User login
NIOSH Releases Virtual Toolkit for Emergency Responders
When first responders arrive at a scene where illicit drugs may be present, they could be at risk of dangerous exposure. They might inhale drugs; they can have contact through mucous membranes or through needlesticks.
A major concern is exposure to fentanyl or its analogues, which can lead to symptoms, including rapid onset of life-threatening respiratory depression. The exception is skin contact, which is not expected to have toxic effects if the visible contamination is removed promptly.
To help EMS providers and other responders protect themselves, the National Institute for Occupational Safety and Health (NIOSH) has released a new virtual toolkit with videos, infographics, and postcards based on NIOSH safety recommendations.
The resources highlight how best to assess the scene for hazards that may indicate the presence of illicit drugs and what to do—for example, use soap and water, not hand sanitizer (it doesn’t remove illicit drugs and may increase exposure), and don’t eat, drink, smoke, or use the bathroom in the affected area. The infographics also show how to decontaminate and prevent “take-home exposure” to protect responders’ families. The guidelines extend to procedures for protecting working dogs exposed to the drugs.
NIOSH notes that it has no occupational exposure data on fentanyl or its analogues for emergency responders. The recommendations are based on the reported toxicity and the chemical and physical properties of fentanyl and its analogues, NIOSH guidance for similar chemicals, recommendations from previous NIOSH health hazard evaluation reports, and “the basic principles of industrial hygiene.” As new research becomes available, NIOSH says, the recommendations will be updated.
The toolkit resources are shareable and available for disseminating via print, social media, text, and more. The kit is accessible at https://www.cdc.gov/niosh/topics/fentanyl/toolkit.html.
When first responders arrive at a scene where illicit drugs may be present, they could be at risk of dangerous exposure. They might inhale drugs; they can have contact through mucous membranes or through needlesticks.
A major concern is exposure to fentanyl or its analogues, which can lead to symptoms, including rapid onset of life-threatening respiratory depression. The exception is skin contact, which is not expected to have toxic effects if the visible contamination is removed promptly.
To help EMS providers and other responders protect themselves, the National Institute for Occupational Safety and Health (NIOSH) has released a new virtual toolkit with videos, infographics, and postcards based on NIOSH safety recommendations.
The resources highlight how best to assess the scene for hazards that may indicate the presence of illicit drugs and what to do—for example, use soap and water, not hand sanitizer (it doesn’t remove illicit drugs and may increase exposure), and don’t eat, drink, smoke, or use the bathroom in the affected area. The infographics also show how to decontaminate and prevent “take-home exposure” to protect responders’ families. The guidelines extend to procedures for protecting working dogs exposed to the drugs.
NIOSH notes that it has no occupational exposure data on fentanyl or its analogues for emergency responders. The recommendations are based on the reported toxicity and the chemical and physical properties of fentanyl and its analogues, NIOSH guidance for similar chemicals, recommendations from previous NIOSH health hazard evaluation reports, and “the basic principles of industrial hygiene.” As new research becomes available, NIOSH says, the recommendations will be updated.
The toolkit resources are shareable and available for disseminating via print, social media, text, and more. The kit is accessible at https://www.cdc.gov/niosh/topics/fentanyl/toolkit.html.
When first responders arrive at a scene where illicit drugs may be present, they could be at risk of dangerous exposure. They might inhale drugs; they can have contact through mucous membranes or through needlesticks.
A major concern is exposure to fentanyl or its analogues, which can lead to symptoms, including rapid onset of life-threatening respiratory depression. The exception is skin contact, which is not expected to have toxic effects if the visible contamination is removed promptly.
To help EMS providers and other responders protect themselves, the National Institute for Occupational Safety and Health (NIOSH) has released a new virtual toolkit with videos, infographics, and postcards based on NIOSH safety recommendations.
The resources highlight how best to assess the scene for hazards that may indicate the presence of illicit drugs and what to do—for example, use soap and water, not hand sanitizer (it doesn’t remove illicit drugs and may increase exposure), and don’t eat, drink, smoke, or use the bathroom in the affected area. The infographics also show how to decontaminate and prevent “take-home exposure” to protect responders’ families. The guidelines extend to procedures for protecting working dogs exposed to the drugs.
NIOSH notes that it has no occupational exposure data on fentanyl or its analogues for emergency responders. The recommendations are based on the reported toxicity and the chemical and physical properties of fentanyl and its analogues, NIOSH guidance for similar chemicals, recommendations from previous NIOSH health hazard evaluation reports, and “the basic principles of industrial hygiene.” As new research becomes available, NIOSH says, the recommendations will be updated.
The toolkit resources are shareable and available for disseminating via print, social media, text, and more. The kit is accessible at https://www.cdc.gov/niosh/topics/fentanyl/toolkit.html.
High maternal lead levels linked to children’s obesity
Children born to mothers with high blood levels of lead have an increased risk of being overweight or obese, particularly if their mothers are also overweight, according to new research.

Adequate maternal plasma levels of folate, however, mitigated this risk.
“When considered simultaneously, maternal lead exposure, rather than early childhood lead exposure, contributed to overweight/obesity risk in a dose-response fashion across multiple developmental stages (preschool age, school age and early adolescence) and amplified intergenerational overweight/obesity risk (additively with maternal overweight/obesity),” Guoying Wang, MD, PhD, of Johns Hopkins Bloomberg School of Public Health, Baltimore, and associates, reported in JAMA Network Open.
“These findings support the hypothesis that the obesity epidemic could be related to environmental chemical exposures in utero and raise the possibility that optimal maternal folate supplementation may help counteract the adverse effects of environmental lead exposure,” the authors wrote.
The prospective urban, low-income cohort study, which ran from 2002 to 2013, involved 1,442 mother-child pairs who joined the study when the children were born and attended follow-up visits at Boston Medical Center. The mean age of the mothers was 29 years, and the children were, on average, 8 years old at follow-up. Half the children were male; 67% of mothers were black, and 20% were Latina.
The researchers collected maternal blood samples within 24-72 hours after birth to measure red blood cell lead levels and plasma folate levels. Children’s whole-blood lead levels were measured during the first lead screening of their well child visits, at a median 10 months of age. Researchers tracked children’s body mass index Z-score and defined overweight/obesity as exceeding the 85th national percentile for their age and sex.
Detectable lead was present in all the mothers’ blood samples. The median maternal red blood cell lead level was 2.5 mcg/dL, although black mothers tended to have higher lead exposure than that of other racial groups. Median maternal plasma folate level was 32 nmol/L. Children’s blood lead levels were a median 1.4 mcg/dL, and their median BMI Z-score was 0.78.
Children whose mothers had red blood cell lead levels of 5.0 mcg/dL or greater (16%) had 65% greater odds of being overweight or obese compared with children whose mothers’ lead level was less than 2 mcg/dL, after adjustment for maternal education, race/ethnicity, smoking status, parity, diabetes, hypertensive disorder, preterm birth, fetal growth, and breastfeeding status (odds ratio [OR], 1.65; 95% confidence internal [CI], 1.18-2.32). Only 5.2% of children had whole-blood lead levels of 5 mcg/dL or greater.
“Mothers with the highest red blood cell lead levels were older and multiparous, were more likely to be black and nonsmokers, had lower plasma folate levels and were more likely to have prepregnancy overweight/obesity and diabetes,” the authors reported.
The dose-response association did not lose significance when the researchers adjusted for children’s blood lead levels, maternal age, cesarean delivery, term births only, and black race. Nor did it change in a subset of children when the researchers adjusted for children’s physical activity.
The strength of the association increased when mothers also had a BMI greater than the average/healthy range. Children were more than four times more likely to be overweight or obese if their mothers were overweight or obese and had lead levels greater than 5.0 mcg/dL, compared with nonoverweight mothers with levels below 2 mcg/dL (OR, 4.24; 95% CI, 2.64-6.82).
Among children whose mothers were overweight/obese and had high blood lead levels, however, high folate levels appeared protective against obesity. These children had a 41% lower risk of being overweight or obese, compared with others in their group, if their mothers had plasma folate levels of at least 20 nmol/L (OR, 0.59 CI, 0.36-0.95; P = .03).
According to an invited commentary, “approximately 140,000 new chemicals and pesticides have appeared since 1950,” with “universal human exposure to approximately 5,000 of those,” wrote Marco Sanchez-Guerra, PhD, of the National Institute of Perinatology in Mexico City, and coauthors Andres Cardenas, PhD, of the University of California, Berkeley, and Citlalli Osorio-Yáñez, PhD, of the National Autonomous University of Mexico in Mexico City. Yet fewer than half of those chemicals have been tested for safety or toxic effect, the editorialists wrote, and scientists know little of their potential reproductive harm.
Dr. Sanchez-Guerra, Dr. Cardenas, and Dr. Osorio-Yáñez agreed with the study authors that elevated lead exposures, especially from gasoline before lead was removed in the United States in 1975, may partly explain the current epidemic of obesity.
“Identifying preventable prenatal causes of obesity is a cornerstone in the fight against the obesity epidemic,” the editorialists said. While most recommendations center on changes to diet and physical activity, environmental factors during pregnancy could be involved in childhood obesity as well.
“The study by Wang et al. opens the door to new questions about whether adequate folate intake might modify the adverse effects of other chemical exposures,” they continued, noting other research suggesting a protective effect from folate against health effects of air pollution exposure. “These efforts could yield substantial public health benefits and represent novel tools in fighting the obesity epidemic,” they concluded.
The research was funded by the National Institutes of Health and the U.S. Department of Health and Human Services. Neither the study authors nor the editorialists had industry financial disclosures.
SOURCES: Wang G et al. JAMA Netw Open. 2019;2(10):e1912343. doi: 10.1001/jamanetworkopen.2019.12343; Sanchez-Guerra M et al. JAMA Netw Open. 2019;2(10):e1912334. doi: 10.1001/jamanetworkopen.2019.12334.
Children born to mothers with high blood levels of lead have an increased risk of being overweight or obese, particularly if their mothers are also overweight, according to new research.
Adequate maternal plasma levels of folate, however, mitigated this risk.
“When considered simultaneously, maternal lead exposure, rather than early childhood lead exposure, contributed to overweight/obesity risk in a dose-response fashion across multiple developmental stages (preschool age, school age and early adolescence) and amplified intergenerational overweight/obesity risk (additively with maternal overweight/obesity),” Guoying Wang, MD, PhD, of Johns Hopkins Bloomberg School of Public Health, Baltimore, and associates, reported in JAMA Network Open.
“These findings support the hypothesis that the obesity epidemic could be related to environmental chemical exposures in utero and raise the possibility that optimal maternal folate supplementation may help counteract the adverse effects of environmental lead exposure,” the authors wrote.
The prospective urban, low-income cohort study, which ran from 2002 to 2013, involved 1,442 mother-child pairs who joined the study when the children were born and attended follow-up visits at Boston Medical Center. The mean age of the mothers was 29 years, and the children were, on average, 8 years old at follow-up. Half the children were male; 67% of mothers were black, and 20% were Latina.
The researchers collected maternal blood samples within 24-72 hours after birth to measure red blood cell lead levels and plasma folate levels. Children’s whole-blood lead levels were measured during the first lead screening of their well child visits, at a median 10 months of age. Researchers tracked children’s body mass index Z-score and defined overweight/obesity as exceeding the 85th national percentile for their age and sex.
Detectable lead was present in all the mothers’ blood samples. The median maternal red blood cell lead level was 2.5 mcg/dL, although black mothers tended to have higher lead exposure than that of other racial groups. Median maternal plasma folate level was 32 nmol/L. Children’s blood lead levels were a median 1.4 mcg/dL, and their median BMI Z-score was 0.78.
Children whose mothers had red blood cell lead levels of 5.0 mcg/dL or greater (16%) had 65% greater odds of being overweight or obese compared with children whose mothers’ lead level was less than 2 mcg/dL, after adjustment for maternal education, race/ethnicity, smoking status, parity, diabetes, hypertensive disorder, preterm birth, fetal growth, and breastfeeding status (odds ratio [OR], 1.65; 95% confidence internal [CI], 1.18-2.32). Only 5.2% of children had whole-blood lead levels of 5 mcg/dL or greater.
“Mothers with the highest red blood cell lead levels were older and multiparous, were more likely to be black and nonsmokers, had lower plasma folate levels and were more likely to have prepregnancy overweight/obesity and diabetes,” the authors reported.
The dose-response association did not lose significance when the researchers adjusted for children’s blood lead levels, maternal age, cesarean delivery, term births only, and black race. Nor did it change in a subset of children when the researchers adjusted for children’s physical activity.
The strength of the association increased when mothers also had a BMI greater than the average/healthy range. Children were more than four times more likely to be overweight or obese if their mothers were overweight or obese and had lead levels greater than 5.0 mcg/dL, compared with nonoverweight mothers with levels below 2 mcg/dL (OR, 4.24; 95% CI, 2.64-6.82).
Among children whose mothers were overweight/obese and had high blood lead levels, however, high folate levels appeared protective against obesity. These children had a 41% lower risk of being overweight or obese, compared with others in their group, if their mothers had plasma folate levels of at least 20 nmol/L (OR, 0.59 CI, 0.36-0.95; P = .03).
According to an invited commentary, “approximately 140,000 new chemicals and pesticides have appeared since 1950,” with “universal human exposure to approximately 5,000 of those,” wrote Marco Sanchez-Guerra, PhD, of the National Institute of Perinatology in Mexico City, and coauthors Andres Cardenas, PhD, of the University of California, Berkeley, and Citlalli Osorio-Yáñez, PhD, of the National Autonomous University of Mexico in Mexico City. Yet fewer than half of those chemicals have been tested for safety or toxic effect, the editorialists wrote, and scientists know little of their potential reproductive harm.
Dr. Sanchez-Guerra, Dr. Cardenas, and Dr. Osorio-Yáñez agreed with the study authors that elevated lead exposures, especially from gasoline before lead was removed in the United States in 1975, may partly explain the current epidemic of obesity.
“Identifying preventable prenatal causes of obesity is a cornerstone in the fight against the obesity epidemic,” the editorialists said. While most recommendations center on changes to diet and physical activity, environmental factors during pregnancy could be involved in childhood obesity as well.
“The study by Wang et al. opens the door to new questions about whether adequate folate intake might modify the adverse effects of other chemical exposures,” they continued, noting other research suggesting a protective effect from folate against health effects of air pollution exposure. “These efforts could yield substantial public health benefits and represent novel tools in fighting the obesity epidemic,” they concluded.
The research was funded by the National Institutes of Health and the U.S. Department of Health and Human Services. Neither the study authors nor the editorialists had industry financial disclosures.
SOURCES: Wang G et al. JAMA Netw Open. 2019;2(10):e1912343. doi: 10.1001/jamanetworkopen.2019.12343; Sanchez-Guerra M et al. JAMA Netw Open. 2019;2(10):e1912334. doi: 10.1001/jamanetworkopen.2019.12334.
Children born to mothers with high blood levels of lead have an increased risk of being overweight or obese, particularly if their mothers are also overweight, according to new research.
Adequate maternal plasma levels of folate, however, mitigated this risk.
“When considered simultaneously, maternal lead exposure, rather than early childhood lead exposure, contributed to overweight/obesity risk in a dose-response fashion across multiple developmental stages (preschool age, school age and early adolescence) and amplified intergenerational overweight/obesity risk (additively with maternal overweight/obesity),” Guoying Wang, MD, PhD, of Johns Hopkins Bloomberg School of Public Health, Baltimore, and associates, reported in JAMA Network Open.
“These findings support the hypothesis that the obesity epidemic could be related to environmental chemical exposures in utero and raise the possibility that optimal maternal folate supplementation may help counteract the adverse effects of environmental lead exposure,” the authors wrote.
The prospective urban, low-income cohort study, which ran from 2002 to 2013, involved 1,442 mother-child pairs who joined the study when the children were born and attended follow-up visits at Boston Medical Center. The mean age of the mothers was 29 years, and the children were, on average, 8 years old at follow-up. Half the children were male; 67% of mothers were black, and 20% were Latina.
The researchers collected maternal blood samples within 24-72 hours after birth to measure red blood cell lead levels and plasma folate levels. Children’s whole-blood lead levels were measured during the first lead screening of their well child visits, at a median 10 months of age. Researchers tracked children’s body mass index Z-score and defined overweight/obesity as exceeding the 85th national percentile for their age and sex.
Detectable lead was present in all the mothers’ blood samples. The median maternal red blood cell lead level was 2.5 mcg/dL, although black mothers tended to have higher lead exposure than that of other racial groups. Median maternal plasma folate level was 32 nmol/L. Children’s blood lead levels were a median 1.4 mcg/dL, and their median BMI Z-score was 0.78.
Children whose mothers had red blood cell lead levels of 5.0 mcg/dL or greater (16%) had 65% greater odds of being overweight or obese compared with children whose mothers’ lead level was less than 2 mcg/dL, after adjustment for maternal education, race/ethnicity, smoking status, parity, diabetes, hypertensive disorder, preterm birth, fetal growth, and breastfeeding status (odds ratio [OR], 1.65; 95% confidence internal [CI], 1.18-2.32). Only 5.2% of children had whole-blood lead levels of 5 mcg/dL or greater.
“Mothers with the highest red blood cell lead levels were older and multiparous, were more likely to be black and nonsmokers, had lower plasma folate levels and were more likely to have prepregnancy overweight/obesity and diabetes,” the authors reported.
The dose-response association did not lose significance when the researchers adjusted for children’s blood lead levels, maternal age, cesarean delivery, term births only, and black race. Nor did it change in a subset of children when the researchers adjusted for children’s physical activity.
The strength of the association increased when mothers also had a BMI greater than the average/healthy range. Children were more than four times more likely to be overweight or obese if their mothers were overweight or obese and had lead levels greater than 5.0 mcg/dL, compared with nonoverweight mothers with levels below 2 mcg/dL (OR, 4.24; 95% CI, 2.64-6.82).
Among children whose mothers were overweight/obese and had high blood lead levels, however, high folate levels appeared protective against obesity. These children had a 41% lower risk of being overweight or obese, compared with others in their group, if their mothers had plasma folate levels of at least 20 nmol/L (OR, 0.59 CI, 0.36-0.95; P = .03).
According to an invited commentary, “approximately 140,000 new chemicals and pesticides have appeared since 1950,” with “universal human exposure to approximately 5,000 of those,” wrote Marco Sanchez-Guerra, PhD, of the National Institute of Perinatology in Mexico City, and coauthors Andres Cardenas, PhD, of the University of California, Berkeley, and Citlalli Osorio-Yáñez, PhD, of the National Autonomous University of Mexico in Mexico City. Yet fewer than half of those chemicals have been tested for safety or toxic effect, the editorialists wrote, and scientists know little of their potential reproductive harm.
Dr. Sanchez-Guerra, Dr. Cardenas, and Dr. Osorio-Yáñez agreed with the study authors that elevated lead exposures, especially from gasoline before lead was removed in the United States in 1975, may partly explain the current epidemic of obesity.
“Identifying preventable prenatal causes of obesity is a cornerstone in the fight against the obesity epidemic,” the editorialists said. While most recommendations center on changes to diet and physical activity, environmental factors during pregnancy could be involved in childhood obesity as well.
“The study by Wang et al. opens the door to new questions about whether adequate folate intake might modify the adverse effects of other chemical exposures,” they continued, noting other research suggesting a protective effect from folate against health effects of air pollution exposure. “These efforts could yield substantial public health benefits and represent novel tools in fighting the obesity epidemic,” they concluded.
The research was funded by the National Institutes of Health and the U.S. Department of Health and Human Services. Neither the study authors nor the editorialists had industry financial disclosures.
SOURCES: Wang G et al. JAMA Netw Open. 2019;2(10):e1912343. doi: 10.1001/jamanetworkopen.2019.12343; Sanchez-Guerra M et al. JAMA Netw Open. 2019;2(10):e1912334. doi: 10.1001/jamanetworkopen.2019.12334.
FROM JAMA NETWORK OPEN
Epidemiology and costs of sepsis in the United States
Background: Sepsis is responsible for an increasingly disproportionate fraction of health care burden. Delays in diagnosis of sepsis are associated with worse outcomes.

Study design: Retrospective observational study.
Setting: Premier Healthcare database, including 20% of U.S. private/academic hospitals.
Synopsis: With use of the Premier Healthcare database, researchers identified 2,566,689 cases of sepsis using ICD-9 and MS-DRG codes between Jan. 1, 2010, and Sept. 30, 2016. Increasing severity of sepsis was associated with increasing mortality and cost, but there was a large discrepancy in cost in patients with sepsis present at admission versus those without it at admission ($18,023 vs. $51,022) and was associated with increases in both mean hospital length of stay and mortality rate across all levels of sepsis severity.
Bottom line: Early identification of sepsis (at admission vs. later in the stay) may be important as a factor to reduce its overall burden on the health care system.
Citation: Paoli CJ et al. Epidemiology and costs of sepsis in the United States – An analysis based on timing of diagnosis and severity level. Crit Care Med. 2018 Dec;46(12):1889-97.
Dr. Ho is an assistant professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
Background: Sepsis is responsible for an increasingly disproportionate fraction of health care burden. Delays in diagnosis of sepsis are associated with worse outcomes.
Study design: Retrospective observational study.
Setting: Premier Healthcare database, including 20% of U.S. private/academic hospitals.
Synopsis: With use of the Premier Healthcare database, researchers identified 2,566,689 cases of sepsis using ICD-9 and MS-DRG codes between Jan. 1, 2010, and Sept. 30, 2016. Increasing severity of sepsis was associated with increasing mortality and cost, but there was a large discrepancy in cost in patients with sepsis present at admission versus those without it at admission ($18,023 vs. $51,022) and was associated with increases in both mean hospital length of stay and mortality rate across all levels of sepsis severity.
Bottom line: Early identification of sepsis (at admission vs. later in the stay) may be important as a factor to reduce its overall burden on the health care system.
Citation: Paoli CJ et al. Epidemiology and costs of sepsis in the United States – An analysis based on timing of diagnosis and severity level. Crit Care Med. 2018 Dec;46(12):1889-97.
Dr. Ho is an assistant professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
Background: Sepsis is responsible for an increasingly disproportionate fraction of health care burden. Delays in diagnosis of sepsis are associated with worse outcomes.
Study design: Retrospective observational study.
Setting: Premier Healthcare database, including 20% of U.S. private/academic hospitals.
Synopsis: With use of the Premier Healthcare database, researchers identified 2,566,689 cases of sepsis using ICD-9 and MS-DRG codes between Jan. 1, 2010, and Sept. 30, 2016. Increasing severity of sepsis was associated with increasing mortality and cost, but there was a large discrepancy in cost in patients with sepsis present at admission versus those without it at admission ($18,023 vs. $51,022) and was associated with increases in both mean hospital length of stay and mortality rate across all levels of sepsis severity.
Bottom line: Early identification of sepsis (at admission vs. later in the stay) may be important as a factor to reduce its overall burden on the health care system.
Citation: Paoli CJ et al. Epidemiology and costs of sepsis in the United States – An analysis based on timing of diagnosis and severity level. Crit Care Med. 2018 Dec;46(12):1889-97.
Dr. Ho is an assistant professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
Few antidepressant adverse effects backed by convincing evidence
Relatively few of the adverse health outcomes attributed to antidepressants are supported by convincing evidence, reported the authors of a systematic review of 45 meta-analyses.
The authors did find convincing evidence linking the use of antidepressants and suicide attempt or completion among people under age 19 years and use of the medication and autism risk among offspring. “However, the few [studies] with convincing evidence associations did not reflect causality, and none of them remained at the convincing evidence level after accounting for confounding by indication,” wrote Elena Dragioti, PhD, of the Pain and Rehabilitation Centre at Linköping (Sweden) University and coauthors. The study was published in JAMA Psychiatry.
Dr. Dragioti and coauthors undertook a systematic “umbrella review” grading the evidence from the 45 meta-analyses of 695 observational studies into the association between antidepressant use and the risk of adverse health outcomes. All the meta-analyses included a control group not exposed to antidepressants, with the exception of one that compared the risk of gastrointestinal bleeding between two classes of antidepressants.
They found 120 possible adverse health associations described in the meta-analyses, 61.7% of which related to maternal and pregnancy-related adverse health outcomes. Two-thirds of the adverse health outcome associations involved selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs).
However, among the 120 adverse health associations, only three (2.5%) were supported by “convincing” evidence. One was the association between SSRIs and increased risk of suicide attempts and completion in children and adolescents. Convincing evidence also was found between any antidepressant use before pregnancy and autism spectrum disorder and between SSRI use during pregnancy and autism spectrum disorder. The evidence for the association with suicide risk was deemed high quality, but the two associations with autism spectrum disorder were only of moderate quality.
The authors commented that these findings needed to be considered when prescribing antidepressants in adolescents and children, particularly as another networked meta-analysis had found fluoxetine was the only antidepressant that worked better than placebo in children and adolescents. “In addition, they wrote.
The review found that 11 adverse health outcomes (9.2%) had “highly suggestive” evidence linking them to antidepressant use. These were ADHD in children, cataract development, severe bleeding at any site, upper gastrointestinal tract bleeding, postpartum hemorrhage, preterm birth, lower Apgar score at 5 minutes, osteoporotic fracture, and hip fracture.
Seven of those – ADHD in children, lower Apgar score, severe bleeding at any site, cataract development, osteoporotic features, preterm birth, and upper GI bleeding – had moderate-quality evidence. However, the authors noted that the effect sizes were small and had low prevalence.
The study also found highly suggestive evidence linking antidepressant use to a decreased risk of suicide attempts or completion in adults.
The authors said several of those adverse events in adults, such as GI bleeding and osteoporotic fractures, could be prevented with medication, so the advantages of antidepressant use in adults could outweigh the disadvantage of those preventable safety issues.
Twenty-one adverse health outcomes showed either suggestive, weak, or no evidence for their association with antidepressant use.
They also conducted a sensitivity analysis that limited the analysis to cohort studies, prospective cohort studies, studies that controlled for confounding by the treatment indication, and studies from North America. This showed that none of the associations for which there was originally deemed to be convincing evidence retained that same rank.
“Overall, the results showed that the association between antidepressant use and adverse health outcomes was not supported by robust evidence and that the underlying disease likely inflated the findings in a relevant way,” the authors wrote.
However, when they looked solely at prospective cohort studies, the association between preterm birth and use of any antidepressant was upgraded to having convincing evidence.
When the analysis focused on SSRIs only, the association with lower Apgar scores at 5 minutes also was upgraded to having convincing evidence. Similarly, the evidence for an association with preterm birth also was found to be convincing when the analysis was limited to other or mixed antidepressants.
Dr. Dragioti and coauthors cited several limitations, including the inability of some randomized, controlled trials to address adverse outcomes.
“Antidepressant use appears to be safe for the treatment of psychiatric disorders, but more studies matching for underlying disease are needed to clarify the degree of confounding by indication and other biases,” the authors wrote.
The study was funded by several entities, including the National Institute for Health Research’s Biomedical Research Centre at South London and Maudsley NHS Foundation Trust. Dr. Dragioti reported no disclosures. Four authors declared funding, consultancies, personal fees, royalties, or shares in the pharmaceutical sector. No other conflicts of interest were declared.
SOURCE: Dragioti E et al. JAMA Psychiatry. 2019 Oct 2. doi: 10.1001/jamapsychiatry.2019.2859.
Relatively few of the adverse health outcomes attributed to antidepressants are supported by convincing evidence, reported the authors of a systematic review of 45 meta-analyses.
The authors did find convincing evidence linking the use of antidepressants and suicide attempt or completion among people under age 19 years and use of the medication and autism risk among offspring. “However, the few [studies] with convincing evidence associations did not reflect causality, and none of them remained at the convincing evidence level after accounting for confounding by indication,” wrote Elena Dragioti, PhD, of the Pain and Rehabilitation Centre at Linköping (Sweden) University and coauthors. The study was published in JAMA Psychiatry.
Dr. Dragioti and coauthors undertook a systematic “umbrella review” grading the evidence from the 45 meta-analyses of 695 observational studies into the association between antidepressant use and the risk of adverse health outcomes. All the meta-analyses included a control group not exposed to antidepressants, with the exception of one that compared the risk of gastrointestinal bleeding between two classes of antidepressants.
They found 120 possible adverse health associations described in the meta-analyses, 61.7% of which related to maternal and pregnancy-related adverse health outcomes. Two-thirds of the adverse health outcome associations involved selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs).
However, among the 120 adverse health associations, only three (2.5%) were supported by “convincing” evidence. One was the association between SSRIs and increased risk of suicide attempts and completion in children and adolescents. Convincing evidence also was found between any antidepressant use before pregnancy and autism spectrum disorder and between SSRI use during pregnancy and autism spectrum disorder. The evidence for the association with suicide risk was deemed high quality, but the two associations with autism spectrum disorder were only of moderate quality.
The authors commented that these findings needed to be considered when prescribing antidepressants in adolescents and children, particularly as another networked meta-analysis had found fluoxetine was the only antidepressant that worked better than placebo in children and adolescents. “In addition, they wrote.
The review found that 11 adverse health outcomes (9.2%) had “highly suggestive” evidence linking them to antidepressant use. These were ADHD in children, cataract development, severe bleeding at any site, upper gastrointestinal tract bleeding, postpartum hemorrhage, preterm birth, lower Apgar score at 5 minutes, osteoporotic fracture, and hip fracture.
Seven of those – ADHD in children, lower Apgar score, severe bleeding at any site, cataract development, osteoporotic features, preterm birth, and upper GI bleeding – had moderate-quality evidence. However, the authors noted that the effect sizes were small and had low prevalence.
The study also found highly suggestive evidence linking antidepressant use to a decreased risk of suicide attempts or completion in adults.
The authors said several of those adverse events in adults, such as GI bleeding and osteoporotic fractures, could be prevented with medication, so the advantages of antidepressant use in adults could outweigh the disadvantage of those preventable safety issues.
Twenty-one adverse health outcomes showed either suggestive, weak, or no evidence for their association with antidepressant use.
They also conducted a sensitivity analysis that limited the analysis to cohort studies, prospective cohort studies, studies that controlled for confounding by the treatment indication, and studies from North America. This showed that none of the associations for which there was originally deemed to be convincing evidence retained that same rank.
“Overall, the results showed that the association between antidepressant use and adverse health outcomes was not supported by robust evidence and that the underlying disease likely inflated the findings in a relevant way,” the authors wrote.
However, when they looked solely at prospective cohort studies, the association between preterm birth and use of any antidepressant was upgraded to having convincing evidence.
When the analysis focused on SSRIs only, the association with lower Apgar scores at 5 minutes also was upgraded to having convincing evidence. Similarly, the evidence for an association with preterm birth also was found to be convincing when the analysis was limited to other or mixed antidepressants.
Dr. Dragioti and coauthors cited several limitations, including the inability of some randomized, controlled trials to address adverse outcomes.
“Antidepressant use appears to be safe for the treatment of psychiatric disorders, but more studies matching for underlying disease are needed to clarify the degree of confounding by indication and other biases,” the authors wrote.
The study was funded by several entities, including the National Institute for Health Research’s Biomedical Research Centre at South London and Maudsley NHS Foundation Trust. Dr. Dragioti reported no disclosures. Four authors declared funding, consultancies, personal fees, royalties, or shares in the pharmaceutical sector. No other conflicts of interest were declared.
SOURCE: Dragioti E et al. JAMA Psychiatry. 2019 Oct 2. doi: 10.1001/jamapsychiatry.2019.2859.
Relatively few of the adverse health outcomes attributed to antidepressants are supported by convincing evidence, reported the authors of a systematic review of 45 meta-analyses.
The authors did find convincing evidence linking the use of antidepressants and suicide attempt or completion among people under age 19 years and use of the medication and autism risk among offspring. “However, the few [studies] with convincing evidence associations did not reflect causality, and none of them remained at the convincing evidence level after accounting for confounding by indication,” wrote Elena Dragioti, PhD, of the Pain and Rehabilitation Centre at Linköping (Sweden) University and coauthors. The study was published in JAMA Psychiatry.
Dr. Dragioti and coauthors undertook a systematic “umbrella review” grading the evidence from the 45 meta-analyses of 695 observational studies into the association between antidepressant use and the risk of adverse health outcomes. All the meta-analyses included a control group not exposed to antidepressants, with the exception of one that compared the risk of gastrointestinal bleeding between two classes of antidepressants.
They found 120 possible adverse health associations described in the meta-analyses, 61.7% of which related to maternal and pregnancy-related adverse health outcomes. Two-thirds of the adverse health outcome associations involved selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs).
However, among the 120 adverse health associations, only three (2.5%) were supported by “convincing” evidence. One was the association between SSRIs and increased risk of suicide attempts and completion in children and adolescents. Convincing evidence also was found between any antidepressant use before pregnancy and autism spectrum disorder and between SSRI use during pregnancy and autism spectrum disorder. The evidence for the association with suicide risk was deemed high quality, but the two associations with autism spectrum disorder were only of moderate quality.
The authors commented that these findings needed to be considered when prescribing antidepressants in adolescents and children, particularly as another networked meta-analysis had found fluoxetine was the only antidepressant that worked better than placebo in children and adolescents. “In addition, they wrote.
The review found that 11 adverse health outcomes (9.2%) had “highly suggestive” evidence linking them to antidepressant use. These were ADHD in children, cataract development, severe bleeding at any site, upper gastrointestinal tract bleeding, postpartum hemorrhage, preterm birth, lower Apgar score at 5 minutes, osteoporotic fracture, and hip fracture.
Seven of those – ADHD in children, lower Apgar score, severe bleeding at any site, cataract development, osteoporotic features, preterm birth, and upper GI bleeding – had moderate-quality evidence. However, the authors noted that the effect sizes were small and had low prevalence.
The study also found highly suggestive evidence linking antidepressant use to a decreased risk of suicide attempts or completion in adults.
The authors said several of those adverse events in adults, such as GI bleeding and osteoporotic fractures, could be prevented with medication, so the advantages of antidepressant use in adults could outweigh the disadvantage of those preventable safety issues.
Twenty-one adverse health outcomes showed either suggestive, weak, or no evidence for their association with antidepressant use.
They also conducted a sensitivity analysis that limited the analysis to cohort studies, prospective cohort studies, studies that controlled for confounding by the treatment indication, and studies from North America. This showed that none of the associations for which there was originally deemed to be convincing evidence retained that same rank.
“Overall, the results showed that the association between antidepressant use and adverse health outcomes was not supported by robust evidence and that the underlying disease likely inflated the findings in a relevant way,” the authors wrote.
However, when they looked solely at prospective cohort studies, the association between preterm birth and use of any antidepressant was upgraded to having convincing evidence.
When the analysis focused on SSRIs only, the association with lower Apgar scores at 5 minutes also was upgraded to having convincing evidence. Similarly, the evidence for an association with preterm birth also was found to be convincing when the analysis was limited to other or mixed antidepressants.
Dr. Dragioti and coauthors cited several limitations, including the inability of some randomized, controlled trials to address adverse outcomes.
“Antidepressant use appears to be safe for the treatment of psychiatric disorders, but more studies matching for underlying disease are needed to clarify the degree of confounding by indication and other biases,” the authors wrote.
The study was funded by several entities, including the National Institute for Health Research’s Biomedical Research Centre at South London and Maudsley NHS Foundation Trust. Dr. Dragioti reported no disclosures. Four authors declared funding, consultancies, personal fees, royalties, or shares in the pharmaceutical sector. No other conflicts of interest were declared.
SOURCE: Dragioti E et al. JAMA Psychiatry. 2019 Oct 2. doi: 10.1001/jamapsychiatry.2019.2859.
FROM JAMA PSYCHIATRY
Key clinical point: “More studies [of antidepressants] matching for underlying disease are needed to clarify the degree of confounding by indication and other biases.”
Major finding: Increased suicide risk in children and adolescents is one of the few adverse health outcomes of antidepressants that is backed by evidence.
Study details: Systematic umbrella review of 45 meta-analyses of 695 observational studies.
Disclosures: The study was funded by several entities, including the National Institute for Health Research’s Biomedical Research Centre at South London and Maudsley NHS Foundation Trust. Dr. Dragioti reported no disclosures. Four authors declared funding, consultancies, personal fees, royalties, or shares in the pharmaceutical sector. No other conflicts of interest were declared.
Source: Dragioti E et al. JAMA Psychiatry. 2019 Oct 2. doi: 10.1001/jamapsychiatry.2019.2859.
Higher teen pregnancy risk in girls with ADHD
Teenage girls with ADHD may be at greater risk of pregnancy than their unaffected peers, which suggests they may benefit from targeted interventions to prevent teen pregnancy.

A Swedish nationwide cohort study published in JAMA Network Open examined data from 384,103 nulliparous women and girls who gave birth between 2007-2014, of whom, 6,410 (1.7%) had received treatment for ADHD.
While the overall rate of teenage births was 3%, the rate among women and girls with ADHD was 15.3%, which represents a greater than sixfold higher odds of giving birth below the age of 20 years (odds ratio, 6.23; 95% confidence interval, 5.80-6.68).
“Becoming a mother at such early age is associated with long-term adverse outcomes for both women and their children,” wrote Charlotte Skoglund, PhD, of the department of clinical neuroscience at the Karolinska Institute in Stockholm and coauthors. “Consequently, our findings argue for an improvement in the standard of care for women and girls with ADHD, including active efforts to prevent teenage pregnancies and address comorbid medical and psychiatric conditions.”
The study also found women and girls with ADHD were significantly more likely to be underweight (OR, 1.29; 95% CI, 1.12-1.49) or have a body mass index greater than 40 kg/m2 (OR, 2.01; 95% CI, 1.60-2.52) when compared with those without ADHD.
They were also six times more likely to smoke, were nearly seven times more likely to continue smoking into their third trimester of pregnancy, and had a 20-fold higher odds of alcohol and substance use disorder. Among individuals who had been diagnosed with ADHD, 7.6% continued to use stimulant and nonstimulant ADHD medication during pregnancy, and 16.4% used antidepressants during pregnancy.
Psychiatric comorbidities were also significantly more common among individuals with ADHD in the year preceding pregnancy, compared with those without ADHD. The authors saw a 17-fold higher odds of receiving a diagnosis of bipolar disorder, nearly 8-fold higher odds of a diagnosis of schizophrenia or other psychotic disorder, and 22-fold higher odds of being diagnosed with emotionally unstable personality disorder among women and girls with ADHD versus those without.
The authors commented that antenatal care should focus on trying to reduce such obstetric risk factors in these women, but also pointed out that ADHD in women and girls was still underdiagnosed and undertreated.
Commenting on the association between ADHD and teenage pregnancy, the authors noted that women and girls with ADHD may be less likely to receive adequate contraceptive counseling and less likely to access, respond to, and act on counseling. They may also experience more adverse effects from hormonal contraceptives.
While Swedish youth clinics enable easier and low-cost access to counseling and contraception, the authors called for greater collaboration between psychiatric care clinics and specialized youth clinics to provide adequate care for women and girls with ADHD.
Three authors declared advisory board positions, grants, personal fees, and speakers’ fees from the pharmaceutical sector. No other conflicts of interest were declared.
SOURCE: Skoglund C et al. JAMA Netw Open. 2019 Oct 2. doi: 10.1001/jamanetworkopen.2019.12463
Teenage girls with ADHD may be at greater risk of pregnancy than their unaffected peers, which suggests they may benefit from targeted interventions to prevent teen pregnancy.
A Swedish nationwide cohort study published in JAMA Network Open examined data from 384,103 nulliparous women and girls who gave birth between 2007-2014, of whom, 6,410 (1.7%) had received treatment for ADHD.
While the overall rate of teenage births was 3%, the rate among women and girls with ADHD was 15.3%, which represents a greater than sixfold higher odds of giving birth below the age of 20 years (odds ratio, 6.23; 95% confidence interval, 5.80-6.68).
“Becoming a mother at such early age is associated with long-term adverse outcomes for both women and their children,” wrote Charlotte Skoglund, PhD, of the department of clinical neuroscience at the Karolinska Institute in Stockholm and coauthors. “Consequently, our findings argue for an improvement in the standard of care for women and girls with ADHD, including active efforts to prevent teenage pregnancies and address comorbid medical and psychiatric conditions.”
The study also found women and girls with ADHD were significantly more likely to be underweight (OR, 1.29; 95% CI, 1.12-1.49) or have a body mass index greater than 40 kg/m2 (OR, 2.01; 95% CI, 1.60-2.52) when compared with those without ADHD.
They were also six times more likely to smoke, were nearly seven times more likely to continue smoking into their third trimester of pregnancy, and had a 20-fold higher odds of alcohol and substance use disorder. Among individuals who had been diagnosed with ADHD, 7.6% continued to use stimulant and nonstimulant ADHD medication during pregnancy, and 16.4% used antidepressants during pregnancy.
Psychiatric comorbidities were also significantly more common among individuals with ADHD in the year preceding pregnancy, compared with those without ADHD. The authors saw a 17-fold higher odds of receiving a diagnosis of bipolar disorder, nearly 8-fold higher odds of a diagnosis of schizophrenia or other psychotic disorder, and 22-fold higher odds of being diagnosed with emotionally unstable personality disorder among women and girls with ADHD versus those without.
The authors commented that antenatal care should focus on trying to reduce such obstetric risk factors in these women, but also pointed out that ADHD in women and girls was still underdiagnosed and undertreated.
Commenting on the association between ADHD and teenage pregnancy, the authors noted that women and girls with ADHD may be less likely to receive adequate contraceptive counseling and less likely to access, respond to, and act on counseling. They may also experience more adverse effects from hormonal contraceptives.
While Swedish youth clinics enable easier and low-cost access to counseling and contraception, the authors called for greater collaboration between psychiatric care clinics and specialized youth clinics to provide adequate care for women and girls with ADHD.
Three authors declared advisory board positions, grants, personal fees, and speakers’ fees from the pharmaceutical sector. No other conflicts of interest were declared.
SOURCE: Skoglund C et al. JAMA Netw Open. 2019 Oct 2. doi: 10.1001/jamanetworkopen.2019.12463
Teenage girls with ADHD may be at greater risk of pregnancy than their unaffected peers, which suggests they may benefit from targeted interventions to prevent teen pregnancy.
A Swedish nationwide cohort study published in JAMA Network Open examined data from 384,103 nulliparous women and girls who gave birth between 2007-2014, of whom, 6,410 (1.7%) had received treatment for ADHD.
While the overall rate of teenage births was 3%, the rate among women and girls with ADHD was 15.3%, which represents a greater than sixfold higher odds of giving birth below the age of 20 years (odds ratio, 6.23; 95% confidence interval, 5.80-6.68).
“Becoming a mother at such early age is associated with long-term adverse outcomes for both women and their children,” wrote Charlotte Skoglund, PhD, of the department of clinical neuroscience at the Karolinska Institute in Stockholm and coauthors. “Consequently, our findings argue for an improvement in the standard of care for women and girls with ADHD, including active efforts to prevent teenage pregnancies and address comorbid medical and psychiatric conditions.”
The study also found women and girls with ADHD were significantly more likely to be underweight (OR, 1.29; 95% CI, 1.12-1.49) or have a body mass index greater than 40 kg/m2 (OR, 2.01; 95% CI, 1.60-2.52) when compared with those without ADHD.
They were also six times more likely to smoke, were nearly seven times more likely to continue smoking into their third trimester of pregnancy, and had a 20-fold higher odds of alcohol and substance use disorder. Among individuals who had been diagnosed with ADHD, 7.6% continued to use stimulant and nonstimulant ADHD medication during pregnancy, and 16.4% used antidepressants during pregnancy.
Psychiatric comorbidities were also significantly more common among individuals with ADHD in the year preceding pregnancy, compared with those without ADHD. The authors saw a 17-fold higher odds of receiving a diagnosis of bipolar disorder, nearly 8-fold higher odds of a diagnosis of schizophrenia or other psychotic disorder, and 22-fold higher odds of being diagnosed with emotionally unstable personality disorder among women and girls with ADHD versus those without.
The authors commented that antenatal care should focus on trying to reduce such obstetric risk factors in these women, but also pointed out that ADHD in women and girls was still underdiagnosed and undertreated.
Commenting on the association between ADHD and teenage pregnancy, the authors noted that women and girls with ADHD may be less likely to receive adequate contraceptive counseling and less likely to access, respond to, and act on counseling. They may also experience more adverse effects from hormonal contraceptives.
While Swedish youth clinics enable easier and low-cost access to counseling and contraception, the authors called for greater collaboration between psychiatric care clinics and specialized youth clinics to provide adequate care for women and girls with ADHD.
Three authors declared advisory board positions, grants, personal fees, and speakers’ fees from the pharmaceutical sector. No other conflicts of interest were declared.
SOURCE: Skoglund C et al. JAMA Netw Open. 2019 Oct 2. doi: 10.1001/jamanetworkopen.2019.12463
FROM JAMA NETWORK OPEN

Two studies reveal preneoplastic links between H. pylori and gastric cancer
Molecular pathways linked with CD44 variant 9 (CD44v9), a cell surface glycoprotein tied to aggressive gastric cancer after Helicobacter pylori infection, may open doors to stop cancer before it starts, according to two recent studies.
Findings from the first study suggest that persistent inflammation after eradication therapy may continue to drive cancer risk after infection, while the second study revealed a potential therapeutic target related to preneoplastic changes.
The first study, conducted by lead author Hitoshi Tsugawa, PhD, of Keio University, Tokyo, and colleagues, aimed to determine the origin of CD44v9-positive cancer stem-like cells.
“These cells strongly contribute to the development and recurrence of gastric cancer,” the investigators wrote. Their report is in Cellular and Molecular Gastroenterology and Hepatology. “However, the origin of CD44v9-positive cells is uncertain.”
The association between H. pylori infection and gastric cancer has been documented, along with a high risk of cancer when gastric epithelial cells overexpress capping actin protein of muscle Z-line alpha subunit 1 (CAPZA1), the researchers noted. Although it has also been shown that CAPZA1 overexpression leads to intracellular accumulations of the H. pylori–derived oncoprotein cytotoxin-associated gene A (CagA), just how these phenomena were connected remained unknown.
Through in vitro analyses of human cells, and in vitro and in vivo experiments involving Mongolian gerbils, the investigators uncovered a chain of events between H. pylori infection and CD44v9 expression. First, the investigators showed that expression levels of CD44v9 and CAPZA1 were directly correlated in five human cases of gastric cancer. Next, several experiments revealed that H. pylori–related oxidative stress drives overexpression of CAPZA1, which, in combination with high levels of beta-catenin, ESRP1, and CagA, promotes expression of CD44v9.
Most directly relevant to future therapies, the investigators compared levels of CAPZA1 between five active cases of H. pylori infection versus five cases successfully treated with eradication therapy. After eradication therapy, CAPZA1 overexpression decreased, but not to a significant degree.
“Our findings suggest that CAPZA1-overexpressing cells remaining in the gastric mucosa after eradication therapy increase the risk of metachronous gastric cancer and that reduction of CAPZA1 expression by amelioration of chronic inflammation after eradication therapy is important to prevent the development of gastric cancer,” the investigators concluded.
The second study, by lead author Anne R. Meyer, a graduate student at Vanderbilt University, Nashville, Tenn., and colleagues, evaluated how zymogenic chief cells are reprogrammed into spasmolytic polypeptide-expressing metaplasia (SPEM), a precursor to dysplasia and gastric cancer.
It had been previously shown that reprogramming to SPEM is promoted and maintained by epithelial cell damage, such as that caused by H. pylori infection, but underlying processes remained unclear, until recent studies suggested a link between SPEM transition and upregulation of CD44v9. Knowing that CD44v9 stabilizes the cystine/glutamate antiporter xCT, the investigators homed in on xCT for a closer look, questioning what role it had in chief cell reprogramming. Again, oxidative stress was identified as the inciting pathophysiologic driver.
“The oxidative stress response, including upregulation of nutrient transporters, plays an important role in many biological processes and the pathogenesis of a variety of diseases,” the investigators wrote in their report, published in Cellular and Molecular Gastroenterology and Hepatology. “Perturbations to the CD44v9-xCT system often result in redox imbalance.”
Using a combination of mouse and human cell lines, and a mouse model, the investigators demonstrated that xCT was upregulated during the initial stages of chief cell programming. Blocking xCT with sulfasalazine after acute gastric injury limited SPEM transition by more than 80%, an effect that was further supported by xCT siRNA knockdown and observations in xCT knockout mice. Reduction in chief cell reprogramming was not observed in the presence of sulfasalazine metabolites, suggesting that the anti-inflammatory properties of sulfasalazine were not responsible for downregulation of reprogramming.
“Targeting xCT may prove an effective tool for arresting metaplasia development in the stomach as well as mucous metaplasia in other epithelial tissues for the analysis of cellular plasticity and oxidative stress response,” the investigators concluded.
The study by Tsugawa and colleagues was funded by Grants-in-Aid for Scientific Research; the Yakult Bio-Science Foundation; the Ministry of Education, Culture, Sports, Science and Technology (MEXT)-supported program for the Strategic Research Foundation at Private Universities; and Keio Gijuku Academic Development Funds. Dr. Suzuki disclosed relationships with Daiichi-Sankyo Co, EA Pharma Co, Otsuka Pharmaceutical Co Ltd, and others. The study by Meyer and colleagues was funded by the National Institutes of Health, the American Association of Cancer Research, the Department of Defense, and others, with no relevant conflicts of interest.
SOURCES: Meyer et al. CMGH. 2019 May 6. doi: 10.1016/j.jcmgh.2019.04.015; Tsugawa et al. CMGH. 2019 May 27. doi: 10.1016/j.jcmgh.2019.05.008.
The mechanisms by which injured cells respond to stress rely in part on their ability to reprogram themselves in the setting of injury. This cellular reprogramming involves sensing and regulating intracellular metabolic cues that dictate survival, organization of secretory and degradative machinery, and proliferation. Meyer et al. and Tsugawa et al. illustrate two distinct mechanisms by which gastric epithelial cells handle oxidative stress during injury.
Meyer et al. focus on the xCT subunit of the cystine/glutamate antiporter as a rheostat for intracellular glutathione stores. Pharmacologic inhibition of xCT activity using sulfasalazine hampers the ability of injured gastric epithelial cells to adequately deal with reactive oxygen species. Importantly, these cells do not appropriately reprogram during injury and instead undergo apoptosis. Tsugawa et al. provide mechanistic insight into how oxidative stress may promote precancerous changes in gastric epithelium. Following H. pylori infection, an intracellular oxidative environment that is characterized by an overexpression of the actin filament capping protein CAPZA1, beta-catenin, and the alternative splicing factor ESRP1, promotes expression of CD44 variant 9 (CD44v9), a cell surface glycoprotein that correlates with gastric cancer. Interestingly, this oxidative milieu promotes accumulation of a critical H. pylori virulence factor, CagA, within infected cells.
Taken together, the ability to manage oxidative stress during cellular injury has significant implications for cell fate. It seems likely that the mechanisms for regulating intracellular oxidative stress are not unique to gastric epithelium and instead underlie a conserved injury response that has correlates in other gastrointestinal organs.
José B. Sáenz, MD, PhD, is an investigator and instructor of medicine in the gastroenterology division, John T. Milliken Department of Internal Medicine at the Washington University in St. Louis School of Medicine. He has no conflicts of interest.
The mechanisms by which injured cells respond to stress rely in part on their ability to reprogram themselves in the setting of injury. This cellular reprogramming involves sensing and regulating intracellular metabolic cues that dictate survival, organization of secretory and degradative machinery, and proliferation. Meyer et al. and Tsugawa et al. illustrate two distinct mechanisms by which gastric epithelial cells handle oxidative stress during injury.
Meyer et al. focus on the xCT subunit of the cystine/glutamate antiporter as a rheostat for intracellular glutathione stores. Pharmacologic inhibition of xCT activity using sulfasalazine hampers the ability of injured gastric epithelial cells to adequately deal with reactive oxygen species. Importantly, these cells do not appropriately reprogram during injury and instead undergo apoptosis. Tsugawa et al. provide mechanistic insight into how oxidative stress may promote precancerous changes in gastric epithelium. Following H. pylori infection, an intracellular oxidative environment that is characterized by an overexpression of the actin filament capping protein CAPZA1, beta-catenin, and the alternative splicing factor ESRP1, promotes expression of CD44 variant 9 (CD44v9), a cell surface glycoprotein that correlates with gastric cancer. Interestingly, this oxidative milieu promotes accumulation of a critical H. pylori virulence factor, CagA, within infected cells.
Taken together, the ability to manage oxidative stress during cellular injury has significant implications for cell fate. It seems likely that the mechanisms for regulating intracellular oxidative stress are not unique to gastric epithelium and instead underlie a conserved injury response that has correlates in other gastrointestinal organs.
José B. Sáenz, MD, PhD, is an investigator and instructor of medicine in the gastroenterology division, John T. Milliken Department of Internal Medicine at the Washington University in St. Louis School of Medicine. He has no conflicts of interest.
The mechanisms by which injured cells respond to stress rely in part on their ability to reprogram themselves in the setting of injury. This cellular reprogramming involves sensing and regulating intracellular metabolic cues that dictate survival, organization of secretory and degradative machinery, and proliferation. Meyer et al. and Tsugawa et al. illustrate two distinct mechanisms by which gastric epithelial cells handle oxidative stress during injury.
Meyer et al. focus on the xCT subunit of the cystine/glutamate antiporter as a rheostat for intracellular glutathione stores. Pharmacologic inhibition of xCT activity using sulfasalazine hampers the ability of injured gastric epithelial cells to adequately deal with reactive oxygen species. Importantly, these cells do not appropriately reprogram during injury and instead undergo apoptosis. Tsugawa et al. provide mechanistic insight into how oxidative stress may promote precancerous changes in gastric epithelium. Following H. pylori infection, an intracellular oxidative environment that is characterized by an overexpression of the actin filament capping protein CAPZA1, beta-catenin, and the alternative splicing factor ESRP1, promotes expression of CD44 variant 9 (CD44v9), a cell surface glycoprotein that correlates with gastric cancer. Interestingly, this oxidative milieu promotes accumulation of a critical H. pylori virulence factor, CagA, within infected cells.
Taken together, the ability to manage oxidative stress during cellular injury has significant implications for cell fate. It seems likely that the mechanisms for regulating intracellular oxidative stress are not unique to gastric epithelium and instead underlie a conserved injury response that has correlates in other gastrointestinal organs.
José B. Sáenz, MD, PhD, is an investigator and instructor of medicine in the gastroenterology division, John T. Milliken Department of Internal Medicine at the Washington University in St. Louis School of Medicine. He has no conflicts of interest.
Molecular pathways linked with CD44 variant 9 (CD44v9), a cell surface glycoprotein tied to aggressive gastric cancer after Helicobacter pylori infection, may open doors to stop cancer before it starts, according to two recent studies.
Findings from the first study suggest that persistent inflammation after eradication therapy may continue to drive cancer risk after infection, while the second study revealed a potential therapeutic target related to preneoplastic changes.
The first study, conducted by lead author Hitoshi Tsugawa, PhD, of Keio University, Tokyo, and colleagues, aimed to determine the origin of CD44v9-positive cancer stem-like cells.
“These cells strongly contribute to the development and recurrence of gastric cancer,” the investigators wrote. Their report is in Cellular and Molecular Gastroenterology and Hepatology. “However, the origin of CD44v9-positive cells is uncertain.”
The association between H. pylori infection and gastric cancer has been documented, along with a high risk of cancer when gastric epithelial cells overexpress capping actin protein of muscle Z-line alpha subunit 1 (CAPZA1), the researchers noted. Although it has also been shown that CAPZA1 overexpression leads to intracellular accumulations of the H. pylori–derived oncoprotein cytotoxin-associated gene A (CagA), just how these phenomena were connected remained unknown.
Through in vitro analyses of human cells, and in vitro and in vivo experiments involving Mongolian gerbils, the investigators uncovered a chain of events between H. pylori infection and CD44v9 expression. First, the investigators showed that expression levels of CD44v9 and CAPZA1 were directly correlated in five human cases of gastric cancer. Next, several experiments revealed that H. pylori–related oxidative stress drives overexpression of CAPZA1, which, in combination with high levels of beta-catenin, ESRP1, and CagA, promotes expression of CD44v9.
Most directly relevant to future therapies, the investigators compared levels of CAPZA1 between five active cases of H. pylori infection versus five cases successfully treated with eradication therapy. After eradication therapy, CAPZA1 overexpression decreased, but not to a significant degree.
“Our findings suggest that CAPZA1-overexpressing cells remaining in the gastric mucosa after eradication therapy increase the risk of metachronous gastric cancer and that reduction of CAPZA1 expression by amelioration of chronic inflammation after eradication therapy is important to prevent the development of gastric cancer,” the investigators concluded.
The second study, by lead author Anne R. Meyer, a graduate student at Vanderbilt University, Nashville, Tenn., and colleagues, evaluated how zymogenic chief cells are reprogrammed into spasmolytic polypeptide-expressing metaplasia (SPEM), a precursor to dysplasia and gastric cancer.
It had been previously shown that reprogramming to SPEM is promoted and maintained by epithelial cell damage, such as that caused by H. pylori infection, but underlying processes remained unclear, until recent studies suggested a link between SPEM transition and upregulation of CD44v9. Knowing that CD44v9 stabilizes the cystine/glutamate antiporter xCT, the investigators homed in on xCT for a closer look, questioning what role it had in chief cell reprogramming. Again, oxidative stress was identified as the inciting pathophysiologic driver.
“The oxidative stress response, including upregulation of nutrient transporters, plays an important role in many biological processes and the pathogenesis of a variety of diseases,” the investigators wrote in their report, published in Cellular and Molecular Gastroenterology and Hepatology. “Perturbations to the CD44v9-xCT system often result in redox imbalance.”
Using a combination of mouse and human cell lines, and a mouse model, the investigators demonstrated that xCT was upregulated during the initial stages of chief cell programming. Blocking xCT with sulfasalazine after acute gastric injury limited SPEM transition by more than 80%, an effect that was further supported by xCT siRNA knockdown and observations in xCT knockout mice. Reduction in chief cell reprogramming was not observed in the presence of sulfasalazine metabolites, suggesting that the anti-inflammatory properties of sulfasalazine were not responsible for downregulation of reprogramming.
“Targeting xCT may prove an effective tool for arresting metaplasia development in the stomach as well as mucous metaplasia in other epithelial tissues for the analysis of cellular plasticity and oxidative stress response,” the investigators concluded.
The study by Tsugawa and colleagues was funded by Grants-in-Aid for Scientific Research; the Yakult Bio-Science Foundation; the Ministry of Education, Culture, Sports, Science and Technology (MEXT)-supported program for the Strategic Research Foundation at Private Universities; and Keio Gijuku Academic Development Funds. Dr. Suzuki disclosed relationships with Daiichi-Sankyo Co, EA Pharma Co, Otsuka Pharmaceutical Co Ltd, and others. The study by Meyer and colleagues was funded by the National Institutes of Health, the American Association of Cancer Research, the Department of Defense, and others, with no relevant conflicts of interest.
SOURCES: Meyer et al. CMGH. 2019 May 6. doi: 10.1016/j.jcmgh.2019.04.015; Tsugawa et al. CMGH. 2019 May 27. doi: 10.1016/j.jcmgh.2019.05.008.
Molecular pathways linked with CD44 variant 9 (CD44v9), a cell surface glycoprotein tied to aggressive gastric cancer after Helicobacter pylori infection, may open doors to stop cancer before it starts, according to two recent studies.
Findings from the first study suggest that persistent inflammation after eradication therapy may continue to drive cancer risk after infection, while the second study revealed a potential therapeutic target related to preneoplastic changes.
The first study, conducted by lead author Hitoshi Tsugawa, PhD, of Keio University, Tokyo, and colleagues, aimed to determine the origin of CD44v9-positive cancer stem-like cells.
“These cells strongly contribute to the development and recurrence of gastric cancer,” the investigators wrote. Their report is in Cellular and Molecular Gastroenterology and Hepatology. “However, the origin of CD44v9-positive cells is uncertain.”
The association between H. pylori infection and gastric cancer has been documented, along with a high risk of cancer when gastric epithelial cells overexpress capping actin protein of muscle Z-line alpha subunit 1 (CAPZA1), the researchers noted. Although it has also been shown that CAPZA1 overexpression leads to intracellular accumulations of the H. pylori–derived oncoprotein cytotoxin-associated gene A (CagA), just how these phenomena were connected remained unknown.
Through in vitro analyses of human cells, and in vitro and in vivo experiments involving Mongolian gerbils, the investigators uncovered a chain of events between H. pylori infection and CD44v9 expression. First, the investigators showed that expression levels of CD44v9 and CAPZA1 were directly correlated in five human cases of gastric cancer. Next, several experiments revealed that H. pylori–related oxidative stress drives overexpression of CAPZA1, which, in combination with high levels of beta-catenin, ESRP1, and CagA, promotes expression of CD44v9.
Most directly relevant to future therapies, the investigators compared levels of CAPZA1 between five active cases of H. pylori infection versus five cases successfully treated with eradication therapy. After eradication therapy, CAPZA1 overexpression decreased, but not to a significant degree.
“Our findings suggest that CAPZA1-overexpressing cells remaining in the gastric mucosa after eradication therapy increase the risk of metachronous gastric cancer and that reduction of CAPZA1 expression by amelioration of chronic inflammation after eradication therapy is important to prevent the development of gastric cancer,” the investigators concluded.
The second study, by lead author Anne R. Meyer, a graduate student at Vanderbilt University, Nashville, Tenn., and colleagues, evaluated how zymogenic chief cells are reprogrammed into spasmolytic polypeptide-expressing metaplasia (SPEM), a precursor to dysplasia and gastric cancer.
It had been previously shown that reprogramming to SPEM is promoted and maintained by epithelial cell damage, such as that caused by H. pylori infection, but underlying processes remained unclear, until recent studies suggested a link between SPEM transition and upregulation of CD44v9. Knowing that CD44v9 stabilizes the cystine/glutamate antiporter xCT, the investigators homed in on xCT for a closer look, questioning what role it had in chief cell reprogramming. Again, oxidative stress was identified as the inciting pathophysiologic driver.
“The oxidative stress response, including upregulation of nutrient transporters, plays an important role in many biological processes and the pathogenesis of a variety of diseases,” the investigators wrote in their report, published in Cellular and Molecular Gastroenterology and Hepatology. “Perturbations to the CD44v9-xCT system often result in redox imbalance.”
Using a combination of mouse and human cell lines, and a mouse model, the investigators demonstrated that xCT was upregulated during the initial stages of chief cell programming. Blocking xCT with sulfasalazine after acute gastric injury limited SPEM transition by more than 80%, an effect that was further supported by xCT siRNA knockdown and observations in xCT knockout mice. Reduction in chief cell reprogramming was not observed in the presence of sulfasalazine metabolites, suggesting that the anti-inflammatory properties of sulfasalazine were not responsible for downregulation of reprogramming.
“Targeting xCT may prove an effective tool for arresting metaplasia development in the stomach as well as mucous metaplasia in other epithelial tissues for the analysis of cellular plasticity and oxidative stress response,” the investigators concluded.
The study by Tsugawa and colleagues was funded by Grants-in-Aid for Scientific Research; the Yakult Bio-Science Foundation; the Ministry of Education, Culture, Sports, Science and Technology (MEXT)-supported program for the Strategic Research Foundation at Private Universities; and Keio Gijuku Academic Development Funds. Dr. Suzuki disclosed relationships with Daiichi-Sankyo Co, EA Pharma Co, Otsuka Pharmaceutical Co Ltd, and others. The study by Meyer and colleagues was funded by the National Institutes of Health, the American Association of Cancer Research, the Department of Defense, and others, with no relevant conflicts of interest.
SOURCES: Meyer et al. CMGH. 2019 May 6. doi: 10.1016/j.jcmgh.2019.04.015; Tsugawa et al. CMGH. 2019 May 27. doi: 10.1016/j.jcmgh.2019.05.008.
FROM CELLULAR AND MOLECULAR GASTROENTEROLOGY AND HEPATOLOGY
Acantholytic Anaplastic Extramammary Paget Disease
To the Editor:
Extramammary Paget disease (EMPD) is a rare intraepidermal neoplasm with glandular differentiation that is classically known as a mimicker of Bowen disease (squamous cell carcinoma in situ of the skin) due to their histologic similarities.1,2 However, acantholytic anaplastic EMPD (AAEMPD) is a rare variant that can pose a particularly difficult diagnostic challenge because of its histologic similarity to benign acantholytic disorders and other malignant neoplasms. Major histologic features suggestive of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The differential diagnosis of EMPD typically includes Bowen disease and pagetoid Bowen disease, but the acantholytic anaplastic variant more often is confused with intraepidermal acantholytic lesions such as acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, and acantholytic Bowen disease. Immunohistochemistry (IHC) studies to assist in the definitive diagnosis of AAEMPD are strongly advised because of these difficulties in diagnosis.4 Cases of EMPD with an acantholytic appearance have rarely been reported in the literature.5-7
A 78-year-old man with a history of arthritis, heart disease, hypertension, and gastrointestinal disease presented for evaluation of a tender lesion of the right genitocrural crease of 5 years’ duration. He had no history of cutaneous or internal malignancy. Previously the lesion had been treated by dermatology with a variety of topical products including antifungal and antibiotic creams with no improvement. Physical examination revealed a well-defined, 7×5-cm, tender, erythematous, macerated plaque on the right upper inner thigh adjacent to the scrotum with an odor possibly due to secondary infection (Figure 1).
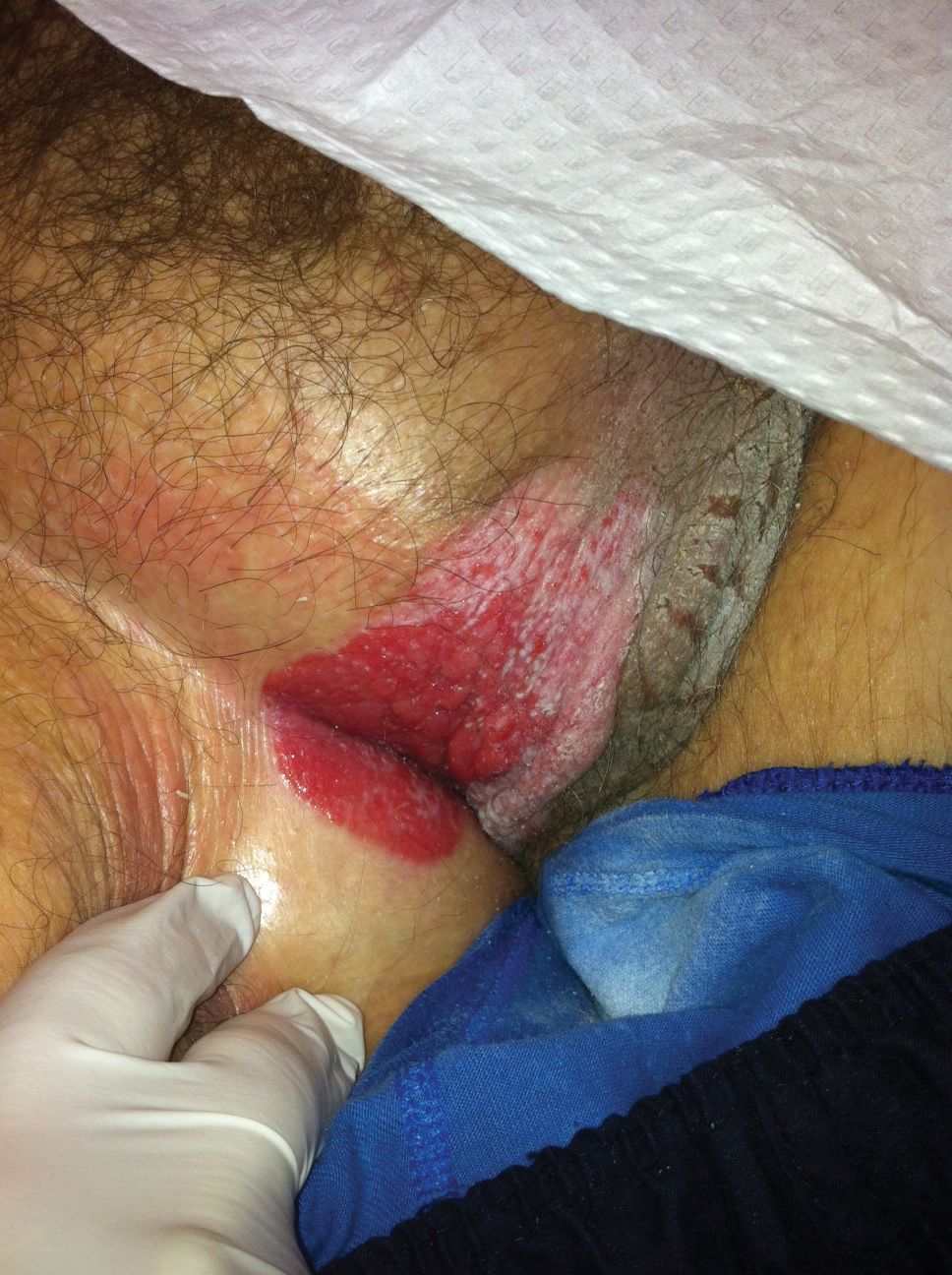
plaque on the right upper inner thigh adjacent to the scrotum.
A biopsy of the lesion was performed, and the specimen was submitted for pathologic examination. Bacterial cultures taken at the time of biopsy revealed polybacterial colonization with Acinetobacter, Morganella, and mixed skin flora. The patient was treated with a 10-day course of oral sulfamethoxazole 800 mg and trimethoprim 160 mg twice daily once culture results returned. The biopsy results were communicated to the patient; however, he subsequently relocated, assumed care at another facility, and has since been lost to follow-up.
The biopsy specimen was examined grossly, serially sectioned, and submitted for routine processing with hematoxylin and eosin, periodic acid–Schiff, and Hale colloidal iron staining. Routine IHC was performed with antibodies to cytokeratin (CK) 7, CK20, carcinoembryonic antigen (CEA), pancytokeratin (CKAE1/AE3), and low- molecular-weight cytokeratin (LMWCK).
Pathologic examination of the biopsy showed prominent acanthosis of the epidermis composed of a proliferation of epithelial cells with associated full-thickness suprabasal acantholysis (Figure 2A). On inspection at higher magnification, the neoplastic cells demonstrated anaplasia as cytologic atypia with prominent and frequently multiple nucleoli, scant cytoplasm, and a high nuclear to cytoplasmic ratio (Figure 2B). There was a marked increase in mitotic activity with as many as 5 mitotic figures per high-power field. A fairly dense mixed inflammatory infiltrate comprised of lymphocytes, plasma cells, neutrophils, and eosinophils was present in the dermis. No fungal elements were observed on periodic acid–Schiff staining. The vast majority of tumor cells demonstrated moderate to abundant cytoplasmic mucin on Hale colloidal iron staining (Figure 3).
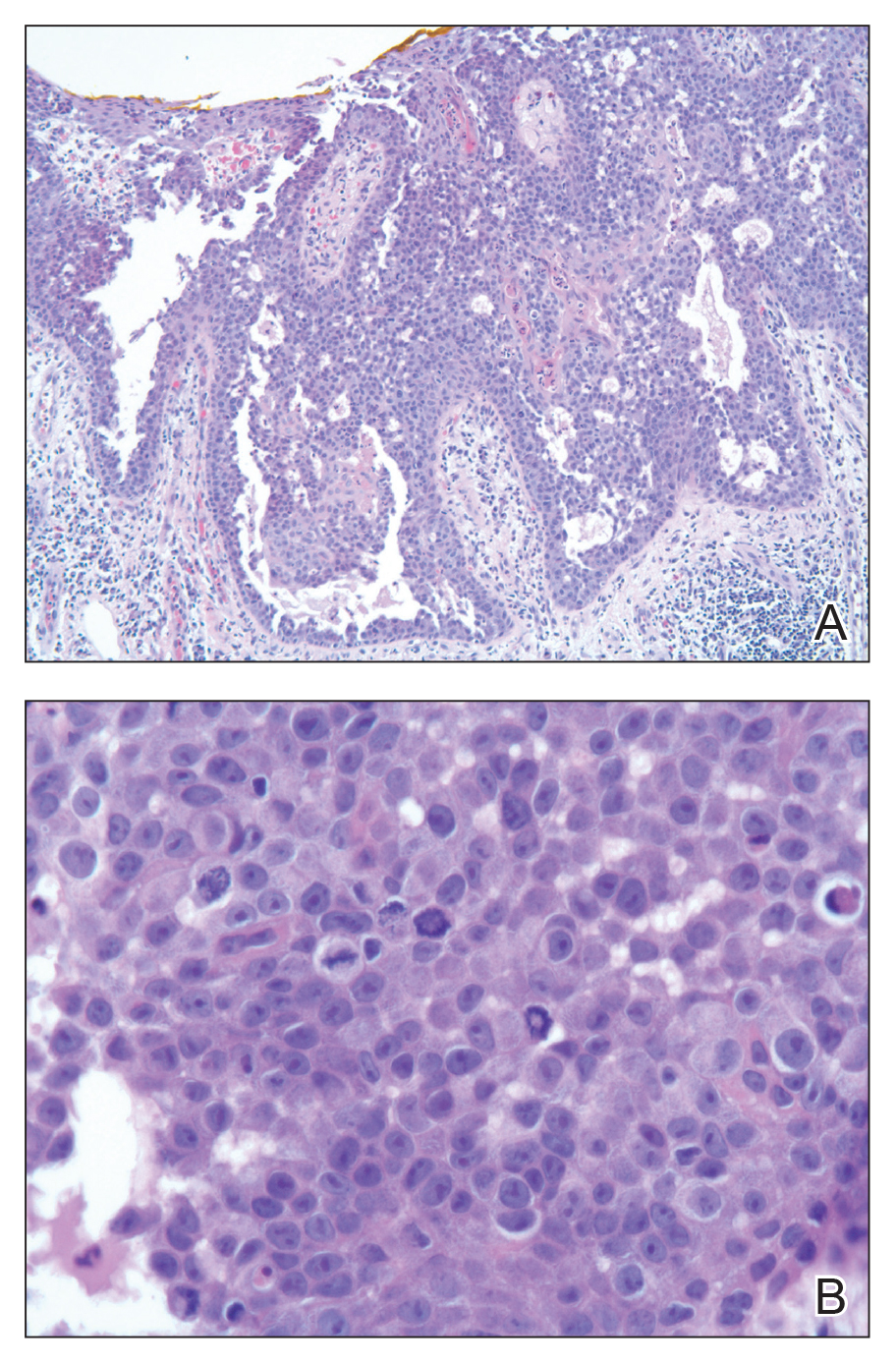
(H&E, original magnification ×400).
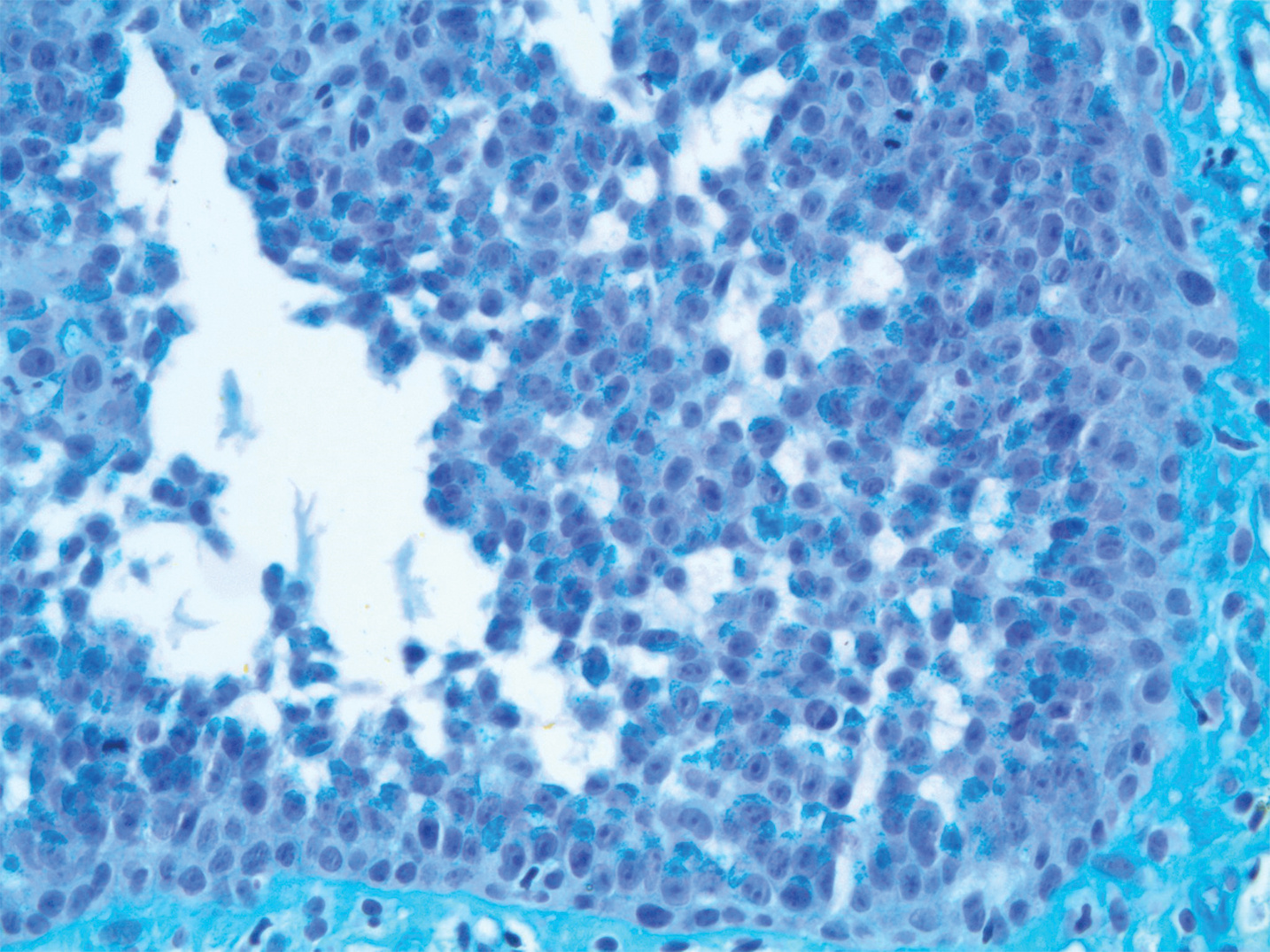
Immunohistochemistry staining of tumor cells was positive for CK7, CEA, pancytokeratin (CKAE1/AE3), and LMWCK. The tumor cells were negative for CK20. On the basis of the histopathologic and IHC findings, the patient was diagnosed with AAEMPD.
Extramammary Paget disease is a rare intraepidermal neoplasm with glandular differentiation. The most commonly involved sites are the anogenital areas including the vulvar, perianal, perineal, scrotal, and penile regions, as well as other areas rich in apocrine glands such as the axillae.8 Extramammary Paget disease most commonly originates as a primary intraepidermal neoplasm (type 1 EMPD), but an underlying malignant neoplasm that spreads intraepithelially is seen in a minority of cases (types 2 and 3 EMPD). In the vulva, type 1a refers to cutaneous noninvasive Paget disease, type 1b refers to dermal invasion of Paget disease, type 1c refers to vulvar adenocarcinoma–associated Paget disease, type 2 refers to rectal/anal adenocarcinoma–associated Paget disease, and type 3 refers to urogenital neoplasia–associated Paget disease.9
The acantholytic anaplastic variant of EMPD can be challenging to diagnose because of its similarities to many other lesions, including acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, Bowen disease, pagetoid Bowen disease, and acantholytic Bowen disease. Major histologic features of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The acantholytic anaplastic variant of EMPD can be differentiated from other diagnoses using IHC studies, with findings indicative of AAEMPD outlined below.
The proliferative neoplastic cell in EMPD is the Paget cell, which can be identified as a large round cell located in the epidermis with pale-staining cytoplasm, a large nucleus, and sometimes a prominent nucleolus. Paget cells can be distributed singly or in clusters, nests, or glandular structures within the epidermis and adjacent to adnexal structures.10 Extramammary Paget disease can have many patterns, including glandular, acantholysis-like, upper nest, tall nest, budding, and sheetlike.11
Immunohistochemically, Paget cells in EMPD typically express pancytokeratins (CKAE1/AE3), low-molecular-weight/simple epithelial type keratins (CK7, CAM 5.2), sweat gland antigens (epithelial membrane antigen, CEA, gross cystic disease fluid protein 15 [GCDFP15]), mucin 5AC (MUC5AC), and often androgen receptor.12-18 Paget cells contain cytoplasmic mucin and demonstrate prominent cytoplasmic staining with Hale colloidal iron.17 Paget cells typically do not express high-molecular-weight cytokeratin (eg, CK5/6), melanocytic antigens, estrogen receptor, or progesterone receptor.15,18
Immunohistochemical staining has been shown to differ between primary cutaneous (type 1) and secondary (types 2 and 3) EMPD. Primary cutaneous EMPD typically expresses sweat gland markers (CK7+, CK20−, GCDFP15+). Secondary EMPD typically expresses an endodermal phenotype (CK7+, CK20+, GCDFP15−).12
Acantholytic dyskeratosis of the genitocrural area is a rare lesion included in the spectrum of focal acantholytic dyskeratoses described by Ackerman.19 It also has been referred to as papular acantholytic dyskeratosis of the vulva, though histologically similar lesions also have been reported in men.20-22 Histologically, acantholytic dyskeratosis of the genitocrural area has prominent acantholysis and dyskeratosis with corps ronds and grains.19 Familial benign pemphigus (Hailey-Hailey disease) is caused by mutations of the ATP2C1 gene, which encodes for a secretory pathway Ca2+/Mn2+-ATPase pump type 1 (SPCA1) in the Golgi apparatus in keratinocytes.23 Familial benign pemphigus has a histologic appearance similar to acantholytic dyskeratosis of the genitocrural area, but a positive family history of familial benign pemphigus can be used to differentiate the 2 entities from each other due to the autosomal-dominant inheritance pattern of familial benign pemphigus. Both of these disorders can appear similar to AAEMPD because of their extensive intraepidermal acantholysis, but they differ in the lack of Paget cells, intraepidermal atypia, and increased mitotic activity.
Acantholytic Bowen disease is a histologic variant that can be difficult to distinguish from AAEMPD on hematoxylin and eosin–stained sections because of their similar histologic features but can be differentiated by IHC stains.5 Acantholytic Bowen disease expresses high-molecular-weight cytokeratin (eg, CK5/6) but is negative for CK7, CAM 5.2, and CEA. Extramammary Paget disease generally has the opposite pattern: positive staining for CK7, CAM 5.2, and CEA, but negative for high-molecular-weight cytokeratin.13,14,24
Primary cutaneous adenosquamous carcinoma is a rare malignancy of squamous and glandular differentiation known for being locally aggressive and metastatic.25 Histologically, cutaneous adenosquamous carcinoma shows infiltrating nests of neoplastic cells with both squamous and glandular features. It differs notably from AAEMPD in that cutaneous adenosquamous carcinomas tend to arise in the head and arm regions, and their histologic morphology is different. The IHC profiles are similar, with positive staining for CEA, CK7, and mucin; however, they differ in that AAEMPD is negative for high-molecular-weight keratin while cutaneous adenosquamous carcinoma is positive.25
Verrucous carcinoma is an uncommon variant of squamous cell carcinoma with well-differentiated keratinocytes and a blunt pushing border.24 Similar to AAEMPD, this neoplasm can arise in the genital and perineal areas; however, the 2 entities differ considerably in morphology on histologic examination.
Pemphigus vulgaris is an autoimmune intraepidermal blistering disorder of the skin and mucous membranes of which pemphigus vegetans is a subtype.26,27 Pemphigus vulgaris is another diagnosis that can possibly be mimicked by AAEMPD.28 Histologic features of pemphigus vulgaris include intraepidermal acantholysis of keratinocytes immediately above the basal layer of the epidermis. Pemphigus vegetans is similar with the addition of papillomatosis, hyperkeratosis, and an eosinophilic infiltrate.26,27 Immunofluorescence typically demonstrates intercellular C3 and IgG deposits.26 These diseases mimic AAEMPD histologically but differ in their relative lack of atypia and Paget cells.
In summary, we report a case of AAEMPD in a 78-year-old man in whom routine histologic specimens showed marked intraepidermal acantholysis and atypical tumor cells with increased mitoses. The latter finding prompted IHC studies that revealed positive CK7, CEA, pancytokeratin, and LMWCK staining with negative CK20 staining. Hale colloidal iron staining showed moderate to abundant cytoplasmic mucin. The patient was diagnosed with AAEMPD. It is imperative to maintain clinical suspicion for AAEMPD and to examine acantholytic disorders with scrutiny. When there is evidence of atypia or mitoses, use of IHC stains can assist in fully characterizing the lesion.
- Bowen JT. Precancerous dermatosis: a study of two cases of chronic atypical epithelial proliferation. J Cutan Dis. 1912;30:241-255.
- Jones RE Jr, Austin C, Ackerman AB. Extramammary Paget’s disease: a critical reexamination. Am J Dermatopathol. 1979;1:101-132.
- Rayne SC, Santa Cruz DJ. Anaplastic Paget’s disease. Am J Surg Pathol. 1992;16:1085-1091.
- Wang EC, Kwah YC, Tan WP, et al. Extramammary Paget disease: immunohistochemistry is critical to distinguish potential mimickers. Dermatol Online J. 2012;18:4.
- Du X, Yin X, Zhou N, et al. Extramammary Paget’s disease mimicking acantholytic squamous cell carcinoma in situ: a case report. J Cutan Pathol. 2010;37:683.
- Mobini N. Acantholytic anaplastic Paget’s disease. J Cutan Pathol. 2009;36:374-380.
- Oh YJ, Lew BL, Sim WY. Acantholytic anaplastic extramammary Paget’s disease: a case report and review of the literature. Ann Dermatol. 2011;23:226-230.
- Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
- Wilkinson EJ, Brown HM. Vulvar Paget disease of urothelial origin: a report of three cases and a proposed classification of vulvar Paget disease. Hum Pathol. 2002;33:549-554.
- Lam C, Funaro D. Extramammary Paget’s disease: summary of current knowledge. Dermatol Clin. 2010;28:807-826.
- Shiomi T, Yoshida Y, Shomori K, et al. Extramammary Paget’s disease: evaluation of the histopathological patterns of Paget cell proliferation in the epidermis. J Dermatol. 2011;38:1054-1057.
- Goldblum JR, Hart WR. Vulvar Paget’s disease: a clinicopathologic and immunohistochemical study of 19 cases. Am J Surg Pathol. 1997;21:1178-1187.
- Alhumaidi A. Practical immunohistochemistry of epithelial skin tumor. Indian J Dermatol Venerol Leprol. 2012;78:698-708.
- Battles O, Page D, Johnson J. Cytokeratins, CEA and mucin histochemistry in the diagnosis and characterization of extramammary Paget’s disease. Am J Clin Pathol. 1997;108:6-12.
- Kanitakis J. Mammary and extramammary Paget’s disease. J Eur Acad Dermatol Venereol. 2007;21:581-590.
- Krishna M. Diagnosis of metastatic neoplasms: an immunohistochemical approach. Arch Pathol Lab Med. 2010;134:207-215.
- Helm KF, Goellner JR, Peters MS. Immunohistochemical stain in extramammary Paget’s disease. Am J Dermatopathol. 1992;14:402-407.
- Liegl B, Horn L, Moinfar F. Androgen receptors are frequently expressed in mammary and extramammary Paget’s disease. Mod Pathol. 2005;18:1283-288.
- Ackerman AB. Focal acantholytic dyskeratosis. Arch Derm. 1972;106:702-706.
- Dittmer CJ, Hornemann A, Rose C, et al. Successful laser therapy of a papular acantholytic dyskeratosis of the vulva: case report and review of literature. Arch Gynecol Obstet. 2010;291:723-725.
- Roh MR, Choi YJ, Lee KG. Papular acantholytic dyskeratosis of the vulva. J Dermatol. 2009;36:427-429.
- Wong KT, Mihm MC Jr. Acantholytic dermatosis localized to genitalia and crural areas of male patients: a report of three cases. J Cutan Pathol. 1994;21:27-32.
- Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat Genet. 2000; 24:61-65.
- Elston DM. Malignant tumors of the epidermis. In: Elston DM, Ferringer T, eds. Requisites in Dermatology: Dermatopathology. Philadelphia, PA: Elsevier Limited; 2012:53-68.
- Fu JM, McCalmont T, Yu SS. Adenosquamous carcinoma of the skin: a case series. Arch Dermatol. 2009;145:1152-1158.
- Becker BA, Gaspari AA. Pemphigus vulgaris and vegetans. Dermatol Clin. 1993;11:429-452.
- Rados J. Autoimmune blistering diseases: histologic meaning. Clin Dermatol. 2011;29:377-388.
- Kohler S, Smoller BR. A case of extramammary Paget’s disease mimicking pemphigus vulgaris on histologic examination. Dermatology. 1997;195:54-56.
To the Editor:
Extramammary Paget disease (EMPD) is a rare intraepidermal neoplasm with glandular differentiation that is classically known as a mimicker of Bowen disease (squamous cell carcinoma in situ of the skin) due to their histologic similarities.1,2 However, acantholytic anaplastic EMPD (AAEMPD) is a rare variant that can pose a particularly difficult diagnostic challenge because of its histologic similarity to benign acantholytic disorders and other malignant neoplasms. Major histologic features suggestive of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The differential diagnosis of EMPD typically includes Bowen disease and pagetoid Bowen disease, but the acantholytic anaplastic variant more often is confused with intraepidermal acantholytic lesions such as acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, and acantholytic Bowen disease. Immunohistochemistry (IHC) studies to assist in the definitive diagnosis of AAEMPD are strongly advised because of these difficulties in diagnosis.4 Cases of EMPD with an acantholytic appearance have rarely been reported in the literature.5-7
A 78-year-old man with a history of arthritis, heart disease, hypertension, and gastrointestinal disease presented for evaluation of a tender lesion of the right genitocrural crease of 5 years’ duration. He had no history of cutaneous or internal malignancy. Previously the lesion had been treated by dermatology with a variety of topical products including antifungal and antibiotic creams with no improvement. Physical examination revealed a well-defined, 7×5-cm, tender, erythematous, macerated plaque on the right upper inner thigh adjacent to the scrotum with an odor possibly due to secondary infection (Figure 1).
plaque on the right upper inner thigh adjacent to the scrotum.
A biopsy of the lesion was performed, and the specimen was submitted for pathologic examination. Bacterial cultures taken at the time of biopsy revealed polybacterial colonization with Acinetobacter, Morganella, and mixed skin flora. The patient was treated with a 10-day course of oral sulfamethoxazole 800 mg and trimethoprim 160 mg twice daily once culture results returned. The biopsy results were communicated to the patient; however, he subsequently relocated, assumed care at another facility, and has since been lost to follow-up.
The biopsy specimen was examined grossly, serially sectioned, and submitted for routine processing with hematoxylin and eosin, periodic acid–Schiff, and Hale colloidal iron staining. Routine IHC was performed with antibodies to cytokeratin (CK) 7, CK20, carcinoembryonic antigen (CEA), pancytokeratin (CKAE1/AE3), and low- molecular-weight cytokeratin (LMWCK).
Pathologic examination of the biopsy showed prominent acanthosis of the epidermis composed of a proliferation of epithelial cells with associated full-thickness suprabasal acantholysis (Figure 2A). On inspection at higher magnification, the neoplastic cells demonstrated anaplasia as cytologic atypia with prominent and frequently multiple nucleoli, scant cytoplasm, and a high nuclear to cytoplasmic ratio (Figure 2B). There was a marked increase in mitotic activity with as many as 5 mitotic figures per high-power field. A fairly dense mixed inflammatory infiltrate comprised of lymphocytes, plasma cells, neutrophils, and eosinophils was present in the dermis. No fungal elements were observed on periodic acid–Schiff staining. The vast majority of tumor cells demonstrated moderate to abundant cytoplasmic mucin on Hale colloidal iron staining (Figure 3).
(H&E, original magnification ×400).
Immunohistochemistry staining of tumor cells was positive for CK7, CEA, pancytokeratin (CKAE1/AE3), and LMWCK. The tumor cells were negative for CK20. On the basis of the histopathologic and IHC findings, the patient was diagnosed with AAEMPD.
Extramammary Paget disease is a rare intraepidermal neoplasm with glandular differentiation. The most commonly involved sites are the anogenital areas including the vulvar, perianal, perineal, scrotal, and penile regions, as well as other areas rich in apocrine glands such as the axillae.8 Extramammary Paget disease most commonly originates as a primary intraepidermal neoplasm (type 1 EMPD), but an underlying malignant neoplasm that spreads intraepithelially is seen in a minority of cases (types 2 and 3 EMPD). In the vulva, type 1a refers to cutaneous noninvasive Paget disease, type 1b refers to dermal invasion of Paget disease, type 1c refers to vulvar adenocarcinoma–associated Paget disease, type 2 refers to rectal/anal adenocarcinoma–associated Paget disease, and type 3 refers to urogenital neoplasia–associated Paget disease.9
The acantholytic anaplastic variant of EMPD can be challenging to diagnose because of its similarities to many other lesions, including acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, Bowen disease, pagetoid Bowen disease, and acantholytic Bowen disease. Major histologic features of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The acantholytic anaplastic variant of EMPD can be differentiated from other diagnoses using IHC studies, with findings indicative of AAEMPD outlined below.
The proliferative neoplastic cell in EMPD is the Paget cell, which can be identified as a large round cell located in the epidermis with pale-staining cytoplasm, a large nucleus, and sometimes a prominent nucleolus. Paget cells can be distributed singly or in clusters, nests, or glandular structures within the epidermis and adjacent to adnexal structures.10 Extramammary Paget disease can have many patterns, including glandular, acantholysis-like, upper nest, tall nest, budding, and sheetlike.11
Immunohistochemically, Paget cells in EMPD typically express pancytokeratins (CKAE1/AE3), low-molecular-weight/simple epithelial type keratins (CK7, CAM 5.2), sweat gland antigens (epithelial membrane antigen, CEA, gross cystic disease fluid protein 15 [GCDFP15]), mucin 5AC (MUC5AC), and often androgen receptor.12-18 Paget cells contain cytoplasmic mucin and demonstrate prominent cytoplasmic staining with Hale colloidal iron.17 Paget cells typically do not express high-molecular-weight cytokeratin (eg, CK5/6), melanocytic antigens, estrogen receptor, or progesterone receptor.15,18
Immunohistochemical staining has been shown to differ between primary cutaneous (type 1) and secondary (types 2 and 3) EMPD. Primary cutaneous EMPD typically expresses sweat gland markers (CK7+, CK20−, GCDFP15+). Secondary EMPD typically expresses an endodermal phenotype (CK7+, CK20+, GCDFP15−).12
Acantholytic dyskeratosis of the genitocrural area is a rare lesion included in the spectrum of focal acantholytic dyskeratoses described by Ackerman.19 It also has been referred to as papular acantholytic dyskeratosis of the vulva, though histologically similar lesions also have been reported in men.20-22 Histologically, acantholytic dyskeratosis of the genitocrural area has prominent acantholysis and dyskeratosis with corps ronds and grains.19 Familial benign pemphigus (Hailey-Hailey disease) is caused by mutations of the ATP2C1 gene, which encodes for a secretory pathway Ca2+/Mn2+-ATPase pump type 1 (SPCA1) in the Golgi apparatus in keratinocytes.23 Familial benign pemphigus has a histologic appearance similar to acantholytic dyskeratosis of the genitocrural area, but a positive family history of familial benign pemphigus can be used to differentiate the 2 entities from each other due to the autosomal-dominant inheritance pattern of familial benign pemphigus. Both of these disorders can appear similar to AAEMPD because of their extensive intraepidermal acantholysis, but they differ in the lack of Paget cells, intraepidermal atypia, and increased mitotic activity.
Acantholytic Bowen disease is a histologic variant that can be difficult to distinguish from AAEMPD on hematoxylin and eosin–stained sections because of their similar histologic features but can be differentiated by IHC stains.5 Acantholytic Bowen disease expresses high-molecular-weight cytokeratin (eg, CK5/6) but is negative for CK7, CAM 5.2, and CEA. Extramammary Paget disease generally has the opposite pattern: positive staining for CK7, CAM 5.2, and CEA, but negative for high-molecular-weight cytokeratin.13,14,24
Primary cutaneous adenosquamous carcinoma is a rare malignancy of squamous and glandular differentiation known for being locally aggressive and metastatic.25 Histologically, cutaneous adenosquamous carcinoma shows infiltrating nests of neoplastic cells with both squamous and glandular features. It differs notably from AAEMPD in that cutaneous adenosquamous carcinomas tend to arise in the head and arm regions, and their histologic morphology is different. The IHC profiles are similar, with positive staining for CEA, CK7, and mucin; however, they differ in that AAEMPD is negative for high-molecular-weight keratin while cutaneous adenosquamous carcinoma is positive.25
Verrucous carcinoma is an uncommon variant of squamous cell carcinoma with well-differentiated keratinocytes and a blunt pushing border.24 Similar to AAEMPD, this neoplasm can arise in the genital and perineal areas; however, the 2 entities differ considerably in morphology on histologic examination.
Pemphigus vulgaris is an autoimmune intraepidermal blistering disorder of the skin and mucous membranes of which pemphigus vegetans is a subtype.26,27 Pemphigus vulgaris is another diagnosis that can possibly be mimicked by AAEMPD.28 Histologic features of pemphigus vulgaris include intraepidermal acantholysis of keratinocytes immediately above the basal layer of the epidermis. Pemphigus vegetans is similar with the addition of papillomatosis, hyperkeratosis, and an eosinophilic infiltrate.26,27 Immunofluorescence typically demonstrates intercellular C3 and IgG deposits.26 These diseases mimic AAEMPD histologically but differ in their relative lack of atypia and Paget cells.
In summary, we report a case of AAEMPD in a 78-year-old man in whom routine histologic specimens showed marked intraepidermal acantholysis and atypical tumor cells with increased mitoses. The latter finding prompted IHC studies that revealed positive CK7, CEA, pancytokeratin, and LMWCK staining with negative CK20 staining. Hale colloidal iron staining showed moderate to abundant cytoplasmic mucin. The patient was diagnosed with AAEMPD. It is imperative to maintain clinical suspicion for AAEMPD and to examine acantholytic disorders with scrutiny. When there is evidence of atypia or mitoses, use of IHC stains can assist in fully characterizing the lesion.
To the Editor:
Extramammary Paget disease (EMPD) is a rare intraepidermal neoplasm with glandular differentiation that is classically known as a mimicker of Bowen disease (squamous cell carcinoma in situ of the skin) due to their histologic similarities.1,2 However, acantholytic anaplastic EMPD (AAEMPD) is a rare variant that can pose a particularly difficult diagnostic challenge because of its histologic similarity to benign acantholytic disorders and other malignant neoplasms. Major histologic features suggestive of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The differential diagnosis of EMPD typically includes Bowen disease and pagetoid Bowen disease, but the acantholytic anaplastic variant more often is confused with intraepidermal acantholytic lesions such as acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, and acantholytic Bowen disease. Immunohistochemistry (IHC) studies to assist in the definitive diagnosis of AAEMPD are strongly advised because of these difficulties in diagnosis.4 Cases of EMPD with an acantholytic appearance have rarely been reported in the literature.5-7
A 78-year-old man with a history of arthritis, heart disease, hypertension, and gastrointestinal disease presented for evaluation of a tender lesion of the right genitocrural crease of 5 years’ duration. He had no history of cutaneous or internal malignancy. Previously the lesion had been treated by dermatology with a variety of topical products including antifungal and antibiotic creams with no improvement. Physical examination revealed a well-defined, 7×5-cm, tender, erythematous, macerated plaque on the right upper inner thigh adjacent to the scrotum with an odor possibly due to secondary infection (Figure 1).
plaque on the right upper inner thigh adjacent to the scrotum.
A biopsy of the lesion was performed, and the specimen was submitted for pathologic examination. Bacterial cultures taken at the time of biopsy revealed polybacterial colonization with Acinetobacter, Morganella, and mixed skin flora. The patient was treated with a 10-day course of oral sulfamethoxazole 800 mg and trimethoprim 160 mg twice daily once culture results returned. The biopsy results were communicated to the patient; however, he subsequently relocated, assumed care at another facility, and has since been lost to follow-up.
The biopsy specimen was examined grossly, serially sectioned, and submitted for routine processing with hematoxylin and eosin, periodic acid–Schiff, and Hale colloidal iron staining. Routine IHC was performed with antibodies to cytokeratin (CK) 7, CK20, carcinoembryonic antigen (CEA), pancytokeratin (CKAE1/AE3), and low- molecular-weight cytokeratin (LMWCK).
Pathologic examination of the biopsy showed prominent acanthosis of the epidermis composed of a proliferation of epithelial cells with associated full-thickness suprabasal acantholysis (Figure 2A). On inspection at higher magnification, the neoplastic cells demonstrated anaplasia as cytologic atypia with prominent and frequently multiple nucleoli, scant cytoplasm, and a high nuclear to cytoplasmic ratio (Figure 2B). There was a marked increase in mitotic activity with as many as 5 mitotic figures per high-power field. A fairly dense mixed inflammatory infiltrate comprised of lymphocytes, plasma cells, neutrophils, and eosinophils was present in the dermis. No fungal elements were observed on periodic acid–Schiff staining. The vast majority of tumor cells demonstrated moderate to abundant cytoplasmic mucin on Hale colloidal iron staining (Figure 3).
(H&E, original magnification ×400).
Immunohistochemistry staining of tumor cells was positive for CK7, CEA, pancytokeratin (CKAE1/AE3), and LMWCK. The tumor cells were negative for CK20. On the basis of the histopathologic and IHC findings, the patient was diagnosed with AAEMPD.
Extramammary Paget disease is a rare intraepidermal neoplasm with glandular differentiation. The most commonly involved sites are the anogenital areas including the vulvar, perianal, perineal, scrotal, and penile regions, as well as other areas rich in apocrine glands such as the axillae.8 Extramammary Paget disease most commonly originates as a primary intraepidermal neoplasm (type 1 EMPD), but an underlying malignant neoplasm that spreads intraepithelially is seen in a minority of cases (types 2 and 3 EMPD). In the vulva, type 1a refers to cutaneous noninvasive Paget disease, type 1b refers to dermal invasion of Paget disease, type 1c refers to vulvar adenocarcinoma–associated Paget disease, type 2 refers to rectal/anal adenocarcinoma–associated Paget disease, and type 3 refers to urogenital neoplasia–associated Paget disease.9
The acantholytic anaplastic variant of EMPD can be challenging to diagnose because of its similarities to many other lesions, including acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, Bowen disease, pagetoid Bowen disease, and acantholytic Bowen disease. Major histologic features of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The acantholytic anaplastic variant of EMPD can be differentiated from other diagnoses using IHC studies, with findings indicative of AAEMPD outlined below.
The proliferative neoplastic cell in EMPD is the Paget cell, which can be identified as a large round cell located in the epidermis with pale-staining cytoplasm, a large nucleus, and sometimes a prominent nucleolus. Paget cells can be distributed singly or in clusters, nests, or glandular structures within the epidermis and adjacent to adnexal structures.10 Extramammary Paget disease can have many patterns, including glandular, acantholysis-like, upper nest, tall nest, budding, and sheetlike.11
Immunohistochemically, Paget cells in EMPD typically express pancytokeratins (CKAE1/AE3), low-molecular-weight/simple epithelial type keratins (CK7, CAM 5.2), sweat gland antigens (epithelial membrane antigen, CEA, gross cystic disease fluid protein 15 [GCDFP15]), mucin 5AC (MUC5AC), and often androgen receptor.12-18 Paget cells contain cytoplasmic mucin and demonstrate prominent cytoplasmic staining with Hale colloidal iron.17 Paget cells typically do not express high-molecular-weight cytokeratin (eg, CK5/6), melanocytic antigens, estrogen receptor, or progesterone receptor.15,18
Immunohistochemical staining has been shown to differ between primary cutaneous (type 1) and secondary (types 2 and 3) EMPD. Primary cutaneous EMPD typically expresses sweat gland markers (CK7+, CK20−, GCDFP15+). Secondary EMPD typically expresses an endodermal phenotype (CK7+, CK20+, GCDFP15−).12
Acantholytic dyskeratosis of the genitocrural area is a rare lesion included in the spectrum of focal acantholytic dyskeratoses described by Ackerman.19 It also has been referred to as papular acantholytic dyskeratosis of the vulva, though histologically similar lesions also have been reported in men.20-22 Histologically, acantholytic dyskeratosis of the genitocrural area has prominent acantholysis and dyskeratosis with corps ronds and grains.19 Familial benign pemphigus (Hailey-Hailey disease) is caused by mutations of the ATP2C1 gene, which encodes for a secretory pathway Ca2+/Mn2+-ATPase pump type 1 (SPCA1) in the Golgi apparatus in keratinocytes.23 Familial benign pemphigus has a histologic appearance similar to acantholytic dyskeratosis of the genitocrural area, but a positive family history of familial benign pemphigus can be used to differentiate the 2 entities from each other due to the autosomal-dominant inheritance pattern of familial benign pemphigus. Both of these disorders can appear similar to AAEMPD because of their extensive intraepidermal acantholysis, but they differ in the lack of Paget cells, intraepidermal atypia, and increased mitotic activity.
Acantholytic Bowen disease is a histologic variant that can be difficult to distinguish from AAEMPD on hematoxylin and eosin–stained sections because of their similar histologic features but can be differentiated by IHC stains.5 Acantholytic Bowen disease expresses high-molecular-weight cytokeratin (eg, CK5/6) but is negative for CK7, CAM 5.2, and CEA. Extramammary Paget disease generally has the opposite pattern: positive staining for CK7, CAM 5.2, and CEA, but negative for high-molecular-weight cytokeratin.13,14,24
Primary cutaneous adenosquamous carcinoma is a rare malignancy of squamous and glandular differentiation known for being locally aggressive and metastatic.25 Histologically, cutaneous adenosquamous carcinoma shows infiltrating nests of neoplastic cells with both squamous and glandular features. It differs notably from AAEMPD in that cutaneous adenosquamous carcinomas tend to arise in the head and arm regions, and their histologic morphology is different. The IHC profiles are similar, with positive staining for CEA, CK7, and mucin; however, they differ in that AAEMPD is negative for high-molecular-weight keratin while cutaneous adenosquamous carcinoma is positive.25
Verrucous carcinoma is an uncommon variant of squamous cell carcinoma with well-differentiated keratinocytes and a blunt pushing border.24 Similar to AAEMPD, this neoplasm can arise in the genital and perineal areas; however, the 2 entities differ considerably in morphology on histologic examination.
Pemphigus vulgaris is an autoimmune intraepidermal blistering disorder of the skin and mucous membranes of which pemphigus vegetans is a subtype.26,27 Pemphigus vulgaris is another diagnosis that can possibly be mimicked by AAEMPD.28 Histologic features of pemphigus vulgaris include intraepidermal acantholysis of keratinocytes immediately above the basal layer of the epidermis. Pemphigus vegetans is similar with the addition of papillomatosis, hyperkeratosis, and an eosinophilic infiltrate.26,27 Immunofluorescence typically demonstrates intercellular C3 and IgG deposits.26 These diseases mimic AAEMPD histologically but differ in their relative lack of atypia and Paget cells.
In summary, we report a case of AAEMPD in a 78-year-old man in whom routine histologic specimens showed marked intraepidermal acantholysis and atypical tumor cells with increased mitoses. The latter finding prompted IHC studies that revealed positive CK7, CEA, pancytokeratin, and LMWCK staining with negative CK20 staining. Hale colloidal iron staining showed moderate to abundant cytoplasmic mucin. The patient was diagnosed with AAEMPD. It is imperative to maintain clinical suspicion for AAEMPD and to examine acantholytic disorders with scrutiny. When there is evidence of atypia or mitoses, use of IHC stains can assist in fully characterizing the lesion.
- Bowen JT. Precancerous dermatosis: a study of two cases of chronic atypical epithelial proliferation. J Cutan Dis. 1912;30:241-255.
- Jones RE Jr, Austin C, Ackerman AB. Extramammary Paget’s disease: a critical reexamination. Am J Dermatopathol. 1979;1:101-132.
- Rayne SC, Santa Cruz DJ. Anaplastic Paget’s disease. Am J Surg Pathol. 1992;16:1085-1091.
- Wang EC, Kwah YC, Tan WP, et al. Extramammary Paget disease: immunohistochemistry is critical to distinguish potential mimickers. Dermatol Online J. 2012;18:4.
- Du X, Yin X, Zhou N, et al. Extramammary Paget’s disease mimicking acantholytic squamous cell carcinoma in situ: a case report. J Cutan Pathol. 2010;37:683.
- Mobini N. Acantholytic anaplastic Paget’s disease. J Cutan Pathol. 2009;36:374-380.
- Oh YJ, Lew BL, Sim WY. Acantholytic anaplastic extramammary Paget’s disease: a case report and review of the literature. Ann Dermatol. 2011;23:226-230.
- Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
- Wilkinson EJ, Brown HM. Vulvar Paget disease of urothelial origin: a report of three cases and a proposed classification of vulvar Paget disease. Hum Pathol. 2002;33:549-554.
- Lam C, Funaro D. Extramammary Paget’s disease: summary of current knowledge. Dermatol Clin. 2010;28:807-826.
- Shiomi T, Yoshida Y, Shomori K, et al. Extramammary Paget’s disease: evaluation of the histopathological patterns of Paget cell proliferation in the epidermis. J Dermatol. 2011;38:1054-1057.
- Goldblum JR, Hart WR. Vulvar Paget’s disease: a clinicopathologic and immunohistochemical study of 19 cases. Am J Surg Pathol. 1997;21:1178-1187.
- Alhumaidi A. Practical immunohistochemistry of epithelial skin tumor. Indian J Dermatol Venerol Leprol. 2012;78:698-708.
- Battles O, Page D, Johnson J. Cytokeratins, CEA and mucin histochemistry in the diagnosis and characterization of extramammary Paget’s disease. Am J Clin Pathol. 1997;108:6-12.
- Kanitakis J. Mammary and extramammary Paget’s disease. J Eur Acad Dermatol Venereol. 2007;21:581-590.
- Krishna M. Diagnosis of metastatic neoplasms: an immunohistochemical approach. Arch Pathol Lab Med. 2010;134:207-215.
- Helm KF, Goellner JR, Peters MS. Immunohistochemical stain in extramammary Paget’s disease. Am J Dermatopathol. 1992;14:402-407.
- Liegl B, Horn L, Moinfar F. Androgen receptors are frequently expressed in mammary and extramammary Paget’s disease. Mod Pathol. 2005;18:1283-288.
- Ackerman AB. Focal acantholytic dyskeratosis. Arch Derm. 1972;106:702-706.
- Dittmer CJ, Hornemann A, Rose C, et al. Successful laser therapy of a papular acantholytic dyskeratosis of the vulva: case report and review of literature. Arch Gynecol Obstet. 2010;291:723-725.
- Roh MR, Choi YJ, Lee KG. Papular acantholytic dyskeratosis of the vulva. J Dermatol. 2009;36:427-429.
- Wong KT, Mihm MC Jr. Acantholytic dermatosis localized to genitalia and crural areas of male patients: a report of three cases. J Cutan Pathol. 1994;21:27-32.
- Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat Genet. 2000; 24:61-65.
- Elston DM. Malignant tumors of the epidermis. In: Elston DM, Ferringer T, eds. Requisites in Dermatology: Dermatopathology. Philadelphia, PA: Elsevier Limited; 2012:53-68.
- Fu JM, McCalmont T, Yu SS. Adenosquamous carcinoma of the skin: a case series. Arch Dermatol. 2009;145:1152-1158.
- Becker BA, Gaspari AA. Pemphigus vulgaris and vegetans. Dermatol Clin. 1993;11:429-452.
- Rados J. Autoimmune blistering diseases: histologic meaning. Clin Dermatol. 2011;29:377-388.
- Kohler S, Smoller BR. A case of extramammary Paget’s disease mimicking pemphigus vulgaris on histologic examination. Dermatology. 1997;195:54-56.
- Bowen JT. Precancerous dermatosis: a study of two cases of chronic atypical epithelial proliferation. J Cutan Dis. 1912;30:241-255.
- Jones RE Jr, Austin C, Ackerman AB. Extramammary Paget’s disease: a critical reexamination. Am J Dermatopathol. 1979;1:101-132.
- Rayne SC, Santa Cruz DJ. Anaplastic Paget’s disease. Am J Surg Pathol. 1992;16:1085-1091.
- Wang EC, Kwah YC, Tan WP, et al. Extramammary Paget disease: immunohistochemistry is critical to distinguish potential mimickers. Dermatol Online J. 2012;18:4.
- Du X, Yin X, Zhou N, et al. Extramammary Paget’s disease mimicking acantholytic squamous cell carcinoma in situ: a case report. J Cutan Pathol. 2010;37:683.
- Mobini N. Acantholytic anaplastic Paget’s disease. J Cutan Pathol. 2009;36:374-380.
- Oh YJ, Lew BL, Sim WY. Acantholytic anaplastic extramammary Paget’s disease: a case report and review of the literature. Ann Dermatol. 2011;23:226-230.
- Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
- Wilkinson EJ, Brown HM. Vulvar Paget disease of urothelial origin: a report of three cases and a proposed classification of vulvar Paget disease. Hum Pathol. 2002;33:549-554.
- Lam C, Funaro D. Extramammary Paget’s disease: summary of current knowledge. Dermatol Clin. 2010;28:807-826.
- Shiomi T, Yoshida Y, Shomori K, et al. Extramammary Paget’s disease: evaluation of the histopathological patterns of Paget cell proliferation in the epidermis. J Dermatol. 2011;38:1054-1057.
- Goldblum JR, Hart WR. Vulvar Paget’s disease: a clinicopathologic and immunohistochemical study of 19 cases. Am J Surg Pathol. 1997;21:1178-1187.
- Alhumaidi A. Practical immunohistochemistry of epithelial skin tumor. Indian J Dermatol Venerol Leprol. 2012;78:698-708.
- Battles O, Page D, Johnson J. Cytokeratins, CEA and mucin histochemistry in the diagnosis and characterization of extramammary Paget’s disease. Am J Clin Pathol. 1997;108:6-12.
- Kanitakis J. Mammary and extramammary Paget’s disease. J Eur Acad Dermatol Venereol. 2007;21:581-590.
- Krishna M. Diagnosis of metastatic neoplasms: an immunohistochemical approach. Arch Pathol Lab Med. 2010;134:207-215.
- Helm KF, Goellner JR, Peters MS. Immunohistochemical stain in extramammary Paget’s disease. Am J Dermatopathol. 1992;14:402-407.
- Liegl B, Horn L, Moinfar F. Androgen receptors are frequently expressed in mammary and extramammary Paget’s disease. Mod Pathol. 2005;18:1283-288.
- Ackerman AB. Focal acantholytic dyskeratosis. Arch Derm. 1972;106:702-706.
- Dittmer CJ, Hornemann A, Rose C, et al. Successful laser therapy of a papular acantholytic dyskeratosis of the vulva: case report and review of literature. Arch Gynecol Obstet. 2010;291:723-725.
- Roh MR, Choi YJ, Lee KG. Papular acantholytic dyskeratosis of the vulva. J Dermatol. 2009;36:427-429.
- Wong KT, Mihm MC Jr. Acantholytic dermatosis localized to genitalia and crural areas of male patients: a report of three cases. J Cutan Pathol. 1994;21:27-32.
- Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat Genet. 2000; 24:61-65.
- Elston DM. Malignant tumors of the epidermis. In: Elston DM, Ferringer T, eds. Requisites in Dermatology: Dermatopathology. Philadelphia, PA: Elsevier Limited; 2012:53-68.
- Fu JM, McCalmont T, Yu SS. Adenosquamous carcinoma of the skin: a case series. Arch Dermatol. 2009;145:1152-1158.
- Becker BA, Gaspari AA. Pemphigus vulgaris and vegetans. Dermatol Clin. 1993;11:429-452.
- Rados J. Autoimmune blistering diseases: histologic meaning. Clin Dermatol. 2011;29:377-388.
- Kohler S, Smoller BR. A case of extramammary Paget’s disease mimicking pemphigus vulgaris on histologic examination. Dermatology. 1997;195:54-56.
Practice Points
- The acantholytic anaplastic variant of extramammary Paget disease (EMPD) can be mimicked by many other entities including Bowen disease, acantholytic dyskeratosis of the genitocrural area, and pemphigus vulgaris.
- A good immunohistochemical panel to evaluate for EMPD includes cytokeratin (CK) 7, pancytokeratin (CKAE1/AE3), CK20, and carcinoembryonic antigen.
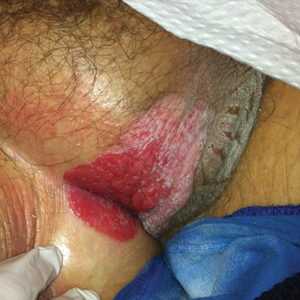
Multicenter trial backs pirfenidone for unclassifiable interstitial lung disease
MADRID – according to results of a late breaker, placebo-controlled, multinational trial presented at the annual congress of the European Respiratory Society.

For preservation of lung function as monitored with forced vital capacity (FVC), pirfenidone provided a large and highly statistically significant advantage over placebo in a phase 2 trial that randomized 253 uILD patients to 2,403 mg pirfenidone or placebo, according to Toby M. Maher, MD, head of the Fibrosis Research Group for the National Heart and Lung Institute, Imperial College, London.
At 24 weeks, FVC lung function declined by just 17.8 mL in the pirfenidone group vs. 113 mL in the placebo group (P = .002). The results, published simultaneously with Dr. Maher’s ERS presentation in The Lancet Respiratory Medicine, are particularly encouraging because there are no currently approved treatments for uILD, according to Dr. Maher.
However, the data from this study, even though it was double blind and involved 70 participating centers in 14 countries, come with an asterisk. The significant FVC advantage was documented with in-hospital measurements, but this was a secondary, not the primary, endpoint. Measurements with hand-held spirometry, which was the primary endpoint, proved to be uninterpretable due to intra-individual variability.
“We had hoped that daily home spirometry would give us more information of the patient’s trajectory over time,” said Dr. Maher, who blames himself for selecting hand-held device measurements as the primary endpoint. In the end, the variability in the home hand-held spirometry data prevented the planned statistical testing.
“There were issues with the hand-held devices we had not anticipated,” Dr. Maher reported. However, hospital-based measurement, which has long been the “regulatory standard” in ILD trials “supports the conclusion that pirfenidone was effective.”
The conclusion is also supported by other secondary outcomes and analyses. For example, the categorical declines in FVC of greater than 5% (37.0% vs. 58.7%; P = .001) and greater than 10% (14.2% vs. 27.9%; P = .011) both favored pirfenidone. There were no between-group differences in progression-free survival at 24 weeks, but events were low in both study arms over this time period.
There was evidence of functional benefit for pirfenidone relative to placebo, such as a smaller decline in the 6-minute walk test (–2 vs. –26.7 M, P = .04). Treatment favoring pirfenidone over placebo was observed across subgroups defined by age, gender, baseline lung function, and presence or absence of interstitial pneumonia with autoimmune features.
Pirfenidone was generally well tolerated with side effects similar to those reported in other studies. The rate of treatment-related discontinuation was 12.6% on pirfenidone versus 0.8% on placebo. The most frequent adverse events, all of which were more common in the pirfenidone group, were gastrointestinal complaints (47.2% vs. 25.8%), rash (10.2% vs. 7.3%), and dizziness (7.9% vs. 0.8%). Rates of photosensitivity were higher in the experimental arm (7.9% vs. 1.8%), but low relative to previous studies, potentially because of greater emphasis on sun protection, Dr. Maher reported.
About 10%-15% of patients with ILD have an unclassifiable type, he noted. Although it is possible for uILD to be a missed diagnosis of an established ILD type, Dr. Maher reported that participating centers for this study were specifically selected for their expertise in ILD. He noted that more than 45% of patients were deemed uILD on the basis of biopsy.
The ERS-invited discussant of this trial, Martin Kolb, MD, professor of respirology, McMaster University, Hamilton, Ont., called the data “strong.” He suggested the data are particularly encouraging in the context of the lack of approved therapies for uILD.
Despite the fact that benefit of pirfenidone was not established on the primary endpoint, Dr. Maher contended that this is a positive study that can be used to design future investigations. “When we use the normal standard endpoint for the study, we see a clear benefit of pirfenidone over placebo.”
Dr. Maher reported no potential conflicts of interest.
SOURCE: Maher TM et al. Lancet Respir Med. 2019 Sep 29. doi: 10.1016/S2213-2600(19)30341-8.
MADRID – according to results of a late breaker, placebo-controlled, multinational trial presented at the annual congress of the European Respiratory Society.
For preservation of lung function as monitored with forced vital capacity (FVC), pirfenidone provided a large and highly statistically significant advantage over placebo in a phase 2 trial that randomized 253 uILD patients to 2,403 mg pirfenidone or placebo, according to Toby M. Maher, MD, head of the Fibrosis Research Group for the National Heart and Lung Institute, Imperial College, London.
At 24 weeks, FVC lung function declined by just 17.8 mL in the pirfenidone group vs. 113 mL in the placebo group (P = .002). The results, published simultaneously with Dr. Maher’s ERS presentation in The Lancet Respiratory Medicine, are particularly encouraging because there are no currently approved treatments for uILD, according to Dr. Maher.
However, the data from this study, even though it was double blind and involved 70 participating centers in 14 countries, come with an asterisk. The significant FVC advantage was documented with in-hospital measurements, but this was a secondary, not the primary, endpoint. Measurements with hand-held spirometry, which was the primary endpoint, proved to be uninterpretable due to intra-individual variability.
“We had hoped that daily home spirometry would give us more information of the patient’s trajectory over time,” said Dr. Maher, who blames himself for selecting hand-held device measurements as the primary endpoint. In the end, the variability in the home hand-held spirometry data prevented the planned statistical testing.
“There were issues with the hand-held devices we had not anticipated,” Dr. Maher reported. However, hospital-based measurement, which has long been the “regulatory standard” in ILD trials “supports the conclusion that pirfenidone was effective.”
The conclusion is also supported by other secondary outcomes and analyses. For example, the categorical declines in FVC of greater than 5% (37.0% vs. 58.7%; P = .001) and greater than 10% (14.2% vs. 27.9%; P = .011) both favored pirfenidone. There were no between-group differences in progression-free survival at 24 weeks, but events were low in both study arms over this time period.
There was evidence of functional benefit for pirfenidone relative to placebo, such as a smaller decline in the 6-minute walk test (–2 vs. –26.7 M, P = .04). Treatment favoring pirfenidone over placebo was observed across subgroups defined by age, gender, baseline lung function, and presence or absence of interstitial pneumonia with autoimmune features.
Pirfenidone was generally well tolerated with side effects similar to those reported in other studies. The rate of treatment-related discontinuation was 12.6% on pirfenidone versus 0.8% on placebo. The most frequent adverse events, all of which were more common in the pirfenidone group, were gastrointestinal complaints (47.2% vs. 25.8%), rash (10.2% vs. 7.3%), and dizziness (7.9% vs. 0.8%). Rates of photosensitivity were higher in the experimental arm (7.9% vs. 1.8%), but low relative to previous studies, potentially because of greater emphasis on sun protection, Dr. Maher reported.
About 10%-15% of patients with ILD have an unclassifiable type, he noted. Although it is possible for uILD to be a missed diagnosis of an established ILD type, Dr. Maher reported that participating centers for this study were specifically selected for their expertise in ILD. He noted that more than 45% of patients were deemed uILD on the basis of biopsy.
The ERS-invited discussant of this trial, Martin Kolb, MD, professor of respirology, McMaster University, Hamilton, Ont., called the data “strong.” He suggested the data are particularly encouraging in the context of the lack of approved therapies for uILD.
Despite the fact that benefit of pirfenidone was not established on the primary endpoint, Dr. Maher contended that this is a positive study that can be used to design future investigations. “When we use the normal standard endpoint for the study, we see a clear benefit of pirfenidone over placebo.”
Dr. Maher reported no potential conflicts of interest.
SOURCE: Maher TM et al. Lancet Respir Med. 2019 Sep 29. doi: 10.1016/S2213-2600(19)30341-8.
MADRID – according to results of a late breaker, placebo-controlled, multinational trial presented at the annual congress of the European Respiratory Society.
For preservation of lung function as monitored with forced vital capacity (FVC), pirfenidone provided a large and highly statistically significant advantage over placebo in a phase 2 trial that randomized 253 uILD patients to 2,403 mg pirfenidone or placebo, according to Toby M. Maher, MD, head of the Fibrosis Research Group for the National Heart and Lung Institute, Imperial College, London.
At 24 weeks, FVC lung function declined by just 17.8 mL in the pirfenidone group vs. 113 mL in the placebo group (P = .002). The results, published simultaneously with Dr. Maher’s ERS presentation in The Lancet Respiratory Medicine, are particularly encouraging because there are no currently approved treatments for uILD, according to Dr. Maher.
However, the data from this study, even though it was double blind and involved 70 participating centers in 14 countries, come with an asterisk. The significant FVC advantage was documented with in-hospital measurements, but this was a secondary, not the primary, endpoint. Measurements with hand-held spirometry, which was the primary endpoint, proved to be uninterpretable due to intra-individual variability.
“We had hoped that daily home spirometry would give us more information of the patient’s trajectory over time,” said Dr. Maher, who blames himself for selecting hand-held device measurements as the primary endpoint. In the end, the variability in the home hand-held spirometry data prevented the planned statistical testing.
“There were issues with the hand-held devices we had not anticipated,” Dr. Maher reported. However, hospital-based measurement, which has long been the “regulatory standard” in ILD trials “supports the conclusion that pirfenidone was effective.”
The conclusion is also supported by other secondary outcomes and analyses. For example, the categorical declines in FVC of greater than 5% (37.0% vs. 58.7%; P = .001) and greater than 10% (14.2% vs. 27.9%; P = .011) both favored pirfenidone. There were no between-group differences in progression-free survival at 24 weeks, but events were low in both study arms over this time period.
There was evidence of functional benefit for pirfenidone relative to placebo, such as a smaller decline in the 6-minute walk test (–2 vs. –26.7 M, P = .04). Treatment favoring pirfenidone over placebo was observed across subgroups defined by age, gender, baseline lung function, and presence or absence of interstitial pneumonia with autoimmune features.
Pirfenidone was generally well tolerated with side effects similar to those reported in other studies. The rate of treatment-related discontinuation was 12.6% on pirfenidone versus 0.8% on placebo. The most frequent adverse events, all of which were more common in the pirfenidone group, were gastrointestinal complaints (47.2% vs. 25.8%), rash (10.2% vs. 7.3%), and dizziness (7.9% vs. 0.8%). Rates of photosensitivity were higher in the experimental arm (7.9% vs. 1.8%), but low relative to previous studies, potentially because of greater emphasis on sun protection, Dr. Maher reported.
About 10%-15% of patients with ILD have an unclassifiable type, he noted. Although it is possible for uILD to be a missed diagnosis of an established ILD type, Dr. Maher reported that participating centers for this study were specifically selected for their expertise in ILD. He noted that more than 45% of patients were deemed uILD on the basis of biopsy.
The ERS-invited discussant of this trial, Martin Kolb, MD, professor of respirology, McMaster University, Hamilton, Ont., called the data “strong.” He suggested the data are particularly encouraging in the context of the lack of approved therapies for uILD.
Despite the fact that benefit of pirfenidone was not established on the primary endpoint, Dr. Maher contended that this is a positive study that can be used to design future investigations. “When we use the normal standard endpoint for the study, we see a clear benefit of pirfenidone over placebo.”
Dr. Maher reported no potential conflicts of interest.
SOURCE: Maher TM et al. Lancet Respir Med. 2019 Sep 29. doi: 10.1016/S2213-2600(19)30341-8.
REPORTING FROM ERS 2019
Barber’s Sinus Between the Toes of a Female Hairdresser
To the Editor:
Barber’s sinus, or interdigital pilonidal sinus, is an occupational dermatosis with a pathognomonic clinical picture. Nearly all reports of barber’s sinus in the literature have involved the hands of male barbers and hairdressers. We present an uncommon case of barber’s sinus between the toes of a female hairdresser. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. Clinicians should advise patients with an occupational risk of barber’s sinus to wear protective footwear and maintain hygiene in the interdigital spaces.
A 23-year-old female hairdresser was referred to our outpatient dermatology clinic by general surgery for evaluation of an asymptomatic interdigital toe lesion of several months’ duration. She was otherwise healthy. Physical examination revealed a 3-mm sinus in the interdigital web space between the fourth and fifth digits of the left foot, creating a partial fistula terminating in an umbilicated pink papule on the dorsal aspect of the interdigital space (Figure). While at work, the patient reported that she usually wore open-toed flip-flops. A diagnosis of barber’s sinus was made clinically. She returned for follow-up to the referring surgeon within 2 months and was offered surgical debridement, but the patient declined treatment, instead opting to wait and monitor for any potential complications. The lesion showed no change in clinical appearance and remained asymptomatic.
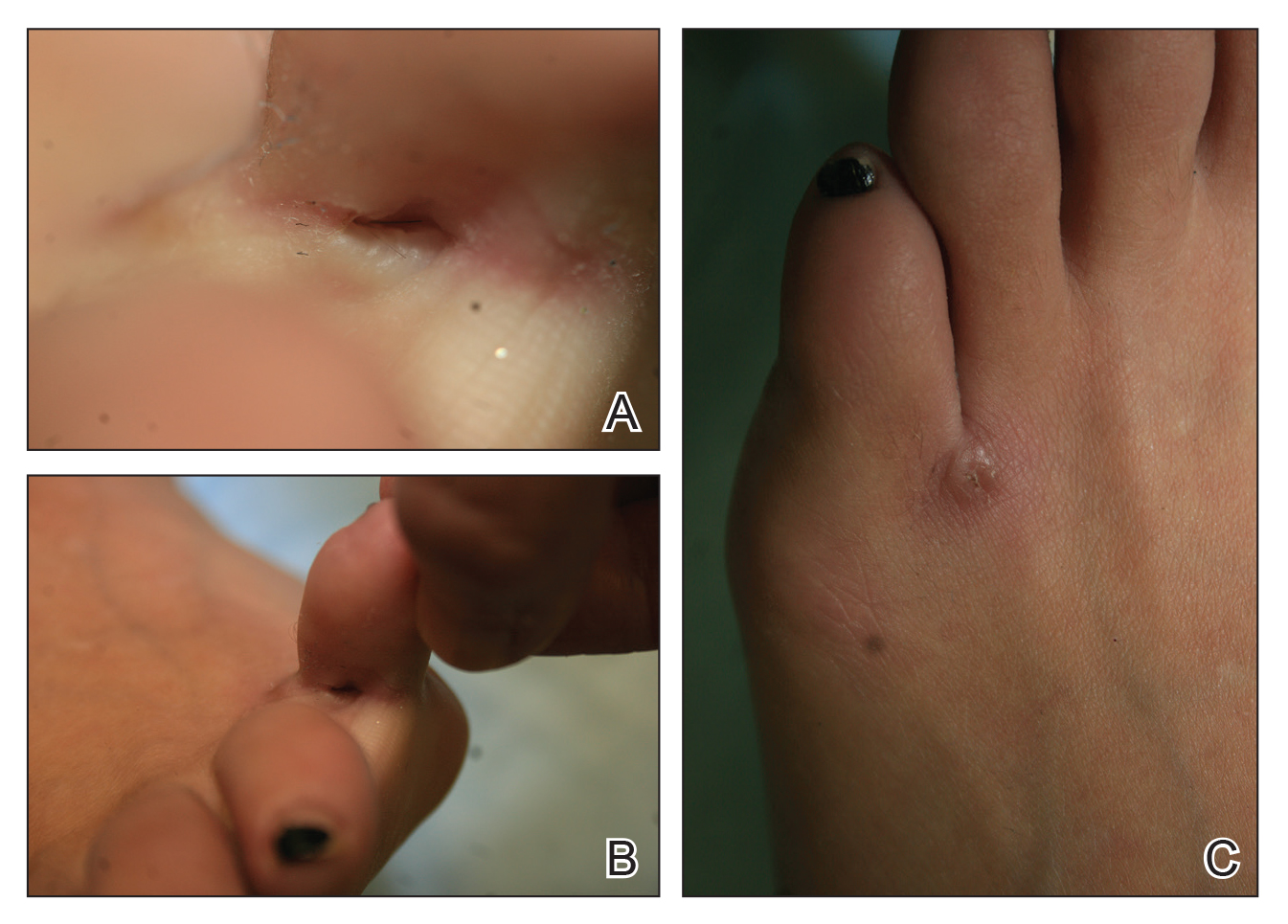
Barber’s sinus is caused by sharp fragments of clipped hair that penetrate the fragile interdigital skin and cause a foreign-body reaction. Males are almost exclusively contributory to the reported cases of barber’s sinus in the literature.1,2
The clinical picture of barber’s sinus is pathognomonic, as demonstrated in our case. Other potential diagnoses to consider include atypical mycobacterial infection, deep fungal infection, other foreign-body granuloma, and erosio interdigitalis blastomycetica. Although thorough removal of embedded hair fragments may be curative, most cases require surgical excision, often by curette, and subsequent skin closure. Pathology shows a foreign-body granulomatous reaction to hair fragments. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. This lesion is preventable by maintaining hygiene of the interdigital spaces, use of barrier creams, and wearing protective footwear.3,4
- Efthimiadis C, Kosmidis C, Anthimidis G, et al. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational disease: a case report. Cases J. 2008;1:214.
- O’Neill AC, Purcell EM, Regan PJ. Interdigital pilonidal sinus of the foot [published online May 31, 2009]. Foot (Edinb). 2009;19:227-228.
- Schröder CM, Merk HF, Frank J. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational dermatosis. J Eur Acad Dermatol Venereol. 2006;20:209-211.
- Joseph HL, Gifford H. Barber’s interdigital pilonidal sinus: the incidence, pathology, and pathogenesis. AMA Arch Derm Syphilol. 1954;70:616-624.
To the Editor:
Barber’s sinus, or interdigital pilonidal sinus, is an occupational dermatosis with a pathognomonic clinical picture. Nearly all reports of barber’s sinus in the literature have involved the hands of male barbers and hairdressers. We present an uncommon case of barber’s sinus between the toes of a female hairdresser. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. Clinicians should advise patients with an occupational risk of barber’s sinus to wear protective footwear and maintain hygiene in the interdigital spaces.
A 23-year-old female hairdresser was referred to our outpatient dermatology clinic by general surgery for evaluation of an asymptomatic interdigital toe lesion of several months’ duration. She was otherwise healthy. Physical examination revealed a 3-mm sinus in the interdigital web space between the fourth and fifth digits of the left foot, creating a partial fistula terminating in an umbilicated pink papule on the dorsal aspect of the interdigital space (Figure). While at work, the patient reported that she usually wore open-toed flip-flops. A diagnosis of barber’s sinus was made clinically. She returned for follow-up to the referring surgeon within 2 months and was offered surgical debridement, but the patient declined treatment, instead opting to wait and monitor for any potential complications. The lesion showed no change in clinical appearance and remained asymptomatic.
Barber’s sinus is caused by sharp fragments of clipped hair that penetrate the fragile interdigital skin and cause a foreign-body reaction. Males are almost exclusively contributory to the reported cases of barber’s sinus in the literature.1,2
The clinical picture of barber’s sinus is pathognomonic, as demonstrated in our case. Other potential diagnoses to consider include atypical mycobacterial infection, deep fungal infection, other foreign-body granuloma, and erosio interdigitalis blastomycetica. Although thorough removal of embedded hair fragments may be curative, most cases require surgical excision, often by curette, and subsequent skin closure. Pathology shows a foreign-body granulomatous reaction to hair fragments. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. This lesion is preventable by maintaining hygiene of the interdigital spaces, use of barrier creams, and wearing protective footwear.3,4
To the Editor:
Barber’s sinus, or interdigital pilonidal sinus, is an occupational dermatosis with a pathognomonic clinical picture. Nearly all reports of barber’s sinus in the literature have involved the hands of male barbers and hairdressers. We present an uncommon case of barber’s sinus between the toes of a female hairdresser. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. Clinicians should advise patients with an occupational risk of barber’s sinus to wear protective footwear and maintain hygiene in the interdigital spaces.
A 23-year-old female hairdresser was referred to our outpatient dermatology clinic by general surgery for evaluation of an asymptomatic interdigital toe lesion of several months’ duration. She was otherwise healthy. Physical examination revealed a 3-mm sinus in the interdigital web space between the fourth and fifth digits of the left foot, creating a partial fistula terminating in an umbilicated pink papule on the dorsal aspect of the interdigital space (Figure). While at work, the patient reported that she usually wore open-toed flip-flops. A diagnosis of barber’s sinus was made clinically. She returned for follow-up to the referring surgeon within 2 months and was offered surgical debridement, but the patient declined treatment, instead opting to wait and monitor for any potential complications. The lesion showed no change in clinical appearance and remained asymptomatic.
Barber’s sinus is caused by sharp fragments of clipped hair that penetrate the fragile interdigital skin and cause a foreign-body reaction. Males are almost exclusively contributory to the reported cases of barber’s sinus in the literature.1,2
The clinical picture of barber’s sinus is pathognomonic, as demonstrated in our case. Other potential diagnoses to consider include atypical mycobacterial infection, deep fungal infection, other foreign-body granuloma, and erosio interdigitalis blastomycetica. Although thorough removal of embedded hair fragments may be curative, most cases require surgical excision, often by curette, and subsequent skin closure. Pathology shows a foreign-body granulomatous reaction to hair fragments. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. This lesion is preventable by maintaining hygiene of the interdigital spaces, use of barrier creams, and wearing protective footwear.3,4
- Efthimiadis C, Kosmidis C, Anthimidis G, et al. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational disease: a case report. Cases J. 2008;1:214.
- O’Neill AC, Purcell EM, Regan PJ. Interdigital pilonidal sinus of the foot [published online May 31, 2009]. Foot (Edinb). 2009;19:227-228.
- Schröder CM, Merk HF, Frank J. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational dermatosis. J Eur Acad Dermatol Venereol. 2006;20:209-211.
- Joseph HL, Gifford H. Barber’s interdigital pilonidal sinus: the incidence, pathology, and pathogenesis. AMA Arch Derm Syphilol. 1954;70:616-624.
- Efthimiadis C, Kosmidis C, Anthimidis G, et al. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational disease: a case report. Cases J. 2008;1:214.
- O’Neill AC, Purcell EM, Regan PJ. Interdigital pilonidal sinus of the foot [published online May 31, 2009]. Foot (Edinb). 2009;19:227-228.
- Schröder CM, Merk HF, Frank J. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational dermatosis. J Eur Acad Dermatol Venereol. 2006;20:209-211.
- Joseph HL, Gifford H. Barber’s interdigital pilonidal sinus: the incidence, pathology, and pathogenesis. AMA Arch Derm Syphilol. 1954;70:616-624.
Practice Points
- This case illustrates a disease in which a medical history and simple clinical examination can lead to the diagnosis.
- Patients may value a diagnosis without treatment. A patient with barber’s sinus may be satisfied with watchful waiting.
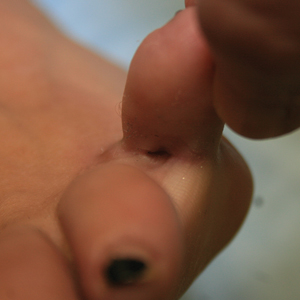
No difference between ipilimumab/nivolumab combo and immunotherapy plus VEGF for metastatic RCC
There is no significant difference in response or survival rates between the combination of ipilimumab/nivolumab (ipi-nivo) and immuno-oncology plus vascular endothelial growth factor inhibition (IOVE) for patients with metastatic renal cell carcinoma.
Therefore, the treatment should probably be directed by patient preferences, among other things, Shaan Dudani, MD, and colleagues wrote in European Oncology.
“Given the current lack of evidence to suggest a difference in efficacy between treatment strategies, patients, clinicians and policy makers are likely to take into account other considerations, such as toxicity, cost, logistics, prognostic categories, and patient preferences in deciding between the various front-line [immuno-oncology] combination regimens,” wrote Dr. Dudani, of the University of Calgary, and coauthors.
The team examined response rates among 263 patients with metastatic renal cell carcinoma from the International Metastatic Renal-Cell Carcinoma Database Consortium (IMDC) dataset. Patients treated with any first-line IOVE combination (n = 113) were compared with those treated with ipi-nivo (n = 75). Patients were about 62 years old. The most common sites of metastasis were liver (about 20%) and bone (about 33%), and about 20% had sarcomatoid features (about 20%). Most (about 75%) had multiple metastatic sites.
Thirty percent of those in the IOVE group and 40% in the ipi-nivo group had received second-line treatments. These included axitinib, levantinib plus severolimus, nivolumab alone, pazopanib, and sunitinib, as well as other treatments.
At a mean follow-up of 11.7 months, the response rates were 33% for IOVE and 40% for ipi-nivo. This difference was not statistically significant (between group difference, 7%; 95% confidence interval, –8% to 22%; P = .4). Complete response occurred in 2% in IOVE and 5% of the ipi-nivo group.
The time to treatment failure was 14.3 months for IOVE and 10.2 months for ipi-nivo – again not a significant difference (P = .2). Time to next treatment also was not significantly different (19.7 vs. 17.9 months; P = .4). Neither group met the study’s overall survival goal.
After adjustment for IMDC risk score, hazard ratios for death were 0.71 for IOVE and 1.74 for ipi-nivo. There were no significant between-group differences when comparing intermediate- and poor-risk patients or when the analysis was restricted only to favorable-risk patients. Among 55 who received second-line therapy, there was also no significant difference in time to treatment failure.
“It was interesting, though not surprising, to observe that the majority [88%] of second-line therapies in this cohort were VEGF-based following ipi-nivo vs. IOVE combinations,” the authors noted. “The higher response rates observed in patients receiving second-line VEGF combinations is noteworthy and thought provoking. Biologically, it is plausible that VEGF-based second-line therapy would be more likely to be effective in the VEGF-naive ipi-nivo cohort. It remains to be seen whether the numerical difference in time to treatment failure becomes significant with increased sample size and further follow-up, and whether this contributes to differences in overall survival, which ultimately impacts treatment selections in the first-line setting.”
Dr. Dudani had no financial disclosures.
SOURCE: Dudani S et al. Euro Onc. 2019 Aug 22. doi: 10.1016/j.eururo.2019.07.048.
There is no significant difference in response or survival rates between the combination of ipilimumab/nivolumab (ipi-nivo) and immuno-oncology plus vascular endothelial growth factor inhibition (IOVE) for patients with metastatic renal cell carcinoma.
Therefore, the treatment should probably be directed by patient preferences, among other things, Shaan Dudani, MD, and colleagues wrote in European Oncology.
“Given the current lack of evidence to suggest a difference in efficacy between treatment strategies, patients, clinicians and policy makers are likely to take into account other considerations, such as toxicity, cost, logistics, prognostic categories, and patient preferences in deciding between the various front-line [immuno-oncology] combination regimens,” wrote Dr. Dudani, of the University of Calgary, and coauthors.
The team examined response rates among 263 patients with metastatic renal cell carcinoma from the International Metastatic Renal-Cell Carcinoma Database Consortium (IMDC) dataset. Patients treated with any first-line IOVE combination (n = 113) were compared with those treated with ipi-nivo (n = 75). Patients were about 62 years old. The most common sites of metastasis were liver (about 20%) and bone (about 33%), and about 20% had sarcomatoid features (about 20%). Most (about 75%) had multiple metastatic sites.
Thirty percent of those in the IOVE group and 40% in the ipi-nivo group had received second-line treatments. These included axitinib, levantinib plus severolimus, nivolumab alone, pazopanib, and sunitinib, as well as other treatments.
At a mean follow-up of 11.7 months, the response rates were 33% for IOVE and 40% for ipi-nivo. This difference was not statistically significant (between group difference, 7%; 95% confidence interval, –8% to 22%; P = .4). Complete response occurred in 2% in IOVE and 5% of the ipi-nivo group.
The time to treatment failure was 14.3 months for IOVE and 10.2 months for ipi-nivo – again not a significant difference (P = .2). Time to next treatment also was not significantly different (19.7 vs. 17.9 months; P = .4). Neither group met the study’s overall survival goal.
After adjustment for IMDC risk score, hazard ratios for death were 0.71 for IOVE and 1.74 for ipi-nivo. There were no significant between-group differences when comparing intermediate- and poor-risk patients or when the analysis was restricted only to favorable-risk patients. Among 55 who received second-line therapy, there was also no significant difference in time to treatment failure.
“It was interesting, though not surprising, to observe that the majority [88%] of second-line therapies in this cohort were VEGF-based following ipi-nivo vs. IOVE combinations,” the authors noted. “The higher response rates observed in patients receiving second-line VEGF combinations is noteworthy and thought provoking. Biologically, it is plausible that VEGF-based second-line therapy would be more likely to be effective in the VEGF-naive ipi-nivo cohort. It remains to be seen whether the numerical difference in time to treatment failure becomes significant with increased sample size and further follow-up, and whether this contributes to differences in overall survival, which ultimately impacts treatment selections in the first-line setting.”
Dr. Dudani had no financial disclosures.
SOURCE: Dudani S et al. Euro Onc. 2019 Aug 22. doi: 10.1016/j.eururo.2019.07.048.
There is no significant difference in response or survival rates between the combination of ipilimumab/nivolumab (ipi-nivo) and immuno-oncology plus vascular endothelial growth factor inhibition (IOVE) for patients with metastatic renal cell carcinoma.
Therefore, the treatment should probably be directed by patient preferences, among other things, Shaan Dudani, MD, and colleagues wrote in European Oncology.
“Given the current lack of evidence to suggest a difference in efficacy between treatment strategies, patients, clinicians and policy makers are likely to take into account other considerations, such as toxicity, cost, logistics, prognostic categories, and patient preferences in deciding between the various front-line [immuno-oncology] combination regimens,” wrote Dr. Dudani, of the University of Calgary, and coauthors.
The team examined response rates among 263 patients with metastatic renal cell carcinoma from the International Metastatic Renal-Cell Carcinoma Database Consortium (IMDC) dataset. Patients treated with any first-line IOVE combination (n = 113) were compared with those treated with ipi-nivo (n = 75). Patients were about 62 years old. The most common sites of metastasis were liver (about 20%) and bone (about 33%), and about 20% had sarcomatoid features (about 20%). Most (about 75%) had multiple metastatic sites.
Thirty percent of those in the IOVE group and 40% in the ipi-nivo group had received second-line treatments. These included axitinib, levantinib plus severolimus, nivolumab alone, pazopanib, and sunitinib, as well as other treatments.
At a mean follow-up of 11.7 months, the response rates were 33% for IOVE and 40% for ipi-nivo. This difference was not statistically significant (between group difference, 7%; 95% confidence interval, –8% to 22%; P = .4). Complete response occurred in 2% in IOVE and 5% of the ipi-nivo group.
The time to treatment failure was 14.3 months for IOVE and 10.2 months for ipi-nivo – again not a significant difference (P = .2). Time to next treatment also was not significantly different (19.7 vs. 17.9 months; P = .4). Neither group met the study’s overall survival goal.
After adjustment for IMDC risk score, hazard ratios for death were 0.71 for IOVE and 1.74 for ipi-nivo. There were no significant between-group differences when comparing intermediate- and poor-risk patients or when the analysis was restricted only to favorable-risk patients. Among 55 who received second-line therapy, there was also no significant difference in time to treatment failure.
“It was interesting, though not surprising, to observe that the majority [88%] of second-line therapies in this cohort were VEGF-based following ipi-nivo vs. IOVE combinations,” the authors noted. “The higher response rates observed in patients receiving second-line VEGF combinations is noteworthy and thought provoking. Biologically, it is plausible that VEGF-based second-line therapy would be more likely to be effective in the VEGF-naive ipi-nivo cohort. It remains to be seen whether the numerical difference in time to treatment failure becomes significant with increased sample size and further follow-up, and whether this contributes to differences in overall survival, which ultimately impacts treatment selections in the first-line setting.”
Dr. Dudani had no financial disclosures.
SOURCE: Dudani S et al. Euro Onc. 2019 Aug 22. doi: 10.1016/j.eururo.2019.07.048.
FROM EUROPEAN ONCOLOGY