| Test | Score | Results | Suggests dementia |
| MMSE | 26 | ≥24 within normal limits10 | No |
| COWAT | 16 | >15 within normal limits25 | No |
| Trail Making A | 80 secs | 50th percentile32 | No |
| Trail Making B | 196 secs | 50th percentile32 | No |
| Decision: Negative for dementia | |||
|
|
|
|
|
| Version 2 – Applied age and education corrections for MMSE and COWAT; current Trail Making norms | |||
| Test | Score | Results | Suggests dementia |
| MMSE | 26 | Expected = 2822 | Yes |
| COWAT | 16 | 38th percentile28 | Yes |
| Trail Making A | 80 secs | <1st percentile31 | Yes |
| Trail Making B | 196 secs | <2nd percentile31 | Yes |
| Decision: Positive for dementia | |||
COWAT, Controlled Oral Word Association Task; MMSE, Mini-Mental State Examination.
Patients deserve an accurate assessment
A diagnosis of dementia profoundly affects patients and families. Progressive dementia such as Alzheimer’s disease means an individual will spend the rest of his or her life (usually 8-10 years) with decreasing cognitive capacity and quality of life.33-35 It also means families will spend years providing or arranging for care, and watching their family member deteriorate. Early detection can afford affected individuals and families the opportunity to make plans for fulfilling wishes and dreams before increased impairment makes such plans unattainable. The importance of rigor in assessment is therefore essential.
Optimizing accuracy in screening for dementia also can enable physicians to reasonably reassure patients that they likely do not suffer from a dementia at the present time, or to at least recommend that they be further assessed by a specialist. Without rigor, time and resources are wasted and the important question that triggered the referral is neither satisfactorily—nor accurately—addressed. Thus, there is a need to use not just simple cutoff scores but to apply the most current age and education normative data, and adhere to administrative instructions verbatim.
CORRESPONDENCE
Lindy A. Kilik, PhD, Geriatric Psychiatry Program, Providence Care Mental Health Services, PO Bag 603, Kingston, Ontario, Canada K7L 4X3; [email protected]
› Use age- and education-corrected normative data when using dementia screening tools. C
› Use verbatim instructions and the same size stimuli and response pages provided in a test’s manual. C
› Ensure that norms used for comparisons are current. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Treatment options for dementia are expanding and improving, giving extra impetus to detecting this progressive disease as early as possible. For example, research on the cholinesterase inhibitor donepezil has shown it can delay cognitive decline by 6 months or more compared with controls1,2 and possibly postpone institutionalization. With the number of elderly individuals and cases of dementia projected to grow significantly over the next 20 years,3 primary care physicians will increasingly be screening for cognitive impairment. Given the time constraints and patient loads in today’s practices, it’s not surprising that physicians tend to use evaluation tools that are brief and simple to administer. However, there are also serious pitfalls in the use of these tools.
When to screen. Many health-related organizations address screening for dementia4,5 and offer screening criteria (eg, the Alzheimer’s Association,6 the US Preventive Services Task Force7). Our experience suggests that specific behavioral changes are reasonable indicators of suspected dementia that should prompt cognitive screening. Using the Kingston Standardized Behavioural Assessment,8 we demonstrated a consistent pattern of earliest behavior change in a community-dwelling group with dementia.9 Meaningful clues are a decreased ability to engage in specific functional activities (including participation in favorite pastimes, ability to eat properly if left to prepare one’s own food, handling of personal finances, word finding, and reading) and unsteadiness. These specific behavioral changes reported by family or a caregiver suggest the need for cognitive screening.
Pitfalls associated with common screening tools, if not taken into account, can seriously limit the usefulness of information gained during assessment and potentially lead to a wrong conclusion. Screening tools are just that: a means of detecting the possible existence of a condition. Results are based on probability and subject to error. Therefore, a single test score is insufficient to render a diagnosis of dementia, and is one variable in a set of diagnostic criteria.
The purpose of this article is to review some of the most commonly used tools and procedures for dementia screening, identify procedural or interpretive errors made in everyday clinical practice, and suggest practical yet simple strategies to address these problems and improve the accuracy of assessments. We illustrate key points with clinical examples and vignettes using the Mini-Mental State Examination (MMSE),10 an Animal Naming Task, and the Trail Making Test.11
Common error #1: Reliance on simple, single cutoff scores
There are a number of important considerations to keep in mind when trying to make sense of scores from the many available cognitive tests.
The range of normal test results is wide. The normal range for most physiologic measures, such as glucose levels or hemoglobin counts, is relatively narrow. However, human cognitive functions can naturally differ from person to person, and the range of normal can be extremely large.
A single, all-purpose cutoff score ignores critical factors. Very often, clinicians have dealt with the issue of wide variance in cognition scores by establishing a general cutoff point to serve as a pass-fail mark. But this practice can result in both under- and overidentification of dementia, and it ignores the 2 components that chiefly determine how individuals differ cognitively: age and intelligence.
Practical fix: Use age-, intelligence-corrected normative data
Level of cognitive performance can be revealing when adjustments are made for age and intelligence. Not taking these factors into account can lead to many errors in clinical decision making.
Age matters. Many cognitive capacities decline as part of normal aging even in otherwise healthy individuals (eg, reaction time, spatial abilities, flexibility in novel problem solving).12 With this in mind, psychologists often have made the distinction between “hold” tests (remaining stable or even improving with age) and “no-hold” tests (declining with age).13 Therefore it is critical to ask, “What is normal, given a particular patient’s age?” If normative data corrected for age are available for a given test, use them.
Intelligence is a factor, too. Intelligence, like most human qualities, is distributed along a bell-shaped curve of normal distribution, wherein most people fall somewhere in the middle and a smaller number will be at the lower and higher tails of the curve. Not all of us fall into the average range of intelligence; indeed, psychometrically, only half of us do. The other half are found somewhere in the more extreme ends. In evaluating a person for dementia, it is critical to compare test results with those found in the appropriate intellectual group. But how does the physician looking for a brief assessment strategy determine a patient’s premorbid level of intellectual functioning?
A widely used and accepted heuristic for gauging intelligence is “years of education.” Of course, education is not perfectly correlated with intelligence, particularly as those who are now elderly may have been denied the opportunity to attend school due to the Great Depression, war, or other life events. Nevertheless, with these limitations in mind, level of education is a reasonable approximation of intelligence. In practical application, premorbid intellectual level is determined by using education-corrected normative data.
Typically with cognitive tests, cutoff scores and score ranges are defined for general levels of education (eg, less than grade 12 or more than grade 12; elementary school, high school, post-secondary, etc). Adjusted norms for age and education are usually determined by taking large samples of subjects and stratifying the distribution by subgroups—eg, 5-year age groups; levels of education such as elementary school or high school—and then statistically analyzing each group and noting the relative differences between them.
Illustration: MMSE. Although not designed for the overall measurement of cognitive impairment in dementia, the MMSE10 has become widely used for that purpose. It is fairly insensitive to cognitive changes associated with earlier stages of dementia,14 and is intended only as a means of identifying patients in need of more comprehensive assessment. However, the MMSE is increasingly used to make a diagnosis of dementia.15 In some areas (eg, Ontario, Canada), it is used to justify paying for treatment with cognitive enhancers.
The universal cutoff score proves inadequate. Although several dementia cutoff scores for the MMSE have been proposed, it is common practice to use an MMSE score ≥24 to rule out dementia.16 In our clinical practice, however, many patients who ultimately are diagnosed with early dementia often perform well on the MMSE, although rather poorly on other dementia screens, such as the Kingston Standardized Cognitive Assessment-Revised (KSCAr)17 or the mini-KSCAr.18
Recently, we reviewed cases of >70 individuals from our outpatient clinic who were given the MMSE and were also diagnosed as having dementia by both DSM-IV (Diagnostic and Statistical Manual of Mental Disorders)19 and the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association20 criteria. Over three-quarters (78%) of these cases had an MMSE score of ≥24. Based on MMSE scores alone, these individuals would have been declared “not demented.”17
Correcting for age and intelligence increases accuracy. Published age and education norms are available for the MMSE.21 In applying these norms to our sample described above, the number of misidentified patients drops to approximately one-third (35.7%). This means that instead of misidentifying 2 out of 3 cases, the age and education corrections reduced this to about one out of 3, thereby increasing sensitivity and specificity. While this is still an unacceptably high rate of false negatives, it shows the considerable value of using age and education corrections.
The challenge of optimizing sensitivity and specificity of dementia screening tools is ongoing. As a matter of interest, we include TABLE 1,4,18,22-24 which shows calculated sensitivities and specificities of some commonly used screening tests.
Another practical fix: Use distributions and percentile-based normative data
Instead of simple cutoff scores, test scores can be, and often are, translated into percentiles to provide a meaningful context for evaluation and to make it easier to compare scores between patients. Someone with a score at the 70th percentile has performed as well as or better than 70% of others in the group who have taken the test. Usually, the average range of a normal population is defined as being between the 25th to 75th percentiles, encompassing 50% of that population. In general, percentiles make interpreting performance easier. Percentile-based test norms can also help determine with increased accuracy if there has been a decline over time.
Illustration: Animal naming task. In a common version of this task, patients are asked to name as many animals as they can in 60 seconds. This task has its roots in neuro- psychological tests of verbal fluency, such as the Controlled Oral Word Association Task.25 Verbal fluency tasks such as naming animals tap verbal generativity/problem-solving and self-monitoring, but are also highly dependent on vocabulary (word knowledge), a cognitive ability that is quite stable and even improves as one ages until individuals are well into their 80s.26
It is common practice with this procedure to consider a cutoff score of 15 as a minimally acceptable level of performance.27 Here again, there are potentially great differences in expected performance based on age and intelligence. TABLE 2 shows the effect of age and education on verbal fluency, expressed as percentiles, using a raw score of 15.28 For an individual in their early 60s who has a university degree, naming just 15 animals puts their performance at the 12th percentile (below average). The same performance for someone in their 90s who has only 8 years of education puts them in the 79th percentile (above the average range of 25th-75th percentiles). This performance would indicate impairment for the 60-year-old university-educated individual, but strong cognitive function for the 90-year-old.
Common error #2: Deviating from standardized procedures
While clinicians specifically trained in cognitive measurement are familiar with the rigor by which tests are constructed, those with less training are often unaware that even seemingly minor deviations in procedure can contaminate results as surely as using nonsterile containers in biologic testing, leading to inaccurate interpretations of cognition.
Practical fix: Administer tests using verbatim instructions
Failing to follow instructions can significantly bias acquired data, particularly when using performance tests that are timed.
Illustration: Trail Making Test. Trail Making is an old 2-part test developed for the United States Army in the 1940s,11 and used in the Halstead-Reitan neuropsychological battery. Part A is a timed measure of an individual’s ability to join up a series of numbered circles in ascending order. Part B measures the ability to alternately switch between 2 related tasks: namely, alternately joining numbered and lettered circles, in ascending order. This is considered a measure of complex attention, which is often disrupted in early dementia.29
The test uses a specific standardized set of instructions, and Part B’s interpretation depends on having first administered Part A. Anecdotally, we have increasingly seen clinician reports using only Part B. Eliminating Part A removes a significant opportunity for patients to become familiar with the task’s demands, placing them at a considerable disadvantage on Part B and thereby invalidating the normative data.
In addition, follow the exact phrasing of the instructions and use stimuli and response pages that are the same size as those provided in the manual. If a patient errs at any point, it’s important that the test administrator reads, verbatim, the provided correction statements because these statements influence the amount of time spent correcting an error and therefore the final score.
Common error #3: Using outdated normative data
Neglecting to use updated norms that reflect current cohort differences can compromise screening accuracy.
Practical fix: Ensure current norms are used for comparisons
Societal influences—computers and other technologies, nutrition, etc—have led to steady improvements in cognitive and physical abilities. In basic psychology, this pattern of improving cognition, documented as an approximate increase of 3 IQ points per decade, is referred to as the Flynn effect.30 Therefore, not only do age and education need to be controlled for, but normative data must be current.
Cognitive screening tools are usually published with norms compiled at the time of the test’s development. However, scores are periodically “re-normed” to reflect current levels of ability. These updated norms are readily available in published journal articles or online. (Current norms for each of the tests used as examples in this article are provided in the references).21,28,31
Illustration: Trail Making Test. The normative data for this test are not only age- and education-sensitive, but are also highly sensitive to cohort effects. Early norms such as those of Davies,32 while often still quoted in literature and even in some training initiatives, are now seriously outdated and should not be used for interpretation. TABLE 3 shows how an average individual (ie, 50th percentile) in the 1960s, in one of 2 age groups, would compare in speed to an individual of similar age today.31 A time score that was at the 50th percentile in 1968 is now at or below the 1st percentile. More recent norms are also usually corrected for education, as are those provided by Tombaugh.31
In “A 'case' for using optimal procedures” (below), TABLE 4 shows the results of using outdated Trail Making norms vs current Trail Making norms.
George is a 77-year-old retired school teacher with >15 years of education who was referred to us for complaints of memory loss and suspicion of progressive cognitive deficits. On cognitive screening he scored 26/30 on the Mini-Mental State Examination, generated 16 animal names in 60 seconds, and completed Parts A and B of the Trail Making test in 80 seconds and 196 seconds, respectively. TABLE 4 summarizes test scores and interpretation with and without appropriate corrections.
George’s case dramatically illustrates the clinical impact of using (or not using) optimal interpretive procedures—ie, age and education corrections and current (not outdated) norms. Using the basic cutoff scores without corrections, George’s performance is within acceptable limits on all 3 screening tests, and he is sent home with the comforting news that his performance was within normal limits. However, by using appropriate comparative data, the same scores on all 3 screens indicate impairment. A likely next step would be referral for specialized testing. Monitoring for progressive deterioration is advisable, and perhaps initiation of medication.
TABLE 4
Trail Making: Outdated norms vs current norms
| Version 1 – No corrections for age or education for MMSE or COWAT; outdated Trail Making norms | |||
| Test | Score | Results | Suggests dementia |
| MMSE | 26 | ≥24 within normal limits10 | No |
| COWAT | 16 | >15 within normal limits25 | No |
| Trail Making A | 80 secs | 50th percentile32 | No |
| Trail Making B | 196 secs | 50th percentile32 | No |
| Decision: Negative for dementia | |||
|
|
|
|
|
| Version 2 – Applied age and education corrections for MMSE and COWAT; current Trail Making norms | |||
| Test | Score | Results | Suggests dementia |
| MMSE | 26 | Expected = 2822 | Yes |
| COWAT | 16 | 38th percentile28 | Yes |
| Trail Making A | 80 secs | <1st percentile31 | Yes |
| Trail Making B | 196 secs | <2nd percentile31 | Yes |
| Decision: Positive for dementia | |||
COWAT, Controlled Oral Word Association Task; MMSE, Mini-Mental State Examination.
Patients deserve an accurate assessment
A diagnosis of dementia profoundly affects patients and families. Progressive dementia such as Alzheimer’s disease means an individual will spend the rest of his or her life (usually 8-10 years) with decreasing cognitive capacity and quality of life.33-35 It also means families will spend years providing or arranging for care, and watching their family member deteriorate. Early detection can afford affected individuals and families the opportunity to make plans for fulfilling wishes and dreams before increased impairment makes such plans unattainable. The importance of rigor in assessment is therefore essential.
Optimizing accuracy in screening for dementia also can enable physicians to reasonably reassure patients that they likely do not suffer from a dementia at the present time, or to at least recommend that they be further assessed by a specialist. Without rigor, time and resources are wasted and the important question that triggered the referral is neither satisfactorily—nor accurately—addressed. Thus, there is a need to use not just simple cutoff scores but to apply the most current age and education normative data, and adhere to administrative instructions verbatim.
CORRESPONDENCE
Lindy A. Kilik, PhD, Geriatric Psychiatry Program, Providence Care Mental Health Services, PO Bag 603, Kingston, Ontario, Canada K7L 4X3; [email protected]
1. Loveman E, Green C, Kirby J, et al. The clinical and cost-effectiveness of donepezil, rivastigmine, galantamine and memantine for Alzheimer’s disease. Health Technol Assess. 2006;10:iii-iv,ix- xi,1-160.
2. Medical Care Corporation. Delaying the onset and progression of Alzheimer’s disease. Prevent AD Web site. Available at: http://www.preventad.com/pdf/support/article/DelayingADProgression.pdf. Accessed June 18, 2014.
3. Hopkins RW. Dementia projections for the counties, regional municipalities and districts of Ontario. Geriatric Psychiatry Unit Clinical/Research Bulletin, No. 16. Providence Care Web site. Available at: http://www.providencecare.ca/clinical-tools/Documents/Ontario-Dementia-Projections-2010.pdf. Accessed June 18, 2014.
4. Simmons BB, Hartmann B, Dejoseph D. Evaluation of suspected dementia. Am Fam Physician. 2011;84:895-902.
5. McCarten JR, Borson S. Should family physicians routinely screen patients for cognitive impairment? Yes: screening is the first step toward improving care. Am Fam Physician. 2014;89: 861-862.
6. Alzheimer’s Association. Health Care Professionals and Alzheimer’s. Alzheimer’s Association Web site. Available at: http://www.alz.org/health-care-professionals/cognitive-tests-patient-assessment.asp. Accessed June 18, 2014.
7. US Preventive Services Task Force. Screening for cognitive impairment in older adults. US Preventive Services Task Force Web site. Available at: http://www.uspreventiveservicestaskforce.org/uspstf/uspsdeme.htm. Accessed June 18, 2014.
8. Hopkins RW, Kilik LA, Day D, et al. Kingston Standardized Behavioural Assessment. Am J Alzheimers Dis Other Demen. 2006;21:339-346.
9. Kilik LA, Hopkins RW, Day D, et al. The progression of behaviour in dementia: an in-office guide for clinicians. Am J Alzheimers Dis Other Demen. 2008;23:242-249.
10. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189-198.
11. Army Individual Test Battery. Manual of Directions and Scoring. Washington, DC: War Department, Adjutant General’s Office; 1944.
12. Wechsler D. The Measurement and Appraisal of Adult Intelligence. 4th ed. The Williams & Wilkins Company: Baltimore, MD; 1958.
13. Larrabee GJ, Largen JW, Levin HS. Sensitivity of age-decline resistant (“hold”) WAIS subtests to Alzheimer’s disease. J Clin Exp Neuropsychol. 1985;7:497-504.
14. Herndon RM. Assessment of the elderly with dementia. In: Handbook of Neurologic Rating Scales. 2nd ed. Demos Medical Publishing LLC: New York, NY; 2006:199.
15. Brugnolo A, Nobili F, Barbieri MP, et al. The factorial structure of the mini mental state examination (MMSE) in Alzheimer’s disease. Arch Gerontol Geriatr. 2009;49:180-185.
16. Folstein M, Anthony JC, Parhad I, et al. The meaning of cognitive impairment in the elderly. J Am Geriatr Soc. 1985;33:228-235.
17. Hopkins RW, Kilik LA, Day DJ, et al. The Revised Kingston Standardized Cognitive Assessment. Int J Geriatr Psychiatry. 2004;19:320-326.
18. Hopkins R, Kilik L. The mini-Kingston Standardized Cognitive Assessment. Kingston Scales Web site. Available at: http://www.kingstonscales.org. Accessed June 18, 2014.
19. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: American Psychiatric Association; 1994.
20. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34: 939-944.
21. Crum RM, Anthony JC, Bassett SS, et al. Population-based norms for the Mini-Mental State Examination by age and educational level. JAMA. 1993;269:2386-2391.
22. O’Bryant SE, Humphreys JD, Smith GE, et al. Detecting dementia with the mini-mental state examination in highly educated individuals. Arch Neurol. 2008;65:963-967.
23. O’Sullivan M, Morris RG, Markus HS. Brief cognitive assessment for patients with cerebral small vessel disease. J Neurol Neurosurg Psychiatry. 2005;76:1140-1145.
24. Nasreddine ZS, Phillips NA, Bédirian V, et al. The Montreal Cognitive Assessment (MoCA): a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53:695-699.
25. Benton AL, Hamsher K. Multilingual Aphasia Examination. 2nd ed. Iowa City, OA: AJA Associates, Inc; 1976.
26. Wechsler D. WAIS-III Administration and Scoring Manual. San Antonio, TX: The Psychological Corporation; 1997.
27. Morris JC, Heyman A, Mohs RC, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part I. Clinical and neuropsychological assesment of Alzheimer’s disease. Neurology. 1989;39:1159-1165.
28. Gladsjo JA, Miller SW, Heaton RK. Norms for Letter and Category Fluency: Demographic Corrections for Age, Education and Ethnicity. Odessa, FL: Psychological Assessment Resources; 1999.
29. Perry R, Hodges J. Attention and executive deficits in Alzheimer’s disease. A critical review. Brain. 1999;122(pt 3):383-404.
30. Flynn JR. The mean IQ of Americans: Massive gains 1932 to 1978. Psychol Bull. 1984;95:29-51.
31. Tombaugh TN. Trail Making Test A and B: normative data stratified by age and education. Arch Clin Neuropsychol. 2004;19: 203-214.
32. Davies A. The influence of age on trail making test performance. J Clin Psychol. 1968;24:96-98.
33. Bianchetti A, Trabucch M. Clinical aspects of Alzheimer’s disease. Aging (Milano). 2001;13:221-230.
34. Kay D, Forster DP, Newens AJ. Long-term survival, place of death, and death certification in clinically diagnosed pre-senile dementia in northern England. Follow-up after 8-12 years. Br J Psychiatry. 2000;177:156-162.
35. Chaussalet T, Thompson WA. Data requirements in a model of the natural history of Alzheimer’s disease. Health Care Manag Sci. 2001;4:13-19.
1. Loveman E, Green C, Kirby J, et al. The clinical and cost-effectiveness of donepezil, rivastigmine, galantamine and memantine for Alzheimer’s disease. Health Technol Assess. 2006;10:iii-iv,ix- xi,1-160.
2. Medical Care Corporation. Delaying the onset and progression of Alzheimer’s disease. Prevent AD Web site. Available at: http://www.preventad.com/pdf/support/article/DelayingADProgression.pdf. Accessed June 18, 2014.
3. Hopkins RW. Dementia projections for the counties, regional municipalities and districts of Ontario. Geriatric Psychiatry Unit Clinical/Research Bulletin, No. 16. Providence Care Web site. Available at: http://www.providencecare.ca/clinical-tools/Documents/Ontario-Dementia-Projections-2010.pdf. Accessed June 18, 2014.
4. Simmons BB, Hartmann B, Dejoseph D. Evaluation of suspected dementia. Am Fam Physician. 2011;84:895-902.
5. McCarten JR, Borson S. Should family physicians routinely screen patients for cognitive impairment? Yes: screening is the first step toward improving care. Am Fam Physician. 2014;89: 861-862.
6. Alzheimer’s Association. Health Care Professionals and Alzheimer’s. Alzheimer’s Association Web site. Available at: http://www.alz.org/health-care-professionals/cognitive-tests-patient-assessment.asp. Accessed June 18, 2014.
7. US Preventive Services Task Force. Screening for cognitive impairment in older adults. US Preventive Services Task Force Web site. Available at: http://www.uspreventiveservicestaskforce.org/uspstf/uspsdeme.htm. Accessed June 18, 2014.
8. Hopkins RW, Kilik LA, Day D, et al. Kingston Standardized Behavioural Assessment. Am J Alzheimers Dis Other Demen. 2006;21:339-346.
9. Kilik LA, Hopkins RW, Day D, et al. The progression of behaviour in dementia: an in-office guide for clinicians. Am J Alzheimers Dis Other Demen. 2008;23:242-249.
10. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189-198.
11. Army Individual Test Battery. Manual of Directions and Scoring. Washington, DC: War Department, Adjutant General’s Office; 1944.
12. Wechsler D. The Measurement and Appraisal of Adult Intelligence. 4th ed. The Williams & Wilkins Company: Baltimore, MD; 1958.
13. Larrabee GJ, Largen JW, Levin HS. Sensitivity of age-decline resistant (“hold”) WAIS subtests to Alzheimer’s disease. J Clin Exp Neuropsychol. 1985;7:497-504.
14. Herndon RM. Assessment of the elderly with dementia. In: Handbook of Neurologic Rating Scales. 2nd ed. Demos Medical Publishing LLC: New York, NY; 2006:199.
15. Brugnolo A, Nobili F, Barbieri MP, et al. The factorial structure of the mini mental state examination (MMSE) in Alzheimer’s disease. Arch Gerontol Geriatr. 2009;49:180-185.
16. Folstein M, Anthony JC, Parhad I, et al. The meaning of cognitive impairment in the elderly. J Am Geriatr Soc. 1985;33:228-235.
17. Hopkins RW, Kilik LA, Day DJ, et al. The Revised Kingston Standardized Cognitive Assessment. Int J Geriatr Psychiatry. 2004;19:320-326.
18. Hopkins R, Kilik L. The mini-Kingston Standardized Cognitive Assessment. Kingston Scales Web site. Available at: http://www.kingstonscales.org. Accessed June 18, 2014.
19. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: American Psychiatric Association; 1994.
20. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34: 939-944.
21. Crum RM, Anthony JC, Bassett SS, et al. Population-based norms for the Mini-Mental State Examination by age and educational level. JAMA. 1993;269:2386-2391.
22. O’Bryant SE, Humphreys JD, Smith GE, et al. Detecting dementia with the mini-mental state examination in highly educated individuals. Arch Neurol. 2008;65:963-967.
23. O’Sullivan M, Morris RG, Markus HS. Brief cognitive assessment for patients with cerebral small vessel disease. J Neurol Neurosurg Psychiatry. 2005;76:1140-1145.
24. Nasreddine ZS, Phillips NA, Bédirian V, et al. The Montreal Cognitive Assessment (MoCA): a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53:695-699.
25. Benton AL, Hamsher K. Multilingual Aphasia Examination. 2nd ed. Iowa City, OA: AJA Associates, Inc; 1976.
26. Wechsler D. WAIS-III Administration and Scoring Manual. San Antonio, TX: The Psychological Corporation; 1997.
27. Morris JC, Heyman A, Mohs RC, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part I. Clinical and neuropsychological assesment of Alzheimer’s disease. Neurology. 1989;39:1159-1165.
28. Gladsjo JA, Miller SW, Heaton RK. Norms for Letter and Category Fluency: Demographic Corrections for Age, Education and Ethnicity. Odessa, FL: Psychological Assessment Resources; 1999.
29. Perry R, Hodges J. Attention and executive deficits in Alzheimer’s disease. A critical review. Brain. 1999;122(pt 3):383-404.
30. Flynn JR. The mean IQ of Americans: Massive gains 1932 to 1978. Psychol Bull. 1984;95:29-51.
31. Tombaugh TN. Trail Making Test A and B: normative data stratified by age and education. Arch Clin Neuropsychol. 2004;19: 203-214.
32. Davies A. The influence of age on trail making test performance. J Clin Psychol. 1968;24:96-98.
33. Bianchetti A, Trabucch M. Clinical aspects of Alzheimer’s disease. Aging (Milano). 2001;13:221-230.
34. Kay D, Forster DP, Newens AJ. Long-term survival, place of death, and death certification in clinically diagnosed pre-senile dementia in northern England. Follow-up after 8-12 years. Br J Psychiatry. 2000;177:156-162.
35. Chaussalet T, Thompson WA. Data requirements in a model of the natural history of Alzheimer’s disease. Health Care Manag Sci. 2001;4:13-19.
