User login
VIDEO: The surgical treatment of pelvic congestion
BY CHARLES E. MILLER, MD
Chronic pelvic pain is described as the presence of lower abdominal or pelvic pain for longer than 6 months. It is believed to affect approximately one in six women and 12%-15% of women of reproductive age. The diagnosis and treatment of chronic pelvic pain adds as much as a $2 billion burden to our health system annually.
It was first described clinically in the literature in 1857, while the existence of pelvic varicosities wasn’t documented for nearly another 100 years. Pelvic congestion syndrome (PCS) accounts for 30%-70% of cases presenting with chronic pelvic pain. PCS can be due to pelvic venous insufficiency, characterized by reflux into pelvic veins leading to pelvic varicosities or alternative venous pathways secondary to varicose veins of the leg.
Other etiologies of PCS include nutcracker syndrome (left renal vein compressed between the aorta and the superior mesenteric artery), May-Thurner syndrome (compression of the left common iliac vein by the right common iliac artery) or, less likely, tumor thrombosis of the inferior vena cava, portal vein thrombosis, renal cell carcinoma, left renal thrombosis, or left kidney arterial-venous fistula.

While there appears to be significant literature indicating a long-term success rate of greater than 80% in patients treated by percutaneous endovascular procedures (embolization, stenting), there is far less information on the postsurgical success of blocking the varicose gonadal vein. Nevertheless, our long-term results with gonadal vein clipping is virtually the same as that of our radiological colleagues.
It is a pleasure to welcome Courtney Steller, DO, to this edition of the Master Class in Gynecologic Surgery to discuss the diagnosis and treatment of PCS, with an emphasis on surgical correction.
Dr. Steller is a recent graduate of the AAGL/SRS Fellowship in Minimally Invasive Gynecologic Surgery at Advocate Lutheran General Hospital, Park Ridge, Ill. She is currently in private practice and is an associate at the Family Health Centers of San Diego, Calif.
Dr. Miller is clinical associate professor at the University of Illinois at Chicago, and past president of the AAGL and the International Society for Gynecologic Endoscopy. He is a reproductive endocrinologist and minimally invasive gynecologic surgeon in private practice in Naperville and Schaumburg, Ill.; director of minimally invasive gynecologic surgery and the director of the AAGL/SRS Fellowship in Minimally Invasive Gynecologic Surgery at Advocate Lutheran General Hospital, Park Ridge, Ill.; and the medical editor of this column, Master Class. He reported having no financial disclosures relevant to this column. Email him at [email protected].
Pelvic congestion syndrome: A treatable cause of pain
BY COURTNEY STELLER, DO
Pelvic congestion syndrome is a poorly understood and underdiagnosed disease. Yet, over the last decade, the syndrome has become less controversial as the etiology has become better understood and as the diagnostic approach has become more specific. Through these advances, treatments have also become increasingly more successful.
This is an important shift, because the chronic pelvic pain experienced by patients with pelvic congestion significantly impacts their quality of life and well-being. As the pain persists, it can become exceedingly difficult to manage. Many patients we have ultimately treated for pelvic congestion syndrome have had years of various work-ups, significant diagnostic investigations, and trials of different treatments without having any cause of their pain identified or achieving any lasting symptom relief.

The pelvic pain in patients with pelvic congestion syndrome (PCS) can be noncyclical or cyclical. It is present most of the time but tends to get worse at the end of the day and after long periods of standing and/or sitting. The pain also may worsen with intercourse, largely afterward. The syndrome tends to occur in premenopausal and multiparous women, but it’s important to appreciate that this is not always the case; we have diagnosed and treated PCS in several young, nulliparous patients as well.
Features and diagnosis
PCS is a disorder of pelvic venous circulation that predominantly affects the ovarian veins. It is sometimes referred to as pelvic vein incompetence or pelvic vascular dysfunction. Just as veins in the legs can enlarge and become varicose, the ovarian veins – and sometimes the internal iliac veins – can become incompetent and unable to effectively return blood back to the heart.
Pregnancy may predispose patients to developing the abnormally dilated and refluxing veins that characterize PCS, as the increase in pelvic vein capacity and uterine compression can lead to significant stasis of blood in the pelvis and subsequent damage to the veins and the venous valves. There also is believed to be an estrogen component to the development of PCS, because estrogen is known to act as a vasodilator. Moreover, a congenital absence and incompetence of venous valves in some cases has been reported.
In a recent study looking at pelvic vein incompetence and symptoms of chronic pelvic pain, these women were reported to have a distinctive symptom profile, with the “most notable” features being the presence of dull pelvic pain that radiates to the upper thighs and is aggravated by prolonged standing and walking – symptoms that are similar to the leg symptoms experienced by patients with severe varicose veins (Eur J Obstet Gynecol Reprod Biol. 2016 Jan;196:21-5).
Other investigators have similarly described the pelvic pain related to PCS as a dull ache or heaviness sensation that is most severe at the end of the day and that is lessened with supine positioning (though not necessarily immediately) and often exacerbated with sexual intercourse, especially post coitus. These descriptions are in line with my experience with PCS. There is usually exquisite tenderness on pelvic exam, especially localized to the adnexa. Patients will often have varicose veins on their upper legs or labia.
Interestingly, it has been repeatedly shown that many women have dilated and incompetent pelvic veins without also having such pathognomonic pain. We therefore cannot treat women based solely on the finding of abnormal veins.
On the other hand we must determine which patients with chronic pelvic pain have PCS. The differential diagnosis for PCS includes endometriosis, adenomyosis chronic pelvic inflammatory disease, adhesive disease, adnexal masses, adnexal torsion, and several nongynecologic diseases including interstitial cystitis and irritable bowel syndrome.
Venography has become the gold standard for diagnosing pelvic congestion. The procedure involves catheterization of the ovarian veins through a femoral or jugular approach. In our experience, the common femoral vein is the more frequently used access point. Using a contrast injection, the interventional radiologist can assess the degree of venous dilation and reflux in the pelvis.

There currently is no consensus on a cutoff for vein diameter or on any validated measures for congestion. According to one report on PCS authored by interventional radiologists, the diagnosis of PCS is confirmed with the venographic findings of ovarian vein diameter greater than 6 mm, retrograde ovarian or pelvic venous flow, presence of several tortuous collateral pelvic venous pathways, and delayed or stagnant clearance on contrast (Semin Intervent Radiol. 2008 Dec;25[4]:361-8).
The criteria vary, however. A recent literature review on pelvic congestion syndrome by Chiara Borghi, MD, and Lucio Dell’Atti, MD, states that incompetent pelvic veins are defined as more than 5-10 mm in diameter (Arch Gynecol Obstet. 2016 Feb;293[2]:291-301).
To more accurately diagnose PCS, our patients undergo tilt-table venography. The patient is placed into a reverse-Trendelenburg upright or semi-upright position to potentially exacerbate any venous reflux or dilation.
Other methods of identifying and diagnosing pelvic congestion have included transabdominal and transvaginal ultrasound, CT, and MRI. While CT and MRI both offer an overview of the pelvic vasculature and are helpful for ruling out other causes of chronic pelvic pain, they have low specificity for pelvic varices, according to the Italian review.
Sonography performed in the supine position, on the other hand, appears to be increasingly viewed as an acceptable screening tool for determining which patients may ultimately benefit from venography. It is also important in evaluation to rule out other pathologies not yet excluded. However, it should not be used for diagnosis of PCS.
Treating PCS
There are two main approaches to treating PCS: venous ligation (a gynecologic surgical approach) and percutaneous transcatheter embolization (performed by interventional radiologists).
The literature and evidence base is still in its infancy, but is growing. In our experience, both approaches lead to good resolution of symptoms over time in the majority of patients, and appear superior to the medical therapies that have been proposed for treating PCS, such as progestins and gonadotropin-releasing hormone agonists. Success rates with medical therapy are more variable and appear to be more short lived.
A review published this year on the effectiveness of embolization of pelvic veins for reducing chronic pelvic pain showed that 75% of women undergoing embolization had symptomatic relief that generally increased over time and was sustained. The authors concluded that embolization appears to be effective for the majority of women, and is safe, although they also noted that the quality of the evidence is low (J Vasc Interv Radiol. 2016 Oct;27[10]:1478-86.e8). Their review was based almost entirely on prospective case series.
Dr. Borghi and Dr. Dell’Atti offered a similar assessment of embolization for PCS, stating in their review article that clinical success has been reported in 70%-85% of patients. They also report nearly equivalent success rates of up to 75% with treatment via surgical ligation of ovarian and/or pelvic vasculature. These findings are from mostly observational data and case series.
Decisions about which approach to take should be individualized. If there are no differences with respect to insurance coverage for the patient, then embolization may be the preferred approach because it is the most minimally invasive technique and can potentially be performed at the time of diagnostic venography, negating the need for a second procedure. A skilled interventional radiologist familiar with the disease and the treatment is necessary. Various embolic agents are utilized, including coils, glues, foams, and other agents that cause sclerosis of the abnormal veins.
In other cases, venous ligation is preferred, especially when an additional gynecologic surgery, such as a cystectomy or myomectomy, is required.
Surgical ligation of ovarian veins was initially performed via laparotomy using a traditional retroperitoneal approach. The surgical goal is to isolate the ovarian vein significantly above the pelvic brim and before the vein becomes substantially dilated. Laparotomy therefore requires a vertical mid-line incision to provide adequate access to the appropriate portion of the ovarian vessels, leading to potentially high morbidity and poor cosmesis.
More recently, gynecologic surgeons skilled in laparoscopy have successfully managed PCS transperitoneally. A few small series of bilateral laparoscopic transperitoneal ligation of ovarian veins have been reported, including one by Tigellio Gargiulo, MD, who clipped both veins in their upper third, near their distal ends at the inferior vena cava (right) and the renal vein (left) (J Am Assoc Gynecol Laparosc. 2003 Nov;10[4]:501-4).
We prefer a robot-assisted laparoscopic approach for most of our patients. Not only does the improved dexterity help while working with sensitive vasculature, but more importantly we are able to use Firefly fluorescence.
The procedure generally is as follows. The uterine adnexa on the affected side is grasped and placed on tension so that the infundibulopelvic (IP) ligament can be visualized as it courses up and above the pelvic brim. The peritoneum immediately over the IP ligament is gently grasped and tented upward, and a small incision is made into the peritoneum, providing access into the retroperitoneum. The ureter should be visualized medial to this dissection.
The peritoneal tissue is then gently dissected off the ovarian vessels. Once the vessels are freed from the peritoneal tissue, the dilated ovarian vein is often clearly visualized. It is important to note that if no venous dilation is seen during laparoscopy, the procedure should not be aborted. Due to the Trendelenburg position that is utilized in gynecologic – and especially laparoscopic – surgery, the venous system sometimes appears falsely “normal” at this time.
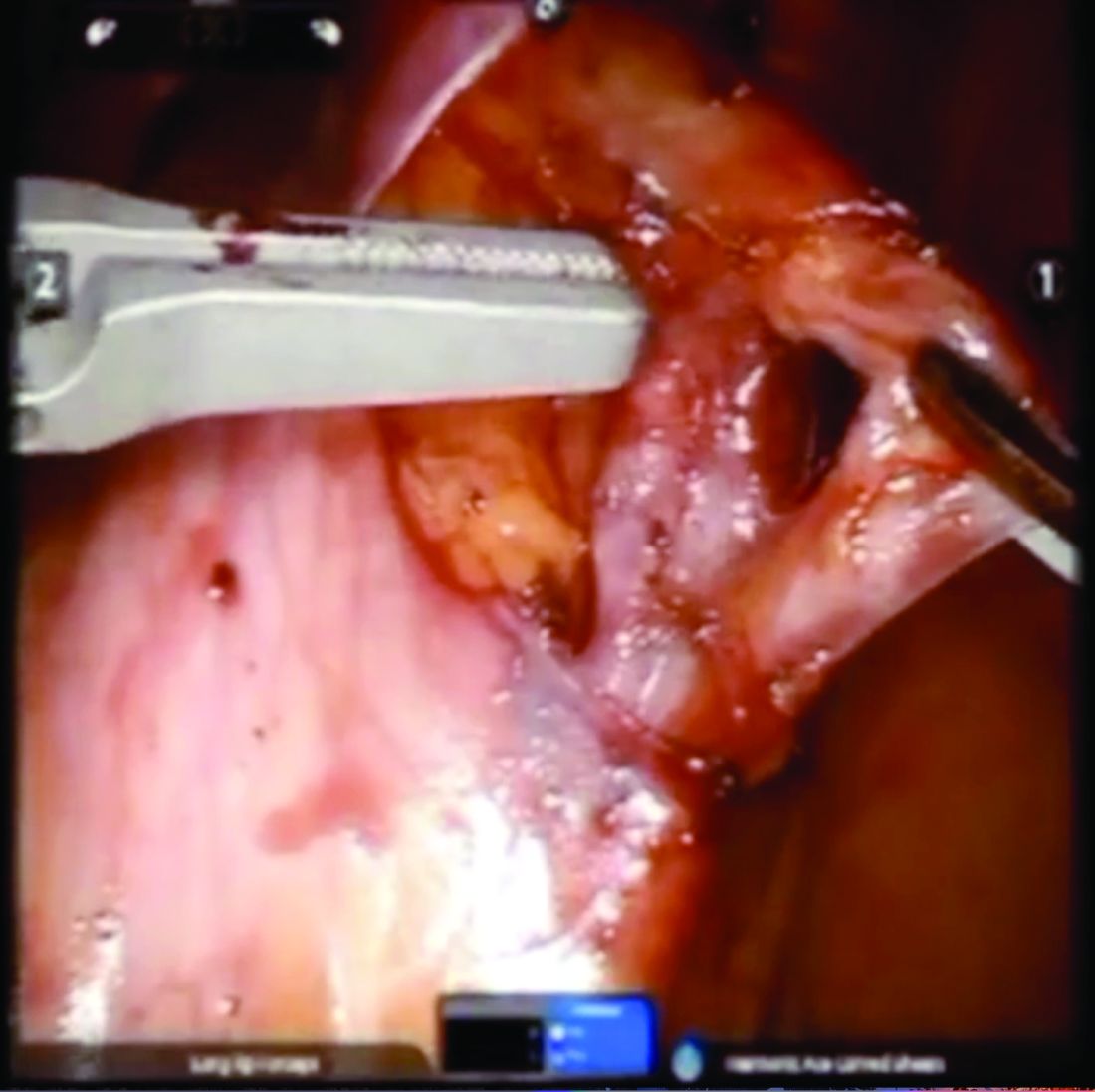
Once the ovarian vessels have been isolated, the arteries must be separated from the veins. The adventitial tissue is dissected until the vessels are separated. Great care should be taken to ensure that all movements run parallel to the vessels and not perpendicular, therefore decreasing the risk of bleeding.
This process can be challenging. The surgeon is working with delicate vasculature. Often there are several branches from the vein that have formed due to the abnormal venous system. The best way to approach it is to identify planes and separate those planes in order to isolate individual vessels. If difficulties are still encountered, the surgeon should restart the dissection higher.
Once the dilated ovarian vein is isolated, one to two clips are placed.
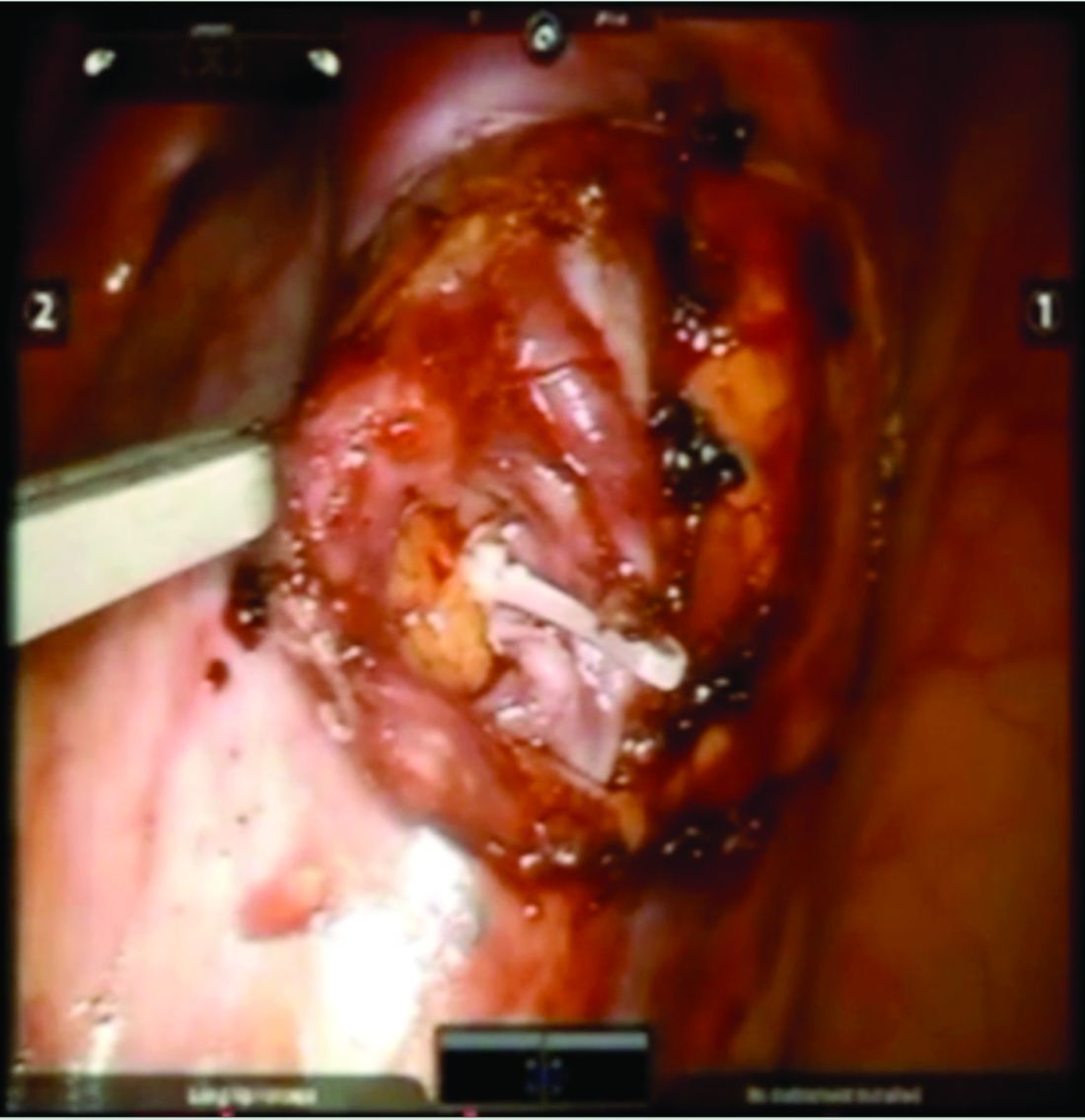
Usually the artery is clearly distinct from the vein as it is smaller, more elastic, and can be seen pulsing. However, occasionally it is difficult to distinguish. In these cases, assistance with the da Vinci surgical system is useful: Indocyanine green (ICG) dye can be injected intravenously and visualized with a near-infrared light on the da Vinci platform. The dye is then seen glowing green as it first courses through the artery and then the vein.
For patients who have been found on venography to have bilateral disease, we perform the ligation procedure bilaterally. Once ligation is complete, the more competent collateral veins in the pelvis will assume more of the venous circulation.
In our experience, patients have ultimately noted substantial pain relief after these procedures, both with the endoscopic embolization and the surgical ligation. Patients are counseled that it can take several months to notice a relief in the pain.
In rare cases, pelvic congestion is related to extrinsic compression. For instance, the left renal vein can become compressed between the aorta and the superior mesenteric artery (the nutcracker syndrome), or the left common iliac vein can be compressed between the overlying right internal iliac artery and the underlying vertebral body (May-Thurner syndrome). Both of these conditions can lead to secondary PCS.
Such complex conditions are usually treated by vascular surgeons. May-Thurner syndrome is treated via stenting, while nutcracker syndrome can be treated with stenting or transposition of the renal vein to the distal vena cava.
Dr. Steller is an associate at the Family Health Centers of San Diego. She reported having no relevant financial disclosures.
BY CHARLES E. MILLER, MD
Chronic pelvic pain is described as the presence of lower abdominal or pelvic pain for longer than 6 months. It is believed to affect approximately one in six women and 12%-15% of women of reproductive age. The diagnosis and treatment of chronic pelvic pain adds as much as a $2 billion burden to our health system annually.
It was first described clinically in the literature in 1857, while the existence of pelvic varicosities wasn’t documented for nearly another 100 years. Pelvic congestion syndrome (PCS) accounts for 30%-70% of cases presenting with chronic pelvic pain. PCS can be due to pelvic venous insufficiency, characterized by reflux into pelvic veins leading to pelvic varicosities or alternative venous pathways secondary to varicose veins of the leg.
Other etiologies of PCS include nutcracker syndrome (left renal vein compressed between the aorta and the superior mesenteric artery), May-Thurner syndrome (compression of the left common iliac vein by the right common iliac artery) or, less likely, tumor thrombosis of the inferior vena cava, portal vein thrombosis, renal cell carcinoma, left renal thrombosis, or left kidney arterial-venous fistula.
While there appears to be significant literature indicating a long-term success rate of greater than 80% in patients treated by percutaneous endovascular procedures (embolization, stenting), there is far less information on the postsurgical success of blocking the varicose gonadal vein. Nevertheless, our long-term results with gonadal vein clipping is virtually the same as that of our radiological colleagues.
It is a pleasure to welcome Courtney Steller, DO, to this edition of the Master Class in Gynecologic Surgery to discuss the diagnosis and treatment of PCS, with an emphasis on surgical correction.
Dr. Steller is a recent graduate of the AAGL/SRS Fellowship in Minimally Invasive Gynecologic Surgery at Advocate Lutheran General Hospital, Park Ridge, Ill. She is currently in private practice and is an associate at the Family Health Centers of San Diego, Calif.
Dr. Miller is clinical associate professor at the University of Illinois at Chicago, and past president of the AAGL and the International Society for Gynecologic Endoscopy. He is a reproductive endocrinologist and minimally invasive gynecologic surgeon in private practice in Naperville and Schaumburg, Ill.; director of minimally invasive gynecologic surgery and the director of the AAGL/SRS Fellowship in Minimally Invasive Gynecologic Surgery at Advocate Lutheran General Hospital, Park Ridge, Ill.; and the medical editor of this column, Master Class. He reported having no financial disclosures relevant to this column. Email him at [email protected].
Pelvic congestion syndrome: A treatable cause of pain
BY COURTNEY STELLER, DO
Pelvic congestion syndrome is a poorly understood and underdiagnosed disease. Yet, over the last decade, the syndrome has become less controversial as the etiology has become better understood and as the diagnostic approach has become more specific. Through these advances, treatments have also become increasingly more successful.
This is an important shift, because the chronic pelvic pain experienced by patients with pelvic congestion significantly impacts their quality of life and well-being. As the pain persists, it can become exceedingly difficult to manage. Many patients we have ultimately treated for pelvic congestion syndrome have had years of various work-ups, significant diagnostic investigations, and trials of different treatments without having any cause of their pain identified or achieving any lasting symptom relief.
The pelvic pain in patients with pelvic congestion syndrome (PCS) can be noncyclical or cyclical. It is present most of the time but tends to get worse at the end of the day and after long periods of standing and/or sitting. The pain also may worsen with intercourse, largely afterward. The syndrome tends to occur in premenopausal and multiparous women, but it’s important to appreciate that this is not always the case; we have diagnosed and treated PCS in several young, nulliparous patients as well.
Features and diagnosis
PCS is a disorder of pelvic venous circulation that predominantly affects the ovarian veins. It is sometimes referred to as pelvic vein incompetence or pelvic vascular dysfunction. Just as veins in the legs can enlarge and become varicose, the ovarian veins – and sometimes the internal iliac veins – can become incompetent and unable to effectively return blood back to the heart.
Pregnancy may predispose patients to developing the abnormally dilated and refluxing veins that characterize PCS, as the increase in pelvic vein capacity and uterine compression can lead to significant stasis of blood in the pelvis and subsequent damage to the veins and the venous valves. There also is believed to be an estrogen component to the development of PCS, because estrogen is known to act as a vasodilator. Moreover, a congenital absence and incompetence of venous valves in some cases has been reported.
In a recent study looking at pelvic vein incompetence and symptoms of chronic pelvic pain, these women were reported to have a distinctive symptom profile, with the “most notable” features being the presence of dull pelvic pain that radiates to the upper thighs and is aggravated by prolonged standing and walking – symptoms that are similar to the leg symptoms experienced by patients with severe varicose veins (Eur J Obstet Gynecol Reprod Biol. 2016 Jan;196:21-5).
Other investigators have similarly described the pelvic pain related to PCS as a dull ache or heaviness sensation that is most severe at the end of the day and that is lessened with supine positioning (though not necessarily immediately) and often exacerbated with sexual intercourse, especially post coitus. These descriptions are in line with my experience with PCS. There is usually exquisite tenderness on pelvic exam, especially localized to the adnexa. Patients will often have varicose veins on their upper legs or labia.
Interestingly, it has been repeatedly shown that many women have dilated and incompetent pelvic veins without also having such pathognomonic pain. We therefore cannot treat women based solely on the finding of abnormal veins.
On the other hand we must determine which patients with chronic pelvic pain have PCS. The differential diagnosis for PCS includes endometriosis, adenomyosis chronic pelvic inflammatory disease, adhesive disease, adnexal masses, adnexal torsion, and several nongynecologic diseases including interstitial cystitis and irritable bowel syndrome.
Venography has become the gold standard for diagnosing pelvic congestion. The procedure involves catheterization of the ovarian veins through a femoral or jugular approach. In our experience, the common femoral vein is the more frequently used access point. Using a contrast injection, the interventional radiologist can assess the degree of venous dilation and reflux in the pelvis.
There currently is no consensus on a cutoff for vein diameter or on any validated measures for congestion. According to one report on PCS authored by interventional radiologists, the diagnosis of PCS is confirmed with the venographic findings of ovarian vein diameter greater than 6 mm, retrograde ovarian or pelvic venous flow, presence of several tortuous collateral pelvic venous pathways, and delayed or stagnant clearance on contrast (Semin Intervent Radiol. 2008 Dec;25[4]:361-8).
The criteria vary, however. A recent literature review on pelvic congestion syndrome by Chiara Borghi, MD, and Lucio Dell’Atti, MD, states that incompetent pelvic veins are defined as more than 5-10 mm in diameter (Arch Gynecol Obstet. 2016 Feb;293[2]:291-301).
To more accurately diagnose PCS, our patients undergo tilt-table venography. The patient is placed into a reverse-Trendelenburg upright or semi-upright position to potentially exacerbate any venous reflux or dilation.
Other methods of identifying and diagnosing pelvic congestion have included transabdominal and transvaginal ultrasound, CT, and MRI. While CT and MRI both offer an overview of the pelvic vasculature and are helpful for ruling out other causes of chronic pelvic pain, they have low specificity for pelvic varices, according to the Italian review.
Sonography performed in the supine position, on the other hand, appears to be increasingly viewed as an acceptable screening tool for determining which patients may ultimately benefit from venography. It is also important in evaluation to rule out other pathologies not yet excluded. However, it should not be used for diagnosis of PCS.
Treating PCS
There are two main approaches to treating PCS: venous ligation (a gynecologic surgical approach) and percutaneous transcatheter embolization (performed by interventional radiologists).
The literature and evidence base is still in its infancy, but is growing. In our experience, both approaches lead to good resolution of symptoms over time in the majority of patients, and appear superior to the medical therapies that have been proposed for treating PCS, such as progestins and gonadotropin-releasing hormone agonists. Success rates with medical therapy are more variable and appear to be more short lived.
A review published this year on the effectiveness of embolization of pelvic veins for reducing chronic pelvic pain showed that 75% of women undergoing embolization had symptomatic relief that generally increased over time and was sustained. The authors concluded that embolization appears to be effective for the majority of women, and is safe, although they also noted that the quality of the evidence is low (J Vasc Interv Radiol. 2016 Oct;27[10]:1478-86.e8). Their review was based almost entirely on prospective case series.
Dr. Borghi and Dr. Dell’Atti offered a similar assessment of embolization for PCS, stating in their review article that clinical success has been reported in 70%-85% of patients. They also report nearly equivalent success rates of up to 75% with treatment via surgical ligation of ovarian and/or pelvic vasculature. These findings are from mostly observational data and case series.
Decisions about which approach to take should be individualized. If there are no differences with respect to insurance coverage for the patient, then embolization may be the preferred approach because it is the most minimally invasive technique and can potentially be performed at the time of diagnostic venography, negating the need for a second procedure. A skilled interventional radiologist familiar with the disease and the treatment is necessary. Various embolic agents are utilized, including coils, glues, foams, and other agents that cause sclerosis of the abnormal veins.
In other cases, venous ligation is preferred, especially when an additional gynecologic surgery, such as a cystectomy or myomectomy, is required.
Surgical ligation of ovarian veins was initially performed via laparotomy using a traditional retroperitoneal approach. The surgical goal is to isolate the ovarian vein significantly above the pelvic brim and before the vein becomes substantially dilated. Laparotomy therefore requires a vertical mid-line incision to provide adequate access to the appropriate portion of the ovarian vessels, leading to potentially high morbidity and poor cosmesis.
More recently, gynecologic surgeons skilled in laparoscopy have successfully managed PCS transperitoneally. A few small series of bilateral laparoscopic transperitoneal ligation of ovarian veins have been reported, including one by Tigellio Gargiulo, MD, who clipped both veins in their upper third, near their distal ends at the inferior vena cava (right) and the renal vein (left) (J Am Assoc Gynecol Laparosc. 2003 Nov;10[4]:501-4).
We prefer a robot-assisted laparoscopic approach for most of our patients. Not only does the improved dexterity help while working with sensitive vasculature, but more importantly we are able to use Firefly fluorescence.
The procedure generally is as follows. The uterine adnexa on the affected side is grasped and placed on tension so that the infundibulopelvic (IP) ligament can be visualized as it courses up and above the pelvic brim. The peritoneum immediately over the IP ligament is gently grasped and tented upward, and a small incision is made into the peritoneum, providing access into the retroperitoneum. The ureter should be visualized medial to this dissection.
The peritoneal tissue is then gently dissected off the ovarian vessels. Once the vessels are freed from the peritoneal tissue, the dilated ovarian vein is often clearly visualized. It is important to note that if no venous dilation is seen during laparoscopy, the procedure should not be aborted. Due to the Trendelenburg position that is utilized in gynecologic – and especially laparoscopic – surgery, the venous system sometimes appears falsely “normal” at this time.
Once the ovarian vessels have been isolated, the arteries must be separated from the veins. The adventitial tissue is dissected until the vessels are separated. Great care should be taken to ensure that all movements run parallel to the vessels and not perpendicular, therefore decreasing the risk of bleeding.
This process can be challenging. The surgeon is working with delicate vasculature. Often there are several branches from the vein that have formed due to the abnormal venous system. The best way to approach it is to identify planes and separate those planes in order to isolate individual vessels. If difficulties are still encountered, the surgeon should restart the dissection higher.
Once the dilated ovarian vein is isolated, one to two clips are placed.
Usually the artery is clearly distinct from the vein as it is smaller, more elastic, and can be seen pulsing. However, occasionally it is difficult to distinguish. In these cases, assistance with the da Vinci surgical system is useful: Indocyanine green (ICG) dye can be injected intravenously and visualized with a near-infrared light on the da Vinci platform. The dye is then seen glowing green as it first courses through the artery and then the vein.
For patients who have been found on venography to have bilateral disease, we perform the ligation procedure bilaterally. Once ligation is complete, the more competent collateral veins in the pelvis will assume more of the venous circulation.
In our experience, patients have ultimately noted substantial pain relief after these procedures, both with the endoscopic embolization and the surgical ligation. Patients are counseled that it can take several months to notice a relief in the pain.
In rare cases, pelvic congestion is related to extrinsic compression. For instance, the left renal vein can become compressed between the aorta and the superior mesenteric artery (the nutcracker syndrome), or the left common iliac vein can be compressed between the overlying right internal iliac artery and the underlying vertebral body (May-Thurner syndrome). Both of these conditions can lead to secondary PCS.
Such complex conditions are usually treated by vascular surgeons. May-Thurner syndrome is treated via stenting, while nutcracker syndrome can be treated with stenting or transposition of the renal vein to the distal vena cava.
Dr. Steller is an associate at the Family Health Centers of San Diego. She reported having no relevant financial disclosures.
BY CHARLES E. MILLER, MD
Chronic pelvic pain is described as the presence of lower abdominal or pelvic pain for longer than 6 months. It is believed to affect approximately one in six women and 12%-15% of women of reproductive age. The diagnosis and treatment of chronic pelvic pain adds as much as a $2 billion burden to our health system annually.
It was first described clinically in the literature in 1857, while the existence of pelvic varicosities wasn’t documented for nearly another 100 years. Pelvic congestion syndrome (PCS) accounts for 30%-70% of cases presenting with chronic pelvic pain. PCS can be due to pelvic venous insufficiency, characterized by reflux into pelvic veins leading to pelvic varicosities or alternative venous pathways secondary to varicose veins of the leg.
Other etiologies of PCS include nutcracker syndrome (left renal vein compressed between the aorta and the superior mesenteric artery), May-Thurner syndrome (compression of the left common iliac vein by the right common iliac artery) or, less likely, tumor thrombosis of the inferior vena cava, portal vein thrombosis, renal cell carcinoma, left renal thrombosis, or left kidney arterial-venous fistula.
While there appears to be significant literature indicating a long-term success rate of greater than 80% in patients treated by percutaneous endovascular procedures (embolization, stenting), there is far less information on the postsurgical success of blocking the varicose gonadal vein. Nevertheless, our long-term results with gonadal vein clipping is virtually the same as that of our radiological colleagues.
It is a pleasure to welcome Courtney Steller, DO, to this edition of the Master Class in Gynecologic Surgery to discuss the diagnosis and treatment of PCS, with an emphasis on surgical correction.
Dr. Steller is a recent graduate of the AAGL/SRS Fellowship in Minimally Invasive Gynecologic Surgery at Advocate Lutheran General Hospital, Park Ridge, Ill. She is currently in private practice and is an associate at the Family Health Centers of San Diego, Calif.
Dr. Miller is clinical associate professor at the University of Illinois at Chicago, and past president of the AAGL and the International Society for Gynecologic Endoscopy. He is a reproductive endocrinologist and minimally invasive gynecologic surgeon in private practice in Naperville and Schaumburg, Ill.; director of minimally invasive gynecologic surgery and the director of the AAGL/SRS Fellowship in Minimally Invasive Gynecologic Surgery at Advocate Lutheran General Hospital, Park Ridge, Ill.; and the medical editor of this column, Master Class. He reported having no financial disclosures relevant to this column. Email him at [email protected].
Pelvic congestion syndrome: A treatable cause of pain
BY COURTNEY STELLER, DO
Pelvic congestion syndrome is a poorly understood and underdiagnosed disease. Yet, over the last decade, the syndrome has become less controversial as the etiology has become better understood and as the diagnostic approach has become more specific. Through these advances, treatments have also become increasingly more successful.
This is an important shift, because the chronic pelvic pain experienced by patients with pelvic congestion significantly impacts their quality of life and well-being. As the pain persists, it can become exceedingly difficult to manage. Many patients we have ultimately treated for pelvic congestion syndrome have had years of various work-ups, significant diagnostic investigations, and trials of different treatments without having any cause of their pain identified or achieving any lasting symptom relief.
The pelvic pain in patients with pelvic congestion syndrome (PCS) can be noncyclical or cyclical. It is present most of the time but tends to get worse at the end of the day and after long periods of standing and/or sitting. The pain also may worsen with intercourse, largely afterward. The syndrome tends to occur in premenopausal and multiparous women, but it’s important to appreciate that this is not always the case; we have diagnosed and treated PCS in several young, nulliparous patients as well.
Features and diagnosis
PCS is a disorder of pelvic venous circulation that predominantly affects the ovarian veins. It is sometimes referred to as pelvic vein incompetence or pelvic vascular dysfunction. Just as veins in the legs can enlarge and become varicose, the ovarian veins – and sometimes the internal iliac veins – can become incompetent and unable to effectively return blood back to the heart.
Pregnancy may predispose patients to developing the abnormally dilated and refluxing veins that characterize PCS, as the increase in pelvic vein capacity and uterine compression can lead to significant stasis of blood in the pelvis and subsequent damage to the veins and the venous valves. There also is believed to be an estrogen component to the development of PCS, because estrogen is known to act as a vasodilator. Moreover, a congenital absence and incompetence of venous valves in some cases has been reported.
In a recent study looking at pelvic vein incompetence and symptoms of chronic pelvic pain, these women were reported to have a distinctive symptom profile, with the “most notable” features being the presence of dull pelvic pain that radiates to the upper thighs and is aggravated by prolonged standing and walking – symptoms that are similar to the leg symptoms experienced by patients with severe varicose veins (Eur J Obstet Gynecol Reprod Biol. 2016 Jan;196:21-5).
Other investigators have similarly described the pelvic pain related to PCS as a dull ache or heaviness sensation that is most severe at the end of the day and that is lessened with supine positioning (though not necessarily immediately) and often exacerbated with sexual intercourse, especially post coitus. These descriptions are in line with my experience with PCS. There is usually exquisite tenderness on pelvic exam, especially localized to the adnexa. Patients will often have varicose veins on their upper legs or labia.
Interestingly, it has been repeatedly shown that many women have dilated and incompetent pelvic veins without also having such pathognomonic pain. We therefore cannot treat women based solely on the finding of abnormal veins.
On the other hand we must determine which patients with chronic pelvic pain have PCS. The differential diagnosis for PCS includes endometriosis, adenomyosis chronic pelvic inflammatory disease, adhesive disease, adnexal masses, adnexal torsion, and several nongynecologic diseases including interstitial cystitis and irritable bowel syndrome.
Venography has become the gold standard for diagnosing pelvic congestion. The procedure involves catheterization of the ovarian veins through a femoral or jugular approach. In our experience, the common femoral vein is the more frequently used access point. Using a contrast injection, the interventional radiologist can assess the degree of venous dilation and reflux in the pelvis.
There currently is no consensus on a cutoff for vein diameter or on any validated measures for congestion. According to one report on PCS authored by interventional radiologists, the diagnosis of PCS is confirmed with the venographic findings of ovarian vein diameter greater than 6 mm, retrograde ovarian or pelvic venous flow, presence of several tortuous collateral pelvic venous pathways, and delayed or stagnant clearance on contrast (Semin Intervent Radiol. 2008 Dec;25[4]:361-8).
The criteria vary, however. A recent literature review on pelvic congestion syndrome by Chiara Borghi, MD, and Lucio Dell’Atti, MD, states that incompetent pelvic veins are defined as more than 5-10 mm in diameter (Arch Gynecol Obstet. 2016 Feb;293[2]:291-301).
To more accurately diagnose PCS, our patients undergo tilt-table venography. The patient is placed into a reverse-Trendelenburg upright or semi-upright position to potentially exacerbate any venous reflux or dilation.
Other methods of identifying and diagnosing pelvic congestion have included transabdominal and transvaginal ultrasound, CT, and MRI. While CT and MRI both offer an overview of the pelvic vasculature and are helpful for ruling out other causes of chronic pelvic pain, they have low specificity for pelvic varices, according to the Italian review.
Sonography performed in the supine position, on the other hand, appears to be increasingly viewed as an acceptable screening tool for determining which patients may ultimately benefit from venography. It is also important in evaluation to rule out other pathologies not yet excluded. However, it should not be used for diagnosis of PCS.
Treating PCS
There are two main approaches to treating PCS: venous ligation (a gynecologic surgical approach) and percutaneous transcatheter embolization (performed by interventional radiologists).
The literature and evidence base is still in its infancy, but is growing. In our experience, both approaches lead to good resolution of symptoms over time in the majority of patients, and appear superior to the medical therapies that have been proposed for treating PCS, such as progestins and gonadotropin-releasing hormone agonists. Success rates with medical therapy are more variable and appear to be more short lived.
A review published this year on the effectiveness of embolization of pelvic veins for reducing chronic pelvic pain showed that 75% of women undergoing embolization had symptomatic relief that generally increased over time and was sustained. The authors concluded that embolization appears to be effective for the majority of women, and is safe, although they also noted that the quality of the evidence is low (J Vasc Interv Radiol. 2016 Oct;27[10]:1478-86.e8). Their review was based almost entirely on prospective case series.
Dr. Borghi and Dr. Dell’Atti offered a similar assessment of embolization for PCS, stating in their review article that clinical success has been reported in 70%-85% of patients. They also report nearly equivalent success rates of up to 75% with treatment via surgical ligation of ovarian and/or pelvic vasculature. These findings are from mostly observational data and case series.
Decisions about which approach to take should be individualized. If there are no differences with respect to insurance coverage for the patient, then embolization may be the preferred approach because it is the most minimally invasive technique and can potentially be performed at the time of diagnostic venography, negating the need for a second procedure. A skilled interventional radiologist familiar with the disease and the treatment is necessary. Various embolic agents are utilized, including coils, glues, foams, and other agents that cause sclerosis of the abnormal veins.
In other cases, venous ligation is preferred, especially when an additional gynecologic surgery, such as a cystectomy or myomectomy, is required.
Surgical ligation of ovarian veins was initially performed via laparotomy using a traditional retroperitoneal approach. The surgical goal is to isolate the ovarian vein significantly above the pelvic brim and before the vein becomes substantially dilated. Laparotomy therefore requires a vertical mid-line incision to provide adequate access to the appropriate portion of the ovarian vessels, leading to potentially high morbidity and poor cosmesis.
More recently, gynecologic surgeons skilled in laparoscopy have successfully managed PCS transperitoneally. A few small series of bilateral laparoscopic transperitoneal ligation of ovarian veins have been reported, including one by Tigellio Gargiulo, MD, who clipped both veins in their upper third, near their distal ends at the inferior vena cava (right) and the renal vein (left) (J Am Assoc Gynecol Laparosc. 2003 Nov;10[4]:501-4).
We prefer a robot-assisted laparoscopic approach for most of our patients. Not only does the improved dexterity help while working with sensitive vasculature, but more importantly we are able to use Firefly fluorescence.
The procedure generally is as follows. The uterine adnexa on the affected side is grasped and placed on tension so that the infundibulopelvic (IP) ligament can be visualized as it courses up and above the pelvic brim. The peritoneum immediately over the IP ligament is gently grasped and tented upward, and a small incision is made into the peritoneum, providing access into the retroperitoneum. The ureter should be visualized medial to this dissection.
The peritoneal tissue is then gently dissected off the ovarian vessels. Once the vessels are freed from the peritoneal tissue, the dilated ovarian vein is often clearly visualized. It is important to note that if no venous dilation is seen during laparoscopy, the procedure should not be aborted. Due to the Trendelenburg position that is utilized in gynecologic – and especially laparoscopic – surgery, the venous system sometimes appears falsely “normal” at this time.
Once the ovarian vessels have been isolated, the arteries must be separated from the veins. The adventitial tissue is dissected until the vessels are separated. Great care should be taken to ensure that all movements run parallel to the vessels and not perpendicular, therefore decreasing the risk of bleeding.
This process can be challenging. The surgeon is working with delicate vasculature. Often there are several branches from the vein that have formed due to the abnormal venous system. The best way to approach it is to identify planes and separate those planes in order to isolate individual vessels. If difficulties are still encountered, the surgeon should restart the dissection higher.
Once the dilated ovarian vein is isolated, one to two clips are placed.
Usually the artery is clearly distinct from the vein as it is smaller, more elastic, and can be seen pulsing. However, occasionally it is difficult to distinguish. In these cases, assistance with the da Vinci surgical system is useful: Indocyanine green (ICG) dye can be injected intravenously and visualized with a near-infrared light on the da Vinci platform. The dye is then seen glowing green as it first courses through the artery and then the vein.
For patients who have been found on venography to have bilateral disease, we perform the ligation procedure bilaterally. Once ligation is complete, the more competent collateral veins in the pelvis will assume more of the venous circulation.
In our experience, patients have ultimately noted substantial pain relief after these procedures, both with the endoscopic embolization and the surgical ligation. Patients are counseled that it can take several months to notice a relief in the pain.
In rare cases, pelvic congestion is related to extrinsic compression. For instance, the left renal vein can become compressed between the aorta and the superior mesenteric artery (the nutcracker syndrome), or the left common iliac vein can be compressed between the overlying right internal iliac artery and the underlying vertebral body (May-Thurner syndrome). Both of these conditions can lead to secondary PCS.
Such complex conditions are usually treated by vascular surgeons. May-Thurner syndrome is treated via stenting, while nutcracker syndrome can be treated with stenting or transposition of the renal vein to the distal vena cava.
Dr. Steller is an associate at the Family Health Centers of San Diego. She reported having no relevant financial disclosures.

Some Patients With Diabetes Aren’t Getting Needed Weight Advice
About two-thirds of overweight and obese patients are not getting the advice they need from their health care providers (HCPs) about weight management, according to a study by Arizona State researchers, reported in the CDC’s Preventing Chronic Disease.
The researchers conducted a phone survey of 1,109 overweight or obese adults, asking whether a HCP had in the previous 12 months given them advice about their weight. A “concerning” finding, the researchers say: Only 35% of the respondents reported getting advice.
Related: Confronting the Diabetes Epidemic
As body mass index (BMI) increased, so did the likelihood of receiving weight-loss advice: 22% of those with a BMI of 25.0 to 29.9 received advice vs 63% of those with a BMI of 40.0 or higher. Hispanics were the most likely of the 3 racial/ethnic groups to report receiving advice from a HCP. The researchers say other studies have suggested that a higher prevalence of weight problems among African Americans and Hispanics draws more attention from HCPs for counseling.
High-risk patients, such as the extremely obese or those with comorbidities, are most likely to receive weight-loss advice, the researchers say. But demographic factors also come into play: People with high levels of education are more likely than those with low levels to receive advice, and middle-aged people are more likely to get advice than are younger or older patients.
Related: Diabetes Report: The News Isn’t Good
Patients in the lowest-income groups had significantly lower odds of receiving weight-loss advice compared with those in higher income groups. Adjusting for health insurance did not change the results. That finding is “problematic,” the researchers say, because people with the lowest incomes tend to have poorer health outcomes than that of those with higher incomes.
About two-thirds of overweight and obese patients are not getting the advice they need from their health care providers (HCPs) about weight management, according to a study by Arizona State researchers, reported in the CDC’s Preventing Chronic Disease.
The researchers conducted a phone survey of 1,109 overweight or obese adults, asking whether a HCP had in the previous 12 months given them advice about their weight. A “concerning” finding, the researchers say: Only 35% of the respondents reported getting advice.
Related: Confronting the Diabetes Epidemic
As body mass index (BMI) increased, so did the likelihood of receiving weight-loss advice: 22% of those with a BMI of 25.0 to 29.9 received advice vs 63% of those with a BMI of 40.0 or higher. Hispanics were the most likely of the 3 racial/ethnic groups to report receiving advice from a HCP. The researchers say other studies have suggested that a higher prevalence of weight problems among African Americans and Hispanics draws more attention from HCPs for counseling.
High-risk patients, such as the extremely obese or those with comorbidities, are most likely to receive weight-loss advice, the researchers say. But demographic factors also come into play: People with high levels of education are more likely than those with low levels to receive advice, and middle-aged people are more likely to get advice than are younger or older patients.
Related: Diabetes Report: The News Isn’t Good
Patients in the lowest-income groups had significantly lower odds of receiving weight-loss advice compared with those in higher income groups. Adjusting for health insurance did not change the results. That finding is “problematic,” the researchers say, because people with the lowest incomes tend to have poorer health outcomes than that of those with higher incomes.
About two-thirds of overweight and obese patients are not getting the advice they need from their health care providers (HCPs) about weight management, according to a study by Arizona State researchers, reported in the CDC’s Preventing Chronic Disease.
The researchers conducted a phone survey of 1,109 overweight or obese adults, asking whether a HCP had in the previous 12 months given them advice about their weight. A “concerning” finding, the researchers say: Only 35% of the respondents reported getting advice.
Related: Confronting the Diabetes Epidemic
As body mass index (BMI) increased, so did the likelihood of receiving weight-loss advice: 22% of those with a BMI of 25.0 to 29.9 received advice vs 63% of those with a BMI of 40.0 or higher. Hispanics were the most likely of the 3 racial/ethnic groups to report receiving advice from a HCP. The researchers say other studies have suggested that a higher prevalence of weight problems among African Americans and Hispanics draws more attention from HCPs for counseling.
High-risk patients, such as the extremely obese or those with comorbidities, are most likely to receive weight-loss advice, the researchers say. But demographic factors also come into play: People with high levels of education are more likely than those with low levels to receive advice, and middle-aged people are more likely to get advice than are younger or older patients.
Related: Diabetes Report: The News Isn’t Good
Patients in the lowest-income groups had significantly lower odds of receiving weight-loss advice compared with those in higher income groups. Adjusting for health insurance did not change the results. That finding is “problematic,” the researchers say, because people with the lowest incomes tend to have poorer health outcomes than that of those with higher incomes.
Use Whiteboards to Enhance Patient-Provider Communication
Editor’s note: “Everything We Say and Do” is an informational series developed by SHM’s Patient Experience Committee to provide readers with thoughtful and actionable communication tactics that have great potential to positively impact patients’ experience of care. Each article will focus on how the contributor applies one or more of the “key communication” tactics in practice to maintain provider accountability for “everything we say and do that affects our patients’ thoughts, feelings, and well-being.”
View a chart outlining key communication tactics
What I Say and Do

With my team, I use whiteboards as a tool to enhance communication: 1) I introduce myself and my team members, then write our names on the whiteboard paired with an explanation of my role as the attending physician for the hospital medicine service; 2) on a daily basis, I ask the patient and family/support what their primary concerns and goals are and write those on the whiteboard; and 3) I invite the patient and family/support to use the whiteboard to write additional concerns or questions as they arise.
Why I Do It
Hospitals are confusing places. One of our key roles as hospitalists is to coordinate and clarify all of the moving pieces and to communicate clearly to patients and their family that there is someone doing that work on their behalf. The whiteboard can help to accomplish that and to visually indicate “reflective listening.” If I ask patients what their concerns and goals are on a daily basis, I can better address them, and writing those on the whiteboard is a way to visually let patients know I have heard them—and heard them accurately. Finally, as we know from experience at our institution, when patients are invited to write on the whiteboard, they are likely to do so and will often write important information that hasn’t come up during routine rounding.
How I Do It
The key to effectiveness is to build whiteboard use into the clinical workflow and patient conversation rather than create an extra task to complete. I have developed a routine using the whiteboard that is more or less the same for every patient.
Also, whiteboard design can influence the use of the whiteboard as a communication tool. I favor designs that have few prescriptive boxes and more space for writing. I have found whiteboards labeled with a “What are your goals?” section to be helpful.
Patrick Kneeland, MD, is medical director for patient and provider experience and director of the Excellence in Communication Curriculum, University of Colorado Hospital and Clinics.
Editor’s note: “Everything We Say and Do” is an informational series developed by SHM’s Patient Experience Committee to provide readers with thoughtful and actionable communication tactics that have great potential to positively impact patients’ experience of care. Each article will focus on how the contributor applies one or more of the “key communication” tactics in practice to maintain provider accountability for “everything we say and do that affects our patients’ thoughts, feelings, and well-being.”
View a chart outlining key communication tactics
What I Say and Do
With my team, I use whiteboards as a tool to enhance communication: 1) I introduce myself and my team members, then write our names on the whiteboard paired with an explanation of my role as the attending physician for the hospital medicine service; 2) on a daily basis, I ask the patient and family/support what their primary concerns and goals are and write those on the whiteboard; and 3) I invite the patient and family/support to use the whiteboard to write additional concerns or questions as they arise.
Why I Do It
Hospitals are confusing places. One of our key roles as hospitalists is to coordinate and clarify all of the moving pieces and to communicate clearly to patients and their family that there is someone doing that work on their behalf. The whiteboard can help to accomplish that and to visually indicate “reflective listening.” If I ask patients what their concerns and goals are on a daily basis, I can better address them, and writing those on the whiteboard is a way to visually let patients know I have heard them—and heard them accurately. Finally, as we know from experience at our institution, when patients are invited to write on the whiteboard, they are likely to do so and will often write important information that hasn’t come up during routine rounding.
How I Do It
The key to effectiveness is to build whiteboard use into the clinical workflow and patient conversation rather than create an extra task to complete. I have developed a routine using the whiteboard that is more or less the same for every patient.
Also, whiteboard design can influence the use of the whiteboard as a communication tool. I favor designs that have few prescriptive boxes and more space for writing. I have found whiteboards labeled with a “What are your goals?” section to be helpful.
Patrick Kneeland, MD, is medical director for patient and provider experience and director of the Excellence in Communication Curriculum, University of Colorado Hospital and Clinics.
Editor’s note: “Everything We Say and Do” is an informational series developed by SHM’s Patient Experience Committee to provide readers with thoughtful and actionable communication tactics that have great potential to positively impact patients’ experience of care. Each article will focus on how the contributor applies one or more of the “key communication” tactics in practice to maintain provider accountability for “everything we say and do that affects our patients’ thoughts, feelings, and well-being.”
View a chart outlining key communication tactics
What I Say and Do
With my team, I use whiteboards as a tool to enhance communication: 1) I introduce myself and my team members, then write our names on the whiteboard paired with an explanation of my role as the attending physician for the hospital medicine service; 2) on a daily basis, I ask the patient and family/support what their primary concerns and goals are and write those on the whiteboard; and 3) I invite the patient and family/support to use the whiteboard to write additional concerns or questions as they arise.
Why I Do It
Hospitals are confusing places. One of our key roles as hospitalists is to coordinate and clarify all of the moving pieces and to communicate clearly to patients and their family that there is someone doing that work on their behalf. The whiteboard can help to accomplish that and to visually indicate “reflective listening.” If I ask patients what their concerns and goals are on a daily basis, I can better address them, and writing those on the whiteboard is a way to visually let patients know I have heard them—and heard them accurately. Finally, as we know from experience at our institution, when patients are invited to write on the whiteboard, they are likely to do so and will often write important information that hasn’t come up during routine rounding.
How I Do It
The key to effectiveness is to build whiteboard use into the clinical workflow and patient conversation rather than create an extra task to complete. I have developed a routine using the whiteboard that is more or less the same for every patient.
Also, whiteboard design can influence the use of the whiteboard as a communication tool. I favor designs that have few prescriptive boxes and more space for writing. I have found whiteboards labeled with a “What are your goals?” section to be helpful.
Patrick Kneeland, MD, is medical director for patient and provider experience and director of the Excellence in Communication Curriculum, University of Colorado Hospital and Clinics.
New SHM Members – November 2016
F. W. Erdman, Alabama
S. Baquai, MD, California
J. Bullock, California
A. Chong, MD, California
J. Decolongon, California
J. Do, MD, California
T. Farmer, ACNP, California
T. Holden, MD, California
M. Khare, California
P. Lally, MD, California
B. Lee, MD, California
J. Lee, MD, California
A. Mannan, MD, California
N. Pandher, California
E. Park, CaliforniaB. Patel, USA, California
N. Patel, California
S. Singh, MD, California
M. Vakili, California
A. Zandpour, AHIP, California
L. Chong, MD, FACP, Connecticut
G. Cudjoe, MBBS, Connecticut
S. Gazi, MD, Connecticut
S. Pattisapu, Connecticut
M. Rai, Connecticut
S. Roshan, MD, Connecticut
A. Seye, MD, Connecticut
L. Zheng, Connecticut
S. Raghavan, MD, PhD, Colorado
M. Altieri, MHSc, PA-C, Florida
M. Bishai, DO, Florida
B. Burns, Florida
A. Chan, MD, Florida
M. Cuk, Florida
S. Epps, DO, Florida
N. Fedotova, PhD, Florida
A. Lee, MD, Florida
M. Ruiz, Florida
D. Britt, Georgia
W. Futch, Georgia
B. Kruszewski, Georgia
I. Lowell, MD, MBA, Georgia
Y. Patel, MD, Georgia
G. Polk-Seldon, FNP, Georgia
T. Truong, MD, Georgia
J. Walker, DO, Georgia
S. Wang, MBA, Georgia
S. Del Mundo, Hawaii
T. Hiura, MD, Hawaii
D. Orchard, Idaho
S. Pontickio, MD, Idaho
R. Antoine, MD, Illinois
A. Baid, Illinois
C. Boyle, MD, Illinois
M. Delibasic, MD, Illinois
J. Gemson, Illinois
J. Mechurova, MD, Illinois
T. Morales, Illinois
H. Omar, Illinois
B. Pathak, MD, Illinois
S. Rao, MD, Illinois
Z. Ritchey, Illinois
A. Veerabahu, Illinois
T. Barley, MD, Indiana
M. Meyer, ACNP, MSN, Indiana
D. Ross, MA, Indiana
K. Sorg, MD, Indiana
A. Evans, Iowa
R. Miller, MD, Louisiana
D. Pollet, MD, Louisiana
J. Goldberg, MD, Maine
S. Gutierrez, MD, Maine
K. Osborne, FNP, Maine
V. Pramanik, MD, Maine
C. Sherpa, Maine
H. Abinader, Maryland
I. Pena, Maryland
D. Press, MD, Maryland
D. Soffer, Maryland
J. Bennett, FAAP, Massachusetts
N. Howe, ACNP, Massachusetts
J. Hudspeth, MD, Massachusetts
I. Ismail, MD, Massachusetts
B. Lall, MD, Massachusetts
M. Lawler, MD, Massachusetts
E. O’Fallon, MD, Massachusetts
J. Ringwala, Massachusetts
M. Soe, MD, Massachusetts
E. Sweatt, MD, Massachusetts
M. Trivedi, Massachusetts
E. Chappe, MD, Michigan
A. Goyal, MD, Michigan
E. Hunter, DO, Michigan
S. Malewskim ACNP, MSN, RN, Michigan
W. Ladner, Minnesota
J. Purdy, Minnesota
T. VanLith, PA-C, Minnesota
P. Acharya, MBBS, Mississippi
A. Ladd, CNP, Mississippi
S. Morris, DO, MBA, Mississippi
R. Walters, DO, Mississippi
R. Allen, DO, Missouri
L. Andrea, MissouriA. Arnaud, Missouri
M. Board, Missouri
S. Njagi, MD, FAAFP, MBchB, Missouri
J. Patel, Missouri
A. Roman, MD, Missouri
R. Singh, Missouri
F. Wang, Missouri
S. Byington, MD, Montana
T. Lloyd, Montana
B. Bulian, MD, Nebraska
S. Adagi, New Jersey
C. Cristescu, MeD, New Jersey
A. Malhotra, MD, New Jersey
K. Patel, DO, New Jersey
S. Terner, MD, New Jersey
H. Alqam, New Mexico
A. Attreya, DO, New Mexico
K. Avila, New Mexico
K. Chan, New Mexico
S. Montano, New Mexico
C. Morales, New Mexico
A. Stecker, New Mexico
D. Varela, New Mexico
M. Ahmed, MD, New York
G. Apergis, MD, New York
J. Dekhtyar, MD, New York
J. Dillon, New York
N. Jaglall, MD, New YorkL. Kruzhkov, New York
R. Malhan, MD, New York
J. Mathew, New YorkV. Miro, New York
B. Gautam, North Carolina
K. Gold, MD, North Carolina P. Greene, MD, North Carolina
S. Grotzke, North Carolina
S. Hester, MD, North Carolina
P. Le, MD, North Carolina
J. McClung, MD, North Carolina
J. Ramsey, MD, North Carolina
J. Sullivan, DO, North Carolina
C. Chadwell, Ohio
R. Dash, MD, Ohio
E. Ofungwu, USAR, Ohio
R. Raj, MD, Ohio
I. Rawal, Ohio
N. Beach, DO, Oregon
M. Christensen, ACNP, Oregon
K. Haugen, MD, Oregon
J. Luty, MD, Oregon
K. Andersen, Pennsylvania
O. Ball, MD, Pennsylvania
S. Harris, DO, Pennsylvania
R. Koubek, Pennsylvania
B. Krug, MHA, Pennsylvania
A. Levin, MD, Pennsylvania
A. Marwah, Pennsylvania
D. McAllister, FNP, Pennsylvania
C. Sakosky, FNP, Pennsylvania
J. Steffl, PA-C, Pennsylvania
A. Sukits, MS, PA-C, Pennsylvania
S. Clemens, Rhode Island
C. Drasny, MD, South Carolina
J. Harris, MD, South Carolina
D. Head, MD, South Carolina
S. Johnson, South Carolina
A. Evjen, MD, South Dakota
P. Pate, Tennessee
J. Patterson, ACNP-BC, MSN, Tennessee
A. Seth, MD, Tennessee
P. Boeckmann, FACHE, Texas
M. Gupta, Texas
J. Jain, MD, Texas
K. Roberts, Texas
C. Romero, MD, PhD, Texas
D. Buzanoski, MD, Utah
N. Whitaker, FACP, Utah
E. Greenberger, MD, Vermont
R. McEntee, MD, Vermont
C. Rickman, FACP, Vermont
W. Austin, MSHA, Virginia
E. Orshansky, MD, Virginia
G. Psarros, MD, Virginia
N. Trivedi, Virginia
E. Addison, Washington
J. Gifford, PA-C, Washington
V. Johnson, ARNP, CFNP, MHSc, Washington
C. Wang, MD, Washington
M. Brown, West Virginia
V. Raina, MD, Wisconsin
F. Germa, MD, CCP(EM), FCEP (C), Canada
J. Podavin, Canada
K. Slatkovsky, Canada
M. Kitamura, Japan
M. Rafei, Oman
F. W. Erdman, Alabama
S. Baquai, MD, California
J. Bullock, California
A. Chong, MD, California
J. Decolongon, California
J. Do, MD, California
T. Farmer, ACNP, California
T. Holden, MD, California
M. Khare, California
P. Lally, MD, California
B. Lee, MD, California
J. Lee, MD, California
A. Mannan, MD, California
N. Pandher, California
E. Park, CaliforniaB. Patel, USA, California
N. Patel, California
S. Singh, MD, California
M. Vakili, California
A. Zandpour, AHIP, California
L. Chong, MD, FACP, Connecticut
G. Cudjoe, MBBS, Connecticut
S. Gazi, MD, Connecticut
S. Pattisapu, Connecticut
M. Rai, Connecticut
S. Roshan, MD, Connecticut
A. Seye, MD, Connecticut
L. Zheng, Connecticut
S. Raghavan, MD, PhD, Colorado
M. Altieri, MHSc, PA-C, Florida
M. Bishai, DO, Florida
B. Burns, Florida
A. Chan, MD, Florida
M. Cuk, Florida
S. Epps, DO, Florida
N. Fedotova, PhD, Florida
A. Lee, MD, Florida
M. Ruiz, Florida
D. Britt, Georgia
W. Futch, Georgia
B. Kruszewski, Georgia
I. Lowell, MD, MBA, Georgia
Y. Patel, MD, Georgia
G. Polk-Seldon, FNP, Georgia
T. Truong, MD, Georgia
J. Walker, DO, Georgia
S. Wang, MBA, Georgia
S. Del Mundo, Hawaii
T. Hiura, MD, Hawaii
D. Orchard, Idaho
S. Pontickio, MD, Idaho
R. Antoine, MD, Illinois
A. Baid, Illinois
C. Boyle, MD, Illinois
M. Delibasic, MD, Illinois
J. Gemson, Illinois
J. Mechurova, MD, Illinois
T. Morales, Illinois
H. Omar, Illinois
B. Pathak, MD, Illinois
S. Rao, MD, Illinois
Z. Ritchey, Illinois
A. Veerabahu, Illinois
T. Barley, MD, Indiana
M. Meyer, ACNP, MSN, Indiana
D. Ross, MA, Indiana
K. Sorg, MD, Indiana
A. Evans, Iowa
R. Miller, MD, Louisiana
D. Pollet, MD, Louisiana
J. Goldberg, MD, Maine
S. Gutierrez, MD, Maine
K. Osborne, FNP, Maine
V. Pramanik, MD, Maine
C. Sherpa, Maine
H. Abinader, Maryland
I. Pena, Maryland
D. Press, MD, Maryland
D. Soffer, Maryland
J. Bennett, FAAP, Massachusetts
N. Howe, ACNP, Massachusetts
J. Hudspeth, MD, Massachusetts
I. Ismail, MD, Massachusetts
B. Lall, MD, Massachusetts
M. Lawler, MD, Massachusetts
E. O’Fallon, MD, Massachusetts
J. Ringwala, Massachusetts
M. Soe, MD, Massachusetts
E. Sweatt, MD, Massachusetts
M. Trivedi, Massachusetts
E. Chappe, MD, Michigan
A. Goyal, MD, Michigan
E. Hunter, DO, Michigan
S. Malewskim ACNP, MSN, RN, Michigan
W. Ladner, Minnesota
J. Purdy, Minnesota
T. VanLith, PA-C, Minnesota
P. Acharya, MBBS, Mississippi
A. Ladd, CNP, Mississippi
S. Morris, DO, MBA, Mississippi
R. Walters, DO, Mississippi
R. Allen, DO, Missouri
L. Andrea, MissouriA. Arnaud, Missouri
M. Board, Missouri
S. Njagi, MD, FAAFP, MBchB, Missouri
J. Patel, Missouri
A. Roman, MD, Missouri
R. Singh, Missouri
F. Wang, Missouri
S. Byington, MD, Montana
T. Lloyd, Montana
B. Bulian, MD, Nebraska
S. Adagi, New Jersey
C. Cristescu, MeD, New Jersey
A. Malhotra, MD, New Jersey
K. Patel, DO, New Jersey
S. Terner, MD, New Jersey
H. Alqam, New Mexico
A. Attreya, DO, New Mexico
K. Avila, New Mexico
K. Chan, New Mexico
S. Montano, New Mexico
C. Morales, New Mexico
A. Stecker, New Mexico
D. Varela, New Mexico
M. Ahmed, MD, New York
G. Apergis, MD, New York
J. Dekhtyar, MD, New York
J. Dillon, New York
N. Jaglall, MD, New YorkL. Kruzhkov, New York
R. Malhan, MD, New York
J. Mathew, New YorkV. Miro, New York
B. Gautam, North Carolina
K. Gold, MD, North Carolina P. Greene, MD, North Carolina
S. Grotzke, North Carolina
S. Hester, MD, North Carolina
P. Le, MD, North Carolina
J. McClung, MD, North Carolina
J. Ramsey, MD, North Carolina
J. Sullivan, DO, North Carolina
C. Chadwell, Ohio
R. Dash, MD, Ohio
E. Ofungwu, USAR, Ohio
R. Raj, MD, Ohio
I. Rawal, Ohio
N. Beach, DO, Oregon
M. Christensen, ACNP, Oregon
K. Haugen, MD, Oregon
J. Luty, MD, Oregon
K. Andersen, Pennsylvania
O. Ball, MD, Pennsylvania
S. Harris, DO, Pennsylvania
R. Koubek, Pennsylvania
B. Krug, MHA, Pennsylvania
A. Levin, MD, Pennsylvania
A. Marwah, Pennsylvania
D. McAllister, FNP, Pennsylvania
C. Sakosky, FNP, Pennsylvania
J. Steffl, PA-C, Pennsylvania
A. Sukits, MS, PA-C, Pennsylvania
S. Clemens, Rhode Island
C. Drasny, MD, South Carolina
J. Harris, MD, South Carolina
D. Head, MD, South Carolina
S. Johnson, South Carolina
A. Evjen, MD, South Dakota
P. Pate, Tennessee
J. Patterson, ACNP-BC, MSN, Tennessee
A. Seth, MD, Tennessee
P. Boeckmann, FACHE, Texas
M. Gupta, Texas
J. Jain, MD, Texas
K. Roberts, Texas
C. Romero, MD, PhD, Texas
D. Buzanoski, MD, Utah
N. Whitaker, FACP, Utah
E. Greenberger, MD, Vermont
R. McEntee, MD, Vermont
C. Rickman, FACP, Vermont
W. Austin, MSHA, Virginia
E. Orshansky, MD, Virginia
G. Psarros, MD, Virginia
N. Trivedi, Virginia
E. Addison, Washington
J. Gifford, PA-C, Washington
V. Johnson, ARNP, CFNP, MHSc, Washington
C. Wang, MD, Washington
M. Brown, West Virginia
V. Raina, MD, Wisconsin
F. Germa, MD, CCP(EM), FCEP (C), Canada
J. Podavin, Canada
K. Slatkovsky, Canada
M. Kitamura, Japan
M. Rafei, Oman
F. W. Erdman, Alabama
S. Baquai, MD, California
J. Bullock, California
A. Chong, MD, California
J. Decolongon, California
J. Do, MD, California
T. Farmer, ACNP, California
T. Holden, MD, California
M. Khare, California
P. Lally, MD, California
B. Lee, MD, California
J. Lee, MD, California
A. Mannan, MD, California
N. Pandher, California
E. Park, CaliforniaB. Patel, USA, California
N. Patel, California
S. Singh, MD, California
M. Vakili, California
A. Zandpour, AHIP, California
L. Chong, MD, FACP, Connecticut
G. Cudjoe, MBBS, Connecticut
S. Gazi, MD, Connecticut
S. Pattisapu, Connecticut
M. Rai, Connecticut
S. Roshan, MD, Connecticut
A. Seye, MD, Connecticut
L. Zheng, Connecticut
S. Raghavan, MD, PhD, Colorado
M. Altieri, MHSc, PA-C, Florida
M. Bishai, DO, Florida
B. Burns, Florida
A. Chan, MD, Florida
M. Cuk, Florida
S. Epps, DO, Florida
N. Fedotova, PhD, Florida
A. Lee, MD, Florida
M. Ruiz, Florida
D. Britt, Georgia
W. Futch, Georgia
B. Kruszewski, Georgia
I. Lowell, MD, MBA, Georgia
Y. Patel, MD, Georgia
G. Polk-Seldon, FNP, Georgia
T. Truong, MD, Georgia
J. Walker, DO, Georgia
S. Wang, MBA, Georgia
S. Del Mundo, Hawaii
T. Hiura, MD, Hawaii
D. Orchard, Idaho
S. Pontickio, MD, Idaho
R. Antoine, MD, Illinois
A. Baid, Illinois
C. Boyle, MD, Illinois
M. Delibasic, MD, Illinois
J. Gemson, Illinois
J. Mechurova, MD, Illinois
T. Morales, Illinois
H. Omar, Illinois
B. Pathak, MD, Illinois
S. Rao, MD, Illinois
Z. Ritchey, Illinois
A. Veerabahu, Illinois
T. Barley, MD, Indiana
M. Meyer, ACNP, MSN, Indiana
D. Ross, MA, Indiana
K. Sorg, MD, Indiana
A. Evans, Iowa
R. Miller, MD, Louisiana
D. Pollet, MD, Louisiana
J. Goldberg, MD, Maine
S. Gutierrez, MD, Maine
K. Osborne, FNP, Maine
V. Pramanik, MD, Maine
C. Sherpa, Maine
H. Abinader, Maryland
I. Pena, Maryland
D. Press, MD, Maryland
D. Soffer, Maryland
J. Bennett, FAAP, Massachusetts
N. Howe, ACNP, Massachusetts
J. Hudspeth, MD, Massachusetts
I. Ismail, MD, Massachusetts
B. Lall, MD, Massachusetts
M. Lawler, MD, Massachusetts
E. O’Fallon, MD, Massachusetts
J. Ringwala, Massachusetts
M. Soe, MD, Massachusetts
E. Sweatt, MD, Massachusetts
M. Trivedi, Massachusetts
E. Chappe, MD, Michigan
A. Goyal, MD, Michigan
E. Hunter, DO, Michigan
S. Malewskim ACNP, MSN, RN, Michigan
W. Ladner, Minnesota
J. Purdy, Minnesota
T. VanLith, PA-C, Minnesota
P. Acharya, MBBS, Mississippi
A. Ladd, CNP, Mississippi
S. Morris, DO, MBA, Mississippi
R. Walters, DO, Mississippi
R. Allen, DO, Missouri
L. Andrea, MissouriA. Arnaud, Missouri
M. Board, Missouri
S. Njagi, MD, FAAFP, MBchB, Missouri
J. Patel, Missouri
A. Roman, MD, Missouri
R. Singh, Missouri
F. Wang, Missouri
S. Byington, MD, Montana
T. Lloyd, Montana
B. Bulian, MD, Nebraska
S. Adagi, New Jersey
C. Cristescu, MeD, New Jersey
A. Malhotra, MD, New Jersey
K. Patel, DO, New Jersey
S. Terner, MD, New Jersey
H. Alqam, New Mexico
A. Attreya, DO, New Mexico
K. Avila, New Mexico
K. Chan, New Mexico
S. Montano, New Mexico
C. Morales, New Mexico
A. Stecker, New Mexico
D. Varela, New Mexico
M. Ahmed, MD, New York
G. Apergis, MD, New York
J. Dekhtyar, MD, New York
J. Dillon, New York
N. Jaglall, MD, New YorkL. Kruzhkov, New York
R. Malhan, MD, New York
J. Mathew, New YorkV. Miro, New York
B. Gautam, North Carolina
K. Gold, MD, North Carolina P. Greene, MD, North Carolina
S. Grotzke, North Carolina
S. Hester, MD, North Carolina
P. Le, MD, North Carolina
J. McClung, MD, North Carolina
J. Ramsey, MD, North Carolina
J. Sullivan, DO, North Carolina
C. Chadwell, Ohio
R. Dash, MD, Ohio
E. Ofungwu, USAR, Ohio
R. Raj, MD, Ohio
I. Rawal, Ohio
N. Beach, DO, Oregon
M. Christensen, ACNP, Oregon
K. Haugen, MD, Oregon
J. Luty, MD, Oregon
K. Andersen, Pennsylvania
O. Ball, MD, Pennsylvania
S. Harris, DO, Pennsylvania
R. Koubek, Pennsylvania
B. Krug, MHA, Pennsylvania
A. Levin, MD, Pennsylvania
A. Marwah, Pennsylvania
D. McAllister, FNP, Pennsylvania
C. Sakosky, FNP, Pennsylvania
J. Steffl, PA-C, Pennsylvania
A. Sukits, MS, PA-C, Pennsylvania
S. Clemens, Rhode Island
C. Drasny, MD, South Carolina
J. Harris, MD, South Carolina
D. Head, MD, South Carolina
S. Johnson, South Carolina
A. Evjen, MD, South Dakota
P. Pate, Tennessee
J. Patterson, ACNP-BC, MSN, Tennessee
A. Seth, MD, Tennessee
P. Boeckmann, FACHE, Texas
M. Gupta, Texas
J. Jain, MD, Texas
K. Roberts, Texas
C. Romero, MD, PhD, Texas
D. Buzanoski, MD, Utah
N. Whitaker, FACP, Utah
E. Greenberger, MD, Vermont
R. McEntee, MD, Vermont
C. Rickman, FACP, Vermont
W. Austin, MSHA, Virginia
E. Orshansky, MD, Virginia
G. Psarros, MD, Virginia
N. Trivedi, Virginia
E. Addison, Washington
J. Gifford, PA-C, Washington
V. Johnson, ARNP, CFNP, MHSc, Washington
C. Wang, MD, Washington
M. Brown, West Virginia
V. Raina, MD, Wisconsin
F. Germa, MD, CCP(EM), FCEP (C), Canada
J. Podavin, Canada
K. Slatkovsky, Canada
M. Kitamura, Japan
M. Rafei, Oman
Preventive Treatment for Posttraumatic Stress Disorder
Identifying people who might be at risk for posttraumatic stress disorder (PTSD) before the trauma—and teaching them preventive coping skills—could reduce or prevent long-term effects, according to University of Oxford in Oxford, United Kingdom, and King’s College London, United Kingdom, researchers.
They assessed 453 newly recruited paramedics every 4 months for 2 years. Of those, 386 paramedics participated in follow-up interviews.
Related: Let’s Dance: A Holistic Approach to Treating Veterans With Posttraumatic Stress Disorder
Over the 2 years, 32 participants (8.3%) had an episode of PTSD, and 41 participants had (10.6%) an episode of major depression (MD). Most of the episodes were moderate and short lived. In most cases, the participant had recovered by the next 4-month assessment. However, at 2 years, those who had experienced episodes of PTSD or MD during the follow-up period reported more days off work, poorer sleep, poorer quality of life, and greater burn out as well as weight gain (mean gain, 6.9 kg) for those with P
Ten participants who developed PTSD received treatment during follow-up, as did 12 participants who developed MD. Five of 9 participants who had recurrent P
Related: Telehealth for Native Americans With PTSD
The researchers tested a number of possible pretrauma predictors of PTSD and MD. They correlated several: cognitive style (eg, suppression, rumination, intentional numbing), coping style (eg, avoidant styles, such as wishful thinking), and psychological traits (eg, neuroticism). However, they found rumination about memories of stressful events uniquely predicted an episode of PTSD. Perceived resilience uniquely predicted an episode of MD.
Interestingly, about 42% of the study participants had a psychiatric history before training—more than the general population. That might be a factor that draws them to emergency work, the researchers suggest.
Some predictors, such as psychiatric history, are fixed, the researchers note. But others, such as cognitive styles, can be modified or taught. Studies have shown that rumination can be redirected through training in concrete thinking, for instance, and psychoeducation and cognitive behavioral techniques (eg, modifying interpretations of stressful events) have been used to strengthen resilience. The predictors they identified in their study could serve as targets, the researchers suggest, for modifying future resilience programs.
Source:
Wild J, Smith KV, Thompson E, Béar F, Lommen MJ, Ehlers A. Psychol Med. 2016;46(12):2571-2582. doi: 10.1017/S0033291716000532.
Identifying people who might be at risk for posttraumatic stress disorder (PTSD) before the trauma—and teaching them preventive coping skills—could reduce or prevent long-term effects, according to University of Oxford in Oxford, United Kingdom, and King’s College London, United Kingdom, researchers.
They assessed 453 newly recruited paramedics every 4 months for 2 years. Of those, 386 paramedics participated in follow-up interviews.
Related: Let’s Dance: A Holistic Approach to Treating Veterans With Posttraumatic Stress Disorder
Over the 2 years, 32 participants (8.3%) had an episode of PTSD, and 41 participants had (10.6%) an episode of major depression (MD). Most of the episodes were moderate and short lived. In most cases, the participant had recovered by the next 4-month assessment. However, at 2 years, those who had experienced episodes of PTSD or MD during the follow-up period reported more days off work, poorer sleep, poorer quality of life, and greater burn out as well as weight gain (mean gain, 6.9 kg) for those with P
Ten participants who developed PTSD received treatment during follow-up, as did 12 participants who developed MD. Five of 9 participants who had recurrent P
Related: Telehealth for Native Americans With PTSD
The researchers tested a number of possible pretrauma predictors of PTSD and MD. They correlated several: cognitive style (eg, suppression, rumination, intentional numbing), coping style (eg, avoidant styles, such as wishful thinking), and psychological traits (eg, neuroticism). However, they found rumination about memories of stressful events uniquely predicted an episode of PTSD. Perceived resilience uniquely predicted an episode of MD.
Interestingly, about 42% of the study participants had a psychiatric history before training—more than the general population. That might be a factor that draws them to emergency work, the researchers suggest.
Some predictors, such as psychiatric history, are fixed, the researchers note. But others, such as cognitive styles, can be modified or taught. Studies have shown that rumination can be redirected through training in concrete thinking, for instance, and psychoeducation and cognitive behavioral techniques (eg, modifying interpretations of stressful events) have been used to strengthen resilience. The predictors they identified in their study could serve as targets, the researchers suggest, for modifying future resilience programs.
Source:
Wild J, Smith KV, Thompson E, Béar F, Lommen MJ, Ehlers A. Psychol Med. 2016;46(12):2571-2582. doi: 10.1017/S0033291716000532.
Identifying people who might be at risk for posttraumatic stress disorder (PTSD) before the trauma—and teaching them preventive coping skills—could reduce or prevent long-term effects, according to University of Oxford in Oxford, United Kingdom, and King’s College London, United Kingdom, researchers.
They assessed 453 newly recruited paramedics every 4 months for 2 years. Of those, 386 paramedics participated in follow-up interviews.
Related: Let’s Dance: A Holistic Approach to Treating Veterans With Posttraumatic Stress Disorder
Over the 2 years, 32 participants (8.3%) had an episode of PTSD, and 41 participants had (10.6%) an episode of major depression (MD). Most of the episodes were moderate and short lived. In most cases, the participant had recovered by the next 4-month assessment. However, at 2 years, those who had experienced episodes of PTSD or MD during the follow-up period reported more days off work, poorer sleep, poorer quality of life, and greater burn out as well as weight gain (mean gain, 6.9 kg) for those with P
Ten participants who developed PTSD received treatment during follow-up, as did 12 participants who developed MD. Five of 9 participants who had recurrent P
Related: Telehealth for Native Americans With PTSD
The researchers tested a number of possible pretrauma predictors of PTSD and MD. They correlated several: cognitive style (eg, suppression, rumination, intentional numbing), coping style (eg, avoidant styles, such as wishful thinking), and psychological traits (eg, neuroticism). However, they found rumination about memories of stressful events uniquely predicted an episode of PTSD. Perceived resilience uniquely predicted an episode of MD.
Interestingly, about 42% of the study participants had a psychiatric history before training—more than the general population. That might be a factor that draws them to emergency work, the researchers suggest.
Some predictors, such as psychiatric history, are fixed, the researchers note. But others, such as cognitive styles, can be modified or taught. Studies have shown that rumination can be redirected through training in concrete thinking, for instance, and psychoeducation and cognitive behavioral techniques (eg, modifying interpretations of stressful events) have been used to strengthen resilience. The predictors they identified in their study could serve as targets, the researchers suggest, for modifying future resilience programs.
Source:
Wild J, Smith KV, Thompson E, Béar F, Lommen MJ, Ehlers A. Psychol Med. 2016;46(12):2571-2582. doi: 10.1017/S0033291716000532.
Improving cognitive function in cancer survivors

chemotherapy
Photo by Rhoda Baer
A new study suggests a computer program known as InsightTM may help improve cognitive function and overall well-being in cancer survivors.
The study included subjects who reported persistent problems with concentration and/or memory after receiving chemotherapy.
Using the Insight program significantly improved the subjects’ self-reported cognitive function and lowered their levels of anxiety, depression, fatigue, and stress.
However, there was no significant difference in the results of objective neuropsychological function tests between subjects who used the Insight program and subjects who received standard care.
These results were published in the Journal of Clinical Oncology.
“To the best of our knowledge, this is the largest cognitive intervention study that has shown a benefit for patients who are reporting persistent cognitive symptoms following chemotherapy,” said study author Victoria J. Bray, MD, of the University of Sydney in Australia.
About the study
Dr Bray and her colleagues enrolled 242 adult cancer survivors in Australia who had completed at least 3 cycles of chemotherapy in the prior 6 months to 60 months and reported persistent cognitive symptoms.
The subjects’ median age was 53 (range, 23 to 74). Nearly all were women (95%), and 89% had survived breast cancer.
At the beginning of the study, all subjects received a personalized, 30-minute telephone consultation that provided tips and strategies for coping with cognitive problems in daily life.
Subjects were then randomized to Insight (used at home) or standard oncology care per their treating physician.
The primary outcome of the study was self-reported cognitive function, which was assessed using a validated questionnaire known as FACT-COG. It evaluates perceived cognitive impairments, perceived cognitive abilities, and the impact of perceived cognitive impairment on quality of life.
Separate measures were used to evaluate objective neuropsychological function, anxiety/depression, fatigue, and stress.
About the intervention
Dr Bray and her colleagues described Insight as a neurocognitive learning program that uses exercises intended to improve cognition through speed and accuracy of information processing.
The program targets visual precision, divided attention, working memory, field of view, and visual processing speed, which are frequently affected in patients with cancer.
Subjects received the Insight program in disc form and were advised to use the program for 40 minutes 4 times a week for 15 weeks (40 hours total). The program had a built-in measure that could determine subjects’ compliance.
Key findings
Self-reported cognitive function was significantly better in the Insight group than the standard care group, both at the end of the 15-week program and 6 months later.
In addition, Insight participants had significantly lower levels of anxiety, depression, and fatigue immediately after the intervention, significant improvements in quality of life at 6 months, and significant improvements in stress at both time points.
Results of objective neuropsychological function tests were not significantly different between the Insight group and the standard care group, either immediately after the intervention or at 6 months.
Next steps
The researchers said longer follow-up is needed to determine if the effects of the Insight program are long-lasting. And there are still a number of other unanswered questions to be addressed in future research.
For one, it is unclear which method of delivering cognitive rehabilitation is better. A self-directed program such as this one may be suitable for some cancer survivors, while a group-based program may work better for others. It’s also unclear what the ideal duration and “dose” of cognitive training should be.
“If we could identify patients who are at risk of cognitive impairment, we could intervene earlier and possibly achieve even better results,” Dr Bray said. “We would also like to explore whether there is added benefit from combining cognitive training with physical exercise.” ![]()
chemotherapy
Photo by Rhoda Baer
A new study suggests a computer program known as InsightTM may help improve cognitive function and overall well-being in cancer survivors.
The study included subjects who reported persistent problems with concentration and/or memory after receiving chemotherapy.
Using the Insight program significantly improved the subjects’ self-reported cognitive function and lowered their levels of anxiety, depression, fatigue, and stress.
However, there was no significant difference in the results of objective neuropsychological function tests between subjects who used the Insight program and subjects who received standard care.
These results were published in the Journal of Clinical Oncology.
“To the best of our knowledge, this is the largest cognitive intervention study that has shown a benefit for patients who are reporting persistent cognitive symptoms following chemotherapy,” said study author Victoria J. Bray, MD, of the University of Sydney in Australia.
About the study
Dr Bray and her colleagues enrolled 242 adult cancer survivors in Australia who had completed at least 3 cycles of chemotherapy in the prior 6 months to 60 months and reported persistent cognitive symptoms.
The subjects’ median age was 53 (range, 23 to 74). Nearly all were women (95%), and 89% had survived breast cancer.
At the beginning of the study, all subjects received a personalized, 30-minute telephone consultation that provided tips and strategies for coping with cognitive problems in daily life.
Subjects were then randomized to Insight (used at home) or standard oncology care per their treating physician.
The primary outcome of the study was self-reported cognitive function, which was assessed using a validated questionnaire known as FACT-COG. It evaluates perceived cognitive impairments, perceived cognitive abilities, and the impact of perceived cognitive impairment on quality of life.
Separate measures were used to evaluate objective neuropsychological function, anxiety/depression, fatigue, and stress.
About the intervention
Dr Bray and her colleagues described Insight as a neurocognitive learning program that uses exercises intended to improve cognition through speed and accuracy of information processing.
The program targets visual precision, divided attention, working memory, field of view, and visual processing speed, which are frequently affected in patients with cancer.
Subjects received the Insight program in disc form and were advised to use the program for 40 minutes 4 times a week for 15 weeks (40 hours total). The program had a built-in measure that could determine subjects’ compliance.
Key findings
Self-reported cognitive function was significantly better in the Insight group than the standard care group, both at the end of the 15-week program and 6 months later.
In addition, Insight participants had significantly lower levels of anxiety, depression, and fatigue immediately after the intervention, significant improvements in quality of life at 6 months, and significant improvements in stress at both time points.
Results of objective neuropsychological function tests were not significantly different between the Insight group and the standard care group, either immediately after the intervention or at 6 months.
Next steps
The researchers said longer follow-up is needed to determine if the effects of the Insight program are long-lasting. And there are still a number of other unanswered questions to be addressed in future research.
For one, it is unclear which method of delivering cognitive rehabilitation is better. A self-directed program such as this one may be suitable for some cancer survivors, while a group-based program may work better for others. It’s also unclear what the ideal duration and “dose” of cognitive training should be.
“If we could identify patients who are at risk of cognitive impairment, we could intervene earlier and possibly achieve even better results,” Dr Bray said. “We would also like to explore whether there is added benefit from combining cognitive training with physical exercise.” ![]()
chemotherapy
Photo by Rhoda Baer
A new study suggests a computer program known as InsightTM may help improve cognitive function and overall well-being in cancer survivors.
The study included subjects who reported persistent problems with concentration and/or memory after receiving chemotherapy.
Using the Insight program significantly improved the subjects’ self-reported cognitive function and lowered their levels of anxiety, depression, fatigue, and stress.
However, there was no significant difference in the results of objective neuropsychological function tests between subjects who used the Insight program and subjects who received standard care.
These results were published in the Journal of Clinical Oncology.
“To the best of our knowledge, this is the largest cognitive intervention study that has shown a benefit for patients who are reporting persistent cognitive symptoms following chemotherapy,” said study author Victoria J. Bray, MD, of the University of Sydney in Australia.
About the study
Dr Bray and her colleagues enrolled 242 adult cancer survivors in Australia who had completed at least 3 cycles of chemotherapy in the prior 6 months to 60 months and reported persistent cognitive symptoms.
The subjects’ median age was 53 (range, 23 to 74). Nearly all were women (95%), and 89% had survived breast cancer.
At the beginning of the study, all subjects received a personalized, 30-minute telephone consultation that provided tips and strategies for coping with cognitive problems in daily life.
Subjects were then randomized to Insight (used at home) or standard oncology care per their treating physician.
The primary outcome of the study was self-reported cognitive function, which was assessed using a validated questionnaire known as FACT-COG. It evaluates perceived cognitive impairments, perceived cognitive abilities, and the impact of perceived cognitive impairment on quality of life.
Separate measures were used to evaluate objective neuropsychological function, anxiety/depression, fatigue, and stress.
About the intervention
Dr Bray and her colleagues described Insight as a neurocognitive learning program that uses exercises intended to improve cognition through speed and accuracy of information processing.
The program targets visual precision, divided attention, working memory, field of view, and visual processing speed, which are frequently affected in patients with cancer.
Subjects received the Insight program in disc form and were advised to use the program for 40 minutes 4 times a week for 15 weeks (40 hours total). The program had a built-in measure that could determine subjects’ compliance.
Key findings
Self-reported cognitive function was significantly better in the Insight group than the standard care group, both at the end of the 15-week program and 6 months later.
In addition, Insight participants had significantly lower levels of anxiety, depression, and fatigue immediately after the intervention, significant improvements in quality of life at 6 months, and significant improvements in stress at both time points.
Results of objective neuropsychological function tests were not significantly different between the Insight group and the standard care group, either immediately after the intervention or at 6 months.
Next steps
The researchers said longer follow-up is needed to determine if the effects of the Insight program are long-lasting. And there are still a number of other unanswered questions to be addressed in future research.
For one, it is unclear which method of delivering cognitive rehabilitation is better. A self-directed program such as this one may be suitable for some cancer survivors, while a group-based program may work better for others. It’s also unclear what the ideal duration and “dose” of cognitive training should be.
“If we could identify patients who are at risk of cognitive impairment, we could intervene earlier and possibly achieve even better results,” Dr Bray said. “We would also like to explore whether there is added benefit from combining cognitive training with physical exercise.” ![]()
Team maps genomic landscape of CBF-AML

whole-genome sequencing
Photo courtesy of the US
Food and Drug Administration
Whole-genome and whole-exome sequencing has provided new insight into the pathogenesis and development of core-binding factor acute myeloid leukemia (CBF-AML), according to researchers.
The team said their work has revealed “dramatic” differences in the genomic landscape of CBF-AMLs that contribute to the diversity of this disease.
The researchers reported their findings in Nature Genetics.
“We set out to understand the genetic variations that contribute to the development of CBF-AML using whole-exome and whole-genome sequencing,” said study author Jeffery Klco, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Our goal was to define a detailed mutational landscape to understand better the genetic changes that contribute to disease.”
Dr Klco and his colleagues sequenced samples from 87 children and 78 adults with CBF-AML. Eighty-five of the patients had the RUNX1-RUNX1T1 subtype, and 80 had the CBFB-MYH11 subtype.
Development and relapse
The researchers identified several genes with mutations that may contribute to CBF-AML development, including CCND2, DHX15, ASXL2, ZBTB7A, and MGA.
“Many of the mutations we identified interfered with molecular signaling or epigenetic factors,” said study author Jinghui Zhang, PhD, of St. Jude Children’s Research Hospital.
“Some of the mutations, like ASXL2, are epigenetic regulators that modify the local state of chromatin,” Dr Klco noted. “Others, like ZBTB7A, appear to act like tumor suppressors.”
The researchers also compared mutations present at diagnosis and relapse in an attempt to understand how CBF-AML changes over time.
Their results suggested that KMT2C mutations are associated with relapse. Of the 4 patients in this study who had KMT2C mutations, 3 relapsed in less than 12 months, and the fourth had residual disease after a course of remission-induction therapy.
Similarities and differences
The researchers found a similar mutational landscape in adults and children with CBF-AML but differences between patients with the RUNX1-RUNX1T1 and CBFB-MYH11 subtypes.
NRAS was the most frequently mutated gene in CBF-AMLs, but NRAS mutations were more common in CBFB-MYH11 AML than RUNX1-RUNX1T1 AML. The same was true for mutations in NF1 and WT1.
Patients with both subtypes of CBF-AML had mutations in NRAS, KIT, NF1, WT1, FLT3, KRAS, MGA, TTN, CCND2, KDM6A, PHIP, TET2, HCN1, KMT2C, and SETD2.
But only patients with CBFB-MYH11 AML had mutations in PTPN11.
Only patients with RUNX1-RUNX1T1 AML had mutations in ASXL2, ZBTB7A, EZH2, SMC1A, DHX15, RAD21, CBL, DNM2, CSF3R, GIGYF2, SMC3, and ZNF687.
The researchers said their findings suggest a range of mutations may play roles in CBF-AML, but additional research is needed to confirm their precise function in the disease.
Further studies are already underway to fully evaluate the contributions of the different genes as well as the roles of the newly identified genetic alterations in CBF-AML. ![]()
whole-genome sequencing
Photo courtesy of the US
Food and Drug Administration
Whole-genome and whole-exome sequencing has provided new insight into the pathogenesis and development of core-binding factor acute myeloid leukemia (CBF-AML), according to researchers.
The team said their work has revealed “dramatic” differences in the genomic landscape of CBF-AMLs that contribute to the diversity of this disease.
The researchers reported their findings in Nature Genetics.
“We set out to understand the genetic variations that contribute to the development of CBF-AML using whole-exome and whole-genome sequencing,” said study author Jeffery Klco, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Our goal was to define a detailed mutational landscape to understand better the genetic changes that contribute to disease.”
Dr Klco and his colleagues sequenced samples from 87 children and 78 adults with CBF-AML. Eighty-five of the patients had the RUNX1-RUNX1T1 subtype, and 80 had the CBFB-MYH11 subtype.
Development and relapse
The researchers identified several genes with mutations that may contribute to CBF-AML development, including CCND2, DHX15, ASXL2, ZBTB7A, and MGA.
“Many of the mutations we identified interfered with molecular signaling or epigenetic factors,” said study author Jinghui Zhang, PhD, of St. Jude Children’s Research Hospital.
“Some of the mutations, like ASXL2, are epigenetic regulators that modify the local state of chromatin,” Dr Klco noted. “Others, like ZBTB7A, appear to act like tumor suppressors.”
The researchers also compared mutations present at diagnosis and relapse in an attempt to understand how CBF-AML changes over time.
Their results suggested that KMT2C mutations are associated with relapse. Of the 4 patients in this study who had KMT2C mutations, 3 relapsed in less than 12 months, and the fourth had residual disease after a course of remission-induction therapy.
Similarities and differences
The researchers found a similar mutational landscape in adults and children with CBF-AML but differences between patients with the RUNX1-RUNX1T1 and CBFB-MYH11 subtypes.
NRAS was the most frequently mutated gene in CBF-AMLs, but NRAS mutations were more common in CBFB-MYH11 AML than RUNX1-RUNX1T1 AML. The same was true for mutations in NF1 and WT1.
Patients with both subtypes of CBF-AML had mutations in NRAS, KIT, NF1, WT1, FLT3, KRAS, MGA, TTN, CCND2, KDM6A, PHIP, TET2, HCN1, KMT2C, and SETD2.
But only patients with CBFB-MYH11 AML had mutations in PTPN11.
Only patients with RUNX1-RUNX1T1 AML had mutations in ASXL2, ZBTB7A, EZH2, SMC1A, DHX15, RAD21, CBL, DNM2, CSF3R, GIGYF2, SMC3, and ZNF687.
The researchers said their findings suggest a range of mutations may play roles in CBF-AML, but additional research is needed to confirm their precise function in the disease.
Further studies are already underway to fully evaluate the contributions of the different genes as well as the roles of the newly identified genetic alterations in CBF-AML. ![]()
whole-genome sequencing
Photo courtesy of the US
Food and Drug Administration
Whole-genome and whole-exome sequencing has provided new insight into the pathogenesis and development of core-binding factor acute myeloid leukemia (CBF-AML), according to researchers.
The team said their work has revealed “dramatic” differences in the genomic landscape of CBF-AMLs that contribute to the diversity of this disease.
The researchers reported their findings in Nature Genetics.
“We set out to understand the genetic variations that contribute to the development of CBF-AML using whole-exome and whole-genome sequencing,” said study author Jeffery Klco, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Our goal was to define a detailed mutational landscape to understand better the genetic changes that contribute to disease.”
Dr Klco and his colleagues sequenced samples from 87 children and 78 adults with CBF-AML. Eighty-five of the patients had the RUNX1-RUNX1T1 subtype, and 80 had the CBFB-MYH11 subtype.
Development and relapse
The researchers identified several genes with mutations that may contribute to CBF-AML development, including CCND2, DHX15, ASXL2, ZBTB7A, and MGA.
“Many of the mutations we identified interfered with molecular signaling or epigenetic factors,” said study author Jinghui Zhang, PhD, of St. Jude Children’s Research Hospital.
“Some of the mutations, like ASXL2, are epigenetic regulators that modify the local state of chromatin,” Dr Klco noted. “Others, like ZBTB7A, appear to act like tumor suppressors.”
The researchers also compared mutations present at diagnosis and relapse in an attempt to understand how CBF-AML changes over time.
Their results suggested that KMT2C mutations are associated with relapse. Of the 4 patients in this study who had KMT2C mutations, 3 relapsed in less than 12 months, and the fourth had residual disease after a course of remission-induction therapy.
Similarities and differences
The researchers found a similar mutational landscape in adults and children with CBF-AML but differences between patients with the RUNX1-RUNX1T1 and CBFB-MYH11 subtypes.
NRAS was the most frequently mutated gene in CBF-AMLs, but NRAS mutations were more common in CBFB-MYH11 AML than RUNX1-RUNX1T1 AML. The same was true for mutations in NF1 and WT1.
Patients with both subtypes of CBF-AML had mutations in NRAS, KIT, NF1, WT1, FLT3, KRAS, MGA, TTN, CCND2, KDM6A, PHIP, TET2, HCN1, KMT2C, and SETD2.
But only patients with CBFB-MYH11 AML had mutations in PTPN11.
Only patients with RUNX1-RUNX1T1 AML had mutations in ASXL2, ZBTB7A, EZH2, SMC1A, DHX15, RAD21, CBL, DNM2, CSF3R, GIGYF2, SMC3, and ZNF687.
The researchers said their findings suggest a range of mutations may play roles in CBF-AML, but additional research is needed to confirm their precise function in the disease.
Further studies are already underway to fully evaluate the contributions of the different genes as well as the roles of the newly identified genetic alterations in CBF-AML. ![]()
Continuous Rd should be standard of care, group says

Photo courtesy of Celgene
Updated trial results support continuous treatment with lenalidomide and low-dose dexamethasone (Rd) as a standard of care for patients of all ages who have newly diagnosed multiple myeloma (MM) and are ineligible for stem cell transplant, according to researchers.
In this phase 3 trial, patients who received continuous Rd (until disease progression) had better progression-free survival (PFS) and overall survival (OS) than patients who received 18 cycles of Rd (Rd18) or a combination of melphalan, prednisone, and thalidomide (MPT).
Updated results from this study, known as the FIRST trial, were published in the Journal of Clinical Oncology.
Results were previously published in NEJM in 2014. The study was supported by Intergroupe Francophone du Myélome and Celgene Corporation, the makers of lenalidomide.
Thierry Facon, MD, of Centre Hospitalier Regional Universitaire de Lille in France, and his colleagues enrolled 1623 patients on this trial. They were newly diagnosed with MM and not eligible for stem cell transplant.
Patients were randomized to receive Rd in 28-day cycles until disease progression (n=535), Rd18 for 72 weeks (n=541), or MPT for 72 weeks (n=547).
Response
In the intent-to-treat population, the overall response rate was 81% for the continuous Rd group, 79% for the Rd18 group, and 67% in the MPT group. The complete response rates were 21%, 20%, and 12%, respectively.
The median duration of response was 32 months (range, 26-37) in the continuous Rd group, 22 months (range, 19-23) in the Rd18 group, and 22 months (range, 20-25) in the MPT group.
PFS and OS
The median PFS was 26.0 months in the continuous Rd group, 21.0 months in the Rd18 group, and 21.9 months in the MPT group. At 4 years, the PFS rate was 33%, 14%, and 13%, respectively.
The hazard ratio (HR) for continuous Rd vs MPT was 0.69. The HR for continuous Rd vs Rd18 was 0.71. And the HR for Rd18 vs MPT was 0.99.
The median OS was 58.9 months in the continuous Rd group, 56.7 months in the Rd18 group, and 48.5 months in the MPT group. At 4 years, the OS rate was 60%, 57%, and 51%, respectively.
The HR for continuous Rd vs MPT was 0.75. The HR for continuous Rd vs Rd18 was 0.91. And the HR for Rd18 vs MPT was 0.83.
Safety
The most frequent grade 3/4 hematologic treatment-emergent adverse events were neutropenia and anemia. The rate of grade 3/4 neutropenia was higher in the MPT group than the continuous Rd or Rd18 groups.
Infections were the most common grade 3/4 non-hematologic treatment-emergent adverse events. The rate of grade 3/4 infections was higher in the Rd groups than the MPT group.
Grade 3/4 peripheral sensory neuropathy was less frequent in the continuous Rd and Rd18 groups than the MPT group.
The incidence of invasive second primary malignancy was 4% in the continuous Rd group, 6% in the Rd18 group, and 6% in the MPT group. ![]()
Photo courtesy of Celgene
Updated trial results support continuous treatment with lenalidomide and low-dose dexamethasone (Rd) as a standard of care for patients of all ages who have newly diagnosed multiple myeloma (MM) and are ineligible for stem cell transplant, according to researchers.
In this phase 3 trial, patients who received continuous Rd (until disease progression) had better progression-free survival (PFS) and overall survival (OS) than patients who received 18 cycles of Rd (Rd18) or a combination of melphalan, prednisone, and thalidomide (MPT).
Updated results from this study, known as the FIRST trial, were published in the Journal of Clinical Oncology.
Results were previously published in NEJM in 2014. The study was supported by Intergroupe Francophone du Myélome and Celgene Corporation, the makers of lenalidomide.
Thierry Facon, MD, of Centre Hospitalier Regional Universitaire de Lille in France, and his colleagues enrolled 1623 patients on this trial. They were newly diagnosed with MM and not eligible for stem cell transplant.
Patients were randomized to receive Rd in 28-day cycles until disease progression (n=535), Rd18 for 72 weeks (n=541), or MPT for 72 weeks (n=547).
Response
In the intent-to-treat population, the overall response rate was 81% for the continuous Rd group, 79% for the Rd18 group, and 67% in the MPT group. The complete response rates were 21%, 20%, and 12%, respectively.
The median duration of response was 32 months (range, 26-37) in the continuous Rd group, 22 months (range, 19-23) in the Rd18 group, and 22 months (range, 20-25) in the MPT group.
PFS and OS
The median PFS was 26.0 months in the continuous Rd group, 21.0 months in the Rd18 group, and 21.9 months in the MPT group. At 4 years, the PFS rate was 33%, 14%, and 13%, respectively.
The hazard ratio (HR) for continuous Rd vs MPT was 0.69. The HR for continuous Rd vs Rd18 was 0.71. And the HR for Rd18 vs MPT was 0.99.
The median OS was 58.9 months in the continuous Rd group, 56.7 months in the Rd18 group, and 48.5 months in the MPT group. At 4 years, the OS rate was 60%, 57%, and 51%, respectively.
The HR for continuous Rd vs MPT was 0.75. The HR for continuous Rd vs Rd18 was 0.91. And the HR for Rd18 vs MPT was 0.83.
Safety
The most frequent grade 3/4 hematologic treatment-emergent adverse events were neutropenia and anemia. The rate of grade 3/4 neutropenia was higher in the MPT group than the continuous Rd or Rd18 groups.
Infections were the most common grade 3/4 non-hematologic treatment-emergent adverse events. The rate of grade 3/4 infections was higher in the Rd groups than the MPT group.
Grade 3/4 peripheral sensory neuropathy was less frequent in the continuous Rd and Rd18 groups than the MPT group.
The incidence of invasive second primary malignancy was 4% in the continuous Rd group, 6% in the Rd18 group, and 6% in the MPT group. ![]()
Photo courtesy of Celgene
Updated trial results support continuous treatment with lenalidomide and low-dose dexamethasone (Rd) as a standard of care for patients of all ages who have newly diagnosed multiple myeloma (MM) and are ineligible for stem cell transplant, according to researchers.
In this phase 3 trial, patients who received continuous Rd (until disease progression) had better progression-free survival (PFS) and overall survival (OS) than patients who received 18 cycles of Rd (Rd18) or a combination of melphalan, prednisone, and thalidomide (MPT).
Updated results from this study, known as the FIRST trial, were published in the Journal of Clinical Oncology.
Results were previously published in NEJM in 2014. The study was supported by Intergroupe Francophone du Myélome and Celgene Corporation, the makers of lenalidomide.
Thierry Facon, MD, of Centre Hospitalier Regional Universitaire de Lille in France, and his colleagues enrolled 1623 patients on this trial. They were newly diagnosed with MM and not eligible for stem cell transplant.
Patients were randomized to receive Rd in 28-day cycles until disease progression (n=535), Rd18 for 72 weeks (n=541), or MPT for 72 weeks (n=547).
Response
In the intent-to-treat population, the overall response rate was 81% for the continuous Rd group, 79% for the Rd18 group, and 67% in the MPT group. The complete response rates were 21%, 20%, and 12%, respectively.
The median duration of response was 32 months (range, 26-37) in the continuous Rd group, 22 months (range, 19-23) in the Rd18 group, and 22 months (range, 20-25) in the MPT group.
PFS and OS
The median PFS was 26.0 months in the continuous Rd group, 21.0 months in the Rd18 group, and 21.9 months in the MPT group. At 4 years, the PFS rate was 33%, 14%, and 13%, respectively.
The hazard ratio (HR) for continuous Rd vs MPT was 0.69. The HR for continuous Rd vs Rd18 was 0.71. And the HR for Rd18 vs MPT was 0.99.
The median OS was 58.9 months in the continuous Rd group, 56.7 months in the Rd18 group, and 48.5 months in the MPT group. At 4 years, the OS rate was 60%, 57%, and 51%, respectively.
The HR for continuous Rd vs MPT was 0.75. The HR for continuous Rd vs Rd18 was 0.91. And the HR for Rd18 vs MPT was 0.83.
Safety
The most frequent grade 3/4 hematologic treatment-emergent adverse events were neutropenia and anemia. The rate of grade 3/4 neutropenia was higher in the MPT group than the continuous Rd or Rd18 groups.
Infections were the most common grade 3/4 non-hematologic treatment-emergent adverse events. The rate of grade 3/4 infections was higher in the Rd groups than the MPT group.
Grade 3/4 peripheral sensory neuropathy was less frequent in the continuous Rd and Rd18 groups than the MPT group.
The incidence of invasive second primary malignancy was 4% in the continuous Rd group, 6% in the Rd18 group, and 6% in the MPT group. ![]()
Results support continued study of CMV vaccine

CMV infection
A vaccine designed to control cytomegalovirus (CMV) has produced favorable results in a phase 1 trial of healthy volunteers.
Investigators said the vaccine, known as Triplex, was well-tolerated at multiple dose levels.
The vaccine also generated “robust” and “durable” virus-specific immunity in subjects who were previously infected with CMV and those who were not.
The results of this study were published in Blood.
Triplex is a universal (non-HLA-restricted), recombinant modified vaccinia ankara viral vector vaccine engineered to induce a virus-specific T-cell response to 3 immuno-dominant proteins (UL83 [pp65], UL123 [IE1], and UL122 [IE2]) linked to CMV complications in the post-transplant setting.
Helocyte Inc. is developing the vaccine for control of CMV in recipients of allogeneic hematopoietic stem cell transplant (HSCT) and solid organ transplant.
This study was not funded by Helocyte. However, investigator Don J. Diamond, PhD, of City of Hope Comprehensive Cancer Center in Duarte, California is chair of Helocyte’s scientific advisory board and receives personal service fees from the company.
In this trial, Dr Diamond and his colleagues studied the response to Triplex in 24 healthy volunteers.
Subjects were divided into 3 groups of 8 receiving 3 different doses of the vaccine. The first dose level was 10xE7 plaque-forming units (pfu), the second was 5x10E7 pfu, and the third was 5x10E8 pfu.
The subjects received the vaccine in a volume of 1 mL by intramuscular injection in the upper arm and an identical booster injection 28 days later.
Safety
The investigators said Triplex was well tolerated in most subjects at all dose levels. There were no dose-limiting toxicities and no serious adverse events attributed to the vaccine.
One subject experienced a grade 3 injection site adverse event (erythema), which resolved in 1 day without treatment. In addition, there were 3 mild-to-moderate cutaneous reactions.
The investigators said the most common systemic reaction was mild-to-moderate flu-like symptoms. Most subjects in the highest dose group experienced these symptoms, as did a few subjects from the lower dose groups. All of these events were transient, self-limiting, and resolved.
Immunogenicity
The investigators reported “robust, functional, and durable” expansion of CMV-specific T cells after Triplex vaccination, in subjects with and without prior CMV infection.
At day 42, subjects had experienced a significant increase in pp65-specific T cells from baseline. The P values were 0.0003 for pp65-specific CD137+ CD8+ T cells and 0.001 for CD137+ CD4+ T cells.
Expansion remained above baseline levels until at least day 360 for pp65-specific CD137+ CD8+ T cells and at least until day 482 for pp65-specific CD137+ CD4+ T cells.
IE1-exon4- and IE2-exon5-specific T-cell expansions occurred as well, although the increase from baseline was not significant for IE1-exon4-specific T cells.
The median concentrations of IE2-exon5-specific T cells had increased significantly from baseline at day 42—for both CD137+ CD8+ T cells (P=0.014) and CD137+ CD4+ T cells (P=0.003).
The investigators noted that there was no significant difference in the responses to all 3 CMV libraries according to Triplex dose level, previous smallpox vaccination, or CMV-serostatus.
Next, the team found that Triplex vaccination induced significant expansion of pp65-specific IFN-γ+ CD8+ T cells. They said this suggests the vaccine was able to expand a functional subset of CMV-specific T cells, even in the absence of CMV viremia.
The median concentration of pp65-specific IFN-γ + CD8+ T cells increased significantly from baseline to day 28 (P=0.024), day 56 (P=0.003), day 100 (P=0.011), and day 360 (P=0.085).
There was no significant increase for pp65-specific IFN-γ + CD4+ T cells, IE1-exon4-specific IFN-γ+ T cells, or IE2-exon5-specific IFN-γ + T cells.
The investigators also said the Triplex vaccine induced significant vaccinia-specific T-cell increases by day 42 (P=0.0005), with an estimated decline to pre-vaccination levels at day 274.
These data supported the initiation of an ongoing phase 2 trial in which investigators are evaluating Triplex in patients undergoing allogeneic HSCT (NCT02506933).
“After years of work, it is very gratifying that we are making advancements in helping people worldwide achieve better health outcomes after a transplant procedure,” said Dr Diamond, who led the team that developed Triplex.
“Furthermore, Triplex’s favorable safety and immunogenicity may make the vaccine an ideal therapeutic platform to combat significant complications in many disease areas, like solid organ transplant and glioblastoma.” ![]()
CMV infection
A vaccine designed to control cytomegalovirus (CMV) has produced favorable results in a phase 1 trial of healthy volunteers.
Investigators said the vaccine, known as Triplex, was well-tolerated at multiple dose levels.
The vaccine also generated “robust” and “durable” virus-specific immunity in subjects who were previously infected with CMV and those who were not.
The results of this study were published in Blood.
Triplex is a universal (non-HLA-restricted), recombinant modified vaccinia ankara viral vector vaccine engineered to induce a virus-specific T-cell response to 3 immuno-dominant proteins (UL83 [pp65], UL123 [IE1], and UL122 [IE2]) linked to CMV complications in the post-transplant setting.
Helocyte Inc. is developing the vaccine for control of CMV in recipients of allogeneic hematopoietic stem cell transplant (HSCT) and solid organ transplant.
This study was not funded by Helocyte. However, investigator Don J. Diamond, PhD, of City of Hope Comprehensive Cancer Center in Duarte, California is chair of Helocyte’s scientific advisory board and receives personal service fees from the company.
In this trial, Dr Diamond and his colleagues studied the response to Triplex in 24 healthy volunteers.
Subjects were divided into 3 groups of 8 receiving 3 different doses of the vaccine. The first dose level was 10xE7 plaque-forming units (pfu), the second was 5x10E7 pfu, and the third was 5x10E8 pfu.
The subjects received the vaccine in a volume of 1 mL by intramuscular injection in the upper arm and an identical booster injection 28 days later.
Safety
The investigators said Triplex was well tolerated in most subjects at all dose levels. There were no dose-limiting toxicities and no serious adverse events attributed to the vaccine.
One subject experienced a grade 3 injection site adverse event (erythema), which resolved in 1 day without treatment. In addition, there were 3 mild-to-moderate cutaneous reactions.
The investigators said the most common systemic reaction was mild-to-moderate flu-like symptoms. Most subjects in the highest dose group experienced these symptoms, as did a few subjects from the lower dose groups. All of these events were transient, self-limiting, and resolved.
Immunogenicity
The investigators reported “robust, functional, and durable” expansion of CMV-specific T cells after Triplex vaccination, in subjects with and without prior CMV infection.
At day 42, subjects had experienced a significant increase in pp65-specific T cells from baseline. The P values were 0.0003 for pp65-specific CD137+ CD8+ T cells and 0.001 for CD137+ CD4+ T cells.
Expansion remained above baseline levels until at least day 360 for pp65-specific CD137+ CD8+ T cells and at least until day 482 for pp65-specific CD137+ CD4+ T cells.
IE1-exon4- and IE2-exon5-specific T-cell expansions occurred as well, although the increase from baseline was not significant for IE1-exon4-specific T cells.
The median concentrations of IE2-exon5-specific T cells had increased significantly from baseline at day 42—for both CD137+ CD8+ T cells (P=0.014) and CD137+ CD4+ T cells (P=0.003).
The investigators noted that there was no significant difference in the responses to all 3 CMV libraries according to Triplex dose level, previous smallpox vaccination, or CMV-serostatus.
Next, the team found that Triplex vaccination induced significant expansion of pp65-specific IFN-γ+ CD8+ T cells. They said this suggests the vaccine was able to expand a functional subset of CMV-specific T cells, even in the absence of CMV viremia.
The median concentration of pp65-specific IFN-γ + CD8+ T cells increased significantly from baseline to day 28 (P=0.024), day 56 (P=0.003), day 100 (P=0.011), and day 360 (P=0.085).
There was no significant increase for pp65-specific IFN-γ + CD4+ T cells, IE1-exon4-specific IFN-γ+ T cells, or IE2-exon5-specific IFN-γ + T cells.
The investigators also said the Triplex vaccine induced significant vaccinia-specific T-cell increases by day 42 (P=0.0005), with an estimated decline to pre-vaccination levels at day 274.
These data supported the initiation of an ongoing phase 2 trial in which investigators are evaluating Triplex in patients undergoing allogeneic HSCT (NCT02506933).
“After years of work, it is very gratifying that we are making advancements in helping people worldwide achieve better health outcomes after a transplant procedure,” said Dr Diamond, who led the team that developed Triplex.
“Furthermore, Triplex’s favorable safety and immunogenicity may make the vaccine an ideal therapeutic platform to combat significant complications in many disease areas, like solid organ transplant and glioblastoma.” ![]()
CMV infection
A vaccine designed to control cytomegalovirus (CMV) has produced favorable results in a phase 1 trial of healthy volunteers.
Investigators said the vaccine, known as Triplex, was well-tolerated at multiple dose levels.
The vaccine also generated “robust” and “durable” virus-specific immunity in subjects who were previously infected with CMV and those who were not.
The results of this study were published in Blood.
Triplex is a universal (non-HLA-restricted), recombinant modified vaccinia ankara viral vector vaccine engineered to induce a virus-specific T-cell response to 3 immuno-dominant proteins (UL83 [pp65], UL123 [IE1], and UL122 [IE2]) linked to CMV complications in the post-transplant setting.
Helocyte Inc. is developing the vaccine for control of CMV in recipients of allogeneic hematopoietic stem cell transplant (HSCT) and solid organ transplant.
This study was not funded by Helocyte. However, investigator Don J. Diamond, PhD, of City of Hope Comprehensive Cancer Center in Duarte, California is chair of Helocyte’s scientific advisory board and receives personal service fees from the company.
In this trial, Dr Diamond and his colleagues studied the response to Triplex in 24 healthy volunteers.
Subjects were divided into 3 groups of 8 receiving 3 different doses of the vaccine. The first dose level was 10xE7 plaque-forming units (pfu), the second was 5x10E7 pfu, and the third was 5x10E8 pfu.
The subjects received the vaccine in a volume of 1 mL by intramuscular injection in the upper arm and an identical booster injection 28 days later.
Safety
The investigators said Triplex was well tolerated in most subjects at all dose levels. There were no dose-limiting toxicities and no serious adverse events attributed to the vaccine.
One subject experienced a grade 3 injection site adverse event (erythema), which resolved in 1 day without treatment. In addition, there were 3 mild-to-moderate cutaneous reactions.
The investigators said the most common systemic reaction was mild-to-moderate flu-like symptoms. Most subjects in the highest dose group experienced these symptoms, as did a few subjects from the lower dose groups. All of these events were transient, self-limiting, and resolved.
Immunogenicity
The investigators reported “robust, functional, and durable” expansion of CMV-specific T cells after Triplex vaccination, in subjects with and without prior CMV infection.
At day 42, subjects had experienced a significant increase in pp65-specific T cells from baseline. The P values were 0.0003 for pp65-specific CD137+ CD8+ T cells and 0.001 for CD137+ CD4+ T cells.
Expansion remained above baseline levels until at least day 360 for pp65-specific CD137+ CD8+ T cells and at least until day 482 for pp65-specific CD137+ CD4+ T cells.
IE1-exon4- and IE2-exon5-specific T-cell expansions occurred as well, although the increase from baseline was not significant for IE1-exon4-specific T cells.
The median concentrations of IE2-exon5-specific T cells had increased significantly from baseline at day 42—for both CD137+ CD8+ T cells (P=0.014) and CD137+ CD4+ T cells (P=0.003).
The investigators noted that there was no significant difference in the responses to all 3 CMV libraries according to Triplex dose level, previous smallpox vaccination, or CMV-serostatus.
Next, the team found that Triplex vaccination induced significant expansion of pp65-specific IFN-γ+ CD8+ T cells. They said this suggests the vaccine was able to expand a functional subset of CMV-specific T cells, even in the absence of CMV viremia.
The median concentration of pp65-specific IFN-γ + CD8+ T cells increased significantly from baseline to day 28 (P=0.024), day 56 (P=0.003), day 100 (P=0.011), and day 360 (P=0.085).
There was no significant increase for pp65-specific IFN-γ + CD4+ T cells, IE1-exon4-specific IFN-γ+ T cells, or IE2-exon5-specific IFN-γ + T cells.
The investigators also said the Triplex vaccine induced significant vaccinia-specific T-cell increases by day 42 (P=0.0005), with an estimated decline to pre-vaccination levels at day 274.
These data supported the initiation of an ongoing phase 2 trial in which investigators are evaluating Triplex in patients undergoing allogeneic HSCT (NCT02506933).
“After years of work, it is very gratifying that we are making advancements in helping people worldwide achieve better health outcomes after a transplant procedure,” said Dr Diamond, who led the team that developed Triplex.
“Furthermore, Triplex’s favorable safety and immunogenicity may make the vaccine an ideal therapeutic platform to combat significant complications in many disease areas, like solid organ transplant and glioblastoma.” ![]()
A girl repeatedly jabs her finger up her nose: Compulsion or self-injury?
CASE Anxious and self-injurious
A, age 6, is forcibly inserting her finger into her nose repeatedly until she bleeds profusely, as many as 20 times per day. She is not nose-picking but is jabbing her finger into her nose as far as possible in a repetitive ramming motion. Less frequently, she inserts her finger into her vagina, resulting in chronic urinary tract infections (UTIs). She has bedtime checking rituals; worries that her parents will die; has a fear of vomiting to the point where she stopped eating normally and lost 5 lb in 6 months; intense fear of storms; refusal to use public bathrooms; and involuntary throat clearing, facial grimacing, and lip twitches.
A’s symptoms began at age 3. There is no history of physical or sexual abuse. She does well in school, but these behaviors have had a significant impact on her social functioning. She is not taking any medications and has been in weekly cognitive-behavioral therapy (CBT) for the last year. A has had several UTIs but otherwise is physically healthy.
Which diagnosis best describes A’s condition?
a) non-suicidal self-injury (NSSI)
b) generalized anxiety disorder (GAD)
c) obsessive-compulsive disorder (OCD)
d) Tourette’s disorder (TD)
The authors’ observations
A is causing herself to bleed and says she wants to stop this behavior. Onset of NSSI typically is age 12 to 14 and could be accompanied by traits of cluster B personality disorders.1 In A’s case, her age and absence of any stated desire to relieve stress or intense negative affective states rules out NSSI.
Because A has multiple and frequent fears, worries, and anxieties that have been present for years and have caused significant functional impairment, a diagnosis of GAD is warranted. Because she has had both motor and vocal tics for more than 1 year, she also meets diagnostic criteria for TD (Table 1).

In young children, OCD manifests primarily with compulsive behavior, such as excessive hand washing, counting, and ordering, that interferes with functioning. Although A has bedtime checking rituals, she has no significant functional impairment from these rituals alone. A’s finger-insertion behavior could be interpreted as a complex motor tic or as a compulsion, in which case impairment was significant enough to justify a diagnosis of OCD.
Many individuals with OCD report the need to engage in compulsive behavior to decrease anxiety or until they experience a “just right” feeling.2 However, neither A nor her mother reported the need for the “just right” feeling. The child recognized the urge to put her finger in her nose and did experience relief of anxiety after drawing blood. Although A said that she was unable to control her hands, she was observed frequently touching the side of her nose in an attempt to avoid inserting her finger in her nose.
Compulsive behavior that results in self-injury typically is not seen in OCD except in children with severe neurologic complications, low intellectual functioning, psychosis, or autism.3
It often is difficult to determine if complex motor or vocal tics are compulsions (Table 2). Indeed, the same biologic mechanisms are thought to be implicated in TD and OCD.4 A significant percentage of children with OCD have tics, and patients often report that they are unable to distinguish between compulsions and complex tics.5 Therefore, we thought that a reasonable differential included both TD and OCD, but more careful assessment over time was required.

Treatment options
A has been receiving CBT for more than 1 year but her symptoms were worsening, which prompted her parents to seek evaluation in our clinic. Because of the level of interference with daily functioning and significant distress, our priority was developing a treatment plan that has the best chance of quickly reducing symptom severity and frequency. The results of the large-scale Pediatric OCD Treatment Study (POTS), which evaluated children age 7 to 17, and the Child/Adolescent Multimodal Anxiety Study, which evaluated children age <12, indicated that the combination of CBT with a selective serotonin reuptake inhibitor (SSRI) reduced OCD symptoms more than either modality alone.6,7 Considerations for using SSRIs in this age group include:
- the risk of behavioral activation
- poor tolerability
- lack of an evidence base for dosage optimization.
The American Academy of Child and Adolescent Psychiatry’s Preschool Psychopharmacology Working Group’s guidelines for treating anxiety in preschoolers state that pharmacotherapeutic intervention can be considered when symptoms are intolerable and adequate psychotherapy interventions have been tried.8 In A’s case, she had been receiving CBT for a year without improvement in symptoms; therefore, initiating medication was indicated, as well as an examination of therapeutic modalities being used.
Treatment Next steps
A is started on liquid fluoxetine, 20 mg/5 mL, 1 mL (4 mg) daily, because of her inability to swallow pills and her young age. According to her mother, a week later A is sleeping better and seems happier. The entire family seems less stressed. During the third week, A successfully goes on a camping trip with her family and is starting to eat better. Her finger-in-nose insertions still are occurring but, according to her mother, she is not putting her finger in her vagina. In session, she is not observed putting her finger in her nose or touching her nose, which she had done frequently during the initial evaluation. Fluoxetine seems to be well tolerated and the dosage is increased to 2 mL (8 mg) per day.
Although A has weekly scheduled appointments, she is not brought in again until a month later. At that time her mother reports an approximately 40% improvement in overall symptoms, including less frequent nose-insertion behaviors.
What type of psychotherapy would you employ for A?
a) CBT
b) behavioral therapy
c) habit reversal training (HRT)
d) pharmacotherapy alone
The authors’ observations
The treatment team planned to begin psychotherapy after A showed a decrease in anxiety and frequency of problem behaviors to a point where she could benefit. Evidence-based treatment for compulsions and tics is CBT and/or HRT.9 However, clinicians frequently encounter special challenges in helping young children (age 5 to 8) who have OCD. Factors such as family functioning, parental accommodation to the child’s symptoms, and the child’s ability to understand symptoms, exposure and response prevention, and willingness to tolerate discomfort should be considered if treatment is to be effective.
Research has shown that including parents when treating anxious children—especially young children—can facilitate gains and hasten positive outcomes.10,11 The POTS Jr study showed the relative efficacy of a family-based CBT model for young children with OCD that emphasizes consistent involvement of parents in all phases of treatment.12 In this case, A and her mother were seen together for psychotherapy, with an initial focus on learning more about the antecedents and consequences of the child’s behaviors.
OUTCOME Inconsistencies
Treatment was initiated during the summer. With the upcoming start of the school year, A begins to complain of daily headache, stomachache, and anxiety related to the start of school. Fluoxetine is increased to 3 mL/d (12 mg/d). After school starts, her mother stops going to work and begins attending school daily with A to relieve both her and the child’s anxiety.
The following week, the mother pages the psychiatrist, hysterical and crying because she thought the child was “pulling her hair out so much she looks like a cancer survivor.” Both parents blame the increase in fluoxetine for the heightened anxiety. At the next visit, the treatment team does not notice any evidence of unusual hair loss on the child. A has not attended school for several weeks, and her mother has not returned to work. Her parents report that the finger-to-nose behavior has increased, although it is not observed during the session, and fluoxetine is tapered as her parents requested.
At the next session, her mother notes a significant increase in finger-to-nose behavior and requests that the child be put back on fluoxetine, saying, “I would give anything to have the child I had on Prozac back.”
How would you proceed?
a) confront the mother’s inconsistencies
b) restart fluoxetine and continue psychotherapy
c) refer A to another clinic or therapist
d) refer A to inpatient care
The authors’ observations
The treatment team identified several barriers to successful treatment in our clinic. The level of functional interference caused by A’s symptoms indicated sessions more often than once a week, but the parents felt that the distance from our clinic to their home made this too difficult. The mother’s anxiety and obvious distress over her daughter’s symptoms precluded working closely with child. Parental anxiety is correlated with the child’s anxiety and can moderate treatment outcome.11 In response to the suffering of their anxious children, especially young ones, parents often will become anxious and accommodate to the child’s symptoms, which we strongly suspected was happening with A’s mother.
Parents’ concerns about A’s symptoms and response to treatment were addressed during a family meeting. Recognizing that the level of care needed by this family was higher than could be provided in our clinic, we recommended referral to a specialty clinic. A was brought to another clinic, and treatment at our facility was terminated.
1. Klonsky ED. The functions of deliberate self-injury: a review of the evidence. Clin Psychol Rev. 2007;27(2):226-239.
2. Miguel EC, do Rosário-Campos MC, Prado HS, et al. Sensory phenomena in obsessive-compulsive disorder and Tourette’s disorder. J Clin Psychiatry. 2000;61(2):150-156.
3. Nock MK, Favazza A. Non-suicidal self-injury: definition and classification. In: Nock MK, ed. Understanding nonsuicidal self-injury: origins, assessment, and treatment. Washington, DC: American Psychological Association; 2009:9-18.
4. Goodman WK, Storch EA, Geffken GR, et al. Obsessive-compulsive disorder in Tourette syndrome. J Child Neurol. 2006;21(8):704-714.
5. Garcia AM, Freeman JB, Himle MB, et al. Phenomenology of early childhood onset obsessive-compulsive disorder. J Psychopathol Behav Assess. 2009;31(2):104-111.
6. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
7. Piacentini JC, Bennett S, Compton SN, et al. 24- and 36-week outcomes for the Child/Adolescent Anxiety Multimodal Study (CAMS). J Am Acad Child Adolesc Psychiatry. 2014;53(3):297-310.
8. Gleason MM, Egger HL, Emslie GJ, et al. Psychopharmacological treatment for very young children: contexts and guidelines. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1532-1572.
9. Abramowitz JS, Whiteside SP, Deacon BJ. The effectiveness of treatment for pediatric obsessive-compulsive disorder: a meta-analysis. Behavior Therapy. 2005;36(1):55-63.
10. Barmish AJ, Kendall PC. Should parents be co-clients in cognitive-behavioral therapy for anxious youth. J Clin Child Adolesc Psychol. 2005;34(3):569-581.
11. Drake KL, Ginsburg GS. Family factors in the development, treatment, and prevention of childhood anxiety disorders. Clin Child Fam Psychol Rev. 2012;15(2):144-162.
12. Freeman J, Sapyta J, Garcia A, et al. Family-based treatment of early childhood obsessive-compulsive disorder: the Pediatric Obsessive-Compulsive Disorder Treatment Study for Young Children (POTS Jr)—a randomized clinical trial. JAMA Psychiatry. 2014;71(6):689-698.
CASE Anxious and self-injurious
A, age 6, is forcibly inserting her finger into her nose repeatedly until she bleeds profusely, as many as 20 times per day. She is not nose-picking but is jabbing her finger into her nose as far as possible in a repetitive ramming motion. Less frequently, she inserts her finger into her vagina, resulting in chronic urinary tract infections (UTIs). She has bedtime checking rituals; worries that her parents will die; has a fear of vomiting to the point where she stopped eating normally and lost 5 lb in 6 months; intense fear of storms; refusal to use public bathrooms; and involuntary throat clearing, facial grimacing, and lip twitches.
A’s symptoms began at age 3. There is no history of physical or sexual abuse. She does well in school, but these behaviors have had a significant impact on her social functioning. She is not taking any medications and has been in weekly cognitive-behavioral therapy (CBT) for the last year. A has had several UTIs but otherwise is physically healthy.
Which diagnosis best describes A’s condition?
a) non-suicidal self-injury (NSSI)
b) generalized anxiety disorder (GAD)
c) obsessive-compulsive disorder (OCD)
d) Tourette’s disorder (TD)
The authors’ observations
A is causing herself to bleed and says she wants to stop this behavior. Onset of NSSI typically is age 12 to 14 and could be accompanied by traits of cluster B personality disorders.1 In A’s case, her age and absence of any stated desire to relieve stress or intense negative affective states rules out NSSI.
Because A has multiple and frequent fears, worries, and anxieties that have been present for years and have caused significant functional impairment, a diagnosis of GAD is warranted. Because she has had both motor and vocal tics for more than 1 year, she also meets diagnostic criteria for TD (Table 1).
In young children, OCD manifests primarily with compulsive behavior, such as excessive hand washing, counting, and ordering, that interferes with functioning. Although A has bedtime checking rituals, she has no significant functional impairment from these rituals alone. A’s finger-insertion behavior could be interpreted as a complex motor tic or as a compulsion, in which case impairment was significant enough to justify a diagnosis of OCD.
Many individuals with OCD report the need to engage in compulsive behavior to decrease anxiety or until they experience a “just right” feeling.2 However, neither A nor her mother reported the need for the “just right” feeling. The child recognized the urge to put her finger in her nose and did experience relief of anxiety after drawing blood. Although A said that she was unable to control her hands, she was observed frequently touching the side of her nose in an attempt to avoid inserting her finger in her nose.
Compulsive behavior that results in self-injury typically is not seen in OCD except in children with severe neurologic complications, low intellectual functioning, psychosis, or autism.3
It often is difficult to determine if complex motor or vocal tics are compulsions (Table 2). Indeed, the same biologic mechanisms are thought to be implicated in TD and OCD.4 A significant percentage of children with OCD have tics, and patients often report that they are unable to distinguish between compulsions and complex tics.5 Therefore, we thought that a reasonable differential included both TD and OCD, but more careful assessment over time was required.
Treatment options
A has been receiving CBT for more than 1 year but her symptoms were worsening, which prompted her parents to seek evaluation in our clinic. Because of the level of interference with daily functioning and significant distress, our priority was developing a treatment plan that has the best chance of quickly reducing symptom severity and frequency. The results of the large-scale Pediatric OCD Treatment Study (POTS), which evaluated children age 7 to 17, and the Child/Adolescent Multimodal Anxiety Study, which evaluated children age <12, indicated that the combination of CBT with a selective serotonin reuptake inhibitor (SSRI) reduced OCD symptoms more than either modality alone.6,7 Considerations for using SSRIs in this age group include:
- the risk of behavioral activation
- poor tolerability
- lack of an evidence base for dosage optimization.
The American Academy of Child and Adolescent Psychiatry’s Preschool Psychopharmacology Working Group’s guidelines for treating anxiety in preschoolers state that pharmacotherapeutic intervention can be considered when symptoms are intolerable and adequate psychotherapy interventions have been tried.8 In A’s case, she had been receiving CBT for a year without improvement in symptoms; therefore, initiating medication was indicated, as well as an examination of therapeutic modalities being used.
Treatment Next steps
A is started on liquid fluoxetine, 20 mg/5 mL, 1 mL (4 mg) daily, because of her inability to swallow pills and her young age. According to her mother, a week later A is sleeping better and seems happier. The entire family seems less stressed. During the third week, A successfully goes on a camping trip with her family and is starting to eat better. Her finger-in-nose insertions still are occurring but, according to her mother, she is not putting her finger in her vagina. In session, she is not observed putting her finger in her nose or touching her nose, which she had done frequently during the initial evaluation. Fluoxetine seems to be well tolerated and the dosage is increased to 2 mL (8 mg) per day.
Although A has weekly scheduled appointments, she is not brought in again until a month later. At that time her mother reports an approximately 40% improvement in overall symptoms, including less frequent nose-insertion behaviors.
What type of psychotherapy would you employ for A?
a) CBT
b) behavioral therapy
c) habit reversal training (HRT)
d) pharmacotherapy alone
The authors’ observations
The treatment team planned to begin psychotherapy after A showed a decrease in anxiety and frequency of problem behaviors to a point where she could benefit. Evidence-based treatment for compulsions and tics is CBT and/or HRT.9 However, clinicians frequently encounter special challenges in helping young children (age 5 to 8) who have OCD. Factors such as family functioning, parental accommodation to the child’s symptoms, and the child’s ability to understand symptoms, exposure and response prevention, and willingness to tolerate discomfort should be considered if treatment is to be effective.
Research has shown that including parents when treating anxious children—especially young children—can facilitate gains and hasten positive outcomes.10,11 The POTS Jr study showed the relative efficacy of a family-based CBT model for young children with OCD that emphasizes consistent involvement of parents in all phases of treatment.12 In this case, A and her mother were seen together for psychotherapy, with an initial focus on learning more about the antecedents and consequences of the child’s behaviors.
OUTCOME Inconsistencies
Treatment was initiated during the summer. With the upcoming start of the school year, A begins to complain of daily headache, stomachache, and anxiety related to the start of school. Fluoxetine is increased to 3 mL/d (12 mg/d). After school starts, her mother stops going to work and begins attending school daily with A to relieve both her and the child’s anxiety.
The following week, the mother pages the psychiatrist, hysterical and crying because she thought the child was “pulling her hair out so much she looks like a cancer survivor.” Both parents blame the increase in fluoxetine for the heightened anxiety. At the next visit, the treatment team does not notice any evidence of unusual hair loss on the child. A has not attended school for several weeks, and her mother has not returned to work. Her parents report that the finger-to-nose behavior has increased, although it is not observed during the session, and fluoxetine is tapered as her parents requested.
At the next session, her mother notes a significant increase in finger-to-nose behavior and requests that the child be put back on fluoxetine, saying, “I would give anything to have the child I had on Prozac back.”
How would you proceed?
a) confront the mother’s inconsistencies
b) restart fluoxetine and continue psychotherapy
c) refer A to another clinic or therapist
d) refer A to inpatient care
The authors’ observations
The treatment team identified several barriers to successful treatment in our clinic. The level of functional interference caused by A’s symptoms indicated sessions more often than once a week, but the parents felt that the distance from our clinic to their home made this too difficult. The mother’s anxiety and obvious distress over her daughter’s symptoms precluded working closely with child. Parental anxiety is correlated with the child’s anxiety and can moderate treatment outcome.11 In response to the suffering of their anxious children, especially young ones, parents often will become anxious and accommodate to the child’s symptoms, which we strongly suspected was happening with A’s mother.
Parents’ concerns about A’s symptoms and response to treatment were addressed during a family meeting. Recognizing that the level of care needed by this family was higher than could be provided in our clinic, we recommended referral to a specialty clinic. A was brought to another clinic, and treatment at our facility was terminated.
CASE Anxious and self-injurious
A, age 6, is forcibly inserting her finger into her nose repeatedly until she bleeds profusely, as many as 20 times per day. She is not nose-picking but is jabbing her finger into her nose as far as possible in a repetitive ramming motion. Less frequently, she inserts her finger into her vagina, resulting in chronic urinary tract infections (UTIs). She has bedtime checking rituals; worries that her parents will die; has a fear of vomiting to the point where she stopped eating normally and lost 5 lb in 6 months; intense fear of storms; refusal to use public bathrooms; and involuntary throat clearing, facial grimacing, and lip twitches.
A’s symptoms began at age 3. There is no history of physical or sexual abuse. She does well in school, but these behaviors have had a significant impact on her social functioning. She is not taking any medications and has been in weekly cognitive-behavioral therapy (CBT) for the last year. A has had several UTIs but otherwise is physically healthy.
Which diagnosis best describes A’s condition?
a) non-suicidal self-injury (NSSI)
b) generalized anxiety disorder (GAD)
c) obsessive-compulsive disorder (OCD)
d) Tourette’s disorder (TD)
The authors’ observations
A is causing herself to bleed and says she wants to stop this behavior. Onset of NSSI typically is age 12 to 14 and could be accompanied by traits of cluster B personality disorders.1 In A’s case, her age and absence of any stated desire to relieve stress or intense negative affective states rules out NSSI.
Because A has multiple and frequent fears, worries, and anxieties that have been present for years and have caused significant functional impairment, a diagnosis of GAD is warranted. Because she has had both motor and vocal tics for more than 1 year, she also meets diagnostic criteria for TD (Table 1).
In young children, OCD manifests primarily with compulsive behavior, such as excessive hand washing, counting, and ordering, that interferes with functioning. Although A has bedtime checking rituals, she has no significant functional impairment from these rituals alone. A’s finger-insertion behavior could be interpreted as a complex motor tic or as a compulsion, in which case impairment was significant enough to justify a diagnosis of OCD.
Many individuals with OCD report the need to engage in compulsive behavior to decrease anxiety or until they experience a “just right” feeling.2 However, neither A nor her mother reported the need for the “just right” feeling. The child recognized the urge to put her finger in her nose and did experience relief of anxiety after drawing blood. Although A said that she was unable to control her hands, she was observed frequently touching the side of her nose in an attempt to avoid inserting her finger in her nose.
Compulsive behavior that results in self-injury typically is not seen in OCD except in children with severe neurologic complications, low intellectual functioning, psychosis, or autism.3
It often is difficult to determine if complex motor or vocal tics are compulsions (Table 2). Indeed, the same biologic mechanisms are thought to be implicated in TD and OCD.4 A significant percentage of children with OCD have tics, and patients often report that they are unable to distinguish between compulsions and complex tics.5 Therefore, we thought that a reasonable differential included both TD and OCD, but more careful assessment over time was required.
Treatment options
A has been receiving CBT for more than 1 year but her symptoms were worsening, which prompted her parents to seek evaluation in our clinic. Because of the level of interference with daily functioning and significant distress, our priority was developing a treatment plan that has the best chance of quickly reducing symptom severity and frequency. The results of the large-scale Pediatric OCD Treatment Study (POTS), which evaluated children age 7 to 17, and the Child/Adolescent Multimodal Anxiety Study, which evaluated children age <12, indicated that the combination of CBT with a selective serotonin reuptake inhibitor (SSRI) reduced OCD symptoms more than either modality alone.6,7 Considerations for using SSRIs in this age group include:
- the risk of behavioral activation
- poor tolerability
- lack of an evidence base for dosage optimization.
The American Academy of Child and Adolescent Psychiatry’s Preschool Psychopharmacology Working Group’s guidelines for treating anxiety in preschoolers state that pharmacotherapeutic intervention can be considered when symptoms are intolerable and adequate psychotherapy interventions have been tried.8 In A’s case, she had been receiving CBT for a year without improvement in symptoms; therefore, initiating medication was indicated, as well as an examination of therapeutic modalities being used.
Treatment Next steps
A is started on liquid fluoxetine, 20 mg/5 mL, 1 mL (4 mg) daily, because of her inability to swallow pills and her young age. According to her mother, a week later A is sleeping better and seems happier. The entire family seems less stressed. During the third week, A successfully goes on a camping trip with her family and is starting to eat better. Her finger-in-nose insertions still are occurring but, according to her mother, she is not putting her finger in her vagina. In session, she is not observed putting her finger in her nose or touching her nose, which she had done frequently during the initial evaluation. Fluoxetine seems to be well tolerated and the dosage is increased to 2 mL (8 mg) per day.
Although A has weekly scheduled appointments, she is not brought in again until a month later. At that time her mother reports an approximately 40% improvement in overall symptoms, including less frequent nose-insertion behaviors.
What type of psychotherapy would you employ for A?
a) CBT
b) behavioral therapy
c) habit reversal training (HRT)
d) pharmacotherapy alone
The authors’ observations
The treatment team planned to begin psychotherapy after A showed a decrease in anxiety and frequency of problem behaviors to a point where she could benefit. Evidence-based treatment for compulsions and tics is CBT and/or HRT.9 However, clinicians frequently encounter special challenges in helping young children (age 5 to 8) who have OCD. Factors such as family functioning, parental accommodation to the child’s symptoms, and the child’s ability to understand symptoms, exposure and response prevention, and willingness to tolerate discomfort should be considered if treatment is to be effective.
Research has shown that including parents when treating anxious children—especially young children—can facilitate gains and hasten positive outcomes.10,11 The POTS Jr study showed the relative efficacy of a family-based CBT model for young children with OCD that emphasizes consistent involvement of parents in all phases of treatment.12 In this case, A and her mother were seen together for psychotherapy, with an initial focus on learning more about the antecedents and consequences of the child’s behaviors.
OUTCOME Inconsistencies
Treatment was initiated during the summer. With the upcoming start of the school year, A begins to complain of daily headache, stomachache, and anxiety related to the start of school. Fluoxetine is increased to 3 mL/d (12 mg/d). After school starts, her mother stops going to work and begins attending school daily with A to relieve both her and the child’s anxiety.
The following week, the mother pages the psychiatrist, hysterical and crying because she thought the child was “pulling her hair out so much she looks like a cancer survivor.” Both parents blame the increase in fluoxetine for the heightened anxiety. At the next visit, the treatment team does not notice any evidence of unusual hair loss on the child. A has not attended school for several weeks, and her mother has not returned to work. Her parents report that the finger-to-nose behavior has increased, although it is not observed during the session, and fluoxetine is tapered as her parents requested.
At the next session, her mother notes a significant increase in finger-to-nose behavior and requests that the child be put back on fluoxetine, saying, “I would give anything to have the child I had on Prozac back.”
How would you proceed?
a) confront the mother’s inconsistencies
b) restart fluoxetine and continue psychotherapy
c) refer A to another clinic or therapist
d) refer A to inpatient care
The authors’ observations
The treatment team identified several barriers to successful treatment in our clinic. The level of functional interference caused by A’s symptoms indicated sessions more often than once a week, but the parents felt that the distance from our clinic to their home made this too difficult. The mother’s anxiety and obvious distress over her daughter’s symptoms precluded working closely with child. Parental anxiety is correlated with the child’s anxiety and can moderate treatment outcome.11 In response to the suffering of their anxious children, especially young ones, parents often will become anxious and accommodate to the child’s symptoms, which we strongly suspected was happening with A’s mother.
Parents’ concerns about A’s symptoms and response to treatment were addressed during a family meeting. Recognizing that the level of care needed by this family was higher than could be provided in our clinic, we recommended referral to a specialty clinic. A was brought to another clinic, and treatment at our facility was terminated.
1. Klonsky ED. The functions of deliberate self-injury: a review of the evidence. Clin Psychol Rev. 2007;27(2):226-239.
2. Miguel EC, do Rosário-Campos MC, Prado HS, et al. Sensory phenomena in obsessive-compulsive disorder and Tourette’s disorder. J Clin Psychiatry. 2000;61(2):150-156.
3. Nock MK, Favazza A. Non-suicidal self-injury: definition and classification. In: Nock MK, ed. Understanding nonsuicidal self-injury: origins, assessment, and treatment. Washington, DC: American Psychological Association; 2009:9-18.
4. Goodman WK, Storch EA, Geffken GR, et al. Obsessive-compulsive disorder in Tourette syndrome. J Child Neurol. 2006;21(8):704-714.
5. Garcia AM, Freeman JB, Himle MB, et al. Phenomenology of early childhood onset obsessive-compulsive disorder. J Psychopathol Behav Assess. 2009;31(2):104-111.
6. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
7. Piacentini JC, Bennett S, Compton SN, et al. 24- and 36-week outcomes for the Child/Adolescent Anxiety Multimodal Study (CAMS). J Am Acad Child Adolesc Psychiatry. 2014;53(3):297-310.
8. Gleason MM, Egger HL, Emslie GJ, et al. Psychopharmacological treatment for very young children: contexts and guidelines. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1532-1572.
9. Abramowitz JS, Whiteside SP, Deacon BJ. The effectiveness of treatment for pediatric obsessive-compulsive disorder: a meta-analysis. Behavior Therapy. 2005;36(1):55-63.
10. Barmish AJ, Kendall PC. Should parents be co-clients in cognitive-behavioral therapy for anxious youth. J Clin Child Adolesc Psychol. 2005;34(3):569-581.
11. Drake KL, Ginsburg GS. Family factors in the development, treatment, and prevention of childhood anxiety disorders. Clin Child Fam Psychol Rev. 2012;15(2):144-162.
12. Freeman J, Sapyta J, Garcia A, et al. Family-based treatment of early childhood obsessive-compulsive disorder: the Pediatric Obsessive-Compulsive Disorder Treatment Study for Young Children (POTS Jr)—a randomized clinical trial. JAMA Psychiatry. 2014;71(6):689-698.
1. Klonsky ED. The functions of deliberate self-injury: a review of the evidence. Clin Psychol Rev. 2007;27(2):226-239.
2. Miguel EC, do Rosário-Campos MC, Prado HS, et al. Sensory phenomena in obsessive-compulsive disorder and Tourette’s disorder. J Clin Psychiatry. 2000;61(2):150-156.
3. Nock MK, Favazza A. Non-suicidal self-injury: definition and classification. In: Nock MK, ed. Understanding nonsuicidal self-injury: origins, assessment, and treatment. Washington, DC: American Psychological Association; 2009:9-18.
4. Goodman WK, Storch EA, Geffken GR, et al. Obsessive-compulsive disorder in Tourette syndrome. J Child Neurol. 2006;21(8):704-714.
5. Garcia AM, Freeman JB, Himle MB, et al. Phenomenology of early childhood onset obsessive-compulsive disorder. J Psychopathol Behav Assess. 2009;31(2):104-111.
6. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
7. Piacentini JC, Bennett S, Compton SN, et al. 24- and 36-week outcomes for the Child/Adolescent Anxiety Multimodal Study (CAMS). J Am Acad Child Adolesc Psychiatry. 2014;53(3):297-310.
8. Gleason MM, Egger HL, Emslie GJ, et al. Psychopharmacological treatment for very young children: contexts and guidelines. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1532-1572.
9. Abramowitz JS, Whiteside SP, Deacon BJ. The effectiveness of treatment for pediatric obsessive-compulsive disorder: a meta-analysis. Behavior Therapy. 2005;36(1):55-63.
10. Barmish AJ, Kendall PC. Should parents be co-clients in cognitive-behavioral therapy for anxious youth. J Clin Child Adolesc Psychol. 2005;34(3):569-581.
11. Drake KL, Ginsburg GS. Family factors in the development, treatment, and prevention of childhood anxiety disorders. Clin Child Fam Psychol Rev. 2012;15(2):144-162.
12. Freeman J, Sapyta J, Garcia A, et al. Family-based treatment of early childhood obsessive-compulsive disorder: the Pediatric Obsessive-Compulsive Disorder Treatment Study for Young Children (POTS Jr)—a randomized clinical trial. JAMA Psychiatry. 2014;71(6):689-698.