User login
When primary anti-TNF fails in axial spondyloarthritis, consider comorbidities or second anti-TNF
Most of the minority of axial spondyloarthritis patients who do not respond to their first tumor necrosis factor inhibitor still meet classification criteria for the condition 5-10 years later, but more than half respond to a second TNFi and most also have comorbidities that could affect the spondyloarthritis evaluation, according to findings from a single-center cohort study.
The study’s finding of TNFi primary inefficacy in 27 (12%) of 222 patients with axial spondyloarthritis (axSpA) who were given their first TNFi during 2004-2009 is in line with previous results of about 5%-15% primary inefficacy for a first TNFi in axSpA. However, the French investigators, led by Sandra Kossi of Cochin Hospital in Paris, noted that the difficulty of making an axSpA diagnosis and the presence of certain comorbidities “could interfere either with the activity of SpA (falsely heightened disease activity) or with the response to TNFi (falsely heightened inefficacy).” They sought to report the characteristics of axSpA patients with primary inefficacy after their first TNFi in the 5- to 10-year period after their prescription (Rheumatology [Oxford]. 2017 Jan 9. doi: 10.1093/rheumatology/kew456).
A total of 25 of the patients with primary inefficacy underwent re-evaluation 5-10 years (mean of 6 years) later. The investigators defined primary inefficacy as “treatment interruption 3-4 months after treatment onset, with a rheumatologist assessment in the medical file citing lack of efficacy as primary reason for drug interruption.”
The patients with primary TNFi inefficacy had a mean age of 53 years at the time of follow-up, and about half were female. All the patients still had symptoms and back pain, but symptoms were moderate based on a mean Bath Ankylosing Spondylitis Disease Activity Index score of 42. Overall, nine were taking a TNFi and nine were taking nonsteroidal anti-inflammatory drugs, while others used analgesics or nonpharmacologic measures.
Primary TNFi inefficacy occurred significantly more often occurred among females (48% vs. 27%; P = .04), older aged patients at first TNFi use (45 vs. 39 years; P = .04), patients with higher mean Bath Ankylosing Spondylitis Functional Index scores (68 vs. 42; P = .03), and in those who did not have an abnormally elevated C-reactive protein level (33% vs. 63%; P = .02). Patients with primary TNFi inefficacy had lower rates of HLAB27 positivity (56% vs. 72%) or radiographic sacroiliitis (67% vs. 81%), but the differences were not statistically significant.
At follow-up, 21 (84%) patients met Assessment of Spondyloarthritis International Society classification criteria for axSpA, and 20 (80%) met the axSpA diagnosis according to the rheumatologists’ opinion, with 17 (68%) fulfilling both.
A total of 18 (72%) had at least one of the following comorbidities: widespread pain syndrome, osteoarthritis, or depression. Five patients – all females – had widespread pain syndrome according to the Fibromyalgia Rapid Screening Tool questionnaire. Another 10 patients had osteoarthritis of lower-limb peripheral joints or of the spine, while an additional 8 had self-declared depression, for which 3 were taking antidepressants.
By the time of follow-up, 16 (64%) had switched to another TNFi, including 9 who had received two TNFi drugs and 7 who had received three or more. The second TNFi was considered efficacious in nine patients. The retention rate of the second TNFi at 1 year among those with primary inefficacy was 50%, and overall, nine patients were still prescribed a TNFi at the time of follow-up.
The fact that most of the patients with primary inefficacy to their first TNFi had confirmed axSpA but also had comorbidities that could affect axSpA evaluation “is important because practitioners might consider that primary inefficacy to TNFi leads to reconsidering the diagnosis of SpA (i.e. the notion of a TNFi prescription being used as the diagnostic test). We suggest here that primary inefficacy should not be considered as equivalent to a diagnostic error, and that a second prescription of TNFi may be of use in such patients, although painful comorbidities should certainly be screened for and taken into account.”
The study was funded by the Assistance Publique des Hôpitaux de Paris. The investigators had no conflicts of interest to declare.
Most of the minority of axial spondyloarthritis patients who do not respond to their first tumor necrosis factor inhibitor still meet classification criteria for the condition 5-10 years later, but more than half respond to a second TNFi and most also have comorbidities that could affect the spondyloarthritis evaluation, according to findings from a single-center cohort study.
The study’s finding of TNFi primary inefficacy in 27 (12%) of 222 patients with axial spondyloarthritis (axSpA) who were given their first TNFi during 2004-2009 is in line with previous results of about 5%-15% primary inefficacy for a first TNFi in axSpA. However, the French investigators, led by Sandra Kossi of Cochin Hospital in Paris, noted that the difficulty of making an axSpA diagnosis and the presence of certain comorbidities “could interfere either with the activity of SpA (falsely heightened disease activity) or with the response to TNFi (falsely heightened inefficacy).” They sought to report the characteristics of axSpA patients with primary inefficacy after their first TNFi in the 5- to 10-year period after their prescription (Rheumatology [Oxford]. 2017 Jan 9. doi: 10.1093/rheumatology/kew456).
A total of 25 of the patients with primary inefficacy underwent re-evaluation 5-10 years (mean of 6 years) later. The investigators defined primary inefficacy as “treatment interruption 3-4 months after treatment onset, with a rheumatologist assessment in the medical file citing lack of efficacy as primary reason for drug interruption.”
The patients with primary TNFi inefficacy had a mean age of 53 years at the time of follow-up, and about half were female. All the patients still had symptoms and back pain, but symptoms were moderate based on a mean Bath Ankylosing Spondylitis Disease Activity Index score of 42. Overall, nine were taking a TNFi and nine were taking nonsteroidal anti-inflammatory drugs, while others used analgesics or nonpharmacologic measures.
Primary TNFi inefficacy occurred significantly more often occurred among females (48% vs. 27%; P = .04), older aged patients at first TNFi use (45 vs. 39 years; P = .04), patients with higher mean Bath Ankylosing Spondylitis Functional Index scores (68 vs. 42; P = .03), and in those who did not have an abnormally elevated C-reactive protein level (33% vs. 63%; P = .02). Patients with primary TNFi inefficacy had lower rates of HLAB27 positivity (56% vs. 72%) or radiographic sacroiliitis (67% vs. 81%), but the differences were not statistically significant.
At follow-up, 21 (84%) patients met Assessment of Spondyloarthritis International Society classification criteria for axSpA, and 20 (80%) met the axSpA diagnosis according to the rheumatologists’ opinion, with 17 (68%) fulfilling both.
A total of 18 (72%) had at least one of the following comorbidities: widespread pain syndrome, osteoarthritis, or depression. Five patients – all females – had widespread pain syndrome according to the Fibromyalgia Rapid Screening Tool questionnaire. Another 10 patients had osteoarthritis of lower-limb peripheral joints or of the spine, while an additional 8 had self-declared depression, for which 3 were taking antidepressants.
By the time of follow-up, 16 (64%) had switched to another TNFi, including 9 who had received two TNFi drugs and 7 who had received three or more. The second TNFi was considered efficacious in nine patients. The retention rate of the second TNFi at 1 year among those with primary inefficacy was 50%, and overall, nine patients were still prescribed a TNFi at the time of follow-up.
The fact that most of the patients with primary inefficacy to their first TNFi had confirmed axSpA but also had comorbidities that could affect axSpA evaluation “is important because practitioners might consider that primary inefficacy to TNFi leads to reconsidering the diagnosis of SpA (i.e. the notion of a TNFi prescription being used as the diagnostic test). We suggest here that primary inefficacy should not be considered as equivalent to a diagnostic error, and that a second prescription of TNFi may be of use in such patients, although painful comorbidities should certainly be screened for and taken into account.”
The study was funded by the Assistance Publique des Hôpitaux de Paris. The investigators had no conflicts of interest to declare.
Most of the minority of axial spondyloarthritis patients who do not respond to their first tumor necrosis factor inhibitor still meet classification criteria for the condition 5-10 years later, but more than half respond to a second TNFi and most also have comorbidities that could affect the spondyloarthritis evaluation, according to findings from a single-center cohort study.
The study’s finding of TNFi primary inefficacy in 27 (12%) of 222 patients with axial spondyloarthritis (axSpA) who were given their first TNFi during 2004-2009 is in line with previous results of about 5%-15% primary inefficacy for a first TNFi in axSpA. However, the French investigators, led by Sandra Kossi of Cochin Hospital in Paris, noted that the difficulty of making an axSpA diagnosis and the presence of certain comorbidities “could interfere either with the activity of SpA (falsely heightened disease activity) or with the response to TNFi (falsely heightened inefficacy).” They sought to report the characteristics of axSpA patients with primary inefficacy after their first TNFi in the 5- to 10-year period after their prescription (Rheumatology [Oxford]. 2017 Jan 9. doi: 10.1093/rheumatology/kew456).
A total of 25 of the patients with primary inefficacy underwent re-evaluation 5-10 years (mean of 6 years) later. The investigators defined primary inefficacy as “treatment interruption 3-4 months after treatment onset, with a rheumatologist assessment in the medical file citing lack of efficacy as primary reason for drug interruption.”
The patients with primary TNFi inefficacy had a mean age of 53 years at the time of follow-up, and about half were female. All the patients still had symptoms and back pain, but symptoms were moderate based on a mean Bath Ankylosing Spondylitis Disease Activity Index score of 42. Overall, nine were taking a TNFi and nine were taking nonsteroidal anti-inflammatory drugs, while others used analgesics or nonpharmacologic measures.
Primary TNFi inefficacy occurred significantly more often occurred among females (48% vs. 27%; P = .04), older aged patients at first TNFi use (45 vs. 39 years; P = .04), patients with higher mean Bath Ankylosing Spondylitis Functional Index scores (68 vs. 42; P = .03), and in those who did not have an abnormally elevated C-reactive protein level (33% vs. 63%; P = .02). Patients with primary TNFi inefficacy had lower rates of HLAB27 positivity (56% vs. 72%) or radiographic sacroiliitis (67% vs. 81%), but the differences were not statistically significant.
At follow-up, 21 (84%) patients met Assessment of Spondyloarthritis International Society classification criteria for axSpA, and 20 (80%) met the axSpA diagnosis according to the rheumatologists’ opinion, with 17 (68%) fulfilling both.
A total of 18 (72%) had at least one of the following comorbidities: widespread pain syndrome, osteoarthritis, or depression. Five patients – all females – had widespread pain syndrome according to the Fibromyalgia Rapid Screening Tool questionnaire. Another 10 patients had osteoarthritis of lower-limb peripheral joints or of the spine, while an additional 8 had self-declared depression, for which 3 were taking antidepressants.
By the time of follow-up, 16 (64%) had switched to another TNFi, including 9 who had received two TNFi drugs and 7 who had received three or more. The second TNFi was considered efficacious in nine patients. The retention rate of the second TNFi at 1 year among those with primary inefficacy was 50%, and overall, nine patients were still prescribed a TNFi at the time of follow-up.
The fact that most of the patients with primary inefficacy to their first TNFi had confirmed axSpA but also had comorbidities that could affect axSpA evaluation “is important because practitioners might consider that primary inefficacy to TNFi leads to reconsidering the diagnosis of SpA (i.e. the notion of a TNFi prescription being used as the diagnostic test). We suggest here that primary inefficacy should not be considered as equivalent to a diagnostic error, and that a second prescription of TNFi may be of use in such patients, although painful comorbidities should certainly be screened for and taken into account.”
The study was funded by the Assistance Publique des Hôpitaux de Paris. The investigators had no conflicts of interest to declare.
Key clinical point:
Major finding: A total of 18 (72%) of 25 patients with primary TNFi inefficacy had at least one of the following comorbidities: widespread pain syndrome, osteoarthritis, or depression.
Data source: A retrospective and prospective cohort study of 222 patients who received a first TNFi after being diagnosed with axial spondyloarthritis during 2004-2009.
Disclosures: The study was funded by the Assistance Publique des Hôpitaux de Paris. The investigators had no conflicts of interest to declare.
New data signal paradigm shift in FMD and arterial disease
CHICAGO – New data have shown that fibromuscular dysplasia is associated with high rates of dissection and/or aneurysm, and emerging recommendations call for routine imaging early on in the diagnosis of FMD to monitor for these vascular events, a researcher who developed those recommendations reported at a symposium on vascular surgery sponsored by Northwestern University.
“Given the very high rate of aneurysms in this population, it is now recommended that all patients with FMD should undergo at least one-time head-to pelvis imaging with CT angiography or MR angiography to screen for the presence of an aneurysm or to identify other areas of FMD involvement,” said Daniella Kadian-Dodov, MD, of Icahn School of Medicine at Mount Sinai in New York (J Am Coll Cardiol. 2016;68:176-85).

First described in 1938, FMD is a non-atherosclerotic, noninflammatory disease that had been thought to be a rare cause of renovascular hypertension with a classic “string-of-beads” appearance upon imaging, Dr. Kadian-Dodov noted. However, recent data from the Fibromuscular Dysplasia Society of America–sponsored U.S. registry has changed that thinking. “We now know it occurs more frequently in the carotid and renal arteries, although it has been observed in almost every artery,” she said. “The pathogenesis is still unknown but up to 10% of cases are familial.”
And manifestations of disease now extend beyond the “string-of-beads” appearance to include aneurysm, dissection, and arterial tortuosity, she said (Circulation. 2012;125:3182-90; Circulation. 2014;129:1048-78; J Am Coll Cardiol. 2016;68:176-85). The classification system for FMD has also undergone a recent change, according to Dr. Kadian-Dodov. “Traditionally, a histopathologic scheme was used to classify FMD,” she said. “Nowadays, fewer and fewer patients are undergoing surgical procedures, so the classification has changed to an angiographic system,” the most common of which is the American Heart Association system adopted in 2014 that distinguishes between multifocal, characterized by the classic “string-of-beads” appearance, and focal FMD with a single area of stenosis.
But the diagnosis of either variant of FMD is not exclusive. “Patients may have multiple areas of disease involvement and the same patient may have both focal and multifocal FMD findings,” Dr. Kadian-Dodov said.
The U.S. registry has helped clarify the thinking on FMD, Dr. Kadian-Dodov said. More than 1,400 patients are in the registry, 90% of whom are women with multifocal disease. The average age of onset of symptoms is 47 years, but 52 is the average age for diagnosis. “So these patients are experiencing several years delay to FMD diagnosis,” she said.
Manifestations depend on the vascular bed involved. “In the case of cervical artery FMD, headaches and pulsatile tinnitus are commonly reported, whereas with renal artery involvement hypertension is the most common symptom,” she said. A recent analysis showed 41.7% of the FMD population have either aneurysm and dissection or both (J Am Coll Cardiol. 2016;68:176-185).
But no specific guidelines for treatment of FMD yet exist, Dr. Kadian-Dodov said. “General guidelines should be applied for the management of dissection and aneurysm in patients with FMD,” she said. For patients with arterial dissection, that means conservative therapy comprising either anticoagulation or antiplatelet agents for 3-6 months followed by daily low-dose aspirin therapy. “Revascularization is rarely required for these patients,” she said. “Endovascular or surgical modalities should be reserved for those with continued ischemia despite conservative management or more complicated pseudoaneurysm formations.”
Daily aspirin therapy is likewise the recommendation for patients with cervical artery multifocal or focal FMD involvement without dissection or aneurysms. “We follow up with imaging every 6 months for 2 years,” Dr. Kadian-Dodov said. “If they’re stable, we switch over to annual surveillance; and if the patient has an aneurysm or dissection, that might alter the imaging and surveillance program.”
During angioplasty, determining the severity of stenosis upon visual inspection is difficult, especially in multifocal FMD. She advised measuring the gradient across the area of FMD involvement with a pressure wire to determine if angioplasty has adequately treated the lesion. “You should see obliteration of the gradient with successful treatment; you don’t have to target your therapy to a perfect angiographic result,” she said.
In patients with FMD and hypertension, she recommended renal artery angioplasty for hypertension of less than 5 years duration or in resistant or labile hypertension. “In this setting, stents are only reserved for complicated or refractory cases; angioplasty alone is sufficient,” Dr. Kadian-Dodov said. Cure rates decline with age, and hypertension in focal disease has a higher cure rate than does multifocal disease, she said (Hypertension. 2010;56:525-32).
Dr. Kadian-Dodov had no relevant financial relationships to disclose.
CHICAGO – New data have shown that fibromuscular dysplasia is associated with high rates of dissection and/or aneurysm, and emerging recommendations call for routine imaging early on in the diagnosis of FMD to monitor for these vascular events, a researcher who developed those recommendations reported at a symposium on vascular surgery sponsored by Northwestern University.
“Given the very high rate of aneurysms in this population, it is now recommended that all patients with FMD should undergo at least one-time head-to pelvis imaging with CT angiography or MR angiography to screen for the presence of an aneurysm or to identify other areas of FMD involvement,” said Daniella Kadian-Dodov, MD, of Icahn School of Medicine at Mount Sinai in New York (J Am Coll Cardiol. 2016;68:176-85).
First described in 1938, FMD is a non-atherosclerotic, noninflammatory disease that had been thought to be a rare cause of renovascular hypertension with a classic “string-of-beads” appearance upon imaging, Dr. Kadian-Dodov noted. However, recent data from the Fibromuscular Dysplasia Society of America–sponsored U.S. registry has changed that thinking. “We now know it occurs more frequently in the carotid and renal arteries, although it has been observed in almost every artery,” she said. “The pathogenesis is still unknown but up to 10% of cases are familial.”
And manifestations of disease now extend beyond the “string-of-beads” appearance to include aneurysm, dissection, and arterial tortuosity, she said (Circulation. 2012;125:3182-90; Circulation. 2014;129:1048-78; J Am Coll Cardiol. 2016;68:176-85). The classification system for FMD has also undergone a recent change, according to Dr. Kadian-Dodov. “Traditionally, a histopathologic scheme was used to classify FMD,” she said. “Nowadays, fewer and fewer patients are undergoing surgical procedures, so the classification has changed to an angiographic system,” the most common of which is the American Heart Association system adopted in 2014 that distinguishes between multifocal, characterized by the classic “string-of-beads” appearance, and focal FMD with a single area of stenosis.
But the diagnosis of either variant of FMD is not exclusive. “Patients may have multiple areas of disease involvement and the same patient may have both focal and multifocal FMD findings,” Dr. Kadian-Dodov said.
The U.S. registry has helped clarify the thinking on FMD, Dr. Kadian-Dodov said. More than 1,400 patients are in the registry, 90% of whom are women with multifocal disease. The average age of onset of symptoms is 47 years, but 52 is the average age for diagnosis. “So these patients are experiencing several years delay to FMD diagnosis,” she said.
Manifestations depend on the vascular bed involved. “In the case of cervical artery FMD, headaches and pulsatile tinnitus are commonly reported, whereas with renal artery involvement hypertension is the most common symptom,” she said. A recent analysis showed 41.7% of the FMD population have either aneurysm and dissection or both (J Am Coll Cardiol. 2016;68:176-185).
But no specific guidelines for treatment of FMD yet exist, Dr. Kadian-Dodov said. “General guidelines should be applied for the management of dissection and aneurysm in patients with FMD,” she said. For patients with arterial dissection, that means conservative therapy comprising either anticoagulation or antiplatelet agents for 3-6 months followed by daily low-dose aspirin therapy. “Revascularization is rarely required for these patients,” she said. “Endovascular or surgical modalities should be reserved for those with continued ischemia despite conservative management or more complicated pseudoaneurysm formations.”
Daily aspirin therapy is likewise the recommendation for patients with cervical artery multifocal or focal FMD involvement without dissection or aneurysms. “We follow up with imaging every 6 months for 2 years,” Dr. Kadian-Dodov said. “If they’re stable, we switch over to annual surveillance; and if the patient has an aneurysm or dissection, that might alter the imaging and surveillance program.”
During angioplasty, determining the severity of stenosis upon visual inspection is difficult, especially in multifocal FMD. She advised measuring the gradient across the area of FMD involvement with a pressure wire to determine if angioplasty has adequately treated the lesion. “You should see obliteration of the gradient with successful treatment; you don’t have to target your therapy to a perfect angiographic result,” she said.
In patients with FMD and hypertension, she recommended renal artery angioplasty for hypertension of less than 5 years duration or in resistant or labile hypertension. “In this setting, stents are only reserved for complicated or refractory cases; angioplasty alone is sufficient,” Dr. Kadian-Dodov said. Cure rates decline with age, and hypertension in focal disease has a higher cure rate than does multifocal disease, she said (Hypertension. 2010;56:525-32).
Dr. Kadian-Dodov had no relevant financial relationships to disclose.
CHICAGO – New data have shown that fibromuscular dysplasia is associated with high rates of dissection and/or aneurysm, and emerging recommendations call for routine imaging early on in the diagnosis of FMD to monitor for these vascular events, a researcher who developed those recommendations reported at a symposium on vascular surgery sponsored by Northwestern University.
“Given the very high rate of aneurysms in this population, it is now recommended that all patients with FMD should undergo at least one-time head-to pelvis imaging with CT angiography or MR angiography to screen for the presence of an aneurysm or to identify other areas of FMD involvement,” said Daniella Kadian-Dodov, MD, of Icahn School of Medicine at Mount Sinai in New York (J Am Coll Cardiol. 2016;68:176-85).
First described in 1938, FMD is a non-atherosclerotic, noninflammatory disease that had been thought to be a rare cause of renovascular hypertension with a classic “string-of-beads” appearance upon imaging, Dr. Kadian-Dodov noted. However, recent data from the Fibromuscular Dysplasia Society of America–sponsored U.S. registry has changed that thinking. “We now know it occurs more frequently in the carotid and renal arteries, although it has been observed in almost every artery,” she said. “The pathogenesis is still unknown but up to 10% of cases are familial.”
And manifestations of disease now extend beyond the “string-of-beads” appearance to include aneurysm, dissection, and arterial tortuosity, she said (Circulation. 2012;125:3182-90; Circulation. 2014;129:1048-78; J Am Coll Cardiol. 2016;68:176-85). The classification system for FMD has also undergone a recent change, according to Dr. Kadian-Dodov. “Traditionally, a histopathologic scheme was used to classify FMD,” she said. “Nowadays, fewer and fewer patients are undergoing surgical procedures, so the classification has changed to an angiographic system,” the most common of which is the American Heart Association system adopted in 2014 that distinguishes between multifocal, characterized by the classic “string-of-beads” appearance, and focal FMD with a single area of stenosis.
But the diagnosis of either variant of FMD is not exclusive. “Patients may have multiple areas of disease involvement and the same patient may have both focal and multifocal FMD findings,” Dr. Kadian-Dodov said.
The U.S. registry has helped clarify the thinking on FMD, Dr. Kadian-Dodov said. More than 1,400 patients are in the registry, 90% of whom are women with multifocal disease. The average age of onset of symptoms is 47 years, but 52 is the average age for diagnosis. “So these patients are experiencing several years delay to FMD diagnosis,” she said.
Manifestations depend on the vascular bed involved. “In the case of cervical artery FMD, headaches and pulsatile tinnitus are commonly reported, whereas with renal artery involvement hypertension is the most common symptom,” she said. A recent analysis showed 41.7% of the FMD population have either aneurysm and dissection or both (J Am Coll Cardiol. 2016;68:176-185).
But no specific guidelines for treatment of FMD yet exist, Dr. Kadian-Dodov said. “General guidelines should be applied for the management of dissection and aneurysm in patients with FMD,” she said. For patients with arterial dissection, that means conservative therapy comprising either anticoagulation or antiplatelet agents for 3-6 months followed by daily low-dose aspirin therapy. “Revascularization is rarely required for these patients,” she said. “Endovascular or surgical modalities should be reserved for those with continued ischemia despite conservative management or more complicated pseudoaneurysm formations.”
Daily aspirin therapy is likewise the recommendation for patients with cervical artery multifocal or focal FMD involvement without dissection or aneurysms. “We follow up with imaging every 6 months for 2 years,” Dr. Kadian-Dodov said. “If they’re stable, we switch over to annual surveillance; and if the patient has an aneurysm or dissection, that might alter the imaging and surveillance program.”
During angioplasty, determining the severity of stenosis upon visual inspection is difficult, especially in multifocal FMD. She advised measuring the gradient across the area of FMD involvement with a pressure wire to determine if angioplasty has adequately treated the lesion. “You should see obliteration of the gradient with successful treatment; you don’t have to target your therapy to a perfect angiographic result,” she said.
In patients with FMD and hypertension, she recommended renal artery angioplasty for hypertension of less than 5 years duration or in resistant or labile hypertension. “In this setting, stents are only reserved for complicated or refractory cases; angioplasty alone is sufficient,” Dr. Kadian-Dodov said. Cure rates decline with age, and hypertension in focal disease has a higher cure rate than does multifocal disease, she said (Hypertension. 2010;56:525-32).
Dr. Kadian-Dodov had no relevant financial relationships to disclose.
Key clinical point: Fibromuscular dysplasia was thought to be a rare cause of renovascular hypertension, but new data has challenged this thinking.
Major finding: FMD accounts for 15%-20% of patients with spontaneous carotid or vertebral artery dissection and 45%-86% of patients with spontaneous coronary artery dissection.
Data source: U.S. Registry for FMD maintained by the Fibromuscular Dystrophy Society of America.
Disclosures: Dr. Kadian-Dodov reported having no financial disclosures.
Oral, liquid supplement improves clinical outcomes in lactose-intolerant adults
Adults with self-reported lactose intolerance were shown to have significant improvement in their clinical outcomes, including abdominal pain, after consuming an oral, liquid supplement intended to increase lactose-fermenting gut bacteria, M. Andrea Azcarate-Peril, PhD, assistant professor of medicine at the University of North Carolina, Chapel Hill, and her colleagues have shown in a small phase IIa study (Proc Nat Acad Sci. doi: 10.1073/pnas.1606722113).
In a placebo-controlled, double-blind trial, randomly assigned in a 2:1 ratio and conducted at two U.S. sites, highly purified (more than 95%) short-chain galactooligosaccharide (GOS) was given to 42 adults with a self-reported history of lactose intolerance, confirmed by a hydrogen breath test administered after a 25-g lactose challenge. The 20 controls were given a corn syrup mixture formulated according to the same sweetness and consistency as the test drug. Each study arm was started on its regimen at 1.5 g daily, with incremental increases in dose every 5 days until reaching 15 g. Beginning with their first dose at day 1, through day 35, all participants avoided consumption of dairy foods. Stool samples were collected from both groups at days 0 and 36. After day 36, all participants were asked to resume eating dairy foods. At day 66, stool samples were once again collected. Changes in the microbiome at all endpoints were measured by testing the stools via polymerase chain reaction.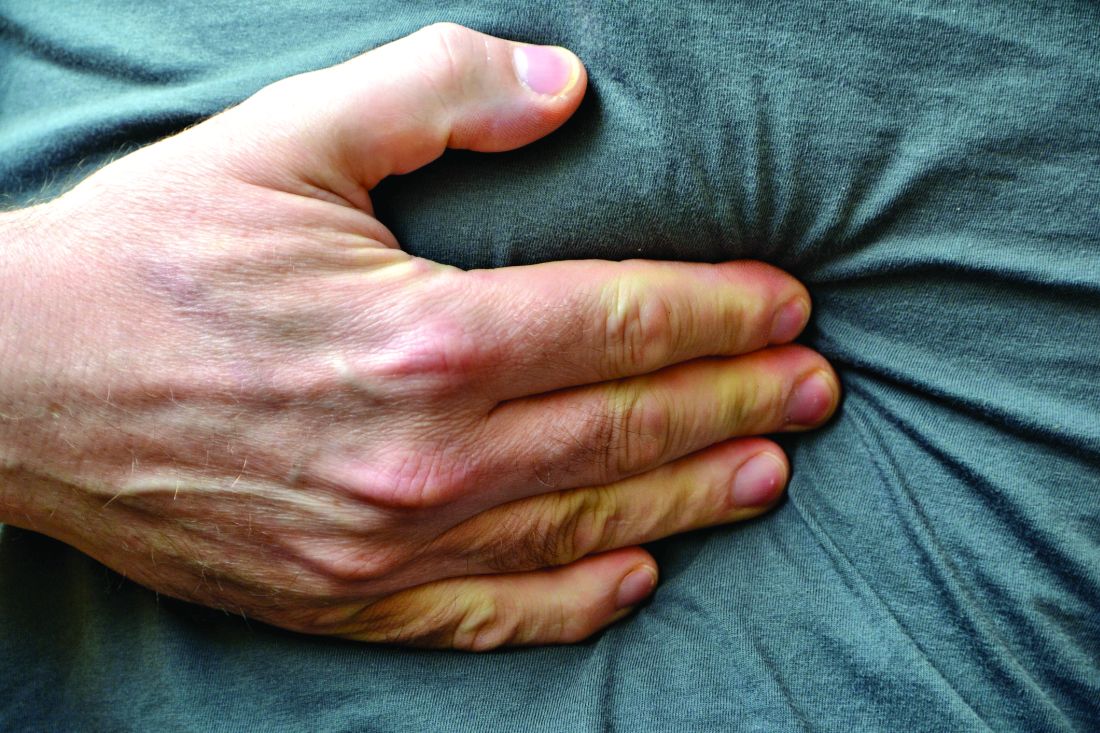
Of the 30 study arm participants for whom complete stool samples were available, 27 were found to have had a bifidobacterial response at day 36, including a significant increase in the lactose-fermenting Bifidobacterium, Faecalibacterium, and Lactobacillus species. The remaining three participants in the study arm were considered nonresponders.
In an interview, Andrew Ritter, whose company, Ritter Pharmaceuticals, sponsored the trial, reported that of the 36 study arm participants who had reported abdominal pain pretreatment, 18 said they no longer had the pain at either endpoint, day 36 or day 66 (P = .019); three of 19 in the placebo group reported they no longer had abdominal pain at either endpoint. The study group was also six times more likely to report lactose tolerance at day 66 compared with their pretreatment levels (P = .0389); 28% of the placebo arm reported lactose tolerance at the endpoints. These results were previously published in Nutrition Journal in 2013. [doi: 10.1186/1475-2891-12-160]
“We’re super excited about these results,” said Mr. Ritter. “This is really one of the first clinical studies in a lactose-intolerant population that shows changes in the microbiome.” As to how long before the treatment will be ready for the Food and Drug Administration approval process, Mr. Ritter said, “We’re probably just a couple of years away.”
Two coauthors are advisers to Ritter Pharmaceuticals, which provided the highly purified GOS used in the study. The North Carolina Agriculture Foundation also provided funding for the study.
[email protected]
On Twitter @whitneymcknight
Adults with self-reported lactose intolerance were shown to have significant improvement in their clinical outcomes, including abdominal pain, after consuming an oral, liquid supplement intended to increase lactose-fermenting gut bacteria, M. Andrea Azcarate-Peril, PhD, assistant professor of medicine at the University of North Carolina, Chapel Hill, and her colleagues have shown in a small phase IIa study (Proc Nat Acad Sci. doi: 10.1073/pnas.1606722113).
In a placebo-controlled, double-blind trial, randomly assigned in a 2:1 ratio and conducted at two U.S. sites, highly purified (more than 95%) short-chain galactooligosaccharide (GOS) was given to 42 adults with a self-reported history of lactose intolerance, confirmed by a hydrogen breath test administered after a 25-g lactose challenge. The 20 controls were given a corn syrup mixture formulated according to the same sweetness and consistency as the test drug. Each study arm was started on its regimen at 1.5 g daily, with incremental increases in dose every 5 days until reaching 15 g. Beginning with their first dose at day 1, through day 35, all participants avoided consumption of dairy foods. Stool samples were collected from both groups at days 0 and 36. After day 36, all participants were asked to resume eating dairy foods. At day 66, stool samples were once again collected. Changes in the microbiome at all endpoints were measured by testing the stools via polymerase chain reaction.
Of the 30 study arm participants for whom complete stool samples were available, 27 were found to have had a bifidobacterial response at day 36, including a significant increase in the lactose-fermenting Bifidobacterium, Faecalibacterium, and Lactobacillus species. The remaining three participants in the study arm were considered nonresponders.
In an interview, Andrew Ritter, whose company, Ritter Pharmaceuticals, sponsored the trial, reported that of the 36 study arm participants who had reported abdominal pain pretreatment, 18 said they no longer had the pain at either endpoint, day 36 or day 66 (P = .019); three of 19 in the placebo group reported they no longer had abdominal pain at either endpoint. The study group was also six times more likely to report lactose tolerance at day 66 compared with their pretreatment levels (P = .0389); 28% of the placebo arm reported lactose tolerance at the endpoints. These results were previously published in Nutrition Journal in 2013. [doi: 10.1186/1475-2891-12-160]
“We’re super excited about these results,” said Mr. Ritter. “This is really one of the first clinical studies in a lactose-intolerant population that shows changes in the microbiome.” As to how long before the treatment will be ready for the Food and Drug Administration approval process, Mr. Ritter said, “We’re probably just a couple of years away.”
Two coauthors are advisers to Ritter Pharmaceuticals, which provided the highly purified GOS used in the study. The North Carolina Agriculture Foundation also provided funding for the study.
[email protected]
On Twitter @whitneymcknight
Adults with self-reported lactose intolerance were shown to have significant improvement in their clinical outcomes, including abdominal pain, after consuming an oral, liquid supplement intended to increase lactose-fermenting gut bacteria, M. Andrea Azcarate-Peril, PhD, assistant professor of medicine at the University of North Carolina, Chapel Hill, and her colleagues have shown in a small phase IIa study (Proc Nat Acad Sci. doi: 10.1073/pnas.1606722113).
In a placebo-controlled, double-blind trial, randomly assigned in a 2:1 ratio and conducted at two U.S. sites, highly purified (more than 95%) short-chain galactooligosaccharide (GOS) was given to 42 adults with a self-reported history of lactose intolerance, confirmed by a hydrogen breath test administered after a 25-g lactose challenge. The 20 controls were given a corn syrup mixture formulated according to the same sweetness and consistency as the test drug. Each study arm was started on its regimen at 1.5 g daily, with incremental increases in dose every 5 days until reaching 15 g. Beginning with their first dose at day 1, through day 35, all participants avoided consumption of dairy foods. Stool samples were collected from both groups at days 0 and 36. After day 36, all participants were asked to resume eating dairy foods. At day 66, stool samples were once again collected. Changes in the microbiome at all endpoints were measured by testing the stools via polymerase chain reaction.
Of the 30 study arm participants for whom complete stool samples were available, 27 were found to have had a bifidobacterial response at day 36, including a significant increase in the lactose-fermenting Bifidobacterium, Faecalibacterium, and Lactobacillus species. The remaining three participants in the study arm were considered nonresponders.
In an interview, Andrew Ritter, whose company, Ritter Pharmaceuticals, sponsored the trial, reported that of the 36 study arm participants who had reported abdominal pain pretreatment, 18 said they no longer had the pain at either endpoint, day 36 or day 66 (P = .019); three of 19 in the placebo group reported they no longer had abdominal pain at either endpoint. The study group was also six times more likely to report lactose tolerance at day 66 compared with their pretreatment levels (P = .0389); 28% of the placebo arm reported lactose tolerance at the endpoints. These results were previously published in Nutrition Journal in 2013. [doi: 10.1186/1475-2891-12-160]
“We’re super excited about these results,” said Mr. Ritter. “This is really one of the first clinical studies in a lactose-intolerant population that shows changes in the microbiome.” As to how long before the treatment will be ready for the Food and Drug Administration approval process, Mr. Ritter said, “We’re probably just a couple of years away.”
Two coauthors are advisers to Ritter Pharmaceuticals, which provided the highly purified GOS used in the study. The North Carolina Agriculture Foundation also provided funding for the study.
[email protected]
On Twitter @whitneymcknight
FROM THE PROCEEDINGS OF THE NATIONAL ACADEMIES OF SCIENCE
Key clinical point:
Major finding: A clinically significant response was seen in patients with lactose intolerance who were given an oral, liquid supplement intended to increase lactose-fermenting bacteria.
Data source: Phase IIa trial of 62 adults with lactose intolerance incrementally dosed with an oral, highly purified (more than 95%) short-chain galactooligosaccharide while dietary dairy was restricted.
Disclosures: Ritter Pharmaceuticals, owned by study coauthor Andrew J. Ritter, funded the study and provided the highly purified GOS used in the study. The North Carolina Agriculture Foundation also provided funding. Two coauthors are advisers to Ritter Pharmaceuticals.
AGA Clinical Practice Update: Endoscope reprocessing guidelines are an improvement
While the 2016 Multi-Society Task Force Endoscope Reprocessing Guidelines are an improvement over the 2011 guidelines, some of its minor changes are unlikely to guarantee against prevention of future outbreaks, according to Susan Hutfless, PhD, and Anthony N. Kalloo, MD.*
“The prevention of future outbreaks is left to the manufacturers to modify their protocols and the endoscopy units to adopt the protocols rapidly,” the authors, both from Johns Hopkins University, Baltimore, wrote in a commentary about the 2016 guidelines, which contain 41 recommendations and were endorsed by the AGA. “If followed, the guidelines will make it possible to better track the source of future outbreaks if the tracking and monitoring suggested is performed.” They added that the current cleaning paradigm for duodenoscopes “is ineffective and these guidelines reflect changes to contain, rather than prevent, future outbreaks.”

Some of the specific changes to the 2016 guidelines include recommendation no. 5, which has been revised to recommend “strict adherence” to manufacturer guidance. “The expectation is that all personnel will remain up to date with the manufacturer guidelines and that there will be documentation of the training,” Dr. Hutfless and Dr. Kalloo wrote. The 2016 guidelines specifically state that a “single standard work process within one institution may be insufficient, given differences among manufacturers’ instructions and varied instrument designs.” However, Dr. Hutfless and Dr. Kalloo point out that “an individual or group of individuals may need to be identified to keep up with the FDA, CDC, manufacturer and professional societies in order to modify and implement the changes to the cleaning and training protocols and update the training of all individuals in the unit. It is unclear from the guidelines what the minimum time should be between change in recommendations and updated training.”
Recommendation no. 24 is new and includes a suggestion consistent with the 2015 FDA endoscope reprocessing communications. “Beyond the reprocessing steps discussed in these recommendations, no validated methods for additional duodenoscope reprocessing currently exist,” the guidelines state. “However, units should review and consider the feasibility and appropriateness for their practice of employing one or more of the additional modalities suggested by the FDA for duodenoscopes: intermittent or per procedure culture surveillance of reprocessing outcomes, sterilization with ethylene oxide gas, repeat application of standard high level disinfection, or use of a liquid chemical germicide.” For their part, Dr. Hutfless and Dr. Kalloo pointed out the limitations of these additional modalities. For example, they wrote, “the per procedure culture surveillance modality suggested by the FDA is not cost-effective unless the unit’s transmission probability of carbapenem-resistant Enterobacteriaceae is 24% or greater. Sterilization with ethylene oxide is problematic because a unit that used this approach still encountered an endoscope with carbapenem-resistant Enterobacteriaceae detected by culture. This unit also incurred extra costs to purchase additional scopes due to the longer reprocessing time for sterilization and had a greater number of endoscopes with damage, although the damage was not directly attributable to sterilization” (Gastrointest Endosc. 2016 Aug;84:259-62).
In 2016, the FDA approved the first disposable colonoscope, a product that is expected to be available in the United States in early 2017. Dr. Hutfless and Dr. Kalloo ended their commentary by suggesting that a disposable endoscope with an elevator mechanism, though not currently available, could be a solution to several of the unresolved issues that were present in the 2003, 2011, and 2016 guidelines. “These unresolved issues include interval of storage after reprocessing, microbiologic surveillance, and endoscope durability and longevity,” they wrote. “If the outbreaks persist after the use of disposable endoscopes it is possible that it is some other product or procedure within the endoscopic procedure that is the source of the infectious transmission.”
*This story was update on Jan. 26, 2017.
While the 2016 Multi-Society Task Force Endoscope Reprocessing Guidelines are an improvement over the 2011 guidelines, some of its minor changes are unlikely to guarantee against prevention of future outbreaks, according to Susan Hutfless, PhD, and Anthony N. Kalloo, MD.*
“The prevention of future outbreaks is left to the manufacturers to modify their protocols and the endoscopy units to adopt the protocols rapidly,” the authors, both from Johns Hopkins University, Baltimore, wrote in a commentary about the 2016 guidelines, which contain 41 recommendations and were endorsed by the AGA. “If followed, the guidelines will make it possible to better track the source of future outbreaks if the tracking and monitoring suggested is performed.” They added that the current cleaning paradigm for duodenoscopes “is ineffective and these guidelines reflect changes to contain, rather than prevent, future outbreaks.”
Some of the specific changes to the 2016 guidelines include recommendation no. 5, which has been revised to recommend “strict adherence” to manufacturer guidance. “The expectation is that all personnel will remain up to date with the manufacturer guidelines and that there will be documentation of the training,” Dr. Hutfless and Dr. Kalloo wrote. The 2016 guidelines specifically state that a “single standard work process within one institution may be insufficient, given differences among manufacturers’ instructions and varied instrument designs.” However, Dr. Hutfless and Dr. Kalloo point out that “an individual or group of individuals may need to be identified to keep up with the FDA, CDC, manufacturer and professional societies in order to modify and implement the changes to the cleaning and training protocols and update the training of all individuals in the unit. It is unclear from the guidelines what the minimum time should be between change in recommendations and updated training.”
Recommendation no. 24 is new and includes a suggestion consistent with the 2015 FDA endoscope reprocessing communications. “Beyond the reprocessing steps discussed in these recommendations, no validated methods for additional duodenoscope reprocessing currently exist,” the guidelines state. “However, units should review and consider the feasibility and appropriateness for their practice of employing one or more of the additional modalities suggested by the FDA for duodenoscopes: intermittent or per procedure culture surveillance of reprocessing outcomes, sterilization with ethylene oxide gas, repeat application of standard high level disinfection, or use of a liquid chemical germicide.” For their part, Dr. Hutfless and Dr. Kalloo pointed out the limitations of these additional modalities. For example, they wrote, “the per procedure culture surveillance modality suggested by the FDA is not cost-effective unless the unit’s transmission probability of carbapenem-resistant Enterobacteriaceae is 24% or greater. Sterilization with ethylene oxide is problematic because a unit that used this approach still encountered an endoscope with carbapenem-resistant Enterobacteriaceae detected by culture. This unit also incurred extra costs to purchase additional scopes due to the longer reprocessing time for sterilization and had a greater number of endoscopes with damage, although the damage was not directly attributable to sterilization” (Gastrointest Endosc. 2016 Aug;84:259-62).
In 2016, the FDA approved the first disposable colonoscope, a product that is expected to be available in the United States in early 2017. Dr. Hutfless and Dr. Kalloo ended their commentary by suggesting that a disposable endoscope with an elevator mechanism, though not currently available, could be a solution to several of the unresolved issues that were present in the 2003, 2011, and 2016 guidelines. “These unresolved issues include interval of storage after reprocessing, microbiologic surveillance, and endoscope durability and longevity,” they wrote. “If the outbreaks persist after the use of disposable endoscopes it is possible that it is some other product or procedure within the endoscopic procedure that is the source of the infectious transmission.”
*This story was update on Jan. 26, 2017.
While the 2016 Multi-Society Task Force Endoscope Reprocessing Guidelines are an improvement over the 2011 guidelines, some of its minor changes are unlikely to guarantee against prevention of future outbreaks, according to Susan Hutfless, PhD, and Anthony N. Kalloo, MD.*
“The prevention of future outbreaks is left to the manufacturers to modify their protocols and the endoscopy units to adopt the protocols rapidly,” the authors, both from Johns Hopkins University, Baltimore, wrote in a commentary about the 2016 guidelines, which contain 41 recommendations and were endorsed by the AGA. “If followed, the guidelines will make it possible to better track the source of future outbreaks if the tracking and monitoring suggested is performed.” They added that the current cleaning paradigm for duodenoscopes “is ineffective and these guidelines reflect changes to contain, rather than prevent, future outbreaks.”
Some of the specific changes to the 2016 guidelines include recommendation no. 5, which has been revised to recommend “strict adherence” to manufacturer guidance. “The expectation is that all personnel will remain up to date with the manufacturer guidelines and that there will be documentation of the training,” Dr. Hutfless and Dr. Kalloo wrote. The 2016 guidelines specifically state that a “single standard work process within one institution may be insufficient, given differences among manufacturers’ instructions and varied instrument designs.” However, Dr. Hutfless and Dr. Kalloo point out that “an individual or group of individuals may need to be identified to keep up with the FDA, CDC, manufacturer and professional societies in order to modify and implement the changes to the cleaning and training protocols and update the training of all individuals in the unit. It is unclear from the guidelines what the minimum time should be between change in recommendations and updated training.”
Recommendation no. 24 is new and includes a suggestion consistent with the 2015 FDA endoscope reprocessing communications. “Beyond the reprocessing steps discussed in these recommendations, no validated methods for additional duodenoscope reprocessing currently exist,” the guidelines state. “However, units should review and consider the feasibility and appropriateness for their practice of employing one or more of the additional modalities suggested by the FDA for duodenoscopes: intermittent or per procedure culture surveillance of reprocessing outcomes, sterilization with ethylene oxide gas, repeat application of standard high level disinfection, or use of a liquid chemical germicide.” For their part, Dr. Hutfless and Dr. Kalloo pointed out the limitations of these additional modalities. For example, they wrote, “the per procedure culture surveillance modality suggested by the FDA is not cost-effective unless the unit’s transmission probability of carbapenem-resistant Enterobacteriaceae is 24% or greater. Sterilization with ethylene oxide is problematic because a unit that used this approach still encountered an endoscope with carbapenem-resistant Enterobacteriaceae detected by culture. This unit also incurred extra costs to purchase additional scopes due to the longer reprocessing time for sterilization and had a greater number of endoscopes with damage, although the damage was not directly attributable to sterilization” (Gastrointest Endosc. 2016 Aug;84:259-62).
In 2016, the FDA approved the first disposable colonoscope, a product that is expected to be available in the United States in early 2017. Dr. Hutfless and Dr. Kalloo ended their commentary by suggesting that a disposable endoscope with an elevator mechanism, though not currently available, could be a solution to several of the unresolved issues that were present in the 2003, 2011, and 2016 guidelines. “These unresolved issues include interval of storage after reprocessing, microbiologic surveillance, and endoscope durability and longevity,” they wrote. “If the outbreaks persist after the use of disposable endoscopes it is possible that it is some other product or procedure within the endoscopic procedure that is the source of the infectious transmission.”
*This story was update on Jan. 26, 2017.
FROM GASTROENTEROLOGY
Promising pipeline for chronic pruritus
VIENNA – Help is on the way for physicians stymied in their efforts to treat patients with severe chronic itch, Sonja Ständer, MD, declared at the annual congress of the European Academy of Dermatology and Venereology.
Now working their way through the developmental pipeline are two promising novel classes of drugs designed to target the physiologic mechanisms underlying this common and challenging problem: neurokinin 1 (NK1) receptor antagonists and selective opioid receptor agonists, explained Dr. Ständer, professor of dermatology and head of the Center for Chronic Pruritus at the University of Münster (Germany).
She led a landmark study that outlined the previously unappreciated dimensions of chronic pruritus as a clinical issue. This was a cross-sectional study of nearly 12,000 German employees in various industries that demonstrated the prevalence of chronic pruritus was 16.8%. One-quarter of affected individuals had chronic pruritus for longer than 5 years. The prevalence climbed with age, from 12% in workers up to age 30 years to 20% in 61- to 70-year-olds (Dermatology. 2010;221[3]:229-35).
One in three patients who present to dermatologists’ offices have chronic pruritus, she added.
Chronic pruritus can have a multitude of different causes: not only chronic skin diseases, but incurable renal and liver diseases, psychiatric conditions, neurologic disorders, drug side effects, and various forms of cancer, with hematologic malignancies figuring prominently. The quality of life impact is huge. And even though a plethora of topical and systemic therapies are available for itchy skin, they often are ineffective in patients with severe chronic pruritus. Thus, there is a significant unmet need for new and effective therapies, Dr. Ständer continued.
NK1 receptor antagonists: Substance P is a neuropeptide which plays a major role in the induction and maintenance of pruritus. The NK1 receptor, which is abundantly expressed in the skin and CNS, is substance P’s receptor – and therefore a logical target for novel anti-itch therapy. Bind that receptor and a key signaling pathway in pruritus is disrupted.
Aprepitant is an oral NK1 receptor antagonist approved as Emend more than a decade ago for prevention of chemotherapy-induced nausea and vomiting. Dr. Ständer was lead author of an early single-arm pilot study showing aprepitant is also dramatically effective for severe refractory chronic pruritus (PLoS One. 2010 Jun 4;5[6]:e10968).
Aprepitant is approved only for 3 days of use for its licensed indication. However, Dr. Ständer said she and other dermatologists have used it off-label for as long as 4 weeks in patients with chronic pruritus and found it to be safe, with mild nonlimiting side effects and relief that is often long lasting after treatment discontinuation.
Oral aprepitant is under study in several ongoing phase II clinical trials for treatment of severe pruritus resulting from targeted biologic therapies for various cancers. In addition, Leo Pharma is developing a topical gel formulation of aprepitant for chronic pruritus which is now in phase II studies.
Serlopitant is a once-daily oral NK1 receptor antagonist under development specifically for treatment of severe chronic pruritus. In a recent as-yet unpublished multicenter, double-blind, placebo-controlled phase II clinical trial involving 257 patients with severe refractory chronic pruritus of various etiologies, 6 weeks of serlopitant at 1 or 5 mg/day was markedly more effective than placebo in reducing itch intensity. Based upon these favorable results, Menlo Therapeutics has begun two new phase II randomized, placebo-controlled studies of the drug: ATOMIK, a roughly 450-patient, 40 U.S.-site study in patients with atopic dermatitis; and AUBURN, involving roughly 150 burn patients with chronic pruritus at 20 centers.
Selective opioid receptor agonists: These agents target kappa- and/or mu-opioid receptors on peripheral pain-sensing neurons in order to inhibit itch without activating other opioid receptors linked to classic opioid side effects such as respiratory depression, constipation, and addiction. These selective agents are essentially designed to be nonnarcotic opioids.
One such agent is nalfurafine, an oral kappa-opioid receptor agonist marketed in Japan for treatment of uremic pruritus in patients with chronic kidney disease undergoing hemodialysis and for refractory pruritus in chronic liver disease. The drug is in phase II studies in the United States.
Nalbuphine, a dual kappa-opiod agonist and partial mu-opioid antagonist, is Food and Drug Administration–approved as an injectable agent, known as Nubain, for moderate to severe pain. An investigational extended-release tablet formulation has successfully completed a U.S. multicenter, double-blind, placebo-controlled phase II/III clinical trial in hemodialysis patients with severe chronic uremic pruritus and a phase II study in patients with prurigo nodularis. Because the drug was effective in two conditions having very different sources of itch, it is likely to be of benefit in many forms of severe chronic pruritus, Dr. Ständer said.
She reported having no financial conflicts of interest.
VIENNA – Help is on the way for physicians stymied in their efforts to treat patients with severe chronic itch, Sonja Ständer, MD, declared at the annual congress of the European Academy of Dermatology and Venereology.
Now working their way through the developmental pipeline are two promising novel classes of drugs designed to target the physiologic mechanisms underlying this common and challenging problem: neurokinin 1 (NK1) receptor antagonists and selective opioid receptor agonists, explained Dr. Ständer, professor of dermatology and head of the Center for Chronic Pruritus at the University of Münster (Germany).
She led a landmark study that outlined the previously unappreciated dimensions of chronic pruritus as a clinical issue. This was a cross-sectional study of nearly 12,000 German employees in various industries that demonstrated the prevalence of chronic pruritus was 16.8%. One-quarter of affected individuals had chronic pruritus for longer than 5 years. The prevalence climbed with age, from 12% in workers up to age 30 years to 20% in 61- to 70-year-olds (Dermatology. 2010;221[3]:229-35).
One in three patients who present to dermatologists’ offices have chronic pruritus, she added.
Chronic pruritus can have a multitude of different causes: not only chronic skin diseases, but incurable renal and liver diseases, psychiatric conditions, neurologic disorders, drug side effects, and various forms of cancer, with hematologic malignancies figuring prominently. The quality of life impact is huge. And even though a plethora of topical and systemic therapies are available for itchy skin, they often are ineffective in patients with severe chronic pruritus. Thus, there is a significant unmet need for new and effective therapies, Dr. Ständer continued.
NK1 receptor antagonists: Substance P is a neuropeptide which plays a major role in the induction and maintenance of pruritus. The NK1 receptor, which is abundantly expressed in the skin and CNS, is substance P’s receptor – and therefore a logical target for novel anti-itch therapy. Bind that receptor and a key signaling pathway in pruritus is disrupted.
Aprepitant is an oral NK1 receptor antagonist approved as Emend more than a decade ago for prevention of chemotherapy-induced nausea and vomiting. Dr. Ständer was lead author of an early single-arm pilot study showing aprepitant is also dramatically effective for severe refractory chronic pruritus (PLoS One. 2010 Jun 4;5[6]:e10968).
Aprepitant is approved only for 3 days of use for its licensed indication. However, Dr. Ständer said she and other dermatologists have used it off-label for as long as 4 weeks in patients with chronic pruritus and found it to be safe, with mild nonlimiting side effects and relief that is often long lasting after treatment discontinuation.
Oral aprepitant is under study in several ongoing phase II clinical trials for treatment of severe pruritus resulting from targeted biologic therapies for various cancers. In addition, Leo Pharma is developing a topical gel formulation of aprepitant for chronic pruritus which is now in phase II studies.
Serlopitant is a once-daily oral NK1 receptor antagonist under development specifically for treatment of severe chronic pruritus. In a recent as-yet unpublished multicenter, double-blind, placebo-controlled phase II clinical trial involving 257 patients with severe refractory chronic pruritus of various etiologies, 6 weeks of serlopitant at 1 or 5 mg/day was markedly more effective than placebo in reducing itch intensity. Based upon these favorable results, Menlo Therapeutics has begun two new phase II randomized, placebo-controlled studies of the drug: ATOMIK, a roughly 450-patient, 40 U.S.-site study in patients with atopic dermatitis; and AUBURN, involving roughly 150 burn patients with chronic pruritus at 20 centers.
Selective opioid receptor agonists: These agents target kappa- and/or mu-opioid receptors on peripheral pain-sensing neurons in order to inhibit itch without activating other opioid receptors linked to classic opioid side effects such as respiratory depression, constipation, and addiction. These selective agents are essentially designed to be nonnarcotic opioids.
One such agent is nalfurafine, an oral kappa-opioid receptor agonist marketed in Japan for treatment of uremic pruritus in patients with chronic kidney disease undergoing hemodialysis and for refractory pruritus in chronic liver disease. The drug is in phase II studies in the United States.
Nalbuphine, a dual kappa-opiod agonist and partial mu-opioid antagonist, is Food and Drug Administration–approved as an injectable agent, known as Nubain, for moderate to severe pain. An investigational extended-release tablet formulation has successfully completed a U.S. multicenter, double-blind, placebo-controlled phase II/III clinical trial in hemodialysis patients with severe chronic uremic pruritus and a phase II study in patients with prurigo nodularis. Because the drug was effective in two conditions having very different sources of itch, it is likely to be of benefit in many forms of severe chronic pruritus, Dr. Ständer said.
She reported having no financial conflicts of interest.
VIENNA – Help is on the way for physicians stymied in their efforts to treat patients with severe chronic itch, Sonja Ständer, MD, declared at the annual congress of the European Academy of Dermatology and Venereology.
Now working their way through the developmental pipeline are two promising novel classes of drugs designed to target the physiologic mechanisms underlying this common and challenging problem: neurokinin 1 (NK1) receptor antagonists and selective opioid receptor agonists, explained Dr. Ständer, professor of dermatology and head of the Center for Chronic Pruritus at the University of Münster (Germany).
She led a landmark study that outlined the previously unappreciated dimensions of chronic pruritus as a clinical issue. This was a cross-sectional study of nearly 12,000 German employees in various industries that demonstrated the prevalence of chronic pruritus was 16.8%. One-quarter of affected individuals had chronic pruritus for longer than 5 years. The prevalence climbed with age, from 12% in workers up to age 30 years to 20% in 61- to 70-year-olds (Dermatology. 2010;221[3]:229-35).
One in three patients who present to dermatologists’ offices have chronic pruritus, she added.
Chronic pruritus can have a multitude of different causes: not only chronic skin diseases, but incurable renal and liver diseases, psychiatric conditions, neurologic disorders, drug side effects, and various forms of cancer, with hematologic malignancies figuring prominently. The quality of life impact is huge. And even though a plethora of topical and systemic therapies are available for itchy skin, they often are ineffective in patients with severe chronic pruritus. Thus, there is a significant unmet need for new and effective therapies, Dr. Ständer continued.
NK1 receptor antagonists: Substance P is a neuropeptide which plays a major role in the induction and maintenance of pruritus. The NK1 receptor, which is abundantly expressed in the skin and CNS, is substance P’s receptor – and therefore a logical target for novel anti-itch therapy. Bind that receptor and a key signaling pathway in pruritus is disrupted.
Aprepitant is an oral NK1 receptor antagonist approved as Emend more than a decade ago for prevention of chemotherapy-induced nausea and vomiting. Dr. Ständer was lead author of an early single-arm pilot study showing aprepitant is also dramatically effective for severe refractory chronic pruritus (PLoS One. 2010 Jun 4;5[6]:e10968).
Aprepitant is approved only for 3 days of use for its licensed indication. However, Dr. Ständer said she and other dermatologists have used it off-label for as long as 4 weeks in patients with chronic pruritus and found it to be safe, with mild nonlimiting side effects and relief that is often long lasting after treatment discontinuation.
Oral aprepitant is under study in several ongoing phase II clinical trials for treatment of severe pruritus resulting from targeted biologic therapies for various cancers. In addition, Leo Pharma is developing a topical gel formulation of aprepitant for chronic pruritus which is now in phase II studies.
Serlopitant is a once-daily oral NK1 receptor antagonist under development specifically for treatment of severe chronic pruritus. In a recent as-yet unpublished multicenter, double-blind, placebo-controlled phase II clinical trial involving 257 patients with severe refractory chronic pruritus of various etiologies, 6 weeks of serlopitant at 1 or 5 mg/day was markedly more effective than placebo in reducing itch intensity. Based upon these favorable results, Menlo Therapeutics has begun two new phase II randomized, placebo-controlled studies of the drug: ATOMIK, a roughly 450-patient, 40 U.S.-site study in patients with atopic dermatitis; and AUBURN, involving roughly 150 burn patients with chronic pruritus at 20 centers.
Selective opioid receptor agonists: These agents target kappa- and/or mu-opioid receptors on peripheral pain-sensing neurons in order to inhibit itch without activating other opioid receptors linked to classic opioid side effects such as respiratory depression, constipation, and addiction. These selective agents are essentially designed to be nonnarcotic opioids.
One such agent is nalfurafine, an oral kappa-opioid receptor agonist marketed in Japan for treatment of uremic pruritus in patients with chronic kidney disease undergoing hemodialysis and for refractory pruritus in chronic liver disease. The drug is in phase II studies in the United States.
Nalbuphine, a dual kappa-opiod agonist and partial mu-opioid antagonist, is Food and Drug Administration–approved as an injectable agent, known as Nubain, for moderate to severe pain. An investigational extended-release tablet formulation has successfully completed a U.S. multicenter, double-blind, placebo-controlled phase II/III clinical trial in hemodialysis patients with severe chronic uremic pruritus and a phase II study in patients with prurigo nodularis. Because the drug was effective in two conditions having very different sources of itch, it is likely to be of benefit in many forms of severe chronic pruritus, Dr. Ständer said.
She reported having no financial conflicts of interest.
EXPERT ANALYSIS FROM THE EADV
Rhinovirus most often caused HA-VRIs in two hospitals
Health care–associated viral respiratory infections (HA-VRIs) were common in two pediatric hospitals, with rhinovirus the most frequent cause of the infections in a 3-year analysis.
The incidence rate of laboratory-confirmed HA-VRIs was 1.29/1,000 patient-days in an examination of the hospitals’ patient data. Forty-eight percent of all 323 HA-VRI cases were caused by rhinovirus, with an overall incidence rate of 0.72/1,000 patient-days. Additionally, rhinovirus was the most frequently identified virus in cases of HA-VRI in all units of both hospitals, followed by parainfluenza virus and respiratory syncytial virus. An exception was the medical/surgical ward of Steven and Alexandra Cohen Children’s Medical Center (CCMC) of New York; in this unit of the CCMC, the incidence rate of parainfluenza virus was higher than that of rhinovirus (0.21/1,000 patient-days vs. 0.15/1,000 patient-days) (J Ped Inf Dis. 2016. doi: 10.1093/jpids/piw072).
The researchers used infection prevention and control surveillance databases from Montreal Children’s Hospital (MCH) in Quebec and the CCMC to identify HA-VRIs that occurred between April 1, 2010, and March 31, 2013, In both hospitals, HAIs were attributed to the unit to which the patient was admitted at the time of transmission. Both hospitals used a multiplex nucleic acid amplification test for respiratory virus detection on nasopharyngeal swabs or aspirates.
“An HA-VRI with an onset of symptoms after hospital discharge would be detected and included only for patients who presented to the emergency department or were readmitted for VRI and tested,” according to Caroline Quach, MD, of the Montreal Children’s Hospital, McGill University Health Centre, Quebec, and her colleagues.
The HA-VRI rate was 1.91/1,000 patient-days at Montreal Children’s Hospital, compared with 0.80/1,000 patient-days at the CCMC (P less than .0001). At the CCMC, the HA-VRI incidence rate was lowest in the neonatal ICU, but at Montgomery Children’s Hospital, the hematology/oncology ward had the lowest rate of HA-VRI.
Having less than 50% single rooms in a given unit was associated with a statistically significantly higher rate of HA-VRI, after the investigators adjusted for unit type and took the correlation of HA-VRI rates within a hospital into consideration. The study authors’ model predicted that units with less than 50% single rooms have 1.33 times higher HA-VRI rates than units with at least 50% single rooms, regardless of unit type.
Dr. Quach has received funding from GlaxoSmithKline, Pfizer, Sage, and AbbVie for an unrelated research project, while the other authors disclosed no financial relationships.
Health care–associated viral respiratory infections (HA-VRIs) were common in two pediatric hospitals, with rhinovirus the most frequent cause of the infections in a 3-year analysis.
The incidence rate of laboratory-confirmed HA-VRIs was 1.29/1,000 patient-days in an examination of the hospitals’ patient data. Forty-eight percent of all 323 HA-VRI cases were caused by rhinovirus, with an overall incidence rate of 0.72/1,000 patient-days. Additionally, rhinovirus was the most frequently identified virus in cases of HA-VRI in all units of both hospitals, followed by parainfluenza virus and respiratory syncytial virus. An exception was the medical/surgical ward of Steven and Alexandra Cohen Children’s Medical Center (CCMC) of New York; in this unit of the CCMC, the incidence rate of parainfluenza virus was higher than that of rhinovirus (0.21/1,000 patient-days vs. 0.15/1,000 patient-days) (J Ped Inf Dis. 2016. doi: 10.1093/jpids/piw072).
The researchers used infection prevention and control surveillance databases from Montreal Children’s Hospital (MCH) in Quebec and the CCMC to identify HA-VRIs that occurred between April 1, 2010, and March 31, 2013, In both hospitals, HAIs were attributed to the unit to which the patient was admitted at the time of transmission. Both hospitals used a multiplex nucleic acid amplification test for respiratory virus detection on nasopharyngeal swabs or aspirates.
“An HA-VRI with an onset of symptoms after hospital discharge would be detected and included only for patients who presented to the emergency department or were readmitted for VRI and tested,” according to Caroline Quach, MD, of the Montreal Children’s Hospital, McGill University Health Centre, Quebec, and her colleagues.
The HA-VRI rate was 1.91/1,000 patient-days at Montreal Children’s Hospital, compared with 0.80/1,000 patient-days at the CCMC (P less than .0001). At the CCMC, the HA-VRI incidence rate was lowest in the neonatal ICU, but at Montgomery Children’s Hospital, the hematology/oncology ward had the lowest rate of HA-VRI.
Having less than 50% single rooms in a given unit was associated with a statistically significantly higher rate of HA-VRI, after the investigators adjusted for unit type and took the correlation of HA-VRI rates within a hospital into consideration. The study authors’ model predicted that units with less than 50% single rooms have 1.33 times higher HA-VRI rates than units with at least 50% single rooms, regardless of unit type.
Dr. Quach has received funding from GlaxoSmithKline, Pfizer, Sage, and AbbVie for an unrelated research project, while the other authors disclosed no financial relationships.
Health care–associated viral respiratory infections (HA-VRIs) were common in two pediatric hospitals, with rhinovirus the most frequent cause of the infections in a 3-year analysis.
The incidence rate of laboratory-confirmed HA-VRIs was 1.29/1,000 patient-days in an examination of the hospitals’ patient data. Forty-eight percent of all 323 HA-VRI cases were caused by rhinovirus, with an overall incidence rate of 0.72/1,000 patient-days. Additionally, rhinovirus was the most frequently identified virus in cases of HA-VRI in all units of both hospitals, followed by parainfluenza virus and respiratory syncytial virus. An exception was the medical/surgical ward of Steven and Alexandra Cohen Children’s Medical Center (CCMC) of New York; in this unit of the CCMC, the incidence rate of parainfluenza virus was higher than that of rhinovirus (0.21/1,000 patient-days vs. 0.15/1,000 patient-days) (J Ped Inf Dis. 2016. doi: 10.1093/jpids/piw072).
The researchers used infection prevention and control surveillance databases from Montreal Children’s Hospital (MCH) in Quebec and the CCMC to identify HA-VRIs that occurred between April 1, 2010, and March 31, 2013, In both hospitals, HAIs were attributed to the unit to which the patient was admitted at the time of transmission. Both hospitals used a multiplex nucleic acid amplification test for respiratory virus detection on nasopharyngeal swabs or aspirates.
“An HA-VRI with an onset of symptoms after hospital discharge would be detected and included only for patients who presented to the emergency department or were readmitted for VRI and tested,” according to Caroline Quach, MD, of the Montreal Children’s Hospital, McGill University Health Centre, Quebec, and her colleagues.
The HA-VRI rate was 1.91/1,000 patient-days at Montreal Children’s Hospital, compared with 0.80/1,000 patient-days at the CCMC (P less than .0001). At the CCMC, the HA-VRI incidence rate was lowest in the neonatal ICU, but at Montgomery Children’s Hospital, the hematology/oncology ward had the lowest rate of HA-VRI.
Having less than 50% single rooms in a given unit was associated with a statistically significantly higher rate of HA-VRI, after the investigators adjusted for unit type and took the correlation of HA-VRI rates within a hospital into consideration. The study authors’ model predicted that units with less than 50% single rooms have 1.33 times higher HA-VRI rates than units with at least 50% single rooms, regardless of unit type.
Dr. Quach has received funding from GlaxoSmithKline, Pfizer, Sage, and AbbVie for an unrelated research project, while the other authors disclosed no financial relationships.
FROM THE JOURNAL OF THE PEDIATRIC INFECTIOUS DISEASES SOCIETY
Key clinical point:
Major finding: The incidence rate of HA-VRIs was 1.29/1,000 patient-days in an examination of two pediatric hospitals’ patient data between April 1, 2010, and March 31, 2013.
Data source: A retrospective comparison of two hospitals’ 3 years of infection prevention and control surveillance data.
Disclosures: Dr. Quach has received funding from GlaxoSmithKline, Pfizer, Sage, and AbbVie for an unrelated research project, while the other authors disclosed no relevant financial relationships.
Vent bundles and ventilator-associated pneumonia outcomes
Clinical question: Are the components of the ventilator bundles (VBs) associated with better outcomes for patients?
Background: VBs have been shown to prevent ventilator-associated pneumonia (VAP). However, most of the studies have analyzed outcomes based on the whole bundle without considering each individual component.
Study design: Retrospective cohort study.
Setting: Brigham and Women’s Hospital in Boston.
Spontaneous breathing trials were associated with lower hazards for VAEs (HR, 0.55; 95% CI, 0.40-0.76; P less than .001) and infection-related ventilator-associated complications (IVACs) (HR, 0.60; 95% CI, 0.37-1.00; P = .05). Head-of-bed elevation (HR, 1.38; 95% CI, 1.14-1.68; P = 0.001) and thromboembolism prophylaxis (HR, 2.57; 95% CI, 1.80-3.66; P less than .001) were associated with less time to extubation.
Oral care with chlorhexidine was associated with lower hazards for IVACs (HR, 0.60; 95% CI 0.36-1.00; P = .05) and for VAPs (HR, 0.55; 95% CI, 0.27-1.14; P = .11) but an increased risk for ventilator mortality (HR, 1.63; 95% CI, 1.15-2.31; P = .006). Stress ulcer prophylaxis was associated with higher risk for VAP (HR, 7.69; 95% CI, 1.44-41.10; P = .02).
Bottom line: Standard VB components merit revision to increase emphasis on beneficial components and eliminate potentially harmful ones.
Citation: Klompas M, Li L, Kleinman K, Szumita PM, Massaro AF. Association between ventilator bundle components and outcomes. JAMA Intern Med. 2016;176(9):1277-1283.
Dr. Mosetti is an assistant professor at the University of Miami Miller School of Medicine and a hospitalist at University of Miami Hospital and Jackson Memorial Hospital.
Clinical question: Are the components of the ventilator bundles (VBs) associated with better outcomes for patients?
Background: VBs have been shown to prevent ventilator-associated pneumonia (VAP). However, most of the studies have analyzed outcomes based on the whole bundle without considering each individual component.
Study design: Retrospective cohort study.
Setting: Brigham and Women’s Hospital in Boston.
Spontaneous breathing trials were associated with lower hazards for VAEs (HR, 0.55; 95% CI, 0.40-0.76; P less than .001) and infection-related ventilator-associated complications (IVACs) (HR, 0.60; 95% CI, 0.37-1.00; P = .05). Head-of-bed elevation (HR, 1.38; 95% CI, 1.14-1.68; P = 0.001) and thromboembolism prophylaxis (HR, 2.57; 95% CI, 1.80-3.66; P less than .001) were associated with less time to extubation.
Oral care with chlorhexidine was associated with lower hazards for IVACs (HR, 0.60; 95% CI 0.36-1.00; P = .05) and for VAPs (HR, 0.55; 95% CI, 0.27-1.14; P = .11) but an increased risk for ventilator mortality (HR, 1.63; 95% CI, 1.15-2.31; P = .006). Stress ulcer prophylaxis was associated with higher risk for VAP (HR, 7.69; 95% CI, 1.44-41.10; P = .02).
Bottom line: Standard VB components merit revision to increase emphasis on beneficial components and eliminate potentially harmful ones.
Citation: Klompas M, Li L, Kleinman K, Szumita PM, Massaro AF. Association between ventilator bundle components and outcomes. JAMA Intern Med. 2016;176(9):1277-1283.
Dr. Mosetti is an assistant professor at the University of Miami Miller School of Medicine and a hospitalist at University of Miami Hospital and Jackson Memorial Hospital.
Clinical question: Are the components of the ventilator bundles (VBs) associated with better outcomes for patients?
Background: VBs have been shown to prevent ventilator-associated pneumonia (VAP). However, most of the studies have analyzed outcomes based on the whole bundle without considering each individual component.
Study design: Retrospective cohort study.
Setting: Brigham and Women’s Hospital in Boston.
Spontaneous breathing trials were associated with lower hazards for VAEs (HR, 0.55; 95% CI, 0.40-0.76; P less than .001) and infection-related ventilator-associated complications (IVACs) (HR, 0.60; 95% CI, 0.37-1.00; P = .05). Head-of-bed elevation (HR, 1.38; 95% CI, 1.14-1.68; P = 0.001) and thromboembolism prophylaxis (HR, 2.57; 95% CI, 1.80-3.66; P less than .001) were associated with less time to extubation.
Oral care with chlorhexidine was associated with lower hazards for IVACs (HR, 0.60; 95% CI 0.36-1.00; P = .05) and for VAPs (HR, 0.55; 95% CI, 0.27-1.14; P = .11) but an increased risk for ventilator mortality (HR, 1.63; 95% CI, 1.15-2.31; P = .006). Stress ulcer prophylaxis was associated with higher risk for VAP (HR, 7.69; 95% CI, 1.44-41.10; P = .02).
Bottom line: Standard VB components merit revision to increase emphasis on beneficial components and eliminate potentially harmful ones.
Citation: Klompas M, Li L, Kleinman K, Szumita PM, Massaro AF. Association between ventilator bundle components and outcomes. JAMA Intern Med. 2016;176(9):1277-1283.
Dr. Mosetti is an assistant professor at the University of Miami Miller School of Medicine and a hospitalist at University of Miami Hospital and Jackson Memorial Hospital.
Overnight extubations associated with worse outcomes
Clinical question: Are overnight extubations in intensive care units associated with higher mortality rate?
Background: Little is known about the frequency, safety, and effectiveness of overnight extubations in the ICU.
Study design: Retrospective cohort study.
Setting: One-hundred sixty-five ICUs in the United States.
Synopsis: Using the Project IMPACT database, 97,844 adults undergoing mechanical ventilation (MV) admitted to ICUs were studied. Overnight extubation was defined as occurring between 7 p.m. and 6:59 a.m. Primary outcome was reintubation; secondary outcomes were ICU and hospital mortality and ICU and hospital length of stay.
Only one-fifth of patients with MV underwent overnight extubations. For MV duration of at least 12 hours, rates of reintubation were higher for patients undergoing overnight extubation (14.6% vs. 12.4%; P less than .001). Mortality was significantly higher for patients undergoing overnight versus daytime extubation in the ICU (11.2% vs. 6.1%; P less than.001) and in the hospital (16.0% vs. 11.1%; P less than .001). Length of ICU and hospital stays did not differ.
Bottom line: Overnight extubations occur in one of five patients in U.S. ICUs and are associated with worse outcomes, compared with daytime extubations.
Citation: Gershengorn HB, Scales DC, Kramer A, Wunsch H. Association between overnight extubations and outcomes in the intensive care unit. JAMA Intern Med. 2016;176(11):1651-1660.
Dr. Mosetti is an assistant professor at the University of Miami Miller School of Medicine and a hospitalist at University of Miami Hospital and Jackson Memorial Hospital.
Clinical question: Are overnight extubations in intensive care units associated with higher mortality rate?
Background: Little is known about the frequency, safety, and effectiveness of overnight extubations in the ICU.
Study design: Retrospective cohort study.
Setting: One-hundred sixty-five ICUs in the United States.
Synopsis: Using the Project IMPACT database, 97,844 adults undergoing mechanical ventilation (MV) admitted to ICUs were studied. Overnight extubation was defined as occurring between 7 p.m. and 6:59 a.m. Primary outcome was reintubation; secondary outcomes were ICU and hospital mortality and ICU and hospital length of stay.
Only one-fifth of patients with MV underwent overnight extubations. For MV duration of at least 12 hours, rates of reintubation were higher for patients undergoing overnight extubation (14.6% vs. 12.4%; P less than .001). Mortality was significantly higher for patients undergoing overnight versus daytime extubation in the ICU (11.2% vs. 6.1%; P less than.001) and in the hospital (16.0% vs. 11.1%; P less than .001). Length of ICU and hospital stays did not differ.
Bottom line: Overnight extubations occur in one of five patients in U.S. ICUs and are associated with worse outcomes, compared with daytime extubations.
Citation: Gershengorn HB, Scales DC, Kramer A, Wunsch H. Association between overnight extubations and outcomes in the intensive care unit. JAMA Intern Med. 2016;176(11):1651-1660.
Dr. Mosetti is an assistant professor at the University of Miami Miller School of Medicine and a hospitalist at University of Miami Hospital and Jackson Memorial Hospital.
Clinical question: Are overnight extubations in intensive care units associated with higher mortality rate?
Background: Little is known about the frequency, safety, and effectiveness of overnight extubations in the ICU.
Study design: Retrospective cohort study.
Setting: One-hundred sixty-five ICUs in the United States.
Synopsis: Using the Project IMPACT database, 97,844 adults undergoing mechanical ventilation (MV) admitted to ICUs were studied. Overnight extubation was defined as occurring between 7 p.m. and 6:59 a.m. Primary outcome was reintubation; secondary outcomes were ICU and hospital mortality and ICU and hospital length of stay.
Only one-fifth of patients with MV underwent overnight extubations. For MV duration of at least 12 hours, rates of reintubation were higher for patients undergoing overnight extubation (14.6% vs. 12.4%; P less than .001). Mortality was significantly higher for patients undergoing overnight versus daytime extubation in the ICU (11.2% vs. 6.1%; P less than.001) and in the hospital (16.0% vs. 11.1%; P less than .001). Length of ICU and hospital stays did not differ.
Bottom line: Overnight extubations occur in one of five patients in U.S. ICUs and are associated with worse outcomes, compared with daytime extubations.
Citation: Gershengorn HB, Scales DC, Kramer A, Wunsch H. Association between overnight extubations and outcomes in the intensive care unit. JAMA Intern Med. 2016;176(11):1651-1660.
Dr. Mosetti is an assistant professor at the University of Miami Miller School of Medicine and a hospitalist at University of Miami Hospital and Jackson Memorial Hospital.
High consumption of red meat increases diverticulitis risk in men
Men who consume higher quantities of red meat are at an increased risk of developing diverticulitis, especially if they’re eating unprocessed red meat, according to a new study published in Gut.
“In our prior analysis from a large prospective cohort study, the Health Professionals Follow-Up Study (HPFS), we found that red meat intake, independent of fiber, may be associated with a composite outcome of symptomatic diverticular disease, which included 385 incident cases over 4 years of follow-up,” wrote the authors, led by Andrew T. Chan, MD, of Massachusetts General Hospital, Boston. Dr. Chan added that “in the present study, we updated this analysis, which allowed us to prospectively examine the association between consumption of meat (total red meat, red unprocessed meat, red processed meat, poultry, and fish) with risk of incident diverticulitis in 764 cases over 26 years of follow-up.”
Dr. Chan and his coinvestigators conducted a prospective cohort study using subjects from the ongoing HPFS. Men who already had a diagnosis of diverticulitis, associated complications, inflammatory bowel disease, or a GI-related cancer at baseline were excluded from this analysis, leaving 46,461 eligible subjects. Of those, 764 developed diverticulitis.
The entirety of the follow-up period constituted 651,970 person-years. Average servings of total red meat per week were 1.2 in quintile 1, compared to 5.3 in quintile 3 and 13.5 in quintile 5. Those in the highest quintile had a multivariable risk ratio of 1.58 (95% CI, 1.19-2.11; P = .01), indicating a significantly higher risk for developing diverticulitis. In terms of unprocessed red meat, the average number of servings per week were 0.8 for the lower quintile, 3.2 for quintile 3, and 8.6 for quintile 5, yielding a risk ratio of 1.51 (95% CI, 1.12-2.03, P = .03) when comparing the highest and lowest cohorts. The increase in risk, however, leveled off after about 6 servings of red meat per week, and was found to be nonlinear (P = .002). Those who ate more servings of poultry or fish did not have a higher risk of diverticulitis.
“We also observed that unprocessed red meat, but not processed red meat, was the primary driver for the association between total red meat and risk of diverticulitis,” the authors explained. “Compared with processed meat, unprocessed meat (e.g., steak) is usually consumed in larger portions, which could lead to a larger undigested piece in the large bowel and induce different changes in colonic microbiota [and] higher cooking temperatures used in the preparation of unprocessed meat may influence bacterial composition or proinflammatory mediators in the colon.”
Although medical information and self-reports were validated, there are inherent possible limitations to self-reported data, such as misremembering the amount of meat consumed or reporting incorrect amounts. Residual confounding may have occurred despite adjustment of the data to account for it.
The National Institutes of Health funded the study. The authors reported no conflicts of interest.
Men who consume higher quantities of red meat are at an increased risk of developing diverticulitis, especially if they’re eating unprocessed red meat, according to a new study published in Gut.
“In our prior analysis from a large prospective cohort study, the Health Professionals Follow-Up Study (HPFS), we found that red meat intake, independent of fiber, may be associated with a composite outcome of symptomatic diverticular disease, which included 385 incident cases over 4 years of follow-up,” wrote the authors, led by Andrew T. Chan, MD, of Massachusetts General Hospital, Boston. Dr. Chan added that “in the present study, we updated this analysis, which allowed us to prospectively examine the association between consumption of meat (total red meat, red unprocessed meat, red processed meat, poultry, and fish) with risk of incident diverticulitis in 764 cases over 26 years of follow-up.”
Dr. Chan and his coinvestigators conducted a prospective cohort study using subjects from the ongoing HPFS. Men who already had a diagnosis of diverticulitis, associated complications, inflammatory bowel disease, or a GI-related cancer at baseline were excluded from this analysis, leaving 46,461 eligible subjects. Of those, 764 developed diverticulitis.
The entirety of the follow-up period constituted 651,970 person-years. Average servings of total red meat per week were 1.2 in quintile 1, compared to 5.3 in quintile 3 and 13.5 in quintile 5. Those in the highest quintile had a multivariable risk ratio of 1.58 (95% CI, 1.19-2.11; P = .01), indicating a significantly higher risk for developing diverticulitis. In terms of unprocessed red meat, the average number of servings per week were 0.8 for the lower quintile, 3.2 for quintile 3, and 8.6 for quintile 5, yielding a risk ratio of 1.51 (95% CI, 1.12-2.03, P = .03) when comparing the highest and lowest cohorts. The increase in risk, however, leveled off after about 6 servings of red meat per week, and was found to be nonlinear (P = .002). Those who ate more servings of poultry or fish did not have a higher risk of diverticulitis.
“We also observed that unprocessed red meat, but not processed red meat, was the primary driver for the association between total red meat and risk of diverticulitis,” the authors explained. “Compared with processed meat, unprocessed meat (e.g., steak) is usually consumed in larger portions, which could lead to a larger undigested piece in the large bowel and induce different changes in colonic microbiota [and] higher cooking temperatures used in the preparation of unprocessed meat may influence bacterial composition or proinflammatory mediators in the colon.”
Although medical information and self-reports were validated, there are inherent possible limitations to self-reported data, such as misremembering the amount of meat consumed or reporting incorrect amounts. Residual confounding may have occurred despite adjustment of the data to account for it.
The National Institutes of Health funded the study. The authors reported no conflicts of interest.
Men who consume higher quantities of red meat are at an increased risk of developing diverticulitis, especially if they’re eating unprocessed red meat, according to a new study published in Gut.
“In our prior analysis from a large prospective cohort study, the Health Professionals Follow-Up Study (HPFS), we found that red meat intake, independent of fiber, may be associated with a composite outcome of symptomatic diverticular disease, which included 385 incident cases over 4 years of follow-up,” wrote the authors, led by Andrew T. Chan, MD, of Massachusetts General Hospital, Boston. Dr. Chan added that “in the present study, we updated this analysis, which allowed us to prospectively examine the association between consumption of meat (total red meat, red unprocessed meat, red processed meat, poultry, and fish) with risk of incident diverticulitis in 764 cases over 26 years of follow-up.”
Dr. Chan and his coinvestigators conducted a prospective cohort study using subjects from the ongoing HPFS. Men who already had a diagnosis of diverticulitis, associated complications, inflammatory bowel disease, or a GI-related cancer at baseline were excluded from this analysis, leaving 46,461 eligible subjects. Of those, 764 developed diverticulitis.
The entirety of the follow-up period constituted 651,970 person-years. Average servings of total red meat per week were 1.2 in quintile 1, compared to 5.3 in quintile 3 and 13.5 in quintile 5. Those in the highest quintile had a multivariable risk ratio of 1.58 (95% CI, 1.19-2.11; P = .01), indicating a significantly higher risk for developing diverticulitis. In terms of unprocessed red meat, the average number of servings per week were 0.8 for the lower quintile, 3.2 for quintile 3, and 8.6 for quintile 5, yielding a risk ratio of 1.51 (95% CI, 1.12-2.03, P = .03) when comparing the highest and lowest cohorts. The increase in risk, however, leveled off after about 6 servings of red meat per week, and was found to be nonlinear (P = .002). Those who ate more servings of poultry or fish did not have a higher risk of diverticulitis.
“We also observed that unprocessed red meat, but not processed red meat, was the primary driver for the association between total red meat and risk of diverticulitis,” the authors explained. “Compared with processed meat, unprocessed meat (e.g., steak) is usually consumed in larger portions, which could lead to a larger undigested piece in the large bowel and induce different changes in colonic microbiota [and] higher cooking temperatures used in the preparation of unprocessed meat may influence bacterial composition or proinflammatory mediators in the colon.”
Although medical information and self-reports were validated, there are inherent possible limitations to self-reported data, such as misremembering the amount of meat consumed or reporting incorrect amounts. Residual confounding may have occurred despite adjustment of the data to account for it.
The National Institutes of Health funded the study. The authors reported no conflicts of interest.
Key clinical point:
Major finding: Men with the highest consumption of red meat per week had a risk ratio of 1.58 (95% CI, 1.19-2.11, P = .01) compared to those with the lowest consumption, with an RR of 1.51 (95% CI, 1.12-2.03, P = .03) when comparing unprocessed red meat consumption.
Data source: Prospective cohort study of 51,529 men aged 40-75 years, in the United States.
Disclosures: The National Institutes of Health funded the study. The authors reported no conflicts of interest.
Macitentan boosts quality of life in PAH patients
Macitentan, a recent addition to the drugs that treat pulmonary arterial hypertension (PAH), improves and stabilizes quality of life for patients with the condition, according to an industry-funded study.
Macitentan (Opsumit) remains tremendously expensive, costing as much as $100,000 per year in the United States, and the study provides little in the way of direct comparison to other drugs in its class. Still, the drug’s effects on quality of life are dramatic, said study lead author Sanjay Mehta, MD, FRCPC, FCCP, professor of medicine at the University of Western Ontario and director of the Southwest Ontario Pulmonary Hypertension Clinic at the London Health Sciences Center in London, Ont.
Researchers found that those who took the 10-mg dose, versus placebo, reported significant improvement in seven of eight quality-of-life domains, and in physical and mental components scores, as measured by the 36-item Short Form Health Survey (SF-36). In addition, the study linked 10-mg doses, versus placebo, to a lower risk of a decline of three points or more in the physical component score (hazard ratio [HR], 0.60; 95% CI, 0.47-0.76; P less than .0001] and the mental component scores (HR, 0.76; 95% CI, 0.61-0.95; P = .0173) until end of treatment.
“The drug has shown stability in patients’ quality of life over 6 months and 12 months,” Dr. Mehta said in an interview. “I can’t cure anybody, and they’ll get worse at some point, but I can improve them. They physically feel better, they’re less short of breath with less body pain, and they feel better psychologically.”
Macitentan, an endothelin receptor antagonist, received Food and Drug Administration approval in 2013 following a study that year (N Engl J Med. 2013 Aug 29;369[9]:809-18) that linked 10-mg doses to a significantly lower risk of death and various complications, compared with placebo and the 3-mg dose. The new study (Chest. 2017 Jan;151[1]:106-18), is an analysis of data from the 2013 study.
The PAH patients were randomly assigned to one of three groups: macitentan 10 mg once daily (234), macitentan 3 mg (237), and placebo (239). The study examined responses from 710 patients (76.9% were female, 55.2% were white, mean age was 45.5) to the SF-36 at baseline, 6 months, 12 months, and end of treatment.
Dr. Mehta noted that macitentan has not been clinically compared to the other drugs. The study, however, notes that it is the first PAH treatment to show improvement in seven of eight domains in the quality-of-life survey.
The new study was funded by Actelion Pharmaceuticals, maker of macitentan. Dr. Mehta has received consulting and speaking fees and institutional support for clinical trials from Actelion, among other drug companies. The other authors report various disclosures, including relationships with Actelion.
Macitentan, a recent addition to the drugs that treat pulmonary arterial hypertension (PAH), improves and stabilizes quality of life for patients with the condition, according to an industry-funded study.
Macitentan (Opsumit) remains tremendously expensive, costing as much as $100,000 per year in the United States, and the study provides little in the way of direct comparison to other drugs in its class. Still, the drug’s effects on quality of life are dramatic, said study lead author Sanjay Mehta, MD, FRCPC, FCCP, professor of medicine at the University of Western Ontario and director of the Southwest Ontario Pulmonary Hypertension Clinic at the London Health Sciences Center in London, Ont.
Researchers found that those who took the 10-mg dose, versus placebo, reported significant improvement in seven of eight quality-of-life domains, and in physical and mental components scores, as measured by the 36-item Short Form Health Survey (SF-36). In addition, the study linked 10-mg doses, versus placebo, to a lower risk of a decline of three points or more in the physical component score (hazard ratio [HR], 0.60; 95% CI, 0.47-0.76; P less than .0001] and the mental component scores (HR, 0.76; 95% CI, 0.61-0.95; P = .0173) until end of treatment.
“The drug has shown stability in patients’ quality of life over 6 months and 12 months,” Dr. Mehta said in an interview. “I can’t cure anybody, and they’ll get worse at some point, but I can improve them. They physically feel better, they’re less short of breath with less body pain, and they feel better psychologically.”
Macitentan, an endothelin receptor antagonist, received Food and Drug Administration approval in 2013 following a study that year (N Engl J Med. 2013 Aug 29;369[9]:809-18) that linked 10-mg doses to a significantly lower risk of death and various complications, compared with placebo and the 3-mg dose. The new study (Chest. 2017 Jan;151[1]:106-18), is an analysis of data from the 2013 study.
The PAH patients were randomly assigned to one of three groups: macitentan 10 mg once daily (234), macitentan 3 mg (237), and placebo (239). The study examined responses from 710 patients (76.9% were female, 55.2% were white, mean age was 45.5) to the SF-36 at baseline, 6 months, 12 months, and end of treatment.
Dr. Mehta noted that macitentan has not been clinically compared to the other drugs. The study, however, notes that it is the first PAH treatment to show improvement in seven of eight domains in the quality-of-life survey.
The new study was funded by Actelion Pharmaceuticals, maker of macitentan. Dr. Mehta has received consulting and speaking fees and institutional support for clinical trials from Actelion, among other drug companies. The other authors report various disclosures, including relationships with Actelion.
Macitentan, a recent addition to the drugs that treat pulmonary arterial hypertension (PAH), improves and stabilizes quality of life for patients with the condition, according to an industry-funded study.
Macitentan (Opsumit) remains tremendously expensive, costing as much as $100,000 per year in the United States, and the study provides little in the way of direct comparison to other drugs in its class. Still, the drug’s effects on quality of life are dramatic, said study lead author Sanjay Mehta, MD, FRCPC, FCCP, professor of medicine at the University of Western Ontario and director of the Southwest Ontario Pulmonary Hypertension Clinic at the London Health Sciences Center in London, Ont.
Researchers found that those who took the 10-mg dose, versus placebo, reported significant improvement in seven of eight quality-of-life domains, and in physical and mental components scores, as measured by the 36-item Short Form Health Survey (SF-36). In addition, the study linked 10-mg doses, versus placebo, to a lower risk of a decline of three points or more in the physical component score (hazard ratio [HR], 0.60; 95% CI, 0.47-0.76; P less than .0001] and the mental component scores (HR, 0.76; 95% CI, 0.61-0.95; P = .0173) until end of treatment.
“The drug has shown stability in patients’ quality of life over 6 months and 12 months,” Dr. Mehta said in an interview. “I can’t cure anybody, and they’ll get worse at some point, but I can improve them. They physically feel better, they’re less short of breath with less body pain, and they feel better psychologically.”
Macitentan, an endothelin receptor antagonist, received Food and Drug Administration approval in 2013 following a study that year (N Engl J Med. 2013 Aug 29;369[9]:809-18) that linked 10-mg doses to a significantly lower risk of death and various complications, compared with placebo and the 3-mg dose. The new study (Chest. 2017 Jan;151[1]:106-18), is an analysis of data from the 2013 study.
The PAH patients were randomly assigned to one of three groups: macitentan 10 mg once daily (234), macitentan 3 mg (237), and placebo (239). The study examined responses from 710 patients (76.9% were female, 55.2% were white, mean age was 45.5) to the SF-36 at baseline, 6 months, 12 months, and end of treatment.
Dr. Mehta noted that macitentan has not been clinically compared to the other drugs. The study, however, notes that it is the first PAH treatment to show improvement in seven of eight domains in the quality-of-life survey.
The new study was funded by Actelion Pharmaceuticals, maker of macitentan. Dr. Mehta has received consulting and speaking fees and institutional support for clinical trials from Actelion, among other drug companies. The other authors report various disclosures, including relationships with Actelion.
FROM CHEST
Key clinical point: Macitentan improves and stabilizes quality of life in patients with pulmonary arterial hypertension.
Major finding: Patients who took 10 mg daily macitentan improved in seven of eight quality-of-life domains and in combined physical and mental health measures.
Data source: Multicenter, double-blind, placebo-controlled, randomized phase III study of 710 patients (76.9% female, 55.2% white, mean age 45.5) assigned to placebo, macitentan 3 mg, or macitentan 10 mg once daily.
Disclosures: Actelion Pharmaceuticals, maker of macitentan, funded the study. The authors disclosed ties with Actelion.