User login
Women with ACS showed improved outcomes, but remain underrepresented in trials
Though acute coronary syndromes are the leading cause of death in U.S. women, an analysis of clinical trials showed that enrollment among women remained disproportionately low from 1994 to 2010, reported Dr. Kristian Kragholm and coauthors at Duke Clinical Research Institute.
An analysis of data in 76,148 non–ST-segment elevation acute coronary syndrome patients from 11 phase III clinical trials found that women comprised just 33.3% of participants, which did not change significantly over the 17-year period. Women consistently had higher incidence of diabetes, hypertension, and heart failure, the authors reported.
Use of ACE inhibitors/angiotensin II receptor blockers, thienopyridines, beta-blockers, and lipid-lowering drugs significantly increased over time for both sexes, as did use of coronary angiography and percutaneous coronary intervention. Observed in-hospital, 30-day, and 6-month mortality decreased significantly in both men and women, Dr. Kragholm and colleagues said.
The findings suggest that “current efforts to representatively enroll women in NSTE ACS trials are insufficient,” the authors wrote. “Because safety and efficacy findings may differ according to sex, this disparity could undermine generalizability of clinical trial results to treatment of the overall NSTE ACS population.”
Read the full article here.
Though acute coronary syndromes are the leading cause of death in U.S. women, an analysis of clinical trials showed that enrollment among women remained disproportionately low from 1994 to 2010, reported Dr. Kristian Kragholm and coauthors at Duke Clinical Research Institute.
An analysis of data in 76,148 non–ST-segment elevation acute coronary syndrome patients from 11 phase III clinical trials found that women comprised just 33.3% of participants, which did not change significantly over the 17-year period. Women consistently had higher incidence of diabetes, hypertension, and heart failure, the authors reported.
Use of ACE inhibitors/angiotensin II receptor blockers, thienopyridines, beta-blockers, and lipid-lowering drugs significantly increased over time for both sexes, as did use of coronary angiography and percutaneous coronary intervention. Observed in-hospital, 30-day, and 6-month mortality decreased significantly in both men and women, Dr. Kragholm and colleagues said.
The findings suggest that “current efforts to representatively enroll women in NSTE ACS trials are insufficient,” the authors wrote. “Because safety and efficacy findings may differ according to sex, this disparity could undermine generalizability of clinical trial results to treatment of the overall NSTE ACS population.”
Read the full article here.
Though acute coronary syndromes are the leading cause of death in U.S. women, an analysis of clinical trials showed that enrollment among women remained disproportionately low from 1994 to 2010, reported Dr. Kristian Kragholm and coauthors at Duke Clinical Research Institute.
An analysis of data in 76,148 non–ST-segment elevation acute coronary syndrome patients from 11 phase III clinical trials found that women comprised just 33.3% of participants, which did not change significantly over the 17-year period. Women consistently had higher incidence of diabetes, hypertension, and heart failure, the authors reported.
Use of ACE inhibitors/angiotensin II receptor blockers, thienopyridines, beta-blockers, and lipid-lowering drugs significantly increased over time for both sexes, as did use of coronary angiography and percutaneous coronary intervention. Observed in-hospital, 30-day, and 6-month mortality decreased significantly in both men and women, Dr. Kragholm and colleagues said.
The findings suggest that “current efforts to representatively enroll women in NSTE ACS trials are insufficient,” the authors wrote. “Because safety and efficacy findings may differ according to sex, this disparity could undermine generalizability of clinical trial results to treatment of the overall NSTE ACS population.”
Read the full article here.
Three Cheers for B3?
At the recent American Society of Clinical Oncology Annual Meeting, Martin et al presented data from the Australian Oral Nicotinamide to Reduce Actinic Cancer (ONTRAC) study. This prospective double-blind, randomized, controlled trial examined 386 immunocompetent patients with 2 or more nonmelanoma skin cancers (NMSCs) in the last 5 years (average, 8). The patients were randomized to receive oral nicotinamide 500 mg twice daily or placebo for 1 year, resulting in significant reduction of new NMSCs (average rate 1.77 vs 2.42; relative rate reduction, 23%; P=.02), with similar results for basal and squamous cell carcinomas. Actinic keratosis counts were reduced throughout the year by up to 20%, peaking at 9 months. No differences in adverse events were noted between the treatment and placebo groups.
What’s the issue?
High-risk NMSC patients present a challenge to dermatologists, as their need for constant surveillance, field therapy for actinic keratoses, and revolving visits between skin examinations and procedural modalities such as Mohs micrographic surgery can be staggering. Chemopreventive strategies pose difficulties, especially for elderly patients, due to tolerability and adherence, skin irritation and cosmetic limitations of topical therapies such as 5-fluorouracil, and inadequacy or financial inaccessibility of oral therapies such as acitretin.
Nicotinamide is a confusing supplement, as it is also called niacinamide. One of the 2 forms of vitamin B3, nicotinic acid (or niacin) is the other form and can be converted to nicotinamide in the body. It has cholesterol and vasodilatory/flushing effects that nicotinamide itself does not. Therefore, these supplement subtypes are not generally interchangeable.
Nicotinamide is postulated to enhance DNA repair and reverse UV immunosuppression in NMSC patients and is a well-tolerated and inexpensive supplement (approximately $10 a month for the dosage in this study). Although the decrease in skin cancer number per year seems modest in this study, my patients would likely welcome at least 1 fewer surgery per year and much less cryotherapy or 5-fluorouracil cream, especially if it is as simple as buying the supplement at the grocery store as they do for their fish oil capsules and probiotics. Does vitamin B3 hold promise for your high-risk NMSC patients?
At the recent American Society of Clinical Oncology Annual Meeting, Martin et al presented data from the Australian Oral Nicotinamide to Reduce Actinic Cancer (ONTRAC) study. This prospective double-blind, randomized, controlled trial examined 386 immunocompetent patients with 2 or more nonmelanoma skin cancers (NMSCs) in the last 5 years (average, 8). The patients were randomized to receive oral nicotinamide 500 mg twice daily or placebo for 1 year, resulting in significant reduction of new NMSCs (average rate 1.77 vs 2.42; relative rate reduction, 23%; P=.02), with similar results for basal and squamous cell carcinomas. Actinic keratosis counts were reduced throughout the year by up to 20%, peaking at 9 months. No differences in adverse events were noted between the treatment and placebo groups.
What’s the issue?
High-risk NMSC patients present a challenge to dermatologists, as their need for constant surveillance, field therapy for actinic keratoses, and revolving visits between skin examinations and procedural modalities such as Mohs micrographic surgery can be staggering. Chemopreventive strategies pose difficulties, especially for elderly patients, due to tolerability and adherence, skin irritation and cosmetic limitations of topical therapies such as 5-fluorouracil, and inadequacy or financial inaccessibility of oral therapies such as acitretin.
Nicotinamide is a confusing supplement, as it is also called niacinamide. One of the 2 forms of vitamin B3, nicotinic acid (or niacin) is the other form and can be converted to nicotinamide in the body. It has cholesterol and vasodilatory/flushing effects that nicotinamide itself does not. Therefore, these supplement subtypes are not generally interchangeable.
Nicotinamide is postulated to enhance DNA repair and reverse UV immunosuppression in NMSC patients and is a well-tolerated and inexpensive supplement (approximately $10 a month for the dosage in this study). Although the decrease in skin cancer number per year seems modest in this study, my patients would likely welcome at least 1 fewer surgery per year and much less cryotherapy or 5-fluorouracil cream, especially if it is as simple as buying the supplement at the grocery store as they do for their fish oil capsules and probiotics. Does vitamin B3 hold promise for your high-risk NMSC patients?
At the recent American Society of Clinical Oncology Annual Meeting, Martin et al presented data from the Australian Oral Nicotinamide to Reduce Actinic Cancer (ONTRAC) study. This prospective double-blind, randomized, controlled trial examined 386 immunocompetent patients with 2 or more nonmelanoma skin cancers (NMSCs) in the last 5 years (average, 8). The patients were randomized to receive oral nicotinamide 500 mg twice daily or placebo for 1 year, resulting in significant reduction of new NMSCs (average rate 1.77 vs 2.42; relative rate reduction, 23%; P=.02), with similar results for basal and squamous cell carcinomas. Actinic keratosis counts were reduced throughout the year by up to 20%, peaking at 9 months. No differences in adverse events were noted between the treatment and placebo groups.
What’s the issue?
High-risk NMSC patients present a challenge to dermatologists, as their need for constant surveillance, field therapy for actinic keratoses, and revolving visits between skin examinations and procedural modalities such as Mohs micrographic surgery can be staggering. Chemopreventive strategies pose difficulties, especially for elderly patients, due to tolerability and adherence, skin irritation and cosmetic limitations of topical therapies such as 5-fluorouracil, and inadequacy or financial inaccessibility of oral therapies such as acitretin.
Nicotinamide is a confusing supplement, as it is also called niacinamide. One of the 2 forms of vitamin B3, nicotinic acid (or niacin) is the other form and can be converted to nicotinamide in the body. It has cholesterol and vasodilatory/flushing effects that nicotinamide itself does not. Therefore, these supplement subtypes are not generally interchangeable.
Nicotinamide is postulated to enhance DNA repair and reverse UV immunosuppression in NMSC patients and is a well-tolerated and inexpensive supplement (approximately $10 a month for the dosage in this study). Although the decrease in skin cancer number per year seems modest in this study, my patients would likely welcome at least 1 fewer surgery per year and much less cryotherapy or 5-fluorouracil cream, especially if it is as simple as buying the supplement at the grocery store as they do for their fish oil capsules and probiotics. Does vitamin B3 hold promise for your high-risk NMSC patients?

Heroin use up across demographic groups from 2002 to 2013
Heroin use is rising across all demographic groups in the United States, and is gaining traction among groups that previously have been associated with lower use, doubling among women and more than doubling among non-Hispanic whites, Dr. Thomas Frieden, director of the Centers for Disease Control and Prevention, said in a July 7 telebriefing.
Between 2002 and 2013, the rate of heroin-related overdose deaths nearly quadrupled, and more than 8,200 people died in 2013, according to the latest Vital Signs report, a combined project from the CDC and the Food and Drug Administration that analyzed data from the 2002-2013 National Survey on Drug Use and Health.

In addition, the gaps between men and women, low and higher incomes, and people with Medicaid and private insurance have narrowed in the past decade, although the most at-risk groups are still non-Hispanic whites, men, 18- to 25-year-olds, people with an annual household incomes of less than $20,000, Medicaid recipients, and the uninsured.
Although people who are addicted to prescription opioid painkillers were 40 times more likely to abuse heroin, the general idea that people gravitating to heroin abuse are doing so because opiates have become harder to get is not true, Dr. Frieden noted. The study identified two factors that were likely responsible for the increase in heroin users – the combination of the increased supply of heroin and higher demand, as well as the number of people already addicted to opioids who are “primed” for a heroin addiction.
However, state agencies have a central role to play in curbing heroin abuse, and will need to increase support for drug monitoring and surveillance programs to make tracking opiate abusers easier and more efficient, Dr. Frieden said.
The CDC is addressing the epidemic by helping to create federal guidelines for pain management, and supporting research and development for less addictive pain medications, he said.
“Improving prescribing practices is part of the solution, not part of the cause,” Dr. Frieden said.
Individual health care providers can help by following best practices for responsible painkiller prescribing to reduce opioid painkiller addiction, and by providing training for ways to adequately and comprehensively address pain beyond simply prescribing painkillers.
“Opiates are very good at curbing severe pain ... But for chronic, noncancer pain, you really need to look at the risks and benefits,” said Christopher M. Jones, Pharm.D., the study’s coauthor and a senior adviser at the FDA.
Heroin use is rising across all demographic groups in the United States, and is gaining traction among groups that previously have been associated with lower use, doubling among women and more than doubling among non-Hispanic whites, Dr. Thomas Frieden, director of the Centers for Disease Control and Prevention, said in a July 7 telebriefing.
Between 2002 and 2013, the rate of heroin-related overdose deaths nearly quadrupled, and more than 8,200 people died in 2013, according to the latest Vital Signs report, a combined project from the CDC and the Food and Drug Administration that analyzed data from the 2002-2013 National Survey on Drug Use and Health.
In addition, the gaps between men and women, low and higher incomes, and people with Medicaid and private insurance have narrowed in the past decade, although the most at-risk groups are still non-Hispanic whites, men, 18- to 25-year-olds, people with an annual household incomes of less than $20,000, Medicaid recipients, and the uninsured.
Although people who are addicted to prescription opioid painkillers were 40 times more likely to abuse heroin, the general idea that people gravitating to heroin abuse are doing so because opiates have become harder to get is not true, Dr. Frieden noted. The study identified two factors that were likely responsible for the increase in heroin users – the combination of the increased supply of heroin and higher demand, as well as the number of people already addicted to opioids who are “primed” for a heroin addiction.
However, state agencies have a central role to play in curbing heroin abuse, and will need to increase support for drug monitoring and surveillance programs to make tracking opiate abusers easier and more efficient, Dr. Frieden said.
The CDC is addressing the epidemic by helping to create federal guidelines for pain management, and supporting research and development for less addictive pain medications, he said.
“Improving prescribing practices is part of the solution, not part of the cause,” Dr. Frieden said.
Individual health care providers can help by following best practices for responsible painkiller prescribing to reduce opioid painkiller addiction, and by providing training for ways to adequately and comprehensively address pain beyond simply prescribing painkillers.
“Opiates are very good at curbing severe pain ... But for chronic, noncancer pain, you really need to look at the risks and benefits,” said Christopher M. Jones, Pharm.D., the study’s coauthor and a senior adviser at the FDA.
Heroin use is rising across all demographic groups in the United States, and is gaining traction among groups that previously have been associated with lower use, doubling among women and more than doubling among non-Hispanic whites, Dr. Thomas Frieden, director of the Centers for Disease Control and Prevention, said in a July 7 telebriefing.
Between 2002 and 2013, the rate of heroin-related overdose deaths nearly quadrupled, and more than 8,200 people died in 2013, according to the latest Vital Signs report, a combined project from the CDC and the Food and Drug Administration that analyzed data from the 2002-2013 National Survey on Drug Use and Health.
In addition, the gaps between men and women, low and higher incomes, and people with Medicaid and private insurance have narrowed in the past decade, although the most at-risk groups are still non-Hispanic whites, men, 18- to 25-year-olds, people with an annual household incomes of less than $20,000, Medicaid recipients, and the uninsured.
Although people who are addicted to prescription opioid painkillers were 40 times more likely to abuse heroin, the general idea that people gravitating to heroin abuse are doing so because opiates have become harder to get is not true, Dr. Frieden noted. The study identified two factors that were likely responsible for the increase in heroin users – the combination of the increased supply of heroin and higher demand, as well as the number of people already addicted to opioids who are “primed” for a heroin addiction.
However, state agencies have a central role to play in curbing heroin abuse, and will need to increase support for drug monitoring and surveillance programs to make tracking opiate abusers easier and more efficient, Dr. Frieden said.
The CDC is addressing the epidemic by helping to create federal guidelines for pain management, and supporting research and development for less addictive pain medications, he said.
“Improving prescribing practices is part of the solution, not part of the cause,” Dr. Frieden said.
Individual health care providers can help by following best practices for responsible painkiller prescribing to reduce opioid painkiller addiction, and by providing training for ways to adequately and comprehensively address pain beyond simply prescribing painkillers.
“Opiates are very good at curbing severe pain ... But for chronic, noncancer pain, you really need to look at the risks and benefits,” said Christopher M. Jones, Pharm.D., the study’s coauthor and a senior adviser at the FDA.
FROM A CDC TELEBRIEFING

Extended warfarin delays return of unprovoked pulmonary embolism
Adding an extra 18 months of warfarin therapy to the standard 6 months of anticoagulation delays the recurrence of venous thrombosis in patients who have a first episode of unprovoked pulmonary embolism – but the risk of recurrence resumes as soon as the warfarin is discontinued, according to a report published online July 7 in JAMA.
“Our results suggest that patients such as those who participated in our study require long-term secondary prophylaxis measures. Whether these should include systematic treatment with vitamin K antagonists, new anticoagulants, or aspirin, or be tailored according to patient risk factors (including elevated D-dimer levels) needs further investigation,” said Dr. Francis Couturaud of the department of internal medicine and chest diseases, University of Brest (France) Hospital, and his associates (JAMA 2015;314:31-40).
Adults with a first episode of unprovoked VT are at much greater risk of recurrence when the standard 6 months of anticoagulation runs out, compared with those whose VT is provoked by a known, transient risk factor such as lengthy surgery, trauma with immobilization of the lower limbs, or bed rest extending longer than 72 hours.
Some experts have advocated extending anticoagulation further in such patients; but whether this is actually beneficial remains uncertain, the investigators said, because most studies have not pursued follow-up beyond the end of treatment.
The researchers performed a multicenter, double-blind trial in which 371 consecutive patients with a first episode of unprovoked PE completed 6 months of anticoagulation and then were randomly assigned to a further 18 months on either warfarin or matching placebo.
During this 18-month treatment period, the primary outcome – a composite of recurrent VT (including PE) and major bleeding – occurred in 3.3% of the warfarin group and 13.5% of the placebo group. That significant difference translated to a 78% reduction in favor of warfarin (hazard ratio, 0.22), Dr. Couturaud and his associates said.
However, after the treatment period ended, the composite outcome occurred in 17.7% of the warfarin group and 10.3% of the placebo group. Thus, the risk of recurrence returned to its normal high level once warfarin was discontinued, the study authors noted.
The study was supported by the Programme Hospitalier de Recherche Clinique (the French Department of Health) and the University Hospital of Brest (France). Dr. Couturaud reported receiving research grants, honoraria, and travel pay from Actelion, AstraZeneca, Bayer, Daiichi Sankyo, Intermune, Leo Pharma, and Pfizer, and his associates reported ties to numerous industry sources.
Related Information
- Computed tomographic pulmonary angiography (CTPA) may be useful in the diagnosis of suspected PE, wrote Dr. Gregoire Le Gal and co-authors from the University of Ottawa. Alternately, a V/Q scan may be performed. The complete accompanying article on diagnostic testing methods for suspected pulmonary embolism can be found here.
- The recently approved anticoagulant edoxaban is similar to warfarin in its ability to treat acute VTE, according to a report published in the Medical Letter on Drugs and Therapeutics in the same issue. However, further study is needed to evaluate its safety and efficacy compared with dabigatran, rivaroxaban, and apixaban, the three other oral anticoagulant drugs currently FDA-approved for acute VTE.
- A meta-analysis of 3,716 patients with VTE found that long-term treatment with Vitamin K antagonists was associated with lower rates of thromboembolic events (relative risk = 0.20) and higher rates of bleeding complications (RR = 3.44), compared with short-term therapy, Dr. Saskia Middeldorp and Dr. Barbara A. Hutten of the University of Amsterdam reported in the same issue. There was no difference in mortality between the two groups.
- Currently, recommended treatment duration for PE can range from three months to lifelong treatment, wrote Dr. Jill Jin in a clinical synopsis for patients published with the study.
- Read the full article and listen to the related podcast: http://jama.jamanetwork.com/article.aspx?doi=10.1001/jama.2015.7046
Madhu Rajaraman contributed to this report.
Adding an extra 18 months of warfarin therapy to the standard 6 months of anticoagulation delays the recurrence of venous thrombosis in patients who have a first episode of unprovoked pulmonary embolism – but the risk of recurrence resumes as soon as the warfarin is discontinued, according to a report published online July 7 in JAMA.
“Our results suggest that patients such as those who participated in our study require long-term secondary prophylaxis measures. Whether these should include systematic treatment with vitamin K antagonists, new anticoagulants, or aspirin, or be tailored according to patient risk factors (including elevated D-dimer levels) needs further investigation,” said Dr. Francis Couturaud of the department of internal medicine and chest diseases, University of Brest (France) Hospital, and his associates (JAMA 2015;314:31-40).
Adults with a first episode of unprovoked VT are at much greater risk of recurrence when the standard 6 months of anticoagulation runs out, compared with those whose VT is provoked by a known, transient risk factor such as lengthy surgery, trauma with immobilization of the lower limbs, or bed rest extending longer than 72 hours.
Some experts have advocated extending anticoagulation further in such patients; but whether this is actually beneficial remains uncertain, the investigators said, because most studies have not pursued follow-up beyond the end of treatment.
The researchers performed a multicenter, double-blind trial in which 371 consecutive patients with a first episode of unprovoked PE completed 6 months of anticoagulation and then were randomly assigned to a further 18 months on either warfarin or matching placebo.
During this 18-month treatment period, the primary outcome – a composite of recurrent VT (including PE) and major bleeding – occurred in 3.3% of the warfarin group and 13.5% of the placebo group. That significant difference translated to a 78% reduction in favor of warfarin (hazard ratio, 0.22), Dr. Couturaud and his associates said.
However, after the treatment period ended, the composite outcome occurred in 17.7% of the warfarin group and 10.3% of the placebo group. Thus, the risk of recurrence returned to its normal high level once warfarin was discontinued, the study authors noted.
The study was supported by the Programme Hospitalier de Recherche Clinique (the French Department of Health) and the University Hospital of Brest (France). Dr. Couturaud reported receiving research grants, honoraria, and travel pay from Actelion, AstraZeneca, Bayer, Daiichi Sankyo, Intermune, Leo Pharma, and Pfizer, and his associates reported ties to numerous industry sources.
Related Information
- Computed tomographic pulmonary angiography (CTPA) may be useful in the diagnosis of suspected PE, wrote Dr. Gregoire Le Gal and co-authors from the University of Ottawa. Alternately, a V/Q scan may be performed. The complete accompanying article on diagnostic testing methods for suspected pulmonary embolism can be found here.
- The recently approved anticoagulant edoxaban is similar to warfarin in its ability to treat acute VTE, according to a report published in the Medical Letter on Drugs and Therapeutics in the same issue. However, further study is needed to evaluate its safety and efficacy compared with dabigatran, rivaroxaban, and apixaban, the three other oral anticoagulant drugs currently FDA-approved for acute VTE.
- A meta-analysis of 3,716 patients with VTE found that long-term treatment with Vitamin K antagonists was associated with lower rates of thromboembolic events (relative risk = 0.20) and higher rates of bleeding complications (RR = 3.44), compared with short-term therapy, Dr. Saskia Middeldorp and Dr. Barbara A. Hutten of the University of Amsterdam reported in the same issue. There was no difference in mortality between the two groups.
- Currently, recommended treatment duration for PE can range from three months to lifelong treatment, wrote Dr. Jill Jin in a clinical synopsis for patients published with the study.
- Read the full article and listen to the related podcast: http://jama.jamanetwork.com/article.aspx?doi=10.1001/jama.2015.7046
Madhu Rajaraman contributed to this report.
Adding an extra 18 months of warfarin therapy to the standard 6 months of anticoagulation delays the recurrence of venous thrombosis in patients who have a first episode of unprovoked pulmonary embolism – but the risk of recurrence resumes as soon as the warfarin is discontinued, according to a report published online July 7 in JAMA.
“Our results suggest that patients such as those who participated in our study require long-term secondary prophylaxis measures. Whether these should include systematic treatment with vitamin K antagonists, new anticoagulants, or aspirin, or be tailored according to patient risk factors (including elevated D-dimer levels) needs further investigation,” said Dr. Francis Couturaud of the department of internal medicine and chest diseases, University of Brest (France) Hospital, and his associates (JAMA 2015;314:31-40).
Adults with a first episode of unprovoked VT are at much greater risk of recurrence when the standard 6 months of anticoagulation runs out, compared with those whose VT is provoked by a known, transient risk factor such as lengthy surgery, trauma with immobilization of the lower limbs, or bed rest extending longer than 72 hours.
Some experts have advocated extending anticoagulation further in such patients; but whether this is actually beneficial remains uncertain, the investigators said, because most studies have not pursued follow-up beyond the end of treatment.
The researchers performed a multicenter, double-blind trial in which 371 consecutive patients with a first episode of unprovoked PE completed 6 months of anticoagulation and then were randomly assigned to a further 18 months on either warfarin or matching placebo.
During this 18-month treatment period, the primary outcome – a composite of recurrent VT (including PE) and major bleeding – occurred in 3.3% of the warfarin group and 13.5% of the placebo group. That significant difference translated to a 78% reduction in favor of warfarin (hazard ratio, 0.22), Dr. Couturaud and his associates said.
However, after the treatment period ended, the composite outcome occurred in 17.7% of the warfarin group and 10.3% of the placebo group. Thus, the risk of recurrence returned to its normal high level once warfarin was discontinued, the study authors noted.
The study was supported by the Programme Hospitalier de Recherche Clinique (the French Department of Health) and the University Hospital of Brest (France). Dr. Couturaud reported receiving research grants, honoraria, and travel pay from Actelion, AstraZeneca, Bayer, Daiichi Sankyo, Intermune, Leo Pharma, and Pfizer, and his associates reported ties to numerous industry sources.
Related Information
- Computed tomographic pulmonary angiography (CTPA) may be useful in the diagnosis of suspected PE, wrote Dr. Gregoire Le Gal and co-authors from the University of Ottawa. Alternately, a V/Q scan may be performed. The complete accompanying article on diagnostic testing methods for suspected pulmonary embolism can be found here.
- The recently approved anticoagulant edoxaban is similar to warfarin in its ability to treat acute VTE, according to a report published in the Medical Letter on Drugs and Therapeutics in the same issue. However, further study is needed to evaluate its safety and efficacy compared with dabigatran, rivaroxaban, and apixaban, the three other oral anticoagulant drugs currently FDA-approved for acute VTE.
- A meta-analysis of 3,716 patients with VTE found that long-term treatment with Vitamin K antagonists was associated with lower rates of thromboembolic events (relative risk = 0.20) and higher rates of bleeding complications (RR = 3.44), compared with short-term therapy, Dr. Saskia Middeldorp and Dr. Barbara A. Hutten of the University of Amsterdam reported in the same issue. There was no difference in mortality between the two groups.
- Currently, recommended treatment duration for PE can range from three months to lifelong treatment, wrote Dr. Jill Jin in a clinical synopsis for patients published with the study.
- Read the full article and listen to the related podcast: http://jama.jamanetwork.com/article.aspx?doi=10.1001/jama.2015.7046
Madhu Rajaraman contributed to this report.
FROM JAMA
Key clinical point: Eighteen additional months of warfarin therapy delays the recurrence of unprovoked pulmonary embolism.
Major finding: During treatment, the primary outcome – a composite of recurrent venous thromboembolism and major bleeding – occurred in 3.3% of the warfarin group and 13.5% of the placebo group, a significant difference that translated to a 78% reduction in favor of warfarin (hazard ratio, 0.22).
Data source: A multicenter, randomized, double-blind, placebo-controlled clinical trial involving 371 patients followed for a mean of 41 months.
Disclosures: This study was supported by the Programme Hospitalier de Recherche Clinique (the French Department of Health) and the University Hospital of Brest (France). Dr. Couturaud reported receiving research grants, honoraria, and travel pay from Actelion, AstraZeneca, Bayer, Daiichi Sankyo, Intermune, Leo Pharma, and Pfizer, and his associates reported ties to numerous industry sources.
Madelung Deformity and Extensor Tendon Rupture
Extensor tendon rupture in chronic Madelung deformity, as a result of tendon attrition on the dislocated distal ulna, occurs infrequently. However, it is often seen in patients with rheumatoid arthritis. This issue has been reported in only a few English-language case reports. Here we report a case of multiple tendon ruptures in a previously undiagnosed Madelung deformity. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 56-year-old active woman presented with 50 days’ inability to extend the fourth and fifth fingers of her dominant right hand. The loss of finger extension progressed, over several weeks, to involve the third finger as well. The first 2 tendon ruptures had been triggered by lifting a light grocery bag, when she noticed a sharp sudden pain and “pop.” The third rupture occurred spontaneously with a snapping sound the night before surgery.
The patient had observed some prominence on the ulnar side of her right wrist since childhood but had never experienced any pain or functional disability. There was neither history of trauma, inflammatory disease, diabetes mellitus, or infection, nor positive family history of similar wrist deformity.
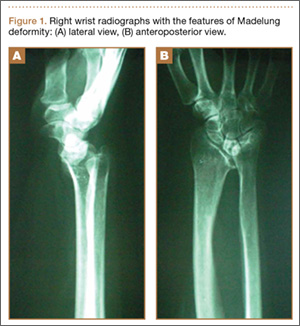
The physical examination showed a dorsally subluxated distal radioulnar joint, prominent ulnar styloid, and mild ulnar and volar deviation of the wrist along with limitation of wrist dorsiflexion. Complete loss of active extension of the 3 ulnar fingers was demonstrated, while neurovascular status and all other hand evaluations were normal. The wrist radiographs confirmed the typical findings of Madelung deformity (Figure 1).
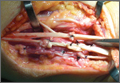
Repair of the ruptured tendons and resection of the prominent distal ulna (Darrach procedure) was planned. (Given the patient’s age and evidence of degenerative changes in the radiocarpal joint, correction of the Madelung deformity did not seem necessary). At time of surgery, the recently ruptured third finger extensor tendon was easily found and approximated, and end-to-end repair was performed. The fourth and fifth fingers, however, had to be fished out more proximally from dense granulation tissue. After the distal ulna was resected for a distance of 1.5 cm, meticulous repair of the ulnar collateral ligament and the capsule and periosteum over the end of the ulna was performed. Then, for grafting of the ruptured tendons, the extensor indicis proprius tendon was isolated and transected at the second metacarpophalangeal joint level. A piece of this tendon was used as interpositional graft for the fourth extensor tendon, and the main tendon unit was transferred to the fifth finger extensor. The extensor digiti quinti tendon, which was about to rupture, was further reinforced by suturing it side to side to the muscle and tendon of the extensor indicis proprius (Figure 2).

Postoperatively, the wrist was kept in extension in a cast for 3 weeks while the fingers were free for active movement. A removable wrist splint was used for an additional month. At 3-month follow-up, the patient had regained full and strong finger extension and wrist motion.
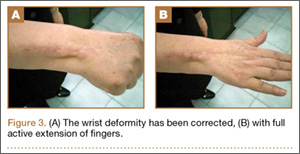
At 3-year follow-up, the patient was pain-free, and had full extension of all fingers, full forearm rotation, and near-normal motion (better than her preoperative motion). The grip power on the operated right hand was 215 N, and pinch power was 93 N. (The values for the left side were 254 N and 83 N, respectively, using the Jamar hydraulic hand dynamometer [Patterson Medical].) The patient has had no additional tendon rupture (Figure 3).
Discussion
Madelung deformity was first described by Madelung in 1878 and several cases have reported this deformity. However, extensor tendon rupture caused by Madelung deformity is very rare, reported in few cases.1
Extensor tendon rupture caused by chronic Madelung deformity has been reported few times in the English literature. Goodwin1 apparently published the first report of such an occurrence in 1979. Ducloyer and colleagues2 from France reported 6 cases of extensor tendon rupture as a result of inferior distal radioulnar joint deformity of Madelung. Jebson and colleagues3 reported bilateral spontaneous extensor tendon ruptures in Madelung deformity in 1992.
The mechanism of tendon rupture seems to be mechanical, resulting from continuous rubbing and erosion of tendons over the deformed ulnar head, which has a rough irregular surface4 and leads to fraying of the tendons and eventual rupture and retraction of the severed tendon ends. This rupture usually progresses stepwise from more medial to the lateral tendons.2 Older patients are, therefore, subject to chronic repetitive attritional trauma leading to tendon rupture.
Tendons may rupture as a result of a variety of conditions, such as chronic synovitis in rheumatoid arthritis, systemic lupus erythematosus, mixed connective tissue disease, or crystal deposition in gout.5-8 Some other metabolic or endocrine conditions that involve tendon ruptures include diabetes mellitus, chronic renal failure, and hyperparathyroidism. Steroid injection into the tendons also has a detrimental effect on tendon integrity and may cause tendon tear.9 Mechanical factors, such as erosion on bony prominences, are well-known etiologies for tendon rupture, as commonly seen in rheumatoid arthritis, and have been reported in Kienböck disease,10 thumb carpometacarpal arthritis,11 Colles fracture, scaphoid fracture nonunion,12 and Madelung deformity.
Conclusion
Our case reflects the usual middle-aged female presentation of such a tendon rupture. The tendon ruptures were spontaneous in the reported order of ulnar to radial, beginning with the little and ring fingers, and progressed radially. The patient had isolated Madelung deformity with no other sign of dyschondrosteosis13 or dwarfism, conditions commonly mentioned in association with Madelung deformity. This case report should raise awareness about possible tendon rupture in any chronic case of Madelung deformity.
1. Goodwin DR, Michels CH, Weissman SL. Spontaneous rupture of extensor tendons in Madelung’s deformity. Hand. 1979;11(1):72-75.
2. Ducloyer P, Leclercq C, Lisfrance R, Saffar P. Spontaneous rupture of the extensor tendons of the fingers in Madelung’s deformity. J Hand Surg Br. 1991;16(3):329-333.
3. Jebson PJ, Blair WF. Bilateral spontaneous extensor tendon ruptures in Madelung’s deformity. J Hand Surg Am. 1992;17(2):277-280.
4. Schulstad I. Madelung’s deformity with extensor tendon rupture. Case report. Scand J Plast Reconstr Surg. 1971;5(2):153-155.
5. Gong HS, Lee JO, Baek GH, et al. Extensor tendon rupture in rheumatoid arthritis: a survey of patients between 2005 and 2010 at five Korean hospitals. Hand Surg. 2012;17(1):43-47.
6. Oishi H, Oda R, Morisaki S, Fujiwara H, Tokunaga D, Kubo T. Spontaneous tendon rupture of the extensor digitrum communis in systemic lupus erythematosus. Mod Rheumatol. 2013;23(3);608-610.
7. Kobayashi A, Futami T, Tadano I, Fujita M. Spontaneous rupture of extensor tendons at the wrist in a patient with mixed connective tissue disease. Mod Rheumatol. 2002;12(3):256-258.
8. Iwamoto T, Toki H, Ikari K, Yamanaka H, Momohara S. Multiple extensor tendon ruptures caused by tophaceous gout. Mod Rheumatol. 2010;20(2):210-212.
9. Nquyen ML, Jones NF. Rupture of both abductor pollicis longus and extensor pollicis brevis tendon after steroid injection for de quervain tenosynovitis. Plast Reconstr Surg. 2012;129(5):883e-886e.
10. Hernández-Cortés P, Pajares-López M, Gómez-Sánchez R, Garrido-Gómez, Lara-Garcia F. Rupture of extensor tendon secondary to previously undiagnosed Kienböck disease. J Plast Surg Hand Surg. 2012;46(3-4):291-293.
11. Apard T, Marcucci L, Jarriges J. Spontaneous rupture of extensor pollicis longus in isolated trapeziometacarpal arthritis. Chir Main. 2011;30(5):349-351.
12. Harvey FJ, Harvey PM. Three rare causes of extensor tendon rupture. J Hand Surg Am. 1989;14(6):957-962.
13. Duro EA, Prado GS. Clinical variations in Léri-Weill dyschondrosteosis. An Esp Pediatr. 1990;33(5):461-463.
Extensor tendon rupture in chronic Madelung deformity, as a result of tendon attrition on the dislocated distal ulna, occurs infrequently. However, it is often seen in patients with rheumatoid arthritis. This issue has been reported in only a few English-language case reports. Here we report a case of multiple tendon ruptures in a previously undiagnosed Madelung deformity. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 56-year-old active woman presented with 50 days’ inability to extend the fourth and fifth fingers of her dominant right hand. The loss of finger extension progressed, over several weeks, to involve the third finger as well. The first 2 tendon ruptures had been triggered by lifting a light grocery bag, when she noticed a sharp sudden pain and “pop.” The third rupture occurred spontaneously with a snapping sound the night before surgery.
The patient had observed some prominence on the ulnar side of her right wrist since childhood but had never experienced any pain or functional disability. There was neither history of trauma, inflammatory disease, diabetes mellitus, or infection, nor positive family history of similar wrist deformity.
The physical examination showed a dorsally subluxated distal radioulnar joint, prominent ulnar styloid, and mild ulnar and volar deviation of the wrist along with limitation of wrist dorsiflexion. Complete loss of active extension of the 3 ulnar fingers was demonstrated, while neurovascular status and all other hand evaluations were normal. The wrist radiographs confirmed the typical findings of Madelung deformity (Figure 1).
Repair of the ruptured tendons and resection of the prominent distal ulna (Darrach procedure) was planned. (Given the patient’s age and evidence of degenerative changes in the radiocarpal joint, correction of the Madelung deformity did not seem necessary). At time of surgery, the recently ruptured third finger extensor tendon was easily found and approximated, and end-to-end repair was performed. The fourth and fifth fingers, however, had to be fished out more proximally from dense granulation tissue. After the distal ulna was resected for a distance of 1.5 cm, meticulous repair of the ulnar collateral ligament and the capsule and periosteum over the end of the ulna was performed. Then, for grafting of the ruptured tendons, the extensor indicis proprius tendon was isolated and transected at the second metacarpophalangeal joint level. A piece of this tendon was used as interpositional graft for the fourth extensor tendon, and the main tendon unit was transferred to the fifth finger extensor. The extensor digiti quinti tendon, which was about to rupture, was further reinforced by suturing it side to side to the muscle and tendon of the extensor indicis proprius (Figure 2).
Postoperatively, the wrist was kept in extension in a cast for 3 weeks while the fingers were free for active movement. A removable wrist splint was used for an additional month. At 3-month follow-up, the patient had regained full and strong finger extension and wrist motion.
At 3-year follow-up, the patient was pain-free, and had full extension of all fingers, full forearm rotation, and near-normal motion (better than her preoperative motion). The grip power on the operated right hand was 215 N, and pinch power was 93 N. (The values for the left side were 254 N and 83 N, respectively, using the Jamar hydraulic hand dynamometer [Patterson Medical].) The patient has had no additional tendon rupture (Figure 3).
Discussion
Madelung deformity was first described by Madelung in 1878 and several cases have reported this deformity. However, extensor tendon rupture caused by Madelung deformity is very rare, reported in few cases.1
Extensor tendon rupture caused by chronic Madelung deformity has been reported few times in the English literature. Goodwin1 apparently published the first report of such an occurrence in 1979. Ducloyer and colleagues2 from France reported 6 cases of extensor tendon rupture as a result of inferior distal radioulnar joint deformity of Madelung. Jebson and colleagues3 reported bilateral spontaneous extensor tendon ruptures in Madelung deformity in 1992.
The mechanism of tendon rupture seems to be mechanical, resulting from continuous rubbing and erosion of tendons over the deformed ulnar head, which has a rough irregular surface4 and leads to fraying of the tendons and eventual rupture and retraction of the severed tendon ends. This rupture usually progresses stepwise from more medial to the lateral tendons.2 Older patients are, therefore, subject to chronic repetitive attritional trauma leading to tendon rupture.
Tendons may rupture as a result of a variety of conditions, such as chronic synovitis in rheumatoid arthritis, systemic lupus erythematosus, mixed connective tissue disease, or crystal deposition in gout.5-8 Some other metabolic or endocrine conditions that involve tendon ruptures include diabetes mellitus, chronic renal failure, and hyperparathyroidism. Steroid injection into the tendons also has a detrimental effect on tendon integrity and may cause tendon tear.9 Mechanical factors, such as erosion on bony prominences, are well-known etiologies for tendon rupture, as commonly seen in rheumatoid arthritis, and have been reported in Kienböck disease,10 thumb carpometacarpal arthritis,11 Colles fracture, scaphoid fracture nonunion,12 and Madelung deformity.
Conclusion
Our case reflects the usual middle-aged female presentation of such a tendon rupture. The tendon ruptures were spontaneous in the reported order of ulnar to radial, beginning with the little and ring fingers, and progressed radially. The patient had isolated Madelung deformity with no other sign of dyschondrosteosis13 or dwarfism, conditions commonly mentioned in association with Madelung deformity. This case report should raise awareness about possible tendon rupture in any chronic case of Madelung deformity.
Extensor tendon rupture in chronic Madelung deformity, as a result of tendon attrition on the dislocated distal ulna, occurs infrequently. However, it is often seen in patients with rheumatoid arthritis. This issue has been reported in only a few English-language case reports. Here we report a case of multiple tendon ruptures in a previously undiagnosed Madelung deformity. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 56-year-old active woman presented with 50 days’ inability to extend the fourth and fifth fingers of her dominant right hand. The loss of finger extension progressed, over several weeks, to involve the third finger as well. The first 2 tendon ruptures had been triggered by lifting a light grocery bag, when she noticed a sharp sudden pain and “pop.” The third rupture occurred spontaneously with a snapping sound the night before surgery.
The patient had observed some prominence on the ulnar side of her right wrist since childhood but had never experienced any pain or functional disability. There was neither history of trauma, inflammatory disease, diabetes mellitus, or infection, nor positive family history of similar wrist deformity.
The physical examination showed a dorsally subluxated distal radioulnar joint, prominent ulnar styloid, and mild ulnar and volar deviation of the wrist along with limitation of wrist dorsiflexion. Complete loss of active extension of the 3 ulnar fingers was demonstrated, while neurovascular status and all other hand evaluations were normal. The wrist radiographs confirmed the typical findings of Madelung deformity (Figure 1).
Repair of the ruptured tendons and resection of the prominent distal ulna (Darrach procedure) was planned. (Given the patient’s age and evidence of degenerative changes in the radiocarpal joint, correction of the Madelung deformity did not seem necessary). At time of surgery, the recently ruptured third finger extensor tendon was easily found and approximated, and end-to-end repair was performed. The fourth and fifth fingers, however, had to be fished out more proximally from dense granulation tissue. After the distal ulna was resected for a distance of 1.5 cm, meticulous repair of the ulnar collateral ligament and the capsule and periosteum over the end of the ulna was performed. Then, for grafting of the ruptured tendons, the extensor indicis proprius tendon was isolated and transected at the second metacarpophalangeal joint level. A piece of this tendon was used as interpositional graft for the fourth extensor tendon, and the main tendon unit was transferred to the fifth finger extensor. The extensor digiti quinti tendon, which was about to rupture, was further reinforced by suturing it side to side to the muscle and tendon of the extensor indicis proprius (Figure 2).
Postoperatively, the wrist was kept in extension in a cast for 3 weeks while the fingers were free for active movement. A removable wrist splint was used for an additional month. At 3-month follow-up, the patient had regained full and strong finger extension and wrist motion.
At 3-year follow-up, the patient was pain-free, and had full extension of all fingers, full forearm rotation, and near-normal motion (better than her preoperative motion). The grip power on the operated right hand was 215 N, and pinch power was 93 N. (The values for the left side were 254 N and 83 N, respectively, using the Jamar hydraulic hand dynamometer [Patterson Medical].) The patient has had no additional tendon rupture (Figure 3).
Discussion
Madelung deformity was first described by Madelung in 1878 and several cases have reported this deformity. However, extensor tendon rupture caused by Madelung deformity is very rare, reported in few cases.1
Extensor tendon rupture caused by chronic Madelung deformity has been reported few times in the English literature. Goodwin1 apparently published the first report of such an occurrence in 1979. Ducloyer and colleagues2 from France reported 6 cases of extensor tendon rupture as a result of inferior distal radioulnar joint deformity of Madelung. Jebson and colleagues3 reported bilateral spontaneous extensor tendon ruptures in Madelung deformity in 1992.
The mechanism of tendon rupture seems to be mechanical, resulting from continuous rubbing and erosion of tendons over the deformed ulnar head, which has a rough irregular surface4 and leads to fraying of the tendons and eventual rupture and retraction of the severed tendon ends. This rupture usually progresses stepwise from more medial to the lateral tendons.2 Older patients are, therefore, subject to chronic repetitive attritional trauma leading to tendon rupture.
Tendons may rupture as a result of a variety of conditions, such as chronic synovitis in rheumatoid arthritis, systemic lupus erythematosus, mixed connective tissue disease, or crystal deposition in gout.5-8 Some other metabolic or endocrine conditions that involve tendon ruptures include diabetes mellitus, chronic renal failure, and hyperparathyroidism. Steroid injection into the tendons also has a detrimental effect on tendon integrity and may cause tendon tear.9 Mechanical factors, such as erosion on bony prominences, are well-known etiologies for tendon rupture, as commonly seen in rheumatoid arthritis, and have been reported in Kienböck disease,10 thumb carpometacarpal arthritis,11 Colles fracture, scaphoid fracture nonunion,12 and Madelung deformity.
Conclusion
Our case reflects the usual middle-aged female presentation of such a tendon rupture. The tendon ruptures were spontaneous in the reported order of ulnar to radial, beginning with the little and ring fingers, and progressed radially. The patient had isolated Madelung deformity with no other sign of dyschondrosteosis13 or dwarfism, conditions commonly mentioned in association with Madelung deformity. This case report should raise awareness about possible tendon rupture in any chronic case of Madelung deformity.
1. Goodwin DR, Michels CH, Weissman SL. Spontaneous rupture of extensor tendons in Madelung’s deformity. Hand. 1979;11(1):72-75.
2. Ducloyer P, Leclercq C, Lisfrance R, Saffar P. Spontaneous rupture of the extensor tendons of the fingers in Madelung’s deformity. J Hand Surg Br. 1991;16(3):329-333.
3. Jebson PJ, Blair WF. Bilateral spontaneous extensor tendon ruptures in Madelung’s deformity. J Hand Surg Am. 1992;17(2):277-280.
4. Schulstad I. Madelung’s deformity with extensor tendon rupture. Case report. Scand J Plast Reconstr Surg. 1971;5(2):153-155.
5. Gong HS, Lee JO, Baek GH, et al. Extensor tendon rupture in rheumatoid arthritis: a survey of patients between 2005 and 2010 at five Korean hospitals. Hand Surg. 2012;17(1):43-47.
6. Oishi H, Oda R, Morisaki S, Fujiwara H, Tokunaga D, Kubo T. Spontaneous tendon rupture of the extensor digitrum communis in systemic lupus erythematosus. Mod Rheumatol. 2013;23(3);608-610.
7. Kobayashi A, Futami T, Tadano I, Fujita M. Spontaneous rupture of extensor tendons at the wrist in a patient with mixed connective tissue disease. Mod Rheumatol. 2002;12(3):256-258.
8. Iwamoto T, Toki H, Ikari K, Yamanaka H, Momohara S. Multiple extensor tendon ruptures caused by tophaceous gout. Mod Rheumatol. 2010;20(2):210-212.
9. Nquyen ML, Jones NF. Rupture of both abductor pollicis longus and extensor pollicis brevis tendon after steroid injection for de quervain tenosynovitis. Plast Reconstr Surg. 2012;129(5):883e-886e.
10. Hernández-Cortés P, Pajares-López M, Gómez-Sánchez R, Garrido-Gómez, Lara-Garcia F. Rupture of extensor tendon secondary to previously undiagnosed Kienböck disease. J Plast Surg Hand Surg. 2012;46(3-4):291-293.
11. Apard T, Marcucci L, Jarriges J. Spontaneous rupture of extensor pollicis longus in isolated trapeziometacarpal arthritis. Chir Main. 2011;30(5):349-351.
12. Harvey FJ, Harvey PM. Three rare causes of extensor tendon rupture. J Hand Surg Am. 1989;14(6):957-962.
13. Duro EA, Prado GS. Clinical variations in Léri-Weill dyschondrosteosis. An Esp Pediatr. 1990;33(5):461-463.
1. Goodwin DR, Michels CH, Weissman SL. Spontaneous rupture of extensor tendons in Madelung’s deformity. Hand. 1979;11(1):72-75.
2. Ducloyer P, Leclercq C, Lisfrance R, Saffar P. Spontaneous rupture of the extensor tendons of the fingers in Madelung’s deformity. J Hand Surg Br. 1991;16(3):329-333.
3. Jebson PJ, Blair WF. Bilateral spontaneous extensor tendon ruptures in Madelung’s deformity. J Hand Surg Am. 1992;17(2):277-280.
4. Schulstad I. Madelung’s deformity with extensor tendon rupture. Case report. Scand J Plast Reconstr Surg. 1971;5(2):153-155.
5. Gong HS, Lee JO, Baek GH, et al. Extensor tendon rupture in rheumatoid arthritis: a survey of patients between 2005 and 2010 at five Korean hospitals. Hand Surg. 2012;17(1):43-47.
6. Oishi H, Oda R, Morisaki S, Fujiwara H, Tokunaga D, Kubo T. Spontaneous tendon rupture of the extensor digitrum communis in systemic lupus erythematosus. Mod Rheumatol. 2013;23(3);608-610.
7. Kobayashi A, Futami T, Tadano I, Fujita M. Spontaneous rupture of extensor tendons at the wrist in a patient with mixed connective tissue disease. Mod Rheumatol. 2002;12(3):256-258.
8. Iwamoto T, Toki H, Ikari K, Yamanaka H, Momohara S. Multiple extensor tendon ruptures caused by tophaceous gout. Mod Rheumatol. 2010;20(2):210-212.
9. Nquyen ML, Jones NF. Rupture of both abductor pollicis longus and extensor pollicis brevis tendon after steroid injection for de quervain tenosynovitis. Plast Reconstr Surg. 2012;129(5):883e-886e.
10. Hernández-Cortés P, Pajares-López M, Gómez-Sánchez R, Garrido-Gómez, Lara-Garcia F. Rupture of extensor tendon secondary to previously undiagnosed Kienböck disease. J Plast Surg Hand Surg. 2012;46(3-4):291-293.
11. Apard T, Marcucci L, Jarriges J. Spontaneous rupture of extensor pollicis longus in isolated trapeziometacarpal arthritis. Chir Main. 2011;30(5):349-351.
12. Harvey FJ, Harvey PM. Three rare causes of extensor tendon rupture. J Hand Surg Am. 1989;14(6):957-962.
13. Duro EA, Prado GS. Clinical variations in Léri-Weill dyschondrosteosis. An Esp Pediatr. 1990;33(5):461-463.
Septic Arthritis and Osteomyelitis Caused by Pasteurella multocida
A few days after an incidental cat bite, a patient presented to the emergency department for treatment of poison sumac exposure. He was discharged with oral methylprednisolone for the dermatitis and returned 1 week later with symptoms, examination findings, and laboratory results consistent with sepsis and bilateral upper extremity necrotizing soft-tissue infections. After administering multiple irrigation and débridement procedures, hyperbaric oxygen treatments, and an antibiotic regimen, the patient’s status greatly improved. However, the patient returned 1 month later with a new sternoclavicular joint prominence that was associated with painful crepitus. Additionally, he noted that his wrists were gradually becoming more swollen and painful. Imaging studies showed a lytic destruction of the sternoclavicular joint and erosive changes throughout the carpus and radiocarpal joint bilaterally, consistent with osteomyelitis. The patient was treated with ertapenem for 6 weeks, and his polyarthropathy resolved. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 73-year-old, right-hand–dominant man with no notable medical history presented to the emergency department for treatment of poison sumac exposure, incidentally, a few days after being bitten by a cat on the bilateral distal upper extremities. He was prescribed a course of oral methylprednisolone for dermatitis. A week later, the patient returned to the emergency department with altered mental status, fevers, diaphoresis, lethargy, and polyarthralgia. At the time of presentation, the patient’s vital signs were labile, and he was found to have extensive bilateral upper extremity erythema, blistering, petechiae, purpuric lesions, and exquisite pain with passive range of motion of his fingers and wrists. His leukocyte count was 25.1 × 109/L, and he had elevated C-reactive protein level and erythrocyte sedimentation rate of 150 mg/L and 120 mm/h, respectively. He was admitted for management of sepsis and presumed bilateral upper extremity necrotizing soft-tissue infection.
Broad-spectrum intravenous (IV) antibiotics (vancomycin, piperacillin, tazobactam) were initiated after blood cultures were obtained, and the patient was taken emergently to the operating theatre for irrigation and débridement of his hands and wrists bilaterally. Arthrotomy of the wrist and débridement of the distal extensor compartment and its tenosynovium were performed on the right forearm, in addition to a decompressive fasciotomy of the left forearm. Postoperatively, the patient’s mental status improved and his vital signs gradually normalized. He received multiple hyperbaric oxygen treatments and underwent several additional operative débridement procedures with eventual closure of his wounds. At initial presentation, the differential diagnosis for the severe soft-tissue infection included necrotizing fasciitis or myositis caused by any of a variety of bacterial pathogens. Most notably, it was important to elicit the history of a cat bite to include and consider Pasteurella multocida as a potential pathogen. Initial cultures supported the diagnosis of acute P multocida necrotizing skin and soft-tissue infection, in addition to septic arthritis. The patient’s blood and intraoperative wound cultures grew P multocida. The antibiotic treatment was tailored initially to ampicillin and sulbactam and then to a final regimen of orally administered ciprofloxacin (750 mg twice a day), once susceptibility testing was performed on the cultures. On hospital day 10, the patient was discharged home, receiving a 6-week course of ciprofloxacin to complete the 8-week course of treatment.
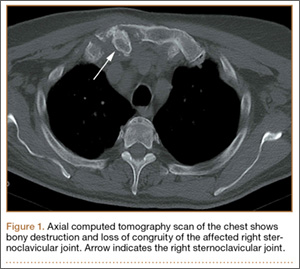
At follow-up, approximately 1 month after discharge, the patient noted that he had developed a new right sternoclavicular joint prominence that was associated with painful crepitus. He also noted that his wrists were gradually becoming more swollen and painful bilaterally. Computed tomography scans of the chest were obtained to evaluate the sternoclavicular joint (Figure 1). Repeat radiographs of the wrists were also obtained (Figure 2). Imaging showed lytic destruction of the sternoclavicular joint and erosive changes throughout the carpus and radiocarpal joint, consistent with osteomyelitis. The C-reactive protein level and erythrocyte sedimentation rate at this time were 34 mg/L and 124 mm/h, respectively.
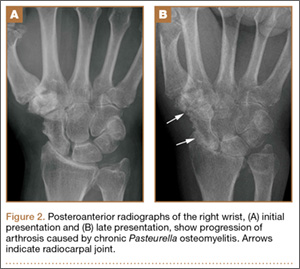
The patient returned to the operating room for débridement and biopsy of the right sternoclavicular joint and left wrist. This patient’s delayed presentation was characterized by a subacute worsening of isolated musculoskeletal complaints. The differential diagnosis then included infection with the same bacterial pathogen versus reactive or inflammatory arthritis. Several intraoperative cultures failed to grow any bacteria, including P multocida, although P multocida was the presumptive cause of the erosive polyarthropathy, considering that symptoms eventually resolved with a repeated course of IV-administered ertapenem for 6 weeks. The patient experienced complete resolution of his joint pain and swelling. He was able to resume his activities of daily living and had no further recurrence of symptoms at follow-up 3 months later.
Discussion
Cat bites often are the source of Pasteurella species infections because the bacteria are carried by more than 90% of cats.1 These types of infections can cause septic arthritis, osteomyelitis, and deep subcutaneous and myofascial infections because of the sharp and narrow morphology of cat teeth. The infections can progress to necrotizing fasciitis and myositis if not recognized early, as was the case with our patient. Prophylactic antibiotic administration for animal bites is controversial and is not a universal practice.1,2Pasteurella bacteremia is an atypical progression that occurs more often in patients with pneumonia, septic arthritis, or meningitis. Cases of Pasteurella sepsis, necrotizing fasciitis, and septic arthritis have been reported.3-7 However, associated progressive septic arthritis and osteomyelitis, despite initial clinical improvement, have not been reported. Severe infection (ie, sepsis and septic shock) can occur in infants, pregnant women, and other immunocompromised patients.7 Immune suppression of our patient with steroid medication for poison sumac dermatitis likely contributed to the progression and systemic spread of an initially benign cat bite. Before prescribing steroids, it is imperative to ask about exposures and encourage patients to seek prompt medical attention with worsening or new symptoms. Healthy individuals rarely develop bacteremia; however, in these cases, mortality remains high at approximately 25%.4,6
The clinical course of this case emphasizes the need for vigilance and thoroughness in obtaining histories from patients presenting with seemingly benign complaints, especially in vulnerable populations, such as infants, pregnant women, and immunocompromised adults. In this case, the progression of symptoms might have been avoided if the patient’s dermatitis had been treated conservatively or with topical rather than systemic steroids.
1. Esposito S, Picciolli I, Semino M, Principi N. Dog and cat bite-associated infections in children. Eur J Clin Microbiol Infect Dis. 2013;32(8):971-976.
2. Medeiros I, Saconato H. Antibiotic prophylaxis for mammalian bites. Cochrane Database Syst Rev. 2001;(2):CD001738.
3. Haybaeck J, Schindler C, Braza P, Willinger B, Drlicek M. Rapidly progressive and lethal septicemia due to infection with Pasteurella multocida in an infant. Wien Klin Wochenschr. 2009;121(5-6):216-219.
4. Migliore E, Serraino C, Brignone C, et al. Pasteurella multocida infection in a cirrhotic patient: case report, microbiological aspects and a review of the literature. Adv Med Sci. 2009;54(1):109-112.
5. Mugambi SM, Ullian ME. Bacteremia, sepsis, and peritonitis with Pasteurella multocida in a peritoneal dialysis patient. Perit Dial Int. 2010;30(3):381-383.
6. Weber DJ, Wolfson JS, Swartz MN, Hooper DC. Pasteurella multocida infections. Report of 34 cases and review of the literature. Medicine (Baltimore). 1984;63(3):133-154.
7. Oehler RL, Velez AP, Mizrachi M, Lamarche J, Gompf S. Bite-related and septic syndromes caused by cats and dogs. Lancet Infect Dis. 2009;9(7):439-447.
A few days after an incidental cat bite, a patient presented to the emergency department for treatment of poison sumac exposure. He was discharged with oral methylprednisolone for the dermatitis and returned 1 week later with symptoms, examination findings, and laboratory results consistent with sepsis and bilateral upper extremity necrotizing soft-tissue infections. After administering multiple irrigation and débridement procedures, hyperbaric oxygen treatments, and an antibiotic regimen, the patient’s status greatly improved. However, the patient returned 1 month later with a new sternoclavicular joint prominence that was associated with painful crepitus. Additionally, he noted that his wrists were gradually becoming more swollen and painful. Imaging studies showed a lytic destruction of the sternoclavicular joint and erosive changes throughout the carpus and radiocarpal joint bilaterally, consistent with osteomyelitis. The patient was treated with ertapenem for 6 weeks, and his polyarthropathy resolved. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 73-year-old, right-hand–dominant man with no notable medical history presented to the emergency department for treatment of poison sumac exposure, incidentally, a few days after being bitten by a cat on the bilateral distal upper extremities. He was prescribed a course of oral methylprednisolone for dermatitis. A week later, the patient returned to the emergency department with altered mental status, fevers, diaphoresis, lethargy, and polyarthralgia. At the time of presentation, the patient’s vital signs were labile, and he was found to have extensive bilateral upper extremity erythema, blistering, petechiae, purpuric lesions, and exquisite pain with passive range of motion of his fingers and wrists. His leukocyte count was 25.1 × 109/L, and he had elevated C-reactive protein level and erythrocyte sedimentation rate of 150 mg/L and 120 mm/h, respectively. He was admitted for management of sepsis and presumed bilateral upper extremity necrotizing soft-tissue infection.
Broad-spectrum intravenous (IV) antibiotics (vancomycin, piperacillin, tazobactam) were initiated after blood cultures were obtained, and the patient was taken emergently to the operating theatre for irrigation and débridement of his hands and wrists bilaterally. Arthrotomy of the wrist and débridement of the distal extensor compartment and its tenosynovium were performed on the right forearm, in addition to a decompressive fasciotomy of the left forearm. Postoperatively, the patient’s mental status improved and his vital signs gradually normalized. He received multiple hyperbaric oxygen treatments and underwent several additional operative débridement procedures with eventual closure of his wounds. At initial presentation, the differential diagnosis for the severe soft-tissue infection included necrotizing fasciitis or myositis caused by any of a variety of bacterial pathogens. Most notably, it was important to elicit the history of a cat bite to include and consider Pasteurella multocida as a potential pathogen. Initial cultures supported the diagnosis of acute P multocida necrotizing skin and soft-tissue infection, in addition to septic arthritis. The patient’s blood and intraoperative wound cultures grew P multocida. The antibiotic treatment was tailored initially to ampicillin and sulbactam and then to a final regimen of orally administered ciprofloxacin (750 mg twice a day), once susceptibility testing was performed on the cultures. On hospital day 10, the patient was discharged home, receiving a 6-week course of ciprofloxacin to complete the 8-week course of treatment.
At follow-up, approximately 1 month after discharge, the patient noted that he had developed a new right sternoclavicular joint prominence that was associated with painful crepitus. He also noted that his wrists were gradually becoming more swollen and painful bilaterally. Computed tomography scans of the chest were obtained to evaluate the sternoclavicular joint (Figure 1). Repeat radiographs of the wrists were also obtained (Figure 2). Imaging showed lytic destruction of the sternoclavicular joint and erosive changes throughout the carpus and radiocarpal joint, consistent with osteomyelitis. The C-reactive protein level and erythrocyte sedimentation rate at this time were 34 mg/L and 124 mm/h, respectively.
The patient returned to the operating room for débridement and biopsy of the right sternoclavicular joint and left wrist. This patient’s delayed presentation was characterized by a subacute worsening of isolated musculoskeletal complaints. The differential diagnosis then included infection with the same bacterial pathogen versus reactive or inflammatory arthritis. Several intraoperative cultures failed to grow any bacteria, including P multocida, although P multocida was the presumptive cause of the erosive polyarthropathy, considering that symptoms eventually resolved with a repeated course of IV-administered ertapenem for 6 weeks. The patient experienced complete resolution of his joint pain and swelling. He was able to resume his activities of daily living and had no further recurrence of symptoms at follow-up 3 months later.
Discussion
Cat bites often are the source of Pasteurella species infections because the bacteria are carried by more than 90% of cats.1 These types of infections can cause septic arthritis, osteomyelitis, and deep subcutaneous and myofascial infections because of the sharp and narrow morphology of cat teeth. The infections can progress to necrotizing fasciitis and myositis if not recognized early, as was the case with our patient. Prophylactic antibiotic administration for animal bites is controversial and is not a universal practice.1,2Pasteurella bacteremia is an atypical progression that occurs more often in patients with pneumonia, septic arthritis, or meningitis. Cases of Pasteurella sepsis, necrotizing fasciitis, and septic arthritis have been reported.3-7 However, associated progressive septic arthritis and osteomyelitis, despite initial clinical improvement, have not been reported. Severe infection (ie, sepsis and septic shock) can occur in infants, pregnant women, and other immunocompromised patients.7 Immune suppression of our patient with steroid medication for poison sumac dermatitis likely contributed to the progression and systemic spread of an initially benign cat bite. Before prescribing steroids, it is imperative to ask about exposures and encourage patients to seek prompt medical attention with worsening or new symptoms. Healthy individuals rarely develop bacteremia; however, in these cases, mortality remains high at approximately 25%.4,6
The clinical course of this case emphasizes the need for vigilance and thoroughness in obtaining histories from patients presenting with seemingly benign complaints, especially in vulnerable populations, such as infants, pregnant women, and immunocompromised adults. In this case, the progression of symptoms might have been avoided if the patient’s dermatitis had been treated conservatively or with topical rather than systemic steroids.
A few days after an incidental cat bite, a patient presented to the emergency department for treatment of poison sumac exposure. He was discharged with oral methylprednisolone for the dermatitis and returned 1 week later with symptoms, examination findings, and laboratory results consistent with sepsis and bilateral upper extremity necrotizing soft-tissue infections. After administering multiple irrigation and débridement procedures, hyperbaric oxygen treatments, and an antibiotic regimen, the patient’s status greatly improved. However, the patient returned 1 month later with a new sternoclavicular joint prominence that was associated with painful crepitus. Additionally, he noted that his wrists were gradually becoming more swollen and painful. Imaging studies showed a lytic destruction of the sternoclavicular joint and erosive changes throughout the carpus and radiocarpal joint bilaterally, consistent with osteomyelitis. The patient was treated with ertapenem for 6 weeks, and his polyarthropathy resolved. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 73-year-old, right-hand–dominant man with no notable medical history presented to the emergency department for treatment of poison sumac exposure, incidentally, a few days after being bitten by a cat on the bilateral distal upper extremities. He was prescribed a course of oral methylprednisolone for dermatitis. A week later, the patient returned to the emergency department with altered mental status, fevers, diaphoresis, lethargy, and polyarthralgia. At the time of presentation, the patient’s vital signs were labile, and he was found to have extensive bilateral upper extremity erythema, blistering, petechiae, purpuric lesions, and exquisite pain with passive range of motion of his fingers and wrists. His leukocyte count was 25.1 × 109/L, and he had elevated C-reactive protein level and erythrocyte sedimentation rate of 150 mg/L and 120 mm/h, respectively. He was admitted for management of sepsis and presumed bilateral upper extremity necrotizing soft-tissue infection.
Broad-spectrum intravenous (IV) antibiotics (vancomycin, piperacillin, tazobactam) were initiated after blood cultures were obtained, and the patient was taken emergently to the operating theatre for irrigation and débridement of his hands and wrists bilaterally. Arthrotomy of the wrist and débridement of the distal extensor compartment and its tenosynovium were performed on the right forearm, in addition to a decompressive fasciotomy of the left forearm. Postoperatively, the patient’s mental status improved and his vital signs gradually normalized. He received multiple hyperbaric oxygen treatments and underwent several additional operative débridement procedures with eventual closure of his wounds. At initial presentation, the differential diagnosis for the severe soft-tissue infection included necrotizing fasciitis or myositis caused by any of a variety of bacterial pathogens. Most notably, it was important to elicit the history of a cat bite to include and consider Pasteurella multocida as a potential pathogen. Initial cultures supported the diagnosis of acute P multocida necrotizing skin and soft-tissue infection, in addition to septic arthritis. The patient’s blood and intraoperative wound cultures grew P multocida. The antibiotic treatment was tailored initially to ampicillin and sulbactam and then to a final regimen of orally administered ciprofloxacin (750 mg twice a day), once susceptibility testing was performed on the cultures. On hospital day 10, the patient was discharged home, receiving a 6-week course of ciprofloxacin to complete the 8-week course of treatment.
At follow-up, approximately 1 month after discharge, the patient noted that he had developed a new right sternoclavicular joint prominence that was associated with painful crepitus. He also noted that his wrists were gradually becoming more swollen and painful bilaterally. Computed tomography scans of the chest were obtained to evaluate the sternoclavicular joint (Figure 1). Repeat radiographs of the wrists were also obtained (Figure 2). Imaging showed lytic destruction of the sternoclavicular joint and erosive changes throughout the carpus and radiocarpal joint, consistent with osteomyelitis. The C-reactive protein level and erythrocyte sedimentation rate at this time were 34 mg/L and 124 mm/h, respectively.
The patient returned to the operating room for débridement and biopsy of the right sternoclavicular joint and left wrist. This patient’s delayed presentation was characterized by a subacute worsening of isolated musculoskeletal complaints. The differential diagnosis then included infection with the same bacterial pathogen versus reactive or inflammatory arthritis. Several intraoperative cultures failed to grow any bacteria, including P multocida, although P multocida was the presumptive cause of the erosive polyarthropathy, considering that symptoms eventually resolved with a repeated course of IV-administered ertapenem for 6 weeks. The patient experienced complete resolution of his joint pain and swelling. He was able to resume his activities of daily living and had no further recurrence of symptoms at follow-up 3 months later.
Discussion
Cat bites often are the source of Pasteurella species infections because the bacteria are carried by more than 90% of cats.1 These types of infections can cause septic arthritis, osteomyelitis, and deep subcutaneous and myofascial infections because of the sharp and narrow morphology of cat teeth. The infections can progress to necrotizing fasciitis and myositis if not recognized early, as was the case with our patient. Prophylactic antibiotic administration for animal bites is controversial and is not a universal practice.1,2Pasteurella bacteremia is an atypical progression that occurs more often in patients with pneumonia, septic arthritis, or meningitis. Cases of Pasteurella sepsis, necrotizing fasciitis, and septic arthritis have been reported.3-7 However, associated progressive septic arthritis and osteomyelitis, despite initial clinical improvement, have not been reported. Severe infection (ie, sepsis and septic shock) can occur in infants, pregnant women, and other immunocompromised patients.7 Immune suppression of our patient with steroid medication for poison sumac dermatitis likely contributed to the progression and systemic spread of an initially benign cat bite. Before prescribing steroids, it is imperative to ask about exposures and encourage patients to seek prompt medical attention with worsening or new symptoms. Healthy individuals rarely develop bacteremia; however, in these cases, mortality remains high at approximately 25%.4,6
The clinical course of this case emphasizes the need for vigilance and thoroughness in obtaining histories from patients presenting with seemingly benign complaints, especially in vulnerable populations, such as infants, pregnant women, and immunocompromised adults. In this case, the progression of symptoms might have been avoided if the patient’s dermatitis had been treated conservatively or with topical rather than systemic steroids.
1. Esposito S, Picciolli I, Semino M, Principi N. Dog and cat bite-associated infections in children. Eur J Clin Microbiol Infect Dis. 2013;32(8):971-976.
2. Medeiros I, Saconato H. Antibiotic prophylaxis for mammalian bites. Cochrane Database Syst Rev. 2001;(2):CD001738.
3. Haybaeck J, Schindler C, Braza P, Willinger B, Drlicek M. Rapidly progressive and lethal septicemia due to infection with Pasteurella multocida in an infant. Wien Klin Wochenschr. 2009;121(5-6):216-219.
4. Migliore E, Serraino C, Brignone C, et al. Pasteurella multocida infection in a cirrhotic patient: case report, microbiological aspects and a review of the literature. Adv Med Sci. 2009;54(1):109-112.
5. Mugambi SM, Ullian ME. Bacteremia, sepsis, and peritonitis with Pasteurella multocida in a peritoneal dialysis patient. Perit Dial Int. 2010;30(3):381-383.
6. Weber DJ, Wolfson JS, Swartz MN, Hooper DC. Pasteurella multocida infections. Report of 34 cases and review of the literature. Medicine (Baltimore). 1984;63(3):133-154.
7. Oehler RL, Velez AP, Mizrachi M, Lamarche J, Gompf S. Bite-related and septic syndromes caused by cats and dogs. Lancet Infect Dis. 2009;9(7):439-447.
1. Esposito S, Picciolli I, Semino M, Principi N. Dog and cat bite-associated infections in children. Eur J Clin Microbiol Infect Dis. 2013;32(8):971-976.
2. Medeiros I, Saconato H. Antibiotic prophylaxis for mammalian bites. Cochrane Database Syst Rev. 2001;(2):CD001738.
3. Haybaeck J, Schindler C, Braza P, Willinger B, Drlicek M. Rapidly progressive and lethal septicemia due to infection with Pasteurella multocida in an infant. Wien Klin Wochenschr. 2009;121(5-6):216-219.
4. Migliore E, Serraino C, Brignone C, et al. Pasteurella multocida infection in a cirrhotic patient: case report, microbiological aspects and a review of the literature. Adv Med Sci. 2009;54(1):109-112.
5. Mugambi SM, Ullian ME. Bacteremia, sepsis, and peritonitis with Pasteurella multocida in a peritoneal dialysis patient. Perit Dial Int. 2010;30(3):381-383.
6. Weber DJ, Wolfson JS, Swartz MN, Hooper DC. Pasteurella multocida infections. Report of 34 cases and review of the literature. Medicine (Baltimore). 1984;63(3):133-154.
7. Oehler RL, Velez AP, Mizrachi M, Lamarche J, Gompf S. Bite-related and septic syndromes caused by cats and dogs. Lancet Infect Dis. 2009;9(7):439-447.
Interim PET provides limited prognostic value for diffuse large B-cell lymphoma
Results from positron emission tomography after 2 and 4 weeks of standardized, dose-dense chemotherapy in patients with diffuse large B-cell lymphoma (DLBCL) failed to provide sufficient prognostic accuracy to guide therapeutic decisions, according to a report published online in the Journal of Clinical Oncology.
“Our data demonstrate that interim PET-CT fails to deliver a PPV [positive predictive value] or NPV [negative predictive value] that reflects the clinical outcome for patients with DLBCL treated with R-CHOP-14 with sufficient accuracy to guide therapeutic decisions such as treatment intensification/deintensification at the present stage,” wrote Dr. Chrisoph Mamot of the division of haematology/oncology, Cantonal Hospital of Aarau, Switzerland, and colleagues (J. Clin. Onc. 2015 July 6 [doi:10.1200/JCO.2014.58.9846]).
Although the likelihood of 2-year event-free survival (EFS) was significantly lower among patients who had positive PET results after 2 weeks of therapy (PET-2-positive), compared with PET-2-negative patients (48% vs. 74%; P = .004), a large proportion of interim PET-positive patients had favorable outcomes. This may be due to the dose-dense regimen, Dr. Mamot and colleagues said.
Patients underwent six cycles of R-CHOP (rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2, vincristine 1.4 mg/m2, and prednisone 100 mg/m2 for 5 days) every 14 days, followed by two cycles of rituximab.
Patients who had a positive PET-2 result received another PET scan after completion of four chemotherapy cycles. The 2-year EFS for PET-4-positive and PET-4-negative patients were not significantly different.
“Our original hypothesis that an interim PET/CT at a later time point might be able to better separate patients with good or poor prognosis was not confirmed,” the authors wrote. The end-of-treatment PET provided the best prediction of good vs. poor prognosis, but this time point is too late to adapt treatment. The authors concluded that interim PET/CT in patients with DLBCL presently is not accurate enough to guide treatment decisions in individual patients.
The multicenter, prospective study evaluated 138 patients, median age 58.5 years, who had untreated DLBCL, Ann Arbor stage I (12%), II (34%), III (23%), or IV (30%).
The study was supported by grants from Amgen (Switzerland) and OncoSuisse. Dr. Christoph Mamot reported consulting or advisory roles with Roche, Novartis, Amgen, and Boehringer-Ingelheim. Several of his coauthors reported ties to industry sources.
Results from positron emission tomography after 2 and 4 weeks of standardized, dose-dense chemotherapy in patients with diffuse large B-cell lymphoma (DLBCL) failed to provide sufficient prognostic accuracy to guide therapeutic decisions, according to a report published online in the Journal of Clinical Oncology.
“Our data demonstrate that interim PET-CT fails to deliver a PPV [positive predictive value] or NPV [negative predictive value] that reflects the clinical outcome for patients with DLBCL treated with R-CHOP-14 with sufficient accuracy to guide therapeutic decisions such as treatment intensification/deintensification at the present stage,” wrote Dr. Chrisoph Mamot of the division of haematology/oncology, Cantonal Hospital of Aarau, Switzerland, and colleagues (J. Clin. Onc. 2015 July 6 [doi:10.1200/JCO.2014.58.9846]).
Although the likelihood of 2-year event-free survival (EFS) was significantly lower among patients who had positive PET results after 2 weeks of therapy (PET-2-positive), compared with PET-2-negative patients (48% vs. 74%; P = .004), a large proportion of interim PET-positive patients had favorable outcomes. This may be due to the dose-dense regimen, Dr. Mamot and colleagues said.
Patients underwent six cycles of R-CHOP (rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2, vincristine 1.4 mg/m2, and prednisone 100 mg/m2 for 5 days) every 14 days, followed by two cycles of rituximab.
Patients who had a positive PET-2 result received another PET scan after completion of four chemotherapy cycles. The 2-year EFS for PET-4-positive and PET-4-negative patients were not significantly different.
“Our original hypothesis that an interim PET/CT at a later time point might be able to better separate patients with good or poor prognosis was not confirmed,” the authors wrote. The end-of-treatment PET provided the best prediction of good vs. poor prognosis, but this time point is too late to adapt treatment. The authors concluded that interim PET/CT in patients with DLBCL presently is not accurate enough to guide treatment decisions in individual patients.
The multicenter, prospective study evaluated 138 patients, median age 58.5 years, who had untreated DLBCL, Ann Arbor stage I (12%), II (34%), III (23%), or IV (30%).
The study was supported by grants from Amgen (Switzerland) and OncoSuisse. Dr. Christoph Mamot reported consulting or advisory roles with Roche, Novartis, Amgen, and Boehringer-Ingelheim. Several of his coauthors reported ties to industry sources.
Results from positron emission tomography after 2 and 4 weeks of standardized, dose-dense chemotherapy in patients with diffuse large B-cell lymphoma (DLBCL) failed to provide sufficient prognostic accuracy to guide therapeutic decisions, according to a report published online in the Journal of Clinical Oncology.
“Our data demonstrate that interim PET-CT fails to deliver a PPV [positive predictive value] or NPV [negative predictive value] that reflects the clinical outcome for patients with DLBCL treated with R-CHOP-14 with sufficient accuracy to guide therapeutic decisions such as treatment intensification/deintensification at the present stage,” wrote Dr. Chrisoph Mamot of the division of haematology/oncology, Cantonal Hospital of Aarau, Switzerland, and colleagues (J. Clin. Onc. 2015 July 6 [doi:10.1200/JCO.2014.58.9846]).
Although the likelihood of 2-year event-free survival (EFS) was significantly lower among patients who had positive PET results after 2 weeks of therapy (PET-2-positive), compared with PET-2-negative patients (48% vs. 74%; P = .004), a large proportion of interim PET-positive patients had favorable outcomes. This may be due to the dose-dense regimen, Dr. Mamot and colleagues said.
Patients underwent six cycles of R-CHOP (rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2, vincristine 1.4 mg/m2, and prednisone 100 mg/m2 for 5 days) every 14 days, followed by two cycles of rituximab.
Patients who had a positive PET-2 result received another PET scan after completion of four chemotherapy cycles. The 2-year EFS for PET-4-positive and PET-4-negative patients were not significantly different.
“Our original hypothesis that an interim PET/CT at a later time point might be able to better separate patients with good or poor prognosis was not confirmed,” the authors wrote. The end-of-treatment PET provided the best prediction of good vs. poor prognosis, but this time point is too late to adapt treatment. The authors concluded that interim PET/CT in patients with DLBCL presently is not accurate enough to guide treatment decisions in individual patients.
The multicenter, prospective study evaluated 138 patients, median age 58.5 years, who had untreated DLBCL, Ann Arbor stage I (12%), II (34%), III (23%), or IV (30%).
The study was supported by grants from Amgen (Switzerland) and OncoSuisse. Dr. Christoph Mamot reported consulting or advisory roles with Roche, Novartis, Amgen, and Boehringer-Ingelheim. Several of his coauthors reported ties to industry sources.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: After two cycles of standardized, dose-dense chemotherapy in patients with diffuse large B-cell lymphoma (DLBCL), positive results from PET signaled worse 2-year event-free survival (EFS) than negative interim PET results, but overall survival was not significantly different.
Major finding: After two cycles of chemotherapy, 2-year EFS for PET-positive, compared with PET-negative patients was 48.2% vs. 74.2% (P = .004). Overall survival was 87.7% vs. 90.6% (P = .6).
Data source: The multicenter, prospective study evaluated 138 patients, median age 58.5 years, who had untreated DLBCL, Ann Arbor stage I (12%), II (34%), III (23%), or IV (30%).
Disclosures: The study was supported by grants from Amgen (Switzerland) and OncoSuisse. Dr. Christoph Mamot reported consulting or advisory roles with Roche, Novartis, Amgen, and Boehringer-Ingelheim. Several of his coauthors reported ties to industry sources.
Enoxaparin and Warfarin for Venous Thromboembolism Prophylaxis in Total Hip Arthroplasty: To Bridge or Not to Bridge?
According to the literature, the rate of deep venous thrombosis after total hip arthroplasty (THA) can be high (45%-63%) without prophylactic anticoagulation.1-6 A meta-analysis of 13 studies found a rate of 51%.7 As lower extremity deep venous thrombi are the initial source of symptomatic pulmonary emboli in about 90% of cases,8 THA patients are usually given medication postoperatively focused on prevention of these thromboembolic events.9 Chemoprophylaxis may involve warfarin, enoxaparin, or their combination in an anticoagulation bridge. Enoxaparin is one of many low-molecular-weight heparins (LMWHs). All LMWHs exert their anticoagulant effect by binding to antithrombin III.10 The binding of LMWH to antithrombin III catalyzes the inhibition of factor Xa by antithrombin III, disrupting clot formation.11
In its hydroquinone form, vitamin K is essential as a cofactor for carboxylation of the glutamic acid residues of the amino-terminals of the coagulation proteins II, VII, IX, and X, leading to their activation. Anticoagulation by warfarin is achieved by the inhibition of the reductase enzymes that produce vitamin K hydroquinone in the liver from vitamin K epoxide.12 This inhibition prevents activation of the clotting proteins.12,13 Prophylaxis with enoxaparin or warfarin can reduce the rate of venous thromboembolic disease to 3.6% and 3.7%, respectively.2 However, these medications inhibit the clotting cascade, and their use risks prolonging the healing process.9 The delay increases the risk for wound infection,14 which can lead to a longer hospital stay and therefore higher costs.
We conducted a study to compare patients who received warfarin only with patients who received warfarin bridged with enoxaparin as antithrombotic chemoprophylaxis after THA. Outcomes of interest were number of days until a dry wound was observed and length of hospital stay. We hypothesized that, compared with warfarin-only therapy, bridged therapy would increase the risk for prolonged wound healing and result in longer hospital stays.
Materials and Methods
At our 746-bed academic medical center, 121 THAs were performed between January 1, 2008 and December 31, 2009. This study was approved by the center’s Office for Human Subjects Protections institutional review board (IRB). The research involved collecting or studying existing data, documents, and records recorded anonymously by the investigator in such a manner that subjects could not be identified, directly or through identifiers linked to the subjects, and therefore patient consent was not needed. Therefore, the IRB waived the need for consent. Relevant data included in this study were extracted from patient medical records, given within 35 days of surgery. For each patient, discharge notes provided data on the hospital course, and nurses’ notes provided data on wound status after THA.
Propensity Score Matching
For accurate analysis, it was important to consider confounding factors in both patient groups. Some covariates that may influence accurate analysis are age,15 diabetes,16 sex,15,17 hypertension,18 and body mass index.15,19Propensity score, defined as the conditional probability of receiving treatment, given the observed background covariates, was initially defined by Rosenbaum20 and Rubin.21 The motivation behind propensity scores can be understood by considering an idealized situation in which the 2 groups are similar on all background characteristics. In nonexperimental studies, researchers aim to find for each treated individual a comparison individual who looks exactly the same as the treated individual with respect to observed pretreatment covariates. Thus, assuming no hidden bias, any difference in outcomes within these pairs can be attributed to the variable of interest and not to any other differences between the treated and comparison individuals. Our study is a typical nonexperimental retrospective study in which the 2 groups being compared are patients receiving warfarin only or warfarin bridged with enoxaparin. To minimize the influence of background covariates, we used matching procedures and present our results both with and without the use of matching techniques.
Data and Results
There are different matching algorithms aimed at matching groups. In our study, the optimal matching procedure alone could not produce adequately matched data, so we used both optimal matching20 and genetic matching.22,23 Genetic matching procedure with replacement22 can produce well-matched data—it matched each patient in the warfarin-only group with a patient in the bridged-therapy group and allowed different patients to be matched with 1 similar patient in the control group. However, as the same patients in the bridged-therapy group might be matched multiple times, it would complicate the after-matching analysis. We therefore used a 2-step matching procedure to obtain well-matched data, and a simplified analysis procedure after matching. In the first step, we implemented genetic matching with replacement, as introduced by Abadie and Imbens,22 to match each warfarin-only patient with 1 bridged-therapy patient. In the second step, we applied optimal matching to the 2 groups. This 2-step matching turned out to produce better matched pairs, as denoted by Rubin.21 Both matching steps were implemented using the MatchIt function in R.24
The balance of matching is checked using criteria suggested by Rubin21: (1) standardized difference of means of propensity score, (2) ratio of variances in propensity score in treated and control groups, and (3) for each covariate, ratio of variance in residuals orthogonal to propensity score in treated and control groups.
Table 1 lists the means of the background covariates for each group before and after matching. Table 2 lists the balance check results suggested by Rubin.21 After matching, all standardized differences of means are smaller than 0.25, and the variance ratios are between 0.5 and 2, which are the standards suggested21 for regression adjustment to be valid after matching.
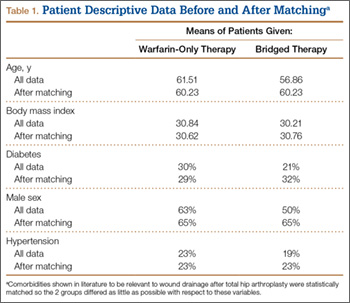
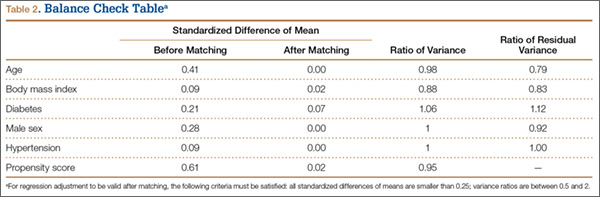
After genetic matching, 31 bridged-therapy patients and 57 warfarin-only patients remained. After optimal matching, there were 31 patients in each group. Poisson regressions of datasets before and after matching adjustment were fitted.
Results
Wounds of bridged-therapy patients took longer to heal than wounds of warfarin-only patients both before (odds ratio, 2.16; P < .05) and after matching data (odds ratio, 2.39; P < .05) with respect to confounding factors. In addition, bridged-therapy patients had longer hospital stays both before (odds ratio 1.20; P < .05) and after matching data (odds ratio, 1.27; P < .05) with respect to confounding factors. Figures 1 and 2 are histograms displaying the 2 groups and their outcomes.
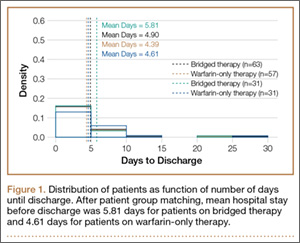
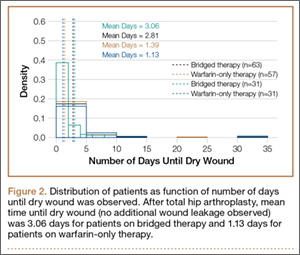
Discussion
For patients undergoing THA procedures, several important considerations should be taken into account. Colwell and colleagues2 showed that, compared with warfarin, enoxaparin offered a 0.1% higher rate of protection against venous thromboembolic disease after THA. However, patients given enoxaparin may face increased risks.25 Hallevi and colleagues26 demonstrated that, compared with warfarin, enoxaparin bridging increased the risk for serious bleeding in patients with cardioembolic stroke. In our review of the literature, we learned that the benefits of bridge therapy in thromboembolic disease have yet to be investigated in THA.
At our academic hospital, the extra costs associated with bridge therapy can be as much as about $200027 per day per patient. These costs can go much higher, depending on type of patient and types of resources used. Over the 2-year period covered by our study, the costs of using enoxaparin amounted to about $151,200 ($2000 × 1.2 days per patient). If bridging offers no significant protection against thromboembolic disease, then it would be more cost-effective to use a single anticoagulant, particularly enoxaparin, for high-risk patients.
There are significant risk factors associated with prolonged healing of surgical wounds. Protocols outlining these factors may help reduce costs. In addition, when deciding on the use of aggressive anticoagulation therapy, surgeons must consider the risks for prolonged leakage and infection in addition to the risk for thromboembolic disease. Protocols may aid in this process as well. Our study results showed that, compared with warfarin-only therapy, bridged therapy (enoxaparin and warfarin) was associated with longer hospital stays. Further research should examine whether there are advantages that justify the higher risks of delayed wound healing and subsequent infection. Improving our understanding of risk factors associated with anticoagulation therapy will make orthopedic surgery safer for patients.
1. Bergqvist D, Benoni G, Björgell O, et al. Low-molecular-weight heparin (enoxaparin) as prophylaxis against venous thromboembolism after total hip replacement. N Engl J Med. 1996;335(10):696-700.
2. Colwell CW Jr, Collis DK, Paulson R, et al. Comparison of enoxaparin and warfarin for the prevention of venous thromboembolic disease after total hip arthroplasty. Evaluation during hospitalization and three months after discharge. J Bone Joint Surg Am. 1999;81(7):932-940.
3. Haake DA, Berkman SA. Venous thromboembolic disease after hip surgery. Risk factors, prophylaxis, and diagnosis. Clin Orthop Relat Res. 1989;(242):212-231.
4. Johnson R, Carmichael JH, Almond HG, Loynes RP. Deep venous thrombosis following Charnley arthroplasty. Clin Orthop Relat Res. 1978;(132):24-30.
5. Stamatakis JD, Kakkar VV, Sagar S, Lawrence D, Nairn D, Bentley PG. Femoral vein thrombosis and total hip replacement. Br Med J. 1977;2(6081):223-225.
6. Turpie AG, Levine MN, Hirsh J, et al. A randomized controlled trial of a low-molecular-weight heparin (enoxaparin) to prevent deep-vein thrombosis in patients undergoing elective hip surgery. N Engl J Med. 1986;315(15):925-929.
7. Clagett GP, Anderson FA Jr, Heit J, Levine MN, Wheeler HB. Prevention of venous thromboembolism. Chest. 1995;108(4 suppl):312S-334S.
8. Westrich GH, Sánchez PM. Prevention and treatment of thromboembolic disease: an overview. Instr Course Lect. 2002;51:471-480.
9. Colwell CW Jr, Froimson MI, Mont MA, et al. Thrombosis prevention after total hip arthroplasty: a prospective, randomized trial comparing a mobile compression device with low-molecular-weight heparin. J Bone Joint Surg Am. 2010;92(3):527-535.
10. Fareed J, Jeske W, Hoppensteadt D, Clarizio R, Walenga JM. Low-molecular-weight heparins: pharmacologic profile and product differentiation. Am J Cardiol. 1998;82(5B):3L-10L.
11. Gerlach AT, Pickworth KK, Seth SK, Tanna SB, Barnes JF. Enoxaparin and bleeding complications: a review in patients with and without renal insufficiency. Pharmacotherapy. 2000;20(7):771-775.
12. Kamali F, Wood P, Ward A. Vitamin K deficiency amplifies anticoagulation response to ximelagatran: possible implications for direct thrombin inhibitors and their clinical safety. Ann Hematol. 2009;88(2):141-149.
13. Choonara IA, Malia RG, Haynes BP, et al. The relationship between inhibition of vitamin K1 2,3-epoxide reductase and reduction of clotting factor activity with warfarin. Br J Clin Pharmacol. 1988;25(1):1-7.
14. Saleh K, Olson M, Resig S, et al. Predictors of wound infection in hip and knee joint replacement: results from a 20 year surveillance program. J Orthop Res. 2002;20(3):506-515.
15. Ridgeway S, Wilson J, Charlet A, Kafatos G, Pearson A, Coello R. Infection of the surgical site after arthroplasty of the hip. J Bone Joint Surg Br. 2005;87(6):844-850.
16. Lai K, Bohm ER, Burnell C, Hedden DR. Presence of medical comorbidities in patients with infected primary hip or knee arthroplasties. J Arthroplasty. 2007;22(5):651-656.
17. Kurtz SM, Lau E, Schmier J, Ong KL, Zhao K, Parvizi J. Infection burden for hip and knee arthroplasty in the United States. J Arthroplasty. 2008;23(7):984-991.
18. Ahmed AA, Mooar PA, Kleiner M, Torg JS, Miyamoto CT. Hypertensive patients show delayed wound healing following total hip arthroplasty. PLoS One. 2011;6(8):e23224.
19. Lübbeke A, Stern R, Garavaglia G, Zurcher L, Hoffmeyer P. Differences in outcomes of obese women and men undergoing primary total hip arthroplasty. Arthritis Rheum. 2007;57(2):327-334.
20. Rosenbaum PR. A characterization of optimal designs for observational studies. J R Stat Soc Ser B. 1991;53(3):597-610.
21. Rubin DB. Using propensity scores to help design observational studies: application to the tobacco litigation. Health Serv Outcomes Res Methodol. 2001;2(1):169-188.
22. Abadie A, Imbens GW. Simple and Bias-Corrected Matching Estimators for Average Treatment Effects. Berkeley, CA: Department of Economics, University of California; 2002.
23. Diamond A, Sekhon J. Genetic matching for estimating causal effects: a new method of achieving balance in observational studies. Paper presented at: Annual Meeting of the Midwest Political Science Association; April 2005; Chicago, IL.
24. Imai K, King G, Lau O. logit: logistic regression for dichotomous dependent variables. In: Imai K, King G, Lau O. Zelig: Everyone’s Statistical Software. 2011; 238-244. http://gking.harvard.edu/zelig. Accessed May 26, 2015.
25. Patel VP, Walsh M, Sehgal B, Preston C, DeWal H, Di Cesare PE. Factors associated with prolonged wound drainage after primary total hip and knee arthroplasty. J Bone Joint Surg Am. 2007;89(1):33-38.
26. Hallevi H, Albright KC, Martin-Schild S, et al. Anticoagulation after cardioembolic stroke: to bridge or not to bridge? Arch Neurol. 2008;65(9):1169-1173.
27. Henry J. Kaiser Family Foundation. Hospital adjusted expenses per inpatient day [2010]. http://kff.org/other/state-indicator/expenses-per-inpatient-day/#table. Accessed May 26, 2015.
According to the literature, the rate of deep venous thrombosis after total hip arthroplasty (THA) can be high (45%-63%) without prophylactic anticoagulation.1-6 A meta-analysis of 13 studies found a rate of 51%.7 As lower extremity deep venous thrombi are the initial source of symptomatic pulmonary emboli in about 90% of cases,8 THA patients are usually given medication postoperatively focused on prevention of these thromboembolic events.9 Chemoprophylaxis may involve warfarin, enoxaparin, or their combination in an anticoagulation bridge. Enoxaparin is one of many low-molecular-weight heparins (LMWHs). All LMWHs exert their anticoagulant effect by binding to antithrombin III.10 The binding of LMWH to antithrombin III catalyzes the inhibition of factor Xa by antithrombin III, disrupting clot formation.11
In its hydroquinone form, vitamin K is essential as a cofactor for carboxylation of the glutamic acid residues of the amino-terminals of the coagulation proteins II, VII, IX, and X, leading to their activation. Anticoagulation by warfarin is achieved by the inhibition of the reductase enzymes that produce vitamin K hydroquinone in the liver from vitamin K epoxide.12 This inhibition prevents activation of the clotting proteins.12,13 Prophylaxis with enoxaparin or warfarin can reduce the rate of venous thromboembolic disease to 3.6% and 3.7%, respectively.2 However, these medications inhibit the clotting cascade, and their use risks prolonging the healing process.9 The delay increases the risk for wound infection,14 which can lead to a longer hospital stay and therefore higher costs.
We conducted a study to compare patients who received warfarin only with patients who received warfarin bridged with enoxaparin as antithrombotic chemoprophylaxis after THA. Outcomes of interest were number of days until a dry wound was observed and length of hospital stay. We hypothesized that, compared with warfarin-only therapy, bridged therapy would increase the risk for prolonged wound healing and result in longer hospital stays.
Materials and Methods
At our 746-bed academic medical center, 121 THAs were performed between January 1, 2008 and December 31, 2009. This study was approved by the center’s Office for Human Subjects Protections institutional review board (IRB). The research involved collecting or studying existing data, documents, and records recorded anonymously by the investigator in such a manner that subjects could not be identified, directly or through identifiers linked to the subjects, and therefore patient consent was not needed. Therefore, the IRB waived the need for consent. Relevant data included in this study were extracted from patient medical records, given within 35 days of surgery. For each patient, discharge notes provided data on the hospital course, and nurses’ notes provided data on wound status after THA.
Propensity Score Matching
For accurate analysis, it was important to consider confounding factors in both patient groups. Some covariates that may influence accurate analysis are age,15 diabetes,16 sex,15,17 hypertension,18 and body mass index.15,19Propensity score, defined as the conditional probability of receiving treatment, given the observed background covariates, was initially defined by Rosenbaum20 and Rubin.21 The motivation behind propensity scores can be understood by considering an idealized situation in which the 2 groups are similar on all background characteristics. In nonexperimental studies, researchers aim to find for each treated individual a comparison individual who looks exactly the same as the treated individual with respect to observed pretreatment covariates. Thus, assuming no hidden bias, any difference in outcomes within these pairs can be attributed to the variable of interest and not to any other differences between the treated and comparison individuals. Our study is a typical nonexperimental retrospective study in which the 2 groups being compared are patients receiving warfarin only or warfarin bridged with enoxaparin. To minimize the influence of background covariates, we used matching procedures and present our results both with and without the use of matching techniques.
Data and Results
There are different matching algorithms aimed at matching groups. In our study, the optimal matching procedure alone could not produce adequately matched data, so we used both optimal matching20 and genetic matching.22,23 Genetic matching procedure with replacement22 can produce well-matched data—it matched each patient in the warfarin-only group with a patient in the bridged-therapy group and allowed different patients to be matched with 1 similar patient in the control group. However, as the same patients in the bridged-therapy group might be matched multiple times, it would complicate the after-matching analysis. We therefore used a 2-step matching procedure to obtain well-matched data, and a simplified analysis procedure after matching. In the first step, we implemented genetic matching with replacement, as introduced by Abadie and Imbens,22 to match each warfarin-only patient with 1 bridged-therapy patient. In the second step, we applied optimal matching to the 2 groups. This 2-step matching turned out to produce better matched pairs, as denoted by Rubin.21 Both matching steps were implemented using the MatchIt function in R.24
The balance of matching is checked using criteria suggested by Rubin21: (1) standardized difference of means of propensity score, (2) ratio of variances in propensity score in treated and control groups, and (3) for each covariate, ratio of variance in residuals orthogonal to propensity score in treated and control groups.
Table 1 lists the means of the background covariates for each group before and after matching. Table 2 lists the balance check results suggested by Rubin.21 After matching, all standardized differences of means are smaller than 0.25, and the variance ratios are between 0.5 and 2, which are the standards suggested21 for regression adjustment to be valid after matching.
After genetic matching, 31 bridged-therapy patients and 57 warfarin-only patients remained. After optimal matching, there were 31 patients in each group. Poisson regressions of datasets before and after matching adjustment were fitted.
Results
Wounds of bridged-therapy patients took longer to heal than wounds of warfarin-only patients both before (odds ratio, 2.16; P < .05) and after matching data (odds ratio, 2.39; P < .05) with respect to confounding factors. In addition, bridged-therapy patients had longer hospital stays both before (odds ratio 1.20; P < .05) and after matching data (odds ratio, 1.27; P < .05) with respect to confounding factors. Figures 1 and 2 are histograms displaying the 2 groups and their outcomes.
Discussion
For patients undergoing THA procedures, several important considerations should be taken into account. Colwell and colleagues2 showed that, compared with warfarin, enoxaparin offered a 0.1% higher rate of protection against venous thromboembolic disease after THA. However, patients given enoxaparin may face increased risks.25 Hallevi and colleagues26 demonstrated that, compared with warfarin, enoxaparin bridging increased the risk for serious bleeding in patients with cardioembolic stroke. In our review of the literature, we learned that the benefits of bridge therapy in thromboembolic disease have yet to be investigated in THA.
At our academic hospital, the extra costs associated with bridge therapy can be as much as about $200027 per day per patient. These costs can go much higher, depending on type of patient and types of resources used. Over the 2-year period covered by our study, the costs of using enoxaparin amounted to about $151,200 ($2000 × 1.2 days per patient). If bridging offers no significant protection against thromboembolic disease, then it would be more cost-effective to use a single anticoagulant, particularly enoxaparin, for high-risk patients.
There are significant risk factors associated with prolonged healing of surgical wounds. Protocols outlining these factors may help reduce costs. In addition, when deciding on the use of aggressive anticoagulation therapy, surgeons must consider the risks for prolonged leakage and infection in addition to the risk for thromboembolic disease. Protocols may aid in this process as well. Our study results showed that, compared with warfarin-only therapy, bridged therapy (enoxaparin and warfarin) was associated with longer hospital stays. Further research should examine whether there are advantages that justify the higher risks of delayed wound healing and subsequent infection. Improving our understanding of risk factors associated with anticoagulation therapy will make orthopedic surgery safer for patients.
According to the literature, the rate of deep venous thrombosis after total hip arthroplasty (THA) can be high (45%-63%) without prophylactic anticoagulation.1-6 A meta-analysis of 13 studies found a rate of 51%.7 As lower extremity deep venous thrombi are the initial source of symptomatic pulmonary emboli in about 90% of cases,8 THA patients are usually given medication postoperatively focused on prevention of these thromboembolic events.9 Chemoprophylaxis may involve warfarin, enoxaparin, or their combination in an anticoagulation bridge. Enoxaparin is one of many low-molecular-weight heparins (LMWHs). All LMWHs exert their anticoagulant effect by binding to antithrombin III.10 The binding of LMWH to antithrombin III catalyzes the inhibition of factor Xa by antithrombin III, disrupting clot formation.11
In its hydroquinone form, vitamin K is essential as a cofactor for carboxylation of the glutamic acid residues of the amino-terminals of the coagulation proteins II, VII, IX, and X, leading to their activation. Anticoagulation by warfarin is achieved by the inhibition of the reductase enzymes that produce vitamin K hydroquinone in the liver from vitamin K epoxide.12 This inhibition prevents activation of the clotting proteins.12,13 Prophylaxis with enoxaparin or warfarin can reduce the rate of venous thromboembolic disease to 3.6% and 3.7%, respectively.2 However, these medications inhibit the clotting cascade, and their use risks prolonging the healing process.9 The delay increases the risk for wound infection,14 which can lead to a longer hospital stay and therefore higher costs.
We conducted a study to compare patients who received warfarin only with patients who received warfarin bridged with enoxaparin as antithrombotic chemoprophylaxis after THA. Outcomes of interest were number of days until a dry wound was observed and length of hospital stay. We hypothesized that, compared with warfarin-only therapy, bridged therapy would increase the risk for prolonged wound healing and result in longer hospital stays.
Materials and Methods
At our 746-bed academic medical center, 121 THAs were performed between January 1, 2008 and December 31, 2009. This study was approved by the center’s Office for Human Subjects Protections institutional review board (IRB). The research involved collecting or studying existing data, documents, and records recorded anonymously by the investigator in such a manner that subjects could not be identified, directly or through identifiers linked to the subjects, and therefore patient consent was not needed. Therefore, the IRB waived the need for consent. Relevant data included in this study were extracted from patient medical records, given within 35 days of surgery. For each patient, discharge notes provided data on the hospital course, and nurses’ notes provided data on wound status after THA.
Propensity Score Matching
For accurate analysis, it was important to consider confounding factors in both patient groups. Some covariates that may influence accurate analysis are age,15 diabetes,16 sex,15,17 hypertension,18 and body mass index.15,19Propensity score, defined as the conditional probability of receiving treatment, given the observed background covariates, was initially defined by Rosenbaum20 and Rubin.21 The motivation behind propensity scores can be understood by considering an idealized situation in which the 2 groups are similar on all background characteristics. In nonexperimental studies, researchers aim to find for each treated individual a comparison individual who looks exactly the same as the treated individual with respect to observed pretreatment covariates. Thus, assuming no hidden bias, any difference in outcomes within these pairs can be attributed to the variable of interest and not to any other differences between the treated and comparison individuals. Our study is a typical nonexperimental retrospective study in which the 2 groups being compared are patients receiving warfarin only or warfarin bridged with enoxaparin. To minimize the influence of background covariates, we used matching procedures and present our results both with and without the use of matching techniques.
Data and Results
There are different matching algorithms aimed at matching groups. In our study, the optimal matching procedure alone could not produce adequately matched data, so we used both optimal matching20 and genetic matching.22,23 Genetic matching procedure with replacement22 can produce well-matched data—it matched each patient in the warfarin-only group with a patient in the bridged-therapy group and allowed different patients to be matched with 1 similar patient in the control group. However, as the same patients in the bridged-therapy group might be matched multiple times, it would complicate the after-matching analysis. We therefore used a 2-step matching procedure to obtain well-matched data, and a simplified analysis procedure after matching. In the first step, we implemented genetic matching with replacement, as introduced by Abadie and Imbens,22 to match each warfarin-only patient with 1 bridged-therapy patient. In the second step, we applied optimal matching to the 2 groups. This 2-step matching turned out to produce better matched pairs, as denoted by Rubin.21 Both matching steps were implemented using the MatchIt function in R.24
The balance of matching is checked using criteria suggested by Rubin21: (1) standardized difference of means of propensity score, (2) ratio of variances in propensity score in treated and control groups, and (3) for each covariate, ratio of variance in residuals orthogonal to propensity score in treated and control groups.
Table 1 lists the means of the background covariates for each group before and after matching. Table 2 lists the balance check results suggested by Rubin.21 After matching, all standardized differences of means are smaller than 0.25, and the variance ratios are between 0.5 and 2, which are the standards suggested21 for regression adjustment to be valid after matching.
After genetic matching, 31 bridged-therapy patients and 57 warfarin-only patients remained. After optimal matching, there were 31 patients in each group. Poisson regressions of datasets before and after matching adjustment were fitted.
Results
Wounds of bridged-therapy patients took longer to heal than wounds of warfarin-only patients both before (odds ratio, 2.16; P < .05) and after matching data (odds ratio, 2.39; P < .05) with respect to confounding factors. In addition, bridged-therapy patients had longer hospital stays both before (odds ratio 1.20; P < .05) and after matching data (odds ratio, 1.27; P < .05) with respect to confounding factors. Figures 1 and 2 are histograms displaying the 2 groups and their outcomes.
Discussion
For patients undergoing THA procedures, several important considerations should be taken into account. Colwell and colleagues2 showed that, compared with warfarin, enoxaparin offered a 0.1% higher rate of protection against venous thromboembolic disease after THA. However, patients given enoxaparin may face increased risks.25 Hallevi and colleagues26 demonstrated that, compared with warfarin, enoxaparin bridging increased the risk for serious bleeding in patients with cardioembolic stroke. In our review of the literature, we learned that the benefits of bridge therapy in thromboembolic disease have yet to be investigated in THA.
At our academic hospital, the extra costs associated with bridge therapy can be as much as about $200027 per day per patient. These costs can go much higher, depending on type of patient and types of resources used. Over the 2-year period covered by our study, the costs of using enoxaparin amounted to about $151,200 ($2000 × 1.2 days per patient). If bridging offers no significant protection against thromboembolic disease, then it would be more cost-effective to use a single anticoagulant, particularly enoxaparin, for high-risk patients.
There are significant risk factors associated with prolonged healing of surgical wounds. Protocols outlining these factors may help reduce costs. In addition, when deciding on the use of aggressive anticoagulation therapy, surgeons must consider the risks for prolonged leakage and infection in addition to the risk for thromboembolic disease. Protocols may aid in this process as well. Our study results showed that, compared with warfarin-only therapy, bridged therapy (enoxaparin and warfarin) was associated with longer hospital stays. Further research should examine whether there are advantages that justify the higher risks of delayed wound healing and subsequent infection. Improving our understanding of risk factors associated with anticoagulation therapy will make orthopedic surgery safer for patients.
1. Bergqvist D, Benoni G, Björgell O, et al. Low-molecular-weight heparin (enoxaparin) as prophylaxis against venous thromboembolism after total hip replacement. N Engl J Med. 1996;335(10):696-700.
2. Colwell CW Jr, Collis DK, Paulson R, et al. Comparison of enoxaparin and warfarin for the prevention of venous thromboembolic disease after total hip arthroplasty. Evaluation during hospitalization and three months after discharge. J Bone Joint Surg Am. 1999;81(7):932-940.
3. Haake DA, Berkman SA. Venous thromboembolic disease after hip surgery. Risk factors, prophylaxis, and diagnosis. Clin Orthop Relat Res. 1989;(242):212-231.
4. Johnson R, Carmichael JH, Almond HG, Loynes RP. Deep venous thrombosis following Charnley arthroplasty. Clin Orthop Relat Res. 1978;(132):24-30.
5. Stamatakis JD, Kakkar VV, Sagar S, Lawrence D, Nairn D, Bentley PG. Femoral vein thrombosis and total hip replacement. Br Med J. 1977;2(6081):223-225.
6. Turpie AG, Levine MN, Hirsh J, et al. A randomized controlled trial of a low-molecular-weight heparin (enoxaparin) to prevent deep-vein thrombosis in patients undergoing elective hip surgery. N Engl J Med. 1986;315(15):925-929.
7. Clagett GP, Anderson FA Jr, Heit J, Levine MN, Wheeler HB. Prevention of venous thromboembolism. Chest. 1995;108(4 suppl):312S-334S.
8. Westrich GH, Sánchez PM. Prevention and treatment of thromboembolic disease: an overview. Instr Course Lect. 2002;51:471-480.
9. Colwell CW Jr, Froimson MI, Mont MA, et al. Thrombosis prevention after total hip arthroplasty: a prospective, randomized trial comparing a mobile compression device with low-molecular-weight heparin. J Bone Joint Surg Am. 2010;92(3):527-535.
10. Fareed J, Jeske W, Hoppensteadt D, Clarizio R, Walenga JM. Low-molecular-weight heparins: pharmacologic profile and product differentiation. Am J Cardiol. 1998;82(5B):3L-10L.
11. Gerlach AT, Pickworth KK, Seth SK, Tanna SB, Barnes JF. Enoxaparin and bleeding complications: a review in patients with and without renal insufficiency. Pharmacotherapy. 2000;20(7):771-775.
12. Kamali F, Wood P, Ward A. Vitamin K deficiency amplifies anticoagulation response to ximelagatran: possible implications for direct thrombin inhibitors and their clinical safety. Ann Hematol. 2009;88(2):141-149.
13. Choonara IA, Malia RG, Haynes BP, et al. The relationship between inhibition of vitamin K1 2,3-epoxide reductase and reduction of clotting factor activity with warfarin. Br J Clin Pharmacol. 1988;25(1):1-7.
14. Saleh K, Olson M, Resig S, et al. Predictors of wound infection in hip and knee joint replacement: results from a 20 year surveillance program. J Orthop Res. 2002;20(3):506-515.
15. Ridgeway S, Wilson J, Charlet A, Kafatos G, Pearson A, Coello R. Infection of the surgical site after arthroplasty of the hip. J Bone Joint Surg Br. 2005;87(6):844-850.
16. Lai K, Bohm ER, Burnell C, Hedden DR. Presence of medical comorbidities in patients with infected primary hip or knee arthroplasties. J Arthroplasty. 2007;22(5):651-656.
17. Kurtz SM, Lau E, Schmier J, Ong KL, Zhao K, Parvizi J. Infection burden for hip and knee arthroplasty in the United States. J Arthroplasty. 2008;23(7):984-991.
18. Ahmed AA, Mooar PA, Kleiner M, Torg JS, Miyamoto CT. Hypertensive patients show delayed wound healing following total hip arthroplasty. PLoS One. 2011;6(8):e23224.
19. Lübbeke A, Stern R, Garavaglia G, Zurcher L, Hoffmeyer P. Differences in outcomes of obese women and men undergoing primary total hip arthroplasty. Arthritis Rheum. 2007;57(2):327-334.
20. Rosenbaum PR. A characterization of optimal designs for observational studies. J R Stat Soc Ser B. 1991;53(3):597-610.
21. Rubin DB. Using propensity scores to help design observational studies: application to the tobacco litigation. Health Serv Outcomes Res Methodol. 2001;2(1):169-188.
22. Abadie A, Imbens GW. Simple and Bias-Corrected Matching Estimators for Average Treatment Effects. Berkeley, CA: Department of Economics, University of California; 2002.
23. Diamond A, Sekhon J. Genetic matching for estimating causal effects: a new method of achieving balance in observational studies. Paper presented at: Annual Meeting of the Midwest Political Science Association; April 2005; Chicago, IL.
24. Imai K, King G, Lau O. logit: logistic regression for dichotomous dependent variables. In: Imai K, King G, Lau O. Zelig: Everyone’s Statistical Software. 2011; 238-244. http://gking.harvard.edu/zelig. Accessed May 26, 2015.
25. Patel VP, Walsh M, Sehgal B, Preston C, DeWal H, Di Cesare PE. Factors associated with prolonged wound drainage after primary total hip and knee arthroplasty. J Bone Joint Surg Am. 2007;89(1):33-38.
26. Hallevi H, Albright KC, Martin-Schild S, et al. Anticoagulation after cardioembolic stroke: to bridge or not to bridge? Arch Neurol. 2008;65(9):1169-1173.
27. Henry J. Kaiser Family Foundation. Hospital adjusted expenses per inpatient day [2010]. http://kff.org/other/state-indicator/expenses-per-inpatient-day/#table. Accessed May 26, 2015.
1. Bergqvist D, Benoni G, Björgell O, et al. Low-molecular-weight heparin (enoxaparin) as prophylaxis against venous thromboembolism after total hip replacement. N Engl J Med. 1996;335(10):696-700.
2. Colwell CW Jr, Collis DK, Paulson R, et al. Comparison of enoxaparin and warfarin for the prevention of venous thromboembolic disease after total hip arthroplasty. Evaluation during hospitalization and three months after discharge. J Bone Joint Surg Am. 1999;81(7):932-940.
3. Haake DA, Berkman SA. Venous thromboembolic disease after hip surgery. Risk factors, prophylaxis, and diagnosis. Clin Orthop Relat Res. 1989;(242):212-231.
4. Johnson R, Carmichael JH, Almond HG, Loynes RP. Deep venous thrombosis following Charnley arthroplasty. Clin Orthop Relat Res. 1978;(132):24-30.
5. Stamatakis JD, Kakkar VV, Sagar S, Lawrence D, Nairn D, Bentley PG. Femoral vein thrombosis and total hip replacement. Br Med J. 1977;2(6081):223-225.
6. Turpie AG, Levine MN, Hirsh J, et al. A randomized controlled trial of a low-molecular-weight heparin (enoxaparin) to prevent deep-vein thrombosis in patients undergoing elective hip surgery. N Engl J Med. 1986;315(15):925-929.
7. Clagett GP, Anderson FA Jr, Heit J, Levine MN, Wheeler HB. Prevention of venous thromboembolism. Chest. 1995;108(4 suppl):312S-334S.
8. Westrich GH, Sánchez PM. Prevention and treatment of thromboembolic disease: an overview. Instr Course Lect. 2002;51:471-480.
9. Colwell CW Jr, Froimson MI, Mont MA, et al. Thrombosis prevention after total hip arthroplasty: a prospective, randomized trial comparing a mobile compression device with low-molecular-weight heparin. J Bone Joint Surg Am. 2010;92(3):527-535.
10. Fareed J, Jeske W, Hoppensteadt D, Clarizio R, Walenga JM. Low-molecular-weight heparins: pharmacologic profile and product differentiation. Am J Cardiol. 1998;82(5B):3L-10L.
11. Gerlach AT, Pickworth KK, Seth SK, Tanna SB, Barnes JF. Enoxaparin and bleeding complications: a review in patients with and without renal insufficiency. Pharmacotherapy. 2000;20(7):771-775.
12. Kamali F, Wood P, Ward A. Vitamin K deficiency amplifies anticoagulation response to ximelagatran: possible implications for direct thrombin inhibitors and their clinical safety. Ann Hematol. 2009;88(2):141-149.
13. Choonara IA, Malia RG, Haynes BP, et al. The relationship between inhibition of vitamin K1 2,3-epoxide reductase and reduction of clotting factor activity with warfarin. Br J Clin Pharmacol. 1988;25(1):1-7.
14. Saleh K, Olson M, Resig S, et al. Predictors of wound infection in hip and knee joint replacement: results from a 20 year surveillance program. J Orthop Res. 2002;20(3):506-515.
15. Ridgeway S, Wilson J, Charlet A, Kafatos G, Pearson A, Coello R. Infection of the surgical site after arthroplasty of the hip. J Bone Joint Surg Br. 2005;87(6):844-850.
16. Lai K, Bohm ER, Burnell C, Hedden DR. Presence of medical comorbidities in patients with infected primary hip or knee arthroplasties. J Arthroplasty. 2007;22(5):651-656.
17. Kurtz SM, Lau E, Schmier J, Ong KL, Zhao K, Parvizi J. Infection burden for hip and knee arthroplasty in the United States. J Arthroplasty. 2008;23(7):984-991.
18. Ahmed AA, Mooar PA, Kleiner M, Torg JS, Miyamoto CT. Hypertensive patients show delayed wound healing following total hip arthroplasty. PLoS One. 2011;6(8):e23224.
19. Lübbeke A, Stern R, Garavaglia G, Zurcher L, Hoffmeyer P. Differences in outcomes of obese women and men undergoing primary total hip arthroplasty. Arthritis Rheum. 2007;57(2):327-334.
20. Rosenbaum PR. A characterization of optimal designs for observational studies. J R Stat Soc Ser B. 1991;53(3):597-610.
21. Rubin DB. Using propensity scores to help design observational studies: application to the tobacco litigation. Health Serv Outcomes Res Methodol. 2001;2(1):169-188.
22. Abadie A, Imbens GW. Simple and Bias-Corrected Matching Estimators for Average Treatment Effects. Berkeley, CA: Department of Economics, University of California; 2002.
23. Diamond A, Sekhon J. Genetic matching for estimating causal effects: a new method of achieving balance in observational studies. Paper presented at: Annual Meeting of the Midwest Political Science Association; April 2005; Chicago, IL.
24. Imai K, King G, Lau O. logit: logistic regression for dichotomous dependent variables. In: Imai K, King G, Lau O. Zelig: Everyone’s Statistical Software. 2011; 238-244. http://gking.harvard.edu/zelig. Accessed May 26, 2015.
25. Patel VP, Walsh M, Sehgal B, Preston C, DeWal H, Di Cesare PE. Factors associated with prolonged wound drainage after primary total hip and knee arthroplasty. J Bone Joint Surg Am. 2007;89(1):33-38.
26. Hallevi H, Albright KC, Martin-Schild S, et al. Anticoagulation after cardioembolic stroke: to bridge or not to bridge? Arch Neurol. 2008;65(9):1169-1173.
27. Henry J. Kaiser Family Foundation. Hospital adjusted expenses per inpatient day [2010]. http://kff.org/other/state-indicator/expenses-per-inpatient-day/#table. Accessed May 26, 2015.

Evaluation of 3 Fixation Devices for Tibial-Sided Anterior Cruciate Ligament Graft Backup Fixation
Restoration of stability with return to activity is generally expected after anterior cruciate ligament (ACL) reconstruction; long-term success rates range from 75% to 95%.1 However, graft failure occurs most frequently with soft-tissue grafts fixated only with interference screws.2,3 Fixation failure also occurs more frequently at the tibial site.2 This failure has been attributed to extensive graft slippage in cases of soft-tissue fixation with interference screws.2 Interference screw fixation alone, with a double-looped hamstring tendon graft, fails at 350 N in young human tibiae.4,5 However, failure is limited with use of a bone–tendon–bone graft or with backup fixation, particularly at the tibial site.3 The superiority of bicortical fixation has also been proven.5-7
In addition, as shown in a goat model, ACL graft fixation is a major cause of failure in the immediate postoperative period, before biological incorporation of the graft.8 Fixation techniques for soft-tissue grafts must withstand stresses during the healing period (grafts may take up to 12 weeks to incorporate).9 Failures may result from forces exerted on the graft—forces that may be as high as 450 to 700 N during daily activities.10,11 Within the tibial tunnel, various fixation devices are used, including interference screws, staples, pins, buttons, and interference screw/sheath constructs.12,13 Primary fixation is commonly achieved with interference screws because of their ease of insertion and greater stiffness. However, fixation of the soft-tissue graft is influenced by several variables, including bone density, insertion torque, thread geometry, and interference screw material.14-16 Many of these variables, which are a source of inconsistency and concern during the immediate postoperative period, have led surgeons to seek alternative methods of backup fixation at the tibial site. Nevertheless, good clinical and subjective results have been found after ACL reconstruction with a 4-stranded semitendinosus tendon at 10-year follow-up.17
An anchor used in rotator cuff repair is the SwiveLock system (Arthrex). Major advantages of this system include ease and speed of insertion, good strength, and reduced need for later hardware removal.
We conducted a study to biomechanically evaluate 3 methods of tibial-sided fixation for ACL reconstruction: fully threaded interference screw only, interference screw backed with 4.75-mm SwiveLock anchor, and fully threaded bio-interference screw backed with 4.5-mm bicortical screw. We hypothesized that a fully threaded bio-interference screw backed with a 4.75-mm SwiveLock anchor would provide mechanical strength no different from that provided by backup fixation with a bicortical post at the tibial site. We further hypothesized that SwiveLock backup fixation would provide more strength than fixation with bio-interference screw alone.
Materials and Methods
The design of this study was adapted from one used by Walsh and colleagues,3 who compared 3 fixation methods: retrograde interference screw, suture button, and combined fixation. Tibiae inspected before selection showed no signs of injury or abnormality. Bovine extensor tendons, which lacked any defects along their entire length, were stored in saline 0.9% solution. Both the tibiae and the extensor tendons were stored at –20°C before completion of the tibial-sided ACL reconstruction. Thirty fresh-frozen, skeletally mature porcine proximal tibiae were selected and thawed at 4°C before preparation. Specimens were prepared by potting the diaphysis in fiberglass resin, and a tunnel 9 mm in diameter was drilled through the anteromedial aspect of the tibia.
For consistency, one author (CAV) prepared all 30 specimens. Both tails of all 30 bovine extensor tendons were whip-stitched with No. 2 FiberLoop (Arthrex) 9 mm in diameter. Grafts and tibiae were randomly divided into 3 sample groups. The first group was prepared by antegrade graft fixation within the tibial tunnel using a fully threaded 9×28-mm BioComposite interference screw (Arthrex). The second and third groups used the same primary fixation within the tibial tunnel along with 2 types of secondary fixation. These backup fixation groups included a 4.5-mm titanium bicortical post (Arthrex) and a 4.75-mm BioComposite SwiveLock C anchor (Arthrex) (Figure 1). The FiberLoop at the ends of the distal graft tails for backup groups were fixated 1 cm distal to the tibial tunnel and tapped before insertion of backup devices (Figures 2A, 2B). Insertion was completed after 4.5-mm bicortical and 4.75-mm unicortical drilling and tapping of the anteromedial cortices for the titanium posts and SwiveLocks, respectively. The free ends of the whip-stitched No. 2 FiberLoop were tied to the proximal end of the titanium post with a single surgical knot followed by 5 square knots.3 The free ends of the No. 2 FiberLoop were inserted into the eyelet of the 4.75-mm SwiveLock and 1 cm directly inferior to the tibial tunnel. Interference fit of FiberLoop with SwiveLock was achieved within the corticocancellous bone of the tibiae. All samples retained a 30-mm tendon loop superior to the tibial plateau to simulate intra-articular ACL length. Specimens were then stored at –20°C and thawed at 4°C before biomechanical testing.
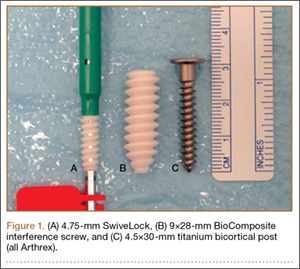
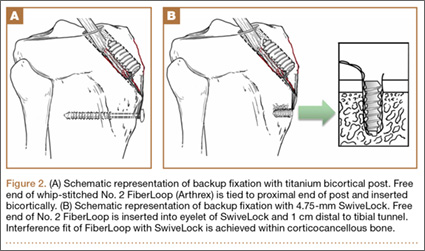
Each of the 30 tibiae was tested once. Each testing group consisted of 10 porcine tibiae. The tendons were kept moist during the entire testing procedure by spraying them thoroughly with saline 0.9% solution. Mechanical testing was performed with an Instron 8871 system with a 5-kN load cell secured to the crosshead. A fixed-angle aperture, attached to the testing surface, was adjusted so that the tendon would be pulled in line with the tibial tunnel. A hook fixture suspended from clevis and dowel was used to secure the tendon to the crosshead (Figure 3). A small preload of 5 N manually applied to each sample was followed by a precycling regimen of 10 cycles between 10 N and 50 N at 1 Hz. Precycling was performed to remove slack from the system. Mechanical testing consisted of 500 cycles between 50 N and 250 N at 1 Hz followed by pull to failure at 20 mm per minute. Load and displacement data were recorded at 500 Hz.
An a priori power analysis was not performed because 6 specimens per group in the study from which the testing protocol was adapted demonstrated sufficient power among 3 testing categories.3 In addition, other studies have demonstrated similar testing protocols using 10 specimens per testing group.7,12,13,18 The data for each sample were analyzed with OriginPro 8.0 software (OriginLab). Ultimate load, yield load, stiffness, and cyclic displacement of the 3 sample groups were compared with 1-way analysis of variance (α = 0.05). Holm-Sidak tests were used for post hoc analysis.19P < .05 was statistically significant.
Results
None of the 30 specimens failed during preloading. Modes of failure were consistent among groups. All 10 specimens in the interference-screw-only group failed by graft slippage past the screw in the tibial tunnel. Nineteen of the 20 specimens in the backup-fixation groups failed by graft slippage past the screw and suture cutout through the distal graft tail. In the bicortical-post backup group, 1 failure was attributed to tendon tearing proximal to whip-stitching. There were no instances of hardware breakage or failure of either titanium screw or SwiveLock anchor.
Mean (SD) cyclic displacement was higher in the interference-screw-only group, 3.5 (2.2) mm, than in the SwiveLock backup group, 2.6 (0.5) mm, and the bicortical-post backup group, 2.1 (0.6) mm; no statistical significance was demonstrated between any 2 of these groups alone (P = .12) (Figure 4). Mean (SD) pullout stiffness was higher in the bicortical-post backup group, 192 (48) N/mm, than in the SwiveLock backup group, 164 (53) N/mm, and the screw-only group, 163 (64) N/mm (P = .42) (Figure 5). Mean (SD) initial load at 5 mm of displacement was higher in the bicortical-post backup group, 482 (156) N, and the SwiveLock backup group, 423 (94) N, than in the screw-only group, 381 (169) N (P = .30).
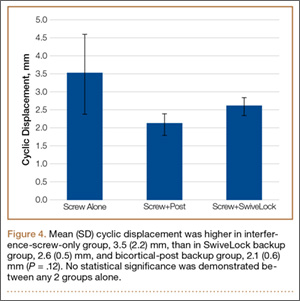
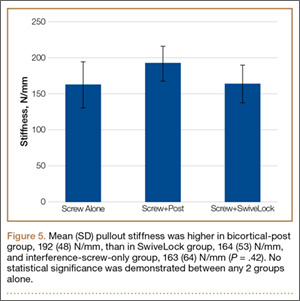
Mean (SD) yield load was higher in the bicortical-post backup group, 829 (253) N, than in the SwiveLock backup group, 642 (172) N, and the interference-screw-only group, 496 (133) N (P = .003). Statistical significance was demonstrated between the screw-only and bicortical-post groups (P = .002) and between the screw-only and SwiveLock groups (P = .048). There was no statistical difference between the bicortical-post and SwiveLock groups (P = .07).
Mean (SD) ultimate load to failure was higher in the bicortical-post backup group, 1148 (186) N, than in the SwiveLock backup group, 1007 (176) N, and the interference-screw-only group, 778 (139) N (Figure 6). The difference was statistically significant, whereby the screw-only group failed at a lower load compared with the bicortical-post group (P < .001) and the SwiveLock group (P = .005). The 2 backup groups were not statistically different (P = .1).
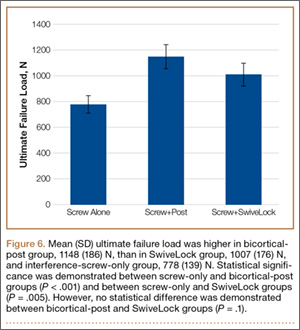
Discussion
We investigated whether a fully threaded bio-interference screw backed with a 4.75-mm SwiveLock anchor would provide mechanically equivalent pullout strength within the tibial tunnel during ACL reconstruction with soft-tissue allografts in comparison either with a fully threaded bio-interference screw backed with a bicortical post or with a fully threaded bio-interference screw without backup fixation. The results of the study support this hypothesis. With SwiveLock used for backup fixation, there was no significant difference in stiffness or cyclic load displacement between the screw-only, SwiveLock, and bicortical-post groups. However, adding backup fixation could particularly help improve fixation consistency. Specifically, although after only 500 cycles there was no statistically significant difference in cyclic displacement, continued cycling may be clinically relevant if graft slippage exceeded limits to allow for healing within the tibial tunnel. Conversely, a significantly larger difference was found between the SwiveLock, bicortical-post, and screw-only groups in yield load and ultimate load to failure. However, there was no significant difference between the SwiveLock and bicortical-post groups.
In this study, interference screw with SwiveLock backup demonstrated a mean (SD) ultimate load to failure of 1007 (176) N, comparable to that found by Walsh and colleagues3 for retrograde bio-interference screw with suture button, 1027 (157.11) N. In a study comparing quadrupled hamstring tibial graft fixation, Intrafix (DePuy Mitek) and an 8×25-mm Bioscrew (Linvatec) demonstrated mean (SD) single-cycle yield loads of 1332 (304) N and 612 (176) N, respectively.13 These results are similar to the ultimate yield loads in the present study: bicortical-post group, 1148 (186) N; SwiveLock group, 1007 (176) N; screw-only group, 778 (139) N. Differences may be attributed to hamstring tendons used in a quadrupled manner in the aforementioned study.12,13 Last, mean (SD) ultimate load to failure in a study that used only a retrograde bio-interference screw (9×20 mm) was 679.00 (109.44) N,3 similar to the 778 (139) N found for interference-screw-only in the present study. The difference is likely attributable to the longer screw (9×28 mm) in our study. Using SwiveLock C in cortical bone, Barber and colleagues18 found mean (SD) loads to failure up to 711.9 (89.1) N.
Clinically, it has been shown that a statistically significant increase in anterior laxity occurred between 4 months and 2 years in 10.7% of patients who underwent hamstring ACL reconstruction.20 The knees were clinically categorized as unstable or severely abnormal. The authors concluded that the clinical outcome was more likely influenced by the methods used to fix the free ends of the graft, specifically with 2 staples or a washer. To simulate early postoperative rehabilitation in the present study, cyclic loading of the graft was performed. Ultimate load to failure was then determined in order to evaluate catastrophic failure strength of the backup fixation devices in comparison with the interference-screw-only group without supplementary fixation.
It has been shown in autologous hamstring ACL reconstruction that a centrally placed polyethylene screw with sheath (Intrafix) performed as well as a standard, eccentrically placed metal interference screw with staple.10 It is therefore logical that backup fixation with use of a similar device (eg, SwiveLock, bicortical post) is necessary to ensure comparable clinical outcomes in relation to a screw/sheath device that has been shown to endure the highest yield loads.2,9,12,13,21-23 Potential benefits of using SwiveLock anchors for backup fixation include a statistically significant increased mean (SD) ultimate yield load of 229 (176) N over interference screw only. These results are similar to those in comparable studies: 218.3 (59.7) N24 and 165 (24.15) N25 in healthy bone with a reported bone mineral density (BMD) of 1.39 g/cm2, similar to that of skeletally mature porcine tibia (1.220-1.675 g/cm²).3 In addition, ease of insertion of this device over a bicortical post was demonstrated. The titanium post required bicortical drilling as well as measurement with a depth gauge to ensure adequate screw length. This process appeared to require more time during specimen preparation and theoretically could prove to be more dangerous clinically.7 However, caution in using a SwiveLock anchor in osteoporotic bone is advised because of reduced pullout strength.26 In this case, bicortical-post backup fixation may be more suitable. Moreover, although not demonstrated in this study, hardware prominence and irritation with a post may cause postoperative morbidity necessitating future removal.20 Hardware removal was the most common reason for additional surgery using hamstring tendons as graft.20 A second surgery for hardware removal was required in 21% of these patients.20 This is unlikely to occur with a SwiveLock, as the anchor is buried within cortical bone.
Limitations
Regarding use of nonhuman tissues in a biomechanical model, porcine tibiae and bovine extensor tendons were used because of availability, consistency among specimens, and cost-effectiveness. However, bovine extensor tendons have been shown to exhibit stiffness and viscoelastic properties similar to those of a human hamstring graft.27 In addition, the BMD of the porcine tibiae used in this study was not tested because of time involved and cost-efficiency. However, it has been shown that average BMD of porcine tibiae, 1.220-1.675 g/cm², is similar to that in a young athletic population, 1.24-1.62 g/cm2.3,28-31 We therefore assumed similarity to a young athletic population and uniformity of BMD of the porcine tibiae used in this study.
In addition, the biomechanical testing protocol did not simulate physiologic loading within the tibial tunnel. Moreover, the testing protocol used loads of only 250 N during cyclic testing for 500 cycles. This simulates only the early rehabilitation period and not the healing period, which may last up to 12 weeks.9 In addition, as previously mentioned, forces on the graft may be as high as 450 to 700 N.11,32 Pullout testing in line with the long axis of the tibia was performed in order to compare mechanical testing results with those of similar studies.3,12,13 Last, the P of .07 for the comparison of ultimate load to failure between the 2 backup fixation groups suggests that this study may have been underpowered.
Conclusion
This study demonstrated an effective, alternative, and equivalent backup fixation device that can help prevent graft slippage within the tibial tunnel during soft-tissue ACL reconstruction. Potential benefits of using SwiveLock anchors for backup fixation include a significantly increased ultimate yield load (229 N) when supplementing an interference screw, ease of insertion compared with a bicortical post, and the improbable need for future hardware removal. We support using SwiveLock for supplementary fixation at the tibial tunnel site when using soft-tissue grafts in ACL reconstruction.
1. Wetzler MJ, Bartolozzi AR, Gillespie MJ, Rubenstein DL, Ciccotti MG, Miller LS. Revision anterior cruciate ligament reconstruction. Oper Tech Orthop. 1996;6(3):181-189.
2. Scheffler SU, Südkamp NP, Göckenjan A, Hoffmann RF, Weiler A. Biomechanical comparison of hamstring and patellar tendon graft anterior cruciate ligament reconstruction techniques: the impact of fixation level and fixation method under cyclic loading. Arthroscopy. 2002;18(3):304-315.
3. Walsh MP, Wijdicks CA, Parker JB, Hapa O, LaPrade RF. A comparison between a retrograde interference screw, suture button, and combined fixation on the tibial side in an all-inside anterior cruciate ligament reconstruction: a biomechanical study in a porcine model. Am J Sports Med. 2009;37(1):160-167.
4. Howell SM, Hull ML. Aggressive rehabilitation using hamstring tendons: graft construct, tibial tunnel placement, fixation properties, and clinical outcome. Am J Knee Surg. 1998;11(2):120-127.
5. Magen HE, Howell SM, Hull ML. Structural properties of six tibial fixation methods for anterior cruciate ligament soft tissue grafts. Am J Sports Med. 1999;27(1):35-43.
6. Beynnon BD, Meriam CM, Ryder SH, Fleming BC, Johnson RJ. The effect of screw insertion torque on tendons fixed with spiked washers. Am J Sports Med. 1998;26(4):536-539.
7. Post WR, King SS. Neurovascular risk of bicortical tibial drilling for screw and spiked washer fixation of soft-tissue anterior cruciate ligament graft. Arthroscopy. 2001;17(3):244-247.
8. Holden JP, Grood ES, Butler DL, et al. Biomechanics of fascia lata ligament replacements: early postoperative changes in the goat. J Orthop Res. 1988;6(5):639-647.
9. Rodeo SA, Arnoczky SP, Torzilli PA, Hidaka C, Warren RF. Tendon-healing in a bone tunnel. A biomechanical and histological study in the dog. J Bone Joint Surg Am. 1993;75(12):1795-1803.
10. Frank CB, Jackson DW. The science of reconstruction of the anterior cruciate ligament. J Bone Joint Surg Am. 1997;79(10):1556-1576.
11. Markolf KL, Willems MJ, Jackson SR, Finerman GA. In situ calibration of miniature sensors implanted into the anterior cruciate ligament. Part I: strain measurements. J Orthop Res. 1998;16(4):455-463.
12. Kousa P, Teppo LN, Jarvinen TL, Vihavainen M, Kannus P, Jarvinen M. The fixation strength of six hamstring tendon graft fixation devices in anterior cruciate ligament reconstruction: I. Femoral site. Am J Sports Med. 2003;3 (2)1:174-181.
13. Kousa P, Jarvinen TL, Vihavainen M, Kannus P, Jarvinen M. The fixation strength of six hamstring tendon graft fixation devices in anterior cruciate ligament reconstruction: II. Tibial site. Am J Sports Med. 2003;31(2):182-188.
14. Brand JC Jr, Pienkowski D, Steenlage E, Hamilton D, Johnson DL, Caborn DN. Interference screw fixation strength of a quadrupled hamstring tendon graft is directly related to bone mineral density and insertion torque. Am J Sports Med. 2000;28(5):705-710.
15. Weiler A, Hoffmann RF, Siepe CJ, Kolbeck SF, Südkamp NP. The influence of screw geometry on hamstring tendon interference fit fixation. Am J Sports Med. 2000;28(3):356-359.
16. Weiler A, Hoffmann RF, Stähelin AC, Bail HJ, Siepe CJ, Südkamp NP. Hamstring tendon fixation using interference screws: a biomechanical study in calf tibial bone. Arthroscopy. 1998;14(1):29-37.
17. Streich NA, Reichenbacher S, Barié A, Buchner M, Schmitt H. Long-term outcome of anterior cruciate ligament reconstruction with an autologous four-strand semitendinosus tendon autograft. Int Orthop. 2013;37(2):279-284.
18. Barber FA, Herbert MA, Beavis C, Barrera Oro F. Suture anchor materials, eyelets, and designs: update 2008. Arthroscopy. 2008;24(8):859-867.
19. Aickin M, Gensler H. Adjusting for multiple testing when reporting research results: the Bonferroni vs Holm methods. Am J Public Health. 1996;86(5):726-728.
20. Howell SM, Deutsch ML. Comparison of endoscopic and two-incision techniques for reconstructing a torn anterior cruciate ligament using hamstring tendons. Arthroscopy. 1999;15(6):594-606.
21. Gwynne-Jones DP, Draffin J, Vane A, Craig R, McMahon S. Failure strengths of concentric and eccentric implants for hamstring graft fixation. ANZ J Surg. 2008;78(3):177-181.
22. Hayes DA, Watts MC, Tevelen GA, Crawford RW. Central versus peripheral tibial screw placement in hamstring anterior cruciate ligament reconstruction: in vitro biomechanics. Arthroscopy. 2005;21(6):703-706.
23. Shino K, Pflaster DS. Comparison of eccentric and concentric screw placement for hamstring graft fixation in the tibial tunnel. Knee Surg Sports Traumatol Arthrosc. 2000;8(2):73-75.
24. Prevrhal S, Fuerst T, Fan B, et al. Quantitative ultrasound of the tibia depends on both cortical density and thickness. Osteoporosis Int. 2001;12(1):28-34.
25. Pietschmann MF, Gülecyüz MF, Fieseler S, et al. Biomechanical stability of knotless suture anchors used in rotator cuff repair in healthy and osteopenic bone. Arthroscopy. 2010;26(8):1035-1044.
26. Burns JP, Snyder SJ, Albritton M. Arthroscopic rotator cuff repair using triple-loaded anchors, suture shuttles, and suture savers. J Am Acad Orthop Surg. 2007;15(7):432-444.
27. Tetsumura S, Fujita A, Nakajima M, Abe M. Biomechanical comparison of different fixation methods on the tibial side in anterior cruciate ligament reconstruction: a biomechanical study in porcine tibial bone. J Orthop Sci. 2006;11(3):278-282.
28. Alfredson H, Nordstrom P, Lorentzon R. Total and regional bone mass in female soccer players. Calcif Tissue Int. 1996;59(6):438-442.
29. Nevill AM, Holder RL, Stewart AD. Modeling elite male athletes’ peripheral bone mass, assessed using regional dual x-ray absorptiometry. Bone. 2003;32(1):62-68.
30. Nordström P, Lorentzon R. Site-specific bone mass differences of the lower extremities in 17-year-old ice hockey players. Calcif Tissue Int. 1996;59(6):4443-4448.
31. Patzer T, Santo G, Olender GD, Wellmann M, Hurschler C, Schofer MD. Suprapectoral or subpectoral position for biceps tenodesis: biomechanical comparison of four different techniques in both positions. J Shoulder Elbow Surg. 2012;21(1):116-125.
32. De Wall M, Scholes CJ, Patel S, Coolican MR, Parker DA. Tibial fixation in anterior cruciate ligament reconstruction: a prospective randomized study comparing metal interference screw and staples with a centrally placed polyethylene screw and sheath. Am J Sports Med. 2011;39(9):1858-1864.
Restoration of stability with return to activity is generally expected after anterior cruciate ligament (ACL) reconstruction; long-term success rates range from 75% to 95%.1 However, graft failure occurs most frequently with soft-tissue grafts fixated only with interference screws.2,3 Fixation failure also occurs more frequently at the tibial site.2 This failure has been attributed to extensive graft slippage in cases of soft-tissue fixation with interference screws.2 Interference screw fixation alone, with a double-looped hamstring tendon graft, fails at 350 N in young human tibiae.4,5 However, failure is limited with use of a bone–tendon–bone graft or with backup fixation, particularly at the tibial site.3 The superiority of bicortical fixation has also been proven.5-7
In addition, as shown in a goat model, ACL graft fixation is a major cause of failure in the immediate postoperative period, before biological incorporation of the graft.8 Fixation techniques for soft-tissue grafts must withstand stresses during the healing period (grafts may take up to 12 weeks to incorporate).9 Failures may result from forces exerted on the graft—forces that may be as high as 450 to 700 N during daily activities.10,11 Within the tibial tunnel, various fixation devices are used, including interference screws, staples, pins, buttons, and interference screw/sheath constructs.12,13 Primary fixation is commonly achieved with interference screws because of their ease of insertion and greater stiffness. However, fixation of the soft-tissue graft is influenced by several variables, including bone density, insertion torque, thread geometry, and interference screw material.14-16 Many of these variables, which are a source of inconsistency and concern during the immediate postoperative period, have led surgeons to seek alternative methods of backup fixation at the tibial site. Nevertheless, good clinical and subjective results have been found after ACL reconstruction with a 4-stranded semitendinosus tendon at 10-year follow-up.17
An anchor used in rotator cuff repair is the SwiveLock system (Arthrex). Major advantages of this system include ease and speed of insertion, good strength, and reduced need for later hardware removal.
We conducted a study to biomechanically evaluate 3 methods of tibial-sided fixation for ACL reconstruction: fully threaded interference screw only, interference screw backed with 4.75-mm SwiveLock anchor, and fully threaded bio-interference screw backed with 4.5-mm bicortical screw. We hypothesized that a fully threaded bio-interference screw backed with a 4.75-mm SwiveLock anchor would provide mechanical strength no different from that provided by backup fixation with a bicortical post at the tibial site. We further hypothesized that SwiveLock backup fixation would provide more strength than fixation with bio-interference screw alone.
Materials and Methods
The design of this study was adapted from one used by Walsh and colleagues,3 who compared 3 fixation methods: retrograde interference screw, suture button, and combined fixation. Tibiae inspected before selection showed no signs of injury or abnormality. Bovine extensor tendons, which lacked any defects along their entire length, were stored in saline 0.9% solution. Both the tibiae and the extensor tendons were stored at –20°C before completion of the tibial-sided ACL reconstruction. Thirty fresh-frozen, skeletally mature porcine proximal tibiae were selected and thawed at 4°C before preparation. Specimens were prepared by potting the diaphysis in fiberglass resin, and a tunnel 9 mm in diameter was drilled through the anteromedial aspect of the tibia.
For consistency, one author (CAV) prepared all 30 specimens. Both tails of all 30 bovine extensor tendons were whip-stitched with No. 2 FiberLoop (Arthrex) 9 mm in diameter. Grafts and tibiae were randomly divided into 3 sample groups. The first group was prepared by antegrade graft fixation within the tibial tunnel using a fully threaded 9×28-mm BioComposite interference screw (Arthrex). The second and third groups used the same primary fixation within the tibial tunnel along with 2 types of secondary fixation. These backup fixation groups included a 4.5-mm titanium bicortical post (Arthrex) and a 4.75-mm BioComposite SwiveLock C anchor (Arthrex) (Figure 1). The FiberLoop at the ends of the distal graft tails for backup groups were fixated 1 cm distal to the tibial tunnel and tapped before insertion of backup devices (Figures 2A, 2B). Insertion was completed after 4.5-mm bicortical and 4.75-mm unicortical drilling and tapping of the anteromedial cortices for the titanium posts and SwiveLocks, respectively. The free ends of the whip-stitched No. 2 FiberLoop were tied to the proximal end of the titanium post with a single surgical knot followed by 5 square knots.3 The free ends of the No. 2 FiberLoop were inserted into the eyelet of the 4.75-mm SwiveLock and 1 cm directly inferior to the tibial tunnel. Interference fit of FiberLoop with SwiveLock was achieved within the corticocancellous bone of the tibiae. All samples retained a 30-mm tendon loop superior to the tibial plateau to simulate intra-articular ACL length. Specimens were then stored at –20°C and thawed at 4°C before biomechanical testing.
Each of the 30 tibiae was tested once. Each testing group consisted of 10 porcine tibiae. The tendons were kept moist during the entire testing procedure by spraying them thoroughly with saline 0.9% solution. Mechanical testing was performed with an Instron 8871 system with a 5-kN load cell secured to the crosshead. A fixed-angle aperture, attached to the testing surface, was adjusted so that the tendon would be pulled in line with the tibial tunnel. A hook fixture suspended from clevis and dowel was used to secure the tendon to the crosshead (Figure 3). A small preload of 5 N manually applied to each sample was followed by a precycling regimen of 10 cycles between 10 N and 50 N at 1 Hz. Precycling was performed to remove slack from the system. Mechanical testing consisted of 500 cycles between 50 N and 250 N at 1 Hz followed by pull to failure at 20 mm per minute. Load and displacement data were recorded at 500 Hz.
An a priori power analysis was not performed because 6 specimens per group in the study from which the testing protocol was adapted demonstrated sufficient power among 3 testing categories.3 In addition, other studies have demonstrated similar testing protocols using 10 specimens per testing group.7,12,13,18 The data for each sample were analyzed with OriginPro 8.0 software (OriginLab). Ultimate load, yield load, stiffness, and cyclic displacement of the 3 sample groups were compared with 1-way analysis of variance (α = 0.05). Holm-Sidak tests were used for post hoc analysis.19P < .05 was statistically significant.
Results
None of the 30 specimens failed during preloading. Modes of failure were consistent among groups. All 10 specimens in the interference-screw-only group failed by graft slippage past the screw in the tibial tunnel. Nineteen of the 20 specimens in the backup-fixation groups failed by graft slippage past the screw and suture cutout through the distal graft tail. In the bicortical-post backup group, 1 failure was attributed to tendon tearing proximal to whip-stitching. There were no instances of hardware breakage or failure of either titanium screw or SwiveLock anchor.
Mean (SD) cyclic displacement was higher in the interference-screw-only group, 3.5 (2.2) mm, than in the SwiveLock backup group, 2.6 (0.5) mm, and the bicortical-post backup group, 2.1 (0.6) mm; no statistical significance was demonstrated between any 2 of these groups alone (P = .12) (Figure 4). Mean (SD) pullout stiffness was higher in the bicortical-post backup group, 192 (48) N/mm, than in the SwiveLock backup group, 164 (53) N/mm, and the screw-only group, 163 (64) N/mm (P = .42) (Figure 5). Mean (SD) initial load at 5 mm of displacement was higher in the bicortical-post backup group, 482 (156) N, and the SwiveLock backup group, 423 (94) N, than in the screw-only group, 381 (169) N (P = .30).
Mean (SD) yield load was higher in the bicortical-post backup group, 829 (253) N, than in the SwiveLock backup group, 642 (172) N, and the interference-screw-only group, 496 (133) N (P = .003). Statistical significance was demonstrated between the screw-only and bicortical-post groups (P = .002) and between the screw-only and SwiveLock groups (P = .048). There was no statistical difference between the bicortical-post and SwiveLock groups (P = .07).
Mean (SD) ultimate load to failure was higher in the bicortical-post backup group, 1148 (186) N, than in the SwiveLock backup group, 1007 (176) N, and the interference-screw-only group, 778 (139) N (Figure 6). The difference was statistically significant, whereby the screw-only group failed at a lower load compared with the bicortical-post group (P < .001) and the SwiveLock group (P = .005). The 2 backup groups were not statistically different (P = .1).
Discussion
We investigated whether a fully threaded bio-interference screw backed with a 4.75-mm SwiveLock anchor would provide mechanically equivalent pullout strength within the tibial tunnel during ACL reconstruction with soft-tissue allografts in comparison either with a fully threaded bio-interference screw backed with a bicortical post or with a fully threaded bio-interference screw without backup fixation. The results of the study support this hypothesis. With SwiveLock used for backup fixation, there was no significant difference in stiffness or cyclic load displacement between the screw-only, SwiveLock, and bicortical-post groups. However, adding backup fixation could particularly help improve fixation consistency. Specifically, although after only 500 cycles there was no statistically significant difference in cyclic displacement, continued cycling may be clinically relevant if graft slippage exceeded limits to allow for healing within the tibial tunnel. Conversely, a significantly larger difference was found between the SwiveLock, bicortical-post, and screw-only groups in yield load and ultimate load to failure. However, there was no significant difference between the SwiveLock and bicortical-post groups.
In this study, interference screw with SwiveLock backup demonstrated a mean (SD) ultimate load to failure of 1007 (176) N, comparable to that found by Walsh and colleagues3 for retrograde bio-interference screw with suture button, 1027 (157.11) N. In a study comparing quadrupled hamstring tibial graft fixation, Intrafix (DePuy Mitek) and an 8×25-mm Bioscrew (Linvatec) demonstrated mean (SD) single-cycle yield loads of 1332 (304) N and 612 (176) N, respectively.13 These results are similar to the ultimate yield loads in the present study: bicortical-post group, 1148 (186) N; SwiveLock group, 1007 (176) N; screw-only group, 778 (139) N. Differences may be attributed to hamstring tendons used in a quadrupled manner in the aforementioned study.12,13 Last, mean (SD) ultimate load to failure in a study that used only a retrograde bio-interference screw (9×20 mm) was 679.00 (109.44) N,3 similar to the 778 (139) N found for interference-screw-only in the present study. The difference is likely attributable to the longer screw (9×28 mm) in our study. Using SwiveLock C in cortical bone, Barber and colleagues18 found mean (SD) loads to failure up to 711.9 (89.1) N.
Clinically, it has been shown that a statistically significant increase in anterior laxity occurred between 4 months and 2 years in 10.7% of patients who underwent hamstring ACL reconstruction.20 The knees were clinically categorized as unstable or severely abnormal. The authors concluded that the clinical outcome was more likely influenced by the methods used to fix the free ends of the graft, specifically with 2 staples or a washer. To simulate early postoperative rehabilitation in the present study, cyclic loading of the graft was performed. Ultimate load to failure was then determined in order to evaluate catastrophic failure strength of the backup fixation devices in comparison with the interference-screw-only group without supplementary fixation.
It has been shown in autologous hamstring ACL reconstruction that a centrally placed polyethylene screw with sheath (Intrafix) performed as well as a standard, eccentrically placed metal interference screw with staple.10 It is therefore logical that backup fixation with use of a similar device (eg, SwiveLock, bicortical post) is necessary to ensure comparable clinical outcomes in relation to a screw/sheath device that has been shown to endure the highest yield loads.2,9,12,13,21-23 Potential benefits of using SwiveLock anchors for backup fixation include a statistically significant increased mean (SD) ultimate yield load of 229 (176) N over interference screw only. These results are similar to those in comparable studies: 218.3 (59.7) N24 and 165 (24.15) N25 in healthy bone with a reported bone mineral density (BMD) of 1.39 g/cm2, similar to that of skeletally mature porcine tibia (1.220-1.675 g/cm²).3 In addition, ease of insertion of this device over a bicortical post was demonstrated. The titanium post required bicortical drilling as well as measurement with a depth gauge to ensure adequate screw length. This process appeared to require more time during specimen preparation and theoretically could prove to be more dangerous clinically.7 However, caution in using a SwiveLock anchor in osteoporotic bone is advised because of reduced pullout strength.26 In this case, bicortical-post backup fixation may be more suitable. Moreover, although not demonstrated in this study, hardware prominence and irritation with a post may cause postoperative morbidity necessitating future removal.20 Hardware removal was the most common reason for additional surgery using hamstring tendons as graft.20 A second surgery for hardware removal was required in 21% of these patients.20 This is unlikely to occur with a SwiveLock, as the anchor is buried within cortical bone.
Limitations
Regarding use of nonhuman tissues in a biomechanical model, porcine tibiae and bovine extensor tendons were used because of availability, consistency among specimens, and cost-effectiveness. However, bovine extensor tendons have been shown to exhibit stiffness and viscoelastic properties similar to those of a human hamstring graft.27 In addition, the BMD of the porcine tibiae used in this study was not tested because of time involved and cost-efficiency. However, it has been shown that average BMD of porcine tibiae, 1.220-1.675 g/cm², is similar to that in a young athletic population, 1.24-1.62 g/cm2.3,28-31 We therefore assumed similarity to a young athletic population and uniformity of BMD of the porcine tibiae used in this study.
In addition, the biomechanical testing protocol did not simulate physiologic loading within the tibial tunnel. Moreover, the testing protocol used loads of only 250 N during cyclic testing for 500 cycles. This simulates only the early rehabilitation period and not the healing period, which may last up to 12 weeks.9 In addition, as previously mentioned, forces on the graft may be as high as 450 to 700 N.11,32 Pullout testing in line with the long axis of the tibia was performed in order to compare mechanical testing results with those of similar studies.3,12,13 Last, the P of .07 for the comparison of ultimate load to failure between the 2 backup fixation groups suggests that this study may have been underpowered.
Conclusion
This study demonstrated an effective, alternative, and equivalent backup fixation device that can help prevent graft slippage within the tibial tunnel during soft-tissue ACL reconstruction. Potential benefits of using SwiveLock anchors for backup fixation include a significantly increased ultimate yield load (229 N) when supplementing an interference screw, ease of insertion compared with a bicortical post, and the improbable need for future hardware removal. We support using SwiveLock for supplementary fixation at the tibial tunnel site when using soft-tissue grafts in ACL reconstruction.
Restoration of stability with return to activity is generally expected after anterior cruciate ligament (ACL) reconstruction; long-term success rates range from 75% to 95%.1 However, graft failure occurs most frequently with soft-tissue grafts fixated only with interference screws.2,3 Fixation failure also occurs more frequently at the tibial site.2 This failure has been attributed to extensive graft slippage in cases of soft-tissue fixation with interference screws.2 Interference screw fixation alone, with a double-looped hamstring tendon graft, fails at 350 N in young human tibiae.4,5 However, failure is limited with use of a bone–tendon–bone graft or with backup fixation, particularly at the tibial site.3 The superiority of bicortical fixation has also been proven.5-7
In addition, as shown in a goat model, ACL graft fixation is a major cause of failure in the immediate postoperative period, before biological incorporation of the graft.8 Fixation techniques for soft-tissue grafts must withstand stresses during the healing period (grafts may take up to 12 weeks to incorporate).9 Failures may result from forces exerted on the graft—forces that may be as high as 450 to 700 N during daily activities.10,11 Within the tibial tunnel, various fixation devices are used, including interference screws, staples, pins, buttons, and interference screw/sheath constructs.12,13 Primary fixation is commonly achieved with interference screws because of their ease of insertion and greater stiffness. However, fixation of the soft-tissue graft is influenced by several variables, including bone density, insertion torque, thread geometry, and interference screw material.14-16 Many of these variables, which are a source of inconsistency and concern during the immediate postoperative period, have led surgeons to seek alternative methods of backup fixation at the tibial site. Nevertheless, good clinical and subjective results have been found after ACL reconstruction with a 4-stranded semitendinosus tendon at 10-year follow-up.17
An anchor used in rotator cuff repair is the SwiveLock system (Arthrex). Major advantages of this system include ease and speed of insertion, good strength, and reduced need for later hardware removal.
We conducted a study to biomechanically evaluate 3 methods of tibial-sided fixation for ACL reconstruction: fully threaded interference screw only, interference screw backed with 4.75-mm SwiveLock anchor, and fully threaded bio-interference screw backed with 4.5-mm bicortical screw. We hypothesized that a fully threaded bio-interference screw backed with a 4.75-mm SwiveLock anchor would provide mechanical strength no different from that provided by backup fixation with a bicortical post at the tibial site. We further hypothesized that SwiveLock backup fixation would provide more strength than fixation with bio-interference screw alone.
Materials and Methods
The design of this study was adapted from one used by Walsh and colleagues,3 who compared 3 fixation methods: retrograde interference screw, suture button, and combined fixation. Tibiae inspected before selection showed no signs of injury or abnormality. Bovine extensor tendons, which lacked any defects along their entire length, were stored in saline 0.9% solution. Both the tibiae and the extensor tendons were stored at –20°C before completion of the tibial-sided ACL reconstruction. Thirty fresh-frozen, skeletally mature porcine proximal tibiae were selected and thawed at 4°C before preparation. Specimens were prepared by potting the diaphysis in fiberglass resin, and a tunnel 9 mm in diameter was drilled through the anteromedial aspect of the tibia.
For consistency, one author (CAV) prepared all 30 specimens. Both tails of all 30 bovine extensor tendons were whip-stitched with No. 2 FiberLoop (Arthrex) 9 mm in diameter. Grafts and tibiae were randomly divided into 3 sample groups. The first group was prepared by antegrade graft fixation within the tibial tunnel using a fully threaded 9×28-mm BioComposite interference screw (Arthrex). The second and third groups used the same primary fixation within the tibial tunnel along with 2 types of secondary fixation. These backup fixation groups included a 4.5-mm titanium bicortical post (Arthrex) and a 4.75-mm BioComposite SwiveLock C anchor (Arthrex) (Figure 1). The FiberLoop at the ends of the distal graft tails for backup groups were fixated 1 cm distal to the tibial tunnel and tapped before insertion of backup devices (Figures 2A, 2B). Insertion was completed after 4.5-mm bicortical and 4.75-mm unicortical drilling and tapping of the anteromedial cortices for the titanium posts and SwiveLocks, respectively. The free ends of the whip-stitched No. 2 FiberLoop were tied to the proximal end of the titanium post with a single surgical knot followed by 5 square knots.3 The free ends of the No. 2 FiberLoop were inserted into the eyelet of the 4.75-mm SwiveLock and 1 cm directly inferior to the tibial tunnel. Interference fit of FiberLoop with SwiveLock was achieved within the corticocancellous bone of the tibiae. All samples retained a 30-mm tendon loop superior to the tibial plateau to simulate intra-articular ACL length. Specimens were then stored at –20°C and thawed at 4°C before biomechanical testing.
Each of the 30 tibiae was tested once. Each testing group consisted of 10 porcine tibiae. The tendons were kept moist during the entire testing procedure by spraying them thoroughly with saline 0.9% solution. Mechanical testing was performed with an Instron 8871 system with a 5-kN load cell secured to the crosshead. A fixed-angle aperture, attached to the testing surface, was adjusted so that the tendon would be pulled in line with the tibial tunnel. A hook fixture suspended from clevis and dowel was used to secure the tendon to the crosshead (Figure 3). A small preload of 5 N manually applied to each sample was followed by a precycling regimen of 10 cycles between 10 N and 50 N at 1 Hz. Precycling was performed to remove slack from the system. Mechanical testing consisted of 500 cycles between 50 N and 250 N at 1 Hz followed by pull to failure at 20 mm per minute. Load and displacement data were recorded at 500 Hz.
An a priori power analysis was not performed because 6 specimens per group in the study from which the testing protocol was adapted demonstrated sufficient power among 3 testing categories.3 In addition, other studies have demonstrated similar testing protocols using 10 specimens per testing group.7,12,13,18 The data for each sample were analyzed with OriginPro 8.0 software (OriginLab). Ultimate load, yield load, stiffness, and cyclic displacement of the 3 sample groups were compared with 1-way analysis of variance (α = 0.05). Holm-Sidak tests were used for post hoc analysis.19P < .05 was statistically significant.
Results
None of the 30 specimens failed during preloading. Modes of failure were consistent among groups. All 10 specimens in the interference-screw-only group failed by graft slippage past the screw in the tibial tunnel. Nineteen of the 20 specimens in the backup-fixation groups failed by graft slippage past the screw and suture cutout through the distal graft tail. In the bicortical-post backup group, 1 failure was attributed to tendon tearing proximal to whip-stitching. There were no instances of hardware breakage or failure of either titanium screw or SwiveLock anchor.
Mean (SD) cyclic displacement was higher in the interference-screw-only group, 3.5 (2.2) mm, than in the SwiveLock backup group, 2.6 (0.5) mm, and the bicortical-post backup group, 2.1 (0.6) mm; no statistical significance was demonstrated between any 2 of these groups alone (P = .12) (Figure 4). Mean (SD) pullout stiffness was higher in the bicortical-post backup group, 192 (48) N/mm, than in the SwiveLock backup group, 164 (53) N/mm, and the screw-only group, 163 (64) N/mm (P = .42) (Figure 5). Mean (SD) initial load at 5 mm of displacement was higher in the bicortical-post backup group, 482 (156) N, and the SwiveLock backup group, 423 (94) N, than in the screw-only group, 381 (169) N (P = .30).
Mean (SD) yield load was higher in the bicortical-post backup group, 829 (253) N, than in the SwiveLock backup group, 642 (172) N, and the interference-screw-only group, 496 (133) N (P = .003). Statistical significance was demonstrated between the screw-only and bicortical-post groups (P = .002) and between the screw-only and SwiveLock groups (P = .048). There was no statistical difference between the bicortical-post and SwiveLock groups (P = .07).
Mean (SD) ultimate load to failure was higher in the bicortical-post backup group, 1148 (186) N, than in the SwiveLock backup group, 1007 (176) N, and the interference-screw-only group, 778 (139) N (Figure 6). The difference was statistically significant, whereby the screw-only group failed at a lower load compared with the bicortical-post group (P < .001) and the SwiveLock group (P = .005). The 2 backup groups were not statistically different (P = .1).
Discussion
We investigated whether a fully threaded bio-interference screw backed with a 4.75-mm SwiveLock anchor would provide mechanically equivalent pullout strength within the tibial tunnel during ACL reconstruction with soft-tissue allografts in comparison either with a fully threaded bio-interference screw backed with a bicortical post or with a fully threaded bio-interference screw without backup fixation. The results of the study support this hypothesis. With SwiveLock used for backup fixation, there was no significant difference in stiffness or cyclic load displacement between the screw-only, SwiveLock, and bicortical-post groups. However, adding backup fixation could particularly help improve fixation consistency. Specifically, although after only 500 cycles there was no statistically significant difference in cyclic displacement, continued cycling may be clinically relevant if graft slippage exceeded limits to allow for healing within the tibial tunnel. Conversely, a significantly larger difference was found between the SwiveLock, bicortical-post, and screw-only groups in yield load and ultimate load to failure. However, there was no significant difference between the SwiveLock and bicortical-post groups.
In this study, interference screw with SwiveLock backup demonstrated a mean (SD) ultimate load to failure of 1007 (176) N, comparable to that found by Walsh and colleagues3 for retrograde bio-interference screw with suture button, 1027 (157.11) N. In a study comparing quadrupled hamstring tibial graft fixation, Intrafix (DePuy Mitek) and an 8×25-mm Bioscrew (Linvatec) demonstrated mean (SD) single-cycle yield loads of 1332 (304) N and 612 (176) N, respectively.13 These results are similar to the ultimate yield loads in the present study: bicortical-post group, 1148 (186) N; SwiveLock group, 1007 (176) N; screw-only group, 778 (139) N. Differences may be attributed to hamstring tendons used in a quadrupled manner in the aforementioned study.12,13 Last, mean (SD) ultimate load to failure in a study that used only a retrograde bio-interference screw (9×20 mm) was 679.00 (109.44) N,3 similar to the 778 (139) N found for interference-screw-only in the present study. The difference is likely attributable to the longer screw (9×28 mm) in our study. Using SwiveLock C in cortical bone, Barber and colleagues18 found mean (SD) loads to failure up to 711.9 (89.1) N.
Clinically, it has been shown that a statistically significant increase in anterior laxity occurred between 4 months and 2 years in 10.7% of patients who underwent hamstring ACL reconstruction.20 The knees were clinically categorized as unstable or severely abnormal. The authors concluded that the clinical outcome was more likely influenced by the methods used to fix the free ends of the graft, specifically with 2 staples or a washer. To simulate early postoperative rehabilitation in the present study, cyclic loading of the graft was performed. Ultimate load to failure was then determined in order to evaluate catastrophic failure strength of the backup fixation devices in comparison with the interference-screw-only group without supplementary fixation.
It has been shown in autologous hamstring ACL reconstruction that a centrally placed polyethylene screw with sheath (Intrafix) performed as well as a standard, eccentrically placed metal interference screw with staple.10 It is therefore logical that backup fixation with use of a similar device (eg, SwiveLock, bicortical post) is necessary to ensure comparable clinical outcomes in relation to a screw/sheath device that has been shown to endure the highest yield loads.2,9,12,13,21-23 Potential benefits of using SwiveLock anchors for backup fixation include a statistically significant increased mean (SD) ultimate yield load of 229 (176) N over interference screw only. These results are similar to those in comparable studies: 218.3 (59.7) N24 and 165 (24.15) N25 in healthy bone with a reported bone mineral density (BMD) of 1.39 g/cm2, similar to that of skeletally mature porcine tibia (1.220-1.675 g/cm²).3 In addition, ease of insertion of this device over a bicortical post was demonstrated. The titanium post required bicortical drilling as well as measurement with a depth gauge to ensure adequate screw length. This process appeared to require more time during specimen preparation and theoretically could prove to be more dangerous clinically.7 However, caution in using a SwiveLock anchor in osteoporotic bone is advised because of reduced pullout strength.26 In this case, bicortical-post backup fixation may be more suitable. Moreover, although not demonstrated in this study, hardware prominence and irritation with a post may cause postoperative morbidity necessitating future removal.20 Hardware removal was the most common reason for additional surgery using hamstring tendons as graft.20 A second surgery for hardware removal was required in 21% of these patients.20 This is unlikely to occur with a SwiveLock, as the anchor is buried within cortical bone.
Limitations
Regarding use of nonhuman tissues in a biomechanical model, porcine tibiae and bovine extensor tendons were used because of availability, consistency among specimens, and cost-effectiveness. However, bovine extensor tendons have been shown to exhibit stiffness and viscoelastic properties similar to those of a human hamstring graft.27 In addition, the BMD of the porcine tibiae used in this study was not tested because of time involved and cost-efficiency. However, it has been shown that average BMD of porcine tibiae, 1.220-1.675 g/cm², is similar to that in a young athletic population, 1.24-1.62 g/cm2.3,28-31 We therefore assumed similarity to a young athletic population and uniformity of BMD of the porcine tibiae used in this study.
In addition, the biomechanical testing protocol did not simulate physiologic loading within the tibial tunnel. Moreover, the testing protocol used loads of only 250 N during cyclic testing for 500 cycles. This simulates only the early rehabilitation period and not the healing period, which may last up to 12 weeks.9 In addition, as previously mentioned, forces on the graft may be as high as 450 to 700 N.11,32 Pullout testing in line with the long axis of the tibia was performed in order to compare mechanical testing results with those of similar studies.3,12,13 Last, the P of .07 for the comparison of ultimate load to failure between the 2 backup fixation groups suggests that this study may have been underpowered.
Conclusion
This study demonstrated an effective, alternative, and equivalent backup fixation device that can help prevent graft slippage within the tibial tunnel during soft-tissue ACL reconstruction. Potential benefits of using SwiveLock anchors for backup fixation include a significantly increased ultimate yield load (229 N) when supplementing an interference screw, ease of insertion compared with a bicortical post, and the improbable need for future hardware removal. We support using SwiveLock for supplementary fixation at the tibial tunnel site when using soft-tissue grafts in ACL reconstruction.
1. Wetzler MJ, Bartolozzi AR, Gillespie MJ, Rubenstein DL, Ciccotti MG, Miller LS. Revision anterior cruciate ligament reconstruction. Oper Tech Orthop. 1996;6(3):181-189.
2. Scheffler SU, Südkamp NP, Göckenjan A, Hoffmann RF, Weiler A. Biomechanical comparison of hamstring and patellar tendon graft anterior cruciate ligament reconstruction techniques: the impact of fixation level and fixation method under cyclic loading. Arthroscopy. 2002;18(3):304-315.
3. Walsh MP, Wijdicks CA, Parker JB, Hapa O, LaPrade RF. A comparison between a retrograde interference screw, suture button, and combined fixation on the tibial side in an all-inside anterior cruciate ligament reconstruction: a biomechanical study in a porcine model. Am J Sports Med. 2009;37(1):160-167.
4. Howell SM, Hull ML. Aggressive rehabilitation using hamstring tendons: graft construct, tibial tunnel placement, fixation properties, and clinical outcome. Am J Knee Surg. 1998;11(2):120-127.
5. Magen HE, Howell SM, Hull ML. Structural properties of six tibial fixation methods for anterior cruciate ligament soft tissue grafts. Am J Sports Med. 1999;27(1):35-43.
6. Beynnon BD, Meriam CM, Ryder SH, Fleming BC, Johnson RJ. The effect of screw insertion torque on tendons fixed with spiked washers. Am J Sports Med. 1998;26(4):536-539.
7. Post WR, King SS. Neurovascular risk of bicortical tibial drilling for screw and spiked washer fixation of soft-tissue anterior cruciate ligament graft. Arthroscopy. 2001;17(3):244-247.
8. Holden JP, Grood ES, Butler DL, et al. Biomechanics of fascia lata ligament replacements: early postoperative changes in the goat. J Orthop Res. 1988;6(5):639-647.
9. Rodeo SA, Arnoczky SP, Torzilli PA, Hidaka C, Warren RF. Tendon-healing in a bone tunnel. A biomechanical and histological study in the dog. J Bone Joint Surg Am. 1993;75(12):1795-1803.
10. Frank CB, Jackson DW. The science of reconstruction of the anterior cruciate ligament. J Bone Joint Surg Am. 1997;79(10):1556-1576.
11. Markolf KL, Willems MJ, Jackson SR, Finerman GA. In situ calibration of miniature sensors implanted into the anterior cruciate ligament. Part I: strain measurements. J Orthop Res. 1998;16(4):455-463.
12. Kousa P, Teppo LN, Jarvinen TL, Vihavainen M, Kannus P, Jarvinen M. The fixation strength of six hamstring tendon graft fixation devices in anterior cruciate ligament reconstruction: I. Femoral site. Am J Sports Med. 2003;3 (2)1:174-181.
13. Kousa P, Jarvinen TL, Vihavainen M, Kannus P, Jarvinen M. The fixation strength of six hamstring tendon graft fixation devices in anterior cruciate ligament reconstruction: II. Tibial site. Am J Sports Med. 2003;31(2):182-188.
14. Brand JC Jr, Pienkowski D, Steenlage E, Hamilton D, Johnson DL, Caborn DN. Interference screw fixation strength of a quadrupled hamstring tendon graft is directly related to bone mineral density and insertion torque. Am J Sports Med. 2000;28(5):705-710.
15. Weiler A, Hoffmann RF, Siepe CJ, Kolbeck SF, Südkamp NP. The influence of screw geometry on hamstring tendon interference fit fixation. Am J Sports Med. 2000;28(3):356-359.
16. Weiler A, Hoffmann RF, Stähelin AC, Bail HJ, Siepe CJ, Südkamp NP. Hamstring tendon fixation using interference screws: a biomechanical study in calf tibial bone. Arthroscopy. 1998;14(1):29-37.
17. Streich NA, Reichenbacher S, Barié A, Buchner M, Schmitt H. Long-term outcome of anterior cruciate ligament reconstruction with an autologous four-strand semitendinosus tendon autograft. Int Orthop. 2013;37(2):279-284.
18. Barber FA, Herbert MA, Beavis C, Barrera Oro F. Suture anchor materials, eyelets, and designs: update 2008. Arthroscopy. 2008;24(8):859-867.
19. Aickin M, Gensler H. Adjusting for multiple testing when reporting research results: the Bonferroni vs Holm methods. Am J Public Health. 1996;86(5):726-728.
20. Howell SM, Deutsch ML. Comparison of endoscopic and two-incision techniques for reconstructing a torn anterior cruciate ligament using hamstring tendons. Arthroscopy. 1999;15(6):594-606.
21. Gwynne-Jones DP, Draffin J, Vane A, Craig R, McMahon S. Failure strengths of concentric and eccentric implants for hamstring graft fixation. ANZ J Surg. 2008;78(3):177-181.
22. Hayes DA, Watts MC, Tevelen GA, Crawford RW. Central versus peripheral tibial screw placement in hamstring anterior cruciate ligament reconstruction: in vitro biomechanics. Arthroscopy. 2005;21(6):703-706.
23. Shino K, Pflaster DS. Comparison of eccentric and concentric screw placement for hamstring graft fixation in the tibial tunnel. Knee Surg Sports Traumatol Arthrosc. 2000;8(2):73-75.
24. Prevrhal S, Fuerst T, Fan B, et al. Quantitative ultrasound of the tibia depends on both cortical density and thickness. Osteoporosis Int. 2001;12(1):28-34.
25. Pietschmann MF, Gülecyüz MF, Fieseler S, et al. Biomechanical stability of knotless suture anchors used in rotator cuff repair in healthy and osteopenic bone. Arthroscopy. 2010;26(8):1035-1044.
26. Burns JP, Snyder SJ, Albritton M. Arthroscopic rotator cuff repair using triple-loaded anchors, suture shuttles, and suture savers. J Am Acad Orthop Surg. 2007;15(7):432-444.
27. Tetsumura S, Fujita A, Nakajima M, Abe M. Biomechanical comparison of different fixation methods on the tibial side in anterior cruciate ligament reconstruction: a biomechanical study in porcine tibial bone. J Orthop Sci. 2006;11(3):278-282.
28. Alfredson H, Nordstrom P, Lorentzon R. Total and regional bone mass in female soccer players. Calcif Tissue Int. 1996;59(6):438-442.
29. Nevill AM, Holder RL, Stewart AD. Modeling elite male athletes’ peripheral bone mass, assessed using regional dual x-ray absorptiometry. Bone. 2003;32(1):62-68.
30. Nordström P, Lorentzon R. Site-specific bone mass differences of the lower extremities in 17-year-old ice hockey players. Calcif Tissue Int. 1996;59(6):4443-4448.
31. Patzer T, Santo G, Olender GD, Wellmann M, Hurschler C, Schofer MD. Suprapectoral or subpectoral position for biceps tenodesis: biomechanical comparison of four different techniques in both positions. J Shoulder Elbow Surg. 2012;21(1):116-125.
32. De Wall M, Scholes CJ, Patel S, Coolican MR, Parker DA. Tibial fixation in anterior cruciate ligament reconstruction: a prospective randomized study comparing metal interference screw and staples with a centrally placed polyethylene screw and sheath. Am J Sports Med. 2011;39(9):1858-1864.
1. Wetzler MJ, Bartolozzi AR, Gillespie MJ, Rubenstein DL, Ciccotti MG, Miller LS. Revision anterior cruciate ligament reconstruction. Oper Tech Orthop. 1996;6(3):181-189.
2. Scheffler SU, Südkamp NP, Göckenjan A, Hoffmann RF, Weiler A. Biomechanical comparison of hamstring and patellar tendon graft anterior cruciate ligament reconstruction techniques: the impact of fixation level and fixation method under cyclic loading. Arthroscopy. 2002;18(3):304-315.
3. Walsh MP, Wijdicks CA, Parker JB, Hapa O, LaPrade RF. A comparison between a retrograde interference screw, suture button, and combined fixation on the tibial side in an all-inside anterior cruciate ligament reconstruction: a biomechanical study in a porcine model. Am J Sports Med. 2009;37(1):160-167.
4. Howell SM, Hull ML. Aggressive rehabilitation using hamstring tendons: graft construct, tibial tunnel placement, fixation properties, and clinical outcome. Am J Knee Surg. 1998;11(2):120-127.
5. Magen HE, Howell SM, Hull ML. Structural properties of six tibial fixation methods for anterior cruciate ligament soft tissue grafts. Am J Sports Med. 1999;27(1):35-43.
6. Beynnon BD, Meriam CM, Ryder SH, Fleming BC, Johnson RJ. The effect of screw insertion torque on tendons fixed with spiked washers. Am J Sports Med. 1998;26(4):536-539.
7. Post WR, King SS. Neurovascular risk of bicortical tibial drilling for screw and spiked washer fixation of soft-tissue anterior cruciate ligament graft. Arthroscopy. 2001;17(3):244-247.
8. Holden JP, Grood ES, Butler DL, et al. Biomechanics of fascia lata ligament replacements: early postoperative changes in the goat. J Orthop Res. 1988;6(5):639-647.
9. Rodeo SA, Arnoczky SP, Torzilli PA, Hidaka C, Warren RF. Tendon-healing in a bone tunnel. A biomechanical and histological study in the dog. J Bone Joint Surg Am. 1993;75(12):1795-1803.
10. Frank CB, Jackson DW. The science of reconstruction of the anterior cruciate ligament. J Bone Joint Surg Am. 1997;79(10):1556-1576.
11. Markolf KL, Willems MJ, Jackson SR, Finerman GA. In situ calibration of miniature sensors implanted into the anterior cruciate ligament. Part I: strain measurements. J Orthop Res. 1998;16(4):455-463.
12. Kousa P, Teppo LN, Jarvinen TL, Vihavainen M, Kannus P, Jarvinen M. The fixation strength of six hamstring tendon graft fixation devices in anterior cruciate ligament reconstruction: I. Femoral site. Am J Sports Med. 2003;3 (2)1:174-181.
13. Kousa P, Jarvinen TL, Vihavainen M, Kannus P, Jarvinen M. The fixation strength of six hamstring tendon graft fixation devices in anterior cruciate ligament reconstruction: II. Tibial site. Am J Sports Med. 2003;31(2):182-188.
14. Brand JC Jr, Pienkowski D, Steenlage E, Hamilton D, Johnson DL, Caborn DN. Interference screw fixation strength of a quadrupled hamstring tendon graft is directly related to bone mineral density and insertion torque. Am J Sports Med. 2000;28(5):705-710.
15. Weiler A, Hoffmann RF, Siepe CJ, Kolbeck SF, Südkamp NP. The influence of screw geometry on hamstring tendon interference fit fixation. Am J Sports Med. 2000;28(3):356-359.
16. Weiler A, Hoffmann RF, Stähelin AC, Bail HJ, Siepe CJ, Südkamp NP. Hamstring tendon fixation using interference screws: a biomechanical study in calf tibial bone. Arthroscopy. 1998;14(1):29-37.
17. Streich NA, Reichenbacher S, Barié A, Buchner M, Schmitt H. Long-term outcome of anterior cruciate ligament reconstruction with an autologous four-strand semitendinosus tendon autograft. Int Orthop. 2013;37(2):279-284.
18. Barber FA, Herbert MA, Beavis C, Barrera Oro F. Suture anchor materials, eyelets, and designs: update 2008. Arthroscopy. 2008;24(8):859-867.
19. Aickin M, Gensler H. Adjusting for multiple testing when reporting research results: the Bonferroni vs Holm methods. Am J Public Health. 1996;86(5):726-728.
20. Howell SM, Deutsch ML. Comparison of endoscopic and two-incision techniques for reconstructing a torn anterior cruciate ligament using hamstring tendons. Arthroscopy. 1999;15(6):594-606.
21. Gwynne-Jones DP, Draffin J, Vane A, Craig R, McMahon S. Failure strengths of concentric and eccentric implants for hamstring graft fixation. ANZ J Surg. 2008;78(3):177-181.
22. Hayes DA, Watts MC, Tevelen GA, Crawford RW. Central versus peripheral tibial screw placement in hamstring anterior cruciate ligament reconstruction: in vitro biomechanics. Arthroscopy. 2005;21(6):703-706.
23. Shino K, Pflaster DS. Comparison of eccentric and concentric screw placement for hamstring graft fixation in the tibial tunnel. Knee Surg Sports Traumatol Arthrosc. 2000;8(2):73-75.
24. Prevrhal S, Fuerst T, Fan B, et al. Quantitative ultrasound of the tibia depends on both cortical density and thickness. Osteoporosis Int. 2001;12(1):28-34.
25. Pietschmann MF, Gülecyüz MF, Fieseler S, et al. Biomechanical stability of knotless suture anchors used in rotator cuff repair in healthy and osteopenic bone. Arthroscopy. 2010;26(8):1035-1044.
26. Burns JP, Snyder SJ, Albritton M. Arthroscopic rotator cuff repair using triple-loaded anchors, suture shuttles, and suture savers. J Am Acad Orthop Surg. 2007;15(7):432-444.
27. Tetsumura S, Fujita A, Nakajima M, Abe M. Biomechanical comparison of different fixation methods on the tibial side in anterior cruciate ligament reconstruction: a biomechanical study in porcine tibial bone. J Orthop Sci. 2006;11(3):278-282.
28. Alfredson H, Nordstrom P, Lorentzon R. Total and regional bone mass in female soccer players. Calcif Tissue Int. 1996;59(6):438-442.
29. Nevill AM, Holder RL, Stewart AD. Modeling elite male athletes’ peripheral bone mass, assessed using regional dual x-ray absorptiometry. Bone. 2003;32(1):62-68.
30. Nordström P, Lorentzon R. Site-specific bone mass differences of the lower extremities in 17-year-old ice hockey players. Calcif Tissue Int. 1996;59(6):4443-4448.
31. Patzer T, Santo G, Olender GD, Wellmann M, Hurschler C, Schofer MD. Suprapectoral or subpectoral position for biceps tenodesis: biomechanical comparison of four different techniques in both positions. J Shoulder Elbow Surg. 2012;21(1):116-125.
32. De Wall M, Scholes CJ, Patel S, Coolican MR, Parker DA. Tibial fixation in anterior cruciate ligament reconstruction: a prospective randomized study comparing metal interference screw and staples with a centrally placed polyethylene screw and sheath. Am J Sports Med. 2011;39(9):1858-1864.
Risk Factors for Thromboembolic Events After Surgery for Ankle Fractures
Venous thromboembolic events (VTEs), encompassing both deep vein thrombosis (DVT) and pulmonary embolism (PE), are potentially fatal events that can occur after orthopedic surgery.1 In patients who do not receive prophylaxis, VTE incidence can be as high as 70% for total hip arthroplasty,2 26% for hip fracture,3 and 5% for ankle fracture.4 Based on the relatively low incidence of VTE after ankle fractures and insufficient evidence for VTE prophylaxis in this population, the American Orthopaedic Foot and Ankle Society and the American College of Chest Physicians do not recommend routine screening or prophylaxis for VTE in patients with ankle fractures.1,5 Nevertheless, certain patients may be at increased risk for VTE after open reduction and internal fixation (ORIF) of an ankle fracture. In such cases, further consideration for prophylaxis may be warranted.
Other studies of VTEs have identified general risk factors of increased age, obesity, prior thromboembolic disease, oral contraceptive use, multitrauma, varicose veins, and prolonged immobilization, among others.1,6,7 In orthopedics, most of this research comes from total joint arthroplasty and hip fracture studies. However, there is relatively limited data for ankle fracture. The best studies directly addressing VTE after ORIF of ankle fractures have had important limitations, including missing patient data and suboptimal capture of VTE occurrences,8-10 possibly leading to underestimates of the incidence of VTEs.
Given the limited data available, we conducted a retrospective national-cohort study to determine the incidence of and independent risk factors for VTEs after ankle fracture ORIF. If patients who are at higher risk for VTE can be identified, they can and should be carefully monitored and be considered for VTE prophylaxis. This information is needed for patient counseling and clinical decision-making.
Materials and Methods
This retrospective study used the American College of Surgeons National Surgical Quality Improvement Program (ACS-NSQIP) database, which captures data from more than 370 participating US hospitals.11 In ACS-NSQIP, 150 patient variables are collected from operative reports, medical records, and patient interviews by trained clinical reviewers.11,12 Patients are identified prospectively and randomly sampled at participating hospitals. Routine auditing is performed to ensure high-quality data. Clinical data are collected for the entire 30-day postoperative period, regardless of discharge status during this time.
Patients who underwent ankle fracture ORIF between 2005 and 2012 were identified in the ACS-NSQIP database. They were initially selected by the postoperative diagnosis of ankle fracture (International Classification of Diseases, Ninth Revision codes 824.0-824.9). Of these patients, only those with primary Current Procedural Terminology codes 27766 (ORIF of medial malleolus fracture), 27769 (ORIF of posterior malleolus fracture), 27792 (ORIF of lateral malleolus fracture), 27814 (ORIF of bimalleollar fracture), and 27822/27823 (ORIF of trimalleollar fracture) were included in the analysis. Patients with incomplete perioperative data were excluded, leaving 4412 patients (out of the initial 4785) for analysis.
Patient characteristics, including sex, age, height, weight, and history of smoking, were collected from the ACS-NSQIP database. Body mass index (BMI) was calculated from each patient’s height and weight. Age was divided into approximately 20-year increments, beginning with age 18 years, in order to compare younger, middle-aged, and elderly groups of patients with ankle fractures. BMI was divided into categories based on the World Health Organization definitions of obesity: under 25 kg/m2 (normal weight), 25 to 30 kg/m2 (overweight), 30 to 35 kg/m2 (class I obesity), and 35 kg/m2 or over (class II and class III obesity).13
Information about medical comorbidities is also available in the ACS-NSQIP database. History of pulmonary disease was defined as a history of dyspnea, severe chronic obstructive pulmonary disease, ventilator-assisted respiration within 48 hours before surgery, or current pneumonia. History of heart disease was defined as a history of congestive heart failure (CHF) or angina within 1 month before admission, myocardial infarction within 6 months before admission, cardiac surgery, or percutaneous coronary intervention. American Society of Anesthesiologists (ASA) classes 3 and above signify severe systemic disease. Steroid use was defined as requiring regular administration of corticosteroid medications within 1 month before surgery. Disseminated cancer was defined as a malignancy that has spread to 1 or more sites besides the primary site.
Functional status was defined as the ability to perform activities of daily living (ADLs) within 30 days before surgery. Best functional status during this period was recorded. ACS-NSQIP defines ADLs as the “activities usually performed in the course of a normal day in a person’s life,” including bathing, feeding, dressing, toileting, and mobility. An independent patient does not require assistance for any ADLs; a partially dependent patient requires assistance for some ADLs; and a totally dependent patient requires assistance in all ADLs. Partially and totally dependent patients were grouped for analysis. Anesthesia type was separated into general and nongeneral, which includes monitored anesthesia care, spinal anesthesia, and regional anesthesia.
ACS-NSQIP also records the occurrence of multiple events up to 30 days after surgery. For our study, VTE was defined as the occurrence of a DVT or a PE during this period. ACS-NSQIP defines DVT as a new blood clot or thrombus identified within a vein—with confirmation by duplex ultrasonography, venogram, or computed tomography (CT)—that required therapy (anticoagulation, placement of vena cava filter, and/or clipping of vena cava). PE is recorded if ventilation/perfusion (VQ) scan, CT examination, transesophageal echocardiogram, pulmonary arteriogram, CT angiogram, or any other definitive modality is positive.
Statistical analyses were performed with Stata Version 11.2 (StataCorp). Demographic and comorbidity variables were tested for association with occurrence of VTE using bivariate and multivariate logistic regression.
Final multivariate models were constructed with a backward stepwise process that initially included all potential variables and sequentially excluded variables with the highest P value until only those with P < .200 remained. Variables with .050 < P < .200 were left in the model to control for potential confounding but are not considered significantly associated with the outcome. Statistical significance was established at a 2-sided α of 0.050 (P < .050). The fitness of the final logistic regression model was assessed with the C statistic and the Hosmer-Lemeshow goodness-of-fit test.
Results
For the 4412 ankle fracture patients who met the inclusion criteria, mean (SD) age was 50.9 (18.2) years, and mean (SD) BMI was 30.4 (7.6) kg/m2. The cohort was 40.4% male. Surgery was performed on 235 patients (5.3%) with medial malleolus fracture, 1143 patients (25.9%) with lateral malleolus fracture, 1705 patients (38.6%) with bimalleollar fracture, and 1329 patients (30.1%) with trimalleollar fracture. Table 1 summarizes the patient characteristics.
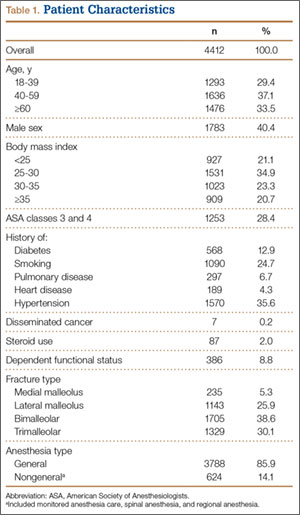
Of the 33 patients (0.8%) with a VTE recorded within the first 30 postoperative days, 16 (0.4% of all patients) had a DVT recorded, 14 (0.3% of all patients) had a PE recorded, and 3 (0.1% of all patients) had both a DVT and a PE recorded. In 13 (39.4%) of the 33 patients with a VTE, the event occurred after discharge. VTEs were reported a mean (SD) of 11.5 (9.6) days after surgery. No patient in this study died of VTE.
Bivariate logistic regressions were performed to test the association of each patient variable with the occurrence of a VTE. Results are listed in Table 2. The bivariate analyses revealed significant associations between VTE after ankle fracture ORIF and the patient variables of age 60 years or older (odds ratio [OR], 2.40; 95% confidence interval [CI], 1.01-5.72), class I obesity (BMI, 30-35 kg/m2: OR, 5.15, 95% CI, 1.14-23.28), class II and class III obesity (BMI, ≥35 kg/m2: OR, 6.33, 95% CI, 1.41-28.38), ASA classes 3 and 4 (OR, 3.05; 95% CI, 1.53-6.08), history of heart disease (OR, 5.10; 95% CI, 2.08-12.49), history of hypertension (OR, 2.81; 95% CI, 1.39-5.66), and dependent functional status (OR, 3.39; 95% CI, 1.52-7.56).
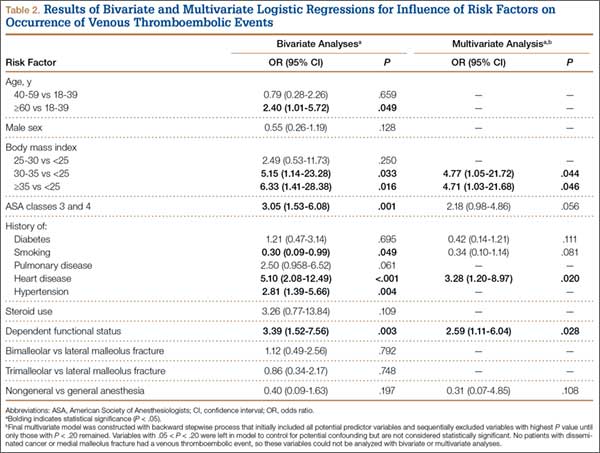
Multivariate logistic regression was used to control for potential confounding variables and determine which factors were independently associated with VTEs. Results of this analysis are listed in Table 2 as well. The multivariate analysis revealed that the patient variables of class I obesity (BMI, 30-35 kg/m2: OR, 4.77; 95% CI, 1.05-21.72; P = .044), class II and class III obesity (BMI, ≥35 kg/m2: OR, 4.71; 95% CI, 1.03-21.68; P = .046), history of heart disease (OR, 3.28; 95% CI, 1.20-8.97; P = .020), and dependent functional status (OR, 2.59; 95% CI, 1.11-6.04; P = .028) were independently associated with an increased rate of VTEs. Of note, anesthesia type was not significantly associated with occurrence of VTE on bivariate or multivariate analysis.
The C statistic of the final multivariate model was 0.76, indicating very good distinguishing ability. The Hosmer-Lemeshow goodness-of-fit test showed no evidence of lack of fit.
Discussion
Citing the lack of conclusive evidence and the low incidence of VTE after ankle fracture surgery, current recommendations are to avoid routine VTE prophylaxis in the postoperative management of patients who undergo this surgery.1,5 However, it is important to identify patients who are at increased risk, as some may benefit from VTE prophylaxis. In the present study, we used the large, high-quality ACS-NSQIP database collecting information from multiple US hospitals to examine risk factors for VTE after ankle fracture ORIF. We identified 4412 patients who underwent ankle fracture ORIF between 2005 and 2012, and found an overall VTE incidence of 0.8%. Multivariate analysis identified obesity, history of heart disease, and dependent functional status as independent risk factors for VTE after ankle fracture ORIF.
This study’s 0.8% incidence of VTE after ankle fracture ORIF is consistent with the range (0.29%-5%) reported in other ankle fracture studies.4,8-10,14-18 We found that VTEs occurred a mean of about 11 days after surgery, and no patient died of VTE.
Obesity (BMI, ≥30 kg/m2) had the strongest association with VTEs in this study. Obesity, which is a growing public health concern, can make postoperative care and mobilization more difficult.19 Obesity has previously been associated with VTEs after ankle fractures, and BMI of over 25 kg/m2 is one of the Caprini criteria for thrombosis risk factor assessment.6,10 In our study, however, BMI of 25 to 30 kg/m2 was not associated with an increased VTE rate, indicating that moderately overweight patients may not be at significantly higher risk for VTE (compared with patients with normal BMI) and may not need VTE prophylaxis. VTE prophylaxis after ankle fracture surgery may be considered in patients with BMI over 30 kg/m2.
History of heart disease was also associated with VTEs in this study. Patients with a history of heart disease were at 3 times the risk for VTE within 30 days of ankle fracture surgery. This association is also consistent with the Caprini criteria, which include acute myocardial infarction and CHF as risk factors for venous thrombosis.6 Other studies have found associations between CHF and VTE and between cardiovascular risk factors and VTE.7,20 The association between cardiovascular disease and VTE may derive from the decreased venous flow rate associated with CHF or an overall vascular disease state. These patients may benefit from heightened surveillance and postoperative prophylaxis for VTE.
Dependent functional status was the final risk factor found to be associated with VTE after ankle fracture ORIF. This association likely derives from an inability to mobilize independently, leading to increased venous stasis. Immobilization has been previously associated with increased risk for VTE after ankle surgery.7,14,16,20 Caretakers should be aware of this increased risk during the postoperative period and diligently monitor these patients for signs and symptoms of VTE. Prophylaxis may also be considered in this patient population.
Several risk factors that were significant on bivariate analysis (increased age; increased ASA class; history of diabetes, pulmonary disease, hypertension) were not significant in the final multivariate model. This finding suggests covariance between these factors and those that were significant in the final multivariate model. In particular, age and increased overall comorbidity (represented by increased ASA class) were not significant in our multivariate model—contrary to findings of other studies.8-10 It is possible that history of heart disease alone was responsible for the association between overall comorbidity and VTE in those studies. In the present study, separating and controlling for individual comorbidities could have allowed this association to be more precisely characterized.
The characteristics of the ACS-NSQIP database limited our study in several ways. First, although ACS-NSQIP makes significant efforts to collect as many patient variables as possible, some information is not captured. Data about additional factors that may affect VTE risk (eg, history of previous VTE, hypercoagulable state, history of malignancy other than disseminated cancer, tourniquet time, patient position in operating room) were not available. Second, data are collected only on those postoperative adverse events that occur within 30 days after surgery; data on VTEs that occur later are not captured. However, it has been shown that the majority of VTEs occur within the first 30 days after lower extremity trauma and surgery,21,22 so this follow-up interval was deemed adequate for capture of VTE data. Third, the database does not include information on the prophylactic regimens used for these patients—which may have weakened the associations between predictor variables and VTE risk and led to an underestimated effect size. VTE incidence, as well as the odds of developing a VTE with one of the identified risk factors, may actually be higher than reported in this study.
Conclusion
VTEs are serious complications that can occur after ORIF of ankle fractures. In this study, the overall incidence of VTE after ankle fracture ORIF was 0.8%. Although the American Orthopaedic Foot and Ankle Society and the American College of Chest Physicians do not recommend routine screening or prophylaxis for VTE in patients with ankle fractures,1,5 the results of this study showed there may be a benefit in emphasizing VTE prophylaxis after ankle fracture ORIF in patients with obesity, history of heart disease, or dependent functional status. At minimum, these patients should be more carefully monitored for development of VTEs.
1. American Orthopaedic Foot and Ankle Society. Position statement: the use of VTED prophylaxis in foot and ankle surgery. http://www.aofas.org/medical-community/health-policy/Documents/VTED-Position-Statement-Approv-7-9-13-FINAL.pdf. Updated 2013. Accessed May 10, 2015.
2. Grady-Benson JC, Oishi CS, Hanson PB, Colwell CW Jr, Otis SM, Walker RH. Routine postoperative duplex ultrasonography screening and monitoring for the detection of deep vein thrombosis. A survey of 110 total hip arthroplasties. Clin Orthop Relat Res. 1994;(307):130-141.
3. Salzman EW, Harris WH, DeSanctis RW. Anticoagulation for prevention of thromboembolism following fractures of the hip. New Engl J Med. 1966;275(3):122-130.
4. Patil S, Gandhi J, Curzon I, Hui AC. Incidence of deep-vein thrombosis in patients with fractures of the ankle treated in a plaster cast. J Bone Joint Surg Br. 2007;89(10):1340-1343.
5. Falck-Ytter Y, Francis CW, Johanson NA, et al; American College of Chest Physicians. Prevention of VTE in orthopedic surgery patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 suppl):e278S-e325S.
6. Caprini JA. Thrombosis risk assessment as a guide to quality patient care. Dis Mon. 2005;51(2-3):70-78.
7. Mayle RE Jr, DiGiovanni CW, Lin SS, Tabrizi P, Chou LB. Current concepts review: venous thromboembolic disease in foot and ankle surgery. Foot Ankle Int. 2007;28(11):1207-1216.
8. Jameson SS, Augustine A, James P, et al. Venous thromboembolic events following foot and ankle surgery in the English National Health Service. J Bone Joint Surg Br. 2011;93(4):490-497.
9. SooHoo NF, Eagan M, Krenek L, Zingmond DS. Incidence and factors predicting pulmonary embolism and deep venous thrombosis following surgical treatment of ankle fractures. Foot Ankle Surg. 2011;17(4):259-262.
10. Shibuya N, Frost CH, Campbell JD, Davis ML, Jupiter DC. Incidence of acute deep vein thrombosis and pulmonary embolism in foot and ankle trauma: analysis of the National Trauma Data Bank. J Foot Ankle Surg. 2012;51(1):63-68.
11. American College of Surgeons National Surgical Quality Improvement Program. User Guide for the 2012 ACS NSQIP Participant Use Data File. http://site.acsnsqip.org/wp-content/uploads/2013/10/ACSNSQIP.PUF_.UserGuide.2012.pdf. Published October 2013. Accessed May 10, 2015.
12. Khuri SF, Henderson WG, Daley J, et al; Principal Investigators of Patient Safety in Surgery Study. Successful implementation of the Department of Veterans Affairs’ National Surgical Quality Improvement Program in the private sector: the Patient Safety in Surgery study. Ann Surg. 2008;248(2):329-336.
13. Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. The disease burden associated with overweight and obesity. JAMA. 1999;282(16):1523-1529.
14. Mizel MS, Temple HT, Michelson JD, et al. Thromboembolism after foot and ankle surgery. A multicenter study. Clin Orthop Relat Res. 1998;(348):180-185.
15. Solis G, Saxby T. Incidence of DVT following surgery of the foot and ankle. Foot Ankle Int. 2002;23(5):411-414.
16. Hanslow SS, Grujic L, Slater HK, Chen D. Thromboembolic disease after foot and ankle surgery. Foot Ankle Int. 2006;27(9):693-695.
17. Pelet S, Roger ME, Belzile EL, Bouchard M. The incidence of thromboembolic events in surgically treated ankle fracture. J Bone Joint Surg Am. 2012;94(6):502-506.
18. Manafi Rasi A, Kazemian G, Emami Moghadam M, et al. Deep vein thrombosis following below knee immobilization: the need for chemoprophylaxis. Trauma Mon. 2013;17(4):367-369.
19. Sabharwal S, Root MZ. Impact of obesity on orthopaedics. J Bone Joint Surg Am. 2012;94(11):1045-1052.
20. Kadous A, Abdelgawad AA, Kanlic E. Deep venous thrombosis and pulmonary embolism after surgical treatment of ankle fractures: a case report and review of literature. J Foot Ankle Surg. 2012;51(4):457-463.
21. Forsythe RM, Peitzman AB, DeCato T, et al. Early lower extremity fracture fixation and the risk of early pulmonary embolus: filter before fixation? J Trauma. 2011;70(6):1381-1388.
22. Bjørnarå BT, Gudmundsen TE, Dahl OE. Frequency and timing of clinical venous thromboembolism after major joint surgery. J Bone Joint Surg Br. 2006;88(3):386-391.
Venous thromboembolic events (VTEs), encompassing both deep vein thrombosis (DVT) and pulmonary embolism (PE), are potentially fatal events that can occur after orthopedic surgery.1 In patients who do not receive prophylaxis, VTE incidence can be as high as 70% for total hip arthroplasty,2 26% for hip fracture,3 and 5% for ankle fracture.4 Based on the relatively low incidence of VTE after ankle fractures and insufficient evidence for VTE prophylaxis in this population, the American Orthopaedic Foot and Ankle Society and the American College of Chest Physicians do not recommend routine screening or prophylaxis for VTE in patients with ankle fractures.1,5 Nevertheless, certain patients may be at increased risk for VTE after open reduction and internal fixation (ORIF) of an ankle fracture. In such cases, further consideration for prophylaxis may be warranted.
Other studies of VTEs have identified general risk factors of increased age, obesity, prior thromboembolic disease, oral contraceptive use, multitrauma, varicose veins, and prolonged immobilization, among others.1,6,7 In orthopedics, most of this research comes from total joint arthroplasty and hip fracture studies. However, there is relatively limited data for ankle fracture. The best studies directly addressing VTE after ORIF of ankle fractures have had important limitations, including missing patient data and suboptimal capture of VTE occurrences,8-10 possibly leading to underestimates of the incidence of VTEs.
Given the limited data available, we conducted a retrospective national-cohort study to determine the incidence of and independent risk factors for VTEs after ankle fracture ORIF. If patients who are at higher risk for VTE can be identified, they can and should be carefully monitored and be considered for VTE prophylaxis. This information is needed for patient counseling and clinical decision-making.
Materials and Methods
This retrospective study used the American College of Surgeons National Surgical Quality Improvement Program (ACS-NSQIP) database, which captures data from more than 370 participating US hospitals.11 In ACS-NSQIP, 150 patient variables are collected from operative reports, medical records, and patient interviews by trained clinical reviewers.11,12 Patients are identified prospectively and randomly sampled at participating hospitals. Routine auditing is performed to ensure high-quality data. Clinical data are collected for the entire 30-day postoperative period, regardless of discharge status during this time.
Patients who underwent ankle fracture ORIF between 2005 and 2012 were identified in the ACS-NSQIP database. They were initially selected by the postoperative diagnosis of ankle fracture (International Classification of Diseases, Ninth Revision codes 824.0-824.9). Of these patients, only those with primary Current Procedural Terminology codes 27766 (ORIF of medial malleolus fracture), 27769 (ORIF of posterior malleolus fracture), 27792 (ORIF of lateral malleolus fracture), 27814 (ORIF of bimalleollar fracture), and 27822/27823 (ORIF of trimalleollar fracture) were included in the analysis. Patients with incomplete perioperative data were excluded, leaving 4412 patients (out of the initial 4785) for analysis.
Patient characteristics, including sex, age, height, weight, and history of smoking, were collected from the ACS-NSQIP database. Body mass index (BMI) was calculated from each patient’s height and weight. Age was divided into approximately 20-year increments, beginning with age 18 years, in order to compare younger, middle-aged, and elderly groups of patients with ankle fractures. BMI was divided into categories based on the World Health Organization definitions of obesity: under 25 kg/m2 (normal weight), 25 to 30 kg/m2 (overweight), 30 to 35 kg/m2 (class I obesity), and 35 kg/m2 or over (class II and class III obesity).13
Information about medical comorbidities is also available in the ACS-NSQIP database. History of pulmonary disease was defined as a history of dyspnea, severe chronic obstructive pulmonary disease, ventilator-assisted respiration within 48 hours before surgery, or current pneumonia. History of heart disease was defined as a history of congestive heart failure (CHF) or angina within 1 month before admission, myocardial infarction within 6 months before admission, cardiac surgery, or percutaneous coronary intervention. American Society of Anesthesiologists (ASA) classes 3 and above signify severe systemic disease. Steroid use was defined as requiring regular administration of corticosteroid medications within 1 month before surgery. Disseminated cancer was defined as a malignancy that has spread to 1 or more sites besides the primary site.
Functional status was defined as the ability to perform activities of daily living (ADLs) within 30 days before surgery. Best functional status during this period was recorded. ACS-NSQIP defines ADLs as the “activities usually performed in the course of a normal day in a person’s life,” including bathing, feeding, dressing, toileting, and mobility. An independent patient does not require assistance for any ADLs; a partially dependent patient requires assistance for some ADLs; and a totally dependent patient requires assistance in all ADLs. Partially and totally dependent patients were grouped for analysis. Anesthesia type was separated into general and nongeneral, which includes monitored anesthesia care, spinal anesthesia, and regional anesthesia.
ACS-NSQIP also records the occurrence of multiple events up to 30 days after surgery. For our study, VTE was defined as the occurrence of a DVT or a PE during this period. ACS-NSQIP defines DVT as a new blood clot or thrombus identified within a vein—with confirmation by duplex ultrasonography, venogram, or computed tomography (CT)—that required therapy (anticoagulation, placement of vena cava filter, and/or clipping of vena cava). PE is recorded if ventilation/perfusion (VQ) scan, CT examination, transesophageal echocardiogram, pulmonary arteriogram, CT angiogram, or any other definitive modality is positive.
Statistical analyses were performed with Stata Version 11.2 (StataCorp). Demographic and comorbidity variables were tested for association with occurrence of VTE using bivariate and multivariate logistic regression.
Final multivariate models were constructed with a backward stepwise process that initially included all potential variables and sequentially excluded variables with the highest P value until only those with P < .200 remained. Variables with .050 < P < .200 were left in the model to control for potential confounding but are not considered significantly associated with the outcome. Statistical significance was established at a 2-sided α of 0.050 (P < .050). The fitness of the final logistic regression model was assessed with the C statistic and the Hosmer-Lemeshow goodness-of-fit test.
Results
For the 4412 ankle fracture patients who met the inclusion criteria, mean (SD) age was 50.9 (18.2) years, and mean (SD) BMI was 30.4 (7.6) kg/m2. The cohort was 40.4% male. Surgery was performed on 235 patients (5.3%) with medial malleolus fracture, 1143 patients (25.9%) with lateral malleolus fracture, 1705 patients (38.6%) with bimalleollar fracture, and 1329 patients (30.1%) with trimalleollar fracture. Table 1 summarizes the patient characteristics.
Of the 33 patients (0.8%) with a VTE recorded within the first 30 postoperative days, 16 (0.4% of all patients) had a DVT recorded, 14 (0.3% of all patients) had a PE recorded, and 3 (0.1% of all patients) had both a DVT and a PE recorded. In 13 (39.4%) of the 33 patients with a VTE, the event occurred after discharge. VTEs were reported a mean (SD) of 11.5 (9.6) days after surgery. No patient in this study died of VTE.
Bivariate logistic regressions were performed to test the association of each patient variable with the occurrence of a VTE. Results are listed in Table 2. The bivariate analyses revealed significant associations between VTE after ankle fracture ORIF and the patient variables of age 60 years or older (odds ratio [OR], 2.40; 95% confidence interval [CI], 1.01-5.72), class I obesity (BMI, 30-35 kg/m2: OR, 5.15, 95% CI, 1.14-23.28), class II and class III obesity (BMI, ≥35 kg/m2: OR, 6.33, 95% CI, 1.41-28.38), ASA classes 3 and 4 (OR, 3.05; 95% CI, 1.53-6.08), history of heart disease (OR, 5.10; 95% CI, 2.08-12.49), history of hypertension (OR, 2.81; 95% CI, 1.39-5.66), and dependent functional status (OR, 3.39; 95% CI, 1.52-7.56).
Multivariate logistic regression was used to control for potential confounding variables and determine which factors were independently associated with VTEs. Results of this analysis are listed in Table 2 as well. The multivariate analysis revealed that the patient variables of class I obesity (BMI, 30-35 kg/m2: OR, 4.77; 95% CI, 1.05-21.72; P = .044), class II and class III obesity (BMI, ≥35 kg/m2: OR, 4.71; 95% CI, 1.03-21.68; P = .046), history of heart disease (OR, 3.28; 95% CI, 1.20-8.97; P = .020), and dependent functional status (OR, 2.59; 95% CI, 1.11-6.04; P = .028) were independently associated with an increased rate of VTEs. Of note, anesthesia type was not significantly associated with occurrence of VTE on bivariate or multivariate analysis.
The C statistic of the final multivariate model was 0.76, indicating very good distinguishing ability. The Hosmer-Lemeshow goodness-of-fit test showed no evidence of lack of fit.
Discussion
Citing the lack of conclusive evidence and the low incidence of VTE after ankle fracture surgery, current recommendations are to avoid routine VTE prophylaxis in the postoperative management of patients who undergo this surgery.1,5 However, it is important to identify patients who are at increased risk, as some may benefit from VTE prophylaxis. In the present study, we used the large, high-quality ACS-NSQIP database collecting information from multiple US hospitals to examine risk factors for VTE after ankle fracture ORIF. We identified 4412 patients who underwent ankle fracture ORIF between 2005 and 2012, and found an overall VTE incidence of 0.8%. Multivariate analysis identified obesity, history of heart disease, and dependent functional status as independent risk factors for VTE after ankle fracture ORIF.
This study’s 0.8% incidence of VTE after ankle fracture ORIF is consistent with the range (0.29%-5%) reported in other ankle fracture studies.4,8-10,14-18 We found that VTEs occurred a mean of about 11 days after surgery, and no patient died of VTE.
Obesity (BMI, ≥30 kg/m2) had the strongest association with VTEs in this study. Obesity, which is a growing public health concern, can make postoperative care and mobilization more difficult.19 Obesity has previously been associated with VTEs after ankle fractures, and BMI of over 25 kg/m2 is one of the Caprini criteria for thrombosis risk factor assessment.6,10 In our study, however, BMI of 25 to 30 kg/m2 was not associated with an increased VTE rate, indicating that moderately overweight patients may not be at significantly higher risk for VTE (compared with patients with normal BMI) and may not need VTE prophylaxis. VTE prophylaxis after ankle fracture surgery may be considered in patients with BMI over 30 kg/m2.
History of heart disease was also associated with VTEs in this study. Patients with a history of heart disease were at 3 times the risk for VTE within 30 days of ankle fracture surgery. This association is also consistent with the Caprini criteria, which include acute myocardial infarction and CHF as risk factors for venous thrombosis.6 Other studies have found associations between CHF and VTE and between cardiovascular risk factors and VTE.7,20 The association between cardiovascular disease and VTE may derive from the decreased venous flow rate associated with CHF or an overall vascular disease state. These patients may benefit from heightened surveillance and postoperative prophylaxis for VTE.
Dependent functional status was the final risk factor found to be associated with VTE after ankle fracture ORIF. This association likely derives from an inability to mobilize independently, leading to increased venous stasis. Immobilization has been previously associated with increased risk for VTE after ankle surgery.7,14,16,20 Caretakers should be aware of this increased risk during the postoperative period and diligently monitor these patients for signs and symptoms of VTE. Prophylaxis may also be considered in this patient population.
Several risk factors that were significant on bivariate analysis (increased age; increased ASA class; history of diabetes, pulmonary disease, hypertension) were not significant in the final multivariate model. This finding suggests covariance between these factors and those that were significant in the final multivariate model. In particular, age and increased overall comorbidity (represented by increased ASA class) were not significant in our multivariate model—contrary to findings of other studies.8-10 It is possible that history of heart disease alone was responsible for the association between overall comorbidity and VTE in those studies. In the present study, separating and controlling for individual comorbidities could have allowed this association to be more precisely characterized.
The characteristics of the ACS-NSQIP database limited our study in several ways. First, although ACS-NSQIP makes significant efforts to collect as many patient variables as possible, some information is not captured. Data about additional factors that may affect VTE risk (eg, history of previous VTE, hypercoagulable state, history of malignancy other than disseminated cancer, tourniquet time, patient position in operating room) were not available. Second, data are collected only on those postoperative adverse events that occur within 30 days after surgery; data on VTEs that occur later are not captured. However, it has been shown that the majority of VTEs occur within the first 30 days after lower extremity trauma and surgery,21,22 so this follow-up interval was deemed adequate for capture of VTE data. Third, the database does not include information on the prophylactic regimens used for these patients—which may have weakened the associations between predictor variables and VTE risk and led to an underestimated effect size. VTE incidence, as well as the odds of developing a VTE with one of the identified risk factors, may actually be higher than reported in this study.
Conclusion
VTEs are serious complications that can occur after ORIF of ankle fractures. In this study, the overall incidence of VTE after ankle fracture ORIF was 0.8%. Although the American Orthopaedic Foot and Ankle Society and the American College of Chest Physicians do not recommend routine screening or prophylaxis for VTE in patients with ankle fractures,1,5 the results of this study showed there may be a benefit in emphasizing VTE prophylaxis after ankle fracture ORIF in patients with obesity, history of heart disease, or dependent functional status. At minimum, these patients should be more carefully monitored for development of VTEs.
Venous thromboembolic events (VTEs), encompassing both deep vein thrombosis (DVT) and pulmonary embolism (PE), are potentially fatal events that can occur after orthopedic surgery.1 In patients who do not receive prophylaxis, VTE incidence can be as high as 70% for total hip arthroplasty,2 26% for hip fracture,3 and 5% for ankle fracture.4 Based on the relatively low incidence of VTE after ankle fractures and insufficient evidence for VTE prophylaxis in this population, the American Orthopaedic Foot and Ankle Society and the American College of Chest Physicians do not recommend routine screening or prophylaxis for VTE in patients with ankle fractures.1,5 Nevertheless, certain patients may be at increased risk for VTE after open reduction and internal fixation (ORIF) of an ankle fracture. In such cases, further consideration for prophylaxis may be warranted.
Other studies of VTEs have identified general risk factors of increased age, obesity, prior thromboembolic disease, oral contraceptive use, multitrauma, varicose veins, and prolonged immobilization, among others.1,6,7 In orthopedics, most of this research comes from total joint arthroplasty and hip fracture studies. However, there is relatively limited data for ankle fracture. The best studies directly addressing VTE after ORIF of ankle fractures have had important limitations, including missing patient data and suboptimal capture of VTE occurrences,8-10 possibly leading to underestimates of the incidence of VTEs.
Given the limited data available, we conducted a retrospective national-cohort study to determine the incidence of and independent risk factors for VTEs after ankle fracture ORIF. If patients who are at higher risk for VTE can be identified, they can and should be carefully monitored and be considered for VTE prophylaxis. This information is needed for patient counseling and clinical decision-making.
Materials and Methods
This retrospective study used the American College of Surgeons National Surgical Quality Improvement Program (ACS-NSQIP) database, which captures data from more than 370 participating US hospitals.11 In ACS-NSQIP, 150 patient variables are collected from operative reports, medical records, and patient interviews by trained clinical reviewers.11,12 Patients are identified prospectively and randomly sampled at participating hospitals. Routine auditing is performed to ensure high-quality data. Clinical data are collected for the entire 30-day postoperative period, regardless of discharge status during this time.
Patients who underwent ankle fracture ORIF between 2005 and 2012 were identified in the ACS-NSQIP database. They were initially selected by the postoperative diagnosis of ankle fracture (International Classification of Diseases, Ninth Revision codes 824.0-824.9). Of these patients, only those with primary Current Procedural Terminology codes 27766 (ORIF of medial malleolus fracture), 27769 (ORIF of posterior malleolus fracture), 27792 (ORIF of lateral malleolus fracture), 27814 (ORIF of bimalleollar fracture), and 27822/27823 (ORIF of trimalleollar fracture) were included in the analysis. Patients with incomplete perioperative data were excluded, leaving 4412 patients (out of the initial 4785) for analysis.
Patient characteristics, including sex, age, height, weight, and history of smoking, were collected from the ACS-NSQIP database. Body mass index (BMI) was calculated from each patient’s height and weight. Age was divided into approximately 20-year increments, beginning with age 18 years, in order to compare younger, middle-aged, and elderly groups of patients with ankle fractures. BMI was divided into categories based on the World Health Organization definitions of obesity: under 25 kg/m2 (normal weight), 25 to 30 kg/m2 (overweight), 30 to 35 kg/m2 (class I obesity), and 35 kg/m2 or over (class II and class III obesity).13
Information about medical comorbidities is also available in the ACS-NSQIP database. History of pulmonary disease was defined as a history of dyspnea, severe chronic obstructive pulmonary disease, ventilator-assisted respiration within 48 hours before surgery, or current pneumonia. History of heart disease was defined as a history of congestive heart failure (CHF) or angina within 1 month before admission, myocardial infarction within 6 months before admission, cardiac surgery, or percutaneous coronary intervention. American Society of Anesthesiologists (ASA) classes 3 and above signify severe systemic disease. Steroid use was defined as requiring regular administration of corticosteroid medications within 1 month before surgery. Disseminated cancer was defined as a malignancy that has spread to 1 or more sites besides the primary site.
Functional status was defined as the ability to perform activities of daily living (ADLs) within 30 days before surgery. Best functional status during this period was recorded. ACS-NSQIP defines ADLs as the “activities usually performed in the course of a normal day in a person’s life,” including bathing, feeding, dressing, toileting, and mobility. An independent patient does not require assistance for any ADLs; a partially dependent patient requires assistance for some ADLs; and a totally dependent patient requires assistance in all ADLs. Partially and totally dependent patients were grouped for analysis. Anesthesia type was separated into general and nongeneral, which includes monitored anesthesia care, spinal anesthesia, and regional anesthesia.
ACS-NSQIP also records the occurrence of multiple events up to 30 days after surgery. For our study, VTE was defined as the occurrence of a DVT or a PE during this period. ACS-NSQIP defines DVT as a new blood clot or thrombus identified within a vein—with confirmation by duplex ultrasonography, venogram, or computed tomography (CT)—that required therapy (anticoagulation, placement of vena cava filter, and/or clipping of vena cava). PE is recorded if ventilation/perfusion (VQ) scan, CT examination, transesophageal echocardiogram, pulmonary arteriogram, CT angiogram, or any other definitive modality is positive.
Statistical analyses were performed with Stata Version 11.2 (StataCorp). Demographic and comorbidity variables were tested for association with occurrence of VTE using bivariate and multivariate logistic regression.
Final multivariate models were constructed with a backward stepwise process that initially included all potential variables and sequentially excluded variables with the highest P value until only those with P < .200 remained. Variables with .050 < P < .200 were left in the model to control for potential confounding but are not considered significantly associated with the outcome. Statistical significance was established at a 2-sided α of 0.050 (P < .050). The fitness of the final logistic regression model was assessed with the C statistic and the Hosmer-Lemeshow goodness-of-fit test.
Results
For the 4412 ankle fracture patients who met the inclusion criteria, mean (SD) age was 50.9 (18.2) years, and mean (SD) BMI was 30.4 (7.6) kg/m2. The cohort was 40.4% male. Surgery was performed on 235 patients (5.3%) with medial malleolus fracture, 1143 patients (25.9%) with lateral malleolus fracture, 1705 patients (38.6%) with bimalleollar fracture, and 1329 patients (30.1%) with trimalleollar fracture. Table 1 summarizes the patient characteristics.
Of the 33 patients (0.8%) with a VTE recorded within the first 30 postoperative days, 16 (0.4% of all patients) had a DVT recorded, 14 (0.3% of all patients) had a PE recorded, and 3 (0.1% of all patients) had both a DVT and a PE recorded. In 13 (39.4%) of the 33 patients with a VTE, the event occurred after discharge. VTEs were reported a mean (SD) of 11.5 (9.6) days after surgery. No patient in this study died of VTE.
Bivariate logistic regressions were performed to test the association of each patient variable with the occurrence of a VTE. Results are listed in Table 2. The bivariate analyses revealed significant associations between VTE after ankle fracture ORIF and the patient variables of age 60 years or older (odds ratio [OR], 2.40; 95% confidence interval [CI], 1.01-5.72), class I obesity (BMI, 30-35 kg/m2: OR, 5.15, 95% CI, 1.14-23.28), class II and class III obesity (BMI, ≥35 kg/m2: OR, 6.33, 95% CI, 1.41-28.38), ASA classes 3 and 4 (OR, 3.05; 95% CI, 1.53-6.08), history of heart disease (OR, 5.10; 95% CI, 2.08-12.49), history of hypertension (OR, 2.81; 95% CI, 1.39-5.66), and dependent functional status (OR, 3.39; 95% CI, 1.52-7.56).
Multivariate logistic regression was used to control for potential confounding variables and determine which factors were independently associated with VTEs. Results of this analysis are listed in Table 2 as well. The multivariate analysis revealed that the patient variables of class I obesity (BMI, 30-35 kg/m2: OR, 4.77; 95% CI, 1.05-21.72; P = .044), class II and class III obesity (BMI, ≥35 kg/m2: OR, 4.71; 95% CI, 1.03-21.68; P = .046), history of heart disease (OR, 3.28; 95% CI, 1.20-8.97; P = .020), and dependent functional status (OR, 2.59; 95% CI, 1.11-6.04; P = .028) were independently associated with an increased rate of VTEs. Of note, anesthesia type was not significantly associated with occurrence of VTE on bivariate or multivariate analysis.
The C statistic of the final multivariate model was 0.76, indicating very good distinguishing ability. The Hosmer-Lemeshow goodness-of-fit test showed no evidence of lack of fit.
Discussion
Citing the lack of conclusive evidence and the low incidence of VTE after ankle fracture surgery, current recommendations are to avoid routine VTE prophylaxis in the postoperative management of patients who undergo this surgery.1,5 However, it is important to identify patients who are at increased risk, as some may benefit from VTE prophylaxis. In the present study, we used the large, high-quality ACS-NSQIP database collecting information from multiple US hospitals to examine risk factors for VTE after ankle fracture ORIF. We identified 4412 patients who underwent ankle fracture ORIF between 2005 and 2012, and found an overall VTE incidence of 0.8%. Multivariate analysis identified obesity, history of heart disease, and dependent functional status as independent risk factors for VTE after ankle fracture ORIF.
This study’s 0.8% incidence of VTE after ankle fracture ORIF is consistent with the range (0.29%-5%) reported in other ankle fracture studies.4,8-10,14-18 We found that VTEs occurred a mean of about 11 days after surgery, and no patient died of VTE.
Obesity (BMI, ≥30 kg/m2) had the strongest association with VTEs in this study. Obesity, which is a growing public health concern, can make postoperative care and mobilization more difficult.19 Obesity has previously been associated with VTEs after ankle fractures, and BMI of over 25 kg/m2 is one of the Caprini criteria for thrombosis risk factor assessment.6,10 In our study, however, BMI of 25 to 30 kg/m2 was not associated with an increased VTE rate, indicating that moderately overweight patients may not be at significantly higher risk for VTE (compared with patients with normal BMI) and may not need VTE prophylaxis. VTE prophylaxis after ankle fracture surgery may be considered in patients with BMI over 30 kg/m2.
History of heart disease was also associated with VTEs in this study. Patients with a history of heart disease were at 3 times the risk for VTE within 30 days of ankle fracture surgery. This association is also consistent with the Caprini criteria, which include acute myocardial infarction and CHF as risk factors for venous thrombosis.6 Other studies have found associations between CHF and VTE and between cardiovascular risk factors and VTE.7,20 The association between cardiovascular disease and VTE may derive from the decreased venous flow rate associated with CHF or an overall vascular disease state. These patients may benefit from heightened surveillance and postoperative prophylaxis for VTE.
Dependent functional status was the final risk factor found to be associated with VTE after ankle fracture ORIF. This association likely derives from an inability to mobilize independently, leading to increased venous stasis. Immobilization has been previously associated with increased risk for VTE after ankle surgery.7,14,16,20 Caretakers should be aware of this increased risk during the postoperative period and diligently monitor these patients for signs and symptoms of VTE. Prophylaxis may also be considered in this patient population.
Several risk factors that were significant on bivariate analysis (increased age; increased ASA class; history of diabetes, pulmonary disease, hypertension) were not significant in the final multivariate model. This finding suggests covariance between these factors and those that were significant in the final multivariate model. In particular, age and increased overall comorbidity (represented by increased ASA class) were not significant in our multivariate model—contrary to findings of other studies.8-10 It is possible that history of heart disease alone was responsible for the association between overall comorbidity and VTE in those studies. In the present study, separating and controlling for individual comorbidities could have allowed this association to be more precisely characterized.
The characteristics of the ACS-NSQIP database limited our study in several ways. First, although ACS-NSQIP makes significant efforts to collect as many patient variables as possible, some information is not captured. Data about additional factors that may affect VTE risk (eg, history of previous VTE, hypercoagulable state, history of malignancy other than disseminated cancer, tourniquet time, patient position in operating room) were not available. Second, data are collected only on those postoperative adverse events that occur within 30 days after surgery; data on VTEs that occur later are not captured. However, it has been shown that the majority of VTEs occur within the first 30 days after lower extremity trauma and surgery,21,22 so this follow-up interval was deemed adequate for capture of VTE data. Third, the database does not include information on the prophylactic regimens used for these patients—which may have weakened the associations between predictor variables and VTE risk and led to an underestimated effect size. VTE incidence, as well as the odds of developing a VTE with one of the identified risk factors, may actually be higher than reported in this study.
Conclusion
VTEs are serious complications that can occur after ORIF of ankle fractures. In this study, the overall incidence of VTE after ankle fracture ORIF was 0.8%. Although the American Orthopaedic Foot and Ankle Society and the American College of Chest Physicians do not recommend routine screening or prophylaxis for VTE in patients with ankle fractures,1,5 the results of this study showed there may be a benefit in emphasizing VTE prophylaxis after ankle fracture ORIF in patients with obesity, history of heart disease, or dependent functional status. At minimum, these patients should be more carefully monitored for development of VTEs.
1. American Orthopaedic Foot and Ankle Society. Position statement: the use of VTED prophylaxis in foot and ankle surgery. http://www.aofas.org/medical-community/health-policy/Documents/VTED-Position-Statement-Approv-7-9-13-FINAL.pdf. Updated 2013. Accessed May 10, 2015.
2. Grady-Benson JC, Oishi CS, Hanson PB, Colwell CW Jr, Otis SM, Walker RH. Routine postoperative duplex ultrasonography screening and monitoring for the detection of deep vein thrombosis. A survey of 110 total hip arthroplasties. Clin Orthop Relat Res. 1994;(307):130-141.
3. Salzman EW, Harris WH, DeSanctis RW. Anticoagulation for prevention of thromboembolism following fractures of the hip. New Engl J Med. 1966;275(3):122-130.
4. Patil S, Gandhi J, Curzon I, Hui AC. Incidence of deep-vein thrombosis in patients with fractures of the ankle treated in a plaster cast. J Bone Joint Surg Br. 2007;89(10):1340-1343.
5. Falck-Ytter Y, Francis CW, Johanson NA, et al; American College of Chest Physicians. Prevention of VTE in orthopedic surgery patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 suppl):e278S-e325S.
6. Caprini JA. Thrombosis risk assessment as a guide to quality patient care. Dis Mon. 2005;51(2-3):70-78.
7. Mayle RE Jr, DiGiovanni CW, Lin SS, Tabrizi P, Chou LB. Current concepts review: venous thromboembolic disease in foot and ankle surgery. Foot Ankle Int. 2007;28(11):1207-1216.
8. Jameson SS, Augustine A, James P, et al. Venous thromboembolic events following foot and ankle surgery in the English National Health Service. J Bone Joint Surg Br. 2011;93(4):490-497.
9. SooHoo NF, Eagan M, Krenek L, Zingmond DS. Incidence and factors predicting pulmonary embolism and deep venous thrombosis following surgical treatment of ankle fractures. Foot Ankle Surg. 2011;17(4):259-262.
10. Shibuya N, Frost CH, Campbell JD, Davis ML, Jupiter DC. Incidence of acute deep vein thrombosis and pulmonary embolism in foot and ankle trauma: analysis of the National Trauma Data Bank. J Foot Ankle Surg. 2012;51(1):63-68.
11. American College of Surgeons National Surgical Quality Improvement Program. User Guide for the 2012 ACS NSQIP Participant Use Data File. http://site.acsnsqip.org/wp-content/uploads/2013/10/ACSNSQIP.PUF_.UserGuide.2012.pdf. Published October 2013. Accessed May 10, 2015.
12. Khuri SF, Henderson WG, Daley J, et al; Principal Investigators of Patient Safety in Surgery Study. Successful implementation of the Department of Veterans Affairs’ National Surgical Quality Improvement Program in the private sector: the Patient Safety in Surgery study. Ann Surg. 2008;248(2):329-336.
13. Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. The disease burden associated with overweight and obesity. JAMA. 1999;282(16):1523-1529.
14. Mizel MS, Temple HT, Michelson JD, et al. Thromboembolism after foot and ankle surgery. A multicenter study. Clin Orthop Relat Res. 1998;(348):180-185.
15. Solis G, Saxby T. Incidence of DVT following surgery of the foot and ankle. Foot Ankle Int. 2002;23(5):411-414.
16. Hanslow SS, Grujic L, Slater HK, Chen D. Thromboembolic disease after foot and ankle surgery. Foot Ankle Int. 2006;27(9):693-695.
17. Pelet S, Roger ME, Belzile EL, Bouchard M. The incidence of thromboembolic events in surgically treated ankle fracture. J Bone Joint Surg Am. 2012;94(6):502-506.
18. Manafi Rasi A, Kazemian G, Emami Moghadam M, et al. Deep vein thrombosis following below knee immobilization: the need for chemoprophylaxis. Trauma Mon. 2013;17(4):367-369.
19. Sabharwal S, Root MZ. Impact of obesity on orthopaedics. J Bone Joint Surg Am. 2012;94(11):1045-1052.
20. Kadous A, Abdelgawad AA, Kanlic E. Deep venous thrombosis and pulmonary embolism after surgical treatment of ankle fractures: a case report and review of literature. J Foot Ankle Surg. 2012;51(4):457-463.
21. Forsythe RM, Peitzman AB, DeCato T, et al. Early lower extremity fracture fixation and the risk of early pulmonary embolus: filter before fixation? J Trauma. 2011;70(6):1381-1388.
22. Bjørnarå BT, Gudmundsen TE, Dahl OE. Frequency and timing of clinical venous thromboembolism after major joint surgery. J Bone Joint Surg Br. 2006;88(3):386-391.
1. American Orthopaedic Foot and Ankle Society. Position statement: the use of VTED prophylaxis in foot and ankle surgery. http://www.aofas.org/medical-community/health-policy/Documents/VTED-Position-Statement-Approv-7-9-13-FINAL.pdf. Updated 2013. Accessed May 10, 2015.
2. Grady-Benson JC, Oishi CS, Hanson PB, Colwell CW Jr, Otis SM, Walker RH. Routine postoperative duplex ultrasonography screening and monitoring for the detection of deep vein thrombosis. A survey of 110 total hip arthroplasties. Clin Orthop Relat Res. 1994;(307):130-141.
3. Salzman EW, Harris WH, DeSanctis RW. Anticoagulation for prevention of thromboembolism following fractures of the hip. New Engl J Med. 1966;275(3):122-130.
4. Patil S, Gandhi J, Curzon I, Hui AC. Incidence of deep-vein thrombosis in patients with fractures of the ankle treated in a plaster cast. J Bone Joint Surg Br. 2007;89(10):1340-1343.
5. Falck-Ytter Y, Francis CW, Johanson NA, et al; American College of Chest Physicians. Prevention of VTE in orthopedic surgery patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 suppl):e278S-e325S.
6. Caprini JA. Thrombosis risk assessment as a guide to quality patient care. Dis Mon. 2005;51(2-3):70-78.
7. Mayle RE Jr, DiGiovanni CW, Lin SS, Tabrizi P, Chou LB. Current concepts review: venous thromboembolic disease in foot and ankle surgery. Foot Ankle Int. 2007;28(11):1207-1216.
8. Jameson SS, Augustine A, James P, et al. Venous thromboembolic events following foot and ankle surgery in the English National Health Service. J Bone Joint Surg Br. 2011;93(4):490-497.
9. SooHoo NF, Eagan M, Krenek L, Zingmond DS. Incidence and factors predicting pulmonary embolism and deep venous thrombosis following surgical treatment of ankle fractures. Foot Ankle Surg. 2011;17(4):259-262.
10. Shibuya N, Frost CH, Campbell JD, Davis ML, Jupiter DC. Incidence of acute deep vein thrombosis and pulmonary embolism in foot and ankle trauma: analysis of the National Trauma Data Bank. J Foot Ankle Surg. 2012;51(1):63-68.
11. American College of Surgeons National Surgical Quality Improvement Program. User Guide for the 2012 ACS NSQIP Participant Use Data File. http://site.acsnsqip.org/wp-content/uploads/2013/10/ACSNSQIP.PUF_.UserGuide.2012.pdf. Published October 2013. Accessed May 10, 2015.
12. Khuri SF, Henderson WG, Daley J, et al; Principal Investigators of Patient Safety in Surgery Study. Successful implementation of the Department of Veterans Affairs’ National Surgical Quality Improvement Program in the private sector: the Patient Safety in Surgery study. Ann Surg. 2008;248(2):329-336.
13. Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. The disease burden associated with overweight and obesity. JAMA. 1999;282(16):1523-1529.
14. Mizel MS, Temple HT, Michelson JD, et al. Thromboembolism after foot and ankle surgery. A multicenter study. Clin Orthop Relat Res. 1998;(348):180-185.
15. Solis G, Saxby T. Incidence of DVT following surgery of the foot and ankle. Foot Ankle Int. 2002;23(5):411-414.
16. Hanslow SS, Grujic L, Slater HK, Chen D. Thromboembolic disease after foot and ankle surgery. Foot Ankle Int. 2006;27(9):693-695.
17. Pelet S, Roger ME, Belzile EL, Bouchard M. The incidence of thromboembolic events in surgically treated ankle fracture. J Bone Joint Surg Am. 2012;94(6):502-506.
18. Manafi Rasi A, Kazemian G, Emami Moghadam M, et al. Deep vein thrombosis following below knee immobilization: the need for chemoprophylaxis. Trauma Mon. 2013;17(4):367-369.
19. Sabharwal S, Root MZ. Impact of obesity on orthopaedics. J Bone Joint Surg Am. 2012;94(11):1045-1052.
20. Kadous A, Abdelgawad AA, Kanlic E. Deep venous thrombosis and pulmonary embolism after surgical treatment of ankle fractures: a case report and review of literature. J Foot Ankle Surg. 2012;51(4):457-463.
21. Forsythe RM, Peitzman AB, DeCato T, et al. Early lower extremity fracture fixation and the risk of early pulmonary embolus: filter before fixation? J Trauma. 2011;70(6):1381-1388.
22. Bjørnarå BT, Gudmundsen TE, Dahl OE. Frequency and timing of clinical venous thromboembolism after major joint surgery. J Bone Joint Surg Br. 2006;88(3):386-391.