User login
FDA approves eribulin for advanced liposarcoma
The Food and Drug Administration has approved eribulin for the treatment of patients with unresectable or metastatic liposarcoma who have received a prior anthracycline-containing regimen.
The approval was based on improved overall survival (OS) in an open-label, randomized, multicenter trial of 446 patients with unresectable, locally advanced or metastatic liposarcoma or leiomyosarcoma who had received at least two prior systemic chemotherapies (one of which must have included an anthracycline) and had experienced disease progression within 6 months of randomization, according to the Jan. 28 statement issued by the FDA.
Patients were randomized to receive either eribulin 1.4 mg/m2 on days 1 and 8 of a 21-day cycle or dacarbazine (at a dose of 850 mg/m2, 1,000 mg/m2, or 1,200 mg/m2 chosen by the investigator prior to randomization) on day 1 of a 21-day cycle.
Eribulin benefit was limited to the subgroup of patients with liposarcoma. The median OS was 15.6 vs. 8.4 months (HR 0.51 [95% CI: 0.35, 0.75]) and the median progression-free survival (PFS) was 2.9 vs. 1.7 months (HR 0.52 [95% CI: 0.35, 0.78]) in patients with liposarcoma treated with eribulin compared to dacarbazine, respectively. There was no evidence of efficacy for eribulin in patients with leiomyosarcoma.
The most common adverse reactions among those who received eribulin in the trial were fatigue, nausea, alopecia, constipation, peripheral neuropathy, abdominal pain, and pyrexia. The most common grade 3-4 laboratory abnormalities were neutropenia, hypokalemia, and hypocalcemia.
The most common serious adverse reactions were neutropenia (4.9%) and pyrexia (4.5%). Febrile neutropenia occurred in 0.9% and fatal neutropenic sepsis in 0.9% of patients treated with eribulin. The most frequent adverse reactions leading to discontinuation were fatigue (0.9%) and thrombocytopenia (0.9%), according to the FDA statement.
Eribulin is marketed as Halaven injection by Eisai. The recommended dose and schedule for eribulin is 1.4 mg/m2 on days 1 and 8 of a 21-day cycle.
On Twitter @nikolaideslaura
The Food and Drug Administration has approved eribulin for the treatment of patients with unresectable or metastatic liposarcoma who have received a prior anthracycline-containing regimen.
The approval was based on improved overall survival (OS) in an open-label, randomized, multicenter trial of 446 patients with unresectable, locally advanced or metastatic liposarcoma or leiomyosarcoma who had received at least two prior systemic chemotherapies (one of which must have included an anthracycline) and had experienced disease progression within 6 months of randomization, according to the Jan. 28 statement issued by the FDA.
Patients were randomized to receive either eribulin 1.4 mg/m2 on days 1 and 8 of a 21-day cycle or dacarbazine (at a dose of 850 mg/m2, 1,000 mg/m2, or 1,200 mg/m2 chosen by the investigator prior to randomization) on day 1 of a 21-day cycle.
Eribulin benefit was limited to the subgroup of patients with liposarcoma. The median OS was 15.6 vs. 8.4 months (HR 0.51 [95% CI: 0.35, 0.75]) and the median progression-free survival (PFS) was 2.9 vs. 1.7 months (HR 0.52 [95% CI: 0.35, 0.78]) in patients with liposarcoma treated with eribulin compared to dacarbazine, respectively. There was no evidence of efficacy for eribulin in patients with leiomyosarcoma.
The most common adverse reactions among those who received eribulin in the trial were fatigue, nausea, alopecia, constipation, peripheral neuropathy, abdominal pain, and pyrexia. The most common grade 3-4 laboratory abnormalities were neutropenia, hypokalemia, and hypocalcemia.
The most common serious adverse reactions were neutropenia (4.9%) and pyrexia (4.5%). Febrile neutropenia occurred in 0.9% and fatal neutropenic sepsis in 0.9% of patients treated with eribulin. The most frequent adverse reactions leading to discontinuation were fatigue (0.9%) and thrombocytopenia (0.9%), according to the FDA statement.
Eribulin is marketed as Halaven injection by Eisai. The recommended dose and schedule for eribulin is 1.4 mg/m2 on days 1 and 8 of a 21-day cycle.
On Twitter @nikolaideslaura
The Food and Drug Administration has approved eribulin for the treatment of patients with unresectable or metastatic liposarcoma who have received a prior anthracycline-containing regimen.
The approval was based on improved overall survival (OS) in an open-label, randomized, multicenter trial of 446 patients with unresectable, locally advanced or metastatic liposarcoma or leiomyosarcoma who had received at least two prior systemic chemotherapies (one of which must have included an anthracycline) and had experienced disease progression within 6 months of randomization, according to the Jan. 28 statement issued by the FDA.
Patients were randomized to receive either eribulin 1.4 mg/m2 on days 1 and 8 of a 21-day cycle or dacarbazine (at a dose of 850 mg/m2, 1,000 mg/m2, or 1,200 mg/m2 chosen by the investigator prior to randomization) on day 1 of a 21-day cycle.
Eribulin benefit was limited to the subgroup of patients with liposarcoma. The median OS was 15.6 vs. 8.4 months (HR 0.51 [95% CI: 0.35, 0.75]) and the median progression-free survival (PFS) was 2.9 vs. 1.7 months (HR 0.52 [95% CI: 0.35, 0.78]) in patients with liposarcoma treated with eribulin compared to dacarbazine, respectively. There was no evidence of efficacy for eribulin in patients with leiomyosarcoma.
The most common adverse reactions among those who received eribulin in the trial were fatigue, nausea, alopecia, constipation, peripheral neuropathy, abdominal pain, and pyrexia. The most common grade 3-4 laboratory abnormalities were neutropenia, hypokalemia, and hypocalcemia.
The most common serious adverse reactions were neutropenia (4.9%) and pyrexia (4.5%). Febrile neutropenia occurred in 0.9% and fatal neutropenic sepsis in 0.9% of patients treated with eribulin. The most frequent adverse reactions leading to discontinuation were fatigue (0.9%) and thrombocytopenia (0.9%), according to the FDA statement.
Eribulin is marketed as Halaven injection by Eisai. The recommended dose and schedule for eribulin is 1.4 mg/m2 on days 1 and 8 of a 21-day cycle.
On Twitter @nikolaideslaura
What Matters: Probiotics for colds
In the midst of the cold and flu season, we should reflect on the fact that our patients are laying down billions of dollars annually on preventions and cures for respiratory tract infections.
An aside: I am frequently turned down on my offer of the influenza vaccine, for which we probably have the best evidence. But $60 per month for a completely unproven preventive/curative agent made in some random factory in some random foreign land with no guarantee of good manufacturing practices (never mind the lack of active ingredients)? Stores can’t keep it in stock.
But I digress.
Our patients may lack the awareness of where to access evidence-based information when seeking answers about efficacy for cold remedies. So, it’s up to us to have at least some sense of where to get reliable information.
Truth be told, I am an enormous fan of safe and effective nonmedication therapies for the treatment and prevention of disease. So, when time permits, I will do a quick PubMed.gov search limiting my articles to randomized trials or systematic reviews on the latest and greatest home remedy.
Probiotics have been around for a while, and I think of them as a cure in search of a disease. The Cochrane Collaboration conducted a systematic review evaluating probiotics for the prevention of upper respiratory tract infection. In this review, 13 randomized, controlled trials were included (Explore [NY]. 2015 Sep-Oct;11[5]:418-20).
Probiotics were observed to be significantly better than placebo for reducing episodes of upper respiratory tract infection, the mean duration of episodes, antibiotic prescription rates, and cold-related absences. The evidence was of moderate to low quality.
Some may wonder how an ingested probiotic helps the respiratory tract stave off or fight infection. The prevailing theory appears to be that probiotics may function by mobilizing cells from the intestine to immunomodulate respiratory mucosa.
As for what probiotic/organism to prescribe? On this issue, there is a lot of smoke and not a lot of heat.
In general, the product should be encapsulated, and the label should include the genus and species of the strains (e.g., Lactobacillus acidophilus), the number of organisms (e.g., 5 billion), storage conditions (e.g., refrigerated or room temperature), and the shelf life. Pharmacy chain house brands may be cheaper. Gummy and chewable products tend to have 92% fewer beneficial bacteria than standard formulations.
How can we ensure purity?
That is tough, because supplements like probiotics are not regulated by the Food and Drug Administration above and beyond the agency’s trying to ensure good manufacturing practices. However, companies such as LabDoor (which generates revenue through affiliate links) test and grade supplements for label accuracy and purity. Websites like this might be the best place to start.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no financial disclosures relevant to this article.
In the midst of the cold and flu season, we should reflect on the fact that our patients are laying down billions of dollars annually on preventions and cures for respiratory tract infections.
An aside: I am frequently turned down on my offer of the influenza vaccine, for which we probably have the best evidence. But $60 per month for a completely unproven preventive/curative agent made in some random factory in some random foreign land with no guarantee of good manufacturing practices (never mind the lack of active ingredients)? Stores can’t keep it in stock.
But I digress.
Our patients may lack the awareness of where to access evidence-based information when seeking answers about efficacy for cold remedies. So, it’s up to us to have at least some sense of where to get reliable information.
Truth be told, I am an enormous fan of safe and effective nonmedication therapies for the treatment and prevention of disease. So, when time permits, I will do a quick PubMed.gov search limiting my articles to randomized trials or systematic reviews on the latest and greatest home remedy.
Probiotics have been around for a while, and I think of them as a cure in search of a disease. The Cochrane Collaboration conducted a systematic review evaluating probiotics for the prevention of upper respiratory tract infection. In this review, 13 randomized, controlled trials were included (Explore [NY]. 2015 Sep-Oct;11[5]:418-20).
Probiotics were observed to be significantly better than placebo for reducing episodes of upper respiratory tract infection, the mean duration of episodes, antibiotic prescription rates, and cold-related absences. The evidence was of moderate to low quality.
Some may wonder how an ingested probiotic helps the respiratory tract stave off or fight infection. The prevailing theory appears to be that probiotics may function by mobilizing cells from the intestine to immunomodulate respiratory mucosa.
As for what probiotic/organism to prescribe? On this issue, there is a lot of smoke and not a lot of heat.
In general, the product should be encapsulated, and the label should include the genus and species of the strains (e.g., Lactobacillus acidophilus), the number of organisms (e.g., 5 billion), storage conditions (e.g., refrigerated or room temperature), and the shelf life. Pharmacy chain house brands may be cheaper. Gummy and chewable products tend to have 92% fewer beneficial bacteria than standard formulations.
How can we ensure purity?
That is tough, because supplements like probiotics are not regulated by the Food and Drug Administration above and beyond the agency’s trying to ensure good manufacturing practices. However, companies such as LabDoor (which generates revenue through affiliate links) test and grade supplements for label accuracy and purity. Websites like this might be the best place to start.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no financial disclosures relevant to this article.
In the midst of the cold and flu season, we should reflect on the fact that our patients are laying down billions of dollars annually on preventions and cures for respiratory tract infections.
An aside: I am frequently turned down on my offer of the influenza vaccine, for which we probably have the best evidence. But $60 per month for a completely unproven preventive/curative agent made in some random factory in some random foreign land with no guarantee of good manufacturing practices (never mind the lack of active ingredients)? Stores can’t keep it in stock.
But I digress.
Our patients may lack the awareness of where to access evidence-based information when seeking answers about efficacy for cold remedies. So, it’s up to us to have at least some sense of where to get reliable information.
Truth be told, I am an enormous fan of safe and effective nonmedication therapies for the treatment and prevention of disease. So, when time permits, I will do a quick PubMed.gov search limiting my articles to randomized trials or systematic reviews on the latest and greatest home remedy.
Probiotics have been around for a while, and I think of them as a cure in search of a disease. The Cochrane Collaboration conducted a systematic review evaluating probiotics for the prevention of upper respiratory tract infection. In this review, 13 randomized, controlled trials were included (Explore [NY]. 2015 Sep-Oct;11[5]:418-20).
Probiotics were observed to be significantly better than placebo for reducing episodes of upper respiratory tract infection, the mean duration of episodes, antibiotic prescription rates, and cold-related absences. The evidence was of moderate to low quality.
Some may wonder how an ingested probiotic helps the respiratory tract stave off or fight infection. The prevailing theory appears to be that probiotics may function by mobilizing cells from the intestine to immunomodulate respiratory mucosa.
As for what probiotic/organism to prescribe? On this issue, there is a lot of smoke and not a lot of heat.
In general, the product should be encapsulated, and the label should include the genus and species of the strains (e.g., Lactobacillus acidophilus), the number of organisms (e.g., 5 billion), storage conditions (e.g., refrigerated or room temperature), and the shelf life. Pharmacy chain house brands may be cheaper. Gummy and chewable products tend to have 92% fewer beneficial bacteria than standard formulations.
How can we ensure purity?
That is tough, because supplements like probiotics are not regulated by the Food and Drug Administration above and beyond the agency’s trying to ensure good manufacturing practices. However, companies such as LabDoor (which generates revenue through affiliate links) test and grade supplements for label accuracy and purity. Websites like this might be the best place to start.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no financial disclosures relevant to this article.

Study reveals subgroups of AYAs more likely to die of HL

patient and her father
Photo by Rhoda Baer
A new study indicates that race, insurance status, and socioeconomic status (SES) impact survival in adolescents and young adults (AYAs) with Hodgkin lymphoma (HL).
Researchers found evidence to suggest that patients diagnosed with HL between the ages of 15 and 39 are less likely to survive the disease if they are black, Hispanic, have no insurance or public health insurance, or live in a neighborhood with low SES.
Theresa H.M. Keegan, PhD, MS, of the UC Davis Comprehensive Cancer Center in Sacramento, California, and her colleagues conducted this research and reported the results in Cancer Epidemiology, Biomarkers & Prevention.
Dr Keegan and her colleagues studied data from 9353 patients in the California Cancer Registry who were between 15 and 39 years old when they were diagnosed with HL between 1988 and 2011.
The team examined the impact of race/ethnicity, neighborhood SES, and health insurance on mortality.
The researchers found that AYAs diagnosed with early stage HL were twice as likely to die if they resided in a lower SES neighborhood.
Subjects were also twice as likely to die from HL if they had public health insurance or were uninsured, regardless of whether they were diagnosed at an early stage or a late stage.
Black AYA patients were 68% more likely to die of HL than non-Hispanic white patients, whether they were diagnosed at an early stage or a late stage.
And Hispanic AYA patients diagnosed at a late stage were 58% more likely than non-Hispanic white patients to die of HL, but there was no significant disparity for Hispanic patients diagnosed at an early stage.
“Identifying and reducing barriers to recommended treatment and surveillance in these AYAs at much higher risk of mortality is essential to ameliorating these survival disparities,” Dr Keegan said.
However, she and her colleagues noted that this study had limitations. The researchers were able to identify the first course of treatment but did not have specific details on the treatment that followed the initial period.
In addition, health insurance status at the time of diagnosis was not available for patients who were diagnosed before 2001, and the researchers did not have information on changes in patients’ insurance status that may have occurred after their initial treatment. ![]()
patient and her father
Photo by Rhoda Baer
A new study indicates that race, insurance status, and socioeconomic status (SES) impact survival in adolescents and young adults (AYAs) with Hodgkin lymphoma (HL).
Researchers found evidence to suggest that patients diagnosed with HL between the ages of 15 and 39 are less likely to survive the disease if they are black, Hispanic, have no insurance or public health insurance, or live in a neighborhood with low SES.
Theresa H.M. Keegan, PhD, MS, of the UC Davis Comprehensive Cancer Center in Sacramento, California, and her colleagues conducted this research and reported the results in Cancer Epidemiology, Biomarkers & Prevention.
Dr Keegan and her colleagues studied data from 9353 patients in the California Cancer Registry who were between 15 and 39 years old when they were diagnosed with HL between 1988 and 2011.
The team examined the impact of race/ethnicity, neighborhood SES, and health insurance on mortality.
The researchers found that AYAs diagnosed with early stage HL were twice as likely to die if they resided in a lower SES neighborhood.
Subjects were also twice as likely to die from HL if they had public health insurance or were uninsured, regardless of whether they were diagnosed at an early stage or a late stage.
Black AYA patients were 68% more likely to die of HL than non-Hispanic white patients, whether they were diagnosed at an early stage or a late stage.
And Hispanic AYA patients diagnosed at a late stage were 58% more likely than non-Hispanic white patients to die of HL, but there was no significant disparity for Hispanic patients diagnosed at an early stage.
“Identifying and reducing barriers to recommended treatment and surveillance in these AYAs at much higher risk of mortality is essential to ameliorating these survival disparities,” Dr Keegan said.
However, she and her colleagues noted that this study had limitations. The researchers were able to identify the first course of treatment but did not have specific details on the treatment that followed the initial period.
In addition, health insurance status at the time of diagnosis was not available for patients who were diagnosed before 2001, and the researchers did not have information on changes in patients’ insurance status that may have occurred after their initial treatment. ![]()
patient and her father
Photo by Rhoda Baer
A new study indicates that race, insurance status, and socioeconomic status (SES) impact survival in adolescents and young adults (AYAs) with Hodgkin lymphoma (HL).
Researchers found evidence to suggest that patients diagnosed with HL between the ages of 15 and 39 are less likely to survive the disease if they are black, Hispanic, have no insurance or public health insurance, or live in a neighborhood with low SES.
Theresa H.M. Keegan, PhD, MS, of the UC Davis Comprehensive Cancer Center in Sacramento, California, and her colleagues conducted this research and reported the results in Cancer Epidemiology, Biomarkers & Prevention.
Dr Keegan and her colleagues studied data from 9353 patients in the California Cancer Registry who were between 15 and 39 years old when they were diagnosed with HL between 1988 and 2011.
The team examined the impact of race/ethnicity, neighborhood SES, and health insurance on mortality.
The researchers found that AYAs diagnosed with early stage HL were twice as likely to die if they resided in a lower SES neighborhood.
Subjects were also twice as likely to die from HL if they had public health insurance or were uninsured, regardless of whether they were diagnosed at an early stage or a late stage.
Black AYA patients were 68% more likely to die of HL than non-Hispanic white patients, whether they were diagnosed at an early stage or a late stage.
And Hispanic AYA patients diagnosed at a late stage were 58% more likely than non-Hispanic white patients to die of HL, but there was no significant disparity for Hispanic patients diagnosed at an early stage.
“Identifying and reducing barriers to recommended treatment and surveillance in these AYAs at much higher risk of mortality is essential to ameliorating these survival disparities,” Dr Keegan said.
However, she and her colleagues noted that this study had limitations. The researchers were able to identify the first course of treatment but did not have specific details on the treatment that followed the initial period.
In addition, health insurance status at the time of diagnosis was not available for patients who were diagnosed before 2001, and the researchers did not have information on changes in patients’ insurance status that may have occurred after their initial treatment. ![]()
Health Canada approves drug for multiple myeloma
Photo courtesy of Amgen
Health Canada has approved the proteasome inhibitor carfilzomib (Kyprolis) for use in combination with lenalidomide and dexamethasone to treat patients with relapsed multiple myeloma (MM) who have received 1 to 3 prior lines of therapy.
Carfilzomib, a product of Amgen Canada, is also approved for use in the US, the European Union, Argentina, Israel, Kuwait, Mexico, Thailand, and Colombia.
Health Canada’s approval is based on results of the phase 3 ASPIRE trial, which were presented at ASH 2014 and published in NEJM.
The trial enrolled 792 patients with relapsed or refractory MM who had received 1 to 3 prior lines of therapy. The patients were randomized (1:1) to receive carfilzomib plus lenalidomide and dexamethasone (KRd) or just lenalidomide and dexamethasone (Rd) for 18 cycles.
Lenalidomide and dexamethasone were continued thereafter until disease progression. There was no planned cross-over from the control arm to treatment with carfilzomib.
The study’s primary endpoint was progression-free survival. The median progression-free survival was significantly longer in the KRd arm than the Rd arm—26.3 months and 17.6 months, respectively (hazard ratio=0.69, P=0.0001).
At the time of analysis, the difference in overall survival did not reach the prespecified boundary for statistical significance.
The overall response rate was 87% in the KRd arm and 67% in the Rd arm. The median duration of response was 28.6 months and 21.2 months, respectively.
The rates of death due to adverse events (AEs) within 30 days of the last dose were similar between the treatment arms.
The most common causes of death not due to progressive disease occurring in patients in the KRd arm and the Rd arm, respectively, were cardiac disorders (3% vs 2%), infection (2% vs 3%), renal events (0% vs less than 1%), and other AEs (2% vs 3%).
Serious AEs were reported in 60% of patients in the KRd arm and 54% in the Rd arm. The most common serious AEs reported in the KRd arm and the Rd arm, respectively, were pneumonia (14% vs 11%), respiratory tract infection (4% vs 2%), pyrexia (4% vs 2%), and pulmonary embolism (3% vs 2%). ![]()
Photo courtesy of Amgen
Health Canada has approved the proteasome inhibitor carfilzomib (Kyprolis) for use in combination with lenalidomide and dexamethasone to treat patients with relapsed multiple myeloma (MM) who have received 1 to 3 prior lines of therapy.
Carfilzomib, a product of Amgen Canada, is also approved for use in the US, the European Union, Argentina, Israel, Kuwait, Mexico, Thailand, and Colombia.
Health Canada’s approval is based on results of the phase 3 ASPIRE trial, which were presented at ASH 2014 and published in NEJM.
The trial enrolled 792 patients with relapsed or refractory MM who had received 1 to 3 prior lines of therapy. The patients were randomized (1:1) to receive carfilzomib plus lenalidomide and dexamethasone (KRd) or just lenalidomide and dexamethasone (Rd) for 18 cycles.
Lenalidomide and dexamethasone were continued thereafter until disease progression. There was no planned cross-over from the control arm to treatment with carfilzomib.
The study’s primary endpoint was progression-free survival. The median progression-free survival was significantly longer in the KRd arm than the Rd arm—26.3 months and 17.6 months, respectively (hazard ratio=0.69, P=0.0001).
At the time of analysis, the difference in overall survival did not reach the prespecified boundary for statistical significance.
The overall response rate was 87% in the KRd arm and 67% in the Rd arm. The median duration of response was 28.6 months and 21.2 months, respectively.
The rates of death due to adverse events (AEs) within 30 days of the last dose were similar between the treatment arms.
The most common causes of death not due to progressive disease occurring in patients in the KRd arm and the Rd arm, respectively, were cardiac disorders (3% vs 2%), infection (2% vs 3%), renal events (0% vs less than 1%), and other AEs (2% vs 3%).
Serious AEs were reported in 60% of patients in the KRd arm and 54% in the Rd arm. The most common serious AEs reported in the KRd arm and the Rd arm, respectively, were pneumonia (14% vs 11%), respiratory tract infection (4% vs 2%), pyrexia (4% vs 2%), and pulmonary embolism (3% vs 2%). ![]()
Photo courtesy of Amgen
Health Canada has approved the proteasome inhibitor carfilzomib (Kyprolis) for use in combination with lenalidomide and dexamethasone to treat patients with relapsed multiple myeloma (MM) who have received 1 to 3 prior lines of therapy.
Carfilzomib, a product of Amgen Canada, is also approved for use in the US, the European Union, Argentina, Israel, Kuwait, Mexico, Thailand, and Colombia.
Health Canada’s approval is based on results of the phase 3 ASPIRE trial, which were presented at ASH 2014 and published in NEJM.
The trial enrolled 792 patients with relapsed or refractory MM who had received 1 to 3 prior lines of therapy. The patients were randomized (1:1) to receive carfilzomib plus lenalidomide and dexamethasone (KRd) or just lenalidomide and dexamethasone (Rd) for 18 cycles.
Lenalidomide and dexamethasone were continued thereafter until disease progression. There was no planned cross-over from the control arm to treatment with carfilzomib.
The study’s primary endpoint was progression-free survival. The median progression-free survival was significantly longer in the KRd arm than the Rd arm—26.3 months and 17.6 months, respectively (hazard ratio=0.69, P=0.0001).
At the time of analysis, the difference in overall survival did not reach the prespecified boundary for statistical significance.
The overall response rate was 87% in the KRd arm and 67% in the Rd arm. The median duration of response was 28.6 months and 21.2 months, respectively.
The rates of death due to adverse events (AEs) within 30 days of the last dose were similar between the treatment arms.
The most common causes of death not due to progressive disease occurring in patients in the KRd arm and the Rd arm, respectively, were cardiac disorders (3% vs 2%), infection (2% vs 3%), renal events (0% vs less than 1%), and other AEs (2% vs 3%).
Serious AEs were reported in 60% of patients in the KRd arm and 54% in the Rd arm. The most common serious AEs reported in the KRd arm and the Rd arm, respectively, were pneumonia (14% vs 11%), respiratory tract infection (4% vs 2%), pyrexia (4% vs 2%), and pulmonary embolism (3% vs 2%). ![]()
Incorporating cultural beliefs into cancer care

Photo by Daniel Sone
Understanding and integrating patients’ cultural beliefs into cancer treatment plans may help improve their acceptance of and adherence to treatment in multicultural settings, according to research published in the Journal of Global Oncology.
Researchers examined traditional Maya healers’ understanding of cancer and found that, although there are key differences between Maya and Western medicine perspectives, they also share many fundamental concepts.
“Maya healers understand cancer in remarkably similar ways to Western doctors,” said lead study author Mónica Berger-González, PhD, of the Institute for Environmental Decisions at ETH Zurich in Switzerland.
“Recognizing this is the first step to bridging the gap between cultures and ultimately providing better, more effective services for indigenous populations.”
Nearly half of the population in Guatemala (approximately 5.4 million people) relies on Maya medicine. Traditional healers have practiced in Guatemala for more than 2000 years, with the healing tradition passed down orally and through apprenticeship.
According to the authors, this is one of the first studies to explore the subject of Maya healers and cancer across several ethno-linguistic groups, and limited data exist on survival outcomes.
Dr Berger-González and her colleagues conducted in-depth interviews with 67 healers across various ethnic and language groups in Guatemala, exploring how its indigenous people define and treat cancer.
Of the Maya healers interviewed, 46% were illiterate. Although only 36% were able to define the word cancer, most (85%) were familiar with the term and identified malignancy as a core characteristic of the disease, explaining the concept of metastasis clearly.
The analysis also revealed that Maya healers understand the origins of cancer in ways that align closely with Western medical concepts.
When asked to identify the physical causes of cancer, 10 of 17 causes provided correlated directly with cancer risk factors as understood in Western medicine. Healers cited causes such as the consumption of harmful foods (46.3%), hereditary conditions (29.6%), and lifestyle factors such as smoking or working with toxic substances (29.6%).
One notable difference identified between the 2 perspectives is that Maya healers’ view of cancer is not limited to the physical body, but rather includes a complex imbalance of the emotions, mind, and spirit.
The Maya treatment of cancer is consequently holistic and seeks to restore that balance. This is achieved through a combination of methods—such as regulating diet, plant therapy, detoxifying baths—as well as social, psychological, and spiritual methods, the latter of which, the authors note, is harder to grasp in Western medicine.
“If healthcare professionals do not understand indigenous peoples’ conception of cancer, these patients are far less likely to accept and adhere to treatment in the public healthcare system,” Dr Berger-González said.
Many indigenous people in Guatemala do not have access to Western medicine services, cannot afford them, or prefer Maya medicine even when Western medical treatment is available. Yet Western medicine practitioners have little to no training in multicultural management or traditional indigenous medicine.
The authors offer 3 key recommendations to help address the challenges of providing care in multicultural settings:
- Adequate training of healthcare professionals on cultural and social perceptions of cancer
- Increasing evidence-based research on traditional medicine
- Establishing national regulations on integrating traditional and Western medicine—following other countries like Peru, Brazil, and Ecuador, which have successfully incorporated these aspects of care.
The authors plan to continue their transdisciplinary research with the goal of providing biomedical evidence that advances different aspects of Maya medicine. ![]()
Photo by Daniel Sone
Understanding and integrating patients’ cultural beliefs into cancer treatment plans may help improve their acceptance of and adherence to treatment in multicultural settings, according to research published in the Journal of Global Oncology.
Researchers examined traditional Maya healers’ understanding of cancer and found that, although there are key differences between Maya and Western medicine perspectives, they also share many fundamental concepts.
“Maya healers understand cancer in remarkably similar ways to Western doctors,” said lead study author Mónica Berger-González, PhD, of the Institute for Environmental Decisions at ETH Zurich in Switzerland.
“Recognizing this is the first step to bridging the gap between cultures and ultimately providing better, more effective services for indigenous populations.”
Nearly half of the population in Guatemala (approximately 5.4 million people) relies on Maya medicine. Traditional healers have practiced in Guatemala for more than 2000 years, with the healing tradition passed down orally and through apprenticeship.
According to the authors, this is one of the first studies to explore the subject of Maya healers and cancer across several ethno-linguistic groups, and limited data exist on survival outcomes.
Dr Berger-González and her colleagues conducted in-depth interviews with 67 healers across various ethnic and language groups in Guatemala, exploring how its indigenous people define and treat cancer.
Of the Maya healers interviewed, 46% were illiterate. Although only 36% were able to define the word cancer, most (85%) were familiar with the term and identified malignancy as a core characteristic of the disease, explaining the concept of metastasis clearly.
The analysis also revealed that Maya healers understand the origins of cancer in ways that align closely with Western medical concepts.
When asked to identify the physical causes of cancer, 10 of 17 causes provided correlated directly with cancer risk factors as understood in Western medicine. Healers cited causes such as the consumption of harmful foods (46.3%), hereditary conditions (29.6%), and lifestyle factors such as smoking or working with toxic substances (29.6%).
One notable difference identified between the 2 perspectives is that Maya healers’ view of cancer is not limited to the physical body, but rather includes a complex imbalance of the emotions, mind, and spirit.
The Maya treatment of cancer is consequently holistic and seeks to restore that balance. This is achieved through a combination of methods—such as regulating diet, plant therapy, detoxifying baths—as well as social, psychological, and spiritual methods, the latter of which, the authors note, is harder to grasp in Western medicine.
“If healthcare professionals do not understand indigenous peoples’ conception of cancer, these patients are far less likely to accept and adhere to treatment in the public healthcare system,” Dr Berger-González said.
Many indigenous people in Guatemala do not have access to Western medicine services, cannot afford them, or prefer Maya medicine even when Western medical treatment is available. Yet Western medicine practitioners have little to no training in multicultural management or traditional indigenous medicine.
The authors offer 3 key recommendations to help address the challenges of providing care in multicultural settings:
- Adequate training of healthcare professionals on cultural and social perceptions of cancer
- Increasing evidence-based research on traditional medicine
- Establishing national regulations on integrating traditional and Western medicine—following other countries like Peru, Brazil, and Ecuador, which have successfully incorporated these aspects of care.
The authors plan to continue their transdisciplinary research with the goal of providing biomedical evidence that advances different aspects of Maya medicine. ![]()
Photo by Daniel Sone
Understanding and integrating patients’ cultural beliefs into cancer treatment plans may help improve their acceptance of and adherence to treatment in multicultural settings, according to research published in the Journal of Global Oncology.
Researchers examined traditional Maya healers’ understanding of cancer and found that, although there are key differences between Maya and Western medicine perspectives, they also share many fundamental concepts.
“Maya healers understand cancer in remarkably similar ways to Western doctors,” said lead study author Mónica Berger-González, PhD, of the Institute for Environmental Decisions at ETH Zurich in Switzerland.
“Recognizing this is the first step to bridging the gap between cultures and ultimately providing better, more effective services for indigenous populations.”
Nearly half of the population in Guatemala (approximately 5.4 million people) relies on Maya medicine. Traditional healers have practiced in Guatemala for more than 2000 years, with the healing tradition passed down orally and through apprenticeship.
According to the authors, this is one of the first studies to explore the subject of Maya healers and cancer across several ethno-linguistic groups, and limited data exist on survival outcomes.
Dr Berger-González and her colleagues conducted in-depth interviews with 67 healers across various ethnic and language groups in Guatemala, exploring how its indigenous people define and treat cancer.
Of the Maya healers interviewed, 46% were illiterate. Although only 36% were able to define the word cancer, most (85%) were familiar with the term and identified malignancy as a core characteristic of the disease, explaining the concept of metastasis clearly.
The analysis also revealed that Maya healers understand the origins of cancer in ways that align closely with Western medical concepts.
When asked to identify the physical causes of cancer, 10 of 17 causes provided correlated directly with cancer risk factors as understood in Western medicine. Healers cited causes such as the consumption of harmful foods (46.3%), hereditary conditions (29.6%), and lifestyle factors such as smoking or working with toxic substances (29.6%).
One notable difference identified between the 2 perspectives is that Maya healers’ view of cancer is not limited to the physical body, but rather includes a complex imbalance of the emotions, mind, and spirit.
The Maya treatment of cancer is consequently holistic and seeks to restore that balance. This is achieved through a combination of methods—such as regulating diet, plant therapy, detoxifying baths—as well as social, psychological, and spiritual methods, the latter of which, the authors note, is harder to grasp in Western medicine.
“If healthcare professionals do not understand indigenous peoples’ conception of cancer, these patients are far less likely to accept and adhere to treatment in the public healthcare system,” Dr Berger-González said.
Many indigenous people in Guatemala do not have access to Western medicine services, cannot afford them, or prefer Maya medicine even when Western medical treatment is available. Yet Western medicine practitioners have little to no training in multicultural management or traditional indigenous medicine.
The authors offer 3 key recommendations to help address the challenges of providing care in multicultural settings:
- Adequate training of healthcare professionals on cultural and social perceptions of cancer
- Increasing evidence-based research on traditional medicine
- Establishing national regulations on integrating traditional and Western medicine—following other countries like Peru, Brazil, and Ecuador, which have successfully incorporated these aspects of care.
The authors plan to continue their transdisciplinary research with the goal of providing biomedical evidence that advances different aspects of Maya medicine. ![]()
Drug nets 3rd breakthrough designation from FDA

Image by Lance Liotta
The US Food and Drug Administration (FDA) has granted a third breakthrough therapy designation for the BCL-2 inhibitor venetoclax (ABT-199).
This time, the designation is for venetoclax in combination with hypomethylating agents to treat patients with treatment-naïve acute myeloid leukemia (AML) who are ineligible for standard induction therapy.
Venetoclax previously received breakthrough designation as a single agent for patients with relapsed or refractory chronic lymphocytic leukemia (CLL) and in combination with rituximab to treat patients with relapsed or refractory CLL and 17p deletion.
Breakthrough therapy designation is designed to accelerate the development and review of medicines that demonstrate early clinical evidence of a substantial improvement over current treatment options for serious diseases.
Venetoclax is currently under investigation in a phase 1/2 trial in combination with low-dose cytarabine for treatment-naïve patients with AML and in a phase 1b study in combination with decitabine or azacitidine for treatment-naïve AML patients.
A phase 2 study of single-agent venetoclax in AML has been completed. The results were presented at ASH 2014.
At that time, the trial had enrolled 32 patients, 30 of whom had relapsed or refractory disease. Patients had a median age of 71 (range, 19 to 84), and half were male.
The overall response rate was 15.5%, with 1 patient achieving a complete response (CR) and 4 patients achieving a CR with incomplete count recovery (CRi).
The researchers noted that 3 of the patients who had a CR/CRi had IDH mutations. Two of these patients also achieved minimal residual disease negativity.
The median bone marrow blast count in evaluable patients decreased 36% after treatment, and 6 patients (19%) had at least a 50% reduction in bone marrow blasts.
Common adverse events following treatment (occurring in at least 25% of patients) included nausea, diarrhea, fatigue, neutropenia, and vomiting.
Grade 3 and 4 adverse events (occurring in 3 or more patients) included febrile neutropenia, anemia, and pneumonia. No patient died as a result of treatment-related adverse events.
Venetoclax is being developed by AbbVie in partnership with Genentech and Roche. ![]()
Image by Lance Liotta
The US Food and Drug Administration (FDA) has granted a third breakthrough therapy designation for the BCL-2 inhibitor venetoclax (ABT-199).
This time, the designation is for venetoclax in combination with hypomethylating agents to treat patients with treatment-naïve acute myeloid leukemia (AML) who are ineligible for standard induction therapy.
Venetoclax previously received breakthrough designation as a single agent for patients with relapsed or refractory chronic lymphocytic leukemia (CLL) and in combination with rituximab to treat patients with relapsed or refractory CLL and 17p deletion.
Breakthrough therapy designation is designed to accelerate the development and review of medicines that demonstrate early clinical evidence of a substantial improvement over current treatment options for serious diseases.
Venetoclax is currently under investigation in a phase 1/2 trial in combination with low-dose cytarabine for treatment-naïve patients with AML and in a phase 1b study in combination with decitabine or azacitidine for treatment-naïve AML patients.
A phase 2 study of single-agent venetoclax in AML has been completed. The results were presented at ASH 2014.
At that time, the trial had enrolled 32 patients, 30 of whom had relapsed or refractory disease. Patients had a median age of 71 (range, 19 to 84), and half were male.
The overall response rate was 15.5%, with 1 patient achieving a complete response (CR) and 4 patients achieving a CR with incomplete count recovery (CRi).
The researchers noted that 3 of the patients who had a CR/CRi had IDH mutations. Two of these patients also achieved minimal residual disease negativity.
The median bone marrow blast count in evaluable patients decreased 36% after treatment, and 6 patients (19%) had at least a 50% reduction in bone marrow blasts.
Common adverse events following treatment (occurring in at least 25% of patients) included nausea, diarrhea, fatigue, neutropenia, and vomiting.
Grade 3 and 4 adverse events (occurring in 3 or more patients) included febrile neutropenia, anemia, and pneumonia. No patient died as a result of treatment-related adverse events.
Venetoclax is being developed by AbbVie in partnership with Genentech and Roche. ![]()
Image by Lance Liotta
The US Food and Drug Administration (FDA) has granted a third breakthrough therapy designation for the BCL-2 inhibitor venetoclax (ABT-199).
This time, the designation is for venetoclax in combination with hypomethylating agents to treat patients with treatment-naïve acute myeloid leukemia (AML) who are ineligible for standard induction therapy.
Venetoclax previously received breakthrough designation as a single agent for patients with relapsed or refractory chronic lymphocytic leukemia (CLL) and in combination with rituximab to treat patients with relapsed or refractory CLL and 17p deletion.
Breakthrough therapy designation is designed to accelerate the development and review of medicines that demonstrate early clinical evidence of a substantial improvement over current treatment options for serious diseases.
Venetoclax is currently under investigation in a phase 1/2 trial in combination with low-dose cytarabine for treatment-naïve patients with AML and in a phase 1b study in combination with decitabine or azacitidine for treatment-naïve AML patients.
A phase 2 study of single-agent venetoclax in AML has been completed. The results were presented at ASH 2014.
At that time, the trial had enrolled 32 patients, 30 of whom had relapsed or refractory disease. Patients had a median age of 71 (range, 19 to 84), and half were male.
The overall response rate was 15.5%, with 1 patient achieving a complete response (CR) and 4 patients achieving a CR with incomplete count recovery (CRi).
The researchers noted that 3 of the patients who had a CR/CRi had IDH mutations. Two of these patients also achieved minimal residual disease negativity.
The median bone marrow blast count in evaluable patients decreased 36% after treatment, and 6 patients (19%) had at least a 50% reduction in bone marrow blasts.
Common adverse events following treatment (occurring in at least 25% of patients) included nausea, diarrhea, fatigue, neutropenia, and vomiting.
Grade 3 and 4 adverse events (occurring in 3 or more patients) included febrile neutropenia, anemia, and pneumonia. No patient died as a result of treatment-related adverse events.
Venetoclax is being developed by AbbVie in partnership with Genentech and Roche. ![]()
Depression and Postdischarge Events
Between 10% and 40% of patients are readmitted after being discharged from the hospital,[1, 2] and as many as another 25% return to the emergency department (ED) within 30 days.[3] This creates a substantial burden on the healthcare system.[2] Various interventions have been tried to improve the quality of discharge transitions and reduce readmission rates, but results thus far have been inconsistent and generally disappointing.[4, 5, 6] Targeted delivery of interventions to those at highest risk might improve the effectiveness of these efforts and reduce costs. However, current readmission risk assessment models are only moderately predictive, suggesting the presence of unrecognized risk factors.[7, 8]
Active depression might represent a potentially modifiable independent predictor of adverse short‐term hospital outcomes that is currently underutilized. Depression occurs in 5% to 58% of hospitalized adults, depending on how cases are defined.[9, 10] Depression is often under‐recognized and undertreated in acute care clinical settings,[11] and relatively few readmission prediction models incorporate mental health related symptoms.[12]
Although several reviews have examined methods of screening for depression in hospitalized patients[9] or the effectiveness of screening in primary care,[13, 14] to our knowledge no systematic review has examined the impact of depression on short‐term prognosis after discharge from acute care. Therefore, the purpose of this systematic review was to summarize all studies that evaluated whether hospitalized medical patients with depressive symptoms are at higher risk of 30‐day all‐cause readmission or all‐cause mortality after being discharged from the hospital.
METHODS
This study followed an a priori protocol developed according to PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses) criteria.[15]
Data Sources and Search Methods
We searched the Cumulative Index to Nursing and Allied Health Literature, Ovid MEDLINE, Ovid Embase, and PsycINFO from inception to January 9, 2015, and the last 5 years of PubMed for full publications with any of the following Medical Subject Headings: depressive disorder, depression, patient readmission, interviews, psychological, inpatients, with restrictions for peer‐reviewed publication, humans, adults aged 18 years, and the English language. Search strategies were developed with a librarian (available upon request). We manually searched reference lists of all included studies and relevant review articles and contacted content experts to identify additional publications.
Eligibility Criteria and Selection of Studies
Two authors (J.L.P. and L.M.W.) independently screened full texts of all relevant articles for inclusion. Disagreements were resolved by consensus or a third reviewer (S.R.M.). We considered any original research that compared readmission or mortality after discharge for hospitalized medical patients (ie, general patients or subgroups thereof) with versus without depression identified by any validated depression measure,[16] including any study design that incorporated at least 30‐day follow‐up postdischarge. We excluded studies that examined patients hospitalized in nonacute care settings or on surgical, psychiatric, obstetric, or intensive care services. We calculated Cohen's coefficient to evaluate inter‐rater agreement on study selection.
Data Extraction
Data were abstracted by 2 authors (J.L.P. and L.M.W.). Disagreements were resolved by consensus or a third reviewer (S.R.M.). We contacted authors of all included studies to obtain missing data. If unavailable, crude data were estimated from published survival curves employing validated techniques in R (version 3.1.2; R Foundation for Statistical Computing, Vienna, Austria) and Digitizeit (
Data Synthesis and Statistical Analysis
Where possible, we calculated the pooled risk ratio (RR) with 95% confidence interval (95% CI) using a random effects models in Review Manager (RevMan) 5.3 (The Nordic Cochrane Centre, Copenhagen, Denmark). The random effects approach that we employed assumes heterogeneity (ie, underlying parameters vary between individual studies) and is distributed around a mean or population average effect, and results in more conservative (wider) confidence intervals, wherein larger cohorts (or studies with smaller standard errors) are given more weight. Heterogeneity was assessed using the I2 statistic, with values of 25%, 25% to 50%, and >50% representing low, moderate, and high heterogeneity.[19] As per the guidance of Higgins et al., we did not a priori define any degree of heterogeneity that would preclude pooling of the data; the expectation would be that heterogeneity would be substantially higher pooling observational studies rather than randomized trials.[19] Statistical significance was considered a 2‐sided P value 0.05.
Quality Assessment and Risk of Bias
We assessed study quality using the 9‐item Newcastle‐Ottawa scale with 0 to 3, 4 to 6, and 7 to 9 stars considered low, moderate, and high quality, respectively, and criteria for external and internal validity, including group selection and comparability, outcome assessment, and adequacy of follow‐up.[20] Adjusted estimates published in individual reports (or obtained directly from authors) were compared wherever possible with unadjusted estimates to assess the degree of confounding. We generated funnel plots in RevMan 5.3 and conducted Egger tests using Stata 13 (StataCorp LP, College Station, TX) to assess for publication bias.[21]
RESULTS
Study Selection
After removing duplicate publications, we identified 4066 reports and reviewed 133 reports in full text (see Supporting Figure 1 in the online version of this article). Despite our broad study inclusion criteria, we found only 35 longitudinal studies addressing this question. All 35 authors were contacted for additional outcomes data and other missing information (response rate of 34%). We had to exclude 17 studies as they did not provide 30 or 90‐day post‐discharge outcomes. Only 4 studies had published crude data for outcomes within 90 days,[22, 23, 24, 25] but after contact with authors, we received unpublished data for a further 7 studies[26, 27, 28, 29, 30, 31, 32] (including individual level data for 2 cohorts).[31, 32] We were able to estimate crude data from Kaplan‐Meier curves for another 3 studies.[33, 34, 35] Another 4 studies did not collect the outcomes we were interested in individually. These studies were included in this systematic review but are not poolable in our models: 3 authors could only provide composite endpoint data,[36, 37, 38] and 1 author provided unadjusted hazard ratios.[39] Inter‐reviewer agreement for inclusion was 80% (Cohen's = 0.60).
Characteristics of Included Studies
The 18 studies ranged in size from 58 to 1418 patients; 13 were cohort studies and 5 included secondary data from randomized control trials.[22, 27, 30, 34, 36] All studies ascertained depressive status by screening during index medical admission with either diagnostic interview or self‐report questionnaires, although a variety of scales and definitions for depression were used (Beck Depression Inventory [BDI] in 6 studies, Geriatric Depression Scale in 5 studies, Patient Health Questionnaire in another 4 studies, Medical Outcomes Study‐Depression Questionnaire in 1 study, and Center for Epidemiologic Studies Depression Scale in another study) (Table 1). Screening interviews were conducted mostly by research assistants or nurses (68%) or self‐administered (21%). Most studies examined specific medical patient subgroups (10 cardiac, 3 pulmonary, and 2 elderly). Major exclusion criteria reported were terminal illness (4 studies), unstable condition (6 studies), severe cognitive impairment (5 studies), and suicidal ideation or known depression (4 studies); 1 study enrolled patients with suspected depression (Table 1). Patient cohorts were on average older (range, 5082 years) (Table 1). Attrition rates for readmission and mortality data were low (average 1% among entire sample of studies). All studies scored at least 5 on the Newcastle‐Ottawa scale and were thus considered of at least moderate quality (see Supporting Table 1 in the online version of this article).
| Author, Date of Publication, Enrollment Period | Setting Country/Region, No. of Hospitals | No. of Inpatients, Clinical Features | Major Exclusion Criteria | Follow‐up, mo | Depression Measure (Cutoff) and Screening Method | Mean Age (SD), y | % Female | Positive Screen, No. (%) | Primary Outcome, Secondary Outcomes |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| Studies that use a scale based on DSM‐III criteria or a diagnostic interview according to DSM‐III criteria | |||||||||
| Frasure‐Smith et al.,[26] 1993, 19911992* | Canada/Quebec, 1 urban teaching | 218, AMI | Terminal noncardiac illness, unstable, not cognitive | 6 | BDI (10); mod DIS by interviewer, after transfer to medicine | 60 (range, 2488) | 22 | 68 (31), 35 (16) | All‐cause mortality |
| Frasure‐Smith et al.,[27] 1999, 19911992,* 19911994 | Canada/Quebec, 1 urban teaching, 10 urban area | 218; 78, AMI | Terminal noncardiac illness, unstable, not cognitive | 12 | BDI (10) by interviewer, after transfer to medicine | 60 (11) | 32 | 290 (32) | Cardiac mortality |
| Freedland et al.,[25] 1991, 1990 | USA/MO, 1 urban teaching | 58, CHF 75 years | Dementia, medically unstable | 3 | Mod DIS by psychiatric residents and interviewer | 78 (6) | 57 | 10 (17) | All‐cause readmission, all‐cause mortality |
| Fulop et al.,[38] 2003, 2002 | USA/NY, 1 urban teaching | 203, CHF 65 years | 1, 6 | GDS (10); SCID‐NP by interviewer, at discharge | 77 (8) | 53 | 73 (36), 44 (22) | Depression, composite PCP, ED, care visits, and readmission | |
| Lesprance et al.,[28] 2000, 19941996 | Canada/Quebec, 1 urban teaching | 430, unstable angina | Terminal noncardiac illness, not cognitive, recent CABG | 12 | BDI (10); mod DIS by interviewer, 5 days after admission | 62 (11) | 29 | 178 (41), 120 (28) | Cardiac death and MI, any death, angina readmission |
| Rumsfeld et al.,[30] 2005, 19992001 | CA, USA, UK, multiple | 634, AMI with CHF | Valvular or congenital heart failure | Up to 32 | MOS‐D (0.06) by interviewer, before discharge | 65 (11) | 28 | 143 (23) | All‐cause death, CVD death and readmission |
| Song et al.,[33] 2009, 2005 | South Korea, 2 urban teaching | 165, HF | If minor criteria for HF attributable to other medical condition | 6 | BDI (10) self‐administer or interviewer, 34 days of admin | 62 (13) | 49 | 131 (79) | HF readmission and all‐cause mortality, HF readmit |
| Papaioannou et al.,[29] 2013, 20092010 | Greece/Athens, 1 urban | 230, AECOPD | Other respiratory illness, known depressed | Monthly up to 12 | BDI‐I (19) self‐administer, first day | 71 (9) | 12 | 91 (40) | All‐cause mortality, AECOPD readmission |
| Studies that use a scale based on or validated against DSM‐IV criteria or a diagnostic interview according to DSM‐IV criteria | |||||||||
| Almagro et al.,[31] 2002, 19961997 | Spain, 1 urban teaching | 130, AECOPD | Other pulmonary disease | July 1999 | GDS‐SF (6) by interviewer, day before discharge | 72 (9) | 8 | 43 (33) | All‐cause mortality |
| Almagro et al.,[32] 2012, 20032004 | Spain, 1 urban teaching | 134, AECOPD | Other pulmonary disease | 1, 36 | GDS‐SF (6) by interviewer | 72 (10) | 5 | 55 (41) | All‐cause mortality, lung function, frailty |
| Bla et al.,[39] 2001, 2000 | Switzerland, 1 urban teaching | 401, medical 75 years | Stay 24 hours, elective/facility transfer, unstable, not cognitive | 6 | GDS‐SF (6) by interviewer, within 2 days of admission | 82 (7599) | 61 | 90 (22) | All‐cause readmission, all‐cause mortality |
| Cancino et al.,[22] 2014, 20062007,* 20082009 | USA/MA, 1 urban tertiary | 680; 738, medical | Nursing home or hospital transfer, isolated, suicidal | 1 | PHQ‐9 (5 or severity) by interviewer, on admin | 50 (14) | 51 | 561 (40) | All‐cause readmission, ED visits, PCP visits |
| Mitchell et al.,[36] 2010, 20062007* | USA/MA, 1 urban tertiary | 738, medical | Nursing home or hospital transfer, isolated, suicidal | 1, 2, 3 | PHQ‐9 (5) by interviewer, on admin | 50 (15) | 50 | 238 (32) | ED visits and all‐cause readmission |
| Covinsky et al.,[34] 1999, 19901992 | USA/OH, 1 urban teaching | 573, medical | ICU, oncology, telemetry, nursing home admissions | 36 | GDS‐SF (6) by interviewer, within 2 days of admission | 80 | 68 | 197 (34) | All‐cause mortality |
| Jiang et al.,[23] 2001, 19971998 | USA/NC, 1 urban teaching | 357 (331 DIS only), CHF | Suicidal, planned surgery, pregnant | 3, 12 | BDI (10) self‐admin; mod DIS (+BDI only) by interviewer | 63 (13) | 33 | 126 (35), 46 (14) | All‐cause mortality, all‐cause readmission |
| Kartha et al.,[24] 2007, 20022004 | USA/MA, 1 urban safety net | 144, medical recently hospitalized | Planned readmission, unable to keep PCP appointments | 3 | PHQ‐9 (algorithm) by interviewer | 55 (16) | 56 | 39 (27) | All‐cause readmission |
| Koenig and Kuchbhatla,[37] 1999, 1997 | USA/NC, 1 urban teaching | 331, medical 60 years | Stay 3 or >7 days, ICU/CCU, severe illness, nursing home transfers | 3, 6, 9, 12 | CES‐D (16) or HAM‐D (11) or DIS by psychiatrist, on or after third day | 70 (7) | 51 | 160 (48) | Depression, composite physical disability, health visits, and all‐cause readmission |
| Rollman et al.,[35] 2012, 20072009 | USA/PA, 4 urban teaching | 471, CHF, suspected depressed | Antidepressants users (excluded from PHQ‐2 group only) | Up to 12 | PHQ‐2; PHQ‐9 (5 in +PHQ‐2), by interviewer, 4 days | 66 (13) | 35 | 371 (79), 351 (74) | All‐cause mortality |
Prevalence and Recognition of Depressive Symptoms
The range of depression prevalence in hospitalized medical patients was 14% to 79%, with a median of 32% (interquartile range, 27%40%) (Table 1). In those studies that used a diagnostic interview, the prevalence tended to be lower for major depression, with a median of 17% (interquartile range, 16%22%) (Table 1). None of the included studies reported frequency of clinically recognized depression (ie, prior to screening for the study). Only 2 studies assessed the persistence of depression after discharge: 1 reported that depression persisted in 53% (by screening questionnaire) and 34% (by diagnostic interview) of patients at 30 days,[38] whereas the other reported 48% persistence at 90 days after discharge according to a combined screening method.[37]
Hospital Readmission
Overall, 8 studies provided readmission data. Among patients discharged from acute care medical wards (4 studies reporting on 5 cohorts), 395 of 2433 (16.2%) patients were readmitted within 30 days (Figure 1). Hospitalized patients with depressive symptoms were more likely to be readmitted within 30 days after discharge (20.4% vs 13.7%, RR: 1.73, 95% CI: 1.16‐2.58, P = 0.007, I2 = 55%) (Figure 1), compared to those without depression. Results were consistent for 90‐day readmissions (39.8% vs 31.0%, RR: 1.68, 95% CI: 1.13‐2.50, P = 0.01, I2 = 76%, n = 1543 patients) (see Supporting Figure 2 in the online version of this article) in 6 studies. One individual study examined readmission within 6 months after discharge, but was not poolable in this model, as it presented only hazard ratios and not raw data; however, it did report a 50% increased risk of readmission in medical inpatients aged 75 years (adjusted hazard ratio: 1.50, 95% CI: 1.03‐2.17, n = 401).[39]
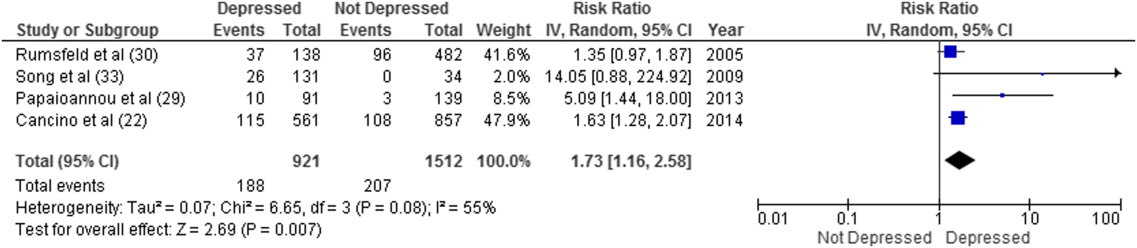
Forest plot presents results of the meta‐analysis in which the size of each data marker indicates the weight assigned to individuals studies. Abbreviations: CI, confidence interval; IV, independent variable.
Mortality After Discharge
Overall, 11 studies provided all‐cause mortality data. Among medical patients discharged from acute care in 9 studies, 69 of 3397 (2.0%) patients died within 30 days (Figure 2). Medical patients discharged with depressive symptoms were more likely to die within 30 days (2.8% vs 1.5%, RR: 2.13, 95% CI: 1.31‐3.44, P = 0.002, I2 = 0%) (Figure 2) compared to those without depression. Similar results were found for 90‐day mortality (7.7% vs 4.1%, RR: 2.01, 95% CI: 1.47‐2.76, P 0.001, I2 = 4%, n = 3784 patients) (see Supporting Figure 3 in the online version of this article) in 11 studies.
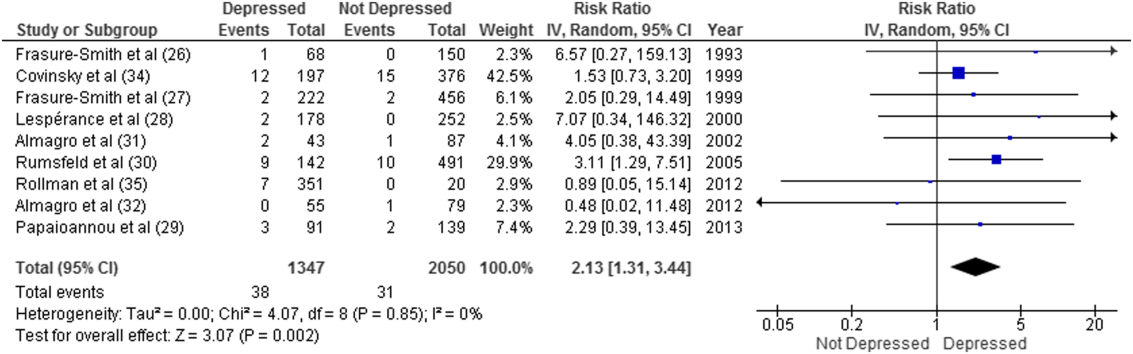
ED and PCP Visits
Four studies examined the use of ED or PCP services within 90 days of discharge, but 3 did not have extractable data for meta‐analysis. All showed increased utilization of health services for depressed compared to nondepressed patients after discharge.[22, 36, 37, 38] Depressed patients were more likely to visit the ED (adjusted incidence rate ratio: 1.73, 95% CI: 1.27‐2.36),[36] had significantly more medical encounters (eg, PCP, ED visits, hospital admissions, laboratory tests, and home care [mean 2.9 vs 2.6, P = 0.05])[38] and had a greater number of ED visits alone (27 vs 15 per 100 patients, P = 0.007)[22] within 30 days of hospital discharge compared to nondepressed patients. Similar results were found at 90 days.[36]
Sensitivity Analyses
All told, most studies reported a positive association between depression and adverse events, and this was true regardless of how much adjustment for potential confounding had been undertaken by the authors. Although all studies were qualitatively in the same direction, the magnitude of the association varied due to methodological and/or clinical heterogeneity. Sensitivity analysis revealed no overall difference in pooled risk ratios or heterogeneity between Mantel‐Haenszel fixed effects versus random effects models or with the addition of 0.5 to cells to permit inclusion of zero‐event data. There was no evidence of publication bias; funnel plots and Egger test results are available upon request. There were no statistically significant differences in the risk associated with depressive symptoms whether studies used Diagnostic and Statistical Manual of Mental Disorders (DSM)‐III or DSM‐IV criteria, whether the study samples were disease specific or unselected general medical cohorts, whether studies were of moderate or high quality, or regardless of the severity of depressive symptoms.
DISCUSSION
Summary of Evidence
We found that depression was common in medical inpatients (about one‐third of all patients) and persisted for at least 30 days in up to half of those patients after discharge. We found strong evidence of an association between depressive symptoms and poor short‐term prognosis after discharge from the hospital: a 73% increased risk of readmission and a 2‐fold risk of death within 30 days compared to patients without depressive symptoms with similar results at 90 days.
Our meta‐analysis complements a recent systematic review that found concomitant depression to be a risk factor for poor prognosis among inpatients and outpatients with acute coronary syndrome,[40] and a meta‐analysis that demonstrated an increased risk of 2‐year mortality among patients with depression after myocardial infarction.[41] To our knowledge, our study is the first to quantify the short‐term postdischarge risks across a diverse group of medical inpatients.
The potential mechanisms underlying the observed relationship between depression and adverse patient outcomes after discharge are likely multiple. We believe there are 2 main possibilities. First, the increased risk associated with depression might be due to residual confounding, even though many of these studies did adjust for extensive lists of comorbidities,[22, 24, 26, 27, 29, 30, 33, 35, 36, 39] including functional status[39] and prior health services utilization.[22, 34, 36] This could occur if other risk factors were not sufficiently adjusted for, such as unrecognized comorbidities or concomitant disability, which are often present among chronically ill patients,[42] or if depression were a marker of psychosocial risk factors such as anxiety,[43] stress or poor resiliency,[44] or low social support,[45] though a few adjusted for psychosocial factors such as social support[26] or anxiety.[35] Confounding could also occur if symptoms of acute illness inflate reports of somatic symptoms of depression on self‐report questionnaires. Recent studies on the BDI, found that scores were higher in postmyocardial infarction patients when compared to outpatient controls,[46] but with no differences between those groups in scores for the BDI‐II,[47] a version with fewer somatic symptom questions.
Second, depression may cause adverse outcomes through indirect or direct pathways. Indirect causation could occur if depression hindered self‐care behaviors such as medication adherence.[42] Depression could also act directly through pathophysiological changes. Some studies have suggested that depression is associated with metabolic abnormalities, including alterations in glucose transport[42, 48] and increased vulnerability to obesity, type 2 diabetes mellitus, and/or diabetic complications, common conditions among hospitalized patients that also adversely affect postdischarge outcomes.[40, 48]
Strengths and Limitations
This review has multiple strengths. We cast a broad search and included studies that examined a wide range of medical patient subgroups, thus increasing the generalizability of our findings. We identified a general scarcity of studies on this topic and obtained additional unpublished data for 10 of the 18 relevant studies, and our response rate of 34% is compatible with the 37% response rate reported for Cochrane reviews when seeking additional data from authors.[49] Whether examined qualitatively (vote counting of the number of studies that showed an association) or quantitatively (via formal meta‐analysis), it seems apparent that there is a clinically important association between depression and postdischarge adverse events, but given the number, quality, and heterogeneity of the studies we examined, there may be some ongoing dispute about exactly how strong this association is and the degree of bias contributed by a couple of large studies of the topic.
There are limitations to our review. First, as we did not have individual‐level patient data, we could not use metaregression to explore sources of heterogeneity (clinical or methodological) or adjust for confounding, and this likely contributes to observed differences between individual estimates. For instance, the included studies had heterogeneous screening measures and cutoffs; thus, all cases of depression in these studies might not be equivalent. Some of the included studies assessed depression early during admission where psychological distress may be greatest; others assessed symptoms closer to discharge. Most studies included patients with specific conditions like heart failure or chronic obstructive pulmonary disease rather than a wide spectrum of medical inpatients. Moreover, few studies adjusted for psychosocial risk factors such as social support, anxiety, and functional status, and only 2 studies assessed the persistence of depressive symptoms after discharge. Second, we did not explore quantitative measures of between‐study variation (eg, I2), because experts question its utility given the expected heterogeneity in meta‐analyses of observational studies.[50] Third, although the included studies were deemed to be of at least moderate quality, they could be at risk for sources of bias that may not be sufficiently appraised by the current version of the Newscastle‐Ottawa scale for observational studies. Finally, we excluded grey literature (eg, conference proceedings or technical reports) that could potentially exclude null findings, although we did contact authors in this field to identify additional unpublished data relevant to this topic.
CONCLUSIONS
We have confirmed that depressive symptoms are common in hospitalized medical patients, frequently persist after discharge, and may predict greater risk of readmission or death after discharge. Thus, depressive symptoms are an additional marker that clinicians can use to help identify patients in acute care medical settings who may be at increased risk for suboptimal transition back to the community and who may require additional resources after discharge. However, future research is required to evaluate whether treatment of individuals who screen positive for depressive symptoms can reduce 30‐day readmission rates, and we are aware of at least 1 relevant ongoing trial (
Acknowledgements
The authors thank the following individuals: Dale Storie, MLIS, Saskatchewan Information and Library Services Consortium, Regina, Saskatchewan, Canada, for assistance in the literature search; James A. Hanley, PhD, Department of Epidemiology and Biostatistics, Faculty of Medicine, McGill University, Montreal, Quebec, Canada, for guidance in data recovery methods; Nancy Frasure‐Smith, PhD, Department of Psychiatry, McGill University, Department of Psychiatry and Research Centre Hospital Centre, University of Montreal, and Montreal Heart Institute Research Centre, Montreal, Quebec, Canada; Andriana I. Papaioannou, MD, 2nd Respiratory Medicine Department, University of Athens Medical School, Athens, Greece; Konstantinos Kostikas, MD, 2nd Respiratory Medicine Department, University of Athens Medical School, Athens, Greece; and Pere Almagro, MD, Servicio de Medicina Interna, Hospital Universitario Mutua de Terrassa, Terrassa, Barcelona, Spain; as well as Philip G. Jones, MS, Saint Luke's Mid America Heart Institute, Kansas City, Missouri; for their retrieval and contribution of unpublished data.
Disclosures
Ms. Pederson affirms that the manuscript is an honest, accurate, and transparent account of the study being reported with no important omissions. All authors had full access to all of the data (including statistical reports and tables) in the study and can take responsibility for the integrity of the data and the accuracy of the data analysis. Design and conduct of the study: Ms. Pederson, Drs. Majumdar and McAlister. Data acquisition: Ms. Pederson, Ms. Warkentin. Analysis and interpretation of the data and drafting of the manuscript: Ms Pederson, Drs. Majumdar and McAlister. Review of the manuscript: all authors. Study supervision: Drs. Majumdar and McAlister. None of the contributors received compensation for their efforts. Salary support for Ms. Pederson was provided by a CRIO grant from Alberta InnovatesHealth Solutions. Drs. McAlister and Majumdar are supported by salary awards from Alberta Innovates‐Health Solutions. Dr. McAlister holds the University of Alberta/Capital Health Chair in Cardiology Outcomes Research. Dr. Majumda holds the University of Alberta Endowed Chair in Patient Health Management. The funding sources had no role in the design or conduct of the study; management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. This work is that of the authors independent of funders. The authors report no conflicts of interest.
- , , . Rehospitalizations among patients in the Medicare fee‐for‐service program. N Engl J Med. 2009;360(14):1418–1428.
- , , , , . Proportion of hospital readmissions deemed avoidable: a systematic review. CMAJ. 2011;183(7):E391–E402.
- , , , . Mental health conditions are associated with increased health care utilization among urban family medicine patients. J Am Board Fam Med. 2008;21(5):398–407.
- , , , , . Interventions to reduce 30‐day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520–528.
- . The elusive quest for quality and cost savings in the Medicare program. JAMA. 2009;301(6):668–670.
- , , , . Effects of care coordination on hospitalization, quality of care, and health care expenditures among Medicare beneficiaries—15 randomized trials. JAMA. 2009;301(6):603–618.
- , , , et al. Unplanned readmissions after hospital discharge among patients identified as being at high risk for readmission using a validated predictive algorithm. Open Med. 2011;5(2):e104–111.
- , , , et al. Risk prediction models for hospital readmission: a systematic review. JAMA. 2011;306(15):1688–1698.
- , , . Depression in older people in the general hospital: a systematic review of screening instruments. Age Ageing. 2012;41(2):148–154.
- , , , et al. Prevalence, correlates and recognition of depression among inpatients of general hospitals in Wuhan, China. Gen Hosp Psychiatry. 2010;32(3):268–275.
- , , , , , . Recognition of depression by non‐psychiatric physicians—a systematic literature review and meta‐analysis. J Gen Intern Med. 2008;23(1):25–36.
- , , , , , . Predicting the risk of unplanned readmission or death within 30 days of discharge after a heart failure hospitalization. Am Heart J. 2012;164(3):365–372.
- , , , et al. Does evidence support the American Heart Association's recommendation to screen patients for depression in cardiovascular care? An updated systematic review. PLoS One. 2013;8(1):e52654.
- , , , et al. Screening for depression: a systematic review and meta‐analysis. CMAJ Open. 2013;1(4):E159–E167.
- , , , et al. The PRISMA statement for reporting systematic reviews and meta‐analyses of studies that evaluate health care interventions: explanation and elaboration. Ann Intern Med. 2009;151(4):W65–94.
- , , , et al. Screening for depression in adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med. 2002;136(10):765–776.
- , , . Recovering the raw data behind a non‐parametric survival curve. Syst Rev. 2014;3:151.
- , , , . Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan‐Meier survival curves. BMC Med Res Methodol. 2012;12(1):9.
- . Commentary: heterogeneity in meta‐analysis should be expected and appropriately quantified. Int J Epidemiol. 2008;37(5):1158–1160.
- , , , et al. The Newcastle‐Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta‐analyses. Available at: http://www.ohri.ca/programs/clinical_epidemiology/oxford.htm. Accessed September 1, 2015.
- , , . The funnel plot. In: Rothstein HR, Sutton AJ, Borenstein M, eds. Publication Bias in Meta‐analysis: Prevention, Assessment and Adjustments. New York, NY: John Wiley 2006:73–98.
- , , , , , . Dose‐response relationship between depressive symptoms and hospital readmission. J Hosp Med. 2014;9(6):358–364.
- , , , et al. Relationship of depression to increased risk of mortality and rehospitalization in patients with congestive heart failure. Arch Intern Med. 2001;161(15):1849–1856.
- , , , et al. Depression is a risk factor for rehospitalization in medical inpatients. Prim Care Companion J Clin Psychiatry. 2007;9(4):256–262.
- , , , et al. Depression in elderly patients with congestive heart failure. J Geriatr Psychiatry. 1991;24(1):59–71.
- , , . Depression following myocardial infarction: impact on 6‐month survival. JAMA. 1993;270(15):1819–1825.
- , , , , . Gender, depression, and one‐year prognosis after myocardial infarction. Psychosom Med. 1999;61(1):26–37.
- , , , . Depression and 1‐year prognosis in unstable angina. Arch Intern Med. 2000;160(9):1354–1360.
- , , , et al. The impact of depressive symptoms on recovery and outcome of hospitalised COPD exacerbations. Eur Respir J. 2013;41(4):815–823.
- , , , et al. Depression predicts mortality and hospitalization in patients with myocardial infarction complicated by heart failure. Am Heart J. 2005;150(5):961–967.
- , , , et al. Mortality after hospitalization for COPD. Chest. 2002;121(5):1441–1448.
- , , , et al. Pseudomonas aeruginosa and mortality after hospital admission for chronic obstructive pulmonary disease. Respiration. 2012;84(1):36–43.
- , , . Depressive symptoms increase risk of rehospitalisation in heart failure patients with preserved systolic function. J Clin Nurs. 2009;18(13):1871–1877.
- , , . Depressive symptoms and 3 year mortality in older hospitalized medical patients. Ann Intern Med. 1999;130(7):563–569.
- , , , et al. A positive 2‐item patient health questionnaire depression screen among hospitalized heart failure patients is associated with elevated 12‐month mortality. J Card Fail. 2012;18(3):238–245.
- , , , et al. Post‐discharge hospital utilization among adult medical inpatients with depressive symptoms. J Hosp Med. 2010;5(7):378–384.
- , . Use of health services by medically ill depressed elderly patients after hospital discharge. Am J Geriatr Psychiatry. 1999;7(1):48–56.
- , , . Congestive heart failure and depression in older adults: clinical course and health services use 6 months after hospitalization. Psychosomatics. 2003;44(5):367–373.
- , , , . Depressive symptoms as a predictor of 6‐month outcomes and services utilization in elderly medical inpatients. Arch Intern Med. 2001;161(21):2609–2615.
- , , , et al. Depression as a risk factor for poor prognosis among patients with acute coronary syndrome: systematic review and recommendations: a scientific statement from the American Heart Association. Circulation. 2014;129(12):1350–1369.
- , , , , , . Prognostic association of depression following myocardial infarction with mortality and cardiovascular events: a meta‐analysis of 25 years of research. Gen Hosp Psychiatry. 2011;33(3):203–216.
- , , , , . Depression and cardiac disease: epidemiology, mechanisms, and diagnosis. Cardiovasc Psychiatry Neurol. 2013;2013:695925.
- , , , et al. Prognostic value of depression, anxiety, and anger in hospitalized cardiovascular disease patients for predicting adverse cardiac outcomes. Am J Cardiol. 2013;111(10):1432–1436.
- , , . The psychobiology of depression and resilience to stress: implications for prevention and treatment. Annu Rev Clin Psychol. 2005;1:255–291.
- , , , et al. Impact of social factors on risk of readmission or mortality in pneumonia and heart failure: systematic review. J Gen Intern Med. 2013;28(2):269–282.
- , , , et al. The influence of somatic symptoms on beck depression inventory scores in hospitalized postmyocardial infarction patients. Can J Psychiatry. 2012;57(12):752–758.
- , , , et al. Somatic symptom overlap in beck depression inventory‐II scores following myocardial infarction. Br J Psychiatry. 2010;197(1):61–66.
- , , , . Relationship of depression to diabetes types 1 and 2: epidemiology, biology, and treatment. Biol Psychiatry. 2003;54(3):317–329.
- , , . Searching for unpublished data for Cochrane reviews: cross sectional study. BMJ. 2013;346:f2231.
- . Comment on: heterogeneity in meta‐analysis should be expected and appropriately quantified. Int J Epidemiol. 2010;39(3):932; author reply 933.
Between 10% and 40% of patients are readmitted after being discharged from the hospital,[1, 2] and as many as another 25% return to the emergency department (ED) within 30 days.[3] This creates a substantial burden on the healthcare system.[2] Various interventions have been tried to improve the quality of discharge transitions and reduce readmission rates, but results thus far have been inconsistent and generally disappointing.[4, 5, 6] Targeted delivery of interventions to those at highest risk might improve the effectiveness of these efforts and reduce costs. However, current readmission risk assessment models are only moderately predictive, suggesting the presence of unrecognized risk factors.[7, 8]
Active depression might represent a potentially modifiable independent predictor of adverse short‐term hospital outcomes that is currently underutilized. Depression occurs in 5% to 58% of hospitalized adults, depending on how cases are defined.[9, 10] Depression is often under‐recognized and undertreated in acute care clinical settings,[11] and relatively few readmission prediction models incorporate mental health related symptoms.[12]
Although several reviews have examined methods of screening for depression in hospitalized patients[9] or the effectiveness of screening in primary care,[13, 14] to our knowledge no systematic review has examined the impact of depression on short‐term prognosis after discharge from acute care. Therefore, the purpose of this systematic review was to summarize all studies that evaluated whether hospitalized medical patients with depressive symptoms are at higher risk of 30‐day all‐cause readmission or all‐cause mortality after being discharged from the hospital.
METHODS
This study followed an a priori protocol developed according to PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses) criteria.[15]
Data Sources and Search Methods
We searched the Cumulative Index to Nursing and Allied Health Literature, Ovid MEDLINE, Ovid Embase, and PsycINFO from inception to January 9, 2015, and the last 5 years of PubMed for full publications with any of the following Medical Subject Headings: depressive disorder, depression, patient readmission, interviews, psychological, inpatients, with restrictions for peer‐reviewed publication, humans, adults aged 18 years, and the English language. Search strategies were developed with a librarian (available upon request). We manually searched reference lists of all included studies and relevant review articles and contacted content experts to identify additional publications.
Eligibility Criteria and Selection of Studies
Two authors (J.L.P. and L.M.W.) independently screened full texts of all relevant articles for inclusion. Disagreements were resolved by consensus or a third reviewer (S.R.M.). We considered any original research that compared readmission or mortality after discharge for hospitalized medical patients (ie, general patients or subgroups thereof) with versus without depression identified by any validated depression measure,[16] including any study design that incorporated at least 30‐day follow‐up postdischarge. We excluded studies that examined patients hospitalized in nonacute care settings or on surgical, psychiatric, obstetric, or intensive care services. We calculated Cohen's coefficient to evaluate inter‐rater agreement on study selection.
Data Extraction
Data were abstracted by 2 authors (J.L.P. and L.M.W.). Disagreements were resolved by consensus or a third reviewer (S.R.M.). We contacted authors of all included studies to obtain missing data. If unavailable, crude data were estimated from published survival curves employing validated techniques in R (version 3.1.2; R Foundation for Statistical Computing, Vienna, Austria) and Digitizeit (
Data Synthesis and Statistical Analysis
Where possible, we calculated the pooled risk ratio (RR) with 95% confidence interval (95% CI) using a random effects models in Review Manager (RevMan) 5.3 (The Nordic Cochrane Centre, Copenhagen, Denmark). The random effects approach that we employed assumes heterogeneity (ie, underlying parameters vary between individual studies) and is distributed around a mean or population average effect, and results in more conservative (wider) confidence intervals, wherein larger cohorts (or studies with smaller standard errors) are given more weight. Heterogeneity was assessed using the I2 statistic, with values of 25%, 25% to 50%, and >50% representing low, moderate, and high heterogeneity.[19] As per the guidance of Higgins et al., we did not a priori define any degree of heterogeneity that would preclude pooling of the data; the expectation would be that heterogeneity would be substantially higher pooling observational studies rather than randomized trials.[19] Statistical significance was considered a 2‐sided P value 0.05.
Quality Assessment and Risk of Bias
We assessed study quality using the 9‐item Newcastle‐Ottawa scale with 0 to 3, 4 to 6, and 7 to 9 stars considered low, moderate, and high quality, respectively, and criteria for external and internal validity, including group selection and comparability, outcome assessment, and adequacy of follow‐up.[20] Adjusted estimates published in individual reports (or obtained directly from authors) were compared wherever possible with unadjusted estimates to assess the degree of confounding. We generated funnel plots in RevMan 5.3 and conducted Egger tests using Stata 13 (StataCorp LP, College Station, TX) to assess for publication bias.[21]
RESULTS
Study Selection
After removing duplicate publications, we identified 4066 reports and reviewed 133 reports in full text (see Supporting Figure 1 in the online version of this article). Despite our broad study inclusion criteria, we found only 35 longitudinal studies addressing this question. All 35 authors were contacted for additional outcomes data and other missing information (response rate of 34%). We had to exclude 17 studies as they did not provide 30 or 90‐day post‐discharge outcomes. Only 4 studies had published crude data for outcomes within 90 days,[22, 23, 24, 25] but after contact with authors, we received unpublished data for a further 7 studies[26, 27, 28, 29, 30, 31, 32] (including individual level data for 2 cohorts).[31, 32] We were able to estimate crude data from Kaplan‐Meier curves for another 3 studies.[33, 34, 35] Another 4 studies did not collect the outcomes we were interested in individually. These studies were included in this systematic review but are not poolable in our models: 3 authors could only provide composite endpoint data,[36, 37, 38] and 1 author provided unadjusted hazard ratios.[39] Inter‐reviewer agreement for inclusion was 80% (Cohen's = 0.60).
Characteristics of Included Studies
The 18 studies ranged in size from 58 to 1418 patients; 13 were cohort studies and 5 included secondary data from randomized control trials.[22, 27, 30, 34, 36] All studies ascertained depressive status by screening during index medical admission with either diagnostic interview or self‐report questionnaires, although a variety of scales and definitions for depression were used (Beck Depression Inventory [BDI] in 6 studies, Geriatric Depression Scale in 5 studies, Patient Health Questionnaire in another 4 studies, Medical Outcomes Study‐Depression Questionnaire in 1 study, and Center for Epidemiologic Studies Depression Scale in another study) (Table 1). Screening interviews were conducted mostly by research assistants or nurses (68%) or self‐administered (21%). Most studies examined specific medical patient subgroups (10 cardiac, 3 pulmonary, and 2 elderly). Major exclusion criteria reported were terminal illness (4 studies), unstable condition (6 studies), severe cognitive impairment (5 studies), and suicidal ideation or known depression (4 studies); 1 study enrolled patients with suspected depression (Table 1). Patient cohorts were on average older (range, 5082 years) (Table 1). Attrition rates for readmission and mortality data were low (average 1% among entire sample of studies). All studies scored at least 5 on the Newcastle‐Ottawa scale and were thus considered of at least moderate quality (see Supporting Table 1 in the online version of this article).
| Author, Date of Publication, Enrollment Period | Setting Country/Region, No. of Hospitals | No. of Inpatients, Clinical Features | Major Exclusion Criteria | Follow‐up, mo | Depression Measure (Cutoff) and Screening Method | Mean Age (SD), y | % Female | Positive Screen, No. (%) | Primary Outcome, Secondary Outcomes |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| Studies that use a scale based on DSM‐III criteria or a diagnostic interview according to DSM‐III criteria | |||||||||
| Frasure‐Smith et al.,[26] 1993, 19911992* | Canada/Quebec, 1 urban teaching | 218, AMI | Terminal noncardiac illness, unstable, not cognitive | 6 | BDI (10); mod DIS by interviewer, after transfer to medicine | 60 (range, 2488) | 22 | 68 (31), 35 (16) | All‐cause mortality |
| Frasure‐Smith et al.,[27] 1999, 19911992,* 19911994 | Canada/Quebec, 1 urban teaching, 10 urban area | 218; 78, AMI | Terminal noncardiac illness, unstable, not cognitive | 12 | BDI (10) by interviewer, after transfer to medicine | 60 (11) | 32 | 290 (32) | Cardiac mortality |
| Freedland et al.,[25] 1991, 1990 | USA/MO, 1 urban teaching | 58, CHF 75 years | Dementia, medically unstable | 3 | Mod DIS by psychiatric residents and interviewer | 78 (6) | 57 | 10 (17) | All‐cause readmission, all‐cause mortality |
| Fulop et al.,[38] 2003, 2002 | USA/NY, 1 urban teaching | 203, CHF 65 years | 1, 6 | GDS (10); SCID‐NP by interviewer, at discharge | 77 (8) | 53 | 73 (36), 44 (22) | Depression, composite PCP, ED, care visits, and readmission | |
| Lesprance et al.,[28] 2000, 19941996 | Canada/Quebec, 1 urban teaching | 430, unstable angina | Terminal noncardiac illness, not cognitive, recent CABG | 12 | BDI (10); mod DIS by interviewer, 5 days after admission | 62 (11) | 29 | 178 (41), 120 (28) | Cardiac death and MI, any death, angina readmission |
| Rumsfeld et al.,[30] 2005, 19992001 | CA, USA, UK, multiple | 634, AMI with CHF | Valvular or congenital heart failure | Up to 32 | MOS‐D (0.06) by interviewer, before discharge | 65 (11) | 28 | 143 (23) | All‐cause death, CVD death and readmission |
| Song et al.,[33] 2009, 2005 | South Korea, 2 urban teaching | 165, HF | If minor criteria for HF attributable to other medical condition | 6 | BDI (10) self‐administer or interviewer, 34 days of admin | 62 (13) | 49 | 131 (79) | HF readmission and all‐cause mortality, HF readmit |
| Papaioannou et al.,[29] 2013, 20092010 | Greece/Athens, 1 urban | 230, AECOPD | Other respiratory illness, known depressed | Monthly up to 12 | BDI‐I (19) self‐administer, first day | 71 (9) | 12 | 91 (40) | All‐cause mortality, AECOPD readmission |
| Studies that use a scale based on or validated against DSM‐IV criteria or a diagnostic interview according to DSM‐IV criteria | |||||||||
| Almagro et al.,[31] 2002, 19961997 | Spain, 1 urban teaching | 130, AECOPD | Other pulmonary disease | July 1999 | GDS‐SF (6) by interviewer, day before discharge | 72 (9) | 8 | 43 (33) | All‐cause mortality |
| Almagro et al.,[32] 2012, 20032004 | Spain, 1 urban teaching | 134, AECOPD | Other pulmonary disease | 1, 36 | GDS‐SF (6) by interviewer | 72 (10) | 5 | 55 (41) | All‐cause mortality, lung function, frailty |
| Bla et al.,[39] 2001, 2000 | Switzerland, 1 urban teaching | 401, medical 75 years | Stay 24 hours, elective/facility transfer, unstable, not cognitive | 6 | GDS‐SF (6) by interviewer, within 2 days of admission | 82 (7599) | 61 | 90 (22) | All‐cause readmission, all‐cause mortality |
| Cancino et al.,[22] 2014, 20062007,* 20082009 | USA/MA, 1 urban tertiary | 680; 738, medical | Nursing home or hospital transfer, isolated, suicidal | 1 | PHQ‐9 (5 or severity) by interviewer, on admin | 50 (14) | 51 | 561 (40) | All‐cause readmission, ED visits, PCP visits |
| Mitchell et al.,[36] 2010, 20062007* | USA/MA, 1 urban tertiary | 738, medical | Nursing home or hospital transfer, isolated, suicidal | 1, 2, 3 | PHQ‐9 (5) by interviewer, on admin | 50 (15) | 50 | 238 (32) | ED visits and all‐cause readmission |
| Covinsky et al.,[34] 1999, 19901992 | USA/OH, 1 urban teaching | 573, medical | ICU, oncology, telemetry, nursing home admissions | 36 | GDS‐SF (6) by interviewer, within 2 days of admission | 80 | 68 | 197 (34) | All‐cause mortality |
| Jiang et al.,[23] 2001, 19971998 | USA/NC, 1 urban teaching | 357 (331 DIS only), CHF | Suicidal, planned surgery, pregnant | 3, 12 | BDI (10) self‐admin; mod DIS (+BDI only) by interviewer | 63 (13) | 33 | 126 (35), 46 (14) | All‐cause mortality, all‐cause readmission |
| Kartha et al.,[24] 2007, 20022004 | USA/MA, 1 urban safety net | 144, medical recently hospitalized | Planned readmission, unable to keep PCP appointments | 3 | PHQ‐9 (algorithm) by interviewer | 55 (16) | 56 | 39 (27) | All‐cause readmission |
| Koenig and Kuchbhatla,[37] 1999, 1997 | USA/NC, 1 urban teaching | 331, medical 60 years | Stay 3 or >7 days, ICU/CCU, severe illness, nursing home transfers | 3, 6, 9, 12 | CES‐D (16) or HAM‐D (11) or DIS by psychiatrist, on or after third day | 70 (7) | 51 | 160 (48) | Depression, composite physical disability, health visits, and all‐cause readmission |
| Rollman et al.,[35] 2012, 20072009 | USA/PA, 4 urban teaching | 471, CHF, suspected depressed | Antidepressants users (excluded from PHQ‐2 group only) | Up to 12 | PHQ‐2; PHQ‐9 (5 in +PHQ‐2), by interviewer, 4 days | 66 (13) | 35 | 371 (79), 351 (74) | All‐cause mortality |
Prevalence and Recognition of Depressive Symptoms
The range of depression prevalence in hospitalized medical patients was 14% to 79%, with a median of 32% (interquartile range, 27%40%) (Table 1). In those studies that used a diagnostic interview, the prevalence tended to be lower for major depression, with a median of 17% (interquartile range, 16%22%) (Table 1). None of the included studies reported frequency of clinically recognized depression (ie, prior to screening for the study). Only 2 studies assessed the persistence of depression after discharge: 1 reported that depression persisted in 53% (by screening questionnaire) and 34% (by diagnostic interview) of patients at 30 days,[38] whereas the other reported 48% persistence at 90 days after discharge according to a combined screening method.[37]
Hospital Readmission
Overall, 8 studies provided readmission data. Among patients discharged from acute care medical wards (4 studies reporting on 5 cohorts), 395 of 2433 (16.2%) patients were readmitted within 30 days (Figure 1). Hospitalized patients with depressive symptoms were more likely to be readmitted within 30 days after discharge (20.4% vs 13.7%, RR: 1.73, 95% CI: 1.16‐2.58, P = 0.007, I2 = 55%) (Figure 1), compared to those without depression. Results were consistent for 90‐day readmissions (39.8% vs 31.0%, RR: 1.68, 95% CI: 1.13‐2.50, P = 0.01, I2 = 76%, n = 1543 patients) (see Supporting Figure 2 in the online version of this article) in 6 studies. One individual study examined readmission within 6 months after discharge, but was not poolable in this model, as it presented only hazard ratios and not raw data; however, it did report a 50% increased risk of readmission in medical inpatients aged 75 years (adjusted hazard ratio: 1.50, 95% CI: 1.03‐2.17, n = 401).[39]
Forest plot presents results of the meta‐analysis in which the size of each data marker indicates the weight assigned to individuals studies. Abbreviations: CI, confidence interval; IV, independent variable.
Mortality After Discharge
Overall, 11 studies provided all‐cause mortality data. Among medical patients discharged from acute care in 9 studies, 69 of 3397 (2.0%) patients died within 30 days (Figure 2). Medical patients discharged with depressive symptoms were more likely to die within 30 days (2.8% vs 1.5%, RR: 2.13, 95% CI: 1.31‐3.44, P = 0.002, I2 = 0%) (Figure 2) compared to those without depression. Similar results were found for 90‐day mortality (7.7% vs 4.1%, RR: 2.01, 95% CI: 1.47‐2.76, P 0.001, I2 = 4%, n = 3784 patients) (see Supporting Figure 3 in the online version of this article) in 11 studies.
ED and PCP Visits
Four studies examined the use of ED or PCP services within 90 days of discharge, but 3 did not have extractable data for meta‐analysis. All showed increased utilization of health services for depressed compared to nondepressed patients after discharge.[22, 36, 37, 38] Depressed patients were more likely to visit the ED (adjusted incidence rate ratio: 1.73, 95% CI: 1.27‐2.36),[36] had significantly more medical encounters (eg, PCP, ED visits, hospital admissions, laboratory tests, and home care [mean 2.9 vs 2.6, P = 0.05])[38] and had a greater number of ED visits alone (27 vs 15 per 100 patients, P = 0.007)[22] within 30 days of hospital discharge compared to nondepressed patients. Similar results were found at 90 days.[36]
Sensitivity Analyses
All told, most studies reported a positive association between depression and adverse events, and this was true regardless of how much adjustment for potential confounding had been undertaken by the authors. Although all studies were qualitatively in the same direction, the magnitude of the association varied due to methodological and/or clinical heterogeneity. Sensitivity analysis revealed no overall difference in pooled risk ratios or heterogeneity between Mantel‐Haenszel fixed effects versus random effects models or with the addition of 0.5 to cells to permit inclusion of zero‐event data. There was no evidence of publication bias; funnel plots and Egger test results are available upon request. There were no statistically significant differences in the risk associated with depressive symptoms whether studies used Diagnostic and Statistical Manual of Mental Disorders (DSM)‐III or DSM‐IV criteria, whether the study samples were disease specific or unselected general medical cohorts, whether studies were of moderate or high quality, or regardless of the severity of depressive symptoms.
DISCUSSION
Summary of Evidence
We found that depression was common in medical inpatients (about one‐third of all patients) and persisted for at least 30 days in up to half of those patients after discharge. We found strong evidence of an association between depressive symptoms and poor short‐term prognosis after discharge from the hospital: a 73% increased risk of readmission and a 2‐fold risk of death within 30 days compared to patients without depressive symptoms with similar results at 90 days.
Our meta‐analysis complements a recent systematic review that found concomitant depression to be a risk factor for poor prognosis among inpatients and outpatients with acute coronary syndrome,[40] and a meta‐analysis that demonstrated an increased risk of 2‐year mortality among patients with depression after myocardial infarction.[41] To our knowledge, our study is the first to quantify the short‐term postdischarge risks across a diverse group of medical inpatients.
The potential mechanisms underlying the observed relationship between depression and adverse patient outcomes after discharge are likely multiple. We believe there are 2 main possibilities. First, the increased risk associated with depression might be due to residual confounding, even though many of these studies did adjust for extensive lists of comorbidities,[22, 24, 26, 27, 29, 30, 33, 35, 36, 39] including functional status[39] and prior health services utilization.[22, 34, 36] This could occur if other risk factors were not sufficiently adjusted for, such as unrecognized comorbidities or concomitant disability, which are often present among chronically ill patients,[42] or if depression were a marker of psychosocial risk factors such as anxiety,[43] stress or poor resiliency,[44] or low social support,[45] though a few adjusted for psychosocial factors such as social support[26] or anxiety.[35] Confounding could also occur if symptoms of acute illness inflate reports of somatic symptoms of depression on self‐report questionnaires. Recent studies on the BDI, found that scores were higher in postmyocardial infarction patients when compared to outpatient controls,[46] but with no differences between those groups in scores for the BDI‐II,[47] a version with fewer somatic symptom questions.
Second, depression may cause adverse outcomes through indirect or direct pathways. Indirect causation could occur if depression hindered self‐care behaviors such as medication adherence.[42] Depression could also act directly through pathophysiological changes. Some studies have suggested that depression is associated with metabolic abnormalities, including alterations in glucose transport[42, 48] and increased vulnerability to obesity, type 2 diabetes mellitus, and/or diabetic complications, common conditions among hospitalized patients that also adversely affect postdischarge outcomes.[40, 48]
Strengths and Limitations
This review has multiple strengths. We cast a broad search and included studies that examined a wide range of medical patient subgroups, thus increasing the generalizability of our findings. We identified a general scarcity of studies on this topic and obtained additional unpublished data for 10 of the 18 relevant studies, and our response rate of 34% is compatible with the 37% response rate reported for Cochrane reviews when seeking additional data from authors.[49] Whether examined qualitatively (vote counting of the number of studies that showed an association) or quantitatively (via formal meta‐analysis), it seems apparent that there is a clinically important association between depression and postdischarge adverse events, but given the number, quality, and heterogeneity of the studies we examined, there may be some ongoing dispute about exactly how strong this association is and the degree of bias contributed by a couple of large studies of the topic.
There are limitations to our review. First, as we did not have individual‐level patient data, we could not use metaregression to explore sources of heterogeneity (clinical or methodological) or adjust for confounding, and this likely contributes to observed differences between individual estimates. For instance, the included studies had heterogeneous screening measures and cutoffs; thus, all cases of depression in these studies might not be equivalent. Some of the included studies assessed depression early during admission where psychological distress may be greatest; others assessed symptoms closer to discharge. Most studies included patients with specific conditions like heart failure or chronic obstructive pulmonary disease rather than a wide spectrum of medical inpatients. Moreover, few studies adjusted for psychosocial risk factors such as social support, anxiety, and functional status, and only 2 studies assessed the persistence of depressive symptoms after discharge. Second, we did not explore quantitative measures of between‐study variation (eg, I2), because experts question its utility given the expected heterogeneity in meta‐analyses of observational studies.[50] Third, although the included studies were deemed to be of at least moderate quality, they could be at risk for sources of bias that may not be sufficiently appraised by the current version of the Newscastle‐Ottawa scale for observational studies. Finally, we excluded grey literature (eg, conference proceedings or technical reports) that could potentially exclude null findings, although we did contact authors in this field to identify additional unpublished data relevant to this topic.
CONCLUSIONS
We have confirmed that depressive symptoms are common in hospitalized medical patients, frequently persist after discharge, and may predict greater risk of readmission or death after discharge. Thus, depressive symptoms are an additional marker that clinicians can use to help identify patients in acute care medical settings who may be at increased risk for suboptimal transition back to the community and who may require additional resources after discharge. However, future research is required to evaluate whether treatment of individuals who screen positive for depressive symptoms can reduce 30‐day readmission rates, and we are aware of at least 1 relevant ongoing trial (
Acknowledgements
The authors thank the following individuals: Dale Storie, MLIS, Saskatchewan Information and Library Services Consortium, Regina, Saskatchewan, Canada, for assistance in the literature search; James A. Hanley, PhD, Department of Epidemiology and Biostatistics, Faculty of Medicine, McGill University, Montreal, Quebec, Canada, for guidance in data recovery methods; Nancy Frasure‐Smith, PhD, Department of Psychiatry, McGill University, Department of Psychiatry and Research Centre Hospital Centre, University of Montreal, and Montreal Heart Institute Research Centre, Montreal, Quebec, Canada; Andriana I. Papaioannou, MD, 2nd Respiratory Medicine Department, University of Athens Medical School, Athens, Greece; Konstantinos Kostikas, MD, 2nd Respiratory Medicine Department, University of Athens Medical School, Athens, Greece; and Pere Almagro, MD, Servicio de Medicina Interna, Hospital Universitario Mutua de Terrassa, Terrassa, Barcelona, Spain; as well as Philip G. Jones, MS, Saint Luke's Mid America Heart Institute, Kansas City, Missouri; for their retrieval and contribution of unpublished data.
Disclosures
Ms. Pederson affirms that the manuscript is an honest, accurate, and transparent account of the study being reported with no important omissions. All authors had full access to all of the data (including statistical reports and tables) in the study and can take responsibility for the integrity of the data and the accuracy of the data analysis. Design and conduct of the study: Ms. Pederson, Drs. Majumdar and McAlister. Data acquisition: Ms. Pederson, Ms. Warkentin. Analysis and interpretation of the data and drafting of the manuscript: Ms Pederson, Drs. Majumdar and McAlister. Review of the manuscript: all authors. Study supervision: Drs. Majumdar and McAlister. None of the contributors received compensation for their efforts. Salary support for Ms. Pederson was provided by a CRIO grant from Alberta InnovatesHealth Solutions. Drs. McAlister and Majumdar are supported by salary awards from Alberta Innovates‐Health Solutions. Dr. McAlister holds the University of Alberta/Capital Health Chair in Cardiology Outcomes Research. Dr. Majumda holds the University of Alberta Endowed Chair in Patient Health Management. The funding sources had no role in the design or conduct of the study; management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. This work is that of the authors independent of funders. The authors report no conflicts of interest.
Between 10% and 40% of patients are readmitted after being discharged from the hospital,[1, 2] and as many as another 25% return to the emergency department (ED) within 30 days.[3] This creates a substantial burden on the healthcare system.[2] Various interventions have been tried to improve the quality of discharge transitions and reduce readmission rates, but results thus far have been inconsistent and generally disappointing.[4, 5, 6] Targeted delivery of interventions to those at highest risk might improve the effectiveness of these efforts and reduce costs. However, current readmission risk assessment models are only moderately predictive, suggesting the presence of unrecognized risk factors.[7, 8]
Active depression might represent a potentially modifiable independent predictor of adverse short‐term hospital outcomes that is currently underutilized. Depression occurs in 5% to 58% of hospitalized adults, depending on how cases are defined.[9, 10] Depression is often under‐recognized and undertreated in acute care clinical settings,[11] and relatively few readmission prediction models incorporate mental health related symptoms.[12]
Although several reviews have examined methods of screening for depression in hospitalized patients[9] or the effectiveness of screening in primary care,[13, 14] to our knowledge no systematic review has examined the impact of depression on short‐term prognosis after discharge from acute care. Therefore, the purpose of this systematic review was to summarize all studies that evaluated whether hospitalized medical patients with depressive symptoms are at higher risk of 30‐day all‐cause readmission or all‐cause mortality after being discharged from the hospital.
METHODS
This study followed an a priori protocol developed according to PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses) criteria.[15]
Data Sources and Search Methods
We searched the Cumulative Index to Nursing and Allied Health Literature, Ovid MEDLINE, Ovid Embase, and PsycINFO from inception to January 9, 2015, and the last 5 years of PubMed for full publications with any of the following Medical Subject Headings: depressive disorder, depression, patient readmission, interviews, psychological, inpatients, with restrictions for peer‐reviewed publication, humans, adults aged 18 years, and the English language. Search strategies were developed with a librarian (available upon request). We manually searched reference lists of all included studies and relevant review articles and contacted content experts to identify additional publications.
Eligibility Criteria and Selection of Studies
Two authors (J.L.P. and L.M.W.) independently screened full texts of all relevant articles for inclusion. Disagreements were resolved by consensus or a third reviewer (S.R.M.). We considered any original research that compared readmission or mortality after discharge for hospitalized medical patients (ie, general patients or subgroups thereof) with versus without depression identified by any validated depression measure,[16] including any study design that incorporated at least 30‐day follow‐up postdischarge. We excluded studies that examined patients hospitalized in nonacute care settings or on surgical, psychiatric, obstetric, or intensive care services. We calculated Cohen's coefficient to evaluate inter‐rater agreement on study selection.
Data Extraction
Data were abstracted by 2 authors (J.L.P. and L.M.W.). Disagreements were resolved by consensus or a third reviewer (S.R.M.). We contacted authors of all included studies to obtain missing data. If unavailable, crude data were estimated from published survival curves employing validated techniques in R (version 3.1.2; R Foundation for Statistical Computing, Vienna, Austria) and Digitizeit (
Data Synthesis and Statistical Analysis
Where possible, we calculated the pooled risk ratio (RR) with 95% confidence interval (95% CI) using a random effects models in Review Manager (RevMan) 5.3 (The Nordic Cochrane Centre, Copenhagen, Denmark). The random effects approach that we employed assumes heterogeneity (ie, underlying parameters vary between individual studies) and is distributed around a mean or population average effect, and results in more conservative (wider) confidence intervals, wherein larger cohorts (or studies with smaller standard errors) are given more weight. Heterogeneity was assessed using the I2 statistic, with values of 25%, 25% to 50%, and >50% representing low, moderate, and high heterogeneity.[19] As per the guidance of Higgins et al., we did not a priori define any degree of heterogeneity that would preclude pooling of the data; the expectation would be that heterogeneity would be substantially higher pooling observational studies rather than randomized trials.[19] Statistical significance was considered a 2‐sided P value 0.05.
Quality Assessment and Risk of Bias
We assessed study quality using the 9‐item Newcastle‐Ottawa scale with 0 to 3, 4 to 6, and 7 to 9 stars considered low, moderate, and high quality, respectively, and criteria for external and internal validity, including group selection and comparability, outcome assessment, and adequacy of follow‐up.[20] Adjusted estimates published in individual reports (or obtained directly from authors) were compared wherever possible with unadjusted estimates to assess the degree of confounding. We generated funnel plots in RevMan 5.3 and conducted Egger tests using Stata 13 (StataCorp LP, College Station, TX) to assess for publication bias.[21]
RESULTS
Study Selection
After removing duplicate publications, we identified 4066 reports and reviewed 133 reports in full text (see Supporting Figure 1 in the online version of this article). Despite our broad study inclusion criteria, we found only 35 longitudinal studies addressing this question. All 35 authors were contacted for additional outcomes data and other missing information (response rate of 34%). We had to exclude 17 studies as they did not provide 30 or 90‐day post‐discharge outcomes. Only 4 studies had published crude data for outcomes within 90 days,[22, 23, 24, 25] but after contact with authors, we received unpublished data for a further 7 studies[26, 27, 28, 29, 30, 31, 32] (including individual level data for 2 cohorts).[31, 32] We were able to estimate crude data from Kaplan‐Meier curves for another 3 studies.[33, 34, 35] Another 4 studies did not collect the outcomes we were interested in individually. These studies were included in this systematic review but are not poolable in our models: 3 authors could only provide composite endpoint data,[36, 37, 38] and 1 author provided unadjusted hazard ratios.[39] Inter‐reviewer agreement for inclusion was 80% (Cohen's = 0.60).
Characteristics of Included Studies
The 18 studies ranged in size from 58 to 1418 patients; 13 were cohort studies and 5 included secondary data from randomized control trials.[22, 27, 30, 34, 36] All studies ascertained depressive status by screening during index medical admission with either diagnostic interview or self‐report questionnaires, although a variety of scales and definitions for depression were used (Beck Depression Inventory [BDI] in 6 studies, Geriatric Depression Scale in 5 studies, Patient Health Questionnaire in another 4 studies, Medical Outcomes Study‐Depression Questionnaire in 1 study, and Center for Epidemiologic Studies Depression Scale in another study) (Table 1). Screening interviews were conducted mostly by research assistants or nurses (68%) or self‐administered (21%). Most studies examined specific medical patient subgroups (10 cardiac, 3 pulmonary, and 2 elderly). Major exclusion criteria reported were terminal illness (4 studies), unstable condition (6 studies), severe cognitive impairment (5 studies), and suicidal ideation or known depression (4 studies); 1 study enrolled patients with suspected depression (Table 1). Patient cohorts were on average older (range, 5082 years) (Table 1). Attrition rates for readmission and mortality data were low (average 1% among entire sample of studies). All studies scored at least 5 on the Newcastle‐Ottawa scale and were thus considered of at least moderate quality (see Supporting Table 1 in the online version of this article).
| Author, Date of Publication, Enrollment Period | Setting Country/Region, No. of Hospitals | No. of Inpatients, Clinical Features | Major Exclusion Criteria | Follow‐up, mo | Depression Measure (Cutoff) and Screening Method | Mean Age (SD), y | % Female | Positive Screen, No. (%) | Primary Outcome, Secondary Outcomes |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| Studies that use a scale based on DSM‐III criteria or a diagnostic interview according to DSM‐III criteria | |||||||||
| Frasure‐Smith et al.,[26] 1993, 19911992* | Canada/Quebec, 1 urban teaching | 218, AMI | Terminal noncardiac illness, unstable, not cognitive | 6 | BDI (10); mod DIS by interviewer, after transfer to medicine | 60 (range, 2488) | 22 | 68 (31), 35 (16) | All‐cause mortality |
| Frasure‐Smith et al.,[27] 1999, 19911992,* 19911994 | Canada/Quebec, 1 urban teaching, 10 urban area | 218; 78, AMI | Terminal noncardiac illness, unstable, not cognitive | 12 | BDI (10) by interviewer, after transfer to medicine | 60 (11) | 32 | 290 (32) | Cardiac mortality |
| Freedland et al.,[25] 1991, 1990 | USA/MO, 1 urban teaching | 58, CHF 75 years | Dementia, medically unstable | 3 | Mod DIS by psychiatric residents and interviewer | 78 (6) | 57 | 10 (17) | All‐cause readmission, all‐cause mortality |
| Fulop et al.,[38] 2003, 2002 | USA/NY, 1 urban teaching | 203, CHF 65 years | 1, 6 | GDS (10); SCID‐NP by interviewer, at discharge | 77 (8) | 53 | 73 (36), 44 (22) | Depression, composite PCP, ED, care visits, and readmission | |
| Lesprance et al.,[28] 2000, 19941996 | Canada/Quebec, 1 urban teaching | 430, unstable angina | Terminal noncardiac illness, not cognitive, recent CABG | 12 | BDI (10); mod DIS by interviewer, 5 days after admission | 62 (11) | 29 | 178 (41), 120 (28) | Cardiac death and MI, any death, angina readmission |
| Rumsfeld et al.,[30] 2005, 19992001 | CA, USA, UK, multiple | 634, AMI with CHF | Valvular or congenital heart failure | Up to 32 | MOS‐D (0.06) by interviewer, before discharge | 65 (11) | 28 | 143 (23) | All‐cause death, CVD death and readmission |
| Song et al.,[33] 2009, 2005 | South Korea, 2 urban teaching | 165, HF | If minor criteria for HF attributable to other medical condition | 6 | BDI (10) self‐administer or interviewer, 34 days of admin | 62 (13) | 49 | 131 (79) | HF readmission and all‐cause mortality, HF readmit |
| Papaioannou et al.,[29] 2013, 20092010 | Greece/Athens, 1 urban | 230, AECOPD | Other respiratory illness, known depressed | Monthly up to 12 | BDI‐I (19) self‐administer, first day | 71 (9) | 12 | 91 (40) | All‐cause mortality, AECOPD readmission |
| Studies that use a scale based on or validated against DSM‐IV criteria or a diagnostic interview according to DSM‐IV criteria | |||||||||
| Almagro et al.,[31] 2002, 19961997 | Spain, 1 urban teaching | 130, AECOPD | Other pulmonary disease | July 1999 | GDS‐SF (6) by interviewer, day before discharge | 72 (9) | 8 | 43 (33) | All‐cause mortality |
| Almagro et al.,[32] 2012, 20032004 | Spain, 1 urban teaching | 134, AECOPD | Other pulmonary disease | 1, 36 | GDS‐SF (6) by interviewer | 72 (10) | 5 | 55 (41) | All‐cause mortality, lung function, frailty |
| Bla et al.,[39] 2001, 2000 | Switzerland, 1 urban teaching | 401, medical 75 years | Stay 24 hours, elective/facility transfer, unstable, not cognitive | 6 | GDS‐SF (6) by interviewer, within 2 days of admission | 82 (7599) | 61 | 90 (22) | All‐cause readmission, all‐cause mortality |
| Cancino et al.,[22] 2014, 20062007,* 20082009 | USA/MA, 1 urban tertiary | 680; 738, medical | Nursing home or hospital transfer, isolated, suicidal | 1 | PHQ‐9 (5 or severity) by interviewer, on admin | 50 (14) | 51 | 561 (40) | All‐cause readmission, ED visits, PCP visits |
| Mitchell et al.,[36] 2010, 20062007* | USA/MA, 1 urban tertiary | 738, medical | Nursing home or hospital transfer, isolated, suicidal | 1, 2, 3 | PHQ‐9 (5) by interviewer, on admin | 50 (15) | 50 | 238 (32) | ED visits and all‐cause readmission |
| Covinsky et al.,[34] 1999, 19901992 | USA/OH, 1 urban teaching | 573, medical | ICU, oncology, telemetry, nursing home admissions | 36 | GDS‐SF (6) by interviewer, within 2 days of admission | 80 | 68 | 197 (34) | All‐cause mortality |
| Jiang et al.,[23] 2001, 19971998 | USA/NC, 1 urban teaching | 357 (331 DIS only), CHF | Suicidal, planned surgery, pregnant | 3, 12 | BDI (10) self‐admin; mod DIS (+BDI only) by interviewer | 63 (13) | 33 | 126 (35), 46 (14) | All‐cause mortality, all‐cause readmission |
| Kartha et al.,[24] 2007, 20022004 | USA/MA, 1 urban safety net | 144, medical recently hospitalized | Planned readmission, unable to keep PCP appointments | 3 | PHQ‐9 (algorithm) by interviewer | 55 (16) | 56 | 39 (27) | All‐cause readmission |
| Koenig and Kuchbhatla,[37] 1999, 1997 | USA/NC, 1 urban teaching | 331, medical 60 years | Stay 3 or >7 days, ICU/CCU, severe illness, nursing home transfers | 3, 6, 9, 12 | CES‐D (16) or HAM‐D (11) or DIS by psychiatrist, on or after third day | 70 (7) | 51 | 160 (48) | Depression, composite physical disability, health visits, and all‐cause readmission |
| Rollman et al.,[35] 2012, 20072009 | USA/PA, 4 urban teaching | 471, CHF, suspected depressed | Antidepressants users (excluded from PHQ‐2 group only) | Up to 12 | PHQ‐2; PHQ‐9 (5 in +PHQ‐2), by interviewer, 4 days | 66 (13) | 35 | 371 (79), 351 (74) | All‐cause mortality |
Prevalence and Recognition of Depressive Symptoms
The range of depression prevalence in hospitalized medical patients was 14% to 79%, with a median of 32% (interquartile range, 27%40%) (Table 1). In those studies that used a diagnostic interview, the prevalence tended to be lower for major depression, with a median of 17% (interquartile range, 16%22%) (Table 1). None of the included studies reported frequency of clinically recognized depression (ie, prior to screening for the study). Only 2 studies assessed the persistence of depression after discharge: 1 reported that depression persisted in 53% (by screening questionnaire) and 34% (by diagnostic interview) of patients at 30 days,[38] whereas the other reported 48% persistence at 90 days after discharge according to a combined screening method.[37]
Hospital Readmission
Overall, 8 studies provided readmission data. Among patients discharged from acute care medical wards (4 studies reporting on 5 cohorts), 395 of 2433 (16.2%) patients were readmitted within 30 days (Figure 1). Hospitalized patients with depressive symptoms were more likely to be readmitted within 30 days after discharge (20.4% vs 13.7%, RR: 1.73, 95% CI: 1.16‐2.58, P = 0.007, I2 = 55%) (Figure 1), compared to those without depression. Results were consistent for 90‐day readmissions (39.8% vs 31.0%, RR: 1.68, 95% CI: 1.13‐2.50, P = 0.01, I2 = 76%, n = 1543 patients) (see Supporting Figure 2 in the online version of this article) in 6 studies. One individual study examined readmission within 6 months after discharge, but was not poolable in this model, as it presented only hazard ratios and not raw data; however, it did report a 50% increased risk of readmission in medical inpatients aged 75 years (adjusted hazard ratio: 1.50, 95% CI: 1.03‐2.17, n = 401).[39]
Forest plot presents results of the meta‐analysis in which the size of each data marker indicates the weight assigned to individuals studies. Abbreviations: CI, confidence interval; IV, independent variable.
Mortality After Discharge
Overall, 11 studies provided all‐cause mortality data. Among medical patients discharged from acute care in 9 studies, 69 of 3397 (2.0%) patients died within 30 days (Figure 2). Medical patients discharged with depressive symptoms were more likely to die within 30 days (2.8% vs 1.5%, RR: 2.13, 95% CI: 1.31‐3.44, P = 0.002, I2 = 0%) (Figure 2) compared to those without depression. Similar results were found for 90‐day mortality (7.7% vs 4.1%, RR: 2.01, 95% CI: 1.47‐2.76, P 0.001, I2 = 4%, n = 3784 patients) (see Supporting Figure 3 in the online version of this article) in 11 studies.
ED and PCP Visits
Four studies examined the use of ED or PCP services within 90 days of discharge, but 3 did not have extractable data for meta‐analysis. All showed increased utilization of health services for depressed compared to nondepressed patients after discharge.[22, 36, 37, 38] Depressed patients were more likely to visit the ED (adjusted incidence rate ratio: 1.73, 95% CI: 1.27‐2.36),[36] had significantly more medical encounters (eg, PCP, ED visits, hospital admissions, laboratory tests, and home care [mean 2.9 vs 2.6, P = 0.05])[38] and had a greater number of ED visits alone (27 vs 15 per 100 patients, P = 0.007)[22] within 30 days of hospital discharge compared to nondepressed patients. Similar results were found at 90 days.[36]
Sensitivity Analyses
All told, most studies reported a positive association between depression and adverse events, and this was true regardless of how much adjustment for potential confounding had been undertaken by the authors. Although all studies were qualitatively in the same direction, the magnitude of the association varied due to methodological and/or clinical heterogeneity. Sensitivity analysis revealed no overall difference in pooled risk ratios or heterogeneity between Mantel‐Haenszel fixed effects versus random effects models or with the addition of 0.5 to cells to permit inclusion of zero‐event data. There was no evidence of publication bias; funnel plots and Egger test results are available upon request. There were no statistically significant differences in the risk associated with depressive symptoms whether studies used Diagnostic and Statistical Manual of Mental Disorders (DSM)‐III or DSM‐IV criteria, whether the study samples were disease specific or unselected general medical cohorts, whether studies were of moderate or high quality, or regardless of the severity of depressive symptoms.
DISCUSSION
Summary of Evidence
We found that depression was common in medical inpatients (about one‐third of all patients) and persisted for at least 30 days in up to half of those patients after discharge. We found strong evidence of an association between depressive symptoms and poor short‐term prognosis after discharge from the hospital: a 73% increased risk of readmission and a 2‐fold risk of death within 30 days compared to patients without depressive symptoms with similar results at 90 days.
Our meta‐analysis complements a recent systematic review that found concomitant depression to be a risk factor for poor prognosis among inpatients and outpatients with acute coronary syndrome,[40] and a meta‐analysis that demonstrated an increased risk of 2‐year mortality among patients with depression after myocardial infarction.[41] To our knowledge, our study is the first to quantify the short‐term postdischarge risks across a diverse group of medical inpatients.
The potential mechanisms underlying the observed relationship between depression and adverse patient outcomes after discharge are likely multiple. We believe there are 2 main possibilities. First, the increased risk associated with depression might be due to residual confounding, even though many of these studies did adjust for extensive lists of comorbidities,[22, 24, 26, 27, 29, 30, 33, 35, 36, 39] including functional status[39] and prior health services utilization.[22, 34, 36] This could occur if other risk factors were not sufficiently adjusted for, such as unrecognized comorbidities or concomitant disability, which are often present among chronically ill patients,[42] or if depression were a marker of psychosocial risk factors such as anxiety,[43] stress or poor resiliency,[44] or low social support,[45] though a few adjusted for psychosocial factors such as social support[26] or anxiety.[35] Confounding could also occur if symptoms of acute illness inflate reports of somatic symptoms of depression on self‐report questionnaires. Recent studies on the BDI, found that scores were higher in postmyocardial infarction patients when compared to outpatient controls,[46] but with no differences between those groups in scores for the BDI‐II,[47] a version with fewer somatic symptom questions.
Second, depression may cause adverse outcomes through indirect or direct pathways. Indirect causation could occur if depression hindered self‐care behaviors such as medication adherence.[42] Depression could also act directly through pathophysiological changes. Some studies have suggested that depression is associated with metabolic abnormalities, including alterations in glucose transport[42, 48] and increased vulnerability to obesity, type 2 diabetes mellitus, and/or diabetic complications, common conditions among hospitalized patients that also adversely affect postdischarge outcomes.[40, 48]
Strengths and Limitations
This review has multiple strengths. We cast a broad search and included studies that examined a wide range of medical patient subgroups, thus increasing the generalizability of our findings. We identified a general scarcity of studies on this topic and obtained additional unpublished data for 10 of the 18 relevant studies, and our response rate of 34% is compatible with the 37% response rate reported for Cochrane reviews when seeking additional data from authors.[49] Whether examined qualitatively (vote counting of the number of studies that showed an association) or quantitatively (via formal meta‐analysis), it seems apparent that there is a clinically important association between depression and postdischarge adverse events, but given the number, quality, and heterogeneity of the studies we examined, there may be some ongoing dispute about exactly how strong this association is and the degree of bias contributed by a couple of large studies of the topic.
There are limitations to our review. First, as we did not have individual‐level patient data, we could not use metaregression to explore sources of heterogeneity (clinical or methodological) or adjust for confounding, and this likely contributes to observed differences between individual estimates. For instance, the included studies had heterogeneous screening measures and cutoffs; thus, all cases of depression in these studies might not be equivalent. Some of the included studies assessed depression early during admission where psychological distress may be greatest; others assessed symptoms closer to discharge. Most studies included patients with specific conditions like heart failure or chronic obstructive pulmonary disease rather than a wide spectrum of medical inpatients. Moreover, few studies adjusted for psychosocial risk factors such as social support, anxiety, and functional status, and only 2 studies assessed the persistence of depressive symptoms after discharge. Second, we did not explore quantitative measures of between‐study variation (eg, I2), because experts question its utility given the expected heterogeneity in meta‐analyses of observational studies.[50] Third, although the included studies were deemed to be of at least moderate quality, they could be at risk for sources of bias that may not be sufficiently appraised by the current version of the Newscastle‐Ottawa scale for observational studies. Finally, we excluded grey literature (eg, conference proceedings or technical reports) that could potentially exclude null findings, although we did contact authors in this field to identify additional unpublished data relevant to this topic.
CONCLUSIONS
We have confirmed that depressive symptoms are common in hospitalized medical patients, frequently persist after discharge, and may predict greater risk of readmission or death after discharge. Thus, depressive symptoms are an additional marker that clinicians can use to help identify patients in acute care medical settings who may be at increased risk for suboptimal transition back to the community and who may require additional resources after discharge. However, future research is required to evaluate whether treatment of individuals who screen positive for depressive symptoms can reduce 30‐day readmission rates, and we are aware of at least 1 relevant ongoing trial (
Acknowledgements
The authors thank the following individuals: Dale Storie, MLIS, Saskatchewan Information and Library Services Consortium, Regina, Saskatchewan, Canada, for assistance in the literature search; James A. Hanley, PhD, Department of Epidemiology and Biostatistics, Faculty of Medicine, McGill University, Montreal, Quebec, Canada, for guidance in data recovery methods; Nancy Frasure‐Smith, PhD, Department of Psychiatry, McGill University, Department of Psychiatry and Research Centre Hospital Centre, University of Montreal, and Montreal Heart Institute Research Centre, Montreal, Quebec, Canada; Andriana I. Papaioannou, MD, 2nd Respiratory Medicine Department, University of Athens Medical School, Athens, Greece; Konstantinos Kostikas, MD, 2nd Respiratory Medicine Department, University of Athens Medical School, Athens, Greece; and Pere Almagro, MD, Servicio de Medicina Interna, Hospital Universitario Mutua de Terrassa, Terrassa, Barcelona, Spain; as well as Philip G. Jones, MS, Saint Luke's Mid America Heart Institute, Kansas City, Missouri; for their retrieval and contribution of unpublished data.
Disclosures
Ms. Pederson affirms that the manuscript is an honest, accurate, and transparent account of the study being reported with no important omissions. All authors had full access to all of the data (including statistical reports and tables) in the study and can take responsibility for the integrity of the data and the accuracy of the data analysis. Design and conduct of the study: Ms. Pederson, Drs. Majumdar and McAlister. Data acquisition: Ms. Pederson, Ms. Warkentin. Analysis and interpretation of the data and drafting of the manuscript: Ms Pederson, Drs. Majumdar and McAlister. Review of the manuscript: all authors. Study supervision: Drs. Majumdar and McAlister. None of the contributors received compensation for their efforts. Salary support for Ms. Pederson was provided by a CRIO grant from Alberta InnovatesHealth Solutions. Drs. McAlister and Majumdar are supported by salary awards from Alberta Innovates‐Health Solutions. Dr. McAlister holds the University of Alberta/Capital Health Chair in Cardiology Outcomes Research. Dr. Majumda holds the University of Alberta Endowed Chair in Patient Health Management. The funding sources had no role in the design or conduct of the study; management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. This work is that of the authors independent of funders. The authors report no conflicts of interest.
- , , . Rehospitalizations among patients in the Medicare fee‐for‐service program. N Engl J Med. 2009;360(14):1418–1428.
- , , , , . Proportion of hospital readmissions deemed avoidable: a systematic review. CMAJ. 2011;183(7):E391–E402.
- , , , . Mental health conditions are associated with increased health care utilization among urban family medicine patients. J Am Board Fam Med. 2008;21(5):398–407.
- , , , , . Interventions to reduce 30‐day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520–528.
- . The elusive quest for quality and cost savings in the Medicare program. JAMA. 2009;301(6):668–670.
- , , , . Effects of care coordination on hospitalization, quality of care, and health care expenditures among Medicare beneficiaries—15 randomized trials. JAMA. 2009;301(6):603–618.
- , , , et al. Unplanned readmissions after hospital discharge among patients identified as being at high risk for readmission using a validated predictive algorithm. Open Med. 2011;5(2):e104–111.
- , , , et al. Risk prediction models for hospital readmission: a systematic review. JAMA. 2011;306(15):1688–1698.
- , , . Depression in older people in the general hospital: a systematic review of screening instruments. Age Ageing. 2012;41(2):148–154.
- , , , et al. Prevalence, correlates and recognition of depression among inpatients of general hospitals in Wuhan, China. Gen Hosp Psychiatry. 2010;32(3):268–275.
- , , , , , . Recognition of depression by non‐psychiatric physicians—a systematic literature review and meta‐analysis. J Gen Intern Med. 2008;23(1):25–36.
- , , , , , . Predicting the risk of unplanned readmission or death within 30 days of discharge after a heart failure hospitalization. Am Heart J. 2012;164(3):365–372.
- , , , et al. Does evidence support the American Heart Association's recommendation to screen patients for depression in cardiovascular care? An updated systematic review. PLoS One. 2013;8(1):e52654.
- , , , et al. Screening for depression: a systematic review and meta‐analysis. CMAJ Open. 2013;1(4):E159–E167.
- , , , et al. The PRISMA statement for reporting systematic reviews and meta‐analyses of studies that evaluate health care interventions: explanation and elaboration. Ann Intern Med. 2009;151(4):W65–94.
- , , , et al. Screening for depression in adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med. 2002;136(10):765–776.
- , , . Recovering the raw data behind a non‐parametric survival curve. Syst Rev. 2014;3:151.
- , , , . Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan‐Meier survival curves. BMC Med Res Methodol. 2012;12(1):9.
- . Commentary: heterogeneity in meta‐analysis should be expected and appropriately quantified. Int J Epidemiol. 2008;37(5):1158–1160.
- , , , et al. The Newcastle‐Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta‐analyses. Available at: http://www.ohri.ca/programs/clinical_epidemiology/oxford.htm. Accessed September 1, 2015.
- , , . The funnel plot. In: Rothstein HR, Sutton AJ, Borenstein M, eds. Publication Bias in Meta‐analysis: Prevention, Assessment and Adjustments. New York, NY: John Wiley 2006:73–98.
- , , , , , . Dose‐response relationship between depressive symptoms and hospital readmission. J Hosp Med. 2014;9(6):358–364.
- , , , et al. Relationship of depression to increased risk of mortality and rehospitalization in patients with congestive heart failure. Arch Intern Med. 2001;161(15):1849–1856.
- , , , et al. Depression is a risk factor for rehospitalization in medical inpatients. Prim Care Companion J Clin Psychiatry. 2007;9(4):256–262.
- , , , et al. Depression in elderly patients with congestive heart failure. J Geriatr Psychiatry. 1991;24(1):59–71.
- , , . Depression following myocardial infarction: impact on 6‐month survival. JAMA. 1993;270(15):1819–1825.
- , , , , . Gender, depression, and one‐year prognosis after myocardial infarction. Psychosom Med. 1999;61(1):26–37.
- , , , . Depression and 1‐year prognosis in unstable angina. Arch Intern Med. 2000;160(9):1354–1360.
- , , , et al. The impact of depressive symptoms on recovery and outcome of hospitalised COPD exacerbations. Eur Respir J. 2013;41(4):815–823.
- , , , et al. Depression predicts mortality and hospitalization in patients with myocardial infarction complicated by heart failure. Am Heart J. 2005;150(5):961–967.
- , , , et al. Mortality after hospitalization for COPD. Chest. 2002;121(5):1441–1448.
- , , , et al. Pseudomonas aeruginosa and mortality after hospital admission for chronic obstructive pulmonary disease. Respiration. 2012;84(1):36–43.
- , , . Depressive symptoms increase risk of rehospitalisation in heart failure patients with preserved systolic function. J Clin Nurs. 2009;18(13):1871–1877.
- , , . Depressive symptoms and 3 year mortality in older hospitalized medical patients. Ann Intern Med. 1999;130(7):563–569.
- , , , et al. A positive 2‐item patient health questionnaire depression screen among hospitalized heart failure patients is associated with elevated 12‐month mortality. J Card Fail. 2012;18(3):238–245.
- , , , et al. Post‐discharge hospital utilization among adult medical inpatients with depressive symptoms. J Hosp Med. 2010;5(7):378–384.
- , . Use of health services by medically ill depressed elderly patients after hospital discharge. Am J Geriatr Psychiatry. 1999;7(1):48–56.
- , , . Congestive heart failure and depression in older adults: clinical course and health services use 6 months after hospitalization. Psychosomatics. 2003;44(5):367–373.
- , , , . Depressive symptoms as a predictor of 6‐month outcomes and services utilization in elderly medical inpatients. Arch Intern Med. 2001;161(21):2609–2615.
- , , , et al. Depression as a risk factor for poor prognosis among patients with acute coronary syndrome: systematic review and recommendations: a scientific statement from the American Heart Association. Circulation. 2014;129(12):1350–1369.
- , , , , , . Prognostic association of depression following myocardial infarction with mortality and cardiovascular events: a meta‐analysis of 25 years of research. Gen Hosp Psychiatry. 2011;33(3):203–216.
- , , , , . Depression and cardiac disease: epidemiology, mechanisms, and diagnosis. Cardiovasc Psychiatry Neurol. 2013;2013:695925.
- , , , et al. Prognostic value of depression, anxiety, and anger in hospitalized cardiovascular disease patients for predicting adverse cardiac outcomes. Am J Cardiol. 2013;111(10):1432–1436.
- , , . The psychobiology of depression and resilience to stress: implications for prevention and treatment. Annu Rev Clin Psychol. 2005;1:255–291.
- , , , et al. Impact of social factors on risk of readmission or mortality in pneumonia and heart failure: systematic review. J Gen Intern Med. 2013;28(2):269–282.
- , , , et al. The influence of somatic symptoms on beck depression inventory scores in hospitalized postmyocardial infarction patients. Can J Psychiatry. 2012;57(12):752–758.
- , , , et al. Somatic symptom overlap in beck depression inventory‐II scores following myocardial infarction. Br J Psychiatry. 2010;197(1):61–66.
- , , , . Relationship of depression to diabetes types 1 and 2: epidemiology, biology, and treatment. Biol Psychiatry. 2003;54(3):317–329.
- , , . Searching for unpublished data for Cochrane reviews: cross sectional study. BMJ. 2013;346:f2231.
- . Comment on: heterogeneity in meta‐analysis should be expected and appropriately quantified. Int J Epidemiol. 2010;39(3):932; author reply 933.
- , , . Rehospitalizations among patients in the Medicare fee‐for‐service program. N Engl J Med. 2009;360(14):1418–1428.
- , , , , . Proportion of hospital readmissions deemed avoidable: a systematic review. CMAJ. 2011;183(7):E391–E402.
- , , , . Mental health conditions are associated with increased health care utilization among urban family medicine patients. J Am Board Fam Med. 2008;21(5):398–407.
- , , , , . Interventions to reduce 30‐day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520–528.
- . The elusive quest for quality and cost savings in the Medicare program. JAMA. 2009;301(6):668–670.
- , , , . Effects of care coordination on hospitalization, quality of care, and health care expenditures among Medicare beneficiaries—15 randomized trials. JAMA. 2009;301(6):603–618.
- , , , et al. Unplanned readmissions after hospital discharge among patients identified as being at high risk for readmission using a validated predictive algorithm. Open Med. 2011;5(2):e104–111.
- , , , et al. Risk prediction models for hospital readmission: a systematic review. JAMA. 2011;306(15):1688–1698.
- , , . Depression in older people in the general hospital: a systematic review of screening instruments. Age Ageing. 2012;41(2):148–154.
- , , , et al. Prevalence, correlates and recognition of depression among inpatients of general hospitals in Wuhan, China. Gen Hosp Psychiatry. 2010;32(3):268–275.
- , , , , , . Recognition of depression by non‐psychiatric physicians—a systematic literature review and meta‐analysis. J Gen Intern Med. 2008;23(1):25–36.
- , , , , , . Predicting the risk of unplanned readmission or death within 30 days of discharge after a heart failure hospitalization. Am Heart J. 2012;164(3):365–372.
- , , , et al. Does evidence support the American Heart Association's recommendation to screen patients for depression in cardiovascular care? An updated systematic review. PLoS One. 2013;8(1):e52654.
- , , , et al. Screening for depression: a systematic review and meta‐analysis. CMAJ Open. 2013;1(4):E159–E167.
- , , , et al. The PRISMA statement for reporting systematic reviews and meta‐analyses of studies that evaluate health care interventions: explanation and elaboration. Ann Intern Med. 2009;151(4):W65–94.
- , , , et al. Screening for depression in adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med. 2002;136(10):765–776.
- , , . Recovering the raw data behind a non‐parametric survival curve. Syst Rev. 2014;3:151.
- , , , . Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan‐Meier survival curves. BMC Med Res Methodol. 2012;12(1):9.
- . Commentary: heterogeneity in meta‐analysis should be expected and appropriately quantified. Int J Epidemiol. 2008;37(5):1158–1160.
- , , , et al. The Newcastle‐Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta‐analyses. Available at: http://www.ohri.ca/programs/clinical_epidemiology/oxford.htm. Accessed September 1, 2015.
- , , . The funnel plot. In: Rothstein HR, Sutton AJ, Borenstein M, eds. Publication Bias in Meta‐analysis: Prevention, Assessment and Adjustments. New York, NY: John Wiley 2006:73–98.
- , , , , , . Dose‐response relationship between depressive symptoms and hospital readmission. J Hosp Med. 2014;9(6):358–364.
- , , , et al. Relationship of depression to increased risk of mortality and rehospitalization in patients with congestive heart failure. Arch Intern Med. 2001;161(15):1849–1856.
- , , , et al. Depression is a risk factor for rehospitalization in medical inpatients. Prim Care Companion J Clin Psychiatry. 2007;9(4):256–262.
- , , , et al. Depression in elderly patients with congestive heart failure. J Geriatr Psychiatry. 1991;24(1):59–71.
- , , . Depression following myocardial infarction: impact on 6‐month survival. JAMA. 1993;270(15):1819–1825.
- , , , , . Gender, depression, and one‐year prognosis after myocardial infarction. Psychosom Med. 1999;61(1):26–37.
- , , , . Depression and 1‐year prognosis in unstable angina. Arch Intern Med. 2000;160(9):1354–1360.
- , , , et al. The impact of depressive symptoms on recovery and outcome of hospitalised COPD exacerbations. Eur Respir J. 2013;41(4):815–823.
- , , , et al. Depression predicts mortality and hospitalization in patients with myocardial infarction complicated by heart failure. Am Heart J. 2005;150(5):961–967.
- , , , et al. Mortality after hospitalization for COPD. Chest. 2002;121(5):1441–1448.
- , , , et al. Pseudomonas aeruginosa and mortality after hospital admission for chronic obstructive pulmonary disease. Respiration. 2012;84(1):36–43.
- , , . Depressive symptoms increase risk of rehospitalisation in heart failure patients with preserved systolic function. J Clin Nurs. 2009;18(13):1871–1877.
- , , . Depressive symptoms and 3 year mortality in older hospitalized medical patients. Ann Intern Med. 1999;130(7):563–569.
- , , , et al. A positive 2‐item patient health questionnaire depression screen among hospitalized heart failure patients is associated with elevated 12‐month mortality. J Card Fail. 2012;18(3):238–245.
- , , , et al. Post‐discharge hospital utilization among adult medical inpatients with depressive symptoms. J Hosp Med. 2010;5(7):378–384.
- , . Use of health services by medically ill depressed elderly patients after hospital discharge. Am J Geriatr Psychiatry. 1999;7(1):48–56.
- , , . Congestive heart failure and depression in older adults: clinical course and health services use 6 months after hospitalization. Psychosomatics. 2003;44(5):367–373.
- , , , . Depressive symptoms as a predictor of 6‐month outcomes and services utilization in elderly medical inpatients. Arch Intern Med. 2001;161(21):2609–2615.
- , , , et al. Depression as a risk factor for poor prognosis among patients with acute coronary syndrome: systematic review and recommendations: a scientific statement from the American Heart Association. Circulation. 2014;129(12):1350–1369.
- , , , , , . Prognostic association of depression following myocardial infarction with mortality and cardiovascular events: a meta‐analysis of 25 years of research. Gen Hosp Psychiatry. 2011;33(3):203–216.
- , , , , . Depression and cardiac disease: epidemiology, mechanisms, and diagnosis. Cardiovasc Psychiatry Neurol. 2013;2013:695925.
- , , , et al. Prognostic value of depression, anxiety, and anger in hospitalized cardiovascular disease patients for predicting adverse cardiac outcomes. Am J Cardiol. 2013;111(10):1432–1436.
- , , . The psychobiology of depression and resilience to stress: implications for prevention and treatment. Annu Rev Clin Psychol. 2005;1:255–291.
- , , , et al. Impact of social factors on risk of readmission or mortality in pneumonia and heart failure: systematic review. J Gen Intern Med. 2013;28(2):269–282.
- , , , et al. The influence of somatic symptoms on beck depression inventory scores in hospitalized postmyocardial infarction patients. Can J Psychiatry. 2012;57(12):752–758.
- , , , et al. Somatic symptom overlap in beck depression inventory‐II scores following myocardial infarction. Br J Psychiatry. 2010;197(1):61–66.
- , , , . Relationship of depression to diabetes types 1 and 2: epidemiology, biology, and treatment. Biol Psychiatry. 2003;54(3):317–329.
- , , . Searching for unpublished data for Cochrane reviews: cross sectional study. BMJ. 2013;346:f2231.
- . Comment on: heterogeneity in meta‐analysis should be expected and appropriately quantified. Int J Epidemiol. 2010;39(3):932; author reply 933.
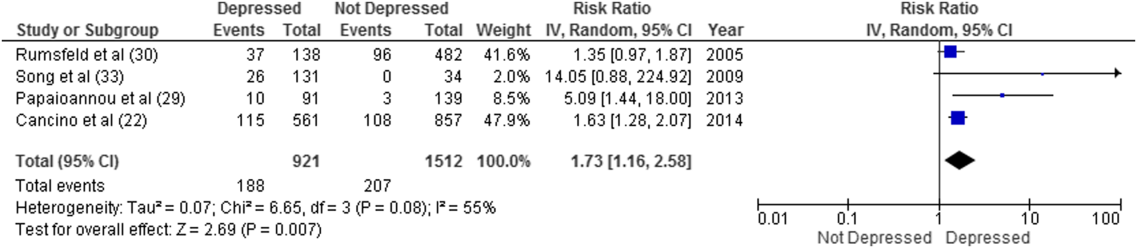
VIDEO: Shorter gap from heart attack to CABG shown safe
PHOENIX – Patients who are stable following a myocardial infarction and need isolated coronary artery bypass surgery (CABG) don’t need to wait 5 or so days for their surgery, a delay that many surgeons and cardiologists often impose.
The operation can safely occur after just a 1- or 2-day gap following either an ST-elevation MI or a non–ST-elevation MI, based on real-world outcomes seen in more than 3,000 patients treated at any of seven U.S. medical centers.
“Waiting an arbitrary 5 days is not important,” Elizabeth L. Nichols said during a video interview and during her report at the annual meeting of the Society of Thoracic Surgeons.
Ms. Nichols and her associates analyzed the in-hospital mortality rates among 3,060 patients who underwent isolated CABG during 2008-2014 at any of the seven medical centers that participate in the Northern New England Cardiovascular Disease Study Group and offer CABG. They included patients who had their surgery within 21 days of their MI, and excluded patients who had their CABG within 6 hours of their MI, had emergency surgery, or those with shock or incomplete data. The study group included 529 patients who had a ST-elevation MI and 2,531 patients with a non-ST-elevation MI.
The analysis divided patients into four groups based on timing of their CABG: 99 patients (3%) had surgery within the first 24 hours, 369 patients (12%) had their surgery 1-2 days after their MI, 1,966 (64%) had their operation 3-7 days following their MI, and 626 (21%) had their surgery 8-21 days after the MI.
The unadjusted mortality rates for these four subgroups were 5.1%, 1.6%, 1.6%, and 2.7%, respectively, reported Ms. Nichols, a health services researcher at the Dartmouth Institute for Health Policy & Clinical Practice, Lebanon, N.H.
After researchers adjusted for several demographic and clinical variables, the mortality rates remained identical for patients who underwent CABG 1 or 2 days following their MI, compared with patients whose surgery was deferred until 3-7 days after the MI. Patients with surgery 8-21 days following the MI had a small but not statistically significant higher rate of in-hospital death.
Patients who had their surgery 7-23 hours following an MI had a statistically significant increased hospital mortality following surgery that ran more than threefold greater than patients who underwent CABG 3-7 days after their MI.
The main message from the analysis is that for the typical, stable MI patient who requires CABG to treat multivessel coronary disease, no need exists to wait several days following an MI to do the surgery, Ms. Nichols explained. A delay of just 1 or 2 days is safe and sufficient, as long as it provides adequate time for any acutely administered antiplatelet or antithrombotic drugs to clear.

The findings “provide a degree of comfort for not waiting the 3-5 days that had previously been thought necessary,” said Dr. Jock N. McCullough, chief of cardiac surgery at Dartmouth-Hitchcock Medical Center in Lebanon and a collaborator on the study.
The findings are not meant to supersede clinical judgment, both Dr. McCullough and Ms. Nichols emphasized. Individual patients might have good reasons to either undergo faster surgery or to wait at least 8 days following their MI.
“The patients who waited 8-21 days had a lot of comorbidities and were sicker patients, and their delay is often warranted” to make sure the patient is stable enough for surgery, Ms. Nichols explained. Other patients might be worsening following their MI and need to undergo their surgery within 24 hours of their MI.
“Clinical judgment is always the trump card,” Ms. Nichols said.
Ms. Nichols and Dr. McCullough had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
PHOENIX – Patients who are stable following a myocardial infarction and need isolated coronary artery bypass surgery (CABG) don’t need to wait 5 or so days for their surgery, a delay that many surgeons and cardiologists often impose.
The operation can safely occur after just a 1- or 2-day gap following either an ST-elevation MI or a non–ST-elevation MI, based on real-world outcomes seen in more than 3,000 patients treated at any of seven U.S. medical centers.
“Waiting an arbitrary 5 days is not important,” Elizabeth L. Nichols said during a video interview and during her report at the annual meeting of the Society of Thoracic Surgeons.
Ms. Nichols and her associates analyzed the in-hospital mortality rates among 3,060 patients who underwent isolated CABG during 2008-2014 at any of the seven medical centers that participate in the Northern New England Cardiovascular Disease Study Group and offer CABG. They included patients who had their surgery within 21 days of their MI, and excluded patients who had their CABG within 6 hours of their MI, had emergency surgery, or those with shock or incomplete data. The study group included 529 patients who had a ST-elevation MI and 2,531 patients with a non-ST-elevation MI.
The analysis divided patients into four groups based on timing of their CABG: 99 patients (3%) had surgery within the first 24 hours, 369 patients (12%) had their surgery 1-2 days after their MI, 1,966 (64%) had their operation 3-7 days following their MI, and 626 (21%) had their surgery 8-21 days after the MI.
The unadjusted mortality rates for these four subgroups were 5.1%, 1.6%, 1.6%, and 2.7%, respectively, reported Ms. Nichols, a health services researcher at the Dartmouth Institute for Health Policy & Clinical Practice, Lebanon, N.H.
After researchers adjusted for several demographic and clinical variables, the mortality rates remained identical for patients who underwent CABG 1 or 2 days following their MI, compared with patients whose surgery was deferred until 3-7 days after the MI. Patients with surgery 8-21 days following the MI had a small but not statistically significant higher rate of in-hospital death.
Patients who had their surgery 7-23 hours following an MI had a statistically significant increased hospital mortality following surgery that ran more than threefold greater than patients who underwent CABG 3-7 days after their MI.
The main message from the analysis is that for the typical, stable MI patient who requires CABG to treat multivessel coronary disease, no need exists to wait several days following an MI to do the surgery, Ms. Nichols explained. A delay of just 1 or 2 days is safe and sufficient, as long as it provides adequate time for any acutely administered antiplatelet or antithrombotic drugs to clear.
The findings “provide a degree of comfort for not waiting the 3-5 days that had previously been thought necessary,” said Dr. Jock N. McCullough, chief of cardiac surgery at Dartmouth-Hitchcock Medical Center in Lebanon and a collaborator on the study.
The findings are not meant to supersede clinical judgment, both Dr. McCullough and Ms. Nichols emphasized. Individual patients might have good reasons to either undergo faster surgery or to wait at least 8 days following their MI.
“The patients who waited 8-21 days had a lot of comorbidities and were sicker patients, and their delay is often warranted” to make sure the patient is stable enough for surgery, Ms. Nichols explained. Other patients might be worsening following their MI and need to undergo their surgery within 24 hours of their MI.
“Clinical judgment is always the trump card,” Ms. Nichols said.
Ms. Nichols and Dr. McCullough had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
PHOENIX – Patients who are stable following a myocardial infarction and need isolated coronary artery bypass surgery (CABG) don’t need to wait 5 or so days for their surgery, a delay that many surgeons and cardiologists often impose.
The operation can safely occur after just a 1- or 2-day gap following either an ST-elevation MI or a non–ST-elevation MI, based on real-world outcomes seen in more than 3,000 patients treated at any of seven U.S. medical centers.
“Waiting an arbitrary 5 days is not important,” Elizabeth L. Nichols said during a video interview and during her report at the annual meeting of the Society of Thoracic Surgeons.
Ms. Nichols and her associates analyzed the in-hospital mortality rates among 3,060 patients who underwent isolated CABG during 2008-2014 at any of the seven medical centers that participate in the Northern New England Cardiovascular Disease Study Group and offer CABG. They included patients who had their surgery within 21 days of their MI, and excluded patients who had their CABG within 6 hours of their MI, had emergency surgery, or those with shock or incomplete data. The study group included 529 patients who had a ST-elevation MI and 2,531 patients with a non-ST-elevation MI.
The analysis divided patients into four groups based on timing of their CABG: 99 patients (3%) had surgery within the first 24 hours, 369 patients (12%) had their surgery 1-2 days after their MI, 1,966 (64%) had their operation 3-7 days following their MI, and 626 (21%) had their surgery 8-21 days after the MI.
The unadjusted mortality rates for these four subgroups were 5.1%, 1.6%, 1.6%, and 2.7%, respectively, reported Ms. Nichols, a health services researcher at the Dartmouth Institute for Health Policy & Clinical Practice, Lebanon, N.H.
After researchers adjusted for several demographic and clinical variables, the mortality rates remained identical for patients who underwent CABG 1 or 2 days following their MI, compared with patients whose surgery was deferred until 3-7 days after the MI. Patients with surgery 8-21 days following the MI had a small but not statistically significant higher rate of in-hospital death.
Patients who had their surgery 7-23 hours following an MI had a statistically significant increased hospital mortality following surgery that ran more than threefold greater than patients who underwent CABG 3-7 days after their MI.
The main message from the analysis is that for the typical, stable MI patient who requires CABG to treat multivessel coronary disease, no need exists to wait several days following an MI to do the surgery, Ms. Nichols explained. A delay of just 1 or 2 days is safe and sufficient, as long as it provides adequate time for any acutely administered antiplatelet or antithrombotic drugs to clear.
The findings “provide a degree of comfort for not waiting the 3-5 days that had previously been thought necessary,” said Dr. Jock N. McCullough, chief of cardiac surgery at Dartmouth-Hitchcock Medical Center in Lebanon and a collaborator on the study.
The findings are not meant to supersede clinical judgment, both Dr. McCullough and Ms. Nichols emphasized. Individual patients might have good reasons to either undergo faster surgery or to wait at least 8 days following their MI.
“The patients who waited 8-21 days had a lot of comorbidities and were sicker patients, and their delay is often warranted” to make sure the patient is stable enough for surgery, Ms. Nichols explained. Other patients might be worsening following their MI and need to undergo their surgery within 24 hours of their MI.
“Clinical judgment is always the trump card,” Ms. Nichols said.
Ms. Nichols and Dr. McCullough had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
AT THE STS ANNUAL MEETING
Key clinical point: Performing coronary artery bypass grafting 1-2 days following an MI was as safe as when surgery was delayed 3-7 days.
Major finding: In-hospital mortality after CABG was identical in patients operated on 1-2 days or 3-7 days following an MI.
Data source: Retrospective analysis of 3,060 patients who underwent CABG within 21 days following an MI at any of seven U.S. centers.
Disclosures: Ms. Nichols and Dr. McCullough had no disclosures.

Telemental health reaches underserved children, builds partnerships
The patient, a 10-year-old girl, was exhibiting defiant and angry behavior at home. Her pediatrician in rural Vermont was not able to support the family optimally, so he referred her to Dr. David C. Rettew.
Dr. Rettew soon learned that the girl blamed herself for her father’s absence, and she interpreted efforts by her mother to set limits as evidence that her mom didn’t love her.

“During the course of the interview, it became clear that the girl had some specific thoughts that were likely fueling her anger,” said Dr. Rettew, director of the Pediatric Psychiatry Clinic at the University of Vermont, Burlington. “She also met criteria clearly for ADHD, which had never been diagnosed. The consultation recommendations not only included some possible medications to try, but also some specific areas that could be addressed for psychotherapy that could really help the relationship between this child and her mother.”
The successful intervention took place via a secure, two-way videoconference. It is one example of how physicians at the University of Vermont are using telemental health to treat children in underserved areas. As part of a state-funded training program, child psychiatry fellows at the university consult with primary care doctors at federally qualified health centers across the state. The primary care physicians discuss patient cases with fellows via phone or email and can refer patients for in-person or telemental health assessments.
The program has been running for about 5 years and so far has yielded countless benefits, said Dr. Rettew, who directs the university’s Child and Adolescent Psychiatry Fellowship Program. “It helps keep the care housed and centered within the primary care home. That can help coordination of services so that care isn’t fragmented around multiple centers. It also allows evaluations to happen that wouldn’t happen otherwise because it’s too much of a hardship for families to travel long distances and come for regular follow-up appointments.”

University of Vermont physicians also use telemedicine to consult with other mental health clinicians across the state. Child psychiatrist Allison Y. Hall provides in-person training to clinicians and then counsels and supervises their efforts through a telemedicine unit.
“For this purpose, it’s great,” said Dr. Hall, who practices at the Vermont Center for Children, Youth, and Families, which is housed within the university’s psychiatry department. “There’s more confidentiality than Skype, for instance. It’s wonderful to be able to reach clinicians at a great distance.”
On a broader scale, telemental health is a promising tool to address the shortage of mental health providers in the United States, said Dr. Robert C. Gunther, a pediatrician at the University of Virginia Health System in Fishersville. Recent research found that one in three children receiving outpatient care for mental health conditions saw only their primary care doctor for care (Pediatrics. 2015 Nov;136;e1178-85).

“There is a tremendous need for pediatric mental health care,” Dr. Gunther said in an interview. “There is a great shortage of child psychiatrists and other child mental health specialists. Telemental health can help in areas where geography or financial barriers exist to accessing care.”
Data from the Children’s ADHD Telemental Health Treatment Study (CATTS) illustrates the impact that telemental health can have on children facing such barriers to care. Researchers randomized 223 children referred by 88 primary care providers in seven underserved communities into two study groups. Children in the first group were seen by child psychiatrists via videoconference six times over 22 weeks; treatment included pharmacotherapy. Their caregivers received behavior training provided in person by community therapists who were supervised remotely. Children in the second group were treated by their primary care physicians and received one telepsychiatry consult.
Children in both groups improved; however, those randomized to the telemental health model improved “significantly more than patients in the augmented primary care arm” (J Am Acad Child Adolesc Psychiatry. 2015 Apr;54[4]:263-74).

“The CATTS trial demonstrated the effectiveness of a telehealth service model to treat ADHD in communities with limited access to specialty mental health services,” investigators concluded.
But Dr. Joshua J. Alexander, chair of the American Academy of Pediatrics Section on Telehealth Care, notes that some mental health conditions fit more smoothly within the telemental health model than others. ADHD is the most common condition treated by telemental health, he said. The model also has shown success in the treatment of childhood adjustment disorders, anxiety, oppositional defiant disorder, mood disorders, anxiety, and depression.
“I think you have to be careful in determining where telemental health would be beneficial to use and in which cases it might not be an appropriate method to deliver care,” Dr. Alexander said in an interview.
Developing trust and rapport with patients through videoconferencing also can be a challenge, added Dr. Alexander, who is director of the TelAbility telehealth program at the University of North Carolina at Chapel Hill. He recommends that specialists use the technology to continue an existing doctor-patient relationship or to provide care in a consultative model in which the child’s primary care doctor is present along with the patient and patient’s family.
The AAP advocates for the use of telemedicine so long as it is conducted within the context of the medical home. Fragmented telemedicine services that could disrupt continuity of care should be avoided, according to a 2015 AAP policy statement (Pediatrics. 2015 Jun. doi: 10.1542/peds.2015-1253). The academy also calls for the expansion of pediatric telemedicine to increase access to care for underserved communities and improve quality of care for children.
More partnerships between mental health specialists and primary care providers are a key step in delivering high quality pediatric telemental care, Dr. Alexander said.
“Some larger pediatric practices already do this by hiring and colocating individuals at their practice site, but other, smaller practices might not have the room, finances, sufficient patient population, or enough local providers to make this happen,” he said. “A telemedicine program, located within the practice, could enable this specialized service to be provided in a convenient, coordinated setting.”
On Twitter @legal_med
The patient, a 10-year-old girl, was exhibiting defiant and angry behavior at home. Her pediatrician in rural Vermont was not able to support the family optimally, so he referred her to Dr. David C. Rettew.
Dr. Rettew soon learned that the girl blamed herself for her father’s absence, and she interpreted efforts by her mother to set limits as evidence that her mom didn’t love her.
“During the course of the interview, it became clear that the girl had some specific thoughts that were likely fueling her anger,” said Dr. Rettew, director of the Pediatric Psychiatry Clinic at the University of Vermont, Burlington. “She also met criteria clearly for ADHD, which had never been diagnosed. The consultation recommendations not only included some possible medications to try, but also some specific areas that could be addressed for psychotherapy that could really help the relationship between this child and her mother.”
The successful intervention took place via a secure, two-way videoconference. It is one example of how physicians at the University of Vermont are using telemental health to treat children in underserved areas. As part of a state-funded training program, child psychiatry fellows at the university consult with primary care doctors at federally qualified health centers across the state. The primary care physicians discuss patient cases with fellows via phone or email and can refer patients for in-person or telemental health assessments.
The program has been running for about 5 years and so far has yielded countless benefits, said Dr. Rettew, who directs the university’s Child and Adolescent Psychiatry Fellowship Program. “It helps keep the care housed and centered within the primary care home. That can help coordination of services so that care isn’t fragmented around multiple centers. It also allows evaluations to happen that wouldn’t happen otherwise because it’s too much of a hardship for families to travel long distances and come for regular follow-up appointments.”
University of Vermont physicians also use telemedicine to consult with other mental health clinicians across the state. Child psychiatrist Allison Y. Hall provides in-person training to clinicians and then counsels and supervises their efforts through a telemedicine unit.
“For this purpose, it’s great,” said Dr. Hall, who practices at the Vermont Center for Children, Youth, and Families, which is housed within the university’s psychiatry department. “There’s more confidentiality than Skype, for instance. It’s wonderful to be able to reach clinicians at a great distance.”
On a broader scale, telemental health is a promising tool to address the shortage of mental health providers in the United States, said Dr. Robert C. Gunther, a pediatrician at the University of Virginia Health System in Fishersville. Recent research found that one in three children receiving outpatient care for mental health conditions saw only their primary care doctor for care (Pediatrics. 2015 Nov;136;e1178-85).
“There is a tremendous need for pediatric mental health care,” Dr. Gunther said in an interview. “There is a great shortage of child psychiatrists and other child mental health specialists. Telemental health can help in areas where geography or financial barriers exist to accessing care.”
Data from the Children’s ADHD Telemental Health Treatment Study (CATTS) illustrates the impact that telemental health can have on children facing such barriers to care. Researchers randomized 223 children referred by 88 primary care providers in seven underserved communities into two study groups. Children in the first group were seen by child psychiatrists via videoconference six times over 22 weeks; treatment included pharmacotherapy. Their caregivers received behavior training provided in person by community therapists who were supervised remotely. Children in the second group were treated by their primary care physicians and received one telepsychiatry consult.
Children in both groups improved; however, those randomized to the telemental health model improved “significantly more than patients in the augmented primary care arm” (J Am Acad Child Adolesc Psychiatry. 2015 Apr;54[4]:263-74).
“The CATTS trial demonstrated the effectiveness of a telehealth service model to treat ADHD in communities with limited access to specialty mental health services,” investigators concluded.
But Dr. Joshua J. Alexander, chair of the American Academy of Pediatrics Section on Telehealth Care, notes that some mental health conditions fit more smoothly within the telemental health model than others. ADHD is the most common condition treated by telemental health, he said. The model also has shown success in the treatment of childhood adjustment disorders, anxiety, oppositional defiant disorder, mood disorders, anxiety, and depression.
“I think you have to be careful in determining where telemental health would be beneficial to use and in which cases it might not be an appropriate method to deliver care,” Dr. Alexander said in an interview.
Developing trust and rapport with patients through videoconferencing also can be a challenge, added Dr. Alexander, who is director of the TelAbility telehealth program at the University of North Carolina at Chapel Hill. He recommends that specialists use the technology to continue an existing doctor-patient relationship or to provide care in a consultative model in which the child’s primary care doctor is present along with the patient and patient’s family.
The AAP advocates for the use of telemedicine so long as it is conducted within the context of the medical home. Fragmented telemedicine services that could disrupt continuity of care should be avoided, according to a 2015 AAP policy statement (Pediatrics. 2015 Jun. doi: 10.1542/peds.2015-1253). The academy also calls for the expansion of pediatric telemedicine to increase access to care for underserved communities and improve quality of care for children.
More partnerships between mental health specialists and primary care providers are a key step in delivering high quality pediatric telemental care, Dr. Alexander said.
“Some larger pediatric practices already do this by hiring and colocating individuals at their practice site, but other, smaller practices might not have the room, finances, sufficient patient population, or enough local providers to make this happen,” he said. “A telemedicine program, located within the practice, could enable this specialized service to be provided in a convenient, coordinated setting.”
On Twitter @legal_med
The patient, a 10-year-old girl, was exhibiting defiant and angry behavior at home. Her pediatrician in rural Vermont was not able to support the family optimally, so he referred her to Dr. David C. Rettew.
Dr. Rettew soon learned that the girl blamed herself for her father’s absence, and she interpreted efforts by her mother to set limits as evidence that her mom didn’t love her.
“During the course of the interview, it became clear that the girl had some specific thoughts that were likely fueling her anger,” said Dr. Rettew, director of the Pediatric Psychiatry Clinic at the University of Vermont, Burlington. “She also met criteria clearly for ADHD, which had never been diagnosed. The consultation recommendations not only included some possible medications to try, but also some specific areas that could be addressed for psychotherapy that could really help the relationship between this child and her mother.”
The successful intervention took place via a secure, two-way videoconference. It is one example of how physicians at the University of Vermont are using telemental health to treat children in underserved areas. As part of a state-funded training program, child psychiatry fellows at the university consult with primary care doctors at federally qualified health centers across the state. The primary care physicians discuss patient cases with fellows via phone or email and can refer patients for in-person or telemental health assessments.
The program has been running for about 5 years and so far has yielded countless benefits, said Dr. Rettew, who directs the university’s Child and Adolescent Psychiatry Fellowship Program. “It helps keep the care housed and centered within the primary care home. That can help coordination of services so that care isn’t fragmented around multiple centers. It also allows evaluations to happen that wouldn’t happen otherwise because it’s too much of a hardship for families to travel long distances and come for regular follow-up appointments.”
University of Vermont physicians also use telemedicine to consult with other mental health clinicians across the state. Child psychiatrist Allison Y. Hall provides in-person training to clinicians and then counsels and supervises their efforts through a telemedicine unit.
“For this purpose, it’s great,” said Dr. Hall, who practices at the Vermont Center for Children, Youth, and Families, which is housed within the university’s psychiatry department. “There’s more confidentiality than Skype, for instance. It’s wonderful to be able to reach clinicians at a great distance.”
On a broader scale, telemental health is a promising tool to address the shortage of mental health providers in the United States, said Dr. Robert C. Gunther, a pediatrician at the University of Virginia Health System in Fishersville. Recent research found that one in three children receiving outpatient care for mental health conditions saw only their primary care doctor for care (Pediatrics. 2015 Nov;136;e1178-85).
“There is a tremendous need for pediatric mental health care,” Dr. Gunther said in an interview. “There is a great shortage of child psychiatrists and other child mental health specialists. Telemental health can help in areas where geography or financial barriers exist to accessing care.”
Data from the Children’s ADHD Telemental Health Treatment Study (CATTS) illustrates the impact that telemental health can have on children facing such barriers to care. Researchers randomized 223 children referred by 88 primary care providers in seven underserved communities into two study groups. Children in the first group were seen by child psychiatrists via videoconference six times over 22 weeks; treatment included pharmacotherapy. Their caregivers received behavior training provided in person by community therapists who were supervised remotely. Children in the second group were treated by their primary care physicians and received one telepsychiatry consult.
Children in both groups improved; however, those randomized to the telemental health model improved “significantly more than patients in the augmented primary care arm” (J Am Acad Child Adolesc Psychiatry. 2015 Apr;54[4]:263-74).
“The CATTS trial demonstrated the effectiveness of a telehealth service model to treat ADHD in communities with limited access to specialty mental health services,” investigators concluded.
But Dr. Joshua J. Alexander, chair of the American Academy of Pediatrics Section on Telehealth Care, notes that some mental health conditions fit more smoothly within the telemental health model than others. ADHD is the most common condition treated by telemental health, he said. The model also has shown success in the treatment of childhood adjustment disorders, anxiety, oppositional defiant disorder, mood disorders, anxiety, and depression.
“I think you have to be careful in determining where telemental health would be beneficial to use and in which cases it might not be an appropriate method to deliver care,” Dr. Alexander said in an interview.
Developing trust and rapport with patients through videoconferencing also can be a challenge, added Dr. Alexander, who is director of the TelAbility telehealth program at the University of North Carolina at Chapel Hill. He recommends that specialists use the technology to continue an existing doctor-patient relationship or to provide care in a consultative model in which the child’s primary care doctor is present along with the patient and patient’s family.
The AAP advocates for the use of telemedicine so long as it is conducted within the context of the medical home. Fragmented telemedicine services that could disrupt continuity of care should be avoided, according to a 2015 AAP policy statement (Pediatrics. 2015 Jun. doi: 10.1542/peds.2015-1253). The academy also calls for the expansion of pediatric telemedicine to increase access to care for underserved communities and improve quality of care for children.
More partnerships between mental health specialists and primary care providers are a key step in delivering high quality pediatric telemental care, Dr. Alexander said.
“Some larger pediatric practices already do this by hiring and colocating individuals at their practice site, but other, smaller practices might not have the room, finances, sufficient patient population, or enough local providers to make this happen,” he said. “A telemedicine program, located within the practice, could enable this specialized service to be provided in a convenient, coordinated setting.”
On Twitter @legal_med

On my own during an employee’s maternity leave
Recently, my secretary was out on maternity leave for 6 weeks.
I run a small practice, and my medical assistant works from home on the far side of town. So I was on my own at the office. My MA and I split things up, and since I was the only one physically in the building, I took over all the front office stuff and she took the back office.
I ran the front desk for the whole time – checking people in and out, taking copays, copying insurance cards, giving referrals to therapy places, sending logs to the billing company, and doing other everyday stuff.

Plenty of people asked why I didn’t hire a temp, obviously not knowing how close to the edge a modern solo practice runs. If I hire a temp, that’s another salary to pay, meaning one of the other three of us here would have to skip a few paychecks. I’m not going to put my secretary on unpaid leave for that time. She’s awesome, has been with me since 2004, and has stuck with me through good and bad years. If I don’t pay her that time, she can’t pay her rent, and I don’t have the heart to do that to her. Maybe a big corporate person wouldn’t lose any sleep about it, but I would. Great people are hard to find, and I want to keep the ones I have.
Besides, if I hired a temp, I’d have to train them from the beginning. I don’t use off-the-shelf medical software, just a system I designed myself. It would take time out of my day to teach them how to use it, where I send patients for tests and referrals, and how to sort documents accurately into the correct e-charts. So, for 6 weeks it just seemed easier to do it myself. I know how I like it done.
It wasn’t easy for my MA as well. She had to take over scheduling appointments, handling billing questions, making reminder calls, and doing other miscellaneous stuff. Even after work was over, I’d be at home catching up on all the dictations I hadn’t had time to do, and we’d be going back and forth by phone and email to settle different issues until 8:00 at night or so. By the end of the 6 weeks, we were both pretty burned out and exhausted.
I’m sure the patients weren’t thrilled, either. During that time, they could only reach a voice mail box telling them to leave a message and we’d get back to them as quickly as possible.
I assumed my practice was the only one dinky (or poor, by medical standards) enough to have to resort to this – until I had a chance conversation with a local family practice doctor, when he mentioned he’d had to do something similar when his secretary retired and he didn’t find a replacement for several weeks. A cardiologist mentioned doing the same thing while we were chatting at the hospital. Like me, they were both in solo practice.
This is, apparently, the nature of a modern small practice. The revenue and expense streams are too tight to allow for an extra salary, so even the doctor pitches in to cover. I’m sure my colleagues in large groups will laugh at the thought, but I don’t care. I have to do what’s right for my practice and to survive in the modern medical climate. And if working the front desk for a few weeks is what’s needed to stay independent, so be it.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Recently, my secretary was out on maternity leave for 6 weeks.
I run a small practice, and my medical assistant works from home on the far side of town. So I was on my own at the office. My MA and I split things up, and since I was the only one physically in the building, I took over all the front office stuff and she took the back office.
I ran the front desk for the whole time – checking people in and out, taking copays, copying insurance cards, giving referrals to therapy places, sending logs to the billing company, and doing other everyday stuff.
Plenty of people asked why I didn’t hire a temp, obviously not knowing how close to the edge a modern solo practice runs. If I hire a temp, that’s another salary to pay, meaning one of the other three of us here would have to skip a few paychecks. I’m not going to put my secretary on unpaid leave for that time. She’s awesome, has been with me since 2004, and has stuck with me through good and bad years. If I don’t pay her that time, she can’t pay her rent, and I don’t have the heart to do that to her. Maybe a big corporate person wouldn’t lose any sleep about it, but I would. Great people are hard to find, and I want to keep the ones I have.
Besides, if I hired a temp, I’d have to train them from the beginning. I don’t use off-the-shelf medical software, just a system I designed myself. It would take time out of my day to teach them how to use it, where I send patients for tests and referrals, and how to sort documents accurately into the correct e-charts. So, for 6 weeks it just seemed easier to do it myself. I know how I like it done.
It wasn’t easy for my MA as well. She had to take over scheduling appointments, handling billing questions, making reminder calls, and doing other miscellaneous stuff. Even after work was over, I’d be at home catching up on all the dictations I hadn’t had time to do, and we’d be going back and forth by phone and email to settle different issues until 8:00 at night or so. By the end of the 6 weeks, we were both pretty burned out and exhausted.
I’m sure the patients weren’t thrilled, either. During that time, they could only reach a voice mail box telling them to leave a message and we’d get back to them as quickly as possible.
I assumed my practice was the only one dinky (or poor, by medical standards) enough to have to resort to this – until I had a chance conversation with a local family practice doctor, when he mentioned he’d had to do something similar when his secretary retired and he didn’t find a replacement for several weeks. A cardiologist mentioned doing the same thing while we were chatting at the hospital. Like me, they were both in solo practice.
This is, apparently, the nature of a modern small practice. The revenue and expense streams are too tight to allow for an extra salary, so even the doctor pitches in to cover. I’m sure my colleagues in large groups will laugh at the thought, but I don’t care. I have to do what’s right for my practice and to survive in the modern medical climate. And if working the front desk for a few weeks is what’s needed to stay independent, so be it.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Recently, my secretary was out on maternity leave for 6 weeks.
I run a small practice, and my medical assistant works from home on the far side of town. So I was on my own at the office. My MA and I split things up, and since I was the only one physically in the building, I took over all the front office stuff and she took the back office.
I ran the front desk for the whole time – checking people in and out, taking copays, copying insurance cards, giving referrals to therapy places, sending logs to the billing company, and doing other everyday stuff.
Plenty of people asked why I didn’t hire a temp, obviously not knowing how close to the edge a modern solo practice runs. If I hire a temp, that’s another salary to pay, meaning one of the other three of us here would have to skip a few paychecks. I’m not going to put my secretary on unpaid leave for that time. She’s awesome, has been with me since 2004, and has stuck with me through good and bad years. If I don’t pay her that time, she can’t pay her rent, and I don’t have the heart to do that to her. Maybe a big corporate person wouldn’t lose any sleep about it, but I would. Great people are hard to find, and I want to keep the ones I have.
Besides, if I hired a temp, I’d have to train them from the beginning. I don’t use off-the-shelf medical software, just a system I designed myself. It would take time out of my day to teach them how to use it, where I send patients for tests and referrals, and how to sort documents accurately into the correct e-charts. So, for 6 weeks it just seemed easier to do it myself. I know how I like it done.
It wasn’t easy for my MA as well. She had to take over scheduling appointments, handling billing questions, making reminder calls, and doing other miscellaneous stuff. Even after work was over, I’d be at home catching up on all the dictations I hadn’t had time to do, and we’d be going back and forth by phone and email to settle different issues until 8:00 at night or so. By the end of the 6 weeks, we were both pretty burned out and exhausted.
I’m sure the patients weren’t thrilled, either. During that time, they could only reach a voice mail box telling them to leave a message and we’d get back to them as quickly as possible.
I assumed my practice was the only one dinky (or poor, by medical standards) enough to have to resort to this – until I had a chance conversation with a local family practice doctor, when he mentioned he’d had to do something similar when his secretary retired and he didn’t find a replacement for several weeks. A cardiologist mentioned doing the same thing while we were chatting at the hospital. Like me, they were both in solo practice.
This is, apparently, the nature of a modern small practice. The revenue and expense streams are too tight to allow for an extra salary, so even the doctor pitches in to cover. I’m sure my colleagues in large groups will laugh at the thought, but I don’t care. I have to do what’s right for my practice and to survive in the modern medical climate. And if working the front desk for a few weeks is what’s needed to stay independent, so be it.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
