User login
Novel models predict Parkinson’s disease motor progression
An ensemble of prediction models developed using genetic data and both baseline molecular and clinical variables from patients with Parkinson’s disease, as well as healthy controls, confirmed established predictors of the disease and identified new ones in a longitudinal cohort study.
Furthermore, the models were shown via simulated, randomized, placebo-controlled trials to have utility for clinical trial design and evaluation, as well as for clinical disease monitoring and treatment, Jeanne C. Latourelle, DSc, of GNS Healthcare, Cambridge, Mass., and her colleagues reported in Lancet Neurology.
“Understanding the causal and physiological factors that contribute to this variability in the evolution of symptoms of Parkinson’s disease is, therefore, a high priority area of Parkinson’s disease research,” they wrote (Lancet Neurol. 2017 Sep 25. doi: 10.1016/S1474-4422[17]30328-9).
The investigators developed the models by applying a Bayesian, machine-learning, multivariate predictive inference platform – known as Reverse Engineering and Forward Simulation – to data from the Parkinson’s Progression Markers Initiative (PPMI) study; they used these models to predict the annual rate of change in combined scores from the Movement Disorder Society–Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) parts II and III. The investigators tested the overall explanatory power of the models using the coefficient of determination (R2); their models also replicated novel findings from an independent clinical cohort from the Longitudinal and Biomarker Study in Parkinson’s disease (LABS-PD).
In 117 healthy age- and sex-matched controls and 312 patients with early-stage Parkinson’s disease from the PPMI study, the model ensemble showed strong performance (fivefold cross-validated Pearson R2, 41%), the investigators said.
In the 317-patient LABS-PD validation cohort, the performance of the models was reduced, but still statistically significant (R2, 9%).
Individual predictive features – including significant replication of higher baseline motor score on MDS-UPDRS, male sex, increased age, and novel Parkinson’s disease–specific epistatic interaction, which all were indicative of faster motor progression in the disease – were confirmed in the LABS-PD cohort, they said. The most useful predictive marker of motor progression was genetic variation (R2, 2.9%), the investigators said, and the cerebrospinal fluid biomarkers at baseline also significantly affected prediction of motor progression, although more modestly (R2, 0.3%).
In 5,000 trial simulations, the incorporation of predicted rates of motor progression into the final models of treatment effect reduced the variability in study outcome, which allowed for the detection of significant differences in sample sizes up to 20% smaller than in trials of drug-naive patients.
The investigators noted that the Bayesian model inference using Reverse Engineering and Forward Simulation was particularly good at predicting Parkinson’s motor progression in early disease stages, which has “immediate relevance toward enabling more effective trial recruitment and clinical disease management.”
This study was supported by grants from the Michael J. Fox Foundation for Parkinson’s Research and the National Institute of Neurological Disorders and Stroke. PPMI is funded by the Michael J. Fox Foundation for Parkinson’s Research, and it also has many industry funding partners. Dr. Latourelle and several of her coauthors are or were employees of GNS Healthcare.
The study by Latourelle and her colleagues adds substantial value to Parkinson’s disease research not only because of the findings but also because it introduces the topic of using multimodal data in disease prediction, an approach that is sure to grow in popularity, and one that is complex in its application and interpretation.
Although the discovery and replication efforts are of somewhat limited power – indicating a need for expansion of such efforts in terms of sample size and length of follow-up – the study marks a movement in the development of methods to predict disease progression and, therefore, assist clinicians, patients with Parkinson’s disease, and their family members with treatment and individualized disease management.
The methodology is promising, but fine-tuning and extension of current efforts are important because, while these endeavors are costly, they are vital for the development of disease-modifying therapies and the treatment of Parkinson’s disease.
Cornelis Blauwendraat, MD, and his colleagues are with the Laboratory of Neurogenetics at the National Institute on Aging. They declared having no competing interests. Their comments are taken from an editorial accompanying Dr. Latourelle and her colleagues’ report (Lancet Neurol. 2017 Sep 25. doi: 10.106/S1474-4422[17]30331-9).
The study by Latourelle and her colleagues adds substantial value to Parkinson’s disease research not only because of the findings but also because it introduces the topic of using multimodal data in disease prediction, an approach that is sure to grow in popularity, and one that is complex in its application and interpretation.
Although the discovery and replication efforts are of somewhat limited power – indicating a need for expansion of such efforts in terms of sample size and length of follow-up – the study marks a movement in the development of methods to predict disease progression and, therefore, assist clinicians, patients with Parkinson’s disease, and their family members with treatment and individualized disease management.
The methodology is promising, but fine-tuning and extension of current efforts are important because, while these endeavors are costly, they are vital for the development of disease-modifying therapies and the treatment of Parkinson’s disease.
Cornelis Blauwendraat, MD, and his colleagues are with the Laboratory of Neurogenetics at the National Institute on Aging. They declared having no competing interests. Their comments are taken from an editorial accompanying Dr. Latourelle and her colleagues’ report (Lancet Neurol. 2017 Sep 25. doi: 10.106/S1474-4422[17]30331-9).
The study by Latourelle and her colleagues adds substantial value to Parkinson’s disease research not only because of the findings but also because it introduces the topic of using multimodal data in disease prediction, an approach that is sure to grow in popularity, and one that is complex in its application and interpretation.
Although the discovery and replication efforts are of somewhat limited power – indicating a need for expansion of such efforts in terms of sample size and length of follow-up – the study marks a movement in the development of methods to predict disease progression and, therefore, assist clinicians, patients with Parkinson’s disease, and their family members with treatment and individualized disease management.
The methodology is promising, but fine-tuning and extension of current efforts are important because, while these endeavors are costly, they are vital for the development of disease-modifying therapies and the treatment of Parkinson’s disease.
Cornelis Blauwendraat, MD, and his colleagues are with the Laboratory of Neurogenetics at the National Institute on Aging. They declared having no competing interests. Their comments are taken from an editorial accompanying Dr. Latourelle and her colleagues’ report (Lancet Neurol. 2017 Sep 25. doi: 10.106/S1474-4422[17]30331-9).
An ensemble of prediction models developed using genetic data and both baseline molecular and clinical variables from patients with Parkinson’s disease, as well as healthy controls, confirmed established predictors of the disease and identified new ones in a longitudinal cohort study.
Furthermore, the models were shown via simulated, randomized, placebo-controlled trials to have utility for clinical trial design and evaluation, as well as for clinical disease monitoring and treatment, Jeanne C. Latourelle, DSc, of GNS Healthcare, Cambridge, Mass., and her colleagues reported in Lancet Neurology.
“Understanding the causal and physiological factors that contribute to this variability in the evolution of symptoms of Parkinson’s disease is, therefore, a high priority area of Parkinson’s disease research,” they wrote (Lancet Neurol. 2017 Sep 25. doi: 10.1016/S1474-4422[17]30328-9).
The investigators developed the models by applying a Bayesian, machine-learning, multivariate predictive inference platform – known as Reverse Engineering and Forward Simulation – to data from the Parkinson’s Progression Markers Initiative (PPMI) study; they used these models to predict the annual rate of change in combined scores from the Movement Disorder Society–Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) parts II and III. The investigators tested the overall explanatory power of the models using the coefficient of determination (R2); their models also replicated novel findings from an independent clinical cohort from the Longitudinal and Biomarker Study in Parkinson’s disease (LABS-PD).
In 117 healthy age- and sex-matched controls and 312 patients with early-stage Parkinson’s disease from the PPMI study, the model ensemble showed strong performance (fivefold cross-validated Pearson R2, 41%), the investigators said.
In the 317-patient LABS-PD validation cohort, the performance of the models was reduced, but still statistically significant (R2, 9%).
Individual predictive features – including significant replication of higher baseline motor score on MDS-UPDRS, male sex, increased age, and novel Parkinson’s disease–specific epistatic interaction, which all were indicative of faster motor progression in the disease – were confirmed in the LABS-PD cohort, they said. The most useful predictive marker of motor progression was genetic variation (R2, 2.9%), the investigators said, and the cerebrospinal fluid biomarkers at baseline also significantly affected prediction of motor progression, although more modestly (R2, 0.3%).
In 5,000 trial simulations, the incorporation of predicted rates of motor progression into the final models of treatment effect reduced the variability in study outcome, which allowed for the detection of significant differences in sample sizes up to 20% smaller than in trials of drug-naive patients.
The investigators noted that the Bayesian model inference using Reverse Engineering and Forward Simulation was particularly good at predicting Parkinson’s motor progression in early disease stages, which has “immediate relevance toward enabling more effective trial recruitment and clinical disease management.”
This study was supported by grants from the Michael J. Fox Foundation for Parkinson’s Research and the National Institute of Neurological Disorders and Stroke. PPMI is funded by the Michael J. Fox Foundation for Parkinson’s Research, and it also has many industry funding partners. Dr. Latourelle and several of her coauthors are or were employees of GNS Healthcare.
An ensemble of prediction models developed using genetic data and both baseline molecular and clinical variables from patients with Parkinson’s disease, as well as healthy controls, confirmed established predictors of the disease and identified new ones in a longitudinal cohort study.
Furthermore, the models were shown via simulated, randomized, placebo-controlled trials to have utility for clinical trial design and evaluation, as well as for clinical disease monitoring and treatment, Jeanne C. Latourelle, DSc, of GNS Healthcare, Cambridge, Mass., and her colleagues reported in Lancet Neurology.
“Understanding the causal and physiological factors that contribute to this variability in the evolution of symptoms of Parkinson’s disease is, therefore, a high priority area of Parkinson’s disease research,” they wrote (Lancet Neurol. 2017 Sep 25. doi: 10.1016/S1474-4422[17]30328-9).
The investigators developed the models by applying a Bayesian, machine-learning, multivariate predictive inference platform – known as Reverse Engineering and Forward Simulation – to data from the Parkinson’s Progression Markers Initiative (PPMI) study; they used these models to predict the annual rate of change in combined scores from the Movement Disorder Society–Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) parts II and III. The investigators tested the overall explanatory power of the models using the coefficient of determination (R2); their models also replicated novel findings from an independent clinical cohort from the Longitudinal and Biomarker Study in Parkinson’s disease (LABS-PD).
In 117 healthy age- and sex-matched controls and 312 patients with early-stage Parkinson’s disease from the PPMI study, the model ensemble showed strong performance (fivefold cross-validated Pearson R2, 41%), the investigators said.
In the 317-patient LABS-PD validation cohort, the performance of the models was reduced, but still statistically significant (R2, 9%).
Individual predictive features – including significant replication of higher baseline motor score on MDS-UPDRS, male sex, increased age, and novel Parkinson’s disease–specific epistatic interaction, which all were indicative of faster motor progression in the disease – were confirmed in the LABS-PD cohort, they said. The most useful predictive marker of motor progression was genetic variation (R2, 2.9%), the investigators said, and the cerebrospinal fluid biomarkers at baseline also significantly affected prediction of motor progression, although more modestly (R2, 0.3%).
In 5,000 trial simulations, the incorporation of predicted rates of motor progression into the final models of treatment effect reduced the variability in study outcome, which allowed for the detection of significant differences in sample sizes up to 20% smaller than in trials of drug-naive patients.
The investigators noted that the Bayesian model inference using Reverse Engineering and Forward Simulation was particularly good at predicting Parkinson’s motor progression in early disease stages, which has “immediate relevance toward enabling more effective trial recruitment and clinical disease management.”
This study was supported by grants from the Michael J. Fox Foundation for Parkinson’s Research and the National Institute of Neurological Disorders and Stroke. PPMI is funded by the Michael J. Fox Foundation for Parkinson’s Research, and it also has many industry funding partners. Dr. Latourelle and several of her coauthors are or were employees of GNS Healthcare.
FROM LANCET NEUROLOGY
Key clinical point:
Major finding: The model ensemble showed strong performance (fivefold cross-validated Pearson R2, 41%) and remained significant in a validation cohort (R2, 9%).
Data source: A longitudinal cohort study of 429 patients and controls, as well as a validation cohort of 317 patients.
Disclosures: This study was supported by grants from the Michael J. Fox Foundation for Parkinson’s Research and the National Institute of Neurological Disorders and Stroke. The Parkinson’s Progression Markers Initiative is funded by the Michael J. Fox Foundation for Parkinson’s Research, and it also has many industry funding partners. Dr. Latourelle and several of her coauthors are or were employees of GNS Healthcare.
Dentist-prescribed antibiotics implicated in community-acquired C. diff
SAN DIEGO – Antibiotic prescriptions written by dentists make a substantial but largely unrecognized contribution to the risk of community-acquired Clostridium difficile (CA-CDI) infections, according to results of a public health initiative that was presented at an annual scientific meeting on infectious diseases.
When nearly 1,000 cases of CA-CDI suspected of being the result of antibiotic exposure were evaluated, it was found that 15% of the prescriptions were written by dentists, often for indications that are counter to current guidelines, reported Maria Bye, MPH, an officer in the Infectious Disease Epidemiology, Prevention, and Control Division of the Minnesota Department of Health, St. Paul.

In another finding from this study relevant to infection control, a substantial proportion of dentist-prescribed antibiotics associated with CA-CDI were not in the medical record. Rather, they were discovered in interviews conducted to isolate risk factors. “This is concerning since clinicians unaware of these exposures will not think about CA-CDI or other complications of antibiotic use, possibly delaying therapy,” Ms. Bye said.
The importance of dental antibiotic prescriptions in CA-CDI was identified in an ongoing infectious disease program managed by the Minnesota Department of Health in collaboration with the Centers for Disease Control and Prevention. In the years 2009-2015, medical records and interviews were conducted to identify risk factors in 1,626 confirmed cases of CA-CDI in five Minnesota counties. All cases were among patients without an overnight stay in a health care facility in the previous 12 weeks, which was an exclusion criterion.
After the review of medical records and patient interviews, 926 (57%) of the CA-CDI cases were deemed likely to be related to antibiotic exposure. Of these antibiotic exposures, 136 (15%) were from prescriptions written by dentists, a figure reached only when patients were interviewed. According to Ms. Bye, 34% of the antibiotics prescribed by dentists were not in the medical record.
There were also notable differences in antibiotic exposures stemming from dentist prescriptions relative to prescriptions from other sources. Perhaps most significantly, dental-related antibiotic prescriptions were far more likely to be for clindamycin (50% vs. 10%; P less than .001), which is commonly associated with increased risk of C. difficile, according to Ms. Bye. Fluoroquinolones (6% vs. 19%; P less than .001) and cephalosporins (7% vs. 30%; P less than .001) were significantly less likely to be prescribed by dentists. Patients who developed CA-CDI associated with antibiotics prescribed by dentists were also significantly older than were those receiving antibiotics from another source (mean age 57 years vs. 45 years; P less than .001).
For the first years of this analysis, information on the indication for dentist-prescribed antibiotics was not collected, but these data were collected beginning in 2015. In the data collected so far, a substantial proportion of prescriptions were written for prophylaxis against systemic infections, including prevention of endocarditis or infection of prosthetic joints. However, few of these prescriptions were indicated.
“The American Dental Association stated that antibiotic prophylaxis is not generally recommended in patients with prosthetic joints,” said Ms. Bye, noting that this is consistent with similar statements issued by the American Academy of Orthopedic Surgery. In 2007, the American Heart Association narrowed its recommendations for prophylactic antibiotics to patients at the highest risk of adverse consequences from endocarditis.
Of the four patients who received prophylactic antibiotics from their dentists for a cardiovascular or orthopedic indication, only one met current criteria, Ms. Bye reported.
The contribution of antibiotic exposures from dentist prescriptions to CA-CDI should not be surprising, according to Ms. Bye. In the United States, dentists prescribe 10% of all outpatient antibiotics. Although there are many valid indications for these prescriptions, Ms. Bye cited a survey that found that the proportion of dentists familiar with current guidelines and the risks of adverse events produced by antibiotics, including CA-CDI, is less than 50%.
“Dentists need to be included in antibiotic stewardship programs,” she advised. This will include educating dentists about the indications for antibiotic prophylaxis for medical conditions. She also suggested that clinicians should routinely ask patients about whether they have received antibiotics from a dentist so that this information gets into the medical record.
The event was the combined annual meetings of the Infectious Diseases Society of America, the Society for Healthcare Epidemiology of America, the HIV Medicine Association, and the Pediatric Infectious Diseases Society.
SAN DIEGO – Antibiotic prescriptions written by dentists make a substantial but largely unrecognized contribution to the risk of community-acquired Clostridium difficile (CA-CDI) infections, according to results of a public health initiative that was presented at an annual scientific meeting on infectious diseases.
When nearly 1,000 cases of CA-CDI suspected of being the result of antibiotic exposure were evaluated, it was found that 15% of the prescriptions were written by dentists, often for indications that are counter to current guidelines, reported Maria Bye, MPH, an officer in the Infectious Disease Epidemiology, Prevention, and Control Division of the Minnesota Department of Health, St. Paul.
In another finding from this study relevant to infection control, a substantial proportion of dentist-prescribed antibiotics associated with CA-CDI were not in the medical record. Rather, they were discovered in interviews conducted to isolate risk factors. “This is concerning since clinicians unaware of these exposures will not think about CA-CDI or other complications of antibiotic use, possibly delaying therapy,” Ms. Bye said.
The importance of dental antibiotic prescriptions in CA-CDI was identified in an ongoing infectious disease program managed by the Minnesota Department of Health in collaboration with the Centers for Disease Control and Prevention. In the years 2009-2015, medical records and interviews were conducted to identify risk factors in 1,626 confirmed cases of CA-CDI in five Minnesota counties. All cases were among patients without an overnight stay in a health care facility in the previous 12 weeks, which was an exclusion criterion.
After the review of medical records and patient interviews, 926 (57%) of the CA-CDI cases were deemed likely to be related to antibiotic exposure. Of these antibiotic exposures, 136 (15%) were from prescriptions written by dentists, a figure reached only when patients were interviewed. According to Ms. Bye, 34% of the antibiotics prescribed by dentists were not in the medical record.
There were also notable differences in antibiotic exposures stemming from dentist prescriptions relative to prescriptions from other sources. Perhaps most significantly, dental-related antibiotic prescriptions were far more likely to be for clindamycin (50% vs. 10%; P less than .001), which is commonly associated with increased risk of C. difficile, according to Ms. Bye. Fluoroquinolones (6% vs. 19%; P less than .001) and cephalosporins (7% vs. 30%; P less than .001) were significantly less likely to be prescribed by dentists. Patients who developed CA-CDI associated with antibiotics prescribed by dentists were also significantly older than were those receiving antibiotics from another source (mean age 57 years vs. 45 years; P less than .001).
For the first years of this analysis, information on the indication for dentist-prescribed antibiotics was not collected, but these data were collected beginning in 2015. In the data collected so far, a substantial proportion of prescriptions were written for prophylaxis against systemic infections, including prevention of endocarditis or infection of prosthetic joints. However, few of these prescriptions were indicated.
“The American Dental Association stated that antibiotic prophylaxis is not generally recommended in patients with prosthetic joints,” said Ms. Bye, noting that this is consistent with similar statements issued by the American Academy of Orthopedic Surgery. In 2007, the American Heart Association narrowed its recommendations for prophylactic antibiotics to patients at the highest risk of adverse consequences from endocarditis.
Of the four patients who received prophylactic antibiotics from their dentists for a cardiovascular or orthopedic indication, only one met current criteria, Ms. Bye reported.
The contribution of antibiotic exposures from dentist prescriptions to CA-CDI should not be surprising, according to Ms. Bye. In the United States, dentists prescribe 10% of all outpatient antibiotics. Although there are many valid indications for these prescriptions, Ms. Bye cited a survey that found that the proportion of dentists familiar with current guidelines and the risks of adverse events produced by antibiotics, including CA-CDI, is less than 50%.
“Dentists need to be included in antibiotic stewardship programs,” she advised. This will include educating dentists about the indications for antibiotic prophylaxis for medical conditions. She also suggested that clinicians should routinely ask patients about whether they have received antibiotics from a dentist so that this information gets into the medical record.
The event was the combined annual meetings of the Infectious Diseases Society of America, the Society for Healthcare Epidemiology of America, the HIV Medicine Association, and the Pediatric Infectious Diseases Society.
SAN DIEGO – Antibiotic prescriptions written by dentists make a substantial but largely unrecognized contribution to the risk of community-acquired Clostridium difficile (CA-CDI) infections, according to results of a public health initiative that was presented at an annual scientific meeting on infectious diseases.
When nearly 1,000 cases of CA-CDI suspected of being the result of antibiotic exposure were evaluated, it was found that 15% of the prescriptions were written by dentists, often for indications that are counter to current guidelines, reported Maria Bye, MPH, an officer in the Infectious Disease Epidemiology, Prevention, and Control Division of the Minnesota Department of Health, St. Paul.
In another finding from this study relevant to infection control, a substantial proportion of dentist-prescribed antibiotics associated with CA-CDI were not in the medical record. Rather, they were discovered in interviews conducted to isolate risk factors. “This is concerning since clinicians unaware of these exposures will not think about CA-CDI or other complications of antibiotic use, possibly delaying therapy,” Ms. Bye said.
The importance of dental antibiotic prescriptions in CA-CDI was identified in an ongoing infectious disease program managed by the Minnesota Department of Health in collaboration with the Centers for Disease Control and Prevention. In the years 2009-2015, medical records and interviews were conducted to identify risk factors in 1,626 confirmed cases of CA-CDI in five Minnesota counties. All cases were among patients without an overnight stay in a health care facility in the previous 12 weeks, which was an exclusion criterion.
After the review of medical records and patient interviews, 926 (57%) of the CA-CDI cases were deemed likely to be related to antibiotic exposure. Of these antibiotic exposures, 136 (15%) were from prescriptions written by dentists, a figure reached only when patients were interviewed. According to Ms. Bye, 34% of the antibiotics prescribed by dentists were not in the medical record.
There were also notable differences in antibiotic exposures stemming from dentist prescriptions relative to prescriptions from other sources. Perhaps most significantly, dental-related antibiotic prescriptions were far more likely to be for clindamycin (50% vs. 10%; P less than .001), which is commonly associated with increased risk of C. difficile, according to Ms. Bye. Fluoroquinolones (6% vs. 19%; P less than .001) and cephalosporins (7% vs. 30%; P less than .001) were significantly less likely to be prescribed by dentists. Patients who developed CA-CDI associated with antibiotics prescribed by dentists were also significantly older than were those receiving antibiotics from another source (mean age 57 years vs. 45 years; P less than .001).
For the first years of this analysis, information on the indication for dentist-prescribed antibiotics was not collected, but these data were collected beginning in 2015. In the data collected so far, a substantial proportion of prescriptions were written for prophylaxis against systemic infections, including prevention of endocarditis or infection of prosthetic joints. However, few of these prescriptions were indicated.
“The American Dental Association stated that antibiotic prophylaxis is not generally recommended in patients with prosthetic joints,” said Ms. Bye, noting that this is consistent with similar statements issued by the American Academy of Orthopedic Surgery. In 2007, the American Heart Association narrowed its recommendations for prophylactic antibiotics to patients at the highest risk of adverse consequences from endocarditis.
Of the four patients who received prophylactic antibiotics from their dentists for a cardiovascular or orthopedic indication, only one met current criteria, Ms. Bye reported.
The contribution of antibiotic exposures from dentist prescriptions to CA-CDI should not be surprising, according to Ms. Bye. In the United States, dentists prescribe 10% of all outpatient antibiotics. Although there are many valid indications for these prescriptions, Ms. Bye cited a survey that found that the proportion of dentists familiar with current guidelines and the risks of adverse events produced by antibiotics, including CA-CDI, is less than 50%.
“Dentists need to be included in antibiotic stewardship programs,” she advised. This will include educating dentists about the indications for antibiotic prophylaxis for medical conditions. She also suggested that clinicians should routinely ask patients about whether they have received antibiotics from a dentist so that this information gets into the medical record.
The event was the combined annual meetings of the Infectious Diseases Society of America, the Society for Healthcare Epidemiology of America, the HIV Medicine Association, and the Pediatric Infectious Diseases Society.
AT ID WEEK 2017
Key clinical point: A public health initiative tracking community-acquired Clostridium difficile infections suggests dental antibiotic prescribing is a significant source.
Major finding: When traced, 15% of the antibiotics related to community-acquired C. difficile infections were from a dentist prescription.
Data source: Population-based surveillance study.
Disclosures: Ms. Bye reported that she has no financial relationships relevant to this topic.
It’s too soon to discard cotesting in cervical cancer
In their new cervical cancer screening draft recommendation statement, the U.S. Preventive Services Task Force recommends screening for cervical cancer every 3 years with cervical cytology alone in women aged 21-29 years, and either continuing 3-year cytology screening or 5-year high-risk human papillomavirus (HPV) screening in women aged 30-65 years.1 They arrived at the conclusion primarily based on an analysis of benefits and harms of each of the currently used screening modalities. Cytology plus HPV cotesting was considered but rejected based on the benefits versus harms calculations. However, some of the assumptions in their modeling are wrong and this has led them to faulty conclusions.
The USPSTF correctly identified CIN-2/3 and CIN-3+ detected and cervical cancer cases and deaths prevented as benefits. But they used the number of colposcopies and the number of tests conducted as their proxy for harms, primarily because these were more easily measured. They identified other harms, such as greater psychological distress related to being told of a positive HPV result (compared with being told of an abnormal cytology or cotest result), but these harms were not numerical and so they could not be incorporated into their modeling. They also did not measure the psychological distress associated with an extended screening interval or HPV-only screening.

The USPSTF also primarily relied on evidence from seven large, randomly controlled trials of primary screening, mostly conducted in Europe.3 Of the seven trials, two of them (SWEDESCREEN and POBASCAM) used HPV testing by a polymerase chain reaction methodology that is not approved by the U.S. Food and Drug Administration, not commercially available in the United States, and has different sensitivity and specificity does than FDA-approved tests.
Most of the studies also were done with conventional cytology that is, for the most part, no longer used in the United States. The age range in some of the studies was also very narrow (SWEDESCREEN evaluated women aged 32-38 years only) while extending to very young ages in others (ARTISTIC evaluated women as young as age 20 years). It may not be possible to extrapolate from these results to the U.S. experience. On the other hand, the USPSTF elected not to use data from very large retrospective U.S. trials, such as the data from more than 1 million women from Kaiser Permanente Northern California, which helped form the foundation of current guidelines.4
Although screening has dramatically reduced the incidence of cervical cancer in the United States, most screening in this country is opportunistic and lacks population-based registries or regular invitations to screening. The USPSTF notes that a sizable proportion of the U.S.-based female population is not routinely screened. In fact, 11.4% of 21- to 65-year-old women have not been screened in the previous 5 years and the percentages are higher among minority and disadvantaged populations. Although the numbers seem relatively small, more than half of women with cervical cancer are found in this population. These women also are at greater risk of being lost to follow-up. Unlike the patients in the European trials, there is no current mechanism in the United States to assure that patients will return in 5 years. In fact, the USPSTF notes this limitation in their evidence, but doesn’t provide a remedy. These women may not get rescreened for many years after the 5-year interval has passed, and there is no evidence that this interval is safe.
Finally, the USPSTF recommendations for HPV primary screening relied in whole or in part on cytology triage of positive results. There is no U.S. experience for the use of cytology within this context, and many experts have argued that the test performance of cytology in a triage setting would be different than in a screening setting and would further be different in a vaccinated population. Since the modeling on which these draft recommendations are based did not account for these changed performance characteristics, their assumptions must be wrong.
Five years have not yet passed since publication of the last set of guidelines, and we have no idea how this extended screening interval functions in our opportunistic screening system. Stated simply, these draft recommendations have gone too far and too fast and should be reconsidered.
Dr. Spitzer is a professor of obstetrics and gynecology at Hofstra Northwell School of Medicine, Hempstead, N.Y., and a past president of the American Society for Colposcopy and Cervical Pathology. He reported receiving royalties from Elsevier, and consulting fees from Hologic, Biop Medical, Illumigen, and Merck.
References
1. Draft Recommendation Statement: Cervical Cancer: Screening. U.S. Preventive Services Task Force. September 2017.
2. Screening for Cervical Cancer in Primary Care: A Decision Analysis for the U.S. Preventive Services Task Force. 2017.
3. Screening for Cervical Cancer With High-Risk Human Papillomavirus Testing: A Systematic Evidence Review for the U.S. Preventive Services Task Force. 2017.
4. J Natl Cancer Inst. 2014 Jul 18;106(8). pii: dju153.
In their new cervical cancer screening draft recommendation statement, the U.S. Preventive Services Task Force recommends screening for cervical cancer every 3 years with cervical cytology alone in women aged 21-29 years, and either continuing 3-year cytology screening or 5-year high-risk human papillomavirus (HPV) screening in women aged 30-65 years.1 They arrived at the conclusion primarily based on an analysis of benefits and harms of each of the currently used screening modalities. Cytology plus HPV cotesting was considered but rejected based on the benefits versus harms calculations. However, some of the assumptions in their modeling are wrong and this has led them to faulty conclusions.
The USPSTF correctly identified CIN-2/3 and CIN-3+ detected and cervical cancer cases and deaths prevented as benefits. But they used the number of colposcopies and the number of tests conducted as their proxy for harms, primarily because these were more easily measured. They identified other harms, such as greater psychological distress related to being told of a positive HPV result (compared with being told of an abnormal cytology or cotest result), but these harms were not numerical and so they could not be incorporated into their modeling. They also did not measure the psychological distress associated with an extended screening interval or HPV-only screening.
The USPSTF also primarily relied on evidence from seven large, randomly controlled trials of primary screening, mostly conducted in Europe.3 Of the seven trials, two of them (SWEDESCREEN and POBASCAM) used HPV testing by a polymerase chain reaction methodology that is not approved by the U.S. Food and Drug Administration, not commercially available in the United States, and has different sensitivity and specificity does than FDA-approved tests.
Most of the studies also were done with conventional cytology that is, for the most part, no longer used in the United States. The age range in some of the studies was also very narrow (SWEDESCREEN evaluated women aged 32-38 years only) while extending to very young ages in others (ARTISTIC evaluated women as young as age 20 years). It may not be possible to extrapolate from these results to the U.S. experience. On the other hand, the USPSTF elected not to use data from very large retrospective U.S. trials, such as the data from more than 1 million women from Kaiser Permanente Northern California, which helped form the foundation of current guidelines.4
Although screening has dramatically reduced the incidence of cervical cancer in the United States, most screening in this country is opportunistic and lacks population-based registries or regular invitations to screening. The USPSTF notes that a sizable proportion of the U.S.-based female population is not routinely screened. In fact, 11.4% of 21- to 65-year-old women have not been screened in the previous 5 years and the percentages are higher among minority and disadvantaged populations. Although the numbers seem relatively small, more than half of women with cervical cancer are found in this population. These women also are at greater risk of being lost to follow-up. Unlike the patients in the European trials, there is no current mechanism in the United States to assure that patients will return in 5 years. In fact, the USPSTF notes this limitation in their evidence, but doesn’t provide a remedy. These women may not get rescreened for many years after the 5-year interval has passed, and there is no evidence that this interval is safe.
Finally, the USPSTF recommendations for HPV primary screening relied in whole or in part on cytology triage of positive results. There is no U.S. experience for the use of cytology within this context, and many experts have argued that the test performance of cytology in a triage setting would be different than in a screening setting and would further be different in a vaccinated population. Since the modeling on which these draft recommendations are based did not account for these changed performance characteristics, their assumptions must be wrong.
Five years have not yet passed since publication of the last set of guidelines, and we have no idea how this extended screening interval functions in our opportunistic screening system. Stated simply, these draft recommendations have gone too far and too fast and should be reconsidered.
Dr. Spitzer is a professor of obstetrics and gynecology at Hofstra Northwell School of Medicine, Hempstead, N.Y., and a past president of the American Society for Colposcopy and Cervical Pathology. He reported receiving royalties from Elsevier, and consulting fees from Hologic, Biop Medical, Illumigen, and Merck.
References
1. Draft Recommendation Statement: Cervical Cancer: Screening. U.S. Preventive Services Task Force. September 2017.
2. Screening for Cervical Cancer in Primary Care: A Decision Analysis for the U.S. Preventive Services Task Force. 2017.
3. Screening for Cervical Cancer With High-Risk Human Papillomavirus Testing: A Systematic Evidence Review for the U.S. Preventive Services Task Force. 2017.
4. J Natl Cancer Inst. 2014 Jul 18;106(8). pii: dju153.
In their new cervical cancer screening draft recommendation statement, the U.S. Preventive Services Task Force recommends screening for cervical cancer every 3 years with cervical cytology alone in women aged 21-29 years, and either continuing 3-year cytology screening or 5-year high-risk human papillomavirus (HPV) screening in women aged 30-65 years.1 They arrived at the conclusion primarily based on an analysis of benefits and harms of each of the currently used screening modalities. Cytology plus HPV cotesting was considered but rejected based on the benefits versus harms calculations. However, some of the assumptions in their modeling are wrong and this has led them to faulty conclusions.
The USPSTF correctly identified CIN-2/3 and CIN-3+ detected and cervical cancer cases and deaths prevented as benefits. But they used the number of colposcopies and the number of tests conducted as their proxy for harms, primarily because these were more easily measured. They identified other harms, such as greater psychological distress related to being told of a positive HPV result (compared with being told of an abnormal cytology or cotest result), but these harms were not numerical and so they could not be incorporated into their modeling. They also did not measure the psychological distress associated with an extended screening interval or HPV-only screening.
The USPSTF also primarily relied on evidence from seven large, randomly controlled trials of primary screening, mostly conducted in Europe.3 Of the seven trials, two of them (SWEDESCREEN and POBASCAM) used HPV testing by a polymerase chain reaction methodology that is not approved by the U.S. Food and Drug Administration, not commercially available in the United States, and has different sensitivity and specificity does than FDA-approved tests.
Most of the studies also were done with conventional cytology that is, for the most part, no longer used in the United States. The age range in some of the studies was also very narrow (SWEDESCREEN evaluated women aged 32-38 years only) while extending to very young ages in others (ARTISTIC evaluated women as young as age 20 years). It may not be possible to extrapolate from these results to the U.S. experience. On the other hand, the USPSTF elected not to use data from very large retrospective U.S. trials, such as the data from more than 1 million women from Kaiser Permanente Northern California, which helped form the foundation of current guidelines.4
Although screening has dramatically reduced the incidence of cervical cancer in the United States, most screening in this country is opportunistic and lacks population-based registries or regular invitations to screening. The USPSTF notes that a sizable proportion of the U.S.-based female population is not routinely screened. In fact, 11.4% of 21- to 65-year-old women have not been screened in the previous 5 years and the percentages are higher among minority and disadvantaged populations. Although the numbers seem relatively small, more than half of women with cervical cancer are found in this population. These women also are at greater risk of being lost to follow-up. Unlike the patients in the European trials, there is no current mechanism in the United States to assure that patients will return in 5 years. In fact, the USPSTF notes this limitation in their evidence, but doesn’t provide a remedy. These women may not get rescreened for many years after the 5-year interval has passed, and there is no evidence that this interval is safe.
Finally, the USPSTF recommendations for HPV primary screening relied in whole or in part on cytology triage of positive results. There is no U.S. experience for the use of cytology within this context, and many experts have argued that the test performance of cytology in a triage setting would be different than in a screening setting and would further be different in a vaccinated population. Since the modeling on which these draft recommendations are based did not account for these changed performance characteristics, their assumptions must be wrong.
Five years have not yet passed since publication of the last set of guidelines, and we have no idea how this extended screening interval functions in our opportunistic screening system. Stated simply, these draft recommendations have gone too far and too fast and should be reconsidered.
Dr. Spitzer is a professor of obstetrics and gynecology at Hofstra Northwell School of Medicine, Hempstead, N.Y., and a past president of the American Society for Colposcopy and Cervical Pathology. He reported receiving royalties from Elsevier, and consulting fees from Hologic, Biop Medical, Illumigen, and Merck.
References
1. Draft Recommendation Statement: Cervical Cancer: Screening. U.S. Preventive Services Task Force. September 2017.
2. Screening for Cervical Cancer in Primary Care: A Decision Analysis for the U.S. Preventive Services Task Force. 2017.
3. Screening for Cervical Cancer With High-Risk Human Papillomavirus Testing: A Systematic Evidence Review for the U.S. Preventive Services Task Force. 2017.
4. J Natl Cancer Inst. 2014 Jul 18;106(8). pii: dju153.
New treatments up the ante against acne
Acne remains “an equal opportunity annoyance,” according to Hilary E. Baldwin, MD, of Rutgers Robert Wood Johnson Medical School, Newark, NJ.
However, acne medications also work equally well across age, gender, and skin type groups, and new systemic and topical options are emerging, said Dr. Baldwin, who serves as medical director of The Acne Treatment and Research Center in Morristown, NJ.
Several products that entered the market in 2016 have demonstrated success, she said in a presentation on acne at the annual Coastal Dermatology Symposium. She cited data on dapsone 7.5% gel (Aczone) applied daily, which showed significant improvements in moderate facial acne and lesion counts compared with vehicle.
A noteworthy new potential acne treatment combines 200 mg doxycycline with topical adapalene 0.3%/benzoyl peroxide 2.5%, Dr. Baldwin said. In a small but promising 12-week open-label study of patients aged 12 years and older with severe facial acne considered candidates for isotretinoin, inflammatory, noninflammatory, and overall total lesion counts reduced significantly from baseline, she said.
Other acne treatments in the pipeline include a nitric oxide gel, a topical sebum inhibitor, and minocycline gel and foam formulations.
Sarecycline, a tetracycline class antibiotic, has generated some excitement after a phase 2 dose ranging study presented at the 2017 American Academy of Dermatology’s annual meeting showed significant improvement in inflammatory lesion counts among acne patients who received 1.5 mg/kg or 3 mg/kg once a day compared with placebo patients, after 12 weeks. Noninflammatory lesion counts were not significantly improved compared with placebo. Potential advantages of sarecycline include improved efficacy with fewer side effects and possibly, a lower risk of antibiotic resistance, Dr. Baldwin said. Phase 3 study results of the 1.5 mg/kg dose are pending.
Data on the potential role of diet in acne continue to evolve, she noted. A recent study of 225 teens with acne suggested that skim and/or low-fat dairy products are associated with acne and that reducing consumption of these products might help (J Am Acad Dermatol. 2016 Aug;75[2]:318-22). Another small study of 64 adults involving a nutritional survey showed that those with moderate to severe acne consumed significantly more carbohydrates than did those without acne, an indication that clinicians could consider recommending that acne patients reduce their carbohydrate intake to see whether it makes a difference.
The symposium was jointly presented by the University of Louisville and Global Academy for Medical Education. This publication and Global Academy for Medical Education are both owned by Frontline Medical News. Dr. Baldwin is a speaker and advisor for Allergan, Galderma, and Valeant; and is an investigator for Dermira, Galderma, Novan, and Valeant.
Acne remains “an equal opportunity annoyance,” according to Hilary E. Baldwin, MD, of Rutgers Robert Wood Johnson Medical School, Newark, NJ.
However, acne medications also work equally well across age, gender, and skin type groups, and new systemic and topical options are emerging, said Dr. Baldwin, who serves as medical director of The Acne Treatment and Research Center in Morristown, NJ.
Several products that entered the market in 2016 have demonstrated success, she said in a presentation on acne at the annual Coastal Dermatology Symposium. She cited data on dapsone 7.5% gel (Aczone) applied daily, which showed significant improvements in moderate facial acne and lesion counts compared with vehicle.
A noteworthy new potential acne treatment combines 200 mg doxycycline with topical adapalene 0.3%/benzoyl peroxide 2.5%, Dr. Baldwin said. In a small but promising 12-week open-label study of patients aged 12 years and older with severe facial acne considered candidates for isotretinoin, inflammatory, noninflammatory, and overall total lesion counts reduced significantly from baseline, she said.
Other acne treatments in the pipeline include a nitric oxide gel, a topical sebum inhibitor, and minocycline gel and foam formulations.
Sarecycline, a tetracycline class antibiotic, has generated some excitement after a phase 2 dose ranging study presented at the 2017 American Academy of Dermatology’s annual meeting showed significant improvement in inflammatory lesion counts among acne patients who received 1.5 mg/kg or 3 mg/kg once a day compared with placebo patients, after 12 weeks. Noninflammatory lesion counts were not significantly improved compared with placebo. Potential advantages of sarecycline include improved efficacy with fewer side effects and possibly, a lower risk of antibiotic resistance, Dr. Baldwin said. Phase 3 study results of the 1.5 mg/kg dose are pending.
Data on the potential role of diet in acne continue to evolve, she noted. A recent study of 225 teens with acne suggested that skim and/or low-fat dairy products are associated with acne and that reducing consumption of these products might help (J Am Acad Dermatol. 2016 Aug;75[2]:318-22). Another small study of 64 adults involving a nutritional survey showed that those with moderate to severe acne consumed significantly more carbohydrates than did those without acne, an indication that clinicians could consider recommending that acne patients reduce their carbohydrate intake to see whether it makes a difference.
The symposium was jointly presented by the University of Louisville and Global Academy for Medical Education. This publication and Global Academy for Medical Education are both owned by Frontline Medical News. Dr. Baldwin is a speaker and advisor for Allergan, Galderma, and Valeant; and is an investigator for Dermira, Galderma, Novan, and Valeant.
Acne remains “an equal opportunity annoyance,” according to Hilary E. Baldwin, MD, of Rutgers Robert Wood Johnson Medical School, Newark, NJ.
However, acne medications also work equally well across age, gender, and skin type groups, and new systemic and topical options are emerging, said Dr. Baldwin, who serves as medical director of The Acne Treatment and Research Center in Morristown, NJ.
Several products that entered the market in 2016 have demonstrated success, she said in a presentation on acne at the annual Coastal Dermatology Symposium. She cited data on dapsone 7.5% gel (Aczone) applied daily, which showed significant improvements in moderate facial acne and lesion counts compared with vehicle.
A noteworthy new potential acne treatment combines 200 mg doxycycline with topical adapalene 0.3%/benzoyl peroxide 2.5%, Dr. Baldwin said. In a small but promising 12-week open-label study of patients aged 12 years and older with severe facial acne considered candidates for isotretinoin, inflammatory, noninflammatory, and overall total lesion counts reduced significantly from baseline, she said.
Other acne treatments in the pipeline include a nitric oxide gel, a topical sebum inhibitor, and minocycline gel and foam formulations.
Sarecycline, a tetracycline class antibiotic, has generated some excitement after a phase 2 dose ranging study presented at the 2017 American Academy of Dermatology’s annual meeting showed significant improvement in inflammatory lesion counts among acne patients who received 1.5 mg/kg or 3 mg/kg once a day compared with placebo patients, after 12 weeks. Noninflammatory lesion counts were not significantly improved compared with placebo. Potential advantages of sarecycline include improved efficacy with fewer side effects and possibly, a lower risk of antibiotic resistance, Dr. Baldwin said. Phase 3 study results of the 1.5 mg/kg dose are pending.
Data on the potential role of diet in acne continue to evolve, she noted. A recent study of 225 teens with acne suggested that skim and/or low-fat dairy products are associated with acne and that reducing consumption of these products might help (J Am Acad Dermatol. 2016 Aug;75[2]:318-22). Another small study of 64 adults involving a nutritional survey showed that those with moderate to severe acne consumed significantly more carbohydrates than did those without acne, an indication that clinicians could consider recommending that acne patients reduce their carbohydrate intake to see whether it makes a difference.
The symposium was jointly presented by the University of Louisville and Global Academy for Medical Education. This publication and Global Academy for Medical Education are both owned by Frontline Medical News. Dr. Baldwin is a speaker and advisor for Allergan, Galderma, and Valeant; and is an investigator for Dermira, Galderma, Novan, and Valeant.
FROM THE COASTAL DERMATOLOGY SYMPOSIUM
PrEP is not main driver in STI epidemic, says expert
SAN DIEGO – Increased use of pre-exposure prophylaxis (PrEP) to prevent HIV transmission has accelerated but is not the main reason for surging rates of sexually transmitted infections in the United States, Kenneth Mayer, MD, said during an oral presentation at an annual scientific meeting on infectious diseases.
Public health officials are seeing unprecedented rises in STIs such as syphilis and gonorrhea in both HIV-negative and HIV-positive individuals, and these trends predate the advent of PrEP, said Dr. Mayer of the Fenway Institute and Harvard University in Boston, Mass. “An overall level of behavioral disinhibition is fueling this epidemic and is not necessarily associated with PrEP,” he said.

Several studies suggest that being on PrEP does not increase the likelihood of acquiring or transmitting an STI, Dr. Mayer emphasized. In the open-label PROUD trial of PrEP in men who have sex with men (MSM), “rates of STI were extremely high and remained so, but did not go up after PrEP was initiated,” he noted. Importantly, the incidence of HIV infection was only 1.6 per 100 person-years when MSM received PrEP immediately but was 9.4 cases per 100 person-years when MSM were randomly assigned to a 1-year wait list (rate ratio, 6.0). In the randomized ANRS IPERGAY trial, 70% of high-risk MSM prescribed PrEP reported engaging in condomless anal intercourse during their most recent sexual encounter, but that proportion remained stable over 24 subsequent months of follow-up.
Providers also should understand that oral PrEP is just as effective at preventing HIV transmission when patients have STIs, Dr. Mayer said. In five recent studies, PrEP was equally efficacious among MSM regardless of whether they had syphilis or other STIs, and bacterial vaginosis in women also did not decrease the efficacy of oral PrEP. “There is no evidence to indicate that the efficacy of PrEP is lower among persons with STIs,” Dr. Mayer said.
Finally, providers should consider screening high-risk individuals for STIs more frequently than every 6 months as recommended by the Centers for Disease Control and Prevention, said Dr. Mayer. “For men who have sex with men, who are sexually active, and are on PrEP, quarterly screening makes exceedingly good sense from a cost-effectiveness standpoint,” he said.
“Screening less frequently than quarterly means that these individuals are having STIs for a longer period of time. When they are sexually active, we have a better chance of interrupting the transmission chain if we detect closer to the time of infection.”
Dr. Mayer disclosed support from the National Institutes of Health and Gilead Sciences, which makes some of the medications used in PrEP regimens.
SAN DIEGO – Increased use of pre-exposure prophylaxis (PrEP) to prevent HIV transmission has accelerated but is not the main reason for surging rates of sexually transmitted infections in the United States, Kenneth Mayer, MD, said during an oral presentation at an annual scientific meeting on infectious diseases.
Public health officials are seeing unprecedented rises in STIs such as syphilis and gonorrhea in both HIV-negative and HIV-positive individuals, and these trends predate the advent of PrEP, said Dr. Mayer of the Fenway Institute and Harvard University in Boston, Mass. “An overall level of behavioral disinhibition is fueling this epidemic and is not necessarily associated with PrEP,” he said.
Several studies suggest that being on PrEP does not increase the likelihood of acquiring or transmitting an STI, Dr. Mayer emphasized. In the open-label PROUD trial of PrEP in men who have sex with men (MSM), “rates of STI were extremely high and remained so, but did not go up after PrEP was initiated,” he noted. Importantly, the incidence of HIV infection was only 1.6 per 100 person-years when MSM received PrEP immediately but was 9.4 cases per 100 person-years when MSM were randomly assigned to a 1-year wait list (rate ratio, 6.0). In the randomized ANRS IPERGAY trial, 70% of high-risk MSM prescribed PrEP reported engaging in condomless anal intercourse during their most recent sexual encounter, but that proportion remained stable over 24 subsequent months of follow-up.
Providers also should understand that oral PrEP is just as effective at preventing HIV transmission when patients have STIs, Dr. Mayer said. In five recent studies, PrEP was equally efficacious among MSM regardless of whether they had syphilis or other STIs, and bacterial vaginosis in women also did not decrease the efficacy of oral PrEP. “There is no evidence to indicate that the efficacy of PrEP is lower among persons with STIs,” Dr. Mayer said.
Finally, providers should consider screening high-risk individuals for STIs more frequently than every 6 months as recommended by the Centers for Disease Control and Prevention, said Dr. Mayer. “For men who have sex with men, who are sexually active, and are on PrEP, quarterly screening makes exceedingly good sense from a cost-effectiveness standpoint,” he said.
“Screening less frequently than quarterly means that these individuals are having STIs for a longer period of time. When they are sexually active, we have a better chance of interrupting the transmission chain if we detect closer to the time of infection.”
Dr. Mayer disclosed support from the National Institutes of Health and Gilead Sciences, which makes some of the medications used in PrEP regimens.
SAN DIEGO – Increased use of pre-exposure prophylaxis (PrEP) to prevent HIV transmission has accelerated but is not the main reason for surging rates of sexually transmitted infections in the United States, Kenneth Mayer, MD, said during an oral presentation at an annual scientific meeting on infectious diseases.
Public health officials are seeing unprecedented rises in STIs such as syphilis and gonorrhea in both HIV-negative and HIV-positive individuals, and these trends predate the advent of PrEP, said Dr. Mayer of the Fenway Institute and Harvard University in Boston, Mass. “An overall level of behavioral disinhibition is fueling this epidemic and is not necessarily associated with PrEP,” he said.
Several studies suggest that being on PrEP does not increase the likelihood of acquiring or transmitting an STI, Dr. Mayer emphasized. In the open-label PROUD trial of PrEP in men who have sex with men (MSM), “rates of STI were extremely high and remained so, but did not go up after PrEP was initiated,” he noted. Importantly, the incidence of HIV infection was only 1.6 per 100 person-years when MSM received PrEP immediately but was 9.4 cases per 100 person-years when MSM were randomly assigned to a 1-year wait list (rate ratio, 6.0). In the randomized ANRS IPERGAY trial, 70% of high-risk MSM prescribed PrEP reported engaging in condomless anal intercourse during their most recent sexual encounter, but that proportion remained stable over 24 subsequent months of follow-up.
Providers also should understand that oral PrEP is just as effective at preventing HIV transmission when patients have STIs, Dr. Mayer said. In five recent studies, PrEP was equally efficacious among MSM regardless of whether they had syphilis or other STIs, and bacterial vaginosis in women also did not decrease the efficacy of oral PrEP. “There is no evidence to indicate that the efficacy of PrEP is lower among persons with STIs,” Dr. Mayer said.
Finally, providers should consider screening high-risk individuals for STIs more frequently than every 6 months as recommended by the Centers for Disease Control and Prevention, said Dr. Mayer. “For men who have sex with men, who are sexually active, and are on PrEP, quarterly screening makes exceedingly good sense from a cost-effectiveness standpoint,” he said.
“Screening less frequently than quarterly means that these individuals are having STIs for a longer period of time. When they are sexually active, we have a better chance of interrupting the transmission chain if we detect closer to the time of infection.”
Dr. Mayer disclosed support from the National Institutes of Health and Gilead Sciences, which makes some of the medications used in PrEP regimens.
AT IDWEEK 2017
HIV antiretroviral resistance can affect more than 10% of pregnant women
SAN DIEGO – HIV antiretroviral resistance can affect more than 10% of pregnant women, even if they are previously treatment naive, results of a case-control study demonstrated.
“Furthermore, if there is an HIV-infected infant who received HIV prophylaxis with zidovudine and nevirapine, the infant may have developed resistance to the nonnucleoside reverse transcriptase inhibitors [NNRTIs] class of medications, and timely antiretroviral-resistant testing is an important step prior to choosing an appropriate regimen,” Nava Yeganeh, MD, said in an interview prior to an annual scientific meeting on infectious diseases.

In all, 140 infants were HIV infected, and 13 had drug-resistant mutations. Of the 606 women who had sufficient nucleic acid amplification for resistance testing, 63 (10.4%) had drug-resistant mutations against one or more classes of antiretrovirals. “These mothers may have been infected with a drug-resistant strain of HIV, which they then may have passed on to their infants,” Dr. Yeganeh said. “We also found that 3 of the 13 HIV-infected infants with drug-resistant mutations against NNRTIs were born to mothers who did not have a resistant strain of HIV. These three infants likely developed resistance because of the infant prophylaxis they received with nevirapine.”
Univariate and multivariate analyses revealed that drug-resistant mutation in mothers was not associated with increased risk of HIV mother-to-child transmission (adjusted odds ratio, 0.79). The only predictors of mother-to-child transmission were log HIV viral load (OR, 1.4) and infant prophylaxis arm with a two-drug regimen (OR, 1.6). In addition, the presence of drug-resistant mutations in mothers who transmitted was strongly associated with presence of drug-resistant mutations in infants (P less than .001).
A key limitation of the trial, Dr. Yeganeh said, was that it was completed in 2011. “Antiretroviral-resistant HIV may be even more common now that antiretrovirals are more available,” she said at the combined annual meetings of the Infectious Diseases Society of America, the Society for Healthcare Epidemiology of America, the HIV Medicine Association, and the Pediatric Infectious Diseases Society. She reported having no financial disclosures.
SAN DIEGO – HIV antiretroviral resistance can affect more than 10% of pregnant women, even if they are previously treatment naive, results of a case-control study demonstrated.
“Furthermore, if there is an HIV-infected infant who received HIV prophylaxis with zidovudine and nevirapine, the infant may have developed resistance to the nonnucleoside reverse transcriptase inhibitors [NNRTIs] class of medications, and timely antiretroviral-resistant testing is an important step prior to choosing an appropriate regimen,” Nava Yeganeh, MD, said in an interview prior to an annual scientific meeting on infectious diseases.
In all, 140 infants were HIV infected, and 13 had drug-resistant mutations. Of the 606 women who had sufficient nucleic acid amplification for resistance testing, 63 (10.4%) had drug-resistant mutations against one or more classes of antiretrovirals. “These mothers may have been infected with a drug-resistant strain of HIV, which they then may have passed on to their infants,” Dr. Yeganeh said. “We also found that 3 of the 13 HIV-infected infants with drug-resistant mutations against NNRTIs were born to mothers who did not have a resistant strain of HIV. These three infants likely developed resistance because of the infant prophylaxis they received with nevirapine.”
Univariate and multivariate analyses revealed that drug-resistant mutation in mothers was not associated with increased risk of HIV mother-to-child transmission (adjusted odds ratio, 0.79). The only predictors of mother-to-child transmission were log HIV viral load (OR, 1.4) and infant prophylaxis arm with a two-drug regimen (OR, 1.6). In addition, the presence of drug-resistant mutations in mothers who transmitted was strongly associated with presence of drug-resistant mutations in infants (P less than .001).
A key limitation of the trial, Dr. Yeganeh said, was that it was completed in 2011. “Antiretroviral-resistant HIV may be even more common now that antiretrovirals are more available,” she said at the combined annual meetings of the Infectious Diseases Society of America, the Society for Healthcare Epidemiology of America, the HIV Medicine Association, and the Pediatric Infectious Diseases Society. She reported having no financial disclosures.
SAN DIEGO – HIV antiretroviral resistance can affect more than 10% of pregnant women, even if they are previously treatment naive, results of a case-control study demonstrated.
“Furthermore, if there is an HIV-infected infant who received HIV prophylaxis with zidovudine and nevirapine, the infant may have developed resistance to the nonnucleoside reverse transcriptase inhibitors [NNRTIs] class of medications, and timely antiretroviral-resistant testing is an important step prior to choosing an appropriate regimen,” Nava Yeganeh, MD, said in an interview prior to an annual scientific meeting on infectious diseases.
In all, 140 infants were HIV infected, and 13 had drug-resistant mutations. Of the 606 women who had sufficient nucleic acid amplification for resistance testing, 63 (10.4%) had drug-resistant mutations against one or more classes of antiretrovirals. “These mothers may have been infected with a drug-resistant strain of HIV, which they then may have passed on to their infants,” Dr. Yeganeh said. “We also found that 3 of the 13 HIV-infected infants with drug-resistant mutations against NNRTIs were born to mothers who did not have a resistant strain of HIV. These three infants likely developed resistance because of the infant prophylaxis they received with nevirapine.”
Univariate and multivariate analyses revealed that drug-resistant mutation in mothers was not associated with increased risk of HIV mother-to-child transmission (adjusted odds ratio, 0.79). The only predictors of mother-to-child transmission were log HIV viral load (OR, 1.4) and infant prophylaxis arm with a two-drug regimen (OR, 1.6). In addition, the presence of drug-resistant mutations in mothers who transmitted was strongly associated with presence of drug-resistant mutations in infants (P less than .001).
A key limitation of the trial, Dr. Yeganeh said, was that it was completed in 2011. “Antiretroviral-resistant HIV may be even more common now that antiretrovirals are more available,” she said at the combined annual meetings of the Infectious Diseases Society of America, the Society for Healthcare Epidemiology of America, the HIV Medicine Association, and the Pediatric Infectious Diseases Society. She reported having no financial disclosures.
AT IDWEEK 2017
Key clinical point:
Major finding: Of 606 women who had sufficient nucleic acid amplification for resistance testing, 63 (10.4%) had drug-resistant mutations against one or more classes of antiretrovirals.
Study details: A case-control study of blood samples from 606 HIV-infected pregnant women and their infants.
Disclosures: Dr. Yeganeh reported having no financial disclosures.
Rosacea: Expert recommends treating erythema, papules/pustules simultaneously
The results of a recently published trial helps answer one of the toughest questions in rosacea treatment: when patients present with both papules/pustules and erythema, which problem do you treat first?
In the past, Hilary Baldwin, MD, tended to target papules and pustules first, usually with ivermectin cream (Soolantra) and, when warranted, anti-inflammatory doses of doxycycline. Going after the erythema first and making the skin paler could make the cherry red inflammatory lesions stand out even more, she said.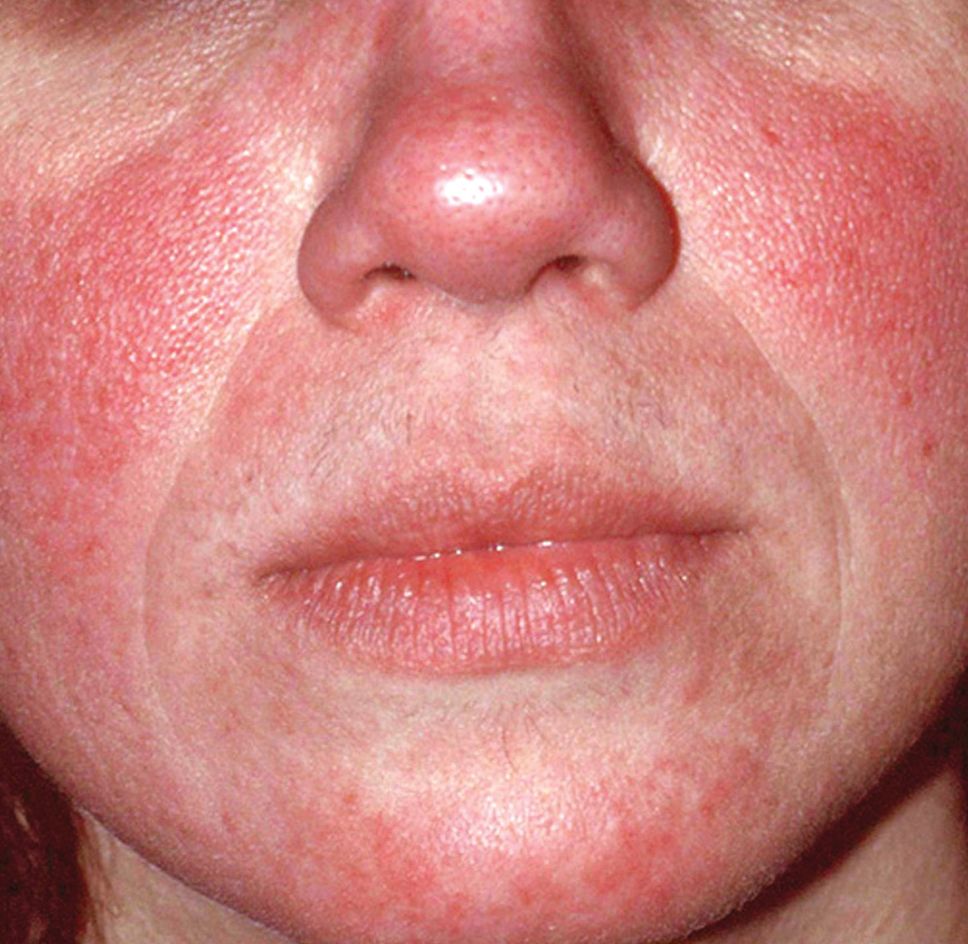
In the study, one group was put on ivermectin for 12 weeks, with the vasoconstrictor brimonidine 0.33% (Mirvaso topical gel) added after 4 weeks to help with the erythema; and another group was treated with both ivermectin 1% cream and brimonidine daily for the entire 12 weeks. The third group received vehicles of both applied every day for 12 weeks (J Drugs Dermatol. 2017 Sep 1;16[9]:909-16).
“What they found was that both the combinations worked better than ivermectin alone,” and that treatment with both agents for the full 12 weeks worked best, with no increased risk of irritation or worsening of erythema than when brimonidine was brought in after 4 weeks of ivermectin, said Dr. Baldwin, a clinical associate professor of dermatology at Rutgers Robert Wood Johnson Medical School, New Brunswick, N.J., said in an interview. The vasoconstriction might have somehow helped with the papules and pustules, she noted.
The lesions might have looked more prominent “for the week or two it took for the ivermectin to kick in, but the patients didn’t care. They were happier campers by virtue of treating both aspects of their rosacea at the same time,” she added.
The new kid on the block for vasoconstriction – oxymetazoline (Rhofade cream) – appears to be gentler than brimonidine. “It takes a little bit longer to reach peak effect, and the peak doesn’t give you quite as much vasoconstriction as brimonidine, which for some people is a good thing,” she said. “Perhaps they’re a little bit too white with brimonidine. For other people who are bright red, oxymetazoline might not be enough. I think there’s a place for both drugs.”
Both vasoconstrictors might actually make erythema temporarily worse; it’s a known side effect. Dr. Baldwin has her patients try them for the first time when they’re at home and don’t have any important impending social engagements, just in case. “I like to give a tube of each one and say, ‘use one on one side of your face and the other on the other side and see which makes you happier.’ ”

Some patients can get away with “a really low dose and be completely cleared,” she commented. “I have some fully controlled on 10 mg twice weekly. As long as there’s no pregnancy risk, there’s no reason you can’t do this almost indefinitely.”
This publication and the Global Academy for Medical Education are both owned by Frontline Medical News. Dr. Baldwin is a speaker and advisor for Allergan, Galderma, and Valeant; and is an investigator for Dermira, Galderma, Novan, and Valeant.
The results of a recently published trial helps answer one of the toughest questions in rosacea treatment: when patients present with both papules/pustules and erythema, which problem do you treat first?
In the past, Hilary Baldwin, MD, tended to target papules and pustules first, usually with ivermectin cream (Soolantra) and, when warranted, anti-inflammatory doses of doxycycline. Going after the erythema first and making the skin paler could make the cherry red inflammatory lesions stand out even more, she said.
In the study, one group was put on ivermectin for 12 weeks, with the vasoconstrictor brimonidine 0.33% (Mirvaso topical gel) added after 4 weeks to help with the erythema; and another group was treated with both ivermectin 1% cream and brimonidine daily for the entire 12 weeks. The third group received vehicles of both applied every day for 12 weeks (J Drugs Dermatol. 2017 Sep 1;16[9]:909-16).
“What they found was that both the combinations worked better than ivermectin alone,” and that treatment with both agents for the full 12 weeks worked best, with no increased risk of irritation or worsening of erythema than when brimonidine was brought in after 4 weeks of ivermectin, said Dr. Baldwin, a clinical associate professor of dermatology at Rutgers Robert Wood Johnson Medical School, New Brunswick, N.J., said in an interview. The vasoconstriction might have somehow helped with the papules and pustules, she noted.
The lesions might have looked more prominent “for the week or two it took for the ivermectin to kick in, but the patients didn’t care. They were happier campers by virtue of treating both aspects of their rosacea at the same time,” she added.
The new kid on the block for vasoconstriction – oxymetazoline (Rhofade cream) – appears to be gentler than brimonidine. “It takes a little bit longer to reach peak effect, and the peak doesn’t give you quite as much vasoconstriction as brimonidine, which for some people is a good thing,” she said. “Perhaps they’re a little bit too white with brimonidine. For other people who are bright red, oxymetazoline might not be enough. I think there’s a place for both drugs.”
Both vasoconstrictors might actually make erythema temporarily worse; it’s a known side effect. Dr. Baldwin has her patients try them for the first time when they’re at home and don’t have any important impending social engagements, just in case. “I like to give a tube of each one and say, ‘use one on one side of your face and the other on the other side and see which makes you happier.’ ”
Some patients can get away with “a really low dose and be completely cleared,” she commented. “I have some fully controlled on 10 mg twice weekly. As long as there’s no pregnancy risk, there’s no reason you can’t do this almost indefinitely.”
This publication and the Global Academy for Medical Education are both owned by Frontline Medical News. Dr. Baldwin is a speaker and advisor for Allergan, Galderma, and Valeant; and is an investigator for Dermira, Galderma, Novan, and Valeant.
The results of a recently published trial helps answer one of the toughest questions in rosacea treatment: when patients present with both papules/pustules and erythema, which problem do you treat first?
In the past, Hilary Baldwin, MD, tended to target papules and pustules first, usually with ivermectin cream (Soolantra) and, when warranted, anti-inflammatory doses of doxycycline. Going after the erythema first and making the skin paler could make the cherry red inflammatory lesions stand out even more, she said.
In the study, one group was put on ivermectin for 12 weeks, with the vasoconstrictor brimonidine 0.33% (Mirvaso topical gel) added after 4 weeks to help with the erythema; and another group was treated with both ivermectin 1% cream and brimonidine daily for the entire 12 weeks. The third group received vehicles of both applied every day for 12 weeks (J Drugs Dermatol. 2017 Sep 1;16[9]:909-16).
“What they found was that both the combinations worked better than ivermectin alone,” and that treatment with both agents for the full 12 weeks worked best, with no increased risk of irritation or worsening of erythema than when brimonidine was brought in after 4 weeks of ivermectin, said Dr. Baldwin, a clinical associate professor of dermatology at Rutgers Robert Wood Johnson Medical School, New Brunswick, N.J., said in an interview. The vasoconstriction might have somehow helped with the papules and pustules, she noted.
The lesions might have looked more prominent “for the week or two it took for the ivermectin to kick in, but the patients didn’t care. They were happier campers by virtue of treating both aspects of their rosacea at the same time,” she added.
The new kid on the block for vasoconstriction – oxymetazoline (Rhofade cream) – appears to be gentler than brimonidine. “It takes a little bit longer to reach peak effect, and the peak doesn’t give you quite as much vasoconstriction as brimonidine, which for some people is a good thing,” she said. “Perhaps they’re a little bit too white with brimonidine. For other people who are bright red, oxymetazoline might not be enough. I think there’s a place for both drugs.”
Both vasoconstrictors might actually make erythema temporarily worse; it’s a known side effect. Dr. Baldwin has her patients try them for the first time when they’re at home and don’t have any important impending social engagements, just in case. “I like to give a tube of each one and say, ‘use one on one side of your face and the other on the other side and see which makes you happier.’ ”
Some patients can get away with “a really low dose and be completely cleared,” she commented. “I have some fully controlled on 10 mg twice weekly. As long as there’s no pregnancy risk, there’s no reason you can’t do this almost indefinitely.”
This publication and the Global Academy for Medical Education are both owned by Frontline Medical News. Dr. Baldwin is a speaker and advisor for Allergan, Galderma, and Valeant; and is an investigator for Dermira, Galderma, Novan, and Valeant.
EXPERT ANALYSIS FROM THE COASTAL DERMATOLOGY SYMPOSIUM
Old and newer systemic therapies benefit patients with chronic eczema
Atopic dermatitis (AD) that becomes chronic and persists into adulthood often becomes less responsive to topical treatment with mid- to high-potency corticosteroids and calcineurin inhibitors, necessitating a different approach.
Joseph F. Fowler Jr., MD, discussed these treatment options at the annual Coastal Dermatology Symposium.
Older systemic medications
These include methotrexate, mycophenolate mofetil, cyclosporine, azathioprine, and retinoids. Methotrexate is predictably effective, and dermatologists generally are comfortable with it. The drug requires monitoring for adverse effects along with other precautions, similar to its use in psoriasis.

Mycophenolate mofetil is useful when the adverse event profiles of azathioprine, methotrexate, and cyclosporin A eliminate them from consideration, but it tends to confer slower improvement and has less efficacy overall.
Cyclosporin A led to successful outcomes in 77% of patients and mild improvement in 16% of patients in one trial, with milder side effects than those commonly seen in transplant patients. There was no increased risk of nephrotoxicity or hypertension over 6 months of treatment. The drug is useful for short-term control of flares and in contact dermatitis when corticosteroids are contraindicated, according to Dr. Fowler of the department of dermatology and director of occupational dermatitis at the University of Louisville (Ky.). It is the only drug other than corticosteroids that offers rapid improvement.
Newer drug options
One is dupilumab (Dupixent), an antibody that blocks interleukin (IL)–4 and IL-13. It received Food and Drug Administration approval in March 2017 for moderate to severe atopic dermatitis that doesn’t respond to topical treatment. Most patients get at least some benefit from the injectable drug, and some get a strong benefit, according to Dr. Fowler, although he pointed out that it can take 12 weeks before it achieves maximum effect. The initial dose is 600 mg administered subcutaneously, followed by 300 mg every 2 weeks. At 16 weeks, it reduced Eczema Area and Severity Index (EASI) scores by about 75% in patients taking a 300 mg dose every other week, compared with about a 20% decline in placebo.
The IL12/23 inhibitor ustekinumab (Stelara), approved for psoriasis and psoriatic arthritis, was effective in a case series of three patients. It led to a greater than 50% reduction in EASI score at week 16, following doses with 45 mg at weeks 0, 4, and 12. Biopsies revealed reductions in Th22 cells and cytokine levels. But, as a caveat, Dr. Fowler reported personal communication with two eczema experts who said they had seen little or no effect with ustekinumab in 10 patients.
The Janus kinase inhibitor tofacitinib, approved for rheumatoid arthritis, also is under investigation for AD. A trial in six patients who had not achieved adequate control with methotrexate or azathioprine showed a 67% improvement in the SCORAD (Scoring Atopic Dermatitis) index at doses of 5 mg of tofacitinib twice per day in five patients and 5 mg every other day in one patient, he said.
The PDE-4 inhibitor apremilast, approved for psoriasis and psoriatic arthritis, has been reported to improve AD symptoms in individual patients. Celgene demonstrated some improvement in a clinical trial of apremilast at doses of 30 or 40 mg twice per day, but the company isn’t pursuing AD as an indication, he noted.
Dr. Fowler has consulted for Abbvie, IntraDerm, and SmartPractice. He is on the speaker’s bureaus of SmartPractice and Regeneron/Sanofi. He has been a research investigator for Abbvie, Allergan, Amgen, Bayer, Dow, Galderma, Genentech, InnovaDerm, Johnson & Johnson, Lilly, Merck, Novartis, Pfizer, Precision Dermatology, Regeneron, SmartPractice, Taro, and Valeant. This publication and the Global Academy for Medical Education are owned by Frontline Medical News.
Atopic dermatitis (AD) that becomes chronic and persists into adulthood often becomes less responsive to topical treatment with mid- to high-potency corticosteroids and calcineurin inhibitors, necessitating a different approach.
Joseph F. Fowler Jr., MD, discussed these treatment options at the annual Coastal Dermatology Symposium.
Older systemic medications
These include methotrexate, mycophenolate mofetil, cyclosporine, azathioprine, and retinoids. Methotrexate is predictably effective, and dermatologists generally are comfortable with it. The drug requires monitoring for adverse effects along with other precautions, similar to its use in psoriasis.
Mycophenolate mofetil is useful when the adverse event profiles of azathioprine, methotrexate, and cyclosporin A eliminate them from consideration, but it tends to confer slower improvement and has less efficacy overall.
Cyclosporin A led to successful outcomes in 77% of patients and mild improvement in 16% of patients in one trial, with milder side effects than those commonly seen in transplant patients. There was no increased risk of nephrotoxicity or hypertension over 6 months of treatment. The drug is useful for short-term control of flares and in contact dermatitis when corticosteroids are contraindicated, according to Dr. Fowler of the department of dermatology and director of occupational dermatitis at the University of Louisville (Ky.). It is the only drug other than corticosteroids that offers rapid improvement.
Newer drug options
One is dupilumab (Dupixent), an antibody that blocks interleukin (IL)–4 and IL-13. It received Food and Drug Administration approval in March 2017 for moderate to severe atopic dermatitis that doesn’t respond to topical treatment. Most patients get at least some benefit from the injectable drug, and some get a strong benefit, according to Dr. Fowler, although he pointed out that it can take 12 weeks before it achieves maximum effect. The initial dose is 600 mg administered subcutaneously, followed by 300 mg every 2 weeks. At 16 weeks, it reduced Eczema Area and Severity Index (EASI) scores by about 75% in patients taking a 300 mg dose every other week, compared with about a 20% decline in placebo.
The IL12/23 inhibitor ustekinumab (Stelara), approved for psoriasis and psoriatic arthritis, was effective in a case series of three patients. It led to a greater than 50% reduction in EASI score at week 16, following doses with 45 mg at weeks 0, 4, and 12. Biopsies revealed reductions in Th22 cells and cytokine levels. But, as a caveat, Dr. Fowler reported personal communication with two eczema experts who said they had seen little or no effect with ustekinumab in 10 patients.
The Janus kinase inhibitor tofacitinib, approved for rheumatoid arthritis, also is under investigation for AD. A trial in six patients who had not achieved adequate control with methotrexate or azathioprine showed a 67% improvement in the SCORAD (Scoring Atopic Dermatitis) index at doses of 5 mg of tofacitinib twice per day in five patients and 5 mg every other day in one patient, he said.
The PDE-4 inhibitor apremilast, approved for psoriasis and psoriatic arthritis, has been reported to improve AD symptoms in individual patients. Celgene demonstrated some improvement in a clinical trial of apremilast at doses of 30 or 40 mg twice per day, but the company isn’t pursuing AD as an indication, he noted.
Dr. Fowler has consulted for Abbvie, IntraDerm, and SmartPractice. He is on the speaker’s bureaus of SmartPractice and Regeneron/Sanofi. He has been a research investigator for Abbvie, Allergan, Amgen, Bayer, Dow, Galderma, Genentech, InnovaDerm, Johnson & Johnson, Lilly, Merck, Novartis, Pfizer, Precision Dermatology, Regeneron, SmartPractice, Taro, and Valeant. This publication and the Global Academy for Medical Education are owned by Frontline Medical News.
Atopic dermatitis (AD) that becomes chronic and persists into adulthood often becomes less responsive to topical treatment with mid- to high-potency corticosteroids and calcineurin inhibitors, necessitating a different approach.
Joseph F. Fowler Jr., MD, discussed these treatment options at the annual Coastal Dermatology Symposium.
Older systemic medications
These include methotrexate, mycophenolate mofetil, cyclosporine, azathioprine, and retinoids. Methotrexate is predictably effective, and dermatologists generally are comfortable with it. The drug requires monitoring for adverse effects along with other precautions, similar to its use in psoriasis.
Mycophenolate mofetil is useful when the adverse event profiles of azathioprine, methotrexate, and cyclosporin A eliminate them from consideration, but it tends to confer slower improvement and has less efficacy overall.
Cyclosporin A led to successful outcomes in 77% of patients and mild improvement in 16% of patients in one trial, with milder side effects than those commonly seen in transplant patients. There was no increased risk of nephrotoxicity or hypertension over 6 months of treatment. The drug is useful for short-term control of flares and in contact dermatitis when corticosteroids are contraindicated, according to Dr. Fowler of the department of dermatology and director of occupational dermatitis at the University of Louisville (Ky.). It is the only drug other than corticosteroids that offers rapid improvement.
Newer drug options
One is dupilumab (Dupixent), an antibody that blocks interleukin (IL)–4 and IL-13. It received Food and Drug Administration approval in March 2017 for moderate to severe atopic dermatitis that doesn’t respond to topical treatment. Most patients get at least some benefit from the injectable drug, and some get a strong benefit, according to Dr. Fowler, although he pointed out that it can take 12 weeks before it achieves maximum effect. The initial dose is 600 mg administered subcutaneously, followed by 300 mg every 2 weeks. At 16 weeks, it reduced Eczema Area and Severity Index (EASI) scores by about 75% in patients taking a 300 mg dose every other week, compared with about a 20% decline in placebo.
The IL12/23 inhibitor ustekinumab (Stelara), approved for psoriasis and psoriatic arthritis, was effective in a case series of three patients. It led to a greater than 50% reduction in EASI score at week 16, following doses with 45 mg at weeks 0, 4, and 12. Biopsies revealed reductions in Th22 cells and cytokine levels. But, as a caveat, Dr. Fowler reported personal communication with two eczema experts who said they had seen little or no effect with ustekinumab in 10 patients.
The Janus kinase inhibitor tofacitinib, approved for rheumatoid arthritis, also is under investigation for AD. A trial in six patients who had not achieved adequate control with methotrexate or azathioprine showed a 67% improvement in the SCORAD (Scoring Atopic Dermatitis) index at doses of 5 mg of tofacitinib twice per day in five patients and 5 mg every other day in one patient, he said.
The PDE-4 inhibitor apremilast, approved for psoriasis and psoriatic arthritis, has been reported to improve AD symptoms in individual patients. Celgene demonstrated some improvement in a clinical trial of apremilast at doses of 30 or 40 mg twice per day, but the company isn’t pursuing AD as an indication, he noted.
Dr. Fowler has consulted for Abbvie, IntraDerm, and SmartPractice. He is on the speaker’s bureaus of SmartPractice and Regeneron/Sanofi. He has been a research investigator for Abbvie, Allergan, Amgen, Bayer, Dow, Galderma, Genentech, InnovaDerm, Johnson & Johnson, Lilly, Merck, Novartis, Pfizer, Precision Dermatology, Regeneron, SmartPractice, Taro, and Valeant. This publication and the Global Academy for Medical Education are owned by Frontline Medical News.
EXPERT ANALYSIS FROM THE COASTAL DERMATOLOGY SYMPOSIUM
Surgical left atrial appendage closure slashes stroke risk
BARCELONA – Routine surgical closure of the left atrial appendage during open heart surgery provides long-term protection against cerebral ischemic events, according to the findings of the first-ever randomized controlled trial to address the issue.
“I think we can say, based on our study, that it would be advisable to routinely add surgical closure of the left atrial appendage to planned open heart surgery,” Jesper Park-Hansen, MD, said at the annual congress of the European Society of Cardiology.

In light of the demonstrated success of percutaneous closure of the LAA using the Watchman and other devices for stroke prevention in patients with atrial fibrillation, Dr. Park-Hansen and his coinvestigators at the University of Copenhagen organized LAACS (the Left Atrial Appendage Closure Study). The goal was to generate solid, randomized trial evidence as to whether preemptive routine surgical closure of the LAA at the time of cardiac surgery is of benefit. Some cardiac surgeons already do this routinely; many others don’t because of the lack of Level 1 supporting evidence.
LAACS included 141 patients randomized to surgical LAA closure or not at the point of first-time open heart surgery. The study population included patients with and without a history of atrial fibrillation. LAA closure was accomplished via a purse string closure with a silk string around the neck of the appendage backed up by an additional single running suture. Transesophageal echocardiography performed in 10 patients a mean of 520 days post closure showed no signs of leakage or incomplete closure.
The primary composite outcome was comprised of clinical stroke or transient ischemic attack diagnosed by a neurologist, or a silent cerebral infarct detected on MRI performed 2-4 weeks post discharge and again at least 6 months later. At a mean follow-up of 3.7 years and a maximum of 6 years, this outcome had occurred in 6.3% of the LAAC group, significantly lower than the 18.3% rate in controls. All but one patient with a cerebral ischemic event in the control group had atrial fibrillation. The risk of an event was unrelated to whether or not a patient had a history of atrial fibrillation prior to surgery or to CHA2DS2-VASc score.
Dr. Park-Hansen emphasized that he and his coinvestigators don’t consider LAACS to be the final word on routine prophylactic appendage closure.
“This is the first randomized study. We are eager to move on to another randomized study on a larger scale. That is the next step for us,” he said.
“The challenge now – and what we will be discussing with our surgeons – is to agree on a feasible safe and effective means of left atrial appendage closure. My personal opinion is the Lariat suture delivery device or some other easily reproducible method of closure could be a good way to go,” Dr. Park-Hansen added.
The research group’s cardiac surgeons already have ruled out excision and stapling because of concerns about bleeding risk and the additional cost imposed by stapling.
Discussant Volkmar Falk, MD, commented that LAACS was too small, probably severely underpowered, should have included a preoperative MRI so investigators could reliably capture perioperative silent cerebral infarcts, and the double suture purse string is “probably not the best method” to occlude the LAA.
“LAACS addresses an important question, but alas, it does not provide the answer,” declared Dr. Falk, professor and director of the department of cardiothoracic and vascular surgery at Charité Medical University in Berlin.
Dr. Park-Hansen and Dr. Falk reported having no financial conflicts of interest.
BARCELONA – Routine surgical closure of the left atrial appendage during open heart surgery provides long-term protection against cerebral ischemic events, according to the findings of the first-ever randomized controlled trial to address the issue.
“I think we can say, based on our study, that it would be advisable to routinely add surgical closure of the left atrial appendage to planned open heart surgery,” Jesper Park-Hansen, MD, said at the annual congress of the European Society of Cardiology.
In light of the demonstrated success of percutaneous closure of the LAA using the Watchman and other devices for stroke prevention in patients with atrial fibrillation, Dr. Park-Hansen and his coinvestigators at the University of Copenhagen organized LAACS (the Left Atrial Appendage Closure Study). The goal was to generate solid, randomized trial evidence as to whether preemptive routine surgical closure of the LAA at the time of cardiac surgery is of benefit. Some cardiac surgeons already do this routinely; many others don’t because of the lack of Level 1 supporting evidence.
LAACS included 141 patients randomized to surgical LAA closure or not at the point of first-time open heart surgery. The study population included patients with and without a history of atrial fibrillation. LAA closure was accomplished via a purse string closure with a silk string around the neck of the appendage backed up by an additional single running suture. Transesophageal echocardiography performed in 10 patients a mean of 520 days post closure showed no signs of leakage or incomplete closure.
The primary composite outcome was comprised of clinical stroke or transient ischemic attack diagnosed by a neurologist, or a silent cerebral infarct detected on MRI performed 2-4 weeks post discharge and again at least 6 months later. At a mean follow-up of 3.7 years and a maximum of 6 years, this outcome had occurred in 6.3% of the LAAC group, significantly lower than the 18.3% rate in controls. All but one patient with a cerebral ischemic event in the control group had atrial fibrillation. The risk of an event was unrelated to whether or not a patient had a history of atrial fibrillation prior to surgery or to CHA2DS2-VASc score.
Dr. Park-Hansen emphasized that he and his coinvestigators don’t consider LAACS to be the final word on routine prophylactic appendage closure.
“This is the first randomized study. We are eager to move on to another randomized study on a larger scale. That is the next step for us,” he said.
“The challenge now – and what we will be discussing with our surgeons – is to agree on a feasible safe and effective means of left atrial appendage closure. My personal opinion is the Lariat suture delivery device or some other easily reproducible method of closure could be a good way to go,” Dr. Park-Hansen added.
The research group’s cardiac surgeons already have ruled out excision and stapling because of concerns about bleeding risk and the additional cost imposed by stapling.
Discussant Volkmar Falk, MD, commented that LAACS was too small, probably severely underpowered, should have included a preoperative MRI so investigators could reliably capture perioperative silent cerebral infarcts, and the double suture purse string is “probably not the best method” to occlude the LAA.
“LAACS addresses an important question, but alas, it does not provide the answer,” declared Dr. Falk, professor and director of the department of cardiothoracic and vascular surgery at Charité Medical University in Berlin.
Dr. Park-Hansen and Dr. Falk reported having no financial conflicts of interest.
BARCELONA – Routine surgical closure of the left atrial appendage during open heart surgery provides long-term protection against cerebral ischemic events, according to the findings of the first-ever randomized controlled trial to address the issue.
“I think we can say, based on our study, that it would be advisable to routinely add surgical closure of the left atrial appendage to planned open heart surgery,” Jesper Park-Hansen, MD, said at the annual congress of the European Society of Cardiology.
In light of the demonstrated success of percutaneous closure of the LAA using the Watchman and other devices for stroke prevention in patients with atrial fibrillation, Dr. Park-Hansen and his coinvestigators at the University of Copenhagen organized LAACS (the Left Atrial Appendage Closure Study). The goal was to generate solid, randomized trial evidence as to whether preemptive routine surgical closure of the LAA at the time of cardiac surgery is of benefit. Some cardiac surgeons already do this routinely; many others don’t because of the lack of Level 1 supporting evidence.
LAACS included 141 patients randomized to surgical LAA closure or not at the point of first-time open heart surgery. The study population included patients with and without a history of atrial fibrillation. LAA closure was accomplished via a purse string closure with a silk string around the neck of the appendage backed up by an additional single running suture. Transesophageal echocardiography performed in 10 patients a mean of 520 days post closure showed no signs of leakage or incomplete closure.
The primary composite outcome was comprised of clinical stroke or transient ischemic attack diagnosed by a neurologist, or a silent cerebral infarct detected on MRI performed 2-4 weeks post discharge and again at least 6 months later. At a mean follow-up of 3.7 years and a maximum of 6 years, this outcome had occurred in 6.3% of the LAAC group, significantly lower than the 18.3% rate in controls. All but one patient with a cerebral ischemic event in the control group had atrial fibrillation. The risk of an event was unrelated to whether or not a patient had a history of atrial fibrillation prior to surgery or to CHA2DS2-VASc score.
Dr. Park-Hansen emphasized that he and his coinvestigators don’t consider LAACS to be the final word on routine prophylactic appendage closure.
“This is the first randomized study. We are eager to move on to another randomized study on a larger scale. That is the next step for us,” he said.
“The challenge now – and what we will be discussing with our surgeons – is to agree on a feasible safe and effective means of left atrial appendage closure. My personal opinion is the Lariat suture delivery device or some other easily reproducible method of closure could be a good way to go,” Dr. Park-Hansen added.
The research group’s cardiac surgeons already have ruled out excision and stapling because of concerns about bleeding risk and the additional cost imposed by stapling.
Discussant Volkmar Falk, MD, commented that LAACS was too small, probably severely underpowered, should have included a preoperative MRI so investigators could reliably capture perioperative silent cerebral infarcts, and the double suture purse string is “probably not the best method” to occlude the LAA.
“LAACS addresses an important question, but alas, it does not provide the answer,” declared Dr. Falk, professor and director of the department of cardiothoracic and vascular surgery at Charité Medical University in Berlin.
Dr. Park-Hansen and Dr. Falk reported having no financial conflicts of interest.
AT THE ESC CONGRESS 2017
Key clinical point:
Major finding: The composite rate of clinical stroke, TIA, or silent cerebral infarct in the years following open heart surgery was threefold higher in patients randomized to no prophylactic surgical closure of the left atrial appendage, compared with patients who got appendage closure during their surgery.
Data source: A randomized trial in which 141 patients undergoing first-time open heart surgery were assigned to prophylactic surgical closure of the left atrial appendage or not.
Disclosures: The study was conducted free of commercial support. The presenter reported having no financial conflicts of interest.
Obinutuzumab edges out rituximab for PFS in follicular lymphoma
In a head-to-head trial of anti-CD20 monoclonal antibodies in first-line therapy for follicular lymphoma, obinutuzumab-based chemotherapy was associated with slightly but significantly better progression-free survival than rituximab-based therapy, but at the cost of higher toxicities, including severe adverse events.
Among 1,202 patients with follicular lymphoma followed for a median of 34.5 months, the estimated 3-year rate of progression-free survival (PFS) for patients randomized to obinutuzumab-based chemotherapy and maintenance was 80%, compared with 73.3% for patients randomized to rituximab chemotherapy and maintenance. Response rates and overall survival were similar between the treatment groups, Robert Marcus, MB, BS, of King’s College Hospital, London, and his coinvestigators reported in the GALLIUM trial.

They acknowledged, however, that there were substantial differences between the treatment groups in the cumulative doses of obinutuzumab (Gazyva) and rituximab (Rituxan and others), which could have affected the relative efficacy of each regimen.
In addition, while patients were randomly assigned to one monoclonal antibody or the other, the choice of chemotherapy regimens, while standardized, was left to the discretion of investigators at each treatment site, another factor that might have influenced outcomes.
The investigators reported the results of a preplanned interim efficacy analysis. They compared obinutuzumab or rituximab plus chemotherapy in patients with indolent non-Hodgkin lymphoma, but the trial was powered to detect a PFS difference only in patients with follicular lymphoma. Patients who had a clinical response to induction therapy went on to maintenance therapy with the same monoclonal antibody.
In all, 1,202 patients with follicular lymphoma were enrolled and randomized, 601 in each arm, to receive induction with either intravenous obinutuzumab 1,000 mg on days 1, 8, and 15 of cycle 1 and on day 1 of subsequent cycles, or rituximab 375 mg/m2 on day 1 of each cycle for six or eight cycles, depending on the accompanying chemotherapy regimen. The regimens used were either CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), or bendamustine.
Patients with partial or complete responses were then maintained on the same monoclonal antibody they had received during induction, either obinutuzumab 1,000 mg or rituximab 375 mg/m2 every 2 months for 2 years, or until disease progression. Patients were not allowed to be crossed over to the other maintenance therapy.
Patients with stable disease after induction continued to be followed, but did not receive maintenance therapy.
The interim analysis was performed after 245 of 370 anticipated events (disease progression, relapse, or death) had occurred. At that time, the independent data and safety monitoring committee recommended full analysis of the trial data, and the sponsor agreed.
After a median follow-up of 34.5 months, an intention-to-treat analysis showed that the investigator-assessed, estimated 3-year rate of PFS was 80.0% in the obinutuzumab arm, compared with 73.3%; in the rituximab arm. This translated into a hazard ratio (HR) for progression, relapse, or death of 0.66 (P = .001). An independent review committee calculated a HR favoring obinutuzumab of 0.71 (P = .01).
Estimated 3-year overall survival rates were not significantly different at 94% and 92.1%, respectively.
Overall response rates were similar between the groups, at 88.5% with obinutuzumab group and 86.9% with rituximab, a difference that was not significant.
Obinutuzumab was associated with a higher rate of prespecified events of special interest, including infections, cardiac events, second neoplasms, infusion-related events, neutropenia, and thrombocytopenia.
Adverse events deemed to be related to the antibodies occurred in 59.3% of patients on obinutuzumab, and 48.9% of patients on rituximab.
There were more frequent grade 3 or 4 adverse events and deaths with obinutuzumab, occurring in 74.6% of patients vs. 67.8% on rituximab. Fatal adverse events occurred in 4% and 3.4% of patients, respectively.
A total of 81 patients died during the trial, including 35 in the obinutuzumab group and 46 in the rituximab group.
F. Hoffmann–La Roche supported the trial. Dr. Marcus disclosed consulting fees and lecture fees from Takeda Pharmaceuticals and travel support, consulting fees, and lecture fees from Roche. The majority of coauthors disclosed similar relationships.
Should obinutuzumab replace rituximab as the standard antibody in the treatment of patients receiving chemoimmunotherapy regimens for follicular lymphoma? Results from this trial would suggest that there might be no advantage for an obinutuzumab-containing chemoimmunotherapy regimen if maintenance treatment was not planned. Even with maintenance therapy, there is no evidence from this trial of an overall survival benefit with obinutuzumab. These findings, combined with the higher rate of toxic effects and, presumably, the higher cost of obinutuzumab, raise important questions regarding the advantage of its use. This issue is complicated further because it is possible that giving rituximab at a dose of 1,000 mg might reduce or eliminate any difference in progression-free survival – that is, if the difference is primarily a dose effect.
When the data on minimal residual disease are made available, the case in favor of obinutuzumab may appear to be more compelling if indeed a higher proportion of patients who received obinutuzumab have minimal residual disease status at some point in treatment and remain in remission longer than those who received rituximab. At the moment, the competition between these agents looks too close to call.
These comments are excerpted from an editorial (N Engl J Med. 2017 Oct 5;377;14:1389-90) by James O. Armitage, MD, University of Nebraska, Omaha, and Dan L. Longo, MD, Dana-Farber Cancer Institute, Boston. Dr. Armitage reported personal fees from Conatus, Samus Therapeutics, and Tesaro. Dr. Longo reported no relevant disclosures. He is deputy editor of The New England Journal of Medicine.
Should obinutuzumab replace rituximab as the standard antibody in the treatment of patients receiving chemoimmunotherapy regimens for follicular lymphoma? Results from this trial would suggest that there might be no advantage for an obinutuzumab-containing chemoimmunotherapy regimen if maintenance treatment was not planned. Even with maintenance therapy, there is no evidence from this trial of an overall survival benefit with obinutuzumab. These findings, combined with the higher rate of toxic effects and, presumably, the higher cost of obinutuzumab, raise important questions regarding the advantage of its use. This issue is complicated further because it is possible that giving rituximab at a dose of 1,000 mg might reduce or eliminate any difference in progression-free survival – that is, if the difference is primarily a dose effect.
When the data on minimal residual disease are made available, the case in favor of obinutuzumab may appear to be more compelling if indeed a higher proportion of patients who received obinutuzumab have minimal residual disease status at some point in treatment and remain in remission longer than those who received rituximab. At the moment, the competition between these agents looks too close to call.
These comments are excerpted from an editorial (N Engl J Med. 2017 Oct 5;377;14:1389-90) by James O. Armitage, MD, University of Nebraska, Omaha, and Dan L. Longo, MD, Dana-Farber Cancer Institute, Boston. Dr. Armitage reported personal fees from Conatus, Samus Therapeutics, and Tesaro. Dr. Longo reported no relevant disclosures. He is deputy editor of The New England Journal of Medicine.
Should obinutuzumab replace rituximab as the standard antibody in the treatment of patients receiving chemoimmunotherapy regimens for follicular lymphoma? Results from this trial would suggest that there might be no advantage for an obinutuzumab-containing chemoimmunotherapy regimen if maintenance treatment was not planned. Even with maintenance therapy, there is no evidence from this trial of an overall survival benefit with obinutuzumab. These findings, combined with the higher rate of toxic effects and, presumably, the higher cost of obinutuzumab, raise important questions regarding the advantage of its use. This issue is complicated further because it is possible that giving rituximab at a dose of 1,000 mg might reduce or eliminate any difference in progression-free survival – that is, if the difference is primarily a dose effect.
When the data on minimal residual disease are made available, the case in favor of obinutuzumab may appear to be more compelling if indeed a higher proportion of patients who received obinutuzumab have minimal residual disease status at some point in treatment and remain in remission longer than those who received rituximab. At the moment, the competition between these agents looks too close to call.
These comments are excerpted from an editorial (N Engl J Med. 2017 Oct 5;377;14:1389-90) by James O. Armitage, MD, University of Nebraska, Omaha, and Dan L. Longo, MD, Dana-Farber Cancer Institute, Boston. Dr. Armitage reported personal fees from Conatus, Samus Therapeutics, and Tesaro. Dr. Longo reported no relevant disclosures. He is deputy editor of The New England Journal of Medicine.
In a head-to-head trial of anti-CD20 monoclonal antibodies in first-line therapy for follicular lymphoma, obinutuzumab-based chemotherapy was associated with slightly but significantly better progression-free survival than rituximab-based therapy, but at the cost of higher toxicities, including severe adverse events.
Among 1,202 patients with follicular lymphoma followed for a median of 34.5 months, the estimated 3-year rate of progression-free survival (PFS) for patients randomized to obinutuzumab-based chemotherapy and maintenance was 80%, compared with 73.3% for patients randomized to rituximab chemotherapy and maintenance. Response rates and overall survival were similar between the treatment groups, Robert Marcus, MB, BS, of King’s College Hospital, London, and his coinvestigators reported in the GALLIUM trial.
They acknowledged, however, that there were substantial differences between the treatment groups in the cumulative doses of obinutuzumab (Gazyva) and rituximab (Rituxan and others), which could have affected the relative efficacy of each regimen.
In addition, while patients were randomly assigned to one monoclonal antibody or the other, the choice of chemotherapy regimens, while standardized, was left to the discretion of investigators at each treatment site, another factor that might have influenced outcomes.
The investigators reported the results of a preplanned interim efficacy analysis. They compared obinutuzumab or rituximab plus chemotherapy in patients with indolent non-Hodgkin lymphoma, but the trial was powered to detect a PFS difference only in patients with follicular lymphoma. Patients who had a clinical response to induction therapy went on to maintenance therapy with the same monoclonal antibody.
In all, 1,202 patients with follicular lymphoma were enrolled and randomized, 601 in each arm, to receive induction with either intravenous obinutuzumab 1,000 mg on days 1, 8, and 15 of cycle 1 and on day 1 of subsequent cycles, or rituximab 375 mg/m2 on day 1 of each cycle for six or eight cycles, depending on the accompanying chemotherapy regimen. The regimens used were either CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), or bendamustine.
Patients with partial or complete responses were then maintained on the same monoclonal antibody they had received during induction, either obinutuzumab 1,000 mg or rituximab 375 mg/m2 every 2 months for 2 years, or until disease progression. Patients were not allowed to be crossed over to the other maintenance therapy.
Patients with stable disease after induction continued to be followed, but did not receive maintenance therapy.
The interim analysis was performed after 245 of 370 anticipated events (disease progression, relapse, or death) had occurred. At that time, the independent data and safety monitoring committee recommended full analysis of the trial data, and the sponsor agreed.
After a median follow-up of 34.5 months, an intention-to-treat analysis showed that the investigator-assessed, estimated 3-year rate of PFS was 80.0% in the obinutuzumab arm, compared with 73.3%; in the rituximab arm. This translated into a hazard ratio (HR) for progression, relapse, or death of 0.66 (P = .001). An independent review committee calculated a HR favoring obinutuzumab of 0.71 (P = .01).
Estimated 3-year overall survival rates were not significantly different at 94% and 92.1%, respectively.
Overall response rates were similar between the groups, at 88.5% with obinutuzumab group and 86.9% with rituximab, a difference that was not significant.
Obinutuzumab was associated with a higher rate of prespecified events of special interest, including infections, cardiac events, second neoplasms, infusion-related events, neutropenia, and thrombocytopenia.
Adverse events deemed to be related to the antibodies occurred in 59.3% of patients on obinutuzumab, and 48.9% of patients on rituximab.
There were more frequent grade 3 or 4 adverse events and deaths with obinutuzumab, occurring in 74.6% of patients vs. 67.8% on rituximab. Fatal adverse events occurred in 4% and 3.4% of patients, respectively.
A total of 81 patients died during the trial, including 35 in the obinutuzumab group and 46 in the rituximab group.
F. Hoffmann–La Roche supported the trial. Dr. Marcus disclosed consulting fees and lecture fees from Takeda Pharmaceuticals and travel support, consulting fees, and lecture fees from Roche. The majority of coauthors disclosed similar relationships.
In a head-to-head trial of anti-CD20 monoclonal antibodies in first-line therapy for follicular lymphoma, obinutuzumab-based chemotherapy was associated with slightly but significantly better progression-free survival than rituximab-based therapy, but at the cost of higher toxicities, including severe adverse events.
Among 1,202 patients with follicular lymphoma followed for a median of 34.5 months, the estimated 3-year rate of progression-free survival (PFS) for patients randomized to obinutuzumab-based chemotherapy and maintenance was 80%, compared with 73.3% for patients randomized to rituximab chemotherapy and maintenance. Response rates and overall survival were similar between the treatment groups, Robert Marcus, MB, BS, of King’s College Hospital, London, and his coinvestigators reported in the GALLIUM trial.
They acknowledged, however, that there were substantial differences between the treatment groups in the cumulative doses of obinutuzumab (Gazyva) and rituximab (Rituxan and others), which could have affected the relative efficacy of each regimen.
In addition, while patients were randomly assigned to one monoclonal antibody or the other, the choice of chemotherapy regimens, while standardized, was left to the discretion of investigators at each treatment site, another factor that might have influenced outcomes.
The investigators reported the results of a preplanned interim efficacy analysis. They compared obinutuzumab or rituximab plus chemotherapy in patients with indolent non-Hodgkin lymphoma, but the trial was powered to detect a PFS difference only in patients with follicular lymphoma. Patients who had a clinical response to induction therapy went on to maintenance therapy with the same monoclonal antibody.
In all, 1,202 patients with follicular lymphoma were enrolled and randomized, 601 in each arm, to receive induction with either intravenous obinutuzumab 1,000 mg on days 1, 8, and 15 of cycle 1 and on day 1 of subsequent cycles, or rituximab 375 mg/m2 on day 1 of each cycle for six or eight cycles, depending on the accompanying chemotherapy regimen. The regimens used were either CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), or bendamustine.
Patients with partial or complete responses were then maintained on the same monoclonal antibody they had received during induction, either obinutuzumab 1,000 mg or rituximab 375 mg/m2 every 2 months for 2 years, or until disease progression. Patients were not allowed to be crossed over to the other maintenance therapy.
Patients with stable disease after induction continued to be followed, but did not receive maintenance therapy.
The interim analysis was performed after 245 of 370 anticipated events (disease progression, relapse, or death) had occurred. At that time, the independent data and safety monitoring committee recommended full analysis of the trial data, and the sponsor agreed.
After a median follow-up of 34.5 months, an intention-to-treat analysis showed that the investigator-assessed, estimated 3-year rate of PFS was 80.0% in the obinutuzumab arm, compared with 73.3%; in the rituximab arm. This translated into a hazard ratio (HR) for progression, relapse, or death of 0.66 (P = .001). An independent review committee calculated a HR favoring obinutuzumab of 0.71 (P = .01).
Estimated 3-year overall survival rates were not significantly different at 94% and 92.1%, respectively.
Overall response rates were similar between the groups, at 88.5% with obinutuzumab group and 86.9% with rituximab, a difference that was not significant.
Obinutuzumab was associated with a higher rate of prespecified events of special interest, including infections, cardiac events, second neoplasms, infusion-related events, neutropenia, and thrombocytopenia.
Adverse events deemed to be related to the antibodies occurred in 59.3% of patients on obinutuzumab, and 48.9% of patients on rituximab.
There were more frequent grade 3 or 4 adverse events and deaths with obinutuzumab, occurring in 74.6% of patients vs. 67.8% on rituximab. Fatal adverse events occurred in 4% and 3.4% of patients, respectively.
A total of 81 patients died during the trial, including 35 in the obinutuzumab group and 46 in the rituximab group.
F. Hoffmann–La Roche supported the trial. Dr. Marcus disclosed consulting fees and lecture fees from Takeda Pharmaceuticals and travel support, consulting fees, and lecture fees from Roche. The majority of coauthors disclosed similar relationships.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Obinutuzumab-based chemotherapy and maintenance was associated with better progression-free survival, but not overall survival, compared with rituximab-based chemotherapy and maintenance.
Major finding: Three-year progression-free survival was 80% with obinutuzumab, vs. 73.3% with rituximab.
Data source: Interim analysis of a randomized phase 3, open-label trial of 1,202 patients with follicular lymphoma.
Disclosures: F. Hoffmann–La Roche supported the trial. Dr. Marcus disclosed consulting fees and lecture fees from Takeda Pharmaceuticals and travel support, consulting fees, and lecture fees from Roche. The majority of coauthors disclosed similar relationships.