User login
EC approves reversal agent for dabigatran

to prevent thrombosis after
knee replacement surgery
© Boehringer Ingelheim
The European Commission (EC) has granted marketing authorization for idarucizumab (Praxbind), a humanized antibody fragment designed to reverse the anticoagulant effects of dabigatran etexilate (Pradaxa) in cases of emergency surgery/urgent procedures or in situations of life-threatening or uncontrolled bleeding.
The EC’s decision applies to the 28 countries of the European Union (EU) as well as Iceland, Lichtenstein, and Norway.
Idarucizumab is the first specific reversal agent for a non-vitamin K antagonist oral anticoagulant to be approved for use in the EU. The drug was approved in the US last month.
The EC’s decision to approve idarucizumab was based on results in healthy volunteers and an interim analysis of the phase 3 RE-VERSE AD trial.
In the study of healthy volunteers, the pharmacokinetic profile of idarucizumab met the requirement for rapid peak exposure and rapid elimination, with no effect on pharmacodynamic parameters. And researchers said the drug was well-tolerated.
In RE-VERSE AD, researchers evaluated idarucizumab in emergency settings. The drug normalized diluted thrombin time and ecarin clotting time in a majority of dabigatran-treated patients with uncontrolled or life-threatening bleeding complications and most patients who required emergency surgery or an invasive procedure.
The researchers said there were no safety concerns related to idarucizumab. However, 23% of patients in this trial experienced serious adverse events, 20% of patients died, and several patients had thrombotic or bleeding events.
Boehringer Ingelheim, the company developing idarucizumab, said it is committed to making the drug available as widely as possible and will launch idarucizumab in European countries as soon as national requirements allow.
Boehringer Ingelheim is also the maker of dabigatran, which is currently approved in the EU for the following indications:
- Prevention of stroke and systemic embolism in patients with non-valvular atrial fibrillation and a risk factor for stroke
- Primary prevention of venous thromboembolic events in patients undergoing elective total hip replacement surgery or total knee replacement surgery
- Treatment of deep vein thrombosis and pulmonary embolism and the prevention of recurrent deep vein thrombosis and pulmonary embolism in adults.

to prevent thrombosis after
knee replacement surgery
© Boehringer Ingelheim
The European Commission (EC) has granted marketing authorization for idarucizumab (Praxbind), a humanized antibody fragment designed to reverse the anticoagulant effects of dabigatran etexilate (Pradaxa) in cases of emergency surgery/urgent procedures or in situations of life-threatening or uncontrolled bleeding.
The EC’s decision applies to the 28 countries of the European Union (EU) as well as Iceland, Lichtenstein, and Norway.
Idarucizumab is the first specific reversal agent for a non-vitamin K antagonist oral anticoagulant to be approved for use in the EU. The drug was approved in the US last month.
The EC’s decision to approve idarucizumab was based on results in healthy volunteers and an interim analysis of the phase 3 RE-VERSE AD trial.
In the study of healthy volunteers, the pharmacokinetic profile of idarucizumab met the requirement for rapid peak exposure and rapid elimination, with no effect on pharmacodynamic parameters. And researchers said the drug was well-tolerated.
In RE-VERSE AD, researchers evaluated idarucizumab in emergency settings. The drug normalized diluted thrombin time and ecarin clotting time in a majority of dabigatran-treated patients with uncontrolled or life-threatening bleeding complications and most patients who required emergency surgery or an invasive procedure.
The researchers said there were no safety concerns related to idarucizumab. However, 23% of patients in this trial experienced serious adverse events, 20% of patients died, and several patients had thrombotic or bleeding events.
Boehringer Ingelheim, the company developing idarucizumab, said it is committed to making the drug available as widely as possible and will launch idarucizumab in European countries as soon as national requirements allow.
Boehringer Ingelheim is also the maker of dabigatran, which is currently approved in the EU for the following indications:
- Prevention of stroke and systemic embolism in patients with non-valvular atrial fibrillation and a risk factor for stroke
- Primary prevention of venous thromboembolic events in patients undergoing elective total hip replacement surgery or total knee replacement surgery
- Treatment of deep vein thrombosis and pulmonary embolism and the prevention of recurrent deep vein thrombosis and pulmonary embolism in adults.
to prevent thrombosis after
knee replacement surgery
© Boehringer Ingelheim
The European Commission (EC) has granted marketing authorization for idarucizumab (Praxbind), a humanized antibody fragment designed to reverse the anticoagulant effects of dabigatran etexilate (Pradaxa) in cases of emergency surgery/urgent procedures or in situations of life-threatening or uncontrolled bleeding.
The EC’s decision applies to the 28 countries of the European Union (EU) as well as Iceland, Lichtenstein, and Norway.
Idarucizumab is the first specific reversal agent for a non-vitamin K antagonist oral anticoagulant to be approved for use in the EU. The drug was approved in the US last month.
The EC’s decision to approve idarucizumab was based on results in healthy volunteers and an interim analysis of the phase 3 RE-VERSE AD trial.
In the study of healthy volunteers, the pharmacokinetic profile of idarucizumab met the requirement for rapid peak exposure and rapid elimination, with no effect on pharmacodynamic parameters. And researchers said the drug was well-tolerated.
In RE-VERSE AD, researchers evaluated idarucizumab in emergency settings. The drug normalized diluted thrombin time and ecarin clotting time in a majority of dabigatran-treated patients with uncontrolled or life-threatening bleeding complications and most patients who required emergency surgery or an invasive procedure.
The researchers said there were no safety concerns related to idarucizumab. However, 23% of patients in this trial experienced serious adverse events, 20% of patients died, and several patients had thrombotic or bleeding events.
Boehringer Ingelheim, the company developing idarucizumab, said it is committed to making the drug available as widely as possible and will launch idarucizumab in European countries as soon as national requirements allow.
Boehringer Ingelheim is also the maker of dabigatran, which is currently approved in the EU for the following indications:
- Prevention of stroke and systemic embolism in patients with non-valvular atrial fibrillation and a risk factor for stroke
- Primary prevention of venous thromboembolic events in patients undergoing elective total hip replacement surgery or total knee replacement surgery
- Treatment of deep vein thrombosis and pulmonary embolism and the prevention of recurrent deep vein thrombosis and pulmonary embolism in adults.
Prognostic test for MM granted CE-IVD mark

Photo courtesy of SkylineDx
The MMprofiler™, a test used to risk-stratify patients with multiple myeloma (MM), has received the CE-IVD mark.
The CE-IVD mark indicates that an in vitro device complies with the European In Vitro Diagnostics Directive and may be legally commercialized and distributed in the European Union.
The MMprofiler classifies patients into high- or standard-risk groups based on the activity of 92 genes considered to be related to MM.
In a 2012 Leukemia paper, researchers described a gene-expression signature, now known as SKY92, that maps the activity of the 92 genes. MM patients who were defined as high-risk by the SKY92 gene signature (called “the EMC92 gene signature” in the paper) had inferior overall survival.
Subsequent studies, including one recently published in Blood, have shown that patients whose plasma cells are SKY92-positive have, on average, 4-times shorter survival and relapse more quickly after treatment than patients whose cells are SKY92-negative.
The intention of the MMprofiler is to classify MM patients according to the SKY92 gene signature and help healthcare professionals select appropriate treatment on the basis of existing guidelines. Healthcare professionals may also decide to monitor patients differently according to their risk group.
The MMprofiler is a product of SkylineDx. The test is run on the company’s proprietary Profiler™ platform, which makes centralized data analysis possible for labs that have the necessary equipment.
To run the MMprofiler test, labs must install a software module on their Affymetrix DX2 Gene scanning equipment and establish a secured gateway connection with the Profiler platform. Then, the lab can process a patient sample and generate the raw genetic data.
The patient’s bone marrow sample is purified, processed, applied to a biochip, and analyzed in the scanner. This is how the MMprofiler determines the active properties of the various genes in the plasma cells of each patient.
The data is then processed via the software system, which reveals the genetic subtype of the plasma cells. Knowing the SKY92 genetic subtype can help healthcare professionals distinguish between high-risk and standard-risk patients.
For more information on the test, visit www.mmprofiler.com. ![]()
Photo courtesy of SkylineDx
The MMprofiler™, a test used to risk-stratify patients with multiple myeloma (MM), has received the CE-IVD mark.
The CE-IVD mark indicates that an in vitro device complies with the European In Vitro Diagnostics Directive and may be legally commercialized and distributed in the European Union.
The MMprofiler classifies patients into high- or standard-risk groups based on the activity of 92 genes considered to be related to MM.
In a 2012 Leukemia paper, researchers described a gene-expression signature, now known as SKY92, that maps the activity of the 92 genes. MM patients who were defined as high-risk by the SKY92 gene signature (called “the EMC92 gene signature” in the paper) had inferior overall survival.
Subsequent studies, including one recently published in Blood, have shown that patients whose plasma cells are SKY92-positive have, on average, 4-times shorter survival and relapse more quickly after treatment than patients whose cells are SKY92-negative.
The intention of the MMprofiler is to classify MM patients according to the SKY92 gene signature and help healthcare professionals select appropriate treatment on the basis of existing guidelines. Healthcare professionals may also decide to monitor patients differently according to their risk group.
The MMprofiler is a product of SkylineDx. The test is run on the company’s proprietary Profiler™ platform, which makes centralized data analysis possible for labs that have the necessary equipment.
To run the MMprofiler test, labs must install a software module on their Affymetrix DX2 Gene scanning equipment and establish a secured gateway connection with the Profiler platform. Then, the lab can process a patient sample and generate the raw genetic data.
The patient’s bone marrow sample is purified, processed, applied to a biochip, and analyzed in the scanner. This is how the MMprofiler determines the active properties of the various genes in the plasma cells of each patient.
The data is then processed via the software system, which reveals the genetic subtype of the plasma cells. Knowing the SKY92 genetic subtype can help healthcare professionals distinguish between high-risk and standard-risk patients.
For more information on the test, visit www.mmprofiler.com. ![]()
Photo courtesy of SkylineDx
The MMprofiler™, a test used to risk-stratify patients with multiple myeloma (MM), has received the CE-IVD mark.
The CE-IVD mark indicates that an in vitro device complies with the European In Vitro Diagnostics Directive and may be legally commercialized and distributed in the European Union.
The MMprofiler classifies patients into high- or standard-risk groups based on the activity of 92 genes considered to be related to MM.
In a 2012 Leukemia paper, researchers described a gene-expression signature, now known as SKY92, that maps the activity of the 92 genes. MM patients who were defined as high-risk by the SKY92 gene signature (called “the EMC92 gene signature” in the paper) had inferior overall survival.
Subsequent studies, including one recently published in Blood, have shown that patients whose plasma cells are SKY92-positive have, on average, 4-times shorter survival and relapse more quickly after treatment than patients whose cells are SKY92-negative.
The intention of the MMprofiler is to classify MM patients according to the SKY92 gene signature and help healthcare professionals select appropriate treatment on the basis of existing guidelines. Healthcare professionals may also decide to monitor patients differently according to their risk group.
The MMprofiler is a product of SkylineDx. The test is run on the company’s proprietary Profiler™ platform, which makes centralized data analysis possible for labs that have the necessary equipment.
To run the MMprofiler test, labs must install a software module on their Affymetrix DX2 Gene scanning equipment and establish a secured gateway connection with the Profiler platform. Then, the lab can process a patient sample and generate the raw genetic data.
The patient’s bone marrow sample is purified, processed, applied to a biochip, and analyzed in the scanner. This is how the MMprofiler determines the active properties of the various genes in the plasma cells of each patient.
The data is then processed via the software system, which reveals the genetic subtype of the plasma cells. Knowing the SKY92 genetic subtype can help healthcare professionals distinguish between high-risk and standard-risk patients.
For more information on the test, visit www.mmprofiler.com. ![]()
EC grants conditional approval for blinatumomab

and solution for infusion
Photo courtesy of Amgen
The European Commission (EC) has granted conditional marketing authorization for blinatumomab (Blincyto) to treat adults with Philadelphia chromosome-negative (Ph-) relapsed or refractory B-precursor acute lymphoblastic leukemia (ALL).
This applies to the 28 member countries of the European Union as well as Iceland, Lichtenstein, and Norway.
Conditional marketing authorizations are valid for 1 year, on a renewable basis. The holder is required to complete ongoing studies or conduct new studies with the goal of confirming that a drug’s benefit-risk balance is positive.
Conditional marketing authorization is converted to a full authorization once these commitments have been fulfilled.
The EC granted conditional marketing authorization for blinatumomab based on a pair of phase 2 trials—Study ‘211 and Study ‘206.
Study ‘211
Results of Study ‘211 were presented at EHA 2014. The trial included 189 patients with Ph- relapsed or refractory B-precursor ALL.
The primary endpoint was complete remission or complete remission with partial hematologic recovery (CR/CRh). About 43% of patients achieved this endpoint within 2 cycles of therapy.
According to researchers, the most serious adverse events in this study were infection (31.7%), neurologic events (16.4%), neutropenia/febrile neutropenia (15.3%), cytokine release syndrome (CRS, 0.5%), and tumor lysis syndrome (0.5%).
Study ‘206
Results of Study ‘206 were presented at ASCO 2012. The trial included 36 patients with relapsed or refractory B-precursor ALL.
In this trial, the CR/CRh rate was 69.4% (25/36), with 15 patients achieving a CR (41.7%), and 10 patients achieving CRh (27.8%).
The “medically important” adverse events in this study, according to researchers, were CRS (n=3), central nervous system (CNS) events (3 seizures and 3 cases of encephalopathy), and fungal infection resulting in death (n=1).
However, the researchers found they could prevent CRS with dexamethasone. In addition, the CNS events were reversible, and blinatumomab could be reintroduced in 4 of the 6 patients with CNS events.
About blinatumomab
Blinatumomab is a bispecific T-cell engager (BiTE®) antibody construct that binds to CD19 on the surface of B cells and CD3 on the surface of T cells.
BiTE antibody constructs are intended to help the immune system detect and target malignant cells. The modified antibodies are designed to engage 2 different targets simultaneously, thereby juxtaposing T cells to cancer cells.
BiTE antibody constructs help place the T cells within reach of the targeted cells, with the intent of allowing T cells to inject toxins and trigger apoptosis in the cancer cells.
Blinatumomab was granted orphan drug designation by the European Medicines Agency in 2009 for the treatment of ALL.
The drug was granted breakthrough therapy designation and priority review by the US Food and Drug Administration.
Blinatumomab has conditional approval in the US to treat patients with relapsed or refractory Ph- B-precursor ALL. Continued approval for this indication may be contingent upon verification of clinical benefit in subsequent trials.
Blinatumomab is being developed by Amgen. ![]()
and solution for infusion
Photo courtesy of Amgen
The European Commission (EC) has granted conditional marketing authorization for blinatumomab (Blincyto) to treat adults with Philadelphia chromosome-negative (Ph-) relapsed or refractory B-precursor acute lymphoblastic leukemia (ALL).
This applies to the 28 member countries of the European Union as well as Iceland, Lichtenstein, and Norway.
Conditional marketing authorizations are valid for 1 year, on a renewable basis. The holder is required to complete ongoing studies or conduct new studies with the goal of confirming that a drug’s benefit-risk balance is positive.
Conditional marketing authorization is converted to a full authorization once these commitments have been fulfilled.
The EC granted conditional marketing authorization for blinatumomab based on a pair of phase 2 trials—Study ‘211 and Study ‘206.
Study ‘211
Results of Study ‘211 were presented at EHA 2014. The trial included 189 patients with Ph- relapsed or refractory B-precursor ALL.
The primary endpoint was complete remission or complete remission with partial hematologic recovery (CR/CRh). About 43% of patients achieved this endpoint within 2 cycles of therapy.
According to researchers, the most serious adverse events in this study were infection (31.7%), neurologic events (16.4%), neutropenia/febrile neutropenia (15.3%), cytokine release syndrome (CRS, 0.5%), and tumor lysis syndrome (0.5%).
Study ‘206
Results of Study ‘206 were presented at ASCO 2012. The trial included 36 patients with relapsed or refractory B-precursor ALL.
In this trial, the CR/CRh rate was 69.4% (25/36), with 15 patients achieving a CR (41.7%), and 10 patients achieving CRh (27.8%).
The “medically important” adverse events in this study, according to researchers, were CRS (n=3), central nervous system (CNS) events (3 seizures and 3 cases of encephalopathy), and fungal infection resulting in death (n=1).
However, the researchers found they could prevent CRS with dexamethasone. In addition, the CNS events were reversible, and blinatumomab could be reintroduced in 4 of the 6 patients with CNS events.
About blinatumomab
Blinatumomab is a bispecific T-cell engager (BiTE®) antibody construct that binds to CD19 on the surface of B cells and CD3 on the surface of T cells.
BiTE antibody constructs are intended to help the immune system detect and target malignant cells. The modified antibodies are designed to engage 2 different targets simultaneously, thereby juxtaposing T cells to cancer cells.
BiTE antibody constructs help place the T cells within reach of the targeted cells, with the intent of allowing T cells to inject toxins and trigger apoptosis in the cancer cells.
Blinatumomab was granted orphan drug designation by the European Medicines Agency in 2009 for the treatment of ALL.
The drug was granted breakthrough therapy designation and priority review by the US Food and Drug Administration.
Blinatumomab has conditional approval in the US to treat patients with relapsed or refractory Ph- B-precursor ALL. Continued approval for this indication may be contingent upon verification of clinical benefit in subsequent trials.
Blinatumomab is being developed by Amgen. ![]()
and solution for infusion
Photo courtesy of Amgen
The European Commission (EC) has granted conditional marketing authorization for blinatumomab (Blincyto) to treat adults with Philadelphia chromosome-negative (Ph-) relapsed or refractory B-precursor acute lymphoblastic leukemia (ALL).
This applies to the 28 member countries of the European Union as well as Iceland, Lichtenstein, and Norway.
Conditional marketing authorizations are valid for 1 year, on a renewable basis. The holder is required to complete ongoing studies or conduct new studies with the goal of confirming that a drug’s benefit-risk balance is positive.
Conditional marketing authorization is converted to a full authorization once these commitments have been fulfilled.
The EC granted conditional marketing authorization for blinatumomab based on a pair of phase 2 trials—Study ‘211 and Study ‘206.
Study ‘211
Results of Study ‘211 were presented at EHA 2014. The trial included 189 patients with Ph- relapsed or refractory B-precursor ALL.
The primary endpoint was complete remission or complete remission with partial hematologic recovery (CR/CRh). About 43% of patients achieved this endpoint within 2 cycles of therapy.
According to researchers, the most serious adverse events in this study were infection (31.7%), neurologic events (16.4%), neutropenia/febrile neutropenia (15.3%), cytokine release syndrome (CRS, 0.5%), and tumor lysis syndrome (0.5%).
Study ‘206
Results of Study ‘206 were presented at ASCO 2012. The trial included 36 patients with relapsed or refractory B-precursor ALL.
In this trial, the CR/CRh rate was 69.4% (25/36), with 15 patients achieving a CR (41.7%), and 10 patients achieving CRh (27.8%).
The “medically important” adverse events in this study, according to researchers, were CRS (n=3), central nervous system (CNS) events (3 seizures and 3 cases of encephalopathy), and fungal infection resulting in death (n=1).
However, the researchers found they could prevent CRS with dexamethasone. In addition, the CNS events were reversible, and blinatumomab could be reintroduced in 4 of the 6 patients with CNS events.
About blinatumomab
Blinatumomab is a bispecific T-cell engager (BiTE®) antibody construct that binds to CD19 on the surface of B cells and CD3 on the surface of T cells.
BiTE antibody constructs are intended to help the immune system detect and target malignant cells. The modified antibodies are designed to engage 2 different targets simultaneously, thereby juxtaposing T cells to cancer cells.
BiTE antibody constructs help place the T cells within reach of the targeted cells, with the intent of allowing T cells to inject toxins and trigger apoptosis in the cancer cells.
Blinatumomab was granted orphan drug designation by the European Medicines Agency in 2009 for the treatment of ALL.
The drug was granted breakthrough therapy designation and priority review by the US Food and Drug Administration.
Blinatumomab has conditional approval in the US to treat patients with relapsed or refractory Ph- B-precursor ALL. Continued approval for this indication may be contingent upon verification of clinical benefit in subsequent trials.
Blinatumomab is being developed by Amgen. ![]()
AHA: New emphasis on percent LDL reduction on-treatment
ORLANDO – The individual variability in percent reduction in LDL cholesterol levels in response to high-intensity statin therapy is far greater than generally appreciated, and this has important implications for clinical practice, Dr. Paul M. Ridker said at the American Heart Association scientific sessions.
A new secondary analysis from the landmark JUPITER trial highlighted this substantial variability in percent reduction in LDL cholesterol on 20 mg/day of rosuvastatin (Crestor). Moreover, it showed that the size of this reduction was directly related to the magnitude of reduction in cardiovascular events.

“These data provide general support for the concept of introducing percent reduction in LDL cholesterol into broader clinical practice,” said Dr. Ridker, director of the Center for Cardiovascular Disease Prevention at Brigham and Women’s Hospital and professor of medicine at Harvard Medical School, Boston.
The concept of percent LDL reduction as a treatment target is already widely embedded in the ACC/AHA, European Society of Cardiology, and Canadian Cardiovascular Society cholesterol management guidelines, he noted.
For example, the 2013 ACC/AHA guidelines state that lower-risk individuals who qualify for statin therapy should receive a moderate-intensity statin regimen capable of reducing LDL by 30%-50% from baseline, while higher-risk patients should be placed on a high-intensity statin, described as an agent that gives a 50% or greater reduction in LDL. The new JUPITER analysis makes the case for featuring percent LDL reduction more prominently as an explicit personalized treatment target in the guidelines, Dr. Ridker continued.
In JUPITER, 17,802 apparently healthy subjects with an LDL cholesterol level below 130 mg/dL were randomized to rosuvastatin or placebo. The trial was halted early, after a median of 1.9 years, because the rosuvastatin group showed a compelling 44% reduction in the composite endpoint of MI, stroke, unstable angina treated by revascularization, or cardiovascular death (N Engl J Med. 2008 Nov 20;359[21]:2195-320).
In JUPITER, rosuvastatin reduced LDL cholesterol by an average of 50% in the 7,856 treated patients. But as the new analysis demonstrates, individual variability in response was huge, ranging from no LDL reduction at all to a greater than 85% reduction. And cardiovascular event rates varied accordingly: from 11.2 events per 1,000 person-years with placebo to 9.2 in rosuvastatin-treated patients with no LDL reduction, 6.7 in those with less than a 50% drop in LDL, and 4.8 events per 1,000 person-years in subjects with a greater than 50% reduction in LDL on rosuvastatin. Thus, the one-half of rosuvastatin-treated patients who had more than a 50% decrease in LDL had an adjusted 59% reduction in major cardiovascular events, compared with placebo, while those with a drop of less than 50% in LDL had a 39% risk reduction.
The same exceptionally wide individual variability was seen in on-treatment reductions in apolipoprotein B cholesterol and non–HDL cholesterol levels, and once again, the magnitude of the percent reduction in these lipids tracked with the size of the reduction in cardiovascular events.
This new analysis from JUPITER essentially confirms the findings of an earlier meta-analysis of eight randomized controlled trials with more than 38,000 patients assigned to statin therapy. The meta-analysis showed very large interindividual variations in reductions in LDL, non–HDL cholesterol, and apolipoprotein B in response to high-dose statin therapy. Moreover, patients who achieved very low LDL levels on-treatment had a lower risk of cardiovascular events than those who achieved more moderate LDL reductions (J Am Coll Cardiol. 2014 Aug 5;64[5]:485-94).
Dr. Ridker said the new findings from JUPITER and the meta-analysis, in addition to their implications for clinical practice, could also be relevant in the future with regard to treatment decisions regarding when to prescribe proprotein convertase subtilisin–kexin type 9 (PCSK9) inhibitors, assuming ongoing clinical trials ultimately show that this novel and expensive class of superpotent LDL-lowering agents reduces the risk of cardiovascular events.
He noted that 20% of rosuvastatin-treated JUPITER participants had a greater than 60% reduction in LDL. In theory, he explained, this might be the group where the PCSK9 inhibitors would have the least benefit because those patients already have a 70%-80% reduction in LDL on a high-intensity statin.
On the other hand, the 35% of JUPITER participants with an on-treatment LDL reduction ranging from zero to less than 40% would have the greatest theoretic benefit from a PCSK9 inhibitor, while patients who obtained a 40%-60% LDL reduction on rosuvastatin would be expected to derive an intermediate benefit from the new drugs.
Discussant Michael J. Pencina, Ph.D., said the current U.S. cholesterol management guidelines focus heavily on cardiovascular risk as determined by the risk calculator equation. This needs to be balanced by a more explicit focus on assessment of the anticipated benefit of therapy, he added. For this reason, he agreed with Dr. Ridker’s call to incorporate measurement of percent reduction in lipid levels into individualized assessment of therapeutic benefit.
It will be important for the ongoing randomized trials of PCSK9 inhibitors to report results stratified by the percent reduction in LDL cholesterol achieved by background statin therapy. This will be useful, as Dr. Ridker said, in figuring out how best to allocate this new class of lipid-lowering medications, added Dr. Pencina, professor of biostatistics and bioinformatics at Duke University, Durham, N.C.
Dr. Ridker reported receiving research grants from AstraZeneca, Pfizer, Amgen, and the National Institutes of Health.
ORLANDO – The individual variability in percent reduction in LDL cholesterol levels in response to high-intensity statin therapy is far greater than generally appreciated, and this has important implications for clinical practice, Dr. Paul M. Ridker said at the American Heart Association scientific sessions.
A new secondary analysis from the landmark JUPITER trial highlighted this substantial variability in percent reduction in LDL cholesterol on 20 mg/day of rosuvastatin (Crestor). Moreover, it showed that the size of this reduction was directly related to the magnitude of reduction in cardiovascular events.
“These data provide general support for the concept of introducing percent reduction in LDL cholesterol into broader clinical practice,” said Dr. Ridker, director of the Center for Cardiovascular Disease Prevention at Brigham and Women’s Hospital and professor of medicine at Harvard Medical School, Boston.
The concept of percent LDL reduction as a treatment target is already widely embedded in the ACC/AHA, European Society of Cardiology, and Canadian Cardiovascular Society cholesterol management guidelines, he noted.
For example, the 2013 ACC/AHA guidelines state that lower-risk individuals who qualify for statin therapy should receive a moderate-intensity statin regimen capable of reducing LDL by 30%-50% from baseline, while higher-risk patients should be placed on a high-intensity statin, described as an agent that gives a 50% or greater reduction in LDL. The new JUPITER analysis makes the case for featuring percent LDL reduction more prominently as an explicit personalized treatment target in the guidelines, Dr. Ridker continued.
In JUPITER, 17,802 apparently healthy subjects with an LDL cholesterol level below 130 mg/dL were randomized to rosuvastatin or placebo. The trial was halted early, after a median of 1.9 years, because the rosuvastatin group showed a compelling 44% reduction in the composite endpoint of MI, stroke, unstable angina treated by revascularization, or cardiovascular death (N Engl J Med. 2008 Nov 20;359[21]:2195-320).
In JUPITER, rosuvastatin reduced LDL cholesterol by an average of 50% in the 7,856 treated patients. But as the new analysis demonstrates, individual variability in response was huge, ranging from no LDL reduction at all to a greater than 85% reduction. And cardiovascular event rates varied accordingly: from 11.2 events per 1,000 person-years with placebo to 9.2 in rosuvastatin-treated patients with no LDL reduction, 6.7 in those with less than a 50% drop in LDL, and 4.8 events per 1,000 person-years in subjects with a greater than 50% reduction in LDL on rosuvastatin. Thus, the one-half of rosuvastatin-treated patients who had more than a 50% decrease in LDL had an adjusted 59% reduction in major cardiovascular events, compared with placebo, while those with a drop of less than 50% in LDL had a 39% risk reduction.
The same exceptionally wide individual variability was seen in on-treatment reductions in apolipoprotein B cholesterol and non–HDL cholesterol levels, and once again, the magnitude of the percent reduction in these lipids tracked with the size of the reduction in cardiovascular events.
This new analysis from JUPITER essentially confirms the findings of an earlier meta-analysis of eight randomized controlled trials with more than 38,000 patients assigned to statin therapy. The meta-analysis showed very large interindividual variations in reductions in LDL, non–HDL cholesterol, and apolipoprotein B in response to high-dose statin therapy. Moreover, patients who achieved very low LDL levels on-treatment had a lower risk of cardiovascular events than those who achieved more moderate LDL reductions (J Am Coll Cardiol. 2014 Aug 5;64[5]:485-94).
Dr. Ridker said the new findings from JUPITER and the meta-analysis, in addition to their implications for clinical practice, could also be relevant in the future with regard to treatment decisions regarding when to prescribe proprotein convertase subtilisin–kexin type 9 (PCSK9) inhibitors, assuming ongoing clinical trials ultimately show that this novel and expensive class of superpotent LDL-lowering agents reduces the risk of cardiovascular events.
He noted that 20% of rosuvastatin-treated JUPITER participants had a greater than 60% reduction in LDL. In theory, he explained, this might be the group where the PCSK9 inhibitors would have the least benefit because those patients already have a 70%-80% reduction in LDL on a high-intensity statin.
On the other hand, the 35% of JUPITER participants with an on-treatment LDL reduction ranging from zero to less than 40% would have the greatest theoretic benefit from a PCSK9 inhibitor, while patients who obtained a 40%-60% LDL reduction on rosuvastatin would be expected to derive an intermediate benefit from the new drugs.
Discussant Michael J. Pencina, Ph.D., said the current U.S. cholesterol management guidelines focus heavily on cardiovascular risk as determined by the risk calculator equation. This needs to be balanced by a more explicit focus on assessment of the anticipated benefit of therapy, he added. For this reason, he agreed with Dr. Ridker’s call to incorporate measurement of percent reduction in lipid levels into individualized assessment of therapeutic benefit.
It will be important for the ongoing randomized trials of PCSK9 inhibitors to report results stratified by the percent reduction in LDL cholesterol achieved by background statin therapy. This will be useful, as Dr. Ridker said, in figuring out how best to allocate this new class of lipid-lowering medications, added Dr. Pencina, professor of biostatistics and bioinformatics at Duke University, Durham, N.C.
Dr. Ridker reported receiving research grants from AstraZeneca, Pfizer, Amgen, and the National Institutes of Health.
ORLANDO – The individual variability in percent reduction in LDL cholesterol levels in response to high-intensity statin therapy is far greater than generally appreciated, and this has important implications for clinical practice, Dr. Paul M. Ridker said at the American Heart Association scientific sessions.
A new secondary analysis from the landmark JUPITER trial highlighted this substantial variability in percent reduction in LDL cholesterol on 20 mg/day of rosuvastatin (Crestor). Moreover, it showed that the size of this reduction was directly related to the magnitude of reduction in cardiovascular events.
“These data provide general support for the concept of introducing percent reduction in LDL cholesterol into broader clinical practice,” said Dr. Ridker, director of the Center for Cardiovascular Disease Prevention at Brigham and Women’s Hospital and professor of medicine at Harvard Medical School, Boston.
The concept of percent LDL reduction as a treatment target is already widely embedded in the ACC/AHA, European Society of Cardiology, and Canadian Cardiovascular Society cholesterol management guidelines, he noted.
For example, the 2013 ACC/AHA guidelines state that lower-risk individuals who qualify for statin therapy should receive a moderate-intensity statin regimen capable of reducing LDL by 30%-50% from baseline, while higher-risk patients should be placed on a high-intensity statin, described as an agent that gives a 50% or greater reduction in LDL. The new JUPITER analysis makes the case for featuring percent LDL reduction more prominently as an explicit personalized treatment target in the guidelines, Dr. Ridker continued.
In JUPITER, 17,802 apparently healthy subjects with an LDL cholesterol level below 130 mg/dL were randomized to rosuvastatin or placebo. The trial was halted early, after a median of 1.9 years, because the rosuvastatin group showed a compelling 44% reduction in the composite endpoint of MI, stroke, unstable angina treated by revascularization, or cardiovascular death (N Engl J Med. 2008 Nov 20;359[21]:2195-320).
In JUPITER, rosuvastatin reduced LDL cholesterol by an average of 50% in the 7,856 treated patients. But as the new analysis demonstrates, individual variability in response was huge, ranging from no LDL reduction at all to a greater than 85% reduction. And cardiovascular event rates varied accordingly: from 11.2 events per 1,000 person-years with placebo to 9.2 in rosuvastatin-treated patients with no LDL reduction, 6.7 in those with less than a 50% drop in LDL, and 4.8 events per 1,000 person-years in subjects with a greater than 50% reduction in LDL on rosuvastatin. Thus, the one-half of rosuvastatin-treated patients who had more than a 50% decrease in LDL had an adjusted 59% reduction in major cardiovascular events, compared with placebo, while those with a drop of less than 50% in LDL had a 39% risk reduction.
The same exceptionally wide individual variability was seen in on-treatment reductions in apolipoprotein B cholesterol and non–HDL cholesterol levels, and once again, the magnitude of the percent reduction in these lipids tracked with the size of the reduction in cardiovascular events.
This new analysis from JUPITER essentially confirms the findings of an earlier meta-analysis of eight randomized controlled trials with more than 38,000 patients assigned to statin therapy. The meta-analysis showed very large interindividual variations in reductions in LDL, non–HDL cholesterol, and apolipoprotein B in response to high-dose statin therapy. Moreover, patients who achieved very low LDL levels on-treatment had a lower risk of cardiovascular events than those who achieved more moderate LDL reductions (J Am Coll Cardiol. 2014 Aug 5;64[5]:485-94).
Dr. Ridker said the new findings from JUPITER and the meta-analysis, in addition to their implications for clinical practice, could also be relevant in the future with regard to treatment decisions regarding when to prescribe proprotein convertase subtilisin–kexin type 9 (PCSK9) inhibitors, assuming ongoing clinical trials ultimately show that this novel and expensive class of superpotent LDL-lowering agents reduces the risk of cardiovascular events.
He noted that 20% of rosuvastatin-treated JUPITER participants had a greater than 60% reduction in LDL. In theory, he explained, this might be the group where the PCSK9 inhibitors would have the least benefit because those patients already have a 70%-80% reduction in LDL on a high-intensity statin.
On the other hand, the 35% of JUPITER participants with an on-treatment LDL reduction ranging from zero to less than 40% would have the greatest theoretic benefit from a PCSK9 inhibitor, while patients who obtained a 40%-60% LDL reduction on rosuvastatin would be expected to derive an intermediate benefit from the new drugs.
Discussant Michael J. Pencina, Ph.D., said the current U.S. cholesterol management guidelines focus heavily on cardiovascular risk as determined by the risk calculator equation. This needs to be balanced by a more explicit focus on assessment of the anticipated benefit of therapy, he added. For this reason, he agreed with Dr. Ridker’s call to incorporate measurement of percent reduction in lipid levels into individualized assessment of therapeutic benefit.
It will be important for the ongoing randomized trials of PCSK9 inhibitors to report results stratified by the percent reduction in LDL cholesterol achieved by background statin therapy. This will be useful, as Dr. Ridker said, in figuring out how best to allocate this new class of lipid-lowering medications, added Dr. Pencina, professor of biostatistics and bioinformatics at Duke University, Durham, N.C.
Dr. Ridker reported receiving research grants from AstraZeneca, Pfizer, Amgen, and the National Institutes of Health.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: An individual’s percent reduction in LDL cholesterol achieved on statin therapy is a clinically important measurement.
Major finding: The percent LDL cholesterol lowering achieved with high-intensity statin therapy varies widely between individuals and tracks closely with the magnitude of cardiovascular risk reduction.
Data source: A secondary analysis of the JUPITER trial, in which more than 17,000 apparently healthy subjects were randomized to 20 mg/day of rosuvastatin or placebo.
Disclosures: The presenter reported receiving research grants from AstraZeneca, Pfizer, Amgen, and the National Institutes of Health.

Medical Roundtable: Pediatric Non-Hodgkin Lymphoma (NHL) Classification Guidelines - International Pediatric NHL Staging System (IPNHLSS)
Moderator: Catherine Bollard, MD, FRACP, FRCPA1
Discussants: Mitchell S. Cairo, MD2; Eric J. Lowe, MD3; Thomas G. Gross, MD, PhD4
Address for correspondence: Catherine Bollard, MD, FRACP, FRCPA, 111 Michigan Avenue, NW, 5th Floor Main, Suite 5225, Washington, DC 20010
E-mail: [email protected]
Biographical sketch: From The George Washington University, School of Medicine and Health Sciences, Washington, DC1; Westchester Medical Center, New York Medical College, Valhalla, NY2; Children’s Hospital of the King’s Daughters, Norfolk, VA3; Center for Global Health at the National Cancer Institute, Rockville, MD
DR. BOLLARD: My name is Dr. Catherine Bollard. I'm Chief of the Division of Allergy and Immunology at Children's National Health System and the Chair of the NHL Committee of the Children's Oncology Group. I hope that today we can provide some clarity and give you some of our first-hand expertise and experience regarding some of the challenges and controversies of treating pediatric patients with non-Hodgkin lymphoma (NHL). Here with me are Drs. Mitchell Cairo, Chief of Pediatric Hematology/Oncology and Stem Cell Transplantation at New York Medical College in the Maria Fareri Children's Hospital, Westchester Medical Center; Eric Lowe, Division Director for Pediatric Hematology/Oncology at the Children's Hospital of the King's Daughters; and Thomas Gross, Deputy Director for Science at the Center for Global Health at the National Cancer Institute.
I'd like to start the questioning, firstly to Dr. Cairo, who recently published with a group of leaders in the pediatric lymphoma field, new staging and response classifications. Dr. Cairo, I’d like you to highlight how these are different from the current classifications, and what you see are the strengths and the limitations at this time.
DR. CAIRO: Thank you, Cath. The original staging classification was developed in the late 1970s by Dr. Murphy while she was at St. Jude's hospital, and either goes by the name the Murphy Staging Classification or the St. Jude's Classification. That classification I think was quite useful at that time when we recognized really only a couple subtypes of NHL, as well as the capabilities we had in those days both imaging as well as further molecular identification as well as trying to identify sites of spread. As some 35 years have evolved, new pathological entities have been identified, much more precise imaging techniques, new methods of detecting more evidence of minimal disease, and also identifying new organ sites of involvement, allowed the creation of a multidisciplinary international task force to look at how we could enhance the original observations by the St. Jude's group.
As Dr. Bollard pointed out, we eventually, over 9 years of evidence-based review, came up with an enhanced staging classification called the International Pediatric NHL Staging System (IPNHLSS).1 In this new system we account for new histological subtypes, allow for different organ distributions, improve on the new imaging techniques to identify areas of involvement, and also to more molecularly identify extent of disease. I think the advantages are stated above. The disadvantage is that like all staging systems it's a breathing document. It will require international collaboration. As time evolves, this staging system will of course need to be updated as we gain new experience.
Briefly, in terms of the response classification that also came out of the same international multidisciplinary task force that was led by Dr. Sandlund at St. Jude's2; there had never been a response criteria that had been focused entirely in childhood and adolescent NHL. The previous response criteria had been developed by adult NHL investigators, and there was a need to develop the first response criteria for pediatric NHL because of different histologies, different sites of sanctuary disease, and now obviously enhanced imaging capabilities. That also now has been named the International Pediatric Non-Hodgkin Lymphoma Response Criteria (IPNHLRC)—hopefully for harmonizing a response across new studies, but also a breathing document that is going to be limited as we gain new knowledge into how we can better assess response as new techniques are developed.
DR. BOLLARD: I thank you very much for your detailed response. My next question is actually to Dr. Gross, who is currently chairing the international study for upfront diffuse large B-cell lymphoma and Burkitt lymphoma in pediatric young adults. I would like you to speak to a couple of issues, and you can put it in the context of the current randomized trial, looking at rituximab vs no rituximab for this disease. I think firstly it would be useful for you to speak to the implications of this new classification system as we go forward with choosing new therapeutic strategies for these patients, and in particular I'd like to focus on the newly diagnosed diffuse large B-cell lymphoma patients who are in that adolescent/young adult range.
I would also be interested in your opinion regarding how you would manage a patient who is 17 years old but is going to turn 18 tomorrow, and he comes to you with newly diagnosed diffuse large B-cell lymphoma. As you know, the adult oncologists treat diffuse large B-cell lymphoma different to Burkitt lymphoma, and in pediatrics we generally treat these diseases the same. Do you tell this patient that you will treat him today on a pediatric regimen, or do you tell him to go tomorrow, when he's 18, to be treated by an adult oncologist? I would like you to justify your answer please.
DR. GROSS: First to discuss the implications of the new staging as it applies to the current international trial. As Dr. Cairo pointed out, this was developed through a literature review and evidence based analyses, but like any new staging system, the value of staging is to provide us with information that can try to help us to identify patients to improve their outcome. Essentially, staging is to help direct therapy or provide prognosis for outcome, and the only way to do that is to test new systems or classifications in a prospective fashion. Indeed, that is what we are trying to do with this international effort.
This international effort, just as an aside, illustrates one of the challenges of all rare cancers, but particularly pediatrics. In pediatric mature B-cell NHL, both large-cell and Burkitt, we are now at a cure rate of about 90%. To make advances, we don't have enough patients seen in North America and Australia, and it requires international collaboration. This trial, to get 600 patients randomized, it will take 7 years with 14 countries participating—that is one of the challenges, certainly, we have with pediatric NHL. Also, we want to try to gain as much information as possible, not just to the effect of rituximab as Dr. Bollard said, but also to test other questions such as the role or the value in validating this new staging system.
To talk about the controversy of treatment, certainly we know that there is a very different approach in pediatrics. For many years, we have treated diffuse large B-cell lymphoma just like Burkitt. This is a very important delineation when you're seen by a medical oncologist because the treatment for diffuse large B-cell lymphoma is outpatient therapy, ie rituximab, cyclophosphamide, adriamycin, vincristine, and prednisone (R-CHOP). Treatment for Burkitt is inpatient with high doses of methotrexate, but other higher doses of the same agents used to treat diffuse large B-cell lymphoma. The question is, do we really need to treat all the pediatric diffuse large B-cell lymphoma with these aggressive Burkitt regimens? I think one of the things that is encouraging to me as a pediatric oncologist is that we are beginning to learn that the biology is very different. Though the disease looks the same under the microscope or by flow cytometry, when you look at it genetically it's quite different. We know now that the younger the patient is with diffuse large B-cell lymphoma, even though it looks for all intents and purposes like the same disease as seen in adults, when you look at the genetics, many times, as high as 30% of the time, it will be genetically the same as a Burkitt lymphoma. I think when you're talking about young patients we can easily justify treating them both the same because the biology would suggest that a good number of patients would need Burkitt therapy to be cured.
Now, that changes over time, so that it appears that sometime in young adulthood, maybe somewhere between 25 and 35 years of age, you don't see the genetic disease that looks like diffuse large B-cell lymphoma, but is genetically Burkitt lymphoma. As for the 18-year-old patient that Dr. Bollard was posing to me, I've had several patients like this. I go through the pluses and minuses of the therapy, inpatient vs outpatient, but also the potential long-term side effects. The outpatient therapy has potentially more long-term side effects as far as potential infertility and potential heart damage. Every time I have given the choice to the family and the patient, the teenager has always chosen the outpatient therapy that you would get as an adult, and the parents always say they would rather have the inpatient therapy, and that spending a couple of days in the hospital to try to reduce the chance of long-term damage is their choice. It's a very interesting dynamic and I think sometimes the issues that go into choice of treatment are quite variable. My personal opinion is that hopefully in the future we will be able to have a better understanding of biology, so that when we see these patients, be they 18 or 25 years old, we're not looking at what it looks like under the microscope or who they see, and what they're used to giving, but the biology will determine which therapy is more likely to cure them. Right now we don't have that ability in most of the patients.
DR. BOLLARD: Thank you, Dr. Gross. Again, another very comprehensive answer to a difficult question. I'm actually going to push this back to Dr. Cairo and then impose the same soon to be 18-year-old patient to you. This time, he's coming to you with relapsed diffuse large B-cell lymphoma. What are you going to tell him? Are you going to treat him today on pediatric protocols, or will you wait until tomorrow when he could have access to adult protocols?
DR. CAIRO: I think the results are relatively similar, but, in part, the answer to the question is of course based on what their original therapy was. If the original therapy was the pediatric-inspired type of treatment, I think there's a world of experience of what are some of the best pediatric-inspired regimens to use for retrieval. If, however, the original therapy was an adult-inspired regimen, then I think the options are open because the disease may not be as resistant in that setting; therefore, one would want to consider all the adult type of retrieval regimens in that case, because that group of patients—at least in the adult experience—tend to have disease that may be more responsive because they're not as resistant to the higher dose and multi-agent therapy that a pediatric-inspired regimen would have given them had they been treated that way.
DR. BOLLARD: I was also trying to ask you to speak to the access that an 18-year-old might have to novel therapies that a 17-year-old might not. How do you address that issue?
DR. CAIRO: That's an excellent question. I think that for first relapse or first induction failure most of the retrieval regimens, the first line regimens, that are available, either pediatric inspired or adult inspired, probably don't require an investigational agent that an 18-year-old might have access to if he was being treated on an adult type of regimen. However, I would strongly encourage an 18-year-old who failed one retrieval regimen to consider experimental therapy. There I think the access to new agents—if you're 18 or over—are so much greater that I would encourage them to be treated on an adult retrieval regimen, where some of the newer agents may be investigational, are not available to a pediatric program.
DR. BOLLARD: Thank you very much, Dr. Cairo. I have one last question on the B-cell diseases before I move to Dr. Lowe, and the last question goes to Dr. Gross. Would you recommend that a patient with relapsed Burkitt lymphoma—now increasingly rare—be treated with salvage chemotherapy and then autologous transplant or allogeneic stem cell transplant?
DR. GROSS: As the others on this discussion know, we performed an analysis from data in the Center for International Blood and Marrow Transplant Research (CIBMTR), and the problem is that Burkitt lymphoma tends to reoccur so rapidly after transplant. The median time to relapse is 3 months after transplant. We could not find a difference in the outcome between autografts and allografts because of its early reoccurrence. That said, my personal opinion is, since we know that Burkitt lymphoma is a hematologically spread disease, that I always prefer a donor source where I know they're not going to have tumor cells in them, which is an allogeneic donor. I always prefer an allogeneic donor, because I know it's tumor-free, but also it gives us an opportunity, if the disease will stay under control long enough, to potentially get an immune response against any residual tumor. For that reason, I recommend an allogeneic donor if it can be found readily.
DR. BOLLARD: Thank you very much, Dr. Gross. Now, on to Dr. Lowe, and Dr. Lowe's particular area of expertise is in anaplastic large cell lymphoma (ALCL) and T-cell diseases. I was wondering if you could explain to me the difference between T-cell lymphoblastic lymphoma and T-cell acute lymphoblastic leukemia (ALL), specifically since the World Health Organization (WHO) groups these two disease entities together as T-lymphoblastic leukemia/lymphoma. If you could clarify that classification that would be very helpful.
DR. LOWE: As you well know, many physicians believe that T-cell lymphoblastic leukemia and T-cell lymphoblastic lymphoma are very similar diseases, but they are not exactly the same disease although we sometimes treat them very similar. We know that T-cells do not mature in bone marrow but rather they are thymic driven cells. Because of this, there are distinct differences between the leukemia and lymphoma. For example, we know that the genetics between the two—although in limited samples—are not always the same, including in prognostic. Just one example, loss of heterozygosity at 6Q has been shown to be prognostically important in lymphoblastic lymphoma but not in T-cell ALL. I think the real challenge is to figure out what the differences are. I think we could argue that potentially, T-ALL is stage four T-cell lymphoblastic lymphoma.
I think that the WHO classifying the two diseases as one entity with T-lymphoblastic leukemia/lymphoma has hindered a little in the advancement of recognizing the differences in that many people assume that they're the same disease. When you look up from a pathological standpoint and you say, well, they're clearly the same disease because they're listed as a single entity, and when you look up treatment, you say, well they're treated very similar, so they must be the same disease. I think that does us a little disservice in trying to advance the field forward, because I think getting lymphoblastic lymphoma samples, which is challenging, is extremely important to determine the genetic drivers of this disease.
DR. BOLLARD: Thank you very much, Dr. Lowe. I would also like you to discuss how ALCL differs between the pediatric and the adult populations, and how that dictates how you would treat those two patient populations.
DR. LOWE: So, ALCL really has a much shorter span in terms of its description pathologically. It was not described by itself until the mid 1980s. In the mid 1990s it started entering classification schemes. It wasn't until 2008 that the WHO separated out three distinct entities within ALCL. You have anaplastic lymphoma kinase (ALK)-positive ALCL, ALK-negative ALCL, and primary cutaneous ALCL. This is a great example where these three different entities have very different epidemiology, very different treatment strategies, and the fact that they are broken up has really helped move the field forward. For example, ALK-positive ALCL is really a disease of children, adolescents, and young adults. It's the most common ALCL by far in that age group. It's extremely rare to have an ALK-negative ALCL, and the pathological reason for the disease with ALK-positive ALCL is a translocation involving the ALK gene leads directly to oncogenesis.
Because of this, we have started to develop treatments that are designed to target this specific oncogenic driving translocation. This is in direct comparison to an ALK-negative ALCL, which is primarily a disease of older individuals, most commonly in their 50s and 60s. The outcome for this disease is consistently poorer than for ALK-positive disease. The treatment, while sometimes the same, is changing now that we have targets for the tyrosine kinase that is driving the ALK-positive ALCL. I think separating these two out has been a huge advantage in terms of figuring out what to do with pediatric ALCL because the 95 plus percent of ALCLs in pediatrics and young adults are ALK-positive.
Primary cutaneous ALCL is almost a completely different entity in and of itself, although it shares the same name. The primary cutaneous ALCLs are usually not treated on similar studies as the systemic forms of ALCL. The primary cutaneous form has different characteristics in terms of location, age, treatment, and natural course. The vast majority does not develop into systemic disease, and thus the treatment is very different. I think ALCL is a very good example where the different pathological entities have led to very different treatments based on what is driving the cancer.
DR. BOLLARD: Thanks, Dr. Lowe. I think that's very important to emphasize how ALK-positivity is more common in children than in adults, and the successes of crizotinib, even in phase 1 in pediatric patients with ALCL. My question now is given the success of this targeted agent in the relapse setting, even in phase 1, do you still see a role for allogeneic stem cell transplant for those patients who have relapsed after conventional therapy?
DR. LOWE: I do still see a role, but I'm not sure how much that role will shrink over time as we learn more and more about this disease. We know that there are very high risk patients that relapse or progress while receiving traditional chemotherapy. Those patients typically have achieved the best outcome with an allogeneic transplant. That said, I think crizotinib and other ALK inhibitors are changing the landscape of treatment for ALK-positive ALCL very fast. We know that some patients who are refractory to many other treatments go into remission with these drugs, and while I think that the role of allogeneic transplant is still there, I think that it may be changing over time. The other decision that I think will be difficult in terms of allogeneic transplant is for patients who receive ALK-inhibitors, like crizotinib, for initial treatment and then relapse. Many patients in that situation currently will end up having an allogenic transplant. However, one can argue that very much like chronic myeloid leukemia, these patients might be rescued without an allogeneic transplant using a second line ALK inhibitor. All of these things, obviously, we hope to know over time, but at this point in time are unknown.
DR. BOLLARD: Thank you very much. I'll take you out of the hot seat now. All of us talked about the concept of the importance of knowing the biology of what we're treating, and with the advent of novel targeted therapies, this concept of precision medicine is becoming increasingly important, ie, targeting the individual patient's tumor with the appropriate targeted agents for their tumor. This is maybe a question for Dr. Gross first, and then Dr. Cairo. What do you see are the challenges for being able to obtain the tissue from pediatric patients to perform these important and critical tests that will be needed as we move the field forward for the management of pediatric patients with NHL?
DR. GROSS: I think that the number one barrier is, as the technology improves to be able to make the essential diagnosis, we need less and less tissue for the pathologist. It becomes increasingly more of a challenge to obtain extra tissue because the standard of practice is to get just enough to make the diagnosis. Unless we can address this challenge, it's going to be extremely difficult.
DR. BOLLARD: Dr. Cairo, do you want to speak to that, since you recently completed a Children’s Oncology Group trial for Burkitt and diffuse large B-cell lymphoma?
DR. CAIRO: Thank you. I agree with Dr. Gross, and that particular trial, despite it being one of the primary objectives and also many of those patients actually had bone marrow involvement, which is the area that we access the easiest as the acute lymphoblastic leukemia colleagues have taught us. We still only were able to get 11 of some 90 patients entered on study, to have specimens sent. That being said, having just come back from the Fifth International Symposium on Childhood, Adolescent and Young Adult Non-Hodgkin Lymphoma, It appears that the Europeans have been much more successful in obtaining specimens for biology studies in particular, the future precision medicine-based trials. We should try to learn a little bit from our European colleagues, who seem to have a much higher percentage of getting specimens, and we need to make every effort, as Dr. Gross said of encouraging our colleagues, that this is as important as making the diagnosis. We face an uphill battle because of our high cure rate, the biology is often considered a second thought sometimes. Europeans are better than us at obtaining biological specimens and we need to compete to achieve the level that they have achieved in Europe.
DR. GROSS: It's almost a catch-22. We know from other diseases in pediatric oncology, but also in adult oncology, that once we are able to demonstrate that the biology will make a difference in the treatment and outcome of the patient, then we're able to get the tissue needed. I think a good example of that is neuroblastoma. However, we can't make those discoveries unless we get enough tissue to study. We're in this catch-22, we cannot demonstrate that the biology makes a difference, unless we will get the tissue for research.
DR. LOWE: I'd like to add one other point to this. I think the rarity of the diseases and the large number of centers that treat the patients also hinders obtaining pathological samples. Because pediatric NHL is a relatively rare disease, you can’t have a single champion for obtaining biology at one institution that can accomplish anything without many other institutions. It requires a large group effort which is more difficult than a single institution collecting colon cancer samples, for example, where you really only need one institution, one champion, one pathologist, and you have all the samples you need.
DR. BOLLARD: Thank you, Dr. Lowe. I really thank you all for speaking in a very detailed way about the importance of obtaining tumor tissue to perform these critical biologic studies, because I do feel that's an important issue to overcome for the future care of our pediatric patients with NHL. I would like to discuss late effects in our survivors. As Dr. Gross said, survival rates for patients with B-cell lymphomas are generally outstanding. Dr. Gross, do you feel that late effects are not something the NHL group has to worry about now that we have obviated the need for radiation, or not? And what are your feelings about trying to minimize these late effects even further?
DR. GROSS: The good news is that over time, we have been able to come up with regimens that are highly effective but have reduced the agents we know have the highest risk of late effects—radiation being the primary one, but also anthracyclines we have been able to reduce in the vast majority of the patients, and to keep alkylating agents in the vast majority of the patients to a level that most patients do not have infertility. The long-term side effects are becoming pretty minimal, but the question is, how low do they have to be to be acceptable? The goal would be cure without any long-term effects. As I said before, certainly we have paid the price in short-term effects. Our regimens are inpatient, and they can have quite severe short-term side effects such as mucositis. We've made great advances but I think there's still room to go.
DR. CAIRO: I agree, of course, with my colleague Dr. Gross. Again, when we look at large series of chronic health care conditions, certainly children with treated NHL still comes up as one showing over 40% to 50% of patients having one or two serious chronic health care conditions. We know the data are a little antiquated, because they include patients who were treated with different regimens in the 1970s and all of the 1980s. However, I think our goal continues to be to identify the most effective treatment regimen, but with the least toxic long-term complications for our patients. That struggle is very difficult because of the very high success rate we have today, and to identify without hurting that high success rate less toxic therapies will require a collaborative, multidisciplinary, international effort to reach that goal.
DR. BOLLARD: Thank you very much Drs. Cairo and Gross. Dr. Lowe, did you have any closing remarks on the late effects issues for the T-cell mediated diseases in particular?
DR. LOWE: I would absolutely agree with Dr. Gross and Dr. Cairo that this is an important issue. I think we in pediatrics do a good job at following our patients for long-term side effects and creating guidelines for screening for these long-term side effects. That said, I think as we start to talk about better and better therapy and even more and more targeted therapy, what we don't know about some of these targeted therapies is their 15 and 20 year long-term side effects. We obviously hope that there aren't any, and that's why we are moving toward these drugs, but again, surveillance of those long-term side effects will be extremely important, especially when you're talking about medications for young children.
DR. BOLLARD: I'd like to thank you all very much for participating in this expert roundtable discussion today. I think the overarching points are that prognoses at the current time for newly diagnosed pediatric patients with NHL range from 70% to over 90% even for patients with disseminated disease. The challenges that we need to overcome are how we can optimize our up front treatment to prevent relapse in all, because I think we've all reiterated the fact that the outcomes for those few patients who do relapse remains extremely poor. I think there is still controversy about how to manage patients with relapsed disease, and how to temper our therapies against long-term side effects of our surviving patients. Finally, I think with the advent of novel targeted agents, it is incredibly important for the optimal management of our current and future patients that we are able to access tumor tissues and perform the critical biologic studies that are required to develop an effective precision medicine approach for pediatric patients with NHL. I would like to again thank Dr. Cairo, Dr. Gross, and Dr. Lowe for their excellent answers to my, at times, difficult and challenging questions and I would like to thank the organizers of this expert roundtable discussion. I hope that in the next decade that we will see even greater advances for the patient population that we treat. Thank you very much.
References
1. Rosolen RA, Perkins SL, Pinkerton CR, et al. Revised International Pediatric Non-Hodgkin Lymphoma Staging System. J Clin Oncol. 2015;33(18):2112–2118.
2. Sandlund JT, Guillerman RP, Perkins SL, et al. International Pediatric Non-Hodgkin Lymphoma Response Criteria. J Clin Oncol. 2015;33(18)2106-2111.
Moderator: Catherine Bollard, MD, FRACP, FRCPA1
Discussants: Mitchell S. Cairo, MD2; Eric J. Lowe, MD3; Thomas G. Gross, MD, PhD4
Address for correspondence: Catherine Bollard, MD, FRACP, FRCPA, 111 Michigan Avenue, NW, 5th Floor Main, Suite 5225, Washington, DC 20010
E-mail: [email protected]
Biographical sketch: From The George Washington University, School of Medicine and Health Sciences, Washington, DC1; Westchester Medical Center, New York Medical College, Valhalla, NY2; Children’s Hospital of the King’s Daughters, Norfolk, VA3; Center for Global Health at the National Cancer Institute, Rockville, MD
DR. BOLLARD: My name is Dr. Catherine Bollard. I'm Chief of the Division of Allergy and Immunology at Children's National Health System and the Chair of the NHL Committee of the Children's Oncology Group. I hope that today we can provide some clarity and give you some of our first-hand expertise and experience regarding some of the challenges and controversies of treating pediatric patients with non-Hodgkin lymphoma (NHL). Here with me are Drs. Mitchell Cairo, Chief of Pediatric Hematology/Oncology and Stem Cell Transplantation at New York Medical College in the Maria Fareri Children's Hospital, Westchester Medical Center; Eric Lowe, Division Director for Pediatric Hematology/Oncology at the Children's Hospital of the King's Daughters; and Thomas Gross, Deputy Director for Science at the Center for Global Health at the National Cancer Institute.
I'd like to start the questioning, firstly to Dr. Cairo, who recently published with a group of leaders in the pediatric lymphoma field, new staging and response classifications. Dr. Cairo, I’d like you to highlight how these are different from the current classifications, and what you see are the strengths and the limitations at this time.
DR. CAIRO: Thank you, Cath. The original staging classification was developed in the late 1970s by Dr. Murphy while she was at St. Jude's hospital, and either goes by the name the Murphy Staging Classification or the St. Jude's Classification. That classification I think was quite useful at that time when we recognized really only a couple subtypes of NHL, as well as the capabilities we had in those days both imaging as well as further molecular identification as well as trying to identify sites of spread. As some 35 years have evolved, new pathological entities have been identified, much more precise imaging techniques, new methods of detecting more evidence of minimal disease, and also identifying new organ sites of involvement, allowed the creation of a multidisciplinary international task force to look at how we could enhance the original observations by the St. Jude's group.
As Dr. Bollard pointed out, we eventually, over 9 years of evidence-based review, came up with an enhanced staging classification called the International Pediatric NHL Staging System (IPNHLSS).1 In this new system we account for new histological subtypes, allow for different organ distributions, improve on the new imaging techniques to identify areas of involvement, and also to more molecularly identify extent of disease. I think the advantages are stated above. The disadvantage is that like all staging systems it's a breathing document. It will require international collaboration. As time evolves, this staging system will of course need to be updated as we gain new experience.
Briefly, in terms of the response classification that also came out of the same international multidisciplinary task force that was led by Dr. Sandlund at St. Jude's2; there had never been a response criteria that had been focused entirely in childhood and adolescent NHL. The previous response criteria had been developed by adult NHL investigators, and there was a need to develop the first response criteria for pediatric NHL because of different histologies, different sites of sanctuary disease, and now obviously enhanced imaging capabilities. That also now has been named the International Pediatric Non-Hodgkin Lymphoma Response Criteria (IPNHLRC)—hopefully for harmonizing a response across new studies, but also a breathing document that is going to be limited as we gain new knowledge into how we can better assess response as new techniques are developed.
DR. BOLLARD: I thank you very much for your detailed response. My next question is actually to Dr. Gross, who is currently chairing the international study for upfront diffuse large B-cell lymphoma and Burkitt lymphoma in pediatric young adults. I would like you to speak to a couple of issues, and you can put it in the context of the current randomized trial, looking at rituximab vs no rituximab for this disease. I think firstly it would be useful for you to speak to the implications of this new classification system as we go forward with choosing new therapeutic strategies for these patients, and in particular I'd like to focus on the newly diagnosed diffuse large B-cell lymphoma patients who are in that adolescent/young adult range.
I would also be interested in your opinion regarding how you would manage a patient who is 17 years old but is going to turn 18 tomorrow, and he comes to you with newly diagnosed diffuse large B-cell lymphoma. As you know, the adult oncologists treat diffuse large B-cell lymphoma different to Burkitt lymphoma, and in pediatrics we generally treat these diseases the same. Do you tell this patient that you will treat him today on a pediatric regimen, or do you tell him to go tomorrow, when he's 18, to be treated by an adult oncologist? I would like you to justify your answer please.
DR. GROSS: First to discuss the implications of the new staging as it applies to the current international trial. As Dr. Cairo pointed out, this was developed through a literature review and evidence based analyses, but like any new staging system, the value of staging is to provide us with information that can try to help us to identify patients to improve their outcome. Essentially, staging is to help direct therapy or provide prognosis for outcome, and the only way to do that is to test new systems or classifications in a prospective fashion. Indeed, that is what we are trying to do with this international effort.
This international effort, just as an aside, illustrates one of the challenges of all rare cancers, but particularly pediatrics. In pediatric mature B-cell NHL, both large-cell and Burkitt, we are now at a cure rate of about 90%. To make advances, we don't have enough patients seen in North America and Australia, and it requires international collaboration. This trial, to get 600 patients randomized, it will take 7 years with 14 countries participating—that is one of the challenges, certainly, we have with pediatric NHL. Also, we want to try to gain as much information as possible, not just to the effect of rituximab as Dr. Bollard said, but also to test other questions such as the role or the value in validating this new staging system.
To talk about the controversy of treatment, certainly we know that there is a very different approach in pediatrics. For many years, we have treated diffuse large B-cell lymphoma just like Burkitt. This is a very important delineation when you're seen by a medical oncologist because the treatment for diffuse large B-cell lymphoma is outpatient therapy, ie rituximab, cyclophosphamide, adriamycin, vincristine, and prednisone (R-CHOP). Treatment for Burkitt is inpatient with high doses of methotrexate, but other higher doses of the same agents used to treat diffuse large B-cell lymphoma. The question is, do we really need to treat all the pediatric diffuse large B-cell lymphoma with these aggressive Burkitt regimens? I think one of the things that is encouraging to me as a pediatric oncologist is that we are beginning to learn that the biology is very different. Though the disease looks the same under the microscope or by flow cytometry, when you look at it genetically it's quite different. We know now that the younger the patient is with diffuse large B-cell lymphoma, even though it looks for all intents and purposes like the same disease as seen in adults, when you look at the genetics, many times, as high as 30% of the time, it will be genetically the same as a Burkitt lymphoma. I think when you're talking about young patients we can easily justify treating them both the same because the biology would suggest that a good number of patients would need Burkitt therapy to be cured.
Now, that changes over time, so that it appears that sometime in young adulthood, maybe somewhere between 25 and 35 years of age, you don't see the genetic disease that looks like diffuse large B-cell lymphoma, but is genetically Burkitt lymphoma. As for the 18-year-old patient that Dr. Bollard was posing to me, I've had several patients like this. I go through the pluses and minuses of the therapy, inpatient vs outpatient, but also the potential long-term side effects. The outpatient therapy has potentially more long-term side effects as far as potential infertility and potential heart damage. Every time I have given the choice to the family and the patient, the teenager has always chosen the outpatient therapy that you would get as an adult, and the parents always say they would rather have the inpatient therapy, and that spending a couple of days in the hospital to try to reduce the chance of long-term damage is their choice. It's a very interesting dynamic and I think sometimes the issues that go into choice of treatment are quite variable. My personal opinion is that hopefully in the future we will be able to have a better understanding of biology, so that when we see these patients, be they 18 or 25 years old, we're not looking at what it looks like under the microscope or who they see, and what they're used to giving, but the biology will determine which therapy is more likely to cure them. Right now we don't have that ability in most of the patients.
DR. BOLLARD: Thank you, Dr. Gross. Again, another very comprehensive answer to a difficult question. I'm actually going to push this back to Dr. Cairo and then impose the same soon to be 18-year-old patient to you. This time, he's coming to you with relapsed diffuse large B-cell lymphoma. What are you going to tell him? Are you going to treat him today on pediatric protocols, or will you wait until tomorrow when he could have access to adult protocols?
DR. CAIRO: I think the results are relatively similar, but, in part, the answer to the question is of course based on what their original therapy was. If the original therapy was the pediatric-inspired type of treatment, I think there's a world of experience of what are some of the best pediatric-inspired regimens to use for retrieval. If, however, the original therapy was an adult-inspired regimen, then I think the options are open because the disease may not be as resistant in that setting; therefore, one would want to consider all the adult type of retrieval regimens in that case, because that group of patients—at least in the adult experience—tend to have disease that may be more responsive because they're not as resistant to the higher dose and multi-agent therapy that a pediatric-inspired regimen would have given them had they been treated that way.
DR. BOLLARD: I was also trying to ask you to speak to the access that an 18-year-old might have to novel therapies that a 17-year-old might not. How do you address that issue?
DR. CAIRO: That's an excellent question. I think that for first relapse or first induction failure most of the retrieval regimens, the first line regimens, that are available, either pediatric inspired or adult inspired, probably don't require an investigational agent that an 18-year-old might have access to if he was being treated on an adult type of regimen. However, I would strongly encourage an 18-year-old who failed one retrieval regimen to consider experimental therapy. There I think the access to new agents—if you're 18 or over—are so much greater that I would encourage them to be treated on an adult retrieval regimen, where some of the newer agents may be investigational, are not available to a pediatric program.
DR. BOLLARD: Thank you very much, Dr. Cairo. I have one last question on the B-cell diseases before I move to Dr. Lowe, and the last question goes to Dr. Gross. Would you recommend that a patient with relapsed Burkitt lymphoma—now increasingly rare—be treated with salvage chemotherapy and then autologous transplant or allogeneic stem cell transplant?
DR. GROSS: As the others on this discussion know, we performed an analysis from data in the Center for International Blood and Marrow Transplant Research (CIBMTR), and the problem is that Burkitt lymphoma tends to reoccur so rapidly after transplant. The median time to relapse is 3 months after transplant. We could not find a difference in the outcome between autografts and allografts because of its early reoccurrence. That said, my personal opinion is, since we know that Burkitt lymphoma is a hematologically spread disease, that I always prefer a donor source where I know they're not going to have tumor cells in them, which is an allogeneic donor. I always prefer an allogeneic donor, because I know it's tumor-free, but also it gives us an opportunity, if the disease will stay under control long enough, to potentially get an immune response against any residual tumor. For that reason, I recommend an allogeneic donor if it can be found readily.
DR. BOLLARD: Thank you very much, Dr. Gross. Now, on to Dr. Lowe, and Dr. Lowe's particular area of expertise is in anaplastic large cell lymphoma (ALCL) and T-cell diseases. I was wondering if you could explain to me the difference between T-cell lymphoblastic lymphoma and T-cell acute lymphoblastic leukemia (ALL), specifically since the World Health Organization (WHO) groups these two disease entities together as T-lymphoblastic leukemia/lymphoma. If you could clarify that classification that would be very helpful.
DR. LOWE: As you well know, many physicians believe that T-cell lymphoblastic leukemia and T-cell lymphoblastic lymphoma are very similar diseases, but they are not exactly the same disease although we sometimes treat them very similar. We know that T-cells do not mature in bone marrow but rather they are thymic driven cells. Because of this, there are distinct differences between the leukemia and lymphoma. For example, we know that the genetics between the two—although in limited samples—are not always the same, including in prognostic. Just one example, loss of heterozygosity at 6Q has been shown to be prognostically important in lymphoblastic lymphoma but not in T-cell ALL. I think the real challenge is to figure out what the differences are. I think we could argue that potentially, T-ALL is stage four T-cell lymphoblastic lymphoma.
I think that the WHO classifying the two diseases as one entity with T-lymphoblastic leukemia/lymphoma has hindered a little in the advancement of recognizing the differences in that many people assume that they're the same disease. When you look up from a pathological standpoint and you say, well, they're clearly the same disease because they're listed as a single entity, and when you look up treatment, you say, well they're treated very similar, so they must be the same disease. I think that does us a little disservice in trying to advance the field forward, because I think getting lymphoblastic lymphoma samples, which is challenging, is extremely important to determine the genetic drivers of this disease.
DR. BOLLARD: Thank you very much, Dr. Lowe. I would also like you to discuss how ALCL differs between the pediatric and the adult populations, and how that dictates how you would treat those two patient populations.
DR. LOWE: So, ALCL really has a much shorter span in terms of its description pathologically. It was not described by itself until the mid 1980s. In the mid 1990s it started entering classification schemes. It wasn't until 2008 that the WHO separated out three distinct entities within ALCL. You have anaplastic lymphoma kinase (ALK)-positive ALCL, ALK-negative ALCL, and primary cutaneous ALCL. This is a great example where these three different entities have very different epidemiology, very different treatment strategies, and the fact that they are broken up has really helped move the field forward. For example, ALK-positive ALCL is really a disease of children, adolescents, and young adults. It's the most common ALCL by far in that age group. It's extremely rare to have an ALK-negative ALCL, and the pathological reason for the disease with ALK-positive ALCL is a translocation involving the ALK gene leads directly to oncogenesis.
Because of this, we have started to develop treatments that are designed to target this specific oncogenic driving translocation. This is in direct comparison to an ALK-negative ALCL, which is primarily a disease of older individuals, most commonly in their 50s and 60s. The outcome for this disease is consistently poorer than for ALK-positive disease. The treatment, while sometimes the same, is changing now that we have targets for the tyrosine kinase that is driving the ALK-positive ALCL. I think separating these two out has been a huge advantage in terms of figuring out what to do with pediatric ALCL because the 95 plus percent of ALCLs in pediatrics and young adults are ALK-positive.
Primary cutaneous ALCL is almost a completely different entity in and of itself, although it shares the same name. The primary cutaneous ALCLs are usually not treated on similar studies as the systemic forms of ALCL. The primary cutaneous form has different characteristics in terms of location, age, treatment, and natural course. The vast majority does not develop into systemic disease, and thus the treatment is very different. I think ALCL is a very good example where the different pathological entities have led to very different treatments based on what is driving the cancer.
DR. BOLLARD: Thanks, Dr. Lowe. I think that's very important to emphasize how ALK-positivity is more common in children than in adults, and the successes of crizotinib, even in phase 1 in pediatric patients with ALCL. My question now is given the success of this targeted agent in the relapse setting, even in phase 1, do you still see a role for allogeneic stem cell transplant for those patients who have relapsed after conventional therapy?
DR. LOWE: I do still see a role, but I'm not sure how much that role will shrink over time as we learn more and more about this disease. We know that there are very high risk patients that relapse or progress while receiving traditional chemotherapy. Those patients typically have achieved the best outcome with an allogeneic transplant. That said, I think crizotinib and other ALK inhibitors are changing the landscape of treatment for ALK-positive ALCL very fast. We know that some patients who are refractory to many other treatments go into remission with these drugs, and while I think that the role of allogeneic transplant is still there, I think that it may be changing over time. The other decision that I think will be difficult in terms of allogeneic transplant is for patients who receive ALK-inhibitors, like crizotinib, for initial treatment and then relapse. Many patients in that situation currently will end up having an allogenic transplant. However, one can argue that very much like chronic myeloid leukemia, these patients might be rescued without an allogeneic transplant using a second line ALK inhibitor. All of these things, obviously, we hope to know over time, but at this point in time are unknown.
DR. BOLLARD: Thank you very much. I'll take you out of the hot seat now. All of us talked about the concept of the importance of knowing the biology of what we're treating, and with the advent of novel targeted therapies, this concept of precision medicine is becoming increasingly important, ie, targeting the individual patient's tumor with the appropriate targeted agents for their tumor. This is maybe a question for Dr. Gross first, and then Dr. Cairo. What do you see are the challenges for being able to obtain the tissue from pediatric patients to perform these important and critical tests that will be needed as we move the field forward for the management of pediatric patients with NHL?
DR. GROSS: I think that the number one barrier is, as the technology improves to be able to make the essential diagnosis, we need less and less tissue for the pathologist. It becomes increasingly more of a challenge to obtain extra tissue because the standard of practice is to get just enough to make the diagnosis. Unless we can address this challenge, it's going to be extremely difficult.
DR. BOLLARD: Dr. Cairo, do you want to speak to that, since you recently completed a Children’s Oncology Group trial for Burkitt and diffuse large B-cell lymphoma?
DR. CAIRO: Thank you. I agree with Dr. Gross, and that particular trial, despite it being one of the primary objectives and also many of those patients actually had bone marrow involvement, which is the area that we access the easiest as the acute lymphoblastic leukemia colleagues have taught us. We still only were able to get 11 of some 90 patients entered on study, to have specimens sent. That being said, having just come back from the Fifth International Symposium on Childhood, Adolescent and Young Adult Non-Hodgkin Lymphoma, It appears that the Europeans have been much more successful in obtaining specimens for biology studies in particular, the future precision medicine-based trials. We should try to learn a little bit from our European colleagues, who seem to have a much higher percentage of getting specimens, and we need to make every effort, as Dr. Gross said of encouraging our colleagues, that this is as important as making the diagnosis. We face an uphill battle because of our high cure rate, the biology is often considered a second thought sometimes. Europeans are better than us at obtaining biological specimens and we need to compete to achieve the level that they have achieved in Europe.
DR. GROSS: It's almost a catch-22. We know from other diseases in pediatric oncology, but also in adult oncology, that once we are able to demonstrate that the biology will make a difference in the treatment and outcome of the patient, then we're able to get the tissue needed. I think a good example of that is neuroblastoma. However, we can't make those discoveries unless we get enough tissue to study. We're in this catch-22, we cannot demonstrate that the biology makes a difference, unless we will get the tissue for research.
DR. LOWE: I'd like to add one other point to this. I think the rarity of the diseases and the large number of centers that treat the patients also hinders obtaining pathological samples. Because pediatric NHL is a relatively rare disease, you can’t have a single champion for obtaining biology at one institution that can accomplish anything without many other institutions. It requires a large group effort which is more difficult than a single institution collecting colon cancer samples, for example, where you really only need one institution, one champion, one pathologist, and you have all the samples you need.
DR. BOLLARD: Thank you, Dr. Lowe. I really thank you all for speaking in a very detailed way about the importance of obtaining tumor tissue to perform these critical biologic studies, because I do feel that's an important issue to overcome for the future care of our pediatric patients with NHL. I would like to discuss late effects in our survivors. As Dr. Gross said, survival rates for patients with B-cell lymphomas are generally outstanding. Dr. Gross, do you feel that late effects are not something the NHL group has to worry about now that we have obviated the need for radiation, or not? And what are your feelings about trying to minimize these late effects even further?
DR. GROSS: The good news is that over time, we have been able to come up with regimens that are highly effective but have reduced the agents we know have the highest risk of late effects—radiation being the primary one, but also anthracyclines we have been able to reduce in the vast majority of the patients, and to keep alkylating agents in the vast majority of the patients to a level that most patients do not have infertility. The long-term side effects are becoming pretty minimal, but the question is, how low do they have to be to be acceptable? The goal would be cure without any long-term effects. As I said before, certainly we have paid the price in short-term effects. Our regimens are inpatient, and they can have quite severe short-term side effects such as mucositis. We've made great advances but I think there's still room to go.
DR. CAIRO: I agree, of course, with my colleague Dr. Gross. Again, when we look at large series of chronic health care conditions, certainly children with treated NHL still comes up as one showing over 40% to 50% of patients having one or two serious chronic health care conditions. We know the data are a little antiquated, because they include patients who were treated with different regimens in the 1970s and all of the 1980s. However, I think our goal continues to be to identify the most effective treatment regimen, but with the least toxic long-term complications for our patients. That struggle is very difficult because of the very high success rate we have today, and to identify without hurting that high success rate less toxic therapies will require a collaborative, multidisciplinary, international effort to reach that goal.
DR. BOLLARD: Thank you very much Drs. Cairo and Gross. Dr. Lowe, did you have any closing remarks on the late effects issues for the T-cell mediated diseases in particular?
DR. LOWE: I would absolutely agree with Dr. Gross and Dr. Cairo that this is an important issue. I think we in pediatrics do a good job at following our patients for long-term side effects and creating guidelines for screening for these long-term side effects. That said, I think as we start to talk about better and better therapy and even more and more targeted therapy, what we don't know about some of these targeted therapies is their 15 and 20 year long-term side effects. We obviously hope that there aren't any, and that's why we are moving toward these drugs, but again, surveillance of those long-term side effects will be extremely important, especially when you're talking about medications for young children.
DR. BOLLARD: I'd like to thank you all very much for participating in this expert roundtable discussion today. I think the overarching points are that prognoses at the current time for newly diagnosed pediatric patients with NHL range from 70% to over 90% even for patients with disseminated disease. The challenges that we need to overcome are how we can optimize our up front treatment to prevent relapse in all, because I think we've all reiterated the fact that the outcomes for those few patients who do relapse remains extremely poor. I think there is still controversy about how to manage patients with relapsed disease, and how to temper our therapies against long-term side effects of our surviving patients. Finally, I think with the advent of novel targeted agents, it is incredibly important for the optimal management of our current and future patients that we are able to access tumor tissues and perform the critical biologic studies that are required to develop an effective precision medicine approach for pediatric patients with NHL. I would like to again thank Dr. Cairo, Dr. Gross, and Dr. Lowe for their excellent answers to my, at times, difficult and challenging questions and I would like to thank the organizers of this expert roundtable discussion. I hope that in the next decade that we will see even greater advances for the patient population that we treat. Thank you very much.
References
1. Rosolen RA, Perkins SL, Pinkerton CR, et al. Revised International Pediatric Non-Hodgkin Lymphoma Staging System. J Clin Oncol. 2015;33(18):2112–2118.
2. Sandlund JT, Guillerman RP, Perkins SL, et al. International Pediatric Non-Hodgkin Lymphoma Response Criteria. J Clin Oncol. 2015;33(18)2106-2111.
Moderator: Catherine Bollard, MD, FRACP, FRCPA1
Discussants: Mitchell S. Cairo, MD2; Eric J. Lowe, MD3; Thomas G. Gross, MD, PhD4
Address for correspondence: Catherine Bollard, MD, FRACP, FRCPA, 111 Michigan Avenue, NW, 5th Floor Main, Suite 5225, Washington, DC 20010
E-mail: [email protected]
Biographical sketch: From The George Washington University, School of Medicine and Health Sciences, Washington, DC1; Westchester Medical Center, New York Medical College, Valhalla, NY2; Children’s Hospital of the King’s Daughters, Norfolk, VA3; Center for Global Health at the National Cancer Institute, Rockville, MD
DR. BOLLARD: My name is Dr. Catherine Bollard. I'm Chief of the Division of Allergy and Immunology at Children's National Health System and the Chair of the NHL Committee of the Children's Oncology Group. I hope that today we can provide some clarity and give you some of our first-hand expertise and experience regarding some of the challenges and controversies of treating pediatric patients with non-Hodgkin lymphoma (NHL). Here with me are Drs. Mitchell Cairo, Chief of Pediatric Hematology/Oncology and Stem Cell Transplantation at New York Medical College in the Maria Fareri Children's Hospital, Westchester Medical Center; Eric Lowe, Division Director for Pediatric Hematology/Oncology at the Children's Hospital of the King's Daughters; and Thomas Gross, Deputy Director for Science at the Center for Global Health at the National Cancer Institute.
I'd like to start the questioning, firstly to Dr. Cairo, who recently published with a group of leaders in the pediatric lymphoma field, new staging and response classifications. Dr. Cairo, I’d like you to highlight how these are different from the current classifications, and what you see are the strengths and the limitations at this time.
DR. CAIRO: Thank you, Cath. The original staging classification was developed in the late 1970s by Dr. Murphy while she was at St. Jude's hospital, and either goes by the name the Murphy Staging Classification or the St. Jude's Classification. That classification I think was quite useful at that time when we recognized really only a couple subtypes of NHL, as well as the capabilities we had in those days both imaging as well as further molecular identification as well as trying to identify sites of spread. As some 35 years have evolved, new pathological entities have been identified, much more precise imaging techniques, new methods of detecting more evidence of minimal disease, and also identifying new organ sites of involvement, allowed the creation of a multidisciplinary international task force to look at how we could enhance the original observations by the St. Jude's group.
As Dr. Bollard pointed out, we eventually, over 9 years of evidence-based review, came up with an enhanced staging classification called the International Pediatric NHL Staging System (IPNHLSS).1 In this new system we account for new histological subtypes, allow for different organ distributions, improve on the new imaging techniques to identify areas of involvement, and also to more molecularly identify extent of disease. I think the advantages are stated above. The disadvantage is that like all staging systems it's a breathing document. It will require international collaboration. As time evolves, this staging system will of course need to be updated as we gain new experience.
Briefly, in terms of the response classification that also came out of the same international multidisciplinary task force that was led by Dr. Sandlund at St. Jude's2; there had never been a response criteria that had been focused entirely in childhood and adolescent NHL. The previous response criteria had been developed by adult NHL investigators, and there was a need to develop the first response criteria for pediatric NHL because of different histologies, different sites of sanctuary disease, and now obviously enhanced imaging capabilities. That also now has been named the International Pediatric Non-Hodgkin Lymphoma Response Criteria (IPNHLRC)—hopefully for harmonizing a response across new studies, but also a breathing document that is going to be limited as we gain new knowledge into how we can better assess response as new techniques are developed.
DR. BOLLARD: I thank you very much for your detailed response. My next question is actually to Dr. Gross, who is currently chairing the international study for upfront diffuse large B-cell lymphoma and Burkitt lymphoma in pediatric young adults. I would like you to speak to a couple of issues, and you can put it in the context of the current randomized trial, looking at rituximab vs no rituximab for this disease. I think firstly it would be useful for you to speak to the implications of this new classification system as we go forward with choosing new therapeutic strategies for these patients, and in particular I'd like to focus on the newly diagnosed diffuse large B-cell lymphoma patients who are in that adolescent/young adult range.
I would also be interested in your opinion regarding how you would manage a patient who is 17 years old but is going to turn 18 tomorrow, and he comes to you with newly diagnosed diffuse large B-cell lymphoma. As you know, the adult oncologists treat diffuse large B-cell lymphoma different to Burkitt lymphoma, and in pediatrics we generally treat these diseases the same. Do you tell this patient that you will treat him today on a pediatric regimen, or do you tell him to go tomorrow, when he's 18, to be treated by an adult oncologist? I would like you to justify your answer please.
DR. GROSS: First to discuss the implications of the new staging as it applies to the current international trial. As Dr. Cairo pointed out, this was developed through a literature review and evidence based analyses, but like any new staging system, the value of staging is to provide us with information that can try to help us to identify patients to improve their outcome. Essentially, staging is to help direct therapy or provide prognosis for outcome, and the only way to do that is to test new systems or classifications in a prospective fashion. Indeed, that is what we are trying to do with this international effort.
This international effort, just as an aside, illustrates one of the challenges of all rare cancers, but particularly pediatrics. In pediatric mature B-cell NHL, both large-cell and Burkitt, we are now at a cure rate of about 90%. To make advances, we don't have enough patients seen in North America and Australia, and it requires international collaboration. This trial, to get 600 patients randomized, it will take 7 years with 14 countries participating—that is one of the challenges, certainly, we have with pediatric NHL. Also, we want to try to gain as much information as possible, not just to the effect of rituximab as Dr. Bollard said, but also to test other questions such as the role or the value in validating this new staging system.
To talk about the controversy of treatment, certainly we know that there is a very different approach in pediatrics. For many years, we have treated diffuse large B-cell lymphoma just like Burkitt. This is a very important delineation when you're seen by a medical oncologist because the treatment for diffuse large B-cell lymphoma is outpatient therapy, ie rituximab, cyclophosphamide, adriamycin, vincristine, and prednisone (R-CHOP). Treatment for Burkitt is inpatient with high doses of methotrexate, but other higher doses of the same agents used to treat diffuse large B-cell lymphoma. The question is, do we really need to treat all the pediatric diffuse large B-cell lymphoma with these aggressive Burkitt regimens? I think one of the things that is encouraging to me as a pediatric oncologist is that we are beginning to learn that the biology is very different. Though the disease looks the same under the microscope or by flow cytometry, when you look at it genetically it's quite different. We know now that the younger the patient is with diffuse large B-cell lymphoma, even though it looks for all intents and purposes like the same disease as seen in adults, when you look at the genetics, many times, as high as 30% of the time, it will be genetically the same as a Burkitt lymphoma. I think when you're talking about young patients we can easily justify treating them both the same because the biology would suggest that a good number of patients would need Burkitt therapy to be cured.
Now, that changes over time, so that it appears that sometime in young adulthood, maybe somewhere between 25 and 35 years of age, you don't see the genetic disease that looks like diffuse large B-cell lymphoma, but is genetically Burkitt lymphoma. As for the 18-year-old patient that Dr. Bollard was posing to me, I've had several patients like this. I go through the pluses and minuses of the therapy, inpatient vs outpatient, but also the potential long-term side effects. The outpatient therapy has potentially more long-term side effects as far as potential infertility and potential heart damage. Every time I have given the choice to the family and the patient, the teenager has always chosen the outpatient therapy that you would get as an adult, and the parents always say they would rather have the inpatient therapy, and that spending a couple of days in the hospital to try to reduce the chance of long-term damage is their choice. It's a very interesting dynamic and I think sometimes the issues that go into choice of treatment are quite variable. My personal opinion is that hopefully in the future we will be able to have a better understanding of biology, so that when we see these patients, be they 18 or 25 years old, we're not looking at what it looks like under the microscope or who they see, and what they're used to giving, but the biology will determine which therapy is more likely to cure them. Right now we don't have that ability in most of the patients.
DR. BOLLARD: Thank you, Dr. Gross. Again, another very comprehensive answer to a difficult question. I'm actually going to push this back to Dr. Cairo and then impose the same soon to be 18-year-old patient to you. This time, he's coming to you with relapsed diffuse large B-cell lymphoma. What are you going to tell him? Are you going to treat him today on pediatric protocols, or will you wait until tomorrow when he could have access to adult protocols?
DR. CAIRO: I think the results are relatively similar, but, in part, the answer to the question is of course based on what their original therapy was. If the original therapy was the pediatric-inspired type of treatment, I think there's a world of experience of what are some of the best pediatric-inspired regimens to use for retrieval. If, however, the original therapy was an adult-inspired regimen, then I think the options are open because the disease may not be as resistant in that setting; therefore, one would want to consider all the adult type of retrieval regimens in that case, because that group of patients—at least in the adult experience—tend to have disease that may be more responsive because they're not as resistant to the higher dose and multi-agent therapy that a pediatric-inspired regimen would have given them had they been treated that way.
DR. BOLLARD: I was also trying to ask you to speak to the access that an 18-year-old might have to novel therapies that a 17-year-old might not. How do you address that issue?
DR. CAIRO: That's an excellent question. I think that for first relapse or first induction failure most of the retrieval regimens, the first line regimens, that are available, either pediatric inspired or adult inspired, probably don't require an investigational agent that an 18-year-old might have access to if he was being treated on an adult type of regimen. However, I would strongly encourage an 18-year-old who failed one retrieval regimen to consider experimental therapy. There I think the access to new agents—if you're 18 or over—are so much greater that I would encourage them to be treated on an adult retrieval regimen, where some of the newer agents may be investigational, are not available to a pediatric program.
DR. BOLLARD: Thank you very much, Dr. Cairo. I have one last question on the B-cell diseases before I move to Dr. Lowe, and the last question goes to Dr. Gross. Would you recommend that a patient with relapsed Burkitt lymphoma—now increasingly rare—be treated with salvage chemotherapy and then autologous transplant or allogeneic stem cell transplant?
DR. GROSS: As the others on this discussion know, we performed an analysis from data in the Center for International Blood and Marrow Transplant Research (CIBMTR), and the problem is that Burkitt lymphoma tends to reoccur so rapidly after transplant. The median time to relapse is 3 months after transplant. We could not find a difference in the outcome between autografts and allografts because of its early reoccurrence. That said, my personal opinion is, since we know that Burkitt lymphoma is a hematologically spread disease, that I always prefer a donor source where I know they're not going to have tumor cells in them, which is an allogeneic donor. I always prefer an allogeneic donor, because I know it's tumor-free, but also it gives us an opportunity, if the disease will stay under control long enough, to potentially get an immune response against any residual tumor. For that reason, I recommend an allogeneic donor if it can be found readily.
DR. BOLLARD: Thank you very much, Dr. Gross. Now, on to Dr. Lowe, and Dr. Lowe's particular area of expertise is in anaplastic large cell lymphoma (ALCL) and T-cell diseases. I was wondering if you could explain to me the difference between T-cell lymphoblastic lymphoma and T-cell acute lymphoblastic leukemia (ALL), specifically since the World Health Organization (WHO) groups these two disease entities together as T-lymphoblastic leukemia/lymphoma. If you could clarify that classification that would be very helpful.
DR. LOWE: As you well know, many physicians believe that T-cell lymphoblastic leukemia and T-cell lymphoblastic lymphoma are very similar diseases, but they are not exactly the same disease although we sometimes treat them very similar. We know that T-cells do not mature in bone marrow but rather they are thymic driven cells. Because of this, there are distinct differences between the leukemia and lymphoma. For example, we know that the genetics between the two—although in limited samples—are not always the same, including in prognostic. Just one example, loss of heterozygosity at 6Q has been shown to be prognostically important in lymphoblastic lymphoma but not in T-cell ALL. I think the real challenge is to figure out what the differences are. I think we could argue that potentially, T-ALL is stage four T-cell lymphoblastic lymphoma.
I think that the WHO classifying the two diseases as one entity with T-lymphoblastic leukemia/lymphoma has hindered a little in the advancement of recognizing the differences in that many people assume that they're the same disease. When you look up from a pathological standpoint and you say, well, they're clearly the same disease because they're listed as a single entity, and when you look up treatment, you say, well they're treated very similar, so they must be the same disease. I think that does us a little disservice in trying to advance the field forward, because I think getting lymphoblastic lymphoma samples, which is challenging, is extremely important to determine the genetic drivers of this disease.
DR. BOLLARD: Thank you very much, Dr. Lowe. I would also like you to discuss how ALCL differs between the pediatric and the adult populations, and how that dictates how you would treat those two patient populations.
DR. LOWE: So, ALCL really has a much shorter span in terms of its description pathologically. It was not described by itself until the mid 1980s. In the mid 1990s it started entering classification schemes. It wasn't until 2008 that the WHO separated out three distinct entities within ALCL. You have anaplastic lymphoma kinase (ALK)-positive ALCL, ALK-negative ALCL, and primary cutaneous ALCL. This is a great example where these three different entities have very different epidemiology, very different treatment strategies, and the fact that they are broken up has really helped move the field forward. For example, ALK-positive ALCL is really a disease of children, adolescents, and young adults. It's the most common ALCL by far in that age group. It's extremely rare to have an ALK-negative ALCL, and the pathological reason for the disease with ALK-positive ALCL is a translocation involving the ALK gene leads directly to oncogenesis.
Because of this, we have started to develop treatments that are designed to target this specific oncogenic driving translocation. This is in direct comparison to an ALK-negative ALCL, which is primarily a disease of older individuals, most commonly in their 50s and 60s. The outcome for this disease is consistently poorer than for ALK-positive disease. The treatment, while sometimes the same, is changing now that we have targets for the tyrosine kinase that is driving the ALK-positive ALCL. I think separating these two out has been a huge advantage in terms of figuring out what to do with pediatric ALCL because the 95 plus percent of ALCLs in pediatrics and young adults are ALK-positive.
Primary cutaneous ALCL is almost a completely different entity in and of itself, although it shares the same name. The primary cutaneous ALCLs are usually not treated on similar studies as the systemic forms of ALCL. The primary cutaneous form has different characteristics in terms of location, age, treatment, and natural course. The vast majority does not develop into systemic disease, and thus the treatment is very different. I think ALCL is a very good example where the different pathological entities have led to very different treatments based on what is driving the cancer.
DR. BOLLARD: Thanks, Dr. Lowe. I think that's very important to emphasize how ALK-positivity is more common in children than in adults, and the successes of crizotinib, even in phase 1 in pediatric patients with ALCL. My question now is given the success of this targeted agent in the relapse setting, even in phase 1, do you still see a role for allogeneic stem cell transplant for those patients who have relapsed after conventional therapy?
DR. LOWE: I do still see a role, but I'm not sure how much that role will shrink over time as we learn more and more about this disease. We know that there are very high risk patients that relapse or progress while receiving traditional chemotherapy. Those patients typically have achieved the best outcome with an allogeneic transplant. That said, I think crizotinib and other ALK inhibitors are changing the landscape of treatment for ALK-positive ALCL very fast. We know that some patients who are refractory to many other treatments go into remission with these drugs, and while I think that the role of allogeneic transplant is still there, I think that it may be changing over time. The other decision that I think will be difficult in terms of allogeneic transplant is for patients who receive ALK-inhibitors, like crizotinib, for initial treatment and then relapse. Many patients in that situation currently will end up having an allogenic transplant. However, one can argue that very much like chronic myeloid leukemia, these patients might be rescued without an allogeneic transplant using a second line ALK inhibitor. All of these things, obviously, we hope to know over time, but at this point in time are unknown.
DR. BOLLARD: Thank you very much. I'll take you out of the hot seat now. All of us talked about the concept of the importance of knowing the biology of what we're treating, and with the advent of novel targeted therapies, this concept of precision medicine is becoming increasingly important, ie, targeting the individual patient's tumor with the appropriate targeted agents for their tumor. This is maybe a question for Dr. Gross first, and then Dr. Cairo. What do you see are the challenges for being able to obtain the tissue from pediatric patients to perform these important and critical tests that will be needed as we move the field forward for the management of pediatric patients with NHL?
DR. GROSS: I think that the number one barrier is, as the technology improves to be able to make the essential diagnosis, we need less and less tissue for the pathologist. It becomes increasingly more of a challenge to obtain extra tissue because the standard of practice is to get just enough to make the diagnosis. Unless we can address this challenge, it's going to be extremely difficult.
DR. BOLLARD: Dr. Cairo, do you want to speak to that, since you recently completed a Children’s Oncology Group trial for Burkitt and diffuse large B-cell lymphoma?
DR. CAIRO: Thank you. I agree with Dr. Gross, and that particular trial, despite it being one of the primary objectives and also many of those patients actually had bone marrow involvement, which is the area that we access the easiest as the acute lymphoblastic leukemia colleagues have taught us. We still only were able to get 11 of some 90 patients entered on study, to have specimens sent. That being said, having just come back from the Fifth International Symposium on Childhood, Adolescent and Young Adult Non-Hodgkin Lymphoma, It appears that the Europeans have been much more successful in obtaining specimens for biology studies in particular, the future precision medicine-based trials. We should try to learn a little bit from our European colleagues, who seem to have a much higher percentage of getting specimens, and we need to make every effort, as Dr. Gross said of encouraging our colleagues, that this is as important as making the diagnosis. We face an uphill battle because of our high cure rate, the biology is often considered a second thought sometimes. Europeans are better than us at obtaining biological specimens and we need to compete to achieve the level that they have achieved in Europe.
DR. GROSS: It's almost a catch-22. We know from other diseases in pediatric oncology, but also in adult oncology, that once we are able to demonstrate that the biology will make a difference in the treatment and outcome of the patient, then we're able to get the tissue needed. I think a good example of that is neuroblastoma. However, we can't make those discoveries unless we get enough tissue to study. We're in this catch-22, we cannot demonstrate that the biology makes a difference, unless we will get the tissue for research.
DR. LOWE: I'd like to add one other point to this. I think the rarity of the diseases and the large number of centers that treat the patients also hinders obtaining pathological samples. Because pediatric NHL is a relatively rare disease, you can’t have a single champion for obtaining biology at one institution that can accomplish anything without many other institutions. It requires a large group effort which is more difficult than a single institution collecting colon cancer samples, for example, where you really only need one institution, one champion, one pathologist, and you have all the samples you need.
DR. BOLLARD: Thank you, Dr. Lowe. I really thank you all for speaking in a very detailed way about the importance of obtaining tumor tissue to perform these critical biologic studies, because I do feel that's an important issue to overcome for the future care of our pediatric patients with NHL. I would like to discuss late effects in our survivors. As Dr. Gross said, survival rates for patients with B-cell lymphomas are generally outstanding. Dr. Gross, do you feel that late effects are not something the NHL group has to worry about now that we have obviated the need for radiation, or not? And what are your feelings about trying to minimize these late effects even further?
DR. GROSS: The good news is that over time, we have been able to come up with regimens that are highly effective but have reduced the agents we know have the highest risk of late effects—radiation being the primary one, but also anthracyclines we have been able to reduce in the vast majority of the patients, and to keep alkylating agents in the vast majority of the patients to a level that most patients do not have infertility. The long-term side effects are becoming pretty minimal, but the question is, how low do they have to be to be acceptable? The goal would be cure without any long-term effects. As I said before, certainly we have paid the price in short-term effects. Our regimens are inpatient, and they can have quite severe short-term side effects such as mucositis. We've made great advances but I think there's still room to go.
DR. CAIRO: I agree, of course, with my colleague Dr. Gross. Again, when we look at large series of chronic health care conditions, certainly children with treated NHL still comes up as one showing over 40% to 50% of patients having one or two serious chronic health care conditions. We know the data are a little antiquated, because they include patients who were treated with different regimens in the 1970s and all of the 1980s. However, I think our goal continues to be to identify the most effective treatment regimen, but with the least toxic long-term complications for our patients. That struggle is very difficult because of the very high success rate we have today, and to identify without hurting that high success rate less toxic therapies will require a collaborative, multidisciplinary, international effort to reach that goal.
DR. BOLLARD: Thank you very much Drs. Cairo and Gross. Dr. Lowe, did you have any closing remarks on the late effects issues for the T-cell mediated diseases in particular?
DR. LOWE: I would absolutely agree with Dr. Gross and Dr. Cairo that this is an important issue. I think we in pediatrics do a good job at following our patients for long-term side effects and creating guidelines for screening for these long-term side effects. That said, I think as we start to talk about better and better therapy and even more and more targeted therapy, what we don't know about some of these targeted therapies is their 15 and 20 year long-term side effects. We obviously hope that there aren't any, and that's why we are moving toward these drugs, but again, surveillance of those long-term side effects will be extremely important, especially when you're talking about medications for young children.
DR. BOLLARD: I'd like to thank you all very much for participating in this expert roundtable discussion today. I think the overarching points are that prognoses at the current time for newly diagnosed pediatric patients with NHL range from 70% to over 90% even for patients with disseminated disease. The challenges that we need to overcome are how we can optimize our up front treatment to prevent relapse in all, because I think we've all reiterated the fact that the outcomes for those few patients who do relapse remains extremely poor. I think there is still controversy about how to manage patients with relapsed disease, and how to temper our therapies against long-term side effects of our surviving patients. Finally, I think with the advent of novel targeted agents, it is incredibly important for the optimal management of our current and future patients that we are able to access tumor tissues and perform the critical biologic studies that are required to develop an effective precision medicine approach for pediatric patients with NHL. I would like to again thank Dr. Cairo, Dr. Gross, and Dr. Lowe for their excellent answers to my, at times, difficult and challenging questions and I would like to thank the organizers of this expert roundtable discussion. I hope that in the next decade that we will see even greater advances for the patient population that we treat. Thank you very much.
References
1. Rosolen RA, Perkins SL, Pinkerton CR, et al. Revised International Pediatric Non-Hodgkin Lymphoma Staging System. J Clin Oncol. 2015;33(18):2112–2118.
2. Sandlund JT, Guillerman RP, Perkins SL, et al. International Pediatric Non-Hodgkin Lymphoma Response Criteria. J Clin Oncol. 2015;33(18)2106-2111.
Dermatologists Need To “Stand Up” For Themselves

Sedentary living, supported by the number of hours spent sitting at work and at home each day, has been associated with increased risk for cardiovascular disease, diabetes, and premature mortality. Buckley et al (Br J Sports Med. 2015;49:1357-1362) provided guidelines for employers regarding interventions that may promote the avoidance of prolonged periods of sedentary work in a consensus statement that was commissioned by Public Health England and the Active Working Community Interest Company (who launched the Get Britain Standing campaign). The authors recommended initially aiming to accumulate at least 2 hours of standing and light activity (such as walking around the office) during work hours, with the eventual goal of 4 hours per day; however, they also cautioned that this positive adaptive process may lead to musculoskeletal sensations and fatigue in some individuals who may not be accustomed to standing-based work.
Buckley et al also suggested that employers should encourage goal that promote good health, such as improving nutrition and reducing alcohol consumption, smoking, and stress. Additionally, the authors highly recommended adjustable desk stations that allow employees to alternate between standing and seated work. Finally, Buckley et al commented that prolonged static standing postures, similar to prolonged static seated positions, also should be avoided.
Buckley et al also noted that workplaces that have initiated these interventions have seen improvement in cardiometabolic, musculoskeletal, and mental health risks in employees. Additionally, the productivity, quality, and efficiency of the work improved and employees experienced a greater sense of collaboration. Incorporating interventions to eliminate sitting for prolonged periods in the workplace resulted in cost savings for health services for both the employees and the employer in the groups studied by Buckley et al.
What’s the Issue?
Like other physicians and health care providers, dermatologists may be working longer hours, dealing with increased administrative demands that require electronic or hand-written documentation, and spending more time sitting each day to accomplish activities that do not involve direct patient care. Incorporating planned periods of activity (eg, standing breaks) in the workday for both themselves and their office personnel may be an effective intervention for dermatologists to use to avoid sitting for prolonged periods of time. Additionally, changing the ergonomic design of the office and workstations with adjustable desks and counter tops that allow employees to alternate between sitting and standing may encourage staff to not only become less sedentary but possibly more productive as well. Is it time for dermatologists to “stand up” for themselves?
Sedentary living, supported by the number of hours spent sitting at work and at home each day, has been associated with increased risk for cardiovascular disease, diabetes, and premature mortality. Buckley et al (Br J Sports Med. 2015;49:1357-1362) provided guidelines for employers regarding interventions that may promote the avoidance of prolonged periods of sedentary work in a consensus statement that was commissioned by Public Health England and the Active Working Community Interest Company (who launched the Get Britain Standing campaign). The authors recommended initially aiming to accumulate at least 2 hours of standing and light activity (such as walking around the office) during work hours, with the eventual goal of 4 hours per day; however, they also cautioned that this positive adaptive process may lead to musculoskeletal sensations and fatigue in some individuals who may not be accustomed to standing-based work.
Buckley et al also suggested that employers should encourage goal that promote good health, such as improving nutrition and reducing alcohol consumption, smoking, and stress. Additionally, the authors highly recommended adjustable desk stations that allow employees to alternate between standing and seated work. Finally, Buckley et al commented that prolonged static standing postures, similar to prolonged static seated positions, also should be avoided.
Buckley et al also noted that workplaces that have initiated these interventions have seen improvement in cardiometabolic, musculoskeletal, and mental health risks in employees. Additionally, the productivity, quality, and efficiency of the work improved and employees experienced a greater sense of collaboration. Incorporating interventions to eliminate sitting for prolonged periods in the workplace resulted in cost savings for health services for both the employees and the employer in the groups studied by Buckley et al.
What’s the Issue?
Like other physicians and health care providers, dermatologists may be working longer hours, dealing with increased administrative demands that require electronic or hand-written documentation, and spending more time sitting each day to accomplish activities that do not involve direct patient care. Incorporating planned periods of activity (eg, standing breaks) in the workday for both themselves and their office personnel may be an effective intervention for dermatologists to use to avoid sitting for prolonged periods of time. Additionally, changing the ergonomic design of the office and workstations with adjustable desks and counter tops that allow employees to alternate between sitting and standing may encourage staff to not only become less sedentary but possibly more productive as well. Is it time for dermatologists to “stand up” for themselves?
Sedentary living, supported by the number of hours spent sitting at work and at home each day, has been associated with increased risk for cardiovascular disease, diabetes, and premature mortality. Buckley et al (Br J Sports Med. 2015;49:1357-1362) provided guidelines for employers regarding interventions that may promote the avoidance of prolonged periods of sedentary work in a consensus statement that was commissioned by Public Health England and the Active Working Community Interest Company (who launched the Get Britain Standing campaign). The authors recommended initially aiming to accumulate at least 2 hours of standing and light activity (such as walking around the office) during work hours, with the eventual goal of 4 hours per day; however, they also cautioned that this positive adaptive process may lead to musculoskeletal sensations and fatigue in some individuals who may not be accustomed to standing-based work.
Buckley et al also suggested that employers should encourage goal that promote good health, such as improving nutrition and reducing alcohol consumption, smoking, and stress. Additionally, the authors highly recommended adjustable desk stations that allow employees to alternate between standing and seated work. Finally, Buckley et al commented that prolonged static standing postures, similar to prolonged static seated positions, also should be avoided.
Buckley et al also noted that workplaces that have initiated these interventions have seen improvement in cardiometabolic, musculoskeletal, and mental health risks in employees. Additionally, the productivity, quality, and efficiency of the work improved and employees experienced a greater sense of collaboration. Incorporating interventions to eliminate sitting for prolonged periods in the workplace resulted in cost savings for health services for both the employees and the employer in the groups studied by Buckley et al.
What’s the Issue?
Like other physicians and health care providers, dermatologists may be working longer hours, dealing with increased administrative demands that require electronic or hand-written documentation, and spending more time sitting each day to accomplish activities that do not involve direct patient care. Incorporating planned periods of activity (eg, standing breaks) in the workday for both themselves and their office personnel may be an effective intervention for dermatologists to use to avoid sitting for prolonged periods of time. Additionally, changing the ergonomic design of the office and workstations with adjustable desks and counter tops that allow employees to alternate between sitting and standing may encourage staff to not only become less sedentary but possibly more productive as well. Is it time for dermatologists to “stand up” for themselves?

EC approves drug for hemophilia A

The European Commission (EC) has approved the recombinant factor VIII Fc fusion protein efmoroctocog alfa (Elocta) to treat patients with hemophilia A in the European Union (EU) as well as Iceland, Liechtenstein, and Norway.
Efmoroctocog alfa is indicated for both on-demand and prophylactic treatment of hemophilia A in patients of all ages.
Research has shown that efmoroctocog alfa has an extended half-life compared to recombinant factor VIII.
Efmoroctocog alfa will be the first hemophilia A treatment in the EU to offer prolonged protection against bleeding episodes with prophylactic injections every 3 to 5 days.
The product is expected to launch in initial EU countries in early 2016.
The EC’s approval of efmoroctocog alfa was based on data from 2 phase 3 studies: A-LONG and Kids A-LONG.
A-LONG
The A-LONG study included 165 previously treated males 12 years of age and older with severe hemophilia A. Researchers evaluated individualized and weekly prophylaxis to reduce or prevent bleeding episodes and on-demand dosing to treat bleeding episodes.
Prophylaxis with efmoroctocog alfa resulted in low annualized bleeding rates, and a majority of bleeding episodes were controlled with a single injection of efmoroctocog alfa.
None of the patients developed neutralizing antibodies, efmoroctocog alfa was considered well-tolerated, and the product had a prolonged half-life when compared with recombinant factor VIII.
Kids A-LONG
The Kids A-LONG study included 71 boys (younger than 12) with severe hemophilia A who had at least 50 prior exposure days to factor VIII therapies.
The children saw their median annualized bleeding rate decrease with efmoroctocog alfa, and close to half of the children did not have any bleeding episodes while they were receiving efmoroctocog alfa.
None of the patients developed inhibitors, and researchers said adverse events were typical of a pediatric hemophilia population.
ASPIRE
Participants in both the A-LONG and Kids A-LONG trials were able to enroll in ASPIRE, a phase 3 extension study evaluating the long-term safety and efficacy of efmoroctocog alfa.
Interim results of ASPIRE suggested that extended treatment with efmoroctocog alfa was largely safe and effective.
About efmoroctocog alfa
Efmoroctocog alfa was developed by fusing B-domain deleted factor VIII to the Fc portion of immunoglobulin G subclass 1. It is believed that this enables efmoroctocog alfa to utilize a naturally occurring pathway to prolong the time the therapy remains in the body.
Sobi and Biogen are collaborators in the development and commercialization of efmoroctocog alfa for hemophilia A.
Last year, Sobi exercised its opt-in right to assumeefmoroctocog alfa’s final development and commercialization in pre-specified territories, which includes Europe, North Africa, Russia, and certain countries in the Middle East.
Biogen leads development and manufacturing of the product and holds commercialization rights in North America and all other regions in the world outside of the Sobi territories.
Elocta is the trade name for efmoroctocog alfa in Sobi’s territory. The product is approved under the name Eloctate (Antihemophilic Factor [Recombinant], Fc Fusion Protein) in the US, Canada, Australia, New Zealand, and Japan. ![]()
The European Commission (EC) has approved the recombinant factor VIII Fc fusion protein efmoroctocog alfa (Elocta) to treat patients with hemophilia A in the European Union (EU) as well as Iceland, Liechtenstein, and Norway.
Efmoroctocog alfa is indicated for both on-demand and prophylactic treatment of hemophilia A in patients of all ages.
Research has shown that efmoroctocog alfa has an extended half-life compared to recombinant factor VIII.
Efmoroctocog alfa will be the first hemophilia A treatment in the EU to offer prolonged protection against bleeding episodes with prophylactic injections every 3 to 5 days.
The product is expected to launch in initial EU countries in early 2016.
The EC’s approval of efmoroctocog alfa was based on data from 2 phase 3 studies: A-LONG and Kids A-LONG.
A-LONG
The A-LONG study included 165 previously treated males 12 years of age and older with severe hemophilia A. Researchers evaluated individualized and weekly prophylaxis to reduce or prevent bleeding episodes and on-demand dosing to treat bleeding episodes.
Prophylaxis with efmoroctocog alfa resulted in low annualized bleeding rates, and a majority of bleeding episodes were controlled with a single injection of efmoroctocog alfa.
None of the patients developed neutralizing antibodies, efmoroctocog alfa was considered well-tolerated, and the product had a prolonged half-life when compared with recombinant factor VIII.
Kids A-LONG
The Kids A-LONG study included 71 boys (younger than 12) with severe hemophilia A who had at least 50 prior exposure days to factor VIII therapies.
The children saw their median annualized bleeding rate decrease with efmoroctocog alfa, and close to half of the children did not have any bleeding episodes while they were receiving efmoroctocog alfa.
None of the patients developed inhibitors, and researchers said adverse events were typical of a pediatric hemophilia population.
ASPIRE
Participants in both the A-LONG and Kids A-LONG trials were able to enroll in ASPIRE, a phase 3 extension study evaluating the long-term safety and efficacy of efmoroctocog alfa.
Interim results of ASPIRE suggested that extended treatment with efmoroctocog alfa was largely safe and effective.
About efmoroctocog alfa
Efmoroctocog alfa was developed by fusing B-domain deleted factor VIII to the Fc portion of immunoglobulin G subclass 1. It is believed that this enables efmoroctocog alfa to utilize a naturally occurring pathway to prolong the time the therapy remains in the body.
Sobi and Biogen are collaborators in the development and commercialization of efmoroctocog alfa for hemophilia A.
Last year, Sobi exercised its opt-in right to assumeefmoroctocog alfa’s final development and commercialization in pre-specified territories, which includes Europe, North Africa, Russia, and certain countries in the Middle East.
Biogen leads development and manufacturing of the product and holds commercialization rights in North America and all other regions in the world outside of the Sobi territories.
Elocta is the trade name for efmoroctocog alfa in Sobi’s territory. The product is approved under the name Eloctate (Antihemophilic Factor [Recombinant], Fc Fusion Protein) in the US, Canada, Australia, New Zealand, and Japan. ![]()
The European Commission (EC) has approved the recombinant factor VIII Fc fusion protein efmoroctocog alfa (Elocta) to treat patients with hemophilia A in the European Union (EU) as well as Iceland, Liechtenstein, and Norway.
Efmoroctocog alfa is indicated for both on-demand and prophylactic treatment of hemophilia A in patients of all ages.
Research has shown that efmoroctocog alfa has an extended half-life compared to recombinant factor VIII.
Efmoroctocog alfa will be the first hemophilia A treatment in the EU to offer prolonged protection against bleeding episodes with prophylactic injections every 3 to 5 days.
The product is expected to launch in initial EU countries in early 2016.
The EC’s approval of efmoroctocog alfa was based on data from 2 phase 3 studies: A-LONG and Kids A-LONG.
A-LONG
The A-LONG study included 165 previously treated males 12 years of age and older with severe hemophilia A. Researchers evaluated individualized and weekly prophylaxis to reduce or prevent bleeding episodes and on-demand dosing to treat bleeding episodes.
Prophylaxis with efmoroctocog alfa resulted in low annualized bleeding rates, and a majority of bleeding episodes were controlled with a single injection of efmoroctocog alfa.
None of the patients developed neutralizing antibodies, efmoroctocog alfa was considered well-tolerated, and the product had a prolonged half-life when compared with recombinant factor VIII.
Kids A-LONG
The Kids A-LONG study included 71 boys (younger than 12) with severe hemophilia A who had at least 50 prior exposure days to factor VIII therapies.
The children saw their median annualized bleeding rate decrease with efmoroctocog alfa, and close to half of the children did not have any bleeding episodes while they were receiving efmoroctocog alfa.
None of the patients developed inhibitors, and researchers said adverse events were typical of a pediatric hemophilia population.
ASPIRE
Participants in both the A-LONG and Kids A-LONG trials were able to enroll in ASPIRE, a phase 3 extension study evaluating the long-term safety and efficacy of efmoroctocog alfa.
Interim results of ASPIRE suggested that extended treatment with efmoroctocog alfa was largely safe and effective.
About efmoroctocog alfa
Efmoroctocog alfa was developed by fusing B-domain deleted factor VIII to the Fc portion of immunoglobulin G subclass 1. It is believed that this enables efmoroctocog alfa to utilize a naturally occurring pathway to prolong the time the therapy remains in the body.
Sobi and Biogen are collaborators in the development and commercialization of efmoroctocog alfa for hemophilia A.
Last year, Sobi exercised its opt-in right to assumeefmoroctocog alfa’s final development and commercialization in pre-specified territories, which includes Europe, North Africa, Russia, and certain countries in the Middle East.
Biogen leads development and manufacturing of the product and holds commercialization rights in North America and all other regions in the world outside of the Sobi territories.
Elocta is the trade name for efmoroctocog alfa in Sobi’s territory. The product is approved under the name Eloctate (Antihemophilic Factor [Recombinant], Fc Fusion Protein) in the US, Canada, Australia, New Zealand, and Japan. ![]()
Trial of ofatumumab in FL stopped early

Photo courtesy of GSK
Genmab A/S said it is stopping a phase 3 trial of ofatumumab in follicular lymphoma (FL) after a planned interim analysis suggested the drug was unlikely to demonstrate superiority over rituximab.
The trial was designed to compare these 2 drugs as single-agent treatment in FL patients who had relapsed at least 6 months after completing treatment with a rituximab-containing regimen.
An interim analysis performed by an independent data monitoring committee indicated that, if the trial was to be completed as planned, it was unlikely that ofatumumab would improve progression-free survival (PFS) when compared to rituximab.
Genmab noted that this analysis did not reveal any new safety signals associated with ofatumumab, and no other ongoing trials of ofatumumab will be affected by the results of this analysis.
“The outcome of the interim analysis in this study is disappointing, as we had hoped to see superiority of ofatumumab,” said Jan van de Winkel, PhD, chief executive officer of Genmab. “The data from the study will now be prepared so that it can be presented at a future scientific conference.”
The goal of the study was to randomize up to 516 patients to receive ofatumumab (at 1000 mg) or rituximab (at 375 mg/m2) by intravenous infusion for 4 weekly doses.
Patients who had stable or responsive disease would then receive single infusions of ofatumumab or rituximab every 2 months for 4 additional doses—a total of 8 doses over 9 months. The primary endpoint of the study was PFS.
Past disappointments
This is not the first time ofatumumab has fallen short of expectations. Last year, the drug failed to meet the primary endpoint in two phase 3 trials.
In the OMB114242 trial, ofatumumab failed to improve PFS when compared to physicians’ choice in patients with bulky, fludarabine-refractory chronic lymphocytic leukemia (CLL).
In the ORCHARRD trial, ofatumumab plus chemotherapy failed to improve PFS when compared to rituximab plus chemotherapy in patients with relapsed or refractory diffuse large B-cell lymphoma.
About ofatumumab
Ofatumumab is a human monoclonal antibody designed to target the CD20 molecule found on the surface of CLL cells and normal B lymphocytes.
In the US, ofatumumab is approved for use in combination with chlorambucil to treat previously untreated patients with CLL for whom fludarabine-based therapy is considered inappropriate.
In the European Union (EU), ofatumumab is approved for use in combination with chlorambucil or bendamustine to treat patients with CLL who have not received prior therapy and who are not eligible for fludarabine-based therapy.
In more than 50 countries worldwide, including the US and EU member countries, ofatumumab is approved as monotherapy for the treatment of patients with CLL who are refractory after prior treatment with fludarabine and alemtuzumab.
Ofatumumab is marketed as Arzerra under a collaboration agreement between Genmab and Novartis (formerly GSK). ![]()
Photo courtesy of GSK
Genmab A/S said it is stopping a phase 3 trial of ofatumumab in follicular lymphoma (FL) after a planned interim analysis suggested the drug was unlikely to demonstrate superiority over rituximab.
The trial was designed to compare these 2 drugs as single-agent treatment in FL patients who had relapsed at least 6 months after completing treatment with a rituximab-containing regimen.
An interim analysis performed by an independent data monitoring committee indicated that, if the trial was to be completed as planned, it was unlikely that ofatumumab would improve progression-free survival (PFS) when compared to rituximab.
Genmab noted that this analysis did not reveal any new safety signals associated with ofatumumab, and no other ongoing trials of ofatumumab will be affected by the results of this analysis.
“The outcome of the interim analysis in this study is disappointing, as we had hoped to see superiority of ofatumumab,” said Jan van de Winkel, PhD, chief executive officer of Genmab. “The data from the study will now be prepared so that it can be presented at a future scientific conference.”
The goal of the study was to randomize up to 516 patients to receive ofatumumab (at 1000 mg) or rituximab (at 375 mg/m2) by intravenous infusion for 4 weekly doses.
Patients who had stable or responsive disease would then receive single infusions of ofatumumab or rituximab every 2 months for 4 additional doses—a total of 8 doses over 9 months. The primary endpoint of the study was PFS.
Past disappointments
This is not the first time ofatumumab has fallen short of expectations. Last year, the drug failed to meet the primary endpoint in two phase 3 trials.
In the OMB114242 trial, ofatumumab failed to improve PFS when compared to physicians’ choice in patients with bulky, fludarabine-refractory chronic lymphocytic leukemia (CLL).
In the ORCHARRD trial, ofatumumab plus chemotherapy failed to improve PFS when compared to rituximab plus chemotherapy in patients with relapsed or refractory diffuse large B-cell lymphoma.
About ofatumumab
Ofatumumab is a human monoclonal antibody designed to target the CD20 molecule found on the surface of CLL cells and normal B lymphocytes.
In the US, ofatumumab is approved for use in combination with chlorambucil to treat previously untreated patients with CLL for whom fludarabine-based therapy is considered inappropriate.
In the European Union (EU), ofatumumab is approved for use in combination with chlorambucil or bendamustine to treat patients with CLL who have not received prior therapy and who are not eligible for fludarabine-based therapy.
In more than 50 countries worldwide, including the US and EU member countries, ofatumumab is approved as monotherapy for the treatment of patients with CLL who are refractory after prior treatment with fludarabine and alemtuzumab.
Ofatumumab is marketed as Arzerra under a collaboration agreement between Genmab and Novartis (formerly GSK). ![]()
Photo courtesy of GSK
Genmab A/S said it is stopping a phase 3 trial of ofatumumab in follicular lymphoma (FL) after a planned interim analysis suggested the drug was unlikely to demonstrate superiority over rituximab.
The trial was designed to compare these 2 drugs as single-agent treatment in FL patients who had relapsed at least 6 months after completing treatment with a rituximab-containing regimen.
An interim analysis performed by an independent data monitoring committee indicated that, if the trial was to be completed as planned, it was unlikely that ofatumumab would improve progression-free survival (PFS) when compared to rituximab.
Genmab noted that this analysis did not reveal any new safety signals associated with ofatumumab, and no other ongoing trials of ofatumumab will be affected by the results of this analysis.
“The outcome of the interim analysis in this study is disappointing, as we had hoped to see superiority of ofatumumab,” said Jan van de Winkel, PhD, chief executive officer of Genmab. “The data from the study will now be prepared so that it can be presented at a future scientific conference.”
The goal of the study was to randomize up to 516 patients to receive ofatumumab (at 1000 mg) or rituximab (at 375 mg/m2) by intravenous infusion for 4 weekly doses.
Patients who had stable or responsive disease would then receive single infusions of ofatumumab or rituximab every 2 months for 4 additional doses—a total of 8 doses over 9 months. The primary endpoint of the study was PFS.
Past disappointments
This is not the first time ofatumumab has fallen short of expectations. Last year, the drug failed to meet the primary endpoint in two phase 3 trials.
In the OMB114242 trial, ofatumumab failed to improve PFS when compared to physicians’ choice in patients with bulky, fludarabine-refractory chronic lymphocytic leukemia (CLL).
In the ORCHARRD trial, ofatumumab plus chemotherapy failed to improve PFS when compared to rituximab plus chemotherapy in patients with relapsed or refractory diffuse large B-cell lymphoma.
About ofatumumab
Ofatumumab is a human monoclonal antibody designed to target the CD20 molecule found on the surface of CLL cells and normal B lymphocytes.
In the US, ofatumumab is approved for use in combination with chlorambucil to treat previously untreated patients with CLL for whom fludarabine-based therapy is considered inappropriate.
In the European Union (EU), ofatumumab is approved for use in combination with chlorambucil or bendamustine to treat patients with CLL who have not received prior therapy and who are not eligible for fludarabine-based therapy.
In more than 50 countries worldwide, including the US and EU member countries, ofatumumab is approved as monotherapy for the treatment of patients with CLL who are refractory after prior treatment with fludarabine and alemtuzumab.
Ofatumumab is marketed as Arzerra under a collaboration agreement between Genmab and Novartis (formerly GSK). ![]()
Chip can remove nanoparticles from blood

Photo from Jacobs School
of Engineering/UC San Diego
Engineers have developed a chip that uses an oscillating electric field to isolate drug-delivery nanoparticles from blood.
They say the device isolated nanoparticles from plasma quickly and easily, without the need to modify the plasma samples or the nanoparticles.
The group believes this technique could be used to separate and recover nanoparticles from other complex fluids for medical, environmental, and industrial applications.
They described the technique in the journal Small.
Previously, nanoparticles proved difficult to separate from plasma due to their small size and low density.
Traditional methods to remove nanoparticles from plasma samples typically involve diluting the plasma, adding a high-concentration sugar solution to the plasma and spinning it in a centrifuge, or attaching a targeting agent to the surface of the nanoparticles.
These methods either alter the normal behavior of the nanoparticles or cannot be applied to some of the most common nanoparticle types.
“This is the first example of isolating a wide range of nanoparticles out of plasma with a minimum amount of manipulation,” said study author Stuart Ibsen, PhD, of the University of California, San Diego.
“We’ve designed a very versatile technique that can be used to recover nanoparticles in a lot of different processes.”
The device used to isolate the drug-delivery nanoparticles was a dime-sized electric chip manufactured by La Jolla-based Biological Dynamics, which licensed the original technology from the University of California, San Diego.
The chip contains hundreds of tiny electrodes that generate a rapidly oscillating electric field that selectively pulls the nanoparticles out of a plasma sample.
The researchers inserted a drop of plasma spiked with nanoparticles into the electric chip and demonstrated nanoparticle recovery within 7 minutes. The technology worked on different types of drug-delivery nanoparticles that are typically studied in various labs.
The researchers said the breakthrough in the technology relies on designing a chip that can work in the high-salt concentration of plasma. The chip’s ability to pull the nanoparticles out of plasma is based on differences in the material properties between the nanoparticles and plasma components.
When the chip’s electrodes apply an oscillating electric field, the positive and negative charges inside the nanoparticles reorient themselves at a different speed than the charges in the surrounding plasma. This momentary imbalance in the charges creates an attractive force between the nanoparticles and the electrodes.
As the electric field oscillates, the nanoparticles are continually pulled toward the electrodes, leaving the rest of the plasma behind. In addition, the electric field is designed to oscillate at just the right frequency: 15,000 times per second.
“It’s amazing that this method works without any modifications to the plasma samples or to the nanoparticles,” Dr Ibsen said.
He and his colleagues believe this technology will enable researchers to better monitor what happens to nanoparticles circulating in a patient’s bloodstream. Researchers could also use this technology in the clinic to determine if the blood chemistry of a particular patient is compatible with the surfaces of certain drug-delivery nanoparticles. ![]()
Photo from Jacobs School
of Engineering/UC San Diego
Engineers have developed a chip that uses an oscillating electric field to isolate drug-delivery nanoparticles from blood.
They say the device isolated nanoparticles from plasma quickly and easily, without the need to modify the plasma samples or the nanoparticles.
The group believes this technique could be used to separate and recover nanoparticles from other complex fluids for medical, environmental, and industrial applications.
They described the technique in the journal Small.
Previously, nanoparticles proved difficult to separate from plasma due to their small size and low density.
Traditional methods to remove nanoparticles from plasma samples typically involve diluting the plasma, adding a high-concentration sugar solution to the plasma and spinning it in a centrifuge, or attaching a targeting agent to the surface of the nanoparticles.
These methods either alter the normal behavior of the nanoparticles or cannot be applied to some of the most common nanoparticle types.
“This is the first example of isolating a wide range of nanoparticles out of plasma with a minimum amount of manipulation,” said study author Stuart Ibsen, PhD, of the University of California, San Diego.
“We’ve designed a very versatile technique that can be used to recover nanoparticles in a lot of different processes.”
The device used to isolate the drug-delivery nanoparticles was a dime-sized electric chip manufactured by La Jolla-based Biological Dynamics, which licensed the original technology from the University of California, San Diego.
The chip contains hundreds of tiny electrodes that generate a rapidly oscillating electric field that selectively pulls the nanoparticles out of a plasma sample.
The researchers inserted a drop of plasma spiked with nanoparticles into the electric chip and demonstrated nanoparticle recovery within 7 minutes. The technology worked on different types of drug-delivery nanoparticles that are typically studied in various labs.
The researchers said the breakthrough in the technology relies on designing a chip that can work in the high-salt concentration of plasma. The chip’s ability to pull the nanoparticles out of plasma is based on differences in the material properties between the nanoparticles and plasma components.
When the chip’s electrodes apply an oscillating electric field, the positive and negative charges inside the nanoparticles reorient themselves at a different speed than the charges in the surrounding plasma. This momentary imbalance in the charges creates an attractive force between the nanoparticles and the electrodes.
As the electric field oscillates, the nanoparticles are continually pulled toward the electrodes, leaving the rest of the plasma behind. In addition, the electric field is designed to oscillate at just the right frequency: 15,000 times per second.
“It’s amazing that this method works without any modifications to the plasma samples or to the nanoparticles,” Dr Ibsen said.
He and his colleagues believe this technology will enable researchers to better monitor what happens to nanoparticles circulating in a patient’s bloodstream. Researchers could also use this technology in the clinic to determine if the blood chemistry of a particular patient is compatible with the surfaces of certain drug-delivery nanoparticles. ![]()
Photo from Jacobs School
of Engineering/UC San Diego
Engineers have developed a chip that uses an oscillating electric field to isolate drug-delivery nanoparticles from blood.
They say the device isolated nanoparticles from plasma quickly and easily, without the need to modify the plasma samples or the nanoparticles.
The group believes this technique could be used to separate and recover nanoparticles from other complex fluids for medical, environmental, and industrial applications.
They described the technique in the journal Small.
Previously, nanoparticles proved difficult to separate from plasma due to their small size and low density.
Traditional methods to remove nanoparticles from plasma samples typically involve diluting the plasma, adding a high-concentration sugar solution to the plasma and spinning it in a centrifuge, or attaching a targeting agent to the surface of the nanoparticles.
These methods either alter the normal behavior of the nanoparticles or cannot be applied to some of the most common nanoparticle types.
“This is the first example of isolating a wide range of nanoparticles out of plasma with a minimum amount of manipulation,” said study author Stuart Ibsen, PhD, of the University of California, San Diego.
“We’ve designed a very versatile technique that can be used to recover nanoparticles in a lot of different processes.”
The device used to isolate the drug-delivery nanoparticles was a dime-sized electric chip manufactured by La Jolla-based Biological Dynamics, which licensed the original technology from the University of California, San Diego.
The chip contains hundreds of tiny electrodes that generate a rapidly oscillating electric field that selectively pulls the nanoparticles out of a plasma sample.
The researchers inserted a drop of plasma spiked with nanoparticles into the electric chip and demonstrated nanoparticle recovery within 7 minutes. The technology worked on different types of drug-delivery nanoparticles that are typically studied in various labs.
The researchers said the breakthrough in the technology relies on designing a chip that can work in the high-salt concentration of plasma. The chip’s ability to pull the nanoparticles out of plasma is based on differences in the material properties between the nanoparticles and plasma components.
When the chip’s electrodes apply an oscillating electric field, the positive and negative charges inside the nanoparticles reorient themselves at a different speed than the charges in the surrounding plasma. This momentary imbalance in the charges creates an attractive force between the nanoparticles and the electrodes.
As the electric field oscillates, the nanoparticles are continually pulled toward the electrodes, leaving the rest of the plasma behind. In addition, the electric field is designed to oscillate at just the right frequency: 15,000 times per second.
“It’s amazing that this method works without any modifications to the plasma samples or to the nanoparticles,” Dr Ibsen said.
He and his colleagues believe this technology will enable researchers to better monitor what happens to nanoparticles circulating in a patient’s bloodstream. Researchers could also use this technology in the clinic to determine if the blood chemistry of a particular patient is compatible with the surfaces of certain drug-delivery nanoparticles. ![]()
Modified mosquitoes may prevent malaria transmission
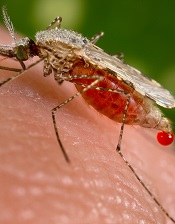
Photo by James Gathany/CDC
Scientists say they have created mosquitoes that can introduce malaria-blocking genes into a mosquito population through their progeny and, ideally, eliminate the insects’ ability to transmit malaria to humans.
The researchers used the CRISPR gene-editing technique to insert a DNA element into the germ line of Anopheles stephensi mosquitoes.
These mosquitoes then passed on genes that may prevent malaria transmission to 99.5% of their offspring.
The scientists said further testing is needed to confirm the efficacy of this approach in preventing malaria transmission, but this is an important first step toward that goal.
“This opens up the real promise that this technique can be adapted for eliminating malaria,” said Anthony James, PhD, of the University of California, Irvine.
Dr James and his colleagues described the technique in PNAS.
In 2012, Dr James and his colleagues showed that antibodies that impair Plasmodium falciparum’s biology could be adapted from the immune systems of mice and introduced into mosquitoes. However, this trait could only be inherited by about half of the progeny.
Earlier this year, Ethan Bier, PhD, and Valentino Gantz, both of the University of California, San Diego, reported a new method for generating mutations in both copies of a gene in fruit flies. This mutagenic chain reaction involved using the CRISPR-associated protein 9 (Cas9) nuclease enzyme and allowed for transmission of mutations through the germ line with an inheritance rate of 95%.
The groups collaborated to fuse Dr Bier and Gantz’s method with Dr James’s mosquitoes. Gantz packaged antimalaria genes with a Cas9 enzyme and a guide RNA to create a genetic “cassette” that, when injected into a mosquito embryo, targeted a highly specific spot on the germ line DNA to insert the antimalaria antibody genes.
To ensure the element carrying the malaria-blocking antibodies had reached the desired DNA site, the researchers included in the cassette a protein that gave the progeny red fluorescence in the eyes. Nearly all offspring—99.5%—exhibited this trait.
Dr James said further testing will be needed to confirm the efficacy of the antibodies, and this could eventually lead to field studies.
“[The current study] is a significant first step,” he said. “We know the gene works. The mosquitoes we created are not the final brand, but we know this technology allows us to efficiently create large populations.” ![]()
Photo by James Gathany/CDC
Scientists say they have created mosquitoes that can introduce malaria-blocking genes into a mosquito population through their progeny and, ideally, eliminate the insects’ ability to transmit malaria to humans.
The researchers used the CRISPR gene-editing technique to insert a DNA element into the germ line of Anopheles stephensi mosquitoes.
These mosquitoes then passed on genes that may prevent malaria transmission to 99.5% of their offspring.
The scientists said further testing is needed to confirm the efficacy of this approach in preventing malaria transmission, but this is an important first step toward that goal.
“This opens up the real promise that this technique can be adapted for eliminating malaria,” said Anthony James, PhD, of the University of California, Irvine.
Dr James and his colleagues described the technique in PNAS.
In 2012, Dr James and his colleagues showed that antibodies that impair Plasmodium falciparum’s biology could be adapted from the immune systems of mice and introduced into mosquitoes. However, this trait could only be inherited by about half of the progeny.
Earlier this year, Ethan Bier, PhD, and Valentino Gantz, both of the University of California, San Diego, reported a new method for generating mutations in both copies of a gene in fruit flies. This mutagenic chain reaction involved using the CRISPR-associated protein 9 (Cas9) nuclease enzyme and allowed for transmission of mutations through the germ line with an inheritance rate of 95%.
The groups collaborated to fuse Dr Bier and Gantz’s method with Dr James’s mosquitoes. Gantz packaged antimalaria genes with a Cas9 enzyme and a guide RNA to create a genetic “cassette” that, when injected into a mosquito embryo, targeted a highly specific spot on the germ line DNA to insert the antimalaria antibody genes.
To ensure the element carrying the malaria-blocking antibodies had reached the desired DNA site, the researchers included in the cassette a protein that gave the progeny red fluorescence in the eyes. Nearly all offspring—99.5%—exhibited this trait.
Dr James said further testing will be needed to confirm the efficacy of the antibodies, and this could eventually lead to field studies.
“[The current study] is a significant first step,” he said. “We know the gene works. The mosquitoes we created are not the final brand, but we know this technology allows us to efficiently create large populations.” ![]()
Photo by James Gathany/CDC
Scientists say they have created mosquitoes that can introduce malaria-blocking genes into a mosquito population through their progeny and, ideally, eliminate the insects’ ability to transmit malaria to humans.
The researchers used the CRISPR gene-editing technique to insert a DNA element into the germ line of Anopheles stephensi mosquitoes.
These mosquitoes then passed on genes that may prevent malaria transmission to 99.5% of their offspring.
The scientists said further testing is needed to confirm the efficacy of this approach in preventing malaria transmission, but this is an important first step toward that goal.
“This opens up the real promise that this technique can be adapted for eliminating malaria,” said Anthony James, PhD, of the University of California, Irvine.
Dr James and his colleagues described the technique in PNAS.
In 2012, Dr James and his colleagues showed that antibodies that impair Plasmodium falciparum’s biology could be adapted from the immune systems of mice and introduced into mosquitoes. However, this trait could only be inherited by about half of the progeny.
Earlier this year, Ethan Bier, PhD, and Valentino Gantz, both of the University of California, San Diego, reported a new method for generating mutations in both copies of a gene in fruit flies. This mutagenic chain reaction involved using the CRISPR-associated protein 9 (Cas9) nuclease enzyme and allowed for transmission of mutations through the germ line with an inheritance rate of 95%.
The groups collaborated to fuse Dr Bier and Gantz’s method with Dr James’s mosquitoes. Gantz packaged antimalaria genes with a Cas9 enzyme and a guide RNA to create a genetic “cassette” that, when injected into a mosquito embryo, targeted a highly specific spot on the germ line DNA to insert the antimalaria antibody genes.
To ensure the element carrying the malaria-blocking antibodies had reached the desired DNA site, the researchers included in the cassette a protein that gave the progeny red fluorescence in the eyes. Nearly all offspring—99.5%—exhibited this trait.
Dr James said further testing will be needed to confirm the efficacy of the antibodies, and this could eventually lead to field studies.
“[The current study] is a significant first step,” he said. “We know the gene works. The mosquitoes we created are not the final brand, but we know this technology allows us to efficiently create large populations.”