User login
Information every surgeon should know about the ACSPA-SurgeonsPAC
Measuring success when it comes to political action, particularly in the current climate, can be challenging. Just as the American College of Surgeons (ACS) strives to represent all of surgery and focuses on an extensive list of priorities, the American College of Surgeons Professional Association Political Action Committee (ACSPA-SurgeonsPAC) works to maintain relationships with numerous lawmakers to ensure the College’s advocacy and health policy agenda remains at the forefront.
Leading up to the November midterm elections, every surgeon should be familiar with some basic facts about ACSPA-SurgeonsPAC fundraising and disbursement efforts.
Fundraising and disbursements
Fundraising is on par with the 2016 election cycle, a record year for “hard” dollars, or personal funds used to support candidates. SurgeonsPAC supports both Democrat and Republican candidates running in U.S. House and Senate races across the nation. SurgeonsPAC has a track record of balanced giving, contributing to candidates and incumbents of both parties who are willing to champion issues of importance to surgeons and surgical patients. All 2017‒2018 election cycle disbursements can be viewed at www.surgeonspac.org/disbursements. Legally, SurgeonsPAC cannot earmark contributions for a particular candidate or based on a single issue.
From January 1, 2017, through May 31, 2018, the ACSPA-SurgeonsPAC reported more than $805,000 in receipts from more than 1,500 College members and staff, and disbursed more than $558,900 to more than 115 congressional candidates, leadership PACs, and political campaign committees. Of the amount given, in line with congressional party ratios, 59 percent was given to Republicans and 41 percent to Democrats.
In addition to raising funds to elect and reelect congressional candidates, SurgeonsPAC hosted several health care industry events in the ACS Washington Office for key members of Congress and political campaign committees, collaborated with other medical community PACs to host fundraising events and physician candidate meet and greets, participated in party committee briefings and additional engagement opportunities, and increased in-district check deliveries and targeted donor events.
To learn more about SurgeonsPAC fundraising or disbursements, visit SurgeonsPAC.org (login required using facs.org username and password) or contact Katie Oehmen, Manager, ACSPA-SurgeonsPAC and Grassroots at 202-672-1503 or [email protected]. For more information about the College’s legislative priorities, visit SurgeonsVoice.org.
Contributions to ACSPA-SurgeonsPAC are not deductible as charitable contributions for federal income tax purposes. Contributions are voluntary, and all members of ACSPA have the right to refuse to contribute without reprisal. Federal law prohibits ACSPA-SurgeonsPAC from accepting contributions from foreign nations. By law, if your contributions are made using a personal check or credit card, ACSPA-SurgeonsPAC may only use your contribution to support candidates in federal elections. All corporate contributions to ACSPA-SurgeonsPAC will be used for educational and administrative fees of ACSPA and other activities permissible under federal law. Federal law requires ACSPA-SurgeonsPAC to use its best efforts to collect and report the name, mailing address, occupation, and the name of the employer of individuals whose contributions exceed $200 in a calendar year. ACSPA-SurgeonsPAC is a program of the ACSPA, which is exempt from federal income tax under section 501c (6) of the Internal Revenue Code.
Measuring success when it comes to political action, particularly in the current climate, can be challenging. Just as the American College of Surgeons (ACS) strives to represent all of surgery and focuses on an extensive list of priorities, the American College of Surgeons Professional Association Political Action Committee (ACSPA-SurgeonsPAC) works to maintain relationships with numerous lawmakers to ensure the College’s advocacy and health policy agenda remains at the forefront.
Leading up to the November midterm elections, every surgeon should be familiar with some basic facts about ACSPA-SurgeonsPAC fundraising and disbursement efforts.
Fundraising and disbursements
Fundraising is on par with the 2016 election cycle, a record year for “hard” dollars, or personal funds used to support candidates. SurgeonsPAC supports both Democrat and Republican candidates running in U.S. House and Senate races across the nation. SurgeonsPAC has a track record of balanced giving, contributing to candidates and incumbents of both parties who are willing to champion issues of importance to surgeons and surgical patients. All 2017‒2018 election cycle disbursements can be viewed at www.surgeonspac.org/disbursements. Legally, SurgeonsPAC cannot earmark contributions for a particular candidate or based on a single issue.
From January 1, 2017, through May 31, 2018, the ACSPA-SurgeonsPAC reported more than $805,000 in receipts from more than 1,500 College members and staff, and disbursed more than $558,900 to more than 115 congressional candidates, leadership PACs, and political campaign committees. Of the amount given, in line with congressional party ratios, 59 percent was given to Republicans and 41 percent to Democrats.
In addition to raising funds to elect and reelect congressional candidates, SurgeonsPAC hosted several health care industry events in the ACS Washington Office for key members of Congress and political campaign committees, collaborated with other medical community PACs to host fundraising events and physician candidate meet and greets, participated in party committee briefings and additional engagement opportunities, and increased in-district check deliveries and targeted donor events.
To learn more about SurgeonsPAC fundraising or disbursements, visit SurgeonsPAC.org (login required using facs.org username and password) or contact Katie Oehmen, Manager, ACSPA-SurgeonsPAC and Grassroots at 202-672-1503 or [email protected]. For more information about the College’s legislative priorities, visit SurgeonsVoice.org.
Contributions to ACSPA-SurgeonsPAC are not deductible as charitable contributions for federal income tax purposes. Contributions are voluntary, and all members of ACSPA have the right to refuse to contribute without reprisal. Federal law prohibits ACSPA-SurgeonsPAC from accepting contributions from foreign nations. By law, if your contributions are made using a personal check or credit card, ACSPA-SurgeonsPAC may only use your contribution to support candidates in federal elections. All corporate contributions to ACSPA-SurgeonsPAC will be used for educational and administrative fees of ACSPA and other activities permissible under federal law. Federal law requires ACSPA-SurgeonsPAC to use its best efforts to collect and report the name, mailing address, occupation, and the name of the employer of individuals whose contributions exceed $200 in a calendar year. ACSPA-SurgeonsPAC is a program of the ACSPA, which is exempt from federal income tax under section 501c (6) of the Internal Revenue Code.
Measuring success when it comes to political action, particularly in the current climate, can be challenging. Just as the American College of Surgeons (ACS) strives to represent all of surgery and focuses on an extensive list of priorities, the American College of Surgeons Professional Association Political Action Committee (ACSPA-SurgeonsPAC) works to maintain relationships with numerous lawmakers to ensure the College’s advocacy and health policy agenda remains at the forefront.
Leading up to the November midterm elections, every surgeon should be familiar with some basic facts about ACSPA-SurgeonsPAC fundraising and disbursement efforts.
Fundraising and disbursements
Fundraising is on par with the 2016 election cycle, a record year for “hard” dollars, or personal funds used to support candidates. SurgeonsPAC supports both Democrat and Republican candidates running in U.S. House and Senate races across the nation. SurgeonsPAC has a track record of balanced giving, contributing to candidates and incumbents of both parties who are willing to champion issues of importance to surgeons and surgical patients. All 2017‒2018 election cycle disbursements can be viewed at www.surgeonspac.org/disbursements. Legally, SurgeonsPAC cannot earmark contributions for a particular candidate or based on a single issue.
From January 1, 2017, through May 31, 2018, the ACSPA-SurgeonsPAC reported more than $805,000 in receipts from more than 1,500 College members and staff, and disbursed more than $558,900 to more than 115 congressional candidates, leadership PACs, and political campaign committees. Of the amount given, in line with congressional party ratios, 59 percent was given to Republicans and 41 percent to Democrats.
In addition to raising funds to elect and reelect congressional candidates, SurgeonsPAC hosted several health care industry events in the ACS Washington Office for key members of Congress and political campaign committees, collaborated with other medical community PACs to host fundraising events and physician candidate meet and greets, participated in party committee briefings and additional engagement opportunities, and increased in-district check deliveries and targeted donor events.
To learn more about SurgeonsPAC fundraising or disbursements, visit SurgeonsPAC.org (login required using facs.org username and password) or contact Katie Oehmen, Manager, ACSPA-SurgeonsPAC and Grassroots at 202-672-1503 or [email protected]. For more information about the College’s legislative priorities, visit SurgeonsVoice.org.
Contributions to ACSPA-SurgeonsPAC are not deductible as charitable contributions for federal income tax purposes. Contributions are voluntary, and all members of ACSPA have the right to refuse to contribute without reprisal. Federal law prohibits ACSPA-SurgeonsPAC from accepting contributions from foreign nations. By law, if your contributions are made using a personal check or credit card, ACSPA-SurgeonsPAC may only use your contribution to support candidates in federal elections. All corporate contributions to ACSPA-SurgeonsPAC will be used for educational and administrative fees of ACSPA and other activities permissible under federal law. Federal law requires ACSPA-SurgeonsPAC to use its best efforts to collect and report the name, mailing address, occupation, and the name of the employer of individuals whose contributions exceed $200 in a calendar year. ACSPA-SurgeonsPAC is a program of the ACSPA, which is exempt from federal income tax under section 501c (6) of the Internal Revenue Code.
Register for ACS Comprehensive General Surgery Review Course, July 26–29
The 2018 American College of Surgeons (ACS) Comprehensive General Surgery Review Course, July 26–29 in Chicago, IL, is an intensive three-and-a-half-day review course that will cover essential content areas in general surgery, including alimentary tract, endocrine and soft tissue, oncology, skin and breast, surgical critical care, trauma, and vascular operations, as well as perioperative care.
Course Chair John A. Weigelt, MD, DVM, MMA, FACS, and a distinguished faculty will use didactic and case-based formats to present a comprehensive and practical review. Dr. Weigelt recently retired from the Medical College of Wisconsin, where he was the Milt & Lidy Lunda/Charles Aprahamian Professor of Trauma Surgery, as well as professor and chief, division of trauma and critical care. He is joining the University of South Dakota and the Sanford Health System, Sioux Falls, this summer as professor of surgery. Dr. Weigelt is Medical Director of the ACS Surgical Education and Self-Assessment Program (also known as SESAP®).
The course offers a pragmatic review designed to focus on practice issues and will offer several special features, such as self-assessment materials, including pre- and posttests. It may be helpful in preparing for examinations. Self-assessment credit will be available.
The ACS is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to provide Continuing Medical Education for physicians.
The ACS designates this live activity for a maximum of 28 AMA PRA Category 1 Credits™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Register today at bit.ly/2s19VtX. Space is limited and registration will be accepted on a first-come, first-served basis. For more information and registration, visit the Comprehensive General Surgery Review Course web page at facs.org/gsreviewcourse or e-mail [email protected] or [email protected].
The 2018 American College of Surgeons (ACS) Comprehensive General Surgery Review Course, July 26–29 in Chicago, IL, is an intensive three-and-a-half-day review course that will cover essential content areas in general surgery, including alimentary tract, endocrine and soft tissue, oncology, skin and breast, surgical critical care, trauma, and vascular operations, as well as perioperative care.
Course Chair John A. Weigelt, MD, DVM, MMA, FACS, and a distinguished faculty will use didactic and case-based formats to present a comprehensive and practical review. Dr. Weigelt recently retired from the Medical College of Wisconsin, where he was the Milt & Lidy Lunda/Charles Aprahamian Professor of Trauma Surgery, as well as professor and chief, division of trauma and critical care. He is joining the University of South Dakota and the Sanford Health System, Sioux Falls, this summer as professor of surgery. Dr. Weigelt is Medical Director of the ACS Surgical Education and Self-Assessment Program (also known as SESAP®).
The course offers a pragmatic review designed to focus on practice issues and will offer several special features, such as self-assessment materials, including pre- and posttests. It may be helpful in preparing for examinations. Self-assessment credit will be available.
The ACS is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to provide Continuing Medical Education for physicians.
The ACS designates this live activity for a maximum of 28 AMA PRA Category 1 Credits™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Register today at bit.ly/2s19VtX. Space is limited and registration will be accepted on a first-come, first-served basis. For more information and registration, visit the Comprehensive General Surgery Review Course web page at facs.org/gsreviewcourse or e-mail [email protected] or [email protected].
The 2018 American College of Surgeons (ACS) Comprehensive General Surgery Review Course, July 26–29 in Chicago, IL, is an intensive three-and-a-half-day review course that will cover essential content areas in general surgery, including alimentary tract, endocrine and soft tissue, oncology, skin and breast, surgical critical care, trauma, and vascular operations, as well as perioperative care.
Course Chair John A. Weigelt, MD, DVM, MMA, FACS, and a distinguished faculty will use didactic and case-based formats to present a comprehensive and practical review. Dr. Weigelt recently retired from the Medical College of Wisconsin, where he was the Milt & Lidy Lunda/Charles Aprahamian Professor of Trauma Surgery, as well as professor and chief, division of trauma and critical care. He is joining the University of South Dakota and the Sanford Health System, Sioux Falls, this summer as professor of surgery. Dr. Weigelt is Medical Director of the ACS Surgical Education and Self-Assessment Program (also known as SESAP®).
The course offers a pragmatic review designed to focus on practice issues and will offer several special features, such as self-assessment materials, including pre- and posttests. It may be helpful in preparing for examinations. Self-assessment credit will be available.
The ACS is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to provide Continuing Medical Education for physicians.
The ACS designates this live activity for a maximum of 28 AMA PRA Category 1 Credits™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Register today at bit.ly/2s19VtX. Space is limited and registration will be accepted on a first-come, first-served basis. For more information and registration, visit the Comprehensive General Surgery Review Course web page at facs.org/gsreviewcourse or e-mail [email protected] or [email protected].
NAPRC awards first accreditation to John Muir Health Rectal Cancer Program
The National Accreditation Program for Rectal Cancer (NAPRC), which the American College of Surgeons (ACS) launched in 2017, has awarded its first accreditation to the John Muir Health Rectal Cancer Program, Walnut Creek and Concord, CA. To earn the voluntary accreditation, the John Muir Health Rectal Cancer Program met 19 standards, including the establishment of a rectal cancer multidisciplin
Thirteen of those standards address clinical services that the program was required to provide, including carcinoembryonic antigen testing, magnetic resonance imagining, and computed tomography imaging for cancer staging, and ensuring a process whereby the patient starts treatment within a defined time frame. One of the most important clinical standards requires all rectal cancer patients to be present at both pre- and post-treatment RC-MDT meetings.![]()
“When a cancer center achieves this type of specialized accreditation, it means that their rectal cancer patients will receive streamlined, modern evaluation and treatment for the disease. Compliance with our standards will assure optimal care for these patients,” said David P. Winchester, MD, FACS, Medical Director, ACS Cancer Programs.
“We have come a long way in the treatment of rectal cancer, but it remains a very complex disease that can be challenging to treat,” said Samuel Oommen, MD, FACS, a colorectal surgeon and medical director of the gastrointestinal oncology program at John Muir Health.
“This accreditation demonstrates to patients that we have an innovative program that is at the forefront of rectal cancer care by following rigorous standards and best practices. Achieving this designation is a recognition of the work done by a dedicated multidisciplinary team providing high quality, patient-centered care to provide superior oncological outcomes while preserving quality of life,” Dr. Oommen said.
The NAPRC was developed through a collaboration between the Optimizing the Surgical Treatment of Rectal Cancer Consortium and the ACS Commission on Cancer. It is based on successful international models that emphasize program structure, patient care processes, performance improvement, and performance measures. Its goal is to ensure that rectal cancer patients receive appropriate care using a multidisciplinary approach.
For more information about the program and instructions on how to apply for accreditation, visit the NAPRC website at facs.org/naprc, or contact [email protected].
The National Accreditation Program for Rectal Cancer (NAPRC), which the American College of Surgeons (ACS) launched in 2017, has awarded its first accreditation to the John Muir Health Rectal Cancer Program, Walnut Creek and Concord, CA. To earn the voluntary accreditation, the John Muir Health Rectal Cancer Program met 19 standards, including the establishment of a rectal cancer multidisciplin
Thirteen of those standards address clinical services that the program was required to provide, including carcinoembryonic antigen testing, magnetic resonance imagining, and computed tomography imaging for cancer staging, and ensuring a process whereby the patient starts treatment within a defined time frame. One of the most important clinical standards requires all rectal cancer patients to be present at both pre- and post-treatment RC-MDT meetings.![]()
“When a cancer center achieves this type of specialized accreditation, it means that their rectal cancer patients will receive streamlined, modern evaluation and treatment for the disease. Compliance with our standards will assure optimal care for these patients,” said David P. Winchester, MD, FACS, Medical Director, ACS Cancer Programs.
“We have come a long way in the treatment of rectal cancer, but it remains a very complex disease that can be challenging to treat,” said Samuel Oommen, MD, FACS, a colorectal surgeon and medical director of the gastrointestinal oncology program at John Muir Health.
“This accreditation demonstrates to patients that we have an innovative program that is at the forefront of rectal cancer care by following rigorous standards and best practices. Achieving this designation is a recognition of the work done by a dedicated multidisciplinary team providing high quality, patient-centered care to provide superior oncological outcomes while preserving quality of life,” Dr. Oommen said.
The NAPRC was developed through a collaboration between the Optimizing the Surgical Treatment of Rectal Cancer Consortium and the ACS Commission on Cancer. It is based on successful international models that emphasize program structure, patient care processes, performance improvement, and performance measures. Its goal is to ensure that rectal cancer patients receive appropriate care using a multidisciplinary approach.
For more information about the program and instructions on how to apply for accreditation, visit the NAPRC website at facs.org/naprc, or contact [email protected].
The National Accreditation Program for Rectal Cancer (NAPRC), which the American College of Surgeons (ACS) launched in 2017, has awarded its first accreditation to the John Muir Health Rectal Cancer Program, Walnut Creek and Concord, CA. To earn the voluntary accreditation, the John Muir Health Rectal Cancer Program met 19 standards, including the establishment of a rectal cancer multidisciplin
Thirteen of those standards address clinical services that the program was required to provide, including carcinoembryonic antigen testing, magnetic resonance imagining, and computed tomography imaging for cancer staging, and ensuring a process whereby the patient starts treatment within a defined time frame. One of the most important clinical standards requires all rectal cancer patients to be present at both pre- and post-treatment RC-MDT meetings.![]()
“When a cancer center achieves this type of specialized accreditation, it means that their rectal cancer patients will receive streamlined, modern evaluation and treatment for the disease. Compliance with our standards will assure optimal care for these patients,” said David P. Winchester, MD, FACS, Medical Director, ACS Cancer Programs.
“We have come a long way in the treatment of rectal cancer, but it remains a very complex disease that can be challenging to treat,” said Samuel Oommen, MD, FACS, a colorectal surgeon and medical director of the gastrointestinal oncology program at John Muir Health.
“This accreditation demonstrates to patients that we have an innovative program that is at the forefront of rectal cancer care by following rigorous standards and best practices. Achieving this designation is a recognition of the work done by a dedicated multidisciplinary team providing high quality, patient-centered care to provide superior oncological outcomes while preserving quality of life,” Dr. Oommen said.
The NAPRC was developed through a collaboration between the Optimizing the Surgical Treatment of Rectal Cancer Consortium and the ACS Commission on Cancer. It is based on successful international models that emphasize program structure, patient care processes, performance improvement, and performance measures. Its goal is to ensure that rectal cancer patients receive appropriate care using a multidisciplinary approach.
For more information about the program and instructions on how to apply for accreditation, visit the NAPRC website at facs.org/naprc, or contact [email protected].
Registration for Clinical Congress 2018 now open
Registration is now open for the American College of Surgeons Clinical Congress 2018, October 21–25 in Boston, MA. The Clinical Congress is one of the largest educational meetings of surgeons in the world and offers outstanding educational opportunities for every stage of your career. The theme of this year’s conference is the Joy and Privilege of a Surgical Career. The scientific program, online registration, travel and hotel reservation links, and previews of other planned events for the conference are available on the Clinical Congress 2018 website at facs.org/clincon2018.
The Clinical Congress 2018 program add
Register now at facs.org/clincon2018/register.
Registration is open to all physicians and individuals in the health care field. To receive the early bird registration fees, be sure to register by 11:59 pm (Central time) Monday, August 27.
Registration is now open for the American College of Surgeons Clinical Congress 2018, October 21–25 in Boston, MA. The Clinical Congress is one of the largest educational meetings of surgeons in the world and offers outstanding educational opportunities for every stage of your career. The theme of this year’s conference is the Joy and Privilege of a Surgical Career. The scientific program, online registration, travel and hotel reservation links, and previews of other planned events for the conference are available on the Clinical Congress 2018 website at facs.org/clincon2018.
The Clinical Congress 2018 program add
Register now at facs.org/clincon2018/register.
Registration is open to all physicians and individuals in the health care field. To receive the early bird registration fees, be sure to register by 11:59 pm (Central time) Monday, August 27.
Registration is now open for the American College of Surgeons Clinical Congress 2018, October 21–25 in Boston, MA. The Clinical Congress is one of the largest educational meetings of surgeons in the world and offers outstanding educational opportunities for every stage of your career. The theme of this year’s conference is the Joy and Privilege of a Surgical Career. The scientific program, online registration, travel and hotel reservation links, and previews of other planned events for the conference are available on the Clinical Congress 2018 website at facs.org/clincon2018.
The Clinical Congress 2018 program add
Register now at facs.org/clincon2018/register.
Registration is open to all physicians and individuals in the health care field. To receive the early bird registration fees, be sure to register by 11:59 pm (Central time) Monday, August 27.
CAR T Therapy: From Bench to Bedside and Back
Release Date: July 15, 2018
Expiration Date: July 14, 2019
Note: This activity is no longer available for credit
Introductory Comments: (Duration: 9 minutes)
Aaron P. Rapoport, MD
Bone Marrow Transplant Program
University of Maryland School of Medicine
Baltimore, MD
Presentation: (Duration: 39 minutes)
Carl H. June, MD
Richard W. Vague Professor in Immunotherapy
Perelman School of Medicine
University of Pennsylvania
Philadelphia, PA
Provided by:
![]()
Learning Objectives
• Review clinical data and individual case studies to determine where CAR T-cell therapy might be appropriate in the treatment of adult and pediatric patients with leukemia, lymphoma, and multiple myeloma.
• Discuss the management of cytotoxicity of CAR T-cell therapy.
Target Audience
Hematologists, oncologists, and other members of the healthcare team who treat or manage patients with hematologic malignancies.
Statement of Need
It is critical that clinicians managing patients with acute leukemia and other hematologic malignancies are cognizant of exciting breakthroughs and are also able to integrate recent progress into practice. However, given the overwhelming influx of data, it is no surprise that many hematology professionals face difficulties in identifying the most relevant findings for clinical practice. Hematologists are unable to stay abreast of the latest evidence on investigational agents. Educational programs are thus crucial to address this important professional practice gap.
Faculty
Carl H. June, MD
Richard W. Vague Professor in Immunotherapy
Perelman School of Medicine
University of Pennsylvania
Philadelphia, PA
Disclosures: Consultant: Novartis; Grant/Research support and royalties/IPR: Novartis
Stockholder: Tmunity Therapeutics, Inc.
Aaron P. Rapoport, MD
Bone Marrow Transplant Program
University of Maryland School of Medicine
Baltimore, Maryland
Disclosures: No relevant financial relationships with a commercial supporter
Permissions
- Slide 3: Complex tumor, host and environmental factors govern the strength and timing of anti-cancer immune responses
- Reprinted from Immunity, Vol 39/No 1, Chen DS, Mellman I, Oncology meets immunology: the cancer-immunity cycle, pp 1-10, 2013, with permission from Elsevier
- Slide 9: Genes differentially expressed in CART19 cellular infusion products from CLL patients
- From Fraietta JA, Lacey SF, Orlando EJ, . . . June CH, Melenhorst JJ. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 2018; 24:563-571
- Slide 10: Characterization of CLL CAR T cells in NSG CLL model
- Same as slide 9
- Slide 15: First adult ALL patient
- Photos originally published in Kaiser Health News/Photo courtesy of Dr Keith Eaton. Available at: https://khn.org/news/cascade-of-costs-could-push-new-gene-therapy-above-1-million-per-patient/
- Slide 21: Efficient trafficking of CTL019 T Cells to CNS in ALL
- From N Engl J Med, Grupp SA, Kalos M, Barrett D, . . V. June CH, Chimeric antigen receptor-modified T cells for acute lymphoid leukemia, Volume No 368, pp 1509-1518. Copyright © 2013 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
- Slide 26: Long-term persistence and expression of CTL019 is associated with durable remission in leukemia: Predictive Biomarker
- From Porter DL, Hwang WT, Frey NV . . . June CH. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 2015; 7(303):303ra139. Reprinted with permission from AAAS.
- Slide 28: Rapid massive expansion of clonal CART cell population in patient #10
- Initially published in Fraietta JA, Nobles CL, Sammons MA, . . . June CH, Melenhors JJ. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018; 558(7709):307-312
- Slide 29: Mapping CAR integration site in Pt #10
- Same as slide 28.
- Slide 31: Long-term stable persistence of TET2-deficient CAR T cells in Pt #10
- Same as slide 28
- Slide 32: Epigenetic and genetic changes uncovered by ATAC-seq in Pt #10
- Same as slide 28.
- Slide 33: TET2 knock down in healthy donor T cells
- Same as slide 28.
- Slide 34: TET2 knock down in healthy donor T cells
- Same as slide 28.
- Slide 36: CAR T for myeloma: BCMA
- From Rickert RC, Jellusova J, Miletic AV. Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunol Rev 2011; 244(1):115-33. Reprinted with permission from John Wiley and Sons.
- Slide 38: CAR T for myeloma: Patient #1
- Photo originally published by UT Southwestern Medical Center. Available at: https://www.utsouthwestern.edu/newsroom/articles/year-2018/wright-car-t.html
- Slide 39: Autoimmunity is the “Achilles’ Heel” of immunotherapy
- First published in June CH, Warshauer JT, and Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med 2017;23(5):540-7
- Slide 41: Multiplex CRISPR /Cas9 editing: Universal T cells TCR, HLA, PD-1, CTLA-4 and Fas
- From Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, Zhao Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 2017; 8:17002-17011.
- Slide 45: CAR T-cell trials for cancer are now global
- From June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science 2018; 359:1361-1365. Reprinted with permission from AAAS.
Disclaimer
The content and views presented in this educational activity are those of the author and do not necessarily reflect those of Hemedicus or Frontline Medical Communications. This material is prepared based upon a review of multiple sources of information, but it is not exhaustive of the subject matter. Therefore, healthcare professionals and other individuals should review and consider other publications and materials on the subject matter before relying solely upon the information contained within this educational activity.
Release Date: July 15, 2018
Expiration Date: July 14, 2019
Note: This activity is no longer available for credit
Introductory Comments: (Duration: 9 minutes)
Aaron P. Rapoport, MD
Bone Marrow Transplant Program
University of Maryland School of Medicine
Baltimore, MD
Presentation: (Duration: 39 minutes)
Carl H. June, MD
Richard W. Vague Professor in Immunotherapy
Perelman School of Medicine
University of Pennsylvania
Philadelphia, PA
Provided by:
![]()
Learning Objectives
• Review clinical data and individual case studies to determine where CAR T-cell therapy might be appropriate in the treatment of adult and pediatric patients with leukemia, lymphoma, and multiple myeloma.
• Discuss the management of cytotoxicity of CAR T-cell therapy.
Target Audience
Hematologists, oncologists, and other members of the healthcare team who treat or manage patients with hematologic malignancies.
Statement of Need
It is critical that clinicians managing patients with acute leukemia and other hematologic malignancies are cognizant of exciting breakthroughs and are also able to integrate recent progress into practice. However, given the overwhelming influx of data, it is no surprise that many hematology professionals face difficulties in identifying the most relevant findings for clinical practice. Hematologists are unable to stay abreast of the latest evidence on investigational agents. Educational programs are thus crucial to address this important professional practice gap.
Faculty
Carl H. June, MD
Richard W. Vague Professor in Immunotherapy
Perelman School of Medicine
University of Pennsylvania
Philadelphia, PA
Disclosures: Consultant: Novartis; Grant/Research support and royalties/IPR: Novartis
Stockholder: Tmunity Therapeutics, Inc.
Aaron P. Rapoport, MD
Bone Marrow Transplant Program
University of Maryland School of Medicine
Baltimore, Maryland
Disclosures: No relevant financial relationships with a commercial supporter
Permissions
- Slide 3: Complex tumor, host and environmental factors govern the strength and timing of anti-cancer immune responses
- Reprinted from Immunity, Vol 39/No 1, Chen DS, Mellman I, Oncology meets immunology: the cancer-immunity cycle, pp 1-10, 2013, with permission from Elsevier
- Slide 9: Genes differentially expressed in CART19 cellular infusion products from CLL patients
- From Fraietta JA, Lacey SF, Orlando EJ, . . . June CH, Melenhorst JJ. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 2018; 24:563-571
- Slide 10: Characterization of CLL CAR T cells in NSG CLL model
- Same as slide 9
- Slide 15: First adult ALL patient
- Photos originally published in Kaiser Health News/Photo courtesy of Dr Keith Eaton. Available at: https://khn.org/news/cascade-of-costs-could-push-new-gene-therapy-above-1-million-per-patient/
- Slide 21: Efficient trafficking of CTL019 T Cells to CNS in ALL
- From N Engl J Med, Grupp SA, Kalos M, Barrett D, . . V. June CH, Chimeric antigen receptor-modified T cells for acute lymphoid leukemia, Volume No 368, pp 1509-1518. Copyright © 2013 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
- Slide 26: Long-term persistence and expression of CTL019 is associated with durable remission in leukemia: Predictive Biomarker
- From Porter DL, Hwang WT, Frey NV . . . June CH. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 2015; 7(303):303ra139. Reprinted with permission from AAAS.
- Slide 28: Rapid massive expansion of clonal CART cell population in patient #10
- Initially published in Fraietta JA, Nobles CL, Sammons MA, . . . June CH, Melenhors JJ. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018; 558(7709):307-312
- Slide 29: Mapping CAR integration site in Pt #10
- Same as slide 28.
- Slide 31: Long-term stable persistence of TET2-deficient CAR T cells in Pt #10
- Same as slide 28
- Slide 32: Epigenetic and genetic changes uncovered by ATAC-seq in Pt #10
- Same as slide 28.
- Slide 33: TET2 knock down in healthy donor T cells
- Same as slide 28.
- Slide 34: TET2 knock down in healthy donor T cells
- Same as slide 28.
- Slide 36: CAR T for myeloma: BCMA
- From Rickert RC, Jellusova J, Miletic AV. Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunol Rev 2011; 244(1):115-33. Reprinted with permission from John Wiley and Sons.
- Slide 38: CAR T for myeloma: Patient #1
- Photo originally published by UT Southwestern Medical Center. Available at: https://www.utsouthwestern.edu/newsroom/articles/year-2018/wright-car-t.html
- Slide 39: Autoimmunity is the “Achilles’ Heel” of immunotherapy
- First published in June CH, Warshauer JT, and Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med 2017;23(5):540-7
- Slide 41: Multiplex CRISPR /Cas9 editing: Universal T cells TCR, HLA, PD-1, CTLA-4 and Fas
- From Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, Zhao Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 2017; 8:17002-17011.
- Slide 45: CAR T-cell trials for cancer are now global
- From June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science 2018; 359:1361-1365. Reprinted with permission from AAAS.
Disclaimer
The content and views presented in this educational activity are those of the author and do not necessarily reflect those of Hemedicus or Frontline Medical Communications. This material is prepared based upon a review of multiple sources of information, but it is not exhaustive of the subject matter. Therefore, healthcare professionals and other individuals should review and consider other publications and materials on the subject matter before relying solely upon the information contained within this educational activity.
Release Date: July 15, 2018
Expiration Date: July 14, 2019
Note: This activity is no longer available for credit
Introductory Comments: (Duration: 9 minutes)
Aaron P. Rapoport, MD
Bone Marrow Transplant Program
University of Maryland School of Medicine
Baltimore, MD
Presentation: (Duration: 39 minutes)
Carl H. June, MD
Richard W. Vague Professor in Immunotherapy
Perelman School of Medicine
University of Pennsylvania
Philadelphia, PA
Provided by:
![]()
Learning Objectives
• Review clinical data and individual case studies to determine where CAR T-cell therapy might be appropriate in the treatment of adult and pediatric patients with leukemia, lymphoma, and multiple myeloma.
• Discuss the management of cytotoxicity of CAR T-cell therapy.
Target Audience
Hematologists, oncologists, and other members of the healthcare team who treat or manage patients with hematologic malignancies.
Statement of Need
It is critical that clinicians managing patients with acute leukemia and other hematologic malignancies are cognizant of exciting breakthroughs and are also able to integrate recent progress into practice. However, given the overwhelming influx of data, it is no surprise that many hematology professionals face difficulties in identifying the most relevant findings for clinical practice. Hematologists are unable to stay abreast of the latest evidence on investigational agents. Educational programs are thus crucial to address this important professional practice gap.
Faculty
Carl H. June, MD
Richard W. Vague Professor in Immunotherapy
Perelman School of Medicine
University of Pennsylvania
Philadelphia, PA
Disclosures: Consultant: Novartis; Grant/Research support and royalties/IPR: Novartis
Stockholder: Tmunity Therapeutics, Inc.
Aaron P. Rapoport, MD
Bone Marrow Transplant Program
University of Maryland School of Medicine
Baltimore, Maryland
Disclosures: No relevant financial relationships with a commercial supporter
Permissions
- Slide 3: Complex tumor, host and environmental factors govern the strength and timing of anti-cancer immune responses
- Reprinted from Immunity, Vol 39/No 1, Chen DS, Mellman I, Oncology meets immunology: the cancer-immunity cycle, pp 1-10, 2013, with permission from Elsevier
- Slide 9: Genes differentially expressed in CART19 cellular infusion products from CLL patients
- From Fraietta JA, Lacey SF, Orlando EJ, . . . June CH, Melenhorst JJ. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 2018; 24:563-571
- Slide 10: Characterization of CLL CAR T cells in NSG CLL model
- Same as slide 9
- Slide 15: First adult ALL patient
- Photos originally published in Kaiser Health News/Photo courtesy of Dr Keith Eaton. Available at: https://khn.org/news/cascade-of-costs-could-push-new-gene-therapy-above-1-million-per-patient/
- Slide 21: Efficient trafficking of CTL019 T Cells to CNS in ALL
- From N Engl J Med, Grupp SA, Kalos M, Barrett D, . . V. June CH, Chimeric antigen receptor-modified T cells for acute lymphoid leukemia, Volume No 368, pp 1509-1518. Copyright © 2013 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
- Slide 26: Long-term persistence and expression of CTL019 is associated with durable remission in leukemia: Predictive Biomarker
- From Porter DL, Hwang WT, Frey NV . . . June CH. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 2015; 7(303):303ra139. Reprinted with permission from AAAS.
- Slide 28: Rapid massive expansion of clonal CART cell population in patient #10
- Initially published in Fraietta JA, Nobles CL, Sammons MA, . . . June CH, Melenhors JJ. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018; 558(7709):307-312
- Slide 29: Mapping CAR integration site in Pt #10
- Same as slide 28.
- Slide 31: Long-term stable persistence of TET2-deficient CAR T cells in Pt #10
- Same as slide 28
- Slide 32: Epigenetic and genetic changes uncovered by ATAC-seq in Pt #10
- Same as slide 28.
- Slide 33: TET2 knock down in healthy donor T cells
- Same as slide 28.
- Slide 34: TET2 knock down in healthy donor T cells
- Same as slide 28.
- Slide 36: CAR T for myeloma: BCMA
- From Rickert RC, Jellusova J, Miletic AV. Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunol Rev 2011; 244(1):115-33. Reprinted with permission from John Wiley and Sons.
- Slide 38: CAR T for myeloma: Patient #1
- Photo originally published by UT Southwestern Medical Center. Available at: https://www.utsouthwestern.edu/newsroom/articles/year-2018/wright-car-t.html
- Slide 39: Autoimmunity is the “Achilles’ Heel” of immunotherapy
- First published in June CH, Warshauer JT, and Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med 2017;23(5):540-7
- Slide 41: Multiplex CRISPR /Cas9 editing: Universal T cells TCR, HLA, PD-1, CTLA-4 and Fas
- From Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, Zhao Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 2017; 8:17002-17011.
- Slide 45: CAR T-cell trials for cancer are now global
- From June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science 2018; 359:1361-1365. Reprinted with permission from AAAS.
Disclaimer
The content and views presented in this educational activity are those of the author and do not necessarily reflect those of Hemedicus or Frontline Medical Communications. This material is prepared based upon a review of multiple sources of information, but it is not exhaustive of the subject matter. Therefore, healthcare professionals and other individuals should review and consider other publications and materials on the subject matter before relying solely upon the information contained within this educational activity.
Migraine treatment insights offered for noninvasive vagus nerve stimulation
SAN FRANCISCO – Now that noninvasive vagus nerve stimulation using a small handheld device has earned clearance from the Food and Drug Administration for acute treatment of migraine attacks, investigators are pouring over data from the major clinical trials to gain post hoc insight into how to optimize use of the gammaCore device in routine clinical practice.
The answer is to use it early in the course of an attack and use it often, Eric Liebler said at the annual meeting of the American Headache Society.

“Our motto here in the U.S. is to get the device and use it as many times as you want during the month. There are no pharmacologic side effects, no drug-drug interactions, and no risk of overusing it that we are aware of at this point. So if you give it to a patient who’s able to say, ‘Wait a second – I don’t feel good, I have premonitory symptoms,’ if they use it, then they’re likely to feel better, with a clinically relevant reduction of at least 1 point when treating a migraine while it’s mild,” according to Mr. Liebler, senior vice president for neurology at electroCore, of Basking Ridge, N.J., which markets the noninvasive vagus nerve stimulation (nVNS) gammaCore device.
The device received FDA clearance for acute treatment of migraine in January 2018 following regulatory approval in the spring of 2017 for treatment of episodic cluster headache.
Mr. Liebler presented key highlights of the recently published pivotal PRESTO trial (Prospective Study of nVNS for the Acute Treatment of Migraine), a randomized, double-blind, multicenter, sham-controlled study of 243 adults under age 50 years who experienced 3-8 migraine attacks per month (Neurology. 2018 Jun 15. doi: 10.1212/WNL.0000000000005857).
“This study provides Class I evidence that, for patients with an episodic migraine, nVNS significantly increases the probability of having mild pain or being pain-free post stimulation,” Mr. Liebler declared.
During the 4-week, double-blind treatment period, 48% of the active nVNS group met the International Headache Society definition of pain relief, which meant at least a 1-point reduction in migraine severity on a 0-3 point pain scale at 120 minutes in at least 50% of treated migraine episodes without using rescue medications. This was a significantly better result than the 32% rate seen in patients in the control group who were given a sham device that emitted a perceptible but ineffective signal.
Similar results were documented during the subsequent 4-week, open-label treatment period, during which the control group was switched over to a functioning gammaCore.
“You can tell your patients they have a reliable chance of having a response more often than not,” according to Mr. Liebler.
What else can physicians tell their migraine patients to expect regarding efficacy? The pain-free responder rate at 120 minutes in PRESTO and other studies is similar to rates reported in meta-analyses of oral triptan therapy but without any drug side effects or limitations on frequency of use. Furthermore, the need for rescue medication at any time during a migraine attack was significantly lower with nVNS than it was with sham treatment, by a margin of 48%-63%.
“If they use this device, they’re often able to save that triptan for when they wake up in the morning and they’re already at a 3 in migraine severity,” he observed.
Also, nVNS is fast-acting: At 30 minutes from onset of pain in the first treated attack, 27% of the nVNS group had experienced pain relief, compared with 19% of controls.
The tolerability profile of the device therapy was outstanding, with no treatment discontinuations in the active treatment arm and only occasional reports of mild transient application site discomfort.
Asked about insurance coverage, Mr. Liebler said broad coverage is coming, but it’s not here yet.
“Insurers don’t really want to pay for anything, and devices confuse them even more than drugs. But we’re getting there because of the quality of the evidence. They were really having a good time telling us we didn’t have any published papers, and now that we’ve given them a class I study published in Neurology, they’ve said they had to review it. CVS will cover it starting Jan. 1,” he continued.
Planning is underway for additional clinical trials of the gammaCore for prevention of episodic migraine, acute treatment of attacks in adolescents, and for use in pregnancy.
Using the nVNS device
This nVNS device produces a proprietary, low-voltage electrical signal with a 24-volt peak voltage and a 60-mA peak output current. In the PRESTO study, patients were instructed to self-administer bilateral 120-second stimulations to the right and left sides of their necks within 20 minutes of pain onset. If the pain hadn’t improved within 15 minutes, they were to repeat the stimulations.
In a post hoc analysis of the PREVA (Prevention and Acute treatment of chronic cluster headache) study (Cephalalgia. 2016;36[6]:534-46), investigators determined that the mean reduction in the number of cluster headache attacks was significantly greater in patients who used the device to treat at least 77% of their attacks. That had a bigger preventive effect than did the number of stimulations a patient applied per day.
The PRESTO and PREVA trials were sponsored by electroCore, where Mr. Liebler is employed.
SAN FRANCISCO – Now that noninvasive vagus nerve stimulation using a small handheld device has earned clearance from the Food and Drug Administration for acute treatment of migraine attacks, investigators are pouring over data from the major clinical trials to gain post hoc insight into how to optimize use of the gammaCore device in routine clinical practice.
The answer is to use it early in the course of an attack and use it often, Eric Liebler said at the annual meeting of the American Headache Society.
“Our motto here in the U.S. is to get the device and use it as many times as you want during the month. There are no pharmacologic side effects, no drug-drug interactions, and no risk of overusing it that we are aware of at this point. So if you give it to a patient who’s able to say, ‘Wait a second – I don’t feel good, I have premonitory symptoms,’ if they use it, then they’re likely to feel better, with a clinically relevant reduction of at least 1 point when treating a migraine while it’s mild,” according to Mr. Liebler, senior vice president for neurology at electroCore, of Basking Ridge, N.J., which markets the noninvasive vagus nerve stimulation (nVNS) gammaCore device.
The device received FDA clearance for acute treatment of migraine in January 2018 following regulatory approval in the spring of 2017 for treatment of episodic cluster headache.
Mr. Liebler presented key highlights of the recently published pivotal PRESTO trial (Prospective Study of nVNS for the Acute Treatment of Migraine), a randomized, double-blind, multicenter, sham-controlled study of 243 adults under age 50 years who experienced 3-8 migraine attacks per month (Neurology. 2018 Jun 15. doi: 10.1212/WNL.0000000000005857).
“This study provides Class I evidence that, for patients with an episodic migraine, nVNS significantly increases the probability of having mild pain or being pain-free post stimulation,” Mr. Liebler declared.
During the 4-week, double-blind treatment period, 48% of the active nVNS group met the International Headache Society definition of pain relief, which meant at least a 1-point reduction in migraine severity on a 0-3 point pain scale at 120 minutes in at least 50% of treated migraine episodes without using rescue medications. This was a significantly better result than the 32% rate seen in patients in the control group who were given a sham device that emitted a perceptible but ineffective signal.
Similar results were documented during the subsequent 4-week, open-label treatment period, during which the control group was switched over to a functioning gammaCore.
“You can tell your patients they have a reliable chance of having a response more often than not,” according to Mr. Liebler.
What else can physicians tell their migraine patients to expect regarding efficacy? The pain-free responder rate at 120 minutes in PRESTO and other studies is similar to rates reported in meta-analyses of oral triptan therapy but without any drug side effects or limitations on frequency of use. Furthermore, the need for rescue medication at any time during a migraine attack was significantly lower with nVNS than it was with sham treatment, by a margin of 48%-63%.
“If they use this device, they’re often able to save that triptan for when they wake up in the morning and they’re already at a 3 in migraine severity,” he observed.
Also, nVNS is fast-acting: At 30 minutes from onset of pain in the first treated attack, 27% of the nVNS group had experienced pain relief, compared with 19% of controls.
The tolerability profile of the device therapy was outstanding, with no treatment discontinuations in the active treatment arm and only occasional reports of mild transient application site discomfort.
Asked about insurance coverage, Mr. Liebler said broad coverage is coming, but it’s not here yet.
“Insurers don’t really want to pay for anything, and devices confuse them even more than drugs. But we’re getting there because of the quality of the evidence. They were really having a good time telling us we didn’t have any published papers, and now that we’ve given them a class I study published in Neurology, they’ve said they had to review it. CVS will cover it starting Jan. 1,” he continued.
Planning is underway for additional clinical trials of the gammaCore for prevention of episodic migraine, acute treatment of attacks in adolescents, and for use in pregnancy.
Using the nVNS device
This nVNS device produces a proprietary, low-voltage electrical signal with a 24-volt peak voltage and a 60-mA peak output current. In the PRESTO study, patients were instructed to self-administer bilateral 120-second stimulations to the right and left sides of their necks within 20 minutes of pain onset. If the pain hadn’t improved within 15 minutes, they were to repeat the stimulations.
In a post hoc analysis of the PREVA (Prevention and Acute treatment of chronic cluster headache) study (Cephalalgia. 2016;36[6]:534-46), investigators determined that the mean reduction in the number of cluster headache attacks was significantly greater in patients who used the device to treat at least 77% of their attacks. That had a bigger preventive effect than did the number of stimulations a patient applied per day.
The PRESTO and PREVA trials were sponsored by electroCore, where Mr. Liebler is employed.
SAN FRANCISCO – Now that noninvasive vagus nerve stimulation using a small handheld device has earned clearance from the Food and Drug Administration for acute treatment of migraine attacks, investigators are pouring over data from the major clinical trials to gain post hoc insight into how to optimize use of the gammaCore device in routine clinical practice.
The answer is to use it early in the course of an attack and use it often, Eric Liebler said at the annual meeting of the American Headache Society.
“Our motto here in the U.S. is to get the device and use it as many times as you want during the month. There are no pharmacologic side effects, no drug-drug interactions, and no risk of overusing it that we are aware of at this point. So if you give it to a patient who’s able to say, ‘Wait a second – I don’t feel good, I have premonitory symptoms,’ if they use it, then they’re likely to feel better, with a clinically relevant reduction of at least 1 point when treating a migraine while it’s mild,” according to Mr. Liebler, senior vice president for neurology at electroCore, of Basking Ridge, N.J., which markets the noninvasive vagus nerve stimulation (nVNS) gammaCore device.
The device received FDA clearance for acute treatment of migraine in January 2018 following regulatory approval in the spring of 2017 for treatment of episodic cluster headache.
Mr. Liebler presented key highlights of the recently published pivotal PRESTO trial (Prospective Study of nVNS for the Acute Treatment of Migraine), a randomized, double-blind, multicenter, sham-controlled study of 243 adults under age 50 years who experienced 3-8 migraine attacks per month (Neurology. 2018 Jun 15. doi: 10.1212/WNL.0000000000005857).
“This study provides Class I evidence that, for patients with an episodic migraine, nVNS significantly increases the probability of having mild pain or being pain-free post stimulation,” Mr. Liebler declared.
During the 4-week, double-blind treatment period, 48% of the active nVNS group met the International Headache Society definition of pain relief, which meant at least a 1-point reduction in migraine severity on a 0-3 point pain scale at 120 minutes in at least 50% of treated migraine episodes without using rescue medications. This was a significantly better result than the 32% rate seen in patients in the control group who were given a sham device that emitted a perceptible but ineffective signal.
Similar results were documented during the subsequent 4-week, open-label treatment period, during which the control group was switched over to a functioning gammaCore.
“You can tell your patients they have a reliable chance of having a response more often than not,” according to Mr. Liebler.
What else can physicians tell their migraine patients to expect regarding efficacy? The pain-free responder rate at 120 minutes in PRESTO and other studies is similar to rates reported in meta-analyses of oral triptan therapy but without any drug side effects or limitations on frequency of use. Furthermore, the need for rescue medication at any time during a migraine attack was significantly lower with nVNS than it was with sham treatment, by a margin of 48%-63%.
“If they use this device, they’re often able to save that triptan for when they wake up in the morning and they’re already at a 3 in migraine severity,” he observed.
Also, nVNS is fast-acting: At 30 minutes from onset of pain in the first treated attack, 27% of the nVNS group had experienced pain relief, compared with 19% of controls.
The tolerability profile of the device therapy was outstanding, with no treatment discontinuations in the active treatment arm and only occasional reports of mild transient application site discomfort.
Asked about insurance coverage, Mr. Liebler said broad coverage is coming, but it’s not here yet.
“Insurers don’t really want to pay for anything, and devices confuse them even more than drugs. But we’re getting there because of the quality of the evidence. They were really having a good time telling us we didn’t have any published papers, and now that we’ve given them a class I study published in Neurology, they’ve said they had to review it. CVS will cover it starting Jan. 1,” he continued.
Planning is underway for additional clinical trials of the gammaCore for prevention of episodic migraine, acute treatment of attacks in adolescents, and for use in pregnancy.
Using the nVNS device
This nVNS device produces a proprietary, low-voltage electrical signal with a 24-volt peak voltage and a 60-mA peak output current. In the PRESTO study, patients were instructed to self-administer bilateral 120-second stimulations to the right and left sides of their necks within 20 minutes of pain onset. If the pain hadn’t improved within 15 minutes, they were to repeat the stimulations.
In a post hoc analysis of the PREVA (Prevention and Acute treatment of chronic cluster headache) study (Cephalalgia. 2016;36[6]:534-46), investigators determined that the mean reduction in the number of cluster headache attacks was significantly greater in patients who used the device to treat at least 77% of their attacks. That had a bigger preventive effect than did the number of stimulations a patient applied per day.
The PRESTO and PREVA trials were sponsored by electroCore, where Mr. Liebler is employed.
EXPERT ANALYSIS FROM THE AHS ANNUAL MEETING
For smokers, the ends may not justify the ENDS
Smokers who used e-cigarettes and other electronic nicotine delivery systems (ENDS) were less likely to quit than were those who did not use such products, according to a 2015 survey and a follow-up conducted a year later.
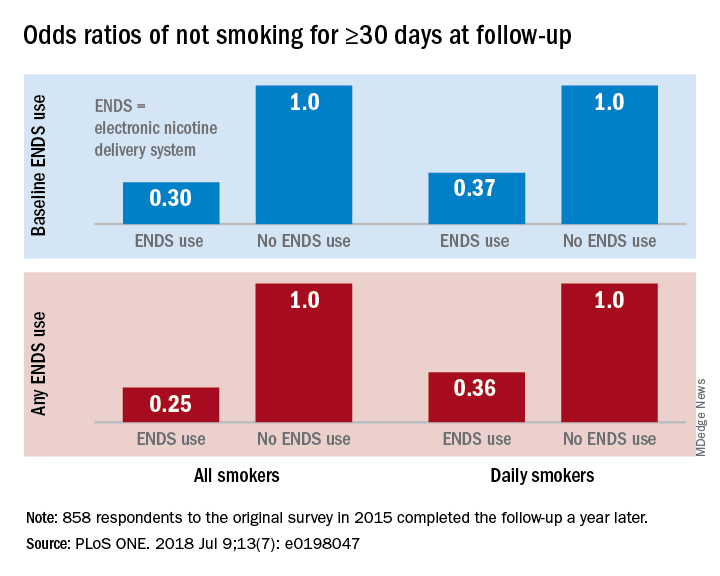
“Under ‘real world’ use and conditions [ENDS] may have suppressed or delayed quitting among some adult smokers,” Scott R. Weaver, PhD, and his associates at Georgia State University, Atlanta, wrote in PLoS One. The original survey, conducted in August and September of 2015, involved 1,284 U.S. adult smokers from the GfK KnowledgePanel, of whom 858 completed the follow-up survey in September 2016.
Smokers who used ENDS at baseline were slightly more likely to attempt to quit (53.7%) than were those who did not (48.6%) but were much less likely to have quit (defined as no smoking for at least 30 days at the time of follow-up): 9.4% vs. 18.9%, for an adjusted odds ratio of 0.30. Those who used ENDS at any time during the study were much more likely than were non-ENDS users to make an attempt (58.5% vs. 44.4%), but they were, again, much less likely to succeed (7.7% vs. 22.2%; AOR, 0.25), the investigators reported.
The results were similar for the subset of respondents who smoked every day: ENDS users were more likely to attempt to quit but less likely to succeed. Odds ratios for quitting were 0.37 for those using ENDS at baseline and 0.36 for those who used ENDS at any time since the first survey, Dr. Weaver and his associates said.
“Use of current ENDS products in real world conditions [does] not seem to improve the chances of quitting for smokers, and, under the current landscape, may not be the disruptive technology that increases the population quit rate and reduces the harm of combustibles,” they wrote.
The study was supported by the National Institute of Drug Abuse and the Food and Drug Administration’s Center for Tobacco Products. One of the investigators has received funding in the form of grant funding from Pfizer and the National Institutes of Health and another has served as a paid consultant to the Centers for Disease Control and Prevention.
SOURCE: Weaver SR et al. PLoS ONE. 2018 Jul 9;13(7): e0198047. doi: 10.1371/journal.pone.0198047.
Smokers who used e-cigarettes and other electronic nicotine delivery systems (ENDS) were less likely to quit than were those who did not use such products, according to a 2015 survey and a follow-up conducted a year later.
“Under ‘real world’ use and conditions [ENDS] may have suppressed or delayed quitting among some adult smokers,” Scott R. Weaver, PhD, and his associates at Georgia State University, Atlanta, wrote in PLoS One. The original survey, conducted in August and September of 2015, involved 1,284 U.S. adult smokers from the GfK KnowledgePanel, of whom 858 completed the follow-up survey in September 2016.
Smokers who used ENDS at baseline were slightly more likely to attempt to quit (53.7%) than were those who did not (48.6%) but were much less likely to have quit (defined as no smoking for at least 30 days at the time of follow-up): 9.4% vs. 18.9%, for an adjusted odds ratio of 0.30. Those who used ENDS at any time during the study were much more likely than were non-ENDS users to make an attempt (58.5% vs. 44.4%), but they were, again, much less likely to succeed (7.7% vs. 22.2%; AOR, 0.25), the investigators reported.
The results were similar for the subset of respondents who smoked every day: ENDS users were more likely to attempt to quit but less likely to succeed. Odds ratios for quitting were 0.37 for those using ENDS at baseline and 0.36 for those who used ENDS at any time since the first survey, Dr. Weaver and his associates said.
“Use of current ENDS products in real world conditions [does] not seem to improve the chances of quitting for smokers, and, under the current landscape, may not be the disruptive technology that increases the population quit rate and reduces the harm of combustibles,” they wrote.
The study was supported by the National Institute of Drug Abuse and the Food and Drug Administration’s Center for Tobacco Products. One of the investigators has received funding in the form of grant funding from Pfizer and the National Institutes of Health and another has served as a paid consultant to the Centers for Disease Control and Prevention.
SOURCE: Weaver SR et al. PLoS ONE. 2018 Jul 9;13(7): e0198047. doi: 10.1371/journal.pone.0198047.
Smokers who used e-cigarettes and other electronic nicotine delivery systems (ENDS) were less likely to quit than were those who did not use such products, according to a 2015 survey and a follow-up conducted a year later.
“Under ‘real world’ use and conditions [ENDS] may have suppressed or delayed quitting among some adult smokers,” Scott R. Weaver, PhD, and his associates at Georgia State University, Atlanta, wrote in PLoS One. The original survey, conducted in August and September of 2015, involved 1,284 U.S. adult smokers from the GfK KnowledgePanel, of whom 858 completed the follow-up survey in September 2016.
Smokers who used ENDS at baseline were slightly more likely to attempt to quit (53.7%) than were those who did not (48.6%) but were much less likely to have quit (defined as no smoking for at least 30 days at the time of follow-up): 9.4% vs. 18.9%, for an adjusted odds ratio of 0.30. Those who used ENDS at any time during the study were much more likely than were non-ENDS users to make an attempt (58.5% vs. 44.4%), but they were, again, much less likely to succeed (7.7% vs. 22.2%; AOR, 0.25), the investigators reported.
The results were similar for the subset of respondents who smoked every day: ENDS users were more likely to attempt to quit but less likely to succeed. Odds ratios for quitting were 0.37 for those using ENDS at baseline and 0.36 for those who used ENDS at any time since the first survey, Dr. Weaver and his associates said.
“Use of current ENDS products in real world conditions [does] not seem to improve the chances of quitting for smokers, and, under the current landscape, may not be the disruptive technology that increases the population quit rate and reduces the harm of combustibles,” they wrote.
The study was supported by the National Institute of Drug Abuse and the Food and Drug Administration’s Center for Tobacco Products. One of the investigators has received funding in the form of grant funding from Pfizer and the National Institutes of Health and another has served as a paid consultant to the Centers for Disease Control and Prevention.
SOURCE: Weaver SR et al. PLoS ONE. 2018 Jul 9;13(7): e0198047. doi: 10.1371/journal.pone.0198047.
FROM PLOS ONE
Fentanyl analogs nearly double their overdose death toll
, according to preliminary data from 10 states.
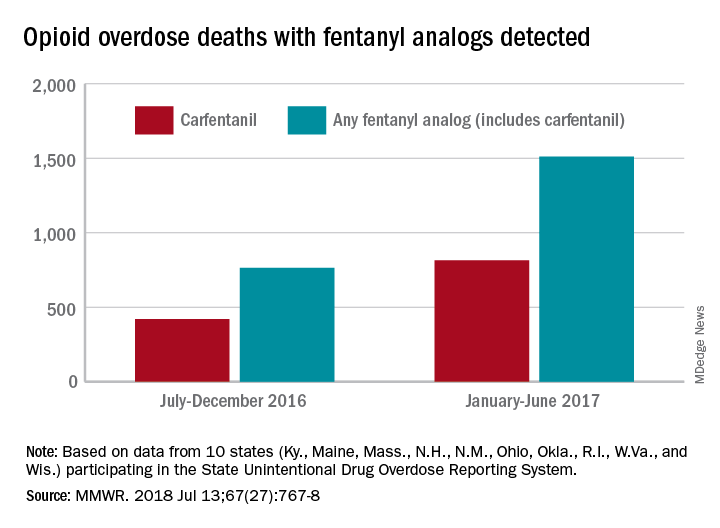
During July 2016 to December 2016, there were 764 opioid overdose deaths that tested positive for any fentanyl analog, with carfentanil being the most common (421 deaths). From January 2017 to June 2017, the respective numbers increased by 98% (1,511) and 94% (815), wrote Julie O’Donnell, PhD, and her associates at the Centers for Disease Control and Prevention’s National Center for Injury Prevention and Control. The report was published in the Morbidity and Mortality Weekly Report.
“The increasing array of fentanyl analogs highlights the need to build forensic toxicological testing capabilities to identify and report emerging threats, and to enhance capacity to rapidly respond to evolving drug trends,” Dr. O’Donnell and her associates said.
Along with carfentanil, 13 other analogs were detected in decedents during the 12-month period: 3-methylfentanyl, 4-fluorobutyrfentanyl, 4-fluorofentanyl, 4-fluoroisobutyrfentanyl, acetylfentanyl, acrylfentanyl, butyrylfentanyl, cyclopropylfentanyl, cyclopentylfentanyl, furanylethylfentanyl, furanylfentanyl, isobutyrylfentanyl, and tetrahydrofuranylfentanyl. Deaths may have involved “more than one analog, as well as ... other opioid and nonopioid substances,” they noted.
The 10 states reporting data to the State Unintentional Drug Overdose Reporting System (SUDORS) were Kentucky, Maine, Massachusetts, New Hampshire, New Mexico, Ohio, Oklahoma, Rhode Island, West Virginia, and Wisconsin. Two other SUDORS-reporting states – Missouri and Pennsylvania – did not have their data ready in time to be included in this analysis.
The increasing availability of fentanyl analogs hit Ohio especially hard: More deaths occurred there than in the other 10 states combined. Of the 421 carfentanil-related deaths in July 2016 to December 2016, nearly 400 were in Ohio, and there were 218 Ohio deaths in April 2017 alone. A look at the bigger picture shows that 3 of the 10 states reported carfentanil-related overdose deaths in the second half of 2016, compared with 7 in the first half of 2017, the investigators said.
Carfentanil, which is the most potent of the 14 fentanyl analogs that have been detected so far, “is intended for sedation of large animals, and is estimated to have 10,000 times the potency of morphine,” Dr. O’Donnell and her associates wrote.
SOURCE: O’Donnell J et al. MMWR. 2018 Jul 13;67(27):767-8.
, according to preliminary data from 10 states.
During July 2016 to December 2016, there were 764 opioid overdose deaths that tested positive for any fentanyl analog, with carfentanil being the most common (421 deaths). From January 2017 to June 2017, the respective numbers increased by 98% (1,511) and 94% (815), wrote Julie O’Donnell, PhD, and her associates at the Centers for Disease Control and Prevention’s National Center for Injury Prevention and Control. The report was published in the Morbidity and Mortality Weekly Report.
“The increasing array of fentanyl analogs highlights the need to build forensic toxicological testing capabilities to identify and report emerging threats, and to enhance capacity to rapidly respond to evolving drug trends,” Dr. O’Donnell and her associates said.
Along with carfentanil, 13 other analogs were detected in decedents during the 12-month period: 3-methylfentanyl, 4-fluorobutyrfentanyl, 4-fluorofentanyl, 4-fluoroisobutyrfentanyl, acetylfentanyl, acrylfentanyl, butyrylfentanyl, cyclopropylfentanyl, cyclopentylfentanyl, furanylethylfentanyl, furanylfentanyl, isobutyrylfentanyl, and tetrahydrofuranylfentanyl. Deaths may have involved “more than one analog, as well as ... other opioid and nonopioid substances,” they noted.
The 10 states reporting data to the State Unintentional Drug Overdose Reporting System (SUDORS) were Kentucky, Maine, Massachusetts, New Hampshire, New Mexico, Ohio, Oklahoma, Rhode Island, West Virginia, and Wisconsin. Two other SUDORS-reporting states – Missouri and Pennsylvania – did not have their data ready in time to be included in this analysis.
The increasing availability of fentanyl analogs hit Ohio especially hard: More deaths occurred there than in the other 10 states combined. Of the 421 carfentanil-related deaths in July 2016 to December 2016, nearly 400 were in Ohio, and there were 218 Ohio deaths in April 2017 alone. A look at the bigger picture shows that 3 of the 10 states reported carfentanil-related overdose deaths in the second half of 2016, compared with 7 in the first half of 2017, the investigators said.
Carfentanil, which is the most potent of the 14 fentanyl analogs that have been detected so far, “is intended for sedation of large animals, and is estimated to have 10,000 times the potency of morphine,” Dr. O’Donnell and her associates wrote.
SOURCE: O’Donnell J et al. MMWR. 2018 Jul 13;67(27):767-8.
, according to preliminary data from 10 states.
During July 2016 to December 2016, there were 764 opioid overdose deaths that tested positive for any fentanyl analog, with carfentanil being the most common (421 deaths). From January 2017 to June 2017, the respective numbers increased by 98% (1,511) and 94% (815), wrote Julie O’Donnell, PhD, and her associates at the Centers for Disease Control and Prevention’s National Center for Injury Prevention and Control. The report was published in the Morbidity and Mortality Weekly Report.
“The increasing array of fentanyl analogs highlights the need to build forensic toxicological testing capabilities to identify and report emerging threats, and to enhance capacity to rapidly respond to evolving drug trends,” Dr. O’Donnell and her associates said.
Along with carfentanil, 13 other analogs were detected in decedents during the 12-month period: 3-methylfentanyl, 4-fluorobutyrfentanyl, 4-fluorofentanyl, 4-fluoroisobutyrfentanyl, acetylfentanyl, acrylfentanyl, butyrylfentanyl, cyclopropylfentanyl, cyclopentylfentanyl, furanylethylfentanyl, furanylfentanyl, isobutyrylfentanyl, and tetrahydrofuranylfentanyl. Deaths may have involved “more than one analog, as well as ... other opioid and nonopioid substances,” they noted.
The 10 states reporting data to the State Unintentional Drug Overdose Reporting System (SUDORS) were Kentucky, Maine, Massachusetts, New Hampshire, New Mexico, Ohio, Oklahoma, Rhode Island, West Virginia, and Wisconsin. Two other SUDORS-reporting states – Missouri and Pennsylvania – did not have their data ready in time to be included in this analysis.
The increasing availability of fentanyl analogs hit Ohio especially hard: More deaths occurred there than in the other 10 states combined. Of the 421 carfentanil-related deaths in July 2016 to December 2016, nearly 400 were in Ohio, and there were 218 Ohio deaths in April 2017 alone. A look at the bigger picture shows that 3 of the 10 states reported carfentanil-related overdose deaths in the second half of 2016, compared with 7 in the first half of 2017, the investigators said.
Carfentanil, which is the most potent of the 14 fentanyl analogs that have been detected so far, “is intended for sedation of large animals, and is estimated to have 10,000 times the potency of morphine,” Dr. O’Donnell and her associates wrote.
SOURCE: O’Donnell J et al. MMWR. 2018 Jul 13;67(27):767-8.
FROM MMWR
New guideline for managing MCL
Rituximab should be included in first-line chemotherapy when treating mantle cell lymphoma, according to a new management guideline from the British Society for Haematology.
The best outcome data is for the R-CHOP regimen (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone) followed by maintenance treatment with rituximab, wrote Pamela McKay, MD, of Beatson West of Scotland Cancer Centre in Glasgow, and her colleagues. The report was published in the British Journal of Haematology. But the combination of rituximab and bendamustine is also effective and a more favorable safety profile, according to the guideline. Single agent rituximab is not recommended.
At relapse, the guideline calls on physicians to take an individualized approach based on age, comorbidities, performance status, and response to prior therapy. Some options to consider include ibrutinib as a single agent or rituximab plus chemotherapy. The authors cautioned that there is little evidence to support maintenance rituximab after relapse treatment.
The guideline also explores the role of autologous stem cell transplantation (ASCT) and allogeneic SCT (alloSCT). The authors recommend that ASCT be considered as consolidation of first-line therapy for patients who are fit for intensive therapy. AlloSCT is a viable option in second remission among fit patients who have an appropriate donor and it may also be effective as a rescue therapy for patients who relapse after ASCT. But alloSCT is appropriate only as a first-line therapy for high-risk patients and is best used as part of a clinical trial, according to the recommendations.
The British Society of Haematology previously issued guidance on mantle cell lymphoma in 2012, but the updated document includes new drug therapeutic options and transplant data. The guideline includes a therapeutic algorithm to assist physicians in choosing first-line therapy, options after first relapse, and management in the case of higher relapse.
The guideline authors reported having no conflicts of interest.
SOURCE: McKay P et al. Br J Haematol. 2018 Jul;182(1):46-62.
Rituximab should be included in first-line chemotherapy when treating mantle cell lymphoma, according to a new management guideline from the British Society for Haematology.
The best outcome data is for the R-CHOP regimen (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone) followed by maintenance treatment with rituximab, wrote Pamela McKay, MD, of Beatson West of Scotland Cancer Centre in Glasgow, and her colleagues. The report was published in the British Journal of Haematology. But the combination of rituximab and bendamustine is also effective and a more favorable safety profile, according to the guideline. Single agent rituximab is not recommended.
At relapse, the guideline calls on physicians to take an individualized approach based on age, comorbidities, performance status, and response to prior therapy. Some options to consider include ibrutinib as a single agent or rituximab plus chemotherapy. The authors cautioned that there is little evidence to support maintenance rituximab after relapse treatment.
The guideline also explores the role of autologous stem cell transplantation (ASCT) and allogeneic SCT (alloSCT). The authors recommend that ASCT be considered as consolidation of first-line therapy for patients who are fit for intensive therapy. AlloSCT is a viable option in second remission among fit patients who have an appropriate donor and it may also be effective as a rescue therapy for patients who relapse after ASCT. But alloSCT is appropriate only as a first-line therapy for high-risk patients and is best used as part of a clinical trial, according to the recommendations.
The British Society of Haematology previously issued guidance on mantle cell lymphoma in 2012, but the updated document includes new drug therapeutic options and transplant data. The guideline includes a therapeutic algorithm to assist physicians in choosing first-line therapy, options after first relapse, and management in the case of higher relapse.
The guideline authors reported having no conflicts of interest.
SOURCE: McKay P et al. Br J Haematol. 2018 Jul;182(1):46-62.
Rituximab should be included in first-line chemotherapy when treating mantle cell lymphoma, according to a new management guideline from the British Society for Haematology.
The best outcome data is for the R-CHOP regimen (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone) followed by maintenance treatment with rituximab, wrote Pamela McKay, MD, of Beatson West of Scotland Cancer Centre in Glasgow, and her colleagues. The report was published in the British Journal of Haematology. But the combination of rituximab and bendamustine is also effective and a more favorable safety profile, according to the guideline. Single agent rituximab is not recommended.
At relapse, the guideline calls on physicians to take an individualized approach based on age, comorbidities, performance status, and response to prior therapy. Some options to consider include ibrutinib as a single agent or rituximab plus chemotherapy. The authors cautioned that there is little evidence to support maintenance rituximab after relapse treatment.
The guideline also explores the role of autologous stem cell transplantation (ASCT) and allogeneic SCT (alloSCT). The authors recommend that ASCT be considered as consolidation of first-line therapy for patients who are fit for intensive therapy. AlloSCT is a viable option in second remission among fit patients who have an appropriate donor and it may also be effective as a rescue therapy for patients who relapse after ASCT. But alloSCT is appropriate only as a first-line therapy for high-risk patients and is best used as part of a clinical trial, according to the recommendations.
The British Society of Haematology previously issued guidance on mantle cell lymphoma in 2012, but the updated document includes new drug therapeutic options and transplant data. The guideline includes a therapeutic algorithm to assist physicians in choosing first-line therapy, options after first relapse, and management in the case of higher relapse.
The guideline authors reported having no conflicts of interest.
SOURCE: McKay P et al. Br J Haematol. 2018 Jul;182(1):46-62.
FROM THE BRITISH JOURNAL OF HAEMATOLOGY
Immunogenicity of two-dose Gardasil 9 persists at 36 months
MALMO, SWEDEN – Robust human papillomavirus antibody responses persist through 36 months in boys and girls who received two doses of the 9-valent HPV vaccine known as Gardasil 9 at age 9-14 years, according to an open-label randomized immunogenicity study conducted in 15 countries.
This is reassuring news supportive of regulatory decisions made in 2016/2017 to license the more convenient two-dose schedule in the United States, European Union, Canada, and other countries, Rosybel Drury, PhD, observed in reporting the results at the annual meeting of the European Society for Paediatric Infectious Diseases.
Moreover, HPV type-specific antibody levels at 36 months post vaccination in boys and girls who received two doses were similar to or greater than in the 36-month follow-up of adolescent and young adult females who received three doses at ages 16-26 years.
“This result supports the bridging of efficacy findings in young women receiving three doses to girls and boys receiving two doses,” according to Dr. Drury, a vaccine scientist at Merck Sharp & Dohme in Lyon, France.

She presented an update of a five-cohort study including roughly 1,500 recipients of either two doses of the 9-valent HPV vaccine given 6 or 12 months apart or three doses administered at 0, 2, and 6 months. Four cohorts were composed of 9- to 14-year-olds. The fifth consisted of adolescent girls and young women who got three doses of Gardasil 9 over the course of 6 months at ages 16-26 years. The previous report from this major study provided only short-term data based upon measurements of immunogenicity obtained 1 month after the last dose of vaccine (JAMA. 2016 Dec. 13;316(22):2411-21).
Anti-HPV geometric mean titers were highest 1 month after completing a two- or three-dose series, dropped off sharply during the next 6-12 months, then declined more slowly through 36 months.
While the demonstration of persistent immunogenicity over 3 years of follow-up was reassuring overall, there was a potential hitch: Significantly lower antibody levels for some HPV types were observed in girls who received two doses of the vaccine than in those who got three. Specifically, levels of anti-HPV antibodies to HPV types 18, 31, 45, and 52 were significantly lower in the girls who got two doses of the 9-valent HPV vaccine than in those who got three doses at the same age. Antibody levels directed against HPV types 6, 11, 16, 33, and 58 were similar in the two patient populations.
“The clinical significance of this finding remains unknown,” Dr. Drury said.
Challenged as to why the study didn’t include a single-dose vaccine arm, which would be the preferred preventive strategy in low-income countries where the burden of anogenital cancers and warts due to HPV is greatest, she replied that while there is great interest in this approach, and some modeling studies suggest it would be beneficial, the study she presented was designed to evaluate immunogenicity over time, not clinical efficacy, which needs to be assessed before single-dose public health programs are implemented.
The study was funded by Merck and presented by a company employee.
MALMO, SWEDEN – Robust human papillomavirus antibody responses persist through 36 months in boys and girls who received two doses of the 9-valent HPV vaccine known as Gardasil 9 at age 9-14 years, according to an open-label randomized immunogenicity study conducted in 15 countries.
This is reassuring news supportive of regulatory decisions made in 2016/2017 to license the more convenient two-dose schedule in the United States, European Union, Canada, and other countries, Rosybel Drury, PhD, observed in reporting the results at the annual meeting of the European Society for Paediatric Infectious Diseases.
Moreover, HPV type-specific antibody levels at 36 months post vaccination in boys and girls who received two doses were similar to or greater than in the 36-month follow-up of adolescent and young adult females who received three doses at ages 16-26 years.
“This result supports the bridging of efficacy findings in young women receiving three doses to girls and boys receiving two doses,” according to Dr. Drury, a vaccine scientist at Merck Sharp & Dohme in Lyon, France.
She presented an update of a five-cohort study including roughly 1,500 recipients of either two doses of the 9-valent HPV vaccine given 6 or 12 months apart or three doses administered at 0, 2, and 6 months. Four cohorts were composed of 9- to 14-year-olds. The fifth consisted of adolescent girls and young women who got three doses of Gardasil 9 over the course of 6 months at ages 16-26 years. The previous report from this major study provided only short-term data based upon measurements of immunogenicity obtained 1 month after the last dose of vaccine (JAMA. 2016 Dec. 13;316(22):2411-21).
Anti-HPV geometric mean titers were highest 1 month after completing a two- or three-dose series, dropped off sharply during the next 6-12 months, then declined more slowly through 36 months.
While the demonstration of persistent immunogenicity over 3 years of follow-up was reassuring overall, there was a potential hitch: Significantly lower antibody levels for some HPV types were observed in girls who received two doses of the vaccine than in those who got three. Specifically, levels of anti-HPV antibodies to HPV types 18, 31, 45, and 52 were significantly lower in the girls who got two doses of the 9-valent HPV vaccine than in those who got three doses at the same age. Antibody levels directed against HPV types 6, 11, 16, 33, and 58 were similar in the two patient populations.
“The clinical significance of this finding remains unknown,” Dr. Drury said.
Challenged as to why the study didn’t include a single-dose vaccine arm, which would be the preferred preventive strategy in low-income countries where the burden of anogenital cancers and warts due to HPV is greatest, she replied that while there is great interest in this approach, and some modeling studies suggest it would be beneficial, the study she presented was designed to evaluate immunogenicity over time, not clinical efficacy, which needs to be assessed before single-dose public health programs are implemented.
The study was funded by Merck and presented by a company employee.
MALMO, SWEDEN – Robust human papillomavirus antibody responses persist through 36 months in boys and girls who received two doses of the 9-valent HPV vaccine known as Gardasil 9 at age 9-14 years, according to an open-label randomized immunogenicity study conducted in 15 countries.
This is reassuring news supportive of regulatory decisions made in 2016/2017 to license the more convenient two-dose schedule in the United States, European Union, Canada, and other countries, Rosybel Drury, PhD, observed in reporting the results at the annual meeting of the European Society for Paediatric Infectious Diseases.
Moreover, HPV type-specific antibody levels at 36 months post vaccination in boys and girls who received two doses were similar to or greater than in the 36-month follow-up of adolescent and young adult females who received three doses at ages 16-26 years.
“This result supports the bridging of efficacy findings in young women receiving three doses to girls and boys receiving two doses,” according to Dr. Drury, a vaccine scientist at Merck Sharp & Dohme in Lyon, France.
She presented an update of a five-cohort study including roughly 1,500 recipients of either two doses of the 9-valent HPV vaccine given 6 or 12 months apart or three doses administered at 0, 2, and 6 months. Four cohorts were composed of 9- to 14-year-olds. The fifth consisted of adolescent girls and young women who got three doses of Gardasil 9 over the course of 6 months at ages 16-26 years. The previous report from this major study provided only short-term data based upon measurements of immunogenicity obtained 1 month after the last dose of vaccine (JAMA. 2016 Dec. 13;316(22):2411-21).
Anti-HPV geometric mean titers were highest 1 month after completing a two- or three-dose series, dropped off sharply during the next 6-12 months, then declined more slowly through 36 months.
While the demonstration of persistent immunogenicity over 3 years of follow-up was reassuring overall, there was a potential hitch: Significantly lower antibody levels for some HPV types were observed in girls who received two doses of the vaccine than in those who got three. Specifically, levels of anti-HPV antibodies to HPV types 18, 31, 45, and 52 were significantly lower in the girls who got two doses of the 9-valent HPV vaccine than in those who got three doses at the same age. Antibody levels directed against HPV types 6, 11, 16, 33, and 58 were similar in the two patient populations.
“The clinical significance of this finding remains unknown,” Dr. Drury said.
Challenged as to why the study didn’t include a single-dose vaccine arm, which would be the preferred preventive strategy in low-income countries where the burden of anogenital cancers and warts due to HPV is greatest, she replied that while there is great interest in this approach, and some modeling studies suggest it would be beneficial, the study she presented was designed to evaluate immunogenicity over time, not clinical efficacy, which needs to be assessed before single-dose public health programs are implemented.
The study was funded by Merck and presented by a company employee.
REPORTING FROM ESPID 2018
Key clinical point:
Major finding: HPV type-specific antibody responses to two doses given at age 9-14 years were as good as or better than in 16- to 26-year-olds who got three doses.
Study details: This prospective open-label immunogenicity study included roughly 1,500 subjects in 15 countries who received either two or three doses of the 9-valent HPV vaccine.
Disclosures: The study was funded by Merck and presented by a company employee.