User login
Contraceptive care best practices
While the unintended pregnancy rate for women ages 15 to 44 years decreased by 18% between 2008 and 2011, almost half of pregnancies in the United States remain unintended.1 On a more positive note, however, women who use birth control consistently and correctly account for only 5% of unintended pregnancies.2 As family physicians (FPs), we can support and facilitate our female patients’ efforts to consistently use highly effective forms of contraception. The 5 initiatives detailed here can help toward that end.
1. Routinely screen patients for their reproductive intentions
All women of reproductive age should be screened routinely for their pregnancy intentions. The American College of Obstetricians and Gynecologists (ACOG) encourages clinicians to ask women about pregnancy intendedness and encourages patients to develop a reproductive life plan, or a set of personal goals about whether or when to have children.3 The Centers for Disease Control and Prevention (CDC) has also developed a reproductive life plan tool for health professionals to encourage women and men to reflect upon their plans.4 So just as we regularly screen and document cigarette use and blood pressure (BP), so too, should we routinely screen women for their reproductive goals.
Ask women this one question. The Oregon Foundation for Reproductive Health launched the One Key Question Initiative, which proposes that the care team ask women ages 18 to 50: “Would you like to become pregnant in the next year?”5 A common workflow includes the medical assistant asking women about pregnancy intentions and providing a preconception and/or contraceptive handout, if appropriate. The physician provides additional counseling as needed. Pilot studies of One Key Question indicate that 30% to 40% of women screened needed follow-up counseling, suggesting the need for clinicians to be proactive in asking about reproductive plans. (Additional information on the Initiative is available on the Foundation’s Web site at http://www.orfrh.org/.)
This approach assumes women feel in control of their reproduction; however, this may not be the reality for many, especially low-income women.6 Additionally, women commonly cite planning a pregnancy as appropriate only when they are in an ideal relationship and when they are living in a financially stable environment—conditions that some women may never achieve.
Another caveat is that women may not have explicit pregnancy intentions, in which case, this particular approach may not be effective. A study of low-income women found only 60% intended to use the method prescribed after contraception counseling, with 37% of those stopping because of adverse effects, 23% saying they wanted another method, and 17% citing method complexity.7
Reproductive coercion from male partners, ranging from pressure to become pregnant to method sabotage, is also common in low-income women.8 Regular conversations that prioritize a woman’s values and experience are needed to promote reproductive autonomy.
2. Decouple provision of contraception from unnecessary exams
Pelvic exams and pap smears should not be required prior to offering patients hormonal contraception, according to the Choosing Wisely campaign of the American Board of Internal Medicine and ACOG.9,10 Hormonal contraception may instead be provided safely based on a medical history and BP assessment. Adolescents, minority groups, obese women, and victims of sexual trauma, in particular may avoid asking about birth control because of anxiety and fear of pain from these exams.11 The American College of Physicians recommends against speculum and bimanual exams in asymptomatic, non-pregnant, adult women.12 Pap smears and sexually transmitted infection (STI) testing should be performed at their normally scheduled intervals as recommended by the US Preventive Services Task Force (USPSTF) and not be tied to contraceptive provision.13
Assess pregnancy status using criteria,rather than a pregnancy text
Use the CDC’s criteria to assess pregnancy status rather than relying on a urine pregnancy test prior to providing contraception. Once you are reasonably sure that a woman is not pregnant (TABLE 114), contraception may be started. Some physicians have traditionally requested that a woman delay starting contraception until the next menses to ensure that she is not already pregnant. However, given the evidence that hormonal contraception does not cause birth defects, such a delay is not warranted and puts the woman at risk of an unintended pregnancy during the gap.15

Furthermore, there is an approximate 2-week window in which a woman could have a negative urine pregnancy test despite being pregnant, so the test alone is not completely reliable. In addition, obese women may experience irregular cycles, further complicating the traditional approach.16
Another largely unnecessary step … The US Selected Practice Recommendations (US SPR) from the CDC notes that additional STI screening prior to an intrauterine device (IUD) insertion is unnecessary for most women if appropriate screening guidelines have been previously followed.14 For those who have not been screened according to guidelines, the CDC recommends same-day screening and IUD insertion. You can then treat an STI without removing the IUD. Women with purulent cervicitis or a current chlamydial or gonorrheal infection should delay IUD insertion until after treatment.
3. Expand long-acting reversible contraception counseling and access
Offer long-acting reversible contraception (LARC), such as IUDs and implants, as first-line options for most women. ACOG endorses LARC as the most effective reversible method for most women, including those who have not given birth and adolescents.17 Unfortunately, a 2012 study found that family physicians were less likely than OB-GYNs to have enough time for contraceptive counseling and fewer than half felt competent inserting IUDs.18 While 79% of OB-GYNs routinely discussed IUDs with their patients, only 47% of family physicians did. In 2014, the American Academy of Pediatrics (AAP) endorsed a LARC-first tiered counseling approach for adolescents.19
A test of LARC-first counseling
The Contraceptive CHOICE project, a St. Louis, Missouri-based initiative, was launched to reduce unintended pregnancies in women ages 14 to 45 years by offering LARC-first counseling and free contraception of their choice.20 This project involved more than 9000 women at high risk for unintended pregnancy. Same-day LARC insertion was available. Seventy-five percent of women chose a LARC method and they reported greater continuation at 12 and 24 months, when compared to women who did not choose a LARC method. LARC users also reported higher satisfaction at one year. Provision of contraception through the project contributed to a reduction in repeat abortions as well as decreased rates of teenage pregnancy, birth, and abortion. Three years after the start of the project, IUDs had continuation rates of nearly 70%, implants of 56%, and non-LARC methods of 31%.21
When counseling women, it’s important to remember that effectiveness may not be the only criterium a woman uses when choosing a method. A 2010 study found that for 91% of women at high risk for unintended pregnancy, no single method possessed all the features they deemed “extremely important.”22 Clinicians should take a patient-centered approach to find birth control that fits each patient’s priorities.
Clinicians need proper training in LARC methods
Only 20% of FPs regularly insert IUDs, and 11% offer contraceptive implants, according to estimates from physicians recertifying with the American Board of Family Medicine in 2014.23 Access to training during residency is a key component to increasing these rates. FPs who practice obstetrics should be trained in postpartum LARC insertion and offer this option prior to hospital discharge as well as during the postpartum office visit.
Performing LARC insertions on the same day as counseling is ideal, and clinics should strive to reduce barriers to same-day procedures. Time constraints may be addressed by shifting tasks among the medical team. In the CHOICE project, contraceptive counselors—half of whom had no clinical experience—were trained to provide tiered counseling to participants. By working with a cross-trained health care team and offering prepared resources, clinicians can save time and improve access.
Physicians may want to incorporate the free online resources Bedsider.org or Stayteen.org to help women learn about contraceptive methods.24 The user-friendly Web sites, operated by the National Campaign to Prevent Teen and Unplanned Pregnancy, describe various forms of contraception and offer text and email reminders. Incorporating Bedsider into the counseling workflow and discussing the various reminder tools available may improve patients’ knowledge and enhance their compliance.
Additional barriers for practices may include high upfront costs associated with stocking devices. Practices that may be unable to sustain the costs surrounding enhanced contraception counseling and provision can collaborate with family planning clinics that are able to offer same-day services. A study of clinics in California found that Title X clinics were more likely to provide on-site LARC services than non-Title X public and private providers.25
4. Follow CDC guidelines for initiating and continuing contraception
Follow the US SPR for guidance on initiating and continuing contraceptive methods.14 The CDC’s Medical Eligibility Criteria for Contraceptive Use is another vital resource, providing recommendations for contraceptive methods to patients who have specific medical conditions or characteristics.26
Utilize the “quick start” method for hormonal contraception, where birth control is started on the same day as its prescription regardless of timing of the menstrual cycle. If you can’t be reasonably certain that a woman is not pregnant based on the criteria listed in TABLE 1,14 conduct a pregnancy test (while recognizing the aforementioned 2-week window of limitations) and counsel the patient to use back-up protection for the first 7 days along with repeating a pregnancy test in 2 weeks’ time.
The quick start method may lead to higher adherence than delayed initiation.27 Differences in continuation rates between women who use the quick start method and those who follow the delayed approach may disappear over time.28
Prescribe and provide a year’s supply of oral contraceptive pills (OCPs) as recommended by the CDC US SPR.14 It is important to note that pharmacists are usually restricted by insurance companies to only fill a one or 3 month’s supply.
In January 2016, Oregon began requiring private and state health insurance providers to reimburse for a year’s supply of prescription contraception; in January 2017, insurers in Washington, DC, were also required to offer women a year’s supply of prescription contraception.29,30 Several other states have followed suit. The California Health Benefits Review Program estimates a savings of $42.8 million a year from fewer office visits and 15,000 fewer unintended pregnancies if their state enacts a similar policy.31
Pharmacist initiatives are worth watching. In January 2016, Oregon pharmacists with additional training were allowed to prescribe OCs and hormonal patches to women 18 years and older.32 In April 2016, a similar law went into effect in California, but without a minimum age requirement and with the additional coverage of vaginal rings and Depo-Provera (depo) injections.33 Pharmacists in both states must review a health questionnaire completed by the woman and can refer to a physician as necessary.
The CDC recommends that clinicians extend the allowed window for repeat depo injections to 15 weeks.14 Common institutional protocol is to give repeat injections every 11 to 13 weeks. If past that window, protocol often dictates the woman abstain from unprotected sex for 2 weeks and then return for a negative pregnancy test (or await menses) before the next injection. However, the CDC notes that depo is effective for longer than the 13-week period.14 No additional birth control or pregnancy testing is needed and the woman can receive the next depo shot if she is up to 15 weeks from the previous shot.
One study found no additional pregnancy risks for those who were up to 4 weeks “late” for their next shot, suggesting there is potential for an even larger grace period.34 The World Health Organization advises allowing a repeat injection up to 4 weeks late.35 We encourage institutions to change their policies to comply with the CDC’s 15-week window.
Another initiative is over-the-counter (OTC) access to OCs, which the American Academy of Family Physicians (AAFP) and ACOG support.36,37 ACOG notes that “no drug or intervention is completely without risk of harm” and that the risk of venous thromboembolism for OC users is lower than the risk of pregnancy.37 Women can successfully self-screen for contraindications using a checklist. Concerns about women potentially being less adherent or less likely to choose LARCs are not reasons to preclude access to other methods. The AAFP supports insurance coverage of OCs, regardless of prescription status.36
5. Routinely counsel about, and advance-prescribe, emergency contraception pills
Physicians should counsel and advance-prescribe emergency contraception pills (ECPs) to women, including adolescents, using less reliable contraception, as recommended by ACOG, AAP, and the CDC.14,37,38 It’s also important to provide information on the copper IUD as the most effective method of emergency contraception, with nearly 100% efficacy if placed within 5 days.39 An easy-to-read patient hand-out in English and Spanish on EC options can be found at http://beyondthepill.ucsf.edu/tools-materials.
Only 3% of respondents participating in the 2006-2010 National Survey of Family Growth received counseling about emergency contraception in the past year.40 ECPs are most effective when used within 24 hours but have some efficacy up to 5 days.37 Due to the Affordable Care Act, most insurance plans will cover ECPs if purchased with a prescription, but coverage varies by state.41 Ulipristal acetate (UPA) ECP is only available with a prescription. Advance prescriptions can alleviate financial burdens on women when they need to access ECPs quickly.
Women should wait at least 5 days before resuming or starting hormonal contraception after taking UPA-based ECP, as it may reduce the ovulation-delaying effect of the ECP.14 For IUDs, implants, and depo, which require a visit to a health care provider, physicians evaluating earlier provision should consider the risks of reduced efficacy against the many barriers to access.
UPA-based ECPs (such as ella) may be more effective for overweight and obese women than levonorgestrel-based ECPs (such as Plan B and Next Choice).14 Consider advance-prescribing UPA ECPs to women with a body mass index (BMI) >25 kg/m2.42 Such considerations are important as the prevalence of obesity in women between 2013 and 2014 was 40.4%.43
In May 2016, the FDA noted that while current data are insufficient regarding whether the effectiveness of levonorgestrel ECPs is reduced in overweight or obese women, there are no safety concerns regarding their use in this population.44 Therefore, a woman with a BMI >25 kg/m2 should use UPA ECPs if available; but if not, she can still use levonorgestrel ECPs. One study, however, has found that UPA ECPs are only as effective as a placebo when BMI is ≥35 kg/m2, at which point a copper IUD may be the only effective form of emergency contraception.45
Transitioning from customary practices to best practices
Following these practical steps, FPs can improve contraceptive care for women. However, to make a significant impact, clinicians must be willing to change customary practices that are based on tradition, routines, or outdated protocols in favor of those based on current evidence.

One good place to start the transition to best practices is to familiarize yourself with the 2016 US Medical Eligibility Criteria for Contraceptive Use26 and Selected Practice Recommendations for Contraceptive Use.14 TABLES 214,26,46,47 and 3 offer additional resources that can enhance contraceptive counseling and further promote access to contraceptive care.

The contraceptive coverage guarantee under the Affordable Care Act has allowed many women to make contraceptive choices based on personal needs and preferences rather than cost. The new contraceptive coverage exemptions issued under the Trump administration will bring cost back as the driving decision factor for women whose employers choose not to provide contraceptive coverage. Providers should be aware of the typical costs associated with the various contraceptive options offered in their practice and community.
CORRESPONDENCE
Jessica Dalby, MD, Department of Family Medicine and Community Health, University of Wisconsin School of Medicine and Public Health, 1102 South Park St, Suite 100, Madison, WI 53715; [email protected].
1. Finer LB, Zolna MR. Declines in unintended pregnancy in the United States, 2008–2011. N Engl J Med. 2016; 374:843-852.
2. Sonfield A, Hasstedt K, Gold RB. Moving Forward: Family Planning in the Era of Health Reform. New York: Guttmacher Institute. 2014. Available at: https://www.guttmacher.org/report/moving-forward-family-planning-era-health-reform. Accessed October 5, 2017.
3. Committee on Health Care for Underserved Women. Reproductive Life Planning to Reduce Unintended Pregnancy: American College of Obstetricians and Gynecologists. 2016. Available at: https://www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Health-Care-for-Underserved-Women/Reproductive-Life-Planning-to-Reduce-Unintended-Pregnancy. Accessed October 5, 2017.
4. Centers for Disease Control and Prevention. Reproductive Life Plan Tool for Health Care Providers. 2016. Available at: http://www.cdc.gov/preconception/rlptool.html. Accessed August 31, 2016.
5. Oregon Health Authority. Effective Contraceptive Use among Women at Risk of Unintended Pregnancy Guidance Document. 2014. Available at: http://www.oregon.gov/oha/HPA/ANALYTICS/CCOData/Effective%20Contraceptive%20Use%20Guidance%20Document.pdf. Accessed October 5, 2017.
6. Borrero S, Nikolajski C, Steinberg JR, et al. “It just happens”: a qualitative study exploring low-income women’s perspectives on pregnancy intention and planning. Contraception. 2015;91:150-156.
7. Yee LM, Farner KC, King E, et al. What do women want? Experiences of low-income women with postpartum contraception and contraceptive counseling. J Pregnancy Child Health. 2015;2.
8. Kalichman SC, Williams EA, Cherry C, et al. Sexual coercion, domestic violence, and negotiating condom use among low-income African American women. J Womens Health. 1998;7:371-378.
9. ABIM Foundation. Pelvic Exams, Pap Tests and Oral Contraceptives. 2016. Available at: http://www.choosingwisely.org/patient-resources/pelvic-exams-pap-tests-and-oral-contraceptives/. Accessed May 31, 2016.
10. Committee on Health Care for Underserved Women. Access to Contraception: American College of Obstetricians and Gynecologists. 2015. Number 615. Available at: https://www.acog.org/-/media/Committee-Opinions/Committee-on-Health-Care-for-Underserved-Women/co615.pdf?dmc=1&ts=201710. Accessed October 5, 2017.
11. Bates CK, Carroll N, Potter J. The challenging pelvic examination. J Gen Intern Med. 2011;26:651-657.
12. Qaseem A, Humphrey LL, Harris R, et al. Screening pelvic examination in adult women: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2014;161:67-72.
13. U.S. Preventive Services Task Force. Cervical Cancer: Screening. 2012. Available at: http://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/cervical-cancer-screening. Accessed May 25, 2016.
14. Curtis KM, Jatlaoui TC, Tepper NK, et al. U.S. selected practice recommendations for contraceptive use, 2016. MMWR Morb Mortal Wkly Rep. 2016;65:1-66.
15. Lesnewski R, Prine L. Initiating hormonal contraception. Am Fam Physician. 2006;74:105-112.
16. Jacobsen BK, Knutsen SF, Oda K, et al. Obesity at age 20 and the risk of miscarriages, irregular periods and reported problems of becoming pregnant: the Adventist Health Study-2. Eur J Epidemiol. 2012; 27:923-931.
17. Committee on Gynecologic Practice. Increasing Access to Contraceptive Implants and Intrauterine Devices to Reduce Unintended Pregnancy: American College of Obstetricians and Gynecologists. 2015. Number 642. Available at: https://www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Gynecologic-Practice/Increasing-Access-to-Contraceptive-Implants-and-Intrauterine-Devices-to-Reduce-Unintended-Pregnancy. Accessed October 5, 2017.
18. Harper CC, Henderson JT, Raine TR, et al. Evidence-based IUD practice: family physicians and obstetrician-gynecologists. Fam Med. 2012;44:637-645.
19. American Academy of Pediatrics, Committee on Adolescence. Policy statement: Contraception for Adolescents. 2014. Available at: http://pediatrics.aappublications.org/content/pediatrics/early/2014/09/24/peds.2014-2299.full.pdf. Accessed October 5, 2017.
20. Birgisson NE, Zhao Q, Secura GM, et al. Preventing unintended pregnancy: The Contraceptive CHOICE Project in review. J Womens Health (Larchmt). 2015;24:349-353.
21. Diedrich JT, Zhao Q, Madden T, et al. Three-year continuation of reversible contraception. Am J Obstet Gynecol. 2015;213:662.e1-e8.
22. Lessard LN, Karasek D, Ma S, et al. Contraceptive features preferred by women at high risk of unintended pregnancy. Perspect Sex Reprod Health. 2012;44:194-200.
23. Nisen MB, Peterson LE, Cochrane A, et al. US family physicians’ intrauterine and implantable contraception provision: results from a national survey. Contraception. 2016;93:432-437.
24. National Campaign to Prevent Teen and Unplanned Pregnancy. Bedsider. Available at: https://bedsider.org/. Accessed June 14, 2016.
25. Park HY, Rodriguez MI, Hulett D, et al. Long-acting reversible contraception method use among Title X providers and non-Title X providers in California. Contraception. 2012;86:557-561.
26. Curtis KM, Tepper NK, Jatlaoui TC, et al. U.S. medical eligibility criteria for contraceptive use, 2016. MMWR Morb Mortal Wkly Rep. 2016;65:1-103.
27. Westhoff C, Kerns J, Morroni C, et al. Quick start: novel oral contraceptive initiation method. Contraception. 2002;66:141-145.
28. Brahmi D, Curtis KM. When can a woman start combined hormonal contraceptives (CHCs)? A systematic review. Contraception. 2013;87:524-538.
29. Lachman S. Oregon To Require Insurers To Cover A Year’s Supply Of Birth Control. Huffington Post. June 11, 2015. Available at: https://www.huffingtonpost.com/2015/06/11/oregon-birth-control-_n_7564712.html. Accessed October 16, 2017.
30. Andrews M. D.C. Women To Get Access To Full Year’s Worth Of Contraceptives. Kaiser Health News. September 25, 2015. Available at: https://khn.org/news/d-c-women-to-get-access-to-full-years-worth-of-contraceptives/. Accessed October 16, 2017.
31. Analysis of California Senate Bill (SB) 999 Contraceptives: Annual Supply: A Report to the 2015-2016 California State Legislature: California Health Benefits Review Program. 2016. Available at: http://chbrp.ucop.edu/index.php?action=read&bill_id=195&doc_type=1000. Accessed October 5, 2017.
32. Frazier A. Pharmacist-prescribed birth control in effect Jan 1. KOIN News. December 30, 2015. Available at: http://koin.com/2015/12/30/pharmacist-provided-birth-control-in-effect-jan-1/. Accessed October 5, 2017.
33. Karlamangla S. Birth control pills without prescriptions, coming soon to California under new law. Los Angeles Times. February 14, 2016. Available at: http://www.latimes.com/health/la-me-birth-control-pharmacies-20160214-story.html. Accessed October 16, 2017.
34. Steiner MJ, Kwok C, Stanback J, et al. Injectable contraception: what should the longest interval be for reinjections? Contraception. 2008;77:410-414.
35. World Health Organization. Family Planning: A Global Handbook for Providers. 2011. Available at: http://apps.who.int/iris/bitstream/10665/44028/1/9780978856373_eng.pdf. Accessed October 5, 2017.
36. American Academy of Family Physicians. Over-the-Counter Oral Contraceptives. 2014; Available at: http://www.aafp.org/about/policies/all/otc-oral-contraceptives.html. Accessed June 2, 2016.
37. Committee on Gynecologic Practice. Over-the-Counter Access to Oral Contraceptives: American College of Obstetricians and Gynecologists. 2012. Number 544. Available at: https://www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Gynecologic-Practice/Over-the-Counter-Access-to-Oral-Contraceptives. Accessed October 5, 2017.
38. Committee on Adolescence. Emergency contraception. Pediatrics. 2012;130:1174-1182.
39. Cleland K, Zhu H, Goldstuck N, et al. The efficacy of intrauterine devices for emergency contraception: a systematic review of 35 years of experience. Hum Reprod. 2012;27:1994-2000.
40. Martinez G, Chandra A, Febo-Vazquez I, et al. Use of Family Planning and Related Medical Services Among Women Aged 15–44 in the United States: National Survey of Family Growth, 2006–2010: National Center for Health Statistics, Centers for Disease Control and Prevention. 2013. Available at: https://www.cdc.gov/nchs/data/nhsr/nhsr068.pdf. Accessed October 5, 2017.
41. Guttmacher Institute. Insurance Coverage of Contraceptives: Guttmacher Institute;2017. Available at: https://www.guttmacher.org/state-policy/explore/insurance-coverage-contraceptives Accessed October 7, 2017.
42. Glasier A, Cameron ST, Blithe D, et al. Can we identify women at risk of pregnancy despite using emergency contraception? Data from randomized trials of ulipristal acetate and levonorgestrel. Contraception. 2011;84:363-367.
43. Flegal KM, Kruszon-Moran D, Carroll MD, et al. Trends in obesity among adults in the United States, 2005 to 2014. JAMA. 2016;315:2284-2291.
44. US Food & Drug Administration. Postmarket Drug Safety Information for Patients and Providers - Plan B (0.75mg levonorgestrel) and Plan B One-Step (1.5 mg levonorgestrel) Tablets Information. 2016; Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm109775.htm. Accessed May 25, 2016.
45. Simmons KB, Edelman AB. Contraception and sexual health in obese women. Best Pract Res Clin Obstet Gynaecol. 2015;29:466-478.
46. Centers for Disease Control and Prevention. Providing quality family planning services: recommendations of CDC and the U.S. Office of Population Affairs. MMWR Recomm Rep. 2014;63:1-29.
47. LARC FIRST. Available at: http://www.larcfirst.com/index.html. Accessed May 2016.
While the unintended pregnancy rate for women ages 15 to 44 years decreased by 18% between 2008 and 2011, almost half of pregnancies in the United States remain unintended.1 On a more positive note, however, women who use birth control consistently and correctly account for only 5% of unintended pregnancies.2 As family physicians (FPs), we can support and facilitate our female patients’ efforts to consistently use highly effective forms of contraception. The 5 initiatives detailed here can help toward that end.
1. Routinely screen patients for their reproductive intentions
All women of reproductive age should be screened routinely for their pregnancy intentions. The American College of Obstetricians and Gynecologists (ACOG) encourages clinicians to ask women about pregnancy intendedness and encourages patients to develop a reproductive life plan, or a set of personal goals about whether or when to have children.3 The Centers for Disease Control and Prevention (CDC) has also developed a reproductive life plan tool for health professionals to encourage women and men to reflect upon their plans.4 So just as we regularly screen and document cigarette use and blood pressure (BP), so too, should we routinely screen women for their reproductive goals.
Ask women this one question. The Oregon Foundation for Reproductive Health launched the One Key Question Initiative, which proposes that the care team ask women ages 18 to 50: “Would you like to become pregnant in the next year?”5 A common workflow includes the medical assistant asking women about pregnancy intentions and providing a preconception and/or contraceptive handout, if appropriate. The physician provides additional counseling as needed. Pilot studies of One Key Question indicate that 30% to 40% of women screened needed follow-up counseling, suggesting the need for clinicians to be proactive in asking about reproductive plans. (Additional information on the Initiative is available on the Foundation’s Web site at http://www.orfrh.org/.)
This approach assumes women feel in control of their reproduction; however, this may not be the reality for many, especially low-income women.6 Additionally, women commonly cite planning a pregnancy as appropriate only when they are in an ideal relationship and when they are living in a financially stable environment—conditions that some women may never achieve.
Another caveat is that women may not have explicit pregnancy intentions, in which case, this particular approach may not be effective. A study of low-income women found only 60% intended to use the method prescribed after contraception counseling, with 37% of those stopping because of adverse effects, 23% saying they wanted another method, and 17% citing method complexity.7
Reproductive coercion from male partners, ranging from pressure to become pregnant to method sabotage, is also common in low-income women.8 Regular conversations that prioritize a woman’s values and experience are needed to promote reproductive autonomy.
2. Decouple provision of contraception from unnecessary exams
Pelvic exams and pap smears should not be required prior to offering patients hormonal contraception, according to the Choosing Wisely campaign of the American Board of Internal Medicine and ACOG.9,10 Hormonal contraception may instead be provided safely based on a medical history and BP assessment. Adolescents, minority groups, obese women, and victims of sexual trauma, in particular may avoid asking about birth control because of anxiety and fear of pain from these exams.11 The American College of Physicians recommends against speculum and bimanual exams in asymptomatic, non-pregnant, adult women.12 Pap smears and sexually transmitted infection (STI) testing should be performed at their normally scheduled intervals as recommended by the US Preventive Services Task Force (USPSTF) and not be tied to contraceptive provision.13
Assess pregnancy status using criteria,rather than a pregnancy text
Use the CDC’s criteria to assess pregnancy status rather than relying on a urine pregnancy test prior to providing contraception. Once you are reasonably sure that a woman is not pregnant (TABLE 114), contraception may be started. Some physicians have traditionally requested that a woman delay starting contraception until the next menses to ensure that she is not already pregnant. However, given the evidence that hormonal contraception does not cause birth defects, such a delay is not warranted and puts the woman at risk of an unintended pregnancy during the gap.15
Furthermore, there is an approximate 2-week window in which a woman could have a negative urine pregnancy test despite being pregnant, so the test alone is not completely reliable. In addition, obese women may experience irregular cycles, further complicating the traditional approach.16
Another largely unnecessary step … The US Selected Practice Recommendations (US SPR) from the CDC notes that additional STI screening prior to an intrauterine device (IUD) insertion is unnecessary for most women if appropriate screening guidelines have been previously followed.14 For those who have not been screened according to guidelines, the CDC recommends same-day screening and IUD insertion. You can then treat an STI without removing the IUD. Women with purulent cervicitis or a current chlamydial or gonorrheal infection should delay IUD insertion until after treatment.
3. Expand long-acting reversible contraception counseling and access
Offer long-acting reversible contraception (LARC), such as IUDs and implants, as first-line options for most women. ACOG endorses LARC as the most effective reversible method for most women, including those who have not given birth and adolescents.17 Unfortunately, a 2012 study found that family physicians were less likely than OB-GYNs to have enough time for contraceptive counseling and fewer than half felt competent inserting IUDs.18 While 79% of OB-GYNs routinely discussed IUDs with their patients, only 47% of family physicians did. In 2014, the American Academy of Pediatrics (AAP) endorsed a LARC-first tiered counseling approach for adolescents.19
A test of LARC-first counseling
The Contraceptive CHOICE project, a St. Louis, Missouri-based initiative, was launched to reduce unintended pregnancies in women ages 14 to 45 years by offering LARC-first counseling and free contraception of their choice.20 This project involved more than 9000 women at high risk for unintended pregnancy. Same-day LARC insertion was available. Seventy-five percent of women chose a LARC method and they reported greater continuation at 12 and 24 months, when compared to women who did not choose a LARC method. LARC users also reported higher satisfaction at one year. Provision of contraception through the project contributed to a reduction in repeat abortions as well as decreased rates of teenage pregnancy, birth, and abortion. Three years after the start of the project, IUDs had continuation rates of nearly 70%, implants of 56%, and non-LARC methods of 31%.21
When counseling women, it’s important to remember that effectiveness may not be the only criterium a woman uses when choosing a method. A 2010 study found that for 91% of women at high risk for unintended pregnancy, no single method possessed all the features they deemed “extremely important.”22 Clinicians should take a patient-centered approach to find birth control that fits each patient’s priorities.
Clinicians need proper training in LARC methods
Only 20% of FPs regularly insert IUDs, and 11% offer contraceptive implants, according to estimates from physicians recertifying with the American Board of Family Medicine in 2014.23 Access to training during residency is a key component to increasing these rates. FPs who practice obstetrics should be trained in postpartum LARC insertion and offer this option prior to hospital discharge as well as during the postpartum office visit.
Performing LARC insertions on the same day as counseling is ideal, and clinics should strive to reduce barriers to same-day procedures. Time constraints may be addressed by shifting tasks among the medical team. In the CHOICE project, contraceptive counselors—half of whom had no clinical experience—were trained to provide tiered counseling to participants. By working with a cross-trained health care team and offering prepared resources, clinicians can save time and improve access.
Physicians may want to incorporate the free online resources Bedsider.org or Stayteen.org to help women learn about contraceptive methods.24 The user-friendly Web sites, operated by the National Campaign to Prevent Teen and Unplanned Pregnancy, describe various forms of contraception and offer text and email reminders. Incorporating Bedsider into the counseling workflow and discussing the various reminder tools available may improve patients’ knowledge and enhance their compliance.
Additional barriers for practices may include high upfront costs associated with stocking devices. Practices that may be unable to sustain the costs surrounding enhanced contraception counseling and provision can collaborate with family planning clinics that are able to offer same-day services. A study of clinics in California found that Title X clinics were more likely to provide on-site LARC services than non-Title X public and private providers.25
4. Follow CDC guidelines for initiating and continuing contraception
Follow the US SPR for guidance on initiating and continuing contraceptive methods.14 The CDC’s Medical Eligibility Criteria for Contraceptive Use is another vital resource, providing recommendations for contraceptive methods to patients who have specific medical conditions or characteristics.26
Utilize the “quick start” method for hormonal contraception, where birth control is started on the same day as its prescription regardless of timing of the menstrual cycle. If you can’t be reasonably certain that a woman is not pregnant based on the criteria listed in TABLE 1,14 conduct a pregnancy test (while recognizing the aforementioned 2-week window of limitations) and counsel the patient to use back-up protection for the first 7 days along with repeating a pregnancy test in 2 weeks’ time.
The quick start method may lead to higher adherence than delayed initiation.27 Differences in continuation rates between women who use the quick start method and those who follow the delayed approach may disappear over time.28
Prescribe and provide a year’s supply of oral contraceptive pills (OCPs) as recommended by the CDC US SPR.14 It is important to note that pharmacists are usually restricted by insurance companies to only fill a one or 3 month’s supply.
In January 2016, Oregon began requiring private and state health insurance providers to reimburse for a year’s supply of prescription contraception; in January 2017, insurers in Washington, DC, were also required to offer women a year’s supply of prescription contraception.29,30 Several other states have followed suit. The California Health Benefits Review Program estimates a savings of $42.8 million a year from fewer office visits and 15,000 fewer unintended pregnancies if their state enacts a similar policy.31
Pharmacist initiatives are worth watching. In January 2016, Oregon pharmacists with additional training were allowed to prescribe OCs and hormonal patches to women 18 years and older.32 In April 2016, a similar law went into effect in California, but without a minimum age requirement and with the additional coverage of vaginal rings and Depo-Provera (depo) injections.33 Pharmacists in both states must review a health questionnaire completed by the woman and can refer to a physician as necessary.
The CDC recommends that clinicians extend the allowed window for repeat depo injections to 15 weeks.14 Common institutional protocol is to give repeat injections every 11 to 13 weeks. If past that window, protocol often dictates the woman abstain from unprotected sex for 2 weeks and then return for a negative pregnancy test (or await menses) before the next injection. However, the CDC notes that depo is effective for longer than the 13-week period.14 No additional birth control or pregnancy testing is needed and the woman can receive the next depo shot if she is up to 15 weeks from the previous shot.
One study found no additional pregnancy risks for those who were up to 4 weeks “late” for their next shot, suggesting there is potential for an even larger grace period.34 The World Health Organization advises allowing a repeat injection up to 4 weeks late.35 We encourage institutions to change their policies to comply with the CDC’s 15-week window.
Another initiative is over-the-counter (OTC) access to OCs, which the American Academy of Family Physicians (AAFP) and ACOG support.36,37 ACOG notes that “no drug or intervention is completely without risk of harm” and that the risk of venous thromboembolism for OC users is lower than the risk of pregnancy.37 Women can successfully self-screen for contraindications using a checklist. Concerns about women potentially being less adherent or less likely to choose LARCs are not reasons to preclude access to other methods. The AAFP supports insurance coverage of OCs, regardless of prescription status.36
5. Routinely counsel about, and advance-prescribe, emergency contraception pills
Physicians should counsel and advance-prescribe emergency contraception pills (ECPs) to women, including adolescents, using less reliable contraception, as recommended by ACOG, AAP, and the CDC.14,37,38 It’s also important to provide information on the copper IUD as the most effective method of emergency contraception, with nearly 100% efficacy if placed within 5 days.39 An easy-to-read patient hand-out in English and Spanish on EC options can be found at http://beyondthepill.ucsf.edu/tools-materials.
Only 3% of respondents participating in the 2006-2010 National Survey of Family Growth received counseling about emergency contraception in the past year.40 ECPs are most effective when used within 24 hours but have some efficacy up to 5 days.37 Due to the Affordable Care Act, most insurance plans will cover ECPs if purchased with a prescription, but coverage varies by state.41 Ulipristal acetate (UPA) ECP is only available with a prescription. Advance prescriptions can alleviate financial burdens on women when they need to access ECPs quickly.
Women should wait at least 5 days before resuming or starting hormonal contraception after taking UPA-based ECP, as it may reduce the ovulation-delaying effect of the ECP.14 For IUDs, implants, and depo, which require a visit to a health care provider, physicians evaluating earlier provision should consider the risks of reduced efficacy against the many barriers to access.
UPA-based ECPs (such as ella) may be more effective for overweight and obese women than levonorgestrel-based ECPs (such as Plan B and Next Choice).14 Consider advance-prescribing UPA ECPs to women with a body mass index (BMI) >25 kg/m2.42 Such considerations are important as the prevalence of obesity in women between 2013 and 2014 was 40.4%.43
In May 2016, the FDA noted that while current data are insufficient regarding whether the effectiveness of levonorgestrel ECPs is reduced in overweight or obese women, there are no safety concerns regarding their use in this population.44 Therefore, a woman with a BMI >25 kg/m2 should use UPA ECPs if available; but if not, she can still use levonorgestrel ECPs. One study, however, has found that UPA ECPs are only as effective as a placebo when BMI is ≥35 kg/m2, at which point a copper IUD may be the only effective form of emergency contraception.45
Transitioning from customary practices to best practices
Following these practical steps, FPs can improve contraceptive care for women. However, to make a significant impact, clinicians must be willing to change customary practices that are based on tradition, routines, or outdated protocols in favor of those based on current evidence.
One good place to start the transition to best practices is to familiarize yourself with the 2016 US Medical Eligibility Criteria for Contraceptive Use26 and Selected Practice Recommendations for Contraceptive Use.14 TABLES 214,26,46,47 and 3 offer additional resources that can enhance contraceptive counseling and further promote access to contraceptive care.
The contraceptive coverage guarantee under the Affordable Care Act has allowed many women to make contraceptive choices based on personal needs and preferences rather than cost. The new contraceptive coverage exemptions issued under the Trump administration will bring cost back as the driving decision factor for women whose employers choose not to provide contraceptive coverage. Providers should be aware of the typical costs associated with the various contraceptive options offered in their practice and community.
CORRESPONDENCE
Jessica Dalby, MD, Department of Family Medicine and Community Health, University of Wisconsin School of Medicine and Public Health, 1102 South Park St, Suite 100, Madison, WI 53715; [email protected].
While the unintended pregnancy rate for women ages 15 to 44 years decreased by 18% between 2008 and 2011, almost half of pregnancies in the United States remain unintended.1 On a more positive note, however, women who use birth control consistently and correctly account for only 5% of unintended pregnancies.2 As family physicians (FPs), we can support and facilitate our female patients’ efforts to consistently use highly effective forms of contraception. The 5 initiatives detailed here can help toward that end.
1. Routinely screen patients for their reproductive intentions
All women of reproductive age should be screened routinely for their pregnancy intentions. The American College of Obstetricians and Gynecologists (ACOG) encourages clinicians to ask women about pregnancy intendedness and encourages patients to develop a reproductive life plan, or a set of personal goals about whether or when to have children.3 The Centers for Disease Control and Prevention (CDC) has also developed a reproductive life plan tool for health professionals to encourage women and men to reflect upon their plans.4 So just as we regularly screen and document cigarette use and blood pressure (BP), so too, should we routinely screen women for their reproductive goals.
Ask women this one question. The Oregon Foundation for Reproductive Health launched the One Key Question Initiative, which proposes that the care team ask women ages 18 to 50: “Would you like to become pregnant in the next year?”5 A common workflow includes the medical assistant asking women about pregnancy intentions and providing a preconception and/or contraceptive handout, if appropriate. The physician provides additional counseling as needed. Pilot studies of One Key Question indicate that 30% to 40% of women screened needed follow-up counseling, suggesting the need for clinicians to be proactive in asking about reproductive plans. (Additional information on the Initiative is available on the Foundation’s Web site at http://www.orfrh.org/.)
This approach assumes women feel in control of their reproduction; however, this may not be the reality for many, especially low-income women.6 Additionally, women commonly cite planning a pregnancy as appropriate only when they are in an ideal relationship and when they are living in a financially stable environment—conditions that some women may never achieve.
Another caveat is that women may not have explicit pregnancy intentions, in which case, this particular approach may not be effective. A study of low-income women found only 60% intended to use the method prescribed after contraception counseling, with 37% of those stopping because of adverse effects, 23% saying they wanted another method, and 17% citing method complexity.7
Reproductive coercion from male partners, ranging from pressure to become pregnant to method sabotage, is also common in low-income women.8 Regular conversations that prioritize a woman’s values and experience are needed to promote reproductive autonomy.
2. Decouple provision of contraception from unnecessary exams
Pelvic exams and pap smears should not be required prior to offering patients hormonal contraception, according to the Choosing Wisely campaign of the American Board of Internal Medicine and ACOG.9,10 Hormonal contraception may instead be provided safely based on a medical history and BP assessment. Adolescents, minority groups, obese women, and victims of sexual trauma, in particular may avoid asking about birth control because of anxiety and fear of pain from these exams.11 The American College of Physicians recommends against speculum and bimanual exams in asymptomatic, non-pregnant, adult women.12 Pap smears and sexually transmitted infection (STI) testing should be performed at their normally scheduled intervals as recommended by the US Preventive Services Task Force (USPSTF) and not be tied to contraceptive provision.13
Assess pregnancy status using criteria,rather than a pregnancy text
Use the CDC’s criteria to assess pregnancy status rather than relying on a urine pregnancy test prior to providing contraception. Once you are reasonably sure that a woman is not pregnant (TABLE 114), contraception may be started. Some physicians have traditionally requested that a woman delay starting contraception until the next menses to ensure that she is not already pregnant. However, given the evidence that hormonal contraception does not cause birth defects, such a delay is not warranted and puts the woman at risk of an unintended pregnancy during the gap.15
Furthermore, there is an approximate 2-week window in which a woman could have a negative urine pregnancy test despite being pregnant, so the test alone is not completely reliable. In addition, obese women may experience irregular cycles, further complicating the traditional approach.16
Another largely unnecessary step … The US Selected Practice Recommendations (US SPR) from the CDC notes that additional STI screening prior to an intrauterine device (IUD) insertion is unnecessary for most women if appropriate screening guidelines have been previously followed.14 For those who have not been screened according to guidelines, the CDC recommends same-day screening and IUD insertion. You can then treat an STI without removing the IUD. Women with purulent cervicitis or a current chlamydial or gonorrheal infection should delay IUD insertion until after treatment.
3. Expand long-acting reversible contraception counseling and access
Offer long-acting reversible contraception (LARC), such as IUDs and implants, as first-line options for most women. ACOG endorses LARC as the most effective reversible method for most women, including those who have not given birth and adolescents.17 Unfortunately, a 2012 study found that family physicians were less likely than OB-GYNs to have enough time for contraceptive counseling and fewer than half felt competent inserting IUDs.18 While 79% of OB-GYNs routinely discussed IUDs with their patients, only 47% of family physicians did. In 2014, the American Academy of Pediatrics (AAP) endorsed a LARC-first tiered counseling approach for adolescents.19
A test of LARC-first counseling
The Contraceptive CHOICE project, a St. Louis, Missouri-based initiative, was launched to reduce unintended pregnancies in women ages 14 to 45 years by offering LARC-first counseling and free contraception of their choice.20 This project involved more than 9000 women at high risk for unintended pregnancy. Same-day LARC insertion was available. Seventy-five percent of women chose a LARC method and they reported greater continuation at 12 and 24 months, when compared to women who did not choose a LARC method. LARC users also reported higher satisfaction at one year. Provision of contraception through the project contributed to a reduction in repeat abortions as well as decreased rates of teenage pregnancy, birth, and abortion. Three years after the start of the project, IUDs had continuation rates of nearly 70%, implants of 56%, and non-LARC methods of 31%.21
When counseling women, it’s important to remember that effectiveness may not be the only criterium a woman uses when choosing a method. A 2010 study found that for 91% of women at high risk for unintended pregnancy, no single method possessed all the features they deemed “extremely important.”22 Clinicians should take a patient-centered approach to find birth control that fits each patient’s priorities.
Clinicians need proper training in LARC methods
Only 20% of FPs regularly insert IUDs, and 11% offer contraceptive implants, according to estimates from physicians recertifying with the American Board of Family Medicine in 2014.23 Access to training during residency is a key component to increasing these rates. FPs who practice obstetrics should be trained in postpartum LARC insertion and offer this option prior to hospital discharge as well as during the postpartum office visit.
Performing LARC insertions on the same day as counseling is ideal, and clinics should strive to reduce barriers to same-day procedures. Time constraints may be addressed by shifting tasks among the medical team. In the CHOICE project, contraceptive counselors—half of whom had no clinical experience—were trained to provide tiered counseling to participants. By working with a cross-trained health care team and offering prepared resources, clinicians can save time and improve access.
Physicians may want to incorporate the free online resources Bedsider.org or Stayteen.org to help women learn about contraceptive methods.24 The user-friendly Web sites, operated by the National Campaign to Prevent Teen and Unplanned Pregnancy, describe various forms of contraception and offer text and email reminders. Incorporating Bedsider into the counseling workflow and discussing the various reminder tools available may improve patients’ knowledge and enhance their compliance.
Additional barriers for practices may include high upfront costs associated with stocking devices. Practices that may be unable to sustain the costs surrounding enhanced contraception counseling and provision can collaborate with family planning clinics that are able to offer same-day services. A study of clinics in California found that Title X clinics were more likely to provide on-site LARC services than non-Title X public and private providers.25
4. Follow CDC guidelines for initiating and continuing contraception
Follow the US SPR for guidance on initiating and continuing contraceptive methods.14 The CDC’s Medical Eligibility Criteria for Contraceptive Use is another vital resource, providing recommendations for contraceptive methods to patients who have specific medical conditions or characteristics.26
Utilize the “quick start” method for hormonal contraception, where birth control is started on the same day as its prescription regardless of timing of the menstrual cycle. If you can’t be reasonably certain that a woman is not pregnant based on the criteria listed in TABLE 1,14 conduct a pregnancy test (while recognizing the aforementioned 2-week window of limitations) and counsel the patient to use back-up protection for the first 7 days along with repeating a pregnancy test in 2 weeks’ time.
The quick start method may lead to higher adherence than delayed initiation.27 Differences in continuation rates between women who use the quick start method and those who follow the delayed approach may disappear over time.28
Prescribe and provide a year’s supply of oral contraceptive pills (OCPs) as recommended by the CDC US SPR.14 It is important to note that pharmacists are usually restricted by insurance companies to only fill a one or 3 month’s supply.
In January 2016, Oregon began requiring private and state health insurance providers to reimburse for a year’s supply of prescription contraception; in January 2017, insurers in Washington, DC, were also required to offer women a year’s supply of prescription contraception.29,30 Several other states have followed suit. The California Health Benefits Review Program estimates a savings of $42.8 million a year from fewer office visits and 15,000 fewer unintended pregnancies if their state enacts a similar policy.31
Pharmacist initiatives are worth watching. In January 2016, Oregon pharmacists with additional training were allowed to prescribe OCs and hormonal patches to women 18 years and older.32 In April 2016, a similar law went into effect in California, but without a minimum age requirement and with the additional coverage of vaginal rings and Depo-Provera (depo) injections.33 Pharmacists in both states must review a health questionnaire completed by the woman and can refer to a physician as necessary.
The CDC recommends that clinicians extend the allowed window for repeat depo injections to 15 weeks.14 Common institutional protocol is to give repeat injections every 11 to 13 weeks. If past that window, protocol often dictates the woman abstain from unprotected sex for 2 weeks and then return for a negative pregnancy test (or await menses) before the next injection. However, the CDC notes that depo is effective for longer than the 13-week period.14 No additional birth control or pregnancy testing is needed and the woman can receive the next depo shot if she is up to 15 weeks from the previous shot.
One study found no additional pregnancy risks for those who were up to 4 weeks “late” for their next shot, suggesting there is potential for an even larger grace period.34 The World Health Organization advises allowing a repeat injection up to 4 weeks late.35 We encourage institutions to change their policies to comply with the CDC’s 15-week window.
Another initiative is over-the-counter (OTC) access to OCs, which the American Academy of Family Physicians (AAFP) and ACOG support.36,37 ACOG notes that “no drug or intervention is completely without risk of harm” and that the risk of venous thromboembolism for OC users is lower than the risk of pregnancy.37 Women can successfully self-screen for contraindications using a checklist. Concerns about women potentially being less adherent or less likely to choose LARCs are not reasons to preclude access to other methods. The AAFP supports insurance coverage of OCs, regardless of prescription status.36
5. Routinely counsel about, and advance-prescribe, emergency contraception pills
Physicians should counsel and advance-prescribe emergency contraception pills (ECPs) to women, including adolescents, using less reliable contraception, as recommended by ACOG, AAP, and the CDC.14,37,38 It’s also important to provide information on the copper IUD as the most effective method of emergency contraception, with nearly 100% efficacy if placed within 5 days.39 An easy-to-read patient hand-out in English and Spanish on EC options can be found at http://beyondthepill.ucsf.edu/tools-materials.
Only 3% of respondents participating in the 2006-2010 National Survey of Family Growth received counseling about emergency contraception in the past year.40 ECPs are most effective when used within 24 hours but have some efficacy up to 5 days.37 Due to the Affordable Care Act, most insurance plans will cover ECPs if purchased with a prescription, but coverage varies by state.41 Ulipristal acetate (UPA) ECP is only available with a prescription. Advance prescriptions can alleviate financial burdens on women when they need to access ECPs quickly.
Women should wait at least 5 days before resuming or starting hormonal contraception after taking UPA-based ECP, as it may reduce the ovulation-delaying effect of the ECP.14 For IUDs, implants, and depo, which require a visit to a health care provider, physicians evaluating earlier provision should consider the risks of reduced efficacy against the many barriers to access.
UPA-based ECPs (such as ella) may be more effective for overweight and obese women than levonorgestrel-based ECPs (such as Plan B and Next Choice).14 Consider advance-prescribing UPA ECPs to women with a body mass index (BMI) >25 kg/m2.42 Such considerations are important as the prevalence of obesity in women between 2013 and 2014 was 40.4%.43
In May 2016, the FDA noted that while current data are insufficient regarding whether the effectiveness of levonorgestrel ECPs is reduced in overweight or obese women, there are no safety concerns regarding their use in this population.44 Therefore, a woman with a BMI >25 kg/m2 should use UPA ECPs if available; but if not, she can still use levonorgestrel ECPs. One study, however, has found that UPA ECPs are only as effective as a placebo when BMI is ≥35 kg/m2, at which point a copper IUD may be the only effective form of emergency contraception.45
Transitioning from customary practices to best practices
Following these practical steps, FPs can improve contraceptive care for women. However, to make a significant impact, clinicians must be willing to change customary practices that are based on tradition, routines, or outdated protocols in favor of those based on current evidence.
One good place to start the transition to best practices is to familiarize yourself with the 2016 US Medical Eligibility Criteria for Contraceptive Use26 and Selected Practice Recommendations for Contraceptive Use.14 TABLES 214,26,46,47 and 3 offer additional resources that can enhance contraceptive counseling and further promote access to contraceptive care.
The contraceptive coverage guarantee under the Affordable Care Act has allowed many women to make contraceptive choices based on personal needs and preferences rather than cost. The new contraceptive coverage exemptions issued under the Trump administration will bring cost back as the driving decision factor for women whose employers choose not to provide contraceptive coverage. Providers should be aware of the typical costs associated with the various contraceptive options offered in their practice and community.
CORRESPONDENCE
Jessica Dalby, MD, Department of Family Medicine and Community Health, University of Wisconsin School of Medicine and Public Health, 1102 South Park St, Suite 100, Madison, WI 53715; [email protected].
1. Finer LB, Zolna MR. Declines in unintended pregnancy in the United States, 2008–2011. N Engl J Med. 2016; 374:843-852.
2. Sonfield A, Hasstedt K, Gold RB. Moving Forward: Family Planning in the Era of Health Reform. New York: Guttmacher Institute. 2014. Available at: https://www.guttmacher.org/report/moving-forward-family-planning-era-health-reform. Accessed October 5, 2017.
3. Committee on Health Care for Underserved Women. Reproductive Life Planning to Reduce Unintended Pregnancy: American College of Obstetricians and Gynecologists. 2016. Available at: https://www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Health-Care-for-Underserved-Women/Reproductive-Life-Planning-to-Reduce-Unintended-Pregnancy. Accessed October 5, 2017.
4. Centers for Disease Control and Prevention. Reproductive Life Plan Tool for Health Care Providers. 2016. Available at: http://www.cdc.gov/preconception/rlptool.html. Accessed August 31, 2016.
5. Oregon Health Authority. Effective Contraceptive Use among Women at Risk of Unintended Pregnancy Guidance Document. 2014. Available at: http://www.oregon.gov/oha/HPA/ANALYTICS/CCOData/Effective%20Contraceptive%20Use%20Guidance%20Document.pdf. Accessed October 5, 2017.
6. Borrero S, Nikolajski C, Steinberg JR, et al. “It just happens”: a qualitative study exploring low-income women’s perspectives on pregnancy intention and planning. Contraception. 2015;91:150-156.
7. Yee LM, Farner KC, King E, et al. What do women want? Experiences of low-income women with postpartum contraception and contraceptive counseling. J Pregnancy Child Health. 2015;2.
8. Kalichman SC, Williams EA, Cherry C, et al. Sexual coercion, domestic violence, and negotiating condom use among low-income African American women. J Womens Health. 1998;7:371-378.
9. ABIM Foundation. Pelvic Exams, Pap Tests and Oral Contraceptives. 2016. Available at: http://www.choosingwisely.org/patient-resources/pelvic-exams-pap-tests-and-oral-contraceptives/. Accessed May 31, 2016.
10. Committee on Health Care for Underserved Women. Access to Contraception: American College of Obstetricians and Gynecologists. 2015. Number 615. Available at: https://www.acog.org/-/media/Committee-Opinions/Committee-on-Health-Care-for-Underserved-Women/co615.pdf?dmc=1&ts=201710. Accessed October 5, 2017.
11. Bates CK, Carroll N, Potter J. The challenging pelvic examination. J Gen Intern Med. 2011;26:651-657.
12. Qaseem A, Humphrey LL, Harris R, et al. Screening pelvic examination in adult women: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2014;161:67-72.
13. U.S. Preventive Services Task Force. Cervical Cancer: Screening. 2012. Available at: http://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/cervical-cancer-screening. Accessed May 25, 2016.
14. Curtis KM, Jatlaoui TC, Tepper NK, et al. U.S. selected practice recommendations for contraceptive use, 2016. MMWR Morb Mortal Wkly Rep. 2016;65:1-66.
15. Lesnewski R, Prine L. Initiating hormonal contraception. Am Fam Physician. 2006;74:105-112.
16. Jacobsen BK, Knutsen SF, Oda K, et al. Obesity at age 20 and the risk of miscarriages, irregular periods and reported problems of becoming pregnant: the Adventist Health Study-2. Eur J Epidemiol. 2012; 27:923-931.
17. Committee on Gynecologic Practice. Increasing Access to Contraceptive Implants and Intrauterine Devices to Reduce Unintended Pregnancy: American College of Obstetricians and Gynecologists. 2015. Number 642. Available at: https://www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Gynecologic-Practice/Increasing-Access-to-Contraceptive-Implants-and-Intrauterine-Devices-to-Reduce-Unintended-Pregnancy. Accessed October 5, 2017.
18. Harper CC, Henderson JT, Raine TR, et al. Evidence-based IUD practice: family physicians and obstetrician-gynecologists. Fam Med. 2012;44:637-645.
19. American Academy of Pediatrics, Committee on Adolescence. Policy statement: Contraception for Adolescents. 2014. Available at: http://pediatrics.aappublications.org/content/pediatrics/early/2014/09/24/peds.2014-2299.full.pdf. Accessed October 5, 2017.
20. Birgisson NE, Zhao Q, Secura GM, et al. Preventing unintended pregnancy: The Contraceptive CHOICE Project in review. J Womens Health (Larchmt). 2015;24:349-353.
21. Diedrich JT, Zhao Q, Madden T, et al. Three-year continuation of reversible contraception. Am J Obstet Gynecol. 2015;213:662.e1-e8.
22. Lessard LN, Karasek D, Ma S, et al. Contraceptive features preferred by women at high risk of unintended pregnancy. Perspect Sex Reprod Health. 2012;44:194-200.
23. Nisen MB, Peterson LE, Cochrane A, et al. US family physicians’ intrauterine and implantable contraception provision: results from a national survey. Contraception. 2016;93:432-437.
24. National Campaign to Prevent Teen and Unplanned Pregnancy. Bedsider. Available at: https://bedsider.org/. Accessed June 14, 2016.
25. Park HY, Rodriguez MI, Hulett D, et al. Long-acting reversible contraception method use among Title X providers and non-Title X providers in California. Contraception. 2012;86:557-561.
26. Curtis KM, Tepper NK, Jatlaoui TC, et al. U.S. medical eligibility criteria for contraceptive use, 2016. MMWR Morb Mortal Wkly Rep. 2016;65:1-103.
27. Westhoff C, Kerns J, Morroni C, et al. Quick start: novel oral contraceptive initiation method. Contraception. 2002;66:141-145.
28. Brahmi D, Curtis KM. When can a woman start combined hormonal contraceptives (CHCs)? A systematic review. Contraception. 2013;87:524-538.
29. Lachman S. Oregon To Require Insurers To Cover A Year’s Supply Of Birth Control. Huffington Post. June 11, 2015. Available at: https://www.huffingtonpost.com/2015/06/11/oregon-birth-control-_n_7564712.html. Accessed October 16, 2017.
30. Andrews M. D.C. Women To Get Access To Full Year’s Worth Of Contraceptives. Kaiser Health News. September 25, 2015. Available at: https://khn.org/news/d-c-women-to-get-access-to-full-years-worth-of-contraceptives/. Accessed October 16, 2017.
31. Analysis of California Senate Bill (SB) 999 Contraceptives: Annual Supply: A Report to the 2015-2016 California State Legislature: California Health Benefits Review Program. 2016. Available at: http://chbrp.ucop.edu/index.php?action=read&bill_id=195&doc_type=1000. Accessed October 5, 2017.
32. Frazier A. Pharmacist-prescribed birth control in effect Jan 1. KOIN News. December 30, 2015. Available at: http://koin.com/2015/12/30/pharmacist-provided-birth-control-in-effect-jan-1/. Accessed October 5, 2017.
33. Karlamangla S. Birth control pills without prescriptions, coming soon to California under new law. Los Angeles Times. February 14, 2016. Available at: http://www.latimes.com/health/la-me-birth-control-pharmacies-20160214-story.html. Accessed October 16, 2017.
34. Steiner MJ, Kwok C, Stanback J, et al. Injectable contraception: what should the longest interval be for reinjections? Contraception. 2008;77:410-414.
35. World Health Organization. Family Planning: A Global Handbook for Providers. 2011. Available at: http://apps.who.int/iris/bitstream/10665/44028/1/9780978856373_eng.pdf. Accessed October 5, 2017.
36. American Academy of Family Physicians. Over-the-Counter Oral Contraceptives. 2014; Available at: http://www.aafp.org/about/policies/all/otc-oral-contraceptives.html. Accessed June 2, 2016.
37. Committee on Gynecologic Practice. Over-the-Counter Access to Oral Contraceptives: American College of Obstetricians and Gynecologists. 2012. Number 544. Available at: https://www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Gynecologic-Practice/Over-the-Counter-Access-to-Oral-Contraceptives. Accessed October 5, 2017.
38. Committee on Adolescence. Emergency contraception. Pediatrics. 2012;130:1174-1182.
39. Cleland K, Zhu H, Goldstuck N, et al. The efficacy of intrauterine devices for emergency contraception: a systematic review of 35 years of experience. Hum Reprod. 2012;27:1994-2000.
40. Martinez G, Chandra A, Febo-Vazquez I, et al. Use of Family Planning and Related Medical Services Among Women Aged 15–44 in the United States: National Survey of Family Growth, 2006–2010: National Center for Health Statistics, Centers for Disease Control and Prevention. 2013. Available at: https://www.cdc.gov/nchs/data/nhsr/nhsr068.pdf. Accessed October 5, 2017.
41. Guttmacher Institute. Insurance Coverage of Contraceptives: Guttmacher Institute;2017. Available at: https://www.guttmacher.org/state-policy/explore/insurance-coverage-contraceptives Accessed October 7, 2017.
42. Glasier A, Cameron ST, Blithe D, et al. Can we identify women at risk of pregnancy despite using emergency contraception? Data from randomized trials of ulipristal acetate and levonorgestrel. Contraception. 2011;84:363-367.
43. Flegal KM, Kruszon-Moran D, Carroll MD, et al. Trends in obesity among adults in the United States, 2005 to 2014. JAMA. 2016;315:2284-2291.
44. US Food & Drug Administration. Postmarket Drug Safety Information for Patients and Providers - Plan B (0.75mg levonorgestrel) and Plan B One-Step (1.5 mg levonorgestrel) Tablets Information. 2016; Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm109775.htm. Accessed May 25, 2016.
45. Simmons KB, Edelman AB. Contraception and sexual health in obese women. Best Pract Res Clin Obstet Gynaecol. 2015;29:466-478.
46. Centers for Disease Control and Prevention. Providing quality family planning services: recommendations of CDC and the U.S. Office of Population Affairs. MMWR Recomm Rep. 2014;63:1-29.
47. LARC FIRST. Available at: http://www.larcfirst.com/index.html. Accessed May 2016.
1. Finer LB, Zolna MR. Declines in unintended pregnancy in the United States, 2008–2011. N Engl J Med. 2016; 374:843-852.
2. Sonfield A, Hasstedt K, Gold RB. Moving Forward: Family Planning in the Era of Health Reform. New York: Guttmacher Institute. 2014. Available at: https://www.guttmacher.org/report/moving-forward-family-planning-era-health-reform. Accessed October 5, 2017.
3. Committee on Health Care for Underserved Women. Reproductive Life Planning to Reduce Unintended Pregnancy: American College of Obstetricians and Gynecologists. 2016. Available at: https://www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Health-Care-for-Underserved-Women/Reproductive-Life-Planning-to-Reduce-Unintended-Pregnancy. Accessed October 5, 2017.
4. Centers for Disease Control and Prevention. Reproductive Life Plan Tool for Health Care Providers. 2016. Available at: http://www.cdc.gov/preconception/rlptool.html. Accessed August 31, 2016.
5. Oregon Health Authority. Effective Contraceptive Use among Women at Risk of Unintended Pregnancy Guidance Document. 2014. Available at: http://www.oregon.gov/oha/HPA/ANALYTICS/CCOData/Effective%20Contraceptive%20Use%20Guidance%20Document.pdf. Accessed October 5, 2017.
6. Borrero S, Nikolajski C, Steinberg JR, et al. “It just happens”: a qualitative study exploring low-income women’s perspectives on pregnancy intention and planning. Contraception. 2015;91:150-156.
7. Yee LM, Farner KC, King E, et al. What do women want? Experiences of low-income women with postpartum contraception and contraceptive counseling. J Pregnancy Child Health. 2015;2.
8. Kalichman SC, Williams EA, Cherry C, et al. Sexual coercion, domestic violence, and negotiating condom use among low-income African American women. J Womens Health. 1998;7:371-378.
9. ABIM Foundation. Pelvic Exams, Pap Tests and Oral Contraceptives. 2016. Available at: http://www.choosingwisely.org/patient-resources/pelvic-exams-pap-tests-and-oral-contraceptives/. Accessed May 31, 2016.
10. Committee on Health Care for Underserved Women. Access to Contraception: American College of Obstetricians and Gynecologists. 2015. Number 615. Available at: https://www.acog.org/-/media/Committee-Opinions/Committee-on-Health-Care-for-Underserved-Women/co615.pdf?dmc=1&ts=201710. Accessed October 5, 2017.
11. Bates CK, Carroll N, Potter J. The challenging pelvic examination. J Gen Intern Med. 2011;26:651-657.
12. Qaseem A, Humphrey LL, Harris R, et al. Screening pelvic examination in adult women: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2014;161:67-72.
13. U.S. Preventive Services Task Force. Cervical Cancer: Screening. 2012. Available at: http://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/cervical-cancer-screening. Accessed May 25, 2016.
14. Curtis KM, Jatlaoui TC, Tepper NK, et al. U.S. selected practice recommendations for contraceptive use, 2016. MMWR Morb Mortal Wkly Rep. 2016;65:1-66.
15. Lesnewski R, Prine L. Initiating hormonal contraception. Am Fam Physician. 2006;74:105-112.
16. Jacobsen BK, Knutsen SF, Oda K, et al. Obesity at age 20 and the risk of miscarriages, irregular periods and reported problems of becoming pregnant: the Adventist Health Study-2. Eur J Epidemiol. 2012; 27:923-931.
17. Committee on Gynecologic Practice. Increasing Access to Contraceptive Implants and Intrauterine Devices to Reduce Unintended Pregnancy: American College of Obstetricians and Gynecologists. 2015. Number 642. Available at: https://www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Gynecologic-Practice/Increasing-Access-to-Contraceptive-Implants-and-Intrauterine-Devices-to-Reduce-Unintended-Pregnancy. Accessed October 5, 2017.
18. Harper CC, Henderson JT, Raine TR, et al. Evidence-based IUD practice: family physicians and obstetrician-gynecologists. Fam Med. 2012;44:637-645.
19. American Academy of Pediatrics, Committee on Adolescence. Policy statement: Contraception for Adolescents. 2014. Available at: http://pediatrics.aappublications.org/content/pediatrics/early/2014/09/24/peds.2014-2299.full.pdf. Accessed October 5, 2017.
20. Birgisson NE, Zhao Q, Secura GM, et al. Preventing unintended pregnancy: The Contraceptive CHOICE Project in review. J Womens Health (Larchmt). 2015;24:349-353.
21. Diedrich JT, Zhao Q, Madden T, et al. Three-year continuation of reversible contraception. Am J Obstet Gynecol. 2015;213:662.e1-e8.
22. Lessard LN, Karasek D, Ma S, et al. Contraceptive features preferred by women at high risk of unintended pregnancy. Perspect Sex Reprod Health. 2012;44:194-200.
23. Nisen MB, Peterson LE, Cochrane A, et al. US family physicians’ intrauterine and implantable contraception provision: results from a national survey. Contraception. 2016;93:432-437.
24. National Campaign to Prevent Teen and Unplanned Pregnancy. Bedsider. Available at: https://bedsider.org/. Accessed June 14, 2016.
25. Park HY, Rodriguez MI, Hulett D, et al. Long-acting reversible contraception method use among Title X providers and non-Title X providers in California. Contraception. 2012;86:557-561.
26. Curtis KM, Tepper NK, Jatlaoui TC, et al. U.S. medical eligibility criteria for contraceptive use, 2016. MMWR Morb Mortal Wkly Rep. 2016;65:1-103.
27. Westhoff C, Kerns J, Morroni C, et al. Quick start: novel oral contraceptive initiation method. Contraception. 2002;66:141-145.
28. Brahmi D, Curtis KM. When can a woman start combined hormonal contraceptives (CHCs)? A systematic review. Contraception. 2013;87:524-538.
29. Lachman S. Oregon To Require Insurers To Cover A Year’s Supply Of Birth Control. Huffington Post. June 11, 2015. Available at: https://www.huffingtonpost.com/2015/06/11/oregon-birth-control-_n_7564712.html. Accessed October 16, 2017.
30. Andrews M. D.C. Women To Get Access To Full Year’s Worth Of Contraceptives. Kaiser Health News. September 25, 2015. Available at: https://khn.org/news/d-c-women-to-get-access-to-full-years-worth-of-contraceptives/. Accessed October 16, 2017.
31. Analysis of California Senate Bill (SB) 999 Contraceptives: Annual Supply: A Report to the 2015-2016 California State Legislature: California Health Benefits Review Program. 2016. Available at: http://chbrp.ucop.edu/index.php?action=read&bill_id=195&doc_type=1000. Accessed October 5, 2017.
32. Frazier A. Pharmacist-prescribed birth control in effect Jan 1. KOIN News. December 30, 2015. Available at: http://koin.com/2015/12/30/pharmacist-provided-birth-control-in-effect-jan-1/. Accessed October 5, 2017.
33. Karlamangla S. Birth control pills without prescriptions, coming soon to California under new law. Los Angeles Times. February 14, 2016. Available at: http://www.latimes.com/health/la-me-birth-control-pharmacies-20160214-story.html. Accessed October 16, 2017.
34. Steiner MJ, Kwok C, Stanback J, et al. Injectable contraception: what should the longest interval be for reinjections? Contraception. 2008;77:410-414.
35. World Health Organization. Family Planning: A Global Handbook for Providers. 2011. Available at: http://apps.who.int/iris/bitstream/10665/44028/1/9780978856373_eng.pdf. Accessed October 5, 2017.
36. American Academy of Family Physicians. Over-the-Counter Oral Contraceptives. 2014; Available at: http://www.aafp.org/about/policies/all/otc-oral-contraceptives.html. Accessed June 2, 2016.
37. Committee on Gynecologic Practice. Over-the-Counter Access to Oral Contraceptives: American College of Obstetricians and Gynecologists. 2012. Number 544. Available at: https://www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Gynecologic-Practice/Over-the-Counter-Access-to-Oral-Contraceptives. Accessed October 5, 2017.
38. Committee on Adolescence. Emergency contraception. Pediatrics. 2012;130:1174-1182.
39. Cleland K, Zhu H, Goldstuck N, et al. The efficacy of intrauterine devices for emergency contraception: a systematic review of 35 years of experience. Hum Reprod. 2012;27:1994-2000.
40. Martinez G, Chandra A, Febo-Vazquez I, et al. Use of Family Planning and Related Medical Services Among Women Aged 15–44 in the United States: National Survey of Family Growth, 2006–2010: National Center for Health Statistics, Centers for Disease Control and Prevention. 2013. Available at: https://www.cdc.gov/nchs/data/nhsr/nhsr068.pdf. Accessed October 5, 2017.
41. Guttmacher Institute. Insurance Coverage of Contraceptives: Guttmacher Institute;2017. Available at: https://www.guttmacher.org/state-policy/explore/insurance-coverage-contraceptives Accessed October 7, 2017.
42. Glasier A, Cameron ST, Blithe D, et al. Can we identify women at risk of pregnancy despite using emergency contraception? Data from randomized trials of ulipristal acetate and levonorgestrel. Contraception. 2011;84:363-367.
43. Flegal KM, Kruszon-Moran D, Carroll MD, et al. Trends in obesity among adults in the United States, 2005 to 2014. JAMA. 2016;315:2284-2291.
44. US Food & Drug Administration. Postmarket Drug Safety Information for Patients and Providers - Plan B (0.75mg levonorgestrel) and Plan B One-Step (1.5 mg levonorgestrel) Tablets Information. 2016; Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm109775.htm. Accessed May 25, 2016.
45. Simmons KB, Edelman AB. Contraception and sexual health in obese women. Best Pract Res Clin Obstet Gynaecol. 2015;29:466-478.
46. Centers for Disease Control and Prevention. Providing quality family planning services: recommendations of CDC and the U.S. Office of Population Affairs. MMWR Recomm Rep. 2014;63:1-29.
47. LARC FIRST. Available at: http://www.larcfirst.com/index.html. Accessed May 2016.

Managing atraumatic meniscal tears in middle-aged patients
Meniscectomy is the most common orthopedic procedure performed in the United States with 700,000 meniscectomies performed every year.1 More than half of these procedures are performed in patients ≥45 years of age,2 giving rise to the question: Does arthroscopic surgery have a role in the treatment of patients who may have osteoarthritis (OA) and another knee condition, such as a symptomatic meniscal tear? Determining the answer is especially important when you consider that the number of true and incidental tears diagnosed on magnetic resonance imaging (MRI) has been on the rise3—a result of the routine use of MRIs to identify the cause of patients’ chronic knee pain.
At a cost of roughly $5,000 per procedure, some experts have suggested that at least a portion of the approximately $4 billion annual direct medical costs associated with meniscectomy could be put to better use.4,5 This prompted us to wonder what the literature tells us about the management of degenerative meniscus tears in middle-aged patients with OA and whether these patients would benefit from nonoperative management with optimized physical and medical therapy as a first-line approach. Our findings follow.
But first, a word about the connection between OA and meniscal tears.
What we’ve learned about meniscal damage
Research has shown that over one-third of individuals >50 years of age and three-quarters of people with knee OA have degenerative meniscal tears.6 In the past, the relative paucity of epidemiologic data on the prevalence of meniscal tears in the general population made it difficult to interpret the diagnostic information provided by MRI.
More recently, experts found that meniscal damage is especially prevalent among individuals with OA, and they began treating with arthroscopic partial meniscectomy (APM), as the meniscal damage was thought to be the anatomical foundation for the complaint of knee pain.7
However, researchers then began realizing that many patients with findings of a meniscal tear visualized on MRI reported no knee symptoms. In one study, adults in a large community-based sample found to have a meniscal tear on MRI were no more likely to have knee pain than subjects without a meniscal tear.6 Similarly, subjects with a meniscal tear and OA had no more severe pain than subjects with OA and no meniscal tear.6
In addition, the landmark Fairbank study from 19488 and others since have shown that meniscectomy can lead to other problems. Removal of meniscal tissue decreases the contact stress area, which increases stress on the articular cartilage, and inevitably leads to degeneration of the involved joint.9 Researchers have shown that even partial meniscectomy produces late articular cartilage changes.10
Which interventions and when?
We conducted an in-depth literature review to determine which approaches were best for the treatment of OA and meniscal tears, and summarize our findings below, according to OA severity. (For details of how the literature review was conducted and an at-a-glance summary of the key findings, see the TABLE.4,5,11-17) Of note: All of the studies reviewed here included patients with chronic knee pain and excluded patients with sudden onset pain from a single physical event.
The findings: Early OA and meniscal tears
The first 2 studies we identified in the literature, both published by Herrlin et al,11,12 examined the efficacy of APM in middle-aged patients with early OA (≤grade 1), according to the Ahlbäck classification.
In the first Herrlin study, a 6-month prospective randomized trial, 90 middle-aged patients with a medial meniscal tear (without traumatic history) were assigned to either APM followed by supervised exercise or 8 weeks of supervised exercise alone.11 Exercise consisted of activities for improving muscle strength, endurance, and flexibility, as well as for balance and proprioception. The authors concluded that a combination of APM and supervised exercise did not lead to greater improvements in knee function compared with supervised exercise alone.11
In the second Herrlin study, a prospective randomized study involving 96 middle-aged patients with an MRI-verified medial meniscal tear and radiographic OA, the authors concluded that arthroscopic surgery followed by exercise therapy was not superior to the same exercise therapy alone.12 The results were gleaned from both patient-reported outcomes and radiographic assessment at 2 and 5 years. Both groups reported significant improvements at 5 years, but participants did not reach the level of fitness and quality of life of similarly-aged healthy controls.
Perhaps one of the most interesting aspects of this study was that approximately one-third of patients from the exercise-only group still had disabling knee symptoms after exercise therapy, but improved to the same level as the rest of the patients after crossing over and undergoing APM.12 Part of the observed benefit of arthroscopy in these patients has the potential to be explained by the placebo effect, especially given that invasive procedures have a stronger placebo effect than do noninvasive ones, and due to the lack of blinding.18 Additionally, limitations of the above studies include small sample sizes, lack of a control group, and short-term follow-up.
Next, a 2013 study by Yim et al looked specifically at APM vs nonoperative treatment with strengthening exercises.13 A total of 102 patients with an average age of 53.8 years, a Kellgren-Lawrence Classification of Osteoarthritis of <2, and an MRI-confirmed degenerative horizontal tear of the posterior horn of the medial meniscus were randomized to the 2 intervention groups. The 2 groups were highly comparable, giving the study high internal validity. These patients were then assessed at 3 months, one year, and at 2 years after treatment.
Although most patients at the outset of the study had intense knee pain with mechanical symptoms, both groups reported a decrease in experienced knee pain, improved function, and a high level of satisfaction with their prescribed treatment, with no significant difference in any of these values after 2 years of follow-up.13 A limitation of the study was that it used subjective questionnaires to assess pain, swelling, and activities of daily living (ADLs).
A fourth study, a 2013 multicenter, randomized, sham-controlled trial, looked at 146 patients ages 35 to 65 years who had knee symptoms consistent with a degenerative medial meniscus tear (confirmed by MRI and arthroscopic evaluation) and no knee arthritis.4 The subjects were assigned to either APM or sham surgery (skin incisions only). The results showed that APM was not superior to sham surgery with regard to outcomes assessed during a 12-month follow-up period.4
Most recently (2016), Kise et al14 published the results of a randomized controlled superiority trial conducted in Norway comparing 12 weeks of supervised exercise therapy with APM for patients with degenerative meniscus tears. Their study included 140 patients ages 36 to 60 years. Notably, most (96%), but not all, of their patients had no radiographic evidence of OA.
At the 2-year follow-up, there were no differences in patient-reported pain or functional outcomes, with all patients improving significantly from baseline. Muscle strength was also measured and found to be significantly greater at 3 and 12 months in the exercise group. Limitations of this study were a lack of patient blinding and a 19% crossover from the exercise to the APM group.
The findings: Mild to moderate OA and meniscal tears
The authors of a 2002 double-blind, placebo-controlled trial randomly assigned 180 patients with degenerative meniscus tears and knee OA to either arthroscopic debridement, arthroscopic lavage, or placebo surgery consisting of superficial skin incisions without insertion of an arthroscope.5 Patients were eligible if they were ≤75 years of age and had not undergone arthroscopy of the knee within the previous 2 years. Arthritis was graded utilizing the Kellgren-Lawrence scale (0-4) and by calculating a severity score (0-12) by adding the individual scores for each of the 3 compartments of the knee; patients were excluded if they had a severity grade ≥9. One-quarter of the participants had severe arthritis, with scores of 7 or 8. Outcomes were measured over a 24-month period. At no point did either of the intervention groups report less pain or better function than the sham-surgery group.5
Additionally, in a single-center RCT involving 178 patients with mild to severe OA, subjects were randomly assigned to surgical lavage and arthroscopic debridement together with optimized physical and medical therapy or to treatment with physical and medical therapy alone.15 Patients were excluded if they had “bucket handle” meniscal tears detected by physical exam or by MRI, previous arthroscopic surgery, previous major knee trauma, or steroid injections in the last 3 months. All participants were required to have Kellgren-Lawrence grade 2, 3, or 4 arthritis.
The researchers used 2 validated outcome measures to evaluate pain, symptoms, and functional ability and followed the patients for 2 years after the initiation of treatment. This study failed to show that arthroscopic surgery provided any additional benefit to optimized physical and medical therapy.15
A 2013 multicenter, RCT randomly assigned 351 patients ≥45 years with a meniscal tear and radiographic evidence of mild to moderate OA to either surgery and postoperative physical therapy or to a standardized physical therapy regimen.16 Patients were required to have at least one symptom that was consistent with a meniscal tear for approximately one month. About 30% of the patients who were assigned to physical therapy alone underwent surgery within 6 months. There were no significant differences between the 2 groups in the magnitude of improvement in functional status or pain at 6 or 12 months.
Finally, a prospective Scottish study consisting of 270 patients with everything from no signs of OA to advanced OA who underwent APM were sent preoperative and 6-month postop questionnaires evaluating ADLs, pain, symptoms, quality of life, and body mass index (BMI).17 Their OA was graded via preop MRI or radiographs and confirmed by the operating surgeon. The investigators were unable to demonstrate any significant benefit associated with arthroscopic meniscectomy, and, therefore, could not recommend the procedure for patients with moderate to advanced OA.
However, in their analysis, 3 subgroup populations were found to benefit from APM: those with greater body habitus (BMI >30 kg/m2), those without signs of OA, and those with early OA. Limitations of this study included the lack of randomization, blinding, a control, and long-term follow up, and that the authors didn’t use established OA grading criteria.17
The bottom line: Nonoperative treatment benefits most patients
Physical therapy is an appropriate first-line treatment for degenerative meniscus tears in middle-aged patients. In fact, a trial of nonoperative treatment is likely to benefit the majority of patients. In addition, avoiding surgery eliminates surgical complications and decreases overall health care costs.
Reserve APM for those patients without significant OA who fail to improve after physical therapy, who have mechanical symptoms, or who have intra-articular loose bodies.
In addition, exercise therapy is an effective treatment for patients with knee OA. It improves function and limits joint pain in both acute arthritic flares and more long-term, chronic situations. There is strong evidence that strengthening plays a critical role in reducing symptoms and improving muscle strength, physical ability, and quality of life.19 It has been suggested that physical exercise 3 times a week for 4 months could lead to >35% improvement of knee function.20 In contrast, other studies reported that while 91% of patients 11.5 years after APM considered their knees “normal or almost normal,” patients actually experienced a reduction in postoperative physical activity and quality of life.21
The most recent American Academy of Orthopaedic Surgeons (AAOS) guidelines22 do not recommend for or against arthroscopic partial meniscectomy in patients with knee OA and a torn meniscus. In middle-aged patients, MRI abnormalities of the meniscus do not consistently correlate with symptoms. Many meniscus lesions are asymptomatic or not the primary source of pain in the setting of OA.3
Potential harms and considerations. Deep venous thrombosis is the most frequently reported adverse event of arthroscopic surgery, with an approximate incidence of 4.13 per 1000 procedures, followed by less frequent complications such as infection, pulmonary embolism, and death.18
It is important to note that the degenerative meniscus tears that occur in middle age and that are associated with OA are not the same as acute, traumatic meniscus tears. All of the studies discussed here included patients with chronic knee pain and excluded patients with sudden onset pain from a single physical event. Many of the studies excluded patients with bucket-handle tears or severe mechanical symptoms (ie, locking). APM may be indicated for these meniscus tears regardless of age or OA status.
Making a sensible choice. Ultimately, physicians and their patients must use the best evidence available to make sensible clinical decisions. The ability to retain native meniscal tissue is of utmost importance to maintaining the longevity of their knee. According to previous studies, OA progression is more likely to occur after meniscectomy than after nonoperative treatment.23
CORRESPONDENCE
William Bassett, MD, Department of Orthopedics, Rutgers-Robert Wood Johnson Medical School, 51 French St., PO Box 19, New Brunswick, NJ 08903; [email protected].
1. Cullen KA, Hall MJ, Golosinskiy A. Ambulatory surgery in the United States, 2006. Natl Health Stat Rep. 2009;11:1-25.
2. Hall MJ, Lawrence L. Ambulatory surgery in the United States, 1996. Advance data from vital and health statistics of the Centers for Disease Control and Prevention/National Center for Health Statistics. No. 300. August 12, 1998. Available at: https://cdc.gov/nchs/data/ad/ad300.pdf. Accessed October 11, 2017.
3. Englund M, Guermazi A, Gale D, et al. Incidental meniscal findings on knee MRI in middle-aged and elderly persons. N Engl J Med. 2008;359:1108-1115.
4. Sihvonen R, Paavola M, Malmivaara A, et al. Arthroscopic partial meniscectomy versus sham surgery for a degenerative meniscal tear. N Engl J Med. 2013;369:2515-2524.
5. Moseley JB, O’Malley KO, Petersen NJ, et al. A controlled trial of arthroscopic surgery for osteoarthritis of the knee. N Engl J Med. 2002;347:81-88.
6. Bhattacharyya T, Gale D, Dewire P, et al. The clinical importance of meniscal tears demonstrated by magnetic resonance imaging in osteoarthritis of the knee. J Bone Joint Surg. 2003;85:4-9.
7. Paxton ES, Stock MV, Brophy RH. Meniscal repair versus partial meniscectomy: a systematic review comparing reoperation rates and clinical outcomes. Arthroscopy. 2011;27:1275-1288.
8. Fairbank TJ. Knee joint changes after meniscectomy. J Bone Joint Surg Br. 1948;30B:664-670.
9. Arnoczky SP, Warren RF. Microvasculature of the human meniscus. Am J Sports Med. 1982;10:90-95.
10. Morgan CD, Wojtys EM, Casscells CD, et al. Arthroscopic meniscal repair evaluated by second-look arthroscopy. Am J Sports Med. 1991;19:632-638.
11. Herrlin S, Hållander M, Wange P, et al. Arthroscopic or conservative treatment of degenerative medial meniscal tears: a prospective randomised trial. Knee Surg Sports Traumatol Arthrosc. 2007;15:393-401.
12. Herrlin SV, Wange PO, Lapidus G, et al. Is arthroscopic surgery beneficial in treating non-traumatic, degenerative medial meniscal tears? A five-year follow-up. Knee Surg Sports Traumatol Arthrosc. 2013;21:358-364.
13. Yim JH, Seon JK, Song EK, et al. A comparative study of meniscectomy and nonoperative treatment for degenerative horizontal tears of the medial meniscus. Am J Sports Med. 2013;41:1565-1570.
14. Kise NJ, Risberg MA, Stensrud S, et al. Exercise therapy versus arthroscopic partial meniscectomy for degenerative meniscal tear in middle aged patients: randomised controlled trial with two year follow-up. BMJ. 2016;354:i3740.
15. Kirkley A, Birmingham TB, Litchfield RB, et al. A randomized trial of arthroscopic surgery for osteoarthritis of the knee. N Engl J Med. 2008;359:1097-1107.
16. Katz JN, Brophy RH, Chaisson CE, et al. Surgery versus physical therapy for a meniscal tear and osteoarthritis. N Engl J Med. 2013;368:1675-1684.
17. Bailey O, Gronkowski K, Leach WJ. Effect of body mass index and osteoarthritis on outcomes following arthroscopic meniscectomy: A prospective nationwide study. The Knee. 2015;22:95-99.
18. Thorlund JB, Juhl CB, Roos EM, et al. Arthroscopic surgery for degenerative knee: systematic review and meta-analysis of benefits and harms. BMJ. 2015;350:h2747.
19. Pedersen BK, Saltin B. Evidence for prescribing exercise as therapy in chronic disease. Scan J Med Sci Sports. 2006;16(Suppl1):3-63.
20. Mangione KK, McCully K, Gloviak A, et al. The effects of high-intensity and low-intensity cycle ergometry in older adults with knee osteoarthritis. J Gerontol A Biol Sci Med Sci. 1999;54:M184-190.
21. Chatain F, Robinson AHN, Adeleine P, et al. The natural history of the knee following arthroscopic medial meniscectomy. Knee Surg Sports Traumatol Arthrosc. 2001;9:15-18.
22. American Academy of Orthopaedic Surgeons. Treatment of Osteoarthritis of the Knee. Evidence-based guideline. 2nd ed. May 18, 2013. Available at: https://www.aaos.org/research/guidelines/TreatmentofOsteoarthritisoftheKneeGuideline.pdf. Accessed October 11, 2017.
23. Lohmander LS, Thorlund JB, Roos EM. Routine knee arthroscopic surgery for the painful knee in middle-aged and old patients—time to abandon ship. Acta Orthop. 2016;87:2-4.
Meniscectomy is the most common orthopedic procedure performed in the United States with 700,000 meniscectomies performed every year.1 More than half of these procedures are performed in patients ≥45 years of age,2 giving rise to the question: Does arthroscopic surgery have a role in the treatment of patients who may have osteoarthritis (OA) and another knee condition, such as a symptomatic meniscal tear? Determining the answer is especially important when you consider that the number of true and incidental tears diagnosed on magnetic resonance imaging (MRI) has been on the rise3—a result of the routine use of MRIs to identify the cause of patients’ chronic knee pain.
At a cost of roughly $5,000 per procedure, some experts have suggested that at least a portion of the approximately $4 billion annual direct medical costs associated with meniscectomy could be put to better use.4,5 This prompted us to wonder what the literature tells us about the management of degenerative meniscus tears in middle-aged patients with OA and whether these patients would benefit from nonoperative management with optimized physical and medical therapy as a first-line approach. Our findings follow.
But first, a word about the connection between OA and meniscal tears.
What we’ve learned about meniscal damage
Research has shown that over one-third of individuals >50 years of age and three-quarters of people with knee OA have degenerative meniscal tears.6 In the past, the relative paucity of epidemiologic data on the prevalence of meniscal tears in the general population made it difficult to interpret the diagnostic information provided by MRI.
More recently, experts found that meniscal damage is especially prevalent among individuals with OA, and they began treating with arthroscopic partial meniscectomy (APM), as the meniscal damage was thought to be the anatomical foundation for the complaint of knee pain.7
However, researchers then began realizing that many patients with findings of a meniscal tear visualized on MRI reported no knee symptoms. In one study, adults in a large community-based sample found to have a meniscal tear on MRI were no more likely to have knee pain than subjects without a meniscal tear.6 Similarly, subjects with a meniscal tear and OA had no more severe pain than subjects with OA and no meniscal tear.6
In addition, the landmark Fairbank study from 19488 and others since have shown that meniscectomy can lead to other problems. Removal of meniscal tissue decreases the contact stress area, which increases stress on the articular cartilage, and inevitably leads to degeneration of the involved joint.9 Researchers have shown that even partial meniscectomy produces late articular cartilage changes.10
Which interventions and when?
We conducted an in-depth literature review to determine which approaches were best for the treatment of OA and meniscal tears, and summarize our findings below, according to OA severity. (For details of how the literature review was conducted and an at-a-glance summary of the key findings, see the TABLE.4,5,11-17) Of note: All of the studies reviewed here included patients with chronic knee pain and excluded patients with sudden onset pain from a single physical event.
The findings: Early OA and meniscal tears
The first 2 studies we identified in the literature, both published by Herrlin et al,11,12 examined the efficacy of APM in middle-aged patients with early OA (≤grade 1), according to the Ahlbäck classification.
In the first Herrlin study, a 6-month prospective randomized trial, 90 middle-aged patients with a medial meniscal tear (without traumatic history) were assigned to either APM followed by supervised exercise or 8 weeks of supervised exercise alone.11 Exercise consisted of activities for improving muscle strength, endurance, and flexibility, as well as for balance and proprioception. The authors concluded that a combination of APM and supervised exercise did not lead to greater improvements in knee function compared with supervised exercise alone.11
In the second Herrlin study, a prospective randomized study involving 96 middle-aged patients with an MRI-verified medial meniscal tear and radiographic OA, the authors concluded that arthroscopic surgery followed by exercise therapy was not superior to the same exercise therapy alone.12 The results were gleaned from both patient-reported outcomes and radiographic assessment at 2 and 5 years. Both groups reported significant improvements at 5 years, but participants did not reach the level of fitness and quality of life of similarly-aged healthy controls.
Perhaps one of the most interesting aspects of this study was that approximately one-third of patients from the exercise-only group still had disabling knee symptoms after exercise therapy, but improved to the same level as the rest of the patients after crossing over and undergoing APM.12 Part of the observed benefit of arthroscopy in these patients has the potential to be explained by the placebo effect, especially given that invasive procedures have a stronger placebo effect than do noninvasive ones, and due to the lack of blinding.18 Additionally, limitations of the above studies include small sample sizes, lack of a control group, and short-term follow-up.
Next, a 2013 study by Yim et al looked specifically at APM vs nonoperative treatment with strengthening exercises.13 A total of 102 patients with an average age of 53.8 years, a Kellgren-Lawrence Classification of Osteoarthritis of <2, and an MRI-confirmed degenerative horizontal tear of the posterior horn of the medial meniscus were randomized to the 2 intervention groups. The 2 groups were highly comparable, giving the study high internal validity. These patients were then assessed at 3 months, one year, and at 2 years after treatment.
Although most patients at the outset of the study had intense knee pain with mechanical symptoms, both groups reported a decrease in experienced knee pain, improved function, and a high level of satisfaction with their prescribed treatment, with no significant difference in any of these values after 2 years of follow-up.13 A limitation of the study was that it used subjective questionnaires to assess pain, swelling, and activities of daily living (ADLs).
A fourth study, a 2013 multicenter, randomized, sham-controlled trial, looked at 146 patients ages 35 to 65 years who had knee symptoms consistent with a degenerative medial meniscus tear (confirmed by MRI and arthroscopic evaluation) and no knee arthritis.4 The subjects were assigned to either APM or sham surgery (skin incisions only). The results showed that APM was not superior to sham surgery with regard to outcomes assessed during a 12-month follow-up period.4
Most recently (2016), Kise et al14 published the results of a randomized controlled superiority trial conducted in Norway comparing 12 weeks of supervised exercise therapy with APM for patients with degenerative meniscus tears. Their study included 140 patients ages 36 to 60 years. Notably, most (96%), but not all, of their patients had no radiographic evidence of OA.
At the 2-year follow-up, there were no differences in patient-reported pain or functional outcomes, with all patients improving significantly from baseline. Muscle strength was also measured and found to be significantly greater at 3 and 12 months in the exercise group. Limitations of this study were a lack of patient blinding and a 19% crossover from the exercise to the APM group.
The findings: Mild to moderate OA and meniscal tears
The authors of a 2002 double-blind, placebo-controlled trial randomly assigned 180 patients with degenerative meniscus tears and knee OA to either arthroscopic debridement, arthroscopic lavage, or placebo surgery consisting of superficial skin incisions without insertion of an arthroscope.5 Patients were eligible if they were ≤75 years of age and had not undergone arthroscopy of the knee within the previous 2 years. Arthritis was graded utilizing the Kellgren-Lawrence scale (0-4) and by calculating a severity score (0-12) by adding the individual scores for each of the 3 compartments of the knee; patients were excluded if they had a severity grade ≥9. One-quarter of the participants had severe arthritis, with scores of 7 or 8. Outcomes were measured over a 24-month period. At no point did either of the intervention groups report less pain or better function than the sham-surgery group.5
Additionally, in a single-center RCT involving 178 patients with mild to severe OA, subjects were randomly assigned to surgical lavage and arthroscopic debridement together with optimized physical and medical therapy or to treatment with physical and medical therapy alone.15 Patients were excluded if they had “bucket handle” meniscal tears detected by physical exam or by MRI, previous arthroscopic surgery, previous major knee trauma, or steroid injections in the last 3 months. All participants were required to have Kellgren-Lawrence grade 2, 3, or 4 arthritis.
The researchers used 2 validated outcome measures to evaluate pain, symptoms, and functional ability and followed the patients for 2 years after the initiation of treatment. This study failed to show that arthroscopic surgery provided any additional benefit to optimized physical and medical therapy.15
A 2013 multicenter, RCT randomly assigned 351 patients ≥45 years with a meniscal tear and radiographic evidence of mild to moderate OA to either surgery and postoperative physical therapy or to a standardized physical therapy regimen.16 Patients were required to have at least one symptom that was consistent with a meniscal tear for approximately one month. About 30% of the patients who were assigned to physical therapy alone underwent surgery within 6 months. There were no significant differences between the 2 groups in the magnitude of improvement in functional status or pain at 6 or 12 months.
Finally, a prospective Scottish study consisting of 270 patients with everything from no signs of OA to advanced OA who underwent APM were sent preoperative and 6-month postop questionnaires evaluating ADLs, pain, symptoms, quality of life, and body mass index (BMI).17 Their OA was graded via preop MRI or radiographs and confirmed by the operating surgeon. The investigators were unable to demonstrate any significant benefit associated with arthroscopic meniscectomy, and, therefore, could not recommend the procedure for patients with moderate to advanced OA.
However, in their analysis, 3 subgroup populations were found to benefit from APM: those with greater body habitus (BMI >30 kg/m2), those without signs of OA, and those with early OA. Limitations of this study included the lack of randomization, blinding, a control, and long-term follow up, and that the authors didn’t use established OA grading criteria.17
The bottom line: Nonoperative treatment benefits most patients
Physical therapy is an appropriate first-line treatment for degenerative meniscus tears in middle-aged patients. In fact, a trial of nonoperative treatment is likely to benefit the majority of patients. In addition, avoiding surgery eliminates surgical complications and decreases overall health care costs.
Reserve APM for those patients without significant OA who fail to improve after physical therapy, who have mechanical symptoms, or who have intra-articular loose bodies.
In addition, exercise therapy is an effective treatment for patients with knee OA. It improves function and limits joint pain in both acute arthritic flares and more long-term, chronic situations. There is strong evidence that strengthening plays a critical role in reducing symptoms and improving muscle strength, physical ability, and quality of life.19 It has been suggested that physical exercise 3 times a week for 4 months could lead to >35% improvement of knee function.20 In contrast, other studies reported that while 91% of patients 11.5 years after APM considered their knees “normal or almost normal,” patients actually experienced a reduction in postoperative physical activity and quality of life.21
The most recent American Academy of Orthopaedic Surgeons (AAOS) guidelines22 do not recommend for or against arthroscopic partial meniscectomy in patients with knee OA and a torn meniscus. In middle-aged patients, MRI abnormalities of the meniscus do not consistently correlate with symptoms. Many meniscus lesions are asymptomatic or not the primary source of pain in the setting of OA.3
Potential harms and considerations. Deep venous thrombosis is the most frequently reported adverse event of arthroscopic surgery, with an approximate incidence of 4.13 per 1000 procedures, followed by less frequent complications such as infection, pulmonary embolism, and death.18
It is important to note that the degenerative meniscus tears that occur in middle age and that are associated with OA are not the same as acute, traumatic meniscus tears. All of the studies discussed here included patients with chronic knee pain and excluded patients with sudden onset pain from a single physical event. Many of the studies excluded patients with bucket-handle tears or severe mechanical symptoms (ie, locking). APM may be indicated for these meniscus tears regardless of age or OA status.
Making a sensible choice. Ultimately, physicians and their patients must use the best evidence available to make sensible clinical decisions. The ability to retain native meniscal tissue is of utmost importance to maintaining the longevity of their knee. According to previous studies, OA progression is more likely to occur after meniscectomy than after nonoperative treatment.23
CORRESPONDENCE
William Bassett, MD, Department of Orthopedics, Rutgers-Robert Wood Johnson Medical School, 51 French St., PO Box 19, New Brunswick, NJ 08903; [email protected].
Meniscectomy is the most common orthopedic procedure performed in the United States with 700,000 meniscectomies performed every year.1 More than half of these procedures are performed in patients ≥45 years of age,2 giving rise to the question: Does arthroscopic surgery have a role in the treatment of patients who may have osteoarthritis (OA) and another knee condition, such as a symptomatic meniscal tear? Determining the answer is especially important when you consider that the number of true and incidental tears diagnosed on magnetic resonance imaging (MRI) has been on the rise3—a result of the routine use of MRIs to identify the cause of patients’ chronic knee pain.
At a cost of roughly $5,000 per procedure, some experts have suggested that at least a portion of the approximately $4 billion annual direct medical costs associated with meniscectomy could be put to better use.4,5 This prompted us to wonder what the literature tells us about the management of degenerative meniscus tears in middle-aged patients with OA and whether these patients would benefit from nonoperative management with optimized physical and medical therapy as a first-line approach. Our findings follow.
But first, a word about the connection between OA and meniscal tears.
What we’ve learned about meniscal damage
Research has shown that over one-third of individuals >50 years of age and three-quarters of people with knee OA have degenerative meniscal tears.6 In the past, the relative paucity of epidemiologic data on the prevalence of meniscal tears in the general population made it difficult to interpret the diagnostic information provided by MRI.
More recently, experts found that meniscal damage is especially prevalent among individuals with OA, and they began treating with arthroscopic partial meniscectomy (APM), as the meniscal damage was thought to be the anatomical foundation for the complaint of knee pain.7
However, researchers then began realizing that many patients with findings of a meniscal tear visualized on MRI reported no knee symptoms. In one study, adults in a large community-based sample found to have a meniscal tear on MRI were no more likely to have knee pain than subjects without a meniscal tear.6 Similarly, subjects with a meniscal tear and OA had no more severe pain than subjects with OA and no meniscal tear.6
In addition, the landmark Fairbank study from 19488 and others since have shown that meniscectomy can lead to other problems. Removal of meniscal tissue decreases the contact stress area, which increases stress on the articular cartilage, and inevitably leads to degeneration of the involved joint.9 Researchers have shown that even partial meniscectomy produces late articular cartilage changes.10
Which interventions and when?
We conducted an in-depth literature review to determine which approaches were best for the treatment of OA and meniscal tears, and summarize our findings below, according to OA severity. (For details of how the literature review was conducted and an at-a-glance summary of the key findings, see the TABLE.4,5,11-17) Of note: All of the studies reviewed here included patients with chronic knee pain and excluded patients with sudden onset pain from a single physical event.
The findings: Early OA and meniscal tears
The first 2 studies we identified in the literature, both published by Herrlin et al,11,12 examined the efficacy of APM in middle-aged patients with early OA (≤grade 1), according to the Ahlbäck classification.
In the first Herrlin study, a 6-month prospective randomized trial, 90 middle-aged patients with a medial meniscal tear (without traumatic history) were assigned to either APM followed by supervised exercise or 8 weeks of supervised exercise alone.11 Exercise consisted of activities for improving muscle strength, endurance, and flexibility, as well as for balance and proprioception. The authors concluded that a combination of APM and supervised exercise did not lead to greater improvements in knee function compared with supervised exercise alone.11
In the second Herrlin study, a prospective randomized study involving 96 middle-aged patients with an MRI-verified medial meniscal tear and radiographic OA, the authors concluded that arthroscopic surgery followed by exercise therapy was not superior to the same exercise therapy alone.12 The results were gleaned from both patient-reported outcomes and radiographic assessment at 2 and 5 years. Both groups reported significant improvements at 5 years, but participants did not reach the level of fitness and quality of life of similarly-aged healthy controls.
Perhaps one of the most interesting aspects of this study was that approximately one-third of patients from the exercise-only group still had disabling knee symptoms after exercise therapy, but improved to the same level as the rest of the patients after crossing over and undergoing APM.12 Part of the observed benefit of arthroscopy in these patients has the potential to be explained by the placebo effect, especially given that invasive procedures have a stronger placebo effect than do noninvasive ones, and due to the lack of blinding.18 Additionally, limitations of the above studies include small sample sizes, lack of a control group, and short-term follow-up.
Next, a 2013 study by Yim et al looked specifically at APM vs nonoperative treatment with strengthening exercises.13 A total of 102 patients with an average age of 53.8 years, a Kellgren-Lawrence Classification of Osteoarthritis of <2, and an MRI-confirmed degenerative horizontal tear of the posterior horn of the medial meniscus were randomized to the 2 intervention groups. The 2 groups were highly comparable, giving the study high internal validity. These patients were then assessed at 3 months, one year, and at 2 years after treatment.
Although most patients at the outset of the study had intense knee pain with mechanical symptoms, both groups reported a decrease in experienced knee pain, improved function, and a high level of satisfaction with their prescribed treatment, with no significant difference in any of these values after 2 years of follow-up.13 A limitation of the study was that it used subjective questionnaires to assess pain, swelling, and activities of daily living (ADLs).
A fourth study, a 2013 multicenter, randomized, sham-controlled trial, looked at 146 patients ages 35 to 65 years who had knee symptoms consistent with a degenerative medial meniscus tear (confirmed by MRI and arthroscopic evaluation) and no knee arthritis.4 The subjects were assigned to either APM or sham surgery (skin incisions only). The results showed that APM was not superior to sham surgery with regard to outcomes assessed during a 12-month follow-up period.4
Most recently (2016), Kise et al14 published the results of a randomized controlled superiority trial conducted in Norway comparing 12 weeks of supervised exercise therapy with APM for patients with degenerative meniscus tears. Their study included 140 patients ages 36 to 60 years. Notably, most (96%), but not all, of their patients had no radiographic evidence of OA.
At the 2-year follow-up, there were no differences in patient-reported pain or functional outcomes, with all patients improving significantly from baseline. Muscle strength was also measured and found to be significantly greater at 3 and 12 months in the exercise group. Limitations of this study were a lack of patient blinding and a 19% crossover from the exercise to the APM group.
The findings: Mild to moderate OA and meniscal tears
The authors of a 2002 double-blind, placebo-controlled trial randomly assigned 180 patients with degenerative meniscus tears and knee OA to either arthroscopic debridement, arthroscopic lavage, or placebo surgery consisting of superficial skin incisions without insertion of an arthroscope.5 Patients were eligible if they were ≤75 years of age and had not undergone arthroscopy of the knee within the previous 2 years. Arthritis was graded utilizing the Kellgren-Lawrence scale (0-4) and by calculating a severity score (0-12) by adding the individual scores for each of the 3 compartments of the knee; patients were excluded if they had a severity grade ≥9. One-quarter of the participants had severe arthritis, with scores of 7 or 8. Outcomes were measured over a 24-month period. At no point did either of the intervention groups report less pain or better function than the sham-surgery group.5
Additionally, in a single-center RCT involving 178 patients with mild to severe OA, subjects were randomly assigned to surgical lavage and arthroscopic debridement together with optimized physical and medical therapy or to treatment with physical and medical therapy alone.15 Patients were excluded if they had “bucket handle” meniscal tears detected by physical exam or by MRI, previous arthroscopic surgery, previous major knee trauma, or steroid injections in the last 3 months. All participants were required to have Kellgren-Lawrence grade 2, 3, or 4 arthritis.
The researchers used 2 validated outcome measures to evaluate pain, symptoms, and functional ability and followed the patients for 2 years after the initiation of treatment. This study failed to show that arthroscopic surgery provided any additional benefit to optimized physical and medical therapy.15
A 2013 multicenter, RCT randomly assigned 351 patients ≥45 years with a meniscal tear and radiographic evidence of mild to moderate OA to either surgery and postoperative physical therapy or to a standardized physical therapy regimen.16 Patients were required to have at least one symptom that was consistent with a meniscal tear for approximately one month. About 30% of the patients who were assigned to physical therapy alone underwent surgery within 6 months. There were no significant differences between the 2 groups in the magnitude of improvement in functional status or pain at 6 or 12 months.
Finally, a prospective Scottish study consisting of 270 patients with everything from no signs of OA to advanced OA who underwent APM were sent preoperative and 6-month postop questionnaires evaluating ADLs, pain, symptoms, quality of life, and body mass index (BMI).17 Their OA was graded via preop MRI or radiographs and confirmed by the operating surgeon. The investigators were unable to demonstrate any significant benefit associated with arthroscopic meniscectomy, and, therefore, could not recommend the procedure for patients with moderate to advanced OA.
However, in their analysis, 3 subgroup populations were found to benefit from APM: those with greater body habitus (BMI >30 kg/m2), those without signs of OA, and those with early OA. Limitations of this study included the lack of randomization, blinding, a control, and long-term follow up, and that the authors didn’t use established OA grading criteria.17
The bottom line: Nonoperative treatment benefits most patients
Physical therapy is an appropriate first-line treatment for degenerative meniscus tears in middle-aged patients. In fact, a trial of nonoperative treatment is likely to benefit the majority of patients. In addition, avoiding surgery eliminates surgical complications and decreases overall health care costs.
Reserve APM for those patients without significant OA who fail to improve after physical therapy, who have mechanical symptoms, or who have intra-articular loose bodies.
In addition, exercise therapy is an effective treatment for patients with knee OA. It improves function and limits joint pain in both acute arthritic flares and more long-term, chronic situations. There is strong evidence that strengthening plays a critical role in reducing symptoms and improving muscle strength, physical ability, and quality of life.19 It has been suggested that physical exercise 3 times a week for 4 months could lead to >35% improvement of knee function.20 In contrast, other studies reported that while 91% of patients 11.5 years after APM considered their knees “normal or almost normal,” patients actually experienced a reduction in postoperative physical activity and quality of life.21
The most recent American Academy of Orthopaedic Surgeons (AAOS) guidelines22 do not recommend for or against arthroscopic partial meniscectomy in patients with knee OA and a torn meniscus. In middle-aged patients, MRI abnormalities of the meniscus do not consistently correlate with symptoms. Many meniscus lesions are asymptomatic or not the primary source of pain in the setting of OA.3
Potential harms and considerations. Deep venous thrombosis is the most frequently reported adverse event of arthroscopic surgery, with an approximate incidence of 4.13 per 1000 procedures, followed by less frequent complications such as infection, pulmonary embolism, and death.18
It is important to note that the degenerative meniscus tears that occur in middle age and that are associated with OA are not the same as acute, traumatic meniscus tears. All of the studies discussed here included patients with chronic knee pain and excluded patients with sudden onset pain from a single physical event. Many of the studies excluded patients with bucket-handle tears or severe mechanical symptoms (ie, locking). APM may be indicated for these meniscus tears regardless of age or OA status.
Making a sensible choice. Ultimately, physicians and their patients must use the best evidence available to make sensible clinical decisions. The ability to retain native meniscal tissue is of utmost importance to maintaining the longevity of their knee. According to previous studies, OA progression is more likely to occur after meniscectomy than after nonoperative treatment.23
CORRESPONDENCE
William Bassett, MD, Department of Orthopedics, Rutgers-Robert Wood Johnson Medical School, 51 French St., PO Box 19, New Brunswick, NJ 08903; [email protected].
1. Cullen KA, Hall MJ, Golosinskiy A. Ambulatory surgery in the United States, 2006. Natl Health Stat Rep. 2009;11:1-25.
2. Hall MJ, Lawrence L. Ambulatory surgery in the United States, 1996. Advance data from vital and health statistics of the Centers for Disease Control and Prevention/National Center for Health Statistics. No. 300. August 12, 1998. Available at: https://cdc.gov/nchs/data/ad/ad300.pdf. Accessed October 11, 2017.
3. Englund M, Guermazi A, Gale D, et al. Incidental meniscal findings on knee MRI in middle-aged and elderly persons. N Engl J Med. 2008;359:1108-1115.
4. Sihvonen R, Paavola M, Malmivaara A, et al. Arthroscopic partial meniscectomy versus sham surgery for a degenerative meniscal tear. N Engl J Med. 2013;369:2515-2524.
5. Moseley JB, O’Malley KO, Petersen NJ, et al. A controlled trial of arthroscopic surgery for osteoarthritis of the knee. N Engl J Med. 2002;347:81-88.
6. Bhattacharyya T, Gale D, Dewire P, et al. The clinical importance of meniscal tears demonstrated by magnetic resonance imaging in osteoarthritis of the knee. J Bone Joint Surg. 2003;85:4-9.
7. Paxton ES, Stock MV, Brophy RH. Meniscal repair versus partial meniscectomy: a systematic review comparing reoperation rates and clinical outcomes. Arthroscopy. 2011;27:1275-1288.
8. Fairbank TJ. Knee joint changes after meniscectomy. J Bone Joint Surg Br. 1948;30B:664-670.
9. Arnoczky SP, Warren RF. Microvasculature of the human meniscus. Am J Sports Med. 1982;10:90-95.
10. Morgan CD, Wojtys EM, Casscells CD, et al. Arthroscopic meniscal repair evaluated by second-look arthroscopy. Am J Sports Med. 1991;19:632-638.
11. Herrlin S, Hållander M, Wange P, et al. Arthroscopic or conservative treatment of degenerative medial meniscal tears: a prospective randomised trial. Knee Surg Sports Traumatol Arthrosc. 2007;15:393-401.
12. Herrlin SV, Wange PO, Lapidus G, et al. Is arthroscopic surgery beneficial in treating non-traumatic, degenerative medial meniscal tears? A five-year follow-up. Knee Surg Sports Traumatol Arthrosc. 2013;21:358-364.
13. Yim JH, Seon JK, Song EK, et al. A comparative study of meniscectomy and nonoperative treatment for degenerative horizontal tears of the medial meniscus. Am J Sports Med. 2013;41:1565-1570.
14. Kise NJ, Risberg MA, Stensrud S, et al. Exercise therapy versus arthroscopic partial meniscectomy for degenerative meniscal tear in middle aged patients: randomised controlled trial with two year follow-up. BMJ. 2016;354:i3740.
15. Kirkley A, Birmingham TB, Litchfield RB, et al. A randomized trial of arthroscopic surgery for osteoarthritis of the knee. N Engl J Med. 2008;359:1097-1107.
16. Katz JN, Brophy RH, Chaisson CE, et al. Surgery versus physical therapy for a meniscal tear and osteoarthritis. N Engl J Med. 2013;368:1675-1684.
17. Bailey O, Gronkowski K, Leach WJ. Effect of body mass index and osteoarthritis on outcomes following arthroscopic meniscectomy: A prospective nationwide study. The Knee. 2015;22:95-99.
18. Thorlund JB, Juhl CB, Roos EM, et al. Arthroscopic surgery for degenerative knee: systematic review and meta-analysis of benefits and harms. BMJ. 2015;350:h2747.
19. Pedersen BK, Saltin B. Evidence for prescribing exercise as therapy in chronic disease. Scan J Med Sci Sports. 2006;16(Suppl1):3-63.
20. Mangione KK, McCully K, Gloviak A, et al. The effects of high-intensity and low-intensity cycle ergometry in older adults with knee osteoarthritis. J Gerontol A Biol Sci Med Sci. 1999;54:M184-190.
21. Chatain F, Robinson AHN, Adeleine P, et al. The natural history of the knee following arthroscopic medial meniscectomy. Knee Surg Sports Traumatol Arthrosc. 2001;9:15-18.
22. American Academy of Orthopaedic Surgeons. Treatment of Osteoarthritis of the Knee. Evidence-based guideline. 2nd ed. May 18, 2013. Available at: https://www.aaos.org/research/guidelines/TreatmentofOsteoarthritisoftheKneeGuideline.pdf. Accessed October 11, 2017.
23. Lohmander LS, Thorlund JB, Roos EM. Routine knee arthroscopic surgery for the painful knee in middle-aged and old patients—time to abandon ship. Acta Orthop. 2016;87:2-4.
1. Cullen KA, Hall MJ, Golosinskiy A. Ambulatory surgery in the United States, 2006. Natl Health Stat Rep. 2009;11:1-25.
2. Hall MJ, Lawrence L. Ambulatory surgery in the United States, 1996. Advance data from vital and health statistics of the Centers for Disease Control and Prevention/National Center for Health Statistics. No. 300. August 12, 1998. Available at: https://cdc.gov/nchs/data/ad/ad300.pdf. Accessed October 11, 2017.
3. Englund M, Guermazi A, Gale D, et al. Incidental meniscal findings on knee MRI in middle-aged and elderly persons. N Engl J Med. 2008;359:1108-1115.
4. Sihvonen R, Paavola M, Malmivaara A, et al. Arthroscopic partial meniscectomy versus sham surgery for a degenerative meniscal tear. N Engl J Med. 2013;369:2515-2524.
5. Moseley JB, O’Malley KO, Petersen NJ, et al. A controlled trial of arthroscopic surgery for osteoarthritis of the knee. N Engl J Med. 2002;347:81-88.
6. Bhattacharyya T, Gale D, Dewire P, et al. The clinical importance of meniscal tears demonstrated by magnetic resonance imaging in osteoarthritis of the knee. J Bone Joint Surg. 2003;85:4-9.
7. Paxton ES, Stock MV, Brophy RH. Meniscal repair versus partial meniscectomy: a systematic review comparing reoperation rates and clinical outcomes. Arthroscopy. 2011;27:1275-1288.
8. Fairbank TJ. Knee joint changes after meniscectomy. J Bone Joint Surg Br. 1948;30B:664-670.
9. Arnoczky SP, Warren RF. Microvasculature of the human meniscus. Am J Sports Med. 1982;10:90-95.
10. Morgan CD, Wojtys EM, Casscells CD, et al. Arthroscopic meniscal repair evaluated by second-look arthroscopy. Am J Sports Med. 1991;19:632-638.
11. Herrlin S, Hållander M, Wange P, et al. Arthroscopic or conservative treatment of degenerative medial meniscal tears: a prospective randomised trial. Knee Surg Sports Traumatol Arthrosc. 2007;15:393-401.
12. Herrlin SV, Wange PO, Lapidus G, et al. Is arthroscopic surgery beneficial in treating non-traumatic, degenerative medial meniscal tears? A five-year follow-up. Knee Surg Sports Traumatol Arthrosc. 2013;21:358-364.
13. Yim JH, Seon JK, Song EK, et al. A comparative study of meniscectomy and nonoperative treatment for degenerative horizontal tears of the medial meniscus. Am J Sports Med. 2013;41:1565-1570.
14. Kise NJ, Risberg MA, Stensrud S, et al. Exercise therapy versus arthroscopic partial meniscectomy for degenerative meniscal tear in middle aged patients: randomised controlled trial with two year follow-up. BMJ. 2016;354:i3740.
15. Kirkley A, Birmingham TB, Litchfield RB, et al. A randomized trial of arthroscopic surgery for osteoarthritis of the knee. N Engl J Med. 2008;359:1097-1107.
16. Katz JN, Brophy RH, Chaisson CE, et al. Surgery versus physical therapy for a meniscal tear and osteoarthritis. N Engl J Med. 2013;368:1675-1684.
17. Bailey O, Gronkowski K, Leach WJ. Effect of body mass index and osteoarthritis on outcomes following arthroscopic meniscectomy: A prospective nationwide study. The Knee. 2015;22:95-99.
18. Thorlund JB, Juhl CB, Roos EM, et al. Arthroscopic surgery for degenerative knee: systematic review and meta-analysis of benefits and harms. BMJ. 2015;350:h2747.
19. Pedersen BK, Saltin B. Evidence for prescribing exercise as therapy in chronic disease. Scan J Med Sci Sports. 2006;16(Suppl1):3-63.
20. Mangione KK, McCully K, Gloviak A, et al. The effects of high-intensity and low-intensity cycle ergometry in older adults with knee osteoarthritis. J Gerontol A Biol Sci Med Sci. 1999;54:M184-190.
21. Chatain F, Robinson AHN, Adeleine P, et al. The natural history of the knee following arthroscopic medial meniscectomy. Knee Surg Sports Traumatol Arthrosc. 2001;9:15-18.
22. American Academy of Orthopaedic Surgeons. Treatment of Osteoarthritis of the Knee. Evidence-based guideline. 2nd ed. May 18, 2013. Available at: https://www.aaos.org/research/guidelines/TreatmentofOsteoarthritisoftheKneeGuideline.pdf. Accessed October 11, 2017.
23. Lohmander LS, Thorlund JB, Roos EM. Routine knee arthroscopic surgery for the painful knee in middle-aged and old patients—time to abandon ship. Acta Orthop. 2016;87:2-4.
From The Journal of Family Practice | 2017;66(11):E1-E6.
PRACTICE RECOMMENDATIONS
› Start middle-aged patients with knee pain and a degenerative meniscal tear on a regimen of strengthening-based physical therapy. A
› Limit meniscectomy to patients who either have no preoperative osteoarthritis (OA) or early-stage OA, unless there is evidence of mechanical locking or intra-articular loose bodies. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Anorectal Evaluations: Diagnosing & Treating Benign Conditions
CE/CME No: CR-1711
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Understand basic anorectal anatomy and how to perform a thorough anorectal exam.
• Describe the physical exam findings of common benign anorectal conditions.
• Discuss the different treatment options for benign anorectal conditions.
• Differentiate between common benign anorectal symptoms and red flags that should prompt referral to a colorectal specialist.
FACULTY
Priscilla Marsicovetere is an Assistant Professor of Medical Education and of Surgery at the Geisel School of Medicine at Dartmouth in Hanover, New Hampshire; Program Director for the Franklin Pierce University PA Program in Lebanon, New Hampshire; and practices with Emergency Services of New England at Springfield Hospital in Vermont. Srinivas Joga Ivatury is an Assistant Professor of Surgery at the Geisel School of Medicine at Dartmouth and practices in the Department of Surgery at the Dartmouth Hitchcock Medical Center in Lebanon, New Hampshire.
The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through October 31, 2018.
Article begins on next page >>
Anorectal pain and discomfort can stem from several possible etiologies, most of which are benign. As such, many patients with anorectal complaints who present in the primary care setting can be adequately evaluated, diagnosed, and treated without referral to a colorectal specialist. However, the clinician must be able to differentiate between common benign anorectal symptoms and red flags that should prompt referral.
Anorectal disorders are common in the United States and result in numerous primary care visits each year. Presentations range from pain and itching to bleeding and lesions. Common anorectal conditions include hemorrhoids, perianal skin tags, fissures, pruritus ani, perianal abscess, and condyloma. Most are benign and can be managed in the primary care setting.
Before a provider can competently diagnose and treat anorectal conditions, however, a comprehensive history and physical examination must be conducted. Grucela and colleagues documented that physicians’ diagnostic accuracy with anorectal conditions is about 50%—highlighting the need for providers to become more familiar with the history and clinical elements associated with anorectal complaints.1
This article reviews the assessment of the anorectum, diagnosis of common disorders and their recommended treatments, and red flags for referral to a colorectal specialist.
ANORECTAL ANATOMY
The beginning of the anal canal is demarcated by its moist, hairless appearance. Just inside the anal opening are two palpable circular muscles, the internal and external anal sphincters, separated by an intersphincteric groove. The sphincters are firmly closed in the resting state, which helps maintain continence.
The anal canal is generally 3 to 4 cm long and ends at the dentate line, a series of crypts in the anal mucosa.2 The crypts are openings into the anal glands, which are mucus-secreting structures in the anus. The dentate line is easily identified on anoscopy as a discrete change in the appearance of the mucosa. The dentate line is an important landmark because it delineates the boundary between somatic and visceral nerve supplies.3 Tissue proximal to the dentate line is innervated by visceral nerves and is insensate, and thus usually not a cause of pain; tissue distal to the dentate line, however, is highly innervated by somatic nerves and can be intensely painful.2
The anorectal canal is lined by three fibrovascular cushions, located in the left lateral, right posterior, and right anterior positions.4 Inside each cushion is a venous structure, called a hemorrhoid, which allows the cushion to enlarge and help maintain continence.5
Proximal to the anus is the rectum, the 12- to 15-cm long terminus of the colon. Anorectal examination in the primary care setting will typically not progress beyond the last 2 to 3 cm of the rectum.
TAKING THE HISTORY
A thorough history will provide clues about potential underlying anorectal pathology. Patients may not be forthcoming about symptoms due to embarrassment, fear of a cancer diagnosis, or cultural customs or habits. A thorough history should elicit information about all of the patient’s symptoms (see Table 1), including bleeding, change in bowel habits, and unintended weight loss.

PHYSICAL EXAM
Positioning the patient
Undergoing an anorectal examination can be embarrassing, whether it be from exposure of sensitive body parts or the less-than-desirable prone jackknife positioning. Patients often have preconceived notions that the exam will be humiliating and/or painful. Care should be taken to minimize any embarrassment and discomfort.
Positioning of the patient is a matter of provider preference. Options include the left lateral decubitus, prone jackknife, or lithotomy positions.
Positioning should always be done with draping. Regardless of position, ensure the draping exposes only the perineum. This can be achieved by encircling the patient’s bare bottom with a plain white sheet that exposes only the anus and surrounding skin, keeping the lower back, lateral buttocks, and thighs covered.
Interestingly, data on patient preference for positioning during anorectal exams are limited. In a 2009 study of 178 patients undergoing anorectal exam, more than half of patients (up to 71.4%) expecting to or having already had a proctologic exam reported that no specific type of positioning (eg, Sims, lithotomy with lifted legs, knee-chest, knee-chest with patient’s body bent forward) was most embarrassing to them.6 The report revealed that while most patients would favor the Sims position if they had a choice, they deferred to their examiner to choose the position that seemed most suitable to get a reliable diagnosis.6
Inspection of the perineum
Once the patient is properly positioned and draped, inspection of the perineum can occur. Begin by gently spreading the buttocks. Describe any abnormality seen (eg, ulcer, lesion, dermatitis, prolapsing tissue, or blood), including size, color, and location.
A common pitfall is to describe the location of abnormalities using a clock face, such as “at 4 o’clock.” This is misleading and should be avoided, because depending on patient position, the clock face can point to different locations (eg, if the patient is in the lithotomy versus prone jackknife position).
A better approach is to divide the perianal area into four anatomic quadrants: right anterior, right posterior, left anterior, left posterior. Using this schematic, the patient's position is irrelevant, and accurate documentation of lesion location is assured.
Digital rectal exam
After visual inspection of the perianal skin, a digital rectal exam (DRE) should be performed. Slowly insert a gloved, lubricated index finger into the anus and lower rectum. Note the tone of the anus at rest (eg, excessively tight vs lax). Palpate the circumference of the anus, sweeping side to side while assessing for any tenderness, mass, or induration—if present, note the anatomic quadrant. If a mass is felt, note whether it is firm or soft, fixed or mobile, and broad-based or pedunculated. When the lubricated finger is removed from the anus, note whether blood is present.
Anoscopy
After DRE, visually inspect the anorectum. The instrument used varies from anoscope to rigid proctoscope to flexible sigmoidoscope. In a primary care setting, the most likely available instrument is an anoscope. The average anoscope is about 7 cm long and 2 cm in diameter, with a beveled tip and an obturator (see Figure 1), and allows a 360° view of the anal canal.7
Examination of the anal canal is accomplished by dividing the canal into the four anatomic quadrants described earlier and inserting the lubricated anoscope for inspection of each of the four quadrants. Observe the rectal mucosa and the anus as the scope is slowly withdrawn. If abnormalities are seen, note the location, size, shape, and any other descriptive features.
It is not necessary to perform a Hemoccult test after examination of the anorectum, as the presence of minor blood may be the direct result of the exam itself and thus provides no useful information to the examiner.
COMMON PATHOLOGIES
Once the history and physical exam are complete, a differential diagnosis can be formulated. Most anorectal disorders are benign conditions that pose no immediate health threat and can be managed in the primary care setting. Others, however, can be more serious and should prompt referral to a colorectal specialist for further evaluation. Knowing the difference can spare a patient unnecessary anxiety and referral; it can also lead to prompt, lifesaving interventions if red flags are recognized.
Hemorrhoids
Hemorrhoids are a common anorectal complaint.8,9 It is estimated that up to 75% of the population will experience symptoms of hemorrhoids during their lifetime.5,8 Whether internal or external, in their normal, nonpathologic, quiescent state, hemorrhoids are asymptomatic. Hemorrhoids become symptomatic when the supporting structures of hemorrhoidal tissue (ie, the anal cushions) deteriorate, resulting in venous dilation, inflammation, and thrombosis, which in turn lead to swelling, bright red bleeding, and/or prolapse.2,10 The most common causes of hemorrhoidal disease are chronic constipation and prolonged straining with bowel movements, though chronic diarrhea and pregnancy have also been identified as risk factors.2,8,11
External hemorrhoids, which are located distal to the dentate line, are typically only visible when they become thrombosed or swollen. In this state, they may manifest as acute-onset, exquisitely painful, large, purple-to-blue bulges at the anal outlet (see Figure 2). The number and size of the lesions can vary. The patient may report pain when sitting or wiping, as well as bleeding from the lesion.12,13 The pain is typically severe in the first couple of days, then slowly starts to subside.2,12
For internal hemorrhoids, which are located proximal to the dentate line, the main symptom is usually painless bright red blood per rectum.8,11,12 Patients may also report a sensation of rectal fullness or experience prolapse of the hemorrhoid through the anus. Prolapse typically occurs with defecation; in more severe cases, it can also occur between bowel movements, usually with any activity that increases intra-abdominal pressure (eg, coughing, heavy lifting, pregnancy, portal hypertension). The prolapse may reduce spontaneously, or may have to be manually reduced. If it cannot be reduced, there is a risk for incarceration or strangulation, potentially leading to gangrene.
The presence of bleeding and/or prolapse determines the classification of internal hemorrhoids (see Table 2). Dietary and lifestyle modification are used in the management of all grades of hemorrhoids. In addition, for grade 1 and 2 lesions, topical medication (eg, anti-inflammatory cream) can be used, whereas grade 3 (and selected grade 2) lesions respond well to rubber band ligation. Given the severity of grade 4 lesions, surgical intervention (eg, hemorrhoidectomy) is usually indicated.10

About a third of patients with symptomatic hemorrhoids seek clinical treatment.14 Most are hemodynamically stable and require no imaging and usually no labs (unless anemia is suspected).2 Management depends on the location and degree of symptoms (eg, internal vs external, prolapse, or thrombosis). In the event of an acutely thrombosed external hemorrhoid, clot excision for pain relief is appropriate if symptoms have been present for less than 48 to 72 hours; after that amount of time, the pain from the procedure will likely exceed the degree of relief provided, and conservative management should instead be recommended.2,8,11
Firstline treatment consists of lifestyle modification with a high-fiber diet and daily fiber supplement to ensure stool is soft and easy to pass.2,8 A meta-analysis of seven clinical trials with a total of 378 patients with hemorrhoids showed that fiber supplementation resulted in a 50% decrease in bleeding risk from internal hemorrhoids.15
Adequate hydration, preferably with noncaffeinated liquids, is also recommended. This will prevent constipation and the need to strain or spend excessive time on the toilet. Sitz baths can help alleviate pain and discomfort.
Several OTC topical medications are marketed for hemorrhoid relief. Many of these preparations contain steroids for their anti-inflammatory effects or astringents to address skin irritation that can result from anal leakage if prolapsing hemorrhoids prevent the anal outlet from closing. Steroid use should be limited to five to seven days, due to atrophic effects on the skin. While OTC preparations may temporarily alleviate discomfort, they will not address the underlying cause of symptoms.
Indications for referral to a colorectal specialist for symptomatic hemorrhoids include failure to improve with conservative management, persistent patient discomfort, and prolapse, as these indicate potential need for more invasive treatment.
Perianal skin tags
Perianal skin tags, while a nuisance, are not pathologic in most instances and pose no threat to health. They are an outgrowth of normal skin, appearing as loose, flesh-colored perianal tissues (see Figure 3). Tags range in size from a few millimeters to a centimeter long and can occur alone or in multiples.
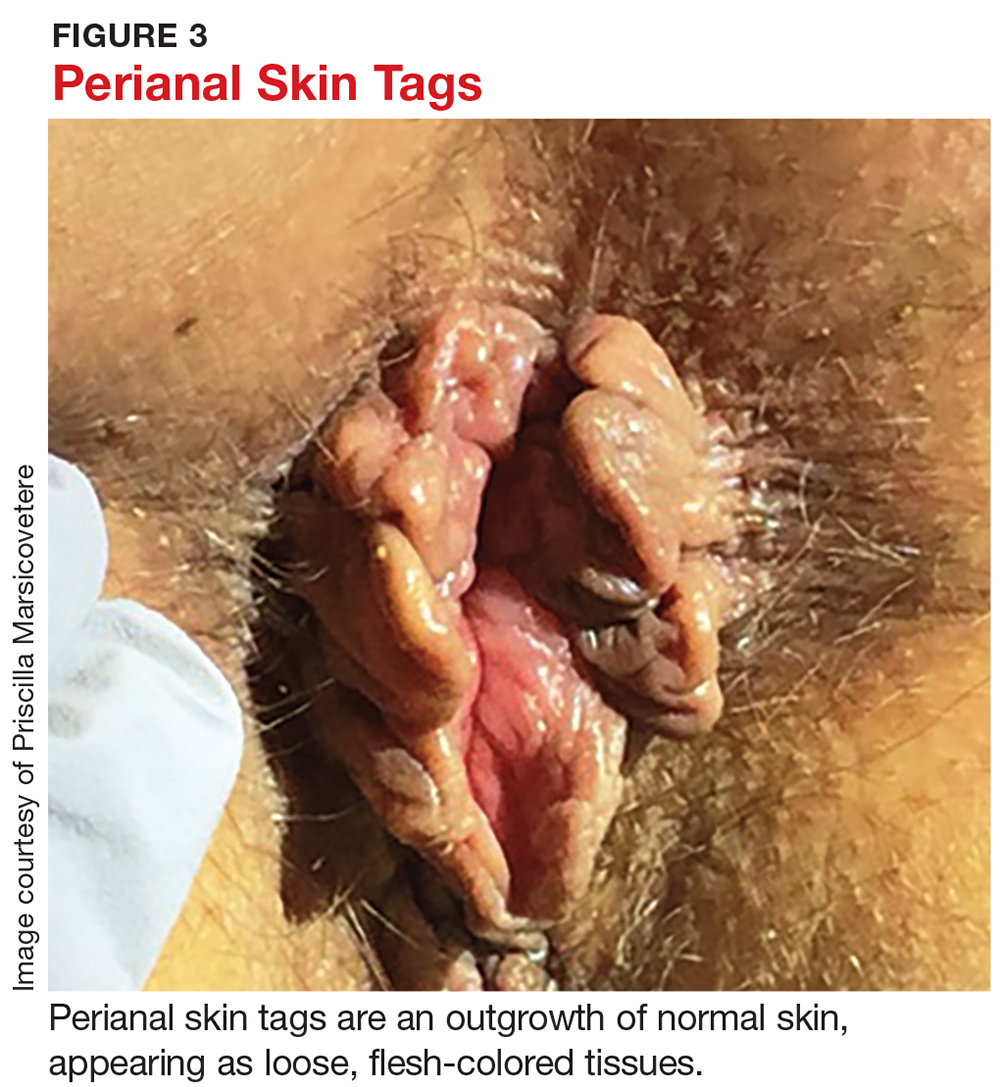
Perianal skin tags are diagnosed clinically and require no labwork or imaging. Visual inspection is typically sufficient to distinguish tags from pathologic lesions such as condyloma or abscess. If there is uncertainty, however, biopsy or referral to a specialist is warranted.
Certain medical conditions can predispose a patient to development of perianal skin tags. They can be sequelae of thrombosed external hemorrhoids.8,11 They are also common in patients with Crohn disease.11 Perianal skin tags are not, however, the result of anal intercourse or sexually transmitted infections.
Treatment is usually not indicated for perianal skin tags. If the tags interfere with hygiene or cause perianal discomfort or significantly decreased quality of life, however, patients may seek removal. These patients should be referred to a colorectal specialist for evaluation for excision.
Anal fissures (fissure in ano)
The most common cause of severe anorectal pain is fissure.4 A fissure is an elliptical tear, or split, in the lining of the anal canal that causes spasm of the anal sphincters. The tear is distal to the dentate line and thus intensely painful.2,5 Common cited causes of fissures are trauma from passage of large, hard stools, straining, or diarrhea.16
Fissures can usually be visualized by spreading the posterior anus apart (see Figure 4). They are most commonly located in the posterior or anterior midline, though they can occur anywhere around the anus.2,4 Often, a sentinel tag—appearing as a taut, flesh-colored skin tag—is present at the external pole of the fissure.5,11 DRE and anoscopy should be avoided, as they will trigger intense pain and spasm of the sphincters.
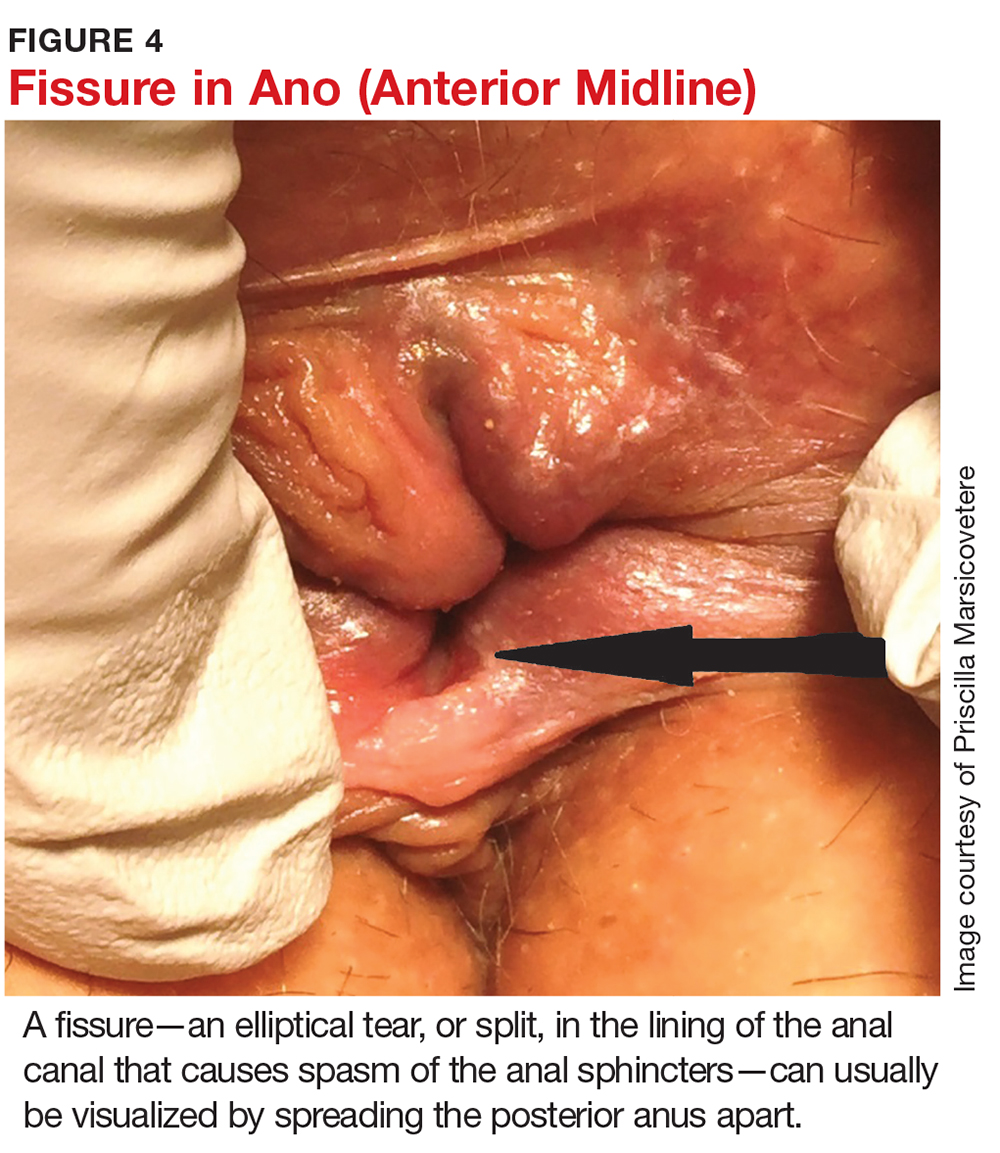
Fissures are characterized as acute (present ≤ 3 months) or chronic (> 3 months).9,11 Visually, acute fissures typically have clean edges, with the appearance of a paper cut to the mucosa, while chronic fissures have indurated, heaped-up edges, often with exposure of the underlying sphincter muscle.17
They tend to be exquisitely painful, as the mucosa distal to the dentate line is highly innervated. Patients report pain akin to “passing shards of broken glass” with bowel movements, which is often accompanied by a fear of defecation and bright red blood on the toilet paper or dripping into the water.11 The pain, caused by spasm of the sphincters, typically starts during a bowel movement and lasts minutes to hours afterward.
Initial treatment is aimed at relaxing the sphincters, as well as softening stool to prevent further trauma and allow the fissure to heal. Patients should be educated about the importance of adequate fiber intake to prevent constipation and straining. A daily bulk fiber supplement, in addition to a high-fiber diet (20-25 g/d), has been shown to result in healing of 87% of acute fissures.16 Sitz baths in plain warm water, three to four times a day, can encourage relaxation of the sphincters and increase local blood flow, both of which help with fissure healing.16 Topical medications can also be prescribed. These include compounded nitroglycerin 0.2% or nifedipine 2.0%, which act to reduce the spasm by relaxing smooth muscle, as well as increase blood flow to the lesion.12,14
Most acute fissures will heal with the regimen of high fiber intake, sitz baths, and topical medication. For refractory or chronic fissures, referral to a colorectal specialist for more invasive treatment is appropriate. Additionally, fissures that are not located in the typical posterior or anterior midline might indicate an atypical etiology, such as Crohn disease, tuberculosis, leukemia, or HIV, and thus patients who present with fissures in these locations should also be referred to a colorectal specialist.2,4,11,18
Pruritus ani
Pruritus ani, known as perianal dermatitis, is a benign condition that presents with intense perianal itching and burning. It is the second most common anorectal condition after hemorrhoids and affects up to 5% of the US population.2,9,11,19
Pruritus ani often develops secondary to local irritation of the skin (eg, from prolonged exposure to moisture), leading to an inflammatory response within the superficial skin layers. The irritation causes patients to scratch the skin, resulting in trauma, excoriation, and ulcer formation and leading to a cycle of further inflammation, exacerbation of symptoms, and persistent scratching.
Physical exam may reveal circumferential erythematous and irritated perianal skin (see Figure 5). Linear or deep, punched-out excoriations may be present. Chronically, patients may develop lichenification with thick, whitened patches of skin.11 In the absence of red flags such as unintentional weight loss, anemia, rectal bleeding, or a family history of colon cancer, no additional evaluation is required during the initial visit, though anoscopy can be used to rule out associated anorectal pathology.
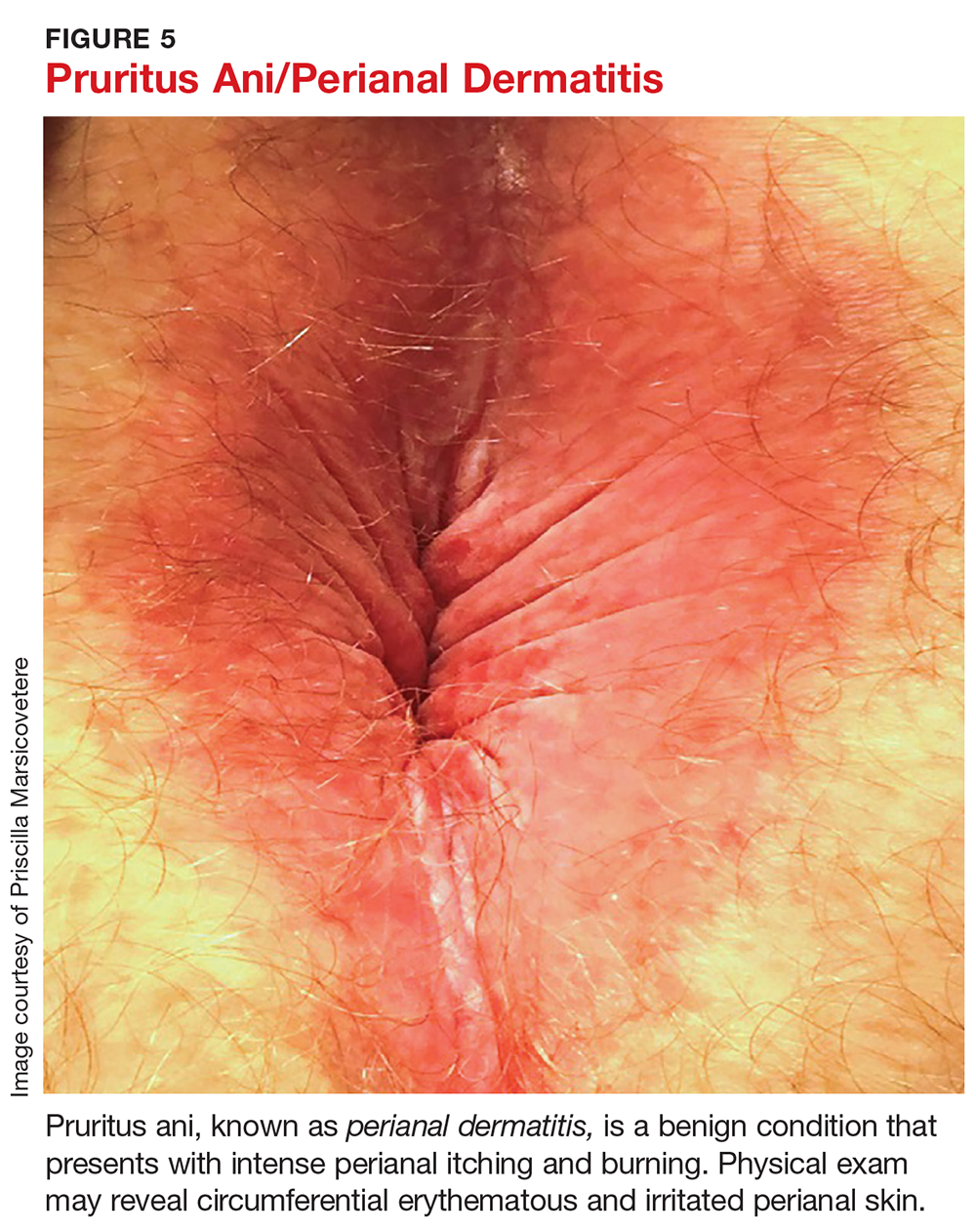
Many different causes of pruritus ani have been reported (see Table 3). In the case of an identifiable cause, symptoms tend to resolve once the offending agent is eliminated. Up to a quarter of cases, however, are idiopathic, with no identifiable trigger.20

Symptom management is thus key. Patients should be educated about lifestyle modification and informed that scratching will irritate the skin further and aggravate symptoms.5,11 If incontinence or diarrhea is thought to cause symptoms, dietary modifications, a fiber supplement, dysmotility agents, and Kegel exercises to strengthen the sphincters and decrease anal leakage can be recommended.
The affected skin should be kept clean and dry at all times. Aggressive wiping and overzealous hygiene should be avoided. Sitz baths can help with hygiene. A topical astringent such as witch hazel can help remove excess moisture from the skin. A layer of protective skin barrier cream with zinc oxide, when applied over dry skin, can help protect the skin from leakage throughout the day.
A sedating antihistamine can reduce scratching during sleep. Topical hydrocortisone 1% cream is effective for itch relief; this should be limited to five to seven days of consecutive use, however, as it can lead to pathologic thinning of the perianal skin. Topical capsaicin 0.006% cream has been shown to help alleviate intractable pruritus.2,21
The goal of these measures is to break the cycle of irritation and inflammation and give the skin an opportunity to heal. If the patient fails to improve, referral to a colorectal specialist or, alternatively, a dermatologist, is warranted for perianal skin biopsy and more invasive treatment options.
Perianal abscess
A perianal abscess is an infected cavity filled with pus under pressure, located near the anus or rectum. It most often results from an acute infection of the anorectal glands located at the dentate line that tracks outward to the perianal skin.12 Abscesses can also result from another disease process, such as Crohn disease, diabetes, or rectal trauma.2,11
Localized pain, swelling, and drainage are common presenting symptoms of an abscess.2,11 Systemic symptoms such as fever and chills may present later in the course but are rare.2
Treatment of the acute process is incision and drainage (I&D).11,12 This can be accomplished in the office with injection of local anesthesia, followed by excision of an ellipse of skin overlying the cavity large enough to allow full drainage of the abscess. Use of drains and packing of the wound are usually not necessary. There is also no role for antibiotics unless the patient is diabetic or immunosuppressed or cellulitis is present.11 In patients with systemic signs of illness, imaging such as CT or MRI of the pelvis can be used to assess for a deeper infection.2,12
For most patients, I&D resolves the process. However, up to half of perianal abscesses progress to form a fistula, a tunnel connecting the infected anal gland to the external skin.2,11,22 Indeed, abscesses and fistulae are part of the same infectious process, with the abscess representing the acute phase of infection and the fistula representing the chronic phase. On physical exam, in addition to the abscess site on the skin, a fistula may manifest as a palpable cord beneath the skin between the anus and the abscess opening.9 Additionally, the patient may report the abscess has been recurrent in nature, cyclically increasing in size and pain, then spontaneously draining.
A fistula requires surgical intervention for definitive treatment and should therefore prompt referral to a colorectal specialist. In the absence of fistula, however, a simple perianal abscess can be treated with I&D in the primary care setting.
Condyloma acuminatum
Condyloma acuminatum, also known as genital warts, is the most common sexually transmitted infection in the United States and a frequent anorectal complaint.23-25 More than 6 million new infections occur annually.25
Condyloma is caused by the human papillomavirus (HPV).24 More than 100 HPV subtypes have been identified.25 Types 6 and 11 are associated with typical condyloma acuminatum, while types 16 and 18 are found more commonly with dysplasia and malignant transformation.3,26
The lesions are spread by direct skin-to-skin or mucosa-to-mucosa contact, including anal intercourse. They appear as tiny finger-like projections on the perianal or genital skin, often with a cauliflower-like appearance, in clusters or as single entities, and ranging in size from a few millimeters to several centimeters (see Figure 6).
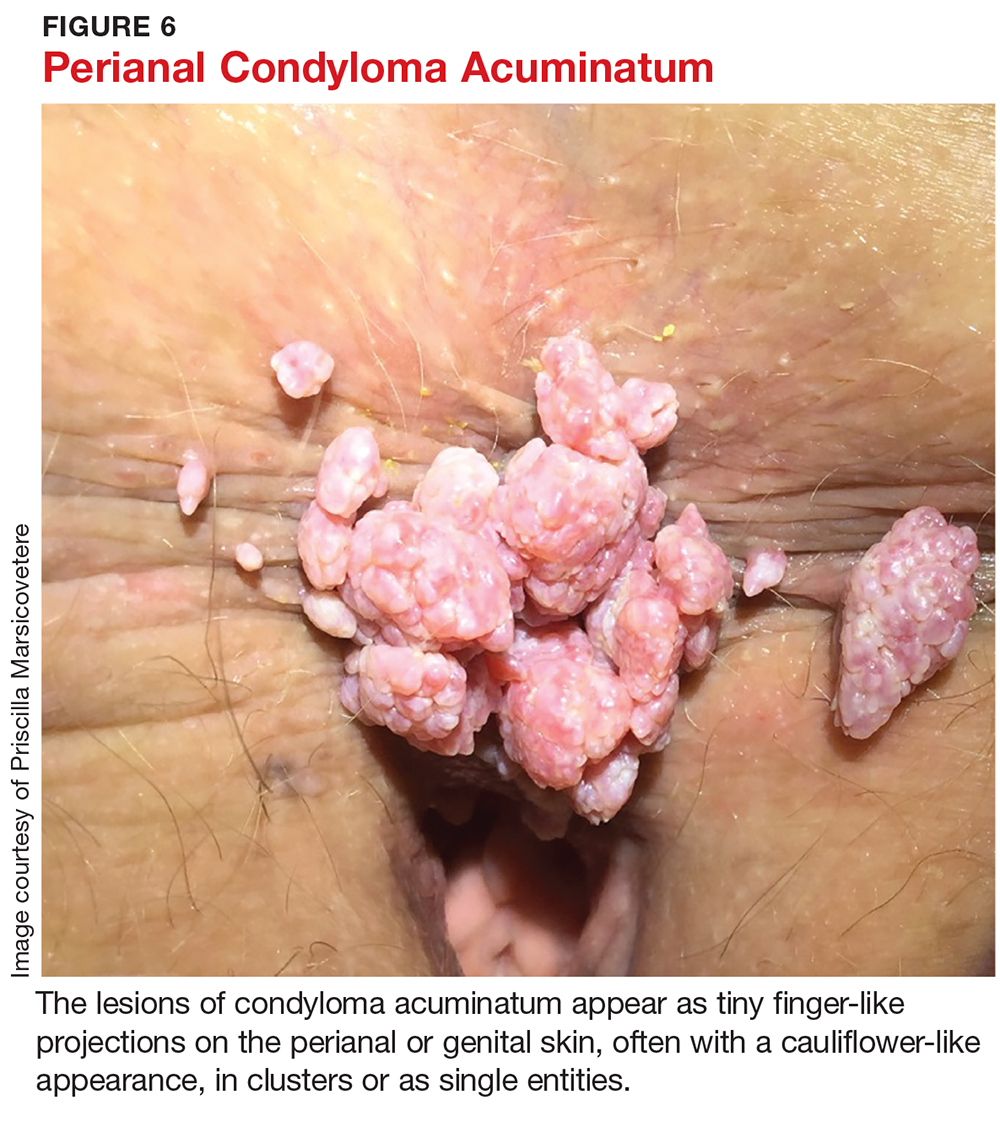
The patient may be asymptomatic or may complain of bleeding with defecation or leakage from the rectum between bowel movements. In up to 90% of perianal condyloma, concomitant anorectal lesions are present.3 Anoscopy is therefore indicated whenever perianal lesions are seen.
Treatment of genital warts varies depending on the number and location of lesions. The goal is lesion removal, but none of the available treatments are curative.24 If only a few small (diameter < 2 mm) warts are seen in the pe
Trichloroacetic acid (TCA), which is considered a form of chemical cautery, has been deemed more effective for tiny warts (diameter 1-2 mm) and better tolerated than podophyllin.24 Unlike podophyllin, however, TCA must be applied in clinic by a specialist, not at home by the patient. It works by chemically burning, cauterizing, and eroding skin and mucosa, thereby destroying old infected cells and allowing for growth of new, healthy cells.24 Using a solution with a higher concentration of TCA (80%-90%) has been shown to be more effective than lower concentration solutions (30%).24 The recurrence rate is high, generally about 36%.27 Due to its low risk for systemic absorption, TCA is safe for use during pregnancy.24
Local application of an immunomodulator, such as imiquimod 2%, 3.75%, or 5% cream, can also be used. Imiquimod works by upregulating tumor necrosis factor, leading to decreased viral replication and subsequent regression of warts.24 The most common side effects of imiquimod are erythema, erosions, burning, and pruritus.24 The higher concentration is associated with increased clearance rates as well as increased adverse effects.27 Therefore, the 3.75% cream is the preferred concentration of most providers.27 Recurrence rates range from 13% to 19%.28
In 2006, the FDA approved polyphenon E, a botanical ointment, to treat anogenital warts. The formulation is composed of eight different sinecatechins extracted from green tea leaves.29 Green tea sinecatechins have been shown to have antioxidant, antiproliferative, and antiviral properties, though the specific mechanisms of action in inhibiting anogenital wart growth have not been well studied to date. In a multicenter, randomized, double-blind, vehicle-controlled, three-arm parallel group phase 3 trial of 495 patients with anogenital warts, sinecatechins ointment 10% or 15% applied three times daily for 16 weeks resulted in complete clearance of warts in 57.2% and 56.3% of patients, respectively, compared to 33.7% clearance in the vehicle patients.30 Partial clearance (≥ 50%) of warts was observed in 78.4% of patients who used the 15% ointment, 74.0% of those who used the 10% ointment, and 51.5% of the vehicle patients. Rates of recurrence were 6.5% and 8.3% with use of the 10% and 15% sinecatechins ointment, respectively, which are much lower than recurrence rates observed with other topical applications (eg, imiquimod and podophyllotoxin).30
For larger warts or when there are anal lesions present, surgical excision and fulguration is required and referral to a colorectal specialist is warranted.
Warts recur from 4.6% to more than 70% of the time, most commonly due to activation of latent virus.23 Therefore, whether initially treated in an office or operating room, condyloma patients require follow-up on a regular basis, generally every six to 12 months, to assess for recurrent anal lesions so that treatment can be promptly initiated.
In the absence of a cure for anogenital warts, prevention through an HPV vaccine is important. The quadrivalent HPV vaccine (Gardasil) was approved by the FDA in 2006 for prophylactic vaccination of females ages 9 to 26. The vaccine triggers host formation of antibodies against four common subtypes of HPV: 6, 11, 16, and 18.31 In 2009, the FDA expanded the use of Gardasil to include males ages 9 to 26; subsequently, Gardasil 9 (which protects against nine types of HPV) was approved for males ages 9 to 15.32 In a group of 4,065 healthy males between the ages of 16 and 26, Gardasil reduced the incidence of external anogenital warts by 90.4%.33
HPV vaccine is now recommended for females through age 26 and males through age 21. The CDC also recommends HPV vaccine for the following individuals through age 26 if they did not get vaccinated when they were younger:
- Young men who have sex with men
- Young men who identify as gay or bisexual or who intend to have sex with men
- Young adults who are transgender
- Young adults with immunocompromising conditions (including HIV).34
CONCLUSION
Anorectal conditions are a common presentation in the primary care setting. A thorough history and physical exam will usually determine the etiology, and most benign pathologies can be successfully treated in the primary care clinic. Knowing how to perform an anorectal exam and having a thorough understanding of the most common anorectal pathologies can help alleviate examiner discomfort, while ensuring that patients receive prompt and adequate care. At the same time, recognizing the red flags can expedite referral for colorectal specialty evaluation when appropriate. Red flags that should prompt immediate referral to a colorectal specialist include older age, unintentional weight loss, iron deficiency anemia, family history of inflammatory bowel disease or colorectal cancer, and persistent anorectal bleeding or persistent symptoms despite adequate treatment of a suspected benign condition.
1. Grucela A, Salinas H, Khaitov S, et al. Prospective analysis of clinical accuracy in the diagnosis of benign anal pathology, comparison across specialties and years of experience. Dis Colon Rectum. 2010;53(1):47-52.
2. Tupe CL, Pham TV. Anorectal complaints in the emergency department. Emerg Med Clin North Am. 2016;34(2):251-270.
3. Billingham RP, Isler JT, Kimmins MH, et al. The diagnosis and management of common anorectal disorders. Curr Probl Surg. 2004;41(7):586-645.
4. Schubert MC, Sridhar S, Schade RR, Wexner SD. What every gastroenterologist needs to know about common anorectal disorders. World J Gastroenterol. 2009;15(26):3201-3209.
5. Henderson PK, Cash BD. Common anorectal conditions: evaluation and treatment. Curr Gastroenterol Rep. 2014; 16(10):408.
6. Gebbensleben O, Hilger Y, Rohde H. Patients’ view of medical positioning for proctologic examination. Clin Exp Gastroenterol. 2009;2:133-138.
7. Gopal DV. Diseases of the rectum and anus: a clinical approach to common disorders. Clin Cornerstone. 2002;4(4): 34-48.
8. Lohsiriwat V. Treatment of hemorrhoids: a coloproctologist’s view. World J Gastroenterol. 2015;21(31):9245-9252.
9. Fargo MV, Latimer K. Evaluation and management of common anorectal conditions. Am Fam Physician. 2012;85(6):624-630.
10. Lohsiriwat V. Hemorrhoids: from basic pathophysiology to clinical management. World J Gastroenterol. 2012;18(17): 2009-2017.
11. Foxx-Orenstein AE, Umar SB, Crowell MD. Common anorectal disorders. Gastroenterol Hepatol. 2014;10(5):294-301.
12. Lohsiriwat V. Anorectal emergencies. World J Gastroenterol. 2016;22(26):5867-5878.
13. Greenspon J, Williams SB, Young HA, Orkin BA. Thrombosed external hemorrhoids: outcome after conservative treatment or surgical management. Dis Colon Rectum. 2004(9);47:1493-1498.
14. Summers A. Assessment and treatment of three common anorectal conditions. Emerg Nurse. 2013;21(2):28-33.
15. Alonso-Coello P, Mills E, Heels-Ansdell D, et al. Fiber for the treatment of hemorrhoids complications: a systematic review and meta-analysis. Am J Gastroenterol. 2006;101(1):181-188.
16. Medhi B, Rao RS, Prakash A, et al. Recent advances in the pharmacotherapy of chronic anal fissure: an update. Asian J Surg. 2008;31(3):154-163.
17. Nelson RL. Anal fissure (chronic). BMJ Clin Evid. 2014;2014. pii: 0407.
18. deRosa M, Cestaro G, Vitiello C, et al. Conservative versus surgical treatment for chronic idiopathic anal fissure: a prospective randomized trial. Updates Surg. 2013;65(3):197-200.
19. Lacy B, Weiser K. Common anorectal disorders: diagnosis and treatment. Curr Gastroenterol Rep. 2009;11(5):413-419.
20. Siddiqi S, Vijay V, Ward M, et al. Pruritus ani. Ann R Coll Surg Engl. 2008;90:457-463.
21. Lysy J, Sistiery-Ittah M, Israelit Y, et al. Topical capsaicin—a novel and effective treatment for idiopathic intractable pruritus ani: a randomized, placebo controlled, crossover study. Gut. 2003;52(9):1323-1326.
22. Burnstein M. Managing anorectal emergencies. Can Fam Physician. 1993;39:1782-1785.
23. Sasaki A, Nakajima T, Egashira H, et al. Condyloma acuminatum of the anal canal, treated with endoscopic submucosal dissection. World J Gastroenterol. 2016;22(8):2636-2641.
24. Kollipara R, Ekhlassi E, Downing C, et al. Advancements in pharmacotherapy for noncancerous manifestations of HPV. J Clin Med. 2015;4(5):832-846.
25. CDC. Manual for the Surveillance of Vaccine-preventable Diseases. Chapter 5: Human papillomavirus (HPV). www.cdc.gov/vaccines/pubs/surv-manual/chpt05-hpv.html. Accessed October 18, 2017.
26. Leszczyszyn J, Lebski I, Lysenko L, et al. Anal warts (condyloma acuminatum)—current issues and treatment modalities. Adv Clin Exp Med. 2014;23(2):307-311.
27. Baker DA, Ferris DG, Martens MG, et al. Imiquimod 3.75% cream applied daily to treat anogenital warts: combined results from women in two randomized, placebo-controlled studies. Infect Dis Obstet Gynecol. 2011;2011:806105.
28. Yan J, Chen SL, Wang HN, et al. Meta-analysis of 5% imiquimod and 0.5% podophyllotoxin in the treatment of condylomata acuminate. Dermatology. 2006;213(3):218-223.
29. Hoy SM. Polyphenon E 10% ointment: in immunocompetent adults with external genital and perianal warts. Am J Clin Dermatol. 2012;13(4):275-281.
30. Tatti S, Swinehart JM, Thielert C, et al. Sinecatechins, a defined green tea extract, in the treatment of external anogenital warts. Obstet Gynecol. 2008;111(6):1371-1379.
31. CDC. Human papillomavirus (HPV). www.cdc.gov/hpv/. Accessed October 18, 2017.
32. National Institutes of Health, National Cancer Institute. Human papillomavirus (HPV) vaccine. www.cancer.gov/about-cancer/causes-prevention/risk/infectious-agents/hpv-vaccine-fact-sheet#q5. Accessed October 18, 2017.
33. Giuliano AR, Palefsky JM, Goldstone S, et al. Efficacy of quadrivalent HPV vaccine against HPV infection and disease in males. N Engl J Med. 2011;364:401-411.
34. CDC. HPV vaccines: vaccinating your preteen or teen. www.cdc.gov/hpv/parents/vaccine.html. Accessed October 18, 2017.
CE/CME No: CR-1711
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Understand basic anorectal anatomy and how to perform a thorough anorectal exam.
• Describe the physical exam findings of common benign anorectal conditions.
• Discuss the different treatment options for benign anorectal conditions.
• Differentiate between common benign anorectal symptoms and red flags that should prompt referral to a colorectal specialist.
FACULTY
Priscilla Marsicovetere is an Assistant Professor of Medical Education and of Surgery at the Geisel School of Medicine at Dartmouth in Hanover, New Hampshire; Program Director for the Franklin Pierce University PA Program in Lebanon, New Hampshire; and practices with Emergency Services of New England at Springfield Hospital in Vermont. Srinivas Joga Ivatury is an Assistant Professor of Surgery at the Geisel School of Medicine at Dartmouth and practices in the Department of Surgery at the Dartmouth Hitchcock Medical Center in Lebanon, New Hampshire.
The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through October 31, 2018.
Article begins on next page >>
Anorectal pain and discomfort can stem from several possible etiologies, most of which are benign. As such, many patients with anorectal complaints who present in the primary care setting can be adequately evaluated, diagnosed, and treated without referral to a colorectal specialist. However, the clinician must be able to differentiate between common benign anorectal symptoms and red flags that should prompt referral.
Anorectal disorders are common in the United States and result in numerous primary care visits each year. Presentations range from pain and itching to bleeding and lesions. Common anorectal conditions include hemorrhoids, perianal skin tags, fissures, pruritus ani, perianal abscess, and condyloma. Most are benign and can be managed in the primary care setting.
Before a provider can competently diagnose and treat anorectal conditions, however, a comprehensive history and physical examination must be conducted. Grucela and colleagues documented that physicians’ diagnostic accuracy with anorectal conditions is about 50%—highlighting the need for providers to become more familiar with the history and clinical elements associated with anorectal complaints.1
This article reviews the assessment of the anorectum, diagnosis of common disorders and their recommended treatments, and red flags for referral to a colorectal specialist.
ANORECTAL ANATOMY
The beginning of the anal canal is demarcated by its moist, hairless appearance. Just inside the anal opening are two palpable circular muscles, the internal and external anal sphincters, separated by an intersphincteric groove. The sphincters are firmly closed in the resting state, which helps maintain continence.
The anal canal is generally 3 to 4 cm long and ends at the dentate line, a series of crypts in the anal mucosa.2 The crypts are openings into the anal glands, which are mucus-secreting structures in the anus. The dentate line is easily identified on anoscopy as a discrete change in the appearance of the mucosa. The dentate line is an important landmark because it delineates the boundary between somatic and visceral nerve supplies.3 Tissue proximal to the dentate line is innervated by visceral nerves and is insensate, and thus usually not a cause of pain; tissue distal to the dentate line, however, is highly innervated by somatic nerves and can be intensely painful.2
The anorectal canal is lined by three fibrovascular cushions, located in the left lateral, right posterior, and right anterior positions.4 Inside each cushion is a venous structure, called a hemorrhoid, which allows the cushion to enlarge and help maintain continence.5
Proximal to the anus is the rectum, the 12- to 15-cm long terminus of the colon. Anorectal examination in the primary care setting will typically not progress beyond the last 2 to 3 cm of the rectum.
TAKING THE HISTORY
A thorough history will provide clues about potential underlying anorectal pathology. Patients may not be forthcoming about symptoms due to embarrassment, fear of a cancer diagnosis, or cultural customs or habits. A thorough history should elicit information about all of the patient’s symptoms (see Table 1), including bleeding, change in bowel habits, and unintended weight loss.
PHYSICAL EXAM
Positioning the patient
Undergoing an anorectal examination can be embarrassing, whether it be from exposure of sensitive body parts or the less-than-desirable prone jackknife positioning. Patients often have preconceived notions that the exam will be humiliating and/or painful. Care should be taken to minimize any embarrassment and discomfort.
Positioning of the patient is a matter of provider preference. Options include the left lateral decubitus, prone jackknife, or lithotomy positions.
Positioning should always be done with draping. Regardless of position, ensure the draping exposes only the perineum. This can be achieved by encircling the patient’s bare bottom with a plain white sheet that exposes only the anus and surrounding skin, keeping the lower back, lateral buttocks, and thighs covered.
Interestingly, data on patient preference for positioning during anorectal exams are limited. In a 2009 study of 178 patients undergoing anorectal exam, more than half of patients (up to 71.4%) expecting to or having already had a proctologic exam reported that no specific type of positioning (eg, Sims, lithotomy with lifted legs, knee-chest, knee-chest with patient’s body bent forward) was most embarrassing to them.6 The report revealed that while most patients would favor the Sims position if they had a choice, they deferred to their examiner to choose the position that seemed most suitable to get a reliable diagnosis.6
Inspection of the perineum
Once the patient is properly positioned and draped, inspection of the perineum can occur. Begin by gently spreading the buttocks. Describe any abnormality seen (eg, ulcer, lesion, dermatitis, prolapsing tissue, or blood), including size, color, and location.
A common pitfall is to describe the location of abnormalities using a clock face, such as “at 4 o’clock.” This is misleading and should be avoided, because depending on patient position, the clock face can point to different locations (eg, if the patient is in the lithotomy versus prone jackknife position).
A better approach is to divide the perianal area into four anatomic quadrants: right anterior, right posterior, left anterior, left posterior. Using this schematic, the patient's position is irrelevant, and accurate documentation of lesion location is assured.
Digital rectal exam
After visual inspection of the perianal skin, a digital rectal exam (DRE) should be performed. Slowly insert a gloved, lubricated index finger into the anus and lower rectum. Note the tone of the anus at rest (eg, excessively tight vs lax). Palpate the circumference of the anus, sweeping side to side while assessing for any tenderness, mass, or induration—if present, note the anatomic quadrant. If a mass is felt, note whether it is firm or soft, fixed or mobile, and broad-based or pedunculated. When the lubricated finger is removed from the anus, note whether blood is present.
Anoscopy
After DRE, visually inspect the anorectum. The instrument used varies from anoscope to rigid proctoscope to flexible sigmoidoscope. In a primary care setting, the most likely available instrument is an anoscope. The average anoscope is about 7 cm long and 2 cm in diameter, with a beveled tip and an obturator (see Figure 1), and allows a 360° view of the anal canal.7
Examination of the anal canal is accomplished by dividing the canal into the four anatomic quadrants described earlier and inserting the lubricated anoscope for inspection of each of the four quadrants. Observe the rectal mucosa and the anus as the scope is slowly withdrawn. If abnormalities are seen, note the location, size, shape, and any other descriptive features.
It is not necessary to perform a Hemoccult test after examination of the anorectum, as the presence of minor blood may be the direct result of the exam itself and thus provides no useful information to the examiner.
COMMON PATHOLOGIES
Once the history and physical exam are complete, a differential diagnosis can be formulated. Most anorectal disorders are benign conditions that pose no immediate health threat and can be managed in the primary care setting. Others, however, can be more serious and should prompt referral to a colorectal specialist for further evaluation. Knowing the difference can spare a patient unnecessary anxiety and referral; it can also lead to prompt, lifesaving interventions if red flags are recognized.
Hemorrhoids
Hemorrhoids are a common anorectal complaint.8,9 It is estimated that up to 75% of the population will experience symptoms of hemorrhoids during their lifetime.5,8 Whether internal or external, in their normal, nonpathologic, quiescent state, hemorrhoids are asymptomatic. Hemorrhoids become symptomatic when the supporting structures of hemorrhoidal tissue (ie, the anal cushions) deteriorate, resulting in venous dilation, inflammation, and thrombosis, which in turn lead to swelling, bright red bleeding, and/or prolapse.2,10 The most common causes of hemorrhoidal disease are chronic constipation and prolonged straining with bowel movements, though chronic diarrhea and pregnancy have also been identified as risk factors.2,8,11
External hemorrhoids, which are located distal to the dentate line, are typically only visible when they become thrombosed or swollen. In this state, they may manifest as acute-onset, exquisitely painful, large, purple-to-blue bulges at the anal outlet (see Figure 2). The number and size of the lesions can vary. The patient may report pain when sitting or wiping, as well as bleeding from the lesion.12,13 The pain is typically severe in the first couple of days, then slowly starts to subside.2,12
For internal hemorrhoids, which are located proximal to the dentate line, the main symptom is usually painless bright red blood per rectum.8,11,12 Patients may also report a sensation of rectal fullness or experience prolapse of the hemorrhoid through the anus. Prolapse typically occurs with defecation; in more severe cases, it can also occur between bowel movements, usually with any activity that increases intra-abdominal pressure (eg, coughing, heavy lifting, pregnancy, portal hypertension). The prolapse may reduce spontaneously, or may have to be manually reduced. If it cannot be reduced, there is a risk for incarceration or strangulation, potentially leading to gangrene.
The presence of bleeding and/or prolapse determines the classification of internal hemorrhoids (see Table 2). Dietary and lifestyle modification are used in the management of all grades of hemorrhoids. In addition, for grade 1 and 2 lesions, topical medication (eg, anti-inflammatory cream) can be used, whereas grade 3 (and selected grade 2) lesions respond well to rubber band ligation. Given the severity of grade 4 lesions, surgical intervention (eg, hemorrhoidectomy) is usually indicated.10
About a third of patients with symptomatic hemorrhoids seek clinical treatment.14 Most are hemodynamically stable and require no imaging and usually no labs (unless anemia is suspected).2 Management depends on the location and degree of symptoms (eg, internal vs external, prolapse, or thrombosis). In the event of an acutely thrombosed external hemorrhoid, clot excision for pain relief is appropriate if symptoms have been present for less than 48 to 72 hours; after that amount of time, the pain from the procedure will likely exceed the degree of relief provided, and conservative management should instead be recommended.2,8,11
Firstline treatment consists of lifestyle modification with a high-fiber diet and daily fiber supplement to ensure stool is soft and easy to pass.2,8 A meta-analysis of seven clinical trials with a total of 378 patients with hemorrhoids showed that fiber supplementation resulted in a 50% decrease in bleeding risk from internal hemorrhoids.15
Adequate hydration, preferably with noncaffeinated liquids, is also recommended. This will prevent constipation and the need to strain or spend excessive time on the toilet. Sitz baths can help alleviate pain and discomfort.
Several OTC topical medications are marketed for hemorrhoid relief. Many of these preparations contain steroids for their anti-inflammatory effects or astringents to address skin irritation that can result from anal leakage if prolapsing hemorrhoids prevent the anal outlet from closing. Steroid use should be limited to five to seven days, due to atrophic effects on the skin. While OTC preparations may temporarily alleviate discomfort, they will not address the underlying cause of symptoms.
Indications for referral to a colorectal specialist for symptomatic hemorrhoids include failure to improve with conservative management, persistent patient discomfort, and prolapse, as these indicate potential need for more invasive treatment.
Perianal skin tags
Perianal skin tags, while a nuisance, are not pathologic in most instances and pose no threat to health. They are an outgrowth of normal skin, appearing as loose, flesh-colored perianal tissues (see Figure 3). Tags range in size from a few millimeters to a centimeter long and can occur alone or in multiples.
Perianal skin tags are diagnosed clinically and require no labwork or imaging. Visual inspection is typically sufficient to distinguish tags from pathologic lesions such as condyloma or abscess. If there is uncertainty, however, biopsy or referral to a specialist is warranted.
Certain medical conditions can predispose a patient to development of perianal skin tags. They can be sequelae of thrombosed external hemorrhoids.8,11 They are also common in patients with Crohn disease.11 Perianal skin tags are not, however, the result of anal intercourse or sexually transmitted infections.
Treatment is usually not indicated for perianal skin tags. If the tags interfere with hygiene or cause perianal discomfort or significantly decreased quality of life, however, patients may seek removal. These patients should be referred to a colorectal specialist for evaluation for excision.
Anal fissures (fissure in ano)
The most common cause of severe anorectal pain is fissure.4 A fissure is an elliptical tear, or split, in the lining of the anal canal that causes spasm of the anal sphincters. The tear is distal to the dentate line and thus intensely painful.2,5 Common cited causes of fissures are trauma from passage of large, hard stools, straining, or diarrhea.16
Fissures can usually be visualized by spreading the posterior anus apart (see Figure 4). They are most commonly located in the posterior or anterior midline, though they can occur anywhere around the anus.2,4 Often, a sentinel tag—appearing as a taut, flesh-colored skin tag—is present at the external pole of the fissure.5,11 DRE and anoscopy should be avoided, as they will trigger intense pain and spasm of the sphincters.
Fissures are characterized as acute (present ≤ 3 months) or chronic (> 3 months).9,11 Visually, acute fissures typically have clean edges, with the appearance of a paper cut to the mucosa, while chronic fissures have indurated, heaped-up edges, often with exposure of the underlying sphincter muscle.17
They tend to be exquisitely painful, as the mucosa distal to the dentate line is highly innervated. Patients report pain akin to “passing shards of broken glass” with bowel movements, which is often accompanied by a fear of defecation and bright red blood on the toilet paper or dripping into the water.11 The pain, caused by spasm of the sphincters, typically starts during a bowel movement and lasts minutes to hours afterward.
Initial treatment is aimed at relaxing the sphincters, as well as softening stool to prevent further trauma and allow the fissure to heal. Patients should be educated about the importance of adequate fiber intake to prevent constipation and straining. A daily bulk fiber supplement, in addition to a high-fiber diet (20-25 g/d), has been shown to result in healing of 87% of acute fissures.16 Sitz baths in plain warm water, three to four times a day, can encourage relaxation of the sphincters and increase local blood flow, both of which help with fissure healing.16 Topical medications can also be prescribed. These include compounded nitroglycerin 0.2% or nifedipine 2.0%, which act to reduce the spasm by relaxing smooth muscle, as well as increase blood flow to the lesion.12,14
Most acute fissures will heal with the regimen of high fiber intake, sitz baths, and topical medication. For refractory or chronic fissures, referral to a colorectal specialist for more invasive treatment is appropriate. Additionally, fissures that are not located in the typical posterior or anterior midline might indicate an atypical etiology, such as Crohn disease, tuberculosis, leukemia, or HIV, and thus patients who present with fissures in these locations should also be referred to a colorectal specialist.2,4,11,18
Pruritus ani
Pruritus ani, known as perianal dermatitis, is a benign condition that presents with intense perianal itching and burning. It is the second most common anorectal condition after hemorrhoids and affects up to 5% of the US population.2,9,11,19
Pruritus ani often develops secondary to local irritation of the skin (eg, from prolonged exposure to moisture), leading to an inflammatory response within the superficial skin layers. The irritation causes patients to scratch the skin, resulting in trauma, excoriation, and ulcer formation and leading to a cycle of further inflammation, exacerbation of symptoms, and persistent scratching.
Physical exam may reveal circumferential erythematous and irritated perianal skin (see Figure 5). Linear or deep, punched-out excoriations may be present. Chronically, patients may develop lichenification with thick, whitened patches of skin.11 In the absence of red flags such as unintentional weight loss, anemia, rectal bleeding, or a family history of colon cancer, no additional evaluation is required during the initial visit, though anoscopy can be used to rule out associated anorectal pathology.
Many different causes of pruritus ani have been reported (see Table 3). In the case of an identifiable cause, symptoms tend to resolve once the offending agent is eliminated. Up to a quarter of cases, however, are idiopathic, with no identifiable trigger.20
Symptom management is thus key. Patients should be educated about lifestyle modification and informed that scratching will irritate the skin further and aggravate symptoms.5,11 If incontinence or diarrhea is thought to cause symptoms, dietary modifications, a fiber supplement, dysmotility agents, and Kegel exercises to strengthen the sphincters and decrease anal leakage can be recommended.
The affected skin should be kept clean and dry at all times. Aggressive wiping and overzealous hygiene should be avoided. Sitz baths can help with hygiene. A topical astringent such as witch hazel can help remove excess moisture from the skin. A layer of protective skin barrier cream with zinc oxide, when applied over dry skin, can help protect the skin from leakage throughout the day.
A sedating antihistamine can reduce scratching during sleep. Topical hydrocortisone 1% cream is effective for itch relief; this should be limited to five to seven days of consecutive use, however, as it can lead to pathologic thinning of the perianal skin. Topical capsaicin 0.006% cream has been shown to help alleviate intractable pruritus.2,21
The goal of these measures is to break the cycle of irritation and inflammation and give the skin an opportunity to heal. If the patient fails to improve, referral to a colorectal specialist or, alternatively, a dermatologist, is warranted for perianal skin biopsy and more invasive treatment options.
Perianal abscess
A perianal abscess is an infected cavity filled with pus under pressure, located near the anus or rectum. It most often results from an acute infection of the anorectal glands located at the dentate line that tracks outward to the perianal skin.12 Abscesses can also result from another disease process, such as Crohn disease, diabetes, or rectal trauma.2,11
Localized pain, swelling, and drainage are common presenting symptoms of an abscess.2,11 Systemic symptoms such as fever and chills may present later in the course but are rare.2
Treatment of the acute process is incision and drainage (I&D).11,12 This can be accomplished in the office with injection of local anesthesia, followed by excision of an ellipse of skin overlying the cavity large enough to allow full drainage of the abscess. Use of drains and packing of the wound are usually not necessary. There is also no role for antibiotics unless the patient is diabetic or immunosuppressed or cellulitis is present.11 In patients with systemic signs of illness, imaging such as CT or MRI of the pelvis can be used to assess for a deeper infection.2,12
For most patients, I&D resolves the process. However, up to half of perianal abscesses progress to form a fistula, a tunnel connecting the infected anal gland to the external skin.2,11,22 Indeed, abscesses and fistulae are part of the same infectious process, with the abscess representing the acute phase of infection and the fistula representing the chronic phase. On physical exam, in addition to the abscess site on the skin, a fistula may manifest as a palpable cord beneath the skin between the anus and the abscess opening.9 Additionally, the patient may report the abscess has been recurrent in nature, cyclically increasing in size and pain, then spontaneously draining.
A fistula requires surgical intervention for definitive treatment and should therefore prompt referral to a colorectal specialist. In the absence of fistula, however, a simple perianal abscess can be treated with I&D in the primary care setting.
Condyloma acuminatum
Condyloma acuminatum, also known as genital warts, is the most common sexually transmitted infection in the United States and a frequent anorectal complaint.23-25 More than 6 million new infections occur annually.25
Condyloma is caused by the human papillomavirus (HPV).24 More than 100 HPV subtypes have been identified.25 Types 6 and 11 are associated with typical condyloma acuminatum, while types 16 and 18 are found more commonly with dysplasia and malignant transformation.3,26
The lesions are spread by direct skin-to-skin or mucosa-to-mucosa contact, including anal intercourse. They appear as tiny finger-like projections on the perianal or genital skin, often with a cauliflower-like appearance, in clusters or as single entities, and ranging in size from a few millimeters to several centimeters (see Figure 6).
The patient may be asymptomatic or may complain of bleeding with defecation or leakage from the rectum between bowel movements. In up to 90% of perianal condyloma, concomitant anorectal lesions are present.3 Anoscopy is therefore indicated whenever perianal lesions are seen.
Treatment of genital warts varies depending on the number and location of lesions. The goal is lesion removal, but none of the available treatments are curative.24 If only a few small (diameter < 2 mm) warts are seen in the pe
Trichloroacetic acid (TCA), which is considered a form of chemical cautery, has been deemed more effective for tiny warts (diameter 1-2 mm) and better tolerated than podophyllin.24 Unlike podophyllin, however, TCA must be applied in clinic by a specialist, not at home by the patient. It works by chemically burning, cauterizing, and eroding skin and mucosa, thereby destroying old infected cells and allowing for growth of new, healthy cells.24 Using a solution with a higher concentration of TCA (80%-90%) has been shown to be more effective than lower concentration solutions (30%).24 The recurrence rate is high, generally about 36%.27 Due to its low risk for systemic absorption, TCA is safe for use during pregnancy.24
Local application of an immunomodulator, such as imiquimod 2%, 3.75%, or 5% cream, can also be used. Imiquimod works by upregulating tumor necrosis factor, leading to decreased viral replication and subsequent regression of warts.24 The most common side effects of imiquimod are erythema, erosions, burning, and pruritus.24 The higher concentration is associated with increased clearance rates as well as increased adverse effects.27 Therefore, the 3.75% cream is the preferred concentration of most providers.27 Recurrence rates range from 13% to 19%.28
In 2006, the FDA approved polyphenon E, a botanical ointment, to treat anogenital warts. The formulation is composed of eight different sinecatechins extracted from green tea leaves.29 Green tea sinecatechins have been shown to have antioxidant, antiproliferative, and antiviral properties, though the specific mechanisms of action in inhibiting anogenital wart growth have not been well studied to date. In a multicenter, randomized, double-blind, vehicle-controlled, three-arm parallel group phase 3 trial of 495 patients with anogenital warts, sinecatechins ointment 10% or 15% applied three times daily for 16 weeks resulted in complete clearance of warts in 57.2% and 56.3% of patients, respectively, compared to 33.7% clearance in the vehicle patients.30 Partial clearance (≥ 50%) of warts was observed in 78.4% of patients who used the 15% ointment, 74.0% of those who used the 10% ointment, and 51.5% of the vehicle patients. Rates of recurrence were 6.5% and 8.3% with use of the 10% and 15% sinecatechins ointment, respectively, which are much lower than recurrence rates observed with other topical applications (eg, imiquimod and podophyllotoxin).30
For larger warts or when there are anal lesions present, surgical excision and fulguration is required and referral to a colorectal specialist is warranted.
Warts recur from 4.6% to more than 70% of the time, most commonly due to activation of latent virus.23 Therefore, whether initially treated in an office or operating room, condyloma patients require follow-up on a regular basis, generally every six to 12 months, to assess for recurrent anal lesions so that treatment can be promptly initiated.
In the absence of a cure for anogenital warts, prevention through an HPV vaccine is important. The quadrivalent HPV vaccine (Gardasil) was approved by the FDA in 2006 for prophylactic vaccination of females ages 9 to 26. The vaccine triggers host formation of antibodies against four common subtypes of HPV: 6, 11, 16, and 18.31 In 2009, the FDA expanded the use of Gardasil to include males ages 9 to 26; subsequently, Gardasil 9 (which protects against nine types of HPV) was approved for males ages 9 to 15.32 In a group of 4,065 healthy males between the ages of 16 and 26, Gardasil reduced the incidence of external anogenital warts by 90.4%.33
HPV vaccine is now recommended for females through age 26 and males through age 21. The CDC also recommends HPV vaccine for the following individuals through age 26 if they did not get vaccinated when they were younger:
- Young men who have sex with men
- Young men who identify as gay or bisexual or who intend to have sex with men
- Young adults who are transgender
- Young adults with immunocompromising conditions (including HIV).34
CONCLUSION
Anorectal conditions are a common presentation in the primary care setting. A thorough history and physical exam will usually determine the etiology, and most benign pathologies can be successfully treated in the primary care clinic. Knowing how to perform an anorectal exam and having a thorough understanding of the most common anorectal pathologies can help alleviate examiner discomfort, while ensuring that patients receive prompt and adequate care. At the same time, recognizing the red flags can expedite referral for colorectal specialty evaluation when appropriate. Red flags that should prompt immediate referral to a colorectal specialist include older age, unintentional weight loss, iron deficiency anemia, family history of inflammatory bowel disease or colorectal cancer, and persistent anorectal bleeding or persistent symptoms despite adequate treatment of a suspected benign condition.
CE/CME No: CR-1711
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Understand basic anorectal anatomy and how to perform a thorough anorectal exam.
• Describe the physical exam findings of common benign anorectal conditions.
• Discuss the different treatment options for benign anorectal conditions.
• Differentiate between common benign anorectal symptoms and red flags that should prompt referral to a colorectal specialist.
FACULTY
Priscilla Marsicovetere is an Assistant Professor of Medical Education and of Surgery at the Geisel School of Medicine at Dartmouth in Hanover, New Hampshire; Program Director for the Franklin Pierce University PA Program in Lebanon, New Hampshire; and practices with Emergency Services of New England at Springfield Hospital in Vermont. Srinivas Joga Ivatury is an Assistant Professor of Surgery at the Geisel School of Medicine at Dartmouth and practices in the Department of Surgery at the Dartmouth Hitchcock Medical Center in Lebanon, New Hampshire.
The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through October 31, 2018.
Article begins on next page >>
Anorectal pain and discomfort can stem from several possible etiologies, most of which are benign. As such, many patients with anorectal complaints who present in the primary care setting can be adequately evaluated, diagnosed, and treated without referral to a colorectal specialist. However, the clinician must be able to differentiate between common benign anorectal symptoms and red flags that should prompt referral.
Anorectal disorders are common in the United States and result in numerous primary care visits each year. Presentations range from pain and itching to bleeding and lesions. Common anorectal conditions include hemorrhoids, perianal skin tags, fissures, pruritus ani, perianal abscess, and condyloma. Most are benign and can be managed in the primary care setting.
Before a provider can competently diagnose and treat anorectal conditions, however, a comprehensive history and physical examination must be conducted. Grucela and colleagues documented that physicians’ diagnostic accuracy with anorectal conditions is about 50%—highlighting the need for providers to become more familiar with the history and clinical elements associated with anorectal complaints.1
This article reviews the assessment of the anorectum, diagnosis of common disorders and their recommended treatments, and red flags for referral to a colorectal specialist.
ANORECTAL ANATOMY
The beginning of the anal canal is demarcated by its moist, hairless appearance. Just inside the anal opening are two palpable circular muscles, the internal and external anal sphincters, separated by an intersphincteric groove. The sphincters are firmly closed in the resting state, which helps maintain continence.
The anal canal is generally 3 to 4 cm long and ends at the dentate line, a series of crypts in the anal mucosa.2 The crypts are openings into the anal glands, which are mucus-secreting structures in the anus. The dentate line is easily identified on anoscopy as a discrete change in the appearance of the mucosa. The dentate line is an important landmark because it delineates the boundary between somatic and visceral nerve supplies.3 Tissue proximal to the dentate line is innervated by visceral nerves and is insensate, and thus usually not a cause of pain; tissue distal to the dentate line, however, is highly innervated by somatic nerves and can be intensely painful.2
The anorectal canal is lined by three fibrovascular cushions, located in the left lateral, right posterior, and right anterior positions.4 Inside each cushion is a venous structure, called a hemorrhoid, which allows the cushion to enlarge and help maintain continence.5
Proximal to the anus is the rectum, the 12- to 15-cm long terminus of the colon. Anorectal examination in the primary care setting will typically not progress beyond the last 2 to 3 cm of the rectum.
TAKING THE HISTORY
A thorough history will provide clues about potential underlying anorectal pathology. Patients may not be forthcoming about symptoms due to embarrassment, fear of a cancer diagnosis, or cultural customs or habits. A thorough history should elicit information about all of the patient’s symptoms (see Table 1), including bleeding, change in bowel habits, and unintended weight loss.
PHYSICAL EXAM
Positioning the patient
Undergoing an anorectal examination can be embarrassing, whether it be from exposure of sensitive body parts or the less-than-desirable prone jackknife positioning. Patients often have preconceived notions that the exam will be humiliating and/or painful. Care should be taken to minimize any embarrassment and discomfort.
Positioning of the patient is a matter of provider preference. Options include the left lateral decubitus, prone jackknife, or lithotomy positions.
Positioning should always be done with draping. Regardless of position, ensure the draping exposes only the perineum. This can be achieved by encircling the patient’s bare bottom with a plain white sheet that exposes only the anus and surrounding skin, keeping the lower back, lateral buttocks, and thighs covered.
Interestingly, data on patient preference for positioning during anorectal exams are limited. In a 2009 study of 178 patients undergoing anorectal exam, more than half of patients (up to 71.4%) expecting to or having already had a proctologic exam reported that no specific type of positioning (eg, Sims, lithotomy with lifted legs, knee-chest, knee-chest with patient’s body bent forward) was most embarrassing to them.6 The report revealed that while most patients would favor the Sims position if they had a choice, they deferred to their examiner to choose the position that seemed most suitable to get a reliable diagnosis.6
Inspection of the perineum
Once the patient is properly positioned and draped, inspection of the perineum can occur. Begin by gently spreading the buttocks. Describe any abnormality seen (eg, ulcer, lesion, dermatitis, prolapsing tissue, or blood), including size, color, and location.
A common pitfall is to describe the location of abnormalities using a clock face, such as “at 4 o’clock.” This is misleading and should be avoided, because depending on patient position, the clock face can point to different locations (eg, if the patient is in the lithotomy versus prone jackknife position).
A better approach is to divide the perianal area into four anatomic quadrants: right anterior, right posterior, left anterior, left posterior. Using this schematic, the patient's position is irrelevant, and accurate documentation of lesion location is assured.
Digital rectal exam
After visual inspection of the perianal skin, a digital rectal exam (DRE) should be performed. Slowly insert a gloved, lubricated index finger into the anus and lower rectum. Note the tone of the anus at rest (eg, excessively tight vs lax). Palpate the circumference of the anus, sweeping side to side while assessing for any tenderness, mass, or induration—if present, note the anatomic quadrant. If a mass is felt, note whether it is firm or soft, fixed or mobile, and broad-based or pedunculated. When the lubricated finger is removed from the anus, note whether blood is present.
Anoscopy
After DRE, visually inspect the anorectum. The instrument used varies from anoscope to rigid proctoscope to flexible sigmoidoscope. In a primary care setting, the most likely available instrument is an anoscope. The average anoscope is about 7 cm long and 2 cm in diameter, with a beveled tip and an obturator (see Figure 1), and allows a 360° view of the anal canal.7
Examination of the anal canal is accomplished by dividing the canal into the four anatomic quadrants described earlier and inserting the lubricated anoscope for inspection of each of the four quadrants. Observe the rectal mucosa and the anus as the scope is slowly withdrawn. If abnormalities are seen, note the location, size, shape, and any other descriptive features.
It is not necessary to perform a Hemoccult test after examination of the anorectum, as the presence of minor blood may be the direct result of the exam itself and thus provides no useful information to the examiner.
COMMON PATHOLOGIES
Once the history and physical exam are complete, a differential diagnosis can be formulated. Most anorectal disorders are benign conditions that pose no immediate health threat and can be managed in the primary care setting. Others, however, can be more serious and should prompt referral to a colorectal specialist for further evaluation. Knowing the difference can spare a patient unnecessary anxiety and referral; it can also lead to prompt, lifesaving interventions if red flags are recognized.
Hemorrhoids
Hemorrhoids are a common anorectal complaint.8,9 It is estimated that up to 75% of the population will experience symptoms of hemorrhoids during their lifetime.5,8 Whether internal or external, in their normal, nonpathologic, quiescent state, hemorrhoids are asymptomatic. Hemorrhoids become symptomatic when the supporting structures of hemorrhoidal tissue (ie, the anal cushions) deteriorate, resulting in venous dilation, inflammation, and thrombosis, which in turn lead to swelling, bright red bleeding, and/or prolapse.2,10 The most common causes of hemorrhoidal disease are chronic constipation and prolonged straining with bowel movements, though chronic diarrhea and pregnancy have also been identified as risk factors.2,8,11
External hemorrhoids, which are located distal to the dentate line, are typically only visible when they become thrombosed or swollen. In this state, they may manifest as acute-onset, exquisitely painful, large, purple-to-blue bulges at the anal outlet (see Figure 2). The number and size of the lesions can vary. The patient may report pain when sitting or wiping, as well as bleeding from the lesion.12,13 The pain is typically severe in the first couple of days, then slowly starts to subside.2,12
For internal hemorrhoids, which are located proximal to the dentate line, the main symptom is usually painless bright red blood per rectum.8,11,12 Patients may also report a sensation of rectal fullness or experience prolapse of the hemorrhoid through the anus. Prolapse typically occurs with defecation; in more severe cases, it can also occur between bowel movements, usually with any activity that increases intra-abdominal pressure (eg, coughing, heavy lifting, pregnancy, portal hypertension). The prolapse may reduce spontaneously, or may have to be manually reduced. If it cannot be reduced, there is a risk for incarceration or strangulation, potentially leading to gangrene.
The presence of bleeding and/or prolapse determines the classification of internal hemorrhoids (see Table 2). Dietary and lifestyle modification are used in the management of all grades of hemorrhoids. In addition, for grade 1 and 2 lesions, topical medication (eg, anti-inflammatory cream) can be used, whereas grade 3 (and selected grade 2) lesions respond well to rubber band ligation. Given the severity of grade 4 lesions, surgical intervention (eg, hemorrhoidectomy) is usually indicated.10
About a third of patients with symptomatic hemorrhoids seek clinical treatment.14 Most are hemodynamically stable and require no imaging and usually no labs (unless anemia is suspected).2 Management depends on the location and degree of symptoms (eg, internal vs external, prolapse, or thrombosis). In the event of an acutely thrombosed external hemorrhoid, clot excision for pain relief is appropriate if symptoms have been present for less than 48 to 72 hours; after that amount of time, the pain from the procedure will likely exceed the degree of relief provided, and conservative management should instead be recommended.2,8,11
Firstline treatment consists of lifestyle modification with a high-fiber diet and daily fiber supplement to ensure stool is soft and easy to pass.2,8 A meta-analysis of seven clinical trials with a total of 378 patients with hemorrhoids showed that fiber supplementation resulted in a 50% decrease in bleeding risk from internal hemorrhoids.15
Adequate hydration, preferably with noncaffeinated liquids, is also recommended. This will prevent constipation and the need to strain or spend excessive time on the toilet. Sitz baths can help alleviate pain and discomfort.
Several OTC topical medications are marketed for hemorrhoid relief. Many of these preparations contain steroids for their anti-inflammatory effects or astringents to address skin irritation that can result from anal leakage if prolapsing hemorrhoids prevent the anal outlet from closing. Steroid use should be limited to five to seven days, due to atrophic effects on the skin. While OTC preparations may temporarily alleviate discomfort, they will not address the underlying cause of symptoms.
Indications for referral to a colorectal specialist for symptomatic hemorrhoids include failure to improve with conservative management, persistent patient discomfort, and prolapse, as these indicate potential need for more invasive treatment.
Perianal skin tags
Perianal skin tags, while a nuisance, are not pathologic in most instances and pose no threat to health. They are an outgrowth of normal skin, appearing as loose, flesh-colored perianal tissues (see Figure 3). Tags range in size from a few millimeters to a centimeter long and can occur alone or in multiples.
Perianal skin tags are diagnosed clinically and require no labwork or imaging. Visual inspection is typically sufficient to distinguish tags from pathologic lesions such as condyloma or abscess. If there is uncertainty, however, biopsy or referral to a specialist is warranted.
Certain medical conditions can predispose a patient to development of perianal skin tags. They can be sequelae of thrombosed external hemorrhoids.8,11 They are also common in patients with Crohn disease.11 Perianal skin tags are not, however, the result of anal intercourse or sexually transmitted infections.
Treatment is usually not indicated for perianal skin tags. If the tags interfere with hygiene or cause perianal discomfort or significantly decreased quality of life, however, patients may seek removal. These patients should be referred to a colorectal specialist for evaluation for excision.
Anal fissures (fissure in ano)
The most common cause of severe anorectal pain is fissure.4 A fissure is an elliptical tear, or split, in the lining of the anal canal that causes spasm of the anal sphincters. The tear is distal to the dentate line and thus intensely painful.2,5 Common cited causes of fissures are trauma from passage of large, hard stools, straining, or diarrhea.16
Fissures can usually be visualized by spreading the posterior anus apart (see Figure 4). They are most commonly located in the posterior or anterior midline, though they can occur anywhere around the anus.2,4 Often, a sentinel tag—appearing as a taut, flesh-colored skin tag—is present at the external pole of the fissure.5,11 DRE and anoscopy should be avoided, as they will trigger intense pain and spasm of the sphincters.
Fissures are characterized as acute (present ≤ 3 months) or chronic (> 3 months).9,11 Visually, acute fissures typically have clean edges, with the appearance of a paper cut to the mucosa, while chronic fissures have indurated, heaped-up edges, often with exposure of the underlying sphincter muscle.17
They tend to be exquisitely painful, as the mucosa distal to the dentate line is highly innervated. Patients report pain akin to “passing shards of broken glass” with bowel movements, which is often accompanied by a fear of defecation and bright red blood on the toilet paper or dripping into the water.11 The pain, caused by spasm of the sphincters, typically starts during a bowel movement and lasts minutes to hours afterward.
Initial treatment is aimed at relaxing the sphincters, as well as softening stool to prevent further trauma and allow the fissure to heal. Patients should be educated about the importance of adequate fiber intake to prevent constipation and straining. A daily bulk fiber supplement, in addition to a high-fiber diet (20-25 g/d), has been shown to result in healing of 87% of acute fissures.16 Sitz baths in plain warm water, three to four times a day, can encourage relaxation of the sphincters and increase local blood flow, both of which help with fissure healing.16 Topical medications can also be prescribed. These include compounded nitroglycerin 0.2% or nifedipine 2.0%, which act to reduce the spasm by relaxing smooth muscle, as well as increase blood flow to the lesion.12,14
Most acute fissures will heal with the regimen of high fiber intake, sitz baths, and topical medication. For refractory or chronic fissures, referral to a colorectal specialist for more invasive treatment is appropriate. Additionally, fissures that are not located in the typical posterior or anterior midline might indicate an atypical etiology, such as Crohn disease, tuberculosis, leukemia, or HIV, and thus patients who present with fissures in these locations should also be referred to a colorectal specialist.2,4,11,18
Pruritus ani
Pruritus ani, known as perianal dermatitis, is a benign condition that presents with intense perianal itching and burning. It is the second most common anorectal condition after hemorrhoids and affects up to 5% of the US population.2,9,11,19
Pruritus ani often develops secondary to local irritation of the skin (eg, from prolonged exposure to moisture), leading to an inflammatory response within the superficial skin layers. The irritation causes patients to scratch the skin, resulting in trauma, excoriation, and ulcer formation and leading to a cycle of further inflammation, exacerbation of symptoms, and persistent scratching.
Physical exam may reveal circumferential erythematous and irritated perianal skin (see Figure 5). Linear or deep, punched-out excoriations may be present. Chronically, patients may develop lichenification with thick, whitened patches of skin.11 In the absence of red flags such as unintentional weight loss, anemia, rectal bleeding, or a family history of colon cancer, no additional evaluation is required during the initial visit, though anoscopy can be used to rule out associated anorectal pathology.
Many different causes of pruritus ani have been reported (see Table 3). In the case of an identifiable cause, symptoms tend to resolve once the offending agent is eliminated. Up to a quarter of cases, however, are idiopathic, with no identifiable trigger.20
Symptom management is thus key. Patients should be educated about lifestyle modification and informed that scratching will irritate the skin further and aggravate symptoms.5,11 If incontinence or diarrhea is thought to cause symptoms, dietary modifications, a fiber supplement, dysmotility agents, and Kegel exercises to strengthen the sphincters and decrease anal leakage can be recommended.
The affected skin should be kept clean and dry at all times. Aggressive wiping and overzealous hygiene should be avoided. Sitz baths can help with hygiene. A topical astringent such as witch hazel can help remove excess moisture from the skin. A layer of protective skin barrier cream with zinc oxide, when applied over dry skin, can help protect the skin from leakage throughout the day.
A sedating antihistamine can reduce scratching during sleep. Topical hydrocortisone 1% cream is effective for itch relief; this should be limited to five to seven days of consecutive use, however, as it can lead to pathologic thinning of the perianal skin. Topical capsaicin 0.006% cream has been shown to help alleviate intractable pruritus.2,21
The goal of these measures is to break the cycle of irritation and inflammation and give the skin an opportunity to heal. If the patient fails to improve, referral to a colorectal specialist or, alternatively, a dermatologist, is warranted for perianal skin biopsy and more invasive treatment options.
Perianal abscess
A perianal abscess is an infected cavity filled with pus under pressure, located near the anus or rectum. It most often results from an acute infection of the anorectal glands located at the dentate line that tracks outward to the perianal skin.12 Abscesses can also result from another disease process, such as Crohn disease, diabetes, or rectal trauma.2,11
Localized pain, swelling, and drainage are common presenting symptoms of an abscess.2,11 Systemic symptoms such as fever and chills may present later in the course but are rare.2
Treatment of the acute process is incision and drainage (I&D).11,12 This can be accomplished in the office with injection of local anesthesia, followed by excision of an ellipse of skin overlying the cavity large enough to allow full drainage of the abscess. Use of drains and packing of the wound are usually not necessary. There is also no role for antibiotics unless the patient is diabetic or immunosuppressed or cellulitis is present.11 In patients with systemic signs of illness, imaging such as CT or MRI of the pelvis can be used to assess for a deeper infection.2,12
For most patients, I&D resolves the process. However, up to half of perianal abscesses progress to form a fistula, a tunnel connecting the infected anal gland to the external skin.2,11,22 Indeed, abscesses and fistulae are part of the same infectious process, with the abscess representing the acute phase of infection and the fistula representing the chronic phase. On physical exam, in addition to the abscess site on the skin, a fistula may manifest as a palpable cord beneath the skin between the anus and the abscess opening.9 Additionally, the patient may report the abscess has been recurrent in nature, cyclically increasing in size and pain, then spontaneously draining.
A fistula requires surgical intervention for definitive treatment and should therefore prompt referral to a colorectal specialist. In the absence of fistula, however, a simple perianal abscess can be treated with I&D in the primary care setting.
Condyloma acuminatum
Condyloma acuminatum, also known as genital warts, is the most common sexually transmitted infection in the United States and a frequent anorectal complaint.23-25 More than 6 million new infections occur annually.25
Condyloma is caused by the human papillomavirus (HPV).24 More than 100 HPV subtypes have been identified.25 Types 6 and 11 are associated with typical condyloma acuminatum, while types 16 and 18 are found more commonly with dysplasia and malignant transformation.3,26
The lesions are spread by direct skin-to-skin or mucosa-to-mucosa contact, including anal intercourse. They appear as tiny finger-like projections on the perianal or genital skin, often with a cauliflower-like appearance, in clusters or as single entities, and ranging in size from a few millimeters to several centimeters (see Figure 6).
The patient may be asymptomatic or may complain of bleeding with defecation or leakage from the rectum between bowel movements. In up to 90% of perianal condyloma, concomitant anorectal lesions are present.3 Anoscopy is therefore indicated whenever perianal lesions are seen.
Treatment of genital warts varies depending on the number and location of lesions. The goal is lesion removal, but none of the available treatments are curative.24 If only a few small (diameter < 2 mm) warts are seen in the pe
Trichloroacetic acid (TCA), which is considered a form of chemical cautery, has been deemed more effective for tiny warts (diameter 1-2 mm) and better tolerated than podophyllin.24 Unlike podophyllin, however, TCA must be applied in clinic by a specialist, not at home by the patient. It works by chemically burning, cauterizing, and eroding skin and mucosa, thereby destroying old infected cells and allowing for growth of new, healthy cells.24 Using a solution with a higher concentration of TCA (80%-90%) has been shown to be more effective than lower concentration solutions (30%).24 The recurrence rate is high, generally about 36%.27 Due to its low risk for systemic absorption, TCA is safe for use during pregnancy.24
Local application of an immunomodulator, such as imiquimod 2%, 3.75%, or 5% cream, can also be used. Imiquimod works by upregulating tumor necrosis factor, leading to decreased viral replication and subsequent regression of warts.24 The most common side effects of imiquimod are erythema, erosions, burning, and pruritus.24 The higher concentration is associated with increased clearance rates as well as increased adverse effects.27 Therefore, the 3.75% cream is the preferred concentration of most providers.27 Recurrence rates range from 13% to 19%.28
In 2006, the FDA approved polyphenon E, a botanical ointment, to treat anogenital warts. The formulation is composed of eight different sinecatechins extracted from green tea leaves.29 Green tea sinecatechins have been shown to have antioxidant, antiproliferative, and antiviral properties, though the specific mechanisms of action in inhibiting anogenital wart growth have not been well studied to date. In a multicenter, randomized, double-blind, vehicle-controlled, three-arm parallel group phase 3 trial of 495 patients with anogenital warts, sinecatechins ointment 10% or 15% applied three times daily for 16 weeks resulted in complete clearance of warts in 57.2% and 56.3% of patients, respectively, compared to 33.7% clearance in the vehicle patients.30 Partial clearance (≥ 50%) of warts was observed in 78.4% of patients who used the 15% ointment, 74.0% of those who used the 10% ointment, and 51.5% of the vehicle patients. Rates of recurrence were 6.5% and 8.3% with use of the 10% and 15% sinecatechins ointment, respectively, which are much lower than recurrence rates observed with other topical applications (eg, imiquimod and podophyllotoxin).30
For larger warts or when there are anal lesions present, surgical excision and fulguration is required and referral to a colorectal specialist is warranted.
Warts recur from 4.6% to more than 70% of the time, most commonly due to activation of latent virus.23 Therefore, whether initially treated in an office or operating room, condyloma patients require follow-up on a regular basis, generally every six to 12 months, to assess for recurrent anal lesions so that treatment can be promptly initiated.
In the absence of a cure for anogenital warts, prevention through an HPV vaccine is important. The quadrivalent HPV vaccine (Gardasil) was approved by the FDA in 2006 for prophylactic vaccination of females ages 9 to 26. The vaccine triggers host formation of antibodies against four common subtypes of HPV: 6, 11, 16, and 18.31 In 2009, the FDA expanded the use of Gardasil to include males ages 9 to 26; subsequently, Gardasil 9 (which protects against nine types of HPV) was approved for males ages 9 to 15.32 In a group of 4,065 healthy males between the ages of 16 and 26, Gardasil reduced the incidence of external anogenital warts by 90.4%.33
HPV vaccine is now recommended for females through age 26 and males through age 21. The CDC also recommends HPV vaccine for the following individuals through age 26 if they did not get vaccinated when they were younger:
- Young men who have sex with men
- Young men who identify as gay or bisexual or who intend to have sex with men
- Young adults who are transgender
- Young adults with immunocompromising conditions (including HIV).34
CONCLUSION
Anorectal conditions are a common presentation in the primary care setting. A thorough history and physical exam will usually determine the etiology, and most benign pathologies can be successfully treated in the primary care clinic. Knowing how to perform an anorectal exam and having a thorough understanding of the most common anorectal pathologies can help alleviate examiner discomfort, while ensuring that patients receive prompt and adequate care. At the same time, recognizing the red flags can expedite referral for colorectal specialty evaluation when appropriate. Red flags that should prompt immediate referral to a colorectal specialist include older age, unintentional weight loss, iron deficiency anemia, family history of inflammatory bowel disease or colorectal cancer, and persistent anorectal bleeding or persistent symptoms despite adequate treatment of a suspected benign condition.
1. Grucela A, Salinas H, Khaitov S, et al. Prospective analysis of clinical accuracy in the diagnosis of benign anal pathology, comparison across specialties and years of experience. Dis Colon Rectum. 2010;53(1):47-52.
2. Tupe CL, Pham TV. Anorectal complaints in the emergency department. Emerg Med Clin North Am. 2016;34(2):251-270.
3. Billingham RP, Isler JT, Kimmins MH, et al. The diagnosis and management of common anorectal disorders. Curr Probl Surg. 2004;41(7):586-645.
4. Schubert MC, Sridhar S, Schade RR, Wexner SD. What every gastroenterologist needs to know about common anorectal disorders. World J Gastroenterol. 2009;15(26):3201-3209.
5. Henderson PK, Cash BD. Common anorectal conditions: evaluation and treatment. Curr Gastroenterol Rep. 2014; 16(10):408.
6. Gebbensleben O, Hilger Y, Rohde H. Patients’ view of medical positioning for proctologic examination. Clin Exp Gastroenterol. 2009;2:133-138.
7. Gopal DV. Diseases of the rectum and anus: a clinical approach to common disorders. Clin Cornerstone. 2002;4(4): 34-48.
8. Lohsiriwat V. Treatment of hemorrhoids: a coloproctologist’s view. World J Gastroenterol. 2015;21(31):9245-9252.
9. Fargo MV, Latimer K. Evaluation and management of common anorectal conditions. Am Fam Physician. 2012;85(6):624-630.
10. Lohsiriwat V. Hemorrhoids: from basic pathophysiology to clinical management. World J Gastroenterol. 2012;18(17): 2009-2017.
11. Foxx-Orenstein AE, Umar SB, Crowell MD. Common anorectal disorders. Gastroenterol Hepatol. 2014;10(5):294-301.
12. Lohsiriwat V. Anorectal emergencies. World J Gastroenterol. 2016;22(26):5867-5878.
13. Greenspon J, Williams SB, Young HA, Orkin BA. Thrombosed external hemorrhoids: outcome after conservative treatment or surgical management. Dis Colon Rectum. 2004(9);47:1493-1498.
14. Summers A. Assessment and treatment of three common anorectal conditions. Emerg Nurse. 2013;21(2):28-33.
15. Alonso-Coello P, Mills E, Heels-Ansdell D, et al. Fiber for the treatment of hemorrhoids complications: a systematic review and meta-analysis. Am J Gastroenterol. 2006;101(1):181-188.
16. Medhi B, Rao RS, Prakash A, et al. Recent advances in the pharmacotherapy of chronic anal fissure: an update. Asian J Surg. 2008;31(3):154-163.
17. Nelson RL. Anal fissure (chronic). BMJ Clin Evid. 2014;2014. pii: 0407.
18. deRosa M, Cestaro G, Vitiello C, et al. Conservative versus surgical treatment for chronic idiopathic anal fissure: a prospective randomized trial. Updates Surg. 2013;65(3):197-200.
19. Lacy B, Weiser K. Common anorectal disorders: diagnosis and treatment. Curr Gastroenterol Rep. 2009;11(5):413-419.
20. Siddiqi S, Vijay V, Ward M, et al. Pruritus ani. Ann R Coll Surg Engl. 2008;90:457-463.
21. Lysy J, Sistiery-Ittah M, Israelit Y, et al. Topical capsaicin—a novel and effective treatment for idiopathic intractable pruritus ani: a randomized, placebo controlled, crossover study. Gut. 2003;52(9):1323-1326.
22. Burnstein M. Managing anorectal emergencies. Can Fam Physician. 1993;39:1782-1785.
23. Sasaki A, Nakajima T, Egashira H, et al. Condyloma acuminatum of the anal canal, treated with endoscopic submucosal dissection. World J Gastroenterol. 2016;22(8):2636-2641.
24. Kollipara R, Ekhlassi E, Downing C, et al. Advancements in pharmacotherapy for noncancerous manifestations of HPV. J Clin Med. 2015;4(5):832-846.
25. CDC. Manual for the Surveillance of Vaccine-preventable Diseases. Chapter 5: Human papillomavirus (HPV). www.cdc.gov/vaccines/pubs/surv-manual/chpt05-hpv.html. Accessed October 18, 2017.
26. Leszczyszyn J, Lebski I, Lysenko L, et al. Anal warts (condyloma acuminatum)—current issues and treatment modalities. Adv Clin Exp Med. 2014;23(2):307-311.
27. Baker DA, Ferris DG, Martens MG, et al. Imiquimod 3.75% cream applied daily to treat anogenital warts: combined results from women in two randomized, placebo-controlled studies. Infect Dis Obstet Gynecol. 2011;2011:806105.
28. Yan J, Chen SL, Wang HN, et al. Meta-analysis of 5% imiquimod and 0.5% podophyllotoxin in the treatment of condylomata acuminate. Dermatology. 2006;213(3):218-223.
29. Hoy SM. Polyphenon E 10% ointment: in immunocompetent adults with external genital and perianal warts. Am J Clin Dermatol. 2012;13(4):275-281.
30. Tatti S, Swinehart JM, Thielert C, et al. Sinecatechins, a defined green tea extract, in the treatment of external anogenital warts. Obstet Gynecol. 2008;111(6):1371-1379.
31. CDC. Human papillomavirus (HPV). www.cdc.gov/hpv/. Accessed October 18, 2017.
32. National Institutes of Health, National Cancer Institute. Human papillomavirus (HPV) vaccine. www.cancer.gov/about-cancer/causes-prevention/risk/infectious-agents/hpv-vaccine-fact-sheet#q5. Accessed October 18, 2017.
33. Giuliano AR, Palefsky JM, Goldstone S, et al. Efficacy of quadrivalent HPV vaccine against HPV infection and disease in males. N Engl J Med. 2011;364:401-411.
34. CDC. HPV vaccines: vaccinating your preteen or teen. www.cdc.gov/hpv/parents/vaccine.html. Accessed October 18, 2017.
1. Grucela A, Salinas H, Khaitov S, et al. Prospective analysis of clinical accuracy in the diagnosis of benign anal pathology, comparison across specialties and years of experience. Dis Colon Rectum. 2010;53(1):47-52.
2. Tupe CL, Pham TV. Anorectal complaints in the emergency department. Emerg Med Clin North Am. 2016;34(2):251-270.
3. Billingham RP, Isler JT, Kimmins MH, et al. The diagnosis and management of common anorectal disorders. Curr Probl Surg. 2004;41(7):586-645.
4. Schubert MC, Sridhar S, Schade RR, Wexner SD. What every gastroenterologist needs to know about common anorectal disorders. World J Gastroenterol. 2009;15(26):3201-3209.
5. Henderson PK, Cash BD. Common anorectal conditions: evaluation and treatment. Curr Gastroenterol Rep. 2014; 16(10):408.
6. Gebbensleben O, Hilger Y, Rohde H. Patients’ view of medical positioning for proctologic examination. Clin Exp Gastroenterol. 2009;2:133-138.
7. Gopal DV. Diseases of the rectum and anus: a clinical approach to common disorders. Clin Cornerstone. 2002;4(4): 34-48.
8. Lohsiriwat V. Treatment of hemorrhoids: a coloproctologist’s view. World J Gastroenterol. 2015;21(31):9245-9252.
9. Fargo MV, Latimer K. Evaluation and management of common anorectal conditions. Am Fam Physician. 2012;85(6):624-630.
10. Lohsiriwat V. Hemorrhoids: from basic pathophysiology to clinical management. World J Gastroenterol. 2012;18(17): 2009-2017.
11. Foxx-Orenstein AE, Umar SB, Crowell MD. Common anorectal disorders. Gastroenterol Hepatol. 2014;10(5):294-301.
12. Lohsiriwat V. Anorectal emergencies. World J Gastroenterol. 2016;22(26):5867-5878.
13. Greenspon J, Williams SB, Young HA, Orkin BA. Thrombosed external hemorrhoids: outcome after conservative treatment or surgical management. Dis Colon Rectum. 2004(9);47:1493-1498.
14. Summers A. Assessment and treatment of three common anorectal conditions. Emerg Nurse. 2013;21(2):28-33.
15. Alonso-Coello P, Mills E, Heels-Ansdell D, et al. Fiber for the treatment of hemorrhoids complications: a systematic review and meta-analysis. Am J Gastroenterol. 2006;101(1):181-188.
16. Medhi B, Rao RS, Prakash A, et al. Recent advances in the pharmacotherapy of chronic anal fissure: an update. Asian J Surg. 2008;31(3):154-163.
17. Nelson RL. Anal fissure (chronic). BMJ Clin Evid. 2014;2014. pii: 0407.
18. deRosa M, Cestaro G, Vitiello C, et al. Conservative versus surgical treatment for chronic idiopathic anal fissure: a prospective randomized trial. Updates Surg. 2013;65(3):197-200.
19. Lacy B, Weiser K. Common anorectal disorders: diagnosis and treatment. Curr Gastroenterol Rep. 2009;11(5):413-419.
20. Siddiqi S, Vijay V, Ward M, et al. Pruritus ani. Ann R Coll Surg Engl. 2008;90:457-463.
21. Lysy J, Sistiery-Ittah M, Israelit Y, et al. Topical capsaicin—a novel and effective treatment for idiopathic intractable pruritus ani: a randomized, placebo controlled, crossover study. Gut. 2003;52(9):1323-1326.
22. Burnstein M. Managing anorectal emergencies. Can Fam Physician. 1993;39:1782-1785.
23. Sasaki A, Nakajima T, Egashira H, et al. Condyloma acuminatum of the anal canal, treated with endoscopic submucosal dissection. World J Gastroenterol. 2016;22(8):2636-2641.
24. Kollipara R, Ekhlassi E, Downing C, et al. Advancements in pharmacotherapy for noncancerous manifestations of HPV. J Clin Med. 2015;4(5):832-846.
25. CDC. Manual for the Surveillance of Vaccine-preventable Diseases. Chapter 5: Human papillomavirus (HPV). www.cdc.gov/vaccines/pubs/surv-manual/chpt05-hpv.html. Accessed October 18, 2017.
26. Leszczyszyn J, Lebski I, Lysenko L, et al. Anal warts (condyloma acuminatum)—current issues and treatment modalities. Adv Clin Exp Med. 2014;23(2):307-311.
27. Baker DA, Ferris DG, Martens MG, et al. Imiquimod 3.75% cream applied daily to treat anogenital warts: combined results from women in two randomized, placebo-controlled studies. Infect Dis Obstet Gynecol. 2011;2011:806105.
28. Yan J, Chen SL, Wang HN, et al. Meta-analysis of 5% imiquimod and 0.5% podophyllotoxin in the treatment of condylomata acuminate. Dermatology. 2006;213(3):218-223.
29. Hoy SM. Polyphenon E 10% ointment: in immunocompetent adults with external genital and perianal warts. Am J Clin Dermatol. 2012;13(4):275-281.
30. Tatti S, Swinehart JM, Thielert C, et al. Sinecatechins, a defined green tea extract, in the treatment of external anogenital warts. Obstet Gynecol. 2008;111(6):1371-1379.
31. CDC. Human papillomavirus (HPV). www.cdc.gov/hpv/. Accessed October 18, 2017.
32. National Institutes of Health, National Cancer Institute. Human papillomavirus (HPV) vaccine. www.cancer.gov/about-cancer/causes-prevention/risk/infectious-agents/hpv-vaccine-fact-sheet#q5. Accessed October 18, 2017.
33. Giuliano AR, Palefsky JM, Goldstone S, et al. Efficacy of quadrivalent HPV vaccine against HPV infection and disease in males. N Engl J Med. 2011;364:401-411.
34. CDC. HPV vaccines: vaccinating your preteen or teen. www.cdc.gov/hpv/parents/vaccine.html. Accessed October 18, 2017.
AGA Clinical Practice Update: Best practices for POEM in achalasia
Peroral endoscopic myotomy, or POEM, should be considered as primary therapy for type III achalasia and as a treatment option comparable with laparoscopic Heller myotomy for any of the achalasia syndromes – but only when physicians with expertise are available, according to a clinical practice update from the American Gastroenterological Association.
Further, post-POEM patients should be considered at high risk of developing reflux esophagitis and should be advised of the management considerations, including potential indefinite proton pump inhibitor therapy and/or surveillance endoscopy, prior to undergoing the procedure, Peter J. Kahrilas, MD, of Northwestern University, Chicago, and his colleagues wrote in the update, which is published in the November issue of Gastroenterology (2017. doi: 10.1053/j.gastro.2017.10.001).
In an effort to describe the place for POEM among the currently available robust treatments for achalasia, the authors conducted a literature review – their “best practice” recommendations are based on the findings from relevant publications and on expert opinion.

Additionally, they said POEM should be performed by experienced physicians in high-volume centers since the procedure is complex and an estimated 20-30 procedures are needed to achieve competence.
The update and these proposed best practices follow the evolution of POEM over the last decade: it began as an exciting concept and is now a mainstream treatment option for achalasia, the authors said.
“Uncontrolled outcome data have been very promising comparing POEM with the standard surgical treatment for achalasia, laparoscopic Heller myotomy (LHM). However, concerns remain regarding post-POEM reflux, the durability of the procedure, and the learning curve for endoscopists adopting the technique,” they wrote, which, when coupled with recent randomized controlled study data showing excellent and equivalent 5-year outcomes with pneumatic dilation and LHM, make the role of POEM somewhat controversial.
As part of the review, they considered the strengths and weaknesses of both POEM and LHM. The data comparing POEM with LHM or pneumatic dilation remain very limited, but based on those that do exist, the authors concluded that “POEM appears to be a safe, effective, and minimally invasive management option in achalasia in the short term.”
Long-term durability data are not yet available, they noted.
Dr. Kahrilas received funding from the U.S. Public Health Service.
Peroral endoscopic myotomy, or POEM, should be considered as primary therapy for type III achalasia and as a treatment option comparable with laparoscopic Heller myotomy for any of the achalasia syndromes – but only when physicians with expertise are available, according to a clinical practice update from the American Gastroenterological Association.
Further, post-POEM patients should be considered at high risk of developing reflux esophagitis and should be advised of the management considerations, including potential indefinite proton pump inhibitor therapy and/or surveillance endoscopy, prior to undergoing the procedure, Peter J. Kahrilas, MD, of Northwestern University, Chicago, and his colleagues wrote in the update, which is published in the November issue of Gastroenterology (2017. doi: 10.1053/j.gastro.2017.10.001).
In an effort to describe the place for POEM among the currently available robust treatments for achalasia, the authors conducted a literature review – their “best practice” recommendations are based on the findings from relevant publications and on expert opinion.
Additionally, they said POEM should be performed by experienced physicians in high-volume centers since the procedure is complex and an estimated 20-30 procedures are needed to achieve competence.
The update and these proposed best practices follow the evolution of POEM over the last decade: it began as an exciting concept and is now a mainstream treatment option for achalasia, the authors said.
“Uncontrolled outcome data have been very promising comparing POEM with the standard surgical treatment for achalasia, laparoscopic Heller myotomy (LHM). However, concerns remain regarding post-POEM reflux, the durability of the procedure, and the learning curve for endoscopists adopting the technique,” they wrote, which, when coupled with recent randomized controlled study data showing excellent and equivalent 5-year outcomes with pneumatic dilation and LHM, make the role of POEM somewhat controversial.
As part of the review, they considered the strengths and weaknesses of both POEM and LHM. The data comparing POEM with LHM or pneumatic dilation remain very limited, but based on those that do exist, the authors concluded that “POEM appears to be a safe, effective, and minimally invasive management option in achalasia in the short term.”
Long-term durability data are not yet available, they noted.
Dr. Kahrilas received funding from the U.S. Public Health Service.
Peroral endoscopic myotomy, or POEM, should be considered as primary therapy for type III achalasia and as a treatment option comparable with laparoscopic Heller myotomy for any of the achalasia syndromes – but only when physicians with expertise are available, according to a clinical practice update from the American Gastroenterological Association.
Further, post-POEM patients should be considered at high risk of developing reflux esophagitis and should be advised of the management considerations, including potential indefinite proton pump inhibitor therapy and/or surveillance endoscopy, prior to undergoing the procedure, Peter J. Kahrilas, MD, of Northwestern University, Chicago, and his colleagues wrote in the update, which is published in the November issue of Gastroenterology (2017. doi: 10.1053/j.gastro.2017.10.001).
In an effort to describe the place for POEM among the currently available robust treatments for achalasia, the authors conducted a literature review – their “best practice” recommendations are based on the findings from relevant publications and on expert opinion.
Additionally, they said POEM should be performed by experienced physicians in high-volume centers since the procedure is complex and an estimated 20-30 procedures are needed to achieve competence.
The update and these proposed best practices follow the evolution of POEM over the last decade: it began as an exciting concept and is now a mainstream treatment option for achalasia, the authors said.
“Uncontrolled outcome data have been very promising comparing POEM with the standard surgical treatment for achalasia, laparoscopic Heller myotomy (LHM). However, concerns remain regarding post-POEM reflux, the durability of the procedure, and the learning curve for endoscopists adopting the technique,” they wrote, which, when coupled with recent randomized controlled study data showing excellent and equivalent 5-year outcomes with pneumatic dilation and LHM, make the role of POEM somewhat controversial.
As part of the review, they considered the strengths and weaknesses of both POEM and LHM. The data comparing POEM with LHM or pneumatic dilation remain very limited, but based on those that do exist, the authors concluded that “POEM appears to be a safe, effective, and minimally invasive management option in achalasia in the short term.”
Long-term durability data are not yet available, they noted.
Dr. Kahrilas received funding from the U.S. Public Health Service.
FROM GASTROENTEROLOGY

New RA JAK inhibitor, monoclonal antibody trial results scheduled for ACR 2017
New results from several investigational oral Janus kinase inhibitor trials and an antifractalkine monoclonal antibody trial for patients with rheumatoid arthritis will be made available for the first time to attendees of the annual meeting of the American College of Rheumatology on Nov. 6 and 7 in San Diego.
Selective JAK-1 inhibitor upadacitinib
Two double-blind, randomized, controlled phase 3 studies of the oral, selective JAK-1 inhibitor upadacitinib (ABT-494) will be presented at the meeting, both involving once-daily extended-release formulations of the drug at 15 mg or 30 mg. The first trial, to be presented during an ACR abstract session on Monday, Nov. 6, showed that the drug met ACR 20 response criteria at significantly higher rates than did placebo at week 12 (63.8% for 15 mg, 66.2% for 30 mg, and 35.7% for placebo) in 661 patients with active RA and an inadequate response to conventional synthetic disease-modifying antirheumatic drugs (csDMARDs). Patients on the study drug also achieved low disease activity (28-joint Disease Activity Score using C-reactive protein [DAS28-CRP] of 3.2 or less) in higher proportions than did those who received placebo (48.4% for 15 mg, 47.9% for 30 mg, and 17.2% for placebo). More infections occurred in patients on 15 mg and 30 mg upadacitinib (29.0% and 31.5%, respectively), compared with placebo (21.3%), but there were no differences in serious infections. Three of the total four cases of herpes zoster infection were in patients taking upadacitinib.
A second upadacitinib trial, which involved patients on stable csDMARD therapy who could not tolerate or had disease refractory to a biologic DMARD, will be presented during a late-breaking poster session on Tuesday, Nov. 7. The study randomized 499 patients who had severe, refractory disease for a mean duration of 13 years. At 12 weeks, treatment with upadacitinib resulted in ACR 20 responses in 64.6% at 15 mg and 56.4% at 30 mg, compared with 28.4% for placebo. Responses at 12 weeks of 3.2 or less for DAS28-CRP also occurred at significantly higher rates of 43.3% for 15 mg and 42.4% for 30 mg, compared with 14.2% for placebo. Patients who were switched from placebo to active treatment at week 12 also achieved these responses at week 24, and the people who stayed on treatment with upadacitinib maintained or improved their response at week 24. More adverse events and infections tended to occur with upadacitinib at 30 mg than with 15 mg, and there were more herpes zoster infections with 30 mg (n = 4) vs. 15 mg (n = 1) and placebo (n = 1).
Antifractalkine monoclonal antibody
The Monday afternoon RA therapy abstract session will also feature the first-in-patient results at 52 weeks for the antifractalkine monoclonal antibody E6011 in an open-label phase 1/2 study of Japanese RA patients with active disease who had an inadequate response or intolerance to methotrexate or a TNF inhibitor. The results from the small trial of the biologic, which targets the chemokine that regulates chemotaxis and the adhesion of inflammatory cells that express CX3C chemokine receptor 1 (CX3CR1), indicate that the treatment has durable efficacy and is safe and well tolerated. Adverse events (AEs) developed in 84% of 28 patients who participated in the 52-week extension study, including a rate of 48% for treatment-emergent adverse events (TEAEs) and 14% for serious AEs. ACR 20 rates at week 52 ranged from 58% for 100 mg of E6011 to 73% for 200 mg and 60% for 400 mg.
Filgotinib safety in an extension trial
The long-term, open-label results from two phase 2b studies of the JAK-1 inhibitor filgotinib that’s in development for RA also will be presented during the same Monday abstract session. These safety data out to 84 weeks from the long-term extension study of the DARWIN 1 and 2 studies, called DARWIN 3, show that, among patients who received at least one dose of filgotinib 100 mg twice daily or 200 mg once daily in addition to methotrexate, the rate of TEAEs ranged from about 146 to 153 per 100 patient-years of exposure (PYE), including about 41-45 infections per 100 PYE. For filgotinib monotherapy, the rate of TEAEs was 150 per 100 PYE for 200 mg once daily, including 38 infections per 100 PYE.
Total cholesterol of 200-239 mg/dL occurred at a rate of about 79 per 100 PYE and a level of 240 mg/dL or greater was seen in 61-64 per 100 PYE for patients who took filgotinib plus methotrexate. Filgotinib monotherapy at 200 mg once daily gave rates of about 94 and 71 per 100 PYE for those respective total cholesterol levels.
New results from several investigational oral Janus kinase inhibitor trials and an antifractalkine monoclonal antibody trial for patients with rheumatoid arthritis will be made available for the first time to attendees of the annual meeting of the American College of Rheumatology on Nov. 6 and 7 in San Diego.
Selective JAK-1 inhibitor upadacitinib
Two double-blind, randomized, controlled phase 3 studies of the oral, selective JAK-1 inhibitor upadacitinib (ABT-494) will be presented at the meeting, both involving once-daily extended-release formulations of the drug at 15 mg or 30 mg. The first trial, to be presented during an ACR abstract session on Monday, Nov. 6, showed that the drug met ACR 20 response criteria at significantly higher rates than did placebo at week 12 (63.8% for 15 mg, 66.2% for 30 mg, and 35.7% for placebo) in 661 patients with active RA and an inadequate response to conventional synthetic disease-modifying antirheumatic drugs (csDMARDs). Patients on the study drug also achieved low disease activity (28-joint Disease Activity Score using C-reactive protein [DAS28-CRP] of 3.2 or less) in higher proportions than did those who received placebo (48.4% for 15 mg, 47.9% for 30 mg, and 17.2% for placebo). More infections occurred in patients on 15 mg and 30 mg upadacitinib (29.0% and 31.5%, respectively), compared with placebo (21.3%), but there were no differences in serious infections. Three of the total four cases of herpes zoster infection were in patients taking upadacitinib.
A second upadacitinib trial, which involved patients on stable csDMARD therapy who could not tolerate or had disease refractory to a biologic DMARD, will be presented during a late-breaking poster session on Tuesday, Nov. 7. The study randomized 499 patients who had severe, refractory disease for a mean duration of 13 years. At 12 weeks, treatment with upadacitinib resulted in ACR 20 responses in 64.6% at 15 mg and 56.4% at 30 mg, compared with 28.4% for placebo. Responses at 12 weeks of 3.2 or less for DAS28-CRP also occurred at significantly higher rates of 43.3% for 15 mg and 42.4% for 30 mg, compared with 14.2% for placebo. Patients who were switched from placebo to active treatment at week 12 also achieved these responses at week 24, and the people who stayed on treatment with upadacitinib maintained or improved their response at week 24. More adverse events and infections tended to occur with upadacitinib at 30 mg than with 15 mg, and there were more herpes zoster infections with 30 mg (n = 4) vs. 15 mg (n = 1) and placebo (n = 1).
Antifractalkine monoclonal antibody
The Monday afternoon RA therapy abstract session will also feature the first-in-patient results at 52 weeks for the antifractalkine monoclonal antibody E6011 in an open-label phase 1/2 study of Japanese RA patients with active disease who had an inadequate response or intolerance to methotrexate or a TNF inhibitor. The results from the small trial of the biologic, which targets the chemokine that regulates chemotaxis and the adhesion of inflammatory cells that express CX3C chemokine receptor 1 (CX3CR1), indicate that the treatment has durable efficacy and is safe and well tolerated. Adverse events (AEs) developed in 84% of 28 patients who participated in the 52-week extension study, including a rate of 48% for treatment-emergent adverse events (TEAEs) and 14% for serious AEs. ACR 20 rates at week 52 ranged from 58% for 100 mg of E6011 to 73% for 200 mg and 60% for 400 mg.
Filgotinib safety in an extension trial
The long-term, open-label results from two phase 2b studies of the JAK-1 inhibitor filgotinib that’s in development for RA also will be presented during the same Monday abstract session. These safety data out to 84 weeks from the long-term extension study of the DARWIN 1 and 2 studies, called DARWIN 3, show that, among patients who received at least one dose of filgotinib 100 mg twice daily or 200 mg once daily in addition to methotrexate, the rate of TEAEs ranged from about 146 to 153 per 100 patient-years of exposure (PYE), including about 41-45 infections per 100 PYE. For filgotinib monotherapy, the rate of TEAEs was 150 per 100 PYE for 200 mg once daily, including 38 infections per 100 PYE.
Total cholesterol of 200-239 mg/dL occurred at a rate of about 79 per 100 PYE and a level of 240 mg/dL or greater was seen in 61-64 per 100 PYE for patients who took filgotinib plus methotrexate. Filgotinib monotherapy at 200 mg once daily gave rates of about 94 and 71 per 100 PYE for those respective total cholesterol levels.
New results from several investigational oral Janus kinase inhibitor trials and an antifractalkine monoclonal antibody trial for patients with rheumatoid arthritis will be made available for the first time to attendees of the annual meeting of the American College of Rheumatology on Nov. 6 and 7 in San Diego.
Selective JAK-1 inhibitor upadacitinib
Two double-blind, randomized, controlled phase 3 studies of the oral, selective JAK-1 inhibitor upadacitinib (ABT-494) will be presented at the meeting, both involving once-daily extended-release formulations of the drug at 15 mg or 30 mg. The first trial, to be presented during an ACR abstract session on Monday, Nov. 6, showed that the drug met ACR 20 response criteria at significantly higher rates than did placebo at week 12 (63.8% for 15 mg, 66.2% for 30 mg, and 35.7% for placebo) in 661 patients with active RA and an inadequate response to conventional synthetic disease-modifying antirheumatic drugs (csDMARDs). Patients on the study drug also achieved low disease activity (28-joint Disease Activity Score using C-reactive protein [DAS28-CRP] of 3.2 or less) in higher proportions than did those who received placebo (48.4% for 15 mg, 47.9% for 30 mg, and 17.2% for placebo). More infections occurred in patients on 15 mg and 30 mg upadacitinib (29.0% and 31.5%, respectively), compared with placebo (21.3%), but there were no differences in serious infections. Three of the total four cases of herpes zoster infection were in patients taking upadacitinib.
A second upadacitinib trial, which involved patients on stable csDMARD therapy who could not tolerate or had disease refractory to a biologic DMARD, will be presented during a late-breaking poster session on Tuesday, Nov. 7. The study randomized 499 patients who had severe, refractory disease for a mean duration of 13 years. At 12 weeks, treatment with upadacitinib resulted in ACR 20 responses in 64.6% at 15 mg and 56.4% at 30 mg, compared with 28.4% for placebo. Responses at 12 weeks of 3.2 or less for DAS28-CRP also occurred at significantly higher rates of 43.3% for 15 mg and 42.4% for 30 mg, compared with 14.2% for placebo. Patients who were switched from placebo to active treatment at week 12 also achieved these responses at week 24, and the people who stayed on treatment with upadacitinib maintained or improved their response at week 24. More adverse events and infections tended to occur with upadacitinib at 30 mg than with 15 mg, and there were more herpes zoster infections with 30 mg (n = 4) vs. 15 mg (n = 1) and placebo (n = 1).
Antifractalkine monoclonal antibody
The Monday afternoon RA therapy abstract session will also feature the first-in-patient results at 52 weeks for the antifractalkine monoclonal antibody E6011 in an open-label phase 1/2 study of Japanese RA patients with active disease who had an inadequate response or intolerance to methotrexate or a TNF inhibitor. The results from the small trial of the biologic, which targets the chemokine that regulates chemotaxis and the adhesion of inflammatory cells that express CX3C chemokine receptor 1 (CX3CR1), indicate that the treatment has durable efficacy and is safe and well tolerated. Adverse events (AEs) developed in 84% of 28 patients who participated in the 52-week extension study, including a rate of 48% for treatment-emergent adverse events (TEAEs) and 14% for serious AEs. ACR 20 rates at week 52 ranged from 58% for 100 mg of E6011 to 73% for 200 mg and 60% for 400 mg.
Filgotinib safety in an extension trial
The long-term, open-label results from two phase 2b studies of the JAK-1 inhibitor filgotinib that’s in development for RA also will be presented during the same Monday abstract session. These safety data out to 84 weeks from the long-term extension study of the DARWIN 1 and 2 studies, called DARWIN 3, show that, among patients who received at least one dose of filgotinib 100 mg twice daily or 200 mg once daily in addition to methotrexate, the rate of TEAEs ranged from about 146 to 153 per 100 patient-years of exposure (PYE), including about 41-45 infections per 100 PYE. For filgotinib monotherapy, the rate of TEAEs was 150 per 100 PYE for 200 mg once daily, including 38 infections per 100 PYE.
Total cholesterol of 200-239 mg/dL occurred at a rate of about 79 per 100 PYE and a level of 240 mg/dL or greater was seen in 61-64 per 100 PYE for patients who took filgotinib plus methotrexate. Filgotinib monotherapy at 200 mg once daily gave rates of about 94 and 71 per 100 PYE for those respective total cholesterol levels.
FROM ACR 2017
Infections, psoriasis, facial fillers heat up Las Vegas Dermatology Seminar
Updates on vaccines and infections, as well as the latest acne treatments and the Psoriasis Forum are among the hot topics at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
The meeting is cochaired by Linda F. Stein Gold, MD, of Henry Ford Health System in Detroit, Mich., Christopher B. Zachary, MD, of the University of California, Irvine, and Joseph F. Fowler Jr., MD, of the University of Louisville (Ky).

Dr. Stein Gold, along with Julie Harper, MD, will direct a rosacea forum on Thursday to review the latest information on pathology and treatment. The day’s acne forum will include details on sebum-reduction products in the pipeline, as well as data on benzoyl peroxide.
The psoriasis forum on Friday will feature “long-term information on the safety of current drugs,” and also look ahead to more data on IL-23 inhibitors, Dr. Fowler said in an interview.
Aesthetic dermatology highlights from Saturday’s scheduled presentations include Dr. Zachary’s review of options for facial rejuvenation and what clinicians need to know about body contouring. Kristin M. Kelly, MD, of the University of California, Irvine, will discuss devices and how they can be best used to modulate scarring, and Howard Steinman, MD, who is in private practice in Irving, Tex., will share tips on “avoiding the danger zones” when using toxins and fillers.
SDEF and this news organization are owned by the same parent company.
Updates on vaccines and infections, as well as the latest acne treatments and the Psoriasis Forum are among the hot topics at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
The meeting is cochaired by Linda F. Stein Gold, MD, of Henry Ford Health System in Detroit, Mich., Christopher B. Zachary, MD, of the University of California, Irvine, and Joseph F. Fowler Jr., MD, of the University of Louisville (Ky).
Dr. Stein Gold, along with Julie Harper, MD, will direct a rosacea forum on Thursday to review the latest information on pathology and treatment. The day’s acne forum will include details on sebum-reduction products in the pipeline, as well as data on benzoyl peroxide.
The psoriasis forum on Friday will feature “long-term information on the safety of current drugs,” and also look ahead to more data on IL-23 inhibitors, Dr. Fowler said in an interview.
Aesthetic dermatology highlights from Saturday’s scheduled presentations include Dr. Zachary’s review of options for facial rejuvenation and what clinicians need to know about body contouring. Kristin M. Kelly, MD, of the University of California, Irvine, will discuss devices and how they can be best used to modulate scarring, and Howard Steinman, MD, who is in private practice in Irving, Tex., will share tips on “avoiding the danger zones” when using toxins and fillers.
SDEF and this news organization are owned by the same parent company.
Updates on vaccines and infections, as well as the latest acne treatments and the Psoriasis Forum are among the hot topics at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
The meeting is cochaired by Linda F. Stein Gold, MD, of Henry Ford Health System in Detroit, Mich., Christopher B. Zachary, MD, of the University of California, Irvine, and Joseph F. Fowler Jr., MD, of the University of Louisville (Ky).
Dr. Stein Gold, along with Julie Harper, MD, will direct a rosacea forum on Thursday to review the latest information on pathology and treatment. The day’s acne forum will include details on sebum-reduction products in the pipeline, as well as data on benzoyl peroxide.
The psoriasis forum on Friday will feature “long-term information on the safety of current drugs,” and also look ahead to more data on IL-23 inhibitors, Dr. Fowler said in an interview.
Aesthetic dermatology highlights from Saturday’s scheduled presentations include Dr. Zachary’s review of options for facial rejuvenation and what clinicians need to know about body contouring. Kristin M. Kelly, MD, of the University of California, Irvine, will discuss devices and how they can be best used to modulate scarring, and Howard Steinman, MD, who is in private practice in Irving, Tex., will share tips on “avoiding the danger zones” when using toxins and fillers.
SDEF and this news organization are owned by the same parent company.
Cardiogenic shock boosts PAH readmissions 10-fold
TORONTO – Cardiogenic shock, acute kidney injury, and chronic obstructive pulmonary disease were the top drivers of 30-day rehospitalizations in U.S. patients after an index hospitalization for pulmonary artery hypertension, based on an analysis of U.S. national data from 2013.
An episode of cardiogenic shock boosted 30-day rehospitalizations nearly 10-fold in recently discharged pulmonary artery hypertension (PAH) patients. A history of chronic obstructive pulmonary disease (COPD) linked with a threefold higher rehospitalization rate, and acute kidney injury linked with a doubled number of 30-day rehospitalizations, Kshitij Chatterjee, MD, said at the CHEST annual meeting.

The powerful impact of cardiogenic shock in particular suggests that interventions that improve patient compliance with stabilizing treatments following an index PAH hospitalization might be effective at preventing a patient’s quick return to the hospital. Contacting PAH patients a week after their index hospitalization discharge to make sure they are compliant with their diuretic regimen, for example, might help prevent a decompensation that then leads to cardiogenic shock and a return trip to the hospital, Dr. Chatterjee suggested.
Follow-up of PAH patients after an index hospitalization “is probably the single most important thing, because it can help with compliance,” he said in an interview.
The rehospitalizations he studied could be for any cause. His analysis showed that the most common cause of rehospitalization was heart failure, which caused 23% of the rehospitalizations, followed by pulmonary hypertension that caused 20%, and acute kidney injury, responsible for 11% of the 30-day rehospitalizations.
Dr. Chatterjee’s study used data collected during 2013 in the National Readmissions Database, run by the federal Agency for Healthcare Quality and Research. During that period, 776 patients entered a U.S. hospital with a primary diagnosis of PAH. During the 30 days following discharge, 114 (15%) returned to the hospital. During the second hospitalization 8% died, and the median length of stay for those who remained alive was 7 days.
Dr. Chatterjee highlighted that the modest number of index hospitalizations for PAH, as well as 30-day rehospitalizations he found in 2013, make it highly unlikely that PAH rehospitalizations will become a target for Medicare penalties as has been done for heart failure, pneumonia, COPD, and a few other disorders. But he stressed that patients with PAH who need rehospitalization generally have a highly compromised quality of life that potentially could be avoided by better management, which could prevent the need for rehospitalization.
Dr. Chatterjee had no disclosures.
[email protected]
On Twitter @mitchelzoler
TORONTO – Cardiogenic shock, acute kidney injury, and chronic obstructive pulmonary disease were the top drivers of 30-day rehospitalizations in U.S. patients after an index hospitalization for pulmonary artery hypertension, based on an analysis of U.S. national data from 2013.
An episode of cardiogenic shock boosted 30-day rehospitalizations nearly 10-fold in recently discharged pulmonary artery hypertension (PAH) patients. A history of chronic obstructive pulmonary disease (COPD) linked with a threefold higher rehospitalization rate, and acute kidney injury linked with a doubled number of 30-day rehospitalizations, Kshitij Chatterjee, MD, said at the CHEST annual meeting.
The powerful impact of cardiogenic shock in particular suggests that interventions that improve patient compliance with stabilizing treatments following an index PAH hospitalization might be effective at preventing a patient’s quick return to the hospital. Contacting PAH patients a week after their index hospitalization discharge to make sure they are compliant with their diuretic regimen, for example, might help prevent a decompensation that then leads to cardiogenic shock and a return trip to the hospital, Dr. Chatterjee suggested.
Follow-up of PAH patients after an index hospitalization “is probably the single most important thing, because it can help with compliance,” he said in an interview.
The rehospitalizations he studied could be for any cause. His analysis showed that the most common cause of rehospitalization was heart failure, which caused 23% of the rehospitalizations, followed by pulmonary hypertension that caused 20%, and acute kidney injury, responsible for 11% of the 30-day rehospitalizations.
Dr. Chatterjee’s study used data collected during 2013 in the National Readmissions Database, run by the federal Agency for Healthcare Quality and Research. During that period, 776 patients entered a U.S. hospital with a primary diagnosis of PAH. During the 30 days following discharge, 114 (15%) returned to the hospital. During the second hospitalization 8% died, and the median length of stay for those who remained alive was 7 days.
Dr. Chatterjee highlighted that the modest number of index hospitalizations for PAH, as well as 30-day rehospitalizations he found in 2013, make it highly unlikely that PAH rehospitalizations will become a target for Medicare penalties as has been done for heart failure, pneumonia, COPD, and a few other disorders. But he stressed that patients with PAH who need rehospitalization generally have a highly compromised quality of life that potentially could be avoided by better management, which could prevent the need for rehospitalization.
Dr. Chatterjee had no disclosures.
[email protected]
On Twitter @mitchelzoler
TORONTO – Cardiogenic shock, acute kidney injury, and chronic obstructive pulmonary disease were the top drivers of 30-day rehospitalizations in U.S. patients after an index hospitalization for pulmonary artery hypertension, based on an analysis of U.S. national data from 2013.
An episode of cardiogenic shock boosted 30-day rehospitalizations nearly 10-fold in recently discharged pulmonary artery hypertension (PAH) patients. A history of chronic obstructive pulmonary disease (COPD) linked with a threefold higher rehospitalization rate, and acute kidney injury linked with a doubled number of 30-day rehospitalizations, Kshitij Chatterjee, MD, said at the CHEST annual meeting.
The powerful impact of cardiogenic shock in particular suggests that interventions that improve patient compliance with stabilizing treatments following an index PAH hospitalization might be effective at preventing a patient’s quick return to the hospital. Contacting PAH patients a week after their index hospitalization discharge to make sure they are compliant with their diuretic regimen, for example, might help prevent a decompensation that then leads to cardiogenic shock and a return trip to the hospital, Dr. Chatterjee suggested.
Follow-up of PAH patients after an index hospitalization “is probably the single most important thing, because it can help with compliance,” he said in an interview.
The rehospitalizations he studied could be for any cause. His analysis showed that the most common cause of rehospitalization was heart failure, which caused 23% of the rehospitalizations, followed by pulmonary hypertension that caused 20%, and acute kidney injury, responsible for 11% of the 30-day rehospitalizations.
Dr. Chatterjee’s study used data collected during 2013 in the National Readmissions Database, run by the federal Agency for Healthcare Quality and Research. During that period, 776 patients entered a U.S. hospital with a primary diagnosis of PAH. During the 30 days following discharge, 114 (15%) returned to the hospital. During the second hospitalization 8% died, and the median length of stay for those who remained alive was 7 days.
Dr. Chatterjee highlighted that the modest number of index hospitalizations for PAH, as well as 30-day rehospitalizations he found in 2013, make it highly unlikely that PAH rehospitalizations will become a target for Medicare penalties as has been done for heart failure, pneumonia, COPD, and a few other disorders. But he stressed that patients with PAH who need rehospitalization generally have a highly compromised quality of life that potentially could be avoided by better management, which could prevent the need for rehospitalization.
Dr. Chatterjee had no disclosures.
[email protected]
On Twitter @mitchelzoler
AT CHEST 2017
Key clinical point:
Major finding: Patients with cardiogenic shock following PAH hospitalization had a 9.7-fold increased rate of 30-day rehospitalization, compared with patients without shock.
Data source: The National Readmissions Database, which included 776 index U.S. hospitalizations for pulmonary arterial hospitalization during 2013.
Disclosures: Dr. Chatterjee had no disclosures.

DDSEP® 8 Quick quiz - November 2017 Question 2
Answer: C
Rationale: Binge-eating disorder (BED) is a distinct clinical entity, which gastroenterologists are likely to encounter. A substantial fraction of patients evaluated for weight loss therapy will have BED, yet it can be difficult to recognize. Similar to bulimia, BED is characterized by binge-eating episodes, which must occur an average of once a week for at least 3 months. However, there are no recurrent inappropriate behaviors such as purging with laxatives, diuretics, or emetics (e.g., syrup of ipecac) or excessive exercise. BED patients most often do not have any specific symptoms or physical exam findings other than being overweight or obese. Very subtle characteristics include very rapid eating, eating despite satiety, eating alone due to feelings of shame, and a negative emotional context after binge eating. BED will negatively impact any interventions for obesity, unless recognized and addressed.
Nocturnal eating syndrome is similar, yet distinct, in that it is also characterized by binge eating without inappropriate compensatory purging behaviors, but prominent features include morning anorexia, nocturnal hyperphagia, and sleep disturbances. The sleep disturbances are characterized by an average of 3-4 awakenings per night, during which an average of roughly 1,100 calories might be consumed during half the episodes.
Anorexia nervosa is characterized by a restriction in food intake relative to needs which results in an inappropriately low body weight (below BMI of 17.5 kg/m2), a fear of gaining weight or being fat despite being underweight, and inappropriate perception/experience of body image. Roughly half of patient with anorexia nervosa may also engage in binge-eating behaviors or purging behaviors.
Purging disorder is a distinct variant recognized as purging behaviors in the absence of the binge-eating behavior.
Answer: C
Rationale: Binge-eating disorder (BED) is a distinct clinical entity, which gastroenterologists are likely to encounter. A substantial fraction of patients evaluated for weight loss therapy will have BED, yet it can be difficult to recognize. Similar to bulimia, BED is characterized by binge-eating episodes, which must occur an average of once a week for at least 3 months. However, there are no recurrent inappropriate behaviors such as purging with laxatives, diuretics, or emetics (e.g., syrup of ipecac) or excessive exercise. BED patients most often do not have any specific symptoms or physical exam findings other than being overweight or obese. Very subtle characteristics include very rapid eating, eating despite satiety, eating alone due to feelings of shame, and a negative emotional context after binge eating. BED will negatively impact any interventions for obesity, unless recognized and addressed.
Nocturnal eating syndrome is similar, yet distinct, in that it is also characterized by binge eating without inappropriate compensatory purging behaviors, but prominent features include morning anorexia, nocturnal hyperphagia, and sleep disturbances. The sleep disturbances are characterized by an average of 3-4 awakenings per night, during which an average of roughly 1,100 calories might be consumed during half the episodes.
Anorexia nervosa is characterized by a restriction in food intake relative to needs which results in an inappropriately low body weight (below BMI of 17.5 kg/m2), a fear of gaining weight or being fat despite being underweight, and inappropriate perception/experience of body image. Roughly half of patient with anorexia nervosa may also engage in binge-eating behaviors or purging behaviors.
Purging disorder is a distinct variant recognized as purging behaviors in the absence of the binge-eating behavior.
Answer: C
Rationale: Binge-eating disorder (BED) is a distinct clinical entity, which gastroenterologists are likely to encounter. A substantial fraction of patients evaluated for weight loss therapy will have BED, yet it can be difficult to recognize. Similar to bulimia, BED is characterized by binge-eating episodes, which must occur an average of once a week for at least 3 months. However, there are no recurrent inappropriate behaviors such as purging with laxatives, diuretics, or emetics (e.g., syrup of ipecac) or excessive exercise. BED patients most often do not have any specific symptoms or physical exam findings other than being overweight or obese. Very subtle characteristics include very rapid eating, eating despite satiety, eating alone due to feelings of shame, and a negative emotional context after binge eating. BED will negatively impact any interventions for obesity, unless recognized and addressed.
Nocturnal eating syndrome is similar, yet distinct, in that it is also characterized by binge eating without inappropriate compensatory purging behaviors, but prominent features include morning anorexia, nocturnal hyperphagia, and sleep disturbances. The sleep disturbances are characterized by an average of 3-4 awakenings per night, during which an average of roughly 1,100 calories might be consumed during half the episodes.
Anorexia nervosa is characterized by a restriction in food intake relative to needs which results in an inappropriately low body weight (below BMI of 17.5 kg/m2), a fear of gaining weight or being fat despite being underweight, and inappropriate perception/experience of body image. Roughly half of patient with anorexia nervosa may also engage in binge-eating behaviors or purging behaviors.
Purging disorder is a distinct variant recognized as purging behaviors in the absence of the binge-eating behavior.
A 26-year-old woman is evaluated for assistance with weight management. She denies any significant medical or surgical history, but her BMI is 42 kg/m2. She does not understand why she maintains this weight. She insists that she follows a careful diet of healthy foods, noting that she has several servings each day of fruits and vegetables. She does not go to a gym, but walks when she can and takes the stairs more frequently than the elevator. Upon further review, she does admit to “stress eating” episodes just once or twice each week, but dismisses this as noncontributory, since this has been normal behavior for her since high school, when she was not considered to be overweight. She describes these episodes as more typically occurring after a stressful work day, during which she may treat herself to a bag of cookies or a carton of ice cream, usually alone in her apartment in the evening after dinner. She denies nausea, vomiting, or using laxatives. She denies sleep disturbance, is usually well rested in the morning, and eats “a healthy breakfast.” A routine electrolyte profile and complete blood count are unremarkable. She seeks your advice on endoscopic devices for weight loss management.
Government uncertainty drives jump in ACA silver plan insurance premiums
Silver plans on the Affordable Care Act insurance exchanges in 2018 will see an average premium increase of 34% nationwide, according to new research from Avalere Health.
“Plans are raising premiums in 2018 to account for market uncertainty and the federal government’s failure to pay for cost-sharing reductions,” Caroline Pearson, senior vice president at Avalere, said in a statement. “These premium increases may allow insurers to remain in the market and enrollees in all regions to have access to coverage.”
The expected premium changes are highly variable by state. Iowa has the highest change in its silver plans, with an average premium increase of 69% for its silver plans, while at the other end of the spectrum, Alaska is actually seeing a 22% decrease.
“These rates may change prior to open enrollment depending on how states respond to the elimination of CSR [cost-sharing reduction] funding for the 2018 plan year,” Avalere notes in its new analysis, adding that states may allow plans to refile for rate hikes now that CSR funding is likely dead. “In states where this occurs, it is expected that the newly updated rates will be substantially higher for the 2018 plan year.”
There was a glimmer of hope that the CSR payments would resume after a compromise was reached in the Senate Health, Education, Labor & Pensions Committee by Chairman Lamar Alexander (R-Tenn.) and Ranking Member Patty Murray (D-Wash.) that would offer 2 years of funding along with flexibility in the waiver program to allow states to tweak Affordable Care Act requirements. However, Speaker Paul Ryan (R-Wisc.) said the House would not be taking on any more health care action for the remainder of the year.
A spokeswoman from America’s Health Insurance Plans said in an interview that, although the CSR payments are no more, premium tax credits still exist to help lower-income individuals obtain insurance coverage.
Mike Nelson, MD, FCCP, comments: One need not “google” too long to find that the United States performs quite poorly in overall health care when compared with other nations, despite spending more than any of the comparators…we’re 37th this year. This information from Avalere Health portends a further drop in our ranking next year. The privilege of good health is a responsibility of the individual, but the right to affordable health care is a responsibility of the government. It is time for our legislators to stop playing partisan politics and start communicating to propose a workable and affordable solution.

Mike Nelson, MD, FCCP, comments: One need not “google” too long to find that the United States performs quite poorly in overall health care when compared with other nations, despite spending more than any of the comparators…we’re 37th this year. This information from Avalere Health portends a further drop in our ranking next year. The privilege of good health is a responsibility of the individual, but the right to affordable health care is a responsibility of the government. It is time for our legislators to stop playing partisan politics and start communicating to propose a workable and affordable solution.
Mike Nelson, MD, FCCP, comments: One need not “google” too long to find that the United States performs quite poorly in overall health care when compared with other nations, despite spending more than any of the comparators…we’re 37th this year. This information from Avalere Health portends a further drop in our ranking next year. The privilege of good health is a responsibility of the individual, but the right to affordable health care is a responsibility of the government. It is time for our legislators to stop playing partisan politics and start communicating to propose a workable and affordable solution.
Silver plans on the Affordable Care Act insurance exchanges in 2018 will see an average premium increase of 34% nationwide, according to new research from Avalere Health.
“Plans are raising premiums in 2018 to account for market uncertainty and the federal government’s failure to pay for cost-sharing reductions,” Caroline Pearson, senior vice president at Avalere, said in a statement. “These premium increases may allow insurers to remain in the market and enrollees in all regions to have access to coverage.”
The expected premium changes are highly variable by state. Iowa has the highest change in its silver plans, with an average premium increase of 69% for its silver plans, while at the other end of the spectrum, Alaska is actually seeing a 22% decrease.
“These rates may change prior to open enrollment depending on how states respond to the elimination of CSR [cost-sharing reduction] funding for the 2018 plan year,” Avalere notes in its new analysis, adding that states may allow plans to refile for rate hikes now that CSR funding is likely dead. “In states where this occurs, it is expected that the newly updated rates will be substantially higher for the 2018 plan year.”
There was a glimmer of hope that the CSR payments would resume after a compromise was reached in the Senate Health, Education, Labor & Pensions Committee by Chairman Lamar Alexander (R-Tenn.) and Ranking Member Patty Murray (D-Wash.) that would offer 2 years of funding along with flexibility in the waiver program to allow states to tweak Affordable Care Act requirements. However, Speaker Paul Ryan (R-Wisc.) said the House would not be taking on any more health care action for the remainder of the year.
A spokeswoman from America’s Health Insurance Plans said in an interview that, although the CSR payments are no more, premium tax credits still exist to help lower-income individuals obtain insurance coverage.
Silver plans on the Affordable Care Act insurance exchanges in 2018 will see an average premium increase of 34% nationwide, according to new research from Avalere Health.
“Plans are raising premiums in 2018 to account for market uncertainty and the federal government’s failure to pay for cost-sharing reductions,” Caroline Pearson, senior vice president at Avalere, said in a statement. “These premium increases may allow insurers to remain in the market and enrollees in all regions to have access to coverage.”
The expected premium changes are highly variable by state. Iowa has the highest change in its silver plans, with an average premium increase of 69% for its silver plans, while at the other end of the spectrum, Alaska is actually seeing a 22% decrease.
“These rates may change prior to open enrollment depending on how states respond to the elimination of CSR [cost-sharing reduction] funding for the 2018 plan year,” Avalere notes in its new analysis, adding that states may allow plans to refile for rate hikes now that CSR funding is likely dead. “In states where this occurs, it is expected that the newly updated rates will be substantially higher for the 2018 plan year.”
There was a glimmer of hope that the CSR payments would resume after a compromise was reached in the Senate Health, Education, Labor & Pensions Committee by Chairman Lamar Alexander (R-Tenn.) and Ranking Member Patty Murray (D-Wash.) that would offer 2 years of funding along with flexibility in the waiver program to allow states to tweak Affordable Care Act requirements. However, Speaker Paul Ryan (R-Wisc.) said the House would not be taking on any more health care action for the remainder of the year.
A spokeswoman from America’s Health Insurance Plans said in an interview that, although the CSR payments are no more, premium tax credits still exist to help lower-income individuals obtain insurance coverage.
Government uncertainty drives jump in ACA silver plan insurance premiums
Silver plans on the Affordable Care Act insurance exchanges in 2018 will see an average premium increase of 34% nationwide, according to new research from Avalere Health.
“Plans are raising premiums in 2018 to account for market uncertainty and the federal government’s failure to pay for cost-sharing reductions,” Caroline Pearson, senior vice president at Avalere, said in a statement. “These premium increases may allow insurers to remain in the market and enrollees in all regions to have access to coverage.”
The expected premium changes are highly variable by state. Iowa has the highest change in its silver plans, with an average premium increase of 69% for its silver plans, while at the other end of the spectrum, Alaska is actually seeing a 22% decrease.
“These rates may change prior to open enrollment depending on how states respond to the elimination of CSR [cost-sharing reduction] funding for the 2018 plan year,” Avalere notes in its new analysis, adding that states may allow plans to refile for rate hikes now that CSR funding is likely dead. “In states where this occurs, it is expected that the newly updated rates will be substantially higher for the 2018 plan year.”
There was a glimmer of hope that the CSR payments would resume after a compromise was reached in the Senate Health, Education, Labor & Pensions Committee by Chairman Lamar Alexander (R-Tenn.) and Ranking Member Patty Murray (D-Wash.) that would offer 2 years of funding along with flexibility in the waiver program to allow states to tweak Affordable Care Act requirements, which is supported by AGA (http://www.gastro.org/news_items/aga-supports-alexander-murray-agreement-to-stabilize-individual-market). However, Speaker Paul Ryan (R-Wisc.) said the House would not be taking on any more health care action for the remainder of the year.
A spokeswoman from America’s Health Insurance Plans said in an interview that, although the CSR payments are no more, premium tax credits still exist to help lower-income individuals obtain insurance coverage.
Silver plans on the Affordable Care Act insurance exchanges in 2018 will see an average premium increase of 34% nationwide, according to new research from Avalere Health.
“Plans are raising premiums in 2018 to account for market uncertainty and the federal government’s failure to pay for cost-sharing reductions,” Caroline Pearson, senior vice president at Avalere, said in a statement. “These premium increases may allow insurers to remain in the market and enrollees in all regions to have access to coverage.”
The expected premium changes are highly variable by state. Iowa has the highest change in its silver plans, with an average premium increase of 69% for its silver plans, while at the other end of the spectrum, Alaska is actually seeing a 22% decrease.
“These rates may change prior to open enrollment depending on how states respond to the elimination of CSR [cost-sharing reduction] funding for the 2018 plan year,” Avalere notes in its new analysis, adding that states may allow plans to refile for rate hikes now that CSR funding is likely dead. “In states where this occurs, it is expected that the newly updated rates will be substantially higher for the 2018 plan year.”
There was a glimmer of hope that the CSR payments would resume after a compromise was reached in the Senate Health, Education, Labor & Pensions Committee by Chairman Lamar Alexander (R-Tenn.) and Ranking Member Patty Murray (D-Wash.) that would offer 2 years of funding along with flexibility in the waiver program to allow states to tweak Affordable Care Act requirements, which is supported by AGA (http://www.gastro.org/news_items/aga-supports-alexander-murray-agreement-to-stabilize-individual-market). However, Speaker Paul Ryan (R-Wisc.) said the House would not be taking on any more health care action for the remainder of the year.
A spokeswoman from America’s Health Insurance Plans said in an interview that, although the CSR payments are no more, premium tax credits still exist to help lower-income individuals obtain insurance coverage.
Silver plans on the Affordable Care Act insurance exchanges in 2018 will see an average premium increase of 34% nationwide, according to new research from Avalere Health.
“Plans are raising premiums in 2018 to account for market uncertainty and the federal government’s failure to pay for cost-sharing reductions,” Caroline Pearson, senior vice president at Avalere, said in a statement. “These premium increases may allow insurers to remain in the market and enrollees in all regions to have access to coverage.”
The expected premium changes are highly variable by state. Iowa has the highest change in its silver plans, with an average premium increase of 69% for its silver plans, while at the other end of the spectrum, Alaska is actually seeing a 22% decrease.
“These rates may change prior to open enrollment depending on how states respond to the elimination of CSR [cost-sharing reduction] funding for the 2018 plan year,” Avalere notes in its new analysis, adding that states may allow plans to refile for rate hikes now that CSR funding is likely dead. “In states where this occurs, it is expected that the newly updated rates will be substantially higher for the 2018 plan year.”
There was a glimmer of hope that the CSR payments would resume after a compromise was reached in the Senate Health, Education, Labor & Pensions Committee by Chairman Lamar Alexander (R-Tenn.) and Ranking Member Patty Murray (D-Wash.) that would offer 2 years of funding along with flexibility in the waiver program to allow states to tweak Affordable Care Act requirements, which is supported by AGA (http://www.gastro.org/news_items/aga-supports-alexander-murray-agreement-to-stabilize-individual-market). However, Speaker Paul Ryan (R-Wisc.) said the House would not be taking on any more health care action for the remainder of the year.
A spokeswoman from America’s Health Insurance Plans said in an interview that, although the CSR payments are no more, premium tax credits still exist to help lower-income individuals obtain insurance coverage.