User login
Systemic Disease Manifestations of TSC Strongly Associated With Epilepsy
In a study of 1816 patients with tuberous sclerosis complex (TSC), researchers found that specific disease manifestations—cardiac rhabodmyomas, retinal hemartomas, renal cysts, renal angiomyolipipomas, and facial angiofibromas—were associated with a higher likelihood of epilepsy development. The authors posit that this research can help identify patients who will benefit from novel, targeted, preventative treatments.
Jeong A, Wong M. Systemic disease manifestations associated with epilepsy in tuberous sclerosis complex [published online ahead of print July 15, 2016]. Epilepsia. 2016;doi:10.1111/epi.13467.
In a study of 1816 patients with tuberous sclerosis complex (TSC), researchers found that specific disease manifestations—cardiac rhabodmyomas, retinal hemartomas, renal cysts, renal angiomyolipipomas, and facial angiofibromas—were associated with a higher likelihood of epilepsy development. The authors posit that this research can help identify patients who will benefit from novel, targeted, preventative treatments.
Jeong A, Wong M. Systemic disease manifestations associated with epilepsy in tuberous sclerosis complex [published online ahead of print July 15, 2016]. Epilepsia. 2016;doi:10.1111/epi.13467.
In a study of 1816 patients with tuberous sclerosis complex (TSC), researchers found that specific disease manifestations—cardiac rhabodmyomas, retinal hemartomas, renal cysts, renal angiomyolipipomas, and facial angiofibromas—were associated with a higher likelihood of epilepsy development. The authors posit that this research can help identify patients who will benefit from novel, targeted, preventative treatments.
Jeong A, Wong M. Systemic disease manifestations associated with epilepsy in tuberous sclerosis complex [published online ahead of print July 15, 2016]. Epilepsia. 2016;doi:10.1111/epi.13467.
A Second Look at Head MRIs Demonstrates the Value of Re-Review
To determine if patients with epilepsy are appropriate candidates for resective surgery, presurgical conferences are conducted to review magnetic resonance images (MRIs) of the patient’s head. Kenney and associates analyzed repeat reviews of MRIs at presurgical epilepsy conferences to assess their impact on the decision-making process. Among the 233 patients whose charts were re-reviewed, 94 patients (40.3%) had the resective surgery performed, and the analysis revealed that 41 patients (17.6%) had previously undiagnosed findings; 18 of the 41 patients had the surgery. However, among 4 of the 41 patients (9.8%), the re-reviews found abnormalities that did not warrant surgical resection, including autoimmunity and bilateral pathology.
Kenney DL, Kelly-Williams KM, Krecke KN et al. Usefulness of Repeat Review of Head Magnetic Resonance Images During Presurgical Epilepsy Conferences. Epilepsy Res. 2016. In press. http://dx.doi.org/10.1016/j.eplepsyres.2016.06.005.
To determine if patients with epilepsy are appropriate candidates for resective surgery, presurgical conferences are conducted to review magnetic resonance images (MRIs) of the patient’s head. Kenney and associates analyzed repeat reviews of MRIs at presurgical epilepsy conferences to assess their impact on the decision-making process. Among the 233 patients whose charts were re-reviewed, 94 patients (40.3%) had the resective surgery performed, and the analysis revealed that 41 patients (17.6%) had previously undiagnosed findings; 18 of the 41 patients had the surgery. However, among 4 of the 41 patients (9.8%), the re-reviews found abnormalities that did not warrant surgical resection, including autoimmunity and bilateral pathology.
Kenney DL, Kelly-Williams KM, Krecke KN et al. Usefulness of Repeat Review of Head Magnetic Resonance Images During Presurgical Epilepsy Conferences. Epilepsy Res. 2016. In press. http://dx.doi.org/10.1016/j.eplepsyres.2016.06.005.
To determine if patients with epilepsy are appropriate candidates for resective surgery, presurgical conferences are conducted to review magnetic resonance images (MRIs) of the patient’s head. Kenney and associates analyzed repeat reviews of MRIs at presurgical epilepsy conferences to assess their impact on the decision-making process. Among the 233 patients whose charts were re-reviewed, 94 patients (40.3%) had the resective surgery performed, and the analysis revealed that 41 patients (17.6%) had previously undiagnosed findings; 18 of the 41 patients had the surgery. However, among 4 of the 41 patients (9.8%), the re-reviews found abnormalities that did not warrant surgical resection, including autoimmunity and bilateral pathology.
Kenney DL, Kelly-Williams KM, Krecke KN et al. Usefulness of Repeat Review of Head Magnetic Resonance Images During Presurgical Epilepsy Conferences. Epilepsy Res. 2016. In press. http://dx.doi.org/10.1016/j.eplepsyres.2016.06.005.
Stimulation-identified Cortical Naming Sites Pose Unexpected Challenges
Before surgeons perform a resection involving the language-dominant hemisphere of a patient with epilepsy, they may do electrical stimulation mapping to identify a patient’s language-dominant hemisphere. Typically they will ask patients to identify objects to help locate the language cortex and then avoid resection in an area of the brain in which electrical stimulation makes it difficult for patients to name said objects. But because word production involves mechanisms that may be centered in more than one area of the brain, Hamberger et al tested locations that have been identified by stimulation as naming sites to look for disparities. Testing patients with refractory temporal lobe epilepsy who had subdural electrodes implanted, they discovered that stimulating naming sites in the superior temporary lobe was more likely to disrupt phonological processing but did not affect a patient’s ability to process semantic information. Stimulating the inferior temporal naming sites was more likely to impair semantic processing.
Hamberger MJ, Miozzo M, Schevon CA, et al. Functional differences among stimulation-identified cortical naming sites in the temporal region. Epilepsy Behav. 2016;60:124-129.
Before surgeons perform a resection involving the language-dominant hemisphere of a patient with epilepsy, they may do electrical stimulation mapping to identify a patient’s language-dominant hemisphere. Typically they will ask patients to identify objects to help locate the language cortex and then avoid resection in an area of the brain in which electrical stimulation makes it difficult for patients to name said objects. But because word production involves mechanisms that may be centered in more than one area of the brain, Hamberger et al tested locations that have been identified by stimulation as naming sites to look for disparities. Testing patients with refractory temporal lobe epilepsy who had subdural electrodes implanted, they discovered that stimulating naming sites in the superior temporary lobe was more likely to disrupt phonological processing but did not affect a patient’s ability to process semantic information. Stimulating the inferior temporal naming sites was more likely to impair semantic processing.
Hamberger MJ, Miozzo M, Schevon CA, et al. Functional differences among stimulation-identified cortical naming sites in the temporal region. Epilepsy Behav. 2016;60:124-129.
Before surgeons perform a resection involving the language-dominant hemisphere of a patient with epilepsy, they may do electrical stimulation mapping to identify a patient’s language-dominant hemisphere. Typically they will ask patients to identify objects to help locate the language cortex and then avoid resection in an area of the brain in which electrical stimulation makes it difficult for patients to name said objects. But because word production involves mechanisms that may be centered in more than one area of the brain, Hamberger et al tested locations that have been identified by stimulation as naming sites to look for disparities. Testing patients with refractory temporal lobe epilepsy who had subdural electrodes implanted, they discovered that stimulating naming sites in the superior temporary lobe was more likely to disrupt phonological processing but did not affect a patient’s ability to process semantic information. Stimulating the inferior temporal naming sites was more likely to impair semantic processing.
Hamberger MJ, Miozzo M, Schevon CA, et al. Functional differences among stimulation-identified cortical naming sites in the temporal region. Epilepsy Behav. 2016;60:124-129.
Evaluating Alternatives to Open Surgical Resection for Epilepsy
Open surgical resection is still considered the best approach for patients with epilepsy that do not respond well to medical therapy. But despite being considered the gold standard in neurosurgical care, the shortcomings of open surgical resection need to be addressed. McGovern and colleagues do so in a review published in Current Neurology and Neuroscience Reports. They point to the value of stereotactic electroencephalography, which can localize deep epileptic foci. Similarly laser interstitial thermal therapy (LITT) and stereotactic radiosurgery have advantages because they can ablate specific regions of the brain using minimally or non-invasive techniques. In the case of LITT, it can offer clinicians near real-time feedback on its effects. Neurostimulation is also worth consideration in select patients because it can reduce seizure occurrence without the need for ablation or resection
McGovern RA, Banks GP, McKhann GM 2nd. New techniques and progress in epilepsy surgery. Curr Neurol Neurosci Rep. 2016;16(7):65.
Open surgical resection is still considered the best approach for patients with epilepsy that do not respond well to medical therapy. But despite being considered the gold standard in neurosurgical care, the shortcomings of open surgical resection need to be addressed. McGovern and colleagues do so in a review published in Current Neurology and Neuroscience Reports. They point to the value of stereotactic electroencephalography, which can localize deep epileptic foci. Similarly laser interstitial thermal therapy (LITT) and stereotactic radiosurgery have advantages because they can ablate specific regions of the brain using minimally or non-invasive techniques. In the case of LITT, it can offer clinicians near real-time feedback on its effects. Neurostimulation is also worth consideration in select patients because it can reduce seizure occurrence without the need for ablation or resection
McGovern RA, Banks GP, McKhann GM 2nd. New techniques and progress in epilepsy surgery. Curr Neurol Neurosci Rep. 2016;16(7):65.
Open surgical resection is still considered the best approach for patients with epilepsy that do not respond well to medical therapy. But despite being considered the gold standard in neurosurgical care, the shortcomings of open surgical resection need to be addressed. McGovern and colleagues do so in a review published in Current Neurology and Neuroscience Reports. They point to the value of stereotactic electroencephalography, which can localize deep epileptic foci. Similarly laser interstitial thermal therapy (LITT) and stereotactic radiosurgery have advantages because they can ablate specific regions of the brain using minimally or non-invasive techniques. In the case of LITT, it can offer clinicians near real-time feedback on its effects. Neurostimulation is also worth consideration in select patients because it can reduce seizure occurrence without the need for ablation or resection
McGovern RA, Banks GP, McKhann GM 2nd. New techniques and progress in epilepsy surgery. Curr Neurol Neurosci Rep. 2016;16(7):65.
Smartphone app aimed at disrupting first-episode psychosis shows promise
BETHESDA, MD. – What would the world look like for people with first-episode psychosis if they could be identified before they ended up in care? And what if once identified, they could begin receiving treatment immediately?
Those questions are not just hypothetical to Danielle Schlosser, PhD

Using online screening based on proven tools, followed by enrollment in a secured, closed community created through the use of a smartphone app, Dr. Schlosser and her colleagues are delivering remote care to people with first-episode psychosis, rather than making them come to the clinic.
“It’s controversial, but you’re not doing anything meaningful if you don’t stir things up,” Dr. Schlosser said in an interview. “Why do we have to have a one-size-fits-all model?”
Statistics from NIMH show that duration of psychosis before treatment is 1-3 years. People in the early stages of psychosis, typically in their late teens or early 20s, often do not find their way to care until after admittance through an emergency department or a brush with the law, Dr. Schlosser said. Once diagnosed, clinically meaningful improvements in outcomes in this cohort often are impeded by cognitive and motivational impairment, including fear of stigma, or logistical challenges such as finding reliable transportation to a treatment site. According to the World Health Organization, half of all people with schizophrenia globally are not receiving treatment.
“We can do better,” Dr. Schlosser said.
In the PRIME design trials, Dr. Schlosser and her colleagues are evaluating how “user-centered design” might improve treatment delivery. In stage 1 of the study, two design phases, each with a separate group of 10 participants with recent-onset schizophrenia, helped a global design firm called IDEO arrive at a product that Dr. Schlosser and her colleagues described in a study published online as “casual, friendly, and nonstigmatizing, which is in line with the recovery model of psychosis” (JMIR Res Protoc. 2016;5[2]:e77).
The app included short motivational texts from trained therapists, a feature for individualized goal setting in prognostically important psychosocial domains, opportunities for social networking via direct peer-to-peer messaging, and a community “moments feed” aimed at capturing and reinforcing rewarding experiences and achieving goals.
After 12 weeks of using the app, Dr. Schlosser and her coinvestigators found that trial participants, all of whom had been asked to use it at least once a week, had used it an average of once every other day and had actively engaged with its various features with every log-in. Retention and satisfaction was 100% in each group, and levels of engagement from stage 1 to stage 2 increased by what Dr. Schlosser and her coauthors reported as “two- or threefold” in almost each aspect of the platform.
Dr. Schlosser said such impressive results have continued now that the study has entered stage 2, which is being conducted across the United States, Canada, and Mexico. Fifty people have enrolled, Dr. Schlosser said.
“So far, we have a 93% retention in our clinical trial, which is very high for this population,” she said. “We’re also seeing that two-thirds of the population are self-referred. You just don’t see that in early psychosis research.”
She credits those results in part to the study’s online recruitment design, but Dr. Schlosser said the patient-reported outcomes are the most gratifying.
“We got a letter from a mom who said she wished we could have seen her daughter’s face when she saw that everyone in her [online] group looks ‘normal.’ Her daughter was looking for hope, and she found it mirrored back at her,” Dr. Schlosser said. “They are not their illness.”
A prime reason people with recent-onset schizophrenia don’t access formal treatment is the fear they will be stigmatized, Dr. Schlosser said. Furthermore, she said, since most of those affected are young, creative, and “antiestablishment” in their attitudes, they already deal with stigma. The most effective treatment experience needed to be based on those kinds of values, she said.
“They want more control in their lives, and they want to connect with others like them. The idea of seeking care in a traditional setting is a deterrent for these people,” Dr. Schlosser said in the interview. “So, rather than build a new building, we built them a digital platform.”
A few audience members expressed concern that such a treatment model may expose these patients to unnecessary harm. Dr. Schlosser replied in the interview that even in clinical settings, patients prescribed medications still may be noncompliant. “We are not the enforcers of medications. It’s still the patient’s choice. Maybe I am being too provocative, but if people don’t want to take medications, then we try to work with them where they are,” she said.
In addition to the support from the online community, trial participants spend an average of 20 minutes with the coaching staff, making the model a “very low resource” one, according to Dr. Schlosser.
PRIME is expected to complete in the spring 2018, at which time the data collected potentially will be used to refine user-designed apps for use in other mental disorders, Dr. Schlosser said.
“I want to promote a digital system of care so that we can move immediately from when we identify a person is at risk to immediately giving them support. I think we can improve things so that we don’t just have equivalent levels of care but optimal ones,” Dr. Schlosser told the audience. “What we have had as our system of care to date isn’t working.”
Dr. Schlosser did not have any relevant disclosures.
On Twitter @whitneymcknight
One of the most compelling aspects to schizophrenia, the most serious of mental disorders, is its age of onset in late adolescence and early adulthood. Intervening early on has good logic and good sense on its side. Nothing can be more important than giving attention to patients and families early on. However, it still remain unknown what biological processes trigger the disorder, and to date there are no data suggesting that we are able to slow the illness progression. Nevertheless, focusing early will teach us about the illness and set the stage when the next generation of biological treatments emerge.
David Pickar, MD, is a psychiatrist and former (retired) director of intramural research at the National Institute of Mental Health. In addition, Dr. Pickar is adjunct professor of psychiatry at Johns Hopkins University, Baltimore, and at the Uniformed Services University of the Health Sciences, Bethesda, Md.
One of the most compelling aspects to schizophrenia, the most serious of mental disorders, is its age of onset in late adolescence and early adulthood. Intervening early on has good logic and good sense on its side. Nothing can be more important than giving attention to patients and families early on. However, it still remain unknown what biological processes trigger the disorder, and to date there are no data suggesting that we are able to slow the illness progression. Nevertheless, focusing early will teach us about the illness and set the stage when the next generation of biological treatments emerge.
David Pickar, MD, is a psychiatrist and former (retired) director of intramural research at the National Institute of Mental Health. In addition, Dr. Pickar is adjunct professor of psychiatry at Johns Hopkins University, Baltimore, and at the Uniformed Services University of the Health Sciences, Bethesda, Md.
One of the most compelling aspects to schizophrenia, the most serious of mental disorders, is its age of onset in late adolescence and early adulthood. Intervening early on has good logic and good sense on its side. Nothing can be more important than giving attention to patients and families early on. However, it still remain unknown what biological processes trigger the disorder, and to date there are no data suggesting that we are able to slow the illness progression. Nevertheless, focusing early will teach us about the illness and set the stage when the next generation of biological treatments emerge.
David Pickar, MD, is a psychiatrist and former (retired) director of intramural research at the National Institute of Mental Health. In addition, Dr. Pickar is adjunct professor of psychiatry at Johns Hopkins University, Baltimore, and at the Uniformed Services University of the Health Sciences, Bethesda, Md.
BETHESDA, MD. – What would the world look like for people with first-episode psychosis if they could be identified before they ended up in care? And what if once identified, they could begin receiving treatment immediately?
Those questions are not just hypothetical to Danielle Schlosser, PhD
Using online screening based on proven tools, followed by enrollment in a secured, closed community created through the use of a smartphone app, Dr. Schlosser and her colleagues are delivering remote care to people with first-episode psychosis, rather than making them come to the clinic.
“It’s controversial, but you’re not doing anything meaningful if you don’t stir things up,” Dr. Schlosser said in an interview. “Why do we have to have a one-size-fits-all model?”
Statistics from NIMH show that duration of psychosis before treatment is 1-3 years. People in the early stages of psychosis, typically in their late teens or early 20s, often do not find their way to care until after admittance through an emergency department or a brush with the law, Dr. Schlosser said. Once diagnosed, clinically meaningful improvements in outcomes in this cohort often are impeded by cognitive and motivational impairment, including fear of stigma, or logistical challenges such as finding reliable transportation to a treatment site. According to the World Health Organization, half of all people with schizophrenia globally are not receiving treatment.
“We can do better,” Dr. Schlosser said.
In the PRIME design trials, Dr. Schlosser and her colleagues are evaluating how “user-centered design” might improve treatment delivery. In stage 1 of the study, two design phases, each with a separate group of 10 participants with recent-onset schizophrenia, helped a global design firm called IDEO arrive at a product that Dr. Schlosser and her colleagues described in a study published online as “casual, friendly, and nonstigmatizing, which is in line with the recovery model of psychosis” (JMIR Res Protoc. 2016;5[2]:e77).
The app included short motivational texts from trained therapists, a feature for individualized goal setting in prognostically important psychosocial domains, opportunities for social networking via direct peer-to-peer messaging, and a community “moments feed” aimed at capturing and reinforcing rewarding experiences and achieving goals.
After 12 weeks of using the app, Dr. Schlosser and her coinvestigators found that trial participants, all of whom had been asked to use it at least once a week, had used it an average of once every other day and had actively engaged with its various features with every log-in. Retention and satisfaction was 100% in each group, and levels of engagement from stage 1 to stage 2 increased by what Dr. Schlosser and her coauthors reported as “two- or threefold” in almost each aspect of the platform.
Dr. Schlosser said such impressive results have continued now that the study has entered stage 2, which is being conducted across the United States, Canada, and Mexico. Fifty people have enrolled, Dr. Schlosser said.
“So far, we have a 93% retention in our clinical trial, which is very high for this population,” she said. “We’re also seeing that two-thirds of the population are self-referred. You just don’t see that in early psychosis research.”
She credits those results in part to the study’s online recruitment design, but Dr. Schlosser said the patient-reported outcomes are the most gratifying.
“We got a letter from a mom who said she wished we could have seen her daughter’s face when she saw that everyone in her [online] group looks ‘normal.’ Her daughter was looking for hope, and she found it mirrored back at her,” Dr. Schlosser said. “They are not their illness.”
A prime reason people with recent-onset schizophrenia don’t access formal treatment is the fear they will be stigmatized, Dr. Schlosser said. Furthermore, she said, since most of those affected are young, creative, and “antiestablishment” in their attitudes, they already deal with stigma. The most effective treatment experience needed to be based on those kinds of values, she said.
“They want more control in their lives, and they want to connect with others like them. The idea of seeking care in a traditional setting is a deterrent for these people,” Dr. Schlosser said in the interview. “So, rather than build a new building, we built them a digital platform.”
A few audience members expressed concern that such a treatment model may expose these patients to unnecessary harm. Dr. Schlosser replied in the interview that even in clinical settings, patients prescribed medications still may be noncompliant. “We are not the enforcers of medications. It’s still the patient’s choice. Maybe I am being too provocative, but if people don’t want to take medications, then we try to work with them where they are,” she said.
In addition to the support from the online community, trial participants spend an average of 20 minutes with the coaching staff, making the model a “very low resource” one, according to Dr. Schlosser.
PRIME is expected to complete in the spring 2018, at which time the data collected potentially will be used to refine user-designed apps for use in other mental disorders, Dr. Schlosser said.
“I want to promote a digital system of care so that we can move immediately from when we identify a person is at risk to immediately giving them support. I think we can improve things so that we don’t just have equivalent levels of care but optimal ones,” Dr. Schlosser told the audience. “What we have had as our system of care to date isn’t working.”
Dr. Schlosser did not have any relevant disclosures.
On Twitter @whitneymcknight
BETHESDA, MD. – What would the world look like for people with first-episode psychosis if they could be identified before they ended up in care? And what if once identified, they could begin receiving treatment immediately?
Those questions are not just hypothetical to Danielle Schlosser, PhD
Using online screening based on proven tools, followed by enrollment in a secured, closed community created through the use of a smartphone app, Dr. Schlosser and her colleagues are delivering remote care to people with first-episode psychosis, rather than making them come to the clinic.
“It’s controversial, but you’re not doing anything meaningful if you don’t stir things up,” Dr. Schlosser said in an interview. “Why do we have to have a one-size-fits-all model?”
Statistics from NIMH show that duration of psychosis before treatment is 1-3 years. People in the early stages of psychosis, typically in their late teens or early 20s, often do not find their way to care until after admittance through an emergency department or a brush with the law, Dr. Schlosser said. Once diagnosed, clinically meaningful improvements in outcomes in this cohort often are impeded by cognitive and motivational impairment, including fear of stigma, or logistical challenges such as finding reliable transportation to a treatment site. According to the World Health Organization, half of all people with schizophrenia globally are not receiving treatment.
“We can do better,” Dr. Schlosser said.
In the PRIME design trials, Dr. Schlosser and her colleagues are evaluating how “user-centered design” might improve treatment delivery. In stage 1 of the study, two design phases, each with a separate group of 10 participants with recent-onset schizophrenia, helped a global design firm called IDEO arrive at a product that Dr. Schlosser and her colleagues described in a study published online as “casual, friendly, and nonstigmatizing, which is in line with the recovery model of psychosis” (JMIR Res Protoc. 2016;5[2]:e77).
The app included short motivational texts from trained therapists, a feature for individualized goal setting in prognostically important psychosocial domains, opportunities for social networking via direct peer-to-peer messaging, and a community “moments feed” aimed at capturing and reinforcing rewarding experiences and achieving goals.
After 12 weeks of using the app, Dr. Schlosser and her coinvestigators found that trial participants, all of whom had been asked to use it at least once a week, had used it an average of once every other day and had actively engaged with its various features with every log-in. Retention and satisfaction was 100% in each group, and levels of engagement from stage 1 to stage 2 increased by what Dr. Schlosser and her coauthors reported as “two- or threefold” in almost each aspect of the platform.
Dr. Schlosser said such impressive results have continued now that the study has entered stage 2, which is being conducted across the United States, Canada, and Mexico. Fifty people have enrolled, Dr. Schlosser said.
“So far, we have a 93% retention in our clinical trial, which is very high for this population,” she said. “We’re also seeing that two-thirds of the population are self-referred. You just don’t see that in early psychosis research.”
She credits those results in part to the study’s online recruitment design, but Dr. Schlosser said the patient-reported outcomes are the most gratifying.
“We got a letter from a mom who said she wished we could have seen her daughter’s face when she saw that everyone in her [online] group looks ‘normal.’ Her daughter was looking for hope, and she found it mirrored back at her,” Dr. Schlosser said. “They are not their illness.”
A prime reason people with recent-onset schizophrenia don’t access formal treatment is the fear they will be stigmatized, Dr. Schlosser said. Furthermore, she said, since most of those affected are young, creative, and “antiestablishment” in their attitudes, they already deal with stigma. The most effective treatment experience needed to be based on those kinds of values, she said.
“They want more control in their lives, and they want to connect with others like them. The idea of seeking care in a traditional setting is a deterrent for these people,” Dr. Schlosser said in the interview. “So, rather than build a new building, we built them a digital platform.”
A few audience members expressed concern that such a treatment model may expose these patients to unnecessary harm. Dr. Schlosser replied in the interview that even in clinical settings, patients prescribed medications still may be noncompliant. “We are not the enforcers of medications. It’s still the patient’s choice. Maybe I am being too provocative, but if people don’t want to take medications, then we try to work with them where they are,” she said.
In addition to the support from the online community, trial participants spend an average of 20 minutes with the coaching staff, making the model a “very low resource” one, according to Dr. Schlosser.
PRIME is expected to complete in the spring 2018, at which time the data collected potentially will be used to refine user-designed apps for use in other mental disorders, Dr. Schlosser said.
“I want to promote a digital system of care so that we can move immediately from when we identify a person is at risk to immediately giving them support. I think we can improve things so that we don’t just have equivalent levels of care but optimal ones,” Dr. Schlosser told the audience. “What we have had as our system of care to date isn’t working.”
Dr. Schlosser did not have any relevant disclosures.
On Twitter @whitneymcknight
EXPERT ANALYSIS FROM THE 2016 NIMH CONFERENCE ON MENTAL HEALTH SERVICES RESEARCH

Sunscreens safe in babies, children
BOSTON – Despite what some popular online media outlets report, sunscreens are safe in children and can even be used on infants under 6 months of age when sun avoidance – the best approach to protecting babies from the damaging effects of the sun – is not possible, according to Mercedes E. Gonzalez, MD.
A quick Google search reveals numerous, widely-shared articles about the dangers lurking in one’s beach bag, and while many product labels recommend asking a doctor about whether the product is safe for babies under age 6 months, that’s only because most product safety studies didn’t include that age group, Dr. Gonzalez of the University of Miami said at the American Academy of Dermatology summer meeting.
In fact, there is “nothing magical that happens” in infant skin after 6 months that makes sunscreen use safer, she said, explaining that infant skin is structurally and functionally different from adult skin, and that while gradual maturation takes place over time, thereby reducing susceptibility to percutaneous absorption of topically applied products, the risk is minimal even in babies younger than age 6 months.
The skin characteristics that make younger skin more susceptible to percutaneous absorption also make babies and children unusually susceptible to ultraviolet radiation and ultraviolet radiation–induced immunosuppression, for which the consequences are not fully understood, she said.
Among the more commonly cited sunscreen ingredients of concern are oxybenzone, or benzonephenone-3, and nanoparticles, she noted.
However, the overall consensus based on studies of oxybenzone is that aside from causing some cases of allergic and irritant contact dermatitis, the compound is safe; no harmful cause and effect relationship with oxybenzone and systemic side effects in humans have been reported, and periodic reviews by European, Australian and U.S. safety panels all conclude that it is safe.
Numerous studies of nanoparticles – such as nanosized zinc oxide and titanium dioxide – have shown that absorption is confined to the level of stratum corneum – even when skin barrier function has been altered, she said, noting that most are coated with aluminum oxide and SiO2 to minimize contact.
However, the safety of sunscreen shouldn’t be seen as license to ignore sun-exposure recommendations; sunscreen in infants should be considered “the last layer of protection,” used only on exposed areas when adequate clothing and shade are not available, according to a 2011 American Academy of Pediatrics statement (Pediatrics. 2011 Feb. doi: 10.1542/peds.2010-3501).
Efforts should be made to keep babies in the shade when outdoors whenever possible, especially during peak sun hours. Use sun-protective clothing, including hats, sunglasses, and long-sleeved shirts, Dr. Gonzalez advised.
When sunscreen is required, a broad-spectrum water-resistant product with an SPF of more than 30 is preferable.
“But the best sunscreen is the one you and your child will use,” she said.
Mineral-based products are less irritating and thus may be a preferred option for children with atopic dermatitis, she added.
Advise parents to apply sunscreen to all areas not protected by their child’s clothing, paying particular attention to vulnerable areas, including the back of the neck, ears, and dorsal feet. Reapply before going outdoors, and then again every 2 hours, she advised.
“So the overall answer to the parents’ question, ‘Are sunscreens safe?’ ... the overwhelming answer here is yes, and the weight of the evidence shows there is no proven harm from sunscreen use especially when used properly,” she said.
Provide specific guidance for pediatric sunscreen use
In the face of conflicting information about sunscreen safety and efficacy, parents with questions about sunscreen are looking for specific direction, Dr. Gonzalez said.
She said she finds it helpful to teach them about the importance of reading labels. That is, looking at the ingredients, and looking for SPF above 30, broad-spectrum coverage, and water resistance. She also recommends providing a list or images of good options, and circling the specific preferred products.
For babies, she finds stick sunscreens most useful for application.
“I generally don’t recommend sprays, but if they’re going to use a spray – and parents love sprays because they are easy to apply – I recommend the ones that have some zinc oxide in them, so that when they apply them they can see where they’re going on the skin,” Dr. Gonzalez said.
Tell patients to apply sunscreen before leaving the house, she advised, adding that making sunscreen application part of a daily routine helps encourage healthy behaviors, as does allowing children, at the right age, to participate in sunscreen application.
For adolescents, avoid scare tactics such as warning about skin cancer. Rather, focus on benefits of avoiding the sun, help them find a product they like by finding out why they don’t like a particular product and recommending an alternative, then following up on that when they come back in, she suggested.
“I really try to address it at every visit,” Dr. Gonzalez said.
“Finally, the most important message is that sunscreen is really just one part of complete sun protection,” she said, noting that specific information about where to buy sun-protective clothing and hats is also important.
Dr. Gonzalez reported serving as a speaker and/or advisory board member and receiving honoraria from Pierre Fabre Dermatologie, Anacor Pharmaceuticals, Encore Dermatology, and PuraCap Pharmaceutical.
BOSTON – Despite what some popular online media outlets report, sunscreens are safe in children and can even be used on infants under 6 months of age when sun avoidance – the best approach to protecting babies from the damaging effects of the sun – is not possible, according to Mercedes E. Gonzalez, MD.
A quick Google search reveals numerous, widely-shared articles about the dangers lurking in one’s beach bag, and while many product labels recommend asking a doctor about whether the product is safe for babies under age 6 months, that’s only because most product safety studies didn’t include that age group, Dr. Gonzalez of the University of Miami said at the American Academy of Dermatology summer meeting.
In fact, there is “nothing magical that happens” in infant skin after 6 months that makes sunscreen use safer, she said, explaining that infant skin is structurally and functionally different from adult skin, and that while gradual maturation takes place over time, thereby reducing susceptibility to percutaneous absorption of topically applied products, the risk is minimal even in babies younger than age 6 months.
The skin characteristics that make younger skin more susceptible to percutaneous absorption also make babies and children unusually susceptible to ultraviolet radiation and ultraviolet radiation–induced immunosuppression, for which the consequences are not fully understood, she said.
Among the more commonly cited sunscreen ingredients of concern are oxybenzone, or benzonephenone-3, and nanoparticles, she noted.
However, the overall consensus based on studies of oxybenzone is that aside from causing some cases of allergic and irritant contact dermatitis, the compound is safe; no harmful cause and effect relationship with oxybenzone and systemic side effects in humans have been reported, and periodic reviews by European, Australian and U.S. safety panels all conclude that it is safe.
Numerous studies of nanoparticles – such as nanosized zinc oxide and titanium dioxide – have shown that absorption is confined to the level of stratum corneum – even when skin barrier function has been altered, she said, noting that most are coated with aluminum oxide and SiO2 to minimize contact.
However, the safety of sunscreen shouldn’t be seen as license to ignore sun-exposure recommendations; sunscreen in infants should be considered “the last layer of protection,” used only on exposed areas when adequate clothing and shade are not available, according to a 2011 American Academy of Pediatrics statement (Pediatrics. 2011 Feb. doi: 10.1542/peds.2010-3501).
Efforts should be made to keep babies in the shade when outdoors whenever possible, especially during peak sun hours. Use sun-protective clothing, including hats, sunglasses, and long-sleeved shirts, Dr. Gonzalez advised.
When sunscreen is required, a broad-spectrum water-resistant product with an SPF of more than 30 is preferable.
“But the best sunscreen is the one you and your child will use,” she said.
Mineral-based products are less irritating and thus may be a preferred option for children with atopic dermatitis, she added.
Advise parents to apply sunscreen to all areas not protected by their child’s clothing, paying particular attention to vulnerable areas, including the back of the neck, ears, and dorsal feet. Reapply before going outdoors, and then again every 2 hours, she advised.
“So the overall answer to the parents’ question, ‘Are sunscreens safe?’ ... the overwhelming answer here is yes, and the weight of the evidence shows there is no proven harm from sunscreen use especially when used properly,” she said.
Provide specific guidance for pediatric sunscreen use
In the face of conflicting information about sunscreen safety and efficacy, parents with questions about sunscreen are looking for specific direction, Dr. Gonzalez said.
She said she finds it helpful to teach them about the importance of reading labels. That is, looking at the ingredients, and looking for SPF above 30, broad-spectrum coverage, and water resistance. She also recommends providing a list or images of good options, and circling the specific preferred products.
For babies, she finds stick sunscreens most useful for application.
“I generally don’t recommend sprays, but if they’re going to use a spray – and parents love sprays because they are easy to apply – I recommend the ones that have some zinc oxide in them, so that when they apply them they can see where they’re going on the skin,” Dr. Gonzalez said.
Tell patients to apply sunscreen before leaving the house, she advised, adding that making sunscreen application part of a daily routine helps encourage healthy behaviors, as does allowing children, at the right age, to participate in sunscreen application.
For adolescents, avoid scare tactics such as warning about skin cancer. Rather, focus on benefits of avoiding the sun, help them find a product they like by finding out why they don’t like a particular product and recommending an alternative, then following up on that when they come back in, she suggested.
“I really try to address it at every visit,” Dr. Gonzalez said.
“Finally, the most important message is that sunscreen is really just one part of complete sun protection,” she said, noting that specific information about where to buy sun-protective clothing and hats is also important.
Dr. Gonzalez reported serving as a speaker and/or advisory board member and receiving honoraria from Pierre Fabre Dermatologie, Anacor Pharmaceuticals, Encore Dermatology, and PuraCap Pharmaceutical.
BOSTON – Despite what some popular online media outlets report, sunscreens are safe in children and can even be used on infants under 6 months of age when sun avoidance – the best approach to protecting babies from the damaging effects of the sun – is not possible, according to Mercedes E. Gonzalez, MD.
A quick Google search reveals numerous, widely-shared articles about the dangers lurking in one’s beach bag, and while many product labels recommend asking a doctor about whether the product is safe for babies under age 6 months, that’s only because most product safety studies didn’t include that age group, Dr. Gonzalez of the University of Miami said at the American Academy of Dermatology summer meeting.
In fact, there is “nothing magical that happens” in infant skin after 6 months that makes sunscreen use safer, she said, explaining that infant skin is structurally and functionally different from adult skin, and that while gradual maturation takes place over time, thereby reducing susceptibility to percutaneous absorption of topically applied products, the risk is minimal even in babies younger than age 6 months.
The skin characteristics that make younger skin more susceptible to percutaneous absorption also make babies and children unusually susceptible to ultraviolet radiation and ultraviolet radiation–induced immunosuppression, for which the consequences are not fully understood, she said.
Among the more commonly cited sunscreen ingredients of concern are oxybenzone, or benzonephenone-3, and nanoparticles, she noted.
However, the overall consensus based on studies of oxybenzone is that aside from causing some cases of allergic and irritant contact dermatitis, the compound is safe; no harmful cause and effect relationship with oxybenzone and systemic side effects in humans have been reported, and periodic reviews by European, Australian and U.S. safety panels all conclude that it is safe.
Numerous studies of nanoparticles – such as nanosized zinc oxide and titanium dioxide – have shown that absorption is confined to the level of stratum corneum – even when skin barrier function has been altered, she said, noting that most are coated with aluminum oxide and SiO2 to minimize contact.
However, the safety of sunscreen shouldn’t be seen as license to ignore sun-exposure recommendations; sunscreen in infants should be considered “the last layer of protection,” used only on exposed areas when adequate clothing and shade are not available, according to a 2011 American Academy of Pediatrics statement (Pediatrics. 2011 Feb. doi: 10.1542/peds.2010-3501).
Efforts should be made to keep babies in the shade when outdoors whenever possible, especially during peak sun hours. Use sun-protective clothing, including hats, sunglasses, and long-sleeved shirts, Dr. Gonzalez advised.
When sunscreen is required, a broad-spectrum water-resistant product with an SPF of more than 30 is preferable.
“But the best sunscreen is the one you and your child will use,” she said.
Mineral-based products are less irritating and thus may be a preferred option for children with atopic dermatitis, she added.
Advise parents to apply sunscreen to all areas not protected by their child’s clothing, paying particular attention to vulnerable areas, including the back of the neck, ears, and dorsal feet. Reapply before going outdoors, and then again every 2 hours, she advised.
“So the overall answer to the parents’ question, ‘Are sunscreens safe?’ ... the overwhelming answer here is yes, and the weight of the evidence shows there is no proven harm from sunscreen use especially when used properly,” she said.
Provide specific guidance for pediatric sunscreen use
In the face of conflicting information about sunscreen safety and efficacy, parents with questions about sunscreen are looking for specific direction, Dr. Gonzalez said.
She said she finds it helpful to teach them about the importance of reading labels. That is, looking at the ingredients, and looking for SPF above 30, broad-spectrum coverage, and water resistance. She also recommends providing a list or images of good options, and circling the specific preferred products.
For babies, she finds stick sunscreens most useful for application.
“I generally don’t recommend sprays, but if they’re going to use a spray – and parents love sprays because they are easy to apply – I recommend the ones that have some zinc oxide in them, so that when they apply them they can see where they’re going on the skin,” Dr. Gonzalez said.
Tell patients to apply sunscreen before leaving the house, she advised, adding that making sunscreen application part of a daily routine helps encourage healthy behaviors, as does allowing children, at the right age, to participate in sunscreen application.
For adolescents, avoid scare tactics such as warning about skin cancer. Rather, focus on benefits of avoiding the sun, help them find a product they like by finding out why they don’t like a particular product and recommending an alternative, then following up on that when they come back in, she suggested.
“I really try to address it at every visit,” Dr. Gonzalez said.
“Finally, the most important message is that sunscreen is really just one part of complete sun protection,” she said, noting that specific information about where to buy sun-protective clothing and hats is also important.
Dr. Gonzalez reported serving as a speaker and/or advisory board member and receiving honoraria from Pierre Fabre Dermatologie, Anacor Pharmaceuticals, Encore Dermatology, and PuraCap Pharmaceutical.
EXPERT ANALYSIS FROM THE AAD SUMMER ACADEMY 2016
Active surveillance may be feasible for some advanced RCC patients
For metastatic renal cell carcinoma patients with fewer adverse risk factors and fewer metastatic disease sites, initial active surveillance may be a safe and feasible approach to delay the toxicities of systemic therapy, investigators report.
Fifty-two patients with treatment-naive, asymptomatic, metastatic renal-cell carcinoma were enrolled in a prospective phase II trial and radiographically assessed at baseline, every 3 months for year 1, every 4 months for year 2, then every 6 months thereafter. Patients continued on observation until the treating physician and patient made the decision to initiate systemic therapy.

Median follow-up time was 38.1 months and median time on surveillance before treatment initiation – the primary endpoint of the study – was 14.9 months (95% confidence interval, 10.6-25.0), reported Brian Rini, MD, of the Cleveland Clinic Taussig Cancer Institute and his associates (Lancet Oncol. 2016. doi: 10.1016/S1470-2045(16)30196-6).
Forty-three (90%) of the 48 evaluable patients experienced disease progression during the study, median time to progression was 9.4 months, and 22 patients died from renal cell carcinoma. One patient developed brain metastases and died without receiving systemic therapy. In multivariable analysis, only the number of involved organs (P = .0414) and number of International Metastatic Database Consortium risk factors (P = .0403) were independently prognostic.
Using this analysis, Dr. Rini and associates identified two prognostic groups – a favorable group consisting of patients with no or one International Metastatic Database Consortium (IMDC) risk factors and two or fewer organs with metastatic disease, and an unfavorable group consisting of all other patients. The favorable group (n = 22) patients had an estimated median surveillance time of 22.2 months (95% CI, 13.8-33.3), whereas the unfavorable group (n = 19) had an estimated median surveillance time of 8.4 months (3.2-14.1; P = .0056).
Anxiety, depression, and quality of life did not change significantly over the period of surveillance, suggesting that living with untreated cancer did not cause psychological harm to patients in this study.
“Findings from our prospective trial show active surveillance to be a viable initial strategy in some patients with metastatic renal-cell carcinoma. The median surveillance period before start of systematic therapy was greater than 1 year, with no observed adverse effects on quality of life, anxiety and depression,” Dr. Rini and his associates said.
“Appropriate selection of patients and adequate monitoring, which should include CNS surveillance, is crucial in application of this approach,” they added.
This study was unfunded. One investigator reported receiving financial compensation from Pfizer and GlaxoSmithKline. All other investigators reported having no relevant disclosures.
On Twitter @jessnicolecraig
For metastatic renal cell carcinoma patients with fewer adverse risk factors and fewer metastatic disease sites, initial active surveillance may be a safe and feasible approach to delay the toxicities of systemic therapy, investigators report.
Fifty-two patients with treatment-naive, asymptomatic, metastatic renal-cell carcinoma were enrolled in a prospective phase II trial and radiographically assessed at baseline, every 3 months for year 1, every 4 months for year 2, then every 6 months thereafter. Patients continued on observation until the treating physician and patient made the decision to initiate systemic therapy.
Median follow-up time was 38.1 months and median time on surveillance before treatment initiation – the primary endpoint of the study – was 14.9 months (95% confidence interval, 10.6-25.0), reported Brian Rini, MD, of the Cleveland Clinic Taussig Cancer Institute and his associates (Lancet Oncol. 2016. doi: 10.1016/S1470-2045(16)30196-6).
Forty-three (90%) of the 48 evaluable patients experienced disease progression during the study, median time to progression was 9.4 months, and 22 patients died from renal cell carcinoma. One patient developed brain metastases and died without receiving systemic therapy. In multivariable analysis, only the number of involved organs (P = .0414) and number of International Metastatic Database Consortium risk factors (P = .0403) were independently prognostic.
Using this analysis, Dr. Rini and associates identified two prognostic groups – a favorable group consisting of patients with no or one International Metastatic Database Consortium (IMDC) risk factors and two or fewer organs with metastatic disease, and an unfavorable group consisting of all other patients. The favorable group (n = 22) patients had an estimated median surveillance time of 22.2 months (95% CI, 13.8-33.3), whereas the unfavorable group (n = 19) had an estimated median surveillance time of 8.4 months (3.2-14.1; P = .0056).
Anxiety, depression, and quality of life did not change significantly over the period of surveillance, suggesting that living with untreated cancer did not cause psychological harm to patients in this study.
“Findings from our prospective trial show active surveillance to be a viable initial strategy in some patients with metastatic renal-cell carcinoma. The median surveillance period before start of systematic therapy was greater than 1 year, with no observed adverse effects on quality of life, anxiety and depression,” Dr. Rini and his associates said.
“Appropriate selection of patients and adequate monitoring, which should include CNS surveillance, is crucial in application of this approach,” they added.
This study was unfunded. One investigator reported receiving financial compensation from Pfizer and GlaxoSmithKline. All other investigators reported having no relevant disclosures.
On Twitter @jessnicolecraig
For metastatic renal cell carcinoma patients with fewer adverse risk factors and fewer metastatic disease sites, initial active surveillance may be a safe and feasible approach to delay the toxicities of systemic therapy, investigators report.
Fifty-two patients with treatment-naive, asymptomatic, metastatic renal-cell carcinoma were enrolled in a prospective phase II trial and radiographically assessed at baseline, every 3 months for year 1, every 4 months for year 2, then every 6 months thereafter. Patients continued on observation until the treating physician and patient made the decision to initiate systemic therapy.
Median follow-up time was 38.1 months and median time on surveillance before treatment initiation – the primary endpoint of the study – was 14.9 months (95% confidence interval, 10.6-25.0), reported Brian Rini, MD, of the Cleveland Clinic Taussig Cancer Institute and his associates (Lancet Oncol. 2016. doi: 10.1016/S1470-2045(16)30196-6).
Forty-three (90%) of the 48 evaluable patients experienced disease progression during the study, median time to progression was 9.4 months, and 22 patients died from renal cell carcinoma. One patient developed brain metastases and died without receiving systemic therapy. In multivariable analysis, only the number of involved organs (P = .0414) and number of International Metastatic Database Consortium risk factors (P = .0403) were independently prognostic.
Using this analysis, Dr. Rini and associates identified two prognostic groups – a favorable group consisting of patients with no or one International Metastatic Database Consortium (IMDC) risk factors and two or fewer organs with metastatic disease, and an unfavorable group consisting of all other patients. The favorable group (n = 22) patients had an estimated median surveillance time of 22.2 months (95% CI, 13.8-33.3), whereas the unfavorable group (n = 19) had an estimated median surveillance time of 8.4 months (3.2-14.1; P = .0056).
Anxiety, depression, and quality of life did not change significantly over the period of surveillance, suggesting that living with untreated cancer did not cause psychological harm to patients in this study.
“Findings from our prospective trial show active surveillance to be a viable initial strategy in some patients with metastatic renal-cell carcinoma. The median surveillance period before start of systematic therapy was greater than 1 year, with no observed adverse effects on quality of life, anxiety and depression,” Dr. Rini and his associates said.
“Appropriate selection of patients and adequate monitoring, which should include CNS surveillance, is crucial in application of this approach,” they added.
This study was unfunded. One investigator reported receiving financial compensation from Pfizer and GlaxoSmithKline. All other investigators reported having no relevant disclosures.
On Twitter @jessnicolecraig
FROM LANCET ONCOLOGY
Key clinical point: For a subset of patients with metastatic renal cell carcinoma, initial active surveillance of metastasis may be a safe and feasible option.
Major finding: The favorable group (n = 22) patients and had an estimated median surveillance time of 22.2 months (95% CI, 13.8-33.3), whereas the unfavorable group (n = 19) had an estimated median surveillance time of 8.4 months (3.2-14.1; P = .0056)
Data source: A prospective phase II trial involving 52 patients with treatment-naive, metastatic renal cell carcinoma.
Disclosures: This study was unfunded. One investigator reported receiving financial compensation from Pfizer and GlaxoSmithKline. All other investigators reported having no relevant disclosures.

CDC reports two new cases of Zika-related birth defects
Two live-born infants with Zika virus–related birth defects were reported the week ending Aug. 4, 2016, bringing the U.S. total to 17, the Centers for Disease Control and Prevention reported.
One of the two infants was born in the 50 states and the District of Columbia, and the other was the first live-born infant with Zika-related birth defects born in the U.S. territories. State- or territorial-level data are not being reported to protect the privacy of affected women and children, the CDC said.
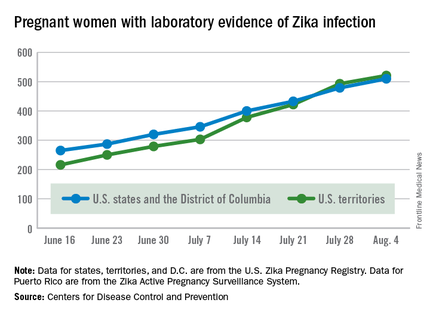
There were no new pregnancy losses among Zika-infected women in the territories, keeping the total at one for the year, but the CDC seems to have adjusted the number of pregnancy losses for the states and D.C., as the most recent total for the year is now five, after six were reported the previous week.
A total of 59 new cases of Zika infection in pregnant women were reported for the week ending Aug. 4: 31 new cases in the states/D.C. and 28 in the territories. For the year, there have been 510 cases of pregnant women with laboratory evidence of Zika infection in the states/D.C. and 521 in the territories, for a U.S. total of 1,031, the CDC reported.
The figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
Zika virus–related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
Two live-born infants with Zika virus–related birth defects were reported the week ending Aug. 4, 2016, bringing the U.S. total to 17, the Centers for Disease Control and Prevention reported.
One of the two infants was born in the 50 states and the District of Columbia, and the other was the first live-born infant with Zika-related birth defects born in the U.S. territories. State- or territorial-level data are not being reported to protect the privacy of affected women and children, the CDC said.
There were no new pregnancy losses among Zika-infected women in the territories, keeping the total at one for the year, but the CDC seems to have adjusted the number of pregnancy losses for the states and D.C., as the most recent total for the year is now five, after six were reported the previous week.
A total of 59 new cases of Zika infection in pregnant women were reported for the week ending Aug. 4: 31 new cases in the states/D.C. and 28 in the territories. For the year, there have been 510 cases of pregnant women with laboratory evidence of Zika infection in the states/D.C. and 521 in the territories, for a U.S. total of 1,031, the CDC reported.
The figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
Zika virus–related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
Two live-born infants with Zika virus–related birth defects were reported the week ending Aug. 4, 2016, bringing the U.S. total to 17, the Centers for Disease Control and Prevention reported.
One of the two infants was born in the 50 states and the District of Columbia, and the other was the first live-born infant with Zika-related birth defects born in the U.S. territories. State- or territorial-level data are not being reported to protect the privacy of affected women and children, the CDC said.
There were no new pregnancy losses among Zika-infected women in the territories, keeping the total at one for the year, but the CDC seems to have adjusted the number of pregnancy losses for the states and D.C., as the most recent total for the year is now five, after six were reported the previous week.
A total of 59 new cases of Zika infection in pregnant women were reported for the week ending Aug. 4: 31 new cases in the states/D.C. and 28 in the territories. For the year, there have been 510 cases of pregnant women with laboratory evidence of Zika infection in the states/D.C. and 521 in the territories, for a U.S. total of 1,031, the CDC reported.
The figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
Zika virus–related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
FDA approves generic version of Tamiflu
The Food and Drug Administration has approved the first generic version of Tamiflu (oseltamivir phosphate), a medication for the treatment of influenza A and B.
The announcement was made Aug. 3, 2016, on the Drugs@FDA website and in an email from the FDA’s Division of Drug Information (DDI). Tamiflu was first approved in 1999.
Oseltamivir phosphate is intended for use in patients 2 weeks of age and older who have had flu symptoms for no more than 48 hours, and for prevention of influenza in patients 1 year of age and older. According to the FDA, the drug does not treat or prevent illness caused by viral infections other than the influenza virus, and does not prevent bacterial infections that may happen with the flu.

Products in the FDA generic approval application submitted by Natco Pharma Ltd., an India-based drug company, include the oral capsule form of the drug, in 30-, 45-, and 75-mg strengths.
The FDA acknowledged in its approval that it does not know if oseltamivir phosphate is effective in patients who start treatment after 2 days of developing symptoms, or have weakened immune systems. The most common side effects reported by patients using oseltamivir phosphate in clinical trials included nausea and vomiting.
For more information on oseltamivir phosphate, see the Tamiflu drug label.
On Twitter @richpizzi
The Food and Drug Administration has approved the first generic version of Tamiflu (oseltamivir phosphate), a medication for the treatment of influenza A and B.
The announcement was made Aug. 3, 2016, on the Drugs@FDA website and in an email from the FDA’s Division of Drug Information (DDI). Tamiflu was first approved in 1999.
Oseltamivir phosphate is intended for use in patients 2 weeks of age and older who have had flu symptoms for no more than 48 hours, and for prevention of influenza in patients 1 year of age and older. According to the FDA, the drug does not treat or prevent illness caused by viral infections other than the influenza virus, and does not prevent bacterial infections that may happen with the flu.
Products in the FDA generic approval application submitted by Natco Pharma Ltd., an India-based drug company, include the oral capsule form of the drug, in 30-, 45-, and 75-mg strengths.
The FDA acknowledged in its approval that it does not know if oseltamivir phosphate is effective in patients who start treatment after 2 days of developing symptoms, or have weakened immune systems. The most common side effects reported by patients using oseltamivir phosphate in clinical trials included nausea and vomiting.
For more information on oseltamivir phosphate, see the Tamiflu drug label.
On Twitter @richpizzi
The Food and Drug Administration has approved the first generic version of Tamiflu (oseltamivir phosphate), a medication for the treatment of influenza A and B.
The announcement was made Aug. 3, 2016, on the Drugs@FDA website and in an email from the FDA’s Division of Drug Information (DDI). Tamiflu was first approved in 1999.
Oseltamivir phosphate is intended for use in patients 2 weeks of age and older who have had flu symptoms for no more than 48 hours, and for prevention of influenza in patients 1 year of age and older. According to the FDA, the drug does not treat or prevent illness caused by viral infections other than the influenza virus, and does not prevent bacterial infections that may happen with the flu.
Products in the FDA generic approval application submitted by Natco Pharma Ltd., an India-based drug company, include the oral capsule form of the drug, in 30-, 45-, and 75-mg strengths.
The FDA acknowledged in its approval that it does not know if oseltamivir phosphate is effective in patients who start treatment after 2 days of developing symptoms, or have weakened immune systems. The most common side effects reported by patients using oseltamivir phosphate in clinical trials included nausea and vomiting.
For more information on oseltamivir phosphate, see the Tamiflu drug label.
On Twitter @richpizzi

Post-TORS neck dissections extend hospital stay
SEATTLE – Oncologic transoral robotic surgery patients spend less time in the hospital if they have neck dissections at the same time, instead of later, according to a review of 441 patients by Stony Brook (N.Y.) University.
The average hospital length of stay (LOS) was 6 days for the 349 patients (79.1%) who had lymphadenectomy neck dissections at the same time as transoral robotic surgery (TORS). The 92 patients (20.9%) who had staged procedures - neck dissections and TORS about a month apart, with TORS usually done first - stayed in the hospital an average of 8 days (P less than .0001). After risk adjustment, LOS was 43% shorter for concurrent dissections.
Cardiac arrhythmias were also more common in staged patients, perhaps because they had general anesthesia twice in a short period or maybe because staged patients were more likely to be obese (18.5% vs. 7.5%; P less than .01).
However, there were no statistically significant outcome differences otherwise, and the investigators concluded that “concurrent and staged procedures are equally safe. It is therefore reasonable to allow operator preference and patient factors to determine surgical logistics.”
Neck dissection timing has been controversial since the advent of TORS several years ago, when surgeons and administrators realized they could fit more cases into the schedule by doing neck dissections, which can take a few hours, at a different time.
Proponents of the staged approach argue, among other things, that it reduces the risk of fistulas and tracheotomies, and allows surgeons a second go at positive margins. Advocates of concurrent procedures counter that fistulas, if found, can be repaired right away, and that same-time surgery saves money, allows for earlier adjuvant therapy, and cuts anesthesia risks.
There hasn’t been much data to settle the debate, and no one has compared LOS before, so it was “important” to look into the matter, lead investigator Catherine Frenkel, MD, a Stony Brook general surgery resident, said at the American Head and Neck Society International Conference on Head and Neck Cancer.
German investigators also recently concluded that it’s pretty much a draw between concurrent and staged dissections. In a study of 41 TORS cases, “the timing of neck dissection did not make a significant difference in the outcomes. We suggest, therefore, that aspiring and established TORS teams do not restrict their appropriate indications due to robotic slot and theatre time constraints, but perform each indicated TORS case as soon as possible within their given systems, even if the neck dissections cannot be done on the same day,” they said (Eur J Surg Oncol. 2015 Jun;41[6]:773-8).
In addition to obese patients, those who had tongue or tonsil lesions were more likely to be staged in the Stony Brook analysis. About half of the surgeons in the study stuck solely to concurrent procedures, while a handful opted for the staged approach, and the rest did both. Perhaps not surprisingly, high-volume surgeons – those who did five or more TORS cases per year – were more likely to stage.
Almost two-thirds of patients had at least one complication, most commonly renal failure, heart problems, extended ventilation, and surgical errors, which included accidental punctures, postop fistulas, hemorrhages, and wound complications. A total of 13% of patients had at least one postop readmission. Apart from arrhythmias, there were no statistically significant differences in complication or 30-day readmission rates between concurrent and staged patients. High-volume surgeons were less likely to have complications.
Postop bleeding was another common problem, and more likely with staged surgeries (12% vs. 7%). Concurrent procedures had a slightly higher rate of new tracheotomies and gastrostomies, but again the differences were not statistically significant, even with pedicle and free-flap reconstruction. There was no outside funding for the work, and the investigators had no relevant conflicts of interest.
SEATTLE – Oncologic transoral robotic surgery patients spend less time in the hospital if they have neck dissections at the same time, instead of later, according to a review of 441 patients by Stony Brook (N.Y.) University.
The average hospital length of stay (LOS) was 6 days for the 349 patients (79.1%) who had lymphadenectomy neck dissections at the same time as transoral robotic surgery (TORS). The 92 patients (20.9%) who had staged procedures - neck dissections and TORS about a month apart, with TORS usually done first - stayed in the hospital an average of 8 days (P less than .0001). After risk adjustment, LOS was 43% shorter for concurrent dissections.
Cardiac arrhythmias were also more common in staged patients, perhaps because they had general anesthesia twice in a short period or maybe because staged patients were more likely to be obese (18.5% vs. 7.5%; P less than .01).
However, there were no statistically significant outcome differences otherwise, and the investigators concluded that “concurrent and staged procedures are equally safe. It is therefore reasonable to allow operator preference and patient factors to determine surgical logistics.”
Neck dissection timing has been controversial since the advent of TORS several years ago, when surgeons and administrators realized they could fit more cases into the schedule by doing neck dissections, which can take a few hours, at a different time.
Proponents of the staged approach argue, among other things, that it reduces the risk of fistulas and tracheotomies, and allows surgeons a second go at positive margins. Advocates of concurrent procedures counter that fistulas, if found, can be repaired right away, and that same-time surgery saves money, allows for earlier adjuvant therapy, and cuts anesthesia risks.
There hasn’t been much data to settle the debate, and no one has compared LOS before, so it was “important” to look into the matter, lead investigator Catherine Frenkel, MD, a Stony Brook general surgery resident, said at the American Head and Neck Society International Conference on Head and Neck Cancer.
German investigators also recently concluded that it’s pretty much a draw between concurrent and staged dissections. In a study of 41 TORS cases, “the timing of neck dissection did not make a significant difference in the outcomes. We suggest, therefore, that aspiring and established TORS teams do not restrict their appropriate indications due to robotic slot and theatre time constraints, but perform each indicated TORS case as soon as possible within their given systems, even if the neck dissections cannot be done on the same day,” they said (Eur J Surg Oncol. 2015 Jun;41[6]:773-8).
In addition to obese patients, those who had tongue or tonsil lesions were more likely to be staged in the Stony Brook analysis. About half of the surgeons in the study stuck solely to concurrent procedures, while a handful opted for the staged approach, and the rest did both. Perhaps not surprisingly, high-volume surgeons – those who did five or more TORS cases per year – were more likely to stage.
Almost two-thirds of patients had at least one complication, most commonly renal failure, heart problems, extended ventilation, and surgical errors, which included accidental punctures, postop fistulas, hemorrhages, and wound complications. A total of 13% of patients had at least one postop readmission. Apart from arrhythmias, there were no statistically significant differences in complication or 30-day readmission rates between concurrent and staged patients. High-volume surgeons were less likely to have complications.
Postop bleeding was another common problem, and more likely with staged surgeries (12% vs. 7%). Concurrent procedures had a slightly higher rate of new tracheotomies and gastrostomies, but again the differences were not statistically significant, even with pedicle and free-flap reconstruction. There was no outside funding for the work, and the investigators had no relevant conflicts of interest.
SEATTLE – Oncologic transoral robotic surgery patients spend less time in the hospital if they have neck dissections at the same time, instead of later, according to a review of 441 patients by Stony Brook (N.Y.) University.
The average hospital length of stay (LOS) was 6 days for the 349 patients (79.1%) who had lymphadenectomy neck dissections at the same time as transoral robotic surgery (TORS). The 92 patients (20.9%) who had staged procedures - neck dissections and TORS about a month apart, with TORS usually done first - stayed in the hospital an average of 8 days (P less than .0001). After risk adjustment, LOS was 43% shorter for concurrent dissections.
Cardiac arrhythmias were also more common in staged patients, perhaps because they had general anesthesia twice in a short period or maybe because staged patients were more likely to be obese (18.5% vs. 7.5%; P less than .01).
However, there were no statistically significant outcome differences otherwise, and the investigators concluded that “concurrent and staged procedures are equally safe. It is therefore reasonable to allow operator preference and patient factors to determine surgical logistics.”
Neck dissection timing has been controversial since the advent of TORS several years ago, when surgeons and administrators realized they could fit more cases into the schedule by doing neck dissections, which can take a few hours, at a different time.
Proponents of the staged approach argue, among other things, that it reduces the risk of fistulas and tracheotomies, and allows surgeons a second go at positive margins. Advocates of concurrent procedures counter that fistulas, if found, can be repaired right away, and that same-time surgery saves money, allows for earlier adjuvant therapy, and cuts anesthesia risks.
There hasn’t been much data to settle the debate, and no one has compared LOS before, so it was “important” to look into the matter, lead investigator Catherine Frenkel, MD, a Stony Brook general surgery resident, said at the American Head and Neck Society International Conference on Head and Neck Cancer.
German investigators also recently concluded that it’s pretty much a draw between concurrent and staged dissections. In a study of 41 TORS cases, “the timing of neck dissection did not make a significant difference in the outcomes. We suggest, therefore, that aspiring and established TORS teams do not restrict their appropriate indications due to robotic slot and theatre time constraints, but perform each indicated TORS case as soon as possible within their given systems, even if the neck dissections cannot be done on the same day,” they said (Eur J Surg Oncol. 2015 Jun;41[6]:773-8).
In addition to obese patients, those who had tongue or tonsil lesions were more likely to be staged in the Stony Brook analysis. About half of the surgeons in the study stuck solely to concurrent procedures, while a handful opted for the staged approach, and the rest did both. Perhaps not surprisingly, high-volume surgeons – those who did five or more TORS cases per year – were more likely to stage.
Almost two-thirds of patients had at least one complication, most commonly renal failure, heart problems, extended ventilation, and surgical errors, which included accidental punctures, postop fistulas, hemorrhages, and wound complications. A total of 13% of patients had at least one postop readmission. Apart from arrhythmias, there were no statistically significant differences in complication or 30-day readmission rates between concurrent and staged patients. High-volume surgeons were less likely to have complications.
Postop bleeding was another common problem, and more likely with staged surgeries (12% vs. 7%). Concurrent procedures had a slightly higher rate of new tracheotomies and gastrostomies, but again the differences were not statistically significant, even with pedicle and free-flap reconstruction. There was no outside funding for the work, and the investigators had no relevant conflicts of interest.
AT THE INTERNATIONAL CONFERENCE ON HEAD AND NECK CANCER
Key clinical point: Oncologic transoral robotic surgery patients spend less time in the hospital if they have neck dissections at the same time, instead of later.
Major finding: The average hospital length of stay was 6 days for the 349 patients (79.1%) who had lymphadenectomy neck dissections at the same time as TORS. The 92 patients (20.9%) who had staged procedures – neck dissections and TORS about a month apart, with TORS usually done first – stayed in the hospital an average of 8 days (P less than .0001).
Data source: Review of 441 TORS patients.
Disclosures: There was no outside funding for the work, and the investigators had no relevant conflicts of interest.