User login
In the wake of a federal lawsuit settlement, can you trust your EHR?
Who watches the watchers?
In a world where the majority of practices depend on electronic health record systems to care for patients, we also depend on the companies that make them. After all, the main (if not only) reason most practices jumped to them was to qualify for meaningful use payments.
In buying them, we’re trusting that the manufacturer is doing its best to keep them updated, operational, and compliant, right? Beyond people’s health, there’s also a lot of money at stake here.
Right? Right.
The alleged bones of the matter is that eClinicalWorks knowingly misrepresented its software to get certification in the EHR incentive program. The U.S. Department of Justice says the program was modified to retrieve only specific drugs and didn’t reliably record certain chart information (such as orders and drug interactions) or allow patient information to transfer to other systems.
I should note that, in settling this matter, eClinicalWorks did not admit wrongdoing. The company just agreed to pay that money to close the lawsuit.
Guess what? If your practice used eClinicalWorks, you’re no longer in compliance. So you could be penalized, too. Fortunately, the Centers for Medicare & Medicaid Services has recognized this and announced that practices won’t be held responsible for the vendor’s failings.
Perhaps eClinicalWorks meant no harm by these things. I understand that. Projects like this are complex. It’s easy for things to fall behind and slip through the cracks. With any software release there are always issues that aren’t recognized until it comes into widespread use. But this is patient health, not the latest version of Flappy Bird.
More worrisome is the other possibility: that eClinicalWorks was aware of the issues and covered them up so as not to affect sales. If this is the case, the company made a conscious decision to choose money over patient safety.
We’ll likely never know.
In its defense, eClinicalWorks states that most of these issues have been fixed, and the others are being actively corrected and tested. The company has agreed to do quality control oversight and to track, publish, and correct problems as they become apparent.
A decision many practices face now is whether or not to stay with the company. Can you trust eClinicalWorks from here on out? If so, how vigilant do you need to be? If not, how much time and money will a new EHR system cost to implement?
Not an easy choice for any practice trying to stay afloat these days.

Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Who watches the watchers?
In a world where the majority of practices depend on electronic health record systems to care for patients, we also depend on the companies that make them. After all, the main (if not only) reason most practices jumped to them was to qualify for meaningful use payments.
In buying them, we’re trusting that the manufacturer is doing its best to keep them updated, operational, and compliant, right? Beyond people’s health, there’s also a lot of money at stake here.
Right? Right.
The alleged bones of the matter is that eClinicalWorks knowingly misrepresented its software to get certification in the EHR incentive program. The U.S. Department of Justice says the program was modified to retrieve only specific drugs and didn’t reliably record certain chart information (such as orders and drug interactions) or allow patient information to transfer to other systems.
I should note that, in settling this matter, eClinicalWorks did not admit wrongdoing. The company just agreed to pay that money to close the lawsuit.
Guess what? If your practice used eClinicalWorks, you’re no longer in compliance. So you could be penalized, too. Fortunately, the Centers for Medicare & Medicaid Services has recognized this and announced that practices won’t be held responsible for the vendor’s failings.
Perhaps eClinicalWorks meant no harm by these things. I understand that. Projects like this are complex. It’s easy for things to fall behind and slip through the cracks. With any software release there are always issues that aren’t recognized until it comes into widespread use. But this is patient health, not the latest version of Flappy Bird.
More worrisome is the other possibility: that eClinicalWorks was aware of the issues and covered them up so as not to affect sales. If this is the case, the company made a conscious decision to choose money over patient safety.
We’ll likely never know.
In its defense, eClinicalWorks states that most of these issues have been fixed, and the others are being actively corrected and tested. The company has agreed to do quality control oversight and to track, publish, and correct problems as they become apparent.
A decision many practices face now is whether or not to stay with the company. Can you trust eClinicalWorks from here on out? If so, how vigilant do you need to be? If not, how much time and money will a new EHR system cost to implement?
Not an easy choice for any practice trying to stay afloat these days.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Who watches the watchers?
In a world where the majority of practices depend on electronic health record systems to care for patients, we also depend on the companies that make them. After all, the main (if not only) reason most practices jumped to them was to qualify for meaningful use payments.
In buying them, we’re trusting that the manufacturer is doing its best to keep them updated, operational, and compliant, right? Beyond people’s health, there’s also a lot of money at stake here.
Right? Right.
The alleged bones of the matter is that eClinicalWorks knowingly misrepresented its software to get certification in the EHR incentive program. The U.S. Department of Justice says the program was modified to retrieve only specific drugs and didn’t reliably record certain chart information (such as orders and drug interactions) or allow patient information to transfer to other systems.
I should note that, in settling this matter, eClinicalWorks did not admit wrongdoing. The company just agreed to pay that money to close the lawsuit.
Guess what? If your practice used eClinicalWorks, you’re no longer in compliance. So you could be penalized, too. Fortunately, the Centers for Medicare & Medicaid Services has recognized this and announced that practices won’t be held responsible for the vendor’s failings.
Perhaps eClinicalWorks meant no harm by these things. I understand that. Projects like this are complex. It’s easy for things to fall behind and slip through the cracks. With any software release there are always issues that aren’t recognized until it comes into widespread use. But this is patient health, not the latest version of Flappy Bird.
More worrisome is the other possibility: that eClinicalWorks was aware of the issues and covered them up so as not to affect sales. If this is the case, the company made a conscious decision to choose money over patient safety.
We’ll likely never know.
In its defense, eClinicalWorks states that most of these issues have been fixed, and the others are being actively corrected and tested. The company has agreed to do quality control oversight and to track, publish, and correct problems as they become apparent.
A decision many practices face now is whether or not to stay with the company. Can you trust eClinicalWorks from here on out? If so, how vigilant do you need to be? If not, how much time and money will a new EHR system cost to implement?
Not an easy choice for any practice trying to stay afloat these days.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Imipramine-Induced Hyperpigmentation
Imipramine is a tricyclic medication uncommonly used to treat depression, anxiety, and other psychiatric illnesses. Although relatively rare, it has been associated with hyperpigmentation of the skin including slate gray discoloration of sun-exposed areas.
We present the case of a 63-year-old woman who had been taking imipramine for more than 20 years when she developed bluish gray discoloration on the face and neck. Histopathology of biopsy specimens showed numerous perivascular and interstitial brown globules in the dermis that were composed of melanin only, as evidenced by positive Fontana-Masson staining and negative Perls Prussian blue staining. A diagnosis of imipramine-induced hyperpigmentation was made based on histopathology and clinical history.
In addition to the case presentation, we provide a review of drugs that commonly cause hyperpigmentation as well as their associated histopathologic staining characteristics.
Case Report
A 63-year-old woman presented with blue-gray discoloration on the face and neck. She first noted the discoloration on the left side of the forehead 3 years prior; it then spread to the right side of the forehead, cheeks, and neck. She denied pruritus, pain, redness, and scaling of the involved areas; any recent changes in medications; or the use of any topical products on the affected areas. Her medical history was remarkable for hypertension, which was inconsistently controlled with lisinopril and hydrochlorothiazide, and depression, which had been managed with oral imipramine.
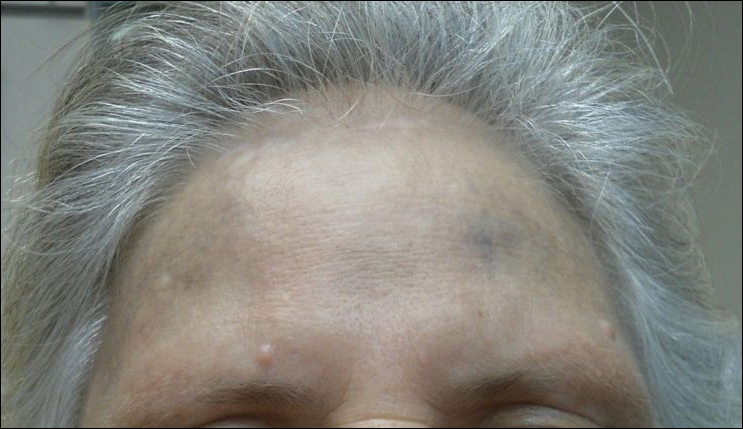
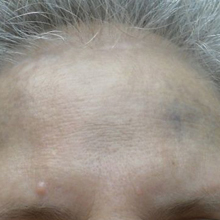
Physical examination disclosed blue-gray hyperpigmented patches with irregular borders on the bilateral forehead, temples, and periorbital skin (Figure 1). Reticulated brown patches were noted on the bilateral cheeks, and the neck displayed diffuse muddy brown patches with sparing of the submental areas.
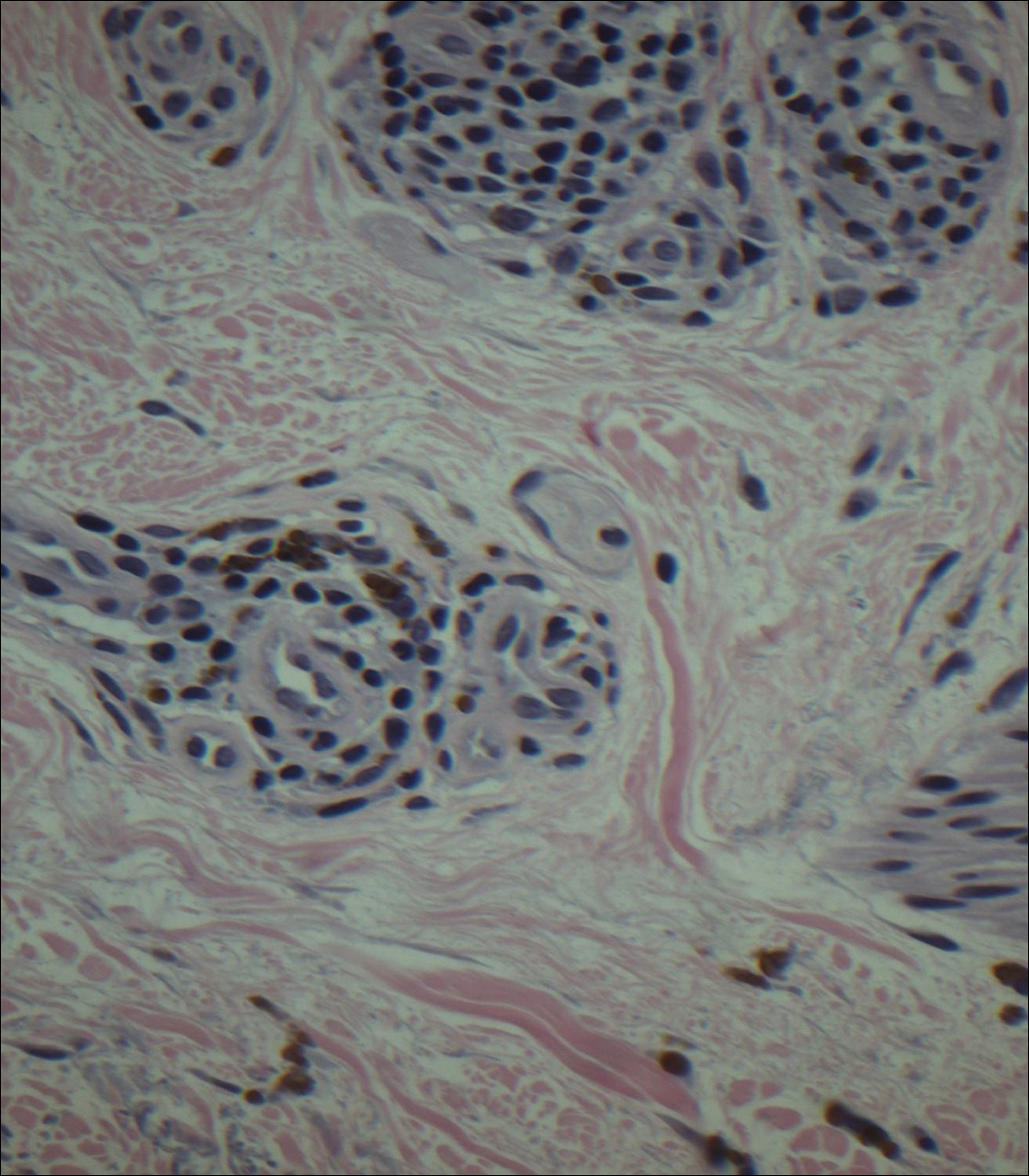
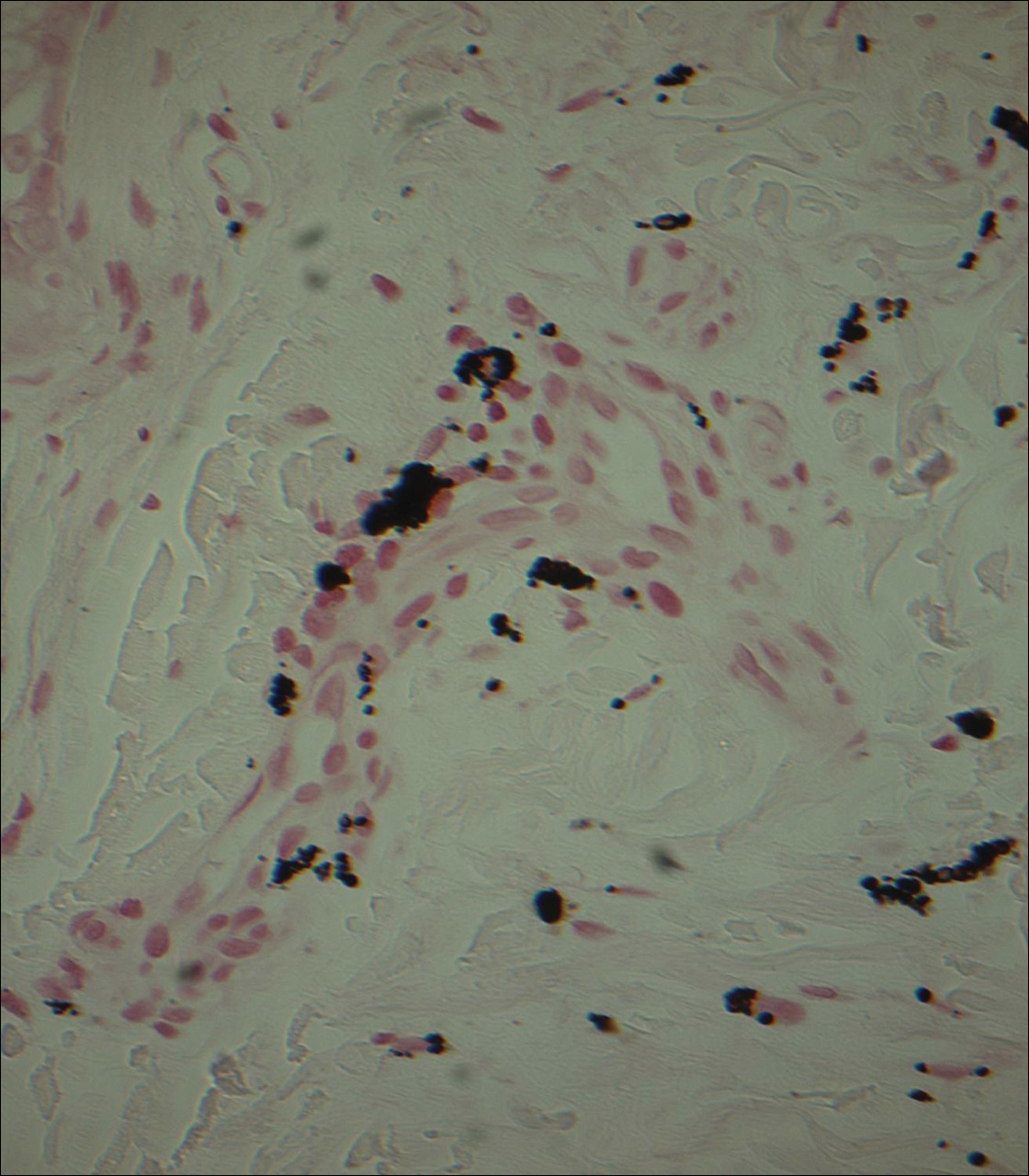
Punch biopsies obtained from the lateral forehead showed an unremarkable epidermis with deposition of numerous golden brown granules in the upper and mid dermis and in perivascular macrophages (Figure 2). The pigmented granules showed positive staining with Fontana-Masson (Figure 3), and a Perls Prussian blue stain for hemosiderin was negative. Based on the clinical history, a diagnosis of imipramine-induced hyperpigmentation was made.
The patient revealed that she had taken imipramine for more than 20 years for depression as prescribed by her mental health professional. She had tried several other antidepressants but none were as effective as imipramine. Therefore, she was not willing to discontinue it despite the likelihood that the hyperpigmentation would persist and could worsen with continued use of the medication. Diligent photoprotection was advised. Additionally, she started taking lisinopril some time after the appearance of the hyperpigmentation presented and had not taken hydrochlorothiazide consistently for several years. Although these drugs are known to cause various cutaneous reactions, it was not considered likely in this case.
Comment
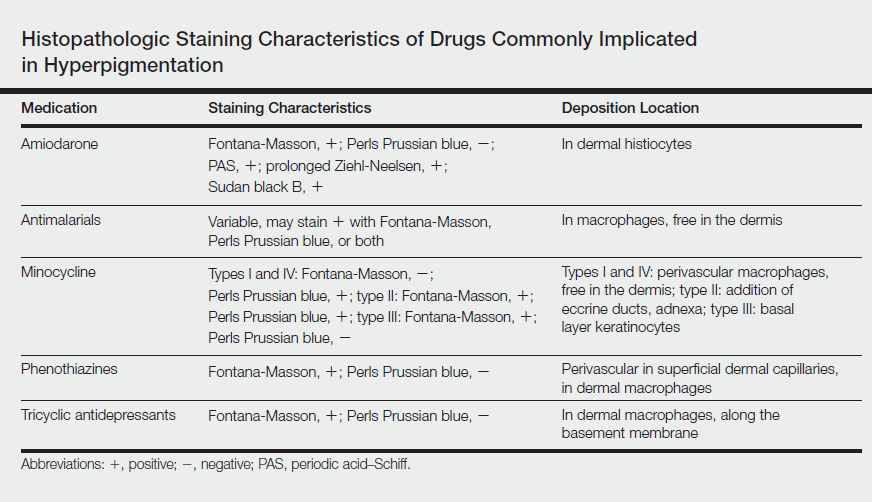
Drug-induced hyperpigmentation accounts for 10% to 20% of all cases of acquired hyperpigmentation.1 Common causative drugs include amiodarone, antimalarials, minocycline, and rarely psychotropics including phenothiazines and tricyclic antidepressants such as imipramine.1-4 Although amiodarone-induced hyperpigmentation is associated with lipofuscin in addition to melanin, most other medications, including imipramine, induce cutaneous effects through deposition of melanin and/or hemosiderin. A review of the histopathologic staining characteristics in pigment anomalies caused by these drugs is summarized in the Table.
Imipramine-induced hyperpigmentation presents as slate gray discrete macules and patches on sun-exposed skin that may appear anywhere from 2 to 22 years after initiating the medication.1-4 Affected areas include the malar cheeks, temples, periorbital areas, hands, forearms, and seldom the iris and sclera.2-4 Although the blue to slate gray coloring is classic, other colors have been described including brown, golden brown, and purple.2
Histopathology of imipramine-induced hyperpigmentation shows golden brown, round to oval granules in the superficial dermis and within dermal macrophages.1,3 Generally, Fontana-Masson staining is positive for melanin and Perls Prussian blue staining is negative for iron.1,2,4
Imipramine-induced hyperpigmentation likely results from photoexcitation of imipramine or one of its metabolites. These compounds activate tyrosinase, increasing melanogenesis and leading to formation of melanin-imipramine or melanin-metabolite complexes.1-3 Complexes are deposited in the dermis and basal layer or are engulfed by dermal macrophages and darkened on sun exposure due to their high melanin content.1 Other possible mechanisms of hyperpigmentation include nonspecific inflammation caused by the drug in the skin, hemosiderin deposition from vessel damage and subsequent erythrocyte extravasation, or deposition of newly formed pigments related to the drug.1
Most patients report satisfactory resolution of imipramine-induced discoloration within 1 year of stopping imipramine or switching to a different antidepressant.1,4 Patients who are unwilling to discontinue imipramine may achieve resolution with alexandrite or Q-switched ruby laser therapy.1,4 Strict sun protective measures are necessary, both to prevent new deposition of melanin and to prevent darkening of existing pigment.
Despite the advent of new psychotropic medications, imipramine remains the antidepressant of choice for many patients. Although rare, it is important to be able to recognize imipramine-induced hyperpigmentation and to encourage patient-psychiatrist communication to determine an antidepressant regimen that avoids unnecessary cutaneous side effects.
- D’Agostino ML, Risser J, Robinson-Bostom L. Imipramine-induced hyperpigmentation: a case report and review of the literature. J Cutan Pathol. 2009;36:799-803.
- Ming ME, Bhawan J, Stefanato CM, et al. Imipramine-induced hyperpigmentation: four cases and a review of the literature. J Am Acad Dermatol. 1999;40(2, pt 1):159-166.
- Sicari MC, Lebwohl M, Baral J, et al. Photoinduced dermal pigmentation in patients taking tricyclic antidepressants: histology, electron microscopy, and energy dispersive spectroscopy. J Am Acad Dermatol.1999;40(2, pt 2):290-293.
- Atkin DH, Fitzpatrick RE. Laser treatment of imipramine-induced hyperpigmentation. J Am Acad Dermatol. 2000;43(1, pt 1):77-80.
Imipramine is a tricyclic medication uncommonly used to treat depression, anxiety, and other psychiatric illnesses. Although relatively rare, it has been associated with hyperpigmentation of the skin including slate gray discoloration of sun-exposed areas.
We present the case of a 63-year-old woman who had been taking imipramine for more than 20 years when she developed bluish gray discoloration on the face and neck. Histopathology of biopsy specimens showed numerous perivascular and interstitial brown globules in the dermis that were composed of melanin only, as evidenced by positive Fontana-Masson staining and negative Perls Prussian blue staining. A diagnosis of imipramine-induced hyperpigmentation was made based on histopathology and clinical history.
In addition to the case presentation, we provide a review of drugs that commonly cause hyperpigmentation as well as their associated histopathologic staining characteristics.
Case Report
A 63-year-old woman presented with blue-gray discoloration on the face and neck. She first noted the discoloration on the left side of the forehead 3 years prior; it then spread to the right side of the forehead, cheeks, and neck. She denied pruritus, pain, redness, and scaling of the involved areas; any recent changes in medications; or the use of any topical products on the affected areas. Her medical history was remarkable for hypertension, which was inconsistently controlled with lisinopril and hydrochlorothiazide, and depression, which had been managed with oral imipramine.
Physical examination disclosed blue-gray hyperpigmented patches with irregular borders on the bilateral forehead, temples, and periorbital skin (Figure 1). Reticulated brown patches were noted on the bilateral cheeks, and the neck displayed diffuse muddy brown patches with sparing of the submental areas.
Punch biopsies obtained from the lateral forehead showed an unremarkable epidermis with deposition of numerous golden brown granules in the upper and mid dermis and in perivascular macrophages (Figure 2). The pigmented granules showed positive staining with Fontana-Masson (Figure 3), and a Perls Prussian blue stain for hemosiderin was negative. Based on the clinical history, a diagnosis of imipramine-induced hyperpigmentation was made.
The patient revealed that she had taken imipramine for more than 20 years for depression as prescribed by her mental health professional. She had tried several other antidepressants but none were as effective as imipramine. Therefore, she was not willing to discontinue it despite the likelihood that the hyperpigmentation would persist and could worsen with continued use of the medication. Diligent photoprotection was advised. Additionally, she started taking lisinopril some time after the appearance of the hyperpigmentation presented and had not taken hydrochlorothiazide consistently for several years. Although these drugs are known to cause various cutaneous reactions, it was not considered likely in this case.
Comment
Drug-induced hyperpigmentation accounts for 10% to 20% of all cases of acquired hyperpigmentation.1 Common causative drugs include amiodarone, antimalarials, minocycline, and rarely psychotropics including phenothiazines and tricyclic antidepressants such as imipramine.1-4 Although amiodarone-induced hyperpigmentation is associated with lipofuscin in addition to melanin, most other medications, including imipramine, induce cutaneous effects through deposition of melanin and/or hemosiderin. A review of the histopathologic staining characteristics in pigment anomalies caused by these drugs is summarized in the Table.
Imipramine-induced hyperpigmentation presents as slate gray discrete macules and patches on sun-exposed skin that may appear anywhere from 2 to 22 years after initiating the medication.1-4 Affected areas include the malar cheeks, temples, periorbital areas, hands, forearms, and seldom the iris and sclera.2-4 Although the blue to slate gray coloring is classic, other colors have been described including brown, golden brown, and purple.2
Histopathology of imipramine-induced hyperpigmentation shows golden brown, round to oval granules in the superficial dermis and within dermal macrophages.1,3 Generally, Fontana-Masson staining is positive for melanin and Perls Prussian blue staining is negative for iron.1,2,4
Imipramine-induced hyperpigmentation likely results from photoexcitation of imipramine or one of its metabolites. These compounds activate tyrosinase, increasing melanogenesis and leading to formation of melanin-imipramine or melanin-metabolite complexes.1-3 Complexes are deposited in the dermis and basal layer or are engulfed by dermal macrophages and darkened on sun exposure due to their high melanin content.1 Other possible mechanisms of hyperpigmentation include nonspecific inflammation caused by the drug in the skin, hemosiderin deposition from vessel damage and subsequent erythrocyte extravasation, or deposition of newly formed pigments related to the drug.1
Most patients report satisfactory resolution of imipramine-induced discoloration within 1 year of stopping imipramine or switching to a different antidepressant.1,4 Patients who are unwilling to discontinue imipramine may achieve resolution with alexandrite or Q-switched ruby laser therapy.1,4 Strict sun protective measures are necessary, both to prevent new deposition of melanin and to prevent darkening of existing pigment.
Despite the advent of new psychotropic medications, imipramine remains the antidepressant of choice for many patients. Although rare, it is important to be able to recognize imipramine-induced hyperpigmentation and to encourage patient-psychiatrist communication to determine an antidepressant regimen that avoids unnecessary cutaneous side effects.
Imipramine is a tricyclic medication uncommonly used to treat depression, anxiety, and other psychiatric illnesses. Although relatively rare, it has been associated with hyperpigmentation of the skin including slate gray discoloration of sun-exposed areas.
We present the case of a 63-year-old woman who had been taking imipramine for more than 20 years when she developed bluish gray discoloration on the face and neck. Histopathology of biopsy specimens showed numerous perivascular and interstitial brown globules in the dermis that were composed of melanin only, as evidenced by positive Fontana-Masson staining and negative Perls Prussian blue staining. A diagnosis of imipramine-induced hyperpigmentation was made based on histopathology and clinical history.
In addition to the case presentation, we provide a review of drugs that commonly cause hyperpigmentation as well as their associated histopathologic staining characteristics.
Case Report
A 63-year-old woman presented with blue-gray discoloration on the face and neck. She first noted the discoloration on the left side of the forehead 3 years prior; it then spread to the right side of the forehead, cheeks, and neck. She denied pruritus, pain, redness, and scaling of the involved areas; any recent changes in medications; or the use of any topical products on the affected areas. Her medical history was remarkable for hypertension, which was inconsistently controlled with lisinopril and hydrochlorothiazide, and depression, which had been managed with oral imipramine.
Physical examination disclosed blue-gray hyperpigmented patches with irregular borders on the bilateral forehead, temples, and periorbital skin (Figure 1). Reticulated brown patches were noted on the bilateral cheeks, and the neck displayed diffuse muddy brown patches with sparing of the submental areas.
Punch biopsies obtained from the lateral forehead showed an unremarkable epidermis with deposition of numerous golden brown granules in the upper and mid dermis and in perivascular macrophages (Figure 2). The pigmented granules showed positive staining with Fontana-Masson (Figure 3), and a Perls Prussian blue stain for hemosiderin was negative. Based on the clinical history, a diagnosis of imipramine-induced hyperpigmentation was made.
The patient revealed that she had taken imipramine for more than 20 years for depression as prescribed by her mental health professional. She had tried several other antidepressants but none were as effective as imipramine. Therefore, she was not willing to discontinue it despite the likelihood that the hyperpigmentation would persist and could worsen with continued use of the medication. Diligent photoprotection was advised. Additionally, she started taking lisinopril some time after the appearance of the hyperpigmentation presented and had not taken hydrochlorothiazide consistently for several years. Although these drugs are known to cause various cutaneous reactions, it was not considered likely in this case.
Comment
Drug-induced hyperpigmentation accounts for 10% to 20% of all cases of acquired hyperpigmentation.1 Common causative drugs include amiodarone, antimalarials, minocycline, and rarely psychotropics including phenothiazines and tricyclic antidepressants such as imipramine.1-4 Although amiodarone-induced hyperpigmentation is associated with lipofuscin in addition to melanin, most other medications, including imipramine, induce cutaneous effects through deposition of melanin and/or hemosiderin. A review of the histopathologic staining characteristics in pigment anomalies caused by these drugs is summarized in the Table.
Imipramine-induced hyperpigmentation presents as slate gray discrete macules and patches on sun-exposed skin that may appear anywhere from 2 to 22 years after initiating the medication.1-4 Affected areas include the malar cheeks, temples, periorbital areas, hands, forearms, and seldom the iris and sclera.2-4 Although the blue to slate gray coloring is classic, other colors have been described including brown, golden brown, and purple.2
Histopathology of imipramine-induced hyperpigmentation shows golden brown, round to oval granules in the superficial dermis and within dermal macrophages.1,3 Generally, Fontana-Masson staining is positive for melanin and Perls Prussian blue staining is negative for iron.1,2,4
Imipramine-induced hyperpigmentation likely results from photoexcitation of imipramine or one of its metabolites. These compounds activate tyrosinase, increasing melanogenesis and leading to formation of melanin-imipramine or melanin-metabolite complexes.1-3 Complexes are deposited in the dermis and basal layer or are engulfed by dermal macrophages and darkened on sun exposure due to their high melanin content.1 Other possible mechanisms of hyperpigmentation include nonspecific inflammation caused by the drug in the skin, hemosiderin deposition from vessel damage and subsequent erythrocyte extravasation, or deposition of newly formed pigments related to the drug.1
Most patients report satisfactory resolution of imipramine-induced discoloration within 1 year of stopping imipramine or switching to a different antidepressant.1,4 Patients who are unwilling to discontinue imipramine may achieve resolution with alexandrite or Q-switched ruby laser therapy.1,4 Strict sun protective measures are necessary, both to prevent new deposition of melanin and to prevent darkening of existing pigment.
Despite the advent of new psychotropic medications, imipramine remains the antidepressant of choice for many patients. Although rare, it is important to be able to recognize imipramine-induced hyperpigmentation and to encourage patient-psychiatrist communication to determine an antidepressant regimen that avoids unnecessary cutaneous side effects.
- D’Agostino ML, Risser J, Robinson-Bostom L. Imipramine-induced hyperpigmentation: a case report and review of the literature. J Cutan Pathol. 2009;36:799-803.
- Ming ME, Bhawan J, Stefanato CM, et al. Imipramine-induced hyperpigmentation: four cases and a review of the literature. J Am Acad Dermatol. 1999;40(2, pt 1):159-166.
- Sicari MC, Lebwohl M, Baral J, et al. Photoinduced dermal pigmentation in patients taking tricyclic antidepressants: histology, electron microscopy, and energy dispersive spectroscopy. J Am Acad Dermatol.1999;40(2, pt 2):290-293.
- Atkin DH, Fitzpatrick RE. Laser treatment of imipramine-induced hyperpigmentation. J Am Acad Dermatol. 2000;43(1, pt 1):77-80.
- D’Agostino ML, Risser J, Robinson-Bostom L. Imipramine-induced hyperpigmentation: a case report and review of the literature. J Cutan Pathol. 2009;36:799-803.
- Ming ME, Bhawan J, Stefanato CM, et al. Imipramine-induced hyperpigmentation: four cases and a review of the literature. J Am Acad Dermatol. 1999;40(2, pt 1):159-166.
- Sicari MC, Lebwohl M, Baral J, et al. Photoinduced dermal pigmentation in patients taking tricyclic antidepressants: histology, electron microscopy, and energy dispersive spectroscopy. J Am Acad Dermatol.1999;40(2, pt 2):290-293.
- Atkin DH, Fitzpatrick RE. Laser treatment of imipramine-induced hyperpigmentation. J Am Acad Dermatol. 2000;43(1, pt 1):77-80.
Practice Points
- Imipramine is a tricyclic medication used for the treatment of depression and mood disorders.
- A rare side effect of treatment with imipramine is a blue-gray discoloration of the skin.
- Thorough medication review is important in patients who present with skin discoloration.
New antibodies, drugs for refractory and relapsed myeloma are effective in patients over 65
MADRID – Patients with refractory/relapsed multiple myeloma had similar rates of progression-free survival (PFS) whether they were younger than age 65 or 65 years and older, based on the results of eight recent phase 3 trials in an analysis presented at the European Society for Medical Oncology Congress.
“The PFS benefit was significant in both younger and older patients with relapsed and refractory multiple myeloma with a cut off at 75 years,” reported Thierry Landre, PharmD, of the department of geriatric oncology, University of Paris 13, France. “Age, by itself, should not be a contraindication to the new myeloma drugs.”
In this meta-analysis, 5,421 patients were evaluated from the CASTOR and POLLUX trials, which evaluated daratumumab; the ELOQUENT-2 trial, which evaluated elotuzumab; the ASPIRE and ENDEAVOR trials, which evaluated carfilzomib; the TOURMALINE-MM trial, which evaluated ixazomib; the PANORAMA trial, which evaluated panobinostat; and the VANTAGE trial, which evaluated vorinostat.
For the analysis, patients were stratified by age younger than 65 years and age 65 years and older. Hazard ratios for benefit were calculated for the experimental and comparator arm for these two age groups. Though the number of patients over age 75 was small, hazard ratios were determined on an exploratory basis for that age group.
For the experimental arm relative to the comparator arm, all hazard ratios were statistically significant for patients less than age 65 with the exception of patients receiving elotuzumab in ELOQUENT-2. In that trial, the hazard ratio slightly exceeded the upward bound of the 95% confidence interval (95% CI, 0.55-1.02).
When the data were combined for the eight trials, the hazard ratio for PFS was 0.62 for newer agents relative to the comparator arms.
Similar results were seen for individual trial and aggregated trial data when the same calculations were done in patients aged 65-75. When the data for the eight trials were combined, the hazard ratio for PFS was 0.67 for newer agents relative to comparators.
The calculations for patients over age 75 were more limited because of the small numbers of trial participants in that age group. Of the hazard ratio estimates that were done, however, they were again of the same general magnitude seen in younger patients.
When PFS was broken down by type of therapy, the hazard ratios for patients younger than age 65 and those aged 65-75 were 0.57 and 0.52, respectively, for monoclonal antibodies. For combined data with the histone deacetylase inhibitors panobinostat and vorinostat, the respective hazard ratios were 0.67 and 0.78, respectively. For the second generation proteasome inhibitors carfilzomib and ixazomib, the respective hazard ratios were 0.61 and 0.70.
The ESMO-invited discussant for this presentation, Evangelos Terpos, MD, PhD, of the University of Athens, called the results reassuring. These data support treating relatively fit elderly patients with the newer agents.
“The data with the monoclonal antibodies suggest that these drugs actually provide their best results in elderly patients,” noted Dr. Terpos, pointing to the numerical advantage for the hazard ratio in older versus younger patients. The efficacy of monoclonal antibodies in older patients was further reinforced by the narrow confidence interval (95% CI, 0.42-0.61).
“There are many new drugs in the refractory setting [of multiple myeloma],” Dr. Terpos remarked. “It is helpful to have some data to show that these are also beneficial in the age group where this disease is most common.”
Dr. Landre agreed, noting that the median age at diagnosis of multiple myeloma is 69. This analysis helps to address the gap of “data available for evaluating efficacy in those older than 65 years and older than 75 years of age.”
MADRID – Patients with refractory/relapsed multiple myeloma had similar rates of progression-free survival (PFS) whether they were younger than age 65 or 65 years and older, based on the results of eight recent phase 3 trials in an analysis presented at the European Society for Medical Oncology Congress.
“The PFS benefit was significant in both younger and older patients with relapsed and refractory multiple myeloma with a cut off at 75 years,” reported Thierry Landre, PharmD, of the department of geriatric oncology, University of Paris 13, France. “Age, by itself, should not be a contraindication to the new myeloma drugs.”
In this meta-analysis, 5,421 patients were evaluated from the CASTOR and POLLUX trials, which evaluated daratumumab; the ELOQUENT-2 trial, which evaluated elotuzumab; the ASPIRE and ENDEAVOR trials, which evaluated carfilzomib; the TOURMALINE-MM trial, which evaluated ixazomib; the PANORAMA trial, which evaluated panobinostat; and the VANTAGE trial, which evaluated vorinostat.
For the analysis, patients were stratified by age younger than 65 years and age 65 years and older. Hazard ratios for benefit were calculated for the experimental and comparator arm for these two age groups. Though the number of patients over age 75 was small, hazard ratios were determined on an exploratory basis for that age group.
For the experimental arm relative to the comparator arm, all hazard ratios were statistically significant for patients less than age 65 with the exception of patients receiving elotuzumab in ELOQUENT-2. In that trial, the hazard ratio slightly exceeded the upward bound of the 95% confidence interval (95% CI, 0.55-1.02).
When the data were combined for the eight trials, the hazard ratio for PFS was 0.62 for newer agents relative to the comparator arms.
Similar results were seen for individual trial and aggregated trial data when the same calculations were done in patients aged 65-75. When the data for the eight trials were combined, the hazard ratio for PFS was 0.67 for newer agents relative to comparators.
The calculations for patients over age 75 were more limited because of the small numbers of trial participants in that age group. Of the hazard ratio estimates that were done, however, they were again of the same general magnitude seen in younger patients.
When PFS was broken down by type of therapy, the hazard ratios for patients younger than age 65 and those aged 65-75 were 0.57 and 0.52, respectively, for monoclonal antibodies. For combined data with the histone deacetylase inhibitors panobinostat and vorinostat, the respective hazard ratios were 0.67 and 0.78, respectively. For the second generation proteasome inhibitors carfilzomib and ixazomib, the respective hazard ratios were 0.61 and 0.70.
The ESMO-invited discussant for this presentation, Evangelos Terpos, MD, PhD, of the University of Athens, called the results reassuring. These data support treating relatively fit elderly patients with the newer agents.
“The data with the monoclonal antibodies suggest that these drugs actually provide their best results in elderly patients,” noted Dr. Terpos, pointing to the numerical advantage for the hazard ratio in older versus younger patients. The efficacy of monoclonal antibodies in older patients was further reinforced by the narrow confidence interval (95% CI, 0.42-0.61).
“There are many new drugs in the refractory setting [of multiple myeloma],” Dr. Terpos remarked. “It is helpful to have some data to show that these are also beneficial in the age group where this disease is most common.”
Dr. Landre agreed, noting that the median age at diagnosis of multiple myeloma is 69. This analysis helps to address the gap of “data available for evaluating efficacy in those older than 65 years and older than 75 years of age.”
MADRID – Patients with refractory/relapsed multiple myeloma had similar rates of progression-free survival (PFS) whether they were younger than age 65 or 65 years and older, based on the results of eight recent phase 3 trials in an analysis presented at the European Society for Medical Oncology Congress.
“The PFS benefit was significant in both younger and older patients with relapsed and refractory multiple myeloma with a cut off at 75 years,” reported Thierry Landre, PharmD, of the department of geriatric oncology, University of Paris 13, France. “Age, by itself, should not be a contraindication to the new myeloma drugs.”
In this meta-analysis, 5,421 patients were evaluated from the CASTOR and POLLUX trials, which evaluated daratumumab; the ELOQUENT-2 trial, which evaluated elotuzumab; the ASPIRE and ENDEAVOR trials, which evaluated carfilzomib; the TOURMALINE-MM trial, which evaluated ixazomib; the PANORAMA trial, which evaluated panobinostat; and the VANTAGE trial, which evaluated vorinostat.
For the analysis, patients were stratified by age younger than 65 years and age 65 years and older. Hazard ratios for benefit were calculated for the experimental and comparator arm for these two age groups. Though the number of patients over age 75 was small, hazard ratios were determined on an exploratory basis for that age group.
For the experimental arm relative to the comparator arm, all hazard ratios were statistically significant for patients less than age 65 with the exception of patients receiving elotuzumab in ELOQUENT-2. In that trial, the hazard ratio slightly exceeded the upward bound of the 95% confidence interval (95% CI, 0.55-1.02).
When the data were combined for the eight trials, the hazard ratio for PFS was 0.62 for newer agents relative to the comparator arms.
Similar results were seen for individual trial and aggregated trial data when the same calculations were done in patients aged 65-75. When the data for the eight trials were combined, the hazard ratio for PFS was 0.67 for newer agents relative to comparators.
The calculations for patients over age 75 were more limited because of the small numbers of trial participants in that age group. Of the hazard ratio estimates that were done, however, they were again of the same general magnitude seen in younger patients.
When PFS was broken down by type of therapy, the hazard ratios for patients younger than age 65 and those aged 65-75 were 0.57 and 0.52, respectively, for monoclonal antibodies. For combined data with the histone deacetylase inhibitors panobinostat and vorinostat, the respective hazard ratios were 0.67 and 0.78, respectively. For the second generation proteasome inhibitors carfilzomib and ixazomib, the respective hazard ratios were 0.61 and 0.70.
The ESMO-invited discussant for this presentation, Evangelos Terpos, MD, PhD, of the University of Athens, called the results reassuring. These data support treating relatively fit elderly patients with the newer agents.
“The data with the monoclonal antibodies suggest that these drugs actually provide their best results in elderly patients,” noted Dr. Terpos, pointing to the numerical advantage for the hazard ratio in older versus younger patients. The efficacy of monoclonal antibodies in older patients was further reinforced by the narrow confidence interval (95% CI, 0.42-0.61).
“There are many new drugs in the refractory setting [of multiple myeloma],” Dr. Terpos remarked. “It is helpful to have some data to show that these are also beneficial in the age group where this disease is most common.”
Dr. Landre agreed, noting that the median age at diagnosis of multiple myeloma is 69. This analysis helps to address the gap of “data available for evaluating efficacy in those older than 65 years and older than 75 years of age.”
AT ESMO 2017
Key clinical point: Recently approved monoclonal antibodies and drugs for treating refractory/relapsed multiple myeloma work well for patients aged 65-75 years.
Major finding: The hazard ratios for progression-free survival were largely similar for patients younger than age 65 years and patients aged 65 and older.
Data source: Meta-analysis of eight phase 3 randomized trials.
Disclosures: Dr. Landre reported having no financial conflicts of interest.
CMS alerts physicians of payment reductions for PQRS noncompliance
Doctors who did not adequately meet Physician Quality Reporting System (PQRS) requirements in 2016 will soon be receiving notification letters alerting them that their Medicare Part B physician fee schedule payments will be reduced by 2%.
Officials from the Centers for Medicare & Medicaid Services said in a statement that “the majority” of eligible professionals “successfully reported to PQRS and avoided the downward payment adjustment,” but did not state how many doctors are expected to receive letters.
The CMS noted that there are no hardship exemptions to avoid the payment reduction for 2018.
Doctors who did not adequately meet Physician Quality Reporting System (PQRS) requirements in 2016 will soon be receiving notification letters alerting them that their Medicare Part B physician fee schedule payments will be reduced by 2%.
Officials from the Centers for Medicare & Medicaid Services said in a statement that “the majority” of eligible professionals “successfully reported to PQRS and avoided the downward payment adjustment,” but did not state how many doctors are expected to receive letters.
The CMS noted that there are no hardship exemptions to avoid the payment reduction for 2018.
Doctors who did not adequately meet Physician Quality Reporting System (PQRS) requirements in 2016 will soon be receiving notification letters alerting them that their Medicare Part B physician fee schedule payments will be reduced by 2%.
Officials from the Centers for Medicare & Medicaid Services said in a statement that “the majority” of eligible professionals “successfully reported to PQRS and avoided the downward payment adjustment,” but did not state how many doctors are expected to receive letters.
The CMS noted that there are no hardship exemptions to avoid the payment reduction for 2018.
Sneak Peek: Journal of Hospital Medicine – Sept. 2017
BACKGROUND: Patient preferences regarding cardiopulmonary resuscitation (CPR) are important, especially during hospitalization when a patient’s health is changing, yet many patients are not adequately informed or involved in the decision-making process.
OBJECTIVES: We examined the effect of an informational video about CPR on hospitalized patients’ code status choices.
DESIGN: This was a prospective, randomized trial conducted at the Veteran’s Affairs Hospital in Minneapolis.
PARTICIPANTS: We enrolled 119 patients who were hospitalized on the general medicine service and at least 65 years old. The majority were men (97%) with a mean age of 75.
INTERVENTION: A video described code status choices: full code (CPR and intubation if required), do not resuscitate (DNR), and do not resuscitate/do not intubate (DNR/DNI). Participants were randomized to watch the video (n = 59) or usual care (n = 60).
MEASUREMENTS: The primary outcome was participants’ code status preferences. Secondary outcomes included a questionnaire designed to evaluate participants’ trust in their health care team and their knowledge and perceptions about CPR.
RESULTS: Participants who viewed the video were less likely to choose full code (37%), compared with participants in the usual-care group (71%), and were more likely to choose DNR/DNI (56% in the video group vs. 17% in the control group) (P < .00001). We did not see a difference in trust in their health care team or knowledge and perceptions about CPR as assessed by our questionnaire.
CONCLUSIONS: Hospitalized patients who watched a video about CPR and code status choices were less likely to choose full code and more likely to choose DNR/DNI.
Also in JHM this month
Influenza season hospitalization trends in Israel: A multi-year comparative analysis 2005/2006 through 2012/2013
AUTHORS: Aharona Glatman-Freedman, MD, MPH, Zalman Kaufman, MS, Yaniv Stein, BS, Hanna Sefty, MS, Hila Zadka, PhD, Barak Gordon, MD, MHA, Jill Meron, BSc, Ethel-Sherry Gordon, PhD, Rita Dichtiar, BSc, Ziona Haklai, MSc, Arnon Afek, MD, Tamy Shohat, MD, MPH
Appropriate reconciliation of cardiovascular medications after elective surgery and postdischarge acute hospital and ambulatory visits
AUTHORS: Jonathan S. Lee, MD, Ralph Gonzales, MD, MSPH, Eric Vittinghoff, PhD, Kitty K. Corbett, PhD, MPH, Kirsten E. Fleischmann, MD, Neil Sehgal, MD, MPH, Andrew D. Auerbach, MD, MPH
Patterns and appropriateness of thrombophilia testing in an academic medical center
AUTHORS: Nicholas Cox, PharmD, Stacy A. Johnson, MD, Sara Vazquez, PharmD, Ryan P. Fleming, PharmD, BCPS, Matthew T. Rondina, MD, David Kaplan, MD, Stephanie Chauv, PharmD, Gabriel V. Fontaine, PharmD, Scott M. Stevens, MD, Scott Woller, MD, Daniel M. Witt, PharmD, BCPS, FCCP
National trends (2007-2013) of Clostridium difficile infection in patients with septic shock: Impact on outcome
AUTHORS: Kshitij Chatterjee, MD, Abhinav Goyal, MD, Aditya Chada, MD, Krishna Siva Sai Kakkera, MD, Howard L Corwin, MD
Blood products provided to patients receiving inappropriate critical care
AUTHORS: Thanh H. Neville, MD, MSHS, Alyssa Ziman, MD, Neil S. Wenger, MD, MPH
BACKGROUND: Patient preferences regarding cardiopulmonary resuscitation (CPR) are important, especially during hospitalization when a patient’s health is changing, yet many patients are not adequately informed or involved in the decision-making process.
OBJECTIVES: We examined the effect of an informational video about CPR on hospitalized patients’ code status choices.
DESIGN: This was a prospective, randomized trial conducted at the Veteran’s Affairs Hospital in Minneapolis.
PARTICIPANTS: We enrolled 119 patients who were hospitalized on the general medicine service and at least 65 years old. The majority were men (97%) with a mean age of 75.
INTERVENTION: A video described code status choices: full code (CPR and intubation if required), do not resuscitate (DNR), and do not resuscitate/do not intubate (DNR/DNI). Participants were randomized to watch the video (n = 59) or usual care (n = 60).
MEASUREMENTS: The primary outcome was participants’ code status preferences. Secondary outcomes included a questionnaire designed to evaluate participants’ trust in their health care team and their knowledge and perceptions about CPR.
RESULTS: Participants who viewed the video were less likely to choose full code (37%), compared with participants in the usual-care group (71%), and were more likely to choose DNR/DNI (56% in the video group vs. 17% in the control group) (P < .00001). We did not see a difference in trust in their health care team or knowledge and perceptions about CPR as assessed by our questionnaire.
CONCLUSIONS: Hospitalized patients who watched a video about CPR and code status choices were less likely to choose full code and more likely to choose DNR/DNI.
Also in JHM this month
Influenza season hospitalization trends in Israel: A multi-year comparative analysis 2005/2006 through 2012/2013
AUTHORS: Aharona Glatman-Freedman, MD, MPH, Zalman Kaufman, MS, Yaniv Stein, BS, Hanna Sefty, MS, Hila Zadka, PhD, Barak Gordon, MD, MHA, Jill Meron, BSc, Ethel-Sherry Gordon, PhD, Rita Dichtiar, BSc, Ziona Haklai, MSc, Arnon Afek, MD, Tamy Shohat, MD, MPH
Appropriate reconciliation of cardiovascular medications after elective surgery and postdischarge acute hospital and ambulatory visits
AUTHORS: Jonathan S. Lee, MD, Ralph Gonzales, MD, MSPH, Eric Vittinghoff, PhD, Kitty K. Corbett, PhD, MPH, Kirsten E. Fleischmann, MD, Neil Sehgal, MD, MPH, Andrew D. Auerbach, MD, MPH
Patterns and appropriateness of thrombophilia testing in an academic medical center
AUTHORS: Nicholas Cox, PharmD, Stacy A. Johnson, MD, Sara Vazquez, PharmD, Ryan P. Fleming, PharmD, BCPS, Matthew T. Rondina, MD, David Kaplan, MD, Stephanie Chauv, PharmD, Gabriel V. Fontaine, PharmD, Scott M. Stevens, MD, Scott Woller, MD, Daniel M. Witt, PharmD, BCPS, FCCP
National trends (2007-2013) of Clostridium difficile infection in patients with septic shock: Impact on outcome
AUTHORS: Kshitij Chatterjee, MD, Abhinav Goyal, MD, Aditya Chada, MD, Krishna Siva Sai Kakkera, MD, Howard L Corwin, MD
Blood products provided to patients receiving inappropriate critical care
AUTHORS: Thanh H. Neville, MD, MSHS, Alyssa Ziman, MD, Neil S. Wenger, MD, MPH
BACKGROUND: Patient preferences regarding cardiopulmonary resuscitation (CPR) are important, especially during hospitalization when a patient’s health is changing, yet many patients are not adequately informed or involved in the decision-making process.
OBJECTIVES: We examined the effect of an informational video about CPR on hospitalized patients’ code status choices.
DESIGN: This was a prospective, randomized trial conducted at the Veteran’s Affairs Hospital in Minneapolis.
PARTICIPANTS: We enrolled 119 patients who were hospitalized on the general medicine service and at least 65 years old. The majority were men (97%) with a mean age of 75.
INTERVENTION: A video described code status choices: full code (CPR and intubation if required), do not resuscitate (DNR), and do not resuscitate/do not intubate (DNR/DNI). Participants were randomized to watch the video (n = 59) or usual care (n = 60).
MEASUREMENTS: The primary outcome was participants’ code status preferences. Secondary outcomes included a questionnaire designed to evaluate participants’ trust in their health care team and their knowledge and perceptions about CPR.
RESULTS: Participants who viewed the video were less likely to choose full code (37%), compared with participants in the usual-care group (71%), and were more likely to choose DNR/DNI (56% in the video group vs. 17% in the control group) (P < .00001). We did not see a difference in trust in their health care team or knowledge and perceptions about CPR as assessed by our questionnaire.
CONCLUSIONS: Hospitalized patients who watched a video about CPR and code status choices were less likely to choose full code and more likely to choose DNR/DNI.
Also in JHM this month
Influenza season hospitalization trends in Israel: A multi-year comparative analysis 2005/2006 through 2012/2013
AUTHORS: Aharona Glatman-Freedman, MD, MPH, Zalman Kaufman, MS, Yaniv Stein, BS, Hanna Sefty, MS, Hila Zadka, PhD, Barak Gordon, MD, MHA, Jill Meron, BSc, Ethel-Sherry Gordon, PhD, Rita Dichtiar, BSc, Ziona Haklai, MSc, Arnon Afek, MD, Tamy Shohat, MD, MPH
Appropriate reconciliation of cardiovascular medications after elective surgery and postdischarge acute hospital and ambulatory visits
AUTHORS: Jonathan S. Lee, MD, Ralph Gonzales, MD, MSPH, Eric Vittinghoff, PhD, Kitty K. Corbett, PhD, MPH, Kirsten E. Fleischmann, MD, Neil Sehgal, MD, MPH, Andrew D. Auerbach, MD, MPH
Patterns and appropriateness of thrombophilia testing in an academic medical center
AUTHORS: Nicholas Cox, PharmD, Stacy A. Johnson, MD, Sara Vazquez, PharmD, Ryan P. Fleming, PharmD, BCPS, Matthew T. Rondina, MD, David Kaplan, MD, Stephanie Chauv, PharmD, Gabriel V. Fontaine, PharmD, Scott M. Stevens, MD, Scott Woller, MD, Daniel M. Witt, PharmD, BCPS, FCCP
National trends (2007-2013) of Clostridium difficile infection in patients with septic shock: Impact on outcome
AUTHORS: Kshitij Chatterjee, MD, Abhinav Goyal, MD, Aditya Chada, MD, Krishna Siva Sai Kakkera, MD, Howard L Corwin, MD
Blood products provided to patients receiving inappropriate critical care
AUTHORS: Thanh H. Neville, MD, MSHS, Alyssa Ziman, MD, Neil S. Wenger, MD, MPH
Clinical trial: Understanding the Genetic Predisposition to the Development of Primary Biliary Cirrhosis
Understanding the Genetic Predisposition to the Development of Primary Biliary Cirrhosis is an observational study recruiting people with a history of PBC and their family members.
The trial will investigate whether or not there is a genetic factor in the development of PBC. Blood and stool samples will be taken from PBC patients and family members of PBC patients and analyzed. If a genetic component is discovered, it will add to current knowledge of how PBC and other adult chronic cholestatic liver diseases develop, as well as suggest new approaches for prevention, diagnosis, and treatment.
Individuals will be included in the study if they are aged 18-90 years old and have a history of PBC or if they are first-degree relatives of someone with PBC. Patients with PBC who have undergone a liver transplant are not excluded.
The primary outcome measure of the study is the mapping of genes that may make patients susceptible to adult chronic cholestatic liver diseases such as PBC.
The study will be completed in December 2025. About 1,500 people are expected to be included in the final analysis.
Find more information at the study page on Clinicaltrials.gov.
Understanding the Genetic Predisposition to the Development of Primary Biliary Cirrhosis is an observational study recruiting people with a history of PBC and their family members.
The trial will investigate whether or not there is a genetic factor in the development of PBC. Blood and stool samples will be taken from PBC patients and family members of PBC patients and analyzed. If a genetic component is discovered, it will add to current knowledge of how PBC and other adult chronic cholestatic liver diseases develop, as well as suggest new approaches for prevention, diagnosis, and treatment.
Individuals will be included in the study if they are aged 18-90 years old and have a history of PBC or if they are first-degree relatives of someone with PBC. Patients with PBC who have undergone a liver transplant are not excluded.
The primary outcome measure of the study is the mapping of genes that may make patients susceptible to adult chronic cholestatic liver diseases such as PBC.
The study will be completed in December 2025. About 1,500 people are expected to be included in the final analysis.
Find more information at the study page on Clinicaltrials.gov.
Understanding the Genetic Predisposition to the Development of Primary Biliary Cirrhosis is an observational study recruiting people with a history of PBC and their family members.
The trial will investigate whether or not there is a genetic factor in the development of PBC. Blood and stool samples will be taken from PBC patients and family members of PBC patients and analyzed. If a genetic component is discovered, it will add to current knowledge of how PBC and other adult chronic cholestatic liver diseases develop, as well as suggest new approaches for prevention, diagnosis, and treatment.
Individuals will be included in the study if they are aged 18-90 years old and have a history of PBC or if they are first-degree relatives of someone with PBC. Patients with PBC who have undergone a liver transplant are not excluded.
The primary outcome measure of the study is the mapping of genes that may make patients susceptible to adult chronic cholestatic liver diseases such as PBC.
The study will be completed in December 2025. About 1,500 people are expected to be included in the final analysis.
Find more information at the study page on Clinicaltrials.gov.
Latest on dupilumab for atopic dermatitis: The who and how
GENEVA – The novel interleukin-4 and IL-13 signaling blocker dupilumab displayed consistently strong efficacy across all patient subgroups in a new analysis from the landmark 52-week CHRONOS trial, Andrew Blauvelt, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“Dupilumab with concomitant topical corticosteroids improved signs and symptoms of atopic dermatitis, compared with placebo injections regardless of age, sex, BMI, or prior history of asthma, allergic rhinitis, or food allergies,” he said. Indeed, he quipped that the biologic proved to be “boring” in its broad effectiveness.

CHRONOS was a 14-country, 1-year, randomized, double-blind, placebo-controlled, phase 3 clinical trial in which 740 adults with moderate to very severe AD were assigned to mid-potency topical corticosteroids along with topical calcineurin inhibitors as needed in steroid-sensitive locations, then randomized 3:1:3 to either subcutaneous dupilumab (Dupixent) at 300 mg once weekly, placebo injections, or dupilumab at 300 mg every 2 weeks on top of the topical therapy.
Dupilumab, a fully human monoclonal antibody, was approved by the Food and Drug Administration in March 2017 at a loading dose of 600 mg, followed by 300 mg every 2 weeks for treatment of adults with moderate to severe AD. The biologic agent is soon expected to be approved across the European Union as well. CHRONOS was the first major trial designed to replicate how the biologic would likely be utilized in real-world clinical practice: that is, in conjunction with rather than as an alternative to topical therapy. Earlier pivotal phase 3 trials were required by regulatory agencies to pit dupilumab monotherapy against placebo.
CHRONOS participants were “a very tough crowd, so to speak,” the dermatologist recalled. Their median body surface area of involvement was a whopping 55%, 60% were men, the median baseline EASI (Eczema Area and Severity Index) score was 30, and fully half of patients had an IGA (Investigator’s Global Assessment) score of 4, indicative of severe disease. Plus, comorbid atopic/allergic diseases were common: half of subjects had a history of asthma, nearly half had a history of allergic rhinitis, and one-third had a history of food allergies.
Dr. Blauvelt previously presented the primary results of CHRONOS without the new subgroup analysis at the American Academy of Dermatology 2017 meeting. The primary results were subsequently published (Lancet. 2017 Jun 10;389[10086]:2287-303). To recap, the bottom line: 39% of dupilumab-treated patients achieved an IGA score of 0/1 – that is, clear or almost clear – coupled with at least a 2-point improvement from baseline at week 16, versus 12% of controls on topical steroids plus placebo injections. That’s a strikingly impressive performance, according to the dermatologist.
“An IGA of 0/1 in AD is a very high bar. It tends to correlated with an EASI 90,” Dr. Blauvelt observed.
Rates of EASI 75 – meaning at least a 75% improvement from baseline EASI scores – were achieved in 64% of patients on weekly dupilumab plus topical steroids, 69% with dupilumab every 2 weeks plus topicals, and 23% on topical steroids plus placebo. These efficacy results were essentially mirrored at week 52, with no significant changes in response rates from week 16 to week 52.
In another new finding from CHRONOS presented by Dr. Blauvelt at the EADV congress, itch also improved sharply across the board in dupilumab-treated patients, regardless of their baseline demographic and other characteristics, meaning there’s no point for clinicians to reserve the biologic for selected subgroups of patients with moderate or severe AD. From a mean baseline score of 7.6 on the 10-point peak pruritus numerical rating scale, at least a 3-point improvement was achieved in 56% of patients on biweekly dupilumab plus topical steroids, 43% on weekly dupilumab and topical steroids, and in 16% on topical steroids plus placebo injections.
Of note, 84% of patients in the two dupilumab arms remained in the CHRONOS trial through the full 52 weeks. That’s a high retention rate for a 1-year study involving a disease with a major adverse quality of life impact. In contrast, only about two-thirds of controls on topical therapy plus placebo injections stuck with the study for the duration.
The high retention rate may have had much to do with dupilumab’s generally favorable safely profile, as seen not only in CHRONOS but in earlier trials. No new safety concerns arose during the 52-week study. The dupilumab and control groups had similar rates of most side effects, with just a few exceptions: nonherpetic skin infections and worsening AD occurred more frequently in controls on topical steroids alone, while mild injection site reactions were twice as common in patients who got dupilumab than in those who received placebo injections.
Also, conjunctivitis occurred in 14% of patients on weekly dupilumab and 19% with biweekly dupilumab, compared with 8% of controls. “To me, conjunctivitis is the only significant side effect for this drug that we’ve seen thus far,” Dr. Blauvelt commented. The mechanism of the conjunctivitis is unknown. It’s clear, however, that the rate goes up with duration on therapy.
“Most of the cases have been mild to moderate and have not interfered with therapy. I’ve treated probably 80 patients with dupilumab, and I’ve had only 1 who had to stop due to her eyes,” the dermatologist recalled. The condition often responds to wetting eye drops, although topical corticosteroid eye drops are required sometimes, he added.

CAFE was a double-blind, randomized, placebo-controlled trial including 325 European patients with moderate or severe AD, all of whom were on a mid-potency topical corticosteroid with or without a topical calcineurin inhibitor. Two-thirds had previously been on cyclosporine with poor results; the drug was contraindicated in the others. Participants were randomized to subcutaneous injections of dupilumab at 300 mg weekly or biweekly or to placebo injections.
The primary outcome – an EASI-75 response at 16 weeks – was achieved in 59% of patients on weekly dupilumab, 63% of those on biweekly dupilumab, and 30% of controls on topical steroids plus placebo injections, according to Dr. De Bruin-Weller of the University Medical Center at Utrecht, the Netherlands.
A clinically meaningful 4-point or greater improvement from baseline in the Dermatology Life Quality Index occurred at 16 weeks in 79% and 88% of patients on dupilumab weekly or biweekly, respectively, compared with 44% of controls.
And among the three-quarters of CAFE participants reporting baseline moderate or severe pain or discomfort from AD, 55% of those on weekly dupilumab had none at all as assessed by the EQ-5D Pain/Discomfort Questionnaire at week 16, as did 64% on dupilumab every 2 weeks and 28% of controls on topical steroids.
At the prestigious joint EADV/AAD session of the European congress, Lawrence F. Eichenfield, MD, hailed dupilumab as evidence that “the systemic therapy revolution in atopic dermatitis is certainly happening,” with a plethora of additional agents targeting various key disease pathways now working their way through the developmental pipeline.
A key remaining question about dupilumab is, what about the safety and efficacy of the drug in the pediatric population, for whom the drug is not currently approved? Separate phase 3 randomized trials addressing this issue are now enrolling 6- to 11-year-olds and 12- to 17-year-olds.
Dr. Eichenfield is optimistic about the outcomes of these ongoing pediatric trials on the basis of an open-label, phase 2a, proof of concept study in 6- to 17-year-olds presented in a late-breaking research session at the 2017 annual meeting of the AAD. The pharmacokinetics were very similar to those seen in adults. Moreover, EASI scores improved by 32%-51% after just a single, weight-based injection of dupilumab and by up to 70% following four consecutive weekly injections.
“This is very, very exciting information. Whether the drug could actually be disease modifying is obviously a big question for us in pediatric dermatology,” observed Dr. Eichenfield, who is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego and professor of dermatology and pediatrics at the University of California, San Diego.
He highlighted two important safety considerations regarding dupilumab: patients on the biologic should not be given any live vaccines, and the same IL-4 and IL-13 cytokine pathways targeted by dupilumab in patients with AD also are key in other atopic and allergic diseases. Indeed, the biologic showed positive outcomes in a recent pivotal phase 3 clinical trial for patients with uncontrolled persistent asthma, and an application for an expanded indication for dupilumab in such patients is expected to be filed with the FDA later in 2017. Dupilumab is also in phase 3 for patients with nasal polyps and in phase 2 studies for eosinophilic esophagitis.
“One of the interesting things about dupilumab is that we in dermatology are going to be in a situation where we’re able to impact diseases other than the single disease that the medication has been approved for,” Dr. Eichenfield said.
However, until such time as dupilumab actually receives an expanded indication for asthma and the details of how best to use the biologic in AD patients with that comorbidity have been worked out, it’s important for dermatologists prescribing dupilumab in those dual-diagnosis patients to discuss with them the necessity of staying on their asthma medications even though their skin is much improved, they’re feeling good, and their asthma seems to be doing better. During the dupilumab clinical trials program for AD, a fatal asthma exacerbation occurred in a patient with comorbid asthma who stopped taking asthma medications, he noted.
The CHRONOS and CAFE trials were funded by Sanofi and Regeneron Pharmaceuticals. Dr. Blauvelt, Dr. De Bruin-Weller, and Dr. Eichenfield reported serving as consultants to and researchers for those pharmaceutical companies and numerous others.
GENEVA – The novel interleukin-4 and IL-13 signaling blocker dupilumab displayed consistently strong efficacy across all patient subgroups in a new analysis from the landmark 52-week CHRONOS trial, Andrew Blauvelt, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“Dupilumab with concomitant topical corticosteroids improved signs and symptoms of atopic dermatitis, compared with placebo injections regardless of age, sex, BMI, or prior history of asthma, allergic rhinitis, or food allergies,” he said. Indeed, he quipped that the biologic proved to be “boring” in its broad effectiveness.
CHRONOS was a 14-country, 1-year, randomized, double-blind, placebo-controlled, phase 3 clinical trial in which 740 adults with moderate to very severe AD were assigned to mid-potency topical corticosteroids along with topical calcineurin inhibitors as needed in steroid-sensitive locations, then randomized 3:1:3 to either subcutaneous dupilumab (Dupixent) at 300 mg once weekly, placebo injections, or dupilumab at 300 mg every 2 weeks on top of the topical therapy.
Dupilumab, a fully human monoclonal antibody, was approved by the Food and Drug Administration in March 2017 at a loading dose of 600 mg, followed by 300 mg every 2 weeks for treatment of adults with moderate to severe AD. The biologic agent is soon expected to be approved across the European Union as well. CHRONOS was the first major trial designed to replicate how the biologic would likely be utilized in real-world clinical practice: that is, in conjunction with rather than as an alternative to topical therapy. Earlier pivotal phase 3 trials were required by regulatory agencies to pit dupilumab monotherapy against placebo.
CHRONOS participants were “a very tough crowd, so to speak,” the dermatologist recalled. Their median body surface area of involvement was a whopping 55%, 60% were men, the median baseline EASI (Eczema Area and Severity Index) score was 30, and fully half of patients had an IGA (Investigator’s Global Assessment) score of 4, indicative of severe disease. Plus, comorbid atopic/allergic diseases were common: half of subjects had a history of asthma, nearly half had a history of allergic rhinitis, and one-third had a history of food allergies.
Dr. Blauvelt previously presented the primary results of CHRONOS without the new subgroup analysis at the American Academy of Dermatology 2017 meeting. The primary results were subsequently published (Lancet. 2017 Jun 10;389[10086]:2287-303). To recap, the bottom line: 39% of dupilumab-treated patients achieved an IGA score of 0/1 – that is, clear or almost clear – coupled with at least a 2-point improvement from baseline at week 16, versus 12% of controls on topical steroids plus placebo injections. That’s a strikingly impressive performance, according to the dermatologist.
“An IGA of 0/1 in AD is a very high bar. It tends to correlated with an EASI 90,” Dr. Blauvelt observed.
Rates of EASI 75 – meaning at least a 75% improvement from baseline EASI scores – were achieved in 64% of patients on weekly dupilumab plus topical steroids, 69% with dupilumab every 2 weeks plus topicals, and 23% on topical steroids plus placebo. These efficacy results were essentially mirrored at week 52, with no significant changes in response rates from week 16 to week 52.
In another new finding from CHRONOS presented by Dr. Blauvelt at the EADV congress, itch also improved sharply across the board in dupilumab-treated patients, regardless of their baseline demographic and other characteristics, meaning there’s no point for clinicians to reserve the biologic for selected subgroups of patients with moderate or severe AD. From a mean baseline score of 7.6 on the 10-point peak pruritus numerical rating scale, at least a 3-point improvement was achieved in 56% of patients on biweekly dupilumab plus topical steroids, 43% on weekly dupilumab and topical steroids, and in 16% on topical steroids plus placebo injections.
Of note, 84% of patients in the two dupilumab arms remained in the CHRONOS trial through the full 52 weeks. That’s a high retention rate for a 1-year study involving a disease with a major adverse quality of life impact. In contrast, only about two-thirds of controls on topical therapy plus placebo injections stuck with the study for the duration.
The high retention rate may have had much to do with dupilumab’s generally favorable safely profile, as seen not only in CHRONOS but in earlier trials. No new safety concerns arose during the 52-week study. The dupilumab and control groups had similar rates of most side effects, with just a few exceptions: nonherpetic skin infections and worsening AD occurred more frequently in controls on topical steroids alone, while mild injection site reactions were twice as common in patients who got dupilumab than in those who received placebo injections.
Also, conjunctivitis occurred in 14% of patients on weekly dupilumab and 19% with biweekly dupilumab, compared with 8% of controls. “To me, conjunctivitis is the only significant side effect for this drug that we’ve seen thus far,” Dr. Blauvelt commented. The mechanism of the conjunctivitis is unknown. It’s clear, however, that the rate goes up with duration on therapy.
“Most of the cases have been mild to moderate and have not interfered with therapy. I’ve treated probably 80 patients with dupilumab, and I’ve had only 1 who had to stop due to her eyes,” the dermatologist recalled. The condition often responds to wetting eye drops, although topical corticosteroid eye drops are required sometimes, he added.
CAFE was a double-blind, randomized, placebo-controlled trial including 325 European patients with moderate or severe AD, all of whom were on a mid-potency topical corticosteroid with or without a topical calcineurin inhibitor. Two-thirds had previously been on cyclosporine with poor results; the drug was contraindicated in the others. Participants were randomized to subcutaneous injections of dupilumab at 300 mg weekly or biweekly or to placebo injections.
The primary outcome – an EASI-75 response at 16 weeks – was achieved in 59% of patients on weekly dupilumab, 63% of those on biweekly dupilumab, and 30% of controls on topical steroids plus placebo injections, according to Dr. De Bruin-Weller of the University Medical Center at Utrecht, the Netherlands.
A clinically meaningful 4-point or greater improvement from baseline in the Dermatology Life Quality Index occurred at 16 weeks in 79% and 88% of patients on dupilumab weekly or biweekly, respectively, compared with 44% of controls.
And among the three-quarters of CAFE participants reporting baseline moderate or severe pain or discomfort from AD, 55% of those on weekly dupilumab had none at all as assessed by the EQ-5D Pain/Discomfort Questionnaire at week 16, as did 64% on dupilumab every 2 weeks and 28% of controls on topical steroids.
At the prestigious joint EADV/AAD session of the European congress, Lawrence F. Eichenfield, MD, hailed dupilumab as evidence that “the systemic therapy revolution in atopic dermatitis is certainly happening,” with a plethora of additional agents targeting various key disease pathways now working their way through the developmental pipeline.
A key remaining question about dupilumab is, what about the safety and efficacy of the drug in the pediatric population, for whom the drug is not currently approved? Separate phase 3 randomized trials addressing this issue are now enrolling 6- to 11-year-olds and 12- to 17-year-olds.
Dr. Eichenfield is optimistic about the outcomes of these ongoing pediatric trials on the basis of an open-label, phase 2a, proof of concept study in 6- to 17-year-olds presented in a late-breaking research session at the 2017 annual meeting of the AAD. The pharmacokinetics were very similar to those seen in adults. Moreover, EASI scores improved by 32%-51% after just a single, weight-based injection of dupilumab and by up to 70% following four consecutive weekly injections.
“This is very, very exciting information. Whether the drug could actually be disease modifying is obviously a big question for us in pediatric dermatology,” observed Dr. Eichenfield, who is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego and professor of dermatology and pediatrics at the University of California, San Diego.
He highlighted two important safety considerations regarding dupilumab: patients on the biologic should not be given any live vaccines, and the same IL-4 and IL-13 cytokine pathways targeted by dupilumab in patients with AD also are key in other atopic and allergic diseases. Indeed, the biologic showed positive outcomes in a recent pivotal phase 3 clinical trial for patients with uncontrolled persistent asthma, and an application for an expanded indication for dupilumab in such patients is expected to be filed with the FDA later in 2017. Dupilumab is also in phase 3 for patients with nasal polyps and in phase 2 studies for eosinophilic esophagitis.
“One of the interesting things about dupilumab is that we in dermatology are going to be in a situation where we’re able to impact diseases other than the single disease that the medication has been approved for,” Dr. Eichenfield said.
However, until such time as dupilumab actually receives an expanded indication for asthma and the details of how best to use the biologic in AD patients with that comorbidity have been worked out, it’s important for dermatologists prescribing dupilumab in those dual-diagnosis patients to discuss with them the necessity of staying on their asthma medications even though their skin is much improved, they’re feeling good, and their asthma seems to be doing better. During the dupilumab clinical trials program for AD, a fatal asthma exacerbation occurred in a patient with comorbid asthma who stopped taking asthma medications, he noted.
The CHRONOS and CAFE trials were funded by Sanofi and Regeneron Pharmaceuticals. Dr. Blauvelt, Dr. De Bruin-Weller, and Dr. Eichenfield reported serving as consultants to and researchers for those pharmaceutical companies and numerous others.
GENEVA – The novel interleukin-4 and IL-13 signaling blocker dupilumab displayed consistently strong efficacy across all patient subgroups in a new analysis from the landmark 52-week CHRONOS trial, Andrew Blauvelt, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“Dupilumab with concomitant topical corticosteroids improved signs and symptoms of atopic dermatitis, compared with placebo injections regardless of age, sex, BMI, or prior history of asthma, allergic rhinitis, or food allergies,” he said. Indeed, he quipped that the biologic proved to be “boring” in its broad effectiveness.
CHRONOS was a 14-country, 1-year, randomized, double-blind, placebo-controlled, phase 3 clinical trial in which 740 adults with moderate to very severe AD were assigned to mid-potency topical corticosteroids along with topical calcineurin inhibitors as needed in steroid-sensitive locations, then randomized 3:1:3 to either subcutaneous dupilumab (Dupixent) at 300 mg once weekly, placebo injections, or dupilumab at 300 mg every 2 weeks on top of the topical therapy.
Dupilumab, a fully human monoclonal antibody, was approved by the Food and Drug Administration in March 2017 at a loading dose of 600 mg, followed by 300 mg every 2 weeks for treatment of adults with moderate to severe AD. The biologic agent is soon expected to be approved across the European Union as well. CHRONOS was the first major trial designed to replicate how the biologic would likely be utilized in real-world clinical practice: that is, in conjunction with rather than as an alternative to topical therapy. Earlier pivotal phase 3 trials were required by regulatory agencies to pit dupilumab monotherapy against placebo.
CHRONOS participants were “a very tough crowd, so to speak,” the dermatologist recalled. Their median body surface area of involvement was a whopping 55%, 60% were men, the median baseline EASI (Eczema Area and Severity Index) score was 30, and fully half of patients had an IGA (Investigator’s Global Assessment) score of 4, indicative of severe disease. Plus, comorbid atopic/allergic diseases were common: half of subjects had a history of asthma, nearly half had a history of allergic rhinitis, and one-third had a history of food allergies.
Dr. Blauvelt previously presented the primary results of CHRONOS without the new subgroup analysis at the American Academy of Dermatology 2017 meeting. The primary results were subsequently published (Lancet. 2017 Jun 10;389[10086]:2287-303). To recap, the bottom line: 39% of dupilumab-treated patients achieved an IGA score of 0/1 – that is, clear or almost clear – coupled with at least a 2-point improvement from baseline at week 16, versus 12% of controls on topical steroids plus placebo injections. That’s a strikingly impressive performance, according to the dermatologist.
“An IGA of 0/1 in AD is a very high bar. It tends to correlated with an EASI 90,” Dr. Blauvelt observed.
Rates of EASI 75 – meaning at least a 75% improvement from baseline EASI scores – were achieved in 64% of patients on weekly dupilumab plus topical steroids, 69% with dupilumab every 2 weeks plus topicals, and 23% on topical steroids plus placebo. These efficacy results were essentially mirrored at week 52, with no significant changes in response rates from week 16 to week 52.
In another new finding from CHRONOS presented by Dr. Blauvelt at the EADV congress, itch also improved sharply across the board in dupilumab-treated patients, regardless of their baseline demographic and other characteristics, meaning there’s no point for clinicians to reserve the biologic for selected subgroups of patients with moderate or severe AD. From a mean baseline score of 7.6 on the 10-point peak pruritus numerical rating scale, at least a 3-point improvement was achieved in 56% of patients on biweekly dupilumab plus topical steroids, 43% on weekly dupilumab and topical steroids, and in 16% on topical steroids plus placebo injections.
Of note, 84% of patients in the two dupilumab arms remained in the CHRONOS trial through the full 52 weeks. That’s a high retention rate for a 1-year study involving a disease with a major adverse quality of life impact. In contrast, only about two-thirds of controls on topical therapy plus placebo injections stuck with the study for the duration.
The high retention rate may have had much to do with dupilumab’s generally favorable safely profile, as seen not only in CHRONOS but in earlier trials. No new safety concerns arose during the 52-week study. The dupilumab and control groups had similar rates of most side effects, with just a few exceptions: nonherpetic skin infections and worsening AD occurred more frequently in controls on topical steroids alone, while mild injection site reactions were twice as common in patients who got dupilumab than in those who received placebo injections.
Also, conjunctivitis occurred in 14% of patients on weekly dupilumab and 19% with biweekly dupilumab, compared with 8% of controls. “To me, conjunctivitis is the only significant side effect for this drug that we’ve seen thus far,” Dr. Blauvelt commented. The mechanism of the conjunctivitis is unknown. It’s clear, however, that the rate goes up with duration on therapy.
“Most of the cases have been mild to moderate and have not interfered with therapy. I’ve treated probably 80 patients with dupilumab, and I’ve had only 1 who had to stop due to her eyes,” the dermatologist recalled. The condition often responds to wetting eye drops, although topical corticosteroid eye drops are required sometimes, he added.
CAFE was a double-blind, randomized, placebo-controlled trial including 325 European patients with moderate or severe AD, all of whom were on a mid-potency topical corticosteroid with or without a topical calcineurin inhibitor. Two-thirds had previously been on cyclosporine with poor results; the drug was contraindicated in the others. Participants were randomized to subcutaneous injections of dupilumab at 300 mg weekly or biweekly or to placebo injections.
The primary outcome – an EASI-75 response at 16 weeks – was achieved in 59% of patients on weekly dupilumab, 63% of those on biweekly dupilumab, and 30% of controls on topical steroids plus placebo injections, according to Dr. De Bruin-Weller of the University Medical Center at Utrecht, the Netherlands.
A clinically meaningful 4-point or greater improvement from baseline in the Dermatology Life Quality Index occurred at 16 weeks in 79% and 88% of patients on dupilumab weekly or biweekly, respectively, compared with 44% of controls.
And among the three-quarters of CAFE participants reporting baseline moderate or severe pain or discomfort from AD, 55% of those on weekly dupilumab had none at all as assessed by the EQ-5D Pain/Discomfort Questionnaire at week 16, as did 64% on dupilumab every 2 weeks and 28% of controls on topical steroids.
At the prestigious joint EADV/AAD session of the European congress, Lawrence F. Eichenfield, MD, hailed dupilumab as evidence that “the systemic therapy revolution in atopic dermatitis is certainly happening,” with a plethora of additional agents targeting various key disease pathways now working their way through the developmental pipeline.
A key remaining question about dupilumab is, what about the safety and efficacy of the drug in the pediatric population, for whom the drug is not currently approved? Separate phase 3 randomized trials addressing this issue are now enrolling 6- to 11-year-olds and 12- to 17-year-olds.
Dr. Eichenfield is optimistic about the outcomes of these ongoing pediatric trials on the basis of an open-label, phase 2a, proof of concept study in 6- to 17-year-olds presented in a late-breaking research session at the 2017 annual meeting of the AAD. The pharmacokinetics were very similar to those seen in adults. Moreover, EASI scores improved by 32%-51% after just a single, weight-based injection of dupilumab and by up to 70% following four consecutive weekly injections.
“This is very, very exciting information. Whether the drug could actually be disease modifying is obviously a big question for us in pediatric dermatology,” observed Dr. Eichenfield, who is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego and professor of dermatology and pediatrics at the University of California, San Diego.
He highlighted two important safety considerations regarding dupilumab: patients on the biologic should not be given any live vaccines, and the same IL-4 and IL-13 cytokine pathways targeted by dupilumab in patients with AD also are key in other atopic and allergic diseases. Indeed, the biologic showed positive outcomes in a recent pivotal phase 3 clinical trial for patients with uncontrolled persistent asthma, and an application for an expanded indication for dupilumab in such patients is expected to be filed with the FDA later in 2017. Dupilumab is also in phase 3 for patients with nasal polyps and in phase 2 studies for eosinophilic esophagitis.
“One of the interesting things about dupilumab is that we in dermatology are going to be in a situation where we’re able to impact diseases other than the single disease that the medication has been approved for,” Dr. Eichenfield said.
However, until such time as dupilumab actually receives an expanded indication for asthma and the details of how best to use the biologic in AD patients with that comorbidity have been worked out, it’s important for dermatologists prescribing dupilumab in those dual-diagnosis patients to discuss with them the necessity of staying on their asthma medications even though their skin is much improved, they’re feeling good, and their asthma seems to be doing better. During the dupilumab clinical trials program for AD, a fatal asthma exacerbation occurred in a patient with comorbid asthma who stopped taking asthma medications, he noted.
The CHRONOS and CAFE trials were funded by Sanofi and Regeneron Pharmaceuticals. Dr. Blauvelt, Dr. De Bruin-Weller, and Dr. Eichenfield reported serving as consultants to and researchers for those pharmaceutical companies and numerous others.
EXPERT ANALYSIS FROM THE EADV CONGRESS
Opioid-Related Hospitalizations Rising Faster Among Women
The crises of opioids and heroin abuse have skyrocketed, but in just 9 years, hospitalizations for women jumped 75%—much higher than the 55% among men and enough to bring hospitalization rates for the 2 sexes neck and neck. According to a report from the Agency for Healthcare Research and Quality, men and women were hospitalized at virtually the same rate in 2014: about 225 hospitalizations per 100,000 people.
The report, which covered data from 44 states and the District of Columbia, highlighted the broad variations in how the epidemic is hitting the U.S. For instance, opioid hospitalization rates among women were highest in West Virginia, Maryland, and Massachusetts (each state reporting rates of more than 350 hospitalizations per 100,000 people). For men, the highest rates were in the District of Columbia, New York, and Maryland (440+ per 100,000). Iowa, Nebraska, Texas, and Wyoming consistently ranked lowest for opioid-related inpatient stays, while Massachusetts consistently had the highest rates.
Men were more likely than women to make opioid-related emergency department (ED) visits, although ED visit rates rose sharply for both sexes between 2005 and 2014. Arkansas and Iowa consistently ranked lowest for opioid-related emergency department visits; Maryland was the highest.
In all states reporting on opioid-related ED visits, the rate was highest among adults aged 25 to 44 years.
The crises of opioids and heroin abuse have skyrocketed, but in just 9 years, hospitalizations for women jumped 75%—much higher than the 55% among men and enough to bring hospitalization rates for the 2 sexes neck and neck. According to a report from the Agency for Healthcare Research and Quality, men and women were hospitalized at virtually the same rate in 2014: about 225 hospitalizations per 100,000 people.
The report, which covered data from 44 states and the District of Columbia, highlighted the broad variations in how the epidemic is hitting the U.S. For instance, opioid hospitalization rates among women were highest in West Virginia, Maryland, and Massachusetts (each state reporting rates of more than 350 hospitalizations per 100,000 people). For men, the highest rates were in the District of Columbia, New York, and Maryland (440+ per 100,000). Iowa, Nebraska, Texas, and Wyoming consistently ranked lowest for opioid-related inpatient stays, while Massachusetts consistently had the highest rates.
Men were more likely than women to make opioid-related emergency department (ED) visits, although ED visit rates rose sharply for both sexes between 2005 and 2014. Arkansas and Iowa consistently ranked lowest for opioid-related emergency department visits; Maryland was the highest.
In all states reporting on opioid-related ED visits, the rate was highest among adults aged 25 to 44 years.
The crises of opioids and heroin abuse have skyrocketed, but in just 9 years, hospitalizations for women jumped 75%—much higher than the 55% among men and enough to bring hospitalization rates for the 2 sexes neck and neck. According to a report from the Agency for Healthcare Research and Quality, men and women were hospitalized at virtually the same rate in 2014: about 225 hospitalizations per 100,000 people.
The report, which covered data from 44 states and the District of Columbia, highlighted the broad variations in how the epidemic is hitting the U.S. For instance, opioid hospitalization rates among women were highest in West Virginia, Maryland, and Massachusetts (each state reporting rates of more than 350 hospitalizations per 100,000 people). For men, the highest rates were in the District of Columbia, New York, and Maryland (440+ per 100,000). Iowa, Nebraska, Texas, and Wyoming consistently ranked lowest for opioid-related inpatient stays, while Massachusetts consistently had the highest rates.
Men were more likely than women to make opioid-related emergency department (ED) visits, although ED visit rates rose sharply for both sexes between 2005 and 2014. Arkansas and Iowa consistently ranked lowest for opioid-related emergency department visits; Maryland was the highest.
In all states reporting on opioid-related ED visits, the rate was highest among adults aged 25 to 44 years.
Team creates guidelines on CAR T-cell-related toxicity

Researchers say they have created guidelines for managing the unique toxicities associated with chimeric antigen receptor (CAR) T-cell therapy.
The guidelines focus on cytokine release syndrome (CRS); neurological toxicity, which the researchers have dubbed “CAR-T-cell-related encephalopathy syndrome (CRES);” and adverse effects related to these syndromes.
“The toxicities are unique, and every member of the care team needs to be trained to recognize them and act accordingly,” said Sattva Neelapu, MD, of University of Texas MD Anderson Cancer Center in Houston.
Dr Neelapu and his colleagues described the toxicities and related recommendations in Nature Reviews Clinical Oncology.
The team’s guidelines include supportive-care considerations for patients receiving CAR T‑cell therapy. For example, they recommend:
- Baseline brain MRI to rule out central nervous system disease
- Cardiac monitoring starting on the day of CAR T‑cell infusion
- Assessing a patient’s vital signs every 4 hours after CAR T-cell infusion
- Assessing and grading CRS at least twice daily and whenever the patient’s status changes
- Assessing and grading CRES at least every 8 hours.
CRS
One section of the guidelines is dedicated to CRS, with subsections on pathophysiology, precautions and supportive care, the use of corticosteroids and IL‑6/IL‑6R antagonists, and grading CRS.
The researchers noted that CRS typically manifests with constitutional symptoms, such as fever, malaise, anorexia, and myalgias. However, CRS can affect any organ system in the body.
The team recommends managing CRS according to grade. For example, patients with grade 1 CRS should typically receive supportive care. However, physicians should consider giving tocilizumab or siltuximab to grade 1 patients who have a refractory fever lasting more than 3 days.
The researchers also noted that CRS can evolve into fulminant hemophagocytic lymphohistiocytosis (HLH), also known as macrophage-activation syndrome (MAS).
The team said HLH/MAS encompasses a group of severe immunological disorders characterized by hyperactivation of macrophages and lymphocytes, proinflammatory cytokine production, lymphohistiocytic tissue infiltration, and immune-mediated multi-organ failure.
The guidelines include diagnostic criteria for CAR T‑cell-related HLH/MAS and recommendations for managing the condition.
CRES
One section of the guidelines is dedicated to the grading and treatment of CRES, which typically manifests as a toxic encephalopathy.
The researchers said the earliest signs of CRES are diminished attention, language disturbance, and impaired handwriting. Other symptoms include confusion, disorientation, agitation, aphasia, somnolence, and tremors.
Patients with severe CRES (grade >2) may experience seizures, motor weakness, incontinence, mental obtundation, increased intracranial pressure, papilledema, and cerebral edema.
Therefore, the guidelines include recommendations for the management of status epilepticus and raised intracranial pressure after CAR T‑cell therapy.
The researchers also devised an algorithm, known as CARTOX-10, for identifying neurotoxicity. (An existing general method didn’t effectively quantify the neurological effects caused by CAR T-cell therapies.)
CARTOX-10 is a 10-point test in which patients are asked to do the following:
- Name the current month (1 point) and year (1 point)
- Name the city (1 point) and hospital they are in (1 point)
- Name the president/prime minister of their home country (1 point)
- Name 3 nearby objects (3 points)
- Write a standard sentence (1 point)
- Count backward from 100 by tens (1 point).
A perfect score indicates normal cognitive function. A patient has mild to severe impairment depending on the number of questions or activities missed.
Dr Neelapu and his colleagues believe their recommendations will be applicable to other types of cell-based immunotherapy as well, including CAR natural killer cells, T-cell receptor engineered T cells, and combination drugs that use an antibody to connect T cells to targets on cancer cells.
Researchers involved in this work have received funding from companies developing/marketing CAR T-cell therapies. ![]()
Researchers say they have created guidelines for managing the unique toxicities associated with chimeric antigen receptor (CAR) T-cell therapy.
The guidelines focus on cytokine release syndrome (CRS); neurological toxicity, which the researchers have dubbed “CAR-T-cell-related encephalopathy syndrome (CRES);” and adverse effects related to these syndromes.
“The toxicities are unique, and every member of the care team needs to be trained to recognize them and act accordingly,” said Sattva Neelapu, MD, of University of Texas MD Anderson Cancer Center in Houston.
Dr Neelapu and his colleagues described the toxicities and related recommendations in Nature Reviews Clinical Oncology.
The team’s guidelines include supportive-care considerations for patients receiving CAR T‑cell therapy. For example, they recommend:
- Baseline brain MRI to rule out central nervous system disease
- Cardiac monitoring starting on the day of CAR T‑cell infusion
- Assessing a patient’s vital signs every 4 hours after CAR T-cell infusion
- Assessing and grading CRS at least twice daily and whenever the patient’s status changes
- Assessing and grading CRES at least every 8 hours.
CRS
One section of the guidelines is dedicated to CRS, with subsections on pathophysiology, precautions and supportive care, the use of corticosteroids and IL‑6/IL‑6R antagonists, and grading CRS.
The researchers noted that CRS typically manifests with constitutional symptoms, such as fever, malaise, anorexia, and myalgias. However, CRS can affect any organ system in the body.
The team recommends managing CRS according to grade. For example, patients with grade 1 CRS should typically receive supportive care. However, physicians should consider giving tocilizumab or siltuximab to grade 1 patients who have a refractory fever lasting more than 3 days.
The researchers also noted that CRS can evolve into fulminant hemophagocytic lymphohistiocytosis (HLH), also known as macrophage-activation syndrome (MAS).
The team said HLH/MAS encompasses a group of severe immunological disorders characterized by hyperactivation of macrophages and lymphocytes, proinflammatory cytokine production, lymphohistiocytic tissue infiltration, and immune-mediated multi-organ failure.
The guidelines include diagnostic criteria for CAR T‑cell-related HLH/MAS and recommendations for managing the condition.
CRES
One section of the guidelines is dedicated to the grading and treatment of CRES, which typically manifests as a toxic encephalopathy.
The researchers said the earliest signs of CRES are diminished attention, language disturbance, and impaired handwriting. Other symptoms include confusion, disorientation, agitation, aphasia, somnolence, and tremors.
Patients with severe CRES (grade >2) may experience seizures, motor weakness, incontinence, mental obtundation, increased intracranial pressure, papilledema, and cerebral edema.
Therefore, the guidelines include recommendations for the management of status epilepticus and raised intracranial pressure after CAR T‑cell therapy.
The researchers also devised an algorithm, known as CARTOX-10, for identifying neurotoxicity. (An existing general method didn’t effectively quantify the neurological effects caused by CAR T-cell therapies.)
CARTOX-10 is a 10-point test in which patients are asked to do the following:
- Name the current month (1 point) and year (1 point)
- Name the city (1 point) and hospital they are in (1 point)
- Name the president/prime minister of their home country (1 point)
- Name 3 nearby objects (3 points)
- Write a standard sentence (1 point)
- Count backward from 100 by tens (1 point).
A perfect score indicates normal cognitive function. A patient has mild to severe impairment depending on the number of questions or activities missed.
Dr Neelapu and his colleagues believe their recommendations will be applicable to other types of cell-based immunotherapy as well, including CAR natural killer cells, T-cell receptor engineered T cells, and combination drugs that use an antibody to connect T cells to targets on cancer cells.
Researchers involved in this work have received funding from companies developing/marketing CAR T-cell therapies. ![]()
Researchers say they have created guidelines for managing the unique toxicities associated with chimeric antigen receptor (CAR) T-cell therapy.
The guidelines focus on cytokine release syndrome (CRS); neurological toxicity, which the researchers have dubbed “CAR-T-cell-related encephalopathy syndrome (CRES);” and adverse effects related to these syndromes.
“The toxicities are unique, and every member of the care team needs to be trained to recognize them and act accordingly,” said Sattva Neelapu, MD, of University of Texas MD Anderson Cancer Center in Houston.
Dr Neelapu and his colleagues described the toxicities and related recommendations in Nature Reviews Clinical Oncology.
The team’s guidelines include supportive-care considerations for patients receiving CAR T‑cell therapy. For example, they recommend:
- Baseline brain MRI to rule out central nervous system disease
- Cardiac monitoring starting on the day of CAR T‑cell infusion
- Assessing a patient’s vital signs every 4 hours after CAR T-cell infusion
- Assessing and grading CRS at least twice daily and whenever the patient’s status changes
- Assessing and grading CRES at least every 8 hours.
CRS
One section of the guidelines is dedicated to CRS, with subsections on pathophysiology, precautions and supportive care, the use of corticosteroids and IL‑6/IL‑6R antagonists, and grading CRS.
The researchers noted that CRS typically manifests with constitutional symptoms, such as fever, malaise, anorexia, and myalgias. However, CRS can affect any organ system in the body.
The team recommends managing CRS according to grade. For example, patients with grade 1 CRS should typically receive supportive care. However, physicians should consider giving tocilizumab or siltuximab to grade 1 patients who have a refractory fever lasting more than 3 days.
The researchers also noted that CRS can evolve into fulminant hemophagocytic lymphohistiocytosis (HLH), also known as macrophage-activation syndrome (MAS).
The team said HLH/MAS encompasses a group of severe immunological disorders characterized by hyperactivation of macrophages and lymphocytes, proinflammatory cytokine production, lymphohistiocytic tissue infiltration, and immune-mediated multi-organ failure.
The guidelines include diagnostic criteria for CAR T‑cell-related HLH/MAS and recommendations for managing the condition.
CRES
One section of the guidelines is dedicated to the grading and treatment of CRES, which typically manifests as a toxic encephalopathy.
The researchers said the earliest signs of CRES are diminished attention, language disturbance, and impaired handwriting. Other symptoms include confusion, disorientation, agitation, aphasia, somnolence, and tremors.
Patients with severe CRES (grade >2) may experience seizures, motor weakness, incontinence, mental obtundation, increased intracranial pressure, papilledema, and cerebral edema.
Therefore, the guidelines include recommendations for the management of status epilepticus and raised intracranial pressure after CAR T‑cell therapy.
The researchers also devised an algorithm, known as CARTOX-10, for identifying neurotoxicity. (An existing general method didn’t effectively quantify the neurological effects caused by CAR T-cell therapies.)
CARTOX-10 is a 10-point test in which patients are asked to do the following:
- Name the current month (1 point) and year (1 point)
- Name the city (1 point) and hospital they are in (1 point)
- Name the president/prime minister of their home country (1 point)
- Name 3 nearby objects (3 points)
- Write a standard sentence (1 point)
- Count backward from 100 by tens (1 point).
A perfect score indicates normal cognitive function. A patient has mild to severe impairment depending on the number of questions or activities missed.
Dr Neelapu and his colleagues believe their recommendations will be applicable to other types of cell-based immunotherapy as well, including CAR natural killer cells, T-cell receptor engineered T cells, and combination drugs that use an antibody to connect T cells to targets on cancer cells.
Researchers involved in this work have received funding from companies developing/marketing CAR T-cell therapies. ![]()
Antibody shows early promise in AML/MDS trial

MADRID—Interim results of a phase 1 study suggest flotetuzumab, a CD123 and CD3 bispecific antibody, may be a feasible treatment option for relapsed or refractory acute myeloid leukemia (AML) or intermediate/high-risk myelodysplastic syndromes (MDS).
Researchers said flotetuzumab demonstrated acceptable tolerability in the dose-escalation portion of the study, with infusion-related reactions (IRRs) and cytokine release syndrome (CRS) being the most common adverse events (AEs).
In addition, flotetuzumab exhibited anti-leukemic activity in 8 of 14 response-evaluable patients, with 6 patients achieving a response.
Norbert Vey, MD, of Institut Paoli-Calmettes in Marseille, France, presented these results at the ESMO 2017 Congress (abstract 995O*). The study is sponsored by MacroGenics, Inc., the company developing flotetuzumab.
Flotetuzumab (MGD006) recognizes CD123 and CD3. The primary mechanism of flotetuzumab is thought to be its ability to redirect T cells to kill CD123-expressing cells. To achieve this, the molecule combines a portion of an antibody recognizing CD3 (an activating molecule expressed by T cells) with an arm that recognizes CD123 on the target cancer cells.
In this ongoing phase 1 study of flotetuzumab, researchers have enrolled 47 patients with a median age of 64 (range, 29-84). About 89% of these patients had AML (n=42), and the rest (n=5) had MDS.
Twenty-four percent had relapsed AML (n=10), 55% had refractory AML (n=23), and 21% had failed treatment with hypomethylating agents (n=9). One patient had intermediate-1-risk MDS, 2 had intermediate-2-risk, and 2 had high-risk MDS.
Treatment
The study began with single patients receiving flotetuzumab at escalating doses—3 ng/kg/day, 10 ng/kg/day, 30 ng/kg/day, and 100 ng/kg/day.
Then, patients received a range of doses on 2 different schedules for cycle 1. One group received treatment 7 days a week. The other had a 4-days-on/3-days-off schedule.
All patients received a lead-in dose during the first week of cycle 1. They received 30 ng/kg/day for 3 days, then 100 ng/kg/day for 4 days.
For the rest of cycle 1, patients in the 4 days/3 days group received doses of 500 ng/kg, 700 ng/kg, 900 ng/kg, or 1000 ng/kg. Patients in the daily dosing group received doses of 300 ng/kg, 500 ng/kg, 700 ng/kg, 900 ng/kg, or 1000 ng/kg.
For cycle 2 and beyond, all patients were on the 4-days-on/3-days-off schedule.
Safety
The maximum tolerated dose and schedule was 500 ng/kg/day for 7 days.
Dose-limiting toxicities occurring at the 700 ng/kg/day dose included grade 2 IRRs/CRS in 2 patients and grade 3 myalgia in 1 patient. There was 1 drug-related central nervous system AE that led to treatment discontinuation.
IRRs/CRS occurred in 77% of patients, with 13% of patients having grade 3 events and 8.5% of patients discontinuing treatment due to IRRs/CRS.
The researchers said they found ways to decrease the incidence and severity of CRS. One is early intervention with tocilizumab. The other is a 2-step lead-in dose during week 1. So patients first receive 30 ng/kg, then 100 ng/kg, and then their target dose.
Other grade 3 AEs occurring in this trial include febrile neutropenia (11%), anemia (11%), and decreases in platelets (13%), white blood cells (11%), and lymphocytes (13%).
Efficacy
The researchers said they observed encouraging anti-leukemic activity in patients treated at 500 ng/kg/day or greater.
As of the data cut-off, 14 patients treated at this dose were evaluable for response. Eight (57%) patients had anti-leukemic activity, with 6 (43%) of these patients experiencing an objective response.
One patient achieved a complete response (CR), 2 had a CR with incomplete count recovery, and 1 had a molecular CR.
In most responders, anti-leukemic activity was observed after a single cycle of therapy.
MacroGenics is currently enrolling patients in dose-expansion cohorts. The company plans to present updated results from this trial at another scientific conference later this year. ![]()
*Slides from this presentation are available on the MacroGenics website at http://ir.macrogenics.com/events.cfm.
MADRID—Interim results of a phase 1 study suggest flotetuzumab, a CD123 and CD3 bispecific antibody, may be a feasible treatment option for relapsed or refractory acute myeloid leukemia (AML) or intermediate/high-risk myelodysplastic syndromes (MDS).
Researchers said flotetuzumab demonstrated acceptable tolerability in the dose-escalation portion of the study, with infusion-related reactions (IRRs) and cytokine release syndrome (CRS) being the most common adverse events (AEs).
In addition, flotetuzumab exhibited anti-leukemic activity in 8 of 14 response-evaluable patients, with 6 patients achieving a response.
Norbert Vey, MD, of Institut Paoli-Calmettes in Marseille, France, presented these results at the ESMO 2017 Congress (abstract 995O*). The study is sponsored by MacroGenics, Inc., the company developing flotetuzumab.
Flotetuzumab (MGD006) recognizes CD123 and CD3. The primary mechanism of flotetuzumab is thought to be its ability to redirect T cells to kill CD123-expressing cells. To achieve this, the molecule combines a portion of an antibody recognizing CD3 (an activating molecule expressed by T cells) with an arm that recognizes CD123 on the target cancer cells.
In this ongoing phase 1 study of flotetuzumab, researchers have enrolled 47 patients with a median age of 64 (range, 29-84). About 89% of these patients had AML (n=42), and the rest (n=5) had MDS.
Twenty-four percent had relapsed AML (n=10), 55% had refractory AML (n=23), and 21% had failed treatment with hypomethylating agents (n=9). One patient had intermediate-1-risk MDS, 2 had intermediate-2-risk, and 2 had high-risk MDS.
Treatment
The study began with single patients receiving flotetuzumab at escalating doses—3 ng/kg/day, 10 ng/kg/day, 30 ng/kg/day, and 100 ng/kg/day.
Then, patients received a range of doses on 2 different schedules for cycle 1. One group received treatment 7 days a week. The other had a 4-days-on/3-days-off schedule.
All patients received a lead-in dose during the first week of cycle 1. They received 30 ng/kg/day for 3 days, then 100 ng/kg/day for 4 days.
For the rest of cycle 1, patients in the 4 days/3 days group received doses of 500 ng/kg, 700 ng/kg, 900 ng/kg, or 1000 ng/kg. Patients in the daily dosing group received doses of 300 ng/kg, 500 ng/kg, 700 ng/kg, 900 ng/kg, or 1000 ng/kg.
For cycle 2 and beyond, all patients were on the 4-days-on/3-days-off schedule.
Safety
The maximum tolerated dose and schedule was 500 ng/kg/day for 7 days.
Dose-limiting toxicities occurring at the 700 ng/kg/day dose included grade 2 IRRs/CRS in 2 patients and grade 3 myalgia in 1 patient. There was 1 drug-related central nervous system AE that led to treatment discontinuation.
IRRs/CRS occurred in 77% of patients, with 13% of patients having grade 3 events and 8.5% of patients discontinuing treatment due to IRRs/CRS.
The researchers said they found ways to decrease the incidence and severity of CRS. One is early intervention with tocilizumab. The other is a 2-step lead-in dose during week 1. So patients first receive 30 ng/kg, then 100 ng/kg, and then their target dose.
Other grade 3 AEs occurring in this trial include febrile neutropenia (11%), anemia (11%), and decreases in platelets (13%), white blood cells (11%), and lymphocytes (13%).
Efficacy
The researchers said they observed encouraging anti-leukemic activity in patients treated at 500 ng/kg/day or greater.
As of the data cut-off, 14 patients treated at this dose were evaluable for response. Eight (57%) patients had anti-leukemic activity, with 6 (43%) of these patients experiencing an objective response.
One patient achieved a complete response (CR), 2 had a CR with incomplete count recovery, and 1 had a molecular CR.
In most responders, anti-leukemic activity was observed after a single cycle of therapy.
MacroGenics is currently enrolling patients in dose-expansion cohorts. The company plans to present updated results from this trial at another scientific conference later this year. ![]()
*Slides from this presentation are available on the MacroGenics website at http://ir.macrogenics.com/events.cfm.
MADRID—Interim results of a phase 1 study suggest flotetuzumab, a CD123 and CD3 bispecific antibody, may be a feasible treatment option for relapsed or refractory acute myeloid leukemia (AML) or intermediate/high-risk myelodysplastic syndromes (MDS).
Researchers said flotetuzumab demonstrated acceptable tolerability in the dose-escalation portion of the study, with infusion-related reactions (IRRs) and cytokine release syndrome (CRS) being the most common adverse events (AEs).
In addition, flotetuzumab exhibited anti-leukemic activity in 8 of 14 response-evaluable patients, with 6 patients achieving a response.
Norbert Vey, MD, of Institut Paoli-Calmettes in Marseille, France, presented these results at the ESMO 2017 Congress (abstract 995O*). The study is sponsored by MacroGenics, Inc., the company developing flotetuzumab.
Flotetuzumab (MGD006) recognizes CD123 and CD3. The primary mechanism of flotetuzumab is thought to be its ability to redirect T cells to kill CD123-expressing cells. To achieve this, the molecule combines a portion of an antibody recognizing CD3 (an activating molecule expressed by T cells) with an arm that recognizes CD123 on the target cancer cells.
In this ongoing phase 1 study of flotetuzumab, researchers have enrolled 47 patients with a median age of 64 (range, 29-84). About 89% of these patients had AML (n=42), and the rest (n=5) had MDS.
Twenty-four percent had relapsed AML (n=10), 55% had refractory AML (n=23), and 21% had failed treatment with hypomethylating agents (n=9). One patient had intermediate-1-risk MDS, 2 had intermediate-2-risk, and 2 had high-risk MDS.
Treatment
The study began with single patients receiving flotetuzumab at escalating doses—3 ng/kg/day, 10 ng/kg/day, 30 ng/kg/day, and 100 ng/kg/day.
Then, patients received a range of doses on 2 different schedules for cycle 1. One group received treatment 7 days a week. The other had a 4-days-on/3-days-off schedule.
All patients received a lead-in dose during the first week of cycle 1. They received 30 ng/kg/day for 3 days, then 100 ng/kg/day for 4 days.
For the rest of cycle 1, patients in the 4 days/3 days group received doses of 500 ng/kg, 700 ng/kg, 900 ng/kg, or 1000 ng/kg. Patients in the daily dosing group received doses of 300 ng/kg, 500 ng/kg, 700 ng/kg, 900 ng/kg, or 1000 ng/kg.
For cycle 2 and beyond, all patients were on the 4-days-on/3-days-off schedule.
Safety
The maximum tolerated dose and schedule was 500 ng/kg/day for 7 days.
Dose-limiting toxicities occurring at the 700 ng/kg/day dose included grade 2 IRRs/CRS in 2 patients and grade 3 myalgia in 1 patient. There was 1 drug-related central nervous system AE that led to treatment discontinuation.
IRRs/CRS occurred in 77% of patients, with 13% of patients having grade 3 events and 8.5% of patients discontinuing treatment due to IRRs/CRS.
The researchers said they found ways to decrease the incidence and severity of CRS. One is early intervention with tocilizumab. The other is a 2-step lead-in dose during week 1. So patients first receive 30 ng/kg, then 100 ng/kg, and then their target dose.
Other grade 3 AEs occurring in this trial include febrile neutropenia (11%), anemia (11%), and decreases in platelets (13%), white blood cells (11%), and lymphocytes (13%).
Efficacy
The researchers said they observed encouraging anti-leukemic activity in patients treated at 500 ng/kg/day or greater.
As of the data cut-off, 14 patients treated at this dose were evaluable for response. Eight (57%) patients had anti-leukemic activity, with 6 (43%) of these patients experiencing an objective response.
One patient achieved a complete response (CR), 2 had a CR with incomplete count recovery, and 1 had a molecular CR.
In most responders, anti-leukemic activity was observed after a single cycle of therapy.
MacroGenics is currently enrolling patients in dose-expansion cohorts. The company plans to present updated results from this trial at another scientific conference later this year. ![]()
*Slides from this presentation are available on the MacroGenics website at http://ir.macrogenics.com/events.cfm.