User login
Accountability and Whistleblowers in VA Spotlight
In an ongoing effort to improve oversight and to protect potential whistleblowers at the VA, President Trump has signed a new executive order creating the Office of Accountability and Whistleblower Protection. The executive order establishes an office that will report directly to VA Secretary David. J. Shulkin, MD. “Accountability is an important issue to us at VA and something that we’re focusing on to make sure that we have employees who work and are committed to the mission of serving our veterans,” Dr. Shulkin explained at an April 26 press conference. “When we find employees that have deviated from those values, we want to make sure that we can move them outside the VA and not have them working at VA.”
The new office is not the first effort at the VA to protect whistleblowers or to expedite the removal of employees. In 2014 the Office of Accountability and Review was established to increase central office scrutiny of senior-level executives at local and regional VA facilities. In contrast Dr. Shulkin noted, “This is a broader office that will be taking a look at all of our employees.” The current VA Whistleblower Office, created just last year will be incorporated into the new office, according to Dr. Shulkin.
Related: VA Secretary Shulkin Calls for New Powers to Fire VA Employees
Not everyone greeted the announcement with praise. “This rush to fire feds faster, first at VA, but with attempts to spread it across government, comes with a serious risk,” argued Washington Post columnist Joe Davidson. “Yes, due process rights can be slow and cumbersome. They protect, however, not just employees, but more importantly, also the public from a politicized system that favors citizens of one political party over another. Reforms must respect civil service protections. They should be acknowledged by government leaders and not be ignored as they were at the signing.”
Officials at the Project on Government Oversight expressed concern that whistleblowers should have an independent channel to report their concerns. Like the current whistleblower office, the new structure “may do far more damage than good,” the organization reported “It is incredibly important that whistleblowers have the ability to go to an independent office to report wrongdoing, since an internal office could be pressured to act in the VA’s interest by covering up problems and silencing whistleblowers.”
Related: VA Launches Investigation into Cincinnati Facility Mismanagement
Dr. Shulkin insisted that the office did not negate the need for the new legislation that he has called for that would speed the process of firing problem employees. Nor will the new office replace the hot line set up by the White House for veteran complaints about VA service. “These are all 3 efforts that are important for us to identify issues that are preventing us from doing the very best job that we can,” he explained. “We’re keeping our employees and our executives accountable to the values, to be able to work at the VA. We are soliciting input from veterans who feel that they have issues that they want to share with us, and that’s what the hotline will be doing.”
While the focus on the effort is on employees malfeasance, Dr. Shulkin cautioned that the VA was still concerned about employee morale and protecting whistleblowers from retaliation. “Our employees have to feel safe, when they see something, to tell us about it,” he explained. “The message is clear that we will not tolerate whistleblower retaliation in the [VA]. And we will take actions if we do determine that retaliation has been imposed upon an employee who has come forth with an issue.”
Related: Deputy Secretary of Veterans Affairs Gibson Defends VA Discipline Guidelines
Dr. Shulkin also announced a new task force that would tackle fraud, waste, and abuse, “to make sure that we are aggressively investigating any issues that might lead to the waste of taxpayer dollars.”
In an ongoing effort to improve oversight and to protect potential whistleblowers at the VA, President Trump has signed a new executive order creating the Office of Accountability and Whistleblower Protection. The executive order establishes an office that will report directly to VA Secretary David. J. Shulkin, MD. “Accountability is an important issue to us at VA and something that we’re focusing on to make sure that we have employees who work and are committed to the mission of serving our veterans,” Dr. Shulkin explained at an April 26 press conference. “When we find employees that have deviated from those values, we want to make sure that we can move them outside the VA and not have them working at VA.”
The new office is not the first effort at the VA to protect whistleblowers or to expedite the removal of employees. In 2014 the Office of Accountability and Review was established to increase central office scrutiny of senior-level executives at local and regional VA facilities. In contrast Dr. Shulkin noted, “This is a broader office that will be taking a look at all of our employees.” The current VA Whistleblower Office, created just last year will be incorporated into the new office, according to Dr. Shulkin.
Related: VA Secretary Shulkin Calls for New Powers to Fire VA Employees
Not everyone greeted the announcement with praise. “This rush to fire feds faster, first at VA, but with attempts to spread it across government, comes with a serious risk,” argued Washington Post columnist Joe Davidson. “Yes, due process rights can be slow and cumbersome. They protect, however, not just employees, but more importantly, also the public from a politicized system that favors citizens of one political party over another. Reforms must respect civil service protections. They should be acknowledged by government leaders and not be ignored as they were at the signing.”
Officials at the Project on Government Oversight expressed concern that whistleblowers should have an independent channel to report their concerns. Like the current whistleblower office, the new structure “may do far more damage than good,” the organization reported “It is incredibly important that whistleblowers have the ability to go to an independent office to report wrongdoing, since an internal office could be pressured to act in the VA’s interest by covering up problems and silencing whistleblowers.”
Related: VA Launches Investigation into Cincinnati Facility Mismanagement
Dr. Shulkin insisted that the office did not negate the need for the new legislation that he has called for that would speed the process of firing problem employees. Nor will the new office replace the hot line set up by the White House for veteran complaints about VA service. “These are all 3 efforts that are important for us to identify issues that are preventing us from doing the very best job that we can,” he explained. “We’re keeping our employees and our executives accountable to the values, to be able to work at the VA. We are soliciting input from veterans who feel that they have issues that they want to share with us, and that’s what the hotline will be doing.”
While the focus on the effort is on employees malfeasance, Dr. Shulkin cautioned that the VA was still concerned about employee morale and protecting whistleblowers from retaliation. “Our employees have to feel safe, when they see something, to tell us about it,” he explained. “The message is clear that we will not tolerate whistleblower retaliation in the [VA]. And we will take actions if we do determine that retaliation has been imposed upon an employee who has come forth with an issue.”
Related: Deputy Secretary of Veterans Affairs Gibson Defends VA Discipline Guidelines
Dr. Shulkin also announced a new task force that would tackle fraud, waste, and abuse, “to make sure that we are aggressively investigating any issues that might lead to the waste of taxpayer dollars.”
In an ongoing effort to improve oversight and to protect potential whistleblowers at the VA, President Trump has signed a new executive order creating the Office of Accountability and Whistleblower Protection. The executive order establishes an office that will report directly to VA Secretary David. J. Shulkin, MD. “Accountability is an important issue to us at VA and something that we’re focusing on to make sure that we have employees who work and are committed to the mission of serving our veterans,” Dr. Shulkin explained at an April 26 press conference. “When we find employees that have deviated from those values, we want to make sure that we can move them outside the VA and not have them working at VA.”
The new office is not the first effort at the VA to protect whistleblowers or to expedite the removal of employees. In 2014 the Office of Accountability and Review was established to increase central office scrutiny of senior-level executives at local and regional VA facilities. In contrast Dr. Shulkin noted, “This is a broader office that will be taking a look at all of our employees.” The current VA Whistleblower Office, created just last year will be incorporated into the new office, according to Dr. Shulkin.
Related: VA Secretary Shulkin Calls for New Powers to Fire VA Employees
Not everyone greeted the announcement with praise. “This rush to fire feds faster, first at VA, but with attempts to spread it across government, comes with a serious risk,” argued Washington Post columnist Joe Davidson. “Yes, due process rights can be slow and cumbersome. They protect, however, not just employees, but more importantly, also the public from a politicized system that favors citizens of one political party over another. Reforms must respect civil service protections. They should be acknowledged by government leaders and not be ignored as they were at the signing.”
Officials at the Project on Government Oversight expressed concern that whistleblowers should have an independent channel to report their concerns. Like the current whistleblower office, the new structure “may do far more damage than good,” the organization reported “It is incredibly important that whistleblowers have the ability to go to an independent office to report wrongdoing, since an internal office could be pressured to act in the VA’s interest by covering up problems and silencing whistleblowers.”
Related: VA Launches Investigation into Cincinnati Facility Mismanagement
Dr. Shulkin insisted that the office did not negate the need for the new legislation that he has called for that would speed the process of firing problem employees. Nor will the new office replace the hot line set up by the White House for veteran complaints about VA service. “These are all 3 efforts that are important for us to identify issues that are preventing us from doing the very best job that we can,” he explained. “We’re keeping our employees and our executives accountable to the values, to be able to work at the VA. We are soliciting input from veterans who feel that they have issues that they want to share with us, and that’s what the hotline will be doing.”
While the focus on the effort is on employees malfeasance, Dr. Shulkin cautioned that the VA was still concerned about employee morale and protecting whistleblowers from retaliation. “Our employees have to feel safe, when they see something, to tell us about it,” he explained. “The message is clear that we will not tolerate whistleblower retaliation in the [VA]. And we will take actions if we do determine that retaliation has been imposed upon an employee who has come forth with an issue.”
Related: Deputy Secretary of Veterans Affairs Gibson Defends VA Discipline Guidelines
Dr. Shulkin also announced a new task force that would tackle fraud, waste, and abuse, “to make sure that we are aggressively investigating any issues that might lead to the waste of taxpayer dollars.”
Up the Creek Without a Provider
ANSWER
The ECG is remarkable for sinus tachycardia and left ventricular hypertrophy. Equal numbers of P and QRS complexes with a consistent PR interval indicate sinus tachycardia. High voltages in the limb leads (R in lead I and S in lead III ≥ 25 mm) or precordial leads (S in lead V1 and R in lead V5 or V6 ≥ 35 mm) constitute left ventricular hypertrophy.
ANSWER
The ECG is remarkable for sinus tachycardia and left ventricular hypertrophy. Equal numbers of P and QRS complexes with a consistent PR interval indicate sinus tachycardia. High voltages in the limb leads (R in lead I and S in lead III ≥ 25 mm) or precordial leads (S in lead V1 and R in lead V5 or V6 ≥ 35 mm) constitute left ventricular hypertrophy.
ANSWER
The ECG is remarkable for sinus tachycardia and left ventricular hypertrophy. Equal numbers of P and QRS complexes with a consistent PR interval indicate sinus tachycardia. High voltages in the limb leads (R in lead I and S in lead III ≥ 25 mm) or precordial leads (S in lead V1 and R in lead V5 or V6 ≥ 35 mm) constitute left ventricular hypertrophy.
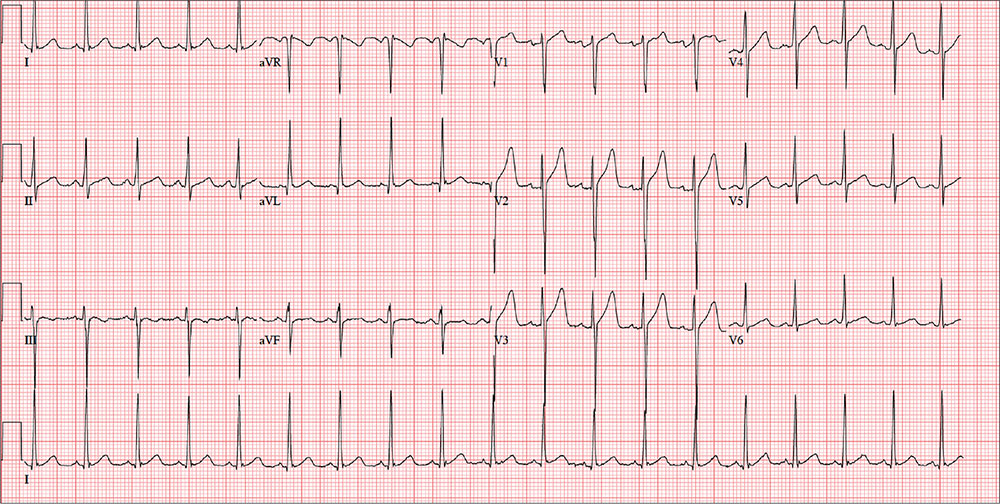
A 40-year-old man presents to establish care with you as his primary care provider; he was recently forced to make this change because his prior network stopped accepting his insurance policy. He works as an electrical engineer and is a competitive rower. He began rowing at age 14; he was on his university’s team and now rows in a competitive, age-matched league.
He has never had any health-related issues, apart from sprained ankles and a fractured right clavicle in childhood. He exercises daily at an exclusive men’s club. He has no history of hypertension, diabetes, hyperlipidemia, chest pain or discomfort, shortness of breath, or exertional dyspnea. He has no surgical history.
The patient is single and
He is not taking any medications and has no known drug allergies. The review of systems is remarkable only for recent rhinitis, which is resolving.
Vital signs include a blood pressure of 124/60 mm Hg; pulse, 110 beats/min; respiratory rate, 14 breaths/min-1; and temperature, 98.4°F. His weight is 194 lb and his height, 75 in.
The physical exam reveals a mildly anxious male in no acute distress. When asked if he is nervous, he says yes, because he’s “used to having someone else examine him.” His HEENT exam is normal, as is his thyroid exam. He has no jugular venous distention. The lungs are clear bilaterally. His heart rate is 110 beats/min and regular with no murmurs, rubs, or gallops. The abdomen is soft and nontender, with no palpable organomegaly. Peripheral pulses are strong and equal bilaterally, and there is no peripheral edema. The neurologic exam is grossly intact.
Routine blood tests are performed, and an HIV titer, ECG, and chest x-ray are ordered. The ECG reveals a heart rate of 112 beats/min; PR interval, 132 ms; QRS duration, 76 ms; QT/QTc interval, 326/444 ms; P axis, 59°; R axis, –8°; T axis, 26°. What is your interpretation of this ECG?
Hospitalists can do better at end-of-life care, expert says
As a 99-year-old friend neared the end of her life, she offered a lesson for the health care world, said Deborah Korenstein, MD, chief of general internal medicine and director of clinical effectiveness at Memorial Sloan Kettering Cancer Center, N.Y., in the Tuesday session “Finding High Value Inpatient Care at the End of Life.”
The woman, nicknamed “Mitch,” had bluntly made her preference clear, Dr. Korenstein said: “She wanted to live independently as long as she could, and then, she wanted to be dead.”
But when a pathology report showed urothelial cancer, that preference didn’t stop an oncology urologist from suggesting that Mitch enter a clinical trial on an unproven therapy. Worse, Mitch initially said “yes” to this idea, seemingly because she thought that’s what she was expected to say.
It was only when Dr. Korenstein spoke with her that she changed her mind, entered inpatient hospice care, and died peacefully.
“I think it’s a cautionary tale about when a patient is crystal clear about their wishes,” she said. “The wave of the medical system kind of pushes them along in a particular direction that may go against their wishes.”
Dr. Korenstein said U.S. health care system does fairly well in some areas – for instance, research shows that about 60% of people die in their preferred location, whether at home or somewhere else. But it does not do so well in others – a 2013 Journal of General Internal Medicine study found that, during 2002-2008, Medicare beneficiaries typically spent $39,000 out of pocket on their medical care, and in 25% of cases, what they spent exceeded the total value of their assets.
As far as individual preferences, these tend to correlate poorly with the care that people actually get, Dr. Korenstein said. Patients often don’t express their wishes, doctors are poor judges of what matters to individual people, and care is largely driven by physician preferences and by the care setting involved, she said.
Given those problems, she said, “we cannot possibly be providing high-value individualized care.”
Hospitalists are well positioned to help patients’ preferences align with care, she added. Sometimes, a sustained relationship with a patient, while generally a positive thing, might lead a provider to become invested in their care in “ways that are not always rational.” So a hospitalist can have a helpful vantage point.
As a 99-year-old friend neared the end of her life, she offered a lesson for the health care world, said Deborah Korenstein, MD, chief of general internal medicine and director of clinical effectiveness at Memorial Sloan Kettering Cancer Center, N.Y., in the Tuesday session “Finding High Value Inpatient Care at the End of Life.”
The woman, nicknamed “Mitch,” had bluntly made her preference clear, Dr. Korenstein said: “She wanted to live independently as long as she could, and then, she wanted to be dead.”
But when a pathology report showed urothelial cancer, that preference didn’t stop an oncology urologist from suggesting that Mitch enter a clinical trial on an unproven therapy. Worse, Mitch initially said “yes” to this idea, seemingly because she thought that’s what she was expected to say.
It was only when Dr. Korenstein spoke with her that she changed her mind, entered inpatient hospice care, and died peacefully.
“I think it’s a cautionary tale about when a patient is crystal clear about their wishes,” she said. “The wave of the medical system kind of pushes them along in a particular direction that may go against their wishes.”
Dr. Korenstein said U.S. health care system does fairly well in some areas – for instance, research shows that about 60% of people die in their preferred location, whether at home or somewhere else. But it does not do so well in others – a 2013 Journal of General Internal Medicine study found that, during 2002-2008, Medicare beneficiaries typically spent $39,000 out of pocket on their medical care, and in 25% of cases, what they spent exceeded the total value of their assets.
As far as individual preferences, these tend to correlate poorly with the care that people actually get, Dr. Korenstein said. Patients often don’t express their wishes, doctors are poor judges of what matters to individual people, and care is largely driven by physician preferences and by the care setting involved, she said.
Given those problems, she said, “we cannot possibly be providing high-value individualized care.”
Hospitalists are well positioned to help patients’ preferences align with care, she added. Sometimes, a sustained relationship with a patient, while generally a positive thing, might lead a provider to become invested in their care in “ways that are not always rational.” So a hospitalist can have a helpful vantage point.
As a 99-year-old friend neared the end of her life, she offered a lesson for the health care world, said Deborah Korenstein, MD, chief of general internal medicine and director of clinical effectiveness at Memorial Sloan Kettering Cancer Center, N.Y., in the Tuesday session “Finding High Value Inpatient Care at the End of Life.”
The woman, nicknamed “Mitch,” had bluntly made her preference clear, Dr. Korenstein said: “She wanted to live independently as long as she could, and then, she wanted to be dead.”
But when a pathology report showed urothelial cancer, that preference didn’t stop an oncology urologist from suggesting that Mitch enter a clinical trial on an unproven therapy. Worse, Mitch initially said “yes” to this idea, seemingly because she thought that’s what she was expected to say.
It was only when Dr. Korenstein spoke with her that she changed her mind, entered inpatient hospice care, and died peacefully.
“I think it’s a cautionary tale about when a patient is crystal clear about their wishes,” she said. “The wave of the medical system kind of pushes them along in a particular direction that may go against their wishes.”
Dr. Korenstein said U.S. health care system does fairly well in some areas – for instance, research shows that about 60% of people die in their preferred location, whether at home or somewhere else. But it does not do so well in others – a 2013 Journal of General Internal Medicine study found that, during 2002-2008, Medicare beneficiaries typically spent $39,000 out of pocket on their medical care, and in 25% of cases, what they spent exceeded the total value of their assets.
As far as individual preferences, these tend to correlate poorly with the care that people actually get, Dr. Korenstein said. Patients often don’t express their wishes, doctors are poor judges of what matters to individual people, and care is largely driven by physician preferences and by the care setting involved, she said.
Given those problems, she said, “we cannot possibly be providing high-value individualized care.”
Hospitalists are well positioned to help patients’ preferences align with care, she added. Sometimes, a sustained relationship with a patient, while generally a positive thing, might lead a provider to become invested in their care in “ways that are not always rational.” So a hospitalist can have a helpful vantage point.
VIDEO: Policy-focused SHM president thinks hospitalists can impact global, systems change
An original member of the Society of Hospital Medicine, new SHM Board President Ron Greeno, MD, MHM, is excited about helping to guide hospitalists into a new era of health system transformation.
The former chair of SHM’s Public Policy Committee, Dr. Greeno believes payment reforms like MACRA will have a “huge impact” on both hospitalists and the hospitals/health systems they work in. He expects hospital medicine, as a field, is well positioned for such changes and can play a vital role in systems change at the global level.
“In order to impact those things, hospitalists have to be ready to help change systems,” he said after his plenary address Tuesday at HM17.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
An original member of the Society of Hospital Medicine, new SHM Board President Ron Greeno, MD, MHM, is excited about helping to guide hospitalists into a new era of health system transformation.
The former chair of SHM’s Public Policy Committee, Dr. Greeno believes payment reforms like MACRA will have a “huge impact” on both hospitalists and the hospitals/health systems they work in. He expects hospital medicine, as a field, is well positioned for such changes and can play a vital role in systems change at the global level.
“In order to impact those things, hospitalists have to be ready to help change systems,” he said after his plenary address Tuesday at HM17.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
An original member of the Society of Hospital Medicine, new SHM Board President Ron Greeno, MD, MHM, is excited about helping to guide hospitalists into a new era of health system transformation.
The former chair of SHM’s Public Policy Committee, Dr. Greeno believes payment reforms like MACRA will have a “huge impact” on both hospitalists and the hospitals/health systems they work in. He expects hospital medicine, as a field, is well positioned for such changes and can play a vital role in systems change at the global level.
“In order to impact those things, hospitalists have to be ready to help change systems,” he said after his plenary address Tuesday at HM17.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Adalimumab strikes out for hand osteoarthritis
LAS VEGAS – Adalimumab proved no better than placebo for the treatment of erosive hand osteoarthritis in the double-blind, placebo-controlled, randomized HUMOR trial, throwing cold water on hopes that a disease-modifying treatment for osteoarthritis might at long last have been found.
“This randomized controlled trial demonstrated that subcutaneous adalimumab at 40 mg every other week was no different from placebo for alleviation of pain, synovitis, or bone marrow lesions in patients with erosive hand osteoarthritis presenting with MRI-detected synovitis. This suggests that pain and inflammation are not responsive to TNF [tumor necrosis factor] inhibition in this patient population,” Dawn Aitken, PhD, declared at the World Congress on Osteoarthritis.

The primary endpoint was change in the visual analog pain score over the course of 12 weeks of therapy. Scores dropped by an average of 3.0 points from a mean baseline of 63.6 with adalimumab and by 0.7 points with placebo. That between-group difference wasn’t statistically significant, nor were those modest reductions clinically meaningful. By convention, a clinically meaningful treatment result requires at least a 15-point improvement on the self-assessed pain scale, noted Dr. Aitken of the University of Tasmania in Hobart, Australia.
It made no difference which treatment arm came first.
“There was absolutely no placebo effect,” she said.
The study proved negative for all prespecified secondary endpoints, too. These included change in the Australian/Canadian Hand OA Index pain, function, and stiffness subscales, as well as change in bone marrow lesions and synovitis. A mere 12% of patients showed improvement in synovitis scores with adalimumab, as did 10% on placebo. Five percent of the adalimumab group and 7% on placebo showed improvement in bone marrow lesion scores.
The proinflammatory cytokine TNF-alpha has been shown to play a key role in OA development and progression. However, several prior studies failed to show a benefit for anti-TNF therapy in terms of reducing pain in hand OA patients, although in one of those negative studies, a post hoc analysis found that adalimumab halted erosive progression in the subset of interphalangeal joints with palpable soft tissue swelling at baseline (Ann Rheum Dis. 2012 Jun;71[6]:891-8).

However, there was a positive signal: In a subgroup analysis confined to the patients with inflammatory joints, the use of etanercept was associated with less structural damage of those joints over time as assessed using the quantitative Ghent University Scoring System, or GUSS, according to Dr. Kloppenburg, professor of rheumatology at Leiden (the Netherlands) University.
The emphatically negative results of the HUMOR trial were greeted with dismay by several disappointed audience members. They raised questions: Might a study featuring more than 12 weeks of treatment have brought positive results? How about a larger study? Why no placebo effect, given that patients were receiving injections? Is this area of investigation of TNF-inhibitor therapy now a dead end? Or could adalimumab and etanercept have differential efficacy in hand OA, even though they are both TNF inhibitors?
Dr. Aitken had an answer for every question.
“I think our study was big enough based on our power calculations to detect a difference of 15 mm on a visual analog scale, which is a clinically important difference. And that’s what we should be chasing in OA, that big effect. Patients are not going to be interested in a treatment that improves their pain by 5 mm,” she said.
As for the possibility that 12 weeks of adalimumab might have been too short to see a treatment effect, Dr. Aitken noted that anti-TNF therapy in rheumatoid arthritis brings a rapid response.
“Ideally, if you did have a disease-modifying drug for osteoarthritis you would want it to have a relatively quick response for pain; if patients aren’t seeing an effect after 3 months they might be less interested in taking it,” she continued.
Also, studies with a crossover design, like HUMOR, are known to have a much smaller placebo effect because patients know that at some point they’re certain to receive the active agent, Dr. Aitken observed.
As for the possibility that etanercept might be effective for erosive hand OA, while adalimumab is not, one physician commented, “That’s really scraping the bottom of the barrel.”
The HUMOR trial was sponsored by AbbVie. Dr. Aitken reported having no financial conflicts.
LAS VEGAS – Adalimumab proved no better than placebo for the treatment of erosive hand osteoarthritis in the double-blind, placebo-controlled, randomized HUMOR trial, throwing cold water on hopes that a disease-modifying treatment for osteoarthritis might at long last have been found.
“This randomized controlled trial demonstrated that subcutaneous adalimumab at 40 mg every other week was no different from placebo for alleviation of pain, synovitis, or bone marrow lesions in patients with erosive hand osteoarthritis presenting with MRI-detected synovitis. This suggests that pain and inflammation are not responsive to TNF [tumor necrosis factor] inhibition in this patient population,” Dawn Aitken, PhD, declared at the World Congress on Osteoarthritis.
The primary endpoint was change in the visual analog pain score over the course of 12 weeks of therapy. Scores dropped by an average of 3.0 points from a mean baseline of 63.6 with adalimumab and by 0.7 points with placebo. That between-group difference wasn’t statistically significant, nor were those modest reductions clinically meaningful. By convention, a clinically meaningful treatment result requires at least a 15-point improvement on the self-assessed pain scale, noted Dr. Aitken of the University of Tasmania in Hobart, Australia.
It made no difference which treatment arm came first.
“There was absolutely no placebo effect,” she said.
The study proved negative for all prespecified secondary endpoints, too. These included change in the Australian/Canadian Hand OA Index pain, function, and stiffness subscales, as well as change in bone marrow lesions and synovitis. A mere 12% of patients showed improvement in synovitis scores with adalimumab, as did 10% on placebo. Five percent of the adalimumab group and 7% on placebo showed improvement in bone marrow lesion scores.
The proinflammatory cytokine TNF-alpha has been shown to play a key role in OA development and progression. However, several prior studies failed to show a benefit for anti-TNF therapy in terms of reducing pain in hand OA patients, although in one of those negative studies, a post hoc analysis found that adalimumab halted erosive progression in the subset of interphalangeal joints with palpable soft tissue swelling at baseline (Ann Rheum Dis. 2012 Jun;71[6]:891-8).
However, there was a positive signal: In a subgroup analysis confined to the patients with inflammatory joints, the use of etanercept was associated with less structural damage of those joints over time as assessed using the quantitative Ghent University Scoring System, or GUSS, according to Dr. Kloppenburg, professor of rheumatology at Leiden (the Netherlands) University.
The emphatically negative results of the HUMOR trial were greeted with dismay by several disappointed audience members. They raised questions: Might a study featuring more than 12 weeks of treatment have brought positive results? How about a larger study? Why no placebo effect, given that patients were receiving injections? Is this area of investigation of TNF-inhibitor therapy now a dead end? Or could adalimumab and etanercept have differential efficacy in hand OA, even though they are both TNF inhibitors?
Dr. Aitken had an answer for every question.
“I think our study was big enough based on our power calculations to detect a difference of 15 mm on a visual analog scale, which is a clinically important difference. And that’s what we should be chasing in OA, that big effect. Patients are not going to be interested in a treatment that improves their pain by 5 mm,” she said.
As for the possibility that 12 weeks of adalimumab might have been too short to see a treatment effect, Dr. Aitken noted that anti-TNF therapy in rheumatoid arthritis brings a rapid response.
“Ideally, if you did have a disease-modifying drug for osteoarthritis you would want it to have a relatively quick response for pain; if patients aren’t seeing an effect after 3 months they might be less interested in taking it,” she continued.
Also, studies with a crossover design, like HUMOR, are known to have a much smaller placebo effect because patients know that at some point they’re certain to receive the active agent, Dr. Aitken observed.
As for the possibility that etanercept might be effective for erosive hand OA, while adalimumab is not, one physician commented, “That’s really scraping the bottom of the barrel.”
The HUMOR trial was sponsored by AbbVie. Dr. Aitken reported having no financial conflicts.
LAS VEGAS – Adalimumab proved no better than placebo for the treatment of erosive hand osteoarthritis in the double-blind, placebo-controlled, randomized HUMOR trial, throwing cold water on hopes that a disease-modifying treatment for osteoarthritis might at long last have been found.
“This randomized controlled trial demonstrated that subcutaneous adalimumab at 40 mg every other week was no different from placebo for alleviation of pain, synovitis, or bone marrow lesions in patients with erosive hand osteoarthritis presenting with MRI-detected synovitis. This suggests that pain and inflammation are not responsive to TNF [tumor necrosis factor] inhibition in this patient population,” Dawn Aitken, PhD, declared at the World Congress on Osteoarthritis.
The primary endpoint was change in the visual analog pain score over the course of 12 weeks of therapy. Scores dropped by an average of 3.0 points from a mean baseline of 63.6 with adalimumab and by 0.7 points with placebo. That between-group difference wasn’t statistically significant, nor were those modest reductions clinically meaningful. By convention, a clinically meaningful treatment result requires at least a 15-point improvement on the self-assessed pain scale, noted Dr. Aitken of the University of Tasmania in Hobart, Australia.
It made no difference which treatment arm came first.
“There was absolutely no placebo effect,” she said.
The study proved negative for all prespecified secondary endpoints, too. These included change in the Australian/Canadian Hand OA Index pain, function, and stiffness subscales, as well as change in bone marrow lesions and synovitis. A mere 12% of patients showed improvement in synovitis scores with adalimumab, as did 10% on placebo. Five percent of the adalimumab group and 7% on placebo showed improvement in bone marrow lesion scores.
The proinflammatory cytokine TNF-alpha has been shown to play a key role in OA development and progression. However, several prior studies failed to show a benefit for anti-TNF therapy in terms of reducing pain in hand OA patients, although in one of those negative studies, a post hoc analysis found that adalimumab halted erosive progression in the subset of interphalangeal joints with palpable soft tissue swelling at baseline (Ann Rheum Dis. 2012 Jun;71[6]:891-8).
However, there was a positive signal: In a subgroup analysis confined to the patients with inflammatory joints, the use of etanercept was associated with less structural damage of those joints over time as assessed using the quantitative Ghent University Scoring System, or GUSS, according to Dr. Kloppenburg, professor of rheumatology at Leiden (the Netherlands) University.
The emphatically negative results of the HUMOR trial were greeted with dismay by several disappointed audience members. They raised questions: Might a study featuring more than 12 weeks of treatment have brought positive results? How about a larger study? Why no placebo effect, given that patients were receiving injections? Is this area of investigation of TNF-inhibitor therapy now a dead end? Or could adalimumab and etanercept have differential efficacy in hand OA, even though they are both TNF inhibitors?
Dr. Aitken had an answer for every question.
“I think our study was big enough based on our power calculations to detect a difference of 15 mm on a visual analog scale, which is a clinically important difference. And that’s what we should be chasing in OA, that big effect. Patients are not going to be interested in a treatment that improves their pain by 5 mm,” she said.
As for the possibility that 12 weeks of adalimumab might have been too short to see a treatment effect, Dr. Aitken noted that anti-TNF therapy in rheumatoid arthritis brings a rapid response.
“Ideally, if you did have a disease-modifying drug for osteoarthritis you would want it to have a relatively quick response for pain; if patients aren’t seeing an effect after 3 months they might be less interested in taking it,” she continued.
Also, studies with a crossover design, like HUMOR, are known to have a much smaller placebo effect because patients know that at some point they’re certain to receive the active agent, Dr. Aitken observed.
As for the possibility that etanercept might be effective for erosive hand OA, while adalimumab is not, one physician commented, “That’s really scraping the bottom of the barrel.”
The HUMOR trial was sponsored by AbbVie. Dr. Aitken reported having no financial conflicts.
FROM OARSI 2017
Key clinical point:
Major finding: Scores dropped by an average of 3.0 points from a mean baseline of 63.6 with adalimumab and by 0.7 points with placebo on the primary endpoint of change in the visual analog pain score over the course of 12 weeks.
Data source: The HUMOR trial was a double-blind, placebo-controlled, randomized, crossover trial in which 43 participants with erosive hand osteoarthritis received 12 weeks of treatment with adalimumab and 12 weeks of placebo.
Disclosures: The study was sponsored by AbbVie. The presenter reported having no financial conflicts.
DeSalvo: HM needs holistic approach to health care
LAS VEGAS – To deliver her message of inclusion Tuesday morning, former acting assistant secretary for health in the U.S. Department of Health and Human Services (HHS) Karen DeSalvo, MD, MPH, MSc, could think of “no finer group” than those assembled before her at HM17.
The thousands of hospitalists gathered to hear her keynote address, “Rethinking Health: The Vital Role of Hospitals and the Hospitalist,” listened as she talked about including more than just the best medical care in HM’s scope of practice. The job must evolve to include a focus on such social issues as economic stability, neighborhood and physical environment, education, and access to healthy options for food.
In other words, Dr. DeSalvo wondered aloud, what good is treating a grandmother’s heart failure over and over if she’s always going to return to the hospital because her home, her neighborhood, or her finances mean she is unable to prevent recurring health issues? [[{"fid":"195561","view_mode":"medstat_image_flush_left","attributes":{"alt":"Dr. Brian Harte conducts an interveiw with Dr. Karen DeSalvo duing the opening plenary Tuesday at HM17.","height":"147","width":"220","class":"media-element file-medstat-image-flush-left","data-delta":"1"},"fields":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Dr. Brian Harte conducts an interview with Dr. Karen DeSalvo during the opening plenary Tuesday at HM17.","field_file_image_credit[und][0][value]":"Darnell Scott","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"1":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Dr. Brian Harte conducts an interview with Dr. Karen DeSalvo during the opening plenary Tuesday at HM17.","field_file_image_credit[und][0][value]":"Darnell Scott"}}}]]
Hospitalists “have been at the center of change, not only in building a new field and showing us that medicine doesn’t have to always be the way it always was,” she said. “You have been at the forefront of seeing that we’re getting better value out of our health care system and, though that work must continue, you must also begin to broaden our thinking and understand that the drivers of health are much more than [just] health care. There are social determinants, social factors.”
Dr. DeSalvo, an internist by training, understands that dealing with social issues may seem like a role for others, but she said that the implications of those factors directly impact hospitalists and their institutions via issues such as readmissions.
“These things … don’t just matter conceptually,” she said. “They [have] direct relationships with mortality and morbidity and cost. They are literally affecting people’s lives in this country every day. When we begin to adjust them, to impact them, you can see that it also affects the health care system.”
On the front lines, Dr. DeSalvo said that hospitalists and others can work to take advantage of their hospital’s existing tools to link their patients to available resources, partner with local public health offices, and push to make their hospitals “anchor institutions to build community capacity to address these social determinants.”
Dr. DeSalvo also praised HM as a field that has already embraced value-based payment (VBP) models. She said that ability to anticipate and adapt to health care’s changing needs positions the field well as the Medicare Access and CHIP Reauthorization Act (MACRA) moves health care from fee-for-service to payment models that seek to manage risk and penalize mistakes.
LAS VEGAS – To deliver her message of inclusion Tuesday morning, former acting assistant secretary for health in the U.S. Department of Health and Human Services (HHS) Karen DeSalvo, MD, MPH, MSc, could think of “no finer group” than those assembled before her at HM17.
The thousands of hospitalists gathered to hear her keynote address, “Rethinking Health: The Vital Role of Hospitals and the Hospitalist,” listened as she talked about including more than just the best medical care in HM’s scope of practice. The job must evolve to include a focus on such social issues as economic stability, neighborhood and physical environment, education, and access to healthy options for food.
In other words, Dr. DeSalvo wondered aloud, what good is treating a grandmother’s heart failure over and over if she’s always going to return to the hospital because her home, her neighborhood, or her finances mean she is unable to prevent recurring health issues? [[{"fid":"195561","view_mode":"medstat_image_flush_left","attributes":{"alt":"Dr. Brian Harte conducts an interveiw with Dr. Karen DeSalvo duing the opening plenary Tuesday at HM17.","height":"147","width":"220","class":"media-element file-medstat-image-flush-left","data-delta":"1"},"fields":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Dr. Brian Harte conducts an interview with Dr. Karen DeSalvo during the opening plenary Tuesday at HM17.","field_file_image_credit[und][0][value]":"Darnell Scott","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"1":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Dr. Brian Harte conducts an interview with Dr. Karen DeSalvo during the opening plenary Tuesday at HM17.","field_file_image_credit[und][0][value]":"Darnell Scott"}}}]]
Hospitalists “have been at the center of change, not only in building a new field and showing us that medicine doesn’t have to always be the way it always was,” she said. “You have been at the forefront of seeing that we’re getting better value out of our health care system and, though that work must continue, you must also begin to broaden our thinking and understand that the drivers of health are much more than [just] health care. There are social determinants, social factors.”
Dr. DeSalvo, an internist by training, understands that dealing with social issues may seem like a role for others, but she said that the implications of those factors directly impact hospitalists and their institutions via issues such as readmissions.
“These things … don’t just matter conceptually,” she said. “They [have] direct relationships with mortality and morbidity and cost. They are literally affecting people’s lives in this country every day. When we begin to adjust them, to impact them, you can see that it also affects the health care system.”
On the front lines, Dr. DeSalvo said that hospitalists and others can work to take advantage of their hospital’s existing tools to link their patients to available resources, partner with local public health offices, and push to make their hospitals “anchor institutions to build community capacity to address these social determinants.”
Dr. DeSalvo also praised HM as a field that has already embraced value-based payment (VBP) models. She said that ability to anticipate and adapt to health care’s changing needs positions the field well as the Medicare Access and CHIP Reauthorization Act (MACRA) moves health care from fee-for-service to payment models that seek to manage risk and penalize mistakes.
LAS VEGAS – To deliver her message of inclusion Tuesday morning, former acting assistant secretary for health in the U.S. Department of Health and Human Services (HHS) Karen DeSalvo, MD, MPH, MSc, could think of “no finer group” than those assembled before her at HM17.
The thousands of hospitalists gathered to hear her keynote address, “Rethinking Health: The Vital Role of Hospitals and the Hospitalist,” listened as she talked about including more than just the best medical care in HM’s scope of practice. The job must evolve to include a focus on such social issues as economic stability, neighborhood and physical environment, education, and access to healthy options for food.
In other words, Dr. DeSalvo wondered aloud, what good is treating a grandmother’s heart failure over and over if she’s always going to return to the hospital because her home, her neighborhood, or her finances mean she is unable to prevent recurring health issues? [[{"fid":"195561","view_mode":"medstat_image_flush_left","attributes":{"alt":"Dr. Brian Harte conducts an interveiw with Dr. Karen DeSalvo duing the opening plenary Tuesday at HM17.","height":"147","width":"220","class":"media-element file-medstat-image-flush-left","data-delta":"1"},"fields":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Dr. Brian Harte conducts an interview with Dr. Karen DeSalvo during the opening plenary Tuesday at HM17.","field_file_image_credit[und][0][value]":"Darnell Scott","field_file_image_caption[und][0][format]":"plain_text","field_file_image_credit[und][0][format]":"plain_text"},"type":"media","field_deltas":{"1":{"format":"medstat_image_flush_left","field_file_image_caption[und][0][value]":"Dr. Brian Harte conducts an interview with Dr. Karen DeSalvo during the opening plenary Tuesday at HM17.","field_file_image_credit[und][0][value]":"Darnell Scott"}}}]]
Hospitalists “have been at the center of change, not only in building a new field and showing us that medicine doesn’t have to always be the way it always was,” she said. “You have been at the forefront of seeing that we’re getting better value out of our health care system and, though that work must continue, you must also begin to broaden our thinking and understand that the drivers of health are much more than [just] health care. There are social determinants, social factors.”
Dr. DeSalvo, an internist by training, understands that dealing with social issues may seem like a role for others, but she said that the implications of those factors directly impact hospitalists and their institutions via issues such as readmissions.
“These things … don’t just matter conceptually,” she said. “They [have] direct relationships with mortality and morbidity and cost. They are literally affecting people’s lives in this country every day. When we begin to adjust them, to impact them, you can see that it also affects the health care system.”
On the front lines, Dr. DeSalvo said that hospitalists and others can work to take advantage of their hospital’s existing tools to link their patients to available resources, partner with local public health offices, and push to make their hospitals “anchor institutions to build community capacity to address these social determinants.”
Dr. DeSalvo also praised HM as a field that has already embraced value-based payment (VBP) models. She said that ability to anticipate and adapt to health care’s changing needs positions the field well as the Medicare Access and CHIP Reauthorization Act (MACRA) moves health care from fee-for-service to payment models that seek to manage risk and penalize mistakes.

Rapid-fire session troubleshoots mechanical ventilation
Troubleshooting problems with mechanical ventilation starts with assessing how much control one has over specific variables, according to an expert at HM17.
“You want to be in charge of everything when you’re dealing with a ventilator, but you have to acknowledge that you only get to be in charge of some stuff,” said Peter Clardy, MD, an assistant professor of medicine at Harvard University in Cambridge, Mass., and its affiliate, Mount Auburn Hospital. He made his remarks during a rapid-fire science session at HM17.
Since successful algorithms for acute mechanical ventilation require control over many independent variables, knowing what is most stable and going from there can allow the physician to develop a workable plan of action, according to Dr. Clardy.
“It’s really good to be explicit about what is dependent and what is independent,” he said. Independent variables might be those specific to the ventilator, but will always include the positive end-expiratory pressure and the fraction of inspired oxygen. Other independent variables will depend on the mode of ventilation – either fully assisted, partially assisted, or noninvasive.
“If you’re in charge of volume, you have to worry about pressure,” he noted. “If you’re in charge of pressure you have to worry about volume.”
Dependent variables also can vary by mode of ventilation. Once the independent and dependent variables are mapped, it is easier to glean more information about the respiratory mechanics of the situation and the physiologic processes, such as the metabolic cost of breathing and whether it can be reduced, what can be done to prevent ventilator-induced lung injury, and how gas exchange can be supported.
Understanding the independent/dependent variable ratio can also help provide valuable clinical information, such as whether reversing hypoxemia and/or hypercarbia is necessary, or if there are signs of respiratory distress or dyspnea. Other clinical indications might include whether there is a need to prevent or reverse atelectasis, or reduce ventilatory muscle fatigue. Additionally, it will be easier to know whether sedation is possible, or if a neuromuscular blockade should be used. Such information can help determine whether to protect the airway.
“Respiratory distress in a patient who is already ventilated is quite common, so having a routinized way to assess these patients and their stability can help you think about what your moves are right there while you’re in the room,” Dr. Clardy explained. “All of that can be incredibly helpful.”
Dr. Clardy had no relevant financial disclosures.
Troubleshooting problems with mechanical ventilation starts with assessing how much control one has over specific variables, according to an expert at HM17.
“You want to be in charge of everything when you’re dealing with a ventilator, but you have to acknowledge that you only get to be in charge of some stuff,” said Peter Clardy, MD, an assistant professor of medicine at Harvard University in Cambridge, Mass., and its affiliate, Mount Auburn Hospital. He made his remarks during a rapid-fire science session at HM17.
Since successful algorithms for acute mechanical ventilation require control over many independent variables, knowing what is most stable and going from there can allow the physician to develop a workable plan of action, according to Dr. Clardy.
“It’s really good to be explicit about what is dependent and what is independent,” he said. Independent variables might be those specific to the ventilator, but will always include the positive end-expiratory pressure and the fraction of inspired oxygen. Other independent variables will depend on the mode of ventilation – either fully assisted, partially assisted, or noninvasive.
“If you’re in charge of volume, you have to worry about pressure,” he noted. “If you’re in charge of pressure you have to worry about volume.”
Dependent variables also can vary by mode of ventilation. Once the independent and dependent variables are mapped, it is easier to glean more information about the respiratory mechanics of the situation and the physiologic processes, such as the metabolic cost of breathing and whether it can be reduced, what can be done to prevent ventilator-induced lung injury, and how gas exchange can be supported.
Understanding the independent/dependent variable ratio can also help provide valuable clinical information, such as whether reversing hypoxemia and/or hypercarbia is necessary, or if there are signs of respiratory distress or dyspnea. Other clinical indications might include whether there is a need to prevent or reverse atelectasis, or reduce ventilatory muscle fatigue. Additionally, it will be easier to know whether sedation is possible, or if a neuromuscular blockade should be used. Such information can help determine whether to protect the airway.
“Respiratory distress in a patient who is already ventilated is quite common, so having a routinized way to assess these patients and their stability can help you think about what your moves are right there while you’re in the room,” Dr. Clardy explained. “All of that can be incredibly helpful.”
Dr. Clardy had no relevant financial disclosures.
Troubleshooting problems with mechanical ventilation starts with assessing how much control one has over specific variables, according to an expert at HM17.
“You want to be in charge of everything when you’re dealing with a ventilator, but you have to acknowledge that you only get to be in charge of some stuff,” said Peter Clardy, MD, an assistant professor of medicine at Harvard University in Cambridge, Mass., and its affiliate, Mount Auburn Hospital. He made his remarks during a rapid-fire science session at HM17.
Since successful algorithms for acute mechanical ventilation require control over many independent variables, knowing what is most stable and going from there can allow the physician to develop a workable plan of action, according to Dr. Clardy.
“It’s really good to be explicit about what is dependent and what is independent,” he said. Independent variables might be those specific to the ventilator, but will always include the positive end-expiratory pressure and the fraction of inspired oxygen. Other independent variables will depend on the mode of ventilation – either fully assisted, partially assisted, or noninvasive.
“If you’re in charge of volume, you have to worry about pressure,” he noted. “If you’re in charge of pressure you have to worry about volume.”
Dependent variables also can vary by mode of ventilation. Once the independent and dependent variables are mapped, it is easier to glean more information about the respiratory mechanics of the situation and the physiologic processes, such as the metabolic cost of breathing and whether it can be reduced, what can be done to prevent ventilator-induced lung injury, and how gas exchange can be supported.
Understanding the independent/dependent variable ratio can also help provide valuable clinical information, such as whether reversing hypoxemia and/or hypercarbia is necessary, or if there are signs of respiratory distress or dyspnea. Other clinical indications might include whether there is a need to prevent or reverse atelectasis, or reduce ventilatory muscle fatigue. Additionally, it will be easier to know whether sedation is possible, or if a neuromuscular blockade should be used. Such information can help determine whether to protect the airway.
“Respiratory distress in a patient who is already ventilated is quite common, so having a routinized way to assess these patients and their stability can help you think about what your moves are right there while you’re in the room,” Dr. Clardy explained. “All of that can be incredibly helpful.”
Dr. Clardy had no relevant financial disclosures.
Clinical staging of depression endorsed
SCOTTSDALE, ARIZ. – Failing to see depression as a chronic condition that needs to be managed has hampered the ability of psychiatry to help patients with the mood disorder, according to the principal investigator of a recently published prospective study on the most refractory of depression cases.
“The majority of research [in our field] has been on how we manage acute episodes of depression. But, these are chronic, often lifelong conditions. We need to pay attention to that and come up with solutions,” said Scott T. Aaronson, MD, program chair of this year’s annual meeting of the American College of Psychiatrists, in an interview.

“There are people who have these terrible depressions, that, even if you could get them 25% better over the course of years instead of 75% better for just a few months, they’d probably have a much better prognosis and a better quality of life. We need to pay attention to that and think of a longer horizon than we currently do,” Dr. Aaronson said.
During a scientific session at the meeting, he presented data from a 5-year, observational registry study, conducted in nearly 800 people with severe treatment-resistant depression – a population for whom no current evidence-based treatments exists – showing that adjunctive vagus nerve stimulation (VNS) had superior outcomes and mortality, compared with treatment as usual (Am J Psychiatry. 2017 Mar 31. doi: 10.1176/appi.ajp.2017.16010034).
Between January 2006 and May 2015, the multicenter study enrolled adults with unremitting unipolar or bipolar depression lasting at least 2 years. It also enrolled adults who had experienced three or more depressive episodes and had failed four or more depression treatments, including electroconvulsive therapy (ECT). People with a history of psychosis or rapid-cycling bipolar disorder were excluded. One cohort came from a patient registry designed as a postmarketing surveillance study stipulated by the Food and Drug Administration for the approval of the refractory depression indication for VNS. Another cohort came from a study that compared patients with refractory depression who received VNS therapy at various doses. People in the registry cohort were seen at 61 U.S. sites in different settings. Patients were assigned to treatment as usual or treatment as usual with adjunctive VNS based on their preference of a treatment arm. Often, patients’ treatment arm depended on whether implantation was available at their site or what their insurance would cover.
In all, 494 patients were in the VNS study arm, and 301 were in the treatment-as-usual arm. The FDA approved use of the pooled data. People in the dose-finding cohort all had VNS implants when they entered the study, and, unless lost to follow-up, all were observed for 60 months, regardless of the point at which they entered the study. About two-thirds of the dose-finding patients remained in the study for all 5 years, as did about half of the registry study cohort. Of those involved, 22 patients exercised their option of switching treatment arms, but their data were censored from the efficacy analysis. At baseline, the mean Montgomery-Asberg Depression Rating Scale (MADRS) score was 29.3 for the treatment-as-usual group and 33.1 for those in the VNS adjunct group. Responders were those who had a 50% or greater reduction in MADRS scores at any point post baseline.
The 5-year cumulative response to treatment rate in the adjunctive VNS group was significantly higher at 67.6%, compared with 40% in the treatment-as-usual arm (P less than .001). The cumulative percentage of first-time responders in the VNS adjunctive arm was nearly double that of the treatment-as-usual group at all follow-up points in the study, and they tended to respond by 1 year, compared with 2 years in the treatment-as-usual group (P less than .001).
A secondary efficacy endpoint was changes in the Clinical Global Impression–Improvement (CGI-I) scores. These also favored the VNS adjunctive group, which had a 75.9% cumulative CGI-I response rate, compared with a 48.6% rate in the treatment-as-usual arm (P less than .001). Scores on the Quick Inventory of Depressive Symptomatology–Self-Report (QIDS-SR) were consistent with these results: The cumulative response rate in the VNS group was 64.7%, compared with 41.7% in the treatment-as-usual arm (P less than .001).
There were 15 deaths during the study, 7 in the VNS arm and 8 in the treatment-as-usual arm. There were two suicides in each arm, meaning the VNS arm, which was larger, experienced a lesser rate of suicides as the treatment-as-usual group. “This is a fairly key point,” Dr. Aaronson said in the interview.
The remission rate also was significantly higher in the adjunctive group at 43.3%, compared with 25.7% in the treatment-as-usual group (P less than .001). Differences in CGI-I and QIDS-SR scores also were statistically significant and were both higher in the VNS adjunct group, with cumulative response rates from baseline CGI-I scores of 49.7% vs. 21.4%, and changes in QIDS-SR scores of 40.4% vs. 25.0% (P less than .001).
Yet, these statistics do not describe the full potential affect of adjunctive VNS on refractory depression, according to Dr. Aaronson. “There were people in this study who did not meet the endpoint but who were no longer suicidal. Some [reported that they] could now just enjoy riding their bicycle,” he said. “If you asked them, ‘Did they think participating was worthwhile?’ they would tell you there was no question about it. Small differences can be incredibly meaningful for these folks. We need to rethink what success means when we treat chronic depression.”
In addition to higher mean depression rating scores at baseline, those in the VNS group also had higher rates of psychiatric hospitalizations and suicide attempts, suggesting more severe illness in this group. However, it is the fact that this group, with its higher response rate overall, also had higher baseline rates of exposure to ECT that excited Dr. Aaronson the most about the study.
A subanalysis showed that 58.7% of the adjunct VNS group and 36.2% of the treatment-as-usual arm had all had at least seven right lateral treatments of ECT, typically an exclusionary criterion in depression treatment trials. For patients in the VNS arm who previously had responded to ECT, the cumulative response rate at 5 years, based on MADRS scores, was 71.3%, compared with 56.9% of those who had responded to ECT in the treatment-as-usual group, a statistically significant difference (P less than .006). Further, a significant difference in response was recorded at 9 months and then sustained throughout the study.
For the ECT nonresponders in the VNS arm, the response rate in this study was 59.6%, compared with 34.1% for the ECT nonresponders who were receiving treatment as usual (P less than .001). Statistical separation of the two arms began after about 2 years and continued throughout the study.
“This is my personal, favorite part,” Dr. Aaronson said in the interview. “We don’t consider ECT very much, but it is, without question, one of the single most effective acute treatments we have in all of psychiatry for depression. The problem is that, for the majority who respond to it, they are sick again within 6 months. The point here is that, if you’ve ever responded to anything, including ECT, we now have a marker for who will respond to VNS.”
In part because VNS is a chronic, and comparatively less expensive, treatment, the study also has implications for patients on maintenance ECT, Dr. Aaronson said. “Wouldn’t it be terrific if I could offer them VNS rather than continuous ECT, which I worry in the long run can be hard on brains and which is expensive and inconvenient?”
More than one-third of people diagnosed with depression have the treatment-resistant type, the standard definition of which is that a person previously has failed two or more treatments. In this patient population, between 10% and 15% will go on to fail at least four treatments, Dr. Aaronson said.
Although previous failure at least 4 previous depression treatment regimens was one of the inclusion criteria in this study, the VNS population had failed an average of 8.2 previous treatments, compared with 7.3 in the treatment-as-usual arm. These data, together with the subanalysis data on ECT responders, make a compelling case for staging depression, Dr. Aaronson said.
He said, he believes that, with more study and differently structured trials, it can be demonstrated that there also should be a clinical diagnosis of “severe” treatment-resistant depression.
Dr. Aaronson and his colleagues are currently seeking funding to conduct a national study that is randomly controlled using VNS or a sham treatment. Once efficacy data are sufficient, making the case for staging depression will be easier, Dr. Aaronson said.
“I am a firm believer that we should look at psychiatric illnesses the same way we do cancers – using levels of severity,” he said. “The neat thing about [these data on] VNS is that it gives me the bully pulpit to start preaching that gospel.”
Dr. Aaronson’s relevant disclosures include Genomind, LivaNova, Neuronetics, Otsuka, Sunovion, and Takeda.
[email protected]
On Twitter @whitneymcknight
SCOTTSDALE, ARIZ. – Failing to see depression as a chronic condition that needs to be managed has hampered the ability of psychiatry to help patients with the mood disorder, according to the principal investigator of a recently published prospective study on the most refractory of depression cases.
“The majority of research [in our field] has been on how we manage acute episodes of depression. But, these are chronic, often lifelong conditions. We need to pay attention to that and come up with solutions,” said Scott T. Aaronson, MD, program chair of this year’s annual meeting of the American College of Psychiatrists, in an interview.
“There are people who have these terrible depressions, that, even if you could get them 25% better over the course of years instead of 75% better for just a few months, they’d probably have a much better prognosis and a better quality of life. We need to pay attention to that and think of a longer horizon than we currently do,” Dr. Aaronson said.
During a scientific session at the meeting, he presented data from a 5-year, observational registry study, conducted in nearly 800 people with severe treatment-resistant depression – a population for whom no current evidence-based treatments exists – showing that adjunctive vagus nerve stimulation (VNS) had superior outcomes and mortality, compared with treatment as usual (Am J Psychiatry. 2017 Mar 31. doi: 10.1176/appi.ajp.2017.16010034).
Between January 2006 and May 2015, the multicenter study enrolled adults with unremitting unipolar or bipolar depression lasting at least 2 years. It also enrolled adults who had experienced three or more depressive episodes and had failed four or more depression treatments, including electroconvulsive therapy (ECT). People with a history of psychosis or rapid-cycling bipolar disorder were excluded. One cohort came from a patient registry designed as a postmarketing surveillance study stipulated by the Food and Drug Administration for the approval of the refractory depression indication for VNS. Another cohort came from a study that compared patients with refractory depression who received VNS therapy at various doses. People in the registry cohort were seen at 61 U.S. sites in different settings. Patients were assigned to treatment as usual or treatment as usual with adjunctive VNS based on their preference of a treatment arm. Often, patients’ treatment arm depended on whether implantation was available at their site or what their insurance would cover.
In all, 494 patients were in the VNS study arm, and 301 were in the treatment-as-usual arm. The FDA approved use of the pooled data. People in the dose-finding cohort all had VNS implants when they entered the study, and, unless lost to follow-up, all were observed for 60 months, regardless of the point at which they entered the study. About two-thirds of the dose-finding patients remained in the study for all 5 years, as did about half of the registry study cohort. Of those involved, 22 patients exercised their option of switching treatment arms, but their data were censored from the efficacy analysis. At baseline, the mean Montgomery-Asberg Depression Rating Scale (MADRS) score was 29.3 for the treatment-as-usual group and 33.1 for those in the VNS adjunct group. Responders were those who had a 50% or greater reduction in MADRS scores at any point post baseline.
The 5-year cumulative response to treatment rate in the adjunctive VNS group was significantly higher at 67.6%, compared with 40% in the treatment-as-usual arm (P less than .001). The cumulative percentage of first-time responders in the VNS adjunctive arm was nearly double that of the treatment-as-usual group at all follow-up points in the study, and they tended to respond by 1 year, compared with 2 years in the treatment-as-usual group (P less than .001).
A secondary efficacy endpoint was changes in the Clinical Global Impression–Improvement (CGI-I) scores. These also favored the VNS adjunctive group, which had a 75.9% cumulative CGI-I response rate, compared with a 48.6% rate in the treatment-as-usual arm (P less than .001). Scores on the Quick Inventory of Depressive Symptomatology–Self-Report (QIDS-SR) were consistent with these results: The cumulative response rate in the VNS group was 64.7%, compared with 41.7% in the treatment-as-usual arm (P less than .001).
There were 15 deaths during the study, 7 in the VNS arm and 8 in the treatment-as-usual arm. There were two suicides in each arm, meaning the VNS arm, which was larger, experienced a lesser rate of suicides as the treatment-as-usual group. “This is a fairly key point,” Dr. Aaronson said in the interview.
The remission rate also was significantly higher in the adjunctive group at 43.3%, compared with 25.7% in the treatment-as-usual group (P less than .001). Differences in CGI-I and QIDS-SR scores also were statistically significant and were both higher in the VNS adjunct group, with cumulative response rates from baseline CGI-I scores of 49.7% vs. 21.4%, and changes in QIDS-SR scores of 40.4% vs. 25.0% (P less than .001).
Yet, these statistics do not describe the full potential affect of adjunctive VNS on refractory depression, according to Dr. Aaronson. “There were people in this study who did not meet the endpoint but who were no longer suicidal. Some [reported that they] could now just enjoy riding their bicycle,” he said. “If you asked them, ‘Did they think participating was worthwhile?’ they would tell you there was no question about it. Small differences can be incredibly meaningful for these folks. We need to rethink what success means when we treat chronic depression.”
In addition to higher mean depression rating scores at baseline, those in the VNS group also had higher rates of psychiatric hospitalizations and suicide attempts, suggesting more severe illness in this group. However, it is the fact that this group, with its higher response rate overall, also had higher baseline rates of exposure to ECT that excited Dr. Aaronson the most about the study.
A subanalysis showed that 58.7% of the adjunct VNS group and 36.2% of the treatment-as-usual arm had all had at least seven right lateral treatments of ECT, typically an exclusionary criterion in depression treatment trials. For patients in the VNS arm who previously had responded to ECT, the cumulative response rate at 5 years, based on MADRS scores, was 71.3%, compared with 56.9% of those who had responded to ECT in the treatment-as-usual group, a statistically significant difference (P less than .006). Further, a significant difference in response was recorded at 9 months and then sustained throughout the study.
For the ECT nonresponders in the VNS arm, the response rate in this study was 59.6%, compared with 34.1% for the ECT nonresponders who were receiving treatment as usual (P less than .001). Statistical separation of the two arms began after about 2 years and continued throughout the study.
“This is my personal, favorite part,” Dr. Aaronson said in the interview. “We don’t consider ECT very much, but it is, without question, one of the single most effective acute treatments we have in all of psychiatry for depression. The problem is that, for the majority who respond to it, they are sick again within 6 months. The point here is that, if you’ve ever responded to anything, including ECT, we now have a marker for who will respond to VNS.”
In part because VNS is a chronic, and comparatively less expensive, treatment, the study also has implications for patients on maintenance ECT, Dr. Aaronson said. “Wouldn’t it be terrific if I could offer them VNS rather than continuous ECT, which I worry in the long run can be hard on brains and which is expensive and inconvenient?”
More than one-third of people diagnosed with depression have the treatment-resistant type, the standard definition of which is that a person previously has failed two or more treatments. In this patient population, between 10% and 15% will go on to fail at least four treatments, Dr. Aaronson said.
Although previous failure at least 4 previous depression treatment regimens was one of the inclusion criteria in this study, the VNS population had failed an average of 8.2 previous treatments, compared with 7.3 in the treatment-as-usual arm. These data, together with the subanalysis data on ECT responders, make a compelling case for staging depression, Dr. Aaronson said.
He said, he believes that, with more study and differently structured trials, it can be demonstrated that there also should be a clinical diagnosis of “severe” treatment-resistant depression.
Dr. Aaronson and his colleagues are currently seeking funding to conduct a national study that is randomly controlled using VNS or a sham treatment. Once efficacy data are sufficient, making the case for staging depression will be easier, Dr. Aaronson said.
“I am a firm believer that we should look at psychiatric illnesses the same way we do cancers – using levels of severity,” he said. “The neat thing about [these data on] VNS is that it gives me the bully pulpit to start preaching that gospel.”
Dr. Aaronson’s relevant disclosures include Genomind, LivaNova, Neuronetics, Otsuka, Sunovion, and Takeda.
[email protected]
On Twitter @whitneymcknight
SCOTTSDALE, ARIZ. – Failing to see depression as a chronic condition that needs to be managed has hampered the ability of psychiatry to help patients with the mood disorder, according to the principal investigator of a recently published prospective study on the most refractory of depression cases.
“The majority of research [in our field] has been on how we manage acute episodes of depression. But, these are chronic, often lifelong conditions. We need to pay attention to that and come up with solutions,” said Scott T. Aaronson, MD, program chair of this year’s annual meeting of the American College of Psychiatrists, in an interview.
“There are people who have these terrible depressions, that, even if you could get them 25% better over the course of years instead of 75% better for just a few months, they’d probably have a much better prognosis and a better quality of life. We need to pay attention to that and think of a longer horizon than we currently do,” Dr. Aaronson said.
During a scientific session at the meeting, he presented data from a 5-year, observational registry study, conducted in nearly 800 people with severe treatment-resistant depression – a population for whom no current evidence-based treatments exists – showing that adjunctive vagus nerve stimulation (VNS) had superior outcomes and mortality, compared with treatment as usual (Am J Psychiatry. 2017 Mar 31. doi: 10.1176/appi.ajp.2017.16010034).
Between January 2006 and May 2015, the multicenter study enrolled adults with unremitting unipolar or bipolar depression lasting at least 2 years. It also enrolled adults who had experienced three or more depressive episodes and had failed four or more depression treatments, including electroconvulsive therapy (ECT). People with a history of psychosis or rapid-cycling bipolar disorder were excluded. One cohort came from a patient registry designed as a postmarketing surveillance study stipulated by the Food and Drug Administration for the approval of the refractory depression indication for VNS. Another cohort came from a study that compared patients with refractory depression who received VNS therapy at various doses. People in the registry cohort were seen at 61 U.S. sites in different settings. Patients were assigned to treatment as usual or treatment as usual with adjunctive VNS based on their preference of a treatment arm. Often, patients’ treatment arm depended on whether implantation was available at their site or what their insurance would cover.
In all, 494 patients were in the VNS study arm, and 301 were in the treatment-as-usual arm. The FDA approved use of the pooled data. People in the dose-finding cohort all had VNS implants when they entered the study, and, unless lost to follow-up, all were observed for 60 months, regardless of the point at which they entered the study. About two-thirds of the dose-finding patients remained in the study for all 5 years, as did about half of the registry study cohort. Of those involved, 22 patients exercised their option of switching treatment arms, but their data were censored from the efficacy analysis. At baseline, the mean Montgomery-Asberg Depression Rating Scale (MADRS) score was 29.3 for the treatment-as-usual group and 33.1 for those in the VNS adjunct group. Responders were those who had a 50% or greater reduction in MADRS scores at any point post baseline.
The 5-year cumulative response to treatment rate in the adjunctive VNS group was significantly higher at 67.6%, compared with 40% in the treatment-as-usual arm (P less than .001). The cumulative percentage of first-time responders in the VNS adjunctive arm was nearly double that of the treatment-as-usual group at all follow-up points in the study, and they tended to respond by 1 year, compared with 2 years in the treatment-as-usual group (P less than .001).
A secondary efficacy endpoint was changes in the Clinical Global Impression–Improvement (CGI-I) scores. These also favored the VNS adjunctive group, which had a 75.9% cumulative CGI-I response rate, compared with a 48.6% rate in the treatment-as-usual arm (P less than .001). Scores on the Quick Inventory of Depressive Symptomatology–Self-Report (QIDS-SR) were consistent with these results: The cumulative response rate in the VNS group was 64.7%, compared with 41.7% in the treatment-as-usual arm (P less than .001).
There were 15 deaths during the study, 7 in the VNS arm and 8 in the treatment-as-usual arm. There were two suicides in each arm, meaning the VNS arm, which was larger, experienced a lesser rate of suicides as the treatment-as-usual group. “This is a fairly key point,” Dr. Aaronson said in the interview.
The remission rate also was significantly higher in the adjunctive group at 43.3%, compared with 25.7% in the treatment-as-usual group (P less than .001). Differences in CGI-I and QIDS-SR scores also were statistically significant and were both higher in the VNS adjunct group, with cumulative response rates from baseline CGI-I scores of 49.7% vs. 21.4%, and changes in QIDS-SR scores of 40.4% vs. 25.0% (P less than .001).
Yet, these statistics do not describe the full potential affect of adjunctive VNS on refractory depression, according to Dr. Aaronson. “There were people in this study who did not meet the endpoint but who were no longer suicidal. Some [reported that they] could now just enjoy riding their bicycle,” he said. “If you asked them, ‘Did they think participating was worthwhile?’ they would tell you there was no question about it. Small differences can be incredibly meaningful for these folks. We need to rethink what success means when we treat chronic depression.”
In addition to higher mean depression rating scores at baseline, those in the VNS group also had higher rates of psychiatric hospitalizations and suicide attempts, suggesting more severe illness in this group. However, it is the fact that this group, with its higher response rate overall, also had higher baseline rates of exposure to ECT that excited Dr. Aaronson the most about the study.
A subanalysis showed that 58.7% of the adjunct VNS group and 36.2% of the treatment-as-usual arm had all had at least seven right lateral treatments of ECT, typically an exclusionary criterion in depression treatment trials. For patients in the VNS arm who previously had responded to ECT, the cumulative response rate at 5 years, based on MADRS scores, was 71.3%, compared with 56.9% of those who had responded to ECT in the treatment-as-usual group, a statistically significant difference (P less than .006). Further, a significant difference in response was recorded at 9 months and then sustained throughout the study.
For the ECT nonresponders in the VNS arm, the response rate in this study was 59.6%, compared with 34.1% for the ECT nonresponders who were receiving treatment as usual (P less than .001). Statistical separation of the two arms began after about 2 years and continued throughout the study.
“This is my personal, favorite part,” Dr. Aaronson said in the interview. “We don’t consider ECT very much, but it is, without question, one of the single most effective acute treatments we have in all of psychiatry for depression. The problem is that, for the majority who respond to it, they are sick again within 6 months. The point here is that, if you’ve ever responded to anything, including ECT, we now have a marker for who will respond to VNS.”
In part because VNS is a chronic, and comparatively less expensive, treatment, the study also has implications for patients on maintenance ECT, Dr. Aaronson said. “Wouldn’t it be terrific if I could offer them VNS rather than continuous ECT, which I worry in the long run can be hard on brains and which is expensive and inconvenient?”
More than one-third of people diagnosed with depression have the treatment-resistant type, the standard definition of which is that a person previously has failed two or more treatments. In this patient population, between 10% and 15% will go on to fail at least four treatments, Dr. Aaronson said.
Although previous failure at least 4 previous depression treatment regimens was one of the inclusion criteria in this study, the VNS population had failed an average of 8.2 previous treatments, compared with 7.3 in the treatment-as-usual arm. These data, together with the subanalysis data on ECT responders, make a compelling case for staging depression, Dr. Aaronson said.
He said, he believes that, with more study and differently structured trials, it can be demonstrated that there also should be a clinical diagnosis of “severe” treatment-resistant depression.
Dr. Aaronson and his colleagues are currently seeking funding to conduct a national study that is randomly controlled using VNS or a sham treatment. Once efficacy data are sufficient, making the case for staging depression will be easier, Dr. Aaronson said.
“I am a firm believer that we should look at psychiatric illnesses the same way we do cancers – using levels of severity,” he said. “The neat thing about [these data on] VNS is that it gives me the bully pulpit to start preaching that gospel.”
Dr. Aaronson’s relevant disclosures include Genomind, LivaNova, Neuronetics, Otsuka, Sunovion, and Takeda.
[email protected]
On Twitter @whitneymcknight
Key clinical point:
Major finding: Adjunctive vagus nerve stimulation had superior outcomes and mortality rates, compared with treatment as usual in people with severe treatment-resistant depression.
Data source: A 5-year, prospective, open-label, nonrandomized, observational, multisite study of 795 adults who had failed four or more depression treatments.
Disclosures: Dr. Aaronson’s relevant disclosures include Genomind, LivaNova, Neuronetics, Otsuka, Sunovion, and Takeda.
Ibrutinib monotherapy data in previously treated MZL is available
Ibrutinib is active in patients with previously treated marginal zone lymphoma (MZL) and has a good safety profile, the results of a multicenter, open-label phase II trial published in Blood suggest.
Nearly half of 63 patients responded to monotherapy with the once-daily oral inhibitor of Bruton tyrosine kinase, and ibrutinib was generally well tolerated, according to the study (Blood. 2017;129:2224-32). The results prompted the U.S. Food and Drug Administration to grant accelerated approval of ibrutinib for patients with MZL who were previously treated with at least one prior anti-CD20–based therapy.
“MZL is frequently linked to chronic infection, which may induce B-cell receptor (BCR) signaling, resulting in aberrant B-cell survival and proliferation,” note the investigators, who were led by Ariela Noy, MD, a hematologic oncologist with the Lymphoma Service at the Memorial Sloan-Kettering Cancer Center, New York. “Single-agent ibrutinib induced durable responses with a favorable benefit–risk profile in patients with previously treated MZL, confirming the role of BCR signaling in this malignancy.”
“As the only approved therapy, ibrutinib provides a treatment option without chemotherapy for MZL,” they maintain. “Future studies will investigate ibrutinib in treatment-naive patients or as combination strategies in relapsed/refractory MZL.”
In the trial, which was funded by Pharmacyclics, an AbbVie Company, patients with MZL of all subtypes who had received at least one prior therapy with an anti-CD20 antibody–containing regimen were treated with 560 mg ibrutinib (Imbruvica) orally once daily. The median number of prior systemic therapies was two, and 63% had received prior chemoimmunotherapy.
The overall response rate, as assessed by an independent review committee using 2007 International Working Group criteria – the trial’s primary endpoint – was 48%, according to the published results. Benefit was similar across patients who differed regarding MZL subtype, number of prior regimen, and previous receipt of chemoimmunotherapy
After a 19.4-month median follow-up, the median duration of response was not reached, and median progression-free survival was 14.2 months.
The most common grade 3 or worse adverse events were anemia (14%), pneumonia (8%), and fatigue (6%). Serious adverse events of any grade affected 44% of patients. The most common was grade 3 or 4 pneumonia. Adverse events led to treatment discontinuation in 17% of patients and dose reductions in 10%.
“Due to evidence of pseudoprogression in our trial … biopsies may be warranted to differentiate between true lymphoma progression and immune-mediated antitumor response,” the investigators note.
Dr. Noy disclosed that she has received research funding from Pharmacyclics.
Putting these results into context, the efficacy of ibrutinib seems similar to that of other single agents evaluated in patients with relapsed marginal zone lymphoma, including other agents that target molecules downstream of the B-cell receptor.
The observed response rate is “modest” compared with that seen when the drug is used to treat other conditions for which it is approved, he notes. Therefore, additional correlative studies to identify biomarkers predicting benefit in marginal zone lymphoma would have been helpful.
Nonetheless, the study team should be congratulated, as the trial demonstrates the ability to investigate rare disease subtypes in multicenter collaborations. The results justify ibrutinib as the first FDA-approved therapy for this disease and form the basis for subsequent trials that combine ibrutinib with anti-CD20 monoclonal antibodies and other targeted agents.
Paul M. Barr, MD, of the Wilmot Cancer Institute at the University of Rochester in New York made his remarks in an accompanying editorial (Blood. 2017;129:2207-08). Dr. Barr disclosed that he has consulted for and received research funding from Pharmacyclics and AbbVie.
Putting these results into context, the efficacy of ibrutinib seems similar to that of other single agents evaluated in patients with relapsed marginal zone lymphoma, including other agents that target molecules downstream of the B-cell receptor.
The observed response rate is “modest” compared with that seen when the drug is used to treat other conditions for which it is approved, he notes. Therefore, additional correlative studies to identify biomarkers predicting benefit in marginal zone lymphoma would have been helpful.
Nonetheless, the study team should be congratulated, as the trial demonstrates the ability to investigate rare disease subtypes in multicenter collaborations. The results justify ibrutinib as the first FDA-approved therapy for this disease and form the basis for subsequent trials that combine ibrutinib with anti-CD20 monoclonal antibodies and other targeted agents.
Paul M. Barr, MD, of the Wilmot Cancer Institute at the University of Rochester in New York made his remarks in an accompanying editorial (Blood. 2017;129:2207-08). Dr. Barr disclosed that he has consulted for and received research funding from Pharmacyclics and AbbVie.
Putting these results into context, the efficacy of ibrutinib seems similar to that of other single agents evaluated in patients with relapsed marginal zone lymphoma, including other agents that target molecules downstream of the B-cell receptor.
The observed response rate is “modest” compared with that seen when the drug is used to treat other conditions for which it is approved, he notes. Therefore, additional correlative studies to identify biomarkers predicting benefit in marginal zone lymphoma would have been helpful.
Nonetheless, the study team should be congratulated, as the trial demonstrates the ability to investigate rare disease subtypes in multicenter collaborations. The results justify ibrutinib as the first FDA-approved therapy for this disease and form the basis for subsequent trials that combine ibrutinib with anti-CD20 monoclonal antibodies and other targeted agents.
Paul M. Barr, MD, of the Wilmot Cancer Institute at the University of Rochester in New York made his remarks in an accompanying editorial (Blood. 2017;129:2207-08). Dr. Barr disclosed that he has consulted for and received research funding from Pharmacyclics and AbbVie.
Ibrutinib is active in patients with previously treated marginal zone lymphoma (MZL) and has a good safety profile, the results of a multicenter, open-label phase II trial published in Blood suggest.
Nearly half of 63 patients responded to monotherapy with the once-daily oral inhibitor of Bruton tyrosine kinase, and ibrutinib was generally well tolerated, according to the study (Blood. 2017;129:2224-32). The results prompted the U.S. Food and Drug Administration to grant accelerated approval of ibrutinib for patients with MZL who were previously treated with at least one prior anti-CD20–based therapy.
“MZL is frequently linked to chronic infection, which may induce B-cell receptor (BCR) signaling, resulting in aberrant B-cell survival and proliferation,” note the investigators, who were led by Ariela Noy, MD, a hematologic oncologist with the Lymphoma Service at the Memorial Sloan-Kettering Cancer Center, New York. “Single-agent ibrutinib induced durable responses with a favorable benefit–risk profile in patients with previously treated MZL, confirming the role of BCR signaling in this malignancy.”
“As the only approved therapy, ibrutinib provides a treatment option without chemotherapy for MZL,” they maintain. “Future studies will investigate ibrutinib in treatment-naive patients or as combination strategies in relapsed/refractory MZL.”
In the trial, which was funded by Pharmacyclics, an AbbVie Company, patients with MZL of all subtypes who had received at least one prior therapy with an anti-CD20 antibody–containing regimen were treated with 560 mg ibrutinib (Imbruvica) orally once daily. The median number of prior systemic therapies was two, and 63% had received prior chemoimmunotherapy.
The overall response rate, as assessed by an independent review committee using 2007 International Working Group criteria – the trial’s primary endpoint – was 48%, according to the published results. Benefit was similar across patients who differed regarding MZL subtype, number of prior regimen, and previous receipt of chemoimmunotherapy
After a 19.4-month median follow-up, the median duration of response was not reached, and median progression-free survival was 14.2 months.
The most common grade 3 or worse adverse events were anemia (14%), pneumonia (8%), and fatigue (6%). Serious adverse events of any grade affected 44% of patients. The most common was grade 3 or 4 pneumonia. Adverse events led to treatment discontinuation in 17% of patients and dose reductions in 10%.
“Due to evidence of pseudoprogression in our trial … biopsies may be warranted to differentiate between true lymphoma progression and immune-mediated antitumor response,” the investigators note.
Dr. Noy disclosed that she has received research funding from Pharmacyclics.
Ibrutinib is active in patients with previously treated marginal zone lymphoma (MZL) and has a good safety profile, the results of a multicenter, open-label phase II trial published in Blood suggest.
Nearly half of 63 patients responded to monotherapy with the once-daily oral inhibitor of Bruton tyrosine kinase, and ibrutinib was generally well tolerated, according to the study (Blood. 2017;129:2224-32). The results prompted the U.S. Food and Drug Administration to grant accelerated approval of ibrutinib for patients with MZL who were previously treated with at least one prior anti-CD20–based therapy.
“MZL is frequently linked to chronic infection, which may induce B-cell receptor (BCR) signaling, resulting in aberrant B-cell survival and proliferation,” note the investigators, who were led by Ariela Noy, MD, a hematologic oncologist with the Lymphoma Service at the Memorial Sloan-Kettering Cancer Center, New York. “Single-agent ibrutinib induced durable responses with a favorable benefit–risk profile in patients with previously treated MZL, confirming the role of BCR signaling in this malignancy.”
“As the only approved therapy, ibrutinib provides a treatment option without chemotherapy for MZL,” they maintain. “Future studies will investigate ibrutinib in treatment-naive patients or as combination strategies in relapsed/refractory MZL.”
In the trial, which was funded by Pharmacyclics, an AbbVie Company, patients with MZL of all subtypes who had received at least one prior therapy with an anti-CD20 antibody–containing regimen were treated with 560 mg ibrutinib (Imbruvica) orally once daily. The median number of prior systemic therapies was two, and 63% had received prior chemoimmunotherapy.
The overall response rate, as assessed by an independent review committee using 2007 International Working Group criteria – the trial’s primary endpoint – was 48%, according to the published results. Benefit was similar across patients who differed regarding MZL subtype, number of prior regimen, and previous receipt of chemoimmunotherapy
After a 19.4-month median follow-up, the median duration of response was not reached, and median progression-free survival was 14.2 months.
The most common grade 3 or worse adverse events were anemia (14%), pneumonia (8%), and fatigue (6%). Serious adverse events of any grade affected 44% of patients. The most common was grade 3 or 4 pneumonia. Adverse events led to treatment discontinuation in 17% of patients and dose reductions in 10%.
“Due to evidence of pseudoprogression in our trial … biopsies may be warranted to differentiate between true lymphoma progression and immune-mediated antitumor response,” the investigators note.
Dr. Noy disclosed that she has received research funding from Pharmacyclics.
Key clinical point:
Major finding: The overall response rate was 48%, and the drug was well tolerated.
Data source: A multicenter, open-label phase II trial of ibrutinib in 63 patients with previously treated marginal zone lymphoma.
Disclosures: Dr. Noy disclosed that she has received research funding from Pharmacyclics. The trial was funded by Pharmacyclics, an AbbVie Company.
VIDEO: Advocacy efforts spur CMS to drop HCAHPS pain domain assessment
How pain management is evaluated in the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey is changing, thanks in part to the advocacy efforts of the Society of Hospital Medicine’s public policy committee.
Based on input from SHM and other organizations, the Centers for Medicare & Medicaid Services decided that the way the survey was worded concerning pain management could be leading to unintended consequences, particularly in light of the opioid epidemic.
In a video interview recorded during HM17, John Biebelhausen, MD, MBA, discussed how SHM worked with the CMS to help “improve the HCAHPS survey to make a better patient satisfaction tool for our assessments and also eliminate some of the competing pressures the physician might face.”
Dr. Biebelhausen is a hospitalist and physician lead for quality reporting at Virginia Mason Hospital in Seattle. He had no relevant disclosures.hospitalist and Physician Lead for Quality Reporting.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
How pain management is evaluated in the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey is changing, thanks in part to the advocacy efforts of the Society of Hospital Medicine’s public policy committee.
Based on input from SHM and other organizations, the Centers for Medicare & Medicaid Services decided that the way the survey was worded concerning pain management could be leading to unintended consequences, particularly in light of the opioid epidemic.
In a video interview recorded during HM17, John Biebelhausen, MD, MBA, discussed how SHM worked with the CMS to help “improve the HCAHPS survey to make a better patient satisfaction tool for our assessments and also eliminate some of the competing pressures the physician might face.”
Dr. Biebelhausen is a hospitalist and physician lead for quality reporting at Virginia Mason Hospital in Seattle. He had no relevant disclosures.hospitalist and Physician Lead for Quality Reporting.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
How pain management is evaluated in the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey is changing, thanks in part to the advocacy efforts of the Society of Hospital Medicine’s public policy committee.
Based on input from SHM and other organizations, the Centers for Medicare & Medicaid Services decided that the way the survey was worded concerning pain management could be leading to unintended consequences, particularly in light of the opioid epidemic.
In a video interview recorded during HM17, John Biebelhausen, MD, MBA, discussed how SHM worked with the CMS to help “improve the HCAHPS survey to make a better patient satisfaction tool for our assessments and also eliminate some of the competing pressures the physician might face.”
Dr. Biebelhausen is a hospitalist and physician lead for quality reporting at Virginia Mason Hospital in Seattle. He had no relevant disclosures.hospitalist and Physician Lead for Quality Reporting.

{kind=link}