User login
February 2018
Gastroenterology:
Living like an academic athlete: How to improve clinical and academic productivity as a gastroenterologist. Benchimol E et al.
2018 Jan;154(1):8-14. doi: 10.1053/j.gastro.2017.11.017.
“Spending your life wisely”: How to create an asset management plan. Adams MA et al.
2017 Dec;153(6):1469-72. doi: 10.1053/j.gastro.2017.10.032.
How to balance clinical work and research in the current era of academic medicine. Katzka DA.
2017 Nov;153(5):1177-80. doi: 10.1053/j.gastro.2017.09.024.
Clin Gastroenterol Hepatol.:
New models of gastroenterology practice. Allen JI et al.
2018 Jan;16(1):3-6. doi: 10.1016/j.cgh.2017.10.003.
Cracking the clinician educator code in gastroenterology. Shapiro JM et al.
2017 Dec;15(12):1828-32. doi: 10.1016/j.cgh.2017.08.040.
Cell Mol Gastroenterol Hepatol.:
Setting up a lab: The early years. Habtezion A.
2017 Nov; 4(3): 445-6. doi: 10.1016/j.jcmgh.2017.08.003.
Gastroenterology:
Living like an academic athlete: How to improve clinical and academic productivity as a gastroenterologist. Benchimol E et al.
2018 Jan;154(1):8-14. doi: 10.1053/j.gastro.2017.11.017.
“Spending your life wisely”: How to create an asset management plan. Adams MA et al.
2017 Dec;153(6):1469-72. doi: 10.1053/j.gastro.2017.10.032.
How to balance clinical work and research in the current era of academic medicine. Katzka DA.
2017 Nov;153(5):1177-80. doi: 10.1053/j.gastro.2017.09.024.
Clin Gastroenterol Hepatol.:
New models of gastroenterology practice. Allen JI et al.
2018 Jan;16(1):3-6. doi: 10.1016/j.cgh.2017.10.003.
Cracking the clinician educator code in gastroenterology. Shapiro JM et al.
2017 Dec;15(12):1828-32. doi: 10.1016/j.cgh.2017.08.040.
Cell Mol Gastroenterol Hepatol.:
Setting up a lab: The early years. Habtezion A.
2017 Nov; 4(3): 445-6. doi: 10.1016/j.jcmgh.2017.08.003.
Gastroenterology:
Living like an academic athlete: How to improve clinical and academic productivity as a gastroenterologist. Benchimol E et al.
2018 Jan;154(1):8-14. doi: 10.1053/j.gastro.2017.11.017.
“Spending your life wisely”: How to create an asset management plan. Adams MA et al.
2017 Dec;153(6):1469-72. doi: 10.1053/j.gastro.2017.10.032.
How to balance clinical work and research in the current era of academic medicine. Katzka DA.
2017 Nov;153(5):1177-80. doi: 10.1053/j.gastro.2017.09.024.
Clin Gastroenterol Hepatol.:
New models of gastroenterology practice. Allen JI et al.
2018 Jan;16(1):3-6. doi: 10.1016/j.cgh.2017.10.003.
Cracking the clinician educator code in gastroenterology. Shapiro JM et al.
2017 Dec;15(12):1828-32. doi: 10.1016/j.cgh.2017.08.040.
Cell Mol Gastroenterol Hepatol.:
Setting up a lab: The early years. Habtezion A.
2017 Nov; 4(3): 445-6. doi: 10.1016/j.jcmgh.2017.08.003.
Impact of Drug Shortages on Patient Safety and Pharmacy Operation Costs
Drug product shortages threaten health care quality and public health by creating barriers to optimal care. The frequency of drug shortages has risen dramatically since 2005 and now influences broad areas of health care practice. More than 400 generic drug products have been affected, forcing institutions to purchase costly brand-name products, substitute alternative therapies, or procure from gray market vendors at increased institutional costs.1 Scarcity and cost have potential to negatively impact patient outcomes and the ability of health care organizations to respond to the needs of their patients.
Background
Although constantly fluctuating, the number of active shortages reached a height of 320 products at the end the third quarter of 2014.2 A 2011 analysis from Premier Healthcare Alliance estimated the added cost of purchasing brand, generic, or alternative drugs due to shortage may have inflated hospital costs by $200 million annually.1 In 2016, the number of active shortages dropped to 176, suggesting a downward trend. However, the drug supply chain remains a concern for pharmacies in the U.S.
Despite creative approaches to shortage management, the variable characteristics of shortages make planning difficult. For example, the drug product in short supply may or may not have an alternative for use in similar clinical scenarios. The impact of shortages of medications lacking an equivalent alternative product has been documented, such as the past shortage of succinylcholine for anesthesia, resulting in surgery cancellations when an alternative paralytic agent was not appropriate.3 In 2016, the Cleveland Clinic reported undertaking “military-style triage” in determining patients who required use of aminocaproic acid during open heart surgery due to its limited supply.4 Decisions to reserve drug supply for emergency use and prefilling syringes under pharmacy supervision to extend stability and shelf life are short-term solutions to larger, systemic issues. Unfortunately, these scenarios have the potential to disrupt patient care and diminish health outcomes.
Shortages of products that have an available therapeutic substitution may seem easily manageable, but additional considerations may be present. Bacillus Calmette-Guérin (BCG) is considered the drug of choice for bladder cancer. In 2011, there was a shortage of the BCG vaccine after mold was discovered in the formulation.5 Providers were forced to choose between reducing or reallocating the dose of BCG, turning away patient, or substituting mitomycin C, which is less effective and costlier. When tamsulosin capsules became difficult to obtain in 2014, some institutions began switching patients to alfuzosin.6 Although alfuzosin is similar in mechanism to tamsulosin, it may prolong the QTc interval. Not only did this substitution present a contraindication for patients with elevated QTc intervals or who were already receiving concomitant medications that prolonged the QTc interval, but also it required additional cost and resources needed to update electrocardiograms.
VA Consolidated Mail Outpatient Pharmacies
The VHA serves nearly 9 million patients at more than 1,200 facilities across the U.S.7 This large patient population results in an estimated 149 million outpatient prescriptions annually.8 About 80% of these are distributed by mail through 7 VA consolidated mail outpatient pharmacies (CMOPs). When drug scarcity impedes the ability of the CMOP to respond to medication demand, the local facility must fill these prescriptions. These rejections sent back to the facility impact workload, patient wait times, and access to medication therapy. Barriers to medication procurement in the VA also stem from regulations based on legislation, including the Trade Agreements Act, Drug Supply Chain Security Act, and the Federal Acquisition Regulation (FAR) (Table). 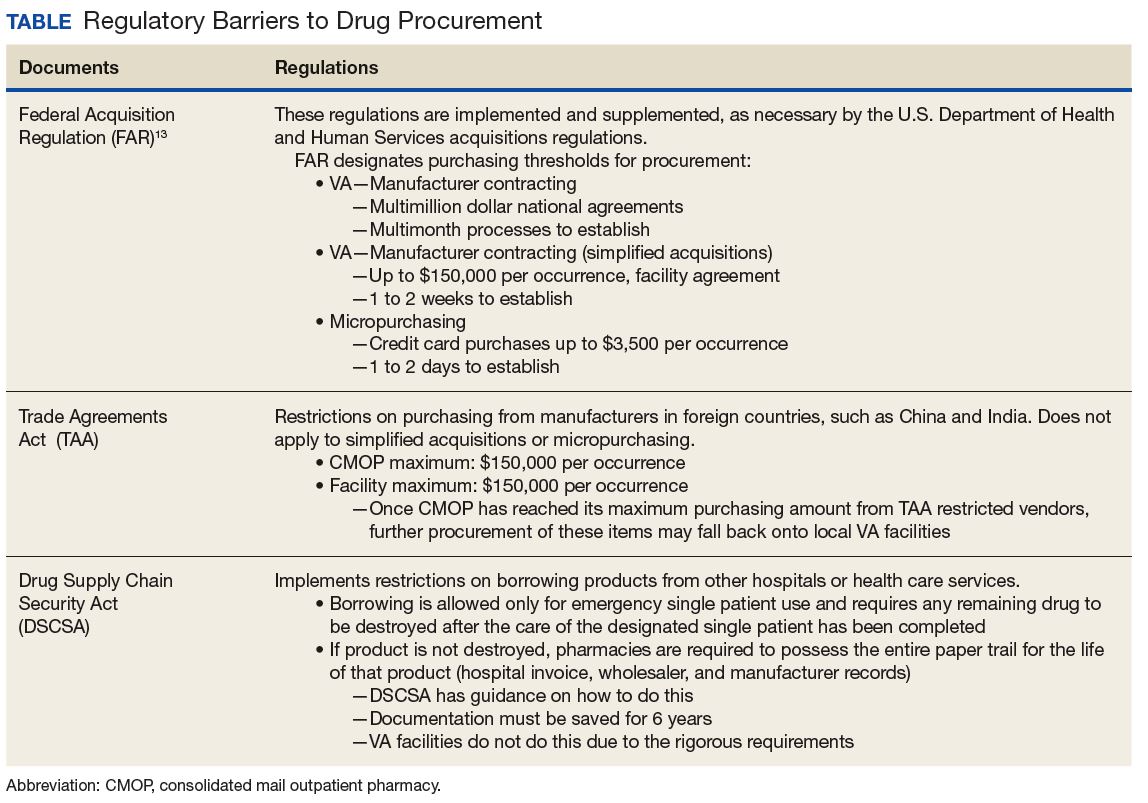
The impact of drug shortages has been described previously in the private sector, particularly for emergency medicine and chemotherapy.9,10 However, the impact of drug shortages on health care provision to veteran populations within the VA has not previously been analyzed. Due to the unique procurement regulations that influence the VA and the importance of continuing to provide optimal health care services to veterans, assessing the impact of drug shortages on patient safety and health care costs is necessary in informing policy decisions and guiding recommendations for mitigation strategies. The purpose of this study was to assess the influence of drug shortages on institutional costs and patient care within VA facilities and formulate recommendations for enhanced mitigation of this issue.
Methods
The primary outcome of this study was to characterize the impact of drug shortages on institutional cost and patient safety events among VHA facilities. Secondary outcomes included subgroup evaluation in reported drug shortage impact among 1a, 1b, and 1c complexity VA facility survey respondents and assessment of drug shortage impact on CMOP prescription order fulfillment and operation cost.
Definitions
The complexity ranking system is a facility grouping method used within the VA to characterize the level of service provision, teaching and research opportunities, patient volume, intensive care unit level, and other factors offered by a VA site. Rankings start from 1 (highest level of services offered) to 3 (lowest level of services offered), with level 1 facilities further divided into a, b, and c subdivisions. A level 1a facility will be larger with more services offered than a 1b, which is larger and offers more services than a 1c facility. The VA facilities are further characterized by regional distribution. Sites are grouped under VISNs of which there are currently 21.
The CMOP program was responsible for dispensing about 119 million outpatient prescriptions in 2016 and includes designated sites for the dispensing of controlled substances and supply items. The VA Pharmacy Benefits Management Service (PBM) oversees formulary management, plans national drug policy, promotes safe and appropriate drug therapy, and delivers high-quality and sustainable pharmacy benefits for veterans.
Study Design
A descriptive study was initiated to characterize the impact of drug shortages among VA facilities. An analysis of administrative medication safety event reporting and institutional costs data at the Denver VAMC in Colorado was done, focusing on predetermined drug products involved in a recent shortage. The analysis was accomplished through a review of the VA adverse drug events reporting system (VA ADERS) reports and a local medication errors quality improvement database and paper procurement records, respectively. Concurrently, a survey was disseminated among qualifying VA facilities across the country that sought to characterize the impact of drug shortages nationally.
Sample Selection
Denver VAMC. The Denver VAMC, where the authors were located, was selected as the local sample site. The intention was to compare the strategies used locally with strategies used among similar (level 1a, 1b, and 1c) facilities. Preselected “cost-impacting” drug products were identified through a review of historic shortages with a significant local impact. These drugs were defined as low cost/high utilization (eg, tamsulosin 0.4-mg capsules and ketorolac solution), medium cost/utilization (eg, piperacillin/tazobactam IV solutions and aminocaproic acid solution), and high cost/low utilization (eg, nitroprusside IV solution and BCG vaccine solution). Additionally, patient safety event data reported internally for quality improvement and locally via VA ADERS were reviewed for preselected “safety impact” drug products and included BCG vaccine, tamsulosin capsules, IV fluid products, calcium gluconate and chloride injections, and aminocaproic acid injection.
National Survey. The authors identified 84 level 1 complexity facilities and used the PBM pharmacy directory to contact the administrative personnel representing each facility. These representatives identified a point of contact to aid in survey completion. A separate survey also was sent to the CMOP facilities (survey outlines available at www.fedprac.com).
Data Collection
Denver VAMC. Financial data were sampled through a manual review of paper procurement records stored by date in the inpatient pharmacy of the Denver VAMC. Variables included units of product used over the period of drug shortage, cost per unit during shortage, and cost per unit before shortage. This information also was supplemented with data from the prescription processing software’s drug file. Patient safety data were gathered through query of the identified event reporting databases for the prespecified drug on shortage. These variables included the type of error and the effect the error had on the patient.
National Survey. Data collection focused on notable drug shortages and patient safety reporting between January 1, 2013 and December 31, 2016. The survey was maintained in a facility-specific spreadsheet. Editing capabilities were disabled for all actions other than responding to questions. Recipients were followed up with a courtesy e-mail after 2 weeks and another 2 times unless a survey was received. Data were de-identified and aggregated for analyses.
Statistical Analyses
Excel 2010 (Microsoft, Redmond, WA) descriptive statistics were used to relay information from this assessment. Extrapolations from procurement cost data and drug product utilization were used to estimate the enhanced direct cost associated with identified drug shortages. Similar extrapolations were used to estimate the cost associated with shortages leading to CMOP rejection and local fill.
Results
Survey completion totaled 20% of invited facilities (n = 17). Good geographic and VISN distribution was noted with representatives from VISNs 2, 4, 8, 9, 10, 12, 15, 16, 21, and 22. VISNs 10 and 12 provided the most representation with 3 participants, each. Level 1a facilities participated most (n = 9), followed by 1b (n = 6) and 1c (n = 2). Participating facilities reported a mean (SD) of 54 (21.5) pharmacists and 34 (15.3) pharmacy technician staff members employed. The most common reason for not participating was lack of personnel resources and competing demands. The CMOP participation was 100% (n = 7) and completed through a coordinated response.
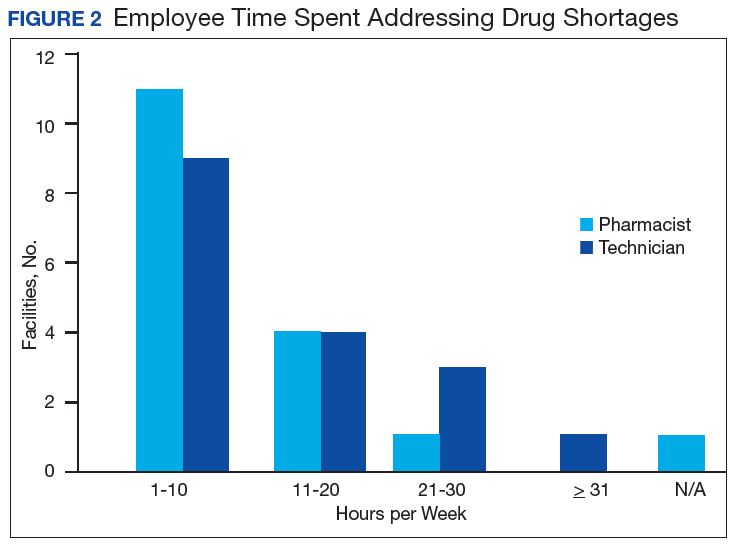
Results of the budgetary increase and staff member time allocation survey assessments are provided (Figures 1 and 2). Five facilities provided an annual estimate of increased cost due to acquisition of drugs on shortage through open market purchases that ranged from about $150,000 to $750,000. Nearly half of the surveyed facilities endorsed having a drug shortage task force (n = 8) to respond to drug shortages and mitigate their impact. 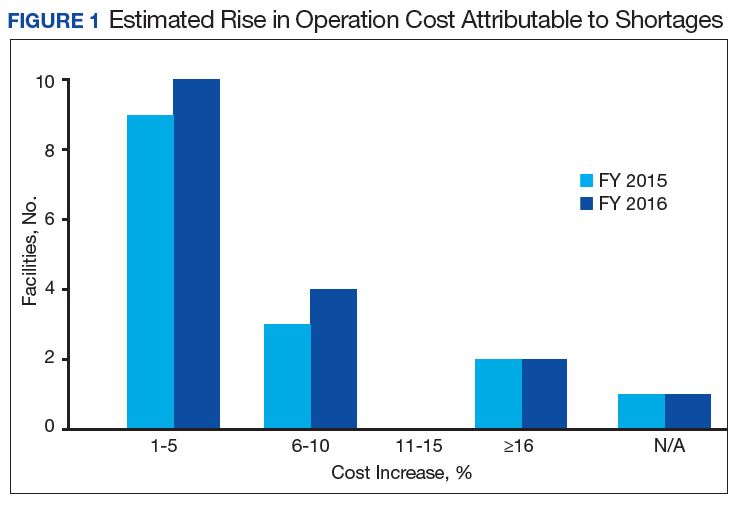
Regarding drug product allocation, only 2 facilities did not have current restrictions for use due to a shortage. Many had between 1 and 10 of these restrictions implemented to conserve supply (n = 11, 64%), 2 facilities reported 11 to 20 restrictions, and 2 facilities noted more than 30 restrictions. Similarly, 3 facilities had not needed to revise any current treatment protocols due to drug shortages. The majority of facilities had revised 1 to 5 current protocols (n = 12, 70%), 1 revised 6 to 10 protocols, and 1 facility revised more than 10 protocols.
In assessing patient safety concerns, 1 facility identified a history of transferring patients to alternative medical sites for the patients to obtain necessary medication impacted by a local shortage. Additionally, during the BCG vaccine shortage, 6 facilities (35.3%) substituted mitomycin C for the treatment of urinary bladder cancer.
Most participants either agreed (n = 8, 47.0%) or strongly agreed (n = 4, 23.5%) that modifications to FAR to increase purchasing opportunities from foreign distributors during drug shortage would help mitigate the impact of such shortages. Similarly, most participants agreed (n = 10, 58.8%) or strongly agreed (n = 3, 17.6%) that PBM guidance on drug shortage management would help efficiently and effectively respond to issues that might arise. The consensus of participants also agreed (n = 13, 76.5%) that organized collaborations or working groups within each VISN might help assist in drug shortage management.
The CMOP facility data revealed that 2 sites did not require dedicated staffing to respond to shortages, and 3 sites had not experienced cost increases because of shortages. Pharmacist use varied between sites, with 2 facilities using 1 to 10 pharmacist h/wk, and 1 facility using 11 to 20 pharmacist h/wk, and 1 facility using 21 to 30 pharmacist h/wk. Technician utilization was more pronounced, with 2 facilities using more than 30 technician h/wk, and 2 facilities using 1 to 10 technician h/wk. Workload and costs may have been influenced in other ways as 3 sites endorsed using overtime pay, shifting product responsibility between CMOPs, prolonging patient wait times, and close monitoring for each. In fiscal year 2015, some sites experienced a 1% to 5% (n = 2) and 6% to 10% (n = 1) increase in operation cost attributable to shortage. Results from fiscal year 2016 showed that some sites continued to see a 1% to 5% (n = 1) and 6% to 10% (n = 2) increase in operation cost attributable to shortage.
Through aggregation of CMOP responses on the number of prescriptions sent back to local facility for fill due to back order, a downward trend in the total number of rejections was seen over the 2.5 fiscal years assessed. This amounted to more than 1 million rejections in fiscal year 2015, about 788,000 rejections in 2016, and about 318,000 rejections through the first 2 quarters of 2017.
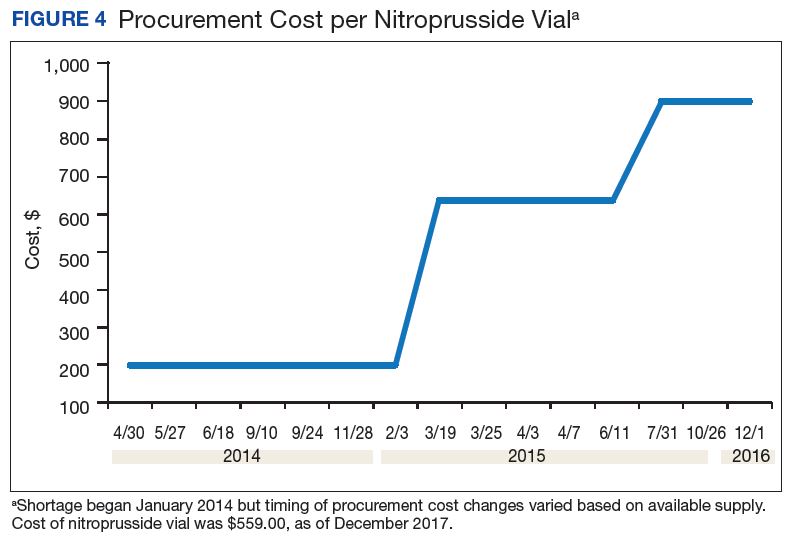
A consistent rise in the medication procurement budget requirement was characterized within the single VA facility review. The quarterly median increase was 2.7% over 2.5 years (min: -1.4%; max: 6.6%) for total outpatient medication costs, excluding hepatitis C antiviral therapies. Procurement cost records were insufficient to characterize historic expenditures for 4 of the prespecified drug products. The data collected on tamsulosin capsule and nitroprusside vial procurement during shortage is provided (Figures 3 and 4). Over the time frame of procurement records found on review, the added costs of nitroprusside vials and tamsulosin capsules were $22,766.09 (+167.9% of base cost) and $17,433.70 (+657.3% of base cost), respectively. No patient safety data were found on review. 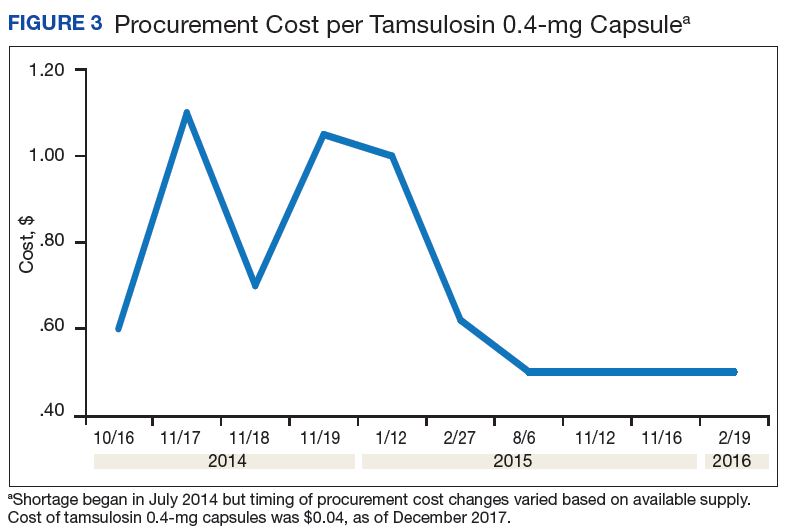
Discussion
Drug product shortages represent a barrier to quality and efficiency across health care institutions. A survey of health system pharmacies in the southeastern U.S. found that the majority of respondents tracking shortage data reported a 300% to 500% markup by alternative or gray market suppliers for hard-to-find medications.11 These reports are similar to the authors’ analyses of the trends in increased procurement expenditures documented during the tamsulosin capsule and nitroprusside vial shortages and indirectly correlate with the survey results indicating that most facilities endorsed a trend in operation cost increase attributable to drugs product shortage. The estimated annual costs for open market purchases further informs the financial burden aggregated by this issue.
Indirect costs from drug shortage further complicated quantifying the impact of shortages. Many facilities acknowledged the indirect influence drug shortages have on staffing and workload due to the implementation of mitigation strategies. Most participants found it necessary to establish restrictions for use in addition to altering protocols. These required the time investment of essential personnel from development through execution and education. Situations also can arise for mass therapeutic substitution. In this example, pharmacy staff may be required to oversee medication transition from the product on shortage to an appropriate alternative. When substitution involves hundreds or thousands of outpatient prescriptions, such as the tamsulosin shortage, the process may be tedious and time consuming, depending on the level of clinical decision making needed to determine patient candidacy for transitioning products.
Improving institutional cost efficiency becomes a significant challenge with persistent drug shortages. Professional advocacy groups, such as the American Society of Health-System Pharmacists (ASHP), help provide guidance to organizations constrained by specific drug shortages.12 Staff knowledgeable in allocation, supply considerations, and product repackaging and stability data also are essential. Other mitigation strategies include automatic substitutions, restrictions for use or inventory control strategies, and open market procurement, or borrowing from other institutions.
Data gathered from the survey of CMOP facilities also helped elucidate strategies used to mitigate drug shortage impacts for those respondents impacted by shortage. Likely, the 2 CMOP facilities without dedicated staff focused on shortages are those whose outpatient prescription fulfillment responsibility were focused on supply items or controlled substances. The impacted CMOP respondents cited overtime pay, shifting product responsibility, and prolonging patient wait times as the most frequently employed mitigation strategies. When these and other strategies fail to manage a shortage, prescriptions are often sent back to the local facility to be filled. Unfortunately for these facilities, the same mitigation strategies used by CMOP are not always feasible. Overtime pay may not be possible given staffing and budgetary resources, sending prescriptions back to facilities in itself prolongs patient wait times, and local medical centers do not have the option of shifting product responsibility between sites or sending the prescription to another facility. Herein lies 1 rationale for the CMOP effort to reduce the volume of prescriptions sent back to local medical centers.
Multiple offices within the FDA have roles in the mitigation of national drug shortages within their regulatory purview. Much of the recent focus stems from provisions enacted under Title X of the FDA Safety and Innovation Act of 2012, which addresses problems in the drug-supply chain.12 Rectifying a shortage involves short- and long-term strategic planning to address supply, distribution, and market reaction to need. Collaboration between the FDA and manufacturers is one method by which demand can be satisfied through the coordination of resources, expedition of inspections, and root cause analysis of the shortage.
Similar collaborations within the VA were viewed favorably by respondents and might yield productive relationships if regional or VISN working groups were to be established. Alternative long-term strategies are executed through regulation, particularly concerning the importation of foreign manufactured drugs and regulatory discretion on supplier vetting. Despite a strong respondent consensus that regulatory modifications of foreign product importation in the setting of a drug shortage may be beneficial, such a change would require a congressional action and is not likely to be timely. Unfortunately, gray market pharmaceutical distribution, driven by wholesaler stockpiling to raise prices, is separate from manufacturer driven shortages and falls outside the FDA’s regulatory purview and institutional mitigation strategies.
Although based on this limited survey, general agreement existed on the importance of greater national collaboration and communication regarding drug shortage management strategies. This could include PBM guidance on specific shortage management opportunities or establishing collaborations by region or VISN. These possibilities may be more realistically attainable in comparison to modifying federal regulations on drug product procurement during active shortages, which requires an act of Congress. Many of the survey participants endorsed a drug shortage task force within their facility. Coordinating interaction between preexisting or newly established task forces or working groups on a monthly or quarterly basis may provide fruitful interactions and the exchange of strategies to reduce shortage impact on institutional cost, efficiency, and patient care.
Limitations
Quantifying the extent of drug shortage impact on patient safety and institutional costs is a difficult task. The procurement records data used for the analysis of a single VAMC were gathered through manual review of stored paper invoices, opening the possibility for missing data. It is also difficult to extrapolate the sum of indirect costs such as process changes, alternative product utilization, and pharmacy staffing resources as additional financial burdens to the affected institution. Any quantifiable cost assessment also is biased by contract terms between the VA and wholesalers in which unavailable products that must be purchased off-contract are subsequently reimbursed through credit or alternative means.
Patient safety events are frequently underreported, leading to underestimation of true safety event incidence. Given that these events are documented by multiple disciplines and that many of these documenters may not be aware consistently of the drug products and volume impacted by shortage, elucidating safety events unfolding in relation to shortage also is difficult to quantify.
The response rate for the survey was low but near the expected rate for this methodology. Feedback from several facilities was received, citing competing demands and workforce shortage as barriers to participation. The survey also was limited by reporting bias and recall bias. As assessment of prespecified past drug shortages may require intimate knowledge of pharmacy department processes and mitigation strategies, the accuracy of question answering may have been limited to the length of time the points of contact had been in their current position.
Conclusion
Drug shortages are a pervasive barrier to patient care within larger facilities of the VA health care system, similar to what has been characterized in the private sector. As a result of these shortages and the mitigation strategies to reduce their burden, many facilities endorsed trends in increasing workload for staff, institutional operation costs, and risk for patient safety and care quality concerns. Due to the demands of shortages, some facilities have implemented drug shortage task forces or equivalent groups to specifically manage these issues. Moving forward, the VA health care system may benefit from similar task forces or working groups at the VISN level, to aid in collaborative efforts to respond to shortage. Support for revising federal regulations on procurement in times of shortage and enhanced PBM drug shortage management guidance also was endorsed.
1. Cherici C, Frazier J, Feldman M, et al. Navigating drug shortages in American healthcare: a premier healthcare alliance analysis. https://www.heartland.org/_template-assets/documents/publications/30103.pdf. Published March 2011. Accessed December 5, 2017.
2. American Society of Health-System Pharmacists. ASHP drug shortage statistics. https://www.ashp.org/Drug-Shortages/Shortage-Resources/Drug-Shortages-Statistics. Updated 2017. Accessed December 5, 2017.
3. Dooren JC. Most hospitals face drug shortages. The Wall Street Journal. http://www.wsj.com/articles/SB10001424052702304584404576442211187884744. Published July 13, 2011. Accessed December 5, 2017.
4. Fink S. Drug shortages forcing hard decisions on rationing treatment. The New York Times. http://www.nytimes.com/2016/01/29/us/drug-shortages-forcing-hard-decisions-on-rationing-treatments.html. Published January 29, 2016. Accessed December 5, 2017.
5. Loftus P. Drug shortages frustrate doctors, patients. The Wall Street Journal. http://www.wsj.com/articles/u-s-drug-shortages-frustrate-doctors-patients-1433125793. Published May, 31, 2015. Accessed December 5, 2017.
6. U.S. Food and Drug Administration. Strategic plan for preventing and mitigating drug shortages. http://www.fda.gov/downloads/Drugs/DrugSafety/DrugShortages/UCM372566.pdf. Published October 2013. Accessed August 22, 2016.
7. U.S. Department of Veteran Affairs, National Center for Veterans Analysis and Statistics. Quick facts. https://www.va.gov/vetdata/Quick_Facts.asp. Updated November 20, 2017. Accessed December 5, 2017.
8. U.S. Department of Veterans Affairs, Office of the Inspector General. Audit of Consolidated Mail Outpatient Pharmacy Program. https://www.va.gov/oig/pubs/VAOIG-15-05255-422.pdf. Accessed December 11, 2017.
9. Mazer-Amirshahi M, Pourmand A, Singer S, Pines JM, van den Anker J. Critical drug shortages: implications for emergency medicine. Acad Emerg Med. 2014;21(6):704-711.
10. McBride A, Holle LM, Westendorf C, et al. National survey on the effect of oncology drug shortages on cancer care. Am J Health Syst Pharm. 2013;70(7):609-617.
11. Caulder CR, Mehta B, Bookstaver PB, Sims LD, Stevenson B; South Carolina Society of Health-System Pharmacists. Impact of drug shortages on health system pharmacies in the southeastern United States. Hosp Pharm. 2015;50(4):279-286.
12. Florida Society of Health-System Pharmacists. Conservation strategies for IV fluids. http://www.fshp.org/news/165998/Conservation-Strategies-for-IV-Fluid.htm. Accessed December 11, 2017.
13. Federal Acquisition Regulation Site. FAR—Part 13 Simplified Acquisition Procedures, 13 CFR §§ 201-302. http://farsite.hill.af.mil/reghtml/regs/far2afmcfars/fardfars/far/13.htm. Updated January 13, 2017. Accessed December 5, 2017.
Drug product shortages threaten health care quality and public health by creating barriers to optimal care. The frequency of drug shortages has risen dramatically since 2005 and now influences broad areas of health care practice. More than 400 generic drug products have been affected, forcing institutions to purchase costly brand-name products, substitute alternative therapies, or procure from gray market vendors at increased institutional costs.1 Scarcity and cost have potential to negatively impact patient outcomes and the ability of health care organizations to respond to the needs of their patients.
Background
Although constantly fluctuating, the number of active shortages reached a height of 320 products at the end the third quarter of 2014.2 A 2011 analysis from Premier Healthcare Alliance estimated the added cost of purchasing brand, generic, or alternative drugs due to shortage may have inflated hospital costs by $200 million annually.1 In 2016, the number of active shortages dropped to 176, suggesting a downward trend. However, the drug supply chain remains a concern for pharmacies in the U.S.
Despite creative approaches to shortage management, the variable characteristics of shortages make planning difficult. For example, the drug product in short supply may or may not have an alternative for use in similar clinical scenarios. The impact of shortages of medications lacking an equivalent alternative product has been documented, such as the past shortage of succinylcholine for anesthesia, resulting in surgery cancellations when an alternative paralytic agent was not appropriate.3 In 2016, the Cleveland Clinic reported undertaking “military-style triage” in determining patients who required use of aminocaproic acid during open heart surgery due to its limited supply.4 Decisions to reserve drug supply for emergency use and prefilling syringes under pharmacy supervision to extend stability and shelf life are short-term solutions to larger, systemic issues. Unfortunately, these scenarios have the potential to disrupt patient care and diminish health outcomes.
Shortages of products that have an available therapeutic substitution may seem easily manageable, but additional considerations may be present. Bacillus Calmette-Guérin (BCG) is considered the drug of choice for bladder cancer. In 2011, there was a shortage of the BCG vaccine after mold was discovered in the formulation.5 Providers were forced to choose between reducing or reallocating the dose of BCG, turning away patient, or substituting mitomycin C, which is less effective and costlier. When tamsulosin capsules became difficult to obtain in 2014, some institutions began switching patients to alfuzosin.6 Although alfuzosin is similar in mechanism to tamsulosin, it may prolong the QTc interval. Not only did this substitution present a contraindication for patients with elevated QTc intervals or who were already receiving concomitant medications that prolonged the QTc interval, but also it required additional cost and resources needed to update electrocardiograms.
VA Consolidated Mail Outpatient Pharmacies
The VHA serves nearly 9 million patients at more than 1,200 facilities across the U.S.7 This large patient population results in an estimated 149 million outpatient prescriptions annually.8 About 80% of these are distributed by mail through 7 VA consolidated mail outpatient pharmacies (CMOPs). When drug scarcity impedes the ability of the CMOP to respond to medication demand, the local facility must fill these prescriptions. These rejections sent back to the facility impact workload, patient wait times, and access to medication therapy. Barriers to medication procurement in the VA also stem from regulations based on legislation, including the Trade Agreements Act, Drug Supply Chain Security Act, and the Federal Acquisition Regulation (FAR) (Table).
The impact of drug shortages has been described previously in the private sector, particularly for emergency medicine and chemotherapy.9,10 However, the impact of drug shortages on health care provision to veteran populations within the VA has not previously been analyzed. Due to the unique procurement regulations that influence the VA and the importance of continuing to provide optimal health care services to veterans, assessing the impact of drug shortages on patient safety and health care costs is necessary in informing policy decisions and guiding recommendations for mitigation strategies. The purpose of this study was to assess the influence of drug shortages on institutional costs and patient care within VA facilities and formulate recommendations for enhanced mitigation of this issue.
Methods
The primary outcome of this study was to characterize the impact of drug shortages on institutional cost and patient safety events among VHA facilities. Secondary outcomes included subgroup evaluation in reported drug shortage impact among 1a, 1b, and 1c complexity VA facility survey respondents and assessment of drug shortage impact on CMOP prescription order fulfillment and operation cost.
Definitions
The complexity ranking system is a facility grouping method used within the VA to characterize the level of service provision, teaching and research opportunities, patient volume, intensive care unit level, and other factors offered by a VA site. Rankings start from 1 (highest level of services offered) to 3 (lowest level of services offered), with level 1 facilities further divided into a, b, and c subdivisions. A level 1a facility will be larger with more services offered than a 1b, which is larger and offers more services than a 1c facility. The VA facilities are further characterized by regional distribution. Sites are grouped under VISNs of which there are currently 21.
The CMOP program was responsible for dispensing about 119 million outpatient prescriptions in 2016 and includes designated sites for the dispensing of controlled substances and supply items. The VA Pharmacy Benefits Management Service (PBM) oversees formulary management, plans national drug policy, promotes safe and appropriate drug therapy, and delivers high-quality and sustainable pharmacy benefits for veterans.
Study Design
A descriptive study was initiated to characterize the impact of drug shortages among VA facilities. An analysis of administrative medication safety event reporting and institutional costs data at the Denver VAMC in Colorado was done, focusing on predetermined drug products involved in a recent shortage. The analysis was accomplished through a review of the VA adverse drug events reporting system (VA ADERS) reports and a local medication errors quality improvement database and paper procurement records, respectively. Concurrently, a survey was disseminated among qualifying VA facilities across the country that sought to characterize the impact of drug shortages nationally.
Sample Selection
Denver VAMC. The Denver VAMC, where the authors were located, was selected as the local sample site. The intention was to compare the strategies used locally with strategies used among similar (level 1a, 1b, and 1c) facilities. Preselected “cost-impacting” drug products were identified through a review of historic shortages with a significant local impact. These drugs were defined as low cost/high utilization (eg, tamsulosin 0.4-mg capsules and ketorolac solution), medium cost/utilization (eg, piperacillin/tazobactam IV solutions and aminocaproic acid solution), and high cost/low utilization (eg, nitroprusside IV solution and BCG vaccine solution). Additionally, patient safety event data reported internally for quality improvement and locally via VA ADERS were reviewed for preselected “safety impact” drug products and included BCG vaccine, tamsulosin capsules, IV fluid products, calcium gluconate and chloride injections, and aminocaproic acid injection.
National Survey. The authors identified 84 level 1 complexity facilities and used the PBM pharmacy directory to contact the administrative personnel representing each facility. These representatives identified a point of contact to aid in survey completion. A separate survey also was sent to the CMOP facilities (survey outlines available at www.fedprac.com).
Data Collection
Denver VAMC. Financial data were sampled through a manual review of paper procurement records stored by date in the inpatient pharmacy of the Denver VAMC. Variables included units of product used over the period of drug shortage, cost per unit during shortage, and cost per unit before shortage. This information also was supplemented with data from the prescription processing software’s drug file. Patient safety data were gathered through query of the identified event reporting databases for the prespecified drug on shortage. These variables included the type of error and the effect the error had on the patient.
National Survey. Data collection focused on notable drug shortages and patient safety reporting between January 1, 2013 and December 31, 2016. The survey was maintained in a facility-specific spreadsheet. Editing capabilities were disabled for all actions other than responding to questions. Recipients were followed up with a courtesy e-mail after 2 weeks and another 2 times unless a survey was received. Data were de-identified and aggregated for analyses.
Statistical Analyses
Excel 2010 (Microsoft, Redmond, WA) descriptive statistics were used to relay information from this assessment. Extrapolations from procurement cost data and drug product utilization were used to estimate the enhanced direct cost associated with identified drug shortages. Similar extrapolations were used to estimate the cost associated with shortages leading to CMOP rejection and local fill.
Results
Survey completion totaled 20% of invited facilities (n = 17). Good geographic and VISN distribution was noted with representatives from VISNs 2, 4, 8, 9, 10, 12, 15, 16, 21, and 22. VISNs 10 and 12 provided the most representation with 3 participants, each. Level 1a facilities participated most (n = 9), followed by 1b (n = 6) and 1c (n = 2). Participating facilities reported a mean (SD) of 54 (21.5) pharmacists and 34 (15.3) pharmacy technician staff members employed. The most common reason for not participating was lack of personnel resources and competing demands. The CMOP participation was 100% (n = 7) and completed through a coordinated response.
Results of the budgetary increase and staff member time allocation survey assessments are provided (Figures 1 and 2). Five facilities provided an annual estimate of increased cost due to acquisition of drugs on shortage through open market purchases that ranged from about $150,000 to $750,000. Nearly half of the surveyed facilities endorsed having a drug shortage task force (n = 8) to respond to drug shortages and mitigate their impact.
Regarding drug product allocation, only 2 facilities did not have current restrictions for use due to a shortage. Many had between 1 and 10 of these restrictions implemented to conserve supply (n = 11, 64%), 2 facilities reported 11 to 20 restrictions, and 2 facilities noted more than 30 restrictions. Similarly, 3 facilities had not needed to revise any current treatment protocols due to drug shortages. The majority of facilities had revised 1 to 5 current protocols (n = 12, 70%), 1 revised 6 to 10 protocols, and 1 facility revised more than 10 protocols.
In assessing patient safety concerns, 1 facility identified a history of transferring patients to alternative medical sites for the patients to obtain necessary medication impacted by a local shortage. Additionally, during the BCG vaccine shortage, 6 facilities (35.3%) substituted mitomycin C for the treatment of urinary bladder cancer.
Most participants either agreed (n = 8, 47.0%) or strongly agreed (n = 4, 23.5%) that modifications to FAR to increase purchasing opportunities from foreign distributors during drug shortage would help mitigate the impact of such shortages. Similarly, most participants agreed (n = 10, 58.8%) or strongly agreed (n = 3, 17.6%) that PBM guidance on drug shortage management would help efficiently and effectively respond to issues that might arise. The consensus of participants also agreed (n = 13, 76.5%) that organized collaborations or working groups within each VISN might help assist in drug shortage management.
The CMOP facility data revealed that 2 sites did not require dedicated staffing to respond to shortages, and 3 sites had not experienced cost increases because of shortages. Pharmacist use varied between sites, with 2 facilities using 1 to 10 pharmacist h/wk, and 1 facility using 11 to 20 pharmacist h/wk, and 1 facility using 21 to 30 pharmacist h/wk. Technician utilization was more pronounced, with 2 facilities using more than 30 technician h/wk, and 2 facilities using 1 to 10 technician h/wk. Workload and costs may have been influenced in other ways as 3 sites endorsed using overtime pay, shifting product responsibility between CMOPs, prolonging patient wait times, and close monitoring for each. In fiscal year 2015, some sites experienced a 1% to 5% (n = 2) and 6% to 10% (n = 1) increase in operation cost attributable to shortage. Results from fiscal year 2016 showed that some sites continued to see a 1% to 5% (n = 1) and 6% to 10% (n = 2) increase in operation cost attributable to shortage.
Through aggregation of CMOP responses on the number of prescriptions sent back to local facility for fill due to back order, a downward trend in the total number of rejections was seen over the 2.5 fiscal years assessed. This amounted to more than 1 million rejections in fiscal year 2015, about 788,000 rejections in 2016, and about 318,000 rejections through the first 2 quarters of 2017.
A consistent rise in the medication procurement budget requirement was characterized within the single VA facility review. The quarterly median increase was 2.7% over 2.5 years (min: -1.4%; max: 6.6%) for total outpatient medication costs, excluding hepatitis C antiviral therapies. Procurement cost records were insufficient to characterize historic expenditures for 4 of the prespecified drug products. The data collected on tamsulosin capsule and nitroprusside vial procurement during shortage is provided (Figures 3 and 4). Over the time frame of procurement records found on review, the added costs of nitroprusside vials and tamsulosin capsules were $22,766.09 (+167.9% of base cost) and $17,433.70 (+657.3% of base cost), respectively. No patient safety data were found on review.
Discussion
Drug product shortages represent a barrier to quality and efficiency across health care institutions. A survey of health system pharmacies in the southeastern U.S. found that the majority of respondents tracking shortage data reported a 300% to 500% markup by alternative or gray market suppliers for hard-to-find medications.11 These reports are similar to the authors’ analyses of the trends in increased procurement expenditures documented during the tamsulosin capsule and nitroprusside vial shortages and indirectly correlate with the survey results indicating that most facilities endorsed a trend in operation cost increase attributable to drugs product shortage. The estimated annual costs for open market purchases further informs the financial burden aggregated by this issue.
Indirect costs from drug shortage further complicated quantifying the impact of shortages. Many facilities acknowledged the indirect influence drug shortages have on staffing and workload due to the implementation of mitigation strategies. Most participants found it necessary to establish restrictions for use in addition to altering protocols. These required the time investment of essential personnel from development through execution and education. Situations also can arise for mass therapeutic substitution. In this example, pharmacy staff may be required to oversee medication transition from the product on shortage to an appropriate alternative. When substitution involves hundreds or thousands of outpatient prescriptions, such as the tamsulosin shortage, the process may be tedious and time consuming, depending on the level of clinical decision making needed to determine patient candidacy for transitioning products.
Improving institutional cost efficiency becomes a significant challenge with persistent drug shortages. Professional advocacy groups, such as the American Society of Health-System Pharmacists (ASHP), help provide guidance to organizations constrained by specific drug shortages.12 Staff knowledgeable in allocation, supply considerations, and product repackaging and stability data also are essential. Other mitigation strategies include automatic substitutions, restrictions for use or inventory control strategies, and open market procurement, or borrowing from other institutions.
Data gathered from the survey of CMOP facilities also helped elucidate strategies used to mitigate drug shortage impacts for those respondents impacted by shortage. Likely, the 2 CMOP facilities without dedicated staff focused on shortages are those whose outpatient prescription fulfillment responsibility were focused on supply items or controlled substances. The impacted CMOP respondents cited overtime pay, shifting product responsibility, and prolonging patient wait times as the most frequently employed mitigation strategies. When these and other strategies fail to manage a shortage, prescriptions are often sent back to the local facility to be filled. Unfortunately for these facilities, the same mitigation strategies used by CMOP are not always feasible. Overtime pay may not be possible given staffing and budgetary resources, sending prescriptions back to facilities in itself prolongs patient wait times, and local medical centers do not have the option of shifting product responsibility between sites or sending the prescription to another facility. Herein lies 1 rationale for the CMOP effort to reduce the volume of prescriptions sent back to local medical centers.
Multiple offices within the FDA have roles in the mitigation of national drug shortages within their regulatory purview. Much of the recent focus stems from provisions enacted under Title X of the FDA Safety and Innovation Act of 2012, which addresses problems in the drug-supply chain.12 Rectifying a shortage involves short- and long-term strategic planning to address supply, distribution, and market reaction to need. Collaboration between the FDA and manufacturers is one method by which demand can be satisfied through the coordination of resources, expedition of inspections, and root cause analysis of the shortage.
Similar collaborations within the VA were viewed favorably by respondents and might yield productive relationships if regional or VISN working groups were to be established. Alternative long-term strategies are executed through regulation, particularly concerning the importation of foreign manufactured drugs and regulatory discretion on supplier vetting. Despite a strong respondent consensus that regulatory modifications of foreign product importation in the setting of a drug shortage may be beneficial, such a change would require a congressional action and is not likely to be timely. Unfortunately, gray market pharmaceutical distribution, driven by wholesaler stockpiling to raise prices, is separate from manufacturer driven shortages and falls outside the FDA’s regulatory purview and institutional mitigation strategies.
Although based on this limited survey, general agreement existed on the importance of greater national collaboration and communication regarding drug shortage management strategies. This could include PBM guidance on specific shortage management opportunities or establishing collaborations by region or VISN. These possibilities may be more realistically attainable in comparison to modifying federal regulations on drug product procurement during active shortages, which requires an act of Congress. Many of the survey participants endorsed a drug shortage task force within their facility. Coordinating interaction between preexisting or newly established task forces or working groups on a monthly or quarterly basis may provide fruitful interactions and the exchange of strategies to reduce shortage impact on institutional cost, efficiency, and patient care.
Limitations
Quantifying the extent of drug shortage impact on patient safety and institutional costs is a difficult task. The procurement records data used for the analysis of a single VAMC were gathered through manual review of stored paper invoices, opening the possibility for missing data. It is also difficult to extrapolate the sum of indirect costs such as process changes, alternative product utilization, and pharmacy staffing resources as additional financial burdens to the affected institution. Any quantifiable cost assessment also is biased by contract terms between the VA and wholesalers in which unavailable products that must be purchased off-contract are subsequently reimbursed through credit or alternative means.
Patient safety events are frequently underreported, leading to underestimation of true safety event incidence. Given that these events are documented by multiple disciplines and that many of these documenters may not be aware consistently of the drug products and volume impacted by shortage, elucidating safety events unfolding in relation to shortage also is difficult to quantify.
The response rate for the survey was low but near the expected rate for this methodology. Feedback from several facilities was received, citing competing demands and workforce shortage as barriers to participation. The survey also was limited by reporting bias and recall bias. As assessment of prespecified past drug shortages may require intimate knowledge of pharmacy department processes and mitigation strategies, the accuracy of question answering may have been limited to the length of time the points of contact had been in their current position.
Conclusion
Drug shortages are a pervasive barrier to patient care within larger facilities of the VA health care system, similar to what has been characterized in the private sector. As a result of these shortages and the mitigation strategies to reduce their burden, many facilities endorsed trends in increasing workload for staff, institutional operation costs, and risk for patient safety and care quality concerns. Due to the demands of shortages, some facilities have implemented drug shortage task forces or equivalent groups to specifically manage these issues. Moving forward, the VA health care system may benefit from similar task forces or working groups at the VISN level, to aid in collaborative efforts to respond to shortage. Support for revising federal regulations on procurement in times of shortage and enhanced PBM drug shortage management guidance also was endorsed.
Drug product shortages threaten health care quality and public health by creating barriers to optimal care. The frequency of drug shortages has risen dramatically since 2005 and now influences broad areas of health care practice. More than 400 generic drug products have been affected, forcing institutions to purchase costly brand-name products, substitute alternative therapies, or procure from gray market vendors at increased institutional costs.1 Scarcity and cost have potential to negatively impact patient outcomes and the ability of health care organizations to respond to the needs of their patients.
Background
Although constantly fluctuating, the number of active shortages reached a height of 320 products at the end the third quarter of 2014.2 A 2011 analysis from Premier Healthcare Alliance estimated the added cost of purchasing brand, generic, or alternative drugs due to shortage may have inflated hospital costs by $200 million annually.1 In 2016, the number of active shortages dropped to 176, suggesting a downward trend. However, the drug supply chain remains a concern for pharmacies in the U.S.
Despite creative approaches to shortage management, the variable characteristics of shortages make planning difficult. For example, the drug product in short supply may or may not have an alternative for use in similar clinical scenarios. The impact of shortages of medications lacking an equivalent alternative product has been documented, such as the past shortage of succinylcholine for anesthesia, resulting in surgery cancellations when an alternative paralytic agent was not appropriate.3 In 2016, the Cleveland Clinic reported undertaking “military-style triage” in determining patients who required use of aminocaproic acid during open heart surgery due to its limited supply.4 Decisions to reserve drug supply for emergency use and prefilling syringes under pharmacy supervision to extend stability and shelf life are short-term solutions to larger, systemic issues. Unfortunately, these scenarios have the potential to disrupt patient care and diminish health outcomes.
Shortages of products that have an available therapeutic substitution may seem easily manageable, but additional considerations may be present. Bacillus Calmette-Guérin (BCG) is considered the drug of choice for bladder cancer. In 2011, there was a shortage of the BCG vaccine after mold was discovered in the formulation.5 Providers were forced to choose between reducing or reallocating the dose of BCG, turning away patient, or substituting mitomycin C, which is less effective and costlier. When tamsulosin capsules became difficult to obtain in 2014, some institutions began switching patients to alfuzosin.6 Although alfuzosin is similar in mechanism to tamsulosin, it may prolong the QTc interval. Not only did this substitution present a contraindication for patients with elevated QTc intervals or who were already receiving concomitant medications that prolonged the QTc interval, but also it required additional cost and resources needed to update electrocardiograms.
VA Consolidated Mail Outpatient Pharmacies
The VHA serves nearly 9 million patients at more than 1,200 facilities across the U.S.7 This large patient population results in an estimated 149 million outpatient prescriptions annually.8 About 80% of these are distributed by mail through 7 VA consolidated mail outpatient pharmacies (CMOPs). When drug scarcity impedes the ability of the CMOP to respond to medication demand, the local facility must fill these prescriptions. These rejections sent back to the facility impact workload, patient wait times, and access to medication therapy. Barriers to medication procurement in the VA also stem from regulations based on legislation, including the Trade Agreements Act, Drug Supply Chain Security Act, and the Federal Acquisition Regulation (FAR) (Table).
The impact of drug shortages has been described previously in the private sector, particularly for emergency medicine and chemotherapy.9,10 However, the impact of drug shortages on health care provision to veteran populations within the VA has not previously been analyzed. Due to the unique procurement regulations that influence the VA and the importance of continuing to provide optimal health care services to veterans, assessing the impact of drug shortages on patient safety and health care costs is necessary in informing policy decisions and guiding recommendations for mitigation strategies. The purpose of this study was to assess the influence of drug shortages on institutional costs and patient care within VA facilities and formulate recommendations for enhanced mitigation of this issue.
Methods
The primary outcome of this study was to characterize the impact of drug shortages on institutional cost and patient safety events among VHA facilities. Secondary outcomes included subgroup evaluation in reported drug shortage impact among 1a, 1b, and 1c complexity VA facility survey respondents and assessment of drug shortage impact on CMOP prescription order fulfillment and operation cost.
Definitions
The complexity ranking system is a facility grouping method used within the VA to characterize the level of service provision, teaching and research opportunities, patient volume, intensive care unit level, and other factors offered by a VA site. Rankings start from 1 (highest level of services offered) to 3 (lowest level of services offered), with level 1 facilities further divided into a, b, and c subdivisions. A level 1a facility will be larger with more services offered than a 1b, which is larger and offers more services than a 1c facility. The VA facilities are further characterized by regional distribution. Sites are grouped under VISNs of which there are currently 21.
The CMOP program was responsible for dispensing about 119 million outpatient prescriptions in 2016 and includes designated sites for the dispensing of controlled substances and supply items. The VA Pharmacy Benefits Management Service (PBM) oversees formulary management, plans national drug policy, promotes safe and appropriate drug therapy, and delivers high-quality and sustainable pharmacy benefits for veterans.
Study Design
A descriptive study was initiated to characterize the impact of drug shortages among VA facilities. An analysis of administrative medication safety event reporting and institutional costs data at the Denver VAMC in Colorado was done, focusing on predetermined drug products involved in a recent shortage. The analysis was accomplished through a review of the VA adverse drug events reporting system (VA ADERS) reports and a local medication errors quality improvement database and paper procurement records, respectively. Concurrently, a survey was disseminated among qualifying VA facilities across the country that sought to characterize the impact of drug shortages nationally.
Sample Selection
Denver VAMC. The Denver VAMC, where the authors were located, was selected as the local sample site. The intention was to compare the strategies used locally with strategies used among similar (level 1a, 1b, and 1c) facilities. Preselected “cost-impacting” drug products were identified through a review of historic shortages with a significant local impact. These drugs were defined as low cost/high utilization (eg, tamsulosin 0.4-mg capsules and ketorolac solution), medium cost/utilization (eg, piperacillin/tazobactam IV solutions and aminocaproic acid solution), and high cost/low utilization (eg, nitroprusside IV solution and BCG vaccine solution). Additionally, patient safety event data reported internally for quality improvement and locally via VA ADERS were reviewed for preselected “safety impact” drug products and included BCG vaccine, tamsulosin capsules, IV fluid products, calcium gluconate and chloride injections, and aminocaproic acid injection.
National Survey. The authors identified 84 level 1 complexity facilities and used the PBM pharmacy directory to contact the administrative personnel representing each facility. These representatives identified a point of contact to aid in survey completion. A separate survey also was sent to the CMOP facilities (survey outlines available at www.fedprac.com).
Data Collection
Denver VAMC. Financial data were sampled through a manual review of paper procurement records stored by date in the inpatient pharmacy of the Denver VAMC. Variables included units of product used over the period of drug shortage, cost per unit during shortage, and cost per unit before shortage. This information also was supplemented with data from the prescription processing software’s drug file. Patient safety data were gathered through query of the identified event reporting databases for the prespecified drug on shortage. These variables included the type of error and the effect the error had on the patient.
National Survey. Data collection focused on notable drug shortages and patient safety reporting between January 1, 2013 and December 31, 2016. The survey was maintained in a facility-specific spreadsheet. Editing capabilities were disabled for all actions other than responding to questions. Recipients were followed up with a courtesy e-mail after 2 weeks and another 2 times unless a survey was received. Data were de-identified and aggregated for analyses.
Statistical Analyses
Excel 2010 (Microsoft, Redmond, WA) descriptive statistics were used to relay information from this assessment. Extrapolations from procurement cost data and drug product utilization were used to estimate the enhanced direct cost associated with identified drug shortages. Similar extrapolations were used to estimate the cost associated with shortages leading to CMOP rejection and local fill.
Results
Survey completion totaled 20% of invited facilities (n = 17). Good geographic and VISN distribution was noted with representatives from VISNs 2, 4, 8, 9, 10, 12, 15, 16, 21, and 22. VISNs 10 and 12 provided the most representation with 3 participants, each. Level 1a facilities participated most (n = 9), followed by 1b (n = 6) and 1c (n = 2). Participating facilities reported a mean (SD) of 54 (21.5) pharmacists and 34 (15.3) pharmacy technician staff members employed. The most common reason for not participating was lack of personnel resources and competing demands. The CMOP participation was 100% (n = 7) and completed through a coordinated response.
Results of the budgetary increase and staff member time allocation survey assessments are provided (Figures 1 and 2). Five facilities provided an annual estimate of increased cost due to acquisition of drugs on shortage through open market purchases that ranged from about $150,000 to $750,000. Nearly half of the surveyed facilities endorsed having a drug shortage task force (n = 8) to respond to drug shortages and mitigate their impact.
Regarding drug product allocation, only 2 facilities did not have current restrictions for use due to a shortage. Many had between 1 and 10 of these restrictions implemented to conserve supply (n = 11, 64%), 2 facilities reported 11 to 20 restrictions, and 2 facilities noted more than 30 restrictions. Similarly, 3 facilities had not needed to revise any current treatment protocols due to drug shortages. The majority of facilities had revised 1 to 5 current protocols (n = 12, 70%), 1 revised 6 to 10 protocols, and 1 facility revised more than 10 protocols.
In assessing patient safety concerns, 1 facility identified a history of transferring patients to alternative medical sites for the patients to obtain necessary medication impacted by a local shortage. Additionally, during the BCG vaccine shortage, 6 facilities (35.3%) substituted mitomycin C for the treatment of urinary bladder cancer.
Most participants either agreed (n = 8, 47.0%) or strongly agreed (n = 4, 23.5%) that modifications to FAR to increase purchasing opportunities from foreign distributors during drug shortage would help mitigate the impact of such shortages. Similarly, most participants agreed (n = 10, 58.8%) or strongly agreed (n = 3, 17.6%) that PBM guidance on drug shortage management would help efficiently and effectively respond to issues that might arise. The consensus of participants also agreed (n = 13, 76.5%) that organized collaborations or working groups within each VISN might help assist in drug shortage management.
The CMOP facility data revealed that 2 sites did not require dedicated staffing to respond to shortages, and 3 sites had not experienced cost increases because of shortages. Pharmacist use varied between sites, with 2 facilities using 1 to 10 pharmacist h/wk, and 1 facility using 11 to 20 pharmacist h/wk, and 1 facility using 21 to 30 pharmacist h/wk. Technician utilization was more pronounced, with 2 facilities using more than 30 technician h/wk, and 2 facilities using 1 to 10 technician h/wk. Workload and costs may have been influenced in other ways as 3 sites endorsed using overtime pay, shifting product responsibility between CMOPs, prolonging patient wait times, and close monitoring for each. In fiscal year 2015, some sites experienced a 1% to 5% (n = 2) and 6% to 10% (n = 1) increase in operation cost attributable to shortage. Results from fiscal year 2016 showed that some sites continued to see a 1% to 5% (n = 1) and 6% to 10% (n = 2) increase in operation cost attributable to shortage.
Through aggregation of CMOP responses on the number of prescriptions sent back to local facility for fill due to back order, a downward trend in the total number of rejections was seen over the 2.5 fiscal years assessed. This amounted to more than 1 million rejections in fiscal year 2015, about 788,000 rejections in 2016, and about 318,000 rejections through the first 2 quarters of 2017.
A consistent rise in the medication procurement budget requirement was characterized within the single VA facility review. The quarterly median increase was 2.7% over 2.5 years (min: -1.4%; max: 6.6%) for total outpatient medication costs, excluding hepatitis C antiviral therapies. Procurement cost records were insufficient to characterize historic expenditures for 4 of the prespecified drug products. The data collected on tamsulosin capsule and nitroprusside vial procurement during shortage is provided (Figures 3 and 4). Over the time frame of procurement records found on review, the added costs of nitroprusside vials and tamsulosin capsules were $22,766.09 (+167.9% of base cost) and $17,433.70 (+657.3% of base cost), respectively. No patient safety data were found on review.
Discussion
Drug product shortages represent a barrier to quality and efficiency across health care institutions. A survey of health system pharmacies in the southeastern U.S. found that the majority of respondents tracking shortage data reported a 300% to 500% markup by alternative or gray market suppliers for hard-to-find medications.11 These reports are similar to the authors’ analyses of the trends in increased procurement expenditures documented during the tamsulosin capsule and nitroprusside vial shortages and indirectly correlate with the survey results indicating that most facilities endorsed a trend in operation cost increase attributable to drugs product shortage. The estimated annual costs for open market purchases further informs the financial burden aggregated by this issue.
Indirect costs from drug shortage further complicated quantifying the impact of shortages. Many facilities acknowledged the indirect influence drug shortages have on staffing and workload due to the implementation of mitigation strategies. Most participants found it necessary to establish restrictions for use in addition to altering protocols. These required the time investment of essential personnel from development through execution and education. Situations also can arise for mass therapeutic substitution. In this example, pharmacy staff may be required to oversee medication transition from the product on shortage to an appropriate alternative. When substitution involves hundreds or thousands of outpatient prescriptions, such as the tamsulosin shortage, the process may be tedious and time consuming, depending on the level of clinical decision making needed to determine patient candidacy for transitioning products.
Improving institutional cost efficiency becomes a significant challenge with persistent drug shortages. Professional advocacy groups, such as the American Society of Health-System Pharmacists (ASHP), help provide guidance to organizations constrained by specific drug shortages.12 Staff knowledgeable in allocation, supply considerations, and product repackaging and stability data also are essential. Other mitigation strategies include automatic substitutions, restrictions for use or inventory control strategies, and open market procurement, or borrowing from other institutions.
Data gathered from the survey of CMOP facilities also helped elucidate strategies used to mitigate drug shortage impacts for those respondents impacted by shortage. Likely, the 2 CMOP facilities without dedicated staff focused on shortages are those whose outpatient prescription fulfillment responsibility were focused on supply items or controlled substances. The impacted CMOP respondents cited overtime pay, shifting product responsibility, and prolonging patient wait times as the most frequently employed mitigation strategies. When these and other strategies fail to manage a shortage, prescriptions are often sent back to the local facility to be filled. Unfortunately for these facilities, the same mitigation strategies used by CMOP are not always feasible. Overtime pay may not be possible given staffing and budgetary resources, sending prescriptions back to facilities in itself prolongs patient wait times, and local medical centers do not have the option of shifting product responsibility between sites or sending the prescription to another facility. Herein lies 1 rationale for the CMOP effort to reduce the volume of prescriptions sent back to local medical centers.
Multiple offices within the FDA have roles in the mitigation of national drug shortages within their regulatory purview. Much of the recent focus stems from provisions enacted under Title X of the FDA Safety and Innovation Act of 2012, which addresses problems in the drug-supply chain.12 Rectifying a shortage involves short- and long-term strategic planning to address supply, distribution, and market reaction to need. Collaboration between the FDA and manufacturers is one method by which demand can be satisfied through the coordination of resources, expedition of inspections, and root cause analysis of the shortage.
Similar collaborations within the VA were viewed favorably by respondents and might yield productive relationships if regional or VISN working groups were to be established. Alternative long-term strategies are executed through regulation, particularly concerning the importation of foreign manufactured drugs and regulatory discretion on supplier vetting. Despite a strong respondent consensus that regulatory modifications of foreign product importation in the setting of a drug shortage may be beneficial, such a change would require a congressional action and is not likely to be timely. Unfortunately, gray market pharmaceutical distribution, driven by wholesaler stockpiling to raise prices, is separate from manufacturer driven shortages and falls outside the FDA’s regulatory purview and institutional mitigation strategies.
Although based on this limited survey, general agreement existed on the importance of greater national collaboration and communication regarding drug shortage management strategies. This could include PBM guidance on specific shortage management opportunities or establishing collaborations by region or VISN. These possibilities may be more realistically attainable in comparison to modifying federal regulations on drug product procurement during active shortages, which requires an act of Congress. Many of the survey participants endorsed a drug shortage task force within their facility. Coordinating interaction between preexisting or newly established task forces or working groups on a monthly or quarterly basis may provide fruitful interactions and the exchange of strategies to reduce shortage impact on institutional cost, efficiency, and patient care.
Limitations
Quantifying the extent of drug shortage impact on patient safety and institutional costs is a difficult task. The procurement records data used for the analysis of a single VAMC were gathered through manual review of stored paper invoices, opening the possibility for missing data. It is also difficult to extrapolate the sum of indirect costs such as process changes, alternative product utilization, and pharmacy staffing resources as additional financial burdens to the affected institution. Any quantifiable cost assessment also is biased by contract terms between the VA and wholesalers in which unavailable products that must be purchased off-contract are subsequently reimbursed through credit or alternative means.
Patient safety events are frequently underreported, leading to underestimation of true safety event incidence. Given that these events are documented by multiple disciplines and that many of these documenters may not be aware consistently of the drug products and volume impacted by shortage, elucidating safety events unfolding in relation to shortage also is difficult to quantify.
The response rate for the survey was low but near the expected rate for this methodology. Feedback from several facilities was received, citing competing demands and workforce shortage as barriers to participation. The survey also was limited by reporting bias and recall bias. As assessment of prespecified past drug shortages may require intimate knowledge of pharmacy department processes and mitigation strategies, the accuracy of question answering may have been limited to the length of time the points of contact had been in their current position.
Conclusion
Drug shortages are a pervasive barrier to patient care within larger facilities of the VA health care system, similar to what has been characterized in the private sector. As a result of these shortages and the mitigation strategies to reduce their burden, many facilities endorsed trends in increasing workload for staff, institutional operation costs, and risk for patient safety and care quality concerns. Due to the demands of shortages, some facilities have implemented drug shortage task forces or equivalent groups to specifically manage these issues. Moving forward, the VA health care system may benefit from similar task forces or working groups at the VISN level, to aid in collaborative efforts to respond to shortage. Support for revising federal regulations on procurement in times of shortage and enhanced PBM drug shortage management guidance also was endorsed.
1. Cherici C, Frazier J, Feldman M, et al. Navigating drug shortages in American healthcare: a premier healthcare alliance analysis. https://www.heartland.org/_template-assets/documents/publications/30103.pdf. Published March 2011. Accessed December 5, 2017.
2. American Society of Health-System Pharmacists. ASHP drug shortage statistics. https://www.ashp.org/Drug-Shortages/Shortage-Resources/Drug-Shortages-Statistics. Updated 2017. Accessed December 5, 2017.
3. Dooren JC. Most hospitals face drug shortages. The Wall Street Journal. http://www.wsj.com/articles/SB10001424052702304584404576442211187884744. Published July 13, 2011. Accessed December 5, 2017.
4. Fink S. Drug shortages forcing hard decisions on rationing treatment. The New York Times. http://www.nytimes.com/2016/01/29/us/drug-shortages-forcing-hard-decisions-on-rationing-treatments.html. Published January 29, 2016. Accessed December 5, 2017.
5. Loftus P. Drug shortages frustrate doctors, patients. The Wall Street Journal. http://www.wsj.com/articles/u-s-drug-shortages-frustrate-doctors-patients-1433125793. Published May, 31, 2015. Accessed December 5, 2017.
6. U.S. Food and Drug Administration. Strategic plan for preventing and mitigating drug shortages. http://www.fda.gov/downloads/Drugs/DrugSafety/DrugShortages/UCM372566.pdf. Published October 2013. Accessed August 22, 2016.
7. U.S. Department of Veteran Affairs, National Center for Veterans Analysis and Statistics. Quick facts. https://www.va.gov/vetdata/Quick_Facts.asp. Updated November 20, 2017. Accessed December 5, 2017.
8. U.S. Department of Veterans Affairs, Office of the Inspector General. Audit of Consolidated Mail Outpatient Pharmacy Program. https://www.va.gov/oig/pubs/VAOIG-15-05255-422.pdf. Accessed December 11, 2017.
9. Mazer-Amirshahi M, Pourmand A, Singer S, Pines JM, van den Anker J. Critical drug shortages: implications for emergency medicine. Acad Emerg Med. 2014;21(6):704-711.
10. McBride A, Holle LM, Westendorf C, et al. National survey on the effect of oncology drug shortages on cancer care. Am J Health Syst Pharm. 2013;70(7):609-617.
11. Caulder CR, Mehta B, Bookstaver PB, Sims LD, Stevenson B; South Carolina Society of Health-System Pharmacists. Impact of drug shortages on health system pharmacies in the southeastern United States. Hosp Pharm. 2015;50(4):279-286.
12. Florida Society of Health-System Pharmacists. Conservation strategies for IV fluids. http://www.fshp.org/news/165998/Conservation-Strategies-for-IV-Fluid.htm. Accessed December 11, 2017.
13. Federal Acquisition Regulation Site. FAR—Part 13 Simplified Acquisition Procedures, 13 CFR §§ 201-302. http://farsite.hill.af.mil/reghtml/regs/far2afmcfars/fardfars/far/13.htm. Updated January 13, 2017. Accessed December 5, 2017.
1. Cherici C, Frazier J, Feldman M, et al. Navigating drug shortages in American healthcare: a premier healthcare alliance analysis. https://www.heartland.org/_template-assets/documents/publications/30103.pdf. Published March 2011. Accessed December 5, 2017.
2. American Society of Health-System Pharmacists. ASHP drug shortage statistics. https://www.ashp.org/Drug-Shortages/Shortage-Resources/Drug-Shortages-Statistics. Updated 2017. Accessed December 5, 2017.
3. Dooren JC. Most hospitals face drug shortages. The Wall Street Journal. http://www.wsj.com/articles/SB10001424052702304584404576442211187884744. Published July 13, 2011. Accessed December 5, 2017.
4. Fink S. Drug shortages forcing hard decisions on rationing treatment. The New York Times. http://www.nytimes.com/2016/01/29/us/drug-shortages-forcing-hard-decisions-on-rationing-treatments.html. Published January 29, 2016. Accessed December 5, 2017.
5. Loftus P. Drug shortages frustrate doctors, patients. The Wall Street Journal. http://www.wsj.com/articles/u-s-drug-shortages-frustrate-doctors-patients-1433125793. Published May, 31, 2015. Accessed December 5, 2017.
6. U.S. Food and Drug Administration. Strategic plan for preventing and mitigating drug shortages. http://www.fda.gov/downloads/Drugs/DrugSafety/DrugShortages/UCM372566.pdf. Published October 2013. Accessed August 22, 2016.
7. U.S. Department of Veteran Affairs, National Center for Veterans Analysis and Statistics. Quick facts. https://www.va.gov/vetdata/Quick_Facts.asp. Updated November 20, 2017. Accessed December 5, 2017.
8. U.S. Department of Veterans Affairs, Office of the Inspector General. Audit of Consolidated Mail Outpatient Pharmacy Program. https://www.va.gov/oig/pubs/VAOIG-15-05255-422.pdf. Accessed December 11, 2017.
9. Mazer-Amirshahi M, Pourmand A, Singer S, Pines JM, van den Anker J. Critical drug shortages: implications for emergency medicine. Acad Emerg Med. 2014;21(6):704-711.
10. McBride A, Holle LM, Westendorf C, et al. National survey on the effect of oncology drug shortages on cancer care. Am J Health Syst Pharm. 2013;70(7):609-617.
11. Caulder CR, Mehta B, Bookstaver PB, Sims LD, Stevenson B; South Carolina Society of Health-System Pharmacists. Impact of drug shortages on health system pharmacies in the southeastern United States. Hosp Pharm. 2015;50(4):279-286.
12. Florida Society of Health-System Pharmacists. Conservation strategies for IV fluids. http://www.fshp.org/news/165998/Conservation-Strategies-for-IV-Fluid.htm. Accessed December 11, 2017.
13. Federal Acquisition Regulation Site. FAR—Part 13 Simplified Acquisition Procedures, 13 CFR §§ 201-302. http://farsite.hill.af.mil/reghtml/regs/far2afmcfars/fardfars/far/13.htm. Updated January 13, 2017. Accessed December 5, 2017.

How a malaria parasite is evading treatment
New research has revealed mutations that help the malaria parasite Plasmodium falciparum evade treatment.
Researchers used whole-genome analyses and chemogenetics to identify drug targets and resistance genes in cell lines of P falciparum that are resistant to antimalarial compounds.
The group’s work confirmed previously known mutations that contribute to the parasite’s resistance but also revealed new targets that may deepen our understanding of the parasite’s underlying biology.
“This exploration of the P falciparum resistome—the collection of antibiotic resistance genes—and its druggable genome will help guide new drug discovery efforts and advance our understanding of how the malaria parasite evolves to fight back,” said Elizabeth Winzeler, PhD, of the University of California San Diego School of Medicine.
She and her colleagues conducted this research and reported the results in Science.
“A single human [malaria] infection can result in a person containing upwards of a trillion asexual blood-stage parasites,” Dr Winzeler said. “Even with a relatively slow random mutation rate, these numbers confer extraordinary adaptability.”
“In just a few cycles of replication, the P falciparum genome can acquire a random genetic change that may render at least one parasite resistant to the activity of a drug or human-encoded antibody.”
Such rapid evolution can be exploited in vitro to document how the parasite evolves in the presence of antimalarials, and it can be used to reveal new drug targets.
With this in mind, Dr Winzeler and her colleagues performed a genome analysis of 262 P falciparum parasites resistant to 37 groups of compounds.
In 83 genes associated with drug resistance, the researchers identified hundreds of changes that could be mediating the resistance, including 159 gene amplifications and 148 nonsynonymous mutations.
The team then used clones of well-studied P falciparum parasites and exposed them to the compounds over time to induce resistance, monitoring the genetic changes that occurred as resistance developed.
The researchers were able to identify a likely target or resistance gene for every compound.
In addition, the team identified mutations that repeatedly occurred upon individual exposure to a variety of drugs, meaning these mutations are likely mediating resistance to numerous existing treatments.
“Our findings showed and underscored the challenging complexity of evolved drug resistance in P falciparum, but they also identified new drug targets or resistance genes for every compound for which resistant parasites were generated,” Dr Winzeler said.
“It revealed the complicated chemogenetic landscape of P falciparum but also provided a potential guide for designing new small-molecule inhibitors to fight this pathogen.” ![]()
New research has revealed mutations that help the malaria parasite Plasmodium falciparum evade treatment.
Researchers used whole-genome analyses and chemogenetics to identify drug targets and resistance genes in cell lines of P falciparum that are resistant to antimalarial compounds.
The group’s work confirmed previously known mutations that contribute to the parasite’s resistance but also revealed new targets that may deepen our understanding of the parasite’s underlying biology.
“This exploration of the P falciparum resistome—the collection of antibiotic resistance genes—and its druggable genome will help guide new drug discovery efforts and advance our understanding of how the malaria parasite evolves to fight back,” said Elizabeth Winzeler, PhD, of the University of California San Diego School of Medicine.
She and her colleagues conducted this research and reported the results in Science.
“A single human [malaria] infection can result in a person containing upwards of a trillion asexual blood-stage parasites,” Dr Winzeler said. “Even with a relatively slow random mutation rate, these numbers confer extraordinary adaptability.”
“In just a few cycles of replication, the P falciparum genome can acquire a random genetic change that may render at least one parasite resistant to the activity of a drug or human-encoded antibody.”
Such rapid evolution can be exploited in vitro to document how the parasite evolves in the presence of antimalarials, and it can be used to reveal new drug targets.
With this in mind, Dr Winzeler and her colleagues performed a genome analysis of 262 P falciparum parasites resistant to 37 groups of compounds.
In 83 genes associated with drug resistance, the researchers identified hundreds of changes that could be mediating the resistance, including 159 gene amplifications and 148 nonsynonymous mutations.
The team then used clones of well-studied P falciparum parasites and exposed them to the compounds over time to induce resistance, monitoring the genetic changes that occurred as resistance developed.
The researchers were able to identify a likely target or resistance gene for every compound.
In addition, the team identified mutations that repeatedly occurred upon individual exposure to a variety of drugs, meaning these mutations are likely mediating resistance to numerous existing treatments.
“Our findings showed and underscored the challenging complexity of evolved drug resistance in P falciparum, but they also identified new drug targets or resistance genes for every compound for which resistant parasites were generated,” Dr Winzeler said.
“It revealed the complicated chemogenetic landscape of P falciparum but also provided a potential guide for designing new small-molecule inhibitors to fight this pathogen.” ![]()
New research has revealed mutations that help the malaria parasite Plasmodium falciparum evade treatment.
Researchers used whole-genome analyses and chemogenetics to identify drug targets and resistance genes in cell lines of P falciparum that are resistant to antimalarial compounds.
The group’s work confirmed previously known mutations that contribute to the parasite’s resistance but also revealed new targets that may deepen our understanding of the parasite’s underlying biology.
“This exploration of the P falciparum resistome—the collection of antibiotic resistance genes—and its druggable genome will help guide new drug discovery efforts and advance our understanding of how the malaria parasite evolves to fight back,” said Elizabeth Winzeler, PhD, of the University of California San Diego School of Medicine.
She and her colleagues conducted this research and reported the results in Science.
“A single human [malaria] infection can result in a person containing upwards of a trillion asexual blood-stage parasites,” Dr Winzeler said. “Even with a relatively slow random mutation rate, these numbers confer extraordinary adaptability.”
“In just a few cycles of replication, the P falciparum genome can acquire a random genetic change that may render at least one parasite resistant to the activity of a drug or human-encoded antibody.”
Such rapid evolution can be exploited in vitro to document how the parasite evolves in the presence of antimalarials, and it can be used to reveal new drug targets.
With this in mind, Dr Winzeler and her colleagues performed a genome analysis of 262 P falciparum parasites resistant to 37 groups of compounds.
In 83 genes associated with drug resistance, the researchers identified hundreds of changes that could be mediating the resistance, including 159 gene amplifications and 148 nonsynonymous mutations.
The team then used clones of well-studied P falciparum parasites and exposed them to the compounds over time to induce resistance, monitoring the genetic changes that occurred as resistance developed.
The researchers were able to identify a likely target or resistance gene for every compound.
In addition, the team identified mutations that repeatedly occurred upon individual exposure to a variety of drugs, meaning these mutations are likely mediating resistance to numerous existing treatments.
“Our findings showed and underscored the challenging complexity of evolved drug resistance in P falciparum, but they also identified new drug targets or resistance genes for every compound for which resistant parasites were generated,” Dr Winzeler said.
“It revealed the complicated chemogenetic landscape of P falciparum but also provided a potential guide for designing new small-molecule inhibitors to fight this pathogen.” ![]()
Four Fattened Phalanges
1. A 54-year-old Colombian man presents with nail dystrophy of two years’ duration. Physical exam reveals longitudinally banded thickening of the lateral half of the nail plate with yellowish brown discoloration, transverse overcurvature of the nail, longitudinal white lines, and splinter hemorrhages.

Diagnosis: The lesion, diagnosed as onychomatricoma was surgically removed and sent for histopathologic study. Onychomatricoma is a subungual tumor characterized by banded or diffuse thickening, yellowish discoloration, splinter hemorrhages, and transverse overcurvature of the nail plate. Because the condition is not well known, it is often misdiagnosed.
For more information, see “Onychomatricoma: An Often Misdiagnosed Tumor of the Nails.” Cutis. 2015;96(2):121-124.
2. A 21-year-old woman has a slow-growing, asymptomatic nodule on the great toe. She denies antecedent trauma. A firm, flesh-colored, semimobile, nontender, subungual nodule can be seen in the distal lateral nail bed, extending into the adjacent tissue. Radiographic exam shows focal calcification of the nodule, with direct communication to the underlying distal phalanx.

Diagnosis: Subungual exostosis is a relatively uncommon, benign, osteocartilaginous tumor arising from the distal phalanx beneath the nail. It typically appears on the great toe during the second to third decade of life and has an equal incidence in both sexes. Its similarities to other dermatologic disorders involving the nail bed can lead to misdiagnosis, which may result in inadequate or extreme treatment.
For more information, see “Subungual Exostosis.” Cutis. 2012;90(5):241-243.
3. For the past year, a 30-year-old man’s left great toe has had a 3-cm exophytic, yellowish red, subungual nodule that is obliterating the nail plate. Ten years ago, a similar nodule in the same location was removed via laser by a podiatrist. Plain radiographs demonstrate an inferior cortical lucency of the distal phalanx, as well as a lucency over the nail bed with calcification extending to the soft tissues. MRI reveals bone erosion from the overlying mass.
Diagnosis: Digital fibromyxoma is the term used to describe a distinctive, slow-growing, soft-tissue tumor with a predilection for the periungual or subungual regions of the fingers and toes. This benign growth typically presents as a painless or tender nodule in middle-aged adults, with a slight male predominance. In a case series of 124 patients, 36% who had imaging studies showed bone involvement by an erosive or lytic lesion. Soft-tissue invasion of the bone is demonstrated by scalloping on plain radiographs.
For more information, see “Toe Nodule Obliterating the Nail Bed.” Cutis. 2016;97(4):260, 281-282.
4. A 41-year-old man presents with a slowly growing, tender lesion located on the left great hallux. When it first appeared five months ago, it resembled a blister. On exam today, a firm, 3.5-cm, flesh-colored, pedunculated nodule is seen on the lateral aspect of the toe. No lymphadenopathy is found. The patient reports no history of keloids or trauma to the foot.

Diagnosis: Shave biopsy showed findings consistent with dermatofibrosarcoma protuberans. A chest radiograph was unremarkable. Re-excision was performed with negative margins on frozen section but with positive peripheral and deep margins on permanent sections. The patient subsequently underwent amputation of the left great toe and was lost to follow-up after the initial postoperative period.
For more information, see “Dermatofibrosarcoma Protuberans.” Cutis. 2017;100(1):E6-E7.
1. A 54-year-old Colombian man presents with nail dystrophy of two years’ duration. Physical exam reveals longitudinally banded thickening of the lateral half of the nail plate with yellowish brown discoloration, transverse overcurvature of the nail, longitudinal white lines, and splinter hemorrhages.
Diagnosis: The lesion, diagnosed as onychomatricoma was surgically removed and sent for histopathologic study. Onychomatricoma is a subungual tumor characterized by banded or diffuse thickening, yellowish discoloration, splinter hemorrhages, and transverse overcurvature of the nail plate. Because the condition is not well known, it is often misdiagnosed.
For more information, see “Onychomatricoma: An Often Misdiagnosed Tumor of the Nails.” Cutis. 2015;96(2):121-124.
2. A 21-year-old woman has a slow-growing, asymptomatic nodule on the great toe. She denies antecedent trauma. A firm, flesh-colored, semimobile, nontender, subungual nodule can be seen in the distal lateral nail bed, extending into the adjacent tissue. Radiographic exam shows focal calcification of the nodule, with direct communication to the underlying distal phalanx.
Diagnosis: Subungual exostosis is a relatively uncommon, benign, osteocartilaginous tumor arising from the distal phalanx beneath the nail. It typically appears on the great toe during the second to third decade of life and has an equal incidence in both sexes. Its similarities to other dermatologic disorders involving the nail bed can lead to misdiagnosis, which may result in inadequate or extreme treatment.
For more information, see “Subungual Exostosis.” Cutis. 2012;90(5):241-243.
3. For the past year, a 30-year-old man’s left great toe has had a 3-cm exophytic, yellowish red, subungual nodule that is obliterating the nail plate. Ten years ago, a similar nodule in the same location was removed via laser by a podiatrist. Plain radiographs demonstrate an inferior cortical lucency of the distal phalanx, as well as a lucency over the nail bed with calcification extending to the soft tissues. MRI reveals bone erosion from the overlying mass.
Diagnosis: Digital fibromyxoma is the term used to describe a distinctive, slow-growing, soft-tissue tumor with a predilection for the periungual or subungual regions of the fingers and toes. This benign growth typically presents as a painless or tender nodule in middle-aged adults, with a slight male predominance. In a case series of 124 patients, 36% who had imaging studies showed bone involvement by an erosive or lytic lesion. Soft-tissue invasion of the bone is demonstrated by scalloping on plain radiographs.
For more information, see “Toe Nodule Obliterating the Nail Bed.” Cutis. 2016;97(4):260, 281-282.
4. A 41-year-old man presents with a slowly growing, tender lesion located on the left great hallux. When it first appeared five months ago, it resembled a blister. On exam today, a firm, 3.5-cm, flesh-colored, pedunculated nodule is seen on the lateral aspect of the toe. No lymphadenopathy is found. The patient reports no history of keloids or trauma to the foot.
Diagnosis: Shave biopsy showed findings consistent with dermatofibrosarcoma protuberans. A chest radiograph was unremarkable. Re-excision was performed with negative margins on frozen section but with positive peripheral and deep margins on permanent sections. The patient subsequently underwent amputation of the left great toe and was lost to follow-up after the initial postoperative period.
For more information, see “Dermatofibrosarcoma Protuberans.” Cutis. 2017;100(1):E6-E7.
1. A 54-year-old Colombian man presents with nail dystrophy of two years’ duration. Physical exam reveals longitudinally banded thickening of the lateral half of the nail plate with yellowish brown discoloration, transverse overcurvature of the nail, longitudinal white lines, and splinter hemorrhages.
Diagnosis: The lesion, diagnosed as onychomatricoma was surgically removed and sent for histopathologic study. Onychomatricoma is a subungual tumor characterized by banded or diffuse thickening, yellowish discoloration, splinter hemorrhages, and transverse overcurvature of the nail plate. Because the condition is not well known, it is often misdiagnosed.
For more information, see “Onychomatricoma: An Often Misdiagnosed Tumor of the Nails.” Cutis. 2015;96(2):121-124.
2. A 21-year-old woman has a slow-growing, asymptomatic nodule on the great toe. She denies antecedent trauma. A firm, flesh-colored, semimobile, nontender, subungual nodule can be seen in the distal lateral nail bed, extending into the adjacent tissue. Radiographic exam shows focal calcification of the nodule, with direct communication to the underlying distal phalanx.
Diagnosis: Subungual exostosis is a relatively uncommon, benign, osteocartilaginous tumor arising from the distal phalanx beneath the nail. It typically appears on the great toe during the second to third decade of life and has an equal incidence in both sexes. Its similarities to other dermatologic disorders involving the nail bed can lead to misdiagnosis, which may result in inadequate or extreme treatment.
For more information, see “Subungual Exostosis.” Cutis. 2012;90(5):241-243.
3. For the past year, a 30-year-old man’s left great toe has had a 3-cm exophytic, yellowish red, subungual nodule that is obliterating the nail plate. Ten years ago, a similar nodule in the same location was removed via laser by a podiatrist. Plain radiographs demonstrate an inferior cortical lucency of the distal phalanx, as well as a lucency over the nail bed with calcification extending to the soft tissues. MRI reveals bone erosion from the overlying mass.
Diagnosis: Digital fibromyxoma is the term used to describe a distinctive, slow-growing, soft-tissue tumor with a predilection for the periungual or subungual regions of the fingers and toes. This benign growth typically presents as a painless or tender nodule in middle-aged adults, with a slight male predominance. In a case series of 124 patients, 36% who had imaging studies showed bone involvement by an erosive or lytic lesion. Soft-tissue invasion of the bone is demonstrated by scalloping on plain radiographs.
For more information, see “Toe Nodule Obliterating the Nail Bed.” Cutis. 2016;97(4):260, 281-282.
4. A 41-year-old man presents with a slowly growing, tender lesion located on the left great hallux. When it first appeared five months ago, it resembled a blister. On exam today, a firm, 3.5-cm, flesh-colored, pedunculated nodule is seen on the lateral aspect of the toe. No lymphadenopathy is found. The patient reports no history of keloids or trauma to the foot.
Diagnosis: Shave biopsy showed findings consistent with dermatofibrosarcoma protuberans. A chest radiograph was unremarkable. Re-excision was performed with negative margins on frozen section but with positive peripheral and deep margins on permanent sections. The patient subsequently underwent amputation of the left great toe and was lost to follow-up after the initial postoperative period.
For more information, see “Dermatofibrosarcoma Protuberans.” Cutis. 2017;100(1):E6-E7.
Special Populations: New Onset Diabetes in the Elderly
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
This video was filmed at Metabolic & Endocrine Disease Summit (MEDS). Click here to learn more.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
This video was filmed at Metabolic & Endocrine Disease Summit (MEDS). Click here to learn more.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
This video was filmed at Metabolic & Endocrine Disease Summit (MEDS). Click here to learn more.

Emicizumab still available despite legal issues

The Roche Group has issued a statement reassuring the US hemophilia community that legal issues are not affecting patient access to emicizumab (Hemlibra), at least for the time being.
Emicizumab is a bispecific factor IXa- and factor X-directed antibody approved in the US as routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children who have hemophilia A and factor VIII (FVIII) inhibitors.
The Roche Group—which consists of Genentech in the US, Chugai in Japan, and Roche in the rest of the world—said emicizumab is still available for these patients, despite a legal battle with Baxalta, a wholly owned subsidiary of Shire.
In May 2017, Baxalta sued Genentech and Chugai for allegedly infringing upon US Patent No. 7,033,590.
The patent covers factor IX/factor IXa antibodies and antibody derivatives. It was issued to Baxter in April 2006 and assigned to Baxalta in March 2016. The patent is still active.
According to Baxalta, Genentech and Chugai are infringing on the patent by manufacturing, selling, or importing emicizumab. Therefore, Baxalta is seeking judgment in its favor and monetary damages.
The trial for this case is scheduled for September 2019.
However, in December 2017, Baxalta filed a motion for a preliminary injunction that would prevent the sale of emicizumab in the US. If granted, the injunction would prevent the following patients from receiving emicizumab:
- Hemophilia A patients with inhibitors (an inhibitor titer of greater than 5 Bethesda units) who cannot be treated effectively with FVIII replacement therapy, unless (i) they have already started emicizumab before the injunction is granted or (ii) they have previous experience with on-demand or prophylactic bypassing agents and their needs are not being met, as defined by Shire using criteria that include experiencing certain life- or limb-threatening bleeds or venous access issues.
- Hemophilia A patients who have an inhibitor titer less than or equal to 5 Bethesda units or who can be effectively treated with FVIII replacement therapy, regardless of whether they have already started emicizumab.
- Hemophilia A patients without inhibitors, regardless of whether they have already started emicizumab.
The Roche Group said it believes Baxalta’s claim is not valid, emicizumab does not infringe upon the patent, and the group will oppose the injunction.
The court’s decision on the injunction is expected this summer. In the meantime, emicizumab is still available for the aforementioned patients. ![]()
The Roche Group has issued a statement reassuring the US hemophilia community that legal issues are not affecting patient access to emicizumab (Hemlibra), at least for the time being.
Emicizumab is a bispecific factor IXa- and factor X-directed antibody approved in the US as routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children who have hemophilia A and factor VIII (FVIII) inhibitors.
The Roche Group—which consists of Genentech in the US, Chugai in Japan, and Roche in the rest of the world—said emicizumab is still available for these patients, despite a legal battle with Baxalta, a wholly owned subsidiary of Shire.
In May 2017, Baxalta sued Genentech and Chugai for allegedly infringing upon US Patent No. 7,033,590.
The patent covers factor IX/factor IXa antibodies and antibody derivatives. It was issued to Baxter in April 2006 and assigned to Baxalta in March 2016. The patent is still active.
According to Baxalta, Genentech and Chugai are infringing on the patent by manufacturing, selling, or importing emicizumab. Therefore, Baxalta is seeking judgment in its favor and monetary damages.
The trial for this case is scheduled for September 2019.
However, in December 2017, Baxalta filed a motion for a preliminary injunction that would prevent the sale of emicizumab in the US. If granted, the injunction would prevent the following patients from receiving emicizumab:
- Hemophilia A patients with inhibitors (an inhibitor titer of greater than 5 Bethesda units) who cannot be treated effectively with FVIII replacement therapy, unless (i) they have already started emicizumab before the injunction is granted or (ii) they have previous experience with on-demand or prophylactic bypassing agents and their needs are not being met, as defined by Shire using criteria that include experiencing certain life- or limb-threatening bleeds or venous access issues.
- Hemophilia A patients who have an inhibitor titer less than or equal to 5 Bethesda units or who can be effectively treated with FVIII replacement therapy, regardless of whether they have already started emicizumab.
- Hemophilia A patients without inhibitors, regardless of whether they have already started emicizumab.
The Roche Group said it believes Baxalta’s claim is not valid, emicizumab does not infringe upon the patent, and the group will oppose the injunction.
The court’s decision on the injunction is expected this summer. In the meantime, emicizumab is still available for the aforementioned patients. ![]()
The Roche Group has issued a statement reassuring the US hemophilia community that legal issues are not affecting patient access to emicizumab (Hemlibra), at least for the time being.
Emicizumab is a bispecific factor IXa- and factor X-directed antibody approved in the US as routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children who have hemophilia A and factor VIII (FVIII) inhibitors.
The Roche Group—which consists of Genentech in the US, Chugai in Japan, and Roche in the rest of the world—said emicizumab is still available for these patients, despite a legal battle with Baxalta, a wholly owned subsidiary of Shire.
In May 2017, Baxalta sued Genentech and Chugai for allegedly infringing upon US Patent No. 7,033,590.
The patent covers factor IX/factor IXa antibodies and antibody derivatives. It was issued to Baxter in April 2006 and assigned to Baxalta in March 2016. The patent is still active.
According to Baxalta, Genentech and Chugai are infringing on the patent by manufacturing, selling, or importing emicizumab. Therefore, Baxalta is seeking judgment in its favor and monetary damages.
The trial for this case is scheduled for September 2019.
However, in December 2017, Baxalta filed a motion for a preliminary injunction that would prevent the sale of emicizumab in the US. If granted, the injunction would prevent the following patients from receiving emicizumab:
- Hemophilia A patients with inhibitors (an inhibitor titer of greater than 5 Bethesda units) who cannot be treated effectively with FVIII replacement therapy, unless (i) they have already started emicizumab before the injunction is granted or (ii) they have previous experience with on-demand or prophylactic bypassing agents and their needs are not being met, as defined by Shire using criteria that include experiencing certain life- or limb-threatening bleeds or venous access issues.
- Hemophilia A patients who have an inhibitor titer less than or equal to 5 Bethesda units or who can be effectively treated with FVIII replacement therapy, regardless of whether they have already started emicizumab.
- Hemophilia A patients without inhibitors, regardless of whether they have already started emicizumab.
The Roche Group said it believes Baxalta’s claim is not valid, emicizumab does not infringe upon the patent, and the group will oppose the injunction.
The court’s decision on the injunction is expected this summer. In the meantime, emicizumab is still available for the aforementioned patients. ![]()
Generic bortezomib available in US
Fresenius Kabi has introduced its generic version of Velcade, Bortezomib for Injection, to the US market.
This is the first intravenous alternative to Velcade available in the US.
Bortezomib for Injection is available as a single dose vial containing 3.5 mg of lyophilized powder.
The product is approved to treat patients with multiple myeloma and patients with mantle cell lymphoma who have received at least 1 prior therapy.
For details, see the prescribing information for Bortezomib for Injection.
Velcade is a registered trademark of Millennium Pharmaceuticals, Inc. ![]()
Fresenius Kabi has introduced its generic version of Velcade, Bortezomib for Injection, to the US market.
This is the first intravenous alternative to Velcade available in the US.
Bortezomib for Injection is available as a single dose vial containing 3.5 mg of lyophilized powder.
The product is approved to treat patients with multiple myeloma and patients with mantle cell lymphoma who have received at least 1 prior therapy.
For details, see the prescribing information for Bortezomib for Injection.
Velcade is a registered trademark of Millennium Pharmaceuticals, Inc. ![]()
Fresenius Kabi has introduced its generic version of Velcade, Bortezomib for Injection, to the US market.
This is the first intravenous alternative to Velcade available in the US.
Bortezomib for Injection is available as a single dose vial containing 3.5 mg of lyophilized powder.
The product is approved to treat patients with multiple myeloma and patients with mantle cell lymphoma who have received at least 1 prior therapy.
For details, see the prescribing information for Bortezomib for Injection.
Velcade is a registered trademark of Millennium Pharmaceuticals, Inc. ![]()
Young e-cigarette users graduating to the real thing
Children who use noncigarette forms of tobacco are significantly more likely to try cigarettes in the future, according to survey data from over 10,000 young people aged 12-17 years.
An initial survey (wave 1) was conducted as part of the nationally representative Population Assessment of Tobacco and Health (PATH) study, with a follow-up (wave 2) administered to participants a year later. The analysis by Shannon L. Watkins, PhD, of the University of California, San Francisco, and her associates was based on data for 10,384 respondents who reported never smoking a cigarette in wave 1 and whose later cigarette use, which occurred in less than 5% overall, was reported in wave 2.
Those who used multiple noncigarette products were more likely than users of a single product to initiate cigarette use by wave 2. With never use of any tobacco as the reference, one model used by the investigators put the odds ratios of cigarette ever use at 4.98 for e-cigarettes only, 3.57 for combustibles only, and 8.57 for use of multiple products.
This study was supported by grants from the National Cancer Institute, Food and Drug Administration Center for Tobacco Products, National Institute on Drug Abuse, and National Center for Advancing Translational Sciences. No conflicts of interest were reported.
SOURCE: Watkins S et al. JAMA Pediatr. 2018 Jan 2. doi: 10.1001/jamapediatrics.2017.4173.
Children who use noncigarette forms of tobacco are significantly more likely to try cigarettes in the future, according to survey data from over 10,000 young people aged 12-17 years.
An initial survey (wave 1) was conducted as part of the nationally representative Population Assessment of Tobacco and Health (PATH) study, with a follow-up (wave 2) administered to participants a year later. The analysis by Shannon L. Watkins, PhD, of the University of California, San Francisco, and her associates was based on data for 10,384 respondents who reported never smoking a cigarette in wave 1 and whose later cigarette use, which occurred in less than 5% overall, was reported in wave 2.
Those who used multiple noncigarette products were more likely than users of a single product to initiate cigarette use by wave 2. With never use of any tobacco as the reference, one model used by the investigators put the odds ratios of cigarette ever use at 4.98 for e-cigarettes only, 3.57 for combustibles only, and 8.57 for use of multiple products.
This study was supported by grants from the National Cancer Institute, Food and Drug Administration Center for Tobacco Products, National Institute on Drug Abuse, and National Center for Advancing Translational Sciences. No conflicts of interest were reported.
SOURCE: Watkins S et al. JAMA Pediatr. 2018 Jan 2. doi: 10.1001/jamapediatrics.2017.4173.
Children who use noncigarette forms of tobacco are significantly more likely to try cigarettes in the future, according to survey data from over 10,000 young people aged 12-17 years.
An initial survey (wave 1) was conducted as part of the nationally representative Population Assessment of Tobacco and Health (PATH) study, with a follow-up (wave 2) administered to participants a year later. The analysis by Shannon L. Watkins, PhD, of the University of California, San Francisco, and her associates was based on data for 10,384 respondents who reported never smoking a cigarette in wave 1 and whose later cigarette use, which occurred in less than 5% overall, was reported in wave 2.
Those who used multiple noncigarette products were more likely than users of a single product to initiate cigarette use by wave 2. With never use of any tobacco as the reference, one model used by the investigators put the odds ratios of cigarette ever use at 4.98 for e-cigarettes only, 3.57 for combustibles only, and 8.57 for use of multiple products.
This study was supported by grants from the National Cancer Institute, Food and Drug Administration Center for Tobacco Products, National Institute on Drug Abuse, and National Center for Advancing Translational Sciences. No conflicts of interest were reported.
SOURCE: Watkins S et al. JAMA Pediatr. 2018 Jan 2. doi: 10.1001/jamapediatrics.2017.4173.
FROM JAMA PEDIATRICS
Innovative cholecystectomy grading scale could pay off for surgeons
ORLANDO – according to a study presented at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
As payment models shift toward bundled care, providers will be more closely evaluated on their postoperative outcomes, which can vary based on the difficulty of surgery, even for relatively common procedures.
“Gallbladder disease affects roughly 20 million people annually in the United States, with laparoscopic cholecystectomy being one of the most common operations performed by the typical surgeon,” said presenter Tarik Madni, MD, of the department of surgery, University of Texas Southwestern Medical Center, Dallas. “However not all cholecystectomies are created equal; increased inflammation can lead to increased operative times, increased conversion rates, as well as increased risk of complications.”
Given the increased scrutiny of surgical procedures, the current application of modifier 22, which allows surgeons to receive greater reimbursement for a more difficult surgery, is not enough, according to Dr. Madni.
To address this shortfall, investigators developed the Parkland grading scale, a five-tiered grading system that is designed to be easy to remember, limited in the number of grades, and correlated with clinical outcomes.
To determine the grades of the scale, Dr. Madni and his fellow investigators used 200 gallbladder images collected immediately before dissection and analyzed anatomy and inflammatory characteristics.
Gallbladders with a grade 1 would be relatively normal looking, while a grade 5 gallbladder would show perforation, necrosis, or not be clearly visible because of adhesions, according to Dr. Madni.
Between September 2016 and March 2017, investigators asked 11 acute care surgeons to prospectively grade gallbladders they saw before surgery using the Parkland scale and to fill out a questionnaire describing the difficulty of the procedure afterwards.
Of 667 gallbladders graded, 60 were assessed to be grade 1 (19%), 90 were grade 2 (28%), 102 were grade 3 (32%), 28 were grade 4 (9%), and 37 were grade 5 (12%) on the Parkland scale.
Grade 1 gallbladders had a mean procedure difficulty score of 1.43, while grade 5 gallbladders had a mean difficulty of 4.46. Grade 1 gallbladders also corresponded with the shortest mean surgery time of 63.31 minutes, compared with an average of 108.13 minutes for grade 5.
Acute cholecystitis diagnosis also increased by Parkland grade, from 36.7% in grade 1 gallbladders to 83.8% in grade 5 (P less than .0001), as did open conversion rates, from 0% to 21.6% (P less than .0001).
Mean length of stay rose fivefold between grade 1 and grade 5 procedures, from around 8 hours to 36 hours, respectively (P less than .0001).
Discussant Martin Zielinski, MD, FACS, director of medical trauma clinical research at the Mayo Clinic, Rochester, Minn., recognized the importance of having a grading scale but was curious why investigators did not analyze the American Association for the Surgery of Trauma’s (AAST) Emergency General Surgery anatomic grading scale, which is already in place.
“The AAST is a uniform, anatomic grading scale to measure the severity of diseases from the 16 most common [Emergency General Surgery] diseases,” Dr. Madni responded. “Unlike our operative-only finding scale, the AAST scale gives grades 1 through 5 definitions for four categories in each disease, not just operative, but clinical, imaging, operative, and pathologic categories.”
Comparatively, the Parkland scale is less cumbersome and covers a wider range of difficulty variation, according to Dr. Madni.
In the future, Dr. Madni and his colleagues will work to compare the Parkland scale to the AAST scale and look for ways to bridge the two.
Dr. Madni reported no relevant financial disclosures.
SOURCE: Madni T et al. EAST Scientific Assembly 2018 abstract #11.
ORLANDO – according to a study presented at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
As payment models shift toward bundled care, providers will be more closely evaluated on their postoperative outcomes, which can vary based on the difficulty of surgery, even for relatively common procedures.
“Gallbladder disease affects roughly 20 million people annually in the United States, with laparoscopic cholecystectomy being one of the most common operations performed by the typical surgeon,” said presenter Tarik Madni, MD, of the department of surgery, University of Texas Southwestern Medical Center, Dallas. “However not all cholecystectomies are created equal; increased inflammation can lead to increased operative times, increased conversion rates, as well as increased risk of complications.”
Given the increased scrutiny of surgical procedures, the current application of modifier 22, which allows surgeons to receive greater reimbursement for a more difficult surgery, is not enough, according to Dr. Madni.
To address this shortfall, investigators developed the Parkland grading scale, a five-tiered grading system that is designed to be easy to remember, limited in the number of grades, and correlated with clinical outcomes.
To determine the grades of the scale, Dr. Madni and his fellow investigators used 200 gallbladder images collected immediately before dissection and analyzed anatomy and inflammatory characteristics.
Gallbladders with a grade 1 would be relatively normal looking, while a grade 5 gallbladder would show perforation, necrosis, or not be clearly visible because of adhesions, according to Dr. Madni.
Between September 2016 and March 2017, investigators asked 11 acute care surgeons to prospectively grade gallbladders they saw before surgery using the Parkland scale and to fill out a questionnaire describing the difficulty of the procedure afterwards.
Of 667 gallbladders graded, 60 were assessed to be grade 1 (19%), 90 were grade 2 (28%), 102 were grade 3 (32%), 28 were grade 4 (9%), and 37 were grade 5 (12%) on the Parkland scale.
Grade 1 gallbladders had a mean procedure difficulty score of 1.43, while grade 5 gallbladders had a mean difficulty of 4.46. Grade 1 gallbladders also corresponded with the shortest mean surgery time of 63.31 minutes, compared with an average of 108.13 minutes for grade 5.
Acute cholecystitis diagnosis also increased by Parkland grade, from 36.7% in grade 1 gallbladders to 83.8% in grade 5 (P less than .0001), as did open conversion rates, from 0% to 21.6% (P less than .0001).
Mean length of stay rose fivefold between grade 1 and grade 5 procedures, from around 8 hours to 36 hours, respectively (P less than .0001).
Discussant Martin Zielinski, MD, FACS, director of medical trauma clinical research at the Mayo Clinic, Rochester, Minn., recognized the importance of having a grading scale but was curious why investigators did not analyze the American Association for the Surgery of Trauma’s (AAST) Emergency General Surgery anatomic grading scale, which is already in place.
“The AAST is a uniform, anatomic grading scale to measure the severity of diseases from the 16 most common [Emergency General Surgery] diseases,” Dr. Madni responded. “Unlike our operative-only finding scale, the AAST scale gives grades 1 through 5 definitions for four categories in each disease, not just operative, but clinical, imaging, operative, and pathologic categories.”
Comparatively, the Parkland scale is less cumbersome and covers a wider range of difficulty variation, according to Dr. Madni.
In the future, Dr. Madni and his colleagues will work to compare the Parkland scale to the AAST scale and look for ways to bridge the two.
Dr. Madni reported no relevant financial disclosures.
SOURCE: Madni T et al. EAST Scientific Assembly 2018 abstract #11.
ORLANDO – according to a study presented at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
As payment models shift toward bundled care, providers will be more closely evaluated on their postoperative outcomes, which can vary based on the difficulty of surgery, even for relatively common procedures.
“Gallbladder disease affects roughly 20 million people annually in the United States, with laparoscopic cholecystectomy being one of the most common operations performed by the typical surgeon,” said presenter Tarik Madni, MD, of the department of surgery, University of Texas Southwestern Medical Center, Dallas. “However not all cholecystectomies are created equal; increased inflammation can lead to increased operative times, increased conversion rates, as well as increased risk of complications.”
Given the increased scrutiny of surgical procedures, the current application of modifier 22, which allows surgeons to receive greater reimbursement for a more difficult surgery, is not enough, according to Dr. Madni.
To address this shortfall, investigators developed the Parkland grading scale, a five-tiered grading system that is designed to be easy to remember, limited in the number of grades, and correlated with clinical outcomes.
To determine the grades of the scale, Dr. Madni and his fellow investigators used 200 gallbladder images collected immediately before dissection and analyzed anatomy and inflammatory characteristics.
Gallbladders with a grade 1 would be relatively normal looking, while a grade 5 gallbladder would show perforation, necrosis, or not be clearly visible because of adhesions, according to Dr. Madni.
Between September 2016 and March 2017, investigators asked 11 acute care surgeons to prospectively grade gallbladders they saw before surgery using the Parkland scale and to fill out a questionnaire describing the difficulty of the procedure afterwards.
Of 667 gallbladders graded, 60 were assessed to be grade 1 (19%), 90 were grade 2 (28%), 102 were grade 3 (32%), 28 were grade 4 (9%), and 37 were grade 5 (12%) on the Parkland scale.
Grade 1 gallbladders had a mean procedure difficulty score of 1.43, while grade 5 gallbladders had a mean difficulty of 4.46. Grade 1 gallbladders also corresponded with the shortest mean surgery time of 63.31 minutes, compared with an average of 108.13 minutes for grade 5.
Acute cholecystitis diagnosis also increased by Parkland grade, from 36.7% in grade 1 gallbladders to 83.8% in grade 5 (P less than .0001), as did open conversion rates, from 0% to 21.6% (P less than .0001).
Mean length of stay rose fivefold between grade 1 and grade 5 procedures, from around 8 hours to 36 hours, respectively (P less than .0001).
Discussant Martin Zielinski, MD, FACS, director of medical trauma clinical research at the Mayo Clinic, Rochester, Minn., recognized the importance of having a grading scale but was curious why investigators did not analyze the American Association for the Surgery of Trauma’s (AAST) Emergency General Surgery anatomic grading scale, which is already in place.
“The AAST is a uniform, anatomic grading scale to measure the severity of diseases from the 16 most common [Emergency General Surgery] diseases,” Dr. Madni responded. “Unlike our operative-only finding scale, the AAST scale gives grades 1 through 5 definitions for four categories in each disease, not just operative, but clinical, imaging, operative, and pathologic categories.”
Comparatively, the Parkland scale is less cumbersome and covers a wider range of difficulty variation, according to Dr. Madni.
In the future, Dr. Madni and his colleagues will work to compare the Parkland scale to the AAST scale and look for ways to bridge the two.
Dr. Madni reported no relevant financial disclosures.
SOURCE: Madni T et al. EAST Scientific Assembly 2018 abstract #11.
REPORTING FROM EAST SCIENTIFIC ASSEMBLY
Key clinical point: A five-tiered grading system was developed to determine grades of cholecystectomy operative difficulty.
Major finding: Acute cholecystitis diagnosis also increased by Parkland grade, from 36.7% in grade 1 gallbladders to 83.8% in grade 5.
Study details: Eleven acute care surgeons graded gallbladders on initial view and then filled out a postoperative questionnaire.
Disclosures: The investigator reported no relevant financial disclosures.
Source: Madni T et al. EAST Scientifc Assembly 2018 abstract #11.
Behavioral issues, anorexia may presage celiac disease
The clinical challenges of celiac disease go beyond identifying the condition and helping families adjust to a child’s gluten-free diet. Behavioral problems and/or an eating disorder may predate celiac disease, according to Alex R. Kemper, MD, MPH, division chief of ambulatory pediatrics at Nationwide Children’s Hospital, Columbus, Ohio, and deputy editor of Pediatrics.

“We are learning more and more about celiac disease. The presentation and implication of celiac disease can involve more than the gastrointestinal tract,” Dr. Kemper noted. “Figuring out who to screen for celiac disease and how best to do so is complex, and we are always learning more about the best way to provide care after celiac disease is diagnosed.”
Impact of undiagnosed celiac disease on behavior
At the 2017 annual meeting of the American Academy of Pediatrics and, in a later interview, Dr. Kemper discussed a study that explored how behavior and celiac disease might be interrelated, particularly among children whose families don’t yet know their child has the condition.
“It’s challenging to assess the psychological impact of celiac disease autoimmunity when families aren’t aware a child has it, because prospective studies are difficult to do and recall bias can distort findings,” he noted.

Smith et al. used data from a prospective international study, The Environment Determinants of Diabetes in the Young (TEDDY), designed to learn about factors associated with type 1 diabetes and celiac disease over a 15-year follow-up period (Pediatrics. 2017 Mar. doi: 10.1542/peds.2016-2848).
TEDDY tracked 8,676 infants deemed at high risk for celiac autoimmunity based on their human leukocyte antigen (HLA) antigen status at birth. The investigators regularly measured celiac disease autoimmunity based on tissue transglutaminase antibodies (tTGA), beginning at age 2 years. They assessed the children’s behavior at ages 3.5 years and 4.5 years using the Achenbach System of Empirically Based Assessment. If a child was found to have celiac disease, the researchers revisited the earlier behavior scores reported by their mothers before their children’s status were known.
When the children were 3.5 years old, 66 had celiac disease that their mothers were not yet aware of and 440 children had diagnosed celiac disease. The 66 mothers unaware of their child’s condition reported more anxiety, depression, aggression, and sleep problems in their children than did the 440 mothers who knew their child’s diagnosis or the 3,651 mothers of children without celiac disease. The differences were subclinical but statistically significant.
“It is important to recognize that the magnitude of the psychological problems in the 3.5 year olds was small,” Dr. Kemper said in an interview. “Parents might not recognize these symptoms.”
When the researchers looked at child behavior reports only among the mothers who knew their children had celiac disease, no differences existed regardless of the children’s tTGA levels or whether they were following a gluten-free diet. Then, when the children were 4.5 years old and all mothers were aware of their child’s status, no significant differences in mothers’ reporting of child behavior existed across any of the groups.
“Perhaps the knowledge of the child’s celiac disease autoimmunity increases a parent’s sensitivity to physical discomforts of their child while providing an alternative explanation for any psychological symptoms the child exhibits,” the researchers offered.
“Pediatricians should be aware of this association and consider testing young children with a family history of celiac disease if there are concerns,” Dr. Kemper said in an interview. “Because the magnitude of change was subclinical, this study does not suggest the need for more extensive screening of all children.”
Link between celiac disease and anorexia nervosa
The eating disorders study Dr. Kemper discussed examined possible associations between celiac disease and anorexia nervosa (Pediatrics. 2017. doi: 10.1542/peds.2016-4367). Researchers compared 17,959 Swedish females diagnosed with celiac disease between 1969 and 2008, at a median 28 years old, to 89,379 controls matched by sex and age.
Anorexia occurred more often among those with celiac disease than those without: a rate of 27 girls per 100,000 with celiac disease developed anorexia per year, compared with 18 of 100,000 without celiac disease, for a hazard ratio for an anorexia nervosa diagnosis of 1.46 (95% confidence interval, 1.08-1.98). In addition, girls whose celiac disease had not yet been identified had more than double the odds of developing anorexia before diagnosis than did those without celiac disease (odds ratio, 2.13).
Females with celiac disease therefore were more likely to have anorexia both before and after their celiac diagnosis, although the authors noted that surveillance bias may have made it more likely for either of the patients’ conditions to be identified after the first was. Another possible explanation is shared genetic risk factors, the authors wrote.
Dr. Kemper also offered possible reasons, including one related to the child behavior study.
“It could be that girls with celiac disease might develop anorexia because of the need to focus on their diet,” he said in an interview. “Celiac disease has been associated with psychological problems, and so that could contribute.”
Until further research can shed light on the reasons for the associations, physicians simply should be aware of the study’s clinical implications.
“Pediatricians should be aware of the bidirectional association between celiac disease and anorexia nervosa in teens and young adult women, and be prepared to evaluate for celiac disease or treat anorexia,” Dr. Kemper said.
He noted the need for more research to learn “what pediatricians can do to help to either prevent these problems from developing in the first place, or identify and treat celiac disease or anorexia nervosa early to prevent long-term complications.”
Dr. Kemper reported having no relevant financial disclosures and no external funding. Ketil Stordal, MD, PhD, of the anorexia study received funding from the OAK foundation in Switzerland, and Cynthia M. Bulik, PhD, from the same study received funding from the Swedish Research Council, and has consulted for and received a grant from Shire. The remaining authors of the anorexia study had no relevant financial disclosures. The behavioral study was funded by the National Institutes of Health, the Juvenile Diabetes Research Foundation and the Centers for Disease Control and Prevention. The authors from the behavioral study had no relevant financial disclosures.
The clinical challenges of celiac disease go beyond identifying the condition and helping families adjust to a child’s gluten-free diet. Behavioral problems and/or an eating disorder may predate celiac disease, according to Alex R. Kemper, MD, MPH, division chief of ambulatory pediatrics at Nationwide Children’s Hospital, Columbus, Ohio, and deputy editor of Pediatrics.
“We are learning more and more about celiac disease. The presentation and implication of celiac disease can involve more than the gastrointestinal tract,” Dr. Kemper noted. “Figuring out who to screen for celiac disease and how best to do so is complex, and we are always learning more about the best way to provide care after celiac disease is diagnosed.”
Impact of undiagnosed celiac disease on behavior
At the 2017 annual meeting of the American Academy of Pediatrics and, in a later interview, Dr. Kemper discussed a study that explored how behavior and celiac disease might be interrelated, particularly among children whose families don’t yet know their child has the condition.
“It’s challenging to assess the psychological impact of celiac disease autoimmunity when families aren’t aware a child has it, because prospective studies are difficult to do and recall bias can distort findings,” he noted.
Smith et al. used data from a prospective international study, The Environment Determinants of Diabetes in the Young (TEDDY), designed to learn about factors associated with type 1 diabetes and celiac disease over a 15-year follow-up period (Pediatrics. 2017 Mar. doi: 10.1542/peds.2016-2848).
TEDDY tracked 8,676 infants deemed at high risk for celiac autoimmunity based on their human leukocyte antigen (HLA) antigen status at birth. The investigators regularly measured celiac disease autoimmunity based on tissue transglutaminase antibodies (tTGA), beginning at age 2 years. They assessed the children’s behavior at ages 3.5 years and 4.5 years using the Achenbach System of Empirically Based Assessment. If a child was found to have celiac disease, the researchers revisited the earlier behavior scores reported by their mothers before their children’s status were known.
When the children were 3.5 years old, 66 had celiac disease that their mothers were not yet aware of and 440 children had diagnosed celiac disease. The 66 mothers unaware of their child’s condition reported more anxiety, depression, aggression, and sleep problems in their children than did the 440 mothers who knew their child’s diagnosis or the 3,651 mothers of children without celiac disease. The differences were subclinical but statistically significant.
“It is important to recognize that the magnitude of the psychological problems in the 3.5 year olds was small,” Dr. Kemper said in an interview. “Parents might not recognize these symptoms.”
When the researchers looked at child behavior reports only among the mothers who knew their children had celiac disease, no differences existed regardless of the children’s tTGA levels or whether they were following a gluten-free diet. Then, when the children were 4.5 years old and all mothers were aware of their child’s status, no significant differences in mothers’ reporting of child behavior existed across any of the groups.
“Perhaps the knowledge of the child’s celiac disease autoimmunity increases a parent’s sensitivity to physical discomforts of their child while providing an alternative explanation for any psychological symptoms the child exhibits,” the researchers offered.
“Pediatricians should be aware of this association and consider testing young children with a family history of celiac disease if there are concerns,” Dr. Kemper said in an interview. “Because the magnitude of change was subclinical, this study does not suggest the need for more extensive screening of all children.”
Link between celiac disease and anorexia nervosa
The eating disorders study Dr. Kemper discussed examined possible associations between celiac disease and anorexia nervosa (Pediatrics. 2017. doi: 10.1542/peds.2016-4367). Researchers compared 17,959 Swedish females diagnosed with celiac disease between 1969 and 2008, at a median 28 years old, to 89,379 controls matched by sex and age.
Anorexia occurred more often among those with celiac disease than those without: a rate of 27 girls per 100,000 with celiac disease developed anorexia per year, compared with 18 of 100,000 without celiac disease, for a hazard ratio for an anorexia nervosa diagnosis of 1.46 (95% confidence interval, 1.08-1.98). In addition, girls whose celiac disease had not yet been identified had more than double the odds of developing anorexia before diagnosis than did those without celiac disease (odds ratio, 2.13).
Females with celiac disease therefore were more likely to have anorexia both before and after their celiac diagnosis, although the authors noted that surveillance bias may have made it more likely for either of the patients’ conditions to be identified after the first was. Another possible explanation is shared genetic risk factors, the authors wrote.
Dr. Kemper also offered possible reasons, including one related to the child behavior study.
“It could be that girls with celiac disease might develop anorexia because of the need to focus on their diet,” he said in an interview. “Celiac disease has been associated with psychological problems, and so that could contribute.”
Until further research can shed light on the reasons for the associations, physicians simply should be aware of the study’s clinical implications.
“Pediatricians should be aware of the bidirectional association between celiac disease and anorexia nervosa in teens and young adult women, and be prepared to evaluate for celiac disease or treat anorexia,” Dr. Kemper said.
He noted the need for more research to learn “what pediatricians can do to help to either prevent these problems from developing in the first place, or identify and treat celiac disease or anorexia nervosa early to prevent long-term complications.”
Dr. Kemper reported having no relevant financial disclosures and no external funding. Ketil Stordal, MD, PhD, of the anorexia study received funding from the OAK foundation in Switzerland, and Cynthia M. Bulik, PhD, from the same study received funding from the Swedish Research Council, and has consulted for and received a grant from Shire. The remaining authors of the anorexia study had no relevant financial disclosures. The behavioral study was funded by the National Institutes of Health, the Juvenile Diabetes Research Foundation and the Centers for Disease Control and Prevention. The authors from the behavioral study had no relevant financial disclosures.
The clinical challenges of celiac disease go beyond identifying the condition and helping families adjust to a child’s gluten-free diet. Behavioral problems and/or an eating disorder may predate celiac disease, according to Alex R. Kemper, MD, MPH, division chief of ambulatory pediatrics at Nationwide Children’s Hospital, Columbus, Ohio, and deputy editor of Pediatrics.
“We are learning more and more about celiac disease. The presentation and implication of celiac disease can involve more than the gastrointestinal tract,” Dr. Kemper noted. “Figuring out who to screen for celiac disease and how best to do so is complex, and we are always learning more about the best way to provide care after celiac disease is diagnosed.”
Impact of undiagnosed celiac disease on behavior
At the 2017 annual meeting of the American Academy of Pediatrics and, in a later interview, Dr. Kemper discussed a study that explored how behavior and celiac disease might be interrelated, particularly among children whose families don’t yet know their child has the condition.
“It’s challenging to assess the psychological impact of celiac disease autoimmunity when families aren’t aware a child has it, because prospective studies are difficult to do and recall bias can distort findings,” he noted.
Smith et al. used data from a prospective international study, The Environment Determinants of Diabetes in the Young (TEDDY), designed to learn about factors associated with type 1 diabetes and celiac disease over a 15-year follow-up period (Pediatrics. 2017 Mar. doi: 10.1542/peds.2016-2848).
TEDDY tracked 8,676 infants deemed at high risk for celiac autoimmunity based on their human leukocyte antigen (HLA) antigen status at birth. The investigators regularly measured celiac disease autoimmunity based on tissue transglutaminase antibodies (tTGA), beginning at age 2 years. They assessed the children’s behavior at ages 3.5 years and 4.5 years using the Achenbach System of Empirically Based Assessment. If a child was found to have celiac disease, the researchers revisited the earlier behavior scores reported by their mothers before their children’s status were known.
When the children were 3.5 years old, 66 had celiac disease that their mothers were not yet aware of and 440 children had diagnosed celiac disease. The 66 mothers unaware of their child’s condition reported more anxiety, depression, aggression, and sleep problems in their children than did the 440 mothers who knew their child’s diagnosis or the 3,651 mothers of children without celiac disease. The differences were subclinical but statistically significant.
“It is important to recognize that the magnitude of the psychological problems in the 3.5 year olds was small,” Dr. Kemper said in an interview. “Parents might not recognize these symptoms.”
When the researchers looked at child behavior reports only among the mothers who knew their children had celiac disease, no differences existed regardless of the children’s tTGA levels or whether they were following a gluten-free diet. Then, when the children were 4.5 years old and all mothers were aware of their child’s status, no significant differences in mothers’ reporting of child behavior existed across any of the groups.
“Perhaps the knowledge of the child’s celiac disease autoimmunity increases a parent’s sensitivity to physical discomforts of their child while providing an alternative explanation for any psychological symptoms the child exhibits,” the researchers offered.
“Pediatricians should be aware of this association and consider testing young children with a family history of celiac disease if there are concerns,” Dr. Kemper said in an interview. “Because the magnitude of change was subclinical, this study does not suggest the need for more extensive screening of all children.”
Link between celiac disease and anorexia nervosa
The eating disorders study Dr. Kemper discussed examined possible associations between celiac disease and anorexia nervosa (Pediatrics. 2017. doi: 10.1542/peds.2016-4367). Researchers compared 17,959 Swedish females diagnosed with celiac disease between 1969 and 2008, at a median 28 years old, to 89,379 controls matched by sex and age.
Anorexia occurred more often among those with celiac disease than those without: a rate of 27 girls per 100,000 with celiac disease developed anorexia per year, compared with 18 of 100,000 without celiac disease, for a hazard ratio for an anorexia nervosa diagnosis of 1.46 (95% confidence interval, 1.08-1.98). In addition, girls whose celiac disease had not yet been identified had more than double the odds of developing anorexia before diagnosis than did those without celiac disease (odds ratio, 2.13).
Females with celiac disease therefore were more likely to have anorexia both before and after their celiac diagnosis, although the authors noted that surveillance bias may have made it more likely for either of the patients’ conditions to be identified after the first was. Another possible explanation is shared genetic risk factors, the authors wrote.
Dr. Kemper also offered possible reasons, including one related to the child behavior study.
“It could be that girls with celiac disease might develop anorexia because of the need to focus on their diet,” he said in an interview. “Celiac disease has been associated with psychological problems, and so that could contribute.”
Until further research can shed light on the reasons for the associations, physicians simply should be aware of the study’s clinical implications.
“Pediatricians should be aware of the bidirectional association between celiac disease and anorexia nervosa in teens and young adult women, and be prepared to evaluate for celiac disease or treat anorexia,” Dr. Kemper said.
He noted the need for more research to learn “what pediatricians can do to help to either prevent these problems from developing in the first place, or identify and treat celiac disease or anorexia nervosa early to prevent long-term complications.”
Dr. Kemper reported having no relevant financial disclosures and no external funding. Ketil Stordal, MD, PhD, of the anorexia study received funding from the OAK foundation in Switzerland, and Cynthia M. Bulik, PhD, from the same study received funding from the Swedish Research Council, and has consulted for and received a grant from Shire. The remaining authors of the anorexia study had no relevant financial disclosures. The behavioral study was funded by the National Institutes of Health, the Juvenile Diabetes Research Foundation and the Centers for Disease Control and Prevention. The authors from the behavioral study had no relevant financial disclosures.