User login
IHS Funds Zero Suicide Programs
Zero Suicide is a key concept of the 2012 National Strategy for Suicide Prevention. It uses a “programmatic approach” to quality improvement, based on the realization that suicidal individuals often fall through the cracks in a “sometimes fragmented and distracted” health care system.
A task force identified 7 essential elements of care for health and behavioral health care systems to adopt, including promoting a “safety-oriented” culture, training a competent and caring workforce, using evidence-based treatments, and providing continuous contact and support. The program represents a commitment to both patient safety and to the safety and support of clinical staff who care for suicidal patients.
The Zero Suicide tool kit includes readings, videos, webinars, and other resources, such as a Mental Health Guide developed by the VA to ensure a “safe and therapeutically enriching environment” and a checklist to review inpatient mental health units for environmental hazards. The tool kit also provides thoughtful supplements, such as hospital care cards to send to patients after discharge and a “caring letter template” that includes caring phrases in the Puyallup language with English translations.
The 8 facilities receiving grants are Apache Behavioral Health Service in Whiteriver, Arizona; Chinle Comprehensive Healthcare Facility in Arizona; Fort Defiance Indian Hospital Board in Arizona; Gallup Indian Medical Center in New Mexico; Lawton Indian Hospital in Oklahoma; Menominee Indian Tribe of Wisconsin in Keshena; Pueblo of Acoma in New Mexico; and Rocky Boy Health Board, Box Elder in Montana.
Zero Suicide is a key concept of the 2012 National Strategy for Suicide Prevention. It uses a “programmatic approach” to quality improvement, based on the realization that suicidal individuals often fall through the cracks in a “sometimes fragmented and distracted” health care system.
A task force identified 7 essential elements of care for health and behavioral health care systems to adopt, including promoting a “safety-oriented” culture, training a competent and caring workforce, using evidence-based treatments, and providing continuous contact and support. The program represents a commitment to both patient safety and to the safety and support of clinical staff who care for suicidal patients.
The Zero Suicide tool kit includes readings, videos, webinars, and other resources, such as a Mental Health Guide developed by the VA to ensure a “safe and therapeutically enriching environment” and a checklist to review inpatient mental health units for environmental hazards. The tool kit also provides thoughtful supplements, such as hospital care cards to send to patients after discharge and a “caring letter template” that includes caring phrases in the Puyallup language with English translations.
The 8 facilities receiving grants are Apache Behavioral Health Service in Whiteriver, Arizona; Chinle Comprehensive Healthcare Facility in Arizona; Fort Defiance Indian Hospital Board in Arizona; Gallup Indian Medical Center in New Mexico; Lawton Indian Hospital in Oklahoma; Menominee Indian Tribe of Wisconsin in Keshena; Pueblo of Acoma in New Mexico; and Rocky Boy Health Board, Box Elder in Montana.
Zero Suicide is a key concept of the 2012 National Strategy for Suicide Prevention. It uses a “programmatic approach” to quality improvement, based on the realization that suicidal individuals often fall through the cracks in a “sometimes fragmented and distracted” health care system.
A task force identified 7 essential elements of care for health and behavioral health care systems to adopt, including promoting a “safety-oriented” culture, training a competent and caring workforce, using evidence-based treatments, and providing continuous contact and support. The program represents a commitment to both patient safety and to the safety and support of clinical staff who care for suicidal patients.
The Zero Suicide tool kit includes readings, videos, webinars, and other resources, such as a Mental Health Guide developed by the VA to ensure a “safe and therapeutically enriching environment” and a checklist to review inpatient mental health units for environmental hazards. The tool kit also provides thoughtful supplements, such as hospital care cards to send to patients after discharge and a “caring letter template” that includes caring phrases in the Puyallup language with English translations.
The 8 facilities receiving grants are Apache Behavioral Health Service in Whiteriver, Arizona; Chinle Comprehensive Healthcare Facility in Arizona; Fort Defiance Indian Hospital Board in Arizona; Gallup Indian Medical Center in New Mexico; Lawton Indian Hospital in Oklahoma; Menominee Indian Tribe of Wisconsin in Keshena; Pueblo of Acoma in New Mexico; and Rocky Boy Health Board, Box Elder in Montana.
Combo produces responses in R/R Ph+ ALL

ATLANTA—A 2-drug combination has produced a high response rate in a small trial of patients with relapsed/refractory (R/R), Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL).
The combination, inotuzumab ozogamicin and bosutinib, produced an overall response rate of 81% in this ongoing, phase 1/2 trial.
Nitin Jain, MD, of the University of Texas MD Anderson Cancer Center in Houston, presented phase 1 results from the study at the 2017 ASH Annual Meeting (abstract 143*).
He reported results in 16 patients, 14 with R/R, Ph+ ALL and 2 with chronic myeloid leukemia in lymphoid blast phase.
The patients received inotuzumab ozogamicin at 0.8 mg/m2 on day 1, 0.5 mg/m2 on day 8, and 0.5 mg/m2 on day 15 of cycle 1. Patients who achieved a response received inotuzumab ozogamicin at 1 mg/m2 once every 4 weeks for subsequent cycles. Six cycles were planned.
Patients also received bosutinib at 300 mg, 400 mg, or 500 mg once a day for 4-week cycles. The median number of cycles was 2.5 (range, 1-8).
The maximum-tolerated dose has not been established, but there were 2 dose-limiting toxicities (DLTs). One DLT occurred with the 400 mg dose of bosutinib, and 1 occurred with the 500 mg dose. Both DLTs were grade 3 skin rash.
The investigators are continuing accrual with the 500 mg dose of bosutinib for the phase 2 portion of the trial, with 22 additional patients.
Response and survival
The overall response rate was 81% (n=13). This included a complete response (CR) in 8 patients, a CR with incomplete blood count recovery in 3 patients, and a CR with incomplete platelet recovery in 2 patients.
All responses occurred among the patients with ALL.
Twelve responders achieved complete cytogenetic remission, 11 achieved a major molecular response, 8 achieved a complete molecular response, and 9 were negative by flow cytometry.
The median duration of response was 8.8 months.
Of the 13 responders, 6 went on to receive an allogeneic stem cell transplant. Five of these patients are still alive, but 1 died from relapse.
The median overall survival was 10.7 months.
“These data suggest the tolerability and efficacy of inotuzumab ozogamicin and bosutinib in R/R Ph+ ALL,” Dr Jain said. “And we are looking forward to the next phase of this study.”
Dr Jain disclosed receiving research funding from Celgene, Verastem, BMS, Incyte, Pharmacyclics, ADC Therapeutics, Genentech, AbbVie, Pfizer, Astra Zeneca, Janssen, Cellectis, and Seattle Genetics. He disclosed membership on boards of directors/advisory committees for Verastem, Servier, Novimmune, Pharmacyclics, Novartis, ADC Therapeutics, AbbVie, Pfizer, Adaptive Biotechnologies, and Janssen. ![]()
*Data in the presentation differ from the abstract.
ATLANTA—A 2-drug combination has produced a high response rate in a small trial of patients with relapsed/refractory (R/R), Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL).
The combination, inotuzumab ozogamicin and bosutinib, produced an overall response rate of 81% in this ongoing, phase 1/2 trial.
Nitin Jain, MD, of the University of Texas MD Anderson Cancer Center in Houston, presented phase 1 results from the study at the 2017 ASH Annual Meeting (abstract 143*).
He reported results in 16 patients, 14 with R/R, Ph+ ALL and 2 with chronic myeloid leukemia in lymphoid blast phase.
The patients received inotuzumab ozogamicin at 0.8 mg/m2 on day 1, 0.5 mg/m2 on day 8, and 0.5 mg/m2 on day 15 of cycle 1. Patients who achieved a response received inotuzumab ozogamicin at 1 mg/m2 once every 4 weeks for subsequent cycles. Six cycles were planned.
Patients also received bosutinib at 300 mg, 400 mg, or 500 mg once a day for 4-week cycles. The median number of cycles was 2.5 (range, 1-8).
The maximum-tolerated dose has not been established, but there were 2 dose-limiting toxicities (DLTs). One DLT occurred with the 400 mg dose of bosutinib, and 1 occurred with the 500 mg dose. Both DLTs were grade 3 skin rash.
The investigators are continuing accrual with the 500 mg dose of bosutinib for the phase 2 portion of the trial, with 22 additional patients.
Response and survival
The overall response rate was 81% (n=13). This included a complete response (CR) in 8 patients, a CR with incomplete blood count recovery in 3 patients, and a CR with incomplete platelet recovery in 2 patients.
All responses occurred among the patients with ALL.
Twelve responders achieved complete cytogenetic remission, 11 achieved a major molecular response, 8 achieved a complete molecular response, and 9 were negative by flow cytometry.
The median duration of response was 8.8 months.
Of the 13 responders, 6 went on to receive an allogeneic stem cell transplant. Five of these patients are still alive, but 1 died from relapse.
The median overall survival was 10.7 months.
“These data suggest the tolerability and efficacy of inotuzumab ozogamicin and bosutinib in R/R Ph+ ALL,” Dr Jain said. “And we are looking forward to the next phase of this study.”
Dr Jain disclosed receiving research funding from Celgene, Verastem, BMS, Incyte, Pharmacyclics, ADC Therapeutics, Genentech, AbbVie, Pfizer, Astra Zeneca, Janssen, Cellectis, and Seattle Genetics. He disclosed membership on boards of directors/advisory committees for Verastem, Servier, Novimmune, Pharmacyclics, Novartis, ADC Therapeutics, AbbVie, Pfizer, Adaptive Biotechnologies, and Janssen. ![]()
*Data in the presentation differ from the abstract.
ATLANTA—A 2-drug combination has produced a high response rate in a small trial of patients with relapsed/refractory (R/R), Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL).
The combination, inotuzumab ozogamicin and bosutinib, produced an overall response rate of 81% in this ongoing, phase 1/2 trial.
Nitin Jain, MD, of the University of Texas MD Anderson Cancer Center in Houston, presented phase 1 results from the study at the 2017 ASH Annual Meeting (abstract 143*).
He reported results in 16 patients, 14 with R/R, Ph+ ALL and 2 with chronic myeloid leukemia in lymphoid blast phase.
The patients received inotuzumab ozogamicin at 0.8 mg/m2 on day 1, 0.5 mg/m2 on day 8, and 0.5 mg/m2 on day 15 of cycle 1. Patients who achieved a response received inotuzumab ozogamicin at 1 mg/m2 once every 4 weeks for subsequent cycles. Six cycles were planned.
Patients also received bosutinib at 300 mg, 400 mg, or 500 mg once a day for 4-week cycles. The median number of cycles was 2.5 (range, 1-8).
The maximum-tolerated dose has not been established, but there were 2 dose-limiting toxicities (DLTs). One DLT occurred with the 400 mg dose of bosutinib, and 1 occurred with the 500 mg dose. Both DLTs were grade 3 skin rash.
The investigators are continuing accrual with the 500 mg dose of bosutinib for the phase 2 portion of the trial, with 22 additional patients.
Response and survival
The overall response rate was 81% (n=13). This included a complete response (CR) in 8 patients, a CR with incomplete blood count recovery in 3 patients, and a CR with incomplete platelet recovery in 2 patients.
All responses occurred among the patients with ALL.
Twelve responders achieved complete cytogenetic remission, 11 achieved a major molecular response, 8 achieved a complete molecular response, and 9 were negative by flow cytometry.
The median duration of response was 8.8 months.
Of the 13 responders, 6 went on to receive an allogeneic stem cell transplant. Five of these patients are still alive, but 1 died from relapse.
The median overall survival was 10.7 months.
“These data suggest the tolerability and efficacy of inotuzumab ozogamicin and bosutinib in R/R Ph+ ALL,” Dr Jain said. “And we are looking forward to the next phase of this study.”
Dr Jain disclosed receiving research funding from Celgene, Verastem, BMS, Incyte, Pharmacyclics, ADC Therapeutics, Genentech, AbbVie, Pfizer, Astra Zeneca, Janssen, Cellectis, and Seattle Genetics. He disclosed membership on boards of directors/advisory committees for Verastem, Servier, Novimmune, Pharmacyclics, Novartis, ADC Therapeutics, AbbVie, Pfizer, Adaptive Biotechnologies, and Janssen. ![]()
*Data in the presentation differ from the abstract.
Metabolic/bariatric surgery reduces CVD risk in teens
Weight loss caused by metabolic and bariatric surgery (MBS) independently predicts the normalization of dyslipedemia, elevated blood pressure, hyperinsulinemia, diabetes, and elevated high-sensitivity C-reactive protein (hs-CRP) in severely obese adolescents, according to results of a longitudinal, multicenter prospective study.
In the study of 242 severely obese adolescents undergoing MBS between Feb. 28, 2007, and Dec. 30, 2011, Marc Michalsky, MD, of Nationwide Children’s Hospital, Columbus, Ohio, and his colleagues found that, with every 10% increase in weight loss, patients were 24%, 11%, 14%, 13%, and 19% more likely to resolve dyslipidemia, elevated blood pressure, hyperinsulinemia, diabetes, and elevated hs-CRP, respectively.
One of the most important facets of this study is the predictive nature of different patient risk factors on the future remission of cardiovascular disease symptoms.
For example, “the evidence suggests that better long-term outcomes may be anticipated among individuals undergoing MBS at lower BMI levels (i.e., less than 50),” they reported in the journal Pediatrics. “Increasing age at the time of MBS was associated with a reduced likelihood of dyslipidemia remission and normalization of hs-CRP,” which was true even in the narrow age range of this group of adolescents.
“The identification of specific predictors of CVD-RF [cardiovascular disease risk factors] normalization and/or remission on the basis of sex, race, preoperative BMI, and age at surgery may serve to improve future study design and insights regarding the optimization of treatment strategies,” wrote Dr. Michalsky and his colleagues. “Collectively, these data demonstrate a reduction in the risk for development of CVD in adulthood and offer additional, compelling support for MBS in adolescents.”
Dr. Inge has worked as a consultant for Standard Bariatrics, UpToDate, and Independent Medical Expert Consulting Services; all of these companies are unrelated to this research. John B. Dixon, PhD, has received support for his research through a National Health and Medical Research Council research fellowship. Anita Courcoulas, MD, has received grants from various health care groups and companies. All other authors had no relevant financial disclosures. The study was funded by a variety of institutional grants and the National Institutes of Health.
AGA Resource
GIs are uniquely positioned to lead a care team to help patients with obesity achieve a healthy weight. The AGA Obesity Practice Guide provides a comprehensive, multidisciplinary process to personalize innovative obesity care for safe and effective weight management.
SOURCE: Michalsky M et al. Pediatrics. 2018 Jan 8. doi: 10.1542/peds.2017-2485.
Weight loss caused by metabolic and bariatric surgery (MBS) independently predicts the normalization of dyslipedemia, elevated blood pressure, hyperinsulinemia, diabetes, and elevated high-sensitivity C-reactive protein (hs-CRP) in severely obese adolescents, according to results of a longitudinal, multicenter prospective study.
In the study of 242 severely obese adolescents undergoing MBS between Feb. 28, 2007, and Dec. 30, 2011, Marc Michalsky, MD, of Nationwide Children’s Hospital, Columbus, Ohio, and his colleagues found that, with every 10% increase in weight loss, patients were 24%, 11%, 14%, 13%, and 19% more likely to resolve dyslipidemia, elevated blood pressure, hyperinsulinemia, diabetes, and elevated hs-CRP, respectively.
One of the most important facets of this study is the predictive nature of different patient risk factors on the future remission of cardiovascular disease symptoms.
For example, “the evidence suggests that better long-term outcomes may be anticipated among individuals undergoing MBS at lower BMI levels (i.e., less than 50),” they reported in the journal Pediatrics. “Increasing age at the time of MBS was associated with a reduced likelihood of dyslipidemia remission and normalization of hs-CRP,” which was true even in the narrow age range of this group of adolescents.
“The identification of specific predictors of CVD-RF [cardiovascular disease risk factors] normalization and/or remission on the basis of sex, race, preoperative BMI, and age at surgery may serve to improve future study design and insights regarding the optimization of treatment strategies,” wrote Dr. Michalsky and his colleagues. “Collectively, these data demonstrate a reduction in the risk for development of CVD in adulthood and offer additional, compelling support for MBS in adolescents.”
Dr. Inge has worked as a consultant for Standard Bariatrics, UpToDate, and Independent Medical Expert Consulting Services; all of these companies are unrelated to this research. John B. Dixon, PhD, has received support for his research through a National Health and Medical Research Council research fellowship. Anita Courcoulas, MD, has received grants from various health care groups and companies. All other authors had no relevant financial disclosures. The study was funded by a variety of institutional grants and the National Institutes of Health.
AGA Resource
GIs are uniquely positioned to lead a care team to help patients with obesity achieve a healthy weight. The AGA Obesity Practice Guide provides a comprehensive, multidisciplinary process to personalize innovative obesity care for safe and effective weight management.
SOURCE: Michalsky M et al. Pediatrics. 2018 Jan 8. doi: 10.1542/peds.2017-2485.
Weight loss caused by metabolic and bariatric surgery (MBS) independently predicts the normalization of dyslipedemia, elevated blood pressure, hyperinsulinemia, diabetes, and elevated high-sensitivity C-reactive protein (hs-CRP) in severely obese adolescents, according to results of a longitudinal, multicenter prospective study.
In the study of 242 severely obese adolescents undergoing MBS between Feb. 28, 2007, and Dec. 30, 2011, Marc Michalsky, MD, of Nationwide Children’s Hospital, Columbus, Ohio, and his colleagues found that, with every 10% increase in weight loss, patients were 24%, 11%, 14%, 13%, and 19% more likely to resolve dyslipidemia, elevated blood pressure, hyperinsulinemia, diabetes, and elevated hs-CRP, respectively.
One of the most important facets of this study is the predictive nature of different patient risk factors on the future remission of cardiovascular disease symptoms.
For example, “the evidence suggests that better long-term outcomes may be anticipated among individuals undergoing MBS at lower BMI levels (i.e., less than 50),” they reported in the journal Pediatrics. “Increasing age at the time of MBS was associated with a reduced likelihood of dyslipidemia remission and normalization of hs-CRP,” which was true even in the narrow age range of this group of adolescents.
“The identification of specific predictors of CVD-RF [cardiovascular disease risk factors] normalization and/or remission on the basis of sex, race, preoperative BMI, and age at surgery may serve to improve future study design and insights regarding the optimization of treatment strategies,” wrote Dr. Michalsky and his colleagues. “Collectively, these data demonstrate a reduction in the risk for development of CVD in adulthood and offer additional, compelling support for MBS in adolescents.”
Dr. Inge has worked as a consultant for Standard Bariatrics, UpToDate, and Independent Medical Expert Consulting Services; all of these companies are unrelated to this research. John B. Dixon, PhD, has received support for his research through a National Health and Medical Research Council research fellowship. Anita Courcoulas, MD, has received grants from various health care groups and companies. All other authors had no relevant financial disclosures. The study was funded by a variety of institutional grants and the National Institutes of Health.
AGA Resource
GIs are uniquely positioned to lead a care team to help patients with obesity achieve a healthy weight. The AGA Obesity Practice Guide provides a comprehensive, multidisciplinary process to personalize innovative obesity care for safe and effective weight management.
SOURCE: Michalsky M et al. Pediatrics. 2018 Jan 8. doi: 10.1542/peds.2017-2485.
Metabolic and bariatric surgery reduces CVD risk in severely obese adolescents
Weight loss caused by metabolic and bariatric surgery (MBS) independently predicts the normalization of dyslipidemia, elevated blood pressure (EPB), hyperinsulinemia, diabetes, and elevated high-sensitivity C-reactive protein (hs-CRP) in severely obese adolescents, according to results of a longitudinal, multicenter prospective study.
In the study of 242 severely obese adolescents undergoing MBS between Feb. 28, 2007, and Dec. 30, 2011, Marc Michalsky, MD, of Nationwide Children’s Hospital, Columbus, Ohio, and his colleagues found that with every 10% increase in weight loss, patients were 24%, 11%, 14%, 13%, and 19% more likely to resolve dyslipidemia, EBP, hyperinsulinemia, diabetes, and elevated hs-CRP, respectively.
One of the most important facets of this study is the predictive nature of different patient risk factors on the future remission of cardiovascular disease symptoms.
For example, “the evidence suggests that better long-term outcomes may be anticipated among individuals undergoing MBS at lower BMI levels (i.e., less than 50),” they reported in the journal Pediatrics. “Increasing age at the time of MBS was associated with a reduced likelihood of dyslipidemia remission and normalization of hs-CRP,” which was true even in the narrow age range of this group of adolescents.
“The identification of specific predictors of CVD-RF [cardiovascular disease risk factors] normalization and/or remission on the basis of sex, race, preoperative BMI, and age at surgery may serve to improve future study design and insights regarding the optimization of treatment strategies,” wrote Dr. Michalsky and his colleagues. “Collectively, these data demonstrate a reduction in the risk for development of CVD in adulthood and offer additional, compelling support for MBS in adolescents.”
Dr. Inge has worked as a consultant for Standard Bariatrics, UpToDate, and Independent Medical Expert Consulting Services; all of these companies are unrelated to this research. John B. Dixon, PhD, has received support for his research through a National Health and Medical Research Council research fellowship. Anita Courcoulas, MD, has received grants from various health care groups and companies. All other authors had no relevant financial disclosures. The study was funded by a variety of institutional grants and the National Institutes of Health.
SOURCE: M Michalsky et al. Pediatrics. 2018 Jan 8. doi: 10.1542/peds.2017-2485.
Weight loss caused by metabolic and bariatric surgery (MBS) independently predicts the normalization of dyslipidemia, elevated blood pressure (EPB), hyperinsulinemia, diabetes, and elevated high-sensitivity C-reactive protein (hs-CRP) in severely obese adolescents, according to results of a longitudinal, multicenter prospective study.
In the study of 242 severely obese adolescents undergoing MBS between Feb. 28, 2007, and Dec. 30, 2011, Marc Michalsky, MD, of Nationwide Children’s Hospital, Columbus, Ohio, and his colleagues found that with every 10% increase in weight loss, patients were 24%, 11%, 14%, 13%, and 19% more likely to resolve dyslipidemia, EBP, hyperinsulinemia, diabetes, and elevated hs-CRP, respectively.
One of the most important facets of this study is the predictive nature of different patient risk factors on the future remission of cardiovascular disease symptoms.
For example, “the evidence suggests that better long-term outcomes may be anticipated among individuals undergoing MBS at lower BMI levels (i.e., less than 50),” they reported in the journal Pediatrics. “Increasing age at the time of MBS was associated with a reduced likelihood of dyslipidemia remission and normalization of hs-CRP,” which was true even in the narrow age range of this group of adolescents.
“The identification of specific predictors of CVD-RF [cardiovascular disease risk factors] normalization and/or remission on the basis of sex, race, preoperative BMI, and age at surgery may serve to improve future study design and insights regarding the optimization of treatment strategies,” wrote Dr. Michalsky and his colleagues. “Collectively, these data demonstrate a reduction in the risk for development of CVD in adulthood and offer additional, compelling support for MBS in adolescents.”
Dr. Inge has worked as a consultant for Standard Bariatrics, UpToDate, and Independent Medical Expert Consulting Services; all of these companies are unrelated to this research. John B. Dixon, PhD, has received support for his research through a National Health and Medical Research Council research fellowship. Anita Courcoulas, MD, has received grants from various health care groups and companies. All other authors had no relevant financial disclosures. The study was funded by a variety of institutional grants and the National Institutes of Health.
SOURCE: M Michalsky et al. Pediatrics. 2018 Jan 8. doi: 10.1542/peds.2017-2485.
Weight loss caused by metabolic and bariatric surgery (MBS) independently predicts the normalization of dyslipidemia, elevated blood pressure (EPB), hyperinsulinemia, diabetes, and elevated high-sensitivity C-reactive protein (hs-CRP) in severely obese adolescents, according to results of a longitudinal, multicenter prospective study.
In the study of 242 severely obese adolescents undergoing MBS between Feb. 28, 2007, and Dec. 30, 2011, Marc Michalsky, MD, of Nationwide Children’s Hospital, Columbus, Ohio, and his colleagues found that with every 10% increase in weight loss, patients were 24%, 11%, 14%, 13%, and 19% more likely to resolve dyslipidemia, EBP, hyperinsulinemia, diabetes, and elevated hs-CRP, respectively.
One of the most important facets of this study is the predictive nature of different patient risk factors on the future remission of cardiovascular disease symptoms.
For example, “the evidence suggests that better long-term outcomes may be anticipated among individuals undergoing MBS at lower BMI levels (i.e., less than 50),” they reported in the journal Pediatrics. “Increasing age at the time of MBS was associated with a reduced likelihood of dyslipidemia remission and normalization of hs-CRP,” which was true even in the narrow age range of this group of adolescents.
“The identification of specific predictors of CVD-RF [cardiovascular disease risk factors] normalization and/or remission on the basis of sex, race, preoperative BMI, and age at surgery may serve to improve future study design and insights regarding the optimization of treatment strategies,” wrote Dr. Michalsky and his colleagues. “Collectively, these data demonstrate a reduction in the risk for development of CVD in adulthood and offer additional, compelling support for MBS in adolescents.”
Dr. Inge has worked as a consultant for Standard Bariatrics, UpToDate, and Independent Medical Expert Consulting Services; all of these companies are unrelated to this research. John B. Dixon, PhD, has received support for his research through a National Health and Medical Research Council research fellowship. Anita Courcoulas, MD, has received grants from various health care groups and companies. All other authors had no relevant financial disclosures. The study was funded by a variety of institutional grants and the National Institutes of Health.
SOURCE: M Michalsky et al. Pediatrics. 2018 Jan 8. doi: 10.1542/peds.2017-2485.
FROM PEDIATRICS
Key clinical point:
Major finding: With every 10% increase in weight loss, patients were 24%, 11%, 14%, 13%, and 19% more likely to resolve dyslipidemia, elevated BP, hyperinsulinemia, diabetes and elevated high-sensitivity C-reactive protein, respectively.
Study details: This study was a longitudinal, multicenter prospective study of 242 severely obese adolescents undergoing metabolic and bariatric surgery between February 28, 2007 and December 30, 2011.
Disclosures: Dr. Inge has worked as a consultant for Standard Bariatrics, UpToDate, and Independent Medical Expert Consulting Services; all of these companies are unrelated to this research. John B. Dixon, PhD, has received support for his research through a National Health and Medical Research Council research fellowship. Anita Courcoulas, MD, has received grants from various healthcare groups and companies. All other authors had no relevant financial disclosures. The study was funded by a variety of institutional grants and the National Institutes of Health.
Source: M Michalsky et al. Pediatrics. 2018 Jan 8. doi: 10.1542/peds.2017-2485
Diagnosing Multiple Myeloma in Primary Care
IN THIS ARTICLE
- Presenting symptoms
- Diagnostic tests
- Differential diagnostic criteria
Multiple myeloma (MM) is a fatal, malignant neoplasm that originates in the plasma cells of bone marrow. A genetic mutation in the plasma cells creates myeloma cells, which replicate and produce monoclonal protein (M-protein). This accumulation of cells and abnormal protein can result in destruction and eventual marrow failure.1,2
MM’s insidious nature means it often goes undetected or misdiagnosed in its early stages; this delayed diagnosis can cause sequelae that limit quality of life. Furthermore, the five-year survival rate for myeloma varies by stage at which the disease is diagnosed: from 48% for distant (metastasized) myeloma to 71% for localized disease.3 It has also been noted that, in the past two decades, improvements in available treatment options and supportive care have contributed to a doubling of median survival time (from three years to six years).4 It is therefore paramount that providers be aware of MM and its signs to facilitate early diagnosis and treatment.
INCIDENCE AND EPIDEMIOLOGY
MM accounts for 1% of all cancers and about 10% of all hematologic malignancies.5 In 2017, the American Cancer Society estimated that more than 30,000 new cases of MM would be diagnosed in the United States.6 Additionally, MM was expected to cause more than 12,000 deaths last year.6
Median age at diagnosis is 69.3 In fact, 75% of men are older than 75 and 79% of women are older than 70 at diagnosis.1
Apart from age, other risk factors for MM have been identified but not fully explicated. For example, the disease is more common in men than in women (with men comprising two-thirds of new cases per year).3 MM is also two to three times more common in black than in white persons, making it the most common hematologic malignancy in this demographic group.3,7
The possibility of a genetic predisposition has also been studied. Several analyses have indicated an increased risk for MM in patients with a family history of the disease—as much as four times higher in those with an affected first-degree relative. This risk was further elevated in black compared with white patients (odds ratios, 17.4 and 1.5, respectively).7 However, many patients with MM have no relatives with this disorder.6,8
DISEASE PROGRESSION
Almost all patients who develop MM also experience an asymptomatic premalignant stage called monoclonal gammopathy of undetermined significance (MGUS). MGUS is present in 3% to 4% of the general population older than 50 and is often an incidental finding. This stage almost always precedes MM—but because it is asymptomatic, only 10% of individuals diagnosed with MM have a known history of MGUS.8
In some patients, an asymptomatic intermediate stage called smoldering multiple myeloma (SMM) can be identified. SMM progresses to MM at a rate of 10% per year for the first five years; the rate decreases to 3% per year over the following five years, and 1% per year after that.8
MM is not curable, but as noted, the survival rate is steadily increasing due to rapidly evolving treatment regimens. Discussion of treatment is outside the scope of this article, but early diagnosis can improve quality of life and clinical outcomes and prolong life expectancy.
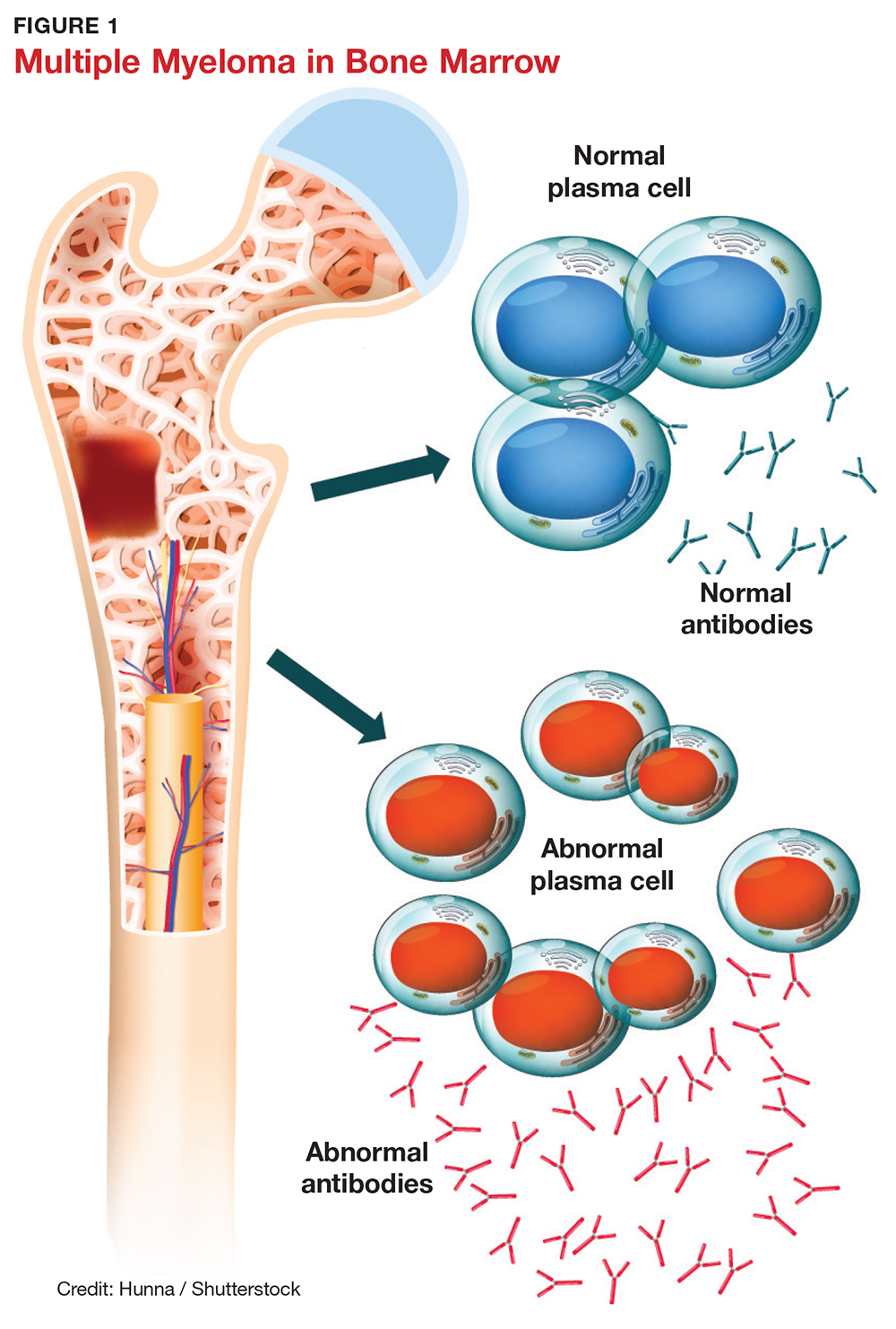
SYMPTOMS
The initial symptoms of MM can be nonspecific and may lead the provider to suspect a host of other conditions.2,6 (Those for advanced disease are also vague but tend to be more pronounced.) These may include fatigue, weakness, easy bruising or bleeding, and bone pain. Other common clinical manifestations of MM are anemia, chronic infection, bone disease, and/or renal failure.1,4 Patients may also experience loss of appetite, nausea, vomiting, increased thirst, and increased urination.9
Recent studies have shown that patients with SMM and/or MGUS also exhibit early signs of bone disease and increased risk for fracture.10 Eighty percent of patients who progress to MM have evidence of pathologic bone fractures.10 It is also possible for bones in the spine to weaken and collapse, pressing on the spinal nerves. This is known as spinal cord compression, which can manifest with sudden, severe back pain or numbness and/or muscle weakness (most often in the legs).6
MM must be included in the differential diagnosis, particularly when symptoms do not point to one specific disease process. Without early diagnosis, disease progression can result in complications such as bone fracture and osteoporosis, reduced kidney function, peripheral neuropathy, chronic anemia, and ultimately, death.2,6 The presence of bone fractures increases mortality risk by 20%.10
DIAGNOSTIC WORKUP
Evidence of MM may be discovered during routine bloodwork and screening tests, while presenting symptoms or subtle changes in lab results can raise suspicion for the disease. Initial bloodwork abnormalities include anemia, elevated calcium levels, renal insufficiency, and/or elevated protein levels.8
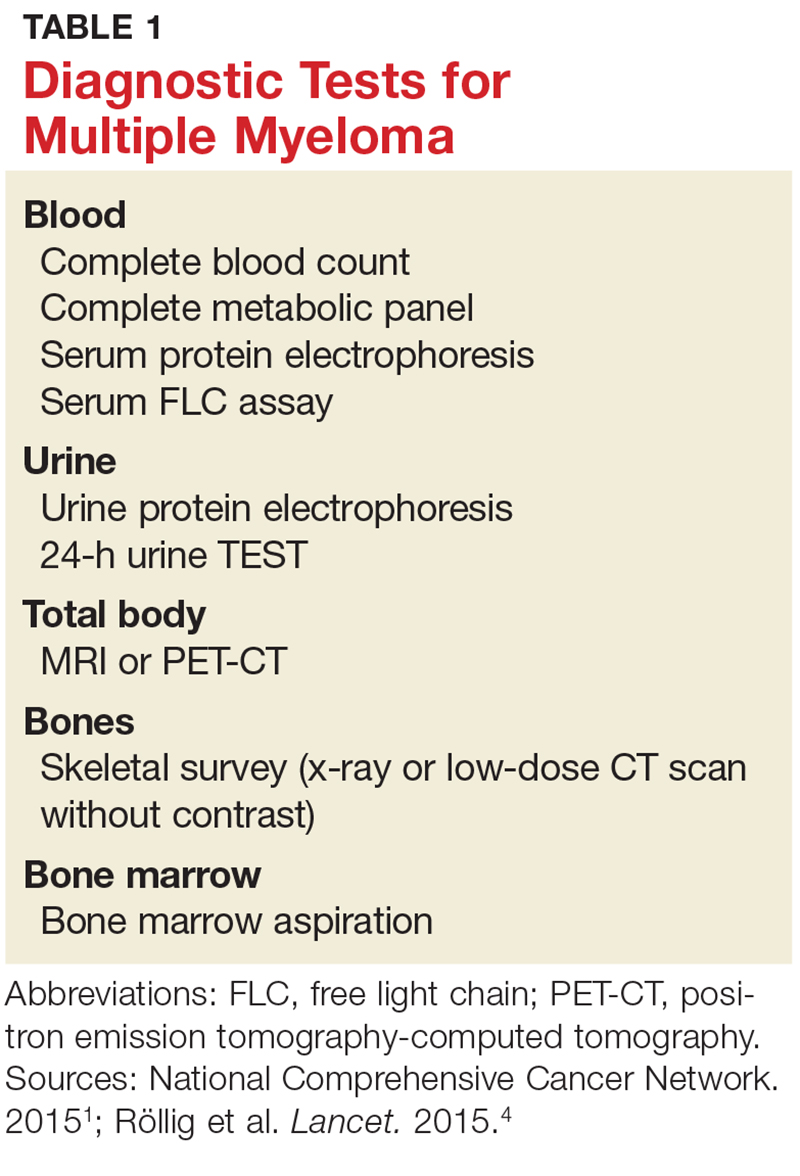
A combination of abnormalities in the complete blood count (CBC) and complete metabolic panel (CMP), along with symptoms, should alert the provider to the possibility of MM, prompting additional workup. Table 1 outlines suggested diagnostic tests; the possible findings are discussed below.
CBC. The CBC may reveal abnormalities including anemia (which occurs in 75% of patients with MM), thrombocytopenia, and leukopenia.1,8 These findings can contribute to fatigue, increased incidence of infection, and abnormal bruising of the skin.2,8
CMP. A CMP may show increases in serum calcium or protein. Hypercalcemia occurs in 15% of patients with MM, leading to symptoms such as loss of appetite, nausea, vomiting, increased urination, weakness, and confusion.8 An increase in protein may alter the albumin/globulin ratio, which should raise suspicion for MM. A decrease in albumin can signify disease severity. Also, the CMP may show worsening renal function and elevated serum creatinine, which occurs in 20% of patients with MM.8
Serum protein electrophoresis (SPEP). Suspicion of MM should prompt the clinician to evaluate proteins via SPEP. This test may be indicated for patients with anemia, hypercalcemia, bone pain, and unexplained neuropathy.9 The electrophoresis separates proteins based on their physical properties. This identifies the presence and amount of M-protein, which can determine the extent of the disease.1 M-protein is identified in approximately 82% of patients with MM using this test.8
Serum free light chain (FLC) assay. This diagnostic test can identify MM in individuals with high clinical suspicion for the disease but no discernible M-protein on SPEP; it increases sensitivity to 97%.8 The serum FLC assay evaluates for presence and ratio of free light chains—proteins produced by plasma cells. This test is also useful for monitoring treatment response and disease progression.1
Urine protein electrophoresis (UPEP). The UPEP separates proteins according to charge, which is helpful for classifying renal injury. Protein patterns are interpreted and may be reported as glomerular, tubular, or mixed. UPEP also tests for M-protein in the urine.1,11
24-hour urine. The 24-h urine test quantifies the amount and type of protein excreted in the urine and helps determine the extent of kidney disease.1
Skeletal survey. MM causes significant bone changes that can be identified with radiographic studies. The most common locations for fractures are the vertebral, pelvic, and clavicular areas.10 Currently, the skeletal survey is the gold standard for detecting fractures and osteolytic lesions associated with MM.10 Radiographic films ordered for other purposes may uncover abnormalities in bones.
Bone mineral density (BMD) test. Most often, BMD testing is used to evaluate treatment and progression of bone involvement. Because it can uncover osteopenia or osteoporosis, however, it can also be used to corroborate the diagnosis of MM.10
Once the presence of M-protein is identified, patients are referred for specialty care. At that time, further workup will include a bone marrow biopsy and imaging studies, such as additional radiographic films, CT scans (without contrast, as contrast dye can damage frail kidneys), and MRI.1,8 These diagnostic tests provide useful information for the classification of the disease and guide initiation of treatment.
CLASSIFICATION OF DISEASE
MM can be classified into three stages—MGUS, SMM, and MM—based on recommendations from the International Myeloma Working Group.12 Table 2 outlines the diagnostic criteria for each stage.
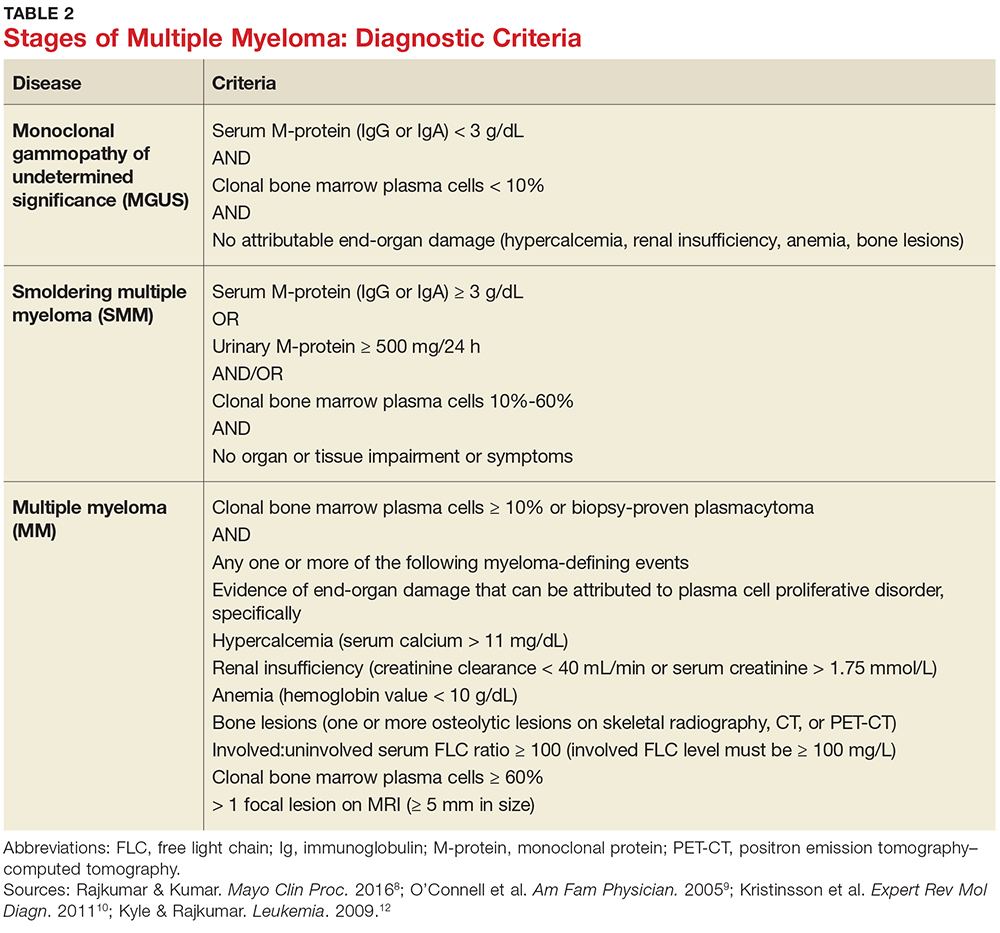
Individuals with MGUS and SMM are considered asymptomatic; guidelines do not recommend treatment for these patients. Those who are diagnosed with MM are referred to oncologists and treated based on current clinical practice guidelines.1
CONCLUSION
Multiple myeloma is a malignant neoplasm without a cure. Presenting symptoms may include anemia, bone pain, elevated creatinine or serum protein, fatigue, and hypercalcemia. Early diagnosis is key to early intervention and treatment, which can improve quality of life and clinical outcomes for those affected. Primary care providers play a major role in recognizing the subtle symptoms and ordering the appropriate diagnostic tests.
1. National Comprehensive Cancer Network. Multiple myeloma. NCCN clinical practice guidelines in oncology version 2.2015.
2. Rajkumar VS. Multiple myeloma symptoms, diagnosis, and staging. www.uptodate.com/contents/clinical-features-laboratory-manifestations-and-diagnosis-of-multiple-myeloma?source=machineLearning&search=multiple+myeloma&selectedTitle=1%7E150§ionRank=1&anchor=H25#H26. Accessed October 16, 2017.
3. National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: myeloma. https://seer.cancer.gov/statfacts/html/mulmy.html. Accessed October 26, 2017.
4. Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet. 2015;385(9983):2197-2208.
5. Moreau P, San Miguel J, Sonneveld M, et al. Multiple myeloma: ESMO clinical practice guidelines. Ann Oncol. 2017;28(4):iv52-iv61.
6. American Cancer Society. Multiple myeloma. www.cancer.org/cancer/multiplemyeloma/detailedguide. Accessed October 16, 2017.
7. Koura DT, Langston AA. Inherited predisposition to multiple myeloma. Ther Adv Hematol. 2013;4(4):291-297.
8. Rajkumar SV, Kumar S. Multiple myeloma: diagnosis and treatment. Mayo Clin Proc. 2016;91:101-119.
9. O’Connell T, Horita TJ, Kasravi B. Understanding and interpreting serum electrophoresis. Am Fam Physician. 2005; 71(1):105-112.
10. Kristinsson SY, Minter AR, Korde N, et al. Bone disease in multiple myeloma and precursor disease; novel diagnostic approaches and implications on clinical management. Expert Rev Mol Diagn. 2011;11(6):593-603.
11. Jacobs D, DeMott W, Oxley D. Laboratory Test Handbook: Concise With Disease Index. Hudson, OH: Lexi-Comp; 2004.
12. Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23(1):3-9.
IN THIS ARTICLE
- Presenting symptoms
- Diagnostic tests
- Differential diagnostic criteria
Multiple myeloma (MM) is a fatal, malignant neoplasm that originates in the plasma cells of bone marrow. A genetic mutation in the plasma cells creates myeloma cells, which replicate and produce monoclonal protein (M-protein). This accumulation of cells and abnormal protein can result in destruction and eventual marrow failure.1,2
MM’s insidious nature means it often goes undetected or misdiagnosed in its early stages; this delayed diagnosis can cause sequelae that limit quality of life. Furthermore, the five-year survival rate for myeloma varies by stage at which the disease is diagnosed: from 48% for distant (metastasized) myeloma to 71% for localized disease.3 It has also been noted that, in the past two decades, improvements in available treatment options and supportive care have contributed to a doubling of median survival time (from three years to six years).4 It is therefore paramount that providers be aware of MM and its signs to facilitate early diagnosis and treatment.
INCIDENCE AND EPIDEMIOLOGY
MM accounts for 1% of all cancers and about 10% of all hematologic malignancies.5 In 2017, the American Cancer Society estimated that more than 30,000 new cases of MM would be diagnosed in the United States.6 Additionally, MM was expected to cause more than 12,000 deaths last year.6
Median age at diagnosis is 69.3 In fact, 75% of men are older than 75 and 79% of women are older than 70 at diagnosis.1
Apart from age, other risk factors for MM have been identified but not fully explicated. For example, the disease is more common in men than in women (with men comprising two-thirds of new cases per year).3 MM is also two to three times more common in black than in white persons, making it the most common hematologic malignancy in this demographic group.3,7
The possibility of a genetic predisposition has also been studied. Several analyses have indicated an increased risk for MM in patients with a family history of the disease—as much as four times higher in those with an affected first-degree relative. This risk was further elevated in black compared with white patients (odds ratios, 17.4 and 1.5, respectively).7 However, many patients with MM have no relatives with this disorder.6,8
DISEASE PROGRESSION
Almost all patients who develop MM also experience an asymptomatic premalignant stage called monoclonal gammopathy of undetermined significance (MGUS). MGUS is present in 3% to 4% of the general population older than 50 and is often an incidental finding. This stage almost always precedes MM—but because it is asymptomatic, only 10% of individuals diagnosed with MM have a known history of MGUS.8
In some patients, an asymptomatic intermediate stage called smoldering multiple myeloma (SMM) can be identified. SMM progresses to MM at a rate of 10% per year for the first five years; the rate decreases to 3% per year over the following five years, and 1% per year after that.8
MM is not curable, but as noted, the survival rate is steadily increasing due to rapidly evolving treatment regimens. Discussion of treatment is outside the scope of this article, but early diagnosis can improve quality of life and clinical outcomes and prolong life expectancy.
SYMPTOMS
The initial symptoms of MM can be nonspecific and may lead the provider to suspect a host of other conditions.2,6 (Those for advanced disease are also vague but tend to be more pronounced.) These may include fatigue, weakness, easy bruising or bleeding, and bone pain. Other common clinical manifestations of MM are anemia, chronic infection, bone disease, and/or renal failure.1,4 Patients may also experience loss of appetite, nausea, vomiting, increased thirst, and increased urination.9
Recent studies have shown that patients with SMM and/or MGUS also exhibit early signs of bone disease and increased risk for fracture.10 Eighty percent of patients who progress to MM have evidence of pathologic bone fractures.10 It is also possible for bones in the spine to weaken and collapse, pressing on the spinal nerves. This is known as spinal cord compression, which can manifest with sudden, severe back pain or numbness and/or muscle weakness (most often in the legs).6
MM must be included in the differential diagnosis, particularly when symptoms do not point to one specific disease process. Without early diagnosis, disease progression can result in complications such as bone fracture and osteoporosis, reduced kidney function, peripheral neuropathy, chronic anemia, and ultimately, death.2,6 The presence of bone fractures increases mortality risk by 20%.10
DIAGNOSTIC WORKUP
Evidence of MM may be discovered during routine bloodwork and screening tests, while presenting symptoms or subtle changes in lab results can raise suspicion for the disease. Initial bloodwork abnormalities include anemia, elevated calcium levels, renal insufficiency, and/or elevated protein levels.8
A combination of abnormalities in the complete blood count (CBC) and complete metabolic panel (CMP), along with symptoms, should alert the provider to the possibility of MM, prompting additional workup. Table 1 outlines suggested diagnostic tests; the possible findings are discussed below.
CBC. The CBC may reveal abnormalities including anemia (which occurs in 75% of patients with MM), thrombocytopenia, and leukopenia.1,8 These findings can contribute to fatigue, increased incidence of infection, and abnormal bruising of the skin.2,8
CMP. A CMP may show increases in serum calcium or protein. Hypercalcemia occurs in 15% of patients with MM, leading to symptoms such as loss of appetite, nausea, vomiting, increased urination, weakness, and confusion.8 An increase in protein may alter the albumin/globulin ratio, which should raise suspicion for MM. A decrease in albumin can signify disease severity. Also, the CMP may show worsening renal function and elevated serum creatinine, which occurs in 20% of patients with MM.8
Serum protein electrophoresis (SPEP). Suspicion of MM should prompt the clinician to evaluate proteins via SPEP. This test may be indicated for patients with anemia, hypercalcemia, bone pain, and unexplained neuropathy.9 The electrophoresis separates proteins based on their physical properties. This identifies the presence and amount of M-protein, which can determine the extent of the disease.1 M-protein is identified in approximately 82% of patients with MM using this test.8
Serum free light chain (FLC) assay. This diagnostic test can identify MM in individuals with high clinical suspicion for the disease but no discernible M-protein on SPEP; it increases sensitivity to 97%.8 The serum FLC assay evaluates for presence and ratio of free light chains—proteins produced by plasma cells. This test is also useful for monitoring treatment response and disease progression.1
Urine protein electrophoresis (UPEP). The UPEP separates proteins according to charge, which is helpful for classifying renal injury. Protein patterns are interpreted and may be reported as glomerular, tubular, or mixed. UPEP also tests for M-protein in the urine.1,11
24-hour urine. The 24-h urine test quantifies the amount and type of protein excreted in the urine and helps determine the extent of kidney disease.1
Skeletal survey. MM causes significant bone changes that can be identified with radiographic studies. The most common locations for fractures are the vertebral, pelvic, and clavicular areas.10 Currently, the skeletal survey is the gold standard for detecting fractures and osteolytic lesions associated with MM.10 Radiographic films ordered for other purposes may uncover abnormalities in bones.
Bone mineral density (BMD) test. Most often, BMD testing is used to evaluate treatment and progression of bone involvement. Because it can uncover osteopenia or osteoporosis, however, it can also be used to corroborate the diagnosis of MM.10
Once the presence of M-protein is identified, patients are referred for specialty care. At that time, further workup will include a bone marrow biopsy and imaging studies, such as additional radiographic films, CT scans (without contrast, as contrast dye can damage frail kidneys), and MRI.1,8 These diagnostic tests provide useful information for the classification of the disease and guide initiation of treatment.
CLASSIFICATION OF DISEASE
MM can be classified into three stages—MGUS, SMM, and MM—based on recommendations from the International Myeloma Working Group.12 Table 2 outlines the diagnostic criteria for each stage.
Individuals with MGUS and SMM are considered asymptomatic; guidelines do not recommend treatment for these patients. Those who are diagnosed with MM are referred to oncologists and treated based on current clinical practice guidelines.1
CONCLUSION
Multiple myeloma is a malignant neoplasm without a cure. Presenting symptoms may include anemia, bone pain, elevated creatinine or serum protein, fatigue, and hypercalcemia. Early diagnosis is key to early intervention and treatment, which can improve quality of life and clinical outcomes for those affected. Primary care providers play a major role in recognizing the subtle symptoms and ordering the appropriate diagnostic tests.
IN THIS ARTICLE
- Presenting symptoms
- Diagnostic tests
- Differential diagnostic criteria
Multiple myeloma (MM) is a fatal, malignant neoplasm that originates in the plasma cells of bone marrow. A genetic mutation in the plasma cells creates myeloma cells, which replicate and produce monoclonal protein (M-protein). This accumulation of cells and abnormal protein can result in destruction and eventual marrow failure.1,2
MM’s insidious nature means it often goes undetected or misdiagnosed in its early stages; this delayed diagnosis can cause sequelae that limit quality of life. Furthermore, the five-year survival rate for myeloma varies by stage at which the disease is diagnosed: from 48% for distant (metastasized) myeloma to 71% for localized disease.3 It has also been noted that, in the past two decades, improvements in available treatment options and supportive care have contributed to a doubling of median survival time (from three years to six years).4 It is therefore paramount that providers be aware of MM and its signs to facilitate early diagnosis and treatment.
INCIDENCE AND EPIDEMIOLOGY
MM accounts for 1% of all cancers and about 10% of all hematologic malignancies.5 In 2017, the American Cancer Society estimated that more than 30,000 new cases of MM would be diagnosed in the United States.6 Additionally, MM was expected to cause more than 12,000 deaths last year.6
Median age at diagnosis is 69.3 In fact, 75% of men are older than 75 and 79% of women are older than 70 at diagnosis.1
Apart from age, other risk factors for MM have been identified but not fully explicated. For example, the disease is more common in men than in women (with men comprising two-thirds of new cases per year).3 MM is also two to three times more common in black than in white persons, making it the most common hematologic malignancy in this demographic group.3,7
The possibility of a genetic predisposition has also been studied. Several analyses have indicated an increased risk for MM in patients with a family history of the disease—as much as four times higher in those with an affected first-degree relative. This risk was further elevated in black compared with white patients (odds ratios, 17.4 and 1.5, respectively).7 However, many patients with MM have no relatives with this disorder.6,8
DISEASE PROGRESSION
Almost all patients who develop MM also experience an asymptomatic premalignant stage called monoclonal gammopathy of undetermined significance (MGUS). MGUS is present in 3% to 4% of the general population older than 50 and is often an incidental finding. This stage almost always precedes MM—but because it is asymptomatic, only 10% of individuals diagnosed with MM have a known history of MGUS.8
In some patients, an asymptomatic intermediate stage called smoldering multiple myeloma (SMM) can be identified. SMM progresses to MM at a rate of 10% per year for the first five years; the rate decreases to 3% per year over the following five years, and 1% per year after that.8
MM is not curable, but as noted, the survival rate is steadily increasing due to rapidly evolving treatment regimens. Discussion of treatment is outside the scope of this article, but early diagnosis can improve quality of life and clinical outcomes and prolong life expectancy.
SYMPTOMS
The initial symptoms of MM can be nonspecific and may lead the provider to suspect a host of other conditions.2,6 (Those for advanced disease are also vague but tend to be more pronounced.) These may include fatigue, weakness, easy bruising or bleeding, and bone pain. Other common clinical manifestations of MM are anemia, chronic infection, bone disease, and/or renal failure.1,4 Patients may also experience loss of appetite, nausea, vomiting, increased thirst, and increased urination.9
Recent studies have shown that patients with SMM and/or MGUS also exhibit early signs of bone disease and increased risk for fracture.10 Eighty percent of patients who progress to MM have evidence of pathologic bone fractures.10 It is also possible for bones in the spine to weaken and collapse, pressing on the spinal nerves. This is known as spinal cord compression, which can manifest with sudden, severe back pain or numbness and/or muscle weakness (most often in the legs).6
MM must be included in the differential diagnosis, particularly when symptoms do not point to one specific disease process. Without early diagnosis, disease progression can result in complications such as bone fracture and osteoporosis, reduced kidney function, peripheral neuropathy, chronic anemia, and ultimately, death.2,6 The presence of bone fractures increases mortality risk by 20%.10
DIAGNOSTIC WORKUP
Evidence of MM may be discovered during routine bloodwork and screening tests, while presenting symptoms or subtle changes in lab results can raise suspicion for the disease. Initial bloodwork abnormalities include anemia, elevated calcium levels, renal insufficiency, and/or elevated protein levels.8
A combination of abnormalities in the complete blood count (CBC) and complete metabolic panel (CMP), along with symptoms, should alert the provider to the possibility of MM, prompting additional workup. Table 1 outlines suggested diagnostic tests; the possible findings are discussed below.
CBC. The CBC may reveal abnormalities including anemia (which occurs in 75% of patients with MM), thrombocytopenia, and leukopenia.1,8 These findings can contribute to fatigue, increased incidence of infection, and abnormal bruising of the skin.2,8
CMP. A CMP may show increases in serum calcium or protein. Hypercalcemia occurs in 15% of patients with MM, leading to symptoms such as loss of appetite, nausea, vomiting, increased urination, weakness, and confusion.8 An increase in protein may alter the albumin/globulin ratio, which should raise suspicion for MM. A decrease in albumin can signify disease severity. Also, the CMP may show worsening renal function and elevated serum creatinine, which occurs in 20% of patients with MM.8
Serum protein electrophoresis (SPEP). Suspicion of MM should prompt the clinician to evaluate proteins via SPEP. This test may be indicated for patients with anemia, hypercalcemia, bone pain, and unexplained neuropathy.9 The electrophoresis separates proteins based on their physical properties. This identifies the presence and amount of M-protein, which can determine the extent of the disease.1 M-protein is identified in approximately 82% of patients with MM using this test.8
Serum free light chain (FLC) assay. This diagnostic test can identify MM in individuals with high clinical suspicion for the disease but no discernible M-protein on SPEP; it increases sensitivity to 97%.8 The serum FLC assay evaluates for presence and ratio of free light chains—proteins produced by plasma cells. This test is also useful for monitoring treatment response and disease progression.1
Urine protein electrophoresis (UPEP). The UPEP separates proteins according to charge, which is helpful for classifying renal injury. Protein patterns are interpreted and may be reported as glomerular, tubular, or mixed. UPEP also tests for M-protein in the urine.1,11
24-hour urine. The 24-h urine test quantifies the amount and type of protein excreted in the urine and helps determine the extent of kidney disease.1
Skeletal survey. MM causes significant bone changes that can be identified with radiographic studies. The most common locations for fractures are the vertebral, pelvic, and clavicular areas.10 Currently, the skeletal survey is the gold standard for detecting fractures and osteolytic lesions associated with MM.10 Radiographic films ordered for other purposes may uncover abnormalities in bones.
Bone mineral density (BMD) test. Most often, BMD testing is used to evaluate treatment and progression of bone involvement. Because it can uncover osteopenia or osteoporosis, however, it can also be used to corroborate the diagnosis of MM.10
Once the presence of M-protein is identified, patients are referred for specialty care. At that time, further workup will include a bone marrow biopsy and imaging studies, such as additional radiographic films, CT scans (without contrast, as contrast dye can damage frail kidneys), and MRI.1,8 These diagnostic tests provide useful information for the classification of the disease and guide initiation of treatment.
CLASSIFICATION OF DISEASE
MM can be classified into three stages—MGUS, SMM, and MM—based on recommendations from the International Myeloma Working Group.12 Table 2 outlines the diagnostic criteria for each stage.
Individuals with MGUS and SMM are considered asymptomatic; guidelines do not recommend treatment for these patients. Those who are diagnosed with MM are referred to oncologists and treated based on current clinical practice guidelines.1
CONCLUSION
Multiple myeloma is a malignant neoplasm without a cure. Presenting symptoms may include anemia, bone pain, elevated creatinine or serum protein, fatigue, and hypercalcemia. Early diagnosis is key to early intervention and treatment, which can improve quality of life and clinical outcomes for those affected. Primary care providers play a major role in recognizing the subtle symptoms and ordering the appropriate diagnostic tests.
1. National Comprehensive Cancer Network. Multiple myeloma. NCCN clinical practice guidelines in oncology version 2.2015.
2. Rajkumar VS. Multiple myeloma symptoms, diagnosis, and staging. www.uptodate.com/contents/clinical-features-laboratory-manifestations-and-diagnosis-of-multiple-myeloma?source=machineLearning&search=multiple+myeloma&selectedTitle=1%7E150§ionRank=1&anchor=H25#H26. Accessed October 16, 2017.
3. National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: myeloma. https://seer.cancer.gov/statfacts/html/mulmy.html. Accessed October 26, 2017.
4. Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet. 2015;385(9983):2197-2208.
5. Moreau P, San Miguel J, Sonneveld M, et al. Multiple myeloma: ESMO clinical practice guidelines. Ann Oncol. 2017;28(4):iv52-iv61.
6. American Cancer Society. Multiple myeloma. www.cancer.org/cancer/multiplemyeloma/detailedguide. Accessed October 16, 2017.
7. Koura DT, Langston AA. Inherited predisposition to multiple myeloma. Ther Adv Hematol. 2013;4(4):291-297.
8. Rajkumar SV, Kumar S. Multiple myeloma: diagnosis and treatment. Mayo Clin Proc. 2016;91:101-119.
9. O’Connell T, Horita TJ, Kasravi B. Understanding and interpreting serum electrophoresis. Am Fam Physician. 2005; 71(1):105-112.
10. Kristinsson SY, Minter AR, Korde N, et al. Bone disease in multiple myeloma and precursor disease; novel diagnostic approaches and implications on clinical management. Expert Rev Mol Diagn. 2011;11(6):593-603.
11. Jacobs D, DeMott W, Oxley D. Laboratory Test Handbook: Concise With Disease Index. Hudson, OH: Lexi-Comp; 2004.
12. Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23(1):3-9.
1. National Comprehensive Cancer Network. Multiple myeloma. NCCN clinical practice guidelines in oncology version 2.2015.
2. Rajkumar VS. Multiple myeloma symptoms, diagnosis, and staging. www.uptodate.com/contents/clinical-features-laboratory-manifestations-and-diagnosis-of-multiple-myeloma?source=machineLearning&search=multiple+myeloma&selectedTitle=1%7E150§ionRank=1&anchor=H25#H26. Accessed October 16, 2017.
3. National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: myeloma. https://seer.cancer.gov/statfacts/html/mulmy.html. Accessed October 26, 2017.
4. Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet. 2015;385(9983):2197-2208.
5. Moreau P, San Miguel J, Sonneveld M, et al. Multiple myeloma: ESMO clinical practice guidelines. Ann Oncol. 2017;28(4):iv52-iv61.
6. American Cancer Society. Multiple myeloma. www.cancer.org/cancer/multiplemyeloma/detailedguide. Accessed October 16, 2017.
7. Koura DT, Langston AA. Inherited predisposition to multiple myeloma. Ther Adv Hematol. 2013;4(4):291-297.
8. Rajkumar SV, Kumar S. Multiple myeloma: diagnosis and treatment. Mayo Clin Proc. 2016;91:101-119.
9. O’Connell T, Horita TJ, Kasravi B. Understanding and interpreting serum electrophoresis. Am Fam Physician. 2005; 71(1):105-112.
10. Kristinsson SY, Minter AR, Korde N, et al. Bone disease in multiple myeloma and precursor disease; novel diagnostic approaches and implications on clinical management. Expert Rev Mol Diagn. 2011;11(6):593-603.
11. Jacobs D, DeMott W, Oxley D. Laboratory Test Handbook: Concise With Disease Index. Hudson, OH: Lexi-Comp; 2004.
12. Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23(1):3-9.
Vascular Risk: What’s Really Important?
This video was filmed at Metabolic & Endocrine Disease Summit (MEDS). Click here to learn more.
This video was filmed at Metabolic & Endocrine Disease Summit (MEDS). Click here to learn more.
This video was filmed at Metabolic & Endocrine Disease Summit (MEDS). Click here to learn more.

Antiviral receives breakthrough designation for CMV

The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to maribavir (SHP620) as a treatment for cytomegalovirus (CMV) infection and disease in transplant recipients who are resistant or refractory to prior therapy.
Maribavir, an antiviral therapy that belongs to a class of drugs called benzimidazole ribosides, is being evaluated in patients who have CMV infection after undergoing hematopoietic stem cell transplant or solid organ transplant.
The drug inhibits the CMV UL97 protein kinase and is thought to affect several critical processes in CMV replication, including viral DNA synthesis, viral gene expression, encapsidation, and egress of mature capsids from the nucleus.
The FDA granted maribavir breakthrough designation based on data from two phase 2 studies. For one of these studies (NCT00223925), data are not yet available.
The other study (NCT01611974) was presented at IDWeek 2016. This study included 120 patients ages 12 and older with CMV infection (≥1000 DNA copies/mL of blood plasma) that was resistant or refractory to (val)ganciclovir or foscarnet.
Forty-seven of the patients had received a hematopoietic stem cell transplant, and 73 had a solid organ transplant.
The patients were randomized to 1 of 3 twice-daily oral doses of maribavir—400 mg, 800 mg, or 1200 mg—for up to 24 weeks of treatment.
The study’s primary efficacy endpoint was the proportion of patients with confirmed undetectable plasma CMV DNA within 6 weeks of treatment. Sixty-seven percent (80/120) of patients met this endpoint. This included 70% (n=28) of patients in the 400 mg group, 63% (n=25) in the 800 mg group, and 67% (n=27) in the 1200 mg group.
CMV infection recurred in 30 patients, including 7 in the 400 mg group, 11 in the 800 mg group, and 12 in the 1200 mg group.
The incidence of treatment-emergent adverse events (AEs) was 78% (n=93) overall, 78% (n=31) in the 400 mg group, 80% (n=32) in the 800 mg group, and 75% (n=30) in the 1200 mg group.
Twenty-seven percent of patients died due to any AE, 1 of which (multi-organ failure) was considered possibly related to maribavir.
Forty-one patients (34%) discontinued treatment with maribavir due to an AE, including 17 patients who discontinued due to CMV infection.
Dysgeusia was the most common treatment-emergent AE and led to treatment discontinuation in 1 patient. Dysgeusia occurred in 65% (n=78) of all patients, including 60% (n=24) in the 400 mg group, 63% (n=25) in the 800 mg group, and 73% (n=29) in the 1200 mg group.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to maribavir (SHP620) as a treatment for cytomegalovirus (CMV) infection and disease in transplant recipients who are resistant or refractory to prior therapy.
Maribavir, an antiviral therapy that belongs to a class of drugs called benzimidazole ribosides, is being evaluated in patients who have CMV infection after undergoing hematopoietic stem cell transplant or solid organ transplant.
The drug inhibits the CMV UL97 protein kinase and is thought to affect several critical processes in CMV replication, including viral DNA synthesis, viral gene expression, encapsidation, and egress of mature capsids from the nucleus.
The FDA granted maribavir breakthrough designation based on data from two phase 2 studies. For one of these studies (NCT00223925), data are not yet available.
The other study (NCT01611974) was presented at IDWeek 2016. This study included 120 patients ages 12 and older with CMV infection (≥1000 DNA copies/mL of blood plasma) that was resistant or refractory to (val)ganciclovir or foscarnet.
Forty-seven of the patients had received a hematopoietic stem cell transplant, and 73 had a solid organ transplant.
The patients were randomized to 1 of 3 twice-daily oral doses of maribavir—400 mg, 800 mg, or 1200 mg—for up to 24 weeks of treatment.
The study’s primary efficacy endpoint was the proportion of patients with confirmed undetectable plasma CMV DNA within 6 weeks of treatment. Sixty-seven percent (80/120) of patients met this endpoint. This included 70% (n=28) of patients in the 400 mg group, 63% (n=25) in the 800 mg group, and 67% (n=27) in the 1200 mg group.
CMV infection recurred in 30 patients, including 7 in the 400 mg group, 11 in the 800 mg group, and 12 in the 1200 mg group.
The incidence of treatment-emergent adverse events (AEs) was 78% (n=93) overall, 78% (n=31) in the 400 mg group, 80% (n=32) in the 800 mg group, and 75% (n=30) in the 1200 mg group.
Twenty-seven percent of patients died due to any AE, 1 of which (multi-organ failure) was considered possibly related to maribavir.
Forty-one patients (34%) discontinued treatment with maribavir due to an AE, including 17 patients who discontinued due to CMV infection.
Dysgeusia was the most common treatment-emergent AE and led to treatment discontinuation in 1 patient. Dysgeusia occurred in 65% (n=78) of all patients, including 60% (n=24) in the 400 mg group, 63% (n=25) in the 800 mg group, and 73% (n=29) in the 1200 mg group.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to maribavir (SHP620) as a treatment for cytomegalovirus (CMV) infection and disease in transplant recipients who are resistant or refractory to prior therapy.
Maribavir, an antiviral therapy that belongs to a class of drugs called benzimidazole ribosides, is being evaluated in patients who have CMV infection after undergoing hematopoietic stem cell transplant or solid organ transplant.
The drug inhibits the CMV UL97 protein kinase and is thought to affect several critical processes in CMV replication, including viral DNA synthesis, viral gene expression, encapsidation, and egress of mature capsids from the nucleus.
The FDA granted maribavir breakthrough designation based on data from two phase 2 studies. For one of these studies (NCT00223925), data are not yet available.
The other study (NCT01611974) was presented at IDWeek 2016. This study included 120 patients ages 12 and older with CMV infection (≥1000 DNA copies/mL of blood plasma) that was resistant or refractory to (val)ganciclovir or foscarnet.
Forty-seven of the patients had received a hematopoietic stem cell transplant, and 73 had a solid organ transplant.
The patients were randomized to 1 of 3 twice-daily oral doses of maribavir—400 mg, 800 mg, or 1200 mg—for up to 24 weeks of treatment.
The study’s primary efficacy endpoint was the proportion of patients with confirmed undetectable plasma CMV DNA within 6 weeks of treatment. Sixty-seven percent (80/120) of patients met this endpoint. This included 70% (n=28) of patients in the 400 mg group, 63% (n=25) in the 800 mg group, and 67% (n=27) in the 1200 mg group.
CMV infection recurred in 30 patients, including 7 in the 400 mg group, 11 in the 800 mg group, and 12 in the 1200 mg group.
The incidence of treatment-emergent adverse events (AEs) was 78% (n=93) overall, 78% (n=31) in the 400 mg group, 80% (n=32) in the 800 mg group, and 75% (n=30) in the 1200 mg group.
Twenty-seven percent of patients died due to any AE, 1 of which (multi-organ failure) was considered possibly related to maribavir.
Forty-one patients (34%) discontinued treatment with maribavir due to an AE, including 17 patients who discontinued due to CMV infection.
Dysgeusia was the most common treatment-emergent AE and led to treatment discontinuation in 1 patient. Dysgeusia occurred in 65% (n=78) of all patients, including 60% (n=24) in the 400 mg group, 63% (n=25) in the 800 mg group, and 73% (n=29) in the 1200 mg group.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need. ![]()
FDA approves denosumab for MM patients
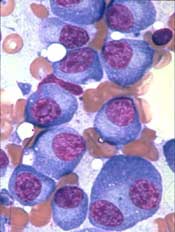
The US Food and Drug Administration (FDA) has approved denosumab (XGEVA®) for use in patients with multiple myeloma (MM).
The drug was previously approved to prevent skeletal-related events in patients with bone metastases from solid tumors.
Now, denosumab is FDA-approved to prevent skeletal-related events in MM patients as well.
Denosumab is a fully human monoclonal antibody that binds to and neutralizes RANK ligand—a protein essential for the formation, function, and survival of osteoclasts—thereby inhibiting osteoclast-mediated bone destruction.
The FDA’s approval of denosumab in MM is based on data from the phase 3 '482 study, which were presented at the 2017 ASCO Annual Meeting last June.
In this trial, researchers compared denosumab to zoledronic acid for the prevention of skeletal-related events in 1718 adults with newly diagnosed MM and bone disease.
Patients were randomized to receive either subcutaneous denosumab at 120 mg and intravenous placebo every 4 weeks (n=859) or intravenous zoledronic acid at 4 mg (adjusted for renal function) and subcutaneous placebo every 4 weeks (n=859).
Denosumab proved non-inferior to zoledronic acid in delaying the time to first on-study skeletal-related event (pathologic fracture, radiation to bone, surgery to bone, or spinal cord compression). The hazard ratio (HR) was 0.98 (95% CI: 0.85, 1.14; P=0.01).
Denosumab was not superior to zoledronic acid in delaying the time to a first skeletal-related event or delaying the time to first-and-subsequent skeletal-related events.
Overall survival was comparable between the treatment arms. The HR was 0.90 (95% CI: 0.70, 1.16; P=0.41).
The median difference in progression-free survival favored denosumab by 10.7 months (HR=0.82, 95% CI: 0.68-0.99; descriptive P=0.036). The median progression-free survival was 46.1 months for denosumab and 35.4 months for zoledronic acid.
The most common adverse events in patients who received denosumab were diarrhea (34%), nausea (32%), anemia (22%), back pain (21%), thrombocytopenia (19%), peripheral edema (17%), hypocalcemia (16%), upper respiratory tract infection (15%), rash (14%) and headache (11%).
The most common adverse event resulting in discontinuation of denosumab was osteonecrosis of the jaw.
In the primary treatment phase of the study, osteonecrosis of the jaw was confirmed in 4.1% of patients in the denosumab arm (median exposure of 16 months; range, 1-50) and 2.8% of those in the zoledronic acid arm (median 15 months; range, 1-45 months). ![]()
The US Food and Drug Administration (FDA) has approved denosumab (XGEVA®) for use in patients with multiple myeloma (MM).
The drug was previously approved to prevent skeletal-related events in patients with bone metastases from solid tumors.
Now, denosumab is FDA-approved to prevent skeletal-related events in MM patients as well.
Denosumab is a fully human monoclonal antibody that binds to and neutralizes RANK ligand—a protein essential for the formation, function, and survival of osteoclasts—thereby inhibiting osteoclast-mediated bone destruction.
The FDA’s approval of denosumab in MM is based on data from the phase 3 '482 study, which were presented at the 2017 ASCO Annual Meeting last June.
In this trial, researchers compared denosumab to zoledronic acid for the prevention of skeletal-related events in 1718 adults with newly diagnosed MM and bone disease.
Patients were randomized to receive either subcutaneous denosumab at 120 mg and intravenous placebo every 4 weeks (n=859) or intravenous zoledronic acid at 4 mg (adjusted for renal function) and subcutaneous placebo every 4 weeks (n=859).
Denosumab proved non-inferior to zoledronic acid in delaying the time to first on-study skeletal-related event (pathologic fracture, radiation to bone, surgery to bone, or spinal cord compression). The hazard ratio (HR) was 0.98 (95% CI: 0.85, 1.14; P=0.01).
Denosumab was not superior to zoledronic acid in delaying the time to a first skeletal-related event or delaying the time to first-and-subsequent skeletal-related events.
Overall survival was comparable between the treatment arms. The HR was 0.90 (95% CI: 0.70, 1.16; P=0.41).
The median difference in progression-free survival favored denosumab by 10.7 months (HR=0.82, 95% CI: 0.68-0.99; descriptive P=0.036). The median progression-free survival was 46.1 months for denosumab and 35.4 months for zoledronic acid.
The most common adverse events in patients who received denosumab were diarrhea (34%), nausea (32%), anemia (22%), back pain (21%), thrombocytopenia (19%), peripheral edema (17%), hypocalcemia (16%), upper respiratory tract infection (15%), rash (14%) and headache (11%).
The most common adverse event resulting in discontinuation of denosumab was osteonecrosis of the jaw.
In the primary treatment phase of the study, osteonecrosis of the jaw was confirmed in 4.1% of patients in the denosumab arm (median exposure of 16 months; range, 1-50) and 2.8% of those in the zoledronic acid arm (median 15 months; range, 1-45 months). ![]()
The US Food and Drug Administration (FDA) has approved denosumab (XGEVA®) for use in patients with multiple myeloma (MM).
The drug was previously approved to prevent skeletal-related events in patients with bone metastases from solid tumors.
Now, denosumab is FDA-approved to prevent skeletal-related events in MM patients as well.
Denosumab is a fully human monoclonal antibody that binds to and neutralizes RANK ligand—a protein essential for the formation, function, and survival of osteoclasts—thereby inhibiting osteoclast-mediated bone destruction.
The FDA’s approval of denosumab in MM is based on data from the phase 3 '482 study, which were presented at the 2017 ASCO Annual Meeting last June.
In this trial, researchers compared denosumab to zoledronic acid for the prevention of skeletal-related events in 1718 adults with newly diagnosed MM and bone disease.
Patients were randomized to receive either subcutaneous denosumab at 120 mg and intravenous placebo every 4 weeks (n=859) or intravenous zoledronic acid at 4 mg (adjusted for renal function) and subcutaneous placebo every 4 weeks (n=859).
Denosumab proved non-inferior to zoledronic acid in delaying the time to first on-study skeletal-related event (pathologic fracture, radiation to bone, surgery to bone, or spinal cord compression). The hazard ratio (HR) was 0.98 (95% CI: 0.85, 1.14; P=0.01).
Denosumab was not superior to zoledronic acid in delaying the time to a first skeletal-related event or delaying the time to first-and-subsequent skeletal-related events.
Overall survival was comparable between the treatment arms. The HR was 0.90 (95% CI: 0.70, 1.16; P=0.41).
The median difference in progression-free survival favored denosumab by 10.7 months (HR=0.82, 95% CI: 0.68-0.99; descriptive P=0.036). The median progression-free survival was 46.1 months for denosumab and 35.4 months for zoledronic acid.
The most common adverse events in patients who received denosumab were diarrhea (34%), nausea (32%), anemia (22%), back pain (21%), thrombocytopenia (19%), peripheral edema (17%), hypocalcemia (16%), upper respiratory tract infection (15%), rash (14%) and headache (11%).
The most common adverse event resulting in discontinuation of denosumab was osteonecrosis of the jaw.
In the primary treatment phase of the study, osteonecrosis of the jaw was confirmed in 4.1% of patients in the denosumab arm (median exposure of 16 months; range, 1-50) and 2.8% of those in the zoledronic acid arm (median 15 months; range, 1-45 months). ![]()
Rituximab tackles relapse of severe, difficult-to-treat pemphigus
, based on data from a case series of 11 patients.
“We found that treatment with rituximab alone, even at a low dose, not only prevented relapse but also maintained complete remission with a better benefit-to-risk ratio than treatment with corticosteroids,” Julia Sanchez, MD, of Reims (France) University Hospital and her colleagues reported in a research letter in JAMA Dermatology.
The study population consisted of patients diagnosed with pemphigus at a single center from Jan. 1, 2014, to Dec. 31, 2014, and treated with at least one cycle of rituximab for corticosteroid dependence, corticosteroid resistance, or adverse events. All the patients were in remission at the time of the first maintenance dose of rituximab.
All patients received a 1-g rituximab infusion every 6 months for 24-67 months; some patients changed to a once-yearly dose after 18 months. Although 5 patients experienced grade 3 or 4 adverse events (1 patient had sepsis; 2, diabetes; 1, hypertension; and 2, endocrine disorders) between the initial therapy cycle and the first rituximab maintenance infusion, no adverse events were reported by any of the 11 patients during the maintenance therapy period.
All 11 patients remained in remission after their last follow-up visit (an average of 78 months after the first cycle), at which point 10 patients had discontinued the therapy.
“A progressive decrease in serum anti-desmoglein autoantibody levels to less than 14 U/mL occurred in all cases along with clinical complete remission even after maintenance therapy cessation,” Dr. Sanchez and her associates noted.
Future research should address questions including the optimal dose and dosing frequency of rituximab, as well as the cost-effectiveness of the treatment and criteria for treatment withdrawal, they said.
The researchers had no relevant financial conflicts disclosures.
SOURCE: JAMA Dermatol. 2017 Jan 3. doi: 10.1001/jamadermatol.2017.5176.
, based on data from a case series of 11 patients.
“We found that treatment with rituximab alone, even at a low dose, not only prevented relapse but also maintained complete remission with a better benefit-to-risk ratio than treatment with corticosteroids,” Julia Sanchez, MD, of Reims (France) University Hospital and her colleagues reported in a research letter in JAMA Dermatology.
The study population consisted of patients diagnosed with pemphigus at a single center from Jan. 1, 2014, to Dec. 31, 2014, and treated with at least one cycle of rituximab for corticosteroid dependence, corticosteroid resistance, or adverse events. All the patients were in remission at the time of the first maintenance dose of rituximab.
All patients received a 1-g rituximab infusion every 6 months for 24-67 months; some patients changed to a once-yearly dose after 18 months. Although 5 patients experienced grade 3 or 4 adverse events (1 patient had sepsis; 2, diabetes; 1, hypertension; and 2, endocrine disorders) between the initial therapy cycle and the first rituximab maintenance infusion, no adverse events were reported by any of the 11 patients during the maintenance therapy period.
All 11 patients remained in remission after their last follow-up visit (an average of 78 months after the first cycle), at which point 10 patients had discontinued the therapy.
“A progressive decrease in serum anti-desmoglein autoantibody levels to less than 14 U/mL occurred in all cases along with clinical complete remission even after maintenance therapy cessation,” Dr. Sanchez and her associates noted.
Future research should address questions including the optimal dose and dosing frequency of rituximab, as well as the cost-effectiveness of the treatment and criteria for treatment withdrawal, they said.
The researchers had no relevant financial conflicts disclosures.
SOURCE: JAMA Dermatol. 2017 Jan 3. doi: 10.1001/jamadermatol.2017.5176.
, based on data from a case series of 11 patients.
“We found that treatment with rituximab alone, even at a low dose, not only prevented relapse but also maintained complete remission with a better benefit-to-risk ratio than treatment with corticosteroids,” Julia Sanchez, MD, of Reims (France) University Hospital and her colleagues reported in a research letter in JAMA Dermatology.
The study population consisted of patients diagnosed with pemphigus at a single center from Jan. 1, 2014, to Dec. 31, 2014, and treated with at least one cycle of rituximab for corticosteroid dependence, corticosteroid resistance, or adverse events. All the patients were in remission at the time of the first maintenance dose of rituximab.
All patients received a 1-g rituximab infusion every 6 months for 24-67 months; some patients changed to a once-yearly dose after 18 months. Although 5 patients experienced grade 3 or 4 adverse events (1 patient had sepsis; 2, diabetes; 1, hypertension; and 2, endocrine disorders) between the initial therapy cycle and the first rituximab maintenance infusion, no adverse events were reported by any of the 11 patients during the maintenance therapy period.
All 11 patients remained in remission after their last follow-up visit (an average of 78 months after the first cycle), at which point 10 patients had discontinued the therapy.
“A progressive decrease in serum anti-desmoglein autoantibody levels to less than 14 U/mL occurred in all cases along with clinical complete remission even after maintenance therapy cessation,” Dr. Sanchez and her associates noted.
Future research should address questions including the optimal dose and dosing frequency of rituximab, as well as the cost-effectiveness of the treatment and criteria for treatment withdrawal, they said.
The researchers had no relevant financial conflicts disclosures.
SOURCE: JAMA Dermatol. 2017 Jan 3. doi: 10.1001/jamadermatol.2017.5176.
FROM JAMA DERMATOLOGY
Key clinical point: Treatment with rituximab prevented relapse and maintained remission in 11 patients with severe, difficult-to-treat pemphigus.
Major finding: All 11 patients treated with a 1-g dose of rituximab given every 6 months maintained remission at an average of 78 months after the first cycle.
Data source: The data come from a single-center, retrospective case series of 11 adults.
Disclosures: The researchers had no relevant financial disclosures.
Source: JAMA Dermatol. 2017 Jan 3. doi: 10.1001/jamadermatol.2017.5176.
FDA cites manufacturer of autologous stem cells for regulatory, manufacturing missteps
for manufacturing processes that may compromise its safety and for failing to toe the regulatory line in marketing.
American CryoStem received an FDA warning letter Jan. 3 demanding that the company comply with best-manufacturing processes and obtain an investigational new drug application if it wishes to continue marketing ATCELL for its currently advertised clinical indications and administration routes. These include intravenous, intrathecal, or aerosol inhalation of the product for anoxic brain injury, Parkinson’s disease, amyotrophic lateral sclerosis, stroke, and multiple sclerosis.
“Please be advised that, to lawfully market a drug that is a biological product, a valid biologics license must be in effect,” noted the letter. “Such licenses are issued only after a showing that the product is safe, pure, and potent. While in the development stage, such products may be distributed for clinical use in humans only if the sponsor has an investigational new drug application (IND) in effect as specified by FDA regulations. ATCELL is not the subject of an approved biologics license application nor is there an IND in effect. Based on this information, we have determined that your actions have violated the Food, Drug, and Cosmetic Act and the Public Health Service Act.”
FDA inspectors conducted a site inspection of American CryoStem in Eatontown, N.J., last summer, during which they “documented evidence of significant deviations from current good manufacturing practice.” The agency then provided the company a chance to respond to these issues. The new warning letter discussed each complaint, noting that some were inadequately addressed, and demanded that the company take action within 15 working days or face potential legal process, including seizure and/or injunction.
American CryoStem is one of the first companies to experience increased scrutiny under FDA’s new commitment to regulate the rapid growth and development of regenerative medicine products, which include novel cellular therapies, with the aim of ensuring their safety and effectiveness.
The new policy is designed to support the potential of cellular rejuvenation medicine, while protecting patients from “unscrupulous actors” who might endanger public health with untested products, according to FDA Commissioner Scott Gottlieb, MD. As enthusiasm for stem cell treatments surges, so are reports of adverse events. The New England Journal of Medicine recently reported on three patients with age-related macular degeneration who were blinded by intravitreal injection of autologous adipose-derived stem cells (N Engl J Med. 2017;376:1047-53).
Under the new policy, cell- and tissue-based products could be exempt from FDA premarket review only if they are removed from and implanted back into the same patient in their original form, or if the products are “minimally manipulated.” ATCELL fulfills neither qualification, the FDA warning letter said.
“You process adipose tissue ... to isolate cellular components of adipose tissue, commonly referred to as stromal vascular fraction [SVF]. Such processing is more than minimal manipulation because [it alters] the original relevant characteristics of the [tissue] relating to its utility for reconstruction, repair, or replacement. Then you process the SVF by expanding it in cell culture to manufacture ATCELL. Such expansion also is more than minimal manipulation because it alters the original relevant characteristics of the tissue.”
Furthermore, the letter noted, at least one of the components used in the clonal expansion process is investigational and not intended for human use. The manufacturer of that component, which was not named, “indicates the following: ‘Unless otherwise stated in our catalog or other company documentation accompanying the product(s), our products are intended for research use only and are not to be used for any other purpose, which includes but is not limited to, unauthorized commercial uses, in vitro diagnostic uses, ex vivo or in vivo therapeutic uses or any type of consumption or application to human and animals.”
The FDA also took exception with several equipment and lab safety issues. ATCELL was being created in areas that had no clean space designation – a serious concern, the letter said.
“American CryoStem’s unvalidated processes, inadequately controlled environment, lack of control of components used in production, and lack of sufficient and validated product testing ... pose a significant risk that ATCELL may be contaminated with microorganisms or have other serious product quality defects ... Because the product is administered to humans by various higher risk routes of administration, including intravenously, intrathecally, and by aerosol inhalation, if contaminated, its use could cause a range of adverse events, from infections to death.”
FDA also expressed concerns over a lack of consistent quality control testing of each batch and questioned whether the company’s method of shipping ATCELL to clinicians had been adequately validated.
Finally, the agency raised concerns that ATCELL, while it is labeled as being for research purposes only, may harm patients indirectly by preventing them from seeking timely treatment with proven therapies.
“ATCELL is intended to treat a variety of serious or life-threatening diseases or conditions, all of which are non-homologous uses,” the warning letter noted. “Such uses raise potential significant safety concerns because there is less basis on which to predict the product’s behavior in the recipient, and use of these unapproved products may cause users to delay or discontinue medical treatments that have been found safe and effective.”
SOURCE: FDA warning letter
for manufacturing processes that may compromise its safety and for failing to toe the regulatory line in marketing.
American CryoStem received an FDA warning letter Jan. 3 demanding that the company comply with best-manufacturing processes and obtain an investigational new drug application if it wishes to continue marketing ATCELL for its currently advertised clinical indications and administration routes. These include intravenous, intrathecal, or aerosol inhalation of the product for anoxic brain injury, Parkinson’s disease, amyotrophic lateral sclerosis, stroke, and multiple sclerosis.
“Please be advised that, to lawfully market a drug that is a biological product, a valid biologics license must be in effect,” noted the letter. “Such licenses are issued only after a showing that the product is safe, pure, and potent. While in the development stage, such products may be distributed for clinical use in humans only if the sponsor has an investigational new drug application (IND) in effect as specified by FDA regulations. ATCELL is not the subject of an approved biologics license application nor is there an IND in effect. Based on this information, we have determined that your actions have violated the Food, Drug, and Cosmetic Act and the Public Health Service Act.”
FDA inspectors conducted a site inspection of American CryoStem in Eatontown, N.J., last summer, during which they “documented evidence of significant deviations from current good manufacturing practice.” The agency then provided the company a chance to respond to these issues. The new warning letter discussed each complaint, noting that some were inadequately addressed, and demanded that the company take action within 15 working days or face potential legal process, including seizure and/or injunction.
American CryoStem is one of the first companies to experience increased scrutiny under FDA’s new commitment to regulate the rapid growth and development of regenerative medicine products, which include novel cellular therapies, with the aim of ensuring their safety and effectiveness.
The new policy is designed to support the potential of cellular rejuvenation medicine, while protecting patients from “unscrupulous actors” who might endanger public health with untested products, according to FDA Commissioner Scott Gottlieb, MD. As enthusiasm for stem cell treatments surges, so are reports of adverse events. The New England Journal of Medicine recently reported on three patients with age-related macular degeneration who were blinded by intravitreal injection of autologous adipose-derived stem cells (N Engl J Med. 2017;376:1047-53).
Under the new policy, cell- and tissue-based products could be exempt from FDA premarket review only if they are removed from and implanted back into the same patient in their original form, or if the products are “minimally manipulated.” ATCELL fulfills neither qualification, the FDA warning letter said.
“You process adipose tissue ... to isolate cellular components of adipose tissue, commonly referred to as stromal vascular fraction [SVF]. Such processing is more than minimal manipulation because [it alters] the original relevant characteristics of the [tissue] relating to its utility for reconstruction, repair, or replacement. Then you process the SVF by expanding it in cell culture to manufacture ATCELL. Such expansion also is more than minimal manipulation because it alters the original relevant characteristics of the tissue.”
Furthermore, the letter noted, at least one of the components used in the clonal expansion process is investigational and not intended for human use. The manufacturer of that component, which was not named, “indicates the following: ‘Unless otherwise stated in our catalog or other company documentation accompanying the product(s), our products are intended for research use only and are not to be used for any other purpose, which includes but is not limited to, unauthorized commercial uses, in vitro diagnostic uses, ex vivo or in vivo therapeutic uses or any type of consumption or application to human and animals.”
The FDA also took exception with several equipment and lab safety issues. ATCELL was being created in areas that had no clean space designation – a serious concern, the letter said.
“American CryoStem’s unvalidated processes, inadequately controlled environment, lack of control of components used in production, and lack of sufficient and validated product testing ... pose a significant risk that ATCELL may be contaminated with microorganisms or have other serious product quality defects ... Because the product is administered to humans by various higher risk routes of administration, including intravenously, intrathecally, and by aerosol inhalation, if contaminated, its use could cause a range of adverse events, from infections to death.”
FDA also expressed concerns over a lack of consistent quality control testing of each batch and questioned whether the company’s method of shipping ATCELL to clinicians had been adequately validated.
Finally, the agency raised concerns that ATCELL, while it is labeled as being for research purposes only, may harm patients indirectly by preventing them from seeking timely treatment with proven therapies.
“ATCELL is intended to treat a variety of serious or life-threatening diseases or conditions, all of which are non-homologous uses,” the warning letter noted. “Such uses raise potential significant safety concerns because there is less basis on which to predict the product’s behavior in the recipient, and use of these unapproved products may cause users to delay or discontinue medical treatments that have been found safe and effective.”
SOURCE: FDA warning letter
for manufacturing processes that may compromise its safety and for failing to toe the regulatory line in marketing.
American CryoStem received an FDA warning letter Jan. 3 demanding that the company comply with best-manufacturing processes and obtain an investigational new drug application if it wishes to continue marketing ATCELL for its currently advertised clinical indications and administration routes. These include intravenous, intrathecal, or aerosol inhalation of the product for anoxic brain injury, Parkinson’s disease, amyotrophic lateral sclerosis, stroke, and multiple sclerosis.
“Please be advised that, to lawfully market a drug that is a biological product, a valid biologics license must be in effect,” noted the letter. “Such licenses are issued only after a showing that the product is safe, pure, and potent. While in the development stage, such products may be distributed for clinical use in humans only if the sponsor has an investigational new drug application (IND) in effect as specified by FDA regulations. ATCELL is not the subject of an approved biologics license application nor is there an IND in effect. Based on this information, we have determined that your actions have violated the Food, Drug, and Cosmetic Act and the Public Health Service Act.”
FDA inspectors conducted a site inspection of American CryoStem in Eatontown, N.J., last summer, during which they “documented evidence of significant deviations from current good manufacturing practice.” The agency then provided the company a chance to respond to these issues. The new warning letter discussed each complaint, noting that some were inadequately addressed, and demanded that the company take action within 15 working days or face potential legal process, including seizure and/or injunction.
American CryoStem is one of the first companies to experience increased scrutiny under FDA’s new commitment to regulate the rapid growth and development of regenerative medicine products, which include novel cellular therapies, with the aim of ensuring their safety and effectiveness.
The new policy is designed to support the potential of cellular rejuvenation medicine, while protecting patients from “unscrupulous actors” who might endanger public health with untested products, according to FDA Commissioner Scott Gottlieb, MD. As enthusiasm for stem cell treatments surges, so are reports of adverse events. The New England Journal of Medicine recently reported on three patients with age-related macular degeneration who were blinded by intravitreal injection of autologous adipose-derived stem cells (N Engl J Med. 2017;376:1047-53).
Under the new policy, cell- and tissue-based products could be exempt from FDA premarket review only if they are removed from and implanted back into the same patient in their original form, or if the products are “minimally manipulated.” ATCELL fulfills neither qualification, the FDA warning letter said.
“You process adipose tissue ... to isolate cellular components of adipose tissue, commonly referred to as stromal vascular fraction [SVF]. Such processing is more than minimal manipulation because [it alters] the original relevant characteristics of the [tissue] relating to its utility for reconstruction, repair, or replacement. Then you process the SVF by expanding it in cell culture to manufacture ATCELL. Such expansion also is more than minimal manipulation because it alters the original relevant characteristics of the tissue.”
Furthermore, the letter noted, at least one of the components used in the clonal expansion process is investigational and not intended for human use. The manufacturer of that component, which was not named, “indicates the following: ‘Unless otherwise stated in our catalog or other company documentation accompanying the product(s), our products are intended for research use only and are not to be used for any other purpose, which includes but is not limited to, unauthorized commercial uses, in vitro diagnostic uses, ex vivo or in vivo therapeutic uses or any type of consumption or application to human and animals.”
The FDA also took exception with several equipment and lab safety issues. ATCELL was being created in areas that had no clean space designation – a serious concern, the letter said.
“American CryoStem’s unvalidated processes, inadequately controlled environment, lack of control of components used in production, and lack of sufficient and validated product testing ... pose a significant risk that ATCELL may be contaminated with microorganisms or have other serious product quality defects ... Because the product is administered to humans by various higher risk routes of administration, including intravenously, intrathecally, and by aerosol inhalation, if contaminated, its use could cause a range of adverse events, from infections to death.”
FDA also expressed concerns over a lack of consistent quality control testing of each batch and questioned whether the company’s method of shipping ATCELL to clinicians had been adequately validated.
Finally, the agency raised concerns that ATCELL, while it is labeled as being for research purposes only, may harm patients indirectly by preventing them from seeking timely treatment with proven therapies.
“ATCELL is intended to treat a variety of serious or life-threatening diseases or conditions, all of which are non-homologous uses,” the warning letter noted. “Such uses raise potential significant safety concerns because there is less basis on which to predict the product’s behavior in the recipient, and use of these unapproved products may cause users to delay or discontinue medical treatments that have been found safe and effective.”
SOURCE: FDA warning letter