User login
Ultrasound’s role in detecting enthesitis in psoriatic arthritis remains debatable
MIAMI – Ultrasound-detected enthesitis was associated with both destructive and bone formation lesions on radiography of peripheral and axial joints in a study of 222 patients with psoriatic arthritis. But its lack of correlation to clinically detected enthesitis in this study and in others has made its clinical usefulness somewhat controversial.
“This is not first time we see this result in the literature,” Ari Polachek, MD, said when presenting the research at the annual meeting of the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA). The findings suggest ultrasound reveals something different than a physical examination does, and because it’s more sensitive and specific, ultrasound is more accurate, he said.

Diagnostic disagreement
Some differing opinions arose during a discussion after Dr. Polachek’s presentation. “When you look at the enthesitis measures [with ultrasound] ... you don’t necessarily measure the same enthesitis points you measure in clinical practice,” said Dafna D. Gladman, MD, a senior scientist at Toronto Western Hospital’s Krembil Research Institute and a rheumatologist at the University of Toronto. “We have to be careful. I don’t think we should immediately cancel the relationship between the ultrasound and clinical exam.”
“Well, I disagree entirely,” said Philip Helliwell, DM, PhD, senior lecturer in rheumatology at the University of Leeds, England, and president of GRAPPA. “We have looked at the Leeds [Enthesitis Index] with ultrasound and MRI and found no significant relationship at all.” Although Dr. Helliwell said there is a possible role for ultrasound to detect enthesitis in the Achilles, “We are fooling ourselves if we are measuring enthesitis as a pathologic entity. I don’t really know what we’re measuring when we do these scores.”
“My message is you do not need to scan every patient,” Dr. Polachek said in an interview. If a physician remains unsure about the physical exam results, it can be useful. Dr. Polachek, who completed his medical training in Israel, added: “In Israel, we say that ultrasound is ‘the final judge.’ ”

Association with radiographic joint findings
The investigators achieved their study aim: demonstrating that the severity of sonographic enthesitis is a marker of radiographic peripheral and axial joint damage in psoriatic arthritis. They found an association for both destructive and bone formation lesions.
“These findings highlight the potential role of enthesitis in the pathogenesis of articular damage in psoriatic arthritis,” said Dr. Polachek, clinical and research fellow at the University of Toronto.
The researchers assessed 12 entheseal sites with ultrasound. They used the Madrid Sonography Enthesitis Index scoring system (MASEI) to determine the global extent of enthesitis in each patient. In addition, they used the modified Steinbrocker score to assess peripheral joint damage, and the modified Stoke Ankylosing Spondylitis Spine Score (mSASSS) to assess spinal damage. Patients also underwent a clinical exam and were asked about their medical history.
Multivariate analysis revealed a significant association between higher MASEI score and joint ankylosis (odds ratio, 2.09; P = .0001) and arthritis mutilans (OR, 1.73; P = .005). The total MASEI score was associated with the modified Steinbrocker score (OR, 9.3; P less than .0001) in a logistic regression analysis; total MASEI also significantly correlated with the mSASSS measure of spinal damage (OR, 1.55; P less than .0001) in a linear regression analysis.

Participants had a mean age of 56 years and a 17-year mean duration of psoriatic arthritis. They presented with a mean of 2.4 tender joints and 1.1 swollen joints. At study entry, their mean scores were 15.6 on MASEI, 18.1 on modified Steinbrocker, and 1.72 on mSASSS.
The strengths of the study included a large number of participants and control of multiple possible confounders (age, sex, body mass index, duration of psoriatic arthritis, and use of disease-modifying antirheumatic drugs and biologics). It is limited by its cross-sectional design, which rules out inferences of causality.
Clinical confirmation
“Traditionally, patients came in with a tender joint, and we might not see anything. Now I can put the sensor down and say they have inflammation,” Dr. Polachek said. In such cases, he may suggest more aggressive treatment. However, if an asymptomatic patient has ultrasound findings, “right now the recommendation is not to do anything. Otherwise, it could be overtreatment.”
“This is the future,” Dr. Polachek said. “I started doing ultrasound myself – it’s a game changer.”
Dr. Polachek has received funding from Janssen and the Krembil Research Institute.
MIAMI – Ultrasound-detected enthesitis was associated with both destructive and bone formation lesions on radiography of peripheral and axial joints in a study of 222 patients with psoriatic arthritis. But its lack of correlation to clinically detected enthesitis in this study and in others has made its clinical usefulness somewhat controversial.
“This is not first time we see this result in the literature,” Ari Polachek, MD, said when presenting the research at the annual meeting of the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA). The findings suggest ultrasound reveals something different than a physical examination does, and because it’s more sensitive and specific, ultrasound is more accurate, he said.
Diagnostic disagreement
Some differing opinions arose during a discussion after Dr. Polachek’s presentation. “When you look at the enthesitis measures [with ultrasound] ... you don’t necessarily measure the same enthesitis points you measure in clinical practice,” said Dafna D. Gladman, MD, a senior scientist at Toronto Western Hospital’s Krembil Research Institute and a rheumatologist at the University of Toronto. “We have to be careful. I don’t think we should immediately cancel the relationship between the ultrasound and clinical exam.”
“Well, I disagree entirely,” said Philip Helliwell, DM, PhD, senior lecturer in rheumatology at the University of Leeds, England, and president of GRAPPA. “We have looked at the Leeds [Enthesitis Index] with ultrasound and MRI and found no significant relationship at all.” Although Dr. Helliwell said there is a possible role for ultrasound to detect enthesitis in the Achilles, “We are fooling ourselves if we are measuring enthesitis as a pathologic entity. I don’t really know what we’re measuring when we do these scores.”
“My message is you do not need to scan every patient,” Dr. Polachek said in an interview. If a physician remains unsure about the physical exam results, it can be useful. Dr. Polachek, who completed his medical training in Israel, added: “In Israel, we say that ultrasound is ‘the final judge.’ ”
Association with radiographic joint findings
The investigators achieved their study aim: demonstrating that the severity of sonographic enthesitis is a marker of radiographic peripheral and axial joint damage in psoriatic arthritis. They found an association for both destructive and bone formation lesions.
“These findings highlight the potential role of enthesitis in the pathogenesis of articular damage in psoriatic arthritis,” said Dr. Polachek, clinical and research fellow at the University of Toronto.
The researchers assessed 12 entheseal sites with ultrasound. They used the Madrid Sonography Enthesitis Index scoring system (MASEI) to determine the global extent of enthesitis in each patient. In addition, they used the modified Steinbrocker score to assess peripheral joint damage, and the modified Stoke Ankylosing Spondylitis Spine Score (mSASSS) to assess spinal damage. Patients also underwent a clinical exam and were asked about their medical history.
Multivariate analysis revealed a significant association between higher MASEI score and joint ankylosis (odds ratio, 2.09; P = .0001) and arthritis mutilans (OR, 1.73; P = .005). The total MASEI score was associated with the modified Steinbrocker score (OR, 9.3; P less than .0001) in a logistic regression analysis; total MASEI also significantly correlated with the mSASSS measure of spinal damage (OR, 1.55; P less than .0001) in a linear regression analysis.
Participants had a mean age of 56 years and a 17-year mean duration of psoriatic arthritis. They presented with a mean of 2.4 tender joints and 1.1 swollen joints. At study entry, their mean scores were 15.6 on MASEI, 18.1 on modified Steinbrocker, and 1.72 on mSASSS.
The strengths of the study included a large number of participants and control of multiple possible confounders (age, sex, body mass index, duration of psoriatic arthritis, and use of disease-modifying antirheumatic drugs and biologics). It is limited by its cross-sectional design, which rules out inferences of causality.
Clinical confirmation
“Traditionally, patients came in with a tender joint, and we might not see anything. Now I can put the sensor down and say they have inflammation,” Dr. Polachek said. In such cases, he may suggest more aggressive treatment. However, if an asymptomatic patient has ultrasound findings, “right now the recommendation is not to do anything. Otherwise, it could be overtreatment.”
“This is the future,” Dr. Polachek said. “I started doing ultrasound myself – it’s a game changer.”
Dr. Polachek has received funding from Janssen and the Krembil Research Institute.
MIAMI – Ultrasound-detected enthesitis was associated with both destructive and bone formation lesions on radiography of peripheral and axial joints in a study of 222 patients with psoriatic arthritis. But its lack of correlation to clinically detected enthesitis in this study and in others has made its clinical usefulness somewhat controversial.
“This is not first time we see this result in the literature,” Ari Polachek, MD, said when presenting the research at the annual meeting of the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA). The findings suggest ultrasound reveals something different than a physical examination does, and because it’s more sensitive and specific, ultrasound is more accurate, he said.
Diagnostic disagreement
Some differing opinions arose during a discussion after Dr. Polachek’s presentation. “When you look at the enthesitis measures [with ultrasound] ... you don’t necessarily measure the same enthesitis points you measure in clinical practice,” said Dafna D. Gladman, MD, a senior scientist at Toronto Western Hospital’s Krembil Research Institute and a rheumatologist at the University of Toronto. “We have to be careful. I don’t think we should immediately cancel the relationship between the ultrasound and clinical exam.”
“Well, I disagree entirely,” said Philip Helliwell, DM, PhD, senior lecturer in rheumatology at the University of Leeds, England, and president of GRAPPA. “We have looked at the Leeds [Enthesitis Index] with ultrasound and MRI and found no significant relationship at all.” Although Dr. Helliwell said there is a possible role for ultrasound to detect enthesitis in the Achilles, “We are fooling ourselves if we are measuring enthesitis as a pathologic entity. I don’t really know what we’re measuring when we do these scores.”
“My message is you do not need to scan every patient,” Dr. Polachek said in an interview. If a physician remains unsure about the physical exam results, it can be useful. Dr. Polachek, who completed his medical training in Israel, added: “In Israel, we say that ultrasound is ‘the final judge.’ ”
Association with radiographic joint findings
The investigators achieved their study aim: demonstrating that the severity of sonographic enthesitis is a marker of radiographic peripheral and axial joint damage in psoriatic arthritis. They found an association for both destructive and bone formation lesions.
“These findings highlight the potential role of enthesitis in the pathogenesis of articular damage in psoriatic arthritis,” said Dr. Polachek, clinical and research fellow at the University of Toronto.
The researchers assessed 12 entheseal sites with ultrasound. They used the Madrid Sonography Enthesitis Index scoring system (MASEI) to determine the global extent of enthesitis in each patient. In addition, they used the modified Steinbrocker score to assess peripheral joint damage, and the modified Stoke Ankylosing Spondylitis Spine Score (mSASSS) to assess spinal damage. Patients also underwent a clinical exam and were asked about their medical history.
Multivariate analysis revealed a significant association between higher MASEI score and joint ankylosis (odds ratio, 2.09; P = .0001) and arthritis mutilans (OR, 1.73; P = .005). The total MASEI score was associated with the modified Steinbrocker score (OR, 9.3; P less than .0001) in a logistic regression analysis; total MASEI also significantly correlated with the mSASSS measure of spinal damage (OR, 1.55; P less than .0001) in a linear regression analysis.
Participants had a mean age of 56 years and a 17-year mean duration of psoriatic arthritis. They presented with a mean of 2.4 tender joints and 1.1 swollen joints. At study entry, their mean scores were 15.6 on MASEI, 18.1 on modified Steinbrocker, and 1.72 on mSASSS.
The strengths of the study included a large number of participants and control of multiple possible confounders (age, sex, body mass index, duration of psoriatic arthritis, and use of disease-modifying antirheumatic drugs and biologics). It is limited by its cross-sectional design, which rules out inferences of causality.
Clinical confirmation
“Traditionally, patients came in with a tender joint, and we might not see anything. Now I can put the sensor down and say they have inflammation,” Dr. Polachek said. In such cases, he may suggest more aggressive treatment. However, if an asymptomatic patient has ultrasound findings, “right now the recommendation is not to do anything. Otherwise, it could be overtreatment.”
“This is the future,” Dr. Polachek said. “I started doing ultrasound myself – it’s a game changer.”
Dr. Polachek has received funding from Janssen and the Krembil Research Institute.
AT 2016 GRAPPA
Key clinical point: Ultrasound can diagnose enthesitis in psoriatic arthritis, but findings differ from physical exam.
Major finding: Multivariate analysis revealed a significant association between higher MASEI score and joint ankylosis (OR, 2.09; P = .0001) and arthritis mutilans (OR, 1.73; P = .005).
Data source: Cross-sectional study of 222 patients with psoriatic arthritis.
Disclosures: Dr. Polachek has received funding from Janssen and the Krembil Research Institute.

Superbug Infections On the Rise With No Antibiotic Success Yet
NEW YORK - After two confirmed U.S. cases of a superbug that thwarts a last-resort antibiotic, infectious disease experts say they expect more cases in coming months because the bacterial gene behind it is likely far more widespread than previously believed.
Army scientists in May reported finding E. coli bacteria that harbor a gene which renders the antibiotic colistin useless. The gene, called mcr-1, was found in a urine sample of a Pennsylvania woman being treated for a urinary tract infection.
On Monday, researchers confirmed preliminary findings that E. coli carrying the same mcr-1 gene were found in a stored bacterial sample of a New York patient who had been treated for an infection last year, as well as in patient samples from nine other countries.
The report came from a global effort called the SENTRY Antimicrobial Surveillance Program, led by Mariana Castanheira of JMI Laboratories based in North Liberty, Iowa.
The mcr-1 superbug has been identified over the past six months in farm animals and people in about 20 countries, including China, Germany and Italy.
The bacteria can be transmitted by fecal contact and poor hygiene, which suggests a far wider likely presence than the documented cases so far, according to leading infectious disease experts.
Health officials fear the mcr-1 gene, carried by a highly mobile piece of DNA called a plasmid, will soon be found in bacteria already resistant to all or virtually all other types of antibiotics, potentially making infections untreatable.
"You can be sure (mcr-1) is already in the guts of people throughout the United States and will continue to spread," said Dr. Brad Spellberg, professor of medicine at the University of Southern California.
Dr. David Van Duin, an infectious disease expert at the University of North Carolina in Chapel Hill, said he expects more documented U.S. cases of mcr-1 in coming months because it is already here and will spread from abroad. "We will see a lot more of this gene."
Colistin causes kidney damage, but doctors have opted for it as other antibiotics increasingly fail. Its overuse, especially in overseas farm animals, has allowed bacteria to develop resistance to it.
PAST AND PRESENT INFECTIONS
To track the mcr-1 gene, U.S. hospitals are working together with state and federal agencies to test bacteria samples of patients that have recently been treated for infections. Many of the largest research hospitals are examining samples of antibiotic-resistant bacteria that have long been stored in their freezers.
Gautam Dantas, associate professor of pathology at Washington University Medical Center in St. Louis, has tested hundreds of U.S. samples of archived bacteria in recent months and has not yet detected mcr-1. But he expects dozens of confirmed cases of the gene will be documented by next year in the country, mostly among current patients.
The concern of many disease experts is that mcr-1 could soon show up in bacteria also resistant to carbapenems, one of the few remaining dependable classes of antibiotics. In that event, with colistin no longer a last-ditch option, some patients would have to rely on their immune systems to fight off infection.
"Within the next two to three years, it's going to be fairly routine for infections to occur in the United States for which we have no (effective) drugs available," Dantas said.
Castanheira also believes mcr-1 will find its way into carbapenem-resistant enterobacteriaceae (CRE).
In an interview, she said the resulting virtually impervious bacterium would likely spread slowly inside the United States because CRE themselves are not yet widespread in the country, giving drugmakers some time to create new antibiotics.
Beginning in August, the U.S. Centers for Disease Control and Prevention will use $21 million to expand surveillance at laboratories operated by all 50 state health departments and seven larger regional labs. The federal funding will help pay for more-sensitive equipment to test for antibiotic resistance in bacteria samples provided by hospitals.
Jean Patel, deputy director of the CDC's Office of Antimicrobial Resistance, said the effort will provide the CDC improved national surveillance of antibiotic-resistance trends, including any spread of mcr-1.
"This is data for action," she said, adding that special procedures to prevent infections from spreading in hospitals could be taken once a patient is identified with mcr-1 related infections or with multidrug-resistant bacteria.
SOURCE: http://bit.ly/29yFekw
Antimicrob Agents Chemother 2016.
NEW YORK - After two confirmed U.S. cases of a superbug that thwarts a last-resort antibiotic, infectious disease experts say they expect more cases in coming months because the bacterial gene behind it is likely far more widespread than previously believed.
Army scientists in May reported finding E. coli bacteria that harbor a gene which renders the antibiotic colistin useless. The gene, called mcr-1, was found in a urine sample of a Pennsylvania woman being treated for a urinary tract infection.
On Monday, researchers confirmed preliminary findings that E. coli carrying the same mcr-1 gene were found in a stored bacterial sample of a New York patient who had been treated for an infection last year, as well as in patient samples from nine other countries.
The report came from a global effort called the SENTRY Antimicrobial Surveillance Program, led by Mariana Castanheira of JMI Laboratories based in North Liberty, Iowa.
The mcr-1 superbug has been identified over the past six months in farm animals and people in about 20 countries, including China, Germany and Italy.
The bacteria can be transmitted by fecal contact and poor hygiene, which suggests a far wider likely presence than the documented cases so far, according to leading infectious disease experts.
Health officials fear the mcr-1 gene, carried by a highly mobile piece of DNA called a plasmid, will soon be found in bacteria already resistant to all or virtually all other types of antibiotics, potentially making infections untreatable.
"You can be sure (mcr-1) is already in the guts of people throughout the United States and will continue to spread," said Dr. Brad Spellberg, professor of medicine at the University of Southern California.
Dr. David Van Duin, an infectious disease expert at the University of North Carolina in Chapel Hill, said he expects more documented U.S. cases of mcr-1 in coming months because it is already here and will spread from abroad. "We will see a lot more of this gene."
Colistin causes kidney damage, but doctors have opted for it as other antibiotics increasingly fail. Its overuse, especially in overseas farm animals, has allowed bacteria to develop resistance to it.
PAST AND PRESENT INFECTIONS
To track the mcr-1 gene, U.S. hospitals are working together with state and federal agencies to test bacteria samples of patients that have recently been treated for infections. Many of the largest research hospitals are examining samples of antibiotic-resistant bacteria that have long been stored in their freezers.
Gautam Dantas, associate professor of pathology at Washington University Medical Center in St. Louis, has tested hundreds of U.S. samples of archived bacteria in recent months and has not yet detected mcr-1. But he expects dozens of confirmed cases of the gene will be documented by next year in the country, mostly among current patients.
The concern of many disease experts is that mcr-1 could soon show up in bacteria also resistant to carbapenems, one of the few remaining dependable classes of antibiotics. In that event, with colistin no longer a last-ditch option, some patients would have to rely on their immune systems to fight off infection.
"Within the next two to three years, it's going to be fairly routine for infections to occur in the United States for which we have no (effective) drugs available," Dantas said.
Castanheira also believes mcr-1 will find its way into carbapenem-resistant enterobacteriaceae (CRE).
In an interview, she said the resulting virtually impervious bacterium would likely spread slowly inside the United States because CRE themselves are not yet widespread in the country, giving drugmakers some time to create new antibiotics.
Beginning in August, the U.S. Centers for Disease Control and Prevention will use $21 million to expand surveillance at laboratories operated by all 50 state health departments and seven larger regional labs. The federal funding will help pay for more-sensitive equipment to test for antibiotic resistance in bacteria samples provided by hospitals.
Jean Patel, deputy director of the CDC's Office of Antimicrobial Resistance, said the effort will provide the CDC improved national surveillance of antibiotic-resistance trends, including any spread of mcr-1.
"This is data for action," she said, adding that special procedures to prevent infections from spreading in hospitals could be taken once a patient is identified with mcr-1 related infections or with multidrug-resistant bacteria.
SOURCE: http://bit.ly/29yFekw
Antimicrob Agents Chemother 2016.
NEW YORK - After two confirmed U.S. cases of a superbug that thwarts a last-resort antibiotic, infectious disease experts say they expect more cases in coming months because the bacterial gene behind it is likely far more widespread than previously believed.
Army scientists in May reported finding E. coli bacteria that harbor a gene which renders the antibiotic colistin useless. The gene, called mcr-1, was found in a urine sample of a Pennsylvania woman being treated for a urinary tract infection.
On Monday, researchers confirmed preliminary findings that E. coli carrying the same mcr-1 gene were found in a stored bacterial sample of a New York patient who had been treated for an infection last year, as well as in patient samples from nine other countries.
The report came from a global effort called the SENTRY Antimicrobial Surveillance Program, led by Mariana Castanheira of JMI Laboratories based in North Liberty, Iowa.
The mcr-1 superbug has been identified over the past six months in farm animals and people in about 20 countries, including China, Germany and Italy.
The bacteria can be transmitted by fecal contact and poor hygiene, which suggests a far wider likely presence than the documented cases so far, according to leading infectious disease experts.
Health officials fear the mcr-1 gene, carried by a highly mobile piece of DNA called a plasmid, will soon be found in bacteria already resistant to all or virtually all other types of antibiotics, potentially making infections untreatable.
"You can be sure (mcr-1) is already in the guts of people throughout the United States and will continue to spread," said Dr. Brad Spellberg, professor of medicine at the University of Southern California.
Dr. David Van Duin, an infectious disease expert at the University of North Carolina in Chapel Hill, said he expects more documented U.S. cases of mcr-1 in coming months because it is already here and will spread from abroad. "We will see a lot more of this gene."
Colistin causes kidney damage, but doctors have opted for it as other antibiotics increasingly fail. Its overuse, especially in overseas farm animals, has allowed bacteria to develop resistance to it.
PAST AND PRESENT INFECTIONS
To track the mcr-1 gene, U.S. hospitals are working together with state and federal agencies to test bacteria samples of patients that have recently been treated for infections. Many of the largest research hospitals are examining samples of antibiotic-resistant bacteria that have long been stored in their freezers.
Gautam Dantas, associate professor of pathology at Washington University Medical Center in St. Louis, has tested hundreds of U.S. samples of archived bacteria in recent months and has not yet detected mcr-1. But he expects dozens of confirmed cases of the gene will be documented by next year in the country, mostly among current patients.
The concern of many disease experts is that mcr-1 could soon show up in bacteria also resistant to carbapenems, one of the few remaining dependable classes of antibiotics. In that event, with colistin no longer a last-ditch option, some patients would have to rely on their immune systems to fight off infection.
"Within the next two to three years, it's going to be fairly routine for infections to occur in the United States for which we have no (effective) drugs available," Dantas said.
Castanheira also believes mcr-1 will find its way into carbapenem-resistant enterobacteriaceae (CRE).
In an interview, she said the resulting virtually impervious bacterium would likely spread slowly inside the United States because CRE themselves are not yet widespread in the country, giving drugmakers some time to create new antibiotics.
Beginning in August, the U.S. Centers for Disease Control and Prevention will use $21 million to expand surveillance at laboratories operated by all 50 state health departments and seven larger regional labs. The federal funding will help pay for more-sensitive equipment to test for antibiotic resistance in bacteria samples provided by hospitals.
Jean Patel, deputy director of the CDC's Office of Antimicrobial Resistance, said the effort will provide the CDC improved national surveillance of antibiotic-resistance trends, including any spread of mcr-1.
"This is data for action," she said, adding that special procedures to prevent infections from spreading in hospitals could be taken once a patient is identified with mcr-1 related infections or with multidrug-resistant bacteria.
SOURCE: http://bit.ly/29yFekw
Antimicrob Agents Chemother 2016.
Aleukemic acute lymphoblastic leukemia with unusual clinical features
Acute lymphoblastic leukemia is a neoplastic proliferation of lymphoblasts in the bone marrow. Normal hematopoiesis is affected, and symptoms from anemia (fatigue, breathlessness), leukopenia (recurrent infections) or thrombocytopenia (easy bruising, mucosal bleeding) are typically described in ALL. Hepatosplenomegaly and B-symptoms (fever, weight loss, and night sweats) are frequently seen. Presence of lymphoblasts in the peripheral smear is indicative of ALL, and a bone marrow biopsy finding of >25% lymphoblasts is confirmatory. Absence of peripheral lymphoblasts in a patient with acute leukemia is known as aleukemic leukemia. Aleukemic leukemia is uncommon, and most cases have described skin lesions from lymphoblast infiltration (leukemia cutis) in addition to bone marrow involvement.1 We report a case of aleukemic ALL in an adult presenting with unusual clinical features including bone pain, osteolytic lesions, hypercalcemia, and normal blood counts. To our knowledge, this is fifth such case ever reported in an adult patient.
Click on the PDF icon at the top of this introduction to read the full article.
Acute lymphoblastic leukemia is a neoplastic proliferation of lymphoblasts in the bone marrow. Normal hematopoiesis is affected, and symptoms from anemia (fatigue, breathlessness), leukopenia (recurrent infections) or thrombocytopenia (easy bruising, mucosal bleeding) are typically described in ALL. Hepatosplenomegaly and B-symptoms (fever, weight loss, and night sweats) are frequently seen. Presence of lymphoblasts in the peripheral smear is indicative of ALL, and a bone marrow biopsy finding of >25% lymphoblasts is confirmatory. Absence of peripheral lymphoblasts in a patient with acute leukemia is known as aleukemic leukemia. Aleukemic leukemia is uncommon, and most cases have described skin lesions from lymphoblast infiltration (leukemia cutis) in addition to bone marrow involvement.1 We report a case of aleukemic ALL in an adult presenting with unusual clinical features including bone pain, osteolytic lesions, hypercalcemia, and normal blood counts. To our knowledge, this is fifth such case ever reported in an adult patient.
Click on the PDF icon at the top of this introduction to read the full article.
Acute lymphoblastic leukemia is a neoplastic proliferation of lymphoblasts in the bone marrow. Normal hematopoiesis is affected, and symptoms from anemia (fatigue, breathlessness), leukopenia (recurrent infections) or thrombocytopenia (easy bruising, mucosal bleeding) are typically described in ALL. Hepatosplenomegaly and B-symptoms (fever, weight loss, and night sweats) are frequently seen. Presence of lymphoblasts in the peripheral smear is indicative of ALL, and a bone marrow biopsy finding of >25% lymphoblasts is confirmatory. Absence of peripheral lymphoblasts in a patient with acute leukemia is known as aleukemic leukemia. Aleukemic leukemia is uncommon, and most cases have described skin lesions from lymphoblast infiltration (leukemia cutis) in addition to bone marrow involvement.1 We report a case of aleukemic ALL in an adult presenting with unusual clinical features including bone pain, osteolytic lesions, hypercalcemia, and normal blood counts. To our knowledge, this is fifth such case ever reported in an adult patient.
Click on the PDF icon at the top of this introduction to read the full article.
The impact of loss of income and medicine costs on the financial burden for cancer patients in Australia
Background The cost of medicines may prove prohibitive for some cancer patients, potentially reducing the ability of a health system to fully deliver best practice care.
Objective To identify nonuse or nonpurchase of cancer-related medicines due to cost, and to describe the perceived financial burden of such medicines and associated patient characteristics.
Methods A cross-sectional pen-and-paper questionnaire was completed by oncology outpatients at 2 hospitals in Australia; 1 in regional New South Wales and 1 in metropolitan Victoria.
Results Almost 1 in 10 study participants had used over-the-counter medicines rather than prescribed medicines for cancer and obtained some but not all of the medicines prescribed in relation to their cancer. 63% of the sample reported some level of financial burden associated with obtaining these medicines, with 34% reporting a moderate or heavy financial burden. 11.8% reported using alternatives to prescribed medicines. People reporting reduced income after being diagnosed with cancer had almost 4 times the odds (OR, 3.73; 95% CI, 1.1-12.1) of reporting a heavy or extreme financial burden associated with prescribed medicines for cancer.
Limitations Study response rate, narrow survey population, self-reported survey used.
Conclusion This study identifies that a number of cancer patients, especially those with a reduced income after their diagnosis, experience financial burden associated with the purchase of medicines and that some go as far as to not use or to not purchase medicines. It seems likely that limiting the cost of medicines for cancer may improve patient ability to fully participate in the intended treatment.
Funding Cancer Council NSW, National Health and Medical Research Council, and Hunter Medical Research Institute, Australia
Click on the PDF icon at the top of this introduction to read the full article.
Background The cost of medicines may prove prohibitive for some cancer patients, potentially reducing the ability of a health system to fully deliver best practice care.
Objective To identify nonuse or nonpurchase of cancer-related medicines due to cost, and to describe the perceived financial burden of such medicines and associated patient characteristics.
Methods A cross-sectional pen-and-paper questionnaire was completed by oncology outpatients at 2 hospitals in Australia; 1 in regional New South Wales and 1 in metropolitan Victoria.
Results Almost 1 in 10 study participants had used over-the-counter medicines rather than prescribed medicines for cancer and obtained some but not all of the medicines prescribed in relation to their cancer. 63% of the sample reported some level of financial burden associated with obtaining these medicines, with 34% reporting a moderate or heavy financial burden. 11.8% reported using alternatives to prescribed medicines. People reporting reduced income after being diagnosed with cancer had almost 4 times the odds (OR, 3.73; 95% CI, 1.1-12.1) of reporting a heavy or extreme financial burden associated with prescribed medicines for cancer.
Limitations Study response rate, narrow survey population, self-reported survey used.
Conclusion This study identifies that a number of cancer patients, especially those with a reduced income after their diagnosis, experience financial burden associated with the purchase of medicines and that some go as far as to not use or to not purchase medicines. It seems likely that limiting the cost of medicines for cancer may improve patient ability to fully participate in the intended treatment.
Funding Cancer Council NSW, National Health and Medical Research Council, and Hunter Medical Research Institute, Australia
Click on the PDF icon at the top of this introduction to read the full article.
Background The cost of medicines may prove prohibitive for some cancer patients, potentially reducing the ability of a health system to fully deliver best practice care.
Objective To identify nonuse or nonpurchase of cancer-related medicines due to cost, and to describe the perceived financial burden of such medicines and associated patient characteristics.
Methods A cross-sectional pen-and-paper questionnaire was completed by oncology outpatients at 2 hospitals in Australia; 1 in regional New South Wales and 1 in metropolitan Victoria.
Results Almost 1 in 10 study participants had used over-the-counter medicines rather than prescribed medicines for cancer and obtained some but not all of the medicines prescribed in relation to their cancer. 63% of the sample reported some level of financial burden associated with obtaining these medicines, with 34% reporting a moderate or heavy financial burden. 11.8% reported using alternatives to prescribed medicines. People reporting reduced income after being diagnosed with cancer had almost 4 times the odds (OR, 3.73; 95% CI, 1.1-12.1) of reporting a heavy or extreme financial burden associated with prescribed medicines for cancer.
Limitations Study response rate, narrow survey population, self-reported survey used.
Conclusion This study identifies that a number of cancer patients, especially those with a reduced income after their diagnosis, experience financial burden associated with the purchase of medicines and that some go as far as to not use or to not purchase medicines. It seems likely that limiting the cost of medicines for cancer may improve patient ability to fully participate in the intended treatment.
Funding Cancer Council NSW, National Health and Medical Research Council, and Hunter Medical Research Institute, Australia
Click on the PDF icon at the top of this introduction to read the full article.
Adolescent and young adult perceptions of cancer survivor care and supportive programming
Background Improvements in cancer therapy have led to an increasing number of adolescent and young adult (AYA) survivors of childhood cancers. Many survivors have ongoing needs for support and information that are not being met.
Objective To conduct a program evaluation to identify AYAs’ perceptions of survivor care services.
Methods Using a community-based approach, 157 AYA childhood cancer survivors (aged 15-30 years) completed a program evaluation survey to assess perceptions of the importance of survivor patient care services and supportive programming using a Likert scale (1, Not At All Important; 2, Of Little Importance; 3, Somewhat Important; 4, Important; 5, Very Important).
Results Receipt of a medical summary was ranked as the most important survivor patient care service (mean, 4.5; SD, 0.91). 70% of respondents reported interest in late-effects education. Informational mailings were the most valued form of supportive programming and were endorsed by 62% of AYAs. Older survivors were more likely to value workshops (P = .01-0.05), whereas those aged 19-22 years valued weekend retreats (P < .01) and social activities (P < .01). Survivors of brain/CNS tumors were more likely to value social activities (P = .03) and support groups (P = .03), compared with leukemia survivors.
Limitations Contact information from the hospital tumor registry was used, which limited the number of correct addresses.
Conclusion The greatest care needs reported by AYA survivors of childhood cancer are services such as generation of a medical summary, late-effects education, and survivor-focused follow-up care, which are provided through cancer survivor programs. Development of additional programming to engage and further educate and encourage AYA survivors will be important to reinforce their adherence with survivor care throughout adulthood.
Funding/Sponsorship LiveStrong Community Based Participatory Research Planning Grant
Click on the PDF icon at the top of this introduction to read the full article.
Background Improvements in cancer therapy have led to an increasing number of adolescent and young adult (AYA) survivors of childhood cancers. Many survivors have ongoing needs for support and information that are not being met.
Objective To conduct a program evaluation to identify AYAs’ perceptions of survivor care services.
Methods Using a community-based approach, 157 AYA childhood cancer survivors (aged 15-30 years) completed a program evaluation survey to assess perceptions of the importance of survivor patient care services and supportive programming using a Likert scale (1, Not At All Important; 2, Of Little Importance; 3, Somewhat Important; 4, Important; 5, Very Important).
Results Receipt of a medical summary was ranked as the most important survivor patient care service (mean, 4.5; SD, 0.91). 70% of respondents reported interest in late-effects education. Informational mailings were the most valued form of supportive programming and were endorsed by 62% of AYAs. Older survivors were more likely to value workshops (P = .01-0.05), whereas those aged 19-22 years valued weekend retreats (P < .01) and social activities (P < .01). Survivors of brain/CNS tumors were more likely to value social activities (P = .03) and support groups (P = .03), compared with leukemia survivors.
Limitations Contact information from the hospital tumor registry was used, which limited the number of correct addresses.
Conclusion The greatest care needs reported by AYA survivors of childhood cancer are services such as generation of a medical summary, late-effects education, and survivor-focused follow-up care, which are provided through cancer survivor programs. Development of additional programming to engage and further educate and encourage AYA survivors will be important to reinforce their adherence with survivor care throughout adulthood.
Funding/Sponsorship LiveStrong Community Based Participatory Research Planning Grant
Click on the PDF icon at the top of this introduction to read the full article.
Background Improvements in cancer therapy have led to an increasing number of adolescent and young adult (AYA) survivors of childhood cancers. Many survivors have ongoing needs for support and information that are not being met.
Objective To conduct a program evaluation to identify AYAs’ perceptions of survivor care services.
Methods Using a community-based approach, 157 AYA childhood cancer survivors (aged 15-30 years) completed a program evaluation survey to assess perceptions of the importance of survivor patient care services and supportive programming using a Likert scale (1, Not At All Important; 2, Of Little Importance; 3, Somewhat Important; 4, Important; 5, Very Important).
Results Receipt of a medical summary was ranked as the most important survivor patient care service (mean, 4.5; SD, 0.91). 70% of respondents reported interest in late-effects education. Informational mailings were the most valued form of supportive programming and were endorsed by 62% of AYAs. Older survivors were more likely to value workshops (P = .01-0.05), whereas those aged 19-22 years valued weekend retreats (P < .01) and social activities (P < .01). Survivors of brain/CNS tumors were more likely to value social activities (P = .03) and support groups (P = .03), compared with leukemia survivors.
Limitations Contact information from the hospital tumor registry was used, which limited the number of correct addresses.
Conclusion The greatest care needs reported by AYA survivors of childhood cancer are services such as generation of a medical summary, late-effects education, and survivor-focused follow-up care, which are provided through cancer survivor programs. Development of additional programming to engage and further educate and encourage AYA survivors will be important to reinforce their adherence with survivor care throughout adulthood.
Funding/Sponsorship LiveStrong Community Based Participatory Research Planning Grant
Click on the PDF icon at the top of this introduction to read the full article.
Hereditary cancer testing in patients with ovarian cancer using a 25-gene panel
Symptoms, unmet need, and quality of life among recent breast cancer survivors
Background Assessing patient quality of life (QoL) apart from symptoms and unmet need may miss important concerns for which remediation is possible. Therapeutic advances have improved survival among breast cancer patients, and 89% can expect to survive for longer than 5 years. However, the price is lasting physical and psychosocial symptoms. Education regarding the value of symptom reduction may be needed for breast cancer survivors and their providers.
Objective To examine the unmet needs for symptom management and the relationships between unmet needs, symptom burden, and patient QoL.
Method Eligibility included nonmetastatic breast cancer survivors who had been treated less than a year before the study and attendance at a survivorship appointment. QoL was assessed using the Medical Outcomes Study Short Form-12 (scale, 0 [Did Not Experience] to 5 [As Bad As Possible]), and 19 symptoms were evaluated. Participants reported unmet need for assistance for each symptom experienced.
Results 164 primarily white, middle-aged, early-stage survivors of breast cancer were recruited. Physical and Mental QoL were similar to national norms. Survivors reported an average of 11.5 symptoms, most commonly Fatigue, Insomnia, Hot Flashes, Joint Pain, but reported unmet need for fewer symptoms (mean, 2.6). Weight Gain, Joint Pain, Numbness were most likely to result in unmet need. Both Physical and Mental QoL were negatively associated with number of symptoms (r = -.46 and -.41, respectively) and unmet needs (r = -.17 and -.41, respectively).
Limitations Cross-sectional sample of consecutive patients from a single clinical site.
Conclusion Symptoms are common among recent survivors of breast cancer, as are unmet needs, but to a lesser extent. Both have a negative impact on Physical and Mental health QoL.
Funding Translational Center of Excellence in Breast Cancer at the Abramson Cancer Center, University of Pennsylvania
Click on the PDF icon at the top of this introduction to read the full article.
Background Assessing patient quality of life (QoL) apart from symptoms and unmet need may miss important concerns for which remediation is possible. Therapeutic advances have improved survival among breast cancer patients, and 89% can expect to survive for longer than 5 years. However, the price is lasting physical and psychosocial symptoms. Education regarding the value of symptom reduction may be needed for breast cancer survivors and their providers.
Objective To examine the unmet needs for symptom management and the relationships between unmet needs, symptom burden, and patient QoL.
Method Eligibility included nonmetastatic breast cancer survivors who had been treated less than a year before the study and attendance at a survivorship appointment. QoL was assessed using the Medical Outcomes Study Short Form-12 (scale, 0 [Did Not Experience] to 5 [As Bad As Possible]), and 19 symptoms were evaluated. Participants reported unmet need for assistance for each symptom experienced.
Results 164 primarily white, middle-aged, early-stage survivors of breast cancer were recruited. Physical and Mental QoL were similar to national norms. Survivors reported an average of 11.5 symptoms, most commonly Fatigue, Insomnia, Hot Flashes, Joint Pain, but reported unmet need for fewer symptoms (mean, 2.6). Weight Gain, Joint Pain, Numbness were most likely to result in unmet need. Both Physical and Mental QoL were negatively associated with number of symptoms (r = -.46 and -.41, respectively) and unmet needs (r = -.17 and -.41, respectively).
Limitations Cross-sectional sample of consecutive patients from a single clinical site.
Conclusion Symptoms are common among recent survivors of breast cancer, as are unmet needs, but to a lesser extent. Both have a negative impact on Physical and Mental health QoL.
Funding Translational Center of Excellence in Breast Cancer at the Abramson Cancer Center, University of Pennsylvania
Click on the PDF icon at the top of this introduction to read the full article.
Background Assessing patient quality of life (QoL) apart from symptoms and unmet need may miss important concerns for which remediation is possible. Therapeutic advances have improved survival among breast cancer patients, and 89% can expect to survive for longer than 5 years. However, the price is lasting physical and psychosocial symptoms. Education regarding the value of symptom reduction may be needed for breast cancer survivors and their providers.
Objective To examine the unmet needs for symptom management and the relationships between unmet needs, symptom burden, and patient QoL.
Method Eligibility included nonmetastatic breast cancer survivors who had been treated less than a year before the study and attendance at a survivorship appointment. QoL was assessed using the Medical Outcomes Study Short Form-12 (scale, 0 [Did Not Experience] to 5 [As Bad As Possible]), and 19 symptoms were evaluated. Participants reported unmet need for assistance for each symptom experienced.
Results 164 primarily white, middle-aged, early-stage survivors of breast cancer were recruited. Physical and Mental QoL were similar to national norms. Survivors reported an average of 11.5 symptoms, most commonly Fatigue, Insomnia, Hot Flashes, Joint Pain, but reported unmet need for fewer symptoms (mean, 2.6). Weight Gain, Joint Pain, Numbness were most likely to result in unmet need. Both Physical and Mental QoL were negatively associated with number of symptoms (r = -.46 and -.41, respectively) and unmet needs (r = -.17 and -.41, respectively).
Limitations Cross-sectional sample of consecutive patients from a single clinical site.
Conclusion Symptoms are common among recent survivors of breast cancer, as are unmet needs, but to a lesser extent. Both have a negative impact on Physical and Mental health QoL.
Funding Translational Center of Excellence in Breast Cancer at the Abramson Cancer Center, University of Pennsylvania
Click on the PDF icon at the top of this introduction to read the full article.
Canada reduces restrictions for blood donation
Photo by Marja Helander
Canadian Blood Services has made several changes to its blood donor policies in an attempt to broaden the pool of eligible donors in the country.
The agency has eliminated the upper age limit for donating blood, and donors over the age of 71 no longer need to have their physician fill out an assessment form before donating.
People with a history of most cancers are now eligible to donate blood if they have been cancer-free for 5 years.
However, this change does not apply to those with a history of hematologic malignancies.
People who have recently received most vaccines, such as a flu shot, will no longer need to wait 2 days before donating blood.
People who were born in or lived in some African countries (Central African Republic, Chad, Congo, Equatorial Guinea, Gabon, Niger, and Nigeria) are now eligible to donate blood. According to Canadian Blood Services, HIV testing performed on blood donors can now detect HIV strains found in these countries.
Canadian Blood Services has also revised geographic deferrals affecting Western Europe based on scientific evidence that indicates the risk of variant Creutzfeldt-Jakob disease has decreased since January 2008.
People who spent 5 years or more in Western Europe since 1980 are deferred from donating blood, but Canadian Blood Services is now including an end date of 2007. People who reached the 5-year limit in Western Europe after 2007 are now eligible to donate.
“Canadian Blood Services regularly reviews the criteria used to determine if someone is eligible to donate blood, including geographic and age restrictions, based on new scientific information,” said Mindy Goldman, medical director of donor and clinical services with Canadian Blood Services.
“These restrictions are no longer necessary. We estimate that about 3000 people who try to donate each year but cannot will now be eligible to donate due to these changes.”
The complete policy changes are available at www.blood.ca/en/blood/recent-changes-donation-criteria. ![]()
Photo by Marja Helander
Canadian Blood Services has made several changes to its blood donor policies in an attempt to broaden the pool of eligible donors in the country.
The agency has eliminated the upper age limit for donating blood, and donors over the age of 71 no longer need to have their physician fill out an assessment form before donating.
People with a history of most cancers are now eligible to donate blood if they have been cancer-free for 5 years.
However, this change does not apply to those with a history of hematologic malignancies.
People who have recently received most vaccines, such as a flu shot, will no longer need to wait 2 days before donating blood.
People who were born in or lived in some African countries (Central African Republic, Chad, Congo, Equatorial Guinea, Gabon, Niger, and Nigeria) are now eligible to donate blood. According to Canadian Blood Services, HIV testing performed on blood donors can now detect HIV strains found in these countries.
Canadian Blood Services has also revised geographic deferrals affecting Western Europe based on scientific evidence that indicates the risk of variant Creutzfeldt-Jakob disease has decreased since January 2008.
People who spent 5 years or more in Western Europe since 1980 are deferred from donating blood, but Canadian Blood Services is now including an end date of 2007. People who reached the 5-year limit in Western Europe after 2007 are now eligible to donate.
“Canadian Blood Services regularly reviews the criteria used to determine if someone is eligible to donate blood, including geographic and age restrictions, based on new scientific information,” said Mindy Goldman, medical director of donor and clinical services with Canadian Blood Services.
“These restrictions are no longer necessary. We estimate that about 3000 people who try to donate each year but cannot will now be eligible to donate due to these changes.”
The complete policy changes are available at www.blood.ca/en/blood/recent-changes-donation-criteria. ![]()
Photo by Marja Helander
Canadian Blood Services has made several changes to its blood donor policies in an attempt to broaden the pool of eligible donors in the country.
The agency has eliminated the upper age limit for donating blood, and donors over the age of 71 no longer need to have their physician fill out an assessment form before donating.
People with a history of most cancers are now eligible to donate blood if they have been cancer-free for 5 years.
However, this change does not apply to those with a history of hematologic malignancies.
People who have recently received most vaccines, such as a flu shot, will no longer need to wait 2 days before donating blood.
People who were born in or lived in some African countries (Central African Republic, Chad, Congo, Equatorial Guinea, Gabon, Niger, and Nigeria) are now eligible to donate blood. According to Canadian Blood Services, HIV testing performed on blood donors can now detect HIV strains found in these countries.
Canadian Blood Services has also revised geographic deferrals affecting Western Europe based on scientific evidence that indicates the risk of variant Creutzfeldt-Jakob disease has decreased since January 2008.
People who spent 5 years or more in Western Europe since 1980 are deferred from donating blood, but Canadian Blood Services is now including an end date of 2007. People who reached the 5-year limit in Western Europe after 2007 are now eligible to donate.
“Canadian Blood Services regularly reviews the criteria used to determine if someone is eligible to donate blood, including geographic and age restrictions, based on new scientific information,” said Mindy Goldman, medical director of donor and clinical services with Canadian Blood Services.
“These restrictions are no longer necessary. We estimate that about 3000 people who try to donate each year but cannot will now be eligible to donate due to these changes.”
The complete policy changes are available at www.blood.ca/en/blood/recent-changes-donation-criteria. ![]()
Delirium in advanced cancer may go undetected

patient and her father
Photo by Rhoda Baer
A new study indicates that delirium is relatively frequent and underdiagnosed in patients with advanced cancer visiting the emergency department.
The research showed that delirium was similarly common among older and younger patients.
According to researchers, this suggests that, in the setting of advanced cancer, all patients should be considered at higher risk for delirium.
The researchers reported their findings in Cancer.
For this study, the team assessed a random sample of 243 advanced cancer patients who presented to the emergency department. They were 19 to 89 years old.
All patients were assessed with 2 methods: the Confusion Assessment Method (CAM) to screen for delirium and the Memorial Delirium Assessment Scale (MDAS) to measure delirium severity (mild ≤15, moderate 16-22, and severe ≥23).
In all, 22 patients (9%) had CAM-positive delirium and a median MDAS score of 14. Among CAM-positive patients, delirium was mild in 18 (82%) and moderate in 4 (18%) according to the MDAS.
Of the 99 patients age 65 and older, 10 (10%) had CAM-positive delirium, compared with 12 (8%) of 144 patients younger than 65.
Emergency department physicians failed to detect delirium in 9 (41%) CAM-positive delirious patients.
“We found evidence of delirium in 1 of every 10 patients with advanced cancer who are treated in the emergency department,” said study author Knox Todd, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“Given that we could only study patients who were able to give consent to enter our study, even 10% is likely to be a low estimate. We also identified many psychoactive medications that could have contributed to delirium, and sharing this information with treating oncologists may help them avoid such complications in the next patient they treat.” ![]()
patient and her father
Photo by Rhoda Baer
A new study indicates that delirium is relatively frequent and underdiagnosed in patients with advanced cancer visiting the emergency department.
The research showed that delirium was similarly common among older and younger patients.
According to researchers, this suggests that, in the setting of advanced cancer, all patients should be considered at higher risk for delirium.
The researchers reported their findings in Cancer.
For this study, the team assessed a random sample of 243 advanced cancer patients who presented to the emergency department. They were 19 to 89 years old.
All patients were assessed with 2 methods: the Confusion Assessment Method (CAM) to screen for delirium and the Memorial Delirium Assessment Scale (MDAS) to measure delirium severity (mild ≤15, moderate 16-22, and severe ≥23).
In all, 22 patients (9%) had CAM-positive delirium and a median MDAS score of 14. Among CAM-positive patients, delirium was mild in 18 (82%) and moderate in 4 (18%) according to the MDAS.
Of the 99 patients age 65 and older, 10 (10%) had CAM-positive delirium, compared with 12 (8%) of 144 patients younger than 65.
Emergency department physicians failed to detect delirium in 9 (41%) CAM-positive delirious patients.
“We found evidence of delirium in 1 of every 10 patients with advanced cancer who are treated in the emergency department,” said study author Knox Todd, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“Given that we could only study patients who were able to give consent to enter our study, even 10% is likely to be a low estimate. We also identified many psychoactive medications that could have contributed to delirium, and sharing this information with treating oncologists may help them avoid such complications in the next patient they treat.” ![]()
patient and her father
Photo by Rhoda Baer
A new study indicates that delirium is relatively frequent and underdiagnosed in patients with advanced cancer visiting the emergency department.
The research showed that delirium was similarly common among older and younger patients.
According to researchers, this suggests that, in the setting of advanced cancer, all patients should be considered at higher risk for delirium.
The researchers reported their findings in Cancer.
For this study, the team assessed a random sample of 243 advanced cancer patients who presented to the emergency department. They were 19 to 89 years old.
All patients were assessed with 2 methods: the Confusion Assessment Method (CAM) to screen for delirium and the Memorial Delirium Assessment Scale (MDAS) to measure delirium severity (mild ≤15, moderate 16-22, and severe ≥23).
In all, 22 patients (9%) had CAM-positive delirium and a median MDAS score of 14. Among CAM-positive patients, delirium was mild in 18 (82%) and moderate in 4 (18%) according to the MDAS.
Of the 99 patients age 65 and older, 10 (10%) had CAM-positive delirium, compared with 12 (8%) of 144 patients younger than 65.
Emergency department physicians failed to detect delirium in 9 (41%) CAM-positive delirious patients.
“We found evidence of delirium in 1 of every 10 patients with advanced cancer who are treated in the emergency department,” said study author Knox Todd, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“Given that we could only study patients who were able to give consent to enter our study, even 10% is likely to be a low estimate. We also identified many psychoactive medications that could have contributed to delirium, and sharing this information with treating oncologists may help them avoid such complications in the next patient they treat.” ![]()
Severe pruritus • crusted lesions affecting face, extremities, and trunk • hepatitis C virus carrier • Dx?
THE CASE
An 85-year-old woman sought care at our outpatient clinic for a 9-month history of severe pruritus and crusted lesions on her face, extremities, and trunk. She had been diagnosed with hepatitis C virus (HCV) infection one year ago and was not taking any medication. The patient, who had been living with her family, had visited various clinics for her complaints and was diagnosed as having contact dermatitis and senile pruritus. She was prescribed topical mometasone furoate and moisturizers.
After 6 months of using this therapy, widespread grey-white plaques and minimal excoriation appeared on her face, scalp, and trunk. This was diagnosed as psoriasis, and the patient was prescribed topical corticosteroids, which she used for 9 months until she came to our clinic. She said the lesions regressed minimally with the topical corticosteroids, but did not fully clear.
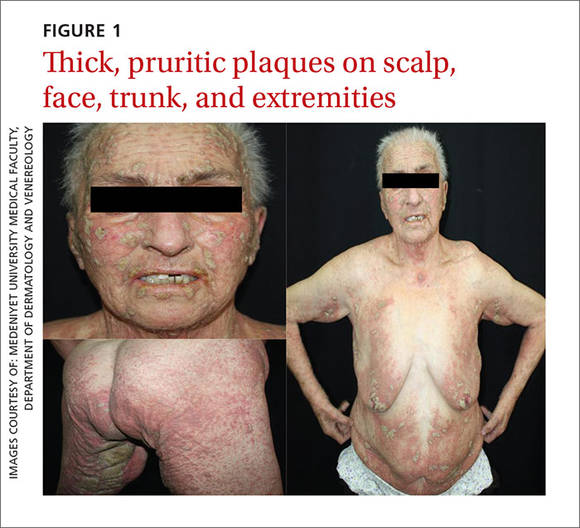
Dermatologic examination revealed widespread erythema and grey-white, cohesive, thick, pruritic plaques on her scalp, face, trunk, and bilateral extremities (FIGURE 1). A punch biopsy specimen was taken from the border of a plaque on her trunk.
THE DIAGNOSIS
A complete blood cell count and wide biochemistry panel, including tumor markers and viral serology for human immunodeficiency virus (HIV), were normal. The patient had lymphadenopathy in her posterior cervical, bilateral preauricular, and bilateral inguinal regions.
Histopathologic examination revealed hyperkeratosis, acanthosis, and spongiotic edema in the epidermis, and vesiculation and mites in the stratum corneum. The dermal changes consisted of perivascular and diffuse cell infiltrates that were mainly mononuclear cells and eosinophilic granulocytes.
Based on the dermatologic examination and the histopathologic findings, we diagnosed the patient with crusted (Norwegian) scabies.
DISCUSSION
Crusted (Norwegian) scabies is a rare, highly contagious form of scabies that is characterized by the presence of millions of Sarcoptes scabiei var hominis mites in the epidermis.1 This variant of scabies can affect individuals of any age, gender, or race.2 It was first described by Boeck and Danielssen in 1848 in Norway and was named Norwegian scabies by von Hebra in 1862.3 In 2010, more than 200 cases of crusted scabies were reported in the literature.4
Crusted scabies is usually seen in immunocompromised patients, such as the elderly, those who’ve had solid organ transplantation, and those with HIV, malignancy, or malnutrition. Crusted scabies may also occur in patients with decreased sensory function (such as those with leprosy) or decreased ability to scratch, intellectual disabilities, and in those who use biologic agents or systemic/topical corticosteroids.4-8
Crusted scabies is associated with increased morbidity and mortality, especially in children and the elderly, because of complications such as secondary bacterial infections and sepsis.1,3 Widespread inflammation may also cause erythroderma, which can lead to metabolic disorders.
Distinguish it from other pruritic papulosquamous diseases
The differential diagnosis for crusted scabies includes psoriasis, eczema, cutaneous lymphoma, Darier disease, and adverse drug reactions.9 Crusted scabies can be differentiated from these other diagnoses by its clinical presentation and histopathological examination.
Crusted scabies is characterized by hyperkeratosis and wart-like crusts that are due to extreme proliferation of mites in the stratum corneum of the epidermis.2 Lesions are usually localized on acral sites (especially the hands), although the entire body, including the face and the scalp, can be involved.1 Psoriasiform or bullous pemphigoid-like eruptions have also been reported in the literature.5,9
Our patient presented with widespread erythema and psoriasiform grey-white crusts on her scalp, face, chest, periareolar region, and extremities. In addition, she did not have an immunosuppressant disease or medication history.
However, the fact that our patient was using topical corticosteroids for so long explained the extent of her condition. Topical corticosteroids have been linked to scabies incognito.10 Topical or systemic corticosteroid use for long periods of time may alter the skin immune system by suppressing cellular immunity, thereby reducing the inflammatory response. This may lead to progression of the regular variant of scabies to crusted scabies, as our patient had.
Topical treatments, oral ivermectin proven to be effective
Topical keratolytics, permethrin 5%, lindane 1%, crotamiton 10%, sulfur ointment (5%-10%), malathion 0.5%, benzyl benzoate (10%-25%), oral ivermectin (2 doses of 200 mcg/kg/dose), and systemic antihistamines are appropriate therapies.3 While oral ivermectin is effective, it is not available in Turkey.
Because of our patient’s hepatic disorder, we opted for a topical, rather than a systemic, treatment and recommended repeated applications of topical permethrin. Repeated treatment with topical permethrin is often sufficient in patients who are unable to take systemic therapy. In fact, Binic et al4 reported a case in which an elderly patient with crusted scabies (who had previously been treated with systemic and topical corticosteroids) responded well to repeated topical treatment with lindane 1%, 25% benzyl benzoate, and 10% precipitated sulfur.
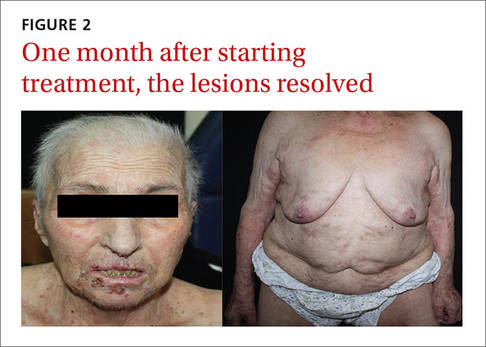
Our patient. We prescribed topical 5% permethrin lotion for our patient to apply to her entire body 4 times a week and advised her to wash her clothing and bed linens at 140° F. She was scheduled for biweekly check-ups. We also advised the patient’s family to use the same topical therapy 2 times per week because crusted scabies is highly contagious. One month later, our patient’s lesions had resolved (FIGURE 2).
THE TAKEAWAY
Early diagnosis and treatment of crusted scabies is important, both for the treatment of the patient and to stop the spread of the disease. Although rare, crusted scabies should be included in the differential diagnosis of long-term pruritic papulosquamous diseases, and the possibility of an atypical presentation in all patients should be considered—whether their immunity is compromised or not. Scabies should also be considered in patients with a positive family history of the disease and in those with chronic pruritus that is unresponsive to topical therapies.
1. Burkhart CN, Burkhart CG, Morrell DS. Infestations. In: Bolognia JL, Jorizzo JL, Schaffer JV, et al, eds. Dermatology. 3rd ed. Philadelphia, PA: Mosby Elsevier; 2012;1423-1426.
2. Subramaniam G, Kaliaperumal K, Duraipandian J, et al. Norwegian scabies in a malnourished young adult: a case report. J Infect Dev Ctries. 2010;4:349-351.
3. Karthikeyan K. Crusted scabies. Indian J Dermatol Venereol Leprol. 2009;75:340-347.
4. Binic I, Jankovic A, Jovanovic D, et al. Crusted (Norwegian) scabies following systemic and topical corticosteroid therapy. J Korean Med Sci. 2010;25:188-191.
5. Ramachandran V, Shankar EM, Devaleenal B, et al. Atypically distributed cutaneous lesions of Norwegian scabies in an HIV-positive man in South India: a case report. J Med Case Rep. 2008;2:82.
6. Lai YC, Teng CJ, Chen PC, et al. Unusual scalp crusted scabies in an adult T-cell leukemia/lymphoma patient. Ups J Med Sci. 2011;116:77-78.
7. Saillard C, Darrieux L, Safa G. Crusted scabies complicates etanercept therapy in a patient with severe psoriasis. J Am Acad Dermatol. 2013;68:e138-e139.
8. Marlière V, Roul S, Labrèze C, et al. Crusted (Norwegian) scabies induced by use of topical corticosteroids and treated successfully with ivermectin. J Pediatr. 1999;135:122-124.
9. Goyal NN, Wong GA. Psoriasis or crusted scabies. Clin Exp Dermatol. 2008;33:211-212.
10. Kim KJ, Roh KH, Choi JH, et al. Scabies incognito presenting as urticaria pigmentosa in an infant. Pediatr Dermatol. 2002;19:409-411.
THE CASE
An 85-year-old woman sought care at our outpatient clinic for a 9-month history of severe pruritus and crusted lesions on her face, extremities, and trunk. She had been diagnosed with hepatitis C virus (HCV) infection one year ago and was not taking any medication. The patient, who had been living with her family, had visited various clinics for her complaints and was diagnosed as having contact dermatitis and senile pruritus. She was prescribed topical mometasone furoate and moisturizers.
After 6 months of using this therapy, widespread grey-white plaques and minimal excoriation appeared on her face, scalp, and trunk. This was diagnosed as psoriasis, and the patient was prescribed topical corticosteroids, which she used for 9 months until she came to our clinic. She said the lesions regressed minimally with the topical corticosteroids, but did not fully clear.
Dermatologic examination revealed widespread erythema and grey-white, cohesive, thick, pruritic plaques on her scalp, face, trunk, and bilateral extremities (FIGURE 1). A punch biopsy specimen was taken from the border of a plaque on her trunk.
THE DIAGNOSIS
A complete blood cell count and wide biochemistry panel, including tumor markers and viral serology for human immunodeficiency virus (HIV), were normal. The patient had lymphadenopathy in her posterior cervical, bilateral preauricular, and bilateral inguinal regions.
Histopathologic examination revealed hyperkeratosis, acanthosis, and spongiotic edema in the epidermis, and vesiculation and mites in the stratum corneum. The dermal changes consisted of perivascular and diffuse cell infiltrates that were mainly mononuclear cells and eosinophilic granulocytes.
Based on the dermatologic examination and the histopathologic findings, we diagnosed the patient with crusted (Norwegian) scabies.
DISCUSSION
Crusted (Norwegian) scabies is a rare, highly contagious form of scabies that is characterized by the presence of millions of Sarcoptes scabiei var hominis mites in the epidermis.1 This variant of scabies can affect individuals of any age, gender, or race.2 It was first described by Boeck and Danielssen in 1848 in Norway and was named Norwegian scabies by von Hebra in 1862.3 In 2010, more than 200 cases of crusted scabies were reported in the literature.4
Crusted scabies is usually seen in immunocompromised patients, such as the elderly, those who’ve had solid organ transplantation, and those with HIV, malignancy, or malnutrition. Crusted scabies may also occur in patients with decreased sensory function (such as those with leprosy) or decreased ability to scratch, intellectual disabilities, and in those who use biologic agents or systemic/topical corticosteroids.4-8
Crusted scabies is associated with increased morbidity and mortality, especially in children and the elderly, because of complications such as secondary bacterial infections and sepsis.1,3 Widespread inflammation may also cause erythroderma, which can lead to metabolic disorders.
Distinguish it from other pruritic papulosquamous diseases
The differential diagnosis for crusted scabies includes psoriasis, eczema, cutaneous lymphoma, Darier disease, and adverse drug reactions.9 Crusted scabies can be differentiated from these other diagnoses by its clinical presentation and histopathological examination.
Crusted scabies is characterized by hyperkeratosis and wart-like crusts that are due to extreme proliferation of mites in the stratum corneum of the epidermis.2 Lesions are usually localized on acral sites (especially the hands), although the entire body, including the face and the scalp, can be involved.1 Psoriasiform or bullous pemphigoid-like eruptions have also been reported in the literature.5,9
Our patient presented with widespread erythema and psoriasiform grey-white crusts on her scalp, face, chest, periareolar region, and extremities. In addition, she did not have an immunosuppressant disease or medication history.
However, the fact that our patient was using topical corticosteroids for so long explained the extent of her condition. Topical corticosteroids have been linked to scabies incognito.10 Topical or systemic corticosteroid use for long periods of time may alter the skin immune system by suppressing cellular immunity, thereby reducing the inflammatory response. This may lead to progression of the regular variant of scabies to crusted scabies, as our patient had.
Topical treatments, oral ivermectin proven to be effective
Topical keratolytics, permethrin 5%, lindane 1%, crotamiton 10%, sulfur ointment (5%-10%), malathion 0.5%, benzyl benzoate (10%-25%), oral ivermectin (2 doses of 200 mcg/kg/dose), and systemic antihistamines are appropriate therapies.3 While oral ivermectin is effective, it is not available in Turkey.
Because of our patient’s hepatic disorder, we opted for a topical, rather than a systemic, treatment and recommended repeated applications of topical permethrin. Repeated treatment with topical permethrin is often sufficient in patients who are unable to take systemic therapy. In fact, Binic et al4 reported a case in which an elderly patient with crusted scabies (who had previously been treated with systemic and topical corticosteroids) responded well to repeated topical treatment with lindane 1%, 25% benzyl benzoate, and 10% precipitated sulfur.
Our patient. We prescribed topical 5% permethrin lotion for our patient to apply to her entire body 4 times a week and advised her to wash her clothing and bed linens at 140° F. She was scheduled for biweekly check-ups. We also advised the patient’s family to use the same topical therapy 2 times per week because crusted scabies is highly contagious. One month later, our patient’s lesions had resolved (FIGURE 2).
THE TAKEAWAY
Early diagnosis and treatment of crusted scabies is important, both for the treatment of the patient and to stop the spread of the disease. Although rare, crusted scabies should be included in the differential diagnosis of long-term pruritic papulosquamous diseases, and the possibility of an atypical presentation in all patients should be considered—whether their immunity is compromised or not. Scabies should also be considered in patients with a positive family history of the disease and in those with chronic pruritus that is unresponsive to topical therapies.
THE CASE
An 85-year-old woman sought care at our outpatient clinic for a 9-month history of severe pruritus and crusted lesions on her face, extremities, and trunk. She had been diagnosed with hepatitis C virus (HCV) infection one year ago and was not taking any medication. The patient, who had been living with her family, had visited various clinics for her complaints and was diagnosed as having contact dermatitis and senile pruritus. She was prescribed topical mometasone furoate and moisturizers.
After 6 months of using this therapy, widespread grey-white plaques and minimal excoriation appeared on her face, scalp, and trunk. This was diagnosed as psoriasis, and the patient was prescribed topical corticosteroids, which she used for 9 months until she came to our clinic. She said the lesions regressed minimally with the topical corticosteroids, but did not fully clear.
Dermatologic examination revealed widespread erythema and grey-white, cohesive, thick, pruritic plaques on her scalp, face, trunk, and bilateral extremities (FIGURE 1). A punch biopsy specimen was taken from the border of a plaque on her trunk.
THE DIAGNOSIS
A complete blood cell count and wide biochemistry panel, including tumor markers and viral serology for human immunodeficiency virus (HIV), were normal. The patient had lymphadenopathy in her posterior cervical, bilateral preauricular, and bilateral inguinal regions.
Histopathologic examination revealed hyperkeratosis, acanthosis, and spongiotic edema in the epidermis, and vesiculation and mites in the stratum corneum. The dermal changes consisted of perivascular and diffuse cell infiltrates that were mainly mononuclear cells and eosinophilic granulocytes.
Based on the dermatologic examination and the histopathologic findings, we diagnosed the patient with crusted (Norwegian) scabies.
DISCUSSION
Crusted (Norwegian) scabies is a rare, highly contagious form of scabies that is characterized by the presence of millions of Sarcoptes scabiei var hominis mites in the epidermis.1 This variant of scabies can affect individuals of any age, gender, or race.2 It was first described by Boeck and Danielssen in 1848 in Norway and was named Norwegian scabies by von Hebra in 1862.3 In 2010, more than 200 cases of crusted scabies were reported in the literature.4
Crusted scabies is usually seen in immunocompromised patients, such as the elderly, those who’ve had solid organ transplantation, and those with HIV, malignancy, or malnutrition. Crusted scabies may also occur in patients with decreased sensory function (such as those with leprosy) or decreased ability to scratch, intellectual disabilities, and in those who use biologic agents or systemic/topical corticosteroids.4-8
Crusted scabies is associated with increased morbidity and mortality, especially in children and the elderly, because of complications such as secondary bacterial infections and sepsis.1,3 Widespread inflammation may also cause erythroderma, which can lead to metabolic disorders.
Distinguish it from other pruritic papulosquamous diseases
The differential diagnosis for crusted scabies includes psoriasis, eczema, cutaneous lymphoma, Darier disease, and adverse drug reactions.9 Crusted scabies can be differentiated from these other diagnoses by its clinical presentation and histopathological examination.
Crusted scabies is characterized by hyperkeratosis and wart-like crusts that are due to extreme proliferation of mites in the stratum corneum of the epidermis.2 Lesions are usually localized on acral sites (especially the hands), although the entire body, including the face and the scalp, can be involved.1 Psoriasiform or bullous pemphigoid-like eruptions have also been reported in the literature.5,9
Our patient presented with widespread erythema and psoriasiform grey-white crusts on her scalp, face, chest, periareolar region, and extremities. In addition, she did not have an immunosuppressant disease or medication history.
However, the fact that our patient was using topical corticosteroids for so long explained the extent of her condition. Topical corticosteroids have been linked to scabies incognito.10 Topical or systemic corticosteroid use for long periods of time may alter the skin immune system by suppressing cellular immunity, thereby reducing the inflammatory response. This may lead to progression of the regular variant of scabies to crusted scabies, as our patient had.
Topical treatments, oral ivermectin proven to be effective
Topical keratolytics, permethrin 5%, lindane 1%, crotamiton 10%, sulfur ointment (5%-10%), malathion 0.5%, benzyl benzoate (10%-25%), oral ivermectin (2 doses of 200 mcg/kg/dose), and systemic antihistamines are appropriate therapies.3 While oral ivermectin is effective, it is not available in Turkey.
Because of our patient’s hepatic disorder, we opted for a topical, rather than a systemic, treatment and recommended repeated applications of topical permethrin. Repeated treatment with topical permethrin is often sufficient in patients who are unable to take systemic therapy. In fact, Binic et al4 reported a case in which an elderly patient with crusted scabies (who had previously been treated with systemic and topical corticosteroids) responded well to repeated topical treatment with lindane 1%, 25% benzyl benzoate, and 10% precipitated sulfur.
Our patient. We prescribed topical 5% permethrin lotion for our patient to apply to her entire body 4 times a week and advised her to wash her clothing and bed linens at 140° F. She was scheduled for biweekly check-ups. We also advised the patient’s family to use the same topical therapy 2 times per week because crusted scabies is highly contagious. One month later, our patient’s lesions had resolved (FIGURE 2).
THE TAKEAWAY
Early diagnosis and treatment of crusted scabies is important, both for the treatment of the patient and to stop the spread of the disease. Although rare, crusted scabies should be included in the differential diagnosis of long-term pruritic papulosquamous diseases, and the possibility of an atypical presentation in all patients should be considered—whether their immunity is compromised or not. Scabies should also be considered in patients with a positive family history of the disease and in those with chronic pruritus that is unresponsive to topical therapies.
1. Burkhart CN, Burkhart CG, Morrell DS. Infestations. In: Bolognia JL, Jorizzo JL, Schaffer JV, et al, eds. Dermatology. 3rd ed. Philadelphia, PA: Mosby Elsevier; 2012;1423-1426.
2. Subramaniam G, Kaliaperumal K, Duraipandian J, et al. Norwegian scabies in a malnourished young adult: a case report. J Infect Dev Ctries. 2010;4:349-351.
3. Karthikeyan K. Crusted scabies. Indian J Dermatol Venereol Leprol. 2009;75:340-347.
4. Binic I, Jankovic A, Jovanovic D, et al. Crusted (Norwegian) scabies following systemic and topical corticosteroid therapy. J Korean Med Sci. 2010;25:188-191.
5. Ramachandran V, Shankar EM, Devaleenal B, et al. Atypically distributed cutaneous lesions of Norwegian scabies in an HIV-positive man in South India: a case report. J Med Case Rep. 2008;2:82.
6. Lai YC, Teng CJ, Chen PC, et al. Unusual scalp crusted scabies in an adult T-cell leukemia/lymphoma patient. Ups J Med Sci. 2011;116:77-78.
7. Saillard C, Darrieux L, Safa G. Crusted scabies complicates etanercept therapy in a patient with severe psoriasis. J Am Acad Dermatol. 2013;68:e138-e139.
8. Marlière V, Roul S, Labrèze C, et al. Crusted (Norwegian) scabies induced by use of topical corticosteroids and treated successfully with ivermectin. J Pediatr. 1999;135:122-124.
9. Goyal NN, Wong GA. Psoriasis or crusted scabies. Clin Exp Dermatol. 2008;33:211-212.
10. Kim KJ, Roh KH, Choi JH, et al. Scabies incognito presenting as urticaria pigmentosa in an infant. Pediatr Dermatol. 2002;19:409-411.
1. Burkhart CN, Burkhart CG, Morrell DS. Infestations. In: Bolognia JL, Jorizzo JL, Schaffer JV, et al, eds. Dermatology. 3rd ed. Philadelphia, PA: Mosby Elsevier; 2012;1423-1426.
2. Subramaniam G, Kaliaperumal K, Duraipandian J, et al. Norwegian scabies in a malnourished young adult: a case report. J Infect Dev Ctries. 2010;4:349-351.
3. Karthikeyan K. Crusted scabies. Indian J Dermatol Venereol Leprol. 2009;75:340-347.
4. Binic I, Jankovic A, Jovanovic D, et al. Crusted (Norwegian) scabies following systemic and topical corticosteroid therapy. J Korean Med Sci. 2010;25:188-191.
5. Ramachandran V, Shankar EM, Devaleenal B, et al. Atypically distributed cutaneous lesions of Norwegian scabies in an HIV-positive man in South India: a case report. J Med Case Rep. 2008;2:82.
6. Lai YC, Teng CJ, Chen PC, et al. Unusual scalp crusted scabies in an adult T-cell leukemia/lymphoma patient. Ups J Med Sci. 2011;116:77-78.
7. Saillard C, Darrieux L, Safa G. Crusted scabies complicates etanercept therapy in a patient with severe psoriasis. J Am Acad Dermatol. 2013;68:e138-e139.
8. Marlière V, Roul S, Labrèze C, et al. Crusted (Norwegian) scabies induced by use of topical corticosteroids and treated successfully with ivermectin. J Pediatr. 1999;135:122-124.
9. Goyal NN, Wong GA. Psoriasis or crusted scabies. Clin Exp Dermatol. 2008;33:211-212.
10. Kim KJ, Roh KH, Choi JH, et al. Scabies incognito presenting as urticaria pigmentosa in an infant. Pediatr Dermatol. 2002;19:409-411.