User login
Impatient patients
Patients are often impatient. They want answers.
To some extent, I can’t blame them. When it’s your disease, you want to know what’s going on and what you can do about it. So I try to keep on top of results as they come in and have my staff contact people to relay the news.
The problem is that medicine (like life) does not provide immediate gratification. It takes time to get routine labs back, and some (such as send-outs) can even take a few weeks.
Radiology reports usually have a 24-hour turnaround, and radiologists will call me if they find something urgent. Yet, it’s amazing how many people will call for results before they even leave that facility.
Did it always used to be like this? Were people always this demanding of immediate answers and test results from their doctors?
We live in a world that gets faster and faster, and people get used to things happening quickly. It’s an age of instant gratification, and having to wait for test results seems silly to laypeople. After all, don’t TV medical shows have results coming back quickly, gleaming advanced scanners, and the machine that goes “ping”? So why doesn’t that happen when you visit a doctor in real life?
Of course, I could get the results faster. I could order everything STAT and abuse the privilege ... but crying wolf only works a few times, and then you can’t do it when you really need it. I could call the radiologists for verbal MRI reads ... but then I’m taking their time away from more urgent cases, and other patients with more concerning issues are affected. So I don’t do that routinely, either.
Even people in slow-moving lines of work can have trouble grasping that medicine is the same way. I tell them we’ll call them when we get results, and try to stay on top of things. I admit sometimes things may slip through, and they’re right to call and ask.
Most patients understand this, and are, well, patient. I just wish more were. It would save a lot of time, effort, and frustration for all involved, including them.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Patients are often impatient. They want answers.
To some extent, I can’t blame them. When it’s your disease, you want to know what’s going on and what you can do about it. So I try to keep on top of results as they come in and have my staff contact people to relay the news.
The problem is that medicine (like life) does not provide immediate gratification. It takes time to get routine labs back, and some (such as send-outs) can even take a few weeks.
Radiology reports usually have a 24-hour turnaround, and radiologists will call me if they find something urgent. Yet, it’s amazing how many people will call for results before they even leave that facility.
Did it always used to be like this? Were people always this demanding of immediate answers and test results from their doctors?
We live in a world that gets faster and faster, and people get used to things happening quickly. It’s an age of instant gratification, and having to wait for test results seems silly to laypeople. After all, don’t TV medical shows have results coming back quickly, gleaming advanced scanners, and the machine that goes “ping”? So why doesn’t that happen when you visit a doctor in real life?
Of course, I could get the results faster. I could order everything STAT and abuse the privilege ... but crying wolf only works a few times, and then you can’t do it when you really need it. I could call the radiologists for verbal MRI reads ... but then I’m taking their time away from more urgent cases, and other patients with more concerning issues are affected. So I don’t do that routinely, either.
Even people in slow-moving lines of work can have trouble grasping that medicine is the same way. I tell them we’ll call them when we get results, and try to stay on top of things. I admit sometimes things may slip through, and they’re right to call and ask.
Most patients understand this, and are, well, patient. I just wish more were. It would save a lot of time, effort, and frustration for all involved, including them.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Patients are often impatient. They want answers.
To some extent, I can’t blame them. When it’s your disease, you want to know what’s going on and what you can do about it. So I try to keep on top of results as they come in and have my staff contact people to relay the news.
The problem is that medicine (like life) does not provide immediate gratification. It takes time to get routine labs back, and some (such as send-outs) can even take a few weeks.
Radiology reports usually have a 24-hour turnaround, and radiologists will call me if they find something urgent. Yet, it’s amazing how many people will call for results before they even leave that facility.
Did it always used to be like this? Were people always this demanding of immediate answers and test results from their doctors?
We live in a world that gets faster and faster, and people get used to things happening quickly. It’s an age of instant gratification, and having to wait for test results seems silly to laypeople. After all, don’t TV medical shows have results coming back quickly, gleaming advanced scanners, and the machine that goes “ping”? So why doesn’t that happen when you visit a doctor in real life?
Of course, I could get the results faster. I could order everything STAT and abuse the privilege ... but crying wolf only works a few times, and then you can’t do it when you really need it. I could call the radiologists for verbal MRI reads ... but then I’m taking their time away from more urgent cases, and other patients with more concerning issues are affected. So I don’t do that routinely, either.
Even people in slow-moving lines of work can have trouble grasping that medicine is the same way. I tell them we’ll call them when we get results, and try to stay on top of things. I admit sometimes things may slip through, and they’re right to call and ask.
Most patients understand this, and are, well, patient. I just wish more were. It would save a lot of time, effort, and frustration for all involved, including them.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.

ACS: No pull-out pneumothorax with ‘party balloon Valsalva’
CHICAGO – Investigators have come up with a simple way to reduce and maybe even eliminate pull-out pneumothoraces during chest tube removal.
Instead of standard inhale or exhale Valsalva maneuvers, they have their patients blow up a party balloon as the tube is pulled.
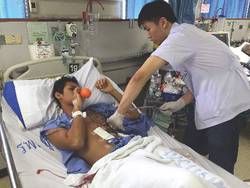
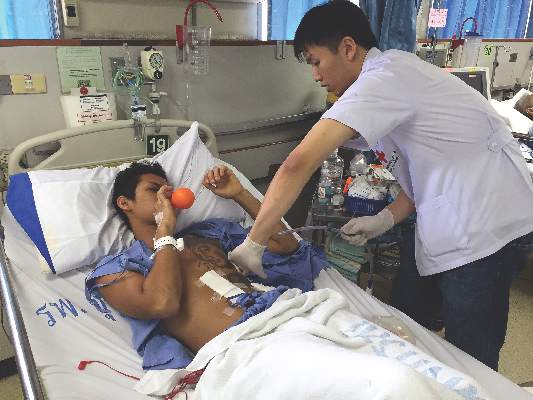
That produces the same Valsalva effects as the standard maneuvers, but with two significant advantages. First, it’s easy to explain and for patients to understand and do – not much more instruction is required than “blow up the balloon” – and, secondly, the inflating balloon is a visual check to make sure patients are doing the maneuver correctly. “It’s easy. Everyone can do it,” said lead investigator Dr. Puwadon Thitivaraporn, who developed the technique with Dr. Kritaya Kritayakirana and colleagues at King Chulalongkorn Memorial Hospital in Bangkok, Thailand.
To see how well it works, the team randomized 10 women and 38 men about equally to four removal techniques: the standard expire Valsalva, the standard inspire Valsalva, and two balloon maneuvers – blowing the balloon up after a deep breath and blowing it up with residual lung volume after an initial exhalation.
The subjects were trauma patients 15-64 years old, with a mean age of 38 years. Lung injuries, rib fractures, and tube suction were a bit more common in the standard maneuver groups. Patients with tracheotomies, chronic lung disease, and Glasgow Coma Scores below 13 were excluded from the study. Hemopneumothorax was the most common indication for tube placement.

Two patients in each of the standard groups (16%) developed a pull-out pneumothorax within 24 hours of tube removal, confirmed by x-ray. One required chest tube reinsertion, and all four ended up spending extra time in the hospital. Similar problems have been reported in American medicine (J Trauma. 2001 Apr;50[4]:674-7).
Meanwhile, not a single balloon patient had a lung collapse when their tube was pulled.
Because of the small number of subjects, the differences weren’t statistically significant, but they came close in a group comparison of standard patients with balloon patients (P = .11). The investigators estimated they would need almost 600 hundred subjects to reach statistical significance.
Even so, the party balloon technique appears to be “easier and safer” than standard maneuvers, as well as “reproducible and cheap, and it can prevent recurrent pneumothorax. It can be used as an alternative to the classic Valsalva,” said Dr. Thitivaraporn, a cardiothoracic surgery resident at the Bangkok hospital.
The balloon method is being used there now in nontrauma patients, as well, but the standard maneuvers are also being used until the balloon technique shows statistically significant benefits, he said.
With manometry, the team found that a party balloon’s internal pressure builds quickly as it’s inflated from a starting diameter of about 4.5 cm to about 9 cm, peaking at about 60 mm Hg; pressure trails off to about 40 mm Hg as inflation continues past 9 cm.
The investigators have no relevant disclosures.
CHICAGO – Investigators have come up with a simple way to reduce and maybe even eliminate pull-out pneumothoraces during chest tube removal.
Instead of standard inhale or exhale Valsalva maneuvers, they have their patients blow up a party balloon as the tube is pulled.
That produces the same Valsalva effects as the standard maneuvers, but with two significant advantages. First, it’s easy to explain and for patients to understand and do – not much more instruction is required than “blow up the balloon” – and, secondly, the inflating balloon is a visual check to make sure patients are doing the maneuver correctly. “It’s easy. Everyone can do it,” said lead investigator Dr. Puwadon Thitivaraporn, who developed the technique with Dr. Kritaya Kritayakirana and colleagues at King Chulalongkorn Memorial Hospital in Bangkok, Thailand.
To see how well it works, the team randomized 10 women and 38 men about equally to four removal techniques: the standard expire Valsalva, the standard inspire Valsalva, and two balloon maneuvers – blowing the balloon up after a deep breath and blowing it up with residual lung volume after an initial exhalation.
The subjects were trauma patients 15-64 years old, with a mean age of 38 years. Lung injuries, rib fractures, and tube suction were a bit more common in the standard maneuver groups. Patients with tracheotomies, chronic lung disease, and Glasgow Coma Scores below 13 were excluded from the study. Hemopneumothorax was the most common indication for tube placement.
Two patients in each of the standard groups (16%) developed a pull-out pneumothorax within 24 hours of tube removal, confirmed by x-ray. One required chest tube reinsertion, and all four ended up spending extra time in the hospital. Similar problems have been reported in American medicine (J Trauma. 2001 Apr;50[4]:674-7).
Meanwhile, not a single balloon patient had a lung collapse when their tube was pulled.
Because of the small number of subjects, the differences weren’t statistically significant, but they came close in a group comparison of standard patients with balloon patients (P = .11). The investigators estimated they would need almost 600 hundred subjects to reach statistical significance.
Even so, the party balloon technique appears to be “easier and safer” than standard maneuvers, as well as “reproducible and cheap, and it can prevent recurrent pneumothorax. It can be used as an alternative to the classic Valsalva,” said Dr. Thitivaraporn, a cardiothoracic surgery resident at the Bangkok hospital.
The balloon method is being used there now in nontrauma patients, as well, but the standard maneuvers are also being used until the balloon technique shows statistically significant benefits, he said.
With manometry, the team found that a party balloon’s internal pressure builds quickly as it’s inflated from a starting diameter of about 4.5 cm to about 9 cm, peaking at about 60 mm Hg; pressure trails off to about 40 mm Hg as inflation continues past 9 cm.
The investigators have no relevant disclosures.
CHICAGO – Investigators have come up with a simple way to reduce and maybe even eliminate pull-out pneumothoraces during chest tube removal.
Instead of standard inhale or exhale Valsalva maneuvers, they have their patients blow up a party balloon as the tube is pulled.
That produces the same Valsalva effects as the standard maneuvers, but with two significant advantages. First, it’s easy to explain and for patients to understand and do – not much more instruction is required than “blow up the balloon” – and, secondly, the inflating balloon is a visual check to make sure patients are doing the maneuver correctly. “It’s easy. Everyone can do it,” said lead investigator Dr. Puwadon Thitivaraporn, who developed the technique with Dr. Kritaya Kritayakirana and colleagues at King Chulalongkorn Memorial Hospital in Bangkok, Thailand.
To see how well it works, the team randomized 10 women and 38 men about equally to four removal techniques: the standard expire Valsalva, the standard inspire Valsalva, and two balloon maneuvers – blowing the balloon up after a deep breath and blowing it up with residual lung volume after an initial exhalation.
The subjects were trauma patients 15-64 years old, with a mean age of 38 years. Lung injuries, rib fractures, and tube suction were a bit more common in the standard maneuver groups. Patients with tracheotomies, chronic lung disease, and Glasgow Coma Scores below 13 were excluded from the study. Hemopneumothorax was the most common indication for tube placement.
Two patients in each of the standard groups (16%) developed a pull-out pneumothorax within 24 hours of tube removal, confirmed by x-ray. One required chest tube reinsertion, and all four ended up spending extra time in the hospital. Similar problems have been reported in American medicine (J Trauma. 2001 Apr;50[4]:674-7).
Meanwhile, not a single balloon patient had a lung collapse when their tube was pulled.
Because of the small number of subjects, the differences weren’t statistically significant, but they came close in a group comparison of standard patients with balloon patients (P = .11). The investigators estimated they would need almost 600 hundred subjects to reach statistical significance.
Even so, the party balloon technique appears to be “easier and safer” than standard maneuvers, as well as “reproducible and cheap, and it can prevent recurrent pneumothorax. It can be used as an alternative to the classic Valsalva,” said Dr. Thitivaraporn, a cardiothoracic surgery resident at the Bangkok hospital.
The balloon method is being used there now in nontrauma patients, as well, but the standard maneuvers are also being used until the balloon technique shows statistically significant benefits, he said.
With manometry, the team found that a party balloon’s internal pressure builds quickly as it’s inflated from a starting diameter of about 4.5 cm to about 9 cm, peaking at about 60 mm Hg; pressure trails off to about 40 mm Hg as inflation continues past 9 cm.
The investigators have no relevant disclosures.
AT THE ACS CLINICAL CONGRESS
Key clinical point: The next time you pull a chest tube, you might want to ask your patient to blow up a balloon.
Major finding: Sixteen percent of patients collapsed a lung with classic inhale/exhale Valsalva maneuvers during chest tube removal, but none did with the balloon technique.
Data source: Randomized, controlled trial of 48 chest tube patients.
Disclosures: The investigators have no relevant disclosures.
Disparity found in PPI risk perception among physicians
HONOLULU – A survey of almost 500 physicians found that primary care physicians (PCPs) are far more concerned about the reported adverse effects of proton pump inhibitors (PPIs) than are gastroenterologists and use them more sparingly. The results of the survey were presented at the 2015 American College of Gastroenterology (ACG) Annual Scientific Meeting and Postgraduate Course.
“We asked physicians about a broad array of adverse effects from long-term use of PPIs and PCPs expressed greater concern for all of them,” reported Dr. Samir Kapadia, division of gastroenterology and hepatology, State University of New York at Stony Brook. “Alternatively, significantly more gastroenterologists responded that they really had no concerns for any of these adverse effects.”

The evidence may be on the side of the gastroenterologists, according to Dr. Kapadia. Although PPIs have been associated with hypomagnesemia, iron deficiency, vitamin B12 deficiency, diarrhea caused by Clostridium difficile infection, and interactions with the platelet inhibitor clopidogrel, Dr. Kapadia noted that few associations have been made on the basis of prospective trials.
“Much of the available literature is observational or based on studies that are heterogeneous and small,” Dr. Kapadia. “Confounding factors in these studies also limit interpretation.”
In this study for which surveys are still being collected, a 19-item questionnaire was distributed to 384 gastroenterologists and 88 PCPs. In addition to demographic information, the surveys were designed to capture opinions about the safety of PPIs as well as elicit information about how these agents are being used in clinical practice.
Of side effects associated with PPIs, significantly more PCPs than gastroenterologists expressed concern about hypomagnesemia (41.7% vs. 6.3%; P less than .001), iron deficiency (33.3% vs. 11.4%; P = .014) and vitamin B12 deficiency (47.6% vs. 17.3%; P = .005). From the other perspective, when asked about their concern for these and other safety issues, the answer was “none of the above” for 26.2% of PCPs and 67.1% of gastroenterologists (P less than .001).
When given specific risk scenarios, PCPs were consistently more prepared to discontinue PPI therapy than were gastroenterologists. For example, in a hypothetical 65-year-old with GERD symptoms expressing concern about risk of hip fracture, 64.5% of PCPs vs. 30.7% of gastroenterologists (P less than .001) responded that they would discontinue the PPI. In a patient of the same age about to start broad-spectrum antibiotics for cellulitis, 16.1% of PCPs, but only 4.3% of gastroenterologists (P = .001) reported that they would discontinue PPIs. Conversely, 68.5% of gastroenterologists vs. 54.2% of PCPs (P = .028) would continue therapy.
For a hypothetical 65-year-old with symptomatic gastroesophageal reflux disease (GERD) initiating clopidogrel, 50% of PCPs vs. 27.6% of gastroenterologists (P = .001) would switch to an H2-receptor antagonist. Only 27.3% of PCPs vs. 46.4% of gastroenterologists (P = .001) would continue the PPI. When the age of the hypothetical patient is raised to 75 years, PCPs, but not gastroenterologists, were even more likely to discontinue PPI therapy.
Using PPIs appropriately is an important goal, Dr. Kapadia emphasized. However, he suggested that many warnings about the risks of PPIs, including those issued by the Food and Drug Administration, are incompletely substantiated and are not being evaluated with an appropriate attention to benefit-to-risk ratio of a drug that not only controls symptoms but may also reduce risk of GI bleeding. Others share this point of view.
“The pendulum has moved too far in regard to the fear of potential side effects,” agreed Dr. Philip Katz, chairman, division of gastroenterology, Albert Einstein Medical Center, Philadelphia. First author of the 2013 ACG guidelines on GERD, which addresses the safety of PPIs (Am J Gastroenterol. 2013;108:308-28), Dr. Katz said in an interview that the data generated by this survey suggest that PCPs are misinterpreting the relative risks and need to be given more information about indications in which benefits are well established.
Making the same point, Dr. Nicholas J. Shaheen, chief, division of gastroenterology and hepatology, University of North Carolina, Chapel Hill, suggested “This may be a failure on our part [as gastroenterologists] to educate our colleagues about the role of these drugs.”
Dr. Kapadia reported no potential conflicts.
HONOLULU – A survey of almost 500 physicians found that primary care physicians (PCPs) are far more concerned about the reported adverse effects of proton pump inhibitors (PPIs) than are gastroenterologists and use them more sparingly. The results of the survey were presented at the 2015 American College of Gastroenterology (ACG) Annual Scientific Meeting and Postgraduate Course.
“We asked physicians about a broad array of adverse effects from long-term use of PPIs and PCPs expressed greater concern for all of them,” reported Dr. Samir Kapadia, division of gastroenterology and hepatology, State University of New York at Stony Brook. “Alternatively, significantly more gastroenterologists responded that they really had no concerns for any of these adverse effects.”
The evidence may be on the side of the gastroenterologists, according to Dr. Kapadia. Although PPIs have been associated with hypomagnesemia, iron deficiency, vitamin B12 deficiency, diarrhea caused by Clostridium difficile infection, and interactions with the platelet inhibitor clopidogrel, Dr. Kapadia noted that few associations have been made on the basis of prospective trials.
“Much of the available literature is observational or based on studies that are heterogeneous and small,” Dr. Kapadia. “Confounding factors in these studies also limit interpretation.”
In this study for which surveys are still being collected, a 19-item questionnaire was distributed to 384 gastroenterologists and 88 PCPs. In addition to demographic information, the surveys were designed to capture opinions about the safety of PPIs as well as elicit information about how these agents are being used in clinical practice.
Of side effects associated with PPIs, significantly more PCPs than gastroenterologists expressed concern about hypomagnesemia (41.7% vs. 6.3%; P less than .001), iron deficiency (33.3% vs. 11.4%; P = .014) and vitamin B12 deficiency (47.6% vs. 17.3%; P = .005). From the other perspective, when asked about their concern for these and other safety issues, the answer was “none of the above” for 26.2% of PCPs and 67.1% of gastroenterologists (P less than .001).
When given specific risk scenarios, PCPs were consistently more prepared to discontinue PPI therapy than were gastroenterologists. For example, in a hypothetical 65-year-old with GERD symptoms expressing concern about risk of hip fracture, 64.5% of PCPs vs. 30.7% of gastroenterologists (P less than .001) responded that they would discontinue the PPI. In a patient of the same age about to start broad-spectrum antibiotics for cellulitis, 16.1% of PCPs, but only 4.3% of gastroenterologists (P = .001) reported that they would discontinue PPIs. Conversely, 68.5% of gastroenterologists vs. 54.2% of PCPs (P = .028) would continue therapy.
For a hypothetical 65-year-old with symptomatic gastroesophageal reflux disease (GERD) initiating clopidogrel, 50% of PCPs vs. 27.6% of gastroenterologists (P = .001) would switch to an H2-receptor antagonist. Only 27.3% of PCPs vs. 46.4% of gastroenterologists (P = .001) would continue the PPI. When the age of the hypothetical patient is raised to 75 years, PCPs, but not gastroenterologists, were even more likely to discontinue PPI therapy.
Using PPIs appropriately is an important goal, Dr. Kapadia emphasized. However, he suggested that many warnings about the risks of PPIs, including those issued by the Food and Drug Administration, are incompletely substantiated and are not being evaluated with an appropriate attention to benefit-to-risk ratio of a drug that not only controls symptoms but may also reduce risk of GI bleeding. Others share this point of view.
“The pendulum has moved too far in regard to the fear of potential side effects,” agreed Dr. Philip Katz, chairman, division of gastroenterology, Albert Einstein Medical Center, Philadelphia. First author of the 2013 ACG guidelines on GERD, which addresses the safety of PPIs (Am J Gastroenterol. 2013;108:308-28), Dr. Katz said in an interview that the data generated by this survey suggest that PCPs are misinterpreting the relative risks and need to be given more information about indications in which benefits are well established.
Making the same point, Dr. Nicholas J. Shaheen, chief, division of gastroenterology and hepatology, University of North Carolina, Chapel Hill, suggested “This may be a failure on our part [as gastroenterologists] to educate our colleagues about the role of these drugs.”
Dr. Kapadia reported no potential conflicts.
HONOLULU – A survey of almost 500 physicians found that primary care physicians (PCPs) are far more concerned about the reported adverse effects of proton pump inhibitors (PPIs) than are gastroenterologists and use them more sparingly. The results of the survey were presented at the 2015 American College of Gastroenterology (ACG) Annual Scientific Meeting and Postgraduate Course.
“We asked physicians about a broad array of adverse effects from long-term use of PPIs and PCPs expressed greater concern for all of them,” reported Dr. Samir Kapadia, division of gastroenterology and hepatology, State University of New York at Stony Brook. “Alternatively, significantly more gastroenterologists responded that they really had no concerns for any of these adverse effects.”
The evidence may be on the side of the gastroenterologists, according to Dr. Kapadia. Although PPIs have been associated with hypomagnesemia, iron deficiency, vitamin B12 deficiency, diarrhea caused by Clostridium difficile infection, and interactions with the platelet inhibitor clopidogrel, Dr. Kapadia noted that few associations have been made on the basis of prospective trials.
“Much of the available literature is observational or based on studies that are heterogeneous and small,” Dr. Kapadia. “Confounding factors in these studies also limit interpretation.”
In this study for which surveys are still being collected, a 19-item questionnaire was distributed to 384 gastroenterologists and 88 PCPs. In addition to demographic information, the surveys were designed to capture opinions about the safety of PPIs as well as elicit information about how these agents are being used in clinical practice.
Of side effects associated with PPIs, significantly more PCPs than gastroenterologists expressed concern about hypomagnesemia (41.7% vs. 6.3%; P less than .001), iron deficiency (33.3% vs. 11.4%; P = .014) and vitamin B12 deficiency (47.6% vs. 17.3%; P = .005). From the other perspective, when asked about their concern for these and other safety issues, the answer was “none of the above” for 26.2% of PCPs and 67.1% of gastroenterologists (P less than .001).
When given specific risk scenarios, PCPs were consistently more prepared to discontinue PPI therapy than were gastroenterologists. For example, in a hypothetical 65-year-old with GERD symptoms expressing concern about risk of hip fracture, 64.5% of PCPs vs. 30.7% of gastroenterologists (P less than .001) responded that they would discontinue the PPI. In a patient of the same age about to start broad-spectrum antibiotics for cellulitis, 16.1% of PCPs, but only 4.3% of gastroenterologists (P = .001) reported that they would discontinue PPIs. Conversely, 68.5% of gastroenterologists vs. 54.2% of PCPs (P = .028) would continue therapy.
For a hypothetical 65-year-old with symptomatic gastroesophageal reflux disease (GERD) initiating clopidogrel, 50% of PCPs vs. 27.6% of gastroenterologists (P = .001) would switch to an H2-receptor antagonist. Only 27.3% of PCPs vs. 46.4% of gastroenterologists (P = .001) would continue the PPI. When the age of the hypothetical patient is raised to 75 years, PCPs, but not gastroenterologists, were even more likely to discontinue PPI therapy.
Using PPIs appropriately is an important goal, Dr. Kapadia emphasized. However, he suggested that many warnings about the risks of PPIs, including those issued by the Food and Drug Administration, are incompletely substantiated and are not being evaluated with an appropriate attention to benefit-to-risk ratio of a drug that not only controls symptoms but may also reduce risk of GI bleeding. Others share this point of view.
“The pendulum has moved too far in regard to the fear of potential side effects,” agreed Dr. Philip Katz, chairman, division of gastroenterology, Albert Einstein Medical Center, Philadelphia. First author of the 2013 ACG guidelines on GERD, which addresses the safety of PPIs (Am J Gastroenterol. 2013;108:308-28), Dr. Katz said in an interview that the data generated by this survey suggest that PCPs are misinterpreting the relative risks and need to be given more information about indications in which benefits are well established.
Making the same point, Dr. Nicholas J. Shaheen, chief, division of gastroenterology and hepatology, University of North Carolina, Chapel Hill, suggested “This may be a failure on our part [as gastroenterologists] to educate our colleagues about the role of these drugs.”
Dr. Kapadia reported no potential conflicts.
FROM THE AMERICAN COLLEGE OF GASTROENTEROLOGY 2015 SCIENTIFIC MEETING AND POSTGRADUATE COURSE
Key clinical point: Primary care physicians used proton pump inhibitors more sparingly, were more concerned about reported adverse effects than were gastroenterologists, but are perhaps too cautious in the cost-benefit analysis.
Major finding: Primary care physicians (PCPs) are far more concerned about the reported adverse effects of proton pump inhibitors than are gastroenterologists.
Data source: A survey of nearly 500 physicians, weighted toward gastroenterologists.
Disclosures: Dr. Kapadia reported no potential conflicts of interest.

MicroRNA may be therapeutic target for MF

A Notch-related microRNA may be a therapeutic target for mycosis fungoides (MF), according to research published in the Journal of Investigative Dermatology.
The Notch pathway has been implicated in the progression of cutaneous T-cell lymphomas, but the mechanisms driving Notch activation has been unclear.
So investigators studied a series of skin samples from patients with MF in tumor phase, focusing on the Notch pathway.
“The purpose of this project has been to research the state of the Notch pathway in a series of samples from patients with mycosis fungoides and compare the results to a control group to discover if Notch activation in tumors is influenced by epigenetic modifications,” said Fernando Gallardo, MD, of Hospital del Mar Investigacions Mèdiques in Barcelona, Spain.
So he and his colleagues looked at methylation patterns in several components of the Notch pathway and confirmed that Notch1 was activated in samples from patients with MF.
They then identified a microRNA, miR-200C, that was epigenetically repressed in the samples. Further investigation revealed that this repression leads to the activation of the Notch pathway.
“The restoration of miR-200C expression, silenced in the tumor cells, could represent a potential therapeutic target for this subtype of lymphomas,” Dr Gallardo concluded. ![]()
A Notch-related microRNA may be a therapeutic target for mycosis fungoides (MF), according to research published in the Journal of Investigative Dermatology.
The Notch pathway has been implicated in the progression of cutaneous T-cell lymphomas, but the mechanisms driving Notch activation has been unclear.
So investigators studied a series of skin samples from patients with MF in tumor phase, focusing on the Notch pathway.
“The purpose of this project has been to research the state of the Notch pathway in a series of samples from patients with mycosis fungoides and compare the results to a control group to discover if Notch activation in tumors is influenced by epigenetic modifications,” said Fernando Gallardo, MD, of Hospital del Mar Investigacions Mèdiques in Barcelona, Spain.
So he and his colleagues looked at methylation patterns in several components of the Notch pathway and confirmed that Notch1 was activated in samples from patients with MF.
They then identified a microRNA, miR-200C, that was epigenetically repressed in the samples. Further investigation revealed that this repression leads to the activation of the Notch pathway.
“The restoration of miR-200C expression, silenced in the tumor cells, could represent a potential therapeutic target for this subtype of lymphomas,” Dr Gallardo concluded. ![]()
A Notch-related microRNA may be a therapeutic target for mycosis fungoides (MF), according to research published in the Journal of Investigative Dermatology.
The Notch pathway has been implicated in the progression of cutaneous T-cell lymphomas, but the mechanisms driving Notch activation has been unclear.
So investigators studied a series of skin samples from patients with MF in tumor phase, focusing on the Notch pathway.
“The purpose of this project has been to research the state of the Notch pathway in a series of samples from patients with mycosis fungoides and compare the results to a control group to discover if Notch activation in tumors is influenced by epigenetic modifications,” said Fernando Gallardo, MD, of Hospital del Mar Investigacions Mèdiques in Barcelona, Spain.
So he and his colleagues looked at methylation patterns in several components of the Notch pathway and confirmed that Notch1 was activated in samples from patients with MF.
They then identified a microRNA, miR-200C, that was epigenetically repressed in the samples. Further investigation revealed that this repression leads to the activation of the Notch pathway.
“The restoration of miR-200C expression, silenced in the tumor cells, could represent a potential therapeutic target for this subtype of lymphomas,” Dr Gallardo concluded. ![]()
Iron chelator tablets may now be crushed

Photo courtesy of the CDC
The US Food and Drug Administration (FDA) has approved a label change for Jadenu, an oral formulation of the iron chelator Exjade (deferasirox).
Jadenu comes in tablet form, and the previous label stated that Jadenu tablets must be swallowed whole.
Now, the medication can also be crushed to help simplify administration for patients who have difficulty swallowing whole tablets.
Jadenu tablets may be crushed and mixed with soft foods, such as yogurt or applesauce, immediately prior to use.
The label notes that commercial crushers with serrated surfaces should be avoided for crushing a single 90 mg tablet. The dose should be consumed immediately and not stored.
Jadenu was granted accelerated approval from the FDA earlier this year.
It is approved to treat patients 2 years of age and older who have chronic iron overload resulting from blood transfusions, as well as to treat chronic iron overload in patients 10 years of age and older who have non-transfusion-dependent thalassemia.
The full prescribing information for Jadenu can be found at http://www.pharma.us.novartis.com/product/pi/pdf/jadenu.pdf. ![]()
Photo courtesy of the CDC
The US Food and Drug Administration (FDA) has approved a label change for Jadenu, an oral formulation of the iron chelator Exjade (deferasirox).
Jadenu comes in tablet form, and the previous label stated that Jadenu tablets must be swallowed whole.
Now, the medication can also be crushed to help simplify administration for patients who have difficulty swallowing whole tablets.
Jadenu tablets may be crushed and mixed with soft foods, such as yogurt or applesauce, immediately prior to use.
The label notes that commercial crushers with serrated surfaces should be avoided for crushing a single 90 mg tablet. The dose should be consumed immediately and not stored.
Jadenu was granted accelerated approval from the FDA earlier this year.
It is approved to treat patients 2 years of age and older who have chronic iron overload resulting from blood transfusions, as well as to treat chronic iron overload in patients 10 years of age and older who have non-transfusion-dependent thalassemia.
The full prescribing information for Jadenu can be found at http://www.pharma.us.novartis.com/product/pi/pdf/jadenu.pdf. ![]()
Photo courtesy of the CDC
The US Food and Drug Administration (FDA) has approved a label change for Jadenu, an oral formulation of the iron chelator Exjade (deferasirox).
Jadenu comes in tablet form, and the previous label stated that Jadenu tablets must be swallowed whole.
Now, the medication can also be crushed to help simplify administration for patients who have difficulty swallowing whole tablets.
Jadenu tablets may be crushed and mixed with soft foods, such as yogurt or applesauce, immediately prior to use.
The label notes that commercial crushers with serrated surfaces should be avoided for crushing a single 90 mg tablet. The dose should be consumed immediately and not stored.
Jadenu was granted accelerated approval from the FDA earlier this year.
It is approved to treat patients 2 years of age and older who have chronic iron overload resulting from blood transfusions, as well as to treat chronic iron overload in patients 10 years of age and older who have non-transfusion-dependent thalassemia.
The full prescribing information for Jadenu can be found at http://www.pharma.us.novartis.com/product/pi/pdf/jadenu.pdf. ![]()
Technique enables SCD detection with a smartphone

Photo by Daniel Sone
Researchers say they’ve developed a simple technique for diagnosing and monitoring sickle cell disease (SCD) that could be used in regions where advanced medical technology and training are scarce.
The team created a 3D-printed box that can be attached to an Android smartphone and used to test a small blood sample.
The testing method involves magnetic levitation, which allows the user to differentiate sickle cells from normal red blood cells with the naked eye.
Savas Tasoglu, PhD, of the University of Connecticut in Storrs, and his colleagues described this technique in Nature Scientific Reports.
First, a clinician takes a blood sample from a patient and mixes it with a common, salt-based solution that draws oxygen out of sickle cells, making them denser and easier to detect via magnetic levitation. The denser sickle cells will float at a lower height than healthy red blood cells, which are not affected by the solution.
The sample is then loaded into a disposable micro-capillary that is inserted into the tester attached to the smartphone. Inside the testing apparatus, the micro-capillary passes between 2 magnets that are aligned so that the same poles face each other, creating a magnetic field.
The capillary is then illuminated with an LED that is filtered through a ground glass diffuser and magnified by an internal lens.
The smartphone’s built-in camera captures the resulting image and presents it digitally on the phone’s external display. The blood cells floating inside the capillary—whether higher-floating healthy red blood cells or lower-floating sickle cells—can be easily observed.
The device also provides clinicians with a digital readout that assigns a numerical value to the sample density to assist with the diagnosis. The entire process takes less than 15 minutes.
“With this device, you’re getting much more specific information about your cells than some other tests,” said Stephanie Knowlton, a graduate student at the University of Connecticut.
“Rather than sending a sample to a lab and waiting 3 days to find out if you have this disease, with this device, you get on-site and portable results right away. We believe a device like this could be very helpful in third-world countries where laboratory resources may be limited.”
Dr Tasoglu’s lab has filed a provisional patent for the device and is working on expanding its capabilities so it can be applied to other diseases. ![]()
Photo by Daniel Sone
Researchers say they’ve developed a simple technique for diagnosing and monitoring sickle cell disease (SCD) that could be used in regions where advanced medical technology and training are scarce.
The team created a 3D-printed box that can be attached to an Android smartphone and used to test a small blood sample.
The testing method involves magnetic levitation, which allows the user to differentiate sickle cells from normal red blood cells with the naked eye.
Savas Tasoglu, PhD, of the University of Connecticut in Storrs, and his colleagues described this technique in Nature Scientific Reports.
First, a clinician takes a blood sample from a patient and mixes it with a common, salt-based solution that draws oxygen out of sickle cells, making them denser and easier to detect via magnetic levitation. The denser sickle cells will float at a lower height than healthy red blood cells, which are not affected by the solution.
The sample is then loaded into a disposable micro-capillary that is inserted into the tester attached to the smartphone. Inside the testing apparatus, the micro-capillary passes between 2 magnets that are aligned so that the same poles face each other, creating a magnetic field.
The capillary is then illuminated with an LED that is filtered through a ground glass diffuser and magnified by an internal lens.
The smartphone’s built-in camera captures the resulting image and presents it digitally on the phone’s external display. The blood cells floating inside the capillary—whether higher-floating healthy red blood cells or lower-floating sickle cells—can be easily observed.
The device also provides clinicians with a digital readout that assigns a numerical value to the sample density to assist with the diagnosis. The entire process takes less than 15 minutes.
“With this device, you’re getting much more specific information about your cells than some other tests,” said Stephanie Knowlton, a graduate student at the University of Connecticut.
“Rather than sending a sample to a lab and waiting 3 days to find out if you have this disease, with this device, you get on-site and portable results right away. We believe a device like this could be very helpful in third-world countries where laboratory resources may be limited.”
Dr Tasoglu’s lab has filed a provisional patent for the device and is working on expanding its capabilities so it can be applied to other diseases. ![]()
Photo by Daniel Sone
Researchers say they’ve developed a simple technique for diagnosing and monitoring sickle cell disease (SCD) that could be used in regions where advanced medical technology and training are scarce.
The team created a 3D-printed box that can be attached to an Android smartphone and used to test a small blood sample.
The testing method involves magnetic levitation, which allows the user to differentiate sickle cells from normal red blood cells with the naked eye.
Savas Tasoglu, PhD, of the University of Connecticut in Storrs, and his colleagues described this technique in Nature Scientific Reports.
First, a clinician takes a blood sample from a patient and mixes it with a common, salt-based solution that draws oxygen out of sickle cells, making them denser and easier to detect via magnetic levitation. The denser sickle cells will float at a lower height than healthy red blood cells, which are not affected by the solution.
The sample is then loaded into a disposable micro-capillary that is inserted into the tester attached to the smartphone. Inside the testing apparatus, the micro-capillary passes between 2 magnets that are aligned so that the same poles face each other, creating a magnetic field.
The capillary is then illuminated with an LED that is filtered through a ground glass diffuser and magnified by an internal lens.
The smartphone’s built-in camera captures the resulting image and presents it digitally on the phone’s external display. The blood cells floating inside the capillary—whether higher-floating healthy red blood cells or lower-floating sickle cells—can be easily observed.
The device also provides clinicians with a digital readout that assigns a numerical value to the sample density to assist with the diagnosis. The entire process takes less than 15 minutes.
“With this device, you’re getting much more specific information about your cells than some other tests,” said Stephanie Knowlton, a graduate student at the University of Connecticut.
“Rather than sending a sample to a lab and waiting 3 days to find out if you have this disease, with this device, you get on-site and portable results right away. We believe a device like this could be very helpful in third-world countries where laboratory resources may be limited.”
Dr Tasoglu’s lab has filed a provisional patent for the device and is working on expanding its capabilities so it can be applied to other diseases. ![]()
Immunosuppressant can treat autoimmune cytopenias

New research suggests the immunosuppressant sirolimus may be a promising treatment option for patients with refractory autoimmune cytopenias.
The drug proved particularly effective in children with autoimmune lymphoproliferative syndrome (ALPS), producing complete responses in all of the ALPS patients studied.
On the other hand, patients with single-lineage autoimmune cytopenias, such as immune thrombocytopenia (ITP), did not fare as well.
David T. Teachey, MD, of The Children’s Hospital of Philadelphia in Pennsylvania, and his colleagues reported these results in Blood.
The group studied sirolimus in 30 patients with refractory autoimmune cytopenias who were 5 to 19 years of age. All of the patients were refractory to or could not tolerate corticosteroids.
Twelve patients had ALPS, 6 had single-lineage autoimmune cytopenias (4 with ITP and 2 with autoimmune hemolytic anemia [AIHA]), and 12 patients had multi-lineage cytopenias secondary to common variable immune deficiency (n=2), Evans syndrome (n=8), or systemic lupus erythematosus (n=2).
The patients received 2 mg/m2 to 2.5 mg/m2 per day of sirolimus in liquid or tablet form for 6 months. After 6 months, those who benefited from the drug were allowed to continue treatment with follow-up appointments to monitor toxicities.
Of the 12 children with ALPS, 11 had complete responses—normalization of blood cell counts—from 1 to 3 months after receiving sirolimus. The remaining patient achieved a complete response after 18 months.
All ALPS patients were successfully weaned off steroids and discontinued all other medications within 1 week to 1 month after starting sirolimus.
The patients with multi-lineage cytopenias also responded well to sirolimus. Eight of the 12 patients had complete responses, although these occurred later than for most ALPS patients (after 3 months).
The 6 patients with single-lineage cytopenias had less robust results—1 complete response and 2 partial responses. One child with ITP achieved a partial response but had to discontinue therapy.
One of the patients with AIHA had a complete response by 6 months and was able to stop taking other medications within a month. The other child with AIHA achieved a partial response.
For all patients, the median time on sirolimus was 2 years (range, 1–4.5 years).
The most common adverse event observed in this study was grade 1-2 mucositis (n=10). Other toxicities included elevated triglycerides and elevated cholesterol (n=2), acne (n=1), sun sensitivity (n=1), and exacerbation of gastro-esophageal reflux disease (n=1).
One patient developed hypertension 2 years after starting sirolimus, but this was temporally related to starting a new psychiatric medication.
Another patient (with Evans syndrome) developed a headache with associated white matter changes (4 different lesions). The changes were attributed to disease-associated vasculitis, and the lesions resolved over a few months with the addition of steroids. The patient was eventually diagnosed with a primary T-cell immune deficiency and underwent hematopoietic stem cell transplant.
“This study demonstrates that sirolimus is an effective and safe alternative to steroids, providing children with an improved quality of life as they continue treatment into adulthood,” Dr Teachey said. “While further studies are needed, sirolimus should be considered an early therapy option for patients with autoimmune blood disorders requiring ongoing therapy.” ![]()
New research suggests the immunosuppressant sirolimus may be a promising treatment option for patients with refractory autoimmune cytopenias.
The drug proved particularly effective in children with autoimmune lymphoproliferative syndrome (ALPS), producing complete responses in all of the ALPS patients studied.
On the other hand, patients with single-lineage autoimmune cytopenias, such as immune thrombocytopenia (ITP), did not fare as well.
David T. Teachey, MD, of The Children’s Hospital of Philadelphia in Pennsylvania, and his colleagues reported these results in Blood.
The group studied sirolimus in 30 patients with refractory autoimmune cytopenias who were 5 to 19 years of age. All of the patients were refractory to or could not tolerate corticosteroids.
Twelve patients had ALPS, 6 had single-lineage autoimmune cytopenias (4 with ITP and 2 with autoimmune hemolytic anemia [AIHA]), and 12 patients had multi-lineage cytopenias secondary to common variable immune deficiency (n=2), Evans syndrome (n=8), or systemic lupus erythematosus (n=2).
The patients received 2 mg/m2 to 2.5 mg/m2 per day of sirolimus in liquid or tablet form for 6 months. After 6 months, those who benefited from the drug were allowed to continue treatment with follow-up appointments to monitor toxicities.
Of the 12 children with ALPS, 11 had complete responses—normalization of blood cell counts—from 1 to 3 months after receiving sirolimus. The remaining patient achieved a complete response after 18 months.
All ALPS patients were successfully weaned off steroids and discontinued all other medications within 1 week to 1 month after starting sirolimus.
The patients with multi-lineage cytopenias also responded well to sirolimus. Eight of the 12 patients had complete responses, although these occurred later than for most ALPS patients (after 3 months).
The 6 patients with single-lineage cytopenias had less robust results—1 complete response and 2 partial responses. One child with ITP achieved a partial response but had to discontinue therapy.
One of the patients with AIHA had a complete response by 6 months and was able to stop taking other medications within a month. The other child with AIHA achieved a partial response.
For all patients, the median time on sirolimus was 2 years (range, 1–4.5 years).
The most common adverse event observed in this study was grade 1-2 mucositis (n=10). Other toxicities included elevated triglycerides and elevated cholesterol (n=2), acne (n=1), sun sensitivity (n=1), and exacerbation of gastro-esophageal reflux disease (n=1).
One patient developed hypertension 2 years after starting sirolimus, but this was temporally related to starting a new psychiatric medication.
Another patient (with Evans syndrome) developed a headache with associated white matter changes (4 different lesions). The changes were attributed to disease-associated vasculitis, and the lesions resolved over a few months with the addition of steroids. The patient was eventually diagnosed with a primary T-cell immune deficiency and underwent hematopoietic stem cell transplant.
“This study demonstrates that sirolimus is an effective and safe alternative to steroids, providing children with an improved quality of life as they continue treatment into adulthood,” Dr Teachey said. “While further studies are needed, sirolimus should be considered an early therapy option for patients with autoimmune blood disorders requiring ongoing therapy.” ![]()
New research suggests the immunosuppressant sirolimus may be a promising treatment option for patients with refractory autoimmune cytopenias.
The drug proved particularly effective in children with autoimmune lymphoproliferative syndrome (ALPS), producing complete responses in all of the ALPS patients studied.
On the other hand, patients with single-lineage autoimmune cytopenias, such as immune thrombocytopenia (ITP), did not fare as well.
David T. Teachey, MD, of The Children’s Hospital of Philadelphia in Pennsylvania, and his colleagues reported these results in Blood.
The group studied sirolimus in 30 patients with refractory autoimmune cytopenias who were 5 to 19 years of age. All of the patients were refractory to or could not tolerate corticosteroids.
Twelve patients had ALPS, 6 had single-lineage autoimmune cytopenias (4 with ITP and 2 with autoimmune hemolytic anemia [AIHA]), and 12 patients had multi-lineage cytopenias secondary to common variable immune deficiency (n=2), Evans syndrome (n=8), or systemic lupus erythematosus (n=2).
The patients received 2 mg/m2 to 2.5 mg/m2 per day of sirolimus in liquid or tablet form for 6 months. After 6 months, those who benefited from the drug were allowed to continue treatment with follow-up appointments to monitor toxicities.
Of the 12 children with ALPS, 11 had complete responses—normalization of blood cell counts—from 1 to 3 months after receiving sirolimus. The remaining patient achieved a complete response after 18 months.
All ALPS patients were successfully weaned off steroids and discontinued all other medications within 1 week to 1 month after starting sirolimus.
The patients with multi-lineage cytopenias also responded well to sirolimus. Eight of the 12 patients had complete responses, although these occurred later than for most ALPS patients (after 3 months).
The 6 patients with single-lineage cytopenias had less robust results—1 complete response and 2 partial responses. One child with ITP achieved a partial response but had to discontinue therapy.
One of the patients with AIHA had a complete response by 6 months and was able to stop taking other medications within a month. The other child with AIHA achieved a partial response.
For all patients, the median time on sirolimus was 2 years (range, 1–4.5 years).
The most common adverse event observed in this study was grade 1-2 mucositis (n=10). Other toxicities included elevated triglycerides and elevated cholesterol (n=2), acne (n=1), sun sensitivity (n=1), and exacerbation of gastro-esophageal reflux disease (n=1).
One patient developed hypertension 2 years after starting sirolimus, but this was temporally related to starting a new psychiatric medication.
Another patient (with Evans syndrome) developed a headache with associated white matter changes (4 different lesions). The changes were attributed to disease-associated vasculitis, and the lesions resolved over a few months with the addition of steroids. The patient was eventually diagnosed with a primary T-cell immune deficiency and underwent hematopoietic stem cell transplant.
“This study demonstrates that sirolimus is an effective and safe alternative to steroids, providing children with an improved quality of life as they continue treatment into adulthood,” Dr Teachey said. “While further studies are needed, sirolimus should be considered an early therapy option for patients with autoimmune blood disorders requiring ongoing therapy.” ![]()
Hospital Evidence‐Based Practice Centers
Hospital evidence‐based practice centers (EPCs) are structures with the potential to facilitate the integration of evidence into institutional decision making to close knowing‐doing gaps[1, 2, 3, 4, 5, 6]; in the process, they can support the evolution of their parent institutions into learning healthcare systems.[7] The potential of hospital EPCs stems from their ability to identify and adapt national evidence‐based guidelines and systematic reviews for the local setting,[8] create local evidence‐based guidelines in the absence of national guidelines, use local data to help define problems and assess the impact of solutions,[9] and implement evidence into practice through computerized clinical decision support (CDS) interventions and other quality‐improvement (QI) initiatives.[9, 10] As such, hospital EPCs have the potential to strengthen relationships and understanding between clinicians and administrators[11]; foster a culture of evidence‐based practice; and improve the quality, safety, and value of care provided.[10]
Formal hospital EPCs remain uncommon in the United States,[10, 11, 12] though their numbers have expanded worldwide.[13, 14] This growth is due not to any reduced role for national EPCs, such as the National Institute for Health and Clinical Excellence[15] in the United Kingdom, or the 13 EPCs funded by the Agency for Healthcare Research and Quality (AHRQ)[16, 17] in the United States. Rather, this growth is fueled by the heightened awareness that the value of healthcare interventions often needs to be assessed locally, and that clinical guidelines that consider local context have a greater potential to improve quality and efficiency.[9, 18, 19]
Despite the increasing number of hospital EPCs globally, their impact on administrative and clinical decision making has rarely been examined,[13, 20] especially for hospital EPCs in the United States. The few studies that have assessed the impact of hospital EPCs on institutional decision making have done so in the context of technology acquisition, neglecting the role hospital EPCs may play in the integration of evidence into clinical practice. For example, the Technology Assessment Unit at McGill University Health Center found that of the 27 reviews commissioned in their first 5 years, 25 were implemented, with 6 (24%) recommending investments in new technologies and 19 (76%) recommending rejection, for a reported net hospital savings of $10 million.[21] Understanding the activities and impact of hospital EPCs is particularly critical for hospitalist leaders, who could leverage hospital EPCs to inform efforts to support the quality, safety, and value of care provided, or who may choose to establish or lead such infrastructure. The availability of such opportunities could also support hospitalist recruitment and retention.
In 2006, the University of Pennsylvania Health System (UPHS) created the Center for Evidence‐based Practice (CEP) to support the integration of evidence into practice to strengthen quality, safety, and value.[10] Cofounded by hospitalists with formal training in clinical epidemiology, the CEP performs rapid systematic reviews of the scientific literature to inform local practice and policy. In this article, we describe the first 8 years of the CEP's evidence synthesis activities and examine its impact on decision making across the health system.
METHODS
Setting
The UPHS includes 3 acute care hospitals, and inpatient facilities specializing in acute rehabilitation, skilled nursing, long‐term acute care, and hospice, with a capacity of more than 1800 beds and 75,000 annual admissions, as well as primary care and specialty clinics with more than 2 million annual outpatient visits. The CEP is funded by and organized within the Office of the UPHS Chief Medical Officer, serves all UPHS facilities, has an annual budget of approximately $1 million, and is currently staffed by a hospitalist director, 3 research analysts, 6 physician and nurse liaisons, a health economist, biostatistician, administrator, and librarians, totaling 5.5 full time equivalents.
The mission of the CEP is to support the quality, safety, and value of care at Penn through evidence‐based practice. To accomplish this mission, the CEP performs rapid systematic reviews, translates evidence into practice through the use of CDS interventions and clinical pathways, and offers education in evidence‐based decision making to trainees, staff, and faculty. This study is focused on the CEP's evidence synthesis activities.
Typically, clinical and administrative leaders submit a request to the CEP for an evidence review, the request is discussed and approved at the weekly staff meeting, and a research analyst and clinical liaison are assigned to the request and communicate with the requestor to clearly define the question of interest. Subsequently, the research analyst completes a protocol, a draft search, and a draft report, each reviewed and approved by the clinical liaison and requestor. The final report is posted to the website, disseminated to all key stakeholders across the UPHS as identified by the clinical liaisons, and integrated into decision making through various routes, including in‐person presentations to decision makers, and CDS and QI initiatives.
Study Design
The study included an analysis of an internal database of evidence reviews and a survey of report requestors, and was exempted from institutional review board review. Survey respondents were informed that their responses would be confidential and did not receive incentives.
Internal Database of Reports
Data from the CEP's internal management database were analyzed for its first 8 fiscal years (July 2006June 2014). Variables included requestor characteristics, report characteristics (eg, technology reviewed, clinical specialty examined, completion time, and performance of meta‐analyses and GRADE [Grading of Recommendations Assessment, Development and Evaluation] analyses[22]), report use (eg, integration of report into CDS interventions) and dissemination beyond the UPHS (eg, submission to Center for Reviews and Dissemination [CRD] Health Technology Assessment [HTA] database[23] and to peer‐reviewed journals). Report completion time was defined as the time between the date work began on the report and the date the final report was sent to the requestor. The technology categorization scheme was adapted from that provided by Goodman (2004)[24] and the UK National Institute for Health Research HTA Programme.[25] We systematically assigned the technology reviewed in each report to 1 of 8 mutually exclusive categories. The clinical specialty examined in each report was determined using an algorithm (see Supporting Information, Appendix 1, in the online version of this article).
We compared the report completion times and the proportions of requestor types, technologies reviewed, and clinical specialties examined in the CEP's first 4 fiscal years (July 2006June 2010) to those in the CEP's second 4 fiscal years (July 2010June 2014) using t tests and 2 tests for continuous and categorical variables, respectively.
Survey
We conducted a Web‐based survey (see Supporting Information, Appendix 2, in the online version of this article) of all requestors of the 139 rapid reviews completed in the last 4 fiscal years. Participants who requested multiple reports were surveyed only about the most recent report. Requestors were invited to participate in the survey via e‐mail, and follow‐up e‐mails were sent to nonrespondents at 7, 14, and 16 days. Nonrespondents and respondents were compared with respect to requestor type (physician vs nonphysician) and topic evaluated (traditional HTA topics such as drugs, biologics, and devices vs nontraditional HTA topics such as processes of care). The survey was administered using REDCap[26] electronic data capture tools. The 44‐item questionnaire collected data on the interaction between the requestor and the CEP, report characteristics, report impact, and requestor satisfaction.
Survey results were imported into Microsoft Excel (Microsoft Corp, Redmond, WA) and SPSS (IBM, Armonk, NY) for analysis. Descriptive statistics were generated, and statistical comparisons were conducted using 2 and Fisher exact tests.
RESULTS
Evidence Synthesis Activity
The CEP has produced several different report products since its inception. Evidence reviews (57%, n = 142) consist of a systematic review and analysis of the primary literature. Evidence advisories (32%, n = 79) are summaries of evidence from secondary sources such as guidelines or systematic reviews. Evidence inventories (3%, n = 7) are literature searches that describe the quantity and focus of available evidence, without analysis or synthesis.[27]
The categories of technologies examined, including their definitions and examples, are provided in Table 1. Drugs (24%, n = 60) and devices/equipment/supplies (19%, n = 48) were most commonly examined. The proportion of reports examining technology types traditionally evaluated by HTA organizations significantly decreased when comparing the first 4 years of CEP activity to the second 4 years (62% vs 38%, P < 0.01), whereas reports examining less traditionally reviewed categories increased (38% vs 62%, P < 0.01). The most common clinical specialties represented by the CEP reports were nursing (11%, n = 28), general surgery (11%, n = 28), critical care (10%, n = 24), and general medicine (9%, n = 22) (see Supporting Information, Appendix 3, in the online version of this article). Clinical departments were the most common requestors (29%, n = 72) (Table 2). The proportion of requests from clinical departments significantly increased when comparing the first 4 years to the second 4 years (20% vs 36%, P < 0.01), with requests from purchasing committees significantly decreasing (25% vs 6%, P < 0.01). The overall report completion time was 70 days, and significantly decreased when comparing the first 4 years to the second 4 years (89 days vs 50 days, P < 0.01).
| Category | Definition | Examples | Total | 20072010 | 20112014 | P Value |
|---|---|---|---|---|---|---|
| Total | 249 (100%) | 109 (100%) | 140 (100%) | |||
| Drug | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of a pharmacologic agent | Celecoxib for pain in joint arthroplasty; colchicine for prevention of pericarditis and atrial fibrillation | 60 (24%) | 35 (32%) | 25 (18%) | 0.009 |
| Device, equipment, and supplies | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory that is intended for use in the prevention, diagnosis, or treatment of disease and does not achieve its primary intended purposes though chemical action or metabolism[50] | Thermometers for pediatric use; femoral closure devices for cardiac catheterization | 48 (19%) | 25 (23%) | 23 (16%) | 0.19 |
| Process of care | A report primarily examining a clinical pathway or a clinical practice guideline that significantly involves elements of prevention, diagnosis, and/or treatment or significantly incorporates 2 or more of the other technology categories | Preventing patient falls; prevention and management of delirium | 31 (12%) | 18 (17%) | 13 (9%) | 0.09 |
| Test, scale, or risk factor | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of a test intended to screen for, diagnose, classify, or monitor the progression of a disease | Computed tomography for acute chest pain; urine drug screening in chronic pain patients on opioid therapy | 31 (12%) | 8 (7%) | 23 (16%) | 0.03 |
| Medical/surgical procedure | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of a medical intervention that is not a drug, device, or test or of the application or removal of a device | Biliary drainage for chemotherapy patients; cognitive behavioral therapy for insomnia | 26 (10%) | 8 (7%) | 18 (13%) | 0.16 |
| Policy or organizational/managerial system | A report primarily examining laws or regulations; the organization, financing, or delivery of care, including settings of care; or healthcare providers | Medical care costs and productivity changes associated with smoking; physician training and credentialing for robotic surgery in obstetrics and gynecology | 26 (10%) | 4 (4%) | 22 (16%) | 0.002 |
| Support system | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of an intervention designed to provide a new or improved service to patients or healthcare providers that does not fall into 1 of the other categories | Reconciliation of data from differing electronic medical records; social media, text messaging, and postdischarge communication | 14 (6%) | 3 (3%) | 11 (8%) | 0.09 |
| Biologic | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of a product manufactured in a living system | Recombinant factor VIIa for cardiovascular surgery; osteobiologics for orthopedic fusions | 13 (5%) | 8 (7%) | 5 (4%) | 0.19 |
| Category | Total | 20072010 | 20112014 | P Value |
|---|---|---|---|---|
| ||||
| Total | 249 (100%) | 109 (100%) | 140 (100%) | |
| Clinical department | 72 (29%) | 22 (20%) | 50 (36%) | 0.007 |
| CMO | 47 (19%) | 21 (19%) | 26 (19%) | 0.92 |
| Purchasing committee | 35 (14%) | 27 (25%) | 8 (6%) | <0.001 |
| Formulary committee | 22 (9%) | 12 (11%) | 10 (7%) | 0.54 |
| Quality committee | 21 (8%) | 11 (10%) | 10 (7%) | 0.42 |
| Administrative department | 19 (8%) | 5 (5%) | 14 (10%) | 0.11 |
| Nursing | 14 (6%) | 4 (4%) | 10 (7%) | 0.23 |
| Other* | 19 (8%) | 7 (6%) | 12 (9%) | 0.55 |
Thirty‐seven (15%) reports included meta‐analyses conducted by CEP staff. Seventy‐five reports (30%) contained an evaluation of the quality of the evidence base using GRADE analyses.[22] Of these reports, the highest GRADE of evidence available for any comparison of interest was moderate (35%, n = 26) or high (33%, n = 25) in most cases, followed by very low (19%, n = 14) and low (13%, n = 10).
Reports were disseminated in a variety of ways beyond direct dissemination and presentation to requestors and posting on the center website. Thirty reports (12%) informed CDS interventions, 24 (10%) resulted in peer‐reviewed publications, and 204 (82%) were posted to the CRD HTA database.
Evidence Synthesis Impact
A total of 139 reports were completed between July 2010 and June 2014 for 65 individual requestors. Email invitations to participate in the survey were sent to the 64 requestors employed by the UPHS. The response rate was 72% (46/64). The proportions of physician requestors and traditional HTA topics evaluated were similar across respondents and nonrespondents (43% [20/46] vs 39% [7/18], P = 0.74; and 37% [17/46] vs 44% [8/18], P = 0.58, respectively). Aggregated survey responses are presented for items using a Likert scale in Figure 1, and for items using a yes/no or ordinal scale in Table 3.
| Items | % of Respondents Responding Affirmatively |
|---|---|
| Percentage of Respondents Ranking as First Choice* | |
| |
| Requestor activity | |
| What factors prompted you to request a report from CEP? (Please select all that apply.) | |
| My own time constraints | 28% (13/46) |
| CEP's ability to identify and synthesize evidence | 89% (41/46) |
| CEP's objectivity | 52% (24/46) |
| Recommendation from colleague | 30% (14/46) |
| Did you conduct any of your own literature searches before contacting CEP? | 67% (31/46) |
| Did you obtain and read any of the articles cited in CEP's report? | 63% (29/46) |
| Did you read the following sections of CEP's report? | |
| Evidence summary (at beginning of report) | 100% (45/45) |
| Introduction/background | 93% (42/45) |
| Methods | 84% (38/45) |
| Results | 98% (43/43) |
| Conclusion | 100% (43/43) |
| Report dissemination | |
| Did you share CEP's report with anyone NOT involved in requesting the report or in making the final decision? | 67% (30/45) |
| Did you share CEP's report with anyone outside of Penn? | 7% (3/45) |
| Requestor preferences | |
| Would it be helpful for CEP staff to call you after you receive any future CEP reports to answer any questions you might have? | 55% (24/44) |
| Following any future reports you request from CEP, would you be willing to complete a brief questionnaire? | 100% (44/44) |
| Please rank how you would prefer to receive reports from CEP in the future. | |
| E‐mail containing the report as a PDF attachment | 77% (34/44) |
| E‐mail containing a link to the report on CEP's website | 16% (7/44) |
| In‐person presentation by the CEP analyst writing the report | 18% (8/44) |
| In‐person presentation by the CEP director involved in the report | 16% (7/44) |
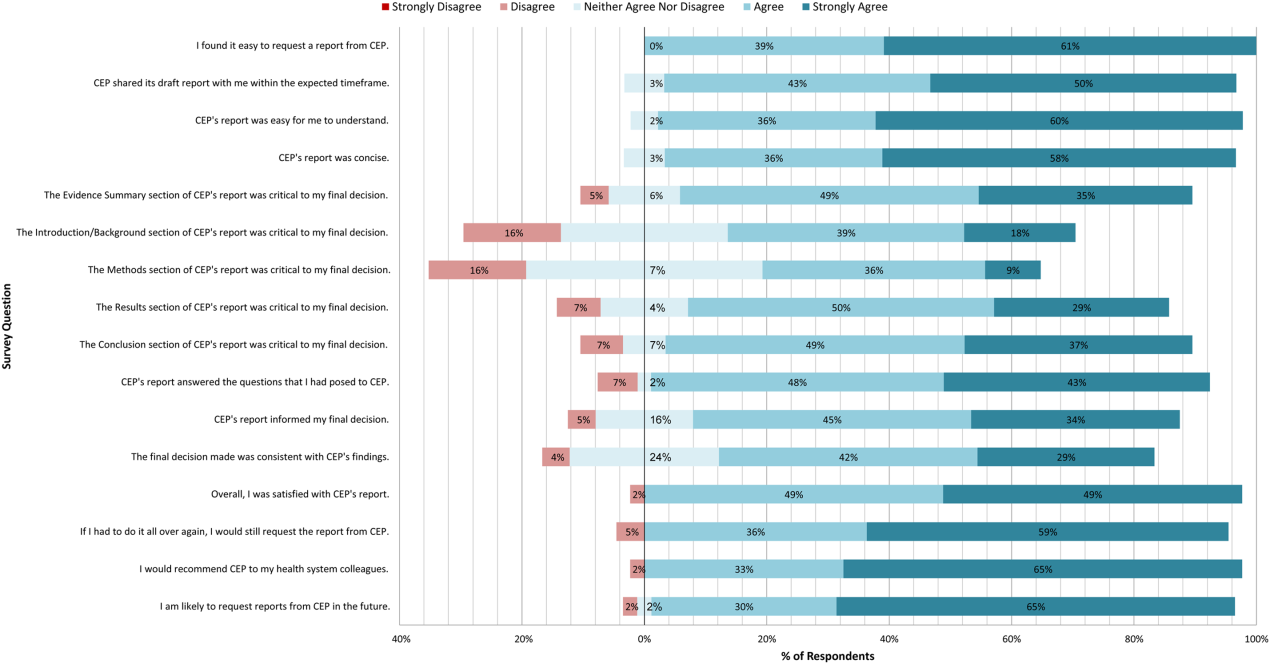
In general, respondents found reports easy to request, easy to use, timely, and relevant, resulting in high requestor satisfaction. In addition, 98% described the scope of content and level of detail as about right. Report impact was rated highly as well, with the evidence summary and conclusions rated as the most critical to decision making. A majority of respondents indicated that reports confirmed their tentative decision (77%, n = 34), whereas some changed their tentative decision (7%, n = 3), and others suggested the report had no effect on their tentative decision (16%, n = 7). Respondents indicated the amount of time that elapsed between receiving reports and making final decisions was 1 to 7 days (5%, n = 2), 8 to 30 days (40%, n = 17), 1 to 3 months (37%, n = 16), 4 to 6 months (9%, n = 4), or greater than 6 months (9%, n = 4). The most common reasons cited for requesting a report were the CEP's evidence synthesis skills and objectivity.
DISCUSSION
To our knowledge, this is the first comprehensive description and assessment of evidence synthesis activity by a hospital EPC in the United States. Our findings suggest that clinical and administrative leaders will request reports from a hospital EPC, and that hospital EPCs can promptly produce reports when requested. Moreover, these syntheses can address a wide range of clinical and policy topics, and can be disseminated through a variety of routes. Lastly, requestors are satisfied by these syntheses, and report that they inform decision making. These results suggest that EPCs may be an effective infrastructure paradigm for promoting evidence‐based decision making within healthcare provider organizations, and are consistent with previous analyses of hospital‐based EPCs.[21, 28, 29]
Over half of report requestors cited CEP's objectivity as a factor in their decision to request a report, underscoring the value of a neutral entity in an environment where clinical departments and hospital committees may have competing interests.[10] This asset was 1 of the primary drivers for establishing our hospital EPC. Concerns by clinical executives about the influence of industry and local politics on institutional decision making, and a desire to have clinical evidence more systematically and objectively integrated into decision making, fueled our center's funding.
The survey results also demonstrate that respondents were satisfied with the reports for many reasons, including readability, concision, timeliness, scope, and content, consistent with the evaluation of the French hospital‐based EPC CEDIT (French Committee for the Assessment and Dissemination of Technological Innovations).[29] Given the importance of readability, concision, and relevance that has been previously described,[16, 28, 30] nearly all CEP reports contain an evidence summary on the first page that highlights key findings in a concise, user‐friendly format.[31] The evidence summaries include bullet points that: (1) reference the most pertinent guideline recommendations along with their strength of recommendation and underlying quality of evidence; (2) organize and summarize study findings using the most critical clinical outcomes, including an assessment of the quality of the underlying evidence for each outcome; and (3) note important limitations of the findings.
Evidence syntheses must be timely to allow decision makers to act on the findings.[28, 32] The primary criticism of CEDIT was the lag between requests and report publication.[29] Rapid reviews, designed to inform urgent decisions, can overcome this challenge.[31, 33, 34] CEP reviews required approximately 2 months to complete on average, consistent with the most rapid timelines reported,[31, 33, 34] and much shorter than standard systematic review timelines, which can take up to 12 to 24 months.[33] Working with requestors to limit the scope of reviews to those issues most critical to a decision, using secondary resources when available, and hiring experienced research analysts help achieve these efficiencies.
The study by Bodeau‐Livinec also argues for the importance of report accessibility to ensure dissemination.[29] This is consistent with the CEP's approach, where all reports are posted on the UPHS internal website. Many also inform QI initiatives, as well as CDS interventions that address topics of general interest to acute care hospitals, such as venous thromboembolism (VTE) prophylaxis,[35] blood product transfusions,[36] sepsis care,[37, 38] and prevention of catheter‐associated urinary tract infections (CAUTI)[39] and hospital readmissions.[40] Most reports are also listed in an international database of rapid reviews,[23] and reports that address topics of general interest, have sufficient evidence to synthesize, and have no prior published systematic reviews are published in the peer‐reviewed literature.[41, 42]
The majority of reports completed by the CEP were evidence reviews, or systematic reviews of primary literature, suggesting that CEP reports often address questions previously unanswered by existing published systematic reviews; however, about a third of reports were evidence advisories, or summaries of evidence from preexisting secondary sources. The relative scarcity of high‐quality evidence bases in those reports where GRADE analyses were conducted might be expected, as requestors may be more likely to seek guidance when the evidence base on a topic is lacking. This was further supported by the small percentage (15%) of reports where adequate data of sufficient homogeneity existed to allow meta‐analyses. The small number of original meta‐analyses performed also reflects our reliance on secondary resources when available.
Only 7% of respondents reported that tentative decisions were changed based on their report. This is not surprising, as evidence reviews infrequently result in clear go or no go recommendations. More commonly, they address or inform complex clinical questions or pathways. In this context, the change/confirm/no effect framework may not completely reflect respondents' use of or benefit from reports. Thus, we included a diverse set of questions in our survey to best estimate the value of our reports. For example, when asked whether the report answered the question posed, informed their final decision, or was consistent with their final decision, 91%, 79%, and 71% agreed or strongly agreed, respectively. When asked whether they would request a report again if they had to do it all over, recommend CEP to their colleagues, and be likely to request reports in the future, at least 95% of survey respondents agreed or strongly agreed. In addition, no respondent indicated that their report was not timely enough to influence their decision. Moreover, only a minority of respondents expressed disappointment that the CEP's report did not provide actionable recommendations due to a lack of published evidence (9%, n = 4). Importantly, the large proportion of requestors indicating that reports confirmed their tentative decisions may be a reflection of hindsight bias.
The most apparent trend in the production of CEP reviews over time is the relative increase in requests by clinical departments, suggesting that the CEP is being increasingly consulted to help define best clinical practices. This is also supported by the relative increase in reports focused on policy or organizational/managerial systems. These findings suggest that hospital EPCs have value beyond the traditional realm of HTA.
This study has a number of limitations. First, not all of the eligible report requestors responded to our survey. Despite this, our response rate of 72% compares favorably with surveys published in medical journals.[43] In addition, nonresponse bias may be less important in physician surveys than surveys of the general population.[44] The similarity in requestor and report characteristics for respondents and nonrespondents supports this. Second, our survey of impact is self‐reported rather than an evaluation of actual decision making or patient outcomes. Thus, the survey relies on the accuracy of the responses. Third, recall bias must be considered, as some respondents were asked to evaluate reports that were greater than 1 year old. To reduce this bias, we asked respondents to consider the most recent report they requested, included that report as an attachment in the survey request, and only surveyed requestors from the most recent 4 of the CEP's 8 fiscal years. Fourth, social desirability bias could have also affected the survey responses, though it was likely minimized by the promise of confidentiality. Fifth, an examination of the impact of the CEP on costs was outside the scope of this evaluation; however, such information may be important to those assessing the sustainability or return on investment of such centers. Simple approaches we have previously used to approximate the value of our activities include: (1) estimating hospital cost savings resulting from decisions supported by our reports, such as the use of technologies like chlorhexidine for surgical site infections[45] or discontinuation of technologies like aprotinin for cardiac surgery[46]; and (2) estimating penalties avoided or rewards attained as a result of center‐led initiatives, such as those to increase VTE prophylaxis,[35] reduce CAUTI rates,[39] and reduce preventable mortality associated with sepsis.[37, 38] Similarly, given the focus of this study on the local evidence synthesis activities of our center, our examination did not include a detailed description of our CDS activities, or teaching activities, including our multidisciplinary workshops for physicians and nurses in evidence‐based QI[47] and our novel evidence‐based practice curriculum for medical students. Our study also did not include a description of our extramural activities, such as those supported by our contract with AHRQ as 1 of their 13 EPCs.[16, 17, 48, 49] A consideration of all of these activities enables a greater appreciation for the potential of such centers. Lastly, we examined a single EPC, which may not be representative of the diversity of hospitals and hospital staff across the United States. However, our EPC serves a diverse array of patient populations, clinical services, and service models throughout our multientity academic healthcare system, which may improve the generalizability of our experience to other settings.
As next steps, we recommend evaluation of other existing hospital EPCs nationally. Such studies could help hospitals and health systems ascertain which of their internal decisions might benefit from locally sourced rapid systematic reviews and determine whether an in‐house EPC could improve the value of care delivered.
In conclusion, our findings suggest that hospital EPCs within academic healthcare systems can efficiently synthesize and disseminate evidence for a variety of stakeholders. Moreover, these syntheses impact decision making in a variety of hospital contexts and clinical specialties. Hospitals and hospitalist leaders seeking to improve the implementation of evidence‐based practice at a systems level might consider establishing such infrastructure locally.
Acknowledgements
The authors thank Fran Barg, PhD (Department of Family Medicine and Community Health, University of Pennsylvania Perelman School of Medicine) and Joel Betesh, MD (University of Pennsylvania Health System) for their contributions to developing the survey. They did not receive any compensation for their contributions.
Disclosures: An earlier version of this work was presented as a poster at the 2014 AMA Research Symposium, November 7, 2014, Dallas, Texas. Mr. Jayakumar reports having received a University of Pennsylvania fellowship as a summer intern at the Center for Evidence‐based Practice. Dr. Umscheid cocreated and directs a hospital evidence‐based practice center, is the Senior Associate Director of an Agency for Healthcare Research and Quality Evidence‐Based Practice Center, and is a past member of the Medicare Evidence Development and Coverage Advisory Committee, which uses evidence reports developed by the Evidence‐based Practice Centers of the Agency for Healthcare Research and Quality. Dr. Umscheid's contribution was supported in part by the National Center for Research Resources, grant UL1RR024134, which is now at the National Center for Advancing Translational Sciences, grant UL1TR000003. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. None of the funders had a role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. Dr. Lavenberg, Dr. Mitchell, and Mr. Leas are employed as research analysts by a hospital evidence‐based practice center. Dr. Doshi is supported in part by a hospital evidence‐based practice center and is an Associate Director of an Agency for Healthcare Research and Quality Evidence‐based Practice Center. Dr. Goldmann is emeritus faculty at Penn, is supported in part by a hospital evidence‐based practice center, and is the Vice President and Chief Quality Assurance Officer in Clinical Solutions, a division of Elsevier, Inc., a global publishing company, and director of the division's Evidence‐based Medicine Center. Dr. Williams cocreated and codirects a hospital evidence‐based practice center. Dr. Brennan has oversight for and helped create a hospital evidence‐based practice center.
- , . “Bench to behavior”: translating comparative effectiveness research into improved clinical practice. Health Aff (Millwood). 2010;29(10):1891–1900.
- , , . Evaluating the status of “translating research into practice” at a major academic healthcare system. Int J Technol Assess Health Care. 2009;25(1):84–89.
- , , , . Five reasons that many comparative effectiveness studies fail to change patient care and clinical practice. Health Aff (Millwood). 2012;31(10):2168–2175.
- , , , , , . Fostering implementation of health services research findings into practice: a consolidated framework for advancing implementation science. Implement Sci. 2009;4(1):50.
- , . From best evidence to best practice: effective implementation of change in patients' care. Lancet. 2003;362(9391):1225–1230.
- , . Incentivizing “structures” over “outcomes” to bridge the knowing‐doing gap. JAMA Intern Med. 2015;175(3):354.
- Olsen L, Aisner D, McGinnis JM, eds. Institute of Medicine (US) Roundtable on Evidence‐Based Medicine. The Learning Healthcare System: Workshop Summary. Washington, DC: National Academies Press; 2007. Available at: http://www.ncbi.nlm.nih.gov/books/NBK53494/. Accessed October 29, 2014.
- , , , . Adapting clinical practice guidelines to local context and assessing barriers to their use. Can Med Assoc J. 2010;182(2):E78–E84.
- , , , . Integrating local data into hospital‐based healthcare technology assessment: two case studies. Int J Technol Assess Health Care. 2010;26(3):294–300.
- , , . Hospital‐based comparative effectiveness centers: translating research into practice to improve the quality, safety and value of patient care. J Gen Intern Med. 2010;25(12):1352–1355.
- , , , , . Health technology assessment at the University of California‐San Francisco. J Healthc Manag Am Coll Healthc Exec. 2011;56(1):15–29; discussion 29–30.
- , . Kaiser Permanente Southern California regional technology management process: evidence‐based medicine operationalized. Perm J. 2006;10(1):38–41.
- . Hospital‐based health technology assessment: developments to date. Pharmacoeconomics. 2014;32(9):819–824.
- , , , . Hospital based health technology assessment world‐wide survey. Available at: http://www.htai.org/fileadmin/HTAi_Files/ISG/HospitalBasedHTA/2008Files/HospitalBasedHTAISGSurveyReport.pdf. Accessed October 11, 2015.
- , . At the center of health care policy making: the use of health technology assessment at NICE. Med Decis Making. 2013;33(3):320–324.
- , , . Better information for better health care: the Evidence‐based Practice Center program and the Agency for Healthcare Research and Quality. Ann Intern Med. 2005;142(12 part 2):1035–1041.
- , . AHRQ's Effective Health Care Program: why comparative effectiveness matters. Am J Med Qual. 2009;24(1):67–70.
- , . Effect of clinical guidelines on medical practice: a systematic review of rigorous evaluations. Lancet. 1993;342(8883):1317–1322.
- , , , et al. Lost in knowledge translation: time for a map? J Contin Educ Health Prof. 2006;26(1):13–24.
- , , , . Effects and repercussions of local/hospital‐based health technology assessment (HTA): a systematic. Syst Rev. 2014;3:129.
- , , , et al. Impact of TAU Reports. McGill University Health Centre. Available at: https://francais.mcgill.ca/files/tau/FINAL_TAU_IMPACT_REPORT_FEB_2008.pdf. Published Feb 1, 2008. Accessed August 19, 2014.
- , , , et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ. 2008;336(7650):924–926.
- , , . Centre for Reviews and Dissemination databases: value, content, and developments. Int J Technol Assess Health Care. 2010;26(4):470–472.
- . HTA 101. Introduction to Health Technology Assessment. Available at: https://www.nlm.nih.gov/nichsr/hta101/ta10103.html. Accessed October 11, 2015.
- National Institute for Health Research. Remit. NIHR HTA Programme. Available at: http://www.nets.nihr.ac.uk/programmes/hta/remit. Accessed August 20, 2014.
- , , , , , . Research Electronic Data Capture (REDCap)—a metadata‐driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381.
- , , , . When the decision is what to decide: Using evidence inventory reports to focus health technology assessments. Int J Technol Assess Health Care. 2011;27(2):127–132.
- , . End‐user involvement in health technology assessment (HTA) development: a way to increase impact. Int J Technol Assess Health Care. 2005;21(02):263–267.
- , , , , . Impact of CEDIT recommendations: an example of health technology assessment in a hospital network. Int J Technol Assess Health Care. 2006;22(2):161–168.
- , , , . Increasing the relevance of research to health care managers: hospital CEO imperatives for improving quality and lowering costs. Health Care Manage Rev. 2007;32(2):150–159.
- , , , , . Evidence summaries: the evolution of a rapid review approach. Syst Rev. 2012;1(1):10.
- , , , . Health care decision makers' use of comparative effectiveness research: report from a series of focus groups. J Manag Care Pharm. 2013;19(9):745–754.
- , , , et al. Rapid reviews versus full systematic reviews: an inventory of current methods and practice in health technology assessment. Int J Technol Assess Health Care. 2008;24(2):133–139.
- , , , et al. EPC Methods: An Exploration of Methods and Context for the Production of Rapid Reviews. Rockville, MD: Agency for Healthcare Research and Quality; 2015. Available at: http://www.ncbi.nlm.nih.gov/books/NBK274092. Accessed March 5, 2015.
- , , , , . Effectiveness of a novel and scalable clinical decision support intervention to improve venous thromboembolism prophylaxis: a quasi‐experimental study. BMC Med Inform Decis Mak. 2012;12:92.
- . Order sets in electronic health records: principles of good practice. Chest. 2013;143(1):228–235.
- , , , et al. Development, implementation, and impact of an automated early warning and response system for sepsis. J Hosp Med. 2015;10(1):26–31.
- , , , et al. Clinician perception of the effectiveness of an automated early warning and response system for sepsis in an academic medical center. Ann Am Thorac Soc. 2015;12(10):1514–1519.
- , , , , , . Usability and impact of a computerized clinical decision support intervention designed to reduce urinary catheter utilization and catheter‐associated urinary tract infections. Infect Control Hosp Epidemiol. 2014;35(9):1147–1155.
- , , , et al. The readmission risk flag: using the electronic health record to automatically identify patients at risk for 30‐day readmission. J Hosp Med. 2013;8(12):689–695.
- , , , , , . A systematic review to inform institutional decisions about the use of extracorporeal membrane oxygenation during the H1N1 influenza pandemic. Crit Care Med. 2010;38(6):1398–1404.
- , , , . Heparin flushing and other interventions to maintain patency of central venous catheters: a systematic review. J Adv Nurs. 2009;65(10):2007–2021.
- , , . Response rates to mail surveys published in medical journals. J Clin Epidemiol. 1997;50(10):1129–1136.
- , . Physician response to surveys: a review of the literature. Am J Prev Med. 2001;20(1):61–67.
- , , , , . Systematic review and cost analysis comparing use of chlorhexidine with use of iodine for preoperative skin antisepsis to prevent surgical site infection. Infect Control Hosp Epidemiol. 2010;31(12):1219–1229.
- , , . Antifibrinolytic use in adult cardiac surgery. Curr Opin Hematol. 2007;14(5):455–467.
- , , , , . Teaching evidence assimilation for collaborative health care (TEACH) 2009–2014: building evidence‐based capacity within health care provider organizations. EGEMS (Wash DC). 2015;3(2):1165.
- , , , , , . Cleaning hospital room surfaces to prevent health care‐associated infections: a technical brief [published online August 11, 2015]. Ann Intern Med. doi:10.7326/M15‐1192.
- , , , Healthcare Infection Control Practices Advisory Committee. Updating the guideline development methodology of the Healthcare Infection Control Practices Advisory Committee (HICPAC). Am J Infect Control. 2010;38(4):264–273.
- U.S. Food and Drug Administration. FDA basics—What is a medical device? Available at: http://www.fda.gov/AboutFDA/Transparency/Basics/ucm211822.htm. Accessed November 12, 2014.
Hospital evidence‐based practice centers (EPCs) are structures with the potential to facilitate the integration of evidence into institutional decision making to close knowing‐doing gaps[1, 2, 3, 4, 5, 6]; in the process, they can support the evolution of their parent institutions into learning healthcare systems.[7] The potential of hospital EPCs stems from their ability to identify and adapt national evidence‐based guidelines and systematic reviews for the local setting,[8] create local evidence‐based guidelines in the absence of national guidelines, use local data to help define problems and assess the impact of solutions,[9] and implement evidence into practice through computerized clinical decision support (CDS) interventions and other quality‐improvement (QI) initiatives.[9, 10] As such, hospital EPCs have the potential to strengthen relationships and understanding between clinicians and administrators[11]; foster a culture of evidence‐based practice; and improve the quality, safety, and value of care provided.[10]
Formal hospital EPCs remain uncommon in the United States,[10, 11, 12] though their numbers have expanded worldwide.[13, 14] This growth is due not to any reduced role for national EPCs, such as the National Institute for Health and Clinical Excellence[15] in the United Kingdom, or the 13 EPCs funded by the Agency for Healthcare Research and Quality (AHRQ)[16, 17] in the United States. Rather, this growth is fueled by the heightened awareness that the value of healthcare interventions often needs to be assessed locally, and that clinical guidelines that consider local context have a greater potential to improve quality and efficiency.[9, 18, 19]
Despite the increasing number of hospital EPCs globally, their impact on administrative and clinical decision making has rarely been examined,[13, 20] especially for hospital EPCs in the United States. The few studies that have assessed the impact of hospital EPCs on institutional decision making have done so in the context of technology acquisition, neglecting the role hospital EPCs may play in the integration of evidence into clinical practice. For example, the Technology Assessment Unit at McGill University Health Center found that of the 27 reviews commissioned in their first 5 years, 25 were implemented, with 6 (24%) recommending investments in new technologies and 19 (76%) recommending rejection, for a reported net hospital savings of $10 million.[21] Understanding the activities and impact of hospital EPCs is particularly critical for hospitalist leaders, who could leverage hospital EPCs to inform efforts to support the quality, safety, and value of care provided, or who may choose to establish or lead such infrastructure. The availability of such opportunities could also support hospitalist recruitment and retention.
In 2006, the University of Pennsylvania Health System (UPHS) created the Center for Evidence‐based Practice (CEP) to support the integration of evidence into practice to strengthen quality, safety, and value.[10] Cofounded by hospitalists with formal training in clinical epidemiology, the CEP performs rapid systematic reviews of the scientific literature to inform local practice and policy. In this article, we describe the first 8 years of the CEP's evidence synthesis activities and examine its impact on decision making across the health system.
METHODS
Setting
The UPHS includes 3 acute care hospitals, and inpatient facilities specializing in acute rehabilitation, skilled nursing, long‐term acute care, and hospice, with a capacity of more than 1800 beds and 75,000 annual admissions, as well as primary care and specialty clinics with more than 2 million annual outpatient visits. The CEP is funded by and organized within the Office of the UPHS Chief Medical Officer, serves all UPHS facilities, has an annual budget of approximately $1 million, and is currently staffed by a hospitalist director, 3 research analysts, 6 physician and nurse liaisons, a health economist, biostatistician, administrator, and librarians, totaling 5.5 full time equivalents.
The mission of the CEP is to support the quality, safety, and value of care at Penn through evidence‐based practice. To accomplish this mission, the CEP performs rapid systematic reviews, translates evidence into practice through the use of CDS interventions and clinical pathways, and offers education in evidence‐based decision making to trainees, staff, and faculty. This study is focused on the CEP's evidence synthesis activities.
Typically, clinical and administrative leaders submit a request to the CEP for an evidence review, the request is discussed and approved at the weekly staff meeting, and a research analyst and clinical liaison are assigned to the request and communicate with the requestor to clearly define the question of interest. Subsequently, the research analyst completes a protocol, a draft search, and a draft report, each reviewed and approved by the clinical liaison and requestor. The final report is posted to the website, disseminated to all key stakeholders across the UPHS as identified by the clinical liaisons, and integrated into decision making through various routes, including in‐person presentations to decision makers, and CDS and QI initiatives.
Study Design
The study included an analysis of an internal database of evidence reviews and a survey of report requestors, and was exempted from institutional review board review. Survey respondents were informed that their responses would be confidential and did not receive incentives.
Internal Database of Reports
Data from the CEP's internal management database were analyzed for its first 8 fiscal years (July 2006June 2014). Variables included requestor characteristics, report characteristics (eg, technology reviewed, clinical specialty examined, completion time, and performance of meta‐analyses and GRADE [Grading of Recommendations Assessment, Development and Evaluation] analyses[22]), report use (eg, integration of report into CDS interventions) and dissemination beyond the UPHS (eg, submission to Center for Reviews and Dissemination [CRD] Health Technology Assessment [HTA] database[23] and to peer‐reviewed journals). Report completion time was defined as the time between the date work began on the report and the date the final report was sent to the requestor. The technology categorization scheme was adapted from that provided by Goodman (2004)[24] and the UK National Institute for Health Research HTA Programme.[25] We systematically assigned the technology reviewed in each report to 1 of 8 mutually exclusive categories. The clinical specialty examined in each report was determined using an algorithm (see Supporting Information, Appendix 1, in the online version of this article).
We compared the report completion times and the proportions of requestor types, technologies reviewed, and clinical specialties examined in the CEP's first 4 fiscal years (July 2006June 2010) to those in the CEP's second 4 fiscal years (July 2010June 2014) using t tests and 2 tests for continuous and categorical variables, respectively.
Survey
We conducted a Web‐based survey (see Supporting Information, Appendix 2, in the online version of this article) of all requestors of the 139 rapid reviews completed in the last 4 fiscal years. Participants who requested multiple reports were surveyed only about the most recent report. Requestors were invited to participate in the survey via e‐mail, and follow‐up e‐mails were sent to nonrespondents at 7, 14, and 16 days. Nonrespondents and respondents were compared with respect to requestor type (physician vs nonphysician) and topic evaluated (traditional HTA topics such as drugs, biologics, and devices vs nontraditional HTA topics such as processes of care). The survey was administered using REDCap[26] electronic data capture tools. The 44‐item questionnaire collected data on the interaction between the requestor and the CEP, report characteristics, report impact, and requestor satisfaction.
Survey results were imported into Microsoft Excel (Microsoft Corp, Redmond, WA) and SPSS (IBM, Armonk, NY) for analysis. Descriptive statistics were generated, and statistical comparisons were conducted using 2 and Fisher exact tests.
RESULTS
Evidence Synthesis Activity
The CEP has produced several different report products since its inception. Evidence reviews (57%, n = 142) consist of a systematic review and analysis of the primary literature. Evidence advisories (32%, n = 79) are summaries of evidence from secondary sources such as guidelines or systematic reviews. Evidence inventories (3%, n = 7) are literature searches that describe the quantity and focus of available evidence, without analysis or synthesis.[27]
The categories of technologies examined, including their definitions and examples, are provided in Table 1. Drugs (24%, n = 60) and devices/equipment/supplies (19%, n = 48) were most commonly examined. The proportion of reports examining technology types traditionally evaluated by HTA organizations significantly decreased when comparing the first 4 years of CEP activity to the second 4 years (62% vs 38%, P < 0.01), whereas reports examining less traditionally reviewed categories increased (38% vs 62%, P < 0.01). The most common clinical specialties represented by the CEP reports were nursing (11%, n = 28), general surgery (11%, n = 28), critical care (10%, n = 24), and general medicine (9%, n = 22) (see Supporting Information, Appendix 3, in the online version of this article). Clinical departments were the most common requestors (29%, n = 72) (Table 2). The proportion of requests from clinical departments significantly increased when comparing the first 4 years to the second 4 years (20% vs 36%, P < 0.01), with requests from purchasing committees significantly decreasing (25% vs 6%, P < 0.01). The overall report completion time was 70 days, and significantly decreased when comparing the first 4 years to the second 4 years (89 days vs 50 days, P < 0.01).
| Category | Definition | Examples | Total | 20072010 | 20112014 | P Value |
|---|---|---|---|---|---|---|
| Total | 249 (100%) | 109 (100%) | 140 (100%) | |||
| Drug | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of a pharmacologic agent | Celecoxib for pain in joint arthroplasty; colchicine for prevention of pericarditis and atrial fibrillation | 60 (24%) | 35 (32%) | 25 (18%) | 0.009 |
| Device, equipment, and supplies | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory that is intended for use in the prevention, diagnosis, or treatment of disease and does not achieve its primary intended purposes though chemical action or metabolism[50] | Thermometers for pediatric use; femoral closure devices for cardiac catheterization | 48 (19%) | 25 (23%) | 23 (16%) | 0.19 |
| Process of care | A report primarily examining a clinical pathway or a clinical practice guideline that significantly involves elements of prevention, diagnosis, and/or treatment or significantly incorporates 2 or more of the other technology categories | Preventing patient falls; prevention and management of delirium | 31 (12%) | 18 (17%) | 13 (9%) | 0.09 |
| Test, scale, or risk factor | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of a test intended to screen for, diagnose, classify, or monitor the progression of a disease | Computed tomography for acute chest pain; urine drug screening in chronic pain patients on opioid therapy | 31 (12%) | 8 (7%) | 23 (16%) | 0.03 |
| Medical/surgical procedure | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of a medical intervention that is not a drug, device, or test or of the application or removal of a device | Biliary drainage for chemotherapy patients; cognitive behavioral therapy for insomnia | 26 (10%) | 8 (7%) | 18 (13%) | 0.16 |
| Policy or organizational/managerial system | A report primarily examining laws or regulations; the organization, financing, or delivery of care, including settings of care; or healthcare providers | Medical care costs and productivity changes associated with smoking; physician training and credentialing for robotic surgery in obstetrics and gynecology | 26 (10%) | 4 (4%) | 22 (16%) | 0.002 |
| Support system | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of an intervention designed to provide a new or improved service to patients or healthcare providers that does not fall into 1 of the other categories | Reconciliation of data from differing electronic medical records; social media, text messaging, and postdischarge communication | 14 (6%) | 3 (3%) | 11 (8%) | 0.09 |
| Biologic | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of a product manufactured in a living system | Recombinant factor VIIa for cardiovascular surgery; osteobiologics for orthopedic fusions | 13 (5%) | 8 (7%) | 5 (4%) | 0.19 |
| Category | Total | 20072010 | 20112014 | P Value |
|---|---|---|---|---|
| ||||
| Total | 249 (100%) | 109 (100%) | 140 (100%) | |
| Clinical department | 72 (29%) | 22 (20%) | 50 (36%) | 0.007 |
| CMO | 47 (19%) | 21 (19%) | 26 (19%) | 0.92 |
| Purchasing committee | 35 (14%) | 27 (25%) | 8 (6%) | <0.001 |
| Formulary committee | 22 (9%) | 12 (11%) | 10 (7%) | 0.54 |
| Quality committee | 21 (8%) | 11 (10%) | 10 (7%) | 0.42 |
| Administrative department | 19 (8%) | 5 (5%) | 14 (10%) | 0.11 |
| Nursing | 14 (6%) | 4 (4%) | 10 (7%) | 0.23 |
| Other* | 19 (8%) | 7 (6%) | 12 (9%) | 0.55 |
Thirty‐seven (15%) reports included meta‐analyses conducted by CEP staff. Seventy‐five reports (30%) contained an evaluation of the quality of the evidence base using GRADE analyses.[22] Of these reports, the highest GRADE of evidence available for any comparison of interest was moderate (35%, n = 26) or high (33%, n = 25) in most cases, followed by very low (19%, n = 14) and low (13%, n = 10).
Reports were disseminated in a variety of ways beyond direct dissemination and presentation to requestors and posting on the center website. Thirty reports (12%) informed CDS interventions, 24 (10%) resulted in peer‐reviewed publications, and 204 (82%) were posted to the CRD HTA database.
Evidence Synthesis Impact
A total of 139 reports were completed between July 2010 and June 2014 for 65 individual requestors. Email invitations to participate in the survey were sent to the 64 requestors employed by the UPHS. The response rate was 72% (46/64). The proportions of physician requestors and traditional HTA topics evaluated were similar across respondents and nonrespondents (43% [20/46] vs 39% [7/18], P = 0.74; and 37% [17/46] vs 44% [8/18], P = 0.58, respectively). Aggregated survey responses are presented for items using a Likert scale in Figure 1, and for items using a yes/no or ordinal scale in Table 3.
| Items | % of Respondents Responding Affirmatively |
|---|---|
| Percentage of Respondents Ranking as First Choice* | |
| |
| Requestor activity | |
| What factors prompted you to request a report from CEP? (Please select all that apply.) | |
| My own time constraints | 28% (13/46) |
| CEP's ability to identify and synthesize evidence | 89% (41/46) |
| CEP's objectivity | 52% (24/46) |
| Recommendation from colleague | 30% (14/46) |
| Did you conduct any of your own literature searches before contacting CEP? | 67% (31/46) |
| Did you obtain and read any of the articles cited in CEP's report? | 63% (29/46) |
| Did you read the following sections of CEP's report? | |
| Evidence summary (at beginning of report) | 100% (45/45) |
| Introduction/background | 93% (42/45) |
| Methods | 84% (38/45) |
| Results | 98% (43/43) |
| Conclusion | 100% (43/43) |
| Report dissemination | |
| Did you share CEP's report with anyone NOT involved in requesting the report or in making the final decision? | 67% (30/45) |
| Did you share CEP's report with anyone outside of Penn? | 7% (3/45) |
| Requestor preferences | |
| Would it be helpful for CEP staff to call you after you receive any future CEP reports to answer any questions you might have? | 55% (24/44) |
| Following any future reports you request from CEP, would you be willing to complete a brief questionnaire? | 100% (44/44) |
| Please rank how you would prefer to receive reports from CEP in the future. | |
| E‐mail containing the report as a PDF attachment | 77% (34/44) |
| E‐mail containing a link to the report on CEP's website | 16% (7/44) |
| In‐person presentation by the CEP analyst writing the report | 18% (8/44) |
| In‐person presentation by the CEP director involved in the report | 16% (7/44) |
In general, respondents found reports easy to request, easy to use, timely, and relevant, resulting in high requestor satisfaction. In addition, 98% described the scope of content and level of detail as about right. Report impact was rated highly as well, with the evidence summary and conclusions rated as the most critical to decision making. A majority of respondents indicated that reports confirmed their tentative decision (77%, n = 34), whereas some changed their tentative decision (7%, n = 3), and others suggested the report had no effect on their tentative decision (16%, n = 7). Respondents indicated the amount of time that elapsed between receiving reports and making final decisions was 1 to 7 days (5%, n = 2), 8 to 30 days (40%, n = 17), 1 to 3 months (37%, n = 16), 4 to 6 months (9%, n = 4), or greater than 6 months (9%, n = 4). The most common reasons cited for requesting a report were the CEP's evidence synthesis skills and objectivity.
DISCUSSION
To our knowledge, this is the first comprehensive description and assessment of evidence synthesis activity by a hospital EPC in the United States. Our findings suggest that clinical and administrative leaders will request reports from a hospital EPC, and that hospital EPCs can promptly produce reports when requested. Moreover, these syntheses can address a wide range of clinical and policy topics, and can be disseminated through a variety of routes. Lastly, requestors are satisfied by these syntheses, and report that they inform decision making. These results suggest that EPCs may be an effective infrastructure paradigm for promoting evidence‐based decision making within healthcare provider organizations, and are consistent with previous analyses of hospital‐based EPCs.[21, 28, 29]
Over half of report requestors cited CEP's objectivity as a factor in their decision to request a report, underscoring the value of a neutral entity in an environment where clinical departments and hospital committees may have competing interests.[10] This asset was 1 of the primary drivers for establishing our hospital EPC. Concerns by clinical executives about the influence of industry and local politics on institutional decision making, and a desire to have clinical evidence more systematically and objectively integrated into decision making, fueled our center's funding.
The survey results also demonstrate that respondents were satisfied with the reports for many reasons, including readability, concision, timeliness, scope, and content, consistent with the evaluation of the French hospital‐based EPC CEDIT (French Committee for the Assessment and Dissemination of Technological Innovations).[29] Given the importance of readability, concision, and relevance that has been previously described,[16, 28, 30] nearly all CEP reports contain an evidence summary on the first page that highlights key findings in a concise, user‐friendly format.[31] The evidence summaries include bullet points that: (1) reference the most pertinent guideline recommendations along with their strength of recommendation and underlying quality of evidence; (2) organize and summarize study findings using the most critical clinical outcomes, including an assessment of the quality of the underlying evidence for each outcome; and (3) note important limitations of the findings.
Evidence syntheses must be timely to allow decision makers to act on the findings.[28, 32] The primary criticism of CEDIT was the lag between requests and report publication.[29] Rapid reviews, designed to inform urgent decisions, can overcome this challenge.[31, 33, 34] CEP reviews required approximately 2 months to complete on average, consistent with the most rapid timelines reported,[31, 33, 34] and much shorter than standard systematic review timelines, which can take up to 12 to 24 months.[33] Working with requestors to limit the scope of reviews to those issues most critical to a decision, using secondary resources when available, and hiring experienced research analysts help achieve these efficiencies.
The study by Bodeau‐Livinec also argues for the importance of report accessibility to ensure dissemination.[29] This is consistent with the CEP's approach, where all reports are posted on the UPHS internal website. Many also inform QI initiatives, as well as CDS interventions that address topics of general interest to acute care hospitals, such as venous thromboembolism (VTE) prophylaxis,[35] blood product transfusions,[36] sepsis care,[37, 38] and prevention of catheter‐associated urinary tract infections (CAUTI)[39] and hospital readmissions.[40] Most reports are also listed in an international database of rapid reviews,[23] and reports that address topics of general interest, have sufficient evidence to synthesize, and have no prior published systematic reviews are published in the peer‐reviewed literature.[41, 42]
The majority of reports completed by the CEP were evidence reviews, or systematic reviews of primary literature, suggesting that CEP reports often address questions previously unanswered by existing published systematic reviews; however, about a third of reports were evidence advisories, or summaries of evidence from preexisting secondary sources. The relative scarcity of high‐quality evidence bases in those reports where GRADE analyses were conducted might be expected, as requestors may be more likely to seek guidance when the evidence base on a topic is lacking. This was further supported by the small percentage (15%) of reports where adequate data of sufficient homogeneity existed to allow meta‐analyses. The small number of original meta‐analyses performed also reflects our reliance on secondary resources when available.
Only 7% of respondents reported that tentative decisions were changed based on their report. This is not surprising, as evidence reviews infrequently result in clear go or no go recommendations. More commonly, they address or inform complex clinical questions or pathways. In this context, the change/confirm/no effect framework may not completely reflect respondents' use of or benefit from reports. Thus, we included a diverse set of questions in our survey to best estimate the value of our reports. For example, when asked whether the report answered the question posed, informed their final decision, or was consistent with their final decision, 91%, 79%, and 71% agreed or strongly agreed, respectively. When asked whether they would request a report again if they had to do it all over, recommend CEP to their colleagues, and be likely to request reports in the future, at least 95% of survey respondents agreed or strongly agreed. In addition, no respondent indicated that their report was not timely enough to influence their decision. Moreover, only a minority of respondents expressed disappointment that the CEP's report did not provide actionable recommendations due to a lack of published evidence (9%, n = 4). Importantly, the large proportion of requestors indicating that reports confirmed their tentative decisions may be a reflection of hindsight bias.
The most apparent trend in the production of CEP reviews over time is the relative increase in requests by clinical departments, suggesting that the CEP is being increasingly consulted to help define best clinical practices. This is also supported by the relative increase in reports focused on policy or organizational/managerial systems. These findings suggest that hospital EPCs have value beyond the traditional realm of HTA.
This study has a number of limitations. First, not all of the eligible report requestors responded to our survey. Despite this, our response rate of 72% compares favorably with surveys published in medical journals.[43] In addition, nonresponse bias may be less important in physician surveys than surveys of the general population.[44] The similarity in requestor and report characteristics for respondents and nonrespondents supports this. Second, our survey of impact is self‐reported rather than an evaluation of actual decision making or patient outcomes. Thus, the survey relies on the accuracy of the responses. Third, recall bias must be considered, as some respondents were asked to evaluate reports that were greater than 1 year old. To reduce this bias, we asked respondents to consider the most recent report they requested, included that report as an attachment in the survey request, and only surveyed requestors from the most recent 4 of the CEP's 8 fiscal years. Fourth, social desirability bias could have also affected the survey responses, though it was likely minimized by the promise of confidentiality. Fifth, an examination of the impact of the CEP on costs was outside the scope of this evaluation; however, such information may be important to those assessing the sustainability or return on investment of such centers. Simple approaches we have previously used to approximate the value of our activities include: (1) estimating hospital cost savings resulting from decisions supported by our reports, such as the use of technologies like chlorhexidine for surgical site infections[45] or discontinuation of technologies like aprotinin for cardiac surgery[46]; and (2) estimating penalties avoided or rewards attained as a result of center‐led initiatives, such as those to increase VTE prophylaxis,[35] reduce CAUTI rates,[39] and reduce preventable mortality associated with sepsis.[37, 38] Similarly, given the focus of this study on the local evidence synthesis activities of our center, our examination did not include a detailed description of our CDS activities, or teaching activities, including our multidisciplinary workshops for physicians and nurses in evidence‐based QI[47] and our novel evidence‐based practice curriculum for medical students. Our study also did not include a description of our extramural activities, such as those supported by our contract with AHRQ as 1 of their 13 EPCs.[16, 17, 48, 49] A consideration of all of these activities enables a greater appreciation for the potential of such centers. Lastly, we examined a single EPC, which may not be representative of the diversity of hospitals and hospital staff across the United States. However, our EPC serves a diverse array of patient populations, clinical services, and service models throughout our multientity academic healthcare system, which may improve the generalizability of our experience to other settings.
As next steps, we recommend evaluation of other existing hospital EPCs nationally. Such studies could help hospitals and health systems ascertain which of their internal decisions might benefit from locally sourced rapid systematic reviews and determine whether an in‐house EPC could improve the value of care delivered.
In conclusion, our findings suggest that hospital EPCs within academic healthcare systems can efficiently synthesize and disseminate evidence for a variety of stakeholders. Moreover, these syntheses impact decision making in a variety of hospital contexts and clinical specialties. Hospitals and hospitalist leaders seeking to improve the implementation of evidence‐based practice at a systems level might consider establishing such infrastructure locally.
Acknowledgements
The authors thank Fran Barg, PhD (Department of Family Medicine and Community Health, University of Pennsylvania Perelman School of Medicine) and Joel Betesh, MD (University of Pennsylvania Health System) for their contributions to developing the survey. They did not receive any compensation for their contributions.
Disclosures: An earlier version of this work was presented as a poster at the 2014 AMA Research Symposium, November 7, 2014, Dallas, Texas. Mr. Jayakumar reports having received a University of Pennsylvania fellowship as a summer intern at the Center for Evidence‐based Practice. Dr. Umscheid cocreated and directs a hospital evidence‐based practice center, is the Senior Associate Director of an Agency for Healthcare Research and Quality Evidence‐Based Practice Center, and is a past member of the Medicare Evidence Development and Coverage Advisory Committee, which uses evidence reports developed by the Evidence‐based Practice Centers of the Agency for Healthcare Research and Quality. Dr. Umscheid's contribution was supported in part by the National Center for Research Resources, grant UL1RR024134, which is now at the National Center for Advancing Translational Sciences, grant UL1TR000003. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. None of the funders had a role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. Dr. Lavenberg, Dr. Mitchell, and Mr. Leas are employed as research analysts by a hospital evidence‐based practice center. Dr. Doshi is supported in part by a hospital evidence‐based practice center and is an Associate Director of an Agency for Healthcare Research and Quality Evidence‐based Practice Center. Dr. Goldmann is emeritus faculty at Penn, is supported in part by a hospital evidence‐based practice center, and is the Vice President and Chief Quality Assurance Officer in Clinical Solutions, a division of Elsevier, Inc., a global publishing company, and director of the division's Evidence‐based Medicine Center. Dr. Williams cocreated and codirects a hospital evidence‐based practice center. Dr. Brennan has oversight for and helped create a hospital evidence‐based practice center.
Hospital evidence‐based practice centers (EPCs) are structures with the potential to facilitate the integration of evidence into institutional decision making to close knowing‐doing gaps[1, 2, 3, 4, 5, 6]; in the process, they can support the evolution of their parent institutions into learning healthcare systems.[7] The potential of hospital EPCs stems from their ability to identify and adapt national evidence‐based guidelines and systematic reviews for the local setting,[8] create local evidence‐based guidelines in the absence of national guidelines, use local data to help define problems and assess the impact of solutions,[9] and implement evidence into practice through computerized clinical decision support (CDS) interventions and other quality‐improvement (QI) initiatives.[9, 10] As such, hospital EPCs have the potential to strengthen relationships and understanding between clinicians and administrators[11]; foster a culture of evidence‐based practice; and improve the quality, safety, and value of care provided.[10]
Formal hospital EPCs remain uncommon in the United States,[10, 11, 12] though their numbers have expanded worldwide.[13, 14] This growth is due not to any reduced role for national EPCs, such as the National Institute for Health and Clinical Excellence[15] in the United Kingdom, or the 13 EPCs funded by the Agency for Healthcare Research and Quality (AHRQ)[16, 17] in the United States. Rather, this growth is fueled by the heightened awareness that the value of healthcare interventions often needs to be assessed locally, and that clinical guidelines that consider local context have a greater potential to improve quality and efficiency.[9, 18, 19]
Despite the increasing number of hospital EPCs globally, their impact on administrative and clinical decision making has rarely been examined,[13, 20] especially for hospital EPCs in the United States. The few studies that have assessed the impact of hospital EPCs on institutional decision making have done so in the context of technology acquisition, neglecting the role hospital EPCs may play in the integration of evidence into clinical practice. For example, the Technology Assessment Unit at McGill University Health Center found that of the 27 reviews commissioned in their first 5 years, 25 were implemented, with 6 (24%) recommending investments in new technologies and 19 (76%) recommending rejection, for a reported net hospital savings of $10 million.[21] Understanding the activities and impact of hospital EPCs is particularly critical for hospitalist leaders, who could leverage hospital EPCs to inform efforts to support the quality, safety, and value of care provided, or who may choose to establish or lead such infrastructure. The availability of such opportunities could also support hospitalist recruitment and retention.
In 2006, the University of Pennsylvania Health System (UPHS) created the Center for Evidence‐based Practice (CEP) to support the integration of evidence into practice to strengthen quality, safety, and value.[10] Cofounded by hospitalists with formal training in clinical epidemiology, the CEP performs rapid systematic reviews of the scientific literature to inform local practice and policy. In this article, we describe the first 8 years of the CEP's evidence synthesis activities and examine its impact on decision making across the health system.
METHODS
Setting
The UPHS includes 3 acute care hospitals, and inpatient facilities specializing in acute rehabilitation, skilled nursing, long‐term acute care, and hospice, with a capacity of more than 1800 beds and 75,000 annual admissions, as well as primary care and specialty clinics with more than 2 million annual outpatient visits. The CEP is funded by and organized within the Office of the UPHS Chief Medical Officer, serves all UPHS facilities, has an annual budget of approximately $1 million, and is currently staffed by a hospitalist director, 3 research analysts, 6 physician and nurse liaisons, a health economist, biostatistician, administrator, and librarians, totaling 5.5 full time equivalents.
The mission of the CEP is to support the quality, safety, and value of care at Penn through evidence‐based practice. To accomplish this mission, the CEP performs rapid systematic reviews, translates evidence into practice through the use of CDS interventions and clinical pathways, and offers education in evidence‐based decision making to trainees, staff, and faculty. This study is focused on the CEP's evidence synthesis activities.
Typically, clinical and administrative leaders submit a request to the CEP for an evidence review, the request is discussed and approved at the weekly staff meeting, and a research analyst and clinical liaison are assigned to the request and communicate with the requestor to clearly define the question of interest. Subsequently, the research analyst completes a protocol, a draft search, and a draft report, each reviewed and approved by the clinical liaison and requestor. The final report is posted to the website, disseminated to all key stakeholders across the UPHS as identified by the clinical liaisons, and integrated into decision making through various routes, including in‐person presentations to decision makers, and CDS and QI initiatives.
Study Design
The study included an analysis of an internal database of evidence reviews and a survey of report requestors, and was exempted from institutional review board review. Survey respondents were informed that their responses would be confidential and did not receive incentives.
Internal Database of Reports
Data from the CEP's internal management database were analyzed for its first 8 fiscal years (July 2006June 2014). Variables included requestor characteristics, report characteristics (eg, technology reviewed, clinical specialty examined, completion time, and performance of meta‐analyses and GRADE [Grading of Recommendations Assessment, Development and Evaluation] analyses[22]), report use (eg, integration of report into CDS interventions) and dissemination beyond the UPHS (eg, submission to Center for Reviews and Dissemination [CRD] Health Technology Assessment [HTA] database[23] and to peer‐reviewed journals). Report completion time was defined as the time between the date work began on the report and the date the final report was sent to the requestor. The technology categorization scheme was adapted from that provided by Goodman (2004)[24] and the UK National Institute for Health Research HTA Programme.[25] We systematically assigned the technology reviewed in each report to 1 of 8 mutually exclusive categories. The clinical specialty examined in each report was determined using an algorithm (see Supporting Information, Appendix 1, in the online version of this article).
We compared the report completion times and the proportions of requestor types, technologies reviewed, and clinical specialties examined in the CEP's first 4 fiscal years (July 2006June 2010) to those in the CEP's second 4 fiscal years (July 2010June 2014) using t tests and 2 tests for continuous and categorical variables, respectively.
Survey
We conducted a Web‐based survey (see Supporting Information, Appendix 2, in the online version of this article) of all requestors of the 139 rapid reviews completed in the last 4 fiscal years. Participants who requested multiple reports were surveyed only about the most recent report. Requestors were invited to participate in the survey via e‐mail, and follow‐up e‐mails were sent to nonrespondents at 7, 14, and 16 days. Nonrespondents and respondents were compared with respect to requestor type (physician vs nonphysician) and topic evaluated (traditional HTA topics such as drugs, biologics, and devices vs nontraditional HTA topics such as processes of care). The survey was administered using REDCap[26] electronic data capture tools. The 44‐item questionnaire collected data on the interaction between the requestor and the CEP, report characteristics, report impact, and requestor satisfaction.
Survey results were imported into Microsoft Excel (Microsoft Corp, Redmond, WA) and SPSS (IBM, Armonk, NY) for analysis. Descriptive statistics were generated, and statistical comparisons were conducted using 2 and Fisher exact tests.
RESULTS
Evidence Synthesis Activity
The CEP has produced several different report products since its inception. Evidence reviews (57%, n = 142) consist of a systematic review and analysis of the primary literature. Evidence advisories (32%, n = 79) are summaries of evidence from secondary sources such as guidelines or systematic reviews. Evidence inventories (3%, n = 7) are literature searches that describe the quantity and focus of available evidence, without analysis or synthesis.[27]
The categories of technologies examined, including their definitions and examples, are provided in Table 1. Drugs (24%, n = 60) and devices/equipment/supplies (19%, n = 48) were most commonly examined. The proportion of reports examining technology types traditionally evaluated by HTA organizations significantly decreased when comparing the first 4 years of CEP activity to the second 4 years (62% vs 38%, P < 0.01), whereas reports examining less traditionally reviewed categories increased (38% vs 62%, P < 0.01). The most common clinical specialties represented by the CEP reports were nursing (11%, n = 28), general surgery (11%, n = 28), critical care (10%, n = 24), and general medicine (9%, n = 22) (see Supporting Information, Appendix 3, in the online version of this article). Clinical departments were the most common requestors (29%, n = 72) (Table 2). The proportion of requests from clinical departments significantly increased when comparing the first 4 years to the second 4 years (20% vs 36%, P < 0.01), with requests from purchasing committees significantly decreasing (25% vs 6%, P < 0.01). The overall report completion time was 70 days, and significantly decreased when comparing the first 4 years to the second 4 years (89 days vs 50 days, P < 0.01).
| Category | Definition | Examples | Total | 20072010 | 20112014 | P Value |
|---|---|---|---|---|---|---|
| Total | 249 (100%) | 109 (100%) | 140 (100%) | |||
| Drug | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of a pharmacologic agent | Celecoxib for pain in joint arthroplasty; colchicine for prevention of pericarditis and atrial fibrillation | 60 (24%) | 35 (32%) | 25 (18%) | 0.009 |
| Device, equipment, and supplies | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory that is intended for use in the prevention, diagnosis, or treatment of disease and does not achieve its primary intended purposes though chemical action or metabolism[50] | Thermometers for pediatric use; femoral closure devices for cardiac catheterization | 48 (19%) | 25 (23%) | 23 (16%) | 0.19 |
| Process of care | A report primarily examining a clinical pathway or a clinical practice guideline that significantly involves elements of prevention, diagnosis, and/or treatment or significantly incorporates 2 or more of the other technology categories | Preventing patient falls; prevention and management of delirium | 31 (12%) | 18 (17%) | 13 (9%) | 0.09 |
| Test, scale, or risk factor | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of a test intended to screen for, diagnose, classify, or monitor the progression of a disease | Computed tomography for acute chest pain; urine drug screening in chronic pain patients on opioid therapy | 31 (12%) | 8 (7%) | 23 (16%) | 0.03 |
| Medical/surgical procedure | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of a medical intervention that is not a drug, device, or test or of the application or removal of a device | Biliary drainage for chemotherapy patients; cognitive behavioral therapy for insomnia | 26 (10%) | 8 (7%) | 18 (13%) | 0.16 |
| Policy or organizational/managerial system | A report primarily examining laws or regulations; the organization, financing, or delivery of care, including settings of care; or healthcare providers | Medical care costs and productivity changes associated with smoking; physician training and credentialing for robotic surgery in obstetrics and gynecology | 26 (10%) | 4 (4%) | 22 (16%) | 0.002 |
| Support system | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of an intervention designed to provide a new or improved service to patients or healthcare providers that does not fall into 1 of the other categories | Reconciliation of data from differing electronic medical records; social media, text messaging, and postdischarge communication | 14 (6%) | 3 (3%) | 11 (8%) | 0.09 |
| Biologic | A report primarily examining the efficacy/effectiveness, safety, appropriate use, or cost of a product manufactured in a living system | Recombinant factor VIIa for cardiovascular surgery; osteobiologics for orthopedic fusions | 13 (5%) | 8 (7%) | 5 (4%) | 0.19 |
| Category | Total | 20072010 | 20112014 | P Value |
|---|---|---|---|---|
| ||||
| Total | 249 (100%) | 109 (100%) | 140 (100%) | |
| Clinical department | 72 (29%) | 22 (20%) | 50 (36%) | 0.007 |
| CMO | 47 (19%) | 21 (19%) | 26 (19%) | 0.92 |
| Purchasing committee | 35 (14%) | 27 (25%) | 8 (6%) | <0.001 |
| Formulary committee | 22 (9%) | 12 (11%) | 10 (7%) | 0.54 |
| Quality committee | 21 (8%) | 11 (10%) | 10 (7%) | 0.42 |
| Administrative department | 19 (8%) | 5 (5%) | 14 (10%) | 0.11 |
| Nursing | 14 (6%) | 4 (4%) | 10 (7%) | 0.23 |
| Other* | 19 (8%) | 7 (6%) | 12 (9%) | 0.55 |
Thirty‐seven (15%) reports included meta‐analyses conducted by CEP staff. Seventy‐five reports (30%) contained an evaluation of the quality of the evidence base using GRADE analyses.[22] Of these reports, the highest GRADE of evidence available for any comparison of interest was moderate (35%, n = 26) or high (33%, n = 25) in most cases, followed by very low (19%, n = 14) and low (13%, n = 10).
Reports were disseminated in a variety of ways beyond direct dissemination and presentation to requestors and posting on the center website. Thirty reports (12%) informed CDS interventions, 24 (10%) resulted in peer‐reviewed publications, and 204 (82%) were posted to the CRD HTA database.
Evidence Synthesis Impact
A total of 139 reports were completed between July 2010 and June 2014 for 65 individual requestors. Email invitations to participate in the survey were sent to the 64 requestors employed by the UPHS. The response rate was 72% (46/64). The proportions of physician requestors and traditional HTA topics evaluated were similar across respondents and nonrespondents (43% [20/46] vs 39% [7/18], P = 0.74; and 37% [17/46] vs 44% [8/18], P = 0.58, respectively). Aggregated survey responses are presented for items using a Likert scale in Figure 1, and for items using a yes/no or ordinal scale in Table 3.
| Items | % of Respondents Responding Affirmatively |
|---|---|
| Percentage of Respondents Ranking as First Choice* | |
| |
| Requestor activity | |
| What factors prompted you to request a report from CEP? (Please select all that apply.) | |
| My own time constraints | 28% (13/46) |
| CEP's ability to identify and synthesize evidence | 89% (41/46) |
| CEP's objectivity | 52% (24/46) |
| Recommendation from colleague | 30% (14/46) |
| Did you conduct any of your own literature searches before contacting CEP? | 67% (31/46) |
| Did you obtain and read any of the articles cited in CEP's report? | 63% (29/46) |
| Did you read the following sections of CEP's report? | |
| Evidence summary (at beginning of report) | 100% (45/45) |
| Introduction/background | 93% (42/45) |
| Methods | 84% (38/45) |
| Results | 98% (43/43) |
| Conclusion | 100% (43/43) |
| Report dissemination | |
| Did you share CEP's report with anyone NOT involved in requesting the report or in making the final decision? | 67% (30/45) |
| Did you share CEP's report with anyone outside of Penn? | 7% (3/45) |
| Requestor preferences | |
| Would it be helpful for CEP staff to call you after you receive any future CEP reports to answer any questions you might have? | 55% (24/44) |
| Following any future reports you request from CEP, would you be willing to complete a brief questionnaire? | 100% (44/44) |
| Please rank how you would prefer to receive reports from CEP in the future. | |
| E‐mail containing the report as a PDF attachment | 77% (34/44) |
| E‐mail containing a link to the report on CEP's website | 16% (7/44) |
| In‐person presentation by the CEP analyst writing the report | 18% (8/44) |
| In‐person presentation by the CEP director involved in the report | 16% (7/44) |
In general, respondents found reports easy to request, easy to use, timely, and relevant, resulting in high requestor satisfaction. In addition, 98% described the scope of content and level of detail as about right. Report impact was rated highly as well, with the evidence summary and conclusions rated as the most critical to decision making. A majority of respondents indicated that reports confirmed their tentative decision (77%, n = 34), whereas some changed their tentative decision (7%, n = 3), and others suggested the report had no effect on their tentative decision (16%, n = 7). Respondents indicated the amount of time that elapsed between receiving reports and making final decisions was 1 to 7 days (5%, n = 2), 8 to 30 days (40%, n = 17), 1 to 3 months (37%, n = 16), 4 to 6 months (9%, n = 4), or greater than 6 months (9%, n = 4). The most common reasons cited for requesting a report were the CEP's evidence synthesis skills and objectivity.
DISCUSSION
To our knowledge, this is the first comprehensive description and assessment of evidence synthesis activity by a hospital EPC in the United States. Our findings suggest that clinical and administrative leaders will request reports from a hospital EPC, and that hospital EPCs can promptly produce reports when requested. Moreover, these syntheses can address a wide range of clinical and policy topics, and can be disseminated through a variety of routes. Lastly, requestors are satisfied by these syntheses, and report that they inform decision making. These results suggest that EPCs may be an effective infrastructure paradigm for promoting evidence‐based decision making within healthcare provider organizations, and are consistent with previous analyses of hospital‐based EPCs.[21, 28, 29]
Over half of report requestors cited CEP's objectivity as a factor in their decision to request a report, underscoring the value of a neutral entity in an environment where clinical departments and hospital committees may have competing interests.[10] This asset was 1 of the primary drivers for establishing our hospital EPC. Concerns by clinical executives about the influence of industry and local politics on institutional decision making, and a desire to have clinical evidence more systematically and objectively integrated into decision making, fueled our center's funding.
The survey results also demonstrate that respondents were satisfied with the reports for many reasons, including readability, concision, timeliness, scope, and content, consistent with the evaluation of the French hospital‐based EPC CEDIT (French Committee for the Assessment and Dissemination of Technological Innovations).[29] Given the importance of readability, concision, and relevance that has been previously described,[16, 28, 30] nearly all CEP reports contain an evidence summary on the first page that highlights key findings in a concise, user‐friendly format.[31] The evidence summaries include bullet points that: (1) reference the most pertinent guideline recommendations along with their strength of recommendation and underlying quality of evidence; (2) organize and summarize study findings using the most critical clinical outcomes, including an assessment of the quality of the underlying evidence for each outcome; and (3) note important limitations of the findings.
Evidence syntheses must be timely to allow decision makers to act on the findings.[28, 32] The primary criticism of CEDIT was the lag between requests and report publication.[29] Rapid reviews, designed to inform urgent decisions, can overcome this challenge.[31, 33, 34] CEP reviews required approximately 2 months to complete on average, consistent with the most rapid timelines reported,[31, 33, 34] and much shorter than standard systematic review timelines, which can take up to 12 to 24 months.[33] Working with requestors to limit the scope of reviews to those issues most critical to a decision, using secondary resources when available, and hiring experienced research analysts help achieve these efficiencies.
The study by Bodeau‐Livinec also argues for the importance of report accessibility to ensure dissemination.[29] This is consistent with the CEP's approach, where all reports are posted on the UPHS internal website. Many also inform QI initiatives, as well as CDS interventions that address topics of general interest to acute care hospitals, such as venous thromboembolism (VTE) prophylaxis,[35] blood product transfusions,[36] sepsis care,[37, 38] and prevention of catheter‐associated urinary tract infections (CAUTI)[39] and hospital readmissions.[40] Most reports are also listed in an international database of rapid reviews,[23] and reports that address topics of general interest, have sufficient evidence to synthesize, and have no prior published systematic reviews are published in the peer‐reviewed literature.[41, 42]
The majority of reports completed by the CEP were evidence reviews, or systematic reviews of primary literature, suggesting that CEP reports often address questions previously unanswered by existing published systematic reviews; however, about a third of reports were evidence advisories, or summaries of evidence from preexisting secondary sources. The relative scarcity of high‐quality evidence bases in those reports where GRADE analyses were conducted might be expected, as requestors may be more likely to seek guidance when the evidence base on a topic is lacking. This was further supported by the small percentage (15%) of reports where adequate data of sufficient homogeneity existed to allow meta‐analyses. The small number of original meta‐analyses performed also reflects our reliance on secondary resources when available.
Only 7% of respondents reported that tentative decisions were changed based on their report. This is not surprising, as evidence reviews infrequently result in clear go or no go recommendations. More commonly, they address or inform complex clinical questions or pathways. In this context, the change/confirm/no effect framework may not completely reflect respondents' use of or benefit from reports. Thus, we included a diverse set of questions in our survey to best estimate the value of our reports. For example, when asked whether the report answered the question posed, informed their final decision, or was consistent with their final decision, 91%, 79%, and 71% agreed or strongly agreed, respectively. When asked whether they would request a report again if they had to do it all over, recommend CEP to their colleagues, and be likely to request reports in the future, at least 95% of survey respondents agreed or strongly agreed. In addition, no respondent indicated that their report was not timely enough to influence their decision. Moreover, only a minority of respondents expressed disappointment that the CEP's report did not provide actionable recommendations due to a lack of published evidence (9%, n = 4). Importantly, the large proportion of requestors indicating that reports confirmed their tentative decisions may be a reflection of hindsight bias.
The most apparent trend in the production of CEP reviews over time is the relative increase in requests by clinical departments, suggesting that the CEP is being increasingly consulted to help define best clinical practices. This is also supported by the relative increase in reports focused on policy or organizational/managerial systems. These findings suggest that hospital EPCs have value beyond the traditional realm of HTA.
This study has a number of limitations. First, not all of the eligible report requestors responded to our survey. Despite this, our response rate of 72% compares favorably with surveys published in medical journals.[43] In addition, nonresponse bias may be less important in physician surveys than surveys of the general population.[44] The similarity in requestor and report characteristics for respondents and nonrespondents supports this. Second, our survey of impact is self‐reported rather than an evaluation of actual decision making or patient outcomes. Thus, the survey relies on the accuracy of the responses. Third, recall bias must be considered, as some respondents were asked to evaluate reports that were greater than 1 year old. To reduce this bias, we asked respondents to consider the most recent report they requested, included that report as an attachment in the survey request, and only surveyed requestors from the most recent 4 of the CEP's 8 fiscal years. Fourth, social desirability bias could have also affected the survey responses, though it was likely minimized by the promise of confidentiality. Fifth, an examination of the impact of the CEP on costs was outside the scope of this evaluation; however, such information may be important to those assessing the sustainability or return on investment of such centers. Simple approaches we have previously used to approximate the value of our activities include: (1) estimating hospital cost savings resulting from decisions supported by our reports, such as the use of technologies like chlorhexidine for surgical site infections[45] or discontinuation of technologies like aprotinin for cardiac surgery[46]; and (2) estimating penalties avoided or rewards attained as a result of center‐led initiatives, such as those to increase VTE prophylaxis,[35] reduce CAUTI rates,[39] and reduce preventable mortality associated with sepsis.[37, 38] Similarly, given the focus of this study on the local evidence synthesis activities of our center, our examination did not include a detailed description of our CDS activities, or teaching activities, including our multidisciplinary workshops for physicians and nurses in evidence‐based QI[47] and our novel evidence‐based practice curriculum for medical students. Our study also did not include a description of our extramural activities, such as those supported by our contract with AHRQ as 1 of their 13 EPCs.[16, 17, 48, 49] A consideration of all of these activities enables a greater appreciation for the potential of such centers. Lastly, we examined a single EPC, which may not be representative of the diversity of hospitals and hospital staff across the United States. However, our EPC serves a diverse array of patient populations, clinical services, and service models throughout our multientity academic healthcare system, which may improve the generalizability of our experience to other settings.
As next steps, we recommend evaluation of other existing hospital EPCs nationally. Such studies could help hospitals and health systems ascertain which of their internal decisions might benefit from locally sourced rapid systematic reviews and determine whether an in‐house EPC could improve the value of care delivered.
In conclusion, our findings suggest that hospital EPCs within academic healthcare systems can efficiently synthesize and disseminate evidence for a variety of stakeholders. Moreover, these syntheses impact decision making in a variety of hospital contexts and clinical specialties. Hospitals and hospitalist leaders seeking to improve the implementation of evidence‐based practice at a systems level might consider establishing such infrastructure locally.
Acknowledgements
The authors thank Fran Barg, PhD (Department of Family Medicine and Community Health, University of Pennsylvania Perelman School of Medicine) and Joel Betesh, MD (University of Pennsylvania Health System) for their contributions to developing the survey. They did not receive any compensation for their contributions.
Disclosures: An earlier version of this work was presented as a poster at the 2014 AMA Research Symposium, November 7, 2014, Dallas, Texas. Mr. Jayakumar reports having received a University of Pennsylvania fellowship as a summer intern at the Center for Evidence‐based Practice. Dr. Umscheid cocreated and directs a hospital evidence‐based practice center, is the Senior Associate Director of an Agency for Healthcare Research and Quality Evidence‐Based Practice Center, and is a past member of the Medicare Evidence Development and Coverage Advisory Committee, which uses evidence reports developed by the Evidence‐based Practice Centers of the Agency for Healthcare Research and Quality. Dr. Umscheid's contribution was supported in part by the National Center for Research Resources, grant UL1RR024134, which is now at the National Center for Advancing Translational Sciences, grant UL1TR000003. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. None of the funders had a role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. Dr. Lavenberg, Dr. Mitchell, and Mr. Leas are employed as research analysts by a hospital evidence‐based practice center. Dr. Doshi is supported in part by a hospital evidence‐based practice center and is an Associate Director of an Agency for Healthcare Research and Quality Evidence‐based Practice Center. Dr. Goldmann is emeritus faculty at Penn, is supported in part by a hospital evidence‐based practice center, and is the Vice President and Chief Quality Assurance Officer in Clinical Solutions, a division of Elsevier, Inc., a global publishing company, and director of the division's Evidence‐based Medicine Center. Dr. Williams cocreated and codirects a hospital evidence‐based practice center. Dr. Brennan has oversight for and helped create a hospital evidence‐based practice center.
- , . “Bench to behavior”: translating comparative effectiveness research into improved clinical practice. Health Aff (Millwood). 2010;29(10):1891–1900.
- , , . Evaluating the status of “translating research into practice” at a major academic healthcare system. Int J Technol Assess Health Care. 2009;25(1):84–89.
- , , , . Five reasons that many comparative effectiveness studies fail to change patient care and clinical practice. Health Aff (Millwood). 2012;31(10):2168–2175.
- , , , , , . Fostering implementation of health services research findings into practice: a consolidated framework for advancing implementation science. Implement Sci. 2009;4(1):50.
- , . From best evidence to best practice: effective implementation of change in patients' care. Lancet. 2003;362(9391):1225–1230.
- , . Incentivizing “structures” over “outcomes” to bridge the knowing‐doing gap. JAMA Intern Med. 2015;175(3):354.
- Olsen L, Aisner D, McGinnis JM, eds. Institute of Medicine (US) Roundtable on Evidence‐Based Medicine. The Learning Healthcare System: Workshop Summary. Washington, DC: National Academies Press; 2007. Available at: http://www.ncbi.nlm.nih.gov/books/NBK53494/. Accessed October 29, 2014.
- , , , . Adapting clinical practice guidelines to local context and assessing barriers to their use. Can Med Assoc J. 2010;182(2):E78–E84.
- , , , . Integrating local data into hospital‐based healthcare technology assessment: two case studies. Int J Technol Assess Health Care. 2010;26(3):294–300.
- , , . Hospital‐based comparative effectiveness centers: translating research into practice to improve the quality, safety and value of patient care. J Gen Intern Med. 2010;25(12):1352–1355.
- , , , , . Health technology assessment at the University of California‐San Francisco. J Healthc Manag Am Coll Healthc Exec. 2011;56(1):15–29; discussion 29–30.
- , . Kaiser Permanente Southern California regional technology management process: evidence‐based medicine operationalized. Perm J. 2006;10(1):38–41.
- . Hospital‐based health technology assessment: developments to date. Pharmacoeconomics. 2014;32(9):819–824.
- , , , . Hospital based health technology assessment world‐wide survey. Available at: http://www.htai.org/fileadmin/HTAi_Files/ISG/HospitalBasedHTA/2008Files/HospitalBasedHTAISGSurveyReport.pdf. Accessed October 11, 2015.
- , . At the center of health care policy making: the use of health technology assessment at NICE. Med Decis Making. 2013;33(3):320–324.
- , , . Better information for better health care: the Evidence‐based Practice Center program and the Agency for Healthcare Research and Quality. Ann Intern Med. 2005;142(12 part 2):1035–1041.
- , . AHRQ's Effective Health Care Program: why comparative effectiveness matters. Am J Med Qual. 2009;24(1):67–70.
- , . Effect of clinical guidelines on medical practice: a systematic review of rigorous evaluations. Lancet. 1993;342(8883):1317–1322.
- , , , et al. Lost in knowledge translation: time for a map? J Contin Educ Health Prof. 2006;26(1):13–24.
- , , , . Effects and repercussions of local/hospital‐based health technology assessment (HTA): a systematic. Syst Rev. 2014;3:129.
- , , , et al. Impact of TAU Reports. McGill University Health Centre. Available at: https://francais.mcgill.ca/files/tau/FINAL_TAU_IMPACT_REPORT_FEB_2008.pdf. Published Feb 1, 2008. Accessed August 19, 2014.
- , , , et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ. 2008;336(7650):924–926.
- , , . Centre for Reviews and Dissemination databases: value, content, and developments. Int J Technol Assess Health Care. 2010;26(4):470–472.
- . HTA 101. Introduction to Health Technology Assessment. Available at: https://www.nlm.nih.gov/nichsr/hta101/ta10103.html. Accessed October 11, 2015.
- National Institute for Health Research. Remit. NIHR HTA Programme. Available at: http://www.nets.nihr.ac.uk/programmes/hta/remit. Accessed August 20, 2014.
- , , , , , . Research Electronic Data Capture (REDCap)—a metadata‐driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381.
- , , , . When the decision is what to decide: Using evidence inventory reports to focus health technology assessments. Int J Technol Assess Health Care. 2011;27(2):127–132.
- , . End‐user involvement in health technology assessment (HTA) development: a way to increase impact. Int J Technol Assess Health Care. 2005;21(02):263–267.
- , , , , . Impact of CEDIT recommendations: an example of health technology assessment in a hospital network. Int J Technol Assess Health Care. 2006;22(2):161–168.
- , , , . Increasing the relevance of research to health care managers: hospital CEO imperatives for improving quality and lowering costs. Health Care Manage Rev. 2007;32(2):150–159.
- , , , , . Evidence summaries: the evolution of a rapid review approach. Syst Rev. 2012;1(1):10.
- , , , . Health care decision makers' use of comparative effectiveness research: report from a series of focus groups. J Manag Care Pharm. 2013;19(9):745–754.
- , , , et al. Rapid reviews versus full systematic reviews: an inventory of current methods and practice in health technology assessment. Int J Technol Assess Health Care. 2008;24(2):133–139.
- , , , et al. EPC Methods: An Exploration of Methods and Context for the Production of Rapid Reviews. Rockville, MD: Agency for Healthcare Research and Quality; 2015. Available at: http://www.ncbi.nlm.nih.gov/books/NBK274092. Accessed March 5, 2015.
- , , , , . Effectiveness of a novel and scalable clinical decision support intervention to improve venous thromboembolism prophylaxis: a quasi‐experimental study. BMC Med Inform Decis Mak. 2012;12:92.
- . Order sets in electronic health records: principles of good practice. Chest. 2013;143(1):228–235.
- , , , et al. Development, implementation, and impact of an automated early warning and response system for sepsis. J Hosp Med. 2015;10(1):26–31.
- , , , et al. Clinician perception of the effectiveness of an automated early warning and response system for sepsis in an academic medical center. Ann Am Thorac Soc. 2015;12(10):1514–1519.
- , , , , , . Usability and impact of a computerized clinical decision support intervention designed to reduce urinary catheter utilization and catheter‐associated urinary tract infections. Infect Control Hosp Epidemiol. 2014;35(9):1147–1155.
- , , , et al. The readmission risk flag: using the electronic health record to automatically identify patients at risk for 30‐day readmission. J Hosp Med. 2013;8(12):689–695.
- , , , , , . A systematic review to inform institutional decisions about the use of extracorporeal membrane oxygenation during the H1N1 influenza pandemic. Crit Care Med. 2010;38(6):1398–1404.
- , , , . Heparin flushing and other interventions to maintain patency of central venous catheters: a systematic review. J Adv Nurs. 2009;65(10):2007–2021.
- , , . Response rates to mail surveys published in medical journals. J Clin Epidemiol. 1997;50(10):1129–1136.
- , . Physician response to surveys: a review of the literature. Am J Prev Med. 2001;20(1):61–67.
- , , , , . Systematic review and cost analysis comparing use of chlorhexidine with use of iodine for preoperative skin antisepsis to prevent surgical site infection. Infect Control Hosp Epidemiol. 2010;31(12):1219–1229.
- , , . Antifibrinolytic use in adult cardiac surgery. Curr Opin Hematol. 2007;14(5):455–467.
- , , , , . Teaching evidence assimilation for collaborative health care (TEACH) 2009–2014: building evidence‐based capacity within health care provider organizations. EGEMS (Wash DC). 2015;3(2):1165.
- , , , , , . Cleaning hospital room surfaces to prevent health care‐associated infections: a technical brief [published online August 11, 2015]. Ann Intern Med. doi:10.7326/M15‐1192.
- , , , Healthcare Infection Control Practices Advisory Committee. Updating the guideline development methodology of the Healthcare Infection Control Practices Advisory Committee (HICPAC). Am J Infect Control. 2010;38(4):264–273.
- U.S. Food and Drug Administration. FDA basics—What is a medical device? Available at: http://www.fda.gov/AboutFDA/Transparency/Basics/ucm211822.htm. Accessed November 12, 2014.
- , . “Bench to behavior”: translating comparative effectiveness research into improved clinical practice. Health Aff (Millwood). 2010;29(10):1891–1900.
- , , . Evaluating the status of “translating research into practice” at a major academic healthcare system. Int J Technol Assess Health Care. 2009;25(1):84–89.
- , , , . Five reasons that many comparative effectiveness studies fail to change patient care and clinical practice. Health Aff (Millwood). 2012;31(10):2168–2175.
- , , , , , . Fostering implementation of health services research findings into practice: a consolidated framework for advancing implementation science. Implement Sci. 2009;4(1):50.
- , . From best evidence to best practice: effective implementation of change in patients' care. Lancet. 2003;362(9391):1225–1230.
- , . Incentivizing “structures” over “outcomes” to bridge the knowing‐doing gap. JAMA Intern Med. 2015;175(3):354.
- Olsen L, Aisner D, McGinnis JM, eds. Institute of Medicine (US) Roundtable on Evidence‐Based Medicine. The Learning Healthcare System: Workshop Summary. Washington, DC: National Academies Press; 2007. Available at: http://www.ncbi.nlm.nih.gov/books/NBK53494/. Accessed October 29, 2014.
- , , , . Adapting clinical practice guidelines to local context and assessing barriers to their use. Can Med Assoc J. 2010;182(2):E78–E84.
- , , , . Integrating local data into hospital‐based healthcare technology assessment: two case studies. Int J Technol Assess Health Care. 2010;26(3):294–300.
- , , . Hospital‐based comparative effectiveness centers: translating research into practice to improve the quality, safety and value of patient care. J Gen Intern Med. 2010;25(12):1352–1355.
- , , , , . Health technology assessment at the University of California‐San Francisco. J Healthc Manag Am Coll Healthc Exec. 2011;56(1):15–29; discussion 29–30.
- , . Kaiser Permanente Southern California regional technology management process: evidence‐based medicine operationalized. Perm J. 2006;10(1):38–41.
- . Hospital‐based health technology assessment: developments to date. Pharmacoeconomics. 2014;32(9):819–824.
- , , , . Hospital based health technology assessment world‐wide survey. Available at: http://www.htai.org/fileadmin/HTAi_Files/ISG/HospitalBasedHTA/2008Files/HospitalBasedHTAISGSurveyReport.pdf. Accessed October 11, 2015.
- , . At the center of health care policy making: the use of health technology assessment at NICE. Med Decis Making. 2013;33(3):320–324.
- , , . Better information for better health care: the Evidence‐based Practice Center program and the Agency for Healthcare Research and Quality. Ann Intern Med. 2005;142(12 part 2):1035–1041.
- , . AHRQ's Effective Health Care Program: why comparative effectiveness matters. Am J Med Qual. 2009;24(1):67–70.
- , . Effect of clinical guidelines on medical practice: a systematic review of rigorous evaluations. Lancet. 1993;342(8883):1317–1322.
- , , , et al. Lost in knowledge translation: time for a map? J Contin Educ Health Prof. 2006;26(1):13–24.
- , , , . Effects and repercussions of local/hospital‐based health technology assessment (HTA): a systematic. Syst Rev. 2014;3:129.
- , , , et al. Impact of TAU Reports. McGill University Health Centre. Available at: https://francais.mcgill.ca/files/tau/FINAL_TAU_IMPACT_REPORT_FEB_2008.pdf. Published Feb 1, 2008. Accessed August 19, 2014.
- , , , et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ. 2008;336(7650):924–926.
- , , . Centre for Reviews and Dissemination databases: value, content, and developments. Int J Technol Assess Health Care. 2010;26(4):470–472.
- . HTA 101. Introduction to Health Technology Assessment. Available at: https://www.nlm.nih.gov/nichsr/hta101/ta10103.html. Accessed October 11, 2015.
- National Institute for Health Research. Remit. NIHR HTA Programme. Available at: http://www.nets.nihr.ac.uk/programmes/hta/remit. Accessed August 20, 2014.
- , , , , , . Research Electronic Data Capture (REDCap)—a metadata‐driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381.
- , , , . When the decision is what to decide: Using evidence inventory reports to focus health technology assessments. Int J Technol Assess Health Care. 2011;27(2):127–132.
- , . End‐user involvement in health technology assessment (HTA) development: a way to increase impact. Int J Technol Assess Health Care. 2005;21(02):263–267.
- , , , , . Impact of CEDIT recommendations: an example of health technology assessment in a hospital network. Int J Technol Assess Health Care. 2006;22(2):161–168.
- , , , . Increasing the relevance of research to health care managers: hospital CEO imperatives for improving quality and lowering costs. Health Care Manage Rev. 2007;32(2):150–159.
- , , , , . Evidence summaries: the evolution of a rapid review approach. Syst Rev. 2012;1(1):10.
- , , , . Health care decision makers' use of comparative effectiveness research: report from a series of focus groups. J Manag Care Pharm. 2013;19(9):745–754.
- , , , et al. Rapid reviews versus full systematic reviews: an inventory of current methods and practice in health technology assessment. Int J Technol Assess Health Care. 2008;24(2):133–139.
- , , , et al. EPC Methods: An Exploration of Methods and Context for the Production of Rapid Reviews. Rockville, MD: Agency for Healthcare Research and Quality; 2015. Available at: http://www.ncbi.nlm.nih.gov/books/NBK274092. Accessed March 5, 2015.
- , , , , . Effectiveness of a novel and scalable clinical decision support intervention to improve venous thromboembolism prophylaxis: a quasi‐experimental study. BMC Med Inform Decis Mak. 2012;12:92.
- . Order sets in electronic health records: principles of good practice. Chest. 2013;143(1):228–235.
- , , , et al. Development, implementation, and impact of an automated early warning and response system for sepsis. J Hosp Med. 2015;10(1):26–31.
- , , , et al. Clinician perception of the effectiveness of an automated early warning and response system for sepsis in an academic medical center. Ann Am Thorac Soc. 2015;12(10):1514–1519.
- , , , , , . Usability and impact of a computerized clinical decision support intervention designed to reduce urinary catheter utilization and catheter‐associated urinary tract infections. Infect Control Hosp Epidemiol. 2014;35(9):1147–1155.
- , , , et al. The readmission risk flag: using the electronic health record to automatically identify patients at risk for 30‐day readmission. J Hosp Med. 2013;8(12):689–695.
- , , , , , . A systematic review to inform institutional decisions about the use of extracorporeal membrane oxygenation during the H1N1 influenza pandemic. Crit Care Med. 2010;38(6):1398–1404.
- , , , . Heparin flushing and other interventions to maintain patency of central venous catheters: a systematic review. J Adv Nurs. 2009;65(10):2007–2021.
- , , . Response rates to mail surveys published in medical journals. J Clin Epidemiol. 1997;50(10):1129–1136.
- , . Physician response to surveys: a review of the literature. Am J Prev Med. 2001;20(1):61–67.
- , , , , . Systematic review and cost analysis comparing use of chlorhexidine with use of iodine for preoperative skin antisepsis to prevent surgical site infection. Infect Control Hosp Epidemiol. 2010;31(12):1219–1229.
- , , . Antifibrinolytic use in adult cardiac surgery. Curr Opin Hematol. 2007;14(5):455–467.
- , , , , . Teaching evidence assimilation for collaborative health care (TEACH) 2009–2014: building evidence‐based capacity within health care provider organizations. EGEMS (Wash DC). 2015;3(2):1165.
- , , , , , . Cleaning hospital room surfaces to prevent health care‐associated infections: a technical brief [published online August 11, 2015]. Ann Intern Med. doi:10.7326/M15‐1192.
- , , , Healthcare Infection Control Practices Advisory Committee. Updating the guideline development methodology of the Healthcare Infection Control Practices Advisory Committee (HICPAC). Am J Infect Control. 2010;38(4):264–273.
- U.S. Food and Drug Administration. FDA basics—What is a medical device? Available at: http://www.fda.gov/AboutFDA/Transparency/Basics/ucm211822.htm. Accessed November 12, 2014.
© 2015 Society of Hospital Medicine
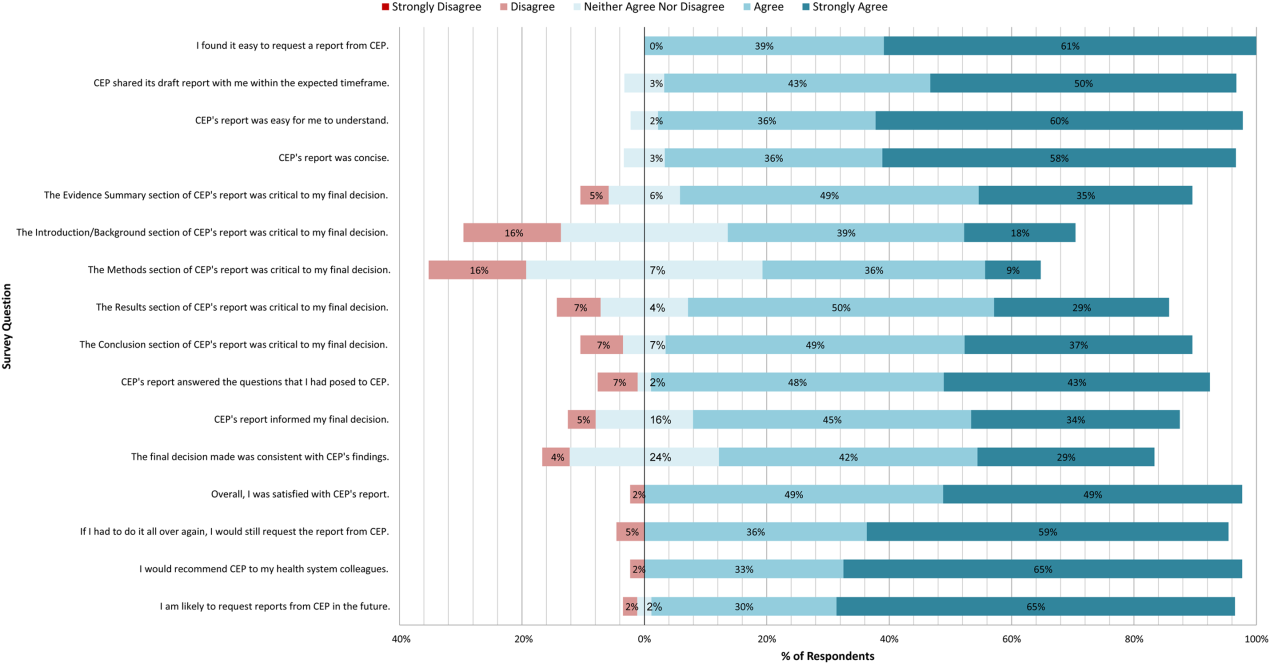
Changeover of Trainee Doctors
In England, the day when trainee doctors start work for the first time in their careers or rotate to a different hospital is the first Wednesday of August. This is often referred to as the Black Wednesday in the National Health Service (NHS), as it is widely perceived that inexperience and nonfamiliarity with the new hospital systems and policies in these first few weeks lead to increased medical errors and mismanagement and may therefore cost lives.[1] However, there is very little evidence in favor of this widely held view in the NHS. A 2009 English study found a small but significant increase of 6% in the odds of death for inpatients admitted in the week following the first Wednesday in August than in the week following the last Wednesday in July, whereas a previous report did not support this.[2, 3] In the United States, the resident trainee doctor's changeover occurs in July, and its negative impact on patient outcomes is often dubbed the July phenomenon.[4] With conflicting reports of the July phenomenon on patient outcomes,[5, 6, 7] Young et al. systematically reviewed 39 studies and concluded that the July phenomenon exists in that there is increased mortality around the changeover period.[4]
It can be hypothesized that glycemic control in inpatients with diabetes would be worse in the immediate period following changeover of trainee doctors for the same reasons mentioned earlier that impact mortality. However, contrary to expectations, a recent single‐hospital study from the United States reported that changeover of resident trainee doctors did not worsen inpatient glycemic control.[8] Although the lack of confidence among trainee doctors in inpatient diabetes management has been clearly demonstrated in England,[9] the impact of August changeover of trainee doctors on inpatient glycemic control is unknown. The aim of this study was to determine whether the August changeover of trainee doctors impacted on glycemic control in inpatients with diabetes in a single English hospital.
MATERIAL AND METHODS
The study setting was a medium‐sized 550‐bed hospital in England that serves a population of approximately 360,000 residents. Capillary blood glucose (CBG) readings for adult inpatients across all wards were downloaded from the Precision Web Point‐of‐Care Data Management System (Abbott Diabetes Care Inc., Alameda, CA), an electronic database where all the CBG readings for inpatients are stored. Patient administration data were used to identify those with diabetes admitted to the hospital for at least 1 day, and only their CBG readings were included in this study. Glucometrics, a term coined by Goldberg et al., refers to standardized glucose performance metrics to assess the quality of inpatient glycemic control.[10] In this study, patient‐day glucometric measures were used, as they are considered the best indicator of inpatient glycemic control compared to other glucometrics.[10] Patient‐day glucometrics were analyzed for 4 weeks before and after Black Wednesday for the years 2012, 2013, and 2014 using Microsoft Excel 2007 (Microsoft Corp., Redmond, WA) and R version 3.1.0 (The R Foundation, Vienna, Austria). Patient‐day glucometrics analyzed were hypoglycemia (any CBG 2.2 mmol/L [40 mg/dL], any CBG 2.9 mmol/L [52 mg/dL], any CBG 3.9 mmol/L [72 mg/dL]), normoglycemia (mean CBGs between 4 and 12 mmol/L [73‐216 mg/dL]), hyperglycemia (any CBG 12.1 mmol/L [218 mg/dL]), and mean CBG. Proportions were compared using the z test, whereas sample means between the groups were compared by nonparametric Mann‐Whitney U tests, as per statistical literature.[11] All P values are 2‐tailed, and 0.05 was considered statistically significant.
Patient characteristics and healthcare professional's workload were identified as potential causes of variation in CBG readings. Regression analysis of covariance was used to identify and adjust for these factors when comparing mean glucose readings. Binomial logistic regression was used to adjust proportions of patients‐days with readings out of range and patient‐days with mean readings within range. Variables tested were length of stay as a proxy for severity of condition, number of patients whose CBG were measured in the hospital in a day as a proxy for the healthcare professional's workload, and location of the patient to account for variation in patient characteristics as the wards were specialty based. Goodness of fit was tested using the R2 value in the linear model, which indicates the proportion of outcome that is explained by the model. For binomial models, McFadden's pseudo R2 (pseudo‐R2McFadden) was used as advised for logistic models. McFadden's pseudo‐R2 ranges from 0 to 1, but unlike R2 in ordinary linear regression, values tend to be significantly lower: McFadden's pseudo R2 values between 0.2 and 0.4 indicate excellent fit.[12]
RESULTS
A total of 16,870 patient‐day CBG measures in 2730 inpatients with diabetes were analyzed. The results of all regressions are presented in Table 1. The coefficients in the first model represent the effect of each covariate on mean patient‐day CBG. For example, each extra day of hospitalization was associated with a 0.02 mmol/L (0.36 mg/dL) increase in mean patient‐day reading, ceteris paribus. The remaining models indicate the change in relative risk (in this case the proportion of patient‐days) associated with the covariates. For example, in patients who were hospitalized for 3 days, the proportion of patient‐days with at least 1 CBG greater than 12 mmol/L (216 mg/dL) was 1.01 times the comparable proportion of patients who were hospitalized for 2 days. Each additional day in the hospital significantly increased the mean CBG by 0.015 mmol/L (0.27 mg/dL) and increased the risk of having at least 1 reading below 3.9 mmol/L (72 mg/dL) or above 12 mmol/L (216 mg/dL). Monitoring more patients in a day also affected outcomes, although the effect was small. Each additional patient monitored reduced mean patient‐day CBG by 0.011 mmol/L (0.198 mg/dL) and increased the proportion of patients with at least 1 reading below 4 mmol/L (72 mg/dL) 1.01 times. Location of the patient also significantly affected CBG readings. This could have been due to either ward or patient characteristics, but lack of data on each ward's healthcare personnel and individual patient characteristics prevented further analysis of this effect, and therefore the results were used for adjustment only. All models have relatively low predictive power, as demonstrated by the low R2 and pseudo‐R2McFadden values. In the linear model that estimated the effect of covariates on mean patient‐day CBG, the R2 is 0.0270, indicating that only 2.70% of results were explained by the covariates in the model. The pseudo‐R2McFadden varied between 0.0146 and 0.0540, as presented in Table 1. Although the pseudo‐R2McFadden generally had lower values than the R2 for the linear models, values of 0.0540 and below are considered to be relatively low.[12]
| Covariate | Outcome | |||||
|---|---|---|---|---|---|---|
| Change in Mean CBG for Each Patient‐Day, mmol/L (mg/dL) | Change in % of Patient‐Days With Any CBG 2.2 mmol/L (40 mg/dL) | Change in % of Patient‐Days With Any CBG 2.9 mmol/L (52 mg/dL) | Change in % of Patient‐Days With Any CBG 3.9 mmol/L (72 mg/dL) | Change in % of Patient‐Days With Mean CBG Between 4 and 12 mmol/L (73216 mg/dL) | Change in % of Patient‐Days With Any CBG >12 mmol/L (218 mg/dL) | |
| ||||||
| Additional day in the hospital | 0.015 (0.27), P 0.001* | 1.00, P = 0.605 | 1.00, P = 0.986 | 1.005, P = 0.004 | 0.99, P 0.001* | 1.01, P 0.001* |
| Additional patients monitored | 0.011 (0.198), P 0.001* | 1.01, P = 0.132 | 1.01, P = 0.084 | 1.01, P = 0.021 | 1.00, P = 0.128 | 0.997, P = 0.011 |
| Ward (range) |
0.5913.68(10.62246.24) |
0.3722.71 | 03.62 | 03.10 | 047,124.14 | 04,094,900 |
| R2/pseudo‐R2McFadden | 0.0247 | 0.0503 | 0.0363 | 0.0270 | 0.0140 | 0.0243 |
Table 2 summarizes outcomes for the 3 years individually. The results suggest that all indices of inpatient glycemic control that were analyzedhypoglycemia, normoglycemia, hyperglycemia, and mean CBGdid not worsen in August compared to July that year. The results are presented after adjustment for variation in the length of stay, number of patients monitored in a day, and location of the patient. Their effect on the difference in proportions of patients with at least 1 reading out of range and mean reading within range were not statistically significant. However, their effect on mean patient‐day CBG measures was statistically significant, although the effect was only a small decrease (0.4 mmol/L or 7.2 mg/dL) in the mean CBG (see Supporting Table 1 in the online version of this article for unadjusted readings).
| 2012 | 2013 | 2014 | ||||
|---|---|---|---|---|---|---|
| Before Changeover | After Changeover | Before Changeover | After Changeover | Before Changeover | After Changeover | |
| ||||||
| No. of inpatients with diabetes whose CBG readings were analyzed | 470 | 482 | 464 | 427 | 440 | 447 |
| No. of patient‐day CBG readings analyzed | 2917 | 3159 | 3097 | 2588 | 2484 | 2625 |
| Mean no. of CBG readings per patient‐day (range) | 2.5 (127) | 2.5 (123), P = 0.676 | 2.6 (121) | 2.4 (118), P = 0.009* | 2.5 (120) | 2.4 (120), P = 0.028 |
| Mean no. of CBG readings per patient‐day (range) in those where at least 1 reading was CBG 3.9 mmol/L (72 mg/dL) or CBG 12.1 mmol/L (218 mg/dL) | 3.8 (127) | 3.8 (123) | 3.7 (121) | 3.5 (118) | 3.2 (120) | 3.5 (120) |
| Mean no. of CBG readings per patient‐day (range) in those where all CBG readings were between 4 and 12 mmol/L (73216mg/dL) | 1.8 (127) | 1.8 (112) | 1.8 (112) | 1.8 (117) | 1.7 (111) | 1.7 (115) |
| % of patient‐days with any CBG 2.2 mmol/L (40 mg/dL) | 0.99% | 1.09%, P = 0.703 | 1.03% | 0.88%, P = 0.544 | 0.84% | 0.87%, P = 0.927 |
| % of patient‐days with any CBG 2.9 mmol/L (52 mg/dL) | 2.53% | 2.68%, P = 0.708 | 2.63% | 1.35%, P = 0.490 | 2.24% | 2.31%, P = 0.874 |
| % of patient‐days with any CBG 3.9 mmol/L (72 mg/dL) | 7.25% | 7.42%, P = 0.792 | 7.56 % | 6.93%, P = 0.361 | 6.55% | 6.70%, P = 0.858 |
| % of patient‐days with mean CBG between 4 and 12 mmol/L (73216 mg/dL) | 79.10% | 79.89%, P = 0.446 | 78.69% | 78.58%, P = 0.924 | 78.65% | 78.61%, P = 0.973 |
| % of patient‐days with any CBG 12.1 mmol/L (218 mg/dL) | 32.32% | 31.40%, P = 0.443 | 32.29% | 32.88%, P = 0.634 | 32.78% | 32.66%, P = 0.928 |
| Median of mean CBG for each patient‐day in mmol/L (mg/dL) | 8.0 (144.6) | 7.8 (140.0) | 8.4 (151.5) | 8.3 (150.2) | 8.9 (159.8) | 8.8 (157.8) |
| Mean of mean CBG for each patient‐day in mmol/L (standard deviation) | 9.1 (4.0) | 8.8 (4.1), P = 0.033+ | 9.4 (4.1) | 9.2 (4.0), P = 0.075 | 9.8 (4.1) | 9.6 (3.8), P = 0.189 |
DISCUSSION
This study shows that contrary to expectation, inpatient glycemic control did not worsen in the 4 weeks following the August changeover of trainee doctors for the years 2012, 2013, and 2014. In fact, inpatient glycemic control was marginally better in the first 4 weeks after changeover each year compared to the preceding 4 weeks before changeover. There may be several reasons for the findings in this study. First, since 2010 in this hospital and since 2012 nationally (further to direction from NHS England Medical Director Sir Bruce Keogh), it has become established practice that newly qualified trainee doctors shadow their colleagues at work a week prior to Black Wednesday.[13, 14] The purpose of this practice, called the preparation for professional practice is to familiarize trainee doctors with the hospital protocols and systems, improve their confidence, and potentially reduce medical errors when starting work. Second, since 2012, this hospital has also implemented the Joint British Diabetes Societies' national guidelines in managing inpatients with diabetes.[15] These guidelines are widely publicized on the changeover day during the trainee doctor's induction program. Finally, since 2012, a diabetes‐specific interactive 1‐hour educational program for trainee doctors devised by this hospital was implemented during the changeover period, which takes them through practical and problem‐solving case scenarios related to inpatient glycemic management, in particular prevention of hypoglycemia and hospital‐acquired diabetic ketoacidosis.[16] Attendance was mandatory, and informal feedback from trainee doctors about the educational program was extremely positive.
There are several limitations in this study. It could be argued that trainee doctors have very little impact on glycemic control in inpatients with diabetes. In NHS hospitals, trainee doctors are often the first port of call for managing glycemic issues in inpatients both in and out of hours, who in turn may or may not call the inpatient diabetes team wherever available. Therefore, trainee doctors' impact on glycemic control in inpatients with diabetes cannot be understated. However, it is acknowledged that in this study, a number of other factors that influence inpatient glycemic control, such as individual patient characteristics, medication errors, and the knowledge and confidence levels of individual trainee doctors, were not accounted for. Nevertheless, such factors are unlikely to have been significantly different over the 3‐year period. A further limitation was the unavailability of hospital‐wide electronic CBG data prior to 2012 to determine whether changeover impacted on inpatient glycemic control prior to this period. Another limitation was the dependence on patient administration data to identify those with diabetes, as it is well recognized that coded data in hospital data management systems can be inaccurate, though this has significantly improved over the years.[17] Finally, the most important limitation is that this is a single‐hospital study, and so the results may not be applicable to other English hospitals. Nevertheless, the finding of this study is similar to the finding in the single‐hospital study from the United States.[8]
The finding that glycemic control in inpatients with diabetes did not worsen in the 4 weeks following changeover of trainee doctors compared to the 4 weeks before changeover each year suggests that appropriate forethought and planning by the deanery foundation school and the inpatient diabetes team has prevented the anticipated deterioration of glycemic control during the August changeover of trainee doctors in this English hospital.
Disclosures: R.R. and G.R. conceived and designed the study. R.R. collected data and drafted the manuscript. R.R., D.J., and G.R. analyzed and interpreted the data. D.J. provided statistical input for analysis of the data. R.R., D.J., and G.R. critically revised the manuscript for intellectual content. All authors have approved the final version. The authors report no conflicts of interest.
- . Black Wednesday: today junior doctors will start work—and cause A4(9):e7103.
- , . The killing season—fact or fiction? BMJ. 1994;309(6970):1690.
- , , , , , . “July effect”: impact of the academic year‐end changeover on patient outcomes: a systematic review. Ann Intern Med. 2011;155(5):309–315.
- , . A July spike in fatal medication errors: a possible effect of new medical residents. J Gen Intern Med. 2010;25(8):774–779.
- , , , et al. Complications and death at the start of the new academic year: is there a July phenomenon? J Trauma. 2010;68(1):19–22.
- , , , . Errors and adverse outcomes on a surgical service: what is the role of residents? J Surg Res. 2004;122(2):162–166.
- , , , . Is There a “July Effect” for inpatient glycemic control? Endocr Pract. 2014;20(19):919–924.
- , , , et al.; TOPDOC Diabetes Study Team. Lack of confidence among trainee doctors in the management of diabetes: the Trainees Own Perception of Delivery of Care (TOPDOC) Diabetes Study. QJM. 2011;104(9):761–766.
- , , , et al. “Glucometrics”—assessing the quality of inpatient glucose management. Diabetes Technol Ther. 2006;8(5):560–569.
- , , . Statistics for Business and Economics. 5th ed. Upper Saddle River, NJ: Prentice Hall; 2002.
- , , . Stated choice methods. New York, NY: Cambridge University Press; 2000.
- Health Education East of England. Preparing for professional practice. Available at: https://heeoe.hee.nhs.uk/foundation_faq. Accessed October 07, 2015.
- Department of Health. Lives will be saved as junior doctors shadow new role 2012. Available at: https://www.gov.uk/government/news/lives‐will‐be‐saved‐as‐junior‐doctors‐shadow‐new‐role. Accessed October 29, 2014.
- Association of British Clinical Diabetologists. Joint British Diabetes Societies for Inpatient Care. Available at: http://www.diabetologists‐abcd.org.uk/JBDS/JBDS.htm. Accessed October 8, 2014.
- , , . An interactive 1‐h educational programme for junior doctors, increases their confidence and improves inpatient diabetes care. Diabet Med. 2012;29(12):1574–1578.
- , , , et al. Systematic review of discharge coding accuracy. J Public Health (Oxf). 2012;34(1):138–148.
In England, the day when trainee doctors start work for the first time in their careers or rotate to a different hospital is the first Wednesday of August. This is often referred to as the Black Wednesday in the National Health Service (NHS), as it is widely perceived that inexperience and nonfamiliarity with the new hospital systems and policies in these first few weeks lead to increased medical errors and mismanagement and may therefore cost lives.[1] However, there is very little evidence in favor of this widely held view in the NHS. A 2009 English study found a small but significant increase of 6% in the odds of death for inpatients admitted in the week following the first Wednesday in August than in the week following the last Wednesday in July, whereas a previous report did not support this.[2, 3] In the United States, the resident trainee doctor's changeover occurs in July, and its negative impact on patient outcomes is often dubbed the July phenomenon.[4] With conflicting reports of the July phenomenon on patient outcomes,[5, 6, 7] Young et al. systematically reviewed 39 studies and concluded that the July phenomenon exists in that there is increased mortality around the changeover period.[4]
It can be hypothesized that glycemic control in inpatients with diabetes would be worse in the immediate period following changeover of trainee doctors for the same reasons mentioned earlier that impact mortality. However, contrary to expectations, a recent single‐hospital study from the United States reported that changeover of resident trainee doctors did not worsen inpatient glycemic control.[8] Although the lack of confidence among trainee doctors in inpatient diabetes management has been clearly demonstrated in England,[9] the impact of August changeover of trainee doctors on inpatient glycemic control is unknown. The aim of this study was to determine whether the August changeover of trainee doctors impacted on glycemic control in inpatients with diabetes in a single English hospital.
MATERIAL AND METHODS
The study setting was a medium‐sized 550‐bed hospital in England that serves a population of approximately 360,000 residents. Capillary blood glucose (CBG) readings for adult inpatients across all wards were downloaded from the Precision Web Point‐of‐Care Data Management System (Abbott Diabetes Care Inc., Alameda, CA), an electronic database where all the CBG readings for inpatients are stored. Patient administration data were used to identify those with diabetes admitted to the hospital for at least 1 day, and only their CBG readings were included in this study. Glucometrics, a term coined by Goldberg et al., refers to standardized glucose performance metrics to assess the quality of inpatient glycemic control.[10] In this study, patient‐day glucometric measures were used, as they are considered the best indicator of inpatient glycemic control compared to other glucometrics.[10] Patient‐day glucometrics were analyzed for 4 weeks before and after Black Wednesday for the years 2012, 2013, and 2014 using Microsoft Excel 2007 (Microsoft Corp., Redmond, WA) and R version 3.1.0 (The R Foundation, Vienna, Austria). Patient‐day glucometrics analyzed were hypoglycemia (any CBG 2.2 mmol/L [40 mg/dL], any CBG 2.9 mmol/L [52 mg/dL], any CBG 3.9 mmol/L [72 mg/dL]), normoglycemia (mean CBGs between 4 and 12 mmol/L [73‐216 mg/dL]), hyperglycemia (any CBG 12.1 mmol/L [218 mg/dL]), and mean CBG. Proportions were compared using the z test, whereas sample means between the groups were compared by nonparametric Mann‐Whitney U tests, as per statistical literature.[11] All P values are 2‐tailed, and 0.05 was considered statistically significant.
Patient characteristics and healthcare professional's workload were identified as potential causes of variation in CBG readings. Regression analysis of covariance was used to identify and adjust for these factors when comparing mean glucose readings. Binomial logistic regression was used to adjust proportions of patients‐days with readings out of range and patient‐days with mean readings within range. Variables tested were length of stay as a proxy for severity of condition, number of patients whose CBG were measured in the hospital in a day as a proxy for the healthcare professional's workload, and location of the patient to account for variation in patient characteristics as the wards were specialty based. Goodness of fit was tested using the R2 value in the linear model, which indicates the proportion of outcome that is explained by the model. For binomial models, McFadden's pseudo R2 (pseudo‐R2McFadden) was used as advised for logistic models. McFadden's pseudo‐R2 ranges from 0 to 1, but unlike R2 in ordinary linear regression, values tend to be significantly lower: McFadden's pseudo R2 values between 0.2 and 0.4 indicate excellent fit.[12]
RESULTS
A total of 16,870 patient‐day CBG measures in 2730 inpatients with diabetes were analyzed. The results of all regressions are presented in Table 1. The coefficients in the first model represent the effect of each covariate on mean patient‐day CBG. For example, each extra day of hospitalization was associated with a 0.02 mmol/L (0.36 mg/dL) increase in mean patient‐day reading, ceteris paribus. The remaining models indicate the change in relative risk (in this case the proportion of patient‐days) associated with the covariates. For example, in patients who were hospitalized for 3 days, the proportion of patient‐days with at least 1 CBG greater than 12 mmol/L (216 mg/dL) was 1.01 times the comparable proportion of patients who were hospitalized for 2 days. Each additional day in the hospital significantly increased the mean CBG by 0.015 mmol/L (0.27 mg/dL) and increased the risk of having at least 1 reading below 3.9 mmol/L (72 mg/dL) or above 12 mmol/L (216 mg/dL). Monitoring more patients in a day also affected outcomes, although the effect was small. Each additional patient monitored reduced mean patient‐day CBG by 0.011 mmol/L (0.198 mg/dL) and increased the proportion of patients with at least 1 reading below 4 mmol/L (72 mg/dL) 1.01 times. Location of the patient also significantly affected CBG readings. This could have been due to either ward or patient characteristics, but lack of data on each ward's healthcare personnel and individual patient characteristics prevented further analysis of this effect, and therefore the results were used for adjustment only. All models have relatively low predictive power, as demonstrated by the low R2 and pseudo‐R2McFadden values. In the linear model that estimated the effect of covariates on mean patient‐day CBG, the R2 is 0.0270, indicating that only 2.70% of results were explained by the covariates in the model. The pseudo‐R2McFadden varied between 0.0146 and 0.0540, as presented in Table 1. Although the pseudo‐R2McFadden generally had lower values than the R2 for the linear models, values of 0.0540 and below are considered to be relatively low.[12]
| Covariate | Outcome | |||||
|---|---|---|---|---|---|---|
| Change in Mean CBG for Each Patient‐Day, mmol/L (mg/dL) | Change in % of Patient‐Days With Any CBG 2.2 mmol/L (40 mg/dL) | Change in % of Patient‐Days With Any CBG 2.9 mmol/L (52 mg/dL) | Change in % of Patient‐Days With Any CBG 3.9 mmol/L (72 mg/dL) | Change in % of Patient‐Days With Mean CBG Between 4 and 12 mmol/L (73216 mg/dL) | Change in % of Patient‐Days With Any CBG >12 mmol/L (218 mg/dL) | |
| ||||||
| Additional day in the hospital | 0.015 (0.27), P 0.001* | 1.00, P = 0.605 | 1.00, P = 0.986 | 1.005, P = 0.004 | 0.99, P 0.001* | 1.01, P 0.001* |
| Additional patients monitored | 0.011 (0.198), P 0.001* | 1.01, P = 0.132 | 1.01, P = 0.084 | 1.01, P = 0.021 | 1.00, P = 0.128 | 0.997, P = 0.011 |
| Ward (range) |
0.5913.68(10.62246.24) |
0.3722.71 | 03.62 | 03.10 | 047,124.14 | 04,094,900 |
| R2/pseudo‐R2McFadden | 0.0247 | 0.0503 | 0.0363 | 0.0270 | 0.0140 | 0.0243 |
Table 2 summarizes outcomes for the 3 years individually. The results suggest that all indices of inpatient glycemic control that were analyzedhypoglycemia, normoglycemia, hyperglycemia, and mean CBGdid not worsen in August compared to July that year. The results are presented after adjustment for variation in the length of stay, number of patients monitored in a day, and location of the patient. Their effect on the difference in proportions of patients with at least 1 reading out of range and mean reading within range were not statistically significant. However, their effect on mean patient‐day CBG measures was statistically significant, although the effect was only a small decrease (0.4 mmol/L or 7.2 mg/dL) in the mean CBG (see Supporting Table 1 in the online version of this article for unadjusted readings).
| 2012 | 2013 | 2014 | ||||
|---|---|---|---|---|---|---|
| Before Changeover | After Changeover | Before Changeover | After Changeover | Before Changeover | After Changeover | |
| ||||||
| No. of inpatients with diabetes whose CBG readings were analyzed | 470 | 482 | 464 | 427 | 440 | 447 |
| No. of patient‐day CBG readings analyzed | 2917 | 3159 | 3097 | 2588 | 2484 | 2625 |
| Mean no. of CBG readings per patient‐day (range) | 2.5 (127) | 2.5 (123), P = 0.676 | 2.6 (121) | 2.4 (118), P = 0.009* | 2.5 (120) | 2.4 (120), P = 0.028 |
| Mean no. of CBG readings per patient‐day (range) in those where at least 1 reading was CBG 3.9 mmol/L (72 mg/dL) or CBG 12.1 mmol/L (218 mg/dL) | 3.8 (127) | 3.8 (123) | 3.7 (121) | 3.5 (118) | 3.2 (120) | 3.5 (120) |
| Mean no. of CBG readings per patient‐day (range) in those where all CBG readings were between 4 and 12 mmol/L (73216mg/dL) | 1.8 (127) | 1.8 (112) | 1.8 (112) | 1.8 (117) | 1.7 (111) | 1.7 (115) |
| % of patient‐days with any CBG 2.2 mmol/L (40 mg/dL) | 0.99% | 1.09%, P = 0.703 | 1.03% | 0.88%, P = 0.544 | 0.84% | 0.87%, P = 0.927 |
| % of patient‐days with any CBG 2.9 mmol/L (52 mg/dL) | 2.53% | 2.68%, P = 0.708 | 2.63% | 1.35%, P = 0.490 | 2.24% | 2.31%, P = 0.874 |
| % of patient‐days with any CBG 3.9 mmol/L (72 mg/dL) | 7.25% | 7.42%, P = 0.792 | 7.56 % | 6.93%, P = 0.361 | 6.55% | 6.70%, P = 0.858 |
| % of patient‐days with mean CBG between 4 and 12 mmol/L (73216 mg/dL) | 79.10% | 79.89%, P = 0.446 | 78.69% | 78.58%, P = 0.924 | 78.65% | 78.61%, P = 0.973 |
| % of patient‐days with any CBG 12.1 mmol/L (218 mg/dL) | 32.32% | 31.40%, P = 0.443 | 32.29% | 32.88%, P = 0.634 | 32.78% | 32.66%, P = 0.928 |
| Median of mean CBG for each patient‐day in mmol/L (mg/dL) | 8.0 (144.6) | 7.8 (140.0) | 8.4 (151.5) | 8.3 (150.2) | 8.9 (159.8) | 8.8 (157.8) |
| Mean of mean CBG for each patient‐day in mmol/L (standard deviation) | 9.1 (4.0) | 8.8 (4.1), P = 0.033+ | 9.4 (4.1) | 9.2 (4.0), P = 0.075 | 9.8 (4.1) | 9.6 (3.8), P = 0.189 |
DISCUSSION
This study shows that contrary to expectation, inpatient glycemic control did not worsen in the 4 weeks following the August changeover of trainee doctors for the years 2012, 2013, and 2014. In fact, inpatient glycemic control was marginally better in the first 4 weeks after changeover each year compared to the preceding 4 weeks before changeover. There may be several reasons for the findings in this study. First, since 2010 in this hospital and since 2012 nationally (further to direction from NHS England Medical Director Sir Bruce Keogh), it has become established practice that newly qualified trainee doctors shadow their colleagues at work a week prior to Black Wednesday.[13, 14] The purpose of this practice, called the preparation for professional practice is to familiarize trainee doctors with the hospital protocols and systems, improve their confidence, and potentially reduce medical errors when starting work. Second, since 2012, this hospital has also implemented the Joint British Diabetes Societies' national guidelines in managing inpatients with diabetes.[15] These guidelines are widely publicized on the changeover day during the trainee doctor's induction program. Finally, since 2012, a diabetes‐specific interactive 1‐hour educational program for trainee doctors devised by this hospital was implemented during the changeover period, which takes them through practical and problem‐solving case scenarios related to inpatient glycemic management, in particular prevention of hypoglycemia and hospital‐acquired diabetic ketoacidosis.[16] Attendance was mandatory, and informal feedback from trainee doctors about the educational program was extremely positive.
There are several limitations in this study. It could be argued that trainee doctors have very little impact on glycemic control in inpatients with diabetes. In NHS hospitals, trainee doctors are often the first port of call for managing glycemic issues in inpatients both in and out of hours, who in turn may or may not call the inpatient diabetes team wherever available. Therefore, trainee doctors' impact on glycemic control in inpatients with diabetes cannot be understated. However, it is acknowledged that in this study, a number of other factors that influence inpatient glycemic control, such as individual patient characteristics, medication errors, and the knowledge and confidence levels of individual trainee doctors, were not accounted for. Nevertheless, such factors are unlikely to have been significantly different over the 3‐year period. A further limitation was the unavailability of hospital‐wide electronic CBG data prior to 2012 to determine whether changeover impacted on inpatient glycemic control prior to this period. Another limitation was the dependence on patient administration data to identify those with diabetes, as it is well recognized that coded data in hospital data management systems can be inaccurate, though this has significantly improved over the years.[17] Finally, the most important limitation is that this is a single‐hospital study, and so the results may not be applicable to other English hospitals. Nevertheless, the finding of this study is similar to the finding in the single‐hospital study from the United States.[8]
The finding that glycemic control in inpatients with diabetes did not worsen in the 4 weeks following changeover of trainee doctors compared to the 4 weeks before changeover each year suggests that appropriate forethought and planning by the deanery foundation school and the inpatient diabetes team has prevented the anticipated deterioration of glycemic control during the August changeover of trainee doctors in this English hospital.
Disclosures: R.R. and G.R. conceived and designed the study. R.R. collected data and drafted the manuscript. R.R., D.J., and G.R. analyzed and interpreted the data. D.J. provided statistical input for analysis of the data. R.R., D.J., and G.R. critically revised the manuscript for intellectual content. All authors have approved the final version. The authors report no conflicts of interest.
In England, the day when trainee doctors start work for the first time in their careers or rotate to a different hospital is the first Wednesday of August. This is often referred to as the Black Wednesday in the National Health Service (NHS), as it is widely perceived that inexperience and nonfamiliarity with the new hospital systems and policies in these first few weeks lead to increased medical errors and mismanagement and may therefore cost lives.[1] However, there is very little evidence in favor of this widely held view in the NHS. A 2009 English study found a small but significant increase of 6% in the odds of death for inpatients admitted in the week following the first Wednesday in August than in the week following the last Wednesday in July, whereas a previous report did not support this.[2, 3] In the United States, the resident trainee doctor's changeover occurs in July, and its negative impact on patient outcomes is often dubbed the July phenomenon.[4] With conflicting reports of the July phenomenon on patient outcomes,[5, 6, 7] Young et al. systematically reviewed 39 studies and concluded that the July phenomenon exists in that there is increased mortality around the changeover period.[4]
It can be hypothesized that glycemic control in inpatients with diabetes would be worse in the immediate period following changeover of trainee doctors for the same reasons mentioned earlier that impact mortality. However, contrary to expectations, a recent single‐hospital study from the United States reported that changeover of resident trainee doctors did not worsen inpatient glycemic control.[8] Although the lack of confidence among trainee doctors in inpatient diabetes management has been clearly demonstrated in England,[9] the impact of August changeover of trainee doctors on inpatient glycemic control is unknown. The aim of this study was to determine whether the August changeover of trainee doctors impacted on glycemic control in inpatients with diabetes in a single English hospital.
MATERIAL AND METHODS
The study setting was a medium‐sized 550‐bed hospital in England that serves a population of approximately 360,000 residents. Capillary blood glucose (CBG) readings for adult inpatients across all wards were downloaded from the Precision Web Point‐of‐Care Data Management System (Abbott Diabetes Care Inc., Alameda, CA), an electronic database where all the CBG readings for inpatients are stored. Patient administration data were used to identify those with diabetes admitted to the hospital for at least 1 day, and only their CBG readings were included in this study. Glucometrics, a term coined by Goldberg et al., refers to standardized glucose performance metrics to assess the quality of inpatient glycemic control.[10] In this study, patient‐day glucometric measures were used, as they are considered the best indicator of inpatient glycemic control compared to other glucometrics.[10] Patient‐day glucometrics were analyzed for 4 weeks before and after Black Wednesday for the years 2012, 2013, and 2014 using Microsoft Excel 2007 (Microsoft Corp., Redmond, WA) and R version 3.1.0 (The R Foundation, Vienna, Austria). Patient‐day glucometrics analyzed were hypoglycemia (any CBG 2.2 mmol/L [40 mg/dL], any CBG 2.9 mmol/L [52 mg/dL], any CBG 3.9 mmol/L [72 mg/dL]), normoglycemia (mean CBGs between 4 and 12 mmol/L [73‐216 mg/dL]), hyperglycemia (any CBG 12.1 mmol/L [218 mg/dL]), and mean CBG. Proportions were compared using the z test, whereas sample means between the groups were compared by nonparametric Mann‐Whitney U tests, as per statistical literature.[11] All P values are 2‐tailed, and 0.05 was considered statistically significant.
Patient characteristics and healthcare professional's workload were identified as potential causes of variation in CBG readings. Regression analysis of covariance was used to identify and adjust for these factors when comparing mean glucose readings. Binomial logistic regression was used to adjust proportions of patients‐days with readings out of range and patient‐days with mean readings within range. Variables tested were length of stay as a proxy for severity of condition, number of patients whose CBG were measured in the hospital in a day as a proxy for the healthcare professional's workload, and location of the patient to account for variation in patient characteristics as the wards were specialty based. Goodness of fit was tested using the R2 value in the linear model, which indicates the proportion of outcome that is explained by the model. For binomial models, McFadden's pseudo R2 (pseudo‐R2McFadden) was used as advised for logistic models. McFadden's pseudo‐R2 ranges from 0 to 1, but unlike R2 in ordinary linear regression, values tend to be significantly lower: McFadden's pseudo R2 values between 0.2 and 0.4 indicate excellent fit.[12]
RESULTS
A total of 16,870 patient‐day CBG measures in 2730 inpatients with diabetes were analyzed. The results of all regressions are presented in Table 1. The coefficients in the first model represent the effect of each covariate on mean patient‐day CBG. For example, each extra day of hospitalization was associated with a 0.02 mmol/L (0.36 mg/dL) increase in mean patient‐day reading, ceteris paribus. The remaining models indicate the change in relative risk (in this case the proportion of patient‐days) associated with the covariates. For example, in patients who were hospitalized for 3 days, the proportion of patient‐days with at least 1 CBG greater than 12 mmol/L (216 mg/dL) was 1.01 times the comparable proportion of patients who were hospitalized for 2 days. Each additional day in the hospital significantly increased the mean CBG by 0.015 mmol/L (0.27 mg/dL) and increased the risk of having at least 1 reading below 3.9 mmol/L (72 mg/dL) or above 12 mmol/L (216 mg/dL). Monitoring more patients in a day also affected outcomes, although the effect was small. Each additional patient monitored reduced mean patient‐day CBG by 0.011 mmol/L (0.198 mg/dL) and increased the proportion of patients with at least 1 reading below 4 mmol/L (72 mg/dL) 1.01 times. Location of the patient also significantly affected CBG readings. This could have been due to either ward or patient characteristics, but lack of data on each ward's healthcare personnel and individual patient characteristics prevented further analysis of this effect, and therefore the results were used for adjustment only. All models have relatively low predictive power, as demonstrated by the low R2 and pseudo‐R2McFadden values. In the linear model that estimated the effect of covariates on mean patient‐day CBG, the R2 is 0.0270, indicating that only 2.70% of results were explained by the covariates in the model. The pseudo‐R2McFadden varied between 0.0146 and 0.0540, as presented in Table 1. Although the pseudo‐R2McFadden generally had lower values than the R2 for the linear models, values of 0.0540 and below are considered to be relatively low.[12]
| Covariate | Outcome | |||||
|---|---|---|---|---|---|---|
| Change in Mean CBG for Each Patient‐Day, mmol/L (mg/dL) | Change in % of Patient‐Days With Any CBG 2.2 mmol/L (40 mg/dL) | Change in % of Patient‐Days With Any CBG 2.9 mmol/L (52 mg/dL) | Change in % of Patient‐Days With Any CBG 3.9 mmol/L (72 mg/dL) | Change in % of Patient‐Days With Mean CBG Between 4 and 12 mmol/L (73216 mg/dL) | Change in % of Patient‐Days With Any CBG >12 mmol/L (218 mg/dL) | |
| ||||||
| Additional day in the hospital | 0.015 (0.27), P 0.001* | 1.00, P = 0.605 | 1.00, P = 0.986 | 1.005, P = 0.004 | 0.99, P 0.001* | 1.01, P 0.001* |
| Additional patients monitored | 0.011 (0.198), P 0.001* | 1.01, P = 0.132 | 1.01, P = 0.084 | 1.01, P = 0.021 | 1.00, P = 0.128 | 0.997, P = 0.011 |
| Ward (range) |
0.5913.68(10.62246.24) |
0.3722.71 | 03.62 | 03.10 | 047,124.14 | 04,094,900 |
| R2/pseudo‐R2McFadden | 0.0247 | 0.0503 | 0.0363 | 0.0270 | 0.0140 | 0.0243 |
Table 2 summarizes outcomes for the 3 years individually. The results suggest that all indices of inpatient glycemic control that were analyzedhypoglycemia, normoglycemia, hyperglycemia, and mean CBGdid not worsen in August compared to July that year. The results are presented after adjustment for variation in the length of stay, number of patients monitored in a day, and location of the patient. Their effect on the difference in proportions of patients with at least 1 reading out of range and mean reading within range were not statistically significant. However, their effect on mean patient‐day CBG measures was statistically significant, although the effect was only a small decrease (0.4 mmol/L or 7.2 mg/dL) in the mean CBG (see Supporting Table 1 in the online version of this article for unadjusted readings).
| 2012 | 2013 | 2014 | ||||
|---|---|---|---|---|---|---|
| Before Changeover | After Changeover | Before Changeover | After Changeover | Before Changeover | After Changeover | |
| ||||||
| No. of inpatients with diabetes whose CBG readings were analyzed | 470 | 482 | 464 | 427 | 440 | 447 |
| No. of patient‐day CBG readings analyzed | 2917 | 3159 | 3097 | 2588 | 2484 | 2625 |
| Mean no. of CBG readings per patient‐day (range) | 2.5 (127) | 2.5 (123), P = 0.676 | 2.6 (121) | 2.4 (118), P = 0.009* | 2.5 (120) | 2.4 (120), P = 0.028 |
| Mean no. of CBG readings per patient‐day (range) in those where at least 1 reading was CBG 3.9 mmol/L (72 mg/dL) or CBG 12.1 mmol/L (218 mg/dL) | 3.8 (127) | 3.8 (123) | 3.7 (121) | 3.5 (118) | 3.2 (120) | 3.5 (120) |
| Mean no. of CBG readings per patient‐day (range) in those where all CBG readings were between 4 and 12 mmol/L (73216mg/dL) | 1.8 (127) | 1.8 (112) | 1.8 (112) | 1.8 (117) | 1.7 (111) | 1.7 (115) |
| % of patient‐days with any CBG 2.2 mmol/L (40 mg/dL) | 0.99% | 1.09%, P = 0.703 | 1.03% | 0.88%, P = 0.544 | 0.84% | 0.87%, P = 0.927 |
| % of patient‐days with any CBG 2.9 mmol/L (52 mg/dL) | 2.53% | 2.68%, P = 0.708 | 2.63% | 1.35%, P = 0.490 | 2.24% | 2.31%, P = 0.874 |
| % of patient‐days with any CBG 3.9 mmol/L (72 mg/dL) | 7.25% | 7.42%, P = 0.792 | 7.56 % | 6.93%, P = 0.361 | 6.55% | 6.70%, P = 0.858 |
| % of patient‐days with mean CBG between 4 and 12 mmol/L (73216 mg/dL) | 79.10% | 79.89%, P = 0.446 | 78.69% | 78.58%, P = 0.924 | 78.65% | 78.61%, P = 0.973 |
| % of patient‐days with any CBG 12.1 mmol/L (218 mg/dL) | 32.32% | 31.40%, P = 0.443 | 32.29% | 32.88%, P = 0.634 | 32.78% | 32.66%, P = 0.928 |
| Median of mean CBG for each patient‐day in mmol/L (mg/dL) | 8.0 (144.6) | 7.8 (140.0) | 8.4 (151.5) | 8.3 (150.2) | 8.9 (159.8) | 8.8 (157.8) |
| Mean of mean CBG for each patient‐day in mmol/L (standard deviation) | 9.1 (4.0) | 8.8 (4.1), P = 0.033+ | 9.4 (4.1) | 9.2 (4.0), P = 0.075 | 9.8 (4.1) | 9.6 (3.8), P = 0.189 |
DISCUSSION
This study shows that contrary to expectation, inpatient glycemic control did not worsen in the 4 weeks following the August changeover of trainee doctors for the years 2012, 2013, and 2014. In fact, inpatient glycemic control was marginally better in the first 4 weeks after changeover each year compared to the preceding 4 weeks before changeover. There may be several reasons for the findings in this study. First, since 2010 in this hospital and since 2012 nationally (further to direction from NHS England Medical Director Sir Bruce Keogh), it has become established practice that newly qualified trainee doctors shadow their colleagues at work a week prior to Black Wednesday.[13, 14] The purpose of this practice, called the preparation for professional practice is to familiarize trainee doctors with the hospital protocols and systems, improve their confidence, and potentially reduce medical errors when starting work. Second, since 2012, this hospital has also implemented the Joint British Diabetes Societies' national guidelines in managing inpatients with diabetes.[15] These guidelines are widely publicized on the changeover day during the trainee doctor's induction program. Finally, since 2012, a diabetes‐specific interactive 1‐hour educational program for trainee doctors devised by this hospital was implemented during the changeover period, which takes them through practical and problem‐solving case scenarios related to inpatient glycemic management, in particular prevention of hypoglycemia and hospital‐acquired diabetic ketoacidosis.[16] Attendance was mandatory, and informal feedback from trainee doctors about the educational program was extremely positive.
There are several limitations in this study. It could be argued that trainee doctors have very little impact on glycemic control in inpatients with diabetes. In NHS hospitals, trainee doctors are often the first port of call for managing glycemic issues in inpatients both in and out of hours, who in turn may or may not call the inpatient diabetes team wherever available. Therefore, trainee doctors' impact on glycemic control in inpatients with diabetes cannot be understated. However, it is acknowledged that in this study, a number of other factors that influence inpatient glycemic control, such as individual patient characteristics, medication errors, and the knowledge and confidence levels of individual trainee doctors, were not accounted for. Nevertheless, such factors are unlikely to have been significantly different over the 3‐year period. A further limitation was the unavailability of hospital‐wide electronic CBG data prior to 2012 to determine whether changeover impacted on inpatient glycemic control prior to this period. Another limitation was the dependence on patient administration data to identify those with diabetes, as it is well recognized that coded data in hospital data management systems can be inaccurate, though this has significantly improved over the years.[17] Finally, the most important limitation is that this is a single‐hospital study, and so the results may not be applicable to other English hospitals. Nevertheless, the finding of this study is similar to the finding in the single‐hospital study from the United States.[8]
The finding that glycemic control in inpatients with diabetes did not worsen in the 4 weeks following changeover of trainee doctors compared to the 4 weeks before changeover each year suggests that appropriate forethought and planning by the deanery foundation school and the inpatient diabetes team has prevented the anticipated deterioration of glycemic control during the August changeover of trainee doctors in this English hospital.
Disclosures: R.R. and G.R. conceived and designed the study. R.R. collected data and drafted the manuscript. R.R., D.J., and G.R. analyzed and interpreted the data. D.J. provided statistical input for analysis of the data. R.R., D.J., and G.R. critically revised the manuscript for intellectual content. All authors have approved the final version. The authors report no conflicts of interest.
- . Black Wednesday: today junior doctors will start work—and cause A4(9):e7103.
- , . The killing season—fact or fiction? BMJ. 1994;309(6970):1690.
- , , , , , . “July effect”: impact of the academic year‐end changeover on patient outcomes: a systematic review. Ann Intern Med. 2011;155(5):309–315.
- , . A July spike in fatal medication errors: a possible effect of new medical residents. J Gen Intern Med. 2010;25(8):774–779.
- , , , et al. Complications and death at the start of the new academic year: is there a July phenomenon? J Trauma. 2010;68(1):19–22.
- , , , . Errors and adverse outcomes on a surgical service: what is the role of residents? J Surg Res. 2004;122(2):162–166.
- , , , . Is There a “July Effect” for inpatient glycemic control? Endocr Pract. 2014;20(19):919–924.
- , , , et al.; TOPDOC Diabetes Study Team. Lack of confidence among trainee doctors in the management of diabetes: the Trainees Own Perception of Delivery of Care (TOPDOC) Diabetes Study. QJM. 2011;104(9):761–766.
- , , , et al. “Glucometrics”—assessing the quality of inpatient glucose management. Diabetes Technol Ther. 2006;8(5):560–569.
- , , . Statistics for Business and Economics. 5th ed. Upper Saddle River, NJ: Prentice Hall; 2002.
- , , . Stated choice methods. New York, NY: Cambridge University Press; 2000.
- Health Education East of England. Preparing for professional practice. Available at: https://heeoe.hee.nhs.uk/foundation_faq. Accessed October 07, 2015.
- Department of Health. Lives will be saved as junior doctors shadow new role 2012. Available at: https://www.gov.uk/government/news/lives‐will‐be‐saved‐as‐junior‐doctors‐shadow‐new‐role. Accessed October 29, 2014.
- Association of British Clinical Diabetologists. Joint British Diabetes Societies for Inpatient Care. Available at: http://www.diabetologists‐abcd.org.uk/JBDS/JBDS.htm. Accessed October 8, 2014.
- , , . An interactive 1‐h educational programme for junior doctors, increases their confidence and improves inpatient diabetes care. Diabet Med. 2012;29(12):1574–1578.
- , , , et al. Systematic review of discharge coding accuracy. J Public Health (Oxf). 2012;34(1):138–148.
- . Black Wednesday: today junior doctors will start work—and cause A4(9):e7103.
- , . The killing season—fact or fiction? BMJ. 1994;309(6970):1690.
- , , , , , . “July effect”: impact of the academic year‐end changeover on patient outcomes: a systematic review. Ann Intern Med. 2011;155(5):309–315.
- , . A July spike in fatal medication errors: a possible effect of new medical residents. J Gen Intern Med. 2010;25(8):774–779.
- , , , et al. Complications and death at the start of the new academic year: is there a July phenomenon? J Trauma. 2010;68(1):19–22.
- , , , . Errors and adverse outcomes on a surgical service: what is the role of residents? J Surg Res. 2004;122(2):162–166.
- , , , . Is There a “July Effect” for inpatient glycemic control? Endocr Pract. 2014;20(19):919–924.
- , , , et al.; TOPDOC Diabetes Study Team. Lack of confidence among trainee doctors in the management of diabetes: the Trainees Own Perception of Delivery of Care (TOPDOC) Diabetes Study. QJM. 2011;104(9):761–766.
- , , , et al. “Glucometrics”—assessing the quality of inpatient glucose management. Diabetes Technol Ther. 2006;8(5):560–569.
- , , . Statistics for Business and Economics. 5th ed. Upper Saddle River, NJ: Prentice Hall; 2002.
- , , . Stated choice methods. New York, NY: Cambridge University Press; 2000.
- Health Education East of England. Preparing for professional practice. Available at: https://heeoe.hee.nhs.uk/foundation_faq. Accessed October 07, 2015.
- Department of Health. Lives will be saved as junior doctors shadow new role 2012. Available at: https://www.gov.uk/government/news/lives‐will‐be‐saved‐as‐junior‐doctors‐shadow‐new‐role. Accessed October 29, 2014.
- Association of British Clinical Diabetologists. Joint British Diabetes Societies for Inpatient Care. Available at: http://www.diabetologists‐abcd.org.uk/JBDS/JBDS.htm. Accessed October 8, 2014.
- , , . An interactive 1‐h educational programme for junior doctors, increases their confidence and improves inpatient diabetes care. Diabet Med. 2012;29(12):1574–1578.
- , , , et al. Systematic review of discharge coding accuracy. J Public Health (Oxf). 2012;34(1):138–148.
Letter to the Editor
We thank Mr. Zilm and colleagues for their interest in our work.[1] Certainly, we did not intend to imply that well‐designed buildings have little value in the efficient and patient‐centered delivery of healthcare. Our main goal was to highlight (1) that patients can distinguish between facility features and actual care delivery, and poor facilities alone should not be an excuse for poor patient satisfaction; and (2) that global evaluations are more dependent on perceived quality of care than on facility features. Furthermore, we agree with many of the points raised. Certainly, patient satisfaction is but 1 measure of successful facility design, and the delivery of modern healthcare requires updated facilities. However, based on our results, we think that healthcare administrators and designers should consider the return on investment on the costly features that are incorporated purely to improve patient satisfaction rather than for safety and staff effectiveness.
Referral patterns and patient expectations are likely very different for a tertiary care hospital like ours. A different relationship between facility design and patient satisfaction may indeed exist for community hospitals. However, we would caution against making this assumption without supportive evidence. Furthermore, it is difficult to attribute lack of improvement of physician scores in our study because of a ceiling effect. The baseline scores were certainly not exemplary, and there was plenty of room for improvement.
We agree that there is a need for high‐quality research to better understand the broader impact of healthcare design on meaningful outcomes. However, we are not impressed with the quality of much of the existing research tying physical facilities with patient stress or shorter length of stay, as mentioned by Mr. Zilm and colleagues. Evidence supporting investment in expensive facilities should be evaluated with the same high standards and rigor as for other healthcare decisions.
- , , , , . Changes in patient satisfaction related to hospital renovation: experience with a new clinical building. J Hosp Med. 2015;10(3):165–171.
We thank Mr. Zilm and colleagues for their interest in our work.[1] Certainly, we did not intend to imply that well‐designed buildings have little value in the efficient and patient‐centered delivery of healthcare. Our main goal was to highlight (1) that patients can distinguish between facility features and actual care delivery, and poor facilities alone should not be an excuse for poor patient satisfaction; and (2) that global evaluations are more dependent on perceived quality of care than on facility features. Furthermore, we agree with many of the points raised. Certainly, patient satisfaction is but 1 measure of successful facility design, and the delivery of modern healthcare requires updated facilities. However, based on our results, we think that healthcare administrators and designers should consider the return on investment on the costly features that are incorporated purely to improve patient satisfaction rather than for safety and staff effectiveness.
Referral patterns and patient expectations are likely very different for a tertiary care hospital like ours. A different relationship between facility design and patient satisfaction may indeed exist for community hospitals. However, we would caution against making this assumption without supportive evidence. Furthermore, it is difficult to attribute lack of improvement of physician scores in our study because of a ceiling effect. The baseline scores were certainly not exemplary, and there was plenty of room for improvement.
We agree that there is a need for high‐quality research to better understand the broader impact of healthcare design on meaningful outcomes. However, we are not impressed with the quality of much of the existing research tying physical facilities with patient stress or shorter length of stay, as mentioned by Mr. Zilm and colleagues. Evidence supporting investment in expensive facilities should be evaluated with the same high standards and rigor as for other healthcare decisions.
We thank Mr. Zilm and colleagues for their interest in our work.[1] Certainly, we did not intend to imply that well‐designed buildings have little value in the efficient and patient‐centered delivery of healthcare. Our main goal was to highlight (1) that patients can distinguish between facility features and actual care delivery, and poor facilities alone should not be an excuse for poor patient satisfaction; and (2) that global evaluations are more dependent on perceived quality of care than on facility features. Furthermore, we agree with many of the points raised. Certainly, patient satisfaction is but 1 measure of successful facility design, and the delivery of modern healthcare requires updated facilities. However, based on our results, we think that healthcare administrators and designers should consider the return on investment on the costly features that are incorporated purely to improve patient satisfaction rather than for safety and staff effectiveness.
Referral patterns and patient expectations are likely very different for a tertiary care hospital like ours. A different relationship between facility design and patient satisfaction may indeed exist for community hospitals. However, we would caution against making this assumption without supportive evidence. Furthermore, it is difficult to attribute lack of improvement of physician scores in our study because of a ceiling effect. The baseline scores were certainly not exemplary, and there was plenty of room for improvement.
We agree that there is a need for high‐quality research to better understand the broader impact of healthcare design on meaningful outcomes. However, we are not impressed with the quality of much of the existing research tying physical facilities with patient stress or shorter length of stay, as mentioned by Mr. Zilm and colleagues. Evidence supporting investment in expensive facilities should be evaluated with the same high standards and rigor as for other healthcare decisions.
- , , , , . Changes in patient satisfaction related to hospital renovation: experience with a new clinical building. J Hosp Med. 2015;10(3):165–171.
- , , , , . Changes in patient satisfaction related to hospital renovation: experience with a new clinical building. J Hosp Med. 2015;10(3):165–171.