User login
Top-selling drugs going to patients with diabetes
, and more than half of the 20 biggest-selling drugs for the year are regularly prescribed to patients with diabetes, according to the Agency for Healthcare Research and Quality.
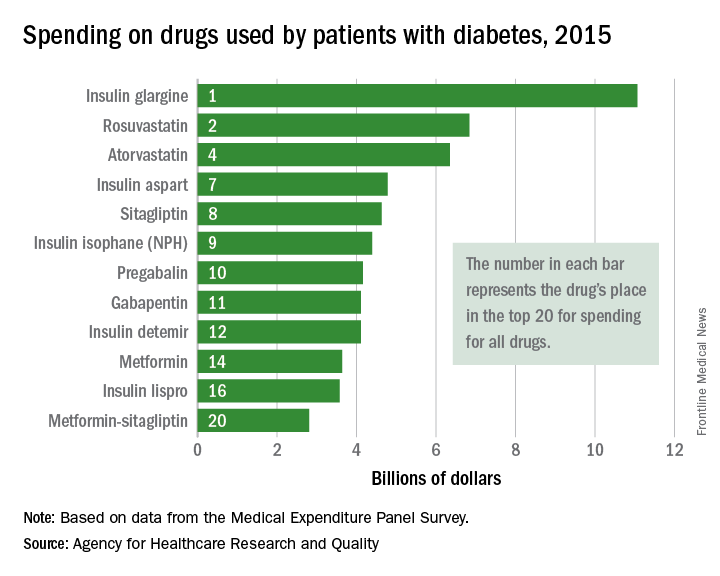
Drugs used by patients with diabetes also took up half of the next 10 spots in the list: gabapentin was 11th, insulin detemir was 12th, metformin was 14th, insulin lispro was 16th, and metformin-sitagliptin was 20th, according to the MEPS data.
The drugs in the MEPS top 10 for 2015 – the most recent year for which data are available – that are not commonly prescribed for diabetes were the asthma/chronic obstructive pulmonary drug fluticasone-salmeterol (third at $6.7 billion), the gastroesophageal reflux disease drug esomeprazole (fifth at $5.3 billion), and aripiprazole (sixth at $5.2 billion), which is used to treat schizophrenia and bipolar disorder.
, and more than half of the 20 biggest-selling drugs for the year are regularly prescribed to patients with diabetes, according to the Agency for Healthcare Research and Quality.
Drugs used by patients with diabetes also took up half of the next 10 spots in the list: gabapentin was 11th, insulin detemir was 12th, metformin was 14th, insulin lispro was 16th, and metformin-sitagliptin was 20th, according to the MEPS data.
The drugs in the MEPS top 10 for 2015 – the most recent year for which data are available – that are not commonly prescribed for diabetes were the asthma/chronic obstructive pulmonary drug fluticasone-salmeterol (third at $6.7 billion), the gastroesophageal reflux disease drug esomeprazole (fifth at $5.3 billion), and aripiprazole (sixth at $5.2 billion), which is used to treat schizophrenia and bipolar disorder.
, and more than half of the 20 biggest-selling drugs for the year are regularly prescribed to patients with diabetes, according to the Agency for Healthcare Research and Quality.
Drugs used by patients with diabetes also took up half of the next 10 spots in the list: gabapentin was 11th, insulin detemir was 12th, metformin was 14th, insulin lispro was 16th, and metformin-sitagliptin was 20th, according to the MEPS data.
The drugs in the MEPS top 10 for 2015 – the most recent year for which data are available – that are not commonly prescribed for diabetes were the asthma/chronic obstructive pulmonary drug fluticasone-salmeterol (third at $6.7 billion), the gastroesophageal reflux disease drug esomeprazole (fifth at $5.3 billion), and aripiprazole (sixth at $5.2 billion), which is used to treat schizophrenia and bipolar disorder.
Debunking Atopic Dermatitis Myths: Does Eczema Limit Patients' Daily Activities?
Myth: Eczema has a minimal impact on patients’ daily activities
Although eczema may be considered a relatively mild skin condition, the effects of the disease can be debilitating for many patients. Low quality of life (QoL) due to eczema flares and trigger avoidance can lead to decreased productivity in this population, and patients often report interference of the disease with participation in daily life, including activities at work and school.
In a series of patient satisfaction surveys administered by the National Eczema Foundation, 75% of adults with eczema said their disease interferes with their job and household chores. A study of 380 adult atopic dermatitis (AD) patients assessing the impact of the disease on QoL showed similar results, as 39.0% of participants said AD impacted shopping, home, and garden activities a lot or very much and 36.8% said it impacted these activities a little. Additionally, reports from caregivers of children with AD indicated that nearly 50% of children miss at least 1 day of the school year due to their disease, and 17% miss 5 or more days. Over time, these limitations can have a serious psychological impact in eczema patients of any age.
In the National Eczema Foundation survey, 71% of respondents said their disease also gets in the way of participating in sports or hobbies. Eczema patients may miss out on the physical and mental health benefits of activities associated with increased body temperature or prolonged contact with sports equipment, which can exacerbate symptoms. In general, outdoor activities in all seasons can be particularly troublesome in this population, as eczema flares can be triggered by cold or hot temperatures, humidity, wind, dry air, sun exposure, pollution, and contact with allergens like pollen or mold.
The impact of eczema treatments on patients’ daily activities also should be considered when evaluating QoL in this population, as it frequently takes considerable time out of a patient’s day to manage his/her disease. Control of symptoms often requires a multistep daily regimen involving medication, bathing, moisturizing, applying wet compresses, ridding the house of allergens, and/or cleaning sheets and clothing. According to the National Eczema Foundation survey, 1 in 3 respondents said it takes 1 or more hours per day to treat their disease. To encourage adherence and ensure optimal outcomes, physicians should work with patients to develop an eczema treatment plan that is both effective and manageable in terms of their daily routines.
Ultimately, the disease burden in eczema patients is multidimensional, extending beyond only cutaneous symptoms; therefore, it is important for clinicians to consider the impact on QoL when choosing treatments for patients with eczema and to initiate appropriate therapy at the onset of disease presentation to mitigate the effects of the condition on patients’ daily lives. Eczema management strategies should include QoL screening to ensure the disease has a minimal impact on patients' daily lives and preferred activities.
Atopic dermatitis affects all ages. https://www.aad.org/media/news-releases/adult-atopic-dermatitis. American Academy of Dermatology website. Posted July 27, 2017. Accessed March 12, 2018.
Impacts of eczema on exercise, social life and hobbies. HealthTalkOnline website. http://www.healthtalk.org/young-peoples-experiences/eczema/impacts-eczema-exercise-social-life-and-hobbies. Accessed March 12, 2018.
In your words. National Eczema Foundation website. https://nationaleczema.org/in-your-words-survey-series. Accessed March 11, 2018.
Simpson EL, Bieber T, Eckert L, et al. Patient burden of moderate to severe atopic dermatitis (AD): Insights from a phase 2b clinical trial of dupilumab in adults [published online January 14, 2016]. J Am Acad Dermatol. 2016;74:491-498.
Myth: Eczema has a minimal impact on patients’ daily activities
Although eczema may be considered a relatively mild skin condition, the effects of the disease can be debilitating for many patients. Low quality of life (QoL) due to eczema flares and trigger avoidance can lead to decreased productivity in this population, and patients often report interference of the disease with participation in daily life, including activities at work and school.
In a series of patient satisfaction surveys administered by the National Eczema Foundation, 75% of adults with eczema said their disease interferes with their job and household chores. A study of 380 adult atopic dermatitis (AD) patients assessing the impact of the disease on QoL showed similar results, as 39.0% of participants said AD impacted shopping, home, and garden activities a lot or very much and 36.8% said it impacted these activities a little. Additionally, reports from caregivers of children with AD indicated that nearly 50% of children miss at least 1 day of the school year due to their disease, and 17% miss 5 or more days. Over time, these limitations can have a serious psychological impact in eczema patients of any age.
In the National Eczema Foundation survey, 71% of respondents said their disease also gets in the way of participating in sports or hobbies. Eczema patients may miss out on the physical and mental health benefits of activities associated with increased body temperature or prolonged contact with sports equipment, which can exacerbate symptoms. In general, outdoor activities in all seasons can be particularly troublesome in this population, as eczema flares can be triggered by cold or hot temperatures, humidity, wind, dry air, sun exposure, pollution, and contact with allergens like pollen or mold.
The impact of eczema treatments on patients’ daily activities also should be considered when evaluating QoL in this population, as it frequently takes considerable time out of a patient’s day to manage his/her disease. Control of symptoms often requires a multistep daily regimen involving medication, bathing, moisturizing, applying wet compresses, ridding the house of allergens, and/or cleaning sheets and clothing. According to the National Eczema Foundation survey, 1 in 3 respondents said it takes 1 or more hours per day to treat their disease. To encourage adherence and ensure optimal outcomes, physicians should work with patients to develop an eczema treatment plan that is both effective and manageable in terms of their daily routines.
Ultimately, the disease burden in eczema patients is multidimensional, extending beyond only cutaneous symptoms; therefore, it is important for clinicians to consider the impact on QoL when choosing treatments for patients with eczema and to initiate appropriate therapy at the onset of disease presentation to mitigate the effects of the condition on patients’ daily lives. Eczema management strategies should include QoL screening to ensure the disease has a minimal impact on patients' daily lives and preferred activities.
Myth: Eczema has a minimal impact on patients’ daily activities
Although eczema may be considered a relatively mild skin condition, the effects of the disease can be debilitating for many patients. Low quality of life (QoL) due to eczema flares and trigger avoidance can lead to decreased productivity in this population, and patients often report interference of the disease with participation in daily life, including activities at work and school.
In a series of patient satisfaction surveys administered by the National Eczema Foundation, 75% of adults with eczema said their disease interferes with their job and household chores. A study of 380 adult atopic dermatitis (AD) patients assessing the impact of the disease on QoL showed similar results, as 39.0% of participants said AD impacted shopping, home, and garden activities a lot or very much and 36.8% said it impacted these activities a little. Additionally, reports from caregivers of children with AD indicated that nearly 50% of children miss at least 1 day of the school year due to their disease, and 17% miss 5 or more days. Over time, these limitations can have a serious psychological impact in eczema patients of any age.
In the National Eczema Foundation survey, 71% of respondents said their disease also gets in the way of participating in sports or hobbies. Eczema patients may miss out on the physical and mental health benefits of activities associated with increased body temperature or prolonged contact with sports equipment, which can exacerbate symptoms. In general, outdoor activities in all seasons can be particularly troublesome in this population, as eczema flares can be triggered by cold or hot temperatures, humidity, wind, dry air, sun exposure, pollution, and contact with allergens like pollen or mold.
The impact of eczema treatments on patients’ daily activities also should be considered when evaluating QoL in this population, as it frequently takes considerable time out of a patient’s day to manage his/her disease. Control of symptoms often requires a multistep daily regimen involving medication, bathing, moisturizing, applying wet compresses, ridding the house of allergens, and/or cleaning sheets and clothing. According to the National Eczema Foundation survey, 1 in 3 respondents said it takes 1 or more hours per day to treat their disease. To encourage adherence and ensure optimal outcomes, physicians should work with patients to develop an eczema treatment plan that is both effective and manageable in terms of their daily routines.
Ultimately, the disease burden in eczema patients is multidimensional, extending beyond only cutaneous symptoms; therefore, it is important for clinicians to consider the impact on QoL when choosing treatments for patients with eczema and to initiate appropriate therapy at the onset of disease presentation to mitigate the effects of the condition on patients’ daily lives. Eczema management strategies should include QoL screening to ensure the disease has a minimal impact on patients' daily lives and preferred activities.
Atopic dermatitis affects all ages. https://www.aad.org/media/news-releases/adult-atopic-dermatitis. American Academy of Dermatology website. Posted July 27, 2017. Accessed March 12, 2018.
Impacts of eczema on exercise, social life and hobbies. HealthTalkOnline website. http://www.healthtalk.org/young-peoples-experiences/eczema/impacts-eczema-exercise-social-life-and-hobbies. Accessed March 12, 2018.
In your words. National Eczema Foundation website. https://nationaleczema.org/in-your-words-survey-series. Accessed March 11, 2018.
Simpson EL, Bieber T, Eckert L, et al. Patient burden of moderate to severe atopic dermatitis (AD): Insights from a phase 2b clinical trial of dupilumab in adults [published online January 14, 2016]. J Am Acad Dermatol. 2016;74:491-498.
Atopic dermatitis affects all ages. https://www.aad.org/media/news-releases/adult-atopic-dermatitis. American Academy of Dermatology website. Posted July 27, 2017. Accessed March 12, 2018.
Impacts of eczema on exercise, social life and hobbies. HealthTalkOnline website. http://www.healthtalk.org/young-peoples-experiences/eczema/impacts-eczema-exercise-social-life-and-hobbies. Accessed March 12, 2018.
In your words. National Eczema Foundation website. https://nationaleczema.org/in-your-words-survey-series. Accessed March 11, 2018.
Simpson EL, Bieber T, Eckert L, et al. Patient burden of moderate to severe atopic dermatitis (AD): Insights from a phase 2b clinical trial of dupilumab in adults [published online January 14, 2016]. J Am Acad Dermatol. 2016;74:491-498.

Career development: One of many new focal points at HM 2018
Editor’s note: Each month, the Society of Hospital Medicine puts the spotlight on some of our most active members who are making substantial contributions to hospital medicine. Visit www.hospitalmedicine.org for more information on how you can lend your expertise to help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Kathleen Finn, MD, M. Phil, FACP, FHM, the inpatient associate program director of the internal medicine residency program at Massachusetts General Hospital and an assistant professor of medicine at Harvard Medical School, both in Boston. Dr. Finn has been a member of the Society of Hospital Medicine’s Annual Conference Committee for the past 8 years and is the course director for Hospital Medicine 2018 (HM18), to be held April 8-11 in Orlando.
When did you become a member of SHM, and how did you initially become involved with the Annual Conference Committee?

I became involved with the Annual Conference Committee 8 years ago because of my interest in education. Being a founding member of the SHM Boston Chapter, I gained experience planning the quarterly local chapter meetings. As a clinical educator and hospitalist, I was involved in planning conferences for faculty at my hospital. I found I really enjoyed developing educational conferences and curriculum, so when I heard about the Annual Conference Committee, I thought it would be a perfect fit.
It’s been a great experience getting to know committee members from all over the country and hearing their thoughts about the annual conference. It’s always exciting to brainstorm topic ideas and think about what would interest conference attendees.
Describe your role as course director.
My job as course director is to challenge committee members to be as creative as possible and help focus the discussion around the needs of SHM members while keeping to a schedule. I led a team of 23 amazing committee members through the planning stages for HM18 this past summer. With the help of Brittany Evans, SHM’s Education and Meetings Project Manager, and Dustin Smith, MD, FHM, the cocourse director, the committee reviewed prior conference agendas and feedback from attendees and from other SHM committees. Using that information, we discussed, brainstormed, voted on, and planned this year’s clinical content talks, workshops, and many of the specialty tracks.
What are you most looking forward to at HM18?
I am looking forward to the entire meeting! First, the location is exciting since this is our first time in Orlando. I’m curious to see what the facility is like, and I am hoping attendees use the location as a reason to bring their families and visit the theme parks. In recognition of our Orlando location, the committee got creative with titles for the conference. For example, geriatrics became “The Tale as Old as Time.” I hope some of the titles put a smile on the attendees’ faces.
I am also eagerly anticipating the nationally recognized speakers. We invited the best speakers we know from both subspecialty backgrounds and fellow hospitalists, and given the Orlando location, we tried to feature the best speakers from the Southeast. Finally, I am looking forward to the diversity of topics. The committee really thought broadly about relevant topics to today’s practicing hospitalists.
What will be new and different for attendees at HM18 in comparison to previous annual conferences?
There are many new things this year. Given the field of hospital medicine is now more than 20 years old, the committee thought it was important to focus on career development – not just for new hospitalists, but midcareer hospitalists as well. How do you make hospital medicine a lifelong, enjoyable, and engaging career? To explore and answer these questions, the Annual Conference Committee created several new tracks for HM18.
We created a Seasoning Your Career track that offers ideas on how to change your role midcareer – how to advance to a leadership position, how to use emotional intelligence to achieve success, how to prevent burnout, and, best of all, how to consider and change your hospitalist group’s work schedule, which rules our lives and our families’ lives. We also added financial planning advice to help you prepare for retirement.
Another new track at HM18 is the Career Development Workshops track, which includes a diversity of workshops meant to help build leadership skills, develop presentation/communication skills, encourage peers to give each other feedback, promote women in hospital medicine, prevent burnout, and turn ideas into clinical research. The Medical Education track also has a session on how to break into educational roles, especially if you want to expand your career into a leadership position in medical education.
In addition to Seasoning Your Career and Career Development Workshops, we have three other new tracks: Palliative Care, NP/PA, and The Great Debate. The Great Debate track uses the popular format of the perioperative debate given every year at the annual conference to tackle topics in infectious disease and pulmonary medicine. We ask very talented, opinionated, and humorous speakers to debate with each other over clinical content; it will be a great “smack down!”
Other new things for HM18 include:
- An interventional radiologist will speak about the latest procedures and when to refer your patients.
- A few surgeons will talk about managing surgical patients on your service and about decubitus ulcers.
- An oncologist will discuss the complications of the latest advanced agents on the wards.
- A rheumatologist will discuss the complications of new biologic agents.
- A rehab specialist will discuss the benefits and limitations of physical/occupational therapists and physiatrists.
- A speaker will discussing vulnerable populations, focusing on the social determinants of health, which last year’s HM17 plenary speaker Karen DeSalvo, MD, MPH, MSc raised as an important issue.
- There will be an “Updates in Addiction Medicine” lecture.
- There will be a new cardiology precourse and an expanded infectious disease precourse, which will also focus on sepsis.
How has the committee worked to ensure the course content is refreshed and current?
The reason the Annual Conference Committee is large is to ensure that there is a diversity of voices and talents from all over the country. There are both academic and community hospitalists on the committee; its members represent internal medicine, family medicine, pediatrics, and subspecialists, as well as administrators and hospitalist leaders. The annual meetings are planned over 3-4 months via weekly calls. In between calls, committee members are encouraged to discuss topics with their colleagues at home for opinions and advice.
The best ideas from the committee come from the group discussion and brainstorming. Someone mentions a topic, which leads someone else to add to it, and so on. Within the hour, we have some fantastic suggestions that the committee can run with. We also rely on input from SHM members: For example, many of the workshops’ topics are chosen from hundreds of submissions from members; speaker and content suggestions are submitted by hospitalist leaders from around the country and thereby provide insight into current topics. Combined, these offer a richness of ideas, which allows the committee to stay up to date and refresh old ideas.
What advice can you offer to early career hospitalists looking to get involved with the Annual Conference Committee or other conference planning roles?
My advice for early career hospitalists is to start locally. Join your local SHM chapter, or start one. In trying to plan local conferences, you begin to figure out which content areas interest hospitalists and how they can best be delivered. You might offer to give a talk at your local chapter or at your hospital and develop presentation skills. Developing a network of fellow hospitalists through your local chapter is important. The more local hospitalists you connect with, the more likely it is that they will think of you when they are planning a conference. At the national level, consider submitting a workshop or submitting an idea for content. Workshops are a great way to get recognized at the national level.
The Annual Conference Committee takes applications every year. Once you have some experience planning conferences or coordinating speakers, it would definitely be worth applying. You may not be selected your first year, but do not let that discourage you! Demonstrating interest and perseverance goes a long way. There are also many other national SHM committees to join and other ways to get involved. Your willingness to provide some of your time makes the society – and the specialty – what it is.
Ms. Steele is the marketing communications specialist at the Society of Hospital Medicine.
Editor’s note: Each month, the Society of Hospital Medicine puts the spotlight on some of our most active members who are making substantial contributions to hospital medicine. Visit www.hospitalmedicine.org for more information on how you can lend your expertise to help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Kathleen Finn, MD, M. Phil, FACP, FHM, the inpatient associate program director of the internal medicine residency program at Massachusetts General Hospital and an assistant professor of medicine at Harvard Medical School, both in Boston. Dr. Finn has been a member of the Society of Hospital Medicine’s Annual Conference Committee for the past 8 years and is the course director for Hospital Medicine 2018 (HM18), to be held April 8-11 in Orlando.
When did you become a member of SHM, and how did you initially become involved with the Annual Conference Committee?
I became involved with the Annual Conference Committee 8 years ago because of my interest in education. Being a founding member of the SHM Boston Chapter, I gained experience planning the quarterly local chapter meetings. As a clinical educator and hospitalist, I was involved in planning conferences for faculty at my hospital. I found I really enjoyed developing educational conferences and curriculum, so when I heard about the Annual Conference Committee, I thought it would be a perfect fit.
It’s been a great experience getting to know committee members from all over the country and hearing their thoughts about the annual conference. It’s always exciting to brainstorm topic ideas and think about what would interest conference attendees.
Describe your role as course director.
My job as course director is to challenge committee members to be as creative as possible and help focus the discussion around the needs of SHM members while keeping to a schedule. I led a team of 23 amazing committee members through the planning stages for HM18 this past summer. With the help of Brittany Evans, SHM’s Education and Meetings Project Manager, and Dustin Smith, MD, FHM, the cocourse director, the committee reviewed prior conference agendas and feedback from attendees and from other SHM committees. Using that information, we discussed, brainstormed, voted on, and planned this year’s clinical content talks, workshops, and many of the specialty tracks.
What are you most looking forward to at HM18?
I am looking forward to the entire meeting! First, the location is exciting since this is our first time in Orlando. I’m curious to see what the facility is like, and I am hoping attendees use the location as a reason to bring their families and visit the theme parks. In recognition of our Orlando location, the committee got creative with titles for the conference. For example, geriatrics became “The Tale as Old as Time.” I hope some of the titles put a smile on the attendees’ faces.
I am also eagerly anticipating the nationally recognized speakers. We invited the best speakers we know from both subspecialty backgrounds and fellow hospitalists, and given the Orlando location, we tried to feature the best speakers from the Southeast. Finally, I am looking forward to the diversity of topics. The committee really thought broadly about relevant topics to today’s practicing hospitalists.
What will be new and different for attendees at HM18 in comparison to previous annual conferences?
There are many new things this year. Given the field of hospital medicine is now more than 20 years old, the committee thought it was important to focus on career development – not just for new hospitalists, but midcareer hospitalists as well. How do you make hospital medicine a lifelong, enjoyable, and engaging career? To explore and answer these questions, the Annual Conference Committee created several new tracks for HM18.
We created a Seasoning Your Career track that offers ideas on how to change your role midcareer – how to advance to a leadership position, how to use emotional intelligence to achieve success, how to prevent burnout, and, best of all, how to consider and change your hospitalist group’s work schedule, which rules our lives and our families’ lives. We also added financial planning advice to help you prepare for retirement.
Another new track at HM18 is the Career Development Workshops track, which includes a diversity of workshops meant to help build leadership skills, develop presentation/communication skills, encourage peers to give each other feedback, promote women in hospital medicine, prevent burnout, and turn ideas into clinical research. The Medical Education track also has a session on how to break into educational roles, especially if you want to expand your career into a leadership position in medical education.
In addition to Seasoning Your Career and Career Development Workshops, we have three other new tracks: Palliative Care, NP/PA, and The Great Debate. The Great Debate track uses the popular format of the perioperative debate given every year at the annual conference to tackle topics in infectious disease and pulmonary medicine. We ask very talented, opinionated, and humorous speakers to debate with each other over clinical content; it will be a great “smack down!”
Other new things for HM18 include:
- An interventional radiologist will speak about the latest procedures and when to refer your patients.
- A few surgeons will talk about managing surgical patients on your service and about decubitus ulcers.
- An oncologist will discuss the complications of the latest advanced agents on the wards.
- A rheumatologist will discuss the complications of new biologic agents.
- A rehab specialist will discuss the benefits and limitations of physical/occupational therapists and physiatrists.
- A speaker will discussing vulnerable populations, focusing on the social determinants of health, which last year’s HM17 plenary speaker Karen DeSalvo, MD, MPH, MSc raised as an important issue.
- There will be an “Updates in Addiction Medicine” lecture.
- There will be a new cardiology precourse and an expanded infectious disease precourse, which will also focus on sepsis.
How has the committee worked to ensure the course content is refreshed and current?
The reason the Annual Conference Committee is large is to ensure that there is a diversity of voices and talents from all over the country. There are both academic and community hospitalists on the committee; its members represent internal medicine, family medicine, pediatrics, and subspecialists, as well as administrators and hospitalist leaders. The annual meetings are planned over 3-4 months via weekly calls. In between calls, committee members are encouraged to discuss topics with their colleagues at home for opinions and advice.
The best ideas from the committee come from the group discussion and brainstorming. Someone mentions a topic, which leads someone else to add to it, and so on. Within the hour, we have some fantastic suggestions that the committee can run with. We also rely on input from SHM members: For example, many of the workshops’ topics are chosen from hundreds of submissions from members; speaker and content suggestions are submitted by hospitalist leaders from around the country and thereby provide insight into current topics. Combined, these offer a richness of ideas, which allows the committee to stay up to date and refresh old ideas.
What advice can you offer to early career hospitalists looking to get involved with the Annual Conference Committee or other conference planning roles?
My advice for early career hospitalists is to start locally. Join your local SHM chapter, or start one. In trying to plan local conferences, you begin to figure out which content areas interest hospitalists and how they can best be delivered. You might offer to give a talk at your local chapter or at your hospital and develop presentation skills. Developing a network of fellow hospitalists through your local chapter is important. The more local hospitalists you connect with, the more likely it is that they will think of you when they are planning a conference. At the national level, consider submitting a workshop or submitting an idea for content. Workshops are a great way to get recognized at the national level.
The Annual Conference Committee takes applications every year. Once you have some experience planning conferences or coordinating speakers, it would definitely be worth applying. You may not be selected your first year, but do not let that discourage you! Demonstrating interest and perseverance goes a long way. There are also many other national SHM committees to join and other ways to get involved. Your willingness to provide some of your time makes the society – and the specialty – what it is.
Ms. Steele is the marketing communications specialist at the Society of Hospital Medicine.
Editor’s note: Each month, the Society of Hospital Medicine puts the spotlight on some of our most active members who are making substantial contributions to hospital medicine. Visit www.hospitalmedicine.org for more information on how you can lend your expertise to help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Kathleen Finn, MD, M. Phil, FACP, FHM, the inpatient associate program director of the internal medicine residency program at Massachusetts General Hospital and an assistant professor of medicine at Harvard Medical School, both in Boston. Dr. Finn has been a member of the Society of Hospital Medicine’s Annual Conference Committee for the past 8 years and is the course director for Hospital Medicine 2018 (HM18), to be held April 8-11 in Orlando.
When did you become a member of SHM, and how did you initially become involved with the Annual Conference Committee?
I became involved with the Annual Conference Committee 8 years ago because of my interest in education. Being a founding member of the SHM Boston Chapter, I gained experience planning the quarterly local chapter meetings. As a clinical educator and hospitalist, I was involved in planning conferences for faculty at my hospital. I found I really enjoyed developing educational conferences and curriculum, so when I heard about the Annual Conference Committee, I thought it would be a perfect fit.
It’s been a great experience getting to know committee members from all over the country and hearing their thoughts about the annual conference. It’s always exciting to brainstorm topic ideas and think about what would interest conference attendees.
Describe your role as course director.
My job as course director is to challenge committee members to be as creative as possible and help focus the discussion around the needs of SHM members while keeping to a schedule. I led a team of 23 amazing committee members through the planning stages for HM18 this past summer. With the help of Brittany Evans, SHM’s Education and Meetings Project Manager, and Dustin Smith, MD, FHM, the cocourse director, the committee reviewed prior conference agendas and feedback from attendees and from other SHM committees. Using that information, we discussed, brainstormed, voted on, and planned this year’s clinical content talks, workshops, and many of the specialty tracks.
What are you most looking forward to at HM18?
I am looking forward to the entire meeting! First, the location is exciting since this is our first time in Orlando. I’m curious to see what the facility is like, and I am hoping attendees use the location as a reason to bring their families and visit the theme parks. In recognition of our Orlando location, the committee got creative with titles for the conference. For example, geriatrics became “The Tale as Old as Time.” I hope some of the titles put a smile on the attendees’ faces.
I am also eagerly anticipating the nationally recognized speakers. We invited the best speakers we know from both subspecialty backgrounds and fellow hospitalists, and given the Orlando location, we tried to feature the best speakers from the Southeast. Finally, I am looking forward to the diversity of topics. The committee really thought broadly about relevant topics to today’s practicing hospitalists.
What will be new and different for attendees at HM18 in comparison to previous annual conferences?
There are many new things this year. Given the field of hospital medicine is now more than 20 years old, the committee thought it was important to focus on career development – not just for new hospitalists, but midcareer hospitalists as well. How do you make hospital medicine a lifelong, enjoyable, and engaging career? To explore and answer these questions, the Annual Conference Committee created several new tracks for HM18.
We created a Seasoning Your Career track that offers ideas on how to change your role midcareer – how to advance to a leadership position, how to use emotional intelligence to achieve success, how to prevent burnout, and, best of all, how to consider and change your hospitalist group’s work schedule, which rules our lives and our families’ lives. We also added financial planning advice to help you prepare for retirement.
Another new track at HM18 is the Career Development Workshops track, which includes a diversity of workshops meant to help build leadership skills, develop presentation/communication skills, encourage peers to give each other feedback, promote women in hospital medicine, prevent burnout, and turn ideas into clinical research. The Medical Education track also has a session on how to break into educational roles, especially if you want to expand your career into a leadership position in medical education.
In addition to Seasoning Your Career and Career Development Workshops, we have three other new tracks: Palliative Care, NP/PA, and The Great Debate. The Great Debate track uses the popular format of the perioperative debate given every year at the annual conference to tackle topics in infectious disease and pulmonary medicine. We ask very talented, opinionated, and humorous speakers to debate with each other over clinical content; it will be a great “smack down!”
Other new things for HM18 include:
- An interventional radiologist will speak about the latest procedures and when to refer your patients.
- A few surgeons will talk about managing surgical patients on your service and about decubitus ulcers.
- An oncologist will discuss the complications of the latest advanced agents on the wards.
- A rheumatologist will discuss the complications of new biologic agents.
- A rehab specialist will discuss the benefits and limitations of physical/occupational therapists and physiatrists.
- A speaker will discussing vulnerable populations, focusing on the social determinants of health, which last year’s HM17 plenary speaker Karen DeSalvo, MD, MPH, MSc raised as an important issue.
- There will be an “Updates in Addiction Medicine” lecture.
- There will be a new cardiology precourse and an expanded infectious disease precourse, which will also focus on sepsis.
How has the committee worked to ensure the course content is refreshed and current?
The reason the Annual Conference Committee is large is to ensure that there is a diversity of voices and talents from all over the country. There are both academic and community hospitalists on the committee; its members represent internal medicine, family medicine, pediatrics, and subspecialists, as well as administrators and hospitalist leaders. The annual meetings are planned over 3-4 months via weekly calls. In between calls, committee members are encouraged to discuss topics with their colleagues at home for opinions and advice.
The best ideas from the committee come from the group discussion and brainstorming. Someone mentions a topic, which leads someone else to add to it, and so on. Within the hour, we have some fantastic suggestions that the committee can run with. We also rely on input from SHM members: For example, many of the workshops’ topics are chosen from hundreds of submissions from members; speaker and content suggestions are submitted by hospitalist leaders from around the country and thereby provide insight into current topics. Combined, these offer a richness of ideas, which allows the committee to stay up to date and refresh old ideas.
What advice can you offer to early career hospitalists looking to get involved with the Annual Conference Committee or other conference planning roles?
My advice for early career hospitalists is to start locally. Join your local SHM chapter, or start one. In trying to plan local conferences, you begin to figure out which content areas interest hospitalists and how they can best be delivered. You might offer to give a talk at your local chapter or at your hospital and develop presentation skills. Developing a network of fellow hospitalists through your local chapter is important. The more local hospitalists you connect with, the more likely it is that they will think of you when they are planning a conference. At the national level, consider submitting a workshop or submitting an idea for content. Workshops are a great way to get recognized at the national level.
The Annual Conference Committee takes applications every year. Once you have some experience planning conferences or coordinating speakers, it would definitely be worth applying. You may not be selected your first year, but do not let that discourage you! Demonstrating interest and perseverance goes a long way. There are also many other national SHM committees to join and other ways to get involved. Your willingness to provide some of your time makes the society – and the specialty – what it is.
Ms. Steele is the marketing communications specialist at the Society of Hospital Medicine.
OSA may provide cardioprotection
, according to researchers.
In a study of 127 patients presenting with acute coronary syndromes (ACS), median peak cardiac troponin-I (cTn-I) values were significantly higher in patients without obstructive sleep apnea, compared with OSA patients (10.7; interquartile range: 1.78-40.1, vs. 3.79; IQR: 0.37-24.3, respectively; P = .04 ). The findings were published Feb. 5 in the journal CHEST®.
The study comprised 89 OSA patients and 38 non-OSA patients who were admitted to a hospital for acute coronary syndromes. The OSA group had a median apnea-hypopnea index (AHI) of 32, while the non-OSA group had a median AHI of 4.8. There was no significant difference between the two groups in gender, age, or cardiovascular risk factors such as hypertension, diabetes mellitus, body mass index, dyslipidemia, and smoking.
The cohort was part of the Continuous Positive Airway Pressure (CPAP) in Patients With Acute Coronary Syndrome and Obstructive Sleep Apnea (ISAACC) study, a prior randomized, controlled trial that evaluated the effect of CPAP treatment on new cardiovascular events in patients with an episode of ACS and OSA, reported Alicia Sánchez-de-la-Torre, PhD, of the respiratory department at Hospital Universitari Arnau de Vilanova and Santa Maria in Catalonia, Spain, and her coauthors.
Respiratory polygraphy was performed in the first 24-72 hours after hospital admission, and patients with an AHI of at least 15 events per hour were considered to have OSA. Those with an AHI less than 15 events per hour were included in the non-OSA group.
The OSA patients were randomized to conservative or CPAP treatment. An obstructive apnea “episode” was defined as a complete cessation of airflow for 10 seconds or longer, and an episode of hypopnea was defined as a reduction in airflow for at least 10 seconds associated with a greater than 4% decrease in arterial oxygen saturation.
Blood samples were collected from patients every 6 hours until two consecutive cTn-I measurements showed a decrease, with the highest measurement considered the peak cTn-I value.
Peak cTn-I value was significantly higher in non-OSA patients than in OSA patients. Median infarct size, measured by calculating the area under the cTn-I curve, was significantly different between the two groups (451 for non-OSA patients vs. 143 in OSA patients; P = .049), wrote Dr. Sánchez-de-la-Torre and her colleagues.
As cTn-I levels decreased, there was a trend toward increased OSA severity (P = .058). In the multivariable linear regression model used to assess OSA severity, patients with severe OSA had 61% lower cTn-I levels than non-OSA patients, the authors noted.
“The effects of chronic hypoxia in individual organ systems are not well understood. While chronic sustained hypoxia as seen with COPD may lead to pulmonary hypertension, chronic intermittent hypoxia (CIH) as seen predominantly in sleep apnea has been attributed to causing widespread effects ranging from systemic hypertension to metabolic dysfunction and systemic inflammation,” noted Krishna Sundar, MD, FCCP. “Despite these associations, an increased risk of major cardiovascular events from untreated OSA is yet to be definitively established.”

In this article, a protective effect from OSA on myocardial ischemic events is demonstrated in a group of 127 consecutively admitted patients with acute coronary syndrome (ACS). While it is interesting that a high proportion of those admitted for ACS had OSA, there were no significant differences in the age, sex, BMI, usage of antihypertensive or antiplatelet agents, presence of hypertension, DM, dyslipidemia or smoking status between those with and without OSA. “OSA appeared to confer a protective effect on the size of myocardial injury with those having higher AHI values demonstrating lower peak cardiac troponin values,” said Dr. Sundar, who is an associate clinical professor of pulmonary, critical care and sleep medicine at the University of Utah.“An effect of age (mean age in this study being 64 years) and BMI (mean being 27) on the occurrence of preconditioning effects of OSA is not excluded given deleterious effects of untreated OSA on infarct size in other studies on obese or younger patients with ACS. Further understanding of molecular effects of chronic hypoxia exposure (high altitude, chronic lung disease, OSA) is required before the complex and often contradictory effects of chronic hypoxia can be affirmed as being protective or deleterious,” added Dr. Sundar, who is also medical director of the Sleep-Wake Center at the University of Utah and a member of CHEST Physician’s editorial advisory board.
According to the study’s authors, their findings “suggest that patients with higher AHI are significantly more likely to have low cTn-I levels than patients without evidence of OSA, which could imply that patients with elevated AHI, particularly those with severe OSA, may experience less severe myocardial injury.”Limitations of the study include exclusion of patients with severe ACS, exclusion of sleepy subjects, and assessment of myocardial injury using cTn-I as a biomarker, without further data to determine infarct size.
“The possible role of OSA in cardioprotection should be explored in future studies,” the authors concluded.
The authors disclosed relationships with ResMed, Spanish Ministry of Health, Spanish Respiratory Society, Catalonian Cardiology Society, and ALLER. No other disclosures were reported.
SOURCE: Chest. 2018 Feb 5;153[2]:329-38. doi: 10.1016/j.chest.2017.06.046.
Although this study cannot definitively establish a clinically meaningful protective effect, it does provide important “preliminary evidence supporting the concept of OSA-induced cardioprotection” and challenges existing research, according to an editorial by Doron Aronson, MD, of the department of cardiology at Rambam Medical Center, Haifa, Israel, and coauthors (CHEST. 2018 Feb 153[2]:295-7. doi: 10.1016/j.chest.2017.07.036).
The results should be interpreted with caution, especially since accurate assessment of infarct size poses a challenge, they wrote.
“Myocardial infarct size is highly variable and is influenced by the duration of coronary occlusion, ST-segment elevation or non–ST elevation myocardial infarction, infarct location, residual antegrade infarct-related artery flow, collateral flow, the presence of non–culprit vessel coronary artery disease and myocardial metabolic demand,” they wrote. “Without accounting for these variables in a small study, results may be affected by variation in the characteristics of the patients.”
Though further study is needed, the findings may have “profound clinical implications regarding our therapeutic approach to patients with sleep apnea” if confirmed, the authors concluded.
Although this study cannot definitively establish a clinically meaningful protective effect, it does provide important “preliminary evidence supporting the concept of OSA-induced cardioprotection” and challenges existing research, according to an editorial by Doron Aronson, MD, of the department of cardiology at Rambam Medical Center, Haifa, Israel, and coauthors (CHEST. 2018 Feb 153[2]:295-7. doi: 10.1016/j.chest.2017.07.036).
The results should be interpreted with caution, especially since accurate assessment of infarct size poses a challenge, they wrote.
“Myocardial infarct size is highly variable and is influenced by the duration of coronary occlusion, ST-segment elevation or non–ST elevation myocardial infarction, infarct location, residual antegrade infarct-related artery flow, collateral flow, the presence of non–culprit vessel coronary artery disease and myocardial metabolic demand,” they wrote. “Without accounting for these variables in a small study, results may be affected by variation in the characteristics of the patients.”
Though further study is needed, the findings may have “profound clinical implications regarding our therapeutic approach to patients with sleep apnea” if confirmed, the authors concluded.
Although this study cannot definitively establish a clinically meaningful protective effect, it does provide important “preliminary evidence supporting the concept of OSA-induced cardioprotection” and challenges existing research, according to an editorial by Doron Aronson, MD, of the department of cardiology at Rambam Medical Center, Haifa, Israel, and coauthors (CHEST. 2018 Feb 153[2]:295-7. doi: 10.1016/j.chest.2017.07.036).
The results should be interpreted with caution, especially since accurate assessment of infarct size poses a challenge, they wrote.
“Myocardial infarct size is highly variable and is influenced by the duration of coronary occlusion, ST-segment elevation or non–ST elevation myocardial infarction, infarct location, residual antegrade infarct-related artery flow, collateral flow, the presence of non–culprit vessel coronary artery disease and myocardial metabolic demand,” they wrote. “Without accounting for these variables in a small study, results may be affected by variation in the characteristics of the patients.”
Though further study is needed, the findings may have “profound clinical implications regarding our therapeutic approach to patients with sleep apnea” if confirmed, the authors concluded.
, according to researchers.
In a study of 127 patients presenting with acute coronary syndromes (ACS), median peak cardiac troponin-I (cTn-I) values were significantly higher in patients without obstructive sleep apnea, compared with OSA patients (10.7; interquartile range: 1.78-40.1, vs. 3.79; IQR: 0.37-24.3, respectively; P = .04 ). The findings were published Feb. 5 in the journal CHEST®.
The study comprised 89 OSA patients and 38 non-OSA patients who were admitted to a hospital for acute coronary syndromes. The OSA group had a median apnea-hypopnea index (AHI) of 32, while the non-OSA group had a median AHI of 4.8. There was no significant difference between the two groups in gender, age, or cardiovascular risk factors such as hypertension, diabetes mellitus, body mass index, dyslipidemia, and smoking.
The cohort was part of the Continuous Positive Airway Pressure (CPAP) in Patients With Acute Coronary Syndrome and Obstructive Sleep Apnea (ISAACC) study, a prior randomized, controlled trial that evaluated the effect of CPAP treatment on new cardiovascular events in patients with an episode of ACS and OSA, reported Alicia Sánchez-de-la-Torre, PhD, of the respiratory department at Hospital Universitari Arnau de Vilanova and Santa Maria in Catalonia, Spain, and her coauthors.
Respiratory polygraphy was performed in the first 24-72 hours after hospital admission, and patients with an AHI of at least 15 events per hour were considered to have OSA. Those with an AHI less than 15 events per hour were included in the non-OSA group.
The OSA patients were randomized to conservative or CPAP treatment. An obstructive apnea “episode” was defined as a complete cessation of airflow for 10 seconds or longer, and an episode of hypopnea was defined as a reduction in airflow for at least 10 seconds associated with a greater than 4% decrease in arterial oxygen saturation.
Blood samples were collected from patients every 6 hours until two consecutive cTn-I measurements showed a decrease, with the highest measurement considered the peak cTn-I value.
Peak cTn-I value was significantly higher in non-OSA patients than in OSA patients. Median infarct size, measured by calculating the area under the cTn-I curve, was significantly different between the two groups (451 for non-OSA patients vs. 143 in OSA patients; P = .049), wrote Dr. Sánchez-de-la-Torre and her colleagues.
As cTn-I levels decreased, there was a trend toward increased OSA severity (P = .058). In the multivariable linear regression model used to assess OSA severity, patients with severe OSA had 61% lower cTn-I levels than non-OSA patients, the authors noted.
“The effects of chronic hypoxia in individual organ systems are not well understood. While chronic sustained hypoxia as seen with COPD may lead to pulmonary hypertension, chronic intermittent hypoxia (CIH) as seen predominantly in sleep apnea has been attributed to causing widespread effects ranging from systemic hypertension to metabolic dysfunction and systemic inflammation,” noted Krishna Sundar, MD, FCCP. “Despite these associations, an increased risk of major cardiovascular events from untreated OSA is yet to be definitively established.”
In this article, a protective effect from OSA on myocardial ischemic events is demonstrated in a group of 127 consecutively admitted patients with acute coronary syndrome (ACS). While it is interesting that a high proportion of those admitted for ACS had OSA, there were no significant differences in the age, sex, BMI, usage of antihypertensive or antiplatelet agents, presence of hypertension, DM, dyslipidemia or smoking status between those with and without OSA. “OSA appeared to confer a protective effect on the size of myocardial injury with those having higher AHI values demonstrating lower peak cardiac troponin values,” said Dr. Sundar, who is an associate clinical professor of pulmonary, critical care and sleep medicine at the University of Utah.“An effect of age (mean age in this study being 64 years) and BMI (mean being 27) on the occurrence of preconditioning effects of OSA is not excluded given deleterious effects of untreated OSA on infarct size in other studies on obese or younger patients with ACS. Further understanding of molecular effects of chronic hypoxia exposure (high altitude, chronic lung disease, OSA) is required before the complex and often contradictory effects of chronic hypoxia can be affirmed as being protective or deleterious,” added Dr. Sundar, who is also medical director of the Sleep-Wake Center at the University of Utah and a member of CHEST Physician’s editorial advisory board.
According to the study’s authors, their findings “suggest that patients with higher AHI are significantly more likely to have low cTn-I levels than patients without evidence of OSA, which could imply that patients with elevated AHI, particularly those with severe OSA, may experience less severe myocardial injury.”Limitations of the study include exclusion of patients with severe ACS, exclusion of sleepy subjects, and assessment of myocardial injury using cTn-I as a biomarker, without further data to determine infarct size.
“The possible role of OSA in cardioprotection should be explored in future studies,” the authors concluded.
The authors disclosed relationships with ResMed, Spanish Ministry of Health, Spanish Respiratory Society, Catalonian Cardiology Society, and ALLER. No other disclosures were reported.
SOURCE: Chest. 2018 Feb 5;153[2]:329-38. doi: 10.1016/j.chest.2017.06.046.
, according to researchers.
In a study of 127 patients presenting with acute coronary syndromes (ACS), median peak cardiac troponin-I (cTn-I) values were significantly higher in patients without obstructive sleep apnea, compared with OSA patients (10.7; interquartile range: 1.78-40.1, vs. 3.79; IQR: 0.37-24.3, respectively; P = .04 ). The findings were published Feb. 5 in the journal CHEST®.
The study comprised 89 OSA patients and 38 non-OSA patients who were admitted to a hospital for acute coronary syndromes. The OSA group had a median apnea-hypopnea index (AHI) of 32, while the non-OSA group had a median AHI of 4.8. There was no significant difference between the two groups in gender, age, or cardiovascular risk factors such as hypertension, diabetes mellitus, body mass index, dyslipidemia, and smoking.
The cohort was part of the Continuous Positive Airway Pressure (CPAP) in Patients With Acute Coronary Syndrome and Obstructive Sleep Apnea (ISAACC) study, a prior randomized, controlled trial that evaluated the effect of CPAP treatment on new cardiovascular events in patients with an episode of ACS and OSA, reported Alicia Sánchez-de-la-Torre, PhD, of the respiratory department at Hospital Universitari Arnau de Vilanova and Santa Maria in Catalonia, Spain, and her coauthors.
Respiratory polygraphy was performed in the first 24-72 hours after hospital admission, and patients with an AHI of at least 15 events per hour were considered to have OSA. Those with an AHI less than 15 events per hour were included in the non-OSA group.
The OSA patients were randomized to conservative or CPAP treatment. An obstructive apnea “episode” was defined as a complete cessation of airflow for 10 seconds or longer, and an episode of hypopnea was defined as a reduction in airflow for at least 10 seconds associated with a greater than 4% decrease in arterial oxygen saturation.
Blood samples were collected from patients every 6 hours until two consecutive cTn-I measurements showed a decrease, with the highest measurement considered the peak cTn-I value.
Peak cTn-I value was significantly higher in non-OSA patients than in OSA patients. Median infarct size, measured by calculating the area under the cTn-I curve, was significantly different between the two groups (451 for non-OSA patients vs. 143 in OSA patients; P = .049), wrote Dr. Sánchez-de-la-Torre and her colleagues.
As cTn-I levels decreased, there was a trend toward increased OSA severity (P = .058). In the multivariable linear regression model used to assess OSA severity, patients with severe OSA had 61% lower cTn-I levels than non-OSA patients, the authors noted.
“The effects of chronic hypoxia in individual organ systems are not well understood. While chronic sustained hypoxia as seen with COPD may lead to pulmonary hypertension, chronic intermittent hypoxia (CIH) as seen predominantly in sleep apnea has been attributed to causing widespread effects ranging from systemic hypertension to metabolic dysfunction and systemic inflammation,” noted Krishna Sundar, MD, FCCP. “Despite these associations, an increased risk of major cardiovascular events from untreated OSA is yet to be definitively established.”
In this article, a protective effect from OSA on myocardial ischemic events is demonstrated in a group of 127 consecutively admitted patients with acute coronary syndrome (ACS). While it is interesting that a high proportion of those admitted for ACS had OSA, there were no significant differences in the age, sex, BMI, usage of antihypertensive or antiplatelet agents, presence of hypertension, DM, dyslipidemia or smoking status between those with and without OSA. “OSA appeared to confer a protective effect on the size of myocardial injury with those having higher AHI values demonstrating lower peak cardiac troponin values,” said Dr. Sundar, who is an associate clinical professor of pulmonary, critical care and sleep medicine at the University of Utah.“An effect of age (mean age in this study being 64 years) and BMI (mean being 27) on the occurrence of preconditioning effects of OSA is not excluded given deleterious effects of untreated OSA on infarct size in other studies on obese or younger patients with ACS. Further understanding of molecular effects of chronic hypoxia exposure (high altitude, chronic lung disease, OSA) is required before the complex and often contradictory effects of chronic hypoxia can be affirmed as being protective or deleterious,” added Dr. Sundar, who is also medical director of the Sleep-Wake Center at the University of Utah and a member of CHEST Physician’s editorial advisory board.
According to the study’s authors, their findings “suggest that patients with higher AHI are significantly more likely to have low cTn-I levels than patients without evidence of OSA, which could imply that patients with elevated AHI, particularly those with severe OSA, may experience less severe myocardial injury.”Limitations of the study include exclusion of patients with severe ACS, exclusion of sleepy subjects, and assessment of myocardial injury using cTn-I as a biomarker, without further data to determine infarct size.
“The possible role of OSA in cardioprotection should be explored in future studies,” the authors concluded.
The authors disclosed relationships with ResMed, Spanish Ministry of Health, Spanish Respiratory Society, Catalonian Cardiology Society, and ALLER. No other disclosures were reported.
SOURCE: Chest. 2018 Feb 5;153[2]:329-38. doi: 10.1016/j.chest.2017.06.046.

Outpatient CAR T infusions feasible using liso-cel
SALT LAKE CITY – A CD19-directed 4-1BB chimeric antigen receptor (CAR) T cell product showed efficacy and a low rate of cytokine release syndrome and neurotoxicity in patients with aggressive lymphomas and poor prognoses, raising the possibility of outpatient administration and fewer hospitalization days in this high-risk group.
A total of 86 patients who received inpatient infusions of lisocabtagene maraleucel (liso-cel, also known as JCAR017) had a mean 15.6 days of hospitalization, compared with 9.3 days for 8 outpatient recipients, said Jeremy Abramson, MD, speaking at a top abstracts session of the combined annual meetings of the Center for International Blood & Marrow Transplant Research and the American Society for Blood and Marrow Transplantation.

As of October 2017, eight patients had received liso-cel infusion as outpatients with at least 28 days of postinfusion data, Dr. Abramson said.
Although all but one required hospital admission, at a median of 5 days postinfusion (range, 4-22 days), there had been no intensive care unit admissions, and no outpatient recipients had experienced severe cytokine release syndrome (CRS) or neurotoxicity. All admitted patients presented with fever.
Among the study population, “Cytokine release syndrome was only seen in 35% of our entire dataset,” with neurologic toxicity seen in 19% of participants, Dr. Abramson said. “The majority of subjects had no CRS and no toxicity,” he said. Severe CRS occurred in 1% of the study population, and severe neurotoxicity in 12%. There were no deaths related to either complication.
Dr. Abramson reported these results from the TRANSCEND NHL 001 trial, a seamless design phase 1 pivotal trial of liso-cel enrolling patients with relapsed and refractory aggressive B cell non-Hodgkin lymphoma (NHL). Liso-cel delivers CD19-directed CD4 and CD8 CAR T cells in a 1:1 ratio, said Dr. Abramson, director of the lymphoma program at the Massachusetts General Hospital Cancer Center, Boston.
A total of 91 patients were randomized to one of the three dose-finding cohorts of the multicenter trial of liso-cel. One cohort received 5 x 107 cells in a single dose; a second cohort received the same number of cells but in two doses administered 14 days apart; the third cohort received a single dose of 1 x 108 cells.
The seamless trial design then moved to dose expansion, using the two single doses established in the dose-finding phase of the study. Ultimately, Dr. Abramson said, the third and pivotal diffuse large B-cell lymphoma (DLBCL) cohort received the higher single dose, since a dose-response relationship was seen in the earlier cohorts. No increase in cytokine release syndrome or neurotoxicity has been seen with the higher dose in patients evaluated to date.
Patients (median age, 61 years) were eligible to participate in the trial if they had relapsed or refractory DLBCL, primary mediastinal B-cell lymphoma, grade 3B follicular lymphoma, or mantle cell lymphoma. Patients with a failed prior allogeneic stem cell transplant or secondary central nervous system involvement were eligible, but all patients had to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2.
As the trial moved to the core pivotal phase, eligibility requirements shifted slightly to include patients with ECOG status 0 or 1, and lymphoma diagnoses narrowed to include only DLBCL not otherwise specified (NOS), transformed follicular lymphoma, and high-grade B-cell lymphoma with double- and triple-hit cytogenetics. The core group was nearing completion of accrual at the time of the presentation, which presented preliminary results from this phase of the trial.
Among the 88 evaluable patients in the initial population with DLBCL receiving any of three dose levels, the best overall response rate (ORR) was 74% (95% confidence interval, 63%-83%); 52% of these patients achieved complete response (CR; 95% CI, 41%-63%).
For patients receiving the higher dose of liso-cel, the ORR was 81% (95% CI, 62%-94%), with a 63% CR rate (95% CI, 42%-81%), bearing out the dose-response rate that had been seen earlier in the trial, Dr. Abramson said.
The median duration of response in all TRANSCEND patients was 9.2 months; the median duration of remission has not been reached, he said. “We see evidence of durable response at 3 months in all our high-risk subsets, and that includes double- and triple-hit lymphomas, double-expresser lymphomas, patients who’ve never achieved prior complete remission, and patients with refractory disease.”
“The overall results are similarly encouraging,” Dr. Abramson said, with 86% of all patients alive at 6 months. Among the complete responders, 94% are alive at the 6-month mark. “The median duration of complete responders has not been reached in this cohort,” he said.
These results are notable, Dr. Abramson said, since about 90% of study participants have at least one disease risk factor that would predict median overall survival of 3-6 months.
During the period after leukapheresis while the CAR T cells were in production, patients could have ongoing treatment, but received PET scans to confirm disease before continuing enrollment in the trial and receiving liso-cel. The time from apheresis to product release for the pivotal cohort is now under 21 days, he said.
The study was supported by Juno Therapeutics, which plans to market liso-cel. Dr. Abramson reported ties with Celgene, Gilead, Seattle Genetics, Novartis, and Genentech.
SOURCE: Abramson J et al. Abstract 5.
SALT LAKE CITY – A CD19-directed 4-1BB chimeric antigen receptor (CAR) T cell product showed efficacy and a low rate of cytokine release syndrome and neurotoxicity in patients with aggressive lymphomas and poor prognoses, raising the possibility of outpatient administration and fewer hospitalization days in this high-risk group.
A total of 86 patients who received inpatient infusions of lisocabtagene maraleucel (liso-cel, also known as JCAR017) had a mean 15.6 days of hospitalization, compared with 9.3 days for 8 outpatient recipients, said Jeremy Abramson, MD, speaking at a top abstracts session of the combined annual meetings of the Center for International Blood & Marrow Transplant Research and the American Society for Blood and Marrow Transplantation.
As of October 2017, eight patients had received liso-cel infusion as outpatients with at least 28 days of postinfusion data, Dr. Abramson said.
Although all but one required hospital admission, at a median of 5 days postinfusion (range, 4-22 days), there had been no intensive care unit admissions, and no outpatient recipients had experienced severe cytokine release syndrome (CRS) or neurotoxicity. All admitted patients presented with fever.
Among the study population, “Cytokine release syndrome was only seen in 35% of our entire dataset,” with neurologic toxicity seen in 19% of participants, Dr. Abramson said. “The majority of subjects had no CRS and no toxicity,” he said. Severe CRS occurred in 1% of the study population, and severe neurotoxicity in 12%. There were no deaths related to either complication.
Dr. Abramson reported these results from the TRANSCEND NHL 001 trial, a seamless design phase 1 pivotal trial of liso-cel enrolling patients with relapsed and refractory aggressive B cell non-Hodgkin lymphoma (NHL). Liso-cel delivers CD19-directed CD4 and CD8 CAR T cells in a 1:1 ratio, said Dr. Abramson, director of the lymphoma program at the Massachusetts General Hospital Cancer Center, Boston.
A total of 91 patients were randomized to one of the three dose-finding cohorts of the multicenter trial of liso-cel. One cohort received 5 x 107 cells in a single dose; a second cohort received the same number of cells but in two doses administered 14 days apart; the third cohort received a single dose of 1 x 108 cells.
The seamless trial design then moved to dose expansion, using the two single doses established in the dose-finding phase of the study. Ultimately, Dr. Abramson said, the third and pivotal diffuse large B-cell lymphoma (DLBCL) cohort received the higher single dose, since a dose-response relationship was seen in the earlier cohorts. No increase in cytokine release syndrome or neurotoxicity has been seen with the higher dose in patients evaluated to date.
Patients (median age, 61 years) were eligible to participate in the trial if they had relapsed or refractory DLBCL, primary mediastinal B-cell lymphoma, grade 3B follicular lymphoma, or mantle cell lymphoma. Patients with a failed prior allogeneic stem cell transplant or secondary central nervous system involvement were eligible, but all patients had to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2.
As the trial moved to the core pivotal phase, eligibility requirements shifted slightly to include patients with ECOG status 0 or 1, and lymphoma diagnoses narrowed to include only DLBCL not otherwise specified (NOS), transformed follicular lymphoma, and high-grade B-cell lymphoma with double- and triple-hit cytogenetics. The core group was nearing completion of accrual at the time of the presentation, which presented preliminary results from this phase of the trial.
Among the 88 evaluable patients in the initial population with DLBCL receiving any of three dose levels, the best overall response rate (ORR) was 74% (95% confidence interval, 63%-83%); 52% of these patients achieved complete response (CR; 95% CI, 41%-63%).
For patients receiving the higher dose of liso-cel, the ORR was 81% (95% CI, 62%-94%), with a 63% CR rate (95% CI, 42%-81%), bearing out the dose-response rate that had been seen earlier in the trial, Dr. Abramson said.
The median duration of response in all TRANSCEND patients was 9.2 months; the median duration of remission has not been reached, he said. “We see evidence of durable response at 3 months in all our high-risk subsets, and that includes double- and triple-hit lymphomas, double-expresser lymphomas, patients who’ve never achieved prior complete remission, and patients with refractory disease.”
“The overall results are similarly encouraging,” Dr. Abramson said, with 86% of all patients alive at 6 months. Among the complete responders, 94% are alive at the 6-month mark. “The median duration of complete responders has not been reached in this cohort,” he said.
These results are notable, Dr. Abramson said, since about 90% of study participants have at least one disease risk factor that would predict median overall survival of 3-6 months.
During the period after leukapheresis while the CAR T cells were in production, patients could have ongoing treatment, but received PET scans to confirm disease before continuing enrollment in the trial and receiving liso-cel. The time from apheresis to product release for the pivotal cohort is now under 21 days, he said.
The study was supported by Juno Therapeutics, which plans to market liso-cel. Dr. Abramson reported ties with Celgene, Gilead, Seattle Genetics, Novartis, and Genentech.
SOURCE: Abramson J et al. Abstract 5.
SALT LAKE CITY – A CD19-directed 4-1BB chimeric antigen receptor (CAR) T cell product showed efficacy and a low rate of cytokine release syndrome and neurotoxicity in patients with aggressive lymphomas and poor prognoses, raising the possibility of outpatient administration and fewer hospitalization days in this high-risk group.
A total of 86 patients who received inpatient infusions of lisocabtagene maraleucel (liso-cel, also known as JCAR017) had a mean 15.6 days of hospitalization, compared with 9.3 days for 8 outpatient recipients, said Jeremy Abramson, MD, speaking at a top abstracts session of the combined annual meetings of the Center for International Blood & Marrow Transplant Research and the American Society for Blood and Marrow Transplantation.
As of October 2017, eight patients had received liso-cel infusion as outpatients with at least 28 days of postinfusion data, Dr. Abramson said.
Although all but one required hospital admission, at a median of 5 days postinfusion (range, 4-22 days), there had been no intensive care unit admissions, and no outpatient recipients had experienced severe cytokine release syndrome (CRS) or neurotoxicity. All admitted patients presented with fever.
Among the study population, “Cytokine release syndrome was only seen in 35% of our entire dataset,” with neurologic toxicity seen in 19% of participants, Dr. Abramson said. “The majority of subjects had no CRS and no toxicity,” he said. Severe CRS occurred in 1% of the study population, and severe neurotoxicity in 12%. There were no deaths related to either complication.
Dr. Abramson reported these results from the TRANSCEND NHL 001 trial, a seamless design phase 1 pivotal trial of liso-cel enrolling patients with relapsed and refractory aggressive B cell non-Hodgkin lymphoma (NHL). Liso-cel delivers CD19-directed CD4 and CD8 CAR T cells in a 1:1 ratio, said Dr. Abramson, director of the lymphoma program at the Massachusetts General Hospital Cancer Center, Boston.
A total of 91 patients were randomized to one of the three dose-finding cohorts of the multicenter trial of liso-cel. One cohort received 5 x 107 cells in a single dose; a second cohort received the same number of cells but in two doses administered 14 days apart; the third cohort received a single dose of 1 x 108 cells.
The seamless trial design then moved to dose expansion, using the two single doses established in the dose-finding phase of the study. Ultimately, Dr. Abramson said, the third and pivotal diffuse large B-cell lymphoma (DLBCL) cohort received the higher single dose, since a dose-response relationship was seen in the earlier cohorts. No increase in cytokine release syndrome or neurotoxicity has been seen with the higher dose in patients evaluated to date.
Patients (median age, 61 years) were eligible to participate in the trial if they had relapsed or refractory DLBCL, primary mediastinal B-cell lymphoma, grade 3B follicular lymphoma, or mantle cell lymphoma. Patients with a failed prior allogeneic stem cell transplant or secondary central nervous system involvement were eligible, but all patients had to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2.
As the trial moved to the core pivotal phase, eligibility requirements shifted slightly to include patients with ECOG status 0 or 1, and lymphoma diagnoses narrowed to include only DLBCL not otherwise specified (NOS), transformed follicular lymphoma, and high-grade B-cell lymphoma with double- and triple-hit cytogenetics. The core group was nearing completion of accrual at the time of the presentation, which presented preliminary results from this phase of the trial.
Among the 88 evaluable patients in the initial population with DLBCL receiving any of three dose levels, the best overall response rate (ORR) was 74% (95% confidence interval, 63%-83%); 52% of these patients achieved complete response (CR; 95% CI, 41%-63%).
For patients receiving the higher dose of liso-cel, the ORR was 81% (95% CI, 62%-94%), with a 63% CR rate (95% CI, 42%-81%), bearing out the dose-response rate that had been seen earlier in the trial, Dr. Abramson said.
The median duration of response in all TRANSCEND patients was 9.2 months; the median duration of remission has not been reached, he said. “We see evidence of durable response at 3 months in all our high-risk subsets, and that includes double- and triple-hit lymphomas, double-expresser lymphomas, patients who’ve never achieved prior complete remission, and patients with refractory disease.”
“The overall results are similarly encouraging,” Dr. Abramson said, with 86% of all patients alive at 6 months. Among the complete responders, 94% are alive at the 6-month mark. “The median duration of complete responders has not been reached in this cohort,” he said.
These results are notable, Dr. Abramson said, since about 90% of study participants have at least one disease risk factor that would predict median overall survival of 3-6 months.
During the period after leukapheresis while the CAR T cells were in production, patients could have ongoing treatment, but received PET scans to confirm disease before continuing enrollment in the trial and receiving liso-cel. The time from apheresis to product release for the pivotal cohort is now under 21 days, he said.
The study was supported by Juno Therapeutics, which plans to market liso-cel. Dr. Abramson reported ties with Celgene, Gilead, Seattle Genetics, Novartis, and Genentech.
SOURCE: Abramson J et al. Abstract 5.
REPORTING FROM THE 2018 BMT TANDEM MEETINGS
Key clinical point:
Major finding: High-risk lymphoma patients had more than 6 fewer inpatient days with outpatient CAR T infusion.
Study details: Seamless phase 1 trial initially evaluating 91 patients with relapsed/refractory diffuse large B cell lymphoma.
Disclosures: Juno Therapeutics sponsored the study. Dr. Abramson reported ties with Celgene, Gilead, Seattle Genetics, Novartis, and Genentech.
Source: Abramson J et al. Abstract 5.
MDedge Daily News: Improving wearable cardioverter defibrillators
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
, alirocumab cuts deaths in post–acute coronary syndrome patients, the ODYSSEY Outcomes trial redefines secondary cardiovascular prevention, and how walking cuts postmenopausal women’s heart failure risk.
Listen to the MDedge Daily News podcast for all the details on today’s top news.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
, alirocumab cuts deaths in post–acute coronary syndrome patients, the ODYSSEY Outcomes trial redefines secondary cardiovascular prevention, and how walking cuts postmenopausal women’s heart failure risk.
Listen to the MDedge Daily News podcast for all the details on today’s top news.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
, alirocumab cuts deaths in post–acute coronary syndrome patients, the ODYSSEY Outcomes trial redefines secondary cardiovascular prevention, and how walking cuts postmenopausal women’s heart failure risk.
Listen to the MDedge Daily News podcast for all the details on today’s top news.

A Peek at Our March 2018 Issue
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Hospital urine screening reduces TB deaths in HIV+ adults
BOSTON – Urine-based screening for tuberculosis added to standard sputum-based screening can reduce the risk of TB-associated deaths among hospitalized patients with advanced HIV compared with sputum-based screening alone, results of a randomized trial indicate.
Among 2,574 hospitalized HIV-positive patients, the rate of death at 56 days, the primary endpoint, was 21.1% for patients screened with standard-of-care sputum testing using the Xpert MTB/RIF assay, compared with 18.3% for patients screened with sputum testing and urine-sample testing by the Xpert MTB/RIF and the Determine TB-LAM urine dipstick test, reported Ankur Gupta-Wright, MBBS, MRCP, from the London School of Hygiene and Tropical Medicine.

“For every 100 patients who are HIV-positive admitted to hospital, with screening for TB using new urine-based testing in addition to sputum testing we can diagnose approximately seven extra TB case, and save approximately three lives,” he said at a briefing following his presentation of the data in a late-breaking oral abstract session.
“The findings our trial support the use of urine-based TB screening, and we hope to change how we screen for TB in HIV-positive admissions in the hospital in places with high burden of HIV and TB,” he said, speaking on behalf of colleagues in the STAMP Study.
HIV-related TB caused an estimated 400,000 deaths worldwide in 2016, and is responsible for between 32% and 67% of deaths in HIV-positive adults admit to hospitals in Africa. Yet half of all of those cases of TB are undiagnosed at the time of death, he said.
Urine based screens are easily obtained and have good diagnostic yield for disseminated TB, which is common in patients with advanced HIV, he said.
To see whether adding urine screening could increase TB diagnosis and treatment and reduce TB deaths among hospitalized HIV+ patients, the investigators enrolled 2,600 unselected HIV+ adults admitted to hospitals in South Africa and Malawi. Patients less than 18 years of age, those who had been treated for TB with the last 12 months or had received TB prophylaxis with isoniazid within the last 6 months were excluded, leaving a sample size of 2,574.
The patients were randomly assigned to standard-of-care Xpert MTB/RIF sputum screening either alone or with the addition of the two urine-based screens already described.
The overall absolute difference in risk of death at 56 days was -2.8% favoring urine screening (P = .074). As noted, this difference was only significant among patients with CD4 counts below 100/uL. Among these patients, mortality rates for nonurine screened versus screened were 35.7% vs. 28.8%, respectively (P = .036).
Baseline hemoglobin below 8 g/dl and TB suspected at admission were also predictive of better outcomes with the addition of urine-based screening.
An analysis of time to death stratified by baseline CD4 cell count also showed the advantage of urine screening for the sickest patients, with an adjusted hazard ratio for patients with CD4 counts below 100 of 0.77 (P = .047).
The urine-based screening also lead to significantly more microbiologically confirmed and clinically diagnosed TB, and more TB treatment (P less than .001 for each).
A separate cost analysis, presented in a scientific poster (CROI 2018 Abstract 1117LB), showed that the incremental cost-effectiveness ratios for the urine-based intervention were $490 per year of life save in Malawi, and $850 per year of life saved in South Africa.
The study was funded by UK AID, the UK Medical Research Council, and the Wellcome Trust. Dr. Gupta-Wright reported having no conflicts of interest to disclose.
SOURCE: Gupta-Wright, A et al. CROI 2018, Abstract 38LB.
BOSTON – Urine-based screening for tuberculosis added to standard sputum-based screening can reduce the risk of TB-associated deaths among hospitalized patients with advanced HIV compared with sputum-based screening alone, results of a randomized trial indicate.
Among 2,574 hospitalized HIV-positive patients, the rate of death at 56 days, the primary endpoint, was 21.1% for patients screened with standard-of-care sputum testing using the Xpert MTB/RIF assay, compared with 18.3% for patients screened with sputum testing and urine-sample testing by the Xpert MTB/RIF and the Determine TB-LAM urine dipstick test, reported Ankur Gupta-Wright, MBBS, MRCP, from the London School of Hygiene and Tropical Medicine.
“For every 100 patients who are HIV-positive admitted to hospital, with screening for TB using new urine-based testing in addition to sputum testing we can diagnose approximately seven extra TB case, and save approximately three lives,” he said at a briefing following his presentation of the data in a late-breaking oral abstract session.
“The findings our trial support the use of urine-based TB screening, and we hope to change how we screen for TB in HIV-positive admissions in the hospital in places with high burden of HIV and TB,” he said, speaking on behalf of colleagues in the STAMP Study.
HIV-related TB caused an estimated 400,000 deaths worldwide in 2016, and is responsible for between 32% and 67% of deaths in HIV-positive adults admit to hospitals in Africa. Yet half of all of those cases of TB are undiagnosed at the time of death, he said.
Urine based screens are easily obtained and have good diagnostic yield for disseminated TB, which is common in patients with advanced HIV, he said.
To see whether adding urine screening could increase TB diagnosis and treatment and reduce TB deaths among hospitalized HIV+ patients, the investigators enrolled 2,600 unselected HIV+ adults admitted to hospitals in South Africa and Malawi. Patients less than 18 years of age, those who had been treated for TB with the last 12 months or had received TB prophylaxis with isoniazid within the last 6 months were excluded, leaving a sample size of 2,574.
The patients were randomly assigned to standard-of-care Xpert MTB/RIF sputum screening either alone or with the addition of the two urine-based screens already described.
The overall absolute difference in risk of death at 56 days was -2.8% favoring urine screening (P = .074). As noted, this difference was only significant among patients with CD4 counts below 100/uL. Among these patients, mortality rates for nonurine screened versus screened were 35.7% vs. 28.8%, respectively (P = .036).
Baseline hemoglobin below 8 g/dl and TB suspected at admission were also predictive of better outcomes with the addition of urine-based screening.
An analysis of time to death stratified by baseline CD4 cell count also showed the advantage of urine screening for the sickest patients, with an adjusted hazard ratio for patients with CD4 counts below 100 of 0.77 (P = .047).
The urine-based screening also lead to significantly more microbiologically confirmed and clinically diagnosed TB, and more TB treatment (P less than .001 for each).
A separate cost analysis, presented in a scientific poster (CROI 2018 Abstract 1117LB), showed that the incremental cost-effectiveness ratios for the urine-based intervention were $490 per year of life save in Malawi, and $850 per year of life saved in South Africa.
The study was funded by UK AID, the UK Medical Research Council, and the Wellcome Trust. Dr. Gupta-Wright reported having no conflicts of interest to disclose.
SOURCE: Gupta-Wright, A et al. CROI 2018, Abstract 38LB.
BOSTON – Urine-based screening for tuberculosis added to standard sputum-based screening can reduce the risk of TB-associated deaths among hospitalized patients with advanced HIV compared with sputum-based screening alone, results of a randomized trial indicate.
Among 2,574 hospitalized HIV-positive patients, the rate of death at 56 days, the primary endpoint, was 21.1% for patients screened with standard-of-care sputum testing using the Xpert MTB/RIF assay, compared with 18.3% for patients screened with sputum testing and urine-sample testing by the Xpert MTB/RIF and the Determine TB-LAM urine dipstick test, reported Ankur Gupta-Wright, MBBS, MRCP, from the London School of Hygiene and Tropical Medicine.
“For every 100 patients who are HIV-positive admitted to hospital, with screening for TB using new urine-based testing in addition to sputum testing we can diagnose approximately seven extra TB case, and save approximately three lives,” he said at a briefing following his presentation of the data in a late-breaking oral abstract session.
“The findings our trial support the use of urine-based TB screening, and we hope to change how we screen for TB in HIV-positive admissions in the hospital in places with high burden of HIV and TB,” he said, speaking on behalf of colleagues in the STAMP Study.
HIV-related TB caused an estimated 400,000 deaths worldwide in 2016, and is responsible for between 32% and 67% of deaths in HIV-positive adults admit to hospitals in Africa. Yet half of all of those cases of TB are undiagnosed at the time of death, he said.
Urine based screens are easily obtained and have good diagnostic yield for disseminated TB, which is common in patients with advanced HIV, he said.
To see whether adding urine screening could increase TB diagnosis and treatment and reduce TB deaths among hospitalized HIV+ patients, the investigators enrolled 2,600 unselected HIV+ adults admitted to hospitals in South Africa and Malawi. Patients less than 18 years of age, those who had been treated for TB with the last 12 months or had received TB prophylaxis with isoniazid within the last 6 months were excluded, leaving a sample size of 2,574.
The patients were randomly assigned to standard-of-care Xpert MTB/RIF sputum screening either alone or with the addition of the two urine-based screens already described.
The overall absolute difference in risk of death at 56 days was -2.8% favoring urine screening (P = .074). As noted, this difference was only significant among patients with CD4 counts below 100/uL. Among these patients, mortality rates for nonurine screened versus screened were 35.7% vs. 28.8%, respectively (P = .036).
Baseline hemoglobin below 8 g/dl and TB suspected at admission were also predictive of better outcomes with the addition of urine-based screening.
An analysis of time to death stratified by baseline CD4 cell count also showed the advantage of urine screening for the sickest patients, with an adjusted hazard ratio for patients with CD4 counts below 100 of 0.77 (P = .047).
The urine-based screening also lead to significantly more microbiologically confirmed and clinically diagnosed TB, and more TB treatment (P less than .001 for each).
A separate cost analysis, presented in a scientific poster (CROI 2018 Abstract 1117LB), showed that the incremental cost-effectiveness ratios for the urine-based intervention were $490 per year of life save in Malawi, and $850 per year of life saved in South Africa.
The study was funded by UK AID, the UK Medical Research Council, and the Wellcome Trust. Dr. Gupta-Wright reported having no conflicts of interest to disclose.
SOURCE: Gupta-Wright, A et al. CROI 2018, Abstract 38LB.
REPORTING FROM CROI
Key clinical point: Adding urine screening for TB to sputum screening in hospitalized HIV+ patients can reduce risk for death from TB.
Major finding: Among patients with CD4 counts below 100/uL, the 56-day mortality rates was of 35.7% with sputum screening alone vs. 28.8% for combined urine and sputum screening.
Data source: Randomized pragmatic trial in 2,574 HIV+ hospitalized adults in Malawi and South Africa.
Disclosures: The study was funded by UK AID, the UK Medical Research Council, and the Wellcome Trust. Dr. Gupta-Wright reported having no conflicts of interest to disclose.
Source: Gupta-Wright A et al. CROI 2018, Abstract 38LB.
Get Pocket Versions of Practice Guidelines
SVS has partnered with Guidelines Central to create a Pocket Guide version of the new AAA guidelines. Also available are pocket guides on Management of Diabetic Foot, Peripheral Arterial Disease and Venous Leg Ulcers.
SVS members can access the digital versions for free; printed guidelines vary in price. The guidelines also are available as a bundled set. Slide sets of the guidelines, useful as educational tools, also are available online.
SVS has partnered with Guidelines Central to create a Pocket Guide version of the new AAA guidelines. Also available are pocket guides on Management of Diabetic Foot, Peripheral Arterial Disease and Venous Leg Ulcers.
SVS members can access the digital versions for free; printed guidelines vary in price. The guidelines also are available as a bundled set. Slide sets of the guidelines, useful as educational tools, also are available online.
SVS has partnered with Guidelines Central to create a Pocket Guide version of the new AAA guidelines. Also available are pocket guides on Management of Diabetic Foot, Peripheral Arterial Disease and Venous Leg Ulcers.
SVS members can access the digital versions for free; printed guidelines vary in price. The guidelines also are available as a bundled set. Slide sets of the guidelines, useful as educational tools, also are available online.
Watch for QTc interval prolongation in patients taking antipsychotics
LAS VEGAS – Many potential but rare side effects, primarily arrhythmias, can occur from taking antipsychotics, according to Carrie L. Ernst, MD.
Risk factors for QT prolongation and torsades de pointes (TdP) include medications such as antiarrhythmics, many antibiotics such as macrolides and quinolones, antifungals, antiemetics, antimalarials, methadone, and antipsychotics and other psychotropics. Other risk factors include cardiac disease, electrolyte abnormalities, being female, age 65 and older, being on another medication that inhibits metabolism of the drug, genetic predisposition to prolonged QT, and impaired liver function.

All antipsychotics can cause an increase in the QT interval and can cause TdP, although some likely confer a greater risk than others. “It’s most likely not an idiosyncratic effect but rather a dose-related effect,” she said. “So the higher the dose, the more the risk.” There are some data suggesting that phenothiazines, particularly thioridazine, seem to present the greatest risk for prolonging the QTc interval. , but IV haloperidol has been linked to prolonged QTc and TdP. “It’s difficult to know whether that’s the impact of the drug or a reflection of the fact that patients on IV haloperidol are usually medically complicated patients,” Dr. Ernst noted. “Unfortunately, there is limited direct comparison data between agents. Atypicals are only implicated in TdP in rare case reports, and there is less available data for the newest atypicals.”
Two drugs have the Food and Drug Administration boxed warning for prolonged QTc and TdP: thioridazine and mesoridazine. In addition, six medications carry a warning/precaution about prolonged QTc: ziprasidone, paliperidone, IV haloperidol, iloperidone, quetiapine, and asenapine. “Even though haloperidol is not approved for IV administration, FDA created a label warning for IV haloperidol knowing that many clinicians are using it,” she said. “The label advises clinicians to use particular caution in treating patients who have high risk for QTc prolongation and TdP.”
All antipsychotics prolong the QTc by some amount, but differences exist between agents. One prospective, randomized trial showed that ziprasidone and thioridazine prolonged the QTc more than four other antipsychotic drugs (J Clin Psychopharmacol. 2004;24:62-9). “I think the important point is that nobody had a QTc over 500 milliseconds and nobody had TdP,” Dr. Ernst said. “Some data estimate that even if the QTc is 600 milliseconds, the rate of a life-threatening event is probably no more than one in 4,000, so it’s very rare.”
In a more recent meta-analysis, researchers compared 15 antipsychotics among 43,049 adults participating in 212 clinical trials (Lancet. 2013;382:951-62). They found that the following drugs were not associated with QTc prolongation, compared with placebo: lurasidone, aripiprazole, paliperidone, and asenapine. A separate meta-analysis comparing numerous different antipsychotics in children found that ziprasidone was the only one to be associated with an increased risk of QTc prolongation (J Am Acad Child Adolesc Psychiatry. 2015:54:25-36).
There are also data to suggest an association between antipsychotic use and an increased risk of ventricular arrhythmia or sudden cardiac death. The relationship between prolonged QTc and sudden cardiac death is unclear. One large retrospective cohort study found that all antipsychotics carried a 2.5-fold increased risk of sudden cardiac death, compared with the general population not taking those agents (N Eng J Med. 2009;360[3]:225-35). “Risk seemed to be dose related,” Dr. Ernst said. All antipsychotics carry a boxed warning for increased mortality in elderly patients with dementia-related psychosis. Most of the deaths reported appear related to cardiovascular and infectious causes.
For patients with cardiac disease or some of the risk factors for prolonged QTc, Dr. Ernst recommends that psychiatrists order a baseline and steady state EKG, with intermittent QTc monitoring if warranted. “Closely weigh the risks and benefits and consider avoiding the antipsychotic if the baseline QTc is over 500 milliseconds,” she said. “Avoid low-potency typicals, IV haloperidol, and ziprasidone.”
For patients on IV haloperidol, she recommends obtaining a baseline and at least daily QTc, as well as electrolyte monitoring. Some guidelines suggest continuous EKG monitoring for those with baseline QTc of more than 500 milliseconds, risk factors, and/or high dose requirements. “If the QTc increases beyond 500 milliseconds, check and replete electrolytes, review the medication regimen for other agents that prolong the QTc, consider holding until the QTc is less than 500 milliseconds, consider alternative agents, and perform frequent EKG monitoring and a cardiology consult if you decide to continue,” she said.
In patients without any risk factors for prolonged QTc, there is no consensus on obtaining a baseline QTc or doing serial QTc monitoring. “Do a careful risk assessment, and if the QTc increases beyond 500 milliseconds or greater than 60 milliseconds from baseline during treatment, use the same approach as in high-risk patients,” Dr. Ernst said. “Maintain an updated medication list, and if QT prolonging medications are added or new pharmacokinetic interactions occur, approach them the same as you would a high-risk patient.”
She reported having no financial disclosures.
LAS VEGAS – Many potential but rare side effects, primarily arrhythmias, can occur from taking antipsychotics, according to Carrie L. Ernst, MD.
Risk factors for QT prolongation and torsades de pointes (TdP) include medications such as antiarrhythmics, many antibiotics such as macrolides and quinolones, antifungals, antiemetics, antimalarials, methadone, and antipsychotics and other psychotropics. Other risk factors include cardiac disease, electrolyte abnormalities, being female, age 65 and older, being on another medication that inhibits metabolism of the drug, genetic predisposition to prolonged QT, and impaired liver function.
All antipsychotics can cause an increase in the QT interval and can cause TdP, although some likely confer a greater risk than others. “It’s most likely not an idiosyncratic effect but rather a dose-related effect,” she said. “So the higher the dose, the more the risk.” There are some data suggesting that phenothiazines, particularly thioridazine, seem to present the greatest risk for prolonging the QTc interval. , but IV haloperidol has been linked to prolonged QTc and TdP. “It’s difficult to know whether that’s the impact of the drug or a reflection of the fact that patients on IV haloperidol are usually medically complicated patients,” Dr. Ernst noted. “Unfortunately, there is limited direct comparison data between agents. Atypicals are only implicated in TdP in rare case reports, and there is less available data for the newest atypicals.”
Two drugs have the Food and Drug Administration boxed warning for prolonged QTc and TdP: thioridazine and mesoridazine. In addition, six medications carry a warning/precaution about prolonged QTc: ziprasidone, paliperidone, IV haloperidol, iloperidone, quetiapine, and asenapine. “Even though haloperidol is not approved for IV administration, FDA created a label warning for IV haloperidol knowing that many clinicians are using it,” she said. “The label advises clinicians to use particular caution in treating patients who have high risk for QTc prolongation and TdP.”
All antipsychotics prolong the QTc by some amount, but differences exist between agents. One prospective, randomized trial showed that ziprasidone and thioridazine prolonged the QTc more than four other antipsychotic drugs (J Clin Psychopharmacol. 2004;24:62-9). “I think the important point is that nobody had a QTc over 500 milliseconds and nobody had TdP,” Dr. Ernst said. “Some data estimate that even if the QTc is 600 milliseconds, the rate of a life-threatening event is probably no more than one in 4,000, so it’s very rare.”
In a more recent meta-analysis, researchers compared 15 antipsychotics among 43,049 adults participating in 212 clinical trials (Lancet. 2013;382:951-62). They found that the following drugs were not associated with QTc prolongation, compared with placebo: lurasidone, aripiprazole, paliperidone, and asenapine. A separate meta-analysis comparing numerous different antipsychotics in children found that ziprasidone was the only one to be associated with an increased risk of QTc prolongation (J Am Acad Child Adolesc Psychiatry. 2015:54:25-36).
There are also data to suggest an association between antipsychotic use and an increased risk of ventricular arrhythmia or sudden cardiac death. The relationship between prolonged QTc and sudden cardiac death is unclear. One large retrospective cohort study found that all antipsychotics carried a 2.5-fold increased risk of sudden cardiac death, compared with the general population not taking those agents (N Eng J Med. 2009;360[3]:225-35). “Risk seemed to be dose related,” Dr. Ernst said. All antipsychotics carry a boxed warning for increased mortality in elderly patients with dementia-related psychosis. Most of the deaths reported appear related to cardiovascular and infectious causes.
For patients with cardiac disease or some of the risk factors for prolonged QTc, Dr. Ernst recommends that psychiatrists order a baseline and steady state EKG, with intermittent QTc monitoring if warranted. “Closely weigh the risks and benefits and consider avoiding the antipsychotic if the baseline QTc is over 500 milliseconds,” she said. “Avoid low-potency typicals, IV haloperidol, and ziprasidone.”
For patients on IV haloperidol, she recommends obtaining a baseline and at least daily QTc, as well as electrolyte monitoring. Some guidelines suggest continuous EKG monitoring for those with baseline QTc of more than 500 milliseconds, risk factors, and/or high dose requirements. “If the QTc increases beyond 500 milliseconds, check and replete electrolytes, review the medication regimen for other agents that prolong the QTc, consider holding until the QTc is less than 500 milliseconds, consider alternative agents, and perform frequent EKG monitoring and a cardiology consult if you decide to continue,” she said.
In patients without any risk factors for prolonged QTc, there is no consensus on obtaining a baseline QTc or doing serial QTc monitoring. “Do a careful risk assessment, and if the QTc increases beyond 500 milliseconds or greater than 60 milliseconds from baseline during treatment, use the same approach as in high-risk patients,” Dr. Ernst said. “Maintain an updated medication list, and if QT prolonging medications are added or new pharmacokinetic interactions occur, approach them the same as you would a high-risk patient.”
She reported having no financial disclosures.
LAS VEGAS – Many potential but rare side effects, primarily arrhythmias, can occur from taking antipsychotics, according to Carrie L. Ernst, MD.
Risk factors for QT prolongation and torsades de pointes (TdP) include medications such as antiarrhythmics, many antibiotics such as macrolides and quinolones, antifungals, antiemetics, antimalarials, methadone, and antipsychotics and other psychotropics. Other risk factors include cardiac disease, electrolyte abnormalities, being female, age 65 and older, being on another medication that inhibits metabolism of the drug, genetic predisposition to prolonged QT, and impaired liver function.
All antipsychotics can cause an increase in the QT interval and can cause TdP, although some likely confer a greater risk than others. “It’s most likely not an idiosyncratic effect but rather a dose-related effect,” she said. “So the higher the dose, the more the risk.” There are some data suggesting that phenothiazines, particularly thioridazine, seem to present the greatest risk for prolonging the QTc interval. , but IV haloperidol has been linked to prolonged QTc and TdP. “It’s difficult to know whether that’s the impact of the drug or a reflection of the fact that patients on IV haloperidol are usually medically complicated patients,” Dr. Ernst noted. “Unfortunately, there is limited direct comparison data between agents. Atypicals are only implicated in TdP in rare case reports, and there is less available data for the newest atypicals.”
Two drugs have the Food and Drug Administration boxed warning for prolonged QTc and TdP: thioridazine and mesoridazine. In addition, six medications carry a warning/precaution about prolonged QTc: ziprasidone, paliperidone, IV haloperidol, iloperidone, quetiapine, and asenapine. “Even though haloperidol is not approved for IV administration, FDA created a label warning for IV haloperidol knowing that many clinicians are using it,” she said. “The label advises clinicians to use particular caution in treating patients who have high risk for QTc prolongation and TdP.”
All antipsychotics prolong the QTc by some amount, but differences exist between agents. One prospective, randomized trial showed that ziprasidone and thioridazine prolonged the QTc more than four other antipsychotic drugs (J Clin Psychopharmacol. 2004;24:62-9). “I think the important point is that nobody had a QTc over 500 milliseconds and nobody had TdP,” Dr. Ernst said. “Some data estimate that even if the QTc is 600 milliseconds, the rate of a life-threatening event is probably no more than one in 4,000, so it’s very rare.”
In a more recent meta-analysis, researchers compared 15 antipsychotics among 43,049 adults participating in 212 clinical trials (Lancet. 2013;382:951-62). They found that the following drugs were not associated with QTc prolongation, compared with placebo: lurasidone, aripiprazole, paliperidone, and asenapine. A separate meta-analysis comparing numerous different antipsychotics in children found that ziprasidone was the only one to be associated with an increased risk of QTc prolongation (J Am Acad Child Adolesc Psychiatry. 2015:54:25-36).
There are also data to suggest an association between antipsychotic use and an increased risk of ventricular arrhythmia or sudden cardiac death. The relationship between prolonged QTc and sudden cardiac death is unclear. One large retrospective cohort study found that all antipsychotics carried a 2.5-fold increased risk of sudden cardiac death, compared with the general population not taking those agents (N Eng J Med. 2009;360[3]:225-35). “Risk seemed to be dose related,” Dr. Ernst said. All antipsychotics carry a boxed warning for increased mortality in elderly patients with dementia-related psychosis. Most of the deaths reported appear related to cardiovascular and infectious causes.
For patients with cardiac disease or some of the risk factors for prolonged QTc, Dr. Ernst recommends that psychiatrists order a baseline and steady state EKG, with intermittent QTc monitoring if warranted. “Closely weigh the risks and benefits and consider avoiding the antipsychotic if the baseline QTc is over 500 milliseconds,” she said. “Avoid low-potency typicals, IV haloperidol, and ziprasidone.”
For patients on IV haloperidol, she recommends obtaining a baseline and at least daily QTc, as well as electrolyte monitoring. Some guidelines suggest continuous EKG monitoring for those with baseline QTc of more than 500 milliseconds, risk factors, and/or high dose requirements. “If the QTc increases beyond 500 milliseconds, check and replete electrolytes, review the medication regimen for other agents that prolong the QTc, consider holding until the QTc is less than 500 milliseconds, consider alternative agents, and perform frequent EKG monitoring and a cardiology consult if you decide to continue,” she said.
In patients without any risk factors for prolonged QTc, there is no consensus on obtaining a baseline QTc or doing serial QTc monitoring. “Do a careful risk assessment, and if the QTc increases beyond 500 milliseconds or greater than 60 milliseconds from baseline during treatment, use the same approach as in high-risk patients,” Dr. Ernst said. “Maintain an updated medication list, and if QT prolonging medications are added or new pharmacokinetic interactions occur, approach them the same as you would a high-risk patient.”
She reported having no financial disclosures.
REPORTING FROM NPA 2018