User login
New type of CAR T cells can produce responses in NHL

Image courtesy of NIAID
Results of a phase 1 study suggest that chimeric antigen receptor T cells specific for the κ light chain (κ.CAR T cells) can produce responses in patients with relapsed or refractory B-cell malignancies, largely without side effects.
The therapy induced complete and partial responses in some patients with non-Hodgkin lymphoma (NHL), and it allowed other patients with chronic lymphocytic leukemia (CLL) or multiple myeloma (MM) to maintain stable disease.
There was 1 adverse event considered possibly related to the treatment.
The researchers reported these results in The Journal of Clinical Investigation.
The κ.CAR T cells were designed to recognize κ-restricted cells and spare normal B cells expressing the nontargeted λ light chain.
“We reasoned that targeting the light chain expressed by malignant B cells should efficiently kill tumor cells while sparing normal B cells expressing the other type of light chain,” said study author Carlos Ramos, MD, of Baylor College of Medicine in Houston, Texas.
He and his colleagues tested the κ.CAR T cells in 16 patients with relapsed or refractory κ+ NHL (n=7), CLL (n=2), or MM (n=7).
The team isolated T cells from these patients and modified the cells so they could target the κ light chain on the surface of malignant B cells. The modified T cells were infused back into the patients, and each patient was monitored for disease progression and side effects.
Eleven patients stopped receiving other treatments at least 4 weeks prior to T-cell infusion. Six patients without lymphopenia received cyclophosphamide at 12.5 mg/kg 4 days before κ.CAR T-cell infusion (0.2×108 to 2×108 κ.CAR T cells/m2).
“We found the treatment to be feasible and safe at all the dose levels studied,” Dr Ramos said.
One MM patient had grade 3 lymphopenia that was deemed possibly related to treatment, but none of the other adverse events were thought to result from the κ.CAR T cells.
Two patients with NHL achieved a complete response to treatment, 1 lasting more than 32 months and the other lasting 6 weeks. A third NHL patient had a partial response lasting 3 months, and a CLL patient had stable disease lasting 6 weeks.
Five of the 7 MM patients had stable disease, 3 lasting 6 weeks, 1 lasting 17 months, and 1 lasting 24 months.
“Our approach, although we are still optimizing it, offers a new possibility for patients in whom other treatments have not been successful,” Dr Ramos concluded. ![]()
Image courtesy of NIAID
Results of a phase 1 study suggest that chimeric antigen receptor T cells specific for the κ light chain (κ.CAR T cells) can produce responses in patients with relapsed or refractory B-cell malignancies, largely without side effects.
The therapy induced complete and partial responses in some patients with non-Hodgkin lymphoma (NHL), and it allowed other patients with chronic lymphocytic leukemia (CLL) or multiple myeloma (MM) to maintain stable disease.
There was 1 adverse event considered possibly related to the treatment.
The researchers reported these results in The Journal of Clinical Investigation.
The κ.CAR T cells were designed to recognize κ-restricted cells and spare normal B cells expressing the nontargeted λ light chain.
“We reasoned that targeting the light chain expressed by malignant B cells should efficiently kill tumor cells while sparing normal B cells expressing the other type of light chain,” said study author Carlos Ramos, MD, of Baylor College of Medicine in Houston, Texas.
He and his colleagues tested the κ.CAR T cells in 16 patients with relapsed or refractory κ+ NHL (n=7), CLL (n=2), or MM (n=7).
The team isolated T cells from these patients and modified the cells so they could target the κ light chain on the surface of malignant B cells. The modified T cells were infused back into the patients, and each patient was monitored for disease progression and side effects.
Eleven patients stopped receiving other treatments at least 4 weeks prior to T-cell infusion. Six patients without lymphopenia received cyclophosphamide at 12.5 mg/kg 4 days before κ.CAR T-cell infusion (0.2×108 to 2×108 κ.CAR T cells/m2).
“We found the treatment to be feasible and safe at all the dose levels studied,” Dr Ramos said.
One MM patient had grade 3 lymphopenia that was deemed possibly related to treatment, but none of the other adverse events were thought to result from the κ.CAR T cells.
Two patients with NHL achieved a complete response to treatment, 1 lasting more than 32 months and the other lasting 6 weeks. A third NHL patient had a partial response lasting 3 months, and a CLL patient had stable disease lasting 6 weeks.
Five of the 7 MM patients had stable disease, 3 lasting 6 weeks, 1 lasting 17 months, and 1 lasting 24 months.
“Our approach, although we are still optimizing it, offers a new possibility for patients in whom other treatments have not been successful,” Dr Ramos concluded. ![]()
Image courtesy of NIAID
Results of a phase 1 study suggest that chimeric antigen receptor T cells specific for the κ light chain (κ.CAR T cells) can produce responses in patients with relapsed or refractory B-cell malignancies, largely without side effects.
The therapy induced complete and partial responses in some patients with non-Hodgkin lymphoma (NHL), and it allowed other patients with chronic lymphocytic leukemia (CLL) or multiple myeloma (MM) to maintain stable disease.
There was 1 adverse event considered possibly related to the treatment.
The researchers reported these results in The Journal of Clinical Investigation.
The κ.CAR T cells were designed to recognize κ-restricted cells and spare normal B cells expressing the nontargeted λ light chain.
“We reasoned that targeting the light chain expressed by malignant B cells should efficiently kill tumor cells while sparing normal B cells expressing the other type of light chain,” said study author Carlos Ramos, MD, of Baylor College of Medicine in Houston, Texas.
He and his colleagues tested the κ.CAR T cells in 16 patients with relapsed or refractory κ+ NHL (n=7), CLL (n=2), or MM (n=7).
The team isolated T cells from these patients and modified the cells so they could target the κ light chain on the surface of malignant B cells. The modified T cells were infused back into the patients, and each patient was monitored for disease progression and side effects.
Eleven patients stopped receiving other treatments at least 4 weeks prior to T-cell infusion. Six patients without lymphopenia received cyclophosphamide at 12.5 mg/kg 4 days before κ.CAR T-cell infusion (0.2×108 to 2×108 κ.CAR T cells/m2).
“We found the treatment to be feasible and safe at all the dose levels studied,” Dr Ramos said.
One MM patient had grade 3 lymphopenia that was deemed possibly related to treatment, but none of the other adverse events were thought to result from the κ.CAR T cells.
Two patients with NHL achieved a complete response to treatment, 1 lasting more than 32 months and the other lasting 6 weeks. A third NHL patient had a partial response lasting 3 months, and a CLL patient had stable disease lasting 6 weeks.
Five of the 7 MM patients had stable disease, 3 lasting 6 weeks, 1 lasting 17 months, and 1 lasting 24 months.
“Our approach, although we are still optimizing it, offers a new possibility for patients in whom other treatments have not been successful,” Dr Ramos concluded. ![]()
Health Canada approves mAb for MM

Photo courtesy of Janssen
Health Canada has granted conditional approval, or a Notice of Compliance with Conditions (NOC/c), for daratumumab (Darzalex), a monoclonal antibody (mAb) targeting CD38.
The mAb is now approved to treat patients with multiple myeloma (MM) who have received at least 3 prior lines of therapy, including a proteasome inhibitor (PI) and an immunomodulatory drug (IMiD), or MM patients who are refractory to both a PI and an IMiD.
An NOC/c is authorization to market a drug with the condition that the sponsor—in this case, Janssen Inc.—undertake additional studies to verify a clinical benefit.
The NOC/c policy is designed to provide access to:
- Drugs that can treat serious, life-threatening, or severely debilitating diseases
- Drugs that can treat conditions for which no drug is currently marketed in Canada
- Drugs that provide a significant increase in efficacy or significant decrease in risk when compared to existing drugs marketed in Canada.
Studies of daratumumab
The NOC/c for daratumumab was based on a review of data from the phase 2 SIRIUS study, the phase 1/2 GEN501 study, and additional supportive studies.
The GEN501 study enrolled 102 patients with relapsed MM or relapsed MM that was refractory to 2 or more prior lines of therapy. The patients received daratumumab at a range of doses and on a number of different schedules.
The results suggested daratumumab is most effective at a dose of 16 mg/kg. At this dose, the overall response rate was 36%. Most adverse events in this study were grade 1 or 2, although serious events did occur.
The SIRIUS study enrolled 124 MM patients who had received 3 or more prior lines of therapy. They received daratumumab at different doses and on different schedules, but 106 patients received the drug at 16 mg/kg.
Twenty-nine percent of the 106 patients responded to treatment, and the median duration of response was 7 months. Thirty percent of patients experienced serious adverse events.
Findings from a combined efficacy analysis of the GEN501 and SIRIUS trials demonstrated that, after a mean follow-up of 14.8 months, the estimated median overall survival for patients who received single-agent daratumumab at 16 mg/kg was 20 months.
Five phase 3 clinical studies with daratumumab in MM patients—in relapsed and frontline settings—are ongoing. Additional studies are ongoing or planned to assess the mAb’s potential in other malignant and pre-malignant diseases in which CD38 is expressed.
Janssen has exclusive worldwide rights to the development, manufacturing, and commercialization of daratumumab for all potential indications. The company licensed daratumumab from Genmab A/S in August 2012. ![]()
Photo courtesy of Janssen
Health Canada has granted conditional approval, or a Notice of Compliance with Conditions (NOC/c), for daratumumab (Darzalex), a monoclonal antibody (mAb) targeting CD38.
The mAb is now approved to treat patients with multiple myeloma (MM) who have received at least 3 prior lines of therapy, including a proteasome inhibitor (PI) and an immunomodulatory drug (IMiD), or MM patients who are refractory to both a PI and an IMiD.
An NOC/c is authorization to market a drug with the condition that the sponsor—in this case, Janssen Inc.—undertake additional studies to verify a clinical benefit.
The NOC/c policy is designed to provide access to:
- Drugs that can treat serious, life-threatening, or severely debilitating diseases
- Drugs that can treat conditions for which no drug is currently marketed in Canada
- Drugs that provide a significant increase in efficacy or significant decrease in risk when compared to existing drugs marketed in Canada.
Studies of daratumumab
The NOC/c for daratumumab was based on a review of data from the phase 2 SIRIUS study, the phase 1/2 GEN501 study, and additional supportive studies.
The GEN501 study enrolled 102 patients with relapsed MM or relapsed MM that was refractory to 2 or more prior lines of therapy. The patients received daratumumab at a range of doses and on a number of different schedules.
The results suggested daratumumab is most effective at a dose of 16 mg/kg. At this dose, the overall response rate was 36%. Most adverse events in this study were grade 1 or 2, although serious events did occur.
The SIRIUS study enrolled 124 MM patients who had received 3 or more prior lines of therapy. They received daratumumab at different doses and on different schedules, but 106 patients received the drug at 16 mg/kg.
Twenty-nine percent of the 106 patients responded to treatment, and the median duration of response was 7 months. Thirty percent of patients experienced serious adverse events.
Findings from a combined efficacy analysis of the GEN501 and SIRIUS trials demonstrated that, after a mean follow-up of 14.8 months, the estimated median overall survival for patients who received single-agent daratumumab at 16 mg/kg was 20 months.
Five phase 3 clinical studies with daratumumab in MM patients—in relapsed and frontline settings—are ongoing. Additional studies are ongoing or planned to assess the mAb’s potential in other malignant and pre-malignant diseases in which CD38 is expressed.
Janssen has exclusive worldwide rights to the development, manufacturing, and commercialization of daratumumab for all potential indications. The company licensed daratumumab from Genmab A/S in August 2012. ![]()
Photo courtesy of Janssen
Health Canada has granted conditional approval, or a Notice of Compliance with Conditions (NOC/c), for daratumumab (Darzalex), a monoclonal antibody (mAb) targeting CD38.
The mAb is now approved to treat patients with multiple myeloma (MM) who have received at least 3 prior lines of therapy, including a proteasome inhibitor (PI) and an immunomodulatory drug (IMiD), or MM patients who are refractory to both a PI and an IMiD.
An NOC/c is authorization to market a drug with the condition that the sponsor—in this case, Janssen Inc.—undertake additional studies to verify a clinical benefit.
The NOC/c policy is designed to provide access to:
- Drugs that can treat serious, life-threatening, or severely debilitating diseases
- Drugs that can treat conditions for which no drug is currently marketed in Canada
- Drugs that provide a significant increase in efficacy or significant decrease in risk when compared to existing drugs marketed in Canada.
Studies of daratumumab
The NOC/c for daratumumab was based on a review of data from the phase 2 SIRIUS study, the phase 1/2 GEN501 study, and additional supportive studies.
The GEN501 study enrolled 102 patients with relapsed MM or relapsed MM that was refractory to 2 or more prior lines of therapy. The patients received daratumumab at a range of doses and on a number of different schedules.
The results suggested daratumumab is most effective at a dose of 16 mg/kg. At this dose, the overall response rate was 36%. Most adverse events in this study were grade 1 or 2, although serious events did occur.
The SIRIUS study enrolled 124 MM patients who had received 3 or more prior lines of therapy. They received daratumumab at different doses and on different schedules, but 106 patients received the drug at 16 mg/kg.
Twenty-nine percent of the 106 patients responded to treatment, and the median duration of response was 7 months. Thirty percent of patients experienced serious adverse events.
Findings from a combined efficacy analysis of the GEN501 and SIRIUS trials demonstrated that, after a mean follow-up of 14.8 months, the estimated median overall survival for patients who received single-agent daratumumab at 16 mg/kg was 20 months.
Five phase 3 clinical studies with daratumumab in MM patients—in relapsed and frontline settings—are ongoing. Additional studies are ongoing or planned to assess the mAb’s potential in other malignant and pre-malignant diseases in which CD38 is expressed.
Janssen has exclusive worldwide rights to the development, manufacturing, and commercialization of daratumumab for all potential indications. The company licensed daratumumab from Genmab A/S in August 2012. ![]()
July 2016 Digital Edition
Click here to access the July 2016 Digital Edition.
Table of Contents
- What is the VA? The Largest Educator of Health Care Professionals in the U.S.
- The Relationship Between Sustained Gripping and the Development of Carpal Tunnel Syndrome
- Efficacy and Safety Outcomes for Patients Taking Warfarin Who Were Switched From Face-to-Face to Telephone Anticoagulation Clinic
- The Most Expensive Drug in the World: To Continue or Discontinue, That Is the Question
- The Unique Value of Externships to Nursing Education and Health Care Organizations
- The Cost of Oncology Drugs: A Pharmacy Perspective, Part 2
- Diabetic Peripheral Neuropathy: The Learning Curve
- Let’s Dance: A Holistic Approach to Treating Veterans With Posttraumatic Stress Disorder

Click here to access the July 2016 Digital Edition.
Table of Contents
- What is the VA? The Largest Educator of Health Care Professionals in the U.S.
- The Relationship Between Sustained Gripping and the Development of Carpal Tunnel Syndrome
- Efficacy and Safety Outcomes for Patients Taking Warfarin Who Were Switched From Face-to-Face to Telephone Anticoagulation Clinic
- The Most Expensive Drug in the World: To Continue or Discontinue, That Is the Question
- The Unique Value of Externships to Nursing Education and Health Care Organizations
- The Cost of Oncology Drugs: A Pharmacy Perspective, Part 2
- Diabetic Peripheral Neuropathy: The Learning Curve
- Let’s Dance: A Holistic Approach to Treating Veterans With Posttraumatic Stress Disorder
Click here to access the July 2016 Digital Edition.
Table of Contents
- What is the VA? The Largest Educator of Health Care Professionals in the U.S.
- The Relationship Between Sustained Gripping and the Development of Carpal Tunnel Syndrome
- Efficacy and Safety Outcomes for Patients Taking Warfarin Who Were Switched From Face-to-Face to Telephone Anticoagulation Clinic
- The Most Expensive Drug in the World: To Continue or Discontinue, That Is the Question
- The Unique Value of Externships to Nursing Education and Health Care Organizations
- The Cost of Oncology Drugs: A Pharmacy Perspective, Part 2
- Diabetic Peripheral Neuropathy: The Learning Curve
- Let’s Dance: A Holistic Approach to Treating Veterans With Posttraumatic Stress Disorder

Psychological Therapies Eased IBS for at Least 6-12 Months
Adults with irritable bowel syndrome (IBS) who underwent psychotherapy improved more than about 75% of controls, and the effect “remained significant and medium in magnitude” for at least 6-12 months, according to a meta-analysis of 41 randomized, controlled trials reported in the July issue of Clinical Gastroenterology and Hepatology.
The finding “is particularly noteworthy because of the typically recurrent, persistent nature of IBS symptoms,” said Kelsey Laird, a doctoral student of clinical psychology at Vanderbilt University in Nashville, Tenn., together with her associates there. “Future research is needed to compare the longevity of treatment effects for psychotherapy with pharmacologic therapies, such as antidepressants,” they added. “Although it is beyond the scope of this review, it is also important to consider the mechanisms by which psychotherapies improve GI symptoms and to determine the ‘active ingredients’ responsible for this effect.”

Up to 16% of individuals in the United States have IBS, and treating it costs anywhere from $950 million to $1.35 billion per year, the researchers noted. Other meta-analyses found psychotherapy about as effective as antidepressants for treating IBS-related GI symptoms over the short term, but its long-term efficacy was unknown, they added. Therefore, they searched PubMed, PsycINFO, Science Direct, and ProQuest through Aug. 15, 2015, identifying randomized controlled trials of psychological therapy and active or nonactive comparators. Psychotherapy included not only traditional psychodynamic and cognitive-behavioral therapies, but also mind-body approaches, such as relaxation training, biofeedback, and yoga, “which can be conceptualized as mindful movement,” Ms. Laird and her associates said. Comparators included support groups, education, sham treatments for hypnosis or biofeedback, online discussion forums, enhanced medical care, treatment as usual, symptom monitoring, and being wait-listed for psychological treatment (Clin Gastroenterol Hepatol. 2016 Jan 21. doi: 10.1016/j.cgh.2015.11.020). The 41 trials included 2,290 patients, comprising 1,183 assigned to the psychological modalities and 1,107 assigned to the various comparators. Taken together, the psychological modalities were associated with greater improvements in GI symptoms immediately after treatment, as compared with the grouped comparators. The Cohen’s d value was 0.69 (95% confidence interval, 0.52-0.86; P less than .001), indicating a medium effect size, the researchers said. Moreover, Cohen’s d values were 0.76 and 0.73, respectively, at short-term follow-up (1-6 months) and long-term follow-up (6-12 months). “On average, individuals who received psychotherapy had a greater reduction in GI symptoms after treatment than 75% of individuals assigned to a control condition,” the researchers concluded.
Effect sizes were similar among cognitive, cognitive-behavioral, and relaxation and hypnosis interventions, and between interventions delivered online and in person, the investigators also reported. Furthermore, longer durations or sessions of psychotherapy did not appear to further improve symptoms.
Study limitations included substantial variability between trials, and the fact that none of the 41 trials could be seen as having a low risk of bias in every domain assessed, the investigators said. “This was partially a result of the difficulty in blinding participants in psychological trials,” they noted. “However, even after excluding this domain, only nine trials were rated as low risk of bias in all remaining domains. Future studies should follow the CONSORT guidelines for [randomized controlled trials], use [intention-to-treat] designs, use active control conditions to control for nonspecific treatment effects, and assess treatment credibility and expectancy.”
The authors reported no funding sources and had no disclosures.
It is well established that psychological therapy is efficacious in managing irritable bowel syndrome (IBS), and it has an associated number needed to treat of four (Am J Gastroenterol. 2014 Sep;109:1350-65). A new meta-analysis from Laird and her colleagues revealed that the positive impact of psychotherapy on IBS symptoms persisted even 1 year after treatment.

|
| Dr. Christopher Almario |
While these findings are impressive and continue to support the use of psychotherapy in IBS, important issues remain. First, these results are based on data gathered in the highly controlled environment of randomized controlled trials (RCTs), and it is unclear whether they will translate to the “real world.” RCT participants may be more willing to complete psychotherapy because they know they are being observed by research staff (referred to as the Hawthorne, or observer, effect). However, in real clinical practice, patients with IBS not subject to the Hawthorne effect may be less compliant with such therapies.
Other issues relate to the current limited adoption of psychotherapy in clinical practice. Factors contributing to the low uptake include variable third-party reimbursement and poor patient and provider acceptance (JAMA. 2015 Mar;313:949-58). Another factor is limited access to qualified psychotherapists. This is an area where telehealth and mobile apps can widen access, especially as Internet-delivered psychotherapy has been shown to be effective (Am J Gastroenterol. 2011;106:1481-91).
Given the high prevalence of IBS, along with the proven, persistent efficacy of psychological therapies in reducing IBS symptoms, efforts to increase both use of and access to these therapies in clinical practice are needed.
Dr. Christopher V. Almario, division of gastroenterology, Cedars-Sinai Medical Center, Los Angeles. He has no relevant conflicts of interest to declare.
It is well established that psychological therapy is efficacious in managing irritable bowel syndrome (IBS), and it has an associated number needed to treat of four (Am J Gastroenterol. 2014 Sep;109:1350-65). A new meta-analysis from Laird and her colleagues revealed that the positive impact of psychotherapy on IBS symptoms persisted even 1 year after treatment.
|
|
| Dr. Christopher Almario |
While these findings are impressive and continue to support the use of psychotherapy in IBS, important issues remain. First, these results are based on data gathered in the highly controlled environment of randomized controlled trials (RCTs), and it is unclear whether they will translate to the “real world.” RCT participants may be more willing to complete psychotherapy because they know they are being observed by research staff (referred to as the Hawthorne, or observer, effect). However, in real clinical practice, patients with IBS not subject to the Hawthorne effect may be less compliant with such therapies.
Other issues relate to the current limited adoption of psychotherapy in clinical practice. Factors contributing to the low uptake include variable third-party reimbursement and poor patient and provider acceptance (JAMA. 2015 Mar;313:949-58). Another factor is limited access to qualified psychotherapists. This is an area where telehealth and mobile apps can widen access, especially as Internet-delivered psychotherapy has been shown to be effective (Am J Gastroenterol. 2011;106:1481-91).
Given the high prevalence of IBS, along with the proven, persistent efficacy of psychological therapies in reducing IBS symptoms, efforts to increase both use of and access to these therapies in clinical practice are needed.
Dr. Christopher V. Almario, division of gastroenterology, Cedars-Sinai Medical Center, Los Angeles. He has no relevant conflicts of interest to declare.
It is well established that psychological therapy is efficacious in managing irritable bowel syndrome (IBS), and it has an associated number needed to treat of four (Am J Gastroenterol. 2014 Sep;109:1350-65). A new meta-analysis from Laird and her colleagues revealed that the positive impact of psychotherapy on IBS symptoms persisted even 1 year after treatment.
|
|
| Dr. Christopher Almario |
While these findings are impressive and continue to support the use of psychotherapy in IBS, important issues remain. First, these results are based on data gathered in the highly controlled environment of randomized controlled trials (RCTs), and it is unclear whether they will translate to the “real world.” RCT participants may be more willing to complete psychotherapy because they know they are being observed by research staff (referred to as the Hawthorne, or observer, effect). However, in real clinical practice, patients with IBS not subject to the Hawthorne effect may be less compliant with such therapies.
Other issues relate to the current limited adoption of psychotherapy in clinical practice. Factors contributing to the low uptake include variable third-party reimbursement and poor patient and provider acceptance (JAMA. 2015 Mar;313:949-58). Another factor is limited access to qualified psychotherapists. This is an area where telehealth and mobile apps can widen access, especially as Internet-delivered psychotherapy has been shown to be effective (Am J Gastroenterol. 2011;106:1481-91).
Given the high prevalence of IBS, along with the proven, persistent efficacy of psychological therapies in reducing IBS symptoms, efforts to increase both use of and access to these therapies in clinical practice are needed.
Dr. Christopher V. Almario, division of gastroenterology, Cedars-Sinai Medical Center, Los Angeles. He has no relevant conflicts of interest to declare.
Adults with irritable bowel syndrome (IBS) who underwent psychotherapy improved more than about 75% of controls, and the effect “remained significant and medium in magnitude” for at least 6-12 months, according to a meta-analysis of 41 randomized, controlled trials reported in the July issue of Clinical Gastroenterology and Hepatology.
The finding “is particularly noteworthy because of the typically recurrent, persistent nature of IBS symptoms,” said Kelsey Laird, a doctoral student of clinical psychology at Vanderbilt University in Nashville, Tenn., together with her associates there. “Future research is needed to compare the longevity of treatment effects for psychotherapy with pharmacologic therapies, such as antidepressants,” they added. “Although it is beyond the scope of this review, it is also important to consider the mechanisms by which psychotherapies improve GI symptoms and to determine the ‘active ingredients’ responsible for this effect.”
Up to 16% of individuals in the United States have IBS, and treating it costs anywhere from $950 million to $1.35 billion per year, the researchers noted. Other meta-analyses found psychotherapy about as effective as antidepressants for treating IBS-related GI symptoms over the short term, but its long-term efficacy was unknown, they added. Therefore, they searched PubMed, PsycINFO, Science Direct, and ProQuest through Aug. 15, 2015, identifying randomized controlled trials of psychological therapy and active or nonactive comparators. Psychotherapy included not only traditional psychodynamic and cognitive-behavioral therapies, but also mind-body approaches, such as relaxation training, biofeedback, and yoga, “which can be conceptualized as mindful movement,” Ms. Laird and her associates said. Comparators included support groups, education, sham treatments for hypnosis or biofeedback, online discussion forums, enhanced medical care, treatment as usual, symptom monitoring, and being wait-listed for psychological treatment (Clin Gastroenterol Hepatol. 2016 Jan 21. doi: 10.1016/j.cgh.2015.11.020). The 41 trials included 2,290 patients, comprising 1,183 assigned to the psychological modalities and 1,107 assigned to the various comparators. Taken together, the psychological modalities were associated with greater improvements in GI symptoms immediately after treatment, as compared with the grouped comparators. The Cohen’s d value was 0.69 (95% confidence interval, 0.52-0.86; P less than .001), indicating a medium effect size, the researchers said. Moreover, Cohen’s d values were 0.76 and 0.73, respectively, at short-term follow-up (1-6 months) and long-term follow-up (6-12 months). “On average, individuals who received psychotherapy had a greater reduction in GI symptoms after treatment than 75% of individuals assigned to a control condition,” the researchers concluded.
Effect sizes were similar among cognitive, cognitive-behavioral, and relaxation and hypnosis interventions, and between interventions delivered online and in person, the investigators also reported. Furthermore, longer durations or sessions of psychotherapy did not appear to further improve symptoms.
Study limitations included substantial variability between trials, and the fact that none of the 41 trials could be seen as having a low risk of bias in every domain assessed, the investigators said. “This was partially a result of the difficulty in blinding participants in psychological trials,” they noted. “However, even after excluding this domain, only nine trials were rated as low risk of bias in all remaining domains. Future studies should follow the CONSORT guidelines for [randomized controlled trials], use [intention-to-treat] designs, use active control conditions to control for nonspecific treatment effects, and assess treatment credibility and expectancy.”
The authors reported no funding sources and had no disclosures.
Adults with irritable bowel syndrome (IBS) who underwent psychotherapy improved more than about 75% of controls, and the effect “remained significant and medium in magnitude” for at least 6-12 months, according to a meta-analysis of 41 randomized, controlled trials reported in the July issue of Clinical Gastroenterology and Hepatology.
The finding “is particularly noteworthy because of the typically recurrent, persistent nature of IBS symptoms,” said Kelsey Laird, a doctoral student of clinical psychology at Vanderbilt University in Nashville, Tenn., together with her associates there. “Future research is needed to compare the longevity of treatment effects for psychotherapy with pharmacologic therapies, such as antidepressants,” they added. “Although it is beyond the scope of this review, it is also important to consider the mechanisms by which psychotherapies improve GI symptoms and to determine the ‘active ingredients’ responsible for this effect.”
Up to 16% of individuals in the United States have IBS, and treating it costs anywhere from $950 million to $1.35 billion per year, the researchers noted. Other meta-analyses found psychotherapy about as effective as antidepressants for treating IBS-related GI symptoms over the short term, but its long-term efficacy was unknown, they added. Therefore, they searched PubMed, PsycINFO, Science Direct, and ProQuest through Aug. 15, 2015, identifying randomized controlled trials of psychological therapy and active or nonactive comparators. Psychotherapy included not only traditional psychodynamic and cognitive-behavioral therapies, but also mind-body approaches, such as relaxation training, biofeedback, and yoga, “which can be conceptualized as mindful movement,” Ms. Laird and her associates said. Comparators included support groups, education, sham treatments for hypnosis or biofeedback, online discussion forums, enhanced medical care, treatment as usual, symptom monitoring, and being wait-listed for psychological treatment (Clin Gastroenterol Hepatol. 2016 Jan 21. doi: 10.1016/j.cgh.2015.11.020). The 41 trials included 2,290 patients, comprising 1,183 assigned to the psychological modalities and 1,107 assigned to the various comparators. Taken together, the psychological modalities were associated with greater improvements in GI symptoms immediately after treatment, as compared with the grouped comparators. The Cohen’s d value was 0.69 (95% confidence interval, 0.52-0.86; P less than .001), indicating a medium effect size, the researchers said. Moreover, Cohen’s d values were 0.76 and 0.73, respectively, at short-term follow-up (1-6 months) and long-term follow-up (6-12 months). “On average, individuals who received psychotherapy had a greater reduction in GI symptoms after treatment than 75% of individuals assigned to a control condition,” the researchers concluded.
Effect sizes were similar among cognitive, cognitive-behavioral, and relaxation and hypnosis interventions, and between interventions delivered online and in person, the investigators also reported. Furthermore, longer durations or sessions of psychotherapy did not appear to further improve symptoms.
Study limitations included substantial variability between trials, and the fact that none of the 41 trials could be seen as having a low risk of bias in every domain assessed, the investigators said. “This was partially a result of the difficulty in blinding participants in psychological trials,” they noted. “However, even after excluding this domain, only nine trials were rated as low risk of bias in all remaining domains. Future studies should follow the CONSORT guidelines for [randomized controlled trials], use [intention-to-treat] designs, use active control conditions to control for nonspecific treatment effects, and assess treatment credibility and expectancy.”
The authors reported no funding sources and had no disclosures.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY

Inhibiting integrin-mediated activation might help treat, reverse chronic pancreatitis
Integrins that bind arginine-glycine-aspartic acid (RGD) activated stellate cells in the pancreas, inducing pancreatic fibrogenesis in mice, researchers reported in the July issue of Cellular and Molecular Gastroenterology and Hepatology.
“Small-molecule antagonists of this interaction might be developed for the treatment of pancreatic fibrotic diseases,” wrote Dr. Barbara Ulmasov and her associates at Saint Louis University.
Cytokine transforming growth factor beta, or TGFB, plays a “central” role in the activation of pancreatic stellate cells and the promotion of fibrogenesis, both in the pancreas and in other organs, the investigators noted. Because latent TGFB is “abundantly present” in the pancreas and most other tissues, it might be more important to control the activation of TGFB than its expression, they added. Studies of other organs have shown that TGFB is activated when integrins of the av family bind the RGD sequence, but no one had determined whether this was true for pancreatic fibrogenesis, Dr. Ulmasov and her associates asserted (Cell Mol Gastroenterol Hepatol. 2016 Mar 16. doi: 10.1016/j.jcmgh.2016.03.004).
Therefore, they repeatedly injected female C57BL/6 mice with cerulein to induce pancreatic fibrogenesis, and gave a group of control mice sterile saline instead. The mice then received continuous infusions of a small molecule called CWHM-12, which is an antagonist of RGD-integrin that is known to prevent both pulmonary and hepatic fibrosis in mice. After euthanizing the mice, the researchers measured pancreatic parenchymal atrophy, fibrosis, and activation of pancreatic stellate cells. They also studied TGFB activation in an established line of pancreatic stellate cells from rats.
Pancreatic stellate cells expressed messenger RNAs encoding RGD-binding integrins, the investigators found. The mice that received cerulein had higher levels of these integrins than the mice that received saline, and the cerulein group also had more disrupted acinar cell architecture, tubular complexes, and infiltrations of inflammatory cells. Mice that received prophylactic CWHM-12 had only somewhat less acinar cell loss and atrophy than the control mice, and had similar levels of inflammatory cell infiltration, but had “dramatically” lower levels of pancreatic fibrosis, the researchers said. Even if mice received CWHM-12 several days after starting cerulein, they still had less fibrosis and activation of TGFB than if they received saline, they noted.
The established line of pancreatic stellate cells “could robustly activate endogenously produced TGFB,” the investigators also reported. Furthermore, CWHM-12 “potently blocked TGFB activation,” unlike the control compound. Taken together, the findings illustrate the “critical role of RGD-binding integrins in chronic pancreatitis, and the promising potential to arrest or possibly even reverse pancreatic fibrosis using a pharmacologic approach to inhibiting integrin-mediated TGFB activation,” the researchers concluded.
The National Pancreas Foundation and the Frank R. Burton Memorial Fund supported the study. Dr. Ulmasov had no disclosures. Three coinvestigators reported being consultants and/or holding equity in Integrin Therapeutics, Nimbus Therapeutics, Bristol-Myers Squibb, Janssen, Mitsubishi Tanabe, Conatus, and Scholar Rock.
Integrins are transmembrane proteins that organize epithelial cells and transmit signals from the tissue matrix. These proteins consist of two subunits that partner to form more than 20 specific combinations, are induced upon tissue injury, and act as signaling molecules that mediate inflammatory and wound-healing responses. The utility of targeting integrins has been established by drugs such as vedolizumab, which targets specific integrins to dampen injury in inflammatory bowel disease.

|
Dr. Chuhan Chung |
Specific integrins also mediate profibrotic responses by activating TGF-beta, the major fibrogenic cytokine. An arginine-glycine-aspartic acid (RGD) integrin-binding motif found on the TGF-beta molecule triggers this activation upon interaction with specific integrins. Blocking the RGD-integrin interaction reduces fibrosis in multiple organs including the lung, liver, and kidney.
In the current issue of Cellular and Molecular Gastroenterology and Hepatology, Ulmasov et al. report on the use of a synthetic peptide (CWHM-12) in a model of chronic pancreatitis. This peptide mimetic antagonizes the RGD interaction with integrins, thereby limiting TGF-beta activation. Using a cerulein-induced pancreatic fibrosis model, the authors demonstrated that CWHM-12 inhibits pancreatic stellate cell (PSC) activation and generation of active TGF-beta. CWHM-12 suppressed fibrosis when administered prior to cerulein injection, and to a lesser extent, during the course of generating fibrosis. The alpha-v-beta1 integrin was identified as a critical integrin and was expressed at high levels in the murine pancreas, primary PSCs, and further in cerulein-induced pancreatitis.

|
Dr. Fred Gorelick |
Limitations of the current study warrant comment. The most obvious is that this model does not generate true chronic pancreatitis, because unlike chronic pancreatitis, fibrosis resolves spontaneously. Proof of effect in more robust chronic pancreatitis models is needed. Off-target effects are also suggested by the finding that serum white blood counts were significantly higher with CWHM-12. Finally, the chronicity and unpredictability of human chronic pancreatitis make this preclinical study an early starting point for determining whether CWHM-12 has true “clinical legs.”
Dr. Chuhan Chung and Dr. Fred Gorelick are in the department of medicine, Yale University, New Haven, Conn., and the VA Connecticut Healthcare System, West Haven.
Integrins are transmembrane proteins that organize epithelial cells and transmit signals from the tissue matrix. These proteins consist of two subunits that partner to form more than 20 specific combinations, are induced upon tissue injury, and act as signaling molecules that mediate inflammatory and wound-healing responses. The utility of targeting integrins has been established by drugs such as vedolizumab, which targets specific integrins to dampen injury in inflammatory bowel disease.
|
|
Dr. Chuhan Chung |
Specific integrins also mediate profibrotic responses by activating TGF-beta, the major fibrogenic cytokine. An arginine-glycine-aspartic acid (RGD) integrin-binding motif found on the TGF-beta molecule triggers this activation upon interaction with specific integrins. Blocking the RGD-integrin interaction reduces fibrosis in multiple organs including the lung, liver, and kidney.
In the current issue of Cellular and Molecular Gastroenterology and Hepatology, Ulmasov et al. report on the use of a synthetic peptide (CWHM-12) in a model of chronic pancreatitis. This peptide mimetic antagonizes the RGD interaction with integrins, thereby limiting TGF-beta activation. Using a cerulein-induced pancreatic fibrosis model, the authors demonstrated that CWHM-12 inhibits pancreatic stellate cell (PSC) activation and generation of active TGF-beta. CWHM-12 suppressed fibrosis when administered prior to cerulein injection, and to a lesser extent, during the course of generating fibrosis. The alpha-v-beta1 integrin was identified as a critical integrin and was expressed at high levels in the murine pancreas, primary PSCs, and further in cerulein-induced pancreatitis.
|
|
Dr. Fred Gorelick |
Limitations of the current study warrant comment. The most obvious is that this model does not generate true chronic pancreatitis, because unlike chronic pancreatitis, fibrosis resolves spontaneously. Proof of effect in more robust chronic pancreatitis models is needed. Off-target effects are also suggested by the finding that serum white blood counts were significantly higher with CWHM-12. Finally, the chronicity and unpredictability of human chronic pancreatitis make this preclinical study an early starting point for determining whether CWHM-12 has true “clinical legs.”
Dr. Chuhan Chung and Dr. Fred Gorelick are in the department of medicine, Yale University, New Haven, Conn., and the VA Connecticut Healthcare System, West Haven.
Integrins are transmembrane proteins that organize epithelial cells and transmit signals from the tissue matrix. These proteins consist of two subunits that partner to form more than 20 specific combinations, are induced upon tissue injury, and act as signaling molecules that mediate inflammatory and wound-healing responses. The utility of targeting integrins has been established by drugs such as vedolizumab, which targets specific integrins to dampen injury in inflammatory bowel disease.
|
|
Dr. Chuhan Chung |
Specific integrins also mediate profibrotic responses by activating TGF-beta, the major fibrogenic cytokine. An arginine-glycine-aspartic acid (RGD) integrin-binding motif found on the TGF-beta molecule triggers this activation upon interaction with specific integrins. Blocking the RGD-integrin interaction reduces fibrosis in multiple organs including the lung, liver, and kidney.
In the current issue of Cellular and Molecular Gastroenterology and Hepatology, Ulmasov et al. report on the use of a synthetic peptide (CWHM-12) in a model of chronic pancreatitis. This peptide mimetic antagonizes the RGD interaction with integrins, thereby limiting TGF-beta activation. Using a cerulein-induced pancreatic fibrosis model, the authors demonstrated that CWHM-12 inhibits pancreatic stellate cell (PSC) activation and generation of active TGF-beta. CWHM-12 suppressed fibrosis when administered prior to cerulein injection, and to a lesser extent, during the course of generating fibrosis. The alpha-v-beta1 integrin was identified as a critical integrin and was expressed at high levels in the murine pancreas, primary PSCs, and further in cerulein-induced pancreatitis.
|
|
Dr. Fred Gorelick |
Limitations of the current study warrant comment. The most obvious is that this model does not generate true chronic pancreatitis, because unlike chronic pancreatitis, fibrosis resolves spontaneously. Proof of effect in more robust chronic pancreatitis models is needed. Off-target effects are also suggested by the finding that serum white blood counts were significantly higher with CWHM-12. Finally, the chronicity and unpredictability of human chronic pancreatitis make this preclinical study an early starting point for determining whether CWHM-12 has true “clinical legs.”
Dr. Chuhan Chung and Dr. Fred Gorelick are in the department of medicine, Yale University, New Haven, Conn., and the VA Connecticut Healthcare System, West Haven.
Integrins that bind arginine-glycine-aspartic acid (RGD) activated stellate cells in the pancreas, inducing pancreatic fibrogenesis in mice, researchers reported in the July issue of Cellular and Molecular Gastroenterology and Hepatology.
“Small-molecule antagonists of this interaction might be developed for the treatment of pancreatic fibrotic diseases,” wrote Dr. Barbara Ulmasov and her associates at Saint Louis University.
Cytokine transforming growth factor beta, or TGFB, plays a “central” role in the activation of pancreatic stellate cells and the promotion of fibrogenesis, both in the pancreas and in other organs, the investigators noted. Because latent TGFB is “abundantly present” in the pancreas and most other tissues, it might be more important to control the activation of TGFB than its expression, they added. Studies of other organs have shown that TGFB is activated when integrins of the av family bind the RGD sequence, but no one had determined whether this was true for pancreatic fibrogenesis, Dr. Ulmasov and her associates asserted (Cell Mol Gastroenterol Hepatol. 2016 Mar 16. doi: 10.1016/j.jcmgh.2016.03.004).
Therefore, they repeatedly injected female C57BL/6 mice with cerulein to induce pancreatic fibrogenesis, and gave a group of control mice sterile saline instead. The mice then received continuous infusions of a small molecule called CWHM-12, which is an antagonist of RGD-integrin that is known to prevent both pulmonary and hepatic fibrosis in mice. After euthanizing the mice, the researchers measured pancreatic parenchymal atrophy, fibrosis, and activation of pancreatic stellate cells. They also studied TGFB activation in an established line of pancreatic stellate cells from rats.
Pancreatic stellate cells expressed messenger RNAs encoding RGD-binding integrins, the investigators found. The mice that received cerulein had higher levels of these integrins than the mice that received saline, and the cerulein group also had more disrupted acinar cell architecture, tubular complexes, and infiltrations of inflammatory cells. Mice that received prophylactic CWHM-12 had only somewhat less acinar cell loss and atrophy than the control mice, and had similar levels of inflammatory cell infiltration, but had “dramatically” lower levels of pancreatic fibrosis, the researchers said. Even if mice received CWHM-12 several days after starting cerulein, they still had less fibrosis and activation of TGFB than if they received saline, they noted.
The established line of pancreatic stellate cells “could robustly activate endogenously produced TGFB,” the investigators also reported. Furthermore, CWHM-12 “potently blocked TGFB activation,” unlike the control compound. Taken together, the findings illustrate the “critical role of RGD-binding integrins in chronic pancreatitis, and the promising potential to arrest or possibly even reverse pancreatic fibrosis using a pharmacologic approach to inhibiting integrin-mediated TGFB activation,” the researchers concluded.
The National Pancreas Foundation and the Frank R. Burton Memorial Fund supported the study. Dr. Ulmasov had no disclosures. Three coinvestigators reported being consultants and/or holding equity in Integrin Therapeutics, Nimbus Therapeutics, Bristol-Myers Squibb, Janssen, Mitsubishi Tanabe, Conatus, and Scholar Rock.
Integrins that bind arginine-glycine-aspartic acid (RGD) activated stellate cells in the pancreas, inducing pancreatic fibrogenesis in mice, researchers reported in the July issue of Cellular and Molecular Gastroenterology and Hepatology.
“Small-molecule antagonists of this interaction might be developed for the treatment of pancreatic fibrotic diseases,” wrote Dr. Barbara Ulmasov and her associates at Saint Louis University.
Cytokine transforming growth factor beta, or TGFB, plays a “central” role in the activation of pancreatic stellate cells and the promotion of fibrogenesis, both in the pancreas and in other organs, the investigators noted. Because latent TGFB is “abundantly present” in the pancreas and most other tissues, it might be more important to control the activation of TGFB than its expression, they added. Studies of other organs have shown that TGFB is activated when integrins of the av family bind the RGD sequence, but no one had determined whether this was true for pancreatic fibrogenesis, Dr. Ulmasov and her associates asserted (Cell Mol Gastroenterol Hepatol. 2016 Mar 16. doi: 10.1016/j.jcmgh.2016.03.004).
Therefore, they repeatedly injected female C57BL/6 mice with cerulein to induce pancreatic fibrogenesis, and gave a group of control mice sterile saline instead. The mice then received continuous infusions of a small molecule called CWHM-12, which is an antagonist of RGD-integrin that is known to prevent both pulmonary and hepatic fibrosis in mice. After euthanizing the mice, the researchers measured pancreatic parenchymal atrophy, fibrosis, and activation of pancreatic stellate cells. They also studied TGFB activation in an established line of pancreatic stellate cells from rats.
Pancreatic stellate cells expressed messenger RNAs encoding RGD-binding integrins, the investigators found. The mice that received cerulein had higher levels of these integrins than the mice that received saline, and the cerulein group also had more disrupted acinar cell architecture, tubular complexes, and infiltrations of inflammatory cells. Mice that received prophylactic CWHM-12 had only somewhat less acinar cell loss and atrophy than the control mice, and had similar levels of inflammatory cell infiltration, but had “dramatically” lower levels of pancreatic fibrosis, the researchers said. Even if mice received CWHM-12 several days after starting cerulein, they still had less fibrosis and activation of TGFB than if they received saline, they noted.
The established line of pancreatic stellate cells “could robustly activate endogenously produced TGFB,” the investigators also reported. Furthermore, CWHM-12 “potently blocked TGFB activation,” unlike the control compound. Taken together, the findings illustrate the “critical role of RGD-binding integrins in chronic pancreatitis, and the promising potential to arrest or possibly even reverse pancreatic fibrosis using a pharmacologic approach to inhibiting integrin-mediated TGFB activation,” the researchers concluded.
The National Pancreas Foundation and the Frank R. Burton Memorial Fund supported the study. Dr. Ulmasov had no disclosures. Three coinvestigators reported being consultants and/or holding equity in Integrin Therapeutics, Nimbus Therapeutics, Bristol-Myers Squibb, Janssen, Mitsubishi Tanabe, Conatus, and Scholar Rock.
FROM CELLULAR AND MOLECULAR GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: Small molecules that inhibit the interaction between integrins and RGD (arginine-glycine-aspartic acid) might effectively treat chronic fibrosing pancreatic diseases.
Major finding: Continuous infusion with an active RGD peptidomimetic reduced atrophy and loss of pancreatic acinar cells and helped prevent pancreatic fibrosis, activation of pancreatic stellate cells, and expression of genes regulated by cytokine transforming growth factor beta.
Data source: A study of C57BL/6 female mice with chronic pancreatitis induced by repeated administration of cerulein.
Disclosures: The National Pancreas Foundation and the Frank R. Burton Memorial Fund supported the study. Dr. Ulmasov had no disclosures. Three coinvestigators reported being consultants and/or holding equity in Integrin Therapeutics, Nimbus Therapeutics, Bristol Myers Squibb, Janssen, Mitsubishi Tanabe, Conatus, and Scholar Rock.

Early sustained viral response linked to better outcomes among HCV patients
Among patients with hepatitis C virus infection who were in compensated cirrhosis, sustained viral response was associated with significantly lower rates of liver decompensation, hepatocellular carcinoma, and liver-related death, even in the presence of clinically meaningful portal hypertension.
But patients in stage 2 cirrhosis were more likely than were stage 1 patients to develop liver decompensation and to die of hepatocellular carcinoma, regardless of SVR, said Dr. Vito Di Marco and his associates at the University of Palermo, Italy. “The available evidence, including our own, suggests that it is opportune to treat HCV as early as possible, in order to reduce progression to stages of cirrhosis in which a viral cure may be less likely lead to ultimate achievement of a major benefit,” they wrote. The report is in the July issue of Gastroenterology.
Clinically significant portal hypertension causes esophageal varices, a “landmark” indicator of stage 2 cirrhosis, the researchers noted. To parse the effects of SVR by cirrhosis stage, they followed all 444 HCV patients with compensated cirrhosis who were treated with pegylated interferon a2b and ribavirin at their tertiary liver centers between 2001 and 2009 (Gastroenterology 2016 March 31. doi: 10.1053/j.gastro.2016.03.036).
The cohort included 218 patients without esophageal varices (that is, patients in stage 1 cirrhosis) and 226 patients with esophageal varices (stage 2 cirrhosis), the researchers said. The groups were demographically similar at baseline, and had similar body mass indices and rates of comorbidities. Rates of SVR were 31% for patients in stage 1 cirrhosis and 18% for patients in stage 2 cirrhosis (P = .003).
Among stage 1 patients, achieving SVR was associated with a lower risk of liver decompensation (0% versus 7%; P = .009), a lower annual rate of hepatocellular carcinoma (0.7% versus 2.9%; P = .002), and a lower risk of liver-related death (3% versus 12%; P = .03). Stage 1 cirrhosis patients also were about 75% less likely to develop esophageal varices if they achieved SVR than if they did not (hazard ratio [HR], 0.23; 95% confidence interval [CI], 0.11 to 0.48; P less than .001).
Eradicating HCV infection also was associated with better clinical outcomes among patients in stage 2 cirrhosis, according to the investigators. For example, those who achieved SVR had a 0.9% annual rate of hepatocellular carcinoma, compared with 3.6% for those who did not (P = .002). And only 2% of stage 2 patients who achieved SVR died from hepatocellular carcinoma, compared to 18% of those who did not (P = .003).
But regardless of SVR, patients with stage 2 cirrhosis were nearly three times more likely to develop liver compensation (HR, 2.8; 95% CI, 1.73 to 4.59) and about 1.75 times more likely to die of liver-related causes (HR, 1.77; 95% CI, 1.12 to 2.80) compared with patients in stage 1 cirrhosis, the investigators emphasized. Also, achieving SVR did not prevent stage 2 patients from developing more esophageal varices (HR, 1.58; 95% CI, 0.33 to 1.03).
The researchers acknowledged several study limitations. “We have performed many comparisons with respect to multiple endpoints, which could have increased the likelihood of inference errors that can result from multiple comparisons,” they wrote. “Furthermore, we cannot exclude the lack of power as an explanation for some of our results.” In addition, pegylated interferon–based therapies have low rates of SVR, and the study was limited to patients who could tolerate the regimen, they added. Nonetheless, the results indicate that eradicating HCV is clinically beneficial even in advanced compensated cirrhosis, but that early treatment helps maximize these outcomes, they concluded. “Pegylated interferon–free direct-acting antiviral combo regimens will ultimately increase the rate of virological cure to almost universal effectiveness,” they added. “It remains to be assessed whether the effectiveness of these new regimens will be automatically translated into a universal clinical benefit.”
The Italian Ministry of Health funded the study. The investigators had no disclosures.
Among patients with hepatitis C virus infection who were in compensated cirrhosis, sustained viral response was associated with significantly lower rates of liver decompensation, hepatocellular carcinoma, and liver-related death, even in the presence of clinically meaningful portal hypertension.
But patients in stage 2 cirrhosis were more likely than were stage 1 patients to develop liver decompensation and to die of hepatocellular carcinoma, regardless of SVR, said Dr. Vito Di Marco and his associates at the University of Palermo, Italy. “The available evidence, including our own, suggests that it is opportune to treat HCV as early as possible, in order to reduce progression to stages of cirrhosis in which a viral cure may be less likely lead to ultimate achievement of a major benefit,” they wrote. The report is in the July issue of Gastroenterology.
Clinically significant portal hypertension causes esophageal varices, a “landmark” indicator of stage 2 cirrhosis, the researchers noted. To parse the effects of SVR by cirrhosis stage, they followed all 444 HCV patients with compensated cirrhosis who were treated with pegylated interferon a2b and ribavirin at their tertiary liver centers between 2001 and 2009 (Gastroenterology 2016 March 31. doi: 10.1053/j.gastro.2016.03.036).
The cohort included 218 patients without esophageal varices (that is, patients in stage 1 cirrhosis) and 226 patients with esophageal varices (stage 2 cirrhosis), the researchers said. The groups were demographically similar at baseline, and had similar body mass indices and rates of comorbidities. Rates of SVR were 31% for patients in stage 1 cirrhosis and 18% for patients in stage 2 cirrhosis (P = .003).
Among stage 1 patients, achieving SVR was associated with a lower risk of liver decompensation (0% versus 7%; P = .009), a lower annual rate of hepatocellular carcinoma (0.7% versus 2.9%; P = .002), and a lower risk of liver-related death (3% versus 12%; P = .03). Stage 1 cirrhosis patients also were about 75% less likely to develop esophageal varices if they achieved SVR than if they did not (hazard ratio [HR], 0.23; 95% confidence interval [CI], 0.11 to 0.48; P less than .001).
Eradicating HCV infection also was associated with better clinical outcomes among patients in stage 2 cirrhosis, according to the investigators. For example, those who achieved SVR had a 0.9% annual rate of hepatocellular carcinoma, compared with 3.6% for those who did not (P = .002). And only 2% of stage 2 patients who achieved SVR died from hepatocellular carcinoma, compared to 18% of those who did not (P = .003).
But regardless of SVR, patients with stage 2 cirrhosis were nearly three times more likely to develop liver compensation (HR, 2.8; 95% CI, 1.73 to 4.59) and about 1.75 times more likely to die of liver-related causes (HR, 1.77; 95% CI, 1.12 to 2.80) compared with patients in stage 1 cirrhosis, the investigators emphasized. Also, achieving SVR did not prevent stage 2 patients from developing more esophageal varices (HR, 1.58; 95% CI, 0.33 to 1.03).
The researchers acknowledged several study limitations. “We have performed many comparisons with respect to multiple endpoints, which could have increased the likelihood of inference errors that can result from multiple comparisons,” they wrote. “Furthermore, we cannot exclude the lack of power as an explanation for some of our results.” In addition, pegylated interferon–based therapies have low rates of SVR, and the study was limited to patients who could tolerate the regimen, they added. Nonetheless, the results indicate that eradicating HCV is clinically beneficial even in advanced compensated cirrhosis, but that early treatment helps maximize these outcomes, they concluded. “Pegylated interferon–free direct-acting antiviral combo regimens will ultimately increase the rate of virological cure to almost universal effectiveness,” they added. “It remains to be assessed whether the effectiveness of these new regimens will be automatically translated into a universal clinical benefit.”
The Italian Ministry of Health funded the study. The investigators had no disclosures.
Among patients with hepatitis C virus infection who were in compensated cirrhosis, sustained viral response was associated with significantly lower rates of liver decompensation, hepatocellular carcinoma, and liver-related death, even in the presence of clinically meaningful portal hypertension.
But patients in stage 2 cirrhosis were more likely than were stage 1 patients to develop liver decompensation and to die of hepatocellular carcinoma, regardless of SVR, said Dr. Vito Di Marco and his associates at the University of Palermo, Italy. “The available evidence, including our own, suggests that it is opportune to treat HCV as early as possible, in order to reduce progression to stages of cirrhosis in which a viral cure may be less likely lead to ultimate achievement of a major benefit,” they wrote. The report is in the July issue of Gastroenterology.
Clinically significant portal hypertension causes esophageal varices, a “landmark” indicator of stage 2 cirrhosis, the researchers noted. To parse the effects of SVR by cirrhosis stage, they followed all 444 HCV patients with compensated cirrhosis who were treated with pegylated interferon a2b and ribavirin at their tertiary liver centers between 2001 and 2009 (Gastroenterology 2016 March 31. doi: 10.1053/j.gastro.2016.03.036).
The cohort included 218 patients without esophageal varices (that is, patients in stage 1 cirrhosis) and 226 patients with esophageal varices (stage 2 cirrhosis), the researchers said. The groups were demographically similar at baseline, and had similar body mass indices and rates of comorbidities. Rates of SVR were 31% for patients in stage 1 cirrhosis and 18% for patients in stage 2 cirrhosis (P = .003).
Among stage 1 patients, achieving SVR was associated with a lower risk of liver decompensation (0% versus 7%; P = .009), a lower annual rate of hepatocellular carcinoma (0.7% versus 2.9%; P = .002), and a lower risk of liver-related death (3% versus 12%; P = .03). Stage 1 cirrhosis patients also were about 75% less likely to develop esophageal varices if they achieved SVR than if they did not (hazard ratio [HR], 0.23; 95% confidence interval [CI], 0.11 to 0.48; P less than .001).
Eradicating HCV infection also was associated with better clinical outcomes among patients in stage 2 cirrhosis, according to the investigators. For example, those who achieved SVR had a 0.9% annual rate of hepatocellular carcinoma, compared with 3.6% for those who did not (P = .002). And only 2% of stage 2 patients who achieved SVR died from hepatocellular carcinoma, compared to 18% of those who did not (P = .003).
But regardless of SVR, patients with stage 2 cirrhosis were nearly three times more likely to develop liver compensation (HR, 2.8; 95% CI, 1.73 to 4.59) and about 1.75 times more likely to die of liver-related causes (HR, 1.77; 95% CI, 1.12 to 2.80) compared with patients in stage 1 cirrhosis, the investigators emphasized. Also, achieving SVR did not prevent stage 2 patients from developing more esophageal varices (HR, 1.58; 95% CI, 0.33 to 1.03).
The researchers acknowledged several study limitations. “We have performed many comparisons with respect to multiple endpoints, which could have increased the likelihood of inference errors that can result from multiple comparisons,” they wrote. “Furthermore, we cannot exclude the lack of power as an explanation for some of our results.” In addition, pegylated interferon–based therapies have low rates of SVR, and the study was limited to patients who could tolerate the regimen, they added. Nonetheless, the results indicate that eradicating HCV is clinically beneficial even in advanced compensated cirrhosis, but that early treatment helps maximize these outcomes, they concluded. “Pegylated interferon–free direct-acting antiviral combo regimens will ultimately increase the rate of virological cure to almost universal effectiveness,” they added. “It remains to be assessed whether the effectiveness of these new regimens will be automatically translated into a universal clinical benefit.”
The Italian Ministry of Health funded the study. The investigators had no disclosures.
FROM GASTROENTEROLOGY
Key clinical point: Patients with hepatitis C virus infections may have the best long-term outcomes if they achieve sustained viral response before they develop clinically significant portal hypertension.
Major finding: Patients with stage 2 cirrhosis, regardless of SVR, were at significantly greater risk of liver decompensation (hazard ratio, 2.8; P less than .001) and liver-related death (HR, 1.77; P = .015) compared with stage 1 cirrhosis patients.
Data source: A single-center prospective observational study of 444 patients with HCV and compensated cirrhosis, including 218 with stage 1 disease and 226 with stage 2 disease.
Disclosures: The Italian Ministry of Health funded the study. The investigators had no disclosures.
Immunosuppressive regimens did not affect risk of cancer recurrence in meta-analysis
Among patients with immune-mediated diseases and a history of malignancy, cancer recurrence rates were similar regardless of whether they received tumor necrosis factor inhibitors, traditional immunosuppressants, or no immunosuppression, according to a meta-analysis of 16 cohort and case-control studies.
“However, there is a need for larger studies to prospectively monitor for cancer recurrence and new malignancies in this population to better inform our practice,” wrote Dr. Edward Shelton of Massachusetts General Hospital in Boston, and his associates. The report is in the July issue of Gastroenterology.
Patients with immune-mediated diseases often have a history of malignancy, raising questions about the effects of therapies that target the immune system, the researchers noted. However, relevant studies have been “small with few events, preventing robust estimates of risk,” they said. They searched Medline, Embase, and conference proceedings through April 2015 for studies of the risk of primary or recurrent cancer among patients with a history of malignancy who were exposed to thiopurines, methotrexate, or anti-TNF agents, or no immunosuppression. Among the 16 studies meeting these criteria, seven included patients with rheumatoid arthritis, seven included patients with inflammatory bowel disease, one included both diseases, and one included patients with psoriasis. Twelve were cohort studies, one was a case-control study, and three were case series. The resulting meta-analysis comprised 11,702 patients who contributed a total of 31,258 person-years of follow-up (Gastroenterology 2016 May 13. doi: 10.1053/j.gastro.2016.03.037).
Rates of cancer recurrence were statistically similar among patients who received no immunosuppression, anti-TNF therapy, traditional immune modulators, or combination regimens (P greater than .1 for differences among groups). Numerically, however, the risk of recurrent cancer was highest with combination immunotherapy (54.5 cases per 1,000 person-years of follow-up), compared with 37.5 cases per 1,000 person-years for patients who did not receive immunosuppressive treatment, 36.2 cases per 1,000 person-years for patients who received traditional immunomodulator monotherapy, and 33.8 cases per 1,000 person-years for patients who received anti-TNF agents. Analyses of subgroups of patients with new or recurrent cancers, or various types of therapies, and of specific immune-mediated diseases yielded “similar results, with no increase in risk,” said the researchers. Furthermore, rates of cancer recurrence did not statistically differ depending on whether patients started immunosuppression less than or more than 6 years after their index cancer (P = .43).
“Treatment decisions after a cancer diagnosis are complex, and must take into account the natural history of cancer, histologic type and stage, time from diagnosis, and course of underlying chronic inflammatory disease,” the researchers noted. “Our findings suggest that anti-TNF therapy, conventional immunosuppressant therapy, or combination immunosuppression are not associated with an increased risk of cancer recurrence in this population.”
The researchers cited several limitations. Notably, three of the studies included only 20 patients, and the studies included variable index cancers and methods for detecting recurrent cancers. “It also is possible, and likely, that patients who were at high risk for recurrence were not recommenced on immunosuppression, and consequently our findings may be more applicable to a population in which the patient and physicians were comfortable with re-initiation of therapy,” they said.
The National Institutes of Health supported several of the study investigators. Dr. Shelton had no disclosures. Five coinvestigators disclosed relationships with a number of pharmaceutical companies.
As the proportion of inflammatory bowel disease (IBD) patients who are elderly rises, so does the occurrence of coexisting cancers accompanying advanced age. Experience from the posttransplant population has prompted concerns that use of immunosuppression may heighten the risk of new or recurrent cancers in individuals who have a prior history of cancer.

|
| Dr. Geoffrey Nguyen |
This apprehension may partly explain why elderly IBD patients are much less likely to be treated with immunosuppressive therapies than their younger counterparts are. Prior studies in IBD populations have not shown any association between use of immunosuppressants and recurrent cancer in individuals with prior cancer. However, these studies may have been underpowered to detect such associations.
Dr. Shelton and his colleagues performed a meta-analysis that draws upon more than 16 studies of patients with immune-mediated diseases with prior history of cancer. The investigators confirmed no association between immunosuppression and recurrent or new cancers. Despite some methodologic limitations, the study’s findings support a body of literature that suggests no increased risk of recurrent malignancy with immunosuppression in immune-mediated diseases, including IBD.
The findings also provide additional support for recent ECCO clinical guidelines, which recommend that immunosuppressive therapy can be initiated in patients with cancer after a 2- to 5-year cancer-free interval, depending on the risk of recurrence associated with specific types of cancers. A key point is that the decision to use immunosuppressants in IBD patients with prior cancer should be made with input from an oncologist and be individualized by taking into account disease course, surgical alternatives, and risk of cancer recurrence.
Dr. Geoffrey Nguyen, Mount Sinai Centre for Inflammatory Bowel Disease, University of Toronto. He has no conflicts of interest.
As the proportion of inflammatory bowel disease (IBD) patients who are elderly rises, so does the occurrence of coexisting cancers accompanying advanced age. Experience from the posttransplant population has prompted concerns that use of immunosuppression may heighten the risk of new or recurrent cancers in individuals who have a prior history of cancer.
|
|
| Dr. Geoffrey Nguyen |
This apprehension may partly explain why elderly IBD patients are much less likely to be treated with immunosuppressive therapies than their younger counterparts are. Prior studies in IBD populations have not shown any association between use of immunosuppressants and recurrent cancer in individuals with prior cancer. However, these studies may have been underpowered to detect such associations.
Dr. Shelton and his colleagues performed a meta-analysis that draws upon more than 16 studies of patients with immune-mediated diseases with prior history of cancer. The investigators confirmed no association between immunosuppression and recurrent or new cancers. Despite some methodologic limitations, the study’s findings support a body of literature that suggests no increased risk of recurrent malignancy with immunosuppression in immune-mediated diseases, including IBD.
The findings also provide additional support for recent ECCO clinical guidelines, which recommend that immunosuppressive therapy can be initiated in patients with cancer after a 2- to 5-year cancer-free interval, depending on the risk of recurrence associated with specific types of cancers. A key point is that the decision to use immunosuppressants in IBD patients with prior cancer should be made with input from an oncologist and be individualized by taking into account disease course, surgical alternatives, and risk of cancer recurrence.
Dr. Geoffrey Nguyen, Mount Sinai Centre for Inflammatory Bowel Disease, University of Toronto. He has no conflicts of interest.
As the proportion of inflammatory bowel disease (IBD) patients who are elderly rises, so does the occurrence of coexisting cancers accompanying advanced age. Experience from the posttransplant population has prompted concerns that use of immunosuppression may heighten the risk of new or recurrent cancers in individuals who have a prior history of cancer.
|
|
| Dr. Geoffrey Nguyen |
This apprehension may partly explain why elderly IBD patients are much less likely to be treated with immunosuppressive therapies than their younger counterparts are. Prior studies in IBD populations have not shown any association between use of immunosuppressants and recurrent cancer in individuals with prior cancer. However, these studies may have been underpowered to detect such associations.
Dr. Shelton and his colleagues performed a meta-analysis that draws upon more than 16 studies of patients with immune-mediated diseases with prior history of cancer. The investigators confirmed no association between immunosuppression and recurrent or new cancers. Despite some methodologic limitations, the study’s findings support a body of literature that suggests no increased risk of recurrent malignancy with immunosuppression in immune-mediated diseases, including IBD.
The findings also provide additional support for recent ECCO clinical guidelines, which recommend that immunosuppressive therapy can be initiated in patients with cancer after a 2- to 5-year cancer-free interval, depending on the risk of recurrence associated with specific types of cancers. A key point is that the decision to use immunosuppressants in IBD patients with prior cancer should be made with input from an oncologist and be individualized by taking into account disease course, surgical alternatives, and risk of cancer recurrence.
Dr. Geoffrey Nguyen, Mount Sinai Centre for Inflammatory Bowel Disease, University of Toronto. He has no conflicts of interest.
Among patients with immune-mediated diseases and a history of malignancy, cancer recurrence rates were similar regardless of whether they received tumor necrosis factor inhibitors, traditional immunosuppressants, or no immunosuppression, according to a meta-analysis of 16 cohort and case-control studies.
“However, there is a need for larger studies to prospectively monitor for cancer recurrence and new malignancies in this population to better inform our practice,” wrote Dr. Edward Shelton of Massachusetts General Hospital in Boston, and his associates. The report is in the July issue of Gastroenterology.
Patients with immune-mediated diseases often have a history of malignancy, raising questions about the effects of therapies that target the immune system, the researchers noted. However, relevant studies have been “small with few events, preventing robust estimates of risk,” they said. They searched Medline, Embase, and conference proceedings through April 2015 for studies of the risk of primary or recurrent cancer among patients with a history of malignancy who were exposed to thiopurines, methotrexate, or anti-TNF agents, or no immunosuppression. Among the 16 studies meeting these criteria, seven included patients with rheumatoid arthritis, seven included patients with inflammatory bowel disease, one included both diseases, and one included patients with psoriasis. Twelve were cohort studies, one was a case-control study, and three were case series. The resulting meta-analysis comprised 11,702 patients who contributed a total of 31,258 person-years of follow-up (Gastroenterology 2016 May 13. doi: 10.1053/j.gastro.2016.03.037).
Rates of cancer recurrence were statistically similar among patients who received no immunosuppression, anti-TNF therapy, traditional immune modulators, or combination regimens (P greater than .1 for differences among groups). Numerically, however, the risk of recurrent cancer was highest with combination immunotherapy (54.5 cases per 1,000 person-years of follow-up), compared with 37.5 cases per 1,000 person-years for patients who did not receive immunosuppressive treatment, 36.2 cases per 1,000 person-years for patients who received traditional immunomodulator monotherapy, and 33.8 cases per 1,000 person-years for patients who received anti-TNF agents. Analyses of subgroups of patients with new or recurrent cancers, or various types of therapies, and of specific immune-mediated diseases yielded “similar results, with no increase in risk,” said the researchers. Furthermore, rates of cancer recurrence did not statistically differ depending on whether patients started immunosuppression less than or more than 6 years after their index cancer (P = .43).
“Treatment decisions after a cancer diagnosis are complex, and must take into account the natural history of cancer, histologic type and stage, time from diagnosis, and course of underlying chronic inflammatory disease,” the researchers noted. “Our findings suggest that anti-TNF therapy, conventional immunosuppressant therapy, or combination immunosuppression are not associated with an increased risk of cancer recurrence in this population.”
The researchers cited several limitations. Notably, three of the studies included only 20 patients, and the studies included variable index cancers and methods for detecting recurrent cancers. “It also is possible, and likely, that patients who were at high risk for recurrence were not recommenced on immunosuppression, and consequently our findings may be more applicable to a population in which the patient and physicians were comfortable with re-initiation of therapy,” they said.
The National Institutes of Health supported several of the study investigators. Dr. Shelton had no disclosures. Five coinvestigators disclosed relationships with a number of pharmaceutical companies.
Among patients with immune-mediated diseases and a history of malignancy, cancer recurrence rates were similar regardless of whether they received tumor necrosis factor inhibitors, traditional immunosuppressants, or no immunosuppression, according to a meta-analysis of 16 cohort and case-control studies.
“However, there is a need for larger studies to prospectively monitor for cancer recurrence and new malignancies in this population to better inform our practice,” wrote Dr. Edward Shelton of Massachusetts General Hospital in Boston, and his associates. The report is in the July issue of Gastroenterology.
Patients with immune-mediated diseases often have a history of malignancy, raising questions about the effects of therapies that target the immune system, the researchers noted. However, relevant studies have been “small with few events, preventing robust estimates of risk,” they said. They searched Medline, Embase, and conference proceedings through April 2015 for studies of the risk of primary or recurrent cancer among patients with a history of malignancy who were exposed to thiopurines, methotrexate, or anti-TNF agents, or no immunosuppression. Among the 16 studies meeting these criteria, seven included patients with rheumatoid arthritis, seven included patients with inflammatory bowel disease, one included both diseases, and one included patients with psoriasis. Twelve were cohort studies, one was a case-control study, and three were case series. The resulting meta-analysis comprised 11,702 patients who contributed a total of 31,258 person-years of follow-up (Gastroenterology 2016 May 13. doi: 10.1053/j.gastro.2016.03.037).
Rates of cancer recurrence were statistically similar among patients who received no immunosuppression, anti-TNF therapy, traditional immune modulators, or combination regimens (P greater than .1 for differences among groups). Numerically, however, the risk of recurrent cancer was highest with combination immunotherapy (54.5 cases per 1,000 person-years of follow-up), compared with 37.5 cases per 1,000 person-years for patients who did not receive immunosuppressive treatment, 36.2 cases per 1,000 person-years for patients who received traditional immunomodulator monotherapy, and 33.8 cases per 1,000 person-years for patients who received anti-TNF agents. Analyses of subgroups of patients with new or recurrent cancers, or various types of therapies, and of specific immune-mediated diseases yielded “similar results, with no increase in risk,” said the researchers. Furthermore, rates of cancer recurrence did not statistically differ depending on whether patients started immunosuppression less than or more than 6 years after their index cancer (P = .43).
“Treatment decisions after a cancer diagnosis are complex, and must take into account the natural history of cancer, histologic type and stage, time from diagnosis, and course of underlying chronic inflammatory disease,” the researchers noted. “Our findings suggest that anti-TNF therapy, conventional immunosuppressant therapy, or combination immunosuppression are not associated with an increased risk of cancer recurrence in this population.”
The researchers cited several limitations. Notably, three of the studies included only 20 patients, and the studies included variable index cancers and methods for detecting recurrent cancers. “It also is possible, and likely, that patients who were at high risk for recurrence were not recommenced on immunosuppression, and consequently our findings may be more applicable to a population in which the patient and physicians were comfortable with re-initiation of therapy,” they said.
The National Institutes of Health supported several of the study investigators. Dr. Shelton had no disclosures. Five coinvestigators disclosed relationships with a number of pharmaceutical companies.
FROM GASTROENTEROLOGY
Key clinical point: Among patients with immune-mediated diseases and a history of cancer, rates of cancer recurrence did not statistically differ according to whether or not they received immunosuppression regimens, or regimen type.
Major finding: Numerically, the rate was highest with combination immunotherapy (54.5 cases per 1,000 person-years of follow-up), vs. 37.5 cases per 1,000 person-years for no immunosuppression, 36.2 cases per 1,000 person-years for traditional immunomodulator monotherapy, and 33.8 cases per 1,000 person-years for anti-TNF agents.
Data source: A systematic review and meta-analysis of 16 studies and 11,702 patients with rheumatoid arthritis, inflammatory bowel disease, and psoriasis.
Disclosures: The National Institutes of Health supported several of the study investigators. Dr. Shelton had no disclosures. Five coinvestigators disclosed relationships with a number of pharmaceutical companies.

Psychological therapies eased IBS for at least 6-12 months
Adults with irritable bowel syndrome (IBS) who underwent psychotherapy improved more than about 75% of controls, and the effect “remained significant and medium in magnitude” for at least 6-12 months, according to a meta-analysis of 41 randomized, controlled trials reported in the July issue of Clinical Gastroenterology and Hepatology.
The finding “is particularly noteworthy because of the typically recurrent, persistent nature of IBS symptoms,” said Kelsey Laird, a doctoral student of clinical psychology at Vanderbilt University in Nashville, Tenn., together with her associates there. “Future research is needed to compare the longevity of treatment effects for psychotherapy with pharmacologic therapies, such as antidepressants,” they added. “Although it is beyond the scope of this review, it is also important to consider the mechanisms by which psychotherapies improve GI symptoms and to determine the ‘active ingredients’ responsible for this effect.”

Up to 16% of individuals in the United States have IBS, and treating it costs anywhere from $950 million to $1.35 billion per year, the researchers noted. Other meta-analyses found psychotherapy about as effective as antidepressants for treating IBS-related GI symptoms over the short term, but its long-term efficacy was unknown, they added. Therefore, they searched PubMed, PsycINFO, Science Direct, and ProQuest through Aug. 15, 2015, identifying randomized controlled trials of psychological therapy and active or nonactive comparators. Psychotherapy included not only traditional psychodynamic and cognitive-behavioral therapies, but also mind-body approaches, such as relaxation training, biofeedback, and yoga, “which can be conceptualized as mindful movement,” Ms. Laird and her associates said. Comparators included support groups, education, sham treatments for hypnosis or biofeedback, online discussion forums, enhanced medical care, treatment as usual, symptom monitoring, and being wait-listed for psychological treatment (Clin Gastroenterol Hepatol. 2016 Jan 21. doi: 10.1016/j.cgh.2015.11.020). The 41 trials included 2,290 patients, comprising 1,183 assigned to the psychological modalities and 1,107 assigned to the various comparators. Taken together, the psychological modalities were associated with greater improvements in GI symptoms immediately after treatment, as compared with the grouped comparators. The Cohen’s d value was 0.69 (95% confidence interval, 0.52-0.86; P less than .001), indicating a medium effect size, the researchers said. Moreover, Cohen’s d values were 0.76 and 0.73, respectively, at short-term follow-up (1-6 months) and long-term follow-up (6-12 months). “On average, individuals who received psychotherapy had a greater reduction in GI symptoms after treatment than 75% of individuals assigned to a control condition,” the researchers concluded.
Effect sizes were similar among cognitive, cognitive-behavioral, and relaxation and hypnosis interventions, and between interventions delivered online and in person, the investigators also reported. Furthermore, longer durations or sessions of psychotherapy did not appear to further improve symptoms.
Study limitations included substantial variability between trials, and the fact that none of the 41 trials could be seen as having a low risk of bias in every domain assessed, the investigators said. “This was partially a result of the difficulty in blinding participants in psychological trials,” they noted. “However, even after excluding this domain, only nine trials were rated as low risk of bias in all remaining domains. Future studies should follow the CONSORT guidelines for [randomized controlled trials], use [intention-to-treat] designs, use active control conditions to control for nonspecific treatment effects, and assess treatment credibility and expectancy.”
The authors reported no funding sources and had no disclosures.
It is well established that psychological therapy is efficacious in managing irritable bowel syndrome (IBS), and it has an associated number needed to treat of four (Am J Gastroenterol. 2014 Sep;109:1350-65). A new meta-analysis from Laird and her colleagues revealed that the positive impact of psychotherapy on IBS symptoms persisted even 1 year after treatment.
|
|
| Dr. Christopher Almario |
While these findings are impressive and continue to support the use of psychotherapy in IBS, important issues remain. First, these results are based on data gathered in the highly controlled environment of randomized controlled trials (RCTs), and it is unclear whether they will translate to the “real world.” RCT participants may be more willing to complete psychotherapy because they know they are being observed by research staff (referred to as the Hawthorne, or observer, effect). However, in real clinical practice, patients with IBS not subject to the Hawthorne effect may be less compliant with such therapies.
Other issues relate to the current limited adoption of psychotherapy in clinical practice. Factors contributing to the low uptake include variable third-party reimbursement and poor patient and provider acceptance (JAMA. 2015 Mar;313:949-58). Another factor is limited access to qualified psychotherapists. This is an area where telehealth and mobile apps can widen access, especially as Internet-delivered psychotherapy has been shown to be effective (Am J Gastroenterol. 2011;106:1481-91).
Given the high prevalence of IBS, along with the proven, persistent efficacy of psychological therapies in reducing IBS symptoms, efforts to increase both use of and access to these therapies in clinical practice are needed.
Dr. Christopher V. Almario, division of gastroenterology, Cedars-Sinai Medical Center, Los Angeles. He has no relevant conflicts of interest to declare.
It is well established that psychological therapy is efficacious in managing irritable bowel syndrome (IBS), and it has an associated number needed to treat of four (Am J Gastroenterol. 2014 Sep;109:1350-65). A new meta-analysis from Laird and her colleagues revealed that the positive impact of psychotherapy on IBS symptoms persisted even 1 year after treatment.
|
|
| Dr. Christopher Almario |
While these findings are impressive and continue to support the use of psychotherapy in IBS, important issues remain. First, these results are based on data gathered in the highly controlled environment of randomized controlled trials (RCTs), and it is unclear whether they will translate to the “real world.” RCT participants may be more willing to complete psychotherapy because they know they are being observed by research staff (referred to as the Hawthorne, or observer, effect). However, in real clinical practice, patients with IBS not subject to the Hawthorne effect may be less compliant with such therapies.
Other issues relate to the current limited adoption of psychotherapy in clinical practice. Factors contributing to the low uptake include variable third-party reimbursement and poor patient and provider acceptance (JAMA. 2015 Mar;313:949-58). Another factor is limited access to qualified psychotherapists. This is an area where telehealth and mobile apps can widen access, especially as Internet-delivered psychotherapy has been shown to be effective (Am J Gastroenterol. 2011;106:1481-91).
Given the high prevalence of IBS, along with the proven, persistent efficacy of psychological therapies in reducing IBS symptoms, efforts to increase both use of and access to these therapies in clinical practice are needed.
Dr. Christopher V. Almario, division of gastroenterology, Cedars-Sinai Medical Center, Los Angeles. He has no relevant conflicts of interest to declare.
It is well established that psychological therapy is efficacious in managing irritable bowel syndrome (IBS), and it has an associated number needed to treat of four (Am J Gastroenterol. 2014 Sep;109:1350-65). A new meta-analysis from Laird and her colleagues revealed that the positive impact of psychotherapy on IBS symptoms persisted even 1 year after treatment.
|
|
| Dr. Christopher Almario |
While these findings are impressive and continue to support the use of psychotherapy in IBS, important issues remain. First, these results are based on data gathered in the highly controlled environment of randomized controlled trials (RCTs), and it is unclear whether they will translate to the “real world.” RCT participants may be more willing to complete psychotherapy because they know they are being observed by research staff (referred to as the Hawthorne, or observer, effect). However, in real clinical practice, patients with IBS not subject to the Hawthorne effect may be less compliant with such therapies.
Other issues relate to the current limited adoption of psychotherapy in clinical practice. Factors contributing to the low uptake include variable third-party reimbursement and poor patient and provider acceptance (JAMA. 2015 Mar;313:949-58). Another factor is limited access to qualified psychotherapists. This is an area where telehealth and mobile apps can widen access, especially as Internet-delivered psychotherapy has been shown to be effective (Am J Gastroenterol. 2011;106:1481-91).
Given the high prevalence of IBS, along with the proven, persistent efficacy of psychological therapies in reducing IBS symptoms, efforts to increase both use of and access to these therapies in clinical practice are needed.
Dr. Christopher V. Almario, division of gastroenterology, Cedars-Sinai Medical Center, Los Angeles. He has no relevant conflicts of interest to declare.
Adults with irritable bowel syndrome (IBS) who underwent psychotherapy improved more than about 75% of controls, and the effect “remained significant and medium in magnitude” for at least 6-12 months, according to a meta-analysis of 41 randomized, controlled trials reported in the July issue of Clinical Gastroenterology and Hepatology.
The finding “is particularly noteworthy because of the typically recurrent, persistent nature of IBS symptoms,” said Kelsey Laird, a doctoral student of clinical psychology at Vanderbilt University in Nashville, Tenn., together with her associates there. “Future research is needed to compare the longevity of treatment effects for psychotherapy with pharmacologic therapies, such as antidepressants,” they added. “Although it is beyond the scope of this review, it is also important to consider the mechanisms by which psychotherapies improve GI symptoms and to determine the ‘active ingredients’ responsible for this effect.”
Up to 16% of individuals in the United States have IBS, and treating it costs anywhere from $950 million to $1.35 billion per year, the researchers noted. Other meta-analyses found psychotherapy about as effective as antidepressants for treating IBS-related GI symptoms over the short term, but its long-term efficacy was unknown, they added. Therefore, they searched PubMed, PsycINFO, Science Direct, and ProQuest through Aug. 15, 2015, identifying randomized controlled trials of psychological therapy and active or nonactive comparators. Psychotherapy included not only traditional psychodynamic and cognitive-behavioral therapies, but also mind-body approaches, such as relaxation training, biofeedback, and yoga, “which can be conceptualized as mindful movement,” Ms. Laird and her associates said. Comparators included support groups, education, sham treatments for hypnosis or biofeedback, online discussion forums, enhanced medical care, treatment as usual, symptom monitoring, and being wait-listed for psychological treatment (Clin Gastroenterol Hepatol. 2016 Jan 21. doi: 10.1016/j.cgh.2015.11.020). The 41 trials included 2,290 patients, comprising 1,183 assigned to the psychological modalities and 1,107 assigned to the various comparators. Taken together, the psychological modalities were associated with greater improvements in GI symptoms immediately after treatment, as compared with the grouped comparators. The Cohen’s d value was 0.69 (95% confidence interval, 0.52-0.86; P less than .001), indicating a medium effect size, the researchers said. Moreover, Cohen’s d values were 0.76 and 0.73, respectively, at short-term follow-up (1-6 months) and long-term follow-up (6-12 months). “On average, individuals who received psychotherapy had a greater reduction in GI symptoms after treatment than 75% of individuals assigned to a control condition,” the researchers concluded.
Effect sizes were similar among cognitive, cognitive-behavioral, and relaxation and hypnosis interventions, and between interventions delivered online and in person, the investigators also reported. Furthermore, longer durations or sessions of psychotherapy did not appear to further improve symptoms.
Study limitations included substantial variability between trials, and the fact that none of the 41 trials could be seen as having a low risk of bias in every domain assessed, the investigators said. “This was partially a result of the difficulty in blinding participants in psychological trials,” they noted. “However, even after excluding this domain, only nine trials were rated as low risk of bias in all remaining domains. Future studies should follow the CONSORT guidelines for [randomized controlled trials], use [intention-to-treat] designs, use active control conditions to control for nonspecific treatment effects, and assess treatment credibility and expectancy.”
The authors reported no funding sources and had no disclosures.
Adults with irritable bowel syndrome (IBS) who underwent psychotherapy improved more than about 75% of controls, and the effect “remained significant and medium in magnitude” for at least 6-12 months, according to a meta-analysis of 41 randomized, controlled trials reported in the July issue of Clinical Gastroenterology and Hepatology.
The finding “is particularly noteworthy because of the typically recurrent, persistent nature of IBS symptoms,” said Kelsey Laird, a doctoral student of clinical psychology at Vanderbilt University in Nashville, Tenn., together with her associates there. “Future research is needed to compare the longevity of treatment effects for psychotherapy with pharmacologic therapies, such as antidepressants,” they added. “Although it is beyond the scope of this review, it is also important to consider the mechanisms by which psychotherapies improve GI symptoms and to determine the ‘active ingredients’ responsible for this effect.”
Up to 16% of individuals in the United States have IBS, and treating it costs anywhere from $950 million to $1.35 billion per year, the researchers noted. Other meta-analyses found psychotherapy about as effective as antidepressants for treating IBS-related GI symptoms over the short term, but its long-term efficacy was unknown, they added. Therefore, they searched PubMed, PsycINFO, Science Direct, and ProQuest through Aug. 15, 2015, identifying randomized controlled trials of psychological therapy and active or nonactive comparators. Psychotherapy included not only traditional psychodynamic and cognitive-behavioral therapies, but also mind-body approaches, such as relaxation training, biofeedback, and yoga, “which can be conceptualized as mindful movement,” Ms. Laird and her associates said. Comparators included support groups, education, sham treatments for hypnosis or biofeedback, online discussion forums, enhanced medical care, treatment as usual, symptom monitoring, and being wait-listed for psychological treatment (Clin Gastroenterol Hepatol. 2016 Jan 21. doi: 10.1016/j.cgh.2015.11.020). The 41 trials included 2,290 patients, comprising 1,183 assigned to the psychological modalities and 1,107 assigned to the various comparators. Taken together, the psychological modalities were associated with greater improvements in GI symptoms immediately after treatment, as compared with the grouped comparators. The Cohen’s d value was 0.69 (95% confidence interval, 0.52-0.86; P less than .001), indicating a medium effect size, the researchers said. Moreover, Cohen’s d values were 0.76 and 0.73, respectively, at short-term follow-up (1-6 months) and long-term follow-up (6-12 months). “On average, individuals who received psychotherapy had a greater reduction in GI symptoms after treatment than 75% of individuals assigned to a control condition,” the researchers concluded.
Effect sizes were similar among cognitive, cognitive-behavioral, and relaxation and hypnosis interventions, and between interventions delivered online and in person, the investigators also reported. Furthermore, longer durations or sessions of psychotherapy did not appear to further improve symptoms.
Study limitations included substantial variability between trials, and the fact that none of the 41 trials could be seen as having a low risk of bias in every domain assessed, the investigators said. “This was partially a result of the difficulty in blinding participants in psychological trials,” they noted. “However, even after excluding this domain, only nine trials were rated as low risk of bias in all remaining domains. Future studies should follow the CONSORT guidelines for [randomized controlled trials], use [intention-to-treat] designs, use active control conditions to control for nonspecific treatment effects, and assess treatment credibility and expectancy.”
The authors reported no funding sources and had no disclosures.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: A meta-analysis found that psychotherapy improved gastrointestinal symptoms in irritable bowel syndrome, with the effects persisting for at least 6-12 months.
Major finding: Immediately after treatment, the Cohen’s d value was 0.69 (P less than .001), indicating a medium effect size. Cohen’s d values were 0.76 and 0.73, respectively, at 1- to 6-month follow-up and at 6- to 12-month follow-up.
Data source: A systematic review and meta-analysis of 41 randomized controlled trials that included 2,290 patients with IBS.
Disclosures: The authors reported no funding sources and had no disclosures.

VIDEO: Nearly half of Medicaid patients denied antivirals for HCV
State Medicaid programs denied nearly half of requests to cover direct-acting antiviral drugs for patients with chronic hepatitis C virus (HCV) infection, according to a prospective study of beneficiaries in Delaware, Maryland, New Jersey, and Pennsylvania reported in the July issue of Clinical Gastroenterology and Hepatology.
In contrast, only 5% of Medicare patients and 10% of privately insured patients were denied coverage, said Dr. Vincent Lo Re III of the University of Pennsylvania, Philadelphia, together with his associates. “Notably, nearly one-quarter of Medicaid recipients with cirrhosis experienced treatment denial. Medicaid patients from these states also experienced a longer time to prescription fill than those with Medicare or commercial insurance,” the researchers said.

Their study included 2,321 HCV patients prescribed a direct-acting antiviral regimen between November 1, 2014 and April 30, 2015. All prescriptions were submitted to a specialty pharmacy that serves HCV patients in Maryland, Pennsylvania, Delaware, and New Jersey, the investigators noted. They focused on “absolute denial,” meaning that the prescription was never filled because the insurer denied coverage, regardless of appeals. “Data are lacking on the incidence of absolute denial of direct-acting antiviral prescriptions and factors associated with this outcome,” the investigators said. “These data are important because absolute denial of HCV treatment by insurers might have adverse outcomes on patients and could harm patient-provider relationships” (Clin Gastroenterol Hepatol. 2016 Apr 5. doi: 10.1016/j.cgh.2016.03.040).
Source: American Gastroenterological Association
A total of 1,023 study patients were privately insured, 795 were enrolled in Medicare, and 503 were Medicaid patients, according to the researchers. In all, 377 patients (16%) received absolute denials, most often because of “insufficient information to assess medical need” (36% of denials) and “lack of medical necessity” (35%). Medicaid patients faced an absolute denial rate of 46% – significantly higher than rates for private insurers (10.5%) or Medicare (5%; P less than .001 for both comparisons). After adjusting for potential confounders, Medicaid patients were more than four times as likely to be denied coverage for direct-acting antivirals, compared with privately insured patients (relative risk, 4.1; 95% confidence interval, 3.4-5.1). Delaware’s Medicaid program had the highest rate of absolute denial (57%), followed by Pennsylvania (48%), Maryland (47%), and New Jersey (37%).
Medicaid programs even refused to cover direct-acting antiviral prescriptions for 25% of patients who had cirrhosis, compared with absolute denial rates of only 1% for Medicare and 3% of private insurers (P less than .001 for both comparisons), according to the researchers. The implications of these denials “remain unknown,” but lack of treatment increases the risk of end-stage liver disease, hepatocellular carcinoma, extrahepatic disease, HCV transmission, and “anxiety and stress about HCV disease progression, [which can] provoke distrust among patients of the health care system and their providers,” they added. “Clinicians then are challenged to explain the denial, and important opportunities for patient engagement, education, and cure could be irrevocably lost.”
Medicaid programs also initially denied about 25% of prescriptions before eventually approving them, despite the fact that “patients had complete prior authorization requests that should have contained the materials needed to justify approval,” said the investigators. Furthermore, denial letters usually did not specify the information that was needed, “making it difficult [for clinicians] to appeal the decision.” In contrast, only about 8% of privately insured patients and 13% of Medicare enrollees were initially denied coverage.
Prescriptions as a whole were more likely to be filled in the last 3 months of the study than earlier, perhaps because insurers are starting to relax their reimbursement criteria, said the investigators. Indeed, in October 2015, the American Association for the Study of Liver Diseases and the Infectious Disease Society of America stopped triaging groups of patients for direct-acting antiviral therapy, they noted.
The research was supported by the Penn Center for AIDS Research, which is funded by the National Institutes of Health. Dr. Lo Re received investigator-initiated research support from AstraZeneca. Five coinvestigators reported relationships with a number of pharmaceutical companies. Four coinvestigators reported employment by the study site, Burman’s Specialty Pharmacy. The other four coauthors had no disclosures.
State Medicaid programs denied nearly half of requests to cover direct-acting antiviral drugs for patients with chronic hepatitis C virus (HCV) infection, according to a prospective study of beneficiaries in Delaware, Maryland, New Jersey, and Pennsylvania reported in the July issue of Clinical Gastroenterology and Hepatology.
In contrast, only 5% of Medicare patients and 10% of privately insured patients were denied coverage, said Dr. Vincent Lo Re III of the University of Pennsylvania, Philadelphia, together with his associates. “Notably, nearly one-quarter of Medicaid recipients with cirrhosis experienced treatment denial. Medicaid patients from these states also experienced a longer time to prescription fill than those with Medicare or commercial insurance,” the researchers said.
Their study included 2,321 HCV patients prescribed a direct-acting antiviral regimen between November 1, 2014 and April 30, 2015. All prescriptions were submitted to a specialty pharmacy that serves HCV patients in Maryland, Pennsylvania, Delaware, and New Jersey, the investigators noted. They focused on “absolute denial,” meaning that the prescription was never filled because the insurer denied coverage, regardless of appeals. “Data are lacking on the incidence of absolute denial of direct-acting antiviral prescriptions and factors associated with this outcome,” the investigators said. “These data are important because absolute denial of HCV treatment by insurers might have adverse outcomes on patients and could harm patient-provider relationships” (Clin Gastroenterol Hepatol. 2016 Apr 5. doi: 10.1016/j.cgh.2016.03.040).
Source: American Gastroenterological Association
A total of 1,023 study patients were privately insured, 795 were enrolled in Medicare, and 503 were Medicaid patients, according to the researchers. In all, 377 patients (16%) received absolute denials, most often because of “insufficient information to assess medical need” (36% of denials) and “lack of medical necessity” (35%). Medicaid patients faced an absolute denial rate of 46% – significantly higher than rates for private insurers (10.5%) or Medicare (5%; P less than .001 for both comparisons). After adjusting for potential confounders, Medicaid patients were more than four times as likely to be denied coverage for direct-acting antivirals, compared with privately insured patients (relative risk, 4.1; 95% confidence interval, 3.4-5.1). Delaware’s Medicaid program had the highest rate of absolute denial (57%), followed by Pennsylvania (48%), Maryland (47%), and New Jersey (37%).
Medicaid programs even refused to cover direct-acting antiviral prescriptions for 25% of patients who had cirrhosis, compared with absolute denial rates of only 1% for Medicare and 3% of private insurers (P less than .001 for both comparisons), according to the researchers. The implications of these denials “remain unknown,” but lack of treatment increases the risk of end-stage liver disease, hepatocellular carcinoma, extrahepatic disease, HCV transmission, and “anxiety and stress about HCV disease progression, [which can] provoke distrust among patients of the health care system and their providers,” they added. “Clinicians then are challenged to explain the denial, and important opportunities for patient engagement, education, and cure could be irrevocably lost.”
Medicaid programs also initially denied about 25% of prescriptions before eventually approving them, despite the fact that “patients had complete prior authorization requests that should have contained the materials needed to justify approval,” said the investigators. Furthermore, denial letters usually did not specify the information that was needed, “making it difficult [for clinicians] to appeal the decision.” In contrast, only about 8% of privately insured patients and 13% of Medicare enrollees were initially denied coverage.
Prescriptions as a whole were more likely to be filled in the last 3 months of the study than earlier, perhaps because insurers are starting to relax their reimbursement criteria, said the investigators. Indeed, in October 2015, the American Association for the Study of Liver Diseases and the Infectious Disease Society of America stopped triaging groups of patients for direct-acting antiviral therapy, they noted.
The research was supported by the Penn Center for AIDS Research, which is funded by the National Institutes of Health. Dr. Lo Re received investigator-initiated research support from AstraZeneca. Five coinvestigators reported relationships with a number of pharmaceutical companies. Four coinvestigators reported employment by the study site, Burman’s Specialty Pharmacy. The other four coauthors had no disclosures.
State Medicaid programs denied nearly half of requests to cover direct-acting antiviral drugs for patients with chronic hepatitis C virus (HCV) infection, according to a prospective study of beneficiaries in Delaware, Maryland, New Jersey, and Pennsylvania reported in the July issue of Clinical Gastroenterology and Hepatology.
In contrast, only 5% of Medicare patients and 10% of privately insured patients were denied coverage, said Dr. Vincent Lo Re III of the University of Pennsylvania, Philadelphia, together with his associates. “Notably, nearly one-quarter of Medicaid recipients with cirrhosis experienced treatment denial. Medicaid patients from these states also experienced a longer time to prescription fill than those with Medicare or commercial insurance,” the researchers said.
Their study included 2,321 HCV patients prescribed a direct-acting antiviral regimen between November 1, 2014 and April 30, 2015. All prescriptions were submitted to a specialty pharmacy that serves HCV patients in Maryland, Pennsylvania, Delaware, and New Jersey, the investigators noted. They focused on “absolute denial,” meaning that the prescription was never filled because the insurer denied coverage, regardless of appeals. “Data are lacking on the incidence of absolute denial of direct-acting antiviral prescriptions and factors associated with this outcome,” the investigators said. “These data are important because absolute denial of HCV treatment by insurers might have adverse outcomes on patients and could harm patient-provider relationships” (Clin Gastroenterol Hepatol. 2016 Apr 5. doi: 10.1016/j.cgh.2016.03.040).
Source: American Gastroenterological Association
A total of 1,023 study patients were privately insured, 795 were enrolled in Medicare, and 503 were Medicaid patients, according to the researchers. In all, 377 patients (16%) received absolute denials, most often because of “insufficient information to assess medical need” (36% of denials) and “lack of medical necessity” (35%). Medicaid patients faced an absolute denial rate of 46% – significantly higher than rates for private insurers (10.5%) or Medicare (5%; P less than .001 for both comparisons). After adjusting for potential confounders, Medicaid patients were more than four times as likely to be denied coverage for direct-acting antivirals, compared with privately insured patients (relative risk, 4.1; 95% confidence interval, 3.4-5.1). Delaware’s Medicaid program had the highest rate of absolute denial (57%), followed by Pennsylvania (48%), Maryland (47%), and New Jersey (37%).
Medicaid programs even refused to cover direct-acting antiviral prescriptions for 25% of patients who had cirrhosis, compared with absolute denial rates of only 1% for Medicare and 3% of private insurers (P less than .001 for both comparisons), according to the researchers. The implications of these denials “remain unknown,” but lack of treatment increases the risk of end-stage liver disease, hepatocellular carcinoma, extrahepatic disease, HCV transmission, and “anxiety and stress about HCV disease progression, [which can] provoke distrust among patients of the health care system and their providers,” they added. “Clinicians then are challenged to explain the denial, and important opportunities for patient engagement, education, and cure could be irrevocably lost.”
Medicaid programs also initially denied about 25% of prescriptions before eventually approving them, despite the fact that “patients had complete prior authorization requests that should have contained the materials needed to justify approval,” said the investigators. Furthermore, denial letters usually did not specify the information that was needed, “making it difficult [for clinicians] to appeal the decision.” In contrast, only about 8% of privately insured patients and 13% of Medicare enrollees were initially denied coverage.
Prescriptions as a whole were more likely to be filled in the last 3 months of the study than earlier, perhaps because insurers are starting to relax their reimbursement criteria, said the investigators. Indeed, in October 2015, the American Association for the Study of Liver Diseases and the Infectious Disease Society of America stopped triaging groups of patients for direct-acting antiviral therapy, they noted.
The research was supported by the Penn Center for AIDS Research, which is funded by the National Institutes of Health. Dr. Lo Re received investigator-initiated research support from AstraZeneca. Five coinvestigators reported relationships with a number of pharmaceutical companies. Four coinvestigators reported employment by the study site, Burman’s Specialty Pharmacy. The other four coauthors had no disclosures.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: Nearly half of Medicaid patients with chronic hepatitis C infections were denied coverage for direct-acting antivirals in a prospective study.
Major finding: In all, 46% of Medicaid patients received absolute denials (46%), compared with 5% of Medicare patients and 10% of privately insured patients (P less than .001 for both comparisons).
Data source: A prospective cohort study of 2,321 patients who submitted prescriptions for direct-acting antivirals to a specialty pharmacy serving the HCV populations of Delaware, Maryland, New Jersey, and Pennsylvania.
Disclosures: The research was supported by the Penn Center for AIDS Research, which is funded by the National Institutes of Health. Dr. Lo Re received investigator-initiated research support from AstraZeneca. Five coinvestigators reported relationships with a number of pharmaceutical companies. Four coinvestigators reported employment by the study site, Burman’s Specialty Pharmacy. The other four coauthors had no disclosures.

Detecting cancer: Pearls for the primary care physician
According to the Surveillance, Epidemiology, and End Results database, 5-year overall survival rates have improved for nearly all tumor types during the past 40 years.1 This has been accomplished with better treatment and earlier detection of the most common cancers, as well as the uncommon but highly curable tumor types.
Primary care physicians play a vital role in detecting cancers at earlier stages and synthesizing information from a patient’s presentation, vital signs, physical examination, and results of laboratory and radiographic testing. Yet cancers can be easily overlooked, and highly curable cancers such as Hodgkin lymphoma and testicular cancer, with 5-year survival rates above 85%, can have unusual presentations. Aside from the obvious health consequences, missed cancer diagnoses are often the subject of malpractice suits.
This paper reviews cancers that are easily missed and provides clinically relevant pearls from an oncologic perspective for primary care physicians, who are generally the first point of contact for patients.
BREAST CANCER DETECTION AND SCREENING
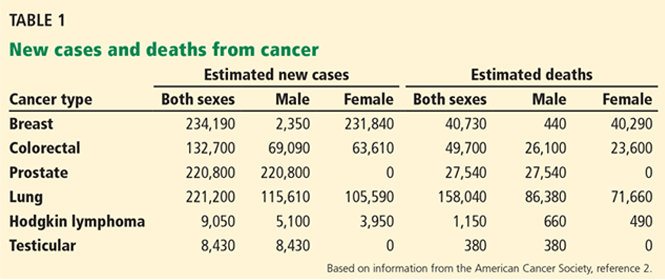
Breast cancer is the second most common cause of cancer death in US women and the most common cause of death in US women ages 20 to 59 (Table 1).2–4
Screening mammography has had a significant impact on early detection rates, and this has translated into a 20% to 30% decrease in the breast cancer mortality rate.5,6 But despite national screening guidelines, up to 15% of cases are diagnosed on the basis of a palpable breast mass not detected on mammography, and 30% are diagnosed with a breast mass during the interval between mammograms.5,6 Moreover, delay in breast cancer diagnosis is one of the most common reasons for malpractice suits.7,8
Warning signs
Breast cancer can present clinically as a single, dominant, indurated mass with irregular borders. The mass can have associated ecchymosis, erythema, nipple discharge, nipple retraction, and nipple eczema.9,10 Pay close attention to any history of breast trauma, pain, signs or symptoms suggestive of local infection, and the lesion’s relationship to the patient’s menstrual cycle. Locally advanced disease typically presents with axillary adenopathy, as well as skin findings such as erythema, thickening, and dimpling.
Initial imaging workup for a breast mass
Women presenting with a breast mass should undergo breast imaging, followed by core needle biopsy of any suspicious abnormality. Depending on the clinical breast examination and the interpretation of the mammogram, as reported as a Breast Imaging Reporting and Data System (BIRADS) score, ultrasonography, magnetic resonance imaging, or biopsy may be the next course of action. Ultrasonography is recommended in evaluating masses in women who are under age 30 (who are more likely to have dense breasts that make standard mammography difficult to interpret) or who are pregnant (because it does not involve radiation).
For patients with a borderline or indeterminate clinical examination (eg, asymmetric skin-thickening or discoloration, nipple discharge or inversion, nodularity, finding on imaging [ie, BIRADS 3 lesion]), closer follow-up with repeat or additional imaging or biopsy, or both, is strongly recommended.
Screening recommendations vary
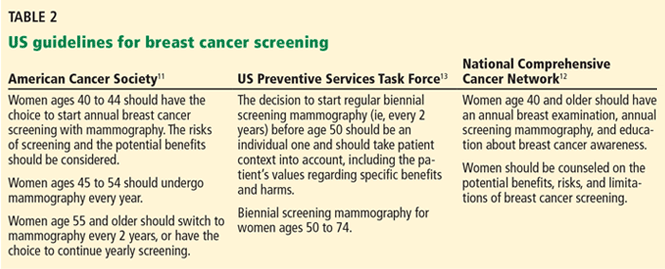
The age at which to start breast cancer screening has been a matter of debate in recent years, and different organizations have different recommendations (Table 2).11–13 According to the American Cancer Society (ACS), women should begin screening mammography at age 45 and should continue it indefinitely as long as they are in good health.11 This guideline is in line with those of the National Comprehensive Cancer Network (NCCN)12 but differs from those of the US Preventive Services Task Force (USPSTF).13
One reason for the controversy is that although starting screening at a younger age may allow for earlier detection, it also leads to overdiagnosis and to unnecessary tests and procedures. However, the NCCN noted limitations in studies looking at the overdiagnosis of breast cancer, including their use of incidence data from the 1970s, which not only underestimated the annual incidence of breast cancer in the United States, but also neglected to differentiate invasive cancer from ductal carcinoma in situ.12 Additionally, by detecting breast cancer lesions 2 years before they are discovered by clinical breast examination, mammography has been found to reduce the mortality rate from breast cancer.14
The frequency of mammography should be individualized and should involve not only an assessment of the patient’s risk factors (eg, age, family history, genetic predisposition, history of precancerous lesions, history of radiation exposure) but also a discussion of the benefits, limitations, and potential harms of screening. Both the ACS and the NCCN recommend yearly mammography for women ages 45 to 54. For those age 55 and older, the ACS recommends screening mammography every 2 years until the patient’s life expectancy is less than 10 years, whereas the NCCN recommends yearly screening mammography indefinitely. Meanwhile, the USPSTF recommends mammograms every 2 years for women ages 50 to 74.
Pearls
- Pay close attention to a history of breast trauma, pain, and signs of infection.
- Consider ultrasonography for women under age 30, who are more likely to have dense breasts.
COLORECTAL CANCER
With an estimated annual incidence of 132,700 cases diagnosed in the United States in 2015, colorectal cancer is the third most common cancer.
National guidelines that recommend colonoscopy (starting at age 50 for people at standard risk) have had a significant impact on early detection rates and have translated into a significant decrease in mortality rates.2,15,16 However, a missed diagnosis of colorectal cancer is one of the most common reasons for malpractice suits, typically because the patient was not referred for colonoscopy according to national guidelines.17–19
Symptoms depend on tumor location
In symptomatic cases, clinical manifestations differ depending on tumor location.
Left-sided tumors can present with hematochezia, colicky abdominal pain, and a change in bowel habits. And because the descending (left) colon has a smaller lumen than the right and tumors typically are annular in shape, left-sided cancers may present with abdominal distention with or without bowel obstruction or nausea and vomiting.
Right-sided tumors typically present with iron deficiency anemia from unrecognized blood loss.
Tumors near the rectum can cause tenesmus, rectal pain, and diminished caliber of stools.
In the United States, 20% of colorectal cancer patients have distant metastases at the time of diagnosis, and the most common sites are the lymph nodes, liver, lungs, and peritoneum.17
Uncommon presentations of colorectal cancer include pneumaturia, fecaluria or recurrent urinary tract infection from a fistula, bacteremia with Streptococcus bovis or Clostridium septicum, and intra-abdominal abscess from a localized bowel perforation.20,21
Initial workup
Once cancer is suspected, colonoscopy is the most accurate and versatile diagnostic test. It not only permits localization and biopsy of lesions throughout the large bowel, but also detects synchronous neoplasms and permits removal of polyps. Computed tomographic (CT) colonography is an alternative if colonoscopy is contraindicated, but it can only detect larger (ie, > 6-mm) tumors.22
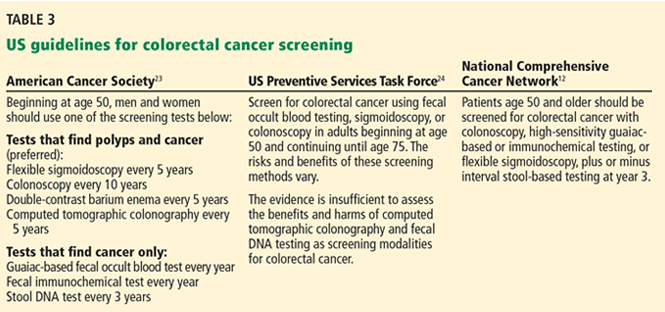
According to the ACS,23 men and women at average risk should undergo colorectal cancer screening beginning at age 50. ACS screening recommendations for polyps and colorectal cancer include flexible sigmoidoscopy every 5 years, colonoscopy every 10 years, double-contrast barium enema every 5 years, or CT colonography every 5 years. Tests that detect cancer but not polyps include guaiac-based fecal occult blood test (every year), fecal immunochemical test (every year), stool DNA test (every 3 years). These recommendations are fairly consistent with those of the NCCN12 and USPSTF24 (Table 3).12,23,24
Pearls
- Uncommon presentations include urinary tract problems and intra-abdominal abscess.
- CT colonography can only detect larger tumors.
PROSTATE CANCER
With an estimated 220,800 cases and 27,540 deaths in 2015, prostate cancer is the most common cancer and the second most common cause of cancer-related death in US men.2 Widespread use of serum prostate-specific antigen (PSA) testing has increased the rate of detection of prostate cancer.
Signs and symptoms
Most men with early-stage prostate cancer have no symptoms directly attributable to the disease.
Obstructive symptoms such as hesitancy, decreased stream, retention, and nocturia are common but are usually related to concomitant benign prostatic hypertrophy. As in prostatitis, patients with prostate cancer may present with irritative symptoms such as urinary frequency, dysuria, and urgency.
Patients who present with locally advanced prostate cancer may have symptoms secondary to local invasion, such as hematuria, hematospermia, and new-onset erectile dysfunction.
Prostate cancer usually metastasizes to bone, most commonly to the vertebrae and sternum, and the associated pain can be acute or insidious.
Diagnosis
Prostate cancer is most often diagnosed after biopsy prompted by an elevated PSA level or an abnormal digital rectal examination. The most common abnormal laboratory findings in patients with metastatic prostate cancer are an elevated serum PSA level (typically > 10 ng/mL), an elevated serum alkaline phosphatase level, and anemia, which are all proportional to the extent of bone involvement.
Screening is still controversial
There has been considerable controversy in recent years with regard to PSA screening because of the lack of significant benefit and the potential for harm to the patient, with an overdiagnosis rate ranging from 23% to 42%.25
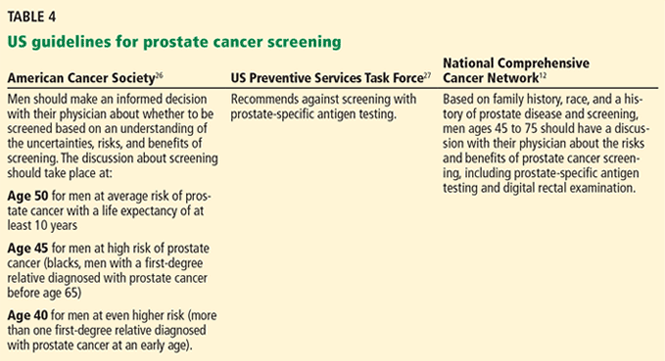
According to the ACS,26 certain groups of men should make an informed decision with their physician about whether to undergo screening: men over age 50 at average risk of prostate cancer and with at least a 10-year life expectancy, men over age 45 at high risk, and men over age 40 at an even higher risk. These ACS guidelines are consistent with those of the NCCN12 but differ from those of the USPSTF27 (Table 4).12,26,27
The patient should fully understand the risks and benefits of prostate cancer screening, as well as why it is controversial: ie, while the lifetime risk of being diagnosed with prostate cancer has increased, the lifetime risk of dying from it has remained the same after the advent of PSA testing.
Adverse effects of biopsy and treatment
Prostate biopsy is associated with infectious and bleeding complications, in addition to anxiety and physical discomfort.28 Treatment-related adverse effects include urinary incontinence, sexual dysfunction, and bowel problems.
Could these potential harms be overstated and the benefit be greater than currently thought? The NCCN12 noted that some of the landmark prostate cancer screening studies found a potential benefit in screening high-risk patients such as black men. Moreover, the studies used the sextant prostate biopsy technique, whereas now the extended core biopsy technique is the standard of care. And the studies may have underestimated the benefit of screening because the trial patients were relatively old (age 60) when their first PSA measurement was done, they were screened at long intervals (every 4 years), and the treatment options available at the time were not as good as those available today.12
Pearls
- Laboratory findings in metastatic prostate cancer are proportional to the extent of bone involvement.
- Most men with early-stage prostate cancer have no symptoms attributable to the disease.
LUNG CANCER
Lung cancer is the second most common type of cancer in men and women but has the highest mortality rate. In the United States, in 2015, an estimated 221,200 new cases of lung cancer and 158,040 deaths were expected.2 Lung cancer deaths have begun to decline in both men and women, and this is due to the decline in smoking. The impact of lung cancer screening may not be seen for another 5 to 10 years.29
A wide range of symptoms, presentations
Many patients with squamous cell carcinoma and small-cell lung carcinoma present with symptoms related to tumor involvement of the central airways,30 including cough, hemoptysis, and postobstructive pneumonia. Partial obstruction of a bronchus may cause localized wheezing, heard by the patient or by the clinician on auscultation, whereas obstruction of larger airways can cause stridor.
Patients with advanced disease present with dull, aching, persistent chest pain from mediastinal, pleural, or chest wall extension, dyspnea from lymphangitic tumor spread, tumor emboli, pneumothorax, pleural effusion, or pericardial effusion with tamponade. Less commonly, patients may present with unilateral paralysis of the diaphragm from phrenic nerve damage or with hoarseness from recurrent laryngeal nerve compression.31
Bronchorrhea—production of large volumes of thin, mucoid secretions resulting in cough—may be a feature of bronchoalveolar cell carcinoma, a rare subtype of non-small-cell lung carcinoma.
Patients uncommonly present with superior vena cava syndrome, an oncologic emergency that most often causes facial and arm swelling, dyspnea, cough, and headache.
Non-small-cell lung carcinoma arising in the superior sulcus may in rare cases cause Pancoast syndrome (manifested by shoulder pain and atrophy of the hand muscles from brachial plexus involvement), Horner syndrome (manifested by ptosis, miosis, and anhidrosis), or rib destruction.
If metastasis occurs, lung cancer commonly metastasizes to the liver and adrenal glands. At the time of diagnosis, 20% to 30% of patients with small-cell lung carcinoma have symptoms of central nervous system metastasis.
The screening controversy

Lung cancer screening is controversial because previous large studies have failed to show a clinical benefit (ie, improved survival rates) of CT screening in smokers. However, based on the results of a later large randomized trial,32 the ACS33 now recommends that patients ages 55 to 74 who are in fairly good health, have at least a 30-pack-year smoking history, and are currently smoking or have quit smoking within the last 15 years should discuss with their physician the benefits, limitations, and potential harms of lung cancer screening. These recommendations are similar to those of the NCCN12,34 and USPSTF35 (Table 5).12,33–35 The ACS guidelines also emphasize that screening should be done only at facilities with extensive experience with low-dose CT.
Follow-up evaluation
If imaging detects a lung nodule, its size and consistency are crucial in determining the course of action.33 If an endobronchial growth or solid nodule larger than 8 mm is discovered, the primary care physician should consider ordering either a repeat low-dose CT scan after 1 month or a positron-emission tomography CT scan.34 The diagnosis should be confirmed by biopsy or by surgical removal of the nodule if localized and accessible, with sites of metastasis typically taking priority.
Pearl
- At diagnosis, 20% to 30% of patients with small-cell lung cancer have symptoms of central nervous system metastasis.
HIGHLY CURABLE CANCERS WITH UNUSUAL PRESENTATIONS
Hodgkin lymphoma
With 9,190 new cases in the United States annually and a 5-year overall survival rate over 85%, Hodgkin lymphoma is one of the least common but most curable cancers.1,2 In the United States, there are two diagnostic peaks, one around age 20 and one around age 65.36 In patients with human immunodeficiency virus infection, the rate is 15 to 30 times higher than in the general population, regardless of disease status or compliance with highly active retroviral therapy.37
Hodgkin lymphoma typically presents as a nontender painless mass with rubbery consistency. The involved lymph node is typically cervical or supraclavicular. Although not detectable on physical examination, enlarged mediastinal nodes and retroperitoneal nodes are often present. Less commonly, patients may present with enlarged axillary and inguinal nodes.38
A second common presentation is the discovery of a mediastinal mass on routine chest radiography. A large percentage of patients present with at least one systemic symptom, which may include fever, night sweats, and unintentional weight loss. Generalized pruritus occurs early in the disease course in 10% to 15% of patients and is occasionally severe enough to cause intense scratching and excoriations.
A more unusual presentation of Hodgkin lymphoma is severe pain at areas of involvement after alcohol ingestion.
Most patients present with overt disease, but the presenting symptoms and signs may be relatively nonspecific and subtle and more consistent with an infectious process.
Hodgkin disease has a variable tempo, but overt symptoms typically occur after several months rather than years. As a general rule, it starts at a single site within the lymphatic system, usually a lymph node, and then spreads to adjacent nodes via lymphatic channels before disseminating to distant nonadjacent sites and organs. With this in mind, it is unusual to have bilateral axillary involvement without disease in the lower neck, and extremely unusual to have hepatic or bone marrow infiltration without disease in the spleen.
The diagnosis is established by whole lymph node tissue biopsy. Due to the high rate of inflammation in the area, inguinal nodes should not be biopsied if other equally suspicious peripheral nodes are present elsewhere. When the diagnosis of Hodgkin lymphoma is made from biopsy of an extranodal site, such as the stomach, spleen, Waldeyer ring, central nervous system, lung, bone, or skin, lymph node biopsy is also desirable for diagnostic confirmation.
Testicular cancer
Although accounting for only about 1% of all cancers in men, testicular cancer is the most common solid tumor affecting males between ages 15 and 35.1,2 With a 5-year survival rate of over 95%, testicular cancer is also one of the most curable cancers.
Testicular tumors usually present as a painless nodule or swelling of one testicle. Uncommonly, patients have metastatic disease at diagnosis, with the most common sites being lymph nodes, lung, bone, and the brain. Gynecomastia, associated with the production of human chorionic gonadotropin, occurs in about 5% of men with testicular germ cell tumors and 20% to 30% of men with Leydig cell tumors.39 Rarely, patients may present with paraneoplastic hyperthyroidism, which is secondary to thyroid-stimulating hormone and human chorionic gonadotropin sharing a common homologous alpha and beta subunit.40
Prompt diagnosis and treatment of testicular cancer provides the best opportunity for cure. Therefore, any testicular mass, even a painful scrotal lesion, should be evaluated as if it is testicular cancer until it is proven otherwise. The diagnostic evaluation of suspected testicular cancer includes scrotal ultrasonography. Radiographic testing, as deemed clinically necessary by the consulting urologist and medical oncologist, may include chest radiography, CT (chest, abdomen, pelvis), brain magnetic resonance imaging, or bone scan.
The primary care laboratory evaluation should include a complete metabolic profile and measurements of lactate dehydrogenase and serum tumor markers such as alpha fetoprotein and human chorionic gonadotropin. In nonseminomatous germ cell tumors, alpha fetoprotein or human chorionic gonadotropin, or both, can be elevated in 80% to 85% of patients. However, in seminoma, alpha fetoprotein is never elevated, and the serum human chorionic gonadotropin is elevated in only 20% to 25% of patients.41
Patients with a suspicious testicular mass should be referred promptly to a urologist for consideration of radical inguinal orchiectomy and, in some cases, retroperitoneal lymph node dissection. Testicular biopsy is not part of the evaluation as it may result in tumor seeding into the scrotal sac or metastatic spread of tumor to the inguinal nodes. Inguinal biopsy of the contralateral testis is considered if ultrasonography raises suspicion of an intratesticular abnormality, cryptorchid testis, or marked testicular atrophy. Discussing sperm banking with the patient is part of the diagnostic workup, as cumulative cisplatin doses greater than 400 mg/m2 can result in permanent infertility in 50% of men.42
Pearls
- In Hodgkin lymphoma, bilateral axillary involvement without disease in the lower neck is unusual.
- Discussing sperm banking is part of the diagnostic workup for testicular cancer.
- National Cancer Institute (NIH). Surveillance, Epidemiology and End Results (SEER) Program. SEER Cancer Statistics Review, 1975–2010. http://seer.cancer.gov/csr/1975_2010/. Accessed May 9, 2016.
- American Cancer Society. Cancer Facts & Figures 2015.
www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2015/. Accessed May 9, 2016. - Tabár L, Vitak B, Chen HH, Yen MF, Duffy SW, Smith RA. Beyond randomized controlled trials: organized mammographic screening substantially reduces breast carcinoma mortality. Cancer 2001; 91:1724–1731.
- Tabar L, Fagerberg G, Chen HH, et al. Efficacy of breast cancer screening by age. New results from the Swedish two-county trial. Cancer 1995; 75:2507–2517.
- Humphrey LL, Helfand M, Chan BK, Woolf SH. Breast cancer screening: a summary of the evidence for the US Preventive Services Task Force. Ann Intern Med 2002; 137:347–360.
- Esserman LJ, Shieh Y, Rutgers EJ, et al. Impact of mammographic screening on the detection of good and poor prognosis breast cancers. Breast Cancer Res Treat 2011; 130:725–734.
- Wallace E, Lowry J, Smith SM, Fahey T. The epidemiology of malpractice claims in primary care: a systematic review. BMJ Open 2013; 3:pii:e002929.
- Gandhi TK, Kachalia A, Thomas EJ, et al. Missed and delayed diagnoses in the ambulatory setting: a study of closed malpractice claims. Ann Intern Med 2006; 145:488–496.
- Morrow M. The evaluation of common breast problems. Am Fam Physician 2000; 61:2371–2378, 2385.
- Santen RJ, Mansel R. Benign breast disorders. N Engl J Med 2005; 353:275–285.
- American Cancer Society. Breast cancer prevention and early detection. www.cancer.org/cancer/breastcancer/moreinformation/breastcancerearlydetection/breast-cancer-early-detection-acs-recs. Accessed May 17, 2016.
- National Comprehensive Cancer Network (NCCN). NCCN Guidelines. www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed May 17, 2016.
- US Preventive Services Task Force (USPSTF). Breast cancers Screening. www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/breast-cancer-screening. Accessed May 17, 2016.
- Mandelblatt JS, Cronin KA, Bailey S, et al; Breast Cancer Working Group of the Cancer Intervention and Surveillance Modeling Network. Effects of mammography screening under different screening schedules: model estimates of potential benefits and harms. Ann Intern Med 2009; 151:738–747.
- Newcomb PA, Norfleet RG, Storer BE, Surawicz TS, Marcus PM. Screening sigmoidoscopy and colorectal cancer mortality. J Natl Cancer Inst 1992; 84:1572–1575.
- Bressler B, Paszat LF, Chen Z, Rothwell DM, Vinden C, Rabeneck L. Rates of new or missed colorectal cancers after colonoscopy and their risk factors: a population-based analysis. Gastroenterology 2007; 132:96–102.
- Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9–29.
- Feld AD. Malpractice risks associated with colon cancer and inflammatory bowel disease. J Gastroenterol 2004; 99:1641–1644.
- Goodman D, Irvin TT. Delay in the diagnosis and prognosis of carcinoma of the right colon. Br J Surg 1993; 80:1327–1329.
- Alvarez JA, Baldonedo RF, Bear IG, Alvarez P, Jorge JL. Anaerobic liver abscesses as initial presentation of silent colonic cancer. HPB (Oxford) 2004; 6:41–42.
- Tsai HL, Hsieh JS, Yu FJ, et al. Perforated colonic cancer presenting as intra-abdominal abscess. Int J Colorectal Dis 2007; 22:15–19.
- Levin B, Lieberman DA, McFarland B, et al; American Cancer Society Colorectal Cancer Advisory Group; US Multi-Society Task Force; American College of Radiology Colon Cancer Committee. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. Gastroenterology 2008; 134:1570–1595.
- American Cancer Society. Colorectal cancer prevention and early detection. www.cancer.org/cancer/colonandrectumcancer/moreinformation/colonandrectumcancerearlydetection/colorectal-cancer-early-detection-acs-recommendations. Accessed May 17, 2016.
- US Preventive Services Task Force (USPSTF). Colorectal cancer: screening. www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/colorectal-cancer-screening. Accessed May 17, 2016.
- Schröder FH, Hugosson J, Roobol MJ, et al; ERSPC Investigators. Screening and prostate cancer mortality: results of the European Randomised Study of Screening for Prostate Cancer (ERSPC) at 13 years of follow-up. Lancet 2014; 384:2027–2035.
- American Cancer Society. Prostate cancer prevention and early detection. www.cancer.org/cancer/prostatecancer/moreinformation/prostatecancerearlydetection/index. Accessed May 17, 2016.
- US Preventive Services Task Force (USPSTF). Prostate cancer: screening. www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/prostate-cancer-screening. Accessed June 1, 2016.
- Essink-Bot ML, de Koning HJ, Nijs HG, Kirkels WJ, van der Maas PJ, Schröder FH. Short-term effects of population-based screening for prostate cancer on health-related quality of life. J Natl Cancer Inst 1998; 90:925–931.
- Peto R, Darby S, Deo H, Silcocks P, Whitley E, Doll R. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ (Clin Res Ed) 2000; 321:323–329.
- Chute CG, Greenberg ER, Baron J, Korson R, Baker J, Yates J. Presenting conditions of 1,539 population-based lung cancer patients by cell type and stage in New Hampshire and Vermont. Cancer 1985; 56:2107–2111.
- Ramadan HH, Wax MK, Avery S. Outcome and changing cause of unilateral vocal cord paralysis. Otolaryngol Head Neck Surg 1998;118:199–202.
- Church TR, Black WC, Aberle DR, et al. Results of initial low-dose computed tomographic screening for lung cancer. N Engl J Med 2013; 368:1980–1991.
- American Cancer Society. Lung cancer prevention and early detection. www.cancer.org/cancer/lungcancer-non-smallcell/moreinformation/lungcancerpreventionandearlydetection/index. Accessed May 17, 2016.
- Lung Cancer Screening. National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology. www.nccn.org/professionals/physician_gls/pdf/lung_screening.pdf. Accessed May 17, 2016.
- US Preventive Services Task Force (USPSTF). Lung cancer: screening. www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/lung-cancer-screening. Accessed May 17, 2016.
- National Cancer Institute (NIH). Surveillance, Epidemiology, and End Results (SEER) Program. SEER Cancer Statistics Review: 1973-1994. http://seer.cancer.gov/archive/csr/1973_1994/. Accessed May 17, 2016.
- Mauch PM, Kalish LA, Kadin M, Coleman CN, Osteen R, Hellman S. Patterns of presentation of Hodgkin disease. Implications for etiology and pathogenesis. Cancer 1993; 71:2062–2071.
- Gobbi PG, Cavalli C, Gendarini A, et al. Reevaluation of prognostic significance of symptoms in Hodgkin’s disease. Cancer 1985; 56:2874–2880.
- Einhorn LH. Treatment of testicular cancer: a new and improved model. J Clin Oncol 1990; 8:1777–1781.
- Tseng A Jr, Horning SJ, Freiha FS, Resser KJ, Hannigan JF Jr, Torti FM. Gynecomastia in testicular cancer patients. Prognostic and therapeutic implications. Cancer 1985; 56:2534–2538.
- Gilligan TD, Seidenfeld J, Basch EM, et al; American Society of Clinical Oncology. American Society of Clinical Oncology clinical practice guideline on uses of serum tumor markers in adult males with germ cell tumors. J Clin Oncol 2010; 28:3388–3404.
- Brydøy M, Fosså SD, Klepp O, et al. Paternity following treatment for testicular cancer. J Natl Cancer Inst 2005; 97:1580–1588.
According to the Surveillance, Epidemiology, and End Results database, 5-year overall survival rates have improved for nearly all tumor types during the past 40 years.1 This has been accomplished with better treatment and earlier detection of the most common cancers, as well as the uncommon but highly curable tumor types.
Primary care physicians play a vital role in detecting cancers at earlier stages and synthesizing information from a patient’s presentation, vital signs, physical examination, and results of laboratory and radiographic testing. Yet cancers can be easily overlooked, and highly curable cancers such as Hodgkin lymphoma and testicular cancer, with 5-year survival rates above 85%, can have unusual presentations. Aside from the obvious health consequences, missed cancer diagnoses are often the subject of malpractice suits.
This paper reviews cancers that are easily missed and provides clinically relevant pearls from an oncologic perspective for primary care physicians, who are generally the first point of contact for patients.
BREAST CANCER DETECTION AND SCREENING
Breast cancer is the second most common cause of cancer death in US women and the most common cause of death in US women ages 20 to 59 (Table 1).2–4
Screening mammography has had a significant impact on early detection rates, and this has translated into a 20% to 30% decrease in the breast cancer mortality rate.5,6 But despite national screening guidelines, up to 15% of cases are diagnosed on the basis of a palpable breast mass not detected on mammography, and 30% are diagnosed with a breast mass during the interval between mammograms.5,6 Moreover, delay in breast cancer diagnosis is one of the most common reasons for malpractice suits.7,8
Warning signs
Breast cancer can present clinically as a single, dominant, indurated mass with irregular borders. The mass can have associated ecchymosis, erythema, nipple discharge, nipple retraction, and nipple eczema.9,10 Pay close attention to any history of breast trauma, pain, signs or symptoms suggestive of local infection, and the lesion’s relationship to the patient’s menstrual cycle. Locally advanced disease typically presents with axillary adenopathy, as well as skin findings such as erythema, thickening, and dimpling.
Initial imaging workup for a breast mass
Women presenting with a breast mass should undergo breast imaging, followed by core needle biopsy of any suspicious abnormality. Depending on the clinical breast examination and the interpretation of the mammogram, as reported as a Breast Imaging Reporting and Data System (BIRADS) score, ultrasonography, magnetic resonance imaging, or biopsy may be the next course of action. Ultrasonography is recommended in evaluating masses in women who are under age 30 (who are more likely to have dense breasts that make standard mammography difficult to interpret) or who are pregnant (because it does not involve radiation).
For patients with a borderline or indeterminate clinical examination (eg, asymmetric skin-thickening or discoloration, nipple discharge or inversion, nodularity, finding on imaging [ie, BIRADS 3 lesion]), closer follow-up with repeat or additional imaging or biopsy, or both, is strongly recommended.
Screening recommendations vary
The age at which to start breast cancer screening has been a matter of debate in recent years, and different organizations have different recommendations (Table 2).11–13 According to the American Cancer Society (ACS), women should begin screening mammography at age 45 and should continue it indefinitely as long as they are in good health.11 This guideline is in line with those of the National Comprehensive Cancer Network (NCCN)12 but differs from those of the US Preventive Services Task Force (USPSTF).13
One reason for the controversy is that although starting screening at a younger age may allow for earlier detection, it also leads to overdiagnosis and to unnecessary tests and procedures. However, the NCCN noted limitations in studies looking at the overdiagnosis of breast cancer, including their use of incidence data from the 1970s, which not only underestimated the annual incidence of breast cancer in the United States, but also neglected to differentiate invasive cancer from ductal carcinoma in situ.12 Additionally, by detecting breast cancer lesions 2 years before they are discovered by clinical breast examination, mammography has been found to reduce the mortality rate from breast cancer.14
The frequency of mammography should be individualized and should involve not only an assessment of the patient’s risk factors (eg, age, family history, genetic predisposition, history of precancerous lesions, history of radiation exposure) but also a discussion of the benefits, limitations, and potential harms of screening. Both the ACS and the NCCN recommend yearly mammography for women ages 45 to 54. For those age 55 and older, the ACS recommends screening mammography every 2 years until the patient’s life expectancy is less than 10 years, whereas the NCCN recommends yearly screening mammography indefinitely. Meanwhile, the USPSTF recommends mammograms every 2 years for women ages 50 to 74.
Pearls
- Pay close attention to a history of breast trauma, pain, and signs of infection.
- Consider ultrasonography for women under age 30, who are more likely to have dense breasts.
COLORECTAL CANCER
With an estimated annual incidence of 132,700 cases diagnosed in the United States in 2015, colorectal cancer is the third most common cancer.
National guidelines that recommend colonoscopy (starting at age 50 for people at standard risk) have had a significant impact on early detection rates and have translated into a significant decrease in mortality rates.2,15,16 However, a missed diagnosis of colorectal cancer is one of the most common reasons for malpractice suits, typically because the patient was not referred for colonoscopy according to national guidelines.17–19
Symptoms depend on tumor location
In symptomatic cases, clinical manifestations differ depending on tumor location.
Left-sided tumors can present with hematochezia, colicky abdominal pain, and a change in bowel habits. And because the descending (left) colon has a smaller lumen than the right and tumors typically are annular in shape, left-sided cancers may present with abdominal distention with or without bowel obstruction or nausea and vomiting.
Right-sided tumors typically present with iron deficiency anemia from unrecognized blood loss.
Tumors near the rectum can cause tenesmus, rectal pain, and diminished caliber of stools.
In the United States, 20% of colorectal cancer patients have distant metastases at the time of diagnosis, and the most common sites are the lymph nodes, liver, lungs, and peritoneum.17
Uncommon presentations of colorectal cancer include pneumaturia, fecaluria or recurrent urinary tract infection from a fistula, bacteremia with Streptococcus bovis or Clostridium septicum, and intra-abdominal abscess from a localized bowel perforation.20,21
Initial workup
Once cancer is suspected, colonoscopy is the most accurate and versatile diagnostic test. It not only permits localization and biopsy of lesions throughout the large bowel, but also detects synchronous neoplasms and permits removal of polyps. Computed tomographic (CT) colonography is an alternative if colonoscopy is contraindicated, but it can only detect larger (ie, > 6-mm) tumors.22
According to the ACS,23 men and women at average risk should undergo colorectal cancer screening beginning at age 50. ACS screening recommendations for polyps and colorectal cancer include flexible sigmoidoscopy every 5 years, colonoscopy every 10 years, double-contrast barium enema every 5 years, or CT colonography every 5 years. Tests that detect cancer but not polyps include guaiac-based fecal occult blood test (every year), fecal immunochemical test (every year), stool DNA test (every 3 years). These recommendations are fairly consistent with those of the NCCN12 and USPSTF24 (Table 3).12,23,24
Pearls
- Uncommon presentations include urinary tract problems and intra-abdominal abscess.
- CT colonography can only detect larger tumors.
PROSTATE CANCER
With an estimated 220,800 cases and 27,540 deaths in 2015, prostate cancer is the most common cancer and the second most common cause of cancer-related death in US men.2 Widespread use of serum prostate-specific antigen (PSA) testing has increased the rate of detection of prostate cancer.
Signs and symptoms
Most men with early-stage prostate cancer have no symptoms directly attributable to the disease.
Obstructive symptoms such as hesitancy, decreased stream, retention, and nocturia are common but are usually related to concomitant benign prostatic hypertrophy. As in prostatitis, patients with prostate cancer may present with irritative symptoms such as urinary frequency, dysuria, and urgency.
Patients who present with locally advanced prostate cancer may have symptoms secondary to local invasion, such as hematuria, hematospermia, and new-onset erectile dysfunction.
Prostate cancer usually metastasizes to bone, most commonly to the vertebrae and sternum, and the associated pain can be acute or insidious.
Diagnosis
Prostate cancer is most often diagnosed after biopsy prompted by an elevated PSA level or an abnormal digital rectal examination. The most common abnormal laboratory findings in patients with metastatic prostate cancer are an elevated serum PSA level (typically > 10 ng/mL), an elevated serum alkaline phosphatase level, and anemia, which are all proportional to the extent of bone involvement.
Screening is still controversial
There has been considerable controversy in recent years with regard to PSA screening because of the lack of significant benefit and the potential for harm to the patient, with an overdiagnosis rate ranging from 23% to 42%.25
According to the ACS,26 certain groups of men should make an informed decision with their physician about whether to undergo screening: men over age 50 at average risk of prostate cancer and with at least a 10-year life expectancy, men over age 45 at high risk, and men over age 40 at an even higher risk. These ACS guidelines are consistent with those of the NCCN12 but differ from those of the USPSTF27 (Table 4).12,26,27
The patient should fully understand the risks and benefits of prostate cancer screening, as well as why it is controversial: ie, while the lifetime risk of being diagnosed with prostate cancer has increased, the lifetime risk of dying from it has remained the same after the advent of PSA testing.
Adverse effects of biopsy and treatment
Prostate biopsy is associated with infectious and bleeding complications, in addition to anxiety and physical discomfort.28 Treatment-related adverse effects include urinary incontinence, sexual dysfunction, and bowel problems.
Could these potential harms be overstated and the benefit be greater than currently thought? The NCCN12 noted that some of the landmark prostate cancer screening studies found a potential benefit in screening high-risk patients such as black men. Moreover, the studies used the sextant prostate biopsy technique, whereas now the extended core biopsy technique is the standard of care. And the studies may have underestimated the benefit of screening because the trial patients were relatively old (age 60) when their first PSA measurement was done, they were screened at long intervals (every 4 years), and the treatment options available at the time were not as good as those available today.12
Pearls
- Laboratory findings in metastatic prostate cancer are proportional to the extent of bone involvement.
- Most men with early-stage prostate cancer have no symptoms attributable to the disease.
LUNG CANCER
Lung cancer is the second most common type of cancer in men and women but has the highest mortality rate. In the United States, in 2015, an estimated 221,200 new cases of lung cancer and 158,040 deaths were expected.2 Lung cancer deaths have begun to decline in both men and women, and this is due to the decline in smoking. The impact of lung cancer screening may not be seen for another 5 to 10 years.29
A wide range of symptoms, presentations
Many patients with squamous cell carcinoma and small-cell lung carcinoma present with symptoms related to tumor involvement of the central airways,30 including cough, hemoptysis, and postobstructive pneumonia. Partial obstruction of a bronchus may cause localized wheezing, heard by the patient or by the clinician on auscultation, whereas obstruction of larger airways can cause stridor.
Patients with advanced disease present with dull, aching, persistent chest pain from mediastinal, pleural, or chest wall extension, dyspnea from lymphangitic tumor spread, tumor emboli, pneumothorax, pleural effusion, or pericardial effusion with tamponade. Less commonly, patients may present with unilateral paralysis of the diaphragm from phrenic nerve damage or with hoarseness from recurrent laryngeal nerve compression.31
Bronchorrhea—production of large volumes of thin, mucoid secretions resulting in cough—may be a feature of bronchoalveolar cell carcinoma, a rare subtype of non-small-cell lung carcinoma.
Patients uncommonly present with superior vena cava syndrome, an oncologic emergency that most often causes facial and arm swelling, dyspnea, cough, and headache.
Non-small-cell lung carcinoma arising in the superior sulcus may in rare cases cause Pancoast syndrome (manifested by shoulder pain and atrophy of the hand muscles from brachial plexus involvement), Horner syndrome (manifested by ptosis, miosis, and anhidrosis), or rib destruction.
If metastasis occurs, lung cancer commonly metastasizes to the liver and adrenal glands. At the time of diagnosis, 20% to 30% of patients with small-cell lung carcinoma have symptoms of central nervous system metastasis.
The screening controversy
Lung cancer screening is controversial because previous large studies have failed to show a clinical benefit (ie, improved survival rates) of CT screening in smokers. However, based on the results of a later large randomized trial,32 the ACS33 now recommends that patients ages 55 to 74 who are in fairly good health, have at least a 30-pack-year smoking history, and are currently smoking or have quit smoking within the last 15 years should discuss with their physician the benefits, limitations, and potential harms of lung cancer screening. These recommendations are similar to those of the NCCN12,34 and USPSTF35 (Table 5).12,33–35 The ACS guidelines also emphasize that screening should be done only at facilities with extensive experience with low-dose CT.
Follow-up evaluation
If imaging detects a lung nodule, its size and consistency are crucial in determining the course of action.33 If an endobronchial growth or solid nodule larger than 8 mm is discovered, the primary care physician should consider ordering either a repeat low-dose CT scan after 1 month or a positron-emission tomography CT scan.34 The diagnosis should be confirmed by biopsy or by surgical removal of the nodule if localized and accessible, with sites of metastasis typically taking priority.
Pearl
- At diagnosis, 20% to 30% of patients with small-cell lung cancer have symptoms of central nervous system metastasis.
HIGHLY CURABLE CANCERS WITH UNUSUAL PRESENTATIONS
Hodgkin lymphoma
With 9,190 new cases in the United States annually and a 5-year overall survival rate over 85%, Hodgkin lymphoma is one of the least common but most curable cancers.1,2 In the United States, there are two diagnostic peaks, one around age 20 and one around age 65.36 In patients with human immunodeficiency virus infection, the rate is 15 to 30 times higher than in the general population, regardless of disease status or compliance with highly active retroviral therapy.37
Hodgkin lymphoma typically presents as a nontender painless mass with rubbery consistency. The involved lymph node is typically cervical or supraclavicular. Although not detectable on physical examination, enlarged mediastinal nodes and retroperitoneal nodes are often present. Less commonly, patients may present with enlarged axillary and inguinal nodes.38
A second common presentation is the discovery of a mediastinal mass on routine chest radiography. A large percentage of patients present with at least one systemic symptom, which may include fever, night sweats, and unintentional weight loss. Generalized pruritus occurs early in the disease course in 10% to 15% of patients and is occasionally severe enough to cause intense scratching and excoriations.
A more unusual presentation of Hodgkin lymphoma is severe pain at areas of involvement after alcohol ingestion.
Most patients present with overt disease, but the presenting symptoms and signs may be relatively nonspecific and subtle and more consistent with an infectious process.
Hodgkin disease has a variable tempo, but overt symptoms typically occur after several months rather than years. As a general rule, it starts at a single site within the lymphatic system, usually a lymph node, and then spreads to adjacent nodes via lymphatic channels before disseminating to distant nonadjacent sites and organs. With this in mind, it is unusual to have bilateral axillary involvement without disease in the lower neck, and extremely unusual to have hepatic or bone marrow infiltration without disease in the spleen.
The diagnosis is established by whole lymph node tissue biopsy. Due to the high rate of inflammation in the area, inguinal nodes should not be biopsied if other equally suspicious peripheral nodes are present elsewhere. When the diagnosis of Hodgkin lymphoma is made from biopsy of an extranodal site, such as the stomach, spleen, Waldeyer ring, central nervous system, lung, bone, or skin, lymph node biopsy is also desirable for diagnostic confirmation.
Testicular cancer
Although accounting for only about 1% of all cancers in men, testicular cancer is the most common solid tumor affecting males between ages 15 and 35.1,2 With a 5-year survival rate of over 95%, testicular cancer is also one of the most curable cancers.
Testicular tumors usually present as a painless nodule or swelling of one testicle. Uncommonly, patients have metastatic disease at diagnosis, with the most common sites being lymph nodes, lung, bone, and the brain. Gynecomastia, associated with the production of human chorionic gonadotropin, occurs in about 5% of men with testicular germ cell tumors and 20% to 30% of men with Leydig cell tumors.39 Rarely, patients may present with paraneoplastic hyperthyroidism, which is secondary to thyroid-stimulating hormone and human chorionic gonadotropin sharing a common homologous alpha and beta subunit.40
Prompt diagnosis and treatment of testicular cancer provides the best opportunity for cure. Therefore, any testicular mass, even a painful scrotal lesion, should be evaluated as if it is testicular cancer until it is proven otherwise. The diagnostic evaluation of suspected testicular cancer includes scrotal ultrasonography. Radiographic testing, as deemed clinically necessary by the consulting urologist and medical oncologist, may include chest radiography, CT (chest, abdomen, pelvis), brain magnetic resonance imaging, or bone scan.
The primary care laboratory evaluation should include a complete metabolic profile and measurements of lactate dehydrogenase and serum tumor markers such as alpha fetoprotein and human chorionic gonadotropin. In nonseminomatous germ cell tumors, alpha fetoprotein or human chorionic gonadotropin, or both, can be elevated in 80% to 85% of patients. However, in seminoma, alpha fetoprotein is never elevated, and the serum human chorionic gonadotropin is elevated in only 20% to 25% of patients.41
Patients with a suspicious testicular mass should be referred promptly to a urologist for consideration of radical inguinal orchiectomy and, in some cases, retroperitoneal lymph node dissection. Testicular biopsy is not part of the evaluation as it may result in tumor seeding into the scrotal sac or metastatic spread of tumor to the inguinal nodes. Inguinal biopsy of the contralateral testis is considered if ultrasonography raises suspicion of an intratesticular abnormality, cryptorchid testis, or marked testicular atrophy. Discussing sperm banking with the patient is part of the diagnostic workup, as cumulative cisplatin doses greater than 400 mg/m2 can result in permanent infertility in 50% of men.42
Pearls
- In Hodgkin lymphoma, bilateral axillary involvement without disease in the lower neck is unusual.
- Discussing sperm banking is part of the diagnostic workup for testicular cancer.
According to the Surveillance, Epidemiology, and End Results database, 5-year overall survival rates have improved for nearly all tumor types during the past 40 years.1 This has been accomplished with better treatment and earlier detection of the most common cancers, as well as the uncommon but highly curable tumor types.
Primary care physicians play a vital role in detecting cancers at earlier stages and synthesizing information from a patient’s presentation, vital signs, physical examination, and results of laboratory and radiographic testing. Yet cancers can be easily overlooked, and highly curable cancers such as Hodgkin lymphoma and testicular cancer, with 5-year survival rates above 85%, can have unusual presentations. Aside from the obvious health consequences, missed cancer diagnoses are often the subject of malpractice suits.
This paper reviews cancers that are easily missed and provides clinically relevant pearls from an oncologic perspective for primary care physicians, who are generally the first point of contact for patients.
BREAST CANCER DETECTION AND SCREENING
Breast cancer is the second most common cause of cancer death in US women and the most common cause of death in US women ages 20 to 59 (Table 1).2–4
Screening mammography has had a significant impact on early detection rates, and this has translated into a 20% to 30% decrease in the breast cancer mortality rate.5,6 But despite national screening guidelines, up to 15% of cases are diagnosed on the basis of a palpable breast mass not detected on mammography, and 30% are diagnosed with a breast mass during the interval between mammograms.5,6 Moreover, delay in breast cancer diagnosis is one of the most common reasons for malpractice suits.7,8
Warning signs
Breast cancer can present clinically as a single, dominant, indurated mass with irregular borders. The mass can have associated ecchymosis, erythema, nipple discharge, nipple retraction, and nipple eczema.9,10 Pay close attention to any history of breast trauma, pain, signs or symptoms suggestive of local infection, and the lesion’s relationship to the patient’s menstrual cycle. Locally advanced disease typically presents with axillary adenopathy, as well as skin findings such as erythema, thickening, and dimpling.
Initial imaging workup for a breast mass
Women presenting with a breast mass should undergo breast imaging, followed by core needle biopsy of any suspicious abnormality. Depending on the clinical breast examination and the interpretation of the mammogram, as reported as a Breast Imaging Reporting and Data System (BIRADS) score, ultrasonography, magnetic resonance imaging, or biopsy may be the next course of action. Ultrasonography is recommended in evaluating masses in women who are under age 30 (who are more likely to have dense breasts that make standard mammography difficult to interpret) or who are pregnant (because it does not involve radiation).
For patients with a borderline or indeterminate clinical examination (eg, asymmetric skin-thickening or discoloration, nipple discharge or inversion, nodularity, finding on imaging [ie, BIRADS 3 lesion]), closer follow-up with repeat or additional imaging or biopsy, or both, is strongly recommended.
Screening recommendations vary
The age at which to start breast cancer screening has been a matter of debate in recent years, and different organizations have different recommendations (Table 2).11–13 According to the American Cancer Society (ACS), women should begin screening mammography at age 45 and should continue it indefinitely as long as they are in good health.11 This guideline is in line with those of the National Comprehensive Cancer Network (NCCN)12 but differs from those of the US Preventive Services Task Force (USPSTF).13
One reason for the controversy is that although starting screening at a younger age may allow for earlier detection, it also leads to overdiagnosis and to unnecessary tests and procedures. However, the NCCN noted limitations in studies looking at the overdiagnosis of breast cancer, including their use of incidence data from the 1970s, which not only underestimated the annual incidence of breast cancer in the United States, but also neglected to differentiate invasive cancer from ductal carcinoma in situ.12 Additionally, by detecting breast cancer lesions 2 years before they are discovered by clinical breast examination, mammography has been found to reduce the mortality rate from breast cancer.14
The frequency of mammography should be individualized and should involve not only an assessment of the patient’s risk factors (eg, age, family history, genetic predisposition, history of precancerous lesions, history of radiation exposure) but also a discussion of the benefits, limitations, and potential harms of screening. Both the ACS and the NCCN recommend yearly mammography for women ages 45 to 54. For those age 55 and older, the ACS recommends screening mammography every 2 years until the patient’s life expectancy is less than 10 years, whereas the NCCN recommends yearly screening mammography indefinitely. Meanwhile, the USPSTF recommends mammograms every 2 years for women ages 50 to 74.
Pearls
- Pay close attention to a history of breast trauma, pain, and signs of infection.
- Consider ultrasonography for women under age 30, who are more likely to have dense breasts.
COLORECTAL CANCER
With an estimated annual incidence of 132,700 cases diagnosed in the United States in 2015, colorectal cancer is the third most common cancer.
National guidelines that recommend colonoscopy (starting at age 50 for people at standard risk) have had a significant impact on early detection rates and have translated into a significant decrease in mortality rates.2,15,16 However, a missed diagnosis of colorectal cancer is one of the most common reasons for malpractice suits, typically because the patient was not referred for colonoscopy according to national guidelines.17–19
Symptoms depend on tumor location
In symptomatic cases, clinical manifestations differ depending on tumor location.
Left-sided tumors can present with hematochezia, colicky abdominal pain, and a change in bowel habits. And because the descending (left) colon has a smaller lumen than the right and tumors typically are annular in shape, left-sided cancers may present with abdominal distention with or without bowel obstruction or nausea and vomiting.
Right-sided tumors typically present with iron deficiency anemia from unrecognized blood loss.
Tumors near the rectum can cause tenesmus, rectal pain, and diminished caliber of stools.
In the United States, 20% of colorectal cancer patients have distant metastases at the time of diagnosis, and the most common sites are the lymph nodes, liver, lungs, and peritoneum.17
Uncommon presentations of colorectal cancer include pneumaturia, fecaluria or recurrent urinary tract infection from a fistula, bacteremia with Streptococcus bovis or Clostridium septicum, and intra-abdominal abscess from a localized bowel perforation.20,21
Initial workup
Once cancer is suspected, colonoscopy is the most accurate and versatile diagnostic test. It not only permits localization and biopsy of lesions throughout the large bowel, but also detects synchronous neoplasms and permits removal of polyps. Computed tomographic (CT) colonography is an alternative if colonoscopy is contraindicated, but it can only detect larger (ie, > 6-mm) tumors.22
According to the ACS,23 men and women at average risk should undergo colorectal cancer screening beginning at age 50. ACS screening recommendations for polyps and colorectal cancer include flexible sigmoidoscopy every 5 years, colonoscopy every 10 years, double-contrast barium enema every 5 years, or CT colonography every 5 years. Tests that detect cancer but not polyps include guaiac-based fecal occult blood test (every year), fecal immunochemical test (every year), stool DNA test (every 3 years). These recommendations are fairly consistent with those of the NCCN12 and USPSTF24 (Table 3).12,23,24
Pearls
- Uncommon presentations include urinary tract problems and intra-abdominal abscess.
- CT colonography can only detect larger tumors.
PROSTATE CANCER
With an estimated 220,800 cases and 27,540 deaths in 2015, prostate cancer is the most common cancer and the second most common cause of cancer-related death in US men.2 Widespread use of serum prostate-specific antigen (PSA) testing has increased the rate of detection of prostate cancer.
Signs and symptoms
Most men with early-stage prostate cancer have no symptoms directly attributable to the disease.
Obstructive symptoms such as hesitancy, decreased stream, retention, and nocturia are common but are usually related to concomitant benign prostatic hypertrophy. As in prostatitis, patients with prostate cancer may present with irritative symptoms such as urinary frequency, dysuria, and urgency.
Patients who present with locally advanced prostate cancer may have symptoms secondary to local invasion, such as hematuria, hematospermia, and new-onset erectile dysfunction.
Prostate cancer usually metastasizes to bone, most commonly to the vertebrae and sternum, and the associated pain can be acute or insidious.
Diagnosis
Prostate cancer is most often diagnosed after biopsy prompted by an elevated PSA level or an abnormal digital rectal examination. The most common abnormal laboratory findings in patients with metastatic prostate cancer are an elevated serum PSA level (typically > 10 ng/mL), an elevated serum alkaline phosphatase level, and anemia, which are all proportional to the extent of bone involvement.
Screening is still controversial
There has been considerable controversy in recent years with regard to PSA screening because of the lack of significant benefit and the potential for harm to the patient, with an overdiagnosis rate ranging from 23% to 42%.25
According to the ACS,26 certain groups of men should make an informed decision with their physician about whether to undergo screening: men over age 50 at average risk of prostate cancer and with at least a 10-year life expectancy, men over age 45 at high risk, and men over age 40 at an even higher risk. These ACS guidelines are consistent with those of the NCCN12 but differ from those of the USPSTF27 (Table 4).12,26,27
The patient should fully understand the risks and benefits of prostate cancer screening, as well as why it is controversial: ie, while the lifetime risk of being diagnosed with prostate cancer has increased, the lifetime risk of dying from it has remained the same after the advent of PSA testing.
Adverse effects of biopsy and treatment
Prostate biopsy is associated with infectious and bleeding complications, in addition to anxiety and physical discomfort.28 Treatment-related adverse effects include urinary incontinence, sexual dysfunction, and bowel problems.
Could these potential harms be overstated and the benefit be greater than currently thought? The NCCN12 noted that some of the landmark prostate cancer screening studies found a potential benefit in screening high-risk patients such as black men. Moreover, the studies used the sextant prostate biopsy technique, whereas now the extended core biopsy technique is the standard of care. And the studies may have underestimated the benefit of screening because the trial patients were relatively old (age 60) when their first PSA measurement was done, they were screened at long intervals (every 4 years), and the treatment options available at the time were not as good as those available today.12
Pearls
- Laboratory findings in metastatic prostate cancer are proportional to the extent of bone involvement.
- Most men with early-stage prostate cancer have no symptoms attributable to the disease.
LUNG CANCER
Lung cancer is the second most common type of cancer in men and women but has the highest mortality rate. In the United States, in 2015, an estimated 221,200 new cases of lung cancer and 158,040 deaths were expected.2 Lung cancer deaths have begun to decline in both men and women, and this is due to the decline in smoking. The impact of lung cancer screening may not be seen for another 5 to 10 years.29
A wide range of symptoms, presentations
Many patients with squamous cell carcinoma and small-cell lung carcinoma present with symptoms related to tumor involvement of the central airways,30 including cough, hemoptysis, and postobstructive pneumonia. Partial obstruction of a bronchus may cause localized wheezing, heard by the patient or by the clinician on auscultation, whereas obstruction of larger airways can cause stridor.
Patients with advanced disease present with dull, aching, persistent chest pain from mediastinal, pleural, or chest wall extension, dyspnea from lymphangitic tumor spread, tumor emboli, pneumothorax, pleural effusion, or pericardial effusion with tamponade. Less commonly, patients may present with unilateral paralysis of the diaphragm from phrenic nerve damage or with hoarseness from recurrent laryngeal nerve compression.31
Bronchorrhea—production of large volumes of thin, mucoid secretions resulting in cough—may be a feature of bronchoalveolar cell carcinoma, a rare subtype of non-small-cell lung carcinoma.
Patients uncommonly present with superior vena cava syndrome, an oncologic emergency that most often causes facial and arm swelling, dyspnea, cough, and headache.
Non-small-cell lung carcinoma arising in the superior sulcus may in rare cases cause Pancoast syndrome (manifested by shoulder pain and atrophy of the hand muscles from brachial plexus involvement), Horner syndrome (manifested by ptosis, miosis, and anhidrosis), or rib destruction.
If metastasis occurs, lung cancer commonly metastasizes to the liver and adrenal glands. At the time of diagnosis, 20% to 30% of patients with small-cell lung carcinoma have symptoms of central nervous system metastasis.
The screening controversy
Lung cancer screening is controversial because previous large studies have failed to show a clinical benefit (ie, improved survival rates) of CT screening in smokers. However, based on the results of a later large randomized trial,32 the ACS33 now recommends that patients ages 55 to 74 who are in fairly good health, have at least a 30-pack-year smoking history, and are currently smoking or have quit smoking within the last 15 years should discuss with their physician the benefits, limitations, and potential harms of lung cancer screening. These recommendations are similar to those of the NCCN12,34 and USPSTF35 (Table 5).12,33–35 The ACS guidelines also emphasize that screening should be done only at facilities with extensive experience with low-dose CT.
Follow-up evaluation
If imaging detects a lung nodule, its size and consistency are crucial in determining the course of action.33 If an endobronchial growth or solid nodule larger than 8 mm is discovered, the primary care physician should consider ordering either a repeat low-dose CT scan after 1 month or a positron-emission tomography CT scan.34 The diagnosis should be confirmed by biopsy or by surgical removal of the nodule if localized and accessible, with sites of metastasis typically taking priority.
Pearl
- At diagnosis, 20% to 30% of patients with small-cell lung cancer have symptoms of central nervous system metastasis.
HIGHLY CURABLE CANCERS WITH UNUSUAL PRESENTATIONS
Hodgkin lymphoma
With 9,190 new cases in the United States annually and a 5-year overall survival rate over 85%, Hodgkin lymphoma is one of the least common but most curable cancers.1,2 In the United States, there are two diagnostic peaks, one around age 20 and one around age 65.36 In patients with human immunodeficiency virus infection, the rate is 15 to 30 times higher than in the general population, regardless of disease status or compliance with highly active retroviral therapy.37
Hodgkin lymphoma typically presents as a nontender painless mass with rubbery consistency. The involved lymph node is typically cervical or supraclavicular. Although not detectable on physical examination, enlarged mediastinal nodes and retroperitoneal nodes are often present. Less commonly, patients may present with enlarged axillary and inguinal nodes.38
A second common presentation is the discovery of a mediastinal mass on routine chest radiography. A large percentage of patients present with at least one systemic symptom, which may include fever, night sweats, and unintentional weight loss. Generalized pruritus occurs early in the disease course in 10% to 15% of patients and is occasionally severe enough to cause intense scratching and excoriations.
A more unusual presentation of Hodgkin lymphoma is severe pain at areas of involvement after alcohol ingestion.
Most patients present with overt disease, but the presenting symptoms and signs may be relatively nonspecific and subtle and more consistent with an infectious process.
Hodgkin disease has a variable tempo, but overt symptoms typically occur after several months rather than years. As a general rule, it starts at a single site within the lymphatic system, usually a lymph node, and then spreads to adjacent nodes via lymphatic channels before disseminating to distant nonadjacent sites and organs. With this in mind, it is unusual to have bilateral axillary involvement without disease in the lower neck, and extremely unusual to have hepatic or bone marrow infiltration without disease in the spleen.
The diagnosis is established by whole lymph node tissue biopsy. Due to the high rate of inflammation in the area, inguinal nodes should not be biopsied if other equally suspicious peripheral nodes are present elsewhere. When the diagnosis of Hodgkin lymphoma is made from biopsy of an extranodal site, such as the stomach, spleen, Waldeyer ring, central nervous system, lung, bone, or skin, lymph node biopsy is also desirable for diagnostic confirmation.
Testicular cancer
Although accounting for only about 1% of all cancers in men, testicular cancer is the most common solid tumor affecting males between ages 15 and 35.1,2 With a 5-year survival rate of over 95%, testicular cancer is also one of the most curable cancers.
Testicular tumors usually present as a painless nodule or swelling of one testicle. Uncommonly, patients have metastatic disease at diagnosis, with the most common sites being lymph nodes, lung, bone, and the brain. Gynecomastia, associated with the production of human chorionic gonadotropin, occurs in about 5% of men with testicular germ cell tumors and 20% to 30% of men with Leydig cell tumors.39 Rarely, patients may present with paraneoplastic hyperthyroidism, which is secondary to thyroid-stimulating hormone and human chorionic gonadotropin sharing a common homologous alpha and beta subunit.40
Prompt diagnosis and treatment of testicular cancer provides the best opportunity for cure. Therefore, any testicular mass, even a painful scrotal lesion, should be evaluated as if it is testicular cancer until it is proven otherwise. The diagnostic evaluation of suspected testicular cancer includes scrotal ultrasonography. Radiographic testing, as deemed clinically necessary by the consulting urologist and medical oncologist, may include chest radiography, CT (chest, abdomen, pelvis), brain magnetic resonance imaging, or bone scan.
The primary care laboratory evaluation should include a complete metabolic profile and measurements of lactate dehydrogenase and serum tumor markers such as alpha fetoprotein and human chorionic gonadotropin. In nonseminomatous germ cell tumors, alpha fetoprotein or human chorionic gonadotropin, or both, can be elevated in 80% to 85% of patients. However, in seminoma, alpha fetoprotein is never elevated, and the serum human chorionic gonadotropin is elevated in only 20% to 25% of patients.41
Patients with a suspicious testicular mass should be referred promptly to a urologist for consideration of radical inguinal orchiectomy and, in some cases, retroperitoneal lymph node dissection. Testicular biopsy is not part of the evaluation as it may result in tumor seeding into the scrotal sac or metastatic spread of tumor to the inguinal nodes. Inguinal biopsy of the contralateral testis is considered if ultrasonography raises suspicion of an intratesticular abnormality, cryptorchid testis, or marked testicular atrophy. Discussing sperm banking with the patient is part of the diagnostic workup, as cumulative cisplatin doses greater than 400 mg/m2 can result in permanent infertility in 50% of men.42
Pearls
- In Hodgkin lymphoma, bilateral axillary involvement without disease in the lower neck is unusual.
- Discussing sperm banking is part of the diagnostic workup for testicular cancer.
- National Cancer Institute (NIH). Surveillance, Epidemiology and End Results (SEER) Program. SEER Cancer Statistics Review, 1975–2010. http://seer.cancer.gov/csr/1975_2010/. Accessed May 9, 2016.
- American Cancer Society. Cancer Facts & Figures 2015.
www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2015/. Accessed May 9, 2016. - Tabár L, Vitak B, Chen HH, Yen MF, Duffy SW, Smith RA. Beyond randomized controlled trials: organized mammographic screening substantially reduces breast carcinoma mortality. Cancer 2001; 91:1724–1731.
- Tabar L, Fagerberg G, Chen HH, et al. Efficacy of breast cancer screening by age. New results from the Swedish two-county trial. Cancer 1995; 75:2507–2517.
- Humphrey LL, Helfand M, Chan BK, Woolf SH. Breast cancer screening: a summary of the evidence for the US Preventive Services Task Force. Ann Intern Med 2002; 137:347–360.
- Esserman LJ, Shieh Y, Rutgers EJ, et al. Impact of mammographic screening on the detection of good and poor prognosis breast cancers. Breast Cancer Res Treat 2011; 130:725–734.
- Wallace E, Lowry J, Smith SM, Fahey T. The epidemiology of malpractice claims in primary care: a systematic review. BMJ Open 2013; 3:pii:e002929.
- Gandhi TK, Kachalia A, Thomas EJ, et al. Missed and delayed diagnoses in the ambulatory setting: a study of closed malpractice claims. Ann Intern Med 2006; 145:488–496.
- Morrow M. The evaluation of common breast problems. Am Fam Physician 2000; 61:2371–2378, 2385.
- Santen RJ, Mansel R. Benign breast disorders. N Engl J Med 2005; 353:275–285.
- American Cancer Society. Breast cancer prevention and early detection. www.cancer.org/cancer/breastcancer/moreinformation/breastcancerearlydetection/breast-cancer-early-detection-acs-recs. Accessed May 17, 2016.
- National Comprehensive Cancer Network (NCCN). NCCN Guidelines. www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed May 17, 2016.
- US Preventive Services Task Force (USPSTF). Breast cancers Screening. www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/breast-cancer-screening. Accessed May 17, 2016.
- Mandelblatt JS, Cronin KA, Bailey S, et al; Breast Cancer Working Group of the Cancer Intervention and Surveillance Modeling Network. Effects of mammography screening under different screening schedules: model estimates of potential benefits and harms. Ann Intern Med 2009; 151:738–747.
- Newcomb PA, Norfleet RG, Storer BE, Surawicz TS, Marcus PM. Screening sigmoidoscopy and colorectal cancer mortality. J Natl Cancer Inst 1992; 84:1572–1575.
- Bressler B, Paszat LF, Chen Z, Rothwell DM, Vinden C, Rabeneck L. Rates of new or missed colorectal cancers after colonoscopy and their risk factors: a population-based analysis. Gastroenterology 2007; 132:96–102.
- Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9–29.
- Feld AD. Malpractice risks associated with colon cancer and inflammatory bowel disease. J Gastroenterol 2004; 99:1641–1644.
- Goodman D, Irvin TT. Delay in the diagnosis and prognosis of carcinoma of the right colon. Br J Surg 1993; 80:1327–1329.
- Alvarez JA, Baldonedo RF, Bear IG, Alvarez P, Jorge JL. Anaerobic liver abscesses as initial presentation of silent colonic cancer. HPB (Oxford) 2004; 6:41–42.
- Tsai HL, Hsieh JS, Yu FJ, et al. Perforated colonic cancer presenting as intra-abdominal abscess. Int J Colorectal Dis 2007; 22:15–19.
- Levin B, Lieberman DA, McFarland B, et al; American Cancer Society Colorectal Cancer Advisory Group; US Multi-Society Task Force; American College of Radiology Colon Cancer Committee. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. Gastroenterology 2008; 134:1570–1595.
- American Cancer Society. Colorectal cancer prevention and early detection. www.cancer.org/cancer/colonandrectumcancer/moreinformation/colonandrectumcancerearlydetection/colorectal-cancer-early-detection-acs-recommendations. Accessed May 17, 2016.
- US Preventive Services Task Force (USPSTF). Colorectal cancer: screening. www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/colorectal-cancer-screening. Accessed May 17, 2016.
- Schröder FH, Hugosson J, Roobol MJ, et al; ERSPC Investigators. Screening and prostate cancer mortality: results of the European Randomised Study of Screening for Prostate Cancer (ERSPC) at 13 years of follow-up. Lancet 2014; 384:2027–2035.
- American Cancer Society. Prostate cancer prevention and early detection. www.cancer.org/cancer/prostatecancer/moreinformation/prostatecancerearlydetection/index. Accessed May 17, 2016.
- US Preventive Services Task Force (USPSTF). Prostate cancer: screening. www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/prostate-cancer-screening. Accessed June 1, 2016.
- Essink-Bot ML, de Koning HJ, Nijs HG, Kirkels WJ, van der Maas PJ, Schröder FH. Short-term effects of population-based screening for prostate cancer on health-related quality of life. J Natl Cancer Inst 1998; 90:925–931.
- Peto R, Darby S, Deo H, Silcocks P, Whitley E, Doll R. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ (Clin Res Ed) 2000; 321:323–329.
- Chute CG, Greenberg ER, Baron J, Korson R, Baker J, Yates J. Presenting conditions of 1,539 population-based lung cancer patients by cell type and stage in New Hampshire and Vermont. Cancer 1985; 56:2107–2111.
- Ramadan HH, Wax MK, Avery S. Outcome and changing cause of unilateral vocal cord paralysis. Otolaryngol Head Neck Surg 1998;118:199–202.
- Church TR, Black WC, Aberle DR, et al. Results of initial low-dose computed tomographic screening for lung cancer. N Engl J Med 2013; 368:1980–1991.
- American Cancer Society. Lung cancer prevention and early detection. www.cancer.org/cancer/lungcancer-non-smallcell/moreinformation/lungcancerpreventionandearlydetection/index. Accessed May 17, 2016.
- Lung Cancer Screening. National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology. www.nccn.org/professionals/physician_gls/pdf/lung_screening.pdf. Accessed May 17, 2016.
- US Preventive Services Task Force (USPSTF). Lung cancer: screening. www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/lung-cancer-screening. Accessed May 17, 2016.
- National Cancer Institute (NIH). Surveillance, Epidemiology, and End Results (SEER) Program. SEER Cancer Statistics Review: 1973-1994. http://seer.cancer.gov/archive/csr/1973_1994/. Accessed May 17, 2016.
- Mauch PM, Kalish LA, Kadin M, Coleman CN, Osteen R, Hellman S. Patterns of presentation of Hodgkin disease. Implications for etiology and pathogenesis. Cancer 1993; 71:2062–2071.
- Gobbi PG, Cavalli C, Gendarini A, et al. Reevaluation of prognostic significance of symptoms in Hodgkin’s disease. Cancer 1985; 56:2874–2880.
- Einhorn LH. Treatment of testicular cancer: a new and improved model. J Clin Oncol 1990; 8:1777–1781.
- Tseng A Jr, Horning SJ, Freiha FS, Resser KJ, Hannigan JF Jr, Torti FM. Gynecomastia in testicular cancer patients. Prognostic and therapeutic implications. Cancer 1985; 56:2534–2538.
- Gilligan TD, Seidenfeld J, Basch EM, et al; American Society of Clinical Oncology. American Society of Clinical Oncology clinical practice guideline on uses of serum tumor markers in adult males with germ cell tumors. J Clin Oncol 2010; 28:3388–3404.
- Brydøy M, Fosså SD, Klepp O, et al. Paternity following treatment for testicular cancer. J Natl Cancer Inst 2005; 97:1580–1588.
- National Cancer Institute (NIH). Surveillance, Epidemiology and End Results (SEER) Program. SEER Cancer Statistics Review, 1975–2010. http://seer.cancer.gov/csr/1975_2010/. Accessed May 9, 2016.
- American Cancer Society. Cancer Facts & Figures 2015.
www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2015/. Accessed May 9, 2016. - Tabár L, Vitak B, Chen HH, Yen MF, Duffy SW, Smith RA. Beyond randomized controlled trials: organized mammographic screening substantially reduces breast carcinoma mortality. Cancer 2001; 91:1724–1731.
- Tabar L, Fagerberg G, Chen HH, et al. Efficacy of breast cancer screening by age. New results from the Swedish two-county trial. Cancer 1995; 75:2507–2517.
- Humphrey LL, Helfand M, Chan BK, Woolf SH. Breast cancer screening: a summary of the evidence for the US Preventive Services Task Force. Ann Intern Med 2002; 137:347–360.
- Esserman LJ, Shieh Y, Rutgers EJ, et al. Impact of mammographic screening on the detection of good and poor prognosis breast cancers. Breast Cancer Res Treat 2011; 130:725–734.
- Wallace E, Lowry J, Smith SM, Fahey T. The epidemiology of malpractice claims in primary care: a systematic review. BMJ Open 2013; 3:pii:e002929.
- Gandhi TK, Kachalia A, Thomas EJ, et al. Missed and delayed diagnoses in the ambulatory setting: a study of closed malpractice claims. Ann Intern Med 2006; 145:488–496.
- Morrow M. The evaluation of common breast problems. Am Fam Physician 2000; 61:2371–2378, 2385.
- Santen RJ, Mansel R. Benign breast disorders. N Engl J Med 2005; 353:275–285.
- American Cancer Society. Breast cancer prevention and early detection. www.cancer.org/cancer/breastcancer/moreinformation/breastcancerearlydetection/breast-cancer-early-detection-acs-recs. Accessed May 17, 2016.
- National Comprehensive Cancer Network (NCCN). NCCN Guidelines. www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed May 17, 2016.
- US Preventive Services Task Force (USPSTF). Breast cancers Screening. www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/breast-cancer-screening. Accessed May 17, 2016.
- Mandelblatt JS, Cronin KA, Bailey S, et al; Breast Cancer Working Group of the Cancer Intervention and Surveillance Modeling Network. Effects of mammography screening under different screening schedules: model estimates of potential benefits and harms. Ann Intern Med 2009; 151:738–747.
- Newcomb PA, Norfleet RG, Storer BE, Surawicz TS, Marcus PM. Screening sigmoidoscopy and colorectal cancer mortality. J Natl Cancer Inst 1992; 84:1572–1575.
- Bressler B, Paszat LF, Chen Z, Rothwell DM, Vinden C, Rabeneck L. Rates of new or missed colorectal cancers after colonoscopy and their risk factors: a population-based analysis. Gastroenterology 2007; 132:96–102.
- Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9–29.
- Feld AD. Malpractice risks associated with colon cancer and inflammatory bowel disease. J Gastroenterol 2004; 99:1641–1644.
- Goodman D, Irvin TT. Delay in the diagnosis and prognosis of carcinoma of the right colon. Br J Surg 1993; 80:1327–1329.
- Alvarez JA, Baldonedo RF, Bear IG, Alvarez P, Jorge JL. Anaerobic liver abscesses as initial presentation of silent colonic cancer. HPB (Oxford) 2004; 6:41–42.
- Tsai HL, Hsieh JS, Yu FJ, et al. Perforated colonic cancer presenting as intra-abdominal abscess. Int J Colorectal Dis 2007; 22:15–19.
- Levin B, Lieberman DA, McFarland B, et al; American Cancer Society Colorectal Cancer Advisory Group; US Multi-Society Task Force; American College of Radiology Colon Cancer Committee. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. Gastroenterology 2008; 134:1570–1595.
- American Cancer Society. Colorectal cancer prevention and early detection. www.cancer.org/cancer/colonandrectumcancer/moreinformation/colonandrectumcancerearlydetection/colorectal-cancer-early-detection-acs-recommendations. Accessed May 17, 2016.
- US Preventive Services Task Force (USPSTF). Colorectal cancer: screening. www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/colorectal-cancer-screening. Accessed May 17, 2016.
- Schröder FH, Hugosson J, Roobol MJ, et al; ERSPC Investigators. Screening and prostate cancer mortality: results of the European Randomised Study of Screening for Prostate Cancer (ERSPC) at 13 years of follow-up. Lancet 2014; 384:2027–2035.
- American Cancer Society. Prostate cancer prevention and early detection. www.cancer.org/cancer/prostatecancer/moreinformation/prostatecancerearlydetection/index. Accessed May 17, 2016.
- US Preventive Services Task Force (USPSTF). Prostate cancer: screening. www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/prostate-cancer-screening. Accessed June 1, 2016.
- Essink-Bot ML, de Koning HJ, Nijs HG, Kirkels WJ, van der Maas PJ, Schröder FH. Short-term effects of population-based screening for prostate cancer on health-related quality of life. J Natl Cancer Inst 1998; 90:925–931.
- Peto R, Darby S, Deo H, Silcocks P, Whitley E, Doll R. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ (Clin Res Ed) 2000; 321:323–329.
- Chute CG, Greenberg ER, Baron J, Korson R, Baker J, Yates J. Presenting conditions of 1,539 population-based lung cancer patients by cell type and stage in New Hampshire and Vermont. Cancer 1985; 56:2107–2111.
- Ramadan HH, Wax MK, Avery S. Outcome and changing cause of unilateral vocal cord paralysis. Otolaryngol Head Neck Surg 1998;118:199–202.
- Church TR, Black WC, Aberle DR, et al. Results of initial low-dose computed tomographic screening for lung cancer. N Engl J Med 2013; 368:1980–1991.
- American Cancer Society. Lung cancer prevention and early detection. www.cancer.org/cancer/lungcancer-non-smallcell/moreinformation/lungcancerpreventionandearlydetection/index. Accessed May 17, 2016.
- Lung Cancer Screening. National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology. www.nccn.org/professionals/physician_gls/pdf/lung_screening.pdf. Accessed May 17, 2016.
- US Preventive Services Task Force (USPSTF). Lung cancer: screening. www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/lung-cancer-screening. Accessed May 17, 2016.
- National Cancer Institute (NIH). Surveillance, Epidemiology, and End Results (SEER) Program. SEER Cancer Statistics Review: 1973-1994. http://seer.cancer.gov/archive/csr/1973_1994/. Accessed May 17, 2016.
- Mauch PM, Kalish LA, Kadin M, Coleman CN, Osteen R, Hellman S. Patterns of presentation of Hodgkin disease. Implications for etiology and pathogenesis. Cancer 1993; 71:2062–2071.
- Gobbi PG, Cavalli C, Gendarini A, et al. Reevaluation of prognostic significance of symptoms in Hodgkin’s disease. Cancer 1985; 56:2874–2880.
- Einhorn LH. Treatment of testicular cancer: a new and improved model. J Clin Oncol 1990; 8:1777–1781.
- Tseng A Jr, Horning SJ, Freiha FS, Resser KJ, Hannigan JF Jr, Torti FM. Gynecomastia in testicular cancer patients. Prognostic and therapeutic implications. Cancer 1985; 56:2534–2538.
- Gilligan TD, Seidenfeld J, Basch EM, et al; American Society of Clinical Oncology. American Society of Clinical Oncology clinical practice guideline on uses of serum tumor markers in adult males with germ cell tumors. J Clin Oncol 2010; 28:3388–3404.
- Brydøy M, Fosså SD, Klepp O, et al. Paternity following treatment for testicular cancer. J Natl Cancer Inst 2005; 97:1580–1588.
KEY POINTS
- By detecting breast cancer lesions 2 years before they are discovered by clinical breast examination, mammography has been found to reduce the mortality rate from breast cancer.
- In the United States, 20% of colorectal cancer patients have distant metastases at the time of diagnosis. The most common sites are the lymph nodes, liver, lungs, and peritoneum.
- The patient should fully understand the risks and benefits of prostate-specific antigen (PSA) screening and that it is controversial because, since the advent of PSA testing, the lifetime risk of being diagnosed with prostate cancer has increased, but the lifetime risk of dying from it has remained the same.